User login
The mammography controversy: When should you screen?
Breast cancer is the second most common cause of cancer death in US women,1,2 and screening mammography has been shown to decrease mortality.3,4 But the age at which to start screening, the intervals between mammograms, and the extent of the benefits (and harmful effects) of mammography are still hotly debated.
The clash between those who favor greater use of mammography and those who prefer less frequent and delayed screening heated up in July, when the American College of Obstetricians and Gynecologists (ACOG) released its new breast cancer screening guidelines.5 ACOG now recommends annual mammography starting at age 40; its previous guidelines called for mammograms every 1 to 2 years for women in their 40s and annual screening beginning at age 50.5
The US Preventive Services Task Force (USPSTF) issued updated breast cancer screening guidelines in November 2009 (TABLE 1).5,6 The new guidelines oppose routine screening for women ages 40 to 49 and recommend biennial, rather than annual, mammography for women ages 50 through 74. The decision to initiate screening before age 50 should be an individual one, based on the patient’s values as well as her individual risk factors, the USPSTF maintains. The Task Force, which previously recommended mammography every 1 to 2 years for all women ages 40 and older, does not recommend breast self-examination and finds insufficient evidence to assess the benefits of clinical breast exams.7
Both organizations have prominent medical groups in their camp: The American Cancer Society, National Comprehensive Cancer Network, American College of Surgeons, and American College of Radiology, among others, echo ACOG’s call for annual screening starting at age 40, while the American Academy of Family Physicians, American College of Physicians, National Breast Cancer Coalition, and World Health Organization (WHO) support the USPSTF’s position.8-10
Where does this leave you and your female patients? A look at the rationale behind these divergent recommendations and the latest evidence of the benefits and risks associated with screening mammography will help you cut through the controversy.
TABLE 1
Breast cancer screening: Divergent views5,6
| Organization | Age (years) | BSE | CBE | Mammography |
|---|---|---|---|---|
| ACOG | ≥40 | Encourages breast self-awareness | Annually | Annually |
| USPSTF | 40-49 50-74 | Recommends against teaching (D) | Insufficient evidence (I) | Not routinely recommended (C) Every 2 y (B) |
| USPSTF grades | ||||
| A: Recommended (high certainty of substantial benefit) | ||||
| B: Recommended (moderate or high certainty of moderate benefit or moderate certainty of substantial benefit) | ||||
| C: Not routinely recommended (at least moderate certainty that benefit is small) | ||||
| D: Not recommended (moderate or high certainty of no benefit or that harms outweigh benefits) | ||||
| I: Evidence is insufficient to assess benefits and harms | ||||
| ACOG, American College of Obstetricians and Gynecologists; BSE, breast self-examination; CBE, clinical breast examination; USPSTF, United States Preventive Services Task Force. Source: USPSTF. Grade definitions. May 2008. Available at: http://www.uspreventiveservicestaskforce.org/uspstf/grades.htm. Accessed August 19, 2011. | ||||
Same facts, different conclusions
The recommendations of the USPSTF are based on a systematic review of randomized clinical trials and data from the Cancer Intervention and Surveillance Modeling Network (CISNET) that allowed the researchers to assess various screening parameters.6,8,11 ACOG, too, based its guidelines on an evidence review,12 including the same data used by the USPSTF. Each organization interpreted the findings differently, however, particularly with regard to the benefits and potential harms associated with screening mammography.

The USPSTF points out that screening leads to the greatest absolute reduction in breast cancer mortality in women older than 50. For women ages 39 to 49, the USPSTF analysis revealed, it would take 1904 mammograms to prevent one breast cancer death. For women ages 50 to 59, the number of mammograms needed to prevent a single breast cancer death is 1339; and for women in their 60s, the number needed to screen is just 377.8
The USPSTF notes that false-positive results can lead to additional medical visits and unnecessary treatment, as well as potential psychological harm.7,8
ACOG focused more on cancer growth. Although women in their 40s have a lower probability of breast cancer (1 in 69) than their older counterparts (1 in 42 for women in their 50s and 1 in 29 for women in their 60s) (TABLE 2),2,5,8,12 tumors tend to grow faster in the younger women. That fact played a key role in shaping ACOG’s new guidelines. The average “sojourn time” (the interval between the time a breast tumor can be detected by mammogram and the time at which it has grown enough to become symptomatic) is 2 to 2.4 years for women in their 40s, compared with 4 to 4.1 years for women ages 70 to 74, ACOG estimates. Annual mammograms starting at age 40 provide a better chance of finding and treating breast cancer in an early stage.5,12
The reduction in breast cancer deaths associated with annual screening is about the same for both groups, according to ACOG—16% for women in their 40s, and 15% for women 50 and older.12 The 5-year survival rate for women whose breast tumors are discovered before they’re palpable and before the cancer has spread is 98%.13
ACOG also interpreted the potential harms associated with screening differently. The organization acknowledges that false-positive findings are a continuing concern, but has determined that the benefits of annual screening outweigh the risks.12
TABLE 2
Breast cancer and mammography: How age affects outcomes
| Breast cancer | |
|---|---|
| Age range (y) | Probability (%)2,12 |
| 40-49 | 1 in 69 (1.4) |
| 50-59 | 1 in 42 (2.4) |
| 60-69 | 1 in 29 (3.5) |
| Sojourn time*5,12 | |
| 40-49 | 2-2.4 y |
| ≥70 | 4-4.1 y |
| NNS to prevent 1 breast cancer death8 | |
| 40-49 | 1904 |
| 50-59 | 1339 |
| 60-69 | 377 |
| NNS, number needed to screen. *Interval between the time a breast tumor is detectable by mammography and it becomes symptomatic. | |
Recent studies hit the headlines, but fail to lend clarity
Norwegian cohort study. One study examining the effect of mammography on breast cancer mortality in a large cohort of Norwegian women found that patients ages 50 to 69 who were screened biennially had a 10% reduction in breast cancer death.14 However, further analysis suggested that screening in and of itself accounted for only about one-third of the reduction—an absolute risk reduction of 2.4 deaths per 100,000 person-years. (The rest was attributed to other factors, such as advances in breast cancer awareness and treatment.14) The study was published in the New England Journal of Medicine along with an editorial suggesting that it might be time to consider the rather small effects of screening mammography.15
Swedish cohort study. A study involving a large cohort of Swedish women found that mammography screening was associated with a 29% reduction in breast cancer mortality for women between the ages of 40 and 49.16 Notably, however, the difference in relative risk (RR) for women who were invited to be screened (0.74; 95% confidence interval [CI], 0.66-0.83) vs those who underwent regular screening (0.71; 95% CI, 0.62-0.80) was small.
CISNET modeling study. In a study in the American Journal of Roentgenology, researchers used the same data and CISNET modeling as the USPSTF, but compared lives saved with biennial screening mammography starting at age 50 vs annual screening starting at 40. The researchers reported that for women ages 40 to 84 years, approximately 12 lives per 1000 women screened annually would be saved; for women between the ages of 50 to 74 years screened biennially, 7 lives per 1000 people screened would be saved. That translates into 71% more lives saved with annual, rather than biennial, screening—a reduction of approximately 23%.17
There was a downside, however: The researchers estimated that, on average, women who initiated annual mammography at age 40 would receive a false-positive result every 10 years, and be recalled for imaging every 12 years. Other potential (albeit rare) harms identified by the researchers: one false-positive biopsy (every 149 years), one missed case of breast cancer (every 1000 years), and one fatal radiation-induced breast cancer (every 76,000-79,000 years). 17
2011 Cochrane review. In an update of a 2006 meta-analysis, Cochrane reviewers estimated that screening mammography results in a 15% decrease in breast cancer deaths (an absolute risk reduction of 0.05%).18 But screening also led to a 30% increase in overdiagnosis and overtreatment (an increase in absolute risk of 0.5%). That finding, which prompted the reviewers to conclude that it is not clear whether screening mammography does more good than harm, means that over the course of 10 years, for every 2000 women screened, 10 healthy women can expect to undergo unnecessary diagnostic procedures and receive unnecessary treatment.18
European trend analysis. A retrospective trend analysis published in the British Medical Journal in July 2011 is the latest assessment of the benefits of screening mammography.19 The researchers used WHO data to evaluate breast cancer mortality in several European countries, comparing nations with similar demographics and access to care but different levels of breast cancer screening. Their findings? From 1989 to 2006, reductions in breast cancer mortality were about the same in countries with similarities in levels of health care and demographics, regardless of mammography screening.19
How best to meet your patient’s needs
Where does this leave you? Supporters of the USPSTF’s recommendations have argued that they offer an evidence-based approach to mammography screening for women at average risk, and will help decrease excessive screening and the overdiagnosis, overtreatment, and psychological stress that often result. Critics maintain that trying to fit all women into a single model of breast cancer screening continues to be a problem—one that neither the USPSTF or ACOG has adequately addressed. The risks of breast cancer among various minority groups, for example, have not been taken into account.
Poll finds that patients and providers don’t see eye to eye
In February 2010, Annals of Internal Medicine conducted a Web-based survey relating to the USPSTF’s new screening guidelines. Of the 651 respondents, more than half (54%) were physicians, 9% were nonphysician health care providers, and 37% were potential patients. The findings suggest that health care providers and those they treat do not always see eye-to-eye when it comes to breast cancer screening.20
Two-thirds of the health care professionals surveyed said they would stop offering routine mammograms to women ages 40 to 49, in accordance with the USPSTF’s recommendation, and 62% would advise women ages 50 to 74 to have biennial, rather than annual, mammograms. In addition, 54% of clinicians indicated that they would stop recommending routine screening mammography to women who are 75 or older—a group for whom the USPSTF has stated that evidence is insufficient to assess the benefits and harms of screening. In contrast, 71% of the women said they were unlikely to forego routine mammography in their 40s—and less than 20% said they would wait until age 50 to begin screening or opt for biennial, rather than annual, screenings.20 Although the women’s views may be similar to those held by many of your female patients, the American Cancer Society estimates that about half of US women who are eligible for screening do not get mammograms.21
FIGURE
A digital mammogram showing normal but dense breast tissue
What is your patient’s level of risk?
Individual risk assessment, as stated earlier, is a key factor in determining whether to initiate screening for women younger than 50. It’s important to keep in mind, however, that only half of all breast cancers occur in women with well-established risk factors, including family history, a variety of reproductive risk factors, a high body mass index, and exposure to exogenous estrogen. Fully 50% of women who develop breast cancer are not at elevated risk.13
New models to aid in the shared decision-making process and risk assessment are being developed. One example is a Web-based interactive tool developed by researchers at the University of Sydney to give women in their 40s the information they need to make an informed decision about whether to start screening before age 50 (http://www.mammogram.med.usyd.edu.au/).22 This decision tool answers 2 key questions for women who are not at elevated risk for breast cancer:
| Q: | How many 40-year-old women who start having screening mammograms every 2 years will die from breast cancer in the next 10 years? |
| A: | Out of 1000 40-year-old women who start having screening mammograms every 2 years for the next 10 years, 2 women will die of breast cancer. |
| Q: | How many 40-year-old women who do not have screening mammograms will die from breast cancer in the next 10 years? |
| A: | Out of 1000 40-year-old women who do not have screening mammograms every 2 years for the next 10 years, 2.5 women will die of breast cancer.22,23 |
To our knowledge, there is no such patient-focused decision aid intended for use in the United States. There are assessment tools recommended for use by health care professionals, however. The interactive Breast Cancer Risk Assessment Tool, also known as the Gail Model (http://www.cancer.gov/bcrisktool), provides a population-based, rather than an individualized, estimate of a woman’s risk of developing invasive breast cancer in the next 5 years, as well as her lifetime risk. It incorporates current age, age at menarche, age at parity, number of first-degree maternal relatives with breast cancer, number of breast biopsies, and history of atypical hyperplasia. However, the Gail Model has a C-statistic (a measure of how well a clinical prediction tool correctly ranks patient risk) of just 0.5 to 0.6, which is slightly better than chance. The addition of breast density as a risk criterion in an attempt to boost the tool’s predictive value resulted in minimal improvement. 24,25
A novel approach. In the absence of ideal screening methodology or risk assessment tools, the authors of a recent cost-effectiveness analysis suggest a novel approach: They recommend that all women have a screening mammogram at the age of 40. The primary purpose is to assess breast density.26 That assessment should be key in making decisions about future screenings, as increased breast density is associated with a 4-fold increase in breast cancer risk.27,28
Faced with 2 very different recommendations for breast cancer screening from 2 very reputable organizations, JFP asked these physicians how they handle the mammography controversy, and what they recommend that primary care physicians do.
 |  |  |
| Andrew M. Kaunitz, MD | Jane L. Murray, MD | Cheryl Iglesia, MD, FACOG |
Andrew M. Kaunitz, MD, a professor of obstetrics and gynecology at the University of Florida College of Medicine and a member of the editorial board of OBG Management, says he continues to recommend mammography to all women ages 50 and older, regardless of risk. He has stopped “nagging” women to get screened, however, and—in the absence of elevated risk—has become more flexible about the frequency of mammograms and the age at which to initiate screening.
Dr. Kaunitz encourages women in their 40s to be screened if they have a history of breast cancer, a high body mass index, or other risk factors. If a woman in her 40s is not at elevated risk but is more comfortable being screened, he says, “I’ll order a mammogram for her, too. I’m certainly not going to stand in the way.”
Most women in their 50s prefer annual mammography, Dr. Kaunitz has found, although some appreciate his flexibility. “We recently moved to an office with imaging facilities and I often tell women they can wait until their next visit to be screened—which may be 3 months, 6 months, 9 months, or more.” Others are “aghast” if their physician does not recommend an annual mammogram.
Jane L. Murray, MD, founder of the Sastun Center of Integrative Health Care in Overland Park, Kan, and a member of the editorial board of The Journal of Family Practice, maintains a similar approach.
“I tell patients that the latest guidelines from an unbiased group [USPSTF] state that low-risk women—women who have no family history of breast cancer and are not taking hormones—can begin screening at age 50 and have mammograms every other year,” Dr. Murray says. “I recommend imaging if there is any suspicion at all.”
About two-thirds of her patients are happy to hear that an annual mammogram is no longer necessary. Some patients insist on annual screening—”‘My best friend got breast cancer,’ they often say.”
Dr. Murray’s approach to screening for patients at low risk for breast cancer is to explain that mammograms aren’t perfect and can miss some tumors and overdiagnose others. “Nonetheless, they’re the best we’ve got,” she tells patients, adding, “I recommend screening, but you decide for yourself. “If I thought mammography was a perfect test, I’d be a lot more adamant,” she says.
Cheryl Iglesia, MD, FACOG, is director, section of female pelvic medicine and reconstructive surgery at Washington (DC) Hospital Center, and a member of the board of OBG Management. Dr. Iglesia was chair of ACOG’s gynecologic practice committee and helped to develop the organization’s new guidelines, and has a different view.
“After reviewing all the data, I think that the most important thing that came out of it is that in women ages 40 through 49, breast cancers are more aggressive than they are in older, post-menopausal women.” Thus, she recommends routine screening for women in this age group. “A practice that delays screening until age 50,” she observes, “may be missing the boat.”
Dr. Iglesia also recommends that women in their 40s receive annual mammograms—a practice that’s in line with the recommendations of the American Cancer Society and one that she herself adheres to. The interval between when a cancer is detectable on mammography and the time it becomes symptomatic—known as the “sojourn time”—is about 2 years for women ages 40 through 49, she explains, and more frequent screening would be more likely to catch breast cancer in the preclinical phase. “That’s what a screening test is supposed to do.”
Helen Lippman, Managing Editor
CORRESPONDENCE
Sandhya Pruthi, MD, Mayo Clinic, 200 First Street SW, Rochester, MN 55905; pruthi.sandhya@mayo.edu
1. Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA-Cancer J Clin. 2009;59:225-249.
2. National Cancer Institute. Fact sheet. Probability of breast cancer in American women. Available at: http://www.cancer.gov/cancertopics/factsheet/detection/probability-breast-cancer. Accessed August 23, 2011.
3. Berry DA, Cronin KA, Plevritis SK, et al. Effect of screening and adjuvant therapy on mortality from breast cancer. N Engl J Med. 2005;353:1784-1792.
4. Humphrey LL, Helfand M, Chan BK, et al. Breast cancer screening: a summary of the evidence for the US Preventive Services Task Force. Ann Intern Med. 2002;137(5 part 1):347-360.
5. American College of Obstetricians and Gynecologists. Annual mammograms now recommended for women beginning at age 40. July 20, 2011. Available at: http://www.acog.org/from_home/publications/press_releases/nr07-20-11-2.cfm. Accessed August 15, 2011.
6. US Preventive Services Task Force. Screening for breast cancer. July 2010. Available at: http://www.uspreventiveservicestaskforce.org/uspstf/uspsbrca.htm. Accessed August 15, 2011.
7. Nelson HD, Tyne K, Naik A, et al. Screening for breast cancer:. an update for the US Preventive Services Task Force. Ann Intern Med. 2009;151:727–737, W237–742.
8. US Preventive Services Task Force. Screening for breast cancer. US Preventive Services Task Force recommendation statement. Ann Intern Med. 2009;151:716-726.
9. Smith RA, Cokkinides V, Brooks D, et al. Cancer screening in the United States, 2010: a review of current American Cancer Society guidelines and issues in cancer screening. CA-Cancer J Clin. 2010;60:99-119.
10. Lee CH, Dershaw DD, Kopans D, et al. Breast cancer screening with imaging: recommendations from the Society of Breast Imaging and the ACR on the use of mammography, breast MRI, breast ultrasound, and other technologies for the detection of clinically occult breast cancer. J Am Coll Radiol. 2010;7:18-27.
11. Mandelblatt JS, Cronin KA, Bailey S, et al. Effects of mammography screening under different screening schedules: model estimates of potential benefits and harms. Ann Intern Med. 2009;151:738-747.
12. American College of Obstetricians and Gynecologists. Practice bulletin no. 122: breast cancer screening. Obstet Gynecol. 2011;118:372-382.
13. Madigan MP, Ziegler RG, Benichou J, et al. Proportion of breast cancer cases in the United States explained by well-established risk factors. J Natl Cancer Inst. 1995;87:1681-1685.
14. Kalager M, Zelen M, Langmark F, et al. Effect of screening mammography on breast-cancer mortality in Norway. N Engl J Med. 2010;363:1203-1210.
15. Welch HG. Screening mammography—a long run for a short slide? N Engl J Med. 2010;363:1276-1278.
16. Hellquist BN, Duffy SW, Abdsaleh S, et al. Effectiveness of population-based service screening with mammography for women ages 40 to 49 years: evaluation of the Swedish Mammography Screening in Young Women (SCRY) cohort. Cancer. 2010;117:714-722.
17. Hendrick RE, Helvie MA. United States Preventive Services Task Force screening mammography recommendations: science ignored. AJR Am J Roentgenol. 2011;196:W112-W116.
18. Gotzche PC, Nielsen M. Screening for breast cancer with mammography. Cochrane Database Syst Rev. 2011;(1):CD001877.-
19. Autier P, Boniol M, Gavin A, et al. Breast cancer mortality in neighbouring European countries with different levels of screening but similar access to treatment: trend analysis of WHO mortality database. BMJ. 2011;343:d4411.-
20. When evidence collides with anecdote, politics, and emotion: breast cancer screening [editorial]. Ann Intern Med. 2010;152:531-532.
21. American Cancer Society. Cancer prevention and early detection facts and figures 2009. Available at: http://www.cancer.org/Research/CancerFactsFigures/CancerPreventionEarlyDetectionFactsFigures/index. Accessed August 15, 2011.
22. Australian Screening Mammography Decision Aid Trial. Available at: http://www.mammogram.med.usyd.edu.au/. Accessed August 15, 2011.
23. Barratt A, Howard K, Irwig L, et al. Model of outcomes of screening mammography: information to support informed choices. BMJ. 2005;330:936.-
24. Chen J, Pee D, Ayyagari R, et al. Projecting absolute invasive breast cancer risk in women with a model that includes mammographic density. J Natl Cancer Inst. 2006;98:1215-1226.
25. Tice JA, Cummings SR, Ziv E, et al. Mammographic breast density and the Gail Model for breast cancer risk prediction in a screening population. Breast Cancer Res Treat. 2005;94:115-122.
26. Schousboe JT, Kerlikowske K, Loh A, et al. Personalizing mammography by breast density and other risk factors for breast cancer: analysis of health benefits and cost-effectiveness. Ann Intern Med. 2011;155:10-20.
27. Boyd NF, Byng JW, Jong RA, et al. Quantitative classification of mammographic densities and breast cancer risk: results from the Canadian National Breast Screening Study. J Natl Cancer Inst. 1995;87:670-675.
28. Tamimi R, Byrne C, Colditz EG, et al. Endogenous hormone levels, mammographic density, and subsequent risk of breast cancer in postmenopausal women. J Natl Cancer Inst. 2007;99:1178-1187.
Breast cancer is the second most common cause of cancer death in US women,1,2 and screening mammography has been shown to decrease mortality.3,4 But the age at which to start screening, the intervals between mammograms, and the extent of the benefits (and harmful effects) of mammography are still hotly debated.
The clash between those who favor greater use of mammography and those who prefer less frequent and delayed screening heated up in July, when the American College of Obstetricians and Gynecologists (ACOG) released its new breast cancer screening guidelines.5 ACOG now recommends annual mammography starting at age 40; its previous guidelines called for mammograms every 1 to 2 years for women in their 40s and annual screening beginning at age 50.5
The US Preventive Services Task Force (USPSTF) issued updated breast cancer screening guidelines in November 2009 (TABLE 1).5,6 The new guidelines oppose routine screening for women ages 40 to 49 and recommend biennial, rather than annual, mammography for women ages 50 through 74. The decision to initiate screening before age 50 should be an individual one, based on the patient’s values as well as her individual risk factors, the USPSTF maintains. The Task Force, which previously recommended mammography every 1 to 2 years for all women ages 40 and older, does not recommend breast self-examination and finds insufficient evidence to assess the benefits of clinical breast exams.7
Both organizations have prominent medical groups in their camp: The American Cancer Society, National Comprehensive Cancer Network, American College of Surgeons, and American College of Radiology, among others, echo ACOG’s call for annual screening starting at age 40, while the American Academy of Family Physicians, American College of Physicians, National Breast Cancer Coalition, and World Health Organization (WHO) support the USPSTF’s position.8-10
Where does this leave you and your female patients? A look at the rationale behind these divergent recommendations and the latest evidence of the benefits and risks associated with screening mammography will help you cut through the controversy.
TABLE 1
Breast cancer screening: Divergent views5,6
| Organization | Age (years) | BSE | CBE | Mammography |
|---|---|---|---|---|
| ACOG | ≥40 | Encourages breast self-awareness | Annually | Annually |
| USPSTF | 40-49 50-74 | Recommends against teaching (D) | Insufficient evidence (I) | Not routinely recommended (C) Every 2 y (B) |
| USPSTF grades | ||||
| A: Recommended (high certainty of substantial benefit) | ||||
| B: Recommended (moderate or high certainty of moderate benefit or moderate certainty of substantial benefit) | ||||
| C: Not routinely recommended (at least moderate certainty that benefit is small) | ||||
| D: Not recommended (moderate or high certainty of no benefit or that harms outweigh benefits) | ||||
| I: Evidence is insufficient to assess benefits and harms | ||||
| ACOG, American College of Obstetricians and Gynecologists; BSE, breast self-examination; CBE, clinical breast examination; USPSTF, United States Preventive Services Task Force. Source: USPSTF. Grade definitions. May 2008. Available at: http://www.uspreventiveservicestaskforce.org/uspstf/grades.htm. Accessed August 19, 2011. | ||||
Same facts, different conclusions
The recommendations of the USPSTF are based on a systematic review of randomized clinical trials and data from the Cancer Intervention and Surveillance Modeling Network (CISNET) that allowed the researchers to assess various screening parameters.6,8,11 ACOG, too, based its guidelines on an evidence review,12 including the same data used by the USPSTF. Each organization interpreted the findings differently, however, particularly with regard to the benefits and potential harms associated with screening mammography.
The USPSTF points out that screening leads to the greatest absolute reduction in breast cancer mortality in women older than 50. For women ages 39 to 49, the USPSTF analysis revealed, it would take 1904 mammograms to prevent one breast cancer death. For women ages 50 to 59, the number of mammograms needed to prevent a single breast cancer death is 1339; and for women in their 60s, the number needed to screen is just 377.8
The USPSTF notes that false-positive results can lead to additional medical visits and unnecessary treatment, as well as potential psychological harm.7,8
ACOG focused more on cancer growth. Although women in their 40s have a lower probability of breast cancer (1 in 69) than their older counterparts (1 in 42 for women in their 50s and 1 in 29 for women in their 60s) (TABLE 2),2,5,8,12 tumors tend to grow faster in the younger women. That fact played a key role in shaping ACOG’s new guidelines. The average “sojourn time” (the interval between the time a breast tumor can be detected by mammogram and the time at which it has grown enough to become symptomatic) is 2 to 2.4 years for women in their 40s, compared with 4 to 4.1 years for women ages 70 to 74, ACOG estimates. Annual mammograms starting at age 40 provide a better chance of finding and treating breast cancer in an early stage.5,12
The reduction in breast cancer deaths associated with annual screening is about the same for both groups, according to ACOG—16% for women in their 40s, and 15% for women 50 and older.12 The 5-year survival rate for women whose breast tumors are discovered before they’re palpable and before the cancer has spread is 98%.13
ACOG also interpreted the potential harms associated with screening differently. The organization acknowledges that false-positive findings are a continuing concern, but has determined that the benefits of annual screening outweigh the risks.12
TABLE 2
Breast cancer and mammography: How age affects outcomes
| Breast cancer | |
|---|---|
| Age range (y) | Probability (%)2,12 |
| 40-49 | 1 in 69 (1.4) |
| 50-59 | 1 in 42 (2.4) |
| 60-69 | 1 in 29 (3.5) |
| Sojourn time*5,12 | |
| 40-49 | 2-2.4 y |
| ≥70 | 4-4.1 y |
| NNS to prevent 1 breast cancer death8 | |
| 40-49 | 1904 |
| 50-59 | 1339 |
| 60-69 | 377 |
| NNS, number needed to screen. *Interval between the time a breast tumor is detectable by mammography and it becomes symptomatic. | |
Recent studies hit the headlines, but fail to lend clarity
Norwegian cohort study. One study examining the effect of mammography on breast cancer mortality in a large cohort of Norwegian women found that patients ages 50 to 69 who were screened biennially had a 10% reduction in breast cancer death.14 However, further analysis suggested that screening in and of itself accounted for only about one-third of the reduction—an absolute risk reduction of 2.4 deaths per 100,000 person-years. (The rest was attributed to other factors, such as advances in breast cancer awareness and treatment.14) The study was published in the New England Journal of Medicine along with an editorial suggesting that it might be time to consider the rather small effects of screening mammography.15
Swedish cohort study. A study involving a large cohort of Swedish women found that mammography screening was associated with a 29% reduction in breast cancer mortality for women between the ages of 40 and 49.16 Notably, however, the difference in relative risk (RR) for women who were invited to be screened (0.74; 95% confidence interval [CI], 0.66-0.83) vs those who underwent regular screening (0.71; 95% CI, 0.62-0.80) was small.
CISNET modeling study. In a study in the American Journal of Roentgenology, researchers used the same data and CISNET modeling as the USPSTF, but compared lives saved with biennial screening mammography starting at age 50 vs annual screening starting at 40. The researchers reported that for women ages 40 to 84 years, approximately 12 lives per 1000 women screened annually would be saved; for women between the ages of 50 to 74 years screened biennially, 7 lives per 1000 people screened would be saved. That translates into 71% more lives saved with annual, rather than biennial, screening—a reduction of approximately 23%.17
There was a downside, however: The researchers estimated that, on average, women who initiated annual mammography at age 40 would receive a false-positive result every 10 years, and be recalled for imaging every 12 years. Other potential (albeit rare) harms identified by the researchers: one false-positive biopsy (every 149 years), one missed case of breast cancer (every 1000 years), and one fatal radiation-induced breast cancer (every 76,000-79,000 years). 17
2011 Cochrane review. In an update of a 2006 meta-analysis, Cochrane reviewers estimated that screening mammography results in a 15% decrease in breast cancer deaths (an absolute risk reduction of 0.05%).18 But screening also led to a 30% increase in overdiagnosis and overtreatment (an increase in absolute risk of 0.5%). That finding, which prompted the reviewers to conclude that it is not clear whether screening mammography does more good than harm, means that over the course of 10 years, for every 2000 women screened, 10 healthy women can expect to undergo unnecessary diagnostic procedures and receive unnecessary treatment.18
European trend analysis. A retrospective trend analysis published in the British Medical Journal in July 2011 is the latest assessment of the benefits of screening mammography.19 The researchers used WHO data to evaluate breast cancer mortality in several European countries, comparing nations with similar demographics and access to care but different levels of breast cancer screening. Their findings? From 1989 to 2006, reductions in breast cancer mortality were about the same in countries with similarities in levels of health care and demographics, regardless of mammography screening.19
How best to meet your patient’s needs
Where does this leave you? Supporters of the USPSTF’s recommendations have argued that they offer an evidence-based approach to mammography screening for women at average risk, and will help decrease excessive screening and the overdiagnosis, overtreatment, and psychological stress that often result. Critics maintain that trying to fit all women into a single model of breast cancer screening continues to be a problem—one that neither the USPSTF or ACOG has adequately addressed. The risks of breast cancer among various minority groups, for example, have not been taken into account.
Poll finds that patients and providers don’t see eye to eye
In February 2010, Annals of Internal Medicine conducted a Web-based survey relating to the USPSTF’s new screening guidelines. Of the 651 respondents, more than half (54%) were physicians, 9% were nonphysician health care providers, and 37% were potential patients. The findings suggest that health care providers and those they treat do not always see eye-to-eye when it comes to breast cancer screening.20
Two-thirds of the health care professionals surveyed said they would stop offering routine mammograms to women ages 40 to 49, in accordance with the USPSTF’s recommendation, and 62% would advise women ages 50 to 74 to have biennial, rather than annual, mammograms. In addition, 54% of clinicians indicated that they would stop recommending routine screening mammography to women who are 75 or older—a group for whom the USPSTF has stated that evidence is insufficient to assess the benefits and harms of screening. In contrast, 71% of the women said they were unlikely to forego routine mammography in their 40s—and less than 20% said they would wait until age 50 to begin screening or opt for biennial, rather than annual, screenings.20 Although the women’s views may be similar to those held by many of your female patients, the American Cancer Society estimates that about half of US women who are eligible for screening do not get mammograms.21
FIGURE
A digital mammogram showing normal but dense breast tissue
What is your patient’s level of risk?
Individual risk assessment, as stated earlier, is a key factor in determining whether to initiate screening for women younger than 50. It’s important to keep in mind, however, that only half of all breast cancers occur in women with well-established risk factors, including family history, a variety of reproductive risk factors, a high body mass index, and exposure to exogenous estrogen. Fully 50% of women who develop breast cancer are not at elevated risk.13
New models to aid in the shared decision-making process and risk assessment are being developed. One example is a Web-based interactive tool developed by researchers at the University of Sydney to give women in their 40s the information they need to make an informed decision about whether to start screening before age 50 (http://www.mammogram.med.usyd.edu.au/).22 This decision tool answers 2 key questions for women who are not at elevated risk for breast cancer:
| Q: | How many 40-year-old women who start having screening mammograms every 2 years will die from breast cancer in the next 10 years? |
| A: | Out of 1000 40-year-old women who start having screening mammograms every 2 years for the next 10 years, 2 women will die of breast cancer. |
| Q: | How many 40-year-old women who do not have screening mammograms will die from breast cancer in the next 10 years? |
| A: | Out of 1000 40-year-old women who do not have screening mammograms every 2 years for the next 10 years, 2.5 women will die of breast cancer.22,23 |
To our knowledge, there is no such patient-focused decision aid intended for use in the United States. There are assessment tools recommended for use by health care professionals, however. The interactive Breast Cancer Risk Assessment Tool, also known as the Gail Model (http://www.cancer.gov/bcrisktool), provides a population-based, rather than an individualized, estimate of a woman’s risk of developing invasive breast cancer in the next 5 years, as well as her lifetime risk. It incorporates current age, age at menarche, age at parity, number of first-degree maternal relatives with breast cancer, number of breast biopsies, and history of atypical hyperplasia. However, the Gail Model has a C-statistic (a measure of how well a clinical prediction tool correctly ranks patient risk) of just 0.5 to 0.6, which is slightly better than chance. The addition of breast density as a risk criterion in an attempt to boost the tool’s predictive value resulted in minimal improvement. 24,25
A novel approach. In the absence of ideal screening methodology or risk assessment tools, the authors of a recent cost-effectiveness analysis suggest a novel approach: They recommend that all women have a screening mammogram at the age of 40. The primary purpose is to assess breast density.26 That assessment should be key in making decisions about future screenings, as increased breast density is associated with a 4-fold increase in breast cancer risk.27,28
Faced with 2 very different recommendations for breast cancer screening from 2 very reputable organizations, JFP asked these physicians how they handle the mammography controversy, and what they recommend that primary care physicians do.
| | |
| Andrew M. Kaunitz, MD | Jane L. Murray, MD | Cheryl Iglesia, MD, FACOG |
Andrew M. Kaunitz, MD, a professor of obstetrics and gynecology at the University of Florida College of Medicine and a member of the editorial board of OBG Management, says he continues to recommend mammography to all women ages 50 and older, regardless of risk. He has stopped “nagging” women to get screened, however, and—in the absence of elevated risk—has become more flexible about the frequency of mammograms and the age at which to initiate screening.
Dr. Kaunitz encourages women in their 40s to be screened if they have a history of breast cancer, a high body mass index, or other risk factors. If a woman in her 40s is not at elevated risk but is more comfortable being screened, he says, “I’ll order a mammogram for her, too. I’m certainly not going to stand in the way.”
Most women in their 50s prefer annual mammography, Dr. Kaunitz has found, although some appreciate his flexibility. “We recently moved to an office with imaging facilities and I often tell women they can wait until their next visit to be screened—which may be 3 months, 6 months, 9 months, or more.” Others are “aghast” if their physician does not recommend an annual mammogram.
Jane L. Murray, MD, founder of the Sastun Center of Integrative Health Care in Overland Park, Kan, and a member of the editorial board of The Journal of Family Practice, maintains a similar approach.
“I tell patients that the latest guidelines from an unbiased group [USPSTF] state that low-risk women—women who have no family history of breast cancer and are not taking hormones—can begin screening at age 50 and have mammograms every other year,” Dr. Murray says. “I recommend imaging if there is any suspicion at all.”
About two-thirds of her patients are happy to hear that an annual mammogram is no longer necessary. Some patients insist on annual screening—”‘My best friend got breast cancer,’ they often say.”
Dr. Murray’s approach to screening for patients at low risk for breast cancer is to explain that mammograms aren’t perfect and can miss some tumors and overdiagnose others. “Nonetheless, they’re the best we’ve got,” she tells patients, adding, “I recommend screening, but you decide for yourself. “If I thought mammography was a perfect test, I’d be a lot more adamant,” she says.
Cheryl Iglesia, MD, FACOG, is director, section of female pelvic medicine and reconstructive surgery at Washington (DC) Hospital Center, and a member of the board of OBG Management. Dr. Iglesia was chair of ACOG’s gynecologic practice committee and helped to develop the organization’s new guidelines, and has a different view.
“After reviewing all the data, I think that the most important thing that came out of it is that in women ages 40 through 49, breast cancers are more aggressive than they are in older, post-menopausal women.” Thus, she recommends routine screening for women in this age group. “A practice that delays screening until age 50,” she observes, “may be missing the boat.”
Dr. Iglesia also recommends that women in their 40s receive annual mammograms—a practice that’s in line with the recommendations of the American Cancer Society and one that she herself adheres to. The interval between when a cancer is detectable on mammography and the time it becomes symptomatic—known as the “sojourn time”—is about 2 years for women ages 40 through 49, she explains, and more frequent screening would be more likely to catch breast cancer in the preclinical phase. “That’s what a screening test is supposed to do.”
Helen Lippman, Managing Editor
CORRESPONDENCE
Sandhya Pruthi, MD, Mayo Clinic, 200 First Street SW, Rochester, MN 55905; pruthi.sandhya@mayo.edu
Breast cancer is the second most common cause of cancer death in US women,1,2 and screening mammography has been shown to decrease mortality.3,4 But the age at which to start screening, the intervals between mammograms, and the extent of the benefits (and harmful effects) of mammography are still hotly debated.
The clash between those who favor greater use of mammography and those who prefer less frequent and delayed screening heated up in July, when the American College of Obstetricians and Gynecologists (ACOG) released its new breast cancer screening guidelines.5 ACOG now recommends annual mammography starting at age 40; its previous guidelines called for mammograms every 1 to 2 years for women in their 40s and annual screening beginning at age 50.5
The US Preventive Services Task Force (USPSTF) issued updated breast cancer screening guidelines in November 2009 (TABLE 1).5,6 The new guidelines oppose routine screening for women ages 40 to 49 and recommend biennial, rather than annual, mammography for women ages 50 through 74. The decision to initiate screening before age 50 should be an individual one, based on the patient’s values as well as her individual risk factors, the USPSTF maintains. The Task Force, which previously recommended mammography every 1 to 2 years for all women ages 40 and older, does not recommend breast self-examination and finds insufficient evidence to assess the benefits of clinical breast exams.7
Both organizations have prominent medical groups in their camp: The American Cancer Society, National Comprehensive Cancer Network, American College of Surgeons, and American College of Radiology, among others, echo ACOG’s call for annual screening starting at age 40, while the American Academy of Family Physicians, American College of Physicians, National Breast Cancer Coalition, and World Health Organization (WHO) support the USPSTF’s position.8-10
Where does this leave you and your female patients? A look at the rationale behind these divergent recommendations and the latest evidence of the benefits and risks associated with screening mammography will help you cut through the controversy.
TABLE 1
Breast cancer screening: Divergent views5,6
| Organization | Age (years) | BSE | CBE | Mammography |
|---|---|---|---|---|
| ACOG | ≥40 | Encourages breast self-awareness | Annually | Annually |
| USPSTF | 40-49 50-74 | Recommends against teaching (D) | Insufficient evidence (I) | Not routinely recommended (C) Every 2 y (B) |
| USPSTF grades | ||||
| A: Recommended (high certainty of substantial benefit) | ||||
| B: Recommended (moderate or high certainty of moderate benefit or moderate certainty of substantial benefit) | ||||
| C: Not routinely recommended (at least moderate certainty that benefit is small) | ||||
| D: Not recommended (moderate or high certainty of no benefit or that harms outweigh benefits) | ||||
| I: Evidence is insufficient to assess benefits and harms | ||||
| ACOG, American College of Obstetricians and Gynecologists; BSE, breast self-examination; CBE, clinical breast examination; USPSTF, United States Preventive Services Task Force. Source: USPSTF. Grade definitions. May 2008. Available at: http://www.uspreventiveservicestaskforce.org/uspstf/grades.htm. Accessed August 19, 2011. | ||||
Same facts, different conclusions
The recommendations of the USPSTF are based on a systematic review of randomized clinical trials and data from the Cancer Intervention and Surveillance Modeling Network (CISNET) that allowed the researchers to assess various screening parameters.6,8,11 ACOG, too, based its guidelines on an evidence review,12 including the same data used by the USPSTF. Each organization interpreted the findings differently, however, particularly with regard to the benefits and potential harms associated with screening mammography.
The USPSTF points out that screening leads to the greatest absolute reduction in breast cancer mortality in women older than 50. For women ages 39 to 49, the USPSTF analysis revealed, it would take 1904 mammograms to prevent one breast cancer death. For women ages 50 to 59, the number of mammograms needed to prevent a single breast cancer death is 1339; and for women in their 60s, the number needed to screen is just 377.8
The USPSTF notes that false-positive results can lead to additional medical visits and unnecessary treatment, as well as potential psychological harm.7,8
ACOG focused more on cancer growth. Although women in their 40s have a lower probability of breast cancer (1 in 69) than their older counterparts (1 in 42 for women in their 50s and 1 in 29 for women in their 60s) (TABLE 2),2,5,8,12 tumors tend to grow faster in the younger women. That fact played a key role in shaping ACOG’s new guidelines. The average “sojourn time” (the interval between the time a breast tumor can be detected by mammogram and the time at which it has grown enough to become symptomatic) is 2 to 2.4 years for women in their 40s, compared with 4 to 4.1 years for women ages 70 to 74, ACOG estimates. Annual mammograms starting at age 40 provide a better chance of finding and treating breast cancer in an early stage.5,12
The reduction in breast cancer deaths associated with annual screening is about the same for both groups, according to ACOG—16% for women in their 40s, and 15% for women 50 and older.12 The 5-year survival rate for women whose breast tumors are discovered before they’re palpable and before the cancer has spread is 98%.13
ACOG also interpreted the potential harms associated with screening differently. The organization acknowledges that false-positive findings are a continuing concern, but has determined that the benefits of annual screening outweigh the risks.12
TABLE 2
Breast cancer and mammography: How age affects outcomes
| Breast cancer | |
|---|---|
| Age range (y) | Probability (%)2,12 |
| 40-49 | 1 in 69 (1.4) |
| 50-59 | 1 in 42 (2.4) |
| 60-69 | 1 in 29 (3.5) |
| Sojourn time*5,12 | |
| 40-49 | 2-2.4 y |
| ≥70 | 4-4.1 y |
| NNS to prevent 1 breast cancer death8 | |
| 40-49 | 1904 |
| 50-59 | 1339 |
| 60-69 | 377 |
| NNS, number needed to screen. *Interval between the time a breast tumor is detectable by mammography and it becomes symptomatic. | |
Recent studies hit the headlines, but fail to lend clarity
Norwegian cohort study. One study examining the effect of mammography on breast cancer mortality in a large cohort of Norwegian women found that patients ages 50 to 69 who were screened biennially had a 10% reduction in breast cancer death.14 However, further analysis suggested that screening in and of itself accounted for only about one-third of the reduction—an absolute risk reduction of 2.4 deaths per 100,000 person-years. (The rest was attributed to other factors, such as advances in breast cancer awareness and treatment.14) The study was published in the New England Journal of Medicine along with an editorial suggesting that it might be time to consider the rather small effects of screening mammography.15
Swedish cohort study. A study involving a large cohort of Swedish women found that mammography screening was associated with a 29% reduction in breast cancer mortality for women between the ages of 40 and 49.16 Notably, however, the difference in relative risk (RR) for women who were invited to be screened (0.74; 95% confidence interval [CI], 0.66-0.83) vs those who underwent regular screening (0.71; 95% CI, 0.62-0.80) was small.
CISNET modeling study. In a study in the American Journal of Roentgenology, researchers used the same data and CISNET modeling as the USPSTF, but compared lives saved with biennial screening mammography starting at age 50 vs annual screening starting at 40. The researchers reported that for women ages 40 to 84 years, approximately 12 lives per 1000 women screened annually would be saved; for women between the ages of 50 to 74 years screened biennially, 7 lives per 1000 people screened would be saved. That translates into 71% more lives saved with annual, rather than biennial, screening—a reduction of approximately 23%.17
There was a downside, however: The researchers estimated that, on average, women who initiated annual mammography at age 40 would receive a false-positive result every 10 years, and be recalled for imaging every 12 years. Other potential (albeit rare) harms identified by the researchers: one false-positive biopsy (every 149 years), one missed case of breast cancer (every 1000 years), and one fatal radiation-induced breast cancer (every 76,000-79,000 years). 17
2011 Cochrane review. In an update of a 2006 meta-analysis, Cochrane reviewers estimated that screening mammography results in a 15% decrease in breast cancer deaths (an absolute risk reduction of 0.05%).18 But screening also led to a 30% increase in overdiagnosis and overtreatment (an increase in absolute risk of 0.5%). That finding, which prompted the reviewers to conclude that it is not clear whether screening mammography does more good than harm, means that over the course of 10 years, for every 2000 women screened, 10 healthy women can expect to undergo unnecessary diagnostic procedures and receive unnecessary treatment.18
European trend analysis. A retrospective trend analysis published in the British Medical Journal in July 2011 is the latest assessment of the benefits of screening mammography.19 The researchers used WHO data to evaluate breast cancer mortality in several European countries, comparing nations with similar demographics and access to care but different levels of breast cancer screening. Their findings? From 1989 to 2006, reductions in breast cancer mortality were about the same in countries with similarities in levels of health care and demographics, regardless of mammography screening.19
How best to meet your patient’s needs
Where does this leave you? Supporters of the USPSTF’s recommendations have argued that they offer an evidence-based approach to mammography screening for women at average risk, and will help decrease excessive screening and the overdiagnosis, overtreatment, and psychological stress that often result. Critics maintain that trying to fit all women into a single model of breast cancer screening continues to be a problem—one that neither the USPSTF or ACOG has adequately addressed. The risks of breast cancer among various minority groups, for example, have not been taken into account.
Poll finds that patients and providers don’t see eye to eye
In February 2010, Annals of Internal Medicine conducted a Web-based survey relating to the USPSTF’s new screening guidelines. Of the 651 respondents, more than half (54%) were physicians, 9% were nonphysician health care providers, and 37% were potential patients. The findings suggest that health care providers and those they treat do not always see eye-to-eye when it comes to breast cancer screening.20
Two-thirds of the health care professionals surveyed said they would stop offering routine mammograms to women ages 40 to 49, in accordance with the USPSTF’s recommendation, and 62% would advise women ages 50 to 74 to have biennial, rather than annual, mammograms. In addition, 54% of clinicians indicated that they would stop recommending routine screening mammography to women who are 75 or older—a group for whom the USPSTF has stated that evidence is insufficient to assess the benefits and harms of screening. In contrast, 71% of the women said they were unlikely to forego routine mammography in their 40s—and less than 20% said they would wait until age 50 to begin screening or opt for biennial, rather than annual, screenings.20 Although the women’s views may be similar to those held by many of your female patients, the American Cancer Society estimates that about half of US women who are eligible for screening do not get mammograms.21
FIGURE
A digital mammogram showing normal but dense breast tissue
What is your patient’s level of risk?
Individual risk assessment, as stated earlier, is a key factor in determining whether to initiate screening for women younger than 50. It’s important to keep in mind, however, that only half of all breast cancers occur in women with well-established risk factors, including family history, a variety of reproductive risk factors, a high body mass index, and exposure to exogenous estrogen. Fully 50% of women who develop breast cancer are not at elevated risk.13
New models to aid in the shared decision-making process and risk assessment are being developed. One example is a Web-based interactive tool developed by researchers at the University of Sydney to give women in their 40s the information they need to make an informed decision about whether to start screening before age 50 (http://www.mammogram.med.usyd.edu.au/).22 This decision tool answers 2 key questions for women who are not at elevated risk for breast cancer:
| Q: | How many 40-year-old women who start having screening mammograms every 2 years will die from breast cancer in the next 10 years? |
| A: | Out of 1000 40-year-old women who start having screening mammograms every 2 years for the next 10 years, 2 women will die of breast cancer. |
| Q: | How many 40-year-old women who do not have screening mammograms will die from breast cancer in the next 10 years? |
| A: | Out of 1000 40-year-old women who do not have screening mammograms every 2 years for the next 10 years, 2.5 women will die of breast cancer.22,23 |
To our knowledge, there is no such patient-focused decision aid intended for use in the United States. There are assessment tools recommended for use by health care professionals, however. The interactive Breast Cancer Risk Assessment Tool, also known as the Gail Model (http://www.cancer.gov/bcrisktool), provides a population-based, rather than an individualized, estimate of a woman’s risk of developing invasive breast cancer in the next 5 years, as well as her lifetime risk. It incorporates current age, age at menarche, age at parity, number of first-degree maternal relatives with breast cancer, number of breast biopsies, and history of atypical hyperplasia. However, the Gail Model has a C-statistic (a measure of how well a clinical prediction tool correctly ranks patient risk) of just 0.5 to 0.6, which is slightly better than chance. The addition of breast density as a risk criterion in an attempt to boost the tool’s predictive value resulted in minimal improvement. 24,25
A novel approach. In the absence of ideal screening methodology or risk assessment tools, the authors of a recent cost-effectiveness analysis suggest a novel approach: They recommend that all women have a screening mammogram at the age of 40. The primary purpose is to assess breast density.26 That assessment should be key in making decisions about future screenings, as increased breast density is associated with a 4-fold increase in breast cancer risk.27,28
Faced with 2 very different recommendations for breast cancer screening from 2 very reputable organizations, JFP asked these physicians how they handle the mammography controversy, and what they recommend that primary care physicians do.
| | |
| Andrew M. Kaunitz, MD | Jane L. Murray, MD | Cheryl Iglesia, MD, FACOG |
Andrew M. Kaunitz, MD, a professor of obstetrics and gynecology at the University of Florida College of Medicine and a member of the editorial board of OBG Management, says he continues to recommend mammography to all women ages 50 and older, regardless of risk. He has stopped “nagging” women to get screened, however, and—in the absence of elevated risk—has become more flexible about the frequency of mammograms and the age at which to initiate screening.
Dr. Kaunitz encourages women in their 40s to be screened if they have a history of breast cancer, a high body mass index, or other risk factors. If a woman in her 40s is not at elevated risk but is more comfortable being screened, he says, “I’ll order a mammogram for her, too. I’m certainly not going to stand in the way.”
Most women in their 50s prefer annual mammography, Dr. Kaunitz has found, although some appreciate his flexibility. “We recently moved to an office with imaging facilities and I often tell women they can wait until their next visit to be screened—which may be 3 months, 6 months, 9 months, or more.” Others are “aghast” if their physician does not recommend an annual mammogram.
Jane L. Murray, MD, founder of the Sastun Center of Integrative Health Care in Overland Park, Kan, and a member of the editorial board of The Journal of Family Practice, maintains a similar approach.
“I tell patients that the latest guidelines from an unbiased group [USPSTF] state that low-risk women—women who have no family history of breast cancer and are not taking hormones—can begin screening at age 50 and have mammograms every other year,” Dr. Murray says. “I recommend imaging if there is any suspicion at all.”
About two-thirds of her patients are happy to hear that an annual mammogram is no longer necessary. Some patients insist on annual screening—”‘My best friend got breast cancer,’ they often say.”
Dr. Murray’s approach to screening for patients at low risk for breast cancer is to explain that mammograms aren’t perfect and can miss some tumors and overdiagnose others. “Nonetheless, they’re the best we’ve got,” she tells patients, adding, “I recommend screening, but you decide for yourself. “If I thought mammography was a perfect test, I’d be a lot more adamant,” she says.
Cheryl Iglesia, MD, FACOG, is director, section of female pelvic medicine and reconstructive surgery at Washington (DC) Hospital Center, and a member of the board of OBG Management. Dr. Iglesia was chair of ACOG’s gynecologic practice committee and helped to develop the organization’s new guidelines, and has a different view.
“After reviewing all the data, I think that the most important thing that came out of it is that in women ages 40 through 49, breast cancers are more aggressive than they are in older, post-menopausal women.” Thus, she recommends routine screening for women in this age group. “A practice that delays screening until age 50,” she observes, “may be missing the boat.”
Dr. Iglesia also recommends that women in their 40s receive annual mammograms—a practice that’s in line with the recommendations of the American Cancer Society and one that she herself adheres to. The interval between when a cancer is detectable on mammography and the time it becomes symptomatic—known as the “sojourn time”—is about 2 years for women ages 40 through 49, she explains, and more frequent screening would be more likely to catch breast cancer in the preclinical phase. “That’s what a screening test is supposed to do.”
Helen Lippman, Managing Editor
CORRESPONDENCE
Sandhya Pruthi, MD, Mayo Clinic, 200 First Street SW, Rochester, MN 55905; pruthi.sandhya@mayo.edu
1. Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA-Cancer J Clin. 2009;59:225-249.
2. National Cancer Institute. Fact sheet. Probability of breast cancer in American women. Available at: http://www.cancer.gov/cancertopics/factsheet/detection/probability-breast-cancer. Accessed August 23, 2011.
3. Berry DA, Cronin KA, Plevritis SK, et al. Effect of screening and adjuvant therapy on mortality from breast cancer. N Engl J Med. 2005;353:1784-1792.
4. Humphrey LL, Helfand M, Chan BK, et al. Breast cancer screening: a summary of the evidence for the US Preventive Services Task Force. Ann Intern Med. 2002;137(5 part 1):347-360.
5. American College of Obstetricians and Gynecologists. Annual mammograms now recommended for women beginning at age 40. July 20, 2011. Available at: http://www.acog.org/from_home/publications/press_releases/nr07-20-11-2.cfm. Accessed August 15, 2011.
6. US Preventive Services Task Force. Screening for breast cancer. July 2010. Available at: http://www.uspreventiveservicestaskforce.org/uspstf/uspsbrca.htm. Accessed August 15, 2011.
7. Nelson HD, Tyne K, Naik A, et al. Screening for breast cancer:. an update for the US Preventive Services Task Force. Ann Intern Med. 2009;151:727–737, W237–742.
8. US Preventive Services Task Force. Screening for breast cancer. US Preventive Services Task Force recommendation statement. Ann Intern Med. 2009;151:716-726.
9. Smith RA, Cokkinides V, Brooks D, et al. Cancer screening in the United States, 2010: a review of current American Cancer Society guidelines and issues in cancer screening. CA-Cancer J Clin. 2010;60:99-119.
10. Lee CH, Dershaw DD, Kopans D, et al. Breast cancer screening with imaging: recommendations from the Society of Breast Imaging and the ACR on the use of mammography, breast MRI, breast ultrasound, and other technologies for the detection of clinically occult breast cancer. J Am Coll Radiol. 2010;7:18-27.
11. Mandelblatt JS, Cronin KA, Bailey S, et al. Effects of mammography screening under different screening schedules: model estimates of potential benefits and harms. Ann Intern Med. 2009;151:738-747.
12. American College of Obstetricians and Gynecologists. Practice bulletin no. 122: breast cancer screening. Obstet Gynecol. 2011;118:372-382.
13. Madigan MP, Ziegler RG, Benichou J, et al. Proportion of breast cancer cases in the United States explained by well-established risk factors. J Natl Cancer Inst. 1995;87:1681-1685.
14. Kalager M, Zelen M, Langmark F, et al. Effect of screening mammography on breast-cancer mortality in Norway. N Engl J Med. 2010;363:1203-1210.
15. Welch HG. Screening mammography—a long run for a short slide? N Engl J Med. 2010;363:1276-1278.
16. Hellquist BN, Duffy SW, Abdsaleh S, et al. Effectiveness of population-based service screening with mammography for women ages 40 to 49 years: evaluation of the Swedish Mammography Screening in Young Women (SCRY) cohort. Cancer. 2010;117:714-722.
17. Hendrick RE, Helvie MA. United States Preventive Services Task Force screening mammography recommendations: science ignored. AJR Am J Roentgenol. 2011;196:W112-W116.
18. Gotzche PC, Nielsen M. Screening for breast cancer with mammography. Cochrane Database Syst Rev. 2011;(1):CD001877.-
19. Autier P, Boniol M, Gavin A, et al. Breast cancer mortality in neighbouring European countries with different levels of screening but similar access to treatment: trend analysis of WHO mortality database. BMJ. 2011;343:d4411.-
20. When evidence collides with anecdote, politics, and emotion: breast cancer screening [editorial]. Ann Intern Med. 2010;152:531-532.
21. American Cancer Society. Cancer prevention and early detection facts and figures 2009. Available at: http://www.cancer.org/Research/CancerFactsFigures/CancerPreventionEarlyDetectionFactsFigures/index. Accessed August 15, 2011.
22. Australian Screening Mammography Decision Aid Trial. Available at: http://www.mammogram.med.usyd.edu.au/. Accessed August 15, 2011.
23. Barratt A, Howard K, Irwig L, et al. Model of outcomes of screening mammography: information to support informed choices. BMJ. 2005;330:936.-
24. Chen J, Pee D, Ayyagari R, et al. Projecting absolute invasive breast cancer risk in women with a model that includes mammographic density. J Natl Cancer Inst. 2006;98:1215-1226.
25. Tice JA, Cummings SR, Ziv E, et al. Mammographic breast density and the Gail Model for breast cancer risk prediction in a screening population. Breast Cancer Res Treat. 2005;94:115-122.
26. Schousboe JT, Kerlikowske K, Loh A, et al. Personalizing mammography by breast density and other risk factors for breast cancer: analysis of health benefits and cost-effectiveness. Ann Intern Med. 2011;155:10-20.
27. Boyd NF, Byng JW, Jong RA, et al. Quantitative classification of mammographic densities and breast cancer risk: results from the Canadian National Breast Screening Study. J Natl Cancer Inst. 1995;87:670-675.
28. Tamimi R, Byrne C, Colditz EG, et al. Endogenous hormone levels, mammographic density, and subsequent risk of breast cancer in postmenopausal women. J Natl Cancer Inst. 2007;99:1178-1187.
1. Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA-Cancer J Clin. 2009;59:225-249.
2. National Cancer Institute. Fact sheet. Probability of breast cancer in American women. Available at: http://www.cancer.gov/cancertopics/factsheet/detection/probability-breast-cancer. Accessed August 23, 2011.
3. Berry DA, Cronin KA, Plevritis SK, et al. Effect of screening and adjuvant therapy on mortality from breast cancer. N Engl J Med. 2005;353:1784-1792.
4. Humphrey LL, Helfand M, Chan BK, et al. Breast cancer screening: a summary of the evidence for the US Preventive Services Task Force. Ann Intern Med. 2002;137(5 part 1):347-360.
5. American College of Obstetricians and Gynecologists. Annual mammograms now recommended for women beginning at age 40. July 20, 2011. Available at: http://www.acog.org/from_home/publications/press_releases/nr07-20-11-2.cfm. Accessed August 15, 2011.
6. US Preventive Services Task Force. Screening for breast cancer. July 2010. Available at: http://www.uspreventiveservicestaskforce.org/uspstf/uspsbrca.htm. Accessed August 15, 2011.
7. Nelson HD, Tyne K, Naik A, et al. Screening for breast cancer:. an update for the US Preventive Services Task Force. Ann Intern Med. 2009;151:727–737, W237–742.
8. US Preventive Services Task Force. Screening for breast cancer. US Preventive Services Task Force recommendation statement. Ann Intern Med. 2009;151:716-726.
9. Smith RA, Cokkinides V, Brooks D, et al. Cancer screening in the United States, 2010: a review of current American Cancer Society guidelines and issues in cancer screening. CA-Cancer J Clin. 2010;60:99-119.
10. Lee CH, Dershaw DD, Kopans D, et al. Breast cancer screening with imaging: recommendations from the Society of Breast Imaging and the ACR on the use of mammography, breast MRI, breast ultrasound, and other technologies for the detection of clinically occult breast cancer. J Am Coll Radiol. 2010;7:18-27.
11. Mandelblatt JS, Cronin KA, Bailey S, et al. Effects of mammography screening under different screening schedules: model estimates of potential benefits and harms. Ann Intern Med. 2009;151:738-747.
12. American College of Obstetricians and Gynecologists. Practice bulletin no. 122: breast cancer screening. Obstet Gynecol. 2011;118:372-382.
13. Madigan MP, Ziegler RG, Benichou J, et al. Proportion of breast cancer cases in the United States explained by well-established risk factors. J Natl Cancer Inst. 1995;87:1681-1685.
14. Kalager M, Zelen M, Langmark F, et al. Effect of screening mammography on breast-cancer mortality in Norway. N Engl J Med. 2010;363:1203-1210.
15. Welch HG. Screening mammography—a long run for a short slide? N Engl J Med. 2010;363:1276-1278.
16. Hellquist BN, Duffy SW, Abdsaleh S, et al. Effectiveness of population-based service screening with mammography for women ages 40 to 49 years: evaluation of the Swedish Mammography Screening in Young Women (SCRY) cohort. Cancer. 2010;117:714-722.
17. Hendrick RE, Helvie MA. United States Preventive Services Task Force screening mammography recommendations: science ignored. AJR Am J Roentgenol. 2011;196:W112-W116.
18. Gotzche PC, Nielsen M. Screening for breast cancer with mammography. Cochrane Database Syst Rev. 2011;(1):CD001877.-
19. Autier P, Boniol M, Gavin A, et al. Breast cancer mortality in neighbouring European countries with different levels of screening but similar access to treatment: trend analysis of WHO mortality database. BMJ. 2011;343:d4411.-
20. When evidence collides with anecdote, politics, and emotion: breast cancer screening [editorial]. Ann Intern Med. 2010;152:531-532.
21. American Cancer Society. Cancer prevention and early detection facts and figures 2009. Available at: http://www.cancer.org/Research/CancerFactsFigures/CancerPreventionEarlyDetectionFactsFigures/index. Accessed August 15, 2011.
22. Australian Screening Mammography Decision Aid Trial. Available at: http://www.mammogram.med.usyd.edu.au/. Accessed August 15, 2011.
23. Barratt A, Howard K, Irwig L, et al. Model of outcomes of screening mammography: information to support informed choices. BMJ. 2005;330:936.-
24. Chen J, Pee D, Ayyagari R, et al. Projecting absolute invasive breast cancer risk in women with a model that includes mammographic density. J Natl Cancer Inst. 2006;98:1215-1226.
25. Tice JA, Cummings SR, Ziv E, et al. Mammographic breast density and the Gail Model for breast cancer risk prediction in a screening population. Breast Cancer Res Treat. 2005;94:115-122.
26. Schousboe JT, Kerlikowske K, Loh A, et al. Personalizing mammography by breast density and other risk factors for breast cancer: analysis of health benefits and cost-effectiveness. Ann Intern Med. 2011;155:10-20.
27. Boyd NF, Byng JW, Jong RA, et al. Quantitative classification of mammographic densities and breast cancer risk: results from the Canadian National Breast Screening Study. J Natl Cancer Inst. 1995;87:670-675.
28. Tamimi R, Byrne C, Colditz EG, et al. Endogenous hormone levels, mammographic density, and subsequent risk of breast cancer in postmenopausal women. J Natl Cancer Inst. 2007;99:1178-1187.
Aspirin for CV prevention—for which patients?
• Calculate a patient’s 10-year global risk of cardiovascular events using a risk-assessment tool before recommending aspirin for primary prevention. A
• Keep in mind that diabetes is not an indication for aspirin as primary cardiovascular protection, unless the patient’s calculated 10-year risk is >10%. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Among individuals at high risk (≥10%) for coronary heart disease (CHD) within 10 years, only 44% are taking aspirin.1 In addition, for patients at high risk for CHD events, estimated aspirin use varies among ethnic groups: 53% for whites, 43% for African Americans, 38% for Hispanics, and 28% for Chinese Americans.1
In contrast to this underuse of aspirin by those who need it, patients who do not need aspirin have been told otherwise,2 following widespread publicity of US Preventive Services Task Force (USPSTF) recommendations from 2002 (that have since been updated). Overuse of aspirin is also likely among individuals whose CHD risk has never been formally assessed but who take it on their own, based on direct-to-consumer advertising about the cardiovascular (CV) benefits of aspirin. Also, the American Diabetes Association (ADA) once recommended aspirin for all patients with diabetes. But it now advises avoiding the use of aspirin for primary prevention of CV events unless a patient’s calculated CV risk over 10 years is >10%.3
Our review summarizes the latest evidence on the use of aspirin for primary prevention of CV events, including the determination of benefit vs harm, the variability in aspirin responsiveness among individuals, and the efficacy of aspirin treatment in men vs women and in those with diabetes.
When does benefit outweigh risk?
In 2002, the USPSTF concluded that patients with a 5-year risk of coronary events ≥3% had the most favorable benefit-to-risk ratio with aspirin use.4 It based its recommendation on 5 randomized, controlled primary prevention studies with aspirin that demonstrated a reduction in the risk of a first myocardial infarction (MI) in men.5-9 In 2009, the USPSTF updated its recommendations regarding the risks and benefits of aspirin for primary prevention of CHD,10 in part to include data from the Women’s Health Study11 that demonstrated a 24% relative risk (RR) reduction of ischemic stroke without reducing the risk of MI.
The USPSTF now recommends aspirin for men ages 45 to 79 to prevent a first MI, and for women ages 55 to 79 to prevent an ischemic stroke when the potential benefit outweighs the increased risk of gastrointestinal (GI) hemorrhage.10 Evidence does not support the use of aspirin for primary CHD prevention in men younger than 45 years or women younger than 55. Evidence is insufficient to recommend aspirin for primary prevention of CHD for individuals ≥80 years of age in the absence of other compelling indications such as atrial fibrillation.
Calculating benefit. The American Heart Association (AHA) recommends low-dose aspirin for primary prevention of CV events in all individuals with a calculated 10-year CHD risk of ≥10%, while cautioning about its use in patients at increased risk for GI bleeding and hemorrhagic stroke.12 The Framingham risk score13 is available online at http://hp2010.nhlbihin.net/atpiii/calculator.asp?usertype=prof to estimate an individual’s 10-year CHD risk (TABLE).14
Judging risk. There are no validated tools for assessing the long-term risk of intracranial or GI hemorrhage with low-dose aspirin. The risk factors for GI bleeding with nonsteroidal anti-inflammatory drugs (NSAIDs) are well known,15 but less data exist for low-dose aspirin. Likely risk factors include a history of peptic ulcer disease, concomitant NSAID therapy, high-dose corticosteroids or anticoagulants, dual antiplatelet therapy, age >60 years, and male sex.16 Although proton-pump inhibitors prevent recurrent peptic ulcers secondary to low-dose aspirin use, little data exist on their value or cost effectiveness for this purpose.17
Why the AHA recommendation makes sense. The 2009 USPSTF recommendations still identify different tiers of risk according to 3 age brackets within the range of 45 (or 55) to 79 years. Since then, however, further studies seem to favor a less aggressive approach to aspirin use, more in keeping with the AHA recommendation.
The Antithrombotic Trialists’ (ATT) Collaboration18 published a meta-analysis using individual participant data from the same studies that served as the basis of the USPSTF recommendations.5-9,11 It found that aspirin did not reduce the risk of death due to CHD, stroke, or other vascular causes. The risk of nonfatal stroke also did not decline. Aspirin use decreased the risk of nonfatal MI (RR=0.77; 99% confidence interval [CI], 0.67-0.89), any major coronary event (RR=0.82; 95% CI, 0.75-0.90), and serious vascular events (RR=0.88; 95% CI, 0.82-0.94). The risk of extracranial hemorrhage, including GI bleeding, increased (RR=1.54; 95% CI, 1.30-1.82). Based on this analysis, the absolute reduction in serious ischemic events was partially offset by a small increase in serious bleeding. However, long-term disability from a nonfatal extracranial hemorrhage is likely less than that from a nonfatal stroke or MI.18
In the ATT Collaboration18 analysis, the 5-year risk of bleeding with low-dose aspirin increased with the predicted 5-year CHD risk. Patients with the lowest CHD risk (<5%) demonstrated a 0.4% risk of bleeding vs 2.7% among patients having the highest CHD risk (>10%). However, the high-risk patients also had the largest benefit with low-dose aspirin therapy. According to the ATT Collaboration data, using aspirin alone vs placebo, the estimated number needed to treat (NNT) to prevent 1 serious vascular event (defined as vascular death, nonfatal MI, or stroke) was 50 patients for 5 years. When aspirin was added to other therapies such as statins, the NNT was 100 patients for 5 years. To cause 1 nonfatal extracranial bleeding event with aspirin in the same high-risk patients, the estimated number needed to harm (NNH) was also 100 patients for 5 years. A meta-analysis of 22 trials estimated a NNH to cause 1 additional major bleeding event with aspirin per year was 769 patients (95% CI, 500-1250).19
The Aspirin for Asymptomatic Atherosclerosis Trial (AAAT)20 involved 3350 men and women ages 50 to 75 years with low ankle-brachial index and no history of CV disease (CVD). Participants were randomized to receive 100 mg enteric-coated aspirin or placebo daily and were followed for a mean of 8.2 years. The primary and secondary end points, which included fatal and nonfatal MI or stroke, were similar in the 2 groups, as were all-cause mortality and total adverse events. A difference in the incidence of major hemorrhage did not reach statistical significance—34 patients in the aspirin arm vs 20 in the placebo arm (hazard ratio [HR]=1.71; 95% CI, 0.99-2.97). One caution: the relative lack of benefit from aspirin reported in the AAAT may be due to the fact that it was powered to detect a 25% reduction in the event rate between groups, whereas the ATT Collaboration study18 found a 12% risk reduction in MI among those taking aspirin.
