User login
Implant Designs in Revision Total Knee Arthroplasty
Before 1990, a considerable number of revisions were performed, largely for implant-associated failures, in the first few years after index primary knee arthroplasties.1,2 Since then, surgeons, manufacturers, and hospitals have collaborated to improve implant designs, techniques, and care guidelines.3,4 Despite the substantial improvements in designs, which led to implant longevity of more than 15 years in many cases, these devices still have limited life spans. Large studies have estimated that the risk for revision required after primary knee arthroplasty ranges from as low as 5% at 15 years to up to 9% at 10 years.4,5
The surgical goals of revision total knee arthroplasty (TKA) are to obtain stable fixation of the prosthesis to host bone, to obtain a stable range of motion compatible with the patient’s activities of daily living, and to achieve these goals while using the smallest amount of prosthetic augments and constraint so that the soft tissues may share in load transfer.6 As prosthetic constraint increases, the soft tissues participate less in load sharing, and increasing stresses are put on the implant–bone interface, which further increases the risk for early implant loosening.7 Hence, as characteristics of a revision implant become more constrained, there is often a higher rate of aseptic loosening expected.8
Controversy remains regarding the ideal implant type for revision TKA. To ensure the success of revision surgery and to reduce the risks for postoperative dissatisfaction, complications, and re-revision, orthopedists must understand the types of revision implant designs available, particularly as each has its own indications and potential complications.
In this article, we review the classification systems used for revision TKA as well as the types of prosthetic designs that can be used: posterior stabilized, nonlinked constrained, rotating hinge, and modular segmental.
1. Classification of bone loss and soft-tissue integrity
To further understand revision TKA, we must consider the complexity level of these cases, particularly by evaluating degree of bone loss and soft-tissue deficiency. The most accepted way to assess bone loss both before and during surgery is to use the AORI (Anderson Orthopaedic Research Institute) classification system.9 Bone loss can be classified into 3 types: I, in which metaphyseal bone is intact and small bone defects do not compromise component stability; II, in which metaphyseal bone is damaged and cancellous bone loss requires cement fill, augments, or bone graft; and III, in which metaphyseal bone is deficient, and lost bone comprises a major portion of condyle or plateau and occasionally requires bone grafts or custom implants (Table 1). These patterns of bone loss are occasionally associated with detachment of the collateral ligament or patellar tendon.
![]()
In addition to understanding bone loss in revision TKA, surgeons must be aware of soft-tissue deficiencies (eg, collateral ligaments, extensor mechanism), which also influence type and amount of prosthesis constraint. Specifically, constraint choice depends on amount of bone loss and on the condition of stabilizing tissues, such as the collateral ligaments. Under conditions of minimal bone loss and intact peripheral ligaments, a less constrained device, such as a primary posterior stabilized system, can be considered. When ligaments are present but insufficient, a semiconstrained device is recommended. In the presence of medial collateral ligament attenuation or complete medial or lateral collateral ligament dysfunction, a fully constrained prosthesis is required.8 Therefore, amount of bone loss or soft-tissue deficiency often dictates which prosthesis to use.
For radiographic classification, the Knee Society roentgenographic evaluation and scoring system10 has been implemented to allow for uniform reporting of radiographic results and to ensure adequate preoperative planning and postoperative assessment of component alignment. This system incorporates the evaluation of alignment in the coronal, sagittal, and patellofemoral planes and assesses radiolucency using zones dividing the implant–bone interface into segments to allow for easier classification of areas of lucency. More recently, a modified version of the Knee Society system was constructed.11 This modification simplifies zone classifications and accommodates more complex revision knee designs and stem extensions.
2. Posterior stabilized designs
Cruciate-retaining prostheses are seldom applicable in the revision TKA setting because of frequent damage to the posterior cruciate ligament, except in the case of simple polyethylene exchanges or, potentially, revisions of failed unicompartmental TKAs. Thus, posterior stabilized designs are the first-line choice for revision TKA (Figure 1). These prostheses are indicated only when the posterior cruciate ligament is incompetent and in the setting of adequate flexion and extension and medial and lateral collateral ligament balancing.
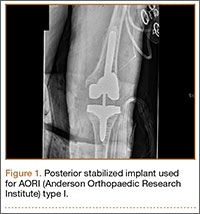
However, studies have shown that posterior stabilized TKAs have a limited role in revision TKAs, as the amount of ligamentous and bony damage is often underestimated in these patients, and use of a primary implant in a revision setting often requires additional augments, all of which may have contributed to the high failure rate. Thus, this design should be used only when the patient has adequate bone stock (AORI type I) and collateral ligament tension. This situation further emphasizes the importance of performing intraoperative testing for ligamentous balance and bone deficit evaluation in order to determine the most appropriate implant (Table 2).
3. Nonlinked constrained designs
Nonlinked constrained (condylar constrained) designs are the devices most commonly used for revision TKAs (>50% of revision knees). These prostheses provide increased articular constraint, which is required in patients with persistent instability, despite appropriate soft-tissue balancing. Increased articular constraint allows for more knee stability by providing progressive varus-valgus, coronal, and rotational stability with the aid of taller and wider tibial posts.12 Specifically, these implants incorporate a tibial post that fits closely between the femoral condyles, allowing for less motion compared with a standard posterior stabilized design.12
In addition, these designs may be used with augments, stems, and allografts when bone loss is more substantial. In particular, stem extensions allow for load distribution to the diaphyseal regions of the tibia and femur and thereby aid in reducing the increased stress at the bone–implant interface, which is a common concern with these implants. However, these extensions cost more, require intramedullary invasion, and are associated with higher rates of leg and thigh pain.12
These prostheses are often implicated in cases involving a high degree of bone loss (eg, AORI type II or III). They are ideally used in cases in which complete revision of both tibial and femoral components is needed and are indicated in cases of incompetent posterior cruciate ligament, partial functional loss of medial or lateral collateral ligaments, or flexion-extension mismatch.13 Furthermore, use of a constrained prosthesis is recommended in the setting of varus or valgus instability, or repeated dislocations of a posterior stabilized design (Table 2).
Ten-year survivorship ranges from 85% to 96%, but this is substantially lower than the 95% to 96% for condylar constrained prostheses used in primary TKAs.14-17 Moreover, the large discrepancy between survivorship of primary TKA and revision TKA with a constrained prosthesis further affirms that the complexity of revision surgery, rather than the prosthesis used, may have more deleterious effects on outcomes. However, surgeons must be aware that increased constraint leads to increased stress on the prosthetic interfaces with associated aseptic loosening and early failure, and this continues to be a legitimate concern.
4. Rotating hinge designs
Many patients who undergo revision TKA can be managed with a posterior stabilizing or nonlinked constrained design. However, in patients who present with severe ligamentous instability and bone loss (AORI type II or III), a rotating hinge prostheses, or highly constrained device, is often recommended (Figure 2).18 By using a rotating mobile-bearing platform, this prosthesis permits axial rotation through a metal-reinforced polyethylene-post articulation in the tibial tray. In addition, it involves use of modular diaphyseal-engaging stems and diaphyseal sleeves, which allow for the bypass of bony defects and areas of bone loss (Table 2).
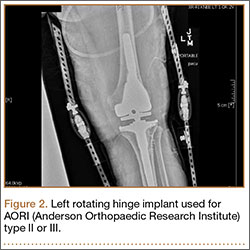
However, the rigid biomechanics of hinged prostheses is associated with increased risk for aseptic loosening (aseptic 10-year survival, 60%-80%), imparted by the transfer of stresses across the bone. The higher risk for early loosening, osteolysis, and excessive wear—caused by the highly restricted biomechanics of early generations of fixed hinged designs—has led to the development of new devices with mobile mechanics. Prosthetic designs have been improved with an added rotational axis to reduce torsional stress, a patellar resurfacing option, and better stem fixation and patellofemoral kinematics. Overall, these are aimed to improve rates of instability and aseptic loosening, with promising results demonstrated in the literature.
5. Modular segmental arthroplasty designs
Segmental arthroplasty prostheses, which typically are end-of-the-line revision TKA options, are applicable only in cases of extensive bone loss (more than can be treated with allografts or augments; AORI type 3), complete ligamentous disruption/absence, loss of periprosthetic soft tissue, and multiple previous revision procedures (Figure 3). Despite the limited indications for these prostheses, they yield quick return to function without graft nonunion or resorption, and they augment ingrowth/ongrowth. Furthermore, the next surgical option could be fusion or amputation. When failures were specifically evaluated for aseptic loosening across 4 studies, the survival rate ranged from 83% to 99.5%, with the most frequent complication being infection (up to 33% in one series).6,19-21
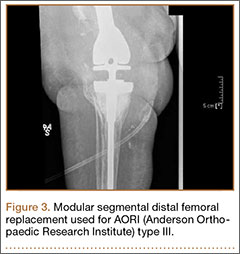
The major roles for segmental arthroplasty prostheses in primary TKAs are in the setting of oncologic conditions that require bony excision, or unreconstuctable fractures about the knee. Used after ancillary metastatic disease, these prostheses demonstrate positive results, according to several reports.22,23 In the setting of revision TKA, however, these prostheses should be used only when other surgical options are unfeasible, given the high risk for infection and the re-revision rates. Currently, revision TKAs with tumor prostheses have a high failure rate (up to 50%) because of the extensive surgery and the lack of bony and soft-tissue support (Table 2).
Conclusion
Orthopedists performing revision TKAs must consider bone stock and remaining ligament stability. In particular, they should choose implants for least constraint and adequate knee stability, as these are essential in minimizing the stresses on the implant–bone interface. Ultimately, functional outcomes, survivorship, and postoperative satisfaction determine the success of these designs. However, predictors of outcomes of revision surgery are often multifactorial, and surgeons must also consider procedure complexity and patient-specific characteristics.
1. Fehring TK, Odum S, Griffin WL, Mason JB, Nadaud M. Early failures in total knee arthroplasty. Clin Orthop Relat Res. 2001;392:315-318.
2. Sharkey PF, Hozack WJ, Rothman RH, Shastri S, Jacoby SM. Insall Award paper. Why are total knee arthroplasties failing today? Clin Orthop Relat Res. 2002;404:7-13.
3. Schroer WC, Berend KR, Lombardi AV, et al. Why are total knees failing today? Etiology of total knee revision in 2010 and 2011. J Arthroplasty. 2013;28(8 suppl):116-119.
4. Kim TK. CORR Insights(®): risk factors for revision within 10 years of total knee arthroplasty. Clin Orthop Relat Res. 2014;472(4):1208-1209.
5. Sheng PY, Jämsen E, Lehto MU, Konttinen YT, Pajamäki J, Halonen P. Revision total knee arthroplasty with the Total Condylar III system in inflammatory arthritis. J Bone Joint Surg Br. 2005;87(9):1222-1224.
6. Haas SB, Insall JN, Montgomery W 3rd, Windsor RE. Revision total knee arthroplasty with use of modular components with stems inserted without cement. J Bone Joint Surg Am. 1995;77(11):1700-1707.
7. Dennis DA. A stepwise approach to revision total knee arthroplasty. J Arthroplasty. 2007;22(4 suppl 1):32-38.
8. Vasso M, Beaufils P, Schiavone Panni A. Constraint choice in revision knee arthroplasty. Int Orthop. 2013;37(7):1279-1284.
9. Engh GA, Ammeen DJ. Bone loss with revision total knee arthroplasty: defect classification and alternatives for reconstruction. Instr Course Lect. 1999;48:167-175.
10. Ewald FC. The Knee Society total knee arthroplasty roentgenographic evaluation and scoring system. Clin Orthop Relat Res. 1989;248:9-12.
11. Meneghini RM, Mont MA, Backstein DB, Bourne RB, Dennis DA, Scuderi GR. Development of a modern Knee Society radiographic evaluation system and methodology for total knee arthroplasty. J Arthroplasty. 2015;30(12):2311-2314.
12. Nam D, Umunna BP, Cross MB, Reinhardt KR, Duggal S, Cornell CN. Clinical results and failure mechanisms of a nonmodular constrained knee without stem extensions. HSS J. 2012;8(2):96-102.
13. Lombardi AV Jr, Berend KR. The role of implant constraint in revision TKA: striking the balance. Orthopedics. 2006;29(9):847-849.
14. Lachiewicz PF, Soileau ES. Results of a second-generation constrained condylar prosthesis in primary total knee arthroplasty. J Arthroplasty. 2011;26(8):1228-1231.
15. Bae DK, Song SJ, Heo DB, Lee SH, Song WJ. Long-term survival rate of implants and modes of failure after revision total knee arthroplasty by a single surgeon. J Arthroplasty. 2013;28(7):1130-1134.
16. Wilke BK, Wagner ER, Trousdale RT. Long-term survival of semi-constrained total knee arthroplasty for revision surgery. J Arthroplasty. 2014;29(5):1005-1008.
17. Lachiewicz PF, Soileau ES. Ten-year survival and clinical results of constrained components in primary total knee arthroplasty. J Arthroplasty. 2006;21(6):803-808.
18. Jones RE. Total knee arthroplasty with modular rotating-platform hinge. Orthopedics. 2006;29(9 suppl):S80-S82.
19. Korim MT, Esler CN, Reddy VR, Ashford RU. A systematic review of endoprosthetic replacement for non-tumour indications around the knee joint. The Knee. 2013;20:367-375.
20. Hofmann AA, Goldberg T, Tanner AM, Kurtin SM. Treatment of infected total knee arthroplasty using an articulating spacer: 2- to 12-year experience. Clin Orthop Relat Res. 2005;(430):125-131.
21. Peters CL, Erickson J, Kloepper RG, Mohr RA. Revision total knee arthroplasty with modular components inserted with metaphyseal cement and stems without cement. J Arthroplasty. 2005;20:302-308.
22. Pala E, Trovarelli G, Calabro T, Angelini A, Abati CN, Ruggieri P. Survival of modern knee tumor megaprostheses: failures, functional results, and a comparative statistical analysis. Clinical Orthop Relat Res. 2015;473:891-899.
23. Angelini A, Henderson E, Trovarelli G, Ruggieri P. Is there a role for knee arthrodesis with modular endoprostheses for tumor and revision of failed endoprostheses? Clin Orthop Relat Res. 2013;471(10):3326-3335.
Before 1990, a considerable number of revisions were performed, largely for implant-associated failures, in the first few years after index primary knee arthroplasties.1,2 Since then, surgeons, manufacturers, and hospitals have collaborated to improve implant designs, techniques, and care guidelines.3,4 Despite the substantial improvements in designs, which led to implant longevity of more than 15 years in many cases, these devices still have limited life spans. Large studies have estimated that the risk for revision required after primary knee arthroplasty ranges from as low as 5% at 15 years to up to 9% at 10 years.4,5
The surgical goals of revision total knee arthroplasty (TKA) are to obtain stable fixation of the prosthesis to host bone, to obtain a stable range of motion compatible with the patient’s activities of daily living, and to achieve these goals while using the smallest amount of prosthetic augments and constraint so that the soft tissues may share in load transfer.6 As prosthetic constraint increases, the soft tissues participate less in load sharing, and increasing stresses are put on the implant–bone interface, which further increases the risk for early implant loosening.7 Hence, as characteristics of a revision implant become more constrained, there is often a higher rate of aseptic loosening expected.8
Controversy remains regarding the ideal implant type for revision TKA. To ensure the success of revision surgery and to reduce the risks for postoperative dissatisfaction, complications, and re-revision, orthopedists must understand the types of revision implant designs available, particularly as each has its own indications and potential complications.
In this article, we review the classification systems used for revision TKA as well as the types of prosthetic designs that can be used: posterior stabilized, nonlinked constrained, rotating hinge, and modular segmental.
1. Classification of bone loss and soft-tissue integrity
To further understand revision TKA, we must consider the complexity level of these cases, particularly by evaluating degree of bone loss and soft-tissue deficiency. The most accepted way to assess bone loss both before and during surgery is to use the AORI (Anderson Orthopaedic Research Institute) classification system.9 Bone loss can be classified into 3 types: I, in which metaphyseal bone is intact and small bone defects do not compromise component stability; II, in which metaphyseal bone is damaged and cancellous bone loss requires cement fill, augments, or bone graft; and III, in which metaphyseal bone is deficient, and lost bone comprises a major portion of condyle or plateau and occasionally requires bone grafts or custom implants (Table 1). These patterns of bone loss are occasionally associated with detachment of the collateral ligament or patellar tendon.
![]()
In addition to understanding bone loss in revision TKA, surgeons must be aware of soft-tissue deficiencies (eg, collateral ligaments, extensor mechanism), which also influence type and amount of prosthesis constraint. Specifically, constraint choice depends on amount of bone loss and on the condition of stabilizing tissues, such as the collateral ligaments. Under conditions of minimal bone loss and intact peripheral ligaments, a less constrained device, such as a primary posterior stabilized system, can be considered. When ligaments are present but insufficient, a semiconstrained device is recommended. In the presence of medial collateral ligament attenuation or complete medial or lateral collateral ligament dysfunction, a fully constrained prosthesis is required.8 Therefore, amount of bone loss or soft-tissue deficiency often dictates which prosthesis to use.
For radiographic classification, the Knee Society roentgenographic evaluation and scoring system10 has been implemented to allow for uniform reporting of radiographic results and to ensure adequate preoperative planning and postoperative assessment of component alignment. This system incorporates the evaluation of alignment in the coronal, sagittal, and patellofemoral planes and assesses radiolucency using zones dividing the implant–bone interface into segments to allow for easier classification of areas of lucency. More recently, a modified version of the Knee Society system was constructed.11 This modification simplifies zone classifications and accommodates more complex revision knee designs and stem extensions.
2. Posterior stabilized designs
Cruciate-retaining prostheses are seldom applicable in the revision TKA setting because of frequent damage to the posterior cruciate ligament, except in the case of simple polyethylene exchanges or, potentially, revisions of failed unicompartmental TKAs. Thus, posterior stabilized designs are the first-line choice for revision TKA (Figure 1). These prostheses are indicated only when the posterior cruciate ligament is incompetent and in the setting of adequate flexion and extension and medial and lateral collateral ligament balancing.
However, studies have shown that posterior stabilized TKAs have a limited role in revision TKAs, as the amount of ligamentous and bony damage is often underestimated in these patients, and use of a primary implant in a revision setting often requires additional augments, all of which may have contributed to the high failure rate. Thus, this design should be used only when the patient has adequate bone stock (AORI type I) and collateral ligament tension. This situation further emphasizes the importance of performing intraoperative testing for ligamentous balance and bone deficit evaluation in order to determine the most appropriate implant (Table 2).
3. Nonlinked constrained designs
Nonlinked constrained (condylar constrained) designs are the devices most commonly used for revision TKAs (>50% of revision knees). These prostheses provide increased articular constraint, which is required in patients with persistent instability, despite appropriate soft-tissue balancing. Increased articular constraint allows for more knee stability by providing progressive varus-valgus, coronal, and rotational stability with the aid of taller and wider tibial posts.12 Specifically, these implants incorporate a tibial post that fits closely between the femoral condyles, allowing for less motion compared with a standard posterior stabilized design.12
In addition, these designs may be used with augments, stems, and allografts when bone loss is more substantial. In particular, stem extensions allow for load distribution to the diaphyseal regions of the tibia and femur and thereby aid in reducing the increased stress at the bone–implant interface, which is a common concern with these implants. However, these extensions cost more, require intramedullary invasion, and are associated with higher rates of leg and thigh pain.12
These prostheses are often implicated in cases involving a high degree of bone loss (eg, AORI type II or III). They are ideally used in cases in which complete revision of both tibial and femoral components is needed and are indicated in cases of incompetent posterior cruciate ligament, partial functional loss of medial or lateral collateral ligaments, or flexion-extension mismatch.13 Furthermore, use of a constrained prosthesis is recommended in the setting of varus or valgus instability, or repeated dislocations of a posterior stabilized design (Table 2).
Ten-year survivorship ranges from 85% to 96%, but this is substantially lower than the 95% to 96% for condylar constrained prostheses used in primary TKAs.14-17 Moreover, the large discrepancy between survivorship of primary TKA and revision TKA with a constrained prosthesis further affirms that the complexity of revision surgery, rather than the prosthesis used, may have more deleterious effects on outcomes. However, surgeons must be aware that increased constraint leads to increased stress on the prosthetic interfaces with associated aseptic loosening and early failure, and this continues to be a legitimate concern.
4. Rotating hinge designs
Many patients who undergo revision TKA can be managed with a posterior stabilizing or nonlinked constrained design. However, in patients who present with severe ligamentous instability and bone loss (AORI type II or III), a rotating hinge prostheses, or highly constrained device, is often recommended (Figure 2).18 By using a rotating mobile-bearing platform, this prosthesis permits axial rotation through a metal-reinforced polyethylene-post articulation in the tibial tray. In addition, it involves use of modular diaphyseal-engaging stems and diaphyseal sleeves, which allow for the bypass of bony defects and areas of bone loss (Table 2).
However, the rigid biomechanics of hinged prostheses is associated with increased risk for aseptic loosening (aseptic 10-year survival, 60%-80%), imparted by the transfer of stresses across the bone. The higher risk for early loosening, osteolysis, and excessive wear—caused by the highly restricted biomechanics of early generations of fixed hinged designs—has led to the development of new devices with mobile mechanics. Prosthetic designs have been improved with an added rotational axis to reduce torsional stress, a patellar resurfacing option, and better stem fixation and patellofemoral kinematics. Overall, these are aimed to improve rates of instability and aseptic loosening, with promising results demonstrated in the literature.
5. Modular segmental arthroplasty designs
Segmental arthroplasty prostheses, which typically are end-of-the-line revision TKA options, are applicable only in cases of extensive bone loss (more than can be treated with allografts or augments; AORI type 3), complete ligamentous disruption/absence, loss of periprosthetic soft tissue, and multiple previous revision procedures (Figure 3). Despite the limited indications for these prostheses, they yield quick return to function without graft nonunion or resorption, and they augment ingrowth/ongrowth. Furthermore, the next surgical option could be fusion or amputation. When failures were specifically evaluated for aseptic loosening across 4 studies, the survival rate ranged from 83% to 99.5%, with the most frequent complication being infection (up to 33% in one series).6,19-21
The major roles for segmental arthroplasty prostheses in primary TKAs are in the setting of oncologic conditions that require bony excision, or unreconstuctable fractures about the knee. Used after ancillary metastatic disease, these prostheses demonstrate positive results, according to several reports.22,23 In the setting of revision TKA, however, these prostheses should be used only when other surgical options are unfeasible, given the high risk for infection and the re-revision rates. Currently, revision TKAs with tumor prostheses have a high failure rate (up to 50%) because of the extensive surgery and the lack of bony and soft-tissue support (Table 2).
Conclusion
Orthopedists performing revision TKAs must consider bone stock and remaining ligament stability. In particular, they should choose implants for least constraint and adequate knee stability, as these are essential in minimizing the stresses on the implant–bone interface. Ultimately, functional outcomes, survivorship, and postoperative satisfaction determine the success of these designs. However, predictors of outcomes of revision surgery are often multifactorial, and surgeons must also consider procedure complexity and patient-specific characteristics.
Before 1990, a considerable number of revisions were performed, largely for implant-associated failures, in the first few years after index primary knee arthroplasties.1,2 Since then, surgeons, manufacturers, and hospitals have collaborated to improve implant designs, techniques, and care guidelines.3,4 Despite the substantial improvements in designs, which led to implant longevity of more than 15 years in many cases, these devices still have limited life spans. Large studies have estimated that the risk for revision required after primary knee arthroplasty ranges from as low as 5% at 15 years to up to 9% at 10 years.4,5
The surgical goals of revision total knee arthroplasty (TKA) are to obtain stable fixation of the prosthesis to host bone, to obtain a stable range of motion compatible with the patient’s activities of daily living, and to achieve these goals while using the smallest amount of prosthetic augments and constraint so that the soft tissues may share in load transfer.6 As prosthetic constraint increases, the soft tissues participate less in load sharing, and increasing stresses are put on the implant–bone interface, which further increases the risk for early implant loosening.7 Hence, as characteristics of a revision implant become more constrained, there is often a higher rate of aseptic loosening expected.8
Controversy remains regarding the ideal implant type for revision TKA. To ensure the success of revision surgery and to reduce the risks for postoperative dissatisfaction, complications, and re-revision, orthopedists must understand the types of revision implant designs available, particularly as each has its own indications and potential complications.
In this article, we review the classification systems used for revision TKA as well as the types of prosthetic designs that can be used: posterior stabilized, nonlinked constrained, rotating hinge, and modular segmental.
1. Classification of bone loss and soft-tissue integrity
To further understand revision TKA, we must consider the complexity level of these cases, particularly by evaluating degree of bone loss and soft-tissue deficiency. The most accepted way to assess bone loss both before and during surgery is to use the AORI (Anderson Orthopaedic Research Institute) classification system.9 Bone loss can be classified into 3 types: I, in which metaphyseal bone is intact and small bone defects do not compromise component stability; II, in which metaphyseal bone is damaged and cancellous bone loss requires cement fill, augments, or bone graft; and III, in which metaphyseal bone is deficient, and lost bone comprises a major portion of condyle or plateau and occasionally requires bone grafts or custom implants (Table 1). These patterns of bone loss are occasionally associated with detachment of the collateral ligament or patellar tendon.
![]()
In addition to understanding bone loss in revision TKA, surgeons must be aware of soft-tissue deficiencies (eg, collateral ligaments, extensor mechanism), which also influence type and amount of prosthesis constraint. Specifically, constraint choice depends on amount of bone loss and on the condition of stabilizing tissues, such as the collateral ligaments. Under conditions of minimal bone loss and intact peripheral ligaments, a less constrained device, such as a primary posterior stabilized system, can be considered. When ligaments are present but insufficient, a semiconstrained device is recommended. In the presence of medial collateral ligament attenuation or complete medial or lateral collateral ligament dysfunction, a fully constrained prosthesis is required.8 Therefore, amount of bone loss or soft-tissue deficiency often dictates which prosthesis to use.
For radiographic classification, the Knee Society roentgenographic evaluation and scoring system10 has been implemented to allow for uniform reporting of radiographic results and to ensure adequate preoperative planning and postoperative assessment of component alignment. This system incorporates the evaluation of alignment in the coronal, sagittal, and patellofemoral planes and assesses radiolucency using zones dividing the implant–bone interface into segments to allow for easier classification of areas of lucency. More recently, a modified version of the Knee Society system was constructed.11 This modification simplifies zone classifications and accommodates more complex revision knee designs and stem extensions.
2. Posterior stabilized designs
Cruciate-retaining prostheses are seldom applicable in the revision TKA setting because of frequent damage to the posterior cruciate ligament, except in the case of simple polyethylene exchanges or, potentially, revisions of failed unicompartmental TKAs. Thus, posterior stabilized designs are the first-line choice for revision TKA (Figure 1). These prostheses are indicated only when the posterior cruciate ligament is incompetent and in the setting of adequate flexion and extension and medial and lateral collateral ligament balancing.
However, studies have shown that posterior stabilized TKAs have a limited role in revision TKAs, as the amount of ligamentous and bony damage is often underestimated in these patients, and use of a primary implant in a revision setting often requires additional augments, all of which may have contributed to the high failure rate. Thus, this design should be used only when the patient has adequate bone stock (AORI type I) and collateral ligament tension. This situation further emphasizes the importance of performing intraoperative testing for ligamentous balance and bone deficit evaluation in order to determine the most appropriate implant (Table 2).
3. Nonlinked constrained designs
Nonlinked constrained (condylar constrained) designs are the devices most commonly used for revision TKAs (>50% of revision knees). These prostheses provide increased articular constraint, which is required in patients with persistent instability, despite appropriate soft-tissue balancing. Increased articular constraint allows for more knee stability by providing progressive varus-valgus, coronal, and rotational stability with the aid of taller and wider tibial posts.12 Specifically, these implants incorporate a tibial post that fits closely between the femoral condyles, allowing for less motion compared with a standard posterior stabilized design.12
In addition, these designs may be used with augments, stems, and allografts when bone loss is more substantial. In particular, stem extensions allow for load distribution to the diaphyseal regions of the tibia and femur and thereby aid in reducing the increased stress at the bone–implant interface, which is a common concern with these implants. However, these extensions cost more, require intramedullary invasion, and are associated with higher rates of leg and thigh pain.12
These prostheses are often implicated in cases involving a high degree of bone loss (eg, AORI type II or III). They are ideally used in cases in which complete revision of both tibial and femoral components is needed and are indicated in cases of incompetent posterior cruciate ligament, partial functional loss of medial or lateral collateral ligaments, or flexion-extension mismatch.13 Furthermore, use of a constrained prosthesis is recommended in the setting of varus or valgus instability, or repeated dislocations of a posterior stabilized design (Table 2).
Ten-year survivorship ranges from 85% to 96%, but this is substantially lower than the 95% to 96% for condylar constrained prostheses used in primary TKAs.14-17 Moreover, the large discrepancy between survivorship of primary TKA and revision TKA with a constrained prosthesis further affirms that the complexity of revision surgery, rather than the prosthesis used, may have more deleterious effects on outcomes. However, surgeons must be aware that increased constraint leads to increased stress on the prosthetic interfaces with associated aseptic loosening and early failure, and this continues to be a legitimate concern.
4. Rotating hinge designs
Many patients who undergo revision TKA can be managed with a posterior stabilizing or nonlinked constrained design. However, in patients who present with severe ligamentous instability and bone loss (AORI type II or III), a rotating hinge prostheses, or highly constrained device, is often recommended (Figure 2).18 By using a rotating mobile-bearing platform, this prosthesis permits axial rotation through a metal-reinforced polyethylene-post articulation in the tibial tray. In addition, it involves use of modular diaphyseal-engaging stems and diaphyseal sleeves, which allow for the bypass of bony defects and areas of bone loss (Table 2).
However, the rigid biomechanics of hinged prostheses is associated with increased risk for aseptic loosening (aseptic 10-year survival, 60%-80%), imparted by the transfer of stresses across the bone. The higher risk for early loosening, osteolysis, and excessive wear—caused by the highly restricted biomechanics of early generations of fixed hinged designs—has led to the development of new devices with mobile mechanics. Prosthetic designs have been improved with an added rotational axis to reduce torsional stress, a patellar resurfacing option, and better stem fixation and patellofemoral kinematics. Overall, these are aimed to improve rates of instability and aseptic loosening, with promising results demonstrated in the literature.
5. Modular segmental arthroplasty designs
Segmental arthroplasty prostheses, which typically are end-of-the-line revision TKA options, are applicable only in cases of extensive bone loss (more than can be treated with allografts or augments; AORI type 3), complete ligamentous disruption/absence, loss of periprosthetic soft tissue, and multiple previous revision procedures (Figure 3). Despite the limited indications for these prostheses, they yield quick return to function without graft nonunion or resorption, and they augment ingrowth/ongrowth. Furthermore, the next surgical option could be fusion or amputation. When failures were specifically evaluated for aseptic loosening across 4 studies, the survival rate ranged from 83% to 99.5%, with the most frequent complication being infection (up to 33% in one series).6,19-21
The major roles for segmental arthroplasty prostheses in primary TKAs are in the setting of oncologic conditions that require bony excision, or unreconstuctable fractures about the knee. Used after ancillary metastatic disease, these prostheses demonstrate positive results, according to several reports.22,23 In the setting of revision TKA, however, these prostheses should be used only when other surgical options are unfeasible, given the high risk for infection and the re-revision rates. Currently, revision TKAs with tumor prostheses have a high failure rate (up to 50%) because of the extensive surgery and the lack of bony and soft-tissue support (Table 2).
Conclusion
Orthopedists performing revision TKAs must consider bone stock and remaining ligament stability. In particular, they should choose implants for least constraint and adequate knee stability, as these are essential in minimizing the stresses on the implant–bone interface. Ultimately, functional outcomes, survivorship, and postoperative satisfaction determine the success of these designs. However, predictors of outcomes of revision surgery are often multifactorial, and surgeons must also consider procedure complexity and patient-specific characteristics.
1. Fehring TK, Odum S, Griffin WL, Mason JB, Nadaud M. Early failures in total knee arthroplasty. Clin Orthop Relat Res. 2001;392:315-318.
2. Sharkey PF, Hozack WJ, Rothman RH, Shastri S, Jacoby SM. Insall Award paper. Why are total knee arthroplasties failing today? Clin Orthop Relat Res. 2002;404:7-13.
3. Schroer WC, Berend KR, Lombardi AV, et al. Why are total knees failing today? Etiology of total knee revision in 2010 and 2011. J Arthroplasty. 2013;28(8 suppl):116-119.
4. Kim TK. CORR Insights(®): risk factors for revision within 10 years of total knee arthroplasty. Clin Orthop Relat Res. 2014;472(4):1208-1209.
5. Sheng PY, Jämsen E, Lehto MU, Konttinen YT, Pajamäki J, Halonen P. Revision total knee arthroplasty with the Total Condylar III system in inflammatory arthritis. J Bone Joint Surg Br. 2005;87(9):1222-1224.
6. Haas SB, Insall JN, Montgomery W 3rd, Windsor RE. Revision total knee arthroplasty with use of modular components with stems inserted without cement. J Bone Joint Surg Am. 1995;77(11):1700-1707.
7. Dennis DA. A stepwise approach to revision total knee arthroplasty. J Arthroplasty. 2007;22(4 suppl 1):32-38.
8. Vasso M, Beaufils P, Schiavone Panni A. Constraint choice in revision knee arthroplasty. Int Orthop. 2013;37(7):1279-1284.
9. Engh GA, Ammeen DJ. Bone loss with revision total knee arthroplasty: defect classification and alternatives for reconstruction. Instr Course Lect. 1999;48:167-175.
10. Ewald FC. The Knee Society total knee arthroplasty roentgenographic evaluation and scoring system. Clin Orthop Relat Res. 1989;248:9-12.
11. Meneghini RM, Mont MA, Backstein DB, Bourne RB, Dennis DA, Scuderi GR. Development of a modern Knee Society radiographic evaluation system and methodology for total knee arthroplasty. J Arthroplasty. 2015;30(12):2311-2314.
12. Nam D, Umunna BP, Cross MB, Reinhardt KR, Duggal S, Cornell CN. Clinical results and failure mechanisms of a nonmodular constrained knee without stem extensions. HSS J. 2012;8(2):96-102.
13. Lombardi AV Jr, Berend KR. The role of implant constraint in revision TKA: striking the balance. Orthopedics. 2006;29(9):847-849.
14. Lachiewicz PF, Soileau ES. Results of a second-generation constrained condylar prosthesis in primary total knee arthroplasty. J Arthroplasty. 2011;26(8):1228-1231.
15. Bae DK, Song SJ, Heo DB, Lee SH, Song WJ. Long-term survival rate of implants and modes of failure after revision total knee arthroplasty by a single surgeon. J Arthroplasty. 2013;28(7):1130-1134.
16. Wilke BK, Wagner ER, Trousdale RT. Long-term survival of semi-constrained total knee arthroplasty for revision surgery. J Arthroplasty. 2014;29(5):1005-1008.
17. Lachiewicz PF, Soileau ES. Ten-year survival and clinical results of constrained components in primary total knee arthroplasty. J Arthroplasty. 2006;21(6):803-808.
18. Jones RE. Total knee arthroplasty with modular rotating-platform hinge. Orthopedics. 2006;29(9 suppl):S80-S82.
19. Korim MT, Esler CN, Reddy VR, Ashford RU. A systematic review of endoprosthetic replacement for non-tumour indications around the knee joint. The Knee. 2013;20:367-375.
20. Hofmann AA, Goldberg T, Tanner AM, Kurtin SM. Treatment of infected total knee arthroplasty using an articulating spacer: 2- to 12-year experience. Clin Orthop Relat Res. 2005;(430):125-131.
21. Peters CL, Erickson J, Kloepper RG, Mohr RA. Revision total knee arthroplasty with modular components inserted with metaphyseal cement and stems without cement. J Arthroplasty. 2005;20:302-308.
22. Pala E, Trovarelli G, Calabro T, Angelini A, Abati CN, Ruggieri P. Survival of modern knee tumor megaprostheses: failures, functional results, and a comparative statistical analysis. Clinical Orthop Relat Res. 2015;473:891-899.
23. Angelini A, Henderson E, Trovarelli G, Ruggieri P. Is there a role for knee arthrodesis with modular endoprostheses for tumor and revision of failed endoprostheses? Clin Orthop Relat Res. 2013;471(10):3326-3335.
1. Fehring TK, Odum S, Griffin WL, Mason JB, Nadaud M. Early failures in total knee arthroplasty. Clin Orthop Relat Res. 2001;392:315-318.
2. Sharkey PF, Hozack WJ, Rothman RH, Shastri S, Jacoby SM. Insall Award paper. Why are total knee arthroplasties failing today? Clin Orthop Relat Res. 2002;404:7-13.
3. Schroer WC, Berend KR, Lombardi AV, et al. Why are total knees failing today? Etiology of total knee revision in 2010 and 2011. J Arthroplasty. 2013;28(8 suppl):116-119.
4. Kim TK. CORR Insights(®): risk factors for revision within 10 years of total knee arthroplasty. Clin Orthop Relat Res. 2014;472(4):1208-1209.
5. Sheng PY, Jämsen E, Lehto MU, Konttinen YT, Pajamäki J, Halonen P. Revision total knee arthroplasty with the Total Condylar III system in inflammatory arthritis. J Bone Joint Surg Br. 2005;87(9):1222-1224.
6. Haas SB, Insall JN, Montgomery W 3rd, Windsor RE. Revision total knee arthroplasty with use of modular components with stems inserted without cement. J Bone Joint Surg Am. 1995;77(11):1700-1707.
7. Dennis DA. A stepwise approach to revision total knee arthroplasty. J Arthroplasty. 2007;22(4 suppl 1):32-38.
8. Vasso M, Beaufils P, Schiavone Panni A. Constraint choice in revision knee arthroplasty. Int Orthop. 2013;37(7):1279-1284.
9. Engh GA, Ammeen DJ. Bone loss with revision total knee arthroplasty: defect classification and alternatives for reconstruction. Instr Course Lect. 1999;48:167-175.
10. Ewald FC. The Knee Society total knee arthroplasty roentgenographic evaluation and scoring system. Clin Orthop Relat Res. 1989;248:9-12.
11. Meneghini RM, Mont MA, Backstein DB, Bourne RB, Dennis DA, Scuderi GR. Development of a modern Knee Society radiographic evaluation system and methodology for total knee arthroplasty. J Arthroplasty. 2015;30(12):2311-2314.
12. Nam D, Umunna BP, Cross MB, Reinhardt KR, Duggal S, Cornell CN. Clinical results and failure mechanisms of a nonmodular constrained knee without stem extensions. HSS J. 2012;8(2):96-102.
13. Lombardi AV Jr, Berend KR. The role of implant constraint in revision TKA: striking the balance. Orthopedics. 2006;29(9):847-849.
14. Lachiewicz PF, Soileau ES. Results of a second-generation constrained condylar prosthesis in primary total knee arthroplasty. J Arthroplasty. 2011;26(8):1228-1231.
15. Bae DK, Song SJ, Heo DB, Lee SH, Song WJ. Long-term survival rate of implants and modes of failure after revision total knee arthroplasty by a single surgeon. J Arthroplasty. 2013;28(7):1130-1134.
16. Wilke BK, Wagner ER, Trousdale RT. Long-term survival of semi-constrained total knee arthroplasty for revision surgery. J Arthroplasty. 2014;29(5):1005-1008.
17. Lachiewicz PF, Soileau ES. Ten-year survival and clinical results of constrained components in primary total knee arthroplasty. J Arthroplasty. 2006;21(6):803-808.
18. Jones RE. Total knee arthroplasty with modular rotating-platform hinge. Orthopedics. 2006;29(9 suppl):S80-S82.
19. Korim MT, Esler CN, Reddy VR, Ashford RU. A systematic review of endoprosthetic replacement for non-tumour indications around the knee joint. The Knee. 2013;20:367-375.
20. Hofmann AA, Goldberg T, Tanner AM, Kurtin SM. Treatment of infected total knee arthroplasty using an articulating spacer: 2- to 12-year experience. Clin Orthop Relat Res. 2005;(430):125-131.
21. Peters CL, Erickson J, Kloepper RG, Mohr RA. Revision total knee arthroplasty with modular components inserted with metaphyseal cement and stems without cement. J Arthroplasty. 2005;20:302-308.
22. Pala E, Trovarelli G, Calabro T, Angelini A, Abati CN, Ruggieri P. Survival of modern knee tumor megaprostheses: failures, functional results, and a comparative statistical analysis. Clinical Orthop Relat Res. 2015;473:891-899.
23. Angelini A, Henderson E, Trovarelli G, Ruggieri P. Is there a role for knee arthrodesis with modular endoprostheses for tumor and revision of failed endoprostheses? Clin Orthop Relat Res. 2013;471(10):3326-3335.

Nonoperative Treatment of Rotator Cuff Tears
Rotator cuff disease is extremely common, yet indications for surgery are not well established. Unfortunately, data on the natural history of patients with rotator cuff disease are lacking, as are high-level studies evaluating the effectiveness of rotator cuff repair. This deficit is highlighted by the recent American Academy of Orthopaedic Surgeons clinical practice guideline on optimizing the management of rotator cuff problems,1 in which none of the position statements were based on high-level evidence, and 22 of 25 statements were inconclusive or based on weak evidence or represented the panel’s consensus opinion. Although the traditional teaching is that rotator cuff tears (RCTs) should be surgically repaired, the present article reviews the evidence supporting physical therapy as a treatment for atraumatic full-thickness RCTs.
1. Less than 5% of people with RCTs undergo surgery
Studies on symptomatic and asymptomatic patients have found a high incidence of RCTs in the population at large.2,3 By conservative estimate, 10% of people older than 65 years have full-thickness RCTs. Therefore, the 2010 US Census4 finding of 57 million people over age 65 years translates to 5.7 million with full-thickness RCTs. In the United States, about 275,000 rotator cuff surgeries are performed annually.5 That is, less than 5% of people with RCTs undergo surgery each year.
2. Symptoms do not correlate well with RCT severity
Pain is statistically more likely in patients who experience RCT progression than in those who do not.6-8 However, RCTs may progress without pain, or there may be pain without progression, making pain a poor sign of RCT progression.9 The Multicenter Orthopaedic Outcome Network (MOON) Shoulder Group, studying a cohort of patients with atraumatic full-thickness RCTs, found no relationship between RCT severity and pain,10 symptom duration,11 or activity level,12 suggesting the relationship between RCTs and symptoms is not robust.
3. The high failure rates of surgical repairs do not affect patient-reported outcomes
Postoperative imaging has demonstrated high failure rates for rotator cuff repairs, yet patient-reported outcome scores do not differ between cases of intact and failed repairs.13,14 Strength is better, however, in intact repairs.14
4. Physical therapy is effective in treating atraumatic RCTs
The MOON Shoulder Group conducted a prospective cohort study to determine the predictors of failed physical therapy for atraumatic full-thickness RCTs and to help define the indications for rotator cuff surgery.15 All enrolled patients started with a well-defined physical therapy program, and they could opt out and have surgery at any time. The physical therapy program, derived from a systematic review of the literature, was found to be effective in more than 80% of patients with follow-up of 2 years or longer.15 The most important predictor of failed nonoperative treatment was patient expectations: For a patient who thought physical therapy would work, it worked; for a patient who thought it would not work, surgery was the more likely choice. No measure of pain or RCT severity predicted the need for surgery.16 For 2 randomized trials that compared surgery and physical therapy, the success of nonoperative treatment was similar: 76% (Moosmayer and colleagues17) and 92% (Kukkonen and colleagues18).
5. What are the indications for surgery?
These data suggest that physical therapy is reasonable for patients with atraumatic RCTs. Some data suggest that traumatic RCTs should be treated with surgery and that it should be performed early.19 Other data suggest strength is better after rotator cuff repair.13,14 What, then, are the indications for surgery? Patients with acute tears probably should have surgery; patients concerned about weakness should consider surgery but should keep in mind that its benefit depends on an intact rotator cuff repair; and patients with low expectations about the effectiveness of physical therapy probably should consider surgery.
When discussing options with a patient, you might approach informed consent as follows:
“Mr. Smith, you have a rotator cuff tear. So do at least 6 million other Americans over age 60 years. Only 5% of those undergo surgery. If your problem is weakness or functional loss, you should have surgery, though there is about a 30% chance the repair will fail. I don’t know how to predict the outcome of repair yet, but I worry your atraumatic tear is at risk for repair failure.
“If your problem is pain, you have an 80% chance of improving with physical therapy, and pain relief seems to last at least 2 years. If you go with physical therapy, however, there is a risk your tear could progress and start causing symptoms. I don’t yet know how likely it is your tear will progress or, if it does progress, how likely it is the tear will cause symptoms. I wish we had better information to help you make your decision.”
1. Pedowitz RA, Yamaguchi K, Ahmad CS, et al. American Academy of Orthopaedic Surgeons clinical practice guideline on: optimizing the management of rotator cuff problems. J Bone Joint Surg Am. 2012;94(2):163-167.
2. Reilly P, Macleod I, Macfarlane R, Windley J, Emery RJ. Dead men and radiologists don’t lie: a review of cadaveric and radiological studies of rotator cuff tear prevalence. Ann R Coll Surg Engl. 2006;88(2):116-121.
3. Teunis, T, Lubberts B, Reilly BT, Ring D. A systematic review and pooled analysis of the prevalence of rotator cuff pathology with increasing age. J Shoulder Elbow Surg. 2014;23(12):1913-1921.
4. Werner CA. The older population: 2010 (2010 Census briefs). US Census Bureau website. http://www.census.gov/prod/cen2010/briefs/c2010br-09.pdf. Published November 2011. Accessed December 13, 2015.
5. Colvin AC, Egorova N, Harrison AK, Moskowitz A, Flatow EL. National trends in rotator cuff repair. J Bone Joint Surg Am. 2012;94(3):227-233.
6. Mall NA, Kim HM, Keener JD, et al. Symptomatic progression of asymptomatic rotator cuff tears: a prospective study of clinical and sonographic variables. J Bone Joint Surg Am. 2010;92(16):2623-2633.
7. Moosmayer S, Tariq R, Stiris M, Smith HJ. The natural history of asymptomatic rotator cuff tears: a three-year follow-up of fifty cases. J Bone Joint Surg Am. 2013;95(14):1249-1255.
8. Safran O, Schroeder J, Bloom R, Weil Y, Milgrom C. Natural history of nonoperatively treated symptomatic rotator cuff tears in patients 60 years old or younger. Am J Sports Med. 2011;39(4):710-714.
9. Kuhn JE. Are atraumatic rotator cuff tears painful? A model to describe the relationship between pain and rotator cuff tears. Minerva Orthop Traumatol. 2015;66:51-61.
10. Dunn WR, Kuhn JE, Sanders R, et al. Symptoms of pain do not correlate with rotator cuff tear severity: a cross-sectional study of 393 patients with a symptomatic atraumatic full-thickness rotator cuff tear. J Bone Joint Surg Am. 2014;96(10):793-800.
11. MOON Shoulder Group: Unruh KP, Kuhn JE, Sanders R, et al. The duration of symptoms does not correlate with rotator cuff tear severity or other patient-related features: a cross-sectional study of patients with atraumatic, full-thickness rotator cuff tears. J Shoulder Elbow Surg. 2014;23(7):1052-1058.
12. Brophy RH, Dunn WR, Kuhn JE; MOON Shoulder Group. Shoulder activity level is not associated with the severity of symptomatic, atraumatic rotator cuff tears in patients electing nonoperative treatment. Am J Sports Med. 2014;42(5):1150-1154.
13. Slabaugh MA, Nho SJ, Grumet RC, et al. Does the literature confirm superior clinical results in radiographically healed rotator cuffs after rotator cuff repair? Arthroscopy. 2010;26(3):393-403.
14. Russell RD, Knight JR, Mulligan E, Khazzam MS. Structural integrity after rotator cuff repair does not correlate with patient function and pain: a meta-analysis. J Bone Joint Surg Am. 2014;96(4):265-271.
15. Kuhn JE, Dunn WR, Sanders R, et al; MOON Shoulder Group. Effectiveness of physical therapy in treating atraumatic full-thickness rotator cuff tears: a multicenter prospective cohort study. J Shoulder Elbow Surg. 2013;22(10):1371-1379.
16. Dunn WR, Kuhn JE, Sanders R, et al. Defining indications for rotator cuff repair: predictors of failure of nonoperative treatment of chronic, symptomatic full-thickness rotator cuff tears. Paper presented at: Open Meeting of the American Shoulder and Elbow Surgeons; March 23, 2013; Chicago, IL.
17. Moosmayer S, Lund G, Seljom US, et al. Tendon repair compared with physiotherapy in the treatment of rotator cuff tears: a randomized controlled study in 103 cases with a five-year follow-up. J Bone Joint Surg Am. 2014;96(18):1504-1514.
18. Kukkonen J, Joukainen A, Lehtinen J, et al. Treatment of non-traumatic rotator cuff tears: a randomised controlled trial with one-year clinical results. Bone Joint J Br. 2014;96(1):75-81.
19. Oh LS, Wolf BR, Hall MP, Levy BA, Marx RG. Indications for rotator cuff repair: a systematic review. Clin Orthop Relat Res. 2007;(455):52-63.
Rotator cuff disease is extremely common, yet indications for surgery are not well established. Unfortunately, data on the natural history of patients with rotator cuff disease are lacking, as are high-level studies evaluating the effectiveness of rotator cuff repair. This deficit is highlighted by the recent American Academy of Orthopaedic Surgeons clinical practice guideline on optimizing the management of rotator cuff problems,1 in which none of the position statements were based on high-level evidence, and 22 of 25 statements were inconclusive or based on weak evidence or represented the panel’s consensus opinion. Although the traditional teaching is that rotator cuff tears (RCTs) should be surgically repaired, the present article reviews the evidence supporting physical therapy as a treatment for atraumatic full-thickness RCTs.
1. Less than 5% of people with RCTs undergo surgery
Studies on symptomatic and asymptomatic patients have found a high incidence of RCTs in the population at large.2,3 By conservative estimate, 10% of people older than 65 years have full-thickness RCTs. Therefore, the 2010 US Census4 finding of 57 million people over age 65 years translates to 5.7 million with full-thickness RCTs. In the United States, about 275,000 rotator cuff surgeries are performed annually.5 That is, less than 5% of people with RCTs undergo surgery each year.
2. Symptoms do not correlate well with RCT severity
Pain is statistically more likely in patients who experience RCT progression than in those who do not.6-8 However, RCTs may progress without pain, or there may be pain without progression, making pain a poor sign of RCT progression.9 The Multicenter Orthopaedic Outcome Network (MOON) Shoulder Group, studying a cohort of patients with atraumatic full-thickness RCTs, found no relationship between RCT severity and pain,10 symptom duration,11 or activity level,12 suggesting the relationship between RCTs and symptoms is not robust.
3. The high failure rates of surgical repairs do not affect patient-reported outcomes
Postoperative imaging has demonstrated high failure rates for rotator cuff repairs, yet patient-reported outcome scores do not differ between cases of intact and failed repairs.13,14 Strength is better, however, in intact repairs.14
4. Physical therapy is effective in treating atraumatic RCTs
The MOON Shoulder Group conducted a prospective cohort study to determine the predictors of failed physical therapy for atraumatic full-thickness RCTs and to help define the indications for rotator cuff surgery.15 All enrolled patients started with a well-defined physical therapy program, and they could opt out and have surgery at any time. The physical therapy program, derived from a systematic review of the literature, was found to be effective in more than 80% of patients with follow-up of 2 years or longer.15 The most important predictor of failed nonoperative treatment was patient expectations: For a patient who thought physical therapy would work, it worked; for a patient who thought it would not work, surgery was the more likely choice. No measure of pain or RCT severity predicted the need for surgery.16 For 2 randomized trials that compared surgery and physical therapy, the success of nonoperative treatment was similar: 76% (Moosmayer and colleagues17) and 92% (Kukkonen and colleagues18).
5. What are the indications for surgery?
These data suggest that physical therapy is reasonable for patients with atraumatic RCTs. Some data suggest that traumatic RCTs should be treated with surgery and that it should be performed early.19 Other data suggest strength is better after rotator cuff repair.13,14 What, then, are the indications for surgery? Patients with acute tears probably should have surgery; patients concerned about weakness should consider surgery but should keep in mind that its benefit depends on an intact rotator cuff repair; and patients with low expectations about the effectiveness of physical therapy probably should consider surgery.
When discussing options with a patient, you might approach informed consent as follows:
“Mr. Smith, you have a rotator cuff tear. So do at least 6 million other Americans over age 60 years. Only 5% of those undergo surgery. If your problem is weakness or functional loss, you should have surgery, though there is about a 30% chance the repair will fail. I don’t know how to predict the outcome of repair yet, but I worry your atraumatic tear is at risk for repair failure.
“If your problem is pain, you have an 80% chance of improving with physical therapy, and pain relief seems to last at least 2 years. If you go with physical therapy, however, there is a risk your tear could progress and start causing symptoms. I don’t yet know how likely it is your tear will progress or, if it does progress, how likely it is the tear will cause symptoms. I wish we had better information to help you make your decision.”
Rotator cuff disease is extremely common, yet indications for surgery are not well established. Unfortunately, data on the natural history of patients with rotator cuff disease are lacking, as are high-level studies evaluating the effectiveness of rotator cuff repair. This deficit is highlighted by the recent American Academy of Orthopaedic Surgeons clinical practice guideline on optimizing the management of rotator cuff problems,1 in which none of the position statements were based on high-level evidence, and 22 of 25 statements were inconclusive or based on weak evidence or represented the panel’s consensus opinion. Although the traditional teaching is that rotator cuff tears (RCTs) should be surgically repaired, the present article reviews the evidence supporting physical therapy as a treatment for atraumatic full-thickness RCTs.
1. Less than 5% of people with RCTs undergo surgery
Studies on symptomatic and asymptomatic patients have found a high incidence of RCTs in the population at large.2,3 By conservative estimate, 10% of people older than 65 years have full-thickness RCTs. Therefore, the 2010 US Census4 finding of 57 million people over age 65 years translates to 5.7 million with full-thickness RCTs. In the United States, about 275,000 rotator cuff surgeries are performed annually.5 That is, less than 5% of people with RCTs undergo surgery each year.
2. Symptoms do not correlate well with RCT severity
Pain is statistically more likely in patients who experience RCT progression than in those who do not.6-8 However, RCTs may progress without pain, or there may be pain without progression, making pain a poor sign of RCT progression.9 The Multicenter Orthopaedic Outcome Network (MOON) Shoulder Group, studying a cohort of patients with atraumatic full-thickness RCTs, found no relationship between RCT severity and pain,10 symptom duration,11 or activity level,12 suggesting the relationship between RCTs and symptoms is not robust.
3. The high failure rates of surgical repairs do not affect patient-reported outcomes
Postoperative imaging has demonstrated high failure rates for rotator cuff repairs, yet patient-reported outcome scores do not differ between cases of intact and failed repairs.13,14 Strength is better, however, in intact repairs.14
4. Physical therapy is effective in treating atraumatic RCTs
The MOON Shoulder Group conducted a prospective cohort study to determine the predictors of failed physical therapy for atraumatic full-thickness RCTs and to help define the indications for rotator cuff surgery.15 All enrolled patients started with a well-defined physical therapy program, and they could opt out and have surgery at any time. The physical therapy program, derived from a systematic review of the literature, was found to be effective in more than 80% of patients with follow-up of 2 years or longer.15 The most important predictor of failed nonoperative treatment was patient expectations: For a patient who thought physical therapy would work, it worked; for a patient who thought it would not work, surgery was the more likely choice. No measure of pain or RCT severity predicted the need for surgery.16 For 2 randomized trials that compared surgery and physical therapy, the success of nonoperative treatment was similar: 76% (Moosmayer and colleagues17) and 92% (Kukkonen and colleagues18).
5. What are the indications for surgery?
These data suggest that physical therapy is reasonable for patients with atraumatic RCTs. Some data suggest that traumatic RCTs should be treated with surgery and that it should be performed early.19 Other data suggest strength is better after rotator cuff repair.13,14 What, then, are the indications for surgery? Patients with acute tears probably should have surgery; patients concerned about weakness should consider surgery but should keep in mind that its benefit depends on an intact rotator cuff repair; and patients with low expectations about the effectiveness of physical therapy probably should consider surgery.
When discussing options with a patient, you might approach informed consent as follows:
“Mr. Smith, you have a rotator cuff tear. So do at least 6 million other Americans over age 60 years. Only 5% of those undergo surgery. If your problem is weakness or functional loss, you should have surgery, though there is about a 30% chance the repair will fail. I don’t know how to predict the outcome of repair yet, but I worry your atraumatic tear is at risk for repair failure.
“If your problem is pain, you have an 80% chance of improving with physical therapy, and pain relief seems to last at least 2 years. If you go with physical therapy, however, there is a risk your tear could progress and start causing symptoms. I don’t yet know how likely it is your tear will progress or, if it does progress, how likely it is the tear will cause symptoms. I wish we had better information to help you make your decision.”
1. Pedowitz RA, Yamaguchi K, Ahmad CS, et al. American Academy of Orthopaedic Surgeons clinical practice guideline on: optimizing the management of rotator cuff problems. J Bone Joint Surg Am. 2012;94(2):163-167.
2. Reilly P, Macleod I, Macfarlane R, Windley J, Emery RJ. Dead men and radiologists don’t lie: a review of cadaveric and radiological studies of rotator cuff tear prevalence. Ann R Coll Surg Engl. 2006;88(2):116-121.
3. Teunis, T, Lubberts B, Reilly BT, Ring D. A systematic review and pooled analysis of the prevalence of rotator cuff pathology with increasing age. J Shoulder Elbow Surg. 2014;23(12):1913-1921.
4. Werner CA. The older population: 2010 (2010 Census briefs). US Census Bureau website. http://www.census.gov/prod/cen2010/briefs/c2010br-09.pdf. Published November 2011. Accessed December 13, 2015.
5. Colvin AC, Egorova N, Harrison AK, Moskowitz A, Flatow EL. National trends in rotator cuff repair. J Bone Joint Surg Am. 2012;94(3):227-233.
6. Mall NA, Kim HM, Keener JD, et al. Symptomatic progression of asymptomatic rotator cuff tears: a prospective study of clinical and sonographic variables. J Bone Joint Surg Am. 2010;92(16):2623-2633.
7. Moosmayer S, Tariq R, Stiris M, Smith HJ. The natural history of asymptomatic rotator cuff tears: a three-year follow-up of fifty cases. J Bone Joint Surg Am. 2013;95(14):1249-1255.
8. Safran O, Schroeder J, Bloom R, Weil Y, Milgrom C. Natural history of nonoperatively treated symptomatic rotator cuff tears in patients 60 years old or younger. Am J Sports Med. 2011;39(4):710-714.
9. Kuhn JE. Are atraumatic rotator cuff tears painful? A model to describe the relationship between pain and rotator cuff tears. Minerva Orthop Traumatol. 2015;66:51-61.
10. Dunn WR, Kuhn JE, Sanders R, et al. Symptoms of pain do not correlate with rotator cuff tear severity: a cross-sectional study of 393 patients with a symptomatic atraumatic full-thickness rotator cuff tear. J Bone Joint Surg Am. 2014;96(10):793-800.
11. MOON Shoulder Group: Unruh KP, Kuhn JE, Sanders R, et al. The duration of symptoms does not correlate with rotator cuff tear severity or other patient-related features: a cross-sectional study of patients with atraumatic, full-thickness rotator cuff tears. J Shoulder Elbow Surg. 2014;23(7):1052-1058.
12. Brophy RH, Dunn WR, Kuhn JE; MOON Shoulder Group. Shoulder activity level is not associated with the severity of symptomatic, atraumatic rotator cuff tears in patients electing nonoperative treatment. Am J Sports Med. 2014;42(5):1150-1154.
13. Slabaugh MA, Nho SJ, Grumet RC, et al. Does the literature confirm superior clinical results in radiographically healed rotator cuffs after rotator cuff repair? Arthroscopy. 2010;26(3):393-403.
14. Russell RD, Knight JR, Mulligan E, Khazzam MS. Structural integrity after rotator cuff repair does not correlate with patient function and pain: a meta-analysis. J Bone Joint Surg Am. 2014;96(4):265-271.
15. Kuhn JE, Dunn WR, Sanders R, et al; MOON Shoulder Group. Effectiveness of physical therapy in treating atraumatic full-thickness rotator cuff tears: a multicenter prospective cohort study. J Shoulder Elbow Surg. 2013;22(10):1371-1379.
16. Dunn WR, Kuhn JE, Sanders R, et al. Defining indications for rotator cuff repair: predictors of failure of nonoperative treatment of chronic, symptomatic full-thickness rotator cuff tears. Paper presented at: Open Meeting of the American Shoulder and Elbow Surgeons; March 23, 2013; Chicago, IL.
17. Moosmayer S, Lund G, Seljom US, et al. Tendon repair compared with physiotherapy in the treatment of rotator cuff tears: a randomized controlled study in 103 cases with a five-year follow-up. J Bone Joint Surg Am. 2014;96(18):1504-1514.
18. Kukkonen J, Joukainen A, Lehtinen J, et al. Treatment of non-traumatic rotator cuff tears: a randomised controlled trial with one-year clinical results. Bone Joint J Br. 2014;96(1):75-81.
19. Oh LS, Wolf BR, Hall MP, Levy BA, Marx RG. Indications for rotator cuff repair: a systematic review. Clin Orthop Relat Res. 2007;(455):52-63.
1. Pedowitz RA, Yamaguchi K, Ahmad CS, et al. American Academy of Orthopaedic Surgeons clinical practice guideline on: optimizing the management of rotator cuff problems. J Bone Joint Surg Am. 2012;94(2):163-167.
2. Reilly P, Macleod I, Macfarlane R, Windley J, Emery RJ. Dead men and radiologists don’t lie: a review of cadaveric and radiological studies of rotator cuff tear prevalence. Ann R Coll Surg Engl. 2006;88(2):116-121.
3. Teunis, T, Lubberts B, Reilly BT, Ring D. A systematic review and pooled analysis of the prevalence of rotator cuff pathology with increasing age. J Shoulder Elbow Surg. 2014;23(12):1913-1921.
4. Werner CA. The older population: 2010 (2010 Census briefs). US Census Bureau website. http://www.census.gov/prod/cen2010/briefs/c2010br-09.pdf. Published November 2011. Accessed December 13, 2015.
5. Colvin AC, Egorova N, Harrison AK, Moskowitz A, Flatow EL. National trends in rotator cuff repair. J Bone Joint Surg Am. 2012;94(3):227-233.
6. Mall NA, Kim HM, Keener JD, et al. Symptomatic progression of asymptomatic rotator cuff tears: a prospective study of clinical and sonographic variables. J Bone Joint Surg Am. 2010;92(16):2623-2633.
7. Moosmayer S, Tariq R, Stiris M, Smith HJ. The natural history of asymptomatic rotator cuff tears: a three-year follow-up of fifty cases. J Bone Joint Surg Am. 2013;95(14):1249-1255.
8. Safran O, Schroeder J, Bloom R, Weil Y, Milgrom C. Natural history of nonoperatively treated symptomatic rotator cuff tears in patients 60 years old or younger. Am J Sports Med. 2011;39(4):710-714.
9. Kuhn JE. Are atraumatic rotator cuff tears painful? A model to describe the relationship between pain and rotator cuff tears. Minerva Orthop Traumatol. 2015;66:51-61.
10. Dunn WR, Kuhn JE, Sanders R, et al. Symptoms of pain do not correlate with rotator cuff tear severity: a cross-sectional study of 393 patients with a symptomatic atraumatic full-thickness rotator cuff tear. J Bone Joint Surg Am. 2014;96(10):793-800.
11. MOON Shoulder Group: Unruh KP, Kuhn JE, Sanders R, et al. The duration of symptoms does not correlate with rotator cuff tear severity or other patient-related features: a cross-sectional study of patients with atraumatic, full-thickness rotator cuff tears. J Shoulder Elbow Surg. 2014;23(7):1052-1058.
12. Brophy RH, Dunn WR, Kuhn JE; MOON Shoulder Group. Shoulder activity level is not associated with the severity of symptomatic, atraumatic rotator cuff tears in patients electing nonoperative treatment. Am J Sports Med. 2014;42(5):1150-1154.
13. Slabaugh MA, Nho SJ, Grumet RC, et al. Does the literature confirm superior clinical results in radiographically healed rotator cuffs after rotator cuff repair? Arthroscopy. 2010;26(3):393-403.
14. Russell RD, Knight JR, Mulligan E, Khazzam MS. Structural integrity after rotator cuff repair does not correlate with patient function and pain: a meta-analysis. J Bone Joint Surg Am. 2014;96(4):265-271.
15. Kuhn JE, Dunn WR, Sanders R, et al; MOON Shoulder Group. Effectiveness of physical therapy in treating atraumatic full-thickness rotator cuff tears: a multicenter prospective cohort study. J Shoulder Elbow Surg. 2013;22(10):1371-1379.
16. Dunn WR, Kuhn JE, Sanders R, et al. Defining indications for rotator cuff repair: predictors of failure of nonoperative treatment of chronic, symptomatic full-thickness rotator cuff tears. Paper presented at: Open Meeting of the American Shoulder and Elbow Surgeons; March 23, 2013; Chicago, IL.
17. Moosmayer S, Lund G, Seljom US, et al. Tendon repair compared with physiotherapy in the treatment of rotator cuff tears: a randomized controlled study in 103 cases with a five-year follow-up. J Bone Joint Surg Am. 2014;96(18):1504-1514.
18. Kukkonen J, Joukainen A, Lehtinen J, et al. Treatment of non-traumatic rotator cuff tears: a randomised controlled trial with one-year clinical results. Bone Joint J Br. 2014;96(1):75-81.
19. Oh LS, Wolf BR, Hall MP, Levy BA, Marx RG. Indications for rotator cuff repair: a systematic review. Clin Orthop Relat Res. 2007;(455):52-63.

Adipose Flap Versus Fascial Sling for Anterior Subcutaneous Transposition of the Ulnar Nerve
Compression of the ulnar nerve at the elbow, also referred to as cubital tunnel syndrome (CuTS), is the second most common peripheral nerve compression syndrome in the upper extremity.1,2 Although the ulnar nerve can be compressed at 5 different sites, including arcade of Struthers, medial intermuscular septum, medial epicondyle, and deep flexor aponeurosis, the cubital tunnel is most commonly affected.3 Patients typically present with paresthesias in the fourth and fifth digits and weakness of hand muscle intrinsics. Activity-related pain or pain at the medial elbow can also occur in more advanced pathology.4 It is estimated that conservative therapy fails and surgical intervention is required in up to 30% of patients with CuTS.1 Surgical approaches range from in situ decompression to transposition techniques, but there is no consensus in the orthopedic community as to which technique offers the best results. In a 2008 meta-analysis, Macadam and colleagues5 found no statistical differences in outcomes among the various surgical approaches. Nevertheless, subcutaneous transposition of the ulnar nerve at the elbow is a popular option.6
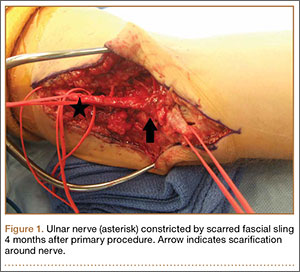
Despite the widespread success of surgical intervention for CuTS, persistent or recurrent pain occurs in 9.9% to 21.0% of cases.7-10 In addition, several investigators have cited perineural scarring as a major cause of recurrent symptoms after primary surgery.11-14 Filippi and colleagues11 noted that patients who required reoperation after primary anterior transposition had “serious epineural fibrosis and fibrosis around the transposed ulnar nerve.” At our institution, we have similarly found that scarring of the fascial sling around the ulnar nerve led to recurrence of CuTS within 4 months after initial surgery (Figure 1).
We therefore prefer to use a vascularized adipose flap to secure the anteriorly transposed ulnar nerve. This flap provides a pliable, vascularized adipose environment for the nerve, which helps reduce nerve adherence and may enhance nerve recovery.15 In the study reported here, we retrospectively reviewed the long-term outcomes of ulnar nerve anterior subcutaneous transposition secured with either an adipose flap or a fascial sling. We hypothesized that patients in the 2 groups (adipose flap, fascial sling) would have equivalent outcomes.
Materials and Methods
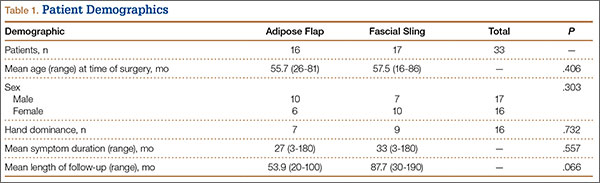
After obtaining institutional review board approval, we reviewed the medical and surgical records of 104 patients (107 limbs) who underwent transposition of the ulnar nerve secured with either an adipose flap (27 limbs) or a fascial sling (80 limbs) over a 14-year period. The fascial sling cohort was used as a comparison group, matched to the adipose flap cohort by sex, age at time of surgery, hand dominance, symptom duration, and length of follow-up (Table 1). Patients were indicated for surgery and were included in the study if they had a history and physical examination consistent with primary CuTS, symptom duration longer than 1 year, and failed conservative management, including activity modification, night splinting, elbow pads, occupational therapy, and home exercise regimen. Electrodiagnostic testing was used at the discretion of the attending surgeon when the diagnosis was not clear from the history and physical examination. All fascial sling procedures were performed at our institution by 1 of 3 fellowship-trained hand surgeons, including Dr. Rosenwasser. The adipose flap modification was performed only by Dr. Rosenwasser. Of the 27 patients in the adipose flap group, 23 underwent surgery for primary CuTS and were included in the study; the other 4 (revision cases) were excluded; 1 patient subsequently died of a cause unrelated to the surgical procedure, and 6 were lost to follow-up. Of the 80 patients in the fascial sling group, 30 underwent surgery for primary CuTS; 5 died before follow-up, and 8 declined to participate.
Thirty-three patients (16 adipose flap, 17 fascial sling) met the inclusion criteria. Of the 16 adipose flap patients, 15 underwent the physical examination and completed the questionnaire, and 1 was interviewed by telephone. Similarly, of the 17 fascial sling patients, 15 underwent the physical examination and completed the questionnaire, and 2 were interviewed by telephone. There were no bilateral cases. Conservative management (activity modification, night splinting, elbow pads, occupational therapy, home exercise) failed in all cases.
