User login
Point of Care Ultrasound (POCUS) for Small Bowel Obstruction in the ED
Small bowel obstruction (SBO) accounts for 2% of all cases of abdominal pain presenting to the ED and 15% of abdominal pain admissions to surgical units from the ED.1,2 SBO can be a difficult diagnosis; the most common symptoms include nausea, vomiting, abdominal pain, obstipation, and constipation. The symptomatology depends on multiple factors: the area of the blockage, length of obstruction, and degree of the obstruction (either partial or complete).3 An upper gastrointestinal (GI) blockage classically presents with nausea and vomiting, while a lower GI blockage often presents with abdominal pain, constipation, and obstipation. Complications of obstruction range from significant morbidity—such as bowel strangulation (23%) and sepsis (31%)—to mortality (9%).4 ED POCUS allows for rapid and accurate diagnosis of SBO.
CASE
A 60-year-old female with a past medical history of peptic ulcer disease and multiple abdominal surgeries, including umbilical hernia repair, appendectomy, and total abdominal hysterectomy, presented to the ED with an 8-hour history of nausea and vomiting. She reported that her abdomen felt bloated. She had experienced non-bloody, watery stools for the prior 3 weeks. She also reported three to four weeks of epigastric abdominal pain similar to her previous “ulcer pain.” Of note, she was evaluated in GI clinic one day prior to her ED visit for dysphagia, abdominal distention, and diarrhea and was scheduled for an outpatient upper endoscopy. Initial vitals were significant for a heart rate of 100 beats/min. Physical exam was significant for a mildly distended abdomen, tender to palpation at epigastrium without rebound or guarding. Labs showed a white blood cell count of 11.8 K/uL and otherwise unremarkable complete blood count, basic metabolic panel, liver function tests, and lactate measurement. Given the patient’s history of multiple abdominal surgeries and clinical presentation, POCUS was performed to evaluate for SBO. Dilated loops of small bowel were visualized in the lower abdomen gas, suggestive of SBO.
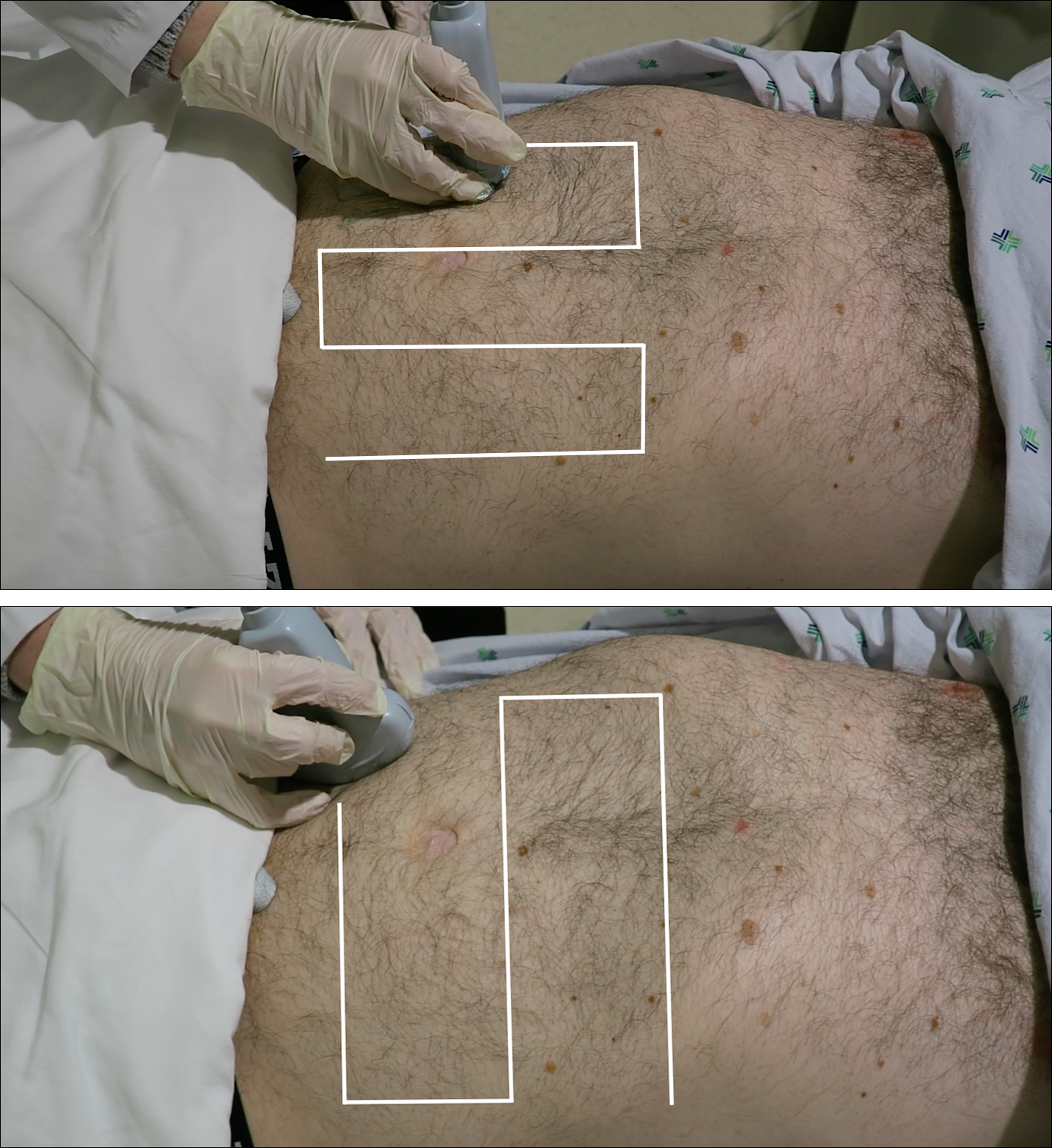
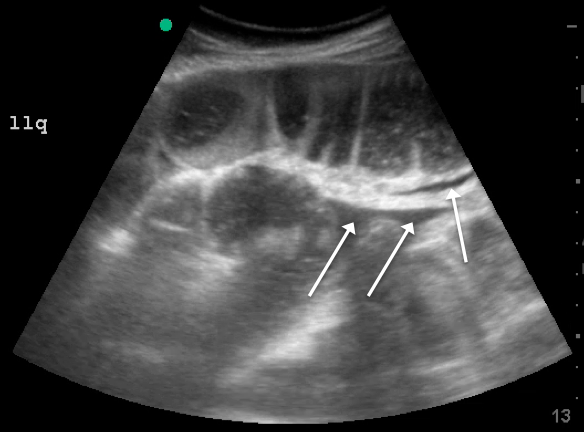
Since the small bowel encompasses a large portion of the abdomen, to fully evaluate for SBO, multiple views are necessary. These include the epigastrium, bilateral colic gutters, and suprapubic regions.5 Use the low-frequency curvilinear transducer to obtain these views, scanning in the transverse and sagittal planes (see Figures 1 and 2). Scan while moving the transducer in columns (ie, “mowing the lawn”), making sure to cover the entire abdomen. To assure that you are evaluating the small bowel, and not the large bowel, look for the characteristic plicae circularis of the small bowel (shown in Figure 3). In children and very slender adults, the high-frequency linear probe may provide enough depth to obtain adequate views.
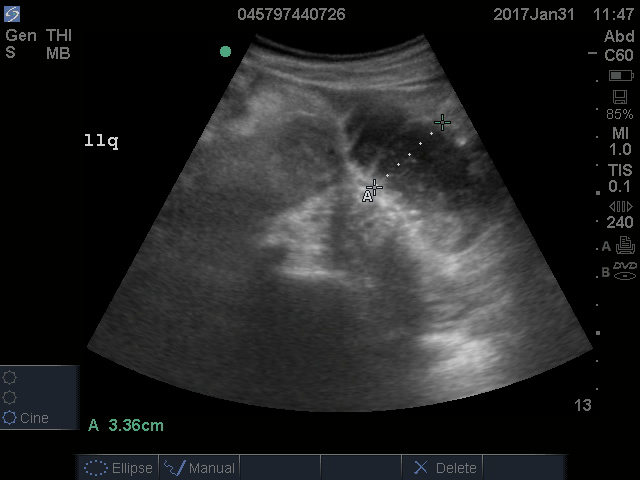
A fluid-filled small intestinal segment >2.5 cm is consistent with a diagnosis of SBO. Measuring the diameter of the small bowel is both the most sensitive and specific sign; a measurement of greater than 2.5 cm is diagnostic, with a sensitivity of 97% and specificity of 91% (see Figure 4).6 This can be somewhat difficult to visualize, as bowel loops are multidirectional and diameters can mistakenly be taken on an indirect cut; to avoid over- or underestimation of bowel diameter, you may want to measure in the short axis using a transverse cross-sectional view of the bowel.
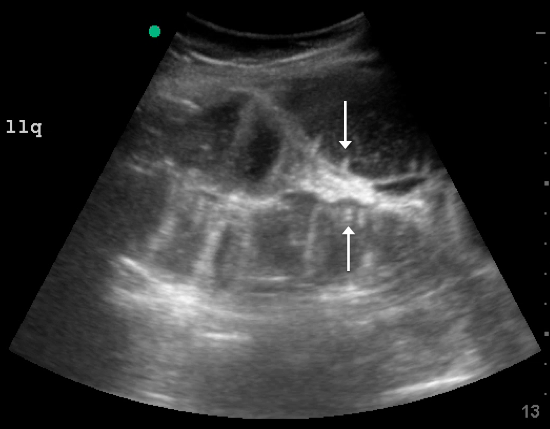
Lack of peristalsis is suggestive of a closed-loop obstruction. However, this finding may be more difficult to visualize, as it requires several continuous minutes of scanning or repeated exams to truly establish absent peristalsis. In prolonged courses of SBO, the bowel wall can measure >3 mm, which suggests necrosis, warranting accelerated surgical intervention. In addition, the detection of extraluminal peritoneal fluid can help determine the severity of the SBO, and small versus large fluid amounts can help determine whether medical or surgical management is warranted (see Figure 5).7
DISCUSSION
Increased time to diagnosis of SBO can lead to prolonged patient suffering and greater complication rates. The gold standard for diagnosing SBO—CT with intravenous and oral contrast—can take hours, requiring patients, who are often nauseated, to ingest and tolerate oral contrast. In the past, an “obstructive series” of x-rays would have been used early in the work-up of possible SBO.6
Recent literature suggests that POCUS is not only faster, more cost effective, and advantageous (involving no ionizing radiation), but also more accurate than x-rays. Specifically, a meta-analysis by Taylor et al showed pooled estimates for obstructive series x-rays have a sensitivity (Sn) of 75%, a specificity (Sp) of 66%, a positive likelihood ratio (+LR) of 1.6, and a negative likelihood ratio (-LR) of 0.43.1 On the other hand, pooled results from ED studies of emergency medicine (EM) residents performing POCUS in patients with signs and symptoms suspicious for SBO showed POCUS had a Sn of 97%, Sp of 90%, +LR of 9.5, and a -LR of 0.04.1,5,8 While detractors point to the operator-dependent nature of POCUS, literature suggests that with EM residents novice to POCUS for SBO (defined as less than 5 previous scans for SBO) were given a 10-minute didactic session and yielded Sn 94%, Sp 81%, +LR 5.0, -LR 0.07.5 Unluer et al trained novice EM residents for 6 hours and found them to yield Sn 98%, Sp 95%, +LR 19.5, and -LR 0.02.8 Thus, while it is no surprise that those with more training attain better results, both studies show it does not take much time for EM providers to surpass the accuracy of x-rays with POCUS.
CASE CONCLUSION
The findings on POCUS highly suggested the diagnosis of an SBO. A CT scan of the abdomen and pelvis with intravenous and oral contrast was ordered to further evaluate obstruction, transition point, and possible complications, including signs of ischemia per surgical request. CT demonstrated dilated loops of small bowel with transition point in the right lower quadrant, with a small amount of mesenteric fluid consistent with SBO with possible early bowel compromise due to ischemia. General surgery admitted the patient; conservative treatment with serial abdominal exams, nasogastric tube, NPO and bowel rest was ordered. The patient’s diet was gradually advanced, and she was discharged on the eleventh day of hospitalization.
SUMMARY
POCUS is a useful non-invasive tool that can accurately diagnose SBO. POCUS has increased sensitivity and specificity when compared to abdominal X-rays. This bedside imaging will not only give the ED provider rapid diagnostic information but also lead to expedited surgical intervention.
- Taylor MR, Lalani N. Adult small bowel obstruction. Acad Emerg Med. 2013;20(6):528-544.
- Hastings RS, Powers RD. Abdominal pain in the ED: a 35-year retrospective. Am J Emerg Med.2011;29:711-716.
- Markogiannakis H, Messaris E, Dardamanis D, et al. Acute mechanical bowel obstruction: clinical presentation, etiology, management and outcome. World J Gastroenterol. 2007;13:432.
- Bickell N, Federman A, Aufses A. Influence of time on risk of bowel resection in complete small bowel obstruction. J Am Coll Surg. 2005;201(6):847-854.
- Jang TB, Chandler D, Kaji AH. Bedside ultrasonography for the detection of small bowel obstruction in the emergency department. Emerg Med J. 2011;28:676-678.
- Carpenter CR, Pines JM. The end of X-rays for suspected small bowel obstruction? Using evidence-based diagnostics to inform best practices in emergency medicine. Acad Emerg Med. 2013;20:618-20.
- Grassi R, Romano S, D’Amario F, et al. The relevance of free fluid between intestinal loops detected by sonography in the clinical assessment of small bowel obstruction in adults. Eur J Radiol. 2004;50(1):5-14.
- Unlüer E, Yavaşi O, Eroğlu O, Yilmaz C, Akarca F. Ultrasonography by emergency medicine and radiology residents for the diagnosis of small bowel obstruction. Eur J Emerg Med. 2010;17(5):260-264.
Small bowel obstruction (SBO) accounts for 2% of all cases of abdominal pain presenting to the ED and 15% of abdominal pain admissions to surgical units from the ED.1,2 SBO can be a difficult diagnosis; the most common symptoms include nausea, vomiting, abdominal pain, obstipation, and constipation. The symptomatology depends on multiple factors: the area of the blockage, length of obstruction, and degree of the obstruction (either partial or complete).3 An upper gastrointestinal (GI) blockage classically presents with nausea and vomiting, while a lower GI blockage often presents with abdominal pain, constipation, and obstipation. Complications of obstruction range from significant morbidity—such as bowel strangulation (23%) and sepsis (31%)—to mortality (9%).4 ED POCUS allows for rapid and accurate diagnosis of SBO.
CASE
A 60-year-old female with a past medical history of peptic ulcer disease and multiple abdominal surgeries, including umbilical hernia repair, appendectomy, and total abdominal hysterectomy, presented to the ED with an 8-hour history of nausea and vomiting. She reported that her abdomen felt bloated. She had experienced non-bloody, watery stools for the prior 3 weeks. She also reported three to four weeks of epigastric abdominal pain similar to her previous “ulcer pain.” Of note, she was evaluated in GI clinic one day prior to her ED visit for dysphagia, abdominal distention, and diarrhea and was scheduled for an outpatient upper endoscopy. Initial vitals were significant for a heart rate of 100 beats/min. Physical exam was significant for a mildly distended abdomen, tender to palpation at epigastrium without rebound or guarding. Labs showed a white blood cell count of 11.8 K/uL and otherwise unremarkable complete blood count, basic metabolic panel, liver function tests, and lactate measurement. Given the patient’s history of multiple abdominal surgeries and clinical presentation, POCUS was performed to evaluate for SBO. Dilated loops of small bowel were visualized in the lower abdomen gas, suggestive of SBO.
Since the small bowel encompasses a large portion of the abdomen, to fully evaluate for SBO, multiple views are necessary. These include the epigastrium, bilateral colic gutters, and suprapubic regions.5 Use the low-frequency curvilinear transducer to obtain these views, scanning in the transverse and sagittal planes (see Figures 1 and 2). Scan while moving the transducer in columns (ie, “mowing the lawn”), making sure to cover the entire abdomen. To assure that you are evaluating the small bowel, and not the large bowel, look for the characteristic plicae circularis of the small bowel (shown in Figure 3). In children and very slender adults, the high-frequency linear probe may provide enough depth to obtain adequate views.
A fluid-filled small intestinal segment >2.5 cm is consistent with a diagnosis of SBO. Measuring the diameter of the small bowel is both the most sensitive and specific sign; a measurement of greater than 2.5 cm is diagnostic, with a sensitivity of 97% and specificity of 91% (see Figure 4).6 This can be somewhat difficult to visualize, as bowel loops are multidirectional and diameters can mistakenly be taken on an indirect cut; to avoid over- or underestimation of bowel diameter, you may want to measure in the short axis using a transverse cross-sectional view of the bowel.
Lack of peristalsis is suggestive of a closed-loop obstruction. However, this finding may be more difficult to visualize, as it requires several continuous minutes of scanning or repeated exams to truly establish absent peristalsis. In prolonged courses of SBO, the bowel wall can measure >3 mm, which suggests necrosis, warranting accelerated surgical intervention. In addition, the detection of extraluminal peritoneal fluid can help determine the severity of the SBO, and small versus large fluid amounts can help determine whether medical or surgical management is warranted (see Figure 5).7
DISCUSSION
Increased time to diagnosis of SBO can lead to prolonged patient suffering and greater complication rates. The gold standard for diagnosing SBO—CT with intravenous and oral contrast—can take hours, requiring patients, who are often nauseated, to ingest and tolerate oral contrast. In the past, an “obstructive series” of x-rays would have been used early in the work-up of possible SBO.6
Recent literature suggests that POCUS is not only faster, more cost effective, and advantageous (involving no ionizing radiation), but also more accurate than x-rays. Specifically, a meta-analysis by Taylor et al showed pooled estimates for obstructive series x-rays have a sensitivity (Sn) of 75%, a specificity (Sp) of 66%, a positive likelihood ratio (+LR) of 1.6, and a negative likelihood ratio (-LR) of 0.43.1 On the other hand, pooled results from ED studies of emergency medicine (EM) residents performing POCUS in patients with signs and symptoms suspicious for SBO showed POCUS had a Sn of 97%, Sp of 90%, +LR of 9.5, and a -LR of 0.04.1,5,8 While detractors point to the operator-dependent nature of POCUS, literature suggests that with EM residents novice to POCUS for SBO (defined as less than 5 previous scans for SBO) were given a 10-minute didactic session and yielded Sn 94%, Sp 81%, +LR 5.0, -LR 0.07.5 Unluer et al trained novice EM residents for 6 hours and found them to yield Sn 98%, Sp 95%, +LR 19.5, and -LR 0.02.8 Thus, while it is no surprise that those with more training attain better results, both studies show it does not take much time for EM providers to surpass the accuracy of x-rays with POCUS.
CASE CONCLUSION
The findings on POCUS highly suggested the diagnosis of an SBO. A CT scan of the abdomen and pelvis with intravenous and oral contrast was ordered to further evaluate obstruction, transition point, and possible complications, including signs of ischemia per surgical request. CT demonstrated dilated loops of small bowel with transition point in the right lower quadrant, with a small amount of mesenteric fluid consistent with SBO with possible early bowel compromise due to ischemia. General surgery admitted the patient; conservative treatment with serial abdominal exams, nasogastric tube, NPO and bowel rest was ordered. The patient’s diet was gradually advanced, and she was discharged on the eleventh day of hospitalization.
SUMMARY
POCUS is a useful non-invasive tool that can accurately diagnose SBO. POCUS has increased sensitivity and specificity when compared to abdominal X-rays. This bedside imaging will not only give the ED provider rapid diagnostic information but also lead to expedited surgical intervention.
Small bowel obstruction (SBO) accounts for 2% of all cases of abdominal pain presenting to the ED and 15% of abdominal pain admissions to surgical units from the ED.1,2 SBO can be a difficult diagnosis; the most common symptoms include nausea, vomiting, abdominal pain, obstipation, and constipation. The symptomatology depends on multiple factors: the area of the blockage, length of obstruction, and degree of the obstruction (either partial or complete).3 An upper gastrointestinal (GI) blockage classically presents with nausea and vomiting, while a lower GI blockage often presents with abdominal pain, constipation, and obstipation. Complications of obstruction range from significant morbidity—such as bowel strangulation (23%) and sepsis (31%)—to mortality (9%).4 ED POCUS allows for rapid and accurate diagnosis of SBO.
CASE
A 60-year-old female with a past medical history of peptic ulcer disease and multiple abdominal surgeries, including umbilical hernia repair, appendectomy, and total abdominal hysterectomy, presented to the ED with an 8-hour history of nausea and vomiting. She reported that her abdomen felt bloated. She had experienced non-bloody, watery stools for the prior 3 weeks. She also reported three to four weeks of epigastric abdominal pain similar to her previous “ulcer pain.” Of note, she was evaluated in GI clinic one day prior to her ED visit for dysphagia, abdominal distention, and diarrhea and was scheduled for an outpatient upper endoscopy. Initial vitals were significant for a heart rate of 100 beats/min. Physical exam was significant for a mildly distended abdomen, tender to palpation at epigastrium without rebound or guarding. Labs showed a white blood cell count of 11.8 K/uL and otherwise unremarkable complete blood count, basic metabolic panel, liver function tests, and lactate measurement. Given the patient’s history of multiple abdominal surgeries and clinical presentation, POCUS was performed to evaluate for SBO. Dilated loops of small bowel were visualized in the lower abdomen gas, suggestive of SBO.
Since the small bowel encompasses a large portion of the abdomen, to fully evaluate for SBO, multiple views are necessary. These include the epigastrium, bilateral colic gutters, and suprapubic regions.5 Use the low-frequency curvilinear transducer to obtain these views, scanning in the transverse and sagittal planes (see Figures 1 and 2). Scan while moving the transducer in columns (ie, “mowing the lawn”), making sure to cover the entire abdomen. To assure that you are evaluating the small bowel, and not the large bowel, look for the characteristic plicae circularis of the small bowel (shown in Figure 3). In children and very slender adults, the high-frequency linear probe may provide enough depth to obtain adequate views.
A fluid-filled small intestinal segment >2.5 cm is consistent with a diagnosis of SBO. Measuring the diameter of the small bowel is both the most sensitive and specific sign; a measurement of greater than 2.5 cm is diagnostic, with a sensitivity of 97% and specificity of 91% (see Figure 4).6 This can be somewhat difficult to visualize, as bowel loops are multidirectional and diameters can mistakenly be taken on an indirect cut; to avoid over- or underestimation of bowel diameter, you may want to measure in the short axis using a transverse cross-sectional view of the bowel.
Lack of peristalsis is suggestive of a closed-loop obstruction. However, this finding may be more difficult to visualize, as it requires several continuous minutes of scanning or repeated exams to truly establish absent peristalsis. In prolonged courses of SBO, the bowel wall can measure >3 mm, which suggests necrosis, warranting accelerated surgical intervention. In addition, the detection of extraluminal peritoneal fluid can help determine the severity of the SBO, and small versus large fluid amounts can help determine whether medical or surgical management is warranted (see Figure 5).7
DISCUSSION
Increased time to diagnosis of SBO can lead to prolonged patient suffering and greater complication rates. The gold standard for diagnosing SBO—CT with intravenous and oral contrast—can take hours, requiring patients, who are often nauseated, to ingest and tolerate oral contrast. In the past, an “obstructive series” of x-rays would have been used early in the work-up of possible SBO.6
Recent literature suggests that POCUS is not only faster, more cost effective, and advantageous (involving no ionizing radiation), but also more accurate than x-rays. Specifically, a meta-analysis by Taylor et al showed pooled estimates for obstructive series x-rays have a sensitivity (Sn) of 75%, a specificity (Sp) of 66%, a positive likelihood ratio (+LR) of 1.6, and a negative likelihood ratio (-LR) of 0.43.1 On the other hand, pooled results from ED studies of emergency medicine (EM) residents performing POCUS in patients with signs and symptoms suspicious for SBO showed POCUS had a Sn of 97%, Sp of 90%, +LR of 9.5, and a -LR of 0.04.1,5,8 While detractors point to the operator-dependent nature of POCUS, literature suggests that with EM residents novice to POCUS for SBO (defined as less than 5 previous scans for SBO) were given a 10-minute didactic session and yielded Sn 94%, Sp 81%, +LR 5.0, -LR 0.07.5 Unluer et al trained novice EM residents for 6 hours and found them to yield Sn 98%, Sp 95%, +LR 19.5, and -LR 0.02.8 Thus, while it is no surprise that those with more training attain better results, both studies show it does not take much time for EM providers to surpass the accuracy of x-rays with POCUS.