TABLE
Should you recommend aspirin? See how these patients “scored”*
| Patient | Risk score | Prophylaxis |
|---|---|---|
| 53-year-old woman, HTN on medication, SBP 152, nonsmoker, Chol 260, HDL 50 | 4% | No |
| 48-year-old man, HTN on medication, SBP 138, nonsmoker, Chol 220, HDL 41 | 7% | No† |
| 68-year-old woman, HTN on medication, SBP 152, nonsmoker, Chol 260, HDL 50 | 11% | Yes |
| 58-year-old man, HTN on medication, SBP 138, nonsmoker, Chol 220, HDL 41 | 14% | Yes |
| 51-year-old man, HTN on medication, SBP 140, nonsmoker, Chol 180, HDL 41, Diabetes | 6% | No† |
| *Score for 10-year risk of CHD calculated with Framingham Heart Study data.14 †Under 2009 USPSTF recommendations, may consider aspirin therapy.10 Chol, total cholesterol; HDL, high-density lipoprotein; HTN, hypertension; SBP, systolic blood pressure. | ||
Test for aspirin resistance? It’s still too soon
Patients receiving aspirin therapy may demonstrate residual platelet reactivity (laboratory resistance) or recurrent ischemic CV events (clinical resistance).21 Estimates of the prevalence of aspirin resistance vary widely.22 And available assays of residual platelet activity yield different results. Higher estimates of aspirin resistance may occur with assays that use an agonist other than arachidonic acid, such as collagen or adenosine diphosphate platelet aggregation, the whole blood platelet function analyzer (PFA-100), or urinary 11-dehydro-thromboxane B2.23
Several secondary prevention studies have demonstrated a positive association with laboratory resistance and adverse CV events, regardless of methods and assays used.24 However, prospective primary prevention studies of this association are lacking. A meta-analysis of 20 clinical studies reported an increased risk of recurrent CV events including graft failure, acute coronary syndrome (ACS), and death among patients who exhibited aspirin resistance (odds ratio [OR]=3.85; 95% CI, 3.08-4.80). The authors identified a high level of heterogeneity among the studies, with 9 of the 20 failing to demonstrate an increased risk of events.25
Using the PFA-100 assay, a prospective cohort study verified the presence or absence of aspirin resistance in 140 patients who presented to the emergency department with a non-ST–elevation ACS and who reported using aspirin daily for at least 7 days before the event.26 Fifty-three patients (37.8%) were found to have aspirin resistance. Baseline characteristics of patients with and without aspirin resistance were similar except for an older age (mean 63.8 vs 58.3 years, respectively) and higher cardiac troponin values (mean 1.11 vs 0.41 ng/mL). Both groups were monitored for an average of 20 months; 45 patients with aspirin resistance and 79 without resistance completed follow-up. The presence of aspirin resistance increased the risk of MI (HR=3.02; 95% CI, 1.15-7.95) and decreased the risk of event-free survival (HR=2.46; 95% CI, 1.18-5.13). Adjusted for age, platelet count, cardiac troponin values, and coronary artery disease severity scores, the presence of aspirin resistance was associated with a 3-fold increased risk of CV events (HR=3.03; 95% CI, 1.06-8.62).
Mechanisms for aspirin resistance may involve an inability of aspirin to partially or completely inhibit the cyclo-oxygenase-1 (COX-1) enzyme leading to thromboxane A2 production, or factors independent of the COX-1 pathway such as elevated levels of C-reactive protein.27 COX-1-related factors include aspirin nonadherence, reduced aspirin bioavailability, competitive inhibition by NSAIDs, inadequate aspirin dosage, genetic COX-1 polymorphisms, and increased platelet turnover.27,28 A subgroup analysis of the Physicians’ Health Study29 suggests that nonadherence with aspirin therapy or concomitant NSAID use negated the benefit of aspirin. In a small cohort study (n=18), patients who took ibuprofen or naproxen and aspirin did not demonstrate inhibition of platelet aggregation and had a 72% rate of recurrent ischemic events despite aspirin therapy.30
Until clinical trials can demonstrate benefit and cost effectiveness of empiric laboratory testing for aspirin resistance in patients without a history of CVD, emphasize adherence to the prescribed antiplatelet therapy and warn against concomitant NSAID use for patients at risk for CHD events.
Aspirin for patients with diabetes: Only when CVD risk is high
In 2010, the ADA revised its clinical practice recommendations to reflect the results of 2 studies that questioned the value of aspirin for primary prevention of CVD events in patients with diabetes.3 Instead of a global statement to use low-dose aspirin, the ADA guideline now recommends its use only in patients with diabetes who have a 10-year risk >10%. This includes men over the age of 50 and women over the age of 60, with at least one major risk factor in addition to diabetes. The studies driving this change were the Prevention of Progression of Arterial Disease and Diabetes (POPADAD)31 and the Japanese Primary Prevention of Atherosclerosis with Aspirin for Diabetes (JPAD).32
The POPADAD study enrolled 1276 patients over the age of 40 with type 1 or type 2 diabetes who also had asymptomatic peripheral arterial disease but without symptomatic CHD. Participants were randomized to take aspirin 100 mg daily or placebo (POPADAD also included a study of antioxidants vs placebo). The participants had diabetes for a mean of 6.3 years. The study had 2 primary composite end points: death from CHD or stroke, nonfatal MI or stroke, or amputation above the ankle for critical ischemia; and death from CHD or stroke. The aspirin and placebo groups were similar at baseline in terms of demographic characteristics and use of statins, beta-blockers, and angiotensin-converting enzyme (ACE) inhibitors among other treatments. The composite end point of death from CHD or stroke was similar in the 2 groups. Nonfatal MI and nonfatal stroke were also similar in the 2 groups.31
The JPAD study enrolled individuals with type 2 diabetes who were over the age of 30 and had no evidence of CVD. Participants were randomized to receive either 81 mg aspirin or placebo daily. The composite end point was sudden death; death from CHD, stroke, or aortic causes; nonfatal MI; nonfatal stroke; unstable angina; transient ischemic attack; or nonfatal peripheral vascular disease. The 2 groups were similar in terms of the composite end point, nonfatal MI, and nonfatal stroke. The risk of death from MI and stroke was lower in the aspirin group.32
The authors of a 2010 consensus report from the ADA, the AHA, and the American College of Cardiology (ACC) evaluated the findings of individual placebo-controlled aspirin studies as well as those included in prior meta-analyses.33 They also conducted a separate meta-analysis, which indicated that aspirin decreased the risk of CHD in patients with diabetes by 9% (RR=0.91; 95% CI, 0.79-1.05), but the reduction was not statistically significant. If the findings of the Early Treatment of Diabetic Retinopathy Study, which included some individuals with prior CVD events, had been excluded from this meta-analysis, the risk reduction due to aspirin would have been smaller.
Results of this meta-analysis are mitigated by certain factors. The 9 studies analyzed were published between 1989 and 2008, and the use of drugs such as statins, beta-blockers, and ACE inhibitors increased over this 20-year period. Also, the age of study participants at enrollment varied, as did the presence of subclinical CVD. The rates of CHD in the placebo groups of the studies also varied significantly.
Accounting for differences between the sexes
A person’s sex in part determines the importance of certain CV risk factors, the prevalence of CV and related comorbid diseases, and the frequency of adverse drug effects. Women with diabetes have a 50% increased relative risk of CVD than men with diabetes, in part because they are often older and have more risk factors.34
A 2011 AHA update on the prevention of CVD in women indicates that women ≥65 years may use aspirin, 81 mg daily or 100 mg every other day, if the benefit in reducing CHD or ischemic stroke is not outweighed by the potential risk of GI bleeding or hemorrhagic stroke. It also deems aspirin an option that women younger than 65 could consider with their physicians for prevention of ischemic stroke, and recommends aspirin 75 to 325 mg daily for women with diabetes.35
The study populations in the ADA/AHA/ACC meta-analysis of aspirin for primary prevention in patients with diabetes varied in the percentage of women enrolled. Three trials did not include women, while one study enrolled women exclusively. The remaining studies had similar numbers of men and women. Aspirin decreased the risk of CHD events in men (RR=0.77; 95% CI, 0.67-0.89) and stroke in women (RR=0.77; 95% CI, 0.59-0.99). The consensus report acknowledged that the findings of the Women’s Health Study strongly influenced this difference in outcomes for men and women.33
CORRESPONDENCE
Anita N. Jackson, PharmD, University of Rhode Island, College of Pharmacy, 41 Lower College Road, Fogarty Hall, Kingston, RI 02881; anitaj@uri.edu
1. Sanchez DR, Diez Roux AV, Michos ED, et al. Comparison of the racial/ethnic prevalence of regular aspirin use for the primary prevention of coronary heart disease from the multi-ethnic study of atherosclerosis. Am J Cardiol. 2011;107:41-46.
2. Dwivedi G, Ball MC, Dilworth MP, et al. Use and misuse of aspirin in the hypertension clinic [letter]. BMJ. May 3, 2010. Available at: http://www.bmj.com/content/340/bmj.c1805.full/reply#bmj_el_235118. Accessed October 15, 2010.
3. American Diabetes Association. Standards of medical care in diabetes–2010. Diabetes Care. 2010;33(suppl 1):S11-S61.
4. US Preventive Services Task Force. Aspirin for the primary prevention of cardiovascular events: recommendation and rationale. Ann Intern Med. 2002;136:157-160.
5. Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. The Medical Research Council’s General Practice Research Framework. Lancet. 1998;351:233-241.
6. Peto R, Gray R, Collins R. Randomised trial of prophylactic daily aspirin in British male doctors. BMJ. 1988;296:313-316.
7. Final report on the aspirin component of the ongoing Physicians’ Health Study. Steering Committee of the Physicians’ Health Study Research Group. N Engl J Med. 1989;321:129-135.
8. Hansson L, Zanchetti A, Carruthers SG. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. Lancet. 1998;351:1755-1762.
9. de Gaetano G. Collaborative Group of the Primary Prevention Project. Low-dose aspirin and vitamin E in people at cardiovascular risk: a randomised trial in general practice. Lancet. 2001;357:89-95.
10. Wolff T, Miller T, Ko S. Aspirin for the prevention of cardiovascular disease: an update of the evidence for the US Preventive Services Task Force. Ann Intern Med. 2009;150:405-410.
11. Ridker PM, Cook NR, Lee IM. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med. 2005;352:1293-1304.
12. Pearson TA, Blair SN, Daniels SR, et al. AHA guidelines for primary prevention of cardiovascular disease and stroke: 2002 update: consensus panel guide to comprehensive risk reduction for adult patients without coronary or other atherosclerotic vascular diseases. Circulation. 2002;106:388-391.
13. Wilson PW, D’Agostino RB, Levy D, et al. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837-1847.
14. National Cholesterol Education Program. Risk assessment tool for estimating 10-year risk of developing hard CHD. Available at: http://hp2010.nhlbihin.net/atpiii/calculator.asp?usertype=prof. Accessed October 15, 2010.
15. Bhatt DL, Scheiman J, Abraham NS, et al. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use. Circulation. 2008;118:1894-1909.
16. Garcia Rodriguez LA, Lin KJ, Hernandez-Diaz S, et al. Risk of upper gastrointestinal bleeding with low-dose acetylsalicylic acid alone and in combination with clopidogrel and other medications. Circulation. 2011;123:1108-1115.
17. Lai KC, Lam SK, Chu Km, et al. Lansoprazole for the prevention of recurrences of ulcer complications from long-term low-dose aspirin use. N Engl J Med. 2002;346:2033-2038.
18. Antithrombotic Trialists’ (ATT) Collaboration. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373:1849-1860.
19. McQuaid KR, Laine L. Systematic review and meta-analysis of adverse events of low-dose aspirin and clopidogrel in randomized controlled trials. Am J Med. 2006;119:624-638.
20. Fowkes FG, Price JF, Stewart MCW, et al. Aspirin for the prevention of cardiovascular events in a general population screened for a low ankle brachial index: a randomized controlled trial. JAMA. 2010;303:841-848.
21. Kuliczkowski W, Witkowski A, Polonski L, et al. Interindividual variability in the response to oral antiplatelet drugs: a position paper of the working group on antiplatelet drugs resistance appointed by the section of cardiovascular interventions of the Polish Cardiac Society, endorsed by the working group on thrombosis of the European Society of Cardiology. Eur Heart J. 2009;30:426-435.
22. Hovens MM, Snoep JD, Eidenboom JC. Prevalence of persistent platelet reactivity despite use of aspirin: a systematic review. Am Heart J. 2007;153:175-181.
23. Gurbel PA, Bliden KP, DiChiara JD, et al. Evaluation of dose-related effects of aspirin on platelet function: results from the Aspirin-Induced Platelet Effect (ASPECT) study. Circulation. 2007;115:3156-3164.
24. Feher G, Geher A, Pusch G, et al. Clinical importance of aspirin and clopidogrel resistance. World J Cardiol. 2010;2:171-186.
25. Krasopoulos G, Brister SJ, Beattie WS, et al. Aspirin “resistance” and risk of cardiovascular morbidity: systematic review and meta-analysis. BMJ. 2008;336:195-198.
26. Hobikoglu GF, Norgaz T, Aksu H, et al. The effect of acetylsalicylic acid resistance on prognosis of patients who have developed acute coronary syndrome during acetylsalicylic acid therapy. Can J Cardiol. 2007;23:201-206.
27. Gasparyan AY, Watson T, Lip GYH. The role of aspirin in cardiovascular prevention: implications of aspirin resistance. J Am Coll Cardiol. 2008;51:1829-1843.
28. Arazi HC, Doiny DG, Torcivia RS, et al. Impaired anti-platelet effect of aspirin, inflammation and platelet turnover in cardiac surgery. Interact Cardiovasc Thorac Surg. 2010;10:863-867.
29. Hennekens CH, Schneider WR, Hebert PR, et al. Hypothesis formulation from subgroup analyses: nonadherence or nonsteroidal anti-inflammatory drug use explains the lack of clinical benefit of aspirin on first myocardial infarction attributed to “aspirin resistance.” Am Heart J. 2010;159:744-748.
30. Gengo FM, Rubin L, Robson M, et al. Effects of ibuprofen on the magnitude and duration of aspirin’s inhibition of platelet aggregation; clinical consequences in stroke prophylaxis. J Clin Pharmacol. 2008;48:117-122.
31. Belch J, MacCuish A, Campbell I, et al. The prevention of progression of arterial disease and diabetes (POPADAD) trial: factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ. 2008;337:a1840.-
32. Ogawa H, Nakayama M, Morimoto T, et al. Low dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: a randomized controlled trial. JAMA. 2008;300:2134-2141.
33. Pignone M, Alberts MJ, Colwell JA, et al. Aspirin for primary prevention of cardiovascular events in people with diabetes. A position statement of the American Diabetes Association, a scientific statement of the American Heart Association, and an expert consensus document of the American College of Cardiology Foundation. Circulation. 2010;121:2694-2701.
34. Barrett-Connor E, Giardina EG, Gitt AK, et al. Women and heart disease: the role of diabetes and hyperglycemia. Arch Intern Med. 2004;164:934-942.
35. Mosca L, Benjamin EJ, Berra K, et al. Effectiveness-based guidelines for the prevention of cardiovascular disease in women—2011 update. Circulation. 2011;123:1243-1262.
• Calculate a patient’s 10-year global risk of cardiovascular events using a risk-assessment tool before recommending aspirin for primary prevention. A
• Keep in mind that diabetes is not an indication for aspirin as primary cardiovascular protection, unless the patient’s calculated 10-year risk is >10%. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Among individuals at high risk (≥10%) for coronary heart disease (CHD) within 10 years, only 44% are taking aspirin.1 In addition, for patients at high risk for CHD events, estimated aspirin use varies among ethnic groups: 53% for whites, 43% for African Americans, 38% for Hispanics, and 28% for Chinese Americans.1
In contrast to this underuse of aspirin by those who need it, patients who do not need aspirin have been told otherwise,2 following widespread publicity of US Preventive Services Task Force (USPSTF) recommendations from 2002 (that have since been updated). Overuse of aspirin is also likely among individuals whose CHD risk has never been formally assessed but who take it on their own, based on direct-to-consumer advertising about the cardiovascular (CV) benefits of aspirin. Also, the American Diabetes Association (ADA) once recommended aspirin for all patients with diabetes. But it now advises avoiding the use of aspirin for primary prevention of CV events unless a patient’s calculated CV risk over 10 years is >10%.3
Our review summarizes the latest evidence on the use of aspirin for primary prevention of CV events, including the determination of benefit vs harm, the variability in aspirin responsiveness among individuals, and the efficacy of aspirin treatment in men vs women and in those with diabetes.
When does benefit outweigh risk?
In 2002, the USPSTF concluded that patients with a 5-year risk of coronary events ≥3% had the most favorable benefit-to-risk ratio with aspirin use.4 It based its recommendation on 5 randomized, controlled primary prevention studies with aspirin that demonstrated a reduction in the risk of a first myocardial infarction (MI) in men.5-9 In 2009, the USPSTF updated its recommendations regarding the risks and benefits of aspirin for primary prevention of CHD,10 in part to include data from the Women’s Health Study11 that demonstrated a 24% relative risk (RR) reduction of ischemic stroke without reducing the risk of MI.
The USPSTF now recommends aspirin for men ages 45 to 79 to prevent a first MI, and for women ages 55 to 79 to prevent an ischemic stroke when the potential benefit outweighs the increased risk of gastrointestinal (GI) hemorrhage.10 Evidence does not support the use of aspirin for primary CHD prevention in men younger than 45 years or women younger than 55. Evidence is insufficient to recommend aspirin for primary prevention of CHD for individuals ≥80 years of age in the absence of other compelling indications such as atrial fibrillation.
Calculating benefit. The American Heart Association (AHA) recommends low-dose aspirin for primary prevention of CV events in all individuals with a calculated 10-year CHD risk of ≥10%, while cautioning about its use in patients at increased risk for GI bleeding and hemorrhagic stroke.12 The Framingham risk score13 is available online at http://hp2010.nhlbihin.net/atpiii/calculator.asp?usertype=prof to estimate an individual’s 10-year CHD risk (TABLE).14
Judging risk. There are no validated tools for assessing the long-term risk of intracranial or GI hemorrhage with low-dose aspirin. The risk factors for GI bleeding with nonsteroidal anti-inflammatory drugs (NSAIDs) are well known,15 but less data exist for low-dose aspirin. Likely risk factors include a history of peptic ulcer disease, concomitant NSAID therapy, high-dose corticosteroids or anticoagulants, dual antiplatelet therapy, age >60 years, and male sex.16 Although proton-pump inhibitors prevent recurrent peptic ulcers secondary to low-dose aspirin use, little data exist on their value or cost effectiveness for this purpose.17
Why the AHA recommendation makes sense. The 2009 USPSTF recommendations still identify different tiers of risk according to 3 age brackets within the range of 45 (or 55) to 79 years. Since then, however, further studies seem to favor a less aggressive approach to aspirin use, more in keeping with the AHA recommendation.
The Antithrombotic Trialists’ (ATT) Collaboration18 published a meta-analysis using individual participant data from the same studies that served as the basis of the USPSTF recommendations.5-9,11 It found that aspirin did not reduce the risk of death due to CHD, stroke, or other vascular causes. The risk of nonfatal stroke also did not decline. Aspirin use decreased the risk of nonfatal MI (RR=0.77; 99% confidence interval [CI], 0.67-0.89), any major coronary event (RR=0.82; 95% CI, 0.75-0.90), and serious vascular events (RR=0.88; 95% CI, 0.82-0.94). The risk of extracranial hemorrhage, including GI bleeding, increased (RR=1.54; 95% CI, 1.30-1.82). Based on this analysis, the absolute reduction in serious ischemic events was partially offset by a small increase in serious bleeding. However, long-term disability from a nonfatal extracranial hemorrhage is likely less than that from a nonfatal stroke or MI.18
In the ATT Collaboration18 analysis, the 5-year risk of bleeding with low-dose aspirin increased with the predicted 5-year CHD risk. Patients with the lowest CHD risk (<5%) demonstrated a 0.4% risk of bleeding vs 2.7% among patients having the highest CHD risk (>10%). However, the high-risk patients also had the largest benefit with low-dose aspirin therapy. According to the ATT Collaboration data, using aspirin alone vs placebo, the estimated number needed to treat (NNT) to prevent 1 serious vascular event (defined as vascular death, nonfatal MI, or stroke) was 50 patients for 5 years. When aspirin was added to other therapies such as statins, the NNT was 100 patients for 5 years. To cause 1 nonfatal extracranial bleeding event with aspirin in the same high-risk patients, the estimated number needed to harm (NNH) was also 100 patients for 5 years. A meta-analysis of 22 trials estimated a NNH to cause 1 additional major bleeding event with aspirin per year was 769 patients (95% CI, 500-1250).19
The Aspirin for Asymptomatic Atherosclerosis Trial (AAAT)20 involved 3350 men and women ages 50 to 75 years with low ankle-brachial index and no history of CV disease (CVD). Participants were randomized to receive 100 mg enteric-coated aspirin or placebo daily and were followed for a mean of 8.2 years. The primary and secondary end points, which included fatal and nonfatal MI or stroke, were similar in the 2 groups, as were all-cause mortality and total adverse events. A difference in the incidence of major hemorrhage did not reach statistical significance—34 patients in the aspirin arm vs 20 in the placebo arm (hazard ratio [HR]=1.71; 95% CI, 0.99-2.97). One caution: the relative lack of benefit from aspirin reported in the AAAT may be due to the fact that it was powered to detect a 25% reduction in the event rate between groups, whereas the ATT Collaboration study18 found a 12% risk reduction in MI among those taking aspirin.
TABLE
Should you recommend aspirin? See how these patients “scored”*
| Patient | Risk score | Prophylaxis |
|---|---|---|
| 53-year-old woman, HTN on medication, SBP 152, nonsmoker, Chol 260, HDL 50 | 4% | No |
| 48-year-old man, HTN on medication, SBP 138, nonsmoker, Chol 220, HDL 41 | 7% | No† |
| 68-year-old woman, HTN on medication, SBP 152, nonsmoker, Chol 260, HDL 50 | 11% | Yes |
| 58-year-old man, HTN on medication, SBP 138, nonsmoker, Chol 220, HDL 41 | 14% | Yes |
| 51-year-old man, HTN on medication, SBP 140, nonsmoker, Chol 180, HDL 41, Diabetes | 6% | No† |
| *Score for 10-year risk of CHD calculated with Framingham Heart Study data.14 †Under 2009 USPSTF recommendations, may consider aspirin therapy.10 Chol, total cholesterol; HDL, high-density lipoprotein; HTN, hypertension; SBP, systolic blood pressure. | ||
Test for aspirin resistance? It’s still too soon
Patients receiving aspirin therapy may demonstrate residual platelet reactivity (laboratory resistance) or recurrent ischemic CV events (clinical resistance).21 Estimates of the prevalence of aspirin resistance vary widely.22 And available assays of residual platelet activity yield different results. Higher estimates of aspirin resistance may occur with assays that use an agonist other than arachidonic acid, such as collagen or adenosine diphosphate platelet aggregation, the whole blood platelet function analyzer (PFA-100), or urinary 11-dehydro-thromboxane B2.23
Several secondary prevention studies have demonstrated a positive association with laboratory resistance and adverse CV events, regardless of methods and assays used.24 However, prospective primary prevention studies of this association are lacking. A meta-analysis of 20 clinical studies reported an increased risk of recurrent CV events including graft failure, acute coronary syndrome (ACS), and death among patients who exhibited aspirin resistance (odds ratio [OR]=3.85; 95% CI, 3.08-4.80). The authors identified a high level of heterogeneity among the studies, with 9 of the 20 failing to demonstrate an increased risk of events.25
Using the PFA-100 assay, a prospective cohort study verified the presence or absence of aspirin resistance in 140 patients who presented to the emergency department with a non-ST–elevation ACS and who reported using aspirin daily for at least 7 days before the event.26 Fifty-three patients (37.8%) were found to have aspirin resistance. Baseline characteristics of patients with and without aspirin resistance were similar except for an older age (mean 63.8 vs 58.3 years, respectively) and higher cardiac troponin values (mean 1.11 vs 0.41 ng/mL). Both groups were monitored for an average of 20 months; 45 patients with aspirin resistance and 79 without resistance completed follow-up. The presence of aspirin resistance increased the risk of MI (HR=3.02; 95% CI, 1.15-7.95) and decreased the risk of event-free survival (HR=2.46; 95% CI, 1.18-5.13). Adjusted for age, platelet count, cardiac troponin values, and coronary artery disease severity scores, the presence of aspirin resistance was associated with a 3-fold increased risk of CV events (HR=3.03; 95% CI, 1.06-8.62).
Mechanisms for aspirin resistance may involve an inability of aspirin to partially or completely inhibit the cyclo-oxygenase-1 (COX-1) enzyme leading to thromboxane A2 production, or factors independent of the COX-1 pathway such as elevated levels of C-reactive protein.27 COX-1-related factors include aspirin nonadherence, reduced aspirin bioavailability, competitive inhibition by NSAIDs, inadequate aspirin dosage, genetic COX-1 polymorphisms, and increased platelet turnover.27,28 A subgroup analysis of the Physicians’ Health Study29 suggests that nonadherence with aspirin therapy or concomitant NSAID use negated the benefit of aspirin. In a small cohort study (n=18), patients who took ibuprofen or naproxen and aspirin did not demonstrate inhibition of platelet aggregation and had a 72% rate of recurrent ischemic events despite aspirin therapy.30
Until clinical trials can demonstrate benefit and cost effectiveness of empiric laboratory testing for aspirin resistance in patients without a history of CVD, emphasize adherence to the prescribed antiplatelet therapy and warn against concomitant NSAID use for patients at risk for CHD events.
Aspirin for patients with diabetes: Only when CVD risk is high
In 2010, the ADA revised its clinical practice recommendations to reflect the results of 2 studies that questioned the value of aspirin for primary prevention of CVD events in patients with diabetes.3 Instead of a global statement to use low-dose aspirin, the ADA guideline now recommends its use only in patients with diabetes who have a 10-year risk >10%. This includes men over the age of 50 and women over the age of 60, with at least one major risk factor in addition to diabetes. The studies driving this change were the Prevention of Progression of Arterial Disease and Diabetes (POPADAD)31 and the Japanese Primary Prevention of Atherosclerosis with Aspirin for Diabetes (JPAD).32
The POPADAD study enrolled 1276 patients over the age of 40 with type 1 or type 2 diabetes who also had asymptomatic peripheral arterial disease but without symptomatic CHD. Participants were randomized to take aspirin 100 mg daily or placebo (POPADAD also included a study of antioxidants vs placebo). The participants had diabetes for a mean of 6.3 years. The study had 2 primary composite end points: death from CHD or stroke, nonfatal MI or stroke, or amputation above the ankle for critical ischemia; and death from CHD or stroke. The aspirin and placebo groups were similar at baseline in terms of demographic characteristics and use of statins, beta-blockers, and angiotensin-converting enzyme (ACE) inhibitors among other treatments. The composite end point of death from CHD or stroke was similar in the 2 groups. Nonfatal MI and nonfatal stroke were also similar in the 2 groups.31
The JPAD study enrolled individuals with type 2 diabetes who were over the age of 30 and had no evidence of CVD. Participants were randomized to receive either 81 mg aspirin or placebo daily. The composite end point was sudden death; death from CHD, stroke, or aortic causes; nonfatal MI; nonfatal stroke; unstable angina; transient ischemic attack; or nonfatal peripheral vascular disease. The 2 groups were similar in terms of the composite end point, nonfatal MI, and nonfatal stroke. The risk of death from MI and stroke was lower in the aspirin group.32
The authors of a 2010 consensus report from the ADA, the AHA, and the American College of Cardiology (ACC) evaluated the findings of individual placebo-controlled aspirin studies as well as those included in prior meta-analyses.33 They also conducted a separate meta-analysis, which indicated that aspirin decreased the risk of CHD in patients with diabetes by 9% (RR=0.91; 95% CI, 0.79-1.05), but the reduction was not statistically significant. If the findings of the Early Treatment of Diabetic Retinopathy Study, which included some individuals with prior CVD events, had been excluded from this meta-analysis, the risk reduction due to aspirin would have been smaller.
Results of this meta-analysis are mitigated by certain factors. The 9 studies analyzed were published between 1989 and 2008, and the use of drugs such as statins, beta-blockers, and ACE inhibitors increased over this 20-year period. Also, the age of study participants at enrollment varied, as did the presence of subclinical CVD. The rates of CHD in the placebo groups of the studies also varied significantly.
Accounting for differences between the sexes
A person’s sex in part determines the importance of certain CV risk factors, the prevalence of CV and related comorbid diseases, and the frequency of adverse drug effects. Women with diabetes have a 50% increased relative risk of CVD than men with diabetes, in part because they are often older and have more risk factors.34
A 2011 AHA update on the prevention of CVD in women indicates that women ≥65 years may use aspirin, 81 mg daily or 100 mg every other day, if the benefit in reducing CHD or ischemic stroke is not outweighed by the potential risk of GI bleeding or hemorrhagic stroke. It also deems aspirin an option that women younger than 65 could consider with their physicians for prevention of ischemic stroke, and recommends aspirin 75 to 325 mg daily for women with diabetes.35
The study populations in the ADA/AHA/ACC meta-analysis of aspirin for primary prevention in patients with diabetes varied in the percentage of women enrolled. Three trials did not include women, while one study enrolled women exclusively. The remaining studies had similar numbers of men and women. Aspirin decreased the risk of CHD events in men (RR=0.77; 95% CI, 0.67-0.89) and stroke in women (RR=0.77; 95% CI, 0.59-0.99). The consensus report acknowledged that the findings of the Women’s Health Study strongly influenced this difference in outcomes for men and women.33
CORRESPONDENCE
Anita N. Jackson, PharmD, University of Rhode Island, College of Pharmacy, 41 Lower College Road, Fogarty Hall, Kingston, RI 02881; anitaj@uri.edu
• Calculate a patient’s 10-year global risk of cardiovascular events using a risk-assessment tool before recommending aspirin for primary prevention. A
• Keep in mind that diabetes is not an indication for aspirin as primary cardiovascular protection, unless the patient’s calculated 10-year risk is >10%. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Among individuals at high risk (≥10%) for coronary heart disease (CHD) within 10 years, only 44% are taking aspirin.1 In addition, for patients at high risk for CHD events, estimated aspirin use varies among ethnic groups: 53% for whites, 43% for African Americans, 38% for Hispanics, and 28% for Chinese Americans.1
In contrast to this underuse of aspirin by those who need it, patients who do not need aspirin have been told otherwise,2 following widespread publicity of US Preventive Services Task Force (USPSTF) recommendations from 2002 (that have since been updated). Overuse of aspirin is also likely among individuals whose CHD risk has never been formally assessed but who take it on their own, based on direct-to-consumer advertising about the cardiovascular (CV) benefits of aspirin. Also, the American Diabetes Association (ADA) once recommended aspirin for all patients with diabetes. But it now advises avoiding the use of aspirin for primary prevention of CV events unless a patient’s calculated CV risk over 10 years is >10%.3
Our review summarizes the latest evidence on the use of aspirin for primary prevention of CV events, including the determination of benefit vs harm, the variability in aspirin responsiveness among individuals, and the efficacy of aspirin treatment in men vs women and in those with diabetes.
When does benefit outweigh risk?
In 2002, the USPSTF concluded that patients with a 5-year risk of coronary events ≥3% had the most favorable benefit-to-risk ratio with aspirin use.4 It based its recommendation on 5 randomized, controlled primary prevention studies with aspirin that demonstrated a reduction in the risk of a first myocardial infarction (MI) in men.5-9 In 2009, the USPSTF updated its recommendations regarding the risks and benefits of aspirin for primary prevention of CHD,10 in part to include data from the Women’s Health Study11 that demonstrated a 24% relative risk (RR) reduction of ischemic stroke without reducing the risk of MI.
The USPSTF now recommends aspirin for men ages 45 to 79 to prevent a first MI, and for women ages 55 to 79 to prevent an ischemic stroke when the potential benefit outweighs the increased risk of gastrointestinal (GI) hemorrhage.10 Evidence does not support the use of aspirin for primary CHD prevention in men younger than 45 years or women younger than 55. Evidence is insufficient to recommend aspirin for primary prevention of CHD for individuals ≥80 years of age in the absence of other compelling indications such as atrial fibrillation.
Calculating benefit. The American Heart Association (AHA) recommends low-dose aspirin for primary prevention of CV events in all individuals with a calculated 10-year CHD risk of ≥10%, while cautioning about its use in patients at increased risk for GI bleeding and hemorrhagic stroke.12 The Framingham risk score13 is available online at http://hp2010.nhlbihin.net/atpiii/calculator.asp?usertype=prof to estimate an individual’s 10-year CHD risk (TABLE).14
Judging risk. There are no validated tools for assessing the long-term risk of intracranial or GI hemorrhage with low-dose aspirin. The risk factors for GI bleeding with nonsteroidal anti-inflammatory drugs (NSAIDs) are well known,15 but less data exist for low-dose aspirin. Likely risk factors include a history of peptic ulcer disease, concomitant NSAID therapy, high-dose corticosteroids or anticoagulants, dual antiplatelet therapy, age >60 years, and male sex.16 Although proton-pump inhibitors prevent recurrent peptic ulcers secondary to low-dose aspirin use, little data exist on their value or cost effectiveness for this purpose.17
Why the AHA recommendation makes sense. The 2009 USPSTF recommendations still identify different tiers of risk according to 3 age brackets within the range of 45 (or 55) to 79 years. Since then, however, further studies seem to favor a less aggressive approach to aspirin use, more in keeping with the AHA recommendation.
The Antithrombotic Trialists’ (ATT) Collaboration18 published a meta-analysis using individual participant data from the same studies that served as the basis of the USPSTF recommendations.5-9,11 It found that aspirin did not reduce the risk of death due to CHD, stroke, or other vascular causes. The risk of nonfatal stroke also did not decline. Aspirin use decreased the risk of nonfatal MI (RR=0.77; 99% confidence interval [CI], 0.67-0.89), any major coronary event (RR=0.82; 95% CI, 0.75-0.90), and serious vascular events (RR=0.88; 95% CI, 0.82-0.94). The risk of extracranial hemorrhage, including GI bleeding, increased (RR=1.54; 95% CI, 1.30-1.82). Based on this analysis, the absolute reduction in serious ischemic events was partially offset by a small increase in serious bleeding. However, long-term disability from a nonfatal extracranial hemorrhage is likely less than that from a nonfatal stroke or MI.18
In the ATT Collaboration18 analysis, the 5-year risk of bleeding with low-dose aspirin increased with the predicted 5-year CHD risk. Patients with the lowest CHD risk (<5%) demonstrated a 0.4% risk of bleeding vs 2.7% among patients having the highest CHD risk (>10%). However, the high-risk patients also had the largest benefit with low-dose aspirin therapy. According to the ATT Collaboration data, using aspirin alone vs placebo, the estimated number needed to treat (NNT) to prevent 1 serious vascular event (defined as vascular death, nonfatal MI, or stroke) was 50 patients for 5 years. When aspirin was added to other therapies such as statins, the NNT was 100 patients for 5 years. To cause 1 nonfatal extracranial bleeding event with aspirin in the same high-risk patients, the estimated number needed to harm (NNH) was also 100 patients for 5 years. A meta-analysis of 22 trials estimated a NNH to cause 1 additional major bleeding event with aspirin per year was 769 patients (95% CI, 500-1250).19
The Aspirin for Asymptomatic Atherosclerosis Trial (AAAT)20 involved 3350 men and women ages 50 to 75 years with low ankle-brachial index and no history of CV disease (CVD). Participants were randomized to receive 100 mg enteric-coated aspirin or placebo daily and were followed for a mean of 8.2 years. The primary and secondary end points, which included fatal and nonfatal MI or stroke, were similar in the 2 groups, as were all-cause mortality and total adverse events. A difference in the incidence of major hemorrhage did not reach statistical significance—34 patients in the aspirin arm vs 20 in the placebo arm (hazard ratio [HR]=1.71; 95% CI, 0.99-2.97). One caution: the relative lack of benefit from aspirin reported in the AAAT may be due to the fact that it was powered to detect a 25% reduction in the event rate between groups, whereas the ATT Collaboration study18 found a 12% risk reduction in MI among those taking aspirin.
TABLE
Should you recommend aspirin? See how these patients “scored”*
| Patient | Risk score | Prophylaxis |
|---|---|---|
| 53-year-old woman, HTN on medication, SBP 152, nonsmoker, Chol 260, HDL 50 | 4% | No |
| 48-year-old man, HTN on medication, SBP 138, nonsmoker, Chol 220, HDL 41 | 7% | No† |
| 68-year-old woman, HTN on medication, SBP 152, nonsmoker, Chol 260, HDL 50 | 11% | Yes |
| 58-year-old man, HTN on medication, SBP 138, nonsmoker, Chol 220, HDL 41 | 14% | Yes |
| 51-year-old man, HTN on medication, SBP 140, nonsmoker, Chol 180, HDL 41, Diabetes | 6% | No† |
| *Score for 10-year risk of CHD calculated with Framingham Heart Study data.14 †Under 2009 USPSTF recommendations, may consider aspirin therapy.10 Chol, total cholesterol; HDL, high-density lipoprotein; HTN, hypertension; SBP, systolic blood pressure. | ||
Test for aspirin resistance? It’s still too soon
Patients receiving aspirin therapy may demonstrate residual platelet reactivity (laboratory resistance) or recurrent ischemic CV events (clinical resistance).21 Estimates of the prevalence of aspirin resistance vary widely.22 And available assays of residual platelet activity yield different results. Higher estimates of aspirin resistance may occur with assays that use an agonist other than arachidonic acid, such as collagen or adenosine diphosphate platelet aggregation, the whole blood platelet function analyzer (PFA-100), or urinary 11-dehydro-thromboxane B2.23
Several secondary prevention studies have demonstrated a positive association with laboratory resistance and adverse CV events, regardless of methods and assays used.24 However, prospective primary prevention studies of this association are lacking. A meta-analysis of 20 clinical studies reported an increased risk of recurrent CV events including graft failure, acute coronary syndrome (ACS), and death among patients who exhibited aspirin resistance (odds ratio [OR]=3.85; 95% CI, 3.08-4.80). The authors identified a high level of heterogeneity among the studies, with 9 of the 20 failing to demonstrate an increased risk of events.25
Using the PFA-100 assay, a prospective cohort study verified the presence or absence of aspirin resistance in 140 patients who presented to the emergency department with a non-ST–elevation ACS and who reported using aspirin daily for at least 7 days before the event.26 Fifty-three patients (37.8%) were found to have aspirin resistance. Baseline characteristics of patients with and without aspirin resistance were similar except for an older age (mean 63.8 vs 58.3 years, respectively) and higher cardiac troponin values (mean 1.11 vs 0.41 ng/mL). Both groups were monitored for an average of 20 months; 45 patients with aspirin resistance and 79 without resistance completed follow-up. The presence of aspirin resistance increased the risk of MI (HR=3.02; 95% CI, 1.15-7.95) and decreased the risk of event-free survival (HR=2.46; 95% CI, 1.18-5.13). Adjusted for age, platelet count, cardiac troponin values, and coronary artery disease severity scores, the presence of aspirin resistance was associated with a 3-fold increased risk of CV events (HR=3.03; 95% CI, 1.06-8.62).
Mechanisms for aspirin resistance may involve an inability of aspirin to partially or completely inhibit the cyclo-oxygenase-1 (COX-1) enzyme leading to thromboxane A2 production, or factors independent of the COX-1 pathway such as elevated levels of C-reactive protein.27 COX-1-related factors include aspirin nonadherence, reduced aspirin bioavailability, competitive inhibition by NSAIDs, inadequate aspirin dosage, genetic COX-1 polymorphisms, and increased platelet turnover.27,28 A subgroup analysis of the Physicians’ Health Study29 suggests that nonadherence with aspirin therapy or concomitant NSAID use negated the benefit of aspirin. In a small cohort study (n=18), patients who took ibuprofen or naproxen and aspirin did not demonstrate inhibition of platelet aggregation and had a 72% rate of recurrent ischemic events despite aspirin therapy.30
Until clinical trials can demonstrate benefit and cost effectiveness of empiric laboratory testing for aspirin resistance in patients without a history of CVD, emphasize adherence to the prescribed antiplatelet therapy and warn against concomitant NSAID use for patients at risk for CHD events.
Aspirin for patients with diabetes: Only when CVD risk is high
In 2010, the ADA revised its clinical practice recommendations to reflect the results of 2 studies that questioned the value of aspirin for primary prevention of CVD events in patients with diabetes.3 Instead of a global statement to use low-dose aspirin, the ADA guideline now recommends its use only in patients with diabetes who have a 10-year risk >10%. This includes men over the age of 50 and women over the age of 60, with at least one major risk factor in addition to diabetes. The studies driving this change were the Prevention of Progression of Arterial Disease and Diabetes (POPADAD)31 and the Japanese Primary Prevention of Atherosclerosis with Aspirin for Diabetes (JPAD).32
The POPADAD study enrolled 1276 patients over the age of 40 with type 1 or type 2 diabetes who also had asymptomatic peripheral arterial disease but without symptomatic CHD. Participants were randomized to take aspirin 100 mg daily or placebo (POPADAD also included a study of antioxidants vs placebo). The participants had diabetes for a mean of 6.3 years. The study had 2 primary composite end points: death from CHD or stroke, nonfatal MI or stroke, or amputation above the ankle for critical ischemia; and death from CHD or stroke. The aspirin and placebo groups were similar at baseline in terms of demographic characteristics and use of statins, beta-blockers, and angiotensin-converting enzyme (ACE) inhibitors among other treatments. The composite end point of death from CHD or stroke was similar in the 2 groups. Nonfatal MI and nonfatal stroke were also similar in the 2 groups.31
The JPAD study enrolled individuals with type 2 diabetes who were over the age of 30 and had no evidence of CVD. Participants were randomized to receive either 81 mg aspirin or placebo daily. The composite end point was sudden death; death from CHD, stroke, or aortic causes; nonfatal MI; nonfatal stroke; unstable angina; transient ischemic attack; or nonfatal peripheral vascular disease. The 2 groups were similar in terms of the composite end point, nonfatal MI, and nonfatal stroke. The risk of death from MI and stroke was lower in the aspirin group.32
The authors of a 2010 consensus report from the ADA, the AHA, and the American College of Cardiology (ACC) evaluated the findings of individual placebo-controlled aspirin studies as well as those included in prior meta-analyses.33 They also conducted a separate meta-analysis, which indicated that aspirin decreased the risk of CHD in patients with diabetes by 9% (RR=0.91; 95% CI, 0.79-1.05), but the reduction was not statistically significant. If the findings of the Early Treatment of Diabetic Retinopathy Study, which included some individuals with prior CVD events, had been excluded from this meta-analysis, the risk reduction due to aspirin would have been smaller.
Results of this meta-analysis are mitigated by certain factors. The 9 studies analyzed were published between 1989 and 2008, and the use of drugs such as statins, beta-blockers, and ACE inhibitors increased over this 20-year period. Also, the age of study participants at enrollment varied, as did the presence of subclinical CVD. The rates of CHD in the placebo groups of the studies also varied significantly.
Accounting for differences between the sexes
A person’s sex in part determines the importance of certain CV risk factors, the prevalence of CV and related comorbid diseases, and the frequency of adverse drug effects. Women with diabetes have a 50% increased relative risk of CVD than men with diabetes, in part because they are often older and have more risk factors.34
A 2011 AHA update on the prevention of CVD in women indicates that women ≥65 years may use aspirin, 81 mg daily or 100 mg every other day, if the benefit in reducing CHD or ischemic stroke is not outweighed by the potential risk of GI bleeding or hemorrhagic stroke. It also deems aspirin an option that women younger than 65 could consider with their physicians for prevention of ischemic stroke, and recommends aspirin 75 to 325 mg daily for women with diabetes.35
The study populations in the ADA/AHA/ACC meta-analysis of aspirin for primary prevention in patients with diabetes varied in the percentage of women enrolled. Three trials did not include women, while one study enrolled women exclusively. The remaining studies had similar numbers of men and women. Aspirin decreased the risk of CHD events in men (RR=0.77; 95% CI, 0.67-0.89) and stroke in women (RR=0.77; 95% CI, 0.59-0.99). The consensus report acknowledged that the findings of the Women’s Health Study strongly influenced this difference in outcomes for men and women.33
CORRESPONDENCE
Anita N. Jackson, PharmD, University of Rhode Island, College of Pharmacy, 41 Lower College Road, Fogarty Hall, Kingston, RI 02881; anitaj@uri.edu
1. Sanchez DR, Diez Roux AV, Michos ED, et al. Comparison of the racial/ethnic prevalence of regular aspirin use for the primary prevention of coronary heart disease from the multi-ethnic study of atherosclerosis. Am J Cardiol. 2011;107:41-46.
2. Dwivedi G, Ball MC, Dilworth MP, et al. Use and misuse of aspirin in the hypertension clinic [letter]. BMJ. May 3, 2010. Available at: http://www.bmj.com/content/340/bmj.c1805.full/reply#bmj_el_235118. Accessed October 15, 2010.
3. American Diabetes Association. Standards of medical care in diabetes–2010. Diabetes Care. 2010;33(suppl 1):S11-S61.
4. US Preventive Services Task Force. Aspirin for the primary prevention of cardiovascular events: recommendation and rationale. Ann Intern Med. 2002;136:157-160.
5. Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. The Medical Research Council’s General Practice Research Framework. Lancet. 1998;351:233-241.
6. Peto R, Gray R, Collins R. Randomised trial of prophylactic daily aspirin in British male doctors. BMJ. 1988;296:313-316.
7. Final report on the aspirin component of the ongoing Physicians’ Health Study. Steering Committee of the Physicians’ Health Study Research Group. N Engl J Med. 1989;321:129-135.
8. Hansson L, Zanchetti A, Carruthers SG. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. Lancet. 1998;351:1755-1762.
9. de Gaetano G. Collaborative Group of the Primary Prevention Project. Low-dose aspirin and vitamin E in people at cardiovascular risk: a randomised trial in general practice. Lancet. 2001;357:89-95.
10. Wolff T, Miller T, Ko S. Aspirin for the prevention of cardiovascular disease: an update of the evidence for the US Preventive Services Task Force. Ann Intern Med. 2009;150:405-410.
11. Ridker PM, Cook NR, Lee IM. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med. 2005;352:1293-1304.
12. Pearson TA, Blair SN, Daniels SR, et al. AHA guidelines for primary prevention of cardiovascular disease and stroke: 2002 update: consensus panel guide to comprehensive risk reduction for adult patients without coronary or other atherosclerotic vascular diseases. Circulation. 2002;106:388-391.
13. Wilson PW, D’Agostino RB, Levy D, et al. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837-1847.
14. National Cholesterol Education Program. Risk assessment tool for estimating 10-year risk of developing hard CHD. Available at: http://hp2010.nhlbihin.net/atpiii/calculator.asp?usertype=prof. Accessed October 15, 2010.
15. Bhatt DL, Scheiman J, Abraham NS, et al. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use. Circulation. 2008;118:1894-1909.
16. Garcia Rodriguez LA, Lin KJ, Hernandez-Diaz S, et al. Risk of upper gastrointestinal bleeding with low-dose acetylsalicylic acid alone and in combination with clopidogrel and other medications. Circulation. 2011;123:1108-1115.
17. Lai KC, Lam SK, Chu Km, et al. Lansoprazole for the prevention of recurrences of ulcer complications from long-term low-dose aspirin use. N Engl J Med. 2002;346:2033-2038.
18. Antithrombotic Trialists’ (ATT) Collaboration. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373:1849-1860.
19. McQuaid KR, Laine L. Systematic review and meta-analysis of adverse events of low-dose aspirin and clopidogrel in randomized controlled trials. Am J Med. 2006;119:624-638.
20. Fowkes FG, Price JF, Stewart MCW, et al. Aspirin for the prevention of cardiovascular events in a general population screened for a low ankle brachial index: a randomized controlled trial. JAMA. 2010;303:841-848.
21. Kuliczkowski W, Witkowski A, Polonski L, et al. Interindividual variability in the response to oral antiplatelet drugs: a position paper of the working group on antiplatelet drugs resistance appointed by the section of cardiovascular interventions of the Polish Cardiac Society, endorsed by the working group on thrombosis of the European Society of Cardiology. Eur Heart J. 2009;30:426-435.
22. Hovens MM, Snoep JD, Eidenboom JC. Prevalence of persistent platelet reactivity despite use of aspirin: a systematic review. Am Heart J. 2007;153:175-181.
23. Gurbel PA, Bliden KP, DiChiara JD, et al. Evaluation of dose-related effects of aspirin on platelet function: results from the Aspirin-Induced Platelet Effect (ASPECT) study. Circulation. 2007;115:3156-3164.
24. Feher G, Geher A, Pusch G, et al. Clinical importance of aspirin and clopidogrel resistance. World J Cardiol. 2010;2:171-186.
25. Krasopoulos G, Brister SJ, Beattie WS, et al. Aspirin “resistance” and risk of cardiovascular morbidity: systematic review and meta-analysis. BMJ. 2008;336:195-198.
26. Hobikoglu GF, Norgaz T, Aksu H, et al. The effect of acetylsalicylic acid resistance on prognosis of patients who have developed acute coronary syndrome during acetylsalicylic acid therapy. Can J Cardiol. 2007;23:201-206.
27. Gasparyan AY, Watson T, Lip GYH. The role of aspirin in cardiovascular prevention: implications of aspirin resistance. J Am Coll Cardiol. 2008;51:1829-1843.
28. Arazi HC, Doiny DG, Torcivia RS, et al. Impaired anti-platelet effect of aspirin, inflammation and platelet turnover in cardiac surgery. Interact Cardiovasc Thorac Surg. 2010;10:863-867.
29. Hennekens CH, Schneider WR, Hebert PR, et al. Hypothesis formulation from subgroup analyses: nonadherence or nonsteroidal anti-inflammatory drug use explains the lack of clinical benefit of aspirin on first myocardial infarction attributed to “aspirin resistance.” Am Heart J. 2010;159:744-748.
30. Gengo FM, Rubin L, Robson M, et al. Effects of ibuprofen on the magnitude and duration of aspirin’s inhibition of platelet aggregation; clinical consequences in stroke prophylaxis. J Clin Pharmacol. 2008;48:117-122.
31. Belch J, MacCuish A, Campbell I, et al. The prevention of progression of arterial disease and diabetes (POPADAD) trial: factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ. 2008;337:a1840.-
32. Ogawa H, Nakayama M, Morimoto T, et al. Low dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: a randomized controlled trial. JAMA. 2008;300:2134-2141.
33. Pignone M, Alberts MJ, Colwell JA, et al. Aspirin for primary prevention of cardiovascular events in people with diabetes. A position statement of the American Diabetes Association, a scientific statement of the American Heart Association, and an expert consensus document of the American College of Cardiology Foundation. Circulation. 2010;121:2694-2701.
34. Barrett-Connor E, Giardina EG, Gitt AK, et al. Women and heart disease: the role of diabetes and hyperglycemia. Arch Intern Med. 2004;164:934-942.
35. Mosca L, Benjamin EJ, Berra K, et al. Effectiveness-based guidelines for the prevention of cardiovascular disease in women—2011 update. Circulation. 2011;123:1243-1262.
1. Sanchez DR, Diez Roux AV, Michos ED, et al. Comparison of the racial/ethnic prevalence of regular aspirin use for the primary prevention of coronary heart disease from the multi-ethnic study of atherosclerosis. Am J Cardiol. 2011;107:41-46.
2. Dwivedi G, Ball MC, Dilworth MP, et al. Use and misuse of aspirin in the hypertension clinic [letter]. BMJ. May 3, 2010. Available at: http://www.bmj.com/content/340/bmj.c1805.full/reply#bmj_el_235118. Accessed October 15, 2010.
3. American Diabetes Association. Standards of medical care in diabetes–2010. Diabetes Care. 2010;33(suppl 1):S11-S61.
4. US Preventive Services Task Force. Aspirin for the primary prevention of cardiovascular events: recommendation and rationale. Ann Intern Med. 2002;136:157-160.
5. Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. The Medical Research Council’s General Practice Research Framework. Lancet. 1998;351:233-241.
6. Peto R, Gray R, Collins R. Randomised trial of prophylactic daily aspirin in British male doctors. BMJ. 1988;296:313-316.
7. Final report on the aspirin component of the ongoing Physicians’ Health Study. Steering Committee of the Physicians’ Health Study Research Group. N Engl J Med. 1989;321:129-135.
8. Hansson L, Zanchetti A, Carruthers SG. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. Lancet. 1998;351:1755-1762.
9. de Gaetano G. Collaborative Group of the Primary Prevention Project. Low-dose aspirin and vitamin E in people at cardiovascular risk: a randomised trial in general practice. Lancet. 2001;357:89-95.
10. Wolff T, Miller T, Ko S. Aspirin for the prevention of cardiovascular disease: an update of the evidence for the US Preventive Services Task Force. Ann Intern Med. 2009;150:405-410.
11. Ridker PM, Cook NR, Lee IM. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med. 2005;352:1293-1304.
12. Pearson TA, Blair SN, Daniels SR, et al. AHA guidelines for primary prevention of cardiovascular disease and stroke: 2002 update: consensus panel guide to comprehensive risk reduction for adult patients without coronary or other atherosclerotic vascular diseases. Circulation. 2002;106:388-391.
13. Wilson PW, D’Agostino RB, Levy D, et al. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837-1847.
14. National Cholesterol Education Program. Risk assessment tool for estimating 10-year risk of developing hard CHD. Available at: http://hp2010.nhlbihin.net/atpiii/calculator.asp?usertype=prof. Accessed October 15, 2010.
15. Bhatt DL, Scheiman J, Abraham NS, et al. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use. Circulation. 2008;118:1894-1909.
16. Garcia Rodriguez LA, Lin KJ, Hernandez-Diaz S, et al. Risk of upper gastrointestinal bleeding with low-dose acetylsalicylic acid alone and in combination with clopidogrel and other medications. Circulation. 2011;123:1108-1115.
17. Lai KC, Lam SK, Chu Km, et al. Lansoprazole for the prevention of recurrences of ulcer complications from long-term low-dose aspirin use. N Engl J Med. 2002;346:2033-2038.
18. Antithrombotic Trialists’ (ATT) Collaboration. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373:1849-1860.
19. McQuaid KR, Laine L. Systematic review and meta-analysis of adverse events of low-dose aspirin and clopidogrel in randomized controlled trials. Am J Med. 2006;119:624-638.
20. Fowkes FG, Price JF, Stewart MCW, et al. Aspirin for the prevention of cardiovascular events in a general population screened for a low ankle brachial index: a randomized controlled trial. JAMA. 2010;303:841-848.
21. Kuliczkowski W, Witkowski A, Polonski L, et al. Interindividual variability in the response to oral antiplatelet drugs: a position paper of the working group on antiplatelet drugs resistance appointed by the section of cardiovascular interventions of the Polish Cardiac Society, endorsed by the working group on thrombosis of the European Society of Cardiology. Eur Heart J. 2009;30:426-435.
22. Hovens MM, Snoep JD, Eidenboom JC. Prevalence of persistent platelet reactivity despite use of aspirin: a systematic review. Am Heart J. 2007;153:175-181.
23. Gurbel PA, Bliden KP, DiChiara JD, et al. Evaluation of dose-related effects of aspirin on platelet function: results from the Aspirin-Induced Platelet Effect (ASPECT) study. Circulation. 2007;115:3156-3164.
24. Feher G, Geher A, Pusch G, et al. Clinical importance of aspirin and clopidogrel resistance. World J Cardiol. 2010;2:171-186.
25. Krasopoulos G, Brister SJ, Beattie WS, et al. Aspirin “resistance” and risk of cardiovascular morbidity: systematic review and meta-analysis. BMJ. 2008;336:195-198.
26. Hobikoglu GF, Norgaz T, Aksu H, et al. The effect of acetylsalicylic acid resistance on prognosis of patients who have developed acute coronary syndrome during acetylsalicylic acid therapy. Can J Cardiol. 2007;23:201-206.
27. Gasparyan AY, Watson T, Lip GYH. The role of aspirin in cardiovascular prevention: implications of aspirin resistance. J Am Coll Cardiol. 2008;51:1829-1843.
28. Arazi HC, Doiny DG, Torcivia RS, et al. Impaired anti-platelet effect of aspirin, inflammation and platelet turnover in cardiac surgery. Interact Cardiovasc Thorac Surg. 2010;10:863-867.
29. Hennekens CH, Schneider WR, Hebert PR, et al. Hypothesis formulation from subgroup analyses: nonadherence or nonsteroidal anti-inflammatory drug use explains the lack of clinical benefit of aspirin on first myocardial infarction attributed to “aspirin resistance.” Am Heart J. 2010;159:744-748.
30. Gengo FM, Rubin L, Robson M, et al. Effects of ibuprofen on the magnitude and duration of aspirin’s inhibition of platelet aggregation; clinical consequences in stroke prophylaxis. J Clin Pharmacol. 2008;48:117-122.
31. Belch J, MacCuish A, Campbell I, et al. The prevention of progression of arterial disease and diabetes (POPADAD) trial: factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ. 2008;337:a1840.-
32. Ogawa H, Nakayama M, Morimoto T, et al. Low dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: a randomized controlled trial. JAMA. 2008;300:2134-2141.
33. Pignone M, Alberts MJ, Colwell JA, et al. Aspirin for primary prevention of cardiovascular events in people with diabetes. A position statement of the American Diabetes Association, a scientific statement of the American Heart Association, and an expert consensus document of the American College of Cardiology Foundation. Circulation. 2010;121:2694-2701.
34. Barrett-Connor E, Giardina EG, Gitt AK, et al. Women and heart disease: the role of diabetes and hyperglycemia. Arch Intern Med. 2004;164:934-942.
35. Mosca L, Benjamin EJ, Berra K, et al. Effectiveness-based guidelines for the prevention of cardiovascular disease in women—2011 update. Circulation. 2011;123:1243-1262.
A stroke—or something else?
A 54-year-old white woman with a history of a cerebrovascular accident (CVA) a year earlier sought care at the local emergency department for numbness and weakness in her right foot. She reported no other neurologic symptoms. She had mild weakness in her right leg and a mildly unsteady gait. Her neurologic examination was otherwise normal.
Initial testing included a complete blood count (CBC), renal profile, and thyroid-stimulating hormone measurement. All results were normal. A noncontrast computed tomography (CT) scan of the head was normal. We admitted her for further evaluation of probable acute ischemic stroke.
By the following day, the patient’s leg weakness and unsteadiness had worsened. A magnetic resonance imaging (MRI) scan of her head showed a prior left pontine infarct, but no new findings. She developed right arm weakness, and an MRI scan of her spine (FIGURE 1) showed multiple intradural lesions. A lumbar puncture showed elevated protein and oligoclonal bands. CT scans of the chest, abdomen, and pelvis were unremarkable. Two lumbar punctures for cytology and culture evaluations yielded negative results. A full-body positron-emission tomography (PET) scan showed diffuse small inguinal adenopathy bilaterally, suggestive of metastatic disease or lymphoma.
FIGURE 1
MRI of the spine
This MRI scan with contrast of the patient’s spine shows diffuse thoracic extramedullary, intradural lesions.
What is your presumptive diagnosis?
Diagnosis: Sarcoidosis
Findings from the full-body PET scan (FIGURE 2) prompted a biopsy of a right inguinal node, which showed a noncaseating granuloma—a hallmark finding of sarcoidosis.
Sarcoidosis is a multisystem disease of unknown cause. The exact prevalence in the general population is estimated at 10 to 20 cases per 100,000.1 A higher incidence occurs in blacks in the United States, with a 2.4% lifetime risk compared with 0.85% of whites.2 Sarcoidosis usually appears in patients ages 20 to 40 years, and although this systemic disease usually affects the lungs, 5% to 10% of patients will have nervous system involvement.3,4
FIGURE 2
Full-body PET scan
This PET scan shows diffuse hypermetabolic adenopathy with bilateral iliac adenopathy, small hypermetabolic bilateral cervical lymph nodes, a hypermetabolic left axillary node, and a large hypermetabolic portacaval node.
What you’ll see
The most common presenting symptoms of systemic sarcoidosis are chronic cough, shortness of breath, and chest pain. Fatigue, weight loss, and myalgias are also frequently part of the initial presentation.
Patients with sarcoidosis can present with neurologic symptoms suggestive of many diseases (TABLE 1), and in the absence of systemic symptoms the diagnosis of neurosarcoidosis is easily confused with CVA. Most patients with neurosarcoidosis have cranial nerve involvement (50%-75%).1 Other common presentations include seizures, meningitis, psychiatric symptoms, mass lesions, or endocrine abnormalities.
TABLE 1
Differential diagnosis of an acute neurologic event
| Infectious Encephalitis Helminthic infection HIV Lyme disease Meningitis Progressive multifocal leukoencephalopathy Syphilis Tuberculosis |
| Neoplastic CNS lymphoma Meningioma/glioma Metastatic disease |
| Neurologic CNS vasculitis Cranial nerve palsy Ischemic or hemorrhagic stroke Meningitis/encephalitis Multiple sclerosis Neurosarcoidosis Peripheral neuropathy Seizure |
| Psychiatric Depression Malingering Pseudoseizures Somatoform disorder |
| Rheumatologic Lupus erythematosus |
| CNS, central nervous system; HIV, human immunodeficiency virus |
Useful studies in the clinical evaluation
Consider a diagnosis of sarcoidosis involving the nervous system when an initial work-up for CVA is negative. In addition to asking about systemic symptoms, perform a complete neurologic exam and skin exam, search for lymphadenopathy, and conduct an ophthalmologic evaluation. After the initial evaluation, a neurology consult will likely be needed to guide further testing.
Choice of serum studies will vary depending on presenting symptoms, but they usually include tests for infection (CBC, cultures, Lyme titers, rapid plasma reagin, tuberculin skin test), rheumatologic disorders (antinuclear antibodies, erythrocyte sedimentation rate, C-reactive protein), and neoplastic diseases (lactate dehydrogenase, peripheral smear).5 Serum angiotensin-converting enzyme (ACE) may be useful in the diagnosis of systemic sarcoidosis, with positive results seen in approximately 75% of cases.3
Examination of cerebrospinal fluid often reveals an elevated total protein with oligoclonal bands, normal to low glucose, and possibly mild pleocytosis of monocytic or lymphocytic predominance.3 Spinal fluid ACE is neither sensitive nor specific for neurosarcoidosis, as it may be elevated in infectious or malignant processes.3
Imaging studies should include contrast-enhanced brain MRI, which may reveal multiple white matter lesions.6 Although the specificity of PET for neurosarcoidosis is poor—with positive results being seen also in infectious and neoplastic processes—the scan may help in identifying extraneural sites for biopsy. Histology will generally show the classic noncaseating granuloma with surrounding lymphocytes, plasma cells, and mast cells.
Treat with high-dose steroids
The mainstay of treatment, based largely on expert opinion, is high-dose steroids that are gradually tapered over weeks (TABLE 2). Other agents may be added if the condition is poorly controlled with steroids alone, or may be given if symptoms recur while tapering the steroid dose. Recurrence of sarcoidosis is common after doses of <10 to 20 mg/d. Prophylactic measures to counteract the adverse effects of long-term steroid use include weight-bearing exercise programs; administration of calcium, vitamin D, and bisphosphonates; and resorting to a stress-dose steroid regimen in times of illness.
The prognosis with sarcoidosis can vary widely. Case studies show that two-thirds of patients may have a nonrecurring illness. Among the remaining one-third, the disease course may be relapsing-remitting or progressive. When confronted with an acute neurologic event, consider recurrent sarcoidosis and coordinate care between specialists. Also, take steps to prevent complications related to prolonged steroid use.
TABLE 2
Treatment of neurosarcoidosis3
| Medication* | Side effects | Comments |
|---|---|---|
| Methylprednisolone | Hyperglycemia | |
| Prednisone | Osteoporosis, hyperglycemia, hypertension, diabetes, glaucoma, cataracts, psychosis, Cushing’s syndrome | Taper as able. Concomitant use of cytotoxic agents may facilitate taper. Monitor glucose and give calcium/vitamin D prophylaxis |
| Methotrexate | Anemia, neutropenia, liver damage | Weekly dosing well tolerated. Give folic acid 1 mg/d. Monitor liver function tests periodically |
| Cyclosporine | Renal insufficiency, hypertension | |
| Azathioprine | Anemia, neutropenia, liver damage | |
| Cyclophosphamide | Cystitis, neutropenia | Monitor urine monthly for microscopic hematuria |
| Hydroxychloroquine | Retinopathy, hypoglycemia, ototoxicity, myopathy, cardiomyopathy, neuropathy | Refer for eye exams every 3-6 months. May be useful to counteract hyperglycemic effect of steroids |
| Infliximab | Fever, headache, dizziness, flushing, abdominal pain, dyspepsia, myalgia, arthralgia, polyneuropathy | Screen for tuberculosis before starting treatment. Contraindicated in patients with congestive heart failure |
| *For dosing details, consult a neurologist or rheumatologist | ||
Improvement for our patient
Based on cerebrospinal fluid study results, a positive peripheral lymph node biopsy, and the exclusion of other diagnoses, we regarded the diagnosis of sarcoidosis as highly probable and initiated high-dose intravenous corticosteroids. Over several weeks, our patient gradually improved with physical therapy and was walking unassisted at the time of discharge from a hospital-based rehabilitation unit. Repeat MRI scans showed a reduction in the size of her intradural lesions, and we slowly tapered her steroids.
CORRESPONDENCE
Hillary R. Mount, MD, 2123 Auburn Avenue,#340, Cincinnati, OH 45219; hillary.mount@thechristhospital.com
1. Joseph FG, Scolding NJ. Sarcoidosis of the nervous system. Pract Neurol. 2007;7:234-244.
2. Burns TM. Neurosarcoidosis. Arch Neurol. 2003;60:1166-1168.
3. Hoitsma E, Drent M, Sharma OP. A pragmatic approach to diagnosing and treating neurosarcoidosis in the 21st century. Curr Opin Pulm Med. 2010;16:472-479.
4. Habersberger J, Manins V, Taylor AJ. Cardiac sarcoidosis. Intern Med J. 2008;38:270-277.
5. Vargas DL, Stern BJ. Neurosarcoidosis: diagnosis and management. Semin Respir Crit Care Med. 2010;31:419-427.
6. Cavazza A, Harari S, Caminati A, et al. The histology of pulmonary sarcoidosis: a review with particular emphasis on unusual and underrecognized features. Int J Surg Pathol. 2009;17:219-230.
A 54-year-old white woman with a history of a cerebrovascular accident (CVA) a year earlier sought care at the local emergency department for numbness and weakness in her right foot. She reported no other neurologic symptoms. She had mild weakness in her right leg and a mildly unsteady gait. Her neurologic examination was otherwise normal.
Initial testing included a complete blood count (CBC), renal profile, and thyroid-stimulating hormone measurement. All results were normal. A noncontrast computed tomography (CT) scan of the head was normal. We admitted her for further evaluation of probable acute ischemic stroke.
By the following day, the patient’s leg weakness and unsteadiness had worsened. A magnetic resonance imaging (MRI) scan of her head showed a prior left pontine infarct, but no new findings. She developed right arm weakness, and an MRI scan of her spine (FIGURE 1) showed multiple intradural lesions. A lumbar puncture showed elevated protein and oligoclonal bands. CT scans of the chest, abdomen, and pelvis were unremarkable. Two lumbar punctures for cytology and culture evaluations yielded negative results. A full-body positron-emission tomography (PET) scan showed diffuse small inguinal adenopathy bilaterally, suggestive of metastatic disease or lymphoma.
FIGURE 1
MRI of the spine
This MRI scan with contrast of the patient’s spine shows diffuse thoracic extramedullary, intradural lesions.
What is your presumptive diagnosis?
Diagnosis: Sarcoidosis
Findings from the full-body PET scan (FIGURE 2) prompted a biopsy of a right inguinal node, which showed a noncaseating granuloma—a hallmark finding of sarcoidosis.
Sarcoidosis is a multisystem disease of unknown cause. The exact prevalence in the general population is estimated at 10 to 20 cases per 100,000.1 A higher incidence occurs in blacks in the United States, with a 2.4% lifetime risk compared with 0.85% of whites.2 Sarcoidosis usually appears in patients ages 20 to 40 years, and although this systemic disease usually affects the lungs, 5% to 10% of patients will have nervous system involvement.3,4
FIGURE 2
Full-body PET scan
This PET scan shows diffuse hypermetabolic adenopathy with bilateral iliac adenopathy, small hypermetabolic bilateral cervical lymph nodes, a hypermetabolic left axillary node, and a large hypermetabolic portacaval node.
What you’ll see
The most common presenting symptoms of systemic sarcoidosis are chronic cough, shortness of breath, and chest pain. Fatigue, weight loss, and myalgias are also frequently part of the initial presentation.
Patients with sarcoidosis can present with neurologic symptoms suggestive of many diseases (TABLE 1), and in the absence of systemic symptoms the diagnosis of neurosarcoidosis is easily confused with CVA. Most patients with neurosarcoidosis have cranial nerve involvement (50%-75%).1 Other common presentations include seizures, meningitis, psychiatric symptoms, mass lesions, or endocrine abnormalities.
TABLE 1
Differential diagnosis of an acute neurologic event
| Infectious Encephalitis Helminthic infection HIV Lyme disease Meningitis Progressive multifocal leukoencephalopathy Syphilis Tuberculosis |
| Neoplastic CNS lymphoma Meningioma/glioma Metastatic disease |
| Neurologic CNS vasculitis Cranial nerve palsy Ischemic or hemorrhagic stroke Meningitis/encephalitis Multiple sclerosis Neurosarcoidosis Peripheral neuropathy Seizure |
| Psychiatric Depression Malingering Pseudoseizures Somatoform disorder |
| Rheumatologic Lupus erythematosus |
| CNS, central nervous system; HIV, human immunodeficiency virus |
Useful studies in the clinical evaluation
Consider a diagnosis of sarcoidosis involving the nervous system when an initial work-up for CVA is negative. In addition to asking about systemic symptoms, perform a complete neurologic exam and skin exam, search for lymphadenopathy, and conduct an ophthalmologic evaluation. After the initial evaluation, a neurology consult will likely be needed to guide further testing.
Choice of serum studies will vary depending on presenting symptoms, but they usually include tests for infection (CBC, cultures, Lyme titers, rapid plasma reagin, tuberculin skin test), rheumatologic disorders (antinuclear antibodies, erythrocyte sedimentation rate, C-reactive protein), and neoplastic diseases (lactate dehydrogenase, peripheral smear).5 Serum angiotensin-converting enzyme (ACE) may be useful in the diagnosis of systemic sarcoidosis, with positive results seen in approximately 75% of cases.3
Examination of cerebrospinal fluid often reveals an elevated total protein with oligoclonal bands, normal to low glucose, and possibly mild pleocytosis of monocytic or lymphocytic predominance.3 Spinal fluid ACE is neither sensitive nor specific for neurosarcoidosis, as it may be elevated in infectious or malignant processes.3
Imaging studies should include contrast-enhanced brain MRI, which may reveal multiple white matter lesions.6 Although the specificity of PET for neurosarcoidosis is poor—with positive results being seen also in infectious and neoplastic processes—the scan may help in identifying extraneural sites for biopsy. Histology will generally show the classic noncaseating granuloma with surrounding lymphocytes, plasma cells, and mast cells.
