User login
Depression and Postdischarge Events
Between 10% and 40% of patients are readmitted after being discharged from the hospital,[1, 2] and as many as another 25% return to the emergency department (ED) within 30 days.[3] This creates a substantial burden on the healthcare system.[2] Various interventions have been tried to improve the quality of discharge transitions and reduce readmission rates, but results thus far have been inconsistent and generally disappointing.[4, 5, 6] Targeted delivery of interventions to those at highest risk might improve the effectiveness of these efforts and reduce costs. However, current readmission risk assessment models are only moderately predictive, suggesting the presence of unrecognized risk factors.[7, 8]
Active depression might represent a potentially modifiable independent predictor of adverse short‐term hospital outcomes that is currently underutilized. Depression occurs in 5% to 58% of hospitalized adults, depending on how cases are defined.[9, 10] Depression is often under‐recognized and undertreated in acute care clinical settings,[11] and relatively few readmission prediction models incorporate mental health related symptoms.[12]
Although several reviews have examined methods of screening for depression in hospitalized patients[9] or the effectiveness of screening in primary care,[13, 14] to our knowledge no systematic review has examined the impact of depression on short‐term prognosis after discharge from acute care. Therefore, the purpose of this systematic review was to summarize all studies that evaluated whether hospitalized medical patients with depressive symptoms are at higher risk of 30‐day all‐cause readmission or all‐cause mortality after being discharged from the hospital.
METHODS
This study followed an a priori protocol developed according to PRISMA (Preferred Reporting Items for Systematic Reviews and Meta‐Analyses) criteria.[15]
Data Sources and Search Methods
We searched the Cumulative Index to Nursing and Allied Health Literature, Ovid MEDLINE, Ovid Embase, and PsycINFO from inception to January 9, 2015, and the last 5 years of PubMed for full publications with any of the following Medical Subject Headings: depressive disorder, depression, patient readmission, interviews, psychological, inpatients, with restrictions for peer‐reviewed publication, humans, adults aged 18 years, and the English language. Search strategies were developed with a librarian (available upon request). We manually searched reference lists of all included studies and relevant review articles and contacted content experts to identify additional publications.
Eligibility Criteria and Selection of Studies
Two authors (J.L.P. and L.M.W.) independently screened full texts of all relevant articles for inclusion. Disagreements were resolved by consensus or a third reviewer (S.R.M.). We considered any original research that compared readmission or mortality after discharge for hospitalized medical patients (ie, general patients or subgroups thereof) with versus without depression identified by any validated depression measure,[16] including any study design that incorporated at least 30‐day follow‐up postdischarge. We excluded studies that examined patients hospitalized in nonacute care settings or on surgical, psychiatric, obstetric, or intensive care services. We calculated Cohen's coefficient to evaluate inter‐rater agreement on study selection.
Data Extraction
Data were abstracted by 2 authors (J.L.P. and L.M.W.). Disagreements were resolved by consensus or a third reviewer (S.R.M.). We contacted authors of all included studies to obtain missing data. If unavailable, crude data were estimated from published survival curves employing validated techniques in R (version 3.1.2; R Foundation for Statistical Computing, Vienna, Austria) and Digitizeit (
Data Synthesis and Statistical Analysis
Where possible, we calculated the pooled risk ratio (RR) with 95% confidence interval (95% CI) using a random effects models in Review Manager (RevMan) 5.3 (The Nordic Cochrane Centre, Copenhagen, Denmark). The random effects approach that we employed assumes heterogeneity (ie, underlying parameters vary between individual studies) and is distributed around a mean or population average effect, and results in more conservative (wider) confidence intervals, wherein larger cohorts (or studies with smaller standard errors) are given more weight. Heterogeneity was assessed using the I2 statistic, with values of <25%, 25% to 50%, and >50% representing low, moderate, and high heterogeneity.[19] As per the guidance of Higgins et al., we did not a priori define any degree of heterogeneity that would preclude pooling of the data; the expectation would be that heterogeneity would be substantially higher pooling observational studies rather than randomized trials.[19] Statistical significance was considered a 2‐sided P value 0.05.
Quality Assessment and Risk of Bias
We assessed study quality using the 9‐item Newcastle‐Ottawa scale with 0 to 3, 4 to 6, and 7 to 9 stars considered low, moderate, and high quality, respectively, and criteria for external and internal validity, including group selection and comparability, outcome assessment, and adequacy of follow‐up.[20] Adjusted estimates published in individual reports (or obtained directly from authors) were compared wherever possible with unadjusted estimates to assess the degree of confounding. We generated funnel plots in RevMan 5.3 and conducted Egger tests using Stata 13 (StataCorp LP, College Station, TX) to assess for publication bias.[21]
RESULTS
Study Selection
After removing duplicate publications, we identified 4066 reports and reviewed 133 reports in full text (see Supporting Figure 1 in the online version of this article). Despite our broad study inclusion criteria, we found only 35 longitudinal studies addressing this question. All 35 authors were contacted for additional outcomes data and other missing information (response rate of 34%). We had to exclude 17 studies as they did not provide 30 or 90‐day post‐discharge outcomes. Only 4 studies had published crude data for outcomes within 90 days,[22, 23, 24, 25] but after contact with authors, we received unpublished data for a further 7 studies[26, 27, 28, 29, 30, 31, 32] (including individual level data for 2 cohorts).[31, 32] We were able to estimate crude data from Kaplan‐Meier curves for another 3 studies.[33, 34, 35] Another 4 studies did not collect the outcomes we were interested in individually. These studies were included in this systematic review but are not poolable in our models: 3 authors could only provide composite endpoint data,[36, 37, 38] and 1 author provided unadjusted hazard ratios.[39] Inter‐reviewer agreement for inclusion was 80% (Cohen's = 0.60).
Characteristics of Included Studies
The 18 studies ranged in size from 58 to 1418 patients; 13 were cohort studies and 5 included secondary data from randomized control trials.[22, 27, 30, 34, 36] All studies ascertained depressive status by screening during index medical admission with either diagnostic interview or self‐report questionnaires, although a variety of scales and definitions for depression were used (Beck Depression Inventory [BDI] in 6 studies, Geriatric Depression Scale in 5 studies, Patient Health Questionnaire in another 4 studies, Medical Outcomes Study‐Depression Questionnaire in 1 study, and Center for Epidemiologic Studies Depression Scale in another study) (Table 1). Screening interviews were conducted mostly by research assistants or nurses (68%) or self‐administered (21%). Most studies examined specific medical patient subgroups (10 cardiac, 3 pulmonary, and 2 elderly). Major exclusion criteria reported were terminal illness (4 studies), unstable condition (6 studies), severe cognitive impairment (5 studies), and suicidal ideation or known depression (4 studies); 1 study enrolled patients with suspected depression (Table 1). Patient cohorts were on average older (range, 5082 years) (Table 1). Attrition rates for readmission and mortality data were low (average <1% among entire sample of studies). All studies scored at least 5 on the Newcastle‐Ottawa scale and were thus considered of at least moderate quality (see Supporting Table 1 in the online version of this article).
| Author, Date of Publication, Enrollment Period | Setting Country/Region, No. of Hospitals | No. of Inpatients, Clinical Features | Major Exclusion Criteria | Follow‐up, mo | Depression Measure (Cutoff) and Screening Method | Mean Age (SD), y | % Female | Positive Screen, No. (%) | Primary Outcome, Secondary Outcomes |
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| Studies that use a scale based on DSM‐III criteria or a diagnostic interview according to DSM‐III criteria | |||||||||
| Frasure‐Smith et al.,[26] 1993, 19911992* | Canada/Quebec, 1 urban teaching | 218, AMI | Terminal noncardiac illness, unstable, not cognitive | 6 | BDI (10); mod DIS by interviewer, after transfer to medicine | 60 (range, 2488) | 22 | 68 (31), 35 (16) | All‐cause mortality |
| Frasure‐Smith et al.,[27] 1999, 19911992,* 19911994 | Canada/Quebec, 1 urban teaching, 10 urban area | 218; 78, AMI | Terminal noncardiac illness, unstable, not cognitive | 12 | BDI (10) by interviewer, after transfer to medicine | 60 (11) | 32 | 290 (32) | Cardiac mortality |
| Freedland et al.,[25] 1991, 1990 | USA/MO, 1 urban teaching | 58, CHF 75 years | Dementia, medically unstable | 3 | Mod DIS by psychiatric residents and interviewer | 78 (6) | 57 | 10 (17) | All‐cause readmission, all‐cause mortality |
| Fulop et al.,[38] 2003, 2002 | USA/NY, 1 urban teaching | 203, CHF 65 years | 1, 6 | GDS (10); SCID‐NP by interviewer, at discharge | 77 (8) | 53 | 73 (36), 44 (22) | Depression, composite PCP, ED, care visits, and readmission | |
| Lesprance et al.,[28] 2000, 19941996 | Canada/Quebec, 1 urban teaching | 430, unstable angina | Terminal noncardiac illness, not cognitive, recent CABG | 12 | BDI (10); mod DIS by interviewer, 5 days after admission | 62 (11) | 29 | 178 (41), 120 (28) | Cardiac death and MI, any death, angina readmission |
| Rumsfeld et al.,[30] 2005, 19992001 | CA, USA, UK, multiple | 634, AMI with CHF | Valvular or congenital heart failure | Up to 32 | MOS‐D (0.06) by interviewer, before discharge | 65 (11) | 28 | 143 (23) | All‐cause death, CVD death and readmission |
| Song et al.,[33] 2009, 2005 | South Korea, 2 urban teaching | 165, HF | If minor criteria for HF attributable to other medical condition | 6 | BDI (10) self‐administer or interviewer, 34 days of admin | 62 (13) | 49 | 131 (79) | HF readmission and all‐cause mortality, HF readmit |
| Papaioannou et al.,[29] 2013, 20092010 | Greece/Athens, 1 urban | 230, AECOPD | Other respiratory illness, known depressed | Monthly up to 12 | BDI‐I (19) self‐administer, first day | 71 (9) | 12 | 91 (40) | All‐cause mortality, AECOPD readmission |
| Studies that use a scale based on or validated against DSM‐IV criteria or a diagnostic interview according to DSM‐IV criteria | |||||||||
| Almagro et al.,[31] 2002, 19961997 | Spain, 1 urban teaching | 130, AECOPD | Other pulmonary disease | July 1999 | GDS‐SF (6) by interviewer, day before discharge | 72 (9) | 8 | 43 (33) | All‐cause mortality |
| Almagro et al.,[32] 2012, 20032004 | Spain, 1 urban teaching | 134, AECOPD | Other pulmonary disease | 1, 36 | GDS‐SF (6) by interviewer | 72 (10) | 5 | 55 (41) | All‐cause mortality, lung function, frailty |
| Bla et al.,[39] 2001, 2000 | Switzerland, 1 urban teaching | 401, medical 75 years | Stay <24 hours, elective/facility transfer, unstable, not cognitive | 6 | GDS‐SF (6) by interviewer, within 2 days of admission | 82 (7599) | 61 | 90 (22) | All‐cause readmission, all‐cause mortality |
| Cancino et al.,[22] 2014, 20062007,* 20082009 | USA/MA, 1 urban tertiary | 680; 738, medical | Nursing home or hospital transfer, isolated, suicidal | 1 | PHQ‐9 (5 or severity) by interviewer, on admin | 50 (14) | 51 | 561 (40) | All‐cause readmission, ED visits, PCP visits |
| Mitchell et al.,[36] 2010, 20062007* | USA/MA, 1 urban tertiary | 738, medical | Nursing home or hospital transfer, isolated, suicidal | 1, 2, 3 | PHQ‐9 (5) by interviewer, on admin | 50 (15) | 50 | 238 (32) | ED visits and all‐cause readmission |
| Covinsky et al.,[34] 1999, 19901992 | USA/OH, 1 urban teaching | 573, medical | ICU, oncology, telemetry, nursing home admissions | 36 | GDS‐SF (6) by interviewer, within 2 days of admission | 80 | 68 | 197 (34) | All‐cause mortality |
| Jiang et al.,[23] 2001, 19971998 | USA/NC, 1 urban teaching | 357 (331 DIS only), CHF | Suicidal, planned surgery, pregnant | 3, 12 | BDI (10) self‐admin; mod DIS (+BDI only) by interviewer | 63 (13) | 33 | 126 (35), 46 (14) | All‐cause mortality, all‐cause readmission |
| Kartha et al.,[24] 2007, 20022004 | USA/MA, 1 urban safety net | 144, medical recently hospitalized | Planned readmission, unable to keep PCP appointments | 3 | PHQ‐9 (algorithm) by interviewer | 55 (16) | 56 | 39 (27) | All‐cause readmission |
| Koenig and Kuchbhatla,[37] 1999, 1997 | USA/NC, 1 urban teaching | 331, medical 60 years | Stay <3 or >7 days, ICU/CCU, severe illness, nursing home transfers | 3, 6, 9, 12 | CES‐D (16) or HAM‐D (11) or DIS by psychiatrist, on or after third day | 70 (7) | 51 | 160 (48) | Depression, composite physical disability, health visits, and all‐cause readmission |
| Rollman et al.,[35] 2012, 20072009 | USA/PA, 4 urban teaching | 471, CHF, suspected depressed | Antidepressants users (excluded from PHQ‐2 group only) | Up to 12 | PHQ‐2; PHQ‐9 (5 in +PHQ‐2), by interviewer, 4 days | 66 (13) | 35 | 371 (79), 351 (74) | All‐cause mortality |
Prevalence and Recognition of Depressive Symptoms
The range of depression prevalence in hospitalized medical patients was 14% to 79%, with a median of 32% (interquartile range, 27%40%) (Table 1). In those studies that used a diagnostic interview, the prevalence tended to be lower for major depression, with a median of 17% (interquartile range, 16%22%) (Table 1). None of the included studies reported frequency of clinically recognized depression (ie, prior to screening for the study). Only 2 studies assessed the persistence of depression after discharge: 1 reported that depression persisted in 53% (by screening questionnaire) and 34% (by diagnostic interview) of patients at 30 days,[38] whereas the other reported 48% persistence at 90 days after discharge according to a combined screening method.[37]
Hospital Readmission
Overall, 8 studies provided readmission data. Among patients discharged from acute care medical wards (4 studies reporting on 5 cohorts), 395 of 2433 (16.2%) patients were readmitted within 30 days (Figure 1). Hospitalized patients with depressive symptoms were more likely to be readmitted within 30 days after discharge (20.4% vs 13.7%, RR: 1.73, 95% CI: 1.16‐2.58, P = 0.007, I2 = 55%) (Figure 1), compared to those without depression. Results were consistent for 90‐day readmissions (39.8% vs 31.0%, RR: 1.68, 95% CI: 1.13‐2.50, P = 0.01, I2 = 76%, n = 1543 patients) (see Supporting Figure 2 in the online version of this article) in 6 studies. One individual study examined readmission within 6 months after discharge, but was not poolable in this model, as it presented only hazard ratios and not raw data; however, it did report a 50% increased risk of readmission in medical inpatients aged 75 years (adjusted hazard ratio: 1.50, 95% CI: 1.03‐2.17, n = 401).[39]
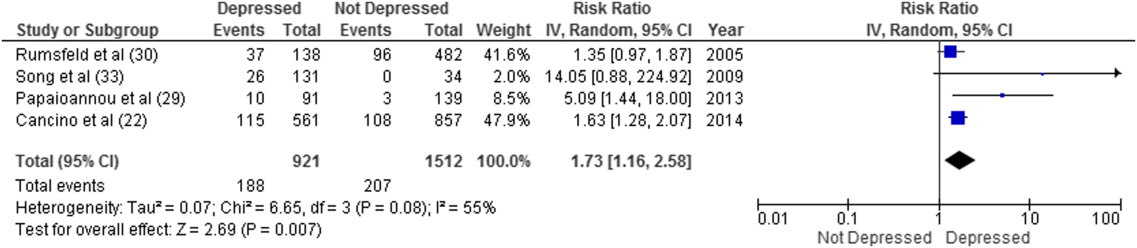
Forest plot presents results of the meta‐analysis in which the size of each data marker indicates the weight assigned to individuals studies. Abbreviations: CI, confidence interval; IV, independent variable.
Mortality After Discharge
Overall, 11 studies provided all‐cause mortality data. Among medical patients discharged from acute care in 9 studies, 69 of 3397 (2.0%) patients died within 30 days (Figure 2). Medical patients discharged with depressive symptoms were more likely to die within 30 days (2.8% vs 1.5%, RR: 2.13, 95% CI: 1.31‐3.44, P = 0.002, I2 = 0%) (Figure 2) compared to those without depression. Similar results were found for 90‐day mortality (7.7% vs 4.1%, RR: 2.01, 95% CI: 1.47‐2.76, P < 0.001, I2 = 4%, n = 3784 patients) (see Supporting Figure 3 in the online version of this article) in 11 studies.
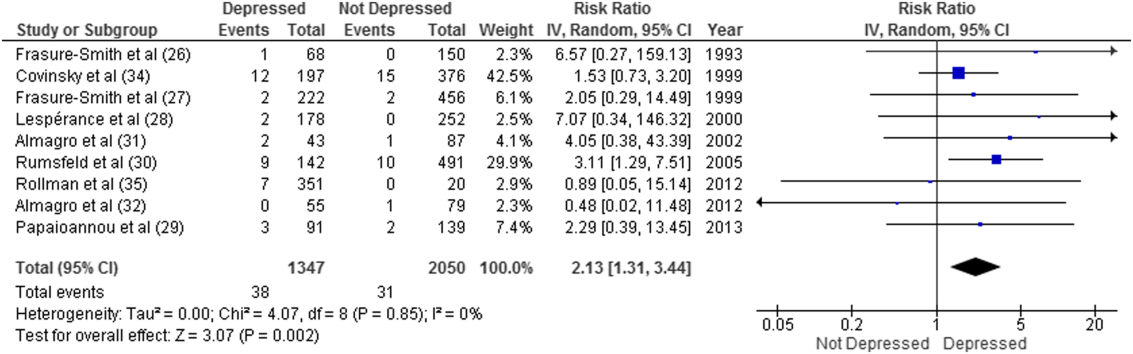
ED and PCP Visits
Four studies examined the use of ED or PCP services within 90 days of discharge, but 3 did not have extractable data for meta‐analysis. All showed increased utilization of health services for depressed compared to nondepressed patients after discharge.[22, 36, 37, 38] Depressed patients were more likely to visit the ED (adjusted incidence rate ratio: 1.73, 95% CI: 1.27‐2.36),[36] had significantly more medical encounters (eg, PCP, ED visits, hospital admissions, laboratory tests, and home care [mean 2.9 vs 2.6, P = 0.05])[38] and had a greater number of ED visits alone (27 vs 15 per 100 patients, P = 0.007)[22] within 30 days of hospital discharge compared to nondepressed patients. Similar results were found at 90 days.[36]
Sensitivity Analyses
All told, most studies reported a positive association between depression and adverse events, and this was true regardless of how much adjustment for potential confounding had been undertaken by the authors. Although all studies were qualitatively in the same direction, the magnitude of the association varied due to methodological and/or clinical heterogeneity. Sensitivity analysis revealed no overall difference in pooled risk ratios or heterogeneity between Mantel‐Haenszel fixed effects versus random effects models or with the addition of 0.5 to cells to permit inclusion of zero‐event data. There was no evidence of publication bias; funnel plots and Egger test results are available upon request. There were no statistically significant differences in the risk associated with depressive symptoms whether studies used Diagnostic and Statistical Manual of Mental Disorders (DSM)‐III or DSM‐IV criteria, whether the study samples were disease specific or unselected general medical cohorts, whether studies were of moderate or high quality, or regardless of the severity of depressive symptoms.
DISCUSSION
Summary of Evidence
We found that depression was common in medical inpatients (about one‐third of all patients) and persisted for at least 30 days in up to half of those patients after discharge. We found strong evidence of an association between depressive symptoms and poor short‐term prognosis after discharge from the hospital: a 73% increased risk of readmission and a 2‐fold risk of death within 30 days compared to patients without depressive symptoms with similar results at 90 days.
Our meta‐analysis complements a recent systematic review that found concomitant depression to be a risk factor for poor prognosis among inpatients and outpatients with acute coronary syndrome,[40] and a meta‐analysis that demonstrated an increased risk of 2‐year mortality among patients with depression after myocardial infarction.[41] To our knowledge, our study is the first to quantify the short‐term postdischarge risks across a diverse group of medical inpatients.
The potential mechanisms underlying the observed relationship between depression and adverse patient outcomes after discharge are likely multiple. We believe there are 2 main possibilities. First, the increased risk associated with depression might be due to residual confounding, even though many of these studies did adjust for extensive lists of comorbidities,[22, 24, 26, 27, 29, 30, 33, 35, 36, 39] including functional status[39] and prior health services utilization.[22, 34, 36] This could occur if other risk factors were not sufficiently adjusted for, such as unrecognized comorbidities or concomitant disability, which are often present among chronically ill patients,[42] or if depression were a marker of psychosocial risk factors such as anxiety,[43] stress or poor resiliency,[44] or low social support,[45] though a few adjusted for psychosocial factors such as social support[26] or anxiety.[35] Confounding could also occur if symptoms of acute illness inflate reports of somatic symptoms of depression on self‐report questionnaires. Recent studies on the BDI, found that scores were higher in postmyocardial infarction patients when compared to outpatient controls,[46] but with no differences between those groups in scores for the BDI‐II,[47] a version with fewer somatic symptom questions.
Second, depression may cause adverse outcomes through indirect or direct pathways. Indirect causation could occur if depression hindered self‐care behaviors such as medication adherence.[42] Depression could also act directly through pathophysiological changes. Some studies have suggested that depression is associated with metabolic abnormalities, including alterations in glucose transport[42, 48] and increased vulnerability to obesity, type 2 diabetes mellitus, and/or diabetic complications, common conditions among hospitalized patients that also adversely affect postdischarge outcomes.[40, 48]
Strengths and Limitations
This review has multiple strengths. We cast a broad search and included studies that examined a wide range of medical patient subgroups, thus increasing the generalizability of our findings. We identified a general scarcity of studies on this topic and obtained additional unpublished data for 10 of the 18 relevant studies, and our response rate of 34% is compatible with the 37% response rate reported for Cochrane reviews when seeking additional data from authors.[49] Whether examined qualitatively (vote counting of the number of studies that showed an association) or quantitatively (via formal meta‐analysis), it seems apparent that there is a clinically important association between depression and postdischarge adverse events, but given the number, quality, and heterogeneity of the studies we examined, there may be some ongoing dispute about exactly how strong this association is and the degree of bias contributed by a couple of large studies of the topic.
There are limitations to our review. First, as we did not have individual‐level patient data, we could not use metaregression to explore sources of heterogeneity (clinical or methodological) or adjust for confounding, and this likely contributes to observed differences between individual estimates. For instance, the included studies had heterogeneous screening measures and cutoffs; thus, all cases of depression in these studies might not be equivalent. Some of the included studies assessed depression early during admission where psychological distress may be greatest; others assessed symptoms closer to discharge. Most studies included patients with specific conditions like heart failure or chronic obstructive pulmonary disease rather than a wide spectrum of medical inpatients. Moreover, few studies adjusted for psychosocial risk factors such as social support, anxiety, and functional status, and only 2 studies assessed the persistence of depressive symptoms after discharge. Second, we did not explore quantitative measures of between‐study variation (eg, I2), because experts question its utility given the expected heterogeneity in meta‐analyses of observational studies.[50] Third, although the included studies were deemed to be of at least moderate quality, they could be at risk for sources of bias that may not be sufficiently appraised by the current version of the Newscastle‐Ottawa scale for observational studies. Finally, we excluded grey literature (eg, conference proceedings or technical reports) that could potentially exclude null findings, although we did contact authors in this field to identify additional unpublished data relevant to this topic.
CONCLUSIONS
We have confirmed that depressive symptoms are common in hospitalized medical patients, frequently persist after discharge, and may predict greater risk of readmission or death after discharge. Thus, depressive symptoms are an additional marker that clinicians can use to help identify patients in acute care medical settings who may be at increased risk for suboptimal transition back to the community and who may require additional resources after discharge. However, future research is required to evaluate whether treatment of individuals who screen positive for depressive symptoms can reduce 30‐day readmission rates, and we are aware of at least 1 relevant ongoing trial (
Acknowledgements
The authors thank the following individuals: Dale Storie, MLIS, Saskatchewan Information and Library Services Consortium, Regina, Saskatchewan, Canada, for assistance in the literature search; James A. Hanley, PhD, Department of Epidemiology and Biostatistics, Faculty of Medicine, McGill University, Montreal, Quebec, Canada, for guidance in data recovery methods; Nancy Frasure‐Smith, PhD, Department of Psychiatry, McGill University, Department of Psychiatry and Research Centre Hospital Centre, University of Montreal, and Montreal Heart Institute Research Centre, Montreal, Quebec, Canada; Andriana I. Papaioannou, MD, 2nd Respiratory Medicine Department, University of Athens Medical School, Athens, Greece; Konstantinos Kostikas, MD, 2nd Respiratory Medicine Department, University of Athens Medical School, Athens, Greece; and Pere Almagro, MD, Servicio de Medicina Interna, Hospital Universitario Mutua de Terrassa, Terrassa, Barcelona, Spain; as well as Philip G. Jones, MS, Saint Luke's Mid America Heart Institute, Kansas City, Missouri; for their retrieval and contribution of unpublished data.
Disclosures
Ms. Pederson affirms that the manuscript is an honest, accurate, and transparent account of the study being reported with no important omissions. All authors had full access to all of the data (including statistical reports and tables) in the study and can take responsibility for the integrity of the data and the accuracy of the data analysis. Design and conduct of the study: Ms. Pederson, Drs. Majumdar and McAlister. Data acquisition: Ms. Pederson, Ms. Warkentin. Analysis and interpretation of the data and drafting of the manuscript: Ms Pederson, Drs. Majumdar and McAlister. Review of the manuscript: all authors. Study supervision: Drs. Majumdar and McAlister. None of the contributors received compensation for their efforts. Salary support for Ms. Pederson was provided by a CRIO grant from Alberta InnovatesHealth Solutions. Drs. McAlister and Majumdar are supported by salary awards from Alberta Innovates‐Health Solutions. Dr. McAlister holds the University of Alberta/Capital Health Chair in Cardiology Outcomes Research. Dr. Majumda holds the University of Alberta Endowed Chair in Patient Health Management. The funding sources had no role in the design or conduct of the study; management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. This work is that of the authors independent of funders. The authors report no conflicts of interest.
- , , . Rehospitalizations among patients in the Medicare fee‐for‐service program. N Engl J Med. 2009;360(14):1418–1428.
- , , , , . Proportion of hospital readmissions deemed avoidable: a systematic review. CMAJ. 2011;183(7):E391–E402.
- , , , . Mental health conditions are associated with increased health care utilization among urban family medicine patients. J Am Board Fam Med. 2008;21(5):398–407.
- , , , , . Interventions to reduce 30‐day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520–528.
- . The elusive quest for quality and cost savings in the Medicare program. JAMA. 2009;301(6):668–670.
- , , , . Effects of care coordination on hospitalization, quality of care, and health care expenditures among Medicare beneficiaries—15 randomized trials. JAMA. 2009;301(6):603–618.
- , , , et al. Unplanned readmissions after hospital discharge among patients identified as being at high risk for readmission using a validated predictive algorithm. Open Med. 2011;5(2):e104–111.
- , , , et al. Risk prediction models for hospital readmission: a systematic review. JAMA. 2011;306(15):1688–1698.
- , , . Depression in older people in the general hospital: a systematic review of screening instruments. Age Ageing. 2012;41(2):148–154.
- , , , et al. Prevalence, correlates and recognition of depression among inpatients of general hospitals in Wuhan, China. Gen Hosp Psychiatry. 2010;32(3):268–275.
- , , , , , . Recognition of depression by non‐psychiatric physicians—a systematic literature review and meta‐analysis. J Gen Intern Med. 2008;23(1):25–36.
- , , , , , . Predicting the risk of unplanned readmission or death within 30 days of discharge after a heart failure hospitalization. Am Heart J. 2012;164(3):365–372.
- , , , et al. Does evidence support the American Heart Association's recommendation to screen patients for depression in cardiovascular care? An updated systematic review. PLoS One. 2013;8(1):e52654.
- , , , et al. Screening for depression: a systematic review and meta‐analysis. CMAJ Open. 2013;1(4):E159–E167.
- , , , et al. The PRISMA statement for reporting systematic reviews and meta‐analyses of studies that evaluate health care interventions: explanation and elaboration. Ann Intern Med. 2009;151(4):W65–94.
- , , , et al. Screening for depression in adults: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med. 2002;136(10):765–776.
- , , . Recovering the raw data behind a non‐parametric survival curve. Syst Rev. 2014;3:151.
- , , , . Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan‐Meier survival curves. BMC Med Res Methodol. 2012;12(1):9.
- . Commentary: heterogeneity in meta‐analysis should be expected and appropriately quantified. Int J Epidemiol. 2008;37(5):1158–1160.
- , , , et al. The Newcastle‐Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta‐analyses. Available at: http://www.ohri.ca/programs/clinical_epidemiology/oxford.htm. Accessed September 1, 2015.
- , , . The funnel plot. In: Rothstein HR, Sutton AJ, Borenstein M, eds. Publication Bias in Meta‐analysis: Prevention, Assessment and Adjustments. New York, NY: John Wiley 2006:73–98.
- , , , , , . Dose‐response relationship between depressive symptoms and hospital readmission. J Hosp Med. 2014;9(6):358–364.
- , , , et al. Relationship of depression to increased risk of mortality and rehospitalization in patients with congestive heart failure. Arch Intern Med. 2001;161(15):1849–1856.
- , , , et al. Depression is a risk factor for rehospitalization in medical inpatients. Prim Care Companion J Clin Psychiatry. 2007;9(4):256–262.
- , , , et al. Depression in elderly patients with congestive heart failure. J Geriatr Psychiatry. 1991;24(1):59–71.
- , , . Depression following myocardial infarction: impact on 6‐month survival. JAMA. 1993;270(15):1819–1825.
- , , , , . Gender, depression, and one‐year prognosis after myocardial infarction. Psychosom Med. 1999;61(1):26–37.
- , , , . Depression and 1‐year prognosis in unstable angina. Arch Intern Med. 2000;160(9):1354–1360.
- , , , et al. The impact of depressive symptoms on recovery and outcome of hospitalised COPD exacerbations. Eur Respir J. 2013;41(4):815–823.
- , , , et al. Depression predicts mortality and hospitalization in patients with myocardial infarction complicated by heart failure. Am Heart J. 2005;150(5):961–967.
- , , , et al. Mortality after hospitalization for COPD. Chest. 2002;121(5):1441–1448.
- , , , et al. Pseudomonas aeruginosa and mortality after hospital admission for chronic obstructive pulmonary disease. Respiration. 2012;84(1):36–43.
- , , . Depressive symptoms increase risk of rehospitalisation in heart failure patients with preserved systolic function. J Clin Nurs. 2009;18(13):1871–1877.
- , , . Depressive symptoms and 3 year mortality in older hospitalized medical patients. Ann Intern Med. 1999;130(7):563–569.
- , , , et al. A positive 2‐item patient health questionnaire depression screen among hospitalized heart failure patients is associated with elevated 12‐month mortality. J Card Fail. 2012;18(3):238–245.
- , , , et al. Post‐discharge hospital utilization among adult medical inpatients with depressive symptoms. J Hosp Med. 2010;5(7):378–384.
- , . Use of health services by medically ill depressed elderly patients after hospital discharge. Am J Geriatr Psychiatry. 1999;7(1):48–56.
- , , . Congestive heart failure and depression in older adults: clinical course and health services use 6 months after hospitalization. Psychosomatics. 2003;44(5):367–373.
- , , , . Depressive symptoms as a predictor of 6‐month outcomes and services utilization in elderly medical inpatients. Arch Intern Med. 2001;161(21):2609–2615.
- , , , et al. Depression as a risk factor for poor prognosis among patients with acute coronary syndrome: systematic review and recommendations: a scientific statement from the American Heart Association. Circulation. 2014;129(12):1350–1369.
- , , , , , . Prognostic association of depression following myocardial infarction with mortality and cardiovascular events: a meta‐analysis of 25 years of research. Gen Hosp Psychiatry. 2011;33(3):203–216.
- , , , , . Depression and cardiac disease: epidemiology, mechanisms, and diagnosis. Cardiovasc Psychiatry Neurol. 2013;2013:695925.
- , , , et al. Prognostic value of depression, anxiety, and anger in hospitalized cardiovascular disease patients for predicting adverse cardiac outcomes. Am J Cardiol. 2013;111(10):1432–1436.
- , , . The psychobiology of depression and resilience to stress: implications for prevention and treatment. Annu Rev Clin Psychol. 2005;1:255–291.
- , , , et al. Impact of social factors on risk of readmission or mortality in pneumonia and heart failure: systematic review. J Gen Intern Med. 2013;28(2):269–282.
- , , , et al. The influence of somatic symptoms on beck depression inventory scores in hospitalized postmyocardial infarction patients. Can J Psychiatry. 2012;57(12):752–758.
- , , , et al. Somatic symptom overlap in beck depression inventory‐II scores following myocardial infarction. Br J Psychiatry. 2010;197(1):61–66.
- , , , . Relationship of depression to diabetes types 1 and 2: epidemiology, biology, and treatment. Biol Psychiatry. 2003;54(3):317–329.
- , , . Searching for unpublished data for Cochrane reviews: cross sectional study. BMJ. 2013;346:f2231.
- . Comment on: heterogeneity in meta‐analysis should be expected and appropriately quantified. Int J Epidemiol. 2010;39(3):932; author reply 933.
Between 10% and 40% of patients are readmitted after being discharged from the hospital,[1, 2] and as many as another 25% return to the emergency department (ED) within 30 days.[3] This creates a substantial burden on the healthcare system.[2] Various interventions have been tried to improve the quality of discharge transitions and reduce readmission rates, but results thus far have been inconsistent and generally disappointing.[4, 5, 6] Targeted delivery of interventions to those at highest risk might improve the effectiveness of these efforts and reduce costs. However, current readmission risk assessment models are only moderately predictive, suggesting the presence of unrecognized risk factors.[7, 8]
Active depression might represent a potentially modifiable independent predictor of adverse short‐term hospital outcomes that is currently underutilized. Depression occurs in 5% to 58% of hospitalized adults, depending on how cases are defined.[9, 10] Depression is often under‐recognized and undertreated in acute care clinical settings,[11] and relatively few readmission prediction models incorporate mental health related symptoms.[12]
Although several reviews have examined methods of screening for depression in hospitalized patients[9] or the effectiveness of screening in primary care,[13, 14] to our knowledge no systematic review has examined the impact of depression on short‐term prognosis after discharge from acute care. Therefore, the purpose of this systematic review was to summarize all studies that evaluated whether hospitalized medical patients with depressive symptoms are at higher risk of 30‐day all‐cause readmission or all‐cause mortality after being discharged from the hospital.
METHODS
This study followed an a priori protocol developed according to PRISMA (Preferred Reporting Items for Systematic Reviews and Meta‐Analyses) criteria.[15]
Data Sources and Search Methods
We searched the Cumulative Index to Nursing and Allied Health Literature, Ovid MEDLINE, Ovid Embase, and PsycINFO from inception to January 9, 2015, and the last 5 years of PubMed for full publications with any of the following Medical Subject Headings: depressive disorder, depression, patient readmission, interviews, psychological, inpatients, with restrictions for peer‐reviewed publication, humans, adults aged 18 years, and the English language. Search strategies were developed with a librarian (available upon request). We manually searched reference lists of all included studies and relevant review articles and contacted content experts to identify additional publications.
Eligibility Criteria and Selection of Studies
Two authors (J.L.P. and L.M.W.) independently screened full texts of all relevant articles for inclusion. Disagreements were resolved by consensus or a third reviewer (S.R.M.). We considered any original research that compared readmission or mortality after discharge for hospitalized medical patients (ie, general patients or subgroups thereof) with versus without depression identified by any validated depression measure,[16] including any study design that incorporated at least 30‐day follow‐up postdischarge. We excluded studies that examined patients hospitalized in nonacute care settings or on surgical, psychiatric, obstetric, or intensive care services. We calculated Cohen's coefficient to evaluate inter‐rater agreement on study selection.
Data Extraction
Data were abstracted by 2 authors (J.L.P. and L.M.W.). Disagreements were resolved by consensus or a third reviewer (S.R.M.). We contacted authors of all included studies to obtain missing data. If unavailable, crude data were estimated from published survival curves employing validated techniques in R (version 3.1.2; R Foundation for Statistical Computing, Vienna, Austria) and Digitizeit (
Data Synthesis and Statistical Analysis
Where possible, we calculated the pooled risk ratio (RR) with 95% confidence interval (95% CI) using a random effects models in Review Manager (RevMan) 5.3 (The Nordic Cochrane Centre, Copenhagen, Denmark). The random effects approach that we employed assumes heterogeneity (ie, underlying parameters vary between individual studies) and is distributed around a mean or population average effect, and results in more conservative (wider) confidence intervals, wherein larger cohorts (or studies with smaller standard errors) are given more weight. Heterogeneity was assessed using the I2 statistic, with values of <25%, 25% to 50%, and >50% representing low, moderate, and high heterogeneity.[19] As per the guidance of Higgins et al., we did not a priori define any degree of heterogeneity that would preclude pooling of the data; the expectation would be that heterogeneity would be substantially higher pooling observational studies rather than randomized trials.[19] Statistical significance was considered a 2‐sided P value 0.05.
Quality Assessment and Risk of Bias
We assessed study quality using the 9‐item Newcastle‐Ottawa scale with 0 to 3, 4 to 6, and 7 to 9 stars considered low, moderate, and high quality, respectively, and criteria for external and internal validity, including group selection and comparability, outcome assessment, and adequacy of follow‐up.[20] Adjusted estimates published in individual reports (or obtained directly from authors) were compared wherever possible with unadjusted estimates to assess the degree of confounding. We generated funnel plots in RevMan 5.3 and conducted Egger tests using Stata 13 (StataCorp LP, College Station, TX) to assess for publication bias.[21]
RESULTS
Study Selection
After removing duplicate publications, we identified 4066 reports and reviewed 133 reports in full text (see Supporting Figure 1 in the online version of this article). Despite our broad study inclusion criteria, we found only 35 longitudinal studies addressing this question. All 35 authors were contacted for additional outcomes data and other missing information (response rate of 34%). We had to exclude 17 studies as they did not provide 30 or 90‐day post‐discharge outcomes. Only 4 studies had published crude data for outcomes within 90 days,[22, 23, 24, 25] but after contact with authors, we received unpublished data for a further 7 studies[26, 27, 28, 29, 30, 31, 32] (including individual level data for 2 cohorts).[31, 32] We were able to estimate crude data from Kaplan‐Meier curves for another 3 studies.[33, 34, 35] Another 4 studies did not collect the outcomes we were interested in individually. These studies were included in this systematic review but are not poolable in our models: 3 authors could only provide composite endpoint data,[36, 37, 38] and 1 author provided unadjusted hazard ratios.[39] Inter‐reviewer agreement for inclusion was 80% (Cohen's = 0.60).
Characteristics of Included Studies
The 18 studies ranged in size from 58 to 1418 patients; 13 were cohort studies and 5 included secondary data from randomized control trials.[22, 27, 30, 34, 36] All studies ascertained depressive status by screening during index medical admission with either diagnostic interview or self‐report questionnaires, although a variety of scales and definitions for depression were used (Beck Depression Inventory [BDI] in 6 studies, Geriatric Depression Scale in 5 studies, Patient Health Questionnaire in another 4 studies, Medical Outcomes Study‐Depression Questionnaire in 1 study, and Center for Epidemiologic Studies Depression Scale in another study) (Table 1). Screening interviews were conducted mostly by research assistants or nurses (68%) or self‐administered (21%). Most studies examined specific medical patient subgroups (10 cardiac, 3 pulmonary, and 2 elderly). Major exclusion criteria reported were terminal illness (4 studies), unstable condition (6 studies), severe cognitive impairment (5 studies), and suicidal ideation or known depression (4 studies); 1 study enrolled patients with suspected depression (Table 1). Patient cohorts were on average older (range, 5082 years) (Table 1). Attrition rates for readmission and mortality data were low (average <1% among entire sample of studies). All studies scored at least 5 on the Newcastle‐Ottawa scale and were thus considered of at least moderate quality (see Supporting Table 1 in the online version of this article).
| Author, Date of Publication, Enrollment Period | Setting Country/Region, No. of Hospitals | No. of Inpatients, Clinical Features | Major Exclusion Criteria | Follow‐up, mo | Depression Measure (Cutoff) and Screening Method | Mean Age (SD), y | % Female | Positive Screen, No. (%) | Primary Outcome, Secondary Outcomes |
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| Studies that use a scale based on DSM‐III criteria or a diagnostic interview according to DSM‐III criteria | |||||||||
| Frasure‐Smith et al.,[26] 1993, 19911992* | Canada/Quebec, 1 urban teaching | 218, AMI | Terminal noncardiac illness, unstable, not cognitive | 6 | BDI (10); mod DIS by interviewer, after transfer to medicine | 60 (range, 2488) | 22 | 68 (31), 35 (16) | All‐cause mortality |
| Frasure‐Smith et al.,[27] 1999, 19911992,* 19911994 | Canada/Quebec, 1 urban teaching, 10 urban area | 218; 78, AMI | Terminal noncardiac illness, unstable, not cognitive | 12 | BDI (10) by interviewer, after transfer to medicine | 60 (11) | 32 | 290 (32) | Cardiac mortality |
| Freedland et al.,[25] 1991, 1990 | USA/MO, 1 urban teaching | 58, CHF 75 years | Dementia, medically unstable | 3 | Mod DIS by psychiatric residents and interviewer | 78 (6) | 57 | 10 (17) | All‐cause readmission, all‐cause mortality |
| Fulop et al.,[38] 2003, 2002 | USA/NY, 1 urban teaching | 203, CHF 65 years | 1, 6 | GDS (10); SCID‐NP by interviewer, at discharge | 77 (8) | 53 | 73 (36), 44 (22) | Depression, composite PCP, ED, care visits, and readmission | |
| Lesprance et al.,[28] 2000, 19941996 | Canada/Quebec, 1 urban teaching | 430, unstable angina | Terminal noncardiac illness, not cognitive, recent CABG | 12 | BDI (10); mod DIS by interviewer, 5 days after admission | 62 (11) | 29 | 178 (41), 120 (28) | Cardiac death and MI, any death, angina readmission |
| Rumsfeld et al.,[30] 2005, 19992001 | CA, USA, UK, multiple | 634, AMI with CHF | Valvular or congenital heart failure | Up to 32 | MOS‐D (0.06) by interviewer, before discharge | 65 (11) | 28 | 143 (23) | All‐cause death, CVD death and readmission |
| Song et al.,[33] 2009, 2005 | South Korea, 2 urban teaching | 165, HF | If minor criteria for HF attributable to other medical condition | 6 | BDI (10) self‐administer or interviewer, 34 days of admin | 62 (13) | 49 | 131 (79) | HF readmission and all‐cause mortality, HF readmit |
| Papaioannou et al.,[29] 2013, 20092010 | Greece/Athens, 1 urban | 230, AECOPD | Other respiratory illness, known depressed | Monthly up to 12 | BDI‐I (19) self‐administer, first day | 71 (9) | 12 | 91 (40) | All‐cause mortality, AECOPD readmission |
| Studies that use a scale based on or validated against DSM‐IV criteria or a diagnostic interview according to DSM‐IV criteria | |||||||||
| Almagro et al.,[31] 2002, 19961997 | Spain, 1 urban teaching | 130, AECOPD | Other pulmonary disease | July 1999 | GDS‐SF (6) by interviewer, day before discharge | 72 (9) | 8 | 43 (33) | All‐cause mortality |
| Almagro et al.,[32] 2012, 20032004 | Spain, 1 urban teaching | 134, AECOPD | Other pulmonary disease | 1, 36 | GDS‐SF (6) by interviewer | 72 (10) | 5 | 55 (41) | All‐cause mortality, lung function, frailty |
| Bla et al.,[39] 2001, 2000 | Switzerland, 1 urban teaching | 401, medical 75 years | Stay <24 hours, elective/facility transfer, unstable, not cognitive | 6 | GDS‐SF (6) by interviewer, within 2 days of admission | 82 (7599) | 61 | 90 (22) | All‐cause readmission, all‐cause mortality |
| Cancino et al.,[22] 2014, 20062007,* 20082009 | USA/MA, 1 urban tertiary | 680; 738, medical | Nursing home or hospital transfer, isolated, suicidal | 1 | PHQ‐9 (5 or severity) by interviewer, on admin | 50 (14) | 51 | 561 (40) | All‐cause readmission, ED visits, PCP visits |
| Mitchell et al.,[36] 2010, 20062007* | USA/MA, 1 urban tertiary | 738, medical | Nursing home or hospital transfer, isolated, suicidal | 1, 2, 3 | PHQ‐9 (5) by interviewer, on admin | 50 (15) | 50 | 238 (32) | ED visits and all‐cause readmission |
| Covinsky et al.,[34] 1999, 19901992 | USA/OH, 1 urban teaching | 573, medical | ICU, oncology, telemetry, nursing home admissions | 36 | GDS‐SF (6) by interviewer, within 2 days of admission | 80 | 68 | 197 (34) | All‐cause mortality |
| Jiang et al.,[23] 2001, 19971998 | USA/NC, 1 urban teaching | 357 (331 DIS only), CHF | Suicidal, planned surgery, pregnant | 3, 12 | BDI (10) self‐admin; mod DIS (+BDI only) by interviewer | 63 (13) | 33 | 126 (35), 46 (14) | All‐cause mortality, all‐cause readmission |
| Kartha et al.,[24] 2007, 20022004 | USA/MA, 1 urban safety net | 144, medical recently hospitalized | Planned readmission, unable to keep PCP appointments | 3 | PHQ‐9 (algorithm) by interviewer | 55 (16) | 56 | 39 (27) | All‐cause readmission |
| Koenig and Kuchbhatla,[37] 1999, 1997 | USA/NC, 1 urban teaching | 331, medical 60 years | Stay <3 or >7 days, ICU/CCU, severe illness, nursing home transfers | 3, 6, 9, 12 | CES‐D (16) or HAM‐D (11) or DIS by psychiatrist, on or after third day | 70 (7) | 51 | 160 (48) | Depression, composite physical disability, health visits, and all‐cause readmission |
| Rollman et al.,[35] 2012, 20072009 | USA/PA, 4 urban teaching | 471, CHF, suspected depressed | Antidepressants users (excluded from PHQ‐2 group only) | Up to 12 | PHQ‐2; PHQ‐9 (5 in +PHQ‐2), by interviewer, 4 days | 66 (13) | 35 | 371 (79), 351 (74) | All‐cause mortality |
Prevalence and Recognition of Depressive Symptoms
The range of depression prevalence in hospitalized medical patients was 14% to 79%, with a median of 32% (interquartile range, 27%40%) (Table 1). In those studies that used a diagnostic interview, the prevalence tended to be lower for major depression, with a median of 17% (interquartile range, 16%22%) (Table 1). None of the included studies reported frequency of clinically recognized depression (ie, prior to screening for the study). Only 2 studies assessed the persistence of depression after discharge: 1 reported that depression persisted in 53% (by screening questionnaire) and 34% (by diagnostic interview) of patients at 30 days,[38] whereas the other reported 48% persistence at 90 days after discharge according to a combined screening method.[37]
Hospital Readmission
Overall, 8 studies provided readmission data. Among patients discharged from acute care medical wards (4 studies reporting on 5 cohorts), 395 of 2433 (16.2%) patients were readmitted within 30 days (Figure 1). Hospitalized patients with depressive symptoms were more likely to be readmitted within 30 days after discharge (20.4% vs 13.7%, RR: 1.73, 95% CI: 1.16‐2.58, P = 0.007, I2 = 55%) (Figure 1), compared to those without depression. Results were consistent for 90‐day readmissions (39.8% vs 31.0%, RR: 1.68, 95% CI: 1.13‐2.50, P = 0.01, I2 = 76%, n = 1543 patients) (see Supporting Figure 2 in the online version of this article) in 6 studies. One individual study examined readmission within 6 months after discharge, but was not poolable in this model, as it presented only hazard ratios and not raw data; however, it did report a 50% increased risk of readmission in medical inpatients aged 75 years (adjusted hazard ratio: 1.50, 95% CI: 1.03‐2.17, n = 401).[39]
Forest plot presents results of the meta‐analysis in which the size of each data marker indicates the weight assigned to individuals studies. Abbreviations: CI, confidence interval; IV, independent variable.
Mortality After Discharge
Overall, 11 studies provided all‐cause mortality data. Among medical patients discharged from acute care in 9 studies, 69 of 3397 (2.0%) patients died within 30 days (Figure 2). Medical patients discharged with depressive symptoms were more likely to die within 30 days (2.8% vs 1.5%, RR: 2.13, 95% CI: 1.31‐3.44, P = 0.002, I2 = 0%) (Figure 2) compared to those without depression. Similar results were found for 90‐day mortality (7.7% vs 4.1%, RR: 2.01, 95% CI: 1.47‐2.76, P < 0.001, I2 = 4%, n = 3784 patients) (see Supporting Figure 3 in the online version of this article) in 11 studies.
ED and PCP Visits
Four studies examined the use of ED or PCP services within 90 days of discharge, but 3 did not have extractable data for meta‐analysis. All showed increased utilization of health services for depressed compared to nondepressed patients after discharge.[22, 36, 37, 38] Depressed patients were more likely to visit the ED (adjusted incidence rate ratio: 1.73, 95% CI: 1.27‐2.36),[36] had significantly more medical encounters (eg, PCP, ED visits, hospital admissions, laboratory tests, and home care [mean 2.9 vs 2.6, P = 0.05])[38] and had a greater number of ED visits alone (27 vs 15 per 100 patients, P = 0.007)[22] within 30 days of hospital discharge compared to nondepressed patients. Similar results were found at 90 days.[36]
Sensitivity Analyses
All told, most studies reported a positive association between depression and adverse events, and this was true regardless of how much adjustment for potential confounding had been undertaken by the authors. Although all studies were qualitatively in the same direction, the magnitude of the association varied due to methodological and/or clinical heterogeneity. Sensitivity analysis revealed no overall difference in pooled risk ratios or heterogeneity between Mantel‐Haenszel fixed effects versus random effects models or with the addition of 0.5 to cells to permit inclusion of zero‐event data. There was no evidence of publication bias; funnel plots and Egger test results are available upon request. There were no statistically significant differences in the risk associated with depressive symptoms whether studies used Diagnostic and Statistical Manual of Mental Disorders (DSM)‐III or DSM‐IV criteria, whether the study samples were disease specific or unselected general medical cohorts, whether studies were of moderate or high quality, or regardless of the severity of depressive symptoms.
DISCUSSION
Summary of Evidence
We found that depression was common in medical inpatients (about one‐third of all patients) and persisted for at least 30 days in up to half of those patients after discharge. We found strong evidence of an association between depressive symptoms and poor short‐term prognosis after discharge from the hospital: a 73% increased risk of readmission and a 2‐fold risk of death within 30 days compared to patients without depressive symptoms with similar results at 90 days.
Our meta‐analysis complements a recent systematic review that found concomitant depression to be a risk factor for poor prognosis among inpatients and outpatients with acute coronary syndrome,[40] and a meta‐analysis that demonstrated an increased risk of 2‐year mortality among patients with depression after myocardial infarction.[41] To our knowledge, our study is the first to quantify the short‐term postdischarge risks across a diverse group of medical inpatients.
The potential mechanisms underlying the observed relationship between depression and adverse patient outcomes after discharge are likely multiple. We believe there are 2 main possibilities. First, the increased risk associated with depression might be due to residual confounding, even though many of these studies did adjust for extensive lists of comorbidities,[22, 24, 26, 27, 29, 30, 33, 35, 36, 39] including functional status[39] and prior health services utilization.[22, 34, 36] This could occur if other risk factors were not sufficiently adjusted for, such as unrecognized comorbidities or concomitant disability, which are often present among chronically ill patients,[42] or if depression were a marker of psychosocial risk factors such as anxiety,[43] stress or poor resiliency,[44] or low social support,[45] though a few adjusted for psychosocial factors such as social support[26] or anxiety.[35] Confounding could also occur if symptoms of acute illness inflate reports of somatic symptoms of depression on self‐report questionnaires. Recent studies on the BDI, found that scores were higher in postmyocardial infarction patients when compared to outpatient controls,[46] but with no differences between those groups in scores for the BDI‐II,[47] a version with fewer somatic symptom questions.
Second, depression may cause adverse outcomes through indirect or direct pathways. Indirect causation could occur if depression hindered self‐care behaviors such as medication adherence.[42] Depression could also act directly through pathophysiological changes. Some studies have suggested that depression is associated with metabolic abnormalities, including alterations in glucose transport[42, 48] and increased vulnerability to obesity, type 2 diabetes mellitus, and/or diabetic complications, common conditions among hospitalized patients that also adversely affect postdischarge outcomes.[40, 48]
Strengths and Limitations
This review has multiple strengths. We cast a broad search and included studies that examined a wide range of medical patient subgroups, thus increasing the generalizability of our findings. We identified a general scarcity of studies on this topic and obtained additional unpublished data for 10 of the 18 relevant studies, and our response rate of 34% is compatible with the 37% response rate reported for Cochrane reviews when seeking additional data from authors.[49] Whether examined qualitatively (vote counting of the number of studies that showed an association) or quantitatively (via formal meta‐analysis), it seems apparent that there is a clinically important association between depression and postdischarge adverse events, but given the number, quality, and heterogeneity of the studies we examined, there may be some ongoing dispute about exactly how strong this association is and the degree of bias contributed by a couple of large studies of the topic.
There are limitations to our review. First, as we did not have individual‐level patient data, we could not use metaregression to explore sources of heterogeneity (clinical or methodological) or adjust for confounding, and this likely contributes to observed differences between individual estimates. For instance, the included studies had heterogeneous screening measures and cutoffs; thus, all cases of depression in these studies might not be equivalent. Some of the included studies assessed depression early during admission where psychological distress may be greatest; others assessed symptoms closer to discharge. Most studies included patients with specific conditions like heart failure or chronic obstructive pulmonary disease rather than a wide spectrum of medical inpatients. Moreover, few studies adjusted for psychosocial risk factors such as social support, anxiety, and functional status, and only 2 studies assessed the persistence of depressive symptoms after discharge. Second, we did not explore quantitative measures of between‐study variation (eg, I2), because experts question its utility given the expected heterogeneity in meta‐analyses of observational studies.[50] Third, although the included studies were deemed to be of at least moderate quality, they could be at risk for sources of bias that may not be sufficiently appraised by the current version of the Newscastle‐Ottawa scale for observational studies. Finally, we excluded grey literature (eg, conference proceedings or technical reports) that could potentially exclude null findings, although we did contact authors in this field to identify additional unpublished data relevant to this topic.
CONCLUSIONS
We have confirmed that depressive symptoms are common in hospitalized medical patients, frequently persist after discharge, and may predict greater risk of readmission or death after discharge. Thus, depressive symptoms are an additional marker that clinicians can use to help identify patients in acute care medical settings who may be at increased risk for suboptimal transition back to the community and who may require additional resources after discharge. However, future research is required to evaluate whether treatment of individuals who screen positive for depressive symptoms can reduce 30‐day readmission rates, and we are aware of at least 1 relevant ongoing trial (
Acknowledgements
The authors thank the following individuals: Dale Storie, MLIS, Saskatchewan Information and Library Services Consortium, Regina, Saskatchewan, Canada, for assistance in the literature search; James A. Hanley, PhD, Department of Epidemiology and Biostatistics, Faculty of Medicine, McGill University, Montreal, Quebec, Canada, for guidance in data recovery methods; Nancy Frasure‐Smith, PhD, Department of Psychiatry, McGill University, Department of Psychiatry and Research Centre Hospital Centre, University of Montreal, and Montreal Heart Institute Research Centre, Montreal, Quebec, Canada; Andriana I. Papaioannou, MD, 2nd Respiratory Medicine Department, University of Athens Medical School, Athens, Greece; Konstantinos Kostikas, MD, 2nd Respiratory Medicine Department, University of Athens Medical School, Athens, Greece; and Pere Almagro, MD, Servicio de Medicina Interna, Hospital Universitario Mutua de Terrassa, Terrassa, Barcelona, Spain; as well as Philip G. Jones, MS, Saint Luke's Mid America Heart Institute, Kansas City, Missouri; for their retrieval and contribution of unpublished data.
Disclosures
Ms. Pederson affirms that the manuscript is an honest, accurate, and transparent account of the study being reported with no important omissions. All authors had full access to all of the data (including statistical reports and tables) in the study and can take responsibility for the integrity of the data and the accuracy of the data analysis. Design and conduct of the study: Ms. Pederson, Drs. Majumdar and McAlister. Data acquisition: Ms. Pederson, Ms. Warkentin. Analysis and interpretation of the data and drafting of the manuscript: Ms Pederson, Drs. Majumdar and McAlister. Review of the manuscript: all authors. Study supervision: Drs. Majumdar and McAlister. None of the contributors received compensation for their efforts. Salary support for Ms. Pederson was provided by a CRIO grant from Alberta InnovatesHealth Solutions. Drs. McAlister and Majumdar are supported by salary awards from Alberta Innovates‐Health Solutions. Dr. McAlister holds the University of Alberta/Capital Health Chair in Cardiology Outcomes Research. Dr. Majumda holds the University of Alberta Endowed Chair in Patient Health Management. The funding sources had no role in the design or conduct of the study; management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. This work is that of the authors independent of funders. The authors report no conflicts of interest.
Between 10% and 40% of patients are readmitted after being discharged from the hospital,[1, 2] and as many as another 25% return to the emergency department (ED) within 30 days.[3] This creates a substantial burden on the healthcare system.[2] Various interventions have been tried to improve the quality of discharge transitions and reduce readmission rates, but results thus far have been inconsistent and generally disappointing.[4, 5, 6] Targeted delivery of interventions to those at highest risk might improve the effectiveness of these efforts and reduce costs. However, current readmission risk assessment models are only moderately predictive, suggesting the presence of unrecognized risk factors.[7, 8]
Active depression might represent a potentially modifiable independent predictor of adverse short‐term hospital outcomes that is currently underutilized. Depression occurs in 5% to 58% of hospitalized adults, depending on how cases are defined.[9, 10] Depression is often under‐recognized and undertreated in acute care clinical settings,[11] and relatively few readmission prediction models incorporate mental health related symptoms.[12]
Although several reviews have examined methods of screening for depression in hospitalized patients[9] or the effectiveness of screening in primary care,[13, 14] to our knowledge no systematic review has examined the impact of depression on short‐term prognosis after discharge from acute care. Therefore, the purpose of this systematic review was to summarize all studies that evaluated whether hospitalized medical patients with depressive symptoms are at higher risk of 30‐day all‐cause readmission or all‐cause mortality after being discharged from the hospital.
METHODS
This study followed an a priori protocol developed according to PRISMA (Preferred Reporting Items for Systematic Reviews and Meta‐Analyses) criteria.[15]
Data Sources and Search Methods
We searched the Cumulative Index to Nursing and Allied Health Literature, Ovid MEDLINE, Ovid Embase, and PsycINFO from inception to January 9, 2015, and the last 5 years of PubMed for full publications with any of the following Medical Subject Headings: depressive disorder, depression, patient readmission, interviews, psychological, inpatients, with restrictions for peer‐reviewed publication, humans, adults aged 18 years, and the English language. Search strategies were developed with a librarian (available upon request). We manually searched reference lists of all included studies and relevant review articles and contacted content experts to identify additional publications.
Eligibility Criteria and Selection of Studies
Two authors (J.L.P. and L.M.W.) independently screened full texts of all relevant articles for inclusion. Disagreements were resolved by consensus or a third reviewer (S.R.M.). We considered any original research that compared readmission or mortality after discharge for hospitalized medical patients (ie, general patients or subgroups thereof) with versus without depression identified by any validated depression measure,[16] including any study design that incorporated at least 30‐day follow‐up postdischarge. We excluded studies that examined patients hospitalized in nonacute care settings or on surgical, psychiatric, obstetric, or intensive care services. We calculated Cohen's coefficient to evaluate inter‐rater agreement on study selection.
Data Extraction
Data were abstracted by 2 authors (J.L.P. and L.M.W.). Disagreements were resolved by consensus or a third reviewer (S.R.M.). We contacted authors of all included studies to obtain missing data. If unavailable, crude data were estimated from published survival curves employing validated techniques in R (version 3.1.2; R Foundation for Statistical Computing, Vienna, Austria) and Digitizeit (