A trained study team member who was not part of the surgical team performed follow-up evaluations using objective outcome measures and subjective questionnaires. Patients were assessed at a mean follow-up of 5.6 years (range, 1.6-15.9 years). Patients completed the DASH (Disabilities of the Arm, Shoulder, and Hand) questionnaire16 and visual analog scales (VASs) for pain, numbness, tingling, and weakness in the ulnar nerve distribution. They also rated the presence of night symptoms that were interfering with sleep. The Modified Bishop Rating Scale (MBRS) was used to quantify patient self-reported data17,18 (Figure 2). The MBRS measures overall satisfaction, symptom improvement, presence of residual symptoms, ability to engage in activities, work capability, and subjective changes in strength and sensibility.
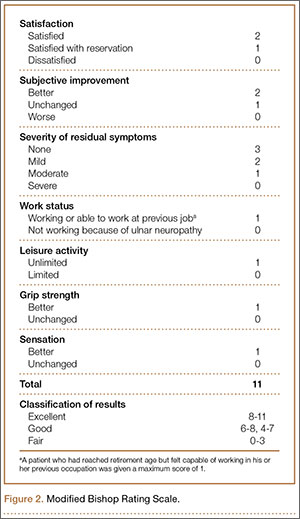
In the physical examinations, we tested for Tinel, Wartenberg, and Froment signs; performed an elbow flexion test; and measured elbow range of motion for flexion and extension as well as forearm pronation and supination. We also evaluated lateral pinch strength and grip strength, using a Jamar hydraulic pinch gauge and a Jamar dynamometer (Therapeutic Equipment Corp) and taking the average of 3 assessments. Fifth-digit abduction strength was graded on a standard muscle strength scale. Two-point discrimination was measured at the middle, ring, and small digits of the operated and contralateral hands.19
Surgical Technique
Standard ulnar nerve decompression with anterior subcutaneous transposition and the following modifications were performed on all patients.20 A posteromedial incision parallel to the intermuscular septum was developed and the ulnar nerve identified. Minimizing stripping of the vascular mesentery, the dissection continued along the course of the nerve, and the medial intermuscular septum was excised to prevent secondary compression after transposition. The ulnar nerve was mobilized and transposed anterior to the medial epicondyle (Figure 3). For patients who received the fascial sling, a fascial sleeve was elevated from the flexor-pronator mass and sutured to the edge of the retinaculum securing the nerve. For patients who received the adipose flap, the flap with its vascular pedicle intact was elevated from the subcutaneous tissue of the anterior skin overlying the transposed nerve. The adipose tissue was sharply dissected in half while sufficient subcutaneous tissue was kept between the skin and the flap. A plane was developed based on an anterior adipose pedicle, which included a cutaneous artery and a vein that would supply the vascularized adipose flap. The flap was elevated and wrapped around the nerve without tension while the ulnar nerve was protected from being kinked by the construct. The flap was sutured to the anterior subcutaneous tissue to create a tunnel of adipose tissue surrounding the nerve along its length (Figure 4). The elbow was then flexed and extended to ensure free nerve gliding before wound closure.
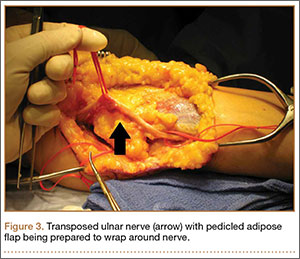
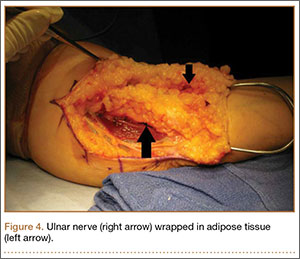
The patient was allowed to move the elbow within the bulky dressings immediately after surgery. After 2 weeks, sutures were removed. Formal occupational therapy is not needed for these patients, except in the presence of significant weakness.
Results
As mentioned, the 2 groups were matched on demographics: age at time of surgery, sex, symptom duration, and length of follow-up (Table 1).
For the 16 adipose flap patients (Table 2), mean DASH score was 19.9 (range, 0-71.7). Seven of these patients reported upper extremity pain with a mean VAS score of 1.7 (range, 0-8); 4 patients reported pain in the wrist and fourth and fifth digits; only 1 patient reported pain that occasionally woke the patient from sleep. Constant numbness was present in 6 patients. Four patients reported constant mild tingling in the hand, and 11 reported intermittent tingling. Eleven patients (68.7%) reported operated-arm weakness with a mean VAS score of 3.4 (range, 0-8). In patients who had a physical examination, mean elbow flexion–extension arc of motion was 134° (range, 95°-150°), representing 99% of the motion of the contralateral arm. Mean pronation–supination arc was 174° (range, 150°-180°), accounting for 104% of the contralateral arm. Mean lateral pinch strength was 73% of the contralateral arm, and mean grip strength was 114% of the contralateral arm. The Tinel sign was present in 2 patients, the Froment sign was present in 3 patients, and the elbow flexion test was positive in 2 patients. No patient had a positive Wartenberg sign. On the MBRS, 10 patients had an excellent score, and 6 had a good score.
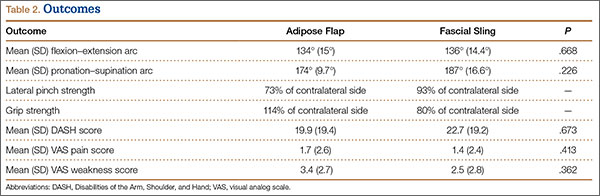
For the 17 fascial sling patients (Table 2), mean DASH score was 22.7 (range, 0-63.3). Three patients reported upper extremity pain with a mean VAS score of 1.4 (range, 0-7); 3 patients reported pain that occasionally woke them from sleep. Seven patients had constant numbness in the distribution of the ulnar nerve. Two patients had constant paresthesias, and 7 had intermittent paresthesias. Nine patients (52.9%) reported arm weakness with a mean VAS score of 2.5 (range, 0-8). Mean elbow flexion–extension arc of motion was 136° (range, 100°-150°), representing 100% of the contralateral arm. Mean pronation–supination arc was 187° (range, 155°-225°), accounting for 102% of the contralateral arm. Mean lateral pinch strength was 93% of the contralateral arm, and mean grip strength was 80% of the contralateral arm. The Tinel sign was present in 6 patients, the Froment sign in 3 patients, and the Wartenberg sign in 2 patients. The elbow flexion test was positive in 4 patients. On the MBRS, 10 patients had an excellent score, and 7 had a good score.
There was no recurrence of CuTS in either group. One adipose flap patient developed a wound infection that required reoperation.
Discussion
Ulnar neuropathy was described by Magee and Phalen21 in 1949 and termed cubital tunnel syndrome by Feindel and Stratford22 in 1958. Since then, numerous procedures, including in situ decompression, medial epicondylectomy, and endoscopic decompression,23,24 have been advocated for the treatment of this condition. In addition, anterior transposition, which involves securing the ulnar nerve in a submuscular, intramuscular, or subcutaneous sleeve,6 remains a popular option. Despite more than half a century of surgical treatment for this condition, there is no consensus about which procedure offers the best outcomes. Bartels and colleagues8 retrospectively reviewed surgical treatments for CuTS, examining 3148 arms over a 27-year period. They found simple decompression and anterior intramuscular transposition had the best results, followed by medial epicondylectomy and anterior subcutaneous transposition, with anterior submuscular transposition yielding the poorest outcomes. Despite these findings, the operative groups’ recurrence rates remained significant. These results were challenged in a 2008 meta-analysis5 that found no significant difference among simple decompression, subcutaneous transposition, and submuscular transposition and instead demonstrated trends toward better outcomes with anterior transposition. Osterman and Davis7 reported a 5% to 15% rate of unsatisfactory outcomes with anterior subcutaneous transposition, a popular technique used by surgeons at our institution.
The causes for failure or recurrence of ulnar neuropathy after surgical intervention are multifactorial and include preexisting medical conditions and improper operative technique. It is well established that failure to excise all 5 anatomical points of entrapment, or creation of new points of tension during surgery, leads to poor outcomes.12 Nevertheless, the contribution of perineural scarring to postoperative recurrent ulnar neuropathy is currently being recognized: Gabel and Amadio13 described postoperative fibrosis in one-third of their patients with surgically treated recurrent CuTS, Rogers and colleagues14 noted dense perineural fibrosis after intramuscular and subcutaneous transposition procedures, Filippi and colleagues11 cited serious epineural fibrosis and fibrosis around the ulnar nerve as the main findings in their study of 22 patients with recurrent ulnar neuropathy, and Vogel and colleagues12 found that 88% of their patients with persistent CuTS after surgery exhibited perineural scarring.
We think that use of a scar tissue barrier during ulnar nerve transposition reduces the incidence of cicatrix and produces better outcomes—a position largely echoed by the orthopedic community, as fascial, fasciocutaneous, free, and venous flaps have all been used for such purposes.25,26 Vein wrapping has demonstrated good recovery of a nerve after perineural scarring.27 Advocates of intramuscular transposition argue that their technique provides the nerve with a vascularized tunnel, as segmental vascular stripping is an inevitability in transposition. However, this technique increases the incidence of scarring and potential muscle damage.28,29 We think the pedicled adipofascial flap benefits the peripheral nerve by providing a scar tissue barrier and an optimal milieu for vascular regeneration. Kilic and colleagues15 demonstrated the regenerative effects of adipose tissue flaps on peripheral nerves after crush injuries in a rat model, and Strickland and colleagues30 retrospectively examined the effects of hypothenar fat flaps on recalcitrant carpal tunnel syndrome, showing excellent results for this procedure. It is hypothesized that adipose tissue provides not only adipose-derived stem cells but also a rich vascular bed on which nerves will regenerate.
For all patients in the present study, symptoms improved, though the adipose flap and fascial sling groups were not significantly different in their outcomes. We used the MBRS to quantify and compare the groups’ patient-rated outcomes. No statistically significant difference was found between the adipose flap and fascial sling groups. On the MBRS, excellent and good outcomes were reported by 62.5% and 37.5% of the adipose flap patients, respectively, and 59% and 41% of the fascial sling patients (Table 3). Likewise, objective measurements did not show a significant difference between the 2 interventions—indicating that, compared with the current standard of care, adipose flaps are more efficacious in securing the anteriorly transposed nerve.
![]()
Complications of the adipose flap technique are consistent with those reported for other techniques for anterior transposition of the ulnar nerve. The most common complication is hematoma, which can be avoided with meticulous hemostasis. Damage of the medial antebrachial cutaneous nerve or motor branches to the flexor carpi ulnaris has been reported for the fascial technique (we have not had such outcomes at our institution). Contraindications to the adipofascial technique include insufficient subcutaneous adipose tissue for covering the ulnar nerve.
This study was limited by its retrospective setup, which reduced access to preoperative objective and subjective data. The small sample size also limited our ability to demonstrate the advantageous effects of an adipofascial flap in preventing postoperative perineural scarring.
The adipose flap technique is a viable option for securing the anteriorly transposed ulnar nerve. Outcomes in this study demonstrated an efficacy comparable to that of the fascial sling technique. Symptoms resolve or improve, and the majority of patients are satisfied with long-term surgical outcomes. The adipofascial flap may have additional advantages, as it provides a pliable, vascular fat envelope mimicking the natural fatty environment of peripheral nerves.
1. Latinovic R, Gulliford MC, Hughes RA. Incidence of common compressive neuropathies in primary care. J Neurol Neurosurg Psychiatry. 2006;77(2):263-265.
2. Robertson C, Saratsiotis J. A review of compression ulnar neuropathy at the elbow. J Manipulative Physiol Ther. 2005;28(5):345.
3. Posner MA. Compressive ulnar neuropathies at the elbow: I. Etiology and diagnosis. J Am Acad Orthop Surg. 1998;6(5):282-288.
4. Piligian G, Herbert R, Hearns M, Dropkin J, Landsbergis P, Cherniack M. Evaluation and management of chronic work-related musculoskeletal disorders of the distal upper extremity. Am J Ind Med. 2000;37(1):75-93.
5. Macadam SA, Gandhi R, Bezuhly M, Lefaivre KA. Simple decompression versus anterior subcutaneous and submuscular transposition of the ulnar nerve for cubital tunnel syndrome: a meta-analysis. J Hand Surg Am. 2008;33(8):1314.e1-e12.
6. Soltani AM, Best MJ, Francis CS, Allan BJ, Panthaki ZJ. Trends in the surgical treatment of cubital tunnel syndrome: an analysis of the National Survey of Ambulatory Surgery database. J Hand Surg Am. 2013;38(8):1551-1556.
7. Osterman AL, Davis CA. Subcutaneous transposition of the ulnar nerve for treatment of cubital tunnel syndrome. Hand Clin. 1996;12(2):421-433.
8. Bartels RH, Menovsky T, Van Overbeeke JJ, Verhagen WI. Surgical management of ulnar nerve compression at the elbow: an analysis of the literature. J Neurosurg. 1998;89(5):722-727.
9. Seradge H, Owen W. Cubital tunnel release with medial epicondylectomy factors influencing the outcome. J Hand Surg Am. 1998;23(3):483-491.
10. Schnabl SM, Kisslinger F, Schramm A, et al. Subjective outcome, neurophysiological investigations, postoperative complications and recurrence rate of partial medial epicondylectomy in cubital tunnel syndrome. Arch Orthop Trauma Surg. 2011;131(8):1027-1033.
11. Filippi R, Charalampaki P, Reisch R, Koch D, Grunert P. Recurrent cubital tunnel syndrome. Etiology and treatment. Minim Invasive Neurosurg. 2001;44(4):197-201.
12. Vogel RB, Nossaman BC, Rayan GM. Revision anterior submuscular transposition of the ulnar nerve for failed subcutaneous transposition. Br J Plast Surg. 2004;57(4):311-316.
13. Gabel GT, Amadio PC. Reoperation for failed decompression of the ulnar nerve in the region of the elbow. J Bone Joint Surg Am. 1990;72(2):213-219.
14. Rogers MR, Bergfield TG, Aulicino PL. The failed ulnar nerve transposition. Etiology and treatment. Clin Orthop. 1991;269:193-200.
15. Kilic A, Ojo B, Rajfer RA, et al. Effect of white adipose tissue flap and insulin-like growth factor-1 on nerve regeneration in rats. Microsurgery. 2013;33(5):367-375.
16. Ebersole GC, Davidge K, Damiano M, Mackinnon SE. Validity and responsiveness of the DASH questionnaire as an outcome measure following ulnar nerve transposition for cubital tunnel syndrome. Plast Reconstr Surg. 2013;132(1):81e-90e.
17. Kleinman WB, Bishop AT. Anterior intramuscular transposition of the ulnar nerve. J Hand Surg Am. 1989;14(6):972-979.
18. Dützmann S, Martin KD, Sobottka S, et al. Open vs retractor-endoscopic in situ decompression of the ulnar nerve in cubital tunnel syndrome: a retrospective cohort study. Neurosurgery. 2013;72(4):605-616.
19. Dellon AL, Mackinnon SE, Crosby PM. Reliability of two-point discrimination measurements. J Hand Surg Am. 1987;12(5 pt 1):693-696.
20. Danoff JR, Lombardi JM, Rosenwasser MP. Use of a pedicled adipose flap as a sling for anterior subcutaneous transposition of the ulnar nerve. J Hand Surg Am. 2014;39(3):552-555.
21. Magee RB, Phalen GS. Tardy ulnar palsy. Am J Surg. 1949;78(4):470-474.
22. Feindel W, Stratford J. Cubital tunnel compression in tardy ulnar palsy. Can Med Assoc J. 1958;78(5):351-353.
23. Tsai TM, Bonczar M, Tsuruta T, Syed SA. A new operative technique: cubital tunnel decompression with endoscopic assistance. Hand Clin. 1995;11(1):71-80.
24. Hoffmann R, Siemionow M. The endoscopic management of cubital tunnel syndrome. J Hand Surg Br. 2006;31(1):23-29.
25. Luchetti R, Riccio M, Papini Zorli I, Fairplay T. Protective coverage of the median nerve using fascial, fasciocutaneous or island flaps. Handchir Mikrochir Plast Chir. 2006;38(5):317-330.
26. Kokkalis ZT, Jain S, Sotereanos DG. Vein wrapping at cubital tunnel for ulnar nerve problems. J Shoulder Elbow Surg. 2010;19(2):91-97.
27. Masear VR, Colgin S. The treatment of epineural scarring with allograft vein wrapping. Hand Clin. 1996;12(4):773-779.
28. Kleinman WB, Bishop AT. Anterior intramuscular transposition of the ulnar nerve. J Hand Surg Am. 1989;14(6):972-979.
29. Lundborg G. Surgical treatment for ulnar nerve entrapment at the elbow. J Hand Surg Br. 1992;17(3):245-247.
30. Strickland JW, Idler RS, Lourie GM, Plancher KD. The hypothenar fat pad flap for management of recalcitrant carpal tunnel syndrome. J Hand Surg Am. 1996;21(5):840-848.
Compression of the ulnar nerve at the elbow, also referred to as cubital tunnel syndrome (CuTS), is the second most common peripheral nerve compression syndrome in the upper extremity.1,2 Although the ulnar nerve can be compressed at 5 different sites, including arcade of Struthers, medial intermuscular septum, medial epicondyle, and deep flexor aponeurosis, the cubital tunnel is most commonly affected.3 Patients typically present with paresthesias in the fourth and fifth digits and weakness of hand muscle intrinsics. Activity-related pain or pain at the medial elbow can also occur in more advanced pathology.4 It is estimated that conservative therapy fails and surgical intervention is required in up to 30% of patients with CuTS.1 Surgical approaches range from in situ decompression to transposition techniques, but there is no consensus in the orthopedic community as to which technique offers the best results. In a 2008 meta-analysis, Macadam and colleagues5 found no statistical differences in outcomes among the various surgical approaches. Nevertheless, subcutaneous transposition of the ulnar nerve at the elbow is a popular option.6
Despite the widespread success of surgical intervention for CuTS, persistent or recurrent pain occurs in 9.9% to 21.0% of cases.7-10 In addition, several investigators have cited perineural scarring as a major cause of recurrent symptoms after primary surgery.11-14 Filippi and colleagues11 noted that patients who required reoperation after primary anterior transposition had “serious epineural fibrosis and fibrosis around the transposed ulnar nerve.” At our institution, we have similarly found that scarring of the fascial sling around the ulnar nerve led to recurrence of CuTS within 4 months after initial surgery (Figure 1).
We therefore prefer to use a vascularized adipose flap to secure the anteriorly transposed ulnar nerve. This flap provides a pliable, vascularized adipose environment for the nerve, which helps reduce nerve adherence and may enhance nerve recovery.15 In the study reported here, we retrospectively reviewed the long-term outcomes of ulnar nerve anterior subcutaneous transposition secured with either an adipose flap or a fascial sling. We hypothesized that patients in the 2 groups (adipose flap, fascial sling) would have equivalent outcomes.
Materials and Methods
After obtaining institutional review board approval, we reviewed the medical and surgical records of 104 patients (107 limbs) who underwent transposition of the ulnar nerve secured with either an adipose flap (27 limbs) or a fascial sling (80 limbs) over a 14-year period. The fascial sling cohort was used as a comparison group, matched to the adipose flap cohort by sex, age at time of surgery, hand dominance, symptom duration, and length of follow-up (Table 1). Patients were indicated for surgery and were included in the study if they had a history and physical examination consistent with primary CuTS, symptom duration longer than 1 year, and failed conservative management, including activity modification, night splinting, elbow pads, occupational therapy, and home exercise regimen. Electrodiagnostic testing was used at the discretion of the attending surgeon when the diagnosis was not clear from the history and physical examination. All fascial sling procedures were performed at our institution by 1 of 3 fellowship-trained hand surgeons, including Dr. Rosenwasser. The adipose flap modification was performed only by Dr. Rosenwasser. Of the 27 patients in the adipose flap group, 23 underwent surgery for primary CuTS and were included in the study; the other 4 (revision cases) were excluded; 1 patient subsequently died of a cause unrelated to the surgical procedure, and 6 were lost to follow-up. Of the 80 patients in the fascial sling group, 30 underwent surgery for primary CuTS; 5 died before follow-up, and 8 declined to participate.
Thirty-three patients (16 adipose flap, 17 fascial sling) met the inclusion criteria. Of the 16 adipose flap patients, 15 underwent the physical examination and completed the questionnaire, and 1 was interviewed by telephone. Similarly, of the 17 fascial sling patients, 15 underwent the physical examination and completed the questionnaire, and 2 were interviewed by telephone. There were no bilateral cases. Conservative management (activity modification, night splinting, elbow pads, occupational therapy, home exercise) failed in all cases.
A trained study team member who was not part of the surgical team performed follow-up evaluations using objective outcome measures and subjective questionnaires. Patients were assessed at a mean follow-up of 5.6 years (range, 1.6-15.9 years). Patients completed the DASH (Disabilities of the Arm, Shoulder, and Hand) questionnaire16 and visual analog scales (VASs) for pain, numbness, tingling, and weakness in the ulnar nerve distribution. They also rated the presence of night symptoms that were interfering with sleep. The Modified Bishop Rating Scale (MBRS) was used to quantify patient self-reported data17,18 (Figure 2). The MBRS measures overall satisfaction, symptom improvement, presence of residual symptoms, ability to engage in activities, work capability, and subjective changes in strength and sensibility.
In the physical examinations, we tested for Tinel, Wartenberg, and Froment signs; performed an elbow flexion test; and measured elbow range of motion for flexion and extension as well as forearm pronation and supination. We also evaluated lateral pinch strength and grip strength, using a Jamar hydraulic pinch gauge and a Jamar dynamometer (Therapeutic Equipment Corp) and taking the average of 3 assessments. Fifth-digit abduction strength was graded on a standard muscle strength scale. Two-point discrimination was measured at the middle, ring, and small digits of the operated and contralateral hands.19
Surgical Technique
Standard ulnar nerve decompression with anterior subcutaneous transposition and the following modifications were performed on all patients.20 A posteromedial incision parallel to the intermuscular septum was developed and the ulnar nerve identified. Minimizing stripping of the vascular mesentery, the dissection continued along the course of the nerve, and the medial intermuscular septum was excised to prevent secondary compression after transposition. The ulnar nerve was mobilized and transposed anterior to the medial epicondyle (Figure 3). For patients who received the fascial sling, a fascial sleeve was elevated from the flexor-pronator mass and sutured to the edge of the retinaculum securing the nerve. For patients who received the adipose flap, the flap with its vascular pedicle intact was elevated from the subcutaneous tissue of the anterior skin overlying the transposed nerve. The adipose tissue was sharply dissected in half while sufficient subcutaneous tissue was kept between the skin and the flap. A plane was developed based on an anterior adipose pedicle, which included a cutaneous artery and a vein that would supply the vascularized adipose flap. The flap was elevated and wrapped around the nerve without tension while the ulnar nerve was protected from being kinked by the construct. The flap was sutured to the anterior subcutaneous tissue to create a tunnel of adipose tissue surrounding the nerve along its length (Figure 4). The elbow was then flexed and extended to ensure free nerve gliding before wound closure.
The patient was allowed to move the elbow within the bulky dressings immediately after surgery. After 2 weeks, sutures were removed. Formal occupational therapy is not needed for these patients, except in the presence of significant weakness.
Results
As mentioned, the 2 groups were matched on demographics: age at time of surgery, sex, symptom duration, and length of follow-up (Table 1).
For the 16 adipose flap patients (Table 2), mean DASH score was 19.9 (range, 0-71.7). Seven of these patients reported upper extremity pain with a mean VAS score of 1.7 (range, 0-8); 4 patients reported pain in the wrist and fourth and fifth digits; only 1 patient reported pain that occasionally woke the patient from sleep. Constant numbness was present in 6 patients. Four patients reported constant mild tingling in the hand, and 11 reported intermittent tingling. Eleven patients (68.7%) reported operated-arm weakness with a mean VAS score of 3.4 (range, 0-8). In patients who had a physical examination, mean elbow flexion–extension arc of motion was 134° (range, 95°-150°), representing 99% of the motion of the contralateral arm. Mean pronation–supination arc was 174° (range, 150°-180°), accounting for 104% of the contralateral arm. Mean lateral pinch strength was 73% of the contralateral arm, and mean grip strength was 114% of the contralateral arm. The Tinel sign was present in 2 patients, the Froment sign was present in 3 patients, and the elbow flexion test was positive in 2 patients. No patient had a positive Wartenberg sign. On the MBRS, 10 patients had an excellent score, and 6 had a good score.
For the 17 fascial sling patients (Table 2), mean DASH score was 22.7 (range, 0-63.3). Three patients reported upper extremity pain with a mean VAS score of 1.4 (range, 0-7); 3 patients reported pain that occasionally woke them from sleep. Seven patients had constant numbness in the distribution of the ulnar nerve. Two patients had constant paresthesias, and 7 had intermittent paresthesias. Nine patients (52.9%) reported arm weakness with a mean VAS score of 2.5 (range, 0-8). Mean elbow flexion–extension arc of motion was 136° (range, 100°-150°), representing 100% of the contralateral arm. Mean pronation–supination arc was 187° (range, 155°-225°), accounting for 102% of the contralateral arm. Mean lateral pinch strength was 93% of the contralateral arm, and mean grip strength was 80% of the contralateral arm. The Tinel sign was present in 6 patients, the Froment sign in 3 patients, and the Wartenberg sign in 2 patients. The elbow flexion test was positive in 4 patients. On the MBRS, 10 patients had an excellent score, and 7 had a good score.
There was no recurrence of CuTS in either group. One adipose flap patient developed a wound infection that required reoperation.
Discussion
Ulnar neuropathy was described by Magee and Phalen21 in 1949 and termed cubital tunnel syndrome by Feindel and Stratford22 in 1958. Since then, numerous procedures, including in situ decompression, medial epicondylectomy, and endoscopic decompression,23,24 have been advocated for the treatment of this condition. In addition, anterior transposition, which involves securing the ulnar nerve in a submuscular, intramuscular, or subcutaneous sleeve,6 remains a popular option. Despite more than half a century of surgical treatment for this condition, there is no consensus about which procedure offers the best outcomes. Bartels and colleagues8 retrospectively reviewed surgical treatments for CuTS, examining 3148 arms over a 27-year period. They found simple decompression and anterior intramuscular transposition had the best results, followed by medial epicondylectomy and anterior subcutaneous transposition, with anterior submuscular transposition yielding the poorest outcomes. Despite these findings, the operative groups’ recurrence rates remained significant. These results were challenged in a 2008 meta-analysis5 that found no significant difference among simple decompression, subcutaneous transposition, and submuscular transposition and instead demonstrated trends toward better outcomes with anterior transposition. Osterman and Davis7 reported a 5% to 15% rate of unsatisfactory outcomes with anterior subcutaneous transposition, a popular technique used by surgeons at our institution.
The causes for failure or recurrence of ulnar neuropathy after surgical intervention are multifactorial and include preexisting medical conditions and improper operative technique. It is well established that failure to excise all 5 anatomical points of entrapment, or creation of new points of tension during surgery, leads to poor outcomes.12 Nevertheless, the contribution of perineural scarring to postoperative recurrent ulnar neuropathy is currently being recognized: Gabel and Amadio13 described postoperative fibrosis in one-third of their patients with surgically treated recurrent CuTS, Rogers and colleagues14 noted dense perineural fibrosis after intramuscular and subcutaneous transposition procedures, Filippi and colleagues11 cited serious epineural fibrosis and fibrosis around the ulnar nerve as the main findings in their study of 22 patients with recurrent ulnar neuropathy, and Vogel and colleagues12 found that 88% of their patients with persistent CuTS after surgery exhibited perineural scarring.
We think that use of a scar tissue barrier during ulnar nerve transposition reduces the incidence of cicatrix and produces better outcomes—a position largely echoed by the orthopedic community, as fascial, fasciocutaneous, free, and venous flaps have all been used for such purposes.25,26 Vein wrapping has demonstrated good recovery of a nerve after perineural scarring.27 Advocates of intramuscular transposition argue that their technique provides the nerve with a vascularized tunnel, as segmental vascular stripping is an inevitability in transposition. However, this technique increases the incidence of scarring and potential muscle damage.28,29 We think the pedicled adipofascial flap benefits the peripheral nerve by providing a scar tissue barrier and an optimal milieu for vascular regeneration. Kilic and colleagues15 demonstrated the regenerative effects of adipose tissue flaps on peripheral nerves after crush injuries in a rat model, and Strickland and colleagues30 retrospectively examined the effects of hypothenar fat flaps on recalcitrant carpal tunnel syndrome, showing excellent results for this procedure. It is hypothesized that adipose tissue provides not only adipose-derived stem cells but also a rich vascular bed on which nerves will regenerate.
For all patients in the present study, symptoms improved, though the adipose flap and fascial sling groups were not significantly different in their outcomes. We used the MBRS to quantify and compare the groups’ patient-rated outcomes. No statistically significant difference was found between the adipose flap and fascial sling groups. On the MBRS, excellent and good outcomes were reported by 62.5% and 37.5% of the adipose flap patients, respectively, and 59% and 41% of the fascial sling patients (Table 3). Likewise, objective measurements did not show a significant difference between the 2 interventions—indicating that, compared with the current standard of care, adipose flaps are more efficacious in securing the anteriorly transposed nerve.
![]()
Complications of the adipose flap technique are consistent with those reported for other techniques for anterior transposition of the ulnar nerve. The most common complication is hematoma, which can be avoided with meticulous hemostasis. Damage of the medial antebrachial cutaneous nerve or motor branches to the flexor carpi ulnaris has been reported for the fascial technique (we have not had such outcomes at our institution). Contraindications to the adipofascial technique include insufficient subcutaneous adipose tissue for covering the ulnar nerve.
This study was limited by its retrospective setup, which reduced access to preoperative objective and subjective data. The small sample size also limited our ability to demonstrate the advantageous effects of an adipofascial flap in preventing postoperative perineural scarring.
The adipose flap technique is a viable option for securing the anteriorly transposed ulnar nerve. Outcomes in this study demonstrated an efficacy comparable to that of the fascial sling technique. Symptoms resolve or improve, and the majority of patients are satisfied with long-term surgical outcomes. The adipofascial flap may have additional advantages, as it provides a pliable, vascular fat envelope mimicking the natural fatty environment of peripheral nerves.
Compression of the ulnar nerve at the elbow, also referred to as cubital tunnel syndrome (CuTS), is the second most common peripheral nerve compression syndrome in the upper extremity.1,2 Although the ulnar nerve can be compressed at 5 different sites, including arcade of Struthers, medial intermuscular septum, medial epicondyle, and deep flexor aponeurosis, the cubital tunnel is most commonly affected.3 Patients typically present with paresthesias in the fourth and fifth digits and weakness of hand muscle intrinsics. Activity-related pain or pain at the medial elbow can also occur in more advanced pathology.4 It is estimated that conservative therapy fails and surgical intervention is required in up to 30% of patients with CuTS.1 Surgical approaches range from in situ decompression to transposition techniques, but there is no consensus in the orthopedic community as to which technique offers the best results. In a 2008 meta-analysis, Macadam and colleagues5 found no statistical differences in outcomes among the various surgical approaches. Nevertheless, subcutaneous transposition of the ulnar nerve at the elbow is a popular option.6
Despite the widespread success of surgical intervention for CuTS, persistent or recurrent pain occurs in 9.9% to 21.0% of cases.7-10 In addition, several investigators have cited perineural scarring as a major cause of recurrent symptoms after primary surgery.11-14 Filippi and colleagues11 noted that patients who required reoperation after primary anterior transposition had “serious epineural fibrosis and fibrosis around the transposed ulnar nerve.” At our institution, we have similarly found that scarring of the fascial sling around the ulnar nerve led to recurrence of CuTS within 4 months after initial surgery (Figure 1).
We therefore prefer to use a vascularized adipose flap to secure the anteriorly transposed ulnar nerve. This flap provides a pliable, vascularized adipose environment for the nerve, which helps reduce nerve adherence and may enhance nerve recovery.15 In the study reported here, we retrospectively reviewed the long-term outcomes of ulnar nerve anterior subcutaneous transposition secured with either an adipose flap or a fascial sling. We hypothesized that patients in the 2 groups (adipose flap, fascial sling) would have equivalent outcomes.
Materials and Methods
After obtaining institutional review board approval, we reviewed the medical and surgical records of 104 patients (107 limbs) who underwent transposition of the ulnar nerve secured with either an adipose flap (27 limbs) or a fascial sling (80 limbs) over a 14-year period. The fascial sling cohort was used as a comparison group, matched to the adipose flap cohort by sex, age at time of surgery, hand dominance, symptom duration, and length of follow-up (Table 1). Patients were indicated for surgery and were included in the study if they had a history and physical examination consistent with primary CuTS, symptom duration longer than 1 year, and failed conservative management, including activity modification, night splinting, elbow pads, occupational therapy, and home exercise regimen. Electrodiagnostic testing was used at the discretion of the attending surgeon when the diagnosis was not clear from the history and physical examination. All fascial sling procedures were performed at our institution by 1 of 3 fellowship-trained hand surgeons, including Dr. Rosenwasser. The adipose flap modification was performed only by Dr. Rosenwasser. Of the 27 patients in the adipose flap group, 23 underwent surgery for primary CuTS and were included in the study; the other 4 (revision cases) were excluded; 1 patient subsequently died of a cause unrelated to the surgical procedure, and 6 were lost to follow-up. Of the 80 patients in the fascial sling group, 30 underwent surgery for primary CuTS; 5 died before follow-up, and 8 declined to participate.
Thirty-three patients (16 adipose flap, 17 fascial sling) met the inclusion criteria. Of the 16 adipose flap patients, 15 underwent the physical examination and completed the questionnaire, and 1 was interviewed by telephone. Similarly, of the 17 fascial sling patients, 15 underwent the physical examination and completed the questionnaire, and 2 were interviewed by telephone. There were no bilateral cases. Conservative management (activity modification, night splinting, elbow pads, occupational therapy, home exercise) failed in all cases.
A trained study team member who was not part of the surgical team performed follow-up evaluations using objective outcome measures and subjective questionnaires. Patients were assessed at a mean follow-up of 5.6 years (range, 1.6-15.9 years). Patients completed the DASH (Disabilities of the Arm, Shoulder, and Hand) questionnaire16 and visual analog scales (VASs) for pain, numbness, tingling, and weakness in the ulnar nerve distribution. They also rated the presence of night symptoms that were interfering with sleep. The Modified Bishop Rating Scale (MBRS) was used to quantify patient self-reported data17,18 (Figure 2). The MBRS measures overall satisfaction, symptom improvement, presence of residual symptoms, ability to engage in activities, work capability, and subjective changes in strength and sensibility.
In the physical examinations, we tested for Tinel, Wartenberg, and Froment signs; performed an elbow flexion test; and measured elbow range of motion for flexion and extension as well as forearm pronation and supination. We also evaluated lateral pinch strength and grip strength, using a Jamar hydraulic pinch gauge and a Jamar dynamometer (Therapeutic Equipment Corp) and taking the average of 3 assessments. Fifth-digit abduction strength was graded on a standard muscle strength scale. Two-point discrimination was measured at the middle, ring, and small digits of the operated and contralateral hands.19
Surgical Technique
Standard ulnar nerve decompression with anterior subcutaneous transposition and the following modifications were performed on all patients.20 A posteromedial incision parallel to the intermuscular septum was developed and the ulnar nerve identified. Minimizing stripping of the vascular mesentery, the dissection continued along the course of the nerve, and the medial intermuscular septum was excised to prevent secondary compression after transposition. The ulnar nerve was mobilized and transposed anterior to the medial epicondyle (Figure 3). For patients who received the fascial sling, a fascial sleeve was elevated from the flexor-pronator mass and sutured to the edge of the retinaculum securing the nerve. For patients who received the adipose flap, the flap with its vascular pedicle intact was elevated from the subcutaneous tissue of the anterior skin overlying the transposed nerve. The adipose tissue was sharply dissected in half while sufficient subcutaneous tissue was kept between the skin and the flap. A plane was developed based on an anterior adipose pedicle, which included a cutaneous artery and a vein that would supply the vascularized adipose flap. The flap was elevated and wrapped around the nerve without tension while the ulnar nerve was protected from being kinked by the construct. The flap was sutured to the anterior subcutaneous tissue to create a tunnel of adipose tissue surrounding the nerve along its length (Figure 4). The elbow was then flexed and extended to ensure free nerve gliding before wound closure.
The patient was allowed to move the elbow within the bulky dressings immediately after surgery. After 2 weeks, sutures were removed. Formal occupational therapy is not needed for these patients, except in the presence of significant weakness.
Results
As mentioned, the 2 groups were matched on demographics: age at time of surgery, sex, symptom duration, and length of follow-up (Table 1).
For the 16 adipose flap patients (Table 2), mean DASH score was 19.9 (range, 0-71.7). Seven of these patients reported upper extremity pain with a mean VAS score of 1.7 (range, 0-8); 4 patients reported pain in the wrist and fourth and fifth digits; only 1 patient reported pain that occasionally woke the patient from sleep. Constant numbness was present in 6 patients. Four patients reported constant mild tingling in the hand, and 11 reported intermittent tingling. Eleven patients (68.7%) reported operated-arm weakness with a mean VAS score of 3.4 (range, 0-8). In patients who had a physical examination, mean elbow flexion–extension arc of motion was 134° (range, 95°-150°), representing 99% of the motion of the contralateral arm. Mean pronation–supination arc was 174° (range, 150°-180°), accounting for 104% of the contralateral arm. Mean lateral pinch strength was 73% of the contralateral arm, and mean grip strength was 114% of the contralateral arm. The Tinel sign was present in 2 patients, the Froment sign was present in 3 patients, and the elbow flexion test was positive in 2 patients. No patient had a positive Wartenberg sign. On the MBRS, 10 patients had an excellent score, and 6 had a good score.
For the 17 fascial sling patients (Table 2), mean DASH score was 22.7 (range, 0-63.3). Three patients reported upper extremity pain with a mean VAS score of 1.4 (range, 0-7); 3 patients reported pain that occasionally woke them from sleep. Seven patients had constant numbness in the distribution of the ulnar nerve. Two patients had constant paresthesias, and 7 had intermittent paresthesias. Nine patients (52.9%) reported arm weakness with a mean VAS score of 2.5 (range, 0-8). Mean elbow flexion–extension arc of motion was 136° (range, 100°-150°), representing 100% of the contralateral arm. Mean pronation–supination arc was 187° (range, 155°-225°), accounting for 102% of the contralateral arm. Mean lateral pinch strength was 93% of the contralateral arm, and mean grip strength was 80% of the contralateral arm. The Tinel sign was present in 6 patients, the Froment sign in 3 patients, and the Wartenberg sign in 2 patients. The elbow flexion test was positive in 4 patients. On the MBRS, 10 patients had an excellent score, and 7 had a good score.
There was no recurrence of CuTS in either group. One adipose flap patient developed a wound infection that required reoperation.
Discussion
Ulnar neuropathy was described by Magee and Phalen21 in 1949 and termed cubital tunnel syndrome by Feindel and Stratford22 in 1958. Since then, numerous procedures, including in situ decompression, medial epicondylectomy, and endoscopic decompression,23,24 have been advocated for the treatment of this condition. In addition, anterior transposition, which involves securing the ulnar nerve in a submuscular, intramuscular, or subcutaneous sleeve,6 remains a popular option. Despite more than half a century of surgical treatment for this condition, there is no consensus about which procedure offers the best outcomes. Bartels and colleagues8 retrospectively reviewed surgical treatments for CuTS, examining 3148 arms over a 27-year period. They found simple decompression and anterior intramuscular transposition had the best results, followed by medial epicondylectomy and anterior subcutaneous transposition, with anterior submuscular transposition yielding the poorest outcomes. Despite these findings, the operative groups’ recurrence rates remained significant. These results were challenged in a 2008 meta-analysis5 that found no significant difference among simple decompression, subcutaneous transposition, and submuscular transposition and instead demonstrated trends toward better outcomes with anterior transposition. Osterman and Davis7 reported a 5% to 15% rate of unsatisfactory outcomes with anterior subcutaneous transposition, a popular technique used by surgeons at our institution.
The causes for failure or recurrence of ulnar neuropathy after surgical intervention are multifactorial and include preexisting medical conditions and improper operative technique. It is well established that failure to excise all 5 anatomical points of entrapment, or creation of new points of tension during surgery, leads to poor outcomes.12 Nevertheless, the contribution of perineural scarring to postoperative recurrent ulnar neuropathy is currently being recognized: Gabel and Amadio13 described postoperative fibrosis in one-third of their patients with surgically treated recurrent CuTS, Rogers and colleagues14 noted dense perineural fibrosis after intramuscular and subcutaneous transposition procedures, Filippi and colleagues11 cited serious epineural fibrosis and fibrosis around the ulnar nerve as the main findings in their study of 22 patients with recurrent ulnar neuropathy, and Vogel and colleagues12 found that 88% of their patients with persistent CuTS after surgery exhibited perineural scarring.
We think that use of a scar tissue barrier during ulnar nerve transposition reduces the incidence of cicatrix and produces better outcomes—a position largely echoed by the orthopedic community, as fascial, fasciocutaneous, free, and venous flaps have all been used for such purposes.25,26 Vein wrapping has demonstrated good recovery of a nerve after perineural scarring.27 Advocates of intramuscular transposition argue that their technique provides the nerve with a vascularized tunnel, as segmental vascular stripping is an inevitability in transposition. However, this technique increases the incidence of scarring and potential muscle damage.28,29 We think the pedicled adipofascial flap benefits the peripheral nerve by providing a scar tissue barrier and an optimal milieu for vascular regeneration. Kilic and colleagues15 demonstrated the regenerative effects of adipose tissue flaps on peripheral nerves after crush injuries in a rat model, and Strickland and colleagues30 retrospectively examined the effects of hypothenar fat flaps on recalcitrant carpal tunnel syndrome, showing excellent results for this procedure. It is hypothesized that adipose tissue provides not only adipose-derived stem cells but also a rich vascular bed on which nerves will regenerate.
For all patients in the present study, symptoms improved, though the adipose flap and fascial sling groups were not significantly different in their outcomes. We used the MBRS to quantify and compare the groups’ patient-rated outcomes. No statistically significant difference was found between the adipose flap and fascial sling groups. On the MBRS, excellent and good outcomes were reported by 62.5% and 37.5% of the adipose flap patients, respectively, and 59% and 41% of the fascial sling patients (Table 3). Likewise, objective measurements did not show a significant difference between the 2 interventions—indicating that, compared with the current standard of care, adipose flaps are more efficacious in securing the anteriorly transposed nerve.
![]()
Complications of the adipose flap technique are consistent with those reported for other techniques for anterior transposition of the ulnar nerve. The most common complication is hematoma, which can be avoided with meticulous hemostasis. Damage of the medial antebrachial cutaneous nerve or motor branches to the flexor carpi ulnaris has been reported for the fascial technique (we have not had such outcomes at our institution). Contraindications to the adipofascial technique include insufficient subcutaneous adipose tissue for covering the ulnar nerve.
This study was limited by its retrospective setup, which reduced access to preoperative objective and subjective data. The small sample size also limited our ability to demonstrate the advantageous effects of an adipofascial flap in preventing postoperative perineural scarring.
The adipose flap technique is a viable option for securing the anteriorly transposed ulnar nerve. Outcomes in this study demonstrated an efficacy comparable to that of the fascial sling technique. Symptoms resolve or improve, and the majority of patients are satisfied with long-term surgical outcomes. The adipofascial flap may have additional advantages, as it provides a pliable, vascular fat envelope mimicking the natural fatty environment of peripheral nerves.
1. Latinovic R, Gulliford MC, Hughes RA. Incidence of common compressive neuropathies in primary care. J Neurol Neurosurg Psychiatry. 2006;77(2):263-265.
2. Robertson C, Saratsiotis J. A review of compression ulnar neuropathy at the elbow. J Manipulative Physiol Ther. 2005;28(5):345.
3. Posner MA. Compressive ulnar neuropathies at the elbow: I. Etiology and diagnosis. J Am Acad Orthop Surg. 1998;6(5):282-288.
4. Piligian G, Herbert R, Hearns M, Dropkin J, Landsbergis P, Cherniack M. Evaluation and management of chronic work-related musculoskeletal disorders of the distal upper extremity. Am J Ind Med. 2000;37(1):75-93.
5. Macadam SA, Gandhi R, Bezuhly M, Lefaivre KA. Simple decompression versus anterior subcutaneous and submuscular transposition of the ulnar nerve for cubital tunnel syndrome: a meta-analysis. J Hand Surg Am. 2008;33(8):1314.e1-e12.
6. Soltani AM, Best MJ, Francis CS, Allan BJ, Panthaki ZJ. Trends in the surgical treatment of cubital tunnel syndrome: an analysis of the National Survey of Ambulatory Surgery database. J Hand Surg Am. 2013;38(8):1551-1556.
7. Osterman AL, Davis CA. Subcutaneous transposition of the ulnar nerve for treatment of cubital tunnel syndrome. Hand Clin. 1996;12(2):421-433.
8. Bartels RH, Menovsky T, Van Overbeeke JJ, Verhagen WI. Surgical management of ulnar nerve compression at the elbow: an analysis of the literature. J Neurosurg. 1998;89(5):722-727.
9. Seradge H, Owen W. Cubital tunnel release with medial epicondylectomy factors influencing the outcome. J Hand Surg Am. 1998;23(3):483-491.
10. Schnabl SM, Kisslinger F, Schramm A, et al. Subjective outcome, neurophysiological investigations, postoperative complications and recurrence rate of partial medial epicondylectomy in cubital tunnel syndrome. Arch Orthop Trauma Surg. 2011;131(8):1027-1033.
11. Filippi R, Charalampaki P, Reisch R, Koch D, Grunert P. Recurrent cubital tunnel syndrome. Etiology and treatment. Minim Invasive Neurosurg. 2001;44(4):197-201.
12. Vogel RB, Nossaman BC, Rayan GM. Revision anterior submuscular transposition of the ulnar nerve for failed subcutaneous transposition. Br J Plast Surg. 2004;57(4):311-316.
13. Gabel GT, Amadio PC. Reoperation for failed decompression of the ulnar nerve in the region of the elbow. J Bone Joint Surg Am. 1990;72(2):213-219.
14. Rogers MR, Bergfield TG, Aulicino PL. The failed ulnar nerve transposition. Etiology and treatment. Clin Orthop. 1991;269:193-200.
15. Kilic A, Ojo B, Rajfer RA, et al. Effect of white adipose tissue flap and insulin-like growth factor-1 on nerve regeneration in rats. Microsurgery. 2013;33(5):367-375.
16. Ebersole GC, Davidge K, Damiano M, Mackinnon SE. Validity and responsiveness of the DASH questionnaire as an outcome measure following ulnar nerve transposition for cubital tunnel syndrome. Plast Reconstr Surg. 2013;132(1):81e-90e.
17. Kleinman WB, Bishop AT. Anterior intramuscular transposition of the ulnar nerve. J Hand Surg Am. 1989;14(6):972-979.
18. Dützmann S, Martin KD, Sobottka S, et al. Open vs retractor-endoscopic in situ decompression of the ulnar nerve in cubital tunnel syndrome: a retrospective cohort study. Neurosurgery. 2013;72(4):605-616.
19. Dellon AL, Mackinnon SE, Crosby PM. Reliability of two-point discrimination measurements. J Hand Surg Am. 1987;12(5 pt 1):693-696.
20. Danoff JR, Lombardi JM, Rosenwasser MP. Use of a pedicled adipose flap as a sling for anterior subcutaneous transposition of the ulnar nerve. J Hand Surg Am. 2014;39(3):552-555.
21. Magee RB, Phalen GS. Tardy ulnar palsy. Am J Surg. 1949;78(4):470-474.
22. Feindel W, Stratford J. Cubital tunnel compression in tardy ulnar palsy. Can Med Assoc J. 1958;78(5):351-353.
23. Tsai TM, Bonczar M, Tsuruta T, Syed SA. A new operative technique: cubital tunnel decompression with endoscopic assistance. Hand Clin. 1995;11(1):71-80.
24. Hoffmann R, Siemionow M. The endoscopic management of cubital tunnel syndrome. J Hand Surg Br. 2006;31(1):23-29.
25. Luchetti R, Riccio M, Papini Zorli I, Fairplay T. Protective coverage of the median nerve using fascial, fasciocutaneous or island flaps. Handchir Mikrochir Plast Chir. 2006;38(5):317-330.
26. Kokkalis ZT, Jain S, Sotereanos DG. Vein wrapping at cubital tunnel for ulnar nerve problems. J Shoulder Elbow Surg. 2010;19(2):91-97.
27. Masear VR, Colgin S. The treatment of epineural scarring with allograft vein wrapping. Hand Clin. 1996;12(4):773-779.
28. Kleinman WB, Bishop AT. Anterior intramuscular transposition of the ulnar nerve. J Hand Surg Am. 1989;14(6):972-979.
29. Lundborg G. Surgical treatment for ulnar nerve entrapment at the elbow. J Hand Surg Br. 1992;17(3):245-247.
30. Strickland JW, Idler RS, Lourie GM, Plancher KD. The hypothenar fat pad flap for management of recalcitrant carpal tunnel syndrome. J Hand Surg Am. 1996;21(5):840-848.
1. Latinovic R, Gulliford MC, Hughes RA. Incidence of common compressive neuropathies in primary care. J Neurol Neurosurg Psychiatry. 2006;77(2):263-265.
2. Robertson C, Saratsiotis J. A review of compression ulnar neuropathy at the elbow. J Manipulative Physiol Ther. 2005;28(5):345.
3. Posner MA. Compressive ulnar neuropathies at the elbow: I. Etiology and diagnosis. J Am Acad Orthop Surg. 1998;6(5):282-288.
4. Piligian G, Herbert R, Hearns M, Dropkin J, Landsbergis P, Cherniack M. Evaluation and management of chronic work-related musculoskeletal disorders of the distal upper extremity. Am J Ind Med. 2000;37(1):75-93.
5. Macadam SA, Gandhi R, Bezuhly M, Lefaivre KA. Simple decompression versus anterior subcutaneous and submuscular transposition of the ulnar nerve for cubital tunnel syndrome: a meta-analysis. J Hand Surg Am. 2008;33(8):1314.e1-e12.
6. Soltani AM, Best MJ, Francis CS, Allan BJ, Panthaki ZJ. Trends in the surgical treatment of cubital tunnel syndrome: an analysis of the National Survey of Ambulatory Surgery database. J Hand Surg Am. 2013;38(8):1551-1556.
7. Osterman AL, Davis CA. Subcutaneous transposition of the ulnar nerve for treatment of cubital tunnel syndrome. Hand Clin. 1996;12(2):421-433.
8. Bartels RH, Menovsky T, Van Overbeeke JJ, Verhagen WI. Surgical management of ulnar nerve compression at the elbow: an analysis of the literature. J Neurosurg. 1998;89(5):722-727.
9. Seradge H, Owen W. Cubital tunnel release with medial epicondylectomy factors influencing the outcome. J Hand Surg Am. 1998;23(3):483-491.
10. Schnabl SM, Kisslinger F, Schramm A, et al. Subjective outcome, neurophysiological investigations, postoperative complications and recurrence rate of partial medial epicondylectomy in cubital tunnel syndrome. Arch Orthop Trauma Surg. 2011;131(8):1027-1033.
11. Filippi R, Charalampaki P, Reisch R, Koch D, Grunert P. Recurrent cubital tunnel syndrome. Etiology and treatment. Minim Invasive Neurosurg. 2001;44(4):197-201.
12. Vogel RB, Nossaman BC, Rayan GM. Revision anterior submuscular transposition of the ulnar nerve for failed subcutaneous transposition. Br J Plast Surg. 2004;57(4):311-316.
13. Gabel GT, Amadio PC. Reoperation for failed decompression of the ulnar nerve in the region of the elbow. J Bone Joint Surg Am. 1990;72(2):213-219.
14. Rogers MR, Bergfield TG, Aulicino PL. The failed ulnar nerve transposition. Etiology and treatment. Clin Orthop. 1991;269:193-200.
15. Kilic A, Ojo B, Rajfer RA, et al. Effect of white adipose tissue flap and insulin-like growth factor-1 on nerve regeneration in rats. Microsurgery. 2013;33(5):367-375.
16. Ebersole GC, Davidge K, Damiano M, Mackinnon SE. Validity and responsiveness of the DASH questionnaire as an outcome measure following ulnar nerve transposition for cubital tunnel syndrome. Plast Reconstr Surg. 2013;132(1):81e-90e.
17. Kleinman WB, Bishop AT. Anterior intramuscular transposition of the ulnar nerve. J Hand Surg Am. 1989;14(6):972-979.
18. Dützmann S, Martin KD, Sobottka S, et al. Open vs retractor-endoscopic in situ decompression of the ulnar nerve in cubital tunnel syndrome: a retrospective cohort study. Neurosurgery. 2013;72(4):605-616.
19. Dellon AL, Mackinnon SE, Crosby PM. Reliability of two-point discrimination measurements. J Hand Surg Am. 1987;12(5 pt 1):693-696.
20. Danoff JR, Lombardi JM, Rosenwasser MP. Use of a pedicled adipose flap as a sling for anterior subcutaneous transposition of the ulnar nerve. J Hand Surg Am. 2014;39(3):552-555.
21. Magee RB, Phalen GS. Tardy ulnar palsy. Am J Surg. 1949;78(4):470-474.
22. Feindel W, Stratford J. Cubital tunnel compression in tardy ulnar palsy. Can Med Assoc J. 1958;78(5):351-353.
23. Tsai TM, Bonczar M, Tsuruta T, Syed SA. A new operative technique: cubital tunnel decompression with endoscopic assistance. Hand Clin. 1995;11(1):71-80.
24. Hoffmann R, Siemionow M. The endoscopic management of cubital tunnel syndrome. J Hand Surg Br. 2006;31(1):23-29.
25. Luchetti R, Riccio M, Papini Zorli I, Fairplay T. Protective coverage of the median nerve using fascial, fasciocutaneous or island flaps. Handchir Mikrochir Plast Chir. 2006;38(5):317-330.
26. Kokkalis ZT, Jain S, Sotereanos DG. Vein wrapping at cubital tunnel for ulnar nerve problems. J Shoulder Elbow Surg. 2010;19(2):91-97.
27. Masear VR, Colgin S. The treatment of epineural scarring with allograft vein wrapping. Hand Clin. 1996;12(4):773-779.
28. Kleinman WB, Bishop AT. Anterior intramuscular transposition of the ulnar nerve. J Hand Surg Am. 1989;14(6):972-979.
29. Lundborg G. Surgical treatment for ulnar nerve entrapment at the elbow. J Hand Surg Br. 1992;17(3):245-247.
30. Strickland JW, Idler RS, Lourie GM, Plancher KD. The hypothenar fat pad flap for management of recalcitrant carpal tunnel syndrome. J Hand Surg Am. 1996;21(5):840-848.
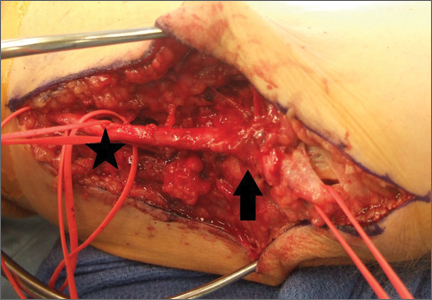
Obesity Management: Clinical Review and Update of the Pharmacologic Treatment Options
Over the past decade the prevalence of obesity as defined by a body mass index (BMI) ≥ 30 kg/m2 has significantly increased. In the U.S. more than 78 million adults are estimated to be obese.1 The World Health Organization projects that by 2025 up to half the U.S. population will be obese. Cardiovascular disease (CVD) and diabetes mellitus (DM) are the main comorbid conditions that are complicated by obesity. Initial weight loss of 5% to 10% of total body weight reduces CVD risk factors, prevents or delays the development of type 2 DM (T2DM) and improves the health consequences of obesity.2
To date, public health initiatives that have focused on obesity prevention and lifestyle intervention have had marginal success. In recent years, anti-obesity drug therapies have had a limited role in clinical treatment algorithms. In 2013, the American Medical Association acknowledged obesity as a disease. In turn, this acknowledgement allowed the recognition of anti-obesity drugs as acceptable therapeutic adjuncts to intensive lifestyle intervention that could address the growing obesity endemic.
In the past, medications for weight reduction were limited. Several that were FDA approved had to be removed from the market due to safety concerns. With few approved options, clinicians often had to resort to off-label use of medications. However, the landscape has changed with 4 new medications gaining recent FDA approval. This review covers older available medications and the newer medications that are now available.
Sympathomimetics
Sympathomimetic drugs have been approved for use as a pharmacological method to lose weight since 1960. Of the many versions of this drug class that have been available since then, there are 4 major versions available today. These include diethylpropion3 and benzphetamine,4 both approved in 1960; phendimetrazine, approved in 1976;5 phentermine, approved in 1980;6 and phentermine hydrochloride, approved in 2012.7 Despite the existence of several other classes of drugs to treat obesity, phentermine remains the most often prescribed weight loss drug in the U.S.8
Although the mechanism of action (MOA) of sympathomimetic drugs is not particularly clear, weight loss from these medications is believed to be due to the increase in the release of biogenic amines (mainly norepinephrine, but also possibly dopamine), from storage sites in nerve terminals. It is possible that these drugs slow catecholamine metabolism by inhibiting the actions of monoamine oxidase. The resulting increase in amine availability, particularly in the lateral hypothalamic feeding center, is associated with reduced food intake. Interestingly, injection of these drugs into the ventromedial satiety center dooes not seem to suppress food intake, and the effects of biogenic amines on increasing metabolism does not seem to play a significant role in weight loss in patients on these medications.9
Each of these drugs is rapidly absorbed from the gastrointestinal (GI) tract except for phentermine hydrochloride, the newest of the medications in this class. Phentermine hydrochloride is a sublingual tablet that is readily absorbed through the buccal mucosa.5 All of the drugs in this class are excreted through the kidneys, with varying rates. Each drug’s excretion is highly dependent on the pH of the urine—more alkaline conditions result in less excretion and more acidic conditions result in more excretion. As a result, these drugs should be used with caution in patients with renal impairment; however, there are no specific contraindications listed for patients with poor renal function.
The adverse effects (AEs) for this drug class are to be expected from an increase in the release of biogenic amines in the central nervous system (CNS). The most common AEs include palpitations, tremors, restlessness, insomnia, dry mouth, constipation, diaphoresis, changes in libido, and irritability. The more dangerous AEs that have been observed include arrhythmias, hypertension, dependency/abuse, convulsions, acute transient ischemic colitis, and acute urinary retention secondary to increased bladder sphincter tone, transient hyperthyroxemia, and paranoia.10
Several contraindications exist for sympathomimetics, including the presence of advanced arteriosclerosis, symptomatic CVD, moderate to severe hypertension, hyperthyroidism, glaucoma, patients in an agitated state, or those with a history of amphetamine abuse. The warnings for prescribers include pulmonary hypertension and cardiomyopathy secondary to chronic use of sympathomimetics, and valvular heart disease secondary to use of sympathomimetics with additional anorectic agents.
Additional precautions should be considered in those with a history of anxiety/psychosis, those who operate machinery and motor vehicles, and even those with mild hypertension. The data surrounding the effects of sympathomimetics on blood pressure (BP) appears to be conflicting and the relationship does not seem to have been significantly studied in depth to warrant any definitive conclusions. The MOA of this drug class itself is enough to urge caution to prescribers.11 Special attention should be given to patients with diabetes when using sympathomimetics. A reduction of insulin dose or oral hypoglycemic dose may be necessary in some people with diabetes.
Only diethylpropion is pregnancy category B, whereas the others drugs in this class are pregnancy category X. It has been demonstrated that diethylpropion and benzphetamine are secreted into breastmilk; insufficient data exist to suggest whether or not phentermine and phendimetrazine are present in breastmilk. All drugs in this class should be used in caution with breastfeeding mothers.
Although all 4 drugs are registered as controlled substances, benzphetamine and phendimetrazine are schedule III and phentermine and diethylpropion are schedule IV, despite evidence suggesting the potential for abuse to be extremely low.12,13 Phentermine has been approved for adults aged > 18 years, phendimetrazine has been approved for those aged > 17 years, diethylpropion has been approved for those aged > 16 years, and benzphetamine has been approved for those aged > 12 years.
There is a wealth of literature surrounding the effectiveness of this drug class for weight loss. One of the longest trials of phentermine was recently conducted as part of the initial component of a FDA study for the newly approved topiramate-phentermine combination. Weight loss at 6 months in the phentermine-only group was significantly higher at -5.8% compared with -1.5% with the placebo group in the last observation carried forward-Intent to treat (LOCF-ITT) analysis.14 Similarly, a long-term study looking at diethylpropion examined the use of diethylpropion for up to a year vs placebo. Participants administered diethylpropion lost a mean 9.8% of original weight vs 3.7% in the placebo group in the first 6 months alone.15
Several meta-analyses and review papers have been authored that examine and analyze the published data on this drug class overall and comparatively within this class. Haddock and colleagues in 2002 reviewed the numerous clinical trials associated with each drug in this class, in addition to several other classes, and found that although each drug demonstrated a significant advantage vs placebo in weight loss, there was not a specific drug that was significantly superior to any of the others.16
These results seem to be in relative agreement with additional studies like that published by Suplicy and colleagues, which demonstrated that several sympathomimetics were better than placebo in weight loss, and that there was little difference between the specific drugs in the class.17 However, it should be noted that as highlighted in a review by Ioannides-Demos and colleagues in 2005, the vast majority of studies that had been performed on this drug class focused on short-term use (< 16 weeks) and none of the sympathomimetics listed here have been approved for long-term use.18
Orlistat
Orlistat 120 mg was approved in 1999 as a reversible inhibitor of GI lipases that specifically reduced the absorption of dietary fat due to the inhibition of triglyceride hydrolysis.19 Orlistat was later approved in 2007 for release in a reduced dosage form (60 mg) for over-the-counter sales.20
Orlistat forms a covalent bond with the active serine residue site of gastric and pancreatic lipases in the lumen of the stomach and small intestine. The inhibition of these enzymes causes dietary fat to remain undigested as triglycerides, which cannot be converted to absorbable free fatty acids and monoglycerides, leading to decreased calorie absorption. Orlistat is not systemically absorbed and is eliminated mainly through feces. Some metabolism occurs in the GI wall.21Orlistat is most known for its GI AEs. Because it is most active in the lumen of the GI system and reduces the absorption of triglycerides, many AEs are related to malabsorption. The most common issues 1 year after starting the drug were oily spotting (26.6% vs 1.3% placebo); flatus with discharge (23.9% vs 1.4% placebo); fecal urgency (22.1% vs 6.7% placebo); fatty/oily stool (20% vs 2.9% placebo); increased defecation (10.8% vs 4.1% placebo); and fecal incontinence (7.7% vs 0.9% placebo) (Table 1). Most of these AEs were greatly reduced after taking the drug for 2 years. Orlistat also has more serious AEs noted, including abdominal pain/discomfort; nausea; infectious diarrhea; rectal pain/discomfort; tooth disorder; gingival disorder; vomiting; upper respiratory infection; lower respiratory infection; ear, nose and throat symptoms; back pain; arthritis; myalgia; joint disorders; tendonitis; headache; dizziness; fatigue; sleep disorders; rash; dry skin; menstrual irregularity; vaginitis; urinary tract infection; and psychiatric disorders, although these did not differ markedly from placebo.
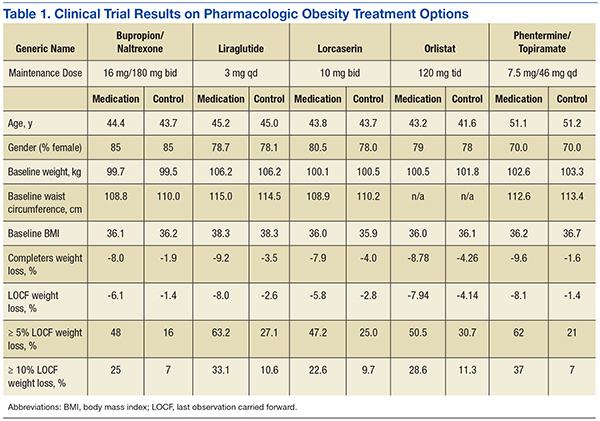
One of the most serious AEs reported was fulminate hepatic failure, though this AE is rare. Thirteen cases of liver injury were reported with the 120-mg prescription dose of orlistat and 1 case report in the U.S. involved the 60-mg over-the-counter dosage of orlistat.21,22 The FDA suggests that patients talk to their physicians about risks of liver failure, and that physicians should educate their patients about signs and symptoms of liver failure so that patients can stop taking orlistat and seek immediate medical help if symptoms occur.
One of the first published trials was the European Multicentre Orlistat Study Group, which included 743 participants with BMI between 28 kg/m2 and 47 kg/m2 from 15 different European centers. To test adherence, a 4-week single blind placebo lead-in was started with a hypocaloric diet. The first stage was completed by 688 patients who then proceeded to the double blind randomized control trial portion with a hypocaloric diet. From the start of lead-in to the end of year 1, the orlistat group weight decreased 10.2% (10.3 kg) vs 6.1% (6.1 kg) in the placebo group. The placebo subtracted difference between the groups was 3.9 kg (P < .001).23
A U.S.-based randomized double-blind placebo-controlled multicenter study included 796 obese patients with BMI between 30 kg/m2 and 44 kg/m2. Patients were assigned to 1 of 3 groups: placebo, orlistat 60 mg 3 times daily, or orlistat 120 mg 3 times daily. All groups were given a reduced energy diet. Patients in the orlistat 120 mg group lost significantly more weight than did the placebo group, -8.78% vs -4.26% respectively in year 1 in the completer analysis (P = .001). More participants who were treated with orlistat 120 mg lost 5% or more of their initial weight in year 1 compared with placebo, 50.5% vs 30.7% respectively (P < .001).24
In the XENDOS study the primary outcome measurement was time to onset of T2DM. Eligible participants were aged 30 to 60 years, with a BMI > 30 kg/m2. All patients had a 75-g oral glucose tolerance test and were required to have normal glucose tolerance or impaired glucose tolerance, but not T2DM. The double-blind randomized controlled trial included 3,305 subjects and compared a group taking 120 mg orlistat 3 times daily vs placebo. All patients were prescribed a reduced-calorie diet (800 kcal/d deficit) containing 30% of calories from fat. Patients were also encouraged to walk at least 1 kilometer daily in addition to their usual physical activity. Incidence of T2DM after 4 years was 6.2% in the orlistat group and 9.0% in the placebo group, reflecting a 37.3% risk reduction in the orlistat group (P = .0032).25,26
Lorcaserin
In 2012, lorcaserin HCl was FDA approved as a schedule IV drug for use as a weight loss medication as an adjunct to a reduced-calorie diet and increased physical activity. Lorcaserin is thought to act on 5-hydroxytryptamine-2c (5HT2c) receptors on the pro-opiomelanocortin (POMC) neurons in the arcuate nucleus, causing release of alpha-melanocortin-stimulating hormone (alpha-MSH), which in turn acts on melanocortin-4 receptors in the paraventricular nucleus to suppress appetite. At the maximum suggested dose of 10 mg twice daily, lorcaserin binds with 15 to 100 times greater affinity to 5HT2c receptors compared with 5HT2a and 5HT2b receptors respectively.
Indications for lorcaserin include patients with BMI ≥ 30 kg/m2 or ≥ 27 kg/m2 or greater with a weight-related comorbid condition such as hypertension, dyslipidemia, cardiovascular disease, impaired glucose tolerance, or sleep apnea.
The efficacy of lorcaserin for weight loss has been evaluated in 3 separate trials. The trials were randomized, double blinded and placebo controlled. The BLOOM trial, which included 3,182 patients with a mean BMI of 36.2 kg/m2, evaluated the efficacy of lorcaserin as a weight loss adjunct.27 Patients with pre-existing valvular disease, uncontrolled hypertension, or a major psychiatric condition were excluded. After initial randomization, patients were assigned to receive either lorcaserin 10 mg twice daily or a placebo. The primary endpoint was a 5% weight reduction from baseline by the end of 2 years. At 1 year, 47.5% of patients in the lorcaserin group and 20.3% in the placebo group had lost ≥ 5% of their body weight (P <.001). The average loss for the lorcaserin group was 5.8 ± 0.2 kg and 2.2 ± 0.1 kg for the placebo group at 1 year (P < .001).
The BLOSSOM trial was a 1-year study of 4,008 patients aged 18 to 65 years. The trial evaluated the effects of lorcaserin on body weight, CVD risk factors, and safety in obese and overweight patients.28 Patients were randomized in a 2:1:2 ratio to receive lorcaserin 10 mg twice daily, lorcaserin 10 mg once daily, or placebo. The primary endpoint was the proportion of patients achieving at least 5% reduction in body weight. Completer analysis showed weight reduction in the placebo group was 4.0% and 7.9% in the lorcaserin group (P < .001). In the modified intent-to-treat/last observation carried forward analysis (MITT/LOCF), a statistically significant 47.2% of patients receiving lorcaserin 10 mg twice daily and 40.2% of patients receiving lorcaserin 10 mg once daily lost at least 5% of baseline body weight; compared with 25% of patients receiving placebo (P < .001). Weight loss of at least 10% was achieved by 22.6% of patients receiving lorcaserin 10 mg twice daily, and 17.4% of patients receiving 10 mg daily compared with 9.7% of patients in the placebo group (P < .001).
The most common AEs noted were headache, nausea, and dizziness. Echocardiographic evidence of valvulopathy occurred in 2% of patients taking lorcaserin 10 mg twice daily and those taking the placebo. Lorcaserin administered in conjunction with a diet and exercise program was associated with an overall reduction in baseline BMI when compared with placebo over the year.
The BLOOM-DM study evaluated efficacy and safety of lorcaserin for weight loss in 604 patients with T2DM over the course of 1 year.29 Patients had a hemoglobin A1c (A1c) of 7% to 10% and were treated with metformin, a sulfonylurea, or both. The primary endpoint was a 5% weight reduction from baseline at the end of 1 year. Patients were randomized into 3 groups: 1 group received lorcaserin 10 mg twice daily, 1 group took lorcaserin 10 mg daily, and 1 group received the placebo. A statistically significant 37.5% of patients taking lorcaserin 10 mg twice daily achieved > 5% body weight reduction, compared with 44.7% in the lorcaserin 10 mg daily group, and 16.1% in the placebo group. Overall reductions in A1c and fasting glucose were observed in both lorcaserin groups taking as compared with placebo. Patient A1c decreased 0.9 ± 0.06 with lorcaserin 10 mg bid, 1.0 ± 0.09 with lorcaserin 10 mg qd, and 0.4 ± 0.06 with the placebo (P < .001). Fasting glucose in the lorcaserin bid, lorcaserin qd, and placebo groups decreased 27.4 ± 2.5 mg/dL, 28.4 ± 3.8 mg/dL, and 11.9 ± 2.5 mg/dL, respectively (P < .001). Symptomatic hypoglycemia occurred in 7.4% of patients on lorcaserin bid, 10.5% on lorcaserin qd, and 6.3% on placebo. Headache, back pain, nasopharyngitis, and nausea were among the most commonly reported AEs.