Treat with high-dose steroids
The mainstay of treatment, based largely on expert opinion, is high-dose steroids that are gradually tapered over weeks (TABLE 2). Other agents may be added if the condition is poorly controlled with steroids alone, or may be given if symptoms recur while tapering the steroid dose. Recurrence of sarcoidosis is common after doses of <10 to 20 mg/d. Prophylactic measures to counteract the adverse effects of long-term steroid use include weight-bearing exercise programs; administration of calcium, vitamin D, and bisphosphonates; and resorting to a stress-dose steroid regimen in times of illness.
The prognosis with sarcoidosis can vary widely. Case studies show that two-thirds of patients may have a nonrecurring illness. Among the remaining one-third, the disease course may be relapsing-remitting or progressive. When confronted with an acute neurologic event, consider recurrent sarcoidosis and coordinate care between specialists. Also, take steps to prevent complications related to prolonged steroid use.
TABLE 2
Treatment of neurosarcoidosis3
| Medication* | Side effects | Comments |
|---|---|---|
| Methylprednisolone | Hyperglycemia | |
| Prednisone | Osteoporosis, hyperglycemia, hypertension, diabetes, glaucoma, cataracts, psychosis, Cushing’s syndrome | Taper as able. Concomitant use of cytotoxic agents may facilitate taper. Monitor glucose and give calcium/vitamin D prophylaxis |
| Methotrexate | Anemia, neutropenia, liver damage | Weekly dosing well tolerated. Give folic acid 1 mg/d. Monitor liver function tests periodically |
| Cyclosporine | Renal insufficiency, hypertension | |
| Azathioprine | Anemia, neutropenia, liver damage | |
| Cyclophosphamide | Cystitis, neutropenia | Monitor urine monthly for microscopic hematuria |
| Hydroxychloroquine | Retinopathy, hypoglycemia, ototoxicity, myopathy, cardiomyopathy, neuropathy | Refer for eye exams every 3-6 months. May be useful to counteract hyperglycemic effect of steroids |
| Infliximab | Fever, headache, dizziness, flushing, abdominal pain, dyspepsia, myalgia, arthralgia, polyneuropathy | Screen for tuberculosis before starting treatment. Contraindicated in patients with congestive heart failure |
| *For dosing details, consult a neurologist or rheumatologist | ||
Improvement for our patient
Based on cerebrospinal fluid study results, a positive peripheral lymph node biopsy, and the exclusion of other diagnoses, we regarded the diagnosis of sarcoidosis as highly probable and initiated high-dose intravenous corticosteroids. Over several weeks, our patient gradually improved with physical therapy and was walking unassisted at the time of discharge from a hospital-based rehabilitation unit. Repeat MRI scans showed a reduction in the size of her intradural lesions, and we slowly tapered her steroids.
CORRESPONDENCE
Hillary R. Mount, MD, 2123 Auburn Avenue,#340, Cincinnati, OH 45219; hillary.mount@thechristhospital.com
A 54-year-old white woman with a history of a cerebrovascular accident (CVA) a year earlier sought care at the local emergency department for numbness and weakness in her right foot. She reported no other neurologic symptoms. She had mild weakness in her right leg and a mildly unsteady gait. Her neurologic examination was otherwise normal.
Initial testing included a complete blood count (CBC), renal profile, and thyroid-stimulating hormone measurement. All results were normal. A noncontrast computed tomography (CT) scan of the head was normal. We admitted her for further evaluation of probable acute ischemic stroke.
By the following day, the patient’s leg weakness and unsteadiness had worsened. A magnetic resonance imaging (MRI) scan of her head showed a prior left pontine infarct, but no new findings. She developed right arm weakness, and an MRI scan of her spine (FIGURE 1) showed multiple intradural lesions. A lumbar puncture showed elevated protein and oligoclonal bands. CT scans of the chest, abdomen, and pelvis were unremarkable. Two lumbar punctures for cytology and culture evaluations yielded negative results. A full-body positron-emission tomography (PET) scan showed diffuse small inguinal adenopathy bilaterally, suggestive of metastatic disease or lymphoma.
FIGURE 1
MRI of the spine
This MRI scan with contrast of the patient’s spine shows diffuse thoracic extramedullary, intradural lesions.
What is your presumptive diagnosis?
Diagnosis: Sarcoidosis
Findings from the full-body PET scan (FIGURE 2) prompted a biopsy of a right inguinal node, which showed a noncaseating granuloma—a hallmark finding of sarcoidosis.
Sarcoidosis is a multisystem disease of unknown cause. The exact prevalence in the general population is estimated at 10 to 20 cases per 100,000.1 A higher incidence occurs in blacks in the United States, with a 2.4% lifetime risk compared with 0.85% of whites.2 Sarcoidosis usually appears in patients ages 20 to 40 years, and although this systemic disease usually affects the lungs, 5% to 10% of patients will have nervous system involvement.3,4
FIGURE 2
Full-body PET scan
This PET scan shows diffuse hypermetabolic adenopathy with bilateral iliac adenopathy, small hypermetabolic bilateral cervical lymph nodes, a hypermetabolic left axillary node, and a large hypermetabolic portacaval node.
What you’ll see
The most common presenting symptoms of systemic sarcoidosis are chronic cough, shortness of breath, and chest pain. Fatigue, weight loss, and myalgias are also frequently part of the initial presentation.
Patients with sarcoidosis can present with neurologic symptoms suggestive of many diseases (TABLE 1), and in the absence of systemic symptoms the diagnosis of neurosarcoidosis is easily confused with CVA. Most patients with neurosarcoidosis have cranial nerve involvement (50%-75%).1 Other common presentations include seizures, meningitis, psychiatric symptoms, mass lesions, or endocrine abnormalities.
TABLE 1
Differential diagnosis of an acute neurologic event
| Infectious Encephalitis Helminthic infection HIV Lyme disease Meningitis Progressive multifocal leukoencephalopathy Syphilis Tuberculosis |
| Neoplastic CNS lymphoma Meningioma/glioma Metastatic disease |
| Neurologic CNS vasculitis Cranial nerve palsy Ischemic or hemorrhagic stroke Meningitis/encephalitis Multiple sclerosis Neurosarcoidosis Peripheral neuropathy Seizure |
| Psychiatric Depression Malingering Pseudoseizures Somatoform disorder |
| Rheumatologic Lupus erythematosus |
| CNS, central nervous system; HIV, human immunodeficiency virus |
Useful studies in the clinical evaluation
Consider a diagnosis of sarcoidosis involving the nervous system when an initial work-up for CVA is negative. In addition to asking about systemic symptoms, perform a complete neurologic exam and skin exam, search for lymphadenopathy, and conduct an ophthalmologic evaluation. After the initial evaluation, a neurology consult will likely be needed to guide further testing.
Choice of serum studies will vary depending on presenting symptoms, but they usually include tests for infection (CBC, cultures, Lyme titers, rapid plasma reagin, tuberculin skin test), rheumatologic disorders (antinuclear antibodies, erythrocyte sedimentation rate, C-reactive protein), and neoplastic diseases (lactate dehydrogenase, peripheral smear).5 Serum angiotensin-converting enzyme (ACE) may be useful in the diagnosis of systemic sarcoidosis, with positive results seen in approximately 75% of cases.3
Examination of cerebrospinal fluid often reveals an elevated total protein with oligoclonal bands, normal to low glucose, and possibly mild pleocytosis of monocytic or lymphocytic predominance.3 Spinal fluid ACE is neither sensitive nor specific for neurosarcoidosis, as it may be elevated in infectious or malignant processes.3
Imaging studies should include contrast-enhanced brain MRI, which may reveal multiple white matter lesions.6 Although the specificity of PET for neurosarcoidosis is poor—with positive results being seen also in infectious and neoplastic processes—the scan may help in identifying extraneural sites for biopsy. Histology will generally show the classic noncaseating granuloma with surrounding lymphocytes, plasma cells, and mast cells.
Treat with high-dose steroids
The mainstay of treatment, based largely on expert opinion, is high-dose steroids that are gradually tapered over weeks (TABLE 2). Other agents may be added if the condition is poorly controlled with steroids alone, or may be given if symptoms recur while tapering the steroid dose. Recurrence of sarcoidosis is common after doses of <10 to 20 mg/d. Prophylactic measures to counteract the adverse effects of long-term steroid use include weight-bearing exercise programs; administration of calcium, vitamin D, and bisphosphonates; and resorting to a stress-dose steroid regimen in times of illness.
The prognosis with sarcoidosis can vary widely. Case studies show that two-thirds of patients may have a nonrecurring illness. Among the remaining one-third, the disease course may be relapsing-remitting or progressive. When confronted with an acute neurologic event, consider recurrent sarcoidosis and coordinate care between specialists. Also, take steps to prevent complications related to prolonged steroid use.
TABLE 2
Treatment of neurosarcoidosis3
| Medication* | Side effects | Comments |
|---|---|---|
| Methylprednisolone | Hyperglycemia | |
| Prednisone | Osteoporosis, hyperglycemia, hypertension, diabetes, glaucoma, cataracts, psychosis, Cushing’s syndrome | Taper as able. Concomitant use of cytotoxic agents may facilitate taper. Monitor glucose and give calcium/vitamin D prophylaxis |
| Methotrexate | Anemia, neutropenia, liver damage | Weekly dosing well tolerated. Give folic acid 1 mg/d. Monitor liver function tests periodically |
| Cyclosporine | Renal insufficiency, hypertension | |
| Azathioprine | Anemia, neutropenia, liver damage | |
| Cyclophosphamide | Cystitis, neutropenia | Monitor urine monthly for microscopic hematuria |
| Hydroxychloroquine | Retinopathy, hypoglycemia, ototoxicity, myopathy, cardiomyopathy, neuropathy | Refer for eye exams every 3-6 months. May be useful to counteract hyperglycemic effect of steroids |
| Infliximab | Fever, headache, dizziness, flushing, abdominal pain, dyspepsia, myalgia, arthralgia, polyneuropathy | Screen for tuberculosis before starting treatment. Contraindicated in patients with congestive heart failure |
| *For dosing details, consult a neurologist or rheumatologist | ||
Improvement for our patient
Based on cerebrospinal fluid study results, a positive peripheral lymph node biopsy, and the exclusion of other diagnoses, we regarded the diagnosis of sarcoidosis as highly probable and initiated high-dose intravenous corticosteroids. Over several weeks, our patient gradually improved with physical therapy and was walking unassisted at the time of discharge from a hospital-based rehabilitation unit. Repeat MRI scans showed a reduction in the size of her intradural lesions, and we slowly tapered her steroids.
CORRESPONDENCE
Hillary R. Mount, MD, 2123 Auburn Avenue,#340, Cincinnati, OH 45219; hillary.mount@thechristhospital.com
1. Joseph FG, Scolding NJ. Sarcoidosis of the nervous system. Pract Neurol. 2007;7:234-244.
2. Burns TM. Neurosarcoidosis. Arch Neurol. 2003;60:1166-1168.
3. Hoitsma E, Drent M, Sharma OP. A pragmatic approach to diagnosing and treating neurosarcoidosis in the 21st century. Curr Opin Pulm Med. 2010;16:472-479.
4. Habersberger J, Manins V, Taylor AJ. Cardiac sarcoidosis. Intern Med J. 2008;38:270-277.
5. Vargas DL, Stern BJ. Neurosarcoidosis: diagnosis and management. Semin Respir Crit Care Med. 2010;31:419-427.
6. Cavazza A, Harari S, Caminati A, et al. The histology of pulmonary sarcoidosis: a review with particular emphasis on unusual and underrecognized features. Int J Surg Pathol. 2009;17:219-230.
1. Joseph FG, Scolding NJ. Sarcoidosis of the nervous system. Pract Neurol. 2007;7:234-244.
2. Burns TM. Neurosarcoidosis. Arch Neurol. 2003;60:1166-1168.
3. Hoitsma E, Drent M, Sharma OP. A pragmatic approach to diagnosing and treating neurosarcoidosis in the 21st century. Curr Opin Pulm Med. 2010;16:472-479.
4. Habersberger J, Manins V, Taylor AJ. Cardiac sarcoidosis. Intern Med J. 2008;38:270-277.
5. Vargas DL, Stern BJ. Neurosarcoidosis: diagnosis and management. Semin Respir Crit Care Med. 2010;31:419-427.
6. Cavazza A, Harari S, Caminati A, et al. The histology of pulmonary sarcoidosis: a review with particular emphasis on unusual and underrecognized features. Int J Surg Pathol. 2009;17:219-230.
Managing seizures: Achieving control while minimizing risk
• Prescribe an antiepileptic drug (AED) after a first unprovoked seizure only if the seizure was prolonged or there is a risk of recurrence. C
• Use monotherapy whenever possible; if seizures continue and potential adverse effects prevent an increase in dosage, switch to a different AED and taper off the first agent. A
• Consider gradual withdrawal of AEDs from patients who have been seizure-free for 2 to 5 years. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Joe G, a 44-year-old man who has been your patient for years, comes to your office 48 hours after having a seizure. He has no history of seizures, had no warning signs or symptoms, and felt fine all day, but simply collapsed when the seizure occurred. He was transported to the emergency department (ED), and found to be postictal, with no further seizure activity. The ED work-up included a hemogram, comprehensive metabolic panel, and computed tomography brain scan, all of which were normal. An hour later, Joe had a normal neurological exam, then underwent electroencephalography (EEG) and magnetic resonance imaging (MRI) and was discharged home without medication.
How would you treat this patient?
About 10% of Americans will experience a seizure at some point in their lives,1,2 and more than 3 million have epilepsy.3 The incidence ranges from 1% among 20-year-olds to more than 3% by the age of 75.1,2
To adequately care for such patients—whether they have had multiple seizures or only one—you need to know whether they’re at risk for recurrences, when (or if) to prescribe an AED, and which agents provide optimal seizure control with the fewest adverse effects. You also need to know when a referral to an epilepsy specialist is indicated, when or whether it’s safe for patients to stop taking antiseizure medication, and how to address lifestyle issues that patients with epilepsy often need help with.
This review addresses these and other questions.
Is it epilepsy? How to respond to a single seizure
A seizure—a transient occurrence of signs or symptoms due to abnormal excessive or synchronous neural activity in the brain—can be either focal (partial) or generalized. In addition, seizures can be broadly divided into 2 categories, based on etiology:
Provoked seizures are caused by an acute structural, toxic, or metabolic insult to the brain, and, presumably, would not have occurred if the underlying medical condition did not exist. Treating the cause—eg, alcohol withdrawal, hyponatremia, or hypoglycemia—should prevent a recurrence.
Unprovoked seizures have no apparent underlying cause. Epilepsy is defined as a chronic condition characterized by ≥2 unprovoked seizures at least 24 hours apart, and epilepsy syndromes are classified as localization-related or generalized (TABLE 1).1,4,5
Generally, epileptologists do not recommend symptomatic treatment of a first unprovoked seizure6—a consensus based on several randomized controlled trials that found immediate treatment with an AED reduced the risk of a subsequent seizure in the short term, but did not affect long-term outcomes or the development of epilepsy.7
Treatment should begin after a single seizure, however, if the seizure was prolonged or there is an increased risk of recurrence.6 Factors that increase this risk include an abnormal EEG, particularly if the abnormality is epileptiform; the presence of a brain lesion; a localized (focal) seizure; and an abnormal neurologic exam.8 A history of status epilepticus—a single, unremitting seizure lasting ≥5 to 10 minutes or frequent seizures without a return to neurologic baseline in between—or complex febrile seizures, and a family history of epilepsy are risk factors for recurrence, as well.7
When the patient is a child. Prescribing an AED for a child after a first unprovoked seizure is not indicated to prevent the development of epilepsy, but may be considered, as for adults, in circumstances where the benefit of reducing the risk of a second seizure outweighs the risk of pharmacologic and psychosocial adverse effects.9
CASE Joe’s ED records show that his MRI was normal, but his EEG revealed an epileptogenic focus on the right temporal region—a finding that indicates that he has an elevated risk of recurrence and is a candidate for an AED. Before selecting a particular agent, you review his chart.
Joe is taking a thiazide diuretic and a calcium channel blocker for hypertension. He was a heavy drinker until he had an episode of pancreatitis 10 years ago, and has been abstinent ever since. About 5 years ago, he suffered from depression and was treated with sertraline, but the depression resolved and the drug was discontinued 3 years ago. The patient’s mother and brother have type 2 diabetes and his father had a myocardial infarction before the age of 60. Joe was laid off from his sales job 18 months ago and is actively seeking employment. At this point, you consider a broad-spectrum AED that would not interact with his current medications or adversely affect his medical conditions, and would be relatively inexpensive.
TABLE 1
Identifying seizures and types of epilepsy:1,4,5 International League Against Epilepsy classification
| Type of seizure |
Focal
Generalized
|
| Type of epilepsy syndrome* |
Localization related (partial or focal)
Generalized
|
| *This is a partial listing, with selected examples of epilepsy syndromes. |
What to consider in a first-line drug
The number of AEDs on the market has increased sharply in the past few years, giving physicians many medications to choose from. Selecting the optimal drug is particularly important for the initial treatment, as many patients remain on the first AED for years. Second-generation AEDs have been found to be as effective as, and better tolerated than, first-generation antiseizure drugs. But all AEDs carry a warning of a potential increase in suicide risk and the need to monitor patients for behavior changes.10
Before selecting an AED for a particular patient, consider the following questions:
What type of seizure? AEDs are generally classified by spectrum of activity into “narrow-spectrum” and “broad-spectrum.” Narrow-spectrum drugs are more effective for controlling partial seizures, but have the potential to exacerbate generalized seizures; broad-spectrum AEDs can be used for both. (TABLE 211-18 lists indications for first- and second-generation AEDs based on type of epilepsy.) If there’s no definitive diagnosis of the type of epilepsy a patient has, use a broad-spectrum drug.
What other drugs is the patient taking? If the AED will be added to the patient’s current medication regimen, look closely at potential pharmacodynamic drug-drug interactions, and consider whether a dosage adjustment is needed. Determine, too, whether the patient has any comorbidities that could affect his or her response to the AED.
Side effects, such as weight gain or loss, urolithiasis, and hepatic enzyme induction, are key considerations. (TABLE W1,19-24 which details dose, side effects, and costs of first- and second-generation AEDs, can be found at jfponline.com.)
Is the patient elderly? AED clearance is reduced in the elderly, so lower doses are needed. Reduction in serum albumin increases the free or active component of highly protein-bound drugs, increasing the likelihood of adverse effects.
Is the patient female? Some AEDs may have effects on women’s hormonal function, sexuality, bone health, and pregnancy.25 Hepatic enzyme inducers increase the clearance of oral contraceptives, reducing their efficacy. Vitamin D and calcium metabolism can also be affected, which can lead to osteomalacia. Valproate treatment in women is associated with higher levels of insulin, testosterone, and triglycerides.26 Cytochrome P-450-activating AEDs in general are associated with higher testosterone levels and reduced libido.27
Potential pregnancy is another consideration. Women with epilepsy are able to bear healthy children. What’s more, patients whose seizures are controlled with AEDs should be maintained on medication throughout pregnancy, as the risk of fetal harm from seizures generally outweighs the teratogenicity of the drug.28
Although large studies are limited, a study of 1532 infants exposed to AEDs in the first trimester did not find an increase in major birth defects compared with infants without such exposure.29 More recently, a large observational cohort study conducted in more than 40 countries found that the possibility of harm to a developing fetus is not only drug-specific but also dose-related.30 (To learn more, see “Pregnancy and epilepsy—when you’re managing both,” in the December 2010 issue of The Journal of Family Practice.)
Is cost a factor? Finally, consider the cost of the AED you would like to prescribe, and whether the patient has a prescription drug plan or the means to pay for his prescription.
CASE After a discussion of potential side effects, including the potential for suicidal ideation associated with AEDs, you prescribe carbamazepine for Joe as seizure prophylaxis, because it is the least expensive of the broad-spectrum AEDs and is unlikely to exacerbate his previous pancreatitis or interact with his current medications.
TABLE 2
Choosing an AED: What to consider11-18
| Epilepsy type | |||||
| Localization-related (focal/partial) | Idiopathic (generalized) | Nonidiopathic (generalized) | |||
| Anticonvulsant* | Tonic-clonic | Absence | Myoclonic | ||
| First generation | |||||
| Carbamazepine† | √ | √ | |||
| Ethosuximide† | √ | ||||
| Phenobarbital† | √ | √ | √ | ||
| Phenytoin† | √ | √ | √ | ||
| Primidone | √ | √ | √ | ||
| Valproate† | √ | √ | √ | √ | √ |
| Second generation | |||||
| Felbamate | √ | √ | |||
| Gabapentin† | √ | ||||
| Lacosamide | √ | ||||
| Lamotrigine | √† | √ | √‡ | √ | |
| Levetiracetam | √ | √ | √ | ||
| Oxcarbazepine† | √ | ||||
| Pregabalin | √ | ||||
| Rufinamide | √ | √ | |||
| Tiagabine | √ | ||||
| Topiramate | √‡ | √ | √ | ||
| Vigabatrin | √ | √ | |||
| Zonisamide | √ | √ | |||
| *Bold type indicates broad-spectrum antiepileptic drugs. †Supported by American Academy of Neurology (AAN) evidence-based guideline level A or B recommendation for monotherapy in newly diagnosed epilepsy patients. ‡Supported by AAN evidence-based guideline level B recommendation for monotherapy in newly diagnosed absence epilepsy. | |||||
TABLE W1
A closer look at antiepileptic drugs19-24
| Drug name | Maintenance dosage | Adverse effects | Cost (30-day supply)* | |
| Common | Rare/idiosyncratic | |||
| First generation | ||||
| Carbamazepine | 800-1200 mg/d | Dizziness, drowsiness, diplopia, nausea, vomiting, diarrhea, rash, pruritus, SIADH | Aplastic anemia, agranulocytosis, hyponatremia, SJS, hepatic failure, pancreatitis, suicidal ideation | $4-$50 (XR: $200) |
| Ethosuximide | 20 mg/kg per day | Sleep disturbance, drowsiness, hyperactivity, behavior changes, headache, nausea, vomiting, hiccups | Agranulocytosis, aplastic anemia, SJS, hepatic failure, serum sickness, suicidal ideation | $40-150 |
| Phenobarbital | 1-4 mg/kg per day; 120-400 mg/d | Altered sleep cycles, sedation, ataxia, lethargy, behavior changes, hyperactivity, nausea, rash | Agranulocytosis, dermatitis, SJS, hepatic failure, serum sickness, connective tissue disorders, metabolic bone disease, intellect blunting, suicidal ideation | $4-$10 |
| Phenytoin | 300-600 mg/d | Confusion, slurred speech, double vision, ataxia, nystagmus, neuropathy, hirsutism, acne, gingival hyperplasia | Neuropathy, agranulocytosis, SJS, immune reactions/serum sickness, hepatic failure, skin thickening, metabolic bone disease, suicidal ideation | $35 |
| Valproic acid | 60-350 mg/kg per day | Tremor, weight gain, PCOS, nausea, vomiting, alopecia, easy bruising | Hepatic failure, pancreatitis, hearing loss, blood dyscrasias/thrombocytopenia, hyperammonemia, encephalopathy, osteoporosis, suicidal ideation | $40 (ER: $150) |
| Second generation | ||||
| Felbamate | 2400-3600 mg/d | Somnolence, nausea, vomiting, weight loss, anorexia | Aplastic anemia (>13 years), hepatic failure, suicidal ideation | $300-$500† |
| Gabapentin | 900-1800 mg/d | Somnolence, fatigue, weight gain, nystagmus | Pedal edema, suicidal ideation | $4-$100 |
| Lacosamide | 200-400 mg/d | Headache, dizziness, ataxia, nausea, diplopia | Euphoria, prolongation of PR interval, heart block, suicidal ideation | $420† |
| Lamotrigine | 300-500 mg/d | Dizziness, ataxia, nausea, somnolence, rash | SJS, hypersensitivity reactions (renal/hepatic failure), DIC, suicidal ideation | $30-$100 |
| Levetiracetam | 3000 mg/d | Somnolence, dizziness, aggression, agitation, anxiety, weight loss | Infection, pancytopenia, liver failure, suicidal ideation | $30-$100 (XR: $245†) |
| Oxcarbazepine | 1200 mg/d | Somnolence, fatigue, headache, ataxia, nausea, rash | Hyponatremia, SJS, TEN, angioedema | $250-$1000 |
| Pregabalin | 150-600 mg/d | Peripheral edema, dry mouth, dizziness, ataxia, diplopia, weight gain | Angioedema, CK elevation, mild PR interval prolongation, suicidal ideation | $100-$350† |
| Rufinamide | 3200 mg/d | Headache, dizziness, fatigue, nausea | Shortened QT interval, hypersensitivity rash, suicidal ideation | $400-$750† |
| Tiagabine | 32-56 mg/d | Difficulty concentrating, dizziness, headache, somnolence, nervousness | Spike-wave stupor, sudden death, suicidal ideation | $140-$650† |
| Topiramate | 200-400 mg/d | Somnolence, dizziness, fatigue, weight loss, difficulty concentrating, speech problems, paresthesias, diarrhea, nausea | Acute myopia and glaucoma, hyperthermia (children); metabolic acidosis, hyperammonemia, liver failure, oligohydrosis, SJS/TEN, kidney stones, suicidal ideation | $40 - $100 |
| Vigabatrin | 1500 mg/d | Fatigue, somnolence, nystagmus, tremor, weight gain | Vision loss (30% of patients) blurred vision, arthralgia, suicidal ideation | :$50 -$100† |
| Zonisamide | 400- 600 mg/d | Somnolence, difficulty concentrating, anorexia, nausea | SJS, TEN, aplastic anemia, agranulocytosis, nephrolithiasis/, oligohydrosis, acidosis, suicidal ideation | $50-$200 |
| CK, creatine kinase; DIC, disseminated intravascular coagulation; ER, extended release; IV, intravenous; PCOS, polycystic ovarian syndrome; SIADH, syndrome of inappropriate antidiuretic hormone hypersecretion; SJS, Stevens-Johnson syndrome; TEN, toxic epidermal necrolysis, XR, extended release. *Costs from www.drugstore.com, www.savewithgenericdrugs.com, and www.pharmacychecker.com. †No generic available. | ||||
When to add a second AED
Monotherapy is the preferred method of epilepsy treatment, and controls seizures for 70% to 90% of patients.31,32 If seizures continue and potential adverse effects prevent you from increasing the dosage, switching to a different AED, then tapering off the first agent, is recommended.33,34
If the new AED fails to provide adequate seizure control, consider combination therapy. An additional 10% to 15% of patients with epilepsy achieve control with dual therapy.33,34
Many second-generation agents are approved for adjunctive therapy. However, the use of 2 AEDs increases the risk of toxicities and drug interactions, and requires complex dosage adjustments, which should be done slowly and cautiously. Combination therapy also increases costs and may cause a decrease in compliance.33,34
Noncompliance is the single most common reason for treatment failure in patients with epilepsy, occurring at an estimated rate of up to 60%.35,36 The complexity of the drug regimen is the major cause, regardless of patient age, sex, psychomotor development, seizure type, or seizure frequency.35,36
Because of the lack of good clinical trials of combination antiepilepsy therapy, no evidence is available to indicate which AEDs are safe and effective when taken together. There is, however, evidence that certain combinations should be avoided due to the risk of increased adverse effects. These include phenobarbital/valproate, phenytoin/carbamazepine, and carbamazepine/lamotrigine.25
Managing the patient who is seizure-free
After a patient has been seizure-free for 2 to 5 years, consider a reduction in, or a discontinuation of, his or her AED. The relapse rate varies from 10% to 70%, with meta-analyses showing a rate of 25% in the first year and 29% in the second year.19,37 The American Academy of Neurology (AAN) has published an evidence-based guideline for discontinuing AEDs in seizure-free patients, available at www.aan.com/professionals/practice/pdfs/gl0007.pdf.
Withdrawal should be gradual and, for patients on combination therapy, carried out one drug at a time to prevent a recurrence of seizures or status epilepticus. The AAN recommends a 2- to- 3-month withdrawal period for AEDs (and longer for benzodiazepines), although relapse rates have been found to be lower when the medication is withdrawn more slowly, over about 6 months.19,34 If seizures recur after withdrawal, restart the AEDs at previous dosages.19,34,38
Should the patient drive?
For patients with epilepsy, loss of independence related to driving restrictions is a major source of stress. A 10-year follow-up study of Danish patients with epilepsy found a 7-fold increase in motor vehicle accidents (MVAs) in patients with seizure disorders.39 Other studies have shown that the seizure-free interval is the best predictor of involvement in an MVA.40
The risk of driving accidents decreases as the seizure-free interval increases. Unfortunately, however, a decline in patient compliance is also associated with longer seizure-free intervals—creating the potential for recurrence and driving risk. Because of this discrepancy, a consensus statement from the AAN, American Epilepsy Society, and Epilepsy Foundation of America recommends a minimum 3-month seizure-free interval before patients are allowed to drive.41
Use clinical judgment in deciding whether to extend the seizure-free period. State laws vary widely regarding the need to report patients with seizure disorders, limitations on professional drivers, and seizure-free intervals required, so it is important to be familiar with the laws in your state. The Epilepsy Foundation has a helpful online resource with a database detailing individual state statutes (http://www.epilepsyfoundation.org/living/wellness/transportation/driverlicensing.cfm).
The danger of uncontrolled seizures
Overall, AEDs effectively control 70% of 80% of cases; the remaining 20% to 30% are considered medically refractory.38 What’s more, after 2 AED failures, a patient’s chances of achieving full seizure control with additional drugs are no better than 10% to 20%.42 And, as more drugs are tried, the likelihood of full control declines even further.43
Patients with uncontrolled seizures have a cumulative risk of sudden unexpected death in epilepsy (SUDEP) of 0.5% per year.44 Cognitive decline is associated with uncontrolled epilepsy, as well. In children, frequent seizures may significantly alter neuronal networks, affecting cognitive and motor development.
Is your patient a candidate for surgery?
Patients with disabling complex partial seizures that remain uncontrolled after 2 or more AED trials (either as monotherapy or in combination) should be referred to an epilepsy specialty center for evaluation for surgery.45 This should be considered as early as possible to afford the patient the best chance of achieving seizure control.
Successful epilepsy surgery—in which the portion of the brain causing the misfiring that causes the seizures is removed—often results in a better quality of life; it is also cost effective.46 Not everyone with refractory epilepsy is a candidate for surgery, of course. Among those who are, however, 50% to 70% of patients can expect to have improved seizure control.47
Status epilepticus is a medical emergency
A patient who develops status epilepticus is at high risk and requires immediate, and simultaneous, evaluation and treatment. Status epilepticus carries nearly a 20% mortality from the first episode,48 and the 10-year mortality rate after an episode of status epilepticus is as high as 40%.49
Although most of the deaths associated with status epilepticus are due to the underlying pathology, early treatment can prevent or ameliorate complications from rhabdomyolysis and irreversible anoxic neuronal damage.50
A benzodiazepine (typically, a 10-mg IV bolus of diazepam) is the initial treatment for status epilepticus, followed by or concurrent with fosphenytoin (15-18 mg/kg). If status epilepticus remains refractory to first-line drugs (lasting >30 minutes), intubation and transfer to an intensive care setting may be required, and a neurological consult should be obtained.
Pharmacologic treatment of status epilepticus falls into 3 main classes: benzodiazepines, standard AEDs, and general anesthetics such as propofol. Benzodiazepines act very rapidly to control most prolonged seizures, and are the first-line treatment choice. Diazepam has long been the mainstay of treatment, and is usually readily available. However, in both a large systematic review and a head-to-head trial, lorazepam was found to be superior to diazepam in ending seizure activity and maintaining seizure control without the use of other medications51,52—and is now the drug of choice for initial treatment of status epilepticus.
CASE You continue to see Joe every 3 to 4 months to monitor his basic blood work and mood. A year after his seizure, he remains seizure-free and is tolerating the AED without any adverse effects.
CORRESPONDENCE
William J. Geiger, MD, FAAFP, Medical College of Wisconsin, Columbia St. Mary’s Family Medicine Residency, 1121 East North Avenue, Milwaukee, WI 53212; bgeiger@mcw.edu
1. Epilepsy Foundation of America. Epilepsy and seizure statistics. Available at: http://www.epilepsyfoundation.org/about/statistics.cfm. Accessed June 15, 2009.
2. Centers for Disease Control and Prevention (CDC). Prevalence and most common causes of disability among adults—United States, 2005. MMWR Morb Mortal Wkly Rep. 2009;58:421-426. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm5816a2.htm. Accessed June 15, 2009.
3. Hirtz D, Thurman DJ, Gwinn-Hardy K, et al. How common are the “common” neurologic disorders? Neurology. 2007;68:326-337.
4. Engel J Jr. ILAE classification of epilepsy syndromes. Epilepsy Res. 2006;70(suppl 1):S5-S10.
5. Rudzinski LA, Shih JJ. Continuum: lifelong learning in neurology. Epilepsia. 2010;16:15-35.
6. Chaves J, Sander JW. Seizure aggravation in idiopathic generalized epilepsies. Epilepsia. 2005;46(suppl 9):S133-S139.
7. Beghi E. Management of first seizure. General conclusions and recommendations. Epilepsia. 2008;49(suppl 1):S58-S61.
8. Berg A. Risk of recurrence after a first unprovoked seizure. Epilepsia. 2008;49(suppl 1):S13-S18.
9. Hirtz D, Ashwal S, Berg A, et al. Practice parameter: evaluating a first non-febrile seizure in children: report of the Quality Standards Subcommittee of the American Academy of Neurology, the Child Neurology Society, and the American Epilepsy Society. Neurology. 2000;55:616-623.
10. US Food and Drug Administration. Suicidal behavior and ideation and antiepileptic drugs. Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm100190.htm. Updated May 5, 2009. Accessed June 28, 2009.
11. French JA, Kanner AM, Bautista J, et al. Efficacy and tolerability of the new antiepileptic drugs I: treatment of new epilepsy, report of the therapeutic and technology assessment subcommittee and quality standards subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2004;62:1252-1260.
12. French J, Smith M, Faught E, et al. Practice advisory: the use of felbamate in the treatment of patients with intractable epilepsy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 1999;52:1540-1545.
13. Glauser T, Kluger G, Sachdeo R, et al. Rufinamide for generalized seizures associated with Lennox-Gastaut syndrome. Neurology. 2008;70:1950-1958.
14. Suzuki Y, Nagai T, Ono J, et al. Zonisamide monotherapy in newly-diagnosed infantile spasms. Epilepsia. 1997;38:1035-1038.
15. Kochak GM, Page JG, Buchanan RA, et al. Steady-state pharmacokinetics of zonisamide, an antiepileptic agent for treatment of refractory complex partial seizures. J Clin Pharmacol. 1998;38:166-171.
16. Arroyo S, Anhut H, Kugler AR, et al. Pregabalin 1008-011 International Study Group. Pregabalin add-on treatment: a randomized, double-blind, placebo-controlled, dose-response study in adults with partial seizures. Epilepsia. 2004;45:20-27.
17. Brodie MJ, Rosenfeld WE, Vazquez B, et al. Rufinamide for the adjunctive treatment of partial seizures in adults and adolescents: a randomized placebo-controlled trial. Epilepsia. 2009;50:1899-1909.
18. Ben-Menachem E, Biton V, Jatuzis D, et al. Efficacy and safety of oral lacosamide as adjunctive therapy in adults with partial-onset seizures. Epilepsia. 2007;48:1308-1317.
19. Gidal B, Garnett W. Epilepsy. In: Dipiro J, et al, eds. Pharmacotherapy: A Pathophysiologic Approach. 6th ed. New York: McGraw-Hill; 2005:1023-1048.
20. Pellock JM, Treatment of epilepsy in the new millennium. Pharmacotherapy. 2000;20:129S-138S.
21. Schachter S. Pharmacology of antiepileptic drugs. Available at: http://www.utdonline.com/online/content/topic.do?topicKey=epil_eeg/5220. Accessed July 15, 2009.
22. Woelfel J. Comparison of antiepileptic drugs. Pharmacist’s Letter/Prescriber's Letter. July 2009;25:1-24.
23. Wolters Kluwer Health Inc. Anticonvulsants. Drug facts and comparisons online. Available at: http://www.efactsonline.com. Accessed July 10, 2009.
24. US Food and Drug Administration. Information for healthcare professionals. Suicidality and antiepileptic drugs [FDA alert]. Available at: http://www.fda.gov/Drugs/DrugSafety/PublicHealthAdvisories/ucm054709.htm. January 31, 2008. Accessed June 30, 2009.
25. French J. Treatment with antiepileptic drugs, new and old. Continuum. 2007;13:71-90.
26. Sheehan M. Polycystic ovarian syndrome: diagnosis and management. Clin Med Res. 2004;2:13-27.
27. Harden CL. Sexual dysfunction in women with epilepsy. Seizure. 2008;17:131-135.
28. Harden CL, Hopp J, Ting TY, et al. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): obstetrical complications and change in seizure frequency. Neurology. 2009;73:126-132.
29. Molgaard-Nielsen D, Hviid A. Newer-generation antiepileptic drugs and the risk of major birth defects. JAMA. 2011;305:1996-2002.
30. Tomson T, Battino D, Bonizonni E, et al. Dose-dependent risk of malformations with antiepileptic drugs: an analysis of data from the EURAP epilepsy and pregnancy registry. Lancet Neurol. 2011;10:609-617.
31. Callaghan BC, Anand K, Hesdorffer D, et al. Likelihood of seizure remission in an adult population with refractory epilepsy. Ann Neurol. 2007;62:382-389.
32. Luciano AL, Shorvon SD. Results of treatment changes in patients with apparently drug-resistant chronic epilepsy. Ann Neurol. 2007;62:375-381.
33. Abramowicz M, ed. Drugs for epilepsy [treatment guidelines]. The Medical Letter. 2008;70:1-12.
34.Stokes T, Shaw EJ, Juarez-Garcia A, et al. Clinical guidelines and evidence review for the epilepsies: diagnosis and management in adults and children in primary and secondary care. London: Royal College of General Practitioners. Available at: www.nice.org.uk/CG020fullguideline. Published October 2004. Accessed July 10, 2009.
35. Garnett WR. Antiepileptic drug treatment: outcomes and adherence. Pharmacotherapy. 2000;20:191S-199S.
36. Briesacher BA, Andrade SE, Fouayzi H, et al. Comparison of drug adherence rates among patients with seven different medical conditions. Pharmacotherapy. 2008;28:437-443.
37. Shinnar S, Gross-Tsur V. Discontinuing antiepileptic drug therapy. In: Wyllie E, ed. The Treatment of Epilepsy. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2001:811-819.
38. Kwan P, Brodie J. Refractory epilepsy: a progressive, intractable but preventable condition? Seizures. 2002;11:77-84.
39. Lings S. Increased driving accident frequency in Danish patients with epilepsy. Neurology. 2001;57:435-439.
40. Krauss GL, Krumholz A, Carter RC, et al. Risk factors for seizure-related motor vehicle crashes in patients with epilepsy. Neurology. 1999;52:1324-1329.
41. American Academy of Neurology, American Epilepsy Society, and Epilepsy Foundation of America. Consensus statements, sample statutory provisions, and model regulations regarding driver licensing and epilepsy. Epilepsia. 1994;35:696-705.
42. Thadani VM, Taylor J. Surgical treatments for epilepsy. Continuum. 2007;13:152-176.
43. Brodie MJ, Kwan P. Staged approach to epilepsy management. Neurology. 2002;58(8 suppl 5):S2-S8.
44. Sillanpaa M, Jalava M, Kaleva O, et al. Long-term prognosis of seizures with onset in childhood. N Engl J Med. 1998;338:1715-1722.
45. Engel J Jr, Wiebe S, French J, et al. Practice parameter: temporal lobe and localized neocortical resections for epilepsy. Neurology. 2003;60:538-547.
46. Boon P, D'Have M, Van Walleghen P, et al. Direct medical costs of refractory epilepsy incurred by three different treatment modalities: a prospective assessment. Epilepsia. 2002;43:96-102.
47. Passaro EA. Outcome of epilepsy surgery. Available at: http://emedicine.medscape.com/article/1185416-overview. Updated May 16, 2011. Accessed June 28, 2011.
48. DeLorenzo RJ, Pellock JM, Towne AR, et al. Epidemiology of status epilepticus. J Clin Neurophysiol. 1995;12:316-325.
49. Logroscino G, Hesdorffer DC, Cascino GD, et al. Long-term mortality after a first episode of status epilepticus. Neurology. 2002; 58:537-541.
50. Kalviaine R. Treatment of status epilepticus. Essential Evidence Plus. Wiley-Blackwell. Available at: http://www.essentialevidenceplus.com/content/ebmg_ebm/766. Accessed July 15, 2009.
51. Prasad K, Al-Roomi K, Krishnan PR, et al. Anticonvulsant therapy for status epilepticus. Cochrane Database Syst Rev. 2005;(4):CD003723.
52. Treiman DM, Meyers PD, Walton NY, et al. A comparison of four treatments for generalized convulsive status epilepticus. N Engl J Med. 1998;339:792-798.
• Prescribe an antiepileptic drug (AED) after a first unprovoked seizure only if the seizure was prolonged or there is a risk of recurrence. C
• Use monotherapy whenever possible; if seizures continue and potential adverse effects prevent an increase in dosage, switch to a different AED and taper off the first agent. A
• Consider gradual withdrawal of AEDs from patients who have been seizure-free for 2 to 5 years. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Joe G, a 44-year-old man who has been your patient for years, comes to your office 48 hours after having a seizure. He has no history of seizures, had no warning signs or symptoms, and felt fine all day, but simply collapsed when the seizure occurred. He was transported to the emergency department (ED), and found to be postictal, with no further seizure activity. The ED work-up included a hemogram, comprehensive metabolic panel, and computed tomography brain scan, all of which were normal. An hour later, Joe had a normal neurological exam, then underwent electroencephalography (EEG) and magnetic resonance imaging (MRI) and was discharged home without medication.
How would you treat this patient?
About 10% of Americans will experience a seizure at some point in their lives,1,2 and more than 3 million have epilepsy.3 The incidence ranges from 1% among 20-year-olds to more than 3% by the age of 75.1,2
To adequately care for such patients—whether they have had multiple seizures or only one—you need to know whether they’re at risk for recurrences, when (or if) to prescribe an AED, and which agents provide optimal seizure control with the fewest adverse effects. You also need to know when a referral to an epilepsy specialist is indicated, when or whether it’s safe for patients to stop taking antiseizure medication, and how to address lifestyle issues that patients with epilepsy often need help with.
This review addresses these and other questions.
Is it epilepsy? How to respond to a single seizure
A seizure—a transient occurrence of signs or symptoms due to abnormal excessive or synchronous neural activity in the brain—can be either focal (partial) or generalized. In addition, seizures can be broadly divided into 2 categories, based on etiology:
Provoked seizures are caused by an acute structural, toxic, or metabolic insult to the brain, and, presumably, would not have occurred if the underlying medical condition did not exist. Treating the cause—eg, alcohol withdrawal, hyponatremia, or hypoglycemia—should prevent a recurrence.
Unprovoked seizures have no apparent underlying cause. Epilepsy is defined as a chronic condition characterized by ≥2 unprovoked seizures at least 24 hours apart, and epilepsy syndromes are classified as localization-related or generalized (TABLE 1).1,4,5
Generally, epileptologists do not recommend symptomatic treatment of a first unprovoked seizure6—a consensus based on several randomized controlled trials that found immediate treatment with an AED reduced the risk of a subsequent seizure in the short term, but did not affect long-term outcomes or the development of epilepsy.7
Treatment should begin after a single seizure, however, if the seizure was prolonged or there is an increased risk of recurrence.6 Factors that increase this risk include an abnormal EEG, particularly if the abnormality is epileptiform; the presence of a brain lesion; a localized (focal) seizure; and an abnormal neurologic exam.8 A history of status epilepticus—a single, unremitting seizure lasting ≥5 to 10 minutes or frequent seizures without a return to neurologic baseline in between—or complex febrile seizures, and a family history of epilepsy are risk factors for recurrence, as well.7
When the patient is a child. Prescribing an AED for a child after a first unprovoked seizure is not indicated to prevent the development of epilepsy, but may be considered, as for adults, in circumstances where the benefit of reducing the risk of a second seizure outweighs the risk of pharmacologic and psychosocial adverse effects.9
CASE Joe’s ED records show that his MRI was normal, but his EEG revealed an epileptogenic focus on the right temporal region—a finding that indicates that he has an elevated risk of recurrence and is a candidate for an AED. Before selecting a particular agent, you review his chart.
Joe is taking a thiazide diuretic and a calcium channel blocker for hypertension. He was a heavy drinker until he had an episode of pancreatitis 10 years ago, and has been abstinent ever since. About 5 years ago, he suffered from depression and was treated with sertraline, but the depression resolved and the drug was discontinued 3 years ago. The patient’s mother and brother have type 2 diabetes and his father had a myocardial infarction before the age of 60. Joe was laid off from his sales job 18 months ago and is actively seeking employment. At this point, you consider a broad-spectrum AED that would not interact with his current medications or adversely affect his medical conditions, and would be relatively inexpensive.
TABLE 1
Identifying seizures and types of epilepsy:1,4,5 International League Against Epilepsy classification
| Type of seizure |
Focal
Generalized
|
| Type of epilepsy syndrome* |
Localization related (partial or focal)
Generalized
|
| *This is a partial listing, with selected examples of epilepsy syndromes. |
What to consider in a first-line drug
The number of AEDs on the market has increased sharply in the past few years, giving physicians many medications to choose from. Selecting the optimal drug is particularly important for the initial treatment, as many patients remain on the first AED for years. Second-generation AEDs have been found to be as effective as, and better tolerated than, first-generation antiseizure drugs. But all AEDs carry a warning of a potential increase in suicide risk and the need to monitor patients for behavior changes.10
Before selecting an AED for a particular patient, consider the following questions:
What type of seizure? AEDs are generally classified by spectrum of activity into “narrow-spectrum” and “broad-spectrum.” Narrow-spectrum drugs are more effective for controlling partial seizures, but have the potential to exacerbate generalized seizures; broad-spectrum AEDs can be used for both. (TABLE 211-18 lists indications for first- and second-generation AEDs based on type of epilepsy.) If there’s no definitive diagnosis of the type of epilepsy a patient has, use a broad-spectrum drug.
What other drugs is the patient taking? If the AED will be added to the patient’s current medication regimen, look closely at potential pharmacodynamic drug-drug interactions, and consider whether a dosage adjustment is needed. Determine, too, whether the patient has any comorbidities that could affect his or her response to the AED.
Side effects, such as weight gain or loss, urolithiasis, and hepatic enzyme induction, are key considerations. (TABLE W1,19-24 which details dose, side effects, and costs of first- and second-generation AEDs, can be found at jfponline.com.)
Is the patient elderly? AED clearance is reduced in the elderly, so lower doses are needed. Reduction in serum albumin increases the free or active component of highly protein-bound drugs, increasing the likelihood of adverse effects.
Is the patient female? Some AEDs may have effects on women’s hormonal function, sexuality, bone health, and pregnancy.25 Hepatic enzyme inducers increase the clearance of oral contraceptives, reducing their efficacy. Vitamin D and calcium metabolism can also be affected, which can lead to osteomalacia. Valproate treatment in women is associated with higher levels of insulin, testosterone, and triglycerides.26 Cytochrome P-450-activating AEDs in general are associated with higher testosterone levels and reduced libido.27
Potential pregnancy is another consideration. Women with epilepsy are able to bear healthy children. What’s more, patients whose seizures are controlled with AEDs should be maintained on medication throughout pregnancy, as the risk of fetal harm from seizures generally outweighs the teratogenicity of the drug.28
Although large studies are limited, a study of 1532 infants exposed to AEDs in the first trimester did not find an increase in major birth defects compared with infants without such exposure.29 More recently, a large observational cohort study conducted in more than 40 countries found that the possibility of harm to a developing fetus is not only drug-specific but also dose-related.30 (To learn more, see “Pregnancy and epilepsy—when you’re managing both,” in the December 2010 issue of The Journal of Family Practice.)
Is cost a factor? Finally, consider the cost of the AED you would like to prescribe, and whether the patient has a prescription drug plan or the means to pay for his prescription.
CASE After a discussion of potential side effects, including the potential for suicidal ideation associated with AEDs, you prescribe carbamazepine for Joe as seizure prophylaxis, because it is the least expensive of the broad-spectrum AEDs and is unlikely to exacerbate his previous pancreatitis or interact with his current medications.
TABLE 2
Choosing an AED: What to consider11-18
| Epilepsy type | |||||
| Localization-related (focal/partial) | Idiopathic (generalized) | Nonidiopathic (generalized) | |||
| Anticonvulsant* | Tonic-clonic | Absence | Myoclonic | ||
| First generation | |||||
| Carbamazepine† | √ | √ | |||
| Ethosuximide† | √ | ||||
| Phenobarbital† | √ | √ | √ | ||
| Phenytoin† | √ | √ | √ | ||
| Primidone | √ | √ | √ | ||
| Valproate† | √ | √ | √ | √ | √ |
| Second generation | |||||
| Felbamate | √ | √ | |||
| Gabapentin† | √ | ||||
| Lacosamide | √ | ||||
| Lamotrigine | √† | √ | √‡ | √ | |
| Levetiracetam | √ | √ | √ | ||
| Oxcarbazepine† | √ | ||||
| Pregabalin | √ | ||||
| Rufinamide | √ | √ | |||
| Tiagabine | √ | ||||
| Topiramate | √‡ | √ | √ | ||
| Vigabatrin | √ | √ | |||
| Zonisamide | √ | √ | |||
| *Bold type indicates broad-spectrum antiepileptic drugs. †Supported by American Academy of Neurology (AAN) evidence-based guideline level A or B recommendation for monotherapy in newly diagnosed epilepsy patients. ‡Supported by AAN evidence-based guideline level B recommendation for monotherapy in newly diagnosed absence epilepsy. | |||||
TABLE W1
A closer look at antiepileptic drugs19-24
| Drug name | Maintenance dosage | Adverse effects | Cost (30-day supply)* | |
| Common | Rare/idiosyncratic | |||
| First generation | ||||
| Carbamazepine | 800-1200 mg/d | Dizziness, drowsiness, diplopia, nausea, vomiting, diarrhea, rash, pruritus, SIADH | Aplastic anemia, agranulocytosis, hyponatremia, SJS, hepatic failure, pancreatitis, suicidal ideation | $4-$50 (XR: $200) |
| Ethosuximide | 20 mg/kg per day | Sleep disturbance, drowsiness, hyperactivity, behavior changes, headache, nausea, vomiting, hiccups | Agranulocytosis, aplastic anemia, SJS, hepatic failure, serum sickness, suicidal ideation | $40-150 |
| Phenobarbital | 1-4 mg/kg per day; 120-400 mg/d | Altered sleep cycles, sedation, ataxia, lethargy, behavior changes, hyperactivity, nausea, rash | Agranulocytosis, dermatitis, SJS, hepatic failure, serum sickness, connective tissue disorders, metabolic bone disease, intellect blunting, suicidal ideation | $4-$10 |
| Phenytoin | 300-600 mg/d | Confusion, slurred speech, double vision, ataxia, nystagmus, neuropathy, hirsutism, acne, gingival hyperplasia | Neuropathy, agranulocytosis, SJS, immune reactions/serum sickness, hepatic failure, skin thickening, metabolic bone disease, suicidal ideation | $35 |
| Valproic acid | 60-350 mg/kg per day | Tremor, weight gain, PCOS, nausea, vomiting, alopecia, easy bruising | Hepatic failure, pancreatitis, hearing loss, blood dyscrasias/thrombocytopenia, hyperammonemia, encephalopathy, osteoporosis, suicidal ideation | $40 (ER: $150) |
| Second generation | ||||
| Felbamate | 2400-3600 mg/d | Somnolence, nausea, vomiting, weight loss, anorexia | Aplastic anemia (>13 years), hepatic failure, suicidal ideation | $300-$500† |
| Gabapentin | 900-1800 mg/d | Somnolence, fatigue, weight gain, nystagmus | Pedal edema, suicidal ideation | $4-$100 |
| Lacosamide | 200-400 mg/d | Headache, dizziness, ataxia, nausea, diplopia | Euphoria, prolongation of PR interval, heart block, suicidal ideation | $420† |
| Lamotrigine | 300-500 mg/d | Dizziness, ataxia, nausea, somnolence, rash | SJS, hypersensitivity reactions (renal/hepatic failure), DIC, suicidal ideation | $30-$100 |
| Levetiracetam | 3000 mg/d | Somnolence, dizziness, aggression, agitation, anxiety, weight loss | Infection, pancytopenia, liver failure, suicidal ideation | $30-$100 (XR: $245†) |
| Oxcarbazepine | 1200 mg/d | Somnolence, fatigue, headache, ataxia, nausea, rash | Hyponatremia, SJS, TEN, angioedema | $250-$1000 |
| Pregabalin | 150-600 mg/d | Peripheral edema, dry mouth, dizziness, ataxia, diplopia, weight gain | Angioedema, CK elevation, mild PR interval prolongation, suicidal ideation | $100-$350† |
| Rufinamide | 3200 mg/d | Headache, dizziness, fatigue, nausea | Shortened QT interval, hypersensitivity rash, suicidal ideation | $400-$750† |
| Tiagabine | 32-56 mg/d | Difficulty concentrating, dizziness, headache, somnolence, nervousness | Spike-wave stupor, sudden death, suicidal ideation | $140-$650† |
| Topiramate | 200-400 mg/d | Somnolence, dizziness, fatigue, weight loss, difficulty concentrating, speech problems, paresthesias, diarrhea, nausea | Acute myopia and glaucoma, hyperthermia (children); metabolic acidosis, hyperammonemia, liver failure, oligohydrosis, SJS/TEN, kidney stones, suicidal ideation | $40 - $100 |
| Vigabatrin | 1500 mg/d | Fatigue, somnolence, nystagmus, tremor, weight gain | Vision loss (30% of patients) blurred vision, arthralgia, suicidal ideation | :$50 -$100† |
| Zonisamide | 400- 600 mg/d | Somnolence, difficulty concentrating, anorexia, nausea | SJS, TEN, aplastic anemia, agranulocytosis, nephrolithiasis/, oligohydrosis, acidosis, suicidal ideation | $50-$200 |
| CK, creatine kinase; DIC, disseminated intravascular coagulation; ER, extended release; IV, intravenous; PCOS, polycystic ovarian syndrome; SIADH, syndrome of inappropriate antidiuretic hormone hypersecretion; SJS, Stevens-Johnson syndrome; TEN, toxic epidermal necrolysis, XR, extended release. *Costs from www.drugstore.com, www.savewithgenericdrugs.com, and www.pharmacychecker.com. †No generic available. | ||||
When to add a second AED
Monotherapy is the preferred method of epilepsy treatment, and controls seizures for 70% to 90% of patients.31,32 If seizures continue and potential adverse effects prevent you from increasing the dosage, switching to a different AED, then tapering off the first agent, is recommended.33,34
If the new AED fails to provide adequate seizure control, consider combination therapy. An additional 10% to 15% of patients with epilepsy achieve control with dual therapy.33,34
Many second-generation agents are approved for adjunctive therapy. However, the use of 2 AEDs increases the risk of toxicities and drug interactions, and requires complex dosage adjustments, which should be done slowly and cautiously. Combination therapy also increases costs and may cause a decrease in compliance.33,34
Noncompliance is the single most common reason for treatment failure in patients with epilepsy, occurring at an estimated rate of up to 60%.35,36 The complexity of the drug regimen is the major cause, regardless of patient age, sex, psychomotor development, seizure type, or seizure frequency.35,36
Because of the lack of good clinical trials of combination antiepilepsy therapy, no evidence is available to indicate which AEDs are safe and effective when taken together. There is, however, evidence that certain combinations should be avoided due to the risk of increased adverse effects. These include phenobarbital/valproate, phenytoin/carbamazepine, and carbamazepine/lamotrigine.25
Managing the patient who is seizure-free
After a patient has been seizure-free for 2 to 5 years, consider a reduction in, or a discontinuation of, his or her AED. The relapse rate varies from 10% to 70%, with meta-analyses showing a rate of 25% in the first year and 29% in the second year.19,37 The American Academy of Neurology (AAN) has published an evidence-based guideline for discontinuing AEDs in seizure-free patients, available at www.aan.com/professionals/practice/pdfs/gl0007.pdf.
Withdrawal should be gradual and, for patients on combination therapy, carried out one drug at a time to prevent a recurrence of seizures or status epilepticus. The AAN recommends a 2- to- 3-month withdrawal period for AEDs (and longer for benzodiazepines), although relapse rates have been found to be lower when the medication is withdrawn more slowly, over about 6 months.19,34 If seizures recur after withdrawal, restart the AEDs at previous dosages.19,34,38
Should the patient drive?
For patients with epilepsy, loss of independence related to driving restrictions is a major source of stress. A 10-year follow-up study of Danish patients with epilepsy found a 7-fold increase in motor vehicle accidents (MVAs) in patients with seizure disorders.39 Other studies have shown that the seizure-free interval is the best predictor of involvement in an MVA.40
The risk of driving accidents decreases as the seizure-free interval increases. Unfortunately, however, a decline in patient compliance is also associated with longer seizure-free intervals—creating the potential for recurrence and driving risk. Because of this discrepancy, a consensus statement from the AAN, American Epilepsy Society, and Epilepsy Foundation of America recommends a minimum 3-month seizure-free interval before patients are allowed to drive.41
Use clinical judgment in deciding whether to extend the seizure-free period. State laws vary widely regarding the need to report patients with seizure disorders, limitations on professional drivers, and seizure-free intervals required, so it is important to be familiar with the laws in your state. The Epilepsy Foundation has a helpful online resource with a database detailing individual state statutes (http://www.epilepsyfoundation.org/living/wellness/transportation/driverlicensing.cfm).
The danger of uncontrolled seizures
Overall, AEDs effectively control 70% of 80% of cases; the remaining 20% to 30% are considered medically refractory.38 What’s more, after 2 AED failures, a patient’s chances of achieving full seizure control with additional drugs are no better than 10% to 20%.42 And, as more drugs are tried, the likelihood of full control declines even further.43
Patients with uncontrolled seizures have a cumulative risk of sudden unexpected death in epilepsy (SUDEP) of 0.5% per year.44 Cognitive decline is associated with uncontrolled epilepsy, as well. In children, frequent seizures may significantly alter neuronal networks, affecting cognitive and motor development.
Is your patient a candidate for surgery?
Patients with disabling complex partial seizures that remain uncontrolled after 2 or more AED trials (either as monotherapy or in combination) should be referred to an epilepsy specialty center for evaluation for surgery.45 This should be considered as early as possible to afford the patient the best chance of achieving seizure control.
Successful epilepsy surgery—in which the portion of the brain causing the misfiring that causes the seizures is removed—often results in a better quality of life; it is also cost effective.46 Not everyone with refractory epilepsy is a candidate for surgery, of course. Among those who are, however, 50% to 70% of patients can expect to have improved seizure control.47
Status epilepticus is a medical emergency
A patient who develops status epilepticus is at high risk and requires immediate, and simultaneous, evaluation and treatment. Status epilepticus carries nearly a 20% mortality from the first episode,48 and the 10-year mortality rate after an episode of status epilepticus is as high as 40%.49
Although most of the deaths associated with status epilepticus are due to the underlying pathology, early treatment can prevent or ameliorate complications from rhabdomyolysis and irreversible anoxic neuronal damage.50
A benzodiazepine (typically, a 10-mg IV bolus of diazepam) is the initial treatment for status epilepticus, followed by or concurrent with fosphenytoin (15-18 mg/kg). If status epilepticus remains refractory to first-line drugs (lasting >30 minutes), intubation and transfer to an intensive care setting may be required, and a neurological consult should be obtained.
Pharmacologic treatment of status epilepticus falls into 3 main classes: benzodiazepines, standard AEDs, and general anesthetics such as propofol. Benzodiazepines act very rapidly to control most prolonged seizures, and are the first-line treatment choice. Diazepam has long been the mainstay of treatment, and is usually readily available. However, in both a large systematic review and a head-to-head trial, lorazepam was found to be superior to diazepam in ending seizure activity and maintaining seizure control without the use of other medications51,52—and is now the drug of choice for initial treatment of status epilepticus.
CASE You continue to see Joe every 3 to 4 months to monitor his basic blood work and mood. A year after his seizure, he remains seizure-free and is tolerating the AED without any adverse effects.
CORRESPONDENCE
William J. Geiger, MD, FAAFP, Medical College of Wisconsin, Columbia St. Mary’s Family Medicine Residency, 1121 East North Avenue, Milwaukee, WI 53212; bgeiger@mcw.edu
• Prescribe an antiepileptic drug (AED) after a first unprovoked seizure only if the seizure was prolonged or there is a risk of recurrence. C
• Use monotherapy whenever possible; if seizures continue and potential adverse effects prevent an increase in dosage, switch to a different AED and taper off the first agent. A
• Consider gradual withdrawal of AEDs from patients who have been seizure-free for 2 to 5 years. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE Joe G, a 44-year-old man who has been your patient for years, comes to your office 48 hours after having a seizure. He has no history of seizures, had no warning signs or symptoms, and felt fine all day, but simply collapsed when the seizure occurred. He was transported to the emergency department (ED), and found to be postictal, with no further seizure activity. The ED work-up included a hemogram, comprehensive metabolic panel, and computed tomography brain scan, all of which were normal. An hour later, Joe had a normal neurological exam, then underwent electroencephalography (EEG) and magnetic resonance imaging (MRI) and was discharged home without medication.
How would you treat this patient?
About 10% of Americans will experience a seizure at some point in their lives,1,2 and more than 3 million have epilepsy.3 The incidence ranges from 1% among 20-year-olds to more than 3% by the age of 75.1,2
To adequately care for such patients—whether they have had multiple seizures or only one—you need to know whether they’re at risk for recurrences, when (or if) to prescribe an AED, and which agents provide optimal seizure control with the fewest adverse effects. You also need to know when a referral to an epilepsy specialist is indicated, when or whether it’s safe for patients to stop taking antiseizure medication, and how to address lifestyle issues that patients with epilepsy often need help with.
This review addresses these and other questions.
Is it epilepsy? How to respond to a single seizure
A seizure—a transient occurrence of signs or symptoms due to abnormal excessive or synchronous neural activity in the brain—can be either focal (partial) or generalized. In addition, seizures can be broadly divided into 2 categories, based on etiology:
Provoked seizures are caused by an acute structural, toxic, or metabolic insult to the brain, and, presumably, would not have occurred if the underlying medical condition did not exist. Treating the cause—eg, alcohol withdrawal, hyponatremia, or hypoglycemia—should prevent a recurrence.
Unprovoked seizures have no apparent underlying cause. Epilepsy is defined as a chronic condition characterized by ≥2 unprovoked seizures at least 24 hours apart, and epilepsy syndromes are classified as localization-related or generalized (TABLE 1).1,4,5
Generally, epileptologists do not recommend symptomatic treatment of a first unprovoked seizure6—a consensus based on several randomized controlled trials that found immediate treatment with an AED reduced the risk of a subsequent seizure in the short term, but did not affect long-term outcomes or the development of epilepsy.7
Treatment should begin after a single seizure, however, if the seizure was prolonged or there is an increased risk of recurrence.6 Factors that increase this risk include an abnormal EEG, particularly if the abnormality is epileptiform; the presence of a brain lesion; a localized (focal) seizure; and an abnormal neurologic exam.8 A history of status epilepticus—a single, unremitting seizure lasting ≥5 to 10 minutes or frequent seizures without a return to neurologic baseline in between—or complex febrile seizures, and a family history of epilepsy are risk factors for recurrence, as well.7
When the patient is a child. Prescribing an AED for a child after a first unprovoked seizure is not indicated to prevent the development of epilepsy, but may be considered, as for adults, in circumstances where the benefit of reducing the risk of a second seizure outweighs the risk of pharmacologic and psychosocial adverse effects.9
CASE Joe’s ED records show that his MRI was normal, but his EEG revealed an epileptogenic focus on the right temporal region—a finding that indicates that he has an elevated risk of recurrence and is a candidate for an AED. Before selecting a particular agent, you review his chart.
Joe is taking a thiazide diuretic and a calcium channel blocker for hypertension. He was a heavy drinker until he had an episode of pancreatitis 10 years ago, and has been abstinent ever since. About 5 years ago, he suffered from depression and was treated with sertraline, but the depression resolved and the drug was discontinued 3 years ago. The patient’s mother and brother have type 2 diabetes and his father had a myocardial infarction before the age of 60. Joe was laid off from his sales job 18 months ago and is actively seeking employment. At this point, you consider a broad-spectrum AED that would not interact with his current medications or adversely affect his medical conditions, and would be relatively inexpensive.
TABLE 1
Identifying seizures and types of epilepsy:1,4,5 International League Against Epilepsy classification
| Type of seizure |
Focal
Generalized
|
| Type of epilepsy syndrome* |
Localization related (partial or focal)
Generalized
|
| *This is a partial listing, with selected examples of epilepsy syndromes. |
What to consider in a first-line drug
The number of AEDs on the market has increased sharply in the past few years, giving physicians many medications to choose from. Selecting the optimal drug is particularly important for the initial treatment, as many patients remain on the first AED for years. Second-generation AEDs have been found to be as effective as, and better tolerated than, first-generation antiseizure drugs. But all AEDs carry a warning of a potential increase in suicide risk and the need to monitor patients for behavior changes.10
Before selecting an AED for a particular patient, consider the following questions:
What type of seizure? AEDs are generally classified by spectrum of activity into “narrow-spectrum” and “broad-spectrum.” Narrow-spectrum drugs are more effective for controlling partial seizures, but have the potential to exacerbate generalized seizures; broad-spectrum AEDs can be used for both. (TABLE 211-18 lists indications for first- and second-generation AEDs based on type of epilepsy.) If there’s no definitive diagnosis of the type of epilepsy a patient has, use a broad-spectrum drug.
What other drugs is the patient taking? If the AED will be added to the patient’s current medication regimen, look closely at potential pharmacodynamic drug-drug interactions, and consider whether a dosage adjustment is needed. Determine, too, whether the patient has any comorbidities that could affect his or her response to the AED.
Side effects, such as weight gain or loss, urolithiasis, and hepatic enzyme induction, are key considerations. (TABLE W1,19-24 which details dose, side effects, and costs of first- and second-generation AEDs, can be found at jfponline.com.)
Is the patient elderly? AED clearance is reduced in the elderly, so lower doses are needed. Reduction in serum albumin increases the free or active component of highly protein-bound drugs, increasing the likelihood of adverse effects.
Is the patient female? Some AEDs may have effects on women’s hormonal function, sexuality, bone health, and pregnancy.25 Hepatic enzyme inducers increase the clearance of oral contraceptives, reducing their efficacy. Vitamin D and calcium metabolism can also be affected, which can lead to osteomalacia. Valproate treatment in women is associated with higher levels of insulin, testosterone, and triglycerides.26 Cytochrome P-450-activating AEDs in general are associated with higher testosterone levels and reduced libido.27
Potential pregnancy is another consideration. Women with epilepsy are able to bear healthy children. What’s more, patients whose seizures are controlled with AEDs should be maintained on medication throughout pregnancy, as the risk of fetal harm from seizures generally outweighs the teratogenicity of the drug.28
Although large studies are limited, a study of 1532 infants exposed to AEDs in the first trimester did not find an increase in major birth defects compared with infants without such exposure.29 More recently, a large observational cohort study conducted in more than 40 countries found that the possibility of harm to a developing fetus is not only drug-specific but also dose-related.30 (To learn more, see “Pregnancy and epilepsy—when you’re managing both,” in the December 2010 issue of The Journal of Family Practice.)
Is cost a factor? Finally, consider the cost of the AED you would like to prescribe, and whether the patient has a prescription drug plan or the means to pay for his prescription.
CASE After a discussion of potential side effects, including the potential for suicidal ideation associated with AEDs, you prescribe carbamazepine for Joe as seizure prophylaxis, because it is the least expensive of the broad-spectrum AEDs and is unlikely to exacerbate his previous pancreatitis or interact with his current medications.
