User login
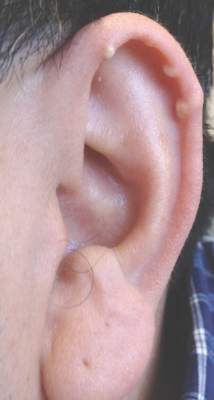
Cyst on the Eyebrow
The best diagnosis is:
a. bronchogenic cyst
b. dermoid cyst
c. epidermal inclusion cyst
d. hidrocystoma
e. steatocystoma
|
|
| H&E, original magnification ×40. |
|
| H&E, original magnification ×100. |
Continue to the next page for the diagnosis >>
Dermoid Cyst
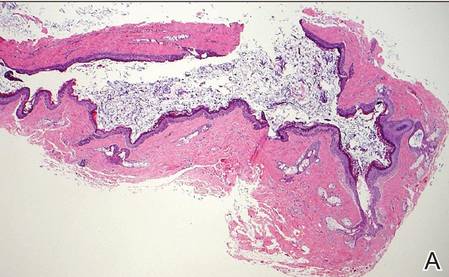
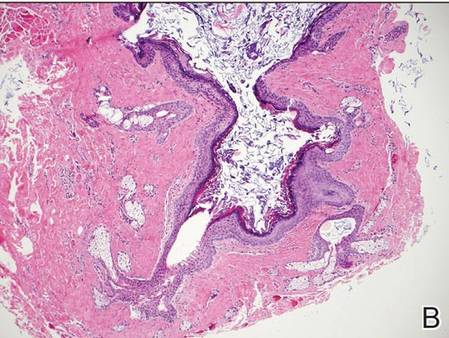
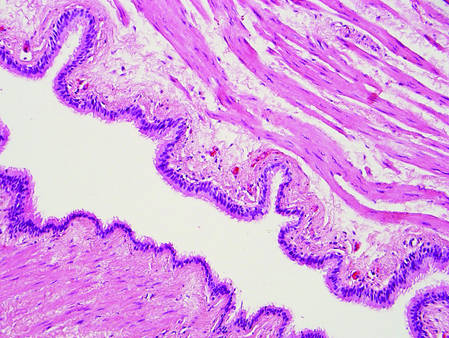
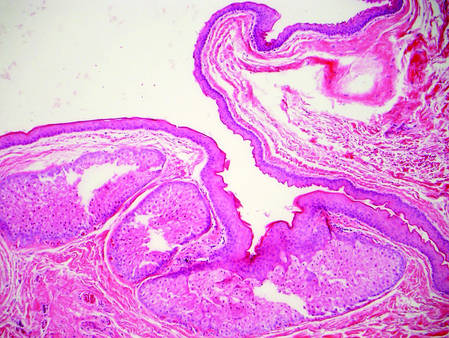
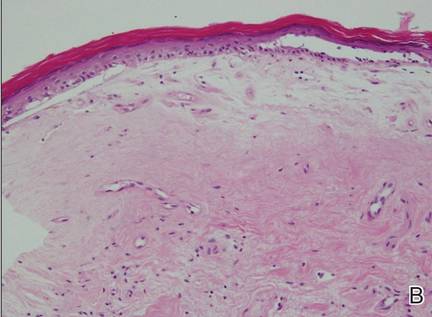
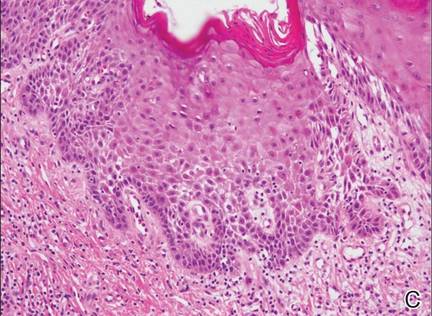
Dermoid cysts often present clinically as firm subcutaneous nodules on the head or neck in young children. They tend to arise along the lateral aspect of the eyebrow but also can occur on the nose, forehead, neck, chest, or scalp.1 Dermoid cysts are thought to arise from the sequestration of ectodermal tissues along the embryonic fusion planes during development.2 As such, they represent congenital defects and often are identified at birth; however, some are not noticed until much later when they enlarge or become inflamed or infected. Midline dermoid cysts may be associated with underlying dysraphism or intracranial extension.3,4 Thus, any midline lesion warrants evaluation that incorporates imaging with computed tomography or magnetic resonance imaging.4,5 Histologically, dermoid cysts are lined by a keratinizing stratified squamous epithelium (quiz image A), but the lining may be brightly eosinophilic and wavy resembling shark teeth.1,3 The wall of a dermoid cyst commonly contains mature adnexal structures such as terminal hair follicles, sebaceous glands, apocrine glands, and/or eccrine glands (quiz image B).1 Smooth muscle also may be seen within the lining; however, bone and cartilage are not commonly reported in dermoid cysts.2 Lamellar keratin is typical of the cyst contents, and terminal hair shafts also are sometimes noted within the cystic space (quiz image B).1,2 Treatment options include excision at the time of diagnosis or close clinical monitoring with subsequent excision if the lesion grows or becomes symptomatic.4,5 Many practitioners opt to excise these cysts at diagnosis, as untreated lesions are at risk for infection and/or inflammation or may be cosmetically deforming.6,7 Surgical resection, including removal of the wall of the cyst, is curative and reoccurrence is rare.5
| |
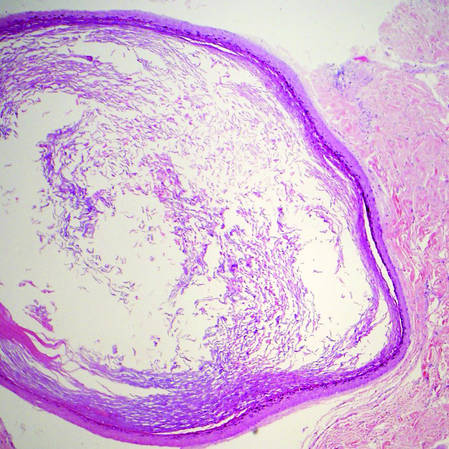
Figure 1. Bronchogenic cyst demonstrating a ciliated pseudostratified epithelial lining encircled by smooth muscle (H&E, original magnification ×200). | |
| |
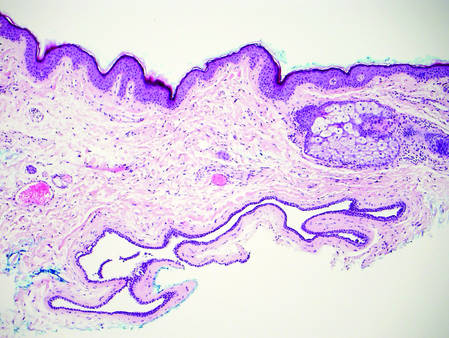
| Figure 2. Epidermal inclusion cyst containing loose lamellar keratin and a lining that closely resembles the surface epidermis (H&E, original magnification ×40). |
|
Bronchogenic cysts demonstrate an epithelial lining that often is pseudostratified cuboidal or columnar as well as ciliated (Figure 1). Goblet cells are present in the lining in approximately 50% of cases. Smooth muscle may be seen circumferentially surrounding the cyst lining, and rare cases also contain cartilage.1 In contrast to dermoid cysts, other types of adnexal structures are not found within the lining. Bronchogenic cysts that arise in the skin are extremely rare.2 These cysts are thought to arise from respiratory epithelium that has been sequestered during embryologic formation of the tracheobronchial tree. They often are seen overlying the suprasternal notch and occasionally are found on the anterior aspect of the neck or chin. These cysts also are present at birth, similar to dermoid cysts.3
Epidermal inclusion cysts have a lining that histologically bears close resemblance to the surface epidermis. These cysts contain loose lamellar keratin, similar to a dermoid cyst. In contrast, the lining of an epidermal inclusion cyst will lack adnexal structures (Figure 2).1 Clinically, epidermal inclusion cysts often present as smooth, dome-shaped papules and nodules with a central punctum. They are classically found on the face, neck, and trunk. These cysts are thought to arise after a traumatic insult to the pilosebaceous unit.2
Hidrocystomas can be apocrine or eccrine.3 Eccrine hidrocystomas are unilocular cysts that are lined by 2 layers of flattened to cuboidal epithelial cells (Figure 3). The cysts are filled with clear fluid and often are found adjacent to normal eccrine glands.1 Apocrine hidrocystomas are unilocular or multilocular cysts that are lined by 1 to several layers of epithelial cells. The lining of an apocrine hidrocystoma will often exhibit luminal decapitation secretion.3 Apocrine and eccrine hidrocystomas are clinically identical and appear as blue translucent papules on the cheeks or eyelids of adults.1-3 They usually occur periorbitally but also can be seen on the trunk, popliteal fossa, external ears, or vulva. Eccrine hidrocystomas can wax and wane in accordance with the amount of sweat produced; thus, they often expand in size during the summer months.2
Steatocystomas, or simple sebaceous duct cysts, histologically demonstrate a characteristically wavy and eosinophilic cuticle resembling shark teeth (Figure 4) similar to the lining of the sebaceous duct where it enters the follicle.1 Sebaceous glands are an almost invariable feature, either present within the lining of the cyst (Figure 4) or in the adjacent tissue.2 In comparison, dermoid cysts may have a red wavy cuticle but also will usually have terminal hair follicles or eccrine or apocrine glands within the wall of the cyst. Steatocystomas typically are collapsed and empty or only contain sebaceous debris (Figure 4) rather than the lamellar keratin seen in dermoid and epidermoid inclusion cysts. Steatocystomas can occur as solitary (steatocystoma simplex) or multiple (steatocystoma multiplex) lesions.1,3 They are clinically comprised of small dome-shaped papules that often are translucent and yellow. These cysts are commonly found on the sternum of males and the axillae or groin of females.2
| 
| |
Figure 3. Eccrine hidrocystoma with clear contents and lined by 2 layers of cuboidal epithelial cells (H&E, original magnification ×100). | Figure 4. Steatocystoma with a red wavy cuticle, sparse sebaceous contents, and sebaceous glands within the lining (H&E, original magnification ×100). |
|
1. Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
2. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin. 4th ed. St Louis, MO: Elsevier/Saunders; 2012.
3. Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
4. Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
5. Sorenson EP, Powel JE, Rozzelle CJ, et al. Scalp dermoids: a review of their anatomy, diagnosis, and treatment. Childs Nerv Syst. 2013;29:375-380.
6. Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolarynol Head Neck Surg. 2005;132:938-942.
7. Abou-Rayyah Y, Rose GE, Konrad H, et al. Clinical, radiological and pathological examination of periocular dermoid cysts: evidence of inflammation from an early age. Eye (Lond). 2002;16:507-512.
The best diagnosis is:
a. bronchogenic cyst
b. dermoid cyst
c. epidermal inclusion cyst
d. hidrocystoma
e. steatocystoma
|
|
|
| H&E, original magnification ×40. |
|
|
| H&E, original magnification ×100. |
Continue to the next page for the diagnosis >>
Dermoid Cyst
Dermoid cysts often present clinically as firm subcutaneous nodules on the head or neck in young children. They tend to arise along the lateral aspect of the eyebrow but also can occur on the nose, forehead, neck, chest, or scalp.1 Dermoid cysts are thought to arise from the sequestration of ectodermal tissues along the embryonic fusion planes during development.2 As such, they represent congenital defects and often are identified at birth; however, some are not noticed until much later when they enlarge or become inflamed or infected. Midline dermoid cysts may be associated with underlying dysraphism or intracranial extension.3,4 Thus, any midline lesion warrants evaluation that incorporates imaging with computed tomography or magnetic resonance imaging.4,5 Histologically, dermoid cysts are lined by a keratinizing stratified squamous epithelium (quiz image A), but the lining may be brightly eosinophilic and wavy resembling shark teeth.1,3 The wall of a dermoid cyst commonly contains mature adnexal structures such as terminal hair follicles, sebaceous glands, apocrine glands, and/or eccrine glands (quiz image B).1 Smooth muscle also may be seen within the lining; however, bone and cartilage are not commonly reported in dermoid cysts.2 Lamellar keratin is typical of the cyst contents, and terminal hair shafts also are sometimes noted within the cystic space (quiz image B).1,2 Treatment options include excision at the time of diagnosis or close clinical monitoring with subsequent excision if the lesion grows or becomes symptomatic.4,5 Many practitioners opt to excise these cysts at diagnosis, as untreated lesions are at risk for infection and/or inflammation or may be cosmetically deforming.6,7 Surgical resection, including removal of the wall of the cyst, is curative and reoccurrence is rare.5
|
| |
Figure 1. Bronchogenic cyst demonstrating a ciliated pseudostratified epithelial lining encircled by smooth muscle (H&E, original magnification ×200). | |
|
| |
| Figure 2. Epidermal inclusion cyst containing loose lamellar keratin and a lining that closely resembles the surface epidermis (H&E, original magnification ×40). |
|
Bronchogenic cysts demonstrate an epithelial lining that often is pseudostratified cuboidal or columnar as well as ciliated (Figure 1). Goblet cells are present in the lining in approximately 50% of cases. Smooth muscle may be seen circumferentially surrounding the cyst lining, and rare cases also contain cartilage.1 In contrast to dermoid cysts, other types of adnexal structures are not found within the lining. Bronchogenic cysts that arise in the skin are extremely rare.2 These cysts are thought to arise from respiratory epithelium that has been sequestered during embryologic formation of the tracheobronchial tree. They often are seen overlying the suprasternal notch and occasionally are found on the anterior aspect of the neck or chin. These cysts also are present at birth, similar to dermoid cysts.3
Epidermal inclusion cysts have a lining that histologically bears close resemblance to the surface epidermis. These cysts contain loose lamellar keratin, similar to a dermoid cyst. In contrast, the lining of an epidermal inclusion cyst will lack adnexal structures (Figure 2).1 Clinically, epidermal inclusion cysts often present as smooth, dome-shaped papules and nodules with a central punctum. They are classically found on the face, neck, and trunk. These cysts are thought to arise after a traumatic insult to the pilosebaceous unit.2
Hidrocystomas can be apocrine or eccrine.3 Eccrine hidrocystomas are unilocular cysts that are lined by 2 layers of flattened to cuboidal epithelial cells (Figure 3). The cysts are filled with clear fluid and often are found adjacent to normal eccrine glands.1 Apocrine hidrocystomas are unilocular or multilocular cysts that are lined by 1 to several layers of epithelial cells. The lining of an apocrine hidrocystoma will often exhibit luminal decapitation secretion.3 Apocrine and eccrine hidrocystomas are clinically identical and appear as blue translucent papules on the cheeks or eyelids of adults.1-3 They usually occur periorbitally but also can be seen on the trunk, popliteal fossa, external ears, or vulva. Eccrine hidrocystomas can wax and wane in accordance with the amount of sweat produced; thus, they often expand in size during the summer months.2
Steatocystomas, or simple sebaceous duct cysts, histologically demonstrate a characteristically wavy and eosinophilic cuticle resembling shark teeth (Figure 4) similar to the lining of the sebaceous duct where it enters the follicle.1 Sebaceous glands are an almost invariable feature, either present within the lining of the cyst (Figure 4) or in the adjacent tissue.2 In comparison, dermoid cysts may have a red wavy cuticle but also will usually have terminal hair follicles or eccrine or apocrine glands within the wall of the cyst. Steatocystomas typically are collapsed and empty or only contain sebaceous debris (Figure 4) rather than the lamellar keratin seen in dermoid and epidermoid inclusion cysts. Steatocystomas can occur as solitary (steatocystoma simplex) or multiple (steatocystoma multiplex) lesions.1,3 They are clinically comprised of small dome-shaped papules that often are translucent and yellow. These cysts are commonly found on the sternum of males and the axillae or groin of females.2
|
|
| |
Figure 3. Eccrine hidrocystoma with clear contents and lined by 2 layers of cuboidal epithelial cells (H&E, original magnification ×100). | Figure 4. Steatocystoma with a red wavy cuticle, sparse sebaceous contents, and sebaceous glands within the lining (H&E, original magnification ×100). |
|
The best diagnosis is:
a. bronchogenic cyst
b. dermoid cyst
c. epidermal inclusion cyst
d. hidrocystoma
e. steatocystoma
|
|
|
| H&E, original magnification ×40. |
|
|
| H&E, original magnification ×100. |
Continue to the next page for the diagnosis >>
Dermoid Cyst
Dermoid cysts often present clinically as firm subcutaneous nodules on the head or neck in young children. They tend to arise along the lateral aspect of the eyebrow but also can occur on the nose, forehead, neck, chest, or scalp.1 Dermoid cysts are thought to arise from the sequestration of ectodermal tissues along the embryonic fusion planes during development.2 As such, they represent congenital defects and often are identified at birth; however, some are not noticed until much later when they enlarge or become inflamed or infected. Midline dermoid cysts may be associated with underlying dysraphism or intracranial extension.3,4 Thus, any midline lesion warrants evaluation that incorporates imaging with computed tomography or magnetic resonance imaging.4,5 Histologically, dermoid cysts are lined by a keratinizing stratified squamous epithelium (quiz image A), but the lining may be brightly eosinophilic and wavy resembling shark teeth.1,3 The wall of a dermoid cyst commonly contains mature adnexal structures such as terminal hair follicles, sebaceous glands, apocrine glands, and/or eccrine glands (quiz image B).1 Smooth muscle also may be seen within the lining; however, bone and cartilage are not commonly reported in dermoid cysts.2 Lamellar keratin is typical of the cyst contents, and terminal hair shafts also are sometimes noted within the cystic space (quiz image B).1,2 Treatment options include excision at the time of diagnosis or close clinical monitoring with subsequent excision if the lesion grows or becomes symptomatic.4,5 Many practitioners opt to excise these cysts at diagnosis, as untreated lesions are at risk for infection and/or inflammation or may be cosmetically deforming.6,7 Surgical resection, including removal of the wall of the cyst, is curative and reoccurrence is rare.5
|
| |
Figure 1. Bronchogenic cyst demonstrating a ciliated pseudostratified epithelial lining encircled by smooth muscle (H&E, original magnification ×200). | |
|
| |
| Figure 2. Epidermal inclusion cyst containing loose lamellar keratin and a lining that closely resembles the surface epidermis (H&E, original magnification ×40). |
|
Bronchogenic cysts demonstrate an epithelial lining that often is pseudostratified cuboidal or columnar as well as ciliated (Figure 1). Goblet cells are present in the lining in approximately 50% of cases. Smooth muscle may be seen circumferentially surrounding the cyst lining, and rare cases also contain cartilage.1 In contrast to dermoid cysts, other types of adnexal structures are not found within the lining. Bronchogenic cysts that arise in the skin are extremely rare.2 These cysts are thought to arise from respiratory epithelium that has been sequestered during embryologic formation of the tracheobronchial tree. They often are seen overlying the suprasternal notch and occasionally are found on the anterior aspect of the neck or chin. These cysts also are present at birth, similar to dermoid cysts.3
Epidermal inclusion cysts have a lining that histologically bears close resemblance to the surface epidermis. These cysts contain loose lamellar keratin, similar to a dermoid cyst. In contrast, the lining of an epidermal inclusion cyst will lack adnexal structures (Figure 2).1 Clinically, epidermal inclusion cysts often present as smooth, dome-shaped papules and nodules with a central punctum. They are classically found on the face, neck, and trunk. These cysts are thought to arise after a traumatic insult to the pilosebaceous unit.2
Hidrocystomas can be apocrine or eccrine.3 Eccrine hidrocystomas are unilocular cysts that are lined by 2 layers of flattened to cuboidal epithelial cells (Figure 3). The cysts are filled with clear fluid and often are found adjacent to normal eccrine glands.1 Apocrine hidrocystomas are unilocular or multilocular cysts that are lined by 1 to several layers of epithelial cells. The lining of an apocrine hidrocystoma will often exhibit luminal decapitation secretion.3 Apocrine and eccrine hidrocystomas are clinically identical and appear as blue translucent papules on the cheeks or eyelids of adults.1-3 They usually occur periorbitally but also can be seen on the trunk, popliteal fossa, external ears, or vulva. Eccrine hidrocystomas can wax and wane in accordance with the amount of sweat produced; thus, they often expand in size during the summer months.2
Steatocystomas, or simple sebaceous duct cysts, histologically demonstrate a characteristically wavy and eosinophilic cuticle resembling shark teeth (Figure 4) similar to the lining of the sebaceous duct where it enters the follicle.1 Sebaceous glands are an almost invariable feature, either present within the lining of the cyst (Figure 4) or in the adjacent tissue.2 In comparison, dermoid cysts may have a red wavy cuticle but also will usually have terminal hair follicles or eccrine or apocrine glands within the wall of the cyst. Steatocystomas typically are collapsed and empty or only contain sebaceous debris (Figure 4) rather than the lamellar keratin seen in dermoid and epidermoid inclusion cysts. Steatocystomas can occur as solitary (steatocystoma simplex) or multiple (steatocystoma multiplex) lesions.1,3 They are clinically comprised of small dome-shaped papules that often are translucent and yellow. These cysts are commonly found on the sternum of males and the axillae or groin of females.2
|
|
| |
Figure 3. Eccrine hidrocystoma with clear contents and lined by 2 layers of cuboidal epithelial cells (H&E, original magnification ×100). | Figure 4. Steatocystoma with a red wavy cuticle, sparse sebaceous contents, and sebaceous glands within the lining (H&E, original magnification ×100). |
|
1. Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
2. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin. 4th ed. St Louis, MO: Elsevier/Saunders; 2012.
3. Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
4. Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
5. Sorenson EP, Powel JE, Rozzelle CJ, et al. Scalp dermoids: a review of their anatomy, diagnosis, and treatment. Childs Nerv Syst. 2013;29:375-380.
6. Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolarynol Head Neck Surg. 2005;132:938-942.
7. Abou-Rayyah Y, Rose GE, Konrad H, et al. Clinical, radiological and pathological examination of periocular dermoid cysts: evidence of inflammation from an early age. Eye (Lond). 2002;16:507-512.
1. Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
2. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin. 4th ed. St Louis, MO: Elsevier/Saunders; 2012.
3. Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012.
4. Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
5. Sorenson EP, Powel JE, Rozzelle CJ, et al. Scalp dermoids: a review of their anatomy, diagnosis, and treatment. Childs Nerv Syst. 2013;29:375-380.
6. Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolarynol Head Neck Surg. 2005;132:938-942.
7. Abou-Rayyah Y, Rose GE, Konrad H, et al. Clinical, radiological and pathological examination of periocular dermoid cysts: evidence of inflammation from an early age. Eye (Lond). 2002;16:507-512.
Onychomatricoma: A Rare Case of Unguioblastic Fibroma of the Fingernail Associated With Trauma
Onychomatricoma (OM) is a rare benign neoplasm of the nail matrix. Even less common is its possible association with both trauma to the nail apparatus and onychomycosis. This case illustrates both of these findings.
Case Report
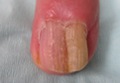
A 72-year-old white man presented to the dermatology clinic with a 26-year history of a thickened nail plate on the right third finger that had developed soon after a baseball injury. The patient reported that the nail was completely normal prior to the trauma. According to the patient, the distal aspect of the finger was directly hit by a baseball and subsequently was wrapped by the patient for a few weeks. The nail then turned black and eventually fell off. When the nail grew back, it appeared abnormal and in its current state. The patient stated the lesion was asymptomatic at the time of presentation.
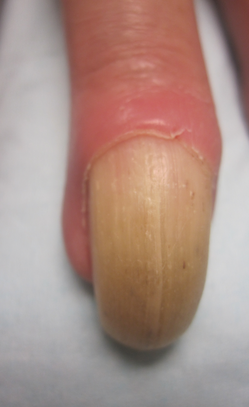
Physical examination revealed thickening, yellow discoloration, and transverse overcurvature of the nail plate on the right third finger with longitudinal ridging (Figure 1). A culture of the nail plate grew Chaetomium species. Application of topical clotrimazole for 3 months followed by a 6-week course of oral terbinafine produced no improvement. The patient then consented to a nail matrix incisional biopsy 6 months after initial presentation. After a digital nerve block was administered and a tourniquet of the proximal digit was applied, a nail avulsion was performed. Subsequently, a 3-mm punch biopsy was taken of the clinically apparent tumor in the nail matrix.
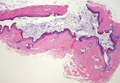
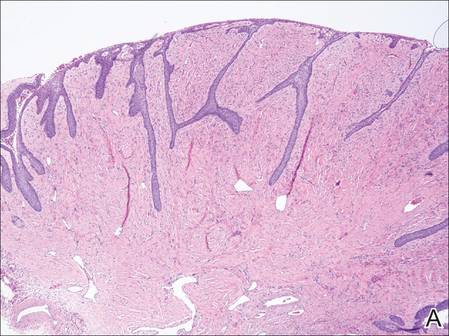
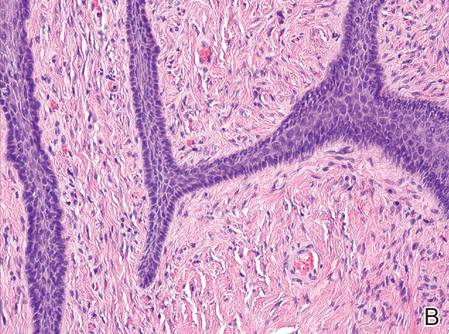
On microscopic examination of the removed tissue, a benign mixed epithelial and stromal proliferative lesion was noted. The basaloid epithelium, lacking a granular layer, arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component, which was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (Figure 2). The stromal component predominated over the epithelial component in this neoplasm. The nail was preserved in formalin and underwent hematoxylin and eosin staining. It was thickened and grossly showed filiform fibrous projections extending into the nail plate. Histologically, the nail displayed prominent oval clear channels. Periodic acid–Schiff staining was negative for fungal organisms.
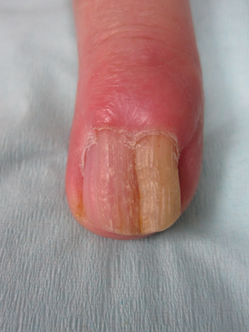
A diagnosis of unguioblastic fibroma–type OM was made. After receiving the diagnosis, expected course, and treatment options, the patient was offered conservative surgical excision but preferred clinical monitoring. At his last visit (6 months after the biopsy), the nail plate distal to the biopsy site had thinning and improvement, while the nail plate distal to the matrix that was not removed continued to show thickening, yellow discoloration, overcurvature, and longitudinal ridging (Figure 3).
| 
| |
Figure 2. The basaloid epithelium arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component (A)(H&E, original magnification ×2). At higher magnification, the stromal component was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (B)(H&E, original magnification ×10). | ||
|
|
Comment
Onychomatricoma is a rare tumor originating from the nail matrix. The tumor was first described by Baran and Kint1 in 1992 using the term onychomatrixoma, but later the term onychomatricoma became more widely used.2 Onychomatricomas are more common in adults (mean age, 48 years) and white individuals with no gender predilection.3,4 Fingernail involvement is twice as common as toenail involvement.3 Onychomatricoma is the only tumor that actively produces a nail plate.4
Clinically, OM presents with yellow discoloration along the entire nail plate and proximal splinter hemorrhages. It has a tendency toward transverse overcurvature of the nail plate with prominent longitudinal ridging.4 Trauma has been associated in at least 3 cases reported in the literature, though the association was sometimes weak.3,4 Xanthonychia and onychodystrophy of the nail are common.3 Pterygium, melanonychia, nail bleeding, and cutaneous horns have been reported but are rare.3-5 The tumor typically is painless with no radiographic bone involvement.3 Onychomycosis can be present,3 which may either be a predisposing factor for the tumor or secondary due to the deformed nail plate.4
When the nail plate is avulsed and the proximal nail fold is turned back, the matrix tumor is exposed. This polypoid and filiform tumor has characteristic fingerlike fibrokeratogenous projections extending from the nail matrix into the nail plate.3
Histologically, the tumor is fibroepithelial or biphasic with stromal and epithelial components. It has a lobulated and papillary growth pattern with 2 distinct areas that correspond to 2 anatomic zones.3 The base of the tumor corresponds to the proximal anatomic zone, which begins at the root of the nail and extends to the cuticle. This area is composed of V-shaped keratinous zones similar to the normal matrix. If the nail is removed prior to excision, these areas can be avulsed, leaving clear clefts. The superficial aspect of the tumor corresponds to the distal anatomic zone, which is located in the region of the lunula. This area is composed of multiple digitate or fingerlike projections with a fibrous core and a thick matrical epithelial covering.3 These digitations extend into small cavities in the nail plate, which can be visualized as clear channels or woodwormlike holes in hematoxylin and eosin–stained specimens. A biphasic fibrous stroma also can be observed with the superficial dermis being cellular with fibrillary collagen and the deep dermis more hypocellular with thicker collagen bundles.3,4
An analysis of keratins in the nail matrix, bed, and isthmus showed that OM has the capacity to recapitulate the entire nail unit with differentiation toward the nail bed and isthmus.6 It appears that the mesenchymal component has an inductive effect that can lead to complete epithelial onychogenic differentiation.6
Due to the histological differences among the described cases of OM in the literature, a new classification based on the spectrum of epithelial to stromal ratio of stromal cellularity and the extent of nuclear pleomorphism was proposed in 2004.7 The prominent feature of the unguioblastoma type of OM is epithelial, while the cellular stroma is the prominent feature in the unguioblastic fibroma type. Atypical unguioblastic fibroma refers to a tumor with increased mitotic activity and nuclear pleomorphism among the stroma.7
Most OM tumors follow a benign clinical course; however, complete excision is advised to include the normal nail matrix proximal to the lesion, which may prevent recurrence and serves as a primary treatment.
Conclusion
Onychomatricoma is a benign neoplasm of the nail matrix that may be triggered by trauma; however, due to the weak association, further observations and studies should be conducted to substantiate this possibility. Patients with the classic clinical presentation possibly may be spared a nail avulsion and biopsy. Onychomycosis occurs in the setting of OM, and culture and treatment are unlikely to change the appearance or course of this nail condition.
1. Baran R, Kint A. Onychomatrixoma. filamentous tufted tumour in the matrix of a funnel-shaped nail: a new entity (report of three cases). Br J Dermatol. 1992;126:510-515.
2. Haneke E, Franken J. Onychomatricoma. Dermatol Surg. 1995;21:984-987.
3. Gaertner EM, Gordon M, Reed T. Onychomatricoma: case report of an unusual subungual tumor with literature review. J Cutan Pathol. 2009;36(suppl 1):66-69.
4. Cañueto J, Santos-Briz Á, García JL, et al. Onychomatricoma: genome-wide analyses of a rare nail matrix tumor. J Am Acad Dermatol. 2011;64:573-578.
5. Perrin C, Baran R. Onychomatricoma with dorsalpterygium: pathogenic mechanisms in 3 cases. J Am Acad Dermatol. 2008;59:990-994.
6. Perrin C, Langbein L, Schweizer J, et al. Onychomatricoma in the light of the microanatomy of the normal nail unit. Am J Dermatopathol. 2011;33:131-139.
7. Ko CJ, Shi L, Barr RJ, et al. Unguioblastoma and unguioblastic fibroma—an expanded spectrum of onychomatricoma. J Cutan Pathol. 2004;31:307-311.
Onychomatricoma (OM) is a rare benign neoplasm of the nail matrix. Even less common is its possible association with both trauma to the nail apparatus and onychomycosis. This case illustrates both of these findings.
Case Report
A 72-year-old white man presented to the dermatology clinic with a 26-year history of a thickened nail plate on the right third finger that had developed soon after a baseball injury. The patient reported that the nail was completely normal prior to the trauma. According to the patient, the distal aspect of the finger was directly hit by a baseball and subsequently was wrapped by the patient for a few weeks. The nail then turned black and eventually fell off. When the nail grew back, it appeared abnormal and in its current state. The patient stated the lesion was asymptomatic at the time of presentation.
Physical examination revealed thickening, yellow discoloration, and transverse overcurvature of the nail plate on the right third finger with longitudinal ridging (Figure 1). A culture of the nail plate grew Chaetomium species. Application of topical clotrimazole for 3 months followed by a 6-week course of oral terbinafine produced no improvement. The patient then consented to a nail matrix incisional biopsy 6 months after initial presentation. After a digital nerve block was administered and a tourniquet of the proximal digit was applied, a nail avulsion was performed. Subsequently, a 3-mm punch biopsy was taken of the clinically apparent tumor in the nail matrix.
On microscopic examination of the removed tissue, a benign mixed epithelial and stromal proliferative lesion was noted. The basaloid epithelium, lacking a granular layer, arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component, which was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (Figure 2). The stromal component predominated over the epithelial component in this neoplasm. The nail was preserved in formalin and underwent hematoxylin and eosin staining. It was thickened and grossly showed filiform fibrous projections extending into the nail plate. Histologically, the nail displayed prominent oval clear channels. Periodic acid–Schiff staining was negative for fungal organisms.
A diagnosis of unguioblastic fibroma–type OM was made. After receiving the diagnosis, expected course, and treatment options, the patient was offered conservative surgical excision but preferred clinical monitoring. At his last visit (6 months after the biopsy), the nail plate distal to the biopsy site had thinning and improvement, while the nail plate distal to the matrix that was not removed continued to show thickening, yellow discoloration, overcurvature, and longitudinal ridging (Figure 3).
|
|
| |
Figure 2. The basaloid epithelium arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component (A)(H&E, original magnification ×2). At higher magnification, the stromal component was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (B)(H&E, original magnification ×10). | ||
|
|
Comment
Onychomatricoma is a rare tumor originating from the nail matrix. The tumor was first described by Baran and Kint1 in 1992 using the term onychomatrixoma, but later the term onychomatricoma became more widely used.2 Onychomatricomas are more common in adults (mean age, 48 years) and white individuals with no gender predilection.3,4 Fingernail involvement is twice as common as toenail involvement.3 Onychomatricoma is the only tumor that actively produces a nail plate.4
Clinically, OM presents with yellow discoloration along the entire nail plate and proximal splinter hemorrhages. It has a tendency toward transverse overcurvature of the nail plate with prominent longitudinal ridging.4 Trauma has been associated in at least 3 cases reported in the literature, though the association was sometimes weak.3,4 Xanthonychia and onychodystrophy of the nail are common.3 Pterygium, melanonychia, nail bleeding, and cutaneous horns have been reported but are rare.3-5 The tumor typically is painless with no radiographic bone involvement.3 Onychomycosis can be present,3 which may either be a predisposing factor for the tumor or secondary due to the deformed nail plate.4
When the nail plate is avulsed and the proximal nail fold is turned back, the matrix tumor is exposed. This polypoid and filiform tumor has characteristic fingerlike fibrokeratogenous projections extending from the nail matrix into the nail plate.3
Histologically, the tumor is fibroepithelial or biphasic with stromal and epithelial components. It has a lobulated and papillary growth pattern with 2 distinct areas that correspond to 2 anatomic zones.3 The base of the tumor corresponds to the proximal anatomic zone, which begins at the root of the nail and extends to the cuticle. This area is composed of V-shaped keratinous zones similar to the normal matrix. If the nail is removed prior to excision, these areas can be avulsed, leaving clear clefts. The superficial aspect of the tumor corresponds to the distal anatomic zone, which is located in the region of the lunula. This area is composed of multiple digitate or fingerlike projections with a fibrous core and a thick matrical epithelial covering.3 These digitations extend into small cavities in the nail plate, which can be visualized as clear channels or woodwormlike holes in hematoxylin and eosin–stained specimens. A biphasic fibrous stroma also can be observed with the superficial dermis being cellular with fibrillary collagen and the deep dermis more hypocellular with thicker collagen bundles.3,4
An analysis of keratins in the nail matrix, bed, and isthmus showed that OM has the capacity to recapitulate the entire nail unit with differentiation toward the nail bed and isthmus.6 It appears that the mesenchymal component has an inductive effect that can lead to complete epithelial onychogenic differentiation.6
Due to the histological differences among the described cases of OM in the literature, a new classification based on the spectrum of epithelial to stromal ratio of stromal cellularity and the extent of nuclear pleomorphism was proposed in 2004.7 The prominent feature of the unguioblastoma type of OM is epithelial, while the cellular stroma is the prominent feature in the unguioblastic fibroma type. Atypical unguioblastic fibroma refers to a tumor with increased mitotic activity and nuclear pleomorphism among the stroma.7
Most OM tumors follow a benign clinical course; however, complete excision is advised to include the normal nail matrix proximal to the lesion, which may prevent recurrence and serves as a primary treatment.