Data Synthesis and Statistical Analysis
Where possible, we calculated the pooled risk ratio (RR) with 95% confidence interval (95% CI) using a random effects models in Review Manager (RevMan) 5.3 (The Nordic Cochrane Centre, Copenhagen, Denmark). The random effects approach that we employed assumes heterogeneity (ie, underlying parameters vary between individual studies) and is distributed around a mean or population average effect, and results in more conservative (wider) confidence intervals, wherein larger cohorts (or studies with smaller standard errors) are given more weight. Heterogeneity was assessed using the I2 statistic, with values of <25%, 25% to 50%, and >50% representing low, moderate, and high heterogeneity.[19] As per the guidance of Higgins et al., we did not a priori define any degree of heterogeneity that would preclude pooling of the data; the expectation would be that heterogeneity would be substantially higher pooling observational studies rather than randomized trials.[19] Statistical significance was considered a 2‐sided P value 0.05.
Quality Assessment and Risk of Bias
We assessed study quality using the 9‐item Newcastle‐Ottawa scale with 0 to 3, 4 to 6, and 7 to 9 stars considered low, moderate, and high quality, respectively, and criteria for external and internal validity, including group selection and comparability, outcome assessment, and adequacy of follow‐up.[20] Adjusted estimates published in individual reports (or obtained directly from authors) were compared wherever possible with unadjusted estimates to assess the degree of confounding. We generated funnel plots in RevMan 5.3 and conducted Egger tests using Stata 13 (StataCorp LP, College Station, TX) to assess for publication bias.[21]
RESULTS
Study Selection
After removing duplicate publications, we identified 4066 reports and reviewed 133 reports in full text (see Supporting Figure 1 in the online version of this article). Despite our broad study inclusion criteria, we found only 35 longitudinal studies addressing this question. All 35 authors were contacted for additional outcomes data and other missing information (response rate of 34%). We had to exclude 17 studies as they did not provide 30 or 90‐day post‐discharge outcomes. Only 4 studies had published crude data for outcomes within 90 days,[22, 23, 24, 25] but after contact with authors, we received unpublished data for a further 7 studies[26, 27, 28, 29, 30, 31, 32] (including individual level data for 2 cohorts).[31, 32] We were able to estimate crude data from Kaplan‐Meier curves for another 3 studies.[33, 34, 35] Another 4 studies did not collect the outcomes we were interested in individually. These studies were included in this systematic review but are not poolable in our models: 3 authors could only provide composite endpoint data,[36, 37, 38] and 1 author provided unadjusted hazard ratios.[39] Inter‐reviewer agreement for inclusion was 80% (Cohen's = 0.60).
Characteristics of Included Studies
The 18 studies ranged in size from 58 to 1418 patients; 13 were cohort studies and 5 included secondary data from randomized control trials.[22, 27, 30, 34, 36] All studies ascertained depressive status by screening during index medical admission with either diagnostic interview or self‐report questionnaires, although a variety of scales and definitions for depression were used (Beck Depression Inventory [BDI] in 6 studies, Geriatric Depression Scale in 5 studies, Patient Health Questionnaire in another 4 studies, Medical Outcomes Study‐Depression Questionnaire in 1 study, and Center for Epidemiologic Studies Depression Scale in another study) (Table 1). Screening interviews were conducted mostly by research assistants or nurses (68%) or self‐administered (21%). Most studies examined specific medical patient subgroups (10 cardiac, 3 pulmonary, and 2 elderly). Major exclusion criteria reported were terminal illness (4 studies), unstable condition (6 studies), severe cognitive impairment (5 studies), and suicidal ideation or known depression (4 studies); 1 study enrolled patients with suspected depression (Table 1). Patient cohorts were on average older (range, 5082 years) (Table 1). Attrition rates for readmission and mortality data were low (average <1% among entire sample of studies). All studies scored at least 5 on the Newcastle‐Ottawa scale and were thus considered of at least moderate quality (see Supporting Table 1 in the online version of this article).
| Author, Date of Publication, Enrollment Period | Setting Country/Region, No. of Hospitals | No. of Inpatients, Clinical Features | Major Exclusion Criteria | Follow‐up, mo | Depression Measure (Cutoff) and Screening Method | Mean Age (SD), y | % Female | Positive Screen, No. (%) | Primary Outcome, Secondary Outcomes |
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| Studies that use a scale based on DSM‐III criteria or a diagnostic interview according to DSM‐III criteria | |||||||||
| Frasure‐Smith et al.,[26] 1993, 19911992* | Canada/Quebec, 1 urban teaching | 218, AMI | Terminal noncardiac illness, unstable, not cognitive | 6 | BDI (10); mod DIS by interviewer, after transfer to medicine | 60 (range, 2488) | 22 | 68 (31), 35 (16) | All‐cause mortality |
| Frasure‐Smith et al.,[27] 1999, 19911992,* 19911994 | Canada/Quebec, 1 urban teaching, 10 urban area | 218; 78, AMI | Terminal noncardiac illness, unstable, not cognitive | 12 | BDI (10) by interviewer, after transfer to medicine | 60 (11) | 32 | 290 (32) | Cardiac mortality |
| Freedland et al.,[25] 1991, 1990 | USA/MO, 1 urban teaching | 58, CHF 75 years | Dementia, medically unstable | 3 | Mod DIS by psychiatric residents and interviewer | 78 (6) | 57 | 10 (17) | All‐cause readmission, all‐cause mortality |
| Fulop et al.,[38] 2003, 2002 | USA/NY, 1 urban teaching | 203, CHF 65 years | 1, 6 | GDS (10); SCID‐NP by interviewer, at discharge | 77 (8) | 53 | 73 (36), 44 (22) | Depression, composite PCP, ED, care visits, and readmission | |
| Lesprance et al.,[28] 2000, 19941996 | Canada/Quebec, 1 urban teaching | 430, unstable angina | Terminal noncardiac illness, not cognitive, recent CABG | 12 | BDI (10); mod DIS by interviewer, 5 days after admission | 62 (11) | 29 | 178 (41), 120 (28) | Cardiac death and MI, any death, angina readmission |
| Rumsfeld et al.,[30] 2005, 19992001 | CA, USA, UK, multiple | 634, AMI with CHF | Valvular or congenital heart failure | Up to 32 | MOS‐D (0.06) by interviewer, before discharge | 65 (11) | 28 | 143 (23) | All‐cause death, CVD death and readmission |
| Song et al.,[33] 2009, 2005 | South Korea, 2 urban teaching | 165, HF | If minor criteria for HF attributable to other medical condition | 6 | BDI (10) self‐administer or interviewer, 34 days of admin | 62 (13) | 49 | 131 (79) | HF readmission and all‐cause mortality, HF readmit |
| Papaioannou et al.,[29] 2013, 20092010 | Greece/Athens, 1 urban | 230, AECOPD | Other respiratory illness, known depressed | Monthly up to 12 | BDI‐I (19) self‐administer, first day | 71 (9) | 12 | 91 (40) | All‐cause mortality, AECOPD readmission |
| Studies that use a scale based on or validated against DSM‐IV criteria or a diagnostic interview according to DSM‐IV criteria | |||||||||
| Almagro et al.,[31] 2002, 19961997 | Spain, 1 urban teaching | 130, AECOPD | Other pulmonary disease | July 1999 | GDS‐SF (6) by interviewer, day before discharge | 72 (9) | 8 | 43 (33) | All‐cause mortality |
| Almagro et al.,[32] 2012, 20032004 | Spain, 1 urban teaching | 134, AECOPD | Other pulmonary disease | 1, 36 | GDS‐SF (6) by interviewer | 72 (10) | 5 | 55 (41) | All‐cause mortality, lung function, frailty |
| Bla et al.,[39] 2001, 2000 | Switzerland, 1 urban teaching | 401, medical 75 years | Stay <24 hours, elective/facility transfer, unstable, not cognitive | 6 | GDS‐SF (6) by interviewer, within 2 days of admission | 82 (7599) | 61 | 90 (22) | All‐cause readmission, all‐cause mortality |
| Cancino et al.,[22] 2014, 20062007,* 20082009 | USA/MA, 1 urban tertiary | 680; 738, medical | Nursing home or hospital transfer, isolated, suicidal | 1 | PHQ‐9 (5 or severity) by interviewer, on admin | 50 (14) | 51 | 561 (40) | All‐cause readmission, ED visits, PCP visits |
| Mitchell et al.,[36] 2010, 20062007* | USA/MA, 1 urban tertiary | 738, medical | Nursing home or hospital transfer, isolated, suicidal | 1, 2, 3 | PHQ‐9 (5) by interviewer, on admin | 50 (15) | 50 | 238 (32) | ED visits and all‐cause readmission |
| Covinsky et al.,[34] 1999, 19901992 | USA/OH, 1 urban teaching | 573, medical | ICU, oncology, telemetry, nursing home admissions | 36 | GDS‐SF (6) by interviewer, within 2 days of admission | 80 | 68 | 197 (34) | All‐cause mortality |
| Jiang et al.,[23] 2001, 19971998 | USA/NC, 1 urban teaching | 357 (331 DIS only), CHF | Suicidal, planned surgery, pregnant | 3, 12 | BDI (10) self‐admin; mod DIS (+BDI only) by interviewer | 63 (13) | 33 | 126 (35), 46 (14) | All‐cause mortality, all‐cause readmission |
| Kartha et al.,[24] 2007, 20022004 | USA/MA, 1 urban safety net | 144, medical recently hospitalized | Planned readmission, unable to keep PCP appointments | 3 | PHQ‐9 (algorithm) by interviewer | 55 (16) | 56 | 39 (27) | All‐cause readmission |
| Koenig and Kuchbhatla,[37] 1999, 1997 | USA/NC, 1 urban teaching | 331, medical 60 years | Stay <3 or >7 days, ICU/CCU, severe illness, nursing home transfers | 3, 6, 9, 12 | CES‐D (16) or HAM‐D (11) or DIS by psychiatrist, on or after third day | 70 (7) | 51 | 160 (48) | Depression, composite physical disability, health visits, and all‐cause readmission |
| Rollman et al.,[35] 2012, 20072009 | USA/PA, 4 urban teaching | 471, CHF, suspected depressed | Antidepressants users (excluded from PHQ‐2 group only) | Up to 12 | PHQ‐2; PHQ‐9 (5 in +PHQ‐2), by interviewer, 4 days | 66 (13) | 35 | 371 (79), 351 (74) | All‐cause mortality |
Prevalence and Recognition of Depressive Symptoms
The range of depression prevalence in hospitalized medical patients was 14% to 79%, with a median of 32% (interquartile range, 27%40%) (Table 1). In those studies that used a diagnostic interview, the prevalence tended to be lower for major depression, with a median of 17% (interquartile range, 16%22%) (Table 1). None of the included studies reported frequency of clinically recognized depression (ie, prior to screening for the study). Only 2 studies assessed the persistence of depression after discharge: 1 reported that depression persisted in 53% (by screening questionnaire) and 34% (by diagnostic interview) of patients at 30 days,[38] whereas the other reported 48% persistence at 90 days after discharge according to a combined screening method.[37]
Hospital Readmission
Overall, 8 studies provided readmission data. Among patients discharged from acute care medical wards (4 studies reporting on 5 cohorts), 395 of 2433 (16.2%) patients were readmitted within 30 days (Figure 1). Hospitalized patients with depressive symptoms were more likely to be readmitted within 30 days after discharge (20.4% vs 13.7%, RR: 1.73, 95% CI: 1.16‐2.58, P = 0.007, I2 = 55%) (Figure 1), compared to those without depression. Results were consistent for 90‐day readmissions (39.8% vs 31.0%, RR: 1.68, 95% CI: 1.13‐2.50, P = 0.01, I2 = 76%, n = 1543 patients) (see Supporting Figure 2 in the online version of this article) in 6 studies. One individual study examined readmission within 6 months after discharge, but was not poolable in this model, as it presented only hazard ratios and not raw data; however, it did report a 50% increased risk of readmission in medical inpatients aged 75 years (adjusted hazard ratio: 1.50, 95% CI: 1.03‐2.17, n = 401).[39]
Forest plot presents results of the meta‐analysis in which the size of each data marker indicates the weight assigned to individuals studies. Abbreviations: CI, confidence interval; IV, independent variable.
Mortality After Discharge
Overall, 11 studies provided all‐cause mortality data. Among medical patients discharged from acute care in 9 studies, 69 of 3397 (2.0%) patients died within 30 days (Figure 2). Medical patients discharged with depressive symptoms were more likely to die within 30 days (2.8% vs 1.5%, RR: 2.13, 95% CI: 1.31‐3.44, P = 0.002, I2 = 0%) (Figure 2) compared to those without depression. Similar results were found for 90‐day mortality (7.7% vs 4.1%, RR: 2.01, 95% CI: 1.47‐2.76, P < 0.001, I2 = 4%, n = 3784 patients) (see Supporting Figure 3 in the online version of this article) in 11 studies.
ED and PCP Visits
Four studies examined the use of ED or PCP services within 90 days of discharge, but 3 did not have extractable data for meta‐analysis. All showed increased utilization of health services for depressed compared to nondepressed patients after discharge.[22, 36, 37, 38] Depressed patients were more likely to visit the ED (adjusted incidence rate ratio: 1.73, 95% CI: 1.27‐2.36),[36] had significantly more medical encounters (eg, PCP, ED visits, hospital admissions, laboratory tests, and home care [mean 2.9 vs 2.6, P = 0.05])[38] and had a greater number of ED visits alone (27 vs 15 per 100 patients, P = 0.007)[22] within 30 days of hospital discharge compared to nondepressed patients. Similar results were found at 90 days.[36]
Sensitivity Analyses
All told, most studies reported a positive association between depression and adverse events, and this was true regardless of how much adjustment for potential confounding had been undertaken by the authors. Although all studies were qualitatively in the same direction, the magnitude of the association varied due to methodological and/or clinical heterogeneity. Sensitivity analysis revealed no overall difference in pooled risk ratios or heterogeneity between Mantel‐Haenszel fixed effects versus random effects models or with the addition of 0.5 to cells to permit inclusion of zero‐event data. There was no evidence of publication bias; funnel plots and Egger test results are available upon request. There were no statistically significant differences in the risk associated with depressive symptoms whether studies used Diagnostic and Statistical Manual of Mental Disorders (DSM)‐III or DSM‐IV criteria, whether the study samples were disease specific or unselected general medical cohorts, whether studies were of moderate or high quality, or regardless of the severity of depressive symptoms.
DISCUSSION
Summary of Evidence
We found that depression was common in medical inpatients (about one‐third of all patients) and persisted for at least 30 days in up to half of those patients after discharge. We found strong evidence of an association between depressive symptoms and poor short‐term prognosis after discharge from the hospital: a 73% increased risk of readmission and a 2‐fold risk of death within 30 days compared to patients without depressive symptoms with similar results at 90 days.
Our meta‐analysis complements a recent systematic review that found concomitant depression to be a risk factor for poor prognosis among inpatients and outpatients with acute coronary syndrome,[40] and a meta‐analysis that demonstrated an increased risk of 2‐year mortality among patients with depression after myocardial infarction.[41] To our knowledge, our study is the first to quantify the short‐term postdischarge risks across a diverse group of medical inpatients.
The potential mechanisms underlying the observed relationship between depression and adverse patient outcomes after discharge are likely multiple. We believe there are 2 main possibilities. First, the increased risk associated with depression might be due to residual confounding, even though many of these studies did adjust for extensive lists of comorbidities,[22, 24, 26, 27, 29, 30, 33, 35, 36, 39] including functional status[39] and prior health services utilization.[22, 34, 36] This could occur if other risk factors were not sufficiently adjusted for, such as unrecognized comorbidities or concomitant disability, which are often present among chronically ill patients,[42] or if depression were a marker of psychosocial risk factors such as anxiety,[43] stress or poor resiliency,[44] or low social support,[45] though a few adjusted for psychosocial factors such as social support[26] or anxiety.[35] Confounding could also occur if symptoms of acute illness inflate reports of somatic symptoms of depression on self‐report questionnaires. Recent studies on the BDI, found that scores were higher in postmyocardial infarction patients when compared to outpatient controls,[46] but with no differences between those groups in scores for the BDI‐II,[47] a version with fewer somatic symptom questions.
Second, depression may cause adverse outcomes through indirect or direct pathways. Indirect causation could occur if depression hindered self‐care behaviors such as medication adherence.[42] Depression could also act directly through pathophysiological changes. Some studies have suggested that depression is associated with metabolic abnormalities, including alterations in glucose transport[42, 48] and increased vulnerability to obesity, type 2 diabetes mellitus, and/or diabetic complications, common conditions among hospitalized patients that also adversely affect postdischarge outcomes.[40, 48]
Strengths and Limitations
This review has multiple strengths. We cast a broad search and included studies that examined a wide range of medical patient subgroups, thus increasing the generalizability of our findings. We identified a general scarcity of studies on this topic and obtained additional unpublished data for 10 of the 18 relevant studies, and our response rate of 34% is compatible with the 37% response rate reported for Cochrane reviews when seeking additional data from authors.[49] Whether examined qualitatively (vote counting of the number of studies that showed an association) or quantitatively (via formal meta‐analysis), it seems apparent that there is a clinically important association between depression and postdischarge adverse events, but given the number, quality, and heterogeneity of the studies we examined, there may be some ongoing dispute about exactly how strong this association is and the degree of bias contributed by a couple of large studies of the topic.
There are limitations to our review. First, as we did not have individual‐level patient data, we could not use metaregression to explore sources of heterogeneity (clinical or methodological) or adjust for confounding, and this likely contributes to observed differences between individual estimates. For instance, the included studies had heterogeneous screening measures and cutoffs; thus, all cases of depression in these studies might not be equivalent. Some of the included studies assessed depression early during admission where psychological distress may be greatest; others assessed symptoms closer to discharge. Most studies included patients with specific conditions like heart failure or chronic obstructive pulmonary disease rather than a wide spectrum of medical inpatients. Moreover, few studies adjusted for psychosocial risk factors such as social support, anxiety, and functional status, and only 2 studies assessed the persistence of depressive symptoms after discharge. Second, we did not explore quantitative measures of between‐study variation (eg, I2), because experts question its utility given the expected heterogeneity in meta‐analyses of observational studies.[50] Third, although the included studies were deemed to be of at least moderate quality, they could be at risk for sources of bias that may not be sufficiently appraised by the current version of the Newscastle‐Ottawa scale for observational studies. Finally, we excluded grey literature (eg, conference proceedings or technical reports) that could potentially exclude null findings, although we did contact authors in this field to identify additional unpublished data relevant to this topic.
CONCLUSIONS
We have confirmed that depressive symptoms are common in hospitalized medical patients, frequently persist after discharge, and may predict greater risk of readmission or death after discharge. Thus, depressive symptoms are an additional marker that clinicians can use to help identify patients in acute care medical settings who may be at increased risk for suboptimal transition back to the community and who may require additional resources after discharge. However, future research is required to evaluate whether treatment of individuals who screen positive for depressive symptoms can reduce 30‐day readmission rates, and we are aware of at least 1 relevant ongoing trial (
Acknowledgements
The authors thank the following individuals: Dale Storie, MLIS, Saskatchewan Information and Library Services Consortium, Regina, Saskatchewan, Canada, for assistance in the literature search; James A. Hanley, PhD, Department of Epidemiology and Biostatistics, Faculty of Medicine, McGill University, Montreal, Quebec, Canada, for guidance in data recovery methods; Nancy Frasure‐Smith, PhD, Department of Psychiatry, McGill University, Department of Psychiatry and Research Centre Hospital Centre, University of Montreal, and Montreal Heart Institute Research Centre, Montreal, Quebec, Canada; Andriana I. Papaioannou, MD, 2nd Respiratory Medicine Department, University of Athens Medical School, Athens, Greece; Konstantinos Kostikas, MD, 2nd Respiratory Medicine Department, University of Athens Medical School, Athens, Greece; and Pere Almagro, MD, Servicio de Medicina Interna, Hospital Universitario Mutua de Terrassa, Terrassa, Barcelona, Spain; as well as Philip G. Jones, MS, Saint Luke's Mid America Heart Institute, Kansas City, Missouri; for their retrieval and contribution of unpublished data.
Disclosures
Ms. Pederson affirms that the manuscript is an honest, accurate, and transparent account of the study being reported with no important omissions. All authors had full access to all of the data (including statistical reports and tables) in the study and can take responsibility for the integrity of the data and the accuracy of the data analysis. Design and conduct of the study: Ms. Pederson, Drs. Majumdar and McAlister. Data acquisition: Ms. Pederson, Ms. Warkentin. Analysis and interpretation of the data and drafting of the manuscript: Ms Pederson, Drs. Majumdar and McAlister. Review of the manuscript: all authors. Study supervision: Drs. Majumdar and McAlister. None of the contributors received compensation for their efforts. Salary support for Ms. Pederson was provided by a CRIO grant from Alberta InnovatesHealth Solutions. Drs. McAlister and Majumdar are supported by salary awards from Alberta Innovates‐Health Solutions. Dr. McAlister holds the University of Alberta/Capital Health Chair in Cardiology Outcomes Research. Dr. Majumda holds the University of Alberta Endowed Chair in Patient Health Management. The funding sources had no role in the design or conduct of the study; management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. This work is that of the authors independent of funders. The authors report no conflicts of interest.
- , , . Rehospitalizations among patients in the Medicare fee‐for‐service program. N Engl J Med. 2009;360(14):1418–1428.
- , , , , . Proportion of hospital readmissions deemed avoidable: a systematic review. CMAJ. 2011;183(7):E391–E402.
- , , , . Mental health conditions are associated with increased health care utilization among urban family medicine patients. J Am Board Fam Med. 2008;21(5):398–407.
- , , , , . Interventions to reduce 30‐day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520–528.
- . The elusive quest for quality and cost savings in the Medicare program. JAMA. 2009;301(6):668–670.
- , , , . Effects of care coordination on hospitalization, quality of care, and health care expenditures among Medicare beneficiaries—15 randomized trials. JAMA. 2009;301(6):603–618.
- , , , et al. Unplanned readmissions after hospital discharge among patients identified as being at high risk for readmission using a validated predictive algorithm. Open Med. 2011;5(2):e104–111.
- , , , et al. Risk prediction models for hospital readmission: a systematic review. JAMA. 2011;306(15):1688–1698.
- , , . Depression in older people in the general hospital: a systematic review of screening instruments. Age Ageing. 2012;41(2):148–154.
- , , , et al. Prevalence, correlates and recognition of depression among inpatients of general hospitals in Wuhan, China. Gen Hosp Psychiatry. 2010;32(3):268–275.
- , , , , , . Recognition of depression by non‐psychiatric physicians—a systematic literature review and meta‐analysis. J Gen Intern Med. 2008;23(1):25–36.
- , , , , , . Predicting the risk of unplanned readmission or death within 30 days of discharge after a heart failure hospitalization. Am Heart J. 2012;164(3):365–372.
- , , , et al. Does evidence support the American Heart Association's recommendation to screen patients for depression in cardiovascular care? An updated systematic review. PLoS One. 2013;8(1):e52654.
- , , , et al. Screening for depression: a systematic review and meta‐analysis. CMAJ Open. 2013;1(4):E159–E167.
- , , , et al. The PRISMA statement for reporting systematic reviews and meta‐analyses of studies that evaluate health care interventions: explanation and elaboration. Ann Intern Med. 2009;151(4):W65–94.
- , , , et al. Screening for depression in adults: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med. 2002;136(10):765–776.
- , , . Recovering the raw data behind a non‐parametric survival curve. Syst Rev. 2014;3:151.
- , , , . Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan‐Meier survival curves. BMC Med Res Methodol. 2012;12(1):9.
- . Commentary: heterogeneity in meta‐analysis should be expected and appropriately quantified. Int J Epidemiol. 2008;37(5):1158–1160.
- , , , et al. The Newcastle‐Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta‐analyses. Available at: http://www.ohri.ca/programs/clinical_epidemiology/oxford.htm. Accessed September 1, 2015.
- , , . The funnel plot. In: Rothstein HR, Sutton AJ, Borenstein M, eds. Publication Bias in Meta‐analysis: Prevention, Assessment and Adjustments. New York, NY: John Wiley 2006:73–98.
- , , , , , . Dose‐response relationship between depressive symptoms and hospital readmission. J Hosp Med. 2014;9(6):358–364.
- , , , et al. Relationship of depression to increased risk of mortality and rehospitalization in patients with congestive heart failure. Arch Intern Med. 2001;161(15):1849–1856.
- , , , et al. Depression is a risk factor for rehospitalization in medical inpatients. Prim Care Companion J Clin Psychiatry. 2007;9(4):256–262.
- , , , et al. Depression in elderly patients with congestive heart failure. J Geriatr Psychiatry. 1991;24(1):59–71.
- , , . Depression following myocardial infarction: impact on 6‐month survival. JAMA. 1993;270(15):1819–1825.
- , , , , . Gender, depression, and one‐year prognosis after myocardial infarction. Psychosom Med. 1999;61(1):26–37.
- , , , . Depression and 1‐year prognosis in unstable angina. Arch Intern Med. 2000;160(9):1354–1360.
- , , , et al. The impact of depressive symptoms on recovery and outcome of hospitalised COPD exacerbations. Eur Respir J. 2013;41(4):815–823.
- , , , et al. Depression predicts mortality and hospitalization in patients with myocardial infarction complicated by heart failure. Am Heart J. 2005;150(5):961–967.
- , , , et al. Mortality after hospitalization for COPD. Chest. 2002;121(5):1441–1448.
- , , , et al. Pseudomonas aeruginosa and mortality after hospital admission for chronic obstructive pulmonary disease. Respiration. 2012;84(1):36–43.
- , , . Depressive symptoms increase risk of rehospitalisation in heart failure patients with preserved systolic function. J Clin Nurs. 2009;18(13):1871–1877.
- , , . Depressive symptoms and 3 year mortality in older hospitalized medical patients. Ann Intern Med. 1999;130(7):563–569.
- , , , et al. A positive 2‐item patient health questionnaire depression screen among hospitalized heart failure patients is associated with elevated 12‐month mortality. J Card Fail. 2012;18(3):238–245.
- , , , et al. Post‐discharge hospital utilization among adult medical inpatients with depressive symptoms. J Hosp Med. 2010;5(7):378–384.
- , . Use of health services by medically ill depressed elderly patients after hospital discharge. Am J Geriatr Psychiatry. 1999;7(1):48–56.
- , , . Congestive heart failure and depression in older adults: clinical course and health services use 6 months after hospitalization. Psychosomatics. 2003;44(5):367–373.
- , , , . Depressive symptoms as a predictor of 6‐month outcomes and services utilization in elderly medical inpatients. Arch Intern Med. 2001;161(21):2609–2615.
- , , , et al. Depression as a risk factor for poor prognosis among patients with acute coronary syndrome: systematic review and recommendations: a scientific statement from the American Heart Association. Circulation. 2014;129(12):1350–1369.
- , , , , , . Prognostic association of depression following myocardial infarction with mortality and cardiovascular events: a meta‐analysis of 25 years of research. Gen Hosp Psychiatry. 2011;33(3):203–216.
- , , , , . Depression and cardiac disease: epidemiology, mechanisms, and diagnosis. Cardiovasc Psychiatry Neurol. 2013;2013:695925.
- , , , et al. Prognostic value of depression, anxiety, and anger in hospitalized cardiovascular disease patients for predicting adverse cardiac outcomes. Am J Cardiol. 2013;111(10):1432–1436.
- , , . The psychobiology of depression and resilience to stress: implications for prevention and treatment. Annu Rev Clin Psychol. 2005;1:255–291.
- , , , et al. Impact of social factors on risk of readmission or mortality in pneumonia and heart failure: systematic review. J Gen Intern Med. 2013;28(2):269–282.
- , , , et al. The influence of somatic symptoms on beck depression inventory scores in hospitalized postmyocardial infarction patients. Can J Psychiatry. 2012;57(12):752–758.
- , , , et al. Somatic symptom overlap in beck depression inventory‐II scores following myocardial infarction. Br J Psychiatry. 2010;197(1):61–66.
- , , , . Relationship of depression to diabetes types 1 and 2: epidemiology, biology, and treatment. Biol Psychiatry. 2003;54(3):317–329.
- , , . Searching for unpublished data for Cochrane reviews: cross sectional study. BMJ. 2013;346:f2231.
- . Comment on: heterogeneity in meta‐analysis should be expected and appropriately quantified. Int J Epidemiol. 2010;39(3):932; author reply 933.
- , , . Rehospitalizations among patients in the Medicare fee‐for‐service program. N Engl J Med. 2009;360(14):1418–1428.
- , , , , . Proportion of hospital readmissions deemed avoidable: a systematic review. CMAJ. 2011;183(7):E391–E402.
- , , , . Mental health conditions are associated with increased health care utilization among urban family medicine patients. J Am Board Fam Med. 2008;21(5):398–407.
- , , , , . Interventions to reduce 30‐day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520–528.
- . The elusive quest for quality and cost savings in the Medicare program. JAMA. 2009;301(6):668–670.
- , , , . Effects of care coordination on hospitalization, quality of care, and health care expenditures among Medicare beneficiaries—15 randomized trials. JAMA. 2009;301(6):603–618.
- , , , et al. Unplanned readmissions after hospital discharge among patients identified as being at high risk for readmission using a validated predictive algorithm. Open Med. 2011;5(2):e104–111.
- , , , et al. Risk prediction models for hospital readmission: a systematic review. JAMA. 2011;306(15):1688–1698.
- , , . Depression in older people in the general hospital: a systematic review of screening instruments. Age Ageing. 2012;41(2):148–154.
- , , , et al. Prevalence, correlates and recognition of depression among inpatients of general hospitals in Wuhan, China. Gen Hosp Psychiatry. 2010;32(3):268–275.
- , , , , , . Recognition of depression by non‐psychiatric physicians—a systematic literature review and meta‐analysis. J Gen Intern Med. 2008;23(1):25–36.
- , , , , , . Predicting the risk of unplanned readmission or death within 30 days of discharge after a heart failure hospitalization. Am Heart J. 2012;164(3):365–372.
- , , , et al. Does evidence support the American Heart Association's recommendation to screen patients for depression in cardiovascular care? An updated systematic review. PLoS One. 2013;8(1):e52654.
- , , , et al. Screening for depression: a systematic review and meta‐analysis. CMAJ Open. 2013;1(4):E159–E167.
- , , , et al. The PRISMA statement for reporting systematic reviews and meta‐analyses of studies that evaluate health care interventions: explanation and elaboration. Ann Intern Med. 2009;151(4):W65–94.
- , , , et al. Screening for depression in adults: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med. 2002;136(10):765–776.
- , , . Recovering the raw data behind a non‐parametric survival curve. Syst Rev. 2014;3:151.
- , , , . Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan‐Meier survival curves. BMC Med Res Methodol. 2012;12(1):9.
- . Commentary: heterogeneity in meta‐analysis should be expected and appropriately quantified. Int J Epidemiol. 2008;37(5):1158–1160.
- , , , et al. The Newcastle‐Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta‐analyses. Available at: http://www.ohri.ca/programs/clinical_epidemiology/oxford.htm. Accessed September 1, 2015.
- , , . The funnel plot. In: Rothstein HR, Sutton AJ, Borenstein M, eds. Publication Bias in Meta‐analysis: Prevention, Assessment and Adjustments. New York, NY: John Wiley 2006:73–98.
- , , , , , . Dose‐response relationship between depressive symptoms and hospital readmission. J Hosp Med. 2014;9(6):358–364.
- , , , et al. Relationship of depression to increased risk of mortality and rehospitalization in patients with congestive heart failure. Arch Intern Med. 2001;161(15):1849–1856.
- , , , et al. Depression is a risk factor for rehospitalization in medical inpatients. Prim Care Companion J Clin Psychiatry. 2007;9(4):256–262.
- , , , et al. Depression in elderly patients with congestive heart failure. J Geriatr Psychiatry. 1991;24(1):59–71.
- , , . Depression following myocardial infarction: impact on 6‐month survival. JAMA. 1993;270(15):1819–1825.
- , , , , . Gender, depression, and one‐year prognosis after myocardial infarction. Psychosom Med. 1999;61(1):26–37.
- , , , . Depression and 1‐year prognosis in unstable angina. Arch Intern Med. 2000;160(9):1354–1360.
- , , , et al. The impact of depressive symptoms on recovery and outcome of hospitalised COPD exacerbations. Eur Respir J. 2013;41(4):815–823.
- , , , et al. Depression predicts mortality and hospitalization in patients with myocardial infarction complicated by heart failure. Am Heart J. 2005;150(5):961–967.
- , , , et al. Mortality after hospitalization for COPD. Chest. 2002;121(5):1441–1448.
- , , , et al. Pseudomonas aeruginosa and mortality after hospital admission for chronic obstructive pulmonary disease. Respiration. 2012;84(1):36–43.
- , , . Depressive symptoms increase risk of rehospitalisation in heart failure patients with preserved systolic function. J Clin Nurs. 2009;18(13):1871–1877.
- , , . Depressive symptoms and 3 year mortality in older hospitalized medical patients. Ann Intern Med. 1999;130(7):563–569.
- , , , et al. A positive 2‐item patient health questionnaire depression screen among hospitalized heart failure patients is associated with elevated 12‐month mortality. J Card Fail. 2012;18(3):238–245.
- , , , et al. Post‐discharge hospital utilization among adult medical inpatients with depressive symptoms. J Hosp Med. 2010;5(7):378–384.
- , . Use of health services by medically ill depressed elderly patients after hospital discharge. Am J Geriatr Psychiatry. 1999;7(1):48–56.
- , , . Congestive heart failure and depression in older adults: clinical course and health services use 6 months after hospitalization. Psychosomatics. 2003;44(5):367–373.
- , , , . Depressive symptoms as a predictor of 6‐month outcomes and services utilization in elderly medical inpatients. Arch Intern Med. 2001;161(21):2609–2615.
- , , , et al. Depression as a risk factor for poor prognosis among patients with acute coronary syndrome: systematic review and recommendations: a scientific statement from the American Heart Association. Circulation. 2014;129(12):1350–1369.
- , , , , , . Prognostic association of depression following myocardial infarction with mortality and cardiovascular events: a meta‐analysis of 25 years of research. Gen Hosp Psychiatry. 2011;33(3):203–216.
- , , , , . Depression and cardiac disease: epidemiology, mechanisms, and diagnosis. Cardiovasc Psychiatry Neurol. 2013;2013:695925.
- , , , et al. Prognostic value of depression, anxiety, and anger in hospitalized cardiovascular disease patients for predicting adverse cardiac outcomes. Am J Cardiol. 2013;111(10):1432–1436.
- , , . The psychobiology of depression and resilience to stress: implications for prevention and treatment. Annu Rev Clin Psychol. 2005;1:255–291.
- , , , et al. Impact of social factors on risk of readmission or mortality in pneumonia and heart failure: systematic review. J Gen Intern Med. 2013;28(2):269–282.
- , , , et al. The influence of somatic symptoms on beck depression inventory scores in hospitalized postmyocardial infarction patients. Can J Psychiatry. 2012;57(12):752–758.
- , , , et al. Somatic symptom overlap in beck depression inventory‐II scores following myocardial infarction. Br J Psychiatry. 2010;197(1):61–66.
- , , , . Relationship of depression to diabetes types 1 and 2: epidemiology, biology, and treatment. Biol Psychiatry. 2003;54(3):317–329.
- , , . Searching for unpublished data for Cochrane reviews: cross sectional study. BMJ. 2013;346:f2231.
- . Comment on: heterogeneity in meta‐analysis should be expected and appropriately quantified. Int J Epidemiol. 2010;39(3):932; author reply 933.
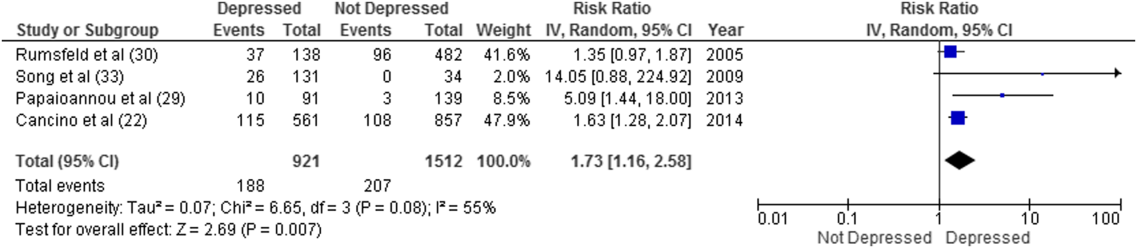
RELAPSE: Answers to why a patient is having a new mood episode
A mood disorder is a chronic illness, associated with episodic recurrence over time1,2; when a patient experiences a new mood episode, explore possible underlying causes of that recurrence. The mnemonic RELAPSE can help you take an informed approach to treatment, instead of making reflexive medication changes (Table).
Rhythm disturbances. Seasonal changes, shift work, jet lag, and sleep irregularity can induce a mood episode in a vulnerable patient. Failure of a patient’s circadian clock to resynchronize itself after such disruption in the dark–light cycle can trigger mood symptoms.
Ending treatment. Intentional or unintentional non-adherence to a prescribed medication or psychotherapy can trigger a mood episode. Likewise, switching from a brand-name medication to a generic equivalent can induce a new episode because the generic drug might be as much as 20% less bioavailable than the brand formulation.3
Life change. Some life events, such as divorce or job loss, can be sufficiently overwhelming—despite medical therapy and psychotherapy—to induce a new episode in a vulnerable patient.
Additional drugs. Opiates, interferon, steroids, reserpine, and other drugs can be depressogenic; on the other hand, steroids, anticholinergic agents, and antidepressants can induce mania. If another physician, or the patient, adds a medication or supplement that causes an interaction with the patient’s current psychotropic prescription, the result might be increased metabolism or clearance of the psychotropic—thus decreasing its efficacy and leading to a new mood episode.
Physical health changes. Neurologic conditions (epilepsy, multiple sclerosis, stroke), autoimmune illnesses (eg, lupus), primary sleep disorders (eg, obstructive sleep apnea), and hormone changes (eg, testosterone, estrogen, and thyroid) that can occur over the lifespan of a patient with a mood disorder can trigger a new episode.
Substance use and withdrawal. Chronic use of alcohol and opiates and withdrawal from cocaine and stimulants in a patient with a mood disorder can induce a depressive episode; use of cocaine, stimulants, and caffeine can induce a manic state.
End of drug response. Some patients experience a loss of drug response over time (tachyphylaxis) or a depressive recurrence while taking an antidepressant.4 These phenomena might be caused by brain changes over time. These are a diagnosis of exclusion after other possibilities have been ruled out.
1. Solomon DA, Keller MB, Leon AC, et al. Multiple recurrences of major depressive disorder. Am J Psychiatry. 2000;157:229-233.
2. Perlis RH, Ostacher MJ, Patel JK, et al. Predictors of recurrence in bipolar disorder: primary outcomes from the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). Am J Psychiatry. 2006;163:217-224.
3. Ellingrod VL. How differences among generics might affect your patient’s response. Current Psychiatry. 2010;9(5):31-32,38.
4. Dunlop BW. Depressive recurrence on antidepressant treatment (DRAT): 4 next-step options. Current Psychiatry. 2013;12:54-55.
A mood disorder is a chronic illness, associated with episodic recurrence over time1,2; when a patient experiences a new mood episode, explore possible underlying causes of that recurrence. The mnemonic RELAPSE can help you take an informed approach to treatment, instead of making reflexive medication changes (Table).
Rhythm disturbances. Seasonal changes, shift work, jet lag, and sleep irregularity can induce a mood episode in a vulnerable patient. Failure of a patient’s circadian clock to resynchronize itself after such disruption in the dark–light cycle can trigger mood symptoms.
Ending treatment. Intentional or unintentional non-adherence to a prescribed medication or psychotherapy can trigger a mood episode. Likewise, switching from a brand-name medication to a generic equivalent can induce a new episode because the generic drug might be as much as 20% less bioavailable than the brand formulation.3
Life change. Some life events, such as divorce or job loss, can be sufficiently overwhelming—despite medical therapy and psychotherapy—to induce a new episode in a vulnerable patient.
Additional drugs. Opiates, interferon, steroids, reserpine, and other drugs can be depressogenic; on the other hand, steroids, anticholinergic agents, and antidepressants can induce mania. If another physician, or the patient, adds a medication or supplement that causes an interaction with the patient’s current psychotropic prescription, the result might be increased metabolism or clearance of the psychotropic—thus decreasing its efficacy and leading to a new mood episode.
Physical health changes. Neurologic conditions (epilepsy, multiple sclerosis, stroke), autoimmune illnesses (eg, lupus), primary sleep disorders (eg, obstructive sleep apnea), and hormone changes (eg, testosterone, estrogen, and thyroid) that can occur over the lifespan of a patient with a mood disorder can trigger a new episode.
Substance use and withdrawal. Chronic use of alcohol and opiates and withdrawal from cocaine and stimulants in a patient with a mood disorder can induce a depressive episode; use of cocaine, stimulants, and caffeine can induce a manic state.
End of drug response. Some patients experience a loss of drug response over time (tachyphylaxis) or a depressive recurrence while taking an antidepressant.4 These phenomena might be caused by brain changes over time. These are a diagnosis of exclusion after other possibilities have been ruled out.
A mood disorder is a chronic illness, associated with episodic recurrence over time1,2; when a patient experiences a new mood episode, explore possible underlying causes of that recurrence. The mnemonic RELAPSE can help you take an informed approach to treatment, instead of making reflexive medication changes (Table).
Rhythm disturbances. Seasonal changes, shift work, jet lag, and sleep irregularity can induce a mood episode in a vulnerable patient. Failure of a patient’s circadian clock to resynchronize itself after such disruption in the dark–light cycle can trigger mood symptoms.
Ending treatment. Intentional or unintentional non-adherence to a prescribed medication or psychotherapy can trigger a mood episode. Likewise, switching from a brand-name medication to a generic equivalent can induce a new episode because the generic drug might be as much as 20% less bioavailable than the brand formulation.3
Life change. Some life events, such as divorce or job loss, can be sufficiently overwhelming—despite medical therapy and psychotherapy—to induce a new episode in a vulnerable patient.
Additional drugs. Opiates, interferon, steroids, reserpine, and other drugs can be depressogenic; on the other hand, steroids, anticholinergic agents, and antidepressants can induce mania. If another physician, or the patient, adds a medication or supplement that causes an interaction with the patient’s current psychotropic prescription, the result might be increased metabolism or clearance of the psychotropic—thus decreasing its efficacy and leading to a new mood episode.
Physical health changes. Neurologic conditions (epilepsy, multiple sclerosis, stroke), autoimmune illnesses (eg, lupus), primary sleep disorders (eg, obstructive sleep apnea), and hormone changes (eg, testosterone, estrogen, and thyroid) that can occur over the lifespan of a patient with a mood disorder can trigger a new episode.
Substance use and withdrawal. Chronic use of alcohol and opiates and withdrawal from cocaine and stimulants in a patient with a mood disorder can induce a depressive episode; use of cocaine, stimulants, and caffeine can induce a manic state.
End of drug response. Some patients experience a loss of drug response over time (tachyphylaxis) or a depressive recurrence while taking an antidepressant.4 These phenomena might be caused by brain changes over time. These are a diagnosis of exclusion after other possibilities have been ruled out.
1. Solomon DA, Keller MB, Leon AC, et al. Multiple recurrences of major depressive disorder. Am J Psychiatry. 2000;157:229-233.
2. Perlis RH, Ostacher MJ, Patel JK, et al. Predictors of recurrence in bipolar disorder: primary outcomes from the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). Am J Psychiatry. 2006;163:217-224.
3. Ellingrod VL. How differences among generics might affect your patient’s response. Current Psychiatry. 2010;9(5):31-32,38.
4. Dunlop BW. Depressive recurrence on antidepressant treatment (DRAT): 4 next-step options. Current Psychiatry. 2013;12:54-55.
1. Solomon DA, Keller MB, Leon AC, et al. Multiple recurrences of major depressive disorder. Am J Psychiatry. 2000;157:229-233.
2. Perlis RH, Ostacher MJ, Patel JK, et al. Predictors of recurrence in bipolar disorder: primary outcomes from the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). Am J Psychiatry. 2006;163:217-224.
3. Ellingrod VL. How differences among generics might affect your patient’s response. Current Psychiatry. 2010;9(5):31-32,38.
4. Dunlop BW. Depressive recurrence on antidepressant treatment (DRAT): 4 next-step options. Current Psychiatry. 2013;12:54-55.
From paranoid fear to completed homicide
A crescendo of paranoid fear sharply increases the likelihood that a person will kill his (her) misperceived persecutor. Persecutory delusions are more likely to lead to homicide than any other psychiatric symptom.1 If people define a delusional situation as real, the situation is real in its consequences.
Based on my experience performing more than 100 insanity evaluations of paranoid persons charged with murder, I have identified 4 paranoid motives for homicide.
Self-defense. The most common paranoid motive for murder is the misperceived need to defend one’s self.
A steel worker believed that there was a conspiracy to kill him. His wife insisted that he go to a hospital emergency room for an evaluation. He then concluded that his wife was in on the conspiracy and stabbed her to death.
Defense of one’s manhood. Homosexual panic occurs in men who think of themselves as heterosexual.
A man with paranoid schizophrenia developed a delusion that his former high school football coach was having the entire team rape him at night. He shot the coach 6 times in front of 22 witnesses.
Defense of one’s children. A parent may kill to save her (his) children’s souls.
A deeply religious woman developed persecutory delusions that her 9-year-old son and 3-year-old daughter were going to be kidnapped and forced to make child pornography. To save her children’s souls, she stabbed her children more than 100 times.
Defense of the world. Homicide may be seen as a way to protect all humankind.
A woman developed a delusion that her father was Satan and would kill her. She believed that if she could kill her father (Satan) and his family she would save herself and bring about world peace. After killing her father, she thrust the sharp end of a tire iron into her grandmother’s umbilicus and vagina because those body parts were involved in “birthing Satan.”
Questioning to determine risk
I have found that, when evaluating a paranoid, delusional person for potential violence, it is better to present that person with a hypothetical question about encountering his perceived persecutor than with a generic question about homicidality.2 For example, a delusional person who reports that he was afraid of being killed by the Mafia could be asked, “If you were walking down an alley and encountered a man dressed like a Mafia hit man with a bulge in his jacket, what would you do?” One interviewee might reply, “The Mafia has so much power there is nothing I could do.” Another might answer, “As soon as I got close enough I would blow his head off with my .357 Magnum.” Although both people would be reporting honestly that they have no homicidal ideas, the latter has a much lower threshold for killing in misperceived self-defense.
Summing up
Persecutory delusions are more likely than any other psychiatric symptom to lead a psychotic person to commit homicide. The killing might be motivated by misperceived self-defense, defense of one’s manhood, defense of one’s children, or defense of the world.
A crescendo of paranoid fear sharply increases the likelihood that a person will kill his (her) misperceived persecutor. Persecutory delusions are more likely to lead to homicide than any other psychiatric symptom.1 If people define a delusional situation as real, the situation is real in its consequences.
Based on my experience performing more than 100 insanity evaluations of paranoid persons charged with murder, I have identified 4 paranoid motives for homicide.
Self-defense. The most common paranoid motive for murder is the misperceived need to defend one’s self.
A steel worker believed that there was a conspiracy to kill him. His wife insisted that he go to a hospital emergency room for an evaluation. He then concluded that his wife was in on the conspiracy and stabbed her to death.
Defense of one’s manhood. Homosexual panic occurs in men who think of themselves as heterosexual.
A man with paranoid schizophrenia developed a delusion that his former high school football coach was having the entire team rape him at night. He shot the coach 6 times in front of 22 witnesses.
Defense of one’s children. A parent may kill to save her (his) children’s souls.
A deeply religious woman developed persecutory delusions that her 9-year-old son and 3-year-old daughter were going to be kidnapped and forced to make child pornography. To save her children’s souls, she stabbed her children more than 100 times.
Defense of the world. Homicide may be seen as a way to protect all humankind.
A woman developed a delusion that her father was Satan and would kill her. She believed that if she could kill her father (Satan) and his family she would save herself and bring about world peace. After killing her father, she thrust the sharp end of a tire iron into her grandmother’s umbilicus and vagina because those body parts were involved in “birthing Satan.”
Questioning to determine risk
I have found that, when evaluating a paranoid, delusional person for potential violence, it is better to present that person with a hypothetical question about encountering his perceived persecutor than with a generic question about homicidality.2 For example, a delusional person who reports that he was afraid of being killed by the Mafia could be asked, “If you were walking down an alley and encountered a man dressed like a Mafia hit man with a bulge in his jacket, what would you do?” One interviewee might reply, “The Mafia has so much power there is nothing I could do.” Another might answer, “As soon as I got close enough I would blow his head off with my .357 Magnum.” Although both people would be reporting honestly that they have no homicidal ideas, the latter has a much lower threshold for killing in misperceived self-defense.
Summing up
Persecutory delusions are more likely than any other psychiatric symptom to lead a psychotic person to commit homicide. The killing might be motivated by misperceived self-defense, defense of one’s manhood, defense of one’s children, or defense of the world.
A crescendo of paranoid fear sharply increases the likelihood that a person will kill his (her) misperceived persecutor. Persecutory delusions are more likely to lead to homicide than any other psychiatric symptom.1 If people define a delusional situation as real, the situation is real in its consequences.
Based on my experience performing more than 100 insanity evaluations of paranoid persons charged with murder, I have identified 4 paranoid motives for homicide.
Self-defense. The most common paranoid motive for murder is the misperceived need to defend one’s self.
A steel worker believed that there was a conspiracy to kill him. His wife insisted that he go to a hospital emergency room for an evaluation. He then concluded that his wife was in on the conspiracy and stabbed her to death.
Defense of one’s manhood. Homosexual panic occurs in men who think of themselves as heterosexual.
A man with paranoid schizophrenia developed a delusion that his former high school football coach was having the entire team rape him at night. He shot the coach 6 times in front of 22 witnesses.
Defense of one’s children. A parent may kill to save her (his) children’s souls.
A deeply religious woman developed persecutory delusions that her 9-year-old son and 3-year-old daughter were going to be kidnapped and forced to make child pornography. To save her children’s souls, she stabbed her children more than 100 times.
Defense of the world. Homicide may be seen as a way to protect all humankind.
A woman developed a delusion that her father was Satan and would kill her. She believed that if she could kill her father (Satan) and his family she would save herself and bring about world peace. After killing her father, she thrust the sharp end of a tire iron into her grandmother’s umbilicus and vagina because those body parts were involved in “birthing Satan.”
Questioning to determine risk
I have found that, when evaluating a paranoid, delusional person for potential violence, it is better to present that person with a hypothetical question about encountering his perceived persecutor than with a generic question about homicidality.2 For example, a delusional person who reports that he was afraid of being killed by the Mafia could be asked, “If you were walking down an alley and encountered a man dressed like a Mafia hit man with a bulge in his jacket, what would you do?” One interviewee might reply, “The Mafia has so much power there is nothing I could do.” Another might answer, “As soon as I got close enough I would blow his head off with my .357 Magnum.” Although both people would be reporting honestly that they have no homicidal ideas, the latter has a much lower threshold for killing in misperceived self-defense.
Summing up
Persecutory delusions are more likely than any other psychiatric symptom to lead a psychotic person to commit homicide. The killing might be motivated by misperceived self-defense, defense of one’s manhood, defense of one’s children, or defense of the world.
Chronic pain and depression: Understanding 2 culprits in common
Any discussion of the relationship between major depressive disorder (MDD) and chronic pain encounters an obstacle immediately: Neither has a singular pathophysiology. Furthermore, MDD and, to a significant extent, chronic pain are defined more by their symptoms than by a presumed etiology and pathogenesis.
Why does this matter to a busy clinician?
Explicitly or implicitly, we often align our treatment approaches with what we assume is the underlying pathophysiology of the conditions we are addressing. An overview of shared pathophysiology of chronic pain conditions and MDD therefore can be useful in practice.
What is chronic pain? Defined as “pain that persists past the healing phase following an injury,”1 chronic pain often is subdivided into 4 types2,3:
- nociceptive (caused by a lesion or potential tissue damage)
- inflammatory
- neuropathic (spontaneous pain or hypersensitivity to pain related to neurologic illness or injury)
- functional (hypersensitivity to pain due to abnormal central processing of a normal input).
Although fibromyalgia often is categorized as a dysfunctional pain syndrome, persons who suffer from it, much like those who suffer neuropathic pain, commonly report hyperalgesia (augmented sensitivity to painful stimuli), allodynia (abnormal pain response to non-noxious stimuli), and paresthesias. These shared clinical features of fibromyalgia and neuropathic pain are consistent with central sensitization, which suggests overlapping pathophysiology.4
Comorbidity between depression and pain is common. A 30% to 60% co-occurrence rate of MDD and chronic pain has been reported.5 Some subtypes of chronic pain, such as fibromyalgia, are so commonly comorbid with psychiatric conditions that they have spawned a scientific debate as to whether the conditions are most parsimoniously considered (1) separate illnesses with high comorbidity or (2) different symptomatic manifestations of a single underlying condition.6 Moreover, cumulative evidence suggests that chronic pain and depression do not just co-occur; each one facilitates development of the other, such that chronic pain is a strong predictor of subsequent onset of MDD, and vice versa.
When pain and depression are comorbid, they also tend to make treatment of each condition more difficult. For example, pain presents (1) a major obstacle to achieving remission when treating depression7,8 and (2) significant risk of relapse.9 A 3-year longitudinal study showed that painful symptoms substantially reduced the chance of recovery in a group of older depressed patients (n = 327). A substantially greater percentage of patients with MDD alone attained recovery (47%), compared with only 9% in whom MDD and painful symptoms were comorbid.10 Furthermore, a higher level of pain can delay remission when treating MDD,11 thus reducing the likelihood of an optimal outcome.12
Understanding shared processes. Recent developments in neuroscience and psycho-immunology point to the fact that comorbid pain and depression might be driven by overlapping pathophysiological processes in the brain and body. In the 2 parts of this article, we (1) review scientific understanding of these shared processes and (2) demonstrate how recent advances in the epidemiology, phenomenology, and etiology of chronic pain and MDD provide important clues for more effective diagnosis (Part 1) and treatment (Part 2, March 2016)—and, therefore, better outcomes. Our focus is primarily on the relationship between MDD and the best-studied comorbid chronic pain conditions: fibromyalgia, neuropathic pain, chronic back pain, and rheumatoid arthritis.
The societal burden of chronic pain conditions is enormous
A recent epidemiological study13 projected that as many as 100 million people in the United States—30.7% of the population—suffer some form of chronic pain, including arthritis and joint pain. A World Health Organization survey yielded a similar (and staggering) 37% prevalence of chronic pain in the population of 10 developed countries.14
Estimates are that various forms of neuropathic pain, including diabetic neuropathy, postherpetic neuralgia, trigeminal neuralgia, spinal cord injury, and radiculopathy, alone afflict as many as 26 million people worldwide, including approximately 1.5% of the U.S. population.15,16
Chronic low back pain is epidemic. With a projected point prevalence of 30%, the condition is the most common cause of activity limitation among people age <45, and the most frequent reason in the United States for visiting a physician.1
Functional somatic syndromes, including fibromyalgia and irritable bowel syndrome, impose an astounding strain on health care: These syndromes account for 25% to 50% of all outpatient visits, or approximately 400 million clinic visits annually in the United States.17
Why should you care about these numbers? The answer is that comorbidity among chronic pain, mood disorders, anxiety disorders, sleep disorders, cognitive impairment, fatigue, and chronic stress presents an enormous clinical challenge because it not only complicates the diagnosis of these conditions but also compromises treatment outcomes and imposes severe limitations on daily functioning and quality of life of those afflicted.5,17-24
A complex relationship and a daunting clinical challenge
Chronic pain enhances the risk of MDD by 2-fold to 5-fold. The risk appears to be mediated by the number of pain conditions rather than by the severity of pain.23 Some authors have noted a kind of dose-response relationship among pain, depression, and anxiety. Among patients who experienced chronic pain that affected 1 body region, the prevalence of generalized anxiety disorder (GAD) and MDD was 30% and 20%, respectively; in patients who experienced pain in ≥2 regions, the prevalence of GAD and MDD was elevated to 54% and 32%.25 Moreover, patients with fibromyalgia were 4.3 times more likely than healthy controls to develop MDD at some point in their lives and 4.7 times more likely to develop an anxiety disorder.26
Although women are more likely to suffer from fibromyalgia, the risk for people of either sex of developing subsequent MDD is comparable once the condition has developed.27 Overall, depression and anxiety are among the most common comorbidities of fibromyalgia, with prevalence ranging from 20% to 80% and 13% to 63.8%, respectively.28
High comorbidity between depression and pain also is relevant for patients with neuropathic pain. A survey from Australia reported depression in 34% and anxiety in 25% of patients with neuropathic pain.29 Pain severity tended to be enduring and associated with significantly impaired functioning. A significant percentage of patients suffering from rheumatoid arthritis and systemic lupus erythematosus tend to manifest anxiety and depression (93% to 94%), cognitive impairment (66%), fatigue (40%), and sleep disorders (72%).22
The relationship between depression and pain appears to be bidirectional. For example, recent studies demonstrate that 30% to 60% of depressed patients also suffer from a painful condition.5
The complex history of patients presenting with concomitant complaints of depression, anxiety, chronic pain, sleep disturbance, cognitive impairment, and fatigue present a daunting diagnostic task. Pain tends to be associated with greater fatigue and sleep disturbance, which in turn depletes a patient’s ability to enjoy life and enhances negative affect.19,20,30 The take-home message might be to screen all chronic pain patients for MDD, anxiety, and sleep disorders, and vice versa.
Furthermore, comorbidity among chronic pain, MDD, anxiety, and sleep disorders can introduce specific intricacies into our treatment approach. Although, in general, comorbidities tend to have a negative impact on treatment outcomes, many pharmacotherapeutic and non-drug interventions targeting chronic pain might ameliorate sleep problems, low energy, anxiety, depression, and anhedonia.18,20,30-32 On the other hand, we should consider that opioid treatment for chronic pain might represent a risk factor for subsequent depression. It is conceivable that chronic opioid treatment and associated sedation can erode self-efficacy and social relationships, thereby compromising sources of support.33,34 It is equally important to keep in mind that, even if we are successful in attaining remission when treating depression and pain, residual pain symptoms might persist, requiring more specific interventions.24
MDD and chronic pain each have, on their own, a well-established association with suicide attempts and completion. Researchers are investigating whether a pathophysiologic suicide-promoting synergy between the 2 disorders exists when they are comorbid (Box35-37).