TABLE 2
Choosing an AED: What to consider11-18
| Epilepsy type | |||||
| Localization-related (focal/partial) | Idiopathic (generalized) | Nonidiopathic (generalized) | |||
| Anticonvulsant* | Tonic-clonic | Absence | Myoclonic | ||
| First generation | |||||
| Carbamazepine† | √ | √ | |||
| Ethosuximide† | √ | ||||
| Phenobarbital† | √ | √ | √ | ||
| Phenytoin† | √ | √ | √ | ||
| Primidone | √ | √ | √ | ||
| Valproate† | √ | √ | √ | √ | √ |
| Second generation | |||||
| Felbamate | √ | √ | |||
| Gabapentin† | √ | ||||
| Lacosamide | √ | ||||
| Lamotrigine | √† | √ | √‡ | √ | |
| Levetiracetam | √ | √ | √ | ||
| Oxcarbazepine† | √ | ||||
| Pregabalin | √ | ||||
| Rufinamide | √ | √ | |||
| Tiagabine | √ | ||||
| Topiramate | √‡ | √ | √ | ||
| Vigabatrin | √ | √ | |||
| Zonisamide | √ | √ | |||
| *Bold type indicates broad-spectrum antiepileptic drugs. †Supported by American Academy of Neurology (AAN) evidence-based guideline level A or B recommendation for monotherapy in newly diagnosed epilepsy patients. ‡Supported by AAN evidence-based guideline level B recommendation for monotherapy in newly diagnosed absence epilepsy. | |||||
TABLE W1
A closer look at antiepileptic drugs19-24
| Drug name | Maintenance dosage | Adverse effects | Cost (30-day supply)* | |
| Common | Rare/idiosyncratic | |||
| First generation | ||||
| Carbamazepine | 800-1200 mg/d | Dizziness, drowsiness, diplopia, nausea, vomiting, diarrhea, rash, pruritus, SIADH | Aplastic anemia, agranulocytosis, hyponatremia, SJS, hepatic failure, pancreatitis, suicidal ideation | $4-$50 (XR: $200) |
| Ethosuximide | 20 mg/kg per day | Sleep disturbance, drowsiness, hyperactivity, behavior changes, headache, nausea, vomiting, hiccups | Agranulocytosis, aplastic anemia, SJS, hepatic failure, serum sickness, suicidal ideation | $40-150 |
| Phenobarbital | 1-4 mg/kg per day; 120-400 mg/d | Altered sleep cycles, sedation, ataxia, lethargy, behavior changes, hyperactivity, nausea, rash | Agranulocytosis, dermatitis, SJS, hepatic failure, serum sickness, connective tissue disorders, metabolic bone disease, intellect blunting, suicidal ideation | $4-$10 |
| Phenytoin | 300-600 mg/d | Confusion, slurred speech, double vision, ataxia, nystagmus, neuropathy, hirsutism, acne, gingival hyperplasia | Neuropathy, agranulocytosis, SJS, immune reactions/serum sickness, hepatic failure, skin thickening, metabolic bone disease, suicidal ideation | $35 |
| Valproic acid | 60-350 mg/kg per day | Tremor, weight gain, PCOS, nausea, vomiting, alopecia, easy bruising | Hepatic failure, pancreatitis, hearing loss, blood dyscrasias/thrombocytopenia, hyperammonemia, encephalopathy, osteoporosis, suicidal ideation | $40 (ER: $150) |
| Second generation | ||||
| Felbamate | 2400-3600 mg/d | Somnolence, nausea, vomiting, weight loss, anorexia | Aplastic anemia (>13 years), hepatic failure, suicidal ideation | $300-$500† |
| Gabapentin | 900-1800 mg/d | Somnolence, fatigue, weight gain, nystagmus | Pedal edema, suicidal ideation | $4-$100 |
| Lacosamide | 200-400 mg/d | Headache, dizziness, ataxia, nausea, diplopia | Euphoria, prolongation of PR interval, heart block, suicidal ideation | $420† |
| Lamotrigine | 300-500 mg/d | Dizziness, ataxia, nausea, somnolence, rash | SJS, hypersensitivity reactions (renal/hepatic failure), DIC, suicidal ideation | $30-$100 |
| Levetiracetam | 3000 mg/d | Somnolence, dizziness, aggression, agitation, anxiety, weight loss | Infection, pancytopenia, liver failure, suicidal ideation | $30-$100 (XR: $245†) |
| Oxcarbazepine | 1200 mg/d | Somnolence, fatigue, headache, ataxia, nausea, rash | Hyponatremia, SJS, TEN, angioedema | $250-$1000 |
| Pregabalin | 150-600 mg/d | Peripheral edema, dry mouth, dizziness, ataxia, diplopia, weight gain | Angioedema, CK elevation, mild PR interval prolongation, suicidal ideation | $100-$350† |
| Rufinamide | 3200 mg/d | Headache, dizziness, fatigue, nausea | Shortened QT interval, hypersensitivity rash, suicidal ideation | $400-$750† |
| Tiagabine | 32-56 mg/d | Difficulty concentrating, dizziness, headache, somnolence, nervousness | Spike-wave stupor, sudden death, suicidal ideation | $140-$650† |
| Topiramate | 200-400 mg/d | Somnolence, dizziness, fatigue, weight loss, difficulty concentrating, speech problems, paresthesias, diarrhea, nausea | Acute myopia and glaucoma, hyperthermia (children); metabolic acidosis, hyperammonemia, liver failure, oligohydrosis, SJS/TEN, kidney stones, suicidal ideation | $40 - $100 |
| Vigabatrin | 1500 mg/d | Fatigue, somnolence, nystagmus, tremor, weight gain | Vision loss (30% of patients) blurred vision, arthralgia, suicidal ideation | :$50 -$100† |
| Zonisamide | 400- 600 mg/d | Somnolence, difficulty concentrating, anorexia, nausea | SJS, TEN, aplastic anemia, agranulocytosis, nephrolithiasis/, oligohydrosis, acidosis, suicidal ideation | $50-$200 |
| CK, creatine kinase; DIC, disseminated intravascular coagulation; ER, extended release; IV, intravenous; PCOS, polycystic ovarian syndrome; SIADH, syndrome of inappropriate antidiuretic hormone hypersecretion; SJS, Stevens-Johnson syndrome; TEN, toxic epidermal necrolysis, XR, extended release. *Costs from www.drugstore.com, www.savewithgenericdrugs.com, and www.pharmacychecker.com. †No generic available. | ||||
When to add a second AED
Monotherapy is the preferred method of epilepsy treatment, and controls seizures for 70% to 90% of patients.31,32 If seizures continue and potential adverse effects prevent you from increasing the dosage, switching to a different AED, then tapering off the first agent, is recommended.33,34
If the new AED fails to provide adequate seizure control, consider combination therapy. An additional 10% to 15% of patients with epilepsy achieve control with dual therapy.33,34
Many second-generation agents are approved for adjunctive therapy. However, the use of 2 AEDs increases the risk of toxicities and drug interactions, and requires complex dosage adjustments, which should be done slowly and cautiously. Combination therapy also increases costs and may cause a decrease in compliance.33,34
Noncompliance is the single most common reason for treatment failure in patients with epilepsy, occurring at an estimated rate of up to 60%.35,36 The complexity of the drug regimen is the major cause, regardless of patient age, sex, psychomotor development, seizure type, or seizure frequency.35,36
Because of the lack of good clinical trials of combination antiepilepsy therapy, no evidence is available to indicate which AEDs are safe and effective when taken together. There is, however, evidence that certain combinations should be avoided due to the risk of increased adverse effects. These include phenobarbital/valproate, phenytoin/carbamazepine, and carbamazepine/lamotrigine.25
Managing the patient who is seizure-free
After a patient has been seizure-free for 2 to 5 years, consider a reduction in, or a discontinuation of, his or her AED. The relapse rate varies from 10% to 70%, with meta-analyses showing a rate of 25% in the first year and 29% in the second year.19,37 The American Academy of Neurology (AAN) has published an evidence-based guideline for discontinuing AEDs in seizure-free patients, available at www.aan.com/professionals/practice/pdfs/gl0007.pdf.
Withdrawal should be gradual and, for patients on combination therapy, carried out one drug at a time to prevent a recurrence of seizures or status epilepticus. The AAN recommends a 2- to- 3-month withdrawal period for AEDs (and longer for benzodiazepines), although relapse rates have been found to be lower when the medication is withdrawn more slowly, over about 6 months.19,34 If seizures recur after withdrawal, restart the AEDs at previous dosages.19,34,38
Should the patient drive?
For patients with epilepsy, loss of independence related to driving restrictions is a major source of stress. A 10-year follow-up study of Danish patients with epilepsy found a 7-fold increase in motor vehicle accidents (MVAs) in patients with seizure disorders.39 Other studies have shown that the seizure-free interval is the best predictor of involvement in an MVA.40
The risk of driving accidents decreases as the seizure-free interval increases. Unfortunately, however, a decline in patient compliance is also associated with longer seizure-free intervals—creating the potential for recurrence and driving risk. Because of this discrepancy, a consensus statement from the AAN, American Epilepsy Society, and Epilepsy Foundation of America recommends a minimum 3-month seizure-free interval before patients are allowed to drive.41
Use clinical judgment in deciding whether to extend the seizure-free period. State laws vary widely regarding the need to report patients with seizure disorders, limitations on professional drivers, and seizure-free intervals required, so it is important to be familiar with the laws in your state. The Epilepsy Foundation has a helpful online resource with a database detailing individual state statutes (http://www.epilepsyfoundation.org/living/wellness/transportation/driverlicensing.cfm).
The danger of uncontrolled seizures
Overall, AEDs effectively control 70% of 80% of cases; the remaining 20% to 30% are considered medically refractory.38 What’s more, after 2 AED failures, a patient’s chances of achieving full seizure control with additional drugs are no better than 10% to 20%.42 And, as more drugs are tried, the likelihood of full control declines even further.43
Patients with uncontrolled seizures have a cumulative risk of sudden unexpected death in epilepsy (SUDEP) of 0.5% per year.44 Cognitive decline is associated with uncontrolled epilepsy, as well. In children, frequent seizures may significantly alter neuronal networks, affecting cognitive and motor development.
Is your patient a candidate for surgery?
Patients with disabling complex partial seizures that remain uncontrolled after 2 or more AED trials (either as monotherapy or in combination) should be referred to an epilepsy specialty center for evaluation for surgery.45 This should be considered as early as possible to afford the patient the best chance of achieving seizure control.
Successful epilepsy surgery—in which the portion of the brain causing the misfiring that causes the seizures is removed—often results in a better quality of life; it is also cost effective.46 Not everyone with refractory epilepsy is a candidate for surgery, of course. Among those who are, however, 50% to 70% of patients can expect to have improved seizure control.47
Status epilepticus is a medical emergency
A patient who develops status epilepticus is at high risk and requires immediate, and simultaneous, evaluation and treatment. Status epilepticus carries nearly a 20% mortality from the first episode,48 and the 10-year mortality rate after an episode of status epilepticus is as high as 40%.49
Although most of the deaths associated with status epilepticus are due to the underlying pathology, early treatment can prevent or ameliorate complications from rhabdomyolysis and irreversible anoxic neuronal damage.50
A benzodiazepine (typically, a 10-mg IV bolus of diazepam) is the initial treatment for status epilepticus, followed by or concurrent with fosphenytoin (15-18 mg/kg). If status epilepticus remains refractory to first-line drugs (lasting >30 minutes), intubation and transfer to an intensive care setting may be required, and a neurological consult should be obtained.
Pharmacologic treatment of status epilepticus falls into 3 main classes: benzodiazepines, standard AEDs, and general anesthetics such as propofol. Benzodiazepines act very rapidly to control most prolonged seizures, and are the first-line treatment choice. Diazepam has long been the mainstay of treatment, and is usually readily available. However, in both a large systematic review and a head-to-head trial, lorazepam was found to be superior to diazepam in ending seizure activity and maintaining seizure control without the use of other medications51,52—and is now the drug of choice for initial treatment of status epilepticus.
CASE You continue to see Joe every 3 to 4 months to monitor his basic blood work and mood. A year after his seizure, he remains seizure-free and is tolerating the AED without any adverse effects.
CORRESPONDENCE
William J. Geiger, MD, FAAFP, Medical College of Wisconsin, Columbia St. Mary’s Family Medicine Residency, 1121 East North Avenue, Milwaukee, WI 53212; bgeiger@mcw.edu
1. Epilepsy Foundation of America. Epilepsy and seizure statistics. Available at: http://www.epilepsyfoundation.org/about/statistics.cfm. Accessed June 15, 2009.
2. Centers for Disease Control and Prevention (CDC). Prevalence and most common causes of disability among adults—United States, 2005. MMWR Morb Mortal Wkly Rep. 2009;58:421-426. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm5816a2.htm. Accessed June 15, 2009.
3. Hirtz D, Thurman DJ, Gwinn-Hardy K, et al. How common are the “common” neurologic disorders? Neurology. 2007;68:326-337.
4. Engel J Jr. ILAE classification of epilepsy syndromes. Epilepsy Res. 2006;70(suppl 1):S5-S10.
5. Rudzinski LA, Shih JJ. Continuum: lifelong learning in neurology. Epilepsia. 2010;16:15-35.
6. Chaves J, Sander JW. Seizure aggravation in idiopathic generalized epilepsies. Epilepsia. 2005;46(suppl 9):S133-S139.
7. Beghi E. Management of first seizure. General conclusions and recommendations. Epilepsia. 2008;49(suppl 1):S58-S61.
8. Berg A. Risk of recurrence after a first unprovoked seizure. Epilepsia. 2008;49(suppl 1):S13-S18.
9. Hirtz D, Ashwal S, Berg A, et al. Practice parameter: evaluating a first non-febrile seizure in children: report of the Quality Standards Subcommittee of the American Academy of Neurology, the Child Neurology Society, and the American Epilepsy Society. Neurology. 2000;55:616-623.
10. US Food and Drug Administration. Suicidal behavior and ideation and antiepileptic drugs. Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm100190.htm. Updated May 5, 2009. Accessed June 28, 2009.
11. French JA, Kanner AM, Bautista J, et al. Efficacy and tolerability of the new antiepileptic drugs I: treatment of new epilepsy, report of the therapeutic and technology assessment subcommittee and quality standards subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2004;62:1252-1260.
12. French J, Smith M, Faught E, et al. Practice advisory: the use of felbamate in the treatment of patients with intractable epilepsy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 1999;52:1540-1545.
13. Glauser T, Kluger G, Sachdeo R, et al. Rufinamide for generalized seizures associated with Lennox-Gastaut syndrome. Neurology. 2008;70:1950-1958.
14. Suzuki Y, Nagai T, Ono J, et al. Zonisamide monotherapy in newly-diagnosed infantile spasms. Epilepsia. 1997;38:1035-1038.
15. Kochak GM, Page JG, Buchanan RA, et al. Steady-state pharmacokinetics of zonisamide, an antiepileptic agent for treatment of refractory complex partial seizures. J Clin Pharmacol. 1998;38:166-171.
16. Arroyo S, Anhut H, Kugler AR, et al. Pregabalin 1008-011 International Study Group. Pregabalin add-on treatment: a randomized, double-blind, placebo-controlled, dose-response study in adults with partial seizures. Epilepsia. 2004;45:20-27.
17. Brodie MJ, Rosenfeld WE, Vazquez B, et al. Rufinamide for the adjunctive treatment of partial seizures in adults and adolescents: a randomized placebo-controlled trial. Epilepsia. 2009;50:1899-1909.
18. Ben-Menachem E, Biton V, Jatuzis D, et al. Efficacy and safety of oral lacosamide as adjunctive therapy in adults with partial-onset seizures. Epilepsia. 2007;48:1308-1317.
19. Gidal B, Garnett W. Epilepsy. In: Dipiro J, et al, eds. Pharmacotherapy: A Pathophysiologic Approach. 6th ed. New York: McGraw-Hill; 2005:1023-1048.
20. Pellock JM, Treatment of epilepsy in the new millennium. Pharmacotherapy. 2000;20:129S-138S.
21. Schachter S. Pharmacology of antiepileptic drugs. Available at: http://www.utdonline.com/online/content/topic.do?topicKey=epil_eeg/5220. Accessed July 15, 2009.
22. Woelfel J. Comparison of antiepileptic drugs. Pharmacist’s Letter/Prescriber's Letter. July 2009;25:1-24.
23. Wolters Kluwer Health Inc. Anticonvulsants. Drug facts and comparisons online. Available at: http://www.efactsonline.com. Accessed July 10, 2009.
24. US Food and Drug Administration. Information for healthcare professionals. Suicidality and antiepileptic drugs [FDA alert]. Available at: http://www.fda.gov/Drugs/DrugSafety/PublicHealthAdvisories/ucm054709.htm. January 31, 2008. Accessed June 30, 2009.
25. French J. Treatment with antiepileptic drugs, new and old. Continuum. 2007;13:71-90.
26. Sheehan M. Polycystic ovarian syndrome: diagnosis and management. Clin Med Res. 2004;2:13-27.
27. Harden CL. Sexual dysfunction in women with epilepsy. Seizure. 2008;17:131-135.
28. Harden CL, Hopp J, Ting TY, et al. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): obstetrical complications and change in seizure frequency. Neurology. 2009;73:126-132.
29. Molgaard-Nielsen D, Hviid A. Newer-generation antiepileptic drugs and the risk of major birth defects. JAMA. 2011;305:1996-2002.
30. Tomson T, Battino D, Bonizonni E, et al. Dose-dependent risk of malformations with antiepileptic drugs: an analysis of data from the EURAP epilepsy and pregnancy registry. Lancet Neurol. 2011;10:609-617.
31. Callaghan BC, Anand K, Hesdorffer D, et al. Likelihood of seizure remission in an adult population with refractory epilepsy. Ann Neurol. 2007;62:382-389.
32. Luciano AL, Shorvon SD. Results of treatment changes in patients with apparently drug-resistant chronic epilepsy. Ann Neurol. 2007;62:375-381.
33. Abramowicz M, ed. Drugs for epilepsy [treatment guidelines]. The Medical Letter. 2008;70:1-12.
34.Stokes T, Shaw EJ, Juarez-Garcia A, et al. Clinical guidelines and evidence review for the epilepsies: diagnosis and management in adults and children in primary and secondary care. London: Royal College of General Practitioners. Available at: www.nice.org.uk/CG020fullguideline. Published October 2004. Accessed July 10, 2009.
35. Garnett WR. Antiepileptic drug treatment: outcomes and adherence. Pharmacotherapy. 2000;20:191S-199S.
36. Briesacher BA, Andrade SE, Fouayzi H, et al. Comparison of drug adherence rates among patients with seven different medical conditions. Pharmacotherapy. 2008;28:437-443.
37. Shinnar S, Gross-Tsur V. Discontinuing antiepileptic drug therapy. In: Wyllie E, ed. The Treatment of Epilepsy. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2001:811-819.
38. Kwan P, Brodie J. Refractory epilepsy: a progressive, intractable but preventable condition? Seizures. 2002;11:77-84.
39. Lings S. Increased driving accident frequency in Danish patients with epilepsy. Neurology. 2001;57:435-439.
40. Krauss GL, Krumholz A, Carter RC, et al. Risk factors for seizure-related motor vehicle crashes in patients with epilepsy. Neurology. 1999;52:1324-1329.
41. American Academy of Neurology, American Epilepsy Society, and Epilepsy Foundation of America. Consensus statements, sample statutory provisions, and model regulations regarding driver licensing and epilepsy. Epilepsia. 1994;35:696-705.
42. Thadani VM, Taylor J. Surgical treatments for epilepsy. Continuum. 2007;13:152-176.
43. Brodie MJ, Kwan P. Staged approach to epilepsy management. Neurology. 2002;58(8 suppl 5):S2-S8.
44. Sillanpaa M, Jalava M, Kaleva O, et al. Long-term prognosis of seizures with onset in childhood. N Engl J Med. 1998;338:1715-1722.
45. Engel J Jr, Wiebe S, French J, et al. Practice parameter: temporal lobe and localized neocortical resections for epilepsy. Neurology. 2003;60:538-547.
46. Boon P, D'Have M, Van Walleghen P, et al. Direct medical costs of refractory epilepsy incurred by three different treatment modalities: a prospective assessment. Epilepsia. 2002;43:96-102.
47. Passaro EA. Outcome of epilepsy surgery. Available at: http://emedicine.medscape.com/article/1185416-overview. Updated May 16, 2011. Accessed June 28, 2011.
48. DeLorenzo RJ, Pellock JM, Towne AR, et al. Epidemiology of status epilepticus. J Clin Neurophysiol. 1995;12:316-325.
49. Logroscino G, Hesdorffer DC, Cascino GD, et al. Long-term mortality after a first episode of status epilepticus. Neurology. 2002; 58:537-541.
50. Kalviaine R. Treatment of status epilepticus. Essential Evidence Plus. Wiley-Blackwell. Available at: http://www.essentialevidenceplus.com/content/ebmg_ebm/766. Accessed July 15, 2009.
51. Prasad K, Al-Roomi K, Krishnan PR, et al. Anticonvulsant therapy for status epilepticus. Cochrane Database Syst Rev. 2005;(4):CD003723.
52. Treiman DM, Meyers PD, Walton NY, et al. A comparison of four treatments for generalized convulsive status epilepticus. N Engl J Med. 1998;339:792-798.
1. Epilepsy Foundation of America. Epilepsy and seizure statistics. Available at: http://www.epilepsyfoundation.org/about/statistics.cfm. Accessed June 15, 2009.
2. Centers for Disease Control and Prevention (CDC). Prevalence and most common causes of disability among adults—United States, 2005. MMWR Morb Mortal Wkly Rep. 2009;58:421-426. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm5816a2.htm. Accessed June 15, 2009.
3. Hirtz D, Thurman DJ, Gwinn-Hardy K, et al. How common are the “common” neurologic disorders? Neurology. 2007;68:326-337.
4. Engel J Jr. ILAE classification of epilepsy syndromes. Epilepsy Res. 2006;70(suppl 1):S5-S10.
5. Rudzinski LA, Shih JJ. Continuum: lifelong learning in neurology. Epilepsia. 2010;16:15-35.
6. Chaves J, Sander JW. Seizure aggravation in idiopathic generalized epilepsies. Epilepsia. 2005;46(suppl 9):S133-S139.
7. Beghi E. Management of first seizure. General conclusions and recommendations. Epilepsia. 2008;49(suppl 1):S58-S61.
8. Berg A. Risk of recurrence after a first unprovoked seizure. Epilepsia. 2008;49(suppl 1):S13-S18.
9. Hirtz D, Ashwal S, Berg A, et al. Practice parameter: evaluating a first non-febrile seizure in children: report of the Quality Standards Subcommittee of the American Academy of Neurology, the Child Neurology Society, and the American Epilepsy Society. Neurology. 2000;55:616-623.
10. US Food and Drug Administration. Suicidal behavior and ideation and antiepileptic drugs. Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm100190.htm. Updated May 5, 2009. Accessed June 28, 2009.
11. French JA, Kanner AM, Bautista J, et al. Efficacy and tolerability of the new antiepileptic drugs I: treatment of new epilepsy, report of the therapeutic and technology assessment subcommittee and quality standards subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2004;62:1252-1260.
12. French J, Smith M, Faught E, et al. Practice advisory: the use of felbamate in the treatment of patients with intractable epilepsy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 1999;52:1540-1545.
13. Glauser T, Kluger G, Sachdeo R, et al. Rufinamide for generalized seizures associated with Lennox-Gastaut syndrome. Neurology. 2008;70:1950-1958.
14. Suzuki Y, Nagai T, Ono J, et al. Zonisamide monotherapy in newly-diagnosed infantile spasms. Epilepsia. 1997;38:1035-1038.
15. Kochak GM, Page JG, Buchanan RA, et al. Steady-state pharmacokinetics of zonisamide, an antiepileptic agent for treatment of refractory complex partial seizures. J Clin Pharmacol. 1998;38:166-171.
16. Arroyo S, Anhut H, Kugler AR, et al. Pregabalin 1008-011 International Study Group. Pregabalin add-on treatment: a randomized, double-blind, placebo-controlled, dose-response study in adults with partial seizures. Epilepsia. 2004;45:20-27.
17. Brodie MJ, Rosenfeld WE, Vazquez B, et al. Rufinamide for the adjunctive treatment of partial seizures in adults and adolescents: a randomized placebo-controlled trial. Epilepsia. 2009;50:1899-1909.
18. Ben-Menachem E, Biton V, Jatuzis D, et al. Efficacy and safety of oral lacosamide as adjunctive therapy in adults with partial-onset seizures. Epilepsia. 2007;48:1308-1317.
19. Gidal B, Garnett W. Epilepsy. In: Dipiro J, et al, eds. Pharmacotherapy: A Pathophysiologic Approach. 6th ed. New York: McGraw-Hill; 2005:1023-1048.
20. Pellock JM, Treatment of epilepsy in the new millennium. Pharmacotherapy. 2000;20:129S-138S.
21. Schachter S. Pharmacology of antiepileptic drugs. Available at: http://www.utdonline.com/online/content/topic.do?topicKey=epil_eeg/5220. Accessed July 15, 2009.
22. Woelfel J. Comparison of antiepileptic drugs. Pharmacist’s Letter/Prescriber's Letter. July 2009;25:1-24.
23. Wolters Kluwer Health Inc. Anticonvulsants. Drug facts and comparisons online. Available at: http://www.efactsonline.com. Accessed July 10, 2009.
24. US Food and Drug Administration. Information for healthcare professionals. Suicidality and antiepileptic drugs [FDA alert]. Available at: http://www.fda.gov/Drugs/DrugSafety/PublicHealthAdvisories/ucm054709.htm. January 31, 2008. Accessed June 30, 2009.
25. French J. Treatment with antiepileptic drugs, new and old. Continuum. 2007;13:71-90.
26. Sheehan M. Polycystic ovarian syndrome: diagnosis and management. Clin Med Res. 2004;2:13-27.
27. Harden CL. Sexual dysfunction in women with epilepsy. Seizure. 2008;17:131-135.
28. Harden CL, Hopp J, Ting TY, et al. Practice parameter update: management issues for women with epilepsy—focus on pregnancy (an evidence-based review): obstetrical complications and change in seizure frequency. Neurology. 2009;73:126-132.
29. Molgaard-Nielsen D, Hviid A. Newer-generation antiepileptic drugs and the risk of major birth defects. JAMA. 2011;305:1996-2002.
30. Tomson T, Battino D, Bonizonni E, et al. Dose-dependent risk of malformations with antiepileptic drugs: an analysis of data from the EURAP epilepsy and pregnancy registry. Lancet Neurol. 2011;10:609-617.
31. Callaghan BC, Anand K, Hesdorffer D, et al. Likelihood of seizure remission in an adult population with refractory epilepsy. Ann Neurol. 2007;62:382-389.
32. Luciano AL, Shorvon SD. Results of treatment changes in patients with apparently drug-resistant chronic epilepsy. Ann Neurol. 2007;62:375-381.
33. Abramowicz M, ed. Drugs for epilepsy [treatment guidelines]. The Medical Letter. 2008;70:1-12.
34.Stokes T, Shaw EJ, Juarez-Garcia A, et al. Clinical guidelines and evidence review for the epilepsies: diagnosis and management in adults and children in primary and secondary care. London: Royal College of General Practitioners. Available at: www.nice.org.uk/CG020fullguideline. Published October 2004. Accessed July 10, 2009.
35. Garnett WR. Antiepileptic drug treatment: outcomes and adherence. Pharmacotherapy. 2000;20:191S-199S.
36. Briesacher BA, Andrade SE, Fouayzi H, et al. Comparison of drug adherence rates among patients with seven different medical conditions. Pharmacotherapy. 2008;28:437-443.
37. Shinnar S, Gross-Tsur V. Discontinuing antiepileptic drug therapy. In: Wyllie E, ed. The Treatment of Epilepsy. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2001:811-819.
38. Kwan P, Brodie J. Refractory epilepsy: a progressive, intractable but preventable condition? Seizures. 2002;11:77-84.
39. Lings S. Increased driving accident frequency in Danish patients with epilepsy. Neurology. 2001;57:435-439.
40. Krauss GL, Krumholz A, Carter RC, et al. Risk factors for seizure-related motor vehicle crashes in patients with epilepsy. Neurology. 1999;52:1324-1329.
41. American Academy of Neurology, American Epilepsy Society, and Epilepsy Foundation of America. Consensus statements, sample statutory provisions, and model regulations regarding driver licensing and epilepsy. Epilepsia. 1994;35:696-705.
42. Thadani VM, Taylor J. Surgical treatments for epilepsy. Continuum. 2007;13:152-176.
43. Brodie MJ, Kwan P. Staged approach to epilepsy management. Neurology. 2002;58(8 suppl 5):S2-S8.
44. Sillanpaa M, Jalava M, Kaleva O, et al. Long-term prognosis of seizures with onset in childhood. N Engl J Med. 1998;338:1715-1722.
45. Engel J Jr, Wiebe S, French J, et al. Practice parameter: temporal lobe and localized neocortical resections for epilepsy. Neurology. 2003;60:538-547.
46. Boon P, D'Have M, Van Walleghen P, et al. Direct medical costs of refractory epilepsy incurred by three different treatment modalities: a prospective assessment. Epilepsia. 2002;43:96-102.
47. Passaro EA. Outcome of epilepsy surgery. Available at: http://emedicine.medscape.com/article/1185416-overview. Updated May 16, 2011. Accessed June 28, 2011.
48. DeLorenzo RJ, Pellock JM, Towne AR, et al. Epidemiology of status epilepticus. J Clin Neurophysiol. 1995;12:316-325.
49. Logroscino G, Hesdorffer DC, Cascino GD, et al. Long-term mortality after a first episode of status epilepticus. Neurology. 2002; 58:537-541.
50. Kalviaine R. Treatment of status epilepticus. Essential Evidence Plus. Wiley-Blackwell. Available at: http://www.essentialevidenceplus.com/content/ebmg_ebm/766. Accessed July 15, 2009.
51. Prasad K, Al-Roomi K, Krishnan PR, et al. Anticonvulsant therapy for status epilepticus. Cochrane Database Syst Rev. 2005;(4):CD003723.
52. Treiman DM, Meyers PD, Walton NY, et al. A comparison of four treatments for generalized convulsive status epilepticus. N Engl J Med. 1998;339:792-798.
Hoarseness and chronic cough: Would you suspect reflux?
• Recommend dietary and behavioral modifications as a first step in treating patients with symptoms suggestive of laryngopharyngeal reflux disease (LPRD). C
• When medications are needed, prescribe a high-dose proton-pump inhibitor, a histamine-2 blocker at bedtime, and prophylactic antacids for reflux-inducing activities, such as exercising and eating. B
• Avoid the rebound effect associated with abrupt cessation of medications prescribed for LPRD with a gradual, 16-week taper. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE When Joan C, a 35-year-old patient whom you’ve known for years, comes in for a physical, you notice that she’s coughing frequently. Upon questioning, Joan says she first noticed the cough several months ago; she also reports that she’s frequently hoarse, but has no other symptoms. Joan is a former smoker, and quit 4 years ago.
If Joan were your patient, would you suspect that she had an upper respiratory infection and prescribe an antibiotic such as azithromycin? Would you include laryngopharyngeal reflux disease in the differential diagnosis?
Laryngopharyngeal reflux disease (LPRD) is a common condition that most primary care physicians encounter frequently. It is also frequently misdiagnosed by clinicians who are unfamiliar with the differences between LPRD and gastroesophageal reflux disease (GERD).
The American Academy of Otolaryngology–Head and Neck Surgery defines laryngopharyngeal reflux as the retrograde movement of gastric contents into the laryngopharynx.1 Common symptoms include hoarseness/dysphonia, chronic throat clearing, dysphagia, globus pharyngeus, and chronic cough, as well as postnasal drip, paroxysmal laryngospasm, odynophagia, excessive throat mucus, and a strange taste in the mouth.2
The diversity and vagueness of these symptoms, as well as the lack of a gold standard diagnostic test for LPRD, make it difficult to estimate its prevalence. In addition, signs of gastroesophageal reflux can be found in the laryngopharynx of up to 86% of healthy individuals, further complicating the clinical picture.3 To avoid missing this often overlooked reflux disease, you need to know how it develops, what signs and symptoms to look for, and which distinguishing features to keep in mind.
Pathophysiology and distinguishing features
The precise way in which LPRD develops is not known, but there are 2 proposed means of laryngeal injury—direct and indirect. In the first case, chemical irritants in the gastric refluxate enter the laryngopharynx and cause local mucosal injury. In the second, gastric reflux irritates the esophageal tissue enough to evoke laryngeal reflexes without ever reaching the larynx—a vagally mediated response associated with symptoms such as chronic cough, throat-clearing sensations, and bronchoconstriction.4
Unlike the esophageal lining, laryngeal epithelium is not protected against chemical injury from gastric acid, as it lacks both the stripping motion of esophageal peristalsis and the neutralizing bicarbonate in saliva.4 Thus, while far smaller amounts of gastric reflux make it into the laryngopharynx, the acid remains there longer and may cause greater injury.5 In some cases, this occurs as often as 50 times a day, although as few as 3 episodes per week have been known to cause LPRD.5
Heartburn is not the rule
Heartburn is a primary complaint of patients with GERD. It is reported by little more than a third (35%) of those with LPRD,5,6 however, (which is why it is sometimes called the “silent” reflux disease). This is because heartburn is caused by esophagitis due to esophageal dysmotility and lower esophageal sphincter dysfunction,3 while most patients with LPRD have normal esophageal motor function and upper esophageal sphincter dysfunction. The fact that only a minimal amount of reflux enters the laryngopharynx may be part of the reason heartburn is less likely in patients with LPRD.
Onset of symptoms. When reflux occurs is another thing that distinguishes LPRD and GERD. Symptoms of GERD typically worsen when the individual is supine, while laryngopharyngeal reflux usually occurs when he or she is upright.7 The frequency with which these 2 conditions overlap is debatable, as there are few studies differentiating LPRD and GERD based on standardized signs and symptoms.7
Making sense of signs and symptoms
Most patients with LPRD seek treatment from their primary care physician, typically reporting symptoms that they don’t associate with gastric reflux, such as hoarseness, a chronic cough or sore throat, or the sensation of a lump in the throat (TABLE 1). Less common manifestations include “water brash”—excessive mucus in the mouth caused by a release of salivary bicarbonate to help neutralize acidity8—otitis media, sinus disease, and dental caries.5
Laryngeal endoscopy may reveal many changes from diffuse irritation. Diffuse erythema, edema, and interarytenoid hypertrophy/cobblestoning are the most useful findings for an LPRD diagnosis.9,10 But in most cases, only a few nonspecific signs with a number of possible causes (infection, environmental irritants, allergies, temperature/climate change, among others) are seen on endoscopic examination, with little correlation with symptom severity. In fact, 74% of otolaryngologists responding to a recent survey said they relied more on patient symptoms than on laryngeal signs for an LPRD diagnosis.10
The Reflux Finding Score (RFS), available at http://www.nature.com/gimo/contents/pt1/fig_tab/gimo46_T3.html, is a clinical tool developed to quantify laryngeal inflammation and standardize objective endoscopic findings. The RFS incorporates the following endolaryngeal signs:
- subglottic edema
- ventricular obliteration
- erythema/hyperemia
- vocal cord edema
- diffuse laryngeal edema
- posterior commissure hypertrophy
- granuloma/granulation tissue
- thick endolaryngeal mucus.
A numeric value is assigned to each, based on whether it is present or absent; partial or complete; local or diffuse; or mild or severe. However, the RFS, too, is an imperfect tool. Clinicians who have used the RFS report that a score higher than 7 identifies LPRD with 95% sensitivity.11 But laryngeal findings may be due to other causes, such as infection, autoimmune reaction, or even allergies, and studies have found the RFS to have poor specificity and inter-rater reliability.12-14
Ambulatory dual probe pH monitoring was considered to be the gold standard test for LPRD at one time, but newer studies have raised questions about its validity and usefulness, especially in patients taking proton-pump inhibitors (PPIs).1,5,7 Newer advanced probes featuring less invasive data collection and greater sensitivity are under development. Ambulatory 24-hour multichannel intraluminal impedance with pH monitoring is the most promising new diagnostic tool, as it can monitor both acidic and nonacidic reflux and distinguish between gas and liquid.15
TABLE 1
When to suspect laryngopharyngeal reflux disease1,5,24
| Finding | Frequency among patients with LPRD (%)* |
|---|---|
| Dysphonia/hoarseness (intermittent) | 71 |
| Chronic cough | 51 |
| Globus pharyngeus | 47 |
| Chronic throat clearing | 42 |
| Dysphagia | 35 |
| Heartburn | 35 |
| *The frequency of other symptoms associated with LPRD is not known. | |
Treatment, like diagnosis, is not clear-cut
LPRD is often called a diagnosis of exclusion, because of the nonspecific nature of its signs and symptoms and the importance of considering a range of other etiologies. The differential diagnosis includes excessive voice use, postnasal drip, upper respiratory infection, habitual throat clearing, allergic rhinitis, environmental irritants, temperature/climate change, chronic or episodic use of alcohol and/or tobacco, and psychological problems related to tics, such as habitual throat clearing or coughing.5
Diagnosis is often based on an empiric trial of high-dose PPIs, with confirmation dependent on symptom relief. Because there have been few placebo-controlled trials with PPIs and those that have been completed had conflicting results, diagnosis based on a combination of medical history and endoscopic laryngeal examination may be a better approach.16,17
Acid suppression therapy with either PPIs or histamine-2 (H2) receptor blockers such as ranitidine or famotidine is the mainstay of treatment for LPRD. But medical societies offer conflicting advice. The American Gastroenterological Association cautions clinicians not to prescribe acid-suppression therapy for patients with LPRD unless they also have GERD.6 The American Academy of Otolaryngology–Head and Neck Surgery recommends twice-daily PPI use for ≥6 months.1,13 The general consensus, based on clinical experience alone, is that patients should be treated with high doses of PPIs (eg, 40 mg omeprazole twice a day) for ≥6 months, with the addition of an H2 receptor blocker to help reduce overnight acid production.1,18 Prophylactic antacid use is also recommended in anticipation of reflux, such as before exercising and right after a meal.
Symptoms should start to improve within 6 to 8 weeks, and patients should be reassessed in about 3 months. To avoid a rebound effect from the abrupt cessation of medications, we suggest a gradual taper over 16 weeks. For the first 8 weeks, the H2 blocker should be discontinued and the PPI decreased from twice a day to once. If symptoms are still controlled, the PPI dose can be reduced to once every other day for another 8 weeks, then stopped if symptoms do not recur.18
Lifestyle and dietary changes (TABLE 2), such as smoking cessation, weight loss, and avoidance of alcohol, are an important part of LPRD treatment, and may be used as a first-line therapy before prescribing medication.19 In fact, some studies have found PPI therapy to be inferior to behavioral/lifestyle modifications.17
Fundoplication surgery, a procedure in which the gastric fundus of the stomach is wrapped around the lower end of the esophagus and stitched in place to prevent reflux, may be an option for patients who do not respond to, or cannot tolerate, aggressive medical treatment for LPRD. A 2006 prospective controlled study found that surgical fundoplication did not consistently relieve laryngeal symptoms.20 But other studies have found that a carefully selected population with medically unresponsive laryngopharyngeal symptoms can benefit from this procedure.21,22 One study showed a significant improvement within one month of fundoplication, with continued improvement observed during a 3-year follow-up.21 In another prospective study, researchers showed that while LPRD-related laryngeal symptoms such as coughing and throat-clearing improved with both medical therapy and laparoscopic fundoplication, voice quality and endoscopic laryngeal/pharyngeal findings improved significantly only with the surgical procedure.23
TABLE 2
Recommend these lifestyle modifications19
| Stop smoking |
Avoid:
|
| Eat smaller, more frequent meals |
| Avoid eating within 3 hours of bedtime |
| Lose weight |
CORRESPONDENCE
Shoib Sana, DO, Detroit Medical Center, Otolaryngology-Head and Neck Surgery, 6533 East Jefferson Avenue, Apartment 316, Detroit, MI 48207; ssana@dmc.org
1. Koufman JA, Aviv JE, Casiano RR, et al. Laryngopharyngeal reflux: position statement of the committee on speech, voice, and swallowing disorders of the American Academy of Otolaryngology-Head and Neck Surgery. Otolaryngol Head Neck Surg. 2002;127:32-35.
2. Papakonstantinou L, Leslie P, Gray J, et al. Laryngopharyngeal reflux: a prospective analysis of a 34 item symptom questionnaire. Clin Otolaryngol. 2009;34:455-459.
3. Hicks DM, Ours TM, Abelson TI, et al. The prevalence of hypopharynx findings associated with gastroesophageal reflux in normal volunteers. J Voice. 2002;16:564.-
4. Johnston N, Bulmer D, Gill GA, et al. Cell biology of laryngeal epithelial defenses in health and disease: further studies. Ann Otol Rhinol Laryngol. 2003;112:481-491.
5. Koufman JA. The otolaryngologic manifestations of gastroesophageal reflux disease (GERD): a clinical investigation of 225 patients using ambulatory 24 hour pH monitoring and an experimental investigation of the role of acid and pepsin in the development of laryngeal injury. Laryngoscope. 1991;101:1-78.
6. Kahrilas PJ, Shaheen NJ, Vaezi M, et al. American Gastroenterological Association Institute (AGAI) medical position statement: management of gastroesophageal reflux disease. Gastroenterology. 2008;135:1383.-
7. Postma GN, Tomek MS, Belafsky PC, et al. Esophageal motor function in laryngopharyngeal reflux is superior to that in classic gastroesophageal reflux disease. Ann Otol Rhinol Laryngol. 2001;111:1114-1116.
8. Helen JF, Dodds WJ, Hogan WJ. Salivary response to esophageal acid in normal subjects and patients with reflux esophagitis. Gastroenterology. 1998;94:1394-1398.
9. Belafsky PC. Abnormal endoscopic pharyngeal and laryngeal findings attributable to reflux. Am J Med 2003;116(suppl 3A):91S-97S.
10. Ahmed TF, Khandwala F, Abelson, et al. Chronic laryngitis associated with gastroesophageal reflux: prospective assessment of differences in practice patterns between gastroenterologists and ENT physicians. Am J Gastroenterol. 2006;102:470-478.
11. Belafsky PC, Postma GN, Koufman JA. The validity and reliability of the reflux finding score (RFS). Laryngoscope. 2001;111:1313-1317.
12. Koufman JA, Sataloff RT, Toohill R. Laryngopharyngeal reflux: consensus conference report. J Voice. 1996;10:215-216.
13. Belafsky PC, Postma GN, Koufman JA. Laryngopharyngeal reflux symptoms improve before changes in physical findings. Laryngoscope 2001;111:979-981.
14. Reichel O, Dressel H, Wiederanders K, et al. Double-blind, placebo-controlled trial with esomeprazole for symptoms and signs associated with laryngopharyngeal reflux. Otolaryngol Head Neck Surg. 2008;139:414-420.
15. Muderris T, Gokcan MK, Yorulmaz I. The clinical value of pharyngeal pH monitoring using a double-probe, triple-sensor catheter in patients with laryngopharyngeal reflux. Arch Otolaryngol Head Neck Surg. 2009;135:163-167.
16. Steward DL, Wilson KM, Kelly DH, et al. Proton pump inhibitor therapy for chronic laryngo-pharyngitis: a randomized placebo-control trial. Otolaryngol Head Neck Surg. 2004;131:342-350.
17. Wo JM, Koopman J, Harrell SP, et al. Double-blind, placebo-controlled trial with single-dose pantoprazole for laryngopharyngeal reflux. Am J Gastroenterol. 2006;101:1972-1978.
18. Park W, Hicks DM, Khandwala F, et al. Laryngopharyngeal reflux: prospective cohort study evaluating optimal dose of proton-pump inhibitor therapy and pretherapy predictors of response. Laryngoscope. 2005;116:1230-1238.
19. Maceri DR, Zim S. Laryngospasm: an atypical manifestation of severe gastroesophageal reflux disease. Laryngoscope. 2001;111:1976-1979.
20. Swoger J, Ponsky J, Hicks DM, et al. Surgical fundoplication in laryngopharyngeal reflux unresponsive to aggressive acid suppression: a controlled study. Clin Gastroenterol Hepatol. 2006;4:433-441.
21. Catania RA, Kavic SM, Roth JS, et al. Laparoscopic Nissen fundoplication effectively relieves symptoms in patients with laryngopharyngeal reflux. J Gastrointest Surg. 2007;11:1579-1587.
22. Ogut F, Ersin S, Engin EZ, et al. The effect of laparoscopic Nissen fundoplication on laryngeal findings and voice quality. Surg Endosc. 2007;21:549-554.
23. Sala E, Salminen P, Simberg S, et al. Laryngopharyngeal reflux disease treated with laparoscopic fundoplication. Dig Dis Sci. 2008;53:2397-2404.
24. Koufman JA, Sataloff RT, Toohill R. Laryngopharyngeal reflux: consensus conference report. J Voice. 1996;10:215-216.
• Recommend dietary and behavioral modifications as a first step in treating patients with symptoms suggestive of laryngopharyngeal reflux disease (LPRD). C
• When medications are needed, prescribe a high-dose proton-pump inhibitor, a histamine-2 blocker at bedtime, and prophylactic antacids for reflux-inducing activities, such as exercising and eating. B
• Avoid the rebound effect associated with abrupt cessation of medications prescribed for LPRD with a gradual, 16-week taper. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE When Joan C, a 35-year-old patient whom you’ve known for years, comes in for a physical, you notice that she’s coughing frequently. Upon questioning, Joan says she first noticed the cough several months ago; she also reports that she’s frequently hoarse, but has no other symptoms. Joan is a former smoker, and quit 4 years ago.
If Joan were your patient, would you suspect that she had an upper respiratory infection and prescribe an antibiotic such as azithromycin? Would you include laryngopharyngeal reflux disease in the differential diagnosis?
Laryngopharyngeal reflux disease (LPRD) is a common condition that most primary care physicians encounter frequently. It is also frequently misdiagnosed by clinicians who are unfamiliar with the differences between LPRD and gastroesophageal reflux disease (GERD).
The American Academy of Otolaryngology–Head and Neck Surgery defines laryngopharyngeal reflux as the retrograde movement of gastric contents into the laryngopharynx.1 Common symptoms include hoarseness/dysphonia, chronic throat clearing, dysphagia, globus pharyngeus, and chronic cough, as well as postnasal drip, paroxysmal laryngospasm, odynophagia, excessive throat mucus, and a strange taste in the mouth.2
The diversity and vagueness of these symptoms, as well as the lack of a gold standard diagnostic test for LPRD, make it difficult to estimate its prevalence. In addition, signs of gastroesophageal reflux can be found in the laryngopharynx of up to 86% of healthy individuals, further complicating the clinical picture.3 To avoid missing this often overlooked reflux disease, you need to know how it develops, what signs and symptoms to look for, and which distinguishing features to keep in mind.
Pathophysiology and distinguishing features
The precise way in which LPRD develops is not known, but there are 2 proposed means of laryngeal injury—direct and indirect. In the first case, chemical irritants in the gastric refluxate enter the laryngopharynx and cause local mucosal injury. In the second, gastric reflux irritates the esophageal tissue enough to evoke laryngeal reflexes without ever reaching the larynx—a vagally mediated response associated with symptoms such as chronic cough, throat-clearing sensations, and bronchoconstriction.4
Unlike the esophageal lining, laryngeal epithelium is not protected against chemical injury from gastric acid, as it lacks both the stripping motion of esophageal peristalsis and the neutralizing bicarbonate in saliva.4 Thus, while far smaller amounts of gastric reflux make it into the laryngopharynx, the acid remains there longer and may cause greater injury.5 In some cases, this occurs as often as 50 times a day, although as few as 3 episodes per week have been known to cause LPRD.5
Heartburn is not the rule
Heartburn is a primary complaint of patients with GERD. It is reported by little more than a third (35%) of those with LPRD,5,6 however, (which is why it is sometimes called the “silent” reflux disease). This is because heartburn is caused by esophagitis due to esophageal dysmotility and lower esophageal sphincter dysfunction,3 while most patients with LPRD have normal esophageal motor function and upper esophageal sphincter dysfunction. The fact that only a minimal amount of reflux enters the laryngopharynx may be part of the reason heartburn is less likely in patients with LPRD.
Onset of symptoms. When reflux occurs is another thing that distinguishes LPRD and GERD. Symptoms of GERD typically worsen when the individual is supine, while laryngopharyngeal reflux usually occurs when he or she is upright.7 The frequency with which these 2 conditions overlap is debatable, as there are few studies differentiating LPRD and GERD based on standardized signs and symptoms.7
Making sense of signs and symptoms
Most patients with LPRD seek treatment from their primary care physician, typically reporting symptoms that they don’t associate with gastric reflux, such as hoarseness, a chronic cough or sore throat, or the sensation of a lump in the throat (TABLE 1). Less common manifestations include “water brash”—excessive mucus in the mouth caused by a release of salivary bicarbonate to help neutralize acidity8—otitis media, sinus disease, and dental caries.5
Laryngeal endoscopy may reveal many changes from diffuse irritation. Diffuse erythema, edema, and interarytenoid hypertrophy/cobblestoning are the most useful findings for an LPRD diagnosis.9,10 But in most cases, only a few nonspecific signs with a number of possible causes (infection, environmental irritants, allergies, temperature/climate change, among others) are seen on endoscopic examination, with little correlation with symptom severity. In fact, 74% of otolaryngologists responding to a recent survey said they relied more on patient symptoms than on laryngeal signs for an LPRD diagnosis.10
The Reflux Finding Score (RFS), available at http://www.nature.com/gimo/contents/pt1/fig_tab/gimo46_T3.html, is a clinical tool developed to quantify laryngeal inflammation and standardize objective endoscopic findings. The RFS incorporates the following endolaryngeal signs:
- subglottic edema
- ventricular obliteration
- erythema/hyperemia
- vocal cord edema
- diffuse laryngeal edema
- posterior commissure hypertrophy
- granuloma/granulation tissue
- thick endolaryngeal mucus.
A numeric value is assigned to each, based on whether it is present or absent; partial or complete; local or diffuse; or mild or severe. However, the RFS, too, is an imperfect tool. Clinicians who have used the RFS report that a score higher than 7 identifies LPRD with 95% sensitivity.11 But laryngeal findings may be due to other causes, such as infection, autoimmune reaction, or even allergies, and studies have found the RFS to have poor specificity and inter-rater reliability.12-14
Ambulatory dual probe pH monitoring was considered to be the gold standard test for LPRD at one time, but newer studies have raised questions about its validity and usefulness, especially in patients taking proton-pump inhibitors (PPIs).1,5,7 Newer advanced probes featuring less invasive data collection and greater sensitivity are under development. Ambulatory 24-hour multichannel intraluminal impedance with pH monitoring is the most promising new diagnostic tool, as it can monitor both acidic and nonacidic reflux and distinguish between gas and liquid.15
TABLE 1
When to suspect laryngopharyngeal reflux disease1,5,24
| Finding | Frequency among patients with LPRD (%)* |
|---|---|
| Dysphonia/hoarseness (intermittent) | 71 |
| Chronic cough | 51 |
| Globus pharyngeus | 47 |
| Chronic throat clearing | 42 |
| Dysphagia | 35 |
| Heartburn | 35 |
| *The frequency of other symptoms associated with LPRD is not known. | |
Treatment, like diagnosis, is not clear-cut
LPRD is often called a diagnosis of exclusion, because of the nonspecific nature of its signs and symptoms and the importance of considering a range of other etiologies. The differential diagnosis includes excessive voice use, postnasal drip, upper respiratory infection, habitual throat clearing, allergic rhinitis, environmental irritants, temperature/climate change, chronic or episodic use of alcohol and/or tobacco, and psychological problems related to tics, such as habitual throat clearing or coughing.5
Diagnosis is often based on an empiric trial of high-dose PPIs, with confirmation dependent on symptom relief. Because there have been few placebo-controlled trials with PPIs and those that have been completed had conflicting results, diagnosis based on a combination of medical history and endoscopic laryngeal examination may be a better approach.16,17
Acid suppression therapy with either PPIs or histamine-2 (H2) receptor blockers such as ranitidine or famotidine is the mainstay of treatment for LPRD. But medical societies offer conflicting advice. The American Gastroenterological Association cautions clinicians not to prescribe acid-suppression therapy for patients with LPRD unless they also have GERD.6 The American Academy of Otolaryngology–Head and Neck Surgery recommends twice-daily PPI use for ≥6 months.1,13 The general consensus, based on clinical experience alone, is that patients should be treated with high doses of PPIs (eg, 40 mg omeprazole twice a day) for ≥6 months, with the addition of an H2 receptor blocker to help reduce overnight acid production.1,18 Prophylactic antacid use is also recommended in anticipation of reflux, such as before exercising and right after a meal.
Symptoms should start to improve within 6 to 8 weeks, and patients should be reassessed in about 3 months. To avoid a rebound effect from the abrupt cessation of medications, we suggest a gradual taper over 16 weeks. For the first 8 weeks, the H2 blocker should be discontinued and the PPI decreased from twice a day to once. If symptoms are still controlled, the PPI dose can be reduced to once every other day for another 8 weeks, then stopped if symptoms do not recur.18
Lifestyle and dietary changes (TABLE 2), such as smoking cessation, weight loss, and avoidance of alcohol, are an important part of LPRD treatment, and may be used as a first-line therapy before prescribing medication.19 In fact, some studies have found PPI therapy to be inferior to behavioral/lifestyle modifications.17
Fundoplication surgery, a procedure in which the gastric fundus of the stomach is wrapped around the lower end of the esophagus and stitched in place to prevent reflux, may be an option for patients who do not respond to, or cannot tolerate, aggressive medical treatment for LPRD. A 2006 prospective controlled study found that surgical fundoplication did not consistently relieve laryngeal symptoms.20 But other studies have found that a carefully selected population with medically unresponsive laryngopharyngeal symptoms can benefit from this procedure.21,22 One study showed a significant improvement within one month of fundoplication, with continued improvement observed during a 3-year follow-up.21 In another prospective study, researchers showed that while LPRD-related laryngeal symptoms such as coughing and throat-clearing improved with both medical therapy and laparoscopic fundoplication, voice quality and endoscopic laryngeal/pharyngeal findings improved significantly only with the surgical procedure.23
TABLE 2
Recommend these lifestyle modifications19
| Stop smoking |
Avoid:
|
| Eat smaller, more frequent meals |
| Avoid eating within 3 hours of bedtime |
| Lose weight |
CORRESPONDENCE
Shoib Sana, DO, Detroit Medical Center, Otolaryngology-Head and Neck Surgery, 6533 East Jefferson Avenue, Apartment 316, Detroit, MI 48207; ssana@dmc.org
• Recommend dietary and behavioral modifications as a first step in treating patients with symptoms suggestive of laryngopharyngeal reflux disease (LPRD). C
• When medications are needed, prescribe a high-dose proton-pump inhibitor, a histamine-2 blocker at bedtime, and prophylactic antacids for reflux-inducing activities, such as exercising and eating. B
• Avoid the rebound effect associated with abrupt cessation of medications prescribed for LPRD with a gradual, 16-week taper. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE When Joan C, a 35-year-old patient whom you’ve known for years, comes in for a physical, you notice that she’s coughing frequently. Upon questioning, Joan says she first noticed the cough several months ago; she also reports that she’s frequently hoarse, but has no other symptoms. Joan is a former smoker, and quit 4 years ago.
If Joan were your patient, would you suspect that she had an upper respiratory infection and prescribe an antibiotic such as azithromycin? Would you include laryngopharyngeal reflux disease in the differential diagnosis?
Laryngopharyngeal reflux disease (LPRD) is a common condition that most primary care physicians encounter frequently. It is also frequently misdiagnosed by clinicians who are unfamiliar with the differences between LPRD and gastroesophageal reflux disease (GERD).
The American Academy of Otolaryngology–Head and Neck Surgery defines laryngopharyngeal reflux as the retrograde movement of gastric contents into the laryngopharynx.1 Common symptoms include hoarseness/dysphonia, chronic throat clearing, dysphagia, globus pharyngeus, and chronic cough, as well as postnasal drip, paroxysmal laryngospasm, odynophagia, excessive throat mucus, and a strange taste in the mouth.2
The diversity and vagueness of these symptoms, as well as the lack of a gold standard diagnostic test for LPRD, make it difficult to estimate its prevalence. In addition, signs of gastroesophageal reflux can be found in the laryngopharynx of up to 86% of healthy individuals, further complicating the clinical picture.3 To avoid missing this often overlooked reflux disease, you need to know how it develops, what signs and symptoms to look for, and which distinguishing features to keep in mind.
Pathophysiology and distinguishing features
The precise way in which LPRD develops is not known, but there are 2 proposed means of laryngeal injury—direct and indirect. In the first case, chemical irritants in the gastric refluxate enter the laryngopharynx and cause local mucosal injury. In the second, gastric reflux irritates the esophageal tissue enough to evoke laryngeal reflexes without ever reaching the larynx—a vagally mediated response associated with symptoms such as chronic cough, throat-clearing sensations, and bronchoconstriction.4
Unlike the esophageal lining, laryngeal epithelium is not protected against chemical injury from gastric acid, as it lacks both the stripping motion of esophageal peristalsis and the neutralizing bicarbonate in saliva.4 Thus, while far smaller amounts of gastric reflux make it into the laryngopharynx, the acid remains there longer and may cause greater injury.5 In some cases, this occurs as often as 50 times a day, although as few as 3 episodes per week have been known to cause LPRD.5
Heartburn is not the rule
Heartburn is a primary complaint of patients with GERD. It is reported by little more than a third (35%) of those with LPRD,5,6 however, (which is why it is sometimes called the “silent” reflux disease). This is because heartburn is caused by esophagitis due to esophageal dysmotility and lower esophageal sphincter dysfunction,3 while most patients with LPRD have normal esophageal motor function and upper esophageal sphincter dysfunction. The fact that only a minimal amount of reflux enters the laryngopharynx may be part of the reason heartburn is less likely in patients with LPRD.
Onset of symptoms. When reflux occurs is another thing that distinguishes LPRD and GERD. Symptoms of GERD typically worsen when the individual is supine, while laryngopharyngeal reflux usually occurs when he or she is upright.7 The frequency with which these 2 conditions overlap is debatable, as there are few studies differentiating LPRD and GERD based on standardized signs and symptoms.7
Making sense of signs and symptoms
Most patients with LPRD seek treatment from their primary care physician, typically reporting symptoms that they don’t associate with gastric reflux, such as hoarseness, a chronic cough or sore throat, or the sensation of a lump in the throat (TABLE 1). Less common manifestations include “water brash”—excessive mucus in the mouth caused by a release of salivary bicarbonate to help neutralize acidity8—otitis media, sinus disease, and dental caries.5
Laryngeal endoscopy may reveal many changes from diffuse irritation. Diffuse erythema, edema, and interarytenoid hypertrophy/cobblestoning are the most useful findings for an LPRD diagnosis.9,10 But in most cases, only a few nonspecific signs with a number of possible causes (infection, environmental irritants, allergies, temperature/climate change, among others) are seen on endoscopic examination, with little correlation with symptom severity. In fact, 74% of otolaryngologists responding to a recent survey said they relied more on patient symptoms than on laryngeal signs for an LPRD diagnosis.10
The Reflux Finding Score (RFS), available at http://www.nature.com/gimo/contents/pt1/fig_tab/gimo46_T3.html, is a clinical tool developed to quantify laryngeal inflammation and standardize objective endoscopic findings. The RFS incorporates the following endolaryngeal signs:
- subglottic edema
- ventricular obliteration
- erythema/hyperemia
- vocal cord edema
- diffuse laryngeal edema
- posterior commissure hypertrophy
- granuloma/granulation tissue
- thick endolaryngeal mucus.
A numeric value is assigned to each, based on whether it is present or absent; partial or complete; local or diffuse; or mild or severe. However, the RFS, too, is an imperfect tool. Clinicians who have used the RFS report that a score higher than 7 identifies LPRD with 95% sensitivity.11 But laryngeal findings may be due to other causes, such as infection, autoimmune reaction, or even allergies, and studies have found the RFS to have poor specificity and inter-rater reliability.12-14
Ambulatory dual probe pH monitoring was considered to be the gold standard test for LPRD at one time, but newer studies have raised questions about its validity and usefulness, especially in patients taking proton-pump inhibitors (PPIs).1,5,7 Newer advanced probes featuring less invasive data collection and greater sensitivity are under development. Ambulatory 24-hour multichannel intraluminal impedance with pH monitoring is the most promising new diagnostic tool, as it can monitor both acidic and nonacidic reflux and distinguish between gas and liquid.15
TABLE 1
When to suspect laryngopharyngeal reflux disease1,5,24
| Finding | Frequency among patients with LPRD (%)* |
|---|---|
| Dysphonia/hoarseness (intermittent) | 71 |
| Chronic cough | 51 |
| Globus pharyngeus | 47 |
| Chronic throat clearing | 42 |
| Dysphagia | 35 |
| Heartburn | 35 |
| *The frequency of other symptoms associated with LPRD is not known. | |
Treatment, like diagnosis, is not clear-cut
LPRD is often called a diagnosis of exclusion, because of the nonspecific nature of its signs and symptoms and the importance of considering a range of other etiologies. The differential diagnosis includes excessive voice use, postnasal drip, upper respiratory infection, habitual throat clearing, allergic rhinitis, environmental irritants, temperature/climate change, chronic or episodic use of alcohol and/or tobacco, and psychological problems related to tics, such as habitual throat clearing or coughing.5
Diagnosis is often based on an empiric trial of high-dose PPIs, with confirmation dependent on symptom relief. Because there have been few placebo-controlled trials with PPIs and those that have been completed had conflicting results, diagnosis based on a combination of medical history and endoscopic laryngeal examination may be a better approach.16,17
Acid suppression therapy with either PPIs or histamine-2 (H2) receptor blockers such as ranitidine or famotidine is the mainstay of treatment for LPRD. But medical societies offer conflicting advice. The American Gastroenterological Association cautions clinicians not to prescribe acid-suppression therapy for patients with LPRD unless they also have GERD.6 The American Academy of Otolaryngology–Head and Neck Surgery recommends twice-daily PPI use for ≥6 months.1,13 The general consensus, based on clinical experience alone, is that patients should be treated with high doses of PPIs (eg, 40 mg omeprazole twice a day) for ≥6 months, with the addition of an H2 receptor blocker to help reduce overnight acid production.1,18 Prophylactic antacid use is also recommended in anticipation of reflux, such as before exercising and right after a meal.
Symptoms should start to improve within 6 to 8 weeks, and patients should be reassessed in about 3 months. To avoid a rebound effect from the abrupt cessation of medications, we suggest a gradual taper over 16 weeks. For the first 8 weeks, the H2 blocker should be discontinued and the PPI decreased from twice a day to once. If symptoms are still controlled, the PPI dose can be reduced to once every other day for another 8 weeks, then stopped if symptoms do not recur.18
Lifestyle and dietary changes (TABLE 2), such as smoking cessation, weight loss, and avoidance of alcohol, are an important part of LPRD treatment, and may be used as a first-line therapy before prescribing medication.19 In fact, some studies have found PPI therapy to be inferior to behavioral/lifestyle modifications.17
Fundoplication surgery, a procedure in which the gastric fundus of the stomach is wrapped around the lower end of the esophagus and stitched in place to prevent reflux, may be an option for patients who do not respond to, or cannot tolerate, aggressive medical treatment for LPRD. A 2006 prospective controlled study found that surgical fundoplication did not consistently relieve laryngeal symptoms.20 But other studies have found that a carefully selected population with medically unresponsive laryngopharyngeal symptoms can benefit from this procedure.21,22 One study showed a significant improvement within one month of fundoplication, with continued improvement observed during a 3-year follow-up.21 In another prospective study, researchers showed that while LPRD-related laryngeal symptoms such as coughing and throat-clearing improved with both medical therapy and laparoscopic fundoplication, voice quality and endoscopic laryngeal/pharyngeal findings improved significantly only with the surgical procedure.23
TABLE 2
Recommend these lifestyle modifications19
| Stop smoking |
Avoid:
|
| Eat smaller, more frequent meals |
| Avoid eating within 3 hours of bedtime |
| Lose weight |
CORRESPONDENCE
Shoib Sana, DO, Detroit Medical Center, Otolaryngology-Head and Neck Surgery, 6533 East Jefferson Avenue, Apartment 316, Detroit, MI 48207; ssana@dmc.org
1. Koufman JA, Aviv JE, Casiano RR, et al. Laryngopharyngeal reflux: position statement of the committee on speech, voice, and swallowing disorders of the American Academy of Otolaryngology-Head and Neck Surgery. Otolaryngol Head Neck Surg. 2002;127:32-35.
2. Papakonstantinou L, Leslie P, Gray J, et al. Laryngopharyngeal reflux: a prospective analysis of a 34 item symptom questionnaire. Clin Otolaryngol. 2009;34:455-459.
3. Hicks DM, Ours TM, Abelson TI, et al. The prevalence of hypopharynx findings associated with gastroesophageal reflux in normal volunteers. J Voice. 2002;16:564.-
4. Johnston N, Bulmer D, Gill GA, et al. Cell biology of laryngeal epithelial defenses in health and disease: further studies. Ann Otol Rhinol Laryngol. 2003;112:481-491.
5. Koufman JA. The otolaryngologic manifestations of gastroesophageal reflux disease (GERD): a clinical investigation of 225 patients using ambulatory 24 hour pH monitoring and an experimental investigation of the role of acid and pepsin in the development of laryngeal injury. Laryngoscope. 1991;101:1-78.
6. Kahrilas PJ, Shaheen NJ, Vaezi M, et al. American Gastroenterological Association Institute (AGAI) medical position statement: management of gastroesophageal reflux disease. Gastroenterology. 2008;135:1383.-
7. Postma GN, Tomek MS, Belafsky PC, et al. Esophageal motor function in laryngopharyngeal reflux is superior to that in classic gastroesophageal reflux disease. Ann Otol Rhinol Laryngol. 2001;111:1114-1116.
8. Helen JF, Dodds WJ, Hogan WJ. Salivary response to esophageal acid in normal subjects and patients with reflux esophagitis. Gastroenterology. 1998;94:1394-1398.
9. Belafsky PC. Abnormal endoscopic pharyngeal and laryngeal findings attributable to reflux. Am J Med 2003;116(suppl 3A):91S-97S.
10. Ahmed TF, Khandwala F, Abelson, et al. Chronic laryngitis associated with gastroesophageal reflux: prospective assessment of differences in practice patterns between gastroenterologists and ENT physicians. Am J Gastroenterol. 2006;102:470-478.
11. Belafsky PC, Postma GN, Koufman JA. The validity and reliability of the reflux finding score (RFS). Laryngoscope. 2001;111:1313-1317.
12. Koufman JA, Sataloff RT, Toohill R. Laryngopharyngeal reflux: consensus conference report. J Voice. 1996;10:215-216.
13. Belafsky PC, Postma GN, Koufman JA. Laryngopharyngeal reflux symptoms improve before changes in physical findings. Laryngoscope 2001;111:979-981.
14. Reichel O, Dressel H, Wiederanders K, et al. Double-blind, placebo-controlled trial with esomeprazole for symptoms and signs associated with laryngopharyngeal reflux. Otolaryngol Head Neck Surg. 2008;139:414-420.
15. Muderris T, Gokcan MK, Yorulmaz I. The clinical value of pharyngeal pH monitoring using a double-probe, triple-sensor catheter in patients with laryngopharyngeal reflux. Arch Otolaryngol Head Neck Surg. 2009;135:163-167.
16. Steward DL, Wilson KM, Kelly DH, et al. Proton pump inhibitor therapy for chronic laryngo-pharyngitis: a randomized placebo-control trial. Otolaryngol Head Neck Surg. 2004;131:342-350.
17. Wo JM, Koopman J, Harrell SP, et al. Double-blind, placebo-controlled trial with single-dose pantoprazole for laryngopharyngeal reflux. Am J Gastroenterol. 2006;101:1972-1978.
18. Park W, Hicks DM, Khandwala F, et al. Laryngopharyngeal reflux: prospective cohort study evaluating optimal dose of proton-pump inhibitor therapy and pretherapy predictors of response. Laryngoscope. 2005;116:1230-1238.
19. Maceri DR, Zim S. Laryngospasm: an atypical manifestation of severe gastroesophageal reflux disease. Laryngoscope. 2001;111:1976-1979.
20. Swoger J, Ponsky J, Hicks DM, et al. Surgical fundoplication in laryngopharyngeal reflux unresponsive to aggressive acid suppression: a controlled study. Clin Gastroenterol Hepatol. 2006;4:433-441.
21. Catania RA, Kavic SM, Roth JS, et al. Laparoscopic Nissen fundoplication effectively relieves symptoms in patients with laryngopharyngeal reflux. J Gastrointest Surg. 2007;11:1579-1587.
22. Ogut F, Ersin S, Engin EZ, et al. The effect of laparoscopic Nissen fundoplication on laryngeal findings and voice quality. Surg Endosc. 2007;21:549-554.
23. Sala E, Salminen P, Simberg S, et al. Laryngopharyngeal reflux disease treated with laparoscopic fundoplication. Dig Dis Sci. 2008;53:2397-2404.
24. Koufman JA, Sataloff RT, Toohill R. Laryngopharyngeal reflux: consensus conference report. J Voice. 1996;10:215-216.
1. Koufman JA, Aviv JE, Casiano RR, et al. Laryngopharyngeal reflux: position statement of the committee on speech, voice, and swallowing disorders of the American Academy of Otolaryngology-Head and Neck Surgery. Otolaryngol Head Neck Surg. 2002;127:32-35.
2. Papakonstantinou L, Leslie P, Gray J, et al. Laryngopharyngeal reflux: a prospective analysis of a 34 item symptom questionnaire. Clin Otolaryngol. 2009;34:455-459.
3. Hicks DM, Ours TM, Abelson TI, et al. The prevalence of hypopharynx findings associated with gastroesophageal reflux in normal volunteers. J Voice. 2002;16:564.-
4. Johnston N, Bulmer D, Gill GA, et al. Cell biology of laryngeal epithelial defenses in health and disease: further studies. Ann Otol Rhinol Laryngol. 2003;112:481-491.
5. Koufman JA. The otolaryngologic manifestations of gastroesophageal reflux disease (GERD): a clinical investigation of 225 patients using ambulatory 24 hour pH monitoring and an experimental investigation of the role of acid and pepsin in the development of laryngeal injury. Laryngoscope. 1991;101:1-78.
6. Kahrilas PJ, Shaheen NJ, Vaezi M, et al. American Gastroenterological Association Institute (AGAI) medical position statement: management of gastroesophageal reflux disease. Gastroenterology. 2008;135:1383.-
7. Postma GN, Tomek MS, Belafsky PC, et al. Esophageal motor function in laryngopharyngeal reflux is superior to that in classic gastroesophageal reflux disease. Ann Otol Rhinol Laryngol. 2001;111:1114-1116.
8. Helen JF, Dodds WJ, Hogan WJ. Salivary response to esophageal acid in normal subjects and patients with reflux esophagitis. Gastroenterology. 1998;94:1394-1398.