As lorcaserin is a serotonergic agonist, potential interactions exist when used with other medications affecting serotonin. Most notably, serotonin syndrome and neuroleptic malignant syndrome-like reactions may occur. Because of this, it is recommended to avoid selective serotonin re-uptake inhibitors (SSRIs), selective norepinephrine reuptake inhibitors, tricyclic antidepressants, bupropion, triptans, monoamine oxidase inhibitors, lithium, dextromethorphan, and dopamine agonists. Lorcaserin seems to be safe in those patient populations with mild hepatic as well as mild renal impairment; however, it is not recommended for those with severe renal impairment. Given the multiple enzymatic pathways used to metabolize lorcaserin, there is a low probability for cytochrome drug interactions. Safety has not been well evaluated in patients aged < 18 years and those that are pregnant (pregnancy category X).
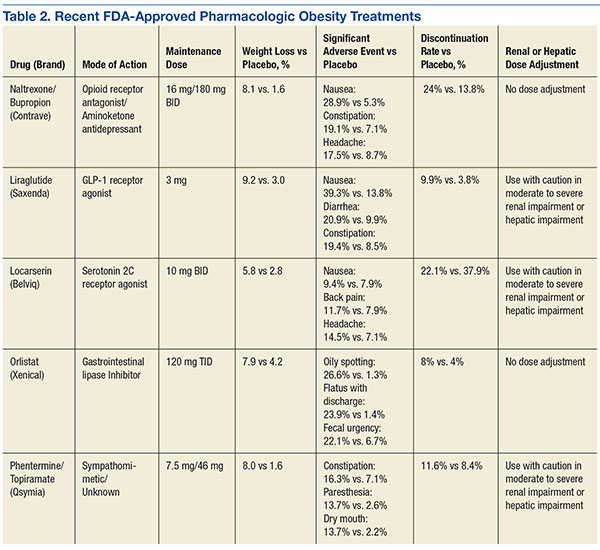
Adverse events include headache, dizziness, fatigue, nausea, and dry mouth. Other notable AEs include nasopharyngitis and URI. Hypoglycemia appeared to be more common in patients with DM taking lorcaserin. Cognitive impairment and psychiatric disorders including euphoria and hallucinations were also reported. Notably, valvular heart disease has been reported in patients who take medications with 5HT2b activity. In a 1-year clinical trial, a small number of patients were found to develop valvular regurgitation. Furthermore, bradycardia, priapism, leucopenia, elevated prolactin, and pulmonary hypertension have also been observed. Caution is recommended if symptoms of any of the aforementioned conditions are noticed.
Qsymia
The schedule IV controlled substance Qsymia (Vivus, Mountain View, CA) is a combination of phentermine, an anorexigenic agent, and topiramate extended-release, an antiepileptic drug. In July of 2012 it was approved for chronic weight management as an addition to a reduced-calorie diet and exercise. The drug is approved for adults with a BMI ≥ 30 kg/m2 or adults with a BMI ≥ 27 kg/m2 who have at least 1 weight-related condition such as hypertension, T2DM, or dyslipidemia.30
In 1996 topiramate was approved by the FDA for the treatment of seizure disorders and was also approved for migraine prophylaxis in 2004. In patients who were treated with topiramate for seizure disorders and migraines, weight loss and a reduction in visceral body fat has been observed.31 The precise MOA of topiramate in regards to weight loss is not fully understood. It may be due to its effects on both appetite suppression and satiety enhancement. Topiramate exhibits a combination of properties including modulatory effects on sodium channels, enhancement of GABA-activated chloride channels, inhibition of excitatory neurotransmission through actions on kainite and AMPA receptors, and inhibition of carbonic anhydrase (CA) isoenzymes in particular CA II and IV.14
The combination of phentermine and topiramate is a once-daily formulation that is designed to provide an immediate release of phentermine and a delayed release of topiramate, allowing a peak exposure of the phentermine in the morning and a peak concentration of topiramate in the evening. It should be taken in the morning in order to avoid the possibility of insomnia that can occur if taken in the evening. It can be taken with or without food. The recommended dose is as follows: Start treatment with Qsymia 3.75 mg/23 mg extended-release daily for 14 days; after 14 days increase to the recommended dose of Qsymia 7.5 mg/46 mg once daily.
Weight loss should be evaluated after 12 weeks at the higher dose. If at least 3% of baseline body weight has not been lost at that time, discontinue or escalate the dose. To escalate the dose: Increase to Qsymia 11.25 mg/69 mg daily for 14 days; followed by Qsymia 15 mg/92 mg daily. Evaluate weight loss following dose escalation to Qsymia 15 mg/92 mg after an additional 12 weeks of treatment. If at least 5% of baseline body weight has not been lost on Qsymia 15 mg/92 mg, discontinue as directed. It is important not to suddenly discontinue, as this may cause seizures. Patients should be slowly titrated off the medication.
In vitro studies of phentermine and topiramate indicate that these drugs are not likely to cause clinically significant interactions with drugs using the cytochrome P450 enzyme pathways, or those involved in plasma protein binding displacement; however there is evidence suggesting that ethinyl estradiol levels may be decreased by 16%, thus raising a concern about the possibility of decreased contraceptive efficacy.31 In patients with moderate (creatine clearance ≥ 30 mL/min to < 50 mL/min) and severe renal dysfunction (< 30 mL/min), the maximum dose of should not exceed 7.5 mg/46 mg.
Qsymia was evaluated in 3 phase 3 trials for its long-term efficacy and safety. In all trials, diet and lifestyle counseling were provided for all patients. The first of these studies was OB-301, a 28-week confirmatory trial with a factorial design involving 7 treatment arms, tested 2 fixed-dose Qsymia combinations—regular dose (7.5 mg/46 mg) and maximum dose (15 mg/92 mg)—as well as regular and maximum doses of the individual constituent drugs vs placebo.32 The study randomized 756 obese patients with a BMI range of 30 kg/m2 to 45 kg/m2 to 1 of the 7 treatment arms for 28 weeks. Patients treated with maximum-dose Qsymia achieved an average weight change of -9.0%, vs -1.5% with placebo (P < .0001). Weight change with regular-dose Qsymia was -8.2%. Weight changes with monotherapies were: -6.1% with topiramate 92 mg, -4.9% with topiramate 46 mg, -5.8% with phentermine 15 mg, and -5.2% with phentermine 7.5 mg.
OB-302 was a 56-week trial that randomized 1,267 morbidly obese patients with a BMI ≥ 35 kg/m2 without significant comorbidities to low-dose Qsymia (3.7 mg/23 mg), maximum-dose Qsymia (15 mg/92 mg), or placebo.33 At baseline, the mean BMI for the entire study cohort was 42 kg/m2. Mean weight changes were -1.6% with placebo, -5.1% with low-dose Qsymia, and -10.9% with maximum-dose Qsymia. The proportions of patients achieving ≥ 5% weight loss were: 17% with placebo, 45% with low-dose Qsymia, and 67% with maximum-dose Qsymia.
CONQUER was the largest of the phase 3 trials. It randomized 2,487 overweight or obese patients with a BMI of 27 kg/m2 to 45 kg/m2 and ≥ 2 obesity-related comorbidities (hypertension, dyslipidemia, T2DM, prediabetes or abdominal obesity) to receive a placebo, regular-dose Qsymia, or maximum-dose Qsymia for 56 weeks.34 In the completer population, mean weight changes in the placebo, regular dose Qsymia, and maximum-dose Qsymia groups were -1.6%, -9.6% (P <.0001), and -12.4% (P < .0001); and weight loss of ≥ 5% was achieved by 21%, 62%, and 70%, respectively. Relative to placebo, there were greater reductions in systolic BP, triglycerides, and fasting insulin with both doses of Qsymia.
Patients should not take Qsymia if they are pregnant, planning to become pregnant, or become pregnant during Qsymia treatment as there is an increased risk of birth defects, namely cleft lip and cleft palate. Women who can become pregnant should have a negative pregnancy test before taking Qsymia and every month while on the medication. They should use effective birth control consistently while taking Qsymia.
Qsymia is contraindicated in patients with glaucoma and patients who have hyperthyroidism. Qsymia can cause an increase in resting heart rate and regular monitoring of resting heart rate is recommended, especially in patients with cardiac or cerebrovascular disease. It has not been studied in patients with recent or unstable cardiac or cerebrovascular disease and therefore use is not recommended.
Qsymia can cause mood disorders such as anxiety and depression and can increase the risk of suicidal thoughts. Patients should be monitored for worsening depression, suicidal thoughts or behavior, or any unusual changes in mood or behavior. It is not recommended in patients with a history of suicidal attempts or active suicidal ideation. Qsymia can cause cognitive dysfunction. It can cause confusion, problems with concentration, attention, memory, or speech. Patients should be cautioned about operating automobiles and hazardous machinery.
Normal anion gap hyperchloremic metabolic acidosis has been reported in patients treated with Qsymia. If this does develop and persists, consideration should be given to either reduce the dose or discontinue Qsymia.
Weight loss may increase the risk of hypoglycemia in patients with T2DM treated with insulin and/or insulin secretagogues (eg, sulfonylureas). Qsymia has not been studied in combination with insulin. A reduction in the dose of antidiabetic medications, which are nonglucose dependent, should be considered to reduce the risk of hypoglycemia.
The most common AEs in controlled clinical studies (≥ 5% and at least 1.5 times placebo) included paraesthesia in the hands, arms, feet or face, dizziness, dysgeusia, insomnia, constipation, and dry mouth.
Contrave
In 2014, the FDA approved Contrave (Takeda, Deerfield, IL) as treatment option for chronic weight management in addition to reduced-calorie diet and physical activity. The combination of naltrexone hydrochloride and bupropion hydrochloride was originally introduced for the treatment of opioid addiction and later expanded to include the treatment of alcoholism. The antidepressant bupropion was approved in the U.S. in 1989. It is structurally different from all other marketed antidepressants (ie, tricyclics, tetracyclics, and SSRIs), but closely resembles the structure of diethylpropion, an appetite depressant with minimal CNS effects.35
This drug is approved for adults with BMI ≥ 30 kg/m2 and for adults with BMI ≥ 27 kg/m2 with at least 1 weight-related risk factors such as hypertension, T2DM, or dyslipidemia. It should be used as an adjunct to diet and exercise and is not approved for use for depression even though it contains bupropion.
Naltrexone is a pure opioid antagonist with high affinity to μ-opioid receptor, which is implicated in eating behavior. Naltrexone is rapidly and nearly completely absorbed from the GI tract after oral administration. The time to peak plasma concentration is about 1 hour. Naltrexone is well absorbed but first pass extraction and metabolism by the liver decreases oral bioavailability to between 5% to 40%. Primary elimination of naltrexone and its metabolites is renal excretion.
Bupropion is a weak inhibitor of neuronal reuptake of dopamine and norepinephrine. This drug is used to treat depression and seasonal affective disorder, and aid in smoking cessation. Bupropion is absorbed rapidly after oral administration, but the absolute oral bioavailability of bupropion is not known because an IV preparation is not available. The time to peak plasma concentrations of bupropion is within 2 hours of oral administration. Bupropion is extensively metabolized by the liver to multiple metabolites. Primary elimination of bupropion is urinary excretion. However, hepatic and renal impairment may affect the elimination of bupropion and its metabolites. Patients with hepatic or renal impairment should use a reduced dosage.
Combination therapy has been found to have complementary actions on CNS to reduce food intake. They are believed to dampen CNS reward pathways, taking away the compulsive feeding behavior and pleasure of feeding, ultimately leading to weight loss. Bupropion stimulates hypothalamic pro-opiomelanocortin neurons (POMC), which results in reduced food intake and increased energy expenditure. Naltrexone blocks opioid-receptor mediated POMC auto-inhibition, blocks the increase in dopamine in nucleus accumbens that occurs when eating, and acting synergistically with bupropion in augmenting POMC firing.
The COR-I and COR-II trials compared Contrave to diet and exercise in patients who did not have DM. The COR-Diabetes trial included the same study design but focused on patients with DM. In all the studies the participants had a 4-week titration to Contrave (naltrexone 8 mg/bupropion 90 mg) to decrease nausea. The first week dosing was 1 tablet in the morning. Week 2 was 1 tablet in morning and 1 tablet in the evening. In week 3, patients took 2 tablets in the morning and 1 tablet in the evening. The final titration step was 2 tablets in the morning and 2 tablets in the evening.
The COR-1 study was a 56-week randomized, double-blind, placebo-controlled study. It compared Contrave 32 mg naltrexone/360 mg bupropion (NB32/360) with an active placebo of diet and exercise.36 To be included adults must be aged 18 to 65 years with a BMI 30 kg/m2 to 45 kg/m2 or a BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia or hypertension. Patients were instructed on a hypocaloric diet that was a 500 kcal per day deficit based on World Health Organization algorithm for calculating metabolic rate and they were urged to increase physical activity.
The completer population results showed 8.0% weight loss in the NB32/360 group and 1.9% weight loss in the placebo group (P < .001). For the NB32/360 and placebo groups, weight loss of ≥ 5% was achieved by 48% and 16% (P <.001), respectively; and weight loss of ≥ 10% by 25% and 7% (P < .001), respectively. The most common AE was nausea—29.8% with NB32/360 vs 5.3% with placebo. Nausea generally occurred early and then diminished and the discontinuation rate from nausea was significantly lower (6.3%) then the overall reported nausea rates.
Contrave was also studied in patients with T2DM. The COR-Diabetes Trial was a 56-week randomized, double blind, placebo-controlled study. The trial compared Contrave 32 mg naltrexone/360 mg bupropion (NB32/360) with an active placebo of diet and exercise.37 Inclusion criteria for the trial were patients aged 18 to 70 years with T2DM and a BMI from 27 kg/m2 to 45 kg/m2, A1c between 7% and 10%, and fasting blood glucose < 270 mg/dL. Participants either were not taking a DM medication or were on stable doses of oral antidiabetes drugs ≥ 3 months prior to randomization. Patients were placed on a 500 kcal hypocaloric diet and advised to increase physical activity.
The results showed 5.0% weight loss in the NB32/360 group and 1.8% weight loss in the placebo group (P < .001). Weight loss of ≥ 5% and ≥ 10% was achieved by 44.5% and 18.5% of the NB32/360 group, respectively, and 18.9% and 5.7%, respectively (P < .001) of the placebo group. The NB32/360 and placebo showed a reduction of A1c of 0.6% and 0.1% respectively (P < .001). The most common AE was nausea (42.3% with NB32/360 vs 7.1% with placebo). Nausea generally occurred early and then diminished and the discontinuation rate from nausea was significantly lower (9.6%) then the overall reported nausea rates.37
Due to potential nausea caused by naltrexone, Contrave should be titrated over 4 weeks as described earlier. At maintenance dose, patients should be evaluated after 12 weeks to determine treatment benefits. If a patient has not lost at least 5% of baseline body weight, Contrave should be discontinued, because it would be unlikely that the patient will achieve and sustain clinically meaningful weight loss with continued treatment. Contrave should not be taken with high-fat meals that may result in significant increase in bupropion and naltrexone systemic exposure.
Since Contrave contains the antidepressant bupropion, it has a boxed warning similar to other antidepressants in its class of increased risk of suicidal thoughts and behaviors, especially in children, adolescents, and young adults.38Contrave can lower the seizure threshold; therefore it should not be used in people with a seizure disorder. It can also raise BP and heart rate; however the clinical significance of hypertension and elevated heart rate observed with Contrave treatment is unclear. Blood pressure rose on average by 1 point during the first 8 weeks of treatment and then returned to baseline.38 The heart rate also increased by about 1.7 beats per minute.38 Patients with uncontrolled hypertension should avoid Contrave.
Contrave should not be taken with products contain bupropion or naltrexone. It should not be taken by patient who are regularly taking opioids or who are opioid dependent, or who are experiencing opiate withdrawal. Pregnant women should also avoid Contrave. In patients with renal impairment the maximum dose is 1 tablet twice a day and in patients with hepatic impairment the maximum dose is 1 tablet a day.
Liraglutide
Liraglutide is the newest weight loss medication to be approved by the FDA for chronic weight management as an adjunct to a reduced calorie diet and increased physical activity in adult patients with BMI ≥ 30 kg/m2 or ≥ 27 kg/m2 with hypertension, diabetes, or dyslipidemia. The recommended dose of liraglutide is 3 mg daily. The initial dose is 0.6 mg daily for the first week, then titrated up by 0.6 each week for 4 weeks, until reaching 3 mg daily.
Liraglutide is an acylated human glucagon-like peptide-1 (GLP-1) receptor agonist, which are expressed in the brain and is involved in the control of appetite. It is also found in the beta cells of the pancreas, where GLP-1 receptors stimulate insulin release in response to elevated blood glucose concentrations and suppress glucagon secretion. Endogenous GLP-1 has a half-life of 1.5 to 2 minutes due to degradation by the DDP-4 enzyme, but liraglutide is stable against degradation by peptidases and has a half- life of 13 hours.
Liraglutide was studied in a 56-week randomized, double-blind, placebo-controlled trial, which compared liraglutide 3 mg with an active placebo of diet and physical activity.39 Inclusion criteria were adults aged ≥ 18 years old with a BMI 30 kg/m2 to 45 kg/m2 or BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia and/or hypertension. Both groups received lifestyle modification counseling. Patients were excluded if they had DM.
In the trial, 3,731 participants enrolled, 2,487 in the liraglutide group and 1,244 in the placebo group; 78.7% of the participants were female and the average age was 45 years. Subjects in the liraglutide group had a weekly titration regimen. The starting dose at week 1 was 0.6 mg, week 2 was 1.2 mg, week 3 was 1.8 mg, week 4 was 2.4 mg, and week 5 was 3.0 kg.
The completer population showed 9.2% weight loss in the liraglutide group and 3.0% weight loss in the control group.39 Weight loss of ≥ 5% was seen in 63.2% and 27.1% of the liraglutide and placebo groups, respectively. Weight loss rates of ≥ 10% was seen by 33.1% and 10.6%, respectively. The most common AEs were nausea, diarrhea, and constipation. Nausea generally occurred early during the titration period and then diminished.
A second clinically relevant study was performed with liraglutide. Often patients are able to lose weight with diet and exercise and then plateau. This study examined participants who lost 5% percent of their initial body weight and then were randomized to liraglutide or placebo.40 Key inclusion criteria were people aged ≥ 18 years old with a BMI 30 kg/m2 to 45 kg/m2 or BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia and/or hypertension. In order to be randomized, participants were required to lose at least 5% of their initial body weight on a 1,200 kcal to 1,400 kcal diet with increased physical activity during a 4 to 12 week run-in period.
Four hundred twenty-two participants were enrolled, 212 in the liraglutide group and 210 in the placebo group. Most of the participants were female (81%). The average BMI in the study was 35.6 kg/m2. Subjects in the liraglutide group had a weekly titration regimen.
After an average weight loss of 6% using a low calorie diet and increased physical activity the participants were randomized to continue diet and increased activity alone (placebo) or with liraglutide. At week 56 the results showed an additional 6.2% weight loss in the liraglutide group and 0.2% weight gain in the placebo group. The liraglutide group had a greater number of participants with ≥ 5% weight loss compared to placebo, 50.5% vs 21.8% (P < .0001).40 In the pooled data set from the registration trials the 3 most common GI AEs were nausea, diarrhea, and constipation occurring in 39.3%, 20.9%, and 19.4% of participants respectively. Discontinuation due to nausea for liraglutide was 2.9%.41
Clinicians should be aware that medications that can cause hypoglycemia such as sulfonylureas and insulin must be tapered as patients lose weight with liraglutide. Documented symptomatic hypoglycemia in patients with T2DM and with sulfonylurea background therapy was 43.6% with liraglutide vs 27.3% with placebo.
In the setting of renal impairment, patients treated with GLP-1 receptor agonists, including liraglutide, have had reports of acute renal failure and worsening of chronic renal failure usually associated with nausea, vomiting, diarrhea, or dehydration. Liraglutide causes thyroid C-cell tumors at clinically relevant exposures in rats and mice. It is unknown whether liraglutide causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans. As the human relevance of liraglutide-induced rodent thyroid C-cell tumors has not been determined liraglutide is contraindicated in patients with a personal or family history of MTC or in patients with multiple endocrine neoplasia syndrome type 2.
Acute pancreatitis, including fatal and nonfatal hemorrhagic or necrotizing pancreatitis, has been observed in patients treated with liraglutide in postmarketing reports. After initiation of liraglutide, observe patients carefully for signs and symptoms of pancreatitis (including persistent severe abdominal pain, sometimes radiating to the back, which may or may not be accompanied by vomiting). If pancreatitis is suspected, liraglutide should promptly be discontinued.
Conclusion
The treatment of obesity and overweight with comorbidities has always been a challenge. In the past there were few FDA-approved drugs and many drugs had to be used off-label. The toolbox of medications available for medical weight management is more robust than ever. The medications have different MOAs and can be used in a variety of patients. There are differences in the classes and some are controlled substances. Phentermine, lorcaserin, and Qsymia (phentermine/topiramate) are controlled substances whereas orlistat, naltrexone/bupropion and liraglutide are not. Other differences exist including duration of use. The sympathomimetic drugs have a limited window of use whereas orlistat, Qsymia (phentermine/topiramate), lorcaserin, naltrexone/bupropion, and liraglutide do not.
The medications that are available have a wide variety of MOAs. Therefore, if a patient fails one medication, then it is very reasonable to try a medication with a different MOA. In addition, there is the potential for weight regain when weight reduction medications are discontinued. As people lose weight their metabolic rate decreases about 15 kcal per pound of weight reduction.42
Another challenge of using these medications is managing patient expectations. The current metric used for FDA approval is a 5% weight loss that is greater in the study group compared with the diet and physical activity active control. However, many clinicians and patients do not find this weight reduction amount consistent with their expectations. In addition weight loss trajectory may also be too slow for patients and cause early discontinuation. Therefore, patient education and a discussion of reasonable expectations for weight reduction medications are necessary.
Clinicians must acknowledge that there are limitations to the use of these medications. Newer agents do have a higher cost and insurance reimbursement is somewhat limited. However, they offer the opportunity to prevent more expensive, protracted conditions such as diabetes and cardiovascular disease. In summary, clinicians now have a wider variety of medication options to be used with dietary and lifestyle changes in order to improve health and prevent chronic diseases.
1. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of obesity in the United States, 2009-2010. NCHS Data Brief. 2012(82):1-8.
2. Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. Circulation. 2014;129(25 suppl 2):S102-S138.
3. U.S. Food and Drug Administration. Drugs@FDA: diethylpropion hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=012546&DrugName=TENUATE%20DOSPAN&ActiveIngred=DIETHYLPROPION%20HYDROCHLORIDE&SponsorApplicant=ACTAVIS%20LABS%20UT%20INC&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
4. U.S. Food and Drug Administration. Drugs@FDA: benzphetamine hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=012427&DrugName=DIDREX&ActiveIngred=BENZPHETAMINE%20HYDROCHLORIDE&SponsorApplicant=PHARMACIA%20AND%20UPJOHN&ProductMktStatus=3&goto=Search.DrugDetails. Accessed December 28, 2015.
5. U.S. Food and Drug Administration. Drugs@ FDA: phendimetrazine tartrate. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=088021&DrugName=BONTRIL&ActiveIngred=PHENDIMETRAZINE%20TARTRATE&SponsorApplicant=VALEANT&ProductMktStatus=3&goto=Search.DrugDetails. Accessed December 28, 2015.
6. U.S. Food and Drug Administration. Drugs@FDA: phentermine. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=085128&DrugName=ADIPEX%2DP&ActiveIngred=PHENTERMINE%20HYDROCHLORIDE&SponsorApplicant=TEVA&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
7. U.S. Food and Drug Administration. Drugs @ FDA: phentermine hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=202088&DrugName=SUPRENZA&ActiveIngred=PHENTERMINE%20HYDROCHLORIDE&SponsorApplicant=CITIUS%20PHARMS&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
8. Hendricks EJ, Rothman RB, Greenway FL. How Physician Obesity Specialists use drugs to treat obesity. Obesity (Silver Spring). 2009;17(9):1730-1735.
9. Gilman AG, Gilman A, Goodman LS, eds. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 8th ed. New York: Pergamon Press; 1990: 211.
10. Gilman AG, Gilman A, Goodman LS, eds. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 8th ed. New York: Pergamon Press; 1990:217.
11. Addy C, Jumes P, Rosko K, et al. Pharmacokinetics, safety, and tolerability of phentermine in health participants receiving taranabant, a novel cannabinoid-1 receptor (CB1R) inverse agonist. J Clin Pharmacol. 2009;49(10):1228-1232.
12. U.S. Department of Justice, Drug Enforcement Administration, Office of Diversion Control. Controlled substances. U.S. Department of Justice Website. http://www.deadiversion.usdoj.gov/schedules/orangebook/c_cs_alpha.pdf. Updated November 12, 2015. Accessed December 16, 2015.
13. Bray GA, Greenway FL. Pharmacological treatment of the overweight patient. Pharmacol Rev. 2007;59(2):151-184.
14. V1-0521 (QNEXA) Advisory committee briefing document. NDA 022580. Endocrinologic and Metabolic Drugs Advisory Committee meeting; June 17, 2010.
15. Cercato C, Roizenblatt VA, Leanca CC, et al. A randomized double-blind placebo-controlled study of the long-term efficacy and safety of diethylpropion in the treatment of obese subjects. Int J Obes (Lond). 2009;33(8):857-865.
16. Haddock CK, Poston WS, Dill PL, Foreyt JP, Ericsson M. Pharmacotherapy for obesity: a quantitative analysis of four decades of published randomized clinical trials. Int J Obes (Lond). 2002;26(2):262-273.
17. Suplicy H, Boquszewski CL, dos Santos CM, do Desterro de Fiqueiredo M, Cunha DR, Radominski R. A comparative study of five centrally acting drugs on pharmacological treatment of obesity. Int J Obes (Lond). 2014;38(8):1097-1103.
18. Ioannides-Demos LL, Proietto J, McNeill JJ. 2005. Pharmacotherapy for obesity. Drugs. 2005;65(10): 1391-1418.
19. Drent ML, van der Veen EA. Lipase inhibition: a novel concept in the treatment of obesity. Int J Obes Relat Metab Disord. 1993;17(4):241-244.
20. Xenical [package insert]. Nutley, NJ: Roche Laboratories Inc.; 1999.
21. U.S. Food and Drug Administration. FDA Drug Safety Communication: Completed safety review of Xenical/Alli and severe liver injury. U.S. Food and Drug Administration Website. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm213038.htm. Updated August 2, 2010. Accessed December 16, 2015.
22. Sall D, Wang J, Rashkin M, Welch M, Droege C, Schauer D. Orlistat-induced fulminant hepatic failure. Clin Obes. 2014;4(6):342-347.
23. Sjöström L, Rissanen A, Andersen T, et al. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. European Multicentre Orlistat Study Group. Lancet. 1998;352(9123):167-172.
24. Hauptman J, Lucas C, Boldrin MN, Collins H, Segal KR. Orlistat in the long-term treatment of obesity in primary care settings. Arch Fam Med. 2000;9(2):160-167.
25. Torgerson JS, Hauptman J, Boldrin MN, Sjöström L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27(1):155-161.
26. Torgerson JS, Arlinger K, Käppi M, Sjöström L. Principles for enhanced recruitment of subjects in a large clinical trial. the XENDOS (XENical in the prevention of Diabetes in Obese Subjects) study experience. Control Clin Trials. 2001;22(5):515-525.
27.Smith SR, Weissman NJ, Anderson CM, et al; Behavioral Modification and Lorcaserin for Overweight and Obesity Management (BLOOM) Study Group. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med. 2010;363(3):245-256.
28. Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011;96(10):3067-3077.
29. O’Neil PM, Smith SR, Weisserman NJ, et al. Randomized placebo-controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM Study. Obesity (Silver Spring). 2012;20(7):1426-1436.
30. U.S. Food and Drug Administration. FDA approves weight-management drug Qsymia. U.S. Food and Drug Administration Website. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm312468.htm. Published July 17, 2012. Accessed December 16, 2015.
31. Shin J, Gadde KM. Clinical utility of phentermine/topiramate (Qsymia™) combination for the treatment of obesity. Diabetes Metab Syndr Obes. 2013;6:131-139.
32. Qsymia [package insert] Mountain View, CA: Vivus, Inc; 2012.
33. Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine/topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring). 2012;20(2):330-342.
34. Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377(9774):1341-1352.
35. Plodkowski RA, Nguyen Q, Sundaram U, Nguyen L, Chau DL, St Jeor S. Bupropion and naltrexone: a review of their use individually and in combination for the treatment of obesity. Expert Opin Pharmacother. 2009;10(6):1069-1081.
36. Greenway FL, Fujioka K, Plodkowski RA, et al; COR-I Study Group. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-1): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595-605.
37. Hollander P, Gupta AK, Plodkowski R, et al; COR-Diabetes Study Group. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care. 2013;36(12):4022-4029.
38. Contrave [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc; 2014.
39. Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Eng J Med. 2015;373(1):11-22.
40. Wadden TA, Hollander P, Klein S, et al; NN8022-1923 Investigators. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet-induced weight loss: the SCALE Maintenance randomized study. Int J Obes (Lond). 2013;37(11):1443-1451
41. Saxenda [package insert]. Novo Nordisk: Plainsboro, NJ; 2015.
42.Schwartz A, Doucet E. Relative changes in resting energy expenditure during weight loss: a systemic review. Obes Rev. 2010;11(7): 531-547. ```````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````
Over the past decade the prevalence of obesity as defined by a body mass index (BMI) ≥ 30 kg/m2 has significantly increased. In the U.S. more than 78 million adults are estimated to be obese.1 The World Health Organization projects that by 2025 up to half the U.S. population will be obese. Cardiovascular disease (CVD) and diabetes mellitus (DM) are the main comorbid conditions that are complicated by obesity. Initial weight loss of 5% to 10% of total body weight reduces CVD risk factors, prevents or delays the development of type 2 DM (T2DM) and improves the health consequences of obesity.2
To date, public health initiatives that have focused on obesity prevention and lifestyle intervention have had marginal success. In recent years, anti-obesity drug therapies have had a limited role in clinical treatment algorithms. In 2013, the American Medical Association acknowledged obesity as a disease. In turn, this acknowledgement allowed the recognition of anti-obesity drugs as acceptable therapeutic adjuncts to intensive lifestyle intervention that could address the growing obesity endemic.
In the past, medications for weight reduction were limited. Several that were FDA approved had to be removed from the market due to safety concerns. With few approved options, clinicians often had to resort to off-label use of medications. However, the landscape has changed with 4 new medications gaining recent FDA approval. This review covers older available medications and the newer medications that are now available.
Sympathomimetics
Sympathomimetic drugs have been approved for use as a pharmacological method to lose weight since 1960. Of the many versions of this drug class that have been available since then, there are 4 major versions available today. These include diethylpropion3 and benzphetamine,4 both approved in 1960; phendimetrazine, approved in 1976;5 phentermine, approved in 1980;6 and phentermine hydrochloride, approved in 2012.7 Despite the existence of several other classes of drugs to treat obesity, phentermine remains the most often prescribed weight loss drug in the U.S.8
Although the mechanism of action (MOA) of sympathomimetic drugs is not particularly clear, weight loss from these medications is believed to be due to the increase in the release of biogenic amines (mainly norepinephrine, but also possibly dopamine), from storage sites in nerve terminals. It is possible that these drugs slow catecholamine metabolism by inhibiting the actions of monoamine oxidase. The resulting increase in amine availability, particularly in the lateral hypothalamic feeding center, is associated with reduced food intake. Interestingly, injection of these drugs into the ventromedial satiety center dooes not seem to suppress food intake, and the effects of biogenic amines on increasing metabolism does not seem to play a significant role in weight loss in patients on these medications.9
Each of these drugs is rapidly absorbed from the gastrointestinal (GI) tract except for phentermine hydrochloride, the newest of the medications in this class. Phentermine hydrochloride is a sublingual tablet that is readily absorbed through the buccal mucosa.5 All of the drugs in this class are excreted through the kidneys, with varying rates. Each drug’s excretion is highly dependent on the pH of the urine—more alkaline conditions result in less excretion and more acidic conditions result in more excretion. As a result, these drugs should be used with caution in patients with renal impairment; however, there are no specific contraindications listed for patients with poor renal function.
The adverse effects (AEs) for this drug class are to be expected from an increase in the release of biogenic amines in the central nervous system (CNS). The most common AEs include palpitations, tremors, restlessness, insomnia, dry mouth, constipation, diaphoresis, changes in libido, and irritability. The more dangerous AEs that have been observed include arrhythmias, hypertension, dependency/abuse, convulsions, acute transient ischemic colitis, and acute urinary retention secondary to increased bladder sphincter tone, transient hyperthyroxemia, and paranoia.10
Several contraindications exist for sympathomimetics, including the presence of advanced arteriosclerosis, symptomatic CVD, moderate to severe hypertension, hyperthyroidism, glaucoma, patients in an agitated state, or those with a history of amphetamine abuse. The warnings for prescribers include pulmonary hypertension and cardiomyopathy secondary to chronic use of sympathomimetics, and valvular heart disease secondary to use of sympathomimetics with additional anorectic agents.
Additional precautions should be considered in those with a history of anxiety/psychosis, those who operate machinery and motor vehicles, and even those with mild hypertension. The data surrounding the effects of sympathomimetics on blood pressure (BP) appears to be conflicting and the relationship does not seem to have been significantly studied in depth to warrant any definitive conclusions. The MOA of this drug class itself is enough to urge caution to prescribers.11 Special attention should be given to patients with diabetes when using sympathomimetics. A reduction of insulin dose or oral hypoglycemic dose may be necessary in some people with diabetes.
Only diethylpropion is pregnancy category B, whereas the others drugs in this class are pregnancy category X. It has been demonstrated that diethylpropion and benzphetamine are secreted into breastmilk; insufficient data exist to suggest whether or not phentermine and phendimetrazine are present in breastmilk. All drugs in this class should be used in caution with breastfeeding mothers.
Although all 4 drugs are registered as controlled substances, benzphetamine and phendimetrazine are schedule III and phentermine and diethylpropion are schedule IV, despite evidence suggesting the potential for abuse to be extremely low.12,13 Phentermine has been approved for adults aged > 18 years, phendimetrazine has been approved for those aged > 17 years, diethylpropion has been approved for those aged > 16 years, and benzphetamine has been approved for those aged > 12 years.
There is a wealth of literature surrounding the effectiveness of this drug class for weight loss. One of the longest trials of phentermine was recently conducted as part of the initial component of a FDA study for the newly approved topiramate-phentermine combination. Weight loss at 6 months in the phentermine-only group was significantly higher at -5.8% compared with -1.5% with the placebo group in the last observation carried forward-Intent to treat (LOCF-ITT) analysis.14 Similarly, a long-term study looking at diethylpropion examined the use of diethylpropion for up to a year vs placebo. Participants administered diethylpropion lost a mean 9.8% of original weight vs 3.7% in the placebo group in the first 6 months alone.15
Several meta-analyses and review papers have been authored that examine and analyze the published data on this drug class overall and comparatively within this class. Haddock and colleagues in 2002 reviewed the numerous clinical trials associated with each drug in this class, in addition to several other classes, and found that although each drug demonstrated a significant advantage vs placebo in weight loss, there was not a specific drug that was significantly superior to any of the others.16
These results seem to be in relative agreement with additional studies like that published by Suplicy and colleagues, which demonstrated that several sympathomimetics were better than placebo in weight loss, and that there was little difference between the specific drugs in the class.17 However, it should be noted that as highlighted in a review by Ioannides-Demos and colleagues in 2005, the vast majority of studies that had been performed on this drug class focused on short-term use (< 16 weeks) and none of the sympathomimetics listed here have been approved for long-term use.18
Orlistat
Orlistat 120 mg was approved in 1999 as a reversible inhibitor of GI lipases that specifically reduced the absorption of dietary fat due to the inhibition of triglyceride hydrolysis.19 Orlistat was later approved in 2007 for release in a reduced dosage form (60 mg) for over-the-counter sales.20
Orlistat forms a covalent bond with the active serine residue site of gastric and pancreatic lipases in the lumen of the stomach and small intestine. The inhibition of these enzymes causes dietary fat to remain undigested as triglycerides, which cannot be converted to absorbable free fatty acids and monoglycerides, leading to decreased calorie absorption. Orlistat is not systemically absorbed and is eliminated mainly through feces. Some metabolism occurs in the GI wall.21Orlistat is most known for its GI AEs. Because it is most active in the lumen of the GI system and reduces the absorption of triglycerides, many AEs are related to malabsorption. The most common issues 1 year after starting the drug were oily spotting (26.6% vs 1.3% placebo); flatus with discharge (23.9% vs 1.4% placebo); fecal urgency (22.1% vs 6.7% placebo); fatty/oily stool (20% vs 2.9% placebo); increased defecation (10.8% vs 4.1% placebo); and fecal incontinence (7.7% vs 0.9% placebo) (Table 1). Most of these AEs were greatly reduced after taking the drug for 2 years. Orlistat also has more serious AEs noted, including abdominal pain/discomfort; nausea; infectious diarrhea; rectal pain/discomfort; tooth disorder; gingival disorder; vomiting; upper respiratory infection; lower respiratory infection; ear, nose and throat symptoms; back pain; arthritis; myalgia; joint disorders; tendonitis; headache; dizziness; fatigue; sleep disorders; rash; dry skin; menstrual irregularity; vaginitis; urinary tract infection; and psychiatric disorders, although these did not differ markedly from placebo.
One of the most serious AEs reported was fulminate hepatic failure, though this AE is rare. Thirteen cases of liver injury were reported with the 120-mg prescription dose of orlistat and 1 case report in the U.S. involved the 60-mg over-the-counter dosage of orlistat.21,22 The FDA suggests that patients talk to their physicians about risks of liver failure, and that physicians should educate their patients about signs and symptoms of liver failure so that patients can stop taking orlistat and seek immediate medical help if symptoms occur.
One of the first published trials was the European Multicentre Orlistat Study Group, which included 743 participants with BMI between 28 kg/m2 and 47 kg/m2 from 15 different European centers. To test adherence, a 4-week single blind placebo lead-in was started with a hypocaloric diet. The first stage was completed by 688 patients who then proceeded to the double blind randomized control trial portion with a hypocaloric diet. From the start of lead-in to the end of year 1, the orlistat group weight decreased 10.2% (10.3 kg) vs 6.1% (6.1 kg) in the placebo group. The placebo subtracted difference between the groups was 3.9 kg (P < .001).23
A U.S.-based randomized double-blind placebo-controlled multicenter study included 796 obese patients with BMI between 30 kg/m2 and 44 kg/m2. Patients were assigned to 1 of 3 groups: placebo, orlistat 60 mg 3 times daily, or orlistat 120 mg 3 times daily. All groups were given a reduced energy diet. Patients in the orlistat 120 mg group lost significantly more weight than did the placebo group, -8.78% vs -4.26% respectively in year 1 in the completer analysis (P = .001). More participants who were treated with orlistat 120 mg lost 5% or more of their initial weight in year 1 compared with placebo, 50.5% vs 30.7% respectively (P < .001).24
In the XENDOS study the primary outcome measurement was time to onset of T2DM. Eligible participants were aged 30 to 60 years, with a BMI > 30 kg/m2. All patients had a 75-g oral glucose tolerance test and were required to have normal glucose tolerance or impaired glucose tolerance, but not T2DM. The double-blind randomized controlled trial included 3,305 subjects and compared a group taking 120 mg orlistat 3 times daily vs placebo. All patients were prescribed a reduced-calorie diet (800 kcal/d deficit) containing 30% of calories from fat. Patients were also encouraged to walk at least 1 kilometer daily in addition to their usual physical activity. Incidence of T2DM after 4 years was 6.2% in the orlistat group and 9.0% in the placebo group, reflecting a 37.3% risk reduction in the orlistat group (P = .0032).25,26
Lorcaserin
In 2012, lorcaserin HCl was FDA approved as a schedule IV drug for use as a weight loss medication as an adjunct to a reduced-calorie diet and increased physical activity. Lorcaserin is thought to act on 5-hydroxytryptamine-2c (5HT2c) receptors on the pro-opiomelanocortin (POMC) neurons in the arcuate nucleus, causing release of alpha-melanocortin-stimulating hormone (alpha-MSH), which in turn acts on melanocortin-4 receptors in the paraventricular nucleus to suppress appetite. At the maximum suggested dose of 10 mg twice daily, lorcaserin binds with 15 to 100 times greater affinity to 5HT2c receptors compared with 5HT2a and 5HT2b receptors respectively.
Indications for lorcaserin include patients with BMI ≥ 30 kg/m2 or ≥ 27 kg/m2 or greater with a weight-related comorbid condition such as hypertension, dyslipidemia, cardiovascular disease, impaired glucose tolerance, or sleep apnea.
The efficacy of lorcaserin for weight loss has been evaluated in 3 separate trials. The trials were randomized, double blinded and placebo controlled. The BLOOM trial, which included 3,182 patients with a mean BMI of 36.2 kg/m2, evaluated the efficacy of lorcaserin as a weight loss adjunct.27 Patients with pre-existing valvular disease, uncontrolled hypertension, or a major psychiatric condition were excluded. After initial randomization, patients were assigned to receive either lorcaserin 10 mg twice daily or a placebo. The primary endpoint was a 5% weight reduction from baseline by the end of 2 years. At 1 year, 47.5% of patients in the lorcaserin group and 20.3% in the placebo group had lost ≥ 5% of their body weight (P <.001). The average loss for the lorcaserin group was 5.8 ± 0.2 kg and 2.2 ± 0.1 kg for the placebo group at 1 year (P < .001).
The BLOSSOM trial was a 1-year study of 4,008 patients aged 18 to 65 years. The trial evaluated the effects of lorcaserin on body weight, CVD risk factors, and safety in obese and overweight patients.28 Patients were randomized in a 2:1:2 ratio to receive lorcaserin 10 mg twice daily, lorcaserin 10 mg once daily, or placebo. The primary endpoint was the proportion of patients achieving at least 5% reduction in body weight. Completer analysis showed weight reduction in the placebo group was 4.0% and 7.9% in the lorcaserin group (P < .001). In the modified intent-to-treat/last observation carried forward analysis (MITT/LOCF), a statistically significant 47.2% of patients receiving lorcaserin 10 mg twice daily and 40.2% of patients receiving lorcaserin 10 mg once daily lost at least 5% of baseline body weight; compared with 25% of patients receiving placebo (P < .001). Weight loss of at least 10% was achieved by 22.6% of patients receiving lorcaserin 10 mg twice daily, and 17.4% of patients receiving 10 mg daily compared with 9.7% of patients in the placebo group (P < .001).
The most common AEs noted were headache, nausea, and dizziness. Echocardiographic evidence of valvulopathy occurred in 2% of patients taking lorcaserin 10 mg twice daily and those taking the placebo. Lorcaserin administered in conjunction with a diet and exercise program was associated with an overall reduction in baseline BMI when compared with placebo over the year.
The BLOOM-DM study evaluated efficacy and safety of lorcaserin for weight loss in 604 patients with T2DM over the course of 1 year.29 Patients had a hemoglobin A1c (A1c) of 7% to 10% and were treated with metformin, a sulfonylurea, or both. The primary endpoint was a 5% weight reduction from baseline at the end of 1 year. Patients were randomized into 3 groups: 1 group received lorcaserin 10 mg twice daily, 1 group took lorcaserin 10 mg daily, and 1 group received the placebo. A statistically significant 37.5% of patients taking lorcaserin 10 mg twice daily achieved > 5% body weight reduction, compared with 44.7% in the lorcaserin 10 mg daily group, and 16.1% in the placebo group. Overall reductions in A1c and fasting glucose were observed in both lorcaserin groups taking as compared with placebo. Patient A1c decreased 0.9 ± 0.06 with lorcaserin 10 mg bid, 1.0 ± 0.09 with lorcaserin 10 mg qd, and 0.4 ± 0.06 with the placebo (P < .001). Fasting glucose in the lorcaserin bid, lorcaserin qd, and placebo groups decreased 27.4 ± 2.5 mg/dL, 28.4 ± 3.8 mg/dL, and 11.9 ± 2.5 mg/dL, respectively (P < .001). Symptomatic hypoglycemia occurred in 7.4% of patients on lorcaserin bid, 10.5% on lorcaserin qd, and 6.3% on placebo. Headache, back pain, nasopharyngitis, and nausea were among the most commonly reported AEs.
As lorcaserin is a serotonergic agonist, potential interactions exist when used with other medications affecting serotonin. Most notably, serotonin syndrome and neuroleptic malignant syndrome-like reactions may occur. Because of this, it is recommended to avoid selective serotonin re-uptake inhibitors (SSRIs), selective norepinephrine reuptake inhibitors, tricyclic antidepressants, bupropion, triptans, monoamine oxidase inhibitors, lithium, dextromethorphan, and dopamine agonists. Lorcaserin seems to be safe in those patient populations with mild hepatic as well as mild renal impairment; however, it is not recommended for those with severe renal impairment. Given the multiple enzymatic pathways used to metabolize lorcaserin, there is a low probability for cytochrome drug interactions. Safety has not been well evaluated in patients aged < 18 years and those that are pregnant (pregnancy category X).
Adverse events include headache, dizziness, fatigue, nausea, and dry mouth. Other notable AEs include nasopharyngitis and URI. Hypoglycemia appeared to be more common in patients with DM taking lorcaserin. Cognitive impairment and psychiatric disorders including euphoria and hallucinations were also reported. Notably, valvular heart disease has been reported in patients who take medications with 5HT2b activity. In a 1-year clinical trial, a small number of patients were found to develop valvular regurgitation. Furthermore, bradycardia, priapism, leucopenia, elevated prolactin, and pulmonary hypertension have also been observed. Caution is recommended if symptoms of any of the aforementioned conditions are noticed.
Qsymia
The schedule IV controlled substance Qsymia (Vivus, Mountain View, CA) is a combination of phentermine, an anorexigenic agent, and topiramate extended-release, an antiepileptic drug. In July of 2012 it was approved for chronic weight management as an addition to a reduced-calorie diet and exercise. The drug is approved for adults with a BMI ≥ 30 kg/m2 or adults with a BMI ≥ 27 kg/m2 who have at least 1 weight-related condition such as hypertension, T2DM, or dyslipidemia.30
In 1996 topiramate was approved by the FDA for the treatment of seizure disorders and was also approved for migraine prophylaxis in 2004. In patients who were treated with topiramate for seizure disorders and migraines, weight loss and a reduction in visceral body fat has been observed.31 The precise MOA of topiramate in regards to weight loss is not fully understood. It may be due to its effects on both appetite suppression and satiety enhancement. Topiramate exhibits a combination of properties including modulatory effects on sodium channels, enhancement of GABA-activated chloride channels, inhibition of excitatory neurotransmission through actions on kainite and AMPA receptors, and inhibition of carbonic anhydrase (CA) isoenzymes in particular CA II and IV.14
The combination of phentermine and topiramate is a once-daily formulation that is designed to provide an immediate release of phentermine and a delayed release of topiramate, allowing a peak exposure of the phentermine in the morning and a peak concentration of topiramate in the evening. It should be taken in the morning in order to avoid the possibility of insomnia that can occur if taken in the evening. It can be taken with or without food. The recommended dose is as follows: Start treatment with Qsymia 3.75 mg/23 mg extended-release daily for 14 days; after 14 days increase to the recommended dose of Qsymia 7.5 mg/46 mg once daily.
Weight loss should be evaluated after 12 weeks at the higher dose. If at least 3% of baseline body weight has not been lost at that time, discontinue or escalate the dose. To escalate the dose: Increase to Qsymia 11.25 mg/69 mg daily for 14 days; followed by Qsymia 15 mg/92 mg daily. Evaluate weight loss following dose escalation to Qsymia 15 mg/92 mg after an additional 12 weeks of treatment. If at least 5% of baseline body weight has not been lost on Qsymia 15 mg/92 mg, discontinue as directed. It is important not to suddenly discontinue, as this may cause seizures. Patients should be slowly titrated off the medication.
In vitro studies of phentermine and topiramate indicate that these drugs are not likely to cause clinically significant interactions with drugs using the cytochrome P450 enzyme pathways, or those involved in plasma protein binding displacement; however there is evidence suggesting that ethinyl estradiol levels may be decreased by 16%, thus raising a concern about the possibility of decreased contraceptive efficacy.31 In patients with moderate (creatine clearance ≥ 30 mL/min to < 50 mL/min) and severe renal dysfunction (< 30 mL/min), the maximum dose of should not exceed 7.5 mg/46 mg.
Qsymia was evaluated in 3 phase 3 trials for its long-term efficacy and safety. In all trials, diet and lifestyle counseling were provided for all patients. The first of these studies was OB-301, a 28-week confirmatory trial with a factorial design involving 7 treatment arms, tested 2 fixed-dose Qsymia combinations—regular dose (7.5 mg/46 mg) and maximum dose (15 mg/92 mg)—as well as regular and maximum doses of the individual constituent drugs vs placebo.32 The study randomized 756 obese patients with a BMI range of 30 kg/m2 to 45 kg/m2 to 1 of the 7 treatment arms for 28 weeks. Patients treated with maximum-dose Qsymia achieved an average weight change of -9.0%, vs -1.5% with placebo (P < .0001). Weight change with regular-dose Qsymia was -8.2%. Weight changes with monotherapies were: -6.1% with topiramate 92 mg, -4.9% with topiramate 46 mg, -5.8% with phentermine 15 mg, and -5.2% with phentermine 7.5 mg.
OB-302 was a 56-week trial that randomized 1,267 morbidly obese patients with a BMI ≥ 35 kg/m2 without significant comorbidities to low-dose Qsymia (3.7 mg/23 mg), maximum-dose Qsymia (15 mg/92 mg), or placebo.33 At baseline, the mean BMI for the entire study cohort was 42 kg/m2. Mean weight changes were -1.6% with placebo, -5.1% with low-dose Qsymia, and -10.9% with maximum-dose Qsymia. The proportions of patients achieving ≥ 5% weight loss were: 17% with placebo, 45% with low-dose Qsymia, and 67% with maximum-dose Qsymia.
CONQUER was the largest of the phase 3 trials. It randomized 2,487 overweight or obese patients with a BMI of 27 kg/m2 to 45 kg/m2 and ≥ 2 obesity-related comorbidities (hypertension, dyslipidemia, T2DM, prediabetes or abdominal obesity) to receive a placebo, regular-dose Qsymia, or maximum-dose Qsymia for 56 weeks.34 In the completer population, mean weight changes in the placebo, regular dose Qsymia, and maximum-dose Qsymia groups were -1.6%, -9.6% (P <.0001), and -12.4% (P < .0001); and weight loss of ≥ 5% was achieved by 21%, 62%, and 70%, respectively. Relative to placebo, there were greater reductions in systolic BP, triglycerides, and fasting insulin with both doses of Qsymia.
Patients should not take Qsymia if they are pregnant, planning to become pregnant, or become pregnant during Qsymia treatment as there is an increased risk of birth defects, namely cleft lip and cleft palate. Women who can become pregnant should have a negative pregnancy test before taking Qsymia and every month while on the medication. They should use effective birth control consistently while taking Qsymia.
Qsymia is contraindicated in patients with glaucoma and patients who have hyperthyroidism. Qsymia can cause an increase in resting heart rate and regular monitoring of resting heart rate is recommended, especially in patients with cardiac or cerebrovascular disease. It has not been studied in patients with recent or unstable cardiac or cerebrovascular disease and therefore use is not recommended.
Qsymia can cause mood disorders such as anxiety and depression and can increase the risk of suicidal thoughts. Patients should be monitored for worsening depression, suicidal thoughts or behavior, or any unusual changes in mood or behavior. It is not recommended in patients with a history of suicidal attempts or active suicidal ideation. Qsymia can cause cognitive dysfunction. It can cause confusion, problems with concentration, attention, memory, or speech. Patients should be cautioned about operating automobiles and hazardous machinery.
Normal anion gap hyperchloremic metabolic acidosis has been reported in patients treated with Qsymia. If this does develop and persists, consideration should be given to either reduce the dose or discontinue Qsymia.
Weight loss may increase the risk of hypoglycemia in patients with T2DM treated with insulin and/or insulin secretagogues (eg, sulfonylureas). Qsymia has not been studied in combination with insulin. A reduction in the dose of antidiabetic medications, which are nonglucose dependent, should be considered to reduce the risk of hypoglycemia.
The most common AEs in controlled clinical studies (≥ 5% and at least 1.5 times placebo) included paraesthesia in the hands, arms, feet or face, dizziness, dysgeusia, insomnia, constipation, and dry mouth.
Contrave
In 2014, the FDA approved Contrave (Takeda, Deerfield, IL) as treatment option for chronic weight management in addition to reduced-calorie diet and physical activity. The combination of naltrexone hydrochloride and bupropion hydrochloride was originally introduced for the treatment of opioid addiction and later expanded to include the treatment of alcoholism. The antidepressant bupropion was approved in the U.S. in 1989. It is structurally different from all other marketed antidepressants (ie, tricyclics, tetracyclics, and SSRIs), but closely resembles the structure of diethylpropion, an appetite depressant with minimal CNS effects.35
This drug is approved for adults with BMI ≥ 30 kg/m2 and for adults with BMI ≥ 27 kg/m2 with at least 1 weight-related risk factors such as hypertension, T2DM, or dyslipidemia. It should be used as an adjunct to diet and exercise and is not approved for use for depression even though it contains bupropion.
Naltrexone is a pure opioid antagonist with high affinity to μ-opioid receptor, which is implicated in eating behavior. Naltrexone is rapidly and nearly completely absorbed from the GI tract after oral administration. The time to peak plasma concentration is about 1 hour. Naltrexone is well absorbed but first pass extraction and metabolism by the liver decreases oral bioavailability to between 5% to 40%. Primary elimination of naltrexone and its metabolites is renal excretion.
Bupropion is a weak inhibitor of neuronal reuptake of dopamine and norepinephrine. This drug is used to treat depression and seasonal affective disorder, and aid in smoking cessation. Bupropion is absorbed rapidly after oral administration, but the absolute oral bioavailability of bupropion is not known because an IV preparation is not available. The time to peak plasma concentrations of bupropion is within 2 hours of oral administration. Bupropion is extensively metabolized by the liver to multiple metabolites. Primary elimination of bupropion is urinary excretion. However, hepatic and renal impairment may affect the elimination of bupropion and its metabolites. Patients with hepatic or renal impairment should use a reduced dosage.
Combination therapy has been found to have complementary actions on CNS to reduce food intake. They are believed to dampen CNS reward pathways, taking away the compulsive feeding behavior and pleasure of feeding, ultimately leading to weight loss. Bupropion stimulates hypothalamic pro-opiomelanocortin neurons (POMC), which results in reduced food intake and increased energy expenditure. Naltrexone blocks opioid-receptor mediated POMC auto-inhibition, blocks the increase in dopamine in nucleus accumbens that occurs when eating, and acting synergistically with bupropion in augmenting POMC firing.
The COR-I and COR-II trials compared Contrave to diet and exercise in patients who did not have DM. The COR-Diabetes trial included the same study design but focused on patients with DM. In all the studies the participants had a 4-week titration to Contrave (naltrexone 8 mg/bupropion 90 mg) to decrease nausea. The first week dosing was 1 tablet in the morning. Week 2 was 1 tablet in morning and 1 tablet in the evening. In week 3, patients took 2 tablets in the morning and 1 tablet in the evening. The final titration step was 2 tablets in the morning and 2 tablets in the evening.
The COR-1 study was a 56-week randomized, double-blind, placebo-controlled study. It compared Contrave 32 mg naltrexone/360 mg bupropion (NB32/360) with an active placebo of diet and exercise.36 To be included adults must be aged 18 to 65 years with a BMI 30 kg/m2 to 45 kg/m2 or a BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia or hypertension. Patients were instructed on a hypocaloric diet that was a 500 kcal per day deficit based on World Health Organization algorithm for calculating metabolic rate and they were urged to increase physical activity.
The completer population results showed 8.0% weight loss in the NB32/360 group and 1.9% weight loss in the placebo group (P < .001). For the NB32/360 and placebo groups, weight loss of ≥ 5% was achieved by 48% and 16% (P <.001), respectively; and weight loss of ≥ 10% by 25% and 7% (P < .001), respectively. The most common AE was nausea—29.8% with NB32/360 vs 5.3% with placebo. Nausea generally occurred early and then diminished and the discontinuation rate from nausea was significantly lower (6.3%) then the overall reported nausea rates.
Contrave was also studied in patients with T2DM. The COR-Diabetes Trial was a 56-week randomized, double blind, placebo-controlled study. The trial compared Contrave 32 mg naltrexone/360 mg bupropion (NB32/360) with an active placebo of diet and exercise.37 Inclusion criteria for the trial were patients aged 18 to 70 years with T2DM and a BMI from 27 kg/m2 to 45 kg/m2, A1c between 7% and 10%, and fasting blood glucose < 270 mg/dL. Participants either were not taking a DM medication or were on stable doses of oral antidiabetes drugs ≥ 3 months prior to randomization. Patients were placed on a 500 kcal hypocaloric diet and advised to increase physical activity.
The results showed 5.0% weight loss in the NB32/360 group and 1.8% weight loss in the placebo group (P < .001). Weight loss of ≥ 5% and ≥ 10% was achieved by 44.5% and 18.5% of the NB32/360 group, respectively, and 18.9% and 5.7%, respectively (P < .001) of the placebo group. The NB32/360 and placebo showed a reduction of A1c of 0.6% and 0.1% respectively (P < .001). The most common AE was nausea (42.3% with NB32/360 vs 7.1% with placebo). Nausea generally occurred early and then diminished and the discontinuation rate from nausea was significantly lower (9.6%) then the overall reported nausea rates.37
Due to potential nausea caused by naltrexone, Contrave should be titrated over 4 weeks as described earlier. At maintenance dose, patients should be evaluated after 12 weeks to determine treatment benefits. If a patient has not lost at least 5% of baseline body weight, Contrave should be discontinued, because it would be unlikely that the patient will achieve and sustain clinically meaningful weight loss with continued treatment. Contrave should not be taken with high-fat meals that may result in significant increase in bupropion and naltrexone systemic exposure.
Since Contrave contains the antidepressant bupropion, it has a boxed warning similar to other antidepressants in its class of increased risk of suicidal thoughts and behaviors, especially in children, adolescents, and young adults.38Contrave can lower the seizure threshold; therefore it should not be used in people with a seizure disorder. It can also raise BP and heart rate; however the clinical significance of hypertension and elevated heart rate observed with Contrave treatment is unclear. Blood pressure rose on average by 1 point during the first 8 weeks of treatment and then returned to baseline.38 The heart rate also increased by about 1.7 beats per minute.38 Patients with uncontrolled hypertension should avoid Contrave.
Contrave should not be taken with products contain bupropion or naltrexone. It should not be taken by patient who are regularly taking opioids or who are opioid dependent, or who are experiencing opiate withdrawal. Pregnant women should also avoid Contrave. In patients with renal impairment the maximum dose is 1 tablet twice a day and in patients with hepatic impairment the maximum dose is 1 tablet a day.
Liraglutide
Liraglutide is the newest weight loss medication to be approved by the FDA for chronic weight management as an adjunct to a reduced calorie diet and increased physical activity in adult patients with BMI ≥ 30 kg/m2 or ≥ 27 kg/m2 with hypertension, diabetes, or dyslipidemia. The recommended dose of liraglutide is 3 mg daily. The initial dose is 0.6 mg daily for the first week, then titrated up by 0.6 each week for 4 weeks, until reaching 3 mg daily.
Liraglutide is an acylated human glucagon-like peptide-1 (GLP-1) receptor agonist, which are expressed in the brain and is involved in the control of appetite. It is also found in the beta cells of the pancreas, where GLP-1 receptors stimulate insulin release in response to elevated blood glucose concentrations and suppress glucagon secretion. Endogenous GLP-1 has a half-life of 1.5 to 2 minutes due to degradation by the DDP-4 enzyme, but liraglutide is stable against degradation by peptidases and has a half- life of 13 hours.
Liraglutide was studied in a 56-week randomized, double-blind, placebo-controlled trial, which compared liraglutide 3 mg with an active placebo of diet and physical activity.39 Inclusion criteria were adults aged ≥ 18 years old with a BMI 30 kg/m2 to 45 kg/m2 or BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia and/or hypertension. Both groups received lifestyle modification counseling. Patients were excluded if they had DM.
In the trial, 3,731 participants enrolled, 2,487 in the liraglutide group and 1,244 in the placebo group; 78.7% of the participants were female and the average age was 45 years. Subjects in the liraglutide group had a weekly titration regimen. The starting dose at week 1 was 0.6 mg, week 2 was 1.2 mg, week 3 was 1.8 mg, week 4 was 2.4 mg, and week 5 was 3.0 kg.
The completer population showed 9.2% weight loss in the liraglutide group and 3.0% weight loss in the control group.39 Weight loss of ≥ 5% was seen in 63.2% and 27.1% of the liraglutide and placebo groups, respectively. Weight loss rates of ≥ 10% was seen by 33.1% and 10.6%, respectively. The most common AEs were nausea, diarrhea, and constipation. Nausea generally occurred early during the titration period and then diminished.
A second clinically relevant study was performed with liraglutide. Often patients are able to lose weight with diet and exercise and then plateau. This study examined participants who lost 5% percent of their initial body weight and then were randomized to liraglutide or placebo.40 Key inclusion criteria were people aged ≥ 18 years old with a BMI 30 kg/m2 to 45 kg/m2 or BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia and/or hypertension. In order to be randomized, participants were required to lose at least 5% of their initial body weight on a 1,200 kcal to 1,400 kcal diet with increased physical activity during a 4 to 12 week run-in period.
Four hundred twenty-two participants were enrolled, 212 in the liraglutide group and 210 in the placebo group. Most of the participants were female (81%). The average BMI in the study was 35.6 kg/m2. Subjects in the liraglutide group had a weekly titration regimen.
After an average weight loss of 6% using a low calorie diet and increased physical activity the participants were randomized to continue diet and increased activity alone (placebo) or with liraglutide. At week 56 the results showed an additional 6.2% weight loss in the liraglutide group and 0.2% weight gain in the placebo group. The liraglutide group had a greater number of participants with ≥ 5% weight loss compared to placebo, 50.5% vs 21.8% (P < .0001).40 In the pooled data set from the registration trials the 3 most common GI AEs were nausea, diarrhea, and constipation occurring in 39.3%, 20.9%, and 19.4% of participants respectively. Discontinuation due to nausea for liraglutide was 2.9%.41
Clinicians should be aware that medications that can cause hypoglycemia such as sulfonylureas and insulin must be tapered as patients lose weight with liraglutide. Documented symptomatic hypoglycemia in patients with T2DM and with sulfonylurea background therapy was 43.6% with liraglutide vs 27.3% with placebo.
In the setting of renal impairment, patients treated with GLP-1 receptor agonists, including liraglutide, have had reports of acute renal failure and worsening of chronic renal failure usually associated with nausea, vomiting, diarrhea, or dehydration. Liraglutide causes thyroid C-cell tumors at clinically relevant exposures in rats and mice. It is unknown whether liraglutide causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans. As the human relevance of liraglutide-induced rodent thyroid C-cell tumors has not been determined liraglutide is contraindicated in patients with a personal or family history of MTC or in patients with multiple endocrine neoplasia syndrome type 2.
Acute pancreatitis, including fatal and nonfatal hemorrhagic or necrotizing pancreatitis, has been observed in patients treated with liraglutide in postmarketing reports. After initiation of liraglutide, observe patients carefully for signs and symptoms of pancreatitis (including persistent severe abdominal pain, sometimes radiating to the back, which may or may not be accompanied by vomiting). If pancreatitis is suspected, liraglutide should promptly be discontinued.
Conclusion
The treatment of obesity and overweight with comorbidities has always been a challenge. In the past there were few FDA-approved drugs and many drugs had to be used off-label. The toolbox of medications available for medical weight management is more robust than ever. The medications have different MOAs and can be used in a variety of patients. There are differences in the classes and some are controlled substances. Phentermine, lorcaserin, and Qsymia (phentermine/topiramate) are controlled substances whereas orlistat, naltrexone/bupropion and liraglutide are not. Other differences exist including duration of use. The sympathomimetic drugs have a limited window of use whereas orlistat, Qsymia (phentermine/topiramate), lorcaserin, naltrexone/bupropion, and liraglutide do not.
The medications that are available have a wide variety of MOAs. Therefore, if a patient fails one medication, then it is very reasonable to try a medication with a different MOA. In addition, there is the potential for weight regain when weight reduction medications are discontinued. As people lose weight their metabolic rate decreases about 15 kcal per pound of weight reduction.42
Another challenge of using these medications is managing patient expectations. The current metric used for FDA approval is a 5% weight loss that is greater in the study group compared with the diet and physical activity active control. However, many clinicians and patients do not find this weight reduction amount consistent with their expectations. In addition weight loss trajectory may also be too slow for patients and cause early discontinuation. Therefore, patient education and a discussion of reasonable expectations for weight reduction medications are necessary.
Clinicians must acknowledge that there are limitations to the use of these medications. Newer agents do have a higher cost and insurance reimbursement is somewhat limited. However, they offer the opportunity to prevent more expensive, protracted conditions such as diabetes and cardiovascular disease. In summary, clinicians now have a wider variety of medication options to be used with dietary and lifestyle changes in order to improve health and prevent chronic diseases.
Over the past decade the prevalence of obesity as defined by a body mass index (BMI) ≥ 30 kg/m2 has significantly increased. In the U.S. more than 78 million adults are estimated to be obese.1 The World Health Organization projects that by 2025 up to half the U.S. population will be obese. Cardiovascular disease (CVD) and diabetes mellitus (DM) are the main comorbid conditions that are complicated by obesity. Initial weight loss of 5% to 10% of total body weight reduces CVD risk factors, prevents or delays the development of type 2 DM (T2DM) and improves the health consequences of obesity.2
To date, public health initiatives that have focused on obesity prevention and lifestyle intervention have had marginal success. In recent years, anti-obesity drug therapies have had a limited role in clinical treatment algorithms. In 2013, the American Medical Association acknowledged obesity as a disease. In turn, this acknowledgement allowed the recognition of anti-obesity drugs as acceptable therapeutic adjuncts to intensive lifestyle intervention that could address the growing obesity endemic.
In the past, medications for weight reduction were limited. Several that were FDA approved had to be removed from the market due to safety concerns. With few approved options, clinicians often had to resort to off-label use of medications. However, the landscape has changed with 4 new medications gaining recent FDA approval. This review covers older available medications and the newer medications that are now available.
Sympathomimetics
Sympathomimetic drugs have been approved for use as a pharmacological method to lose weight since 1960. Of the many versions of this drug class that have been available since then, there are 4 major versions available today. These include diethylpropion3 and benzphetamine,4 both approved in 1960; phendimetrazine, approved in 1976;5 phentermine, approved in 1980;6 and phentermine hydrochloride, approved in 2012.7 Despite the existence of several other classes of drugs to treat obesity, phentermine remains the most often prescribed weight loss drug in the U.S.8
Although the mechanism of action (MOA) of sympathomimetic drugs is not particularly clear, weight loss from these medications is believed to be due to the increase in the release of biogenic amines (mainly norepinephrine, but also possibly dopamine), from storage sites in nerve terminals. It is possible that these drugs slow catecholamine metabolism by inhibiting the actions of monoamine oxidase. The resulting increase in amine availability, particularly in the lateral hypothalamic feeding center, is associated with reduced food intake. Interestingly, injection of these drugs into the ventromedial satiety center dooes not seem to suppress food intake, and the effects of biogenic amines on increasing metabolism does not seem to play a significant role in weight loss in patients on these medications.9
Each of these drugs is rapidly absorbed from the gastrointestinal (GI) tract except for phentermine hydrochloride, the newest of the medications in this class. Phentermine hydrochloride is a sublingual tablet that is readily absorbed through the buccal mucosa.5 All of the drugs in this class are excreted through the kidneys, with varying rates. Each drug’s excretion is highly dependent on the pH of the urine—more alkaline conditions result in less excretion and more acidic conditions result in more excretion. As a result, these drugs should be used with caution in patients with renal impairment; however, there are no specific contraindications listed for patients with poor renal function.
The adverse effects (AEs) for this drug class are to be expected from an increase in the release of biogenic amines in the central nervous system (CNS). The most common AEs include palpitations, tremors, restlessness, insomnia, dry mouth, constipation, diaphoresis, changes in libido, and irritability. The more dangerous AEs that have been observed include arrhythmias, hypertension, dependency/abuse, convulsions, acute transient ischemic colitis, and acute urinary retention secondary to increased bladder sphincter tone, transient hyperthyroxemia, and paranoia.10
Several contraindications exist for sympathomimetics, including the presence of advanced arteriosclerosis, symptomatic CVD, moderate to severe hypertension, hyperthyroidism, glaucoma, patients in an agitated state, or those with a history of amphetamine abuse. The warnings for prescribers include pulmonary hypertension and cardiomyopathy secondary to chronic use of sympathomimetics, and valvular heart disease secondary to use of sympathomimetics with additional anorectic agents.
Additional precautions should be considered in those with a history of anxiety/psychosis, those who operate machinery and motor vehicles, and even those with mild hypertension. The data surrounding the effects of sympathomimetics on blood pressure (BP) appears to be conflicting and the relationship does not seem to have been significantly studied in depth to warrant any definitive conclusions. The MOA of this drug class itself is enough to urge caution to prescribers.11 Special attention should be given to patients with diabetes when using sympathomimetics. A reduction of insulin dose or oral hypoglycemic dose may be necessary in some people with diabetes.
Only diethylpropion is pregnancy category B, whereas the others drugs in this class are pregnancy category X. It has been demonstrated that diethylpropion and benzphetamine are secreted into breastmilk; insufficient data exist to suggest whether or not phentermine and phendimetrazine are present in breastmilk. All drugs in this class should be used in caution with breastfeeding mothers.
Although all 4 drugs are registered as controlled substances, benzphetamine and phendimetrazine are schedule III and phentermine and diethylpropion are schedule IV, despite evidence suggesting the potential for abuse to be extremely low.12,13 Phentermine has been approved for adults aged > 18 years, phendimetrazine has been approved for those aged > 17 years, diethylpropion has been approved for those aged > 16 years, and benzphetamine has been approved for those aged > 12 years.