Shared genetics and pathophysiology
Several candidate genes have been identified as risk genes for chronic pain, depression, and anxiety. One of those studied the most is 5-HTTLPR, involved in regulating synthesis of serotonin transporter. The short form of this gene has been implicated in a diverse set of conditions, including MDD, anxiety disorders, and substance abuse—and fibromyalgia. Other genes associated with the risk of MDD and pain disorders are ones that code for:
- serotonin 5-HT2A and 5-HT1A receptors
- catechol-O-methyltransferase, an enzyme involved in catecholamine metabolism
- dopamine D4 receptor
- proinflammatory cytokines interleukin-1 and interleukin-6.4
Both monoamines and inflammatory cytokines play a role in modulating γ-aminobutyric acid (GABA) and glutamate neurons, as well as glia cells constituting peripheral pain pathways and central circuits that participate in the pain response and regulation of mood.4,17,38
The ‘pain matrix’
Brain circuitry that is involved in processing pain stimuli—often referred to as the pain matrix—shares many structural components with circuitry involved in the stress response and emotional modulation.4 Emerging evidence indicates that the pain matrix might not be pain-specific but, instead, a complex aggregate of interconnected brain structures involved in evoking defensive responses to a number of offending stimuli, including pain, threat, danger, loss, and social rejection or isolation.
It is remarkable, in this regard, that imaging studies show that the dorsal anterior cingulate, central to experiencing negative affect in response to physical pain, also mediates distress in response to the “pain” of social exclusion.39 Emerging functional and structural imaging provides evidence of continuous reorganization of prefrontal cortices as a consequence of enduring chronic pain.1 Of particular interest are findings of (1) a reduction of gray matter in the dorsolateral prefrontal cortex (DLPFC) and (2) functional activation of the medial prefrontal cortex (mPFC), both of which correlate with the duration and experience of chronic back pain.1 It is tempting to speculate that structural decline of the DLPFC, observed in MDD and chronic pain, is linked to cognitive and executive function deficits, which are readily observed in patients with either disorder—given that DLPFC is a “hub” of the so-called “cognitive-executive functional network.”1,4
Likewise, the mPFC is a key component of the default mode network (DMN), a functional network also comprising the posterior cingulate cortex and hippocampus. DMN performs a diverse set of activities, including self-reflection, daydreaming, reminiscing, planning, processing of social information, and creative thinking. Negative neuroplastic changes in the DMN are a common finding in MDD and chronic pain, and might be associated with a tendency toward rumination and catastrophizing—key clinical manifestations of MDD and chronic pain—and linked with pervasive negative affect and sleep disturbance.4,32
Furthermore, functional and structural changes in the amygdala and hippocampus have been described in MDD, fibromyalgia, and neuropathic pain.4 Dysfunction of these limbic formations may be a contributing factor in the disruption of neuroendocrine, autonomic, and immune function, which could further contribute to aggravated mood and pain symptoms.4,17,40
Consequently, excessive hypothalamic-pituitary-adrenal axis and sympathetic activation, combined with elevation of proinflammatory cytokine production and release, likely plays a role in the pathophysiology of MDD and chronic pain disorders.4,17,40 Moreover, at cellular, subcellular, and molecular levels, chronic pain and MDD are associated with:
- perturbed neuron-glia relationships
- altered glutamatergic, GABA, glycine, substance-P, opioid, 5-HT, norepinephrine, and dopamine signaling
- dysfunction of intracellular signaling cascades and neurotrophic signaling.4,20,30,31,38
The Figure that describes how homeostatic function of prefrontal cortical-limbic circuitry is compromised in MDD and chronic pain—thus disrupting autonomic, neuroendocrine, and neuroimmune regulation.
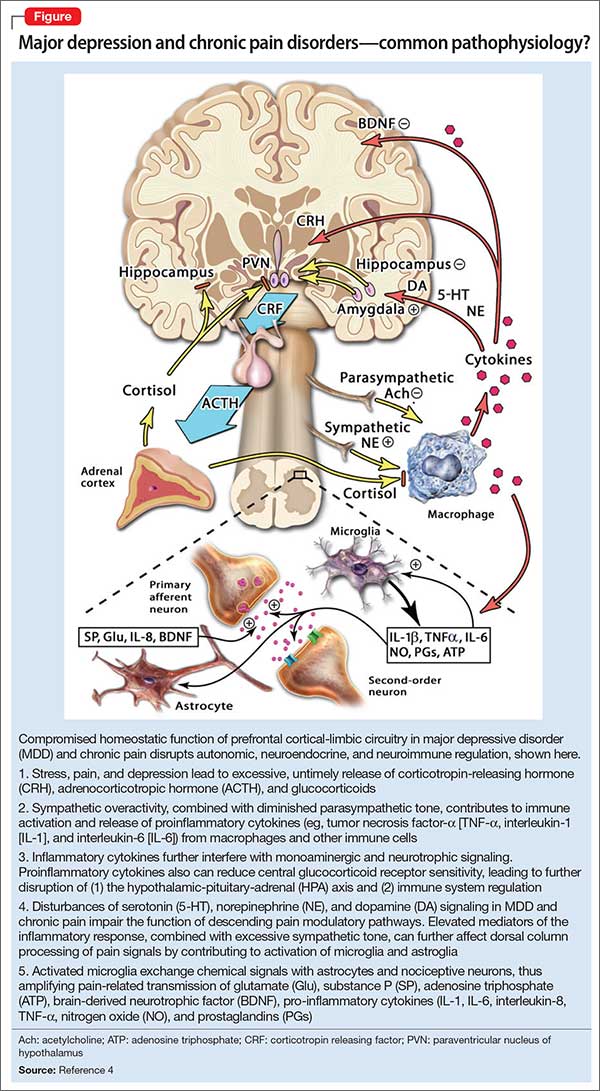
Disturbance in monoamine signaling in chronic pain and MDD might give rise to profound anhedonia, cognitive impairment, anxiety, insomnia, sensitivity to stress, and inadequate functioning of descending pain-regulatory pathways, which primarily use norepinephrine and 5-HT.4,9,20,30,31,38 Using pharmacotherapeutic agents that successfully modulate monoamines, therefore, might ameliorate the function of brain networks innervated by neurotransmitter systems involved in the regulation of pain, mood, cognition, stress response, and sleep. Notably, the same monoamines serve as transmitters in descending pain pathways.
In summary, convergent evidence indicates that MDD and chronic pain states amplify each other, thus contributing to treatment resistance in both disorders.
On the bright side, timely and effective treatment of MDD might optimize the chance of remission and minimize the risk of enduring structural brain changes in MDD and chronic pain.1,4,31,32 The obverse is also true: Emphasizing the importance of the resolution of painful symptoms in the context of MDD, a study reported a significantly greater remission rate of 36.2% in those who had >50% reduction of pain on a visual analogue scale following treatment with a serotonin-norepinephrine reuptake inhibitor, compared with a 17.8% remission rate in persons who experienced <50% pain reduction on the scale.3
Editors’ note: In Part 2 of this article (March 2016), the authors review pharmacotherapeutic and non-drug strategies for managing comorbid chronic pain conditions and MDD.
1. Apkarian AV, Baliki MN, Geha PY. Towards a theory of chronic pain. Prog Neurobiol. 2009;87(2):81-97.
2. Verdu B, Decosterd I, Buclin T, et al. Antidepressants for the treatment of chronic pain. Drugs. 2008;68(18):2611-2632.
3. Woolf CJ; American College of Physicians, American Physiological Society. Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann Intern Med. 2004;140(6):441-451.
4. Maletic V, Raison CL. Neurobiology of depression, fibromyalgia and neuropathic pain. Front Biosci (Landmark Ed). 2009;14:5291-5338.
5. Bair MJ, Wu J, Damush TM, et al. Association of depression and anxiety alone and in combination with chronic musculoskeletal pain in primary care patients. Psychosom Med. 2008;70(8):890-897.
6. Cho HJ, Skowera A, Cleare A, et al. Chronic fatigue syndrome: an update focusing on phenomenology and pathophysiology. Curr Opin Psychiatry. 2006;19(1):67-73.
7. Fava M. Depression with physical symptoms: treating to remission. J Clin Psychiatry. 2003;64(suppl 7):24-28.
8. Bair MJ, Robinson RL, Eckert GJ, et al. Impact of pain on depression treatment response in primary care. Psychosom Med. 2004;66(1):17-22.
9. Ohayon MM. Specific characteristics of the pain/depression association in the general population. J Clin Psychiatry. 2004;65(suppl 12):5-9.
10. Geerlings SW, Twisk JW, Beekman AT, et al. Longitudinal relationship between pain and depression in older adults: sex, age and physical disability. Soc Psychiatry Psychiatr Epidemiol. 2002;37(1):23-30.
11. Karp JF, Scott J, Houck P, et al. Pain predicts longer time to remission during treatment of recurrent depression. J Clin Psychiatry. 2005;66(5):591-597.
12. Spijker J, de Graaf R, Bijl RV, et al. Determinants of persistence of major depressive episodes in the general population. Results from the Netherlands Mental Health Survey and Incidence Study (NEMESIS). J Affect Disord. 2004;81(3):231-240.
13. Johannes CB, Le TK, Zhou X, et al. The prevalence of chronic pain in United States adults: results of an Internet-based survey. J Pain. 2010;11(11):1230-1239.
14. Dzau VJ, Pizzo PA. Relieving pain in America: insights from an Institute of Medicine committee. JAMA. 2014;312(15):1507-1508.
15. Butera JA. Current and emerging targets to treat neuropathic pain. J Med Chem. 2007;50(11):2543-2546.
16. Offenbaecher M, Ackenheil M. Current trends in neuropathic pain treatments with special reference to fibromyalgia. CNS Spectr. 2005;10(4):285-297.
17. Goldenberg DL. Pain/depression dyad: a key to a better understanding and treatment of functional somatic syndromes. Am J Med. 2010;123(8):675-682.
18. Argoff CE. The coexistence of neuropathic pain, sleep, and psychiatric disorders: a novel treatment approach. Clin J Pain. 2007;23(1):15-22.
19. Zautra AJ, Fasman R, Parish BP, et al. Daily fatigue in women with osteoarthritis, rheumatoid arthritis, and fibromyalgia. Pain. 2007;128(1-2):128-135.
20. Finan PH, Smith MT. The comorbidity of insomnia, chronic pain, and depression: dopamine as a putative mechanism. Sleep Med Rev. 2013;17(3):173-183.
21. Senba E. A key to dissect the triad of insomnia, chronic pain, and depression. Neurosci Lett. 2015;589:197-199.
22. Torta R, Pennazio F, Ieraci V. Anxiety and depression in rheumatologic diseases: the relevance of diagnosis and management. Reumatismo. 2014;66(1):92-97.
23. Howe CQ, Robinson JP, Sullivan MD. Psychiatric and psychological perspectives on chronic pain. Phys Med Rehabil Clin N Am. 2015;26(2):283-300.
24. Gerrits MM, van Marwijk HW, van Oppen P, et al. Longitudinal association between pain, and depression and anxiety over four years. J Psychosom Res. 2015;78(1):64-70.
25. Manchikanti L, Pampati V, Beyer C, et al. Do number of pain conditions influence emotional status? Pain Physician. 2002;5(2):200-205.
26. Arnold LM. Biology and therapy of fibromyalgia. New therapies in fibromyalgia. Arthritis Res Ther. 2006;8(4):212.
27. Weir PT, Harlan GA, Nkoy FL, et al. The incidence of fibromyalgia and its associated comorbidities: a population-based retrospective cohort study based on International Classification of Diseases, 9th Revision codes. J Clin Rheumatol. 2006;12(3):124-128.
28. Fietta P, Fietta P, Manganelli P. Fibromyalgia and psychiatric disorders. Acta Biomed. 2007;78(2):88-95.
29. Gustorff B, Dorner T, Likar R, et al. Prevalence of self-reported neuropathic pain and impact on quality of life: a prospective representative survey. Acta Anaesthesiol Scand. 2008;52(1):132-136.
30. Boakye PA, Olechowski C, Rashiq S, et al. A critical review of neurobiological factors involved in the interactions between chronic pain, depression, and sleep disruption [published online May 28, 2015]. Clin J Pain. doi: 10.1097/ AJP.0000000000000260.
31. Jann MW, Slade JH. Antidepressant agents for the treatment of chronic pain and depression. Pharmacotherapy. 2007;27(11):1571-1587.
32. Nekovarova T, Yamamotova A, Vales K, et al. Common mechanisms of pain and depression: are antidepressants also analgesics? Front Behav Neurosci. 2014;8:99.
33. Smith K, Mattick RP, Bruno R, et al. Factors associated with the development of depression in chronic non-cancer pain patients following the onset of opioid treatment for pain. J Affect Disord. 2015;184:72-80.
34. Scherrer JF, Svrakic DM, Freedland KE, et al. Prescription opioid analgesics increase the risk of depression. J Gen Intern Med. 2014;29(3):491-499.
35. Fishbain DA, Lewis JE, Gao J. The pain suicidality association: a narrative review. Pain Med. 2014;15(11):1835-1849.
36. Elman I, Borsook D, Volkow ND. Pain and suicidality: insights from reward and addiction neuroscience. Prog Neurobiol. 2013;109:1-27.
37. Olié E, Guillaume S, Jaussent I, et al. Higher psychological pain during a major depressive episode may be a factor of vulnerability to suicidal ideation and act. J Affect Disord. 2010;120(1-3):226-230.
38. Han C, Pae CU. Pain and depression: a neurobiological perspective of their relationship. Psychiatry Investig. 2015;12(1):1-8.
39. Eisenberger NI, Lieberman MD, Williams KD. Does rejection hurt? An FMRI study of social exclusion. Science. 2003;302(5643):290-292.
40. Gracely RH, Ceko M, Bushnell MC. Fibromyalgia and depression [published online November 19, 2011]. Pain Res Treat. 2012;2012:486590. doi: 10.1155/2012/486590.
Any discussion of the relationship between major depressive disorder (MDD) and chronic pain encounters an obstacle immediately: Neither has a singular pathophysiology. Furthermore, MDD and, to a significant extent, chronic pain are defined more by their symptoms than by a presumed etiology and pathogenesis.
Why does this matter to a busy clinician?
Explicitly or implicitly, we often align our treatment approaches with what we assume is the underlying pathophysiology of the conditions we are addressing. An overview of shared pathophysiology of chronic pain conditions and MDD therefore can be useful in practice.
What is chronic pain? Defined as “pain that persists past the healing phase following an injury,”1 chronic pain often is subdivided into 4 types2,3:
- nociceptive (caused by a lesion or potential tissue damage)
- inflammatory
- neuropathic (spontaneous pain or hypersensitivity to pain related to neurologic illness or injury)
- functional (hypersensitivity to pain due to abnormal central processing of a normal input).
Although fibromyalgia often is categorized as a dysfunctional pain syndrome, persons who suffer from it, much like those who suffer neuropathic pain, commonly report hyperalgesia (augmented sensitivity to painful stimuli), allodynia (abnormal pain response to non-noxious stimuli), and paresthesias. These shared clinical features of fibromyalgia and neuropathic pain are consistent with central sensitization, which suggests overlapping pathophysiology.4
Comorbidity between depression and pain is common. A 30% to 60% co-occurrence rate of MDD and chronic pain has been reported.5 Some subtypes of chronic pain, such as fibromyalgia, are so commonly comorbid with psychiatric conditions that they have spawned a scientific debate as to whether the conditions are most parsimoniously considered (1) separate illnesses with high comorbidity or (2) different symptomatic manifestations of a single underlying condition.6 Moreover, cumulative evidence suggests that chronic pain and depression do not just co-occur; each one facilitates development of the other, such that chronic pain is a strong predictor of subsequent onset of MDD, and vice versa.
When pain and depression are comorbid, they also tend to make treatment of each condition more difficult. For example, pain presents (1) a major obstacle to achieving remission when treating depression7,8 and (2) significant risk of relapse.9 A 3-year longitudinal study showed that painful symptoms substantially reduced the chance of recovery in a group of older depressed patients (n = 327). A substantially greater percentage of patients with MDD alone attained recovery (47%), compared with only 9% in whom MDD and painful symptoms were comorbid.10 Furthermore, a higher level of pain can delay remission when treating MDD,11 thus reducing the likelihood of an optimal outcome.12
Understanding shared processes. Recent developments in neuroscience and psycho-immunology point to the fact that comorbid pain and depression might be driven by overlapping pathophysiological processes in the brain and body. In the 2 parts of this article, we (1) review scientific understanding of these shared processes and (2) demonstrate how recent advances in the epidemiology, phenomenology, and etiology of chronic pain and MDD provide important clues for more effective diagnosis (Part 1) and treatment (Part 2, March 2016)—and, therefore, better outcomes. Our focus is primarily on the relationship between MDD and the best-studied comorbid chronic pain conditions: fibromyalgia, neuropathic pain, chronic back pain, and rheumatoid arthritis.
The societal burden of chronic pain conditions is enormous
A recent epidemiological study13 projected that as many as 100 million people in the United States—30.7% of the population—suffer some form of chronic pain, including arthritis and joint pain. A World Health Organization survey yielded a similar (and staggering) 37% prevalence of chronic pain in the population of 10 developed countries.14
Estimates are that various forms of neuropathic pain, including diabetic neuropathy, postherpetic neuralgia, trigeminal neuralgia, spinal cord injury, and radiculopathy, alone afflict as many as 26 million people worldwide, including approximately 1.5% of the U.S. population.15,16
Chronic low back pain is epidemic. With a projected point prevalence of 30%, the condition is the most common cause of activity limitation among people age <45, and the most frequent reason in the United States for visiting a physician.1
Functional somatic syndromes, including fibromyalgia and irritable bowel syndrome, impose an astounding strain on health care: These syndromes account for 25% to 50% of all outpatient visits, or approximately 400 million clinic visits annually in the United States.17
Why should you care about these numbers? The answer is that comorbidity among chronic pain, mood disorders, anxiety disorders, sleep disorders, cognitive impairment, fatigue, and chronic stress presents an enormous clinical challenge because it not only complicates the diagnosis of these conditions but also compromises treatment outcomes and imposes severe limitations on daily functioning and quality of life of those afflicted.5,17-24
A complex relationship and a daunting clinical challenge
Chronic pain enhances the risk of MDD by 2-fold to 5-fold. The risk appears to be mediated by the number of pain conditions rather than by the severity of pain.23 Some authors have noted a kind of dose-response relationship among pain, depression, and anxiety. Among patients who experienced chronic pain that affected 1 body region, the prevalence of generalized anxiety disorder (GAD) and MDD was 30% and 20%, respectively; in patients who experienced pain in ≥2 regions, the prevalence of GAD and MDD was elevated to 54% and 32%.25 Moreover, patients with fibromyalgia were 4.3 times more likely than healthy controls to develop MDD at some point in their lives and 4.7 times more likely to develop an anxiety disorder.26
Although women are more likely to suffer from fibromyalgia, the risk for people of either sex of developing subsequent MDD is comparable once the condition has developed.27 Overall, depression and anxiety are among the most common comorbidities of fibromyalgia, with prevalence ranging from 20% to 80% and 13% to 63.8%, respectively.28
High comorbidity between depression and pain also is relevant for patients with neuropathic pain. A survey from Australia reported depression in 34% and anxiety in 25% of patients with neuropathic pain.29 Pain severity tended to be enduring and associated with significantly impaired functioning. A significant percentage of patients suffering from rheumatoid arthritis and systemic lupus erythematosus tend to manifest anxiety and depression (93% to 94%), cognitive impairment (66%), fatigue (40%), and sleep disorders (72%).22
The relationship between depression and pain appears to be bidirectional. For example, recent studies demonstrate that 30% to 60% of depressed patients also suffer from a painful condition.5
The complex history of patients presenting with concomitant complaints of depression, anxiety, chronic pain, sleep disturbance, cognitive impairment, and fatigue present a daunting diagnostic task. Pain tends to be associated with greater fatigue and sleep disturbance, which in turn depletes a patient’s ability to enjoy life and enhances negative affect.19,20,30 The take-home message might be to screen all chronic pain patients for MDD, anxiety, and sleep disorders, and vice versa.
Furthermore, comorbidity among chronic pain, MDD, anxiety, and sleep disorders can introduce specific intricacies into our treatment approach. Although, in general, comorbidities tend to have a negative impact on treatment outcomes, many pharmacotherapeutic and non-drug interventions targeting chronic pain might ameliorate sleep problems, low energy, anxiety, depression, and anhedonia.18,20,30-32 On the other hand, we should consider that opioid treatment for chronic pain might represent a risk factor for subsequent depression. It is conceivable that chronic opioid treatment and associated sedation can erode self-efficacy and social relationships, thereby compromising sources of support.33,34 It is equally important to keep in mind that, even if we are successful in attaining remission when treating depression and pain, residual pain symptoms might persist, requiring more specific interventions.24
MDD and chronic pain each have, on their own, a well-established association with suicide attempts and completion. Researchers are investigating whether a pathophysiologic suicide-promoting synergy between the 2 disorders exists when they are comorbid (Box35-37).
Shared genetics and pathophysiology
Several candidate genes have been identified as risk genes for chronic pain, depression, and anxiety. One of those studied the most is 5-HTTLPR, involved in regulating synthesis of serotonin transporter. The short form of this gene has been implicated in a diverse set of conditions, including MDD, anxiety disorders, and substance abuse—and fibromyalgia. Other genes associated with the risk of MDD and pain disorders are ones that code for:
- serotonin 5-HT2A and 5-HT1A receptors
- catechol-O-methyltransferase, an enzyme involved in catecholamine metabolism
- dopamine D4 receptor
- proinflammatory cytokines interleukin-1 and interleukin-6.4
Both monoamines and inflammatory cytokines play a role in modulating γ-aminobutyric acid (GABA) and glutamate neurons, as well as glia cells constituting peripheral pain pathways and central circuits that participate in the pain response and regulation of mood.4,17,38
The ‘pain matrix’
Brain circuitry that is involved in processing pain stimuli—often referred to as the pain matrix—shares many structural components with circuitry involved in the stress response and emotional modulation.4 Emerging evidence indicates that the pain matrix might not be pain-specific but, instead, a complex aggregate of interconnected brain structures involved in evoking defensive responses to a number of offending stimuli, including pain, threat, danger, loss, and social rejection or isolation.
It is remarkable, in this regard, that imaging studies show that the dorsal anterior cingulate, central to experiencing negative affect in response to physical pain, also mediates distress in response to the “pain” of social exclusion.39 Emerging functional and structural imaging provides evidence of continuous reorganization of prefrontal cortices as a consequence of enduring chronic pain.1 Of particular interest are findings of (1) a reduction of gray matter in the dorsolateral prefrontal cortex (DLPFC) and (2) functional activation of the medial prefrontal cortex (mPFC), both of which correlate with the duration and experience of chronic back pain.1 It is tempting to speculate that structural decline of the DLPFC, observed in MDD and chronic pain, is linked to cognitive and executive function deficits, which are readily observed in patients with either disorder—given that DLPFC is a “hub” of the so-called “cognitive-executive functional network.”1,4
Likewise, the mPFC is a key component of the default mode network (DMN), a functional network also comprising the posterior cingulate cortex and hippocampus. DMN performs a diverse set of activities, including self-reflection, daydreaming, reminiscing, planning, processing of social information, and creative thinking. Negative neuroplastic changes in the DMN are a common finding in MDD and chronic pain, and might be associated with a tendency toward rumination and catastrophizing—key clinical manifestations of MDD and chronic pain—and linked with pervasive negative affect and sleep disturbance.4,32
Furthermore, functional and structural changes in the amygdala and hippocampus have been described in MDD, fibromyalgia, and neuropathic pain.4 Dysfunction of these limbic formations may be a contributing factor in the disruption of neuroendocrine, autonomic, and immune function, which could further contribute to aggravated mood and pain symptoms.4,17,40
Consequently, excessive hypothalamic-pituitary-adrenal axis and sympathetic activation, combined with elevation of proinflammatory cytokine production and release, likely plays a role in the pathophysiology of MDD and chronic pain disorders.4,17,40 Moreover, at cellular, subcellular, and molecular levels, chronic pain and MDD are associated with:
- perturbed neuron-glia relationships
- altered glutamatergic, GABA, glycine, substance-P, opioid, 5-HT, norepinephrine, and dopamine signaling
- dysfunction of intracellular signaling cascades and neurotrophic signaling.4,20,30,31,38
The Figure that describes how homeostatic function of prefrontal cortical-limbic circuitry is compromised in MDD and chronic pain—thus disrupting autonomic, neuroendocrine, and neuroimmune regulation.
Disturbance in monoamine signaling in chronic pain and MDD might give rise to profound anhedonia, cognitive impairment, anxiety, insomnia, sensitivity to stress, and inadequate functioning of descending pain-regulatory pathways, which primarily use norepinephrine and 5-HT.4,9,20,30,31,38 Using pharmacotherapeutic agents that successfully modulate monoamines, therefore, might ameliorate the function of brain networks innervated by neurotransmitter systems involved in the regulation of pain, mood, cognition, stress response, and sleep. Notably, the same monoamines serve as transmitters in descending pain pathways.
In summary, convergent evidence indicates that MDD and chronic pain states amplify each other, thus contributing to treatment resistance in both disorders.
On the bright side, timely and effective treatment of MDD might optimize the chance of remission and minimize the risk of enduring structural brain changes in MDD and chronic pain.1,4,31,32 The obverse is also true: Emphasizing the importance of the resolution of painful symptoms in the context of MDD, a study reported a significantly greater remission rate of 36.2% in those who had >50% reduction of pain on a visual analogue scale following treatment with a serotonin-norepinephrine reuptake inhibitor, compared with a 17.8% remission rate in persons who experienced <50% pain reduction on the scale.3
Editors’ note: In Part 2 of this article (March 2016), the authors review pharmacotherapeutic and non-drug strategies for managing comorbid chronic pain conditions and MDD.
Any discussion of the relationship between major depressive disorder (MDD) and chronic pain encounters an obstacle immediately: Neither has a singular pathophysiology. Furthermore, MDD and, to a significant extent, chronic pain are defined more by their symptoms than by a presumed etiology and pathogenesis.
Why does this matter to a busy clinician?
Explicitly or implicitly, we often align our treatment approaches with what we assume is the underlying pathophysiology of the conditions we are addressing. An overview of shared pathophysiology of chronic pain conditions and MDD therefore can be useful in practice.
What is chronic pain? Defined as “pain that persists past the healing phase following an injury,”1 chronic pain often is subdivided into 4 types2,3:
- nociceptive (caused by a lesion or potential tissue damage)
- inflammatory
- neuropathic (spontaneous pain or hypersensitivity to pain related to neurologic illness or injury)
- functional (hypersensitivity to pain due to abnormal central processing of a normal input).
Although fibromyalgia often is categorized as a dysfunctional pain syndrome, persons who suffer from it, much like those who suffer neuropathic pain, commonly report hyperalgesia (augmented sensitivity to painful stimuli), allodynia (abnormal pain response to non-noxious stimuli), and paresthesias. These shared clinical features of fibromyalgia and neuropathic pain are consistent with central sensitization, which suggests overlapping pathophysiology.4
Comorbidity between depression and pain is common. A 30% to 60% co-occurrence rate of MDD and chronic pain has been reported.5 Some subtypes of chronic pain, such as fibromyalgia, are so commonly comorbid with psychiatric conditions that they have spawned a scientific debate as to whether the conditions are most parsimoniously considered (1) separate illnesses with high comorbidity or (2) different symptomatic manifestations of a single underlying condition.6 Moreover, cumulative evidence suggests that chronic pain and depression do not just co-occur; each one facilitates development of the other, such that chronic pain is a strong predictor of subsequent onset of MDD, and vice versa.
When pain and depression are comorbid, they also tend to make treatment of each condition more difficult. For example, pain presents (1) a major obstacle to achieving remission when treating depression7,8 and (2) significant risk of relapse.9 A 3-year longitudinal study showed that painful symptoms substantially reduced the chance of recovery in a group of older depressed patients (n = 327). A substantially greater percentage of patients with MDD alone attained recovery (47%), compared with only 9% in whom MDD and painful symptoms were comorbid.10 Furthermore, a higher level of pain can delay remission when treating MDD,11 thus reducing the likelihood of an optimal outcome.12
Understanding shared processes. Recent developments in neuroscience and psycho-immunology point to the fact that comorbid pain and depression might be driven by overlapping pathophysiological processes in the brain and body. In the 2 parts of this article, we (1) review scientific understanding of these shared processes and (2) demonstrate how recent advances in the epidemiology, phenomenology, and etiology of chronic pain and MDD provide important clues for more effective diagnosis (Part 1) and treatment (Part 2, March 2016)—and, therefore, better outcomes. Our focus is primarily on the relationship between MDD and the best-studied comorbid chronic pain conditions: fibromyalgia, neuropathic pain, chronic back pain, and rheumatoid arthritis.
The societal burden of chronic pain conditions is enormous
A recent epidemiological study13 projected that as many as 100 million people in the United States—30.7% of the population—suffer some form of chronic pain, including arthritis and joint pain. A World Health Organization survey yielded a similar (and staggering) 37% prevalence of chronic pain in the population of 10 developed countries.14
Estimates are that various forms of neuropathic pain, including diabetic neuropathy, postherpetic neuralgia, trigeminal neuralgia, spinal cord injury, and radiculopathy, alone afflict as many as 26 million people worldwide, including approximately 1.5% of the U.S. population.15,16
Chronic low back pain is epidemic. With a projected point prevalence of 30%, the condition is the most common cause of activity limitation among people age <45, and the most frequent reason in the United States for visiting a physician.1
Functional somatic syndromes, including fibromyalgia and irritable bowel syndrome, impose an astounding strain on health care: These syndromes account for 25% to 50% of all outpatient visits, or approximately 400 million clinic visits annually in the United States.17
Why should you care about these numbers? The answer is that comorbidity among chronic pain, mood disorders, anxiety disorders, sleep disorders, cognitive impairment, fatigue, and chronic stress presents an enormous clinical challenge because it not only complicates the diagnosis of these conditions but also compromises treatment outcomes and imposes severe limitations on daily functioning and quality of life of those afflicted.5,17-24
A complex relationship and a daunting clinical challenge
Chronic pain enhances the risk of MDD by 2-fold to 5-fold. The risk appears to be mediated by the number of pain conditions rather than by the severity of pain.23 Some authors have noted a kind of dose-response relationship among pain, depression, and anxiety. Among patients who experienced chronic pain that affected 1 body region, the prevalence of generalized anxiety disorder (GAD) and MDD was 30% and 20%, respectively; in patients who experienced pain in ≥2 regions, the prevalence of GAD and MDD was elevated to 54% and 32%.25 Moreover, patients with fibromyalgia were 4.3 times more likely than healthy controls to develop MDD at some point in their lives and 4.7 times more likely to develop an anxiety disorder.26
Although women are more likely to suffer from fibromyalgia, the risk for people of either sex of developing subsequent MDD is comparable once the condition has developed.27 Overall, depression and anxiety are among the most common comorbidities of fibromyalgia, with prevalence ranging from 20% to 80% and 13% to 63.8%, respectively.28
High comorbidity between depression and pain also is relevant for patients with neuropathic pain. A survey from Australia reported depression in 34% and anxiety in 25% of patients with neuropathic pain.29 Pain severity tended to be enduring and associated with significantly impaired functioning. A significant percentage of patients suffering from rheumatoid arthritis and systemic lupus erythematosus tend to manifest anxiety and depression (93% to 94%), cognitive impairment (66%), fatigue (40%), and sleep disorders (72%).22
The relationship between depression and pain appears to be bidirectional. For example, recent studies demonstrate that 30% to 60% of depressed patients also suffer from a painful condition.5
The complex history of patients presenting with concomitant complaints of depression, anxiety, chronic pain, sleep disturbance, cognitive impairment, and fatigue present a daunting diagnostic task. Pain tends to be associated with greater fatigue and sleep disturbance, which in turn depletes a patient’s ability to enjoy life and enhances negative affect.19,20,30 The take-home message might be to screen all chronic pain patients for MDD, anxiety, and sleep disorders, and vice versa.
Furthermore, comorbidity among chronic pain, MDD, anxiety, and sleep disorders can introduce specific intricacies into our treatment approach. Although, in general, comorbidities tend to have a negative impact on treatment outcomes, many pharmacotherapeutic and non-drug interventions targeting chronic pain might ameliorate sleep problems, low energy, anxiety, depression, and anhedonia.18,20,30-32 On the other hand, we should consider that opioid treatment for chronic pain might represent a risk factor for subsequent depression. It is conceivable that chronic opioid treatment and associated sedation can erode self-efficacy and social relationships, thereby compromising sources of support.33,34 It is equally important to keep in mind that, even if we are successful in attaining remission when treating depression and pain, residual pain symptoms might persist, requiring more specific interventions.24
MDD and chronic pain each have, on their own, a well-established association with suicide attempts and completion. Researchers are investigating whether a pathophysiologic suicide-promoting synergy between the 2 disorders exists when they are comorbid (Box35-37).
Shared genetics and pathophysiology
Several candidate genes have been identified as risk genes for chronic pain, depression, and anxiety. One of those studied the most is 5-HTTLPR, involved in regulating synthesis of serotonin transporter. The short form of this gene has been implicated in a diverse set of conditions, including MDD, anxiety disorders, and substance abuse—and fibromyalgia. Other genes associated with the risk of MDD and pain disorders are ones that code for:
- serotonin 5-HT2A and 5-HT1A receptors
- catechol-O-methyltransferase, an enzyme involved in catecholamine metabolism
- dopamine D4 receptor
- proinflammatory cytokines interleukin-1 and interleukin-6.4
Both monoamines and inflammatory cytokines play a role in modulating γ-aminobutyric acid (GABA) and glutamate neurons, as well as glia cells constituting peripheral pain pathways and central circuits that participate in the pain response and regulation of mood.4,17,38
The ‘pain matrix’
Brain circuitry that is involved in processing pain stimuli—often referred to as the pain matrix—shares many structural components with circuitry involved in the stress response and emotional modulation.4 Emerging evidence indicates that the pain matrix might not be pain-specific but, instead, a complex aggregate of interconnected brain structures involved in evoking defensive responses to a number of offending stimuli, including pain, threat, danger, loss, and social rejection or isolation.
It is remarkable, in this regard, that imaging studies show that the dorsal anterior cingulate, central to experiencing negative affect in response to physical pain, also mediates distress in response to the “pain” of social exclusion.39 Emerging functional and structural imaging provides evidence of continuous reorganization of prefrontal cortices as a consequence of enduring chronic pain.1 Of particular interest are findings of (1) a reduction of gray matter in the dorsolateral prefrontal cortex (DLPFC) and (2) functional activation of the medial prefrontal cortex (mPFC), both of which correlate with the duration and experience of chronic back pain.1 It is tempting to speculate that structural decline of the DLPFC, observed in MDD and chronic pain, is linked to cognitive and executive function deficits, which are readily observed in patients with either disorder—given that DLPFC is a “hub” of the so-called “cognitive-executive functional network.”1,4
Likewise, the mPFC is a key component of the default mode network (DMN), a functional network also comprising the posterior cingulate cortex and hippocampus. DMN performs a diverse set of activities, including self-reflection, daydreaming, reminiscing, planning, processing of social information, and creative thinking. Negative neuroplastic changes in the DMN are a common finding in MDD and chronic pain, and might be associated with a tendency toward rumination and catastrophizing—key clinical manifestations of MDD and chronic pain—and linked with pervasive negative affect and sleep disturbance.4,32
Furthermore, functional and structural changes in the amygdala and hippocampus have been described in MDD, fibromyalgia, and neuropathic pain.4 Dysfunction of these limbic formations may be a contributing factor in the disruption of neuroendocrine, autonomic, and immune function, which could further contribute to aggravated mood and pain symptoms.4,17,40
Consequently, excessive hypothalamic-pituitary-adrenal axis and sympathetic activation, combined with elevation of proinflammatory cytokine production and release, likely plays a role in the pathophysiology of MDD and chronic pain disorders.4,17,40 Moreover, at cellular, subcellular, and molecular levels, chronic pain and MDD are associated with:
- perturbed neuron-glia relationships
- altered glutamatergic, GABA, glycine, substance-P, opioid, 5-HT, norepinephrine, and dopamine signaling
- dysfunction of intracellular signaling cascades and neurotrophic signaling.4,20,30,31,38
The Figure that describes how homeostatic function of prefrontal cortical-limbic circuitry is compromised in MDD and chronic pain—thus disrupting autonomic, neuroendocrine, and neuroimmune regulation.
Disturbance in monoamine signaling in chronic pain and MDD might give rise to profound anhedonia, cognitive impairment, anxiety, insomnia, sensitivity to stress, and inadequate functioning of descending pain-regulatory pathways, which primarily use norepinephrine and 5-HT.4,9,20,30,31,38 Using pharmacotherapeutic agents that successfully modulate monoamines, therefore, might ameliorate the function of brain networks innervated by neurotransmitter systems involved in the regulation of pain, mood, cognition, stress response, and sleep. Notably, the same monoamines serve as transmitters in descending pain pathways.
In summary, convergent evidence indicates that MDD and chronic pain states amplify each other, thus contributing to treatment resistance in both disorders.
On the bright side, timely and effective treatment of MDD might optimize the chance of remission and minimize the risk of enduring structural brain changes in MDD and chronic pain.1,4,31,32 The obverse is also true: Emphasizing the importance of the resolution of painful symptoms in the context of MDD, a study reported a significantly greater remission rate of 36.2% in those who had >50% reduction of pain on a visual analogue scale following treatment with a serotonin-norepinephrine reuptake inhibitor, compared with a 17.8% remission rate in persons who experienced <50% pain reduction on the scale.3
Editors’ note: In Part 2 of this article (March 2016), the authors review pharmacotherapeutic and non-drug strategies for managing comorbid chronic pain conditions and MDD.
1. Apkarian AV, Baliki MN, Geha PY. Towards a theory of chronic pain. Prog Neurobiol. 2009;87(2):81-97.
2. Verdu B, Decosterd I, Buclin T, et al. Antidepressants for the treatment of chronic pain. Drugs. 2008;68(18):2611-2632.
3. Woolf CJ; American College of Physicians, American Physiological Society. Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann Intern Med. 2004;140(6):441-451.
4. Maletic V, Raison CL. Neurobiology of depression, fibromyalgia and neuropathic pain. Front Biosci (Landmark Ed). 2009;14:5291-5338.
5. Bair MJ, Wu J, Damush TM, et al. Association of depression and anxiety alone and in combination with chronic musculoskeletal pain in primary care patients. Psychosom Med. 2008;70(8):890-897.
6. Cho HJ, Skowera A, Cleare A, et al. Chronic fatigue syndrome: an update focusing on phenomenology and pathophysiology. Curr Opin Psychiatry. 2006;19(1):67-73.
7. Fava M. Depression with physical symptoms: treating to remission. J Clin Psychiatry. 2003;64(suppl 7):24-28.
8. Bair MJ, Robinson RL, Eckert GJ, et al. Impact of pain on depression treatment response in primary care. Psychosom Med. 2004;66(1):17-22.
9. Ohayon MM. Specific characteristics of the pain/depression association in the general population. J Clin Psychiatry. 2004;65(suppl 12):5-9.
10. Geerlings SW, Twisk JW, Beekman AT, et al. Longitudinal relationship between pain and depression in older adults: sex, age and physical disability. Soc Psychiatry Psychiatr Epidemiol. 2002;37(1):23-30.
11. Karp JF, Scott J, Houck P, et al. Pain predicts longer time to remission during treatment of recurrent depression. J Clin Psychiatry. 2005;66(5):591-597.
12. Spijker J, de Graaf R, Bijl RV, et al. Determinants of persistence of major depressive episodes in the general population. Results from the Netherlands Mental Health Survey and Incidence Study (NEMESIS). J Affect Disord. 2004;81(3):231-240.
13. Johannes CB, Le TK, Zhou X, et al. The prevalence of chronic pain in United States adults: results of an Internet-based survey. J Pain. 2010;11(11):1230-1239.
14. Dzau VJ, Pizzo PA. Relieving pain in America: insights from an Institute of Medicine committee. JAMA. 2014;312(15):1507-1508.
15. Butera JA. Current and emerging targets to treat neuropathic pain. J Med Chem. 2007;50(11):2543-2546.
16. Offenbaecher M, Ackenheil M. Current trends in neuropathic pain treatments with special reference to fibromyalgia. CNS Spectr. 2005;10(4):285-297.
17. Goldenberg DL. Pain/depression dyad: a key to a better understanding and treatment of functional somatic syndromes. Am J Med. 2010;123(8):675-682.
18. Argoff CE. The coexistence of neuropathic pain, sleep, and psychiatric disorders: a novel treatment approach. Clin J Pain. 2007;23(1):15-22.
19. Zautra AJ, Fasman R, Parish BP, et al. Daily fatigue in women with osteoarthritis, rheumatoid arthritis, and fibromyalgia. Pain. 2007;128(1-2):128-135.
20. Finan PH, Smith MT. The comorbidity of insomnia, chronic pain, and depression: dopamine as a putative mechanism. Sleep Med Rev. 2013;17(3):173-183.
21. Senba E. A key to dissect the triad of insomnia, chronic pain, and depression. Neurosci Lett. 2015;589:197-199.
22. Torta R, Pennazio F, Ieraci V. Anxiety and depression in rheumatologic diseases: the relevance of diagnosis and management. Reumatismo. 2014;66(1):92-97.
23. Howe CQ, Robinson JP, Sullivan MD. Psychiatric and psychological perspectives on chronic pain. Phys Med Rehabil Clin N Am. 2015;26(2):283-300.
24. Gerrits MM, van Marwijk HW, van Oppen P, et al. Longitudinal association between pain, and depression and anxiety over four years. J Psychosom Res. 2015;78(1):64-70.
25. Manchikanti L, Pampati V, Beyer C, et al. Do number of pain conditions influence emotional status? Pain Physician. 2002;5(2):200-205.
26. Arnold LM. Biology and therapy of fibromyalgia. New therapies in fibromyalgia. Arthritis Res Ther. 2006;8(4):212.
27. Weir PT, Harlan GA, Nkoy FL, et al. The incidence of fibromyalgia and its associated comorbidities: a population-based retrospective cohort study based on International Classification of Diseases, 9th Revision codes. J Clin Rheumatol. 2006;12(3):124-128.
28. Fietta P, Fietta P, Manganelli P. Fibromyalgia and psychiatric disorders. Acta Biomed. 2007;78(2):88-95.
29. Gustorff B, Dorner T, Likar R, et al. Prevalence of self-reported neuropathic pain and impact on quality of life: a prospective representative survey. Acta Anaesthesiol Scand. 2008;52(1):132-136.
30. Boakye PA, Olechowski C, Rashiq S, et al. A critical review of neurobiological factors involved in the interactions between chronic pain, depression, and sleep disruption [published online May 28, 2015]. Clin J Pain. doi: 10.1097/ AJP.0000000000000260.
31. Jann MW, Slade JH. Antidepressant agents for the treatment of chronic pain and depression. Pharmacotherapy. 2007;27(11):1571-1587.
32. Nekovarova T, Yamamotova A, Vales K, et al. Common mechanisms of pain and depression: are antidepressants also analgesics? Front Behav Neurosci. 2014;8:99.
33. Smith K, Mattick RP, Bruno R, et al. Factors associated with the development of depression in chronic non-cancer pain patients following the onset of opioid treatment for pain. J Affect Disord. 2015;184:72-80.
34. Scherrer JF, Svrakic DM, Freedland KE, et al. Prescription opioid analgesics increase the risk of depression. J Gen Intern Med. 2014;29(3):491-499.
35. Fishbain DA, Lewis JE, Gao J. The pain suicidality association: a narrative review. Pain Med. 2014;15(11):1835-1849.
36. Elman I, Borsook D, Volkow ND. Pain and suicidality: insights from reward and addiction neuroscience. Prog Neurobiol. 2013;109:1-27.
37. Olié E, Guillaume S, Jaussent I, et al. Higher psychological pain during a major depressive episode may be a factor of vulnerability to suicidal ideation and act. J Affect Disord. 2010;120(1-3):226-230.
38. Han C, Pae CU. Pain and depression: a neurobiological perspective of their relationship. Psychiatry Investig. 2015;12(1):1-8.
39. Eisenberger NI, Lieberman MD, Williams KD. Does rejection hurt? An FMRI study of social exclusion. Science. 2003;302(5643):290-292.
40. Gracely RH, Ceko M, Bushnell MC. Fibromyalgia and depression [published online November 19, 2011]. Pain Res Treat. 2012;2012:486590. doi: 10.1155/2012/486590.
1. Apkarian AV, Baliki MN, Geha PY. Towards a theory of chronic pain. Prog Neurobiol. 2009;87(2):81-97.
2. Verdu B, Decosterd I, Buclin T, et al. Antidepressants for the treatment of chronic pain. Drugs. 2008;68(18):2611-2632.
3. Woolf CJ; American College of Physicians, American Physiological Society. Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann Intern Med. 2004;140(6):441-451.
4. Maletic V, Raison CL. Neurobiology of depression, fibromyalgia and neuropathic pain. Front Biosci (Landmark Ed). 2009;14:5291-5338.
5. Bair MJ, Wu J, Damush TM, et al. Association of depression and anxiety alone and in combination with chronic musculoskeletal pain in primary care patients. Psychosom Med. 2008;70(8):890-897.
6. Cho HJ, Skowera A, Cleare A, et al. Chronic fatigue syndrome: an update focusing on phenomenology and pathophysiology. Curr Opin Psychiatry. 2006;19(1):67-73.
7. Fava M. Depression with physical symptoms: treating to remission. J Clin Psychiatry. 2003;64(suppl 7):24-28.
8. Bair MJ, Robinson RL, Eckert GJ, et al. Impact of pain on depression treatment response in primary care. Psychosom Med. 2004;66(1):17-22.
9. Ohayon MM. Specific characteristics of the pain/depression association in the general population. J Clin Psychiatry. 2004;65(suppl 12):5-9.
10. Geerlings SW, Twisk JW, Beekman AT, et al. Longitudinal relationship between pain and depression in older adults: sex, age and physical disability. Soc Psychiatry Psychiatr Epidemiol. 2002;37(1):23-30.
11. Karp JF, Scott J, Houck P, et al. Pain predicts longer time to remission during treatment of recurrent depression. J Clin Psychiatry. 2005;66(5):591-597.
12. Spijker J, de Graaf R, Bijl RV, et al. Determinants of persistence of major depressive episodes in the general population. Results from the Netherlands Mental Health Survey and Incidence Study (NEMESIS). J Affect Disord. 2004;81(3):231-240.
13. Johannes CB, Le TK, Zhou X, et al. The prevalence of chronic pain in United States adults: results of an Internet-based survey. J Pain. 2010;11(11):1230-1239.
14. Dzau VJ, Pizzo PA. Relieving pain in America: insights from an Institute of Medicine committee. JAMA. 2014;312(15):1507-1508.
15. Butera JA. Current and emerging targets to treat neuropathic pain. J Med Chem. 2007;50(11):2543-2546.
16. Offenbaecher M, Ackenheil M. Current trends in neuropathic pain treatments with special reference to fibromyalgia. CNS Spectr. 2005;10(4):285-297.
17. Goldenberg DL. Pain/depression dyad: a key to a better understanding and treatment of functional somatic syndromes. Am J Med. 2010;123(8):675-682.
18. Argoff CE. The coexistence of neuropathic pain, sleep, and psychiatric disorders: a novel treatment approach. Clin J Pain. 2007;23(1):15-22.
19. Zautra AJ, Fasman R, Parish BP, et al. Daily fatigue in women with osteoarthritis, rheumatoid arthritis, and fibromyalgia. Pain. 2007;128(1-2):128-135.
20. Finan PH, Smith MT. The comorbidity of insomnia, chronic pain, and depression: dopamine as a putative mechanism. Sleep Med Rev. 2013;17(3):173-183.
21. Senba E. A key to dissect the triad of insomnia, chronic pain, and depression. Neurosci Lett. 2015;589:197-199.
22. Torta R, Pennazio F, Ieraci V. Anxiety and depression in rheumatologic diseases: the relevance of diagnosis and management. Reumatismo. 2014;66(1):92-97.
23. Howe CQ, Robinson JP, Sullivan MD. Psychiatric and psychological perspectives on chronic pain. Phys Med Rehabil Clin N Am. 2015;26(2):283-300.
24. Gerrits MM, van Marwijk HW, van Oppen P, et al. Longitudinal association between pain, and depression and anxiety over four years. J Psychosom Res. 2015;78(1):64-70.
25. Manchikanti L, Pampati V, Beyer C, et al. Do number of pain conditions influence emotional status? Pain Physician. 2002;5(2):200-205.
26. Arnold LM. Biology and therapy of fibromyalgia. New therapies in fibromyalgia. Arthritis Res Ther. 2006;8(4):212.
27. Weir PT, Harlan GA, Nkoy FL, et al. The incidence of fibromyalgia and its associated comorbidities: a population-based retrospective cohort study based on International Classification of Diseases, 9th Revision codes. J Clin Rheumatol. 2006;12(3):124-128.
28. Fietta P, Fietta P, Manganelli P. Fibromyalgia and psychiatric disorders. Acta Biomed. 2007;78(2):88-95.
29. Gustorff B, Dorner T, Likar R, et al. Prevalence of self-reported neuropathic pain and impact on quality of life: a prospective representative survey. Acta Anaesthesiol Scand. 2008;52(1):132-136.
30. Boakye PA, Olechowski C, Rashiq S, et al. A critical review of neurobiological factors involved in the interactions between chronic pain, depression, and sleep disruption [published online May 28, 2015]. Clin J Pain. doi: 10.1097/ AJP.0000000000000260.
31. Jann MW, Slade JH. Antidepressant agents for the treatment of chronic pain and depression. Pharmacotherapy. 2007;27(11):1571-1587.
32. Nekovarova T, Yamamotova A, Vales K, et al. Common mechanisms of pain and depression: are antidepressants also analgesics? Front Behav Neurosci. 2014;8:99.
33. Smith K, Mattick RP, Bruno R, et al. Factors associated with the development of depression in chronic non-cancer pain patients following the onset of opioid treatment for pain. J Affect Disord. 2015;184:72-80.
34. Scherrer JF, Svrakic DM, Freedland KE, et al. Prescription opioid analgesics increase the risk of depression. J Gen Intern Med. 2014;29(3):491-499.
35. Fishbain DA, Lewis JE, Gao J. The pain suicidality association: a narrative review. Pain Med. 2014;15(11):1835-1849.
36. Elman I, Borsook D, Volkow ND. Pain and suicidality: insights from reward and addiction neuroscience. Prog Neurobiol. 2013;109:1-27.
37. Olié E, Guillaume S, Jaussent I, et al. Higher psychological pain during a major depressive episode may be a factor of vulnerability to suicidal ideation and act. J Affect Disord. 2010;120(1-3):226-230.
38. Han C, Pae CU. Pain and depression: a neurobiological perspective of their relationship. Psychiatry Investig. 2015;12(1):1-8.
39. Eisenberger NI, Lieberman MD, Williams KD. Does rejection hurt? An FMRI study of social exclusion. Science. 2003;302(5643):290-292.
40. Gracely RH, Ceko M, Bushnell MC. Fibromyalgia and depression [published online November 19, 2011]. Pain Res Treat. 2012;2012:486590. doi: 10.1155/2012/486590.