9. Belafsky PC. Abnormal endoscopic pharyngeal and laryngeal findings attributable to reflux. Am J Med 2003;116(suppl 3A):91S-97S.
10. Ahmed TF, Khandwala F, Abelson, et al. Chronic laryngitis associated with gastroesophageal reflux: prospective assessment of differences in practice patterns between gastroenterologists and ENT physicians. Am J Gastroenterol. 2006;102:470-478.
11. Belafsky PC, Postma GN, Koufman JA. The validity and reliability of the reflux finding score (RFS). Laryngoscope. 2001;111:1313-1317.
12. Koufman JA, Sataloff RT, Toohill R. Laryngopharyngeal reflux: consensus conference report. J Voice. 1996;10:215-216.
13. Belafsky PC, Postma GN, Koufman JA. Laryngopharyngeal reflux symptoms improve before changes in physical findings. Laryngoscope 2001;111:979-981.
14. Reichel O, Dressel H, Wiederanders K, et al. Double-blind, placebo-controlled trial with esomeprazole for symptoms and signs associated with laryngopharyngeal reflux. Otolaryngol Head Neck Surg. 2008;139:414-420.
15. Muderris T, Gokcan MK, Yorulmaz I. The clinical value of pharyngeal pH monitoring using a double-probe, triple-sensor catheter in patients with laryngopharyngeal reflux. Arch Otolaryngol Head Neck Surg. 2009;135:163-167.
16. Steward DL, Wilson KM, Kelly DH, et al. Proton pump inhibitor therapy for chronic laryngo-pharyngitis: a randomized placebo-control trial. Otolaryngol Head Neck Surg. 2004;131:342-350.
17. Wo JM, Koopman J, Harrell SP, et al. Double-blind, placebo-controlled trial with single-dose pantoprazole for laryngopharyngeal reflux. Am J Gastroenterol. 2006;101:1972-1978.
18. Park W, Hicks DM, Khandwala F, et al. Laryngopharyngeal reflux: prospective cohort study evaluating optimal dose of proton-pump inhibitor therapy and pretherapy predictors of response. Laryngoscope. 2005;116:1230-1238.
19. Maceri DR, Zim S. Laryngospasm: an atypical manifestation of severe gastroesophageal reflux disease. Laryngoscope. 2001;111:1976-1979.
20. Swoger J, Ponsky J, Hicks DM, et al. Surgical fundoplication in laryngopharyngeal reflux unresponsive to aggressive acid suppression: a controlled study. Clin Gastroenterol Hepatol. 2006;4:433-441.
21. Catania RA, Kavic SM, Roth JS, et al. Laparoscopic Nissen fundoplication effectively relieves symptoms in patients with laryngopharyngeal reflux. J Gastrointest Surg. 2007;11:1579-1587.
22. Ogut F, Ersin S, Engin EZ, et al. The effect of laparoscopic Nissen fundoplication on laryngeal findings and voice quality. Surg Endosc. 2007;21:549-554.
23. Sala E, Salminen P, Simberg S, et al. Laryngopharyngeal reflux disease treated with laparoscopic fundoplication. Dig Dis Sci. 2008;53:2397-2404.
24. Koufman JA, Sataloff RT, Toohill R. Laryngopharyngeal reflux: consensus conference report. J Voice. 1996;10:215-216.
The dangers of colon cleansing
• Advise patients that colon cleansing has no proven benefits and many adverse effects. B
• Ask patients with otherwise unexplained nausea, vomiting, or diarrhea if they engage in colon cleansing. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1 A 31-year-old African American woman sought treatment at her local emergency department (ED) for nausea, vomiting, and diarrhea. She reported passing more than 6 yellowish-brown, watery, nonbloody stools during the previous 2 days. She felt weak, feverish, and light-headed and showed signs of dehydration.
The patient had Crohn’s disease and had undergone a partial colectomy 5 years earlier. She told the ED physician that 2 days before visiting the ED she had gone to a “cleansing center” for a colonic cleansing, but was unable to complete the process because she developed cramps 15 minutes into the procedure. Less than an hour later, she developed diarrhea, nausea, and vomiting.
In the ED, her serum potassium was 2.9 mEq/L, blood urea nitrogen was 26 mg/dL, and creatinine was 1.9 mg/dL. She was afebrile, with a blood pressure of 135/75 mm Hg and a heart rate of 113 beats per minute. After receiving 2 liters of normal saline and 90 mEq of potassium chloride replacement, the patient felt better and was later discharged from the ED.
Three days later, the patient came to our residency clinic. She described her stools as being loose, but not watery or bloody, and passed in small amounts, about 4 times daily. She still had some abdominal cramping just before passing stool, but bowel movements relieved that. Her vital signs were within normal limits, and her physical exam was benign. The patient was instructed to follow her normal diet, as tolerated, and drink plenty of fluids to maintain good hydration. Her symptoms resolved by the following week.
CASE 2 A 49-year-old African American man came to our community hospital because of vomiting, diarrhea, and abdominal pain he had been experiencing for 4 days. He linked the symptoms to eating a large fast-food breakfast, followed by a big lunch the day before. He described having multiple episodes of nonbloody, nonbilious vomiting, nonbloody watery diarrhea, and “twisting” abdominal pain that was constant but temporarily relieved with a warm compress or positional maneuvers. He had never had a similar episode and had not taken any antibiotics recently.
Upon further questioning, the patient revealed that he had used a colon cleanser a few days earlier. A review showed that he had lost 24 pounds in 10 days. Vitals were within normal limits. Serum potassium was 2.9 mEq/L, and creatinine was 2.1 mg/dL. A computed tomography scan of the abdomen revealed moderate to moderately severe dilatation of multiple small bowel loops with multiple air fluid levels, suggesting an early or partial small bowel obstruction. We obtained a surgical consultation, but surgery was not required. He was discharged after 2 days.
The patient returned to the hospital 3 days later with similar symptoms and severe weakness associated with dizziness. At that time his serum potassium was 2.4 mEq/L and creatinine was 4.0 mg/dL. Aspartate aminotransferase was 29 U/L, alanine aminotransferase was 80 U/L, lipase was 418 U/L, and amylase was 94 U/L.
The patient was readmitted for dehydration, hypokalemia, and pancreatitis and, following a colonoscopy and biopsy that revealed chronic and acute inflammation, a gastroenterologist made a diagnosis of “herbal intoxication.” The patient was hydrated, his electrolytes were replaced, and his diet was slowly returned to normal. He was discharged after 5 days.
An old practice rediscovered
Colon cleansing has been around since ancient times, when its purported benefits were based on the belief that intestinal waste can poison the body (“autointoxication”).1 The procedure became popular in the early 1900s, but in a 1919 paper, the American Medical Association discounted the autointoxication theory and condemned the practice.1 The procedure then fell out of favor, albeit temporarily.2 Colon cleansing has staged a comeback in recent years.
Colon cleansing basics
Colon cleansing, also called colonic irrigation or colonic hydrotherapy, is performed by colonic hygienists or colon therapists, or can be self-administered. The procedure works like an enema. The patient generally lies on a table and water (with or without additional herbs or compounds) is pumped through the rectum via a tube.
Unlike enemas, for which a small amount of fluid is used, however, colon cleansing calls for a large volume of fluid—up to 60 liters—to be introduced into the rectum.3,4 Fluids and waste are expelled through another tube. The procedure may be repeated several times.
Products go by many names
Most colon cleansing products come in the forms of laxatives, teas, powders, and capsules. They can be taken by mouth or inserted into the rectum. They often contain sodium phosphate, coffee, probiotics, enzymes, or any of a variety of herbs.5 Some products contain fiber preparations, including psyllium, flaxseed, and laxatives such as cascara, magnesium oxide, cat’s claw, artichoke leaves, burdock root, licorice, and milk thistle.2
With names such as Nature’s Bounty Colon Cleanser Natural Detox Formula, Health Plus Inc. Colon Cleanse, and 7-Day Miracle Cleanse, as well as endorsements by movie stars, these colon cleansing products are actively promoted as a natural way to enhance one’s well-being. Advertisements promising that colon cleansing will alleviate fatigue, headache, weight gain, and low energy are ubiquitous on the Internet and in newspapers and magazines. The ads tout the safety of “herbal” and “natural” preparations. These materials also provide anecdotal support for claims that colon cleansing improves the immune and circulatory systems, enhances cognitive abilities, and aids weight loss through “detoxification.”6
Individuals who want to cleanse their own colons can choose among home kits, some of which include disposable tubing, while others have components that can be reused if they are sterilized after each use.5,7 But many people turn to a “hydrotherapist” for colon irrigation. The services are also increasingly being offered by practitioners who describe themselves as “colon hygienists.”
These individuals sometimes belong to organizations such as the National Board for Colon Hydrotherapy (NBCH) or the International Association for Colon Hydrotherapy (I-ACT).8,9 These practitioners are not licensed, but they are required to have a high school or equivalent degree plus 3 semesters of postsecondary education and to be certified in cardiopulmonary resuscitation. They also take various seminars and continuing education courses from the NBCH and I-ACT.
How many individuals have used colon cleansing is unclear, although one study suggested that in the United Kingdom, registered practitioners carry out an estimated 5600 procedures every month.10
Where’s the evidence?
Despite colon cleansing’s long history and current popularity, the literature does not support its purported benefits. Historically, colon cleansing was thought to prevent autointoxication from toxins originating in the colon, but the evidence for this claim is limited.11 A search of the literature using the terms “colon cleansing,” “herbal colon cleanse,” “colon detoxification,” and “colon irrigation,” yielded no scientifically robust studies in support of this practice. One study suggested that lymphocytes might migrate from the gut into the circulation after the procedure, which may “improve colon and immune system function.”12
Even though colon cleansing is touted as a commonly used form of holistic, complementary and alternative medicine, the Natural Standard Professional Database concluded in a monograph that there is “limited clinical evidence validating colon therapy as a health promotion practice” and noted a “lack of sufficient evidence” for most of its prescribed uses.13
Adverse effects: From cramping to renal failure
Most reports in the literature note a variety of adverse effects of colon cleansing that range from mild (eg, cramping, abdominal pain, fullness, bloating, nausea, vomiting, perianal irritation, and soreness) to severe (eg, electrolyte imbalance and renal failure).11,14-17 Some herbal preparations have also been associated with aplastic anemia and liver toxicity.18
Case reports also have noted back and pelvic abscesses after colonic hydrotherapy, fatal aeroportia (gas accumulation in the mesenteric veins) with air emboli, rectal perforations, perineal gangrene, acute water intoxication, coffee enema-associated colitis and septicemia, and deaths due to amebiasis.2,3,19-21
The FDA has issued many warning letters
The preparations used for colon cleansing are considered dietary supplements, and the US Food and Drug Administration (FDA) requires that they be labeled as such; the FDA does not preapprove these substances, however. The FDA also requires that colonic hydrotherapy and irrigation system devices meet certain requirements, but the agency has never approved any system for general nonmedical purposes, such as colon cleansing.
The devices have an FDA Class III designation, indicating that if a device is used for purposes beyond what is medically indicated (preparation for radiologic and endoscopic procedures), the manufacturer must obtain premarket approval from the FDA, which is based on evaluation of the safety and effectiveness of the device as shown by available scientific evidence and current regulations.22 During the past decade the FDA has issued numerous warning letters to manufacturers for unapproved use of the devices for colon cleansing.23-26
Raise the issue with patients
Given the current popularity of colon cleansing, it’s important to recognize that some of your patients may engage in, or be thinking about, the practice. (See “4 things to tell patients about colon cleansing”.) Be sure to tell patients about the potential consequences of colon cleansing and to emphasize that there is a lack of evidence to back up supporters’ claims.
- Colon irrigation is not wise—particularly if you have a history of gastrointestinal disease (including diverticulitis, Crohn’s disease, or ulcerative colitis) or a history of colon surgery, severe hemorrhoids, kidney disease, or heart disease. These conditions increase the risk of adverse effects.2,3,11,16
- Side effects of colon cleansing include nausea, vomiting, diarrhea, dizziness, dehydration, electrolyte abnormalities, acute kidney insufficiency, pancreatitis, bowel perforation, heart failure, and infection. 2,3,11,16
- The devices that practitioners use for the procedure are not approved for colon cleansing by the US Food and Drug Administration. Inadequately disinfected or sterilized irrigation machines have been linked to bacterial contamination.2,11,19
- Colon cleansing practitioners are not licensed by a scientifically based organization. Rather, practitioners have undergone a training process structured by an organization that is attempting to institute its own certification and licensing requirements.
CORRESPONDENCE
Ranit Mishori, MD, MHS, Georgetown University School of Medicine, 3900 Reservoir Road, NW, Washington, DC 20007; mishorir@georgetown.edu
1. Ernst E. Colonic irrigation and the theory of autointoxication: a triumph of ignorance over science. J Clin Gastroenterol. 1997;24:196-198.
2. Acosta RD, Cash BD. Clinical effects of colonic cleansing for general health promotion: a systemic review. Am J Gastroenterol. 2009;104:2830-2836.
3. Handley DV, Rieger NA, Rodda DJ. Rectal perforation from colonic irrigation administered by alternative practitioners. Med J Aust. 2004;181:575-576.
4. Seow-Choen F. The physiology of colonic therapy. Colorectal Dis. 2009;11:686-688.
5. Colon cleansing. Med Lett Drugs Ther. 2009;51:39.-
6. Just cleansing. A guide to cleansing and detox. Available at: http://www.justcleansing.com. Accessed November 17, 2010.
7. Home Colonics Company. Available at: http://www.homecolonics.com. Accessed November 17, 2010.
8. National Board for Colon Hydrotherapy. Available at: http://www.nbcht.org. Accessed November 27, 2010.
9. International Association for Colon Hydrotherapy. Available at: http://www.i-act.org. Accessed November 27, 1010.
10. Taffinder NJ, Tan E, Webb IG, et al. Retrograde commercial colonic hydrotherapy. Colorectal Dis. 2004;6:258-260.
11. Richards DG, McMillin DL, Mein EA, et al. Colonic irrigations: a review of the historical controversy and the potential for adverse effects. J Altern Complement Med. 2006;12:389-393.
12. Uchiyama-Tanaka Y. Colon irrigation causes lymphocyte movement from gut-associated lymphatic tissues to peripheral blood. Biomed Res. 2009;30:311-314.
13. Colon therapy/colonic irrigation. Natural Standard Professional Monograph. 2011. Available at: http://naturalstandard.com/databases/hw/colon.asp. Accessed June 21, 2011.
14. Abaskharoun R, Depew W, Vanner S. Changes in renal function following administration of oral sodium phosphate or polyethylene glycol for colon cleansing before colonoscopy. Can J Gastroenterol. 2007;21:227-231.
15. Rex D. Dosing considerations in the use of sodium phosphate bowel preparations for colonoscopy Ann Pharmacother. 2007;41:1466-1475.
16. Dykes C, Cash BD. Key safety issues of bowel preparations for colonoscopy and importance of adequate hydration. Gastroenterology Nurs. 2007;31:30-35.
17. Norlela S, Izham C, Khalid BA. Colonic irrigation-induced hyponatremia. Malays J Pathol. 2004;26:117-118.
18. Smereck J. Aplastic anemia: a possible toxic effect of an herbal “colon cleansing” preparation. J Emerg Med. 2007;11:191-192.
19. Ratnaraja N, Raymond N. Extensive abscesses following colonic hydrotherapy. Lancet Infect Dis. 2005;5:527.-
20. Chen WL, Tsao YT. Fatal aeroportia with systemic air embolism after colon hydrotherapy. J Trauma. 2010;68:247.-
21. Tan MP, Cheong DM. Life-threatening perineal gangrene from rectal perforation following colonic hydrotherapy: a case report. Ann Acad Med Singapore. 1999;28:583-585.
22. US Food and Drug Administration. Premarket approval. Available at: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/PremarketSubmissions/PremarketApprovalPMA/default.htm. Accessed June 21, 2011.
23. US Food and Drug Administration. Device classification. Available at: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Overview/ClassifyYourDevice/default.htm. Accessed December 1, 2010.
24. US Food and Drug Administration. Warning letter to Clearwater Colon Hydrotherapy. Available at: http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2001/ucm178494.htm. Accessed June 21, 2011.
25. US Food and Drug Administration Warning letter to Augustine R. Hoerninger, III, PhD, ND. Available at: http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2003/ucm147378.htm. Accessed June 21, 2011.
26. US Food and Drug Administration. Warning letter to Jimmy J. Girouard. Available at: http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2003/ucm147792.htm. Accessed June 21, 2011.
• Advise patients that colon cleansing has no proven benefits and many adverse effects. B
• Ask patients with otherwise unexplained nausea, vomiting, or diarrhea if they engage in colon cleansing. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1 A 31-year-old African American woman sought treatment at her local emergency department (ED) for nausea, vomiting, and diarrhea. She reported passing more than 6 yellowish-brown, watery, nonbloody stools during the previous 2 days. She felt weak, feverish, and light-headed and showed signs of dehydration.
The patient had Crohn’s disease and had undergone a partial colectomy 5 years earlier. She told the ED physician that 2 days before visiting the ED she had gone to a “cleansing center” for a colonic cleansing, but was unable to complete the process because she developed cramps 15 minutes into the procedure. Less than an hour later, she developed diarrhea, nausea, and vomiting.
In the ED, her serum potassium was 2.9 mEq/L, blood urea nitrogen was 26 mg/dL, and creatinine was 1.9 mg/dL. She was afebrile, with a blood pressure of 135/75 mm Hg and a heart rate of 113 beats per minute. After receiving 2 liters of normal saline and 90 mEq of potassium chloride replacement, the patient felt better and was later discharged from the ED.
Three days later, the patient came to our residency clinic. She described her stools as being loose, but not watery or bloody, and passed in small amounts, about 4 times daily. She still had some abdominal cramping just before passing stool, but bowel movements relieved that. Her vital signs were within normal limits, and her physical exam was benign. The patient was instructed to follow her normal diet, as tolerated, and drink plenty of fluids to maintain good hydration. Her symptoms resolved by the following week.
CASE 2 A 49-year-old African American man came to our community hospital because of vomiting, diarrhea, and abdominal pain he had been experiencing for 4 days. He linked the symptoms to eating a large fast-food breakfast, followed by a big lunch the day before. He described having multiple episodes of nonbloody, nonbilious vomiting, nonbloody watery diarrhea, and “twisting” abdominal pain that was constant but temporarily relieved with a warm compress or positional maneuvers. He had never had a similar episode and had not taken any antibiotics recently.
Upon further questioning, the patient revealed that he had used a colon cleanser a few days earlier. A review showed that he had lost 24 pounds in 10 days. Vitals were within normal limits. Serum potassium was 2.9 mEq/L, and creatinine was 2.1 mg/dL. A computed tomography scan of the abdomen revealed moderate to moderately severe dilatation of multiple small bowel loops with multiple air fluid levels, suggesting an early or partial small bowel obstruction. We obtained a surgical consultation, but surgery was not required. He was discharged after 2 days.
The patient returned to the hospital 3 days later with similar symptoms and severe weakness associated with dizziness. At that time his serum potassium was 2.4 mEq/L and creatinine was 4.0 mg/dL. Aspartate aminotransferase was 29 U/L, alanine aminotransferase was 80 U/L, lipase was 418 U/L, and amylase was 94 U/L.
The patient was readmitted for dehydration, hypokalemia, and pancreatitis and, following a colonoscopy and biopsy that revealed chronic and acute inflammation, a gastroenterologist made a diagnosis of “herbal intoxication.” The patient was hydrated, his electrolytes were replaced, and his diet was slowly returned to normal. He was discharged after 5 days.
An old practice rediscovered
Colon cleansing has been around since ancient times, when its purported benefits were based on the belief that intestinal waste can poison the body (“autointoxication”).1 The procedure became popular in the early 1900s, but in a 1919 paper, the American Medical Association discounted the autointoxication theory and condemned the practice.1 The procedure then fell out of favor, albeit temporarily.2 Colon cleansing has staged a comeback in recent years.
Colon cleansing basics
Colon cleansing, also called colonic irrigation or colonic hydrotherapy, is performed by colonic hygienists or colon therapists, or can be self-administered. The procedure works like an enema. The patient generally lies on a table and water (with or without additional herbs or compounds) is pumped through the rectum via a tube.
Unlike enemas, for which a small amount of fluid is used, however, colon cleansing calls for a large volume of fluid—up to 60 liters—to be introduced into the rectum.3,4 Fluids and waste are expelled through another tube. The procedure may be repeated several times.
Products go by many names
Most colon cleansing products come in the forms of laxatives, teas, powders, and capsules. They can be taken by mouth or inserted into the rectum. They often contain sodium phosphate, coffee, probiotics, enzymes, or any of a variety of herbs.5 Some products contain fiber preparations, including psyllium, flaxseed, and laxatives such as cascara, magnesium oxide, cat’s claw, artichoke leaves, burdock root, licorice, and milk thistle.2
With names such as Nature’s Bounty Colon Cleanser Natural Detox Formula, Health Plus Inc. Colon Cleanse, and 7-Day Miracle Cleanse, as well as endorsements by movie stars, these colon cleansing products are actively promoted as a natural way to enhance one’s well-being. Advertisements promising that colon cleansing will alleviate fatigue, headache, weight gain, and low energy are ubiquitous on the Internet and in newspapers and magazines. The ads tout the safety of “herbal” and “natural” preparations. These materials also provide anecdotal support for claims that colon cleansing improves the immune and circulatory systems, enhances cognitive abilities, and aids weight loss through “detoxification.”6
Individuals who want to cleanse their own colons can choose among home kits, some of which include disposable tubing, while others have components that can be reused if they are sterilized after each use.5,7 But many people turn to a “hydrotherapist” for colon irrigation. The services are also increasingly being offered by practitioners who describe themselves as “colon hygienists.”
These individuals sometimes belong to organizations such as the National Board for Colon Hydrotherapy (NBCH) or the International Association for Colon Hydrotherapy (I-ACT).8,9 These practitioners are not licensed, but they are required to have a high school or equivalent degree plus 3 semesters of postsecondary education and to be certified in cardiopulmonary resuscitation. They also take various seminars and continuing education courses from the NBCH and I-ACT.
How many individuals have used colon cleansing is unclear, although one study suggested that in the United Kingdom, registered practitioners carry out an estimated 5600 procedures every month.10
Where’s the evidence?
Despite colon cleansing’s long history and current popularity, the literature does not support its purported benefits. Historically, colon cleansing was thought to prevent autointoxication from toxins originating in the colon, but the evidence for this claim is limited.11 A search of the literature using the terms “colon cleansing,” “herbal colon cleanse,” “colon detoxification,” and “colon irrigation,” yielded no scientifically robust studies in support of this practice. One study suggested that lymphocytes might migrate from the gut into the circulation after the procedure, which may “improve colon and immune system function.”12
Even though colon cleansing is touted as a commonly used form of holistic, complementary and alternative medicine, the Natural Standard Professional Database concluded in a monograph that there is “limited clinical evidence validating colon therapy as a health promotion practice” and noted a “lack of sufficient evidence” for most of its prescribed uses.13
Adverse effects: From cramping to renal failure
Most reports in the literature note a variety of adverse effects of colon cleansing that range from mild (eg, cramping, abdominal pain, fullness, bloating, nausea, vomiting, perianal irritation, and soreness) to severe (eg, electrolyte imbalance and renal failure).11,14-17 Some herbal preparations have also been associated with aplastic anemia and liver toxicity.18
Case reports also have noted back and pelvic abscesses after colonic hydrotherapy, fatal aeroportia (gas accumulation in the mesenteric veins) with air emboli, rectal perforations, perineal gangrene, acute water intoxication, coffee enema-associated colitis and septicemia, and deaths due to amebiasis.2,3,19-21
The FDA has issued many warning letters
The preparations used for colon cleansing are considered dietary supplements, and the US Food and Drug Administration (FDA) requires that they be labeled as such; the FDA does not preapprove these substances, however. The FDA also requires that colonic hydrotherapy and irrigation system devices meet certain requirements, but the agency has never approved any system for general nonmedical purposes, such as colon cleansing.
The devices have an FDA Class III designation, indicating that if a device is used for purposes beyond what is medically indicated (preparation for radiologic and endoscopic procedures), the manufacturer must obtain premarket approval from the FDA, which is based on evaluation of the safety and effectiveness of the device as shown by available scientific evidence and current regulations.22 During the past decade the FDA has issued numerous warning letters to manufacturers for unapproved use of the devices for colon cleansing.23-26
Raise the issue with patients
Given the current popularity of colon cleansing, it’s important to recognize that some of your patients may engage in, or be thinking about, the practice. (See “4 things to tell patients about colon cleansing”.) Be sure to tell patients about the potential consequences of colon cleansing and to emphasize that there is a lack of evidence to back up supporters’ claims.
- Colon irrigation is not wise—particularly if you have a history of gastrointestinal disease (including diverticulitis, Crohn’s disease, or ulcerative colitis) or a history of colon surgery, severe hemorrhoids, kidney disease, or heart disease. These conditions increase the risk of adverse effects.2,3,11,16
- Side effects of colon cleansing include nausea, vomiting, diarrhea, dizziness, dehydration, electrolyte abnormalities, acute kidney insufficiency, pancreatitis, bowel perforation, heart failure, and infection. 2,3,11,16
- The devices that practitioners use for the procedure are not approved for colon cleansing by the US Food and Drug Administration. Inadequately disinfected or sterilized irrigation machines have been linked to bacterial contamination.2,11,19
- Colon cleansing practitioners are not licensed by a scientifically based organization. Rather, practitioners have undergone a training process structured by an organization that is attempting to institute its own certification and licensing requirements.
CORRESPONDENCE
Ranit Mishori, MD, MHS, Georgetown University School of Medicine, 3900 Reservoir Road, NW, Washington, DC 20007; mishorir@georgetown.edu
• Advise patients that colon cleansing has no proven benefits and many adverse effects. B
• Ask patients with otherwise unexplained nausea, vomiting, or diarrhea if they engage in colon cleansing. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE 1 A 31-year-old African American woman sought treatment at her local emergency department (ED) for nausea, vomiting, and diarrhea. She reported passing more than 6 yellowish-brown, watery, nonbloody stools during the previous 2 days. She felt weak, feverish, and light-headed and showed signs of dehydration.
The patient had Crohn’s disease and had undergone a partial colectomy 5 years earlier. She told the ED physician that 2 days before visiting the ED she had gone to a “cleansing center” for a colonic cleansing, but was unable to complete the process because she developed cramps 15 minutes into the procedure. Less than an hour later, she developed diarrhea, nausea, and vomiting.
In the ED, her serum potassium was 2.9 mEq/L, blood urea nitrogen was 26 mg/dL, and creatinine was 1.9 mg/dL. She was afebrile, with a blood pressure of 135/75 mm Hg and a heart rate of 113 beats per minute. After receiving 2 liters of normal saline and 90 mEq of potassium chloride replacement, the patient felt better and was later discharged from the ED.
Three days later, the patient came to our residency clinic. She described her stools as being loose, but not watery or bloody, and passed in small amounts, about 4 times daily. She still had some abdominal cramping just before passing stool, but bowel movements relieved that. Her vital signs were within normal limits, and her physical exam was benign. The patient was instructed to follow her normal diet, as tolerated, and drink plenty of fluids to maintain good hydration. Her symptoms resolved by the following week.
CASE 2 A 49-year-old African American man came to our community hospital because of vomiting, diarrhea, and abdominal pain he had been experiencing for 4 days. He linked the symptoms to eating a large fast-food breakfast, followed by a big lunch the day before. He described having multiple episodes of nonbloody, nonbilious vomiting, nonbloody watery diarrhea, and “twisting” abdominal pain that was constant but temporarily relieved with a warm compress or positional maneuvers. He had never had a similar episode and had not taken any antibiotics recently.
Upon further questioning, the patient revealed that he had used a colon cleanser a few days earlier. A review showed that he had lost 24 pounds in 10 days. Vitals were within normal limits. Serum potassium was 2.9 mEq/L, and creatinine was 2.1 mg/dL. A computed tomography scan of the abdomen revealed moderate to moderately severe dilatation of multiple small bowel loops with multiple air fluid levels, suggesting an early or partial small bowel obstruction. We obtained a surgical consultation, but surgery was not required. He was discharged after 2 days.
The patient returned to the hospital 3 days later with similar symptoms and severe weakness associated with dizziness. At that time his serum potassium was 2.4 mEq/L and creatinine was 4.0 mg/dL. Aspartate aminotransferase was 29 U/L, alanine aminotransferase was 80 U/L, lipase was 418 U/L, and amylase was 94 U/L.
The patient was readmitted for dehydration, hypokalemia, and pancreatitis and, following a colonoscopy and biopsy that revealed chronic and acute inflammation, a gastroenterologist made a diagnosis of “herbal intoxication.” The patient was hydrated, his electrolytes were replaced, and his diet was slowly returned to normal. He was discharged after 5 days.
An old practice rediscovered
Colon cleansing has been around since ancient times, when its purported benefits were based on the belief that intestinal waste can poison the body (“autointoxication”).1 The procedure became popular in the early 1900s, but in a 1919 paper, the American Medical Association discounted the autointoxication theory and condemned the practice.1 The procedure then fell out of favor, albeit temporarily.2 Colon cleansing has staged a comeback in recent years.
Colon cleansing basics
Colon cleansing, also called colonic irrigation or colonic hydrotherapy, is performed by colonic hygienists or colon therapists, or can be self-administered. The procedure works like an enema. The patient generally lies on a table and water (with or without additional herbs or compounds) is pumped through the rectum via a tube.
Unlike enemas, for which a small amount of fluid is used, however, colon cleansing calls for a large volume of fluid—up to 60 liters—to be introduced into the rectum.3,4 Fluids and waste are expelled through another tube. The procedure may be repeated several times.
Products go by many names
Most colon cleansing products come in the forms of laxatives, teas, powders, and capsules. They can be taken by mouth or inserted into the rectum. They often contain sodium phosphate, coffee, probiotics, enzymes, or any of a variety of herbs.5 Some products contain fiber preparations, including psyllium, flaxseed, and laxatives such as cascara, magnesium oxide, cat’s claw, artichoke leaves, burdock root, licorice, and milk thistle.2
With names such as Nature’s Bounty Colon Cleanser Natural Detox Formula, Health Plus Inc. Colon Cleanse, and 7-Day Miracle Cleanse, as well as endorsements by movie stars, these colon cleansing products are actively promoted as a natural way to enhance one’s well-being. Advertisements promising that colon cleansing will alleviate fatigue, headache, weight gain, and low energy are ubiquitous on the Internet and in newspapers and magazines. The ads tout the safety of “herbal” and “natural” preparations. These materials also provide anecdotal support for claims that colon cleansing improves the immune and circulatory systems, enhances cognitive abilities, and aids weight loss through “detoxification.”6
Individuals who want to cleanse their own colons can choose among home kits, some of which include disposable tubing, while others have components that can be reused if they are sterilized after each use.5,7 But many people turn to a “hydrotherapist” for colon irrigation. The services are also increasingly being offered by practitioners who describe themselves as “colon hygienists.”
These individuals sometimes belong to organizations such as the National Board for Colon Hydrotherapy (NBCH) or the International Association for Colon Hydrotherapy (I-ACT).8,9 These practitioners are not licensed, but they are required to have a high school or equivalent degree plus 3 semesters of postsecondary education and to be certified in cardiopulmonary resuscitation. They also take various seminars and continuing education courses from the NBCH and I-ACT.
How many individuals have used colon cleansing is unclear, although one study suggested that in the United Kingdom, registered practitioners carry out an estimated 5600 procedures every month.10
Where’s the evidence?
Despite colon cleansing’s long history and current popularity, the literature does not support its purported benefits. Historically, colon cleansing was thought to prevent autointoxication from toxins originating in the colon, but the evidence for this claim is limited.11 A search of the literature using the terms “colon cleansing,” “herbal colon cleanse,” “colon detoxification,” and “colon irrigation,” yielded no scientifically robust studies in support of this practice. One study suggested that lymphocytes might migrate from the gut into the circulation after the procedure, which may “improve colon and immune system function.”12
Even though colon cleansing is touted as a commonly used form of holistic, complementary and alternative medicine, the Natural Standard Professional Database concluded in a monograph that there is “limited clinical evidence validating colon therapy as a health promotion practice” and noted a “lack of sufficient evidence” for most of its prescribed uses.13
Adverse effects: From cramping to renal failure
Most reports in the literature note a variety of adverse effects of colon cleansing that range from mild (eg, cramping, abdominal pain, fullness, bloating, nausea, vomiting, perianal irritation, and soreness) to severe (eg, electrolyte imbalance and renal failure).11,14-17 Some herbal preparations have also been associated with aplastic anemia and liver toxicity.18
Case reports also have noted back and pelvic abscesses after colonic hydrotherapy, fatal aeroportia (gas accumulation in the mesenteric veins) with air emboli, rectal perforations, perineal gangrene, acute water intoxication, coffee enema-associated colitis and septicemia, and deaths due to amebiasis.2,3,19-21
The FDA has issued many warning letters
The preparations used for colon cleansing are considered dietary supplements, and the US Food and Drug Administration (FDA) requires that they be labeled as such; the FDA does not preapprove these substances, however. The FDA also requires that colonic hydrotherapy and irrigation system devices meet certain requirements, but the agency has never approved any system for general nonmedical purposes, such as colon cleansing.
The devices have an FDA Class III designation, indicating that if a device is used for purposes beyond what is medically indicated (preparation for radiologic and endoscopic procedures), the manufacturer must obtain premarket approval from the FDA, which is based on evaluation of the safety and effectiveness of the device as shown by available scientific evidence and current regulations.22 During the past decade the FDA has issued numerous warning letters to manufacturers for unapproved use of the devices for colon cleansing.23-26
Raise the issue with patients
Given the current popularity of colon cleansing, it’s important to recognize that some of your patients may engage in, or be thinking about, the practice. (See “4 things to tell patients about colon cleansing”.) Be sure to tell patients about the potential consequences of colon cleansing and to emphasize that there is a lack of evidence to back up supporters’ claims.
- Colon irrigation is not wise—particularly if you have a history of gastrointestinal disease (including diverticulitis, Crohn’s disease, or ulcerative colitis) or a history of colon surgery, severe hemorrhoids, kidney disease, or heart disease. These conditions increase the risk of adverse effects.2,3,11,16
- Side effects of colon cleansing include nausea, vomiting, diarrhea, dizziness, dehydration, electrolyte abnormalities, acute kidney insufficiency, pancreatitis, bowel perforation, heart failure, and infection. 2,3,11,16
- The devices that practitioners use for the procedure are not approved for colon cleansing by the US Food and Drug Administration. Inadequately disinfected or sterilized irrigation machines have been linked to bacterial contamination.2,11,19
- Colon cleansing practitioners are not licensed by a scientifically based organization. Rather, practitioners have undergone a training process structured by an organization that is attempting to institute its own certification and licensing requirements.
CORRESPONDENCE
Ranit Mishori, MD, MHS, Georgetown University School of Medicine, 3900 Reservoir Road, NW, Washington, DC 20007; mishorir@georgetown.edu
1. Ernst E. Colonic irrigation and the theory of autointoxication: a triumph of ignorance over science. J Clin Gastroenterol. 1997;24:196-198.
2. Acosta RD, Cash BD. Clinical effects of colonic cleansing for general health promotion: a systemic review. Am J Gastroenterol. 2009;104:2830-2836.
3. Handley DV, Rieger NA, Rodda DJ. Rectal perforation from colonic irrigation administered by alternative practitioners. Med J Aust. 2004;181:575-576.
4. Seow-Choen F. The physiology of colonic therapy. Colorectal Dis. 2009;11:686-688.
5. Colon cleansing. Med Lett Drugs Ther. 2009;51:39.-
6. Just cleansing. A guide to cleansing and detox. Available at: http://www.justcleansing.com. Accessed November 17, 2010.
7. Home Colonics Company. Available at: http://www.homecolonics.com. Accessed November 17, 2010.
8. National Board for Colon Hydrotherapy. Available at: http://www.nbcht.org. Accessed November 27, 2010.
9. International Association for Colon Hydrotherapy. Available at: http://www.i-act.org. Accessed November 27, 1010.
10. Taffinder NJ, Tan E, Webb IG, et al. Retrograde commercial colonic hydrotherapy. Colorectal Dis. 2004;6:258-260.
11. Richards DG, McMillin DL, Mein EA, et al. Colonic irrigations: a review of the historical controversy and the potential for adverse effects. J Altern Complement Med. 2006;12:389-393.
12. Uchiyama-Tanaka Y. Colon irrigation causes lymphocyte movement from gut-associated lymphatic tissues to peripheral blood. Biomed Res. 2009;30:311-314.
13. Colon therapy/colonic irrigation. Natural Standard Professional Monograph. 2011. Available at: http://naturalstandard.com/databases/hw/colon.asp. Accessed June 21, 2011.
14. Abaskharoun R, Depew W, Vanner S. Changes in renal function following administration of oral sodium phosphate or polyethylene glycol for colon cleansing before colonoscopy. Can J Gastroenterol. 2007;21:227-231.
15. Rex D. Dosing considerations in the use of sodium phosphate bowel preparations for colonoscopy Ann Pharmacother. 2007;41:1466-1475.
16. Dykes C, Cash BD. Key safety issues of bowel preparations for colonoscopy and importance of adequate hydration. Gastroenterology Nurs. 2007;31:30-35.
17. Norlela S, Izham C, Khalid BA. Colonic irrigation-induced hyponatremia. Malays J Pathol. 2004;26:117-118.
18. Smereck J. Aplastic anemia: a possible toxic effect of an herbal “colon cleansing” preparation. J Emerg Med. 2007;11:191-192.
19. Ratnaraja N, Raymond N. Extensive abscesses following colonic hydrotherapy. Lancet Infect Dis. 2005;5:527.-
20. Chen WL, Tsao YT. Fatal aeroportia with systemic air embolism after colon hydrotherapy. J Trauma. 2010;68:247.-
21. Tan MP, Cheong DM. Life-threatening perineal gangrene from rectal perforation following colonic hydrotherapy: a case report. Ann Acad Med Singapore. 1999;28:583-585.
22. US Food and Drug Administration. Premarket approval. Available at: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/PremarketSubmissions/PremarketApprovalPMA/default.htm. Accessed June 21, 2011.
23. US Food and Drug Administration. Device classification. Available at: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Overview/ClassifyYourDevice/default.htm. Accessed December 1, 2010.
24. US Food and Drug Administration. Warning letter to Clearwater Colon Hydrotherapy. Available at: http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2001/ucm178494.htm. Accessed June 21, 2011.
25. US Food and Drug Administration Warning letter to Augustine R. Hoerninger, III, PhD, ND. Available at: http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2003/ucm147378.htm. Accessed June 21, 2011.
26. US Food and Drug Administration. Warning letter to Jimmy J. Girouard. Available at: http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2003/ucm147792.htm. Accessed June 21, 2011.
1. Ernst E. Colonic irrigation and the theory of autointoxication: a triumph of ignorance over science. J Clin Gastroenterol. 1997;24:196-198.
2. Acosta RD, Cash BD. Clinical effects of colonic cleansing for general health promotion: a systemic review. Am J Gastroenterol. 2009;104:2830-2836.
3. Handley DV, Rieger NA, Rodda DJ. Rectal perforation from colonic irrigation administered by alternative practitioners. Med J Aust. 2004;181:575-576.
4. Seow-Choen F. The physiology of colonic therapy. Colorectal Dis. 2009;11:686-688.
5. Colon cleansing. Med Lett Drugs Ther. 2009;51:39.-
6. Just cleansing. A guide to cleansing and detox. Available at: http://www.justcleansing.com. Accessed November 17, 2010.
7. Home Colonics Company. Available at: http://www.homecolonics.com. Accessed November 17, 2010.
8. National Board for Colon Hydrotherapy. Available at: http://www.nbcht.org. Accessed November 27, 2010.
9. International Association for Colon Hydrotherapy. Available at: http://www.i-act.org. Accessed November 27, 1010.
10. Taffinder NJ, Tan E, Webb IG, et al. Retrograde commercial colonic hydrotherapy. Colorectal Dis. 2004;6:258-260.
11. Richards DG, McMillin DL, Mein EA, et al. Colonic irrigations: a review of the historical controversy and the potential for adverse effects. J Altern Complement Med. 2006;12:389-393.
12. Uchiyama-Tanaka Y. Colon irrigation causes lymphocyte movement from gut-associated lymphatic tissues to peripheral blood. Biomed Res. 2009;30:311-314.
13. Colon therapy/colonic irrigation. Natural Standard Professional Monograph. 2011. Available at: http://naturalstandard.com/databases/hw/colon.asp. Accessed June 21, 2011.
14. Abaskharoun R, Depew W, Vanner S. Changes in renal function following administration of oral sodium phosphate or polyethylene glycol for colon cleansing before colonoscopy. Can J Gastroenterol. 2007;21:227-231.
15. Rex D. Dosing considerations in the use of sodium phosphate bowel preparations for colonoscopy Ann Pharmacother. 2007;41:1466-1475.
16. Dykes C, Cash BD. Key safety issues of bowel preparations for colonoscopy and importance of adequate hydration. Gastroenterology Nurs. 2007;31:30-35.
17. Norlela S, Izham C, Khalid BA. Colonic irrigation-induced hyponatremia. Malays J Pathol. 2004;26:117-118.
18. Smereck J. Aplastic anemia: a possible toxic effect of an herbal “colon cleansing” preparation. J Emerg Med. 2007;11:191-192.
19. Ratnaraja N, Raymond N. Extensive abscesses following colonic hydrotherapy. Lancet Infect Dis. 2005;5:527.-
20. Chen WL, Tsao YT. Fatal aeroportia with systemic air embolism after colon hydrotherapy. J Trauma. 2010;68:247.-
21. Tan MP, Cheong DM. Life-threatening perineal gangrene from rectal perforation following colonic hydrotherapy: a case report. Ann Acad Med Singapore. 1999;28:583-585.
22. US Food and Drug Administration. Premarket approval. Available at: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/HowtoMarketYourDevice/PremarketSubmissions/PremarketApprovalPMA/default.htm. Accessed June 21, 2011.
23. US Food and Drug Administration. Device classification. Available at: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Overview/ClassifyYourDevice/default.htm. Accessed December 1, 2010.
24. US Food and Drug Administration. Warning letter to Clearwater Colon Hydrotherapy. Available at: http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2001/ucm178494.htm. Accessed June 21, 2011.
25. US Food and Drug Administration Warning letter to Augustine R. Hoerninger, III, PhD, ND. Available at: http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2003/ucm147378.htm. Accessed June 21, 2011.
26. US Food and Drug Administration. Warning letter to Jimmy J. Girouard. Available at: http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2003/ucm147792.htm. Accessed June 21, 2011.
Beta-blockers for heart failure: Why you should use them more
• Initiate beta-blocker therapy in low doses for patients with heart failure, and increase the dose gradually until the target dosage is achieved. A
• The benefit of beta-blocker therapy for patients with heart failure is proportional to the degree of heart rate reduction. A
• Consider beta-blocker therapy for patients with coexisting chronic obstructive pulmonary disease or decompensated heart failure, although treatment may have to be reduced or temporarily withheld. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The evidence is clear: Beta-blockers reduce mortality and hospitalization in patients with systolic heart failure.1-3 Yet this class of drugs is underutilized by physicians who fear that beta-blocker’s negative inotropic effect will lead to worsening heart failure.4
Our aim in presenting this review is to counter such concerns by detailing the latest evidence. We draw on current research findings to answer questions about beta-blocker selection and dosage and address common misconceptions.
Beta-blockers shown blocking the effects of epinephrine and norepinephrine at the receptor sites.
Do beta-blockers lower mortality rates for patients with heart failure?
Yes. Three beta-blockers—bisoprolol, carvedilol, and metoprolol succinate—have been conclusively shown to reduce morbidity as well as mortality in patients with systolic heart failure (TABLE 1).1-3,5,6 Here’s a look at the studies:
Bisoprolol. The Cardiac Insufficiency Bisoprolol Study (CIBIS II), a randomized controlled trial (RCT) involving 2647 patients with New York Heart Association (NYHA) Class III or IV heart failure and an ejection fraction (EF) ≤35%, found that bisoprolol reduced the primary end point of all-cause mortality (hazard ratio [HR]=0.66; 95% confidence interval [CI], 0.54-0.81; P<.0001) compared with placebo. Cardiovascular mortality rates (HR=0.71; 95% CI, 0.56-0.90; P=.0049) and hospitalization rates (HR=0.80; 95% CI, 0.71-0.91; P=.0006) were significantly reduced, as well.1
Carvedilol. In the Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) trial, an RCT featuring 2289 patients with EF <25%, carvedilol significantly reduced the total death rate (HR=0.65; 95% CI, 0.52-0.81; P=.0014) compared with placebo.2
Metoprolol succinate. The Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure (MERIT-HF), a study of nearly 4000 patients with Class II to IV heart failure and EF ≤40%, found that metoprolol succinate lowered total mortality or all-cause hospitalization (HR=0.81; 95% CI, 0.73-0.90; P<.001) compared with placebo.3
TABLE 1
Beta-blockers for heart failure patients: What the studies show
| Trial | Study group (N) | Mean follow-up | Agent tested | Primary end point | RR; 95% CI; P value |
|---|---|---|---|---|---|
| BEST5 | Class III-IV HF, EF ≤35% (2708) | 2 y | Bucindolol | All-cause death | 0.90; 0.78-1.02; .13 |
| CIBIS II1 | Class III-IV HF, EF ≤35% (2647) | 1.3 y | Bisoprolol | All-cause death | 0.66; 0.54-0.81; <.0001 |
| COPERNICUS2 | HF symptoms, EF ≤25% (2289) | 10.4 mo | Carvedilol | All-cause death | 0.65; 0.52-0.81; .0014 |
| MERIT-HF3 | Class II-IV HF, EF ≤40% (3991) | 1 y | Metoprolol succinate | Composite* | 0.81; 0.73-0.90; <.001 |
| SENIORS6 | Age >70 y and hospitalization for HF or EF ≤35% (2128) | 21 mo | Nebivolol | All-cause death and CVD hospitalization | 0.86; 0.74-0.99; .039 |
| *All-cause mortality and all-cause hospitalization. BEST, Beta-blocker Evaluation of Survival Trial; CI, confidence interval; CIBIS II, Cardiac Insufficiency Bisoprolol Study; COPERNICUS, Carvedilol Prospective Randomized Cumulative Survival; CVD, cardiovascular disease; EF, ejection fraction; HF, heart failure; MERIT-HF, Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure; RR, relative risk; SENIORS, Study of the Effects of Nebivolol Intervention on Outcomes and Rehospitalisation in Seniors with Heart Failure. | |||||
Carvedilol and metoprolol go head-to-head
Although carvedilol and metoprolol have been shown to have similar hemodynamic and heart rate effects, the Carvedilol or Metoprolol European Trial (COMET) found that carvedilol is superior in extending survival. More than 3000 patients with Class II to IV heart failure and an EF <35% were randomized to carvedilol (target dose 25 mg bid) or metoprolol tartrate (target dose 50 mg bid). After 58 months, total mortality was significantly lower in the carvedilol arm (HR=0.83; 95% CI, 0.74-0.93; P=.0017).7
Which metoprolol formulation? While RCTs have found that metoprolol tartrate has a favorable effect on EF and hemodynamic data, it is not approved by the US Food and Drug Administration (FDA) as a treatment for heart failure—and its ability to reduce morbidity and mortality in patients with heart failure has not been established.8,9 Thus, metoprolol succinate, but not metoprolol tartrate, is recommended for heart failure treatment by the American College of Cardiology, American Heart Association, and European Society of Intensive Care Medicine.10,11
These agents lack evidence of efficacy
Not all beta-blockers have therapeutic value for patients with heart failure—or evidence to support them.
Bucindolol. The Beta-blocker Evaluation of Survival Trial (BEST), a trial of 2708 patients with Class III or IV heart failure and an EF ≤35%, found no difference in total mortality between bucindolol and placebo.5 As a result, the drug did not receive FDA approval.12 The FDA has since designated the investigation of bucindolol (trade name Gencaro) for the reduction of cardiovascular hospitalizations and mortality of heart failure patients with a particular genotype as a Fast Track development program.13
Nebivolol. The Study of the Effects of Nebivolol Intervention on Outcomes and Rehospitalisation in Seniors with Heart Failure (SENIORS) randomized 2128 patients older than 70 years with prior hospitalization for heart failure or an EF ≤35% to nebivolol (1.25-10 mg/d) or placebo. Nebivolol (which is not approved for the treatment of heart failure in the United States) reduced the composite end point of all-cause mortality and cardiovascular hospitalization (HR=0.86; 95% CI, 0.74-0.99; P=.039), but did not reduce the total mortality rate.6
Atenolol. Some retrospective analyses have suggested that heart failure patients do as well on atenolol as patients taking metoprolol or carvedilol.14,15 Because no RCTs have established the efficacy of atenolol, however, it is not recommended for the treatment of heart failure.
Is the dose sufficient to reduce heart rate?
The benefit of beta-blocker therapy for patients with heart failure is proportional to the degree of heart rate reduction, so it is important to find the highest tolerable dose.16,17 The COMET study detailed earlier sparked considerable controversy, with some observers contending that the dose of metoprolol used was too small to adequately lower the heart rate.18,19
A subsequent study, the Systolic Heart Failure Treatment with the I(f) Inhibitor Ivabradine Trial (SHIFT), highlights the importance of rate reduction in heart failure outcomes. In this placebo-controlled trial of 6558 patients with EF ≤35%, treatment with the heart rate-reducing agent ivabradine reduced cardiovascular death and hospitalization from heart failure (HR=0.82; 95% CI, 0.75-0.90; P<.0001) compared with placebo.20 A subsequent analysis showed that the primary outcome increased by 16% for every 5 beats-per-minute (BPM) increase.21
Start low, go slow
When initiating and titrating beta-blockers, the major RCTs clearly illustrate the importance of the dictum, “Start low, go slow” (TABLE 2).1-3
In CIBIS II, patients were started on bisoprolol at a dose of 1.25 mg/d. After a week, the dosage was increased by 1.25 mg. Titration continued over a 4-week period until the maximum tolerable dose was reached. Although 43% of patients reached the 10 mg/d target, a third of those studied remained on <5 mg/d.1
In COPERNICUS, carvedilol was started at 3.125 mg twice a day and continued at that dosage for 2 weeks. The dose was then titrated up at 2-week intervals, to 6.25 mg bid, then 12.5 mg bid, before attempting to reach the target dose of 25 mg bid. Ultimately, 66% received the target dose.2
In MERIT-HF, metoprolol succinate was initiated at 12.5 mg daily and doubled every 2 weeks until the target (200 mg/d) was achieved. Nearly two-thirds (64%) of those in the treatment group reached the target dose.3
In COMET, the researchers used the same drug regimen for carvedilol that was used in COPERNICUS (starting at 3.125 mg bid and slowly titrating to reach a 25-mg bid target). Patients on metoprolol tartrate initially received 5 mg bid; the dose was increased every 2 weeks until the target—50 mg bid—was reached. Seventy-five percent of patients reached the targeted carvedilol dose, and 78% reached the metoprolol target.7
TABLE 2
Titrating beta-blocker therapy
| Trial | Agent | Initial dose | Interval on starting dose | Mean dose achieved | Target dose achieved |
|---|---|---|---|---|---|
| CIBIS II1 | Bisoprolol | 1.25 mg/d | 1 week | 8.5 mg/d | 10 mg/d (43%) |
| COPERNICUS2 | Carvedilol | 3.125 mg bid | 2 weeks | 18.5 mg bid | 25 mg bid (66%) |
| MERIT-HF3 | Metoprolol succinate | 12.5 mg/d | 2 weeks | 159 mg/d | 200 mg/d (64%) |
| CIBIS-II, Cardiac Insufficiency Bisoprolol Study; COPERNICUS, Carvedilol Prospective Randomized Cumulative Survival; MERIT-HF, Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure. | |||||
Help beta-blocker therapy succeed
A significant number of patients with heart failure will be unable to tolerate an adequate dose of beta-blockers, at least on the first attempt.22 In such cases, a second attempt on another occasion—eg, after symptomatic bronchospasm or acute heart failure has been controlled—should be made.
In CIBIS II, 15% of the patients randomized to bisoprolol stopped taking it;1 in COPERNICUS, the withdrawal rate from carvedilol was also 15%;2 and in MERIT-HF, 10% of patients taking metoprolol experienced an adverse event that led to drug withdrawal.3 Although withdrawal rates were similar among patients on placebo in all 3 trials, they nonetheless suggest that even with the precautions and scrutiny characteristic of clinical trials, 10% to 15% of patients with heart failure will experience difficulty with beta-blocker treatment. (In a study of patients in one heart failure clinic, the withdrawal rate approached 40%.22)
Considering the benefits of beta-blockers for patients with all levels of heart failure, it is incumbent on physicians to prescribe them for as many of these patients as possible (See “Are beta-blockers contraindicated for these heart failure patients?”) and to attempt to reduce withdrawal rates.
Educate the patient. One way to do this is to provide adequate patient education, stressing the importance of taking the medication exactly as prescribed and, when necessary, showing patients how to divide pills until the target dose is reached.
Respond to adverse effects. Closely monitoring for adverse effects is crucial, as well. The development of symptomatic bradycardia, second or third degree atrioventricular block, or a heart rate <50 BPM suggests that the dosage be reduced or the medication withheld, with this caveat: There is increasing recognition that heart rate and BP readings change throughout the day, and a decision to adjust or to halt beta-blocker therapy should not be based on a single measure.
That said, physicians should watch for clinical evidence of hypoperfusion, such as postural dizziness or decreasing urine output, when systolic BP approaches 80 to 90 mm Hg in patients with heart failure. In such cases, adjusting the dose, increasing the interval between doses, or even discontinuing beta-blocker therapy may be necessary.
Because of the bradyarrhythmic and hypotensive effects of beta-blockers, the major heart failure trials excluded patients with a heart rate of <50 to 68 beats per minute (BPM) or systolic blood pressure <80 to 100 mm Hg (the ranges cited reflect the variation in cut points from one study to another).1-3,6 And in clinical practice, physicians often withhold beta-blocker therapy from heart failure patients who also have chronic obstructive pulmonary disease (COPD) or asthma, hypotension, or metabolic risk factors for diabetes.4 Some avoid prescribing beta-blockers because they believe that the drugs adversely affect patients’ quality of life, despite evidence to the contrary.3,23-25 In all these cases, there is little justification for doing so.
COPD and asthma. Although beta-blockers can worsen and precipitate bronchospasm, recent evidence suggests that patients with COPD and asthma can tolerate them.26-28 In fact, there is reason to believe that bronchospasm is aggravated by excessive stimulation and sensitization of the beta-2 receptors, and that blocking them may even be of therapeutic value.29 Nonetheless, the danger of worsening bronchospasm with a nonselective beta-blocker such as carvedilol remains—particularly for patients with asthma, who tend to have a higher degree of bronchial sensitivity and reactivity. So, while beta-blockers are not contraindicated for patients with COPD, their use in this patient population requires caution.30,31
Metabolic risk factors. Caution is also needed for patients with metabolic risk factors. Although beta-blockers have been found to increase the risk of diabetes, raise triglycerides, and lower high-density lipoprotein cholesterol,32-34 the benefits for patients with heart failure outweigh the risk. Physicians must remember that the mortality rate of heart failure, as well as the rate of progression, is higher than that of metabolic abnormalities, asymptomatic bradycardia, hypotension, or bronchospasm, which are relatively benign. In view of evidence that beta-blockers reduce both mortality and hospitalization rates associated with heart failure, the best approach is to continue beta-blocker therapy and seek control of risk factors and adverse effects.
CORRESPONDENCE
HT Ong FRCP, FACC, FESC, HT Ong Heart Clinic, 251C Burma Road, Penang 103250, Malaysia; ongheanteik@gmail.com
1. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet. 1999;353:9-13.
2. Packer M, Fowler MB, Roecker EB, et al. Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) Study Group. Effect of carvedilol on the morbidity of patients with severe chronic heart failure: results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation. 2002;106:2194-2199.
3. Hjalmarson A, Goldstein S, Fagerberg B, et al. for the MERIT-HF Study Group. Effects of controlled-release metoprolol on total mortality, hospitalization and well-being in patients with heart failure. The Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure (MERIT-HF). JAMA. 2000;283:1295-1302.
4. Mann DL. Management of heart failure patients with reduced ejection fraction. In: Libby P, Bonow RO, Mann DL, et al, eds. Braunwald’s Heart Disease. A Textbook of Cardiovascular Medicine. 8th ed. Philadelphia: Saunders Elsevier; 2008:611-640.
5. Beta-Blocker Evaluation of Survival Trial Investigators. A trial of the beta-blocker bucindolol in patients with advanced chronic heart failure. N Engl J Med. 2001;344:1659-1667.
6. Flather MD, Shibata MC, Coats AJ, et al. SENIORS Investigators. Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur Heart J. 2005;26:215-225.
7. Poole-Wilson PA, Swedberg K, Cleland JG, et al. Carvedilol Or Metoprolol European Trial Investigators. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol Or Metoprolol European Trial (COMET): randomised controlled trial. Lancet. 2003;362:7-13.
8. Waagstein F, Bristow MR, Swedberg K, et al. Beneficial effects of metoprolol in idiopathic dilated cardiomyopathy. Metoprolol in Dilated Cardiomyopathy (MDC) Trial Study Group. Lancet. 1993;342:1441-1446.
9. Waagstein F, Stromblad O, Andersson B, et al. Increased exercise ejection fraction and reversed remodeling after long-term treatment with metoprolol in congestive heart failure: a randomized, stratified, double-blind, placebo-controlled trial in mild to moderate heart failure due to ischemic or idiopathic dilated cardiomyopathy. Eur J Heart Fail. 2003;5:679-691.
10. Dickstein K, Cohen-Solal A, Filippatos G. ESC Committee for Practice Guidelines (CPG). ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the diagnosis and treatment of acute and chronic heart failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur J Heart Fail. 2008;10:933-989.
11. Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation. 2009;119:e391-e479.
12. Complete response letter for Gencaro NDA. Available at: http://www.drugs.com/nda/gencaro_090601.html. June 1, 2009. Accessed July 15, 2011.
13. ARCA Biopharma. ARCA announces Special Protocol Assessment agreement with FDA for bucindolol development in genotype-defined heart failure patients. May 17, 2010. Available at: http://www.advfn.com/news_ARCA-Announces-Special-Protocol-Assessment-Agreement-with-FDA-for-Bucindolol-Dev_42847369. Accessed July 14, 2010.
14. Go AS, Yang J, Gurwitz JH, Hsu J, et al. Comparative effectiveness of different beta-adrenergic antagonists on mortality among adults with heart failure in clinical practice. Arch Intern Med. 2008;168:2415-2421.
15. Kapoor JR, Heidenreich PA. Survival among patients with left ventricular systolic dysfunction treated with atenolol. Congest Heart Fail. 2009;15:213-217.
16. Nishiyama K, Tsutamoto T, Yamaji M, et al. Dose-dependent prognostic effect of carvedilol in patients with chronic heart failure—special reference to transcardiac [corrected] gradient of norepinephrine. Circ J. 2009;73:2270-2275.
17. McAlister FA, Wiebe N, Ezekowitz JA, et al. Meta-analysis: beta-blocker dose, heart rate reduction, and death in patients with heart failure. Ann Intern Med. 2009;150:784-794.
18. Hjalmarson A, Waagstein F. COMET: a proposed mechanism of action to explain the results and concerns about dose. Lancet. 2003;362:1077.-
19. Dargie HJ. Beta blockers in heart failure. Lancet. 2003;362:2-3.
20. Swedberg K, Komajda M, Böhm M, et al. SHIFT Investigators. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet. 2010;376:875-885.
21. Böhm M, Swedberg K, Komajda M, et al. SHIFT Investigators. Heart rate as a risk factor in chronic heart failure (SHIFT): the association between heart rate and outcomes in a randomised placebo-controlled trial. Lancet. 2010;376:886-894.
22. Galatius S, Gustafsson F, Atar D, et al. Tolerability of beta-blocker initiation and titration with bisoprolol and carvedilol in congestive heart failure—a randomized comparison. Cardiology. 2004;102:160-165.
23. Dobre D, van Jaarsveld CH, deJongste MJ, et al. The effect of beta-blocker therapy on quality of life in heart failure patients: a systematic review and meta-analysis. Pharmacoepidemiol Drug Saf. 2007;16:152-159.
24. Tate CW 3rd, Robertson AD, Zolty R, et al. Quality of life and prognosis in heart failure: results of the Beta-Blocker Evaluation of Survival Trial (BEST). J Card Fail. 2007;13:732-737.
25. Belenkov IuN, Skvortsov AA, Mareev VIu, et al. Clinical, hemodynamic and neurohumoral effects of long-term therapy of patients with severe chronic heart failure with beta-adrenoblocker bisoprolol. Kardiologiia. 2003;43:10-21.
26. LeJemtel TH, Padeletti M, Jelic S. Diagnostic and therapeutic challenges in patients with coexistent chronic obstructive pulmonary disease and chronic heart failure. J Am Coll Cardiol. 2007;49:171-180.
27. Mascarenhas J, Azevedo A, Bettencourt P. Coexisting chronic obstructive pulmonary disease and heart failure: implications for treatment, course and mortality. Curr Opin Pulm Med. 2010;16:106-111.
28. Navas EV, Taylor DO. Q: Can patients with COPD or asthma take a beta-blocker? Cleve Clin J Med. 2010;77:498-499.
29. Bond RA, Spina D, Parra S, et al. Getting to the heart of asthma: can “beta blockers” be useful to treat asthma? Pharmacol Ther. 2007;115:360-374.
30. Cazzola M, Matera MG. Beta-blockers are safe in patients with chronic obstructive pulmonary disease, but only with caution. Am J Respir Crit Care Med. 2008;178:661-662.
31. Shaw SM, Hasleton J, Williams SG. Beta-blocker use in heart failure patients with airways disease. Clin Cardiol. 2009;32:393-396.
32. Elliott WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet. 2007;369:249-256.
33. Dammitt SB, Williams PD, Croft KD, et al. Effects of beta-blockers on the concentration and oxidizability of plasma lipids. Clin Sci (Lond). 1998;94:573-578.
34. Kuster GM, Amann FW, Neuenschwander C, Drexel H. High density-lipoprotein subfractions of patients using cardio-selective beta-blockers. Cardiovasc Drugs Ther. 2002;16:127-131.
• Initiate beta-blocker therapy in low doses for patients with heart failure, and increase the dose gradually until the target dosage is achieved. A
• The benefit of beta-blocker therapy for patients with heart failure is proportional to the degree of heart rate reduction. A
• Consider beta-blocker therapy for patients with coexisting chronic obstructive pulmonary disease or decompensated heart failure, although treatment may have to be reduced or temporarily withheld. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The evidence is clear: Beta-blockers reduce mortality and hospitalization in patients with systolic heart failure.1-3 Yet this class of drugs is underutilized by physicians who fear that beta-blocker’s negative inotropic effect will lead to worsening heart failure.4
Our aim in presenting this review is to counter such concerns by detailing the latest evidence. We draw on current research findings to answer questions about beta-blocker selection and dosage and address common misconceptions.
Beta-blockers shown blocking the effects of epinephrine and norepinephrine at the receptor sites.
Do beta-blockers lower mortality rates for patients with heart failure?
Yes. Three beta-blockers—bisoprolol, carvedilol, and metoprolol succinate—have been conclusively shown to reduce morbidity as well as mortality in patients with systolic heart failure (TABLE 1).1-3,5,6 Here’s a look at the studies:
Bisoprolol. The Cardiac Insufficiency Bisoprolol Study (CIBIS II), a randomized controlled trial (RCT) involving 2647 patients with New York Heart Association (NYHA) Class III or IV heart failure and an ejection fraction (EF) ≤35%, found that bisoprolol reduced the primary end point of all-cause mortality (hazard ratio [HR]=0.66; 95% confidence interval [CI], 0.54-0.81; P<.0001) compared with placebo. Cardiovascular mortality rates (HR=0.71; 95% CI, 0.56-0.90; P=.0049) and hospitalization rates (HR=0.80; 95% CI, 0.71-0.91; P=.0006) were significantly reduced, as well.1
Carvedilol. In the Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) trial, an RCT featuring 2289 patients with EF <25%, carvedilol significantly reduced the total death rate (HR=0.65; 95% CI, 0.52-0.81; P=.0014) compared with placebo.2
Metoprolol succinate. The Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure (MERIT-HF), a study of nearly 4000 patients with Class II to IV heart failure and EF ≤40%, found that metoprolol succinate lowered total mortality or all-cause hospitalization (HR=0.81; 95% CI, 0.73-0.90; P<.001) compared with placebo.3
TABLE 1
Beta-blockers for heart failure patients: What the studies show
| Trial | Study group (N) | Mean follow-up | Agent tested | Primary end point | RR; 95% CI; P value |
|---|---|---|---|---|---|
| BEST5 | Class III-IV HF, EF ≤35% (2708) | 2 y | Bucindolol | All-cause death | 0.90; 0.78-1.02; .13 |
| CIBIS II1 | Class III-IV HF, EF ≤35% (2647) | 1.3 y | Bisoprolol | All-cause death | 0.66; 0.54-0.81; <.0001 |
| COPERNICUS2 | HF symptoms, EF ≤25% (2289) | 10.4 mo | Carvedilol | All-cause death | 0.65; 0.52-0.81; .0014 |
| MERIT-HF3 | Class II-IV HF, EF ≤40% (3991) | 1 y | Metoprolol succinate | Composite* | 0.81; 0.73-0.90; <.001 |
| SENIORS6 | Age >70 y and hospitalization for HF or EF ≤35% (2128) | 21 mo | Nebivolol | All-cause death and CVD hospitalization | 0.86; 0.74-0.99; .039 |
| *All-cause mortality and all-cause hospitalization. BEST, Beta-blocker Evaluation of Survival Trial; CI, confidence interval; CIBIS II, Cardiac Insufficiency Bisoprolol Study; COPERNICUS, Carvedilol Prospective Randomized Cumulative Survival; CVD, cardiovascular disease; EF, ejection fraction; HF, heart failure; MERIT-HF, Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure; RR, relative risk; SENIORS, Study of the Effects of Nebivolol Intervention on Outcomes and Rehospitalisation in Seniors with Heart Failure. | |||||
Carvedilol and metoprolol go head-to-head
Although carvedilol and metoprolol have been shown to have similar hemodynamic and heart rate effects, the Carvedilol or Metoprolol European Trial (COMET) found that carvedilol is superior in extending survival. More than 3000 patients with Class II to IV heart failure and an EF <35% were randomized to carvedilol (target dose 25 mg bid) or metoprolol tartrate (target dose 50 mg bid). After 58 months, total mortality was significantly lower in the carvedilol arm (HR=0.83; 95% CI, 0.74-0.93; P=.0017).7
Which metoprolol formulation? While RCTs have found that metoprolol tartrate has a favorable effect on EF and hemodynamic data, it is not approved by the US Food and Drug Administration (FDA) as a treatment for heart failure—and its ability to reduce morbidity and mortality in patients with heart failure has not been established.8,9 Thus, metoprolol succinate, but not metoprolol tartrate, is recommended for heart failure treatment by the American College of Cardiology, American Heart Association, and European Society of Intensive Care Medicine.10,11
These agents lack evidence of efficacy
Not all beta-blockers have therapeutic value for patients with heart failure—or evidence to support them.
Bucindolol. The Beta-blocker Evaluation of Survival Trial (BEST), a trial of 2708 patients with Class III or IV heart failure and an EF ≤35%, found no difference in total mortality between bucindolol and placebo.5 As a result, the drug did not receive FDA approval.12 The FDA has since designated the investigation of bucindolol (trade name Gencaro) for the reduction of cardiovascular hospitalizations and mortality of heart failure patients with a particular genotype as a Fast Track development program.13
Nebivolol. The Study of the Effects of Nebivolol Intervention on Outcomes and Rehospitalisation in Seniors with Heart Failure (SENIORS) randomized 2128 patients older than 70 years with prior hospitalization for heart failure or an EF ≤35% to nebivolol (1.25-10 mg/d) or placebo. Nebivolol (which is not approved for the treatment of heart failure in the United States) reduced the composite end point of all-cause mortality and cardiovascular hospitalization (HR=0.86; 95% CI, 0.74-0.99; P=.039), but did not reduce the total mortality rate.6
Atenolol. Some retrospective analyses have suggested that heart failure patients do as well on atenolol as patients taking metoprolol or carvedilol.14,15 Because no RCTs have established the efficacy of atenolol, however, it is not recommended for the treatment of heart failure.
Is the dose sufficient to reduce heart rate?
