User login
Roundtable Discussion: Anticoagulation Management
Case 1
Tracy Minichiello, MD. The first case we’ll discuss is a 75-year-old man with mild chronic kidney disease (CKD). His calculated creatinine clearance (CrCl) is about 52 mL/min, and he has a remote history of a gastrointestinal (GI) bleeding 3 years previously from a peptic ulcer. He presents with new onset nonvalvular atrial fibrillation (AF), and he’s already on aspirin for his stable coronary artery disease (CAD).
How do we think about anticoagulant selection in this patient? We have a number of new oral anticoagulants and we have warfarin. How do we decide between warfarin vs one of the direct-acting oral anticoagulants (DOACs)? If we choose a DOAC, which one would we select?
David Parra, PharmD. The first step for anticoagulation is to assess a patient’s thromboembolic risk utilizing the CHA2DS2-VASc and bleeding risk using a HAS-BLED score, or something similar. The next question is which oral anticoagulant to use. We have widespread experience with warfarin and can measure the anticoagulant effect easily. Warfarin has a long duration of action, so perhaps it’s more forgiving if you miss a dose. It also has an antidote. Lastly, organ dysfunction doesn’t preclude use of warfarin as you can still monitor the anticoagulant effect. So there still may be patients that may benefit significantly from warfarin vs a DOAC.
On the flip side, DOACs are easier to use and perform quite acceptably in comparison with warfarin in nonvalvular AF. There are some scenarios where a specific DOAC may be preferred over another, such as recent GI bleeding.
Dr. Minichiello. Do you consider renal function, bleeding history, or concomitant antiplatelet therapy?
Geoffrey Barnes, MD, MSc. A couple of factors are relevant. I think we should consider renal function for this gentleman. However, I look at some of the other features as Dr. Parra suggested. What’s the likelihood that this patient is going to take the medicine as prescribed? Is a twice-a-day regimen going to be something that’s particularly challenging? I also look at the real-world vs randomized trial experience.
This patient has a remote GI bleeding history. Some of the real-world data suggest there might be some more GI bleeding with rivaroxaban, but across the board, apixaban (in both the randomized trials and much of the real-world data) seem to have a favorable bleeding risk profile. For a patient who is open and reliable for taking medicine twice a day, apixaban might be a good option as long as we make sure that the dose is appropriate.
Arthur L. Allen, PharmD, CACP. In pivotal trial experience, dabigatran and rivaroxaban demonstrated an increased incidence of GI bleeding compared with warfarin. In some of the real-world studies, rivaroxaban mirrors warfarin with regard to bleeding, whereas dabigatran and apixaban have a lower incidence. In the pivotal trials, apixaban did not have a trigger of increased GI bleeding, but I would let the details of this patient’s GI bleeding history help me determine how important an issue this is at this point.
The other thing that is important to understand when considering choice of agents: As Dr. Parra mentioned, we do have quite a bit of experience with warfarin. But comparing the quality of evidence, the DOACs have been investigated in a far more rigorous fashion and in far more patients than warfarin ever was in its more than 60 years on the market. For example, the RE-LY trial alone enrolled more than 18,000 patients. Each of the DOACs have been studied in tens of thousands of patients for their approved indications. Further, we shouldn’t forget that the risk of intracranial hemorrhage is reduced by roughly 50% by choosing a DOAC over warfarin, which should be a consideration in this elderly gentleman.
Dr. Minichiello. In the veteran population, there is a sense of comfort with warfarin, and some concerns have been raised over a lack of reversibility for the newer agents. We have patients who have trepidation about starting one of the new anticoagulants. However, there is a marked reduction in the risk of the most devastating bleeding complication, namely intracranial hemorrhage, making the use of these agents most compelling. And when they did have bleeding complications, at least in the trials, their outcomes were no worse than they were with warfarin, where there is a reversal agent. In most cases the outcomes were actually better. 
Dr. Barnes. You often have to remind patients that there was no reversal agent in these huge trials where the DOACs showed similar or safer bleeding risk profiles, especially for the most serious bleeding, such as intracranial hemorrhage. I find patients often are reassured by knowing that.
Dr. Allen. I agree that there is concern about the lack of reversibility, but I think it has been completely overplayed. In the pivotal trials, patients who bled on DOAC therapy actually had better outcomes than those that bled on warfarin. This includes intracranial hemorrhage. There was a paper published in Stroke in 2012 that evaluated the subgroup of patients in the RE-LY trial that suffered intracranial hemorrhage. Patients on dabigatran actually fared better despite a lack of a specific reversal agent. When evaluating the available data about reversal of the DOACs, I’m not 100% convinced that we’re significantly impacting outcomes by reversing these agents. We’re certainly running up the bill, but are we treating the patient or treating the providers? As long as the renal function remains intact, the DOACs clear quickly, perhaps more quickly than warfarin can typically be reversed with standard reversal agents.
Dr. Minichiello. Remember that this patient has a history of a GI bleed. We are going to start him on full-dose anticoagulation for stroke prevention for his nonvalvular AF. He’s also on aspirin, and he has stable coronary disease. He does not have any stents in place but he did have a remote non-ST elevation myocardial infarction (MI) a number of years ago. Do we feel that the risk of dual therapy—anticoagulation combined with antiplatelet therapy—outweighs the risks? And how do we approach that risk?
Dr. Barnes. This is an important point to discuss. There has been a lot of discussion in the literature recently. When I start this type of patient on an oral anticoagulant, I try to discontinue the antiplatelet agent because I know how much bleeding risk that brings. The European guidelines (for example, Eur Heart J. 2014;35[45]:3155-3179) have been forward thinking with this for the last couple of years and have highlighted that if there’s an indication for anticoagulation for patients with stable coronary disease, meaning no MI and no stent within the past year, then we should stop the antiplatelet agents after a year in order to reduce the risk of MI. This is based on a lot of older literature where warfarin was compared with aspirin and shown to be protective in coronary patients, but at the risk of bleeding.
It’s important because there have been recent studies that have raised questions, including a recent Swedish article in (Circulation. 2017;136:1183-1192) that suggested discontinuing aspirin led to increased mortality. But it’s important to look at the details. While that was true for most patients, it was not true for the group of patients who were on an oral anticoagulant. Many colleagues ask me questions about that particular paper and its media coverage. I tell them that for our patients on chronic oral anticoagulants, the paper supports the notion that there is not increased mortality when aspirin use is stopped. We know that aspirin plus an anticoagulant leads to increased bleeding, so I try to stop it for patients who have stable CAD but are on long-term anticoagulation.
Dr. Allen. This isn’t a new thought. Back in early 2012, the 9th edition of the American College of Chest Physicians (CHEST) Antithrombotic Guidelines probably gave us the best guidance that we had ever seen to help us address this issue. Since that time the cardiology guidelines have caught up to recommend that we do not need additional antiplatelet therapy for stable CAD, and, in fact, it should be limited even in the setting of acute coronary syndromes and percutaneous coronary intervention (PCI).
Dr. Minichiello. That’s a good point because people are not necessarily clear about when there would be an indication to continue dual therapy and when it is safe to go to monotherapy. Scenarios where benefit of dual therapy may outweigh risk suggested in the CHEST 2012 guidelines include acute coronary syndrome or a recent stent, high-risk mechanical valves, and history of coronary artery bypass surgery.
I think the important thing is to consider each case individually and not to reflexively continue aspirin therapy. Often what we see is once on aspirin—always on aspirin. Being thoughtful about it, we should acknowledge that it likely results in a 2-fold increased risk of bleeding and make sure that we believe that the benefit outweighs the risk.
Dr. Allen. I agree. We probably have better evidence in the CAD population, but what do we do for patients with significant peripheral vascular disease, or those patients with symptomatic carotid stenosis or history of strokes? Some of the European guidance suggests taking a similar approach to CAD, but these are the patients for whom stopping aspirin makes me more nervous.
Dr. Parra. This is a perfect example of where less is more. All too often the reflex is to continue aspirin treatment indefinitely because the patient has a history of acute coronary syndrome or even peripheral arterial disease, when the best thing to do would be to drop the aspirin. It involves an individualized risk assessment and underscores the need to periodically do a risk/benefit assessment in all patients on anticoagulants, whether it’s warfarin or a DOAC.
I’d like to take a moment and step back to the case in the context of the GI bleeding. When we look at patients with a history of GI bleeding, it is important to understand the circumstances that surround it. This individual had a GI bleed 3 years previously and peptic ulcer disease. In these situations I ask whether the patient was taking over-the-counter nonsteroidal anti-inflamitory drugs at the time, had excessive alcohol use, or was successfully treated for Helicobacter pylori. All of these may influence whether or not I think the GI bleed is significant to influence the DOAC choice.
The other thing I consider is that the overall risk of major GI bleeding in those pivotal DOAC trials was quite small, < 1.5% per year with dabigatran 150 mg twice daily and < 1% per year with apixaban. The numbers needed to harm were quite high, over 200 patients per year with dabigatran 150 mg twice daily vs warfarin and over 350 patients per year with edoxaban 60 mg daily vs warfarin. There are no head-to-head comparisons with DOACs, but this small increased risk vs warfarin may still be an important consideration in some patients. In addition, it is important to remember that intracranial hemorrhage and fatal bleeding was less in all the pivotal NVAF trials with the DOACs when compared with warfarin. So that is something we need to reinforce with patients when we discuss treatment regardless of the DOAC selected.
Case 2
Dr. Minichiello. The next case is a 63-year-old man with hypertension, diabetes mellitus, nonvalvular AF, and he is taking dabigatran for stroke prevention. He presents in the emergency department with chest pain, and he is found to have a non-ST elevation MI. He goes to the cath lab and he is found to have a lesion in his left circumflex. The patient receives a newer generation drug-eluting stent. What are we going to do with his anticoagulation? We know he’s going to get some antiplatelet therapy, but what are our thoughts on this?
Dr. Parra. This is something that we run into all too often. I think the estimates are about 20% to 30% of patients who have indications for anticoagulation also end up having ischemic heart disease that requires PCI. The second thing is that we know combining an anticoagulant with antiplatelet therapy is associated with a 4% to 16% risk of fatal and nonfatal bleeding, and we have found out in patients with ischemic heart disease that when they bleed, they also have a higher mortality rate.
We’re trying to find the optimal balance between ischemic and thrombotic risk and bleeding risk. This is where some of the risk assessment tools that we have come into play. First, we need to establish the thrombotic risk by considering the CHA2DS2-VASc score, and the factors associated with increasing bleeding risk and stent thrombosis. You have time to work this out because, initially, all patients that are at sufficiently high thrombotic risk will receive dual antiplatelet therapy and anticoagulation therapy for a given period. This gives providers time to use some of those resources and figure out a long-term plan for the patient.
Dr. Allen. The fear and loathing that this brings up comes back to some historic things that should be considered. What we did for drug-eluting stents or bare-metal stents comes from older data where different stent technology was used. The stents used today are safer with a lower risk of in-stent thrombosis. Historically, we knew what to do for ACS and PCI and we knew what to do for AF, but we didn’t know what to do when the 2 crossed paths. We would put patients on warfarin and say, “Well, for now, target the INR between 2 and 2.5 and good luck with that.” That was all we had. The good news is now we have some evidence to move away from the use of dual antiplatelet plus anticoagulant therapies.
There were 2 DOAC studies recently published: The PIONEER-AF trial used rivaroxaban and more recently the RE-DUAL PCI trial used dabigatran. Each of the studies had some issues, but they were both studied in AF populations and aimed to address this issue of triple therapy. The PIONEER-AF trial looked at a number of different scenarios on different doses of rivaroxaban with either single or dual antiplatelet therapy compared to triple therapy with warfarin. The RE-DUAL PCI trial with dabigatran was less complex. Both studies were powered to look at safety, and they did show that with single antiplatelet plus oral anticoagulation regimens, the incidence of major bleeding complications was reduced.
However, that brings up some issues about how the studies were conducted. Both studied AF populations and in some cases did not study doses approved for AF. Yet at the same time, the studies were not powered to look at stroke outcomes, which raises the question: Are we running the risk of giving up stroke efficacy for reduced bleeding? I don’t think that we’ve fully answered that, certainly not with the PIONEER AF trial.
Dr. Parra. I agree. When we look at those trials, 2 things come to mind. First, the doses of dabigatran used in the RE-DUAL PCI trial were doses that have been shown to be beneficial in the nonvalvular AF population. Second, a take-home point from those trials is the P2Y12 inhibitor that was utilized—close to 90% or more used clopidogrel in the PIONEER-AF trial. Clopidogrel remains the P2Y12 inhibitor of choice. One of the other findings is that the aspirin dosing should be low, < 100 mg daily, and that we need to consider routine use of proton pump inhibitors to protect against the bleeding that can be found with the antiplatelet agents.
Also of interest, the European Society of Cardiology (ESC) recently released a focused update with some excellent recommendations on dual antiplatelet therapy in coronary artery disease in which they incorporated the results from the PIONEER-AF-PCI trial. The RE-DUAL PCI trial had not been published when these came out. If you’re concerned about ischemic risk prevailing, ESC recommendations based upon risk stratification are triple therapy with aspirin, clopidogrel, and an oral anticoagulant for longer than 1 month and up to 6 months and then dual therapy with 1 antiplatelet agent and an oral anticoagulant to complete the 12 months; afterward just oral anticoagulation alone. If the concerns about bleeding prevail, then we have 2 different pathways: one limiting triple therapy to 1 month and then dual therapy with 1 antiplatelet agent and an oral anticoagulant to complete the 12 months. But the ESC also has a second recommendation for patients at high risk of bleeding, which is dual therapy with clopidogrel and an oral anticoagulant at the offset for up to 12 months. I found these guidelines to be particularly helpful in terms of how to put this into practice.
Dr. Allen. There’s still so much concern about in-stent thrombosis. Although a smaller trial, we knew from the WOEST trial that single antiplatelet therapy with warfarin was reasonable. We know from the PIONEER-AF and RE-DUAL PCI trials that we didn’t get significantly more in-stent thrombosis by giving up the second antiplatelet. Whether or not we answered the stroke question is another issue, but the cardiology societies are still hanging on to dual antiplatelet therapy. I question if that’s based on the older data and the older stent technologies.
Dr. Minichiello. This highlights again that often we have to consider these patients’ case-by-case analysis, and that these decisions require multidisciplinary input. It involves coming together and figuring out in this particular patient, which of those 3 options would be best. We have a lot more options than we did just a short year or year and a half ago with at least some data providing comfort that DOACs at effective doses for stroke prevention in nonvalvular AF look like they can be combined with single antiplatelet therapy for post-PCI patients.
Dr. Barnes. Speaking as a cardiologist, this is a question I encounter all the time. I think everything said here is really well taken. I’ll just summarize to say for patients who have acute coronary syndromes and AF. First, I’m okay with using warfarin and now I’m okay using the DOACs, but the anticoagulant needs to continue for stroke prevention. Second, the patient has to be on clopidogrel as a P2Y12 inhibitor because it has the lowest bleeding risk profile. Third, the patient doesn’t always need 1 year of dual antiplatelet therapy the way we used to think of it. With newer generation stents and ongoing anticoagulation, you can get away with shorter courses of your antiplatelets, albeit 3 or 6 months. Providers should have a conversation with patients and think hard about how to balance clotting vs that bleeding profile.
Case 3
Dr. Minichiello. This case involves a 66-year-old man who has nonvalvular AF and is on warfarin. He has CKD, and his CrCl is about 30. He has hypertension, diabetes mellitus, and he is going to go for a colonoscopy. The proceduralist lets you know about the date and wants to know whether he needs to be bridged. He also has a remote history of a transient ischemic attack.
Dr. Allen. Historically, we’ve had detailed guidance on how to risk assess patients with AF, venous thromboembolism (VT), and mechanical heart valves in the periprocedural period. From that risk assessment the guidance helped us determine whether or not we should offer periprocedural bridging with heparin or low-molecularweight heparin.
The issue is that the detailed guidance was always based on expert opinion not hard science and there was no great evidence that we were preventing thrombotic events or that there was a net clinical benefit to bridging. Some retrospective cohort studies started coming out around 2012 that demonstrated an increased incidence of major bleeding events associated with bridging with no reduction in thrombotic complications. Some might argue that this is because thrombotic complications are so rare that you would have to have tens of thousands of patients for adequate power. Nonetheless, these studies were adequately powered to show a significant increase in major bleeding.
The best prospective trial data we have for this population comes from the BRIDGE trial, which randomized AF patients to receive dalteparin bridge therapy vs placebo during periprocedural interruptions in warfarin therapy. It too demonstrated a significant increase in the risk of bleeding complications associated with bridging with no significant reduction in the risk of thromboembolic events. Critics point out that some of the higher risk patients were underrepresented, and the same could be said about some of the other retrospective studies in the VT and mechanical valve populations.
We have waited for quite a while for guidances to catch up with these data. The most recent guidance published would apply to this patient. It was guidance for the AF population published by the American College of Cardiology (ACC) in early 2017
To make a decision to bridge, you not only have to make an assumption that your patient is at such extraordinary thrombotic risk that you would find some reduction in thrombotic events associated with bridging, but also that the benefit is going to be so great that it would overcome this very clear increased risk of bleeding, resulting in a net clinical benefit. Only then would it make sense to bridge. Based on the available data, it is quite possible that no such patient population exists.
Dr. Parra. I agree. We found from the BRIDGE trial, with the caveat that you pointed out, that high or moderately high thrombotic risk individuals weren’t as well represented, that there is at least a 2.5-fold increase in major bleeding. We also know that higher thrombotic-risk patients tend to also have higher bleeding risk. So in high thrombotic-risk patients, while there is uncertainty about whether or not there’s going to be a thrombotic benefit from bridging, we can be confident there will be an increased risk of major bleeding perhaps even more than that seen in the BRIDGE trial.
One thing that really illustrates this is the 2017 American Heart Association/ACC-focused update on the 2014 guideline for the management of patients with valvular heart disease. The guideline has a section on bridging therapy for prosthetic valves. And that was recently downgraded from a Class I recommendation to Class IIA, recognizing that the data are limited in terms of even when to bridge prosthetic valves.
Dr. Allen. Bridging is an area where we continue to do something that we know causes harm because we hope it has some benefit. Despite what some societal guidance still says, I very rarely bridge patients. If I do, it’s because the patient and/or caregivers have heard the facts and have opted to do it.
Dr. Minichiello. Practically speaking, the data—the retrospective data, the observational data, the BRIDGE trial, etc—definitely make us step back and think about bridging and realize that it’s a HIGH RISK of intervention. We really need to be informing patients about that risk. We know that bridging increases the risk of bleeding. We do not have data showing a reduction in the risk of thromboembolic disease with bridge therapy. That’s all based on what we think would happen, what we hope would be the result of bridge therapy, but in truth we do not know if this is the case. The risks of bridge therapy must be weighed heavily each time we consider using it.
In my practice I reserve bridge therapy, which we know is associated with increased harm for patients in whom we think there is the very highest risk of thromboembolism, ie, those with very recent arterial or venous thrombosis, and by recently, I mean within the past 1 to 3 months; patients who have very severe thrombophilia like antitphospholipid antibody syndrome, some cancer patients, and those with mechanical valves in the mitral position or those with high-risk mechanical aortic valves. 
I don’t bridge most AF patients. In fact, I can’t remember the last time I bridged an AF patient. I do know that this is somewhat discordant with the ACC recommendations, but absent the data to support bridge therapy, I’m really concerned that the risk outweighs the benefit. This is particularly true in our VA population where the bleeding risk is high because of CKD or a history of bleeding or thrombocytopenia or concomitant aspirin or something else.
Dr. Barnes. We know from some studies that have been published that the biggest driver of a clinician deciding whether or not a patient should bridge is actually whether or not they’ve had a stroke. The truth is that the BRIDGE trial enrolled a sizeable population; it was about 15% of patients with a prior TIA or stroke. So it gave us some insight. Despite that, our event rates for thromboembolism, arterial thromboembolism, including stroke, were quite low.
I tend to look far more at the collection of risk factors and not just at a history of stroke. Now as you mentioned, Dr. Minichiello, a stroke within the past 1 to 3 months, I ask, “does this procedure even need to happen?” But outside of that, it’s not a history of stroke that’s going to make the decision for me: It’s all the risk factors together.
There’s an ongoing study, the PERIOP 2 study, that is enrolling patients at higher risk for stroke and patients with mechanical valves. This study may give us more insight into exactly what kind of risks these patients are at and whether they get benefit from bridging. But in the meantime, I’m really reserving bridging for my highest risk patients, those with multiple risk factors, CHADS2 scores of 5 and 6 or CHA2DS2-VASc of 7, 8, 9, and those with a recent VT or mechanical mitral valves.
Click here to read the digital edition.
Case 1
Tracy Minichiello, MD. The first case we’ll discuss is a 75-year-old man with mild chronic kidney disease (CKD). His calculated creatinine clearance (CrCl) is about 52 mL/min, and he has a remote history of a gastrointestinal (GI) bleeding 3 years previously from a peptic ulcer. He presents with new onset nonvalvular atrial fibrillation (AF), and he’s already on aspirin for his stable coronary artery disease (CAD).
How do we think about anticoagulant selection in this patient? We have a number of new oral anticoagulants and we have warfarin. How do we decide between warfarin vs one of the direct-acting oral anticoagulants (DOACs)? If we choose a DOAC, which one would we select?
David Parra, PharmD. The first step for anticoagulation is to assess a patient’s thromboembolic risk utilizing the CHA2DS2-VASc and bleeding risk using a HAS-BLED score, or something similar. The next question is which oral anticoagulant to use. We have widespread experience with warfarin and can measure the anticoagulant effect easily. Warfarin has a long duration of action, so perhaps it’s more forgiving if you miss a dose. It also has an antidote. Lastly, organ dysfunction doesn’t preclude use of warfarin as you can still monitor the anticoagulant effect. So there still may be patients that may benefit significantly from warfarin vs a DOAC.
On the flip side, DOACs are easier to use and perform quite acceptably in comparison with warfarin in nonvalvular AF. There are some scenarios where a specific DOAC may be preferred over another, such as recent GI bleeding.
Dr. Minichiello. Do you consider renal function, bleeding history, or concomitant antiplatelet therapy?
Geoffrey Barnes, MD, MSc. A couple of factors are relevant. I think we should consider renal function for this gentleman. However, I look at some of the other features as Dr. Parra suggested. What’s the likelihood that this patient is going to take the medicine as prescribed? Is a twice-a-day regimen going to be something that’s particularly challenging? I also look at the real-world vs randomized trial experience.
This patient has a remote GI bleeding history. Some of the real-world data suggest there might be some more GI bleeding with rivaroxaban, but across the board, apixaban (in both the randomized trials and much of the real-world data) seem to have a favorable bleeding risk profile. For a patient who is open and reliable for taking medicine twice a day, apixaban might be a good option as long as we make sure that the dose is appropriate.
Arthur L. Allen, PharmD, CACP. In pivotal trial experience, dabigatran and rivaroxaban demonstrated an increased incidence of GI bleeding compared with warfarin. In some of the real-world studies, rivaroxaban mirrors warfarin with regard to bleeding, whereas dabigatran and apixaban have a lower incidence. In the pivotal trials, apixaban did not have a trigger of increased GI bleeding, but I would let the details of this patient’s GI bleeding history help me determine how important an issue this is at this point.
The other thing that is important to understand when considering choice of agents: As Dr. Parra mentioned, we do have quite a bit of experience with warfarin. But comparing the quality of evidence, the DOACs have been investigated in a far more rigorous fashion and in far more patients than warfarin ever was in its more than 60 years on the market. For example, the RE-LY trial alone enrolled more than 18,000 patients. Each of the DOACs have been studied in tens of thousands of patients for their approved indications. Further, we shouldn’t forget that the risk of intracranial hemorrhage is reduced by roughly 50% by choosing a DOAC over warfarin, which should be a consideration in this elderly gentleman.
Dr. Minichiello. In the veteran population, there is a sense of comfort with warfarin, and some concerns have been raised over a lack of reversibility for the newer agents. We have patients who have trepidation about starting one of the new anticoagulants. However, there is a marked reduction in the risk of the most devastating bleeding complication, namely intracranial hemorrhage, making the use of these agents most compelling. And when they did have bleeding complications, at least in the trials, their outcomes were no worse than they were with warfarin, where there is a reversal agent. In most cases the outcomes were actually better.
Dr. Barnes. You often have to remind patients that there was no reversal agent in these huge trials where the DOACs showed similar or safer bleeding risk profiles, especially for the most serious bleeding, such as intracranial hemorrhage. I find patients often are reassured by knowing that.
Dr. Allen. I agree that there is concern about the lack of reversibility, but I think it has been completely overplayed. In the pivotal trials, patients who bled on DOAC therapy actually had better outcomes than those that bled on warfarin. This includes intracranial hemorrhage. There was a paper published in Stroke in 2012 that evaluated the subgroup of patients in the RE-LY trial that suffered intracranial hemorrhage. Patients on dabigatran actually fared better despite a lack of a specific reversal agent. When evaluating the available data about reversal of the DOACs, I’m not 100% convinced that we’re significantly impacting outcomes by reversing these agents. We’re certainly running up the bill, but are we treating the patient or treating the providers? As long as the renal function remains intact, the DOACs clear quickly, perhaps more quickly than warfarin can typically be reversed with standard reversal agents.
Dr. Minichiello. Remember that this patient has a history of a GI bleed. We are going to start him on full-dose anticoagulation for stroke prevention for his nonvalvular AF. He’s also on aspirin, and he has stable coronary disease. He does not have any stents in place but he did have a remote non-ST elevation myocardial infarction (MI) a number of years ago. Do we feel that the risk of dual therapy—anticoagulation combined with antiplatelet therapy—outweighs the risks? And how do we approach that risk?
Dr. Barnes. This is an important point to discuss. There has been a lot of discussion in the literature recently. When I start this type of patient on an oral anticoagulant, I try to discontinue the antiplatelet agent because I know how much bleeding risk that brings. The European guidelines (for example, Eur Heart J. 2014;35[45]:3155-3179) have been forward thinking with this for the last couple of years and have highlighted that if there’s an indication for anticoagulation for patients with stable coronary disease, meaning no MI and no stent within the past year, then we should stop the antiplatelet agents after a year in order to reduce the risk of MI. This is based on a lot of older literature where warfarin was compared with aspirin and shown to be protective in coronary patients, but at the risk of bleeding.
It’s important because there have been recent studies that have raised questions, including a recent Swedish article in (Circulation. 2017;136:1183-1192) that suggested discontinuing aspirin led to increased mortality. But it’s important to look at the details. While that was true for most patients, it was not true for the group of patients who were on an oral anticoagulant. Many colleagues ask me questions about that particular paper and its media coverage. I tell them that for our patients on chronic oral anticoagulants, the paper supports the notion that there is not increased mortality when aspirin use is stopped. We know that aspirin plus an anticoagulant leads to increased bleeding, so I try to stop it for patients who have stable CAD but are on long-term anticoagulation.
Dr. Allen. This isn’t a new thought. Back in early 2012, the 9th edition of the American College of Chest Physicians (CHEST) Antithrombotic Guidelines probably gave us the best guidance that we had ever seen to help us address this issue. Since that time the cardiology guidelines have caught up to recommend that we do not need additional antiplatelet therapy for stable CAD, and, in fact, it should be limited even in the setting of acute coronary syndromes and percutaneous coronary intervention (PCI).
Dr. Minichiello. That’s a good point because people are not necessarily clear about when there would be an indication to continue dual therapy and when it is safe to go to monotherapy. Scenarios where benefit of dual therapy may outweigh risk suggested in the CHEST 2012 guidelines include acute coronary syndrome or a recent stent, high-risk mechanical valves, and history of coronary artery bypass surgery.
I think the important thing is to consider each case individually and not to reflexively continue aspirin therapy. Often what we see is once on aspirin—always on aspirin. Being thoughtful about it, we should acknowledge that it likely results in a 2-fold increased risk of bleeding and make sure that we believe that the benefit outweighs the risk.
Dr. Allen. I agree. We probably have better evidence in the CAD population, but what do we do for patients with significant peripheral vascular disease, or those patients with symptomatic carotid stenosis or history of strokes? Some of the European guidance suggests taking a similar approach to CAD, but these are the patients for whom stopping aspirin makes me more nervous.
Dr. Parra. This is a perfect example of where less is more. All too often the reflex is to continue aspirin treatment indefinitely because the patient has a history of acute coronary syndrome or even peripheral arterial disease, when the best thing to do would be to drop the aspirin. It involves an individualized risk assessment and underscores the need to periodically do a risk/benefit assessment in all patients on anticoagulants, whether it’s warfarin or a DOAC.
I’d like to take a moment and step back to the case in the context of the GI bleeding. When we look at patients with a history of GI bleeding, it is important to understand the circumstances that surround it. This individual had a GI bleed 3 years previously and peptic ulcer disease. In these situations I ask whether the patient was taking over-the-counter nonsteroidal anti-inflamitory drugs at the time, had excessive alcohol use, or was successfully treated for Helicobacter pylori. All of these may influence whether or not I think the GI bleed is significant to influence the DOAC choice.
The other thing I consider is that the overall risk of major GI bleeding in those pivotal DOAC trials was quite small, < 1.5% per year with dabigatran 150 mg twice daily and < 1% per year with apixaban. The numbers needed to harm were quite high, over 200 patients per year with dabigatran 150 mg twice daily vs warfarin and over 350 patients per year with edoxaban 60 mg daily vs warfarin. There are no head-to-head comparisons with DOACs, but this small increased risk vs warfarin may still be an important consideration in some patients. In addition, it is important to remember that intracranial hemorrhage and fatal bleeding was less in all the pivotal NVAF trials with the DOACs when compared with warfarin. So that is something we need to reinforce with patients when we discuss treatment regardless of the DOAC selected.
Case 2
Dr. Minichiello. The next case is a 63-year-old man with hypertension, diabetes mellitus, nonvalvular AF, and he is taking dabigatran for stroke prevention. He presents in the emergency department with chest pain, and he is found to have a non-ST elevation MI. He goes to the cath lab and he is found to have a lesion in his left circumflex. The patient receives a newer generation drug-eluting stent. What are we going to do with his anticoagulation? We know he’s going to get some antiplatelet therapy, but what are our thoughts on this?
Dr. Parra. This is something that we run into all too often. I think the estimates are about 20% to 30% of patients who have indications for anticoagulation also end up having ischemic heart disease that requires PCI. The second thing is that we know combining an anticoagulant with antiplatelet therapy is associated with a 4% to 16% risk of fatal and nonfatal bleeding, and we have found out in patients with ischemic heart disease that when they bleed, they also have a higher mortality rate.
We’re trying to find the optimal balance between ischemic and thrombotic risk and bleeding risk. This is where some of the risk assessment tools that we have come into play. First, we need to establish the thrombotic risk by considering the CHA2DS2-VASc score, and the factors associated with increasing bleeding risk and stent thrombosis. You have time to work this out because, initially, all patients that are at sufficiently high thrombotic risk will receive dual antiplatelet therapy and anticoagulation therapy for a given period. This gives providers time to use some of those resources and figure out a long-term plan for the patient.
Dr. Allen. The fear and loathing that this brings up comes back to some historic things that should be considered. What we did for drug-eluting stents or bare-metal stents comes from older data where different stent technology was used. The stents used today are safer with a lower risk of in-stent thrombosis. Historically, we knew what to do for ACS and PCI and we knew what to do for AF, but we didn’t know what to do when the 2 crossed paths. We would put patients on warfarin and say, “Well, for now, target the INR between 2 and 2.5 and good luck with that.” That was all we had. The good news is now we have some evidence to move away from the use of dual antiplatelet plus anticoagulant therapies.
There were 2 DOAC studies recently published: The PIONEER-AF trial used rivaroxaban and more recently the RE-DUAL PCI trial used dabigatran. Each of the studies had some issues, but they were both studied in AF populations and aimed to address this issue of triple therapy. The PIONEER-AF trial looked at a number of different scenarios on different doses of rivaroxaban with either single or dual antiplatelet therapy compared to triple therapy with warfarin. The RE-DUAL PCI trial with dabigatran was less complex. Both studies were powered to look at safety, and they did show that with single antiplatelet plus oral anticoagulation regimens, the incidence of major bleeding complications was reduced.
However, that brings up some issues about how the studies were conducted. Both studied AF populations and in some cases did not study doses approved for AF. Yet at the same time, the studies were not powered to look at stroke outcomes, which raises the question: Are we running the risk of giving up stroke efficacy for reduced bleeding? I don’t think that we’ve fully answered that, certainly not with the PIONEER AF trial.
Dr. Parra. I agree. When we look at those trials, 2 things come to mind. First, the doses of dabigatran used in the RE-DUAL PCI trial were doses that have been shown to be beneficial in the nonvalvular AF population. Second, a take-home point from those trials is the P2Y12 inhibitor that was utilized—close to 90% or more used clopidogrel in the PIONEER-AF trial. Clopidogrel remains the P2Y12 inhibitor of choice. One of the other findings is that the aspirin dosing should be low, < 100 mg daily, and that we need to consider routine use of proton pump inhibitors to protect against the bleeding that can be found with the antiplatelet agents.
Also of interest, the European Society of Cardiology (ESC) recently released a focused update with some excellent recommendations on dual antiplatelet therapy in coronary artery disease in which they incorporated the results from the PIONEER-AF-PCI trial. The RE-DUAL PCI trial had not been published when these came out. If you’re concerned about ischemic risk prevailing, ESC recommendations based upon risk stratification are triple therapy with aspirin, clopidogrel, and an oral anticoagulant for longer than 1 month and up to 6 months and then dual therapy with 1 antiplatelet agent and an oral anticoagulant to complete the 12 months; afterward just oral anticoagulation alone. If the concerns about bleeding prevail, then we have 2 different pathways: one limiting triple therapy to 1 month and then dual therapy with 1 antiplatelet agent and an oral anticoagulant to complete the 12 months. But the ESC also has a second recommendation for patients at high risk of bleeding, which is dual therapy with clopidogrel and an oral anticoagulant at the offset for up to 12 months. I found these guidelines to be particularly helpful in terms of how to put this into practice.
Dr. Allen. There’s still so much concern about in-stent thrombosis. Although a smaller trial, we knew from the WOEST trial that single antiplatelet therapy with warfarin was reasonable. We know from the PIONEER-AF and RE-DUAL PCI trials that we didn’t get significantly more in-stent thrombosis by giving up the second antiplatelet. Whether or not we answered the stroke question is another issue, but the cardiology societies are still hanging on to dual antiplatelet therapy. I question if that’s based on the older data and the older stent technologies.
Dr. Minichiello. This highlights again that often we have to consider these patients’ case-by-case analysis, and that these decisions require multidisciplinary input. It involves coming together and figuring out in this particular patient, which of those 3 options would be best. We have a lot more options than we did just a short year or year and a half ago with at least some data providing comfort that DOACs at effective doses for stroke prevention in nonvalvular AF look like they can be combined with single antiplatelet therapy for post-PCI patients.
Dr. Barnes. Speaking as a cardiologist, this is a question I encounter all the time. I think everything said here is really well taken. I’ll just summarize to say for patients who have acute coronary syndromes and AF. First, I’m okay with using warfarin and now I’m okay using the DOACs, but the anticoagulant needs to continue for stroke prevention. Second, the patient has to be on clopidogrel as a P2Y12 inhibitor because it has the lowest bleeding risk profile. Third, the patient doesn’t always need 1 year of dual antiplatelet therapy the way we used to think of it. With newer generation stents and ongoing anticoagulation, you can get away with shorter courses of your antiplatelets, albeit 3 or 6 months. Providers should have a conversation with patients and think hard about how to balance clotting vs that bleeding profile.
Case 3
Dr. Minichiello. This case involves a 66-year-old man who has nonvalvular AF and is on warfarin. He has CKD, and his CrCl is about 30. He has hypertension, diabetes mellitus, and he is going to go for a colonoscopy. The proceduralist lets you know about the date and wants to know whether he needs to be bridged. He also has a remote history of a transient ischemic attack.
Dr. Allen. Historically, we’ve had detailed guidance on how to risk assess patients with AF, venous thromboembolism (VT), and mechanical heart valves in the periprocedural period. From that risk assessment the guidance helped us determine whether or not we should offer periprocedural bridging with heparin or low-molecularweight heparin.
The issue is that the detailed guidance was always based on expert opinion not hard science and there was no great evidence that we were preventing thrombotic events or that there was a net clinical benefit to bridging. Some retrospective cohort studies started coming out around 2012 that demonstrated an increased incidence of major bleeding events associated with bridging with no reduction in thrombotic complications. Some might argue that this is because thrombotic complications are so rare that you would have to have tens of thousands of patients for adequate power. Nonetheless, these studies were adequately powered to show a significant increase in major bleeding.
The best prospective trial data we have for this population comes from the BRIDGE trial, which randomized AF patients to receive dalteparin bridge therapy vs placebo during periprocedural interruptions in warfarin therapy. It too demonstrated a significant increase in the risk of bleeding complications associated with bridging with no significant reduction in the risk of thromboembolic events. Critics point out that some of the higher risk patients were underrepresented, and the same could be said about some of the other retrospective studies in the VT and mechanical valve populations.
We have waited for quite a while for guidances to catch up with these data. The most recent guidance published would apply to this patient. It was guidance for the AF population published by the American College of Cardiology (ACC) in early 2017
To make a decision to bridge, you not only have to make an assumption that your patient is at such extraordinary thrombotic risk that you would find some reduction in thrombotic events associated with bridging, but also that the benefit is going to be so great that it would overcome this very clear increased risk of bleeding, resulting in a net clinical benefit. Only then would it make sense to bridge. Based on the available data, it is quite possible that no such patient population exists.
Dr. Parra. I agree. We found from the BRIDGE trial, with the caveat that you pointed out, that high or moderately high thrombotic risk individuals weren’t as well represented, that there is at least a 2.5-fold increase in major bleeding. We also know that higher thrombotic-risk patients tend to also have higher bleeding risk. So in high thrombotic-risk patients, while there is uncertainty about whether or not there’s going to be a thrombotic benefit from bridging, we can be confident there will be an increased risk of major bleeding perhaps even more than that seen in the BRIDGE trial.
One thing that really illustrates this is the 2017 American Heart Association/ACC-focused update on the 2014 guideline for the management of patients with valvular heart disease. The guideline has a section on bridging therapy for prosthetic valves. And that was recently downgraded from a Class I recommendation to Class IIA, recognizing that the data are limited in terms of even when to bridge prosthetic valves.
Dr. Allen. Bridging is an area where we continue to do something that we know causes harm because we hope it has some benefit. Despite what some societal guidance still says, I very rarely bridge patients. If I do, it’s because the patient and/or caregivers have heard the facts and have opted to do it.
Dr. Minichiello. Practically speaking, the data—the retrospective data, the observational data, the BRIDGE trial, etc—definitely make us step back and think about bridging and realize that it’s a HIGH RISK of intervention. We really need to be informing patients about that risk. We know that bridging increases the risk of bleeding. We do not have data showing a reduction in the risk of thromboembolic disease with bridge therapy. That’s all based on what we think would happen, what we hope would be the result of bridge therapy, but in truth we do not know if this is the case. The risks of bridge therapy must be weighed heavily each time we consider using it.
In my practice I reserve bridge therapy, which we know is associated with increased harm for patients in whom we think there is the very highest risk of thromboembolism, ie, those with very recent arterial or venous thrombosis, and by recently, I mean within the past 1 to 3 months; patients who have very severe thrombophilia like antitphospholipid antibody syndrome, some cancer patients, and those with mechanical valves in the mitral position or those with high-risk mechanical aortic valves.
I don’t bridge most AF patients. In fact, I can’t remember the last time I bridged an AF patient. I do know that this is somewhat discordant with the ACC recommendations, but absent the data to support bridge therapy, I’m really concerned that the risk outweighs the benefit. This is particularly true in our VA population where the bleeding risk is high because of CKD or a history of bleeding or thrombocytopenia or concomitant aspirin or something else.
Dr. Barnes. We know from some studies that have been published that the biggest driver of a clinician deciding whether or not a patient should bridge is actually whether or not they’ve had a stroke. The truth is that the BRIDGE trial enrolled a sizeable population; it was about 15% of patients with a prior TIA or stroke. So it gave us some insight. Despite that, our event rates for thromboembolism, arterial thromboembolism, including stroke, were quite low.
I tend to look far more at the collection of risk factors and not just at a history of stroke. Now as you mentioned, Dr. Minichiello, a stroke within the past 1 to 3 months, I ask, “does this procedure even need to happen?” But outside of that, it’s not a history of stroke that’s going to make the decision for me: It’s all the risk factors together.
There’s an ongoing study, the PERIOP 2 study, that is enrolling patients at higher risk for stroke and patients with mechanical valves. This study may give us more insight into exactly what kind of risks these patients are at and whether they get benefit from bridging. But in the meantime, I’m really reserving bridging for my highest risk patients, those with multiple risk factors, CHADS2 scores of 5 and 6 or CHA2DS2-VASc of 7, 8, 9, and those with a recent VT or mechanical mitral valves.