There is a wealth of literature surrounding the effectiveness of this drug class for weight loss. One of the longest trials of phentermine was recently conducted as part of the initial component of a FDA study for the newly approved topiramate-phentermine combination. Weight loss at 6 months in the phentermine-only group was significantly higher at -5.8% compared with -1.5% with the placebo group in the last observation carried forward-Intent to treat (LOCF-ITT) analysis.14 Similarly, a long-term study looking at diethylpropion examined the use of diethylpropion for up to a year vs placebo. Participants administered diethylpropion lost a mean 9.8% of original weight vs 3.7% in the placebo group in the first 6 months alone.15
Several meta-analyses and review papers have been authored that examine and analyze the published data on this drug class overall and comparatively within this class. Haddock and colleagues in 2002 reviewed the numerous clinical trials associated with each drug in this class, in addition to several other classes, and found that although each drug demonstrated a significant advantage vs placebo in weight loss, there was not a specific drug that was significantly superior to any of the others.16
These results seem to be in relative agreement with additional studies like that published by Suplicy and colleagues, which demonstrated that several sympathomimetics were better than placebo in weight loss, and that there was little difference between the specific drugs in the class.17 However, it should be noted that as highlighted in a review by Ioannides-Demos and colleagues in 2005, the vast majority of studies that had been performed on this drug class focused on short-term use (< 16 weeks) and none of the sympathomimetics listed here have been approved for long-term use.18
Orlistat
Orlistat 120 mg was approved in 1999 as a reversible inhibitor of GI lipases that specifically reduced the absorption of dietary fat due to the inhibition of triglyceride hydrolysis.19 Orlistat was later approved in 2007 for release in a reduced dosage form (60 mg) for over-the-counter sales.20
Orlistat forms a covalent bond with the active serine residue site of gastric and pancreatic lipases in the lumen of the stomach and small intestine. The inhibition of these enzymes causes dietary fat to remain undigested as triglycerides, which cannot be converted to absorbable free fatty acids and monoglycerides, leading to decreased calorie absorption. Orlistat is not systemically absorbed and is eliminated mainly through feces. Some metabolism occurs in the GI wall.21Orlistat is most known for its GI AEs. Because it is most active in the lumen of the GI system and reduces the absorption of triglycerides, many AEs are related to malabsorption. The most common issues 1 year after starting the drug were oily spotting (26.6% vs 1.3% placebo); flatus with discharge (23.9% vs 1.4% placebo); fecal urgency (22.1% vs 6.7% placebo); fatty/oily stool (20% vs 2.9% placebo); increased defecation (10.8% vs 4.1% placebo); and fecal incontinence (7.7% vs 0.9% placebo) (Table 1). Most of these AEs were greatly reduced after taking the drug for 2 years. Orlistat also has more serious AEs noted, including abdominal pain/discomfort; nausea; infectious diarrhea; rectal pain/discomfort; tooth disorder; gingival disorder; vomiting; upper respiratory infection; lower respiratory infection; ear, nose and throat symptoms; back pain; arthritis; myalgia; joint disorders; tendonitis; headache; dizziness; fatigue; sleep disorders; rash; dry skin; menstrual irregularity; vaginitis; urinary tract infection; and psychiatric disorders, although these did not differ markedly from placebo.
One of the most serious AEs reported was fulminate hepatic failure, though this AE is rare. Thirteen cases of liver injury were reported with the 120-mg prescription dose of orlistat and 1 case report in the U.S. involved the 60-mg over-the-counter dosage of orlistat.21,22 The FDA suggests that patients talk to their physicians about risks of liver failure, and that physicians should educate their patients about signs and symptoms of liver failure so that patients can stop taking orlistat and seek immediate medical help if symptoms occur.
One of the first published trials was the European Multicentre Orlistat Study Group, which included 743 participants with BMI between 28 kg/m2 and 47 kg/m2 from 15 different European centers. To test adherence, a 4-week single blind placebo lead-in was started with a hypocaloric diet. The first stage was completed by 688 patients who then proceeded to the double blind randomized control trial portion with a hypocaloric diet. From the start of lead-in to the end of year 1, the orlistat group weight decreased 10.2% (10.3 kg) vs 6.1% (6.1 kg) in the placebo group. The placebo subtracted difference between the groups was 3.9 kg (P < .001).23
A U.S.-based randomized double-blind placebo-controlled multicenter study included 796 obese patients with BMI between 30 kg/m2 and 44 kg/m2. Patients were assigned to 1 of 3 groups: placebo, orlistat 60 mg 3 times daily, or orlistat 120 mg 3 times daily. All groups were given a reduced energy diet. Patients in the orlistat 120 mg group lost significantly more weight than did the placebo group, -8.78% vs -4.26% respectively in year 1 in the completer analysis (P = .001). More participants who were treated with orlistat 120 mg lost 5% or more of their initial weight in year 1 compared with placebo, 50.5% vs 30.7% respectively (P < .001).24
In the XENDOS study the primary outcome measurement was time to onset of T2DM. Eligible participants were aged 30 to 60 years, with a BMI > 30 kg/m2. All patients had a 75-g oral glucose tolerance test and were required to have normal glucose tolerance or impaired glucose tolerance, but not T2DM. The double-blind randomized controlled trial included 3,305 subjects and compared a group taking 120 mg orlistat 3 times daily vs placebo. All patients were prescribed a reduced-calorie diet (800 kcal/d deficit) containing 30% of calories from fat. Patients were also encouraged to walk at least 1 kilometer daily in addition to their usual physical activity. Incidence of T2DM after 4 years was 6.2% in the orlistat group and 9.0% in the placebo group, reflecting a 37.3% risk reduction in the orlistat group (P = .0032).25,26
Lorcaserin
In 2012, lorcaserin HCl was FDA approved as a schedule IV drug for use as a weight loss medication as an adjunct to a reduced-calorie diet and increased physical activity. Lorcaserin is thought to act on 5-hydroxytryptamine-2c (5HT2c) receptors on the pro-opiomelanocortin (POMC) neurons in the arcuate nucleus, causing release of alpha-melanocortin-stimulating hormone (alpha-MSH), which in turn acts on melanocortin-4 receptors in the paraventricular nucleus to suppress appetite. At the maximum suggested dose of 10 mg twice daily, lorcaserin binds with 15 to 100 times greater affinity to 5HT2c receptors compared with 5HT2a and 5HT2b receptors respectively.
Indications for lorcaserin include patients with BMI ≥ 30 kg/m2 or ≥ 27 kg/m2 or greater with a weight-related comorbid condition such as hypertension, dyslipidemia, cardiovascular disease, impaired glucose tolerance, or sleep apnea.
The efficacy of lorcaserin for weight loss has been evaluated in 3 separate trials. The trials were randomized, double blinded and placebo controlled. The BLOOM trial, which included 3,182 patients with a mean BMI of 36.2 kg/m2, evaluated the efficacy of lorcaserin as a weight loss adjunct.27 Patients with pre-existing valvular disease, uncontrolled hypertension, or a major psychiatric condition were excluded. After initial randomization, patients were assigned to receive either lorcaserin 10 mg twice daily or a placebo. The primary endpoint was a 5% weight reduction from baseline by the end of 2 years. At 1 year, 47.5% of patients in the lorcaserin group and 20.3% in the placebo group had lost ≥ 5% of their body weight (P <.001). The average loss for the lorcaserin group was 5.8 ± 0.2 kg and 2.2 ± 0.1 kg for the placebo group at 1 year (P < .001).
The BLOSSOM trial was a 1-year study of 4,008 patients aged 18 to 65 years. The trial evaluated the effects of lorcaserin on body weight, CVD risk factors, and safety in obese and overweight patients.28 Patients were randomized in a 2:1:2 ratio to receive lorcaserin 10 mg twice daily, lorcaserin 10 mg once daily, or placebo. The primary endpoint was the proportion of patients achieving at least 5% reduction in body weight. Completer analysis showed weight reduction in the placebo group was 4.0% and 7.9% in the lorcaserin group (P < .001). In the modified intent-to-treat/last observation carried forward analysis (MITT/LOCF), a statistically significant 47.2% of patients receiving lorcaserin 10 mg twice daily and 40.2% of patients receiving lorcaserin 10 mg once daily lost at least 5% of baseline body weight; compared with 25% of patients receiving placebo (P < .001). Weight loss of at least 10% was achieved by 22.6% of patients receiving lorcaserin 10 mg twice daily, and 17.4% of patients receiving 10 mg daily compared with 9.7% of patients in the placebo group (P < .001).
The most common AEs noted were headache, nausea, and dizziness. Echocardiographic evidence of valvulopathy occurred in 2% of patients taking lorcaserin 10 mg twice daily and those taking the placebo. Lorcaserin administered in conjunction with a diet and exercise program was associated with an overall reduction in baseline BMI when compared with placebo over the year.
The BLOOM-DM study evaluated efficacy and safety of lorcaserin for weight loss in 604 patients with T2DM over the course of 1 year.29 Patients had a hemoglobin A1c (A1c) of 7% to 10% and were treated with metformin, a sulfonylurea, or both. The primary endpoint was a 5% weight reduction from baseline at the end of 1 year. Patients were randomized into 3 groups: 1 group received lorcaserin 10 mg twice daily, 1 group took lorcaserin 10 mg daily, and 1 group received the placebo. A statistically significant 37.5% of patients taking lorcaserin 10 mg twice daily achieved > 5% body weight reduction, compared with 44.7% in the lorcaserin 10 mg daily group, and 16.1% in the placebo group. Overall reductions in A1c and fasting glucose were observed in both lorcaserin groups taking as compared with placebo. Patient A1c decreased 0.9 ± 0.06 with lorcaserin 10 mg bid, 1.0 ± 0.09 with lorcaserin 10 mg qd, and 0.4 ± 0.06 with the placebo (P < .001). Fasting glucose in the lorcaserin bid, lorcaserin qd, and placebo groups decreased 27.4 ± 2.5 mg/dL, 28.4 ± 3.8 mg/dL, and 11.9 ± 2.5 mg/dL, respectively (P < .001). Symptomatic hypoglycemia occurred in 7.4% of patients on lorcaserin bid, 10.5% on lorcaserin qd, and 6.3% on placebo. Headache, back pain, nasopharyngitis, and nausea were among the most commonly reported AEs.
As lorcaserin is a serotonergic agonist, potential interactions exist when used with other medications affecting serotonin. Most notably, serotonin syndrome and neuroleptic malignant syndrome-like reactions may occur. Because of this, it is recommended to avoid selective serotonin re-uptake inhibitors (SSRIs), selective norepinephrine reuptake inhibitors, tricyclic antidepressants, bupropion, triptans, monoamine oxidase inhibitors, lithium, dextromethorphan, and dopamine agonists. Lorcaserin seems to be safe in those patient populations with mild hepatic as well as mild renal impairment; however, it is not recommended for those with severe renal impairment. Given the multiple enzymatic pathways used to metabolize lorcaserin, there is a low probability for cytochrome drug interactions. Safety has not been well evaluated in patients aged < 18 years and those that are pregnant (pregnancy category X).
Adverse events include headache, dizziness, fatigue, nausea, and dry mouth. Other notable AEs include nasopharyngitis and URI. Hypoglycemia appeared to be more common in patients with DM taking lorcaserin. Cognitive impairment and psychiatric disorders including euphoria and hallucinations were also reported. Notably, valvular heart disease has been reported in patients who take medications with 5HT2b activity. In a 1-year clinical trial, a small number of patients were found to develop valvular regurgitation. Furthermore, bradycardia, priapism, leucopenia, elevated prolactin, and pulmonary hypertension have also been observed. Caution is recommended if symptoms of any of the aforementioned conditions are noticed.
Qsymia
The schedule IV controlled substance Qsymia (Vivus, Mountain View, CA) is a combination of phentermine, an anorexigenic agent, and topiramate extended-release, an antiepileptic drug. In July of 2012 it was approved for chronic weight management as an addition to a reduced-calorie diet and exercise. The drug is approved for adults with a BMI ≥ 30 kg/m2 or adults with a BMI ≥ 27 kg/m2 who have at least 1 weight-related condition such as hypertension, T2DM, or dyslipidemia.30
In 1996 topiramate was approved by the FDA for the treatment of seizure disorders and was also approved for migraine prophylaxis in 2004. In patients who were treated with topiramate for seizure disorders and migraines, weight loss and a reduction in visceral body fat has been observed.31 The precise MOA of topiramate in regards to weight loss is not fully understood. It may be due to its effects on both appetite suppression and satiety enhancement. Topiramate exhibits a combination of properties including modulatory effects on sodium channels, enhancement of GABA-activated chloride channels, inhibition of excitatory neurotransmission through actions on kainite and AMPA receptors, and inhibition of carbonic anhydrase (CA) isoenzymes in particular CA II and IV.14
The combination of phentermine and topiramate is a once-daily formulation that is designed to provide an immediate release of phentermine and a delayed release of topiramate, allowing a peak exposure of the phentermine in the morning and a peak concentration of topiramate in the evening. It should be taken in the morning in order to avoid the possibility of insomnia that can occur if taken in the evening. It can be taken with or without food. The recommended dose is as follows: Start treatment with Qsymia 3.75 mg/23 mg extended-release daily for 14 days; after 14 days increase to the recommended dose of Qsymia 7.5 mg/46 mg once daily.
Weight loss should be evaluated after 12 weeks at the higher dose. If at least 3% of baseline body weight has not been lost at that time, discontinue or escalate the dose. To escalate the dose: Increase to Qsymia 11.25 mg/69 mg daily for 14 days; followed by Qsymia 15 mg/92 mg daily. Evaluate weight loss following dose escalation to Qsymia 15 mg/92 mg after an additional 12 weeks of treatment. If at least 5% of baseline body weight has not been lost on Qsymia 15 mg/92 mg, discontinue as directed. It is important not to suddenly discontinue, as this may cause seizures. Patients should be slowly titrated off the medication.
In vitro studies of phentermine and topiramate indicate that these drugs are not likely to cause clinically significant interactions with drugs using the cytochrome P450 enzyme pathways, or those involved in plasma protein binding displacement; however there is evidence suggesting that ethinyl estradiol levels may be decreased by 16%, thus raising a concern about the possibility of decreased contraceptive efficacy.31 In patients with moderate (creatine clearance ≥ 30 mL/min to < 50 mL/min) and severe renal dysfunction (< 30 mL/min), the maximum dose of should not exceed 7.5 mg/46 mg.
Qsymia was evaluated in 3 phase 3 trials for its long-term efficacy and safety. In all trials, diet and lifestyle counseling were provided for all patients. The first of these studies was OB-301, a 28-week confirmatory trial with a factorial design involving 7 treatment arms, tested 2 fixed-dose Qsymia combinations—regular dose (7.5 mg/46 mg) and maximum dose (15 mg/92 mg)—as well as regular and maximum doses of the individual constituent drugs vs placebo.32 The study randomized 756 obese patients with a BMI range of 30 kg/m2 to 45 kg/m2 to 1 of the 7 treatment arms for 28 weeks. Patients treated with maximum-dose Qsymia achieved an average weight change of -9.0%, vs -1.5% with placebo (P < .0001). Weight change with regular-dose Qsymia was -8.2%. Weight changes with monotherapies were: -6.1% with topiramate 92 mg, -4.9% with topiramate 46 mg, -5.8% with phentermine 15 mg, and -5.2% with phentermine 7.5 mg.
OB-302 was a 56-week trial that randomized 1,267 morbidly obese patients with a BMI ≥ 35 kg/m2 without significant comorbidities to low-dose Qsymia (3.7 mg/23 mg), maximum-dose Qsymia (15 mg/92 mg), or placebo.33 At baseline, the mean BMI for the entire study cohort was 42 kg/m2. Mean weight changes were -1.6% with placebo, -5.1% with low-dose Qsymia, and -10.9% with maximum-dose Qsymia. The proportions of patients achieving ≥ 5% weight loss were: 17% with placebo, 45% with low-dose Qsymia, and 67% with maximum-dose Qsymia.
CONQUER was the largest of the phase 3 trials. It randomized 2,487 overweight or obese patients with a BMI of 27 kg/m2 to 45 kg/m2 and ≥ 2 obesity-related comorbidities (hypertension, dyslipidemia, T2DM, prediabetes or abdominal obesity) to receive a placebo, regular-dose Qsymia, or maximum-dose Qsymia for 56 weeks.34 In the completer population, mean weight changes in the placebo, regular dose Qsymia, and maximum-dose Qsymia groups were -1.6%, -9.6% (P <.0001), and -12.4% (P < .0001); and weight loss of ≥ 5% was achieved by 21%, 62%, and 70%, respectively. Relative to placebo, there were greater reductions in systolic BP, triglycerides, and fasting insulin with both doses of Qsymia.
Patients should not take Qsymia if they are pregnant, planning to become pregnant, or become pregnant during Qsymia treatment as there is an increased risk of birth defects, namely cleft lip and cleft palate. Women who can become pregnant should have a negative pregnancy test before taking Qsymia and every month while on the medication. They should use effective birth control consistently while taking Qsymia.
Qsymia is contraindicated in patients with glaucoma and patients who have hyperthyroidism. Qsymia can cause an increase in resting heart rate and regular monitoring of resting heart rate is recommended, especially in patients with cardiac or cerebrovascular disease. It has not been studied in patients with recent or unstable cardiac or cerebrovascular disease and therefore use is not recommended.
Qsymia can cause mood disorders such as anxiety and depression and can increase the risk of suicidal thoughts. Patients should be monitored for worsening depression, suicidal thoughts or behavior, or any unusual changes in mood or behavior. It is not recommended in patients with a history of suicidal attempts or active suicidal ideation. Qsymia can cause cognitive dysfunction. It can cause confusion, problems with concentration, attention, memory, or speech. Patients should be cautioned about operating automobiles and hazardous machinery.
Normal anion gap hyperchloremic metabolic acidosis has been reported in patients treated with Qsymia. If this does develop and persists, consideration should be given to either reduce the dose or discontinue Qsymia.
Weight loss may increase the risk of hypoglycemia in patients with T2DM treated with insulin and/or insulin secretagogues (eg, sulfonylureas). Qsymia has not been studied in combination with insulin. A reduction in the dose of antidiabetic medications, which are nonglucose dependent, should be considered to reduce the risk of hypoglycemia.
The most common AEs in controlled clinical studies (≥ 5% and at least 1.5 times placebo) included paraesthesia in the hands, arms, feet or face, dizziness, dysgeusia, insomnia, constipation, and dry mouth.
Contrave
In 2014, the FDA approved Contrave (Takeda, Deerfield, IL) as treatment option for chronic weight management in addition to reduced-calorie diet and physical activity. The combination of naltrexone hydrochloride and bupropion hydrochloride was originally introduced for the treatment of opioid addiction and later expanded to include the treatment of alcoholism. The antidepressant bupropion was approved in the U.S. in 1989. It is structurally different from all other marketed antidepressants (ie, tricyclics, tetracyclics, and SSRIs), but closely resembles the structure of diethylpropion, an appetite depressant with minimal CNS effects.35
This drug is approved for adults with BMI ≥ 30 kg/m2 and for adults with BMI ≥ 27 kg/m2 with at least 1 weight-related risk factors such as hypertension, T2DM, or dyslipidemia. It should be used as an adjunct to diet and exercise and is not approved for use for depression even though it contains bupropion.
Naltrexone is a pure opioid antagonist with high affinity to μ-opioid receptor, which is implicated in eating behavior. Naltrexone is rapidly and nearly completely absorbed from the GI tract after oral administration. The time to peak plasma concentration is about 1 hour. Naltrexone is well absorbed but first pass extraction and metabolism by the liver decreases oral bioavailability to between 5% to 40%. Primary elimination of naltrexone and its metabolites is renal excretion.
Bupropion is a weak inhibitor of neuronal reuptake of dopamine and norepinephrine. This drug is used to treat depression and seasonal affective disorder, and aid in smoking cessation. Bupropion is absorbed rapidly after oral administration, but the absolute oral bioavailability of bupropion is not known because an IV preparation is not available. The time to peak plasma concentrations of bupropion is within 2 hours of oral administration. Bupropion is extensively metabolized by the liver to multiple metabolites. Primary elimination of bupropion is urinary excretion. However, hepatic and renal impairment may affect the elimination of bupropion and its metabolites. Patients with hepatic or renal impairment should use a reduced dosage.
Combination therapy has been found to have complementary actions on CNS to reduce food intake. They are believed to dampen CNS reward pathways, taking away the compulsive feeding behavior and pleasure of feeding, ultimately leading to weight loss. Bupropion stimulates hypothalamic pro-opiomelanocortin neurons (POMC), which results in reduced food intake and increased energy expenditure. Naltrexone blocks opioid-receptor mediated POMC auto-inhibition, blocks the increase in dopamine in nucleus accumbens that occurs when eating, and acting synergistically with bupropion in augmenting POMC firing.
The COR-I and COR-II trials compared Contrave to diet and exercise in patients who did not have DM. The COR-Diabetes trial included the same study design but focused on patients with DM. In all the studies the participants had a 4-week titration to Contrave (naltrexone 8 mg/bupropion 90 mg) to decrease nausea. The first week dosing was 1 tablet in the morning. Week 2 was 1 tablet in morning and 1 tablet in the evening. In week 3, patients took 2 tablets in the morning and 1 tablet in the evening. The final titration step was 2 tablets in the morning and 2 tablets in the evening.
The COR-1 study was a 56-week randomized, double-blind, placebo-controlled study. It compared Contrave 32 mg naltrexone/360 mg bupropion (NB32/360) with an active placebo of diet and exercise.36 To be included adults must be aged 18 to 65 years with a BMI 30 kg/m2 to 45 kg/m2 or a BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia or hypertension. Patients were instructed on a hypocaloric diet that was a 500 kcal per day deficit based on World Health Organization algorithm for calculating metabolic rate and they were urged to increase physical activity.
The completer population results showed 8.0% weight loss in the NB32/360 group and 1.9% weight loss in the placebo group (P < .001). For the NB32/360 and placebo groups, weight loss of ≥ 5% was achieved by 48% and 16% (P <.001), respectively; and weight loss of ≥ 10% by 25% and 7% (P < .001), respectively. The most common AE was nausea—29.8% with NB32/360 vs 5.3% with placebo. Nausea generally occurred early and then diminished and the discontinuation rate from nausea was significantly lower (6.3%) then the overall reported nausea rates.
Contrave was also studied in patients with T2DM. The COR-Diabetes Trial was a 56-week randomized, double blind, placebo-controlled study. The trial compared Contrave 32 mg naltrexone/360 mg bupropion (NB32/360) with an active placebo of diet and exercise.37 Inclusion criteria for the trial were patients aged 18 to 70 years with T2DM and a BMI from 27 kg/m2 to 45 kg/m2, A1c between 7% and 10%, and fasting blood glucose < 270 mg/dL. Participants either were not taking a DM medication or were on stable doses of oral antidiabetes drugs ≥ 3 months prior to randomization. Patients were placed on a 500 kcal hypocaloric diet and advised to increase physical activity.
The results showed 5.0% weight loss in the NB32/360 group and 1.8% weight loss in the placebo group (P < .001). Weight loss of ≥ 5% and ≥ 10% was achieved by 44.5% and 18.5% of the NB32/360 group, respectively, and 18.9% and 5.7%, respectively (P < .001) of the placebo group. The NB32/360 and placebo showed a reduction of A1c of 0.6% and 0.1% respectively (P < .001). The most common AE was nausea (42.3% with NB32/360 vs 7.1% with placebo). Nausea generally occurred early and then diminished and the discontinuation rate from nausea was significantly lower (9.6%) then the overall reported nausea rates.37
Due to potential nausea caused by naltrexone, Contrave should be titrated over 4 weeks as described earlier. At maintenance dose, patients should be evaluated after 12 weeks to determine treatment benefits. If a patient has not lost at least 5% of baseline body weight, Contrave should be discontinued, because it would be unlikely that the patient will achieve and sustain clinically meaningful weight loss with continued treatment. Contrave should not be taken with high-fat meals that may result in significant increase in bupropion and naltrexone systemic exposure.
Since Contrave contains the antidepressant bupropion, it has a boxed warning similar to other antidepressants in its class of increased risk of suicidal thoughts and behaviors, especially in children, adolescents, and young adults.38Contrave can lower the seizure threshold; therefore it should not be used in people with a seizure disorder. It can also raise BP and heart rate; however the clinical significance of hypertension and elevated heart rate observed with Contrave treatment is unclear. Blood pressure rose on average by 1 point during the first 8 weeks of treatment and then returned to baseline.38 The heart rate also increased by about 1.7 beats per minute.38 Patients with uncontrolled hypertension should avoid Contrave.
Contrave should not be taken with products contain bupropion or naltrexone. It should not be taken by patient who are regularly taking opioids or who are opioid dependent, or who are experiencing opiate withdrawal. Pregnant women should also avoid Contrave. In patients with renal impairment the maximum dose is 1 tablet twice a day and in patients with hepatic impairment the maximum dose is 1 tablet a day.
Liraglutide
Liraglutide is the newest weight loss medication to be approved by the FDA for chronic weight management as an adjunct to a reduced calorie diet and increased physical activity in adult patients with BMI ≥ 30 kg/m2 or ≥ 27 kg/m2 with hypertension, diabetes, or dyslipidemia. The recommended dose of liraglutide is 3 mg daily. The initial dose is 0.6 mg daily for the first week, then titrated up by 0.6 each week for 4 weeks, until reaching 3 mg daily.
Liraglutide is an acylated human glucagon-like peptide-1 (GLP-1) receptor agonist, which are expressed in the brain and is involved in the control of appetite. It is also found in the beta cells of the pancreas, where GLP-1 receptors stimulate insulin release in response to elevated blood glucose concentrations and suppress glucagon secretion. Endogenous GLP-1 has a half-life of 1.5 to 2 minutes due to degradation by the DDP-4 enzyme, but liraglutide is stable against degradation by peptidases and has a half- life of 13 hours.
Liraglutide was studied in a 56-week randomized, double-blind, placebo-controlled trial, which compared liraglutide 3 mg with an active placebo of diet and physical activity.39 Inclusion criteria were adults aged ≥ 18 years old with a BMI 30 kg/m2 to 45 kg/m2 or BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia and/or hypertension. Both groups received lifestyle modification counseling. Patients were excluded if they had DM.
In the trial, 3,731 participants enrolled, 2,487 in the liraglutide group and 1,244 in the placebo group; 78.7% of the participants were female and the average age was 45 years. Subjects in the liraglutide group had a weekly titration regimen. The starting dose at week 1 was 0.6 mg, week 2 was 1.2 mg, week 3 was 1.8 mg, week 4 was 2.4 mg, and week 5 was 3.0 kg.
The completer population showed 9.2% weight loss in the liraglutide group and 3.0% weight loss in the control group.39 Weight loss of ≥ 5% was seen in 63.2% and 27.1% of the liraglutide and placebo groups, respectively. Weight loss rates of ≥ 10% was seen by 33.1% and 10.6%, respectively. The most common AEs were nausea, diarrhea, and constipation. Nausea generally occurred early during the titration period and then diminished.
A second clinically relevant study was performed with liraglutide. Often patients are able to lose weight with diet and exercise and then plateau. This study examined participants who lost 5% percent of their initial body weight and then were randomized to liraglutide or placebo.40 Key inclusion criteria were people aged ≥ 18 years old with a BMI 30 kg/m2 to 45 kg/m2 or BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia and/or hypertension. In order to be randomized, participants were required to lose at least 5% of their initial body weight on a 1,200 kcal to 1,400 kcal diet with increased physical activity during a 4 to 12 week run-in period.
Four hundred twenty-two participants were enrolled, 212 in the liraglutide group and 210 in the placebo group. Most of the participants were female (81%). The average BMI in the study was 35.6 kg/m2. Subjects in the liraglutide group had a weekly titration regimen.
After an average weight loss of 6% using a low calorie diet and increased physical activity the participants were randomized to continue diet and increased activity alone (placebo) or with liraglutide. At week 56 the results showed an additional 6.2% weight loss in the liraglutide group and 0.2% weight gain in the placebo group. The liraglutide group had a greater number of participants with ≥ 5% weight loss compared to placebo, 50.5% vs 21.8% (P < .0001).40 In the pooled data set from the registration trials the 3 most common GI AEs were nausea, diarrhea, and constipation occurring in 39.3%, 20.9%, and 19.4% of participants respectively. Discontinuation due to nausea for liraglutide was 2.9%.41
Clinicians should be aware that medications that can cause hypoglycemia such as sulfonylureas and insulin must be tapered as patients lose weight with liraglutide. Documented symptomatic hypoglycemia in patients with T2DM and with sulfonylurea background therapy was 43.6% with liraglutide vs 27.3% with placebo.
In the setting of renal impairment, patients treated with GLP-1 receptor agonists, including liraglutide, have had reports of acute renal failure and worsening of chronic renal failure usually associated with nausea, vomiting, diarrhea, or dehydration. Liraglutide causes thyroid C-cell tumors at clinically relevant exposures in rats and mice. It is unknown whether liraglutide causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans. As the human relevance of liraglutide-induced rodent thyroid C-cell tumors has not been determined liraglutide is contraindicated in patients with a personal or family history of MTC or in patients with multiple endocrine neoplasia syndrome type 2.
Acute pancreatitis, including fatal and nonfatal hemorrhagic or necrotizing pancreatitis, has been observed in patients treated with liraglutide in postmarketing reports. After initiation of liraglutide, observe patients carefully for signs and symptoms of pancreatitis (including persistent severe abdominal pain, sometimes radiating to the back, which may or may not be accompanied by vomiting). If pancreatitis is suspected, liraglutide should promptly be discontinued.
Conclusion
The treatment of obesity and overweight with comorbidities has always been a challenge. In the past there were few FDA-approved drugs and many drugs had to be used off-label. The toolbox of medications available for medical weight management is more robust than ever. The medications have different MOAs and can be used in a variety of patients. There are differences in the classes and some are controlled substances. Phentermine, lorcaserin, and Qsymia (phentermine/topiramate) are controlled substances whereas orlistat, naltrexone/bupropion and liraglutide are not. Other differences exist including duration of use. The sympathomimetic drugs have a limited window of use whereas orlistat, Qsymia (phentermine/topiramate), lorcaserin, naltrexone/bupropion, and liraglutide do not.
The medications that are available have a wide variety of MOAs. Therefore, if a patient fails one medication, then it is very reasonable to try a medication with a different MOA. In addition, there is the potential for weight regain when weight reduction medications are discontinued. As people lose weight their metabolic rate decreases about 15 kcal per pound of weight reduction.42
Another challenge of using these medications is managing patient expectations. The current metric used for FDA approval is a 5% weight loss that is greater in the study group compared with the diet and physical activity active control. However, many clinicians and patients do not find this weight reduction amount consistent with their expectations. In addition weight loss trajectory may also be too slow for patients and cause early discontinuation. Therefore, patient education and a discussion of reasonable expectations for weight reduction medications are necessary.
Clinicians must acknowledge that there are limitations to the use of these medications. Newer agents do have a higher cost and insurance reimbursement is somewhat limited. However, they offer the opportunity to prevent more expensive, protracted conditions such as diabetes and cardiovascular disease. In summary, clinicians now have a wider variety of medication options to be used with dietary and lifestyle changes in order to improve health and prevent chronic diseases.
1. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of obesity in the United States, 2009-2010. NCHS Data Brief. 2012(82):1-8.
2. Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. Circulation. 2014;129(25 suppl 2):S102-S138.
3. U.S. Food and Drug Administration. Drugs@FDA: diethylpropion hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=012546&DrugName=TENUATE%20DOSPAN&ActiveIngred=DIETHYLPROPION%20HYDROCHLORIDE&SponsorApplicant=ACTAVIS%20LABS%20UT%20INC&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
4. U.S. Food and Drug Administration. Drugs@FDA: benzphetamine hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=012427&DrugName=DIDREX&ActiveIngred=BENZPHETAMINE%20HYDROCHLORIDE&SponsorApplicant=PHARMACIA%20AND%20UPJOHN&ProductMktStatus=3&goto=Search.DrugDetails. Accessed December 28, 2015.
5. U.S. Food and Drug Administration. Drugs@ FDA: phendimetrazine tartrate. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=088021&DrugName=BONTRIL&ActiveIngred=PHENDIMETRAZINE%20TARTRATE&SponsorApplicant=VALEANT&ProductMktStatus=3&goto=Search.DrugDetails. Accessed December 28, 2015.
6. U.S. Food and Drug Administration. Drugs@FDA: phentermine. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=085128&DrugName=ADIPEX%2DP&ActiveIngred=PHENTERMINE%20HYDROCHLORIDE&SponsorApplicant=TEVA&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
7. U.S. Food and Drug Administration. Drugs @ FDA: phentermine hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=202088&DrugName=SUPRENZA&ActiveIngred=PHENTERMINE%20HYDROCHLORIDE&SponsorApplicant=CITIUS%20PHARMS&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
8. Hendricks EJ, Rothman RB, Greenway FL. How Physician Obesity Specialists use drugs to treat obesity. Obesity (Silver Spring). 2009;17(9):1730-1735.
9. Gilman AG, Gilman A, Goodman LS, eds. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 8th ed. New York: Pergamon Press; 1990: 211.
10. Gilman AG, Gilman A, Goodman LS, eds. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 8th ed. New York: Pergamon Press; 1990:217.
11. Addy C, Jumes P, Rosko K, et al. Pharmacokinetics, safety, and tolerability of phentermine in health participants receiving taranabant, a novel cannabinoid-1 receptor (CB1R) inverse agonist. J Clin Pharmacol. 2009;49(10):1228-1232.
12. U.S. Department of Justice, Drug Enforcement Administration, Office of Diversion Control. Controlled substances. U.S. Department of Justice Website. http://www.deadiversion.usdoj.gov/schedules/orangebook/c_cs_alpha.pdf. Updated November 12, 2015. Accessed December 16, 2015.
13. Bray GA, Greenway FL. Pharmacological treatment of the overweight patient. Pharmacol Rev. 2007;59(2):151-184.
14. V1-0521 (QNEXA) Advisory committee briefing document. NDA 022580. Endocrinologic and Metabolic Drugs Advisory Committee meeting; June 17, 2010.
15. Cercato C, Roizenblatt VA, Leanca CC, et al. A randomized double-blind placebo-controlled study of the long-term efficacy and safety of diethylpropion in the treatment of obese subjects. Int J Obes (Lond). 2009;33(8):857-865.
16. Haddock CK, Poston WS, Dill PL, Foreyt JP, Ericsson M. Pharmacotherapy for obesity: a quantitative analysis of four decades of published randomized clinical trials. Int J Obes (Lond). 2002;26(2):262-273.
17. Suplicy H, Boquszewski CL, dos Santos CM, do Desterro de Fiqueiredo M, Cunha DR, Radominski R. A comparative study of five centrally acting drugs on pharmacological treatment of obesity. Int J Obes (Lond). 2014;38(8):1097-1103.
18. Ioannides-Demos LL, Proietto J, McNeill JJ. 2005. Pharmacotherapy for obesity. Drugs. 2005;65(10): 1391-1418.
19. Drent ML, van der Veen EA. Lipase inhibition: a novel concept in the treatment of obesity. Int J Obes Relat Metab Disord. 1993;17(4):241-244.
20. Xenical [package insert]. Nutley, NJ: Roche Laboratories Inc.; 1999.
21. U.S. Food and Drug Administration. FDA Drug Safety Communication: Completed safety review of Xenical/Alli and severe liver injury. U.S. Food and Drug Administration Website. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm213038.htm. Updated August 2, 2010. Accessed December 16, 2015.
22. Sall D, Wang J, Rashkin M, Welch M, Droege C, Schauer D. Orlistat-induced fulminant hepatic failure. Clin Obes. 2014;4(6):342-347.
23. Sjöström L, Rissanen A, Andersen T, et al. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. European Multicentre Orlistat Study Group. Lancet. 1998;352(9123):167-172.
24. Hauptman J, Lucas C, Boldrin MN, Collins H, Segal KR. Orlistat in the long-term treatment of obesity in primary care settings. Arch Fam Med. 2000;9(2):160-167.
25. Torgerson JS, Hauptman J, Boldrin MN, Sjöström L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27(1):155-161.
26. Torgerson JS, Arlinger K, Käppi M, Sjöström L. Principles for enhanced recruitment of subjects in a large clinical trial. the XENDOS (XENical in the prevention of Diabetes in Obese Subjects) study experience. Control Clin Trials. 2001;22(5):515-525.
27.Smith SR, Weissman NJ, Anderson CM, et al; Behavioral Modification and Lorcaserin for Overweight and Obesity Management (BLOOM) Study Group. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med. 2010;363(3):245-256.
28. Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011;96(10):3067-3077.
29. O’Neil PM, Smith SR, Weisserman NJ, et al. Randomized placebo-controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM Study. Obesity (Silver Spring). 2012;20(7):1426-1436.
30. U.S. Food and Drug Administration. FDA approves weight-management drug Qsymia. U.S. Food and Drug Administration Website. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm312468.htm. Published July 17, 2012. Accessed December 16, 2015.
31. Shin J, Gadde KM. Clinical utility of phentermine/topiramate (Qsymia™) combination for the treatment of obesity. Diabetes Metab Syndr Obes. 2013;6:131-139.
32. Qsymia [package insert] Mountain View, CA: Vivus, Inc; 2012.
33. Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine/topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring). 2012;20(2):330-342.
34. Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377(9774):1341-1352.
35. Plodkowski RA, Nguyen Q, Sundaram U, Nguyen L, Chau DL, St Jeor S. Bupropion and naltrexone: a review of their use individually and in combination for the treatment of obesity. Expert Opin Pharmacother. 2009;10(6):1069-1081.
36. Greenway FL, Fujioka K, Plodkowski RA, et al; COR-I Study Group. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-1): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595-605.
37. Hollander P, Gupta AK, Plodkowski R, et al; COR-Diabetes Study Group. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care. 2013;36(12):4022-4029.
38. Contrave [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc; 2014.
39. Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Eng J Med. 2015;373(1):11-22.
40. Wadden TA, Hollander P, Klein S, et al; NN8022-1923 Investigators. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet-induced weight loss: the SCALE Maintenance randomized study. Int J Obes (Lond). 2013;37(11):1443-1451
41. Saxenda [package insert]. Novo Nordisk: Plainsboro, NJ; 2015.
42.Schwartz A, Doucet E. Relative changes in resting energy expenditure during weight loss: a systemic review. Obes Rev. 2010;11(7): 531-547. ```````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````
1. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of obesity in the United States, 2009-2010. NCHS Data Brief. 2012(82):1-8.
2. Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. Circulation. 2014;129(25 suppl 2):S102-S138.
3. U.S. Food and Drug Administration. Drugs@FDA: diethylpropion hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=012546&DrugName=TENUATE%20DOSPAN&ActiveIngred=DIETHYLPROPION%20HYDROCHLORIDE&SponsorApplicant=ACTAVIS%20LABS%20UT%20INC&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
4. U.S. Food and Drug Administration. Drugs@FDA: benzphetamine hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=012427&DrugName=DIDREX&ActiveIngred=BENZPHETAMINE%20HYDROCHLORIDE&SponsorApplicant=PHARMACIA%20AND%20UPJOHN&ProductMktStatus=3&goto=Search.DrugDetails. Accessed December 28, 2015.
5. U.S. Food and Drug Administration. Drugs@ FDA: phendimetrazine tartrate. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=088021&DrugName=BONTRIL&ActiveIngred=PHENDIMETRAZINE%20TARTRATE&SponsorApplicant=VALEANT&ProductMktStatus=3&goto=Search.DrugDetails. Accessed December 28, 2015.
6. U.S. Food and Drug Administration. Drugs@FDA: phentermine. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=085128&DrugName=ADIPEX%2DP&ActiveIngred=PHENTERMINE%20HYDROCHLORIDE&SponsorApplicant=TEVA&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
7. U.S. Food and Drug Administration. Drugs @ FDA: phentermine hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=202088&DrugName=SUPRENZA&ActiveIngred=PHENTERMINE%20HYDROCHLORIDE&SponsorApplicant=CITIUS%20PHARMS&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
8. Hendricks EJ, Rothman RB, Greenway FL. How Physician Obesity Specialists use drugs to treat obesity. Obesity (Silver Spring). 2009;17(9):1730-1735.
9. Gilman AG, Gilman A, Goodman LS, eds. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 8th ed. New York: Pergamon Press; 1990: 211.
10. Gilman AG, Gilman A, Goodman LS, eds. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 8th ed. New York: Pergamon Press; 1990:217.
11. Addy C, Jumes P, Rosko K, et al. Pharmacokinetics, safety, and tolerability of phentermine in health participants receiving taranabant, a novel cannabinoid-1 receptor (CB1R) inverse agonist. J Clin Pharmacol. 2009;49(10):1228-1232.
12. U.S. Department of Justice, Drug Enforcement Administration, Office of Diversion Control. Controlled substances. U.S. Department of Justice Website. http://www.deadiversion.usdoj.gov/schedules/orangebook/c_cs_alpha.pdf. Updated November 12, 2015. Accessed December 16, 2015.
13. Bray GA, Greenway FL. Pharmacological treatment of the overweight patient. Pharmacol Rev. 2007;59(2):151-184.
14. V1-0521 (QNEXA) Advisory committee briefing document. NDA 022580. Endocrinologic and Metabolic Drugs Advisory Committee meeting; June 17, 2010.
15. Cercato C, Roizenblatt VA, Leanca CC, et al. A randomized double-blind placebo-controlled study of the long-term efficacy and safety of diethylpropion in the treatment of obese subjects. Int J Obes (Lond). 2009;33(8):857-865.
16. Haddock CK, Poston WS, Dill PL, Foreyt JP, Ericsson M. Pharmacotherapy for obesity: a quantitative analysis of four decades of published randomized clinical trials. Int J Obes (Lond). 2002;26(2):262-273.
17. Suplicy H, Boquszewski CL, dos Santos CM, do Desterro de Fiqueiredo M, Cunha DR, Radominski R. A comparative study of five centrally acting drugs on pharmacological treatment of obesity. Int J Obes (Lond). 2014;38(8):1097-1103.
18. Ioannides-Demos LL, Proietto J, McNeill JJ. 2005. Pharmacotherapy for obesity. Drugs. 2005;65(10): 1391-1418.
19. Drent ML, van der Veen EA. Lipase inhibition: a novel concept in the treatment of obesity. Int J Obes Relat Metab Disord. 1993;17(4):241-244.
20. Xenical [package insert]. Nutley, NJ: Roche Laboratories Inc.; 1999.
21. U.S. Food and Drug Administration. FDA Drug Safety Communication: Completed safety review of Xenical/Alli and severe liver injury. U.S. Food and Drug Administration Website. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm213038.htm. Updated August 2, 2010. Accessed December 16, 2015.
22. Sall D, Wang J, Rashkin M, Welch M, Droege C, Schauer D. Orlistat-induced fulminant hepatic failure. Clin Obes. 2014;4(6):342-347.
23. Sjöström L, Rissanen A, Andersen T, et al. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. European Multicentre Orlistat Study Group. Lancet. 1998;352(9123):167-172.
24. Hauptman J, Lucas C, Boldrin MN, Collins H, Segal KR. Orlistat in the long-term treatment of obesity in primary care settings. Arch Fam Med. 2000;9(2):160-167.
25. Torgerson JS, Hauptman J, Boldrin MN, Sjöström L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27(1):155-161.
26. Torgerson JS, Arlinger K, Käppi M, Sjöström L. Principles for enhanced recruitment of subjects in a large clinical trial. the XENDOS (XENical in the prevention of Diabetes in Obese Subjects) study experience. Control Clin Trials. 2001;22(5):515-525.
27.Smith SR, Weissman NJ, Anderson CM, et al; Behavioral Modification and Lorcaserin for Overweight and Obesity Management (BLOOM) Study Group. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med. 2010;363(3):245-256.
28. Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011;96(10):3067-3077.
29. O’Neil PM, Smith SR, Weisserman NJ, et al. Randomized placebo-controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM Study. Obesity (Silver Spring). 2012;20(7):1426-1436.
30. U.S. Food and Drug Administration. FDA approves weight-management drug Qsymia. U.S. Food and Drug Administration Website. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm312468.htm. Published July 17, 2012. Accessed December 16, 2015.
31. Shin J, Gadde KM. Clinical utility of phentermine/topiramate (Qsymia™) combination for the treatment of obesity. Diabetes Metab Syndr Obes. 2013;6:131-139.
32. Qsymia [package insert] Mountain View, CA: Vivus, Inc; 2012.
33. Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine/topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring). 2012;20(2):330-342.
34. Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377(9774):1341-1352.
35. Plodkowski RA, Nguyen Q, Sundaram U, Nguyen L, Chau DL, St Jeor S. Bupropion and naltrexone: a review of their use individually and in combination for the treatment of obesity. Expert Opin Pharmacother. 2009;10(6):1069-1081.
36. Greenway FL, Fujioka K, Plodkowski RA, et al; COR-I Study Group. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-1): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595-605.
37. Hollander P, Gupta AK, Plodkowski R, et al; COR-Diabetes Study Group. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care. 2013;36(12):4022-4029.
38. Contrave [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc; 2014.
39. Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Eng J Med. 2015;373(1):11-22.
40. Wadden TA, Hollander P, Klein S, et al; NN8022-1923 Investigators. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet-induced weight loss: the SCALE Maintenance randomized study. Int J Obes (Lond). 2013;37(11):1443-1451
41. Saxenda [package insert]. Novo Nordisk: Plainsboro, NJ; 2015.
42.Schwartz A, Doucet E. Relative changes in resting energy expenditure during weight loss: a systemic review. Obes Rev. 2010;11(7): 531-547. ```````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````
Lessons Learned From the RACAT Trial: A Comparison of Rheumatoid Arthritis Therapies
Rheumatoid arthritis (RA) is a chronic inflammatory disease of the joints, leading to joint destruction, with significant long-term morbidity and mortality. Over the past quarter century, multiple new therapies and approaches have been introduced, so patients newly diagnosed with RA can realistically expect to be in remission while taking their medications. However, many of the most commonly used medications are costly, making RA care one of the most expensive per patient.1 Early treatment with disease-modifying antirheumatic drugs (DMARDs) and treating all patients to the target of low-disease activity are critical keys to optimal outcomes.
Methotrexate (MTX) is a highly effective and economical first-line DMARD that is recommended as the initial therapy for most patients.2,3 Unfortunately, one-half to two-thirds of patients will not have complete responses and, therefore, require additional therapy. Fortunately, there are more than a dozen therapies that, when added to MTX, have been shown to be better than MTX alone. However, since some of these options use conventional DMARDs and others require biologics, there exist very different economic as well as potential toxicity implications. Understanding how best to treat patients with RA with active disease while on an appropriate dose of MTX is important for both medical and economic reasons.
Despite this being a seminal question for the past 15 years, no blinded trial had addressed this issue before the VA Cooperative Studies Program (CSP) Rheumatoid Arthritis: Comparison of Active Therapies (RACAT) trial. This was true for several reasons, likely including the considerable cost of conducting such a trial and the low priority of this research question for the pharmaceutical industry. Industry-funded trials in RA often focus on new indications, and these studies often fail to address the questions most relevant to the day-to-day care of patients.
For example, it is often not particularly helpful to the clinician that patients placed on “therapy A” are doing better or worse than those placed on “therapy B” after 1 year of the same treatment. Such rigid protocols do not mimic what is done in the clinic: A patient’s treatment program is often changed much earlier than 1 year when it is not working. Therefore, RACAT was designed to more closely mirror clinical practice and to test the strategy of starting conventional therapy before biologic therapy, with the option of changing therapy for nonresponders—similar to what most clinicians would do in practice. This article explores the lessons learned from this landmark trial and highlights the critical role that the VA CSP played.
Trial Background
The RACAT trial, a comparative effectiveness, randomized, double-blind, noninferiority trial, originated as a joint effort of investigators from the VA and the Rheumatology and Arthritis Investigational Network (RAIN) and subsequently involved Canadian enrollment sites. The RACAT results were published in the New England Journal of Medicine in 2013, and its investigative team was awarded the 2014 Lee C. Howley Sr. Prize by the Arthritis Foundation for conducting the most important arthritis research worldwide from the previous year.4
The RACAT originated with a letter of intent to the VA CSP in 2003. The central question to be addressed was whether biologic therapy should be added first in patients with active RA despite MTX or whether clinicians should first add the much less expensive but very effective combination of conventional therapies, including sulfasalazine (SSZ) and hydroxychloroquine to MTX.5,6 This led to the 48-week, binational, multicenter, randomized, double-blind, noninferiority trial comparing the strategy of initially adding hydroxychloroquine and SSZ to MTX (triple therapy group) in patients with active disease despite MTX compared with the strategy of first adding etanercept to MTX.4 Etanercept is among the most commonly used biologic agents approved for RA. Etanercept works by targeting tumor necrosis factor, a pro-inflammatory cytokine central in disease pathogenesis. Both RACAT treatment groups were switched in a blinded fashion to the other therapy at 24 weeks if they did not have a clinically significant improvement. The primary endpoint was a change in the disease activity score (DAS28) from baseline to 48 weeks. An important secondary endpoint was the comparison of radiographic progression of disease at 48 weeks as measured by the validated modified Sharp scoring method. Additionally, and very importantly, economic and functional outcomes were assessed. To conduct the trial, investigators and patients participated from 16 VA sites in addition to 8 Canadian and 12 RAIN sites. The study was sponsored and primarily funded by the VA CSP, VA Office of Research and Development with additional funding coming from the Canadian Institutes of Health Research (CIHR) and from the National Institutes of Health.
Trial Design
To understand the RACAT trial design, one must appreciate the landscape of RA trials conducted in the early-to-mid 2000s. At that time, there had been an explosion of new biologic therapies for RA. Most of the trials were placebo-controlled studies with nonresponders to MTX being placed on placebo vs active drug.7 For ethical and legal reasons, however, clinicians do not treat patients with placebo, especially when highly effective therapies exist, thus limiting the relevance of the classic placebo-controlled trial in RA.8 One of the main tenets of RA therapy in this century has been to use effective therapies to treat patients with active RA with the goal of achieving (and maintaining) either low-disease activity or remission as measured by a composite scoring system, most commonly the DAS28. In order to do this in the framework of a designed research trial, therapies commonly need to be escalated when patients are not doing well, similar to what is done in clinical practice.
The RACAT trial was a comparative effectiveness trial. Comparative effectiveness is not a new idea; in fact, it is precisely how many clinicians practice medicine. It is simply comparing 2 or more treatments to determine which is more effective. Since the inception of the RACAT trial, the American Recovery and Reinvestment Act of 2009 provided $1.1 billion for major expansion of comparative effectiveness research. This changing landscape of federally funded research has highlighted the growing national interest in this type of trial.
This trial design posed several barriers as it applies to the study medications. Methotrexate, hydroxychloroquine, and SSZ are generic medications most often taken orally (MTX is available for parenteral administration). In contrast, etanercept is most often given as a subcutaneous injection and currently is not available in a biosimilar (generic) form in the U.S.; thus, the medication and its delivery device are proprietary. Because this was a double-blind, noninferiority trial, the study required both etanercept-active medication and placebo in identical delivery devices. The makers of etanercept donated placebo etanercept to make blinding possible. The VA, along with CIHR, purchased active etanercept for all trial participants, including those from Canada and the RAIN network. The VA research pharmacy in Albuquerque, New Mexico, was responsible for blinding all active and placebo drugs used in the trial and made these drugs available to all patients, even those not eligible for VA care.In a precedent-setting effort for rheumatology research, RACAT culminated from the collaborations among the private sector, the Canadian health system, and VA. The VA CSP was responsible for the collection of the clinical data, data analyses (Massachusetts Veterans Epidemiology Research and Information Center, VA Boston Healthcare System [VABHS]); the collection of economic data (VA Palo Alto Health Care System); the provision of and payment for the study medications; and the preparation and distribution of active etanercept and placebos (New Mexico VA Health Care System [NMVAHCS]). Through this organizational structure, the trial was successfully completed. In addition to placebo etanercept provided by Amgen (Thousand Oaks, CA), Pharmascience (Montreal, Quebec) provided blinded SSZ and blinded placebo. Neither company was involved in the study design nor did they have an active role in the trial. Hydroxychoroquine and matched placebo were provided by the central pharmacy of the NMVAHCS.
Safety Monitoring
As with any treatment study, patient safety was of paramount importance. Through the aforementioned organizational structure, each participating site had administrative team members who were responsible to the VABHS CSP to ensure research adherence and compliance with best practices. Additionally, an independent data and safety monitoring committee (DSMC) monitored the trial for safety and scientific integrity. At the time that the trial began in 2007, there were questions about the relative efficacy of triple therapy vs MTX plus biologic therapy. Because of this question, the DSMC raised concerns that patients may be placed at a higher risk of joint damage if not placed sooner on biologic therapy. As a response to this concern, the blinded radiographic reviewers were asked to read the hand and feet X-rays as the study progressed, allowing the DSMC to watch for any emerging safety signals. There were none, and in fact, the therapies were essentially identical radiographically.
The consequence of this request was multifold. First, patient safety was maintained; second, the study team was forced to navigate the logistic and technologic challenges posed by the reading and interpretation of the radiographs at an earlier time point than was originally planned; and last, as a result, results became available and were disseminated in a relatively narrow time frame. This third point was very important. One of the major concerns of foregoing biologic therapy was the potential for joint damage. Without the radiographic information, the manuscript could not have been completed in a timely fashion.
Trial Results
The trial was a 48-week, double-blind, noninferiority trial in which 353 patients with RA who had active disease despite MTX therapy were randomized to a triple therapy regimen of DMARDs (baseline MTX, plus SSZ and hydroxychloroquine) or baseline MTX plus etanercept (Figure 1). Patients who did not have a clinically significant improvement at 24 weeks according to a prespecified threshold were switched in a blinded fashion to the other therapy.
The primary endpoint, the change between baseline and 48 weeks in the DAS28, was similar; thus the strategy of first starting triple therapy was not inferior to first starting etanercept (the change in DAS28 was -2.12 and -2.29 respectively, P < .0001, supporting noninferiority, Figure 2). Both groups had significant improvement over the course of the first 24 weeks (P = .001). A total of 27% of participants in each group switched at 24 weeks (Figure 3). Patients in both groups who switched therapies had improvement after switching (P < .001), and the response after switching did not differ significantly between the 2 groups. Importantly, there were no significant between-group differences in radiographic progression (P = .43), pain and health-related quality of life (QOL), or in medication-associated major adverse events (AEs), although there were numerically more serious infections with etanercept-MTX therapy (12 vs 4). Gastrointestinal AEs were numerically more frequent with triple therapy; whereas infections and skin and subcutaneous AEs were more frequent with etanercept-MTX therapy.
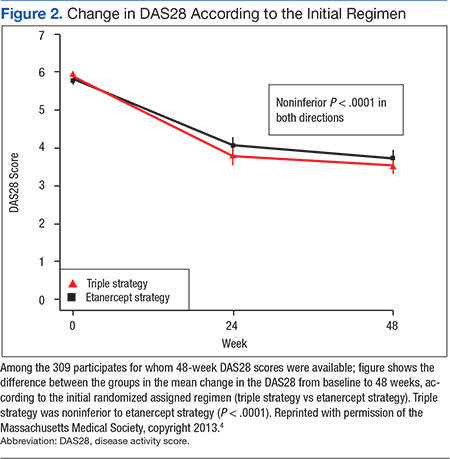
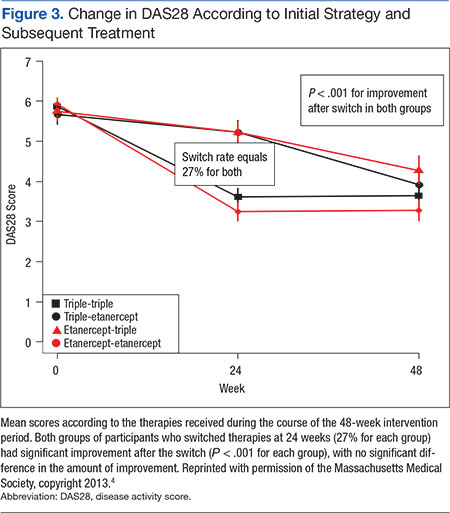
The cost-effectiveness of adding SSZ and hydroxychloroquine to MTX vs adding etanercept to MTX, using a predetermined measurement of QOL, was assessed in this trial.9 These data were initially presented in abstract form at the 2014 American College of Rheumatology national meeting. Considered was the ratio of all the incremental costs between the 2 treatment strategies to the benefits, as measured in quality-adjusted life-years (QALYs), where QOL is measured as an index with 1 being equivalent to full health and 0 being equivalent to death. This incremental cost-effectiveness ratio produces a monetary cost for each QALY, which is an indication of cost-effectiveness and value. Most health care systems currently consider anything that costs < $50,000/QALY to be cost-effective. To be conservative, the trial researchers considered anything up to $100,000 for an additional QALY acceptable.
In the 48-week trial analysis, the use of etanercept first, instead of triple therapy, would incur about $1 million of cost per QALY, far more than the $50,000 to $100,000 deemed to be reasonable value. Biologic therapy use first, had a near-zero chance of being cost-effective and would be cost-effective only after failure of triple therapy; results that were robust to all plausible scenarios.
Economic Implications
As noted in the trial design, economic data were prospectively collected for later analysis. The availability and cost of biologic treatments have become a critical issue. In 2005, it was reported that the U.S. biologics market reached $52 billion and was noted to have an annual growth of 17%.10 In 2011, 8 of the top 20 drugs sold in the U.S. were biologics, and year-to-year biologics spending has grown by 6.6%. In 2013, the top 100 biologics in the U.S. had combined sales of $66.3 billion.11 The researchers analysis demonstrated that using a strategy of triple therapy first could result in health care cost savings in the tens of billions of dollars. Importantly, these savings would occur at the same time as patients were receiving optimal care.
Summary
The major conclusions from the RACAT trial in RA patients with active disease despite MTX are the following: 1. The strategy of first starting the conventional DMARD triple therapy combination is noninferior to first starting etanercept, based on both clinical and radiographic outcomes. 2. The triple therapy group had more minor gastrointestinal events, whereas the etanercept group had more infections. Patients in either group who did not respond well to the initial treatment and switched (27% in both groups) improved significantly after the switch. 3. The economic implications of these findings are significant. The incremental cost differences approached $1 million per QALY.
The VA CSP should be congratulated for supporting and funding this trial, which will inform therapeutic decisions in RA for years to come. These results allow clinicians to provide not only optimal health care for their RA patients, but also maximize the value of their health care.
1. Gavan S, Harrison M, Iglesias C, Barton A, Manca A, Payne K. Economics of stratified medicine in rheumatoid arthritis. Curr Rheumatol Rep. 2014;16(12):468.
2. Singh JA, Furst DE, Bharat A, et al. 2012 update of the 2008 American College of Rheumatology recommendations for the use of disease-modifying anti-rheumatic drugs and biologics in the treatment of rheumatoid arthritis (RA). Arthritis Care Res (Hoboken). 2012;64(5):625-639.
3. Smolen JS, Landewé R, Breedveld FC, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis. 2014;73(3):492-509.
4. O'Dell JR, Mikuls TR, Taylor TH, et al; CSP 551 RACAT Investigators. Therapies for active rheumatoid arthritis after methotrexate failure. N Eng J Med. 2013;369(4):307-318.
5. Moreland LW, O'Dell JR, Paulus HE, et al; TEAR Investigators. A randomized comparative effectiveness study of oral triple therapy versus etanercept plus methotrexate in early aggressive rheumatoid arthritis: the Treatment of Early Aggressive Rheumatoid Arthritis Trial. Arthritis Rheum. 2012;64(9):2824-2835.
6. O'Dell JR, Leff R, Paulsen G, et al. Treatment of rheumatoid arthritis with methotrexate and hydroxychloroquine, methotrexate and sulfasalazine, or a combination of the three medications: results of a two-year, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46(5):1164-1170.
7. Singh JA, Cameron DR. Summary of AHRQ's comparative effectiveness review of drug therapy for rheumatoid arthritis in adults-an update. J Manag Care Pharm. 2012;18(4)(suppl C):S1-S18.
8. Chan TE. Regulating the placebo effect in clinical practice. Med Law Rev. 2014;23(1):1-26.
9. Bansback N, Phibb S, Sun H, et al. Cost effectiveness of adding etanercept to methotrexate therapy versus first adding sulfasalazine and hydroxychloroquine: a randomized noninferiority trial [American College of Rheumatology abstract 2781]. Arthritis Rheum. 2014;66(suppl 11):S1214.
10. Ernst and Young. Beyond borders: global biotechnology report 2005. Ernst and Young Website. https://www2.eycom.ch/publications/items/biotech-report/2005/2005_EY_Global_Biotech_Report.pdf. Accessed December 3, 2015.
11. Mulcahy AW, Predmore Z, Mattke S. The cost savings potential of biosimilar drugs in the United States. Rand Corporation Website. http://www.rand.org/pubs/perspectives/PE127.html. Accessed December 3, 2015.
Rheumatoid arthritis (RA) is a chronic inflammatory disease of the joints, leading to joint destruction, with significant long-term morbidity and mortality. Over the past quarter century, multiple new therapies and approaches have been introduced, so patients newly diagnosed with RA can realistically expect to be in remission while taking their medications. However, many of the most commonly used medications are costly, making RA care one of the most expensive per patient.1 Early treatment with disease-modifying antirheumatic drugs (DMARDs) and treating all patients to the target of low-disease activity are critical keys to optimal outcomes.
Methotrexate (MTX) is a highly effective and economical first-line DMARD that is recommended as the initial therapy for most patients.2,3 Unfortunately, one-half to two-thirds of patients will not have complete responses and, therefore, require additional therapy. Fortunately, there are more than a dozen therapies that, when added to MTX, have been shown to be better than MTX alone. However, since some of these options use conventional DMARDs and others require biologics, there exist very different economic as well as potential toxicity implications. Understanding how best to treat patients with RA with active disease while on an appropriate dose of MTX is important for both medical and economic reasons.
Despite this being a seminal question for the past 15 years, no blinded trial had addressed this issue before the VA Cooperative Studies Program (CSP) Rheumatoid Arthritis: Comparison of Active Therapies (RACAT) trial. This was true for several reasons, likely including the considerable cost of conducting such a trial and the low priority of this research question for the pharmaceutical industry. Industry-funded trials in RA often focus on new indications, and these studies often fail to address the questions most relevant to the day-to-day care of patients.
For example, it is often not particularly helpful to the clinician that patients placed on “therapy A” are doing better or worse than those placed on “therapy B” after 1 year of the same treatment. Such rigid protocols do not mimic what is done in the clinic: A patient’s treatment program is often changed much earlier than 1 year when it is not working. Therefore, RACAT was designed to more closely mirror clinical practice and to test the strategy of starting conventional therapy before biologic therapy, with the option of changing therapy for nonresponders—similar to what most clinicians would do in practice. This article explores the lessons learned from this landmark trial and highlights the critical role that the VA CSP played.
Trial Background
The RACAT trial, a comparative effectiveness, randomized, double-blind, noninferiority trial, originated as a joint effort of investigators from the VA and the Rheumatology and Arthritis Investigational Network (RAIN) and subsequently involved Canadian enrollment sites. The RACAT results were published in the New England Journal of Medicine in 2013, and its investigative team was awarded the 2014 Lee C. Howley Sr. Prize by the Arthritis Foundation for conducting the most important arthritis research worldwide from the previous year.4
The RACAT originated with a letter of intent to the VA CSP in 2003. The central question to be addressed was whether biologic therapy should be added first in patients with active RA despite MTX or whether clinicians should first add the much less expensive but very effective combination of conventional therapies, including sulfasalazine (SSZ) and hydroxychloroquine to MTX.5,6 This led to the 48-week, binational, multicenter, randomized, double-blind, noninferiority trial comparing the strategy of initially adding hydroxychloroquine and SSZ to MTX (triple therapy group) in patients with active disease despite MTX compared with the strategy of first adding etanercept to MTX.4 Etanercept is among the most commonly used biologic agents approved for RA. Etanercept works by targeting tumor necrosis factor, a pro-inflammatory cytokine central in disease pathogenesis. Both RACAT treatment groups were switched in a blinded fashion to the other therapy at 24 weeks if they did not have a clinically significant improvement. The primary endpoint was a change in the disease activity score (DAS28) from baseline to 48 weeks. An important secondary endpoint was the comparison of radiographic progression of disease at 48 weeks as measured by the validated modified Sharp scoring method. Additionally, and very importantly, economic and functional outcomes were assessed. To conduct the trial, investigators and patients participated from 16 VA sites in addition to 8 Canadian and 12 RAIN sites. The study was sponsored and primarily funded by the VA CSP, VA Office of Research and Development with additional funding coming from the Canadian Institutes of Health Research (CIHR) and from the National Institutes of Health.
Trial Design
To understand the RACAT trial design, one must appreciate the landscape of RA trials conducted in the early-to-mid 2000s. At that time, there had been an explosion of new biologic therapies for RA. Most of the trials were placebo-controlled studies with nonresponders to MTX being placed on placebo vs active drug.7 For ethical and legal reasons, however, clinicians do not treat patients with placebo, especially when highly effective therapies exist, thus limiting the relevance of the classic placebo-controlled trial in RA.8 One of the main tenets of RA therapy in this century has been to use effective therapies to treat patients with active RA with the goal of achieving (and maintaining) either low-disease activity or remission as measured by a composite scoring system, most commonly the DAS28. In order to do this in the framework of a designed research trial, therapies commonly need to be escalated when patients are not doing well, similar to what is done in clinical practice.
The RACAT trial was a comparative effectiveness trial. Comparative effectiveness is not a new idea; in fact, it is precisely how many clinicians practice medicine. It is simply comparing 2 or more treatments to determine which is more effective. Since the inception of the RACAT trial, the American Recovery and Reinvestment Act of 2009 provided $1.1 billion for major expansion of comparative effectiveness research. This changing landscape of federally funded research has highlighted the growing national interest in this type of trial.
This trial design posed several barriers as it applies to the study medications. Methotrexate, hydroxychloroquine, and SSZ are generic medications most often taken orally (MTX is available for parenteral administration). In contrast, etanercept is most often given as a subcutaneous injection and currently is not available in a biosimilar (generic) form in the U.S.; thus, the medication and its delivery device are proprietary. Because this was a double-blind, noninferiority trial, the study required both etanercept-active medication and placebo in identical delivery devices. The makers of etanercept donated placebo etanercept to make blinding possible. The VA, along with CIHR, purchased active etanercept for all trial participants, including those from Canada and the RAIN network. The VA research pharmacy in Albuquerque, New Mexico, was responsible for blinding all active and placebo drugs used in the trial and made these drugs available to all patients, even those not eligible for VA care.In a precedent-setting effort for rheumatology research, RACAT culminated from the collaborations among the private sector, the Canadian health system, and VA. The VA CSP was responsible for the collection of the clinical data, data analyses (Massachusetts Veterans Epidemiology Research and Information Center, VA Boston Healthcare System [VABHS]); the collection of economic data (VA Palo Alto Health Care System); the provision of and payment for the study medications; and the preparation and distribution of active etanercept and placebos (New Mexico VA Health Care System [NMVAHCS]). Through this organizational structure, the trial was successfully completed. In addition to placebo etanercept provided by Amgen (Thousand Oaks, CA), Pharmascience (Montreal, Quebec) provided blinded SSZ and blinded placebo. Neither company was involved in the study design nor did they have an active role in the trial. Hydroxychoroquine and matched placebo were provided by the central pharmacy of the NMVAHCS.
Safety Monitoring
As with any treatment study, patient safety was of paramount importance. Through the aforementioned organizational structure, each participating site had administrative team members who were responsible to the VABHS CSP to ensure research adherence and compliance with best practices. Additionally, an independent data and safety monitoring committee (DSMC) monitored the trial for safety and scientific integrity. At the time that the trial began in 2007, there were questions about the relative efficacy of triple therapy vs MTX plus biologic therapy. Because of this question, the DSMC raised concerns that patients may be placed at a higher risk of joint damage if not placed sooner on biologic therapy. As a response to this concern, the blinded radiographic reviewers were asked to read the hand and feet X-rays as the study progressed, allowing the DSMC to watch for any emerging safety signals. There were none, and in fact, the therapies were essentially identical radiographically.
The consequence of this request was multifold. First, patient safety was maintained; second, the study team was forced to navigate the logistic and technologic challenges posed by the reading and interpretation of the radiographs at an earlier time point than was originally planned; and last, as a result, results became available and were disseminated in a relatively narrow time frame. This third point was very important. One of the major concerns of foregoing biologic therapy was the potential for joint damage. Without the radiographic information, the manuscript could not have been completed in a timely fashion.
Trial Results
The trial was a 48-week, double-blind, noninferiority trial in which 353 patients with RA who had active disease despite MTX therapy were randomized to a triple therapy regimen of DMARDs (baseline MTX, plus SSZ and hydroxychloroquine) or baseline MTX plus etanercept (Figure 1). Patients who did not have a clinically significant improvement at 24 weeks according to a prespecified threshold were switched in a blinded fashion to the other therapy.
The primary endpoint, the change between baseline and 48 weeks in the DAS28, was similar; thus the strategy of first starting triple therapy was not inferior to first starting etanercept (the change in DAS28 was -2.12 and -2.29 respectively, P < .0001, supporting noninferiority, Figure 2). Both groups had significant improvement over the course of the first 24 weeks (P = .001). A total of 27% of participants in each group switched at 24 weeks (Figure 3). Patients in both groups who switched therapies had improvement after switching (P < .001), and the response after switching did not differ significantly between the 2 groups. Importantly, there were no significant between-group differences in radiographic progression (P = .43), pain and health-related quality of life (QOL), or in medication-associated major adverse events (AEs), although there were numerically more serious infections with etanercept-MTX therapy (12 vs 4). Gastrointestinal AEs were numerically more frequent with triple therapy; whereas infections and skin and subcutaneous AEs were more frequent with etanercept-MTX therapy.
The cost-effectiveness of adding SSZ and hydroxychloroquine to MTX vs adding etanercept to MTX, using a predetermined measurement of QOL, was assessed in this trial.9 These data were initially presented in abstract form at the 2014 American College of Rheumatology national meeting. Considered was the ratio of all the incremental costs between the 2 treatment strategies to the benefits, as measured in quality-adjusted life-years (QALYs), where QOL is measured as an index with 1 being equivalent to full health and 0 being equivalent to death. This incremental cost-effectiveness ratio produces a monetary cost for each QALY, which is an indication of cost-effectiveness and value. Most health care systems currently consider anything that costs < $50,000/QALY to be cost-effective. To be conservative, the trial researchers considered anything up to $100,000 for an additional QALY acceptable.