CASE CONCLUSION
The findings on POCUS highly suggested the diagnosis of an SBO. A CT scan of the abdomen and pelvis with intravenous and oral contrast was ordered to further evaluate obstruction, transition point, and possible complications, including signs of ischemia per surgical request. CT demonstrated dilated loops of small bowel with transition point in the right lower quadrant, with a small amount of mesenteric fluid consistent with SBO with possible early bowel compromise due to ischemia. General surgery admitted the patient; conservative treatment with serial abdominal exams, nasogastric tube, NPO and bowel rest was ordered. The patient’s diet was gradually advanced, and she was discharged on the eleventh day of hospitalization.
SUMMARY
POCUS is a useful non-invasive tool that can accurately diagnose SBO. POCUS has increased sensitivity and specificity when compared to abdominal X-rays. This bedside imaging will not only give the ED provider rapid diagnostic information but also lead to expedited surgical intervention.
- Taylor MR, Lalani N. Adult small bowel obstruction. Acad Emerg Med. 2013;20(6):528-544.
- Hastings RS, Powers RD. Abdominal pain in the ED: a 35-year retrospective. Am J Emerg Med.2011;29:711-716.
- Markogiannakis H, Messaris E, Dardamanis D, et al. Acute mechanical bowel obstruction: clinical presentation, etiology, management and outcome. World J Gastroenterol. 2007;13:432.
- Bickell N, Federman A, Aufses A. Influence of time on risk of bowel resection in complete small bowel obstruction. J Am Coll Surg. 2005;201(6):847-854.
- Jang TB, Chandler D, Kaji AH. Bedside ultrasonography for the detection of small bowel obstruction in the emergency department. Emerg Med J. 2011;28:676-678.
- Carpenter CR, Pines JM. The end of X-rays for suspected small bowel obstruction? Using evidence-based diagnostics to inform best practices in emergency medicine. Acad Emerg Med. 2013;20:618-20.
- Grassi R, Romano S, D’Amario F, et al. The relevance of free fluid between intestinal loops detected by sonography in the clinical assessment of small bowel obstruction in adults. Eur J Radiol. 2004;50(1):5-14.
- Unlüer E, Yavaşi O, Eroğlu O, Yilmaz C, Akarca F. Ultrasonography by emergency medicine and radiology residents for the diagnosis of small bowel obstruction. Eur J Emerg Med. 2010;17(5):260-264.
- Taylor MR, Lalani N. Adult small bowel obstruction. Acad Emerg Med. 2013;20(6):528-544.
- Hastings RS, Powers RD. Abdominal pain in the ED: a 35-year retrospective. Am J Emerg Med.2011;29:711-716.
- Markogiannakis H, Messaris E, Dardamanis D, et al. Acute mechanical bowel obstruction: clinical presentation, etiology, management and outcome. World J Gastroenterol. 2007;13:432.
- Bickell N, Federman A, Aufses A. Influence of time on risk of bowel resection in complete small bowel obstruction. J Am Coll Surg. 2005;201(6):847-854.
- Jang TB, Chandler D, Kaji AH. Bedside ultrasonography for the detection of small bowel obstruction in the emergency department. Emerg Med J. 2011;28:676-678.
- Carpenter CR, Pines JM. The end of X-rays for suspected small bowel obstruction? Using evidence-based diagnostics to inform best practices in emergency medicine. Acad Emerg Med. 2013;20:618-20.
- Grassi R, Romano S, D’Amario F, et al. The relevance of free fluid between intestinal loops detected by sonography in the clinical assessment of small bowel obstruction in adults. Eur J Radiol. 2004;50(1):5-14.
- Unlüer E, Yavaşi O, Eroğlu O, Yilmaz C, Akarca F. Ultrasonography by emergency medicine and radiology residents for the diagnosis of small bowel obstruction. Eur J Emerg Med. 2010;17(5):260-264.
Malignant olecranon bursitis in the setting of multiple myeloma relapse
Multiple myeloma is the most common plasma cell neoplasm, with an estimated 24,000 cases occurring annually.1 Symptomatic multiple myeloma most commonly presents with one or more of the cardinal CRAB phenomena of hypercalcemia, renal dysfunction, anemia, or lytic bone lesions.2 Less commonly, patients may present with plasmacytomas (focal lesions of malignant plasma cells), which may involve bony or soft tissues.1
Plasma cell neoplasms occasionally involve the joints, including the elbows, typically as plasmacytomas. The elbow is an unusual but reported location of plasmacytomas.3,4 A case of multiple myeloma and amyloid light-chain (AL) amyloidosis has been reported, with manifestations including pseudomyopathy, bone marrow plasmacytosis, and bilateral trochanteric bursitis.5Bursitis is defined as inflammation of the synovial-fluid–containing sacs that lubricate joints. The olecranon bursa is commonly affected. Etiologies include infection, inflammatory disease, trauma, and malignancy. Furthermore, there is an association between bursitis and immunosuppression.6,7 The most common modes of therapy used to treat bursitis are nonsteroidal anti-inflammatory drugs, corticosteroid injections, and surgical management.
Trochanteric bursitis has been attributed to multiple myeloma in one previous case report, but we are not aware of any previous cases of olecranon bursitis caused by multiple myeloma. Here, we present the case of a 46-year-old man with heavily pretreated multiple myeloma and amyloidosis who developed left olecranon bursitis contemporaneously with disease relapse; flow cytometric analysis of the bursal fluid demonstrated an abnormal plasma cell population, establishing the etiology.
Case presentation and summary
A 46-year-old man with a longstanding history of multiple myeloma developed swelling of the left elbow that was initially painless in September 2016. He had been diagnosed with IgA kappa multiple myeloma and AL deposition in 2011. Over the course of his disease, he was treated with the following sequence of therapies: cyclophosphamide, bortezomib, and dexamethasone, followed by melphalan-conditioned autologous peripheral blood stem cell transplant; lenalidomide and dexamethasone; carfilzomib and dexamethasone; pomalidomide, bortezomib, and dexamethasone; and bortezomib, lenalidomide, dexamethasone, doxorubicin, cyclophosphamide, and etoposide, followed by second melphalan-conditioned autologous peripheral blood stem cell transplant. In addition to treatment with numerous novel and chemotherapeutic agents, his disease course was notable for amyloid deposition in the liver, bone marrow, and kidneys, which resulted in dialysis dependence.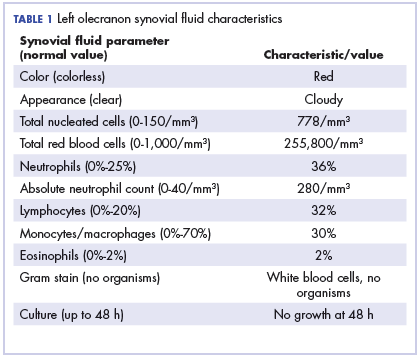
After the second autologous transplant, he achieved a very good partial response and experienced about 9 months of remission, after which laboratory evaluation indicated recurrence of IgA kappa monoclonal protein and free kappa light-chains, which increased slowly over several months without focal symptoms, cytopenias, or decline in organ function (Figure 1).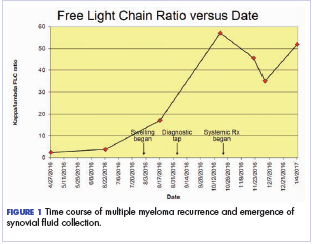
Twelve months after his second transplant, he presented in September 2016 with 4 weeks of left elbow swelling, with the appearance suggesting a fluid collection over the left olecranon process (Figure 2). The fluid collection was not painful unless bumped or pushed. The maximum pain level was 1-2 on a scale of 0-10. His daughter drained the fluid collection on 2 occasions, but it reaccumulated over 2 to 3 days. He reported no fevers, chills, or sweats. He did not have any redness at the site. He did not report any systemic symptoms.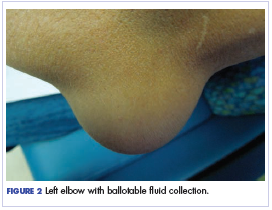
Physical examination of the left elbow demonstrated a ballotable fluid collection associated with the olecranon, with no associated warmth, tenderness, or erythema. Bursal fluid was sampled, yielding orange-colored serous fluid with bland characteristics (Figure 3). Microbiologic studies were negative (Table 1). We did not suspect a malignant cause initially.
The fluid collection persisted despite treatment with nonsteroidal anti-inflammatory drugs and serial drainage procedures approximately twice per week. It became more erythematous and uncomfortable. We repeated diagnostic sampling at 13 months post-transplant. Cytospin revealed scant plasma cells. A multiparametric 8-color flow cytometric analysis was performed on the bursal fluid. It demonstrated the presence of a small abnormal population of plasma cells (0.04%). The abnormal plasma cells showed expression of CD138 and bright CD38 with aberrant expression of CD56, dim CD45, and loss of CD19, CD81 and CD27. They did not express CD117 or CD20 (Figure 4).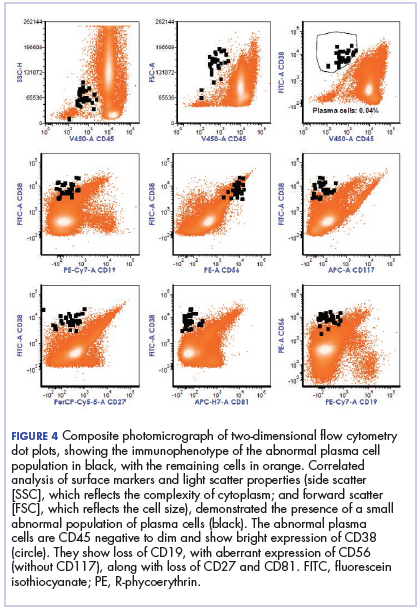
Because of the patient’s discomfort and his history of multidrug-refractory multiple myeloma, we obtained computed tomography imaging of the axial and appendicular skeleton, which demonstrated diffuse small lytic lesions, none larger than 3 mm, including the left elbow joint. The patient began systemic treatment with ixazomib, pomalidomide, and dexamethasone and then received radiation therapy of 20 Gy in 4 fractions to the left olecranon area. The bursal fluid collection remained stable in size but required periodic, though less frequent, drainage procedures. Unfortunately, the patient only tolerated 2 cycles of systemic therapy before experiencing hypercalcemia, exacerbation of hepatic amyloidosis, and a decline in performance status. He died 17 months after the transplant.
Discussion
Our patient experienced left olecranon bursitis simultaneously with relapse of multiple myeloma and AL amyloidosis. Evaluation for infectious causes was negative, and the bursal fluid did not have strongly inflammatory characteristics. Furthermore, a small plasma cell population was isolated from the fluid. Imaging did not reveal an underlying dominant lytic lesion. Although we do not have direct pathologic confirmation, the clinical scenario and flow cytometry findings support our interpretation that the patient’s bursitis was caused by or at least related to underlying multiple myeloma. While reactive plasma cells are also CD38 positive and CD138 positive, they maintain the expression of CD19 and CD45 without aberrant expression of CD56 or CD117 and do not show loss of expression of CD81 or CD27. In this situation, we suspect that either a plasmacytoma involving the soft tissue of the bursa or amyloid infiltration of the synovium may have occurred. Anti-myeloma therapies and radiation therapy did not result in control of the bursitis, though it should be noted that the patient’s highly refractory disease progressed despite treatment with a combination of later-generation immunomodulatory imide and proteasome inhibitor therapies.
Cases of malignant bursitis have been reported several times in the literature, though nearly all of the instances involved connective tissue or metastatic tumors. Tumor histologies include osteochondroma,8,9 malignant fibrous histiocytoma,10 synovial sarcoma,11 and metastatic breast cancer.12
Hematologic malignancies are more rare causes of bursitis; our literature search identified a report of 2 cases of non-Hodgkin lymphoma mimicking rheumatoid arthritis. The joints were the knee and elbow. Synovial fluid from one case was clear and yellow, with leukocytosis with a neutrophilic predominance (similar to our case). In both cases, pathology confirmed lymphomatous infiltration of the synovium.13 Notably, we identified a case of a previously healthy 35-year-old woman with bilateral trochanteric bursitis. Biopsy of tissue from the right trochanteric bursa demonstrated positive birefringence, diagnostic of AL amyloidosis. The patient also had a biclonal paraprotein accompanied by calvarial lytic lesions. She was treated with a corticosteroid pulse and bisphosphonates, followed by autologous hematopoietic stem cell transplant. 5 Our case shares features with the above case, including the relatively young age of the patient and the presence of AL amyloidosis.
Our patient wished to avoid a surgical biopsy procedure, and therefore we utilized flow cytometry of the bursal fluid to establish that the etiology of fluid collection was consistent with his concurrent relapse of multiple myeloma. We believe that we are reporting the second case of multiple myeloma-associated bursitis and the first case associated with multiple myeloma relapse; to our knowledge, it is the first to be diagnosed with the aid of flow cytometry.
Because of our patient’s reliance on hemodialysis beginning one year prior to his presentation with olecranon bursitis, we entertain “dialysis elbow” within the differential diagnosis. Dialysis elbow is a relatively uncommon complication of dialysis, in which patients develop olecranon bursitis on the same side as the hemodialysis access after a prolonged (months to years) duration of hemodialysis. Serositis and mechanical forces are the hypothesized etiologies14; infectious and rheumatologic causes were excluded from the reported cases. Nevertheless, we favor a malignant cause based upon the flow cytometry findings indicating involvement by immunophenotypically abnormal plasma cells.
Our patient was treated initially with serial drainage and nonsteroidals, which had little impact. After diagnosis of a plasma cell population in the fluid, we offered local treatment with radiation and systemic treatment of multiple myeloma, which offered better but suboptimal control. Possible treatments for olecranon bursitis include surgery, corticosteroid injections, anti-inflammatories, and serial drainage. Nonsurgical management may be more effective than surgical management, and corticosteroid injection carries significant risks. On the other hand, serial drainage does not confer additional infection risk in cases with aseptic etiology.15 We combined conservative measures as well as treatment of the underlying disease, but we believe that our patient did not derive significant benefit because of the refractory nature of his disease; he also expressed a preference to avoid surgical intervention.
Conclusion
Bursitis is a rare but thought-provoking potential manifestation of multiple myeloma and AL amyloidosis; we believe that our patient’s bursitis was related to plasma cell neoplasia based upon co-occurrence with disease relapse. His bursitis turned out to be an early indicator of impending systemic relapse. In this particular case, in which the patient wished to avoid surgical intervention, flow cytometry was of great value, and we believe that our case is the first report of malignant bursitis being diagnosed by flow cytometry. Our patient’s case shares similarities with other biopsy-confirmed cases of malignant bursitis, but we were able to avoid the need for surgical biopsy or bursal stripping.
The authors thank Jennifer Wilham MT (ASCP), Pat Byrd MT (ASCP), and Darlene Mann MT (ASCP) for their technical support.
1. Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016;66(6):443-459.
2. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–e548.
3. Gozzetti A, Coviello G, Fabbri A, et al. Unusual localizations of plasmacytoma. Leuk Res. 2011;35(7):e104-e105.
4. Kivioja AH, Karaharju EO, Elomaa I, Böhling TO. Surgical treatment of myeloma of bone. Eur J Cancer. 1992;28(11):1865-1869.
5. Santos MS, Soares B, Mendes O, Carvalho CM, Casimiro RF. Multiple myeloma-amyloidosis presenting as pseudomyopathy. Rev Bras Reumatol. 2011;51(6):651-654. 6. Blackwell JR, Hay BA, Bolt AM, May SM. Olecranon bursitis: a systematic overview. Shoulder Elbow. 2014;6(3):182-190.
7. Reilly D, Kamineni S. Olecranon bursitis. J Shoulder Elbow Surg. 2016;25(1):158-167.
8. De Groote J, Geerts B, Mermuys K, Verstraete K. Osteochondroma of the proximal humerus with frictional bursitis and secondary synovial osteochondromatosis. JBR-BTR. 2015;98(1):45-47. 9. Kumar R, Anjana, Kundan M. Retrocalcaneal bursitis due to rare calcaneal osteochrondroma in adult male: excision and outcome. J Orthop Case Rep. 2016;6(2):16-19.
10. Yoon PW, Jang WY, Yoo JJ, Yoon KS, Kim HJ. Malignant fibrous histiocytoma at the site of an alumina-on-alumina-bearing total hip arthroplasty mimicking infected trochanteric bursitis. J Arthroplasty. 2012;27(2):324.e9-324.e12.
11. Hutchison CW, Kling DH. Malignant synovioma. Am J Cancer. 1940;40(1):8-84.
12. Hutchings C, Hull R. Metastatic bone disease presenting as trochanteric bursitis. J R Soc Med. 1997;90(12):685-686.
13. Dorfman HD, Siegel HL, Perry MC, Oxenhandler R. Non-Hodgkin’s lymphoma of the synovium simulating rheumatoid arthritis. Arthritis Rheum. 1987;30(2):155-161.
14. Chao CT, Wu MS. Dialysis elbow. QJM. 2012;105(5):485-486.
15. Sayegh ET, Strauch RJ. Treatment of olecranon bursitis: a systematic review. Arch Orthop Trauma Surg. 2014;134(11):1517-1536.
Multiple myeloma is the most common plasma cell neoplasm, with an estimated 24,000 cases occurring annually.1 Symptomatic multiple myeloma most commonly presents with one or more of the cardinal CRAB phenomena of hypercalcemia, renal dysfunction, anemia, or lytic bone lesions.2 Less commonly, patients may present with plasmacytomas (focal lesions of malignant plasma cells), which may involve bony or soft tissues.1
Plasma cell neoplasms occasionally involve the joints, including the elbows, typically as plasmacytomas. The elbow is an unusual but reported location of plasmacytomas.3,4 A case of multiple myeloma and amyloid light-chain (AL) amyloidosis has been reported, with manifestations including pseudomyopathy, bone marrow plasmacytosis, and bilateral trochanteric bursitis.5Bursitis is defined as inflammation of the synovial-fluid–containing sacs that lubricate joints. The olecranon bursa is commonly affected. Etiologies include infection, inflammatory disease, trauma, and malignancy. Furthermore, there is an association between bursitis and immunosuppression.6,7 The most common modes of therapy used to treat bursitis are nonsteroidal anti-inflammatory drugs, corticosteroid injections, and surgical management.
Trochanteric bursitis has been attributed to multiple myeloma in one previous case report, but we are not aware of any previous cases of olecranon bursitis caused by multiple myeloma. Here, we present the case of a 46-year-old man with heavily pretreated multiple myeloma and amyloidosis who developed left olecranon bursitis contemporaneously with disease relapse; flow cytometric analysis of the bursal fluid demonstrated an abnormal plasma cell population, establishing the etiology.
Case presentation and summary
A 46-year-old man with a longstanding history of multiple myeloma developed swelling of the left elbow that was initially painless in September 2016. He had been diagnosed with IgA kappa multiple myeloma and AL deposition in 2011. Over the course of his disease, he was treated with the following sequence of therapies: cyclophosphamide, bortezomib, and dexamethasone, followed by melphalan-conditioned autologous peripheral blood stem cell transplant; lenalidomide and dexamethasone; carfilzomib and dexamethasone; pomalidomide, bortezomib, and dexamethasone; and bortezomib, lenalidomide, dexamethasone, doxorubicin, cyclophosphamide, and etoposide, followed by second melphalan-conditioned autologous peripheral blood stem cell transplant. In addition to treatment with numerous novel and chemotherapeutic agents, his disease course was notable for amyloid deposition in the liver, bone marrow, and kidneys, which resulted in dialysis dependence.
After the second autologous transplant, he achieved a very good partial response and experienced about 9 months of remission, after which laboratory evaluation indicated recurrence of IgA kappa monoclonal protein and free kappa light-chains, which increased slowly over several months without focal symptoms, cytopenias, or decline in organ function (Figure 1).
Twelve months after his second transplant, he presented in September 2016 with 4 weeks of left elbow swelling, with the appearance suggesting a fluid collection over the left olecranon process (Figure 2). The fluid collection was not painful unless bumped or pushed. The maximum pain level was 1-2 on a scale of 0-10. His daughter drained the fluid collection on 2 occasions, but it reaccumulated over 2 to 3 days. He reported no fevers, chills, or sweats. He did not have any redness at the site. He did not report any systemic symptoms.
Physical examination of the left elbow demonstrated a ballotable fluid collection associated with the olecranon, with no associated warmth, tenderness, or erythema. Bursal fluid was sampled, yielding orange-colored serous fluid with bland characteristics (Figure 3). Microbiologic studies were negative (Table 1). We did not suspect a malignant cause initially.
The fluid collection persisted despite treatment with nonsteroidal anti-inflammatory drugs and serial drainage procedures approximately twice per week. It became more erythematous and uncomfortable. We repeated diagnostic sampling at 13 months post-transplant. Cytospin revealed scant plasma cells. A multiparametric 8-color flow cytometric analysis was performed on the bursal fluid. It demonstrated the presence of a small abnormal population of plasma cells (0.04%). The abnormal plasma cells showed expression of CD138 and bright CD38 with aberrant expression of CD56, dim CD45, and loss of CD19, CD81 and CD27. They did not express CD117 or CD20 (Figure 4).
Because of the patient’s discomfort and his history of multidrug-refractory multiple myeloma, we obtained computed tomography imaging of the axial and appendicular skeleton, which demonstrated diffuse small lytic lesions, none larger than 3 mm, including the left elbow joint. The patient began systemic treatment with ixazomib, pomalidomide, and dexamethasone and then received radiation therapy of 20 Gy in 4 fractions to the left olecranon area. The bursal fluid collection remained stable in size but required periodic, though less frequent, drainage procedures. Unfortunately, the patient only tolerated 2 cycles of systemic therapy before experiencing hypercalcemia, exacerbation of hepatic amyloidosis, and a decline in performance status. He died 17 months after the transplant.
Discussion
Our patient experienced left olecranon bursitis simultaneously with relapse of multiple myeloma and AL amyloidosis. Evaluation for infectious causes was negative, and the bursal fluid did not have strongly inflammatory characteristics. Furthermore, a small plasma cell population was isolated from the fluid. Imaging did not reveal an underlying dominant lytic lesion. Although we do not have direct pathologic confirmation, the clinical scenario and flow cytometry findings support our interpretation that the patient’s bursitis was caused by or at least related to underlying multiple myeloma. While reactive plasma cells are also CD38 positive and CD138 positive, they maintain the expression of CD19 and CD45 without aberrant expression of CD56 or CD117 and do not show loss of expression of CD81 or CD27. In this situation, we suspect that either a plasmacytoma involving the soft tissue of the bursa or amyloid infiltration of the synovium may have occurred. Anti-myeloma therapies and radiation therapy did not result in control of the bursitis, though it should be noted that the patient’s highly refractory disease progressed despite treatment with a combination of later-generation immunomodulatory imide and proteasome inhibitor therapies.
Cases of malignant bursitis have been reported several times in the literature, though nearly all of the instances involved connective tissue or metastatic tumors. Tumor histologies include osteochondroma,8,9 malignant fibrous histiocytoma,10 synovial sarcoma,11 and metastatic breast cancer.12
Hematologic malignancies are more rare causes of bursitis; our literature search identified a report of 2 cases of non-Hodgkin lymphoma mimicking rheumatoid arthritis. The joints were the knee and elbow. Synovial fluid from one case was clear and yellow, with leukocytosis with a neutrophilic predominance (similar to our case). In both cases, pathology confirmed lymphomatous infiltration of the synovium.13 Notably, we identified a case of a previously healthy 35-year-old woman with bilateral trochanteric bursitis. Biopsy of tissue from the right trochanteric bursa demonstrated positive birefringence, diagnostic of AL amyloidosis. The patient also had a biclonal paraprotein accompanied by calvarial lytic lesions. She was treated with a corticosteroid pulse and bisphosphonates, followed by autologous hematopoietic stem cell transplant. 5 Our case shares features with the above case, including the relatively young age of the patient and the presence of AL amyloidosis.