Click here to read the digital edition.
Case 1
Tracy Minichiello, MD. The first case we’ll discuss is a 75-year-old man with mild chronic kidney disease (CKD). His calculated creatinine clearance (CrCl) is about 52 mL/min, and he has a remote history of a gastrointestinal (GI) bleeding 3 years previously from a peptic ulcer. He presents with new onset nonvalvular atrial fibrillation (AF), and he’s already on aspirin for his stable coronary artery disease (CAD).
How do we think about anticoagulant selection in this patient? We have a number of new oral anticoagulants and we have warfarin. How do we decide between warfarin vs one of the direct-acting oral anticoagulants (DOACs)? If we choose a DOAC, which one would we select?
David Parra, PharmD. The first step for anticoagulation is to assess a patient’s thromboembolic risk utilizing the CHA2DS2-VASc and bleeding risk using a HAS-BLED score, or something similar. The next question is which oral anticoagulant to use. We have widespread experience with warfarin and can measure the anticoagulant effect easily. Warfarin has a long duration of action, so perhaps it’s more forgiving if you miss a dose. It also has an antidote. Lastly, organ dysfunction doesn’t preclude use of warfarin as you can still monitor the anticoagulant effect. So there still may be patients that may benefit significantly from warfarin vs a DOAC.
On the flip side, DOACs are easier to use and perform quite acceptably in comparison with warfarin in nonvalvular AF. There are some scenarios where a specific DOAC may be preferred over another, such as recent GI bleeding.
Dr. Minichiello. Do you consider renal function, bleeding history, or concomitant antiplatelet therapy?
Geoffrey Barnes, MD, MSc. A couple of factors are relevant. I think we should consider renal function for this gentleman. However, I look at some of the other features as Dr. Parra suggested. What’s the likelihood that this patient is going to take the medicine as prescribed? Is a twice-a-day regimen going to be something that’s particularly challenging? I also look at the real-world vs randomized trial experience.
This patient has a remote GI bleeding history. Some of the real-world data suggest there might be some more GI bleeding with rivaroxaban, but across the board, apixaban (in both the randomized trials and much of the real-world data) seem to have a favorable bleeding risk profile. For a patient who is open and reliable for taking medicine twice a day, apixaban might be a good option as long as we make sure that the dose is appropriate.
Arthur L. Allen, PharmD, CACP. In pivotal trial experience, dabigatran and rivaroxaban demonstrated an increased incidence of GI bleeding compared with warfarin. In some of the real-world studies, rivaroxaban mirrors warfarin with regard to bleeding, whereas dabigatran and apixaban have a lower incidence. In the pivotal trials, apixaban did not have a trigger of increased GI bleeding, but I would let the details of this patient’s GI bleeding history help me determine how important an issue this is at this point.
The other thing that is important to understand when considering choice of agents: As Dr. Parra mentioned, we do have quite a bit of experience with warfarin. But comparing the quality of evidence, the DOACs have been investigated in a far more rigorous fashion and in far more patients than warfarin ever was in its more than 60 years on the market. For example, the RE-LY trial alone enrolled more than 18,000 patients. Each of the DOACs have been studied in tens of thousands of patients for their approved indications. Further, we shouldn’t forget that the risk of intracranial hemorrhage is reduced by roughly 50% by choosing a DOAC over warfarin, which should be a consideration in this elderly gentleman.
Dr. Minichiello. In the veteran population, there is a sense of comfort with warfarin, and some concerns have been raised over a lack of reversibility for the newer agents. We have patients who have trepidation about starting one of the new anticoagulants. However, there is a marked reduction in the risk of the most devastating bleeding complication, namely intracranial hemorrhage, making the use of these agents most compelling. And when they did have bleeding complications, at least in the trials, their outcomes were no worse than they were with warfarin, where there is a reversal agent. In most cases the outcomes were actually better.
Dr. Barnes. You often have to remind patients that there was no reversal agent in these huge trials where the DOACs showed similar or safer bleeding risk profiles, especially for the most serious bleeding, such as intracranial hemorrhage. I find patients often are reassured by knowing that.
Dr. Allen. I agree that there is concern about the lack of reversibility, but I think it has been completely overplayed. In the pivotal trials, patients who bled on DOAC therapy actually had better outcomes than those that bled on warfarin. This includes intracranial hemorrhage. There was a paper published in Stroke in 2012 that evaluated the subgroup of patients in the RE-LY trial that suffered intracranial hemorrhage. Patients on dabigatran actually fared better despite a lack of a specific reversal agent. When evaluating the available data about reversal of the DOACs, I’m not 100% convinced that we’re significantly impacting outcomes by reversing these agents. We’re certainly running up the bill, but are we treating the patient or treating the providers? As long as the renal function remains intact, the DOACs clear quickly, perhaps more quickly than warfarin can typically be reversed with standard reversal agents.
Dr. Minichiello. Remember that this patient has a history of a GI bleed. We are going to start him on full-dose anticoagulation for stroke prevention for his nonvalvular AF. He’s also on aspirin, and he has stable coronary disease. He does not have any stents in place but he did have a remote non-ST elevation myocardial infarction (MI) a number of years ago. Do we feel that the risk of dual therapy—anticoagulation combined with antiplatelet therapy—outweighs the risks? And how do we approach that risk?
Dr. Barnes. This is an important point to discuss. There has been a lot of discussion in the literature recently. When I start this type of patient on an oral anticoagulant, I try to discontinue the antiplatelet agent because I know how much bleeding risk that brings. The European guidelines (for example, Eur Heart J. 2014;35[45]:3155-3179) have been forward thinking with this for the last couple of years and have highlighted that if there’s an indication for anticoagulation for patients with stable coronary disease, meaning no MI and no stent within the past year, then we should stop the antiplatelet agents after a year in order to reduce the risk of MI. This is based on a lot of older literature where warfarin was compared with aspirin and shown to be protective in coronary patients, but at the risk of bleeding.
It’s important because there have been recent studies that have raised questions, including a recent Swedish article in (Circulation. 2017;136:1183-1192) that suggested discontinuing aspirin led to increased mortality. But it’s important to look at the details. While that was true for most patients, it was not true for the group of patients who were on an oral anticoagulant. Many colleagues ask me questions about that particular paper and its media coverage. I tell them that for our patients on chronic oral anticoagulants, the paper supports the notion that there is not increased mortality when aspirin use is stopped. We know that aspirin plus an anticoagulant leads to increased bleeding, so I try to stop it for patients who have stable CAD but are on long-term anticoagulation.
Dr. Allen. This isn’t a new thought. Back in early 2012, the 9th edition of the American College of Chest Physicians (CHEST) Antithrombotic Guidelines probably gave us the best guidance that we had ever seen to help us address this issue. Since that time the cardiology guidelines have caught up to recommend that we do not need additional antiplatelet therapy for stable CAD, and, in fact, it should be limited even in the setting of acute coronary syndromes and percutaneous coronary intervention (PCI).
Dr. Minichiello. That’s a good point because people are not necessarily clear about when there would be an indication to continue dual therapy and when it is safe to go to monotherapy. Scenarios where benefit of dual therapy may outweigh risk suggested in the CHEST 2012 guidelines include acute coronary syndrome or a recent stent, high-risk mechanical valves, and history of coronary artery bypass surgery.
I think the important thing is to consider each case individually and not to reflexively continue aspirin therapy. Often what we see is once on aspirin—always on aspirin. Being thoughtful about it, we should acknowledge that it likely results in a 2-fold increased risk of bleeding and make sure that we believe that the benefit outweighs the risk.
Dr. Allen. I agree. We probably have better evidence in the CAD population, but what do we do for patients with significant peripheral vascular disease, or those patients with symptomatic carotid stenosis or history of strokes? Some of the European guidance suggests taking a similar approach to CAD, but these are the patients for whom stopping aspirin makes me more nervous.
Dr. Parra. This is a perfect example of where less is more. All too often the reflex is to continue aspirin treatment indefinitely because the patient has a history of acute coronary syndrome or even peripheral arterial disease, when the best thing to do would be to drop the aspirin. It involves an individualized risk assessment and underscores the need to periodically do a risk/benefit assessment in all patients on anticoagulants, whether it’s warfarin or a DOAC.
I’d like to take a moment and step back to the case in the context of the GI bleeding. When we look at patients with a history of GI bleeding, it is important to understand the circumstances that surround it. This individual had a GI bleed 3 years previously and peptic ulcer disease. In these situations I ask whether the patient was taking over-the-counter nonsteroidal anti-inflamitory drugs at the time, had excessive alcohol use, or was successfully treated for Helicobacter pylori. All of these may influence whether or not I think the GI bleed is significant to influence the DOAC choice.
The other thing I consider is that the overall risk of major GI bleeding in those pivotal DOAC trials was quite small, < 1.5% per year with dabigatran 150 mg twice daily and < 1% per year with apixaban. The numbers needed to harm were quite high, over 200 patients per year with dabigatran 150 mg twice daily vs warfarin and over 350 patients per year with edoxaban 60 mg daily vs warfarin. There are no head-to-head comparisons with DOACs, but this small increased risk vs warfarin may still be an important consideration in some patients. In addition, it is important to remember that intracranial hemorrhage and fatal bleeding was less in all the pivotal NVAF trials with the DOACs when compared with warfarin. So that is something we need to reinforce with patients when we discuss treatment regardless of the DOAC selected.
Case 2
Dr. Minichiello. The next case is a 63-year-old man with hypertension, diabetes mellitus, nonvalvular AF, and he is taking dabigatran for stroke prevention. He presents in the emergency department with chest pain, and he is found to have a non-ST elevation MI. He goes to the cath lab and he is found to have a lesion in his left circumflex. The patient receives a newer generation drug-eluting stent. What are we going to do with his anticoagulation? We know he’s going to get some antiplatelet therapy, but what are our thoughts on this?
Dr. Parra. This is something that we run into all too often. I think the estimates are about 20% to 30% of patients who have indications for anticoagulation also end up having ischemic heart disease that requires PCI. The second thing is that we know combining an anticoagulant with antiplatelet therapy is associated with a 4% to 16% risk of fatal and nonfatal bleeding, and we have found out in patients with ischemic heart disease that when they bleed, they also have a higher mortality rate.
We’re trying to find the optimal balance between ischemic and thrombotic risk and bleeding risk. This is where some of the risk assessment tools that we have come into play. First, we need to establish the thrombotic risk by considering the CHA2DS2-VASc score, and the factors associated with increasing bleeding risk and stent thrombosis. You have time to work this out because, initially, all patients that are at sufficiently high thrombotic risk will receive dual antiplatelet therapy and anticoagulation therapy for a given period. This gives providers time to use some of those resources and figure out a long-term plan for the patient.
Dr. Allen. The fear and loathing that this brings up comes back to some historic things that should be considered. What we did for drug-eluting stents or bare-metal stents comes from older data where different stent technology was used. The stents used today are safer with a lower risk of in-stent thrombosis. Historically, we knew what to do for ACS and PCI and we knew what to do for AF, but we didn’t know what to do when the 2 crossed paths. We would put patients on warfarin and say, “Well, for now, target the INR between 2 and 2.5 and good luck with that.” That was all we had. The good news is now we have some evidence to move away from the use of dual antiplatelet plus anticoagulant therapies.
There were 2 DOAC studies recently published: The PIONEER-AF trial used rivaroxaban and more recently the RE-DUAL PCI trial used dabigatran. Each of the studies had some issues, but they were both studied in AF populations and aimed to address this issue of triple therapy. The PIONEER-AF trial looked at a number of different scenarios on different doses of rivaroxaban with either single or dual antiplatelet therapy compared to triple therapy with warfarin. The RE-DUAL PCI trial with dabigatran was less complex. Both studies were powered to look at safety, and they did show that with single antiplatelet plus oral anticoagulation regimens, the incidence of major bleeding complications was reduced.
However, that brings up some issues about how the studies were conducted. Both studied AF populations and in some cases did not study doses approved for AF. Yet at the same time, the studies were not powered to look at stroke outcomes, which raises the question: Are we running the risk of giving up stroke efficacy for reduced bleeding? I don’t think that we’ve fully answered that, certainly not with the PIONEER AF trial.
Dr. Parra. I agree. When we look at those trials, 2 things come to mind. First, the doses of dabigatran used in the RE-DUAL PCI trial were doses that have been shown to be beneficial in the nonvalvular AF population. Second, a take-home point from those trials is the P2Y12 inhibitor that was utilized—close to 90% or more used clopidogrel in the PIONEER-AF trial. Clopidogrel remains the P2Y12 inhibitor of choice. One of the other findings is that the aspirin dosing should be low, < 100 mg daily, and that we need to consider routine use of proton pump inhibitors to protect against the bleeding that can be found with the antiplatelet agents.
Also of interest, the European Society of Cardiology (ESC) recently released a focused update with some excellent recommendations on dual antiplatelet therapy in coronary artery disease in which they incorporated the results from the PIONEER-AF-PCI trial. The RE-DUAL PCI trial had not been published when these came out. If you’re concerned about ischemic risk prevailing, ESC recommendations based upon risk stratification are triple therapy with aspirin, clopidogrel, and an oral anticoagulant for longer than 1 month and up to 6 months and then dual therapy with 1 antiplatelet agent and an oral anticoagulant to complete the 12 months; afterward just oral anticoagulation alone. If the concerns about bleeding prevail, then we have 2 different pathways: one limiting triple therapy to 1 month and then dual therapy with 1 antiplatelet agent and an oral anticoagulant to complete the 12 months. But the ESC also has a second recommendation for patients at high risk of bleeding, which is dual therapy with clopidogrel and an oral anticoagulant at the offset for up to 12 months. I found these guidelines to be particularly helpful in terms of how to put this into practice.
Dr. Allen. There’s still so much concern about in-stent thrombosis. Although a smaller trial, we knew from the WOEST trial that single antiplatelet therapy with warfarin was reasonable. We know from the PIONEER-AF and RE-DUAL PCI trials that we didn’t get significantly more in-stent thrombosis by giving up the second antiplatelet. Whether or not we answered the stroke question is another issue, but the cardiology societies are still hanging on to dual antiplatelet therapy. I question if that’s based on the older data and the older stent technologies.
Dr. Minichiello. This highlights again that often we have to consider these patients’ case-by-case analysis, and that these decisions require multidisciplinary input. It involves coming together and figuring out in this particular patient, which of those 3 options would be best. We have a lot more options than we did just a short year or year and a half ago with at least some data providing comfort that DOACs at effective doses for stroke prevention in nonvalvular AF look like they can be combined with single antiplatelet therapy for post-PCI patients.
Dr. Barnes. Speaking as a cardiologist, this is a question I encounter all the time. I think everything said here is really well taken. I’ll just summarize to say for patients who have acute coronary syndromes and AF. First, I’m okay with using warfarin and now I’m okay using the DOACs, but the anticoagulant needs to continue for stroke prevention. Second, the patient has to be on clopidogrel as a P2Y12 inhibitor because it has the lowest bleeding risk profile. Third, the patient doesn’t always need 1 year of dual antiplatelet therapy the way we used to think of it. With newer generation stents and ongoing anticoagulation, you can get away with shorter courses of your antiplatelets, albeit 3 or 6 months. Providers should have a conversation with patients and think hard about how to balance clotting vs that bleeding profile.
Case 3
Dr. Minichiello. This case involves a 66-year-old man who has nonvalvular AF and is on warfarin. He has CKD, and his CrCl is about 30. He has hypertension, diabetes mellitus, and he is going to go for a colonoscopy. The proceduralist lets you know about the date and wants to know whether he needs to be bridged. He also has a remote history of a transient ischemic attack.
Dr. Allen. Historically, we’ve had detailed guidance on how to risk assess patients with AF, venous thromboembolism (VT), and mechanical heart valves in the periprocedural period. From that risk assessment the guidance helped us determine whether or not we should offer periprocedural bridging with heparin or low-molecularweight heparin.
The issue is that the detailed guidance was always based on expert opinion not hard science and there was no great evidence that we were preventing thrombotic events or that there was a net clinical benefit to bridging. Some retrospective cohort studies started coming out around 2012 that demonstrated an increased incidence of major bleeding events associated with bridging with no reduction in thrombotic complications. Some might argue that this is because thrombotic complications are so rare that you would have to have tens of thousands of patients for adequate power. Nonetheless, these studies were adequately powered to show a significant increase in major bleeding.
The best prospective trial data we have for this population comes from the BRIDGE trial, which randomized AF patients to receive dalteparin bridge therapy vs placebo during periprocedural interruptions in warfarin therapy. It too demonstrated a significant increase in the risk of bleeding complications associated with bridging with no significant reduction in the risk of thromboembolic events. Critics point out that some of the higher risk patients were underrepresented, and the same could be said about some of the other retrospective studies in the VT and mechanical valve populations.
We have waited for quite a while for guidances to catch up with these data. The most recent guidance published would apply to this patient. It was guidance for the AF population published by the American College of Cardiology (ACC) in early 2017
To make a decision to bridge, you not only have to make an assumption that your patient is at such extraordinary thrombotic risk that you would find some reduction in thrombotic events associated with bridging, but also that the benefit is going to be so great that it would overcome this very clear increased risk of bleeding, resulting in a net clinical benefit. Only then would it make sense to bridge. Based on the available data, it is quite possible that no such patient population exists.
Dr. Parra. I agree. We found from the BRIDGE trial, with the caveat that you pointed out, that high or moderately high thrombotic risk individuals weren’t as well represented, that there is at least a 2.5-fold increase in major bleeding. We also know that higher thrombotic-risk patients tend to also have higher bleeding risk. So in high thrombotic-risk patients, while there is uncertainty about whether or not there’s going to be a thrombotic benefit from bridging, we can be confident there will be an increased risk of major bleeding perhaps even more than that seen in the BRIDGE trial.
One thing that really illustrates this is the 2017 American Heart Association/ACC-focused update on the 2014 guideline for the management of patients with valvular heart disease. The guideline has a section on bridging therapy for prosthetic valves. And that was recently downgraded from a Class I recommendation to Class IIA, recognizing that the data are limited in terms of even when to bridge prosthetic valves.
Dr. Allen. Bridging is an area where we continue to do something that we know causes harm because we hope it has some benefit. Despite what some societal guidance still says, I very rarely bridge patients. If I do, it’s because the patient and/or caregivers have heard the facts and have opted to do it.
Dr. Minichiello. Practically speaking, the data—the retrospective data, the observational data, the BRIDGE trial, etc—definitely make us step back and think about bridging and realize that it’s a HIGH RISK of intervention. We really need to be informing patients about that risk. We know that bridging increases the risk of bleeding. We do not have data showing a reduction in the risk of thromboembolic disease with bridge therapy. That’s all based on what we think would happen, what we hope would be the result of bridge therapy, but in truth we do not know if this is the case. The risks of bridge therapy must be weighed heavily each time we consider using it.
In my practice I reserve bridge therapy, which we know is associated with increased harm for patients in whom we think there is the very highest risk of thromboembolism, ie, those with very recent arterial or venous thrombosis, and by recently, I mean within the past 1 to 3 months; patients who have very severe thrombophilia like antitphospholipid antibody syndrome, some cancer patients, and those with mechanical valves in the mitral position or those with high-risk mechanical aortic valves.
I don’t bridge most AF patients. In fact, I can’t remember the last time I bridged an AF patient. I do know that this is somewhat discordant with the ACC recommendations, but absent the data to support bridge therapy, I’m really concerned that the risk outweighs the benefit. This is particularly true in our VA population where the bleeding risk is high because of CKD or a history of bleeding or thrombocytopenia or concomitant aspirin or something else.
Dr. Barnes. We know from some studies that have been published that the biggest driver of a clinician deciding whether or not a patient should bridge is actually whether or not they’ve had a stroke. The truth is that the BRIDGE trial enrolled a sizeable population; it was about 15% of patients with a prior TIA or stroke. So it gave us some insight. Despite that, our event rates for thromboembolism, arterial thromboembolism, including stroke, were quite low.
I tend to look far more at the collection of risk factors and not just at a history of stroke. Now as you mentioned, Dr. Minichiello, a stroke within the past 1 to 3 months, I ask, “does this procedure even need to happen?” But outside of that, it’s not a history of stroke that’s going to make the decision for me: It’s all the risk factors together.
There’s an ongoing study, the PERIOP 2 study, that is enrolling patients at higher risk for stroke and patients with mechanical valves. This study may give us more insight into exactly what kind of risks these patients are at and whether they get benefit from bridging. But in the meantime, I’m really reserving bridging for my highest risk patients, those with multiple risk factors, CHADS2 scores of 5 and 6 or CHA2DS2-VASc of 7, 8, 9, and those with a recent VT or mechanical mitral valves.
Click here to read the digital edition.
Eruptive Vellus Hair Cysts in Identical Triplets With Dermoscopic Findings
Case Report
Four-year-old identical triplet girls with numerous asymptomatic scattered papules on the chest of 4 months’ duration were referred to a dermatologist by their pediatrician for molluscum contagiosum. The patients’ father reported that there was no history of trauma, irritation, or manipulation to the affected area. Their medical history was notable for prematurity at 32 weeks’ gestation and congenital dermal melanocytosis. Family history was notable for their father having acne and similar papules on the chest during adolescence that resolved with isotretinoin therapy.
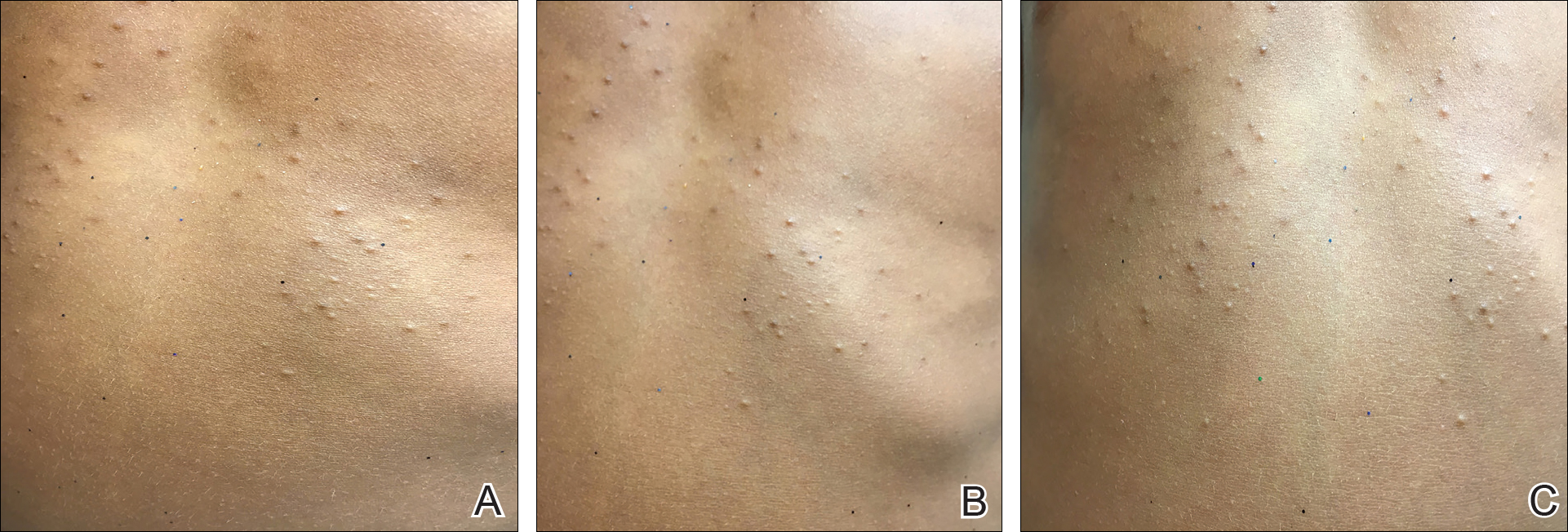
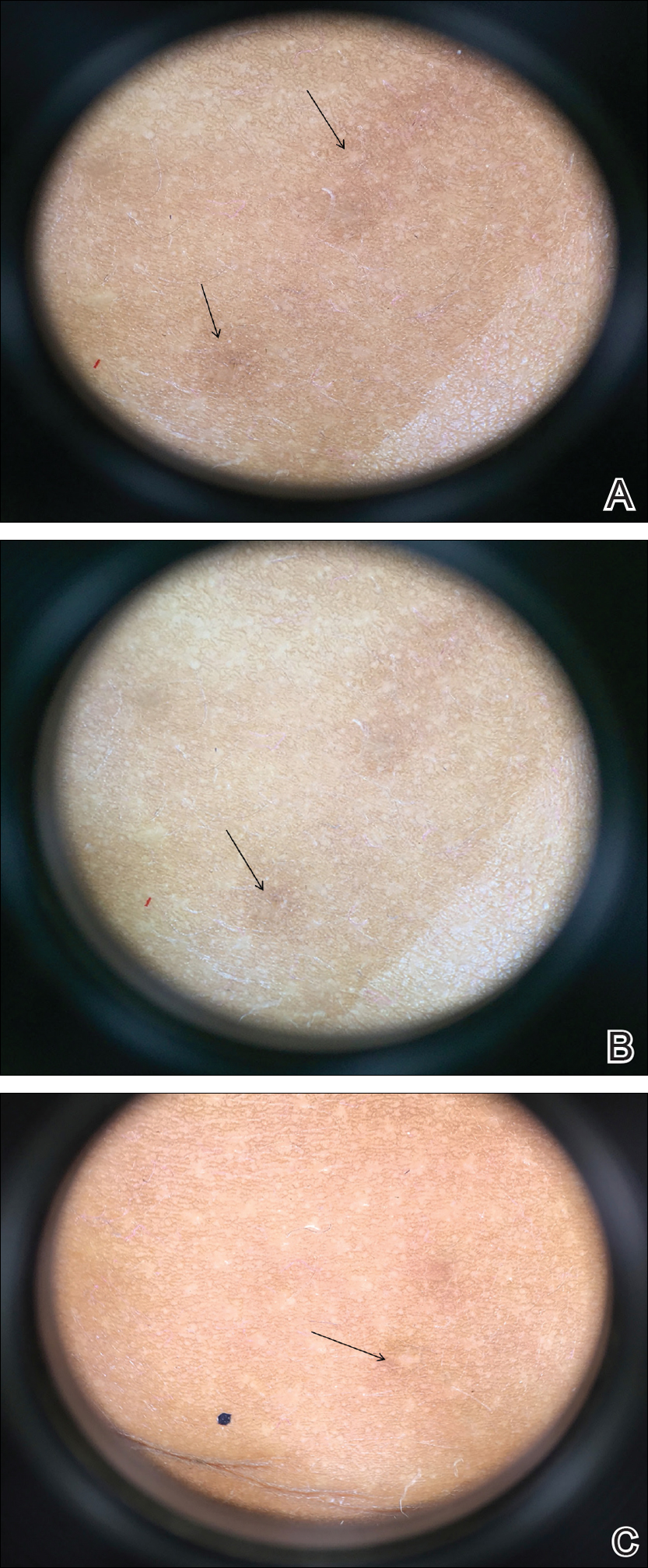
On physical examination there were multiple smooth, hyperpigmented to erythematous, comedonal, 1- to 2-mm papules dispersed on the anterior central chest of all 3 patients (Figure 1). Clinically, these lesions were fairly indistinguishable from other common dermatologic conditions such as acne or milia. Dermoscopic examination revealed homogenous yellow-white areas surrounded by light brown to erythematous halos (Figure 2). Histopathologic examination was not performed given the benign clinical diagnosis and avoidance of biopsy in pediatric populations. Based on dermoscopic features and history, a diagnosis of eruptive vellus hair cysts (EVHCs) in identical triplets was made.
Comment
Pathogenesis
Eruptive vellus hair cysts, first introduced by Esterly et al1 in 1977, are uncommon benign lesions presumed to be caused by an abnormal development of the infundibular portion of the hair follicle.2 They are usually 1- to 3-mm, reddish brown, monomorphous papules overlapping with pilosebaceous and apocrine units.3 Although the lesions typically are located on the chest and extremities, they may occur on the face, abdomen, axillae, buttocks, or genital area.1,3 The inheritance of EVHCs is unclear. The majority of reported cases are sporadic; however, the literature mentions 19 families affected by autosomal-dominant EVHCs based on phylogeny.3 In 2015, EVHCs were reported in identical twins, further supporting the case for a genetic mutation.4 We augment this autosomal-dominant inheritance pattern by presenting a case of identical triplets with EVHCs. The patients’ father reported similar lesions in childhood, further underscoring a genetic basis.
The pathogenesis of EVHC is uncertain, with 2 main theories. Some propose retention of vellus hair and keratin in a cavity formed by an abnormal vellus hair follicle causing infundibular occlusion. Others consider the growth of benign follicular hamartomas that differentiate to become vellus hairs.1
Clinical Presentation
The sporadic form of EVHCs is noted to be more common and clinically presents later, with an average age at onset of 16 years and an average age at diagnosis of 24 years.3 The sporadic form occurs without trauma or manipulation as a precursor. Less commonly, lesions present at birth or in early infancy and may show an autosomal-dominant inheritance pattern with a similar distribution across relatives.3
Other variants of EVHCs have been described. Late-onset EVHC usually occurs at 35 years or older (average age, 57 years), with a female to male predominance of 2.5 to 1.3 This late onset may be attributed to proliferation of ductal follicular keratinocytes or loss of perifollicular elastic fibers exacerbated by exogenous factors such as manipulation, UV rays, or trauma.5
For unilesional EVHC, the average age at diagnosis is 27 years.3 Some of these lesions may be pedunculated and greater than 8 mm. There is a female to male predominance of 2 to 1. Eruptive vellus hair cysts with steatocystoma multiplex can be seen with an average age at onset of 19 years and a female to male predominance of 0.2 to 1. There may be a family history of this subset, as reported in 3 patients with this pattern.3
Diagnosis
The recommended workup for EVHCs varies by patient and age. Eruptive vellus hair cysts present an opportunity to utilize noninvasive diagnostic procedures, especially for the pediatric population, to avoid scarring and pain from manipulation or biopsies. Although many practitioners may comfortably diagnose EVHCs clinically, 6 cases were misdiagnosed as steatocystoma multiplex, keratosis pilaris, or milia prior to histopathology revealing vellus hair cysts.6
Dermoscopy presents as a useful diagnostic aid. Eruptive vellus hair cysts exhibit light yellow homogenous circular structures with a maroon or erythematous halo.2,7 A central gray-blue color point may be seen due to melanin in the pigmented hair shaft.7 A dermoscopy review of EVHCs reported radiating capillaries.2 Occasionally, nonfollicular homogenous blue pigmentation may be seen due to a connection to atrophic hair follicles in the mid dermis and no normal hair follicle around the cysts.8 In comparison, dermoscopic characteristics of molluscum contagiosum demonstrated a polylobular, white-yellow, amorphous structure at the center with a hardened central umbilicated core and a crown of hairpin vessels at the periphery. Additionally, comedonal acne, commonly mistaken for EVHCs, reveals a brown-yellow hard central plug with sparse inflammation under dermoscopy.2 Thus, differentiation of these entities with dermoscopy should be highly prioritized to better aid in the diagnosis of pediatric dermatologic conditions using painless noninvasive techniques.
Treatment
The main indication for treatment of EVHCs is cosmetic concern. Twenty-five percent of EVHCs spontaneously resolve with transepidermal hair elimination or a granulomatous reaction.4,5 A case report of 4 siblings with congenital EVHCs also described a mother with similar lesions that resolved spontaneously in early adulthood,3 as our patients’ father also noted. Treatment modalities including topical keratolytic agents such as urea 10%, retinoic acid 0.05%, tazarotene cream 0.1%, and lactic acid 12%; incision and drainage; CO2 laser; or erbium-doped YAG laser ablation have been tried with minimal improvement.9 Of note, tazarotene cream 0.1% has demonstrated better results than both erbium-doped YAG laser and drainage and incision of EVHCs.4 Additionally, another report evidenced partial improvement with calcipotriene within 2 months with some lesions completely resolved and others flattened, which may be attributed to the antiproliferative and prodifferentiating effects on the ductal follicular keratinocytes by calcipotriene.5 Lastly, an additional study indicated that isotretinoin and vitamin A derivatives were ineffective for clearing EVHCs.10
Conclusion
We presented 3 identical triplets with the classic pediatric onset and dermoscopic findings of EVHCs on the trunk. Although the definitive diagnosis of EVHCs relies on histopathology, we argue that their unique dermoscopic findings combined with a thorough clinical examination is sufficient to recognize this benign condition and avoid painful procedures in the pediatric population.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Alfaro-Castellón P, Mejía-Rodríguez SA, Valencia-Herrera A, et al. Dermoscopy distinction of eruptive vellus hair cysts with molluscum contagiosum and acne lesions. Pediatr Dermatol. 2012;29:772-773.
- Torchia D, Vega J, Schachner LA. Eruptive vellus hair cysts: a systematic review. Am J Clin Dermatol. 2012;13:19-28.
- Pauline G, Alain H, Jean-Jaques R, et al. Eruptive vellus hair cysts: an original case occurring in twins [published online July 11, 2014]. Int J Dermatol. 2015;54:E209-E212.
- Erkek E, Kurtipek GS, Duman D, et al. Eruptive vellus hair cysts: report of a pediatric case with partial response to calcipotriene therapy. Cutis. 2009;84:295-298.
- Shi G, Zhou Y, Cai YX, et al. Clinicopathological features and expression of four keratins (K10, K14, K17 and K19) in six cases of eruptive vellus hair cysts. Clin Exp Dermatol. 2014;39:496-499.
- Panchaprateep R, Tanus A, Tosti A. Clinical, dermoscopic, and histopathologic features of body hair disorders. J Am Acad Dermatol. 2015;72:890-900.
- Takada S, Togawa Y, Wakabayashii S, et al. Dermoscopic findings in eruptive vellus hair cysts: a case report. Austin J Dermatol. 2014;1:1004.
- Khatu S, Vasani R, Amin S. Eruptive vellus hair cyst presenting as asymptomatic follicular papules on extremities. Indian Dermatol Online J. 2013;4:213-215.
- Urbina-Gonzalez F, Aguilar-Martinez A, Cristobal-Gil M, et al. The treatment of eruptive vellus hair cysts with isotretinoin. Br J Dermatol. 1987;116:465-466.
Case Report
Four-year-old identical triplet girls with numerous asymptomatic scattered papules on the chest of 4 months’ duration were referred to a dermatologist by their pediatrician for molluscum contagiosum. The patients’ father reported that there was no history of trauma, irritation, or manipulation to the affected area. Their medical history was notable for prematurity at 32 weeks’ gestation and congenital dermal melanocytosis. Family history was notable for their father having acne and similar papules on the chest during adolescence that resolved with isotretinoin therapy.
On physical examination there were multiple smooth, hyperpigmented to erythematous, comedonal, 1- to 2-mm papules dispersed on the anterior central chest of all 3 patients (Figure 1). Clinically, these lesions were fairly indistinguishable from other common dermatologic conditions such as acne or milia. Dermoscopic examination revealed homogenous yellow-white areas surrounded by light brown to erythematous halos (Figure 2). Histopathologic examination was not performed given the benign clinical diagnosis and avoidance of biopsy in pediatric populations. Based on dermoscopic features and history, a diagnosis of eruptive vellus hair cysts (EVHCs) in identical triplets was made.
Comment
Pathogenesis
Eruptive vellus hair cysts, first introduced by Esterly et al1 in 1977, are uncommon benign lesions presumed to be caused by an abnormal development of the infundibular portion of the hair follicle.2 They are usually 1- to 3-mm, reddish brown, monomorphous papules overlapping with pilosebaceous and apocrine units.3 Although the lesions typically are located on the chest and extremities, they may occur on the face, abdomen, axillae, buttocks, or genital area.1,3 The inheritance of EVHCs is unclear. The majority of reported cases are sporadic; however, the literature mentions 19 families affected by autosomal-dominant EVHCs based on phylogeny.3 In 2015, EVHCs were reported in identical twins, further supporting the case for a genetic mutation.4 We augment this autosomal-dominant inheritance pattern by presenting a case of identical triplets with EVHCs. The patients’ father reported similar lesions in childhood, further underscoring a genetic basis.
The pathogenesis of EVHC is uncertain, with 2 main theories. Some propose retention of vellus hair and keratin in a cavity formed by an abnormal vellus hair follicle causing infundibular occlusion. Others consider the growth of benign follicular hamartomas that differentiate to become vellus hairs.1
Clinical Presentation
The sporadic form of EVHCs is noted to be more common and clinically presents later, with an average age at onset of 16 years and an average age at diagnosis of 24 years.3 The sporadic form occurs without trauma or manipulation as a precursor. Less commonly, lesions present at birth or in early infancy and may show an autosomal-dominant inheritance pattern with a similar distribution across relatives.3
Other variants of EVHCs have been described. Late-onset EVHC usually occurs at 35 years or older (average age, 57 years), with a female to male predominance of 2.5 to 1.3 This late onset may be attributed to proliferation of ductal follicular keratinocytes or loss of perifollicular elastic fibers exacerbated by exogenous factors such as manipulation, UV rays, or trauma.5
For unilesional EVHC, the average age at diagnosis is 27 years.3 Some of these lesions may be pedunculated and greater than 8 mm. There is a female to male predominance of 2 to 1. Eruptive vellus hair cysts with steatocystoma multiplex can be seen with an average age at onset of 19 years and a female to male predominance of 0.2 to 1. There may be a family history of this subset, as reported in 3 patients with this pattern.3
Diagnosis
The recommended workup for EVHCs varies by patient and age. Eruptive vellus hair cysts present an opportunity to utilize noninvasive diagnostic procedures, especially for the pediatric population, to avoid scarring and pain from manipulation or biopsies. Although many practitioners may comfortably diagnose EVHCs clinically, 6 cases were misdiagnosed as steatocystoma multiplex, keratosis pilaris, or milia prior to histopathology revealing vellus hair cysts.6
Dermoscopy presents as a useful diagnostic aid. Eruptive vellus hair cysts exhibit light yellow homogenous circular structures with a maroon or erythematous halo.2,7 A central gray-blue color point may be seen due to melanin in the pigmented hair shaft.7 A dermoscopy review of EVHCs reported radiating capillaries.2 Occasionally, nonfollicular homogenous blue pigmentation may be seen due to a connection to atrophic hair follicles in the mid dermis and no normal hair follicle around the cysts.8 In comparison, dermoscopic characteristics of molluscum contagiosum demonstrated a polylobular, white-yellow, amorphous structure at the center with a hardened central umbilicated core and a crown of hairpin vessels at the periphery. Additionally, comedonal acne, commonly mistaken for EVHCs, reveals a brown-yellow hard central plug with sparse inflammation under dermoscopy.2 Thus, differentiation of these entities with dermoscopy should be highly prioritized to better aid in the diagnosis of pediatric dermatologic conditions using painless noninvasive techniques.
Treatment
The main indication for treatment of EVHCs is cosmetic concern. Twenty-five percent of EVHCs spontaneously resolve with transepidermal hair elimination or a granulomatous reaction.4,5 A case report of 4 siblings with congenital EVHCs also described a mother with similar lesions that resolved spontaneously in early adulthood,3 as our patients’ father also noted. Treatment modalities including topical keratolytic agents such as urea 10%, retinoic acid 0.05%, tazarotene cream 0.1%, and lactic acid 12%; incision and drainage; CO2 laser; or erbium-doped YAG laser ablation have been tried with minimal improvement.9 Of note, tazarotene cream 0.1% has demonstrated better results than both erbium-doped YAG laser and drainage and incision of EVHCs.4 Additionally, another report evidenced partial improvement with calcipotriene within 2 months with some lesions completely resolved and others flattened, which may be attributed to the antiproliferative and prodifferentiating effects on the ductal follicular keratinocytes by calcipotriene.5 Lastly, an additional study indicated that isotretinoin and vitamin A derivatives were ineffective for clearing EVHCs.10
Conclusion
We presented 3 identical triplets with the classic pediatric onset and dermoscopic findings of EVHCs on the trunk. Although the definitive diagnosis of EVHCs relies on histopathology, we argue that their unique dermoscopic findings combined with a thorough clinical examination is sufficient to recognize this benign condition and avoid painful procedures in the pediatric population.
Case Report
Four-year-old identical triplet girls with numerous asymptomatic scattered papules on the chest of 4 months’ duration were referred to a dermatologist by their pediatrician for molluscum contagiosum. The patients’ father reported that there was no history of trauma, irritation, or manipulation to the affected area. Their medical history was notable for prematurity at 32 weeks’ gestation and congenital dermal melanocytosis. Family history was notable for their father having acne and similar papules on the chest during adolescence that resolved with isotretinoin therapy.
On physical examination there were multiple smooth, hyperpigmented to erythematous, comedonal, 1- to 2-mm papules dispersed on the anterior central chest of all 3 patients (Figure 1). Clinically, these lesions were fairly indistinguishable from other common dermatologic conditions such as acne or milia. Dermoscopic examination revealed homogenous yellow-white areas surrounded by light brown to erythematous halos (Figure 2). Histopathologic examination was not performed given the benign clinical diagnosis and avoidance of biopsy in pediatric populations. Based on dermoscopic features and history, a diagnosis of eruptive vellus hair cysts (EVHCs) in identical triplets was made.