The benefit of beta-blocker therapy for patients with heart failure is proportional to the degree of heart rate reduction, so it is important to find the highest tolerable dose.16,17 The COMET study detailed earlier sparked considerable controversy, with some observers contending that the dose of metoprolol used was too small to adequately lower the heart rate.18,19
A subsequent study, the Systolic Heart Failure Treatment with the I(f) Inhibitor Ivabradine Trial (SHIFT), highlights the importance of rate reduction in heart failure outcomes. In this placebo-controlled trial of 6558 patients with EF ≤35%, treatment with the heart rate-reducing agent ivabradine reduced cardiovascular death and hospitalization from heart failure (HR=0.82; 95% CI, 0.75-0.90; P<.0001) compared with placebo.20 A subsequent analysis showed that the primary outcome increased by 16% for every 5 beats-per-minute (BPM) increase.21
Start low, go slow
When initiating and titrating beta-blockers, the major RCTs clearly illustrate the importance of the dictum, “Start low, go slow” (TABLE 2).1-3
In CIBIS II, patients were started on bisoprolol at a dose of 1.25 mg/d. After a week, the dosage was increased by 1.25 mg. Titration continued over a 4-week period until the maximum tolerable dose was reached. Although 43% of patients reached the 10 mg/d target, a third of those studied remained on <5 mg/d.1
In COPERNICUS, carvedilol was started at 3.125 mg twice a day and continued at that dosage for 2 weeks. The dose was then titrated up at 2-week intervals, to 6.25 mg bid, then 12.5 mg bid, before attempting to reach the target dose of 25 mg bid. Ultimately, 66% received the target dose.2
In MERIT-HF, metoprolol succinate was initiated at 12.5 mg daily and doubled every 2 weeks until the target (200 mg/d) was achieved. Nearly two-thirds (64%) of those in the treatment group reached the target dose.3
In COMET, the researchers used the same drug regimen for carvedilol that was used in COPERNICUS (starting at 3.125 mg bid and slowly titrating to reach a 25-mg bid target). Patients on metoprolol tartrate initially received 5 mg bid; the dose was increased every 2 weeks until the target—50 mg bid—was reached. Seventy-five percent of patients reached the targeted carvedilol dose, and 78% reached the metoprolol target.7
TABLE 2
Titrating beta-blocker therapy
| Trial | Agent | Initial dose | Interval on starting dose | Mean dose achieved | Target dose achieved |
|---|---|---|---|---|---|
| CIBIS II1 | Bisoprolol | 1.25 mg/d | 1 week | 8.5 mg/d | 10 mg/d (43%) |
| COPERNICUS2 | Carvedilol | 3.125 mg bid | 2 weeks | 18.5 mg bid | 25 mg bid (66%) |
| MERIT-HF3 | Metoprolol succinate | 12.5 mg/d | 2 weeks | 159 mg/d | 200 mg/d (64%) |
| CIBIS-II, Cardiac Insufficiency Bisoprolol Study; COPERNICUS, Carvedilol Prospective Randomized Cumulative Survival; MERIT-HF, Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure. | |||||
Help beta-blocker therapy succeed
A significant number of patients with heart failure will be unable to tolerate an adequate dose of beta-blockers, at least on the first attempt.22 In such cases, a second attempt on another occasion—eg, after symptomatic bronchospasm or acute heart failure has been controlled—should be made.
In CIBIS II, 15% of the patients randomized to bisoprolol stopped taking it;1 in COPERNICUS, the withdrawal rate from carvedilol was also 15%;2 and in MERIT-HF, 10% of patients taking metoprolol experienced an adverse event that led to drug withdrawal.3 Although withdrawal rates were similar among patients on placebo in all 3 trials, they nonetheless suggest that even with the precautions and scrutiny characteristic of clinical trials, 10% to 15% of patients with heart failure will experience difficulty with beta-blocker treatment. (In a study of patients in one heart failure clinic, the withdrawal rate approached 40%.22)
Considering the benefits of beta-blockers for patients with all levels of heart failure, it is incumbent on physicians to prescribe them for as many of these patients as possible (See “Are beta-blockers contraindicated for these heart failure patients?”) and to attempt to reduce withdrawal rates.
Educate the patient. One way to do this is to provide adequate patient education, stressing the importance of taking the medication exactly as prescribed and, when necessary, showing patients how to divide pills until the target dose is reached.
Respond to adverse effects. Closely monitoring for adverse effects is crucial, as well. The development of symptomatic bradycardia, second or third degree atrioventricular block, or a heart rate <50 BPM suggests that the dosage be reduced or the medication withheld, with this caveat: There is increasing recognition that heart rate and BP readings change throughout the day, and a decision to adjust or to halt beta-blocker therapy should not be based on a single measure.
That said, physicians should watch for clinical evidence of hypoperfusion, such as postural dizziness or decreasing urine output, when systolic BP approaches 80 to 90 mm Hg in patients with heart failure. In such cases, adjusting the dose, increasing the interval between doses, or even discontinuing beta-blocker therapy may be necessary.
Because of the bradyarrhythmic and hypotensive effects of beta-blockers, the major heart failure trials excluded patients with a heart rate of <50 to 68 beats per minute (BPM) or systolic blood pressure <80 to 100 mm Hg (the ranges cited reflect the variation in cut points from one study to another).1-3,6 And in clinical practice, physicians often withhold beta-blocker therapy from heart failure patients who also have chronic obstructive pulmonary disease (COPD) or asthma, hypotension, or metabolic risk factors for diabetes.4 Some avoid prescribing beta-blockers because they believe that the drugs adversely affect patients’ quality of life, despite evidence to the contrary.3,23-25 In all these cases, there is little justification for doing so.
COPD and asthma. Although beta-blockers can worsen and precipitate bronchospasm, recent evidence suggests that patients with COPD and asthma can tolerate them.26-28 In fact, there is reason to believe that bronchospasm is aggravated by excessive stimulation and sensitization of the beta-2 receptors, and that blocking them may even be of therapeutic value.29 Nonetheless, the danger of worsening bronchospasm with a nonselective beta-blocker such as carvedilol remains—particularly for patients with asthma, who tend to have a higher degree of bronchial sensitivity and reactivity. So, while beta-blockers are not contraindicated for patients with COPD, their use in this patient population requires caution.30,31
Metabolic risk factors. Caution is also needed for patients with metabolic risk factors. Although beta-blockers have been found to increase the risk of diabetes, raise triglycerides, and lower high-density lipoprotein cholesterol,32-34 the benefits for patients with heart failure outweigh the risk. Physicians must remember that the mortality rate of heart failure, as well as the rate of progression, is higher than that of metabolic abnormalities, asymptomatic bradycardia, hypotension, or bronchospasm, which are relatively benign. In view of evidence that beta-blockers reduce both mortality and hospitalization rates associated with heart failure, the best approach is to continue beta-blocker therapy and seek control of risk factors and adverse effects.
CORRESPONDENCE
HT Ong FRCP, FACC, FESC, HT Ong Heart Clinic, 251C Burma Road, Penang 103250, Malaysia; ongheanteik@gmail.com
• Initiate beta-blocker therapy in low doses for patients with heart failure, and increase the dose gradually until the target dosage is achieved. A
• The benefit of beta-blocker therapy for patients with heart failure is proportional to the degree of heart rate reduction. A
• Consider beta-blocker therapy for patients with coexisting chronic obstructive pulmonary disease or decompensated heart failure, although treatment may have to be reduced or temporarily withheld. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The evidence is clear: Beta-blockers reduce mortality and hospitalization in patients with systolic heart failure.1-3 Yet this class of drugs is underutilized by physicians who fear that beta-blocker’s negative inotropic effect will lead to worsening heart failure.4
Our aim in presenting this review is to counter such concerns by detailing the latest evidence. We draw on current research findings to answer questions about beta-blocker selection and dosage and address common misconceptions.
Beta-blockers shown blocking the effects of epinephrine and norepinephrine at the receptor sites.
Do beta-blockers lower mortality rates for patients with heart failure?
Yes. Three beta-blockers—bisoprolol, carvedilol, and metoprolol succinate—have been conclusively shown to reduce morbidity as well as mortality in patients with systolic heart failure (TABLE 1).1-3,5,6 Here’s a look at the studies:
Bisoprolol. The Cardiac Insufficiency Bisoprolol Study (CIBIS II), a randomized controlled trial (RCT) involving 2647 patients with New York Heart Association (NYHA) Class III or IV heart failure and an ejection fraction (EF) ≤35%, found that bisoprolol reduced the primary end point of all-cause mortality (hazard ratio [HR]=0.66; 95% confidence interval [CI], 0.54-0.81; P<.0001) compared with placebo. Cardiovascular mortality rates (HR=0.71; 95% CI, 0.56-0.90; P=.0049) and hospitalization rates (HR=0.80; 95% CI, 0.71-0.91; P=.0006) were significantly reduced, as well.1
Carvedilol. In the Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) trial, an RCT featuring 2289 patients with EF <25%, carvedilol significantly reduced the total death rate (HR=0.65; 95% CI, 0.52-0.81; P=.0014) compared with placebo.2
Metoprolol succinate. The Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure (MERIT-HF), a study of nearly 4000 patients with Class II to IV heart failure and EF ≤40%, found that metoprolol succinate lowered total mortality or all-cause hospitalization (HR=0.81; 95% CI, 0.73-0.90; P<.001) compared with placebo.3
TABLE 1
Beta-blockers for heart failure patients: What the studies show
| Trial | Study group (N) | Mean follow-up | Agent tested | Primary end point | RR; 95% CI; P value |
|---|---|---|---|---|---|
| BEST5 | Class III-IV HF, EF ≤35% (2708) | 2 y | Bucindolol | All-cause death | 0.90; 0.78-1.02; .13 |
| CIBIS II1 | Class III-IV HF, EF ≤35% (2647) | 1.3 y | Bisoprolol | All-cause death | 0.66; 0.54-0.81; <.0001 |
| COPERNICUS2 | HF symptoms, EF ≤25% (2289) | 10.4 mo | Carvedilol | All-cause death | 0.65; 0.52-0.81; .0014 |
| MERIT-HF3 | Class II-IV HF, EF ≤40% (3991) | 1 y | Metoprolol succinate | Composite* | 0.81; 0.73-0.90; <.001 |
| SENIORS6 | Age >70 y and hospitalization for HF or EF ≤35% (2128) | 21 mo | Nebivolol | All-cause death and CVD hospitalization | 0.86; 0.74-0.99; .039 |
| *All-cause mortality and all-cause hospitalization. BEST, Beta-blocker Evaluation of Survival Trial; CI, confidence interval; CIBIS II, Cardiac Insufficiency Bisoprolol Study; COPERNICUS, Carvedilol Prospective Randomized Cumulative Survival; CVD, cardiovascular disease; EF, ejection fraction; HF, heart failure; MERIT-HF, Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure; RR, relative risk; SENIORS, Study of the Effects of Nebivolol Intervention on Outcomes and Rehospitalisation in Seniors with Heart Failure. | |||||
Carvedilol and metoprolol go head-to-head
Although carvedilol and metoprolol have been shown to have similar hemodynamic and heart rate effects, the Carvedilol or Metoprolol European Trial (COMET) found that carvedilol is superior in extending survival. More than 3000 patients with Class II to IV heart failure and an EF <35% were randomized to carvedilol (target dose 25 mg bid) or metoprolol tartrate (target dose 50 mg bid). After 58 months, total mortality was significantly lower in the carvedilol arm (HR=0.83; 95% CI, 0.74-0.93; P=.0017).7
Which metoprolol formulation? While RCTs have found that metoprolol tartrate has a favorable effect on EF and hemodynamic data, it is not approved by the US Food and Drug Administration (FDA) as a treatment for heart failure—and its ability to reduce morbidity and mortality in patients with heart failure has not been established.8,9 Thus, metoprolol succinate, but not metoprolol tartrate, is recommended for heart failure treatment by the American College of Cardiology, American Heart Association, and European Society of Intensive Care Medicine.10,11
These agents lack evidence of efficacy
Not all beta-blockers have therapeutic value for patients with heart failure—or evidence to support them.
Bucindolol. The Beta-blocker Evaluation of Survival Trial (BEST), a trial of 2708 patients with Class III or IV heart failure and an EF ≤35%, found no difference in total mortality between bucindolol and placebo.5 As a result, the drug did not receive FDA approval.12 The FDA has since designated the investigation of bucindolol (trade name Gencaro) for the reduction of cardiovascular hospitalizations and mortality of heart failure patients with a particular genotype as a Fast Track development program.13
Nebivolol. The Study of the Effects of Nebivolol Intervention on Outcomes and Rehospitalisation in Seniors with Heart Failure (SENIORS) randomized 2128 patients older than 70 years with prior hospitalization for heart failure or an EF ≤35% to nebivolol (1.25-10 mg/d) or placebo. Nebivolol (which is not approved for the treatment of heart failure in the United States) reduced the composite end point of all-cause mortality and cardiovascular hospitalization (HR=0.86; 95% CI, 0.74-0.99; P=.039), but did not reduce the total mortality rate.6
Atenolol. Some retrospective analyses have suggested that heart failure patients do as well on atenolol as patients taking metoprolol or carvedilol.14,15 Because no RCTs have established the efficacy of atenolol, however, it is not recommended for the treatment of heart failure.
Is the dose sufficient to reduce heart rate?
The benefit of beta-blocker therapy for patients with heart failure is proportional to the degree of heart rate reduction, so it is important to find the highest tolerable dose.16,17 The COMET study detailed earlier sparked considerable controversy, with some observers contending that the dose of metoprolol used was too small to adequately lower the heart rate.18,19
A subsequent study, the Systolic Heart Failure Treatment with the I(f) Inhibitor Ivabradine Trial (SHIFT), highlights the importance of rate reduction in heart failure outcomes. In this placebo-controlled trial of 6558 patients with EF ≤35%, treatment with the heart rate-reducing agent ivabradine reduced cardiovascular death and hospitalization from heart failure (HR=0.82; 95% CI, 0.75-0.90; P<.0001) compared with placebo.20 A subsequent analysis showed that the primary outcome increased by 16% for every 5 beats-per-minute (BPM) increase.21
Start low, go slow
When initiating and titrating beta-blockers, the major RCTs clearly illustrate the importance of the dictum, “Start low, go slow” (TABLE 2).1-3
In CIBIS II, patients were started on bisoprolol at a dose of 1.25 mg/d. After a week, the dosage was increased by 1.25 mg. Titration continued over a 4-week period until the maximum tolerable dose was reached. Although 43% of patients reached the 10 mg/d target, a third of those studied remained on <5 mg/d.1
In COPERNICUS, carvedilol was started at 3.125 mg twice a day and continued at that dosage for 2 weeks. The dose was then titrated up at 2-week intervals, to 6.25 mg bid, then 12.5 mg bid, before attempting to reach the target dose of 25 mg bid. Ultimately, 66% received the target dose.2
In MERIT-HF, metoprolol succinate was initiated at 12.5 mg daily and doubled every 2 weeks until the target (200 mg/d) was achieved. Nearly two-thirds (64%) of those in the treatment group reached the target dose.3
In COMET, the researchers used the same drug regimen for carvedilol that was used in COPERNICUS (starting at 3.125 mg bid and slowly titrating to reach a 25-mg bid target). Patients on metoprolol tartrate initially received 5 mg bid; the dose was increased every 2 weeks until the target—50 mg bid—was reached. Seventy-five percent of patients reached the targeted carvedilol dose, and 78% reached the metoprolol target.7
TABLE 2
Titrating beta-blocker therapy
| Trial | Agent | Initial dose | Interval on starting dose | Mean dose achieved | Target dose achieved |
|---|---|---|---|---|---|
| CIBIS II1 | Bisoprolol | 1.25 mg/d | 1 week | 8.5 mg/d | 10 mg/d (43%) |
| COPERNICUS2 | Carvedilol | 3.125 mg bid | 2 weeks | 18.5 mg bid | 25 mg bid (66%) |
| MERIT-HF3 | Metoprolol succinate | 12.5 mg/d | 2 weeks | 159 mg/d | 200 mg/d (64%) |
| CIBIS-II, Cardiac Insufficiency Bisoprolol Study; COPERNICUS, Carvedilol Prospective Randomized Cumulative Survival; MERIT-HF, Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure. | |||||
Help beta-blocker therapy succeed
A significant number of patients with heart failure will be unable to tolerate an adequate dose of beta-blockers, at least on the first attempt.22 In such cases, a second attempt on another occasion—eg, after symptomatic bronchospasm or acute heart failure has been controlled—should be made.
In CIBIS II, 15% of the patients randomized to bisoprolol stopped taking it;1 in COPERNICUS, the withdrawal rate from carvedilol was also 15%;2 and in MERIT-HF, 10% of patients taking metoprolol experienced an adverse event that led to drug withdrawal.3 Although withdrawal rates were similar among patients on placebo in all 3 trials, they nonetheless suggest that even with the precautions and scrutiny characteristic of clinical trials, 10% to 15% of patients with heart failure will experience difficulty with beta-blocker treatment. (In a study of patients in one heart failure clinic, the withdrawal rate approached 40%.22)
Considering the benefits of beta-blockers for patients with all levels of heart failure, it is incumbent on physicians to prescribe them for as many of these patients as possible (See “Are beta-blockers contraindicated for these heart failure patients?”) and to attempt to reduce withdrawal rates.
Educate the patient. One way to do this is to provide adequate patient education, stressing the importance of taking the medication exactly as prescribed and, when necessary, showing patients how to divide pills until the target dose is reached.
Respond to adverse effects. Closely monitoring for adverse effects is crucial, as well. The development of symptomatic bradycardia, second or third degree atrioventricular block, or a heart rate <50 BPM suggests that the dosage be reduced or the medication withheld, with this caveat: There is increasing recognition that heart rate and BP readings change throughout the day, and a decision to adjust or to halt beta-blocker therapy should not be based on a single measure.
That said, physicians should watch for clinical evidence of hypoperfusion, such as postural dizziness or decreasing urine output, when systolic BP approaches 80 to 90 mm Hg in patients with heart failure. In such cases, adjusting the dose, increasing the interval between doses, or even discontinuing beta-blocker therapy may be necessary.
Because of the bradyarrhythmic and hypotensive effects of beta-blockers, the major heart failure trials excluded patients with a heart rate of <50 to 68 beats per minute (BPM) or systolic blood pressure <80 to 100 mm Hg (the ranges cited reflect the variation in cut points from one study to another).1-3,6 And in clinical practice, physicians often withhold beta-blocker therapy from heart failure patients who also have chronic obstructive pulmonary disease (COPD) or asthma, hypotension, or metabolic risk factors for diabetes.4 Some avoid prescribing beta-blockers because they believe that the drugs adversely affect patients’ quality of life, despite evidence to the contrary.3,23-25 In all these cases, there is little justification for doing so.
COPD and asthma. Although beta-blockers can worsen and precipitate bronchospasm, recent evidence suggests that patients with COPD and asthma can tolerate them.26-28 In fact, there is reason to believe that bronchospasm is aggravated by excessive stimulation and sensitization of the beta-2 receptors, and that blocking them may even be of therapeutic value.29 Nonetheless, the danger of worsening bronchospasm with a nonselective beta-blocker such as carvedilol remains—particularly for patients with asthma, who tend to have a higher degree of bronchial sensitivity and reactivity. So, while beta-blockers are not contraindicated for patients with COPD, their use in this patient population requires caution.30,31
Metabolic risk factors. Caution is also needed for patients with metabolic risk factors. Although beta-blockers have been found to increase the risk of diabetes, raise triglycerides, and lower high-density lipoprotein cholesterol,32-34 the benefits for patients with heart failure outweigh the risk. Physicians must remember that the mortality rate of heart failure, as well as the rate of progression, is higher than that of metabolic abnormalities, asymptomatic bradycardia, hypotension, or bronchospasm, which are relatively benign. In view of evidence that beta-blockers reduce both mortality and hospitalization rates associated with heart failure, the best approach is to continue beta-blocker therapy and seek control of risk factors and adverse effects.
CORRESPONDENCE
HT Ong FRCP, FACC, FESC, HT Ong Heart Clinic, 251C Burma Road, Penang 103250, Malaysia; ongheanteik@gmail.com
1. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet. 1999;353:9-13.
2. Packer M, Fowler MB, Roecker EB, et al. Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) Study Group. Effect of carvedilol on the morbidity of patients with severe chronic heart failure: results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation. 2002;106:2194-2199.
3. Hjalmarson A, Goldstein S, Fagerberg B, et al. for the MERIT-HF Study Group. Effects of controlled-release metoprolol on total mortality, hospitalization and well-being in patients with heart failure. The Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure (MERIT-HF). JAMA. 2000;283:1295-1302.
4. Mann DL. Management of heart failure patients with reduced ejection fraction. In: Libby P, Bonow RO, Mann DL, et al, eds. Braunwald’s Heart Disease. A Textbook of Cardiovascular Medicine. 8th ed. Philadelphia: Saunders Elsevier; 2008:611-640.
5. Beta-Blocker Evaluation of Survival Trial Investigators. A trial of the beta-blocker bucindolol in patients with advanced chronic heart failure. N Engl J Med. 2001;344:1659-1667.
6. Flather MD, Shibata MC, Coats AJ, et al. SENIORS Investigators. Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur Heart J. 2005;26:215-225.
7. Poole-Wilson PA, Swedberg K, Cleland JG, et al. Carvedilol Or Metoprolol European Trial Investigators. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol Or Metoprolol European Trial (COMET): randomised controlled trial. Lancet. 2003;362:7-13.
8. Waagstein F, Bristow MR, Swedberg K, et al. Beneficial effects of metoprolol in idiopathic dilated cardiomyopathy. Metoprolol in Dilated Cardiomyopathy (MDC) Trial Study Group. Lancet. 1993;342:1441-1446.
9. Waagstein F, Stromblad O, Andersson B, et al. Increased exercise ejection fraction and reversed remodeling after long-term treatment with metoprolol in congestive heart failure: a randomized, stratified, double-blind, placebo-controlled trial in mild to moderate heart failure due to ischemic or idiopathic dilated cardiomyopathy. Eur J Heart Fail. 2003;5:679-691.
10. Dickstein K, Cohen-Solal A, Filippatos G. ESC Committee for Practice Guidelines (CPG). ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the diagnosis and treatment of acute and chronic heart failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur J Heart Fail. 2008;10:933-989.
11. Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation. 2009;119:e391-e479.
12. Complete response letter for Gencaro NDA. Available at: http://www.drugs.com/nda/gencaro_090601.html. June 1, 2009. Accessed July 15, 2011.
13. ARCA Biopharma. ARCA announces Special Protocol Assessment agreement with FDA for bucindolol development in genotype-defined heart failure patients. May 17, 2010. Available at: http://www.advfn.com/news_ARCA-Announces-Special-Protocol-Assessment-Agreement-with-FDA-for-Bucindolol-Dev_42847369. Accessed July 14, 2010.
14. Go AS, Yang J, Gurwitz JH, Hsu J, et al. Comparative effectiveness of different beta-adrenergic antagonists on mortality among adults with heart failure in clinical practice. Arch Intern Med. 2008;168:2415-2421.
15. Kapoor JR, Heidenreich PA. Survival among patients with left ventricular systolic dysfunction treated with atenolol. Congest Heart Fail. 2009;15:213-217.
16. Nishiyama K, Tsutamoto T, Yamaji M, et al. Dose-dependent prognostic effect of carvedilol in patients with chronic heart failure—special reference to transcardiac [corrected] gradient of norepinephrine. Circ J. 2009;73:2270-2275.
17. McAlister FA, Wiebe N, Ezekowitz JA, et al. Meta-analysis: beta-blocker dose, heart rate reduction, and death in patients with heart failure. Ann Intern Med. 2009;150:784-794.
18. Hjalmarson A, Waagstein F. COMET: a proposed mechanism of action to explain the results and concerns about dose. Lancet. 2003;362:1077.-
19. Dargie HJ. Beta blockers in heart failure. Lancet. 2003;362:2-3.
20. Swedberg K, Komajda M, Böhm M, et al. SHIFT Investigators. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet. 2010;376:875-885.
21. Böhm M, Swedberg K, Komajda M, et al. SHIFT Investigators. Heart rate as a risk factor in chronic heart failure (SHIFT): the association between heart rate and outcomes in a randomised placebo-controlled trial. Lancet. 2010;376:886-894.
22. Galatius S, Gustafsson F, Atar D, et al. Tolerability of beta-blocker initiation and titration with bisoprolol and carvedilol in congestive heart failure—a randomized comparison. Cardiology. 2004;102:160-165.
23. Dobre D, van Jaarsveld CH, deJongste MJ, et al. The effect of beta-blocker therapy on quality of life in heart failure patients: a systematic review and meta-analysis. Pharmacoepidemiol Drug Saf. 2007;16:152-159.
24. Tate CW 3rd, Robertson AD, Zolty R, et al. Quality of life and prognosis in heart failure: results of the Beta-Blocker Evaluation of Survival Trial (BEST). J Card Fail. 2007;13:732-737.
25. Belenkov IuN, Skvortsov AA, Mareev VIu, et al. Clinical, hemodynamic and neurohumoral effects of long-term therapy of patients with severe chronic heart failure with beta-adrenoblocker bisoprolol. Kardiologiia. 2003;43:10-21.
26. LeJemtel TH, Padeletti M, Jelic S. Diagnostic and therapeutic challenges in patients with coexistent chronic obstructive pulmonary disease and chronic heart failure. J Am Coll Cardiol. 2007;49:171-180.
27. Mascarenhas J, Azevedo A, Bettencourt P. Coexisting chronic obstructive pulmonary disease and heart failure: implications for treatment, course and mortality. Curr Opin Pulm Med. 2010;16:106-111.
28. Navas EV, Taylor DO. Q: Can patients with COPD or asthma take a beta-blocker? Cleve Clin J Med. 2010;77:498-499.
29. Bond RA, Spina D, Parra S, et al. Getting to the heart of asthma: can “beta blockers” be useful to treat asthma? Pharmacol Ther. 2007;115:360-374.
30. Cazzola M, Matera MG. Beta-blockers are safe in patients with chronic obstructive pulmonary disease, but only with caution. Am J Respir Crit Care Med. 2008;178:661-662.
31. Shaw SM, Hasleton J, Williams SG. Beta-blocker use in heart failure patients with airways disease. Clin Cardiol. 2009;32:393-396.
32. Elliott WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet. 2007;369:249-256.
33. Dammitt SB, Williams PD, Croft KD, et al. Effects of beta-blockers on the concentration and oxidizability of plasma lipids. Clin Sci (Lond). 1998;94:573-578.
34. Kuster GM, Amann FW, Neuenschwander C, Drexel H. High density-lipoprotein subfractions of patients using cardio-selective beta-blockers. Cardiovasc Drugs Ther. 2002;16:127-131.
1. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet. 1999;353:9-13.
2. Packer M, Fowler MB, Roecker EB, et al. Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) Study Group. Effect of carvedilol on the morbidity of patients with severe chronic heart failure: results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation. 2002;106:2194-2199.
3. Hjalmarson A, Goldstein S, Fagerberg B, et al. for the MERIT-HF Study Group. Effects of controlled-release metoprolol on total mortality, hospitalization and well-being in patients with heart failure. The Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure (MERIT-HF). JAMA. 2000;283:1295-1302.
4. Mann DL. Management of heart failure patients with reduced ejection fraction. In: Libby P, Bonow RO, Mann DL, et al, eds. Braunwald’s Heart Disease. A Textbook of Cardiovascular Medicine. 8th ed. Philadelphia: Saunders Elsevier; 2008:611-640.
5. Beta-Blocker Evaluation of Survival Trial Investigators. A trial of the beta-blocker bucindolol in patients with advanced chronic heart failure. N Engl J Med. 2001;344:1659-1667.
6. Flather MD, Shibata MC, Coats AJ, et al. SENIORS Investigators. Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur Heart J. 2005;26:215-225.
7. Poole-Wilson PA, Swedberg K, Cleland JG, et al. Carvedilol Or Metoprolol European Trial Investigators. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol Or Metoprolol European Trial (COMET): randomised controlled trial. Lancet. 2003;362:7-13.
8. Waagstein F, Bristow MR, Swedberg K, et al. Beneficial effects of metoprolol in idiopathic dilated cardiomyopathy. Metoprolol in Dilated Cardiomyopathy (MDC) Trial Study Group. Lancet. 1993;342:1441-1446.
9. Waagstein F, Stromblad O, Andersson B, et al. Increased exercise ejection fraction and reversed remodeling after long-term treatment with metoprolol in congestive heart failure: a randomized, stratified, double-blind, placebo-controlled trial in mild to moderate heart failure due to ischemic or idiopathic dilated cardiomyopathy. Eur J Heart Fail. 2003;5:679-691.
10. Dickstein K, Cohen-Solal A, Filippatos G. ESC Committee for Practice Guidelines (CPG). ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the diagnosis and treatment of acute and chronic heart failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur J Heart Fail. 2008;10:933-989.
11. Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation. 2009;119:e391-e479.
12. Complete response letter for Gencaro NDA. Available at: http://www.drugs.com/nda/gencaro_090601.html. June 1, 2009. Accessed July 15, 2011.
13. ARCA Biopharma. ARCA announces Special Protocol Assessment agreement with FDA for bucindolol development in genotype-defined heart failure patients. May 17, 2010. Available at: http://www.advfn.com/news_ARCA-Announces-Special-Protocol-Assessment-Agreement-with-FDA-for-Bucindolol-Dev_42847369. Accessed July 14, 2010.
14. Go AS, Yang J, Gurwitz JH, Hsu J, et al. Comparative effectiveness of different beta-adrenergic antagonists on mortality among adults with heart failure in clinical practice. Arch Intern Med. 2008;168:2415-2421.
15. Kapoor JR, Heidenreich PA. Survival among patients with left ventricular systolic dysfunction treated with atenolol. Congest Heart Fail. 2009;15:213-217.
16. Nishiyama K, Tsutamoto T, Yamaji M, et al. Dose-dependent prognostic effect of carvedilol in patients with chronic heart failure—special reference to transcardiac [corrected] gradient of norepinephrine. Circ J. 2009;73:2270-2275.
17. McAlister FA, Wiebe N, Ezekowitz JA, et al. Meta-analysis: beta-blocker dose, heart rate reduction, and death in patients with heart failure. Ann Intern Med. 2009;150:784-794.
18. Hjalmarson A, Waagstein F. COMET: a proposed mechanism of action to explain the results and concerns about dose. Lancet. 2003;362:1077.-
19. Dargie HJ. Beta blockers in heart failure. Lancet. 2003;362:2-3.
20. Swedberg K, Komajda M, Böhm M, et al. SHIFT Investigators. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet. 2010;376:875-885.
21. Böhm M, Swedberg K, Komajda M, et al. SHIFT Investigators. Heart rate as a risk factor in chronic heart failure (SHIFT): the association between heart rate and outcomes in a randomised placebo-controlled trial. Lancet. 2010;376:886-894.
22. Galatius S, Gustafsson F, Atar D, et al. Tolerability of beta-blocker initiation and titration with bisoprolol and carvedilol in congestive heart failure—a randomized comparison. Cardiology. 2004;102:160-165.
23. Dobre D, van Jaarsveld CH, deJongste MJ, et al. The effect of beta-blocker therapy on quality of life in heart failure patients: a systematic review and meta-analysis. Pharmacoepidemiol Drug Saf. 2007;16:152-159.
24. Tate CW 3rd, Robertson AD, Zolty R, et al. Quality of life and prognosis in heart failure: results of the Beta-Blocker Evaluation of Survival Trial (BEST). J Card Fail. 2007;13:732-737.
25. Belenkov IuN, Skvortsov AA, Mareev VIu, et al. Clinical, hemodynamic and neurohumoral effects of long-term therapy of patients with severe chronic heart failure with beta-adrenoblocker bisoprolol. Kardiologiia. 2003;43:10-21.
26. LeJemtel TH, Padeletti M, Jelic S. Diagnostic and therapeutic challenges in patients with coexistent chronic obstructive pulmonary disease and chronic heart failure. J Am Coll Cardiol. 2007;49:171-180.
27. Mascarenhas J, Azevedo A, Bettencourt P. Coexisting chronic obstructive pulmonary disease and heart failure: implications for treatment, course and mortality. Curr Opin Pulm Med. 2010;16:106-111.
28. Navas EV, Taylor DO. Q: Can patients with COPD or asthma take a beta-blocker? Cleve Clin J Med. 2010;77:498-499.
29. Bond RA, Spina D, Parra S, et al. Getting to the heart of asthma: can “beta blockers” be useful to treat asthma? Pharmacol Ther. 2007;115:360-374.
30. Cazzola M, Matera MG. Beta-blockers are safe in patients with chronic obstructive pulmonary disease, but only with caution. Am J Respir Crit Care Med. 2008;178:661-662.
31. Shaw SM, Hasleton J, Williams SG. Beta-blocker use in heart failure patients with airways disease. Clin Cardiol. 2009;32:393-396.
32. Elliott WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet. 2007;369:249-256.
33. Dammitt SB, Williams PD, Croft KD, et al. Effects of beta-blockers on the concentration and oxidizability of plasma lipids. Clin Sci (Lond). 1998;94:573-578.
34. Kuster GM, Amann FW, Neuenschwander C, Drexel H. High density-lipoprotein subfractions of patients using cardio-selective beta-blockers. Cardiovasc Drugs Ther. 2002;16:127-131.
Did too much Wii cause your patient’s injury?
• Ask patients with repetitive motion injuries (RMIs) whether they use interactive game consoles and, if so, how much time they spend playing virtual sports each day. C
• Be aware that RMIs associated with video game use are similar to injuries associated with the sports they simulate. A
• Advise patients to take the same precautions with virtual sports as they would with any physical activities, including warm-up exercises and moderation. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
This article is an expansion of a poster session presented at the 12th annual Northeastern Ohio Universities College of Medicine Department of Surgery Resident Research Day in May 2009 and at the American College of Preventive Medicine Annual Meeting in February 2010.
The release of the Wii—Nintendo’s 4th generation gaming console—in 2006 revolutionized the video game industry. By March 31, 2010, more than 70 million units had been sold worldwide, earning Wii the title of “fastest-selling game console of all time.”1-3
Today, there are several game consoles that, like Wii, allow the user not only to push buttons or move levers, but to control the game using physical movements (TABLE 1). And the devices and the many sports they simulate—once popular primarily among adolescents—are in widespread use by people of all ages, including the young and fit, out-of-shape “arm chair” athletes, and elderly people in senior housing, rehabilitation centers, and long-term care facilities alike.4
Not surprisingly, simulated sports play has spawned an array of repetitive motion and overuse injuries. To identify and treat them, ask all patients who present with musculoskeletal injuries whether they engage in game console sports activities; if so, identify the type of game and how much time they spend playing it each day. Although injuries associated with specific video games are often given names like “Wii-itis,”5 “Nintendinitis,”6 and “Playstation thumb,”7 the types of injuries caused by playing simulated sports are generally the same as (or similar to) injuries sustained by those engaging in the sport itself.
TABLE 1
Popular motion-controlled games: A partial list
| Type of game console | |||
|---|---|---|---|
| Nintendo: Wii | Microsoft Xbox 360: Kinect | Sony Playstation 3: Move | |
| Motion-control mechanism | Handheld remote | Full body | Handheld remote |
| Games bundled with console | Wii Sports | Kinect Adventures | PS3 Sports Champions |
| Popular games | Wii Fit Wii Play Mario Kart Super Smash Bros Brawl Guitar Hero III: Legends of Rock | Kinect Sports Dance Central Your Shape: Fitness Evolved The Biggest Loser: Kinectimals | Sports Champions Time Crisis: Razing Storm Killzone 3 Little Big Planet 2 Sorcery |
| Manufacturer-recommended game space | ≤6 feet using wireless sensor | ≥6 feet from device | ≥6 feet from device |
| Sources: 1. Nintendo (http://www.nintendo.com/consumer/wiisafety.jsp). 2. Microsoft Xbox 360 (http:www.xbox.com/en-US/Kinect/PrivacyandOnlineSafety). 3. Sony Playstation (http://us.playstation.com/support/answer/index.htm). | |||
Video game pathology is well established
In 1987, Osterman et al published the first report of a musculoskeletal disorder associated with electronic games—a case of volar flexor tenosynovitis (“joystick digit”) trigger finger.8 Several years later, a physician coined the term “Nintendinitis” to describe video game-related overuse syndrome6—acute tendinopathy of the extensor pollicis longus tendon after prolonged play with early versions of the thumb-activated game controller.9,10 In 2002, a child using a vibrating Sony Playstation for up to 7 hours a day received a diagnosis of vibratory syndrome of the hand.11 A few years later, a report of “Playstation thumb,” an overuse syndrome associated with later generations of game consoles, followed.7
Several other reports of game-related injury patterns can be found in medical journals, including pressure ulcer formation (“ulcerative Nintendinitis”),12 the “How!” sign of central palmar blistering,13 “mouse elbow” secondary to epicondylitis,14 and other tendinopathies associated with various gaming consoles.10,15,16 All the reports clearly describe the relationship between video game use and the pathology, and clinical improvement after cessation of the activity.

Many manifestations of Wii-itis
An epidemiologic review of the National Electronic Injury Surveillance System (http://www.cpsc.gov/library/neiss.html) found that in the Wii’s first year, 67% of the musculoskeletal injuries reported (29% were defined as sprains and strains and 38% as overuse injuries) involved the use of the Wii to play simulated sports.17 Overuse syndrome associated with Wii was initially called “acute Wii-itis,”5 a description of acute tendinopathy of the infraspinatus.18 (Infraspinatus tendinopathy is most commonly associated with games involving intense arm activity, including Wii baseball, bowling, and boxing (TABLE 2).5 However, Wii-itis is now widely used to describe any acute inflammatory syndrome associated with use of this popular game console.
Wii knee, for example, refers to an acute patellar dislocation associated with simulated bowling.19 Multiple cases of patellar injury, including associated osteochondral fracture, have been reported in association with a variety of game titles, including Raymond Raving Rabbids and Brunswick Pro Bowling.19 In a review of self-reported Wii injuries, patellar dislocation was the fourth most common injury (hand lacerations were first, followed by periorbital hematoma [“black eye”], and forehead lacerations/ecchymoses).20
Wii shoulder, another variant of Wii-itis, is an acute inflammation of the upper extremity musculature after repetitive motion. This injury is most often associated with games that require swinging of the controller, such as Wii tennis or bowling. Upper extremity magnetic resonance imaging (MRI) of one Wii enthusiast revealed inflammatory swelling of the shoulder joint that extended to the suprascapular region, corresponding to a diagnosis of delayed–onset muscle soreness (DOMS).9
DOMS, which is often associated with acute injury patterns, is a well-accepted diagnosis among patients who play physically interactive sports and, by extension, video games.17 Usually lacking frank deformity on plain radiographs, DOMS is a disorder of the soft tissue that can best be visualized with MRI delineation of tissue planes and musculature compartments. Clinical signs and symptoms of DOMS can include edema of the affected extremity, rubor, and tenderness to palpation during active range of motion. Treatment for DOMS, like all RMIs, includes cessation of the offending activity.
Another recently reported variant of Wii-itis is the acute onset of carpal tunnel syndrome21 after playing Wii bowling for long periods of time. The case involved a 19-year-old woman who presented with swelling over the volar wrist and had positive Tinel and Phalen signs. She received conservative treatment with etodolac, a nightly volar splint, cold compresses, and rest.
Achilles Wii-itis refers to a partial or complete rupture of the Achilles tendon during simulated sports activity.22 This injury has been reported in people using the Wii Fit exercise pad for virtual running and stretching, and is diagnosed clinically with a positive Thompson sign (failure to plantar flex the foot while compressing the gastrocnemius). Complete Achilles rupture requires surgical repair, but less severe cases can be treated conservatively, with cold compresses, lifestyle modification, and nonsteroidal anti-inflammatory drugs (NSAIDs).
TABLE 2
Repetitive motion injuries (and possible causes)*30,36
| Type of injury | Games with potential for injury† | Possible injury sites | Common physical exam findings |
|---|---|---|---|
| Tendinopathy | Guitar Hero III: Legends of Rock The Legend of Zelda: Twilight Princess Wii Fit | Achilles tendon Patella Supraspinatous Forearm Extensors | Pain or stiffness in the local area of the tendon. Progression can lead to redness and swelling at the joint of the inflamed tendon |
| Bursitis | Kinect Sports: Soccer Dance Dance Revolution Star Wars: The Clone Wars Wii Fit | Subacromial bursa Trochanteric bursa Patellar bursa | Burning pain over the joint during and after activity, with delayed-onset joint stiffness due to local inflammation |
| Enthesitis | Wii Sports Sports Champions Kinect Sports | Achilles tendon Tuberosity of the tibia Iliac crest | Pain at joint on palpation or during range-of-motion exam. Calcification or fibrosis can be identified in chronic, nonacute presentations that are generally autoimmune mediated |
| Epicondylitis | Wii: Major League Baseball Grand Slam Tennis Tiger Woods PGA Tour | Olecranon process, lateral epicondyle (tennis elbow) | Point tenderness over the lateral epicondyle with acute pain on arm extension |
| Olecranon process, medial epicondyle (golf elbow) | Point tenderness over the medial epicondyle with acute pain on wrist flexion or resisted forearm pronation | ||
| *The authors have included games that, in their opinion, have the potential for injury based on the biomechanics involved (eg, running, jumping, waving, etc). † Many of these games are bundled and incorporate multiple activities (eg, baseball, bowling, boxing, soccer, track and field, tennis, volleyball). | |||
Categorizing Wii-type injuries
Game-related injuries typically fall into 4 broad categories: tendinopathy, bursitis, enthesitis, and epicondylitis. (See TABLE 2 for a list of games with the potential to cause particular types of injuries.)
Tendinopathy. Overuse tendon injuries, or tendinopathies, account for up to 50% of all sports-related injuries.23 By extrapolation, physically interactive game systems that simulate actual sports can be expected to increase tendon overuse injuries.
Most major tendons are vulnerable to overuse injury, including the Achilles (FIGURE), as noted earlier; and the patellar, rotator cuff, and forearm extensor tendons, among others. Repetitive motion, or strain, injuries to these tendons are often thought to be cumulative, with hypoperfusion, local inflammation, and neuropathy contributing to the degree of tendinopathy. Other risk factors for tendon injury include age and sex (men have a higher relative risk than women; older people, in their fourth and fifth decades of life, also face an increased risk), postmenopausal status, obesity, use of fluoroquinolone antibiotics or corticosteroids, and playing on nonpadded surfaces.24-29
Conservative therapy, with cessation of the offending activity and rest of the affected extremity, is the initial treatment of choice for tendinopathy. Severe cases of compound injuries or tendon reinjury can also be treated with splinting, taping, cryotherapy, electrotherapy, deep tissue tendon massage, pharmaceuticals (NSAIDs and corticosteroid injections), and early rehabilitation.15,30 Surgery may eventually be required to remove fibrotic tissue, modify the vascularity, or reconstruct the tendon.15
Bursitis. Bursitis is characterized by inflammation of the subacromial, olecranon, trochanteric, prepatellar, suprapatellar, infrapatellar, pes anserine, or iliotibial bursa—synovial-lined cavities overlying bony prominences that minimize the friction of movement.31 Patellar and olecranon bursitis are most frequently associated with sports, particularly soccer and golf.
Clinically characterized by pain on flexion, bursitis can also present with localized tenderness, stiffness, and swelling of the affected joint. Bursitis generally responds to RICE (rest, ice, compression, elevation) therapy, but can potentially advance to a chronic disease state if the activity that caused the inflammation continues.31
Enthesitis. Characterized by inflammation of the bony insertions of a tendon or ligament, enthesitis is generally linked to an autoimmune disease such as ankylosing spondylitis or rheumatoid arthritis. But it can also be an acquired condition associated with repetitive motion. Sports-related activity is the most common cause of acquired enthesitis,32 with injury most likely to occur at the Achilles tendon, the insertion point of the tibial tuberosity, or the iliac crest.33 Like most RMIs, acquired enthesitis can usually be treated simply by stopping the offending activity. If not properly recognized or treated, however, permanent injury can occur. 34
Epicondylitis. This RMI results in pain or ipsilateral weakness of the upper extremity due to repetitive strain at the musculotendinous junction and its origin at the epicondyle. Neuropraxia is often associated with epicondylitis due to posterior interosseous nerve, median nerve, or ulnar nerve involvement at either the medial or lateral epicondyles.35
Commonly affecting computer users who perform repetitive motion via mouse manipulation, the term “mouse elbow” was first described in 1992.14 Golfer’s elbow (with involvement of the medial epicondyle), and tennis elbow (involving the lateral epicondyle) are also common, and individuals who frequently play simulated golf or tennis games are at risk.
FIGURE
Achilles tendon injury
An MRI reveals anterior bulging and thickening of the Achilles tendon (arrow)—the type of injury you might see in a patient using the Wii Fit exercise pad for running and stretching.
Tell patients how to prevent injury
Older patients and deconditioned “arm chair” athletes who are unaccustomed to prolonged physical activity face an increased risk for injuries related to video game sports. You can help by pointing out that because simulated activities require a fraction of the strength and endurance required to play the actual sport, people who might normally tire easily are apt to overdo it.
In fact, Nintendo has a dedicated safety page regarding the use of game consoles on its Web site (http://www.nintendo.com/consumer/wiisafety.jsp). The company advises Wii users to take a 10- to 15-minute break every hour, even if they don’t think they need it, to prevent repetitive motion and eyestrain injuries, and to stop playing for several hours if they experience tingling, numbness, burning, or stiffness. Some software titles, including Wii Fit, are programmed to remind users to take a break after they’ve been playing nonstop for 45 minutes to an hour. You can help by reminding patients of all ages that warm-up exercises, moderation, and hydration are crucial, whether the sports they’re engaging in are virtual or real.
Acknowledgement
The authors would like to thank Dan Dunlany for his invaluable research assistance.
CORRESPONDENCE
Lisa M. Coughlin, MD, Department of Surgery, University of Toledo Medical Center, 3065 Arlington Avenue, Toledo, OH 43614; LcoughlinMD@gmail.com
1. NPD Seventh Generation. Wikia. Available at: http://vgsales.wikia.com/wiki/NPD_Seventh_generation#NPD_hardware_sales. Accessed September 1, 2010.
2. Thorsen T. Wii sales near 71 million, DS almost 129 million. May 6, 2010. Gamespot. Available at: http://www.gamespot.com/news/6261400.html. Accessed September 15, 2010.
3. Nintendo Wii is the fastest selling gaming console, beats Xbox 360 sales in less than a year August 25, 2007. TechShout. Available at: http://www.techshout.com/gaming/2007/25/nintendo-wii-is-the-fastest-selling-gaming-console-beats-xbox-360-sales-in-less-than-a-year/. Accessed February 18, 2009.
4. Hsu JK, Thibodeau R, Wong SJ, et al. A “Wii” bit of fun: the effects of adding Nintendo Wii bowling to a standard exercise regimen for residents of long-term care with upper extremity dysfunction. Physiother Theory Pract. 2011;27:185-193.
5. Bonis J. Acute Wiiitis. N Engl J Med. 2007;356:2431-2432.
6. Brasington R. Nintendinitis. N Engl J Med. 1990;322:1473-1474.
7. Vaidya HJ. Playstation thumb. Lancet. 2004;363:1080.-
8. Osterman AL, Weinberg P, Miller G. Joystick digit. JAMA. 1987;257:782.-
9. Nett MP, Collins MS, Sperling JW. Magnetic resonance imaging of acute “wiiitis” of the upper extremity. Skeletal Radiol. 2008;37:481-483.
10. Macgregor DM. Nintendonitis? A case report of repetitive strain injury in a child as a result of playing computer games. Scott Med J. 2000;45:150.-
11. Cleary AG, McKendrick H, Sills JA. Hand-arm vibration syndrome may be associated with prolonged use of vibrating computer games. BMJ. 2000;324:301.-
12. Koh TH. Ulcerative “nintendinitis”: a new kind of repetitive strain injury. Med J Aust. 2000;173:671.-
13. Wood J. The “How! ” sign—a central palmar blister induced by overplaying on a Nintendo console. Arch Dis Child. 2001;84:288.-
14. Mirman MJ, Bonian VG. “Mouse elbow”: a new repetitive stress injury. J Am Osteopath Assoc. 1992;92:701.-
15. Rees JD, Maffulli N, Cook J. Management of tendinopathy. Am J Sports Med. 2009;37:1855-1867.
16. Kujala UM, Taimela S, Viljanen T. Leisure physical activity and various pain symptoms among adolescents. Br J Sports Med. 1999;33:325-328.
17. Jones C, Hammig B. Case report: injuries associated with interactive game consoles: preliminary data. Phys Sports Med. 2009;37:138-140.
18. Hertel R, Ballmer FT, Lombert SM, et al. Lag signs in the diagnosis of rotator cuff rupture. J Shoulder Elbow Surg. 1996;5:307-313.
19. Robinson RJ, Barron DA, Grainger AJ, et al. Wii knee. Emerg Radiol. 2008;15:255-257.
20. Sparks DA, Chase DM, Coughlin LM. Wii have a problem: a review of self-reported Wii related injuries. Inform Prim Care. 2009;17:55-57.
21. Boehm KM, Pugh A. A new variant of Wii-itis. J Emerg Med. 2009;36:80.-
22. Beddy P, Dunne R, de Blacam C. Achilles wiiitis. AJR Am J Roentgenol. 2009;192:W79.-
23. [Herring SA, Nilson KL. Introduction to overuse injuries. Clin Sports Med. 1987;6:225-23.
24. Maffulli N, Waterston SW, Squair J, et al. Changing incidence of Achilles tendon rupture in Scotland: a 15-year study. Clin J Sport Med. 1999;9:157-160.
25. Malliaras P, Cook J. Patellar tendons with normal imaging and pain: change in imaging and pain status over a volleyball season. Clin J Sport Med. 2006;16:388-391.
26. Malliaras PJ, Cook JL, Kent PM. Anthropometric risk factors for patellar tendon injury among volleyball players. Br J Sports Med. 2007;41:259-263.
27. McGarvey WC, Singh D, Trevino SG. Partial Achilles tendon ruptures associated with fluoroquinolone antibiotics: a case report and literature review. Foot Ankle Int. 1996;17:496-498.
28. Ford LT, DeBender J. Tendon rupture after local steroid injection. South Med J. 1979;72:827-830.
29. Hess GW. Achilles tendon rupture: a review of etiology, population, anatomy, risk factors, and injury prevention. Foot Ankle Spec. 2010;3:29-32.
30. Rettig AC. Wrist and hand overuse syndromes. Clin Sports Med. 2001;20:591-611.
31. Huie G. Diagnosing bursitis in the knee. JAAPA. 2002;15:14-16.
32. Jennings F, Lambert E, Fredericson M. Rheumatic diseases presenting as sports-related injuries. Sports Med. 2008;38:917-930.
33. Leppilahti J, Orava S, Karpakka J, et al. Overuse injuries of the Achilles tendon. Ann Chir Gynaecol. 1991;80:202-207.
34. Slobodin G, Rozenbaum M, Boulman N, et al. Varied presentations of enthesopathy. Semin Arthritis Rheum. 2007;37:119-126.
35. Jepsen JR, Thomsen G. A cross-sectional study of the relation between symptoms and physical findings in computer operators. BMC Neurol. 2006;6:40.-
36. Biundo JJ, Jr, Irwin RW, Umpierre E. Sports and soft tissue injuries, tendinitis, bursitis, and occupation-related syndromes. Curr Opin Rheumatol. 2001;13:146-149.
• Ask patients with repetitive motion injuries (RMIs) whether they use interactive game consoles and, if so, how much time they spend playing virtual sports each day. C
• Be aware that RMIs associated with video game use are similar to injuries associated with the sports they simulate. A
• Advise patients to take the same precautions with virtual sports as they would with any physical activities, including warm-up exercises and moderation. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
This article is an expansion of a poster session presented at the 12th annual Northeastern Ohio Universities College of Medicine Department of Surgery Resident Research Day in May 2009 and at the American College of Preventive Medicine Annual Meeting in February 2010.
The release of the Wii—Nintendo’s 4th generation gaming console—in 2006 revolutionized the video game industry. By March 31, 2010, more than 70 million units had been sold worldwide, earning Wii the title of “fastest-selling game console of all time.”1-3
Today, there are several game consoles that, like Wii, allow the user not only to push buttons or move levers, but to control the game using physical movements (TABLE 1). And the devices and the many sports they simulate—once popular primarily among adolescents—are in widespread use by people of all ages, including the young and fit, out-of-shape “arm chair” athletes, and elderly people in senior housing, rehabilitation centers, and long-term care facilities alike.4
Not surprisingly, simulated sports play has spawned an array of repetitive motion and overuse injuries. To identify and treat them, ask all patients who present with musculoskeletal injuries whether they engage in game console sports activities; if so, identify the type of game and how much time they spend playing it each day. Although injuries associated with specific video games are often given names like “Wii-itis,”5 “Nintendinitis,”6 and “Playstation thumb,”7 the types of injuries caused by playing simulated sports are generally the same as (or similar to) injuries sustained by those engaging in the sport itself.
TABLE 1
Popular motion-controlled games: A partial list
| Type of game console | |||
|---|---|---|---|
| Nintendo: Wii | Microsoft Xbox 360: Kinect | Sony Playstation 3: Move | |
| Motion-control mechanism | Handheld remote | Full body | Handheld remote |
| Games bundled with console | Wii Sports | Kinect Adventures | PS3 Sports Champions |
| Popular games | Wii Fit Wii Play Mario Kart Super Smash Bros Brawl Guitar Hero III: Legends of Rock | Kinect Sports Dance Central Your Shape: Fitness Evolved The Biggest Loser: Kinectimals | Sports Champions Time Crisis: Razing Storm Killzone 3 Little Big Planet 2 Sorcery |
| Manufacturer-recommended game space | ≤6 feet using wireless sensor | ≥6 feet from device | ≥6 feet from device |
| Sources: 1. Nintendo (http://www.nintendo.com/consumer/wiisafety.jsp). 2. Microsoft Xbox 360 (http:www.xbox.com/en-US/Kinect/PrivacyandOnlineSafety). 3. Sony Playstation (http://us.playstation.com/support/answer/index.htm). | |||
Video game pathology is well established
In 1987, Osterman et al published the first report of a musculoskeletal disorder associated with electronic games—a case of volar flexor tenosynovitis (“joystick digit”) trigger finger.8 Several years later, a physician coined the term “Nintendinitis” to describe video game-related overuse syndrome6—acute tendinopathy of the extensor pollicis longus tendon after prolonged play with early versions of the thumb-activated game controller.9,10 In 2002, a child using a vibrating Sony Playstation for up to 7 hours a day received a diagnosis of vibratory syndrome of the hand.11 A few years later, a report of “Playstation thumb,” an overuse syndrome associated with later generations of game consoles, followed.7
Several other reports of game-related injury patterns can be found in medical journals, including pressure ulcer formation (“ulcerative Nintendinitis”),12 the “How!” sign of central palmar blistering,13 “mouse elbow” secondary to epicondylitis,14 and other tendinopathies associated with various gaming consoles.10,15,16 All the reports clearly describe the relationship between video game use and the pathology, and clinical improvement after cessation of the activity.
Many manifestations of Wii-itis
An epidemiologic review of the National Electronic Injury Surveillance System (http://www.cpsc.gov/library/neiss.html) found that in the Wii’s first year, 67% of the musculoskeletal injuries reported (29% were defined as sprains and strains and 38% as overuse injuries) involved the use of the Wii to play simulated sports.17 Overuse syndrome associated with Wii was initially called “acute Wii-itis,”5 a description of acute tendinopathy of the infraspinatus.18 (Infraspinatus tendinopathy is most commonly associated with games involving intense arm activity, including Wii baseball, bowling, and boxing (TABLE 2).5 However, Wii-itis is now widely used to describe any acute inflammatory syndrome associated with use of this popular game console.
Wii knee, for example, refers to an acute patellar dislocation associated with simulated bowling.19 Multiple cases of patellar injury, including associated osteochondral fracture, have been reported in association with a variety of game titles, including Raymond Raving Rabbids and Brunswick Pro Bowling.19 In a review of self-reported Wii injuries, patellar dislocation was the fourth most common injury (hand lacerations were first, followed by periorbital hematoma [“black eye”], and forehead lacerations/ecchymoses).20
Wii shoulder, another variant of Wii-itis, is an acute inflammation of the upper extremity musculature after repetitive motion. This injury is most often associated with games that require swinging of the controller, such as Wii tennis or bowling. Upper extremity magnetic resonance imaging (MRI) of one Wii enthusiast revealed inflammatory swelling of the shoulder joint that extended to the suprascapular region, corresponding to a diagnosis of delayed–onset muscle soreness (DOMS).9
DOMS, which is often associated with acute injury patterns, is a well-accepted diagnosis among patients who play physically interactive sports and, by extension, video games.17 Usually lacking frank deformity on plain radiographs, DOMS is a disorder of the soft tissue that can best be visualized with MRI delineation of tissue planes and musculature compartments. Clinical signs and symptoms of DOMS can include edema of the affected extremity, rubor, and tenderness to palpation during active range of motion. Treatment for DOMS, like all RMIs, includes cessation of the offending activity.
Another recently reported variant of Wii-itis is the acute onset of carpal tunnel syndrome21 after playing Wii bowling for long periods of time. The case involved a 19-year-old woman who presented with swelling over the volar wrist and had positive Tinel and Phalen signs. She received conservative treatment with etodolac, a nightly volar splint, cold compresses, and rest.
Achilles Wii-itis refers to a partial or complete rupture of the Achilles tendon during simulated sports activity.22 This injury has been reported in people using the Wii Fit exercise pad for virtual running and stretching, and is diagnosed clinically with a positive Thompson sign (failure to plantar flex the foot while compressing the gastrocnemius). Complete Achilles rupture requires surgical repair, but less severe cases can be treated conservatively, with cold compresses, lifestyle modification, and nonsteroidal anti-inflammatory drugs (NSAIDs).
TABLE 2
Repetitive motion injuries (and possible causes)*30,36
| Type of injury | Games with potential for injury† | Possible injury sites | Common physical exam findings |
|---|---|---|---|
| Tendinopathy | Guitar Hero III: Legends of Rock The Legend of Zelda: Twilight Princess Wii Fit | Achilles tendon Patella Supraspinatous Forearm Extensors | Pain or stiffness in the local area of the tendon. Progression can lead to redness and swelling at the joint of the inflamed tendon |
| Bursitis | Kinect Sports: Soccer Dance Dance Revolution Star Wars: The Clone Wars Wii Fit | Subacromial bursa Trochanteric bursa Patellar bursa | Burning pain over the joint during and after activity, with delayed-onset joint stiffness due to local inflammation |
| Enthesitis | Wii Sports Sports Champions Kinect Sports | Achilles tendon Tuberosity of the tibia Iliac crest | Pain at joint on palpation or during range-of-motion exam. Calcification or fibrosis can be identified in chronic, nonacute presentations that are generally autoimmune mediated |
| Epicondylitis | Wii: Major League Baseball Grand Slam Tennis Tiger Woods PGA Tour | Olecranon process, lateral epicondyle (tennis elbow) | Point tenderness over the lateral epicondyle with acute pain on arm extension |
| Olecranon process, medial epicondyle (golf elbow) | Point tenderness over the medial epicondyle with acute pain on wrist flexion or resisted forearm pronation | ||
| *The authors have included games that, in their opinion, have the potential for injury based on the biomechanics involved (eg, running, jumping, waving, etc). † Many of these games are bundled and incorporate multiple activities (eg, baseball, bowling, boxing, soccer, track and field, tennis, volleyball). | |||
Categorizing Wii-type injuries
Game-related injuries typically fall into 4 broad categories: tendinopathy, bursitis, enthesitis, and epicondylitis. (See TABLE 2 for a list of games with the potential to cause particular types of injuries.)
Tendinopathy. Overuse tendon injuries, or tendinopathies, account for up to 50% of all sports-related injuries.23 By extrapolation, physically interactive game systems that simulate actual sports can be expected to increase tendon overuse injuries.
Most major tendons are vulnerable to overuse injury, including the Achilles (FIGURE), as noted earlier; and the patellar, rotator cuff, and forearm extensor tendons, among others. Repetitive motion, or strain, injuries to these tendons are often thought to be cumulative, with hypoperfusion, local inflammation, and neuropathy contributing to the degree of tendinopathy. Other risk factors for tendon injury include age and sex (men have a higher relative risk than women; older people, in their fourth and fifth decades of life, also face an increased risk), postmenopausal status, obesity, use of fluoroquinolone antibiotics or corticosteroids, and playing on nonpadded surfaces.24-29
Conservative therapy, with cessation of the offending activity and rest of the affected extremity, is the initial treatment of choice for tendinopathy. Severe cases of compound injuries or tendon reinjury can also be treated with splinting, taping, cryotherapy, electrotherapy, deep tissue tendon massage, pharmaceuticals (NSAIDs and corticosteroid injections), and early rehabilitation.15,30 Surgery may eventually be required to remove fibrotic tissue, modify the vascularity, or reconstruct the tendon.15
Bursitis. Bursitis is characterized by inflammation of the subacromial, olecranon, trochanteric, prepatellar, suprapatellar, infrapatellar, pes anserine, or iliotibial bursa—synovial-lined cavities overlying bony prominences that minimize the friction of movement.31 Patellar and olecranon bursitis are most frequently associated with sports, particularly soccer and golf.
Clinically characterized by pain on flexion, bursitis can also present with localized tenderness, stiffness, and swelling of the affected joint. Bursitis generally responds to RICE (rest, ice, compression, elevation) therapy, but can potentially advance to a chronic disease state if the activity that caused the inflammation continues.31
Enthesitis. Characterized by inflammation of the bony insertions of a tendon or ligament, enthesitis is generally linked to an autoimmune disease such as ankylosing spondylitis or rheumatoid arthritis. But it can also be an acquired condition associated with repetitive motion. Sports-related activity is the most common cause of acquired enthesitis,32 with injury most likely to occur at the Achilles tendon, the insertion point of the tibial tuberosity, or the iliac crest.33 Like most RMIs, acquired enthesitis can usually be treated simply by stopping the offending activity. If not properly recognized or treated, however, permanent injury can occur. 34
Epicondylitis. This RMI results in pain or ipsilateral weakness of the upper extremity due to repetitive strain at the musculotendinous junction and its origin at the epicondyle. Neuropraxia is often associated with epicondylitis due to posterior interosseous nerve, median nerve, or ulnar nerve involvement at either the medial or lateral epicondyles.35
Commonly affecting computer users who perform repetitive motion via mouse manipulation, the term “mouse elbow” was first described in 1992.14 Golfer’s elbow (with involvement of the medial epicondyle), and tennis elbow (involving the lateral epicondyle) are also common, and individuals who frequently play simulated golf or tennis games are at risk.
FIGURE
Achilles tendon injury
An MRI reveals anterior bulging and thickening of the Achilles tendon (arrow)—the type of injury you might see in a patient using the Wii Fit exercise pad for running and stretching.
Tell patients how to prevent injury
Older patients and deconditioned “arm chair” athletes who are unaccustomed to prolonged physical activity face an increased risk for injuries related to video game sports. You can help by pointing out that because simulated activities require a fraction of the strength and endurance required to play the actual sport, people who might normally tire easily are apt to overdo it.
In fact, Nintendo has a dedicated safety page regarding the use of game consoles on its Web site (http://www.nintendo.com/consumer/wiisafety.jsp). The company advises Wii users to take a 10- to 15-minute break every hour, even if they don’t think they need it, to prevent repetitive motion and eyestrain injuries, and to stop playing for several hours if they experience tingling, numbness, burning, or stiffness. Some software titles, including Wii Fit, are programmed to remind users to take a break after they’ve been playing nonstop for 45 minutes to an hour. You can help by reminding patients of all ages that warm-up exercises, moderation, and hydration are crucial, whether the sports they’re engaging in are virtual or real.
Acknowledgement
The authors would like to thank Dan Dunlany for his invaluable research assistance.
CORRESPONDENCE
Lisa M. Coughlin, MD, Department of Surgery, University of Toledo Medical Center, 3065 Arlington Avenue, Toledo, OH 43614; LcoughlinMD@gmail.com
• Ask patients with repetitive motion injuries (RMIs) whether they use interactive game consoles and, if so, how much time they spend playing virtual sports each day. C
• Be aware that RMIs associated with video game use are similar to injuries associated with the sports they simulate. A
• Advise patients to take the same precautions with virtual sports as they would with any physical activities, including warm-up exercises and moderation. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
This article is an expansion of a poster session presented at the 12th annual Northeastern Ohio Universities College of Medicine Department of Surgery Resident Research Day in May 2009 and at the American College of Preventive Medicine Annual Meeting in February 2010.
The release of the Wii—Nintendo’s 4th generation gaming console—in 2006 revolutionized the video game industry. By March 31, 2010, more than 70 million units had been sold worldwide, earning Wii the title of “fastest-selling game console of all time.”1-3
Today, there are several game consoles that, like Wii, allow the user not only to push buttons or move levers, but to control the game using physical movements (TABLE 1). And the devices and the many sports they simulate—once popular primarily among adolescents—are in widespread use by people of all ages, including the young and fit, out-of-shape “arm chair” athletes, and elderly people in senior housing, rehabilitation centers, and long-term care facilities alike.4
Not surprisingly, simulated sports play has spawned an array of repetitive motion and overuse injuries. To identify and treat them, ask all patients who present with musculoskeletal injuries whether they engage in game console sports activities; if so, identify the type of game and how much time they spend playing it each day. Although injuries associated with specific video games are often given names like “Wii-itis,”5 “Nintendinitis,”6 and “Playstation thumb,”7 the types of injuries caused by playing simulated sports are generally the same as (or similar to) injuries sustained by those engaging in the sport itself.
TABLE 1
Popular motion-controlled games: A partial list
| Type of game console | |||
|---|---|---|---|
| Nintendo: Wii | Microsoft Xbox 360: Kinect | Sony Playstation 3: Move | |
| Motion-control mechanism | Handheld remote | Full body | Handheld remote |
| Games bundled with console | Wii Sports | Kinect Adventures | PS3 Sports Champions |
| Popular games | Wii Fit Wii Play Mario Kart Super Smash Bros Brawl Guitar Hero III: Legends of Rock | Kinect Sports Dance Central Your Shape: Fitness Evolved The Biggest Loser: Kinectimals | Sports Champions Time Crisis: Razing Storm Killzone 3 Little Big Planet 2 Sorcery |
| Manufacturer-recommended game space | ≤6 feet using wireless sensor | ≥6 feet from device | ≥6 feet from device |
| Sources: 1. Nintendo (http://www.nintendo.com/consumer/wiisafety.jsp). 2. Microsoft Xbox 360 (http:www.xbox.com/en-US/Kinect/PrivacyandOnlineSafety). 3. Sony Playstation (http://us.playstation.com/support/answer/index.htm). | |||
Video game pathology is well established
In 1987, Osterman et al published the first report of a musculoskeletal disorder associated with electronic games—a case of volar flexor tenosynovitis (“joystick digit”) trigger finger.8 Several years later, a physician coined the term “Nintendinitis” to describe video game-related overuse syndrome6—acute tendinopathy of the extensor pollicis longus tendon after prolonged play with early versions of the thumb-activated game controller.9,10 In 2002, a child using a vibrating Sony Playstation for up to 7 hours a day received a diagnosis of vibratory syndrome of the hand.11 A few years later, a report of “Playstation thumb,” an overuse syndrome associated with later generations of game consoles, followed.7
Several other reports of game-related injury patterns can be found in medical journals, including pressure ulcer formation (“ulcerative Nintendinitis”),12 the “How!” sign of central palmar blistering,13 “mouse elbow” secondary to epicondylitis,14 and other tendinopathies associated with various gaming consoles.10,15,16 All the reports clearly describe the relationship between video game use and the pathology, and clinical improvement after cessation of the activity.
Many manifestations of Wii-itis
An epidemiologic review of the National Electronic Injury Surveillance System (http://www.cpsc.gov/library/neiss.html) found that in the Wii’s first year, 67% of the musculoskeletal injuries reported (29% were defined as sprains and strains and 38% as overuse injuries) involved the use of the Wii to play simulated sports.17 Overuse syndrome associated with Wii was initially called “acute Wii-itis,”5 a description of acute tendinopathy of the infraspinatus.18 (Infraspinatus tendinopathy is most commonly associated with games involving intense arm activity, including Wii baseball, bowling, and boxing (TABLE 2).5 However, Wii-itis is now widely used to describe any acute inflammatory syndrome associated with use of this popular game console.
Wii knee, for example, refers to an acute patellar dislocation associated with simulated bowling.19 Multiple cases of patellar injury, including associated osteochondral fracture, have been reported in association with a variety of game titles, including Raymond Raving Rabbids and Brunswick Pro Bowling.19 In a review of self-reported Wii injuries, patellar dislocation was the fourth most common injury (hand lacerations were first, followed by periorbital hematoma [“black eye”], and forehead lacerations/ecchymoses).20
Wii shoulder, another variant of Wii-itis, is an acute inflammation of the upper extremity musculature after repetitive motion. This injury is most often associated with games that require swinging of the controller, such as Wii tennis or bowling. Upper extremity magnetic resonance imaging (MRI) of one Wii enthusiast revealed inflammatory swelling of the shoulder joint that extended to the suprascapular region, corresponding to a diagnosis of delayed–onset muscle soreness (DOMS).9
DOMS, which is often associated with acute injury patterns, is a well-accepted diagnosis among patients who play physically interactive sports and, by extension, video games.17 Usually lacking frank deformity on plain radiographs, DOMS is a disorder of the soft tissue that can best be visualized with MRI delineation of tissue planes and musculature compartments. Clinical signs and symptoms of DOMS can include edema of the affected extremity, rubor, and tenderness to palpation during active range of motion. Treatment for DOMS, like all RMIs, includes cessation of the offending activity.
Another recently reported variant of Wii-itis is the acute onset of carpal tunnel syndrome21 after playing Wii bowling for long periods of time. The case involved a 19-year-old woman who presented with swelling over the volar wrist and had positive Tinel and Phalen signs. She received conservative treatment with etodolac, a nightly volar splint, cold compresses, and rest.
Achilles Wii-itis refers to a partial or complete rupture of the Achilles tendon during simulated sports activity.22 This injury has been reported in people using the Wii Fit exercise pad for virtual running and stretching, and is diagnosed clinically with a positive Thompson sign (failure to plantar flex the foot while compressing the gastrocnemius). Complete Achilles rupture requires surgical repair, but less severe cases can be treated conservatively, with cold compresses, lifestyle modification, and nonsteroidal anti-inflammatory drugs (NSAIDs).