Our patient wished to avoid a surgical biopsy procedure, and therefore we utilized flow cytometry of the bursal fluid to establish that the etiology of fluid collection was consistent with his concurrent relapse of multiple myeloma. We believe that we are reporting the second case of multiple myeloma-associated bursitis and the first case associated with multiple myeloma relapse; to our knowledge, it is the first to be diagnosed with the aid of flow cytometry.
Because of our patient’s reliance on hemodialysis beginning one year prior to his presentation with olecranon bursitis, we entertain “dialysis elbow” within the differential diagnosis. Dialysis elbow is a relatively uncommon complication of dialysis, in which patients develop olecranon bursitis on the same side as the hemodialysis access after a prolonged (months to years) duration of hemodialysis. Serositis and mechanical forces are the hypothesized etiologies14; infectious and rheumatologic causes were excluded from the reported cases. Nevertheless, we favor a malignant cause based upon the flow cytometry findings indicating involvement by immunophenotypically abnormal plasma cells.
Our patient was treated initially with serial drainage and nonsteroidals, which had little impact. After diagnosis of a plasma cell population in the fluid, we offered local treatment with radiation and systemic treatment of multiple myeloma, which offered better but suboptimal control. Possible treatments for olecranon bursitis include surgery, corticosteroid injections, anti-inflammatories, and serial drainage. Nonsurgical management may be more effective than surgical management, and corticosteroid injection carries significant risks. On the other hand, serial drainage does not confer additional infection risk in cases with aseptic etiology.15 We combined conservative measures as well as treatment of the underlying disease, but we believe that our patient did not derive significant benefit because of the refractory nature of his disease; he also expressed a preference to avoid surgical intervention.
Conclusion
Bursitis is a rare but thought-provoking potential manifestation of multiple myeloma and AL amyloidosis; we believe that our patient’s bursitis was related to plasma cell neoplasia based upon co-occurrence with disease relapse. His bursitis turned out to be an early indicator of impending systemic relapse. In this particular case, in which the patient wished to avoid surgical intervention, flow cytometry was of great value, and we believe that our case is the first report of malignant bursitis being diagnosed by flow cytometry. Our patient’s case shares similarities with other biopsy-confirmed cases of malignant bursitis, but we were able to avoid the need for surgical biopsy or bursal stripping.
The authors thank Jennifer Wilham MT (ASCP), Pat Byrd MT (ASCP), and Darlene Mann MT (ASCP) for their technical support.
Multiple myeloma is the most common plasma cell neoplasm, with an estimated 24,000 cases occurring annually.1 Symptomatic multiple myeloma most commonly presents with one or more of the cardinal CRAB phenomena of hypercalcemia, renal dysfunction, anemia, or lytic bone lesions.2 Less commonly, patients may present with plasmacytomas (focal lesions of malignant plasma cells), which may involve bony or soft tissues.1
Plasma cell neoplasms occasionally involve the joints, including the elbows, typically as plasmacytomas. The elbow is an unusual but reported location of plasmacytomas.3,4 A case of multiple myeloma and amyloid light-chain (AL) amyloidosis has been reported, with manifestations including pseudomyopathy, bone marrow plasmacytosis, and bilateral trochanteric bursitis.5Bursitis is defined as inflammation of the synovial-fluid–containing sacs that lubricate joints. The olecranon bursa is commonly affected. Etiologies include infection, inflammatory disease, trauma, and malignancy. Furthermore, there is an association between bursitis and immunosuppression.6,7 The most common modes of therapy used to treat bursitis are nonsteroidal anti-inflammatory drugs, corticosteroid injections, and surgical management.
Trochanteric bursitis has been attributed to multiple myeloma in one previous case report, but we are not aware of any previous cases of olecranon bursitis caused by multiple myeloma. Here, we present the case of a 46-year-old man with heavily pretreated multiple myeloma and amyloidosis who developed left olecranon bursitis contemporaneously with disease relapse; flow cytometric analysis of the bursal fluid demonstrated an abnormal plasma cell population, establishing the etiology.
Case presentation and summary
A 46-year-old man with a longstanding history of multiple myeloma developed swelling of the left elbow that was initially painless in September 2016. He had been diagnosed with IgA kappa multiple myeloma and AL deposition in 2011. Over the course of his disease, he was treated with the following sequence of therapies: cyclophosphamide, bortezomib, and dexamethasone, followed by melphalan-conditioned autologous peripheral blood stem cell transplant; lenalidomide and dexamethasone; carfilzomib and dexamethasone; pomalidomide, bortezomib, and dexamethasone; and bortezomib, lenalidomide, dexamethasone, doxorubicin, cyclophosphamide, and etoposide, followed by second melphalan-conditioned autologous peripheral blood stem cell transplant. In addition to treatment with numerous novel and chemotherapeutic agents, his disease course was notable for amyloid deposition in the liver, bone marrow, and kidneys, which resulted in dialysis dependence.
After the second autologous transplant, he achieved a very good partial response and experienced about 9 months of remission, after which laboratory evaluation indicated recurrence of IgA kappa monoclonal protein and free kappa light-chains, which increased slowly over several months without focal symptoms, cytopenias, or decline in organ function (Figure 1).
Twelve months after his second transplant, he presented in September 2016 with 4 weeks of left elbow swelling, with the appearance suggesting a fluid collection over the left olecranon process (Figure 2). The fluid collection was not painful unless bumped or pushed. The maximum pain level was 1-2 on a scale of 0-10. His daughter drained the fluid collection on 2 occasions, but it reaccumulated over 2 to 3 days. He reported no fevers, chills, or sweats. He did not have any redness at the site. He did not report any systemic symptoms.
Physical examination of the left elbow demonstrated a ballotable fluid collection associated with the olecranon, with no associated warmth, tenderness, or erythema. Bursal fluid was sampled, yielding orange-colored serous fluid with bland characteristics (Figure 3). Microbiologic studies were negative (Table 1). We did not suspect a malignant cause initially.
The fluid collection persisted despite treatment with nonsteroidal anti-inflammatory drugs and serial drainage procedures approximately twice per week. It became more erythematous and uncomfortable. We repeated diagnostic sampling at 13 months post-transplant. Cytospin revealed scant plasma cells. A multiparametric 8-color flow cytometric analysis was performed on the bursal fluid. It demonstrated the presence of a small abnormal population of plasma cells (0.04%). The abnormal plasma cells showed expression of CD138 and bright CD38 with aberrant expression of CD56, dim CD45, and loss of CD19, CD81 and CD27. They did not express CD117 or CD20 (Figure 4).
Because of the patient’s discomfort and his history of multidrug-refractory multiple myeloma, we obtained computed tomography imaging of the axial and appendicular skeleton, which demonstrated diffuse small lytic lesions, none larger than 3 mm, including the left elbow joint. The patient began systemic treatment with ixazomib, pomalidomide, and dexamethasone and then received radiation therapy of 20 Gy in 4 fractions to the left olecranon area. The bursal fluid collection remained stable in size but required periodic, though less frequent, drainage procedures. Unfortunately, the patient only tolerated 2 cycles of systemic therapy before experiencing hypercalcemia, exacerbation of hepatic amyloidosis, and a decline in performance status. He died 17 months after the transplant.
Discussion
Our patient experienced left olecranon bursitis simultaneously with relapse of multiple myeloma and AL amyloidosis. Evaluation for infectious causes was negative, and the bursal fluid did not have strongly inflammatory characteristics. Furthermore, a small plasma cell population was isolated from the fluid. Imaging did not reveal an underlying dominant lytic lesion. Although we do not have direct pathologic confirmation, the clinical scenario and flow cytometry findings support our interpretation that the patient’s bursitis was caused by or at least related to underlying multiple myeloma. While reactive plasma cells are also CD38 positive and CD138 positive, they maintain the expression of CD19 and CD45 without aberrant expression of CD56 or CD117 and do not show loss of expression of CD81 or CD27. In this situation, we suspect that either a plasmacytoma involving the soft tissue of the bursa or amyloid infiltration of the synovium may have occurred. Anti-myeloma therapies and radiation therapy did not result in control of the bursitis, though it should be noted that the patient’s highly refractory disease progressed despite treatment with a combination of later-generation immunomodulatory imide and proteasome inhibitor therapies.
Cases of malignant bursitis have been reported several times in the literature, though nearly all of the instances involved connective tissue or metastatic tumors. Tumor histologies include osteochondroma,8,9 malignant fibrous histiocytoma,10 synovial sarcoma,11 and metastatic breast cancer.12
Hematologic malignancies are more rare causes of bursitis; our literature search identified a report of 2 cases of non-Hodgkin lymphoma mimicking rheumatoid arthritis. The joints were the knee and elbow. Synovial fluid from one case was clear and yellow, with leukocytosis with a neutrophilic predominance (similar to our case). In both cases, pathology confirmed lymphomatous infiltration of the synovium.13 Notably, we identified a case of a previously healthy 35-year-old woman with bilateral trochanteric bursitis. Biopsy of tissue from the right trochanteric bursa demonstrated positive birefringence, diagnostic of AL amyloidosis. The patient also had a biclonal paraprotein accompanied by calvarial lytic lesions. She was treated with a corticosteroid pulse and bisphosphonates, followed by autologous hematopoietic stem cell transplant. 5 Our case shares features with the above case, including the relatively young age of the patient and the presence of AL amyloidosis.
Our patient wished to avoid a surgical biopsy procedure, and therefore we utilized flow cytometry of the bursal fluid to establish that the etiology of fluid collection was consistent with his concurrent relapse of multiple myeloma. We believe that we are reporting the second case of multiple myeloma-associated bursitis and the first case associated with multiple myeloma relapse; to our knowledge, it is the first to be diagnosed with the aid of flow cytometry.
Because of our patient’s reliance on hemodialysis beginning one year prior to his presentation with olecranon bursitis, we entertain “dialysis elbow” within the differential diagnosis. Dialysis elbow is a relatively uncommon complication of dialysis, in which patients develop olecranon bursitis on the same side as the hemodialysis access after a prolonged (months to years) duration of hemodialysis. Serositis and mechanical forces are the hypothesized etiologies14; infectious and rheumatologic causes were excluded from the reported cases. Nevertheless, we favor a malignant cause based upon the flow cytometry findings indicating involvement by immunophenotypically abnormal plasma cells.
Our patient was treated initially with serial drainage and nonsteroidals, which had little impact. After diagnosis of a plasma cell population in the fluid, we offered local treatment with radiation and systemic treatment of multiple myeloma, which offered better but suboptimal control. Possible treatments for olecranon bursitis include surgery, corticosteroid injections, anti-inflammatories, and serial drainage. Nonsurgical management may be more effective than surgical management, and corticosteroid injection carries significant risks. On the other hand, serial drainage does not confer additional infection risk in cases with aseptic etiology.15 We combined conservative measures as well as treatment of the underlying disease, but we believe that our patient did not derive significant benefit because of the refractory nature of his disease; he also expressed a preference to avoid surgical intervention.
Conclusion
Bursitis is a rare but thought-provoking potential manifestation of multiple myeloma and AL amyloidosis; we believe that our patient’s bursitis was related to plasma cell neoplasia based upon co-occurrence with disease relapse. His bursitis turned out to be an early indicator of impending systemic relapse. In this particular case, in which the patient wished to avoid surgical intervention, flow cytometry was of great value, and we believe that our case is the first report of malignant bursitis being diagnosed by flow cytometry. Our patient’s case shares similarities with other biopsy-confirmed cases of malignant bursitis, but we were able to avoid the need for surgical biopsy or bursal stripping.
The authors thank Jennifer Wilham MT (ASCP), Pat Byrd MT (ASCP), and Darlene Mann MT (ASCP) for their technical support.
1. Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016;66(6):443-459.
2. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–e548.
3. Gozzetti A, Coviello G, Fabbri A, et al. Unusual localizations of plasmacytoma. Leuk Res. 2011;35(7):e104-e105.
4. Kivioja AH, Karaharju EO, Elomaa I, Böhling TO. Surgical treatment of myeloma of bone. Eur J Cancer. 1992;28(11):1865-1869.
5. Santos MS, Soares B, Mendes O, Carvalho CM, Casimiro RF. Multiple myeloma-amyloidosis presenting as pseudomyopathy. Rev Bras Reumatol. 2011;51(6):651-654. 6. Blackwell JR, Hay BA, Bolt AM, May SM. Olecranon bursitis: a systematic overview. Shoulder Elbow. 2014;6(3):182-190.
7. Reilly D, Kamineni S. Olecranon bursitis. J Shoulder Elbow Surg. 2016;25(1):158-167.
8. De Groote J, Geerts B, Mermuys K, Verstraete K. Osteochondroma of the proximal humerus with frictional bursitis and secondary synovial osteochondromatosis. JBR-BTR. 2015;98(1):45-47. 9. Kumar R, Anjana, Kundan M. Retrocalcaneal bursitis due to rare calcaneal osteochrondroma in adult male: excision and outcome. J Orthop Case Rep. 2016;6(2):16-19.
10. Yoon PW, Jang WY, Yoo JJ, Yoon KS, Kim HJ. Malignant fibrous histiocytoma at the site of an alumina-on-alumina-bearing total hip arthroplasty mimicking infected trochanteric bursitis. J Arthroplasty. 2012;27(2):324.e9-324.e12.
11. Hutchison CW, Kling DH. Malignant synovioma. Am J Cancer. 1940;40(1):8-84.
12. Hutchings C, Hull R. Metastatic bone disease presenting as trochanteric bursitis. J R Soc Med. 1997;90(12):685-686.
13. Dorfman HD, Siegel HL, Perry MC, Oxenhandler R. Non-Hodgkin’s lymphoma of the synovium simulating rheumatoid arthritis. Arthritis Rheum. 1987;30(2):155-161.
14. Chao CT, Wu MS. Dialysis elbow. QJM. 2012;105(5):485-486.
15. Sayegh ET, Strauch RJ. Treatment of olecranon bursitis: a systematic review. Arch Orthop Trauma Surg. 2014;134(11):1517-1536.
1. Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016;66(6):443-459.
2. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–e548.
3. Gozzetti A, Coviello G, Fabbri A, et al. Unusual localizations of plasmacytoma. Leuk Res. 2011;35(7):e104-e105.
4. Kivioja AH, Karaharju EO, Elomaa I, Böhling TO. Surgical treatment of myeloma of bone. Eur J Cancer. 1992;28(11):1865-1869.
5. Santos MS, Soares B, Mendes O, Carvalho CM, Casimiro RF. Multiple myeloma-amyloidosis presenting as pseudomyopathy. Rev Bras Reumatol. 2011;51(6):651-654. 6. Blackwell JR, Hay BA, Bolt AM, May SM. Olecranon bursitis: a systematic overview. Shoulder Elbow. 2014;6(3):182-190.
7. Reilly D, Kamineni S. Olecranon bursitis. J Shoulder Elbow Surg. 2016;25(1):158-167.
8. De Groote J, Geerts B, Mermuys K, Verstraete K. Osteochondroma of the proximal humerus with frictional bursitis and secondary synovial osteochondromatosis. JBR-BTR. 2015;98(1):45-47. 9. Kumar R, Anjana, Kundan M. Retrocalcaneal bursitis due to rare calcaneal osteochrondroma in adult male: excision and outcome. J Orthop Case Rep. 2016;6(2):16-19.
10. Yoon PW, Jang WY, Yoo JJ, Yoon KS, Kim HJ. Malignant fibrous histiocytoma at the site of an alumina-on-alumina-bearing total hip arthroplasty mimicking infected trochanteric bursitis. J Arthroplasty. 2012;27(2):324.e9-324.e12.
11. Hutchison CW, Kling DH. Malignant synovioma. Am J Cancer. 1940;40(1):8-84.
12. Hutchings C, Hull R. Metastatic bone disease presenting as trochanteric bursitis. J R Soc Med. 1997;90(12):685-686.
13. Dorfman HD, Siegel HL, Perry MC, Oxenhandler R. Non-Hodgkin’s lymphoma of the synovium simulating rheumatoid arthritis. Arthritis Rheum. 1987;30(2):155-161.
14. Chao CT, Wu MS. Dialysis elbow. QJM. 2012;105(5):485-486.
15. Sayegh ET, Strauch RJ. Treatment of olecranon bursitis: a systematic review. Arch Orthop Trauma Surg. 2014;134(11):1517-1536.
Primary Cutaneous Epstein-Barr Virus–Positive Diffuse Large B-Cell Lymphoma: A Rare and Aggressive Cutaneous Lymphoma
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
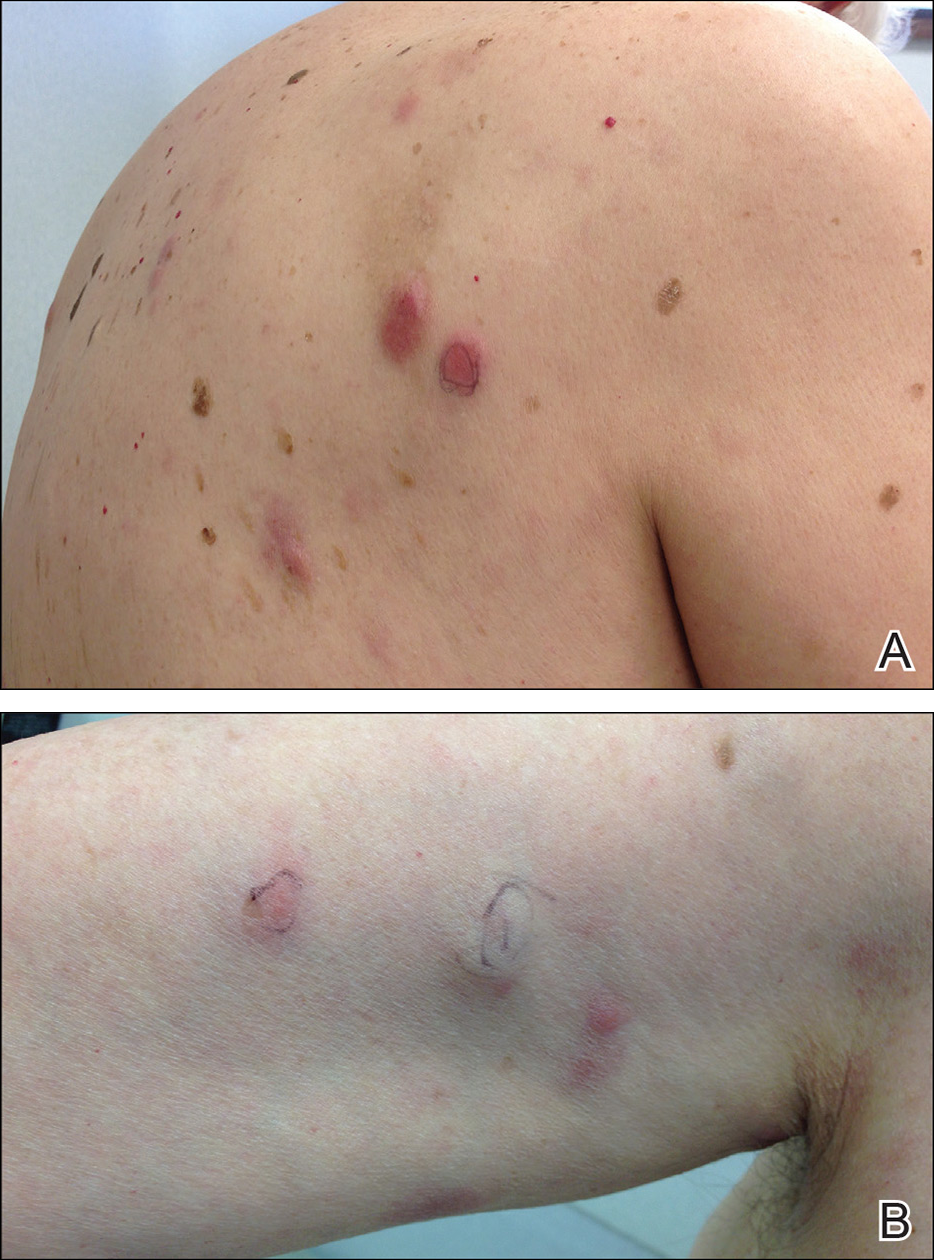
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
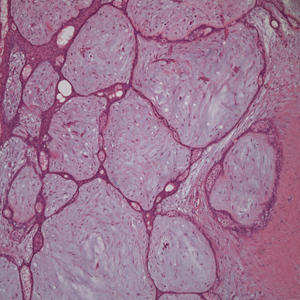
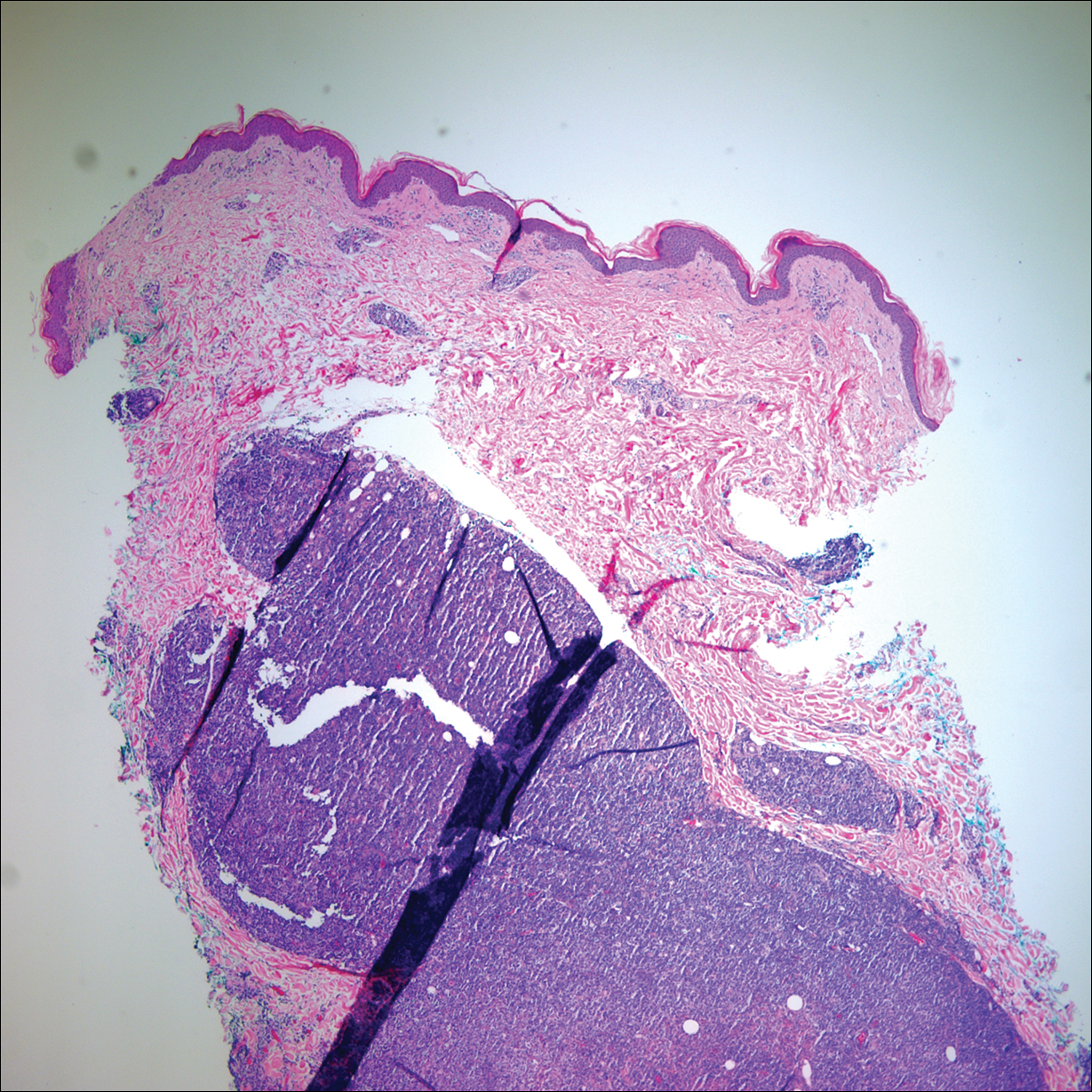
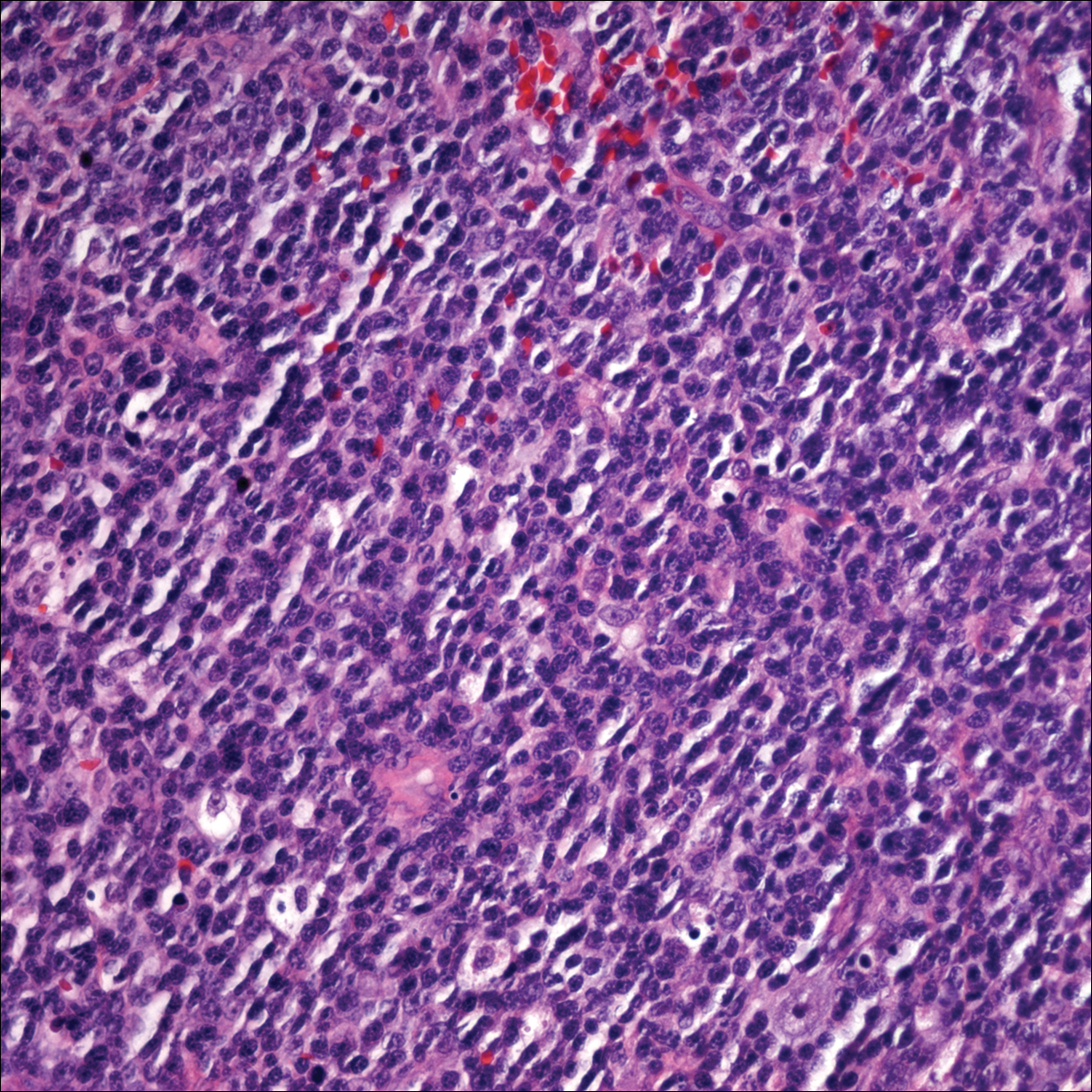
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
Practice Points
- Primary cutaneous lymphomas are malignant lymphomas confined to the skin.