Comment
Pathogenesis
Eruptive vellus hair cysts, first introduced by Esterly et al1 in 1977, are uncommon benign lesions presumed to be caused by an abnormal development of the infundibular portion of the hair follicle.2 They are usually 1- to 3-mm, reddish brown, monomorphous papules overlapping with pilosebaceous and apocrine units.3 Although the lesions typically are located on the chest and extremities, they may occur on the face, abdomen, axillae, buttocks, or genital area.1,3 The inheritance of EVHCs is unclear. The majority of reported cases are sporadic; however, the literature mentions 19 families affected by autosomal-dominant EVHCs based on phylogeny.3 In 2015, EVHCs were reported in identical twins, further supporting the case for a genetic mutation.4 We augment this autosomal-dominant inheritance pattern by presenting a case of identical triplets with EVHCs. The patients’ father reported similar lesions in childhood, further underscoring a genetic basis.
The pathogenesis of EVHC is uncertain, with 2 main theories. Some propose retention of vellus hair and keratin in a cavity formed by an abnormal vellus hair follicle causing infundibular occlusion. Others consider the growth of benign follicular hamartomas that differentiate to become vellus hairs.1
Clinical Presentation
The sporadic form of EVHCs is noted to be more common and clinically presents later, with an average age at onset of 16 years and an average age at diagnosis of 24 years.3 The sporadic form occurs without trauma or manipulation as a precursor. Less commonly, lesions present at birth or in early infancy and may show an autosomal-dominant inheritance pattern with a similar distribution across relatives.3
Other variants of EVHCs have been described. Late-onset EVHC usually occurs at 35 years or older (average age, 57 years), with a female to male predominance of 2.5 to 1.3 This late onset may be attributed to proliferation of ductal follicular keratinocytes or loss of perifollicular elastic fibers exacerbated by exogenous factors such as manipulation, UV rays, or trauma.5
For unilesional EVHC, the average age at diagnosis is 27 years.3 Some of these lesions may be pedunculated and greater than 8 mm. There is a female to male predominance of 2 to 1. Eruptive vellus hair cysts with steatocystoma multiplex can be seen with an average age at onset of 19 years and a female to male predominance of 0.2 to 1. There may be a family history of this subset, as reported in 3 patients with this pattern.3
Diagnosis
The recommended workup for EVHCs varies by patient and age. Eruptive vellus hair cysts present an opportunity to utilize noninvasive diagnostic procedures, especially for the pediatric population, to avoid scarring and pain from manipulation or biopsies. Although many practitioners may comfortably diagnose EVHCs clinically, 6 cases were misdiagnosed as steatocystoma multiplex, keratosis pilaris, or milia prior to histopathology revealing vellus hair cysts.6
Dermoscopy presents as a useful diagnostic aid. Eruptive vellus hair cysts exhibit light yellow homogenous circular structures with a maroon or erythematous halo.2,7 A central gray-blue color point may be seen due to melanin in the pigmented hair shaft.7 A dermoscopy review of EVHCs reported radiating capillaries.2 Occasionally, nonfollicular homogenous blue pigmentation may be seen due to a connection to atrophic hair follicles in the mid dermis and no normal hair follicle around the cysts.8 In comparison, dermoscopic characteristics of molluscum contagiosum demonstrated a polylobular, white-yellow, amorphous structure at the center with a hardened central umbilicated core and a crown of hairpin vessels at the periphery. Additionally, comedonal acne, commonly mistaken for EVHCs, reveals a brown-yellow hard central plug with sparse inflammation under dermoscopy.2 Thus, differentiation of these entities with dermoscopy should be highly prioritized to better aid in the diagnosis of pediatric dermatologic conditions using painless noninvasive techniques.
Treatment
The main indication for treatment of EVHCs is cosmetic concern. Twenty-five percent of EVHCs spontaneously resolve with transepidermal hair elimination or a granulomatous reaction.4,5 A case report of 4 siblings with congenital EVHCs also described a mother with similar lesions that resolved spontaneously in early adulthood,3 as our patients’ father also noted. Treatment modalities including topical keratolytic agents such as urea 10%, retinoic acid 0.05%, tazarotene cream 0.1%, and lactic acid 12%; incision and drainage; CO2 laser; or erbium-doped YAG laser ablation have been tried with minimal improvement.9 Of note, tazarotene cream 0.1% has demonstrated better results than both erbium-doped YAG laser and drainage and incision of EVHCs.4 Additionally, another report evidenced partial improvement with calcipotriene within 2 months with some lesions completely resolved and others flattened, which may be attributed to the antiproliferative and prodifferentiating effects on the ductal follicular keratinocytes by calcipotriene.5 Lastly, an additional study indicated that isotretinoin and vitamin A derivatives were ineffective for clearing EVHCs.10
Conclusion
We presented 3 identical triplets with the classic pediatric onset and dermoscopic findings of EVHCs on the trunk. Although the definitive diagnosis of EVHCs relies on histopathology, we argue that their unique dermoscopic findings combined with a thorough clinical examination is sufficient to recognize this benign condition and avoid painful procedures in the pediatric population.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Alfaro-Castellón P, Mejía-Rodríguez SA, Valencia-Herrera A, et al. Dermoscopy distinction of eruptive vellus hair cysts with molluscum contagiosum and acne lesions. Pediatr Dermatol. 2012;29:772-773.
- Torchia D, Vega J, Schachner LA. Eruptive vellus hair cysts: a systematic review. Am J Clin Dermatol. 2012;13:19-28.
- Pauline G, Alain H, Jean-Jaques R, et al. Eruptive vellus hair cysts: an original case occurring in twins [published online July 11, 2014]. Int J Dermatol. 2015;54:E209-E212.
- Erkek E, Kurtipek GS, Duman D, et al. Eruptive vellus hair cysts: report of a pediatric case with partial response to calcipotriene therapy. Cutis. 2009;84:295-298.
- Shi G, Zhou Y, Cai YX, et al. Clinicopathological features and expression of four keratins (K10, K14, K17 and K19) in six cases of eruptive vellus hair cysts. Clin Exp Dermatol. 2014;39:496-499.
- Panchaprateep R, Tanus A, Tosti A. Clinical, dermoscopic, and histopathologic features of body hair disorders. J Am Acad Dermatol. 2015;72:890-900.
- Takada S, Togawa Y, Wakabayashii S, et al. Dermoscopic findings in eruptive vellus hair cysts: a case report. Austin J Dermatol. 2014;1:1004.
- Khatu S, Vasani R, Amin S. Eruptive vellus hair cyst presenting as asymptomatic follicular papules on extremities. Indian Dermatol Online J. 2013;4:213-215.
- Urbina-Gonzalez F, Aguilar-Martinez A, Cristobal-Gil M, et al. The treatment of eruptive vellus hair cysts with isotretinoin. Br J Dermatol. 1987;116:465-466.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Alfaro-Castellón P, Mejía-Rodríguez SA, Valencia-Herrera A, et al. Dermoscopy distinction of eruptive vellus hair cysts with molluscum contagiosum and acne lesions. Pediatr Dermatol. 2012;29:772-773.
- Torchia D, Vega J, Schachner LA. Eruptive vellus hair cysts: a systematic review. Am J Clin Dermatol. 2012;13:19-28.
- Pauline G, Alain H, Jean-Jaques R, et al. Eruptive vellus hair cysts: an original case occurring in twins [published online July 11, 2014]. Int J Dermatol. 2015;54:E209-E212.
- Erkek E, Kurtipek GS, Duman D, et al. Eruptive vellus hair cysts: report of a pediatric case with partial response to calcipotriene therapy. Cutis. 2009;84:295-298.
- Shi G, Zhou Y, Cai YX, et al. Clinicopathological features and expression of four keratins (K10, K14, K17 and K19) in six cases of eruptive vellus hair cysts. Clin Exp Dermatol. 2014;39:496-499.
- Panchaprateep R, Tanus A, Tosti A. Clinical, dermoscopic, and histopathologic features of body hair disorders. J Am Acad Dermatol. 2015;72:890-900.
- Takada S, Togawa Y, Wakabayashii S, et al. Dermoscopic findings in eruptive vellus hair cysts: a case report. Austin J Dermatol. 2014;1:1004.
- Khatu S, Vasani R, Amin S. Eruptive vellus hair cyst presenting as asymptomatic follicular papules on extremities. Indian Dermatol Online J. 2013;4:213-215.
- Urbina-Gonzalez F, Aguilar-Martinez A, Cristobal-Gil M, et al. The treatment of eruptive vellus hair cysts with isotretinoin. Br J Dermatol. 1987;116:465-466.
Practice Points
- Eruptive vellus hair cysts (EVHCs) are 1- to 3-mm round, dome-shaped, flesh-colored, asymptomatic, benign papules typically occurring on the chest and extremities.
- Pathogenesis and inheritance are unclear. Although the majority of EVHC cases are sporadic, the strong influence of genes is indicated by numerous reports of families in whom 2 or more members were affected.
- Dermoscopy is a noninvasive diagnostic procedure that should be utilized to diagnose EVHCs in the pediatric population; specifically, EVHCs exhibit light yellow, homogenous, circular structures with a maroon or erythematous halo.
- The main indication for treatment of EVHCs is cosmetic concern; however, one-quarter of cases may resolve spontaneously.
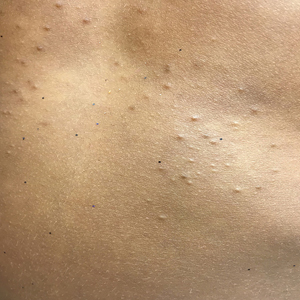
Pediatric Primary Cutaneous Blastomycosis Clinically Responsive to Itraconazole
Blastomycosis is a polymorphic disease caused by the thermally dimorphic fungus Blastomyces dermatitidis, which is naturally occurring worldwide but particularly prominent in the Great Lakes, Mississippi, and Ohio River areas of the United States. The disease was first described by Thomas Caspar Gilchrist in 1894 and historically has been referred to as Gilchrist disease, North American blastomycosis, or Chicago disease.1,2 Cutaneous blastomycosis can occur by dissemination of yeast to the skin from systemic and pulmonary disease or rarely via direct inoculation of the skin resulting in primary cutaneous disease. Clinically, the lesions are polymorphic and may appear as well-demarcated verrucous plaques containing foci of pustules or ulcerations. Lesions typically heal centrifugally with a cribriform scar.3
We describe an adolescent with a unique history of inoculation 2 weeks prior to the development of a biopsy-confirmed lesion of cutaneous blastomycosis on the left chest wall that clinically resolved following 6 months of itraconazole.
Case Report
A 16-year-old adolescent boy with a history of morbid obesity, asthma, and seasonal allergies presented for evaluation of a painful, slowly enlarging skin lesion on the left chest wall of 2 months’ duration. According to the patient, a “small pimple” appeared at the site of impact 2 weeks following a fall into a muddy flowerbed in Madison, Wisconsin. The patient recalled that although he had soiled his clothing, there was no identifiable puncture of the skin. Despite daily application of hydrogen peroxide and a 1-week course of trimethoprim-sulfamethoxazole, the lesion gradually enlarged. Complete review of systems as well as exposure and travel history were otherwise negative.
Physical examination revealed a 5.0×2.5-cm exophytic, firm, well-circumscribed plaque with a papillated crusted surface on the left side of the chest near the posterior axillary line (Figure 1). There was no palpable regional lymphadenopathy. Pulmonary examination was unremarkable. Diagnostic workup, including complete blood cell count with differential, hemoglobin A1c, human immunodeficiency virus antibody/antigen testing, interferon-gamma release assay, and chest radiograph were all within normal limits.
Histologic examination of a biopsy specimen showed pseudoepitheliomatous hyperplasia of the epidermis with a brisk mixed inflammatory infiltrate (Figure 2). Displayed in Figure 3 is the Grocott-Gomori methenamine-silver stain that highlighted the thick double-contoured wall-budding yeasts.
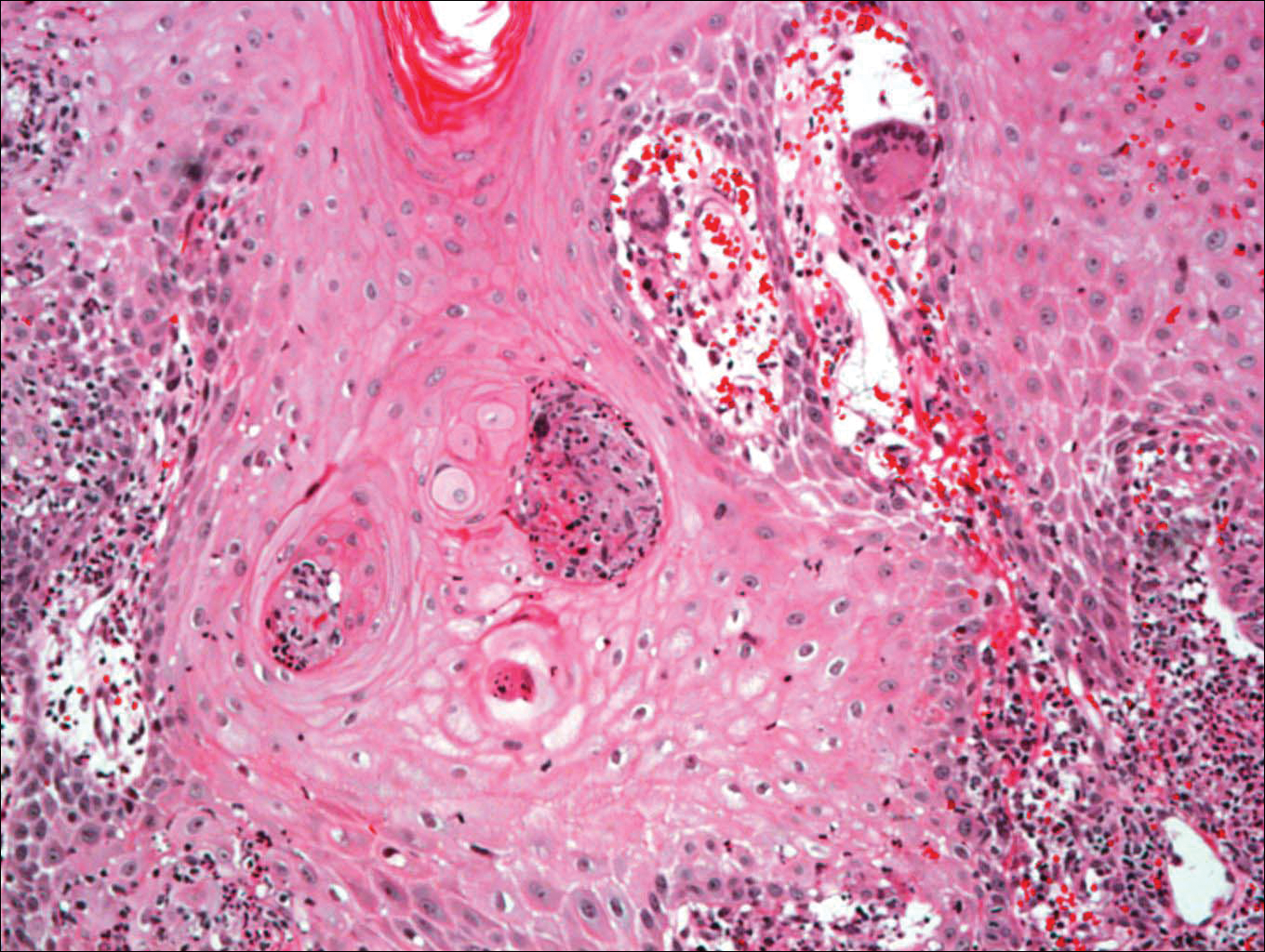
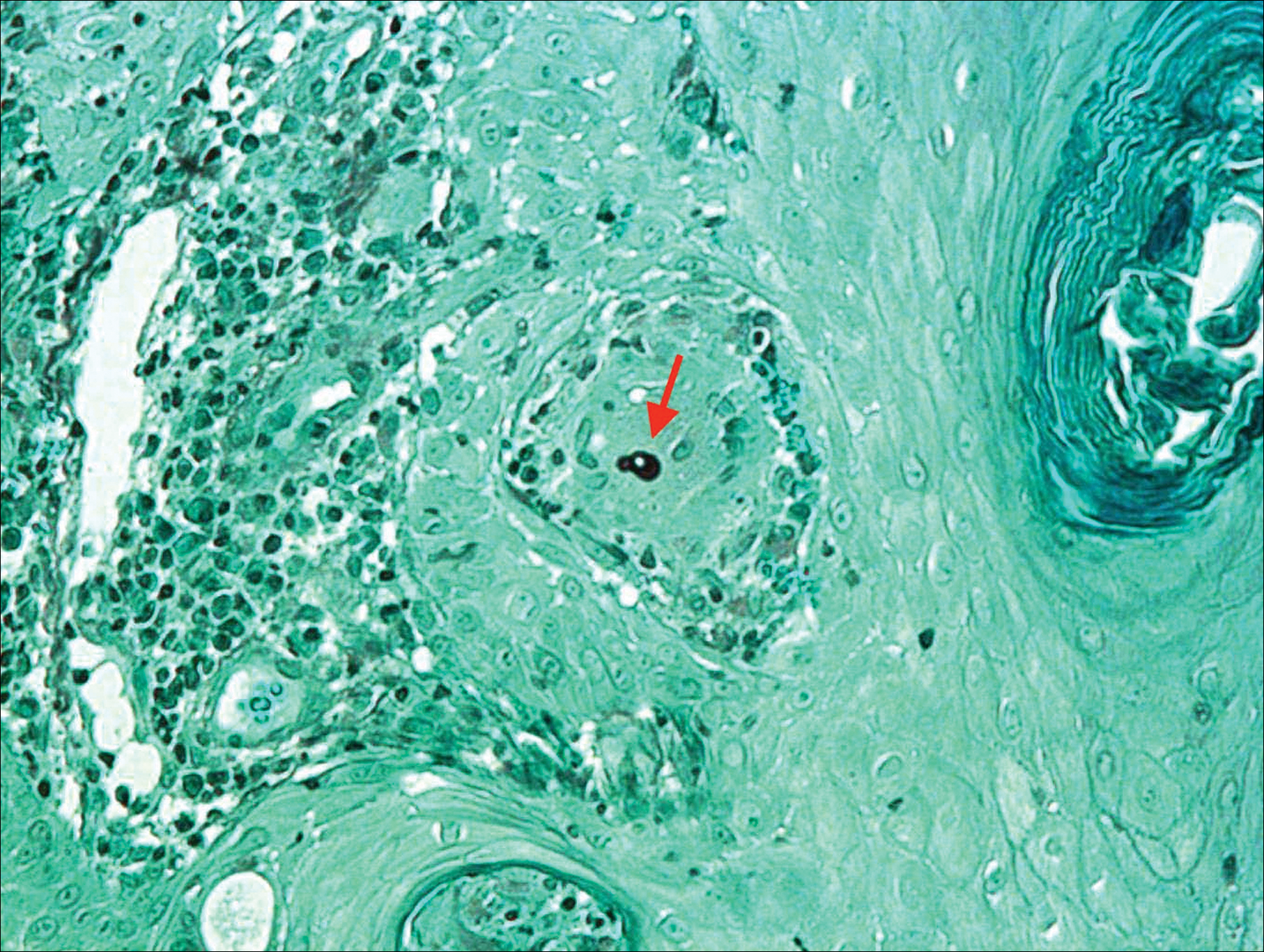
The patient was diagnosed with primary cutaneous blastomycosis. Treatment was initiated with itraconazole 200 mg 3 times daily for 3 days, followed by 200 mg 2 times daily for 6 months. Following 3 months of therapy, the lesion had markedly improved with violaceous dyschromia and no residual surface changes. After 5 months of itraconazole, the patient stopped taking the medication for 2 months due to pharmacy issues and then resumed. After 6 total months of therapy, the lesion healed with only residual dyschromia and itraconazole was discontinued.
Comment
Epidemiology
Blastomycosis is a polymorphic pyogranulomatous disease caused by the dimorphic fungus B dermatitidis, naturally occurring in the soil with a worldwide distribution.4 Individuals affected by the disease often reside in locations where the fungus is endemic, specifically in areas that border the Mississippi and Ohio rivers, the Great Lakes, and Canadian provinces near the Saint Lawrence Seaway. More recently there has been an increased incidence of blastomycosis, with the highest proportion found in Wisconsin and Michigan.1,2 Exposures often are associated with recreational and occupational activities near streams or rivers where there may be decaying vegetation.1 Despite the ubiquitous presence of B dermatitidis in regions where the species is endemic, it is likely that many individuals who are exposed to the organism do not develop infection.
Pathogenesis
The exact pathogenesis for the development of disease in a particular individual remains unclear. Immunosuppression is not a prerequisite for susceptibility, as evidenced by a review of 123 cases of blastomycosis in which a preceding immunodepressive disorder was present in only 25% of patients. The same study found that it was almost equally common as diabetes mellitus and present in 22% of patients.5 The organism is considered a true pathogen given its ability to affect healthy individuals and the presence of a newly identified novel 120-kD glycoprotein antigen (WI-1) on the cell wall that may confer virulence via extracellular matrix and macrophage binding. Intact cell-mediated immunity that prevents the conversion of conidia (the infectious agent) to yeast (the form that exists at body temperature) plays a key role in conferring natural resistance.6,7
Cutaneous infection may occur by either dissemination of yeast to the skin from systemic disease or less commonly via direct inoculation of the skin, resulting in primary cutaneous disease. With respect to systemic disease, infection occurs through inhalation of conidia from moist soil containing organic debris, with an incubation period of 4 to 6 weeks. In the lungs, in a process largely dependent on host cell-mediated immunity, the mold quickly converts to yeast and may then either multiply or be phagocytized.2,6,7 Transmission does not occur from person to person.7 Asymptomatic infection may occur in at least 50% of patients, often leading to a delay in diagnosis. Symptomatic pulmonary disease may range from mild flulike symptoms to overt pneumonia, clinically indistinguishable from community-acquired bacterial pneumonia, tuberculosis, other fungal infections, and cancer. Of patients with primary pulmonary disease, 25% to 80% have been reported to develop secondary organ involvement via lymphohematogenous spread most commonly to the skin, followed respectively by the skeletal, genitourinary, and central nervous systems. Currently, there are 54 documented cases of secondary disseminated cutaneous blastomycosis in children reported in the literature.3,8-14
Presentation
Primary cutaneous disease resulting from direct cutaneous inoculation is rare, especially among children.14 Of 28 cases of isolated cutaneous blastomycosis reported in the literature, 12 (42%) were pediatric.3,8-21 Inoculation blastomycosis typically presents as a papule that expands to a well-demarcated verrucous plaque, often up to several centimeters in diameter, and is located on the skin at the site of contact. The lesion may exhibit a myriad of features ranging from pustules or nodules to focal ulcerations, either present centrally or within raised borders that ultimately may communicate via sinus tracking.7 Lesions that are purely pustular in morphology also have been reported. Healing typically begins centrally and expands centrifugally, often with cribriform scarring.2,4,22 Histologic features of primary and secondary blastomycosis include pseudoepitheliomatous hyperplasia, intraepidermal microabscesses, and dermal suppurative granulomatous inflammation.4 Classically, broad-based budding yeast are identified with a doubly refractile cell wall that is best visualized on periodic acid–Schiff staining.2
Diagnosis
In approximately 50% of patients with cutaneous blastomycosis resulting from secondary spread, there may be an absence of clinically active pulmonary disease, posing a diagnostic dilemma when differentiating from primary cutaneous disease.1,2,4 Furthermore, the skin findings exhibited in primary and secondary cutaneous blastomycosis cannot be distinguished by clinical inspection.19 To fulfill the criteria for diagnosis of primary cutaneous blastomycosis, there must be an identifiable source of infection from the environment, a lesion at the site of contact, a proven absence of systemic infection, and visualization and/or isolation of fungus from the lesion.4,12 The incubation period of lesions is shorter in primary cutaneous disease (2 weeks) and may aid in its differentiation from secondary disease, which typically is longer with lesions presenting 4 to 6 weeks following initial exposure.4
Treatment
Under the current 2015 guidelines from the American Academy of Pediatrics Committee on Infectious Diseases, 6 to 12 months of itraconazole is the treatment recommendation for mild to moderate pulmonary systemic disease without central nervous system involvement.7 Central nervous system disease and moderate to severe pulmonary and systemic disease are treated with intravenous amphotericin B followed by 12 months of oral itraconazole.1,7 Primary cutaneous disease, unlike secondary disease, may self-resolve; however, primary cutaneous disease usually is treated with 6 months of itraconazole, though successful therapy with surgical excision, radiation therapy, and incision and drainage have been reported.19
Unlike secondary cutaneous blastomycosis, primary inoculation disease may be self-limited; however, as treatment with antifungal therapy has become the standard of care, the disease’s propensity to self-resolve has not been well studied.4 Oral itraconazole for 6 to 12 months is the treatment of choice for mild to moderate cutaneous disease.1,22 Effective treatment duration may be difficult to definitively assess because of the self-limited nature of the disease. Our patient showed marked improvement after 3 months and resolution of the skin lesion following 6 months of itraconazole therapy. Our findings support the previously documented observation that systemic therapy might potentially be needed only for the time required to eliminate the clinical evidence of cutaneous disease.19 Our patient received the full 6 months of treatment according to current guidelines. Among a review of 22 cases of primary inoculation blastomycosis, the 5 patients who were treated with an azole agent alone showed disease clearance with an average treatment course of 3.2 months, ranging from 1 to 6 months.19 Further studies that assess the time to clearance with antifungal therapy and subsequent recurrence rates may be warranted.
Conclusion
Pediatric primary cutaneous blastomycosis is a rare cutaneous disease. Identifying sources of probable inoculation from the environment for this patient was unique in that the patient fell into a muddy puddle within a flowerbed. Given the patient’s atopic history, a predominance of humoral over cell-mediated immunity may have placed him at risk. He responded well to 6 months of oral itraconazole and there was no ulceration or scar formation. An increased awareness of this infection, particularly in geographic areas where its reported incidence is on the rise, could be helpful in reducing delays in diagnosis and treatment.
Acknowledgments
We thank Wenhua Liu, MD (Libertyville, Illinois), for reviewing the pathology and Pravin Muniyappa, MD (Chicago, Illinois), for referring the case.
- Chapman SW, Dismukes WE, Proia LA, et al. Clinical practice guidelines for the management of blastomycosis: 2008 update by the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:1801-1812.
- Smith JA, Riddell Jt, Kauffman CA. Cutaneous manifestations of endemic mycoses. Curr Infect Dis Rep. 2013;15:440-449.
- Fisher KR, Baselski V, Beard G, et al. Pustular blastomycosis. J Am Acad Dermatol. 2009;6:355-358.
- Mason AR, Cortes GY, Cook J, et al. Cutaneous blastomycosis: a diagnostic challenge. Int J Dermatol. 2008;47:824-830.
- Lemos LB, Baliga M, Guo M. Blastomycosis: the great pretender can also be an opportunist. initial clinical diagnosis and underlying diseases in 123 patients. Ann Diagn Pathol. 2002;6:194-203.
- Bradsher RW, Chapman SW, Pappas PG. Blastomycosis. Infect Dis Clin North Am. 2003;17:21-40, vii.
- Blastomycosis. In: Kimberlin DW, ed. Red Book: 2015 Report of the Committee on Infectious Diseases. 30th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2015:263-264.
- Brick KE, Drolet BA, Lyon VB, et al. Cutaneous and disseminated blastomycosis: a pediatric case series. Pediatr Dermatol. 2013;30:23-28.
- Fanella S, Skinner S, Trepman E, et al. Blastomycosis in children and adolescents: a 30-year experience from Manitoba. Med Mycol. 2011;49:627-632.
- Frost HM, Anderson J, Ivacic L, et al. Blastomycosis in children: an analysis of clinical, epidemiologic, and genetic features. J Pediatr Infect Dis Soc. 2017;6:49-56.
- Shukla S, Singh S, Jain M, et al. Paediatric cutaneous blastomycosis: a rare case diagnosed on FNAC. Diagn Cytopathol. 2009;37:119-121.
- Smith RJ, Boos MD, Burnham JM, et al. Atypical cutaneous blastomycosis in a child with juvenile idiopathic arthritis on infliximab. Pediatrics. 2015;136:E1386-E1389.
- Wilson JW, Cawley EP, Weidman FD, et al. Primary cutaneous North American blastomycosis. AMA Arch Derm. 1955;71:39-45.
- Zampogna JC, Hoy MJ, Ramos-Caro FA. Primary cutaneous north american blastomycosis in an immunosuppressed child. Pediatr Dermatol. 2003;20:128-130.
- Balasaraswathy P, Theerthanath. Cutaneous blastomycosis presenting as non-healing ulcer and responding to oral ketoconazole. Dermatol Online J. 2003;9:19.
- Bonifaz A, Morales D, Morales N, et al. Cutaneous blastomycosis. an imported case with good response to itraconazole. Rev Iberoam Micol. 2016;33:51-54.
- Clinton TS, Timko AL. Cutaneous blastomycosis without evidence of pulmonary involvement. Mil Med. 2003;168:651-653.
- Dhamija A, D’Souza P, Salgia P, et al. Blastomycosis presenting as solitary nodule: a rare presentation. Indian J Dermatol. 2012;57:133-135.
- Gray NA, Baddour LM. Cutaneous inoculation blastomycosis. Clin Infect Dis. 2002;34:E44-E49.
- Motswaledi HM, Monyemangene FM, Maloba BR, et al. Blastomycosis: a case report and review of the literature. Int J Dermatol. 2012;51:1090-1093.
- Rodríguez-Mena A, Mayorga J, Solís-Ledesma G, et al. Blastomycosis: report of an imported case in Mexico, with only cutaneous lesions [in Spanish]. Rev Iberoam Micol. 2010;27:210-212.
- Saccente M, Woods GL. Clinical and laboratory update on blastomycosis. Clin Microbiol Rev. 2010;23:367-381.
Blastomycosis is a polymorphic disease caused by the thermally dimorphic fungus Blastomyces dermatitidis, which is naturally occurring worldwide but particularly prominent in the Great Lakes, Mississippi, and Ohio River areas of the United States. The disease was first described by Thomas Caspar Gilchrist in 1894 and historically has been referred to as Gilchrist disease, North American blastomycosis, or Chicago disease.1,2 Cutaneous blastomycosis can occur by dissemination of yeast to the skin from systemic and pulmonary disease or rarely via direct inoculation of the skin resulting in primary cutaneous disease. Clinically, the lesions are polymorphic and may appear as well-demarcated verrucous plaques containing foci of pustules or ulcerations. Lesions typically heal centrifugally with a cribriform scar.3
We describe an adolescent with a unique history of inoculation 2 weeks prior to the development of a biopsy-confirmed lesion of cutaneous blastomycosis on the left chest wall that clinically resolved following 6 months of itraconazole.
Case Report
A 16-year-old adolescent boy with a history of morbid obesity, asthma, and seasonal allergies presented for evaluation of a painful, slowly enlarging skin lesion on the left chest wall of 2 months’ duration. According to the patient, a “small pimple” appeared at the site of impact 2 weeks following a fall into a muddy flowerbed in Madison, Wisconsin. The patient recalled that although he had soiled his clothing, there was no identifiable puncture of the skin. Despite daily application of hydrogen peroxide and a 1-week course of trimethoprim-sulfamethoxazole, the lesion gradually enlarged. Complete review of systems as well as exposure and travel history were otherwise negative.
Physical examination revealed a 5.0×2.5-cm exophytic, firm, well-circumscribed plaque with a papillated crusted surface on the left side of the chest near the posterior axillary line (Figure 1). There was no palpable regional lymphadenopathy. Pulmonary examination was unremarkable. Diagnostic workup, including complete blood cell count with differential, hemoglobin A1c, human immunodeficiency virus antibody/antigen testing, interferon-gamma release assay, and chest radiograph were all within normal limits.
Histologic examination of a biopsy specimen showed pseudoepitheliomatous hyperplasia of the epidermis with a brisk mixed inflammatory infiltrate (Figure 2). Displayed in Figure 3 is the Grocott-Gomori methenamine-silver stain that highlighted the thick double-contoured wall-budding yeasts.
The patient was diagnosed with primary cutaneous blastomycosis. Treatment was initiated with itraconazole 200 mg 3 times daily for 3 days, followed by 200 mg 2 times daily for 6 months. Following 3 months of therapy, the lesion had markedly improved with violaceous dyschromia and no residual surface changes. After 5 months of itraconazole, the patient stopped taking the medication for 2 months due to pharmacy issues and then resumed. After 6 total months of therapy, the lesion healed with only residual dyschromia and itraconazole was discontinued.
Comment
Epidemiology
Blastomycosis is a polymorphic pyogranulomatous disease caused by the dimorphic fungus B dermatitidis, naturally occurring in the soil with a worldwide distribution.4 Individuals affected by the disease often reside in locations where the fungus is endemic, specifically in areas that border the Mississippi and Ohio rivers, the Great Lakes, and Canadian provinces near the Saint Lawrence Seaway. More recently there has been an increased incidence of blastomycosis, with the highest proportion found in Wisconsin and Michigan.1,2 Exposures often are associated with recreational and occupational activities near streams or rivers where there may be decaying vegetation.1 Despite the ubiquitous presence of B dermatitidis in regions where the species is endemic, it is likely that many individuals who are exposed to the organism do not develop infection.
Pathogenesis
The exact pathogenesis for the development of disease in a particular individual remains unclear. Immunosuppression is not a prerequisite for susceptibility, as evidenced by a review of 123 cases of blastomycosis in which a preceding immunodepressive disorder was present in only 25% of patients. The same study found that it was almost equally common as diabetes mellitus and present in 22% of patients.5 The organism is considered a true pathogen given its ability to affect healthy individuals and the presence of a newly identified novel 120-kD glycoprotein antigen (WI-1) on the cell wall that may confer virulence via extracellular matrix and macrophage binding. Intact cell-mediated immunity that prevents the conversion of conidia (the infectious agent) to yeast (the form that exists at body temperature) plays a key role in conferring natural resistance.6,7
Cutaneous infection may occur by either dissemination of yeast to the skin from systemic disease or less commonly via direct inoculation of the skin, resulting in primary cutaneous disease. With respect to systemic disease, infection occurs through inhalation of conidia from moist soil containing organic debris, with an incubation period of 4 to 6 weeks. In the lungs, in a process largely dependent on host cell-mediated immunity, the mold quickly converts to yeast and may then either multiply or be phagocytized.2,6,7 Transmission does not occur from person to person.7 Asymptomatic infection may occur in at least 50% of patients, often leading to a delay in diagnosis. Symptomatic pulmonary disease may range from mild flulike symptoms to overt pneumonia, clinically indistinguishable from community-acquired bacterial pneumonia, tuberculosis, other fungal infections, and cancer. Of patients with primary pulmonary disease, 25% to 80% have been reported to develop secondary organ involvement via lymphohematogenous spread most commonly to the skin, followed respectively by the skeletal, genitourinary, and central nervous systems. Currently, there are 54 documented cases of secondary disseminated cutaneous blastomycosis in children reported in the literature.3,8-14
Presentation
Primary cutaneous disease resulting from direct cutaneous inoculation is rare, especially among children.14 Of 28 cases of isolated cutaneous blastomycosis reported in the literature, 12 (42%) were pediatric.3,8-21 Inoculation blastomycosis typically presents as a papule that expands to a well-demarcated verrucous plaque, often up to several centimeters in diameter, and is located on the skin at the site of contact. The lesion may exhibit a myriad of features ranging from pustules or nodules to focal ulcerations, either present centrally or within raised borders that ultimately may communicate via sinus tracking.7 Lesions that are purely pustular in morphology also have been reported. Healing typically begins centrally and expands centrifugally, often with cribriform scarring.2,4,22 Histologic features of primary and secondary blastomycosis include pseudoepitheliomatous hyperplasia, intraepidermal microabscesses, and dermal suppurative granulomatous inflammation.4 Classically, broad-based budding yeast are identified with a doubly refractile cell wall that is best visualized on periodic acid–Schiff staining.2
Diagnosis
In approximately 50% of patients with cutaneous blastomycosis resulting from secondary spread, there may be an absence of clinically active pulmonary disease, posing a diagnostic dilemma when differentiating from primary cutaneous disease.1,2,4 Furthermore, the skin findings exhibited in primary and secondary cutaneous blastomycosis cannot be distinguished by clinical inspection.19 To fulfill the criteria for diagnosis of primary cutaneous blastomycosis, there must be an identifiable source of infection from the environment, a lesion at the site of contact, a proven absence of systemic infection, and visualization and/or isolation of fungus from the lesion.4,12 The incubation period of lesions is shorter in primary cutaneous disease (2 weeks) and may aid in its differentiation from secondary disease, which typically is longer with lesions presenting 4 to 6 weeks following initial exposure.4
Treatment
Under the current 2015 guidelines from the American Academy of Pediatrics Committee on Infectious Diseases, 6 to 12 months of itraconazole is the treatment recommendation for mild to moderate pulmonary systemic disease without central nervous system involvement.7 Central nervous system disease and moderate to severe pulmonary and systemic disease are treated with intravenous amphotericin B followed by 12 months of oral itraconazole.1,7 Primary cutaneous disease, unlike secondary disease, may self-resolve; however, primary cutaneous disease usually is treated with 6 months of itraconazole, though successful therapy with surgical excision, radiation therapy, and incision and drainage have been reported.19
Unlike secondary cutaneous blastomycosis, primary inoculation disease may be self-limited; however, as treatment with antifungal therapy has become the standard of care, the disease’s propensity to self-resolve has not been well studied.4 Oral itraconazole for 6 to 12 months is the treatment of choice for mild to moderate cutaneous disease.1,22 Effective treatment duration may be difficult to definitively assess because of the self-limited nature of the disease. Our patient showed marked improvement after 3 months and resolution of the skin lesion following 6 months of itraconazole therapy. Our findings support the previously documented observation that systemic therapy might potentially be needed only for the time required to eliminate the clinical evidence of cutaneous disease.19 Our patient received the full 6 months of treatment according to current guidelines. Among a review of 22 cases of primary inoculation blastomycosis, the 5 patients who were treated with an azole agent alone showed disease clearance with an average treatment course of 3.2 months, ranging from 1 to 6 months.19 Further studies that assess the time to clearance with antifungal therapy and subsequent recurrence rates may be warranted.
Conclusion
Pediatric primary cutaneous blastomycosis is a rare cutaneous disease. Identifying sources of probable inoculation from the environment for this patient was unique in that the patient fell into a muddy puddle within a flowerbed. Given the patient’s atopic history, a predominance of humoral over cell-mediated immunity may have placed him at risk. He responded well to 6 months of oral itraconazole and there was no ulceration or scar formation. An increased awareness of this infection, particularly in geographic areas where its reported incidence is on the rise, could be helpful in reducing delays in diagnosis and treatment.
Acknowledgments
We thank Wenhua Liu, MD (Libertyville, Illinois), for reviewing the pathology and Pravin Muniyappa, MD (Chicago, Illinois), for referring the case.
Blastomycosis is a polymorphic disease caused by the thermally dimorphic fungus Blastomyces dermatitidis, which is naturally occurring worldwide but particularly prominent in the Great Lakes, Mississippi, and Ohio River areas of the United States. The disease was first described by Thomas Caspar Gilchrist in 1894 and historically has been referred to as Gilchrist disease, North American blastomycosis, or Chicago disease.1,2 Cutaneous blastomycosis can occur by dissemination of yeast to the skin from systemic and pulmonary disease or rarely via direct inoculation of the skin resulting in primary cutaneous disease. Clinically, the lesions are polymorphic and may appear as well-demarcated verrucous plaques containing foci of pustules or ulcerations. Lesions typically heal centrifugally with a cribriform scar.3
We describe an adolescent with a unique history of inoculation 2 weeks prior to the development of a biopsy-confirmed lesion of cutaneous blastomycosis on the left chest wall that clinically resolved following 6 months of itraconazole.
Case Report
A 16-year-old adolescent boy with a history of morbid obesity, asthma, and seasonal allergies presented for evaluation of a painful, slowly enlarging skin lesion on the left chest wall of 2 months’ duration. According to the patient, a “small pimple” appeared at the site of impact 2 weeks following a fall into a muddy flowerbed in Madison, Wisconsin. The patient recalled that although he had soiled his clothing, there was no identifiable puncture of the skin. Despite daily application of hydrogen peroxide and a 1-week course of trimethoprim-sulfamethoxazole, the lesion gradually enlarged. Complete review of systems as well as exposure and travel history were otherwise negative.
Physical examination revealed a 5.0×2.5-cm exophytic, firm, well-circumscribed plaque with a papillated crusted surface on the left side of the chest near the posterior axillary line (Figure 1). There was no palpable regional lymphadenopathy. Pulmonary examination was unremarkable. Diagnostic workup, including complete blood cell count with differential, hemoglobin A1c, human immunodeficiency virus antibody/antigen testing, interferon-gamma release assay, and chest radiograph were all within normal limits.