In the 48-week trial analysis, the use of etanercept first, instead of triple therapy, would incur about $1 million of cost per QALY, far more than the $50,000 to $100,000 deemed to be reasonable value. Biologic therapy use first, had a near-zero chance of being cost-effective and would be cost-effective only after failure of triple therapy; results that were robust to all plausible scenarios.
Economic Implications
As noted in the trial design, economic data were prospectively collected for later analysis. The availability and cost of biologic treatments have become a critical issue. In 2005, it was reported that the U.S. biologics market reached $52 billion and was noted to have an annual growth of 17%.10 In 2011, 8 of the top 20 drugs sold in the U.S. were biologics, and year-to-year biologics spending has grown by 6.6%. In 2013, the top 100 biologics in the U.S. had combined sales of $66.3 billion.11 The researchers analysis demonstrated that using a strategy of triple therapy first could result in health care cost savings in the tens of billions of dollars. Importantly, these savings would occur at the same time as patients were receiving optimal care.
Summary
The major conclusions from the RACAT trial in RA patients with active disease despite MTX are the following: 1. The strategy of first starting the conventional DMARD triple therapy combination is noninferior to first starting etanercept, based on both clinical and radiographic outcomes. 2. The triple therapy group had more minor gastrointestinal events, whereas the etanercept group had more infections. Patients in either group who did not respond well to the initial treatment and switched (27% in both groups) improved significantly after the switch. 3. The economic implications of these findings are significant. The incremental cost differences approached $1 million per QALY.
The VA CSP should be congratulated for supporting and funding this trial, which will inform therapeutic decisions in RA for years to come. These results allow clinicians to provide not only optimal health care for their RA patients, but also maximize the value of their health care.
Rheumatoid arthritis (RA) is a chronic inflammatory disease of the joints, leading to joint destruction, with significant long-term morbidity and mortality. Over the past quarter century, multiple new therapies and approaches have been introduced, so patients newly diagnosed with RA can realistically expect to be in remission while taking their medications. However, many of the most commonly used medications are costly, making RA care one of the most expensive per patient.1 Early treatment with disease-modifying antirheumatic drugs (DMARDs) and treating all patients to the target of low-disease activity are critical keys to optimal outcomes.
Methotrexate (MTX) is a highly effective and economical first-line DMARD that is recommended as the initial therapy for most patients.2,3 Unfortunately, one-half to two-thirds of patients will not have complete responses and, therefore, require additional therapy. Fortunately, there are more than a dozen therapies that, when added to MTX, have been shown to be better than MTX alone. However, since some of these options use conventional DMARDs and others require biologics, there exist very different economic as well as potential toxicity implications. Understanding how best to treat patients with RA with active disease while on an appropriate dose of MTX is important for both medical and economic reasons.
Despite this being a seminal question for the past 15 years, no blinded trial had addressed this issue before the VA Cooperative Studies Program (CSP) Rheumatoid Arthritis: Comparison of Active Therapies (RACAT) trial. This was true for several reasons, likely including the considerable cost of conducting such a trial and the low priority of this research question for the pharmaceutical industry. Industry-funded trials in RA often focus on new indications, and these studies often fail to address the questions most relevant to the day-to-day care of patients.
For example, it is often not particularly helpful to the clinician that patients placed on “therapy A” are doing better or worse than those placed on “therapy B” after 1 year of the same treatment. Such rigid protocols do not mimic what is done in the clinic: A patient’s treatment program is often changed much earlier than 1 year when it is not working. Therefore, RACAT was designed to more closely mirror clinical practice and to test the strategy of starting conventional therapy before biologic therapy, with the option of changing therapy for nonresponders—similar to what most clinicians would do in practice. This article explores the lessons learned from this landmark trial and highlights the critical role that the VA CSP played.
Trial Background
The RACAT trial, a comparative effectiveness, randomized, double-blind, noninferiority trial, originated as a joint effort of investigators from the VA and the Rheumatology and Arthritis Investigational Network (RAIN) and subsequently involved Canadian enrollment sites. The RACAT results were published in the New England Journal of Medicine in 2013, and its investigative team was awarded the 2014 Lee C. Howley Sr. Prize by the Arthritis Foundation for conducting the most important arthritis research worldwide from the previous year.4
The RACAT originated with a letter of intent to the VA CSP in 2003. The central question to be addressed was whether biologic therapy should be added first in patients with active RA despite MTX or whether clinicians should first add the much less expensive but very effective combination of conventional therapies, including sulfasalazine (SSZ) and hydroxychloroquine to MTX.5,6 This led to the 48-week, binational, multicenter, randomized, double-blind, noninferiority trial comparing the strategy of initially adding hydroxychloroquine and SSZ to MTX (triple therapy group) in patients with active disease despite MTX compared with the strategy of first adding etanercept to MTX.4 Etanercept is among the most commonly used biologic agents approved for RA. Etanercept works by targeting tumor necrosis factor, a pro-inflammatory cytokine central in disease pathogenesis. Both RACAT treatment groups were switched in a blinded fashion to the other therapy at 24 weeks if they did not have a clinically significant improvement. The primary endpoint was a change in the disease activity score (DAS28) from baseline to 48 weeks. An important secondary endpoint was the comparison of radiographic progression of disease at 48 weeks as measured by the validated modified Sharp scoring method. Additionally, and very importantly, economic and functional outcomes were assessed. To conduct the trial, investigators and patients participated from 16 VA sites in addition to 8 Canadian and 12 RAIN sites. The study was sponsored and primarily funded by the VA CSP, VA Office of Research and Development with additional funding coming from the Canadian Institutes of Health Research (CIHR) and from the National Institutes of Health.
Trial Design
To understand the RACAT trial design, one must appreciate the landscape of RA trials conducted in the early-to-mid 2000s. At that time, there had been an explosion of new biologic therapies for RA. Most of the trials were placebo-controlled studies with nonresponders to MTX being placed on placebo vs active drug.7 For ethical and legal reasons, however, clinicians do not treat patients with placebo, especially when highly effective therapies exist, thus limiting the relevance of the classic placebo-controlled trial in RA.8 One of the main tenets of RA therapy in this century has been to use effective therapies to treat patients with active RA with the goal of achieving (and maintaining) either low-disease activity or remission as measured by a composite scoring system, most commonly the DAS28. In order to do this in the framework of a designed research trial, therapies commonly need to be escalated when patients are not doing well, similar to what is done in clinical practice.
The RACAT trial was a comparative effectiveness trial. Comparative effectiveness is not a new idea; in fact, it is precisely how many clinicians practice medicine. It is simply comparing 2 or more treatments to determine which is more effective. Since the inception of the RACAT trial, the American Recovery and Reinvestment Act of 2009 provided $1.1 billion for major expansion of comparative effectiveness research. This changing landscape of federally funded research has highlighted the growing national interest in this type of trial.
This trial design posed several barriers as it applies to the study medications. Methotrexate, hydroxychloroquine, and SSZ are generic medications most often taken orally (MTX is available for parenteral administration). In contrast, etanercept is most often given as a subcutaneous injection and currently is not available in a biosimilar (generic) form in the U.S.; thus, the medication and its delivery device are proprietary. Because this was a double-blind, noninferiority trial, the study required both etanercept-active medication and placebo in identical delivery devices. The makers of etanercept donated placebo etanercept to make blinding possible. The VA, along with CIHR, purchased active etanercept for all trial participants, including those from Canada and the RAIN network. The VA research pharmacy in Albuquerque, New Mexico, was responsible for blinding all active and placebo drugs used in the trial and made these drugs available to all patients, even those not eligible for VA care.In a precedent-setting effort for rheumatology research, RACAT culminated from the collaborations among the private sector, the Canadian health system, and VA. The VA CSP was responsible for the collection of the clinical data, data analyses (Massachusetts Veterans Epidemiology Research and Information Center, VA Boston Healthcare System [VABHS]); the collection of economic data (VA Palo Alto Health Care System); the provision of and payment for the study medications; and the preparation and distribution of active etanercept and placebos (New Mexico VA Health Care System [NMVAHCS]). Through this organizational structure, the trial was successfully completed. In addition to placebo etanercept provided by Amgen (Thousand Oaks, CA), Pharmascience (Montreal, Quebec) provided blinded SSZ and blinded placebo. Neither company was involved in the study design nor did they have an active role in the trial. Hydroxychoroquine and matched placebo were provided by the central pharmacy of the NMVAHCS.
Safety Monitoring
As with any treatment study, patient safety was of paramount importance. Through the aforementioned organizational structure, each participating site had administrative team members who were responsible to the VABHS CSP to ensure research adherence and compliance with best practices. Additionally, an independent data and safety monitoring committee (DSMC) monitored the trial for safety and scientific integrity. At the time that the trial began in 2007, there were questions about the relative efficacy of triple therapy vs MTX plus biologic therapy. Because of this question, the DSMC raised concerns that patients may be placed at a higher risk of joint damage if not placed sooner on biologic therapy. As a response to this concern, the blinded radiographic reviewers were asked to read the hand and feet X-rays as the study progressed, allowing the DSMC to watch for any emerging safety signals. There were none, and in fact, the therapies were essentially identical radiographically.
The consequence of this request was multifold. First, patient safety was maintained; second, the study team was forced to navigate the logistic and technologic challenges posed by the reading and interpretation of the radiographs at an earlier time point than was originally planned; and last, as a result, results became available and were disseminated in a relatively narrow time frame. This third point was very important. One of the major concerns of foregoing biologic therapy was the potential for joint damage. Without the radiographic information, the manuscript could not have been completed in a timely fashion.
Trial Results
The trial was a 48-week, double-blind, noninferiority trial in which 353 patients with RA who had active disease despite MTX therapy were randomized to a triple therapy regimen of DMARDs (baseline MTX, plus SSZ and hydroxychloroquine) or baseline MTX plus etanercept (Figure 1). Patients who did not have a clinically significant improvement at 24 weeks according to a prespecified threshold were switched in a blinded fashion to the other therapy.
The primary endpoint, the change between baseline and 48 weeks in the DAS28, was similar; thus the strategy of first starting triple therapy was not inferior to first starting etanercept (the change in DAS28 was -2.12 and -2.29 respectively, P < .0001, supporting noninferiority, Figure 2). Both groups had significant improvement over the course of the first 24 weeks (P = .001). A total of 27% of participants in each group switched at 24 weeks (Figure 3). Patients in both groups who switched therapies had improvement after switching (P < .001), and the response after switching did not differ significantly between the 2 groups. Importantly, there were no significant between-group differences in radiographic progression (P = .43), pain and health-related quality of life (QOL), or in medication-associated major adverse events (AEs), although there were numerically more serious infections with etanercept-MTX therapy (12 vs 4). Gastrointestinal AEs were numerically more frequent with triple therapy; whereas infections and skin and subcutaneous AEs were more frequent with etanercept-MTX therapy.
The cost-effectiveness of adding SSZ and hydroxychloroquine to MTX vs adding etanercept to MTX, using a predetermined measurement of QOL, was assessed in this trial.9 These data were initially presented in abstract form at the 2014 American College of Rheumatology national meeting. Considered was the ratio of all the incremental costs between the 2 treatment strategies to the benefits, as measured in quality-adjusted life-years (QALYs), where QOL is measured as an index with 1 being equivalent to full health and 0 being equivalent to death. This incremental cost-effectiveness ratio produces a monetary cost for each QALY, which is an indication of cost-effectiveness and value. Most health care systems currently consider anything that costs < $50,000/QALY to be cost-effective. To be conservative, the trial researchers considered anything up to $100,000 for an additional QALY acceptable.
In the 48-week trial analysis, the use of etanercept first, instead of triple therapy, would incur about $1 million of cost per QALY, far more than the $50,000 to $100,000 deemed to be reasonable value. Biologic therapy use first, had a near-zero chance of being cost-effective and would be cost-effective only after failure of triple therapy; results that were robust to all plausible scenarios.
Economic Implications
As noted in the trial design, economic data were prospectively collected for later analysis. The availability and cost of biologic treatments have become a critical issue. In 2005, it was reported that the U.S. biologics market reached $52 billion and was noted to have an annual growth of 17%.10 In 2011, 8 of the top 20 drugs sold in the U.S. were biologics, and year-to-year biologics spending has grown by 6.6%. In 2013, the top 100 biologics in the U.S. had combined sales of $66.3 billion.11 The researchers analysis demonstrated that using a strategy of triple therapy first could result in health care cost savings in the tens of billions of dollars. Importantly, these savings would occur at the same time as patients were receiving optimal care.
Summary
The major conclusions from the RACAT trial in RA patients with active disease despite MTX are the following: 1. The strategy of first starting the conventional DMARD triple therapy combination is noninferior to first starting etanercept, based on both clinical and radiographic outcomes. 2. The triple therapy group had more minor gastrointestinal events, whereas the etanercept group had more infections. Patients in either group who did not respond well to the initial treatment and switched (27% in both groups) improved significantly after the switch. 3. The economic implications of these findings are significant. The incremental cost differences approached $1 million per QALY.
The VA CSP should be congratulated for supporting and funding this trial, which will inform therapeutic decisions in RA for years to come. These results allow clinicians to provide not only optimal health care for their RA patients, but also maximize the value of their health care.
1. Gavan S, Harrison M, Iglesias C, Barton A, Manca A, Payne K. Economics of stratified medicine in rheumatoid arthritis. Curr Rheumatol Rep. 2014;16(12):468.
2. Singh JA, Furst DE, Bharat A, et al. 2012 update of the 2008 American College of Rheumatology recommendations for the use of disease-modifying anti-rheumatic drugs and biologics in the treatment of rheumatoid arthritis (RA). Arthritis Care Res (Hoboken). 2012;64(5):625-639.
3. Smolen JS, Landewé R, Breedveld FC, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis. 2014;73(3):492-509.
4. O'Dell JR, Mikuls TR, Taylor TH, et al; CSP 551 RACAT Investigators. Therapies for active rheumatoid arthritis after methotrexate failure. N Eng J Med. 2013;369(4):307-318.
5. Moreland LW, O'Dell JR, Paulus HE, et al; TEAR Investigators. A randomized comparative effectiveness study of oral triple therapy versus etanercept plus methotrexate in early aggressive rheumatoid arthritis: the Treatment of Early Aggressive Rheumatoid Arthritis Trial. Arthritis Rheum. 2012;64(9):2824-2835.
6. O'Dell JR, Leff R, Paulsen G, et al. Treatment of rheumatoid arthritis with methotrexate and hydroxychloroquine, methotrexate and sulfasalazine, or a combination of the three medications: results of a two-year, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46(5):1164-1170.
7. Singh JA, Cameron DR. Summary of AHRQ's comparative effectiveness review of drug therapy for rheumatoid arthritis in adults-an update. J Manag Care Pharm. 2012;18(4)(suppl C):S1-S18.
8. Chan TE. Regulating the placebo effect in clinical practice. Med Law Rev. 2014;23(1):1-26.
9. Bansback N, Phibb S, Sun H, et al. Cost effectiveness of adding etanercept to methotrexate therapy versus first adding sulfasalazine and hydroxychloroquine: a randomized noninferiority trial [American College of Rheumatology abstract 2781]. Arthritis Rheum. 2014;66(suppl 11):S1214.
10. Ernst and Young. Beyond borders: global biotechnology report 2005. Ernst and Young Website. https://www2.eycom.ch/publications/items/biotech-report/2005/2005_EY_Global_Biotech_Report.pdf. Accessed December 3, 2015.
11. Mulcahy AW, Predmore Z, Mattke S. The cost savings potential of biosimilar drugs in the United States. Rand Corporation Website. http://www.rand.org/pubs/perspectives/PE127.html. Accessed December 3, 2015.
1. Gavan S, Harrison M, Iglesias C, Barton A, Manca A, Payne K. Economics of stratified medicine in rheumatoid arthritis. Curr Rheumatol Rep. 2014;16(12):468.
2. Singh JA, Furst DE, Bharat A, et al. 2012 update of the 2008 American College of Rheumatology recommendations for the use of disease-modifying anti-rheumatic drugs and biologics in the treatment of rheumatoid arthritis (RA). Arthritis Care Res (Hoboken). 2012;64(5):625-639.
3. Smolen JS, Landewé R, Breedveld FC, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis. 2014;73(3):492-509.
4. O'Dell JR, Mikuls TR, Taylor TH, et al; CSP 551 RACAT Investigators. Therapies for active rheumatoid arthritis after methotrexate failure. N Eng J Med. 2013;369(4):307-318.
5. Moreland LW, O'Dell JR, Paulus HE, et al; TEAR Investigators. A randomized comparative effectiveness study of oral triple therapy versus etanercept plus methotrexate in early aggressive rheumatoid arthritis: the Treatment of Early Aggressive Rheumatoid Arthritis Trial. Arthritis Rheum. 2012;64(9):2824-2835.
6. O'Dell JR, Leff R, Paulsen G, et al. Treatment of rheumatoid arthritis with methotrexate and hydroxychloroquine, methotrexate and sulfasalazine, or a combination of the three medications: results of a two-year, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46(5):1164-1170.
7. Singh JA, Cameron DR. Summary of AHRQ's comparative effectiveness review of drug therapy for rheumatoid arthritis in adults-an update. J Manag Care Pharm. 2012;18(4)(suppl C):S1-S18.
8. Chan TE. Regulating the placebo effect in clinical practice. Med Law Rev. 2014;23(1):1-26.
9. Bansback N, Phibb S, Sun H, et al. Cost effectiveness of adding etanercept to methotrexate therapy versus first adding sulfasalazine and hydroxychloroquine: a randomized noninferiority trial [American College of Rheumatology abstract 2781]. Arthritis Rheum. 2014;66(suppl 11):S1214.
10. Ernst and Young. Beyond borders: global biotechnology report 2005. Ernst and Young Website. https://www2.eycom.ch/publications/items/biotech-report/2005/2005_EY_Global_Biotech_Report.pdf. Accessed December 3, 2015.
11. Mulcahy AW, Predmore Z, Mattke S. The cost savings potential of biosimilar drugs in the United States. Rand Corporation Website. http://www.rand.org/pubs/perspectives/PE127.html. Accessed December 3, 2015.
Long-Term Surgical Management of Severe Pelvic Injury and Resulting Neurogenic Bladder From an Improvised Explosive Device
More than 52,000 soldiers have been injured and 6,800 have been killed during the wars in Iraq and Afghanistan.1 Blast injuries from improvised explosive devices (IEDs) account for 70% to 79% of combat-related injuries and deaths in these wars.2 Advances in personal body armor, rapid and advanced surgical treatment, and the changing nature of combat in Iraq and Afghanistan have changed injury patterns and survival compared with prior military conflicts such as those in Vietnam and Korea.3
The most common combat-related injuries in the recent wars are extremity, facial, brain, and gastrointestinal injuries. Pelvic and genitourinary injuries are also common, accounting for about 8% of total injuries.2 Pelvic and genitourinary injury can cause long-term disability from nerve injury (neurogenic bladder, neurogenic bowel, sexual dysfunction, urethral injury), as well as general loss of genital structures from blast injuries.
The usual care for bladder dysfunction from pelvic or genitourinary injury ranges from the use of chronic indwelling catheters to reconstructive surgery. However, there is no standard of care for long-term treatment of patients with pelvic or genitourinary injury who experience bladder dysfunction. Reconstructive surgery has the potential to improve quality of life (QOL) and eliminate chronic indwelling catheters, which are prone to cause infection and long-term kidney problems in patients with bladder dysfunction from traumatic injury.
This case report evaluates the efficacy of reconstructive surgery for bladder dysfunction to improve independence and QOL and decrease complications associated with chronic indwelling urinary catheters. The authors hope to raise awareness regarding this option for patients with pelvic, spinal cord, or genitourinary injury who are young and face long-term disability from their injuries.
Case Presentation
A 22-year-old man presented to the George E. Wahlen VAMC Urology Clinic in Salt Lake City, Utah with a complicated history related to combat injuries. During combat operations 3 years earlier, he was injured by an IED blast while on foot patrol. His injuries included bilateral severe extremity injury, perineal and genital blast wounds, a bladder injury, pelvic fracture, colorectal injury, and extensive soft tissue loss. He underwent multiple abdominal explorations, left leg amputation below the knee, multiple skin grafts, soft tissue debridements, left-side orchiectomy, bladder repair, and diverting colostomy. He survived the injuries and was eventually discharged from active military service and returned home.
Upon presentation to the VAMC, the patient had a diverting colostomy, suprapubic bladder catheter, and bladder and bowel function consistent with cauda equina syndrome (pelvic nerve injury). Given the lack of rectal tone, fecal incontinence was likely with colostomy reversal. His bladder had low volume and poor compliance (elasticity). In addition, the patient had no volitional control of urination or defecation.
The patient previously performed intermittent self-catheterization but experienced total urinary incontinence (UI) between catheterizations, due to his bladder dynamics and a lack of urinary sphincter tone. A suprapubic bladder catheter was previously placed to control UI. However, the patient remained incontinent, and urinary leakage, need for diapers, and urinary tract infections (UTIs) negatively impacted QOL. The patient ambulated well and was physically active. His priority was to reduce incontinence and improve QOL.
Catheterizable Ileal Cecocystoplasty
The patient underwent cutaneous catheterizable ileal cecocystoplasty (CCIC) (Figure 1). In this surgery, a segment of the cecum and ascending colon with attached terminal ileum is used to increase the size of the bladder (augmentation cystoplasty) and create a channel for catheterization from the umbilicus. The cecum and colon are detubularized, and a large rectangular plate of large bowel is formed, which is then sewn to the bladder, expanding its volume. About 10 to 15 cm of the terminal ileum is tapered to the diameter of a pencil and brought through the base of the umbilicus, creating a small stoma for intermittent bladder catheterization. The ileocecal valve is tightened and serves as a continence mechanism to prevent urinary leakage through the small stoma in the umbilicus.4
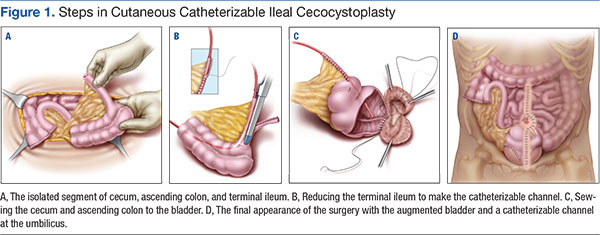
A perineal urethral mesh sling was placed at the time of the patient’s surgery to bolster the deinnervated urinary sphincter and prevent urethral leakage. The goal of reconstructive surgery for this patient was to create a small bowel channel connecting the umbilicus and bladder that could be catheterized every 4 to 6 hours, increase bladder capacity, and increase sphincteric resistance to reduce urethral leakage through the penis. Because there can be damage from passing a catheter through mesh slings and the urethra over time, including stenosis or erosion of the sling, an alternative catheterizable channel was needed in this patient.
The patient recovered after the surgery and was able to self-catheterize without difficulty. However, the urethral mesh sling did not place enough pressure on the urethra to prevent leakage, and he had persistent incontinence from the penis. Three months after the original surgery the patient had exploration of the perineum, which revealed that the mesh sling was loose and exerting inadequate pressure on the urethra. It was likely the sling slipped postoperatively—a known complication of urethral slings. An artificial urinary sphincter (AUS) was placed around the urethra during the second surgery to address the patient’s UI. 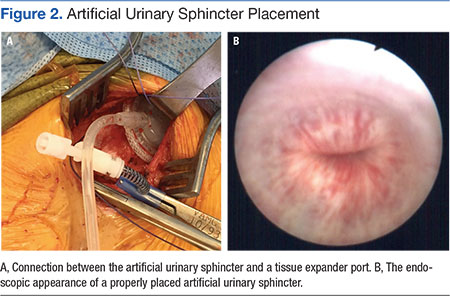
More than 1 year after the original surgery, the patient self-catheterizes about 4 to 5 times daily via the catheterizable channel using a single-use catheter. His bladder holds at least 500 mL. The patient does not have significant leakage from the channel or the penis. He is no longer dependent on a chronic indwelling catheter and is free of the problems associated with severe UI, including foul odor, UTIs, and social isolation.
Discussion
Patients with spinal cord or pelvic nerve injury often develop spastic bladders with low capacities. This is similar to muscle spasticity that may occur with a neurologic injury, below the level of the injury, such as in the lower extremities. The powerful uncontrolled bladder spasms and small bladder capacity most often lead to incontinence. Additionally, neurologic control of the urinary sphincter is affected, leading to either uncontrolled spasms or poor tone. Patients with these injuries have no volitional control of bladder functions and are forced to catheterize intermittently, use a condom-type catheter, or have a chronic indwelling catheter (a Foley catheter or suprapubic catheter).
Intermittent catheterization is the preferred management option for neurogenic bladder. When compared with chronic indwelling catheters, intermittent catheterization is associated with lower rates of UTI and upper tract abnormalities and with the loss of renal function.5 Unfortunately, patients do not often stay on intermittent catheterization. A recent study showed that up to 70% of patients with spinal cord injuries who used clean intermittent catheterization when discharged from acute rehabilitation discontinue use and are subsequently managed by chronic indwelling catheters.6 Although the reasons why intermittent catheterization is discontinued are unclear, patient dissatisfaction with catheterization, anatomic problems, such as urethral scarring, or continued leakage despite medical treatments, such as anticholinergic medicines, may be factors.
Uncontrolled leakage and UI significantly impacts QOL and may cause patients to choose chronic indwelling catheters over intermittent catheterization. Several treatments are available to control incontinence associated with intermittent catheterization. Anticholinergic medications and more recently onabotulinum toxin A may help improve bladder spasticity. In 2011, the FDA approved onabotulinum toxin A for transurethral bladder injections. It has been shown to increase functional bladder capacity and decrease spasticity.7,8 Onabotulinum toxin A treatment will not enlarge a small, contracted bladder.
Onabotulinum toxin A treatment would not be ideal for the patient in this case study. His absolute bladder capacity was 200 mL, and onabotulinum toxin A treatment would not significantly improve capacity or make intermittent catheterization practical. Additionally, the patient had poor urinary sphincter function, and he would continue to leak regardless of improvements in the bladder spasticity or tone.
Augmentation enterocystoplasty is surgical enlargement of the bladder, using a piece of the bowel and is indicated in patients with low bladder volumes. With this procedure the native bladder becomes defunctionalized, and patients experience a dramatic improvement in bladder volumes and a reduction in bladder spasms and leakage. The use of the colon and terminal ileum for bladder augmentation, or CCIC, was first reported by Sarosdy in 2 patients in 1992.9 In 1996, King and colleagues demonstrated successful outcomes with CCIC in a cohort of 8 patients after 34 months of follow-up.10 Seven patients successfully used clean intermittent catheterization, and 1 patient chose an indwelling catheter because of progressive upper extremity weakness. No patients experienced worsened renal function or pyelonephritis suggestive of upper urinary tract deterioration. A single patient had mild stomal stenosis, which was successfully revised under local anesthesia.
In another study, Sutton and colleagues reported at 27 months an improvement of 276 mL in bladder capacity, no metabolic complications, and a 95% continence rate in a cohort of 23 patients with neurogenic bladder who underwent CCIC.4 Sutton and colleagues later reported outcomes for 34 patients with a median of 31 months follow-up.11 The most common complications were recurrent UTIs (12%) and stomal stenosis (12%). Only 3 patients (9%) required surgical revisions for stomal stenosis.
Altered bowel function and metabolic abnormalities are a concern after bowel resection and reconstruction. However, a study has found no subjective change in bowel function following ileal resection of up to 60 cm for urinary diversion for bladder malignancy.12 Rates of hyperchloremic hypokalemic metabolic acidosis are low, and most changes in electrolytes are subclinical.13,14 Long-term vitamin B12 deficiency is seen with larger (> 50 cm) ileal resections but is rare with CCIC, given the small segment used for reconstruction.15 Overall, CCIC is shown to have excellent surgical outcomes in carefully selected patients with neurogenic bladder.
In addition to low bladder capacity, the case study patient also had intrinsic sphincteric deficiency (very low urinary sphincter tone), which is common with pelvic nerve injury but unusual with spinal cord injury. He initially received a suburethral mesh sling that supported and compressed the urethra and buttressed the natural urinary sphincter. However, patients can develop catheterization issues with a suburethral sling due to mechanical compression of the urethra and traversing the compressed area with a urinary catheter. Given the indication for augmentation cystoplasty in this patient, he additionally elected to undergo catheterization channel creation to avoid long-term issues of urethral catheterization through the urethra compressed by the sling.
Unfortunately, this patient had postoperative issues with his suburethral sling, and a modified AUS was inserted rather than a second sling. Normally, an AUS is attached to a pump mechanism in the scrotum. The pump allows the patient to cycle fluid from the sphincter cuff to a reservoir in the abdomen, removing compression on the urethra and allowing normal urination. Because this patient could not effectively urinate from the penis, the authors wanted to obstruct the urethra to prevent leakage without closing it permanently. The AUS was connected to a tissue expander port placed subcutaneously in the lower abdomen rather than to a pump mechanism. This modified approach used fewer mechanical parts compared with the pump mechanism, possibly reducing rates of mechanical failure. Additionally, a lower cuff pressure could be used to obstruct the urethra and prevent leakage, reducing the likelihood of urethral atrophy. Fewer mechanical parts and a lower cuff pressure could theoretically improve longevity of the AUS (Figure 3). This modified method of AUS placement has been described in patients with sphincteric deficiency and spinal cord injury.16
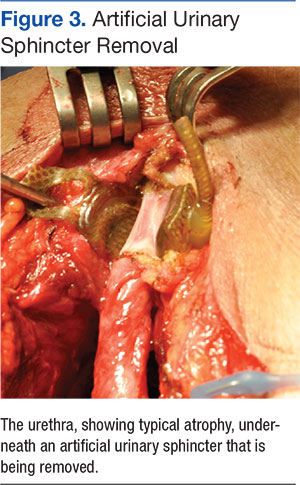
These 2 reconstructive surgeries freed the patient from indwelling catheter dependence and significantly improved his incontinence and QOL. Many patients with spinal cord injury or pelvic injury could benefit from similar reconstructive surgeries if conservative measures such as anticholinergic medications or onabotulinum toxin A treatments do not control incontinence.
Conclusion
Blast injuries in soldiers often cause pelvic and genitourinary injuries. These injuries can lead to chronic urinary problems and profound social and physical disability. These young veterans need innovative, individualized approaches to best manage their long-term urinary issues. Reconstructive surgery may improve QOL and decrease disability from bladder dysfunction for carefully selected patients. Clinicians caring for veterans with pelvic and genitourinary injury should strive to create a system where these options are available when they are appropriate.
1. U.S. Department of Defense. U.S. Casualty Status. U.S. Department of Defense Website. http://www.defense.gov/casualty.pdf. Updated November 3, 2015. Accessed November 4, 2015.
2. Schoenfeld AJ, Dunn JC, Bader JO, Belmont PJ Jr. The nature and extent of war injuries sustained by combat specialty personnel killed and wounded in Afghanistan and Iraq, 2003-2011. J Trauma Acute Care Surg. 2013;75(2):287-291.
3. Pannell D, Brisebois R, Talbot M, et al. Causes of death in Canadian Forces members deployed to Afghanistan and implications on tactical combat casualty care provision. J Trauma. 2011;71(5)(suppl 1):S401-S407.
4. Sutton MA, Hinson JL, Nickell KG, Boone TB. Continent ileocecal augmentation cystoplasty. Spinal Cord. 1998;36(4):246-251.
5. Weld KJ, Wall BM, Mangold TA, Steere EL, Dmochowski RR. Influences on renal function in chronic spinal cord injured patients. J Urol. 2000;164(5):1490-1493.
6. Cameron AP, Wallner LP, Tate DG, Sarma AV, Rodriguez GM, Clemens JQ. Bladder management after spinal cord injury in the United States 1972 to 2005. J Urol. 2010;184(1):213-217.
7. Cruz F, Herschorn S, Aliotta P, et al. Efficacy and safety of onabotulinumtoxinA in patients with urinary incontinence due to neurogenic detrusor overactivity: a randomised, double-blind, placebo-controlled trial. Eur Urol. 2011;60(4):742-750.
8. Ginsberg D, Gousse A, Keppenne V, et al. Phase 3 efficacy and tolerability study of onabotulinumtoxinA for urinary incontinence from neurogenic detrusor overactivity. J Urol. 2012;187(6):2131-2139.
9. Sarosdy MF. Continent urinary diversion using cutaneous ileocecocystoplasty. Urology. 1992;40(2):102-106.
10. King DH, Hlavinka TC, Sarosdy MF. Additional experience with continent urinary diversion using cutaneous ileocecocystoplasty. Urology. 1996;47(4):471-475.
11. Khavari R, Fletcher SG, Liu J, Boone TB. A modification to augmentation cystoplasty with catheterizable stoma for neurogenic patients: technique and long-term results. Urology. 2012;80(2):460-464.
12. Fung B, Kessler TM, Haeni K, Burkhard FC, Studer UE. Bowel function remains subjectively unchanged after ileal resection for construction of continent ileal reservoirs. Eur Urol. 2011;60(3):585-590.
13. Adams RC, Vachha B, Samuelson ML, Keefover-Hicks A, Snodgrass WT. Incidence of new onset metabolic acidosis following enteroplasty for myelomeningocele. J Urol. 2010;183(1):302-305.
14. Hensle TW, Gilbert SM. A review of metabolic consequences and long-term complications of enterocystoplasty in children. Curr Urol Rep. 2007;8(2):157-162.
15. Pannek J, Haupt G, Schulze H, Senge T. Influence of continent ileal urinary diversion on vitamin B12 absorption. J Urol. 1996;155(4):1206-1208.
16. Bersch U, Göcking K, Pannek J. The artificial urinary sphincter in patients with spinal cord lesion: description of a modified technique and clinical results. Eur Urol. 2009;55(3):687-693.
More than 52,000 soldiers have been injured and 6,800 have been killed during the wars in Iraq and Afghanistan.1 Blast injuries from improvised explosive devices (IEDs) account for 70% to 79% of combat-related injuries and deaths in these wars.2 Advances in personal body armor, rapid and advanced surgical treatment, and the changing nature of combat in Iraq and Afghanistan have changed injury patterns and survival compared with prior military conflicts such as those in Vietnam and Korea.3
The most common combat-related injuries in the recent wars are extremity, facial, brain, and gastrointestinal injuries. Pelvic and genitourinary injuries are also common, accounting for about 8% of total injuries.2 Pelvic and genitourinary injury can cause long-term disability from nerve injury (neurogenic bladder, neurogenic bowel, sexual dysfunction, urethral injury), as well as general loss of genital structures from blast injuries.
The usual care for bladder dysfunction from pelvic or genitourinary injury ranges from the use of chronic indwelling catheters to reconstructive surgery. However, there is no standard of care for long-term treatment of patients with pelvic or genitourinary injury who experience bladder dysfunction. Reconstructive surgery has the potential to improve quality of life (QOL) and eliminate chronic indwelling catheters, which are prone to cause infection and long-term kidney problems in patients with bladder dysfunction from traumatic injury.
This case report evaluates the efficacy of reconstructive surgery for bladder dysfunction to improve independence and QOL and decrease complications associated with chronic indwelling urinary catheters. The authors hope to raise awareness regarding this option for patients with pelvic, spinal cord, or genitourinary injury who are young and face long-term disability from their injuries.
Case Presentation
A 22-year-old man presented to the George E. Wahlen VAMC Urology Clinic in Salt Lake City, Utah with a complicated history related to combat injuries. During combat operations 3 years earlier, he was injured by an IED blast while on foot patrol. His injuries included bilateral severe extremity injury, perineal and genital blast wounds, a bladder injury, pelvic fracture, colorectal injury, and extensive soft tissue loss. He underwent multiple abdominal explorations, left leg amputation below the knee, multiple skin grafts, soft tissue debridements, left-side orchiectomy, bladder repair, and diverting colostomy. He survived the injuries and was eventually discharged from active military service and returned home.
Upon presentation to the VAMC, the patient had a diverting colostomy, suprapubic bladder catheter, and bladder and bowel function consistent with cauda equina syndrome (pelvic nerve injury). Given the lack of rectal tone, fecal incontinence was likely with colostomy reversal. His bladder had low volume and poor compliance (elasticity). In addition, the patient had no volitional control of urination or defecation.
The patient previously performed intermittent self-catheterization but experienced total urinary incontinence (UI) between catheterizations, due to his bladder dynamics and a lack of urinary sphincter tone. A suprapubic bladder catheter was previously placed to control UI. However, the patient remained incontinent, and urinary leakage, need for diapers, and urinary tract infections (UTIs) negatively impacted QOL. The patient ambulated well and was physically active. His priority was to reduce incontinence and improve QOL.
Catheterizable Ileal Cecocystoplasty
The patient underwent cutaneous catheterizable ileal cecocystoplasty (CCIC) (Figure 1). In this surgery, a segment of the cecum and ascending colon with attached terminal ileum is used to increase the size of the bladder (augmentation cystoplasty) and create a channel for catheterization from the umbilicus. The cecum and colon are detubularized, and a large rectangular plate of large bowel is formed, which is then sewn to the bladder, expanding its volume. About 10 to 15 cm of the terminal ileum is tapered to the diameter of a pencil and brought through the base of the umbilicus, creating a small stoma for intermittent bladder catheterization. The ileocecal valve is tightened and serves as a continence mechanism to prevent urinary leakage through the small stoma in the umbilicus.4
A perineal urethral mesh sling was placed at the time of the patient’s surgery to bolster the deinnervated urinary sphincter and prevent urethral leakage. The goal of reconstructive surgery for this patient was to create a small bowel channel connecting the umbilicus and bladder that could be catheterized every 4 to 6 hours, increase bladder capacity, and increase sphincteric resistance to reduce urethral leakage through the penis. Because there can be damage from passing a catheter through mesh slings and the urethra over time, including stenosis or erosion of the sling, an alternative catheterizable channel was needed in this patient.
The patient recovered after the surgery and was able to self-catheterize without difficulty. However, the urethral mesh sling did not place enough pressure on the urethra to prevent leakage, and he had persistent incontinence from the penis. Three months after the original surgery the patient had exploration of the perineum, which revealed that the mesh sling was loose and exerting inadequate pressure on the urethra. It was likely the sling slipped postoperatively—a known complication of urethral slings. An artificial urinary sphincter (AUS) was placed around the urethra during the second surgery to address the patient’s UI.
More than 1 year after the original surgery, the patient self-catheterizes about 4 to 5 times daily via the catheterizable channel using a single-use catheter. His bladder holds at least 500 mL. The patient does not have significant leakage from the channel or the penis. He is no longer dependent on a chronic indwelling catheter and is free of the problems associated with severe UI, including foul odor, UTIs, and social isolation.
Discussion
Patients with spinal cord or pelvic nerve injury often develop spastic bladders with low capacities. This is similar to muscle spasticity that may occur with a neurologic injury, below the level of the injury, such as in the lower extremities. The powerful uncontrolled bladder spasms and small bladder capacity most often lead to incontinence. Additionally, neurologic control of the urinary sphincter is affected, leading to either uncontrolled spasms or poor tone. Patients with these injuries have no volitional control of bladder functions and are forced to catheterize intermittently, use a condom-type catheter, or have a chronic indwelling catheter (a Foley catheter or suprapubic catheter).
Intermittent catheterization is the preferred management option for neurogenic bladder. When compared with chronic indwelling catheters, intermittent catheterization is associated with lower rates of UTI and upper tract abnormalities and with the loss of renal function.5 Unfortunately, patients do not often stay on intermittent catheterization. A recent study showed that up to 70% of patients with spinal cord injuries who used clean intermittent catheterization when discharged from acute rehabilitation discontinue use and are subsequently managed by chronic indwelling catheters.6 Although the reasons why intermittent catheterization is discontinued are unclear, patient dissatisfaction with catheterization, anatomic problems, such as urethral scarring, or continued leakage despite medical treatments, such as anticholinergic medicines, may be factors.
Uncontrolled leakage and UI significantly impacts QOL and may cause patients to choose chronic indwelling catheters over intermittent catheterization. Several treatments are available to control incontinence associated with intermittent catheterization. Anticholinergic medications and more recently onabotulinum toxin A may help improve bladder spasticity. In 2011, the FDA approved onabotulinum toxin A for transurethral bladder injections. It has been shown to increase functional bladder capacity and decrease spasticity.7,8 Onabotulinum toxin A treatment will not enlarge a small, contracted bladder.
Onabotulinum toxin A treatment would not be ideal for the patient in this case study. His absolute bladder capacity was 200 mL, and onabotulinum toxin A treatment would not significantly improve capacity or make intermittent catheterization practical. Additionally, the patient had poor urinary sphincter function, and he would continue to leak regardless of improvements in the bladder spasticity or tone.
Augmentation enterocystoplasty is surgical enlargement of the bladder, using a piece of the bowel and is indicated in patients with low bladder volumes. With this procedure the native bladder becomes defunctionalized, and patients experience a dramatic improvement in bladder volumes and a reduction in bladder spasms and leakage. The use of the colon and terminal ileum for bladder augmentation, or CCIC, was first reported by Sarosdy in 2 patients in 1992.9 In 1996, King and colleagues demonstrated successful outcomes with CCIC in a cohort of 8 patients after 34 months of follow-up.10 Seven patients successfully used clean intermittent catheterization, and 1 patient chose an indwelling catheter because of progressive upper extremity weakness. No patients experienced worsened renal function or pyelonephritis suggestive of upper urinary tract deterioration. A single patient had mild stomal stenosis, which was successfully revised under local anesthesia.
In another study, Sutton and colleagues reported at 27 months an improvement of 276 mL in bladder capacity, no metabolic complications, and a 95% continence rate in a cohort of 23 patients with neurogenic bladder who underwent CCIC.4 Sutton and colleagues later reported outcomes for 34 patients with a median of 31 months follow-up.11 The most common complications were recurrent UTIs (12%) and stomal stenosis (12%). Only 3 patients (9%) required surgical revisions for stomal stenosis.
Altered bowel function and metabolic abnormalities are a concern after bowel resection and reconstruction. However, a study has found no subjective change in bowel function following ileal resection of up to 60 cm for urinary diversion for bladder malignancy.12 Rates of hyperchloremic hypokalemic metabolic acidosis are low, and most changes in electrolytes are subclinical.13,14 Long-term vitamin B12 deficiency is seen with larger (> 50 cm) ileal resections but is rare with CCIC, given the small segment used for reconstruction.15 Overall, CCIC is shown to have excellent surgical outcomes in carefully selected patients with neurogenic bladder.
In addition to low bladder capacity, the case study patient also had intrinsic sphincteric deficiency (very low urinary sphincter tone), which is common with pelvic nerve injury but unusual with spinal cord injury. He initially received a suburethral mesh sling that supported and compressed the urethra and buttressed the natural urinary sphincter. However, patients can develop catheterization issues with a suburethral sling due to mechanical compression of the urethra and traversing the compressed area with a urinary catheter. Given the indication for augmentation cystoplasty in this patient, he additionally elected to undergo catheterization channel creation to avoid long-term issues of urethral catheterization through the urethra compressed by the sling.
Unfortunately, this patient had postoperative issues with his suburethral sling, and a modified AUS was inserted rather than a second sling. Normally, an AUS is attached to a pump mechanism in the scrotum. The pump allows the patient to cycle fluid from the sphincter cuff to a reservoir in the abdomen, removing compression on the urethra and allowing normal urination. Because this patient could not effectively urinate from the penis, the authors wanted to obstruct the urethra to prevent leakage without closing it permanently. The AUS was connected to a tissue expander port placed subcutaneously in the lower abdomen rather than to a pump mechanism. This modified approach used fewer mechanical parts compared with the pump mechanism, possibly reducing rates of mechanical failure. Additionally, a lower cuff pressure could be used to obstruct the urethra and prevent leakage, reducing the likelihood of urethral atrophy. Fewer mechanical parts and a lower cuff pressure could theoretically improve longevity of the AUS (Figure 3). This modified method of AUS placement has been described in patients with sphincteric deficiency and spinal cord injury.16
These 2 reconstructive surgeries freed the patient from indwelling catheter dependence and significantly improved his incontinence and QOL. Many patients with spinal cord injury or pelvic injury could benefit from similar reconstructive surgeries if conservative measures such as anticholinergic medications or onabotulinum toxin A treatments do not control incontinence.
Conclusion
Blast injuries in soldiers often cause pelvic and genitourinary injuries. These injuries can lead to chronic urinary problems and profound social and physical disability. These young veterans need innovative, individualized approaches to best manage their long-term urinary issues. Reconstructive surgery may improve QOL and decrease disability from bladder dysfunction for carefully selected patients. Clinicians caring for veterans with pelvic and genitourinary injury should strive to create a system where these options are available when they are appropriate.
More than 52,000 soldiers have been injured and 6,800 have been killed during the wars in Iraq and Afghanistan.1 Blast injuries from improvised explosive devices (IEDs) account for 70% to 79% of combat-related injuries and deaths in these wars.2 Advances in personal body armor, rapid and advanced surgical treatment, and the changing nature of combat in Iraq and Afghanistan have changed injury patterns and survival compared with prior military conflicts such as those in Vietnam and Korea.3
The most common combat-related injuries in the recent wars are extremity, facial, brain, and gastrointestinal injuries. Pelvic and genitourinary injuries are also common, accounting for about 8% of total injuries.2 Pelvic and genitourinary injury can cause long-term disability from nerve injury (neurogenic bladder, neurogenic bowel, sexual dysfunction, urethral injury), as well as general loss of genital structures from blast injuries.
The usual care for bladder dysfunction from pelvic or genitourinary injury ranges from the use of chronic indwelling catheters to reconstructive surgery. However, there is no standard of care for long-term treatment of patients with pelvic or genitourinary injury who experience bladder dysfunction. Reconstructive surgery has the potential to improve quality of life (QOL) and eliminate chronic indwelling catheters, which are prone to cause infection and long-term kidney problems in patients with bladder dysfunction from traumatic injury.
This case report evaluates the efficacy of reconstructive surgery for bladder dysfunction to improve independence and QOL and decrease complications associated with chronic indwelling urinary catheters. The authors hope to raise awareness regarding this option for patients with pelvic, spinal cord, or genitourinary injury who are young and face long-term disability from their injuries.
Case Presentation
A 22-year-old man presented to the George E. Wahlen VAMC Urology Clinic in Salt Lake City, Utah with a complicated history related to combat injuries. During combat operations 3 years earlier, he was injured by an IED blast while on foot patrol. His injuries included bilateral severe extremity injury, perineal and genital blast wounds, a bladder injury, pelvic fracture, colorectal injury, and extensive soft tissue loss. He underwent multiple abdominal explorations, left leg amputation below the knee, multiple skin grafts, soft tissue debridements, left-side orchiectomy, bladder repair, and diverting colostomy. He survived the injuries and was eventually discharged from active military service and returned home.
Upon presentation to the VAMC, the patient had a diverting colostomy, suprapubic bladder catheter, and bladder and bowel function consistent with cauda equina syndrome (pelvic nerve injury). Given the lack of rectal tone, fecal incontinence was likely with colostomy reversal. His bladder had low volume and poor compliance (elasticity). In addition, the patient had no volitional control of urination or defecation.
The patient previously performed intermittent self-catheterization but experienced total urinary incontinence (UI) between catheterizations, due to his bladder dynamics and a lack of urinary sphincter tone. A suprapubic bladder catheter was previously placed to control UI. However, the patient remained incontinent, and urinary leakage, need for diapers, and urinary tract infections (UTIs) negatively impacted QOL. The patient ambulated well and was physically active. His priority was to reduce incontinence and improve QOL.
Catheterizable Ileal Cecocystoplasty
The patient underwent cutaneous catheterizable ileal cecocystoplasty (CCIC) (Figure 1). In this surgery, a segment of the cecum and ascending colon with attached terminal ileum is used to increase the size of the bladder (augmentation cystoplasty) and create a channel for catheterization from the umbilicus. The cecum and colon are detubularized, and a large rectangular plate of large bowel is formed, which is then sewn to the bladder, expanding its volume. About 10 to 15 cm of the terminal ileum is tapered to the diameter of a pencil and brought through the base of the umbilicus, creating a small stoma for intermittent bladder catheterization. The ileocecal valve is tightened and serves as a continence mechanism to prevent urinary leakage through the small stoma in the umbilicus.4
A perineal urethral mesh sling was placed at the time of the patient’s surgery to bolster the deinnervated urinary sphincter and prevent urethral leakage. The goal of reconstructive surgery for this patient was to create a small bowel channel connecting the umbilicus and bladder that could be catheterized every 4 to 6 hours, increase bladder capacity, and increase sphincteric resistance to reduce urethral leakage through the penis. Because there can be damage from passing a catheter through mesh slings and the urethra over time, including stenosis or erosion of the sling, an alternative catheterizable channel was needed in this patient.
The patient recovered after the surgery and was able to self-catheterize without difficulty. However, the urethral mesh sling did not place enough pressure on the urethra to prevent leakage, and he had persistent incontinence from the penis. Three months after the original surgery the patient had exploration of the perineum, which revealed that the mesh sling was loose and exerting inadequate pressure on the urethra. It was likely the sling slipped postoperatively—a known complication of urethral slings. An artificial urinary sphincter (AUS) was placed around the urethra during the second surgery to address the patient’s UI.
More than 1 year after the original surgery, the patient self-catheterizes about 4 to 5 times daily via the catheterizable channel using a single-use catheter. His bladder holds at least 500 mL. The patient does not have significant leakage from the channel or the penis. He is no longer dependent on a chronic indwelling catheter and is free of the problems associated with severe UI, including foul odor, UTIs, and social isolation.
Discussion
Patients with spinal cord or pelvic nerve injury often develop spastic bladders with low capacities. This is similar to muscle spasticity that may occur with a neurologic injury, below the level of the injury, such as in the lower extremities. The powerful uncontrolled bladder spasms and small bladder capacity most often lead to incontinence. Additionally, neurologic control of the urinary sphincter is affected, leading to either uncontrolled spasms or poor tone. Patients with these injuries have no volitional control of bladder functions and are forced to catheterize intermittently, use a condom-type catheter, or have a chronic indwelling catheter (a Foley catheter or suprapubic catheter).
Intermittent catheterization is the preferred management option for neurogenic bladder. When compared with chronic indwelling catheters, intermittent catheterization is associated with lower rates of UTI and upper tract abnormalities and with the loss of renal function.5 Unfortunately, patients do not often stay on intermittent catheterization. A recent study showed that up to 70% of patients with spinal cord injuries who used clean intermittent catheterization when discharged from acute rehabilitation discontinue use and are subsequently managed by chronic indwelling catheters.6 Although the reasons why intermittent catheterization is discontinued are unclear, patient dissatisfaction with catheterization, anatomic problems, such as urethral scarring, or continued leakage despite medical treatments, such as anticholinergic medicines, may be factors.
Uncontrolled leakage and UI significantly impacts QOL and may cause patients to choose chronic indwelling catheters over intermittent catheterization. Several treatments are available to control incontinence associated with intermittent catheterization. Anticholinergic medications and more recently onabotulinum toxin A may help improve bladder spasticity. In 2011, the FDA approved onabotulinum toxin A for transurethral bladder injections. It has been shown to increase functional bladder capacity and decrease spasticity.7,8 Onabotulinum toxin A treatment will not enlarge a small, contracted bladder.
Onabotulinum toxin A treatment would not be ideal for the patient in this case study. His absolute bladder capacity was 200 mL, and onabotulinum toxin A treatment would not significantly improve capacity or make intermittent catheterization practical. Additionally, the patient had poor urinary sphincter function, and he would continue to leak regardless of improvements in the bladder spasticity or tone.
Augmentation enterocystoplasty is surgical enlargement of the bladder, using a piece of the bowel and is indicated in patients with low bladder volumes. With this procedure the native bladder becomes defunctionalized, and patients experience a dramatic improvement in bladder volumes and a reduction in bladder spasms and leakage. The use of the colon and terminal ileum for bladder augmentation, or CCIC, was first reported by Sarosdy in 2 patients in 1992.9 In 1996, King and colleagues demonstrated successful outcomes with CCIC in a cohort of 8 patients after 34 months of follow-up.10 Seven patients successfully used clean intermittent catheterization, and 1 patient chose an indwelling catheter because of progressive upper extremity weakness. No patients experienced worsened renal function or pyelonephritis suggestive of upper urinary tract deterioration. A single patient had mild stomal stenosis, which was successfully revised under local anesthesia.
In another study, Sutton and colleagues reported at 27 months an improvement of 276 mL in bladder capacity, no metabolic complications, and a 95% continence rate in a cohort of 23 patients with neurogenic bladder who underwent CCIC.4 Sutton and colleagues later reported outcomes for 34 patients with a median of 31 months follow-up.11 The most common complications were recurrent UTIs (12%) and stomal stenosis (12%). Only 3 patients (9%) required surgical revisions for stomal stenosis.
Altered bowel function and metabolic abnormalities are a concern after bowel resection and reconstruction. However, a study has found no subjective change in bowel function following ileal resection of up to 60 cm for urinary diversion for bladder malignancy.12 Rates of hyperchloremic hypokalemic metabolic acidosis are low, and most changes in electrolytes are subclinical.13,14 Long-term vitamin B12 deficiency is seen with larger (> 50 cm) ileal resections but is rare with CCIC, given the small segment used for reconstruction.15 Overall, CCIC is shown to have excellent surgical outcomes in carefully selected patients with neurogenic bladder.
In addition to low bladder capacity, the case study patient also had intrinsic sphincteric deficiency (very low urinary sphincter tone), which is common with pelvic nerve injury but unusual with spinal cord injury. He initially received a suburethral mesh sling that supported and compressed the urethra and buttressed the natural urinary sphincter. However, patients can develop catheterization issues with a suburethral sling due to mechanical compression of the urethra and traversing the compressed area with a urinary catheter. Given the indication for augmentation cystoplasty in this patient, he additionally elected to undergo catheterization channel creation to avoid long-term issues of urethral catheterization through the urethra compressed by the sling.
Unfortunately, this patient had postoperative issues with his suburethral sling, and a modified AUS was inserted rather than a second sling. Normally, an AUS is attached to a pump mechanism in the scrotum. The pump allows the patient to cycle fluid from the sphincter cuff to a reservoir in the abdomen, removing compression on the urethra and allowing normal urination. Because this patient could not effectively urinate from the penis, the authors wanted to obstruct the urethra to prevent leakage without closing it permanently. The AUS was connected to a tissue expander port placed subcutaneously in the lower abdomen rather than to a pump mechanism. This modified approach used fewer mechanical parts compared with the pump mechanism, possibly reducing rates of mechanical failure. Additionally, a lower cuff pressure could be used to obstruct the urethra and prevent leakage, reducing the likelihood of urethral atrophy. Fewer mechanical parts and a lower cuff pressure could theoretically improve longevity of the AUS (Figure 3). This modified method of AUS placement has been described in patients with sphincteric deficiency and spinal cord injury.16
These 2 reconstructive surgeries freed the patient from indwelling catheter dependence and significantly improved his incontinence and QOL. Many patients with spinal cord injury or pelvic injury could benefit from similar reconstructive surgeries if conservative measures such as anticholinergic medications or onabotulinum toxin A treatments do not control incontinence.
Conclusion
Blast injuries in soldiers often cause pelvic and genitourinary injuries. These injuries can lead to chronic urinary problems and profound social and physical disability. These young veterans need innovative, individualized approaches to best manage their long-term urinary issues. Reconstructive surgery may improve QOL and decrease disability from bladder dysfunction for carefully selected patients. Clinicians caring for veterans with pelvic and genitourinary injury should strive to create a system where these options are available when they are appropriate.
1. U.S. Department of Defense. U.S. Casualty Status. U.S. Department of Defense Website. http://www.defense.gov/casualty.pdf. Updated November 3, 2015. Accessed November 4, 2015.
2. Schoenfeld AJ, Dunn JC, Bader JO, Belmont PJ Jr. The nature and extent of war injuries sustained by combat specialty personnel killed and wounded in Afghanistan and Iraq, 2003-2011. J Trauma Acute Care Surg. 2013;75(2):287-291.
3. Pannell D, Brisebois R, Talbot M, et al. Causes of death in Canadian Forces members deployed to Afghanistan and implications on tactical combat casualty care provision. J Trauma. 2011;71(5)(suppl 1):S401-S407.
4. Sutton MA, Hinson JL, Nickell KG, Boone TB. Continent ileocecal augmentation cystoplasty. Spinal Cord. 1998;36(4):246-251.
5. Weld KJ, Wall BM, Mangold TA, Steere EL, Dmochowski RR. Influences on renal function in chronic spinal cord injured patients. J Urol. 2000;164(5):1490-1493.
6. Cameron AP, Wallner LP, Tate DG, Sarma AV, Rodriguez GM, Clemens JQ. Bladder management after spinal cord injury in the United States 1972 to 2005. J Urol. 2010;184(1):213-217.
7. Cruz F, Herschorn S, Aliotta P, et al. Efficacy and safety of onabotulinumtoxinA in patients with urinary incontinence due to neurogenic detrusor overactivity: a randomised, double-blind, placebo-controlled trial. Eur Urol. 2011;60(4):742-750.
8. Ginsberg D, Gousse A, Keppenne V, et al. Phase 3 efficacy and tolerability study of onabotulinumtoxinA for urinary incontinence from neurogenic detrusor overactivity. J Urol. 2012;187(6):2131-2139.
9. Sarosdy MF. Continent urinary diversion using cutaneous ileocecocystoplasty. Urology. 1992;40(2):102-106.
10. King DH, Hlavinka TC, Sarosdy MF. Additional experience with continent urinary diversion using cutaneous ileocecocystoplasty. Urology. 1996;47(4):471-475.
11. Khavari R, Fletcher SG, Liu J, Boone TB. A modification to augmentation cystoplasty with catheterizable stoma for neurogenic patients: technique and long-term results. Urology. 2012;80(2):460-464.
12. Fung B, Kessler TM, Haeni K, Burkhard FC, Studer UE. Bowel function remains subjectively unchanged after ileal resection for construction of continent ileal reservoirs. Eur Urol. 2011;60(3):585-590.
13. Adams RC, Vachha B, Samuelson ML, Keefover-Hicks A, Snodgrass WT. Incidence of new onset metabolic acidosis following enteroplasty for myelomeningocele. J Urol. 2010;183(1):302-305.
14. Hensle TW, Gilbert SM. A review of metabolic consequences and long-term complications of enterocystoplasty in children. Curr Urol Rep. 2007;8(2):157-162.
15. Pannek J, Haupt G, Schulze H, Senge T. Influence of continent ileal urinary diversion on vitamin B12 absorption. J Urol. 1996;155(4):1206-1208.
16. Bersch U, Göcking K, Pannek J. The artificial urinary sphincter in patients with spinal cord lesion: description of a modified technique and clinical results. Eur Urol. 2009;55(3):687-693.
1. U.S. Department of Defense. U.S. Casualty Status. U.S. Department of Defense Website. http://www.defense.gov/casualty.pdf. Updated November 3, 2015. Accessed November 4, 2015.
2. Schoenfeld AJ, Dunn JC, Bader JO, Belmont PJ Jr. The nature and extent of war injuries sustained by combat specialty personnel killed and wounded in Afghanistan and Iraq, 2003-2011. J Trauma Acute Care Surg. 2013;75(2):287-291.
3. Pannell D, Brisebois R, Talbot M, et al. Causes of death in Canadian Forces members deployed to Afghanistan and implications on tactical combat casualty care provision. J Trauma. 2011;71(5)(suppl 1):S401-S407.
4. Sutton MA, Hinson JL, Nickell KG, Boone TB. Continent ileocecal augmentation cystoplasty. Spinal Cord. 1998;36(4):246-251.
5. Weld KJ, Wall BM, Mangold TA, Steere EL, Dmochowski RR. Influences on renal function in chronic spinal cord injured patients. J Urol. 2000;164(5):1490-1493.
6. Cameron AP, Wallner LP, Tate DG, Sarma AV, Rodriguez GM, Clemens JQ. Bladder management after spinal cord injury in the United States 1972 to 2005. J Urol. 2010;184(1):213-217.
7. Cruz F, Herschorn S, Aliotta P, et al. Efficacy and safety of onabotulinumtoxinA in patients with urinary incontinence due to neurogenic detrusor overactivity: a randomised, double-blind, placebo-controlled trial. Eur Urol. 2011;60(4):742-750.
8. Ginsberg D, Gousse A, Keppenne V, et al. Phase 3 efficacy and tolerability study of onabotulinumtoxinA for urinary incontinence from neurogenic detrusor overactivity. J Urol. 2012;187(6):2131-2139.
9. Sarosdy MF. Continent urinary diversion using cutaneous ileocecocystoplasty. Urology. 1992;40(2):102-106.
10. King DH, Hlavinka TC, Sarosdy MF. Additional experience with continent urinary diversion using cutaneous ileocecocystoplasty. Urology. 1996;47(4):471-475.
11. Khavari R, Fletcher SG, Liu J, Boone TB. A modification to augmentation cystoplasty with catheterizable stoma for neurogenic patients: technique and long-term results. Urology. 2012;80(2):460-464.
12. Fung B, Kessler TM, Haeni K, Burkhard FC, Studer UE. Bowel function remains subjectively unchanged after ileal resection for construction of continent ileal reservoirs. Eur Urol. 2011;60(3):585-590.
13. Adams RC, Vachha B, Samuelson ML, Keefover-Hicks A, Snodgrass WT. Incidence of new onset metabolic acidosis following enteroplasty for myelomeningocele. J Urol. 2010;183(1):302-305.
14. Hensle TW, Gilbert SM. A review of metabolic consequences and long-term complications of enterocystoplasty in children. Curr Urol Rep. 2007;8(2):157-162.
15. Pannek J, Haupt G, Schulze H, Senge T. Influence of continent ileal urinary diversion on vitamin B12 absorption. J Urol. 1996;155(4):1206-1208.
16. Bersch U, Göcking K, Pannek J. The artificial urinary sphincter in patients with spinal cord lesion: description of a modified technique and clinical results. Eur Urol. 2009;55(3):687-693.
Personalized Health Planning in Primary Care Settings
Health care has become increasingly unaffordable in the U.S., yet it remains ineffective in preventing or effectively treating chronic diseases.1,2 Given the increasing burden of chronic disease on the American health care system, there is an effort to shift the practice of medicine away from its reactive, disease-oriented approach to a more sustainable proactive model.3-5
Personalized health care (PHC) is an approach to the practice of medicine where prediction, prevention, intense patient engagement, shared health care decision making, and coordination of care are essential to cost effectively facilitate better outcomes.3,5-7 Greater collaboration between patient and clinician replaces the traditional clinician-dominated dialogue with more effective patient-clinician partnerships.8,9 Patients’ knowledge, skills, and confidence to manage their health care have been linked to improved health outcomes, lower costs, and greater satisfaction with health care experiences.10,11
Personalized health care has been proposed as a means to achieve better patient engagement as part of an aligned, proactive clinical approach. At the heart of PHC is personalized health planning, wherein the patient and clinician develop shared health-related goals and a plan to achieve them.3
The VHA, the largest integrated health care system in the country, is on the vanguard of incorporating tenets of PHC into its delivery model. In 2011, the Office of Patient Centered Care and Cultural Transformation (OPCC&CT) was founded to “oversee the VHA’s cultural transformation to patient-centered care.”12 This undertaking represents “one of the most massive changes in the philosophy and process for health care delivery ever undertaken by an organized health care system.”13
The primary goal of the VHA’s strategic plan for 2013 to 2018 is to provide veterans personalized, proactive, patient-driven health care.14 The intention of this approach is to engage and inspire veterans to their highest possible level of health and well-being. A personalized approach requires a dynamic customization of care that is specifically relevant to the individual, based on factors such as medical conditions, genome, needs, values, and circumstances. In addition to being personalized, this approach must be proactive, and therefore, preventive and include strategies to strengthen the person’s innate capacity for enhancing health.
The third distinction of this new model health care is that it is patient-driven, rooted in and driven by that which matters most to people in their lives and aligns their health care with their day-to-day and long-term life goals.15 The latter may be the most critical of the 3 tenets, because a personalized, proactive approach that is not driven by an engaged and inspired individual will be unlikely to achieve adherence, let alone the highest level of health and well-being.
The VHA is uniquely positioned to optimize health and well-being for veterans due in part to a systemwide emphasis on training providers to promote and support behavior change through approaches including health coaching and motivational interviewing. These synergistic approaches used widely by clinicians throughout the VHA are influenced by the transtheoretical model (ie, the stages of change theory), which considers patients holistically and helps them identify intrinsic motivation to improve their health behaviors.16,17 The transformation occurring in the VHA is intended to shift the current disease-centric medical model to an approach that optimizes the health of veterans through patient-clinician engagement, health risk assessment (HRA), shared health goal creation, and a coordinated plan to attain them.12,13
Personalized Health Planning
In recognition of the need to deliver care that emphasizes prevention and coordination, the patient-centered medical home, patient-aligned care teams (PACTs), and the chronic care model were developed. All of these embrace concepts of patient engagement, shared decision making, and team-based care. However, none of these approaches have outlined a clinical workflow that systematically and proactively operationalizes these concepts with the creation of a risk-based personalized care approach. Personalized health planning provides a clinical workflow that operationalizes all these features (Figure 1).
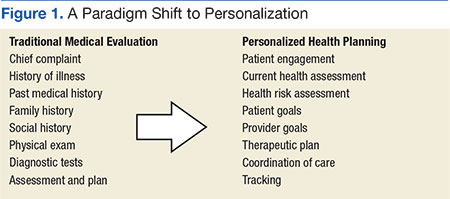
Of central importance is the creation of a personalized health plan (PHP), which the patient and clinician develop collaboratively. The plan serves to organize and coordinate care while engaging the patient in the process of care delivery and appropriate self-management of health.3,5 This approach promotes personalized and proactive care that values the individual and fosters meaningful patient self-awareness and engagement through shared decision making.7
The personalized health planning process is composed of several key components (Figure 2). 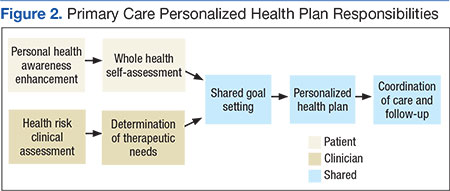
With this information, the clinician develops a preliminary therapeutic plan to meet these goals and discusses this plan with the patient. The next component is the synthesis of the clinician’s goals and/or treatment plan with the priorities of the patient to establish shared clinician-patient goals. This is followed by the establishment of the PHP, which consists of the agreed upon shared clinician and patient goals, a therapeutic and wellness plan to meet them, metrics for tracking progress, consults and referrals, and a time frame for the patient to achieve the health goals.
Related: The Right Care at the Right Time and in the Right Place: The Role of Technology in the VHA
The final component is coordination of care and a formalized follow-up system in which the health care team monitors the patient’s progress and provides support by revisiting or updating the PHP at intervals determined by the provider, based on the level of monitoring required by the patient’s health status. This approach invites the patient to become an empowered member of the care team by creating a patient-clinician partnership and providing a model for delivering personalized, proactive, patient-driven care to individuals with a diverse range of needs.4,5,18
Design and Implementation
This project qualified as exempt through the Duke University Institutional Review Board. The primary aim of this pilot was to examine the feasibility of implementing personalized health planning into primary care settings and to develop a workable process that is scalable and customizable to inpatient and outpatient clinics of varying sizes for different patient populations within the VHA. The pilot included 5 clinics in 2 geographic areas that were selected for their facility’s leadership support and desire to participate.
The VA Boston Healthcare System implemented personalized health planning at 3 primary care clinics: Jamaica Plain Primary Care, Jamaica Plain Women’s Health, and Quincy Primary Care. Three distinct PACTs participated in Boston, each composed of a medical doctor or doctor of osteopathy, a registered nurse (RN) or health technician, and a medical support assistant. The Sam Rayburn Memorial Veterans Center in Bonham, Texas, implemented personalized health planning at 2 clinics: the Hypertension Shared Medical Appointment and the Domiciliary Inpatient Primary Care. At Bonham, a medical doctor, pharmacist, RN, and an integrated mental health provider led the shared medical appointment (SMA) with guest presenters for individual appointments, depending on the topic covered. A RN and social worker implemented personalized health planning in the domiciliary.
After receiving training in the personalized health planning process, each of the clinics’ multidisciplinary PACTs incorporated their custom personalized health planning workflow into patient encounters. During the intake process of the clinic visit, the patient received a Personal Health Inventory (PHI) to determine health care priorities. The OPCC&CT developed the PHI as a whole-health self-assessment tool to help patients reflect on their health and lives, including core values, disparities between current and desired states, and preparedness to make behavioral changes to promote health.
The PHI assisted with framing this whole health approach to clinical care by expanding the definition of health to include more holistic elements of well-being, such as spirituality, personal relationships, emotional health, and personal development. A visual representation of the whole health domains termed The Circle of Health introduced this concept to the patient and assisted with goal setting (Figure 3).12
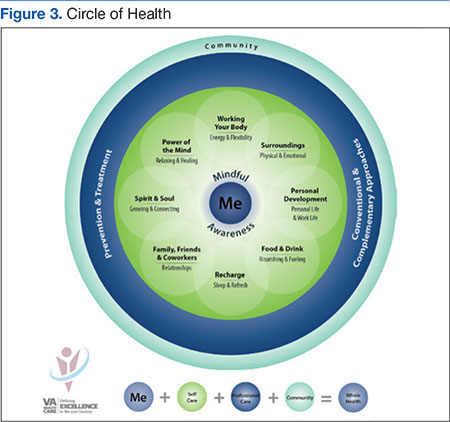
The PHI organized the patient’s input and was provided to the clinician to contribute to the development of the shared therapeutic goals and a final treatment plan. The PHP provided the tool to organize the goals, plans, and care following the visit and to connect the patient to additional resources within the VHA to support goal attainment through skill building and support. Each clinical site developed a mechanism to follow up with these patients either telephonically or with additional clinic appointments. The participating clinics implemented their customized version of personalized health planning for an average of 3 months.
Personalized health planning and accompanying tools were used primarily in routine ambulatory care visits. They were also used in the Bonham domiciliary clinic, which provides care for veterans with mental illnesses or addictive disorders who require additional structure and support. The purpose of the pilot was to determine whether personalized health planning could be used within this population. Given the small sample size and incompleteness of data collected, the Bonham domiciliary group was not included in the participating patient total. Across the other 4 clinics a total of 153 patients participated in the 3-month pilot study by establishing shared health goals and plans to meet them.
Results and Evaluation
Using a structured interview guide, a total of 6 small group interviews with participating clinicians were held (3 in Boston and 3 in Bonham, N = 18). Qualitative methods for research and evaluation were used to capture the depth of responses and to provide complex descriptions of the clinicians’ perspectives on the implementation of the personalized health planning process. Two researchers reviewed the transcripts to identify and code themes, and a third researcher reviewed the transcripts to confirm themes and resolve any discrepancies.