Conclusion
Onychomatricoma is a benign neoplasm of the nail matrix that may be triggered by trauma; however, due to the weak association, further observations and studies should be conducted to substantiate this possibility. Patients with the classic clinical presentation possibly may be spared a nail avulsion and biopsy. Onychomycosis occurs in the setting of OM, and culture and treatment are unlikely to change the appearance or course of this nail condition.
Onychomatricoma (OM) is a rare benign neoplasm of the nail matrix. Even less common is its possible association with both trauma to the nail apparatus and onychomycosis. This case illustrates both of these findings.
Case Report
A 72-year-old white man presented to the dermatology clinic with a 26-year history of a thickened nail plate on the right third finger that had developed soon after a baseball injury. The patient reported that the nail was completely normal prior to the trauma. According to the patient, the distal aspect of the finger was directly hit by a baseball and subsequently was wrapped by the patient for a few weeks. The nail then turned black and eventually fell off. When the nail grew back, it appeared abnormal and in its current state. The patient stated the lesion was asymptomatic at the time of presentation.
Physical examination revealed thickening, yellow discoloration, and transverse overcurvature of the nail plate on the right third finger with longitudinal ridging (Figure 1). A culture of the nail plate grew Chaetomium species. Application of topical clotrimazole for 3 months followed by a 6-week course of oral terbinafine produced no improvement. The patient then consented to a nail matrix incisional biopsy 6 months after initial presentation. After a digital nerve block was administered and a tourniquet of the proximal digit was applied, a nail avulsion was performed. Subsequently, a 3-mm punch biopsy was taken of the clinically apparent tumor in the nail matrix.
On microscopic examination of the removed tissue, a benign mixed epithelial and stromal proliferative lesion was noted. The basaloid epithelium, lacking a granular layer, arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component, which was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (Figure 2). The stromal component predominated over the epithelial component in this neoplasm. The nail was preserved in formalin and underwent hematoxylin and eosin staining. It was thickened and grossly showed filiform fibrous projections extending into the nail plate. Histologically, the nail displayed prominent oval clear channels. Periodic acid–Schiff staining was negative for fungal organisms.
A diagnosis of unguioblastic fibroma–type OM was made. After receiving the diagnosis, expected course, and treatment options, the patient was offered conservative surgical excision but preferred clinical monitoring. At his last visit (6 months after the biopsy), the nail plate distal to the biopsy site had thinning and improvement, while the nail plate distal to the matrix that was not removed continued to show thickening, yellow discoloration, overcurvature, and longitudinal ridging (Figure 3).
|
|
| |
Figure 2. The basaloid epithelium arose from the surface epithelial layer and formed a reticulated pattern extending into the stromal component (A)(H&E, original magnification ×2). At higher magnification, the stromal component was moderately cellular with spindle to fusiform nuclei dissecting between collagen bundles arranged in parallel arrays (B)(H&E, original magnification ×10). | ||
|
|
Comment
Onychomatricoma is a rare tumor originating from the nail matrix. The tumor was first described by Baran and Kint1 in 1992 using the term onychomatrixoma, but later the term onychomatricoma became more widely used.2 Onychomatricomas are more common in adults (mean age, 48 years) and white individuals with no gender predilection.3,4 Fingernail involvement is twice as common as toenail involvement.3 Onychomatricoma is the only tumor that actively produces a nail plate.4
Clinically, OM presents with yellow discoloration along the entire nail plate and proximal splinter hemorrhages. It has a tendency toward transverse overcurvature of the nail plate with prominent longitudinal ridging.4 Trauma has been associated in at least 3 cases reported in the literature, though the association was sometimes weak.3,4 Xanthonychia and onychodystrophy of the nail are common.3 Pterygium, melanonychia, nail bleeding, and cutaneous horns have been reported but are rare.3-5 The tumor typically is painless with no radiographic bone involvement.3 Onychomycosis can be present,3 which may either be a predisposing factor for the tumor or secondary due to the deformed nail plate.4
When the nail plate is avulsed and the proximal nail fold is turned back, the matrix tumor is exposed. This polypoid and filiform tumor has characteristic fingerlike fibrokeratogenous projections extending from the nail matrix into the nail plate.3
Histologically, the tumor is fibroepithelial or biphasic with stromal and epithelial components. It has a lobulated and papillary growth pattern with 2 distinct areas that correspond to 2 anatomic zones.3 The base of the tumor corresponds to the proximal anatomic zone, which begins at the root of the nail and extends to the cuticle. This area is composed of V-shaped keratinous zones similar to the normal matrix. If the nail is removed prior to excision, these areas can be avulsed, leaving clear clefts. The superficial aspect of the tumor corresponds to the distal anatomic zone, which is located in the region of the lunula. This area is composed of multiple digitate or fingerlike projections with a fibrous core and a thick matrical epithelial covering.3 These digitations extend into small cavities in the nail plate, which can be visualized as clear channels or woodwormlike holes in hematoxylin and eosin–stained specimens. A biphasic fibrous stroma also can be observed with the superficial dermis being cellular with fibrillary collagen and the deep dermis more hypocellular with thicker collagen bundles.3,4
An analysis of keratins in the nail matrix, bed, and isthmus showed that OM has the capacity to recapitulate the entire nail unit with differentiation toward the nail bed and isthmus.6 It appears that the mesenchymal component has an inductive effect that can lead to complete epithelial onychogenic differentiation.6
Due to the histological differences among the described cases of OM in the literature, a new classification based on the spectrum of epithelial to stromal ratio of stromal cellularity and the extent of nuclear pleomorphism was proposed in 2004.7 The prominent feature of the unguioblastoma type of OM is epithelial, while the cellular stroma is the prominent feature in the unguioblastic fibroma type. Atypical unguioblastic fibroma refers to a tumor with increased mitotic activity and nuclear pleomorphism among the stroma.7
Most OM tumors follow a benign clinical course; however, complete excision is advised to include the normal nail matrix proximal to the lesion, which may prevent recurrence and serves as a primary treatment.
Conclusion
Onychomatricoma is a benign neoplasm of the nail matrix that may be triggered by trauma; however, due to the weak association, further observations and studies should be conducted to substantiate this possibility. Patients with the classic clinical presentation possibly may be spared a nail avulsion and biopsy. Onychomycosis occurs in the setting of OM, and culture and treatment are unlikely to change the appearance or course of this nail condition.
1. Baran R, Kint A. Onychomatrixoma. filamentous tufted tumour in the matrix of a funnel-shaped nail: a new entity (report of three cases). Br J Dermatol. 1992;126:510-515.
2. Haneke E, Franken J. Onychomatricoma. Dermatol Surg. 1995;21:984-987.
3. Gaertner EM, Gordon M, Reed T. Onychomatricoma: case report of an unusual subungual tumor with literature review. J Cutan Pathol. 2009;36(suppl 1):66-69.
4. Cañueto J, Santos-Briz Á, García JL, et al. Onychomatricoma: genome-wide analyses of a rare nail matrix tumor. J Am Acad Dermatol. 2011;64:573-578.
5. Perrin C, Baran R. Onychomatricoma with dorsalpterygium: pathogenic mechanisms in 3 cases. J Am Acad Dermatol. 2008;59:990-994.
6. Perrin C, Langbein L, Schweizer J, et al. Onychomatricoma in the light of the microanatomy of the normal nail unit. Am J Dermatopathol. 2011;33:131-139.
7. Ko CJ, Shi L, Barr RJ, et al. Unguioblastoma and unguioblastic fibroma—an expanded spectrum of onychomatricoma. J Cutan Pathol. 2004;31:307-311.
1. Baran R, Kint A. Onychomatrixoma. filamentous tufted tumour in the matrix of a funnel-shaped nail: a new entity (report of three cases). Br J Dermatol. 1992;126:510-515.
2. Haneke E, Franken J. Onychomatricoma. Dermatol Surg. 1995;21:984-987.
3. Gaertner EM, Gordon M, Reed T. Onychomatricoma: case report of an unusual subungual tumor with literature review. J Cutan Pathol. 2009;36(suppl 1):66-69.
4. Cañueto J, Santos-Briz Á, García JL, et al. Onychomatricoma: genome-wide analyses of a rare nail matrix tumor. J Am Acad Dermatol. 2011;64:573-578.
5. Perrin C, Baran R. Onychomatricoma with dorsalpterygium: pathogenic mechanisms in 3 cases. J Am Acad Dermatol. 2008;59:990-994.
6. Perrin C, Langbein L, Schweizer J, et al. Onychomatricoma in the light of the microanatomy of the normal nail unit. Am J Dermatopathol. 2011;33:131-139.
7. Ko CJ, Shi L, Barr RJ, et al. Unguioblastoma and unguioblastic fibroma—an expanded spectrum of onychomatricoma. J Cutan Pathol. 2004;31:307-311.
Practice Points
- Onychomatricoma is a rare benign neoplasm of the nail matrix that actively produces a nail plate.
- Onychomatricoma should be in the differential diagnosis of a thickened discolored nail plate with transverse overcurvature.
- Onychomatricoma has been associated with onychomycosis and trauma to the nail apparatus.
Intralymphatic Histiocytosis Associated With an Orthopedic Metal Implant
To the Editor:
|
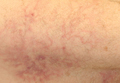
| Figure 1. A 30-cm pink and violaceous, asymmetric, reticulated patch on the lateral aspect of the right thigh. |
|
|
|
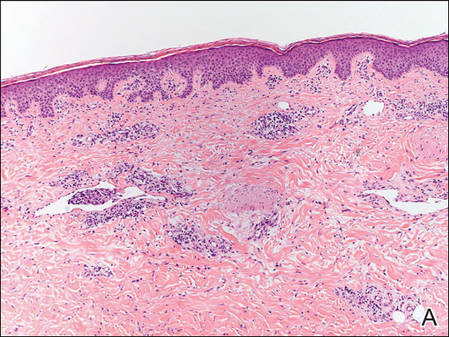
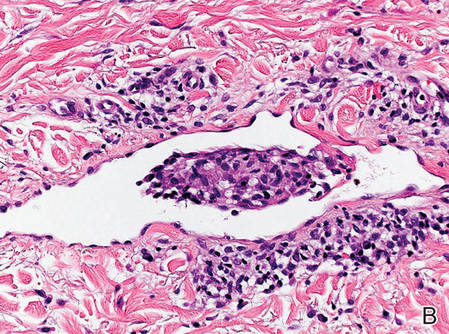
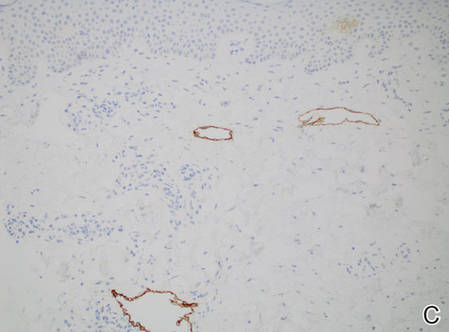
Figure 2. Histopathology revealed widely dilated vascular channels containing collections of histiocytes in the superficial dermis with adjacent features of chronic lymphedema (A)(H&E, original magnification ×10) as well as a collection of histiocytes in a dilated lymphatic channel (B)(H&E, original magnification ×40). D2-40 staining demonstrated ectatic lymphatic vessels in the upper dermins (C)(original magnification ×20).
|
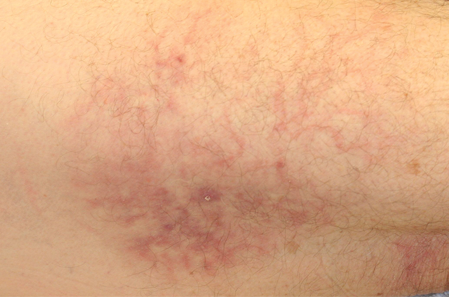
A 70-year-old white man presented with an asymptomatic patch on the lateral aspect of the right thigh of 15 months’ duration. The patient believed the patch correlated with a hip replacement 3 years prior; however, it was 6 inches inferior to the incision site. Physical examination revealed a 30-cm pink and violaceous, asymmetric, reticulated patch (Figure 1). The patch was unresponsive to topical corticosteroids as well as a short course of oral prednisone. The patient’s medical history was notable for type 2 diabetes mellitus. Histopathologic examination revealed widely dilated vascular channels containing collections of histiocytes in the superficial dermis. In addition, adjacent features of chronic lymphedema were present, namely interstitial fibroplasia with dilated lymphatic vessels and a lymphoplasmacytic infiltrate (Figure 2). These findings were consistent with intralymphatic histiocytosis, a rare disease most commonly associated with rheumatoid arthritis. Our patient did not have a history or clinical symptoms of rheumatoid arthritis.
Intralymphatic histiocytosis is a rare cutaneous condition reported by O’Grady et al1 in 1994. This condition has been most frequently associated with rheumatoid arthritis2; however, there has been an emerging association in patients with orthopedic metal implants, with and without a concomitant diagnosis of rheumatoid arthritis. Cases associated with metal implants are rare.2-7
The condition presents as asymptomatic red, brown, or violaceous patches, plaques, papules, or nodules that are ill defined and tend to demonstrate a livedo reticularis–like pattern. The lesions typically are overlying or in close proximity to a joint. Histopathologic findings include dilated vascular structures in the reticular dermis, some with empty lumina and others containing collections of mononuclear histiocytes. There also may be an inflammatory infiltrate in the adjacent dermis composed of a mix of lymphocytes, plasma cells, and/or histiocytes. Endothelial cells lining the dilated lumina express immunoreactivity for CD31, CD34, D2-40, Lyve-1, and Prox-1. Intravascular histiocytes are positive for CD68 and CD31.6
The pathogenesis of intralymphatic histiocytosis remains undefined. Some hypothesize that intralymphatic histiocytosis could be the early stage of reactive angioendotheliomatosis, as these conditions share clinical and histological features.8 Reactive angioendotheliomatosis also is a rare condition that may present as erythematous to violaceous patches or plaques. The lesions are commonly found on the limbs and may be associated with constitutional symptoms. Histologic findings of reactive angioendotheliomatosis include a proliferation of epithelioid, round, or spindle-shaped cells within the lumina of dermal blood vessels, which show positivity for CD31 and CD34.9 Others suggest the lesions of intralymphatic histiocytosis arise from lymphangiectasia; obstruction of lymphatic drainage due to congenital abnormalities; or acquired damage from infection, trauma, surgery, or radiation.2 Due to the common association with rheumatoid arthritis and orthopedic implants, it is likely that lymphatic stasis secondary to chronic inflammation plays a notable role.
Therapies such as topical and systemic corticosteroids, local radiotherapy, cyclophosphamide, pentoxifylline, and arthrocentesis have been attempted without evidence of efficacy.2 Although intralymphatic histiocytosis is chronic and resistant to therapy, patients can be reassured that the condition runs a benign course.
1. O’Grady JT, Shahidullah H, Doherty VR, et al. Intravascular histiocytosis. Histopathology. 1994;24:265-268.
2. Requena L, El-Shabrawi-Caelen L, Walsh SN, et al. Intralymphatic histiocytosis. clinicopathologic study of 16 cases. Am J Dermatopathol. 2009;31:140-151.
3. Saggar S, Lee B, Krivo J, et al. Intralymphatic histiocytosis associated with orthopedic implants. J Drugs Dermatol. 2011;10:1208-1209.
4. Chiu YE, Maloney JE, Bengana C. Erythematous patch overlying a swollen knee—quiz case. intralymphatic histiocytosis. Arch Dermatol. 2010;146:1037-1042.
5. Rossari S, Scatena C, Gori A, et al. Intralymphatic histiocytosis: cutaneous nodules and metal implants. J Cutan Pathol. 2011;38:534-535.
6. Grekin S, Mesfin M, Kang S, et al. Intralymphatic histiocytosis following placement of a metal implant. J Cutan Pathol. 2011;38:351-353.
7. Watanabe T, Yamada N, Yoshida Y, et al. Intralymphatic histiocytosis with granuloma formation associated with orthopaedic metal implants. Br J Dermatol. 2008;158:402-404.
8. Rieger E, Soyer HP, Leboit PE, et al. Reactive angioendotheliomatosis or intravascular histiocytosis? an immunohistochemical and ultrastructural study in two cases of intravascular histiocytic cell proliferation. Br J Dermatol. 1999;140:497-504.
9. Rongioletti F, Rebora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
To the Editor:
|
|
| Figure 1. A 30-cm pink and violaceous, asymmetric, reticulated patch on the lateral aspect of the right thigh. |
|
|
|
|
|
|
Figure 2. Histopathology revealed widely dilated vascular channels containing collections of histiocytes in the superficial dermis with adjacent features of chronic lymphedema (A)(H&E, original magnification ×10) as well as a collection of histiocytes in a dilated lymphatic channel (B)(H&E, original magnification ×40). D2-40 staining demonstrated ectatic lymphatic vessels in the upper dermins (C)(original magnification ×20).
|
A 70-year-old white man presented with an asymptomatic patch on the lateral aspect of the right thigh of 15 months’ duration. The patient believed the patch correlated with a hip replacement 3 years prior; however, it was 6 inches inferior to the incision site. Physical examination revealed a 30-cm pink and violaceous, asymmetric, reticulated patch (Figure 1). The patch was unresponsive to topical corticosteroids as well as a short course of oral prednisone. The patient’s medical history was notable for type 2 diabetes mellitus. Histopathologic examination revealed widely dilated vascular channels containing collections of histiocytes in the superficial dermis. In addition, adjacent features of chronic lymphedema were present, namely interstitial fibroplasia with dilated lymphatic vessels and a lymphoplasmacytic infiltrate (Figure 2). These findings were consistent with intralymphatic histiocytosis, a rare disease most commonly associated with rheumatoid arthritis. Our patient did not have a history or clinical symptoms of rheumatoid arthritis.