Histologic examination of a biopsy specimen showed pseudoepitheliomatous hyperplasia of the epidermis with a brisk mixed inflammatory infiltrate (Figure 2). Displayed in Figure 3 is the Grocott-Gomori methenamine-silver stain that highlighted the thick double-contoured wall-budding yeasts.
The patient was diagnosed with primary cutaneous blastomycosis. Treatment was initiated with itraconazole 200 mg 3 times daily for 3 days, followed by 200 mg 2 times daily for 6 months. Following 3 months of therapy, the lesion had markedly improved with violaceous dyschromia and no residual surface changes. After 5 months of itraconazole, the patient stopped taking the medication for 2 months due to pharmacy issues and then resumed. After 6 total months of therapy, the lesion healed with only residual dyschromia and itraconazole was discontinued.
Comment
Epidemiology
Blastomycosis is a polymorphic pyogranulomatous disease caused by the dimorphic fungus B dermatitidis, naturally occurring in the soil with a worldwide distribution.4 Individuals affected by the disease often reside in locations where the fungus is endemic, specifically in areas that border the Mississippi and Ohio rivers, the Great Lakes, and Canadian provinces near the Saint Lawrence Seaway. More recently there has been an increased incidence of blastomycosis, with the highest proportion found in Wisconsin and Michigan.1,2 Exposures often are associated with recreational and occupational activities near streams or rivers where there may be decaying vegetation.1 Despite the ubiquitous presence of B dermatitidis in regions where the species is endemic, it is likely that many individuals who are exposed to the organism do not develop infection.
Pathogenesis
The exact pathogenesis for the development of disease in a particular individual remains unclear. Immunosuppression is not a prerequisite for susceptibility, as evidenced by a review of 123 cases of blastomycosis in which a preceding immunodepressive disorder was present in only 25% of patients. The same study found that it was almost equally common as diabetes mellitus and present in 22% of patients.5 The organism is considered a true pathogen given its ability to affect healthy individuals and the presence of a newly identified novel 120-kD glycoprotein antigen (WI-1) on the cell wall that may confer virulence via extracellular matrix and macrophage binding. Intact cell-mediated immunity that prevents the conversion of conidia (the infectious agent) to yeast (the form that exists at body temperature) plays a key role in conferring natural resistance.6,7
Cutaneous infection may occur by either dissemination of yeast to the skin from systemic disease or less commonly via direct inoculation of the skin, resulting in primary cutaneous disease. With respect to systemic disease, infection occurs through inhalation of conidia from moist soil containing organic debris, with an incubation period of 4 to 6 weeks. In the lungs, in a process largely dependent on host cell-mediated immunity, the mold quickly converts to yeast and may then either multiply or be phagocytized.2,6,7 Transmission does not occur from person to person.7 Asymptomatic infection may occur in at least 50% of patients, often leading to a delay in diagnosis. Symptomatic pulmonary disease may range from mild flulike symptoms to overt pneumonia, clinically indistinguishable from community-acquired bacterial pneumonia, tuberculosis, other fungal infections, and cancer. Of patients with primary pulmonary disease, 25% to 80% have been reported to develop secondary organ involvement via lymphohematogenous spread most commonly to the skin, followed respectively by the skeletal, genitourinary, and central nervous systems. Currently, there are 54 documented cases of secondary disseminated cutaneous blastomycosis in children reported in the literature.3,8-14
Presentation
Primary cutaneous disease resulting from direct cutaneous inoculation is rare, especially among children.14 Of 28 cases of isolated cutaneous blastomycosis reported in the literature, 12 (42%) were pediatric.3,8-21 Inoculation blastomycosis typically presents as a papule that expands to a well-demarcated verrucous plaque, often up to several centimeters in diameter, and is located on the skin at the site of contact. The lesion may exhibit a myriad of features ranging from pustules or nodules to focal ulcerations, either present centrally or within raised borders that ultimately may communicate via sinus tracking.7 Lesions that are purely pustular in morphology also have been reported. Healing typically begins centrally and expands centrifugally, often with cribriform scarring.2,4,22 Histologic features of primary and secondary blastomycosis include pseudoepitheliomatous hyperplasia, intraepidermal microabscesses, and dermal suppurative granulomatous inflammation.4 Classically, broad-based budding yeast are identified with a doubly refractile cell wall that is best visualized on periodic acid–Schiff staining.2
Diagnosis
In approximately 50% of patients with cutaneous blastomycosis resulting from secondary spread, there may be an absence of clinically active pulmonary disease, posing a diagnostic dilemma when differentiating from primary cutaneous disease.1,2,4 Furthermore, the skin findings exhibited in primary and secondary cutaneous blastomycosis cannot be distinguished by clinical inspection.19 To fulfill the criteria for diagnosis of primary cutaneous blastomycosis, there must be an identifiable source of infection from the environment, a lesion at the site of contact, a proven absence of systemic infection, and visualization and/or isolation of fungus from the lesion.4,12 The incubation period of lesions is shorter in primary cutaneous disease (2 weeks) and may aid in its differentiation from secondary disease, which typically is longer with lesions presenting 4 to 6 weeks following initial exposure.4
Treatment
Under the current 2015 guidelines from the American Academy of Pediatrics Committee on Infectious Diseases, 6 to 12 months of itraconazole is the treatment recommendation for mild to moderate pulmonary systemic disease without central nervous system involvement.7 Central nervous system disease and moderate to severe pulmonary and systemic disease are treated with intravenous amphotericin B followed by 12 months of oral itraconazole.1,7 Primary cutaneous disease, unlike secondary disease, may self-resolve; however, primary cutaneous disease usually is treated with 6 months of itraconazole, though successful therapy with surgical excision, radiation therapy, and incision and drainage have been reported.19
Unlike secondary cutaneous blastomycosis, primary inoculation disease may be self-limited; however, as treatment with antifungal therapy has become the standard of care, the disease’s propensity to self-resolve has not been well studied.4 Oral itraconazole for 6 to 12 months is the treatment of choice for mild to moderate cutaneous disease.1,22 Effective treatment duration may be difficult to definitively assess because of the self-limited nature of the disease. Our patient showed marked improvement after 3 months and resolution of the skin lesion following 6 months of itraconazole therapy. Our findings support the previously documented observation that systemic therapy might potentially be needed only for the time required to eliminate the clinical evidence of cutaneous disease.19 Our patient received the full 6 months of treatment according to current guidelines. Among a review of 22 cases of primary inoculation blastomycosis, the 5 patients who were treated with an azole agent alone showed disease clearance with an average treatment course of 3.2 months, ranging from 1 to 6 months.19 Further studies that assess the time to clearance with antifungal therapy and subsequent recurrence rates may be warranted.
Conclusion
Pediatric primary cutaneous blastomycosis is a rare cutaneous disease. Identifying sources of probable inoculation from the environment for this patient was unique in that the patient fell into a muddy puddle within a flowerbed. Given the patient’s atopic history, a predominance of humoral over cell-mediated immunity may have placed him at risk. He responded well to 6 months of oral itraconazole and there was no ulceration or scar formation. An increased awareness of this infection, particularly in geographic areas where its reported incidence is on the rise, could be helpful in reducing delays in diagnosis and treatment.
Acknowledgments
We thank Wenhua Liu, MD (Libertyville, Illinois), for reviewing the pathology and Pravin Muniyappa, MD (Chicago, Illinois), for referring the case.
- Chapman SW, Dismukes WE, Proia LA, et al. Clinical practice guidelines for the management of blastomycosis: 2008 update by the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:1801-1812.
- Smith JA, Riddell Jt, Kauffman CA. Cutaneous manifestations of endemic mycoses. Curr Infect Dis Rep. 2013;15:440-449.
- Fisher KR, Baselski V, Beard G, et al. Pustular blastomycosis. J Am Acad Dermatol. 2009;6:355-358.
- Mason AR, Cortes GY, Cook J, et al. Cutaneous blastomycosis: a diagnostic challenge. Int J Dermatol. 2008;47:824-830.
- Lemos LB, Baliga M, Guo M. Blastomycosis: the great pretender can also be an opportunist. initial clinical diagnosis and underlying diseases in 123 patients. Ann Diagn Pathol. 2002;6:194-203.
- Bradsher RW, Chapman SW, Pappas PG. Blastomycosis. Infect Dis Clin North Am. 2003;17:21-40, vii.
- Blastomycosis. In: Kimberlin DW, ed. Red Book: 2015 Report of the Committee on Infectious Diseases. 30th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2015:263-264.
- Brick KE, Drolet BA, Lyon VB, et al. Cutaneous and disseminated blastomycosis: a pediatric case series. Pediatr Dermatol. 2013;30:23-28.
- Fanella S, Skinner S, Trepman E, et al. Blastomycosis in children and adolescents: a 30-year experience from Manitoba. Med Mycol. 2011;49:627-632.
- Frost HM, Anderson J, Ivacic L, et al. Blastomycosis in children: an analysis of clinical, epidemiologic, and genetic features. J Pediatr Infect Dis Soc. 2017;6:49-56.
- Shukla S, Singh S, Jain M, et al. Paediatric cutaneous blastomycosis: a rare case diagnosed on FNAC. Diagn Cytopathol. 2009;37:119-121.
- Smith RJ, Boos MD, Burnham JM, et al. Atypical cutaneous blastomycosis in a child with juvenile idiopathic arthritis on infliximab. Pediatrics. 2015;136:E1386-E1389.
- Wilson JW, Cawley EP, Weidman FD, et al. Primary cutaneous North American blastomycosis. AMA Arch Derm. 1955;71:39-45.
- Zampogna JC, Hoy MJ, Ramos-Caro FA. Primary cutaneous north american blastomycosis in an immunosuppressed child. Pediatr Dermatol. 2003;20:128-130.
- Balasaraswathy P, Theerthanath. Cutaneous blastomycosis presenting as non-healing ulcer and responding to oral ketoconazole. Dermatol Online J. 2003;9:19.
- Bonifaz A, Morales D, Morales N, et al. Cutaneous blastomycosis. an imported case with good response to itraconazole. Rev Iberoam Micol. 2016;33:51-54.
- Clinton TS, Timko AL. Cutaneous blastomycosis without evidence of pulmonary involvement. Mil Med. 2003;168:651-653.
- Dhamija A, D’Souza P, Salgia P, et al. Blastomycosis presenting as solitary nodule: a rare presentation. Indian J Dermatol. 2012;57:133-135.
- Gray NA, Baddour LM. Cutaneous inoculation blastomycosis. Clin Infect Dis. 2002;34:E44-E49.
- Motswaledi HM, Monyemangene FM, Maloba BR, et al. Blastomycosis: a case report and review of the literature. Int J Dermatol. 2012;51:1090-1093.
- Rodríguez-Mena A, Mayorga J, Solís-Ledesma G, et al. Blastomycosis: report of an imported case in Mexico, with only cutaneous lesions [in Spanish]. Rev Iberoam Micol. 2010;27:210-212.
- Saccente M, Woods GL. Clinical and laboratory update on blastomycosis. Clin Microbiol Rev. 2010;23:367-381.
- Chapman SW, Dismukes WE, Proia LA, et al. Clinical practice guidelines for the management of blastomycosis: 2008 update by the Infectious Diseases Society of America. Clin Infect Dis. 2008;46:1801-1812.
- Smith JA, Riddell Jt, Kauffman CA. Cutaneous manifestations of endemic mycoses. Curr Infect Dis Rep. 2013;15:440-449.
- Fisher KR, Baselski V, Beard G, et al. Pustular blastomycosis. J Am Acad Dermatol. 2009;6:355-358.
- Mason AR, Cortes GY, Cook J, et al. Cutaneous blastomycosis: a diagnostic challenge. Int J Dermatol. 2008;47:824-830.
- Lemos LB, Baliga M, Guo M. Blastomycosis: the great pretender can also be an opportunist. initial clinical diagnosis and underlying diseases in 123 patients. Ann Diagn Pathol. 2002;6:194-203.
- Bradsher RW, Chapman SW, Pappas PG. Blastomycosis. Infect Dis Clin North Am. 2003;17:21-40, vii.
- Blastomycosis. In: Kimberlin DW, ed. Red Book: 2015 Report of the Committee on Infectious Diseases. 30th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2015:263-264.
- Brick KE, Drolet BA, Lyon VB, et al. Cutaneous and disseminated blastomycosis: a pediatric case series. Pediatr Dermatol. 2013;30:23-28.
- Fanella S, Skinner S, Trepman E, et al. Blastomycosis in children and adolescents: a 30-year experience from Manitoba. Med Mycol. 2011;49:627-632.
- Frost HM, Anderson J, Ivacic L, et al. Blastomycosis in children: an analysis of clinical, epidemiologic, and genetic features. J Pediatr Infect Dis Soc. 2017;6:49-56.
- Shukla S, Singh S, Jain M, et al. Paediatric cutaneous blastomycosis: a rare case diagnosed on FNAC. Diagn Cytopathol. 2009;37:119-121.
- Smith RJ, Boos MD, Burnham JM, et al. Atypical cutaneous blastomycosis in a child with juvenile idiopathic arthritis on infliximab. Pediatrics. 2015;136:E1386-E1389.
- Wilson JW, Cawley EP, Weidman FD, et al. Primary cutaneous North American blastomycosis. AMA Arch Derm. 1955;71:39-45.
- Zampogna JC, Hoy MJ, Ramos-Caro FA. Primary cutaneous north american blastomycosis in an immunosuppressed child. Pediatr Dermatol. 2003;20:128-130.
- Balasaraswathy P, Theerthanath. Cutaneous blastomycosis presenting as non-healing ulcer and responding to oral ketoconazole. Dermatol Online J. 2003;9:19.
- Bonifaz A, Morales D, Morales N, et al. Cutaneous blastomycosis. an imported case with good response to itraconazole. Rev Iberoam Micol. 2016;33:51-54.
- Clinton TS, Timko AL. Cutaneous blastomycosis without evidence of pulmonary involvement. Mil Med. 2003;168:651-653.
- Dhamija A, D’Souza P, Salgia P, et al. Blastomycosis presenting as solitary nodule: a rare presentation. Indian J Dermatol. 2012;57:133-135.
- Gray NA, Baddour LM. Cutaneous inoculation blastomycosis. Clin Infect Dis. 2002;34:E44-E49.
- Motswaledi HM, Monyemangene FM, Maloba BR, et al. Blastomycosis: a case report and review of the literature. Int J Dermatol. 2012;51:1090-1093.
- Rodríguez-Mena A, Mayorga J, Solís-Ledesma G, et al. Blastomycosis: report of an imported case in Mexico, with only cutaneous lesions [in Spanish]. Rev Iberoam Micol. 2010;27:210-212.
- Saccente M, Woods GL. Clinical and laboratory update on blastomycosis. Clin Microbiol Rev. 2010;23:367-381.
Practice Points
- Cutaneous blastomycosis can occur by dissemination of yeast to the skin from systemic and pulmonary disease or rarely via direct inoculation of the skin, resulting in primary cutaneous disease.
- Exposures often are associated with recreational and occupational activities near streams or rivers where there may be decaying vegetation.
- Oral itraconazole for 6 to 12 months is the treatment of choice for mild to moderate cutaneous disease.
- Increased awareness of this rare infection, particularly in geographic areas where its reported incidence is on the rise, could be helpful in reducing delays in diagnosis and treatment.
Acute Hemorrhagic Edema of Infancy: Guide to Prevent Misdiagnosis
Acute hemorrhagic edema of infancy (AHEI) is an uncommon leukocytoclastic vasculitis affecting children aged 6 to 24 months; Henoch-Schönlein purpura (HSP) is the most common misdiagnosis. The 2 entities should be differentiated, as HSP may have renal and gastrointestinal (GI) comorbidities that need serial follow-up, whereas AHEI follows a benign course without systemic sequelae. Patient history and physical examination are the most important factors in differentiating the 2 diseases; histopathologic and direct immunofluorescence (DIF) analyses may lend further diagnostic confidence.
We report the case of a 10-month-old previously healthy boy who presented with acute rash, edema, and low-grade fever in the setting of recent diarrhea. We differentiate between AHEI and HSP to help prevent misdiagnosis by health care providers.
Case Report
A 10-month-old previously healthy boy presented to the emergency department (ED) for evaluation of a rash and swelling of 4 days’ duration. He had nonbloody diarrhea 1 week prior; soon after, he developed bilateral lower leg edema and rash. On evaluation in a different ED, he had a low-grade fever (rectal temperature, 38.0°C) but normal blood work, including complete blood cell count, basic metabolic panel, and coagulation studies. The patient was discharged to outpatient follow-up with his pediatrician who reported normal urinalysis.
Due to progression of the rash, the patient presented to our ED 3 days after his initial ED assessment. Dermatology was consulted. At the time of presentation, he was afebrile but with GI upset and fussiness. His parents denied additional symptoms or blood in urine or stool. Physical examination revealed a nontoxic-appearing infant with scattered palpable, annular, purpuric papules coalescing into plaques on both legs and feet (Figure 1), with sparse petechiae noted on the lower abdomen. The cheeks had scattered purpuric papules and plaques bilaterally, a few with a small central crust (Figure 2), and the right superior helix had a faint purpuric macule. The hands had a few pink edematous coalescing papules.
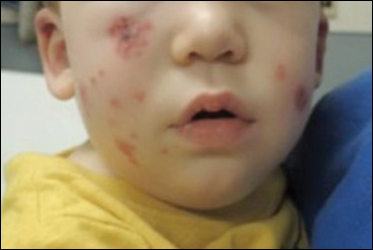
Histopathologic analyses with hematoxylin and eosin staining (Figure 3) and DIF (Figure 4) were performed from within a representative purpuric plaque on the right hip. Direct immunofluorescence was performed to evaluate for an IgA vasculitis versus an alternative type of vasculitis. The hematoxylin and eosin–stained specimen demonstrated a dermal perivascular infiltrate involving superficial and deep vessels with neutrophils, karyorrhexis, and erythrocyte extravasation. The endothelium was intact, with a mild suggestion of fibrinoid change of the blood vessel walls. Direct immunofluorescence revealed granular deposition of IgA, C3, and fibrinogen in multiple dermal blood vessels. Combined, the specimens were interpreted as evolving IgA-associated leukocytoclastic vasculitis.
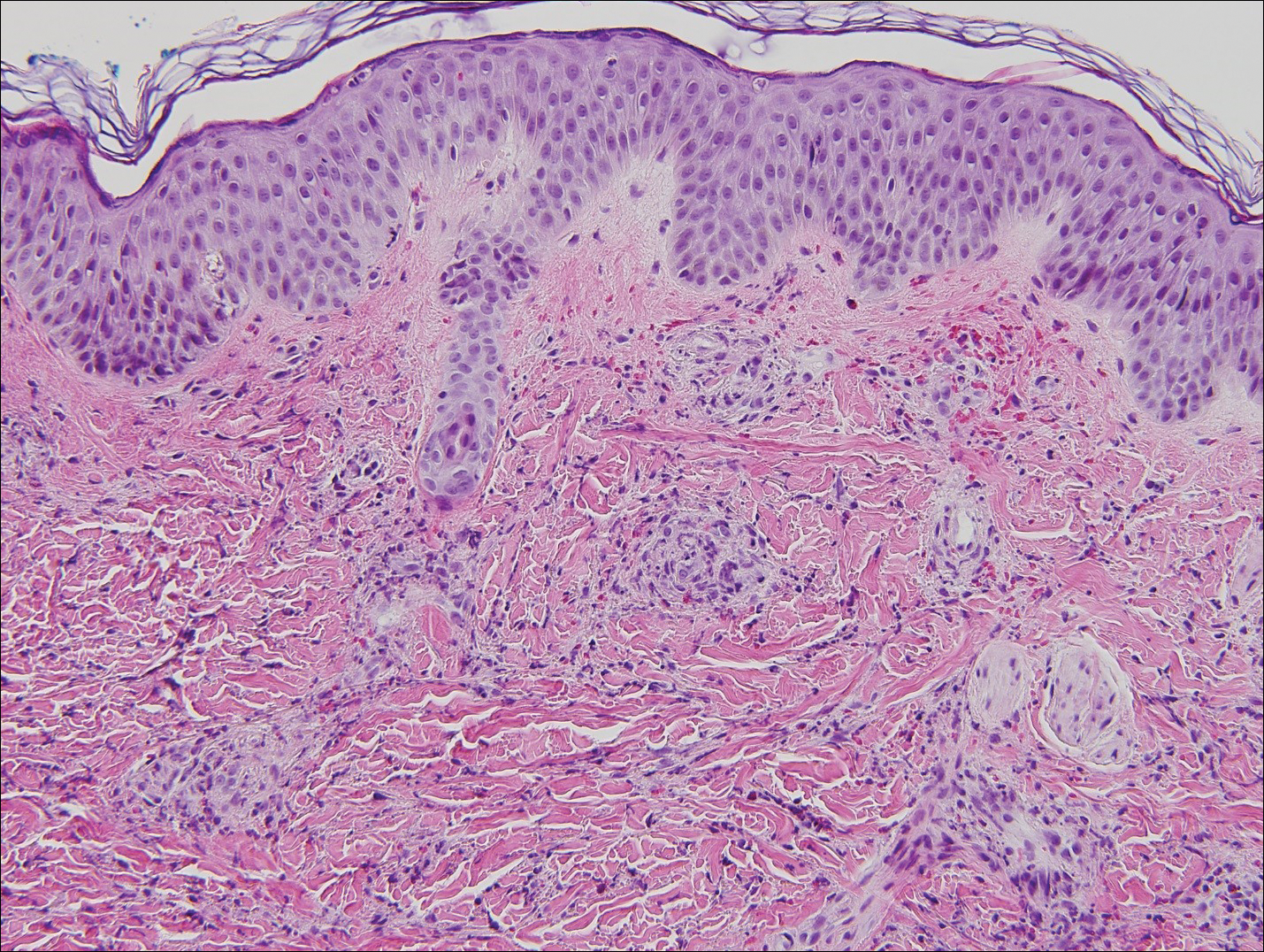
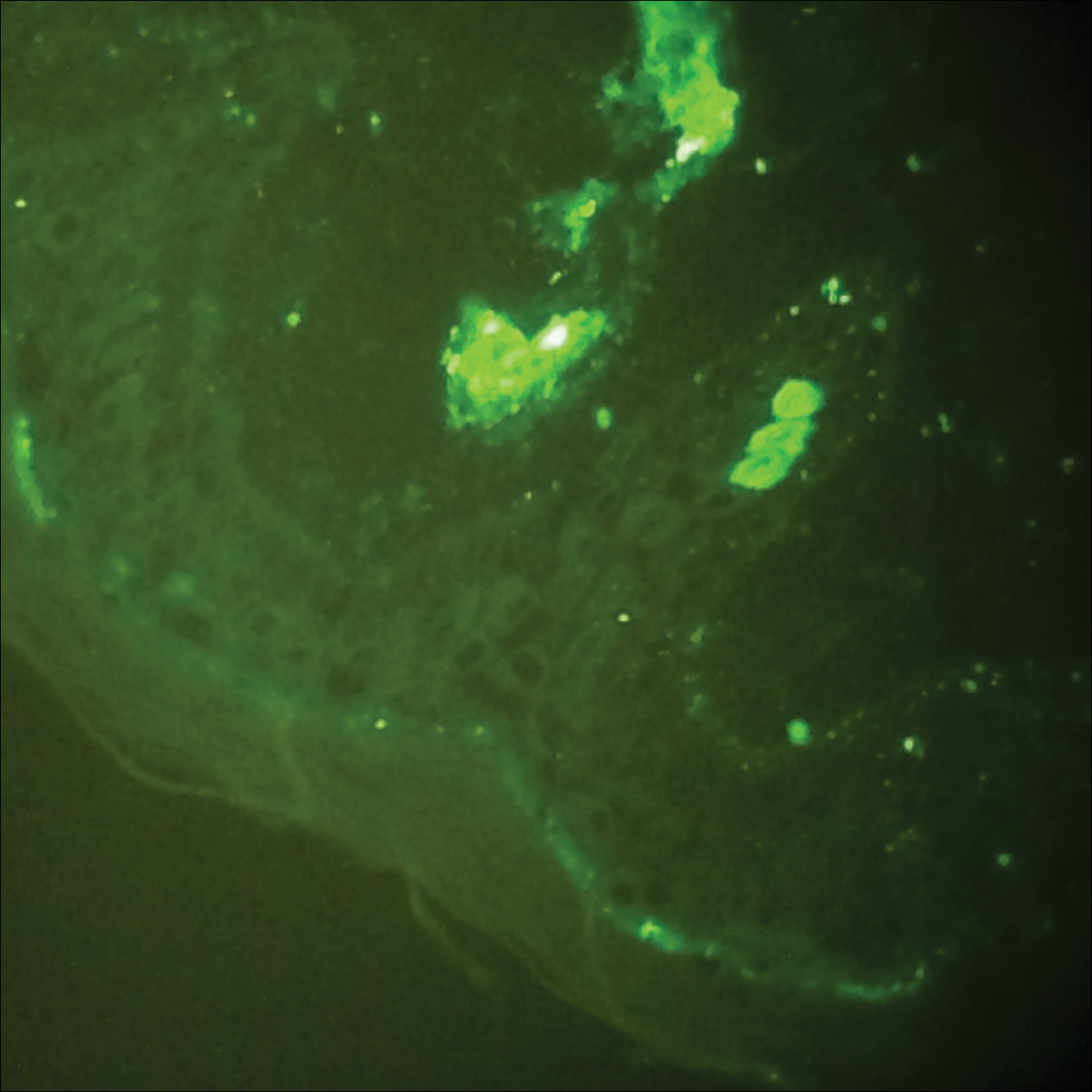
The case was reviewed with our 2 department pediatric dermatologists; a diagnosis of AHEI was made based on the clinical and supportive histopathological presentations. The patient’s parents chose active treatment with a 2-week taper of oral prednisone because of the patient’s discomfort with edema. No GI or adverse renal sequelae, including findings on urinalysis, were reported at 1-month hospital follow-up with dermatology and pediatrics.
Comment
Incidence and Clinical Characteristics
Acute hemorrhagic edema of infancy is an uncommon leukocytoclastic vasculitis first described in the United States by Snow1 in 1913. Other names for the disorder include acute hemorrhagic edema of young children, cockade purpura and edema, Finkelstein disease, and Seidlmayer disease.2 Boys are affected more often than girls, with most children presenting at 6 to 24 months of age. Most affected children experience a prodrome of simple respiratory tract illness (most common), diarrhea (as in our case), or urinary tract infection.2 The exact pathophysiology behind AHEI is unknown, but it is thought to be an immune complex–mediated disease evidenced by the fact that infection, use of medication, or immunization precedes most cases.3,4
Diagnosis
Acute hemorrhagic edema of infancy is diagnosed clinically, with or without the support of skin biopsy. It should be differentiated from HSP because of renal and GI sequelae that HSP portends compared to the benign course of AHEI.2 Notably, some health care providers consider AHEI a benign variant of HSP.2,3
Characteristically, AHEI patients are nontoxic-appearing infants with a low-grade fever who develop relatively large (1–5 cm) targetoid purpuric lesions and indurated nonpitting edema of the extremities.2,5 Purpura in AHEI frequently occurs on the face, ears, and upper and lower extremities, whereas purpura in HSP most commonly presents on the buttocks and extensor legs with sparing of the face. Henoch-Schönlein purpura most often affects children aged 3 to 6 years compared to AHEI’s younger demographic (age <2 years).4,5 Clinically, HSP presents with palpable purpura and 1 or more of the following features: diffuse abdominal pain, arthritis/arthralgia, renal involvement, and skin or renal biopsy showing predominant IgA deposition.2,6
Both AHEI and HSP show leukocytoclastic vasculitis on histopathology.2-4,6,7 Positive perivascular IgA staining on DIF is strongly associated with HSP, but nearly one-quarter of AHEI cases also show this deposition pattern2,4,7; therefore, DIF alone cannot exclude a diagnosis of AHEI.
Differential Diagnosis
Alternative diagnoses to consider with AHEI include drug-induced vasculitis, erythema multiforme, HSP, Kawasaki disease, meningococcemia, nonaccidental skin bruising, Rocky Mountain spotted fever, septic vasculitis, and urticarial vasculitis (Table).2-4,6-8

Treatment
Acute hemorrhagic edema of infancy is self-limited, with only rare reports of extracutaneous involvement. Supportive treatment is indicated because spontaneous recovery without sequelae is expected within 21 days.2,3,6 If edema is symptomatic, as was the case with our patient, corticosteroids may shorten the disease course.3
Conclusion
Our case highlights the need to combine clinical history, physical examination, and histopathologic analysis to differentiate between AHEI and HSP, which is important for 2 reasons: (1) it helps with the decision to undertake active or observational treatment, and (2) it helps the clinician counsel the patient and guardians regarding potential associated renal and GI risks.
- Snow IM. Purpura, urticaria and angioneurotic edema of the hands and feet in a nursing baby. JAMA. 1913;61:18-19.
- Fiore E, Rizzi M, Ragazzi M, et al. Acute hemorrhagic edema of young children (cockade purpura and edema): a case series and systematic review. J Am Acad Dermatol. 2008;59:684-695.
- Freitas P, Bygum A. Visual impairment caused by periorbital edema in an infant with acute hemorrhagic edema of infancy. Pediatr Dermatol. 2013;30:e132-e135.
- Legrain V, Lejean S, Taïeb A, et al. Infantile acute hemorrhagic edema of the skin: study of ten cases. J Am Acad Dermatol. 1991;24:17-22.
- Breda L, Franchini S, Marzetti V, et al. Escherichia coli urinary infection as a cause of acute hemorrhagic edema in infancy. Pediatr Dermatol. 2015;32:e309-e311.
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941.
- Saraclar Y, Tinaztepe K, Adalioğlu G, et al. Acute hemorrhagic edema of infancy (AHEI)—a variant of Henoch-Schönlein purpura or a distinct clinical entity? J Allergy Clin Immunol. 1990;86:473-483.
- Shinkai K, Fox L. Cutaneous vasculitis. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. China: Elsevier Limited; 2012:385-410.
Acute hemorrhagic edema of infancy (AHEI) is an uncommon leukocytoclastic vasculitis affecting children aged 6 to 24 months; Henoch-Schönlein purpura (HSP) is the most common misdiagnosis. The 2 entities should be differentiated, as HSP may have renal and gastrointestinal (GI) comorbidities that need serial follow-up, whereas AHEI follows a benign course without systemic sequelae. Patient history and physical examination are the most important factors in differentiating the 2 diseases; histopathologic and direct immunofluorescence (DIF) analyses may lend further diagnostic confidence.
We report the case of a 10-month-old previously healthy boy who presented with acute rash, edema, and low-grade fever in the setting of recent diarrhea. We differentiate between AHEI and HSP to help prevent misdiagnosis by health care providers.
Case Report
A 10-month-old previously healthy boy presented to the emergency department (ED) for evaluation of a rash and swelling of 4 days’ duration. He had nonbloody diarrhea 1 week prior; soon after, he developed bilateral lower leg edema and rash. On evaluation in a different ED, he had a low-grade fever (rectal temperature, 38.0°C) but normal blood work, including complete blood cell count, basic metabolic panel, and coagulation studies. The patient was discharged to outpatient follow-up with his pediatrician who reported normal urinalysis.
Due to progression of the rash, the patient presented to our ED 3 days after his initial ED assessment. Dermatology was consulted. At the time of presentation, he was afebrile but with GI upset and fussiness. His parents denied additional symptoms or blood in urine or stool. Physical examination revealed a nontoxic-appearing infant with scattered palpable, annular, purpuric papules coalescing into plaques on both legs and feet (Figure 1), with sparse petechiae noted on the lower abdomen. The cheeks had scattered purpuric papules and plaques bilaterally, a few with a small central crust (Figure 2), and the right superior helix had a faint purpuric macule. The hands had a few pink edematous coalescing papules.
Histopathologic analyses with hematoxylin and eosin staining (Figure 3) and DIF (Figure 4) were performed from within a representative purpuric plaque on the right hip. Direct immunofluorescence was performed to evaluate for an IgA vasculitis versus an alternative type of vasculitis. The hematoxylin and eosin–stained specimen demonstrated a dermal perivascular infiltrate involving superficial and deep vessels with neutrophils, karyorrhexis, and erythrocyte extravasation. The endothelium was intact, with a mild suggestion of fibrinoid change of the blood vessel walls. Direct immunofluorescence revealed granular deposition of IgA, C3, and fibrinogen in multiple dermal blood vessels. Combined, the specimens were interpreted as evolving IgA-associated leukocytoclastic vasculitis.
The case was reviewed with our 2 department pediatric dermatologists; a diagnosis of AHEI was made based on the clinical and supportive histopathological presentations. The patient’s parents chose active treatment with a 2-week taper of oral prednisone because of the patient’s discomfort with edema. No GI or adverse renal sequelae, including findings on urinalysis, were reported at 1-month hospital follow-up with dermatology and pediatrics.
Comment
Incidence and Clinical Characteristics
Acute hemorrhagic edema of infancy is an uncommon leukocytoclastic vasculitis first described in the United States by Snow1 in 1913. Other names for the disorder include acute hemorrhagic edema of young children, cockade purpura and edema, Finkelstein disease, and Seidlmayer disease.2 Boys are affected more often than girls, with most children presenting at 6 to 24 months of age. Most affected children experience a prodrome of simple respiratory tract illness (most common), diarrhea (as in our case), or urinary tract infection.2 The exact pathophysiology behind AHEI is unknown, but it is thought to be an immune complex–mediated disease evidenced by the fact that infection, use of medication, or immunization precedes most cases.3,4
Diagnosis
Acute hemorrhagic edema of infancy is diagnosed clinically, with or without the support of skin biopsy. It should be differentiated from HSP because of renal and GI sequelae that HSP portends compared to the benign course of AHEI.2 Notably, some health care providers consider AHEI a benign variant of HSP.2,3
Characteristically, AHEI patients are nontoxic-appearing infants with a low-grade fever who develop relatively large (1–5 cm) targetoid purpuric lesions and indurated nonpitting edema of the extremities.2,5 Purpura in AHEI frequently occurs on the face, ears, and upper and lower extremities, whereas purpura in HSP most commonly presents on the buttocks and extensor legs with sparing of the face. Henoch-Schönlein purpura most often affects children aged 3 to 6 years compared to AHEI’s younger demographic (age <2 years).4,5 Clinically, HSP presents with palpable purpura and 1 or more of the following features: diffuse abdominal pain, arthritis/arthralgia, renal involvement, and skin or renal biopsy showing predominant IgA deposition.2,6
Both AHEI and HSP show leukocytoclastic vasculitis on histopathology.2-4,6,7 Positive perivascular IgA staining on DIF is strongly associated with HSP, but nearly one-quarter of AHEI cases also show this deposition pattern2,4,7; therefore, DIF alone cannot exclude a diagnosis of AHEI.
Differential Diagnosis
Alternative diagnoses to consider with AHEI include drug-induced vasculitis, erythema multiforme, HSP, Kawasaki disease, meningococcemia, nonaccidental skin bruising, Rocky Mountain spotted fever, septic vasculitis, and urticarial vasculitis (Table).2-4,6-8
Treatment
Acute hemorrhagic edema of infancy is self-limited, with only rare reports of extracutaneous involvement. Supportive treatment is indicated because spontaneous recovery without sequelae is expected within 21 days.2,3,6 If edema is symptomatic, as was the case with our patient, corticosteroids may shorten the disease course.3
Conclusion
Our case highlights the need to combine clinical history, physical examination, and histopathologic analysis to differentiate between AHEI and HSP, which is important for 2 reasons: (1) it helps with the decision to undertake active or observational treatment, and (2) it helps the clinician counsel the patient and guardians regarding potential associated renal and GI risks.
Acute hemorrhagic edema of infancy (AHEI) is an uncommon leukocytoclastic vasculitis affecting children aged 6 to 24 months; Henoch-Schönlein purpura (HSP) is the most common misdiagnosis. The 2 entities should be differentiated, as HSP may have renal and gastrointestinal (GI) comorbidities that need serial follow-up, whereas AHEI follows a benign course without systemic sequelae. Patient history and physical examination are the most important factors in differentiating the 2 diseases; histopathologic and direct immunofluorescence (DIF) analyses may lend further diagnostic confidence.
We report the case of a 10-month-old previously healthy boy who presented with acute rash, edema, and low-grade fever in the setting of recent diarrhea. We differentiate between AHEI and HSP to help prevent misdiagnosis by health care providers.
Case Report
A 10-month-old previously healthy boy presented to the emergency department (ED) for evaluation of a rash and swelling of 4 days’ duration. He had nonbloody diarrhea 1 week prior; soon after, he developed bilateral lower leg edema and rash. On evaluation in a different ED, he had a low-grade fever (rectal temperature, 38.0°C) but normal blood work, including complete blood cell count, basic metabolic panel, and coagulation studies. The patient was discharged to outpatient follow-up with his pediatrician who reported normal urinalysis.
Due to progression of the rash, the patient presented to our ED 3 days after his initial ED assessment. Dermatology was consulted. At the time of presentation, he was afebrile but with GI upset and fussiness. His parents denied additional symptoms or blood in urine or stool. Physical examination revealed a nontoxic-appearing infant with scattered palpable, annular, purpuric papules coalescing into plaques on both legs and feet (Figure 1), with sparse petechiae noted on the lower abdomen. The cheeks had scattered purpuric papules and plaques bilaterally, a few with a small central crust (Figure 2), and the right superior helix had a faint purpuric macule. The hands had a few pink edematous coalescing papules.
Histopathologic analyses with hematoxylin and eosin staining (Figure 3) and DIF (Figure 4) were performed from within a representative purpuric plaque on the right hip. Direct immunofluorescence was performed to evaluate for an IgA vasculitis versus an alternative type of vasculitis. The hematoxylin and eosin–stained specimen demonstrated a dermal perivascular infiltrate involving superficial and deep vessels with neutrophils, karyorrhexis, and erythrocyte extravasation. The endothelium was intact, with a mild suggestion of fibrinoid change of the blood vessel walls. Direct immunofluorescence revealed granular deposition of IgA, C3, and fibrinogen in multiple dermal blood vessels. Combined, the specimens were interpreted as evolving IgA-associated leukocytoclastic vasculitis.
The case was reviewed with our 2 department pediatric dermatologists; a diagnosis of AHEI was made based on the clinical and supportive histopathological presentations. The patient’s parents chose active treatment with a 2-week taper of oral prednisone because of the patient’s discomfort with edema. No GI or adverse renal sequelae, including findings on urinalysis, were reported at 1-month hospital follow-up with dermatology and pediatrics.
Comment
Incidence and Clinical Characteristics
Acute hemorrhagic edema of infancy is an uncommon leukocytoclastic vasculitis first described in the United States by Snow1 in 1913. Other names for the disorder include acute hemorrhagic edema of young children, cockade purpura and edema, Finkelstein disease, and Seidlmayer disease.2 Boys are affected more often than girls, with most children presenting at 6 to 24 months of age. Most affected children experience a prodrome of simple respiratory tract illness (most common), diarrhea (as in our case), or urinary tract infection.2 The exact pathophysiology behind AHEI is unknown, but it is thought to be an immune complex–mediated disease evidenced by the fact that infection, use of medication, or immunization precedes most cases.3,4
Diagnosis
Acute hemorrhagic edema of infancy is diagnosed clinically, with or without the support of skin biopsy. It should be differentiated from HSP because of renal and GI sequelae that HSP portends compared to the benign course of AHEI.2 Notably, some health care providers consider AHEI a benign variant of HSP.2,3
Characteristically, AHEI patients are nontoxic-appearing infants with a low-grade fever who develop relatively large (1–5 cm) targetoid purpuric lesions and indurated nonpitting edema of the extremities.2,5 Purpura in AHEI frequently occurs on the face, ears, and upper and lower extremities, whereas purpura in HSP most commonly presents on the buttocks and extensor legs with sparing of the face. Henoch-Schönlein purpura most often affects children aged 3 to 6 years compared to AHEI’s younger demographic (age <2 years).4,5 Clinically, HSP presents with palpable purpura and 1 or more of the following features: diffuse abdominal pain, arthritis/arthralgia, renal involvement, and skin or renal biopsy showing predominant IgA deposition.2,6
Both AHEI and HSP show leukocytoclastic vasculitis on histopathology.2-4,6,7 Positive perivascular IgA staining on DIF is strongly associated with HSP, but nearly one-quarter of AHEI cases also show this deposition pattern2,4,7; therefore, DIF alone cannot exclude a diagnosis of AHEI.