- Complete staging workup is necessary to rule out secondary involvement of the skin from a nodal lymphoma.
- Epstein-Barr virus-positive diffuse large B-cell lymphoma is a rare and aggressive primary cutaneous lymphoma.
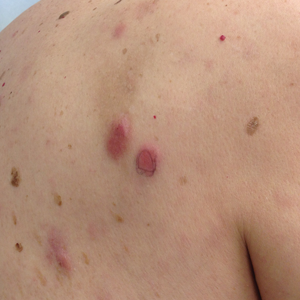
Ichthyosiform Sarcoidosis and Systemic Involvement
Sarcoidosis is a multiorgan, systemic, granulomatous disease that most commonly affects the cutaneous, pulmonary, ocular, and cardiac organ systems. Cutaneous involvement occurs in approximately 20% to 35% of patients, with approximately 25% of patients demonstrating only dermatologic findings.1 Cutaneous sarcoidosis can have a highly variable presentation. Ichthyosiform sarcoidosis (IS) is a rare form of this disease that has been described as presenting as polygonal adherent scales.2 It often is associated with internal organ involvement. We present a case of IS without any organ system involvement at the time of diagnosis. A review of the English-language literature was performed to ascertain the internal organ associations most commonly reported with IS.
Case Report
A 66-year-old black woman presented to dermatology with dark scaly patches noted by her primary care physician to be present on both of the lower extremities. The patient believed they were present for at least 4 years. She described dark spots confined to the lower legs that had gradually increased in size. Review of systems was negative for fever, chills, night sweats, weight loss, vision changes, cough, dyspnea, and joint pains, and there was no history of either personal or familial cutaneous diseases.
Physical examination revealed cutaneous patches of thin white scale with a sharp edge in arciform patterns on the lower extremities. Several of these patches were hyperpigmented and xerotic in appearance (Figure 1). The patches were limited to the lower legs, with no other lesions noted.
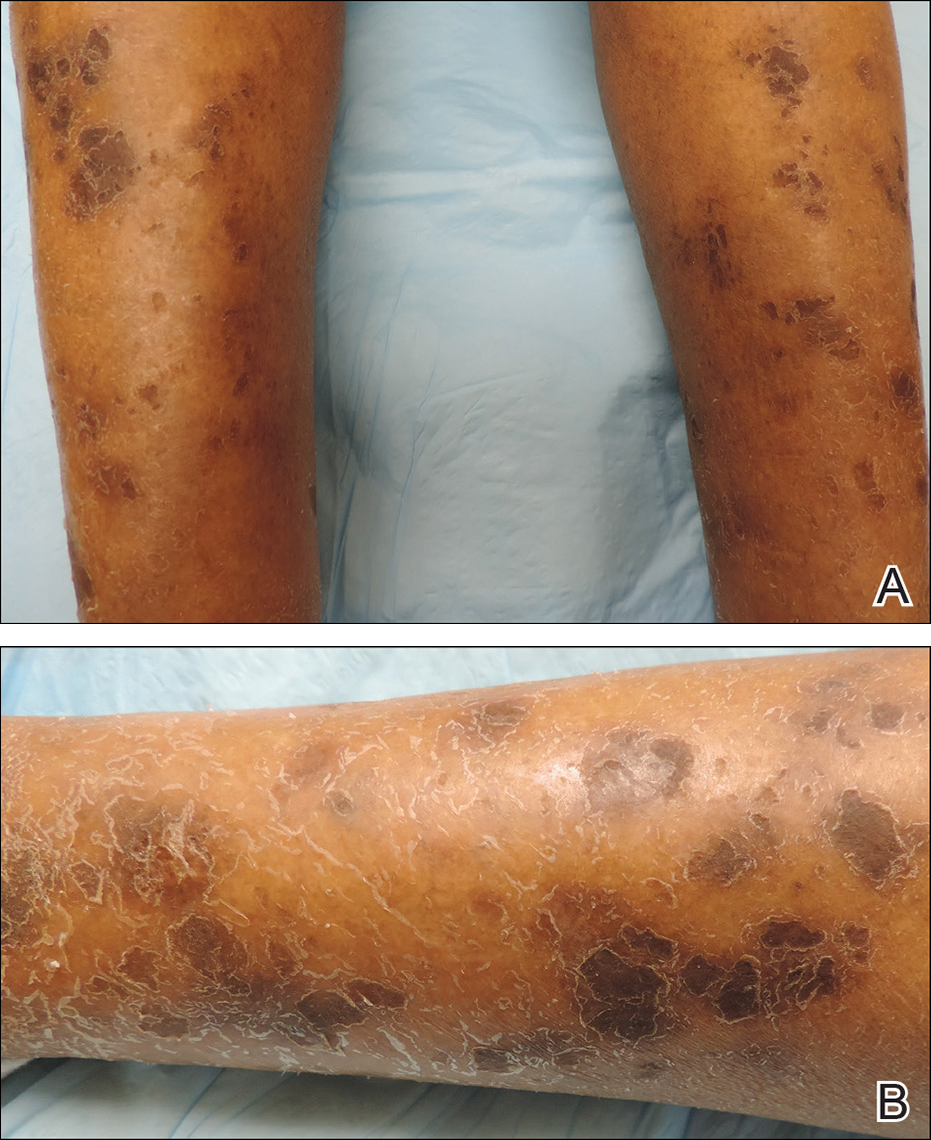
A punch biopsy of the skin on the right lower leg was performed. Histopathologic analysis showed epidermal compact hyperkeratosis with deep granulomatous infiltration into the subcutaneous tissue (Figures 2A and 2B). At high power, these granulomas were noted to be noncaseating naked granulomas composed of epithelioid histiocytes surrounded by sparse lymphocytic inflammation (Figure 2C). Special stains including acid-fast bacilli, Fite, and periodic acid–Schiff were negative. The diagnosis of IS was made based on clinical presentation and primarily by histopathologic analysis.
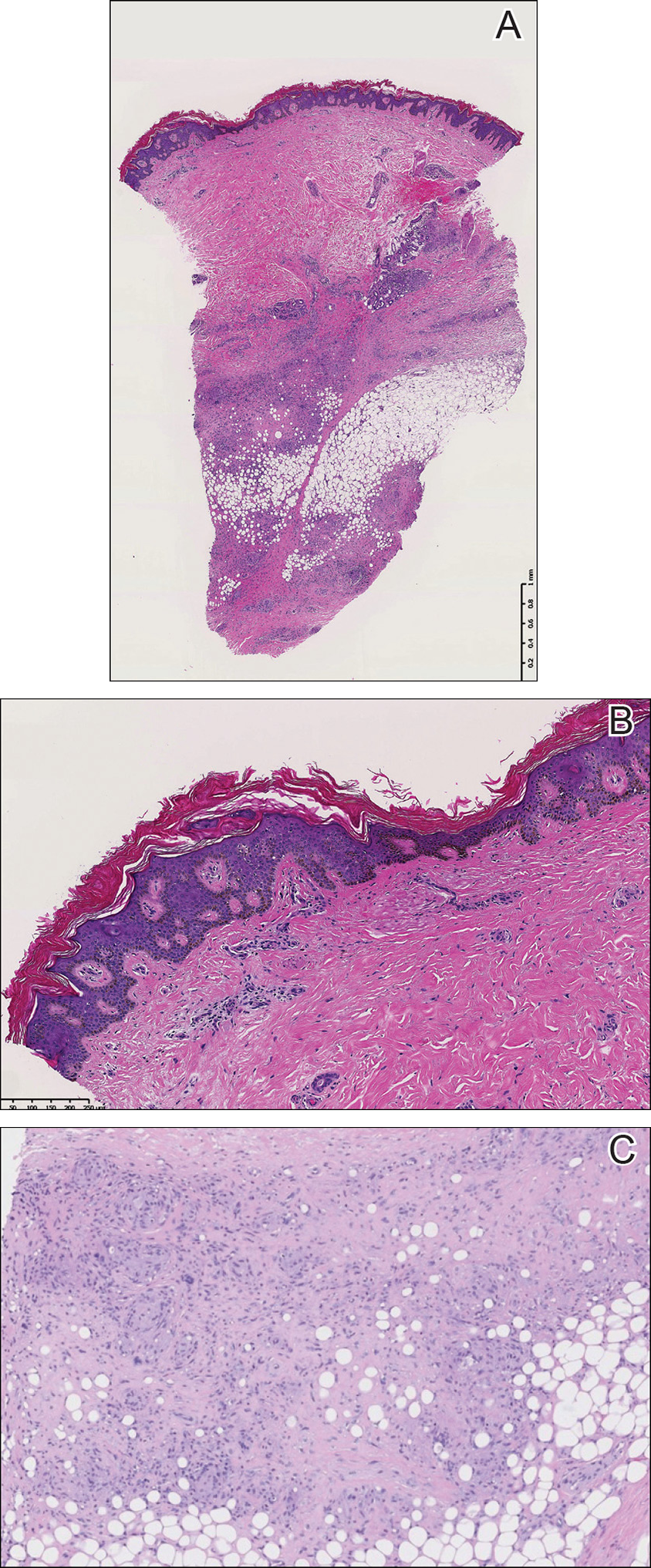
The patient’s cutaneous lesions were treated with fluocinonide ointment 0.05% twice daily. Although she did not notice a dramatic improvement in the plaques, they stabilized in size. Her primary care physician was notified and advised to begin a workup for involvement of other organ systems by sarcoidosis. Her initial evaluation, which included a chest radiograph and electrocardiogram, were unremarkable. Despite multiple attempts to persuade the patient to return for further follow-up, neither dermatology nor her primary care physician were able to complete a full workup.
Comment
Etiology
Although there are several theories regarding the etiology of sarcoidosis, the exact cause remains unknown. The body’s immune response, infectious agents, genetics, and the environment have all been thought to play a role. It has been well established that helper T cell (TH1) production of interferon and increased levels of tumor necrosis factor propagate the inflammatory response seen in sarcoidosis.3 More recently, TH17 cells have been found in cutaneous lesions, bronchoalveolar lavage samples, and the blood of patients with sarcoidosis, especially in those with active disease progression.3 Infectious agents such as mycobacteria and propionibacteria DNA or RNA also have been found in sarcoid samples.4 Several HLA-DRB1 variants have been associated with an increased incidence of sarcoidosis.5
Presentation
Characteristic dermatologic findings of sarcoidosis include macules, papules, nodules, and plaques located on the face, especially the nose, cheeks, and ears, and on the shins or ankles, as well as similar lesions around tattoos or scars. Sarcoid lesions also have been described as angiolupoid, lichenoid, annular, verrucous, ulcerative, and psoriasiform. Here we present an example of the uncommon type, ichthyosiform. Ichthyosiform sarcoidosis is a rare variant described primarily in dark-skinned individuals, a finding supported by both our case and prior reports. Most reported cases have described IS lesions as having a pasted-on appearance, with adherent centers on the extensor surfaces of the lower extremities, head, and/or neck.6 Our case follows this descriptive pattern previously reported with adherent patches limited to the lower extremities.
Histopathology
The key histopathologic finding is the presence of noncaseating granulomas on biopsy. Sarcoid “specific” lesions rest on the identification of the noncaseating granulomas, while “nonspecific” lesions such as erythema nodosum fail to demonstrate this finding.1
Systemic Involvement
The IS type is believed to be an excellent marker for systemic disease, with approximately 95% of reported cases having some form of systemic illness.6 Acquired ichthyosis should warrant further investigation for systemic disease. Early recognition could be beneficial for the patient because the ichthyosiform type is believed to precede the diagnosis of systemic disease in most cases by a median of 3 months.6
The most common site of internal sarcoid involvement is the lungs, but the lymph nodes, eyes, liver, spleen, heart, and central nervous system also can be involved. Patients can present with nonspecific symptoms such as erythema nodosum in the skin, dyspnea, cough, chest pain, vision changes, enlarged lymph nodes, headaches, joint pain, fever, fatigue, weight loss, and malaise. According to a PubMed search of articles indexed for MEDLINE using the term ichthyosiform sarcoidosis, 16 cases have been reported in the English-language literature (eTable).1,6-14 Of these 16 cases, 3 involved men and 13 involved women. The median age of a patient diagnosed with IS was 37 years. The respiratory system was found to be the most common organ system involved (14 of 16 patients), with hilar adenopathy and restrictive lung disease being the most common findings. Neurologic findings and hepatic involvement also were seen in 3 and 3 patients, respectively. Eight of 16 cases had an elevated serum angiotensin-converting enzyme level. Details of systemic involvement in other cases of IS are listed in the eTable.
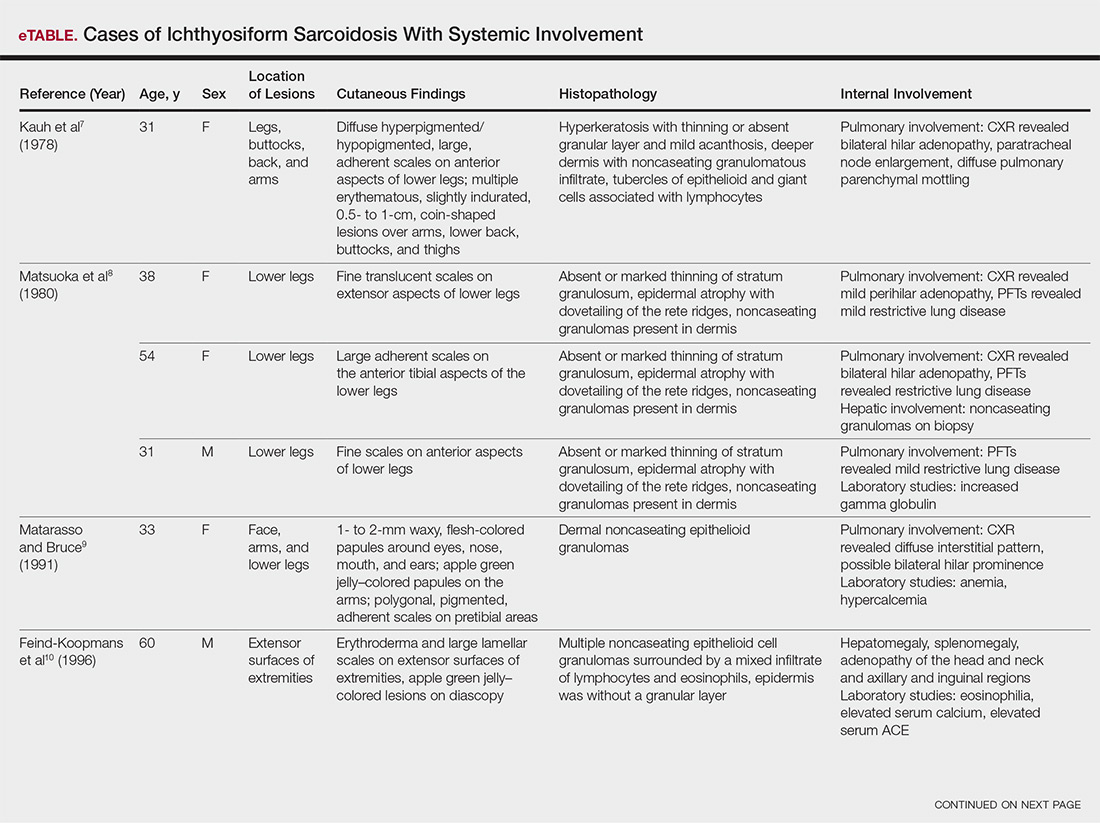
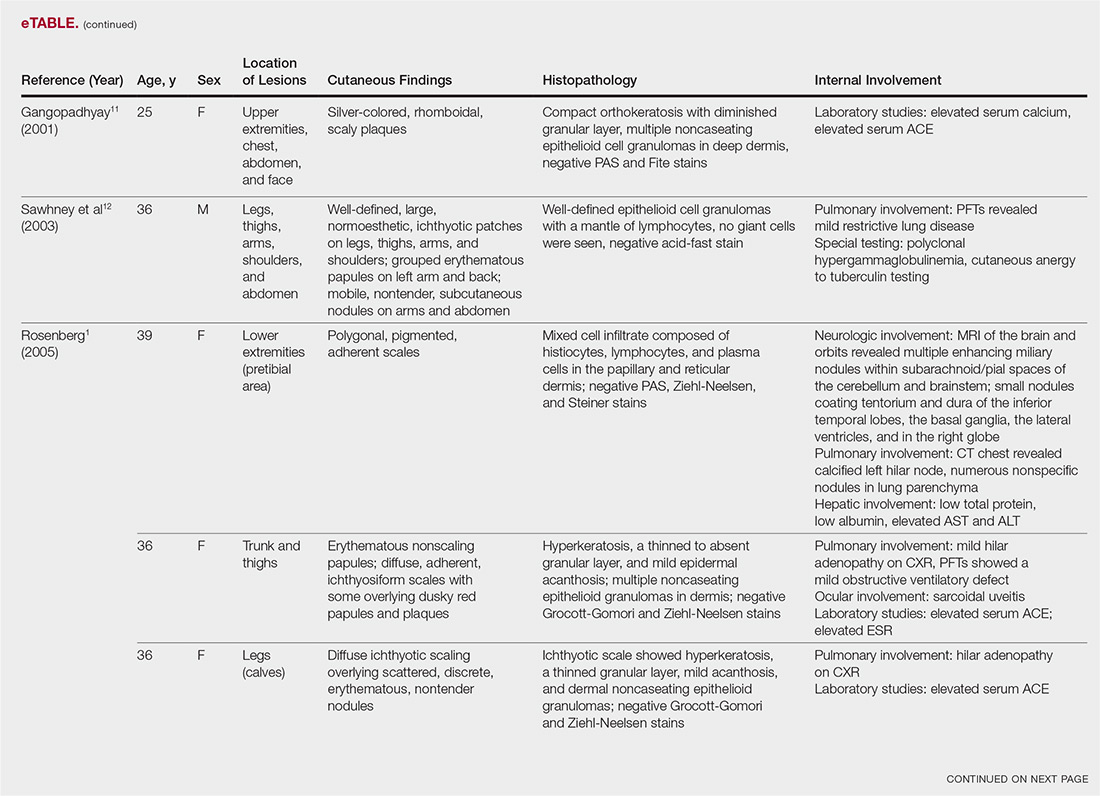
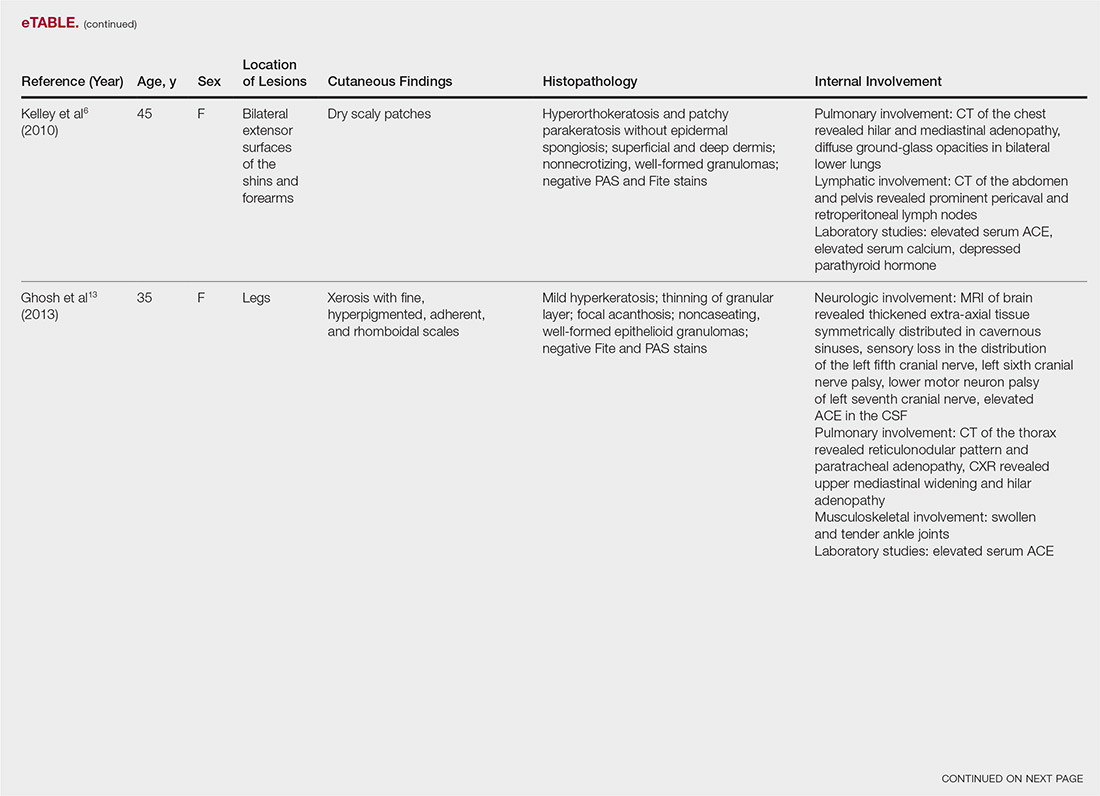
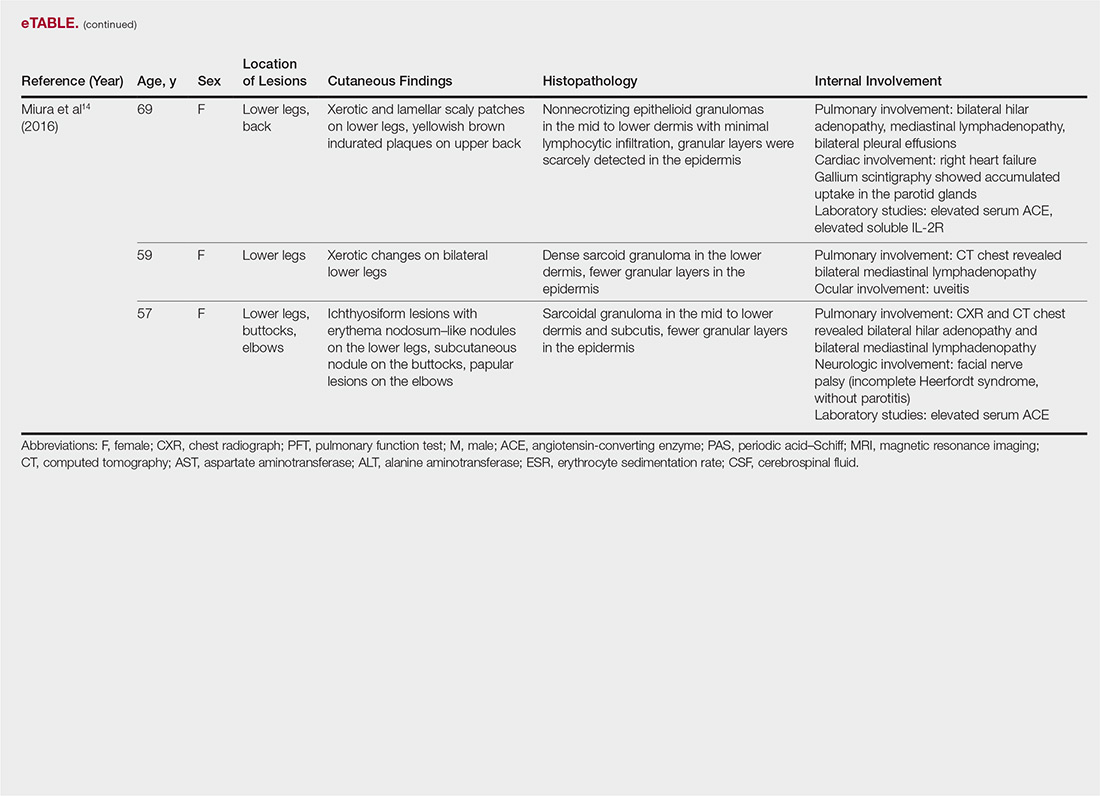
Management
Most patients are given topical corticosteroids for their cutaneous lesions, but patients with systemic involvement will likely need some type of systemic immunosuppressive therapy to control their disease. Systemic therapy often is warranted in IS because of reports of rapid progression. Our case differs from these prior reports in the relative stability of the disease at the last patient encounter. Systemic treatment commonly includes oral corticosteroids such as prednisone. Other options, such as hydroxychloroquine, methotrexate, azathioprine, pentoxifylline, thalidomide, cyclophosphamide, cyclosporine, and infliximab, can be considered if other treatments fail.13 Ichthyosiform sarcoidosis patients should continue to have regular follow-up to monitor for disease progression.