Cariprazine for schizophrenia and bipolar I disorder
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
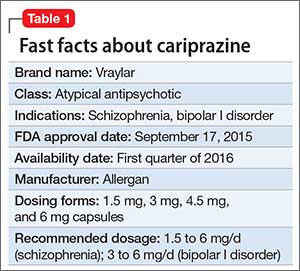
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
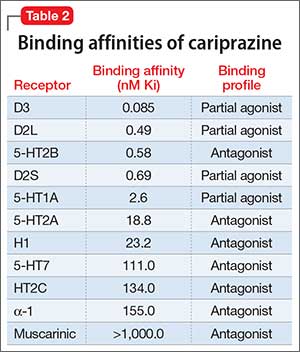
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
Cariprazine for schizophrenia and bipolar I disorder
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
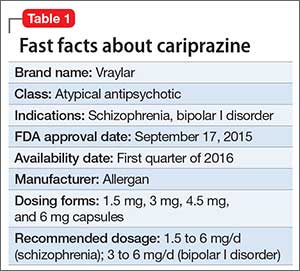
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
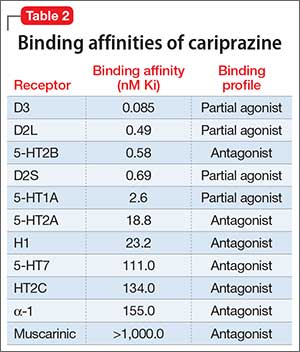
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
Cariprazine is a newly approved (September 2015) dopamine D3/D2 receptor partial agonist with higher affinity for the D3 receptor than for D2. The drug is FDA-indicated for treating schizophrenia and bipolar I disorder (BD I)1,2 (Table 1). In clinical trials, cariprazine alleviated symptoms of schizophrenia and mixed and manic symptoms of BD I, with minimal effect on metabolic parameters, the prolactin level, and cardiac conduction.
Clinical implications
Despite numerous developments in pharmacotherapeutics, people with schizophrenia or bipolar disorder continue to struggle with residual symptoms or endure treatments that produce adverse effects (AEs). In particular, metabolic issues, sedation, and cognitive impairment plague many current treatment options for these disorders.
Receptor blocking. As a dopamine D3-preferring D3/D2 partial agonist, cariprazine offers an alternative to antipsychotics that preferentially modulate D2 receptors. First-generation (typical) antipsychotics block D2 receptors; atypical antipsychotics block D2 receptors and 5-HT2A receptors. Dopamine partial agonists aripiprazole and brexpiprazole are D2-preferring, with minimal D3 effects. In contrast, cariprazine has a 6-fold to 8-fold higher affinity for D3 receptors than for D2 receptors, and has specificity for the D3 receptor that is 3 to 10 times higher than what aripiprazole has for the D3 receptor3-5 (Table 2).
Use in schizophrenia. Recommended dosage range is 1.5 to 6 mg/d. In Phase-III clinical trials, dosages of 3 to 9 mg/d produced significant improvement on the Positive and Negative Symptom Scale (PANSS) and on the Clinical Global Impression scale. Higher dosages (6 to 9 mg/d) showed early separation from placebo—by the end of Week 1—but carried a dosage-related risk of AEs, leading the FDA to recommend 6 mg/d as the maximum dosage.1,6-8
Use in manic or mixed episodes of BD I. Recommended dosage range is 3 to 6 mg/d. In clinical trials, dosages in the range of 3 to 12 mg/d were effective for acute manic or mixed symptoms; significant improvement in the Young Mania Rating Scale (YMRS) score was seen as early as Day 4. Dosages >6 mg/d yielded no additional benefit and were associated with increased risk of AEs.9-12
Pharmacologic profile, adverse effects. Cariprazine has a pharmacologic profile consistent with the generally favorable metabolic profile and lack of anticholinergic effects seen in clinical trials. In short- and long-term trials, the drug had minimal effects on prolactin, blood pressure, and cardiac conduction.13
Across clinical trials for both disorders, akathisia and parkinsonism were among more common AEs of cariprazine. Both AEs were usually mild, resulting in relatively few premature discontinuations from trials. Parkinsonism appeared somewhat dosage-related; akathisia had no clear relationship to dosage.
How it works
The theory behind the use of partial agonists, including cariprazine, is that these agents restore homeostatic balance to neurochemical circuits by:
- decreasing the effects of endogenous neurotransmitters (dopamine tone) in regions of the brain where their transmission is excessive, such as mesolimbic regions in schizophrenia or mania
- simultaneously increasing neurotransmission in regions where transmission of endogenous neurotransmitters is low, such as the prefrontal cortex in schizophrenia
- exerting little effect in regions where neurotransmitter activity is normal, such as the pituitary gland.
- simultaneously
Cariprazine has higher binding affinity for dopamine D3 receptors (Ki 0.085 nM) than for D2L receptors (Ki 0.49 nM) and D2S receptors (Ki 0.69 nM). The drug also has strong affinity for serotonin receptor 5-HT2B; moderate affinity for 5-HT1A; and lower affinity for 5-HT2A, histamine H1, and 5-HT7 receptors. Cariprazine has little or no affinity for adrenergic or cholinergic receptors.14In patients with schizophrenia, as measured on PET scanning, a dosage of 1.5 mg/d yielded 69% to 75% D2/D3 receptor occupancy. A dosage of 3 mg/d yielded >90% occupancy.
Search for an understanding of action continues. The relative contribution of D3 partial agonism, compared with D2 partial agonism, is a subject of ongoing basic scientific and clinical research. D3 is an autoreceptor that (1) controls phasic, but not tonic, activity of dopamine nerve cells and (2) mediates behavioral abnormalities induced by glutamate and N-methyl-D-aspartate receptor antagonists.5,12 In animal studies, D3-preferring agents have been shown to exert pro-cognitive effects and improve anhedonic symptoms.
Pharmacokinetics
Cariprazine is a once-daily medication with a relatively long half-life that can be taken with or without food. Dosages of 3 to 12 mg/d yield a fairly linear, dose-proportional increase in plasma concentration. The peak serum concentration for cariprazine is 3 to 4 hours under fasting conditions; taking the drug with food causes a slight delay in absorption but does not have a significant effect on the area under the curve. Mean half-life for cariprazine is 2 to 5 days over a dosage range of 1.5 to 12.5 mg/d in otherwise healthy adults with schizophrenia.1
Cariprazine is metabolized primarily by cytochrome P450 (CYP) 3A4. It is a weak inhibitor of CYP2D6 and CYP3A4.1 Hepatic metabolism of cariprazine produces 2 active metabolites: desmethyl-cariprazine (DCAR) and didesmethyl-cariprazine (DDCAR), both of which are equipotent to cariprazine. After multiple dose administration, mean cariprazine and DCAR levels reach steady state in 1 to 2 weeks; DDCAR, in 4 to 8 weeks. The systemic exposure and serum levels of DDCAR are roughly 3-fold greater than cariprazine because of the longer elimination half-life of DDCAR.1
Efficacy in schizophrenia
The efficacy of cariprazine in schizophrenia was established by 3 six-week, randomized, placebo-controlled trials. Two trials were fixed-dosage; a third used 2 flexible dosage ranges. The primary efficacy measure was change from baseline in the total score of the PANSS at the end of Week 6, compared with placebo. In all trials, patients were adults (age 18 to 60) who met DSM-IV-TR criteria for schizophrenia and had a PANSS score between 80 and 120 at screening and baseline.
Study 1 (n = 711) compared dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d with placebo.7 All cariprazine dosages and an active control (risperdone) were superior to placebo in reducing symptoms of schizophrenia, as measured by the PANSS. The placebo-subtracted differences on PANSS score at 6 weeks for dosages of 1.5 mg/d, 3 mg/d, and 4.5 mg/d were –7.6, –8.8, –10.4, respectively (significant at 95% CI).
Study 2 (n = 151) compared 3 mg/d and 6 mg/d dosages of cariprazine with placebo.1 Both dosages and an active control (aripiprazole) were superior to placebo in reducing PANSS scores. Placebo-subtracted differences on PANSS score at 6 weeks for dosages of 3 mg/d and 6 mg/day were –6.0, –8.8, respectively (significant at 95% CI).
Study 3 (n = 147) was a fixed-flexible dosage trial comparing cariprazine, 3 to 6 mg/d and 6 to 9 mg/d dosage ranges, to placebo.8 Both ranges were superior to placebo in reducing symptoms on PANSS. Placebo-subtracted differences from placebo on PANSS at 6 weeks for cariprazine 3 to 6 or 6 to 9 mg/d were –6.8, –9.9, respectively (significant at 95% CI).
These trials established the efficacy of cariprazine for acute schizophrenia at dosages ranging from 1.5 to 9 mg/d. Although there was a modest trend toward higher efficacy at higher dosages, there was a dose-related increase in certain adverse reactions (extrapyramidal symptoms [EPS]) at dosages >6 mg/d.1
Efficacy in bipolar disorder
The efficacy of cariprazine for acute treatment of manic or mixed episodes of BD I was established in 3 randomized, placebo-controlled, flexibly dosed 3-week trials. In all trials, patients were adults (age 18 to 65) who met DSM-IV-TR criteria for BD I with manic or mixed episodes and with or without psychotic features (YMRS score, ≥20). The primary efficacy measure in the 3 trials was a change from baseline in the total YMRS score at the end of Week 3, compared with placebo.
Study 1 (n = 492) compared 2 flexibly dosed ranges of cariprazine (3 to 6 mg/d and 6 to 12 mg/d) with placebo.10 Both dosage ranges were superior to placebo in reducing mixed and manic symptoms, as measured by reduction in the total YMRS score. Placebo-subtracted differences in YMRS scores from placebo at Week 3 for cariprazine 3 to 6 mg/d and 6 to 12 mg/d were –6.1, –5.9, respectively (significant at 95% CI). The higher range offered no additional advantage over the lower range.
Study 2 (n = 235) compared flexibly dosed cariprazine, 3 to 12 mg/d, to placebo.11 Cariprazine was superior to placebo in reducing bipolar symptoms as measured by the YMRS. The difference between cariprazine 3 to 12 mg/d and placebo on the YMRS score at Week 3 was –6.1 (significant at 95% CI).
Study 3 (n = 310) compared flexibly dosed cariprazine, 3 to 12 mg/d, with placebo.15 Again, cariprazine was superior to placebo in reducing the YMRS score at Week 3: difference, –4.3 (significant at 95% CI).
These trials establish the efficacy of cariprazine in treating acute mania or mixed BD I episodes at dosages ranging from 3 to 12 mg/d. Dosages >6 mg/d did not offer additional benefit over lower dosages, and resulted in a dosage-related increase in EPS at dosages >6 mg/d.16
Tolerability
Cariprazine generally was well tolerated in short-term trials for schizophrenia and BD I. The only treatment-emergent adverse event reported for at least 1 treatment group in all trials at a rate of ≥10%, and at least twice the rate seen with placebo was akathisia. Adverse events reported at a lower rate than placebo included EPS (particularly parkinsonism), restlessness, headache, insomnia, fatigue, and gastrointestinal distress. The discontinuation rate due to AEs for treatment groups and placebo-treated patients generally was similar. In schizophrenia Study 3, for example, the discontinuation rate due to AEs was 13% for placebo; 14% for cariprazine, 3 to 6 mg/d; and 13% for cariprazine, 6 to 9 mg/d.1 48-Week open-label safety study. Patients with schizophrenia received open-label cariprazine for as long as 48 weeks.7 Serious adverse events were reported in 12.9%, including 1 death (suicide); exacerbation of symptoms of schizophrenia (4.3%); and psychosis (2.2%). Treatment-emergent adverse events reported in at least 10% of patients included akathisia (14.0%), insomnia (14.0%), and weight gain (11.8%). The mean change in laboratory values, blood pressure, pulse rate, and electrocardiographic parameters was clinically insignificant.
Other studies. In a 16-week, open-label extension study of patients with BD I, the major tolerability issue was akathisia. This AE developed in 37% of patients and led to a 5% withdrawal rate.12
In short- and long-term studies for either indication, the effect of the drug on metabolic parameters appears to be small. In studies with active controls, potentially significant weight gain (>7%) was greater for aripiprazole and risperidone than for cariprazine.6,7 The effect on the prolactin level was minimal. There do not appear to be clinically meaningful changes in laboratory values, vital signs, or QT interval.
Unique clinical issues
Preferential binding. Cariprazine is the third dopamine partial agonist approved for use in the United States; unlike the other 2—aripiprazole and brexpiprazole—cariprazine shows preference for D3 receptors over D2 receptors. The exact clinical impact of a preference for D3 and the drug’s partial agonism of 5-HT1A has not been fully elucidated.
EPS, including akathisia and parkinsonism, were among common adverse events. Both were usually mild, with 0.5% of schizophrenia patients and 2% of BD I patients dropping out of trials because of any type of EPS-related AEs.
Why Rx? On a practical medical level, reasons to prescribe cariprazine likely include:
- minimal effect on prolactin
- relative lack of effect on metabolic parameters, including weight (cariprazine showed less weight gain than risperidone or aripiprazole control arms in trials).
Dosing
The recommended dosage of cariprazine for schizophrenia ranges from 1.5 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
The recommended dosages of cariprazine for mixed and manic episodes of BD I range from 3 to 6 mg/d. The recommended starting dosage is 1.5 mg/d, which can be increased to 3 mg on Day 2, with further upward dosage adjustments of 1.5 to 3 mg/d, based on clinical response and tolerability.1
Other key aspects of dosing to keep in mind:
- Because of the long half-life and 2 equipotent active metabolites of cariprazine, any changes made to the dosage will not be reflected fully in the serum level for 2 weeks.
- Administering the drug with food slightly delays, but does not affect, the extent of absorption.
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 inhibitor; the recommended starting dosage of cariprazine is 1.5 mg every other day with a maximum dosage of 3 mg/d when it is administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4 inducer, this practice is not recommended.1
- Because the drug is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a CYP3A4 Because data are not available regarding concomitant use of cariprazine with a strong CYP3A4
Contraindications
Cariprazine carries a FDA black-box warning of increased mortality in older patients who have dementia-related psychosis, as other atypical antipsychotics do. Clinical trials produced few data about the use of cariprazine in geriatric patients; no data exist about use in the pediatric population.1
Metabolic, prolactin, and cardiac concerns about cariprazine appeared favorably minor in Phase-III and long-term safety trials. Concomitant use of cariprazine with any strong inducer of CYP3A4 has not been studied, and is not recommended. Dosage reduction is recommended when using cariprazine concomitantly with a CYP3A4 inhibitor.1
In conclusion
The puzzle in neuropsychiatry has always been to find ways to produce different effects in different brain regions—with a single drug. Cariprazine’s particular binding profile—higher affinity and higher selectivity for D3 receptors than for D2 receptors compared with either aripiprazole or brexpiprazole—may secure a role for it in managing psychosis and mood disorders.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
1. Vraylar [package insert]. Parsippany, NJ: Actavis Pharma, Inc.; 2015.
2. McCormack PL, Cariprazine: first global approval. Drugs. 2015;75(17):2035-2043.
3. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
4. Potkin, S, Keator, D, Mukherjee J, et al. P. 1. E 028 dopamine D3 and D2 receptor occupancy of cariprazine in schizophrenic patients. Eur Neuropsychopharmacology. 2009;19(suppl 3):S316.
5. Veselinovicˇ T, Paulzen M, Gründer G. Cariprazine, a new, orally active dopamine D2/3 receptor partial agonist for the treatment of schizophrenia, bipolar mania and depression. Expert Rev Neurother. 2013;13(11):1141-1159.
6. Cutler A, Mokliatchouk O, Laszlovszky I, et al. Cariprazine in acute schizophrenia: a fixed-dose phase III, randomized, double-blind, placebo- and active-controlled trial. Abstract presented at: 166th Annual Meeting of the American Psychiatric Association; May 18-22, 2013; San Francisco, CA.
7. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
8. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
9. Bose A, Starace A, Lu, K, et al. Cariprazine in the treatment of acute mania in bipolar disorder: a double-blind, placebo-controlled, phase III trial. Poster presented at: 16th Annual Meeting of the College of Psychiatric and Neurologic Pharmacists; April 21-24, 2013; Colorado Springs, CO.
10. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
11. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
12. Ketter, T. A phase III, open-label, 16-week study of flexibly dosed cariprazine in 402 patients with bipolar I disorder. Presented at: 53rd Annual Meeting of the New Clinical Drug Evaluation Unit; May 28-31, 2013; Hollywood, FL.
13. Bose A, Li D, Migliore R. The efficacy and safety of the novel antipsychotic cariprazine in the acute exacerbation of schizophrenia. Poster presented at: 50th Annual Meeting of the New Clinical Drug Evaluation Unit; June 14-17, 2010; Boca Raton, FL.
14. Citrome L. Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol. 2013;9(2):193-206.
15. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
16. Vieta E, Durgam S, Lu K, et al. Effect of cariprazine across the symptoms of mania in bipolar I disorder: analyses of pooled data from phase II/III trials. Eur Neuropsycholpharmacol. 2015;25(11):1882-1891.
Management of epidermal growth factor receptor inhibitor-associated rash: a systematic review
New therapies for antiemetic prophylaxis for chemotherapy
A number of new advances have occurred in the management of chemotherapy-related nausea and vomiting (CINV). A new neurokinin-1 receptor antagonist (NK1RA), netupitant, has been combined with palonosetron in a single oral tablet for treating the effects of moderately emetogenic chemotherapy (MEC) and highly emetogenic chemotherapy (HEC). Rolapitant, another NK1RA, unlike aprepitant, has a long half-life and does not block CYP-3A4 and therefore has fewer drug interactions. Olanzapine reduces nausea more effectively than aprepitant in patients who are receiving HEC and is a better rescue antiemetic than is metoclopramide. Ginger lacks efficacy as an antiemetic agent for CINV. Although there was some evidence in a pilot study of gabapentin as an antiemetic, it was no better in reducing CINV than was placebo. Compliance to guidelines in multiple settings ranges from 50%-60% but is improved by computerized order entry of antiemetics and recommendations displayed with chemotherapy.
Click on the PDF icon at the top of this introduction to read the full article.
A number of new advances have occurred in the management of chemotherapy-related nausea and vomiting (CINV). A new neurokinin-1 receptor antagonist (NK1RA), netupitant, has been combined with palonosetron in a single oral tablet for treating the effects of moderately emetogenic chemotherapy (MEC) and highly emetogenic chemotherapy (HEC). Rolapitant, another NK1RA, unlike aprepitant, has a long half-life and does not block CYP-3A4 and therefore has fewer drug interactions. Olanzapine reduces nausea more effectively than aprepitant in patients who are receiving HEC and is a better rescue antiemetic than is metoclopramide. Ginger lacks efficacy as an antiemetic agent for CINV. Although there was some evidence in a pilot study of gabapentin as an antiemetic, it was no better in reducing CINV than was placebo. Compliance to guidelines in multiple settings ranges from 50%-60% but is improved by computerized order entry of antiemetics and recommendations displayed with chemotherapy.
Click on the PDF icon at the top of this introduction to read the full article.
A number of new advances have occurred in the management of chemotherapy-related nausea and vomiting (CINV). A new neurokinin-1 receptor antagonist (NK1RA), netupitant, has been combined with palonosetron in a single oral tablet for treating the effects of moderately emetogenic chemotherapy (MEC) and highly emetogenic chemotherapy (HEC). Rolapitant, another NK1RA, unlike aprepitant, has a long half-life and does not block CYP-3A4 and therefore has fewer drug interactions. Olanzapine reduces nausea more effectively than aprepitant in patients who are receiving HEC and is a better rescue antiemetic than is metoclopramide. Ginger lacks efficacy as an antiemetic agent for CINV. Although there was some evidence in a pilot study of gabapentin as an antiemetic, it was no better in reducing CINV than was placebo. Compliance to guidelines in multiple settings ranges from 50%-60% but is improved by computerized order entry of antiemetics and recommendations displayed with chemotherapy.
Click on the PDF icon at the top of this introduction to read the full article.
Managing interstitial lung disease detected on CT during lung cancer screening
Primary care physicians are playing a bigger role in evaluating the incidental finding of interstitial lung diseases since the recent publication of guidelines recommending computed tomography (CT) to screen for lung cancer.
In August 2011, the National Cancer Institute published its findings from the National Lung Screening Trial, which demonstrated a 20% reduction in mortality from lung cancer in patients at high risk screened with low-dose CT.1 Based on these results, the American Cancer Society, the American College of Chest Physicians, the American Society of Clinical Oncology, and the National Comprehensive Cancer Network recommended annual screening for lung cancer with low-dose CT in adults ages 55 to 74 who have a 30-pack-year smoking history and who currently smoke or have quit within the past 15 years.2 In December 2013, the US Preventive Services Task Force published similar guidelines but increased the age range to include high-risk patients ages 55 to 80.3
Bach et al4 estimated that, in 2010 in the United States, 8.6 million people met the criteria used in the National Lung Screening Trial for low-dose CT screening. These are the same criteria as in the multisociety recommendations cited above.2 With such large numbers of patients eligible for CT screening, internists and other primary care physicians are undoubtedly encountering the incidental discovery of nonmalignant pulmonary diseases such as interstitial lung disease.
This article reviews the radiographic characteristics of the most common interstitial lung diseases the internist may encounter on screening CT in long-term smokers.
Referral to a specialist has been associated with lower rates of morbidity and death,5 and a diagnosis of interstitial lung disease should be confirmed by a pulmonologist and a radiologist specializing in differentiating the subtypes. But the primary care physician now plays a critical role in recognizing the need for further evaluation.
HOW COMMON IS INTERSTITIAL LUNG DISEASE IN SMOKERS?
Several studies have published data on the prevalence of interstitial lung disease in patients undergoing low-dose CT for lung cancer screening.
A trial at Mayo Clinic in current and former smokers identified “diffuse lung disease” in 9 (0.9%) of 1,049 participants.6
A trial in Ireland identified idiopathic pulmonary fibrosis in 6 (1.3%) of 449 current smokers who underwent low-dose CT screening for lung cancer.7
Sverzellati et al8 evaluated 692 participants in the Multicentric Italian Lung Detection CT screening study and reported a respiratory bronchiolitis pattern in 109 (15.7%), a usual interstitial pneumonia pattern in 2 (0.3%), and other patterns of chronic interstitial pneumonia in 26 (3.8%).
The National Lung Screening Trial reported that the frequency of “clinically significant” incidental findings (including pulmonary fibrosis) in all participants was 7.5%.1 A retrospective analysis of 884 participants at a single site in this trial identified interstitial lung abnormalities in 86 participants (9.7%).9 These abnormalities were further categorized as nonfibrotic in 52 (5.9%) of 884, fibrotic in 19 (2.1%) of 884, and mixed fibrotic and nonfibrotic in 15 (1.7%) of 884.
Follow-up CT at 2 years in this trial demonstrated improvement in 50% and progression in 11% of patients who had nonfibrotic abnormalities, while fibrotic abnormalities improved in no cases and progressed in 37%. Interstitial lung abnormalities were more common in those who currently smoked and in those with more pack-years of cigarette smoking.9
In sum, these trials suggest that low-dose CT screening for lung cancer can detect the most common forms of interstitial lung disease in this at-risk population and can characterize them as fibrotic or nonfibrotic, a distinction important for prognosis and subsequent management.
NONFIBROTIC VS FIBROTIC DISEASE
It is important to distinguish between nonfibrotic and fibrotic interstitial lung disease, as fibrotic disease carries a worse prognosis and is treated differently.
Features of nonfibrotic interstitial lung disease:
- Ground-glass opacities
- Nodules
- Mosaic attenuation or consolidation.
Features of fibrotic interstitial lung disease:
- Combination of ground-glass opacities and reticulation
- Reticulation by itself
- Traction bronchiectasis
- Honeycombing
- Loss of lung volume.
NONFIBROTIC INTERSTITIAL LUNG DISEASES
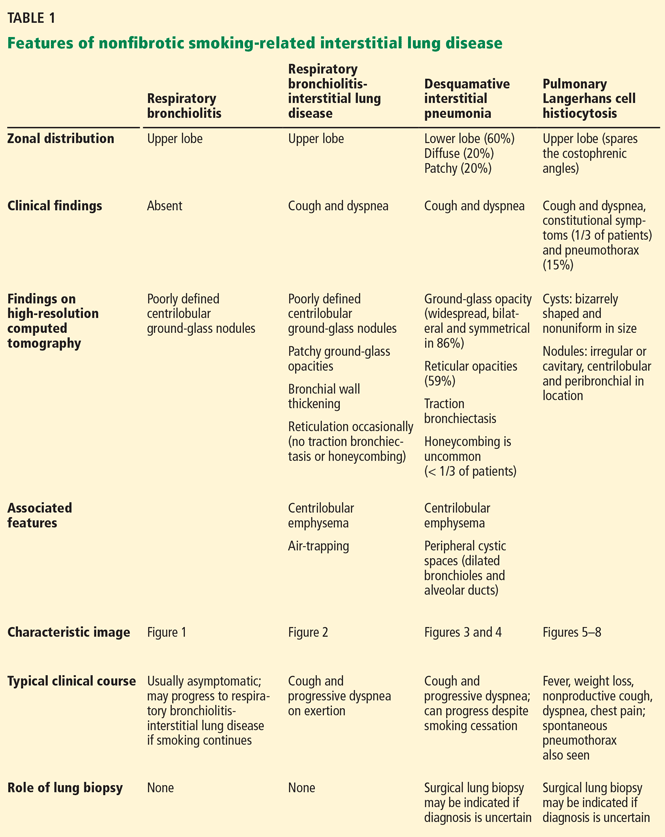
Given the strong likelihood that a patient undergoing screening CT is either a current or former smoker, physicians may encounter, in addition to emphysema and lung cancer, the following smoking-related interstitial lung diseases, which are primarily nonfibrotic and which frequently coexist (Table 1):
- Respiratory bronchiolitis
- Respiratory bronchiolitis-interstitial lung disease
- Desquamative interstitial pneumonia
- Pulmonary Langerhans cell histiocytosis.
Respiratory bronchiolitis
Respiratory bronchiolitis occurs mostly in smokers and does not necessarily lead to respiratory symptoms in all patients.10 It cannot always be identified radiographically but occasionally appears as predominantly upper-lobe, patchy ground-glass opacities or ill-defined centrilobular nodules without evidence of fibrosis (Figure 1).