Differential Diagnosis
Alternative diagnoses to consider with AHEI include drug-induced vasculitis, erythema multiforme, HSP, Kawasaki disease, meningococcemia, nonaccidental skin bruising, Rocky Mountain spotted fever, septic vasculitis, and urticarial vasculitis (Table).2-4,6-8
Treatment
Acute hemorrhagic edema of infancy is self-limited, with only rare reports of extracutaneous involvement. Supportive treatment is indicated because spontaneous recovery without sequelae is expected within 21 days.2,3,6 If edema is symptomatic, as was the case with our patient, corticosteroids may shorten the disease course.3
Conclusion
Our case highlights the need to combine clinical history, physical examination, and histopathologic analysis to differentiate between AHEI and HSP, which is important for 2 reasons: (1) it helps with the decision to undertake active or observational treatment, and (2) it helps the clinician counsel the patient and guardians regarding potential associated renal and GI risks.
- Snow IM. Purpura, urticaria and angioneurotic edema of the hands and feet in a nursing baby. JAMA. 1913;61:18-19.
- Fiore E, Rizzi M, Ragazzi M, et al. Acute hemorrhagic edema of young children (cockade purpura and edema): a case series and systematic review. J Am Acad Dermatol. 2008;59:684-695.
- Freitas P, Bygum A. Visual impairment caused by periorbital edema in an infant with acute hemorrhagic edema of infancy. Pediatr Dermatol. 2013;30:e132-e135.
- Legrain V, Lejean S, Taïeb A, et al. Infantile acute hemorrhagic edema of the skin: study of ten cases. J Am Acad Dermatol. 1991;24:17-22.
- Breda L, Franchini S, Marzetti V, et al. Escherichia coli urinary infection as a cause of acute hemorrhagic edema in infancy. Pediatr Dermatol. 2015;32:e309-e311.
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941.
- Saraclar Y, Tinaztepe K, Adalioğlu G, et al. Acute hemorrhagic edema of infancy (AHEI)—a variant of Henoch-Schönlein purpura or a distinct clinical entity? J Allergy Clin Immunol. 1990;86:473-483.
- Shinkai K, Fox L. Cutaneous vasculitis. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. China: Elsevier Limited; 2012:385-410.
- Snow IM. Purpura, urticaria and angioneurotic edema of the hands and feet in a nursing baby. JAMA. 1913;61:18-19.
- Fiore E, Rizzi M, Ragazzi M, et al. Acute hemorrhagic edema of young children (cockade purpura and edema): a case series and systematic review. J Am Acad Dermatol. 2008;59:684-695.
- Freitas P, Bygum A. Visual impairment caused by periorbital edema in an infant with acute hemorrhagic edema of infancy. Pediatr Dermatol. 2013;30:e132-e135.
- Legrain V, Lejean S, Taïeb A, et al. Infantile acute hemorrhagic edema of the skin: study of ten cases. J Am Acad Dermatol. 1991;24:17-22.
- Breda L, Franchini S, Marzetti V, et al. Escherichia coli urinary infection as a cause of acute hemorrhagic edema in infancy. Pediatr Dermatol. 2015;32:e309-e311.
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941.
- Saraclar Y, Tinaztepe K, Adalioğlu G, et al. Acute hemorrhagic edema of infancy (AHEI)—a variant of Henoch-Schönlein purpura or a distinct clinical entity? J Allergy Clin Immunol. 1990;86:473-483.
- Shinkai K, Fox L. Cutaneous vasculitis. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. China: Elsevier Limited; 2012:385-410.
Practice Points
- Acute hemorrhagic edema of infancy (AHEI) is an uncommon benign leukocytoclastic vasculitis of unknown precise pathophysiology that is thought be immune complex mediated.
- Clinical history, physical examination, and histopathologic analysis combine to allow the important differentiation between AHEI and Henoch-Schönlein purpura (HSP).
- Differentiation between AHEI and HSP determines treatment decisions and indicates the need for counseling on potential associated renal and gastrointestinal risks of HSP.
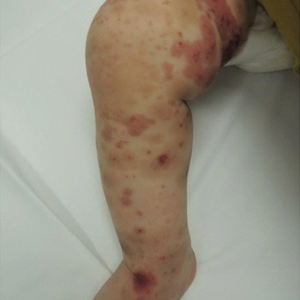
Acrokeratoelastoidosis and Knuckle Pads Coexisting in a Child
Case Report
An 11-year-old boy presented with atraumatic thickening of the skin on the bilateral distal and proximal interphalangeal joints of 1 year’s duration. The patient also noted small bumps of unknown duration across the bilateral palms and soles with prominence on the lateral aspects. The patient previously used over-the-counter topical wart removal treatment and topical salicylic acid with minimal improvement. The patient reported no pertinent medical or surgical history, although there was a family history of Alport syndrome, predominantly in male relatives. The patient’s father and paternal grandfather were noted to have similar lesions on the palms.
On physical examination, multiple pink to flesh-colored hyperkeratotic plaques were noted over the proximal and distal interphalangeal joints of the bilateral hands (Figure 1A). Upon close inspection, there were small flesh-colored and slightly translucent papules in a linear distribution on the palmar surfaces of the hands (Figure 2A) with predominance on the thenar and hypothenar eminences. The flexural creases of the bilateral wrists also revealed linear flesh-colored papules. The same small flesh-colored and translucent papules also were noted on the plantar surfaces of the bilateral feet (Figure 2B).
A biopsy was obtained from one of the small translucent papules on the left palm. Hematoxylin and eosin–stained sections revealed elevated compact orthokeratosis with an underlying central epidermal dell (Figure 3). A diagnosis of marginal papular keratoderma was made and further elastin staining was completed. Elastin stains showed marked thinning of the elastin fibers throughout the reticular dermis. Many elastin fibers in the reticular dermis demonstrated a fine arborizing pattern that normally is only evident in the papillary dermis (Figure 4). Acrokeratoelastoidosis (AKE) was diagnosed histopathologically, and knuckle pads were diagnosed clinically.
Because the patient was asymptomatic, he did not want treatment of AKE. He had marked improvement of the knuckle pads after 1 month with daily application of urea cream 10% (Figure 1B), and intermittent use was required for maintenance.
Comment
Etiology
Acrokeratoelastoidosis was first described in 1953 and is considered a type of palmoplantar marginal papular keratoderma.1 There is overlap within the marginal papular keratodermas that makes precise diagnosis difficult within this group. The marginal papular keratodermas on the palms and soles are a group of disorders that include AKE, focal acral hyperkeratosis (FAH), mosaic acral keratosis, degenerative collagenous plaques on the hands, and digital papular calcific elastosis. These diseases are similar in clinical and histopathological features; some argue these diseases are the same entity.2
Acrokeratoelastoidosis has been hypothesized to originate from altered elastic fiber synthesis from fibroblasts.3 Because AKE is rare, most cases of common knuckle pads do not coexist with AKE; therefore, it is unknown if the underlying etiology remains the same for both entities. Unlike AKE, knuckle pads are often associated with Dupuytren contractures, repetitive trauma, or friction to the area.1,2
Presentation
Acrokeratoelastoidosis is a rare disease with onset in childhood or young adulthood. Childhood cases are inherited in an autosomal-dominant fashion.1 Adulthood onset suggests a sporadic form of inheritance. Acrokeratoelastoidosis has no gender or racial predilection.4 It presents over the thenar and hypothenar eminences, as well as the lateral digits, calcaneal tendon, and dorsal digits.1 Most often, AKE occurs symmetrically along the border separating the ventral and dorsal aspects on the palms and soles. These lesions present as small, firm, translucent papules that align linearly on the ventral-dorsal palmoplantar junction in a pattern resembling paving stones.1 Coalescence of papules into plaques has been reported. Extension of lesions to the dorsal and palmar surfaces can occur. Small circumscribed callosities may develop over the metacarpophalangeal and interphalangeal joints resembling knuckle pads.2
Histopathology
Histopathologically, AKE is distinguished by elastorrhexis—thinning, fragmenting, and rarefaction of elastin fibers—in the epidermis and reticular dermis layers.3 Acrokeratoelastoidosis also presents with orthokeratosis overlying a cuplike epithelial depression and possible epithelial acanthosis.2,5 Many cases exhibit hypergranulosis at the base of the epidermal dell. Dense basophilic granules may be seen in the peripheral cytoplasm of fibroblast cells coming from the hypothesized defect in elastin secretion.1,3,4
Differential Diagnosis
The main differential diagnosis of AKE is FAH. Clinically and histopathologically they appear identical; both diseases have cuplike epidermal depressions with overlying orthohyperkeratosis and prominent hypergranulosis.5 The elastin stains, Verhoeff-van Gieson or acid orcein stain, are imperative for distinguishing these two diseases. Although AKE demonstrates elastorrhexis and reduced elastic fibers, FAH reveals no alteration of elastic fibers. It has been suggested that FAH is a clinical variant of AKE and should be titled AKE without elastorrhexis.1
Treatment
Acrokeratoelastoidosis is asymptomatic except for mild palmoplantar hyperhidrosis and typically does not require treatment4; however, the condition can be of cosmetic concern for patients. Lesions can be treated topically with keratolytics such as tretinoin and salicylic acid. A wide variety of systemic treatments including methotrexate, prednisolone, dapsone, and acitretin have been reported with variable clinical response.2-4 Copresenting knuckle pads can be treated with urea cream, salicylic acid cream, or intralesional corticosteroids.1
- Erkek E, Koçak M, Bozdog˘an O, et al. Focal acral hyperkeratosis: a rare cutaneous disorder within the spectrum are Costa acrokeratoelastoidosis. Pediatr Dermatol. 2004;21:128-130.
- Abulafia J, Vignale R. Degenerative collagenous plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
- Nelson-Adesokan P, Mallory SB, Leonardi CL, et al. Acrokeratoelastoidosis of Costa. Int J Dermatol. 1995;34:431-433.
- Shbaklo Z, Jamaleddine NF, Kibbi AG, et al. Acrokeratoelastoidosis. Int J Dermatol. 1990;29:333-336.
- Ming M. Papules overlying finger joints—diagnosis. Arch Dermatol. 2006;142:235-240.
Case Report
An 11-year-old boy presented with atraumatic thickening of the skin on the bilateral distal and proximal interphalangeal joints of 1 year’s duration. The patient also noted small bumps of unknown duration across the bilateral palms and soles with prominence on the lateral aspects. The patient previously used over-the-counter topical wart removal treatment and topical salicylic acid with minimal improvement. The patient reported no pertinent medical or surgical history, although there was a family history of Alport syndrome, predominantly in male relatives. The patient’s father and paternal grandfather were noted to have similar lesions on the palms.
On physical examination, multiple pink to flesh-colored hyperkeratotic plaques were noted over the proximal and distal interphalangeal joints of the bilateral hands (Figure 1A). Upon close inspection, there were small flesh-colored and slightly translucent papules in a linear distribution on the palmar surfaces of the hands (Figure 2A) with predominance on the thenar and hypothenar eminences. The flexural creases of the bilateral wrists also revealed linear flesh-colored papules. The same small flesh-colored and translucent papules also were noted on the plantar surfaces of the bilateral feet (Figure 2B).
A biopsy was obtained from one of the small translucent papules on the left palm. Hematoxylin and eosin–stained sections revealed elevated compact orthokeratosis with an underlying central epidermal dell (Figure 3). A diagnosis of marginal papular keratoderma was made and further elastin staining was completed. Elastin stains showed marked thinning of the elastin fibers throughout the reticular dermis. Many elastin fibers in the reticular dermis demonstrated a fine arborizing pattern that normally is only evident in the papillary dermis (Figure 4). Acrokeratoelastoidosis (AKE) was diagnosed histopathologically, and knuckle pads were diagnosed clinically.
Because the patient was asymptomatic, he did not want treatment of AKE. He had marked improvement of the knuckle pads after 1 month with daily application of urea cream 10% (Figure 1B), and intermittent use was required for maintenance.
Comment
Etiology
Acrokeratoelastoidosis was first described in 1953 and is considered a type of palmoplantar marginal papular keratoderma.1 There is overlap within the marginal papular keratodermas that makes precise diagnosis difficult within this group. The marginal papular keratodermas on the palms and soles are a group of disorders that include AKE, focal acral hyperkeratosis (FAH), mosaic acral keratosis, degenerative collagenous plaques on the hands, and digital papular calcific elastosis. These diseases are similar in clinical and histopathological features; some argue these diseases are the same entity.2
Acrokeratoelastoidosis has been hypothesized to originate from altered elastic fiber synthesis from fibroblasts.3 Because AKE is rare, most cases of common knuckle pads do not coexist with AKE; therefore, it is unknown if the underlying etiology remains the same for both entities. Unlike AKE, knuckle pads are often associated with Dupuytren contractures, repetitive trauma, or friction to the area.1,2
Presentation
Acrokeratoelastoidosis is a rare disease with onset in childhood or young adulthood. Childhood cases are inherited in an autosomal-dominant fashion.1 Adulthood onset suggests a sporadic form of inheritance. Acrokeratoelastoidosis has no gender or racial predilection.4 It presents over the thenar and hypothenar eminences, as well as the lateral digits, calcaneal tendon, and dorsal digits.1 Most often, AKE occurs symmetrically along the border separating the ventral and dorsal aspects on the palms and soles. These lesions present as small, firm, translucent papules that align linearly on the ventral-dorsal palmoplantar junction in a pattern resembling paving stones.1 Coalescence of papules into plaques has been reported. Extension of lesions to the dorsal and palmar surfaces can occur. Small circumscribed callosities may develop over the metacarpophalangeal and interphalangeal joints resembling knuckle pads.2
Histopathology
Histopathologically, AKE is distinguished by elastorrhexis—thinning, fragmenting, and rarefaction of elastin fibers—in the epidermis and reticular dermis layers.3 Acrokeratoelastoidosis also presents with orthokeratosis overlying a cuplike epithelial depression and possible epithelial acanthosis.2,5 Many cases exhibit hypergranulosis at the base of the epidermal dell. Dense basophilic granules may be seen in the peripheral cytoplasm of fibroblast cells coming from the hypothesized defect in elastin secretion.1,3,4
Differential Diagnosis
The main differential diagnosis of AKE is FAH. Clinically and histopathologically they appear identical; both diseases have cuplike epidermal depressions with overlying orthohyperkeratosis and prominent hypergranulosis.5 The elastin stains, Verhoeff-van Gieson or acid orcein stain, are imperative for distinguishing these two diseases. Although AKE demonstrates elastorrhexis and reduced elastic fibers, FAH reveals no alteration of elastic fibers. It has been suggested that FAH is a clinical variant of AKE and should be titled AKE without elastorrhexis.1
Treatment
Acrokeratoelastoidosis is asymptomatic except for mild palmoplantar hyperhidrosis and typically does not require treatment4; however, the condition can be of cosmetic concern for patients. Lesions can be treated topically with keratolytics such as tretinoin and salicylic acid. A wide variety of systemic treatments including methotrexate, prednisolone, dapsone, and acitretin have been reported with variable clinical response.2-4 Copresenting knuckle pads can be treated with urea cream, salicylic acid cream, or intralesional corticosteroids.1
Case Report
An 11-year-old boy presented with atraumatic thickening of the skin on the bilateral distal and proximal interphalangeal joints of 1 year’s duration. The patient also noted small bumps of unknown duration across the bilateral palms and soles with prominence on the lateral aspects. The patient previously used over-the-counter topical wart removal treatment and topical salicylic acid with minimal improvement. The patient reported no pertinent medical or surgical history, although there was a family history of Alport syndrome, predominantly in male relatives. The patient’s father and paternal grandfather were noted to have similar lesions on the palms.
On physical examination, multiple pink to flesh-colored hyperkeratotic plaques were noted over the proximal and distal interphalangeal joints of the bilateral hands (Figure 1A). Upon close inspection, there were small flesh-colored and slightly translucent papules in a linear distribution on the palmar surfaces of the hands (Figure 2A) with predominance on the thenar and hypothenar eminences. The flexural creases of the bilateral wrists also revealed linear flesh-colored papules. The same small flesh-colored and translucent papules also were noted on the plantar surfaces of the bilateral feet (Figure 2B).
A biopsy was obtained from one of the small translucent papules on the left palm. Hematoxylin and eosin–stained sections revealed elevated compact orthokeratosis with an underlying central epidermal dell (Figure 3). A diagnosis of marginal papular keratoderma was made and further elastin staining was completed. Elastin stains showed marked thinning of the elastin fibers throughout the reticular dermis. Many elastin fibers in the reticular dermis demonstrated a fine arborizing pattern that normally is only evident in the papillary dermis (Figure 4). Acrokeratoelastoidosis (AKE) was diagnosed histopathologically, and knuckle pads were diagnosed clinically.
Because the patient was asymptomatic, he did not want treatment of AKE. He had marked improvement of the knuckle pads after 1 month with daily application of urea cream 10% (Figure 1B), and intermittent use was required for maintenance.
Comment
Etiology
Acrokeratoelastoidosis was first described in 1953 and is considered a type of palmoplantar marginal papular keratoderma.1 There is overlap within the marginal papular keratodermas that makes precise diagnosis difficult within this group. The marginal papular keratodermas on the palms and soles are a group of disorders that include AKE, focal acral hyperkeratosis (FAH), mosaic acral keratosis, degenerative collagenous plaques on the hands, and digital papular calcific elastosis. These diseases are similar in clinical and histopathological features; some argue these diseases are the same entity.2
Acrokeratoelastoidosis has been hypothesized to originate from altered elastic fiber synthesis from fibroblasts.3 Because AKE is rare, most cases of common knuckle pads do not coexist with AKE; therefore, it is unknown if the underlying etiology remains the same for both entities. Unlike AKE, knuckle pads are often associated with Dupuytren contractures, repetitive trauma, or friction to the area.1,2
Presentation
Acrokeratoelastoidosis is a rare disease with onset in childhood or young adulthood. Childhood cases are inherited in an autosomal-dominant fashion.1 Adulthood onset suggests a sporadic form of inheritance. Acrokeratoelastoidosis has no gender or racial predilection.4 It presents over the thenar and hypothenar eminences, as well as the lateral digits, calcaneal tendon, and dorsal digits.1 Most often, AKE occurs symmetrically along the border separating the ventral and dorsal aspects on the palms and soles. These lesions present as small, firm, translucent papules that align linearly on the ventral-dorsal palmoplantar junction in a pattern resembling paving stones.1 Coalescence of papules into plaques has been reported. Extension of lesions to the dorsal and palmar surfaces can occur. Small circumscribed callosities may develop over the metacarpophalangeal and interphalangeal joints resembling knuckle pads.2
Histopathology
Histopathologically, AKE is distinguished by elastorrhexis—thinning, fragmenting, and rarefaction of elastin fibers—in the epidermis and reticular dermis layers.3 Acrokeratoelastoidosis also presents with orthokeratosis overlying a cuplike epithelial depression and possible epithelial acanthosis.2,5 Many cases exhibit hypergranulosis at the base of the epidermal dell. Dense basophilic granules may be seen in the peripheral cytoplasm of fibroblast cells coming from the hypothesized defect in elastin secretion.1,3,4
Differential Diagnosis
The main differential diagnosis of AKE is FAH. Clinically and histopathologically they appear identical; both diseases have cuplike epidermal depressions with overlying orthohyperkeratosis and prominent hypergranulosis.5 The elastin stains, Verhoeff-van Gieson or acid orcein stain, are imperative for distinguishing these two diseases. Although AKE demonstrates elastorrhexis and reduced elastic fibers, FAH reveals no alteration of elastic fibers. It has been suggested that FAH is a clinical variant of AKE and should be titled AKE without elastorrhexis.1
Treatment
Acrokeratoelastoidosis is asymptomatic except for mild palmoplantar hyperhidrosis and typically does not require treatment4; however, the condition can be of cosmetic concern for patients. Lesions can be treated topically with keratolytics such as tretinoin and salicylic acid. A wide variety of systemic treatments including methotrexate, prednisolone, dapsone, and acitretin have been reported with variable clinical response.2-4 Copresenting knuckle pads can be treated with urea cream, salicylic acid cream, or intralesional corticosteroids.1
- Erkek E, Koçak M, Bozdog˘an O, et al. Focal acral hyperkeratosis: a rare cutaneous disorder within the spectrum are Costa acrokeratoelastoidosis. Pediatr Dermatol. 2004;21:128-130.
- Abulafia J, Vignale R. Degenerative collagenous plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
- Nelson-Adesokan P, Mallory SB, Leonardi CL, et al. Acrokeratoelastoidosis of Costa. Int J Dermatol. 1995;34:431-433.
- Shbaklo Z, Jamaleddine NF, Kibbi AG, et al. Acrokeratoelastoidosis. Int J Dermatol. 1990;29:333-336.
- Ming M. Papules overlying finger joints—diagnosis. Arch Dermatol. 2006;142:235-240.
- Erkek E, Koçak M, Bozdog˘an O, et al. Focal acral hyperkeratosis: a rare cutaneous disorder within the spectrum are Costa acrokeratoelastoidosis. Pediatr Dermatol. 2004;21:128-130.
- Abulafia J, Vignale R. Degenerative collagenous plaques of the hands and acrokeratoelastoidosis: pathogenesis and relationship with knuckle pads. Int J Dermatol. 2000;39:424-432.
- Nelson-Adesokan P, Mallory SB, Leonardi CL, et al. Acrokeratoelastoidosis of Costa. Int J Dermatol. 1995;34:431-433.
- Shbaklo Z, Jamaleddine NF, Kibbi AG, et al. Acrokeratoelastoidosis. Int J Dermatol. 1990;29:333-336.
- Ming M. Papules overlying finger joints—diagnosis. Arch Dermatol. 2006;142:235-240.
Practice Points
- Acrokeratoelastoidosis presents as small, firm, translucent, linear papules on the ventral-dorsal palmoplantar junction.
- Acrokeratoelastoidosis does not require treatment but can be treated topically with keratolytics such as tretinoin and salicylic acid.
- Knuckle pads may respond to urea cream, salicylic acid cream, or intralesional corticosteroids.
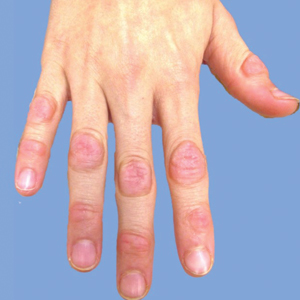
How could improved provider communication have improved the care this patient received?
THE CASE
A 40-year-old white woman presented to clinic with multiple pruritic skin lesions on her abdomen, arms, and legs that had developed over a 2-month period. The patient reported that she’d been feeling tired and had been experiencing psychological stressors in her personal life. Her medical history was significant for psoriasis (which was controlled), and her family history was significant for breast and bone cancer (mother) and asbestos-related lung cancer (maternal grandfather).
A physical examination, which included breast and pelvic exams, was unremarkable apart from the lesions located on her abdomen, arms, and legs. On skin examination, we noted multiple polygonal, planar papules and plaques of varying size with an overlying scale (FIGURE).

THE DIAGNOSIS
The physician obtained a biopsy of one of the skin lesions, and it was sent to a dermatopathologist to evaluate. Unfortunately, though, the patient’s history and a description of the lesion were not included with the initial biopsy requisition form. Based on the biopsy sample alone, the dermatopathologist’s report indicated a diagnosis of seborrheic keratosis.
A search for malignancy. Any case of sudden, extensive seborrheic keratosis is suspected to be a Leser-Trélat sign, which is known to be associated with human immunodeficiency virus or underlying malignancy—especially in the gastrointestinal system. The physician talked to the patient about the possibility of malignancy, and an extensive work-up was performed, including multiple laboratory tests, computed tomography (CT) imaging, an esophagogastroduodenoscopy, a colonoscopy, and mammography. None of the test results showed signs of an underlying malignancy.
In light of the negative findings, the physician reached out to the dermatopathologist to further discuss the case. It was determined that the dermatopathologist did not receive any clinical information (prior to this discussion) from the primary care office. This was surprising to the primary care physician, who was under the assumption that the clinical chart would be sent along with the biopsy sample. With this new information, the dermatopathologist reexamined the slides and diagnosed the lesion as lichen planus, a rather common skin disease not associated with cancer.
[polldaddy:10153197]
DISCUSSION
A root-cause analysis of this case identified multiple system failures, focused mainly on a lack of communication between providers:
- The description of the lesion and of the patient’s history were not included with the initial biopsy requisition form due to a lack of communication between the nurse and the physician performing the procedure.
- The dermatopathologist did not seek additional clinical information from the referring physician after receiving the sample.
- When the various providers did communicate, an accurate diagnosis was reached—but only after extensive investigation (and worry).
Communication is key to an accurate diagnosis
In 2000, it was estimated that health care costs due to preventable adverse events represent more than half of the $37.6 billion spent on health care.1 Since then, considerable effort has been made to address patient safety, misdiagnosis, and cost-effectiveness. Root cause analysis is one of the most popular methods used to evaluate and prevent future serious adverse events.2
Continue to: Diagnostic errors are often unreported...
Diagnostic errors are often unreported or unrecognized, especially in the outpatient setting.3 Studies focused on reducing diagnostic error show that a second review of pathology slides reduces error, controls costs, and improves quality of health care.4
Don’t rely (exclusively) on the health record. Gaps in effective communication between providers are a leading cause of preventable adverse events.5,6 The incorporation of electronic health records has allowed for more streamlined communication between providers. However, the mere presence of patient records in a common system does not guarantee the receipt or communication of information. The next step after entering the information into the record is to communicate it.
Our patient underwent a battery of costly and unnecessary tests and procedures, many of which were unwarranted at her age. In addition to being exposed to harmful radiation, she also experienced significant stress secondary to the tests and anticipation of the results. However, a root cause analysis of the case led to an improved protocol for communication between providers at the outpatient clinic. We now emphasize the necessity of including a clinical history and corresponding physical findings with all biopsies. We also encourage more direct communication between nursing staff, primary care physicians, and specialists.
THE TAKEAWAY
As medical professionals become increasingly reliant on the many emerging studies available to them, we sometimes forget that communication is key to optimal medical care, an accurate diagnosis, and patient safety.
Continue to: In addition, a second review...
In addition, a second review of dermatopathologic slides may be warranted if the pathologic diagnosis is inconsistent with the clinical picture or if the diagnosed condition is resistant to the usual therapies of choice. Incorrect diagnoses are more likely to occur when tests are interpreted in a vacuum without the corresponding clinical correlation. The weight of these mistakes is felt not only by the health care system, but by the patients themselves.
CORRESPONDENCE
Magdalena Pasarica, MD, PhD, University of Central Florida College of Medicine, 6850 Lake Nona Boulevard, Orlando, FL 32827; Magdalena.Pasarica@ucf.edu
1. Kohn LT, Corrigan JM, Donaldson MS. To Err is Human: Building a Safer Health System. Washington, DC: National Academies Press; 2000.
2. U.S. Department of Health and Human Services. Patient safety primer: root cause analysis. https://psnet.ahrq.gov/primers/primer/10/root-cause-analysis. Updated August 2018. Accessed September 27, 2018.
3. Newman-Toker DE, Pronovost PJ. Diagnostic errors-the next frontier for patient safety. JAMA. 2009;301:1060-1062.
4. Kuijpers CC, Burger G, Al-Janabi S, et al. Improved quality of patient care through routine second review of histopathology specimens prior to multidisciplinary meetings. J Clin Pathol. 2016;69:866-871.
5. Leonard M, Graham S, Bonacum D. The human factor: the critical importance of effective teamwork and communication in providing safe care. Qual Saf Health Care. 2004;13:85-90.
6. Robinson NL. Promoting patient safety with perioperative hand-off communication. J Perianesth Nurs. 2016;31:245-253.
THE CASE
A 40-year-old white woman presented to clinic with multiple pruritic skin lesions on her abdomen, arms, and legs that had developed over a 2-month period. The patient reported that she’d been feeling tired and had been experiencing psychological stressors in her personal life. Her medical history was significant for psoriasis (which was controlled), and her family history was significant for breast and bone cancer (mother) and asbestos-related lung cancer (maternal grandfather).
A physical examination, which included breast and pelvic exams, was unremarkable apart from the lesions located on her abdomen, arms, and legs. On skin examination, we noted multiple polygonal, planar papules and plaques of varying size with an overlying scale (FIGURE).
THE DIAGNOSIS
The physician obtained a biopsy of one of the skin lesions, and it was sent to a dermatopathologist to evaluate. Unfortunately, though, the patient’s history and a description of the lesion were not included with the initial biopsy requisition form. Based on the biopsy sample alone, the dermatopathologist’s report indicated a diagnosis of seborrheic keratosis.
A search for malignancy. Any case of sudden, extensive seborrheic keratosis is suspected to be a Leser-Trélat sign, which is known to be associated with human immunodeficiency virus or underlying malignancy—especially in the gastrointestinal system. The physician talked to the patient about the possibility of malignancy, and an extensive work-up was performed, including multiple laboratory tests, computed tomography (CT) imaging, an esophagogastroduodenoscopy, a colonoscopy, and mammography. None of the test results showed signs of an underlying malignancy.
In light of the negative findings, the physician reached out to the dermatopathologist to further discuss the case. It was determined that the dermatopathologist did not receive any clinical information (prior to this discussion) from the primary care office. This was surprising to the primary care physician, who was under the assumption that the clinical chart would be sent along with the biopsy sample. With this new information, the dermatopathologist reexamined the slides and diagnosed the lesion as lichen planus, a rather common skin disease not associated with cancer.
[polldaddy:10153197]
DISCUSSION
A root-cause analysis of this case identified multiple system failures, focused mainly on a lack of communication between providers:
- The description of the lesion and of the patient’s history were not included with the initial biopsy requisition form due to a lack of communication between the nurse and the physician performing the procedure.
- The dermatopathologist did not seek additional clinical information from the referring physician after receiving the sample.
- When the various providers did communicate, an accurate diagnosis was reached—but only after extensive investigation (and worry).
Communication is key to an accurate diagnosis
In 2000, it was estimated that health care costs due to preventable adverse events represent more than half of the $37.6 billion spent on health care.1 Since then, considerable effort has been made to address patient safety, misdiagnosis, and cost-effectiveness. Root cause analysis is one of the most popular methods used to evaluate and prevent future serious adverse events.2
Continue to: Diagnostic errors are often unreported...
Diagnostic errors are often unreported or unrecognized, especially in the outpatient setting.3 Studies focused on reducing diagnostic error show that a second review of pathology slides reduces error, controls costs, and improves quality of health care.4
Don’t rely (exclusively) on the health record. Gaps in effective communication between providers are a leading cause of preventable adverse events.5,6 The incorporation of electronic health records has allowed for more streamlined communication between providers. However, the mere presence of patient records in a common system does not guarantee the receipt or communication of information. The next step after entering the information into the record is to communicate it.
Our patient underwent a battery of costly and unnecessary tests and procedures, many of which were unwarranted at her age. In addition to being exposed to harmful radiation, she also experienced significant stress secondary to the tests and anticipation of the results. However, a root cause analysis of the case led to an improved protocol for communication between providers at the outpatient clinic. We now emphasize the necessity of including a clinical history and corresponding physical findings with all biopsies. We also encourage more direct communication between nursing staff, primary care physicians, and specialists.
THE TAKEAWAY
As medical professionals become increasingly reliant on the many emerging studies available to them, we sometimes forget that communication is key to optimal medical care, an accurate diagnosis, and patient safety.
Continue to: In addition, a second review...
In addition, a second review of dermatopathologic slides may be warranted if the pathologic diagnosis is inconsistent with the clinical picture or if the diagnosed condition is resistant to the usual therapies of choice. Incorrect diagnoses are more likely to occur when tests are interpreted in a vacuum without the corresponding clinical correlation. The weight of these mistakes is felt not only by the health care system, but by the patients themselves.
CORRESPONDENCE
Magdalena Pasarica, MD, PhD, University of Central Florida College of Medicine, 6850 Lake Nona Boulevard, Orlando, FL 32827; Magdalena.Pasarica@ucf.edu
THE CASE
A 40-year-old white woman presented to clinic with multiple pruritic skin lesions on her abdomen, arms, and legs that had developed over a 2-month period. The patient reported that she’d been feeling tired and had been experiencing psychological stressors in her personal life. Her medical history was significant for psoriasis (which was controlled), and her family history was significant for breast and bone cancer (mother) and asbestos-related lung cancer (maternal grandfather).
A physical examination, which included breast and pelvic exams, was unremarkable apart from the lesions located on her abdomen, arms, and legs. On skin examination, we noted multiple polygonal, planar papules and plaques of varying size with an overlying scale (FIGURE).
THE DIAGNOSIS
The physician obtained a biopsy of one of the skin lesions, and it was sent to a dermatopathologist to evaluate. Unfortunately, though, the patient’s history and a description of the lesion were not included with the initial biopsy requisition form. Based on the biopsy sample alone, the dermatopathologist’s report indicated a diagnosis of seborrheic keratosis.
A search for malignancy. Any case of sudden, extensive seborrheic keratosis is suspected to be a Leser-Trélat sign, which is known to be associated with human immunodeficiency virus or underlying malignancy—especially in the gastrointestinal system. The physician talked to the patient about the possibility of malignancy, and an extensive work-up was performed, including multiple laboratory tests, computed tomography (CT) imaging, an esophagogastroduodenoscopy, a colonoscopy, and mammography. None of the test results showed signs of an underlying malignancy.
In light of the negative findings, the physician reached out to the dermatopathologist to further discuss the case. It was determined that the dermatopathologist did not receive any clinical information (prior to this discussion) from the primary care office. This was surprising to the primary care physician, who was under the assumption that the clinical chart would be sent along with the biopsy sample. With this new information, the dermatopathologist reexamined the slides and diagnosed the lesion as lichen planus, a rather common skin disease not associated with cancer.
[polldaddy:10153197]
DISCUSSION
A root-cause analysis of this case identified multiple system failures, focused mainly on a lack of communication between providers:
- The description of the lesion and of the patient’s history were not included with the initial biopsy requisition form due to a lack of communication between the nurse and the physician performing the procedure.
- The dermatopathologist did not seek additional clinical information from the referring physician after receiving the sample.
- When the various providers did communicate, an accurate diagnosis was reached—but only after extensive investigation (and worry).
Communication is key to an accurate diagnosis
In 2000, it was estimated that health care costs due to preventable adverse events represent more than half of the $37.6 billion spent on health care.1 Since then, considerable effort has been made to address patient safety, misdiagnosis, and cost-effectiveness. Root cause analysis is one of the most popular methods used to evaluate and prevent future serious adverse events.2
Continue to: Diagnostic errors are often unreported...
Diagnostic errors are often unreported or unrecognized, especially in the outpatient setting.3 Studies focused on reducing diagnostic error show that a second review of pathology slides reduces error, controls costs, and improves quality of health care.4
Don’t rely (exclusively) on the health record. Gaps in effective communication between providers are a leading cause of preventable adverse events.5,6 The incorporation of electronic health records has allowed for more streamlined communication between providers. However, the mere presence of patient records in a common system does not guarantee the receipt or communication of information. The next step after entering the information into the record is to communicate it.
Our patient underwent a battery of costly and unnecessary tests and procedures, many of which were unwarranted at her age. In addition to being exposed to harmful radiation, she also experienced significant stress secondary to the tests and anticipation of the results. However, a root cause analysis of the case led to an improved protocol for communication between providers at the outpatient clinic. We now emphasize the necessity of including a clinical history and corresponding physical findings with all biopsies. We also encourage more direct communication between nursing staff, primary care physicians, and specialists.
THE TAKEAWAY
As medical professionals become increasingly reliant on the many emerging studies available to them, we sometimes forget that communication is key to optimal medical care, an accurate diagnosis, and patient safety.
Continue to: In addition, a second review...
In addition, a second review of dermatopathologic slides may be warranted if the pathologic diagnosis is inconsistent with the clinical picture or if the diagnosed condition is resistant to the usual therapies of choice. Incorrect diagnoses are more likely to occur when tests are interpreted in a vacuum without the corresponding clinical correlation. The weight of these mistakes is felt not only by the health care system, but by the patients themselves.
CORRESPONDENCE
Magdalena Pasarica, MD, PhD, University of Central Florida College of Medicine, 6850 Lake Nona Boulevard, Orlando, FL 32827; Magdalena.Pasarica@ucf.edu
1. Kohn LT, Corrigan JM, Donaldson MS. To Err is Human: Building a Safer Health System. Washington, DC: National Academies Press; 2000.
2. U.S. Department of Health and Human Services. Patient safety primer: root cause analysis. https://psnet.ahrq.gov/primers/primer/10/root-cause-analysis. Updated August 2018. Accessed September 27, 2018.
3. Newman-Toker DE, Pronovost PJ. Diagnostic errors-the next frontier for patient safety. JAMA. 2009;301:1060-1062.
4. Kuijpers CC, Burger G, Al-Janabi S, et al. Improved quality of patient care through routine second review of histopathology specimens prior to multidisciplinary meetings. J Clin Pathol. 2016;69:866-871.
5. Leonard M, Graham S, Bonacum D. The human factor: the critical importance of effective teamwork and communication in providing safe care. Qual Saf Health Care. 2004;13:85-90.
6. Robinson NL. Promoting patient safety with perioperative hand-off communication. J Perianesth Nurs. 2016;31:245-253.
1. Kohn LT, Corrigan JM, Donaldson MS. To Err is Human: Building a Safer Health System. Washington, DC: National Academies Press; 2000.
2. U.S. Department of Health and Human Services. Patient safety primer: root cause analysis. https://psnet.ahrq.gov/primers/primer/10/root-cause-analysis. Updated August 2018. Accessed September 27, 2018.
3. Newman-Toker DE, Pronovost PJ. Diagnostic errors-the next frontier for patient safety. JAMA. 2009;301:1060-1062.
4. Kuijpers CC, Burger G, Al-Janabi S, et al. Improved quality of patient care through routine second review of histopathology specimens prior to multidisciplinary meetings. J Clin Pathol. 2016;69:866-871.
5. Leonard M, Graham S, Bonacum D. The human factor: the critical importance of effective teamwork and communication in providing safe care. Qual Saf Health Care. 2004;13:85-90.
6. Robinson NL. Promoting patient safety with perioperative hand-off communication. J Perianesth Nurs. 2016;31:245-253.

Prolonged survival in adenocarcinoma of unknown primary treated with chemoradiotherapy
Cancer of unknown primary (CUP) represents 3% to 5% of all cancer malignancies in the world.1 Since 2003, CUP has been divided into 2 subsets – favorable (20% of the cases) and unfavorable (80% of the cases) – based on histopathologic and clinical manifestations.2 The impact of locoregional therapies, such as surgery and radiation, in addition to systemic chemotherapy in adenocarcinomas of unknown primary is not well described in the literature.
Case presentation and summary
The patient was frustrated by the lack of diagnosis and extensive work-up and decided to travel to Bangladesh for several months. Upon her return in May 2015, the patient underwent dilation and curettage at an outside tertiary care center because of her persistently elevated beta-hCG levels (>500 mIU/mL; reference range for nonpregnant woman, <5 mIU/mL) that found no products of conception and excluded a malignant process. Endoscopy and colonoscopy at that time failed to reveal a primary tumor.
She was then referred to our institution. Her level of beta-hCG remained elevated, and another transvaginal ultrasound was performed but failed to reveal any masses or evidence of pregnancy. Mammogram and a breast ultrasound showed left breast lesions. Biopsy of the breast lesions was performed, and the pathology demonstrated fibrocystic changes.
The results of a PET-CT scan in August 2015 showed a lobulated abdominal mass of 5.7 x 3.7 cm, consisting of multiple periportal necrotic lymph nodes with a standardized uptake value (SUV) of 14 (Figure 1A) and a 2.0-cm hypermetabolic retroperitoneal lymph node at the aortic bifurcation level with an SUV of 8.6. The SUV is a ratio of activity per unit volume of a region of interest to the activity per unit whole body volume. An SUV of 2.5 or higher is generally considered to be indicative of malignant tissue. We conducted a detailed review of the lymph node pathologic specimen. Immunohistochemical (IHC) studies were positive for CK7, CDX2, and EMA; focally positive for PR and mammaglobin; and negative for CK20, ER, TTF-1, and WT-1. Nonspecific staining was seen with BRST2, and there was no staining with GATA3. IHC stain for HER2-NEU was equivocal. Molecular analysis did not detect BRAF, KRAS, NRAS, and PIK3CA mutations, but did find a CTNNB1 mutation. The IHC pattern suggested pancreatobiliary origin of the tumor.3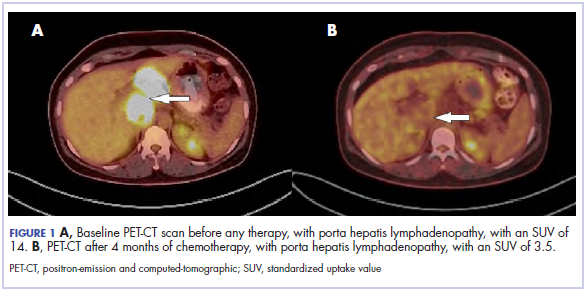
Although serum tumor marker pattern of elevated beta-hCG, AFP, and LDH can be seen in germ cell tumors, the pathology evaluation did not favor a germ cell tumor. No site of origin was evident on radiographic evaluation, and the patient was diagnosed with CUP. Based on tumor metastatic distribution and the elevated beta-hCG level,4 we suspected that an undetected pancreatic primary was possible, and we therefore chose the folinic acid, fluorouracil, irinotecan, oxaliplatin (FOLFIRINOX) chemotherapy regimen for its evidence in prolonging survival in metastatic pancreatic cancer.5 At the initiation of treatment, the patient’s elevated tumor markers were beta-hCG 953.6 mIU/mL (reference for nonpregnant woman, <5 mIU/mL) and AFP 1,800.7 ng/mL (reference range, 0.0-9.0 ng/mL). The patient began FOLFIRINOX chemotherapy in August 2015 and after 1 month of treatment, her beta-hCG and AFP levels declined notably to 1.7 mIU/mL and 11.2 ng/mL, respectively. She completed a total of 8 cycles of FOLFIRINOX in November 2015. After completion of chemotherapy, the PET-CT scan showed a decrease in fluoro-D-glucose (FDG) uptake in the porta hepatis and retroperitoneal lymph nodes (Figure 1B). SUV in the porta hepatis lymph nodes declined from 14 to 3.5. The patient’s case was presented to our institution’s multidisciplinary tumor board, and the members deemed the risk of possible lymph node dissection surgery would outweigh the benefit. It was recommended that we proceed with radiotherapy to the residual lymph node stations.