Differential
When evaluating an acquired ichthyosis, dermatologists can consider other associations such as Hodgkin disease, hypothyroidism, multiple myeloma, carcinomatosis, and chronic malnutrition.1 Skin biopsy demonstrating granuloma formation also is not specific for sarcoidosis. Other etiologies, such as autoimmune diseases, immunodeficiency disorders, infections, foreign body granulomas, neoplasms, and drug reactions, should be considered.15 All patients with acquired ichthyosis should undergo a thorough evaluation for internal involvement.
Conclusion
We presented a case of IS, a rare type of sarcoidosis commonly associated with further internal involvement of the respiratory, nervous, or hepatic organ systems. Recognition of an acquired form of ichthyosis and its potential disease associations, including sarcoidosis, is important to improve early detection of any internal disease, allowing prompt initiation of treatment.
- Rosenberg B. Ichthyosiform sarcoidosis. Dermatol Online J. 2005;11:15.
- Banse-Kupin L, Pelachyk JM. Ichthyosiform sarcoidosis: report of two cases and review of the literature. J Am Acad Dermatol. 1987;17:616-620.
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416.
- Celada LJ, Hawkins C, Drake WP. The etiologic role of infectious antigens in sarcoidosis pathogenesis. Clin Chest Med. 2015;36:561-568.
- Fingerlin TE, Hamzeh N, Maier LA. Genetics of sarcoidosis. Clin Chest Med. 2015;36:569-584.
- Kelley BP, George DE, LeLeux TM, et al. Ichthyosiform sarcoidosis: a case report and review of the literature. Dermatol Online J. 2010;16:5.
- Kauh YC, Goody HE, Luscombe HA. Ichthyosiform sarcoidosis. Arch Dermatol. 1978;114:100-101.
- Matsuoka LY, LeVine M, Glasser S, et al. Ichthyosiform sarcoid. Cutis. 1980;25:188-189.
- Matarasso SL, Bruce S. Ichthyosiform sarcoidosis: report of a case. Cutis. 1991;47:405-408.
- Feind-Koopmans AG, Lucker GP, van de Kerkhof PC. Acquired ichthyosiform erythroderma and sarcoidosis. J Am Acad Dermatol. 1996;35:826-828.
- Gangopadhyay AK. Ichthyosiform sarcoidosis. Indian J Dermatol Venereol Leprol. 2001;67:91-92.
- Sawhney M, Sharma YK, Gera V, et al. Ichthyosiform sarcoidosis following chemotherapy of Hodgkin’s disease. Indian J Dermatol Venereol Leprol. 2003;69:220-222.
- Ghosh UC, Ghosh SK, Hazra K, et al. Ichthyosiform sarcoidosis revisited. Indian J Dermatol Venereol Leprol. 2013;79:795-798.
- Miura T, Kato Y, Yamamoto T. Ichthyosiform sarcoidosis: report of three cases from Japan and literature review. Sarcoidosis Vasc Diffuse Lung Dis. 2016;33:392-397.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
Sarcoidosis is a multiorgan, systemic, granulomatous disease that most commonly affects the cutaneous, pulmonary, ocular, and cardiac organ systems. Cutaneous involvement occurs in approximately 20% to 35% of patients, with approximately 25% of patients demonstrating only dermatologic findings.1 Cutaneous sarcoidosis can have a highly variable presentation. Ichthyosiform sarcoidosis (IS) is a rare form of this disease that has been described as presenting as polygonal adherent scales.2 It often is associated with internal organ involvement. We present a case of IS without any organ system involvement at the time of diagnosis. A review of the English-language literature was performed to ascertain the internal organ associations most commonly reported with IS.
Case Report
A 66-year-old black woman presented to dermatology with dark scaly patches noted by her primary care physician to be present on both of the lower extremities. The patient believed they were present for at least 4 years. She described dark spots confined to the lower legs that had gradually increased in size. Review of systems was negative for fever, chills, night sweats, weight loss, vision changes, cough, dyspnea, and joint pains, and there was no history of either personal or familial cutaneous diseases.
Physical examination revealed cutaneous patches of thin white scale with a sharp edge in arciform patterns on the lower extremities. Several of these patches were hyperpigmented and xerotic in appearance (Figure 1). The patches were limited to the lower legs, with no other lesions noted.
A punch biopsy of the skin on the right lower leg was performed. Histopathologic analysis showed epidermal compact hyperkeratosis with deep granulomatous infiltration into the subcutaneous tissue (Figures 2A and 2B). At high power, these granulomas were noted to be noncaseating naked granulomas composed of epithelioid histiocytes surrounded by sparse lymphocytic inflammation (Figure 2C). Special stains including acid-fast bacilli, Fite, and periodic acid–Schiff were negative. The diagnosis of IS was made based on clinical presentation and primarily by histopathologic analysis.
The patient’s cutaneous lesions were treated with fluocinonide ointment 0.05% twice daily. Although she did not notice a dramatic improvement in the plaques, they stabilized in size. Her primary care physician was notified and advised to begin a workup for involvement of other organ systems by sarcoidosis. Her initial evaluation, which included a chest radiograph and electrocardiogram, were unremarkable. Despite multiple attempts to persuade the patient to return for further follow-up, neither dermatology nor her primary care physician were able to complete a full workup.
Comment
Etiology
Although there are several theories regarding the etiology of sarcoidosis, the exact cause remains unknown. The body’s immune response, infectious agents, genetics, and the environment have all been thought to play a role. It has been well established that helper T cell (TH1) production of interferon and increased levels of tumor necrosis factor propagate the inflammatory response seen in sarcoidosis.3 More recently, TH17 cells have been found in cutaneous lesions, bronchoalveolar lavage samples, and the blood of patients with sarcoidosis, especially in those with active disease progression.3 Infectious agents such as mycobacteria and propionibacteria DNA or RNA also have been found in sarcoid samples.4 Several HLA-DRB1 variants have been associated with an increased incidence of sarcoidosis.5
Presentation
Characteristic dermatologic findings of sarcoidosis include macules, papules, nodules, and plaques located on the face, especially the nose, cheeks, and ears, and on the shins or ankles, as well as similar lesions around tattoos or scars. Sarcoid lesions also have been described as angiolupoid, lichenoid, annular, verrucous, ulcerative, and psoriasiform. Here we present an example of the uncommon type, ichthyosiform. Ichthyosiform sarcoidosis is a rare variant described primarily in dark-skinned individuals, a finding supported by both our case and prior reports. Most reported cases have described IS lesions as having a pasted-on appearance, with adherent centers on the extensor surfaces of the lower extremities, head, and/or neck.6 Our case follows this descriptive pattern previously reported with adherent patches limited to the lower extremities.
Histopathology
The key histopathologic finding is the presence of noncaseating granulomas on biopsy. Sarcoid “specific” lesions rest on the identification of the noncaseating granulomas, while “nonspecific” lesions such as erythema nodosum fail to demonstrate this finding.1
Systemic Involvement
The IS type is believed to be an excellent marker for systemic disease, with approximately 95% of reported cases having some form of systemic illness.6 Acquired ichthyosis should warrant further investigation for systemic disease. Early recognition could be beneficial for the patient because the ichthyosiform type is believed to precede the diagnosis of systemic disease in most cases by a median of 3 months.6
The most common site of internal sarcoid involvement is the lungs, but the lymph nodes, eyes, liver, spleen, heart, and central nervous system also can be involved. Patients can present with nonspecific symptoms such as erythema nodosum in the skin, dyspnea, cough, chest pain, vision changes, enlarged lymph nodes, headaches, joint pain, fever, fatigue, weight loss, and malaise. According to a PubMed search of articles indexed for MEDLINE using the term ichthyosiform sarcoidosis, 16 cases have been reported in the English-language literature (eTable).1,6-14 Of these 16 cases, 3 involved men and 13 involved women. The median age of a patient diagnosed with IS was 37 years. The respiratory system was found to be the most common organ system involved (14 of 16 patients), with hilar adenopathy and restrictive lung disease being the most common findings. Neurologic findings and hepatic involvement also were seen in 3 and 3 patients, respectively. Eight of 16 cases had an elevated serum angiotensin-converting enzyme level. Details of systemic involvement in other cases of IS are listed in the eTable.
Management
Most patients are given topical corticosteroids for their cutaneous lesions, but patients with systemic involvement will likely need some type of systemic immunosuppressive therapy to control their disease. Systemic therapy often is warranted in IS because of reports of rapid progression. Our case differs from these prior reports in the relative stability of the disease at the last patient encounter. Systemic treatment commonly includes oral corticosteroids such as prednisone. Other options, such as hydroxychloroquine, methotrexate, azathioprine, pentoxifylline, thalidomide, cyclophosphamide, cyclosporine, and infliximab, can be considered if other treatments fail.13 Ichthyosiform sarcoidosis patients should continue to have regular follow-up to monitor for disease progression.
Differential
When evaluating an acquired ichthyosis, dermatologists can consider other associations such as Hodgkin disease, hypothyroidism, multiple myeloma, carcinomatosis, and chronic malnutrition.1 Skin biopsy demonstrating granuloma formation also is not specific for sarcoidosis. Other etiologies, such as autoimmune diseases, immunodeficiency disorders, infections, foreign body granulomas, neoplasms, and drug reactions, should be considered.15 All patients with acquired ichthyosis should undergo a thorough evaluation for internal involvement.
Conclusion
We presented a case of IS, a rare type of sarcoidosis commonly associated with further internal involvement of the respiratory, nervous, or hepatic organ systems. Recognition of an acquired form of ichthyosis and its potential disease associations, including sarcoidosis, is important to improve early detection of any internal disease, allowing prompt initiation of treatment.
Sarcoidosis is a multiorgan, systemic, granulomatous disease that most commonly affects the cutaneous, pulmonary, ocular, and cardiac organ systems. Cutaneous involvement occurs in approximately 20% to 35% of patients, with approximately 25% of patients demonstrating only dermatologic findings.1 Cutaneous sarcoidosis can have a highly variable presentation. Ichthyosiform sarcoidosis (IS) is a rare form of this disease that has been described as presenting as polygonal adherent scales.2 It often is associated with internal organ involvement. We present a case of IS without any organ system involvement at the time of diagnosis. A review of the English-language literature was performed to ascertain the internal organ associations most commonly reported with IS.
Case Report
A 66-year-old black woman presented to dermatology with dark scaly patches noted by her primary care physician to be present on both of the lower extremities. The patient believed they were present for at least 4 years. She described dark spots confined to the lower legs that had gradually increased in size. Review of systems was negative for fever, chills, night sweats, weight loss, vision changes, cough, dyspnea, and joint pains, and there was no history of either personal or familial cutaneous diseases.
Physical examination revealed cutaneous patches of thin white scale with a sharp edge in arciform patterns on the lower extremities. Several of these patches were hyperpigmented and xerotic in appearance (Figure 1). The patches were limited to the lower legs, with no other lesions noted.
A punch biopsy of the skin on the right lower leg was performed. Histopathologic analysis showed epidermal compact hyperkeratosis with deep granulomatous infiltration into the subcutaneous tissue (Figures 2A and 2B). At high power, these granulomas were noted to be noncaseating naked granulomas composed of epithelioid histiocytes surrounded by sparse lymphocytic inflammation (Figure 2C). Special stains including acid-fast bacilli, Fite, and periodic acid–Schiff were negative. The diagnosis of IS was made based on clinical presentation and primarily by histopathologic analysis.
The patient’s cutaneous lesions were treated with fluocinonide ointment 0.05% twice daily. Although she did not notice a dramatic improvement in the plaques, they stabilized in size. Her primary care physician was notified and advised to begin a workup for involvement of other organ systems by sarcoidosis. Her initial evaluation, which included a chest radiograph and electrocardiogram, were unremarkable. Despite multiple attempts to persuade the patient to return for further follow-up, neither dermatology nor her primary care physician were able to complete a full workup.
Comment
Etiology
Although there are several theories regarding the etiology of sarcoidosis, the exact cause remains unknown. The body’s immune response, infectious agents, genetics, and the environment have all been thought to play a role. It has been well established that helper T cell (TH1) production of interferon and increased levels of tumor necrosis factor propagate the inflammatory response seen in sarcoidosis.3 More recently, TH17 cells have been found in cutaneous lesions, bronchoalveolar lavage samples, and the blood of patients with sarcoidosis, especially in those with active disease progression.3 Infectious agents such as mycobacteria and propionibacteria DNA or RNA also have been found in sarcoid samples.4 Several HLA-DRB1 variants have been associated with an increased incidence of sarcoidosis.5
Presentation
Characteristic dermatologic findings of sarcoidosis include macules, papules, nodules, and plaques located on the face, especially the nose, cheeks, and ears, and on the shins or ankles, as well as similar lesions around tattoos or scars. Sarcoid lesions also have been described as angiolupoid, lichenoid, annular, verrucous, ulcerative, and psoriasiform. Here we present an example of the uncommon type, ichthyosiform. Ichthyosiform sarcoidosis is a rare variant described primarily in dark-skinned individuals, a finding supported by both our case and prior reports. Most reported cases have described IS lesions as having a pasted-on appearance, with adherent centers on the extensor surfaces of the lower extremities, head, and/or neck.6 Our case follows this descriptive pattern previously reported with adherent patches limited to the lower extremities.
Histopathology
The key histopathologic finding is the presence of noncaseating granulomas on biopsy. Sarcoid “specific” lesions rest on the identification of the noncaseating granulomas, while “nonspecific” lesions such as erythema nodosum fail to demonstrate this finding.1
Systemic Involvement
The IS type is believed to be an excellent marker for systemic disease, with approximately 95% of reported cases having some form of systemic illness.6 Acquired ichthyosis should warrant further investigation for systemic disease. Early recognition could be beneficial for the patient because the ichthyosiform type is believed to precede the diagnosis of systemic disease in most cases by a median of 3 months.6
The most common site of internal sarcoid involvement is the lungs, but the lymph nodes, eyes, liver, spleen, heart, and central nervous system also can be involved. Patients can present with nonspecific symptoms such as erythema nodosum in the skin, dyspnea, cough, chest pain, vision changes, enlarged lymph nodes, headaches, joint pain, fever, fatigue, weight loss, and malaise. According to a PubMed search of articles indexed for MEDLINE using the term ichthyosiform sarcoidosis, 16 cases have been reported in the English-language literature (eTable).1,6-14 Of these 16 cases, 3 involved men and 13 involved women. The median age of a patient diagnosed with IS was 37 years. The respiratory system was found to be the most common organ system involved (14 of 16 patients), with hilar adenopathy and restrictive lung disease being the most common findings. Neurologic findings and hepatic involvement also were seen in 3 and 3 patients, respectively. Eight of 16 cases had an elevated serum angiotensin-converting enzyme level. Details of systemic involvement in other cases of IS are listed in the eTable.
Management
Most patients are given topical corticosteroids for their cutaneous lesions, but patients with systemic involvement will likely need some type of systemic immunosuppressive therapy to control their disease. Systemic therapy often is warranted in IS because of reports of rapid progression. Our case differs from these prior reports in the relative stability of the disease at the last patient encounter. Systemic treatment commonly includes oral corticosteroids such as prednisone. Other options, such as hydroxychloroquine, methotrexate, azathioprine, pentoxifylline, thalidomide, cyclophosphamide, cyclosporine, and infliximab, can be considered if other treatments fail.13 Ichthyosiform sarcoidosis patients should continue to have regular follow-up to monitor for disease progression.
Differential
When evaluating an acquired ichthyosis, dermatologists can consider other associations such as Hodgkin disease, hypothyroidism, multiple myeloma, carcinomatosis, and chronic malnutrition.1 Skin biopsy demonstrating granuloma formation also is not specific for sarcoidosis. Other etiologies, such as autoimmune diseases, immunodeficiency disorders, infections, foreign body granulomas, neoplasms, and drug reactions, should be considered.15 All patients with acquired ichthyosis should undergo a thorough evaluation for internal involvement.
Conclusion
We presented a case of IS, a rare type of sarcoidosis commonly associated with further internal involvement of the respiratory, nervous, or hepatic organ systems. Recognition of an acquired form of ichthyosis and its potential disease associations, including sarcoidosis, is important to improve early detection of any internal disease, allowing prompt initiation of treatment.
- Rosenberg B. Ichthyosiform sarcoidosis. Dermatol Online J. 2005;11:15.
- Banse-Kupin L, Pelachyk JM. Ichthyosiform sarcoidosis: report of two cases and review of the literature. J Am Acad Dermatol. 1987;17:616-620.
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416.
- Celada LJ, Hawkins C, Drake WP. The etiologic role of infectious antigens in sarcoidosis pathogenesis. Clin Chest Med. 2015;36:561-568.
- Fingerlin TE, Hamzeh N, Maier LA. Genetics of sarcoidosis. Clin Chest Med. 2015;36:569-584.
- Kelley BP, George DE, LeLeux TM, et al. Ichthyosiform sarcoidosis: a case report and review of the literature. Dermatol Online J. 2010;16:5.
- Kauh YC, Goody HE, Luscombe HA. Ichthyosiform sarcoidosis. Arch Dermatol. 1978;114:100-101.
- Matsuoka LY, LeVine M, Glasser S, et al. Ichthyosiform sarcoid. Cutis. 1980;25:188-189.
- Matarasso SL, Bruce S. Ichthyosiform sarcoidosis: report of a case. Cutis. 1991;47:405-408.
- Feind-Koopmans AG, Lucker GP, van de Kerkhof PC. Acquired ichthyosiform erythroderma and sarcoidosis. J Am Acad Dermatol. 1996;35:826-828.
- Gangopadhyay AK. Ichthyosiform sarcoidosis. Indian J Dermatol Venereol Leprol. 2001;67:91-92.
- Sawhney M, Sharma YK, Gera V, et al. Ichthyosiform sarcoidosis following chemotherapy of Hodgkin’s disease. Indian J Dermatol Venereol Leprol. 2003;69:220-222.
- Ghosh UC, Ghosh SK, Hazra K, et al. Ichthyosiform sarcoidosis revisited. Indian J Dermatol Venereol Leprol. 2013;79:795-798.
- Miura T, Kato Y, Yamamoto T. Ichthyosiform sarcoidosis: report of three cases from Japan and literature review. Sarcoidosis Vasc Diffuse Lung Dis. 2016;33:392-397.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
- Rosenberg B. Ichthyosiform sarcoidosis. Dermatol Online J. 2005;11:15.
- Banse-Kupin L, Pelachyk JM. Ichthyosiform sarcoidosis: report of two cases and review of the literature. J Am Acad Dermatol. 1987;17:616-620.
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416.
- Celada LJ, Hawkins C, Drake WP. The etiologic role of infectious antigens in sarcoidosis pathogenesis. Clin Chest Med. 2015;36:561-568.
- Fingerlin TE, Hamzeh N, Maier LA. Genetics of sarcoidosis. Clin Chest Med. 2015;36:569-584.
- Kelley BP, George DE, LeLeux TM, et al. Ichthyosiform sarcoidosis: a case report and review of the literature. Dermatol Online J. 2010;16:5.
- Kauh YC, Goody HE, Luscombe HA. Ichthyosiform sarcoidosis. Arch Dermatol. 1978;114:100-101.
- Matsuoka LY, LeVine M, Glasser S, et al. Ichthyosiform sarcoid. Cutis. 1980;25:188-189.
- Matarasso SL, Bruce S. Ichthyosiform sarcoidosis: report of a case. Cutis. 1991;47:405-408.
- Feind-Koopmans AG, Lucker GP, van de Kerkhof PC. Acquired ichthyosiform erythroderma and sarcoidosis. J Am Acad Dermatol. 1996;35:826-828.
- Gangopadhyay AK. Ichthyosiform sarcoidosis. Indian J Dermatol Venereol Leprol. 2001;67:91-92.
- Sawhney M, Sharma YK, Gera V, et al. Ichthyosiform sarcoidosis following chemotherapy of Hodgkin’s disease. Indian J Dermatol Venereol Leprol. 2003;69:220-222.
- Ghosh UC, Ghosh SK, Hazra K, et al. Ichthyosiform sarcoidosis revisited. Indian J Dermatol Venereol Leprol. 2013;79:795-798.
- Miura T, Kato Y, Yamamoto T. Ichthyosiform sarcoidosis: report of three cases from Japan and literature review. Sarcoidosis Vasc Diffuse Lung Dis. 2016;33:392-397.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
Practice Points
- Ichthyosiform sarcoidosis is a rare form of sarcoidosis that presents as polygonal adherent scales.
- Ichthyosiform sarcoidosis is commonly associated with pulmonary, neurologic, and hepatic involvement.