Intralymphatic histiocytosis is a rare cutaneous condition reported by O’Grady et al1 in 1994. This condition has been most frequently associated with rheumatoid arthritis2; however, there has been an emerging association in patients with orthopedic metal implants, with and without a concomitant diagnosis of rheumatoid arthritis. Cases associated with metal implants are rare.2-7
The condition presents as asymptomatic red, brown, or violaceous patches, plaques, papules, or nodules that are ill defined and tend to demonstrate a livedo reticularis–like pattern. The lesions typically are overlying or in close proximity to a joint. Histopathologic findings include dilated vascular structures in the reticular dermis, some with empty lumina and others containing collections of mononuclear histiocytes. There also may be an inflammatory infiltrate in the adjacent dermis composed of a mix of lymphocytes, plasma cells, and/or histiocytes. Endothelial cells lining the dilated lumina express immunoreactivity for CD31, CD34, D2-40, Lyve-1, and Prox-1. Intravascular histiocytes are positive for CD68 and CD31.6
The pathogenesis of intralymphatic histiocytosis remains undefined. Some hypothesize that intralymphatic histiocytosis could be the early stage of reactive angioendotheliomatosis, as these conditions share clinical and histological features.8 Reactive angioendotheliomatosis also is a rare condition that may present as erythematous to violaceous patches or plaques. The lesions are commonly found on the limbs and may be associated with constitutional symptoms. Histologic findings of reactive angioendotheliomatosis include a proliferation of epithelioid, round, or spindle-shaped cells within the lumina of dermal blood vessels, which show positivity for CD31 and CD34.9 Others suggest the lesions of intralymphatic histiocytosis arise from lymphangiectasia; obstruction of lymphatic drainage due to congenital abnormalities; or acquired damage from infection, trauma, surgery, or radiation.2 Due to the common association with rheumatoid arthritis and orthopedic implants, it is likely that lymphatic stasis secondary to chronic inflammation plays a notable role.
Therapies such as topical and systemic corticosteroids, local radiotherapy, cyclophosphamide, pentoxifylline, and arthrocentesis have been attempted without evidence of efficacy.2 Although intralymphatic histiocytosis is chronic and resistant to therapy, patients can be reassured that the condition runs a benign course.
To the Editor:
|
|
| Figure 1. A 30-cm pink and violaceous, asymmetric, reticulated patch on the lateral aspect of the right thigh. |
|
|
|
|
|
|
Figure 2. Histopathology revealed widely dilated vascular channels containing collections of histiocytes in the superficial dermis with adjacent features of chronic lymphedema (A)(H&E, original magnification ×10) as well as a collection of histiocytes in a dilated lymphatic channel (B)(H&E, original magnification ×40). D2-40 staining demonstrated ectatic lymphatic vessels in the upper dermins (C)(original magnification ×20).
|
A 70-year-old white man presented with an asymptomatic patch on the lateral aspect of the right thigh of 15 months’ duration. The patient believed the patch correlated with a hip replacement 3 years prior; however, it was 6 inches inferior to the incision site. Physical examination revealed a 30-cm pink and violaceous, asymmetric, reticulated patch (Figure 1). The patch was unresponsive to topical corticosteroids as well as a short course of oral prednisone. The patient’s medical history was notable for type 2 diabetes mellitus. Histopathologic examination revealed widely dilated vascular channels containing collections of histiocytes in the superficial dermis. In addition, adjacent features of chronic lymphedema were present, namely interstitial fibroplasia with dilated lymphatic vessels and a lymphoplasmacytic infiltrate (Figure 2). These findings were consistent with intralymphatic histiocytosis, a rare disease most commonly associated with rheumatoid arthritis. Our patient did not have a history or clinical symptoms of rheumatoid arthritis.
Intralymphatic histiocytosis is a rare cutaneous condition reported by O’Grady et al1 in 1994. This condition has been most frequently associated with rheumatoid arthritis2; however, there has been an emerging association in patients with orthopedic metal implants, with and without a concomitant diagnosis of rheumatoid arthritis. Cases associated with metal implants are rare.2-7
The condition presents as asymptomatic red, brown, or violaceous patches, plaques, papules, or nodules that are ill defined and tend to demonstrate a livedo reticularis–like pattern. The lesions typically are overlying or in close proximity to a joint. Histopathologic findings include dilated vascular structures in the reticular dermis, some with empty lumina and others containing collections of mononuclear histiocytes. There also may be an inflammatory infiltrate in the adjacent dermis composed of a mix of lymphocytes, plasma cells, and/or histiocytes. Endothelial cells lining the dilated lumina express immunoreactivity for CD31, CD34, D2-40, Lyve-1, and Prox-1. Intravascular histiocytes are positive for CD68 and CD31.6
The pathogenesis of intralymphatic histiocytosis remains undefined. Some hypothesize that intralymphatic histiocytosis could be the early stage of reactive angioendotheliomatosis, as these conditions share clinical and histological features.8 Reactive angioendotheliomatosis also is a rare condition that may present as erythematous to violaceous patches or plaques. The lesions are commonly found on the limbs and may be associated with constitutional symptoms. Histologic findings of reactive angioendotheliomatosis include a proliferation of epithelioid, round, or spindle-shaped cells within the lumina of dermal blood vessels, which show positivity for CD31 and CD34.9 Others suggest the lesions of intralymphatic histiocytosis arise from lymphangiectasia; obstruction of lymphatic drainage due to congenital abnormalities; or acquired damage from infection, trauma, surgery, or radiation.2 Due to the common association with rheumatoid arthritis and orthopedic implants, it is likely that lymphatic stasis secondary to chronic inflammation plays a notable role.
Therapies such as topical and systemic corticosteroids, local radiotherapy, cyclophosphamide, pentoxifylline, and arthrocentesis have been attempted without evidence of efficacy.2 Although intralymphatic histiocytosis is chronic and resistant to therapy, patients can be reassured that the condition runs a benign course.
1. O’Grady JT, Shahidullah H, Doherty VR, et al. Intravascular histiocytosis. Histopathology. 1994;24:265-268.
2. Requena L, El-Shabrawi-Caelen L, Walsh SN, et al. Intralymphatic histiocytosis. clinicopathologic study of 16 cases. Am J Dermatopathol. 2009;31:140-151.
3. Saggar S, Lee B, Krivo J, et al. Intralymphatic histiocytosis associated with orthopedic implants. J Drugs Dermatol. 2011;10:1208-1209.
4. Chiu YE, Maloney JE, Bengana C. Erythematous patch overlying a swollen knee—quiz case. intralymphatic histiocytosis. Arch Dermatol. 2010;146:1037-1042.
5. Rossari S, Scatena C, Gori A, et al. Intralymphatic histiocytosis: cutaneous nodules and metal implants. J Cutan Pathol. 2011;38:534-535.
6. Grekin S, Mesfin M, Kang S, et al. Intralymphatic histiocytosis following placement of a metal implant. J Cutan Pathol. 2011;38:351-353.
7. Watanabe T, Yamada N, Yoshida Y, et al. Intralymphatic histiocytosis with granuloma formation associated with orthopaedic metal implants. Br J Dermatol. 2008;158:402-404.
8. Rieger E, Soyer HP, Leboit PE, et al. Reactive angioendotheliomatosis or intravascular histiocytosis? an immunohistochemical and ultrastructural study in two cases of intravascular histiocytic cell proliferation. Br J Dermatol. 1999;140:497-504.
9. Rongioletti F, Rebora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
1. O’Grady JT, Shahidullah H, Doherty VR, et al. Intravascular histiocytosis. Histopathology. 1994;24:265-268.
2. Requena L, El-Shabrawi-Caelen L, Walsh SN, et al. Intralymphatic histiocytosis. clinicopathologic study of 16 cases. Am J Dermatopathol. 2009;31:140-151.
3. Saggar S, Lee B, Krivo J, et al. Intralymphatic histiocytosis associated with orthopedic implants. J Drugs Dermatol. 2011;10:1208-1209.
4. Chiu YE, Maloney JE, Bengana C. Erythematous patch overlying a swollen knee—quiz case. intralymphatic histiocytosis. Arch Dermatol. 2010;146:1037-1042.
5. Rossari S, Scatena C, Gori A, et al. Intralymphatic histiocytosis: cutaneous nodules and metal implants. J Cutan Pathol. 2011;38:534-535.
6. Grekin S, Mesfin M, Kang S, et al. Intralymphatic histiocytosis following placement of a metal implant. J Cutan Pathol. 2011;38:351-353.
7. Watanabe T, Yamada N, Yoshida Y, et al. Intralymphatic histiocytosis with granuloma formation associated with orthopaedic metal implants. Br J Dermatol. 2008;158:402-404.
8. Rieger E, Soyer HP, Leboit PE, et al. Reactive angioendotheliomatosis or intravascular histiocytosis? an immunohistochemical and ultrastructural study in two cases of intravascular histiocytic cell proliferation. Br J Dermatol. 1999;140:497-504.
9. Rongioletti F, Rebora A. Cutaneous reactive angiomatoses: patterns and classification of reactive vascular proliferation. J Am Acad Dermatol. 2003;49:887-896.
Practice Points
- Consider intralymphatic histiocytosis in the differential diagnosis of an asymptomatic skin lesion overlying a joint, particularly in patients with orthopedic metal implants or rheumatoid arthritis.
- Biopsy is essential for the diagnosis of intralymphatic histiocytosis; special stains highlighting dilated lymphatic vessels and intravascular histiocytes may be necessary.
- Intralymphatic histiocytosis is chronic and resistant to therapy; however, patients can be reassured that the condition runs a benign course.
Transition From Lichen Sclerosus to Squamous Cell Carcinoma in a Single Tissue Section
To the Editor:
Lichen sclerosus (LS) is a chronic inflammatory disorder of unknown etiology that most commonly affects the anogenital region. Progressive sclerosis results in scarring with distortion of the normal epithelial architecture.1,2 The lifetime risk for developing squamous cell carcinoma (SCC) as a complication of long-standing LS has been estimated as 4% to 6%.3,4 However, there is no general agreement concerning the exact relationship between anogenital LS and SCC.1 The coexistence of histologic findings of LS, vulvar intraepithelial neoplasia (VIN), and SCC in the same tissue is rare. We report a case of VIN and SCC developing in a region of preexisting LS.
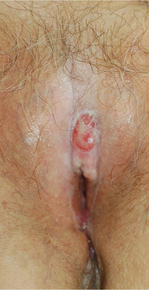
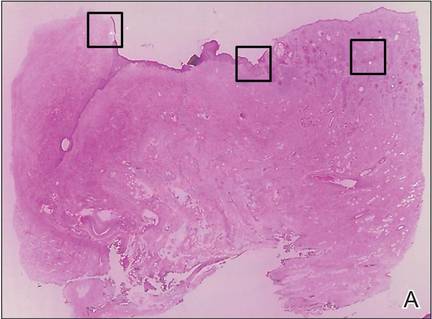
A 76-year-old woman presented with a 7-mm nodule on the clitoris that was surrounded by a pearly white, smooth, glistening area (Figure 1). The patient reported pain and tenderness associated with the nodule. No regional lymphadenopathy was evident. We performed an excisional biopsy of the entire nodule and a small part of the whitish patch (Figure 2A). On histologic examination, the presence of hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was consistent with LS (Figure 2B). The presence of dysplastic changes with mild disturbance of the epithelial architecture as well as acanthosis and dyskeratosis in the same tissue confirmed VIN (Figure 2C). Dermal invasion and transition to SCC were seen in the part of the tissue verified as VIN. The presence of dermal tumor nests and an irregular border between the epidermis and dermis pointed to the existence of fully developed SCC (Figure 2D). To prevent the recurrence of SCC, the patient returned for follow-up periodically. There was no recurrence within 6 months after excision.
|
|
|
|
| Figure 2. An excisional biopsy showed epidermal thinning on the left side and invasion of the dermis by a tumor nest on the right side (A)(H&E, original magnification ×10). Left, center, and right boxes indicate areas shown in Figures 2B, 2C and 2D, respectively. Hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was evident (B)(H&E, original magnification ×200). Dysplactic changes with mild disturbance of the epithelial architecture accompanied by acanthosis and nuclear atypia were seen (C)(H&E, original magnification ×200). Irregular masses of atypical squamous cells spread downward into the dermis representing squamous cell carcinoma of a well-differentiated type (D)(H&E, original magnification ×200). | |

Although LS is considered a premalignant condition, only a small portion of patients with LS ultimately develop vulvar SCC.5 There are a number of reasons for linking LS with the development of vulvar SCC. First, in the majority of cases of vulvar SCC, LS, squamous cell hyperplasia, or VIN is present in the adjacent epithelium. Lichen sclerosus is found in adjacent regions in up to 62% of vulvar SCC cases.6 Second, patients with LS may develop vulvar SCC, as frequently reported. Third, in a series of LS patients who underwent long-term follow-up, 4% to 6% were reported to have developed vulvar SCC.3,4,7
Lichen sclerosus is an inflammatory dermatosis characterized by clinicopathologic persistence and hypocellular fibrosis.2 Changes in the local environment of the keratinocyte, including chronic inflammation and sclerosis, may be responsible for the promotion of carcinogenesis.8 However, no molecular markers have been proven to identify the LS lesions that are at risk for developing into vulvar SCC.9,10 It has been suggested that VIN is the direct precursor of vulvar SCC.11,12
Histologic diagnosis of VIN is difficult. Its identification is hindered by a high degree of cellular differentiation combined with the absence of widespread architectural disorder, nuclear pleomorphism, and diffuse nuclear atypia.13 The atypia in VIN lesions is strictly confined to the basal and parabasal layers of the epithelium.11 Vulvar intraepithelial neoplasia has seldom been diagnosed as a solitary lesion because it appears to have a short intraepithelial lifetime.
Vulvar SCC can be divided into 2 patterns. The first is found in older women, which is unrelated to human papillomavirus (HPV). This type occurs in a background of LS and/or differentiated VIN. The second is predominantly found in younger women, which is related to high-risk HPV. This type of vulvar SCC frequently is associated with the histologic subtypes of warty and basaloid differentiations and is referred to as undifferentiated VIN. There is no association with LS in these cases.2,14,15
It has been suggested that LS and HPV may not be mutually exclusive but may act as cofactors in SCC pathogenesis.16 Infection with HPV is an early event in the multistep process of vulvar carcinogenesis, and HPV integration into host cell genome seems to be related to the progression of vulvar dysplasia.17 Viral integration generally disrupts the E2 region, resulting in enhanced expression of E6 and E7. E6 and E7 have the ability to bind and inactivate the protein p53 and retinoblastoma protein, which promotes rapid progression through the cell cycle without p53-mediated control of DNA integrity.18 However, the exact influence of HPV in vulvar SCC is uncertain, as divergent prevalence rates have been published.
In our case, histologic examination revealed the characteristic findings of LS, VIN, and SCC in succession. This sequence is evidence of progressive transition from LS to VIN and then to SCC. Consequently, this case suggests that vulvar LS may act as both an initiator and a promoter of carcinogenesis and that VIN may be the direct precursor of vulvar SCC. In conclusion, LS has a considerable risk for malignant transformation and requires continuous follow-up in all patients. Early histological detection of invasive lesions is crucial to reduce the risk for vulvar cancer.
1. Bhattacharjee P, Fatteh SM, Lloyd KL. Squamous cell carcinoma arising in long-standing lichen sclerosus et atrophicus. J Am Geriatr Soc. 2004;52:319-320.
2. Funaro D. Lichen sclerosus: a review and practical approach. Dermatol Ther. 2004;17:28-37.
3. Ulrich RH. Lichen sclerosus. In: Wolff K, Goldsmith L, Katz S, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw Hill; 2007:546-550.
4. Heymann WR. Lichen sclerosus. J Am Acad Dermatol. 2007;56:683-684.
5. Cooper SM, Gao XH, Powell JJ, et al. Does treatment of vulvar lichen sclerosus influence its prognosis? Arch Dermatol. 2004;140:702-706.
6. Kagie MJ, Kenter GG, Hermans J, et al. The relevance of various vulvar epithelial changes in the early detection of squamous cell carcinoma of the vulva. Int J Gynecol Cancer. 1997;7:50-57.
7. Thomas RH, Ridley CM, McGibbon DH, et al. Anogenital lichen sclerosus in women. J R Soc Med. 1996;89:694-698.
8. Walkden V, Chia Y, Wojnarowska F. The association of squamous cell carcinoma of the vulva and lichen sclerosus: implications for follow-up. J Obstet Gynaecol. 1997;17:551-553.
9. Tasker GL, Wojnarowska F. Lichen sclerosus. Clin Exp Dermatol. 2003;28:128-133.
10. Wang SH, Chi CC, Wong YW, et al. Genital verrucous carcinoma is associated with lichen sclerosus: a retrospective study and review of the literature. J Eur Acad Dermatol Venereol. 2010;24:815-819.
11. Hart WR. Vulvar intraepithelial neoplasia: historical aspects and current status. Int J Gynecol Pathol. 2001;20:16-30.
12. van de Nieuwenhof HP, Massuger LF, van der Avoort IA, et al. Vulvar squamous cell carcinoma development after diagnosis of VIN increases with age. Eur J Cancer. 2009;45:851-856.
13. Taube JM, Badger J, Kong CS, et al. Differentiated (simplex) vulvar intraepithelial neoplasia: a case report and review of the literature. Am J Dermatopathol. 2011;33:27-30.
14. Derrick EK, Ridley CM, Kobza-Black A, et al. A clinical study of 23 cases of female anogenital carcinoma. Br J Dermatol. 2000;143:1217-1223.
15. Crum C, McLachlin CM, Tate JE, et al. Pathobiology of vulvar squamous neoplasia. Gynecol Oncol Pathol. 1997;9:63-69.
16. Ansink AC, Krul MRL, De Weger RA, et al. Human papillomavirus, lichen sclerosus, and squamous cell carcinoma of the vulva: detection and prognostic significance. Gynecol Oncol. 1994;52:180-184.
17. Hillemanns P, Wang X. Integration of HPV-16 and HPV-18 DNA in vulvar intraepithelial neoplasia. Gynecol Oncol. 2006;100:276-282.
18. Stoler MH. Human papillomaviruses and cervical neoplasia: a model for carcinogenesis. Int J Gynecol Pathol. 2000;19:16-28.