TABLE 2
Repetitive motion injuries (and possible causes)*30,36
| Type of injury | Games with potential for injury† | Possible injury sites | Common physical exam findings |
|---|---|---|---|
| Tendinopathy | Guitar Hero III: Legends of Rock The Legend of Zelda: Twilight Princess Wii Fit | Achilles tendon Patella Supraspinatous Forearm Extensors | Pain or stiffness in the local area of the tendon. Progression can lead to redness and swelling at the joint of the inflamed tendon |
| Bursitis | Kinect Sports: Soccer Dance Dance Revolution Star Wars: The Clone Wars Wii Fit | Subacromial bursa Trochanteric bursa Patellar bursa | Burning pain over the joint during and after activity, with delayed-onset joint stiffness due to local inflammation |
| Enthesitis | Wii Sports Sports Champions Kinect Sports | Achilles tendon Tuberosity of the tibia Iliac crest | Pain at joint on palpation or during range-of-motion exam. Calcification or fibrosis can be identified in chronic, nonacute presentations that are generally autoimmune mediated |
| Epicondylitis | Wii: Major League Baseball Grand Slam Tennis Tiger Woods PGA Tour | Olecranon process, lateral epicondyle (tennis elbow) | Point tenderness over the lateral epicondyle with acute pain on arm extension |
| Olecranon process, medial epicondyle (golf elbow) | Point tenderness over the medial epicondyle with acute pain on wrist flexion or resisted forearm pronation | ||
| *The authors have included games that, in their opinion, have the potential for injury based on the biomechanics involved (eg, running, jumping, waving, etc). † Many of these games are bundled and incorporate multiple activities (eg, baseball, bowling, boxing, soccer, track and field, tennis, volleyball). | |||
Categorizing Wii-type injuries
Game-related injuries typically fall into 4 broad categories: tendinopathy, bursitis, enthesitis, and epicondylitis. (See TABLE 2 for a list of games with the potential to cause particular types of injuries.)
Tendinopathy. Overuse tendon injuries, or tendinopathies, account for up to 50% of all sports-related injuries.23 By extrapolation, physically interactive game systems that simulate actual sports can be expected to increase tendon overuse injuries.
Most major tendons are vulnerable to overuse injury, including the Achilles (FIGURE), as noted earlier; and the patellar, rotator cuff, and forearm extensor tendons, among others. Repetitive motion, or strain, injuries to these tendons are often thought to be cumulative, with hypoperfusion, local inflammation, and neuropathy contributing to the degree of tendinopathy. Other risk factors for tendon injury include age and sex (men have a higher relative risk than women; older people, in their fourth and fifth decades of life, also face an increased risk), postmenopausal status, obesity, use of fluoroquinolone antibiotics or corticosteroids, and playing on nonpadded surfaces.24-29
Conservative therapy, with cessation of the offending activity and rest of the affected extremity, is the initial treatment of choice for tendinopathy. Severe cases of compound injuries or tendon reinjury can also be treated with splinting, taping, cryotherapy, electrotherapy, deep tissue tendon massage, pharmaceuticals (NSAIDs and corticosteroid injections), and early rehabilitation.15,30 Surgery may eventually be required to remove fibrotic tissue, modify the vascularity, or reconstruct the tendon.15
Bursitis. Bursitis is characterized by inflammation of the subacromial, olecranon, trochanteric, prepatellar, suprapatellar, infrapatellar, pes anserine, or iliotibial bursa—synovial-lined cavities overlying bony prominences that minimize the friction of movement.31 Patellar and olecranon bursitis are most frequently associated with sports, particularly soccer and golf.
Clinically characterized by pain on flexion, bursitis can also present with localized tenderness, stiffness, and swelling of the affected joint. Bursitis generally responds to RICE (rest, ice, compression, elevation) therapy, but can potentially advance to a chronic disease state if the activity that caused the inflammation continues.31
Enthesitis. Characterized by inflammation of the bony insertions of a tendon or ligament, enthesitis is generally linked to an autoimmune disease such as ankylosing spondylitis or rheumatoid arthritis. But it can also be an acquired condition associated with repetitive motion. Sports-related activity is the most common cause of acquired enthesitis,32 with injury most likely to occur at the Achilles tendon, the insertion point of the tibial tuberosity, or the iliac crest.33 Like most RMIs, acquired enthesitis can usually be treated simply by stopping the offending activity. If not properly recognized or treated, however, permanent injury can occur. 34
Epicondylitis. This RMI results in pain or ipsilateral weakness of the upper extremity due to repetitive strain at the musculotendinous junction and its origin at the epicondyle. Neuropraxia is often associated with epicondylitis due to posterior interosseous nerve, median nerve, or ulnar nerve involvement at either the medial or lateral epicondyles.35
Commonly affecting computer users who perform repetitive motion via mouse manipulation, the term “mouse elbow” was first described in 1992.14 Golfer’s elbow (with involvement of the medial epicondyle), and tennis elbow (involving the lateral epicondyle) are also common, and individuals who frequently play simulated golf or tennis games are at risk.
FIGURE
Achilles tendon injury
An MRI reveals anterior bulging and thickening of the Achilles tendon (arrow)—the type of injury you might see in a patient using the Wii Fit exercise pad for running and stretching.
Tell patients how to prevent injury
Older patients and deconditioned “arm chair” athletes who are unaccustomed to prolonged physical activity face an increased risk for injuries related to video game sports. You can help by pointing out that because simulated activities require a fraction of the strength and endurance required to play the actual sport, people who might normally tire easily are apt to overdo it.
In fact, Nintendo has a dedicated safety page regarding the use of game consoles on its Web site (http://www.nintendo.com/consumer/wiisafety.jsp). The company advises Wii users to take a 10- to 15-minute break every hour, even if they don’t think they need it, to prevent repetitive motion and eyestrain injuries, and to stop playing for several hours if they experience tingling, numbness, burning, or stiffness. Some software titles, including Wii Fit, are programmed to remind users to take a break after they’ve been playing nonstop for 45 minutes to an hour. You can help by reminding patients of all ages that warm-up exercises, moderation, and hydration are crucial, whether the sports they’re engaging in are virtual or real.
Acknowledgement
The authors would like to thank Dan Dunlany for his invaluable research assistance.
CORRESPONDENCE
Lisa M. Coughlin, MD, Department of Surgery, University of Toledo Medical Center, 3065 Arlington Avenue, Toledo, OH 43614; LcoughlinMD@gmail.com
1. NPD Seventh Generation. Wikia. Available at: http://vgsales.wikia.com/wiki/NPD_Seventh_generation#NPD_hardware_sales. Accessed September 1, 2010.
2. Thorsen T. Wii sales near 71 million, DS almost 129 million. May 6, 2010. Gamespot. Available at: http://www.gamespot.com/news/6261400.html. Accessed September 15, 2010.
3. Nintendo Wii is the fastest selling gaming console, beats Xbox 360 sales in less than a year August 25, 2007. TechShout. Available at: http://www.techshout.com/gaming/2007/25/nintendo-wii-is-the-fastest-selling-gaming-console-beats-xbox-360-sales-in-less-than-a-year/. Accessed February 18, 2009.
4. Hsu JK, Thibodeau R, Wong SJ, et al. A “Wii” bit of fun: the effects of adding Nintendo Wii bowling to a standard exercise regimen for residents of long-term care with upper extremity dysfunction. Physiother Theory Pract. 2011;27:185-193.
5. Bonis J. Acute Wiiitis. N Engl J Med. 2007;356:2431-2432.
6. Brasington R. Nintendinitis. N Engl J Med. 1990;322:1473-1474.
7. Vaidya HJ. Playstation thumb. Lancet. 2004;363:1080.-
8. Osterman AL, Weinberg P, Miller G. Joystick digit. JAMA. 1987;257:782.-
9. Nett MP, Collins MS, Sperling JW. Magnetic resonance imaging of acute “wiiitis” of the upper extremity. Skeletal Radiol. 2008;37:481-483.
10. Macgregor DM. Nintendonitis? A case report of repetitive strain injury in a child as a result of playing computer games. Scott Med J. 2000;45:150.-
11. Cleary AG, McKendrick H, Sills JA. Hand-arm vibration syndrome may be associated with prolonged use of vibrating computer games. BMJ. 2000;324:301.-
12. Koh TH. Ulcerative “nintendinitis”: a new kind of repetitive strain injury. Med J Aust. 2000;173:671.-
13. Wood J. The “How! ” sign—a central palmar blister induced by overplaying on a Nintendo console. Arch Dis Child. 2001;84:288.-
14. Mirman MJ, Bonian VG. “Mouse elbow”: a new repetitive stress injury. J Am Osteopath Assoc. 1992;92:701.-
15. Rees JD, Maffulli N, Cook J. Management of tendinopathy. Am J Sports Med. 2009;37:1855-1867.
16. Kujala UM, Taimela S, Viljanen T. Leisure physical activity and various pain symptoms among adolescents. Br J Sports Med. 1999;33:325-328.
17. Jones C, Hammig B. Case report: injuries associated with interactive game consoles: preliminary data. Phys Sports Med. 2009;37:138-140.
18. Hertel R, Ballmer FT, Lombert SM, et al. Lag signs in the diagnosis of rotator cuff rupture. J Shoulder Elbow Surg. 1996;5:307-313.
19. Robinson RJ, Barron DA, Grainger AJ, et al. Wii knee. Emerg Radiol. 2008;15:255-257.
20. Sparks DA, Chase DM, Coughlin LM. Wii have a problem: a review of self-reported Wii related injuries. Inform Prim Care. 2009;17:55-57.
21. Boehm KM, Pugh A. A new variant of Wii-itis. J Emerg Med. 2009;36:80.-
22. Beddy P, Dunne R, de Blacam C. Achilles wiiitis. AJR Am J Roentgenol. 2009;192:W79.-
23. [Herring SA, Nilson KL. Introduction to overuse injuries. Clin Sports Med. 1987;6:225-23.
24. Maffulli N, Waterston SW, Squair J, et al. Changing incidence of Achilles tendon rupture in Scotland: a 15-year study. Clin J Sport Med. 1999;9:157-160.
25. Malliaras P, Cook J. Patellar tendons with normal imaging and pain: change in imaging and pain status over a volleyball season. Clin J Sport Med. 2006;16:388-391.
26. Malliaras PJ, Cook JL, Kent PM. Anthropometric risk factors for patellar tendon injury among volleyball players. Br J Sports Med. 2007;41:259-263.
27. McGarvey WC, Singh D, Trevino SG. Partial Achilles tendon ruptures associated with fluoroquinolone antibiotics: a case report and literature review. Foot Ankle Int. 1996;17:496-498.
28. Ford LT, DeBender J. Tendon rupture after local steroid injection. South Med J. 1979;72:827-830.
29. Hess GW. Achilles tendon rupture: a review of etiology, population, anatomy, risk factors, and injury prevention. Foot Ankle Spec. 2010;3:29-32.
30. Rettig AC. Wrist and hand overuse syndromes. Clin Sports Med. 2001;20:591-611.
31. Huie G. Diagnosing bursitis in the knee. JAAPA. 2002;15:14-16.
32. Jennings F, Lambert E, Fredericson M. Rheumatic diseases presenting as sports-related injuries. Sports Med. 2008;38:917-930.
33. Leppilahti J, Orava S, Karpakka J, et al. Overuse injuries of the Achilles tendon. Ann Chir Gynaecol. 1991;80:202-207.
34. Slobodin G, Rozenbaum M, Boulman N, et al. Varied presentations of enthesopathy. Semin Arthritis Rheum. 2007;37:119-126.
35. Jepsen JR, Thomsen G. A cross-sectional study of the relation between symptoms and physical findings in computer operators. BMC Neurol. 2006;6:40.-
36. Biundo JJ, Jr, Irwin RW, Umpierre E. Sports and soft tissue injuries, tendinitis, bursitis, and occupation-related syndromes. Curr Opin Rheumatol. 2001;13:146-149.
1. NPD Seventh Generation. Wikia. Available at: http://vgsales.wikia.com/wiki/NPD_Seventh_generation#NPD_hardware_sales. Accessed September 1, 2010.
2. Thorsen T. Wii sales near 71 million, DS almost 129 million. May 6, 2010. Gamespot. Available at: http://www.gamespot.com/news/6261400.html. Accessed September 15, 2010.
3. Nintendo Wii is the fastest selling gaming console, beats Xbox 360 sales in less than a year August 25, 2007. TechShout. Available at: http://www.techshout.com/gaming/2007/25/nintendo-wii-is-the-fastest-selling-gaming-console-beats-xbox-360-sales-in-less-than-a-year/. Accessed February 18, 2009.
4. Hsu JK, Thibodeau R, Wong SJ, et al. A “Wii” bit of fun: the effects of adding Nintendo Wii bowling to a standard exercise regimen for residents of long-term care with upper extremity dysfunction. Physiother Theory Pract. 2011;27:185-193.
5. Bonis J. Acute Wiiitis. N Engl J Med. 2007;356:2431-2432.
6. Brasington R. Nintendinitis. N Engl J Med. 1990;322:1473-1474.
7. Vaidya HJ. Playstation thumb. Lancet. 2004;363:1080.-
8. Osterman AL, Weinberg P, Miller G. Joystick digit. JAMA. 1987;257:782.-
9. Nett MP, Collins MS, Sperling JW. Magnetic resonance imaging of acute “wiiitis” of the upper extremity. Skeletal Radiol. 2008;37:481-483.
10. Macgregor DM. Nintendonitis? A case report of repetitive strain injury in a child as a result of playing computer games. Scott Med J. 2000;45:150.-
11. Cleary AG, McKendrick H, Sills JA. Hand-arm vibration syndrome may be associated with prolonged use of vibrating computer games. BMJ. 2000;324:301.-
12. Koh TH. Ulcerative “nintendinitis”: a new kind of repetitive strain injury. Med J Aust. 2000;173:671.-
13. Wood J. The “How! ” sign—a central palmar blister induced by overplaying on a Nintendo console. Arch Dis Child. 2001;84:288.-
14. Mirman MJ, Bonian VG. “Mouse elbow”: a new repetitive stress injury. J Am Osteopath Assoc. 1992;92:701.-
15. Rees JD, Maffulli N, Cook J. Management of tendinopathy. Am J Sports Med. 2009;37:1855-1867.
16. Kujala UM, Taimela S, Viljanen T. Leisure physical activity and various pain symptoms among adolescents. Br J Sports Med. 1999;33:325-328.
17. Jones C, Hammig B. Case report: injuries associated with interactive game consoles: preliminary data. Phys Sports Med. 2009;37:138-140.
18. Hertel R, Ballmer FT, Lombert SM, et al. Lag signs in the diagnosis of rotator cuff rupture. J Shoulder Elbow Surg. 1996;5:307-313.
19. Robinson RJ, Barron DA, Grainger AJ, et al. Wii knee. Emerg Radiol. 2008;15:255-257.
20. Sparks DA, Chase DM, Coughlin LM. Wii have a problem: a review of self-reported Wii related injuries. Inform Prim Care. 2009;17:55-57.
21. Boehm KM, Pugh A. A new variant of Wii-itis. J Emerg Med. 2009;36:80.-
22. Beddy P, Dunne R, de Blacam C. Achilles wiiitis. AJR Am J Roentgenol. 2009;192:W79.-
23. [Herring SA, Nilson KL. Introduction to overuse injuries. Clin Sports Med. 1987;6:225-23.
24. Maffulli N, Waterston SW, Squair J, et al. Changing incidence of Achilles tendon rupture in Scotland: a 15-year study. Clin J Sport Med. 1999;9:157-160.
25. Malliaras P, Cook J. Patellar tendons with normal imaging and pain: change in imaging and pain status over a volleyball season. Clin J Sport Med. 2006;16:388-391.
26. Malliaras PJ, Cook JL, Kent PM. Anthropometric risk factors for patellar tendon injury among volleyball players. Br J Sports Med. 2007;41:259-263.
27. McGarvey WC, Singh D, Trevino SG. Partial Achilles tendon ruptures associated with fluoroquinolone antibiotics: a case report and literature review. Foot Ankle Int. 1996;17:496-498.
28. Ford LT, DeBender J. Tendon rupture after local steroid injection. South Med J. 1979;72:827-830.
29. Hess GW. Achilles tendon rupture: a review of etiology, population, anatomy, risk factors, and injury prevention. Foot Ankle Spec. 2010;3:29-32.
30. Rettig AC. Wrist and hand overuse syndromes. Clin Sports Med. 2001;20:591-611.
31. Huie G. Diagnosing bursitis in the knee. JAAPA. 2002;15:14-16.
32. Jennings F, Lambert E, Fredericson M. Rheumatic diseases presenting as sports-related injuries. Sports Med. 2008;38:917-930.
33. Leppilahti J, Orava S, Karpakka J, et al. Overuse injuries of the Achilles tendon. Ann Chir Gynaecol. 1991;80:202-207.
34. Slobodin G, Rozenbaum M, Boulman N, et al. Varied presentations of enthesopathy. Semin Arthritis Rheum. 2007;37:119-126.
35. Jepsen JR, Thomsen G. A cross-sectional study of the relation between symptoms and physical findings in computer operators. BMC Neurol. 2006;6:40.-
36. Biundo JJ, Jr, Irwin RW, Umpierre E. Sports and soft tissue injuries, tendinitis, bursitis, and occupation-related syndromes. Curr Opin Rheumatol. 2001;13:146-149.
Looking beyond the D-dimer
A 44-year-old woman sought care at the emergency department (ED) because she was having difficulty breathing and felt faint. She had been fine until that morning. Three days earlier the patient, who had a history of high blood pressure and elevated cholesterol levels, had driven from Connecticut to New York and back, spending a total of 4 hours in her car. The patient indicated that she’d been taking oral contraceptives (OCPs) for several years, but she did not smoke. There was no history of hemoptysis, recent surgery, or trauma. Neither blood clots nor cancer were part of her or her family’s history.
In the ED, the patient did not have any signs or symptoms of a deep venous thrombosis (DVT). She was obese, with a body mass index of 40.3 kg/m2; other vitals were: blood pressure (BP), 134/88 mm Hg; heart rate (HR), 64 beats per minute (bpm); respiratory rate (RR), 12; and O2 saturation, 99% with ambulation.
The ED physician strongly suspected a pulmonary embolism (PE), but the patient’s score on a clinical probability algorithm (using the Wells criteria) was a 3, indicating only “moderate probability“ of a PE (TABLE 1). (She scored a 3 because an “alternative diagnosis [was] less likely than PE.”) In addition, her D-dimer level was 160 ng/mL using the Triage D-Dimer Test by Biosite, Inc (normal <400 ng/mL), which ruled out a PE. (Many ED physicians at our institution are more cautious when using this D-dimer assay and use a lower cutoff value.)
Given these results, the ED physician did not order imaging studies because the expense and radiation exposure outweighed the probability of the patient having a PE. A subsequent coronary work-up was also negative. The patient was discharged to home and advised to follow up with her primary care physician a few days later.
Two days later we saw the patient at our office. Not only had her dyspnea gotten worse while the presyncope remained, but she now had left-sided pleuritic chest pain. She also reported mild pain in her right calf. On examination, the patient’s BP was 126/86 mm Hg, HR was 82 bpm, RR was 12, and O2 saturation was 96% with ambulation. Her Wells score was now 6, still a moderate probability for PE. (She received another 3 points for the new DVT symptoms—“clinically suspected DVT.”)
Although the patient did not also have signs of a DVT, her additional symptoms along with the original symptoms’ persistence and the existence of other risk factors (OCP use and obesity) led us to reconsider a PE diagnosis. These suspicions prompted us to send the patient back to the ED, where a Doppler ultrasound of the right lower extremity was negative, but the D-dimer was positive at 565 ng/mL.
A pulmonary computed tomography angiogram (CTA) showed 2 small pulmonary emboli within the distal left upper lobe pulmonary arteries.
The patient was treated with heparin and warfarin and discharged without complications.
TABLE 1
Calculating and interpreting the Wells score4,5,7,9,10
| Clinical parameter | Points |
|---|---|
| Clinically suspected DVT | 3.0 |
| Alternative diagnosis less likely than PE | 3.0 |
| Tachycardia | 1.5 |
| Immobilization/surgery (within 4 weeks) | 1.5 |
| History of DVT or PE | 1.5 |
| Hemoptysis | 1.0 |
| Malignancy (treatment within 6 months, palliative) | 1.0 |
| TOTAL | |
| Score | Traditional interpretation |
| <2.0 | Low probability of PE |
| 2.0-6.0 | Moderate probability of PE |
| >6.0 | High probability of PE |
| Score | Alternative classification scheme |
| ≤4.0 | PE unlikely |
| >4.0 | PE likely |
| DVT, deep venous thrombosis; PE, pulmonary embolism. | |
Discussion
The incidence of PE in the United States varies significantly: Individuals younger than 40 have a risk of 1 in 10,000 compared with 1 in 100 for those older than 80.1 Mortality associated with undiagnosed PE varies widely, from 9.2% to 51%.2 This percentage is significant given that half of all PEs go undiagnosed.3 In addition, when left untreated, PE will recur in 30% to 50% of patients, with a fatality rate of 10% to 45%.1 Further, up to 4% of patients with acute PE develop chronic PE and subsequent pulmonary hypertension.4,5 Given the consequences of failing to diagnose a PE, clinicians must consider this condition in patients who present with unexplained hypotension, dyspnea, or chest pain.6
Not an easy diagnosis
This case report demonstrates the inherent difficulty in diagnosing a PE. Still, certain clinical symptoms/signs can aid in the decision-making process. Fever, crackles, and wheezes decrease the probability of PE, whereas syncope, hemodynamic shock, leg edema, and hemoptysis increase its likelihood.7 Despite the many commonly reported risk factors for PE, only malignancy, recent surgery, or a history of DVT/PE significantly increase the risk of developing a clot.8
The Wells criteria. This scoring system groups patients according to the probability of having a PE: low (score: <2), moderate (score: 2-6), and high (score: >6).6 An alternative classification scheme divides patients into 2 groups: likely to have a PE (score: >4) or unlikely to have a PE (score: ≤4).8
This case report illustrates a key problem with the Wells criteria—the somewhat subjective nature of the scoring. Some physicians find it questionable to award 3 points for “alternative diagnosis less likely than PE,” for example.4 Similarly, with respect to immobilization, some clinicians might have awarded our patient 1.5 points for her recent car trip to New York. We did not think that riding in a car for 2 uninterrupted hours for each leg of the trip was significant enough. However, awarding this patient 1.5 points could have made an important difference in her clinical management if the alternative classification scheme was used. Instead of having a score of 3, the patient would have had a score of 4.5, placing her in the “likely to have a PE” group and prompting us to perform a CTA sooner (FIGURE).
FIGURE
Diagnostic algorithm for pulmonary embolism6,7,10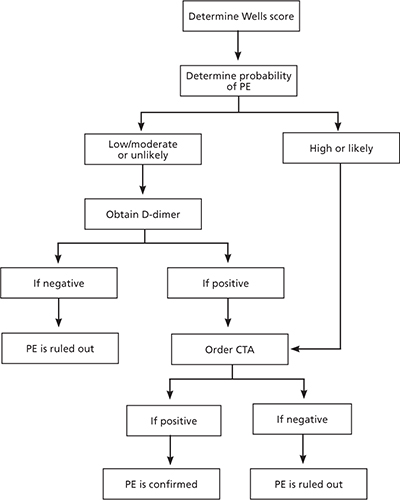
CTA, computed tomography angiogram; PE, pulmonary embolism.
Inappropriate work-ups are common
Some physicians ignore algorithms when working up a PE and simply order a CTA. In fact, a large multicenter trial showed that 43% of patients suspected of having a PE were inappropriately managed diagnostically.9 Similarly, a meta-analysis of 4 studies including 1660 patients found that only 58% of those with a positive D-dimer had the requisite CTA, as did 7% of patients with a negative D-dimer.2
Physicians should not be concerned about ruling out a PE in the setting of a negative D-dimer, as a meta-analysis found that this diagnostic approach has a negative predictive value (NPV) of 99.7%.2 It is important to note that the NPV is significantly affected by the sensitivity of the D-dimer assay used. If the D-dimer assay is highly sensitive, a negative result in combination with a low, moderate, or unlikely probability Wells score rules out the diagnosis of PE. If the assay is moderately sensitive, however, only a low or unlikely probability Wells score rules out PE.10
The inappropriate work-up of this group of patients is significant and extends beyond the ultimate goal of preventing morbidity and mortality. The unnecessary use of pulmonary CTA is extremely expensive, exposes patients to unnecessary radiation, and results in contrast nephrotoxicity in about 4% of patients.9 Although pulmonary CTA is the standard diagnostic test for PE, other imaging modalities are more appropriate in some cases (TABLE 2).
TABLE 2
Alternative imaging modalities for diagnosing PE1,4,7,11
| Modality | Indication |
|---|---|
| Ventilation-perfusion scanning | Patients with contrast allergies or renal failure; test of choice for diagnosing chronic PE due to limited sensitivity of CT |
| Venous compression ultrasonography | Patients with symptoms of PE and signs/symptoms of DVT |
| Pulmonary angiography | Most invasive test. Should be used only in patients with high probability of PE who may need vascular intervention |
| CT, computed tomography; DVT, deep venous thrombosis; PE, pulmonary embolism. | |
The bottom line
This case report illustrates the importance of using sound clinical judgment when diagnosing a PE. Although our patient initially had a moderate probability Wells score and a negative D-dimer, her symptoms persisted. Her history of OCP use, persistent dyspnea, and new symptoms of a DVT prompted us to reinitiate the diagnostic algorithm and eventually diagnose a PE.
It is always essential to treat the patient and not simply react to laboratory values. To avoid unnecessary testing, however, adhering to the algorithm is equally important.
CORRESPONDENCE
Michael S. Kelleher, MD, University of Connecticut School of Medicine, 263 Farmington Avenue, Farmington, CT 06030; mkelleher@student.uchc.edu
1. Meyer G, Roy PM, Gilberg S, et al. Pulmonary embolism. BMJ. 2010;340:1421.-
2. Pasha SM, Kiok FA, Snoep JD, et al. Safety of excluding acute pulmonary embolism based on an unlikely clinical probability by the Wells rule and normal D-dimer concentration: a meta-analysis. Thromb Res. 2010;125:e123-e127.
3. Taira T, Taira BR, Carmen M, et al. Risk of venous thromboembolism in patients with borderline quantitative D-dimer levels. Am J Emerg Med. 2010;28:450-453.
4. Bounameaux H, Perrier A, Righini M. Diagnosis of venous thromboembolism: an update. Vasc Med. 2010;15:399-406.
5. Pengo V, Lensing AW, Prins MH, et al. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med. 2004;350:2257-2264.
6. Agnelli G, Becattini C. Acute pulmonary embolism. N Engl J Med. 2010;363:266-274.
7. Gandara E, Wells PS. Diagnosis: use of clinical probability algorithms. Clin Chest Med. 2010;31:629-639.
8. Drescher FS, Chandrika S, Weir ID, et al. Effectiveness and acceptability of a computerized decision support system using modified wells criteria for evaluation of suspected pulmonary embolism. Ann Emerg Med. 2011;57:613-621.
9. Gimber LH, Travis RI, Takahashi JM, et al. Computed tomography angiography in patients evaluated for acute pulmonary embolism with low serum D-dimer levels: a prospective study. Perm J. 2009;13:4-10.
10. Agency for Healthcare Research and Quality. Guidelines on the diagnosis and management of acute pulmonary embolism. Available at: http://www.guideline.gov/content.aspx?id=13410#Section420. Accessed February 12, 2011.
11. Kim NH. Chronic thromboembolic pulmonary hypertension: diagnosis. Medscape. Available at: http://www.medscape.org/viewarticle/556058_3. Accessed May 9, 2011.
A 44-year-old woman sought care at the emergency department (ED) because she was having difficulty breathing and felt faint. She had been fine until that morning. Three days earlier the patient, who had a history of high blood pressure and elevated cholesterol levels, had driven from Connecticut to New York and back, spending a total of 4 hours in her car. The patient indicated that she’d been taking oral contraceptives (OCPs) for several years, but she did not smoke. There was no history of hemoptysis, recent surgery, or trauma. Neither blood clots nor cancer were part of her or her family’s history.
In the ED, the patient did not have any signs or symptoms of a deep venous thrombosis (DVT). She was obese, with a body mass index of 40.3 kg/m2; other vitals were: blood pressure (BP), 134/88 mm Hg; heart rate (HR), 64 beats per minute (bpm); respiratory rate (RR), 12; and O2 saturation, 99% with ambulation.
The ED physician strongly suspected a pulmonary embolism (PE), but the patient’s score on a clinical probability algorithm (using the Wells criteria) was a 3, indicating only “moderate probability“ of a PE (TABLE 1). (She scored a 3 because an “alternative diagnosis [was] less likely than PE.”) In addition, her D-dimer level was 160 ng/mL using the Triage D-Dimer Test by Biosite, Inc (normal <400 ng/mL), which ruled out a PE. (Many ED physicians at our institution are more cautious when using this D-dimer assay and use a lower cutoff value.)
Given these results, the ED physician did not order imaging studies because the expense and radiation exposure outweighed the probability of the patient having a PE. A subsequent coronary work-up was also negative. The patient was discharged to home and advised to follow up with her primary care physician a few days later.
Two days later we saw the patient at our office. Not only had her dyspnea gotten worse while the presyncope remained, but she now had left-sided pleuritic chest pain. She also reported mild pain in her right calf. On examination, the patient’s BP was 126/86 mm Hg, HR was 82 bpm, RR was 12, and O2 saturation was 96% with ambulation. Her Wells score was now 6, still a moderate probability for PE. (She received another 3 points for the new DVT symptoms—“clinically suspected DVT.”)
Although the patient did not also have signs of a DVT, her additional symptoms along with the original symptoms’ persistence and the existence of other risk factors (OCP use and obesity) led us to reconsider a PE diagnosis. These suspicions prompted us to send the patient back to the ED, where a Doppler ultrasound of the right lower extremity was negative, but the D-dimer was positive at 565 ng/mL.
A pulmonary computed tomography angiogram (CTA) showed 2 small pulmonary emboli within the distal left upper lobe pulmonary arteries.
The patient was treated with heparin and warfarin and discharged without complications.
TABLE 1
Calculating and interpreting the Wells score4,5,7,9,10
| Clinical parameter | Points |
|---|---|
| Clinically suspected DVT | 3.0 |
| Alternative diagnosis less likely than PE | 3.0 |
| Tachycardia | 1.5 |
| Immobilization/surgery (within 4 weeks) | 1.5 |
| History of DVT or PE | 1.5 |
| Hemoptysis | 1.0 |
| Malignancy (treatment within 6 months, palliative) | 1.0 |
| TOTAL | |
| Score | Traditional interpretation |
| <2.0 | Low probability of PE |
| 2.0-6.0 | Moderate probability of PE |
| >6.0 | High probability of PE |
| Score | Alternative classification scheme |
| ≤4.0 | PE unlikely |
| >4.0 | PE likely |
| DVT, deep venous thrombosis; PE, pulmonary embolism. | |
Discussion
The incidence of PE in the United States varies significantly: Individuals younger than 40 have a risk of 1 in 10,000 compared with 1 in 100 for those older than 80.1 Mortality associated with undiagnosed PE varies widely, from 9.2% to 51%.2 This percentage is significant given that half of all PEs go undiagnosed.3 In addition, when left untreated, PE will recur in 30% to 50% of patients, with a fatality rate of 10% to 45%.1 Further, up to 4% of patients with acute PE develop chronic PE and subsequent pulmonary hypertension.4,5 Given the consequences of failing to diagnose a PE, clinicians must consider this condition in patients who present with unexplained hypotension, dyspnea, or chest pain.6
Not an easy diagnosis
This case report demonstrates the inherent difficulty in diagnosing a PE. Still, certain clinical symptoms/signs can aid in the decision-making process. Fever, crackles, and wheezes decrease the probability of PE, whereas syncope, hemodynamic shock, leg edema, and hemoptysis increase its likelihood.7 Despite the many commonly reported risk factors for PE, only malignancy, recent surgery, or a history of DVT/PE significantly increase the risk of developing a clot.8
The Wells criteria. This scoring system groups patients according to the probability of having a PE: low (score: <2), moderate (score: 2-6), and high (score: >6).6 An alternative classification scheme divides patients into 2 groups: likely to have a PE (score: >4) or unlikely to have a PE (score: ≤4).8
This case report illustrates a key problem with the Wells criteria—the somewhat subjective nature of the scoring. Some physicians find it questionable to award 3 points for “alternative diagnosis less likely than PE,” for example.4 Similarly, with respect to immobilization, some clinicians might have awarded our patient 1.5 points for her recent car trip to New York. We did not think that riding in a car for 2 uninterrupted hours for each leg of the trip was significant enough. However, awarding this patient 1.5 points could have made an important difference in her clinical management if the alternative classification scheme was used. Instead of having a score of 3, the patient would have had a score of 4.5, placing her in the “likely to have a PE” group and prompting us to perform a CTA sooner (FIGURE).
FIGURE
Diagnostic algorithm for pulmonary embolism6,7,10
CTA, computed tomography angiogram; PE, pulmonary embolism.
Inappropriate work-ups are common
Some physicians ignore algorithms when working up a PE and simply order a CTA. In fact, a large multicenter trial showed that 43% of patients suspected of having a PE were inappropriately managed diagnostically.9 Similarly, a meta-analysis of 4 studies including 1660 patients found that only 58% of those with a positive D-dimer had the requisite CTA, as did 7% of patients with a negative D-dimer.2
Physicians should not be concerned about ruling out a PE in the setting of a negative D-dimer, as a meta-analysis found that this diagnostic approach has a negative predictive value (NPV) of 99.7%.2 It is important to note that the NPV is significantly affected by the sensitivity of the D-dimer assay used. If the D-dimer assay is highly sensitive, a negative result in combination with a low, moderate, or unlikely probability Wells score rules out the diagnosis of PE. If the assay is moderately sensitive, however, only a low or unlikely probability Wells score rules out PE.10
The inappropriate work-up of this group of patients is significant and extends beyond the ultimate goal of preventing morbidity and mortality. The unnecessary use of pulmonary CTA is extremely expensive, exposes patients to unnecessary radiation, and results in contrast nephrotoxicity in about 4% of patients.9 Although pulmonary CTA is the standard diagnostic test for PE, other imaging modalities are more appropriate in some cases (TABLE 2).
TABLE 2
Alternative imaging modalities for diagnosing PE1,4,7,11
| Modality | Indication |
|---|---|
| Ventilation-perfusion scanning | Patients with contrast allergies or renal failure; test of choice for diagnosing chronic PE due to limited sensitivity of CT |
| Venous compression ultrasonography | Patients with symptoms of PE and signs/symptoms of DVT |
| Pulmonary angiography | Most invasive test. Should be used only in patients with high probability of PE who may need vascular intervention |
| CT, computed tomography; DVT, deep venous thrombosis; PE, pulmonary embolism. | |
The bottom line
This case report illustrates the importance of using sound clinical judgment when diagnosing a PE. Although our patient initially had a moderate probability Wells score and a negative D-dimer, her symptoms persisted. Her history of OCP use, persistent dyspnea, and new symptoms of a DVT prompted us to reinitiate the diagnostic algorithm and eventually diagnose a PE.
It is always essential to treat the patient and not simply react to laboratory values. To avoid unnecessary testing, however, adhering to the algorithm is equally important.
CORRESPONDENCE
Michael S. Kelleher, MD, University of Connecticut School of Medicine, 263 Farmington Avenue, Farmington, CT 06030; mkelleher@student.uchc.edu
A 44-year-old woman sought care at the emergency department (ED) because she was having difficulty breathing and felt faint. She had been fine until that morning. Three days earlier the patient, who had a history of high blood pressure and elevated cholesterol levels, had driven from Connecticut to New York and back, spending a total of 4 hours in her car. The patient indicated that she’d been taking oral contraceptives (OCPs) for several years, but she did not smoke. There was no history of hemoptysis, recent surgery, or trauma. Neither blood clots nor cancer were part of her or her family’s history.
In the ED, the patient did not have any signs or symptoms of a deep venous thrombosis (DVT). She was obese, with a body mass index of 40.3 kg/m2; other vitals were: blood pressure (BP), 134/88 mm Hg; heart rate (HR), 64 beats per minute (bpm); respiratory rate (RR), 12; and O2 saturation, 99% with ambulation.
The ED physician strongly suspected a pulmonary embolism (PE), but the patient’s score on a clinical probability algorithm (using the Wells criteria) was a 3, indicating only “moderate probability“ of a PE (TABLE 1). (She scored a 3 because an “alternative diagnosis [was] less likely than PE.”) In addition, her D-dimer level was 160 ng/mL using the Triage D-Dimer Test by Biosite, Inc (normal <400 ng/mL), which ruled out a PE. (Many ED physicians at our institution are more cautious when using this D-dimer assay and use a lower cutoff value.)
Given these results, the ED physician did not order imaging studies because the expense and radiation exposure outweighed the probability of the patient having a PE. A subsequent coronary work-up was also negative. The patient was discharged to home and advised to follow up with her primary care physician a few days later.
Two days later we saw the patient at our office. Not only had her dyspnea gotten worse while the presyncope remained, but she now had left-sided pleuritic chest pain. She also reported mild pain in her right calf. On examination, the patient’s BP was 126/86 mm Hg, HR was 82 bpm, RR was 12, and O2 saturation was 96% with ambulation. Her Wells score was now 6, still a moderate probability for PE. (She received another 3 points for the new DVT symptoms—“clinically suspected DVT.”)
Although the patient did not also have signs of a DVT, her additional symptoms along with the original symptoms’ persistence and the existence of other risk factors (OCP use and obesity) led us to reconsider a PE diagnosis. These suspicions prompted us to send the patient back to the ED, where a Doppler ultrasound of the right lower extremity was negative, but the D-dimer was positive at 565 ng/mL.
A pulmonary computed tomography angiogram (CTA) showed 2 small pulmonary emboli within the distal left upper lobe pulmonary arteries.
The patient was treated with heparin and warfarin and discharged without complications.
TABLE 1
Calculating and interpreting the Wells score4,5,7,9,10
| Clinical parameter | Points |
|---|---|
| Clinically suspected DVT | 3.0 |
| Alternative diagnosis less likely than PE | 3.0 |
| Tachycardia | 1.5 |
| Immobilization/surgery (within 4 weeks) | 1.5 |
| History of DVT or PE | 1.5 |
| Hemoptysis | 1.0 |
| Malignancy (treatment within 6 months, palliative) | 1.0 |
| TOTAL | |
| Score | Traditional interpretation |
| <2.0 | Low probability of PE |
| 2.0-6.0 | Moderate probability of PE |
| >6.0 | High probability of PE |
| Score | Alternative classification scheme |
| ≤4.0 | PE unlikely |
| >4.0 | PE likely |
| DVT, deep venous thrombosis; PE, pulmonary embolism. | |
Discussion
The incidence of PE in the United States varies significantly: Individuals younger than 40 have a risk of 1 in 10,000 compared with 1 in 100 for those older than 80.1 Mortality associated with undiagnosed PE varies widely, from 9.2% to 51%.2 This percentage is significant given that half of all PEs go undiagnosed.3 In addition, when left untreated, PE will recur in 30% to 50% of patients, with a fatality rate of 10% to 45%.1 Further, up to 4% of patients with acute PE develop chronic PE and subsequent pulmonary hypertension.4,5 Given the consequences of failing to diagnose a PE, clinicians must consider this condition in patients who present with unexplained hypotension, dyspnea, or chest pain.6
Not an easy diagnosis
This case report demonstrates the inherent difficulty in diagnosing a PE. Still, certain clinical symptoms/signs can aid in the decision-making process. Fever, crackles, and wheezes decrease the probability of PE, whereas syncope, hemodynamic shock, leg edema, and hemoptysis increase its likelihood.7 Despite the many commonly reported risk factors for PE, only malignancy, recent surgery, or a history of DVT/PE significantly increase the risk of developing a clot.8
The Wells criteria. This scoring system groups patients according to the probability of having a PE: low (score: <2), moderate (score: 2-6), and high (score: >6).6 An alternative classification scheme divides patients into 2 groups: likely to have a PE (score: >4) or unlikely to have a PE (score: ≤4).8
This case report illustrates a key problem with the Wells criteria—the somewhat subjective nature of the scoring. Some physicians find it questionable to award 3 points for “alternative diagnosis less likely than PE,” for example.4 Similarly, with respect to immobilization, some clinicians might have awarded our patient 1.5 points for her recent car trip to New York. We did not think that riding in a car for 2 uninterrupted hours for each leg of the trip was significant enough. However, awarding this patient 1.5 points could have made an important difference in her clinical management if the alternative classification scheme was used. Instead of having a score of 3, the patient would have had a score of 4.5, placing her in the “likely to have a PE” group and prompting us to perform a CTA sooner (FIGURE).
FIGURE
Diagnostic algorithm for pulmonary embolism6,7,10
CTA, computed tomography angiogram; PE, pulmonary embolism.
Inappropriate work-ups are common
Some physicians ignore algorithms when working up a PE and simply order a CTA. In fact, a large multicenter trial showed that 43% of patients suspected of having a PE were inappropriately managed diagnostically.9 Similarly, a meta-analysis of 4 studies including 1660 patients found that only 58% of those with a positive D-dimer had the requisite CTA, as did 7% of patients with a negative D-dimer.2
Physicians should not be concerned about ruling out a PE in the setting of a negative D-dimer, as a meta-analysis found that this diagnostic approach has a negative predictive value (NPV) of 99.7%.2 It is important to note that the NPV is significantly affected by the sensitivity of the D-dimer assay used. If the D-dimer assay is highly sensitive, a negative result in combination with a low, moderate, or unlikely probability Wells score rules out the diagnosis of PE. If the assay is moderately sensitive, however, only a low or unlikely probability Wells score rules out PE.10
The inappropriate work-up of this group of patients is significant and extends beyond the ultimate goal of preventing morbidity and mortality. The unnecessary use of pulmonary CTA is extremely expensive, exposes patients to unnecessary radiation, and results in contrast nephrotoxicity in about 4% of patients.9 Although pulmonary CTA is the standard diagnostic test for PE, other imaging modalities are more appropriate in some cases (TABLE 2).
TABLE 2
Alternative imaging modalities for diagnosing PE1,4,7,11
| Modality | Indication |
|---|---|
| Ventilation-perfusion scanning | Patients with contrast allergies or renal failure; test of choice for diagnosing chronic PE due to limited sensitivity of CT |
| Venous compression ultrasonography | Patients with symptoms of PE and signs/symptoms of DVT |
| Pulmonary angiography | Most invasive test. Should be used only in patients with high probability of PE who may need vascular intervention |
| CT, computed tomography; DVT, deep venous thrombosis; PE, pulmonary embolism. | |
The bottom line
This case report illustrates the importance of using sound clinical judgment when diagnosing a PE. Although our patient initially had a moderate probability Wells score and a negative D-dimer, her symptoms persisted. Her history of OCP use, persistent dyspnea, and new symptoms of a DVT prompted us to reinitiate the diagnostic algorithm and eventually diagnose a PE.
It is always essential to treat the patient and not simply react to laboratory values. To avoid unnecessary testing, however, adhering to the algorithm is equally important.
CORRESPONDENCE
Michael S. Kelleher, MD, University of Connecticut School of Medicine, 263 Farmington Avenue, Farmington, CT 06030; mkelleher@student.uchc.edu
1. Meyer G, Roy PM, Gilberg S, et al. Pulmonary embolism. BMJ. 2010;340:1421.-
2. Pasha SM, Kiok FA, Snoep JD, et al. Safety of excluding acute pulmonary embolism based on an unlikely clinical probability by the Wells rule and normal D-dimer concentration: a meta-analysis. Thromb Res. 2010;125:e123-e127.
3. Taira T, Taira BR, Carmen M, et al. Risk of venous thromboembolism in patients with borderline quantitative D-dimer levels. Am J Emerg Med. 2010;28:450-453.
4. Bounameaux H, Perrier A, Righini M. Diagnosis of venous thromboembolism: an update. Vasc Med. 2010;15:399-406.
5. Pengo V, Lensing AW, Prins MH, et al. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med. 2004;350:2257-2264.
6. Agnelli G, Becattini C. Acute pulmonary embolism. N Engl J Med. 2010;363:266-274.
7. Gandara E, Wells PS. Diagnosis: use of clinical probability algorithms. Clin Chest Med. 2010;31:629-639.
8. Drescher FS, Chandrika S, Weir ID, et al. Effectiveness and acceptability of a computerized decision support system using modified wells criteria for evaluation of suspected pulmonary embolism. Ann Emerg Med. 2011;57:613-621.
9. Gimber LH, Travis RI, Takahashi JM, et al. Computed tomography angiography in patients evaluated for acute pulmonary embolism with low serum D-dimer levels: a prospective study. Perm J. 2009;13:4-10.
10. Agency for Healthcare Research and Quality. Guidelines on the diagnosis and management of acute pulmonary embolism. Available at: http://www.guideline.gov/content.aspx?id=13410#Section420. Accessed February 12, 2011.
11. Kim NH. Chronic thromboembolic pulmonary hypertension: diagnosis. Medscape. Available at: http://www.medscape.org/viewarticle/556058_3. Accessed May 9, 2011.
1. Meyer G, Roy PM, Gilberg S, et al. Pulmonary embolism. BMJ. 2010;340:1421.-
2. Pasha SM, Kiok FA, Snoep JD, et al. Safety of excluding acute pulmonary embolism based on an unlikely clinical probability by the Wells rule and normal D-dimer concentration: a meta-analysis. Thromb Res. 2010;125:e123-e127.
3. Taira T, Taira BR, Carmen M, et al. Risk of venous thromboembolism in patients with borderline quantitative D-dimer levels. Am J Emerg Med. 2010;28:450-453.
4. Bounameaux H, Perrier A, Righini M. Diagnosis of venous thromboembolism: an update. Vasc Med. 2010;15:399-406.
5. Pengo V, Lensing AW, Prins MH, et al. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med. 2004;350:2257-2264.
6. Agnelli G, Becattini C. Acute pulmonary embolism. N Engl J Med. 2010;363:266-274.
7. Gandara E, Wells PS. Diagnosis: use of clinical probability algorithms. Clin Chest Med. 2010;31:629-639.
8. Drescher FS, Chandrika S, Weir ID, et al. Effectiveness and acceptability of a computerized decision support system using modified wells criteria for evaluation of suspected pulmonary embolism. Ann Emerg Med. 2011;57:613-621.
9. Gimber LH, Travis RI, Takahashi JM, et al. Computed tomography angiography in patients evaluated for acute pulmonary embolism with low serum D-dimer levels: a prospective study. Perm J. 2009;13:4-10.
10. Agency for Healthcare Research and Quality. Guidelines on the diagnosis and management of acute pulmonary embolism. Available at: http://www.guideline.gov/content.aspx?id=13410#Section420. Accessed February 12, 2011.
11. Kim NH. Chronic thromboembolic pulmonary hypertension: diagnosis. Medscape. Available at: http://www.medscape.org/viewarticle/556058_3. Accessed May 9, 2011.
Hyperthyroidism: A stepwise approach to management
• Measure TSH in any patient >60 years presenting with fatigue, atrial fibrillation, weight loss, and shortness of breath. B
• Achieve faster control of symptoms in elderly patients and those with cardiac disease by pursuing the ablative method with radioactive iodine (RAI). This method is also recommended for patients with toxic multinodular goiter and toxic adenoma. A
• Initiate steroid prophylaxis for patients with Graves’ ophthalmopathy undergoing RAI. A
• Opt for a 12- to 18-month course of an antithyroid drug, rather than a 6-month course. The longer course is associated with a lower relapse rate. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 72-year-old man arrives at the clinic with insomnia and fatigue. His medical history is significant for hypertension, hyperlipidemia, and degenerative joint disease, for which he is taking, respectively, metoprolol 25 mg twice daily, simvastatin 20 mg daily, and acetaminophen as needed for joint pain. He has experienced no weight loss, anxiety, or gastrointestinal or urinary symptoms. He does not smoke or drink alcohol. His blood pressure is 140/75 mm Hg, pulse is 85, respiratory rate is 20, and temperature is 97.1°F. The rest of the physical examination is unremarkable except for 1+ lower extremity edema, unchanged since his previous visit. Routine blood work, however, reveals his thyroid-stimulating hormone (TSH) level to be 0.03 mIU/L.
Clues from the clinical presentation
The subtle, "apathetic presentation" with few symptoms, as described in the case above, is typical of older individuals with hyperthyroidism.1 In contrast, younger patients with hyperthyroidism and those with comorbidities can manifest a number of signs and symptoms (TABLE 1).2
Graves’ disease, the most common cause of hyperthyroidism,3 causes such ocular disturbances as exophthalmos, lid lag, lid retraction, and proptosis in 60% of patients with the condition.3 These findings help differentiate Graves’ disease from other causes of hyperthyroidism. (See “Common [and not so common] causes of hyperthyroidism”.) Palmar sweating, pretibial myxedema, and Plummer’s nails (onycholysis) are also unique for Graves’ disease.4
When you suspect hyperthyroidism, assess the thyroid for size, nodularity, and vascularity. Goiter is less prevalent in the elderly, occurring in less than 50% of patients 61 and older, compared with 77% of patients younger than 60 years.5 Diffuse goiter is typical with Graves’ disease, while a mass with multiple nodules suggests possible toxic multinodular goiter. A solitary palpable nodule could mean toxic adenoma. A thyroid that is tender on palpation may point to subacute thyroiditis, particularly if the patient has had a viral illness recently (TABLE 2).
Measuring a patient’s TSH level is warranted with the above findings. Additionally, measure TSH in any patient older than 60 years presenting with fatigue, atrial fibrillation, weight loss, and shortness of breath.5
TABLE 1
Clinical manifestations of hyperthyroidism2
| Acropachy (swelling of the fingers) |
| Bruit (thyroid) |
| Decreased attention span |
| Diarrhea |
| Edema |
| Exertional dyspnea |
| Fatigue |
| Goiter (smooth or nodular) |
| Gynecomastia |
| Hair loss |
| Heat intolerance |
| Hyperactive deep tendon reflex |
| Hypertension |
| Increased appetite |
| Infertility |
| Insomnia |
| Lid lag, proptosis |
| Muscle weakness |
| Nervousness and irritability |
| Oligomenorrhea |
| Palmar erythema |
| Palpitations |
| Paralysis (sudden) |
| Photophobia, eye irritation, diplopia |
| Pretibial myxedema |
| Tachycardia |
| Tremors |
| Warm, moist skin |
| Weight loss |
Graves’ disease—an autoimmune disorder in which antibodies target thyroid tissue and enzymes and activate thyroid hormone synthesis—affects more than 3 million people in the United States and accounts for 60% of hyperthyroidism cases.3 Remission does occur; however, the recurrence rate is as high as 60%.50 Factors associated with recurrence include tobacco use; male sex; young age; large goiter size or increase in goiter size during treatment; elevated TSH receptor antibodies (TRab); presence of Graves’ ophthalmopathy; markedly elevated thyroid hormones, or delayed treatment.51
Toxic multinodular goiter, also known as Plummer’s disease, is the underlying condition in 15% to 20% of hyperthyroidism cases; it is more common in young patients and in iodine-deficient locations (eg, Denmark).52 However, it also occurs in elderly patients with longstanding goiter.
Toxic adenoma causes just 3% to 5% of cases of hyperthyroidism.53 It, too, occurs more commonly in young patients and in iodine-deficient regions. The radioactive iodine uptake test shows a hot nodule, with suppressed uptake in the surrounding thyroid gland.
Subacute thyroiditis, also known as de Quervain’s thyroiditis, is the reason for 15% to 20% of hyperthyroidism cases; it is usually preceded by viral infection and inflammation that lead to destructive release of preformed thyroid hormone. Symptoms—typically fever, malaise, and tender goiter—usually occur more abruptly than symptoms of Graves’ disease.54 Most cases resolve spontaneously within a few months, and relapse is less common than in Graves’ disease. Other lab abnormalities include increased erythrocyte sedimentation rate and low radioiodine uptake.
Postpartum thyroiditis is an autoimmune disease. Prevalence ranges from 1% to 17% of new mothers.55 It is characterized by a thyroid gland that is painless on palpation and low radioiodine uptake.56 Most cases are reversible with treatment.
Factitious or iatrogenic hyperthyroidism is due to an exogenous intake of thyroid hormone, and typically exhibits a normal or low radioactive iodine uptake and a low thyroglobulin level.
Secondary hyperthyroidism, or TSH-mediated hyperthyroidism, is rare. It is always associated with goiter, and approximately 40% of patients have visual field defects.57
TABLE 2
Clinical and laboratory findings associated with common causes of hyperthyroidism51-57
| Mechanism | Thyroid exam | Lab results | Radioactive iodine uptake | |
|---|---|---|---|---|
| Graves’ disease | Antithyroid antibodies | Diffuse goiter | Low TSH; elevated T3 and/or T4; elevated thyroid antibodies | Diffusely increased |
| Toxic multinodular goiter | Iodine deficiency | Goiter with multiple nodules | Low TSH; elevated T3 and/or T4 | Normal/increased uptake; "hot nodules" with suppression of extranodular tissue |
| Toxic adenoma | Benign thyroid hormone?secreting tumor; iodine deficiency | Palpable nodule | Low TSH; elevated T3 and/or T4 | Normal/increased uptake; functioning "hot nodule" on scan with suppression of surrounding thyroid tissue |
| Subacute thyroiditis | Viral | Tender thyroid on palpation | Low TSH; elevated T3 and/or T4; elevated ESR; elevated thyroid antibodies | Low uptake with poor imaging of the thyroid on scan |
| Factitious hyperthyroidism | Excessive intake of exogenous thyroid hormone | Normal exam | Low TSH; elevated T3 and/or T4; low thyroglobulin level | Low or normal uptake |
| Secondary hyperthyroidism | Excessive pituitary TSH | Goiter | Elevated TSH; elevated T3 and/or T4 | Diffusely increased uptake |
| ESR, erythrocyte sedimentation rate; T3, triiodothyronine; T4, thyroxine; TSH, thyroid-stimulating hormone. | ||||
Which laboratory tests to order, and what the results may mean
Rely on second- or third-generation TSH screening (normal=0.5-5 mIU/L), which is more sensitive and specific than measuring free T4 (thyroxine) alone.6
Older patients usually have a higher normal TSH level. In one study, 70% of patients >80 years had a TSH >4.5 mIU/L.7
If the TSH level is low (<0.5 mIU/L), measure free T3 (triiodothyronine) and free T4 levels, which are elevated in hyperthyroidism, and are normal in subclinical hyperthyroidism.
Patients with Graves’ disease tend to have T3 thyrotoxicosis with a T3T4 ratio >20.8 Isolated T4 thyrotoxicosis is more commonly seen with nonthyroidal illness as a result of decreased conversion from T4 to T3, and also in amiodarone-induced hyperthyroidism.9
When further testing is needed (FIGURE). If the underlying cause of hyperthyroidism is not established on the basis of clinical findings (eg, diffuse goiter, myxedema, ophthalmopathy), order a 24-hour radioactive iodine (RAI) uptake test.10 Graves’ disease and toxic multinodular goiter exhibit increased RAI uptake that is diffuse and nodular, respectively. Subacute thyroiditis is associated with low RAI uptake (TABLE 2).
If RAI is contraindicated—eg, in pregnancy—testing for elevated levels of thyroid peroxidase antibodies (TPOab), TSH receptor antibodies (TRab), and thyroglobulin may help to differentiate Graves’ disease from multinodular goiter or uncover another autoimmune thyroid disorder.11 If a patient’s TSH, T4, and T3 levels are all elevated, refer him or her for magnetic resonance imaging of the pituitary gland to look for a TSH-secreting adenoma.
FIGURE
Suspect hyperthyroidism? Order these tests6-11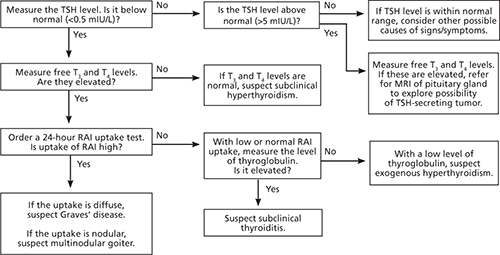
MRI, magnetic resonance imaging; RAI, radioactive iodine; T3, triiodothyronine; T4, thyroxine; TSH, thyroid-stimulating hormone.
Matching treatment to the underlying cause
RAI is usually the treatment of choice for patients without contraindications, although no randomized clinical trials have compared it with antithyroid medications or surgery.12 Each modality has its own risks and benefits (TABLE 3), and treatment selection should be individualized.
TABLE 3
Comparison of treatment modalities for hyperthyroidism12-21
| Antithyroid drugs* | Radioactive iodine* | Surgery | |
|---|---|---|---|
| Recurrence rate | High recurrence rate; no permanent hypothyroidism | Usually permanent hypothyroidism; long-term use of levothyroxine is required | Subtotal thyroidectomy associated with higher rates of recurrence or persistence of hyperthyroidism than total thyroidectomy; permanent hypothyroidism; long-term use of levothyroxine is required |
| Preferred method within treatment modality | MMI is the preferred medication; PTU is used with pregnancy and severe hyperthyroidism not responding to MMI | High ablative dose is preferred in MNG, toxic nodule, cardiac disease, elderly; low calculated dose is preferred in patients with GO | No outcome differences for GO, whether thyroidectomy is total, bilateral subtotal, or unilateral total and contralateral subtotal |
| Setting | Outpatient | Outpatient | Inpatient |
| Risks | No surgical risks | No surgical risks | Reaction to anesthesia, recurrent laryngeal nerve palsy, hypoparathyroidism |
| Adverse effects | ATD adverse effects, including life-threatening agranulocytosis | Worsening of Graves’ ophthalmopathy; transient exacerbation of hyperthyroid symptoms | Permanent hypothyroidism; hypoparathyroidism; anesthesia complications |
| Safety in pregnancy | PTU is used in pregnancy | Contraindicated in pregnancy/lactation | If surgery is indicated in pregnancy, it is best performed in the second trimester |
| ATD, antithyroid drugs; GO, Graves’ ophthalmopathy; MMI, methimazole; MNG, toxic multinodular goiter; PTU, propylthiouracil. *Concomitant use of ATD and RAI is associated with a high failure rate and persistent or recurrent hyperthyroidism. Discontinue ATD 2 weeks before radioactive iodine treatment. | |||
Radioactive iodine
In a 1990 survey, as many as 70% of specialists in the United States used RAI to treat hyperthyroidism, compared with just 22% of specialists in Europe.13 RAI is usually given in a single dose, and its maximal benefit is noted within 3 to 6 months. Two treatment methods are available: the ablative method and the gland-specific dosing method. Both have similar euthyroid state outcomes.14
The ablative method uses a high dose of RAI to achieve permanent hypothyroidism, necessitating lifelong levothyroxine replacement. This method is preferred for the elderly and for patients with cardiac disease, to achieve faster control of symptoms. It is also recommended for patients with toxic multinodular goiter and toxic nodules.
The gland-specific dosing method induces a euthyroid state with a calculated low dose of RAI based on the estimated weight of the patient’s thyroid. The optimal dosage may be difficult to calculate, but it is usually the preferred method for patients with Graves’ ophthalmopathy.
Adverse effects of RAI can include worsening of Graves’ ophthalmopathy and an acute rise in thyroid hormone that increases hyperthyroid symptoms or even causes a thyroid storm associated with increased cardiovascular risk.2 A negative pregnancy test result is a prerequisite for all women of childbearing age before taking RAI, and patients are advised to use contraception for 6 months after RAI administration.
Although RAI is often the initial treatment for hyperthyroidism, in some instances—eg, for older patients with comorbidities—pre-treatment with antithyroid drugs (ATD) is indicated to avoid transient worsening of hyperthyroid symptoms after RAI. However, always discontinue ATD 2 weeks before RAI administration; concomitant use is associated with a higher failure rate and persistent or recurrent hyperthyroidism.15
Antithyroid drugs
Two antithyroid medications are available for use in the United States: propylthiouracil (PTU) and methimazole (MMI). In the United Kingdom, carbimazole is also available.
MMI is the drug of choice.16 Compared with PTU, MMI costs less, has a longer half-life, and causes fewer adverse effects. A starting dose of 15 mg per day for MMI is suitable for mild and moderate hyperthyroidism. For more severe cases, 30 mg per day is the recommended starting dose.16 Reserve PTU for treating hyperthyroidism in pregnancy, during which MMI should be avoided, if possible.
Allergic reactions to ATDs appear in around 5% of patients and usually occur in the first 6 weeks of treatment.17 Agranulocytosis is the main concern, although it occurs in fewer than 1% of patients17 and is reversible by stopping the medication. Measure the leukocyte count 1 week after initiation of treatment and repeat the measurement at 1-month intervals.
Two methods are used to dose these medications: titration and block-and-replace. Titration is as effective as the block-and-replace method and is associated with fewer rashes (6% vs 10% of patients) and less agranulocytosis (0.4 % vs 1.4%). The 2 methods have similar relapse rates (around 50%).18
With titration, MMI is started at a dose of 15 mg per day and titrated upward to the lowest effective dose. Treatment for 12 to 18 months is associated with a lower relapse rate than treatment for 6 months (37% vs 58%).19
The block-and-replace method uses persistently high ATD doses in combination with L-thyroxin replacement to avoid hypothyroidism (MMI 30 mg and levothyroxine 80 mcg).
To monitor effectiveness initially, measure free T4 and T3 levels, because TSH concentration changes slowly and may stay low for a few months. Response to treatment is often temporary.8 More definitive treatment with RAI or surgery is usually necessary.
Surgery
Thyroidectomy creates permanent hypothyroidism, necessitating lifelong thyroxine replacement. In the United States, surgical intervention is reserved for special situations, such as pregnant women with severe disease who are allergic or not responding to antithyroid medications, removal of a clinically suspicious thyroid nodule coexisting with hyperthyroidism, or severe or recurrent Graves’ disease with severe ophthalmopathy.20 Surgical options are total or subtotal thyroidectomy. Hyperthyroidism persists or recurs in 8% of patients with subtotal thyroidectomy.21 Potential complications of thyroidectomy include adverse effects of anesthesia, hypoparathyroidism, and vocal cord paralysis.
Other treatment options
Iodides
Iodides inhibit thyroid hormone release and block conversion of T4 to T3. Use potassium iodide only in combination with ATDs, for patients with severe thyrotoxicosis or as pretreatment for urgent thyroidectomy in patients with Graves’ disease. It has been shown to improve the short-term control of Graves’ hyperthyroidism and is not associated with worsening hyperthyroidism;22 however, potassium iodide should not be used for more than 12 weeks as it can cause paradoxical hyperthyroidism.22
Beta-blockers
Hyperthyroidism is associated with an increased number of beta-adrenergic receptors,23 which explains the symptoms of palpitations, anxiety, and tremors. Nonselective beta-blockers are usually preferred for symptomatic treatment of hyperthyroid symptoms, and propranolol is the most widely used agent.24 If you decide to use a beta-blocker, start it with the ATD and continue it until the patient becomes euthyroid or asymptomatic, then taper it over a period of 4 to 6 weeks. Symptoms may persist, however, and require higher doses of propranolol (80-320 mg/d) given more frequently.
Treating Graves’ ophthalmopathy
Exophthalmos and other eye signs are the hallmark of Graves’ disease and may occur in the absence of hyperthyroidism. Smoking is a significant risk factor for developing ophthalmopathy due to increased orbital connective tissue volume,25 and smoking cessation is recommended.26
Using RAI to treat Graves’ disease increases the risk that ophthalmopathy will develop or worsen. Worsening of Graves’ ophthalmopathy secondary to RAI treatment occurs in 20% of treated patients (transient in 15%; permanent in 5%).27 Steroid prophylaxis is beneficial for patients with ophthalmopathy,28 and prednisone 40 to 80 mg per day tapered over at least 3 months can help reduce the condition.19 In patients with moderate to severe active ophthalmopathy, intravenous corticosteroid therapy has a small but statistically significant advantage over oral therapy and causes significantly fewer adverse events.29
Orbital radiotherapy is also used, and has been shown to decrease diplopia.30 However, the best available evidence recommends combining orbital radiotherapy and oral corticosteroids, which yields efficacy beyond that achievable with either radiotherapy or oral corticosteroids alone.16 Moreover, intravenous methylprednisolone combined with orbital radiotherapy seems to be most efficacious.31 The course of ophthalmopathy is the same whether total or subtotal thyroidectomy is used.32
Prognosis without treatment
Individuals with high-normal thyroid function tests, subclinical hyperthyroidism, and clinical hyperthyroidism are at increased risk for atrial fibrillation.33-35 Hyperthyroidism is also associated with increased risk of heart failure (6% of patients), which might be secondary to coexisting atrial fibrillation or tachycardia-mediated cardiomyopathy.36 Heart failure is usually reversible when the hyperthyroidism is treated.
Patients with overt hyperthyroidism are also at risk for pulmonary hypertension secondary to increased cardiac output and decreased pulmonary vascular resistance.37
In patients with preexisting cardiac disease, hyperthyroidism increases risk of death (hazard ratio [HR]=1.57),38 and might even do so in patients without cardiac disease.39,40 It also increases risk of ischemic stroke (HR=1.44) among adults ages 18 to 44 years.41 Untreated hyperthyroidism also contributes to low bone mineral density and increases the risk of hip fracture.42
Subclinical hyperthyroidism
Subclinical hyperthyroidism occurs in 2% of the US population and is characterized by low serum TSH (<0.1 mIU/L) with normal levels of free T3 and free T4. The causes are similar to overt hyperthyroidism. In addition, it can result from overtreating hypothyroidism with thyroid hormone, thereby inducing a subclinical hyperthyroid state.
The most common endogenous cause of subclinical hyperthyroidism (~60% of patients) is multinodular goiter.43 Subclinical hyperthyroidism carries significant health risks, and yet evidence is lacking on when to treat this condition. Prolonged subclinical hyperthyroidism can lead to atrial fibrillation,24,44 and to systolic and diastolic cardiac dysfunction.45 Subclinical hyperthyroidism is also associated with decreased bone density,46 and an increased risk of dementia.47
The American Association of Clinical Endocrinologists recommends periodic clinical and laboratory assessment for patients with subclinical hyperthyroidism (TSH=0.1-0.5 mIU/mL), including rechecking TSH, free T3 and free T4 at 2- to 4-month intervals.
Treatment of the underlying cause of hyperthyroidism is indicated if serum TSH is <0.1 mIU/mL.
For patients older than 65 years who have persistent subclinical hyperthyroidism, consider treatment in the following scenarios:48
- nodular thyroid disease (due to high conversion rate to overt hyperthyroidism)
- osteopenia or osteoporosis (in women)
- atrial fibrillation
- underlying cardiac disease.
Hyperthyroidism in pregnancy
PTU is the first choice for treating hyperthyroidism in pregnancy. It crosses the placenta less readily than MMI, and is thus less likely to cause fetal hypothyroidism. Additionally, MMI is associated with increased risk of fetal anomalies, such as aplasia cutis and esophageal atresia. MMI may be considered if the patient is intolerant to PTU or fails to become euthyroid while receiving PTU.49 Use the lowest possible dose of either PTU or MMI to maintain thyroid function within the upper limit of normal. The dose of the antithyroid medication is usually decreased as pregnancy progresses and discontinued in the last few weeks, as pregnancy is thought to improve the course of Graves’ disease.
The use of RAI is contraindicated during pregnancy and breastfeeding. Hyperthyroidism symptoms usually resolve after delivery. If symptoms persist, however, the treatment of choice is ATD. Surgery is an option in severe Graves’ disease not responding to ATD.
CORRESPONDENCE
Abdulraouf Ghandour, MD, Department of Family and Community Medicine, University of Missouri-Columbia, One Hospital Drive, Columbia, MO 65212; ghandoura@health.missouri.edu
1. Levy EG. Thyroid disease in the elderly. Med Clin North Am. 1991;75:151-167.
2. Cooper DS. Hyperthyroidism. Lancet. 2003;362:459-468.
3. Weetman AP. Graves’ disease. N Engl J Med. 2000;343:1236-1248.
4. Heymann WR. Cutaneous manifestations of thyroid disease. J Am Acad Dermatol. 1992;26:885-902.
5. Boelaert K, Torlinska B. Older subjects with hyperthyroidism present with a paucity of symptoms and signs: a large cross-sectional study. J Clin Endocrinol Metab. 2010;95:2715-2726.
6. Danese MD, Powe NR, Sawin CT, et al. Screening of mild thyroid failure at the periodic health examination: a decision and cost-effectiveness analysis. JAMA. 1996;276:285-292.
7. Surks MI, Hollowell JG. Age-specific distribution of serum thyrotropin and antithyroid antibodies in the US population: implications for the prevalence of subclinical hypothyroidism. J Clin Endocrinol Metab. 2007;92:4575-4582.
8. Amino N, Yabu Y, Miki T, et al. Serum ratio of triiodothyronine to thyroxine and thyroxine binding globulin and calcitonin concentrations in Graves’ disease and destruction-induced thyrotoxicosis. J Clin Endocrinol Metab. 1981;53:113-116.
9. Bambini G, Aghini-Lombardi F, Rosner W, et al. Serum sex hormone-binding globulin in amiodarone-treated patients. A marker for tissue thyrotoxicosis. Arch Intern Med. 1987;147:1781-1785.
10. Fogelman I, Cooke SG, Maisey MN. The role of thyroid scanning in hyperthyroidism. Eur J Nucl Med. 1986;11:397-400.
11. Costagliola S, Morgenthaler NG, Hoermann R, et al. Second generation assay for thyrotropin receptor antibodies has superior diagnostic sensitivity for Graves’ disease. J Clin Endocrinol Metab. 1999;84:90-97.
12. Streetman DD, Khanderia U. Diagnosis and treatment of Graves’ disease. Ann Pharmacother. 2003;37:1100-1109.
13. Wartofsky L, Glinoer D, Solomon B, et al. Differences and similarities in the diagnosis and treatment of Graves’ disease in Europe, Japan, and the United States. Thyroid. 1991;1:129-135.