Respiratory bronchiolitis-interstitial lung disease
In rare cases, respiratory bronchiolitis leads to peribronchial fibrosis invading the alveolar walls, which is then classified as respiratory bronchiolitis-interstitial lung disease.11 The CT findings in respiratory bronchiolitis-interstitial lung disease are upper-lobe-predominant centrilobular ground-glass nodules, patchy ground-glass opacities, and bronchial wall thickening (Figure 2).10 Occasionally, mild reticulation is noted without honeycombing. Mild air trapping can be seen in the lower lobes, with centrilobular emphysema in the upper lobes.12

The only successful therapy for respiratory bronchiolitis and respiratory bronchiolitis-interstitial lung disease is smoking cessation. Finding either of these diseases should prompt aggressive counseling by the internist and consideration of referral to a specialist in interstitial lung disease.
Desquamative interstitial pneumonia
Although pathologically different from respiratory bronchiolitis-interstitial lung disease, desquamative interstitial pneumonia has a similar clinical and radiographic presentation. Because their features significantly overlap, they are considered a pathomorphologic continuum, representing degrees of severity of the same disease process caused by prolonged tobacco inhalation.10,13
Widespread ground-glass opacities are the predominant CT finding. These are bilateral and symmetric in distribution in 86%, basal and peripheral in 60%, patchy in 20%, and diffuse in 20% (Figure 3).14 Other frequent findings are mild reticulation with traction bronchiectasis and coexistent emphysema (Figure 4).15 The small peripheral cystic spaces noted in this disease most likely represent dilated bronchioles and alveolar ducts rather than honeycombing.16


No additional treatment beyond elimination of smoking has been proven effective for desquamative interstitial pneumonia, and patients who manage to quit smoking generally have a favorable prognosis.17,18
Pulmonary Langerhans cell histiocytosis
The combination of upper-lobe-predominant cysts and nodules in a young heavy smoker are diagnostic of pulmonary Langerhans cell histiocytosis. The cysts are bizarrely shaped, thin- or thick-walled, and nonuniform in size (Figure 5). The irregular cavitary nodules are centrilobular. The disease characteristically spares the costophrenic angles.

Spontaneous pneumothorax is the initial clinical presentation in 15% of patients.16 In the early stages of the disease (nodule-predominant disease without cysts), infection and metastatic disease need to be excluded (Figure 6). In the later stages, the cysts become coalescent, making the distinction between this disease and “burned-out” lymphangioleiomyomatosis or severe emphysema extremely difficult (Figure 7).17 Smoking cessation and corticosteroids are the mainstay of medical therapy for pulmonary Langerhans cell histiocytosis, and about 50% of patients who quit smoking and receive corticosteroids demonstrate partial or complete clearing of the radiographic abnormalities and symptoms (Figure 8).



FIBROTIC INTERSTITIAL LUNG DISEASES
If CT identifies a diffuse fibrotic pattern, the two most common possibilities (Table 2) are:
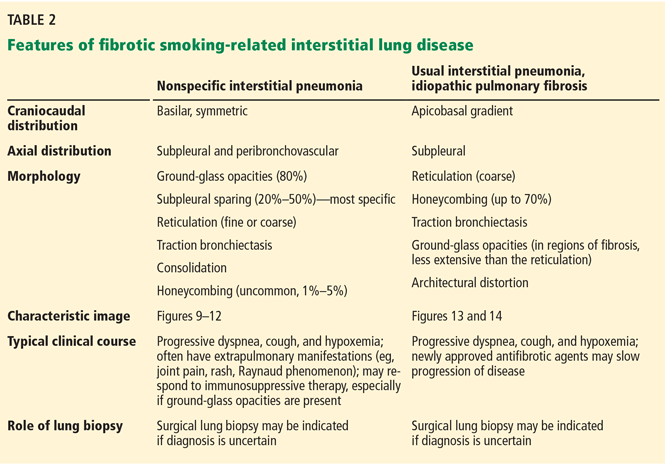
- Nonspecific interstitial pneumonia
- Usual interstitial pneumonia.
As noted above, these carry a worse prognosis than the nonfibrotic interstitial lung diseases.
Nonspecific interstitial pneumonia
While most frequently idiopathic, the nonspecific interstitial pneumonia pattern can often be seen in connective tissue diseases. It has also been associated with chronic hypersensitivity pneumonitis, drug toxicity, and slowly resolving diffuse alveolar damage.19 Although it is not the only pathologic pattern in interstitial lung disease associated with connective tissue disease, it is the most common pattern in systemic sclerosis, systemic lupus erythematosus, dermatomyositis-polymyositis, and mixed connective tissue disease.20
The parenchymal changes are typically subpleural and symmetric in distribution (Figure 9). In about one-third of cases, there is a peribronchovascular distribution of the abnormalities (Figure 10).


Ground-glass opacities are the dominant imaging findings, seen in 80% of cases.18 In advanced disease (also referred to as fibrotic nonspecific interstitial pneumonia), patients have accompanying fine or coarse reticular opacities, traction bronchiectasis, and consolidation (Figure 11). Honeycombing is seen in 1% to 5% of patients.21

The most specific sign of nonspecific interstitial pneumonia is sparing of the immediate subpleural lung, apparent in 30% to 50% of patients (Figure 12).22 Subpleural sparing with a peribronchovascular distribution of abnormalities, absence of lobular areas with decreased attenuation, and lack of honeycombing are imaging features that increase the diagnostic confidence of nonspecific interstitial pneumonia (Table 3).23 Clinically, compared with those who have usual interstitial pneumonia (see below), patients are younger and more of them are female. These patients also present with extrapulmonary manifestations such as joint involvement, rash, and Raynaud phenomenon. Therefore, these associated symptoms on presentation can help distinguish nonspecific interstitial pneumonia or usual interstitial pneumonia related to connective tissue disease from the idiopathic forms.

The first step in managing nonspecific interstitial pneumonia is to remove all potential exposure to inhaled substances or to drugs. Although immunosuppressive therapy has never been studied in a randomized controlled trial in this disease, numerous reports suggest that patients may respond to prednisone and to steroid-sparing immunosuppressants.24

In several studies, survival rates in nonspecific interstitial pneumonia were significantly greater than in usual interstitial pneumonia independent of the treatment strategy. In long-term follow-up of patients with idiopathic nonspecific interstitial pneumonia treated with immunosuppressive therapy, two-thirds remained stable or improved.25–27
Although most connective tissue diseases cause a lung pattern of nonspecific interstitial pneumonia, some (eg, rheumatoid arthritis) may present with a pattern of usual interstitial pneumonia. In these cases and in those of advanced fibrotic nonspecific interstitial pneumonia, the prognosis is worse, as the disease is less responsive to immunosuppressive therapy.20
Usual interstitial pneumonia
Usual interstitial pneumonia is the most severe form of lung fibrosis. Most cases are idiopathic and are termed idiopathic pulmonary fibrosis. Other causes of the usual interstitial pneumonia pattern include domestic and occupational environmental exposures, connective tissue disease, and drug toxicity.28 An epidemiologic association between smoking and usual interstitial pneumonia is well documented.28
Idiopathic pulmonary fibrosis typically affects men ages 50 to 70. Because its risk factors coincide with those of lung cancer, there is a high likelihood of detecting idiopathic pulmonary fibrosis early in this screening population. It has an especially poor prognosis, with a mean survival of 2 to 5 years from the time of diagnosis.18
The distribution of disease in usual interstitial pneumonia is characteristically subpleural and basal. CT features include coarse subpleural reticulation and honeycombing combined with traction bronchiectasis or bronchiolectasis and architectural distortion (Figure 13).18 Honeycombing is the most specific and key diagnostic CT finding for establishing a definitive diagnosis of usual interstitial pneumonia.29 However, ground-glass opacities are present in most patients, typically in the region of interstitial fibrosis, and are always less extensive than the reticulation.30 The findings demonstrate morphologic heterogeneity, with areas of fibrosis adjacent to areas of normal lung (Figure 14).
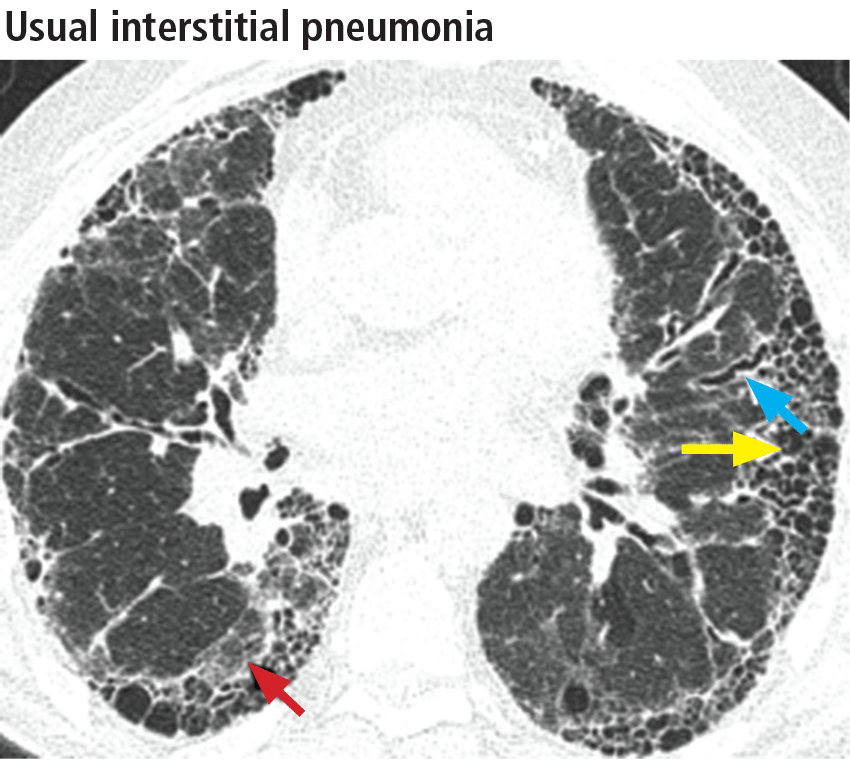

In addition to the aforementioned imaging features, the 2011 American Thoracic Society and European Respiratory Society joint guidelines for the CT diagnosis of usual interstitial pneumonia patterns require the absence of atypical features that suggest an alternative diagnosis, including those seen in nonspecific interstitial pneumonia, such as an upper, midlung, or peribronchovascular distribution and extensive ground-glass attenuation.28 Mild mediastinal lymphadenopathy (usually < 1.5 cm in the short axis) is common in usual interstitial pneumonia.31
Because other chronic interstitial pneumonias that may resemble usual interstitial pneumonia have a more favorable course and may respond to immunosuppressive therapy, establishing an early and accurate diagnosis is of the utmost importance.5 Additionally, the emergence of possible new therapies for idiopathic pulmonary fibrosis makes early referral to a specialist paramount in these cases. Recent studies have demonstrated significant slowing of the progression of disease in idiopathic pulmonary fibrosis with both pirfenidone and nintedanib.32,33
DIAGNOSIS AND MANAGEMENT