- Acquired ichthyosis should warrant further investigation for systemic disease.
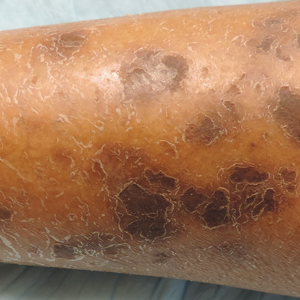
Crizotinib-Induced Lichenoid Drug Eruption in a Patient With Lung Cancer
Crizotinib is a multitargeted tyrosine kinase inhibitor that blocks anaplastic lymphoma kinase (ALK), hepatocyte growth factor receptor (c-Met), and their oncogenic variants ALK fusion proteins or c-Met/hepatocyte growth factor receptor mutant variants.1 Additionally, crizotinib was approved by the US Food and Drug Administration in 2011 for the treatment of patients with non–small cell lung cancer (NSCLC) whose tumors are echinoderm microtubule-associated proteinlike 4 (EML4)/ALK or ROS1 positive.2,3 Among unselected populations of patients with NSCLC, the frequency of EML4/ALK rearrangements ranges from 1.5% to 6.7%.1 Crizotinib is superior to standard chemotherapy in patients with ALK-positive NSCLC.2
In clinical trials, adverse reactions (grades 1 to 4) to crizotinib occurring in at least 25% of patients included visual disturbances, gastrointestinal tract disorders, fatigue, and pitting edema.1,2,4 Adverse reactions (grades 3 and 4) occurring in more than 5% of patients included elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, dyspnea, pneumonia, and neutropenia.1,4 Although the incidence of dermatologic adverse reactions is approximately 11%, substantial progression of drug eruptions rarely has been reported.2,5 We describe a case of lichenoid drug eruption (LDE) that appeared 4 weeks after initiation of crizotinib treatment in a patient with ALK-positive metastatic lung adenocarcinoma.
Case Report
A 61-year-old man presented with a history of ALK-positive NSCLC with lung-to-lung metastasis and pleural seeding treated with a right lower lobectomy and chemotherapy 9 years prior. Chemotherapy was reattempted 5 years later. Targeted therapy with gefitinib was initiated following the lobectomy and 5 years later with erlotinib. The NSCLC was stable, as indicated by computed tomography performed once every 3 or 6 months. After 5 years of treatment, follow-up computed tomography showed slowly growing nodular shadows in the right middle and lower lung fields. Due to this disease progression, treatment with crizotinib (250 mg twice daily) was initiated. Four weeks after the initiation of crizotinib therapy, mild itchy skin eruptions developed on all extremities and the lower lip. He also reported that the skin lesions became more itchy and red with sun exposure. He had no history of drug allergies and denied taking any other medications.
Physical examination revealed multiple brown to violaceous, slightly scaly, flat-topped polygonal papules or plaques on both lower legs (Figure 1A), dorsal hands (Figure 1B), and extensor sites of the elbows, as well as lacelike fine white lines on the lower lip (Figure 1C). There were no nail lesions. The patient’s dermatologic history was unremarkable, except for a few vitiligo lesions on the dorsal hands, extensor sites of the elbows, and mouth angles diagnosed 20 years earlier.
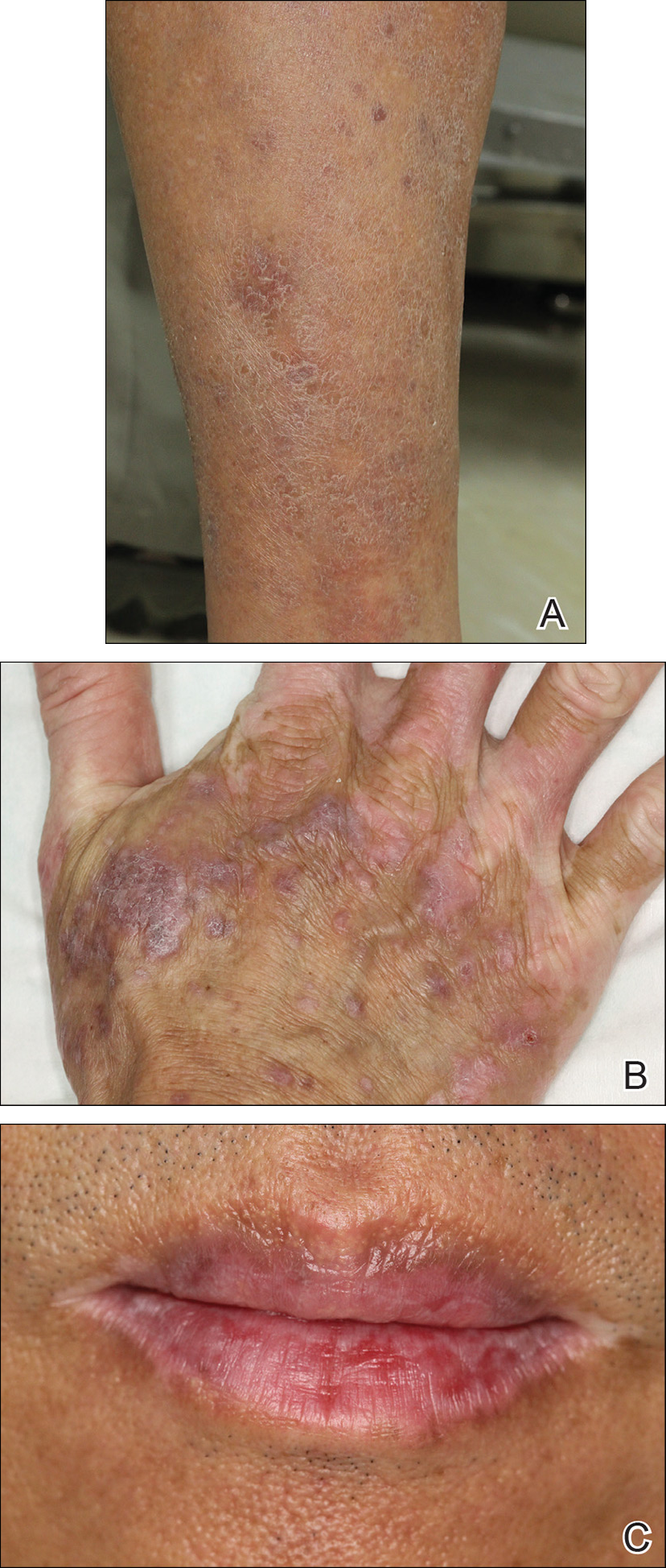
A skin biopsy from the right dorsal hand revealed a lichenoid infiltrate in the superficial dermis composed of lymphocytes, histiocytes and scattered eosinophils, focal parakeratosis, focal hypergranulosis, mild acanthosis, and basal vacuolization (Figure 2A). In addition, some dyskeratotic keratinocytes in the stratum spinosum and granulosum were identified (Figure 2B). The histopathology was consistent with the diagnosis of an LDE. Direct immunofluorescence revealed no globular or cytoid body–like deposits of immunoglobulin, with IgM, IgA, IgG, or C3 in the epidermis, dermis, and basement membrane zone. Routine laboratory studies revealed elevated liver enzymes, including an ALT level of 115 U/L (reference range, 0–40 U/L) and AST level of 60 U/L (reference range, 5–45 U/L). Negative results for the serum hepatitis B surface antigen and anti– hepatitis C virus tests were recorded. The patient had no medical history of alcohol consumption or abnormal liver function tests. The skin lesions were treated with diflucortolone valerate fatty ointment 0.1% twice daily and abnormal liver functions were treated with silymarin (150 mg per cap twice daily). He experienced some improvement.
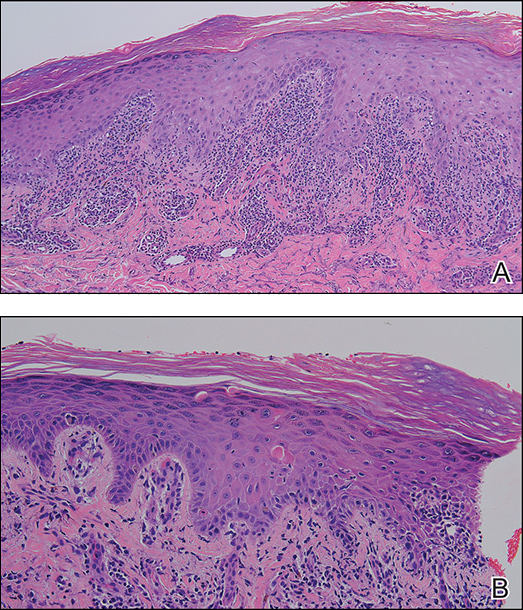
A causality assessment was performed using the Naranjo Adverse Drug Reaction Probability Scale,6,7 and we concluded that crizotinib was the possible cause (Naranjo score, 4) of this adverse drug reaction (Table). Because the skin reaction was tolerable and liver enzymes were mildly elevated (ALT, 50 U/L; AST, 48 U/L), the offending drug was continued to benefit the underlying disease. His NSCLC was stable on computed tomography 3 months later.
Comment
The number of indicated uses of crizotinib, an oral small-molecule ALK tyrosine kinase inhibitor for the treatment of NSCLC, has gradually increased, but only a few cases of cutaneous adverse reactions, such as erythema multiforme and severe photosensitivity dermatitis, have been reported.2,5 Skin toxicity is a common and well-known side effect of other small-molecule tyrosine kinase inhibitors, particularly epidermal growth factor receptor inhibitors.8 However, LDE is not commonly associated with small-molecule tyrosine kinase inhibitors, though it has been described in a few patients taking imatinib for chronic myelogenous leukemia and gastrointestinal tract stromal tumors.9,10
The clinical morphology of LDE may resemble lichen planus, but certain features, such as larger skin lesions, the absence of Wickham striae, and photodistribution, help to differentiate between the two.10 Histologically, some findings are more common in LDE, including focal parakeratosis, cytoid bodies in the cornified and granular layers, and the presence of eosinophils.11
Our patient developed lichenoid rashes after 1 month of crizotinib therapy. The latency period for developing a medication-induced LDE varies from months to 1 year and is dependent on the dosage, host response, prior exposure, and concomitant drug administration. No additional medications had been added to our patient’s regimen after initiating crizotinib therapy, and he did not take any other known medications. Ultimately, based on the time-event relationship, morphology, distribution, and histopathologic findings, we concluded that our patient developed an LDE due to crizotinib.
Our patient also had a history of vitiligo affecting the hands, elbows, and mouth angles for 20 years. Although there are limited reports of a possible causal link between lichen planus or drug-induced lichen planus eruption and vitiligo,12-14 we do not think these conditions were associated in our case because the patient’s vitiligo lesions persisted for many years, did not progress, and remained inactive and stable, and there was a lack of co-localization of LDE and vitiligo.
Our patient reported that the skin eruptions worsened after sun exposure. Oser and Janne5 also reported a patient with ALK-positive metastatic lung adenocarcinoma who developed severe crizotinib-induced photosensitive rashes. Further accumulation of similar cases and pathophysiological studies will be necessary to clarify whether this photosensitivity dermatitis is caused by ALK inhibition itself or mediated through host-immune mechanisms.5
Conclusion
As crizotinib prescriptions for patients with NSCLC are increasing, clinicians should be aware of the possibility of cutaneous LDEs occurring as an adverse effect. Additionally, physicians should treat appropriately to avoid unnecessarily discontinuing a potentially life-saving medication and to improve quality of life for patients with NSCLC who are treated with crizotinib.
- Malik SM, Maher VE, Bijwaard KE, et al. U.S. Food and Drug Administration approval: crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res. 2014;20:2029-2034.
- Sawamura S, Kajihara I, Ichihara A, et al. Crizotinib-associated erythema multiforme in a lung cancer patient. Drug Discov Ther. 2015;9:142-143.
- Liao BC, Lin CC, Shih JY, et al. Treating patients with ALK-positive non-small cell lung cancer: latest evidence and management strategy. Ther Adv Med Oncol. 2015;7:274-290.
- Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011-1019.
- Oser MG, Janne PA. A severe photosensitivity dermatitis caused by crizotinib. J Thorac Oncol. 2014;9:E51-E53.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Zaki SA. Adverse drug reaction and causality assessment scales. Lung India. 2011;28:152-153.
- Aw DC, Tan EH, Chin TM, et al. Management of epidermal growth factor receptor tyrosine kinase inhibitor-related cutaneous and gastrointestinal toxicities. Asia Pac J Clin Oncol. 2018;14:23-31.
- Penn EH, Chung HJ, Keller M. Imatinib mesylate-induced lichenoid drug eruption. Cutis. 2017;99:189-192.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Lage D, Juliano PB, Metze K, et al. Lichen planus and lichenoid drug-induced eruption: a histological and immunohistochemical study. Int J Dermatol. 2012;51:1199-1205.
- Veitch D, Kravvas G, Hughes S, et al. A rare colocalization of lichen planus and vitiligo. Case Rep Dermatol Med. 2015;2015:840193.
- Baghestani S, Moosavi A, Eftekhari T. Familial colocalization of lichen planus and vitiligo on sun exposed areas. Ann Dermatol. 2013;25:223-225.
- Chan WP, Mackey VT, Sun DK. Telmisartan-induced lichen planus eruption manifested on vitiliginous skin. Cutis. 2017;99:E16-E19.
Crizotinib is a multitargeted tyrosine kinase inhibitor that blocks anaplastic lymphoma kinase (ALK), hepatocyte growth factor receptor (c-Met), and their oncogenic variants ALK fusion proteins or c-Met/hepatocyte growth factor receptor mutant variants.1 Additionally, crizotinib was approved by the US Food and Drug Administration in 2011 for the treatment of patients with non–small cell lung cancer (NSCLC) whose tumors are echinoderm microtubule-associated proteinlike 4 (EML4)/ALK or ROS1 positive.2,3 Among unselected populations of patients with NSCLC, the frequency of EML4/ALK rearrangements ranges from 1.5% to 6.7%.1 Crizotinib is superior to standard chemotherapy in patients with ALK-positive NSCLC.2
In clinical trials, adverse reactions (grades 1 to 4) to crizotinib occurring in at least 25% of patients included visual disturbances, gastrointestinal tract disorders, fatigue, and pitting edema.1,2,4 Adverse reactions (grades 3 and 4) occurring in more than 5% of patients included elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, dyspnea, pneumonia, and neutropenia.1,4 Although the incidence of dermatologic adverse reactions is approximately 11%, substantial progression of drug eruptions rarely has been reported.2,5 We describe a case of lichenoid drug eruption (LDE) that appeared 4 weeks after initiation of crizotinib treatment in a patient with ALK-positive metastatic lung adenocarcinoma.
Case Report
A 61-year-old man presented with a history of ALK-positive NSCLC with lung-to-lung metastasis and pleural seeding treated with a right lower lobectomy and chemotherapy 9 years prior. Chemotherapy was reattempted 5 years later. Targeted therapy with gefitinib was initiated following the lobectomy and 5 years later with erlotinib. The NSCLC was stable, as indicated by computed tomography performed once every 3 or 6 months. After 5 years of treatment, follow-up computed tomography showed slowly growing nodular shadows in the right middle and lower lung fields. Due to this disease progression, treatment with crizotinib (250 mg twice daily) was initiated. Four weeks after the initiation of crizotinib therapy, mild itchy skin eruptions developed on all extremities and the lower lip. He also reported that the skin lesions became more itchy and red with sun exposure. He had no history of drug allergies and denied taking any other medications.
Physical examination revealed multiple brown to violaceous, slightly scaly, flat-topped polygonal papules or plaques on both lower legs (Figure 1A), dorsal hands (Figure 1B), and extensor sites of the elbows, as well as lacelike fine white lines on the lower lip (Figure 1C). There were no nail lesions. The patient’s dermatologic history was unremarkable, except for a few vitiligo lesions on the dorsal hands, extensor sites of the elbows, and mouth angles diagnosed 20 years earlier.
A skin biopsy from the right dorsal hand revealed a lichenoid infiltrate in the superficial dermis composed of lymphocytes, histiocytes and scattered eosinophils, focal parakeratosis, focal hypergranulosis, mild acanthosis, and basal vacuolization (Figure 2A). In addition, some dyskeratotic keratinocytes in the stratum spinosum and granulosum were identified (Figure 2B). The histopathology was consistent with the diagnosis of an LDE. Direct immunofluorescence revealed no globular or cytoid body–like deposits of immunoglobulin, with IgM, IgA, IgG, or C3 in the epidermis, dermis, and basement membrane zone. Routine laboratory studies revealed elevated liver enzymes, including an ALT level of 115 U/L (reference range, 0–40 U/L) and AST level of 60 U/L (reference range, 5–45 U/L). Negative results for the serum hepatitis B surface antigen and anti– hepatitis C virus tests were recorded. The patient had no medical history of alcohol consumption or abnormal liver function tests. The skin lesions were treated with diflucortolone valerate fatty ointment 0.1% twice daily and abnormal liver functions were treated with silymarin (150 mg per cap twice daily). He experienced some improvement.
A causality assessment was performed using the Naranjo Adverse Drug Reaction Probability Scale,6,7 and we concluded that crizotinib was the possible cause (Naranjo score, 4) of this adverse drug reaction (Table). Because the skin reaction was tolerable and liver enzymes were mildly elevated (ALT, 50 U/L; AST, 48 U/L), the offending drug was continued to benefit the underlying disease. His NSCLC was stable on computed tomography 3 months later.
Comment
The number of indicated uses of crizotinib, an oral small-molecule ALK tyrosine kinase inhibitor for the treatment of NSCLC, has gradually increased, but only a few cases of cutaneous adverse reactions, such as erythema multiforme and severe photosensitivity dermatitis, have been reported.2,5 Skin toxicity is a common and well-known side effect of other small-molecule tyrosine kinase inhibitors, particularly epidermal growth factor receptor inhibitors.8 However, LDE is not commonly associated with small-molecule tyrosine kinase inhibitors, though it has been described in a few patients taking imatinib for chronic myelogenous leukemia and gastrointestinal tract stromal tumors.9,10
The clinical morphology of LDE may resemble lichen planus, but certain features, such as larger skin lesions, the absence of Wickham striae, and photodistribution, help to differentiate between the two.10 Histologically, some findings are more common in LDE, including focal parakeratosis, cytoid bodies in the cornified and granular layers, and the presence of eosinophils.11
Our patient developed lichenoid rashes after 1 month of crizotinib therapy. The latency period for developing a medication-induced LDE varies from months to 1 year and is dependent on the dosage, host response, prior exposure, and concomitant drug administration. No additional medications had been added to our patient’s regimen after initiating crizotinib therapy, and he did not take any other known medications. Ultimately, based on the time-event relationship, morphology, distribution, and histopathologic findings, we concluded that our patient developed an LDE due to crizotinib.
Our patient also had a history of vitiligo affecting the hands, elbows, and mouth angles for 20 years. Although there are limited reports of a possible causal link between lichen planus or drug-induced lichen planus eruption and vitiligo,12-14 we do not think these conditions were associated in our case because the patient’s vitiligo lesions persisted for many years, did not progress, and remained inactive and stable, and there was a lack of co-localization of LDE and vitiligo.
Our patient reported that the skin eruptions worsened after sun exposure. Oser and Janne5 also reported a patient with ALK-positive metastatic lung adenocarcinoma who developed severe crizotinib-induced photosensitive rashes. Further accumulation of similar cases and pathophysiological studies will be necessary to clarify whether this photosensitivity dermatitis is caused by ALK inhibition itself or mediated through host-immune mechanisms.5
Conclusion
As crizotinib prescriptions for patients with NSCLC are increasing, clinicians should be aware of the possibility of cutaneous LDEs occurring as an adverse effect. Additionally, physicians should treat appropriately to avoid unnecessarily discontinuing a potentially life-saving medication and to improve quality of life for patients with NSCLC who are treated with crizotinib.
Crizotinib is a multitargeted tyrosine kinase inhibitor that blocks anaplastic lymphoma kinase (ALK), hepatocyte growth factor receptor (c-Met), and their oncogenic variants ALK fusion proteins or c-Met/hepatocyte growth factor receptor mutant variants.1 Additionally, crizotinib was approved by the US Food and Drug Administration in 2011 for the treatment of patients with non–small cell lung cancer (NSCLC) whose tumors are echinoderm microtubule-associated proteinlike 4 (EML4)/ALK or ROS1 positive.2,3 Among unselected populations of patients with NSCLC, the frequency of EML4/ALK rearrangements ranges from 1.5% to 6.7%.1 Crizotinib is superior to standard chemotherapy in patients with ALK-positive NSCLC.2
In clinical trials, adverse reactions (grades 1 to 4) to crizotinib occurring in at least 25% of patients included visual disturbances, gastrointestinal tract disorders, fatigue, and pitting edema.1,2,4 Adverse reactions (grades 3 and 4) occurring in more than 5% of patients included elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, dyspnea, pneumonia, and neutropenia.1,4 Although the incidence of dermatologic adverse reactions is approximately 11%, substantial progression of drug eruptions rarely has been reported.2,5 We describe a case of lichenoid drug eruption (LDE) that appeared 4 weeks after initiation of crizotinib treatment in a patient with ALK-positive metastatic lung adenocarcinoma.
Case Report
A 61-year-old man presented with a history of ALK-positive NSCLC with lung-to-lung metastasis and pleural seeding treated with a right lower lobectomy and chemotherapy 9 years prior. Chemotherapy was reattempted 5 years later. Targeted therapy with gefitinib was initiated following the lobectomy and 5 years later with erlotinib. The NSCLC was stable, as indicated by computed tomography performed once every 3 or 6 months. After 5 years of treatment, follow-up computed tomography showed slowly growing nodular shadows in the right middle and lower lung fields. Due to this disease progression, treatment with crizotinib (250 mg twice daily) was initiated. Four weeks after the initiation of crizotinib therapy, mild itchy skin eruptions developed on all extremities and the lower lip. He also reported that the skin lesions became more itchy and red with sun exposure. He had no history of drug allergies and denied taking any other medications.
Physical examination revealed multiple brown to violaceous, slightly scaly, flat-topped polygonal papules or plaques on both lower legs (Figure 1A), dorsal hands (Figure 1B), and extensor sites of the elbows, as well as lacelike fine white lines on the lower lip (Figure 1C). There were no nail lesions. The patient’s dermatologic history was unremarkable, except for a few vitiligo lesions on the dorsal hands, extensor sites of the elbows, and mouth angles diagnosed 20 years earlier.
A skin biopsy from the right dorsal hand revealed a lichenoid infiltrate in the superficial dermis composed of lymphocytes, histiocytes and scattered eosinophils, focal parakeratosis, focal hypergranulosis, mild acanthosis, and basal vacuolization (Figure 2A). In addition, some dyskeratotic keratinocytes in the stratum spinosum and granulosum were identified (Figure 2B). The histopathology was consistent with the diagnosis of an LDE. Direct immunofluorescence revealed no globular or cytoid body–like deposits of immunoglobulin, with IgM, IgA, IgG, or C3 in the epidermis, dermis, and basement membrane zone. Routine laboratory studies revealed elevated liver enzymes, including an ALT level of 115 U/L (reference range, 0–40 U/L) and AST level of 60 U/L (reference range, 5–45 U/L). Negative results for the serum hepatitis B surface antigen and anti– hepatitis C virus tests were recorded. The patient had no medical history of alcohol consumption or abnormal liver function tests. The skin lesions were treated with diflucortolone valerate fatty ointment 0.1% twice daily and abnormal liver functions were treated with silymarin (150 mg per cap twice daily). He experienced some improvement.
A causality assessment was performed using the Naranjo Adverse Drug Reaction Probability Scale,6,7 and we concluded that crizotinib was the possible cause (Naranjo score, 4) of this adverse drug reaction (Table). Because the skin reaction was tolerable and liver enzymes were mildly elevated (ALT, 50 U/L; AST, 48 U/L), the offending drug was continued to benefit the underlying disease. His NSCLC was stable on computed tomography 3 months later.