14. de Rooij A, Vandenbroucke JP. Clinical outcomes after estimated versus calculated activity of radioiodine for the treatment of hyperthyroidism: systematic review and meta-analysis. Eur J Endocrinol. 2009;161:771-777.
15. Walter MA, Briel M, Christ-Crain M, et al. Effects of antithyroid drugs on radioiodine treatment: systematic review and meta-analysis of randomised controlled trials. BMJ. 2007;334:514.-
16. Nakamura H, Noh JY. Comparison of methimazole and propylthiouracil in patients with hyperthyroidism caused by Graves’ disease. J Clin Endocrinol Metab. 2007;92:2157-2162.
17. Cooper DS. Antithyroid drugs. N Engl J Med. 2005;352:905-917.
18. Abraham P, Avenell A. A systematic review of drug therapy for Graves’ hyperthyroidism. Eur J Endocrinol. 2005;153:489-498.
19. Abraham P, Avenell A, McGeoch SC, et al. Antithyroid drug regimen for treating Graves’ hyperthyroidism. Cochrane Database Sys Rev. 2010;(1):CD003420.-
20. Stalberg P, Svensson A. Surgical treatment of Graves’ disease: evidence-based approach. World J Surg. 2008;32:1269-1277.
21. Palit TK, Miller CC, Miltenburg DM. The efficacy of thyroidectomy for Graves’ disease: a meta-analysis. J Surg Res. 2000;90:161-165.
22. Takata K, Amino N, Kubota S. Benefit of short-term iodide supplementation to antithyroid drug treatment of thyrotoxicosis due to Graves’ disease. Clin Endocrinol. 2010;72:845-850.
23. Bilezikian JP, Loeb JN. The influence of hyperthyroidism and hypothyroidism on alpha- and beta-adrenergic receptor systems and adrenergic responsiveness. Endocr Rev. 1983;4:378-388.
24. Jansson S, Lie-Karlsen K, Stenqvist O, et al. Oxygen consumption in patients with hyperthyroidism before and after treatment with beta-blockade versus thyrostatic treatment: a prospective randomized study. Ann Surg. 2001;233:60-64.
25. Zucs-Frkas Z, Toth J, Kollar J, et al. Volume changes in intra- and extraorbital compartments in patients with Graves’ ophthalmopathy: effect of smoking. Thyroid. 2005;15:146-151.
26. Träisk F, Tallstedt L. Thyroid-associated ophthalmopathy after treatment for Graves’ hyperthyroidism with antithyroid drugs or iodine-131. J Clin Endocrinol Metab. 2009;94:3700-3707.
27. Bartalena L, Marcocci C, Bogazzi F, et al. Relation between therapy of hyperthyroidism and the course of Graves’ ophthalmopathy. N Engl J Med. 1998;338:73-78.
28. Acharya SH, Avenell A. Radioiodine therapy (RAI) for Graves’ disease (GD) and the effect on ophthalmopathy: a systematic review. Clin Endocrinol (Oxf). 2008;69:943-950.
29. Stiebel-Kalish H, Robenshtok E. Treatment modalities for Graves’ ophthalmopathy: systematic review and meta-analysis. J Clin Endocrinol Metab. 2009;94:2708-2716.
30. Bradley EA, Gower EW. Orbital radiation for graves ophthalmopathy: a report by the American Academy of Ophthalmology. Ophthalmology. 2008;115:398-409.
31. Wei RL, Cheng JW. The use of orbital radiotherapy for Graves’ ophthalmopathy: quantitative review of the evidence. Ophthalmologica. 2008;222:27-31.
32. Witte J, Goretzki PE, Dotzenrath C, et al. Surgery for Graves’ disease: total versus subtotal thyroidectomy–result of a prospective randomized trial. World J Surg. 2000;24:1303-1311.
33. Heeringa J, Hoogendoorn EH. High-normal thyroid function and risk of atrial fibrillation: the Rotterdam study. Arch Intern Med. 2008;168:2219-2224.
34. Sawin CT, Geller A, Wolf PA, et al. Low serum thyrotropin concentrations as a risk factor for atrial fibrillation in older persons. N Engl J Med. 1994;331:1249-1252.
35. Cappola AR, Fried LP. Thyroid status, cardiovascular risk, and mortality in older adults. JAMA. 2006;295:1033-1041.
36. Siu CW, Yeung CY, Lau CP, et al. Incidence, clinical characteristics and outcome of congestive heart failure as the initial presentation in patients with primary hyperthyroidism. Heart. 2007;93:483-487.
37. Lozano HF, Sharma CN. Reversible pulmonary hypertension, tricuspid regurgitation and right-sided heart failure associated with hyperthyroidism: case report and review of the literature. Cardiol Rev. 2004;12:299-305.
38. Iervasi G, Molinaro S. Association between increased mortality and mild thyroid dysfunction in cardiac patients. Arch Intern Med. 2007;167:1526-1532.
39. Parle JV, Maisonneuve P, Sheppard MC, et al. Prediction of all-cause and cardiovascular mortality in elderly people from one low serum thyrotropin result: a 10-year cohort study. Lancet. 2001;358:861-865.
40. Flynn RW, McDonald TM, Jung RT, et al. Mortality and vascular outcomes in patients treated for thyroid dysfunction. J Clin Endocrinol Metab. 2006;91:2169-2164.
41. Sheu JJ, Kang JH. Hyperthyroidism and risk of ischemic stroke in young adults: a 5-year follow-up study. Stroke. 2010;41:961-966.
42. Vestergaard P, Mosekilde L. Hyperthyroidism, bone mineral, and fracture risk—a meta-analysis. Thyroid. 2003;13:585-593.
43. Diez JJ. Hyperthyroidism in patients older than 55 years: an analysis of the etiology and management. Gerontology. 2003;49:316-323.
44. Sawin CT, Geller A, Wolf PA, et al. Low serum thyrotropin concentrations as a risk factor for atrial fibrillation in older persons. N Engl J Med. 1994;331:1249-1252.
45. Abdulrahman RM, Delgado V. Abnormal cardiac contractility in long-term exogenous subclinical hyperthyroid patients as demonstrated by two-dimensional echocardiography speckle tracking imaging. Eur J Endocrinol. 2010;163:435-441.
46. Faber J, Jensen IW, Petersen L, et al. Normalization of serum thyrotrophin by means of radioiodine treatment in subclinical hyperthyroidism: effect on bone loss in postmenopausal women. Clin Endocrinol (Oxf). 1998;48:285-290.
47. Tan ZS, Beiser A, Vasan RS, et al. Thyroid function and the risk of Alzheimer disease: Framingham study. Arch Intern Med. 2008;168:1514-1520.
48. American Association of Clinical Endocrinologists. Medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. 2006. Available at: https://www.aace.com/sites/default/files/hypo_hyper.pdf. Accessed July 9, 2010.
49. Chattaway JM, Klepser TB. Propylthiouracil versus methimazole in treatment of Grave’s disease during pregnancy. Ann Pharmacother. 2007;41:1018-1022.
50. Lucas A, Salinas I. Medical therapy of Graves’ disease: does thyroxine prevent recurrence of hyperthyroidism? J Clin Endocrinol Metab. 1997;82:2410-2413.
51. Vitti P, Rago T, Chiovato L, et al. Clinical features of patients with Graves’ disease undergoing remission after antithyroid drug treatment. Thyroid. 1997;7:369-375.
52. Laurberg P, Bulow Pedersen I, Pedersen KM, et al. Low incidence rate of overt hypothyroidism compared with hyperthyroidism in an area with moderately low iodine intake. Thyroid. 1999;9:33-38.
53. Siegel RD, Lee SL. Toxic nodular goiter: Toxic adenoma and toxic multinodular goiter. Endocrinol Metab Clin North Am. 1998;27:151-168.
54. Volpe R. Subacute (de Quervain’s) thyroiditis. Clin Endocrinol Metab. 1979;8:81-95.
55. Nicholson WK, Robinson KA, Smallridge RC, et al. Prevalence of postpartum thyroid dysfunction: a quantitative review. Thyroid. 2006;16:573-582.
56. Roti E, Emerson CH. Clinical review 29: postpartum thyroiditis. J Clin Endocrinol Metab. 1992;74:3-5.
57. Beck-Peccoz P, Brucker-Davis F, Persani L, et al. Thyrotropin-secreting pituitary tumors. Endocr Rev. 1996;17:610-638.
• Measure TSH in any patient >60 years presenting with fatigue, atrial fibrillation, weight loss, and shortness of breath. B
• Achieve faster control of symptoms in elderly patients and those with cardiac disease by pursuing the ablative method with radioactive iodine (RAI). This method is also recommended for patients with toxic multinodular goiter and toxic adenoma. A
• Initiate steroid prophylaxis for patients with Graves’ ophthalmopathy undergoing RAI. A
• Opt for a 12- to 18-month course of an antithyroid drug, rather than a 6-month course. The longer course is associated with a lower relapse rate. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 72-year-old man arrives at the clinic with insomnia and fatigue. His medical history is significant for hypertension, hyperlipidemia, and degenerative joint disease, for which he is taking, respectively, metoprolol 25 mg twice daily, simvastatin 20 mg daily, and acetaminophen as needed for joint pain. He has experienced no weight loss, anxiety, or gastrointestinal or urinary symptoms. He does not smoke or drink alcohol. His blood pressure is 140/75 mm Hg, pulse is 85, respiratory rate is 20, and temperature is 97.1°F. The rest of the physical examination is unremarkable except for 1+ lower extremity edema, unchanged since his previous visit. Routine blood work, however, reveals his thyroid-stimulating hormone (TSH) level to be 0.03 mIU/L.
Clues from the clinical presentation
The subtle, "apathetic presentation" with few symptoms, as described in the case above, is typical of older individuals with hyperthyroidism.1 In contrast, younger patients with hyperthyroidism and those with comorbidities can manifest a number of signs and symptoms (TABLE 1).2
Graves’ disease, the most common cause of hyperthyroidism,3 causes such ocular disturbances as exophthalmos, lid lag, lid retraction, and proptosis in 60% of patients with the condition.3 These findings help differentiate Graves’ disease from other causes of hyperthyroidism. (See “Common [and not so common] causes of hyperthyroidism”.) Palmar sweating, pretibial myxedema, and Plummer’s nails (onycholysis) are also unique for Graves’ disease.4
When you suspect hyperthyroidism, assess the thyroid for size, nodularity, and vascularity. Goiter is less prevalent in the elderly, occurring in less than 50% of patients 61 and older, compared with 77% of patients younger than 60 years.5 Diffuse goiter is typical with Graves’ disease, while a mass with multiple nodules suggests possible toxic multinodular goiter. A solitary palpable nodule could mean toxic adenoma. A thyroid that is tender on palpation may point to subacute thyroiditis, particularly if the patient has had a viral illness recently (TABLE 2).
Measuring a patient’s TSH level is warranted with the above findings. Additionally, measure TSH in any patient older than 60 years presenting with fatigue, atrial fibrillation, weight loss, and shortness of breath.5
TABLE 1
Clinical manifestations of hyperthyroidism2
| Acropachy (swelling of the fingers) |
| Bruit (thyroid) |
| Decreased attention span |
| Diarrhea |
| Edema |
| Exertional dyspnea |
| Fatigue |
| Goiter (smooth or nodular) |
| Gynecomastia |
| Hair loss |
| Heat intolerance |
| Hyperactive deep tendon reflex |
| Hypertension |
| Increased appetite |
| Infertility |
| Insomnia |
| Lid lag, proptosis |
| Muscle weakness |
| Nervousness and irritability |
| Oligomenorrhea |
| Palmar erythema |
| Palpitations |
| Paralysis (sudden) |
| Photophobia, eye irritation, diplopia |
| Pretibial myxedema |
| Tachycardia |
| Tremors |
| Warm, moist skin |
| Weight loss |
Graves’ disease—an autoimmune disorder in which antibodies target thyroid tissue and enzymes and activate thyroid hormone synthesis—affects more than 3 million people in the United States and accounts for 60% of hyperthyroidism cases.3 Remission does occur; however, the recurrence rate is as high as 60%.50 Factors associated with recurrence include tobacco use; male sex; young age; large goiter size or increase in goiter size during treatment; elevated TSH receptor antibodies (TRab); presence of Graves’ ophthalmopathy; markedly elevated thyroid hormones, or delayed treatment.51
Toxic multinodular goiter, also known as Plummer’s disease, is the underlying condition in 15% to 20% of hyperthyroidism cases; it is more common in young patients and in iodine-deficient locations (eg, Denmark).52 However, it also occurs in elderly patients with longstanding goiter.
Toxic adenoma causes just 3% to 5% of cases of hyperthyroidism.53 It, too, occurs more commonly in young patients and in iodine-deficient regions. The radioactive iodine uptake test shows a hot nodule, with suppressed uptake in the surrounding thyroid gland.
Subacute thyroiditis, also known as de Quervain’s thyroiditis, is the reason for 15% to 20% of hyperthyroidism cases; it is usually preceded by viral infection and inflammation that lead to destructive release of preformed thyroid hormone. Symptoms—typically fever, malaise, and tender goiter—usually occur more abruptly than symptoms of Graves’ disease.54 Most cases resolve spontaneously within a few months, and relapse is less common than in Graves’ disease. Other lab abnormalities include increased erythrocyte sedimentation rate and low radioiodine uptake.
Postpartum thyroiditis is an autoimmune disease. Prevalence ranges from 1% to 17% of new mothers.55 It is characterized by a thyroid gland that is painless on palpation and low radioiodine uptake.56 Most cases are reversible with treatment.
Factitious or iatrogenic hyperthyroidism is due to an exogenous intake of thyroid hormone, and typically exhibits a normal or low radioactive iodine uptake and a low thyroglobulin level.
Secondary hyperthyroidism, or TSH-mediated hyperthyroidism, is rare. It is always associated with goiter, and approximately 40% of patients have visual field defects.57
TABLE 2
Clinical and laboratory findings associated with common causes of hyperthyroidism51-57
| Mechanism | Thyroid exam | Lab results | Radioactive iodine uptake | |
|---|---|---|---|---|
| Graves’ disease | Antithyroid antibodies | Diffuse goiter | Low TSH; elevated T3 and/or T4; elevated thyroid antibodies | Diffusely increased |
| Toxic multinodular goiter | Iodine deficiency | Goiter with multiple nodules | Low TSH; elevated T3 and/or T4 | Normal/increased uptake; "hot nodules" with suppression of extranodular tissue |
| Toxic adenoma | Benign thyroid hormone?secreting tumor; iodine deficiency | Palpable nodule | Low TSH; elevated T3 and/or T4 | Normal/increased uptake; functioning "hot nodule" on scan with suppression of surrounding thyroid tissue |
| Subacute thyroiditis | Viral | Tender thyroid on palpation | Low TSH; elevated T3 and/or T4; elevated ESR; elevated thyroid antibodies | Low uptake with poor imaging of the thyroid on scan |
| Factitious hyperthyroidism | Excessive intake of exogenous thyroid hormone | Normal exam | Low TSH; elevated T3 and/or T4; low thyroglobulin level | Low or normal uptake |
| Secondary hyperthyroidism | Excessive pituitary TSH | Goiter | Elevated TSH; elevated T3 and/or T4 | Diffusely increased uptake |
| ESR, erythrocyte sedimentation rate; T3, triiodothyronine; T4, thyroxine; TSH, thyroid-stimulating hormone. | ||||
Which laboratory tests to order, and what the results may mean
Rely on second- or third-generation TSH screening (normal=0.5-5 mIU/L), which is more sensitive and specific than measuring free T4 (thyroxine) alone.6
Older patients usually have a higher normal TSH level. In one study, 70% of patients >80 years had a TSH >4.5 mIU/L.7
If the TSH level is low (<0.5 mIU/L), measure free T3 (triiodothyronine) and free T4 levels, which are elevated in hyperthyroidism, and are normal in subclinical hyperthyroidism.
Patients with Graves’ disease tend to have T3 thyrotoxicosis with a T3T4 ratio >20.8 Isolated T4 thyrotoxicosis is more commonly seen with nonthyroidal illness as a result of decreased conversion from T4 to T3, and also in amiodarone-induced hyperthyroidism.9
When further testing is needed (FIGURE). If the underlying cause of hyperthyroidism is not established on the basis of clinical findings (eg, diffuse goiter, myxedema, ophthalmopathy), order a 24-hour radioactive iodine (RAI) uptake test.10 Graves’ disease and toxic multinodular goiter exhibit increased RAI uptake that is diffuse and nodular, respectively. Subacute thyroiditis is associated with low RAI uptake (TABLE 2).
If RAI is contraindicated—eg, in pregnancy—testing for elevated levels of thyroid peroxidase antibodies (TPOab), TSH receptor antibodies (TRab), and thyroglobulin may help to differentiate Graves’ disease from multinodular goiter or uncover another autoimmune thyroid disorder.11 If a patient’s TSH, T4, and T3 levels are all elevated, refer him or her for magnetic resonance imaging of the pituitary gland to look for a TSH-secreting adenoma.
FIGURE
Suspect hyperthyroidism? Order these tests6-11
MRI, magnetic resonance imaging; RAI, radioactive iodine; T3, triiodothyronine; T4, thyroxine; TSH, thyroid-stimulating hormone.
Matching treatment to the underlying cause
RAI is usually the treatment of choice for patients without contraindications, although no randomized clinical trials have compared it with antithyroid medications or surgery.12 Each modality has its own risks and benefits (TABLE 3), and treatment selection should be individualized.
TABLE 3
Comparison of treatment modalities for hyperthyroidism12-21
| Antithyroid drugs* | Radioactive iodine* | Surgery | |
|---|---|---|---|
| Recurrence rate | High recurrence rate; no permanent hypothyroidism | Usually permanent hypothyroidism; long-term use of levothyroxine is required | Subtotal thyroidectomy associated with higher rates of recurrence or persistence of hyperthyroidism than total thyroidectomy; permanent hypothyroidism; long-term use of levothyroxine is required |
| Preferred method within treatment modality | MMI is the preferred medication; PTU is used with pregnancy and severe hyperthyroidism not responding to MMI | High ablative dose is preferred in MNG, toxic nodule, cardiac disease, elderly; low calculated dose is preferred in patients with GO | No outcome differences for GO, whether thyroidectomy is total, bilateral subtotal, or unilateral total and contralateral subtotal |
| Setting | Outpatient | Outpatient | Inpatient |
| Risks | No surgical risks | No surgical risks | Reaction to anesthesia, recurrent laryngeal nerve palsy, hypoparathyroidism |
| Adverse effects | ATD adverse effects, including life-threatening agranulocytosis | Worsening of Graves’ ophthalmopathy; transient exacerbation of hyperthyroid symptoms | Permanent hypothyroidism; hypoparathyroidism; anesthesia complications |
| Safety in pregnancy | PTU is used in pregnancy | Contraindicated in pregnancy/lactation | If surgery is indicated in pregnancy, it is best performed in the second trimester |
| ATD, antithyroid drugs; GO, Graves’ ophthalmopathy; MMI, methimazole; MNG, toxic multinodular goiter; PTU, propylthiouracil. *Concomitant use of ATD and RAI is associated with a high failure rate and persistent or recurrent hyperthyroidism. Discontinue ATD 2 weeks before radioactive iodine treatment. | |||
Radioactive iodine
In a 1990 survey, as many as 70% of specialists in the United States used RAI to treat hyperthyroidism, compared with just 22% of specialists in Europe.13 RAI is usually given in a single dose, and its maximal benefit is noted within 3 to 6 months. Two treatment methods are available: the ablative method and the gland-specific dosing method. Both have similar euthyroid state outcomes.14
The ablative method uses a high dose of RAI to achieve permanent hypothyroidism, necessitating lifelong levothyroxine replacement. This method is preferred for the elderly and for patients with cardiac disease, to achieve faster control of symptoms. It is also recommended for patients with toxic multinodular goiter and toxic nodules.
The gland-specific dosing method induces a euthyroid state with a calculated low dose of RAI based on the estimated weight of the patient’s thyroid. The optimal dosage may be difficult to calculate, but it is usually the preferred method for patients with Graves’ ophthalmopathy.
Adverse effects of RAI can include worsening of Graves’ ophthalmopathy and an acute rise in thyroid hormone that increases hyperthyroid symptoms or even causes a thyroid storm associated with increased cardiovascular risk.2 A negative pregnancy test result is a prerequisite for all women of childbearing age before taking RAI, and patients are advised to use contraception for 6 months after RAI administration.
Although RAI is often the initial treatment for hyperthyroidism, in some instances—eg, for older patients with comorbidities—pre-treatment with antithyroid drugs (ATD) is indicated to avoid transient worsening of hyperthyroid symptoms after RAI. However, always discontinue ATD 2 weeks before RAI administration; concomitant use is associated with a higher failure rate and persistent or recurrent hyperthyroidism.15
Antithyroid drugs
Two antithyroid medications are available for use in the United States: propylthiouracil (PTU) and methimazole (MMI). In the United Kingdom, carbimazole is also available.
MMI is the drug of choice.16 Compared with PTU, MMI costs less, has a longer half-life, and causes fewer adverse effects. A starting dose of 15 mg per day for MMI is suitable for mild and moderate hyperthyroidism. For more severe cases, 30 mg per day is the recommended starting dose.16 Reserve PTU for treating hyperthyroidism in pregnancy, during which MMI should be avoided, if possible.
Allergic reactions to ATDs appear in around 5% of patients and usually occur in the first 6 weeks of treatment.17 Agranulocytosis is the main concern, although it occurs in fewer than 1% of patients17 and is reversible by stopping the medication. Measure the leukocyte count 1 week after initiation of treatment and repeat the measurement at 1-month intervals.
Two methods are used to dose these medications: titration and block-and-replace. Titration is as effective as the block-and-replace method and is associated with fewer rashes (6% vs 10% of patients) and less agranulocytosis (0.4 % vs 1.4%). The 2 methods have similar relapse rates (around 50%).18
With titration, MMI is started at a dose of 15 mg per day and titrated upward to the lowest effective dose. Treatment for 12 to 18 months is associated with a lower relapse rate than treatment for 6 months (37% vs 58%).19
The block-and-replace method uses persistently high ATD doses in combination with L-thyroxin replacement to avoid hypothyroidism (MMI 30 mg and levothyroxine 80 mcg).
To monitor effectiveness initially, measure free T4 and T3 levels, because TSH concentration changes slowly and may stay low for a few months. Response to treatment is often temporary.8 More definitive treatment with RAI or surgery is usually necessary.
Surgery
Thyroidectomy creates permanent hypothyroidism, necessitating lifelong thyroxine replacement. In the United States, surgical intervention is reserved for special situations, such as pregnant women with severe disease who are allergic or not responding to antithyroid medications, removal of a clinically suspicious thyroid nodule coexisting with hyperthyroidism, or severe or recurrent Graves’ disease with severe ophthalmopathy.20 Surgical options are total or subtotal thyroidectomy. Hyperthyroidism persists or recurs in 8% of patients with subtotal thyroidectomy.21 Potential complications of thyroidectomy include adverse effects of anesthesia, hypoparathyroidism, and vocal cord paralysis.
Other treatment options
Iodides
Iodides inhibit thyroid hormone release and block conversion of T4 to T3. Use potassium iodide only in combination with ATDs, for patients with severe thyrotoxicosis or as pretreatment for urgent thyroidectomy in patients with Graves’ disease. It has been shown to improve the short-term control of Graves’ hyperthyroidism and is not associated with worsening hyperthyroidism;22 however, potassium iodide should not be used for more than 12 weeks as it can cause paradoxical hyperthyroidism.22
Beta-blockers
Hyperthyroidism is associated with an increased number of beta-adrenergic receptors,23 which explains the symptoms of palpitations, anxiety, and tremors. Nonselective beta-blockers are usually preferred for symptomatic treatment of hyperthyroid symptoms, and propranolol is the most widely used agent.24 If you decide to use a beta-blocker, start it with the ATD and continue it until the patient becomes euthyroid or asymptomatic, then taper it over a period of 4 to 6 weeks. Symptoms may persist, however, and require higher doses of propranolol (80-320 mg/d) given more frequently.
Treating Graves’ ophthalmopathy
Exophthalmos and other eye signs are the hallmark of Graves’ disease and may occur in the absence of hyperthyroidism. Smoking is a significant risk factor for developing ophthalmopathy due to increased orbital connective tissue volume,25 and smoking cessation is recommended.26
Using RAI to treat Graves’ disease increases the risk that ophthalmopathy will develop or worsen. Worsening of Graves’ ophthalmopathy secondary to RAI treatment occurs in 20% of treated patients (transient in 15%; permanent in 5%).27 Steroid prophylaxis is beneficial for patients with ophthalmopathy,28 and prednisone 40 to 80 mg per day tapered over at least 3 months can help reduce the condition.19 In patients with moderate to severe active ophthalmopathy, intravenous corticosteroid therapy has a small but statistically significant advantage over oral therapy and causes significantly fewer adverse events.29
Orbital radiotherapy is also used, and has been shown to decrease diplopia.30 However, the best available evidence recommends combining orbital radiotherapy and oral corticosteroids, which yields efficacy beyond that achievable with either radiotherapy or oral corticosteroids alone.16 Moreover, intravenous methylprednisolone combined with orbital radiotherapy seems to be most efficacious.31 The course of ophthalmopathy is the same whether total or subtotal thyroidectomy is used.32
Prognosis without treatment
Individuals with high-normal thyroid function tests, subclinical hyperthyroidism, and clinical hyperthyroidism are at increased risk for atrial fibrillation.33-35 Hyperthyroidism is also associated with increased risk of heart failure (6% of patients), which might be secondary to coexisting atrial fibrillation or tachycardia-mediated cardiomyopathy.36 Heart failure is usually reversible when the hyperthyroidism is treated.
Patients with overt hyperthyroidism are also at risk for pulmonary hypertension secondary to increased cardiac output and decreased pulmonary vascular resistance.37
In patients with preexisting cardiac disease, hyperthyroidism increases risk of death (hazard ratio [HR]=1.57),38 and might even do so in patients without cardiac disease.39,40 It also increases risk of ischemic stroke (HR=1.44) among adults ages 18 to 44 years.41 Untreated hyperthyroidism also contributes to low bone mineral density and increases the risk of hip fracture.42
Subclinical hyperthyroidism
Subclinical hyperthyroidism occurs in 2% of the US population and is characterized by low serum TSH (<0.1 mIU/L) with normal levels of free T3 and free T4. The causes are similar to overt hyperthyroidism. In addition, it can result from overtreating hypothyroidism with thyroid hormone, thereby inducing a subclinical hyperthyroid state.
The most common endogenous cause of subclinical hyperthyroidism (~60% of patients) is multinodular goiter.43 Subclinical hyperthyroidism carries significant health risks, and yet evidence is lacking on when to treat this condition. Prolonged subclinical hyperthyroidism can lead to atrial fibrillation,24,44 and to systolic and diastolic cardiac dysfunction.45 Subclinical hyperthyroidism is also associated with decreased bone density,46 and an increased risk of dementia.47
The American Association of Clinical Endocrinologists recommends periodic clinical and laboratory assessment for patients with subclinical hyperthyroidism (TSH=0.1-0.5 mIU/mL), including rechecking TSH, free T3 and free T4 at 2- to 4-month intervals.
Treatment of the underlying cause of hyperthyroidism is indicated if serum TSH is <0.1 mIU/mL.
For patients older than 65 years who have persistent subclinical hyperthyroidism, consider treatment in the following scenarios:48
- nodular thyroid disease (due to high conversion rate to overt hyperthyroidism)
- osteopenia or osteoporosis (in women)
- atrial fibrillation
- underlying cardiac disease.
Hyperthyroidism in pregnancy
PTU is the first choice for treating hyperthyroidism in pregnancy. It crosses the placenta less readily than MMI, and is thus less likely to cause fetal hypothyroidism. Additionally, MMI is associated with increased risk of fetal anomalies, such as aplasia cutis and esophageal atresia. MMI may be considered if the patient is intolerant to PTU or fails to become euthyroid while receiving PTU.49 Use the lowest possible dose of either PTU or MMI to maintain thyroid function within the upper limit of normal. The dose of the antithyroid medication is usually decreased as pregnancy progresses and discontinued in the last few weeks, as pregnancy is thought to improve the course of Graves’ disease.
The use of RAI is contraindicated during pregnancy and breastfeeding. Hyperthyroidism symptoms usually resolve after delivery. If symptoms persist, however, the treatment of choice is ATD. Surgery is an option in severe Graves’ disease not responding to ATD.
CORRESPONDENCE
Abdulraouf Ghandour, MD, Department of Family and Community Medicine, University of Missouri-Columbia, One Hospital Drive, Columbia, MO 65212; ghandoura@health.missouri.edu
• Measure TSH in any patient >60 years presenting with fatigue, atrial fibrillation, weight loss, and shortness of breath. B
• Achieve faster control of symptoms in elderly patients and those with cardiac disease by pursuing the ablative method with radioactive iodine (RAI). This method is also recommended for patients with toxic multinodular goiter and toxic adenoma. A
• Initiate steroid prophylaxis for patients with Graves’ ophthalmopathy undergoing RAI. A
• Opt for a 12- to 18-month course of an antithyroid drug, rather than a 6-month course. The longer course is associated with a lower relapse rate. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
A 72-year-old man arrives at the clinic with insomnia and fatigue. His medical history is significant for hypertension, hyperlipidemia, and degenerative joint disease, for which he is taking, respectively, metoprolol 25 mg twice daily, simvastatin 20 mg daily, and acetaminophen as needed for joint pain. He has experienced no weight loss, anxiety, or gastrointestinal or urinary symptoms. He does not smoke or drink alcohol. His blood pressure is 140/75 mm Hg, pulse is 85, respiratory rate is 20, and temperature is 97.1°F. The rest of the physical examination is unremarkable except for 1+ lower extremity edema, unchanged since his previous visit. Routine blood work, however, reveals his thyroid-stimulating hormone (TSH) level to be 0.03 mIU/L.
Clues from the clinical presentation
The subtle, "apathetic presentation" with few symptoms, as described in the case above, is typical of older individuals with hyperthyroidism.1 In contrast, younger patients with hyperthyroidism and those with comorbidities can manifest a number of signs and symptoms (TABLE 1).2
Graves’ disease, the most common cause of hyperthyroidism,3 causes such ocular disturbances as exophthalmos, lid lag, lid retraction, and proptosis in 60% of patients with the condition.3 These findings help differentiate Graves’ disease from other causes of hyperthyroidism. (See “Common [and not so common] causes of hyperthyroidism”.) Palmar sweating, pretibial myxedema, and Plummer’s nails (onycholysis) are also unique for Graves’ disease.4
When you suspect hyperthyroidism, assess the thyroid for size, nodularity, and vascularity. Goiter is less prevalent in the elderly, occurring in less than 50% of patients 61 and older, compared with 77% of patients younger than 60 years.5 Diffuse goiter is typical with Graves’ disease, while a mass with multiple nodules suggests possible toxic multinodular goiter. A solitary palpable nodule could mean toxic adenoma. A thyroid that is tender on palpation may point to subacute thyroiditis, particularly if the patient has had a viral illness recently (TABLE 2).
Measuring a patient’s TSH level is warranted with the above findings. Additionally, measure TSH in any patient older than 60 years presenting with fatigue, atrial fibrillation, weight loss, and shortness of breath.5
TABLE 1
Clinical manifestations of hyperthyroidism2
| Acropachy (swelling of the fingers) |
| Bruit (thyroid) |
| Decreased attention span |
| Diarrhea |
| Edema |
| Exertional dyspnea |
| Fatigue |
| Goiter (smooth or nodular) |
| Gynecomastia |
| Hair loss |
| Heat intolerance |
| Hyperactive deep tendon reflex |
| Hypertension |
| Increased appetite |
| Infertility |
| Insomnia |
| Lid lag, proptosis |
| Muscle weakness |
| Nervousness and irritability |
| Oligomenorrhea |
| Palmar erythema |
| Palpitations |
| Paralysis (sudden) |
| Photophobia, eye irritation, diplopia |
| Pretibial myxedema |
| Tachycardia |
| Tremors |
| Warm, moist skin |
| Weight loss |
Graves’ disease—an autoimmune disorder in which antibodies target thyroid tissue and enzymes and activate thyroid hormone synthesis—affects more than 3 million people in the United States and accounts for 60% of hyperthyroidism cases.3 Remission does occur; however, the recurrence rate is as high as 60%.50 Factors associated with recurrence include tobacco use; male sex; young age; large goiter size or increase in goiter size during treatment; elevated TSH receptor antibodies (TRab); presence of Graves’ ophthalmopathy; markedly elevated thyroid hormones, or delayed treatment.51
Toxic multinodular goiter, also known as Plummer’s disease, is the underlying condition in 15% to 20% of hyperthyroidism cases; it is more common in young patients and in iodine-deficient locations (eg, Denmark).52 However, it also occurs in elderly patients with longstanding goiter.
Toxic adenoma causes just 3% to 5% of cases of hyperthyroidism.53 It, too, occurs more commonly in young patients and in iodine-deficient regions. The radioactive iodine uptake test shows a hot nodule, with suppressed uptake in the surrounding thyroid gland.
Subacute thyroiditis, also known as de Quervain’s thyroiditis, is the reason for 15% to 20% of hyperthyroidism cases; it is usually preceded by viral infection and inflammation that lead to destructive release of preformed thyroid hormone. Symptoms—typically fever, malaise, and tender goiter—usually occur more abruptly than symptoms of Graves’ disease.54 Most cases resolve spontaneously within a few months, and relapse is less common than in Graves’ disease. Other lab abnormalities include increased erythrocyte sedimentation rate and low radioiodine uptake.
Postpartum thyroiditis is an autoimmune disease. Prevalence ranges from 1% to 17% of new mothers.55 It is characterized by a thyroid gland that is painless on palpation and low radioiodine uptake.56 Most cases are reversible with treatment.
Factitious or iatrogenic hyperthyroidism is due to an exogenous intake of thyroid hormone, and typically exhibits a normal or low radioactive iodine uptake and a low thyroglobulin level.
Secondary hyperthyroidism, or TSH-mediated hyperthyroidism, is rare. It is always associated with goiter, and approximately 40% of patients have visual field defects.57
TABLE 2
Clinical and laboratory findings associated with common causes of hyperthyroidism51-57
| Mechanism | Thyroid exam | Lab results | Radioactive iodine uptake | |
|---|---|---|---|---|
| Graves’ disease | Antithyroid antibodies | Diffuse goiter | Low TSH; elevated T3 and/or T4; elevated thyroid antibodies | Diffusely increased |
| Toxic multinodular goiter | Iodine deficiency | Goiter with multiple nodules | Low TSH; elevated T3 and/or T4 | Normal/increased uptake; "hot nodules" with suppression of extranodular tissue |
| Toxic adenoma | Benign thyroid hormone?secreting tumor; iodine deficiency | Palpable nodule | Low TSH; elevated T3 and/or T4 | Normal/increased uptake; functioning "hot nodule" on scan with suppression of surrounding thyroid tissue |
| Subacute thyroiditis | Viral | Tender thyroid on palpation | Low TSH; elevated T3 and/or T4; elevated ESR; elevated thyroid antibodies | Low uptake with poor imaging of the thyroid on scan |
| Factitious hyperthyroidism | Excessive intake of exogenous thyroid hormone | Normal exam | Low TSH; elevated T3 and/or T4; low thyroglobulin level | Low or normal uptake |
| Secondary hyperthyroidism | Excessive pituitary TSH | Goiter | Elevated TSH; elevated T3 and/or T4 | Diffusely increased uptake |
| ESR, erythrocyte sedimentation rate; T3, triiodothyronine; T4, thyroxine; TSH, thyroid-stimulating hormone. | ||||
Which laboratory tests to order, and what the results may mean
Rely on second- or third-generation TSH screening (normal=0.5-5 mIU/L), which is more sensitive and specific than measuring free T4 (thyroxine) alone.6
Older patients usually have a higher normal TSH level. In one study, 70% of patients >80 years had a TSH >4.5 mIU/L.7
If the TSH level is low (<0.5 mIU/L), measure free T3 (triiodothyronine) and free T4 levels, which are elevated in hyperthyroidism, and are normal in subclinical hyperthyroidism.
Patients with Graves’ disease tend to have T3 thyrotoxicosis with a T3T4 ratio >20.8 Isolated T4 thyrotoxicosis is more commonly seen with nonthyroidal illness as a result of decreased conversion from T4 to T3, and also in amiodarone-induced hyperthyroidism.9
When further testing is needed (FIGURE). If the underlying cause of hyperthyroidism is not established on the basis of clinical findings (eg, diffuse goiter, myxedema, ophthalmopathy), order a 24-hour radioactive iodine (RAI) uptake test.10 Graves’ disease and toxic multinodular goiter exhibit increased RAI uptake that is diffuse and nodular, respectively. Subacute thyroiditis is associated with low RAI uptake (TABLE 2).
If RAI is contraindicated—eg, in pregnancy—testing for elevated levels of thyroid peroxidase antibodies (TPOab), TSH receptor antibodies (TRab), and thyroglobulin may help to differentiate Graves’ disease from multinodular goiter or uncover another autoimmune thyroid disorder.11 If a patient’s TSH, T4, and T3 levels are all elevated, refer him or her for magnetic resonance imaging of the pituitary gland to look for a TSH-secreting adenoma.
FIGURE
Suspect hyperthyroidism? Order these tests6-11
MRI, magnetic resonance imaging; RAI, radioactive iodine; T3, triiodothyronine; T4, thyroxine; TSH, thyroid-stimulating hormone.
Matching treatment to the underlying cause
RAI is usually the treatment of choice for patients without contraindications, although no randomized clinical trials have compared it with antithyroid medications or surgery.12 Each modality has its own risks and benefits (TABLE 3), and treatment selection should be individualized.
TABLE 3
Comparison of treatment modalities for hyperthyroidism12-21
| Antithyroid drugs* | Radioactive iodine* | Surgery | |
|---|---|---|---|
| Recurrence rate | High recurrence rate; no permanent hypothyroidism | Usually permanent hypothyroidism; long-term use of levothyroxine is required | Subtotal thyroidectomy associated with higher rates of recurrence or persistence of hyperthyroidism than total thyroidectomy; permanent hypothyroidism; long-term use of levothyroxine is required |
| Preferred method within treatment modality | MMI is the preferred medication; PTU is used with pregnancy and severe hyperthyroidism not responding to MMI | High ablative dose is preferred in MNG, toxic nodule, cardiac disease, elderly; low calculated dose is preferred in patients with GO | No outcome differences for GO, whether thyroidectomy is total, bilateral subtotal, or unilateral total and contralateral subtotal |
| Setting | Outpatient | Outpatient | Inpatient |
| Risks | No surgical risks | No surgical risks | Reaction to anesthesia, recurrent laryngeal nerve palsy, hypoparathyroidism |
| Adverse effects | ATD adverse effects, including life-threatening agranulocytosis | Worsening of Graves’ ophthalmopathy; transient exacerbation of hyperthyroid symptoms | Permanent hypothyroidism; hypoparathyroidism; anesthesia complications |
| Safety in pregnancy | PTU is used in pregnancy | Contraindicated in pregnancy/lactation | If surgery is indicated in pregnancy, it is best performed in the second trimester |
| ATD, antithyroid drugs; GO, Graves’ ophthalmopathy; MMI, methimazole; MNG, toxic multinodular goiter; PTU, propylthiouracil. *Concomitant use of ATD and RAI is associated with a high failure rate and persistent or recurrent hyperthyroidism. Discontinue ATD 2 weeks before radioactive iodine treatment. | |||
Radioactive iodine
In a 1990 survey, as many as 70% of specialists in the United States used RAI to treat hyperthyroidism, compared with just 22% of specialists in Europe.13 RAI is usually given in a single dose, and its maximal benefit is noted within 3 to 6 months. Two treatment methods are available: the ablative method and the gland-specific dosing method. Both have similar euthyroid state outcomes.14
The ablative method uses a high dose of RAI to achieve permanent hypothyroidism, necessitating lifelong levothyroxine replacement. This method is preferred for the elderly and for patients with cardiac disease, to achieve faster control of symptoms. It is also recommended for patients with toxic multinodular goiter and toxic nodules.
The gland-specific dosing method induces a euthyroid state with a calculated low dose of RAI based on the estimated weight of the patient’s thyroid. The optimal dosage may be difficult to calculate, but it is usually the preferred method for patients with Graves’ ophthalmopathy.
Adverse effects of RAI can include worsening of Graves’ ophthalmopathy and an acute rise in thyroid hormone that increases hyperthyroid symptoms or even causes a thyroid storm associated with increased cardiovascular risk.2 A negative pregnancy test result is a prerequisite for all women of childbearing age before taking RAI, and patients are advised to use contraception for 6 months after RAI administration.
Although RAI is often the initial treatment for hyperthyroidism, in some instances—eg, for older patients with comorbidities—pre-treatment with antithyroid drugs (ATD) is indicated to avoid transient worsening of hyperthyroid symptoms after RAI. However, always discontinue ATD 2 weeks before RAI administration; concomitant use is associated with a higher failure rate and persistent or recurrent hyperthyroidism.15
Antithyroid drugs
Two antithyroid medications are available for use in the United States: propylthiouracil (PTU) and methimazole (MMI). In the United Kingdom, carbimazole is also available.
MMI is the drug of choice.16 Compared with PTU, MMI costs less, has a longer half-life, and causes fewer adverse effects. A starting dose of 15 mg per day for MMI is suitable for mild and moderate hyperthyroidism. For more severe cases, 30 mg per day is the recommended starting dose.16 Reserve PTU for treating hyperthyroidism in pregnancy, during which MMI should be avoided, if possible.
Allergic reactions to ATDs appear in around 5% of patients and usually occur in the first 6 weeks of treatment.17 Agranulocytosis is the main concern, although it occurs in fewer than 1% of patients17 and is reversible by stopping the medication. Measure the leukocyte count 1 week after initiation of treatment and repeat the measurement at 1-month intervals.
Two methods are used to dose these medications: titration and block-and-replace. Titration is as effective as the block-and-replace method and is associated with fewer rashes (6% vs 10% of patients) and less agranulocytosis (0.4 % vs 1.4%). The 2 methods have similar relapse rates (around 50%).18
With titration, MMI is started at a dose of 15 mg per day and titrated upward to the lowest effective dose. Treatment for 12 to 18 months is associated with a lower relapse rate than treatment for 6 months (37% vs 58%).19
The block-and-replace method uses persistently high ATD doses in combination with L-thyroxin replacement to avoid hypothyroidism (MMI 30 mg and levothyroxine 80 mcg).
To monitor effectiveness initially, measure free T4 and T3 levels, because TSH concentration changes slowly and may stay low for a few months. Response to treatment is often temporary.8 More definitive treatment with RAI or surgery is usually necessary.
Surgery
Thyroidectomy creates permanent hypothyroidism, necessitating lifelong thyroxine replacement. In the United States, surgical intervention is reserved for special situations, such as pregnant women with severe disease who are allergic or not responding to antithyroid medications, removal of a clinically suspicious thyroid nodule coexisting with hyperthyroidism, or severe or recurrent Graves’ disease with severe ophthalmopathy.20 Surgical options are total or subtotal thyroidectomy. Hyperthyroidism persists or recurs in 8% of patients with subtotal thyroidectomy.21 Potential complications of thyroidectomy include adverse effects of anesthesia, hypoparathyroidism, and vocal cord paralysis.
Other treatment options
Iodides
Iodides inhibit thyroid hormone release and block conversion of T4 to T3. Use potassium iodide only in combination with ATDs, for patients with severe thyrotoxicosis or as pretreatment for urgent thyroidectomy in patients with Graves’ disease. It has been shown to improve the short-term control of Graves’ hyperthyroidism and is not associated with worsening hyperthyroidism;22 however, potassium iodide should not be used for more than 12 weeks as it can cause paradoxical hyperthyroidism.22
Beta-blockers
Hyperthyroidism is associated with an increased number of beta-adrenergic receptors,23 which explains the symptoms of palpitations, anxiety, and tremors. Nonselective beta-blockers are usually preferred for symptomatic treatment of hyperthyroid symptoms, and propranolol is the most widely used agent.24 If you decide to use a beta-blocker, start it with the ATD and continue it until the patient becomes euthyroid or asymptomatic, then taper it over a period of 4 to 6 weeks. Symptoms may persist, however, and require higher doses of propranolol (80-320 mg/d) given more frequently.
Treating Graves’ ophthalmopathy
Exophthalmos and other eye signs are the hallmark of Graves’ disease and may occur in the absence of hyperthyroidism. Smoking is a significant risk factor for developing ophthalmopathy due to increased orbital connective tissue volume,25 and smoking cessation is recommended.26
Using RAI to treat Graves’ disease increases the risk that ophthalmopathy will develop or worsen. Worsening of Graves’ ophthalmopathy secondary to RAI treatment occurs in 20% of treated patients (transient in 15%; permanent in 5%).27 Steroid prophylaxis is beneficial for patients with ophthalmopathy,28 and prednisone 40 to 80 mg per day tapered over at least 3 months can help reduce the condition.19 In patients with moderate to severe active ophthalmopathy, intravenous corticosteroid therapy has a small but statistically significant advantage over oral therapy and causes significantly fewer adverse events.29
Orbital radiotherapy is also used, and has been shown to decrease diplopia.30 However, the best available evidence recommends combining orbital radiotherapy and oral corticosteroids, which yields efficacy beyond that achievable with either radiotherapy or oral corticosteroids alone.16 Moreover, intravenous methylprednisolone combined with orbital radiotherapy seems to be most efficacious.31 The course of ophthalmopathy is the same whether total or subtotal thyroidectomy is used.32
Prognosis without treatment
Individuals with high-normal thyroid function tests, subclinical hyperthyroidism, and clinical hyperthyroidism are at increased risk for atrial fibrillation.33-35 Hyperthyroidism is also associated with increased risk of heart failure (6% of patients), which might be secondary to coexisting atrial fibrillation or tachycardia-mediated cardiomyopathy.36 Heart failure is usually reversible when the hyperthyroidism is treated.
Patients with overt hyperthyroidism are also at risk for pulmonary hypertension secondary to increased cardiac output and decreased pulmonary vascular resistance.37
In patients with preexisting cardiac disease, hyperthyroidism increases risk of death (hazard ratio [HR]=1.57),38 and might even do so in patients without cardiac disease.39,40 It also increases risk of ischemic stroke (HR=1.44) among adults ages 18 to 44 years.41 Untreated hyperthyroidism also contributes to low bone mineral density and increases the risk of hip fracture.42
Subclinical hyperthyroidism
Subclinical hyperthyroidism occurs in 2% of the US population and is characterized by low serum TSH (<0.1 mIU/L) with normal levels of free T3 and free T4. The causes are similar to overt hyperthyroidism. In addition, it can result from overtreating hypothyroidism with thyroid hormone, thereby inducing a subclinical hyperthyroid state.
The most common endogenous cause of subclinical hyperthyroidism (~60% of patients) is multinodular goiter.43 Subclinical hyperthyroidism carries significant health risks, and yet evidence is lacking on when to treat this condition. Prolonged subclinical hyperthyroidism can lead to atrial fibrillation,24,44 and to systolic and diastolic cardiac dysfunction.45 Subclinical hyperthyroidism is also associated with decreased bone density,46 and an increased risk of dementia.47
The American Association of Clinical Endocrinologists recommends periodic clinical and laboratory assessment for patients with subclinical hyperthyroidism (TSH=0.1-0.5 mIU/mL), including rechecking TSH, free T3 and free T4 at 2- to 4-month intervals.
Treatment of the underlying cause of hyperthyroidism is indicated if serum TSH is <0.1 mIU/mL.
For patients older than 65 years who have persistent subclinical hyperthyroidism, consider treatment in the following scenarios:48
- nodular thyroid disease (due to high conversion rate to overt hyperthyroidism)
- osteopenia or osteoporosis (in women)
- atrial fibrillation
- underlying cardiac disease.
Hyperthyroidism in pregnancy
PTU is the first choice for treating hyperthyroidism in pregnancy. It crosses the placenta less readily than MMI, and is thus less likely to cause fetal hypothyroidism. Additionally, MMI is associated with increased risk of fetal anomalies, such as aplasia cutis and esophageal atresia. MMI may be considered if the patient is intolerant to PTU or fails to become euthyroid while receiving PTU.49 Use the lowest possible dose of either PTU or MMI to maintain thyroid function within the upper limit of normal. The dose of the antithyroid medication is usually decreased as pregnancy progresses and discontinued in the last few weeks, as pregnancy is thought to improve the course of Graves’ disease.
The use of RAI is contraindicated during pregnancy and breastfeeding. Hyperthyroidism symptoms usually resolve after delivery. If symptoms persist, however, the treatment of choice is ATD. Surgery is an option in severe Graves’ disease not responding to ATD.
CORRESPONDENCE
Abdulraouf Ghandour, MD, Department of Family and Community Medicine, University of Missouri-Columbia, One Hospital Drive, Columbia, MO 65212; ghandoura@health.missouri.edu
1. Levy EG. Thyroid disease in the elderly. Med Clin North Am. 1991;75:151-167.
2. Cooper DS. Hyperthyroidism. Lancet. 2003;362:459-468.
3. Weetman AP. Graves’ disease. N Engl J Med. 2000;343:1236-1248.
4. Heymann WR. Cutaneous manifestations of thyroid disease. J Am Acad Dermatol. 1992;26:885-902.
5. Boelaert K, Torlinska B. Older subjects with hyperthyroidism present with a paucity of symptoms and signs: a large cross-sectional study. J Clin Endocrinol Metab. 2010;95:2715-2726.
6. Danese MD, Powe NR, Sawin CT, et al. Screening of mild thyroid failure at the periodic health examination: a decision and cost-effectiveness analysis. JAMA. 1996;276:285-292.
7. Surks MI, Hollowell JG. Age-specific distribution of serum thyrotropin and antithyroid antibodies in the US population: implications for the prevalence of subclinical hypothyroidism. J Clin Endocrinol Metab. 2007;92:4575-4582.
8. Amino N, Yabu Y, Miki T, et al. Serum ratio of triiodothyronine to thyroxine and thyroxine binding globulin and calcitonin concentrations in Graves’ disease and destruction-induced thyrotoxicosis. J Clin Endocrinol Metab. 1981;53:113-116.
9. Bambini G, Aghini-Lombardi F, Rosner W, et al. Serum sex hormone-binding globulin in amiodarone-treated patients. A marker for tissue thyrotoxicosis. Arch Intern Med. 1987;147:1781-1785.
10. Fogelman I, Cooke SG, Maisey MN. The role of thyroid scanning in hyperthyroidism. Eur J Nucl Med. 1986;11:397-400.
11. Costagliola S, Morgenthaler NG, Hoermann R, et al. Second generation assay for thyrotropin receptor antibodies has superior diagnostic sensitivity for Graves’ disease. J Clin Endocrinol Metab. 1999;84:90-97.
12. Streetman DD, Khanderia U. Diagnosis and treatment of Graves’ disease. Ann Pharmacother. 2003;37:1100-1109.
13. Wartofsky L, Glinoer D, Solomon B, et al. Differences and similarities in the diagnosis and treatment of Graves’ disease in Europe, Japan, and the United States. Thyroid. 1991;1:129-135.
14. de Rooij A, Vandenbroucke JP. Clinical outcomes after estimated versus calculated activity of radioiodine for the treatment of hyperthyroidism: systematic review and meta-analysis. Eur J Endocrinol. 2009;161:771-777.
15. Walter MA, Briel M, Christ-Crain M, et al. Effects of antithyroid drugs on radioiodine treatment: systematic review and meta-analysis of randomised controlled trials. BMJ. 2007;334:514.-
16. Nakamura H, Noh JY. Comparison of methimazole and propylthiouracil in patients with hyperthyroidism caused by Graves’ disease. J Clin Endocrinol Metab. 2007;92:2157-2162.
17. Cooper DS. Antithyroid drugs. N Engl J Med. 2005;352:905-917.
18. Abraham P, Avenell A. A systematic review of drug therapy for Graves’ hyperthyroidism. Eur J Endocrinol. 2005;153:489-498.
19. Abraham P, Avenell A, McGeoch SC, et al. Antithyroid drug regimen for treating Graves’ hyperthyroidism. Cochrane Database Sys Rev. 2010;(1):CD003420.-
20. Stalberg P, Svensson A. Surgical treatment of Graves’ disease: evidence-based approach. World J Surg. 2008;32:1269-1277.
21. Palit TK, Miller CC, Miltenburg DM. The efficacy of thyroidectomy for Graves’ disease: a meta-analysis. J Surg Res. 2000;90:161-165.
22. Takata K, Amino N, Kubota S. Benefit of short-term iodide supplementation to antithyroid drug treatment of thyrotoxicosis due to Graves’ disease. Clin Endocrinol. 2010;72:845-850.
23. Bilezikian JP, Loeb JN. The influence of hyperthyroidism and hypothyroidism on alpha- and beta-adrenergic receptor systems and adrenergic responsiveness. Endocr Rev. 1983;4:378-388.
24. Jansson S, Lie-Karlsen K, Stenqvist O, et al. Oxygen consumption in patients with hyperthyroidism before and after treatment with beta-blockade versus thyrostatic treatment: a prospective randomized study. Ann Surg. 2001;233:60-64.
25. Zucs-Frkas Z, Toth J, Kollar J, et al. Volume changes in intra- and extraorbital compartments in patients with Graves’ ophthalmopathy: effect of smoking. Thyroid. 2005;15:146-151.
26. Träisk F, Tallstedt L. Thyroid-associated ophthalmopathy after treatment for Graves’ hyperthyroidism with antithyroid drugs or iodine-131. J Clin Endocrinol Metab. 2009;94:3700-3707.
27. Bartalena L, Marcocci C, Bogazzi F, et al. Relation between therapy of hyperthyroidism and the course of Graves’ ophthalmopathy. N Engl J Med. 1998;338:73-78.
28. Acharya SH, Avenell A. Radioiodine therapy (RAI) for Graves’ disease (GD) and the effect on ophthalmopathy: a systematic review. Clin Endocrinol (Oxf). 2008;69:943-950.
29. Stiebel-Kalish H, Robenshtok E. Treatment modalities for Graves’ ophthalmopathy: systematic review and meta-analysis. J Clin Endocrinol Metab. 2009;94:2708-2716.
30. Bradley EA, Gower EW. Orbital radiation for graves ophthalmopathy: a report by the American Academy of Ophthalmology. Ophthalmology. 2008;115:398-409.
31. Wei RL, Cheng JW. The use of orbital radiotherapy for Graves’ ophthalmopathy: quantitative review of the evidence. Ophthalmologica. 2008;222:27-31.
32. Witte J, Goretzki PE, Dotzenrath C, et al. Surgery for Graves’ disease: total versus subtotal thyroidectomy–result of a prospective randomized trial. World J Surg. 2000;24:1303-1311.
33. Heeringa J, Hoogendoorn EH. High-normal thyroid function and risk of atrial fibrillation: the Rotterdam study. Arch Intern Med. 2008;168:2219-2224.
34. Sawin CT, Geller A, Wolf PA, et al. Low serum thyrotropin concentrations as a risk factor for atrial fibrillation in older persons. N Engl J Med. 1994;331:1249-1252.
35. Cappola AR, Fried LP. Thyroid status, cardiovascular risk, and mortality in older adults. JAMA. 2006;295:1033-1041.
36. Siu CW, Yeung CY, Lau CP, et al. Incidence, clinical characteristics and outcome of congestive heart failure as the initial presentation in patients with primary hyperthyroidism. Heart. 2007;93:483-487.
37. Lozano HF, Sharma CN. Reversible pulmonary hypertension, tricuspid regurgitation and right-sided heart failure associated with hyperthyroidism: case report and review of the literature. Cardiol Rev. 2004;12:299-305.
38. Iervasi G, Molinaro S. Association between increased mortality and mild thyroid dysfunction in cardiac patients. Arch Intern Med. 2007;167:1526-1532.
39. Parle JV, Maisonneuve P, Sheppard MC, et al. Prediction of all-cause and cardiovascular mortality in elderly people from one low serum thyrotropin result: a 10-year cohort study. Lancet. 2001;358:861-865.
40. Flynn RW, McDonald TM, Jung RT, et al. Mortality and vascular outcomes in patients treated for thyroid dysfunction. J Clin Endocrinol Metab. 2006;91:2169-2164.
41. Sheu JJ, Kang JH. Hyperthyroidism and risk of ischemic stroke in young adults: a 5-year follow-up study. Stroke. 2010;41:961-966.
42. Vestergaard P, Mosekilde L. Hyperthyroidism, bone mineral, and fracture risk—a meta-analysis. Thyroid. 2003;13:585-593.
43. Diez JJ. Hyperthyroidism in patients older than 55 years: an analysis of the etiology and management. Gerontology. 2003;49:316-323.
44. Sawin CT, Geller A, Wolf PA, et al. Low serum thyrotropin concentrations as a risk factor for atrial fibrillation in older persons. N Engl J Med. 1994;331:1249-1252.
45. Abdulrahman RM, Delgado V. Abnormal cardiac contractility in long-term exogenous subclinical hyperthyroid patients as demonstrated by two-dimensional echocardiography speckle tracking imaging. Eur J Endocrinol. 2010;163:435-441.
46. Faber J, Jensen IW, Petersen L, et al. Normalization of serum thyrotrophin by means of radioiodine treatment in subclinical hyperthyroidism: effect on bone loss in postmenopausal women. Clin Endocrinol (Oxf). 1998;48:285-290.
47. Tan ZS, Beiser A, Vasan RS, et al. Thyroid function and the risk of Alzheimer disease: Framingham study. Arch Intern Med. 2008;168:1514-1520.
48. American Association of Clinical Endocrinologists. Medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. 2006. Available at: https://www.aace.com/sites/default/files/hypo_hyper.pdf. Accessed July 9, 2010.
49. Chattaway JM, Klepser TB. Propylthiouracil versus methimazole in treatment of Grave’s disease during pregnancy. Ann Pharmacother. 2007;41:1018-1022.
50. Lucas A, Salinas I. Medical therapy of Graves’ disease: does thyroxine prevent recurrence of hyperthyroidism? J Clin Endocrinol Metab. 1997;82:2410-2413.
51. Vitti P, Rago T, Chiovato L, et al. Clinical features of patients with Graves’ disease undergoing remission after antithyroid drug treatment. Thyroid. 1997;7:369-375.
52. Laurberg P, Bulow Pedersen I, Pedersen KM, et al. Low incidence rate of overt hypothyroidism compared with hyperthyroidism in an area with moderately low iodine intake. Thyroid. 1999;9:33-38.
53. Siegel RD, Lee SL. Toxic nodular goiter: Toxic adenoma and toxic multinodular goiter. Endocrinol Metab Clin North Am. 1998;27:151-168.
54. Volpe R. Subacute (de Quervain’s) thyroiditis. Clin Endocrinol Metab. 1979;8:81-95.
55. Nicholson WK, Robinson KA, Smallridge RC, et al. Prevalence of postpartum thyroid dysfunction: a quantitative review. Thyroid. 2006;16:573-582.
56. Roti E, Emerson CH. Clinical review 29: postpartum thyroiditis. J Clin Endocrinol Metab. 1992;74:3-5.
57. Beck-Peccoz P, Brucker-Davis F, Persani L, et al. Thyrotropin-secreting pituitary tumors. Endocr Rev. 1996;17:610-638.
1. Levy EG. Thyroid disease in the elderly. Med Clin North Am. 1991;75:151-167.
2. Cooper DS. Hyperthyroidism. Lancet. 2003;362:459-468.
3. Weetman AP. Graves’ disease. N Engl J Med. 2000;343:1236-1248.
4. Heymann WR. Cutaneous manifestations of thyroid disease. J Am Acad Dermatol. 1992;26:885-902.
5. Boelaert K, Torlinska B. Older subjects with hyperthyroidism present with a paucity of symptoms and signs: a large cross-sectional study. J Clin Endocrinol Metab. 2010;95:2715-2726.
6. Danese MD, Powe NR, Sawin CT, et al. Screening of mild thyroid failure at the periodic health examination: a decision and cost-effectiveness analysis. JAMA. 1996;276:285-292.
7. Surks MI, Hollowell JG. Age-specific distribution of serum thyrotropin and antithyroid antibodies in the US population: implications for the prevalence of subclinical hypothyroidism. J Clin Endocrinol Metab. 2007;92:4575-4582.
8. Amino N, Yabu Y, Miki T, et al. Serum ratio of triiodothyronine to thyroxine and thyroxine binding globulin and calcitonin concentrations in Graves’ disease and destruction-induced thyrotoxicosis. J Clin Endocrinol Metab. 1981;53:113-116.
9. Bambini G, Aghini-Lombardi F, Rosner W, et al. Serum sex hormone-binding globulin in amiodarone-treated patients. A marker for tissue thyrotoxicosis. Arch Intern Med. 1987;147:1781-1785.
10. Fogelman I, Cooke SG, Maisey MN. The role of thyroid scanning in hyperthyroidism. Eur J Nucl Med. 1986;11:397-400.
11. Costagliola S, Morgenthaler NG, Hoermann R, et al. Second generation assay for thyrotropin receptor antibodies has superior diagnostic sensitivity for Graves’ disease. J Clin Endocrinol Metab. 1999;84:90-97.
12. Streetman DD, Khanderia U. Diagnosis and treatment of Graves’ disease. Ann Pharmacother. 2003;37:1100-1109.
13. Wartofsky L, Glinoer D, Solomon B, et al. Differences and similarities in the diagnosis and treatment of Graves’ disease in Europe, Japan, and the United States. Thyroid. 1991;1:129-135.
14. de Rooij A, Vandenbroucke JP. Clinical outcomes after estimated versus calculated activity of radioiodine for the treatment of hyperthyroidism: systematic review and meta-analysis. Eur J Endocrinol. 2009;161:771-777.
15. Walter MA, Briel M, Christ-Crain M, et al. Effects of antithyroid drugs on radioiodine treatment: systematic review and meta-analysis of randomised controlled trials. BMJ. 2007;334:514.-
16. Nakamura H, Noh JY. Comparison of methimazole and propylthiouracil in patients with hyperthyroidism caused by Graves’ disease. J Clin Endocrinol Metab. 2007;92:2157-2162.
17. Cooper DS. Antithyroid drugs. N Engl J Med. 2005;352:905-917.
18. Abraham P, Avenell A. A systematic review of drug therapy for Graves’ hyperthyroidism. Eur J Endocrinol. 2005;153:489-498.
19. Abraham P, Avenell A, McGeoch SC, et al. Antithyroid drug regimen for treating Graves’ hyperthyroidism. Cochrane Database Sys Rev. 2010;(1):CD003420.-
20. Stalberg P, Svensson A. Surgical treatment of Graves’ disease: evidence-based approach. World J Surg. 2008;32:1269-1277.
21. Palit TK, Miller CC, Miltenburg DM. The efficacy of thyroidectomy for Graves’ disease: a meta-analysis. J Surg Res. 2000;90:161-165.
22. Takata K, Amino N, Kubota S. Benefit of short-term iodide supplementation to antithyroid drug treatment of thyrotoxicosis due to Graves’ disease. Clin Endocrinol. 2010;72:845-850.
23. Bilezikian JP, Loeb JN. The influence of hyperthyroidism and hypothyroidism on alpha- and beta-adrenergic receptor systems and adrenergic responsiveness. Endocr Rev. 1983;4:378-388.
24. Jansson S, Lie-Karlsen K, Stenqvist O, et al. Oxygen consumption in patients with hyperthyroidism before and after treatment with beta-blockade versus thyrostatic treatment: a prospective randomized study. Ann Surg. 2001;233:60-64.
25. Zucs-Frkas Z, Toth J, Kollar J, et al. Volume changes in intra- and extraorbital compartments in patients with Graves’ ophthalmopathy: effect of smoking. Thyroid. 2005;15:146-151.
26. Träisk F, Tallstedt L. Thyroid-associated ophthalmopathy after treatment for Graves’ hyperthyroidism with antithyroid drugs or iodine-131. J Clin Endocrinol Metab. 2009;94:3700-3707.
27. Bartalena L, Marcocci C, Bogazzi F, et al. Relation between therapy of hyperthyroidism and the course of Graves’ ophthalmopathy. N Engl J Med. 1998;338:73-78.
28. Acharya SH, Avenell A. Radioiodine therapy (RAI) for Graves’ disease (GD) and the effect on ophthalmopathy: a systematic review. Clin Endocrinol (Oxf). 2008;69:943-950.
29. Stiebel-Kalish H, Robenshtok E. Treatment modalities for Graves’ ophthalmopathy: systematic review and meta-analysis. J Clin Endocrinol Metab. 2009;94:2708-2716.
30. Bradley EA, Gower EW. Orbital radiation for graves ophthalmopathy: a report by the American Academy of Ophthalmology. Ophthalmology. 2008;115:398-409.
31. Wei RL, Cheng JW. The use of orbital radiotherapy for Graves’ ophthalmopathy: quantitative review of the evidence. Ophthalmologica. 2008;222:27-31.
32. Witte J, Goretzki PE, Dotzenrath C, et al. Surgery for Graves’ disease: total versus subtotal thyroidectomy–result of a prospective randomized trial. World J Surg. 2000;24:1303-1311.
33. Heeringa J, Hoogendoorn EH. High-normal thyroid function and risk of atrial fibrillation: the Rotterdam study. Arch Intern Med. 2008;168:2219-2224.
34. Sawin CT, Geller A, Wolf PA, et al. Low serum thyrotropin concentrations as a risk factor for atrial fibrillation in older persons. N Engl J Med. 1994;331:1249-1252.
35. Cappola AR, Fried LP. Thyroid status, cardiovascular risk, and mortality in older adults. JAMA. 2006;295:1033-1041.
36. Siu CW, Yeung CY, Lau CP, et al. Incidence, clinical characteristics and outcome of congestive heart failure as the initial presentation in patients with primary hyperthyroidism. Heart. 2007;93:483-487.
37. Lozano HF, Sharma CN. Reversible pulmonary hypertension, tricuspid regurgitation and right-sided heart failure associated with hyperthyroidism: case report and review of the literature. Cardiol Rev. 2004;12:299-305.
38. Iervasi G, Molinaro S. Association between increased mortality and mild thyroid dysfunction in cardiac patients. Arch Intern Med. 2007;167:1526-1532.
39. Parle JV, Maisonneuve P, Sheppard MC, et al. Prediction of all-cause and cardiovascular mortality in elderly people from one low serum thyrotropin result: a 10-year cohort study. Lancet. 2001;358:861-865.
40. Flynn RW, McDonald TM, Jung RT, et al. Mortality and vascular outcomes in patients treated for thyroid dysfunction. J Clin Endocrinol Metab. 2006;91:2169-2164.
41. Sheu JJ, Kang JH. Hyperthyroidism and risk of ischemic stroke in young adults: a 5-year follow-up study. Stroke. 2010;41:961-966.
42. Vestergaard P, Mosekilde L. Hyperthyroidism, bone mineral, and fracture risk—a meta-analysis. Thyroid. 2003;13:585-593.
43. Diez JJ. Hyperthyroidism in patients older than 55 years: an analysis of the etiology and management. Gerontology. 2003;49:316-323.
44. Sawin CT, Geller A, Wolf PA, et al. Low serum thyrotropin concentrations as a risk factor for atrial fibrillation in older persons. N Engl J Med. 1994;331:1249-1252.
45. Abdulrahman RM, Delgado V. Abnormal cardiac contractility in long-term exogenous subclinical hyperthyroid patients as demonstrated by two-dimensional echocardiography speckle tracking imaging. Eur J Endocrinol. 2010;163:435-441.
46. Faber J, Jensen IW, Petersen L, et al. Normalization of serum thyrotrophin by means of radioiodine treatment in subclinical hyperthyroidism: effect on bone loss in postmenopausal women. Clin Endocrinol (Oxf). 1998;48:285-290.
47. Tan ZS, Beiser A, Vasan RS, et al. Thyroid function and the risk of Alzheimer disease: Framingham study. Arch Intern Med. 2008;168:1514-1520.
48. American Association of Clinical Endocrinologists. Medical guidelines for clinical practice for the evaluation and treatment of hyperthyroidism and hypothyroidism. 2006. Available at: https://www.aace.com/sites/default/files/hypo_hyper.pdf. Accessed July 9, 2010.
49. Chattaway JM, Klepser TB. Propylthiouracil versus methimazole in treatment of Grave’s disease during pregnancy. Ann Pharmacother. 2007;41:1018-1022.
50. Lucas A, Salinas I. Medical therapy of Graves’ disease: does thyroxine prevent recurrence of hyperthyroidism? J Clin Endocrinol Metab. 1997;82:2410-2413.
51. Vitti P, Rago T, Chiovato L, et al. Clinical features of patients with Graves’ disease undergoing remission after antithyroid drug treatment. Thyroid. 1997;7:369-375.
52. Laurberg P, Bulow Pedersen I, Pedersen KM, et al. Low incidence rate of overt hypothyroidism compared with hyperthyroidism in an area with moderately low iodine intake. Thyroid. 1999;9:33-38.
53. Siegel RD, Lee SL. Toxic nodular goiter: Toxic adenoma and toxic multinodular goiter. Endocrinol Metab Clin North Am. 1998;27:151-168.
54. Volpe R. Subacute (de Quervain’s) thyroiditis. Clin Endocrinol Metab. 1979;8:81-95.
55. Nicholson WK, Robinson KA, Smallridge RC, et al. Prevalence of postpartum thyroid dysfunction: a quantitative review. Thyroid. 2006;16:573-582.
56. Roti E, Emerson CH. Clinical review 29: postpartum thyroiditis. J Clin Endocrinol Metab. 1992;74:3-5.
57. Beck-Peccoz P, Brucker-Davis F, Persani L, et al. Thyrotropin-secreting pituitary tumors. Endocr Rev. 1996;17:610-638.