To the Editor:
Lichen sclerosus (LS) is a chronic inflammatory disorder of unknown etiology that most commonly affects the anogenital region. Progressive sclerosis results in scarring with distortion of the normal epithelial architecture.1,2 The lifetime risk for developing squamous cell carcinoma (SCC) as a complication of long-standing LS has been estimated as 4% to 6%.3,4 However, there is no general agreement concerning the exact relationship between anogenital LS and SCC.1 The coexistence of histologic findings of LS, vulvar intraepithelial neoplasia (VIN), and SCC in the same tissue is rare. We report a case of VIN and SCC developing in a region of preexisting LS.
A 76-year-old woman presented with a 7-mm nodule on the clitoris that was surrounded by a pearly white, smooth, glistening area (Figure 1). The patient reported pain and tenderness associated with the nodule. No regional lymphadenopathy was evident. We performed an excisional biopsy of the entire nodule and a small part of the whitish patch (Figure 2A). On histologic examination, the presence of hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was consistent with LS (Figure 2B). The presence of dysplastic changes with mild disturbance of the epithelial architecture as well as acanthosis and dyskeratosis in the same tissue confirmed VIN (Figure 2C). Dermal invasion and transition to SCC were seen in the part of the tissue verified as VIN. The presence of dermal tumor nests and an irregular border between the epidermis and dermis pointed to the existence of fully developed SCC (Figure 2D). To prevent the recurrence of SCC, the patient returned for follow-up periodically. There was no recurrence within 6 months after excision.
|
|
|
|
| Figure 2. An excisional biopsy showed epidermal thinning on the left side and invasion of the dermis by a tumor nest on the right side (A)(H&E, original magnification ×10). Left, center, and right boxes indicate areas shown in Figures 2B, 2C and 2D, respectively. Hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was evident (B)(H&E, original magnification ×200). Dysplactic changes with mild disturbance of the epithelial architecture accompanied by acanthosis and nuclear atypia were seen (C)(H&E, original magnification ×200). Irregular masses of atypical squamous cells spread downward into the dermis representing squamous cell carcinoma of a well-differentiated type (D)(H&E, original magnification ×200). | |
Although LS is considered a premalignant condition, only a small portion of patients with LS ultimately develop vulvar SCC.5 There are a number of reasons for linking LS with the development of vulvar SCC. First, in the majority of cases of vulvar SCC, LS, squamous cell hyperplasia, or VIN is present in the adjacent epithelium. Lichen sclerosus is found in adjacent regions in up to 62% of vulvar SCC cases.6 Second, patients with LS may develop vulvar SCC, as frequently reported. Third, in a series of LS patients who underwent long-term follow-up, 4% to 6% were reported to have developed vulvar SCC.3,4,7
Lichen sclerosus is an inflammatory dermatosis characterized by clinicopathologic persistence and hypocellular fibrosis.2 Changes in the local environment of the keratinocyte, including chronic inflammation and sclerosis, may be responsible for the promotion of carcinogenesis.8 However, no molecular markers have been proven to identify the LS lesions that are at risk for developing into vulvar SCC.9,10 It has been suggested that VIN is the direct precursor of vulvar SCC.11,12
Histologic diagnosis of VIN is difficult. Its identification is hindered by a high degree of cellular differentiation combined with the absence of widespread architectural disorder, nuclear pleomorphism, and diffuse nuclear atypia.13 The atypia in VIN lesions is strictly confined to the basal and parabasal layers of the epithelium.11 Vulvar intraepithelial neoplasia has seldom been diagnosed as a solitary lesion because it appears to have a short intraepithelial lifetime.
Vulvar SCC can be divided into 2 patterns. The first is found in older women, which is unrelated to human papillomavirus (HPV). This type occurs in a background of LS and/or differentiated VIN. The second is predominantly found in younger women, which is related to high-risk HPV. This type of vulvar SCC frequently is associated with the histologic subtypes of warty and basaloid differentiations and is referred to as undifferentiated VIN. There is no association with LS in these cases.2,14,15
It has been suggested that LS and HPV may not be mutually exclusive but may act as cofactors in SCC pathogenesis.16 Infection with HPV is an early event in the multistep process of vulvar carcinogenesis, and HPV integration into host cell genome seems to be related to the progression of vulvar dysplasia.17 Viral integration generally disrupts the E2 region, resulting in enhanced expression of E6 and E7. E6 and E7 have the ability to bind and inactivate the protein p53 and retinoblastoma protein, which promotes rapid progression through the cell cycle without p53-mediated control of DNA integrity.18 However, the exact influence of HPV in vulvar SCC is uncertain, as divergent prevalence rates have been published.
In our case, histologic examination revealed the characteristic findings of LS, VIN, and SCC in succession. This sequence is evidence of progressive transition from LS to VIN and then to SCC. Consequently, this case suggests that vulvar LS may act as both an initiator and a promoter of carcinogenesis and that VIN may be the direct precursor of vulvar SCC. In conclusion, LS has a considerable risk for malignant transformation and requires continuous follow-up in all patients. Early histological detection of invasive lesions is crucial to reduce the risk for vulvar cancer.
To the Editor:
Lichen sclerosus (LS) is a chronic inflammatory disorder of unknown etiology that most commonly affects the anogenital region. Progressive sclerosis results in scarring with distortion of the normal epithelial architecture.1,2 The lifetime risk for developing squamous cell carcinoma (SCC) as a complication of long-standing LS has been estimated as 4% to 6%.3,4 However, there is no general agreement concerning the exact relationship between anogenital LS and SCC.1 The coexistence of histologic findings of LS, vulvar intraepithelial neoplasia (VIN), and SCC in the same tissue is rare. We report a case of VIN and SCC developing in a region of preexisting LS.
A 76-year-old woman presented with a 7-mm nodule on the clitoris that was surrounded by a pearly white, smooth, glistening area (Figure 1). The patient reported pain and tenderness associated with the nodule. No regional lymphadenopathy was evident. We performed an excisional biopsy of the entire nodule and a small part of the whitish patch (Figure 2A). On histologic examination, the presence of hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was consistent with LS (Figure 2B). The presence of dysplastic changes with mild disturbance of the epithelial architecture as well as acanthosis and dyskeratosis in the same tissue confirmed VIN (Figure 2C). Dermal invasion and transition to SCC were seen in the part of the tissue verified as VIN. The presence of dermal tumor nests and an irregular border between the epidermis and dermis pointed to the existence of fully developed SCC (Figure 2D). To prevent the recurrence of SCC, the patient returned for follow-up periodically. There was no recurrence within 6 months after excision.
|
|
|
|
| Figure 2. An excisional biopsy showed epidermal thinning on the left side and invasion of the dermis by a tumor nest on the right side (A)(H&E, original magnification ×10). Left, center, and right boxes indicate areas shown in Figures 2B, 2C and 2D, respectively. Hyperkeratosis, epidermal atrophy, a swollen dermal collagen bundle, and prominent edema was evident (B)(H&E, original magnification ×200). Dysplactic changes with mild disturbance of the epithelial architecture accompanied by acanthosis and nuclear atypia were seen (C)(H&E, original magnification ×200). Irregular masses of atypical squamous cells spread downward into the dermis representing squamous cell carcinoma of a well-differentiated type (D)(H&E, original magnification ×200). | |
Although LS is considered a premalignant condition, only a small portion of patients with LS ultimately develop vulvar SCC.5 There are a number of reasons for linking LS with the development of vulvar SCC. First, in the majority of cases of vulvar SCC, LS, squamous cell hyperplasia, or VIN is present in the adjacent epithelium. Lichen sclerosus is found in adjacent regions in up to 62% of vulvar SCC cases.6 Second, patients with LS may develop vulvar SCC, as frequently reported. Third, in a series of LS patients who underwent long-term follow-up, 4% to 6% were reported to have developed vulvar SCC.3,4,7
Lichen sclerosus is an inflammatory dermatosis characterized by clinicopathologic persistence and hypocellular fibrosis.2 Changes in the local environment of the keratinocyte, including chronic inflammation and sclerosis, may be responsible for the promotion of carcinogenesis.8 However, no molecular markers have been proven to identify the LS lesions that are at risk for developing into vulvar SCC.9,10 It has been suggested that VIN is the direct precursor of vulvar SCC.11,12
Histologic diagnosis of VIN is difficult. Its identification is hindered by a high degree of cellular differentiation combined with the absence of widespread architectural disorder, nuclear pleomorphism, and diffuse nuclear atypia.13 The atypia in VIN lesions is strictly confined to the basal and parabasal layers of the epithelium.11 Vulvar intraepithelial neoplasia has seldom been diagnosed as a solitary lesion because it appears to have a short intraepithelial lifetime.
Vulvar SCC can be divided into 2 patterns. The first is found in older women, which is unrelated to human papillomavirus (HPV). This type occurs in a background of LS and/or differentiated VIN. The second is predominantly found in younger women, which is related to high-risk HPV. This type of vulvar SCC frequently is associated with the histologic subtypes of warty and basaloid differentiations and is referred to as undifferentiated VIN. There is no association with LS in these cases.2,14,15
It has been suggested that LS and HPV may not be mutually exclusive but may act as cofactors in SCC pathogenesis.16 Infection with HPV is an early event in the multistep process of vulvar carcinogenesis, and HPV integration into host cell genome seems to be related to the progression of vulvar dysplasia.17 Viral integration generally disrupts the E2 region, resulting in enhanced expression of E6 and E7. E6 and E7 have the ability to bind and inactivate the protein p53 and retinoblastoma protein, which promotes rapid progression through the cell cycle without p53-mediated control of DNA integrity.18 However, the exact influence of HPV in vulvar SCC is uncertain, as divergent prevalence rates have been published.
In our case, histologic examination revealed the characteristic findings of LS, VIN, and SCC in succession. This sequence is evidence of progressive transition from LS to VIN and then to SCC. Consequently, this case suggests that vulvar LS may act as both an initiator and a promoter of carcinogenesis and that VIN may be the direct precursor of vulvar SCC. In conclusion, LS has a considerable risk for malignant transformation and requires continuous follow-up in all patients. Early histological detection of invasive lesions is crucial to reduce the risk for vulvar cancer.
1. Bhattacharjee P, Fatteh SM, Lloyd KL. Squamous cell carcinoma arising in long-standing lichen sclerosus et atrophicus. J Am Geriatr Soc. 2004;52:319-320.
2. Funaro D. Lichen sclerosus: a review and practical approach. Dermatol Ther. 2004;17:28-37.
3. Ulrich RH. Lichen sclerosus. In: Wolff K, Goldsmith L, Katz S, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw Hill; 2007:546-550.
4. Heymann WR. Lichen sclerosus. J Am Acad Dermatol. 2007;56:683-684.
5. Cooper SM, Gao XH, Powell JJ, et al. Does treatment of vulvar lichen sclerosus influence its prognosis? Arch Dermatol. 2004;140:702-706.
6. Kagie MJ, Kenter GG, Hermans J, et al. The relevance of various vulvar epithelial changes in the early detection of squamous cell carcinoma of the vulva. Int J Gynecol Cancer. 1997;7:50-57.
7. Thomas RH, Ridley CM, McGibbon DH, et al. Anogenital lichen sclerosus in women. J R Soc Med. 1996;89:694-698.
8. Walkden V, Chia Y, Wojnarowska F. The association of squamous cell carcinoma of the vulva and lichen sclerosus: implications for follow-up. J Obstet Gynaecol. 1997;17:551-553.
9. Tasker GL, Wojnarowska F. Lichen sclerosus. Clin Exp Dermatol. 2003;28:128-133.
10. Wang SH, Chi CC, Wong YW, et al. Genital verrucous carcinoma is associated with lichen sclerosus: a retrospective study and review of the literature. J Eur Acad Dermatol Venereol. 2010;24:815-819.
11. Hart WR. Vulvar intraepithelial neoplasia: historical aspects and current status. Int J Gynecol Pathol. 2001;20:16-30.
12. van de Nieuwenhof HP, Massuger LF, van der Avoort IA, et al. Vulvar squamous cell carcinoma development after diagnosis of VIN increases with age. Eur J Cancer. 2009;45:851-856.
13. Taube JM, Badger J, Kong CS, et al. Differentiated (simplex) vulvar intraepithelial neoplasia: a case report and review of the literature. Am J Dermatopathol. 2011;33:27-30.
14. Derrick EK, Ridley CM, Kobza-Black A, et al. A clinical study of 23 cases of female anogenital carcinoma. Br J Dermatol. 2000;143:1217-1223.
15. Crum C, McLachlin CM, Tate JE, et al. Pathobiology of vulvar squamous neoplasia. Gynecol Oncol Pathol. 1997;9:63-69.
16. Ansink AC, Krul MRL, De Weger RA, et al. Human papillomavirus, lichen sclerosus, and squamous cell carcinoma of the vulva: detection and prognostic significance. Gynecol Oncol. 1994;52:180-184.
17. Hillemanns P, Wang X. Integration of HPV-16 and HPV-18 DNA in vulvar intraepithelial neoplasia. Gynecol Oncol. 2006;100:276-282.
18. Stoler MH. Human papillomaviruses and cervical neoplasia: a model for carcinogenesis. Int J Gynecol Pathol. 2000;19:16-28.
1. Bhattacharjee P, Fatteh SM, Lloyd KL. Squamous cell carcinoma arising in long-standing lichen sclerosus et atrophicus. J Am Geriatr Soc. 2004;52:319-320.
2. Funaro D. Lichen sclerosus: a review and practical approach. Dermatol Ther. 2004;17:28-37.
3. Ulrich RH. Lichen sclerosus. In: Wolff K, Goldsmith L, Katz S, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw Hill; 2007:546-550.
4. Heymann WR. Lichen sclerosus. J Am Acad Dermatol. 2007;56:683-684.
5. Cooper SM, Gao XH, Powell JJ, et al. Does treatment of vulvar lichen sclerosus influence its prognosis? Arch Dermatol. 2004;140:702-706.
6. Kagie MJ, Kenter GG, Hermans J, et al. The relevance of various vulvar epithelial changes in the early detection of squamous cell carcinoma of the vulva. Int J Gynecol Cancer. 1997;7:50-57.
7. Thomas RH, Ridley CM, McGibbon DH, et al. Anogenital lichen sclerosus in women. J R Soc Med. 1996;89:694-698.
8. Walkden V, Chia Y, Wojnarowska F. The association of squamous cell carcinoma of the vulva and lichen sclerosus: implications for follow-up. J Obstet Gynaecol. 1997;17:551-553.
9. Tasker GL, Wojnarowska F. Lichen sclerosus. Clin Exp Dermatol. 2003;28:128-133.
10. Wang SH, Chi CC, Wong YW, et al. Genital verrucous carcinoma is associated with lichen sclerosus: a retrospective study and review of the literature. J Eur Acad Dermatol Venereol. 2010;24:815-819.
11. Hart WR. Vulvar intraepithelial neoplasia: historical aspects and current status. Int J Gynecol Pathol. 2001;20:16-30.
12. van de Nieuwenhof HP, Massuger LF, van der Avoort IA, et al. Vulvar squamous cell carcinoma development after diagnosis of VIN increases with age. Eur J Cancer. 2009;45:851-856.
13. Taube JM, Badger J, Kong CS, et al. Differentiated (simplex) vulvar intraepithelial neoplasia: a case report and review of the literature. Am J Dermatopathol. 2011;33:27-30.
14. Derrick EK, Ridley CM, Kobza-Black A, et al. A clinical study of 23 cases of female anogenital carcinoma. Br J Dermatol. 2000;143:1217-1223.
15. Crum C, McLachlin CM, Tate JE, et al. Pathobiology of vulvar squamous neoplasia. Gynecol Oncol Pathol. 1997;9:63-69.
16. Ansink AC, Krul MRL, De Weger RA, et al. Human papillomavirus, lichen sclerosus, and squamous cell carcinoma of the vulva: detection and prognostic significance. Gynecol Oncol. 1994;52:180-184.
17. Hillemanns P, Wang X. Integration of HPV-16 and HPV-18 DNA in vulvar intraepithelial neoplasia. Gynecol Oncol. 2006;100:276-282.
18. Stoler MH. Human papillomaviruses and cervical neoplasia: a model for carcinogenesis. Int J Gynecol Pathol. 2000;19:16-28.
Practice Points
- Lichen sclerosus has a considerable risk for malignant transformation and requires continuous follow-up in all patients.
- Early histological detection of invasive lesions is crucial to reduce the risk for vulvar cancer.
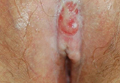
Dome-Shaped Papule With a Bloody Crust
The Diagnosis: Congenital Folliculosebaceous Cystic Hamartoma
Folliculosebaceous cystic hamartoma (FSCH) is a rare skin condition that is either congenital or acquired. It presents as a slow-growing and flesh-colored papulonodular lesion1 that mainly occurs on the head and neck. Involvement of the nipples, perineum, back, forearms, genital areas, and subcutaneous tissue also has been reported but usually indicates a larger lesion.1,2
Histologically, FSCH is considered a hamartoma composed of both ectodermal and mesodermal elements.1 Folliculosebaceous cystic hamartoma is a more complex lesion composed of infundibulocystic structures connected to maloriented folliculosebaceous units surrounded by whorls of highly vascularized fibrous stroma and adipocytes. Clefts between fibroepithelial units and surrounding stroma usually are present.1
Epithelial components contribute to the adnexal and folliculosebaceous cystic proliferations, and mesenchymal elements include vascular tissue, adipose tissue, and fibroblast-rich stroma.1,2 Acquired lesions arising in adults have been described,1-5 but the congenital presentation of FSCH in infancy is rare.
Histopathologically, some variations of FSCH are mainly composed of epithelial components while others are composed of nonepithelial components. Nonepithelial components include neural proliferation, muscle components, vascular proliferation, and mucin deposition.1-4 In some cases, FSCH may coexist with other diseases, such as nevus lipomatosus cutaneous superficialis and neurofibromatosis type I.4,5
In our case, histopathology showed several dermal infundibulocystic structures that were lined by stratified squamous epithelium and contained horny material (Figure 1). Numerous immature sebaceous lobules and rudimentary hair follicles emanated from some of the cyst walls. Mesenchymal changes around the fibroepithelial units included fibrillary bundles of collagen, clusters of adipocytes, and an increased number of small venules (Figure 2). In addition, the stroma adjacent to the malformed perifollicle contained some amount of mucin. Prominent clefts formed between fibroepithelial units and the surrounding altered stroma.