Comment
The number of indicated uses of crizotinib, an oral small-molecule ALK tyrosine kinase inhibitor for the treatment of NSCLC, has gradually increased, but only a few cases of cutaneous adverse reactions, such as erythema multiforme and severe photosensitivity dermatitis, have been reported.2,5 Skin toxicity is a common and well-known side effect of other small-molecule tyrosine kinase inhibitors, particularly epidermal growth factor receptor inhibitors.8 However, LDE is not commonly associated with small-molecule tyrosine kinase inhibitors, though it has been described in a few patients taking imatinib for chronic myelogenous leukemia and gastrointestinal tract stromal tumors.9,10
The clinical morphology of LDE may resemble lichen planus, but certain features, such as larger skin lesions, the absence of Wickham striae, and photodistribution, help to differentiate between the two.10 Histologically, some findings are more common in LDE, including focal parakeratosis, cytoid bodies in the cornified and granular layers, and the presence of eosinophils.11
Our patient developed lichenoid rashes after 1 month of crizotinib therapy. The latency period for developing a medication-induced LDE varies from months to 1 year and is dependent on the dosage, host response, prior exposure, and concomitant drug administration. No additional medications had been added to our patient’s regimen after initiating crizotinib therapy, and he did not take any other known medications. Ultimately, based on the time-event relationship, morphology, distribution, and histopathologic findings, we concluded that our patient developed an LDE due to crizotinib.
Our patient also had a history of vitiligo affecting the hands, elbows, and mouth angles for 20 years. Although there are limited reports of a possible causal link between lichen planus or drug-induced lichen planus eruption and vitiligo,12-14 we do not think these conditions were associated in our case because the patient’s vitiligo lesions persisted for many years, did not progress, and remained inactive and stable, and there was a lack of co-localization of LDE and vitiligo.
Our patient reported that the skin eruptions worsened after sun exposure. Oser and Janne5 also reported a patient with ALK-positive metastatic lung adenocarcinoma who developed severe crizotinib-induced photosensitive rashes. Further accumulation of similar cases and pathophysiological studies will be necessary to clarify whether this photosensitivity dermatitis is caused by ALK inhibition itself or mediated through host-immune mechanisms.5
Conclusion
As crizotinib prescriptions for patients with NSCLC are increasing, clinicians should be aware of the possibility of cutaneous LDEs occurring as an adverse effect. Additionally, physicians should treat appropriately to avoid unnecessarily discontinuing a potentially life-saving medication and to improve quality of life for patients with NSCLC who are treated with crizotinib.
- Malik SM, Maher VE, Bijwaard KE, et al. U.S. Food and Drug Administration approval: crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res. 2014;20:2029-2034.
- Sawamura S, Kajihara I, Ichihara A, et al. Crizotinib-associated erythema multiforme in a lung cancer patient. Drug Discov Ther. 2015;9:142-143.
- Liao BC, Lin CC, Shih JY, et al. Treating patients with ALK-positive non-small cell lung cancer: latest evidence and management strategy. Ther Adv Med Oncol. 2015;7:274-290.
- Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011-1019.
- Oser MG, Janne PA. A severe photosensitivity dermatitis caused by crizotinib. J Thorac Oncol. 2014;9:E51-E53.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Zaki SA. Adverse drug reaction and causality assessment scales. Lung India. 2011;28:152-153.
- Aw DC, Tan EH, Chin TM, et al. Management of epidermal growth factor receptor tyrosine kinase inhibitor-related cutaneous and gastrointestinal toxicities. Asia Pac J Clin Oncol. 2018;14:23-31.
- Penn EH, Chung HJ, Keller M. Imatinib mesylate-induced lichenoid drug eruption. Cutis. 2017;99:189-192.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Lage D, Juliano PB, Metze K, et al. Lichen planus and lichenoid drug-induced eruption: a histological and immunohistochemical study. Int J Dermatol. 2012;51:1199-1205.
- Veitch D, Kravvas G, Hughes S, et al. A rare colocalization of lichen planus and vitiligo. Case Rep Dermatol Med. 2015;2015:840193.
- Baghestani S, Moosavi A, Eftekhari T. Familial colocalization of lichen planus and vitiligo on sun exposed areas. Ann Dermatol. 2013;25:223-225.
- Chan WP, Mackey VT, Sun DK. Telmisartan-induced lichen planus eruption manifested on vitiliginous skin. Cutis. 2017;99:E16-E19.
- Malik SM, Maher VE, Bijwaard KE, et al. U.S. Food and Drug Administration approval: crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res. 2014;20:2029-2034.
- Sawamura S, Kajihara I, Ichihara A, et al. Crizotinib-associated erythema multiforme in a lung cancer patient. Drug Discov Ther. 2015;9:142-143.
- Liao BC, Lin CC, Shih JY, et al. Treating patients with ALK-positive non-small cell lung cancer: latest evidence and management strategy. Ther Adv Med Oncol. 2015;7:274-290.
- Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011-1019.
- Oser MG, Janne PA. A severe photosensitivity dermatitis caused by crizotinib. J Thorac Oncol. 2014;9:E51-E53.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Zaki SA. Adverse drug reaction and causality assessment scales. Lung India. 2011;28:152-153.
- Aw DC, Tan EH, Chin TM, et al. Management of epidermal growth factor receptor tyrosine kinase inhibitor-related cutaneous and gastrointestinal toxicities. Asia Pac J Clin Oncol. 2018;14:23-31.
- Penn EH, Chung HJ, Keller M. Imatinib mesylate-induced lichenoid drug eruption. Cutis. 2017;99:189-192.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Lage D, Juliano PB, Metze K, et al. Lichen planus and lichenoid drug-induced eruption: a histological and immunohistochemical study. Int J Dermatol. 2012;51:1199-1205.
- Veitch D, Kravvas G, Hughes S, et al. A rare colocalization of lichen planus and vitiligo. Case Rep Dermatol Med. 2015;2015:840193.
- Baghestani S, Moosavi A, Eftekhari T. Familial colocalization of lichen planus and vitiligo on sun exposed areas. Ann Dermatol. 2013;25:223-225.
- Chan WP, Mackey VT, Sun DK. Telmisartan-induced lichen planus eruption manifested on vitiliginous skin. Cutis. 2017;99:E16-E19.
Practice Points
- Cutaneous lichenoid drug eruptions (LDEs) and photosensitive rash may be caused by crizotinib.
- The clinical morphology of LDE may resemble lichen planus, but certain features, such as larger skin lesions, the absence of Wickham striae, and photodistribution, help to differentiate between the two.
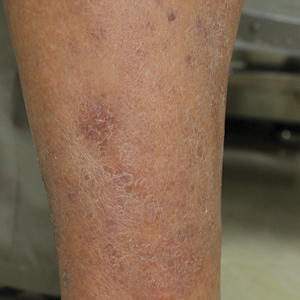
A ‘double-hit’ bone marrow rare co-occurrence of 2 different pathologies
Chronic myeloid leukemia and chronic lymphocytic leukemia are entirely different in terms of pathogenesis, presentation, diagnostic work-up, treatment, and prognosis: CML is a myeloproliferative condition, whereas CLL involves lymphoid population. Here we discuss a very rare case of co-occurrence of CML and CLL in the same patient.
Click on the PDF icon at the top of this introduction to read the full article.
Chronic myeloid leukemia and chronic lymphocytic leukemia are entirely different in terms of pathogenesis, presentation, diagnostic work-up, treatment, and prognosis: CML is a myeloproliferative condition, whereas CLL involves lymphoid population. Here we discuss a very rare case of co-occurrence of CML and CLL in the same patient.
Click on the PDF icon at the top of this introduction to read the full article.
Chronic myeloid leukemia and chronic lymphocytic leukemia are entirely different in terms of pathogenesis, presentation, diagnostic work-up, treatment, and prognosis: CML is a myeloproliferative condition, whereas CLL involves lymphoid population. Here we discuss a very rare case of co-occurrence of CML and CLL in the same patient.
Click on the PDF icon at the top of this introduction to read the full article.
Skin-Colored Papules on the Chest
An otherwise healthy male presents with multiple smooth uniform painless cystic papules scattered across his central chest.
A 25-year-old man presented with multiple sternal cysts that he first noticed when he was aged 18 years and had persisted despite treatment with topical anti-acne agents, including tretinoin. No other medications were used. The patient was unable to express purulent material from the lesions and reported no infection or additional trauma to the affected area. He had no other significant past medical history and no family history of similar skin lesions.
A physical examination revealed an otherwise healthy-appearing male with multiple uniform painless cystic papules scattered across his central chest that were smooth and flesh-colored to slightly yellow-colored, measuring 2 mm to 6 mm in diameter (Figure). A ring of erythema surrounded the lesions that had been recently manipulated by the patient. There were no overlying central puncta, and the remainder of his body was spared.
Related: Mohs Micrographic Surgery in the VHA
- What is your diagnosis?
- How would you treat this patient?
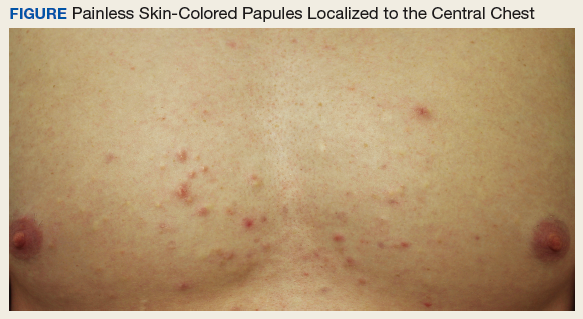
Diagnosis
The patient was diagnosed with steatocystoma multiplex based on his poor response to topical anti-acne agents, the location of his lesions, and histopathology of a biopsy specimen. Steatocystoma multiplex, sometimes termed sebocystomatosis, typically presents between puberty and the third decade of life. Lesions are usually < 2 cm in diameter and occur as multiple smooth skin-colored or yellow-colored painless papules on areas with high concentrations of hormonally sensitive sebaceous glands, especially the chest. Lesions also can be found in the axillae and on the neck.1-3 Solitary lesions can occur and are termed steatocystoma simplex.
The timing and location of presentation can easily be mistaken for acne vulgaris, but steatocystoma lesions are true sebaceous cysts, which are rare, and spontaneous resolution with increasing age does not typically occur. The diagnosis of steatocystoma often goes unreported because the disease is usually asymptomatic and mimics more common benign skin conditions, so an accurate prevalence and incidence are both unknown.
First on the differential diagnosis is acne vulgaris, which also presents at puberty and affects nearly 85% of adolescents. However, acne is less common in people of Asian or African descent and may progress along a continuum of increasingly severe and larger lesions, including the primary comedones and papules followed by pustules, nodules, and pseudocysts. Painful lesions develop from inflammation of pilosebaceous units concentrated on the face, neck, trunk, upper arms, or buttocks and are typically worse in males. Resolution often occurs spontaneously by the third decade of life, but scarring can persist.4
Related: Using Dermoscopy to Identify Melanoma and Improve Diagnostic Discrimination
Eruptive vellus hair cysts present as dozens of skin-colored small (1-4 mm) painless dome-shaped papules, sometimes with erythema and crusting. Typically these appear on the head, trunk, or flexor surfaces of infants (familial cases) or adolescents (sporadic cases) without bias for gender or ethnicity. Although benign and potential mimickers of steatocystoma and acne, these lesions can also be associated with more serious syndromes, like ectodermal dysplasias and pachyonychia congenita.2,3
Epidermoid cysts are common benign solitary skin-colored subcutaneous dome-shaped nodules that contain a central punctum through which cheeselike keratinaceous material can be expressed.4 These benign lesions arising from the dermis can enlarge to several centimeters, and adults of both genders and most ethnicities tend to develop the lesions on the trunk or face, with small cysts on the face termed milia. Ruptured cysts can incite intense inflammation, and multiple epidermoid cysts should raise concern for Gardner syndrome.2,3
About This Condition
Steatocystoma lesions are benign and thought to arise from a mutation in keratin 17. The mutation can be inherited in an autosomal dominant pattern, but sporadic nonheritable cases are more common.5 There are no distinct associations with gender or ethnicity. The dermal cysts arise from the sebaceous ducts of the pilosebaceous unit, and histopathology typically shows numerous mature sebaceous cells encased by a thin wall of stratified squamous epithelium.2 Immunohistochemical staining for the defective keratin can help diagnose biopsy specimens, and histopathology confirmed the diagnosis in this case.
Related: Recurring Bilateral Rash Concomitant With Upper Respiratory Tract Infection in a Healthy Adult Male
Treatment
Steatocystoma is usually asymptomatic, so patients mainly present to physicians for cosmetic reasons. Puncturing the cyst wall within the dermis produces translucent sebum-containing fluid, and ruptured cysts can incite inflammation, pain, and scarring.2 However, prognosis is good, and treatment consists of excision, aspiration and curettage of the cyst wall, oral isotretinoin, or laser therapy. Our patient elected to forego treatment and will consider definitive removal in the future, since the lesions will persist and potentially enlarge. Accurate diagnosis of this rare cause of chest papules improves the timeliness and efficacy of appropriate treatment, favoring good cosmesis.
1. Zuber TJ. Minimal excision technique for epidermoid (sebaceous) cysts. Am Fam Physician. 2002;65(7):1409-1412.
2. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012.
3. Brinster N, Liu V, Diwan AH, McKee PH. High Yield Pathology: Dermatopathology. 1st ed. Philadelphia, PA: Elsevier Saunders; 2011.
4. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York: McGraw-Hill; 2005.
5. Gordon Spratt EA, Kaplan J, Patel RR, Kamino H, Ramachandran SM. Steatocystoma. Dermatol Online J. 2013;19(12):20721.
An otherwise healthy male presents with multiple smooth uniform painless cystic papules scattered across his central chest.
An otherwise healthy male presents with multiple smooth uniform painless cystic papules scattered across his central chest.
A 25-year-old man presented with multiple sternal cysts that he first noticed when he was aged 18 years and had persisted despite treatment with topical anti-acne agents, including tretinoin. No other medications were used. The patient was unable to express purulent material from the lesions and reported no infection or additional trauma to the affected area. He had no other significant past medical history and no family history of similar skin lesions.
A physical examination revealed an otherwise healthy-appearing male with multiple uniform painless cystic papules scattered across his central chest that were smooth and flesh-colored to slightly yellow-colored, measuring 2 mm to 6 mm in diameter (Figure). A ring of erythema surrounded the lesions that had been recently manipulated by the patient. There were no overlying central puncta, and the remainder of his body was spared.
Related: Mohs Micrographic Surgery in the VHA
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
The patient was diagnosed with steatocystoma multiplex based on his poor response to topical anti-acne agents, the location of his lesions, and histopathology of a biopsy specimen. Steatocystoma multiplex, sometimes termed sebocystomatosis, typically presents between puberty and the third decade of life. Lesions are usually < 2 cm in diameter and occur as multiple smooth skin-colored or yellow-colored painless papules on areas with high concentrations of hormonally sensitive sebaceous glands, especially the chest. Lesions also can be found in the axillae and on the neck.1-3 Solitary lesions can occur and are termed steatocystoma simplex.
The timing and location of presentation can easily be mistaken for acne vulgaris, but steatocystoma lesions are true sebaceous cysts, which are rare, and spontaneous resolution with increasing age does not typically occur. The diagnosis of steatocystoma often goes unreported because the disease is usually asymptomatic and mimics more common benign skin conditions, so an accurate prevalence and incidence are both unknown.
First on the differential diagnosis is acne vulgaris, which also presents at puberty and affects nearly 85% of adolescents. However, acne is less common in people of Asian or African descent and may progress along a continuum of increasingly severe and larger lesions, including the primary comedones and papules followed by pustules, nodules, and pseudocysts. Painful lesions develop from inflammation of pilosebaceous units concentrated on the face, neck, trunk, upper arms, or buttocks and are typically worse in males. Resolution often occurs spontaneously by the third decade of life, but scarring can persist.4
Related: Using Dermoscopy to Identify Melanoma and Improve Diagnostic Discrimination
Eruptive vellus hair cysts present as dozens of skin-colored small (1-4 mm) painless dome-shaped papules, sometimes with erythema and crusting. Typically these appear on the head, trunk, or flexor surfaces of infants (familial cases) or adolescents (sporadic cases) without bias for gender or ethnicity. Although benign and potential mimickers of steatocystoma and acne, these lesions can also be associated with more serious syndromes, like ectodermal dysplasias and pachyonychia congenita.2,3
Epidermoid cysts are common benign solitary skin-colored subcutaneous dome-shaped nodules that contain a central punctum through which cheeselike keratinaceous material can be expressed.4 These benign lesions arising from the dermis can enlarge to several centimeters, and adults of both genders and most ethnicities tend to develop the lesions on the trunk or face, with small cysts on the face termed milia. Ruptured cysts can incite intense inflammation, and multiple epidermoid cysts should raise concern for Gardner syndrome.2,3
About This Condition
Steatocystoma lesions are benign and thought to arise from a mutation in keratin 17. The mutation can be inherited in an autosomal dominant pattern, but sporadic nonheritable cases are more common.5 There are no distinct associations with gender or ethnicity. The dermal cysts arise from the sebaceous ducts of the pilosebaceous unit, and histopathology typically shows numerous mature sebaceous cells encased by a thin wall of stratified squamous epithelium.2 Immunohistochemical staining for the defective keratin can help diagnose biopsy specimens, and histopathology confirmed the diagnosis in this case.
Related: Recurring Bilateral Rash Concomitant With Upper Respiratory Tract Infection in a Healthy Adult Male
Treatment
Steatocystoma is usually asymptomatic, so patients mainly present to physicians for cosmetic reasons. Puncturing the cyst wall within the dermis produces translucent sebum-containing fluid, and ruptured cysts can incite inflammation, pain, and scarring.2 However, prognosis is good, and treatment consists of excision, aspiration and curettage of the cyst wall, oral isotretinoin, or laser therapy. Our patient elected to forego treatment and will consider definitive removal in the future, since the lesions will persist and potentially enlarge. Accurate diagnosis of this rare cause of chest papules improves the timeliness and efficacy of appropriate treatment, favoring good cosmesis.
A 25-year-old man presented with multiple sternal cysts that he first noticed when he was aged 18 years and had persisted despite treatment with topical anti-acne agents, including tretinoin. No other medications were used. The patient was unable to express purulent material from the lesions and reported no infection or additional trauma to the affected area. He had no other significant past medical history and no family history of similar skin lesions.
A physical examination revealed an otherwise healthy-appearing male with multiple uniform painless cystic papules scattered across his central chest that were smooth and flesh-colored to slightly yellow-colored, measuring 2 mm to 6 mm in diameter (Figure). A ring of erythema surrounded the lesions that had been recently manipulated by the patient. There were no overlying central puncta, and the remainder of his body was spared.
Related: Mohs Micrographic Surgery in the VHA
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
The patient was diagnosed with steatocystoma multiplex based on his poor response to topical anti-acne agents, the location of his lesions, and histopathology of a biopsy specimen. Steatocystoma multiplex, sometimes termed sebocystomatosis, typically presents between puberty and the third decade of life. Lesions are usually < 2 cm in diameter and occur as multiple smooth skin-colored or yellow-colored painless papules on areas with high concentrations of hormonally sensitive sebaceous glands, especially the chest. Lesions also can be found in the axillae and on the neck.1-3 Solitary lesions can occur and are termed steatocystoma simplex.
The timing and location of presentation can easily be mistaken for acne vulgaris, but steatocystoma lesions are true sebaceous cysts, which are rare, and spontaneous resolution with increasing age does not typically occur. The diagnosis of steatocystoma often goes unreported because the disease is usually asymptomatic and mimics more common benign skin conditions, so an accurate prevalence and incidence are both unknown.
First on the differential diagnosis is acne vulgaris, which also presents at puberty and affects nearly 85% of adolescents. However, acne is less common in people of Asian or African descent and may progress along a continuum of increasingly severe and larger lesions, including the primary comedones and papules followed by pustules, nodules, and pseudocysts. Painful lesions develop from inflammation of pilosebaceous units concentrated on the face, neck, trunk, upper arms, or buttocks and are typically worse in males. Resolution often occurs spontaneously by the third decade of life, but scarring can persist.4
Related: Using Dermoscopy to Identify Melanoma and Improve Diagnostic Discrimination
Eruptive vellus hair cysts present as dozens of skin-colored small (1-4 mm) painless dome-shaped papules, sometimes with erythema and crusting. Typically these appear on the head, trunk, or flexor surfaces of infants (familial cases) or adolescents (sporadic cases) without bias for gender or ethnicity. Although benign and potential mimickers of steatocystoma and acne, these lesions can also be associated with more serious syndromes, like ectodermal dysplasias and pachyonychia congenita.2,3
Epidermoid cysts are common benign solitary skin-colored subcutaneous dome-shaped nodules that contain a central punctum through which cheeselike keratinaceous material can be expressed.4 These benign lesions arising from the dermis can enlarge to several centimeters, and adults of both genders and most ethnicities tend to develop the lesions on the trunk or face, with small cysts on the face termed milia. Ruptured cysts can incite intense inflammation, and multiple epidermoid cysts should raise concern for Gardner syndrome.2,3
About This Condition
Steatocystoma lesions are benign and thought to arise from a mutation in keratin 17. The mutation can be inherited in an autosomal dominant pattern, but sporadic nonheritable cases are more common.5 There are no distinct associations with gender or ethnicity. The dermal cysts arise from the sebaceous ducts of the pilosebaceous unit, and histopathology typically shows numerous mature sebaceous cells encased by a thin wall of stratified squamous epithelium.2 Immunohistochemical staining for the defective keratin can help diagnose biopsy specimens, and histopathology confirmed the diagnosis in this case.
Related: Recurring Bilateral Rash Concomitant With Upper Respiratory Tract Infection in a Healthy Adult Male
Treatment
Steatocystoma is usually asymptomatic, so patients mainly present to physicians for cosmetic reasons. Puncturing the cyst wall within the dermis produces translucent sebum-containing fluid, and ruptured cysts can incite inflammation, pain, and scarring.2 However, prognosis is good, and treatment consists of excision, aspiration and curettage of the cyst wall, oral isotretinoin, or laser therapy. Our patient elected to forego treatment and will consider definitive removal in the future, since the lesions will persist and potentially enlarge. Accurate diagnosis of this rare cause of chest papules improves the timeliness and efficacy of appropriate treatment, favoring good cosmesis.
1. Zuber TJ. Minimal excision technique for epidermoid (sebaceous) cysts. Am Fam Physician. 2002;65(7):1409-1412.
2. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012.
3. Brinster N, Liu V, Diwan AH, McKee PH. High Yield Pathology: Dermatopathology. 1st ed. Philadelphia, PA: Elsevier Saunders; 2011.
4. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York: McGraw-Hill; 2005.
5. Gordon Spratt EA, Kaplan J, Patel RR, Kamino H, Ramachandran SM. Steatocystoma. Dermatol Online J. 2013;19(12):20721.
1. Zuber TJ. Minimal excision technique for epidermoid (sebaceous) cysts. Am Fam Physician. 2002;65(7):1409-1412.
2. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012.
3. Brinster N, Liu V, Diwan AH, McKee PH. High Yield Pathology: Dermatopathology. 1st ed. Philadelphia, PA: Elsevier Saunders; 2011.
4. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York: McGraw-Hill; 2005.
5. Gordon Spratt EA, Kaplan J, Patel RR, Kamino H, Ramachandran SM. Steatocystoma. Dermatol Online J. 2013;19(12):20721.
Concomitant Fibrofolliculoma and Trichodiscoma on the Abdomen
Fibrofolliculomas and trichodiscomas typically present on the head or neck as smooth, flesh-colored, dome-shaped papules. These two entities are considered to constitute two separate time points on a spectrum of histopathologic changes in mantleoma differentiation.1 Histologically, both are benign hamartomas of the pilosebaceous subunit and collectively are known as mantleomas. We present an unusual case of a concomitant fibrofolliculoma and trichodiscoma on the abdomen.
Case Report
An asymptomatic 54-year-old man presented for a routine full-body skin examination. A solitary, 2×1-cm, subcutaneous, doughy, mobile nodule was found on the left side of the abdomen with an overlying 2-mm yellow fleshy papule. The patient declined excision of the lesion, and it was recommended that he return for follow-up 3 months later.