The diagnosis of these nonfibrotic and fibrotic lung diseases is complex. In all cases in which interstitial lung disease is detected on screening CT for lung cancer, the internist should strongly consider further evaluation with dedicated high-resolution CT and early referral to a specialist (Figure 15).
Because smoking cessation is the only recommended treatment for nonfibrotic smoking-related interstitial lung diseases, particular emphasis on smoking cessation counseling is essential.
Referral for bronchoscopy with transbronchial lung biopsy is generally not helpful in the diagnosis of the interstitial lung diseases discussed in this article unless there is a need to rule out infection or neoplasm.34 Referral for surgical lung biopsy may be indicated in some cases of suspected pulmonary Langerhans cell histiocytosis, desquamative interstitial pneumonia, nonspecific interstitial pneumonia, or usual interstitial pneumonia if the diagnosis is uncertain (Tables 1 and 2).35
The American Thoracic Society/European Respiratory Society guidelines suggest a multidisciplinary team approach that includes a pathologist, radiologist, and clinician.35 This approach more readily determines the correct diagnosis and relies less on invasive methods such as surgical biopsy and more on noninvasive methods such as radiology and clinical history. Overall, this will promote earlier access to appropriate therapies, clinical trial enrollment, and in more severe cases, lung transplant.
Currently, 23% of all lung transplants worldwide are performed in patients with idiopathic pulmonary fibrosis. Other forms of pulmonary fibrosis account for 3% to 4% of lung transplants performed.36
Evidence suggests that early referral reduces rates of morbidity and death in these patients. The results of a single-center study37 of patients with idiopathic pulmonary fibrosis demonstrated that a longer delay from the onset of symptoms to evaluation by a specialist at a tertiary care referral center was associated with a higher rate of death from this disease independent of disease severity. Those with the longest delay in referral had a multivariable-adjusted death rate 3.4 times higher than those with the shortest delay.5,37
In summary, with implementation of the new lung cancer screening guidelines, primary care physicians are more often encountering the incidental finding of interstitial lung disease in their patients. Prompt diagnosis of interstitial lung disease helps ensure that patients receive appropriate care and early consideration for clinical trials and lung transplant.
Primary care physicians play a critical role in the initial identification of key characteristics of the interstitial abnormality—namely, whether the pattern is nonfibrotic or fibrotic—and in the correlation of the history and physical findings to expedite the diagnosis. Subsequently, ordering high-resolution CT for more detailed characterization and prompt referral to a specialist in interstitial lung disease allow for a more rapid and accurate diagnosis, specialized therapy, and supportive care.
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365:395–409.
- Detterbeck FC, Lewis SZ, Diekemper R, Addrizzo-Harris D, Alberts WM. Executive summary: diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013; 143(suppl 5):7S–37S.
- Moyer VA; US Preventive Services Task Force. Screening for lung cancer: US Preventive Services Task Force recommendation statement. Ann Intern Med 2014; 160:330–338.
- Bach PB, Mirkin JN, Oliver TK, et al. Benefits and harms of CT screening for lung cancer: a systematic review. JAMA 2012; 307:2418–2429.
- Lamas DJ, Kawut SM, Bagiella E, Philip N, Arcasoy SM, Lederer DJ. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med 2011; 184:842–847.
- Swensen SJ, Jett JR, Hartman TE, et al. Lung cancer screening with CT: Mayo Clinic experience. Radiology 2003; 226:756–761.
- MacRedmond R, Logan PM, Lee M, Kenny D, Foley C, Costello RW. Screening for lung cancer using low dose CT scanning. Thorax 2004; 59:237–241.
- Sverzellati N, Guerci L, Randi G, et al. Interstitial lung diseases in a lung cancer screening trial. Eur Respir J 2011; 38:392–400.
- Jin GY, Lynch D, Chawla A, et al. Interstitial lung abnormalities in a CT lung cancer screening population: prevalence and progression rate. Radiology 2013; 268:563–571.
- Heyneman LE, Ward S, Lynch DA, Remy-Jardin M, Johkoh T, Müller NL. Respiratory bronchiolitis, respiratory bronchiolitis-associated interstitial lung disease, and desquamative interstitial pneumonia: different entities or part of the spectrum of the same disease process? AJR Am J Roentgenol 1999; 173:1617–1622.
- Moon J, du Bois RM, Colby TV, Hansell DM, Nicholson AG. Clinical significance of respiratory bronchiolitis on open lung biopsy and its relationship to smoking related interstitial lung disease. Thorax 1999; 54:1009–1014.
- Holt RM, Schmidt RA, Godwin JD, Raghu G. High resolution CT in respiratory bronchiolitis-associated interstitial lung disease. J Comput Assist Tomogr 1993; 17:46–50.
- Ryu JH, Myers JL, Capizzi SA, Douglas WW, Vassallo R, Decker PA. Desquamative interstitial pneumonia and respiratory bronchiolitis-associated interstitial lung disease. Chest 2005; 127:178–184.
- Hartman TE, Primack SL, Swensen SJ, Hansell D, McGuinness G, Müller NL. Desquamative interstitial pneumonia: thin-section CT findings in 22 patients. Radiology 1993; 187:787–790.
- Akira M, Yamamoto S, Hara H, Sakatani M, Ueda E. Serial computed tomographic evaluation in desquamative interstitial pneumonia. Thorax 1997; 52:333–337.
- Lacronique J, Roth C, Battesti JP, Basset F, Chretien J. Chest radiological features of pulmonary histiocytosis X: a report based on 50 adult cases. Thorax 1982; 37:104–109.
- Remy-Jardin M, Edme JL, Boulenguez C, Remy J, Mastora I, Sobaszek A. Longitudinal follow-up study of smoker’s lung with thin-section CT in correlation with pulmonary function tests. Radiology 2002; 222:261–270.
- Mueller-Mang C, Grosse C, Schmid K, Stiebellehner L, Bankier AA. What every radiologist should know about idiopathic interstitial pneumonias. Radiographics 2007; 27:595–615.
- Katzenstein AL, Fiorelli RF. Nonspecific interstitial pneumonia/fibrosis. Histologic features and clinical significance. Am J Surg Pathol 1994; 18:136–147.
- Bryson T, Sundaram B, Khanna D, Kazerooni EA. Connective tissue disease-associated interstitial pneumonia and idiopathic interstitial pneumonia: similarity and difference. Semin Ultrasound CT MR 2014; 35:29–38.
- Desai SR, Veeraraghavan S, Hansell DM, et al. CT features of lung disease in patients with systemic sclerosis: comparison with idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia. Radiology 2004; 232:560–567.
- Tsubamoto M, Müller NL, Johkoh T, et al. Pathologic subgroups of nonspecific interstitial pneumonia: differential diagnosis from other idiopathic interstitial pneumonias on high-resolution computed tomography. J Comput Assist Tomogr 2005; 29:793–800.
- Silva CI, Müller NL, Lynch DA, et al. Chronic hypersensitivity pneumonitis: differentiation from idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia by using thin-section CT. Radiology 2008; 246:288–297.
- Antin-Ozerkis D, Rubinowitz A. An update on nonspecific interstitial pneumonia. Clin Pulm Med 2010; 17:122–128.
- Daniil ZD, Gilchrist FC, Nicholson AG, et al. A histologic pattern of nonspecific interstitial pneumonia is associated with a better prognosis than usual interstitial pneumonia in patients with cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 1999; 160:899–905.
- Travis WD, Matsui K, Moss J, Ferrans VJ. Idiopathic nonspecific interstitial pneumonia: prognostic significance of cellular and fibrosing patterns: survival comparison with usual interstitial pneumonia and desquamative interstitial pneumonia. Am J Surg Pathol 2000; 24:19–33.
- Riha RL, Duhig EE, Clarke BE, Steele RH, Slaughter RE, Zimmerman PV. Survival of patients with biopsy-proven usual interstitial pneumonia and nonspecific interstitial pneumonia. Eur Respir J 2002; 19:1114–1118.
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183:788–824.
- du Bois RM. An earlier and more confident diagnosis of idiopathic pulmonary fibrosis. Eur Respir Rev 2012; 21:141–146.
- Nishimura K, Kitaichi M, Izumi T, Nagai S, Kanaoka M, Itoh H. Usual interstitial pneumonia: histologic correlation with high-resolution CT. Radiology 1992; 182:337–342.
- Souza CA, Müller NL, Lee KS, Johkoh T, Mitsuhiro H, Chong S. Idiopathic interstitial pneumonias: prevalence of mediastinal lymph node enlargement in 206 patients. AJR Am J Roentgenol 2006; 186:995–999.
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370:2083–2092.
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370:2071–2082.
- Bradley B, Branley HM, Egan JJ, et al; British Thoracic Society Interstitial Lung Disease Guideline Group, British Thoracic Society Standards of Care Committee; Thoracic Society of Australia; New Zealand Thoracic Society; Irish Thoracic Society. Interstitial lung disease guideline: the British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax 2008; 63(suppl 5):v1–v58.
- Travis WD, Costabel U, Hansell DM, et al; ATS/ERS Committee on Idiopathic Interstitial Pneumonias. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013; 188:733–748.
- Stehlik J, Edwards LB, Kucheryavaya AY, et al; International Society of Heart and Lung Transplantation. The Registry of the International Society for Heart and Lung Transplantation: 29th official adult heart transplant report—2012. J Heart Lung Transplant 2012; 31:1052–1064.
- Oldham JM, Noth I. Idiopathic pulmonary fibrosis: early detection and referral. Respir Med 2014; 108:819–829.
Primary care physicians are playing a bigger role in evaluating the incidental finding of interstitial lung diseases since the recent publication of guidelines recommending computed tomography (CT) to screen for lung cancer.
In August 2011, the National Cancer Institute published its findings from the National Lung Screening Trial, which demonstrated a 20% reduction in mortality from lung cancer in patients at high risk screened with low-dose CT.1 Based on these results, the American Cancer Society, the American College of Chest Physicians, the American Society of Clinical Oncology, and the National Comprehensive Cancer Network recommended annual screening for lung cancer with low-dose CT in adults ages 55 to 74 who have a 30-pack-year smoking history and who currently smoke or have quit within the past 15 years.2 In December 2013, the US Preventive Services Task Force published similar guidelines but increased the age range to include high-risk patients ages 55 to 80.3
Bach et al4 estimated that, in 2010 in the United States, 8.6 million people met the criteria used in the National Lung Screening Trial for low-dose CT screening. These are the same criteria as in the multisociety recommendations cited above.2 With such large numbers of patients eligible for CT screening, internists and other primary care physicians are undoubtedly encountering the incidental discovery of nonmalignant pulmonary diseases such as interstitial lung disease.
This article reviews the radiographic characteristics of the most common interstitial lung diseases the internist may encounter on screening CT in long-term smokers.
Referral to a specialist has been associated with lower rates of morbidity and death,5 and a diagnosis of interstitial lung disease should be confirmed by a pulmonologist and a radiologist specializing in differentiating the subtypes. But the primary care physician now plays a critical role in recognizing the need for further evaluation.
HOW COMMON IS INTERSTITIAL LUNG DISEASE IN SMOKERS?
Several studies have published data on the prevalence of interstitial lung disease in patients undergoing low-dose CT for lung cancer screening.
A trial at Mayo Clinic in current and former smokers identified “diffuse lung disease” in 9 (0.9%) of 1,049 participants.6
A trial in Ireland identified idiopathic pulmonary fibrosis in 6 (1.3%) of 449 current smokers who underwent low-dose CT screening for lung cancer.7
Sverzellati et al8 evaluated 692 participants in the Multicentric Italian Lung Detection CT screening study and reported a respiratory bronchiolitis pattern in 109 (15.7%), a usual interstitial pneumonia pattern in 2 (0.3%), and other patterns of chronic interstitial pneumonia in 26 (3.8%).
The National Lung Screening Trial reported that the frequency of “clinically significant” incidental findings (including pulmonary fibrosis) in all participants was 7.5%.1 A retrospective analysis of 884 participants at a single site in this trial identified interstitial lung abnormalities in 86 participants (9.7%).9 These abnormalities were further categorized as nonfibrotic in 52 (5.9%) of 884, fibrotic in 19 (2.1%) of 884, and mixed fibrotic and nonfibrotic in 15 (1.7%) of 884.
Follow-up CT at 2 years in this trial demonstrated improvement in 50% and progression in 11% of patients who had nonfibrotic abnormalities, while fibrotic abnormalities improved in no cases and progressed in 37%. Interstitial lung abnormalities were more common in those who currently smoked and in those with more pack-years of cigarette smoking.9
In sum, these trials suggest that low-dose CT screening for lung cancer can detect the most common forms of interstitial lung disease in this at-risk population and can characterize them as fibrotic or nonfibrotic, a distinction important for prognosis and subsequent management.
NONFIBROTIC VS FIBROTIC DISEASE
It is important to distinguish between nonfibrotic and fibrotic interstitial lung disease, as fibrotic disease carries a worse prognosis and is treated differently.
Features of nonfibrotic interstitial lung disease:
- Ground-glass opacities
- Nodules
- Mosaic attenuation or consolidation.
Features of fibrotic interstitial lung disease:
- Combination of ground-glass opacities and reticulation
- Reticulation by itself
- Traction bronchiectasis
- Honeycombing
- Loss of lung volume.
NONFIBROTIC INTERSTITIAL LUNG DISEASES
Given the strong likelihood that a patient undergoing screening CT is either a current or former smoker, physicians may encounter, in addition to emphysema and lung cancer, the following smoking-related interstitial lung diseases, which are primarily nonfibrotic and which frequently coexist (Table 1):
- Respiratory bronchiolitis
- Respiratory bronchiolitis-interstitial lung disease
- Desquamative interstitial pneumonia
- Pulmonary Langerhans cell histiocytosis.
Respiratory bronchiolitis
Respiratory bronchiolitis occurs mostly in smokers and does not necessarily lead to respiratory symptoms in all patients.10 It cannot always be identified radiographically but occasionally appears as predominantly upper-lobe, patchy ground-glass opacities or ill-defined centrilobular nodules without evidence of fibrosis (Figure 1).
Respiratory bronchiolitis-interstitial lung disease
In rare cases, respiratory bronchiolitis leads to peribronchial fibrosis invading the alveolar walls, which is then classified as respiratory bronchiolitis-interstitial lung disease.11 The CT findings in respiratory bronchiolitis-interstitial lung disease are upper-lobe-predominant centrilobular ground-glass nodules, patchy ground-glass opacities, and bronchial wall thickening (Figure 2).10 Occasionally, mild reticulation is noted without honeycombing. Mild air trapping can be seen in the lower lobes, with centrilobular emphysema in the upper lobes.12
The only successful therapy for respiratory bronchiolitis and respiratory bronchiolitis-interstitial lung disease is smoking cessation. Finding either of these diseases should prompt aggressive counseling by the internist and consideration of referral to a specialist in interstitial lung disease.
Desquamative interstitial pneumonia
Although pathologically different from respiratory bronchiolitis-interstitial lung disease, desquamative interstitial pneumonia has a similar clinical and radiographic presentation. Because their features significantly overlap, they are considered a pathomorphologic continuum, representing degrees of severity of the same disease process caused by prolonged tobacco inhalation.10,13
Widespread ground-glass opacities are the predominant CT finding. These are bilateral and symmetric in distribution in 86%, basal and peripheral in 60%, patchy in 20%, and diffuse in 20% (Figure 3).14 Other frequent findings are mild reticulation with traction bronchiectasis and coexistent emphysema (Figure 4).15 The small peripheral cystic spaces noted in this disease most likely represent dilated bronchioles and alveolar ducts rather than honeycombing.16
No additional treatment beyond elimination of smoking has been proven effective for desquamative interstitial pneumonia, and patients who manage to quit smoking generally have a favorable prognosis.17,18
Pulmonary Langerhans cell histiocytosis
The combination of upper-lobe-predominant cysts and nodules in a young heavy smoker are diagnostic of pulmonary Langerhans cell histiocytosis. The cysts are bizarrely shaped, thin- or thick-walled, and nonuniform in size (Figure 5). The irregular cavitary nodules are centrilobular. The disease characteristically spares the costophrenic angles.
Spontaneous pneumothorax is the initial clinical presentation in 15% of patients.16 In the early stages of the disease (nodule-predominant disease without cysts), infection and metastatic disease need to be excluded (Figure 6). In the later stages, the cysts become coalescent, making the distinction between this disease and “burned-out” lymphangioleiomyomatosis or severe emphysema extremely difficult (Figure 7).17 Smoking cessation and corticosteroids are the mainstay of medical therapy for pulmonary Langerhans cell histiocytosis, and about 50% of patients who quit smoking and receive corticosteroids demonstrate partial or complete clearing of the radiographic abnormalities and symptoms (Figure 8).
FIBROTIC INTERSTITIAL LUNG DISEASES
If CT identifies a diffuse fibrotic pattern, the two most common possibilities (Table 2) are:
- Nonspecific interstitial pneumonia
- Usual interstitial pneumonia.
As noted above, these carry a worse prognosis than the nonfibrotic interstitial lung diseases.
Nonspecific interstitial pneumonia
While most frequently idiopathic, the nonspecific interstitial pneumonia pattern can often be seen in connective tissue diseases. It has also been associated with chronic hypersensitivity pneumonitis, drug toxicity, and slowly resolving diffuse alveolar damage.19 Although it is not the only pathologic pattern in interstitial lung disease associated with connective tissue disease, it is the most common pattern in systemic sclerosis, systemic lupus erythematosus, dermatomyositis-polymyositis, and mixed connective tissue disease.20
The parenchymal changes are typically subpleural and symmetric in distribution (Figure 9). In about one-third of cases, there is a peribronchovascular distribution of the abnormalities (Figure 10).
Ground-glass opacities are the dominant imaging findings, seen in 80% of cases.18 In advanced disease (also referred to as fibrotic nonspecific interstitial pneumonia), patients have accompanying fine or coarse reticular opacities, traction bronchiectasis, and consolidation (Figure 11). Honeycombing is seen in 1% to 5% of patients.21
The most specific sign of nonspecific interstitial pneumonia is sparing of the immediate subpleural lung, apparent in 30% to 50% of patients (Figure 12).22 Subpleural sparing with a peribronchovascular distribution of abnormalities, absence of lobular areas with decreased attenuation, and lack of honeycombing are imaging features that increase the diagnostic confidence of nonspecific interstitial pneumonia (Table 3).23 Clinically, compared with those who have usual interstitial pneumonia (see below), patients are younger and more of them are female. These patients also present with extrapulmonary manifestations such as joint involvement, rash, and Raynaud phenomenon. Therefore, these associated symptoms on presentation can help distinguish nonspecific interstitial pneumonia or usual interstitial pneumonia related to connective tissue disease from the idiopathic forms.
The first step in managing nonspecific interstitial pneumonia is to remove all potential exposure to inhaled substances or to drugs. Although immunosuppressive therapy has never been studied in a randomized controlled trial in this disease, numerous reports suggest that patients may respond to prednisone and to steroid-sparing immunosuppressants.24
In several studies, survival rates in nonspecific interstitial pneumonia were significantly greater than in usual interstitial pneumonia independent of the treatment strategy. In long-term follow-up of patients with idiopathic nonspecific interstitial pneumonia treated with immunosuppressive therapy, two-thirds remained stable or improved.25–27
Although most connective tissue diseases cause a lung pattern of nonspecific interstitial pneumonia, some (eg, rheumatoid arthritis) may present with a pattern of usual interstitial pneumonia. In these cases and in those of advanced fibrotic nonspecific interstitial pneumonia, the prognosis is worse, as the disease is less responsive to immunosuppressive therapy.20
Usual interstitial pneumonia
Usual interstitial pneumonia is the most severe form of lung fibrosis. Most cases are idiopathic and are termed idiopathic pulmonary fibrosis. Other causes of the usual interstitial pneumonia pattern include domestic and occupational environmental exposures, connective tissue disease, and drug toxicity.28 An epidemiologic association between smoking and usual interstitial pneumonia is well documented.28
Idiopathic pulmonary fibrosis typically affects men ages 50 to 70. Because its risk factors coincide with those of lung cancer, there is a high likelihood of detecting idiopathic pulmonary fibrosis early in this screening population. It has an especially poor prognosis, with a mean survival of 2 to 5 years from the time of diagnosis.18
The distribution of disease in usual interstitial pneumonia is characteristically subpleural and basal. CT features include coarse subpleural reticulation and honeycombing combined with traction bronchiectasis or bronchiolectasis and architectural distortion (Figure 13).18 Honeycombing is the most specific and key diagnostic CT finding for establishing a definitive diagnosis of usual interstitial pneumonia.29 However, ground-glass opacities are present in most patients, typically in the region of interstitial fibrosis, and are always less extensive than the reticulation.30 The findings demonstrate morphologic heterogeneity, with areas of fibrosis adjacent to areas of normal lung (Figure 14).
In addition to the aforementioned imaging features, the 2011 American Thoracic Society and European Respiratory Society joint guidelines for the CT diagnosis of usual interstitial pneumonia patterns require the absence of atypical features that suggest an alternative diagnosis, including those seen in nonspecific interstitial pneumonia, such as an upper, midlung, or peribronchovascular distribution and extensive ground-glass attenuation.28 Mild mediastinal lymphadenopathy (usually < 1.5 cm in the short axis) is common in usual interstitial pneumonia.31
Because other chronic interstitial pneumonias that may resemble usual interstitial pneumonia have a more favorable course and may respond to immunosuppressive therapy, establishing an early and accurate diagnosis is of the utmost importance.5 Additionally, the emergence of possible new therapies for idiopathic pulmonary fibrosis makes early referral to a specialist paramount in these cases. Recent studies have demonstrated significant slowing of the progression of disease in idiopathic pulmonary fibrosis with both pirfenidone and nintedanib.32,33
DIAGNOSIS AND MANAGEMENT
The diagnosis of these nonfibrotic and fibrotic lung diseases is complex. In all cases in which interstitial lung disease is detected on screening CT for lung cancer, the internist should strongly consider further evaluation with dedicated high-resolution CT and early referral to a specialist (Figure 15).
Because smoking cessation is the only recommended treatment for nonfibrotic smoking-related interstitial lung diseases, particular emphasis on smoking cessation counseling is essential.
Referral for bronchoscopy with transbronchial lung biopsy is generally not helpful in the diagnosis of the interstitial lung diseases discussed in this article unless there is a need to rule out infection or neoplasm.34 Referral for surgical lung biopsy may be indicated in some cases of suspected pulmonary Langerhans cell histiocytosis, desquamative interstitial pneumonia, nonspecific interstitial pneumonia, or usual interstitial pneumonia if the diagnosis is uncertain (Tables 1 and 2).35
The American Thoracic Society/European Respiratory Society guidelines suggest a multidisciplinary team approach that includes a pathologist, radiologist, and clinician.35 This approach more readily determines the correct diagnosis and relies less on invasive methods such as surgical biopsy and more on noninvasive methods such as radiology and clinical history. Overall, this will promote earlier access to appropriate therapies, clinical trial enrollment, and in more severe cases, lung transplant.
Currently, 23% of all lung transplants worldwide are performed in patients with idiopathic pulmonary fibrosis. Other forms of pulmonary fibrosis account for 3% to 4% of lung transplants performed.36
Evidence suggests that early referral reduces rates of morbidity and death in these patients. The results of a single-center study37 of patients with idiopathic pulmonary fibrosis demonstrated that a longer delay from the onset of symptoms to evaluation by a specialist at a tertiary care referral center was associated with a higher rate of death from this disease independent of disease severity. Those with the longest delay in referral had a multivariable-adjusted death rate 3.4 times higher than those with the shortest delay.5,37
In summary, with implementation of the new lung cancer screening guidelines, primary care physicians are more often encountering the incidental finding of interstitial lung disease in their patients. Prompt diagnosis of interstitial lung disease helps ensure that patients receive appropriate care and early consideration for clinical trials and lung transplant.
Primary care physicians play a critical role in the initial identification of key characteristics of the interstitial abnormality—namely, whether the pattern is nonfibrotic or fibrotic—and in the correlation of the history and physical findings to expedite the diagnosis. Subsequently, ordering high-resolution CT for more detailed characterization and prompt referral to a specialist in interstitial lung disease allow for a more rapid and accurate diagnosis, specialized therapy, and supportive care.
Primary care physicians are playing a bigger role in evaluating the incidental finding of interstitial lung diseases since the recent publication of guidelines recommending computed tomography (CT) to screen for lung cancer.
In August 2011, the National Cancer Institute published its findings from the National Lung Screening Trial, which demonstrated a 20% reduction in mortality from lung cancer in patients at high risk screened with low-dose CT.1 Based on these results, the American Cancer Society, the American College of Chest Physicians, the American Society of Clinical Oncology, and the National Comprehensive Cancer Network recommended annual screening for lung cancer with low-dose CT in adults ages 55 to 74 who have a 30-pack-year smoking history and who currently smoke or have quit within the past 15 years.2 In December 2013, the US Preventive Services Task Force published similar guidelines but increased the age range to include high-risk patients ages 55 to 80.3
Bach et al4 estimated that, in 2010 in the United States, 8.6 million people met the criteria used in the National Lung Screening Trial for low-dose CT screening. These are the same criteria as in the multisociety recommendations cited above.2 With such large numbers of patients eligible for CT screening, internists and other primary care physicians are undoubtedly encountering the incidental discovery of nonmalignant pulmonary diseases such as interstitial lung disease.
This article reviews the radiographic characteristics of the most common interstitial lung diseases the internist may encounter on screening CT in long-term smokers.
Referral to a specialist has been associated with lower rates of morbidity and death,5 and a diagnosis of interstitial lung disease should be confirmed by a pulmonologist and a radiologist specializing in differentiating the subtypes. But the primary care physician now plays a critical role in recognizing the need for further evaluation.
HOW COMMON IS INTERSTITIAL LUNG DISEASE IN SMOKERS?
Several studies have published data on the prevalence of interstitial lung disease in patients undergoing low-dose CT for lung cancer screening.
A trial at Mayo Clinic in current and former smokers identified “diffuse lung disease” in 9 (0.9%) of 1,049 participants.6
A trial in Ireland identified idiopathic pulmonary fibrosis in 6 (1.3%) of 449 current smokers who underwent low-dose CT screening for lung cancer.7
Sverzellati et al8 evaluated 692 participants in the Multicentric Italian Lung Detection CT screening study and reported a respiratory bronchiolitis pattern in 109 (15.7%), a usual interstitial pneumonia pattern in 2 (0.3%), and other patterns of chronic interstitial pneumonia in 26 (3.8%).
The National Lung Screening Trial reported that the frequency of “clinically significant” incidental findings (including pulmonary fibrosis) in all participants was 7.5%.1 A retrospective analysis of 884 participants at a single site in this trial identified interstitial lung abnormalities in 86 participants (9.7%).9 These abnormalities were further categorized as nonfibrotic in 52 (5.9%) of 884, fibrotic in 19 (2.1%) of 884, and mixed fibrotic and nonfibrotic in 15 (1.7%) of 884.
Follow-up CT at 2 years in this trial demonstrated improvement in 50% and progression in 11% of patients who had nonfibrotic abnormalities, while fibrotic abnormalities improved in no cases and progressed in 37%. Interstitial lung abnormalities were more common in those who currently smoked and in those with more pack-years of cigarette smoking.9
In sum, these trials suggest that low-dose CT screening for lung cancer can detect the most common forms of interstitial lung disease in this at-risk population and can characterize them as fibrotic or nonfibrotic, a distinction important for prognosis and subsequent management.
NONFIBROTIC VS FIBROTIC DISEASE
It is important to distinguish between nonfibrotic and fibrotic interstitial lung disease, as fibrotic disease carries a worse prognosis and is treated differently.
Features of nonfibrotic interstitial lung disease:
- Ground-glass opacities
- Nodules
- Mosaic attenuation or consolidation.
Features of fibrotic interstitial lung disease:
- Combination of ground-glass opacities and reticulation
- Reticulation by itself
- Traction bronchiectasis
- Honeycombing
- Loss of lung volume.
NONFIBROTIC INTERSTITIAL LUNG DISEASES
Given the strong likelihood that a patient undergoing screening CT is either a current or former smoker, physicians may encounter, in addition to emphysema and lung cancer, the following smoking-related interstitial lung diseases, which are primarily nonfibrotic and which frequently coexist (Table 1):
- Respiratory bronchiolitis
- Respiratory bronchiolitis-interstitial lung disease
- Desquamative interstitial pneumonia
- Pulmonary Langerhans cell histiocytosis.
Respiratory bronchiolitis
Respiratory bronchiolitis occurs mostly in smokers and does not necessarily lead to respiratory symptoms in all patients.10 It cannot always be identified radiographically but occasionally appears as predominantly upper-lobe, patchy ground-glass opacities or ill-defined centrilobular nodules without evidence of fibrosis (Figure 1).
Respiratory bronchiolitis-interstitial lung disease
In rare cases, respiratory bronchiolitis leads to peribronchial fibrosis invading the alveolar walls, which is then classified as respiratory bronchiolitis-interstitial lung disease.11 The CT findings in respiratory bronchiolitis-interstitial lung disease are upper-lobe-predominant centrilobular ground-glass nodules, patchy ground-glass opacities, and bronchial wall thickening (Figure 2).10 Occasionally, mild reticulation is noted without honeycombing. Mild air trapping can be seen in the lower lobes, with centrilobular emphysema in the upper lobes.12
The only successful therapy for respiratory bronchiolitis and respiratory bronchiolitis-interstitial lung disease is smoking cessation. Finding either of these diseases should prompt aggressive counseling by the internist and consideration of referral to a specialist in interstitial lung disease.
Desquamative interstitial pneumonia
Although pathologically different from respiratory bronchiolitis-interstitial lung disease, desquamative interstitial pneumonia has a similar clinical and radiographic presentation. Because their features significantly overlap, they are considered a pathomorphologic continuum, representing degrees of severity of the same disease process caused by prolonged tobacco inhalation.10,13
Widespread ground-glass opacities are the predominant CT finding. These are bilateral and symmetric in distribution in 86%, basal and peripheral in 60%, patchy in 20%, and diffuse in 20% (Figure 3).14 Other frequent findings are mild reticulation with traction bronchiectasis and coexistent emphysema (Figure 4).15 The small peripheral cystic spaces noted in this disease most likely represent dilated bronchioles and alveolar ducts rather than honeycombing.16
No additional treatment beyond elimination of smoking has been proven effective for desquamative interstitial pneumonia, and patients who manage to quit smoking generally have a favorable prognosis.17,18
Pulmonary Langerhans cell histiocytosis
The combination of upper-lobe-predominant cysts and nodules in a young heavy smoker are diagnostic of pulmonary Langerhans cell histiocytosis. The cysts are bizarrely shaped, thin- or thick-walled, and nonuniform in size (Figure 5). The irregular cavitary nodules are centrilobular. The disease characteristically spares the costophrenic angles.
Spontaneous pneumothorax is the initial clinical presentation in 15% of patients.16 In the early stages of the disease (nodule-predominant disease without cysts), infection and metastatic disease need to be excluded (Figure 6). In the later stages, the cysts become coalescent, making the distinction between this disease and “burned-out” lymphangioleiomyomatosis or severe emphysema extremely difficult (Figure 7).17 Smoking cessation and corticosteroids are the mainstay of medical therapy for pulmonary Langerhans cell histiocytosis, and about 50% of patients who quit smoking and receive corticosteroids demonstrate partial or complete clearing of the radiographic abnormalities and symptoms (Figure 8).
FIBROTIC INTERSTITIAL LUNG DISEASES
If CT identifies a diffuse fibrotic pattern, the two most common possibilities (Table 2) are:
- Nonspecific interstitial pneumonia
- Usual interstitial pneumonia.
As noted above, these carry a worse prognosis than the nonfibrotic interstitial lung diseases.
Nonspecific interstitial pneumonia
While most frequently idiopathic, the nonspecific interstitial pneumonia pattern can often be seen in connective tissue diseases. It has also been associated with chronic hypersensitivity pneumonitis, drug toxicity, and slowly resolving diffuse alveolar damage.19 Although it is not the only pathologic pattern in interstitial lung disease associated with connective tissue disease, it is the most common pattern in systemic sclerosis, systemic lupus erythematosus, dermatomyositis-polymyositis, and mixed connective tissue disease.20
The parenchymal changes are typically subpleural and symmetric in distribution (Figure 9). In about one-third of cases, there is a peribronchovascular distribution of the abnormalities (Figure 10).
Ground-glass opacities are the dominant imaging findings, seen in 80% of cases.18 In advanced disease (also referred to as fibrotic nonspecific interstitial pneumonia), patients have accompanying fine or coarse reticular opacities, traction bronchiectasis, and consolidation (Figure 11). Honeycombing is seen in 1% to 5% of patients.21
The most specific sign of nonspecific interstitial pneumonia is sparing of the immediate subpleural lung, apparent in 30% to 50% of patients (Figure 12).22 Subpleural sparing with a peribronchovascular distribution of abnormalities, absence of lobular areas with decreased attenuation, and lack of honeycombing are imaging features that increase the diagnostic confidence of nonspecific interstitial pneumonia (Table 3).23 Clinically, compared with those who have usual interstitial pneumonia (see below), patients are younger and more of them are female. These patients also present with extrapulmonary manifestations such as joint involvement, rash, and Raynaud phenomenon. Therefore, these associated symptoms on presentation can help distinguish nonspecific interstitial pneumonia or usual interstitial pneumonia related to connective tissue disease from the idiopathic forms.
The first step in managing nonspecific interstitial pneumonia is to remove all potential exposure to inhaled substances or to drugs. Although immunosuppressive therapy has never been studied in a randomized controlled trial in this disease, numerous reports suggest that patients may respond to prednisone and to steroid-sparing immunosuppressants.24
In several studies, survival rates in nonspecific interstitial pneumonia were significantly greater than in usual interstitial pneumonia independent of the treatment strategy. In long-term follow-up of patients with idiopathic nonspecific interstitial pneumonia treated with immunosuppressive therapy, two-thirds remained stable or improved.25–27
Although most connective tissue diseases cause a lung pattern of nonspecific interstitial pneumonia, some (eg, rheumatoid arthritis) may present with a pattern of usual interstitial pneumonia. In these cases and in those of advanced fibrotic nonspecific interstitial pneumonia, the prognosis is worse, as the disease is less responsive to immunosuppressive therapy.20
Usual interstitial pneumonia
Usual interstitial pneumonia is the most severe form of lung fibrosis. Most cases are idiopathic and are termed idiopathic pulmonary fibrosis. Other causes of the usual interstitial pneumonia pattern include domestic and occupational environmental exposures, connective tissue disease, and drug toxicity.28 An epidemiologic association between smoking and usual interstitial pneumonia is well documented.28
Idiopathic pulmonary fibrosis typically affects men ages 50 to 70. Because its risk factors coincide with those of lung cancer, there is a high likelihood of detecting idiopathic pulmonary fibrosis early in this screening population. It has an especially poor prognosis, with a mean survival of 2 to 5 years from the time of diagnosis.18
The distribution of disease in usual interstitial pneumonia is characteristically subpleural and basal. CT features include coarse subpleural reticulation and honeycombing combined with traction bronchiectasis or bronchiolectasis and architectural distortion (Figure 13).18 Honeycombing is the most specific and key diagnostic CT finding for establishing a definitive diagnosis of usual interstitial pneumonia.29 However, ground-glass opacities are present in most patients, typically in the region of interstitial fibrosis, and are always less extensive than the reticulation.30 The findings demonstrate morphologic heterogeneity, with areas of fibrosis adjacent to areas of normal lung (Figure 14).
In addition to the aforementioned imaging features, the 2011 American Thoracic Society and European Respiratory Society joint guidelines for the CT diagnosis of usual interstitial pneumonia patterns require the absence of atypical features that suggest an alternative diagnosis, including those seen in nonspecific interstitial pneumonia, such as an upper, midlung, or peribronchovascular distribution and extensive ground-glass attenuation.28 Mild mediastinal lymphadenopathy (usually < 1.5 cm in the short axis) is common in usual interstitial pneumonia.31
Because other chronic interstitial pneumonias that may resemble usual interstitial pneumonia have a more favorable course and may respond to immunosuppressive therapy, establishing an early and accurate diagnosis is of the utmost importance.5 Additionally, the emergence of possible new therapies for idiopathic pulmonary fibrosis makes early referral to a specialist paramount in these cases. Recent studies have demonstrated significant slowing of the progression of disease in idiopathic pulmonary fibrosis with both pirfenidone and nintedanib.32,33
DIAGNOSIS AND MANAGEMENT
The diagnosis of these nonfibrotic and fibrotic lung diseases is complex. In all cases in which interstitial lung disease is detected on screening CT for lung cancer, the internist should strongly consider further evaluation with dedicated high-resolution CT and early referral to a specialist (Figure 15).
Because smoking cessation is the only recommended treatment for nonfibrotic smoking-related interstitial lung diseases, particular emphasis on smoking cessation counseling is essential.
Referral for bronchoscopy with transbronchial lung biopsy is generally not helpful in the diagnosis of the interstitial lung diseases discussed in this article unless there is a need to rule out infection or neoplasm.34 Referral for surgical lung biopsy may be indicated in some cases of suspected pulmonary Langerhans cell histiocytosis, desquamative interstitial pneumonia, nonspecific interstitial pneumonia, or usual interstitial pneumonia if the diagnosis is uncertain (Tables 1 and 2).35
The American Thoracic Society/European Respiratory Society guidelines suggest a multidisciplinary team approach that includes a pathologist, radiologist, and clinician.35 This approach more readily determines the correct diagnosis and relies less on invasive methods such as surgical biopsy and more on noninvasive methods such as radiology and clinical history. Overall, this will promote earlier access to appropriate therapies, clinical trial enrollment, and in more severe cases, lung transplant.
Currently, 23% of all lung transplants worldwide are performed in patients with idiopathic pulmonary fibrosis. Other forms of pulmonary fibrosis account for 3% to 4% of lung transplants performed.36
Evidence suggests that early referral reduces rates of morbidity and death in these patients. The results of a single-center study37 of patients with idiopathic pulmonary fibrosis demonstrated that a longer delay from the onset of symptoms to evaluation by a specialist at a tertiary care referral center was associated with a higher rate of death from this disease independent of disease severity. Those with the longest delay in referral had a multivariable-adjusted death rate 3.4 times higher than those with the shortest delay.5,37
In summary, with implementation of the new lung cancer screening guidelines, primary care physicians are more often encountering the incidental finding of interstitial lung disease in their patients. Prompt diagnosis of interstitial lung disease helps ensure that patients receive appropriate care and early consideration for clinical trials and lung transplant.
Primary care physicians play a critical role in the initial identification of key characteristics of the interstitial abnormality—namely, whether the pattern is nonfibrotic or fibrotic—and in the correlation of the history and physical findings to expedite the diagnosis. Subsequently, ordering high-resolution CT for more detailed characterization and prompt referral to a specialist in interstitial lung disease allow for a more rapid and accurate diagnosis, specialized therapy, and supportive care.
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365:395–409.
- Detterbeck FC, Lewis SZ, Diekemper R, Addrizzo-Harris D, Alberts WM. Executive summary: diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013; 143(suppl 5):7S–37S.
- Moyer VA; US Preventive Services Task Force. Screening for lung cancer: US Preventive Services Task Force recommendation statement. Ann Intern Med 2014; 160:330–338.
- Bach PB, Mirkin JN, Oliver TK, et al. Benefits and harms of CT screening for lung cancer: a systematic review. JAMA 2012; 307:2418–2429.
- Lamas DJ, Kawut SM, Bagiella E, Philip N, Arcasoy SM, Lederer DJ. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med 2011; 184:842–847.
- Swensen SJ, Jett JR, Hartman TE, et al. Lung cancer screening with CT: Mayo Clinic experience. Radiology 2003; 226:756–761.
- MacRedmond R, Logan PM, Lee M, Kenny D, Foley C, Costello RW. Screening for lung cancer using low dose CT scanning. Thorax 2004; 59:237–241.
- Sverzellati N, Guerci L, Randi G, et al. Interstitial lung diseases in a lung cancer screening trial. Eur Respir J 2011; 38:392–400.
- Jin GY, Lynch D, Chawla A, et al. Interstitial lung abnormalities in a CT lung cancer screening population: prevalence and progression rate. Radiology 2013; 268:563–571.
- Heyneman LE, Ward S, Lynch DA, Remy-Jardin M, Johkoh T, Müller NL. Respiratory bronchiolitis, respiratory bronchiolitis-associated interstitial lung disease, and desquamative interstitial pneumonia: different entities or part of the spectrum of the same disease process? AJR Am J Roentgenol 1999; 173:1617–1622.
- Moon J, du Bois RM, Colby TV, Hansell DM, Nicholson AG. Clinical significance of respiratory bronchiolitis on open lung biopsy and its relationship to smoking related interstitial lung disease. Thorax 1999; 54:1009–1014.
- Holt RM, Schmidt RA, Godwin JD, Raghu G. High resolution CT in respiratory bronchiolitis-associated interstitial lung disease. J Comput Assist Tomogr 1993; 17:46–50.
- Ryu JH, Myers JL, Capizzi SA, Douglas WW, Vassallo R, Decker PA. Desquamative interstitial pneumonia and respiratory bronchiolitis-associated interstitial lung disease. Chest 2005; 127:178–184.
- Hartman TE, Primack SL, Swensen SJ, Hansell D, McGuinness G, Müller NL. Desquamative interstitial pneumonia: thin-section CT findings in 22 patients. Radiology 1993; 187:787–790.
- Akira M, Yamamoto S, Hara H, Sakatani M, Ueda E. Serial computed tomographic evaluation in desquamative interstitial pneumonia. Thorax 1997; 52:333–337.
- Lacronique J, Roth C, Battesti JP, Basset F, Chretien J. Chest radiological features of pulmonary histiocytosis X: a report based on 50 adult cases. Thorax 1982; 37:104–109.
- Remy-Jardin M, Edme JL, Boulenguez C, Remy J, Mastora I, Sobaszek A. Longitudinal follow-up study of smoker’s lung with thin-section CT in correlation with pulmonary function tests. Radiology 2002; 222:261–270.
- Mueller-Mang C, Grosse C, Schmid K, Stiebellehner L, Bankier AA. What every radiologist should know about idiopathic interstitial pneumonias. Radiographics 2007; 27:595–615.
- Katzenstein AL, Fiorelli RF. Nonspecific interstitial pneumonia/fibrosis. Histologic features and clinical significance. Am J Surg Pathol 1994; 18:136–147.
- Bryson T, Sundaram B, Khanna D, Kazerooni EA. Connective tissue disease-associated interstitial pneumonia and idiopathic interstitial pneumonia: similarity and difference. Semin Ultrasound CT MR 2014; 35:29–38.
- Desai SR, Veeraraghavan S, Hansell DM, et al. CT features of lung disease in patients with systemic sclerosis: comparison with idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia. Radiology 2004; 232:560–567.
- Tsubamoto M, Müller NL, Johkoh T, et al. Pathologic subgroups of nonspecific interstitial pneumonia: differential diagnosis from other idiopathic interstitial pneumonias on high-resolution computed tomography. J Comput Assist Tomogr 2005; 29:793–800.
- Silva CI, Müller NL, Lynch DA, et al. Chronic hypersensitivity pneumonitis: differentiation from idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia by using thin-section CT. Radiology 2008; 246:288–297.
- Antin-Ozerkis D, Rubinowitz A. An update on nonspecific interstitial pneumonia. Clin Pulm Med 2010; 17:122–128.
- Daniil ZD, Gilchrist FC, Nicholson AG, et al. A histologic pattern of nonspecific interstitial pneumonia is associated with a better prognosis than usual interstitial pneumonia in patients with cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 1999; 160:899–905.
- Travis WD, Matsui K, Moss J, Ferrans VJ. Idiopathic nonspecific interstitial pneumonia: prognostic significance of cellular and fibrosing patterns: survival comparison with usual interstitial pneumonia and desquamative interstitial pneumonia. Am J Surg Pathol 2000; 24:19–33.
- Riha RL, Duhig EE, Clarke BE, Steele RH, Slaughter RE, Zimmerman PV. Survival of patients with biopsy-proven usual interstitial pneumonia and nonspecific interstitial pneumonia. Eur Respir J 2002; 19:1114–1118.
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183:788–824.
- du Bois RM. An earlier and more confident diagnosis of idiopathic pulmonary fibrosis. Eur Respir Rev 2012; 21:141–146.
- Nishimura K, Kitaichi M, Izumi T, Nagai S, Kanaoka M, Itoh H. Usual interstitial pneumonia: histologic correlation with high-resolution CT. Radiology 1992; 182:337–342.
- Souza CA, Müller NL, Lee KS, Johkoh T, Mitsuhiro H, Chong S. Idiopathic interstitial pneumonias: prevalence of mediastinal lymph node enlargement in 206 patients. AJR Am J Roentgenol 2006; 186:995–999.
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370:2083–2092.
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370:2071–2082.
- Bradley B, Branley HM, Egan JJ, et al; British Thoracic Society Interstitial Lung Disease Guideline Group, British Thoracic Society Standards of Care Committee; Thoracic Society of Australia; New Zealand Thoracic Society; Irish Thoracic Society. Interstitial lung disease guideline: the British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax 2008; 63(suppl 5):v1–v58.
- Travis WD, Costabel U, Hansell DM, et al; ATS/ERS Committee on Idiopathic Interstitial Pneumonias. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013; 188:733–748.
- Stehlik J, Edwards LB, Kucheryavaya AY, et al; International Society of Heart and Lung Transplantation. The Registry of the International Society for Heart and Lung Transplantation: 29th official adult heart transplant report—2012. J Heart Lung Transplant 2012; 31:1052–1064.
- Oldham JM, Noth I. Idiopathic pulmonary fibrosis: early detection and referral. Respir Med 2014; 108:819–829.
- National Lung Screening Trial Research Team; Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365:395–409.
- Detterbeck FC, Lewis SZ, Diekemper R, Addrizzo-Harris D, Alberts WM. Executive summary: diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013; 143(suppl 5):7S–37S.
- Moyer VA; US Preventive Services Task Force. Screening for lung cancer: US Preventive Services Task Force recommendation statement. Ann Intern Med 2014; 160:330–338.
- Bach PB, Mirkin JN, Oliver TK, et al. Benefits and harms of CT screening for lung cancer: a systematic review. JAMA 2012; 307:2418–2429.
- Lamas DJ, Kawut SM, Bagiella E, Philip N, Arcasoy SM, Lederer DJ. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med 2011; 184:842–847.
- Swensen SJ, Jett JR, Hartman TE, et al. Lung cancer screening with CT: Mayo Clinic experience. Radiology 2003; 226:756–761.
- MacRedmond R, Logan PM, Lee M, Kenny D, Foley C, Costello RW. Screening for lung cancer using low dose CT scanning. Thorax 2004; 59:237–241.
- Sverzellati N, Guerci L, Randi G, et al. Interstitial lung diseases in a lung cancer screening trial. Eur Respir J 2011; 38:392–400.
- Jin GY, Lynch D, Chawla A, et al. Interstitial lung abnormalities in a CT lung cancer screening population: prevalence and progression rate. Radiology 2013; 268:563–571.
- Heyneman LE, Ward S, Lynch DA, Remy-Jardin M, Johkoh T, Müller NL. Respiratory bronchiolitis, respiratory bronchiolitis-associated interstitial lung disease, and desquamative interstitial pneumonia: different entities or part of the spectrum of the same disease process? AJR Am J Roentgenol 1999; 173:1617–1622.
- Moon J, du Bois RM, Colby TV, Hansell DM, Nicholson AG. Clinical significance of respiratory bronchiolitis on open lung biopsy and its relationship to smoking related interstitial lung disease. Thorax 1999; 54:1009–1014.
- Holt RM, Schmidt RA, Godwin JD, Raghu G. High resolution CT in respiratory bronchiolitis-associated interstitial lung disease. J Comput Assist Tomogr 1993; 17:46–50.
- Ryu JH, Myers JL, Capizzi SA, Douglas WW, Vassallo R, Decker PA. Desquamative interstitial pneumonia and respiratory bronchiolitis-associated interstitial lung disease. Chest 2005; 127:178–184.
- Hartman TE, Primack SL, Swensen SJ, Hansell D, McGuinness G, Müller NL. Desquamative interstitial pneumonia: thin-section CT findings in 22 patients. Radiology 1993; 187:787–790.
- Akira M, Yamamoto S, Hara H, Sakatani M, Ueda E. Serial computed tomographic evaluation in desquamative interstitial pneumonia. Thorax 1997; 52:333–337.
- Lacronique J, Roth C, Battesti JP, Basset F, Chretien J. Chest radiological features of pulmonary histiocytosis X: a report based on 50 adult cases. Thorax 1982; 37:104–109.
- Remy-Jardin M, Edme JL, Boulenguez C, Remy J, Mastora I, Sobaszek A. Longitudinal follow-up study of smoker’s lung with thin-section CT in correlation with pulmonary function tests. Radiology 2002; 222:261–270.
- Mueller-Mang C, Grosse C, Schmid K, Stiebellehner L, Bankier AA. What every radiologist should know about idiopathic interstitial pneumonias. Radiographics 2007; 27:595–615.
- Katzenstein AL, Fiorelli RF. Nonspecific interstitial pneumonia/fibrosis. Histologic features and clinical significance. Am J Surg Pathol 1994; 18:136–147.
- Bryson T, Sundaram B, Khanna D, Kazerooni EA. Connective tissue disease-associated interstitial pneumonia and idiopathic interstitial pneumonia: similarity and difference. Semin Ultrasound CT MR 2014; 35:29–38.
- Desai SR, Veeraraghavan S, Hansell DM, et al. CT features of lung disease in patients with systemic sclerosis: comparison with idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia. Radiology 2004; 232:560–567.
- Tsubamoto M, Müller NL, Johkoh T, et al. Pathologic subgroups of nonspecific interstitial pneumonia: differential diagnosis from other idiopathic interstitial pneumonias on high-resolution computed tomography. J Comput Assist Tomogr 2005; 29:793–800.
- Silva CI, Müller NL, Lynch DA, et al. Chronic hypersensitivity pneumonitis: differentiation from idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia by using thin-section CT. Radiology 2008; 246:288–297.
- Antin-Ozerkis D, Rubinowitz A. An update on nonspecific interstitial pneumonia. Clin Pulm Med 2010; 17:122–128.
- Daniil ZD, Gilchrist FC, Nicholson AG, et al. A histologic pattern of nonspecific interstitial pneumonia is associated with a better prognosis than usual interstitial pneumonia in patients with cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 1999; 160:899–905.
- Travis WD, Matsui K, Moss J, Ferrans VJ. Idiopathic nonspecific interstitial pneumonia: prognostic significance of cellular and fibrosing patterns: survival comparison with usual interstitial pneumonia and desquamative interstitial pneumonia. Am J Surg Pathol 2000; 24:19–33.
- Riha RL, Duhig EE, Clarke BE, Steele RH, Slaughter RE, Zimmerman PV. Survival of patients with biopsy-proven usual interstitial pneumonia and nonspecific interstitial pneumonia. Eur Respir J 2002; 19:1114–1118.
- Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183:788–824.
- du Bois RM. An earlier and more confident diagnosis of idiopathic pulmonary fibrosis. Eur Respir Rev 2012; 21:141–146.
- Nishimura K, Kitaichi M, Izumi T, Nagai S, Kanaoka M, Itoh H. Usual interstitial pneumonia: histologic correlation with high-resolution CT. Radiology 1992; 182:337–342.
- Souza CA, Müller NL, Lee KS, Johkoh T, Mitsuhiro H, Chong S. Idiopathic interstitial pneumonias: prevalence of mediastinal lymph node enlargement in 206 patients. AJR Am J Roentgenol 2006; 186:995–999.
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370:2083–2092.
- Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370:2071–2082.
- Bradley B, Branley HM, Egan JJ, et al; British Thoracic Society Interstitial Lung Disease Guideline Group, British Thoracic Society Standards of Care Committee; Thoracic Society of Australia; New Zealand Thoracic Society; Irish Thoracic Society. Interstitial lung disease guideline: the British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax 2008; 63(suppl 5):v1–v58.
- Travis WD, Costabel U, Hansell DM, et al; ATS/ERS Committee on Idiopathic Interstitial Pneumonias. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013; 188:733–748.
- Stehlik J, Edwards LB, Kucheryavaya AY, et al; International Society of Heart and Lung Transplantation. The Registry of the International Society for Heart and Lung Transplantation: 29th official adult heart transplant report—2012. J Heart Lung Transplant 2012; 31:1052–1064.
- Oldham JM, Noth I. Idiopathic pulmonary fibrosis: early detection and referral. Respir Med 2014; 108:819–829.
KEY POINTS
- Smoking-related interstitial lung diseases can broadly be categorized as fibrotic or nonfibrotic on the basis of their appearance on CT. Fibrotic disease generally carries a worse prognosis.
- Nonfibrotic interstitial lung diseases include respiratory bronchiolitis, respiratory bronchiolitis-interstitial lung disease, desquamative interstitial pneumonia, and pulmonary Langerhans cell histiocytosis.
- Smoking-related fibrotic interstitial lung diseases include nonspecific interstitial pneumonia and usual interstitial pneumonia. A subset of usual interstitial pneumonia, called idiopathic pulmonary fibrosis, carries the worst prognosis of all.
- If CT detects interstitial lung disease during screening for lung cancer, the clinician should strongly consider further evaluation with dedicated high-resolution CT and early referral to a specialist. Smoking cessation is extremely important.
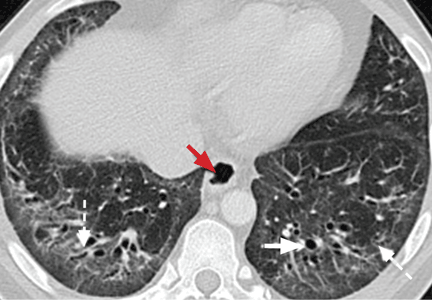
Alcohol withdrawal syndrome in medical patients
Deprived of alcohol while in the hospital, up to 80% of patients who are alcohol-dependent risk developing alcohol withdrawal syndrome,1 a potentially life-threatening condition. Clinicians should anticipate the syndrome and be ready to treat and prevent its complications.
Because alcoholism is common, nearly every provider will encounter its complications and withdrawal symptoms. Each year, an estimated 1.2 million hospital admissions are related to alcohol abuse, and about 500,000 episodes of withdrawal symptoms are severe enough to require clinical attention.1–3 Nearly 50% of patients with alcohol withdrawal syndrome are middle-class, highly functional individuals, making withdrawal difficult to recognize.1
While acute trauma patients or those with alcohol withdrawal delirium are often admitted directly to an intensive care unit (ICU), many others are at risk for or develop alcohol withdrawal syndrome and are managed initially or wholly on the acute medical unit. While specific statistics have not been published on non-ICU patients with alcohol withdrawal syndrome, they are an important group of patients who need to be well managed to prevent the progression of alcohol withdrawal syndrome to alcohol withdrawal delirium, alcohol withdrawal-induced seizure, and other complications.
This article reviews how to identify and manage alcohol withdrawal symptoms in noncritical, acutely ill medical patients, with practical recommendations for diagnosis and management.
CAN LEAD TO DELIRIUM TREMENS
In people who are physiologically dependent on alcohol, symptoms of withdrawal usually occur after abrupt cessation.4 If not addressed early in the hospitalization, alcohol withdrawal syndrome can progress to alcohol withdrawal delirium (also known as delirium tremens or DTs), in which the mortality rate is 5% to 10%.5,6 Potential mechanisms of DTs include increased dopamine release and dopamine receptor activity, hypersensitivity to N-methyl-d-aspartate, and reduced levels of gamma-aminobutyric acid (GABA).7
Long-term changes are thought to occur in neurons after repeated detoxification from alcohol, a phenomenon called “kindling.” After each detoxification, alcohol craving and obsessive thoughts increase,8 and subsequent episodes of alcohol withdrawal tend to be progressively worse.
Withdrawal symptoms
Alcohol withdrawal syndrome encompasses a spectrum of symptoms and conditions, from minor (eg, insomnia, tremulousness) to severe (seizures, DTs).2 The symptoms typically depend on the amount of alcohol consumed, the time since the last drink, and the number of previous detoxifications.9
The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition,1 states that to establish a diagnosis of alcohol withdrawal syndrome, a patient must meet four criteria:
- The patient must have ceased or reduced alcohol intake after heavy or prolonged use.
- Two or more of the following must develop within a few hours to a few days: autonomic hyperactivity (sweating or pulse greater than 100 beats per minute); increased hand tremor; insomnia; nausea or vomiting; transient visual, tactile, or auditory hallucinations or illusions; psychomotor agitation; anxiety; grand mal seizure.
- The above symptoms must cause significant distress or functional impairment.
- The symptoms must not be related to another medical condition.
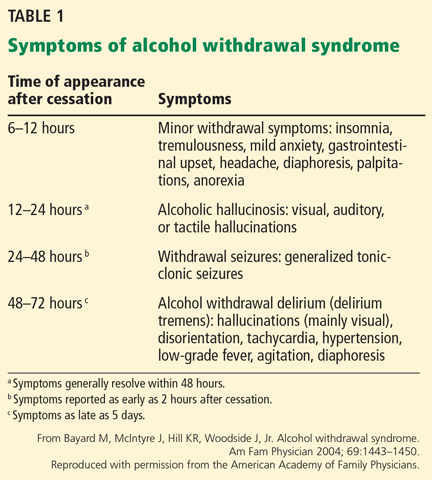
Some of the symptoms described in the second criterion above can occur while the patient still has a measurable blood alcohol level, usually within 6 hours of cessation of drinking.10 Table 1 describes the timetable of onset of symptoms and their severity.2
The elderly may be affected more severely
While the progression of the symptoms described above is commonly used for medical inpatients, the timeline may be different in an elderly patient. Compared with younger patients, elderly patients may have higher blood alcohol concentrations owing to lower total body water, so small amounts of alcohol can produce significant effects.11,12 Brower et al12 found that elderly patients experienced more withdrawal symptoms, especially cognitive impairment, weakness, and high blood pressure, and for 3 days longer.
In the elderly, alcohol may have a greater impact on the central nervous system because of increased permeability of the blood-brain barrier. And importantly, elderly patients tend to have more concomitant diseases and take more medications, all of which can affect alcohol metabolism.
ASSESSMENT SCALES FOR ALCOHOL WITHDRAWAL SYNDROME
A number of clinical scales for evaluating alcohol withdrawal have been developed. Early ones such as the 30-item Total Severity Assessment (TSA) scale and the 11-item Selected Severity Assessment (SSA) scale were limited because they were extremely detailed, burdensome to nursing staff to administer, and contained items such as daily “sleep disturbances” that were not acute enough to meet specific monitoring needs or to guide drug therapy.13,14
The Clinical Institute Withdrawal Assessment for alcohol (CIWA-A) scale, with 15 items, was derived from the SSA scale and includes acute items for assessment as often as every half-hour.15

The CIWA-Ar scale (Table 2) was developed from the CIWA-A scale by Sullivan et al.15 Using both observation and interview, it focuses on 10 areas: nausea and vomiting, tremor, paroxysmal sweats, anxiety, agitation, headache, disorientation, tactile disturbances, auditory disturbances, and visual disturbances. Scores can range from 0 to 67; a higher score indicates worse withdrawal symptoms and outcomes and therefore necessitates escalation of treatment.
The CIWA-Ar scale is now the one most commonly used in clinical trials16–20 and, we believe, in practice. Other scales, including the CIWA-AD and the Alcohol Withdrawal Scale have been validated but are not widely used in practice.14,21
BASELINE ASSESSMENT AND EARLY SUPPORTIVE CARE
A thorough history and physical examination should be performed on admission in patients known to be or suspected of being alcohol-dependent to assess the patient’s affected body systems. The time elapsed since the patient’s last alcohol drink helps predict the onset of withdrawal complications.
Baseline laboratory tests for most patients with suspected alcohol withdrawal syndrome should include a basic blood chemistry panel, complete blood cell count, and possibly an alcohol and toxicology screen, depending on the patient’s history and presentation.
Hydration and nutritional support are important in patients presenting with alcohol withdrawal syndrome. Severe disturbances in electrolytes can lead to serious complications, including cardiac arrhythmia. Close monitoring and electrolyte replacement as needed are recommended for hospitalized alcoholic patients and should follow hospital protocols.22
Thiamine and folic acid status deserve special attention, since long-standing malnutrition is common in alcoholic patients. Thiamine deficiency can result in Wernicke encephalopathy and Korsakoff syndrome, characterized by delirium, ataxia, vision changes, and amnesia. Alcohol withdrawal guidelines recommend giving thiamine intravenously for the first 2 to 5 days after admission.23 In addition, thiamine must be given before any intravenous glucose product, as thiamine is a cofactor in carbohydrate metabolism.23 Folic acid should also be supplemented, as chronic deficiencies may lead to megaloblastic or macrocytic anemia.
CIWA-Ar scale. To provide consistent monitoring and ongoing treatment, clinicians and institutions are encouraged to use a simple assessment scale that detects and quantifies alcohol withdrawal syndrome and that can be used for reassessment after an intervention.21 The CIWA-Ar scale should be used to facilitate “symptom-triggered therapy” in which, depending on the score, the patient receives pharmacologic treatment followed by a scheduled reevaluation.23,24 Most patients with a CIWA-Ar score of 8 or higher benefit from benzodiazepine therapy.16,18,19
PRIMARY DRUG THERAPIES FOR MEDICAL INPATIENTS
Benzodiazepines are the first-line agents
Benzodiazepines are the first-line agents recommended for preventing and treating alcohol withdrawal syndrome.23 Their various pharmacokinetic profiles, wide therapeutic indices, and safety compared with older sedative hypnotics make them the preferred class.23,25 No single benzodiazepine is preferred over the others for treating alcohol withdrawal syndrome: studies have shown benefits using short-acting, intermediate-acting, and long-acting agents. The choice of drug is variable and patient-specific.16,18,26
Benzodiazepines promote and enhance binding of the inhibitory neurotransmitter GABA to GABAA receptors in the central nervous system.27 As a class, benzodiazepines are all structurally related and produce the same effects—namely, sedation, hypnosis, decreased anxiety, muscle relaxation, anterograde amnesia, and anticonvulsant activity.27
The most studied benzodiazepines for treating and preventing alcohol withdrawal syndrome are chlordiazepoxide, oxazepam, and lorazepam,16–20 whereas diazepam was used in older studies.23
Diazepam and chlordiazepoxide are metabolized by oxidation, each sharing the long-acting active metabolite desmethyldiazepam (half-life 72 hours), and short-acting metabolite oxazepam (half-life 8 hours).27 In addition, the parent drugs also have varying pharmacokinetic profiles: diazepam has a half-life of more than 30 hours and chlordiazepoxide a half-life of about 8 hours. Chlordiazepoxide and diazepam’s combination of both long- and short-acting benzodiazepine activity provides long-term efficacy in attenuating withdrawal symptoms, but chlordiazepoxide’s shorter parent half-life allows more frequent dosing.
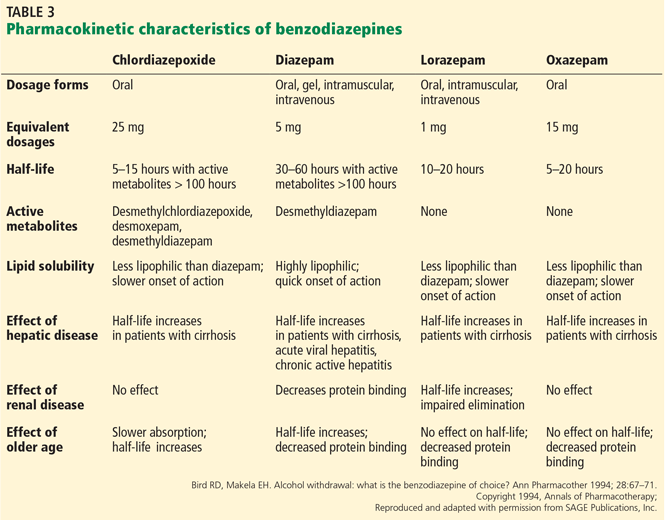
Lorazepam (half-life 10–20 hours) and oxazepam (half-life 5–20 hours) undergo glucuronide conjugation and do not have metabolites.27,28 Table 3 provides a pharmacokinetic summary.27,28
Various dosage regimens are used in giving benzodiazepines, the most common being symptom-triggered therapy, governed by assessment scales, and scheduled around-the-clock therapy.29 Current evidence supports symptom-triggered therapy in most inpatients who are not critically ill, as it can reduce both benzodiazepine use and adverse drug events and can reduce the length of stay.16,19
Trials of symptom-triggered benzodiazepine therapy
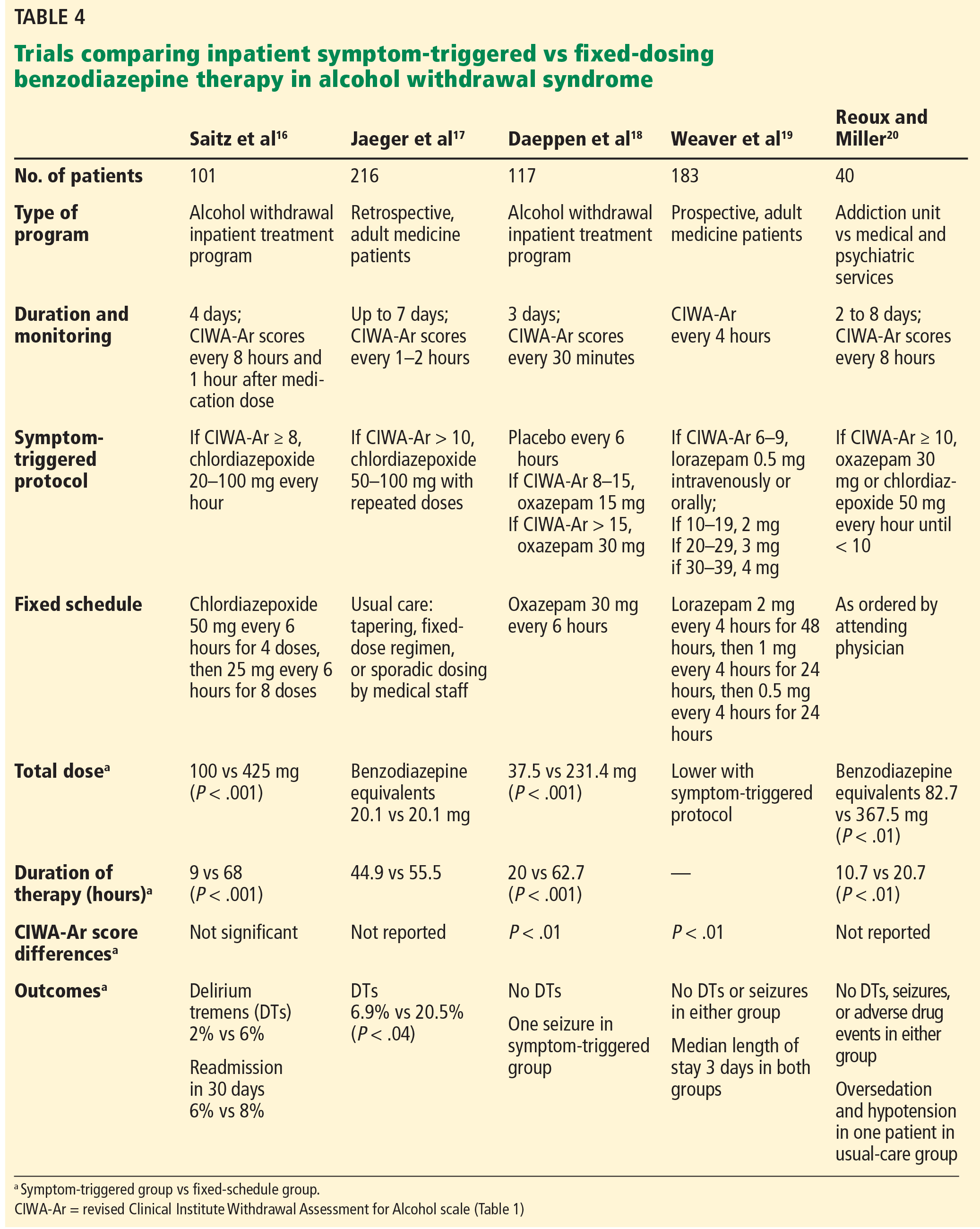
Most inpatient trials of symptom-triggered therapy (Table 4)3,16–20 used the CIWA-Ar scale for monitoring. In some of the studies, benzodiazepines were given if the score was 8 or higher, but others used cut points as high as 15 or higher. Doses:
- Chlordiazepoxide (first dose 25–100 mg)
- Lorazepam (first dose 0.5–2 mg)
- Oxazepam (30 mg).
After each dose, patients were reevaluated at intervals of 30 minutes to 8 hours.
Most of these trials showed no difference in rates of adverse drug events such as seizures, hallucinations, and lethargy with symptom-triggered therapy compared with scheduled therapy.16,18,20 They also found either no difference in the incidence of delirium tremens, or a lower incidence of delirium tremens with symptom-triggered therapy than with scheduled therapy.16–18,20
Weaver et al19 found no difference in length of stay between scheduled therapy and symptom-triggered therapy, but Saitz et al16 reported a median benzodiazepine treatment duration of 9 hours with symptom-triggered therapy vs 68 hours with fixed dosing. Thus, the study by Saitz et al suggests that hospitalization might be shorter with symptom-triggered therapy.
Many of the trials had notable limitations related to the diversity of patients enrolled and the protocols for both symptom-triggered therapy and fixed dosing. Some trials enrolled only inpatients in detoxification programs; others focused on inpatients with acute medical illness. The inpatient alcohol treatment trials16,18 excluded medically ill patients and those with concurrent psychiatric illness,16,18 and one excluded patients with seizures.16 One of the inpatient alcohol treatment program trials16 excluded patients on beta-blockers or clonidine because of concern that these drugs could mask withdrawal symptoms, whereas trials in medically ill patients allowed these same drugs.17,19,20
Most of the patients were men (approximately 75%, but ranging from 74% to 100%), and therefore the study results may not be as applicable to women.16–20 Most participants were middle-aged, with average ages in all studies between 46 and 55. Finally, the studies used a wide range of medications and dosing, with patient monitoring intervals ranging from every 30 minutes to every 8 hours.16–20
In a 2010 Cochrane analysis, Amato et al29 concluded that the limited evidence available favors symptom-triggered regimens over fixed-dosing regimens, but that differences in isolated trials should be interpreted very cautiously.
Therapeutic ethanol
Aside from the lack of evidence to support its use in alcohol withdrawal syndrome, prescribing oral ethanol to alcoholic patients clearly poses an ethical dilemma. However, giving ethanol intravenously has been studied, mostly in surgical trauma patients.30
Early reports described giving intravenous ethanol on a gram-to-gram basis to match the patient’s consumption before admission to prevent alcohol withdrawal syndrome. But later studies reported prevention of alcohol withdrawal syndrome with very small amounts of intravenous ethanol.30,31 While clinical trials have been limited to ICU patients, ethanol infusion at an initial rate of 2.5 to 5 g per hour and titrated up to 10 g per hour has appeared to be safe and effective for preventing alcohol withdrawal syndrome.30,31 The initial infusion rate of 2.5 to 5 g per hour is equivalent to 4 to 10 alcoholic beverages per 24 hours.
Nevertheless, ethanol infusion carries the potential for toxicities (eg, gastric irritation, precipitation of acute hepatic failure, hypoglycemia, pancreatitis, bone marrow suppression, prolonged wound healing) and drug interactions (eg, with anticoagulants and anticonvulsants). Thus, ethanol is neither widely used nor recommended.25,31
ADJUNCTIVE THERAPIES
Many medications are used adjunctively in the acute setting, both for symptoms of alcohol withdrawal syndrome and for agitation.
Haloperidol
No clinical trial has yet examined haloperidol monotherapy in patients with alcohol withdrawal syndrome in either general medical units or intensive care units. Yet haloperidol remains important and is recommended as an adjunct therapy for agitation.23,32 Dosing of haloperidol in protocols for surgical patients ranged from 2 to 5 mg intravenously every 0.5 to 2 hours, with a maximum dosage of 0.5 mg per kg per 24 hours.7,33
Alpha-2 agonists
Alpha-2 agonists are thought to reduce sympathetic overdrive and the autonomic symptoms associated with alcohol withdrawal syndrome, and these agents (primarily clonidine) have been studied in the treatment of alcohol withdrawal syndrome.34,35
Clonidine. In a Swedish study,34 26 men ages 20 to 55 who presented with the tremor, sweating, dysphoria, tension, anxiety, and tachycardia associated with alcohol withdrawal syndrome received clonidine 4 µg per kg twice daily or carbamazepine 200 mg three to four times daily in addition to an antiepileptic. Adjunctive use of a benzodiazepine was allowed at night in both groups. No statistically significant difference in symptom reduction was noted between the two groups, and there was no difference in total benzodiazepine use.
Dexmedetomidine, given intravenously, has been tested as an adjunct to benzodiazepine treatment in severe alcohol withdrawal syndrome. It has been shown to decrease the amount of total benzodiazepine needed compared with benzodiazepine therapy alone, but no differences have been seen in length of hospital stay.36–39 However, research on this drug so far is limited to ICU patients.
Beta-blockers
Beta-blockers have been used in inpatients with alcohol withdrawal syndrome to reduce heart rate and potentially reduce alcohol craving. However, the data are limited and conflicting.
Atenolol 50 to 100 mg daily, in a study in 120 patients, reduced length of stay (4 vs 5 days), reduced benzodiazepine use, and improved vital signs and behavior compared with placebo.40
Propranolol 40 mg every 6 hours reduced arrhythmias but increased hallucinations when used alone in a study in 47 patients.41 When used in combination with chlordiazepoxide, no benefit was seen in arrhythmia reduction.41
Barbiturates and other antiepileptics
Data continue to emerge on antiepileptics as both monotherapy and adjunctive therapy for alcohol withdrawal syndrome. Barbiturates as monotherapy were largely replaced by benzodiazepines in view of the narrow therapeutic index of barbiturates and their full agonist effect on the GABA receptor complex. However, phenobarbital has been evaluated in patients presenting with severe alcohol withdrawal syndrome or resistant alcohol withdrawal (ie, symptoms despite large or repeated doses of benzodiazepines) as an adjunct to benzodiazepines.42,43
In addition, a newer trial44 involved giving a single dose of phenobarbital in the emergency department in combination with a CIWA-Ar–based benzodiazepine protocol, compared with the benzodiazepine protocol alone. The group that received phenobarbital had fewer ICU admissions; its evaluation is ongoing.
The three other medications with the most data are carbamazepine, valproic acid, and gabapentin.45,46 However, the studies were small and the benefit was modest. Although these agents are possible alternatives in protracted alcohol withdrawal syndrome, no definite conclusion can be made regarding their place in therapy.46
RECOMMENDATIONS FOR DRUG THERAPY AND SUPPORTIVE CARE
Which benzodiazepine to use?
No specific benzodiazepine is recommended over the others for managing alcohol withdrawal syndrome, but studies best support the long-acting agent chlordiazepoxide.16,17,20 Other benzodiazepines such as lorazepam and oxazepam have proved to be beneficial, but drugs should be selected on the basis of patient characteristics and drug metabolism.18,19,27
Patients with severe liver dysfunction and the elderly may have slower oxidative metabolism, so the effects of medications that are primarily oxidized, such as chlordiazepoxide and diazepam, may be prolonged. Therefore, lorazepam and oxazepam would be preferred in these groups.47 While most patients with alcohol withdrawal syndrome and liver dysfunction do not have advanced cirrhosis, we recommend liver function testing (serum aspartate aminotransferase, alanine aminotransferase, and alkaline phosphatase levels) and screening for liver disease, given the drug metabolism and package insert caution for use in those with impaired hepatic function.48
Patients with end-stage renal disease (stage 5 chronic kidney disease) or acute kidney injury should not receive parenteral diazepam or lorazepam. The rationale is the potential accumulation of propylene glycol, the solvent used in these formulations.
In the elderly, the Beers list of drugs to avoid in older adults includes benzodiazepines, not differentiating individual benzodiazepines in terms of risk.49 However, chlordiazepoxide may be preferable to diazepam due to its shorter parent half-life and lower lipophilicity.27 Few studies have been done using benzodiazepines in elderly patients with alcohol withdrawal syndrome, but those published have shown either equivalent dosing required compared with younger patients or more severe withdrawal for which they received greater amounts of chlordiazepoxide.9,12 Lorazepam and oxazepam have less potential to accumulate in the elderly compared with the nonelderly due to the drugs’ metabolic profiles; lorazepam is the preferred agent because of its faster onset of action.47 Ultimately, the choice of benzodiazepine in elderly patients with alcohol withdrawal syndrome should be based on patient-specific characteristics.
How should benzodiazepines be dosed?
While the CIWA-Ar thresholds and subsequent dosing of benzodiazepines varied in different studies, we recommend starting benzodiazepine therapy at a CIWA-Ar score of 8 or higher, with subsequent dosing based on score reassessment. Starting doses of benzodiazepines should be chlordiazepoxide 25 to 50 mg, lorazepam 1 to 2 mg, or oxazepam 15 mg.16–20
Subsequent doses should be titrated upward, increasing by 1.5 to 2 times the previous dose and monitored at least every 1 to 2 hours after dose adjustments. Once a patient is stable and the CIWA-Ar score is less than 8, monitoring intervals can be extended to every 4 to 8 hours. If the CIWA-Ar score is more than 20, studies suggest the need for patient reevaluation for transfer to the ICU; however, some health systems have a lower threshold for this intervention.7,14,50