Analysis of the interview data revealed 9 core themes related to the feasibility, effectiveness, and future dissemination of personalized health planning. These themes, described below with exemplar data, include (a) patient engagement; (b) clinical assessment; (c) goal setting; (d) clinical workflow; (e) resources and support for veterans and clinicians; (f) Computerized Patient Record System (CPRS) integration; (g) patient-clinician relationship; (h) clinical outcomes; and (i) patient satisfaction.
The purpose of this study was to evaluate the feasibility of introducing personalized health planning within the workflows of the clinics that were participating. As a consequence of this, interviews were held with clinic staff rather than patients. However, the authors did obtain patient satisfaction data from 10 patients who received care from the hypertension SMA and responded to TruthPoint questions after their visit (Table).
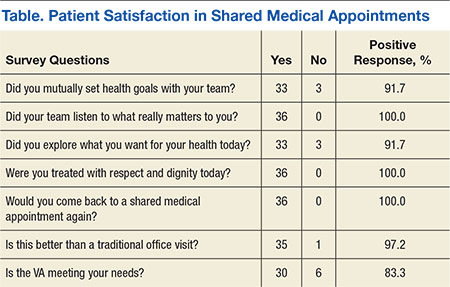
Patient Engagement
A central tenant of personalized health planning is to engage patients in their health and health care. Findings revealed that clinicians at both sites perceived the PHP as an effective tool for integrating patients as robust members of the care team. Clinicians noted that by asking patients what was important to them, the patients felt more empowered to actively engage in the clinical encounter and to take responsibility for their health decisions. One pharmacist noted, “Patients are more empowered…when you change how you’re having your conversation with them that helps people start to recognize that they are an active participant [sic] and they can have an impact and can help with minimizing medicines or trying other things.”
Clinicians reported that including patients as active members in their care created a level of buy-in that motivated behavioral changes, because the patients identified behaviors they wanted to change vs the clinician telling them what they should or should not do. A nurse manager reported, “The key is that (the health goal) is coming from the patient…. Once it comes out of their mouth, they’re thinking about it and it’s not the clinician telling them what they should or shouldn’t do, but it’s helping them…identify something that’s important that will keep them into staying the way they want to, for the reasons that they want to.”
Clinical Assessment
The HRA tools are a vital part of the personalized health planning process, as they focus on preventive strategies that are most important for the patient.4 Clinicians reported using the PHI and additional HRA tools as part of the pilot program. The PHI is a self-assessment tool designed to identify psychosocial, behavioral, and environmental issues that can impact the patient’s care and health status. Most clinicians found that the PHI helped to solicit patients’ input on what was important to them and their health status while introducing the new approach to care. One nurse commented, “I found [PHI] very effective if I could actually sit down and review it with them to see what it was that was truly important to them and explain that this new approach is for a better understanding.”
Clinicians also found that the PHI helped focus the patient’s attention toward self-care areas that facilitated the shared goal setting process with the clinician. It moved the clinical encounter away from the chief health problems and toward identifying what is important to the patient and leveraging his or her intrinsic motivation to support health promotion via lifestyle modification.
Goal Setting
Shared goal setting is a critical component of personalized health planning. The clinician and patient must agree about realistic goals to improve the patient’s health. Clinicians reported that the goal setting stage was most successful when patients were invited to guide the process and offered the goals themselves; ie, when it was not just patient-centered but patient-driven.
“Setting a goal with a patient is pretty easy because people have an idea of what they should be doing and what they want to be doing,” one clinician reported. “They know their goal. So it’s a matter of just listening, really listening, and seeing what they want…. It’s not incongruous to get the medical goal and the patient goal to match.”
Patients were amenable to this collaborative approach to goal setting, and there was often commonality between the clinician’s goals and what was important to the patient. Occasionally, the patient set goals in seemingly unrelated areas that facilitated chronic disease management.
“One of our hypertensive patients wanted to work on things that are external that they felt are stressors that actually caused their blood pressure to be high,” a pharmacist recalled. “At the end of the day they wanted to control their environment better so that they could see if they could then be off of antihypertensives altogether. It appears that may be the case right now. That this individual has been able to accomplish that, which I thought was amazing, and since it’s still new, I’m still a little bit skeptical.... Is that possible? But if at the end of the day that is an outcome that we see from doing this, I think that’s wonderful.”
Clinicians reported that follow-up with the patient was a critical aspect of goal setting, because it improved accountability and helped track progress and health outcomes. However, due to the 3-month time limit of the pilot, there was insufficient time to get uniform data on the formalized follow-up systems developed by each clinic.
Clinical Workflow
Examining the feasibility of creating a process to incorporate personalized health planning into a busy primary care clinic was one of the major aims of this pilot. As such, issues of time, staff responsibilities, and use of the CPRS and other systems to facilitate the process were challenges that each clinic addressed in slightly different ways and with varying success. One method was to leverage the SMA for a group of patients with a common diagnosis to discuss their goals, provide accountability, and improve access to a medical team.
Another innovative approach was utilizing medical support administrators and health technicians to front-load some of the introductory information and patient education in the waiting room or during the intake process. Despite these efforts, some clinicians thought that the personalized health planning process might take longer than the traditional clinical encounter. One nurse manager commented, “I think it did affect the length of visits…it has made them a little bit longer.”
Resources and Support
The VHA has been undergoing a shift from an emphasis on tertiary care to include a greater focus on primary care. Part of this shift has been an investment in complementary and alternative medicine (CAM). The PHP helped clinicians explore what resources existed at their facility to support veterans in accomplishing their goals, including CAM. One nurse reported, “It made us look into other avenues that were actually available at the VHA that we didn’t even know we had…the acupuncture, the qigong, the voluntary services getting the veterans involved.”
Clinicians also identified the need for patient education in the concepts of whole health, personalized care, and patient involvement as necessary for moving the piloted approach forward. A nurse noted that “for [personalized health planning] to work well, [the patients] need more orientation and education upfront systemwide so that when they get into an appointment with us, we’re not starting at explaining the whole world view of partnership and doing things differently.”
Resources for clinicians were just as critical as resources for patients in facilitating the personalized health planning process. Specifically, most clinicians identified their own education and training in techniques to engage the patient in a meaningful way via motivational interviewing and health coaching complemented the personalized approach to care, particularly for shared goal setting,
CPRS Integration
The CPRS is an integrated, electronic patient record system that provides a single interface for clinicians to manage patient care as well as an efficient means for others to access and use patient information.19 The most commonly cited challenge in the pilot was the lack of available staff and time in the patient visit to complete the PHP while completing documentation requirements in CPRS.
One clinician stated, “For clinicians, the barriers…it’s time to get through the reminders and preventatives.” Clinicians reported that the process and the CPRS documentation were misaligned and lacked integration to coordinate care or support health planning. Moreover, clinicians reported that the data being collected did not support patient-centered care.
Patient-Clinician Relationship
A significant strength of personalized health planning was that it fostered a beneficial patient-clinician relationship that promoted greater depth of care. One clinician noted, “I think that it adds a more personalized dimension to the whole patient visit.” In addition to experiencing a deeper relationship with their patients, clinicians also expressed having higher levels of job satisfaction and relished the opportunity to connect with their patients in a more personal way.
Clinicians also reported that patients seemed more satisfied with the experience. One nurse commented, “They really respond to it very well when they figure out that you care about them as a whole… it’s not just about the disease process anymore.”
Clinical Outcomes
Clinicians reported a number of positive health outcomes during the pilot. One physician reported, “I have a patient, he had a follow-up today, a 29-year-old veteran, who was 260 pounds 5 months ago, and he’s 230 pounds today. He comes in monthly to see the nurse to let us know he’s doing it.”
The same physician also shared a similar transformation in a patient as a result of personalized health planning. “We had another one yesterday, 4 months ago his [hemoglobin] A1c was 10.3%. It was 7.2% yesterday, and [his weight was] down 20 pounds.” In addition to positive clinical outcomes, patients made changes in areas of their health that they identified as important through the PHI, although these areas are not typically discussed in a clinical visit.
Patient Satisfaction
Although the overall goal of this pilot was to determine the feasibility of a clinical workflow embracing personalized health planning, data on patient satisfaction were collected from patients receiving care in the Hypertension Shared Medical Appointments Program at Bohnam. Ten patients were seen over the course of 5 visits. At each visit, they were asked to rate their satisfaction (Table).
Overall, patients were highly satisfied with their experience and the care they received: 91.7% reported exploring what they wanted for their health and setting shared goals; 100% reported that their providers truly listened to their needs and treated them with respect and dignity; and 97.2% reported that their experience was better than a traditional office visit. One participating physician noted that higher levels of patient and provider satisfaction are a product of this type of patient engagement. “I also think that looking at patient and provider satisfaction, the visits feel more meaningful, and there’s a better relationship built through this discussion,” he noted. These findings demonstrating increased satisfaction further suggest the benefits of personalized health planning approach.
Discussion
In 2012, the VHA National Leadership Council convened a Strategic Planning Summit to set goals and objectives to help the VHA be at the vanguard of a movement toward a more proactive health care delivery model. The first of 3 goals developed was to provide veterans personalized, proactive, patient-driven health care.13 It is becoming increasingly clear that truly affecting health and health outcomes requires motivated, engaged, and informed patients with a care delivery approach that provides ample opportunities for patient involvement and input in health care decision making.10,11
The OPCC&CT has ongoing initiatives driving innovation, research, education, and deployment across the system to set the stage for personalized, proactive, patient-driven care.20 Some of these innovations include clinician education in the concept of whole health; health coaching; group-based, peer-led approaches; and the expansion of CAM such as mind-body approaches, qi-gong, massage therapy, yoga, and acupuncture.21
The primary aim of the Whole Health in Primary Care Project was to determine the feasibility of using personalized health planning as the operational model to deliver personalized, proactive, patient-driven care. The decision was made to integrate personalized health planning into ongoing clinical operations rather than design clinical pilots de novo. This had the advantage of speed in starting the project but limited the ability to create an optimal workflow from scratch. Given the time and resources available for this study, it was not possible to obtain quantitative data particularly as it related to quantifying clinical outcomes.
Despite these limitations, early indications suggest that the personalized health planning process can serve as the operational clinical working model to enable personalized, proactive, patient-driven care in a variety of primary care settings. As noted by one nurse manager, preparing the personalized health plan made the initial visit “a bit longer.” However, after the first visit, monitoring health risk abatement and goal achievement is akin to what is currently done by reviewing problem lists. Thus, although the personalized health planning experience is just beginning, clinicians noted that it fostered a beneficial patient-clinician relationship. This deeper relationship between the patient and the clinician may be the most powerful signal that the process is worthwhile.
This pilot provided valuable information related to the implementation of a clinical workflow redesign, an initial step toward developing an optimized operational model of the PHP process. Additionally, although it is not yet possible to quantify the clinical impact of the personalized health planning, anecdotal evidence suggests its positive potential. Clinicians reported that patients were successful in managing a multitude of common chronic diseases, including weight loss, high blood pressure management, reduction of A1c, and improved sleep habits.
These findings compare with studies using similar approaches that demonstrated their value in the treatment of congestive heart failure, cardiovascular disease risk, type 2 diabetes, and postpartum weight retention.22-25 A growing body of evidence continues to affirm that a primary care model designed to deliver individualized care focused on improving health and an augmented patient-clinician relationship results in significant savings, primarily from reduced medical expenditures.26
This pilot provided an important opportunity to learn how to improve the effectiveness of personalized health planning and how to scale it. The experiences in Boston and Bonham demonstrated that personalized health planning can be integrated into diverse primary care settings with PACTs. The authors suggest that the knowledge gained from this project should be incorporated into new pilots at various clinical settings to determine the usefulness of the PHP for clinical indications beyond primary care. Specialty care clinics, home-based primary care services, and telehealth programs would be potential clinical applications for such pilots.
New pilots should be designed de novo and be of sufficient length to gain quantitative data on patient activation and clinical outcomes. Furthermore, future studies of personalized health planning should obtain input from the patient using Likert scales, surveys, and focus groups to gauge and quantify patient satisfaction and outcomes with the approach. Since patient engagement and better understanding of patients’ holistic needs are central to development of the PHP, patients need to be educated about this new approach to care and their active role in it.
The choice of the tools, including the HRA instrument, materials for orienting patients to their more active role in their care, the PHI, the PHP template to document shared goals, and other avenues used to engage patients, require refinement to improve their clarity, effectiveness of conveying the intended information, and ease of use. These studies demonstrated the vital need to address the best means to engage patients in understanding the value of their health to them since the clinician visit is likely to be an opportune teaching moment. Initial observations suggested that patients respond with different degrees of enthusiasm when given the opportunity to be more engaged in their care. Future pilots should clarify whether these differences stem from (a) how the invitation is presented; (b) individual differences in personality and preferences; (c) perceived clinical needs; or (d) unfamiliarity with the collaborative personalized health planning process.
The alignment of personalized health planning with outcomes data in the CPRS is essential for widespread adoption. Importantly, incentives and performance metrics will need to be redesigned to support the intended outcomes of using personalized health planning in clinical care. To that end, further investigation into the potential for cost savings associated with personalized health planning use is warranted, especially given studies that suggest high levels of patient engagement result in lower health care utilization expenditures.27
Additionally, wherever personalized health planning is initiated, employees across all levels of the system would benefit from training in patient engagement techniques and other means of attaining behavioral change. This would facilitate more effective use of time during the clinical visit and improve both the patient’s and the clinician’s satisfaction. Indeed, preliminary data indicate that this approach in a SMA setting is greatly valued by the patients.
Conclusions
The Whole Health in Primary Care Project was conducted to determine the feasibility of personalized health planning as the basis for primary care designed to facilitate personalized, proactive, patient-driven care. The pilot demonstrated that personalized health planning could be operational in VHA clinical settings and used to enhance patient-clinician engagement, establish shared health goals, and increase patient satisfaction. The personalized health planning process also provides a framework for the rational introduction of new capabilities to enhance prediction, clinical tracking, coordination of ancillary services, and clinical data collection. Future research should validate the efficacy of personalized health planning within both the VHA and health systems nationwide. Such research has the potential to refine this process so it becomes a key part of a personalized, proactive, patient-driven delivery approach.
Acknowledgements
We gratefully acknowledge the assistance of Cindy Mitchell at Duke University Medical Center with the editing and preparation of this manuscript. We also gratefully acknowledge the participation of the providers and patients at VA Boston Healthcare System and Sam Rayburn Memorial Veterans Center. Funding for this project was provided by VA777-12-C-002 to the Pacific Institute for Research and Evaluation through subcontracts to Ralph Snyderman, MD, and to the Duke University School of Nursing (Simmons PI.)
1. Anderson G. Chronic care: making the case for ongoing care. Princeton, NJ: Robert Wood Johnson Foundation; 2010.
2. Anderson G, Horvath J. The growing burden of chronic disease in America. Public Health Rep. 2004;119(3):263-270.
3. Dinan MA, Simmons LA, Snyderman R. Commentary: Personalized health planning and the Patient Protection and Affordable Care Act: an opportunity for academic medicine to lead health care reform. Acad Med. 2010;85(11):1665-1668.
4. Snyderman R, Yoediono Z. Prospective care: a personalized, preventative approach to medicine. Pharmacogenomics. 2006;7(1):5-9.
5. Snyderman R. Personalized health care: from theory to practice. Biotechnol. 2012;7(8):973-979.
6. Burnette R, Simmons LA, Snyderman R. Personalized health care as a pathway for the adoption of genomic medicine. J Pers Med. 2012;2(4):232-240.
7. Simmons LA, Dinan MA, Robinson TJ, Snyderman R. Personalized medicine is more than genomic medicine: confusion over terminology impedes progress towards personalized healthcare. J Pers Med. 2012;9(1):85-91.
8. Pelletier LR, Stichler JF. Patient-centered care and engagement: nurse leaders' imperative for health reform. J Nurs Adm. 2014;44(9):473-480.
9. Epstein RM, Street RL Jr. The values and value of patient-centered care. Ann Fam Med. 2011;9(2):100-103.
10. Simmons LA, Wolever RQ, Bechard EM, Snyderman R. Patient engagement as a risk factor in personalized health care: a systematic review of the literature on chronic disease. Genome Med. 2014;6(2):16.
11. Greene J, Hibbard JH. Why does patient activation matter? An examination of the relationships between patient activation and health-related outcomes. J Gen Intern Med. 2012;27(5):520-526.
12. U.S. Department of Veterans Affairs. VA Patient Centered Care. U.S. Department of Veterans Affairs Website. http://www.va.gov/patientcenteredcare. Updated October 30, 2015. Accessed December 3, 2015.
13. Gaudet T. The Transformation of Healthcare. Paper presented at: 27th Annual Voluntary Health Leadership Conference; 2014; Tucson, Arizona.
14. U.S. Department of Veterans Affairs. VHA Strategic Plan FY 2013-2018. U.S. Department of Veterans Affairs Website. http://www.va.gov/health/docs/VHA_STRATEGIC_PLAN_FY2013-2018.pdf. Accessed December 3, 2015.
15. U.S. Department of Veterans Affairs. Blueprint for excellence. U.S. Department of Veterans Affairs Website. http://www.va.gov/health/docs/VHA_Blueprint_for_Excellence.pdf. Published September 21, 2014. Accessed December 3, 2015.
16. Prochaska JO, Velicer WF. The transtheoretical model of health behavior change. Am J Health Promot. 1997;12(1):38-48.
17. Simmons LA, Wolever RQ. Integrative health coaching and motivational interviewing: synergistic approaches to behavior change in healthcare. Glob Adv Health Med. 2013;2(4):28-35.
18. Snyderman R, Dinan MA. Improving health by taking it personally. JAMA. 2010;303(4):363-364.
19. U.S. Department of Veterans Affairs. Computerized Patient Record System (CPRS) User Guide: GUI version. U.S Department of Veterans Affairs Website. http://www.va.gov/vdl/documents/Clinical/Comp_Patient_Recrd_Sys_(CPRS)/cprsguium.pdf. Published November 2015. Accessed December 3, 2015.
20. Perlin JB, Kolodner RM, Roswell RH. The Veterans Health Administration: quality, value, accountability, and information as transforming strategies for patient-centered care. Healthc Pap. 2005;5(4):10-24.
21. Denneson LM, Corson K, Dobscha SK. Complementary and alternative medicine use among veterans with chronic noncancer pain. JRRD. 2011;48(9):1119-1128.
22. Whellan DJ, Gaulden L, Gattis WA, et al. The benefit of implementing a heart failure disease management program. Arch Intern Med. 2001;161(18):2223-2228.
23. Edelman D, Oddone EZ, Liebowitz RS, et al. A multidimensional integrative medicine intervention to improve cardiovascular risk. J Gen Intern Med. 2006;21(7):728-734.
24. Wolever RQ, Dreusicke M, Fikkan J, et al. Integrative health coaching for patients with type 2 diabetes: a randomized clinical trial. Diabetes Educ. 2010;36(4):629-639.
25. Yang NY, Wroth S, Parham C, Strait M, Simmons LA. Personalized health planning with integrative health coaching to reduce obesity risk among women gaining excess weight during pregnancy. Glob Adv Health Med. 2013;2(4):72-77.
26. Musich S, Klemes A, Kubica MA, Wang S, Hawkins K. Personalized preventive care reduces healthcare expenditures among Medicare advantage beneficiaries. Am J Manag Care. 2014;20(8):613-620.
27. Hibbard JH, Greene J, Overton V. Patients with lower activation associated with higher costs; delivery systems should know their patients' 'scores.' Health Aff. 2013;32(2):216-222.
Health care has become increasingly unaffordable in the U.S., yet it remains ineffective in preventing or effectively treating chronic diseases.1,2 Given the increasing burden of chronic disease on the American health care system, there is an effort to shift the practice of medicine away from its reactive, disease-oriented approach to a more sustainable proactive model.3-5
Personalized health care (PHC) is an approach to the practice of medicine where prediction, prevention, intense patient engagement, shared health care decision making, and coordination of care are essential to cost effectively facilitate better outcomes.3,5-7 Greater collaboration between patient and clinician replaces the traditional clinician-dominated dialogue with more effective patient-clinician partnerships.8,9 Patients’ knowledge, skills, and confidence to manage their health care have been linked to improved health outcomes, lower costs, and greater satisfaction with health care experiences.10,11
Personalized health care has been proposed as a means to achieve better patient engagement as part of an aligned, proactive clinical approach. At the heart of PHC is personalized health planning, wherein the patient and clinician develop shared health-related goals and a plan to achieve them.3
The VHA, the largest integrated health care system in the country, is on the vanguard of incorporating tenets of PHC into its delivery model. In 2011, the Office of Patient Centered Care and Cultural Transformation (OPCC&CT) was founded to “oversee the VHA’s cultural transformation to patient-centered care.”12 This undertaking represents “one of the most massive changes in the philosophy and process for health care delivery ever undertaken by an organized health care system.”13
The primary goal of the VHA’s strategic plan for 2013 to 2018 is to provide veterans personalized, proactive, patient-driven health care.14 The intention of this approach is to engage and inspire veterans to their highest possible level of health and well-being. A personalized approach requires a dynamic customization of care that is specifically relevant to the individual, based on factors such as medical conditions, genome, needs, values, and circumstances. In addition to being personalized, this approach must be proactive, and therefore, preventive and include strategies to strengthen the person’s innate capacity for enhancing health.
The third distinction of this new model health care is that it is patient-driven, rooted in and driven by that which matters most to people in their lives and aligns their health care with their day-to-day and long-term life goals.15 The latter may be the most critical of the 3 tenets, because a personalized, proactive approach that is not driven by an engaged and inspired individual will be unlikely to achieve adherence, let alone the highest level of health and well-being.
The VHA is uniquely positioned to optimize health and well-being for veterans due in part to a systemwide emphasis on training providers to promote and support behavior change through approaches including health coaching and motivational interviewing. These synergistic approaches used widely by clinicians throughout the VHA are influenced by the transtheoretical model (ie, the stages of change theory), which considers patients holistically and helps them identify intrinsic motivation to improve their health behaviors.16,17 The transformation occurring in the VHA is intended to shift the current disease-centric medical model to an approach that optimizes the health of veterans through patient-clinician engagement, health risk assessment (HRA), shared health goal creation, and a coordinated plan to attain them.12,13
Personalized Health Planning
In recognition of the need to deliver care that emphasizes prevention and coordination, the patient-centered medical home, patient-aligned care teams (PACTs), and the chronic care model were developed. All of these embrace concepts of patient engagement, shared decision making, and team-based care. However, none of these approaches have outlined a clinical workflow that systematically and proactively operationalizes these concepts with the creation of a risk-based personalized care approach. Personalized health planning provides a clinical workflow that operationalizes all these features (Figure 1).
Of central importance is the creation of a personalized health plan (PHP), which the patient and clinician develop collaboratively. The plan serves to organize and coordinate care while engaging the patient in the process of care delivery and appropriate self-management of health.3,5 This approach promotes personalized and proactive care that values the individual and fosters meaningful patient self-awareness and engagement through shared decision making.7
The personalized health planning process is composed of several key components (Figure 2).
With this information, the clinician develops a preliminary therapeutic plan to meet these goals and discusses this plan with the patient. The next component is the synthesis of the clinician’s goals and/or treatment plan with the priorities of the patient to establish shared clinician-patient goals. This is followed by the establishment of the PHP, which consists of the agreed upon shared clinician and patient goals, a therapeutic and wellness plan to meet them, metrics for tracking progress, consults and referrals, and a time frame for the patient to achieve the health goals.
Related: The Right Care at the Right Time and in the Right Place: The Role of Technology in the VHA
The final component is coordination of care and a formalized follow-up system in which the health care team monitors the patient’s progress and provides support by revisiting or updating the PHP at intervals determined by the provider, based on the level of monitoring required by the patient’s health status. This approach invites the patient to become an empowered member of the care team by creating a patient-clinician partnership and providing a model for delivering personalized, proactive, patient-driven care to individuals with a diverse range of needs.4,5,18
Design and Implementation
This project qualified as exempt through the Duke University Institutional Review Board. The primary aim of this pilot was to examine the feasibility of implementing personalized health planning into primary care settings and to develop a workable process that is scalable and customizable to inpatient and outpatient clinics of varying sizes for different patient populations within the VHA. The pilot included 5 clinics in 2 geographic areas that were selected for their facility’s leadership support and desire to participate.
The VA Boston Healthcare System implemented personalized health planning at 3 primary care clinics: Jamaica Plain Primary Care, Jamaica Plain Women’s Health, and Quincy Primary Care. Three distinct PACTs participated in Boston, each composed of a medical doctor or doctor of osteopathy, a registered nurse (RN) or health technician, and a medical support assistant. The Sam Rayburn Memorial Veterans Center in Bonham, Texas, implemented personalized health planning at 2 clinics: the Hypertension Shared Medical Appointment and the Domiciliary Inpatient Primary Care. At Bonham, a medical doctor, pharmacist, RN, and an integrated mental health provider led the shared medical appointment (SMA) with guest presenters for individual appointments, depending on the topic covered. A RN and social worker implemented personalized health planning in the domiciliary.
After receiving training in the personalized health planning process, each of the clinics’ multidisciplinary PACTs incorporated their custom personalized health planning workflow into patient encounters. During the intake process of the clinic visit, the patient received a Personal Health Inventory (PHI) to determine health care priorities. The OPCC&CT developed the PHI as a whole-health self-assessment tool to help patients reflect on their health and lives, including core values, disparities between current and desired states, and preparedness to make behavioral changes to promote health.
The PHI assisted with framing this whole health approach to clinical care by expanding the definition of health to include more holistic elements of well-being, such as spirituality, personal relationships, emotional health, and personal development. A visual representation of the whole health domains termed The Circle of Health introduced this concept to the patient and assisted with goal setting (Figure 3).12
The PHI organized the patient’s input and was provided to the clinician to contribute to the development of the shared therapeutic goals and a final treatment plan. The PHP provided the tool to organize the goals, plans, and care following the visit and to connect the patient to additional resources within the VHA to support goal attainment through skill building and support. Each clinical site developed a mechanism to follow up with these patients either telephonically or with additional clinic appointments. The participating clinics implemented their customized version of personalized health planning for an average of 3 months.
Personalized health planning and accompanying tools were used primarily in routine ambulatory care visits. They were also used in the Bonham domiciliary clinic, which provides care for veterans with mental illnesses or addictive disorders who require additional structure and support. The purpose of the pilot was to determine whether personalized health planning could be used within this population. Given the small sample size and incompleteness of data collected, the Bonham domiciliary group was not included in the participating patient total. Across the other 4 clinics a total of 153 patients participated in the 3-month pilot study by establishing shared health goals and plans to meet them.
Results and Evaluation
Using a structured interview guide, a total of 6 small group interviews with participating clinicians were held (3 in Boston and 3 in Bonham, N = 18). Qualitative methods for research and evaluation were used to capture the depth of responses and to provide complex descriptions of the clinicians’ perspectives on the implementation of the personalized health planning process. Two researchers reviewed the transcripts to identify and code themes, and a third researcher reviewed the transcripts to confirm themes and resolve any discrepancies.
Analysis of the interview data revealed 9 core themes related to the feasibility, effectiveness, and future dissemination of personalized health planning. These themes, described below with exemplar data, include (a) patient engagement; (b) clinical assessment; (c) goal setting; (d) clinical workflow; (e) resources and support for veterans and clinicians; (f) Computerized Patient Record System (CPRS) integration; (g) patient-clinician relationship; (h) clinical outcomes; and (i) patient satisfaction.
The purpose of this study was to evaluate the feasibility of introducing personalized health planning within the workflows of the clinics that were participating. As a consequence of this, interviews were held with clinic staff rather than patients. However, the authors did obtain patient satisfaction data from 10 patients who received care from the hypertension SMA and responded to TruthPoint questions after their visit (Table).
Patient Engagement
A central tenant of personalized health planning is to engage patients in their health and health care. Findings revealed that clinicians at both sites perceived the PHP as an effective tool for integrating patients as robust members of the care team. Clinicians noted that by asking patients what was important to them, the patients felt more empowered to actively engage in the clinical encounter and to take responsibility for their health decisions. One pharmacist noted, “Patients are more empowered…when you change how you’re having your conversation with them that helps people start to recognize that they are an active participant [sic] and they can have an impact and can help with minimizing medicines or trying other things.”
Clinicians reported that including patients as active members in their care created a level of buy-in that motivated behavioral changes, because the patients identified behaviors they wanted to change vs the clinician telling them what they should or should not do. A nurse manager reported, “The key is that (the health goal) is coming from the patient…. Once it comes out of their mouth, they’re thinking about it and it’s not the clinician telling them what they should or shouldn’t do, but it’s helping them…identify something that’s important that will keep them into staying the way they want to, for the reasons that they want to.”
Clinical Assessment
The HRA tools are a vital part of the personalized health planning process, as they focus on preventive strategies that are most important for the patient.4 Clinicians reported using the PHI and additional HRA tools as part of the pilot program. The PHI is a self-assessment tool designed to identify psychosocial, behavioral, and environmental issues that can impact the patient’s care and health status. Most clinicians found that the PHI helped to solicit patients’ input on what was important to them and their health status while introducing the new approach to care. One nurse commented, “I found [PHI] very effective if I could actually sit down and review it with them to see what it was that was truly important to them and explain that this new approach is for a better understanding.”
Clinicians also found that the PHI helped focus the patient’s attention toward self-care areas that facilitated the shared goal setting process with the clinician. It moved the clinical encounter away from the chief health problems and toward identifying what is important to the patient and leveraging his or her intrinsic motivation to support health promotion via lifestyle modification.
Goal Setting
Shared goal setting is a critical component of personalized health planning. The clinician and patient must agree about realistic goals to improve the patient’s health. Clinicians reported that the goal setting stage was most successful when patients were invited to guide the process and offered the goals themselves; ie, when it was not just patient-centered but patient-driven.
“Setting a goal with a patient is pretty easy because people have an idea of what they should be doing and what they want to be doing,” one clinician reported. “They know their goal. So it’s a matter of just listening, really listening, and seeing what they want…. It’s not incongruous to get the medical goal and the patient goal to match.”
Patients were amenable to this collaborative approach to goal setting, and there was often commonality between the clinician’s goals and what was important to the patient. Occasionally, the patient set goals in seemingly unrelated areas that facilitated chronic disease management.
“One of our hypertensive patients wanted to work on things that are external that they felt are stressors that actually caused their blood pressure to be high,” a pharmacist recalled. “At the end of the day they wanted to control their environment better so that they could see if they could then be off of antihypertensives altogether. It appears that may be the case right now. That this individual has been able to accomplish that, which I thought was amazing, and since it’s still new, I’m still a little bit skeptical.... Is that possible? But if at the end of the day that is an outcome that we see from doing this, I think that’s wonderful.”
Clinicians reported that follow-up with the patient was a critical aspect of goal setting, because it improved accountability and helped track progress and health outcomes. However, due to the 3-month time limit of the pilot, there was insufficient time to get uniform data on the formalized follow-up systems developed by each clinic.
Clinical Workflow
Examining the feasibility of creating a process to incorporate personalized health planning into a busy primary care clinic was one of the major aims of this pilot. As such, issues of time, staff responsibilities, and use of the CPRS and other systems to facilitate the process were challenges that each clinic addressed in slightly different ways and with varying success. One method was to leverage the SMA for a group of patients with a common diagnosis to discuss their goals, provide accountability, and improve access to a medical team.
Another innovative approach was utilizing medical support administrators and health technicians to front-load some of the introductory information and patient education in the waiting room or during the intake process. Despite these efforts, some clinicians thought that the personalized health planning process might take longer than the traditional clinical encounter. One nurse manager commented, “I think it did affect the length of visits…it has made them a little bit longer.”
Resources and Support
The VHA has been undergoing a shift from an emphasis on tertiary care to include a greater focus on primary care. Part of this shift has been an investment in complementary and alternative medicine (CAM). The PHP helped clinicians explore what resources existed at their facility to support veterans in accomplishing their goals, including CAM. One nurse reported, “It made us look into other avenues that were actually available at the VHA that we didn’t even know we had…the acupuncture, the qigong, the voluntary services getting the veterans involved.”
Clinicians also identified the need for patient education in the concepts of whole health, personalized care, and patient involvement as necessary for moving the piloted approach forward. A nurse noted that “for [personalized health planning] to work well, [the patients] need more orientation and education upfront systemwide so that when they get into an appointment with us, we’re not starting at explaining the whole world view of partnership and doing things differently.”
Resources for clinicians were just as critical as resources for patients in facilitating the personalized health planning process. Specifically, most clinicians identified their own education and training in techniques to engage the patient in a meaningful way via motivational interviewing and health coaching complemented the personalized approach to care, particularly for shared goal setting,
CPRS Integration
The CPRS is an integrated, electronic patient record system that provides a single interface for clinicians to manage patient care as well as an efficient means for others to access and use patient information.19 The most commonly cited challenge in the pilot was the lack of available staff and time in the patient visit to complete the PHP while completing documentation requirements in CPRS.
One clinician stated, “For clinicians, the barriers…it’s time to get through the reminders and preventatives.” Clinicians reported that the process and the CPRS documentation were misaligned and lacked integration to coordinate care or support health planning. Moreover, clinicians reported that the data being collected did not support patient-centered care.
Patient-Clinician Relationship
A significant strength of personalized health planning was that it fostered a beneficial patient-clinician relationship that promoted greater depth of care. One clinician noted, “I think that it adds a more personalized dimension to the whole patient visit.” In addition to experiencing a deeper relationship with their patients, clinicians also expressed having higher levels of job satisfaction and relished the opportunity to connect with their patients in a more personal way.
Clinicians also reported that patients seemed more satisfied with the experience. One nurse commented, “They really respond to it very well when they figure out that you care about them as a whole… it’s not just about the disease process anymore.”
Clinical Outcomes
Clinicians reported a number of positive health outcomes during the pilot. One physician reported, “I have a patient, he had a follow-up today, a 29-year-old veteran, who was 260 pounds 5 months ago, and he’s 230 pounds today. He comes in monthly to see the nurse to let us know he’s doing it.”
The same physician also shared a similar transformation in a patient as a result of personalized health planning. “We had another one yesterday, 4 months ago his [hemoglobin] A1c was 10.3%. It was 7.2% yesterday, and [his weight was] down 20 pounds.” In addition to positive clinical outcomes, patients made changes in areas of their health that they identified as important through the PHI, although these areas are not typically discussed in a clinical visit.
Patient Satisfaction
Although the overall goal of this pilot was to determine the feasibility of a clinical workflow embracing personalized health planning, data on patient satisfaction were collected from patients receiving care in the Hypertension Shared Medical Appointments Program at Bohnam. Ten patients were seen over the course of 5 visits. At each visit, they were asked to rate their satisfaction (Table).
Overall, patients were highly satisfied with their experience and the care they received: 91.7% reported exploring what they wanted for their health and setting shared goals; 100% reported that their providers truly listened to their needs and treated them with respect and dignity; and 97.2% reported that their experience was better than a traditional office visit. One participating physician noted that higher levels of patient and provider satisfaction are a product of this type of patient engagement. “I also think that looking at patient and provider satisfaction, the visits feel more meaningful, and there’s a better relationship built through this discussion,” he noted. These findings demonstrating increased satisfaction further suggest the benefits of personalized health planning approach.
Discussion
In 2012, the VHA National Leadership Council convened a Strategic Planning Summit to set goals and objectives to help the VHA be at the vanguard of a movement toward a more proactive health care delivery model. The first of 3 goals developed was to provide veterans personalized, proactive, patient-driven health care.13 It is becoming increasingly clear that truly affecting health and health outcomes requires motivated, engaged, and informed patients with a care delivery approach that provides ample opportunities for patient involvement and input in health care decision making.10,11
The OPCC&CT has ongoing initiatives driving innovation, research, education, and deployment across the system to set the stage for personalized, proactive, patient-driven care.20 Some of these innovations include clinician education in the concept of whole health; health coaching; group-based, peer-led approaches; and the expansion of CAM such as mind-body approaches, qi-gong, massage therapy, yoga, and acupuncture.21
The primary aim of the Whole Health in Primary Care Project was to determine the feasibility of using personalized health planning as the operational model to deliver personalized, proactive, patient-driven care. The decision was made to integrate personalized health planning into ongoing clinical operations rather than design clinical pilots de novo. This had the advantage of speed in starting the project but limited the ability to create an optimal workflow from scratch. Given the time and resources available for this study, it was not possible to obtain quantitative data particularly as it related to quantifying clinical outcomes.
Despite these limitations, early indications suggest that the personalized health planning process can serve as the operational clinical working model to enable personalized, proactive, patient-driven care in a variety of primary care settings. As noted by one nurse manager, preparing the personalized health plan made the initial visit “a bit longer.” However, after the first visit, monitoring health risk abatement and goal achievement is akin to what is currently done by reviewing problem lists. Thus, although the personalized health planning experience is just beginning, clinicians noted that it fostered a beneficial patient-clinician relationship. This deeper relationship between the patient and the clinician may be the most powerful signal that the process is worthwhile.
This pilot provided valuable information related to the implementation of a clinical workflow redesign, an initial step toward developing an optimized operational model of the PHP process. Additionally, although it is not yet possible to quantify the clinical impact of the personalized health planning, anecdotal evidence suggests its positive potential. Clinicians reported that patients were successful in managing a multitude of common chronic diseases, including weight loss, high blood pressure management, reduction of A1c, and improved sleep habits.
These findings compare with studies using similar approaches that demonstrated their value in the treatment of congestive heart failure, cardiovascular disease risk, type 2 diabetes, and postpartum weight retention.22-25 A growing body of evidence continues to affirm that a primary care model designed to deliver individualized care focused on improving health and an augmented patient-clinician relationship results in significant savings, primarily from reduced medical expenditures.26
This pilot provided an important opportunity to learn how to improve the effectiveness of personalized health planning and how to scale it. The experiences in Boston and Bonham demonstrated that personalized health planning can be integrated into diverse primary care settings with PACTs. The authors suggest that the knowledge gained from this project should be incorporated into new pilots at various clinical settings to determine the usefulness of the PHP for clinical indications beyond primary care. Specialty care clinics, home-based primary care services, and telehealth programs would be potential clinical applications for such pilots.
New pilots should be designed de novo and be of sufficient length to gain quantitative data on patient activation and clinical outcomes. Furthermore, future studies of personalized health planning should obtain input from the patient using Likert scales, surveys, and focus groups to gauge and quantify patient satisfaction and outcomes with the approach. Since patient engagement and better understanding of patients’ holistic needs are central to development of the PHP, patients need to be educated about this new approach to care and their active role in it.
The choice of the tools, including the HRA instrument, materials for orienting patients to their more active role in their care, the PHI, the PHP template to document shared goals, and other avenues used to engage patients, require refinement to improve their clarity, effectiveness of conveying the intended information, and ease of use. These studies demonstrated the vital need to address the best means to engage patients in understanding the value of their health to them since the clinician visit is likely to be an opportune teaching moment. Initial observations suggested that patients respond with different degrees of enthusiasm when given the opportunity to be more engaged in their care. Future pilots should clarify whether these differences stem from (a) how the invitation is presented; (b) individual differences in personality and preferences; (c) perceived clinical needs; or (d) unfamiliarity with the collaborative personalized health planning process.
The alignment of personalized health planning with outcomes data in the CPRS is essential for widespread adoption. Importantly, incentives and performance metrics will need to be redesigned to support the intended outcomes of using personalized health planning in clinical care. To that end, further investigation into the potential for cost savings associated with personalized health planning use is warranted, especially given studies that suggest high levels of patient engagement result in lower health care utilization expenditures.27
Additionally, wherever personalized health planning is initiated, employees across all levels of the system would benefit from training in patient engagement techniques and other means of attaining behavioral change. This would facilitate more effective use of time during the clinical visit and improve both the patient’s and the clinician’s satisfaction. Indeed, preliminary data indicate that this approach in a SMA setting is greatly valued by the patients.
Conclusions
The Whole Health in Primary Care Project was conducted to determine the feasibility of personalized health planning as the basis for primary care designed to facilitate personalized, proactive, patient-driven care. The pilot demonstrated that personalized health planning could be operational in VHA clinical settings and used to enhance patient-clinician engagement, establish shared health goals, and increase patient satisfaction. The personalized health planning process also provides a framework for the rational introduction of new capabilities to enhance prediction, clinical tracking, coordination of ancillary services, and clinical data collection. Future research should validate the efficacy of personalized health planning within both the VHA and health systems nationwide. Such research has the potential to refine this process so it becomes a key part of a personalized, proactive, patient-driven delivery approach.
Acknowledgements
We gratefully acknowledge the assistance of Cindy Mitchell at Duke University Medical Center with the editing and preparation of this manuscript. We also gratefully acknowledge the participation of the providers and patients at VA Boston Healthcare System and Sam Rayburn Memorial Veterans Center. Funding for this project was provided by VA777-12-C-002 to the Pacific Institute for Research and Evaluation through subcontracts to Ralph Snyderman, MD, and to the Duke University School of Nursing (Simmons PI.)
Health care has become increasingly unaffordable in the U.S., yet it remains ineffective in preventing or effectively treating chronic diseases.1,2 Given the increasing burden of chronic disease on the American health care system, there is an effort to shift the practice of medicine away from its reactive, disease-oriented approach to a more sustainable proactive model.3-5
Personalized health care (PHC) is an approach to the practice of medicine where prediction, prevention, intense patient engagement, shared health care decision making, and coordination of care are essential to cost effectively facilitate better outcomes.3,5-7 Greater collaboration between patient and clinician replaces the traditional clinician-dominated dialogue with more effective patient-clinician partnerships.8,9 Patients’ knowledge, skills, and confidence to manage their health care have been linked to improved health outcomes, lower costs, and greater satisfaction with health care experiences.10,11
Personalized health care has been proposed as a means to achieve better patient engagement as part of an aligned, proactive clinical approach. At the heart of PHC is personalized health planning, wherein the patient and clinician develop shared health-related goals and a plan to achieve them.3
The VHA, the largest integrated health care system in the country, is on the vanguard of incorporating tenets of PHC into its delivery model. In 2011, the Office of Patient Centered Care and Cultural Transformation (OPCC&CT) was founded to “oversee the VHA’s cultural transformation to patient-centered care.”12 This undertaking represents “one of the most massive changes in the philosophy and process for health care delivery ever undertaken by an organized health care system.”13
The primary goal of the VHA’s strategic plan for 2013 to 2018 is to provide veterans personalized, proactive, patient-driven health care.14 The intention of this approach is to engage and inspire veterans to their highest possible level of health and well-being. A personalized approach requires a dynamic customization of care that is specifically relevant to the individual, based on factors such as medical conditions, genome, needs, values, and circumstances. In addition to being personalized, this approach must be proactive, and therefore, preventive and include strategies to strengthen the person’s innate capacity for enhancing health.
The third distinction of this new model health care is that it is patient-driven, rooted in and driven by that which matters most to people in their lives and aligns their health care with their day-to-day and long-term life goals.15 The latter may be the most critical of the 3 tenets, because a personalized, proactive approach that is not driven by an engaged and inspired individual will be unlikely to achieve adherence, let alone the highest level of health and well-being.
The VHA is uniquely positioned to optimize health and well-being for veterans due in part to a systemwide emphasis on training providers to promote and support behavior change through approaches including health coaching and motivational interviewing. These synergistic approaches used widely by clinicians throughout the VHA are influenced by the transtheoretical model (ie, the stages of change theory), which considers patients holistically and helps them identify intrinsic motivation to improve their health behaviors.16,17 The transformation occurring in the VHA is intended to shift the current disease-centric medical model to an approach that optimizes the health of veterans through patient-clinician engagement, health risk assessment (HRA), shared health goal creation, and a coordinated plan to attain them.12,13
Personalized Health Planning
In recognition of the need to deliver care that emphasizes prevention and coordination, the patient-centered medical home, patient-aligned care teams (PACTs), and the chronic care model were developed. All of these embrace concepts of patient engagement, shared decision making, and team-based care. However, none of these approaches have outlined a clinical workflow that systematically and proactively operationalizes these concepts with the creation of a risk-based personalized care approach. Personalized health planning provides a clinical workflow that operationalizes all these features (Figure 1).
Of central importance is the creation of a personalized health plan (PHP), which the patient and clinician develop collaboratively. The plan serves to organize and coordinate care while engaging the patient in the process of care delivery and appropriate self-management of health.3,5 This approach promotes personalized and proactive care that values the individual and fosters meaningful patient self-awareness and engagement through shared decision making.7
The personalized health planning process is composed of several key components (Figure 2).
With this information, the clinician develops a preliminary therapeutic plan to meet these goals and discusses this plan with the patient. The next component is the synthesis of the clinician’s goals and/or treatment plan with the priorities of the patient to establish shared clinician-patient goals. This is followed by the establishment of the PHP, which consists of the agreed upon shared clinician and patient goals, a therapeutic and wellness plan to meet them, metrics for tracking progress, consults and referrals, and a time frame for the patient to achieve the health goals.
Related: The Right Care at the Right Time and in the Right Place: The Role of Technology in the VHA
The final component is coordination of care and a formalized follow-up system in which the health care team monitors the patient’s progress and provides support by revisiting or updating the PHP at intervals determined by the provider, based on the level of monitoring required by the patient’s health status. This approach invites the patient to become an empowered member of the care team by creating a patient-clinician partnership and providing a model for delivering personalized, proactive, patient-driven care to individuals with a diverse range of needs.4,5,18
Design and Implementation
This project qualified as exempt through the Duke University Institutional Review Board. The primary aim of this pilot was to examine the feasibility of implementing personalized health planning into primary care settings and to develop a workable process that is scalable and customizable to inpatient and outpatient clinics of varying sizes for different patient populations within the VHA. The pilot included 5 clinics in 2 geographic areas that were selected for their facility’s leadership support and desire to participate.
The VA Boston Healthcare System implemented personalized health planning at 3 primary care clinics: Jamaica Plain Primary Care, Jamaica Plain Women’s Health, and Quincy Primary Care. Three distinct PACTs participated in Boston, each composed of a medical doctor or doctor of osteopathy, a registered nurse (RN) or health technician, and a medical support assistant. The Sam Rayburn Memorial Veterans Center in Bonham, Texas, implemented personalized health planning at 2 clinics: the Hypertension Shared Medical Appointment and the Domiciliary Inpatient Primary Care. At Bonham, a medical doctor, pharmacist, RN, and an integrated mental health provider led the shared medical appointment (SMA) with guest presenters for individual appointments, depending on the topic covered. A RN and social worker implemented personalized health planning in the domiciliary.
After receiving training in the personalized health planning process, each of the clinics’ multidisciplinary PACTs incorporated their custom personalized health planning workflow into patient encounters. During the intake process of the clinic visit, the patient received a Personal Health Inventory (PHI) to determine health care priorities. The OPCC&CT developed the PHI as a whole-health self-assessment tool to help patients reflect on their health and lives, including core values, disparities between current and desired states, and preparedness to make behavioral changes to promote health.
The PHI assisted with framing this whole health approach to clinical care by expanding the definition of health to include more holistic elements of well-being, such as spirituality, personal relationships, emotional health, and personal development. A visual representation of the whole health domains termed The Circle of Health introduced this concept to the patient and assisted with goal setting (Figure 3).12
The PHI organized the patient’s input and was provided to the clinician to contribute to the development of the shared therapeutic goals and a final treatment plan. The PHP provided the tool to organize the goals, plans, and care following the visit and to connect the patient to additional resources within the VHA to support goal attainment through skill building and support. Each clinical site developed a mechanism to follow up with these patients either telephonically or with additional clinic appointments. The participating clinics implemented their customized version of personalized health planning for an average of 3 months.
Personalized health planning and accompanying tools were used primarily in routine ambulatory care visits. They were also used in the Bonham domiciliary clinic, which provides care for veterans with mental illnesses or addictive disorders who require additional structure and support. The purpose of the pilot was to determine whether personalized health planning could be used within this population. Given the small sample size and incompleteness of data collected, the Bonham domiciliary group was not included in the participating patient total. Across the other 4 clinics a total of 153 patients participated in the 3-month pilot study by establishing shared health goals and plans to meet them.
Results and Evaluation
Using a structured interview guide, a total of 6 small group interviews with participating clinicians were held (3 in Boston and 3 in Bonham, N = 18). Qualitative methods for research and evaluation were used to capture the depth of responses and to provide complex descriptions of the clinicians’ perspectives on the implementation of the personalized health planning process. Two researchers reviewed the transcripts to identify and code themes, and a third researcher reviewed the transcripts to confirm themes and resolve any discrepancies.
Analysis of the interview data revealed 9 core themes related to the feasibility, effectiveness, and future dissemination of personalized health planning. These themes, described below with exemplar data, include (a) patient engagement; (b) clinical assessment; (c) goal setting; (d) clinical workflow; (e) resources and support for veterans and clinicians; (f) Computerized Patient Record System (CPRS) integration; (g) patient-clinician relationship; (h) clinical outcomes; and (i) patient satisfaction.
The purpose of this study was to evaluate the feasibility of introducing personalized health planning within the workflows of the clinics that were participating. As a consequence of this, interviews were held with clinic staff rather than patients. However, the authors did obtain patient satisfaction data from 10 patients who received care from the hypertension SMA and responded to TruthPoint questions after their visit (Table).
Patient Engagement
A central tenant of personalized health planning is to engage patients in their health and health care. Findings revealed that clinicians at both sites perceived the PHP as an effective tool for integrating patients as robust members of the care team. Clinicians noted that by asking patients what was important to them, the patients felt more empowered to actively engage in the clinical encounter and to take responsibility for their health decisions. One pharmacist noted, “Patients are more empowered…when you change how you’re having your conversation with them that helps people start to recognize that they are an active participant [sic] and they can have an impact and can help with minimizing medicines or trying other things.”
Clinicians reported that including patients as active members in their care created a level of buy-in that motivated behavioral changes, because the patients identified behaviors they wanted to change vs the clinician telling them what they should or should not do. A nurse manager reported, “The key is that (the health goal) is coming from the patient…. Once it comes out of their mouth, they’re thinking about it and it’s not the clinician telling them what they should or shouldn’t do, but it’s helping them…identify something that’s important that will keep them into staying the way they want to, for the reasons that they want to.”
Clinical Assessment
The HRA tools are a vital part of the personalized health planning process, as they focus on preventive strategies that are most important for the patient.4 Clinicians reported using the PHI and additional HRA tools as part of the pilot program. The PHI is a self-assessment tool designed to identify psychosocial, behavioral, and environmental issues that can impact the patient’s care and health status. Most clinicians found that the PHI helped to solicit patients’ input on what was important to them and their health status while introducing the new approach to care. One nurse commented, “I found [PHI] very effective if I could actually sit down and review it with them to see what it was that was truly important to them and explain that this new approach is for a better understanding.”
Clinicians also found that the PHI helped focus the patient’s attention toward self-care areas that facilitated the shared goal setting process with the clinician. It moved the clinical encounter away from the chief health problems and toward identifying what is important to the patient and leveraging his or her intrinsic motivation to support health promotion via lifestyle modification.
Goal Setting
Shared goal setting is a critical component of personalized health planning. The clinician and patient must agree about realistic goals to improve the patient’s health. Clinicians reported that the goal setting stage was most successful when patients were invited to guide the process and offered the goals themselves; ie, when it was not just patient-centered but patient-driven.
“Setting a goal with a patient is pretty easy because people have an idea of what they should be doing and what they want to be doing,” one clinician reported. “They know their goal. So it’s a matter of just listening, really listening, and seeing what they want…. It’s not incongruous to get the medical goal and the patient goal to match.”
Patients were amenable to this collaborative approach to goal setting, and there was often commonality between the clinician’s goals and what was important to the patient. Occasionally, the patient set goals in seemingly unrelated areas that facilitated chronic disease management.
“One of our hypertensive patients wanted to work on things that are external that they felt are stressors that actually caused their blood pressure to be high,” a pharmacist recalled. “At the end of the day they wanted to control their environment better so that they could see if they could then be off of antihypertensives altogether. It appears that may be the case right now. That this individual has been able to accomplish that, which I thought was amazing, and since it’s still new, I’m still a little bit skeptical.... Is that possible? But if at the end of the day that is an outcome that we see from doing this, I think that’s wonderful.”
Clinicians reported that follow-up with the patient was a critical aspect of goal setting, because it improved accountability and helped track progress and health outcomes. However, due to the 3-month time limit of the pilot, there was insufficient time to get uniform data on the formalized follow-up systems developed by each clinic.
Clinical Workflow
Examining the feasibility of creating a process to incorporate personalized health planning into a busy primary care clinic was one of the major aims of this pilot. As such, issues of time, staff responsibilities, and use of the CPRS and other systems to facilitate the process were challenges that each clinic addressed in slightly different ways and with varying success. One method was to leverage the SMA for a group of patients with a common diagnosis to discuss their goals, provide accountability, and improve access to a medical team.
Another innovative approach was utilizing medical support administrators and health technicians to front-load some of the introductory information and patient education in the waiting room or during the intake process. Despite these efforts, some clinicians thought that the personalized health planning process might take longer than the traditional clinical encounter. One nurse manager commented, “I think it did affect the length of visits…it has made them a little bit longer.”
Resources and Support
The VHA has been undergoing a shift from an emphasis on tertiary care to include a greater focus on primary care. Part of this shift has been an investment in complementary and alternative medicine (CAM). The PHP helped clinicians explore what resources existed at their facility to support veterans in accomplishing their goals, including CAM. One nurse reported, “It made us look into other avenues that were actually available at the VHA that we didn’t even know we had…the acupuncture, the qigong, the voluntary services getting the veterans involved.”
Clinicians also identified the need for patient education in the concepts of whole health, personalized care, and patient involvement as necessary for moving the piloted approach forward. A nurse noted that “for [personalized health planning] to work well, [the patients] need more orientation and education upfront systemwide so that when they get into an appointment with us, we’re not starting at explaining the whole world view of partnership and doing things differently.”
Resources for clinicians were just as critical as resources for patients in facilitating the personalized health planning process. Specifically, most clinicians identified their own education and training in techniques to engage the patient in a meaningful way via motivational interviewing and health coaching complemented the personalized approach to care, particularly for shared goal setting,
CPRS Integration
The CPRS is an integrated, electronic patient record system that provides a single interface for clinicians to manage patient care as well as an efficient means for others to access and use patient information.19 The most commonly cited challenge in the pilot was the lack of available staff and time in the patient visit to complete the PHP while completing documentation requirements in CPRS.
One clinician stated, “For clinicians, the barriers…it’s time to get through the reminders and preventatives.” Clinicians reported that the process and the CPRS documentation were misaligned and lacked integration to coordinate care or support health planning. Moreover, clinicians reported that the data being collected did not support patient-centered care.
Patient-Clinician Relationship
A significant strength of personalized health planning was that it fostered a beneficial patient-clinician relationship that promoted greater depth of care. One clinician noted, “I think that it adds a more personalized dimension to the whole patient visit.” In addition to experiencing a deeper relationship with their patients, clinicians also expressed having higher levels of job satisfaction and relished the opportunity to connect with their patients in a more personal way.
Clinicians also reported that patients seemed more satisfied with the experience. One nurse commented, “They really respond to it very well when they figure out that you care about them as a whole… it’s not just about the disease process anymore.”
Clinical Outcomes
Clinicians reported a number of positive health outcomes during the pilot. One physician reported, “I have a patient, he had a follow-up today, a 29-year-old veteran, who was 260 pounds 5 months ago, and he’s 230 pounds today. He comes in monthly to see the nurse to let us know he’s doing it.”
The same physician also shared a similar transformation in a patient as a result of personalized health planning. “We had another one yesterday, 4 months ago his [hemoglobin] A1c was 10.3%. It was 7.2% yesterday, and [his weight was] down 20 pounds.” In addition to positive clinical outcomes, patients made changes in areas of their health that they identified as important through the PHI, although these areas are not typically discussed in a clinical visit.
Patient Satisfaction
Although the overall goal of this pilot was to determine the feasibility of a clinical workflow embracing personalized health planning, data on patient satisfaction were collected from patients receiving care in the Hypertension Shared Medical Appointments Program at Bohnam. Ten patients were seen over the course of 5 visits. At each visit, they were asked to rate their satisfaction (Table).
Overall, patients were highly satisfied with their experience and the care they received: 91.7% reported exploring what they wanted for their health and setting shared goals; 100% reported that their providers truly listened to their needs and treated them with respect and dignity; and 97.2% reported that their experience was better than a traditional office visit. One participating physician noted that higher levels of patient and provider satisfaction are a product of this type of patient engagement. “I also think that looking at patient and provider satisfaction, the visits feel more meaningful, and there’s a better relationship built through this discussion,” he noted. These findings demonstrating increased satisfaction further suggest the benefits of personalized health planning approach.
Discussion
In 2012, the VHA National Leadership Council convened a Strategic Planning Summit to set goals and objectives to help the VHA be at the vanguard of a movement toward a more proactive health care delivery model. The first of 3 goals developed was to provide veterans personalized, proactive, patient-driven health care.13 It is becoming increasingly clear that truly affecting health and health outcomes requires motivated, engaged, and informed patients with a care delivery approach that provides ample opportunities for patient involvement and input in health care decision making.10,11
The OPCC&CT has ongoing initiatives driving innovation, research, education, and deployment across the system to set the stage for personalized, proactive, patient-driven care.20 Some of these innovations include clinician education in the concept of whole health; health coaching; group-based, peer-led approaches; and the expansion of CAM such as mind-body approaches, qi-gong, massage therapy, yoga, and acupuncture.21
The primary aim of the Whole Health in Primary Care Project was to determine the feasibility of using personalized health planning as the operational model to deliver personalized, proactive, patient-driven care. The decision was made to integrate personalized health planning into ongoing clinical operations rather than design clinical pilots de novo. This had the advantage of speed in starting the project but limited the ability to create an optimal workflow from scratch. Given the time and resources available for this study, it was not possible to obtain quantitative data particularly as it related to quantifying clinical outcomes.
Despite these limitations, early indications suggest that the personalized health planning process can serve as the operational clinical working model to enable personalized, proactive, patient-driven care in a variety of primary care settings. As noted by one nurse manager, preparing the personalized health plan made the initial visit “a bit longer.” However, after the first visit, monitoring health risk abatement and goal achievement is akin to what is currently done by reviewing problem lists. Thus, although the personalized health planning experience is just beginning, clinicians noted that it fostered a beneficial patient-clinician relationship. This deeper relationship between the patient and the clinician may be the most powerful signal that the process is worthwhile.
This pilot provided valuable information related to the implementation of a clinical workflow redesign, an initial step toward developing an optimized operational model of the PHP process. Additionally, although it is not yet possible to quantify the clinical impact of the personalized health planning, anecdotal evidence suggests its positive potential. Clinicians reported that patients were successful in managing a multitude of common chronic diseases, including weight loss, high blood pressure management, reduction of A1c, and improved sleep habits.
These findings compare with studies using similar approaches that demonstrated their value in the treatment of congestive heart failure, cardiovascular disease risk, type 2 diabetes, and postpartum weight retention.22-25 A growing body of evidence continues to affirm that a primary care model designed to deliver individualized care focused on improving health and an augmented patient-clinician relationship results in significant savings, primarily from reduced medical expenditures.26
This pilot provided an important opportunity to learn how to improve the effectiveness of personalized health planning and how to scale it. The experiences in Boston and Bonham demonstrated that personalized health planning can be integrated into diverse primary care settings with PACTs. The authors suggest that the knowledge gained from this project should be incorporated into new pilots at various clinical settings to determine the usefulness of the PHP for clinical indications beyond primary care. Specialty care clinics, home-based primary care services, and telehealth programs would be potential clinical applications for such pilots.
New pilots should be designed de novo and be of sufficient length to gain quantitative data on patient activation and clinical outcomes. Furthermore, future studies of personalized health planning should obtain input from the patient using Likert scales, surveys, and focus groups to gauge and quantify patient satisfaction and outcomes with the approach. Since patient engagement and better understanding of patients’ holistic needs are central to development of the PHP, patients need to be educated about this new approach to care and their active role in it.
The choice of the tools, including the HRA instrument, materials for orienting patients to their more active role in their care, the PHI, the PHP template to document shared goals, and other avenues used to engage patients, require refinement to improve their clarity, effectiveness of conveying the intended information, and ease of use. These studies demonstrated the vital need to address the best means to engage patients in understanding the value of their health to them since the clinician visit is likely to be an opportune teaching moment. Initial observations suggested that patients respond with different degrees of enthusiasm when given the opportunity to be more engaged in their care. Future pilots should clarify whether these differences stem from (a) how the invitation is presented; (b) individual differences in personality and preferences; (c) perceived clinical needs; or (d) unfamiliarity with the collaborative personalized health planning process.
The alignment of personalized health planning with outcomes data in the CPRS is essential for widespread adoption. Importantly, incentives and performance metrics will need to be redesigned to support the intended outcomes of using personalized health planning in clinical care. To that end, further investigation into the potential for cost savings associated with personalized health planning use is warranted, especially given studies that suggest high levels of patient engagement result in lower health care utilization expenditures.27
Additionally, wherever personalized health planning is initiated, employees across all levels of the system would benefit from training in patient engagement techniques and other means of attaining behavioral change. This would facilitate more effective use of time during the clinical visit and improve both the patient’s and the clinician’s satisfaction. Indeed, preliminary data indicate that this approach in a SMA setting is greatly valued by the patients.
Conclusions
The Whole Health in Primary Care Project was conducted to determine the feasibility of personalized health planning as the basis for primary care designed to facilitate personalized, proactive, patient-driven care. The pilot demonstrated that personalized health planning could be operational in VHA clinical settings and used to enhance patient-clinician engagement, establish shared health goals, and increase patient satisfaction. The personalized health planning process also provides a framework for the rational introduction of new capabilities to enhance prediction, clinical tracking, coordination of ancillary services, and clinical data collection. Future research should validate the efficacy of personalized health planning within both the VHA and health systems nationwide. Such research has the potential to refine this process so it becomes a key part of a personalized, proactive, patient-driven delivery approach.
Acknowledgements
We gratefully acknowledge the assistance of Cindy Mitchell at Duke University Medical Center with the editing and preparation of this manuscript. We also gratefully acknowledge the participation of the providers and patients at VA Boston Healthcare System and Sam Rayburn Memorial Veterans Center. Funding for this project was provided by VA777-12-C-002 to the Pacific Institute for Research and Evaluation through subcontracts to Ralph Snyderman, MD, and to the Duke University School of Nursing (Simmons PI.)
1. Anderson G. Chronic care: making the case for ongoing care. Princeton, NJ: Robert Wood Johnson Foundation; 2010.
2. Anderson G, Horvath J. The growing burden of chronic disease in America. Public Health Rep. 2004;119(3):263-270.
3. Dinan MA, Simmons LA, Snyderman R. Commentary: Personalized health planning and the Patient Protection and Affordable Care Act: an opportunity for academic medicine to lead health care reform. Acad Med. 2010;85(11):1665-1668.
4. Snyderman R, Yoediono Z. Prospective care: a personalized, preventative approach to medicine. Pharmacogenomics. 2006;7(1):5-9.
5. Snyderman R. Personalized health care: from theory to practice. Biotechnol. 2012;7(8):973-979.
6. Burnette R, Simmons LA, Snyderman R. Personalized health care as a pathway for the adoption of genomic medicine. J Pers Med. 2012;2(4):232-240.
7. Simmons LA, Dinan MA, Robinson TJ, Snyderman R. Personalized medicine is more than genomic medicine: confusion over terminology impedes progress towards personalized healthcare. J Pers Med. 2012;9(1):85-91.
8. Pelletier LR, Stichler JF. Patient-centered care and engagement: nurse leaders' imperative for health reform. J Nurs Adm. 2014;44(9):473-480.
9. Epstein RM, Street RL Jr. The values and value of patient-centered care. Ann Fam Med. 2011;9(2):100-103.
10. Simmons LA, Wolever RQ, Bechard EM, Snyderman R. Patient engagement as a risk factor in personalized health care: a systematic review of the literature on chronic disease. Genome Med. 2014;6(2):16.
11. Greene J, Hibbard JH. Why does patient activation matter? An examination of the relationships between patient activation and health-related outcomes. J Gen Intern Med. 2012;27(5):520-526.
12. U.S. Department of Veterans Affairs. VA Patient Centered Care. U.S. Department of Veterans Affairs Website. http://www.va.gov/patientcenteredcare. Updated October 30, 2015. Accessed December 3, 2015.
13. Gaudet T. The Transformation of Healthcare. Paper presented at: 27th Annual Voluntary Health Leadership Conference; 2014; Tucson, Arizona.
14. U.S. Department of Veterans Affairs. VHA Strategic Plan FY 2013-2018. U.S. Department of Veterans Affairs Website. http://www.va.gov/health/docs/VHA_STRATEGIC_PLAN_FY2013-2018.pdf. Accessed December 3, 2015.
15. U.S. Department of Veterans Affairs. Blueprint for excellence. U.S. Department of Veterans Affairs Website. http://www.va.gov/health/docs/VHA_Blueprint_for_Excellence.pdf. Published September 21, 2014. Accessed December 3, 2015.
16. Prochaska JO, Velicer WF. The transtheoretical model of health behavior change. Am J Health Promot. 1997;12(1):38-48.
17. Simmons LA, Wolever RQ. Integrative health coaching and motivational interviewing: synergistic approaches to behavior change in healthcare. Glob Adv Health Med. 2013;2(4):28-35.
18. Snyderman R, Dinan MA. Improving health by taking it personally. JAMA. 2010;303(4):363-364.
19. U.S. Department of Veterans Affairs. Computerized Patient Record System (CPRS) User Guide: GUI version. U.S Department of Veterans Affairs Website. http://www.va.gov/vdl/documents/Clinical/Comp_Patient_Recrd_Sys_(CPRS)/cprsguium.pdf. Published November 2015. Accessed December 3, 2015.
20. Perlin JB, Kolodner RM, Roswell RH. The Veterans Health Administration: quality, value, accountability, and information as transforming strategies for patient-centered care. Healthc Pap. 2005;5(4):10-24.
21. Denneson LM, Corson K, Dobscha SK. Complementary and alternative medicine use among veterans with chronic noncancer pain. JRRD. 2011;48(9):1119-1128.
22. Whellan DJ, Gaulden L, Gattis WA, et al. The benefit of implementing a heart failure disease management program. Arch Intern Med. 2001;161(18):2223-2228.
23. Edelman D, Oddone EZ, Liebowitz RS, et al. A multidimensional integrative medicine intervention to improve cardiovascular risk. J Gen Intern Med. 2006;21(7):728-734.
24. Wolever RQ, Dreusicke M, Fikkan J, et al. Integrative health coaching for patients with type 2 diabetes: a randomized clinical trial. Diabetes Educ. 2010;36(4):629-639.
25. Yang NY, Wroth S, Parham C, Strait M, Simmons LA. Personalized health planning with integrative health coaching to reduce obesity risk among women gaining excess weight during pregnancy. Glob Adv Health Med. 2013;2(4):72-77.
26. Musich S, Klemes A, Kubica MA, Wang S, Hawkins K. Personalized preventive care reduces healthcare expenditures among Medicare advantage beneficiaries. Am J Manag Care. 2014;20(8):613-620.
27. Hibbard JH, Greene J, Overton V. Patients with lower activation associated with higher costs; delivery systems should know their patients' 'scores.' Health Aff. 2013;32(2):216-222.
1. Anderson G. Chronic care: making the case for ongoing care. Princeton, NJ: Robert Wood Johnson Foundation; 2010.
2. Anderson G, Horvath J. The growing burden of chronic disease in America. Public Health Rep. 2004;119(3):263-270.
3. Dinan MA, Simmons LA, Snyderman R. Commentary: Personalized health planning and the Patient Protection and Affordable Care Act: an opportunity for academic medicine to lead health care reform. Acad Med. 2010;85(11):1665-1668.
4. Snyderman R, Yoediono Z. Prospective care: a personalized, preventative approach to medicine. Pharmacogenomics. 2006;7(1):5-9.
5. Snyderman R. Personalized health care: from theory to practice. Biotechnol. 2012;7(8):973-979.
6. Burnette R, Simmons LA, Snyderman R. Personalized health care as a pathway for the adoption of genomic medicine. J Pers Med. 2012;2(4):232-240.
7. Simmons LA, Dinan MA, Robinson TJ, Snyderman R. Personalized medicine is more than genomic medicine: confusion over terminology impedes progress towards personalized healthcare. J Pers Med. 2012;9(1):85-91.
8. Pelletier LR, Stichler JF. Patient-centered care and engagement: nurse leaders' imperative for health reform. J Nurs Adm. 2014;44(9):473-480.
9. Epstein RM, Street RL Jr. The values and value of patient-centered care. Ann Fam Med. 2011;9(2):100-103.
10. Simmons LA, Wolever RQ, Bechard EM, Snyderman R. Patient engagement as a risk factor in personalized health care: a systematic review of the literature on chronic disease. Genome Med. 2014;6(2):16.
11. Greene J, Hibbard JH. Why does patient activation matter? An examination of the relationships between patient activation and health-related outcomes. J Gen Intern Med. 2012;27(5):520-526.
12. U.S. Department of Veterans Affairs. VA Patient Centered Care. U.S. Department of Veterans Affairs Website. http://www.va.gov/patientcenteredcare. Updated October 30, 2015. Accessed December 3, 2015.
13. Gaudet T. The Transformation of Healthcare. Paper presented at: 27th Annual Voluntary Health Leadership Conference; 2014; Tucson, Arizona.
14. U.S. Department of Veterans Affairs. VHA Strategic Plan FY 2013-2018. U.S. Department of Veterans Affairs Website. http://www.va.gov/health/docs/VHA_STRATEGIC_PLAN_FY2013-2018.pdf. Accessed December 3, 2015.
15. U.S. Department of Veterans Affairs. Blueprint for excellence. U.S. Department of Veterans Affairs Website. http://www.va.gov/health/docs/VHA_Blueprint_for_Excellence.pdf. Published September 21, 2014. Accessed December 3, 2015.
16. Prochaska JO, Velicer WF. The transtheoretical model of health behavior change. Am J Health Promot. 1997;12(1):38-48.
17. Simmons LA, Wolever RQ. Integrative health coaching and motivational interviewing: synergistic approaches to behavior change in healthcare. Glob Adv Health Med. 2013;2(4):28-35.
18. Snyderman R, Dinan MA. Improving health by taking it personally. JAMA. 2010;303(4):363-364.
19. U.S. Department of Veterans Affairs. Computerized Patient Record System (CPRS) User Guide: GUI version. U.S Department of Veterans Affairs Website. http://www.va.gov/vdl/documents/Clinical/Comp_Patient_Recrd_Sys_(CPRS)/cprsguium.pdf. Published November 2015. Accessed December 3, 2015.