During December 2015 through February 2016, the patient underwent a course of consolidative chemoradiation therapy to the intra-abdominal lymph nodes to a dose of 5,400 cGy in 30 fractions, with concurrent capecitabine as radiosensitizer, using intensity-modulated radiation therapy. During both chemotherapy and CRT, the patient experienced nausea, vomiting, fatigue, and anorexia, which were treated with antiemetics. She completed therapy without major complications and recovered completely from the adverse effects.
Five weeks after completion of chemoradiation, a restaging PET-CT scan showed a persistent small FDG uptake in the periportal region (SUV, 4.2). After CRT, tumor markers beta-hCG and AFP declined to less than 1.2 mIU/mL and less than 2.0 ng/mL, respectively.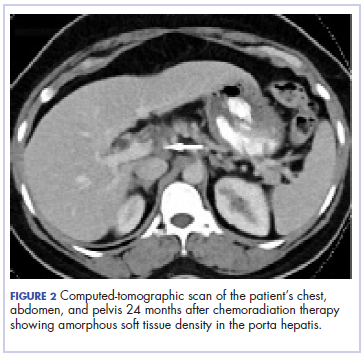
Discussion
CUP is divided into favorable and unfavorable subsets.1 The favorable subset includes women with adenocarcinoma involving axillary lymph nodes, women with papillary adenocarcinoma of peritoneal cavity, and adenocarcinoma with a colon profile. The unfavorable subset includes moderate to poorly differentiated adenocarcinomas (64%) and undifferentiated tumors (36%). It involves the liver in 40% to 50% of the cases, followed by lymph nodes (35%), lungs (31%), bones (28%), and the brain (15%).1,2,6 Although data suggest that CUP with lymph-node–only metastases generally fall into an unfavorable prognosis group, our patient’s survival and progression-free survival have been especially prolonged.
The combined platinum–paclitaxel-based regimens are the treatment of choice in this unfavorable subset of CUP,7,8 with patients showing 16% to 38% response rates and median overall survival times of 6.5 to 13 months.7 Platinum–gemcitabine combinations can also be used as an alternative first-line regimen, with an overall response rate of 55% and a median survival of 8 months.9 The addition of the targeted agents bevacizumab and erlotinib to the carboplatin–paclitaxel combination, followed by bevacizumab and erlotinib maintenance, has been shown to yield a median survival of 12.6 months but was not meaningfully superior to historical studies with chemotherapy alone.10
We chose the FOLFIRINOX regimen for our patient. Conroy and colleagues reported a notably improved survival of 11.1 months with that combination chemotherapy in patients with metastatic pancreatic cancer compared with 6.8 months with gemcitabine alone.5 Given the possible pancreatobiliary site of tumor origin on IHC, the lymph node pattern of spread, and the patient’s young age and robust performance status, we felt that this multiagent systemic therapy would offer the best chance of prolonged survival. FOLFIRINOX includes a platinum agent, oxaliplatin, and platinum agents are recommended to be included in chemotherapy combinations for CUP.9,10 Although there is no data to suggest the superiority of a triplet regimen over a doublet regimen in a CUP, a triplet chemotherapy regimen may be considered in select cases.
There have been only a few reports showing the effectiveness of radiotherapy in the treatment of adenocarcinomas of unknown primary outside of the head and neck. Kubisch and colleagues have reported a case of a woman with hepatic adenocarcinoma of unknown primary that was treated with chemotherapy and surgery. Upon recurrence, the patient was then treated with selective internal radiation therapy (SIRT). She was still alive 3 years after diagnosis, and there had been no tumor relapse 21 months after SIRT.11 Shiota and colleagues have reported a case of a mediastinal lymph node CUP that was treated with docetaxel and cisplatin with concurrent thoracic radiation therapy.12 The patient remained free of symptoms without regrowth of the primary site 22 months after disease onset, and exploration of the body with enhanced and PET-CT scan showed no further abnormalities.
Other reports suggest that locoregional therapy such as surgery and radiation may be of benefit to select patients with CUP. A retrospective study by Löffler and colleagues reported that patients with a limited local involvement who received radical surgery had a median overall survival of 52.7 months compared with those who received radiation (median overall survival, 19.4 months) and those who received chemotherapy alone (median overall survival, 16 months).13 A case of a metastatic undifferentiated CUP also reported a long-term (>5 years), disease-free survivor after pancreaticoduodenectomy and systemic adjuvant chemotherapy.14
Our case further demonstrates that a multidisciplinary approach to CUP may lead to excellent clinical outcomes. Chemotherapy followed by chemoradiation in our patient increased local tumor control and survival.
Adenocarcinomas of unknown primary cases should involve management by a multidisciplinary team. Clinical trials incorporating locoregional therapies for CUP in addition to systemic therapy are warranted.
1. Pavlidis N, Khaled H, Gaafar R. A mini review on cancer of unknown primary site: a clinical puzzle for the oncologists. J Adv Res. 2015;6(3):375-382.
2. Pavlidis N, Briasoulis E, Hainsworth J, Greco FA. Diagnostic and therapeutic management of cancer of an unknown primary. Eur J Cancer. 2003;39(14):1990-2005.
3. Oien KA. Pathologic evaluation of unknown primary cancer. Semin Oncol. 2009;36(1):8-37.
4. Louhimo J, Alfthan H, Stenman UH, Hagland C. Serum HCG beta and CA 72-4 are stronger prognostic factors than CEA, CA 19-9 and CA 242 in pancreatic cancer. Oncology. 2004;66(2):126-131.
5. Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817-1825.
6. Pavlidis N, Pentheroudakis G. Cancer of unknown primary site. Lancet. 2012;379:1428-1435.
7. Bochtler T, Löffler H, Krämer A. Diagnosis and management of metastatic neoplasms with unknown primary. Semin Diagn Pathol. 2017;35(3):199-206.
8. Amela EY, Lauridant-Philippin G, Cousin S, Ryckewaert T, Adenis A, Penel N. Management of 'unfavourable' carcinoma of unknown primary site: synthesis of recent literature. Crit Rev Oncol Hematol. 2012;84(2):213-223.
9. Culine S, Lortholary A, Voigt J-J, et al. Cisplatin in combination with either gemcitabine or irinotecan in carcinomas of unknown primary site: results of a randomized phase II study--trial for the French study group on carcinomas of unknown primary (GEFCAPI 01). J Clin Oncol. 2003;21(18):3479-3482.
10. Hainsworth JD, Spigel DR, Thompson DS, et al. Paclitaxel/carboplatin plus bevacizumab/erlotinib in the first-line treatment of patients with carcinoma of unknown primary site. Oncologist. 2009;14(12):1189-1197.
11. Kubisch CH, Beigel F, Ihrler S, Goke B, Reiser MF, Hoffmann RT. Oesophageal ulceration after selective internal radiation therapy in a patient with carcinoma of unknown primary. Z Gastroenterol. 2010;48(5):546-550.
12. Shiota Y, Imai S, Sasaki N, et al. A case of mediastinal lymph node carcinoma of unknown primary site treated with docetaxel and cisplatin with concurrent thoracic radiation therapy. Acta Med Okayama. 2011;65(6):407-411.
13. Löffler H, Puthenparambil J, Hielscher T, Neben K, Krämer A. Patients with cancer of unknown primary: a retrospective analysis of 223 patients with adenocarcinoma or undifferentiated carcinoma. Dtsch Arztebl Int. 111(27-28):481-487.
14. Nakagawa Y, Todoroki T, Morishita Y, et al. A long-term survivor after pancreaticoduodenectomy for metastatic undifferentiated carcinoma of an unknown primary. Hepatogastroenterology. 2008;55(86-87):1557-1561.
15. Rodríguez-López JL, Toro-Bahamonde AM, Santiago-Méndez RJ, González-Cancel IF, Vélez-Cortés HA. An unusual case of colorectal adenocarcinoma presenting as an anterior mediastinal mass. Clin Colorectal Cancer. 2018;17(1):e115-e119.
Cancer of unknown primary (CUP) represents 3% to 5% of all cancer malignancies in the world.1 Since 2003, CUP has been divided into 2 subsets – favorable (20% of the cases) and unfavorable (80% of the cases) – based on histopathologic and clinical manifestations.2 The impact of locoregional therapies, such as surgery and radiation, in addition to systemic chemotherapy in adenocarcinomas of unknown primary is not well described in the literature.
Case presentation and summary
The patient was frustrated by the lack of diagnosis and extensive work-up and decided to travel to Bangladesh for several months. Upon her return in May 2015, the patient underwent dilation and curettage at an outside tertiary care center because of her persistently elevated beta-hCG levels (>500 mIU/mL; reference range for nonpregnant woman, <5 mIU/mL) that found no products of conception and excluded a malignant process. Endoscopy and colonoscopy at that time failed to reveal a primary tumor.
She was then referred to our institution. Her level of beta-hCG remained elevated, and another transvaginal ultrasound was performed but failed to reveal any masses or evidence of pregnancy. Mammogram and a breast ultrasound showed left breast lesions. Biopsy of the breast lesions was performed, and the pathology demonstrated fibrocystic changes.
The results of a PET-CT scan in August 2015 showed a lobulated abdominal mass of 5.7 x 3.7 cm, consisting of multiple periportal necrotic lymph nodes with a standardized uptake value (SUV) of 14 (Figure 1A) and a 2.0-cm hypermetabolic retroperitoneal lymph node at the aortic bifurcation level with an SUV of 8.6. The SUV is a ratio of activity per unit volume of a region of interest to the activity per unit whole body volume. An SUV of 2.5 or higher is generally considered to be indicative of malignant tissue. We conducted a detailed review of the lymph node pathologic specimen. Immunohistochemical (IHC) studies were positive for CK7, CDX2, and EMA; focally positive for PR and mammaglobin; and negative for CK20, ER, TTF-1, and WT-1. Nonspecific staining was seen with BRST2, and there was no staining with GATA3. IHC stain for HER2-NEU was equivocal. Molecular analysis did not detect BRAF, KRAS, NRAS, and PIK3CA mutations, but did find a CTNNB1 mutation. The IHC pattern suggested pancreatobiliary origin of the tumor.3
Although serum tumor marker pattern of elevated beta-hCG, AFP, and LDH can be seen in germ cell tumors, the pathology evaluation did not favor a germ cell tumor. No site of origin was evident on radiographic evaluation, and the patient was diagnosed with CUP. Based on tumor metastatic distribution and the elevated beta-hCG level,4 we suspected that an undetected pancreatic primary was possible, and we therefore chose the folinic acid, fluorouracil, irinotecan, oxaliplatin (FOLFIRINOX) chemotherapy regimen for its evidence in prolonging survival in metastatic pancreatic cancer.5 At the initiation of treatment, the patient’s elevated tumor markers were beta-hCG 953.6 mIU/mL (reference for nonpregnant woman, <5 mIU/mL) and AFP 1,800.7 ng/mL (reference range, 0.0-9.0 ng/mL). The patient began FOLFIRINOX chemotherapy in August 2015 and after 1 month of treatment, her beta-hCG and AFP levels declined notably to 1.7 mIU/mL and 11.2 ng/mL, respectively. She completed a total of 8 cycles of FOLFIRINOX in November 2015. After completion of chemotherapy, the PET-CT scan showed a decrease in fluoro-D-glucose (FDG) uptake in the porta hepatis and retroperitoneal lymph nodes (Figure 1B). SUV in the porta hepatis lymph nodes declined from 14 to 3.5. The patient’s case was presented to our institution’s multidisciplinary tumor board, and the members deemed the risk of possible lymph node dissection surgery would outweigh the benefit. It was recommended that we proceed with radiotherapy to the residual lymph node stations.
During December 2015 through February 2016, the patient underwent a course of consolidative chemoradiation therapy to the intra-abdominal lymph nodes to a dose of 5,400 cGy in 30 fractions, with concurrent capecitabine as radiosensitizer, using intensity-modulated radiation therapy. During both chemotherapy and CRT, the patient experienced nausea, vomiting, fatigue, and anorexia, which were treated with antiemetics. She completed therapy without major complications and recovered completely from the adverse effects.
Five weeks after completion of chemoradiation, a restaging PET-CT scan showed a persistent small FDG uptake in the periportal region (SUV, 4.2). After CRT, tumor markers beta-hCG and AFP declined to less than 1.2 mIU/mL and less than 2.0 ng/mL, respectively.
Discussion
CUP is divided into favorable and unfavorable subsets.1 The favorable subset includes women with adenocarcinoma involving axillary lymph nodes, women with papillary adenocarcinoma of peritoneal cavity, and adenocarcinoma with a colon profile. The unfavorable subset includes moderate to poorly differentiated adenocarcinomas (64%) and undifferentiated tumors (36%). It involves the liver in 40% to 50% of the cases, followed by lymph nodes (35%), lungs (31%), bones (28%), and the brain (15%).1,2,6 Although data suggest that CUP with lymph-node–only metastases generally fall into an unfavorable prognosis group, our patient’s survival and progression-free survival have been especially prolonged.
The combined platinum–paclitaxel-based regimens are the treatment of choice in this unfavorable subset of CUP,7,8 with patients showing 16% to 38% response rates and median overall survival times of 6.5 to 13 months.7 Platinum–gemcitabine combinations can also be used as an alternative first-line regimen, with an overall response rate of 55% and a median survival of 8 months.9 The addition of the targeted agents bevacizumab and erlotinib to the carboplatin–paclitaxel combination, followed by bevacizumab and erlotinib maintenance, has been shown to yield a median survival of 12.6 months but was not meaningfully superior to historical studies with chemotherapy alone.10
We chose the FOLFIRINOX regimen for our patient. Conroy and colleagues reported a notably improved survival of 11.1 months with that combination chemotherapy in patients with metastatic pancreatic cancer compared with 6.8 months with gemcitabine alone.5 Given the possible pancreatobiliary site of tumor origin on IHC, the lymph node pattern of spread, and the patient’s young age and robust performance status, we felt that this multiagent systemic therapy would offer the best chance of prolonged survival. FOLFIRINOX includes a platinum agent, oxaliplatin, and platinum agents are recommended to be included in chemotherapy combinations for CUP.9,10 Although there is no data to suggest the superiority of a triplet regimen over a doublet regimen in a CUP, a triplet chemotherapy regimen may be considered in select cases.
There have been only a few reports showing the effectiveness of radiotherapy in the treatment of adenocarcinomas of unknown primary outside of the head and neck. Kubisch and colleagues have reported a case of a woman with hepatic adenocarcinoma of unknown primary that was treated with chemotherapy and surgery. Upon recurrence, the patient was then treated with selective internal radiation therapy (SIRT). She was still alive 3 years after diagnosis, and there had been no tumor relapse 21 months after SIRT.11 Shiota and colleagues have reported a case of a mediastinal lymph node CUP that was treated with docetaxel and cisplatin with concurrent thoracic radiation therapy.12 The patient remained free of symptoms without regrowth of the primary site 22 months after disease onset, and exploration of the body with enhanced and PET-CT scan showed no further abnormalities.
Other reports suggest that locoregional therapy such as surgery and radiation may be of benefit to select patients with CUP. A retrospective study by Löffler and colleagues reported that patients with a limited local involvement who received radical surgery had a median overall survival of 52.7 months compared with those who received radiation (median overall survival, 19.4 months) and those who received chemotherapy alone (median overall survival, 16 months).13 A case of a metastatic undifferentiated CUP also reported a long-term (>5 years), disease-free survivor after pancreaticoduodenectomy and systemic adjuvant chemotherapy.14
Our case further demonstrates that a multidisciplinary approach to CUP may lead to excellent clinical outcomes. Chemotherapy followed by chemoradiation in our patient increased local tumor control and survival.
Adenocarcinomas of unknown primary cases should involve management by a multidisciplinary team. Clinical trials incorporating locoregional therapies for CUP in addition to systemic therapy are warranted.
Cancer of unknown primary (CUP) represents 3% to 5% of all cancer malignancies in the world.1 Since 2003, CUP has been divided into 2 subsets – favorable (20% of the cases) and unfavorable (80% of the cases) – based on histopathologic and clinical manifestations.2 The impact of locoregional therapies, such as surgery and radiation, in addition to systemic chemotherapy in adenocarcinomas of unknown primary is not well described in the literature.
Case presentation and summary
The patient was frustrated by the lack of diagnosis and extensive work-up and decided to travel to Bangladesh for several months. Upon her return in May 2015, the patient underwent dilation and curettage at an outside tertiary care center because of her persistently elevated beta-hCG levels (>500 mIU/mL; reference range for nonpregnant woman, <5 mIU/mL) that found no products of conception and excluded a malignant process. Endoscopy and colonoscopy at that time failed to reveal a primary tumor.
She was then referred to our institution. Her level of beta-hCG remained elevated, and another transvaginal ultrasound was performed but failed to reveal any masses or evidence of pregnancy. Mammogram and a breast ultrasound showed left breast lesions. Biopsy of the breast lesions was performed, and the pathology demonstrated fibrocystic changes.
The results of a PET-CT scan in August 2015 showed a lobulated abdominal mass of 5.7 x 3.7 cm, consisting of multiple periportal necrotic lymph nodes with a standardized uptake value (SUV) of 14 (Figure 1A) and a 2.0-cm hypermetabolic retroperitoneal lymph node at the aortic bifurcation level with an SUV of 8.6. The SUV is a ratio of activity per unit volume of a region of interest to the activity per unit whole body volume. An SUV of 2.5 or higher is generally considered to be indicative of malignant tissue. We conducted a detailed review of the lymph node pathologic specimen. Immunohistochemical (IHC) studies were positive for CK7, CDX2, and EMA; focally positive for PR and mammaglobin; and negative for CK20, ER, TTF-1, and WT-1. Nonspecific staining was seen with BRST2, and there was no staining with GATA3. IHC stain for HER2-NEU was equivocal. Molecular analysis did not detect BRAF, KRAS, NRAS, and PIK3CA mutations, but did find a CTNNB1 mutation. The IHC pattern suggested pancreatobiliary origin of the tumor.3
Although serum tumor marker pattern of elevated beta-hCG, AFP, and LDH can be seen in germ cell tumors, the pathology evaluation did not favor a germ cell tumor. No site of origin was evident on radiographic evaluation, and the patient was diagnosed with CUP. Based on tumor metastatic distribution and the elevated beta-hCG level,4 we suspected that an undetected pancreatic primary was possible, and we therefore chose the folinic acid, fluorouracil, irinotecan, oxaliplatin (FOLFIRINOX) chemotherapy regimen for its evidence in prolonging survival in metastatic pancreatic cancer.5 At the initiation of treatment, the patient’s elevated tumor markers were beta-hCG 953.6 mIU/mL (reference for nonpregnant woman, <5 mIU/mL) and AFP 1,800.7 ng/mL (reference range, 0.0-9.0 ng/mL). The patient began FOLFIRINOX chemotherapy in August 2015 and after 1 month of treatment, her beta-hCG and AFP levels declined notably to 1.7 mIU/mL and 11.2 ng/mL, respectively. She completed a total of 8 cycles of FOLFIRINOX in November 2015. After completion of chemotherapy, the PET-CT scan showed a decrease in fluoro-D-glucose (FDG) uptake in the porta hepatis and retroperitoneal lymph nodes (Figure 1B). SUV in the porta hepatis lymph nodes declined from 14 to 3.5. The patient’s case was presented to our institution’s multidisciplinary tumor board, and the members deemed the risk of possible lymph node dissection surgery would outweigh the benefit. It was recommended that we proceed with radiotherapy to the residual lymph node stations.
During December 2015 through February 2016, the patient underwent a course of consolidative chemoradiation therapy to the intra-abdominal lymph nodes to a dose of 5,400 cGy in 30 fractions, with concurrent capecitabine as radiosensitizer, using intensity-modulated radiation therapy. During both chemotherapy and CRT, the patient experienced nausea, vomiting, fatigue, and anorexia, which were treated with antiemetics. She completed therapy without major complications and recovered completely from the adverse effects.
Five weeks after completion of chemoradiation, a restaging PET-CT scan showed a persistent small FDG uptake in the periportal region (SUV, 4.2). After CRT, tumor markers beta-hCG and AFP declined to less than 1.2 mIU/mL and less than 2.0 ng/mL, respectively.
Discussion
CUP is divided into favorable and unfavorable subsets.1 The favorable subset includes women with adenocarcinoma involving axillary lymph nodes, women with papillary adenocarcinoma of peritoneal cavity, and adenocarcinoma with a colon profile. The unfavorable subset includes moderate to poorly differentiated adenocarcinomas (64%) and undifferentiated tumors (36%). It involves the liver in 40% to 50% of the cases, followed by lymph nodes (35%), lungs (31%), bones (28%), and the brain (15%).1,2,6 Although data suggest that CUP with lymph-node–only metastases generally fall into an unfavorable prognosis group, our patient’s survival and progression-free survival have been especially prolonged.
The combined platinum–paclitaxel-based regimens are the treatment of choice in this unfavorable subset of CUP,7,8 with patients showing 16% to 38% response rates and median overall survival times of 6.5 to 13 months.7 Platinum–gemcitabine combinations can also be used as an alternative first-line regimen, with an overall response rate of 55% and a median survival of 8 months.9 The addition of the targeted agents bevacizumab and erlotinib to the carboplatin–paclitaxel combination, followed by bevacizumab and erlotinib maintenance, has been shown to yield a median survival of 12.6 months but was not meaningfully superior to historical studies with chemotherapy alone.10
We chose the FOLFIRINOX regimen for our patient. Conroy and colleagues reported a notably improved survival of 11.1 months with that combination chemotherapy in patients with metastatic pancreatic cancer compared with 6.8 months with gemcitabine alone.5 Given the possible pancreatobiliary site of tumor origin on IHC, the lymph node pattern of spread, and the patient’s young age and robust performance status, we felt that this multiagent systemic therapy would offer the best chance of prolonged survival. FOLFIRINOX includes a platinum agent, oxaliplatin, and platinum agents are recommended to be included in chemotherapy combinations for CUP.9,10 Although there is no data to suggest the superiority of a triplet regimen over a doublet regimen in a CUP, a triplet chemotherapy regimen may be considered in select cases.
There have been only a few reports showing the effectiveness of radiotherapy in the treatment of adenocarcinomas of unknown primary outside of the head and neck. Kubisch and colleagues have reported a case of a woman with hepatic adenocarcinoma of unknown primary that was treated with chemotherapy and surgery. Upon recurrence, the patient was then treated with selective internal radiation therapy (SIRT). She was still alive 3 years after diagnosis, and there had been no tumor relapse 21 months after SIRT.11 Shiota and colleagues have reported a case of a mediastinal lymph node CUP that was treated with docetaxel and cisplatin with concurrent thoracic radiation therapy.12 The patient remained free of symptoms without regrowth of the primary site 22 months after disease onset, and exploration of the body with enhanced and PET-CT scan showed no further abnormalities.
Other reports suggest that locoregional therapy such as surgery and radiation may be of benefit to select patients with CUP. A retrospective study by Löffler and colleagues reported that patients with a limited local involvement who received radical surgery had a median overall survival of 52.7 months compared with those who received radiation (median overall survival, 19.4 months) and those who received chemotherapy alone (median overall survival, 16 months).13 A case of a metastatic undifferentiated CUP also reported a long-term (>5 years), disease-free survivor after pancreaticoduodenectomy and systemic adjuvant chemotherapy.14
Our case further demonstrates that a multidisciplinary approach to CUP may lead to excellent clinical outcomes. Chemotherapy followed by chemoradiation in our patient increased local tumor control and survival.
Adenocarcinomas of unknown primary cases should involve management by a multidisciplinary team. Clinical trials incorporating locoregional therapies for CUP in addition to systemic therapy are warranted.
1. Pavlidis N, Khaled H, Gaafar R. A mini review on cancer of unknown primary site: a clinical puzzle for the oncologists. J Adv Res. 2015;6(3):375-382.
2. Pavlidis N, Briasoulis E, Hainsworth J, Greco FA. Diagnostic and therapeutic management of cancer of an unknown primary. Eur J Cancer. 2003;39(14):1990-2005.
3. Oien KA. Pathologic evaluation of unknown primary cancer. Semin Oncol. 2009;36(1):8-37.
4. Louhimo J, Alfthan H, Stenman UH, Hagland C. Serum HCG beta and CA 72-4 are stronger prognostic factors than CEA, CA 19-9 and CA 242 in pancreatic cancer. Oncology. 2004;66(2):126-131.
5. Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817-1825.
6. Pavlidis N, Pentheroudakis G. Cancer of unknown primary site. Lancet. 2012;379:1428-1435.
7. Bochtler T, Löffler H, Krämer A. Diagnosis and management of metastatic neoplasms with unknown primary. Semin Diagn Pathol. 2017;35(3):199-206.
8. Amela EY, Lauridant-Philippin G, Cousin S, Ryckewaert T, Adenis A, Penel N. Management of 'unfavourable' carcinoma of unknown primary site: synthesis of recent literature. Crit Rev Oncol Hematol. 2012;84(2):213-223.
9. Culine S, Lortholary A, Voigt J-J, et al. Cisplatin in combination with either gemcitabine or irinotecan in carcinomas of unknown primary site: results of a randomized phase II study--trial for the French study group on carcinomas of unknown primary (GEFCAPI 01). J Clin Oncol. 2003;21(18):3479-3482.
10. Hainsworth JD, Spigel DR, Thompson DS, et al. Paclitaxel/carboplatin plus bevacizumab/erlotinib in the first-line treatment of patients with carcinoma of unknown primary site. Oncologist. 2009;14(12):1189-1197.
11. Kubisch CH, Beigel F, Ihrler S, Goke B, Reiser MF, Hoffmann RT. Oesophageal ulceration after selective internal radiation therapy in a patient with carcinoma of unknown primary. Z Gastroenterol. 2010;48(5):546-550.
12. Shiota Y, Imai S, Sasaki N, et al. A case of mediastinal lymph node carcinoma of unknown primary site treated with docetaxel and cisplatin with concurrent thoracic radiation therapy. Acta Med Okayama. 2011;65(6):407-411.
13. Löffler H, Puthenparambil J, Hielscher T, Neben K, Krämer A. Patients with cancer of unknown primary: a retrospective analysis of 223 patients with adenocarcinoma or undifferentiated carcinoma. Dtsch Arztebl Int. 111(27-28):481-487.
14. Nakagawa Y, Todoroki T, Morishita Y, et al. A long-term survivor after pancreaticoduodenectomy for metastatic undifferentiated carcinoma of an unknown primary. Hepatogastroenterology. 2008;55(86-87):1557-1561.
15. Rodríguez-López JL, Toro-Bahamonde AM, Santiago-Méndez RJ, González-Cancel IF, Vélez-Cortés HA. An unusual case of colorectal adenocarcinoma presenting as an anterior mediastinal mass. Clin Colorectal Cancer. 2018;17(1):e115-e119.
1. Pavlidis N, Khaled H, Gaafar R. A mini review on cancer of unknown primary site: a clinical puzzle for the oncologists. J Adv Res. 2015;6(3):375-382.
2. Pavlidis N, Briasoulis E, Hainsworth J, Greco FA. Diagnostic and therapeutic management of cancer of an unknown primary. Eur J Cancer. 2003;39(14):1990-2005.
3. Oien KA. Pathologic evaluation of unknown primary cancer. Semin Oncol. 2009;36(1):8-37.
4. Louhimo J, Alfthan H, Stenman UH, Hagland C. Serum HCG beta and CA 72-4 are stronger prognostic factors than CEA, CA 19-9 and CA 242 in pancreatic cancer. Oncology. 2004;66(2):126-131.
5. Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817-1825.
6. Pavlidis N, Pentheroudakis G. Cancer of unknown primary site. Lancet. 2012;379:1428-1435.
7. Bochtler T, Löffler H, Krämer A. Diagnosis and management of metastatic neoplasms with unknown primary. Semin Diagn Pathol. 2017;35(3):199-206.
8. Amela EY, Lauridant-Philippin G, Cousin S, Ryckewaert T, Adenis A, Penel N. Management of 'unfavourable' carcinoma of unknown primary site: synthesis of recent literature. Crit Rev Oncol Hematol. 2012;84(2):213-223.
9. Culine S, Lortholary A, Voigt J-J, et al. Cisplatin in combination with either gemcitabine or irinotecan in carcinomas of unknown primary site: results of a randomized phase II study--trial for the French study group on carcinomas of unknown primary (GEFCAPI 01). J Clin Oncol. 2003;21(18):3479-3482.
10. Hainsworth JD, Spigel DR, Thompson DS, et al. Paclitaxel/carboplatin plus bevacizumab/erlotinib in the first-line treatment of patients with carcinoma of unknown primary site. Oncologist. 2009;14(12):1189-1197.
11. Kubisch CH, Beigel F, Ihrler S, Goke B, Reiser MF, Hoffmann RT. Oesophageal ulceration after selective internal radiation therapy in a patient with carcinoma of unknown primary. Z Gastroenterol. 2010;48(5):546-550.
12. Shiota Y, Imai S, Sasaki N, et al. A case of mediastinal lymph node carcinoma of unknown primary site treated with docetaxel and cisplatin with concurrent thoracic radiation therapy. Acta Med Okayama. 2011;65(6):407-411.
13. Löffler H, Puthenparambil J, Hielscher T, Neben K, Krämer A. Patients with cancer of unknown primary: a retrospective analysis of 223 patients with adenocarcinoma or undifferentiated carcinoma. Dtsch Arztebl Int. 111(27-28):481-487.
14. Nakagawa Y, Todoroki T, Morishita Y, et al. A long-term survivor after pancreaticoduodenectomy for metastatic undifferentiated carcinoma of an unknown primary. Hepatogastroenterology. 2008;55(86-87):1557-1561.
15. Rodríguez-López JL, Toro-Bahamonde AM, Santiago-Méndez RJ, González-Cancel IF, Vélez-Cortés HA. An unusual case of colorectal adenocarcinoma presenting as an anterior mediastinal mass. Clin Colorectal Cancer. 2018;17(1):e115-e119.
An Imposter Twice Over: A Case of IgG4-Related Disease
Immunoglobulin G4-related disease (IgG4-RD) is an immune-mediated fibroinflammatory condition that involves multiple organs and appears as syndromes that were once thought to be unrelated. This disease leads to mass lesions, fibrosis, and subsequent organ failure if allowed to progress untreated.1 Involvement of gastrointestinal (GI) organs, salivary glands, lacrimal glands, lymph, prostate, pulmonary, and vascular system have all been reported.2 Elevated IgG4 serum levels are common, but about one-third of patients with biopsy-proven IgG4-RD do not manifest this characteristic.3,4
Diagnostic confirmation is with biopsy, and all patients with symptomatic, active IgG4-RD require treatment. Glucocorticoids are first-line treatment and are utilized for relapse of symptoms. In addition to glucocorticoids, steroid-sparing medications, including rituximab, azathioprine, mycophenolate mofetil, tacrolimus, and cyclophosphamide have all been used with successful remission.5,6 Here, the authors discuss a case of IgG4-RD that presented with intrahepatic biliary obstruction (mimicking cholangiocarcinoma) and subsequent development of coronary arteritis despite treatment.
Case Presentation
In June 2015, a 57-year-old Air Force veteran presented to Eglin AFB Hospital with pruritic jaundice and acute abdominal pain. He was found to have elevated bilirubin levels (total bilirubin 10 mg/dL [normal range 0.2-1.3 mg/dL], direct bilirubin 6.6 mg/dL [normal range 0.1-0.4 mg/dL]). Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) also were moderately elevated (147 U/L and 337 U/L, respectively). 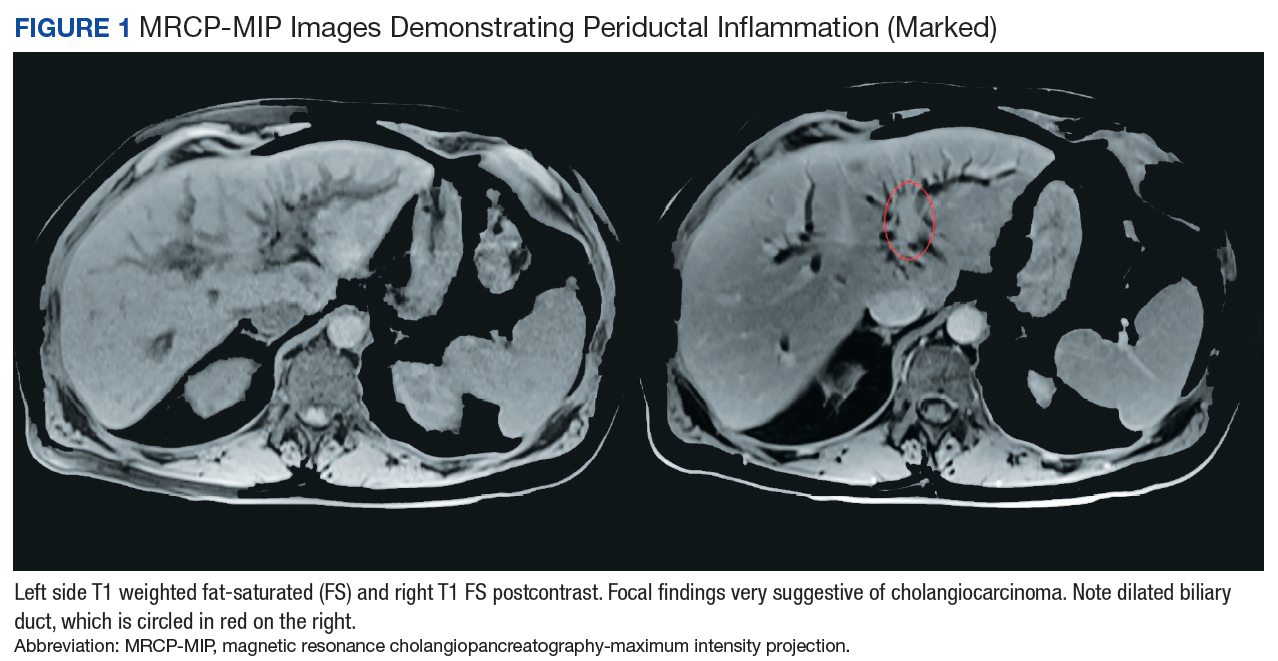
Prior to this presentation, the patient had been in his usual state of health. His past medical history was notable only for minimal change kidney disease (MCD). MCD is defined as effacement of the podocyte seen on electron microscopy, which allows the passage of large amounts of protein. 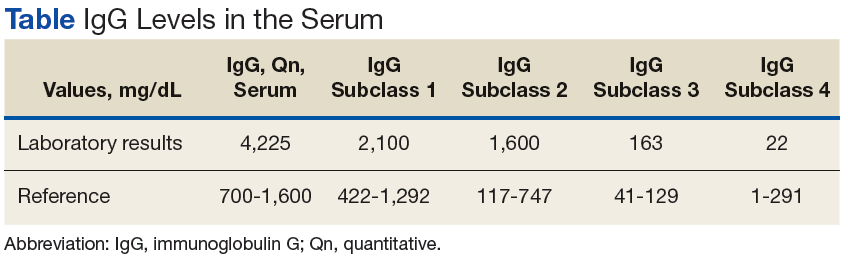
A cholangiogram showed abnormal filling into the left main intrahepatic duct and obvious obstruction at the bifurcation of the bile duct. A biliary drainage catheter was placed, and a repeat cholangiogram 2 days later showed involvement of both right and left intrahepatic ducts. The distal common bile duct appeared uninvolved as did the pancreas. Lymphadenopathy was noted at the liver hilum. Klatskin cholangiocarcinoma (type IIIB) was the presumed diagnosis. Based on these findings, tumor resection was performed 3 weeks later, including left hepatectomy, caudate lobe resection, complete bile duct resection, cholecystectomy, with reconstruction by Roux-en-Y intrahepaticojejunostomy. In addition, portal and hepatic artery lymph node dissection was completed.
Surgical specimens were sent for pathologic evaluation and were found negative for malignancy. Patchy areas of storiform fibrosis, obliterative phlebitis, and lymphoplasmacytic infiltrate were noted. IgG4 immunostain highlighted the presence of IgG4 positive plasma cells with a peak count of 145 IgG4 positive plasma cells/hpf. About 80% of the plasma cells were positive for IgG4. Unusually dense eosinophilic infiltrate with plasma cells and regions of dense fibrosis that strongly contributed to the masslike appearance on CT imaging also were noted. Final histology confirmed the diagnosis of IgG4-RD. Elevated levels of total IgG in the serum were observed without elevation in serum IgG4 (Table).
The patient was started on prednisone 40 mg and azathioprine 150 mg daily, with subsequent taper of prednisone over the next 6 months. After prednisone was discontinued, the patient reported new symptoms of lower extremity pain, neuropathy, and swelling of his face. Laboratory results were notable for elevated erythrocyte sedimentation rate. The patient was restarted on prednisone 40 mg daily. Azathioprine was replaced with a regimen of 4 doses every 6 months of IV rituximab 700 mg q week and mycophenolate mofetil (1,000 mg bid). After remission was induced, the patient was slowly weaned off prednisone again.
Following 6 months of successful discontinuation of prednisone and continued rituximab and mycophenolate mofetil therapy, the patient presented to the emergency department with new onset chest pain and shortness of breath. A CT angiography of the chest showed right upper and middle lobe infiltrate, and he was treated for community acquired pneumonia. Additionally, he was noted to have elevated troponin levels suggestive of myocardial infarction (MI). Initial troponin was 1.23 ng/mL (normal range < 0.015 ng/mL), which trended down over the next 18 hours. A bedside echocardiogram showed a normal left ventricular ejection fraction without wall motion abnormalities. Etiology for his acute MI was presumed to be demand ischemia from fixed atherosclerotic plaque. Further inpatient cardiac risk stratification was changed to the outpatient setting, and he was started on medical management for coronary artery disease with a beta blocker, a statin, and aspirin. He was discharged home on 10 mg prednisone daily, which was subsequently tapered over several weeks.