Dosing algorithms and CIWA-Ar goals may vary slightly from institution to institution, but it has been shown that symptom-triggered therapy works best when hospitals have a protocol for it and staff are adequately trained to assess patients with alcohol withdrawal syndrome.7,50,51 Suggestions for dose ranges and symptom-triggered therapy are shown in Table 5.
In case of benzodiazepine overdose or potential benzodiazepine-induced delirium, flumazenil could be considered.52
Patients who should not receive symptom-triggered therapy include immediate postoperative patients in whom clinicians cannot properly assess withdrawal symptoms and patients with a history of DTs.51 While controversy exists regarding the use of symptom-triggered therapy in patients with complicated medical comorbidities, there are data to support symptom-triggered therapy in some ICU patients, as it has resulted in less benzodiazepine use and reduced mechanical ventilation.53,54
There are limited data to support phenobarbital in treating resistant alcohol withdrawal syndrome, either alone or concurrently with benzodiazepines, in escalating doses ranging from 65 to 260 mg, with a maximum daily dose of 520 mg.42,55,56
Haloperidol
For patients exhibiting agitation despite benzodiazepine therapy, giving haloperidol adjunctively can be beneficial.
Haloperidol can be used in medical patients as an adjunctive therapy for agitation, but caution is advised because of the potential for a lowering of the seizure threshold, extrapyramidal effects, and risk of QTc prolongation leading to arrhythmias. Patients considered at highest risk for torsades de pointes may have a QTc of 500 msec or greater.57
Patients should also be screened for factors that have been shown to be independent predictors of QTc prolongation (female sex, diagnosis of myocardial infarction, septic shock or left ventricular dysfunction, other QT-prolonging drugs, age > 68, baseline QTc ≥ 450 msec, and hypokalemia).58 If combined predictors have been identified, it is recommended that haloperidol be avoided.
If haloperidol is to be given, a baseline electrocardiogram and electrolyte panel should be obtained, with daily electrocardiograms thereafter, as well as ongoing review of the patient’s medications to minimize drug interactions that could further increase the risk for QTc prolongation.
Suggested haloperidol dosing is 2 to 5 mg intravenously every 0.5 to 2 hours with a maximum dose of 0.5 mg/kg/24 hours.8,33 A maximum of 35 mg of intravenous haloperidol should be used in a 24-hour period to avoid QTc prolongation.57
Antihypertensive therapy
Many patients receive symptomatic relief of autonomic hyperreactivity with benzodiazepines. However, some may require additional antihypertensive therapy for cardiac adrenergic symptoms (hypertension, tachycardia) if symptoms do not resolve by treating other medical problems commonly seen in patients with alcohol withdrawal syndrome, such as dehydration and electrolyte imbalances.7
Published protocols suggest giving clonidine 0.1 mg orally every hour up to three times as needed until systolic blood pressure is less than 140 mm Hg (less than 160 mm Hg if the patient is over age 60) and diastolic pressure is less than 90 mm Hg.51 Once the patient is stabilized, the dosing can be scheduled to a maximum of 2.4 mg daily.59 However, we believe that the use of clonidine should be restricted to patients who have a substantial increase in blood pressure over baseline or are nearing a hypertensive urgency or emergency (pressures > 180/120 mm Hg) and should not be used to treat other general symptoms associated with alcohol withdrawal syndrome.42
In addition, based on limited evidence, we recommend using beta-blockers only in patients with symptomatic tachycardia or as an adjunct in hypertension management.40,41
Therapies to avoid in acutely ill medical patients
Ethanol is not recommended. Instead, intravenous benzodiazepines should be given in patients presenting with severe alcohol withdrawal syndrome.
Antiepileptics, including valproic acid, carbamazepine, and pregabalin, lack benefit in these patients either as monotherapy or as adjunctive therapy and so are not recommended.45,60–62
Magnesium supplementation (in patients with normal serum magnesium levels) should not be given, as no clinical benefit has been shown.63
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association, 2013:501.
- Bayard M, McIntyre J, Hill KR, Woodside J Jr. Alcohol withdrawal syndrome. Am Fam Physician 2004; 69:1443–1450.
- Kosten TR, O'Connor PG. Management of drug and alcohol withdrawal. N Engl J Med 2003; 348:1786–1795.
- Isbell H, Fraser HF, Wilker A, Bellevile RE, Eisenman AJ. An experimental study of the etiology of rum fits and delirium tremens. Q J Stud Alcohol 1955; 16:1–33.
- Khan A, Levy P, DeHorn S, Miller W, Compton S. Predictors of mortality in patients with delirium tremens. Acad Emerg Med 2008; 15:788–790.
- Monte R, Rabuñal R, Casariego E, López-Agreda H, Mateos A, Pértega S. Analysis of the factors determining survival of alcoholic withdrawal syndrome patients in a general hospital. Alcohol Alcohol 2010; 45:151–158.
- Stanley KM, Amabile CM, Simpson KN, Couillard D, Norcross ED, Worrall CL. Impact of an alcohol withdrawal syndrome practice guideline on surgical patient outcomes. Pharmacotherapy 2003; 23:843–854.
- Brousse G, Arnaud B, Vorspan F, et al. Alteration of glutamate/GABA balance during acute alcohol withdrawal in emergency department: a prospective analysis. Alcohol Alcohol 2012; 47:501–508.
- Liskow BI, Rinck C, Campbell J, DeSouza C. Alcohol withdrawal in the elderly. J Stud Alcohol 1989; 50:414–421.
- Etherington JM. Emergency management of acute alcohol problems. Part 1: uncomplicated withdrawal. Can Fam Physician 1996; 42:2186–2190.
- Letizia M, Reinbolz M. Identifying and managing acute alcohol withdrawal in the elderly. Geriatr Nurs 2005; 26:176–183.
- Brower KJ, Mudd S, Blow FC, Young JP, Hill EM. Severity and treatment of alcohol withdrawal in elderly versus younger patients. Alcohol Clin Exp Res 1994; 18:196–201.
- Williams D, Lewis J, McBride A. A comparison of rating scales for the alcohol-withdrawal syndrome. Alcohol Alcohol 2001; 36:104–108.
- Reoux JP, Oreskovich MR. A comparison of two versions of the Clinical Institute Withdrawal Assessment for Alcohol: the CIWA-Ar and CIWA-AD. Am J Addict 2006; 15:85–93.
- Sullivan JT, Sykora K, Schneiderman J, Naranjo CA, Sellers EM. Assessment of alcohol withdrawal: the revised Clinical Institute Withdrawal Assessment for alcohol scale (CIWA-Ar). Br J Addict 1989; 84:1353–1357.
- Saitz R, Mayo-Smith MF, Roberts MS, Redmond HA, Bernard DR, Calkins DR. Individualized treatment for alcohol withdrawal. A randomized double-blind controlled trial. JAMA 1994; 272:519–523.
- Jaeger TM, Lohr RH, Pankratz VS. Symptom-triggered therapy for alcohol withdrawal syndrome in medical inpatients. Mayo Clin Proc 2001; 76:695–701.
- Daeppen JB, Gache P, Landry U, et al. Symptom-triggered vs fixed-schedule doses of benzodiazepine for alcohol withdrawal: a randomized treatment trial. Arch Intern Med 2002; 162:1117–1121.
- Weaver MF, Hoffman HJ, Johnson RE, Mauck K. Alcohol withdrawal pharmacotherapy for inpatients with medical comorbidity. J Addict Dis 2006; 25:17–24.
- Reoux JP, Miller K. Routine hospital alcohol detoxification practice compared to symptom triggered management with an Objective Withdrawal Scale (CIWA-Ar). Am J Addict 2000; 9:135–144.
- Wetterling T, Kanitz RD, Besters B, et al. A new rating scale for the assessment of the alcohol-withdrawal syndrome (AWS scale). Alcohol Alcohol 1997; 32:753–760.
- Myrick H, Anton RF. Treatment of alcohol withdrawal. Alcohol Health Res World 1998; 22:38–43.
- Mayo-Smith MF, Beecher LH, Fischer TL, et al; Working Group on the Management of Alcohol Withdrawal Delirium, Practice Guidelines Committee, American Society of Addiction Medicine. Management of alcohol withdrawal delirium. An evidence-based practice guideline. Arch Intern Med 2004; 164:1405–1412.
- Sellers EM, Sullivan JT, Somer G, Sykora K. Characterization of DSM-III-R criteria for uncomplicated alcohol withdrawal provides an empirical basis for DSM-IV. Arch Gen Psychiatry 1991; 48:442–447.
- Sarff M, Gold JA. Alcohol withdrawal syndromes in the intensive care unit. Crit Care Med 2010; 38(suppl):S494–S501.
- Kumar CN, Andrade C, Murthy P. A randomized, double-blind comparison of lorazepam and chlordiazepoxide in patients with uncomplicated alcohol withdrawal. J Stud Alcohol Drugs 2009; 70:467–474.
- Bird RD, Makela EH. Alcohol withdrawal: what is the benzodiazepine of choice? Ann Pharmacother 1994; 28:67–71.
- Perry EC. Inpatient management of acute alcohol withdrawal syndrome. CNS Drugs 2014; 28:401–410.
- Amato L, Minozzi S, Vecchi S, Davoli M. Benzodiazepines for alcohol withdrawal. Cochrane Database Syst Rev 2010; 3:CD005063.
- Weinberg JA, Magnotti LJ, Fischer PE, et al. Comparison of intravenous ethanol versus diazepam for alcohol withdrawal prophylaxis in the trauma ICU: results of a randomized trial. J Trauma 2008; 64:99–104.
- Craft PP, Foil MB, Cunningham PR, Patselas PC, Long-Snyder BM, Collier MS. Intravenous ethanol for alcohol detoxification in trauma patients. South Med J 1994; 87:47–54.
- Ungur LA, Neuner B, John S, Wernecke K, Spies C. Prevention and therapy of alcohol withdrawal on intensive care units: systematic review of controlled trials. Alcohol Clin Exp Res 2013; 37:675–686.
- Lansford CD, Guerriero CH, Kocan MJ, et al. Improved outcomes in patients with head and neck cancer using a standardized care protocol for postoperative alcohol withdrawal. Arch Otolaryngol Head Neck Surg 2008; 134:865–872.
- Walinder J, Balldin J, Bokstrom K, Karlsson I, Lundstrom B, Svensson TH. Clonidine suppression of the alcohol withdrawal syndrome. Drug Alcohol Depend 1981; 8:345–348.
- Muzyk AJ, Fowler JA, Norwood DK, Chilipko A. Role of alpha2-agonists in the treatment of acute alcohol withdrawal. Ann Pharmacother 2011; 45:649–657.
- Crispo AL, Daley MJ, Pepin JL, Harford PH, Brown CV. Comparison of clinical outcomes in nonintubated patients with severe alcohol withdrawal syndrome treated with continuous-infusion sedatives: dexmedetomidine versus benzodiazepines. Pharmacotherapy 2014; 34:910–917.
- VanderWeide LA, Foster CJ, MacLaren R, Kiser TH, Fish DN, Mueller SW. Evaluation of early dexmedetomidine addition to the standard of care for severe alcohol withdrawal in the ICU: a retrospective controlled cohort study. J Intensive Care Med 2014. [Epub ahead of print October 16, 2014]
- Rayner SG, Weinert CR, Peng H, Jepsen S, Broccard AF. Dexmedetomidine as adjunct treatment for severe alcohol withdrawal in the ICU. Ann Intensive Care 2012; 2:12.
- Muzyk AJ, Kerns S, Brudney S, Gagliardi JP. Dexmedetomidine for the treatment of alcohol withdrawal syndrome: rationale and current status of research. CNS Drugs 2013; 27:913–920.
- Kraus ML, Gottlieb LD, Horwitz RI, Anscher M. Randomized clinical trial of atenolol in patients with alcohol withdrawal. N Engl J Med 1985; 313:905–909.
- Zilm DH, Jacob MS, MacLeod SM, Sellers EM, Ti TY. Propranolol and chlordiazepoxide effects on cardiac arrhythmias during alcohol withdrawal. Alcohol Clin Exp Res 1980; 4:400–405.
- Hack JB, Hoffmann RS, Nelson LS. Resistant alcohol withdrawal: does an unexpectedly large sedative requirement identify these patients early? J Med Toxicol 2006; 2:55–60.
- Hayner CE, Wuestefeld NL, Bolton PJ. Phenobarbital treatment in a patient with resistant alcohol withdrawal syndrome. Pharmacotherapy 2009; 29:875–878.
- Rosenson J, Clements C, Simon B, et al. Phenobarbital for acute alcohol withdrawal: a prospective randomized double-blind placebo-controlled study. J Emerg Med 2013; 44:592–598.e2.
- Prince V, Turpin KR. Treatment of alcohol withdrawal syndrome with carbamazepine, gabapentin, and nitrous oxide. Am J Health Syst Pharm 2008; 65:1039–1047.
- Leggio L, Kenna GA, Swift RM. New developments for the pharmacological treatment of alcohol withdrawal syndrome. A focus on non-benzodiazepine GABAergic medications. Prog Neuropsychopharmacol Biol Psychiatry 2008; 32:1106–1117.
- Peppers MP. Benzodiazepines for alcohol withdrawal in the elderly and in patients with liver disease. Pharmacotherapy 1996; 16:49–57.
- Valeant Pharmaceuticals North America LLC. Librium—chlordiazepoxide hydrochloride capsule, gelatin coated. http://dailymed.nlm.nih.gov/dailymed/archives/fdaDrugInfo.cfm?archiveid=125207. Accessed November 20, 2015.
- American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2012; 60:616–631.
- Hecksel KA, Bostwick JM, Jaeger TM, Cha SS. Inappropriate use of symptom-triggered therapy for alcohol withdrawal in the general hospital. Mayo Clin Proc 2008; 83:274–279.
- Manasco A, Chang S, Larriviere J, Hamm LL, Glass M. Alcohol withdrawal. South Med J 2012; 105:607–612.
- Moore PW, Donovan JW, Burkhart KK, et al. Safety and efficacy of flumazenil for reversal of iatrogenic benzodiazepine-associated delirium toxicity during treatment of alcohol withdrawal, a retrospective review at one center. J Med Toxicol 2014; 10:126–132.
- Bostwick JM, Lapid MI. False positives on the clinical institute withdrawal assessment for alcohol-revised: is this scale appropriate for use in the medically ill? Psychosomatics 2004; 45:256–261.
- de Wit M, Jones DG, Sessler CN, Zilberberg MD, Weaver MF. Alcohol-use disorders in the critically ill patient. Chest 2010; 138:994–1003.
- Young GP, Rores C, Murphy C, Dailey RH. Intravenous phenobarbital for alcohol withdrawal and convulsions. Ann Emerg Med 1987; 16:847–850.
- Hendey GW, Dery RA, Barnes RL, Snowden B, Mentler P. A prospective, randomized trial of phenobarbital versus benzodiazepines for acute alcohol withdrawal. Am J Emerg Med 2011; 29:382–385.
- Sharma ND, Rosman HS, Padhi ID, Tisdale JE. Torsades de pointes associated with intravenous haloperidol in critically ill patients. Am J Cardiol 1998; 81:238–240.
- Tisdale JE, Jaynes HA, Kingery JR, et al. Development and validation of a risk score to predict QT interval prolongation in hospitalized patients. Circ Cardiovasc Qual Outcomes 2013; 6:479–487.
- Boehringer Ingelheim Pharmaceuticals, Inc. Product Information: Catapres oral tablets, clonidine HCl oral tablets, 2012.
- Reoux JP, Saxon AJ, Malte CA, Baer JS, Sloan KL. Divalproex sodium in alcohol withdrawal: a randomized double-blind placebo-controlled clinical trial. Alcohol Clin Exp Res 2001; 25:1324–1329.
- Malcolm R, Ballenger JC, Sturgis ET, Anton R. Double-blind controlled trial comparing carbamazepine to oxazepam treatment of alcohol withdrawal. Am J Psychiatry 1989; 146:617–621.
- Förg A, Hein J, Volkmar K, et al. Efficacy and safety of pregabalin in the treatment of alcohol withdrawal syndrome: a randomized placebo-controlled trial. Alcohol Alcohol 2012; 47:149–155.
- Wilson A, Vulcano B. A double-blind, placebo-controlled trial of magnesium sulfate in the ethanol withdrawal syndrome. Alcohol Clin Exp Res 1984; 8:542–545.
Deprived of alcohol while in the hospital, up to 80% of patients who are alcohol-dependent risk developing alcohol withdrawal syndrome,1 a potentially life-threatening condition. Clinicians should anticipate the syndrome and be ready to treat and prevent its complications.
Because alcoholism is common, nearly every provider will encounter its complications and withdrawal symptoms. Each year, an estimated 1.2 million hospital admissions are related to alcohol abuse, and about 500,000 episodes of withdrawal symptoms are severe enough to require clinical attention.1–3 Nearly 50% of patients with alcohol withdrawal syndrome are middle-class, highly functional individuals, making withdrawal difficult to recognize.1
While acute trauma patients or those with alcohol withdrawal delirium are often admitted directly to an intensive care unit (ICU), many others are at risk for or develop alcohol withdrawal syndrome and are managed initially or wholly on the acute medical unit. While specific statistics have not been published on non-ICU patients with alcohol withdrawal syndrome, they are an important group of patients who need to be well managed to prevent the progression of alcohol withdrawal syndrome to alcohol withdrawal delirium, alcohol withdrawal-induced seizure, and other complications.
This article reviews how to identify and manage alcohol withdrawal symptoms in noncritical, acutely ill medical patients, with practical recommendations for diagnosis and management.
CAN LEAD TO DELIRIUM TREMENS
In people who are physiologically dependent on alcohol, symptoms of withdrawal usually occur after abrupt cessation.4 If not addressed early in the hospitalization, alcohol withdrawal syndrome can progress to alcohol withdrawal delirium (also known as delirium tremens or DTs), in which the mortality rate is 5% to 10%.5,6 Potential mechanisms of DTs include increased dopamine release and dopamine receptor activity, hypersensitivity to N-methyl-d-aspartate, and reduced levels of gamma-aminobutyric acid (GABA).7
Long-term changes are thought to occur in neurons after repeated detoxification from alcohol, a phenomenon called “kindling.” After each detoxification, alcohol craving and obsessive thoughts increase,8 and subsequent episodes of alcohol withdrawal tend to be progressively worse.
Withdrawal symptoms
Alcohol withdrawal syndrome encompasses a spectrum of symptoms and conditions, from minor (eg, insomnia, tremulousness) to severe (seizures, DTs).2 The symptoms typically depend on the amount of alcohol consumed, the time since the last drink, and the number of previous detoxifications.9
The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition,1 states that to establish a diagnosis of alcohol withdrawal syndrome, a patient must meet four criteria:
- The patient must have ceased or reduced alcohol intake after heavy or prolonged use.
- Two or more of the following must develop within a few hours to a few days: autonomic hyperactivity (sweating or pulse greater than 100 beats per minute); increased hand tremor; insomnia; nausea or vomiting; transient visual, tactile, or auditory hallucinations or illusions; psychomotor agitation; anxiety; grand mal seizure.
- The above symptoms must cause significant distress or functional impairment.
- The symptoms must not be related to another medical condition.
Some of the symptoms described in the second criterion above can occur while the patient still has a measurable blood alcohol level, usually within 6 hours of cessation of drinking.10 Table 1 describes the timetable of onset of symptoms and their severity.2
The elderly may be affected more severely
While the progression of the symptoms described above is commonly used for medical inpatients, the timeline may be different in an elderly patient. Compared with younger patients, elderly patients may have higher blood alcohol concentrations owing to lower total body water, so small amounts of alcohol can produce significant effects.11,12 Brower et al12 found that elderly patients experienced more withdrawal symptoms, especially cognitive impairment, weakness, and high blood pressure, and for 3 days longer.
In the elderly, alcohol may have a greater impact on the central nervous system because of increased permeability of the blood-brain barrier. And importantly, elderly patients tend to have more concomitant diseases and take more medications, all of which can affect alcohol metabolism.
ASSESSMENT SCALES FOR ALCOHOL WITHDRAWAL SYNDROME
A number of clinical scales for evaluating alcohol withdrawal have been developed. Early ones such as the 30-item Total Severity Assessment (TSA) scale and the 11-item Selected Severity Assessment (SSA) scale were limited because they were extremely detailed, burdensome to nursing staff to administer, and contained items such as daily “sleep disturbances” that were not acute enough to meet specific monitoring needs or to guide drug therapy.13,14
The Clinical Institute Withdrawal Assessment for alcohol (CIWA-A) scale, with 15 items, was derived from the SSA scale and includes acute items for assessment as often as every half-hour.15
The CIWA-Ar scale (Table 2) was developed from the CIWA-A scale by Sullivan et al.15 Using both observation and interview, it focuses on 10 areas: nausea and vomiting, tremor, paroxysmal sweats, anxiety, agitation, headache, disorientation, tactile disturbances, auditory disturbances, and visual disturbances. Scores can range from 0 to 67; a higher score indicates worse withdrawal symptoms and outcomes and therefore necessitates escalation of treatment.
The CIWA-Ar scale is now the one most commonly used in clinical trials16–20 and, we believe, in practice. Other scales, including the CIWA-AD and the Alcohol Withdrawal Scale have been validated but are not widely used in practice.14,21
BASELINE ASSESSMENT AND EARLY SUPPORTIVE CARE
A thorough history and physical examination should be performed on admission in patients known to be or suspected of being alcohol-dependent to assess the patient’s affected body systems. The time elapsed since the patient’s last alcohol drink helps predict the onset of withdrawal complications.
Baseline laboratory tests for most patients with suspected alcohol withdrawal syndrome should include a basic blood chemistry panel, complete blood cell count, and possibly an alcohol and toxicology screen, depending on the patient’s history and presentation.
Hydration and nutritional support are important in patients presenting with alcohol withdrawal syndrome. Severe disturbances in electrolytes can lead to serious complications, including cardiac arrhythmia. Close monitoring and electrolyte replacement as needed are recommended for hospitalized alcoholic patients and should follow hospital protocols.22
Thiamine and folic acid status deserve special attention, since long-standing malnutrition is common in alcoholic patients. Thiamine deficiency can result in Wernicke encephalopathy and Korsakoff syndrome, characterized by delirium, ataxia, vision changes, and amnesia. Alcohol withdrawal guidelines recommend giving thiamine intravenously for the first 2 to 5 days after admission.23 In addition, thiamine must be given before any intravenous glucose product, as thiamine is a cofactor in carbohydrate metabolism.23 Folic acid should also be supplemented, as chronic deficiencies may lead to megaloblastic or macrocytic anemia.
CIWA-Ar scale. To provide consistent monitoring and ongoing treatment, clinicians and institutions are encouraged to use a simple assessment scale that detects and quantifies alcohol withdrawal syndrome and that can be used for reassessment after an intervention.21 The CIWA-Ar scale should be used to facilitate “symptom-triggered therapy” in which, depending on the score, the patient receives pharmacologic treatment followed by a scheduled reevaluation.23,24 Most patients with a CIWA-Ar score of 8 or higher benefit from benzodiazepine therapy.16,18,19
PRIMARY DRUG THERAPIES FOR MEDICAL INPATIENTS
Benzodiazepines are the first-line agents
Benzodiazepines are the first-line agents recommended for preventing and treating alcohol withdrawal syndrome.23 Their various pharmacokinetic profiles, wide therapeutic indices, and safety compared with older sedative hypnotics make them the preferred class.23,25 No single benzodiazepine is preferred over the others for treating alcohol withdrawal syndrome: studies have shown benefits using short-acting, intermediate-acting, and long-acting agents. The choice of drug is variable and patient-specific.16,18,26
Benzodiazepines promote and enhance binding of the inhibitory neurotransmitter GABA to GABAA receptors in the central nervous system.27 As a class, benzodiazepines are all structurally related and produce the same effects—namely, sedation, hypnosis, decreased anxiety, muscle relaxation, anterograde amnesia, and anticonvulsant activity.27
The most studied benzodiazepines for treating and preventing alcohol withdrawal syndrome are chlordiazepoxide, oxazepam, and lorazepam,16–20 whereas diazepam was used in older studies.23
Diazepam and chlordiazepoxide are metabolized by oxidation, each sharing the long-acting active metabolite desmethyldiazepam (half-life 72 hours), and short-acting metabolite oxazepam (half-life 8 hours).27 In addition, the parent drugs also have varying pharmacokinetic profiles: diazepam has a half-life of more than 30 hours and chlordiazepoxide a half-life of about 8 hours. Chlordiazepoxide and diazepam’s combination of both long- and short-acting benzodiazepine activity provides long-term efficacy in attenuating withdrawal symptoms, but chlordiazepoxide’s shorter parent half-life allows more frequent dosing.
Lorazepam (half-life 10–20 hours) and oxazepam (half-life 5–20 hours) undergo glucuronide conjugation and do not have metabolites.27,28 Table 3 provides a pharmacokinetic summary.27,28
Various dosage regimens are used in giving benzodiazepines, the most common being symptom-triggered therapy, governed by assessment scales, and scheduled around-the-clock therapy.29 Current evidence supports symptom-triggered therapy in most inpatients who are not critically ill, as it can reduce both benzodiazepine use and adverse drug events and can reduce the length of stay.16,19
Trials of symptom-triggered benzodiazepine therapy
Most inpatient trials of symptom-triggered therapy (Table 4)3,16–20 used the CIWA-Ar scale for monitoring. In some of the studies, benzodiazepines were given if the score was 8 or higher, but others used cut points as high as 15 or higher. Doses:
- Chlordiazepoxide (first dose 25–100 mg)
- Lorazepam (first dose 0.5–2 mg)
- Oxazepam (30 mg).
After each dose, patients were reevaluated at intervals of 30 minutes to 8 hours.
Most of these trials showed no difference in rates of adverse drug events such as seizures, hallucinations, and lethargy with symptom-triggered therapy compared with scheduled therapy.16,18,20 They also found either no difference in the incidence of delirium tremens, or a lower incidence of delirium tremens with symptom-triggered therapy than with scheduled therapy.16–18,20
Weaver et al19 found no difference in length of stay between scheduled therapy and symptom-triggered therapy, but Saitz et al16 reported a median benzodiazepine treatment duration of 9 hours with symptom-triggered therapy vs 68 hours with fixed dosing. Thus, the study by Saitz et al suggests that hospitalization might be shorter with symptom-triggered therapy.
Many of the trials had notable limitations related to the diversity of patients enrolled and the protocols for both symptom-triggered therapy and fixed dosing. Some trials enrolled only inpatients in detoxification programs; others focused on inpatients with acute medical illness. The inpatient alcohol treatment trials16,18 excluded medically ill patients and those with concurrent psychiatric illness,16,18 and one excluded patients with seizures.16 One of the inpatient alcohol treatment program trials16 excluded patients on beta-blockers or clonidine because of concern that these drugs could mask withdrawal symptoms, whereas trials in medically ill patients allowed these same drugs.17,19,20
Most of the patients were men (approximately 75%, but ranging from 74% to 100%), and therefore the study results may not be as applicable to women.16–20 Most participants were middle-aged, with average ages in all studies between 46 and 55. Finally, the studies used a wide range of medications and dosing, with patient monitoring intervals ranging from every 30 minutes to every 8 hours.16–20
In a 2010 Cochrane analysis, Amato et al29 concluded that the limited evidence available favors symptom-triggered regimens over fixed-dosing regimens, but that differences in isolated trials should be interpreted very cautiously.
Therapeutic ethanol
Aside from the lack of evidence to support its use in alcohol withdrawal syndrome, prescribing oral ethanol to alcoholic patients clearly poses an ethical dilemma. However, giving ethanol intravenously has been studied, mostly in surgical trauma patients.30
Early reports described giving intravenous ethanol on a gram-to-gram basis to match the patient’s consumption before admission to prevent alcohol withdrawal syndrome. But later studies reported prevention of alcohol withdrawal syndrome with very small amounts of intravenous ethanol.30,31 While clinical trials have been limited to ICU patients, ethanol infusion at an initial rate of 2.5 to 5 g per hour and titrated up to 10 g per hour has appeared to be safe and effective for preventing alcohol withdrawal syndrome.30,31 The initial infusion rate of 2.5 to 5 g per hour is equivalent to 4 to 10 alcoholic beverages per 24 hours.
Nevertheless, ethanol infusion carries the potential for toxicities (eg, gastric irritation, precipitation of acute hepatic failure, hypoglycemia, pancreatitis, bone marrow suppression, prolonged wound healing) and drug interactions (eg, with anticoagulants and anticonvulsants). Thus, ethanol is neither widely used nor recommended.25,31
ADJUNCTIVE THERAPIES
Many medications are used adjunctively in the acute setting, both for symptoms of alcohol withdrawal syndrome and for agitation.
Haloperidol
No clinical trial has yet examined haloperidol monotherapy in patients with alcohol withdrawal syndrome in either general medical units or intensive care units. Yet haloperidol remains important and is recommended as an adjunct therapy for agitation.23,32 Dosing of haloperidol in protocols for surgical patients ranged from 2 to 5 mg intravenously every 0.5 to 2 hours, with a maximum dosage of 0.5 mg per kg per 24 hours.7,33
Alpha-2 agonists
Alpha-2 agonists are thought to reduce sympathetic overdrive and the autonomic symptoms associated with alcohol withdrawal syndrome, and these agents (primarily clonidine) have been studied in the treatment of alcohol withdrawal syndrome.34,35
Clonidine. In a Swedish study,34 26 men ages 20 to 55 who presented with the tremor, sweating, dysphoria, tension, anxiety, and tachycardia associated with alcohol withdrawal syndrome received clonidine 4 µg per kg twice daily or carbamazepine 200 mg three to four times daily in addition to an antiepileptic. Adjunctive use of a benzodiazepine was allowed at night in both groups. No statistically significant difference in symptom reduction was noted between the two groups, and there was no difference in total benzodiazepine use.
Dexmedetomidine, given intravenously, has been tested as an adjunct to benzodiazepine treatment in severe alcohol withdrawal syndrome. It has been shown to decrease the amount of total benzodiazepine needed compared with benzodiazepine therapy alone, but no differences have been seen in length of hospital stay.36–39 However, research on this drug so far is limited to ICU patients.
Beta-blockers
Beta-blockers have been used in inpatients with alcohol withdrawal syndrome to reduce heart rate and potentially reduce alcohol craving. However, the data are limited and conflicting.
Atenolol 50 to 100 mg daily, in a study in 120 patients, reduced length of stay (4 vs 5 days), reduced benzodiazepine use, and improved vital signs and behavior compared with placebo.40
Propranolol 40 mg every 6 hours reduced arrhythmias but increased hallucinations when used alone in a study in 47 patients.41 When used in combination with chlordiazepoxide, no benefit was seen in arrhythmia reduction.41
Barbiturates and other antiepileptics
Data continue to emerge on antiepileptics as both monotherapy and adjunctive therapy for alcohol withdrawal syndrome. Barbiturates as monotherapy were largely replaced by benzodiazepines in view of the narrow therapeutic index of barbiturates and their full agonist effect on the GABA receptor complex. However, phenobarbital has been evaluated in patients presenting with severe alcohol withdrawal syndrome or resistant alcohol withdrawal (ie, symptoms despite large or repeated doses of benzodiazepines) as an adjunct to benzodiazepines.42,43
In addition, a newer trial44 involved giving a single dose of phenobarbital in the emergency department in combination with a CIWA-Ar–based benzodiazepine protocol, compared with the benzodiazepine protocol alone. The group that received phenobarbital had fewer ICU admissions; its evaluation is ongoing.
The three other medications with the most data are carbamazepine, valproic acid, and gabapentin.45,46 However, the studies were small and the benefit was modest. Although these agents are possible alternatives in protracted alcohol withdrawal syndrome, no definite conclusion can be made regarding their place in therapy.46
RECOMMENDATIONS FOR DRUG THERAPY AND SUPPORTIVE CARE
Which benzodiazepine to use?
No specific benzodiazepine is recommended over the others for managing alcohol withdrawal syndrome, but studies best support the long-acting agent chlordiazepoxide.16,17,20 Other benzodiazepines such as lorazepam and oxazepam have proved to be beneficial, but drugs should be selected on the basis of patient characteristics and drug metabolism.18,19,27
Patients with severe liver dysfunction and the elderly may have slower oxidative metabolism, so the effects of medications that are primarily oxidized, such as chlordiazepoxide and diazepam, may be prolonged. Therefore, lorazepam and oxazepam would be preferred in these groups.47 While most patients with alcohol withdrawal syndrome and liver dysfunction do not have advanced cirrhosis, we recommend liver function testing (serum aspartate aminotransferase, alanine aminotransferase, and alkaline phosphatase levels) and screening for liver disease, given the drug metabolism and package insert caution for use in those with impaired hepatic function.48
Patients with end-stage renal disease (stage 5 chronic kidney disease) or acute kidney injury should not receive parenteral diazepam or lorazepam. The rationale is the potential accumulation of propylene glycol, the solvent used in these formulations.
In the elderly, the Beers list of drugs to avoid in older adults includes benzodiazepines, not differentiating individual benzodiazepines in terms of risk.49 However, chlordiazepoxide may be preferable to diazepam due to its shorter parent half-life and lower lipophilicity.27 Few studies have been done using benzodiazepines in elderly patients with alcohol withdrawal syndrome, but those published have shown either equivalent dosing required compared with younger patients or more severe withdrawal for which they received greater amounts of chlordiazepoxide.9,12 Lorazepam and oxazepam have less potential to accumulate in the elderly compared with the nonelderly due to the drugs’ metabolic profiles; lorazepam is the preferred agent because of its faster onset of action.47 Ultimately, the choice of benzodiazepine in elderly patients with alcohol withdrawal syndrome should be based on patient-specific characteristics.
How should benzodiazepines be dosed?
While the CIWA-Ar thresholds and subsequent dosing of benzodiazepines varied in different studies, we recommend starting benzodiazepine therapy at a CIWA-Ar score of 8 or higher, with subsequent dosing based on score reassessment. Starting doses of benzodiazepines should be chlordiazepoxide 25 to 50 mg, lorazepam 1 to 2 mg, or oxazepam 15 mg.16–20
Subsequent doses should be titrated upward, increasing by 1.5 to 2 times the previous dose and monitored at least every 1 to 2 hours after dose adjustments. Once a patient is stable and the CIWA-Ar score is less than 8, monitoring intervals can be extended to every 4 to 8 hours. If the CIWA-Ar score is more than 20, studies suggest the need for patient reevaluation for transfer to the ICU; however, some health systems have a lower threshold for this intervention.7,14,50
Dosing algorithms and CIWA-Ar goals may vary slightly from institution to institution, but it has been shown that symptom-triggered therapy works best when hospitals have a protocol for it and staff are adequately trained to assess patients with alcohol withdrawal syndrome.7,50,51 Suggestions for dose ranges and symptom-triggered therapy are shown in Table 5.
In case of benzodiazepine overdose or potential benzodiazepine-induced delirium, flumazenil could be considered.52
Patients who should not receive symptom-triggered therapy include immediate postoperative patients in whom clinicians cannot properly assess withdrawal symptoms and patients with a history of DTs.51 While controversy exists regarding the use of symptom-triggered therapy in patients with complicated medical comorbidities, there are data to support symptom-triggered therapy in some ICU patients, as it has resulted in less benzodiazepine use and reduced mechanical ventilation.53,54
There are limited data to support phenobarbital in treating resistant alcohol withdrawal syndrome, either alone or concurrently with benzodiazepines, in escalating doses ranging from 65 to 260 mg, with a maximum daily dose of 520 mg.42,55,56
Haloperidol
For patients exhibiting agitation despite benzodiazepine therapy, giving haloperidol adjunctively can be beneficial.
Haloperidol can be used in medical patients as an adjunctive therapy for agitation, but caution is advised because of the potential for a lowering of the seizure threshold, extrapyramidal effects, and risk of QTc prolongation leading to arrhythmias. Patients considered at highest risk for torsades de pointes may have a QTc of 500 msec or greater.57
Patients should also be screened for factors that have been shown to be independent predictors of QTc prolongation (female sex, diagnosis of myocardial infarction, septic shock or left ventricular dysfunction, other QT-prolonging drugs, age > 68, baseline QTc ≥ 450 msec, and hypokalemia).58 If combined predictors have been identified, it is recommended that haloperidol be avoided.
If haloperidol is to be given, a baseline electrocardiogram and electrolyte panel should be obtained, with daily electrocardiograms thereafter, as well as ongoing review of the patient’s medications to minimize drug interactions that could further increase the risk for QTc prolongation.
Suggested haloperidol dosing is 2 to 5 mg intravenously every 0.5 to 2 hours with a maximum dose of 0.5 mg/kg/24 hours.8,33 A maximum of 35 mg of intravenous haloperidol should be used in a 24-hour period to avoid QTc prolongation.57
Antihypertensive therapy
Many patients receive symptomatic relief of autonomic hyperreactivity with benzodiazepines. However, some may require additional antihypertensive therapy for cardiac adrenergic symptoms (hypertension, tachycardia) if symptoms do not resolve by treating other medical problems commonly seen in patients with alcohol withdrawal syndrome, such as dehydration and electrolyte imbalances.7
Published protocols suggest giving clonidine 0.1 mg orally every hour up to three times as needed until systolic blood pressure is less than 140 mm Hg (less than 160 mm Hg if the patient is over age 60) and diastolic pressure is less than 90 mm Hg.51 Once the patient is stabilized, the dosing can be scheduled to a maximum of 2.4 mg daily.59 However, we believe that the use of clonidine should be restricted to patients who have a substantial increase in blood pressure over baseline or are nearing a hypertensive urgency or emergency (pressures > 180/120 mm Hg) and should not be used to treat other general symptoms associated with alcohol withdrawal syndrome.42
In addition, based on limited evidence, we recommend using beta-blockers only in patients with symptomatic tachycardia or as an adjunct in hypertension management.40,41
Therapies to avoid in acutely ill medical patients
Ethanol is not recommended. Instead, intravenous benzodiazepines should be given in patients presenting with severe alcohol withdrawal syndrome.
Antiepileptics, including valproic acid, carbamazepine, and pregabalin, lack benefit in these patients either as monotherapy or as adjunctive therapy and so are not recommended.45,60–62
Magnesium supplementation (in patients with normal serum magnesium levels) should not be given, as no clinical benefit has been shown.63
Deprived of alcohol while in the hospital, up to 80% of patients who are alcohol-dependent risk developing alcohol withdrawal syndrome,1 a potentially life-threatening condition. Clinicians should anticipate the syndrome and be ready to treat and prevent its complications.
Because alcoholism is common, nearly every provider will encounter its complications and withdrawal symptoms. Each year, an estimated 1.2 million hospital admissions are related to alcohol abuse, and about 500,000 episodes of withdrawal symptoms are severe enough to require clinical attention.1–3 Nearly 50% of patients with alcohol withdrawal syndrome are middle-class, highly functional individuals, making withdrawal difficult to recognize.1
While acute trauma patients or those with alcohol withdrawal delirium are often admitted directly to an intensive care unit (ICU), many others are at risk for or develop alcohol withdrawal syndrome and are managed initially or wholly on the acute medical unit. While specific statistics have not been published on non-ICU patients with alcohol withdrawal syndrome, they are an important group of patients who need to be well managed to prevent the progression of alcohol withdrawal syndrome to alcohol withdrawal delirium, alcohol withdrawal-induced seizure, and other complications.
This article reviews how to identify and manage alcohol withdrawal symptoms in noncritical, acutely ill medical patients, with practical recommendations for diagnosis and management.
CAN LEAD TO DELIRIUM TREMENS
In people who are physiologically dependent on alcohol, symptoms of withdrawal usually occur after abrupt cessation.4 If not addressed early in the hospitalization, alcohol withdrawal syndrome can progress to alcohol withdrawal delirium (also known as delirium tremens or DTs), in which the mortality rate is 5% to 10%.5,6 Potential mechanisms of DTs include increased dopamine release and dopamine receptor activity, hypersensitivity to N-methyl-d-aspartate, and reduced levels of gamma-aminobutyric acid (GABA).7
Long-term changes are thought to occur in neurons after repeated detoxification from alcohol, a phenomenon called “kindling.” After each detoxification, alcohol craving and obsessive thoughts increase,8 and subsequent episodes of alcohol withdrawal tend to be progressively worse.
Withdrawal symptoms
Alcohol withdrawal syndrome encompasses a spectrum of symptoms and conditions, from minor (eg, insomnia, tremulousness) to severe (seizures, DTs).2 The symptoms typically depend on the amount of alcohol consumed, the time since the last drink, and the number of previous detoxifications.9
The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition,1 states that to establish a diagnosis of alcohol withdrawal syndrome, a patient must meet four criteria:
- The patient must have ceased or reduced alcohol intake after heavy or prolonged use.
- Two or more of the following must develop within a few hours to a few days: autonomic hyperactivity (sweating or pulse greater than 100 beats per minute); increased hand tremor; insomnia; nausea or vomiting; transient visual, tactile, or auditory hallucinations or illusions; psychomotor agitation; anxiety; grand mal seizure.
- The above symptoms must cause significant distress or functional impairment.
- The symptoms must not be related to another medical condition.
Some of the symptoms described in the second criterion above can occur while the patient still has a measurable blood alcohol level, usually within 6 hours of cessation of drinking.10 Table 1 describes the timetable of onset of symptoms and their severity.2
The elderly may be affected more severely