20. Perlin JB, Kolodner RM, Roswell RH. The Veterans Health Administration: quality, value, accountability, and information as transforming strategies for patient-centered care. Healthc Pap. 2005;5(4):10-24.
21. Denneson LM, Corson K, Dobscha SK. Complementary and alternative medicine use among veterans with chronic noncancer pain. JRRD. 2011;48(9):1119-1128.
22. Whellan DJ, Gaulden L, Gattis WA, et al. The benefit of implementing a heart failure disease management program. Arch Intern Med. 2001;161(18):2223-2228.
23. Edelman D, Oddone EZ, Liebowitz RS, et al. A multidimensional integrative medicine intervention to improve cardiovascular risk. J Gen Intern Med. 2006;21(7):728-734.
24. Wolever RQ, Dreusicke M, Fikkan J, et al. Integrative health coaching for patients with type 2 diabetes: a randomized clinical trial. Diabetes Educ. 2010;36(4):629-639.
25. Yang NY, Wroth S, Parham C, Strait M, Simmons LA. Personalized health planning with integrative health coaching to reduce obesity risk among women gaining excess weight during pregnancy. Glob Adv Health Med. 2013;2(4):72-77.
26. Musich S, Klemes A, Kubica MA, Wang S, Hawkins K. Personalized preventive care reduces healthcare expenditures among Medicare advantage beneficiaries. Am J Manag Care. 2014;20(8):613-620.
27. Hibbard JH, Greene J, Overton V. Patients with lower activation associated with higher costs; delivery systems should know their patients' 'scores.' Health Aff. 2013;32(2):216-222.
Treatment Options for Acute Gout
Gout is an extremely painful arthritis initiated by innate immune responses to monosodium urate crystals that accumulate in affected joints and surrounding tissues. As a result, gout is characterized by painful arthritis flares followed by intervening periods of disease quiescence. Over time, gout can lead to chronic pain, disability, and tophi. Nearly 10% of those aged > 65 years report having gout. The overall prevalence in the U.S. population approaches 4%.1
Gout treatment has 2 overarching goals: alleviating the pain and inflammation caused by acute gout attacks and long-term management that is focused on lowering serum urate (sUA) levels to reduce the risk of future attacks. Alleviating the pain and inflammation of an acute attack is often complicated by patient characteristics, namely, other chronic health conditions that frequently accompany gout, such as diabetes mellitus (DM), chronic kidney disease (CKD), hypertension, and cardiovascular disease (CVD).
Patients with gout tend to be older and have multiple comorbidities that require the use of many medications.2 Because the VA patient population tends to be older, acute gout and attendant complications of treatment are an important consideration for VA health care providers (HCPs).
Recently, the American College of Rheumatology (ACR) released management recommendations for gout, including those for the treatment of acute gout.3 The ACR recommends 3 first-line therapies, but limited guidance is provided for deciding among therapies. This article briefly reviews the relevant ACR recommendations and details important comorbidity and concomitant medication considerations in the treatment of acute gout.
Acute Gout Characteristics
Acute gout attacks are characterized by a rapid onset and escalation with joint pain typically peaking within 24 hours of attack onset. An acute attack often begins to remit after 5 to 12 days without intervention, but complete resolution may take longer in some patients.4 In one study, at 24 hours after attack onset, 16% of patients on placebo had > 50% reduction in pain compared with 70% that had no recovery at all.5 By 48 hours, one-third of patients on placebo achieved a 50% reduction in pain.6 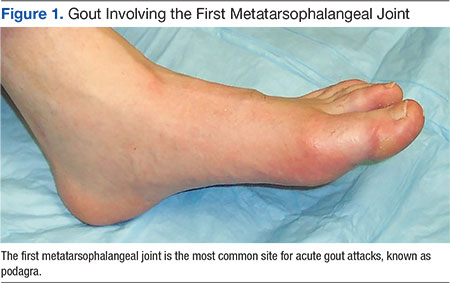
Treatment Recommendations
Therapy for acute gout attacks aims to reduce pain and promote a full, early resolution. The ACR recommends pharmacologic therapy as first-line treatment with adjunctive topical ice and rest as needed.3 Typically, monotherapy is appropriate if the individual is experiencing mild-to-moderate pain affecting ≤ 2 joints of any size. Severe pain or attacks affecting multiple joints may benefit from initial combination therapy. Three first-line therapies are available: nonsteroidal anti-inflammatory drugs (NSAIDs) or cyclooxygenase-2 (COX-2) inhibitors, colchicine, or systemic glucocorticoids (Figure 2).

Few studies compare the efficacy of first-line therapeutic categories. There are no clinical trials directly comparing colchicine with NSAIDs or colchicine with glucocorticoids. No difference in mean reduction of pain and no differences in adverse events (AEs) were shown in a trial that compared glucocorticoids with NSAIDs.8 Thus, without further study, treatment choices made by HCPs are often guided by factors other than the existence of robust evidence.
Treatment with NSAIDs or COX-2 inhibitors should be initiated at the approved dose and continued until the gout attack has completely resolved. In one study, 73% of patients had pain reduction of ≥ 50% when taking NSAIDs relative to only 27% of patients on placebo.8 All available NSAIDs are considered effective, but only 3 NSAIDs are specifically approved for treatment of acute gout (naproxen, indomethacin, and sulindac). There is no evidence supporting one NSAID as being more effective than another; evidence fails to show a meaningful difference.8 Limited evidence indicates that selective COX-2 inhibitors, including celexocib, have similar efficacy as nonselective NSAIDs but may have fewer AEs, driven in part by fewer gastrointestinal (GI) events (6% vs 16% for GI events).8
Colchicine has long been used as prophylaxis for acute gout attacks and has been endorsed for the treatment of acute attacks. Recent evidence suggests that colchicine initially dosed at 1.2 mg followed by a single 0.6-mg dose 1 hour later is as effective with fewer AEs compared with a traditional regimen of 1.2 mg followed by 0.6 mg every hour for up to 6 hours.5 About 40% of patients have 50% pain reduction within 24 hours and a 40% absolute risk reduction in AEs on this low-dose regimen. The efficacy of colchicine relative to other therapies is poorly defined, especially for patients presenting longer after attack onset. The ACR guidelines recommend colchicine only if treatment is initiated within 36 hours of attack onset, but this is based solely on expert consensus. Likewise, the above trial for low-dose colchicine did not provide information about dosing beyond the first 6 hours, leaving little guidance for follow-up treatment of residual pain beyond the 32 hours reported.5 Traditionally, one 0.6-mg dose is provided every 12 to 24 hours.3
Systemic glucocorticoids are also commonly used in treating acute gout.9 There was a small pain reduction benefit for prednisolone, but the difference was not clinically significant in one clinical trial comparing oral prednisolone 30 mg daily for 5 days vs a combination of indomethacin for 5 days and an initial intramuscular injection of diclofenac 75 mg.10 The prednisolone group also had fewer patients with AEs, including abdominal pain (0% vs 30%) and GI bleeding (0% vs 11%). The lower incidence of short-term AEs may be a primary benefit of systemic glucocorticoids.11
Intra-articular glucocorticoids are not suggested first-line therapies but are commonly used by rheumatologists.9 In an uncontrolled study conducted by Fernández and colleagues, intra-articular glucocorticoid injections helped to quickly resolve 20 out of 20 crystal-proven gout attacks.12 However, no randomized controlled trials have examined this approach. Although seemingly efficacious, other considerations are important for this modality. Intra-articular glucocorticoids may not be preferred for polyarticular attacks or attacks in difficult-to-aspirate joints. Additionally, intra-articular glucocorticoids have been anecdotally associated with rebound attacks (ie, attacks that occur shortly after resolution without other interventions). However, the Fernández study had no such attacks occur among participants.12 Finally, septic arthritis must be ruled out as in any case of acute onset monoarticular arthritis.
Biologic agents targeting interleukin-1(IL-1) are not currently approved for gout, although there is burgeoning data suggesting that this strategy may have substantial merit.13 Additionally, there is limited evidence that adrenocorticotropic hormone (ACTH) may provide rapid pain relief when other available therapies are ineffective or contraindicated. However, ACTH studies have not provided robust trial designs, and drug costs remain substantial, thus limiting the widespread use of ACTH in acute gout.14,15 Anti-IL-1 agents and ACTH may both be considered as second-line options if first-line therapies are contraindicated or fail. Careful consideration should be given to AE profiles, patient preferences, and cost.
Comorbidities
Acute gout care, especially in the context of comorbidities, has been identified as a critical treatment concern by an international panel of rheumatologists as part of the 3e (Evidence, Expertise, Exchange) Initiative.16 However, regular clinical trial exclusion criteria have limited data necessary to guide treatment when comorbidities are present. Therefore, studies of acute gout treatment in the context of disease comorbidity represents a major unmet need in understanding and optimizing gout care.
Chronic Kidney Disease
Chronic kidney disease is common in gout; 20% of patients with gout have an estimated glomerular filtration rate (eGFR) of < 30 mL/min.2 Thus, CKD is an important consideration when deciding the best treatment for acute gout. The ACR recommendations do not provide specific guidance on NSAID use in CKD but suggest the potential option of tapering the dose as pain begins to resolve. There is mixed evidence that NSAIDs accelerate CKD progression with the best evidence for high-dose NSAID use.17 When prescribing the concomitant use of NSAIDs with other medications affecting kidney function, HCPs should consider CKD.
For colchicine, current labeling and evidence indicate that no dose adjustments are needed for stage 3 or better CKD (eGFR ≥ 60 mL/min) even among the elderly.18,19 Although labeling indicates that a single unadjusted dose (0.6 mg) can be given once every 2 weeks for those with severe CKD (eGFR < 30 mL/min) or for those who are on dialysis, alternative therapies should be considered, as AEs increase with decreasing renal function.19 Colchicine should not be used in those with eGFR < 10 mL/min.20 All patients who have CKD and are treated with colchicine should be informed of the AEs and closely observed for signs of toxicity, including blood dyscrasias, neuromyopathy, emesis, or diarrhea.
Considering the potential complications for NSAIDs and colchicine, patients with CKD may be good candidates for glucocorticoid therapy, administered either systemically or as an intra-articular injection. Alternatively, second-line agents such as ACTH or IL-1 inhibition may be considered in such patients.
Hypertension
Hypertension is one of the most common comorbidities among patients with gout. It is important for HCP consideration when deciding treatment. Poorly controlled hypertension is a contraindication for both NSAIDs and systemic glucocorticoids. Patients with hypertension in the absence of significant renal impairment may be good candidates for colchicine.
Diabetes and Hyperlipidemia
Glucocorticoids should be avoided if possible in the setting of inadequately controlled type 2 DM (T2DM) or hyperlipidemia. Glucocorticoids exacerbate insulin resistance and stimulate glucose secretion from the liver. This can create substantial and sometimes dangerous fluctuations in circulating glucose concentrations. Additionally, glucocorticoids may increase serum triglycerides and low-density lipoprotein levels. Thus, patients with T2DM or hyperlipidemia may be good candidates for alternative treatments, such as colchicine or NSAIDs.
Cardiovascular Disease
Cardiovascular disease risk has been shown to increase with the use of COX-2 inhibitors. This risk may be present for all NSAIDs. Current FDA labeling suggests limiting NSAID and COX-2 inhibitor use in patients with a history of myocardial infarction (MI), congestive heart failure, or stroke. Given the potential impact on cardiovascular risk factors, including hypertension, T2DM, and hyperlipidemia, glucocorticoids may not be ideal for patients with known CVD or those at high risk.
Recent evidence has shown that colchicine use is associated with a lower risk of MI among patients with gout.21 These results, in addition to a proposed dual role of IL-1 in both gout and CVD, suggest that either colchicine or IL-1 inhibitors may be rational agents in the treatment of acute gout in the context of CVD.22
Hepatic Impairment and GI Bleeding
Patients with cirrhosis should avoid NSAID use due to the potential increased bleeding risk from underlying coagulopathy. Additionally, colchicine clearance may be reduced in patients with severe liver impairment, mandating close surveillance when this agent is used. If hepatic impairment is mild to moderate, judicious use of any of the first-line therapies may be appropriate.
Patients with GI bleeding or a history of peptic ulcer disease should avoid NSAID use because of increased bleeding risk. If an NSAID is used, proton pump inhibitors decrease the risk of NSAID-associated mucosal damage.
Drug Interactions
Colchicine is metabolized by the cytochrome P450 3A4 enzyme (CYP3A4) and is a substrate for P-glycoprotein (P-gp). Therefore, concomitant use of colchicine with potent inhibitors of CYP3A4 or P-gp should be avoided when possible. These agents include macrolide antibiotics (clarithromyocin), calcium channel blockers (verapamil and diltiazem), and cyclosporine (commonly used in transplant patients who are at high risk for gout). New evidence-based dosing recommendations indicate that no dose reduction is required with azithromyocin.23
Nonsteroidal anti-inflammatory drugs are contraindicated with the concomitant use of angiotensin-converting enzyme (ACE) inhibitors and/or diuretics. Prostaglandin production is decreased while using NSAIDs, resulting in increased constriction of afferent renal arterioles and decreased glomerular filtration pressure. This physiologic effect of NSAIDs can be exacerbated when used in combination with ACE inhibitors or diuretics, both of which can also reduce glomerular filtration pressures. Combination therapy with either ACE inhibitors or diuretics increases the risk for NSAID-mediated acute kidney injury. Additionally, NSAID use should be avoided in patients taking anticoagulants such as warfarin or heparin due to increased bleeding risk.
Diagnosis
Diagnosis is a key component of proper treatment of acute gout. A gout diagnosis is usually made based on clinical signs and symptoms, including sudden onset of pain that peaks within 24 hours, past history of acute self-limited attacks of arthritis, first MTP involvement, and an elevated sUA. However, not all these factors must be present. In a study conducted by Janssens and colleagues, these factors plus additional demographics had a sensitivity of 90% and specificity of 65% when compared with crystal diagnosis.24 However, a normal sUA level does not exclude gout as a diagnosis due to the uricosuric effect of the inflammatory process.25 In fact, one observational cohort recorded an average sUA decrease from baseline of 2 mg/dL during an acute gout attack.26
Alternative diagnoses, including septic arthritis, should be considered, particularly in the context of treatment failure (< 50% reduction in pain) within the first 24 to 48 hours. A definitive diagnosis is made by identifying negatively birefringent crystals in the synovial fluid of the affected joint (using polarized microscopy) with negative cultures. In the absence of crystal confirmation, there is an emerging role for imaging in gout diagnosis, including the use of ultrasound and dual-energy computed tomography.27
Long-term Treatment Considerations
During treatment for acute gout attacks, urate-lowering therapy that was initiated before the attack should not be discontinued.28 There is no evidence to suggest that current urate lowering has any AEs during attacks. However, removing treatment may increase sUA levels, precipitating attacks in other joints by “destabilizing” crystals still present. Current recommendations also state that urate-lowering therapy may be started during an attack despite traditionally being deferred until the attack has resolved.28 In a randomized trial comparing a group starting allopurinol 300 mg during an attack vs a placebo group (with all patients receiving anti-inflammatory treatment for the acute attack), there was no difference in pain outcomes.29 Regardless of the chosen timing, lowering and maintaining sUA ≤ 6.0 mg/dL is the primary method for minimizing long-term risk of gout attacks.28
Health care providers should discuss with patients the likely need for indefinite urate-lowering therapy while noting that attacks related to therapy initiation are relatively common.30 Current guidelines recommend starting urate-lowering therapy in low doses (≤ 100 mg/d for allopurinol) and titrating to achieve and maintain the target sUA level. Along with the judicious use of anti-inflammatory prophylaxis, this may minimize attacks related to therapy initiation.3,28 By lowering and maintaining sUA below the target level, monosodium urate crystals will dissolve, thereby eliminating the major inciting factor of acute attacks.
Other day-to-day triggers such as alcohol, meat or seafood consumption, and dehydration exist for some patients with gout. Patients should be informed of these inciting factors, as they could potentially be avoided, reducing the risk of future gout attacks. It is important to recognize, however, that dietary or behavioral interventions have generally yielded only modest sUA reductions. For the majority of patients, therefore, reduction and maintenance of sUA ≤ 6.0 mg/dL requires pharmacologic intervention.
Conclusions
Gout attacks should be treated immediately with pharmacologic treatment when contraindications are absent. First-line treatment options include NSAIDs, colchicine, and systemic glucocorticoids. Use of these modalities can be complicated because of comorbidity and concomitant medication use that is prevalent among patients with gout. Comorbidities commonly limiting treatment choice include hypertension (NSAIDs, glucocorticoids), CKD (NSAIDS, colchicine), CVD (NSAIDs, COX-2 inhibitors, glucocorticoids), T2DM (glucocorticoids), and liver disease (NSAIDs, colchicine). Careful consideration must be given to these comorbidities and contraindications as well as patient preferences.
1. Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63(10):3136-3141.
2. Zhu Y, Pandya BJ, Choi HK. Comorbidities of gout and hyperuricemia in the US general population: NHANES 2007-2008. Am J Med. 2012;125(7):679-687.e1.
3. Khanna D, Khanna PP, Fitzgerald JD, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 2: therapy and antiinflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res (Hoboken). 2012;64(10):1447-1461.
4. Bellamy N, Downie WW, Buchanan WW. Observations on spontaneous improvement in patients with podagra: implications for therapeutic trials of non-steroidal anti-inflammatory drugs. Br J Clin Pharmacol. 1987;24(1):33-36.
5. Terkeltaub RA, Furst DE, Bennett K, Kook KA, Crockett RS, Davis MW. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum. 2010;62(4):1060-1068.
6. Ahern MJ, Reid C, Gordon TP, McCredie M, Brooks PM, Jones M. Does colchicine work? The results of the first controlled study in acute gout. Aust N Z J Med. 1987;17(3):301-304.
7. Puig JG, Michán AD, Jiménez ML, et al. Female gout. Clinical spectrum and uric acid metabolism. Arch Intern Med. 1991;151(4):726-732.
8. van Durme CM, Wechalekar MD, Buchbinder R, Schlesinger N, van der Heijde D, Landewé RB. Non-steroidal anti-inflammatory drugs for acute gout. Cochrane Database Syst Rev. 2014;9:CD010120.
9. Schlesinger N, Moore DF, Sun JD, Schumacher HR Jr. A survey of current evaluation and treatment of gout. J Rheumatol. 2006;33(10):2050-2052.
10. Man CY, Cheung IT, Cameron PA, Rainer TH. Comparison of oral prednisolone/paracetamol and oral indomethacin/paracetamol combination therapy in the treatment of acute goutlike arthritis: a double-blind, randomized, controlled trial. Ann Emerg Med. 2007;49(5):670-677.
11. Janssens HJ, Lucassen PL, Van de Laar FA, Janssen M, Van de Lisdonk EH. Systemic corticosteroids for acute gout. Cochrane Database Syst Rev. 2008;(2):CD005521.
12. Fernández C, Noguera R, González JA, Pascual E. Treatment of acute attacks of gout with a small dose of intraarticular triamcinolone acetonide. J Rheumatol. 1999;26(10):2285-2286.
13. Burns CM, Wortmann RL. Gout therapeutics: new drugs for an old disease. Lancet. 2011;377(9760):165-177.
14. Axelrod D, Preston S. Comparison of parenteral adrenocorticotropic hormone with oral indomethacin in the treatment of acute gout. Arthritis Rheum. 1988;31(6):803-805.
15. Ritter J, Kerr LD, Valeriano-Marcet J, Spiera H. ACTH revisited: effective treatment for acute crystal induced synovitis in patients with multiple medical problems. J Rheumatol. 1994;21(4):696-699.
16. Sivera F, Andrés M, Carmona L, et al. Multinational evidence-based recommendations for the diagnosis and management of gout: integrating systematic literature review and expert opinion of a broad panel of rheumatologists in the 3e initiative. Ann Rheum Dis. 2014;73(2):328-335.
17. Nderitu P, Doos L, Jones PW, Davies SJ, Kadam UT. Non-steroidal anti-inflammatory drugs and chronic kidney disease progression: a systematic review. Fam Pract. 2013;30(3):247-255.
18. Wason S, Faulkner RD, Davis MW. Are dosing adjustments required for colchicine in the elderly compared with younger patients? Adv Ther. 2012;29(6):551-561.
19. Wason S, Mount D, Faulkner R. Single-dose, open-label study of the differences in pharmacokinetics of colchicine in subjects with renal impairment, including end-stage renal disease. Clin Drug Investig. 2014;34(12):845-855.
20. Hanlon JT, Aspinall SL, Semla TP, et al. Consensus guidelines for oral dosing of primarily renally cleared medications in older adults. J Am Geriatr Soc. 2009;57(2):335-340.
21. Crittenden DB, Lehmann RA, Schneck L, et al. Colchicine use is associated with decreased prevalence of myocardial infarction in patients with gout. J Rheumatol. 2012;39(7):1458-1464.
22. Esser N, Paquot N, Scheen AJ. Anti-inflammatory agents to treat or prevent type 2 diabetes, metabolic syndrome and cardiovascular disease. Expert Opin Investig Drugs. 2015;24(3):283-307.
23. Terkeltaub RA, Furst DE, Digiacinto JL, Kook KA, Davis MW. Novel evidence-based colchicine dose-reduction algorithm to predict and prevent colchicine toxicity in the presence of cytochrome P450 3A4/P-glycoprotein inhibitors. Arthritis Rheum. 2011;63(8):2226-2237.
24. Janssens HJ, Fransen J, van de Lisdonk EH, van Riel PL, van Weel C, Janssen M. A diagnostic rule for acute gouty arthritis in primary care without joint fluid analysis. Arch Intern Med. 2010;170(13):1120-1126.
25. Urano W, Yamanaka H, Tsutani H, et al. The inflammatory process in the mechanism of decreased serum uric acid concentrations during acute gouty arthritis. J Rheumatol. 2002;29(9):1950-1953.
26. Logan JA, Morrison E, McGill PE. Serum uric acid in acute gout. Ann Rheum Dis. 1997;56(11):696-697.
27. Ogdie A, Taylor WJ, Weatherall M, et al. Imaging modalities for the classification of gout: systematic literature review and meta-analysis. Ann Rheum Dis. 2015; 74(10):1868-1874.
28. Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012;64(10):1431-1446.
29. Taylor TH, Mecchella JN, Larson RJ, Kerin KD, Mackenzie TA. Initiation of allopurinol at first medical contact for acute attacks of gout: a randomized clinical trial. Am J Med. 2012;125(11):1126-11134.e7.
30. Becker MA, MacDonald PA, Hunt BJ, Lademacher C, Joseph-Ridge N. Determinants of the clinical outcomes of gout during the first year of urate-lowering therapy. Nucleosides Nucleotides Nucleic Acids. 2008;27(6):585-591.
Gout is an extremely painful arthritis initiated by innate immune responses to monosodium urate crystals that accumulate in affected joints and surrounding tissues. As a result, gout is characterized by painful arthritis flares followed by intervening periods of disease quiescence. Over time, gout can lead to chronic pain, disability, and tophi. Nearly 10% of those aged > 65 years report having gout. The overall prevalence in the U.S. population approaches 4%.1
Gout treatment has 2 overarching goals: alleviating the pain and inflammation caused by acute gout attacks and long-term management that is focused on lowering serum urate (sUA) levels to reduce the risk of future attacks. Alleviating the pain and inflammation of an acute attack is often complicated by patient characteristics, namely, other chronic health conditions that frequently accompany gout, such as diabetes mellitus (DM), chronic kidney disease (CKD), hypertension, and cardiovascular disease (CVD).
Patients with gout tend to be older and have multiple comorbidities that require the use of many medications.2 Because the VA patient population tends to be older, acute gout and attendant complications of treatment are an important consideration for VA health care providers (HCPs).
Recently, the American College of Rheumatology (ACR) released management recommendations for gout, including those for the treatment of acute gout.3 The ACR recommends 3 first-line therapies, but limited guidance is provided for deciding among therapies. This article briefly reviews the relevant ACR recommendations and details important comorbidity and concomitant medication considerations in the treatment of acute gout.
Acute Gout Characteristics
Acute gout attacks are characterized by a rapid onset and escalation with joint pain typically peaking within 24 hours of attack onset. An acute attack often begins to remit after 5 to 12 days without intervention, but complete resolution may take longer in some patients.4 In one study, at 24 hours after attack onset, 16% of patients on placebo had > 50% reduction in pain compared with 70% that had no recovery at all.5 By 48 hours, one-third of patients on placebo achieved a 50% reduction in pain.6
Treatment Recommendations
Therapy for acute gout attacks aims to reduce pain and promote a full, early resolution. The ACR recommends pharmacologic therapy as first-line treatment with adjunctive topical ice and rest as needed.3 Typically, monotherapy is appropriate if the individual is experiencing mild-to-moderate pain affecting ≤ 2 joints of any size. Severe pain or attacks affecting multiple joints may benefit from initial combination therapy. Three first-line therapies are available: nonsteroidal anti-inflammatory drugs (NSAIDs) or cyclooxygenase-2 (COX-2) inhibitors, colchicine, or systemic glucocorticoids (Figure 2).
Few studies compare the efficacy of first-line therapeutic categories. There are no clinical trials directly comparing colchicine with NSAIDs or colchicine with glucocorticoids. No difference in mean reduction of pain and no differences in adverse events (AEs) were shown in a trial that compared glucocorticoids with NSAIDs.8 Thus, without further study, treatment choices made by HCPs are often guided by factors other than the existence of robust evidence.
Treatment with NSAIDs or COX-2 inhibitors should be initiated at the approved dose and continued until the gout attack has completely resolved. In one study, 73% of patients had pain reduction of ≥ 50% when taking NSAIDs relative to only 27% of patients on placebo.8 All available NSAIDs are considered effective, but only 3 NSAIDs are specifically approved for treatment of acute gout (naproxen, indomethacin, and sulindac). There is no evidence supporting one NSAID as being more effective than another; evidence fails to show a meaningful difference.8 Limited evidence indicates that selective COX-2 inhibitors, including celexocib, have similar efficacy as nonselective NSAIDs but may have fewer AEs, driven in part by fewer gastrointestinal (GI) events (6% vs 16% for GI events).8
Colchicine has long been used as prophylaxis for acute gout attacks and has been endorsed for the treatment of acute attacks. Recent evidence suggests that colchicine initially dosed at 1.2 mg followed by a single 0.6-mg dose 1 hour later is as effective with fewer AEs compared with a traditional regimen of 1.2 mg followed by 0.6 mg every hour for up to 6 hours.5 About 40% of patients have 50% pain reduction within 24 hours and a 40% absolute risk reduction in AEs on this low-dose regimen. The efficacy of colchicine relative to other therapies is poorly defined, especially for patients presenting longer after attack onset. The ACR guidelines recommend colchicine only if treatment is initiated within 36 hours of attack onset, but this is based solely on expert consensus. Likewise, the above trial for low-dose colchicine did not provide information about dosing beyond the first 6 hours, leaving little guidance for follow-up treatment of residual pain beyond the 32 hours reported.5 Traditionally, one 0.6-mg dose is provided every 12 to 24 hours.3
Systemic glucocorticoids are also commonly used in treating acute gout.9 There was a small pain reduction benefit for prednisolone, but the difference was not clinically significant in one clinical trial comparing oral prednisolone 30 mg daily for 5 days vs a combination of indomethacin for 5 days and an initial intramuscular injection of diclofenac 75 mg.10 The prednisolone group also had fewer patients with AEs, including abdominal pain (0% vs 30%) and GI bleeding (0% vs 11%). The lower incidence of short-term AEs may be a primary benefit of systemic glucocorticoids.11
Intra-articular glucocorticoids are not suggested first-line therapies but are commonly used by rheumatologists.9 In an uncontrolled study conducted by Fernández and colleagues, intra-articular glucocorticoid injections helped to quickly resolve 20 out of 20 crystal-proven gout attacks.12 However, no randomized controlled trials have examined this approach. Although seemingly efficacious, other considerations are important for this modality. Intra-articular glucocorticoids may not be preferred for polyarticular attacks or attacks in difficult-to-aspirate joints. Additionally, intra-articular glucocorticoids have been anecdotally associated with rebound attacks (ie, attacks that occur shortly after resolution without other interventions). However, the Fernández study had no such attacks occur among participants.12 Finally, septic arthritis must be ruled out as in any case of acute onset monoarticular arthritis.
Biologic agents targeting interleukin-1(IL-1) are not currently approved for gout, although there is burgeoning data suggesting that this strategy may have substantial merit.13 Additionally, there is limited evidence that adrenocorticotropic hormone (ACTH) may provide rapid pain relief when other available therapies are ineffective or contraindicated. However, ACTH studies have not provided robust trial designs, and drug costs remain substantial, thus limiting the widespread use of ACTH in acute gout.14,15 Anti-IL-1 agents and ACTH may both be considered as second-line options if first-line therapies are contraindicated or fail. Careful consideration should be given to AE profiles, patient preferences, and cost.
Comorbidities
Acute gout care, especially in the context of comorbidities, has been identified as a critical treatment concern by an international panel of rheumatologists as part of the 3e (Evidence, Expertise, Exchange) Initiative.16 However, regular clinical trial exclusion criteria have limited data necessary to guide treatment when comorbidities are present. Therefore, studies of acute gout treatment in the context of disease comorbidity represents a major unmet need in understanding and optimizing gout care.
Chronic Kidney Disease
Chronic kidney disease is common in gout; 20% of patients with gout have an estimated glomerular filtration rate (eGFR) of < 30 mL/min.2 Thus, CKD is an important consideration when deciding the best treatment for acute gout. The ACR recommendations do not provide specific guidance on NSAID use in CKD but suggest the potential option of tapering the dose as pain begins to resolve. There is mixed evidence that NSAIDs accelerate CKD progression with the best evidence for high-dose NSAID use.17 When prescribing the concomitant use of NSAIDs with other medications affecting kidney function, HCPs should consider CKD.
For colchicine, current labeling and evidence indicate that no dose adjustments are needed for stage 3 or better CKD (eGFR ≥ 60 mL/min) even among the elderly.18,19 Although labeling indicates that a single unadjusted dose (0.6 mg) can be given once every 2 weeks for those with severe CKD (eGFR < 30 mL/min) or for those who are on dialysis, alternative therapies should be considered, as AEs increase with decreasing renal function.19 Colchicine should not be used in those with eGFR < 10 mL/min.20 All patients who have CKD and are treated with colchicine should be informed of the AEs and closely observed for signs of toxicity, including blood dyscrasias, neuromyopathy, emesis, or diarrhea.
Considering the potential complications for NSAIDs and colchicine, patients with CKD may be good candidates for glucocorticoid therapy, administered either systemically or as an intra-articular injection. Alternatively, second-line agents such as ACTH or IL-1 inhibition may be considered in such patients.
Hypertension
Hypertension is one of the most common comorbidities among patients with gout. It is important for HCP consideration when deciding treatment. Poorly controlled hypertension is a contraindication for both NSAIDs and systemic glucocorticoids. Patients with hypertension in the absence of significant renal impairment may be good candidates for colchicine.
Diabetes and Hyperlipidemia
Glucocorticoids should be avoided if possible in the setting of inadequately controlled type 2 DM (T2DM) or hyperlipidemia. Glucocorticoids exacerbate insulin resistance and stimulate glucose secretion from the liver. This can create substantial and sometimes dangerous fluctuations in circulating glucose concentrations. Additionally, glucocorticoids may increase serum triglycerides and low-density lipoprotein levels. Thus, patients with T2DM or hyperlipidemia may be good candidates for alternative treatments, such as colchicine or NSAIDs.
Cardiovascular Disease
Cardiovascular disease risk has been shown to increase with the use of COX-2 inhibitors. This risk may be present for all NSAIDs. Current FDA labeling suggests limiting NSAID and COX-2 inhibitor use in patients with a history of myocardial infarction (MI), congestive heart failure, or stroke. Given the potential impact on cardiovascular risk factors, including hypertension, T2DM, and hyperlipidemia, glucocorticoids may not be ideal for patients with known CVD or those at high risk.
Recent evidence has shown that colchicine use is associated with a lower risk of MI among patients with gout.21 These results, in addition to a proposed dual role of IL-1 in both gout and CVD, suggest that either colchicine or IL-1 inhibitors may be rational agents in the treatment of acute gout in the context of CVD.22
Hepatic Impairment and GI Bleeding
Patients with cirrhosis should avoid NSAID use due to the potential increased bleeding risk from underlying coagulopathy. Additionally, colchicine clearance may be reduced in patients with severe liver impairment, mandating close surveillance when this agent is used. If hepatic impairment is mild to moderate, judicious use of any of the first-line therapies may be appropriate.
Patients with GI bleeding or a history of peptic ulcer disease should avoid NSAID use because of increased bleeding risk. If an NSAID is used, proton pump inhibitors decrease the risk of NSAID-associated mucosal damage.
Drug Interactions
Colchicine is metabolized by the cytochrome P450 3A4 enzyme (CYP3A4) and is a substrate for P-glycoprotein (P-gp). Therefore, concomitant use of colchicine with potent inhibitors of CYP3A4 or P-gp should be avoided when possible. These agents include macrolide antibiotics (clarithromyocin), calcium channel blockers (verapamil and diltiazem), and cyclosporine (commonly used in transplant patients who are at high risk for gout). New evidence-based dosing recommendations indicate that no dose reduction is required with azithromyocin.23
Nonsteroidal anti-inflammatory drugs are contraindicated with the concomitant use of angiotensin-converting enzyme (ACE) inhibitors and/or diuretics. Prostaglandin production is decreased while using NSAIDs, resulting in increased constriction of afferent renal arterioles and decreased glomerular filtration pressure. This physiologic effect of NSAIDs can be exacerbated when used in combination with ACE inhibitors or diuretics, both of which can also reduce glomerular filtration pressures. Combination therapy with either ACE inhibitors or diuretics increases the risk for NSAID-mediated acute kidney injury. Additionally, NSAID use should be avoided in patients taking anticoagulants such as warfarin or heparin due to increased bleeding risk.
Diagnosis
Diagnosis is a key component of proper treatment of acute gout. A gout diagnosis is usually made based on clinical signs and symptoms, including sudden onset of pain that peaks within 24 hours, past history of acute self-limited attacks of arthritis, first MTP involvement, and an elevated sUA. However, not all these factors must be present. In a study conducted by Janssens and colleagues, these factors plus additional demographics had a sensitivity of 90% and specificity of 65% when compared with crystal diagnosis.24 However, a normal sUA level does not exclude gout as a diagnosis due to the uricosuric effect of the inflammatory process.25 In fact, one observational cohort recorded an average sUA decrease from baseline of 2 mg/dL during an acute gout attack.26
Alternative diagnoses, including septic arthritis, should be considered, particularly in the context of treatment failure (< 50% reduction in pain) within the first 24 to 48 hours. A definitive diagnosis is made by identifying negatively birefringent crystals in the synovial fluid of the affected joint (using polarized microscopy) with negative cultures. In the absence of crystal confirmation, there is an emerging role for imaging in gout diagnosis, including the use of ultrasound and dual-energy computed tomography.27
Long-term Treatment Considerations
During treatment for acute gout attacks, urate-lowering therapy that was initiated before the attack should not be discontinued.28 There is no evidence to suggest that current urate lowering has any AEs during attacks. However, removing treatment may increase sUA levels, precipitating attacks in other joints by “destabilizing” crystals still present. Current recommendations also state that urate-lowering therapy may be started during an attack despite traditionally being deferred until the attack has resolved.28 In a randomized trial comparing a group starting allopurinol 300 mg during an attack vs a placebo group (with all patients receiving anti-inflammatory treatment for the acute attack), there was no difference in pain outcomes.29 Regardless of the chosen timing, lowering and maintaining sUA ≤ 6.0 mg/dL is the primary method for minimizing long-term risk of gout attacks.28
Health care providers should discuss with patients the likely need for indefinite urate-lowering therapy while noting that attacks related to therapy initiation are relatively common.30 Current guidelines recommend starting urate-lowering therapy in low doses (≤ 100 mg/d for allopurinol) and titrating to achieve and maintain the target sUA level. Along with the judicious use of anti-inflammatory prophylaxis, this may minimize attacks related to therapy initiation.3,28 By lowering and maintaining sUA below the target level, monosodium urate crystals will dissolve, thereby eliminating the major inciting factor of acute attacks.
Other day-to-day triggers such as alcohol, meat or seafood consumption, and dehydration exist for some patients with gout. Patients should be informed of these inciting factors, as they could potentially be avoided, reducing the risk of future gout attacks. It is important to recognize, however, that dietary or behavioral interventions have generally yielded only modest sUA reductions. For the majority of patients, therefore, reduction and maintenance of sUA ≤ 6.0 mg/dL requires pharmacologic intervention.
Conclusions
Gout attacks should be treated immediately with pharmacologic treatment when contraindications are absent. First-line treatment options include NSAIDs, colchicine, and systemic glucocorticoids. Use of these modalities can be complicated because of comorbidity and concomitant medication use that is prevalent among patients with gout. Comorbidities commonly limiting treatment choice include hypertension (NSAIDs, glucocorticoids), CKD (NSAIDS, colchicine), CVD (NSAIDs, COX-2 inhibitors, glucocorticoids), T2DM (glucocorticoids), and liver disease (NSAIDs, colchicine). Careful consideration must be given to these comorbidities and contraindications as well as patient preferences.
Gout is an extremely painful arthritis initiated by innate immune responses to monosodium urate crystals that accumulate in affected joints and surrounding tissues. As a result, gout is characterized by painful arthritis flares followed by intervening periods of disease quiescence. Over time, gout can lead to chronic pain, disability, and tophi. Nearly 10% of those aged > 65 years report having gout. The overall prevalence in the U.S. population approaches 4%.1
Gout treatment has 2 overarching goals: alleviating the pain and inflammation caused by acute gout attacks and long-term management that is focused on lowering serum urate (sUA) levels to reduce the risk of future attacks. Alleviating the pain and inflammation of an acute attack is often complicated by patient characteristics, namely, other chronic health conditions that frequently accompany gout, such as diabetes mellitus (DM), chronic kidney disease (CKD), hypertension, and cardiovascular disease (CVD).
Patients with gout tend to be older and have multiple comorbidities that require the use of many medications.2 Because the VA patient population tends to be older, acute gout and attendant complications of treatment are an important consideration for VA health care providers (HCPs).
Recently, the American College of Rheumatology (ACR) released management recommendations for gout, including those for the treatment of acute gout.3 The ACR recommends 3 first-line therapies, but limited guidance is provided for deciding among therapies. This article briefly reviews the relevant ACR recommendations and details important comorbidity and concomitant medication considerations in the treatment of acute gout.
Acute Gout Characteristics
Acute gout attacks are characterized by a rapid onset and escalation with joint pain typically peaking within 24 hours of attack onset. An acute attack often begins to remit after 5 to 12 days without intervention, but complete resolution may take longer in some patients.4 In one study, at 24 hours after attack onset, 16% of patients on placebo had > 50% reduction in pain compared with 70% that had no recovery at all.5 By 48 hours, one-third of patients on placebo achieved a 50% reduction in pain.6
Treatment Recommendations
Therapy for acute gout attacks aims to reduce pain and promote a full, early resolution. The ACR recommends pharmacologic therapy as first-line treatment with adjunctive topical ice and rest as needed.3 Typically, monotherapy is appropriate if the individual is experiencing mild-to-moderate pain affecting ≤ 2 joints of any size. Severe pain or attacks affecting multiple joints may benefit from initial combination therapy. Three first-line therapies are available: nonsteroidal anti-inflammatory drugs (NSAIDs) or cyclooxygenase-2 (COX-2) inhibitors, colchicine, or systemic glucocorticoids (Figure 2).
Few studies compare the efficacy of first-line therapeutic categories. There are no clinical trials directly comparing colchicine with NSAIDs or colchicine with glucocorticoids. No difference in mean reduction of pain and no differences in adverse events (AEs) were shown in a trial that compared glucocorticoids with NSAIDs.8 Thus, without further study, treatment choices made by HCPs are often guided by factors other than the existence of robust evidence.
Treatment with NSAIDs or COX-2 inhibitors should be initiated at the approved dose and continued until the gout attack has completely resolved. In one study, 73% of patients had pain reduction of ≥ 50% when taking NSAIDs relative to only 27% of patients on placebo.8 All available NSAIDs are considered effective, but only 3 NSAIDs are specifically approved for treatment of acute gout (naproxen, indomethacin, and sulindac). There is no evidence supporting one NSAID as being more effective than another; evidence fails to show a meaningful difference.8 Limited evidence indicates that selective COX-2 inhibitors, including celexocib, have similar efficacy as nonselective NSAIDs but may have fewer AEs, driven in part by fewer gastrointestinal (GI) events (6% vs 16% for GI events).8
Colchicine has long been used as prophylaxis for acute gout attacks and has been endorsed for the treatment of acute attacks. Recent evidence suggests that colchicine initially dosed at 1.2 mg followed by a single 0.6-mg dose 1 hour later is as effective with fewer AEs compared with a traditional regimen of 1.2 mg followed by 0.6 mg every hour for up to 6 hours.5 About 40% of patients have 50% pain reduction within 24 hours and a 40% absolute risk reduction in AEs on this low-dose regimen. The efficacy of colchicine relative to other therapies is poorly defined, especially for patients presenting longer after attack onset. The ACR guidelines recommend colchicine only if treatment is initiated within 36 hours of attack onset, but this is based solely on expert consensus. Likewise, the above trial for low-dose colchicine did not provide information about dosing beyond the first 6 hours, leaving little guidance for follow-up treatment of residual pain beyond the 32 hours reported.5 Traditionally, one 0.6-mg dose is provided every 12 to 24 hours.3
Systemic glucocorticoids are also commonly used in treating acute gout.9 There was a small pain reduction benefit for prednisolone, but the difference was not clinically significant in one clinical trial comparing oral prednisolone 30 mg daily for 5 days vs a combination of indomethacin for 5 days and an initial intramuscular injection of diclofenac 75 mg.10 The prednisolone group also had fewer patients with AEs, including abdominal pain (0% vs 30%) and GI bleeding (0% vs 11%). The lower incidence of short-term AEs may be a primary benefit of systemic glucocorticoids.11
Intra-articular glucocorticoids are not suggested first-line therapies but are commonly used by rheumatologists.9 In an uncontrolled study conducted by Fernández and colleagues, intra-articular glucocorticoid injections helped to quickly resolve 20 out of 20 crystal-proven gout attacks.12 However, no randomized controlled trials have examined this approach. Although seemingly efficacious, other considerations are important for this modality. Intra-articular glucocorticoids may not be preferred for polyarticular attacks or attacks in difficult-to-aspirate joints. Additionally, intra-articular glucocorticoids have been anecdotally associated with rebound attacks (ie, attacks that occur shortly after resolution without other interventions). However, the Fernández study had no such attacks occur among participants.12 Finally, septic arthritis must be ruled out as in any case of acute onset monoarticular arthritis.
Biologic agents targeting interleukin-1(IL-1) are not currently approved for gout, although there is burgeoning data suggesting that this strategy may have substantial merit.13 Additionally, there is limited evidence that adrenocorticotropic hormone (ACTH) may provide rapid pain relief when other available therapies are ineffective or contraindicated. However, ACTH studies have not provided robust trial designs, and drug costs remain substantial, thus limiting the widespread use of ACTH in acute gout.14,15 Anti-IL-1 agents and ACTH may both be considered as second-line options if first-line therapies are contraindicated or fail. Careful consideration should be given to AE profiles, patient preferences, and cost.
Comorbidities
Acute gout care, especially in the context of comorbidities, has been identified as a critical treatment concern by an international panel of rheumatologists as part of the 3e (Evidence, Expertise, Exchange) Initiative.16 However, regular clinical trial exclusion criteria have limited data necessary to guide treatment when comorbidities are present. Therefore, studies of acute gout treatment in the context of disease comorbidity represents a major unmet need in understanding and optimizing gout care.
Chronic Kidney Disease
Chronic kidney disease is common in gout; 20% of patients with gout have an estimated glomerular filtration rate (eGFR) of < 30 mL/min.2 Thus, CKD is an important consideration when deciding the best treatment for acute gout. The ACR recommendations do not provide specific guidance on NSAID use in CKD but suggest the potential option of tapering the dose as pain begins to resolve. There is mixed evidence that NSAIDs accelerate CKD progression with the best evidence for high-dose NSAID use.17 When prescribing the concomitant use of NSAIDs with other medications affecting kidney function, HCPs should consider CKD.
For colchicine, current labeling and evidence indicate that no dose adjustments are needed for stage 3 or better CKD (eGFR ≥ 60 mL/min) even among the elderly.18,19 Although labeling indicates that a single unadjusted dose (0.6 mg) can be given once every 2 weeks for those with severe CKD (eGFR < 30 mL/min) or for those who are on dialysis, alternative therapies should be considered, as AEs increase with decreasing renal function.19 Colchicine should not be used in those with eGFR < 10 mL/min.20 All patients who have CKD and are treated with colchicine should be informed of the AEs and closely observed for signs of toxicity, including blood dyscrasias, neuromyopathy, emesis, or diarrhea.
Considering the potential complications for NSAIDs and colchicine, patients with CKD may be good candidates for glucocorticoid therapy, administered either systemically or as an intra-articular injection. Alternatively, second-line agents such as ACTH or IL-1 inhibition may be considered in such patients.
Hypertension
Hypertension is one of the most common comorbidities among patients with gout. It is important for HCP consideration when deciding treatment. Poorly controlled hypertension is a contraindication for both NSAIDs and systemic glucocorticoids. Patients with hypertension in the absence of significant renal impairment may be good candidates for colchicine.
Diabetes and Hyperlipidemia
Glucocorticoids should be avoided if possible in the setting of inadequately controlled type 2 DM (T2DM) or hyperlipidemia. Glucocorticoids exacerbate insulin resistance and stimulate glucose secretion from the liver. This can create substantial and sometimes dangerous fluctuations in circulating glucose concentrations. Additionally, glucocorticoids may increase serum triglycerides and low-density lipoprotein levels. Thus, patients with T2DM or hyperlipidemia may be good candidates for alternative treatments, such as colchicine or NSAIDs.
Cardiovascular Disease
Cardiovascular disease risk has been shown to increase with the use of COX-2 inhibitors. This risk may be present for all NSAIDs. Current FDA labeling suggests limiting NSAID and COX-2 inhibitor use in patients with a history of myocardial infarction (MI), congestive heart failure, or stroke. Given the potential impact on cardiovascular risk factors, including hypertension, T2DM, and hyperlipidemia, glucocorticoids may not be ideal for patients with known CVD or those at high risk.
Recent evidence has shown that colchicine use is associated with a lower risk of MI among patients with gout.21 These results, in addition to a proposed dual role of IL-1 in both gout and CVD, suggest that either colchicine or IL-1 inhibitors may be rational agents in the treatment of acute gout in the context of CVD.22
Hepatic Impairment and GI Bleeding
Patients with cirrhosis should avoid NSAID use due to the potential increased bleeding risk from underlying coagulopathy. Additionally, colchicine clearance may be reduced in patients with severe liver impairment, mandating close surveillance when this agent is used. If hepatic impairment is mild to moderate, judicious use of any of the first-line therapies may be appropriate.
Patients with GI bleeding or a history of peptic ulcer disease should avoid NSAID use because of increased bleeding risk. If an NSAID is used, proton pump inhibitors decrease the risk of NSAID-associated mucosal damage.
Drug Interactions
Colchicine is metabolized by the cytochrome P450 3A4 enzyme (CYP3A4) and is a substrate for P-glycoprotein (P-gp). Therefore, concomitant use of colchicine with potent inhibitors of CYP3A4 or P-gp should be avoided when possible. These agents include macrolide antibiotics (clarithromyocin), calcium channel blockers (verapamil and diltiazem), and cyclosporine (commonly used in transplant patients who are at high risk for gout). New evidence-based dosing recommendations indicate that no dose reduction is required with azithromyocin.23
Nonsteroidal anti-inflammatory drugs are contraindicated with the concomitant use of angiotensin-converting enzyme (ACE) inhibitors and/or diuretics. Prostaglandin production is decreased while using NSAIDs, resulting in increased constriction of afferent renal arterioles and decreased glomerular filtration pressure. This physiologic effect of NSAIDs can be exacerbated when used in combination with ACE inhibitors or diuretics, both of which can also reduce glomerular filtration pressures. Combination therapy with either ACE inhibitors or diuretics increases the risk for NSAID-mediated acute kidney injury. Additionally, NSAID use should be avoided in patients taking anticoagulants such as warfarin or heparin due to increased bleeding risk.
Diagnosis
Diagnosis is a key component of proper treatment of acute gout. A gout diagnosis is usually made based on clinical signs and symptoms, including sudden onset of pain that peaks within 24 hours, past history of acute self-limited attacks of arthritis, first MTP involvement, and an elevated sUA. However, not all these factors must be present. In a study conducted by Janssens and colleagues, these factors plus additional demographics had a sensitivity of 90% and specificity of 65% when compared with crystal diagnosis.24 However, a normal sUA level does not exclude gout as a diagnosis due to the uricosuric effect of the inflammatory process.25 In fact, one observational cohort recorded an average sUA decrease from baseline of 2 mg/dL during an acute gout attack.26
Alternative diagnoses, including septic arthritis, should be considered, particularly in the context of treatment failure (< 50% reduction in pain) within the first 24 to 48 hours. A definitive diagnosis is made by identifying negatively birefringent crystals in the synovial fluid of the affected joint (using polarized microscopy) with negative cultures. In the absence of crystal confirmation, there is an emerging role for imaging in gout diagnosis, including the use of ultrasound and dual-energy computed tomography.27
Long-term Treatment Considerations
During treatment for acute gout attacks, urate-lowering therapy that was initiated before the attack should not be discontinued.28 There is no evidence to suggest that current urate lowering has any AEs during attacks. However, removing treatment may increase sUA levels, precipitating attacks in other joints by “destabilizing” crystals still present. Current recommendations also state that urate-lowering therapy may be started during an attack despite traditionally being deferred until the attack has resolved.28 In a randomized trial comparing a group starting allopurinol 300 mg during an attack vs a placebo group (with all patients receiving anti-inflammatory treatment for the acute attack), there was no difference in pain outcomes.29 Regardless of the chosen timing, lowering and maintaining sUA ≤ 6.0 mg/dL is the primary method for minimizing long-term risk of gout attacks.28
Health care providers should discuss with patients the likely need for indefinite urate-lowering therapy while noting that attacks related to therapy initiation are relatively common.30 Current guidelines recommend starting urate-lowering therapy in low doses (≤ 100 mg/d for allopurinol) and titrating to achieve and maintain the target sUA level. Along with the judicious use of anti-inflammatory prophylaxis, this may minimize attacks related to therapy initiation.3,28 By lowering and maintaining sUA below the target level, monosodium urate crystals will dissolve, thereby eliminating the major inciting factor of acute attacks.
Other day-to-day triggers such as alcohol, meat or seafood consumption, and dehydration exist for some patients with gout. Patients should be informed of these inciting factors, as they could potentially be avoided, reducing the risk of future gout attacks. It is important to recognize, however, that dietary or behavioral interventions have generally yielded only modest sUA reductions. For the majority of patients, therefore, reduction and maintenance of sUA ≤ 6.0 mg/dL requires pharmacologic intervention.
Conclusions
Gout attacks should be treated immediately with pharmacologic treatment when contraindications are absent. First-line treatment options include NSAIDs, colchicine, and systemic glucocorticoids. Use of these modalities can be complicated because of comorbidity and concomitant medication use that is prevalent among patients with gout. Comorbidities commonly limiting treatment choice include hypertension (NSAIDs, glucocorticoids), CKD (NSAIDS, colchicine), CVD (NSAIDs, COX-2 inhibitors, glucocorticoids), T2DM (glucocorticoids), and liver disease (NSAIDs, colchicine). Careful consideration must be given to these comorbidities and contraindications as well as patient preferences.
1. Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63(10):3136-3141.
2. Zhu Y, Pandya BJ, Choi HK. Comorbidities of gout and hyperuricemia in the US general population: NHANES 2007-2008. Am J Med. 2012;125(7):679-687.e1.
3. Khanna D, Khanna PP, Fitzgerald JD, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 2: therapy and antiinflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res (Hoboken). 2012;64(10):1447-1461.
4. Bellamy N, Downie WW, Buchanan WW. Observations on spontaneous improvement in patients with podagra: implications for therapeutic trials of non-steroidal anti-inflammatory drugs. Br J Clin Pharmacol. 1987;24(1):33-36.
5. Terkeltaub RA, Furst DE, Bennett K, Kook KA, Crockett RS, Davis MW. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum. 2010;62(4):1060-1068.
6. Ahern MJ, Reid C, Gordon TP, McCredie M, Brooks PM, Jones M. Does colchicine work? The results of the first controlled study in acute gout. Aust N Z J Med. 1987;17(3):301-304.
7. Puig JG, Michán AD, Jiménez ML, et al. Female gout. Clinical spectrum and uric acid metabolism. Arch Intern Med. 1991;151(4):726-732.
8. van Durme CM, Wechalekar MD, Buchbinder R, Schlesinger N, van der Heijde D, Landewé RB. Non-steroidal anti-inflammatory drugs for acute gout. Cochrane Database Syst Rev. 2014;9:CD010120.
9. Schlesinger N, Moore DF, Sun JD, Schumacher HR Jr. A survey of current evaluation and treatment of gout. J Rheumatol. 2006;33(10):2050-2052.
10. Man CY, Cheung IT, Cameron PA, Rainer TH. Comparison of oral prednisolone/paracetamol and oral indomethacin/paracetamol combination therapy in the treatment of acute goutlike arthritis: a double-blind, randomized, controlled trial. Ann Emerg Med. 2007;49(5):670-677.
11. Janssens HJ, Lucassen PL, Van de Laar FA, Janssen M, Van de Lisdonk EH. Systemic corticosteroids for acute gout. Cochrane Database Syst Rev. 2008;(2):CD005521.
12. Fernández C, Noguera R, González JA, Pascual E. Treatment of acute attacks of gout with a small dose of intraarticular triamcinolone acetonide. J Rheumatol. 1999;26(10):2285-2286.
13. Burns CM, Wortmann RL. Gout therapeutics: new drugs for an old disease. Lancet. 2011;377(9760):165-177.
14. Axelrod D, Preston S. Comparison of parenteral adrenocorticotropic hormone with oral indomethacin in the treatment of acute gout. Arthritis Rheum. 1988;31(6):803-805.
15. Ritter J, Kerr LD, Valeriano-Marcet J, Spiera H. ACTH revisited: effective treatment for acute crystal induced synovitis in patients with multiple medical problems. J Rheumatol. 1994;21(4):696-699.
16. Sivera F, Andrés M, Carmona L, et al. Multinational evidence-based recommendations for the diagnosis and management of gout: integrating systematic literature review and expert opinion of a broad panel of rheumatologists in the 3e initiative. Ann Rheum Dis. 2014;73(2):328-335.
17. Nderitu P, Doos L, Jones PW, Davies SJ, Kadam UT. Non-steroidal anti-inflammatory drugs and chronic kidney disease progression: a systematic review. Fam Pract. 2013;30(3):247-255.
18. Wason S, Faulkner RD, Davis MW. Are dosing adjustments required for colchicine in the elderly compared with younger patients? Adv Ther. 2012;29(6):551-561.
19. Wason S, Mount D, Faulkner R. Single-dose, open-label study of the differences in pharmacokinetics of colchicine in subjects with renal impairment, including end-stage renal disease. Clin Drug Investig. 2014;34(12):845-855.
20. Hanlon JT, Aspinall SL, Semla TP, et al. Consensus guidelines for oral dosing of primarily renally cleared medications in older adults. J Am Geriatr Soc. 2009;57(2):335-340.
21. Crittenden DB, Lehmann RA, Schneck L, et al. Colchicine use is associated with decreased prevalence of myocardial infarction in patients with gout. J Rheumatol. 2012;39(7):1458-1464.
22. Esser N, Paquot N, Scheen AJ. Anti-inflammatory agents to treat or prevent type 2 diabetes, metabolic syndrome and cardiovascular disease. Expert Opin Investig Drugs. 2015;24(3):283-307.
23. Terkeltaub RA, Furst DE, Digiacinto JL, Kook KA, Davis MW. Novel evidence-based colchicine dose-reduction algorithm to predict and prevent colchicine toxicity in the presence of cytochrome P450 3A4/P-glycoprotein inhibitors. Arthritis Rheum. 2011;63(8):2226-2237.
24. Janssens HJ, Fransen J, van de Lisdonk EH, van Riel PL, van Weel C, Janssen M. A diagnostic rule for acute gouty arthritis in primary care without joint fluid analysis. Arch Intern Med. 2010;170(13):1120-1126.
25. Urano W, Yamanaka H, Tsutani H, et al. The inflammatory process in the mechanism of decreased serum uric acid concentrations during acute gouty arthritis. J Rheumatol. 2002;29(9):1950-1953.
26. Logan JA, Morrison E, McGill PE. Serum uric acid in acute gout. Ann Rheum Dis. 1997;56(11):696-697.
27. Ogdie A, Taylor WJ, Weatherall M, et al. Imaging modalities for the classification of gout: systematic literature review and meta-analysis. Ann Rheum Dis. 2015; 74(10):1868-1874.
28. Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012;64(10):1431-1446.
29. Taylor TH, Mecchella JN, Larson RJ, Kerin KD, Mackenzie TA. Initiation of allopurinol at first medical contact for acute attacks of gout: a randomized clinical trial. Am J Med. 2012;125(11):1126-11134.e7.
30. Becker MA, MacDonald PA, Hunt BJ, Lademacher C, Joseph-Ridge N. Determinants of the clinical outcomes of gout during the first year of urate-lowering therapy. Nucleosides Nucleotides Nucleic Acids. 2008;27(6):585-591.
1. Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63(10):3136-3141.
2. Zhu Y, Pandya BJ, Choi HK. Comorbidities of gout and hyperuricemia in the US general population: NHANES 2007-2008. Am J Med. 2012;125(7):679-687.e1.
3. Khanna D, Khanna PP, Fitzgerald JD, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 2: therapy and antiinflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res (Hoboken). 2012;64(10):1447-1461.
4. Bellamy N, Downie WW, Buchanan WW. Observations on spontaneous improvement in patients with podagra: implications for therapeutic trials of non-steroidal anti-inflammatory drugs. Br J Clin Pharmacol. 1987;24(1):33-36.
5. Terkeltaub RA, Furst DE, Bennett K, Kook KA, Crockett RS, Davis MW. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum. 2010;62(4):1060-1068.
6. Ahern MJ, Reid C, Gordon TP, McCredie M, Brooks PM, Jones M. Does colchicine work? The results of the first controlled study in acute gout. Aust N Z J Med. 1987;17(3):301-304.
7. Puig JG, Michán AD, Jiménez ML, et al. Female gout. Clinical spectrum and uric acid metabolism. Arch Intern Med. 1991;151(4):726-732.
8. van Durme CM, Wechalekar MD, Buchbinder R, Schlesinger N, van der Heijde D, Landewé RB. Non-steroidal anti-inflammatory drugs for acute gout. Cochrane Database Syst Rev. 2014;9:CD010120.
9. Schlesinger N, Moore DF, Sun JD, Schumacher HR Jr. A survey of current evaluation and treatment of gout. J Rheumatol. 2006;33(10):2050-2052.
10. Man CY, Cheung IT, Cameron PA, Rainer TH. Comparison of oral prednisolone/paracetamol and oral indomethacin/paracetamol combination therapy in the treatment of acute goutlike arthritis: a double-blind, randomized, controlled trial. Ann Emerg Med. 2007;49(5):670-677.
11. Janssens HJ, Lucassen PL, Van de Laar FA, Janssen M, Van de Lisdonk EH. Systemic corticosteroids for acute gout. Cochrane Database Syst Rev. 2008;(2):CD005521.
12. Fernández C, Noguera R, González JA, Pascual E. Treatment of acute attacks of gout with a small dose of intraarticular triamcinolone acetonide. J Rheumatol. 1999;26(10):2285-2286.
13. Burns CM, Wortmann RL. Gout therapeutics: new drugs for an old disease. Lancet. 2011;377(9760):165-177.
14. Axelrod D, Preston S. Comparison of parenteral adrenocorticotropic hormone with oral indomethacin in the treatment of acute gout. Arthritis Rheum. 1988;31(6):803-805.
15. Ritter J, Kerr LD, Valeriano-Marcet J, Spiera H. ACTH revisited: effective treatment for acute crystal induced synovitis in patients with multiple medical problems. J Rheumatol. 1994;21(4):696-699.
16. Sivera F, Andrés M, Carmona L, et al. Multinational evidence-based recommendations for the diagnosis and management of gout: integrating systematic literature review and expert opinion of a broad panel of rheumatologists in the 3e initiative. Ann Rheum Dis. 2014;73(2):328-335.
17. Nderitu P, Doos L, Jones PW, Davies SJ, Kadam UT. Non-steroidal anti-inflammatory drugs and chronic kidney disease progression: a systematic review. Fam Pract. 2013;30(3):247-255.
18. Wason S, Faulkner RD, Davis MW. Are dosing adjustments required for colchicine in the elderly compared with younger patients? Adv Ther. 2012;29(6):551-561.
19. Wason S, Mount D, Faulkner R. Single-dose, open-label study of the differences in pharmacokinetics of colchicine in subjects with renal impairment, including end-stage renal disease. Clin Drug Investig. 2014;34(12):845-855.
20. Hanlon JT, Aspinall SL, Semla TP, et al. Consensus guidelines for oral dosing of primarily renally cleared medications in older adults. J Am Geriatr Soc. 2009;57(2):335-340.
21. Crittenden DB, Lehmann RA, Schneck L, et al. Colchicine use is associated with decreased prevalence of myocardial infarction in patients with gout. J Rheumatol. 2012;39(7):1458-1464.
22. Esser N, Paquot N, Scheen AJ. Anti-inflammatory agents to treat or prevent type 2 diabetes, metabolic syndrome and cardiovascular disease. Expert Opin Investig Drugs. 2015;24(3):283-307.
23. Terkeltaub RA, Furst DE, Digiacinto JL, Kook KA, Davis MW. Novel evidence-based colchicine dose-reduction algorithm to predict and prevent colchicine toxicity in the presence of cytochrome P450 3A4/P-glycoprotein inhibitors. Arthritis Rheum. 2011;63(8):2226-2237.
24. Janssens HJ, Fransen J, van de Lisdonk EH, van Riel PL, van Weel C, Janssen M. A diagnostic rule for acute gouty arthritis in primary care without joint fluid analysis. Arch Intern Med. 2010;170(13):1120-1126.
25. Urano W, Yamanaka H, Tsutani H, et al. The inflammatory process in the mechanism of decreased serum uric acid concentrations during acute gouty arthritis. J Rheumatol. 2002;29(9):1950-1953.
26. Logan JA, Morrison E, McGill PE. Serum uric acid in acute gout. Ann Rheum Dis. 1997;56(11):696-697.
27. Ogdie A, Taylor WJ, Weatherall M, et al. Imaging modalities for the classification of gout: systematic literature review and meta-analysis. Ann Rheum Dis. 2015; 74(10):1868-1874.
28. Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012;64(10):1431-1446.
29. Taylor TH, Mecchella JN, Larson RJ, Kerin KD, Mackenzie TA. Initiation of allopurinol at first medical contact for acute attacks of gout: a randomized clinical trial. Am J Med. 2012;125(11):1126-11134.e7.
30. Becker MA, MacDonald PA, Hunt BJ, Lademacher C, Joseph-Ridge N. Determinants of the clinical outcomes of gout during the first year of urate-lowering therapy. Nucleosides Nucleotides Nucleic Acids. 2008;27(6):585-591.
Minimum 5-Year Results With Duracon Press-Fit Metal-Backed Patellae
The metal-backed patella was originally designed to address the shortcomings of cemented, all-polyethylene patellae: deformation, aseptic loosening, stress fractures of polyethylene, and possible thermal damage from bone cement.1-3 Several long-term studies have found very good outcomes with use of all-polyethylene patellae.4-6 However, complications of using an all-polyethylene patella reportedly accounted for up to half of all knee revisions, and during revision surgery patellar bone stock was often found to have been compromised.7
The intention behind the design of press-fit metal-backed patellae was to address the shortcomings of all-polyethylene patellae by eliminating the need for bone cement and providing stiffness that would help resist polyethylene deformation while decreasing implant–bone interface stresses.8 However, early design iterations of metal-backed patellae demonstrated short-term failures—most commonly, local polyethylene wear damaging the locking mechanism and subsequent dissociation or fracture from the metal baseplate; polyethylene delamination from the metal baseplate; and failure of interface fixation.9,10 On the other hand, good fixation with bony ingrowth was observed in both titanium and cobalt-chromium porous-coated patellae.1,3,9,11-13 Overall, however, negative outcomes reported for metal-backed patellae led many surgeons to abandon these components and return to using cemented all-polyethylene patellae.
Negative outcomes of earlier metal-backed patellae designs have overshadowed reports of positive outcomes achieved with careful attention paid to component design, patellar tracking, and surgical technique.2,3,14 Subsequent design improvements (eg, a third stabilizing peg, thicker polyethylene, improved conformity) produced excellent outcomes.8,12,15 The advantages of using a metal-backed patella (eg, uniform load sharing, decreased polyethylene deformation, potential for biological fixation) may be unjustly outweighed by the fear of patellar component failure.3
Our 30-plus years of experience with metal-backed patellar components reflect the evolving effect of component design on outcome. Much as reported elsewhere, we found earlier component failures were caused by poor locking mechanisms, thin polyethylene, poor tracking, and minimal femur contact. Over the past decade, however, our outcomes with Duracon metal-backed patellae (Stryker) have been encouraging. We think these positive outcomes, seen over minimum 5-year follow-up, are largely attributable to the thicker polyethylene and improved articular conformity of this component relative to earlier designs. We have also found it helpful to adhere to certain criteria when implanting metal-backed patellae, and we think adhering to these criteria, along with improved component design, indicates use of press-fit metal-backed patellae. In this article, we report our failure incidence with use of this device at minimum 5-year follow-up.
Materials and Methods
In this single-center study, we performed clinical and independent radiographic reviews of 88 primary press-fit metal-backed patellae with minimum 5-year follow-up. All components were the same design (Duracon metal-backed patella) from the same manufacturer (Stryker).
This study, which began in September 2003, was reviewed and approved by the Western Institutional Review Board (WIRB). Either the investigator (Dr. Hedley) or the clinical study coordinator gave study candidates a full explanation of the study and answered any questions. Patients who still wanted to participate in the study signed WIRB consent forms after their index surgery but before minimum 5-year follow-up.
Device Description
This Duracon patella has a porous-coated cobalt-chromium metal back intended for press-fit fixation, 3 cobalt-chromium porous-coated pegs, and a preassembled polyethylene anterior surface (Figure 1). Four sizes are available to fit the peripheral shape of the resected patella.
This patella has 3 styles: symmetric, asymmetric, and conversion. In this study, we used only the asymmetric and conversion styles. The design of each style incorporates medial/lateral facets intended to conform to the convex intercondylar radii of the femoral component, thereby allowing the patella to ride deeply in the recessed patellofemoral groove. The asymmetric patella is a resurfacing component with a generous polyethylene thickness (4.6 mm at its thinnest) and a larger lateral facet for more bone coverage. The asymmetric patella naturally medializes component placement. The articulating surface of the conversion patella is identical to that of the asymmetric patella. However, the conversion patella allows for exchange of the polyethylene portion of the implant without revising a stable, well-fixed metal baseplate.
Patient Selection
Candidates were recruited from a group of metal-backed patella patients within Dr. Hedley’s medical practice. All candidates had undergone primary total knee arthroplasty and received a Duracon press-fit metal-backed patella. All recruited patients had undergone primary knee arthroplasty at least 5 years before clinical and radiographic evaluation. Patients were included in the study if they had a diagnosis of noninflammatory degenerative joint disease (eg, osteoarthritis, traumatic arthritis, avascular necrosis). Patients with body mass index higher than 40 were excluded from the study.
Surgical Technique
The patella is everted completely or as much as feasible. Debridement is done circumferentially around the patella. Adherent fat and pseudomeniscus are stripped back until the surgeon sees the entry point of the quadriceps tendon fibers above and the patella tendon fibers below. The cut is then made at this level to remove as much bone as needed to restore the normal height of the patella with the implant in place. The cut is usually made by hand—without guides but with the patella stabilized with a towel clip above and below to prevent any movement during the action.
The desired cut must be absolutely planar, and this should be checked by placing the edge of the blade across the interface. Repeated passes with the saw blade are needed if the cut is not 100% planar. Once the cut is made, the patella is sized with the patella sizers and drill guide. After the appropriate size is selected, the patella is drilled with a bit that is slightly undersized from the size of the pegs (1/32 inch smaller than the bit supplied by the manufacturer).
Once the patella is prepared, the rest of the knee arthroplasty is performed. The patella is press-fit as the last component to be inserted.
Radiologic Review
Radiographic analysis was performed by an independent reviewer according to the current Knee Society total knee arthroplasty roentgenographic evaluation and scoring system (Figure 2).16 The reviewer was an orthopedist specializing in hip and knee surgery. Radiographs the reviewer deemed questionable were shown to another independent hip and knee surgeon for validation. In all cases, the second reviewer confirmed the first reviewer’s initial recorded observations.
KSS (Knee Society Scale), WOMAC (Western Ontario and McMaster Universities Arthritis Index), and SF-36 (36-Item Short Form Health Survey) were also used to evaluate effectiveness in this protocol.
Survivorship Calculations
Kaplan-Meier survivorship was determined for all metal-backed patellae. For survival analysis, only knees with radiographic data were included (74 knees). Mean follow-up was 75.8 months (range, 60-105 months).
Seventy-four patients (88 knees) met the study criteria (Table). At minimum 5-year follow-up, complete data were acquired for 59 patients (72 knees). Of the total group, 14 knees did not have radiographic data. Those knees were categorized as lost to follow-up and were excluded from the survivorship analysis. The status of patients enrolled in the study at minimum 5-year follow-up is shown in the Table.
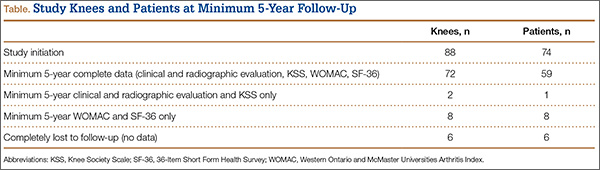
Mann-Whitney U test (nonparametric t test) was used to compare WOMAC and SF-36 scores between the “complete” and the “WOMAC and SF-36 only” data groups.
Statistical Analysis
Kaplan-Meier survivorship probabilities (asymmetric method) were calculated using SAS Version 9.2 (SAS Institute); 95% pointwise confidence limits were used.