In follow-up, a Lexiscan myocardial perfusion imaging was conducted that demonstrated an inferolateral defect and associated wall motion abnormalities (Figures 2 and 3). 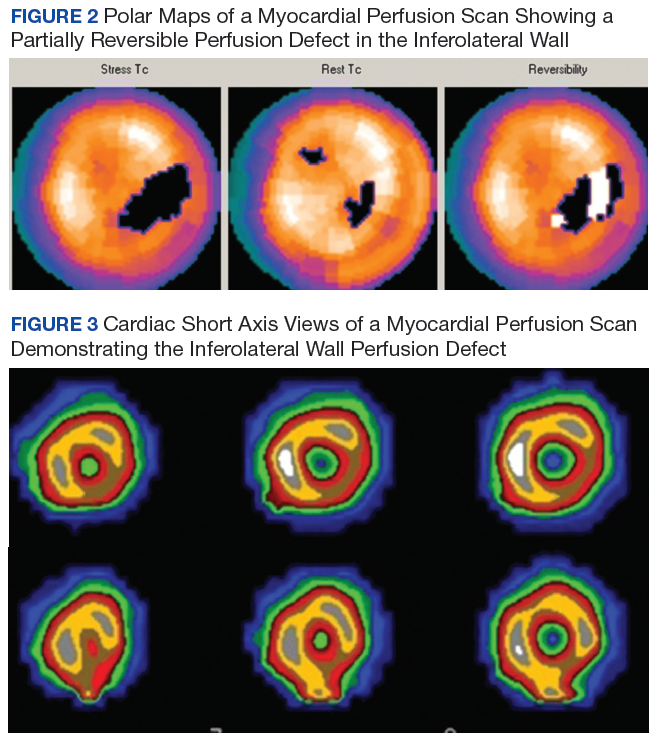
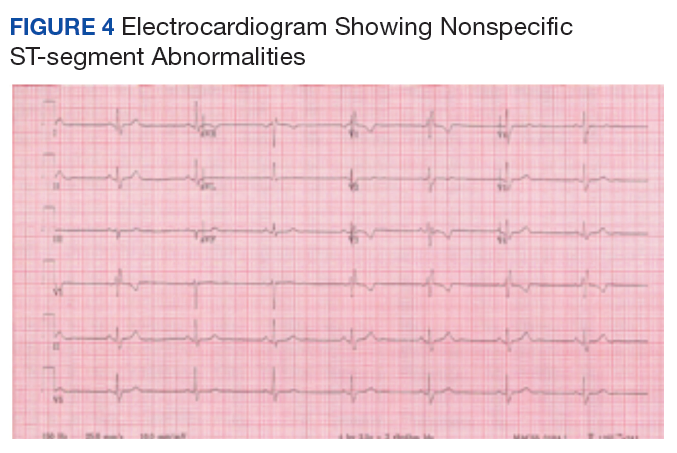
Discussion
IgG4-related disease has been found to be a systemic disorder. Typical characteristics include predominance in men aged > 50 years, elevated IgG4 levels, and findings on histology.1 It has been reported to involve many organs, including pancreas, liver, gallbladder, salivary glands, thyroid, and pleura of the lung.2,5
This case report begins with a presumptive diagnosis of cholangiocarcinoma, which was treated aggressively with extensive surgery. Several case reports of complex tumefactive lesions in the GI area (mostly pancreatic and biliary) have detailed IgG4-RD as both a risk factor for subsequent development of cholangiocarcinoma and as a separate entity of IgG4-related sclerosing cholangitis.7-9 It is hypothesized that the induction of IgG4- positive plasma cells has been intertwined with the development of cholangiocarcinoma. Differentiation between IgG4 reaction that is scattered around cancerous nests and IgG4 sclerosing cholangitis without malignancy is challenging. It has been documented that both elevated IgG4 levels and hilar hepatic lesions that resemble cholangiocarcinoma frequently accompany those cases of IgG4 sclerosing chlolangitis without pancreatic involvement.9 The histologic features of IgG4-RD need to be identified with multiple biopsies and cytology, and superficial biopsy from biliary mucosa cannot reliably exclude cholangiocarcinoma.
Lymphoplasmacytic aortitis and arteritis have been documented in IgG4-RD. In 2017, Barbu and colleagues described how one such case of coronary arteritis presented with typical angina and coronary catheterization revealing coronary artery stenosis.10 However, during coronary artery bypass surgery, the aorta and coronary vessels were noted to be abnormally stiff. A diffuse fibrotic tissue was identified to be causing the significant stenosis without evidence of atherosclerosis. Pathology showed typical findings of IgG4-RD, and there was a rapid response to immunosuppressive therapy. Involvement of coronary arteries has been described in a small number of cases at this time and is associated with progressive fibrotic changes resulting in an MI, aneurysms, and sudden cardiac death.2,10,11
IgG4-RD can be an extensively systemic disease. All presentations of fibrosis or vasculitis should be viewed with heightened suspicion in the future as being a facet of his IgG4-RD. Pleural involvement has been reported in 12% of cases presenting with systemic presentation, kidney involvement in 13%.2,12
Unfortunately, there is no standard laboratory parameter to date that is diagnostic for IgG4-RD. The gold standard remains confirmation of histologic findings with biopsy. According to an international consensus from 2015, 2 out of the 3 major findings need to be present: (1) dense lymphoplasmacytic infiltrate; (2) storiform fibrosis; and (3) obliterative phlebitis in veins and arteries.1,5 Most patients present with symptoms related to either tumefaction or fibrosis of an organ system.1 Peripheral eosinophilia and elevated serum IgE are often present in IgG4-RD.13 Although IgG4 values are elevated in 51% of biopsy-proven cases, flow cytometry of CD19lowCD38+CD20-CD27+ plasmablasts has been explored recently as a correlation with disease flare.3,14 These particular plasmablasts mark a stage between B cells and plasma cells and have been reported to have a sensitivity of 95% and a specificity of 82% in association with actual IgG4-RD.14 Furthermore, blood plasmablast concentrations decrease in response to glucocorticoid treatment, thereby providing a possible quantifiable value by which to measure success of IgG4 treatment.5,12
Treatment for this disease consists of immunosuppressive therapy. There is documentation of successful remission with rituximab and azathioprine, as well as methotrexate.1,5 Both 2015 consensus guidelines and a recent small single-center retrospective study support addition of second-line steroid sparing agents such as mycophenolate mofetil.5,6 For acute flairs, however, glucocorticoids with slow taper are usually utilized. In these cases, they should be tapered as soon as clinically feasible to avoid long-term adverse effects. Untreated IgG4-RD, even asymptomatic, has been shown to progress to fibrosis.5
Conclusion
IgG4-RD is a complicated disease process that requires a high index of suspicion to diagnose. In addition, for patients who are diagnosed with this condition, its ability to mimic other pathologic conditions should be taken into account with manifestation of any new illness. This case emphasizes the ability of this disease to localize in multiple organs over time and the need for lifetime surveillance in patients with IgG4-RD disease.
1. Lang D, Zwerina J, Pieringer H. IgG4-related disease: current challenges and future prospects. Ther Clin Risk Manag. 2016;12:189-199.
2. Brito-Zerón P, Ramos-Casals M, Bosch X, Stone JH. The clinical spectrum of IgG4-related disease. Autoimmun Rev. 2014;13(12):1203-1210.
3. Wallace ZS, Deshpande V, Mattoo H, et al. IgG4-related disease: clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheumatol. 2015;67(9):2466-2475.
4. Carruthers MN, Khosroshahi A, Augustin T, Deshpande V, Stone JH. The diagnostic utility of serum IgG4 concentrations in IgG4-RD. Ann Rheum Dis. 2015;74(1):14-18.
5. Khosroshahi A, Wallace ZS, Crowe JL, et al; Second International Symposium on IgG4-Related Disease. International consensus guidance statement on the management and treatment of IgG4-Related disease. Arthritis Rheumatol. 2015;67(7):1688-1699.
6. Gupta N, Mathew J, Mohan H, et al. Addition of second-line steroid sparing immunosuppressants like mycophenolate mofetil improves outcome of immunoglobulin G4-related disease (IgG4-RD): a series from a tertiary care teaching hospital in South India. Rheumatol Int. 2017;38(2):203-209.
7. Lin HP, Lin KT, Ho WC, Chen CB, Kuo, CY, Lin YC. IgG4-associated cholangitis mimicking cholangiocarcinoma-report of a case. J Intern Med Taiwan. 2013;24:137-141.
8. Douhara A, Mitoro A, Otani E, et al. Cholangiocarcinoma developed in a patient with IgG4-related disease. World J Gastrointest Oncol. 2013;5(8):181-185.
9. Harada K, Nakanuma Y. Cholangiocarcinoma with respect to IgG4 reaction. Int J Hepatol. 2014;2014:803876.
10. Barbu M, Lindström U, Nordborg C, Martinsson A, Dworeck C, Jeppsson A. Sclerosing aortic and coronary arteritis due to IgG4-related disease. Ann Thorac Surg. 2017;103(6):e487-e489.
11. Kim YJ, Park YS, Koo BS, et al. Immunoglobulin G4-related disease with lymphoplasmacytic aortitis mimicking Takayasu arteritis. J Clin Rheumatol. 2011;17(8):451-452.
12. Khosroshahi A, Digumarthy SR, Gibbons FK, Deshpande V. Case 34-2015: A 36-year-old woman with a lung mass, pleural effusion and hip pain. N Engl J Med. 2015;373(18):1762-1772.
13. Della Torre E, Mattoo H, Mahajan VS, Carruthers M, Pillai S, Stone JH. Prevalence of atopy, eosinophilia and IgE elevation in IgG4-related disease. Allergy. 2014;69(2):191-206.
14. Wallace ZS, Mattoo H, Carruthers M, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2015;74(1):190-195.
Immunoglobulin G4-related disease (IgG4-RD) is an immune-mediated fibroinflammatory condition that involves multiple organs and appears as syndromes that were once thought to be unrelated. This disease leads to mass lesions, fibrosis, and subsequent organ failure if allowed to progress untreated.1 Involvement of gastrointestinal (GI) organs, salivary glands, lacrimal glands, lymph, prostate, pulmonary, and vascular system have all been reported.2 Elevated IgG4 serum levels are common, but about one-third of patients with biopsy-proven IgG4-RD do not manifest this characteristic.3,4
Diagnostic confirmation is with biopsy, and all patients with symptomatic, active IgG4-RD require treatment. Glucocorticoids are first-line treatment and are utilized for relapse of symptoms. In addition to glucocorticoids, steroid-sparing medications, including rituximab, azathioprine, mycophenolate mofetil, tacrolimus, and cyclophosphamide have all been used with successful remission.5,6 Here, the authors discuss a case of IgG4-RD that presented with intrahepatic biliary obstruction (mimicking cholangiocarcinoma) and subsequent development of coronary arteritis despite treatment.
Case Presentation
In June 2015, a 57-year-old Air Force veteran presented to Eglin AFB Hospital with pruritic jaundice and acute abdominal pain. He was found to have elevated bilirubin levels (total bilirubin 10 mg/dL [normal range 0.2-1.3 mg/dL], direct bilirubin 6.6 mg/dL [normal range 0.1-0.4 mg/dL]). Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) also were moderately elevated (147 U/L and 337 U/L, respectively).
Prior to this presentation, the patient had been in his usual state of health. His past medical history was notable only for minimal change kidney disease (MCD). MCD is defined as effacement of the podocyte seen on electron microscopy, which allows the passage of large amounts of protein.
A cholangiogram showed abnormal filling into the left main intrahepatic duct and obvious obstruction at the bifurcation of the bile duct. A biliary drainage catheter was placed, and a repeat cholangiogram 2 days later showed involvement of both right and left intrahepatic ducts. The distal common bile duct appeared uninvolved as did the pancreas. Lymphadenopathy was noted at the liver hilum. Klatskin cholangiocarcinoma (type IIIB) was the presumed diagnosis. Based on these findings, tumor resection was performed 3 weeks later, including left hepatectomy, caudate lobe resection, complete bile duct resection, cholecystectomy, with reconstruction by Roux-en-Y intrahepaticojejunostomy. In addition, portal and hepatic artery lymph node dissection was completed.
Surgical specimens were sent for pathologic evaluation and were found negative for malignancy. Patchy areas of storiform fibrosis, obliterative phlebitis, and lymphoplasmacytic infiltrate were noted. IgG4 immunostain highlighted the presence of IgG4 positive plasma cells with a peak count of 145 IgG4 positive plasma cells/hpf. About 80% of the plasma cells were positive for IgG4. Unusually dense eosinophilic infiltrate with plasma cells and regions of dense fibrosis that strongly contributed to the masslike appearance on CT imaging also were noted. Final histology confirmed the diagnosis of IgG4-RD. Elevated levels of total IgG in the serum were observed without elevation in serum IgG4 (Table).
The patient was started on prednisone 40 mg and azathioprine 150 mg daily, with subsequent taper of prednisone over the next 6 months. After prednisone was discontinued, the patient reported new symptoms of lower extremity pain, neuropathy, and swelling of his face. Laboratory results were notable for elevated erythrocyte sedimentation rate. The patient was restarted on prednisone 40 mg daily. Azathioprine was replaced with a regimen of 4 doses every 6 months of IV rituximab 700 mg q week and mycophenolate mofetil (1,000 mg bid). After remission was induced, the patient was slowly weaned off prednisone again.
Following 6 months of successful discontinuation of prednisone and continued rituximab and mycophenolate mofetil therapy, the patient presented to the emergency department with new onset chest pain and shortness of breath. A CT angiography of the chest showed right upper and middle lobe infiltrate, and he was treated for community acquired pneumonia. Additionally, he was noted to have elevated troponin levels suggestive of myocardial infarction (MI). Initial troponin was 1.23 ng/mL (normal range < 0.015 ng/mL), which trended down over the next 18 hours. A bedside echocardiogram showed a normal left ventricular ejection fraction without wall motion abnormalities. Etiology for his acute MI was presumed to be demand ischemia from fixed atherosclerotic plaque. Further inpatient cardiac risk stratification was changed to the outpatient setting, and he was started on medical management for coronary artery disease with a beta blocker, a statin, and aspirin. He was discharged home on 10 mg prednisone daily, which was subsequently tapered over several weeks.
In follow-up, a Lexiscan myocardial perfusion imaging was conducted that demonstrated an inferolateral defect and associated wall motion abnormalities (Figures 2 and 3).
Discussion
IgG4-related disease has been found to be a systemic disorder. Typical characteristics include predominance in men aged > 50 years, elevated IgG4 levels, and findings on histology.1 It has been reported to involve many organs, including pancreas, liver, gallbladder, salivary glands, thyroid, and pleura of the lung.2,5
This case report begins with a presumptive diagnosis of cholangiocarcinoma, which was treated aggressively with extensive surgery. Several case reports of complex tumefactive lesions in the GI area (mostly pancreatic and biliary) have detailed IgG4-RD as both a risk factor for subsequent development of cholangiocarcinoma and as a separate entity of IgG4-related sclerosing cholangitis.7-9 It is hypothesized that the induction of IgG4- positive plasma cells has been intertwined with the development of cholangiocarcinoma. Differentiation between IgG4 reaction that is scattered around cancerous nests and IgG4 sclerosing cholangitis without malignancy is challenging. It has been documented that both elevated IgG4 levels and hilar hepatic lesions that resemble cholangiocarcinoma frequently accompany those cases of IgG4 sclerosing chlolangitis without pancreatic involvement.9 The histologic features of IgG4-RD need to be identified with multiple biopsies and cytology, and superficial biopsy from biliary mucosa cannot reliably exclude cholangiocarcinoma.
Lymphoplasmacytic aortitis and arteritis have been documented in IgG4-RD. In 2017, Barbu and colleagues described how one such case of coronary arteritis presented with typical angina and coronary catheterization revealing coronary artery stenosis.10 However, during coronary artery bypass surgery, the aorta and coronary vessels were noted to be abnormally stiff. A diffuse fibrotic tissue was identified to be causing the significant stenosis without evidence of atherosclerosis. Pathology showed typical findings of IgG4-RD, and there was a rapid response to immunosuppressive therapy. Involvement of coronary arteries has been described in a small number of cases at this time and is associated with progressive fibrotic changes resulting in an MI, aneurysms, and sudden cardiac death.2,10,11
IgG4-RD can be an extensively systemic disease. All presentations of fibrosis or vasculitis should be viewed with heightened suspicion in the future as being a facet of his IgG4-RD. Pleural involvement has been reported in 12% of cases presenting with systemic presentation, kidney involvement in 13%.2,12
Unfortunately, there is no standard laboratory parameter to date that is diagnostic for IgG4-RD. The gold standard remains confirmation of histologic findings with biopsy. According to an international consensus from 2015, 2 out of the 3 major findings need to be present: (1) dense lymphoplasmacytic infiltrate; (2) storiform fibrosis; and (3) obliterative phlebitis in veins and arteries.1,5 Most patients present with symptoms related to either tumefaction or fibrosis of an organ system.1 Peripheral eosinophilia and elevated serum IgE are often present in IgG4-RD.13 Although IgG4 values are elevated in 51% of biopsy-proven cases, flow cytometry of CD19lowCD38+CD20-CD27+ plasmablasts has been explored recently as a correlation with disease flare.3,14 These particular plasmablasts mark a stage between B cells and plasma cells and have been reported to have a sensitivity of 95% and a specificity of 82% in association with actual IgG4-RD.14 Furthermore, blood plasmablast concentrations decrease in response to glucocorticoid treatment, thereby providing a possible quantifiable value by which to measure success of IgG4 treatment.5,12
Treatment for this disease consists of immunosuppressive therapy. There is documentation of successful remission with rituximab and azathioprine, as well as methotrexate.1,5 Both 2015 consensus guidelines and a recent small single-center retrospective study support addition of second-line steroid sparing agents such as mycophenolate mofetil.5,6 For acute flairs, however, glucocorticoids with slow taper are usually utilized. In these cases, they should be tapered as soon as clinically feasible to avoid long-term adverse effects. Untreated IgG4-RD, even asymptomatic, has been shown to progress to fibrosis.5
Conclusion
IgG4-RD is a complicated disease process that requires a high index of suspicion to diagnose. In addition, for patients who are diagnosed with this condition, its ability to mimic other pathologic conditions should be taken into account with manifestation of any new illness. This case emphasizes the ability of this disease to localize in multiple organs over time and the need for lifetime surveillance in patients with IgG4-RD disease.
Immunoglobulin G4-related disease (IgG4-RD) is an immune-mediated fibroinflammatory condition that involves multiple organs and appears as syndromes that were once thought to be unrelated. This disease leads to mass lesions, fibrosis, and subsequent organ failure if allowed to progress untreated.1 Involvement of gastrointestinal (GI) organs, salivary glands, lacrimal glands, lymph, prostate, pulmonary, and vascular system have all been reported.2 Elevated IgG4 serum levels are common, but about one-third of patients with biopsy-proven IgG4-RD do not manifest this characteristic.3,4
Diagnostic confirmation is with biopsy, and all patients with symptomatic, active IgG4-RD require treatment. Glucocorticoids are first-line treatment and are utilized for relapse of symptoms. In addition to glucocorticoids, steroid-sparing medications, including rituximab, azathioprine, mycophenolate mofetil, tacrolimus, and cyclophosphamide have all been used with successful remission.5,6 Here, the authors discuss a case of IgG4-RD that presented with intrahepatic biliary obstruction (mimicking cholangiocarcinoma) and subsequent development of coronary arteritis despite treatment.
Case Presentation
In June 2015, a 57-year-old Air Force veteran presented to Eglin AFB Hospital with pruritic jaundice and acute abdominal pain. He was found to have elevated bilirubin levels (total bilirubin 10 mg/dL [normal range 0.2-1.3 mg/dL], direct bilirubin 6.6 mg/dL [normal range 0.1-0.4 mg/dL]). Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) also were moderately elevated (147 U/L and 337 U/L, respectively).
Prior to this presentation, the patient had been in his usual state of health. His past medical history was notable only for minimal change kidney disease (MCD). MCD is defined as effacement of the podocyte seen on electron microscopy, which allows the passage of large amounts of protein.
A cholangiogram showed abnormal filling into the left main intrahepatic duct and obvious obstruction at the bifurcation of the bile duct. A biliary drainage catheter was placed, and a repeat cholangiogram 2 days later showed involvement of both right and left intrahepatic ducts. The distal common bile duct appeared uninvolved as did the pancreas. Lymphadenopathy was noted at the liver hilum. Klatskin cholangiocarcinoma (type IIIB) was the presumed diagnosis. Based on these findings, tumor resection was performed 3 weeks later, including left hepatectomy, caudate lobe resection, complete bile duct resection, cholecystectomy, with reconstruction by Roux-en-Y intrahepaticojejunostomy. In addition, portal and hepatic artery lymph node dissection was completed.
Surgical specimens were sent for pathologic evaluation and were found negative for malignancy. Patchy areas of storiform fibrosis, obliterative phlebitis, and lymphoplasmacytic infiltrate were noted. IgG4 immunostain highlighted the presence of IgG4 positive plasma cells with a peak count of 145 IgG4 positive plasma cells/hpf. About 80% of the plasma cells were positive for IgG4. Unusually dense eosinophilic infiltrate with plasma cells and regions of dense fibrosis that strongly contributed to the masslike appearance on CT imaging also were noted. Final histology confirmed the diagnosis of IgG4-RD. Elevated levels of total IgG in the serum were observed without elevation in serum IgG4 (Table).
The patient was started on prednisone 40 mg and azathioprine 150 mg daily, with subsequent taper of prednisone over the next 6 months. After prednisone was discontinued, the patient reported new symptoms of lower extremity pain, neuropathy, and swelling of his face. Laboratory results were notable for elevated erythrocyte sedimentation rate. The patient was restarted on prednisone 40 mg daily. Azathioprine was replaced with a regimen of 4 doses every 6 months of IV rituximab 700 mg q week and mycophenolate mofetil (1,000 mg bid). After remission was induced, the patient was slowly weaned off prednisone again.
Following 6 months of successful discontinuation of prednisone and continued rituximab and mycophenolate mofetil therapy, the patient presented to the emergency department with new onset chest pain and shortness of breath. A CT angiography of the chest showed right upper and middle lobe infiltrate, and he was treated for community acquired pneumonia. Additionally, he was noted to have elevated troponin levels suggestive of myocardial infarction (MI). Initial troponin was 1.23 ng/mL (normal range < 0.015 ng/mL), which trended down over the next 18 hours. A bedside echocardiogram showed a normal left ventricular ejection fraction without wall motion abnormalities. Etiology for his acute MI was presumed to be demand ischemia from fixed atherosclerotic plaque. Further inpatient cardiac risk stratification was changed to the outpatient setting, and he was started on medical management for coronary artery disease with a beta blocker, a statin, and aspirin. He was discharged home on 10 mg prednisone daily, which was subsequently tapered over several weeks.
In follow-up, a Lexiscan myocardial perfusion imaging was conducted that demonstrated an inferolateral defect and associated wall motion abnormalities (Figures 2 and 3).
Discussion
IgG4-related disease has been found to be a systemic disorder. Typical characteristics include predominance in men aged > 50 years, elevated IgG4 levels, and findings on histology.1 It has been reported to involve many organs, including pancreas, liver, gallbladder, salivary glands, thyroid, and pleura of the lung.2,5
This case report begins with a presumptive diagnosis of cholangiocarcinoma, which was treated aggressively with extensive surgery. Several case reports of complex tumefactive lesions in the GI area (mostly pancreatic and biliary) have detailed IgG4-RD as both a risk factor for subsequent development of cholangiocarcinoma and as a separate entity of IgG4-related sclerosing cholangitis.7-9 It is hypothesized that the induction of IgG4- positive plasma cells has been intertwined with the development of cholangiocarcinoma. Differentiation between IgG4 reaction that is scattered around cancerous nests and IgG4 sclerosing cholangitis without malignancy is challenging. It has been documented that both elevated IgG4 levels and hilar hepatic lesions that resemble cholangiocarcinoma frequently accompany those cases of IgG4 sclerosing chlolangitis without pancreatic involvement.9 The histologic features of IgG4-RD need to be identified with multiple biopsies and cytology, and superficial biopsy from biliary mucosa cannot reliably exclude cholangiocarcinoma.
Lymphoplasmacytic aortitis and arteritis have been documented in IgG4-RD. In 2017, Barbu and colleagues described how one such case of coronary arteritis presented with typical angina and coronary catheterization revealing coronary artery stenosis.10 However, during coronary artery bypass surgery, the aorta and coronary vessels were noted to be abnormally stiff. A diffuse fibrotic tissue was identified to be causing the significant stenosis without evidence of atherosclerosis. Pathology showed typical findings of IgG4-RD, and there was a rapid response to immunosuppressive therapy. Involvement of coronary arteries has been described in a small number of cases at this time and is associated with progressive fibrotic changes resulting in an MI, aneurysms, and sudden cardiac death.2,10,11
IgG4-RD can be an extensively systemic disease. All presentations of fibrosis or vasculitis should be viewed with heightened suspicion in the future as being a facet of his IgG4-RD. Pleural involvement has been reported in 12% of cases presenting with systemic presentation, kidney involvement in 13%.2,12
Unfortunately, there is no standard laboratory parameter to date that is diagnostic for IgG4-RD. The gold standard remains confirmation of histologic findings with biopsy. According to an international consensus from 2015, 2 out of the 3 major findings need to be present: (1) dense lymphoplasmacytic infiltrate; (2) storiform fibrosis; and (3) obliterative phlebitis in veins and arteries.1,5 Most patients present with symptoms related to either tumefaction or fibrosis of an organ system.1 Peripheral eosinophilia and elevated serum IgE are often present in IgG4-RD.13 Although IgG4 values are elevated in 51% of biopsy-proven cases, flow cytometry of CD19lowCD38+CD20-CD27+ plasmablasts has been explored recently as a correlation with disease flare.3,14 These particular plasmablasts mark a stage between B cells and plasma cells and have been reported to have a sensitivity of 95% and a specificity of 82% in association with actual IgG4-RD.14 Furthermore, blood plasmablast concentrations decrease in response to glucocorticoid treatment, thereby providing a possible quantifiable value by which to measure success of IgG4 treatment.5,12
Treatment for this disease consists of immunosuppressive therapy. There is documentation of successful remission with rituximab and azathioprine, as well as methotrexate.1,5 Both 2015 consensus guidelines and a recent small single-center retrospective study support addition of second-line steroid sparing agents such as mycophenolate mofetil.5,6 For acute flairs, however, glucocorticoids with slow taper are usually utilized. In these cases, they should be tapered as soon as clinically feasible to avoid long-term adverse effects. Untreated IgG4-RD, even asymptomatic, has been shown to progress to fibrosis.5
Conclusion
IgG4-RD is a complicated disease process that requires a high index of suspicion to diagnose. In addition, for patients who are diagnosed with this condition, its ability to mimic other pathologic conditions should be taken into account with manifestation of any new illness. This case emphasizes the ability of this disease to localize in multiple organs over time and the need for lifetime surveillance in patients with IgG4-RD disease.
1. Lang D, Zwerina J, Pieringer H. IgG4-related disease: current challenges and future prospects. Ther Clin Risk Manag. 2016;12:189-199.
2. Brito-Zerón P, Ramos-Casals M, Bosch X, Stone JH. The clinical spectrum of IgG4-related disease. Autoimmun Rev. 2014;13(12):1203-1210.
3. Wallace ZS, Deshpande V, Mattoo H, et al. IgG4-related disease: clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheumatol. 2015;67(9):2466-2475.
4. Carruthers MN, Khosroshahi A, Augustin T, Deshpande V, Stone JH. The diagnostic utility of serum IgG4 concentrations in IgG4-RD. Ann Rheum Dis. 2015;74(1):14-18.
5. Khosroshahi A, Wallace ZS, Crowe JL, et al; Second International Symposium on IgG4-Related Disease. International consensus guidance statement on the management and treatment of IgG4-Related disease. Arthritis Rheumatol. 2015;67(7):1688-1699.
6. Gupta N, Mathew J, Mohan H, et al. Addition of second-line steroid sparing immunosuppressants like mycophenolate mofetil improves outcome of immunoglobulin G4-related disease (IgG4-RD): a series from a tertiary care teaching hospital in South India. Rheumatol Int. 2017;38(2):203-209.
7. Lin HP, Lin KT, Ho WC, Chen CB, Kuo, CY, Lin YC. IgG4-associated cholangitis mimicking cholangiocarcinoma-report of a case. J Intern Med Taiwan. 2013;24:137-141.
8. Douhara A, Mitoro A, Otani E, et al. Cholangiocarcinoma developed in a patient with IgG4-related disease. World J Gastrointest Oncol. 2013;5(8):181-185.
9. Harada K, Nakanuma Y. Cholangiocarcinoma with respect to IgG4 reaction. Int J Hepatol. 2014;2014:803876.
10. Barbu M, Lindström U, Nordborg C, Martinsson A, Dworeck C, Jeppsson A. Sclerosing aortic and coronary arteritis due to IgG4-related disease. Ann Thorac Surg. 2017;103(6):e487-e489.
11. Kim YJ, Park YS, Koo BS, et al. Immunoglobulin G4-related disease with lymphoplasmacytic aortitis mimicking Takayasu arteritis. J Clin Rheumatol. 2011;17(8):451-452.
12. Khosroshahi A, Digumarthy SR, Gibbons FK, Deshpande V. Case 34-2015: A 36-year-old woman with a lung mass, pleural effusion and hip pain. N Engl J Med. 2015;373(18):1762-1772.
13. Della Torre E, Mattoo H, Mahajan VS, Carruthers M, Pillai S, Stone JH. Prevalence of atopy, eosinophilia and IgE elevation in IgG4-related disease. Allergy. 2014;69(2):191-206.
14. Wallace ZS, Mattoo H, Carruthers M, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2015;74(1):190-195.
1. Lang D, Zwerina J, Pieringer H. IgG4-related disease: current challenges and future prospects. Ther Clin Risk Manag. 2016;12:189-199.
2. Brito-Zerón P, Ramos-Casals M, Bosch X, Stone JH. The clinical spectrum of IgG4-related disease. Autoimmun Rev. 2014;13(12):1203-1210.
3. Wallace ZS, Deshpande V, Mattoo H, et al. IgG4-related disease: clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheumatol. 2015;67(9):2466-2475.
4. Carruthers MN, Khosroshahi A, Augustin T, Deshpande V, Stone JH. The diagnostic utility of serum IgG4 concentrations in IgG4-RD. Ann Rheum Dis. 2015;74(1):14-18.
5. Khosroshahi A, Wallace ZS, Crowe JL, et al; Second International Symposium on IgG4-Related Disease. International consensus guidance statement on the management and treatment of IgG4-Related disease. Arthritis Rheumatol. 2015;67(7):1688-1699.
6. Gupta N, Mathew J, Mohan H, et al. Addition of second-line steroid sparing immunosuppressants like mycophenolate mofetil improves outcome of immunoglobulin G4-related disease (IgG4-RD): a series from a tertiary care teaching hospital in South India. Rheumatol Int. 2017;38(2):203-209.
7. Lin HP, Lin KT, Ho WC, Chen CB, Kuo, CY, Lin YC. IgG4-associated cholangitis mimicking cholangiocarcinoma-report of a case. J Intern Med Taiwan. 2013;24:137-141.
8. Douhara A, Mitoro A, Otani E, et al. Cholangiocarcinoma developed in a patient with IgG4-related disease. World J Gastrointest Oncol. 2013;5(8):181-185.
9. Harada K, Nakanuma Y. Cholangiocarcinoma with respect to IgG4 reaction. Int J Hepatol. 2014;2014:803876.
10. Barbu M, Lindström U, Nordborg C, Martinsson A, Dworeck C, Jeppsson A. Sclerosing aortic and coronary arteritis due to IgG4-related disease. Ann Thorac Surg. 2017;103(6):e487-e489.
11. Kim YJ, Park YS, Koo BS, et al. Immunoglobulin G4-related disease with lymphoplasmacytic aortitis mimicking Takayasu arteritis. J Clin Rheumatol. 2011;17(8):451-452.
12. Khosroshahi A, Digumarthy SR, Gibbons FK, Deshpande V. Case 34-2015: A 36-year-old woman with a lung mass, pleural effusion and hip pain. N Engl J Med. 2015;373(18):1762-1772.
13. Della Torre E, Mattoo H, Mahajan VS, Carruthers M, Pillai S, Stone JH. Prevalence of atopy, eosinophilia and IgE elevation in IgG4-related disease. Allergy. 2014;69(2):191-206.
14. Wallace ZS, Mattoo H, Carruthers M, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2015;74(1):190-195.
Primary renal synovial sarcoma – a diagnostic dilemma
Soft tissue sarcomas are rare mesenchymal tumors that comprise 1% of all malignancies. Synovial sarcoma accounts for 5% to 10% of adult soft tissue sarcomas and usually occurs in close association with joint capsules, tendon sheaths, and bursa in the extremities of young and middle-aged adults.1 Synovial sarcomas have been reported in other unusual sites, including the head and neck, thoracic and abdominal wall, retroperitoneum, bone, pleura, and visceral organs such as the lung, prostate, or kidney.2 Primary renal synovial sarcoma is an extremely rare tumor accounting for <2% of all malignant renal tumors.3 To the best of our knowledge, fewer than 50 cases of primary renal synovial sarcoma have been described in the English literature.4 It presents as a diagnostic dilemma because of the dearth of specific clinical and imaging findings and is often confused with benign and malignant tumors. The differential diagnosis includes angiomyolipoma, renal cell carcinoma with sarcomatoid differentiation, metastatic sarcoma, hemangiopericytoma, malignant solitary fibrous tumor, Wilms tumor, and malignant peripheral nerve sheath tumor. Hence, a combination of histomorphologic, immunohistochemical, cytogenetic, and molecular studies that show a unique chromosomal translocation t(X;18) (p11;q11) is imperative in the diagnosis of primary renal synovial sarcoma.4 In the present report, we present the case of a 38-year-old man who was diagnosed with primary renal synovial sarcoma.
Case presentation and summary
A 38-year-old man with a medical history of gastroesophageal reflux disease and Barrett’s esophagus presented to our hospital for the first time with persistent and progressive right-sided flank and abdominal pain that was aggravated after a minor trauma to the back. There was no associated hematuria or dysuria.
Of note is that he had experienced intermittent flank pain for 2 years before this transfer. He had initially been diagnosed at his local hospital close to his home by ultrasound with an angiomyolipoma of 2 × 3 cm arising from the upper pole of his right kidney, which remained stable on repeat sonograms. About 22 months after his initial presentation at his local hospital, the flank pain increased, and a computed-tomographic (CT) scan revealed a perinephric hematoma that was thought to originate from a ruptured angiomyolipoma. He subsequently underwent embolization, but his symptoms recurred soon after. He presented again to his local hospital where CT imaging revealed a significant increase in the size of the retroperitoneal mass, and findings were suggestive of a hematoma. Subsequent angiogram did not reveal active extravasation, so a biopsy was performed.
Before confirmatory pathologic evaluation could be completed, the patient presented to his local hospital again in excruciating pain. A CT scan of his abdomen and pelvis demonstrated a massive subacute on chronic hematoma in the right retroperitoneum measuring 22 × 19 × 18 cm, with calcifications originating from an upper pole right renal neoplasm. The right kidney was displaced antero-inferiorly, and the inferior vena cava was displaced anteriorly and to the left. The preliminary pathology returned with findings suggestive of sarcoma (Figures 1 and 2).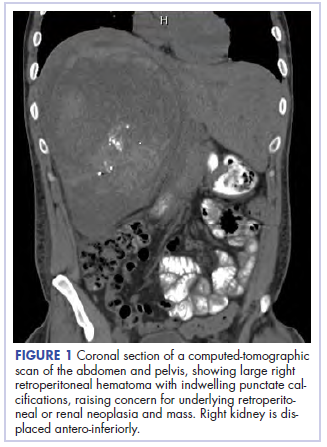
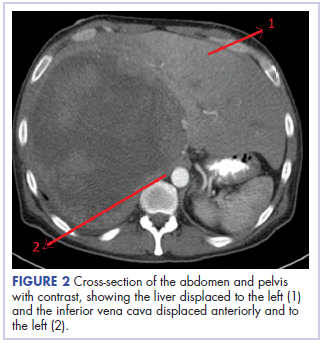
The patient was then transferred to our institution, where he was evaluated by medical and surgical oncology. A CT scan of the chest and magnetic-resonance imaging (MRI) of the brain did not reveal metastatic disease. He underwent exploratory laparotomy that involved the resection of a 22-cm retroperitoneal mass, right nephrectomy, right adrenalectomy, partial right hepatectomy, and a full thickness resection of the right postero-inferior diaphragm followed by mesh repair because of involvement by the tumor.
In its entirety, the specimen was a mass of 26 × 24 × 14 cm. It was sectioned to show extensively necrotic and hemorrhagic variegated white to tan-red parenchyma (Figure 3). Histology revealed a poorly differentiated malignant neoplasm composed of round cells with scant amphophilic cytoplasm arranged in solid, variably sized nests separated by prominent thin-walled branching vascular channels (Figure 4). The mitotic rate was high. It was determined to be a histologically ungraded sarcoma according to the French Federation of Comprehensive Cancer Centers system of grading soft tissue sarcomas; the margins were indeterminate. Immunohistochemistry was positive for EMA, TLE1, and negative for AE1/AE3, S100, STAT6, and Nkx2.2. Molecular pathology fluorescent in situ hybridization (FISH) analysis demonstrated positivity for SS18 gene rearrangement (SS18-SSX1 fusion).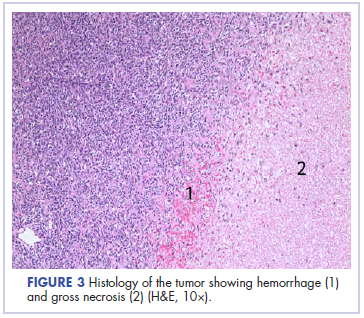
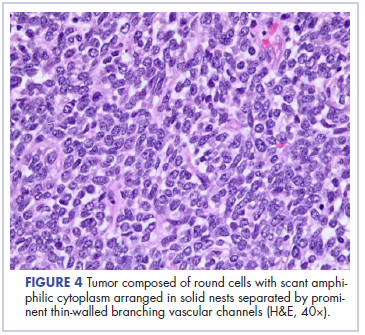
After recovering from surgery, the patient received adjuvant chemotherapy with doxorubicin and ifosfamide. It has been almost 16 months since we first saw this patient. He was started on doxorubicin 20 mg/m2 on days 1 to 4, ifosfamide 2,500 mg on days 1 to 4, and mesna 800 mg on days 1 to 4, for a total of 6 cycles. He did well for the first 5 months, after which he developed disease recurrence in the postoperative nephrectomy bed (a biopsy showed it to be recurrent synovial sarcoma) as well as pulmonary nodules, for which he was started on trabectedin 1.5 mg/m2 every 3 weeks. Two months later, a CT scan showed an increase in the size of his retroperitoneal mass, and the treatment was changed to pazopanib 400 mg daily orally, on which he remained at the time of publication.
Discussion
Synovial sarcoma is the fourth most common type of soft tissue sarcoma, accounting for 2.5% to 10.5% of all primary soft tissue malignancies worldwide. It occurs most frequently in adolescents and young adults, with most patients presenting between the ages of 15 and 40 years. Median age of presentation is 36 years. Despite the nomenclature, synovial sarcoma does not arise in intra-articular locations but typically occurs in proximity to joints in the extremities. Synovial sarcomas are less commonly described in other sites, including the head and neck, mediastinum, intraperitoneum, retroperitoneum, lung, pleura, and kidney.4,5 Renal synovial sarcoma was first described in a published article by Argani and colleagues in 2000.5
Adult renal mesenchymal tumors are classified into benign and malignant tumors on the basis of the histologic features and clinicobiologic behavior.6,7 The benign esenchymal renal tumors include angiomyolipoma, leiomyoma, hemangioma, lymphangioma, juxtaglomerular cell tumor, renomedullary interstitial cell tumor (medullary fibroma), lipoma, solitary fibrous tumor, and schwannoma. Malignant renal tumors of mesenchymal origin include leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, osteosarcoma, fibrosarcoma, malignant fibrous histiocytoma, solitary fibrous tumor, and synovial sarcoma.
Most of these tumor types cause the same nonspecific symptoms in patients – abdominal pain, flank pain, abdominal fullness, a palpable mass, and hematuria – although they can be clinically silent. The average duration of symptoms in synovial sarcoma is 2 to 4 years.8 The long duration of symptoms and initial slow growth of synovial sarcomas may give a false impression of a benign process.
A preoperative radiological diagnosis of primary renal synovial sarcoma may be suspected by analyzing the tumor’s growth patterns on CT scans.9 Renal synovial sarcomas often appear as large, well-defined soft tissue masses that can extend into the renal pelvis or into the perinephric region.9 A CT scan may identify soft tissue calcifications, especially subtle ones in areas where the tumor anatomy is complex. A CT scan may also reveal areas of hemorrhage, necrosis, or cyst formation within the tumor, and can easily confirm bone involvement. Intravenous contrast may help in differentiating the mass from adjacent muscle and neurovascular complex.9,10 On MRI, renal synovial sarcomas are often described as nonspecific heterogeneous masses, although they may also exhibit heterogeneous enhancement of hemorrhagic areas, calcifications, and air-fluid levels (known as “triple sign”) as well as septae. The triple sign may be identified as areas of low, intermediate, and high signal intensity, correlating with areas of hemorrhage, calcification, and air-fluid level.9,10 Signal intensity is about equal to that of skeletal muscle on T1-weighted MRI and higher than that of subcutaneous fat on T2-weighted MRI.
In the present case, the tumor was initially misdiagnosed as an angiomyolipoma, the most common benign tumor of the kidney. Angiomyolipomas are usually solid triphasic tumors arising from the renal cortex and are composed of 3 major elements: dysmorphic blood vessels, smooth muscle components, and adipose tissue. When angiomyolipomas are large enough, they are readily recognized by the identification of macroscopic fat within the tumor, either by CT scan or MRI.11 When they are small, they may be difficult to distinguish from a small cyst on CT because of volume averaging.
On pathology, synovial sarcoma has dual epithelial and mesenchymal differentiation. They are frequently multi-lobulated, and areas of necrosis, hemorrhage, and cyst formation are also common. There are 3 main histologic subtypes of synovial sarcoma: biphasic (20%-30%), monophasic (50%-60%), and poorly differentiated (15%-25%). Poorly differentiated synovial sarcomas are generally epithelioid in morphology, have high mitotic activity (usually 10-20 mitoses/10 high-power field; range is <5 for well differentiated, low-grade tumors), and can be confused with round cell tumors such as Ewing sarcoma. Poorly differentiated synovial sarcomas are high-grade tumors.