While the progression of the symptoms described above is commonly used for medical inpatients, the timeline may be different in an elderly patient. Compared with younger patients, elderly patients may have higher blood alcohol concentrations owing to lower total body water, so small amounts of alcohol can produce significant effects.11,12 Brower et al12 found that elderly patients experienced more withdrawal symptoms, especially cognitive impairment, weakness, and high blood pressure, and for 3 days longer.
In the elderly, alcohol may have a greater impact on the central nervous system because of increased permeability of the blood-brain barrier. And importantly, elderly patients tend to have more concomitant diseases and take more medications, all of which can affect alcohol metabolism.
ASSESSMENT SCALES FOR ALCOHOL WITHDRAWAL SYNDROME
A number of clinical scales for evaluating alcohol withdrawal have been developed. Early ones such as the 30-item Total Severity Assessment (TSA) scale and the 11-item Selected Severity Assessment (SSA) scale were limited because they were extremely detailed, burdensome to nursing staff to administer, and contained items such as daily “sleep disturbances” that were not acute enough to meet specific monitoring needs or to guide drug therapy.13,14
The Clinical Institute Withdrawal Assessment for alcohol (CIWA-A) scale, with 15 items, was derived from the SSA scale and includes acute items for assessment as often as every half-hour.15
The CIWA-Ar scale (Table 2) was developed from the CIWA-A scale by Sullivan et al.15 Using both observation and interview, it focuses on 10 areas: nausea and vomiting, tremor, paroxysmal sweats, anxiety, agitation, headache, disorientation, tactile disturbances, auditory disturbances, and visual disturbances. Scores can range from 0 to 67; a higher score indicates worse withdrawal symptoms and outcomes and therefore necessitates escalation of treatment.
The CIWA-Ar scale is now the one most commonly used in clinical trials16–20 and, we believe, in practice. Other scales, including the CIWA-AD and the Alcohol Withdrawal Scale have been validated but are not widely used in practice.14,21
BASELINE ASSESSMENT AND EARLY SUPPORTIVE CARE
A thorough history and physical examination should be performed on admission in patients known to be or suspected of being alcohol-dependent to assess the patient’s affected body systems. The time elapsed since the patient’s last alcohol drink helps predict the onset of withdrawal complications.
Baseline laboratory tests for most patients with suspected alcohol withdrawal syndrome should include a basic blood chemistry panel, complete blood cell count, and possibly an alcohol and toxicology screen, depending on the patient’s history and presentation.
Hydration and nutritional support are important in patients presenting with alcohol withdrawal syndrome. Severe disturbances in electrolytes can lead to serious complications, including cardiac arrhythmia. Close monitoring and electrolyte replacement as needed are recommended for hospitalized alcoholic patients and should follow hospital protocols.22
Thiamine and folic acid status deserve special attention, since long-standing malnutrition is common in alcoholic patients. Thiamine deficiency can result in Wernicke encephalopathy and Korsakoff syndrome, characterized by delirium, ataxia, vision changes, and amnesia. Alcohol withdrawal guidelines recommend giving thiamine intravenously for the first 2 to 5 days after admission.23 In addition, thiamine must be given before any intravenous glucose product, as thiamine is a cofactor in carbohydrate metabolism.23 Folic acid should also be supplemented, as chronic deficiencies may lead to megaloblastic or macrocytic anemia.
CIWA-Ar scale. To provide consistent monitoring and ongoing treatment, clinicians and institutions are encouraged to use a simple assessment scale that detects and quantifies alcohol withdrawal syndrome and that can be used for reassessment after an intervention.21 The CIWA-Ar scale should be used to facilitate “symptom-triggered therapy” in which, depending on the score, the patient receives pharmacologic treatment followed by a scheduled reevaluation.23,24 Most patients with a CIWA-Ar score of 8 or higher benefit from benzodiazepine therapy.16,18,19
PRIMARY DRUG THERAPIES FOR MEDICAL INPATIENTS
Benzodiazepines are the first-line agents
Benzodiazepines are the first-line agents recommended for preventing and treating alcohol withdrawal syndrome.23 Their various pharmacokinetic profiles, wide therapeutic indices, and safety compared with older sedative hypnotics make them the preferred class.23,25 No single benzodiazepine is preferred over the others for treating alcohol withdrawal syndrome: studies have shown benefits using short-acting, intermediate-acting, and long-acting agents. The choice of drug is variable and patient-specific.16,18,26
Benzodiazepines promote and enhance binding of the inhibitory neurotransmitter GABA to GABAA receptors in the central nervous system.27 As a class, benzodiazepines are all structurally related and produce the same effects—namely, sedation, hypnosis, decreased anxiety, muscle relaxation, anterograde amnesia, and anticonvulsant activity.27
The most studied benzodiazepines for treating and preventing alcohol withdrawal syndrome are chlordiazepoxide, oxazepam, and lorazepam,16–20 whereas diazepam was used in older studies.23
Diazepam and chlordiazepoxide are metabolized by oxidation, each sharing the long-acting active metabolite desmethyldiazepam (half-life 72 hours), and short-acting metabolite oxazepam (half-life 8 hours).27 In addition, the parent drugs also have varying pharmacokinetic profiles: diazepam has a half-life of more than 30 hours and chlordiazepoxide a half-life of about 8 hours. Chlordiazepoxide and diazepam’s combination of both long- and short-acting benzodiazepine activity provides long-term efficacy in attenuating withdrawal symptoms, but chlordiazepoxide’s shorter parent half-life allows more frequent dosing.
Lorazepam (half-life 10–20 hours) and oxazepam (half-life 5–20 hours) undergo glucuronide conjugation and do not have metabolites.27,28 Table 3 provides a pharmacokinetic summary.27,28
Various dosage regimens are used in giving benzodiazepines, the most common being symptom-triggered therapy, governed by assessment scales, and scheduled around-the-clock therapy.29 Current evidence supports symptom-triggered therapy in most inpatients who are not critically ill, as it can reduce both benzodiazepine use and adverse drug events and can reduce the length of stay.16,19
Trials of symptom-triggered benzodiazepine therapy
Most inpatient trials of symptom-triggered therapy (Table 4)3,16–20 used the CIWA-Ar scale for monitoring. In some of the studies, benzodiazepines were given if the score was 8 or higher, but others used cut points as high as 15 or higher. Doses:
- Chlordiazepoxide (first dose 25–100 mg)
- Lorazepam (first dose 0.5–2 mg)
- Oxazepam (30 mg).
After each dose, patients were reevaluated at intervals of 30 minutes to 8 hours.
Most of these trials showed no difference in rates of adverse drug events such as seizures, hallucinations, and lethargy with symptom-triggered therapy compared with scheduled therapy.16,18,20 They also found either no difference in the incidence of delirium tremens, or a lower incidence of delirium tremens with symptom-triggered therapy than with scheduled therapy.16–18,20
Weaver et al19 found no difference in length of stay between scheduled therapy and symptom-triggered therapy, but Saitz et al16 reported a median benzodiazepine treatment duration of 9 hours with symptom-triggered therapy vs 68 hours with fixed dosing. Thus, the study by Saitz et al suggests that hospitalization might be shorter with symptom-triggered therapy.
Many of the trials had notable limitations related to the diversity of patients enrolled and the protocols for both symptom-triggered therapy and fixed dosing. Some trials enrolled only inpatients in detoxification programs; others focused on inpatients with acute medical illness. The inpatient alcohol treatment trials16,18 excluded medically ill patients and those with concurrent psychiatric illness,16,18 and one excluded patients with seizures.16 One of the inpatient alcohol treatment program trials16 excluded patients on beta-blockers or clonidine because of concern that these drugs could mask withdrawal symptoms, whereas trials in medically ill patients allowed these same drugs.17,19,20
Most of the patients were men (approximately 75%, but ranging from 74% to 100%), and therefore the study results may not be as applicable to women.16–20 Most participants were middle-aged, with average ages in all studies between 46 and 55. Finally, the studies used a wide range of medications and dosing, with patient monitoring intervals ranging from every 30 minutes to every 8 hours.16–20
In a 2010 Cochrane analysis, Amato et al29 concluded that the limited evidence available favors symptom-triggered regimens over fixed-dosing regimens, but that differences in isolated trials should be interpreted very cautiously.
Therapeutic ethanol
Aside from the lack of evidence to support its use in alcohol withdrawal syndrome, prescribing oral ethanol to alcoholic patients clearly poses an ethical dilemma. However, giving ethanol intravenously has been studied, mostly in surgical trauma patients.30
Early reports described giving intravenous ethanol on a gram-to-gram basis to match the patient’s consumption before admission to prevent alcohol withdrawal syndrome. But later studies reported prevention of alcohol withdrawal syndrome with very small amounts of intravenous ethanol.30,31 While clinical trials have been limited to ICU patients, ethanol infusion at an initial rate of 2.5 to 5 g per hour and titrated up to 10 g per hour has appeared to be safe and effective for preventing alcohol withdrawal syndrome.30,31 The initial infusion rate of 2.5 to 5 g per hour is equivalent to 4 to 10 alcoholic beverages per 24 hours.
Nevertheless, ethanol infusion carries the potential for toxicities (eg, gastric irritation, precipitation of acute hepatic failure, hypoglycemia, pancreatitis, bone marrow suppression, prolonged wound healing) and drug interactions (eg, with anticoagulants and anticonvulsants). Thus, ethanol is neither widely used nor recommended.25,31
ADJUNCTIVE THERAPIES
Many medications are used adjunctively in the acute setting, both for symptoms of alcohol withdrawal syndrome and for agitation.
Haloperidol
No clinical trial has yet examined haloperidol monotherapy in patients with alcohol withdrawal syndrome in either general medical units or intensive care units. Yet haloperidol remains important and is recommended as an adjunct therapy for agitation.23,32 Dosing of haloperidol in protocols for surgical patients ranged from 2 to 5 mg intravenously every 0.5 to 2 hours, with a maximum dosage of 0.5 mg per kg per 24 hours.7,33
Alpha-2 agonists
Alpha-2 agonists are thought to reduce sympathetic overdrive and the autonomic symptoms associated with alcohol withdrawal syndrome, and these agents (primarily clonidine) have been studied in the treatment of alcohol withdrawal syndrome.34,35
Clonidine. In a Swedish study,34 26 men ages 20 to 55 who presented with the tremor, sweating, dysphoria, tension, anxiety, and tachycardia associated with alcohol withdrawal syndrome received clonidine 4 µg per kg twice daily or carbamazepine 200 mg three to four times daily in addition to an antiepileptic. Adjunctive use of a benzodiazepine was allowed at night in both groups. No statistically significant difference in symptom reduction was noted between the two groups, and there was no difference in total benzodiazepine use.
Dexmedetomidine, given intravenously, has been tested as an adjunct to benzodiazepine treatment in severe alcohol withdrawal syndrome. It has been shown to decrease the amount of total benzodiazepine needed compared with benzodiazepine therapy alone, but no differences have been seen in length of hospital stay.36–39 However, research on this drug so far is limited to ICU patients.
Beta-blockers
Beta-blockers have been used in inpatients with alcohol withdrawal syndrome to reduce heart rate and potentially reduce alcohol craving. However, the data are limited and conflicting.
Atenolol 50 to 100 mg daily, in a study in 120 patients, reduced length of stay (4 vs 5 days), reduced benzodiazepine use, and improved vital signs and behavior compared with placebo.40
Propranolol 40 mg every 6 hours reduced arrhythmias but increased hallucinations when used alone in a study in 47 patients.41 When used in combination with chlordiazepoxide, no benefit was seen in arrhythmia reduction.41
Barbiturates and other antiepileptics
Data continue to emerge on antiepileptics as both monotherapy and adjunctive therapy for alcohol withdrawal syndrome. Barbiturates as monotherapy were largely replaced by benzodiazepines in view of the narrow therapeutic index of barbiturates and their full agonist effect on the GABA receptor complex. However, phenobarbital has been evaluated in patients presenting with severe alcohol withdrawal syndrome or resistant alcohol withdrawal (ie, symptoms despite large or repeated doses of benzodiazepines) as an adjunct to benzodiazepines.42,43
In addition, a newer trial44 involved giving a single dose of phenobarbital in the emergency department in combination with a CIWA-Ar–based benzodiazepine protocol, compared with the benzodiazepine protocol alone. The group that received phenobarbital had fewer ICU admissions; its evaluation is ongoing.
The three other medications with the most data are carbamazepine, valproic acid, and gabapentin.45,46 However, the studies were small and the benefit was modest. Although these agents are possible alternatives in protracted alcohol withdrawal syndrome, no definite conclusion can be made regarding their place in therapy.46
RECOMMENDATIONS FOR DRUG THERAPY AND SUPPORTIVE CARE
Which benzodiazepine to use?
No specific benzodiazepine is recommended over the others for managing alcohol withdrawal syndrome, but studies best support the long-acting agent chlordiazepoxide.16,17,20 Other benzodiazepines such as lorazepam and oxazepam have proved to be beneficial, but drugs should be selected on the basis of patient characteristics and drug metabolism.18,19,27
Patients with severe liver dysfunction and the elderly may have slower oxidative metabolism, so the effects of medications that are primarily oxidized, such as chlordiazepoxide and diazepam, may be prolonged. Therefore, lorazepam and oxazepam would be preferred in these groups.47 While most patients with alcohol withdrawal syndrome and liver dysfunction do not have advanced cirrhosis, we recommend liver function testing (serum aspartate aminotransferase, alanine aminotransferase, and alkaline phosphatase levels) and screening for liver disease, given the drug metabolism and package insert caution for use in those with impaired hepatic function.48
Patients with end-stage renal disease (stage 5 chronic kidney disease) or acute kidney injury should not receive parenteral diazepam or lorazepam. The rationale is the potential accumulation of propylene glycol, the solvent used in these formulations.
In the elderly, the Beers list of drugs to avoid in older adults includes benzodiazepines, not differentiating individual benzodiazepines in terms of risk.49 However, chlordiazepoxide may be preferable to diazepam due to its shorter parent half-life and lower lipophilicity.27 Few studies have been done using benzodiazepines in elderly patients with alcohol withdrawal syndrome, but those published have shown either equivalent dosing required compared with younger patients or more severe withdrawal for which they received greater amounts of chlordiazepoxide.9,12 Lorazepam and oxazepam have less potential to accumulate in the elderly compared with the nonelderly due to the drugs’ metabolic profiles; lorazepam is the preferred agent because of its faster onset of action.47 Ultimately, the choice of benzodiazepine in elderly patients with alcohol withdrawal syndrome should be based on patient-specific characteristics.
How should benzodiazepines be dosed?
While the CIWA-Ar thresholds and subsequent dosing of benzodiazepines varied in different studies, we recommend starting benzodiazepine therapy at a CIWA-Ar score of 8 or higher, with subsequent dosing based on score reassessment. Starting doses of benzodiazepines should be chlordiazepoxide 25 to 50 mg, lorazepam 1 to 2 mg, or oxazepam 15 mg.16–20
Subsequent doses should be titrated upward, increasing by 1.5 to 2 times the previous dose and monitored at least every 1 to 2 hours after dose adjustments. Once a patient is stable and the CIWA-Ar score is less than 8, monitoring intervals can be extended to every 4 to 8 hours. If the CIWA-Ar score is more than 20, studies suggest the need for patient reevaluation for transfer to the ICU; however, some health systems have a lower threshold for this intervention.7,14,50
Dosing algorithms and CIWA-Ar goals may vary slightly from institution to institution, but it has been shown that symptom-triggered therapy works best when hospitals have a protocol for it and staff are adequately trained to assess patients with alcohol withdrawal syndrome.7,50,51 Suggestions for dose ranges and symptom-triggered therapy are shown in Table 5.
In case of benzodiazepine overdose or potential benzodiazepine-induced delirium, flumazenil could be considered.52
Patients who should not receive symptom-triggered therapy include immediate postoperative patients in whom clinicians cannot properly assess withdrawal symptoms and patients with a history of DTs.51 While controversy exists regarding the use of symptom-triggered therapy in patients with complicated medical comorbidities, there are data to support symptom-triggered therapy in some ICU patients, as it has resulted in less benzodiazepine use and reduced mechanical ventilation.53,54
There are limited data to support phenobarbital in treating resistant alcohol withdrawal syndrome, either alone or concurrently with benzodiazepines, in escalating doses ranging from 65 to 260 mg, with a maximum daily dose of 520 mg.42,55,56
Haloperidol
For patients exhibiting agitation despite benzodiazepine therapy, giving haloperidol adjunctively can be beneficial.
Haloperidol can be used in medical patients as an adjunctive therapy for agitation, but caution is advised because of the potential for a lowering of the seizure threshold, extrapyramidal effects, and risk of QTc prolongation leading to arrhythmias. Patients considered at highest risk for torsades de pointes may have a QTc of 500 msec or greater.57
Patients should also be screened for factors that have been shown to be independent predictors of QTc prolongation (female sex, diagnosis of myocardial infarction, septic shock or left ventricular dysfunction, other QT-prolonging drugs, age > 68, baseline QTc ≥ 450 msec, and hypokalemia).58 If combined predictors have been identified, it is recommended that haloperidol be avoided.
If haloperidol is to be given, a baseline electrocardiogram and electrolyte panel should be obtained, with daily electrocardiograms thereafter, as well as ongoing review of the patient’s medications to minimize drug interactions that could further increase the risk for QTc prolongation.
Suggested haloperidol dosing is 2 to 5 mg intravenously every 0.5 to 2 hours with a maximum dose of 0.5 mg/kg/24 hours.8,33 A maximum of 35 mg of intravenous haloperidol should be used in a 24-hour period to avoid QTc prolongation.57
Antihypertensive therapy
Many patients receive symptomatic relief of autonomic hyperreactivity with benzodiazepines. However, some may require additional antihypertensive therapy for cardiac adrenergic symptoms (hypertension, tachycardia) if symptoms do not resolve by treating other medical problems commonly seen in patients with alcohol withdrawal syndrome, such as dehydration and electrolyte imbalances.7
Published protocols suggest giving clonidine 0.1 mg orally every hour up to three times as needed until systolic blood pressure is less than 140 mm Hg (less than 160 mm Hg if the patient is over age 60) and diastolic pressure is less than 90 mm Hg.51 Once the patient is stabilized, the dosing can be scheduled to a maximum of 2.4 mg daily.59 However, we believe that the use of clonidine should be restricted to patients who have a substantial increase in blood pressure over baseline or are nearing a hypertensive urgency or emergency (pressures > 180/120 mm Hg) and should not be used to treat other general symptoms associated with alcohol withdrawal syndrome.42
In addition, based on limited evidence, we recommend using beta-blockers only in patients with symptomatic tachycardia or as an adjunct in hypertension management.40,41
Therapies to avoid in acutely ill medical patients
Ethanol is not recommended. Instead, intravenous benzodiazepines should be given in patients presenting with severe alcohol withdrawal syndrome.
Antiepileptics, including valproic acid, carbamazepine, and pregabalin, lack benefit in these patients either as monotherapy or as adjunctive therapy and so are not recommended.45,60–62
Magnesium supplementation (in patients with normal serum magnesium levels) should not be given, as no clinical benefit has been shown.63
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association, 2013:501.
- Bayard M, McIntyre J, Hill KR, Woodside J Jr. Alcohol withdrawal syndrome. Am Fam Physician 2004; 69:1443–1450.
- Kosten TR, O'Connor PG. Management of drug and alcohol withdrawal. N Engl J Med 2003; 348:1786–1795.
- Isbell H, Fraser HF, Wilker A, Bellevile RE, Eisenman AJ. An experimental study of the etiology of rum fits and delirium tremens. Q J Stud Alcohol 1955; 16:1–33.
- Khan A, Levy P, DeHorn S, Miller W, Compton S. Predictors of mortality in patients with delirium tremens. Acad Emerg Med 2008; 15:788–790.
- Monte R, Rabuñal R, Casariego E, López-Agreda H, Mateos A, Pértega S. Analysis of the factors determining survival of alcoholic withdrawal syndrome patients in a general hospital. Alcohol Alcohol 2010; 45:151–158.
- Stanley KM, Amabile CM, Simpson KN, Couillard D, Norcross ED, Worrall CL. Impact of an alcohol withdrawal syndrome practice guideline on surgical patient outcomes. Pharmacotherapy 2003; 23:843–854.
- Brousse G, Arnaud B, Vorspan F, et al. Alteration of glutamate/GABA balance during acute alcohol withdrawal in emergency department: a prospective analysis. Alcohol Alcohol 2012; 47:501–508.
- Liskow BI, Rinck C, Campbell J, DeSouza C. Alcohol withdrawal in the elderly. J Stud Alcohol 1989; 50:414–421.
- Etherington JM. Emergency management of acute alcohol problems. Part 1: uncomplicated withdrawal. Can Fam Physician 1996; 42:2186–2190.
- Letizia M, Reinbolz M. Identifying and managing acute alcohol withdrawal in the elderly. Geriatr Nurs 2005; 26:176–183.
- Brower KJ, Mudd S, Blow FC, Young JP, Hill EM. Severity and treatment of alcohol withdrawal in elderly versus younger patients. Alcohol Clin Exp Res 1994; 18:196–201.
- Williams D, Lewis J, McBride A. A comparison of rating scales for the alcohol-withdrawal syndrome. Alcohol Alcohol 2001; 36:104–108.
- Reoux JP, Oreskovich MR. A comparison of two versions of the Clinical Institute Withdrawal Assessment for Alcohol: the CIWA-Ar and CIWA-AD. Am J Addict 2006; 15:85–93.
- Sullivan JT, Sykora K, Schneiderman J, Naranjo CA, Sellers EM. Assessment of alcohol withdrawal: the revised Clinical Institute Withdrawal Assessment for alcohol scale (CIWA-Ar). Br J Addict 1989; 84:1353–1357.
- Saitz R, Mayo-Smith MF, Roberts MS, Redmond HA, Bernard DR, Calkins DR. Individualized treatment for alcohol withdrawal. A randomized double-blind controlled trial. JAMA 1994; 272:519–523.
- Jaeger TM, Lohr RH, Pankratz VS. Symptom-triggered therapy for alcohol withdrawal syndrome in medical inpatients. Mayo Clin Proc 2001; 76:695–701.
- Daeppen JB, Gache P, Landry U, et al. Symptom-triggered vs fixed-schedule doses of benzodiazepine for alcohol withdrawal: a randomized treatment trial. Arch Intern Med 2002; 162:1117–1121.
- Weaver MF, Hoffman HJ, Johnson RE, Mauck K. Alcohol withdrawal pharmacotherapy for inpatients with medical comorbidity. J Addict Dis 2006; 25:17–24.
- Reoux JP, Miller K. Routine hospital alcohol detoxification practice compared to symptom triggered management with an Objective Withdrawal Scale (CIWA-Ar). Am J Addict 2000; 9:135–144.
- Wetterling T, Kanitz RD, Besters B, et al. A new rating scale for the assessment of the alcohol-withdrawal syndrome (AWS scale). Alcohol Alcohol 1997; 32:753–760.
- Myrick H, Anton RF. Treatment of alcohol withdrawal. Alcohol Health Res World 1998; 22:38–43.
- Mayo-Smith MF, Beecher LH, Fischer TL, et al; Working Group on the Management of Alcohol Withdrawal Delirium, Practice Guidelines Committee, American Society of Addiction Medicine. Management of alcohol withdrawal delirium. An evidence-based practice guideline. Arch Intern Med 2004; 164:1405–1412.
- Sellers EM, Sullivan JT, Somer G, Sykora K. Characterization of DSM-III-R criteria for uncomplicated alcohol withdrawal provides an empirical basis for DSM-IV. Arch Gen Psychiatry 1991; 48:442–447.
- Sarff M, Gold JA. Alcohol withdrawal syndromes in the intensive care unit. Crit Care Med 2010; 38(suppl):S494–S501.
- Kumar CN, Andrade C, Murthy P. A randomized, double-blind comparison of lorazepam and chlordiazepoxide in patients with uncomplicated alcohol withdrawal. J Stud Alcohol Drugs 2009; 70:467–474.
- Bird RD, Makela EH. Alcohol withdrawal: what is the benzodiazepine of choice? Ann Pharmacother 1994; 28:67–71.
- Perry EC. Inpatient management of acute alcohol withdrawal syndrome. CNS Drugs 2014; 28:401–410.
- Amato L, Minozzi S, Vecchi S, Davoli M. Benzodiazepines for alcohol withdrawal. Cochrane Database Syst Rev 2010; 3:CD005063.
- Weinberg JA, Magnotti LJ, Fischer PE, et al. Comparison of intravenous ethanol versus diazepam for alcohol withdrawal prophylaxis in the trauma ICU: results of a randomized trial. J Trauma 2008; 64:99–104.
- Craft PP, Foil MB, Cunningham PR, Patselas PC, Long-Snyder BM, Collier MS. Intravenous ethanol for alcohol detoxification in trauma patients. South Med J 1994; 87:47–54.
- Ungur LA, Neuner B, John S, Wernecke K, Spies C. Prevention and therapy of alcohol withdrawal on intensive care units: systematic review of controlled trials. Alcohol Clin Exp Res 2013; 37:675–686.
- Lansford CD, Guerriero CH, Kocan MJ, et al. Improved outcomes in patients with head and neck cancer using a standardized care protocol for postoperative alcohol withdrawal. Arch Otolaryngol Head Neck Surg 2008; 134:865–872.
- Walinder J, Balldin J, Bokstrom K, Karlsson I, Lundstrom B, Svensson TH. Clonidine suppression of the alcohol withdrawal syndrome. Drug Alcohol Depend 1981; 8:345–348.
- Muzyk AJ, Fowler JA, Norwood DK, Chilipko A. Role of alpha2-agonists in the treatment of acute alcohol withdrawal. Ann Pharmacother 2011; 45:649–657.
- Crispo AL, Daley MJ, Pepin JL, Harford PH, Brown CV. Comparison of clinical outcomes in nonintubated patients with severe alcohol withdrawal syndrome treated with continuous-infusion sedatives: dexmedetomidine versus benzodiazepines. Pharmacotherapy 2014; 34:910–917.
- VanderWeide LA, Foster CJ, MacLaren R, Kiser TH, Fish DN, Mueller SW. Evaluation of early dexmedetomidine addition to the standard of care for severe alcohol withdrawal in the ICU: a retrospective controlled cohort study. J Intensive Care Med 2014. [Epub ahead of print October 16, 2014]
- Rayner SG, Weinert CR, Peng H, Jepsen S, Broccard AF. Dexmedetomidine as adjunct treatment for severe alcohol withdrawal in the ICU. Ann Intensive Care 2012; 2:12.
- Muzyk AJ, Kerns S, Brudney S, Gagliardi JP. Dexmedetomidine for the treatment of alcohol withdrawal syndrome: rationale and current status of research. CNS Drugs 2013; 27:913–920.
- Kraus ML, Gottlieb LD, Horwitz RI, Anscher M. Randomized clinical trial of atenolol in patients with alcohol withdrawal. N Engl J Med 1985; 313:905–909.
- Zilm DH, Jacob MS, MacLeod SM, Sellers EM, Ti TY. Propranolol and chlordiazepoxide effects on cardiac arrhythmias during alcohol withdrawal. Alcohol Clin Exp Res 1980; 4:400–405.
- Hack JB, Hoffmann RS, Nelson LS. Resistant alcohol withdrawal: does an unexpectedly large sedative requirement identify these patients early? J Med Toxicol 2006; 2:55–60.
- Hayner CE, Wuestefeld NL, Bolton PJ. Phenobarbital treatment in a patient with resistant alcohol withdrawal syndrome. Pharmacotherapy 2009; 29:875–878.
- Rosenson J, Clements C, Simon B, et al. Phenobarbital for acute alcohol withdrawal: a prospective randomized double-blind placebo-controlled study. J Emerg Med 2013; 44:592–598.e2.
- Prince V, Turpin KR. Treatment of alcohol withdrawal syndrome with carbamazepine, gabapentin, and nitrous oxide. Am J Health Syst Pharm 2008; 65:1039–1047.
- Leggio L, Kenna GA, Swift RM. New developments for the pharmacological treatment of alcohol withdrawal syndrome. A focus on non-benzodiazepine GABAergic medications. Prog Neuropsychopharmacol Biol Psychiatry 2008; 32:1106–1117.
- Peppers MP. Benzodiazepines for alcohol withdrawal in the elderly and in patients with liver disease. Pharmacotherapy 1996; 16:49–57.
- Valeant Pharmaceuticals North America LLC. Librium—chlordiazepoxide hydrochloride capsule, gelatin coated. http://dailymed.nlm.nih.gov/dailymed/archives/fdaDrugInfo.cfm?archiveid=125207. Accessed November 20, 2015.
- American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2012; 60:616–631.
- Hecksel KA, Bostwick JM, Jaeger TM, Cha SS. Inappropriate use of symptom-triggered therapy for alcohol withdrawal in the general hospital. Mayo Clin Proc 2008; 83:274–279.
- Manasco A, Chang S, Larriviere J, Hamm LL, Glass M. Alcohol withdrawal. South Med J 2012; 105:607–612.
- Moore PW, Donovan JW, Burkhart KK, et al. Safety and efficacy of flumazenil for reversal of iatrogenic benzodiazepine-associated delirium toxicity during treatment of alcohol withdrawal, a retrospective review at one center. J Med Toxicol 2014; 10:126–132.
- Bostwick JM, Lapid MI. False positives on the clinical institute withdrawal assessment for alcohol-revised: is this scale appropriate for use in the medically ill? Psychosomatics 2004; 45:256–261.
- de Wit M, Jones DG, Sessler CN, Zilberberg MD, Weaver MF. Alcohol-use disorders in the critically ill patient. Chest 2010; 138:994–1003.
- Young GP, Rores C, Murphy C, Dailey RH. Intravenous phenobarbital for alcohol withdrawal and convulsions. Ann Emerg Med 1987; 16:847–850.
- Hendey GW, Dery RA, Barnes RL, Snowden B, Mentler P. A prospective, randomized trial of phenobarbital versus benzodiazepines for acute alcohol withdrawal. Am J Emerg Med 2011; 29:382–385.
- Sharma ND, Rosman HS, Padhi ID, Tisdale JE. Torsades de pointes associated with intravenous haloperidol in critically ill patients. Am J Cardiol 1998; 81:238–240.
- Tisdale JE, Jaynes HA, Kingery JR, et al. Development and validation of a risk score to predict QT interval prolongation in hospitalized patients. Circ Cardiovasc Qual Outcomes 2013; 6:479–487.
- Boehringer Ingelheim Pharmaceuticals, Inc. Product Information: Catapres oral tablets, clonidine HCl oral tablets, 2012.
- Reoux JP, Saxon AJ, Malte CA, Baer JS, Sloan KL. Divalproex sodium in alcohol withdrawal: a randomized double-blind placebo-controlled clinical trial. Alcohol Clin Exp Res 2001; 25:1324–1329.
- Malcolm R, Ballenger JC, Sturgis ET, Anton R. Double-blind controlled trial comparing carbamazepine to oxazepam treatment of alcohol withdrawal. Am J Psychiatry 1989; 146:617–621.
- Förg A, Hein J, Volkmar K, et al. Efficacy and safety of pregabalin in the treatment of alcohol withdrawal syndrome: a randomized placebo-controlled trial. Alcohol Alcohol 2012; 47:149–155.
- Wilson A, Vulcano B. A double-blind, placebo-controlled trial of magnesium sulfate in the ethanol withdrawal syndrome. Alcohol Clin Exp Res 1984; 8:542–545.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association, 2013:501.
- Bayard M, McIntyre J, Hill KR, Woodside J Jr. Alcohol withdrawal syndrome. Am Fam Physician 2004; 69:1443–1450.
- Kosten TR, O'Connor PG. Management of drug and alcohol withdrawal. N Engl J Med 2003; 348:1786–1795.
- Isbell H, Fraser HF, Wilker A, Bellevile RE, Eisenman AJ. An experimental study of the etiology of rum fits and delirium tremens. Q J Stud Alcohol 1955; 16:1–33.
- Khan A, Levy P, DeHorn S, Miller W, Compton S. Predictors of mortality in patients with delirium tremens. Acad Emerg Med 2008; 15:788–790.
- Monte R, Rabuñal R, Casariego E, López-Agreda H, Mateos A, Pértega S. Analysis of the factors determining survival of alcoholic withdrawal syndrome patients in a general hospital. Alcohol Alcohol 2010; 45:151–158.
- Stanley KM, Amabile CM, Simpson KN, Couillard D, Norcross ED, Worrall CL. Impact of an alcohol withdrawal syndrome practice guideline on surgical patient outcomes. Pharmacotherapy 2003; 23:843–854.
- Brousse G, Arnaud B, Vorspan F, et al. Alteration of glutamate/GABA balance during acute alcohol withdrawal in emergency department: a prospective analysis. Alcohol Alcohol 2012; 47:501–508.
- Liskow BI, Rinck C, Campbell J, DeSouza C. Alcohol withdrawal in the elderly. J Stud Alcohol 1989; 50:414–421.
- Etherington JM. Emergency management of acute alcohol problems. Part 1: uncomplicated withdrawal. Can Fam Physician 1996; 42:2186–2190.
- Letizia M, Reinbolz M. Identifying and managing acute alcohol withdrawal in the elderly. Geriatr Nurs 2005; 26:176–183.
- Brower KJ, Mudd S, Blow FC, Young JP, Hill EM. Severity and treatment of alcohol withdrawal in elderly versus younger patients. Alcohol Clin Exp Res 1994; 18:196–201.
- Williams D, Lewis J, McBride A. A comparison of rating scales for the alcohol-withdrawal syndrome. Alcohol Alcohol 2001; 36:104–108.
- Reoux JP, Oreskovich MR. A comparison of two versions of the Clinical Institute Withdrawal Assessment for Alcohol: the CIWA-Ar and CIWA-AD. Am J Addict 2006; 15:85–93.
- Sullivan JT, Sykora K, Schneiderman J, Naranjo CA, Sellers EM. Assessment of alcohol withdrawal: the revised Clinical Institute Withdrawal Assessment for alcohol scale (CIWA-Ar). Br J Addict 1989; 84:1353–1357.
- Saitz R, Mayo-Smith MF, Roberts MS, Redmond HA, Bernard DR, Calkins DR. Individualized treatment for alcohol withdrawal. A randomized double-blind controlled trial. JAMA 1994; 272:519–523.
- Jaeger TM, Lohr RH, Pankratz VS. Symptom-triggered therapy for alcohol withdrawal syndrome in medical inpatients. Mayo Clin Proc 2001; 76:695–701.
- Daeppen JB, Gache P, Landry U, et al. Symptom-triggered vs fixed-schedule doses of benzodiazepine for alcohol withdrawal: a randomized treatment trial. Arch Intern Med 2002; 162:1117–1121.
- Weaver MF, Hoffman HJ, Johnson RE, Mauck K. Alcohol withdrawal pharmacotherapy for inpatients with medical comorbidity. J Addict Dis 2006; 25:17–24.
- Reoux JP, Miller K. Routine hospital alcohol detoxification practice compared to symptom triggered management with an Objective Withdrawal Scale (CIWA-Ar). Am J Addict 2000; 9:135–144.
- Wetterling T, Kanitz RD, Besters B, et al. A new rating scale for the assessment of the alcohol-withdrawal syndrome (AWS scale). Alcohol Alcohol 1997; 32:753–760.
- Myrick H, Anton RF. Treatment of alcohol withdrawal. Alcohol Health Res World 1998; 22:38–43.
- Mayo-Smith MF, Beecher LH, Fischer TL, et al; Working Group on the Management of Alcohol Withdrawal Delirium, Practice Guidelines Committee, American Society of Addiction Medicine. Management of alcohol withdrawal delirium. An evidence-based practice guideline. Arch Intern Med 2004; 164:1405–1412.
- Sellers EM, Sullivan JT, Somer G, Sykora K. Characterization of DSM-III-R criteria for uncomplicated alcohol withdrawal provides an empirical basis for DSM-IV. Arch Gen Psychiatry 1991; 48:442–447.
- Sarff M, Gold JA. Alcohol withdrawal syndromes in the intensive care unit. Crit Care Med 2010; 38(suppl):S494–S501.
- Kumar CN, Andrade C, Murthy P. A randomized, double-blind comparison of lorazepam and chlordiazepoxide in patients with uncomplicated alcohol withdrawal. J Stud Alcohol Drugs 2009; 70:467–474.
- Bird RD, Makela EH. Alcohol withdrawal: what is the benzodiazepine of choice? Ann Pharmacother 1994; 28:67–71.
- Perry EC. Inpatient management of acute alcohol withdrawal syndrome. CNS Drugs 2014; 28:401–410.
- Amato L, Minozzi S, Vecchi S, Davoli M. Benzodiazepines for alcohol withdrawal. Cochrane Database Syst Rev 2010; 3:CD005063.
- Weinberg JA, Magnotti LJ, Fischer PE, et al. Comparison of intravenous ethanol versus diazepam for alcohol withdrawal prophylaxis in the trauma ICU: results of a randomized trial. J Trauma 2008; 64:99–104.
- Craft PP, Foil MB, Cunningham PR, Patselas PC, Long-Snyder BM, Collier MS. Intravenous ethanol for alcohol detoxification in trauma patients. South Med J 1994; 87:47–54.
- Ungur LA, Neuner B, John S, Wernecke K, Spies C. Prevention and therapy of alcohol withdrawal on intensive care units: systematic review of controlled trials. Alcohol Clin Exp Res 2013; 37:675–686.
- Lansford CD, Guerriero CH, Kocan MJ, et al. Improved outcomes in patients with head and neck cancer using a standardized care protocol for postoperative alcohol withdrawal. Arch Otolaryngol Head Neck Surg 2008; 134:865–872.
- Walinder J, Balldin J, Bokstrom K, Karlsson I, Lundstrom B, Svensson TH. Clonidine suppression of the alcohol withdrawal syndrome. Drug Alcohol Depend 1981; 8:345–348.
- Muzyk AJ, Fowler JA, Norwood DK, Chilipko A. Role of alpha2-agonists in the treatment of acute alcohol withdrawal. Ann Pharmacother 2011; 45:649–657.
- Crispo AL, Daley MJ, Pepin JL, Harford PH, Brown CV. Comparison of clinical outcomes in nonintubated patients with severe alcohol withdrawal syndrome treated with continuous-infusion sedatives: dexmedetomidine versus benzodiazepines. Pharmacotherapy 2014; 34:910–917.
- VanderWeide LA, Foster CJ, MacLaren R, Kiser TH, Fish DN, Mueller SW. Evaluation of early dexmedetomidine addition to the standard of care for severe alcohol withdrawal in the ICU: a retrospective controlled cohort study. J Intensive Care Med 2014. [Epub ahead of print October 16, 2014]
- Rayner SG, Weinert CR, Peng H, Jepsen S, Broccard AF. Dexmedetomidine as adjunct treatment for severe alcohol withdrawal in the ICU. Ann Intensive Care 2012; 2:12.
- Muzyk AJ, Kerns S, Brudney S, Gagliardi JP. Dexmedetomidine for the treatment of alcohol withdrawal syndrome: rationale and current status of research. CNS Drugs 2013; 27:913–920.
- Kraus ML, Gottlieb LD, Horwitz RI, Anscher M. Randomized clinical trial of atenolol in patients with alcohol withdrawal. N Engl J Med 1985; 313:905–909.
- Zilm DH, Jacob MS, MacLeod SM, Sellers EM, Ti TY. Propranolol and chlordiazepoxide effects on cardiac arrhythmias during alcohol withdrawal. Alcohol Clin Exp Res 1980; 4:400–405.
- Hack JB, Hoffmann RS, Nelson LS. Resistant alcohol withdrawal: does an unexpectedly large sedative requirement identify these patients early? J Med Toxicol 2006; 2:55–60.
- Hayner CE, Wuestefeld NL, Bolton PJ. Phenobarbital treatment in a patient with resistant alcohol withdrawal syndrome. Pharmacotherapy 2009; 29:875–878.
- Rosenson J, Clements C, Simon B, et al. Phenobarbital for acute alcohol withdrawal: a prospective randomized double-blind placebo-controlled study. J Emerg Med 2013; 44:592–598.e2.
- Prince V, Turpin KR. Treatment of alcohol withdrawal syndrome with carbamazepine, gabapentin, and nitrous oxide. Am J Health Syst Pharm 2008; 65:1039–1047.
- Leggio L, Kenna GA, Swift RM. New developments for the pharmacological treatment of alcohol withdrawal syndrome. A focus on non-benzodiazepine GABAergic medications. Prog Neuropsychopharmacol Biol Psychiatry 2008; 32:1106–1117.
- Peppers MP. Benzodiazepines for alcohol withdrawal in the elderly and in patients with liver disease. Pharmacotherapy 1996; 16:49–57.
- Valeant Pharmaceuticals North America LLC. Librium—chlordiazepoxide hydrochloride capsule, gelatin coated. http://dailymed.nlm.nih.gov/dailymed/archives/fdaDrugInfo.cfm?archiveid=125207. Accessed November 20, 2015.
- American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2012; 60:616–631.
- Hecksel KA, Bostwick JM, Jaeger TM, Cha SS. Inappropriate use of symptom-triggered therapy for alcohol withdrawal in the general hospital. Mayo Clin Proc 2008; 83:274–279.
- Manasco A, Chang S, Larriviere J, Hamm LL, Glass M. Alcohol withdrawal. South Med J 2012; 105:607–612.
- Moore PW, Donovan JW, Burkhart KK, et al. Safety and efficacy of flumazenil for reversal of iatrogenic benzodiazepine-associated delirium toxicity during treatment of alcohol withdrawal, a retrospective review at one center. J Med Toxicol 2014; 10:126–132.
- Bostwick JM, Lapid MI. False positives on the clinical institute withdrawal assessment for alcohol-revised: is this scale appropriate for use in the medically ill? Psychosomatics 2004; 45:256–261.
- de Wit M, Jones DG, Sessler CN, Zilberberg MD, Weaver MF. Alcohol-use disorders in the critically ill patient. Chest 2010; 138:994–1003.
- Young GP, Rores C, Murphy C, Dailey RH. Intravenous phenobarbital for alcohol withdrawal and convulsions. Ann Emerg Med 1987; 16:847–850.
- Hendey GW, Dery RA, Barnes RL, Snowden B, Mentler P. A prospective, randomized trial of phenobarbital versus benzodiazepines for acute alcohol withdrawal. Am J Emerg Med 2011; 29:382–385.
- Sharma ND, Rosman HS, Padhi ID, Tisdale JE. Torsades de pointes associated with intravenous haloperidol in critically ill patients. Am J Cardiol 1998; 81:238–240.
- Tisdale JE, Jaynes HA, Kingery JR, et al. Development and validation of a risk score to predict QT interval prolongation in hospitalized patients. Circ Cardiovasc Qual Outcomes 2013; 6:479–487.
- Boehringer Ingelheim Pharmaceuticals, Inc. Product Information: Catapres oral tablets, clonidine HCl oral tablets, 2012.
- Reoux JP, Saxon AJ, Malte CA, Baer JS, Sloan KL. Divalproex sodium in alcohol withdrawal: a randomized double-blind placebo-controlled clinical trial. Alcohol Clin Exp Res 2001; 25:1324–1329.
- Malcolm R, Ballenger JC, Sturgis ET, Anton R. Double-blind controlled trial comparing carbamazepine to oxazepam treatment of alcohol withdrawal. Am J Psychiatry 1989; 146:617–621.
- Förg A, Hein J, Volkmar K, et al. Efficacy and safety of pregabalin in the treatment of alcohol withdrawal syndrome: a randomized placebo-controlled trial. Alcohol Alcohol 2012; 47:149–155.
- Wilson A, Vulcano B. A double-blind, placebo-controlled trial of magnesium sulfate in the ethanol withdrawal syndrome. Alcohol Clin Exp Res 1984; 8:542–545.
KEY POINTS
- Patients diagnosed with or suspected of having alcohol withdrawal syndrome need a thorough history and physical examination, appropriate laboratory tests, and monitoring using the revised Clinical Institute Withdrawal Assessment for Alcohol scale (CIWA-Ar) or a similar scale.
- For most patients, benzodiazepines should be given in a symptom-triggered fashion, using the CIWA-Ar score as a monitoring tool. Alternatively, scheduled benzodiazepine dosing should be considered for patients with a history of alcohol withdrawal delirium or for patients in whom withdrawal symptoms cannot be easily assessed.
- The choice of benzodiazepine should be individualized, based on the half-life of the drug, comorbid diseases, and monitoring plans.
- Many patients with alcohol withdrawal syndrome require fluid and electrolyte replacement, as well as adjunctive therapies such as haloperidol for delirium and antihypertensives for cardiac or adrenergic symptoms. No standard currently exists for drug dosing, administration, and assessment protocols in these patients. Therefore, clinicians are adapting study designs and assessment scales to meet patients’ individual needs.