The Mann-Whitney U test is a nonparametric analogue to the independent-samples t test. It was used here to compare WOMAC and SF-36 scores of patients with “complete” data with scores of patients with “WOMAC and SF-36 only” data. In either group, for patients who had primary bilateral knee arthroplasty, mean WOMAC and SF-36 scores were used.
Comparisons were made between the unilateral and bilateral knee arthroplasty groups. There were no differences in age, height, or weight (Mann-Whitney U test) or in sex, primary diagnosis, or number of patients lost to follow-up (Fisher exact test). Fisher exact test (vs χ2 test) was used for the contingency table analysis because of small cell sizes (eg, ≤10 females in ‘‘both knees” group), suggesting the unilateral and bilateral patients did not differ in demographics.
For all patient-reported questionnaires, bilateral patients were given the opportunity to note any differences between their knee arthroplasties, but none of these patients made any special notations. We interpreted this to mean that all survey responses from bilateral patients were applicable to both knee arthroplasties.
Results
Seventy-four patients (88 knees) were enrolled in the study: 31 women (41.2%) and 43 men (58.1%). At time of surgery, mean age was 59.7 years (range, 40-86 years), and mean body mass index was 30.6 (range, 19.1-39.6). Eighty-three knees were diagnosed with osteoarthritis, and 5 knees were diagnosed with posttraumatic arthritis. Mean time to follow-up was 74.8 months (range, 60-105 months). Fourteen knees (14 patients) were considered lost to follow-up. However, 8 patients (8 knees) were contacted by telephone about the status of their knee(s), and all 8 completed and returned the minimum 5-year follow-up WOMAC and SF-36 forms; they did not return for their minimum 5-year clinical or radiographic evaluations.
Asymmetric patellae were used in 24 knees, conversion patellae in 64 knees (88 knees total). Forty-nine months after surgery, 1 patella was revised for loosening at its interface with the bone. The 51-year-old active female patient’s asymmetric patella was revised to a conversion patella. The decision to implant another metal-backed device was based on its high density; proper intrusion of acrylic cement would have been questionable. Some early wear was observed on the tibial insert, which was replaced. Sixty-eight months after the revision, the patient was asymptomatic, with a KSS Pain score of 96 and a KSS Function score of 100 (Figure 3). Another revision, for tibial insert exchange only, was performed 48 months after surgery. During this revision, the patella was evaluated and found to be well fixed and functioning normally.
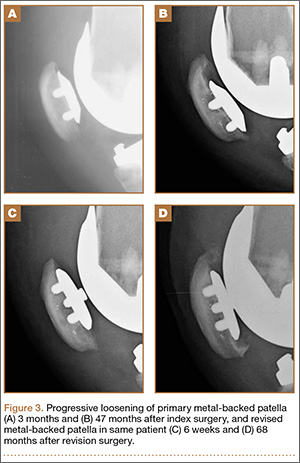
Survivorship of the Duracon metal-backed patella at minimum 5-year follow-up was estimated to be 93.95%, with bounds of 73.61% and 98.74%.
Radiographic analysis revealed no radiolucencies larger than 1 mm (Figure 4). Seventeen 1-mm radiolucencies were recorded: 6 (35.3%) in zone 1, 2 (11.8%) in zone 2, and 9 (52.9%) in zone 4. Twelve (70.6%) of the 17 radiolucencies were in the left knee. Nine radiolucencies were in women and 8 in men. Most (55.6%) of the women’s radiolucencies were in zone 1, and most (75.0%) of the men’s were in zone 4. There were no loose beads other than in the case that was later revised.
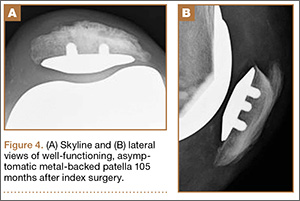
KSS, WOMAC, and SF-36 scores and radiographic reviews were used to evaluate effectiveness in accordance with the protocol. At minimum 5-year follow-up, mean KSS Pain score was 94.10 (range, 55-100), and mean KSS Function score was 92.67 (range, 60-100). Mean WOMAC score was 2.21 (range, 0-19.70), mean SF-36 Physical score was 83.65 (range, 30.70-100), and mean SF-36 Mental score was 89.41 (range, 1.4-100).
The preceding calculations do not include WOMAC and SF-36 data for the 8 patients (8 knees) who were counted as lost to follow-up but who submitted minimum 5-year follow-up data. We compared these 8 patients with the 60 patients (74 knees) who had complete WOMAC and SF-36 data at the end of the study in order to determine whether there were any statistically significant differences between the 2 groups’ mean scores. No statistically significant differences were detected in any WOMAC or SF-36 category (α = 0.05).
Discussion
Metal-backed patellar components were originally designed to address the shortcomings (eg, fracture, deformation, aseptic loosening) of cemented all-polyethylene patellae.1-3 It was thought that the stiffness of the metal could help resist polyethylene deformation and that the press-fit interface with bone might eliminate issues related to bone cement.8 However, short-term failures were reported with early metal-backed designs.9,10 At the same time, good fixation with bone ingrowth was observed in both titanium and cobalt-chromium porous-coated patellae.1,3,9-12,17 Further, reports of poor outcomes with some metal-backed patella designs overshadowed reports of positive outcomes.2,3 In all reports (of both poor and positive outcomes), component design, patellar tracking, and surgical technique were cited as contributing to implant success.2,3,14,17,18 Subsequent design improvements (eg, use of a third stabilizing peg, thicker polyethylene, improved conformity) produced excellent outcomes.8,12,15
Our early results are similar to those reported in the literature, and we observed markedly better outcomes that we think resulted from component design improvements. Over the past decade, this has been particularly true with our use of the Duracon metal-backed patella, which has thicker polyethylene, better articular conformity, and a third stabilizing peg, all of which were previously noted as contributing to a successful metal-backed patellar component.2,12,14,15,19 In our study, all 72 knees radiographically evaluated and independently reviewed at minimum 5-year follow-up had well-fixed press-fit metal-backed patellae. Seventeen patellae had 1-mm radiolucencies; the other 59 had no radiolucencies in any zone around the patella–bone interface.
One of the most important aspects of removing a metal-backed patellar component from a patella is that the remaining bone stock is often far superior to the stock available after revision of a cemented patella. Careful removal should leave an excellent bony bed for reimplantation.
We think that surgeons should adhere to certain indications and contraindications when implanting metal-backed patellae and that doing so can contribute to successful outcomes. Type of bone stock available should be considered, as successful biological fixation relies on a good blood supply. A dense (or thin) patella in which intrusion of acrylic cement is improbable or impossible may favor use of a metal-backed patella. Cement is not an adhesive but a grout, so successful cementation requires intrusion of cement into the interstices of the cancellous bone. As adequate intrusion of cement into dense bone is not possible, cementation may not be the best option. Some patellae have failed because of peg “shear-off,”9 likely caused not by failure of peg strength but by failure of cement fixation at the nonpeg interface.20,21 Polyethylene pegs fail when used as the sole method of fixation (they were never designed for that). In addition, we think younger patients are often indicated for a metal-backed patella because, over the long term, loosening of a cemented patella (and the accompanying stress shielding and osteolysis) may cause severe patellar bone destruction. Last, we have found that abnormally high or small patellae are not good candidates for cement fixation because they tend to work themselves loose riding on and off the superior flange. These types of patellae appear to have a much sturdier and longer lasting interface than cement, once biological fixation has occurred.
In summary, we think the indications for a metal-backed implant are a patella that is dense or sclerotic; a patella that is thin, abnormally high, or small; and a younger patient. In addition, a metal-backed implant is not indicated for soft, osteoporotic bone.
This study had a few limitations. Fourteen knees (14 patients), or 15.9% of all knees in the study, were categorized as lost to follow-up. Comparing the WOMAC and SF-36 scores of 8 patients (8 knees) who completed minimum 5-year follow-up but were not clinically evaluated with the scores of patients who had complete data, we found no statistically significant differences in any category. However, 5-year follow-up clinical data were available for those 8 patients. Nevertheless, 74 knees were available for radiologic evaluation, and during telephone interviews all 8 patients indicated they had their original implant(s) and were asymptomatic.
Our experience with the Duracon metal-backed patella has been encouraging. In the study reported here, there were no failures caused by dissociation of plastic. We think that, because the porous coating is under almost constant compression, biological fixation is likely in most instances, as observed in our minimum 5-year radiologic results. Given our minimum 5-year follow-up results with uncemented metal-backed patellae, we think their use may be a viable alternative to use of all-polyethylene patellae.
1. Firestone TP, Teeny SM, Krackow KA, Hungerford DS. The clinical and roentgenographic results of cementless porous-coated patellar fixation. Clin Orthop Relat Res. 1991;273:184-189.
2. Laskin RS, Bucknell A. The use of metal-backed patellar prostheses in total knee arthroplasty. Clin Orthop Relat Res. 1990;260:52-55.
3. Evanich CJ, Tkach TK, von Glinski S, Camargo MP, Hofmann AA. 6- to 10-year experience using countersunk metal-backed patellas. J Arthroplasty. 1997;12(2):149-154.
4. Schwartz AJ, Della Vale CJ, Rosenberg AG, Jacobs JJ, Berger RA, Galante JO. Cruciate-retaining TKA using a third-generation system with a four-pegged tibial component: a minimum 10-year followup note. Clin Orthop Relat Res. 2010;468(8):2160-2167.
5. Bisschop R, Brouwer RW, Van Raay JJ. Total knee arthroplasty in younger patients: a 13-year follow-up study. Orthopedics. 2010;33(12):876-880.
6. Dixon MC, Brown RR, Parsch D, Scott RD. Modular fixed-bearing total knee arthroplasty with retention of the posterior cruciate ligament. A study of patients followed for a minimum of fifteen years. J Bone Joint Surg Am. 2005;87(3):598-603.
7. Brick GW, Scott RD. The patellofemoral component of total knee arthroplasty. Clin Orthop Relat Res. 1988;231)163-178.
8. Garcia RM, Kraay MJ, Goldberg VM. Isolated all-polyethylene patellar revisions for metal-backed patellar failure. Clin Orthop Relat Res. 2008;466(11):2784-2789.
9. Rosenberg AG, Andriacchi TP, Barden R, Galante JO. Patellar component failure in cementless total knee arthroplasty. Clin Orthop Relat Res. 1988;(236):106-114.
10. Stulberg SD, Stulberg BN, Hamati Y, Tsao A. Failure mechanisms of metal-backed patellar components. Clin Orthop Relat Res. 1988;236:88-105.
11. Sundfeldt M, Johansson CB, Regner L, Albrektsson T, Carlsson LV. Long-term results of a cementless knee prosthesis with a metal-backed patellar component: clinical and radiological follow-up with histology from retrieved components. J Long Term Eff Med Implants. 2003;13(4):341-354.
12. Kraay MJ, Darr OJ, Salata MJ, Goldberg VM. Outcome of metal-backed cementless patellar components: the effect of implant design. Clin Orthop Relat Res. 2001;392:239-244.
13. Jensen LN, Lund B, Gotfredsen K. Bone growth into a revised porous-coated patellar implant. Acta Orthop Scand. 1990;61(3):213-216.
14. Hsu HP, Walker PS. Wear and deformation of patellar components in total knee arthroplasty. Clin Orthop Relat Res. 1989;246:260-265.
15. Jordan LR, Sorrells RB, Jordan LC, Olivo JL. The long-term results of a metal-backed mobile bearing patella. Clin Orthop Relat Res. 2005;436:111-118.
16. Ewald FC. The Knee Society total knee arthroplasty roentgenographic evaluation and scoring system. Clin Orthop Relat Res. 1989;248:9-12.
17. Bayley JC, Scott RD, Ewald FC, Holmes GB Jr. Failure of the metal-backed patellar component after total knee replacement. J Bone Joint Surg Am. 1988;70(5):668-674.
18. Lombardi AV Jr, Engh GA, Volz RG, Albrigo JL, Brainard BJ. Fracture/dissociation of the polyethylene in metal-backed patellar components in total knee arthroplasty. J Bone Joint Surg Am. 1988;70(5):675-679.
19. Moreland JR. Mechanisms of failure in total knee arthroplasty. Clin Orthop Relat Res. 1988;226:49-64.
20. Francke EI, Lachiewicz PF. Failure of a cemented all-polyethylene patellar component of a press-fit condylar total knee arthroplasty. J Arthroplasty. 2000;15(2):234-237.
21. Stulberg BN, Wright TM, Stoller AP, Mimnaugh KL, Mason JJ. Bilateral patellar component shear failure of highly cross-linked polyethylene components: report of a case and laboratory analysis of failure mechanisms. J Arthroplasty. 2012;27(5):789-796.
The metal-backed patella was originally designed to address the shortcomings of cemented, all-polyethylene patellae: deformation, aseptic loosening, stress fractures of polyethylene, and possible thermal damage from bone cement.1-3 Several long-term studies have found very good outcomes with use of all-polyethylene patellae.4-6 However, complications of using an all-polyethylene patella reportedly accounted for up to half of all knee revisions, and during revision surgery patellar bone stock was often found to have been compromised.7
The intention behind the design of press-fit metal-backed patellae was to address the shortcomings of all-polyethylene patellae by eliminating the need for bone cement and providing stiffness that would help resist polyethylene deformation while decreasing implant–bone interface stresses.8 However, early design iterations of metal-backed patellae demonstrated short-term failures—most commonly, local polyethylene wear damaging the locking mechanism and subsequent dissociation or fracture from the metal baseplate; polyethylene delamination from the metal baseplate; and failure of interface fixation.9,10 On the other hand, good fixation with bony ingrowth was observed in both titanium and cobalt-chromium porous-coated patellae.1,3,9,11-13 Overall, however, negative outcomes reported for metal-backed patellae led many surgeons to abandon these components and return to using cemented all-polyethylene patellae.
Negative outcomes of earlier metal-backed patellae designs have overshadowed reports of positive outcomes achieved with careful attention paid to component design, patellar tracking, and surgical technique.2,3,14 Subsequent design improvements (eg, a third stabilizing peg, thicker polyethylene, improved conformity) produced excellent outcomes.8,12,15 The advantages of using a metal-backed patella (eg, uniform load sharing, decreased polyethylene deformation, potential for biological fixation) may be unjustly outweighed by the fear of patellar component failure.3
Our 30-plus years of experience with metal-backed patellar components reflect the evolving effect of component design on outcome. Much as reported elsewhere, we found earlier component failures were caused by poor locking mechanisms, thin polyethylene, poor tracking, and minimal femur contact. Over the past decade, however, our outcomes with Duracon metal-backed patellae (Stryker) have been encouraging. We think these positive outcomes, seen over minimum 5-year follow-up, are largely attributable to the thicker polyethylene and improved articular conformity of this component relative to earlier designs. We have also found it helpful to adhere to certain criteria when implanting metal-backed patellae, and we think adhering to these criteria, along with improved component design, indicates use of press-fit metal-backed patellae. In this article, we report our failure incidence with use of this device at minimum 5-year follow-up.
Materials and Methods
In this single-center study, we performed clinical and independent radiographic reviews of 88 primary press-fit metal-backed patellae with minimum 5-year follow-up. All components were the same design (Duracon metal-backed patella) from the same manufacturer (Stryker).
This study, which began in September 2003, was reviewed and approved by the Western Institutional Review Board (WIRB). Either the investigator (Dr. Hedley) or the clinical study coordinator gave study candidates a full explanation of the study and answered any questions. Patients who still wanted to participate in the study signed WIRB consent forms after their index surgery but before minimum 5-year follow-up.
Device Description
This Duracon patella has a porous-coated cobalt-chromium metal back intended for press-fit fixation, 3 cobalt-chromium porous-coated pegs, and a preassembled polyethylene anterior surface (Figure 1). Four sizes are available to fit the peripheral shape of the resected patella.
This patella has 3 styles: symmetric, asymmetric, and conversion. In this study, we used only the asymmetric and conversion styles. The design of each style incorporates medial/lateral facets intended to conform to the convex intercondylar radii of the femoral component, thereby allowing the patella to ride deeply in the recessed patellofemoral groove. The asymmetric patella is a resurfacing component with a generous polyethylene thickness (4.6 mm at its thinnest) and a larger lateral facet for more bone coverage. The asymmetric patella naturally medializes component placement. The articulating surface of the conversion patella is identical to that of the asymmetric patella. However, the conversion patella allows for exchange of the polyethylene portion of the implant without revising a stable, well-fixed metal baseplate.
Patient Selection
Candidates were recruited from a group of metal-backed patella patients within Dr. Hedley’s medical practice. All candidates had undergone primary total knee arthroplasty and received a Duracon press-fit metal-backed patella. All recruited patients had undergone primary knee arthroplasty at least 5 years before clinical and radiographic evaluation. Patients were included in the study if they had a diagnosis of noninflammatory degenerative joint disease (eg, osteoarthritis, traumatic arthritis, avascular necrosis). Patients with body mass index higher than 40 were excluded from the study.
Surgical Technique
The patella is everted completely or as much as feasible. Debridement is done circumferentially around the patella. Adherent fat and pseudomeniscus are stripped back until the surgeon sees the entry point of the quadriceps tendon fibers above and the patella tendon fibers below. The cut is then made at this level to remove as much bone as needed to restore the normal height of the patella with the implant in place. The cut is usually made by hand—without guides but with the patella stabilized with a towel clip above and below to prevent any movement during the action.
The desired cut must be absolutely planar, and this should be checked by placing the edge of the blade across the interface. Repeated passes with the saw blade are needed if the cut is not 100% planar. Once the cut is made, the patella is sized with the patella sizers and drill guide. After the appropriate size is selected, the patella is drilled with a bit that is slightly undersized from the size of the pegs (1/32 inch smaller than the bit supplied by the manufacturer).
Once the patella is prepared, the rest of the knee arthroplasty is performed. The patella is press-fit as the last component to be inserted.
Radiologic Review
Radiographic analysis was performed by an independent reviewer according to the current Knee Society total knee arthroplasty roentgenographic evaluation and scoring system (Figure 2).16 The reviewer was an orthopedist specializing in hip and knee surgery. Radiographs the reviewer deemed questionable were shown to another independent hip and knee surgeon for validation. In all cases, the second reviewer confirmed the first reviewer’s initial recorded observations.
KSS (Knee Society Scale), WOMAC (Western Ontario and McMaster Universities Arthritis Index), and SF-36 (36-Item Short Form Health Survey) were also used to evaluate effectiveness in this protocol.
Survivorship Calculations
Kaplan-Meier survivorship was determined for all metal-backed patellae. For survival analysis, only knees with radiographic data were included (74 knees). Mean follow-up was 75.8 months (range, 60-105 months).
Seventy-four patients (88 knees) met the study criteria (Table). At minimum 5-year follow-up, complete data were acquired for 59 patients (72 knees). Of the total group, 14 knees did not have radiographic data. Those knees were categorized as lost to follow-up and were excluded from the survivorship analysis. The status of patients enrolled in the study at minimum 5-year follow-up is shown in the Table.
Mann-Whitney U test (nonparametric t test) was used to compare WOMAC and SF-36 scores between the “complete” and the “WOMAC and SF-36 only” data groups.
Statistical Analysis
Kaplan-Meier survivorship probabilities (asymmetric method) were calculated using SAS Version 9.2 (SAS Institute); 95% pointwise confidence limits were used.
The Mann-Whitney U test is a nonparametric analogue to the independent-samples t test. It was used here to compare WOMAC and SF-36 scores of patients with “complete” data with scores of patients with “WOMAC and SF-36 only” data. In either group, for patients who had primary bilateral knee arthroplasty, mean WOMAC and SF-36 scores were used.
Comparisons were made between the unilateral and bilateral knee arthroplasty groups. There were no differences in age, height, or weight (Mann-Whitney U test) or in sex, primary diagnosis, or number of patients lost to follow-up (Fisher exact test). Fisher exact test (vs χ2 test) was used for the contingency table analysis because of small cell sizes (eg, ≤10 females in ‘‘both knees” group), suggesting the unilateral and bilateral patients did not differ in demographics.
For all patient-reported questionnaires, bilateral patients were given the opportunity to note any differences between their knee arthroplasties, but none of these patients made any special notations. We interpreted this to mean that all survey responses from bilateral patients were applicable to both knee arthroplasties.
Results
Seventy-four patients (88 knees) were enrolled in the study: 31 women (41.2%) and 43 men (58.1%). At time of surgery, mean age was 59.7 years (range, 40-86 years), and mean body mass index was 30.6 (range, 19.1-39.6). Eighty-three knees were diagnosed with osteoarthritis, and 5 knees were diagnosed with posttraumatic arthritis. Mean time to follow-up was 74.8 months (range, 60-105 months). Fourteen knees (14 patients) were considered lost to follow-up. However, 8 patients (8 knees) were contacted by telephone about the status of their knee(s), and all 8 completed and returned the minimum 5-year follow-up WOMAC and SF-36 forms; they did not return for their minimum 5-year clinical or radiographic evaluations.
Asymmetric patellae were used in 24 knees, conversion patellae in 64 knees (88 knees total). Forty-nine months after surgery, 1 patella was revised for loosening at its interface with the bone. The 51-year-old active female patient’s asymmetric patella was revised to a conversion patella. The decision to implant another metal-backed device was based on its high density; proper intrusion of acrylic cement would have been questionable. Some early wear was observed on the tibial insert, which was replaced. Sixty-eight months after the revision, the patient was asymptomatic, with a KSS Pain score of 96 and a KSS Function score of 100 (Figure 3). Another revision, for tibial insert exchange only, was performed 48 months after surgery. During this revision, the patella was evaluated and found to be well fixed and functioning normally.
Survivorship of the Duracon metal-backed patella at minimum 5-year follow-up was estimated to be 93.95%, with bounds of 73.61% and 98.74%.
Radiographic analysis revealed no radiolucencies larger than 1 mm (Figure 4). Seventeen 1-mm radiolucencies were recorded: 6 (35.3%) in zone 1, 2 (11.8%) in zone 2, and 9 (52.9%) in zone 4. Twelve (70.6%) of the 17 radiolucencies were in the left knee. Nine radiolucencies were in women and 8 in men. Most (55.6%) of the women’s radiolucencies were in zone 1, and most (75.0%) of the men’s were in zone 4. There were no loose beads other than in the case that was later revised.
KSS, WOMAC, and SF-36 scores and radiographic reviews were used to evaluate effectiveness in accordance with the protocol. At minimum 5-year follow-up, mean KSS Pain score was 94.10 (range, 55-100), and mean KSS Function score was 92.67 (range, 60-100). Mean WOMAC score was 2.21 (range, 0-19.70), mean SF-36 Physical score was 83.65 (range, 30.70-100), and mean SF-36 Mental score was 89.41 (range, 1.4-100).
The preceding calculations do not include WOMAC and SF-36 data for the 8 patients (8 knees) who were counted as lost to follow-up but who submitted minimum 5-year follow-up data. We compared these 8 patients with the 60 patients (74 knees) who had complete WOMAC and SF-36 data at the end of the study in order to determine whether there were any statistically significant differences between the 2 groups’ mean scores. No statistically significant differences were detected in any WOMAC or SF-36 category (α = 0.05).
Discussion
Metal-backed patellar components were originally designed to address the shortcomings (eg, fracture, deformation, aseptic loosening) of cemented all-polyethylene patellae.1-3 It was thought that the stiffness of the metal could help resist polyethylene deformation and that the press-fit interface with bone might eliminate issues related to bone cement.8 However, short-term failures were reported with early metal-backed designs.9,10 At the same time, good fixation with bone ingrowth was observed in both titanium and cobalt-chromium porous-coated patellae.1,3,9-12,17 Further, reports of poor outcomes with some metal-backed patella designs overshadowed reports of positive outcomes.2,3 In all reports (of both poor and positive outcomes), component design, patellar tracking, and surgical technique were cited as contributing to implant success.2,3,14,17,18 Subsequent design improvements (eg, use of a third stabilizing peg, thicker polyethylene, improved conformity) produced excellent outcomes.8,12,15
Our early results are similar to those reported in the literature, and we observed markedly better outcomes that we think resulted from component design improvements. Over the past decade, this has been particularly true with our use of the Duracon metal-backed patella, which has thicker polyethylene, better articular conformity, and a third stabilizing peg, all of which were previously noted as contributing to a successful metal-backed patellar component.2,12,14,15,19 In our study, all 72 knees radiographically evaluated and independently reviewed at minimum 5-year follow-up had well-fixed press-fit metal-backed patellae. Seventeen patellae had 1-mm radiolucencies; the other 59 had no radiolucencies in any zone around the patella–bone interface.
One of the most important aspects of removing a metal-backed patellar component from a patella is that the remaining bone stock is often far superior to the stock available after revision of a cemented patella. Careful removal should leave an excellent bony bed for reimplantation.
We think that surgeons should adhere to certain indications and contraindications when implanting metal-backed patellae and that doing so can contribute to successful outcomes. Type of bone stock available should be considered, as successful biological fixation relies on a good blood supply. A dense (or thin) patella in which intrusion of acrylic cement is improbable or impossible may favor use of a metal-backed patella. Cement is not an adhesive but a grout, so successful cementation requires intrusion of cement into the interstices of the cancellous bone. As adequate intrusion of cement into dense bone is not possible, cementation may not be the best option. Some patellae have failed because of peg “shear-off,”9 likely caused not by failure of peg strength but by failure of cement fixation at the nonpeg interface.20,21 Polyethylene pegs fail when used as the sole method of fixation (they were never designed for that). In addition, we think younger patients are often indicated for a metal-backed patella because, over the long term, loosening of a cemented patella (and the accompanying stress shielding and osteolysis) may cause severe patellar bone destruction. Last, we have found that abnormally high or small patellae are not good candidates for cement fixation because they tend to work themselves loose riding on and off the superior flange. These types of patellae appear to have a much sturdier and longer lasting interface than cement, once biological fixation has occurred.
In summary, we think the indications for a metal-backed implant are a patella that is dense or sclerotic; a patella that is thin, abnormally high, or small; and a younger patient. In addition, a metal-backed implant is not indicated for soft, osteoporotic bone.
This study had a few limitations. Fourteen knees (14 patients), or 15.9% of all knees in the study, were categorized as lost to follow-up. Comparing the WOMAC and SF-36 scores of 8 patients (8 knees) who completed minimum 5-year follow-up but were not clinically evaluated with the scores of patients who had complete data, we found no statistically significant differences in any category. However, 5-year follow-up clinical data were available for those 8 patients. Nevertheless, 74 knees were available for radiologic evaluation, and during telephone interviews all 8 patients indicated they had their original implant(s) and were asymptomatic.
Our experience with the Duracon metal-backed patella has been encouraging. In the study reported here, there were no failures caused by dissociation of plastic. We think that, because the porous coating is under almost constant compression, biological fixation is likely in most instances, as observed in our minimum 5-year radiologic results. Given our minimum 5-year follow-up results with uncemented metal-backed patellae, we think their use may be a viable alternative to use of all-polyethylene patellae.
The metal-backed patella was originally designed to address the shortcomings of cemented, all-polyethylene patellae: deformation, aseptic loosening, stress fractures of polyethylene, and possible thermal damage from bone cement.1-3 Several long-term studies have found very good outcomes with use of all-polyethylene patellae.4-6 However, complications of using an all-polyethylene patella reportedly accounted for up to half of all knee revisions, and during revision surgery patellar bone stock was often found to have been compromised.7
The intention behind the design of press-fit metal-backed patellae was to address the shortcomings of all-polyethylene patellae by eliminating the need for bone cement and providing stiffness that would help resist polyethylene deformation while decreasing implant–bone interface stresses.8 However, early design iterations of metal-backed patellae demonstrated short-term failures—most commonly, local polyethylene wear damaging the locking mechanism and subsequent dissociation or fracture from the metal baseplate; polyethylene delamination from the metal baseplate; and failure of interface fixation.9,10 On the other hand, good fixation with bony ingrowth was observed in both titanium and cobalt-chromium porous-coated patellae.1,3,9,11-13 Overall, however, negative outcomes reported for metal-backed patellae led many surgeons to abandon these components and return to using cemented all-polyethylene patellae.
Negative outcomes of earlier metal-backed patellae designs have overshadowed reports of positive outcomes achieved with careful attention paid to component design, patellar tracking, and surgical technique.2,3,14 Subsequent design improvements (eg, a third stabilizing peg, thicker polyethylene, improved conformity) produced excellent outcomes.8,12,15 The advantages of using a metal-backed patella (eg, uniform load sharing, decreased polyethylene deformation, potential for biological fixation) may be unjustly outweighed by the fear of patellar component failure.3
Our 30-plus years of experience with metal-backed patellar components reflect the evolving effect of component design on outcome. Much as reported elsewhere, we found earlier component failures were caused by poor locking mechanisms, thin polyethylene, poor tracking, and minimal femur contact. Over the past decade, however, our outcomes with Duracon metal-backed patellae (Stryker) have been encouraging. We think these positive outcomes, seen over minimum 5-year follow-up, are largely attributable to the thicker polyethylene and improved articular conformity of this component relative to earlier designs. We have also found it helpful to adhere to certain criteria when implanting metal-backed patellae, and we think adhering to these criteria, along with improved component design, indicates use of press-fit metal-backed patellae. In this article, we report our failure incidence with use of this device at minimum 5-year follow-up.
Materials and Methods
In this single-center study, we performed clinical and independent radiographic reviews of 88 primary press-fit metal-backed patellae with minimum 5-year follow-up. All components were the same design (Duracon metal-backed patella) from the same manufacturer (Stryker).
This study, which began in September 2003, was reviewed and approved by the Western Institutional Review Board (WIRB). Either the investigator (Dr. Hedley) or the clinical study coordinator gave study candidates a full explanation of the study and answered any questions. Patients who still wanted to participate in the study signed WIRB consent forms after their index surgery but before minimum 5-year follow-up.
Device Description
This Duracon patella has a porous-coated cobalt-chromium metal back intended for press-fit fixation, 3 cobalt-chromium porous-coated pegs, and a preassembled polyethylene anterior surface (Figure 1). Four sizes are available to fit the peripheral shape of the resected patella.
This patella has 3 styles: symmetric, asymmetric, and conversion. In this study, we used only the asymmetric and conversion styles. The design of each style incorporates medial/lateral facets intended to conform to the convex intercondylar radii of the femoral component, thereby allowing the patella to ride deeply in the recessed patellofemoral groove. The asymmetric patella is a resurfacing component with a generous polyethylene thickness (4.6 mm at its thinnest) and a larger lateral facet for more bone coverage. The asymmetric patella naturally medializes component placement. The articulating surface of the conversion patella is identical to that of the asymmetric patella. However, the conversion patella allows for exchange of the polyethylene portion of the implant without revising a stable, well-fixed metal baseplate.
Patient Selection
Candidates were recruited from a group of metal-backed patella patients within Dr. Hedley’s medical practice. All candidates had undergone primary total knee arthroplasty and received a Duracon press-fit metal-backed patella. All recruited patients had undergone primary knee arthroplasty at least 5 years before clinical and radiographic evaluation. Patients were included in the study if they had a diagnosis of noninflammatory degenerative joint disease (eg, osteoarthritis, traumatic arthritis, avascular necrosis). Patients with body mass index higher than 40 were excluded from the study.
Surgical Technique
The patella is everted completely or as much as feasible. Debridement is done circumferentially around the patella. Adherent fat and pseudomeniscus are stripped back until the surgeon sees the entry point of the quadriceps tendon fibers above and the patella tendon fibers below. The cut is then made at this level to remove as much bone as needed to restore the normal height of the patella with the implant in place. The cut is usually made by hand—without guides but with the patella stabilized with a towel clip above and below to prevent any movement during the action.
The desired cut must be absolutely planar, and this should be checked by placing the edge of the blade across the interface. Repeated passes with the saw blade are needed if the cut is not 100% planar. Once the cut is made, the patella is sized with the patella sizers and drill guide. After the appropriate size is selected, the patella is drilled with a bit that is slightly undersized from the size of the pegs (1/32 inch smaller than the bit supplied by the manufacturer).
Once the patella is prepared, the rest of the knee arthroplasty is performed. The patella is press-fit as the last component to be inserted.
Radiologic Review
Radiographic analysis was performed by an independent reviewer according to the current Knee Society total knee arthroplasty roentgenographic evaluation and scoring system (Figure 2).16 The reviewer was an orthopedist specializing in hip and knee surgery. Radiographs the reviewer deemed questionable were shown to another independent hip and knee surgeon for validation. In all cases, the second reviewer confirmed the first reviewer’s initial recorded observations.
KSS (Knee Society Scale), WOMAC (Western Ontario and McMaster Universities Arthritis Index), and SF-36 (36-Item Short Form Health Survey) were also used to evaluate effectiveness in this protocol.
Survivorship Calculations
Kaplan-Meier survivorship was determined for all metal-backed patellae. For survival analysis, only knees with radiographic data were included (74 knees). Mean follow-up was 75.8 months (range, 60-105 months).
Seventy-four patients (88 knees) met the study criteria (Table). At minimum 5-year follow-up, complete data were acquired for 59 patients (72 knees). Of the total group, 14 knees did not have radiographic data. Those knees were categorized as lost to follow-up and were excluded from the survivorship analysis. The status of patients enrolled in the study at minimum 5-year follow-up is shown in the Table.
Mann-Whitney U test (nonparametric t test) was used to compare WOMAC and SF-36 scores between the “complete” and the “WOMAC and SF-36 only” data groups.
Statistical Analysis
Kaplan-Meier survivorship probabilities (asymmetric method) were calculated using SAS Version 9.2 (SAS Institute); 95% pointwise confidence limits were used.
The Mann-Whitney U test is a nonparametric analogue to the independent-samples t test. It was used here to compare WOMAC and SF-36 scores of patients with “complete” data with scores of patients with “WOMAC and SF-36 only” data. In either group, for patients who had primary bilateral knee arthroplasty, mean WOMAC and SF-36 scores were used.
Comparisons were made between the unilateral and bilateral knee arthroplasty groups. There were no differences in age, height, or weight (Mann-Whitney U test) or in sex, primary diagnosis, or number of patients lost to follow-up (Fisher exact test). Fisher exact test (vs χ2 test) was used for the contingency table analysis because of small cell sizes (eg, ≤10 females in ‘‘both knees” group), suggesting the unilateral and bilateral patients did not differ in demographics.
For all patient-reported questionnaires, bilateral patients were given the opportunity to note any differences between their knee arthroplasties, but none of these patients made any special notations. We interpreted this to mean that all survey responses from bilateral patients were applicable to both knee arthroplasties.
Results
Seventy-four patients (88 knees) were enrolled in the study: 31 women (41.2%) and 43 men (58.1%). At time of surgery, mean age was 59.7 years (range, 40-86 years), and mean body mass index was 30.6 (range, 19.1-39.6). Eighty-three knees were diagnosed with osteoarthritis, and 5 knees were diagnosed with posttraumatic arthritis. Mean time to follow-up was 74.8 months (range, 60-105 months). Fourteen knees (14 patients) were considered lost to follow-up. However, 8 patients (8 knees) were contacted by telephone about the status of their knee(s), and all 8 completed and returned the minimum 5-year follow-up WOMAC and SF-36 forms; they did not return for their minimum 5-year clinical or radiographic evaluations.
Asymmetric patellae were used in 24 knees, conversion patellae in 64 knees (88 knees total). Forty-nine months after surgery, 1 patella was revised for loosening at its interface with the bone. The 51-year-old active female patient’s asymmetric patella was revised to a conversion patella. The decision to implant another metal-backed device was based on its high density; proper intrusion of acrylic cement would have been questionable. Some early wear was observed on the tibial insert, which was replaced. Sixty-eight months after the revision, the patient was asymptomatic, with a KSS Pain score of 96 and a KSS Function score of 100 (Figure 3). Another revision, for tibial insert exchange only, was performed 48 months after surgery. During this revision, the patella was evaluated and found to be well fixed and functioning normally.
Survivorship of the Duracon metal-backed patella at minimum 5-year follow-up was estimated to be 93.95%, with bounds of 73.61% and 98.74%.
Radiographic analysis revealed no radiolucencies larger than 1 mm (Figure 4). Seventeen 1-mm radiolucencies were recorded: 6 (35.3%) in zone 1, 2 (11.8%) in zone 2, and 9 (52.9%) in zone 4. Twelve (70.6%) of the 17 radiolucencies were in the left knee. Nine radiolucencies were in women and 8 in men. Most (55.6%) of the women’s radiolucencies were in zone 1, and most (75.0%) of the men’s were in zone 4. There were no loose beads other than in the case that was later revised.
KSS, WOMAC, and SF-36 scores and radiographic reviews were used to evaluate effectiveness in accordance with the protocol. At minimum 5-year follow-up, mean KSS Pain score was 94.10 (range, 55-100), and mean KSS Function score was 92.67 (range, 60-100). Mean WOMAC score was 2.21 (range, 0-19.70), mean SF-36 Physical score was 83.65 (range, 30.70-100), and mean SF-36 Mental score was 89.41 (range, 1.4-100).
The preceding calculations do not include WOMAC and SF-36 data for the 8 patients (8 knees) who were counted as lost to follow-up but who submitted minimum 5-year follow-up data. We compared these 8 patients with the 60 patients (74 knees) who had complete WOMAC and SF-36 data at the end of the study in order to determine whether there were any statistically significant differences between the 2 groups’ mean scores. No statistically significant differences were detected in any WOMAC or SF-36 category (α = 0.05).
Discussion
Metal-backed patellar components were originally designed to address the shortcomings (eg, fracture, deformation, aseptic loosening) of cemented all-polyethylene patellae.1-3 It was thought that the stiffness of the metal could help resist polyethylene deformation and that the press-fit interface with bone might eliminate issues related to bone cement.8 However, short-term failures were reported with early metal-backed designs.9,10 At the same time, good fixation with bone ingrowth was observed in both titanium and cobalt-chromium porous-coated patellae.1,3,9-12,17 Further, reports of poor outcomes with some metal-backed patella designs overshadowed reports of positive outcomes.2,3 In all reports (of both poor and positive outcomes), component design, patellar tracking, and surgical technique were cited as contributing to implant success.2,3,14,17,18 Subsequent design improvements (eg, use of a third stabilizing peg, thicker polyethylene, improved conformity) produced excellent outcomes.8,12,15
Our early results are similar to those reported in the literature, and we observed markedly better outcomes that we think resulted from component design improvements. Over the past decade, this has been particularly true with our use of the Duracon metal-backed patella, which has thicker polyethylene, better articular conformity, and a third stabilizing peg, all of which were previously noted as contributing to a successful metal-backed patellar component.2,12,14,15,19 In our study, all 72 knees radiographically evaluated and independently reviewed at minimum 5-year follow-up had well-fixed press-fit metal-backed patellae. Seventeen patellae had 1-mm radiolucencies; the other 59 had no radiolucencies in any zone around the patella–bone interface.
One of the most important aspects of removing a metal-backed patellar component from a patella is that the remaining bone stock is often far superior to the stock available after revision of a cemented patella. Careful removal should leave an excellent bony bed for reimplantation.
We think that surgeons should adhere to certain indications and contraindications when implanting metal-backed patellae and that doing so can contribute to successful outcomes. Type of bone stock available should be considered, as successful biological fixation relies on a good blood supply. A dense (or thin) patella in which intrusion of acrylic cement is improbable or impossible may favor use of a metal-backed patella. Cement is not an adhesive but a grout, so successful cementation requires intrusion of cement into the interstices of the cancellous bone. As adequate intrusion of cement into dense bone is not possible, cementation may not be the best option. Some patellae have failed because of peg “shear-off,”9 likely caused not by failure of peg strength but by failure of cement fixation at the nonpeg interface.20,21 Polyethylene pegs fail when used as the sole method of fixation (they were never designed for that). In addition, we think younger patients are often indicated for a metal-backed patella because, over the long term, loosening of a cemented patella (and the accompanying stress shielding and osteolysis) may cause severe patellar bone destruction. Last, we have found that abnormally high or small patellae are not good candidates for cement fixation because they tend to work themselves loose riding on and off the superior flange. These types of patellae appear to have a much sturdier and longer lasting interface than cement, once biological fixation has occurred.
In summary, we think the indications for a metal-backed implant are a patella that is dense or sclerotic; a patella that is thin, abnormally high, or small; and a younger patient. In addition, a metal-backed implant is not indicated for soft, osteoporotic bone.
This study had a few limitations. Fourteen knees (14 patients), or 15.9% of all knees in the study, were categorized as lost to follow-up. Comparing the WOMAC and SF-36 scores of 8 patients (8 knees) who completed minimum 5-year follow-up but were not clinically evaluated with the scores of patients who had complete data, we found no statistically significant differences in any category. However, 5-year follow-up clinical data were available for those 8 patients. Nevertheless, 74 knees were available for radiologic evaluation, and during telephone interviews all 8 patients indicated they had their original implant(s) and were asymptomatic.
Our experience with the Duracon metal-backed patella has been encouraging. In the study reported here, there were no failures caused by dissociation of plastic. We think that, because the porous coating is under almost constant compression, biological fixation is likely in most instances, as observed in our minimum 5-year radiologic results. Given our minimum 5-year follow-up results with uncemented metal-backed patellae, we think their use may be a viable alternative to use of all-polyethylene patellae.
1. Firestone TP, Teeny SM, Krackow KA, Hungerford DS. The clinical and roentgenographic results of cementless porous-coated patellar fixation. Clin Orthop Relat Res. 1991;273:184-189.
2. Laskin RS, Bucknell A. The use of metal-backed patellar prostheses in total knee arthroplasty. Clin Orthop Relat Res. 1990;260:52-55.
3. Evanich CJ, Tkach TK, von Glinski S, Camargo MP, Hofmann AA. 6- to 10-year experience using countersunk metal-backed patellas. J Arthroplasty. 1997;12(2):149-154.
4. Schwartz AJ, Della Vale CJ, Rosenberg AG, Jacobs JJ, Berger RA, Galante JO. Cruciate-retaining TKA using a third-generation system with a four-pegged tibial component: a minimum 10-year followup note. Clin Orthop Relat Res. 2010;468(8):2160-2167.
5. Bisschop R, Brouwer RW, Van Raay JJ. Total knee arthroplasty in younger patients: a 13-year follow-up study. Orthopedics. 2010;33(12):876-880.
6. Dixon MC, Brown RR, Parsch D, Scott RD. Modular fixed-bearing total knee arthroplasty with retention of the posterior cruciate ligament. A study of patients followed for a minimum of fifteen years. J Bone Joint Surg Am. 2005;87(3):598-603.
7. Brick GW, Scott RD. The patellofemoral component of total knee arthroplasty. Clin Orthop Relat Res. 1988;231)163-178.
8. Garcia RM, Kraay MJ, Goldberg VM. Isolated all-polyethylene patellar revisions for metal-backed patellar failure. Clin Orthop Relat Res. 2008;466(11):2784-2789.
9. Rosenberg AG, Andriacchi TP, Barden R, Galante JO. Patellar component failure in cementless total knee arthroplasty. Clin Orthop Relat Res. 1988;(236):106-114.
10. Stulberg SD, Stulberg BN, Hamati Y, Tsao A. Failure mechanisms of metal-backed patellar components. Clin Orthop Relat Res. 1988;236:88-105.
11. Sundfeldt M, Johansson CB, Regner L, Albrektsson T, Carlsson LV. Long-term results of a cementless knee prosthesis with a metal-backed patellar component: clinical and radiological follow-up with histology from retrieved components. J Long Term Eff Med Implants. 2003;13(4):341-354.
12. Kraay MJ, Darr OJ, Salata MJ, Goldberg VM. Outcome of metal-backed cementless patellar components: the effect of implant design. Clin Orthop Relat Res. 2001;392:239-244.
13. Jensen LN, Lund B, Gotfredsen K. Bone growth into a revised porous-coated patellar implant. Acta Orthop Scand. 1990;61(3):213-216.
14. Hsu HP, Walker PS. Wear and deformation of patellar components in total knee arthroplasty. Clin Orthop Relat Res. 1989;246:260-265.
15. Jordan LR, Sorrells RB, Jordan LC, Olivo JL. The long-term results of a metal-backed mobile bearing patella. Clin Orthop Relat Res. 2005;436:111-118.
16. Ewald FC. The Knee Society total knee arthroplasty roentgenographic evaluation and scoring system. Clin Orthop Relat Res. 1989;248:9-12.
17. Bayley JC, Scott RD, Ewald FC, Holmes GB Jr. Failure of the metal-backed patellar component after total knee replacement. J Bone Joint Surg Am. 1988;70(5):668-674.
18. Lombardi AV Jr, Engh GA, Volz RG, Albrigo JL, Brainard BJ. Fracture/dissociation of the polyethylene in metal-backed patellar components in total knee arthroplasty. J Bone Joint Surg Am. 1988;70(5):675-679.
19. Moreland JR. Mechanisms of failure in total knee arthroplasty. Clin Orthop Relat Res. 1988;226:49-64.
20. Francke EI, Lachiewicz PF. Failure of a cemented all-polyethylene patellar component of a press-fit condylar total knee arthroplasty. J Arthroplasty. 2000;15(2):234-237.
21. Stulberg BN, Wright TM, Stoller AP, Mimnaugh KL, Mason JJ. Bilateral patellar component shear failure of highly cross-linked polyethylene components: report of a case and laboratory analysis of failure mechanisms. J Arthroplasty. 2012;27(5):789-796.
1. Firestone TP, Teeny SM, Krackow KA, Hungerford DS. The clinical and roentgenographic results of cementless porous-coated patellar fixation. Clin Orthop Relat Res. 1991;273:184-189.
2. Laskin RS, Bucknell A. The use of metal-backed patellar prostheses in total knee arthroplasty. Clin Orthop Relat Res. 1990;260:52-55.
3. Evanich CJ, Tkach TK, von Glinski S, Camargo MP, Hofmann AA. 6- to 10-year experience using countersunk metal-backed patellas. J Arthroplasty. 1997;12(2):149-154.
4. Schwartz AJ, Della Vale CJ, Rosenberg AG, Jacobs JJ, Berger RA, Galante JO. Cruciate-retaining TKA using a third-generation system with a four-pegged tibial component: a minimum 10-year followup note. Clin Orthop Relat Res. 2010;468(8):2160-2167.
5. Bisschop R, Brouwer RW, Van Raay JJ. Total knee arthroplasty in younger patients: a 13-year follow-up study. Orthopedics. 2010;33(12):876-880.
6. Dixon MC, Brown RR, Parsch D, Scott RD. Modular fixed-bearing total knee arthroplasty with retention of the posterior cruciate ligament. A study of patients followed for a minimum of fifteen years. J Bone Joint Surg Am. 2005;87(3):598-603.
7. Brick GW, Scott RD. The patellofemoral component of total knee arthroplasty. Clin Orthop Relat Res. 1988;231)163-178.
8. Garcia RM, Kraay MJ, Goldberg VM. Isolated all-polyethylene patellar revisions for metal-backed patellar failure. Clin Orthop Relat Res. 2008;466(11):2784-2789.
9. Rosenberg AG, Andriacchi TP, Barden R, Galante JO. Patellar component failure in cementless total knee arthroplasty. Clin Orthop Relat Res. 1988;(236):106-114.
10. Stulberg SD, Stulberg BN, Hamati Y, Tsao A. Failure mechanisms of metal-backed patellar components. Clin Orthop Relat Res. 1988;236:88-105.
11. Sundfeldt M, Johansson CB, Regner L, Albrektsson T, Carlsson LV. Long-term results of a cementless knee prosthesis with a metal-backed patellar component: clinical and radiological follow-up with histology from retrieved components. J Long Term Eff Med Implants. 2003;13(4):341-354.
12. Kraay MJ, Darr OJ, Salata MJ, Goldberg VM. Outcome of metal-backed cementless patellar components: the effect of implant design. Clin Orthop Relat Res. 2001;392:239-244.
13. Jensen LN, Lund B, Gotfredsen K. Bone growth into a revised porous-coated patellar implant. Acta Orthop Scand. 1990;61(3):213-216.
14. Hsu HP, Walker PS. Wear and deformation of patellar components in total knee arthroplasty. Clin Orthop Relat Res. 1989;246:260-265.
15. Jordan LR, Sorrells RB, Jordan LC, Olivo JL. The long-term results of a metal-backed mobile bearing patella. Clin Orthop Relat Res. 2005;436:111-118.
16. Ewald FC. The Knee Society total knee arthroplasty roentgenographic evaluation and scoring system. Clin Orthop Relat Res. 1989;248:9-12.
17. Bayley JC, Scott RD, Ewald FC, Holmes GB Jr. Failure of the metal-backed patellar component after total knee replacement. J Bone Joint Surg Am. 1988;70(5):668-674.
18. Lombardi AV Jr, Engh GA, Volz RG, Albrigo JL, Brainard BJ. Fracture/dissociation of the polyethylene in metal-backed patellar components in total knee arthroplasty. J Bone Joint Surg Am. 1988;70(5):675-679.
19. Moreland JR. Mechanisms of failure in total knee arthroplasty. Clin Orthop Relat Res. 1988;226:49-64.
20. Francke EI, Lachiewicz PF. Failure of a cemented all-polyethylene patellar component of a press-fit condylar total knee arthroplasty. J Arthroplasty. 2000;15(2):234-237.
21. Stulberg BN, Wright TM, Stoller AP, Mimnaugh KL, Mason JJ. Bilateral patellar component shear failure of highly cross-linked polyethylene components: report of a case and laboratory analysis of failure mechanisms. J Arthroplasty. 2012;27(5):789-796.
Navigating the Alphabet Soup of Labroligamentous Pathology of the Shoulder
The widespread use of eponyms and acronyms to describe labroligamentous findings in the shoulder has made interpretation of shoulder magnetic resonance imaging (MRI) reports challenging. We review and discuss the appearance of these lesions on shoulder MRI to help the orthopedic surgeon understand these entities as imaging findings.
Glenolabral articular disruption (GLAD) occurs secondary to impaction of the humeral head on the glenoid articular cartilage. There is a resultant defect in the glenoid articular cartilage, which extends to the glenoid labrum. A GLAD lesion is diagnosed only if the glenohumeral ligament and scapular periosteum remain intact1 (Figure 1).
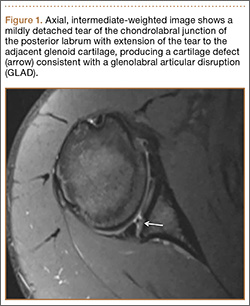
Complete detachment of the anteroinferior labrum with tearing of the anterior glenoid periosteum represents a Bankart lesion. Cartilaginous Bankart lesions are caused by an anterior glenohumeral dislocation with resultant avulsion of the anteroinferior labrum and disruption of the scapular periosteum because of acute traction on the anterior band of the inferior glenohumeral ligament (Figure 2). Anterior instability, caused by disruption of the anterior labroligamentous complex, results. Osseous Bankart lesions occur when the anterior displaced humeral head impacts the anterior inferior glenoid rim, causing a fracture (Figure 3). This loss of the glenoid articular surface area can result in glenohumeral instability. Posterior shoulder dislocations can result in corresponding findings in the posterior inferior glenoid labrum (reverse Bankart lesion) and anterior medial humeral head (reverse Hill-Sachs lesion) (Figure 2).
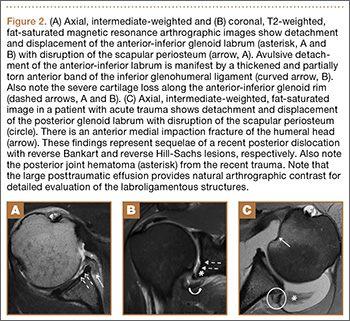
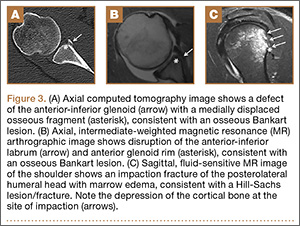
A variant of the Bankart lesion is the anterior labroligamentous periosteal sleeve avulsion (ALPSA). This refers to a medially displaced tear of the anterior labrum with intact periosteal stripping along the medial glenoid2with medial rotation and inferior displacement of the anterior inferior labrum along the scapular neck. An ALPSA lesion can heal via the intact periosteal blood supply. If not repaired, anterior instability will result because of malposition of the labrum, causing a patulous anterior capsule.3 When a corresponding lesion occurs in the posterior labrum because of a posterior dislocation, it is called a posterior labrocapsular periosteal sleeve avulsion (POLPSA) (Figure 4).
Another variant of the Bankart lesion is the Perthes lesion, which is a nondisplaced tear of the anteroinferior labrum with periosteal stripping. This differs from the ALPSA because the detached labrum and periosteum are held in anatomic position, possibly making the lesion difficult to detect on magnetic resonance arthrography (MRA).3 Obtaining images in the abduction external rotation (ABER) position exerts traction on the anterior inferior joint capsule and may make the Perthes lesion more conspicuous.4 When this occurs in the posterior labrum, it is called a reverse Perthes lesion (Figure 5).
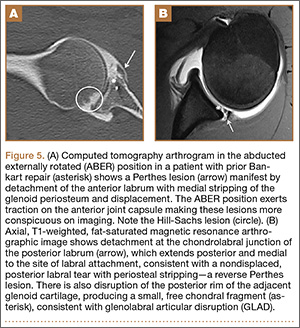
In a patient with anterior glenohumeral instability without a Bankart lesion, pathology of the anterior band of the inferior glenohumeral ligament (IGHL) at its humeral attachment must be suspected. Humeral avulsion of the IGHL (HAGL) or its variants can be overlooked on arthroscopy. HAGL is diagnosed on MRA when the normally U-shaped IGHL takes on a J-shape, and joint fluid extravasates across the torn humeral attachment (Figure 6). If there is an avulsed bony fragment from the medial humeral neck, the lesion is termed a bony HAGL (BHAGL). In addition to the findings of a HAGL, a BHAGL shows the osseous fragment and donor site on MRI. Since a BHAGL is a bony avulsion, it can even be suggested on radiography if a bony fragment is seen adjacent to the medial humeral neck.5 These lesions are highly associated with other shoulder injuries, particularly Hill-Sachs deformities and subscapularis tendon tears, and it is imperative, therefore, to search for additional injuries if a HAGL-type injury is seen.6
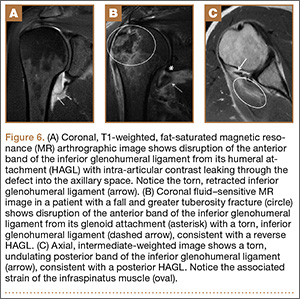
A more uncommon type of HAGL can occur in the setting of posterior capsulolabral injury. A posterior-band IGHL avulsion from the humerus (PHAGL) has similar imaging findings to a HAGL, except that it involves the posterior band of the IGHL. PHAGLs are usually not associated with an acute injury and are thought to be related to repetitive microtrauma, perhaps since the posterior band of the IGHL is the thinnest portion of the IGHL complex.7
A Kim lesion is an arthroscopic finding described in patients with posterior instability as a superficial defect at the undersurface of the posterior labrum and adjacent glenoid cartilage without detachment or extension to the chondrolabral junction.8 It is, by its nature, a concealed finding on routine MRI but can be more conspicuous in FADIR (flexed, adducted, internally rotated) positioning on MRA, which exerts traction on the posterior joint capsule, allowing intra-articular contrast to fill the tear (Figure 7).
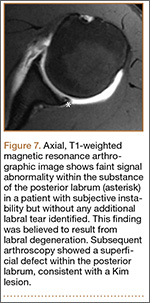
This list describes several of the most commonly encountered acronyms in shoulder MRI. A review of SLAP (superior labrum anterior to posterior) lesions was described in a previous article in the journal’s Imaging Series.9 A thorough understanding of these lesions is helpful in interpreting reports and determining the appropriate treatment for patients with shoulder injuries.
1. Sanders TG, Tirman PF, Linares R, Feller JF, Richardson R. The glenolabral articular disruption lesion: MR arthrography with arthroscopic correlation. AJR Am J Roentgenol. 1999;172(1):171-175.
2. Beltran J, Jbara M, Maimon R. Shoulder: labrum and bicipital tendon. Top Magn Reson Imaging. 2003;14(1):35-50.
3. Waldt S, Burkart A, Imhoff AB, Bruegel M, Rummeny EJ, Woertler K. Anterior shoulder instability: accuracy of MR arthrography in the classification of anteroinferior labroligamentous injuries. Radiology. 2005;237(2):578-583.
4. Schreinemachers SA, van der Hulst VP, Willems J, Bipat S, van der Woude H. Is a single direct MR arthrography series in ABER position as accurate in detecting anteroinferior labroligamentous lesions as conventional MR arthrography? Skeletal Radiol. 2009;38(7):675-683.
5. Bui-Mansfield LT, Taylor DC, Uhorchak JM, Tenuta JT. Humeral avulsions of the glenohumeral ligament: imaging features and a review of the literature. AJR Am J Roentgenol. 2002;179(3):649-655.
6. Magee T. Prevalence of HAGL lesions and associated abnormalities on shoulder MR examination. Skeletal Radiol. 2014;43(3):307-313.
7. Chung CB, Sorenson S, Dwek JR, Resnick D. Humeral avulsion of the posterior band of the inferior glenohumeral ligament: MR arthrography and clinical correlation in 17 patients. AJR Am J Roentgenol. 2004;183(2):355-359.
8. Kim SH, Ha KI, Yoo JC, Noh KC. Kim’s lesion: an incomplete and concealed avulsion of the posteroinferior labrum in posterior or multidirectional posteroinferior instability of the shoulder. Arthroscopy. 2004;20(7):712-720.
9. Grubin J, Maderazo A, Fitzpatrick D. Imaging evaluation of superior labral anteroposterior (SLAP) tears. Am J Orthop. 2015;44(10):476-477.
The widespread use of eponyms and acronyms to describe labroligamentous findings in the shoulder has made interpretation of shoulder magnetic resonance imaging (MRI) reports challenging. We review and discuss the appearance of these lesions on shoulder MRI to help the orthopedic surgeon understand these entities as imaging findings.
Glenolabral articular disruption (GLAD) occurs secondary to impaction of the humeral head on the glenoid articular cartilage. There is a resultant defect in the glenoid articular cartilage, which extends to the glenoid labrum. A GLAD lesion is diagnosed only if the glenohumeral ligament and scapular periosteum remain intact1 (Figure 1).
Complete detachment of the anteroinferior labrum with tearing of the anterior glenoid periosteum represents a Bankart lesion. Cartilaginous Bankart lesions are caused by an anterior glenohumeral dislocation with resultant avulsion of the anteroinferior labrum and disruption of the scapular periosteum because of acute traction on the anterior band of the inferior glenohumeral ligament (Figure 2). Anterior instability, caused by disruption of the anterior labroligamentous complex, results. Osseous Bankart lesions occur when the anterior displaced humeral head impacts the anterior inferior glenoid rim, causing a fracture (Figure 3). This loss of the glenoid articular surface area can result in glenohumeral instability. Posterior shoulder dislocations can result in corresponding findings in the posterior inferior glenoid labrum (reverse Bankart lesion) and anterior medial humeral head (reverse Hill-Sachs lesion) (Figure 2).
A variant of the Bankart lesion is the anterior labroligamentous periosteal sleeve avulsion (ALPSA). This refers to a medially displaced tear of the anterior labrum with intact periosteal stripping along the medial glenoid2with medial rotation and inferior displacement of the anterior inferior labrum along the scapular neck. An ALPSA lesion can heal via the intact periosteal blood supply. If not repaired, anterior instability will result because of malposition of the labrum, causing a patulous anterior capsule.3 When a corresponding lesion occurs in the posterior labrum because of a posterior dislocation, it is called a posterior labrocapsular periosteal sleeve avulsion (POLPSA) (Figure 4).
Another variant of the Bankart lesion is the Perthes lesion, which is a nondisplaced tear of the anteroinferior labrum with periosteal stripping. This differs from the ALPSA because the detached labrum and periosteum are held in anatomic position, possibly making the lesion difficult to detect on magnetic resonance arthrography (MRA).3 Obtaining images in the abduction external rotation (ABER) position exerts traction on the anterior inferior joint capsule and may make the Perthes lesion more conspicuous.4 When this occurs in the posterior labrum, it is called a reverse Perthes lesion (Figure 5).
In a patient with anterior glenohumeral instability without a Bankart lesion, pathology of the anterior band of the inferior glenohumeral ligament (IGHL) at its humeral attachment must be suspected. Humeral avulsion of the IGHL (HAGL) or its variants can be overlooked on arthroscopy. HAGL is diagnosed on MRA when the normally U-shaped IGHL takes on a J-shape, and joint fluid extravasates across the torn humeral attachment (Figure 6). If there is an avulsed bony fragment from the medial humeral neck, the lesion is termed a bony HAGL (BHAGL). In addition to the findings of a HAGL, a BHAGL shows the osseous fragment and donor site on MRI. Since a BHAGL is a bony avulsion, it can even be suggested on radiography if a bony fragment is seen adjacent to the medial humeral neck.5 These lesions are highly associated with other shoulder injuries, particularly Hill-Sachs deformities and subscapularis tendon tears, and it is imperative, therefore, to search for additional injuries if a HAGL-type injury is seen.6
A more uncommon type of HAGL can occur in the setting of posterior capsulolabral injury. A posterior-band IGHL avulsion from the humerus (PHAGL) has similar imaging findings to a HAGL, except that it involves the posterior band of the IGHL. PHAGLs are usually not associated with an acute injury and are thought to be related to repetitive microtrauma, perhaps since the posterior band of the IGHL is the thinnest portion of the IGHL complex.7
A Kim lesion is an arthroscopic finding described in patients with posterior instability as a superficial defect at the undersurface of the posterior labrum and adjacent glenoid cartilage without detachment or extension to the chondrolabral junction.8 It is, by its nature, a concealed finding on routine MRI but can be more conspicuous in FADIR (flexed, adducted, internally rotated) positioning on MRA, which exerts traction on the posterior joint capsule, allowing intra-articular contrast to fill the tear (Figure 7).
This list describes several of the most commonly encountered acronyms in shoulder MRI. A review of SLAP (superior labrum anterior to posterior) lesions was described in a previous article in the journal’s Imaging Series.9 A thorough understanding of these lesions is helpful in interpreting reports and determining the appropriate treatment for patients with shoulder injuries.
The widespread use of eponyms and acronyms to describe labroligamentous findings in the shoulder has made interpretation of shoulder magnetic resonance imaging (MRI) reports challenging. We review and discuss the appearance of these lesions on shoulder MRI to help the orthopedic surgeon understand these entities as imaging findings.
Glenolabral articular disruption (GLAD) occurs secondary to impaction of the humeral head on the glenoid articular cartilage. There is a resultant defect in the glenoid articular cartilage, which extends to the glenoid labrum. A GLAD lesion is diagnosed only if the glenohumeral ligament and scapular periosteum remain intact1 (Figure 1).
Complete detachment of the anteroinferior labrum with tearing of the anterior glenoid periosteum represents a Bankart lesion. Cartilaginous Bankart lesions are caused by an anterior glenohumeral dislocation with resultant avulsion of the anteroinferior labrum and disruption of the scapular periosteum because of acute traction on the anterior band of the inferior glenohumeral ligament (Figure 2). Anterior instability, caused by disruption of the anterior labroligamentous complex, results. Osseous Bankart lesions occur when the anterior displaced humeral head impacts the anterior inferior glenoid rim, causing a fracture (Figure 3). This loss of the glenoid articular surface area can result in glenohumeral instability. Posterior shoulder dislocations can result in corresponding findings in the posterior inferior glenoid labrum (reverse Bankart lesion) and anterior medial humeral head (reverse Hill-Sachs lesion) (Figure 2).
A variant of the Bankart lesion is the anterior labroligamentous periosteal sleeve avulsion (ALPSA). This refers to a medially displaced tear of the anterior labrum with intact periosteal stripping along the medial glenoid2with medial rotation and inferior displacement of the anterior inferior labrum along the scapular neck. An ALPSA lesion can heal via the intact periosteal blood supply. If not repaired, anterior instability will result because of malposition of the labrum, causing a patulous anterior capsule.3 When a corresponding lesion occurs in the posterior labrum because of a posterior dislocation, it is called a posterior labrocapsular periosteal sleeve avulsion (POLPSA) (Figure 4).
Another variant of the Bankart lesion is the Perthes lesion, which is a nondisplaced tear of the anteroinferior labrum with periosteal stripping. This differs from the ALPSA because the detached labrum and periosteum are held in anatomic position, possibly making the lesion difficult to detect on magnetic resonance arthrography (MRA).3 Obtaining images in the abduction external rotation (ABER) position exerts traction on the anterior inferior joint capsule and may make the Perthes lesion more conspicuous.4 When this occurs in the posterior labrum, it is called a reverse Perthes lesion (Figure 5).
In a patient with anterior glenohumeral instability without a Bankart lesion, pathology of the anterior band of the inferior glenohumeral ligament (IGHL) at its humeral attachment must be suspected. Humeral avulsion of the IGHL (HAGL) or its variants can be overlooked on arthroscopy. HAGL is diagnosed on MRA when the normally U-shaped IGHL takes on a J-shape, and joint fluid extravasates across the torn humeral attachment (Figure 6). If there is an avulsed bony fragment from the medial humeral neck, the lesion is termed a bony HAGL (BHAGL). In addition to the findings of a HAGL, a BHAGL shows the osseous fragment and donor site on MRI. Since a BHAGL is a bony avulsion, it can even be suggested on radiography if a bony fragment is seen adjacent to the medial humeral neck.5 These lesions are highly associated with other shoulder injuries, particularly Hill-Sachs deformities and subscapularis tendon tears, and it is imperative, therefore, to search for additional injuries if a HAGL-type injury is seen.6
A more uncommon type of HAGL can occur in the setting of posterior capsulolabral injury. A posterior-band IGHL avulsion from the humerus (PHAGL) has similar imaging findings to a HAGL, except that it involves the posterior band of the IGHL. PHAGLs are usually not associated with an acute injury and are thought to be related to repetitive microtrauma, perhaps since the posterior band of the IGHL is the thinnest portion of the IGHL complex.7
A Kim lesion is an arthroscopic finding described in patients with posterior instability as a superficial defect at the undersurface of the posterior labrum and adjacent glenoid cartilage without detachment or extension to the chondrolabral junction.8 It is, by its nature, a concealed finding on routine MRI but can be more conspicuous in FADIR (flexed, adducted, internally rotated) positioning on MRA, which exerts traction on the posterior joint capsule, allowing intra-articular contrast to fill the tear (Figure 7).
This list describes several of the most commonly encountered acronyms in shoulder MRI. A review of SLAP (superior labrum anterior to posterior) lesions was described in a previous article in the journal’s Imaging Series.9 A thorough understanding of these lesions is helpful in interpreting reports and determining the appropriate treatment for patients with shoulder injuries.
1. Sanders TG, Tirman PF, Linares R, Feller JF, Richardson R. The glenolabral articular disruption lesion: MR arthrography with arthroscopic correlation. AJR Am J Roentgenol. 1999;172(1):171-175.
2. Beltran J, Jbara M, Maimon R. Shoulder: labrum and bicipital tendon. Top Magn Reson Imaging. 2003;14(1):35-50.
3. Waldt S, Burkart A, Imhoff AB, Bruegel M, Rummeny EJ, Woertler K. Anterior shoulder instability: accuracy of MR arthrography in the classification of anteroinferior labroligamentous injuries. Radiology. 2005;237(2):578-583.
4. Schreinemachers SA, van der Hulst VP, Willems J, Bipat S, van der Woude H. Is a single direct MR arthrography series in ABER position as accurate in detecting anteroinferior labroligamentous lesions as conventional MR arthrography? Skeletal Radiol. 2009;38(7):675-683.
5. Bui-Mansfield LT, Taylor DC, Uhorchak JM, Tenuta JT. Humeral avulsions of the glenohumeral ligament: imaging features and a review of the literature. AJR Am J Roentgenol. 2002;179(3):649-655.
6. Magee T. Prevalence of HAGL lesions and associated abnormalities on shoulder MR examination. Skeletal Radiol. 2014;43(3):307-313.
7. Chung CB, Sorenson S, Dwek JR, Resnick D. Humeral avulsion of the posterior band of the inferior glenohumeral ligament: MR arthrography and clinical correlation in 17 patients. AJR Am J Roentgenol. 2004;183(2):355-359.
8. Kim SH, Ha KI, Yoo JC, Noh KC. Kim’s lesion: an incomplete and concealed avulsion of the posteroinferior labrum in posterior or multidirectional posteroinferior instability of the shoulder. Arthroscopy. 2004;20(7):712-720.
9. Grubin J, Maderazo A, Fitzpatrick D. Imaging evaluation of superior labral anteroposterior (SLAP) tears. Am J Orthop. 2015;44(10):476-477.
1. Sanders TG, Tirman PF, Linares R, Feller JF, Richardson R. The glenolabral articular disruption lesion: MR arthrography with arthroscopic correlation. AJR Am J Roentgenol. 1999;172(1):171-175.
2. Beltran J, Jbara M, Maimon R. Shoulder: labrum and bicipital tendon. Top Magn Reson Imaging. 2003;14(1):35-50.
3. Waldt S, Burkart A, Imhoff AB, Bruegel M, Rummeny EJ, Woertler K. Anterior shoulder instability: accuracy of MR arthrography in the classification of anteroinferior labroligamentous injuries. Radiology. 2005;237(2):578-583.
4. Schreinemachers SA, van der Hulst VP, Willems J, Bipat S, van der Woude H. Is a single direct MR arthrography series in ABER position as accurate in detecting anteroinferior labroligamentous lesions as conventional MR arthrography? Skeletal Radiol. 2009;38(7):675-683.
5. Bui-Mansfield LT, Taylor DC, Uhorchak JM, Tenuta JT. Humeral avulsions of the glenohumeral ligament: imaging features and a review of the literature. AJR Am J Roentgenol. 2002;179(3):649-655.
6. Magee T. Prevalence of HAGL lesions and associated abnormalities on shoulder MR examination. Skeletal Radiol. 2014;43(3):307-313.
7. Chung CB, Sorenson S, Dwek JR, Resnick D. Humeral avulsion of the posterior band of the inferior glenohumeral ligament: MR arthrography and clinical correlation in 17 patients. AJR Am J Roentgenol. 2004;183(2):355-359.
8. Kim SH, Ha KI, Yoo JC, Noh KC. Kim’s lesion: an incomplete and concealed avulsion of the posteroinferior labrum in posterior or multidirectional posteroinferior instability of the shoulder. Arthroscopy. 2004;20(7):712-720.
9. Grubin J, Maderazo A, Fitzpatrick D. Imaging evaluation of superior labral anteroposterior (SLAP) tears. Am J Orthop. 2015;44(10):476-477.