Immunohistochemical studies can confirm the pathological diagnosis. Synovial sarcomas usually stain positive for Bcl2, CD99/Mic2, CD56, Vim, and focally for EMA but negatively for desmin, actin, WT1, S-100, CD34, and CD31.5 Currently, the gold standard for diagnosis and hallmark for synovial sarcomas are the t (X;18) translocation and SYT-SSX gene fusion products (SYT-SSX1 in 67% and SYT-SSX2 in 33% of cases). These can be detected either by FISH or reverse-transcription polymerase chain reaction. This genetic alteration is identified in more than 90% of synovial sarcomas and is highly specific.
The role of SYT-SSX gene fusion in the pathogenesis of synovial sarcoma is an active area of investigation. The fusion of SYT with SSX translates into a fusion protein that binds to the transcription activator SMARCA4 that is involved in chromatin remodeling, thus displacing both the wildtype SYT and the tumor suppressor gene SMARCB1. The modified protein complex then binds at several super-enhancer loci, unlocking suppressed genes such as Sox2, which is known to be necessary for synovial sarcoma proliferation. Alterations in SMARCB1 are involved in several cancer types, implicating this event as a driver of these malignancies.12 This results in a global alteration in chromatin remodeling that needs to be better understood to design targeted therapies.
The clinical course of synovial sarcoma, regardless of the tissue of origin, is typically poor. Multiple clinical and pathologic factors, including tumor size, location, patient age, and presence of poorly differentiated areas, are thought to have prognostic significance. A tumor size of more than 5 cm at presentation has the greatest impact on prognosis, with studies showing 5-year survival rates of 64% for patients with tumors smaller than 5 cm and 26% for patients with masses greater than 5 cm.13,14 High-grade synovial sarcoma is favored in tumors that have cystic components, hemorrhage, and fluid levels and the triple sign.
Patients with tumors in the extremities have a more favorable prognosis than those with lesions in the head and neck area or axially, a feature that likely reflects better surgical control available for extremity lesions. Patient age of less than 15 to 20 years is also associated with a better long-term prognosis.15,16 Varela-Duran and Enzinger17 reported that the presence of extensive calcifications suggests improved long-term survival, with 5-year survival rates of 82% and decreased rates of local recurrence (32%) and metastatic disease (29%). The poorly differentiated subtype is associated with a worsened prognosis, with a 5-year survival rate of 20% through 30%.18,19 Other pathologic factors associated with worsened prognosis include presence of rhabdoid cells, extensive tumor necrosis, high nuclear grade, p53 mutations, and high mitotic rate (>10 mitoses/10 high-power field). More recently, the gene fusion type SYT-SSX2 (more common in monophasic lesions) has been associated with an improved prognosis, compared with that for SYT-SSX1, and an 89% metastasis-free survival.20
Although there are no guidelines for the treatment of primary renal synovial sarcoma because of the limited number of cases reported, surgery is considered the first choice. Adjuvant chemotherapy with an anthracycline (doxorubicin or epirubicin) combined with ifosfamide has been the most frequently used regimen in published cases, especially in those in which patients have poor prognostic factors as mentioned above.
Overall, the 5-year survival rate ranges from 36% to 76%.14 The clinical course of synovial sarcoma is characterized by a high rate of local recurrence (30%-50%) and metastatic disease (41%). Most metastases occur within the first 2 to 5 years after treatment cessation. Metastases are present in 16% to 25% of patients at their initial presentation, with the most frequent metastatic site being the lung, followed by the lymph nodes (4%-18%) and bone (8%-11%).
Conclusion
Primary renal synovial sarcoma is extremely rare, and preoperative diagnosis is difficult in the absence of specific clinical or imaging findings. A high index of suspicion combined with pathologic, immunohistochemical, cytogenetic, and molecular studies is essential for accurate diagnosis and subsequent treatment planning. The differential diagnosis of renal synovial sarcoma can be extensive, and our experience with this patient illustrates the diagnostic dilemma associated with renal synovial sarcoma.
1. Majumder A, Dey S, Khandakar B, Medda S, Chandra Paul P. Primary renal synovial sarcoma: a rare tumor with an atypical presentation. Arch Iran Med. 2014;17(10):726-728.
2. Fetsch JF, Meis JM. Synovial sarcoma of the abdominal wall. Cancer. 1993;72(2):469 477.
3. Wang Z, Zhong Z, Zhu L, et al. Primary synovial sarcoma of the kidney: a case report. Oncol Lett. 2015;10(6):3542-3544.
4. Abbas M, Dämmrich ME, Braubach P, et al. Synovial sarcoma of the kidney in a young patient with a review of the literature. Rare tumors. 2014;6(2):5393
5. Argani P, Faria PA, Epstein JI, et al. Primary renal synovial sarcoma: molecular and morphologic delineation of an entity previously included among embryonal sarcomas of the kidney. Am J Surg Pathol. 2000;24(8):1087-1096.
6. Eble JN, Sauter G, Epstein JI, Sesterhenn IA, eds. World Health Organization classification of tumours: pathology and genetics of tumours of the urinary system and male genital organs. Lyon, France: IARC; 2004.
7. Tamboli P, Ro JY, Amin MB, Ligato S, Ayala AG. Benign tumors and tumor-like lesions of the adult kidney. Part II: benign mesenchymal and mixed neoplasms, and tumor-like lesions. Adv Anat Pathol. 2000;7(1):47-66.
8. Weiss SW, Goldblum JR. Malignant soft tissue tumors of uncertain type. In: Weiss SW, Goldblum JR, eds. Enzinger and Weiss’s soft tissue tumors. 4th ed. St. Louis, MO: Mosby, 2001; 1483-1565.
9. Lacovelli R, Altavilla A, Ciardi A, et al. Clinical and pathological features of primary renal synovial sarcoma: analysis of 64 cases from 11 years of medical literature. BJU Int. 2012;110(10):1449-1454.
10. Alhazzani AR, El-Sharkawy MS, Hassan H. Primary retroperitoneal synovial sarcoma in CT and MRI. Urol Ann. 2010;2(1):39-41.
11. Katabathina VS, Vikram R, Nagar AM, Tamboli P, Menias CO, Prasad SR. Mesenchymal neoplasms of the kidney in adults: imaging spectrum with radiologic-pathologic correlation. Radiographics. 2010;30(6):1525-1540.
12. Sápi Z, Papp G, Szendrői M, et al. Epigenetic regulation of SMARCB1 by miR-206, -381 and -671- 5p is evident in a variety of SMARCB1 immunonegative soft tissue sarcomas, while miR-765 appears specific for epithelioid sarcoma. A miRNA study of 223 soft tissue sarcomas. Genes Chromosomes Cancer. 2016;55(10):786-802.
13. Ferrari A, Gronchi A, Casanova M, et al. Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer. 2004;101(3):627-634.
14. Rangheard AS, Vanel D, Viala J, Schwaab G, Casiraghi O, Sigal R. Synovial sarcomas of the head and neck: CT and MR imaging findings of eight patients. Am J Neuroradiol. 2001;22(5):851-857.
15. Oda Y, Hashimoto H, Tsuneyoshi M, Takeshita S. Survival in synovial sarcoma: a multivariate study of prognostic factors with special emphasis on the comparison between early death and long-term survival. Am J Surg Pathol. 1993;17(1):35-44.
16. Raney RB. Synovial sarcoma in young people: background, prognostic factors and therapeutic questions. J Pediatr Hematol Oncol. 2005;27(4):207-211.
17. Varela-Duran J, Enzinger FM. Calcifying synovial sarcoma. Cancer. 1982;50(2):345-352.
18. Cagle LA, Mirra JM, Storm FK, Roe DJ, Eilber FR. Histologic features relating to prognosis in synovial sarcoma. Cancer. 1987;59(10):1810-1814.
19. Skytting B, Meis-Kindblom JM, Larsson O, et al. Synovial sarcoma – identification of favorable and unfavorable histologic types: a Scandinavian sarcoma group study of 104 cases. Acta Orthop Scand. 1999:70(6):543-554.
20. Murphey MD, Gibson MS, Jennings BT, Crespo-Rodríguez AM, Fanburg-Smith J, Gajewski DA. Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics. 2006;26(5):1543-1565.
Soft tissue sarcomas are rare mesenchymal tumors that comprise 1% of all malignancies. Synovial sarcoma accounts for 5% to 10% of adult soft tissue sarcomas and usually occurs in close association with joint capsules, tendon sheaths, and bursa in the extremities of young and middle-aged adults.1 Synovial sarcomas have been reported in other unusual sites, including the head and neck, thoracic and abdominal wall, retroperitoneum, bone, pleura, and visceral organs such as the lung, prostate, or kidney.2 Primary renal synovial sarcoma is an extremely rare tumor accounting for <2% of all malignant renal tumors.3 To the best of our knowledge, fewer than 50 cases of primary renal synovial sarcoma have been described in the English literature.4 It presents as a diagnostic dilemma because of the dearth of specific clinical and imaging findings and is often confused with benign and malignant tumors. The differential diagnosis includes angiomyolipoma, renal cell carcinoma with sarcomatoid differentiation, metastatic sarcoma, hemangiopericytoma, malignant solitary fibrous tumor, Wilms tumor, and malignant peripheral nerve sheath tumor. Hence, a combination of histomorphologic, immunohistochemical, cytogenetic, and molecular studies that show a unique chromosomal translocation t(X;18) (p11;q11) is imperative in the diagnosis of primary renal synovial sarcoma.4 In the present report, we present the case of a 38-year-old man who was diagnosed with primary renal synovial sarcoma.
Case presentation and summary
A 38-year-old man with a medical history of gastroesophageal reflux disease and Barrett’s esophagus presented to our hospital for the first time with persistent and progressive right-sided flank and abdominal pain that was aggravated after a minor trauma to the back. There was no associated hematuria or dysuria.
Of note is that he had experienced intermittent flank pain for 2 years before this transfer. He had initially been diagnosed at his local hospital close to his home by ultrasound with an angiomyolipoma of 2 × 3 cm arising from the upper pole of his right kidney, which remained stable on repeat sonograms. About 22 months after his initial presentation at his local hospital, the flank pain increased, and a computed-tomographic (CT) scan revealed a perinephric hematoma that was thought to originate from a ruptured angiomyolipoma. He subsequently underwent embolization, but his symptoms recurred soon after. He presented again to his local hospital where CT imaging revealed a significant increase in the size of the retroperitoneal mass, and findings were suggestive of a hematoma. Subsequent angiogram did not reveal active extravasation, so a biopsy was performed.
Before confirmatory pathologic evaluation could be completed, the patient presented to his local hospital again in excruciating pain. A CT scan of his abdomen and pelvis demonstrated a massive subacute on chronic hematoma in the right retroperitoneum measuring 22 × 19 × 18 cm, with calcifications originating from an upper pole right renal neoplasm. The right kidney was displaced antero-inferiorly, and the inferior vena cava was displaced anteriorly and to the left. The preliminary pathology returned with findings suggestive of sarcoma (Figures 1 and 2).
The patient was then transferred to our institution, where he was evaluated by medical and surgical oncology. A CT scan of the chest and magnetic-resonance imaging (MRI) of the brain did not reveal metastatic disease. He underwent exploratory laparotomy that involved the resection of a 22-cm retroperitoneal mass, right nephrectomy, right adrenalectomy, partial right hepatectomy, and a full thickness resection of the right postero-inferior diaphragm followed by mesh repair because of involvement by the tumor.
In its entirety, the specimen was a mass of 26 × 24 × 14 cm. It was sectioned to show extensively necrotic and hemorrhagic variegated white to tan-red parenchyma (Figure 3). Histology revealed a poorly differentiated malignant neoplasm composed of round cells with scant amphophilic cytoplasm arranged in solid, variably sized nests separated by prominent thin-walled branching vascular channels (Figure 4). The mitotic rate was high. It was determined to be a histologically ungraded sarcoma according to the French Federation of Comprehensive Cancer Centers system of grading soft tissue sarcomas; the margins were indeterminate. Immunohistochemistry was positive for EMA, TLE1, and negative for AE1/AE3, S100, STAT6, and Nkx2.2. Molecular pathology fluorescent in situ hybridization (FISH) analysis demonstrated positivity for SS18 gene rearrangement (SS18-SSX1 fusion).
After recovering from surgery, the patient received adjuvant chemotherapy with doxorubicin and ifosfamide. It has been almost 16 months since we first saw this patient. He was started on doxorubicin 20 mg/m2 on days 1 to 4, ifosfamide 2,500 mg on days 1 to 4, and mesna 800 mg on days 1 to 4, for a total of 6 cycles. He did well for the first 5 months, after which he developed disease recurrence in the postoperative nephrectomy bed (a biopsy showed it to be recurrent synovial sarcoma) as well as pulmonary nodules, for which he was started on trabectedin 1.5 mg/m2 every 3 weeks. Two months later, a CT scan showed an increase in the size of his retroperitoneal mass, and the treatment was changed to pazopanib 400 mg daily orally, on which he remained at the time of publication.
Discussion
Synovial sarcoma is the fourth most common type of soft tissue sarcoma, accounting for 2.5% to 10.5% of all primary soft tissue malignancies worldwide. It occurs most frequently in adolescents and young adults, with most patients presenting between the ages of 15 and 40 years. Median age of presentation is 36 years. Despite the nomenclature, synovial sarcoma does not arise in intra-articular locations but typically occurs in proximity to joints in the extremities. Synovial sarcomas are less commonly described in other sites, including the head and neck, mediastinum, intraperitoneum, retroperitoneum, lung, pleura, and kidney.4,5 Renal synovial sarcoma was first described in a published article by Argani and colleagues in 2000.5
Adult renal mesenchymal tumors are classified into benign and malignant tumors on the basis of the histologic features and clinicobiologic behavior.6,7 The benign esenchymal renal tumors include angiomyolipoma, leiomyoma, hemangioma, lymphangioma, juxtaglomerular cell tumor, renomedullary interstitial cell tumor (medullary fibroma), lipoma, solitary fibrous tumor, and schwannoma. Malignant renal tumors of mesenchymal origin include leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, osteosarcoma, fibrosarcoma, malignant fibrous histiocytoma, solitary fibrous tumor, and synovial sarcoma.
Most of these tumor types cause the same nonspecific symptoms in patients – abdominal pain, flank pain, abdominal fullness, a palpable mass, and hematuria – although they can be clinically silent. The average duration of symptoms in synovial sarcoma is 2 to 4 years.8 The long duration of symptoms and initial slow growth of synovial sarcomas may give a false impression of a benign process.
A preoperative radiological diagnosis of primary renal synovial sarcoma may be suspected by analyzing the tumor’s growth patterns on CT scans.9 Renal synovial sarcomas often appear as large, well-defined soft tissue masses that can extend into the renal pelvis or into the perinephric region.9 A CT scan may identify soft tissue calcifications, especially subtle ones in areas where the tumor anatomy is complex. A CT scan may also reveal areas of hemorrhage, necrosis, or cyst formation within the tumor, and can easily confirm bone involvement. Intravenous contrast may help in differentiating the mass from adjacent muscle and neurovascular complex.9,10 On MRI, renal synovial sarcomas are often described as nonspecific heterogeneous masses, although they may also exhibit heterogeneous enhancement of hemorrhagic areas, calcifications, and air-fluid levels (known as “triple sign”) as well as septae. The triple sign may be identified as areas of low, intermediate, and high signal intensity, correlating with areas of hemorrhage, calcification, and air-fluid level.9,10 Signal intensity is about equal to that of skeletal muscle on T1-weighted MRI and higher than that of subcutaneous fat on T2-weighted MRI.
In the present case, the tumor was initially misdiagnosed as an angiomyolipoma, the most common benign tumor of the kidney. Angiomyolipomas are usually solid triphasic tumors arising from the renal cortex and are composed of 3 major elements: dysmorphic blood vessels, smooth muscle components, and adipose tissue. When angiomyolipomas are large enough, they are readily recognized by the identification of macroscopic fat within the tumor, either by CT scan or MRI.11 When they are small, they may be difficult to distinguish from a small cyst on CT because of volume averaging.
On pathology, synovial sarcoma has dual epithelial and mesenchymal differentiation. They are frequently multi-lobulated, and areas of necrosis, hemorrhage, and cyst formation are also common. There are 3 main histologic subtypes of synovial sarcoma: biphasic (20%-30%), monophasic (50%-60%), and poorly differentiated (15%-25%). Poorly differentiated synovial sarcomas are generally epithelioid in morphology, have high mitotic activity (usually 10-20 mitoses/10 high-power field; range is <5 for well differentiated, low-grade tumors), and can be confused with round cell tumors such as Ewing sarcoma. Poorly differentiated synovial sarcomas are high-grade tumors.
Immunohistochemical studies can confirm the pathological diagnosis. Synovial sarcomas usually stain positive for Bcl2, CD99/Mic2, CD56, Vim, and focally for EMA but negatively for desmin, actin, WT1, S-100, CD34, and CD31.5 Currently, the gold standard for diagnosis and hallmark for synovial sarcomas are the t (X;18) translocation and SYT-SSX gene fusion products (SYT-SSX1 in 67% and SYT-SSX2 in 33% of cases). These can be detected either by FISH or reverse-transcription polymerase chain reaction. This genetic alteration is identified in more than 90% of synovial sarcomas and is highly specific.
The role of SYT-SSX gene fusion in the pathogenesis of synovial sarcoma is an active area of investigation. The fusion of SYT with SSX translates into a fusion protein that binds to the transcription activator SMARCA4 that is involved in chromatin remodeling, thus displacing both the wildtype SYT and the tumor suppressor gene SMARCB1. The modified protein complex then binds at several super-enhancer loci, unlocking suppressed genes such as Sox2, which is known to be necessary for synovial sarcoma proliferation. Alterations in SMARCB1 are involved in several cancer types, implicating this event as a driver of these malignancies.12 This results in a global alteration in chromatin remodeling that needs to be better understood to design targeted therapies.
The clinical course of synovial sarcoma, regardless of the tissue of origin, is typically poor. Multiple clinical and pathologic factors, including tumor size, location, patient age, and presence of poorly differentiated areas, are thought to have prognostic significance. A tumor size of more than 5 cm at presentation has the greatest impact on prognosis, with studies showing 5-year survival rates of 64% for patients with tumors smaller than 5 cm and 26% for patients with masses greater than 5 cm.13,14 High-grade synovial sarcoma is favored in tumors that have cystic components, hemorrhage, and fluid levels and the triple sign.
Patients with tumors in the extremities have a more favorable prognosis than those with lesions in the head and neck area or axially, a feature that likely reflects better surgical control available for extremity lesions. Patient age of less than 15 to 20 years is also associated with a better long-term prognosis.15,16 Varela-Duran and Enzinger17 reported that the presence of extensive calcifications suggests improved long-term survival, with 5-year survival rates of 82% and decreased rates of local recurrence (32%) and metastatic disease (29%). The poorly differentiated subtype is associated with a worsened prognosis, with a 5-year survival rate of 20% through 30%.18,19 Other pathologic factors associated with worsened prognosis include presence of rhabdoid cells, extensive tumor necrosis, high nuclear grade, p53 mutations, and high mitotic rate (>10 mitoses/10 high-power field). More recently, the gene fusion type SYT-SSX2 (more common in monophasic lesions) has been associated with an improved prognosis, compared with that for SYT-SSX1, and an 89% metastasis-free survival.20
Although there are no guidelines for the treatment of primary renal synovial sarcoma because of the limited number of cases reported, surgery is considered the first choice. Adjuvant chemotherapy with an anthracycline (doxorubicin or epirubicin) combined with ifosfamide has been the most frequently used regimen in published cases, especially in those in which patients have poor prognostic factors as mentioned above.
Overall, the 5-year survival rate ranges from 36% to 76%.14 The clinical course of synovial sarcoma is characterized by a high rate of local recurrence (30%-50%) and metastatic disease (41%). Most metastases occur within the first 2 to 5 years after treatment cessation. Metastases are present in 16% to 25% of patients at their initial presentation, with the most frequent metastatic site being the lung, followed by the lymph nodes (4%-18%) and bone (8%-11%).
Conclusion
Primary renal synovial sarcoma is extremely rare, and preoperative diagnosis is difficult in the absence of specific clinical or imaging findings. A high index of suspicion combined with pathologic, immunohistochemical, cytogenetic, and molecular studies is essential for accurate diagnosis and subsequent treatment planning. The differential diagnosis of renal synovial sarcoma can be extensive, and our experience with this patient illustrates the diagnostic dilemma associated with renal synovial sarcoma.
Soft tissue sarcomas are rare mesenchymal tumors that comprise 1% of all malignancies. Synovial sarcoma accounts for 5% to 10% of adult soft tissue sarcomas and usually occurs in close association with joint capsules, tendon sheaths, and bursa in the extremities of young and middle-aged adults.1 Synovial sarcomas have been reported in other unusual sites, including the head and neck, thoracic and abdominal wall, retroperitoneum, bone, pleura, and visceral organs such as the lung, prostate, or kidney.2 Primary renal synovial sarcoma is an extremely rare tumor accounting for <2% of all malignant renal tumors.3 To the best of our knowledge, fewer than 50 cases of primary renal synovial sarcoma have been described in the English literature.4 It presents as a diagnostic dilemma because of the dearth of specific clinical and imaging findings and is often confused with benign and malignant tumors. The differential diagnosis includes angiomyolipoma, renal cell carcinoma with sarcomatoid differentiation, metastatic sarcoma, hemangiopericytoma, malignant solitary fibrous tumor, Wilms tumor, and malignant peripheral nerve sheath tumor. Hence, a combination of histomorphologic, immunohistochemical, cytogenetic, and molecular studies that show a unique chromosomal translocation t(X;18) (p11;q11) is imperative in the diagnosis of primary renal synovial sarcoma.4 In the present report, we present the case of a 38-year-old man who was diagnosed with primary renal synovial sarcoma.
Case presentation and summary
A 38-year-old man with a medical history of gastroesophageal reflux disease and Barrett’s esophagus presented to our hospital for the first time with persistent and progressive right-sided flank and abdominal pain that was aggravated after a minor trauma to the back. There was no associated hematuria or dysuria.
Of note is that he had experienced intermittent flank pain for 2 years before this transfer. He had initially been diagnosed at his local hospital close to his home by ultrasound with an angiomyolipoma of 2 × 3 cm arising from the upper pole of his right kidney, which remained stable on repeat sonograms. About 22 months after his initial presentation at his local hospital, the flank pain increased, and a computed-tomographic (CT) scan revealed a perinephric hematoma that was thought to originate from a ruptured angiomyolipoma. He subsequently underwent embolization, but his symptoms recurred soon after. He presented again to his local hospital where CT imaging revealed a significant increase in the size of the retroperitoneal mass, and findings were suggestive of a hematoma. Subsequent angiogram did not reveal active extravasation, so a biopsy was performed.
Before confirmatory pathologic evaluation could be completed, the patient presented to his local hospital again in excruciating pain. A CT scan of his abdomen and pelvis demonstrated a massive subacute on chronic hematoma in the right retroperitoneum measuring 22 × 19 × 18 cm, with calcifications originating from an upper pole right renal neoplasm. The right kidney was displaced antero-inferiorly, and the inferior vena cava was displaced anteriorly and to the left. The preliminary pathology returned with findings suggestive of sarcoma (Figures 1 and 2).
The patient was then transferred to our institution, where he was evaluated by medical and surgical oncology. A CT scan of the chest and magnetic-resonance imaging (MRI) of the brain did not reveal metastatic disease. He underwent exploratory laparotomy that involved the resection of a 22-cm retroperitoneal mass, right nephrectomy, right adrenalectomy, partial right hepatectomy, and a full thickness resection of the right postero-inferior diaphragm followed by mesh repair because of involvement by the tumor.
In its entirety, the specimen was a mass of 26 × 24 × 14 cm. It was sectioned to show extensively necrotic and hemorrhagic variegated white to tan-red parenchyma (Figure 3). Histology revealed a poorly differentiated malignant neoplasm composed of round cells with scant amphophilic cytoplasm arranged in solid, variably sized nests separated by prominent thin-walled branching vascular channels (Figure 4). The mitotic rate was high. It was determined to be a histologically ungraded sarcoma according to the French Federation of Comprehensive Cancer Centers system of grading soft tissue sarcomas; the margins were indeterminate. Immunohistochemistry was positive for EMA, TLE1, and negative for AE1/AE3, S100, STAT6, and Nkx2.2. Molecular pathology fluorescent in situ hybridization (FISH) analysis demonstrated positivity for SS18 gene rearrangement (SS18-SSX1 fusion).
After recovering from surgery, the patient received adjuvant chemotherapy with doxorubicin and ifosfamide. It has been almost 16 months since we first saw this patient. He was started on doxorubicin 20 mg/m2 on days 1 to 4, ifosfamide 2,500 mg on days 1 to 4, and mesna 800 mg on days 1 to 4, for a total of 6 cycles. He did well for the first 5 months, after which he developed disease recurrence in the postoperative nephrectomy bed (a biopsy showed it to be recurrent synovial sarcoma) as well as pulmonary nodules, for which he was started on trabectedin 1.5 mg/m2 every 3 weeks. Two months later, a CT scan showed an increase in the size of his retroperitoneal mass, and the treatment was changed to pazopanib 400 mg daily orally, on which he remained at the time of publication.
Discussion
Synovial sarcoma is the fourth most common type of soft tissue sarcoma, accounting for 2.5% to 10.5% of all primary soft tissue malignancies worldwide. It occurs most frequently in adolescents and young adults, with most patients presenting between the ages of 15 and 40 years. Median age of presentation is 36 years. Despite the nomenclature, synovial sarcoma does not arise in intra-articular locations but typically occurs in proximity to joints in the extremities. Synovial sarcomas are less commonly described in other sites, including the head and neck, mediastinum, intraperitoneum, retroperitoneum, lung, pleura, and kidney.4,5 Renal synovial sarcoma was first described in a published article by Argani and colleagues in 2000.5
Adult renal mesenchymal tumors are classified into benign and malignant tumors on the basis of the histologic features and clinicobiologic behavior.6,7 The benign esenchymal renal tumors include angiomyolipoma, leiomyoma, hemangioma, lymphangioma, juxtaglomerular cell tumor, renomedullary interstitial cell tumor (medullary fibroma), lipoma, solitary fibrous tumor, and schwannoma. Malignant renal tumors of mesenchymal origin include leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, osteosarcoma, fibrosarcoma, malignant fibrous histiocytoma, solitary fibrous tumor, and synovial sarcoma.
Most of these tumor types cause the same nonspecific symptoms in patients – abdominal pain, flank pain, abdominal fullness, a palpable mass, and hematuria – although they can be clinically silent. The average duration of symptoms in synovial sarcoma is 2 to 4 years.8 The long duration of symptoms and initial slow growth of synovial sarcomas may give a false impression of a benign process.
A preoperative radiological diagnosis of primary renal synovial sarcoma may be suspected by analyzing the tumor’s growth patterns on CT scans.9 Renal synovial sarcomas often appear as large, well-defined soft tissue masses that can extend into the renal pelvis or into the perinephric region.9 A CT scan may identify soft tissue calcifications, especially subtle ones in areas where the tumor anatomy is complex. A CT scan may also reveal areas of hemorrhage, necrosis, or cyst formation within the tumor, and can easily confirm bone involvement. Intravenous contrast may help in differentiating the mass from adjacent muscle and neurovascular complex.9,10 On MRI, renal synovial sarcomas are often described as nonspecific heterogeneous masses, although they may also exhibit heterogeneous enhancement of hemorrhagic areas, calcifications, and air-fluid levels (known as “triple sign”) as well as septae. The triple sign may be identified as areas of low, intermediate, and high signal intensity, correlating with areas of hemorrhage, calcification, and air-fluid level.9,10 Signal intensity is about equal to that of skeletal muscle on T1-weighted MRI and higher than that of subcutaneous fat on T2-weighted MRI.
In the present case, the tumor was initially misdiagnosed as an angiomyolipoma, the most common benign tumor of the kidney. Angiomyolipomas are usually solid triphasic tumors arising from the renal cortex and are composed of 3 major elements: dysmorphic blood vessels, smooth muscle components, and adipose tissue. When angiomyolipomas are large enough, they are readily recognized by the identification of macroscopic fat within the tumor, either by CT scan or MRI.11 When they are small, they may be difficult to distinguish from a small cyst on CT because of volume averaging.
On pathology, synovial sarcoma has dual epithelial and mesenchymal differentiation. They are frequently multi-lobulated, and areas of necrosis, hemorrhage, and cyst formation are also common. There are 3 main histologic subtypes of synovial sarcoma: biphasic (20%-30%), monophasic (50%-60%), and poorly differentiated (15%-25%). Poorly differentiated synovial sarcomas are generally epithelioid in morphology, have high mitotic activity (usually 10-20 mitoses/10 high-power field; range is <5 for well differentiated, low-grade tumors), and can be confused with round cell tumors such as Ewing sarcoma. Poorly differentiated synovial sarcomas are high-grade tumors.
Immunohistochemical studies can confirm the pathological diagnosis. Synovial sarcomas usually stain positive for Bcl2, CD99/Mic2, CD56, Vim, and focally for EMA but negatively for desmin, actin, WT1, S-100, CD34, and CD31.5 Currently, the gold standard for diagnosis and hallmark for synovial sarcomas are the t (X;18) translocation and SYT-SSX gene fusion products (SYT-SSX1 in 67% and SYT-SSX2 in 33% of cases). These can be detected either by FISH or reverse-transcription polymerase chain reaction. This genetic alteration is identified in more than 90% of synovial sarcomas and is highly specific.
The role of SYT-SSX gene fusion in the pathogenesis of synovial sarcoma is an active area of investigation. The fusion of SYT with SSX translates into a fusion protein that binds to the transcription activator SMARCA4 that is involved in chromatin remodeling, thus displacing both the wildtype SYT and the tumor suppressor gene SMARCB1. The modified protein complex then binds at several super-enhancer loci, unlocking suppressed genes such as Sox2, which is known to be necessary for synovial sarcoma proliferation. Alterations in SMARCB1 are involved in several cancer types, implicating this event as a driver of these malignancies.12 This results in a global alteration in chromatin remodeling that needs to be better understood to design targeted therapies.
The clinical course of synovial sarcoma, regardless of the tissue of origin, is typically poor. Multiple clinical and pathologic factors, including tumor size, location, patient age, and presence of poorly differentiated areas, are thought to have prognostic significance. A tumor size of more than 5 cm at presentation has the greatest impact on prognosis, with studies showing 5-year survival rates of 64% for patients with tumors smaller than 5 cm and 26% for patients with masses greater than 5 cm.13,14 High-grade synovial sarcoma is favored in tumors that have cystic components, hemorrhage, and fluid levels and the triple sign.
Patients with tumors in the extremities have a more favorable prognosis than those with lesions in the head and neck area or axially, a feature that likely reflects better surgical control available for extremity lesions. Patient age of less than 15 to 20 years is also associated with a better long-term prognosis.15,16 Varela-Duran and Enzinger17 reported that the presence of extensive calcifications suggests improved long-term survival, with 5-year survival rates of 82% and decreased rates of local recurrence (32%) and metastatic disease (29%). The poorly differentiated subtype is associated with a worsened prognosis, with a 5-year survival rate of 20% through 30%.18,19 Other pathologic factors associated with worsened prognosis include presence of rhabdoid cells, extensive tumor necrosis, high nuclear grade, p53 mutations, and high mitotic rate (>10 mitoses/10 high-power field). More recently, the gene fusion type SYT-SSX2 (more common in monophasic lesions) has been associated with an improved prognosis, compared with that for SYT-SSX1, and an 89% metastasis-free survival.20
Although there are no guidelines for the treatment of primary renal synovial sarcoma because of the limited number of cases reported, surgery is considered the first choice. Adjuvant chemotherapy with an anthracycline (doxorubicin or epirubicin) combined with ifosfamide has been the most frequently used regimen in published cases, especially in those in which patients have poor prognostic factors as mentioned above.
Overall, the 5-year survival rate ranges from 36% to 76%.14 The clinical course of synovial sarcoma is characterized by a high rate of local recurrence (30%-50%) and metastatic disease (41%). Most metastases occur within the first 2 to 5 years after treatment cessation. Metastases are present in 16% to 25% of patients at their initial presentation, with the most frequent metastatic site being the lung, followed by the lymph nodes (4%-18%) and bone (8%-11%).
Conclusion
Primary renal synovial sarcoma is extremely rare, and preoperative diagnosis is difficult in the absence of specific clinical or imaging findings. A high index of suspicion combined with pathologic, immunohistochemical, cytogenetic, and molecular studies is essential for accurate diagnosis and subsequent treatment planning. The differential diagnosis of renal synovial sarcoma can be extensive, and our experience with this patient illustrates the diagnostic dilemma associated with renal synovial sarcoma.
1. Majumder A, Dey S, Khandakar B, Medda S, Chandra Paul P. Primary renal synovial sarcoma: a rare tumor with an atypical presentation. Arch Iran Med. 2014;17(10):726-728.
2. Fetsch JF, Meis JM. Synovial sarcoma of the abdominal wall. Cancer. 1993;72(2):469 477.
3. Wang Z, Zhong Z, Zhu L, et al. Primary synovial sarcoma of the kidney: a case report. Oncol Lett. 2015;10(6):3542-3544.
4. Abbas M, Dämmrich ME, Braubach P, et al. Synovial sarcoma of the kidney in a young patient with a review of the literature. Rare tumors. 2014;6(2):5393
5. Argani P, Faria PA, Epstein JI, et al. Primary renal synovial sarcoma: molecular and morphologic delineation of an entity previously included among embryonal sarcomas of the kidney. Am J Surg Pathol. 2000;24(8):1087-1096.
6. Eble JN, Sauter G, Epstein JI, Sesterhenn IA, eds. World Health Organization classification of tumours: pathology and genetics of tumours of the urinary system and male genital organs. Lyon, France: IARC; 2004.
7. Tamboli P, Ro JY, Amin MB, Ligato S, Ayala AG. Benign tumors and tumor-like lesions of the adult kidney. Part II: benign mesenchymal and mixed neoplasms, and tumor-like lesions. Adv Anat Pathol. 2000;7(1):47-66.
8. Weiss SW, Goldblum JR. Malignant soft tissue tumors of uncertain type. In: Weiss SW, Goldblum JR, eds. Enzinger and Weiss’s soft tissue tumors. 4th ed. St. Louis, MO: Mosby, 2001; 1483-1565.
9. Lacovelli R, Altavilla A, Ciardi A, et al. Clinical and pathological features of primary renal synovial sarcoma: analysis of 64 cases from 11 years of medical literature. BJU Int. 2012;110(10):1449-1454.
10. Alhazzani AR, El-Sharkawy MS, Hassan H. Primary retroperitoneal synovial sarcoma in CT and MRI. Urol Ann. 2010;2(1):39-41.
11. Katabathina VS, Vikram R, Nagar AM, Tamboli P, Menias CO, Prasad SR. Mesenchymal neoplasms of the kidney in adults: imaging spectrum with radiologic-pathologic correlation. Radiographics. 2010;30(6):1525-1540.
12. Sápi Z, Papp G, Szendrői M, et al. Epigenetic regulation of SMARCB1 by miR-206, -381 and -671- 5p is evident in a variety of SMARCB1 immunonegative soft tissue sarcomas, while miR-765 appears specific for epithelioid sarcoma. A miRNA study of 223 soft tissue sarcomas. Genes Chromosomes Cancer. 2016;55(10):786-802.
13. Ferrari A, Gronchi A, Casanova M, et al. Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer. 2004;101(3):627-634.
14. Rangheard AS, Vanel D, Viala J, Schwaab G, Casiraghi O, Sigal R. Synovial sarcomas of the head and neck: CT and MR imaging findings of eight patients. Am J Neuroradiol. 2001;22(5):851-857.
15. Oda Y, Hashimoto H, Tsuneyoshi M, Takeshita S. Survival in synovial sarcoma: a multivariate study of prognostic factors with special emphasis on the comparison between early death and long-term survival. Am J Surg Pathol. 1993;17(1):35-44.
16. Raney RB. Synovial sarcoma in young people: background, prognostic factors and therapeutic questions. J Pediatr Hematol Oncol. 2005;27(4):207-211.
17. Varela-Duran J, Enzinger FM. Calcifying synovial sarcoma. Cancer. 1982;50(2):345-352.
18. Cagle LA, Mirra JM, Storm FK, Roe DJ, Eilber FR. Histologic features relating to prognosis in synovial sarcoma. Cancer. 1987;59(10):1810-1814.
19. Skytting B, Meis-Kindblom JM, Larsson O, et al. Synovial sarcoma – identification of favorable and unfavorable histologic types: a Scandinavian sarcoma group study of 104 cases. Acta Orthop Scand. 1999:70(6):543-554.
20. Murphey MD, Gibson MS, Jennings BT, Crespo-Rodríguez AM, Fanburg-Smith J, Gajewski DA. Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics. 2006;26(5):1543-1565.
1. Majumder A, Dey S, Khandakar B, Medda S, Chandra Paul P. Primary renal synovial sarcoma: a rare tumor with an atypical presentation. Arch Iran Med. 2014;17(10):726-728.
2. Fetsch JF, Meis JM. Synovial sarcoma of the abdominal wall. Cancer. 1993;72(2):469 477.
3. Wang Z, Zhong Z, Zhu L, et al. Primary synovial sarcoma of the kidney: a case report. Oncol Lett. 2015;10(6):3542-3544.
4. Abbas M, Dämmrich ME, Braubach P, et al. Synovial sarcoma of the kidney in a young patient with a review of the literature. Rare tumors. 2014;6(2):5393
5. Argani P, Faria PA, Epstein JI, et al. Primary renal synovial sarcoma: molecular and morphologic delineation of an entity previously included among embryonal sarcomas of the kidney. Am J Surg Pathol. 2000;24(8):1087-1096.
6. Eble JN, Sauter G, Epstein JI, Sesterhenn IA, eds. World Health Organization classification of tumours: pathology and genetics of tumours of the urinary system and male genital organs. Lyon, France: IARC; 2004.
7. Tamboli P, Ro JY, Amin MB, Ligato S, Ayala AG. Benign tumors and tumor-like lesions of the adult kidney. Part II: benign mesenchymal and mixed neoplasms, and tumor-like lesions. Adv Anat Pathol. 2000;7(1):47-66.
8. Weiss SW, Goldblum JR. Malignant soft tissue tumors of uncertain type. In: Weiss SW, Goldblum JR, eds. Enzinger and Weiss’s soft tissue tumors. 4th ed. St. Louis, MO: Mosby, 2001; 1483-1565.
9. Lacovelli R, Altavilla A, Ciardi A, et al. Clinical and pathological features of primary renal synovial sarcoma: analysis of 64 cases from 11 years of medical literature. BJU Int. 2012;110(10):1449-1454.
10. Alhazzani AR, El-Sharkawy MS, Hassan H. Primary retroperitoneal synovial sarcoma in CT and MRI. Urol Ann. 2010;2(1):39-41.
11. Katabathina VS, Vikram R, Nagar AM, Tamboli P, Menias CO, Prasad SR. Mesenchymal neoplasms of the kidney in adults: imaging spectrum with radiologic-pathologic correlation. Radiographics. 2010;30(6):1525-1540.
12. Sápi Z, Papp G, Szendrői M, et al. Epigenetic regulation of SMARCB1 by miR-206, -381 and -671- 5p is evident in a variety of SMARCB1 immunonegative soft tissue sarcomas, while miR-765 appears specific for epithelioid sarcoma. A miRNA study of 223 soft tissue sarcomas. Genes Chromosomes Cancer. 2016;55(10):786-802.
13. Ferrari A, Gronchi A, Casanova M, et al. Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer. 2004;101(3):627-634.
14. Rangheard AS, Vanel D, Viala J, Schwaab G, Casiraghi O, Sigal R. Synovial sarcomas of the head and neck: CT and MR imaging findings of eight patients. Am J Neuroradiol. 2001;22(5):851-857.
15. Oda Y, Hashimoto H, Tsuneyoshi M, Takeshita S. Survival in synovial sarcoma: a multivariate study of prognostic factors with special emphasis on the comparison between early death and long-term survival. Am J Surg Pathol. 1993;17(1):35-44.
16. Raney RB. Synovial sarcoma in young people: background, prognostic factors and therapeutic questions. J Pediatr Hematol Oncol. 2005;27(4):207-211.
17. Varela-Duran J, Enzinger FM. Calcifying synovial sarcoma. Cancer. 1982;50(2):345-352.
18. Cagle LA, Mirra JM, Storm FK, Roe DJ, Eilber FR. Histologic features relating to prognosis in synovial sarcoma. Cancer. 1987;59(10):1810-1814.
19. Skytting B, Meis-Kindblom JM, Larsson O, et al. Synovial sarcoma – identification of favorable and unfavorable histologic types: a Scandinavian sarcoma group study of 104 cases. Acta Orthop Scand. 1999:70(6):543-554.
20. Murphey MD, Gibson MS, Jennings BT, Crespo-Rodríguez AM, Fanburg-Smith J, Gajewski DA. Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics. 2006;26(5):1543-1565.