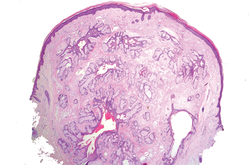
| 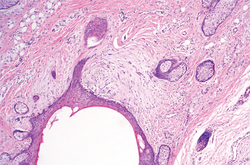
| |
|
The differential diagnosis mainly includes sebaceous trichofolliculoma, molluscum contagiosum, dermoid cysts, pilomatrixoma, Spitz nevus, and nevus lipomatosus superficialis. The differential diagnosis between FSCH and sebaceous trichofolliculoma is challenging. Both lesions show an infundibular cyst and surrounding sebaceous nodules. According to Plewig,6 trichofolliculoma has a wide spectrum ranging from low to high differentiation represented by trichofolliculoma, sebaceous trichofolliculoma, and FSCH, respectively. It is not difficult to distinguish FSCH from other diseases according to its peculiar histopathologic features.
The clinicopathologic features of our case were similar to those of reported FSCH cases, except for the following unique characteristics: congenital lesion, lack of terminal hair, and no sebaceous material extrusion. These features of hair and sebaceous material may be correlated with the patient’s age and hormonal level.1 Androgen may play a key role in sebaceous gland development at puberty, which leads to sebaceous gland hyperplasia and hypertrophy. Therefore, slight pressure from the lesions can make ivory-white sebaceous material discharge. Hence, the dermatologist and pediatrician must be poised and sensitive while making an initial diagnosis of FSCH.
1. Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma: a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
2. Moriki M, Ito T, Hirakawa S, et al. Folliculosebaceous cystic hamartoma presenting as a subcutaneous nodule on the thigh. J Dermatol. 2013;40:483-484.
3. Aloi F, Tomasini C, Pippione M. Folliculosebaceous cystic hamartoma with perifollicular mucinosis. Am J Dermatopathol. 1996;18:58-62.
4. Brasanac D, Boricic I. Giant nevus lipomatosus superficialis with multiple folliculosebaceous cystic hamartomas and dermoid cysts. J Eur Acad Dermatol Venereol. 2005;19:84-86.
5. Noh S, Kwon JE, Lee KG, et al. Folliculosebaceous cystic hamartoma in a patient with neurofibromatosis type I. Ann Dermatol. 2011;23(suppl 2):S185-S187.
6. Plewig G. In discussion of: Leserbrief zu Zheng LQ, Han XC, Huang Y, Li HW. Several acneiform papules and nodules on the neck. diagnosis: folliculosebaceous cystic hamartoma. J Dtsch Dermatol Ges. 2014;12:824-825.
The Diagnosis: Congenital Folliculosebaceous Cystic Hamartoma
Folliculosebaceous cystic hamartoma (FSCH) is a rare skin condition that is either congenital or acquired. It presents as a slow-growing and flesh-colored papulonodular lesion1 that mainly occurs on the head and neck. Involvement of the nipples, perineum, back, forearms, genital areas, and subcutaneous tissue also has been reported but usually indicates a larger lesion.1,2
Histologically, FSCH is considered a hamartoma composed of both ectodermal and mesodermal elements.1 Folliculosebaceous cystic hamartoma is a more complex lesion composed of infundibulocystic structures connected to maloriented folliculosebaceous units surrounded by whorls of highly vascularized fibrous stroma and adipocytes. Clefts between fibroepithelial units and surrounding stroma usually are present.1
Epithelial components contribute to the adnexal and folliculosebaceous cystic proliferations, and mesenchymal elements include vascular tissue, adipose tissue, and fibroblast-rich stroma.1,2 Acquired lesions arising in adults have been described,1-5 but the congenital presentation of FSCH in infancy is rare.
Histopathologically, some variations of FSCH are mainly composed of epithelial components while others are composed of nonepithelial components. Nonepithelial components include neural proliferation, muscle components, vascular proliferation, and mucin deposition.1-4 In some cases, FSCH may coexist with other diseases, such as nevus lipomatosus cutaneous superficialis and neurofibromatosis type I.4,5
In our case, histopathology showed several dermal infundibulocystic structures that were lined by stratified squamous epithelium and contained horny material (Figure 1). Numerous immature sebaceous lobules and rudimentary hair follicles emanated from some of the cyst walls. Mesenchymal changes around the fibroepithelial units included fibrillary bundles of collagen, clusters of adipocytes, and an increased number of small venules (Figure 2). In addition, the stroma adjacent to the malformed perifollicle contained some amount of mucin. Prominent clefts formed between fibroepithelial units and the surrounding altered stroma.
|
|
| |
|
The differential diagnosis mainly includes sebaceous trichofolliculoma, molluscum contagiosum, dermoid cysts, pilomatrixoma, Spitz nevus, and nevus lipomatosus superficialis. The differential diagnosis between FSCH and sebaceous trichofolliculoma is challenging. Both lesions show an infundibular cyst and surrounding sebaceous nodules. According to Plewig,6 trichofolliculoma has a wide spectrum ranging from low to high differentiation represented by trichofolliculoma, sebaceous trichofolliculoma, and FSCH, respectively. It is not difficult to distinguish FSCH from other diseases according to its peculiar histopathologic features.
The clinicopathologic features of our case were similar to those of reported FSCH cases, except for the following unique characteristics: congenital lesion, lack of terminal hair, and no sebaceous material extrusion. These features of hair and sebaceous material may be correlated with the patient’s age and hormonal level.1 Androgen may play a key role in sebaceous gland development at puberty, which leads to sebaceous gland hyperplasia and hypertrophy. Therefore, slight pressure from the lesions can make ivory-white sebaceous material discharge. Hence, the dermatologist and pediatrician must be poised and sensitive while making an initial diagnosis of FSCH.
The Diagnosis: Congenital Folliculosebaceous Cystic Hamartoma
Folliculosebaceous cystic hamartoma (FSCH) is a rare skin condition that is either congenital or acquired. It presents as a slow-growing and flesh-colored papulonodular lesion1 that mainly occurs on the head and neck. Involvement of the nipples, perineum, back, forearms, genital areas, and subcutaneous tissue also has been reported but usually indicates a larger lesion.1,2
Histologically, FSCH is considered a hamartoma composed of both ectodermal and mesodermal elements.1 Folliculosebaceous cystic hamartoma is a more complex lesion composed of infundibulocystic structures connected to maloriented folliculosebaceous units surrounded by whorls of highly vascularized fibrous stroma and adipocytes. Clefts between fibroepithelial units and surrounding stroma usually are present.1
Epithelial components contribute to the adnexal and folliculosebaceous cystic proliferations, and mesenchymal elements include vascular tissue, adipose tissue, and fibroblast-rich stroma.1,2 Acquired lesions arising in adults have been described,1-5 but the congenital presentation of FSCH in infancy is rare.
Histopathologically, some variations of FSCH are mainly composed of epithelial components while others are composed of nonepithelial components. Nonepithelial components include neural proliferation, muscle components, vascular proliferation, and mucin deposition.1-4 In some cases, FSCH may coexist with other diseases, such as nevus lipomatosus cutaneous superficialis and neurofibromatosis type I.4,5
In our case, histopathology showed several dermal infundibulocystic structures that were lined by stratified squamous epithelium and contained horny material (Figure 1). Numerous immature sebaceous lobules and rudimentary hair follicles emanated from some of the cyst walls. Mesenchymal changes around the fibroepithelial units included fibrillary bundles of collagen, clusters of adipocytes, and an increased number of small venules (Figure 2). In addition, the stroma adjacent to the malformed perifollicle contained some amount of mucin. Prominent clefts formed between fibroepithelial units and the surrounding altered stroma.
|
|
| |
|
The differential diagnosis mainly includes sebaceous trichofolliculoma, molluscum contagiosum, dermoid cysts, pilomatrixoma, Spitz nevus, and nevus lipomatosus superficialis. The differential diagnosis between FSCH and sebaceous trichofolliculoma is challenging. Both lesions show an infundibular cyst and surrounding sebaceous nodules. According to Plewig,6 trichofolliculoma has a wide spectrum ranging from low to high differentiation represented by trichofolliculoma, sebaceous trichofolliculoma, and FSCH, respectively. It is not difficult to distinguish FSCH from other diseases according to its peculiar histopathologic features.
The clinicopathologic features of our case were similar to those of reported FSCH cases, except for the following unique characteristics: congenital lesion, lack of terminal hair, and no sebaceous material extrusion. These features of hair and sebaceous material may be correlated with the patient’s age and hormonal level.1 Androgen may play a key role in sebaceous gland development at puberty, which leads to sebaceous gland hyperplasia and hypertrophy. Therefore, slight pressure from the lesions can make ivory-white sebaceous material discharge. Hence, the dermatologist and pediatrician must be poised and sensitive while making an initial diagnosis of FSCH.
1. Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma: a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
2. Moriki M, Ito T, Hirakawa S, et al. Folliculosebaceous cystic hamartoma presenting as a subcutaneous nodule on the thigh. J Dermatol. 2013;40:483-484.
3. Aloi F, Tomasini C, Pippione M. Folliculosebaceous cystic hamartoma with perifollicular mucinosis. Am J Dermatopathol. 1996;18:58-62.
4. Brasanac D, Boricic I. Giant nevus lipomatosus superficialis with multiple folliculosebaceous cystic hamartomas and dermoid cysts. J Eur Acad Dermatol Venereol. 2005;19:84-86.
5. Noh S, Kwon JE, Lee KG, et al. Folliculosebaceous cystic hamartoma in a patient with neurofibromatosis type I. Ann Dermatol. 2011;23(suppl 2):S185-S187.
6. Plewig G. In discussion of: Leserbrief zu Zheng LQ, Han XC, Huang Y, Li HW. Several acneiform papules and nodules on the neck. diagnosis: folliculosebaceous cystic hamartoma. J Dtsch Dermatol Ges. 2014;12:824-825.
1. Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma: a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
2. Moriki M, Ito T, Hirakawa S, et al. Folliculosebaceous cystic hamartoma presenting as a subcutaneous nodule on the thigh. J Dermatol. 2013;40:483-484.
3. Aloi F, Tomasini C, Pippione M. Folliculosebaceous cystic hamartoma with perifollicular mucinosis. Am J Dermatopathol. 1996;18:58-62.
4. Brasanac D, Boricic I. Giant nevus lipomatosus superficialis with multiple folliculosebaceous cystic hamartomas and dermoid cysts. J Eur Acad Dermatol Venereol. 2005;19:84-86.
5. Noh S, Kwon JE, Lee KG, et al. Folliculosebaceous cystic hamartoma in a patient with neurofibromatosis type I. Ann Dermatol. 2011;23(suppl 2):S185-S187.
6. Plewig G. In discussion of: Leserbrief zu Zheng LQ, Han XC, Huang Y, Li HW. Several acneiform papules and nodules on the neck. diagnosis: folliculosebaceous cystic hamartoma. J Dtsch Dermatol Ges. 2014;12:824-825.

A 3-year-old girl was referred to our clinic for a lesion on the face that had been present since birth and had enlarged slowly with slight itching. Physical examination revealed a 1.0×1.0-cm, sessile, flesh-colored, sharply demarcated, and dome-shaped papule with a bloody crust. It was firm and slightly painful to palpation. Dilated hair follicle–like orifices and thick central terminal hair were not found. Sebaceous material was not discharged. There was no notable family history or evidence of systemic disease. The lesion was surgically removed for cosmetic reasons and further histopathologic examination was performed.
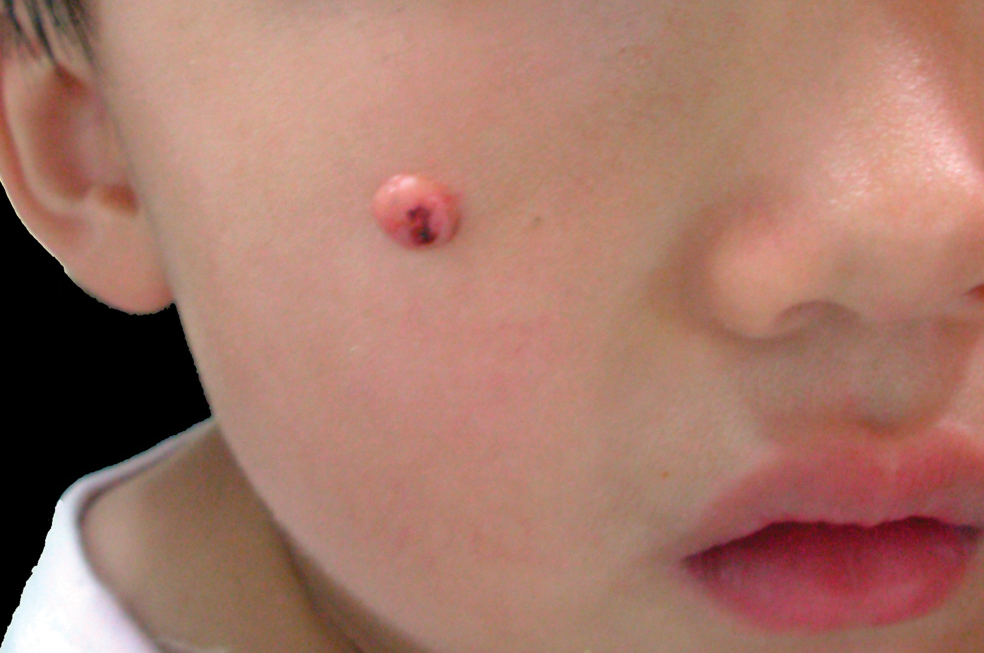
Radiation-Induced Pemphigus or Pemphigoid Disease in 3 Patients With Distinct Underlying Malignancies
A number of adverse cutaneous effects may result from radiation therapy, including radiodermatitis, alopecia, and radiation-induced neoplasms. Radiation therapy rarely induces pemphigus or pemphigoid disease, but awareness of this disorder is of clinical importance because these cutaneous lesions may resemble other skin diseases, including recurrent underlying cancer. We report 3 cases of pemphigus or pemphigoid disease that occurred after radiation therapy for in situ ductal carcinoma of the breast, cervical squamous cell carcinoma, and metastatic squamous cell carcinoma of unknown origin, respectively.
Case Reports
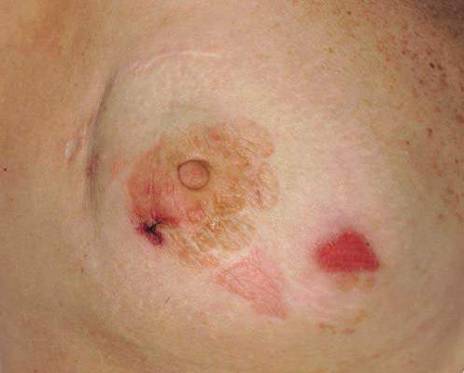
To identify all the patients with radiation-induced pemphigus, pemphigoid diseases, or both diagnosed and treated at Mayo Clinic (Rochester, Minnesota) from 1988 to 2009, we performed a computerized search of dermatology, laboratory medicine, and pathology medical records using the following keywords: radiation, pemphigoid, pemphigus vulgaris, pemphigus foliaceus, pemphigus erythematosus, and blistering disease. Inclusion criteria were a history of radiation therapy and subsequent development of pemphigus or pemphigoid disease within the irradiated fields. Patients with a history of immunobullous disease preceding radiation therapy and patients with a diagnosis of paraneoplastic pemphigus or paraneoplastic autoimmune multiorgan syndrome were excluded. The diagnoses were confirmed by routine pathology as well as direct and indirect immunofluorescence examinations.
We identified 3 patients with severe extensive radiation-associated pemphigus/pemphigoid disease that had developed within 14 months after they received radiation therapy for their underlying cancer. The identified patients’ medical records were reviewed for underlying malignancy, symptoms at the time of diagnosis, treatment course, and follow-up. The protocol was reviewed and approved by the Mayo Clinic institutional review board.
Patient 1—A 58-year-old woman was diagnosed with in situ ductal carcinoma of the right breast and underwent a lumpectomy with subsequent radiation therapy at an outside institution. Fourteen months after the final radiation treatment, she developed localized flaccid blisters and a superficial erosion on the right areola (Figure 1). Routine pathologic and direct immunofluorescence studies performed on shave biopsies in conjunction with serum analysis by indirect immunofluorescence confirmed the diagnosis of pemphigus vulgaris (Figure 2). Additionally, a deeper 4-mm punch biopsy ruled out metastatic breast carcinoma. The patient initially was treated with prednisone 60 mg and azathioprine 50 mg daily. The prednisone was tapered over 4 to 5 months to a dose of 5 mg every other day for another 4 to 5 months. Azathioprine was discontinued after a few months because of increased liver enzyme levels and a rapid clinical response of the pemphigus to this regimen.
Subsequently, she developed oral and ocular erosions that were compatible with pemphigus and were believed to be precipitated by trauma secondary to dental work and to the use of contact lenses. These flares were treated and stabilized with short courses of prednisone at higher doses that were successfully tapered to a maintenance dose of 5 mg every other day to control the pemphigus. With that prednisone dosage, her disease has remained clinically stable.
Patient 2—A 40-year-old woman was diagnosed with stage IIIB cervical squamous carcinoma with para-aortic adenopathy. She was initially treated with primary radiation therapy directed at the pelvis and para-aortic regions using a 4-field approach at our institution, and she received weekly cisplatin chemotherapy at another institution. Nine months later, the patient was admitted to our institution with persistent metastatic cervical carcinoma of the retroperitoneum. She was scheduled for intraoperative radiation therapy as well as aggressive surgical cytoreduction. The day before her surgery she presented to our dermatology clinic with a generalized pruritic rash of 1 month’s duration and occasional blistering without mucosal involvement. Biopsy specimens from the lower back and abdomen were sent for routine histologic studies and direct immunofluorescence. Serum was sent for analysis by indirect immunofluorescence. Pathology results were consistent with a diagnosis of bullous pemphigoid with an infiltrate of eosinophils in the papillary dermis; direct immunofluorescence revealed continuous strong linear deposition of C3, which also was consistent with pemphigoid.