The patient did not present for follow-up until 4.5 years later, at which point the lesion had grown to 3.0×2.5 cm in size. An excision was performed, at which time the lesion was noted to be cystic, extruding an oily, yellow-white liquid. Bacterial culture was negative. Histopathologic sections showed a dome-shaped papule with connection to the overlying epidermis. Epithelial extensions from the infundibular epithelium formed a fenestrated pattern surrounding a fibrous and mucinous stroma (Figure, A and B). The differential diagnosis at this time included an epidermal inclusion cyst, fibroma, intradermal nevus, verruca, hemangioma, angiofibroma, and lipoma.2-4
The same lesion cut in a different plane of sectioning showed an expansile dermal nodule comprising clusters of sebaceous lobules surrounding a fibrous and mucinous stroma. Within the second lesion, fibrous and stromal components predominated over epithelial components (Figure, C). A diagnosis of fibrofolliculoma showing features of a trichodiscoma arising in the unusual location of the abdomen was made.
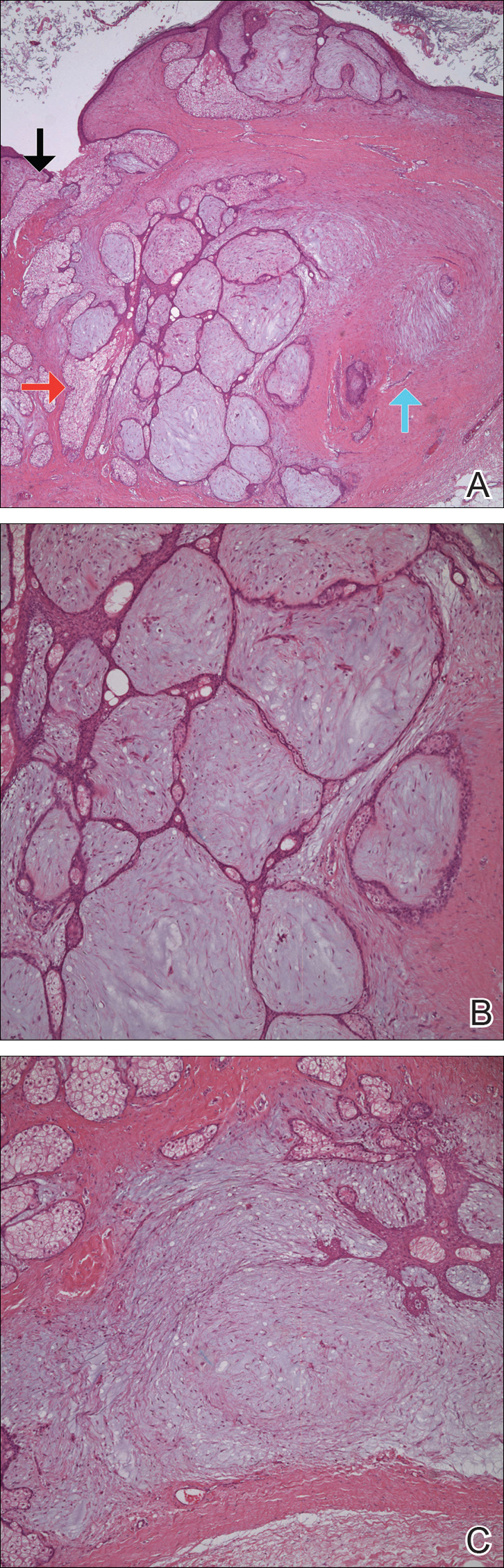
Comment
Solitary fibrofolliculomas and trichodiscomas are flesh-colored, dome-shaped papules that generally present on the face, specifically on the chin, nose, cheeks, ears, and eyebrows without considerable symptoms.2,4,5 Clinically, fibrofolliculomas are indistinguishable from trichodiscomas but demonstrate different features on biopsy.1,5
Fibrofolliculomas and trichodiscomas are well known for their association with Birt-Hogg-Dubé (BHD) syndrome when they present concomitantly and typically arise earlier in the third decade of life than solitary fibrofolliculomas; however, there have been reports of solitary fibrofolliculomas in patients aged 1 to 36 years.4,6 The triad of BHD syndrome consists of multiple fibrofolliculomas, trichodiscomas, and acrochordons, and it is acquired in an autosomal-dominant manner, unlike solitary fibrofolliculomas, which typically are not inherited. Birt-Hogg-Dubé syndrome is caused by a mutation in the FLCN gene that codes for the tumor-suppressor protein folliculin, which when mutated can cause unregulated proliferation of cells.7 Solitary fibrofolliculomas and the multiple fibrofolliculomas seen in BHD syndrome are histologically similar.
Fibrofolliculoma can be clinically indistinguishable from fibroepithelioma of Pinkus, perifollicular fibroma, trichilemmoma, trichodiscoma, trichoepithelioma, and trichofolliculoma. All typically present clinically as flesh-colored papules,1 although histologic distinction can be made (Table).5,8-13
Fibrofolliculoma is a benign hamartoma that arises from the pilosebaceous follicle and consists of an expansion of the fibrous root sheath, which typically surrounds the hair follicle along with proliferating bands or ribbons of perifollicular connective tissue. As such, the hair follicle may be dilated and filled with keratin in the expanded infundibulum.8 Follicles also may be surrounded by a myxoid stroma.2 In contrast, trichodiscoma is characterized by connective tissue with mature sebaceous lobules in the periphery. It has a myxoid stroma, as opposed to the more fibrous stroma seen in fibrofolliculomas.

Reports have examined the staining patterns of fibrofolliculomas, which show characteristics similar to those of other hair follicle hamartomas, including trichodiscomas.10 The connective tissue and epithelial components that constitute a fibrofolliculoma show different staining patterns. The connective tissue component stains positive for CD34 spindle cells, factor XIIIa, and nestin (a marker of angiogenesis). CD117 (c-kit) expression in the stroma, a marker of fibrocytes, is a feature of both fibrofolliculoma and perifollicular fibromas. The epithelial component, consisting of the hair follicle itself, stains positive for CK15. CK15 expression has been reported in undifferentiated sebocytes of the mantle and in the hair follicle.10 Immunohistochemical staining supports the notion that fibrofolliculomas contain connective tissue and epithelial components and helps to compare and contrast them to those of other hair follicle hamartomas.
Ackerman et al1 considered both fibrofolliculomas and trichodiscomas to be hamartomas of the epithelial hair follicle. The exact etiology of each of these hamartomas is unknown, but the undifferentiated epithelial strands protruding from the hair follicle in a fibrofolliculoma lie in close proximity to sebaceous glands. Furthermore, the authors postulated that fibrofolliculomas and trichodiscomas constitute a spectrum that encompasses the differentiation process of a mantleoma, with fibrofolliculoma representing the beginning of mantleoma differentiation and trichodiscoma representing the end. This end stage of follicular differentiation is one in which there is a predominant stroma and the previously undifferentiated epithelium has formed into sebaceous ducts and lobules in the stroma.1
Most cases of fibrofolliculoma and/or trichodiscoma arise in areas of dense sebaceous follicle concentration (eg, face), further supporting the hypothesis that sebaceous gland proliferation contributes to fibrofolliculoma.14 The case described here, with the fibrofolliculoma arising on the abdomen in conjunction with a trichodiscoma, is therefore worth noting because its location differs from what has been observed in previously reported cases.4
There are both surgical and medical options for treatment of fibrofolliculoma. Although surgical excision is an option for a single lesion, patients with multiple fibrofolliculomas or BHD may prefer removal with the combined CO2 laser and erbium-doped YAG laser.15
Conclusion
We present a rare case of concomitant fibrofolliculoma and trichodiscoma arising on the unusual location of the abdomen. This report highlights the histopathologic features of multiple adnexal tumors and emphasizes the importance of biopsy for differentiating fibrofolliculoma and trichodiscoma.
- Ackerman AB, Chongchitnant N, DeViragh P. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
- Scully K, Bargman H, Assaad D. Solitary fibrofolliculoma. J Am Acad Dermatol. 1984;11:361-363.
- Chang JK, Lee DC, Chang MH. A solitary fibrofolliculoma in the eyelid. Korean J Ophthalmol. 2007;21:169-171.
- Starink TM, Brownstein MH. Fibrofolliculoma: solitary and multiple types. J Am Acad Dermatol. 1987;17:493-496.
- Cho EU, Lee JD, Cho SH. A solitary fibrofolliculoma on the concha of the ear. Int J Dermatol. 2012;51:616-628.
- Mo HJ, Park CK, Yi JY. A case of solitary fibrofolliculoma. Korean J Dermatol. 2001;39:602-604.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157-164.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674-1677.
- Foucar K, Rosen TH, Foucar E, et al. Fibrofolliculoma: a clinicopathologic study. Cutis. 1981;28:429-432.
- Misago NO, Kimura TE, Narisawa YU. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol. 2009;36:943-951.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol. 2005;53(2 suppl 1):S108-S111.
- Lee Y, Su H, Chen H. Fibroepithelioma of Pinkus. a case report. Dermatologica Sinica. 2002;20:142-146.
- Nam JH, Min JH, Lee GY, et al. A case of perifollicular fibroma. Ann Dermatol. 2011:23:236-238.
- Vernooij M, Claessens T, Luijten M, et al. Birt-Hogg-Dubé syndrome and the skin. Fam Cancer. 2013;12:381-385.
- Jacob CI, Dover JS. Birt-Hogg-Dubé syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol. 2001;137:98-99.
Fibrofolliculomas and trichodiscomas typically present on the head or neck as smooth, flesh-colored, dome-shaped papules. These two entities are considered to constitute two separate time points on a spectrum of histopathologic changes in mantleoma differentiation.1 Histologically, both are benign hamartomas of the pilosebaceous subunit and collectively are known as mantleomas. We present an unusual case of a concomitant fibrofolliculoma and trichodiscoma on the abdomen.
Case Report
An asymptomatic 54-year-old man presented for a routine full-body skin examination. A solitary, 2×1-cm, subcutaneous, doughy, mobile nodule was found on the left side of the abdomen with an overlying 2-mm yellow fleshy papule. The patient declined excision of the lesion, and it was recommended that he return for follow-up 3 months later.
The patient did not present for follow-up until 4.5 years later, at which point the lesion had grown to 3.0×2.5 cm in size. An excision was performed, at which time the lesion was noted to be cystic, extruding an oily, yellow-white liquid. Bacterial culture was negative. Histopathologic sections showed a dome-shaped papule with connection to the overlying epidermis. Epithelial extensions from the infundibular epithelium formed a fenestrated pattern surrounding a fibrous and mucinous stroma (Figure, A and B). The differential diagnosis at this time included an epidermal inclusion cyst, fibroma, intradermal nevus, verruca, hemangioma, angiofibroma, and lipoma.2-4
The same lesion cut in a different plane of sectioning showed an expansile dermal nodule comprising clusters of sebaceous lobules surrounding a fibrous and mucinous stroma. Within the second lesion, fibrous and stromal components predominated over epithelial components (Figure, C). A diagnosis of fibrofolliculoma showing features of a trichodiscoma arising in the unusual location of the abdomen was made.
Comment
Solitary fibrofolliculomas and trichodiscomas are flesh-colored, dome-shaped papules that generally present on the face, specifically on the chin, nose, cheeks, ears, and eyebrows without considerable symptoms.2,4,5 Clinically, fibrofolliculomas are indistinguishable from trichodiscomas but demonstrate different features on biopsy.1,5
Fibrofolliculomas and trichodiscomas are well known for their association with Birt-Hogg-Dubé (BHD) syndrome when they present concomitantly and typically arise earlier in the third decade of life than solitary fibrofolliculomas; however, there have been reports of solitary fibrofolliculomas in patients aged 1 to 36 years.4,6 The triad of BHD syndrome consists of multiple fibrofolliculomas, trichodiscomas, and acrochordons, and it is acquired in an autosomal-dominant manner, unlike solitary fibrofolliculomas, which typically are not inherited. Birt-Hogg-Dubé syndrome is caused by a mutation in the FLCN gene that codes for the tumor-suppressor protein folliculin, which when mutated can cause unregulated proliferation of cells.7 Solitary fibrofolliculomas and the multiple fibrofolliculomas seen in BHD syndrome are histologically similar.
Fibrofolliculoma can be clinically indistinguishable from fibroepithelioma of Pinkus, perifollicular fibroma, trichilemmoma, trichodiscoma, trichoepithelioma, and trichofolliculoma. All typically present clinically as flesh-colored papules,1 although histologic distinction can be made (Table).5,8-13
Fibrofolliculoma is a benign hamartoma that arises from the pilosebaceous follicle and consists of an expansion of the fibrous root sheath, which typically surrounds the hair follicle along with proliferating bands or ribbons of perifollicular connective tissue. As such, the hair follicle may be dilated and filled with keratin in the expanded infundibulum.8 Follicles also may be surrounded by a myxoid stroma.2 In contrast, trichodiscoma is characterized by connective tissue with mature sebaceous lobules in the periphery. It has a myxoid stroma, as opposed to the more fibrous stroma seen in fibrofolliculomas.
Reports have examined the staining patterns of fibrofolliculomas, which show characteristics similar to those of other hair follicle hamartomas, including trichodiscomas.10 The connective tissue and epithelial components that constitute a fibrofolliculoma show different staining patterns. The connective tissue component stains positive for CD34 spindle cells, factor XIIIa, and nestin (a marker of angiogenesis). CD117 (c-kit) expression in the stroma, a marker of fibrocytes, is a feature of both fibrofolliculoma and perifollicular fibromas. The epithelial component, consisting of the hair follicle itself, stains positive for CK15. CK15 expression has been reported in undifferentiated sebocytes of the mantle and in the hair follicle.10 Immunohistochemical staining supports the notion that fibrofolliculomas contain connective tissue and epithelial components and helps to compare and contrast them to those of other hair follicle hamartomas.
Ackerman et al1 considered both fibrofolliculomas and trichodiscomas to be hamartomas of the epithelial hair follicle. The exact etiology of each of these hamartomas is unknown, but the undifferentiated epithelial strands protruding from the hair follicle in a fibrofolliculoma lie in close proximity to sebaceous glands. Furthermore, the authors postulated that fibrofolliculomas and trichodiscomas constitute a spectrum that encompasses the differentiation process of a mantleoma, with fibrofolliculoma representing the beginning of mantleoma differentiation and trichodiscoma representing the end. This end stage of follicular differentiation is one in which there is a predominant stroma and the previously undifferentiated epithelium has formed into sebaceous ducts and lobules in the stroma.1
Most cases of fibrofolliculoma and/or trichodiscoma arise in areas of dense sebaceous follicle concentration (eg, face), further supporting the hypothesis that sebaceous gland proliferation contributes to fibrofolliculoma.14 The case described here, with the fibrofolliculoma arising on the abdomen in conjunction with a trichodiscoma, is therefore worth noting because its location differs from what has been observed in previously reported cases.4
There are both surgical and medical options for treatment of fibrofolliculoma. Although surgical excision is an option for a single lesion, patients with multiple fibrofolliculomas or BHD may prefer removal with the combined CO2 laser and erbium-doped YAG laser.15
Conclusion
We present a rare case of concomitant fibrofolliculoma and trichodiscoma arising on the unusual location of the abdomen. This report highlights the histopathologic features of multiple adnexal tumors and emphasizes the importance of biopsy for differentiating fibrofolliculoma and trichodiscoma.
Fibrofolliculomas and trichodiscomas typically present on the head or neck as smooth, flesh-colored, dome-shaped papules. These two entities are considered to constitute two separate time points on a spectrum of histopathologic changes in mantleoma differentiation.1 Histologically, both are benign hamartomas of the pilosebaceous subunit and collectively are known as mantleomas. We present an unusual case of a concomitant fibrofolliculoma and trichodiscoma on the abdomen.
Case Report
An asymptomatic 54-year-old man presented for a routine full-body skin examination. A solitary, 2×1-cm, subcutaneous, doughy, mobile nodule was found on the left side of the abdomen with an overlying 2-mm yellow fleshy papule. The patient declined excision of the lesion, and it was recommended that he return for follow-up 3 months later.
The patient did not present for follow-up until 4.5 years later, at which point the lesion had grown to 3.0×2.5 cm in size. An excision was performed, at which time the lesion was noted to be cystic, extruding an oily, yellow-white liquid. Bacterial culture was negative. Histopathologic sections showed a dome-shaped papule with connection to the overlying epidermis. Epithelial extensions from the infundibular epithelium formed a fenestrated pattern surrounding a fibrous and mucinous stroma (Figure, A and B). The differential diagnosis at this time included an epidermal inclusion cyst, fibroma, intradermal nevus, verruca, hemangioma, angiofibroma, and lipoma.2-4
The same lesion cut in a different plane of sectioning showed an expansile dermal nodule comprising clusters of sebaceous lobules surrounding a fibrous and mucinous stroma. Within the second lesion, fibrous and stromal components predominated over epithelial components (Figure, C). A diagnosis of fibrofolliculoma showing features of a trichodiscoma arising in the unusual location of the abdomen was made.
Comment
Solitary fibrofolliculomas and trichodiscomas are flesh-colored, dome-shaped papules that generally present on the face, specifically on the chin, nose, cheeks, ears, and eyebrows without considerable symptoms.2,4,5 Clinically, fibrofolliculomas are indistinguishable from trichodiscomas but demonstrate different features on biopsy.1,5
Fibrofolliculomas and trichodiscomas are well known for their association with Birt-Hogg-Dubé (BHD) syndrome when they present concomitantly and typically arise earlier in the third decade of life than solitary fibrofolliculomas; however, there have been reports of solitary fibrofolliculomas in patients aged 1 to 36 years.4,6 The triad of BHD syndrome consists of multiple fibrofolliculomas, trichodiscomas, and acrochordons, and it is acquired in an autosomal-dominant manner, unlike solitary fibrofolliculomas, which typically are not inherited. Birt-Hogg-Dubé syndrome is caused by a mutation in the FLCN gene that codes for the tumor-suppressor protein folliculin, which when mutated can cause unregulated proliferation of cells.7 Solitary fibrofolliculomas and the multiple fibrofolliculomas seen in BHD syndrome are histologically similar.
Fibrofolliculoma can be clinically indistinguishable from fibroepithelioma of Pinkus, perifollicular fibroma, trichilemmoma, trichodiscoma, trichoepithelioma, and trichofolliculoma. All typically present clinically as flesh-colored papules,1 although histologic distinction can be made (Table).5,8-13
Fibrofolliculoma is a benign hamartoma that arises from the pilosebaceous follicle and consists of an expansion of the fibrous root sheath, which typically surrounds the hair follicle along with proliferating bands or ribbons of perifollicular connective tissue. As such, the hair follicle may be dilated and filled with keratin in the expanded infundibulum.8 Follicles also may be surrounded by a myxoid stroma.2 In contrast, trichodiscoma is characterized by connective tissue with mature sebaceous lobules in the periphery. It has a myxoid stroma, as opposed to the more fibrous stroma seen in fibrofolliculomas.
Reports have examined the staining patterns of fibrofolliculomas, which show characteristics similar to those of other hair follicle hamartomas, including trichodiscomas.10 The connective tissue and epithelial components that constitute a fibrofolliculoma show different staining patterns. The connective tissue component stains positive for CD34 spindle cells, factor XIIIa, and nestin (a marker of angiogenesis). CD117 (c-kit) expression in the stroma, a marker of fibrocytes, is a feature of both fibrofolliculoma and perifollicular fibromas. The epithelial component, consisting of the hair follicle itself, stains positive for CK15. CK15 expression has been reported in undifferentiated sebocytes of the mantle and in the hair follicle.10 Immunohistochemical staining supports the notion that fibrofolliculomas contain connective tissue and epithelial components and helps to compare and contrast them to those of other hair follicle hamartomas.
Ackerman et al1 considered both fibrofolliculomas and trichodiscomas to be hamartomas of the epithelial hair follicle. The exact etiology of each of these hamartomas is unknown, but the undifferentiated epithelial strands protruding from the hair follicle in a fibrofolliculoma lie in close proximity to sebaceous glands. Furthermore, the authors postulated that fibrofolliculomas and trichodiscomas constitute a spectrum that encompasses the differentiation process of a mantleoma, with fibrofolliculoma representing the beginning of mantleoma differentiation and trichodiscoma representing the end. This end stage of follicular differentiation is one in which there is a predominant stroma and the previously undifferentiated epithelium has formed into sebaceous ducts and lobules in the stroma.1
Most cases of fibrofolliculoma and/or trichodiscoma arise in areas of dense sebaceous follicle concentration (eg, face), further supporting the hypothesis that sebaceous gland proliferation contributes to fibrofolliculoma.14 The case described here, with the fibrofolliculoma arising on the abdomen in conjunction with a trichodiscoma, is therefore worth noting because its location differs from what has been observed in previously reported cases.4
There are both surgical and medical options for treatment of fibrofolliculoma. Although surgical excision is an option for a single lesion, patients with multiple fibrofolliculomas or BHD may prefer removal with the combined CO2 laser and erbium-doped YAG laser.15
Conclusion
We present a rare case of concomitant fibrofolliculoma and trichodiscoma arising on the unusual location of the abdomen. This report highlights the histopathologic features of multiple adnexal tumors and emphasizes the importance of biopsy for differentiating fibrofolliculoma and trichodiscoma.
- Ackerman AB, Chongchitnant N, DeViragh P. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
- Scully K, Bargman H, Assaad D. Solitary fibrofolliculoma. J Am Acad Dermatol. 1984;11:361-363.
- Chang JK, Lee DC, Chang MH. A solitary fibrofolliculoma in the eyelid. Korean J Ophthalmol. 2007;21:169-171.
- Starink TM, Brownstein MH. Fibrofolliculoma: solitary and multiple types. J Am Acad Dermatol. 1987;17:493-496.
- Cho EU, Lee JD, Cho SH. A solitary fibrofolliculoma on the concha of the ear. Int J Dermatol. 2012;51:616-628.
- Mo HJ, Park CK, Yi JY. A case of solitary fibrofolliculoma. Korean J Dermatol. 2001;39:602-604.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157-164.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674-1677.
- Foucar K, Rosen TH, Foucar E, et al. Fibrofolliculoma: a clinicopathologic study. Cutis. 1981;28:429-432.
- Misago NO, Kimura TE, Narisawa YU. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol. 2009;36:943-951.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol. 2005;53(2 suppl 1):S108-S111.
- Lee Y, Su H, Chen H. Fibroepithelioma of Pinkus. a case report. Dermatologica Sinica. 2002;20:142-146.
- Nam JH, Min JH, Lee GY, et al. A case of perifollicular fibroma. Ann Dermatol. 2011:23:236-238.
- Vernooij M, Claessens T, Luijten M, et al. Birt-Hogg-Dubé syndrome and the skin. Fam Cancer. 2013;12:381-385.
- Jacob CI, Dover JS. Birt-Hogg-Dubé syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol. 2001;137:98-99.
- Ackerman AB, Chongchitnant N, DeViragh P. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
- Scully K, Bargman H, Assaad D. Solitary fibrofolliculoma. J Am Acad Dermatol. 1984;11:361-363.
- Chang JK, Lee DC, Chang MH. A solitary fibrofolliculoma in the eyelid. Korean J Ophthalmol. 2007;21:169-171.
- Starink TM, Brownstein MH. Fibrofolliculoma: solitary and multiple types. J Am Acad Dermatol. 1987;17:493-496.
- Cho EU, Lee JD, Cho SH. A solitary fibrofolliculoma on the concha of the ear. Int J Dermatol. 2012;51:616-628.
- Mo HJ, Park CK, Yi JY. A case of solitary fibrofolliculoma. Korean J Dermatol. 2001;39:602-604.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157-164.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674-1677.
- Foucar K, Rosen TH, Foucar E, et al. Fibrofolliculoma: a clinicopathologic study. Cutis. 1981;28:429-432.
- Misago NO, Kimura TE, Narisawa YU. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol. 2009;36:943-951.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol. 2005;53(2 suppl 1):S108-S111.
- Lee Y, Su H, Chen H. Fibroepithelioma of Pinkus. a case report. Dermatologica Sinica. 2002;20:142-146.
- Nam JH, Min JH, Lee GY, et al. A case of perifollicular fibroma. Ann Dermatol. 2011:23:236-238.
- Vernooij M, Claessens T, Luijten M, et al. Birt-Hogg-Dubé syndrome and the skin. Fam Cancer. 2013;12:381-385.
- Jacob CI, Dover JS. Birt-Hogg-Dubé syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol. 2001;137:98-99.
Practice Points
- Fibrofolliculoma and trichodiscoma are flesh-colored adnexal tumors that arise from or around hair follicles.
- It is important to recognize these entities, as they can be related to Birt-Hogg-Dubé syndrome.