At that time, we recommended application of topical clobetasol 0.05% twice daily to affected areas before initiating prednisone. Postoperatively, her rash improved dramatically with clobetasol monotherapy. However, 4 months after discharge from our hospital, her local dermatologist called us for a telephone consultation regarding clinical and laboratory evidence of pemphigoid relapse. A direct immunofluorescence study showed both linear IgG and C3 deposition. The patient had healed well from the surgery, and the metastatic cervical carcinoma was quiescent. Prednisone in combination with a second immunosuppressive agent was recommended, pending approval by her local oncologist. No further follow-up information is available at this time.
Patient 3—A 72-year-old woman presented with a blistering eruption that had developed on the neck, the upper part of the chest, and other body sites, including the oral mucosa, 6 months after radiation therapy for metastatic squamous cell carcinoma of unknown origin on the neck. On admission to the local hospital, she received a diagnosis of pemphigoid, although the outside biopsy specimens and reports were not available.
The patient was initially treated with prednisone, which was rapidly tapered because she was diabetic and her blood glucose levels were labile. Consequently, she was switched to azathioprine 50 mg 3 times daily and mycophenolate mofetil 500 mg 3 times daily. The patient was transferred to our institution with mild fatigue, dysphagia, weight loss, and generalized blistering involving the skin and lips. Otolaryngologic consultation and radiographic evaluation revealed no evidence of recurrent carcinoma. A shave biopsy was obtained for routine histologic evaluation and immunofluorescence and confirmed the diagnosis of bullous pemphigoid. The patient, however, also was found to have pancytopenia, most likely induced by the combination of azathioprine and mycophenolate mofetil. Her therapeutic regimen was switched to triamcinolone ointment 0.1% to be applied to the eroded areas twice daily and mupirocin ointment to be applied to the hemorrhagic scabs. Subsequently, her complete blood cell count returned to normal.
She continued to use topical corticosteroid therapy to control pemphigoid symptoms, but 6 months later the patient was found to have a lung mass and died secondary to respiratory failure.
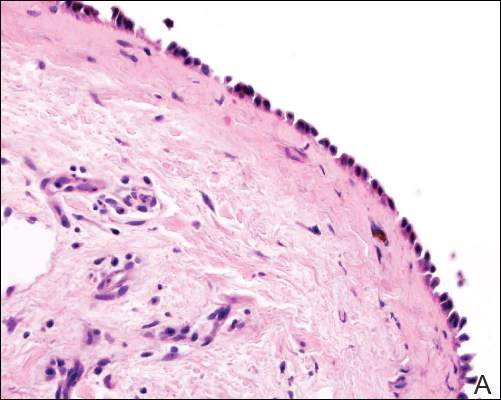
| 
|
| Figure 2. Pathologic and immunofluorescence studies confirmed the diagnosis of pemphigus vulgaris. Intraepidermal acantholysis forming a suprabasal blister with a tombstone appearance was seen along the basal cell layer (A)(H&E, original magnification ×400). Intercellular IgG deposition involving the epidermis was noted with direct immunofluorescence (B)(original magnification ×600). | |
|
|
Comment
A wide range of cutaneous reactions are known to occur in conjunction with radiation therapy. Early or acute adverse effects on the skin, such as erythema, edema, and desquamation, can be observed during radiation therapy and for several weeks thereafter. They are usually followed by hair loss and postinflammatory hyperpigmentation. Pemphigus or pemphigoid disease is a rare complication of radiation therapy and has been reported in case reports and small case series.1-17 These disorders include bullous pemphigoid, pemphigus vulgaris, pemphigus foliaceus, bullous lupus erythematosus, and acquired epidermolysis bullosa.10
The mechanism by which radiation therapy induces pemphigus remains open to speculation. Ionizing radiation may alter the antigenicity of the keratinocyte surface by disrupting the sulfhydryl groups,13 thus changing the immunoreactivity of the desmogleins or unmasking certain epidermal antigens. Another possible explanation is immune surveillance interference by damaged T-suppressor cells, which are preferentially sensitive to radiation.8 Robbins et al12 presented a patient with radiation-induced mucocutaneous pemphigus. They performed immunomapping of perilesional skin for the irradiated field, which illustrated altered expression of desmoglein (Dsg) 1, a commonly targeted antigen in pemphigus. Their study also suggested that radiation changed either the distribution or the expression of Dsg1 in the epidermis.12
Approximately half the reported cases we identified were associated with breast carcinoma,1-4,8,14 as in the case of patient 1. The majority of patients initially experienced blistering confined to the irradiated area followed by a variable degree of dissemination to other sites, probably due to the epitope-spreading phenomenon.12 During the months after radiation therapy, Aguado et al1 documented that their patient, who was initially positive for only anti-Dsg3 antibody, developed anti-Dsg1 antibodies. Therefore, the unusual development of mucosal ulcers, other skin lesions, or both after radiation therapy should raise suspicion for this diagnosis.
Bullous pemphigoid primarily affects elderly patients with blister formation along the dermoepidermal junction. Various causes, such as drugs, trauma, UV light, and ionizing radiation, have been associated with this autoimmune blistering disorder. In a systemic literature review, Mul et al10 discovered 27 case reports of bullous pemphigoid that were associated with radiation. It has been suggested that the alteration of the antigenicity and damaged dermoepidermal junction by radiation is a disease-producing mechanism.15,16 Another explanation is that the patients had subclinical pemphigoid and underwent radiation therapy, which damaged the basal layer sufficiently to produce subepidermal blister formation (triggered pemphigoid).17
The patients in this analysis had clinical presentations similar to those previously reported, with a blistering rash that usually began in the irradiated field, raising the possibility of acute radiation dermatitis. However, unlike acute radiation dermatitis, the lesions extended beyond the radiation fields in all 3 cases with mucosal involvement in patients 1 and 3. Although an onset of pemphigoid was previously observed after a minimum dose of 20 Gy,10 there was no definitive correlation observed between the extent and the severity of the cutaneous eruption and the radiation dose in prior studies. Unfortunately, we could not obtain exact radiation doses in our cases because all 3 patients were treated by radiation oncologists at other institutions. We did not, however, observe in our patients that the eruptions were more severe within the irradiated areas. Our analysis demonstrated that radiation-induced pemphigus or pemphigoid disease does not differ greatly from the endogenous form of the disease in its response to therapy or clinical course.
In summary, radiation-induced pemphigus or pemphigoid disease, a rare but serious adverse effect of radiation therapy, should be considered in patients with new-onset blistering or erosive skin disease who have recently undergone irradiation. The accurate diagnosis of pemphigus or pemphigoid disease is important because such diseases often require long-term immunosuppressive therapy. A thorough history and skin examination must be obtained from all patients who receive radiation therapy and subsequently have blisters or eruptions on the skin, mucous membranes, or both. Appropriate diagnostic studies, including routine biopsy for histologic evaluation and direct immunofluorescence, serum for indirect immunofluorescence, and enzyme-linked immunosorbent assay, should be performed to exclude pemphigus or pemphigoid disease.
1. Aguado L, Marguina M, Pretel M, et al. Lesions of pemphigus vulgaris on irradiated skin [published online January 13, 2009]. Clin Exper Dermatol. 2009;34:e148-e150.
2. Ambay A, Sratman E. Ionizing radiation-induced pemphigus foliaceus. J Am Acad Dermatol. 2005;54(suppl 5):S251-S252.
3. Cianchini G, Lembo L, Colonna L, et al. Pemphigus foliaceus induced by radiotherapy and response to dapsone. J Dermatol Treat. 2006;17:244-246.
4. Correia MP, Santos D, Jorge M, et al. Radiotherapy-induced pemphigus. Acta Med Port. 1998;11:581-583.
5. Delaporte E, Piette F, Bergoend H. Pemphigus vulgaris induced by radiotherapy. Ann Dermatol Venereol. 1991;118:447-451.
6. Girolomoni G, Mazzone E, Zambrunno G. Pemphigus vulgaris following cobalt therapy for bronchial carcinoma. Dermatologica. 1989;178:37-38.
7. Krauze E, Wygledowska-Kania M, Kaminska-Budzinska G, et al. Radiotherapy induced pemphigus vulgaris [in French]. Ann Dermatol Venereol. 2003;130:549-550.
8. Low GJ, Keeling JH. Ionizing radiation-induced pemphigus. case presentations and literature review. Arch Dermatol. 1990;126:1319-1323.
9. Mseddi M, Bouassida S, Khemakhem M, et al. Radiotherapy-induced pemphigus: a case report [published online January 18, 2005]. Cancer Radiother. 2005;9:96-98.
10. Mul VE, van Geest AJ, Pijls-Johannesma MC, et al. Radiation-induced bullous pemphigoid: a systemic review of an unusual radiation side effect [published online December 11, 2006]. Radiother Oncol. 2007;82:5-9.
11. Orion E, Matz H, Wolf R. Pemphigus vulgaris induced by radiotherapy. J Eur Acad Dermatol Venereol. 2004;18:508-509.
12. Robbins AC, Lazarova Z, Janson MM, et al. Pemphigus vulgaris presenting in a radiation portal. J Am Acad Dermatol. 2007;56(suppl 5):S82-S85.
13. Rucco V, Pisani M. Induced pemphigus. Arch Dermatol Res. 1982;274:123-140.
14. Vigna-Taglianti R, Russi EG, Denaro N, et al. Radiation-induced pemphigus vulgaris of the breast [published online April 20, 2011]. Cancer Radiother. 2011;15:334-337.
15. Cliff S, Harland CC, Fallowfield ME, et al. Localised bullous pemphigoid following radiotherapy Acta Derm Venereol. 1997;76:330-331.
16. Ohata C, Shirabe H, Takagi K, et al. Localized bullous pemphigoid after radiation therapy: two cases. Acta Derm Venereol. 1997;77:157.
17. Bernhardt M. Bullous pemphigoid after irradiation therapy. J Am Acad Dermatol. 1989;20:141-142.
A number of adverse cutaneous effects may result from radiation therapy, including radiodermatitis, alopecia, and radiation-induced neoplasms. Radiation therapy rarely induces pemphigus or pemphigoid disease, but awareness of this disorder is of clinical importance because these cutaneous lesions may resemble other skin diseases, including recurrent underlying cancer. We report 3 cases of pemphigus or pemphigoid disease that occurred after radiation therapy for in situ ductal carcinoma of the breast, cervical squamous cell carcinoma, and metastatic squamous cell carcinoma of unknown origin, respectively.
Case Reports
To identify all the patients with radiation-induced pemphigus, pemphigoid diseases, or both diagnosed and treated at Mayo Clinic (Rochester, Minnesota) from 1988 to 2009, we performed a computerized search of dermatology, laboratory medicine, and pathology medical records using the following keywords: radiation, pemphigoid, pemphigus vulgaris, pemphigus foliaceus, pemphigus erythematosus, and blistering disease. Inclusion criteria were a history of radiation therapy and subsequent development of pemphigus or pemphigoid disease within the irradiated fields. Patients with a history of immunobullous disease preceding radiation therapy and patients with a diagnosis of paraneoplastic pemphigus or paraneoplastic autoimmune multiorgan syndrome were excluded. The diagnoses were confirmed by routine pathology as well as direct and indirect immunofluorescence examinations.
We identified 3 patients with severe extensive radiation-associated pemphigus/pemphigoid disease that had developed within 14 months after they received radiation therapy for their underlying cancer. The identified patients’ medical records were reviewed for underlying malignancy, symptoms at the time of diagnosis, treatment course, and follow-up. The protocol was reviewed and approved by the Mayo Clinic institutional review board.
Patient 1—A 58-year-old woman was diagnosed with in situ ductal carcinoma of the right breast and underwent a lumpectomy with subsequent radiation therapy at an outside institution. Fourteen months after the final radiation treatment, she developed localized flaccid blisters and a superficial erosion on the right areola (Figure 1). Routine pathologic and direct immunofluorescence studies performed on shave biopsies in conjunction with serum analysis by indirect immunofluorescence confirmed the diagnosis of pemphigus vulgaris (Figure 2). Additionally, a deeper 4-mm punch biopsy ruled out metastatic breast carcinoma. The patient initially was treated with prednisone 60 mg and azathioprine 50 mg daily. The prednisone was tapered over 4 to 5 months to a dose of 5 mg every other day for another 4 to 5 months. Azathioprine was discontinued after a few months because of increased liver enzyme levels and a rapid clinical response of the pemphigus to this regimen.
Subsequently, she developed oral and ocular erosions that were compatible with pemphigus and were believed to be precipitated by trauma secondary to dental work and to the use of contact lenses. These flares were treated and stabilized with short courses of prednisone at higher doses that were successfully tapered to a maintenance dose of 5 mg every other day to control the pemphigus. With that prednisone dosage, her disease has remained clinically stable.
Patient 2—A 40-year-old woman was diagnosed with stage IIIB cervical squamous carcinoma with para-aortic adenopathy. She was initially treated with primary radiation therapy directed at the pelvis and para-aortic regions using a 4-field approach at our institution, and she received weekly cisplatin chemotherapy at another institution. Nine months later, the patient was admitted to our institution with persistent metastatic cervical carcinoma of the retroperitoneum. She was scheduled for intraoperative radiation therapy as well as aggressive surgical cytoreduction. The day before her surgery she presented to our dermatology clinic with a generalized pruritic rash of 1 month’s duration and occasional blistering without mucosal involvement. Biopsy specimens from the lower back and abdomen were sent for routine histologic studies and direct immunofluorescence. Serum was sent for analysis by indirect immunofluorescence. Pathology results were consistent with a diagnosis of bullous pemphigoid with an infiltrate of eosinophils in the papillary dermis; direct immunofluorescence revealed continuous strong linear deposition of C3, which also was consistent with pemphigoid.
At that time, we recommended application of topical clobetasol 0.05% twice daily to affected areas before initiating prednisone. Postoperatively, her rash improved dramatically with clobetasol monotherapy. However, 4 months after discharge from our hospital, her local dermatologist called us for a telephone consultation regarding clinical and laboratory evidence of pemphigoid relapse. A direct immunofluorescence study showed both linear IgG and C3 deposition. The patient had healed well from the surgery, and the metastatic cervical carcinoma was quiescent. Prednisone in combination with a second immunosuppressive agent was recommended, pending approval by her local oncologist. No further follow-up information is available at this time.
Patient 3—A 72-year-old woman presented with a blistering eruption that had developed on the neck, the upper part of the chest, and other body sites, including the oral mucosa, 6 months after radiation therapy for metastatic squamous cell carcinoma of unknown origin on the neck. On admission to the local hospital, she received a diagnosis of pemphigoid, although the outside biopsy specimens and reports were not available.
The patient was initially treated with prednisone, which was rapidly tapered because she was diabetic and her blood glucose levels were labile. Consequently, she was switched to azathioprine 50 mg 3 times daily and mycophenolate mofetil 500 mg 3 times daily. The patient was transferred to our institution with mild fatigue, dysphagia, weight loss, and generalized blistering involving the skin and lips. Otolaryngologic consultation and radiographic evaluation revealed no evidence of recurrent carcinoma. A shave biopsy was obtained for routine histologic evaluation and immunofluorescence and confirmed the diagnosis of bullous pemphigoid. The patient, however, also was found to have pancytopenia, most likely induced by the combination of azathioprine and mycophenolate mofetil. Her therapeutic regimen was switched to triamcinolone ointment 0.1% to be applied to the eroded areas twice daily and mupirocin ointment to be applied to the hemorrhagic scabs. Subsequently, her complete blood cell count returned to normal.
She continued to use topical corticosteroid therapy to control pemphigoid symptoms, but 6 months later the patient was found to have a lung mass and died secondary to respiratory failure.
|
|
|
| Figure 2. Pathologic and immunofluorescence studies confirmed the diagnosis of pemphigus vulgaris. Intraepidermal acantholysis forming a suprabasal blister with a tombstone appearance was seen along the basal cell layer (A)(H&E, original magnification ×400). Intercellular IgG deposition involving the epidermis was noted with direct immunofluorescence (B)(original magnification ×600). | |
|
|
Comment
A wide range of cutaneous reactions are known to occur in conjunction with radiation therapy. Early or acute adverse effects on the skin, such as erythema, edema, and desquamation, can be observed during radiation therapy and for several weeks thereafter. They are usually followed by hair loss and postinflammatory hyperpigmentation. Pemphigus or pemphigoid disease is a rare complication of radiation therapy and has been reported in case reports and small case series.1-17 These disorders include bullous pemphigoid, pemphigus vulgaris, pemphigus foliaceus, bullous lupus erythematosus, and acquired epidermolysis bullosa.10
The mechanism by which radiation therapy induces pemphigus remains open to speculation. Ionizing radiation may alter the antigenicity of the keratinocyte surface by disrupting the sulfhydryl groups,13 thus changing the immunoreactivity of the desmogleins or unmasking certain epidermal antigens. Another possible explanation is immune surveillance interference by damaged T-suppressor cells, which are preferentially sensitive to radiation.8 Robbins et al12 presented a patient with radiation-induced mucocutaneous pemphigus. They performed immunomapping of perilesional skin for the irradiated field, which illustrated altered expression of desmoglein (Dsg) 1, a commonly targeted antigen in pemphigus. Their study also suggested that radiation changed either the distribution or the expression of Dsg1 in the epidermis.12
Approximately half the reported cases we identified were associated with breast carcinoma,1-4,8,14 as in the case of patient 1. The majority of patients initially experienced blistering confined to the irradiated area followed by a variable degree of dissemination to other sites, probably due to the epitope-spreading phenomenon.12 During the months after radiation therapy, Aguado et al1 documented that their patient, who was initially positive for only anti-Dsg3 antibody, developed anti-Dsg1 antibodies. Therefore, the unusual development of mucosal ulcers, other skin lesions, or both after radiation therapy should raise suspicion for this diagnosis.
Bullous pemphigoid primarily affects elderly patients with blister formation along the dermoepidermal junction. Various causes, such as drugs, trauma, UV light, and ionizing radiation, have been associated with this autoimmune blistering disorder. In a systemic literature review, Mul et al10 discovered 27 case reports of bullous pemphigoid that were associated with radiation. It has been suggested that the alteration of the antigenicity and damaged dermoepidermal junction by radiation is a disease-producing mechanism.15,16 Another explanation is that the patients had subclinical pemphigoid and underwent radiation therapy, which damaged the basal layer sufficiently to produce subepidermal blister formation (triggered pemphigoid).17
The patients in this analysis had clinical presentations similar to those previously reported, with a blistering rash that usually began in the irradiated field, raising the possibility of acute radiation dermatitis. However, unlike acute radiation dermatitis, the lesions extended beyond the radiation fields in all 3 cases with mucosal involvement in patients 1 and 3. Although an onset of pemphigoid was previously observed after a minimum dose of 20 Gy,10 there was no definitive correlation observed between the extent and the severity of the cutaneous eruption and the radiation dose in prior studies. Unfortunately, we could not obtain exact radiation doses in our cases because all 3 patients were treated by radiation oncologists at other institutions. We did not, however, observe in our patients that the eruptions were more severe within the irradiated areas. Our analysis demonstrated that radiation-induced pemphigus or pemphigoid disease does not differ greatly from the endogenous form of the disease in its response to therapy or clinical course.
In summary, radiation-induced pemphigus or pemphigoid disease, a rare but serious adverse effect of radiation therapy, should be considered in patients with new-onset blistering or erosive skin disease who have recently undergone irradiation. The accurate diagnosis of pemphigus or pemphigoid disease is important because such diseases often require long-term immunosuppressive therapy. A thorough history and skin examination must be obtained from all patients who receive radiation therapy and subsequently have blisters or eruptions on the skin, mucous membranes, or both. Appropriate diagnostic studies, including routine biopsy for histologic evaluation and direct immunofluorescence, serum for indirect immunofluorescence, and enzyme-linked immunosorbent assay, should be performed to exclude pemphigus or pemphigoid disease.
A number of adverse cutaneous effects may result from radiation therapy, including radiodermatitis, alopecia, and radiation-induced neoplasms. Radiation therapy rarely induces pemphigus or pemphigoid disease, but awareness of this disorder is of clinical importance because these cutaneous lesions may resemble other skin diseases, including recurrent underlying cancer. We report 3 cases of pemphigus or pemphigoid disease that occurred after radiation therapy for in situ ductal carcinoma of the breast, cervical squamous cell carcinoma, and metastatic squamous cell carcinoma of unknown origin, respectively.
Case Reports
To identify all the patients with radiation-induced pemphigus, pemphigoid diseases, or both diagnosed and treated at Mayo Clinic (Rochester, Minnesota) from 1988 to 2009, we performed a computerized search of dermatology, laboratory medicine, and pathology medical records using the following keywords: radiation, pemphigoid, pemphigus vulgaris, pemphigus foliaceus, pemphigus erythematosus, and blistering disease. Inclusion criteria were a history of radiation therapy and subsequent development of pemphigus or pemphigoid disease within the irradiated fields. Patients with a history of immunobullous disease preceding radiation therapy and patients with a diagnosis of paraneoplastic pemphigus or paraneoplastic autoimmune multiorgan syndrome were excluded. The diagnoses were confirmed by routine pathology as well as direct and indirect immunofluorescence examinations.
We identified 3 patients with severe extensive radiation-associated pemphigus/pemphigoid disease that had developed within 14 months after they received radiation therapy for their underlying cancer. The identified patients’ medical records were reviewed for underlying malignancy, symptoms at the time of diagnosis, treatment course, and follow-up. The protocol was reviewed and approved by the Mayo Clinic institutional review board.
Patient 1—A 58-year-old woman was diagnosed with in situ ductal carcinoma of the right breast and underwent a lumpectomy with subsequent radiation therapy at an outside institution. Fourteen months after the final radiation treatment, she developed localized flaccid blisters and a superficial erosion on the right areola (Figure 1). Routine pathologic and direct immunofluorescence studies performed on shave biopsies in conjunction with serum analysis by indirect immunofluorescence confirmed the diagnosis of pemphigus vulgaris (Figure 2). Additionally, a deeper 4-mm punch biopsy ruled out metastatic breast carcinoma. The patient initially was treated with prednisone 60 mg and azathioprine 50 mg daily. The prednisone was tapered over 4 to 5 months to a dose of 5 mg every other day for another 4 to 5 months. Azathioprine was discontinued after a few months because of increased liver enzyme levels and a rapid clinical response of the pemphigus to this regimen.
Subsequently, she developed oral and ocular erosions that were compatible with pemphigus and were believed to be precipitated by trauma secondary to dental work and to the use of contact lenses. These flares were treated and stabilized with short courses of prednisone at higher doses that were successfully tapered to a maintenance dose of 5 mg every other day to control the pemphigus. With that prednisone dosage, her disease has remained clinically stable.
Patient 2—A 40-year-old woman was diagnosed with stage IIIB cervical squamous carcinoma with para-aortic adenopathy. She was initially treated with primary radiation therapy directed at the pelvis and para-aortic regions using a 4-field approach at our institution, and she received weekly cisplatin chemotherapy at another institution. Nine months later, the patient was admitted to our institution with persistent metastatic cervical carcinoma of the retroperitoneum. She was scheduled for intraoperative radiation therapy as well as aggressive surgical cytoreduction. The day before her surgery she presented to our dermatology clinic with a generalized pruritic rash of 1 month’s duration and occasional blistering without mucosal involvement. Biopsy specimens from the lower back and abdomen were sent for routine histologic studies and direct immunofluorescence. Serum was sent for analysis by indirect immunofluorescence. Pathology results were consistent with a diagnosis of bullous pemphigoid with an infiltrate of eosinophils in the papillary dermis; direct immunofluorescence revealed continuous strong linear deposition of C3, which also was consistent with pemphigoid.
At that time, we recommended application of topical clobetasol 0.05% twice daily to affected areas before initiating prednisone. Postoperatively, her rash improved dramatically with clobetasol monotherapy. However, 4 months after discharge from our hospital, her local dermatologist called us for a telephone consultation regarding clinical and laboratory evidence of pemphigoid relapse. A direct immunofluorescence study showed both linear IgG and C3 deposition. The patient had healed well from the surgery, and the metastatic cervical carcinoma was quiescent. Prednisone in combination with a second immunosuppressive agent was recommended, pending approval by her local oncologist. No further follow-up information is available at this time.
Patient 3—A 72-year-old woman presented with a blistering eruption that had developed on the neck, the upper part of the chest, and other body sites, including the oral mucosa, 6 months after radiation therapy for metastatic squamous cell carcinoma of unknown origin on the neck. On admission to the local hospital, she received a diagnosis of pemphigoid, although the outside biopsy specimens and reports were not available.
The patient was initially treated with prednisone, which was rapidly tapered because she was diabetic and her blood glucose levels were labile. Consequently, she was switched to azathioprine 50 mg 3 times daily and mycophenolate mofetil 500 mg 3 times daily. The patient was transferred to our institution with mild fatigue, dysphagia, weight loss, and generalized blistering involving the skin and lips. Otolaryngologic consultation and radiographic evaluation revealed no evidence of recurrent carcinoma. A shave biopsy was obtained for routine histologic evaluation and immunofluorescence and confirmed the diagnosis of bullous pemphigoid. The patient, however, also was found to have pancytopenia, most likely induced by the combination of azathioprine and mycophenolate mofetil. Her therapeutic regimen was switched to triamcinolone ointment 0.1% to be applied to the eroded areas twice daily and mupirocin ointment to be applied to the hemorrhagic scabs. Subsequently, her complete blood cell count returned to normal.
She continued to use topical corticosteroid therapy to control pemphigoid symptoms, but 6 months later the patient was found to have a lung mass and died secondary to respiratory failure.
|
|
|
| Figure 2. Pathologic and immunofluorescence studies confirmed the diagnosis of pemphigus vulgaris. Intraepidermal acantholysis forming a suprabasal blister with a tombstone appearance was seen along the basal cell layer (A)(H&E, original magnification ×400). Intercellular IgG deposition involving the epidermis was noted with direct immunofluorescence (B)(original magnification ×600). | |
|
|
Comment
A wide range of cutaneous reactions are known to occur in conjunction with radiation therapy. Early or acute adverse effects on the skin, such as erythema, edema, and desquamation, can be observed during radiation therapy and for several weeks thereafter. They are usually followed by hair loss and postinflammatory hyperpigmentation. Pemphigus or pemphigoid disease is a rare complication of radiation therapy and has been reported in case reports and small case series.1-17 These disorders include bullous pemphigoid, pemphigus vulgaris, pemphigus foliaceus, bullous lupus erythematosus, and acquired epidermolysis bullosa.10
The mechanism by which radiation therapy induces pemphigus remains open to speculation. Ionizing radiation may alter the antigenicity of the keratinocyte surface by disrupting the sulfhydryl groups,13 thus changing the immunoreactivity of the desmogleins or unmasking certain epidermal antigens. Another possible explanation is immune surveillance interference by damaged T-suppressor cells, which are preferentially sensitive to radiation.8 Robbins et al12 presented a patient with radiation-induced mucocutaneous pemphigus. They performed immunomapping of perilesional skin for the irradiated field, which illustrated altered expression of desmoglein (Dsg) 1, a commonly targeted antigen in pemphigus. Their study also suggested that radiation changed either the distribution or the expression of Dsg1 in the epidermis.12
Approximately half the reported cases we identified were associated with breast carcinoma,1-4,8,14 as in the case of patient 1. The majority of patients initially experienced blistering confined to the irradiated area followed by a variable degree of dissemination to other sites, probably due to the epitope-spreading phenomenon.12 During the months after radiation therapy, Aguado et al1 documented that their patient, who was initially positive for only anti-Dsg3 antibody, developed anti-Dsg1 antibodies. Therefore, the unusual development of mucosal ulcers, other skin lesions, or both after radiation therapy should raise suspicion for this diagnosis.
Bullous pemphigoid primarily affects elderly patients with blister formation along the dermoepidermal junction. Various causes, such as drugs, trauma, UV light, and ionizing radiation, have been associated with this autoimmune blistering disorder. In a systemic literature review, Mul et al10 discovered 27 case reports of bullous pemphigoid that were associated with radiation. It has been suggested that the alteration of the antigenicity and damaged dermoepidermal junction by radiation is a disease-producing mechanism.15,16 Another explanation is that the patients had subclinical pemphigoid and underwent radiation therapy, which damaged the basal layer sufficiently to produce subepidermal blister formation (triggered pemphigoid).17
The patients in this analysis had clinical presentations similar to those previously reported, with a blistering rash that usually began in the irradiated field, raising the possibility of acute radiation dermatitis. However, unlike acute radiation dermatitis, the lesions extended beyond the radiation fields in all 3 cases with mucosal involvement in patients 1 and 3. Although an onset of pemphigoid was previously observed after a minimum dose of 20 Gy,10 there was no definitive correlation observed between the extent and the severity of the cutaneous eruption and the radiation dose in prior studies. Unfortunately, we could not obtain exact radiation doses in our cases because all 3 patients were treated by radiation oncologists at other institutions. We did not, however, observe in our patients that the eruptions were more severe within the irradiated areas. Our analysis demonstrated that radiation-induced pemphigus or pemphigoid disease does not differ greatly from the endogenous form of the disease in its response to therapy or clinical course.
In summary, radiation-induced pemphigus or pemphigoid disease, a rare but serious adverse effect of radiation therapy, should be considered in patients with new-onset blistering or erosive skin disease who have recently undergone irradiation. The accurate diagnosis of pemphigus or pemphigoid disease is important because such diseases often require long-term immunosuppressive therapy. A thorough history and skin examination must be obtained from all patients who receive radiation therapy and subsequently have blisters or eruptions on the skin, mucous membranes, or both. Appropriate diagnostic studies, including routine biopsy for histologic evaluation and direct immunofluorescence, serum for indirect immunofluorescence, and enzyme-linked immunosorbent assay, should be performed to exclude pemphigus or pemphigoid disease.
1. Aguado L, Marguina M, Pretel M, et al. Lesions of pemphigus vulgaris on irradiated skin [published online January 13, 2009]. Clin Exper Dermatol. 2009;34:e148-e150.
2. Ambay A, Sratman E. Ionizing radiation-induced pemphigus foliaceus. J Am Acad Dermatol. 2005;54(suppl 5):S251-S252.
3. Cianchini G, Lembo L, Colonna L, et al. Pemphigus foliaceus induced by radiotherapy and response to dapsone. J Dermatol Treat. 2006;17:244-246.
4. Correia MP, Santos D, Jorge M, et al. Radiotherapy-induced pemphigus. Acta Med Port. 1998;11:581-583.
5. Delaporte E, Piette F, Bergoend H. Pemphigus vulgaris induced by radiotherapy. Ann Dermatol Venereol. 1991;118:447-451.
6. Girolomoni G, Mazzone E, Zambrunno G. Pemphigus vulgaris following cobalt therapy for bronchial carcinoma. Dermatologica. 1989;178:37-38.
7. Krauze E, Wygledowska-Kania M, Kaminska-Budzinska G, et al. Radiotherapy induced pemphigus vulgaris [in French]. Ann Dermatol Venereol. 2003;130:549-550.
8. Low GJ, Keeling JH. Ionizing radiation-induced pemphigus. case presentations and literature review. Arch Dermatol. 1990;126:1319-1323.
9. Mseddi M, Bouassida S, Khemakhem M, et al. Radiotherapy-induced pemphigus: a case report [published online January 18, 2005]. Cancer Radiother. 2005;9:96-98.
10. Mul VE, van Geest AJ, Pijls-Johannesma MC, et al. Radiation-induced bullous pemphigoid: a systemic review of an unusual radiation side effect [published online December 11, 2006]. Radiother Oncol. 2007;82:5-9.
11. Orion E, Matz H, Wolf R. Pemphigus vulgaris induced by radiotherapy. J Eur Acad Dermatol Venereol. 2004;18:508-509.
12. Robbins AC, Lazarova Z, Janson MM, et al. Pemphigus vulgaris presenting in a radiation portal. J Am Acad Dermatol. 2007;56(suppl 5):S82-S85.
13. Rucco V, Pisani M. Induced pemphigus. Arch Dermatol Res. 1982;274:123-140.
14. Vigna-Taglianti R, Russi EG, Denaro N, et al. Radiation-induced pemphigus vulgaris of the breast [published online April 20, 2011]. Cancer Radiother. 2011;15:334-337.
15. Cliff S, Harland CC, Fallowfield ME, et al. Localised bullous pemphigoid following radiotherapy Acta Derm Venereol. 1997;76:330-331.
16. Ohata C, Shirabe H, Takagi K, et al. Localized bullous pemphigoid after radiation therapy: two cases. Acta Derm Venereol. 1997;77:157.
17. Bernhardt M. Bullous pemphigoid after irradiation therapy. J Am Acad Dermatol. 1989;20:141-142.
1. Aguado L, Marguina M, Pretel M, et al. Lesions of pemphigus vulgaris on irradiated skin [published online January 13, 2009]. Clin Exper Dermatol. 2009;34:e148-e150.
2. Ambay A, Sratman E. Ionizing radiation-induced pemphigus foliaceus. J Am Acad Dermatol. 2005;54(suppl 5):S251-S252.
3. Cianchini G, Lembo L, Colonna L, et al. Pemphigus foliaceus induced by radiotherapy and response to dapsone. J Dermatol Treat. 2006;17:244-246.
4. Correia MP, Santos D, Jorge M, et al. Radiotherapy-induced pemphigus. Acta Med Port. 1998;11:581-583.
5. Delaporte E, Piette F, Bergoend H. Pemphigus vulgaris induced by radiotherapy. Ann Dermatol Venereol. 1991;118:447-451.
6. Girolomoni G, Mazzone E, Zambrunno G. Pemphigus vulgaris following cobalt therapy for bronchial carcinoma. Dermatologica. 1989;178:37-38.
7. Krauze E, Wygledowska-Kania M, Kaminska-Budzinska G, et al. Radiotherapy induced pemphigus vulgaris [in French]. Ann Dermatol Venereol. 2003;130:549-550.
8. Low GJ, Keeling JH. Ionizing radiation-induced pemphigus. case presentations and literature review. Arch Dermatol. 1990;126:1319-1323.
9. Mseddi M, Bouassida S, Khemakhem M, et al. Radiotherapy-induced pemphigus: a case report [published online January 18, 2005]. Cancer Radiother. 2005;9:96-98.
10. Mul VE, van Geest AJ, Pijls-Johannesma MC, et al. Radiation-induced bullous pemphigoid: a systemic review of an unusual radiation side effect [published online December 11, 2006]. Radiother Oncol. 2007;82:5-9.
11. Orion E, Matz H, Wolf R. Pemphigus vulgaris induced by radiotherapy. J Eur Acad Dermatol Venereol. 2004;18:508-509.
12. Robbins AC, Lazarova Z, Janson MM, et al. Pemphigus vulgaris presenting in a radiation portal. J Am Acad Dermatol. 2007;56(suppl 5):S82-S85.
13. Rucco V, Pisani M. Induced pemphigus. Arch Dermatol Res. 1982;274:123-140.
14. Vigna-Taglianti R, Russi EG, Denaro N, et al. Radiation-induced pemphigus vulgaris of the breast [published online April 20, 2011]. Cancer Radiother. 2011;15:334-337.
15. Cliff S, Harland CC, Fallowfield ME, et al. Localised bullous pemphigoid following radiotherapy Acta Derm Venereol. 1997;76:330-331.
16. Ohata C, Shirabe H, Takagi K, et al. Localized bullous pemphigoid after radiation therapy: two cases. Acta Derm Venereol. 1997;77:157.
17. Bernhardt M. Bullous pemphigoid after irradiation therapy. J Am Acad Dermatol. 1989;20:141-142.
Practice Points
- The use of radiation therapy is increasing because of its therapeutic benefit, especially in advanced-stage cancer patients.
- Although there is a wide range of adverse effects associated with radiation therapy, pemphigus or pemphigoid disease is rare and needs to be distinguished from other skin diseases or even recurrent underlying cancer.
- The precise mechanism of radiation-induced pemphigus or pemphigoid disease is unknown, but clinicians should be alert to this potentially serious complication, and all cutaneous eruptions developing during and after radiation therapy should be evaluated with routine histologic examination in conjunction with direct immunofluorescence, serum for indirect immunofluorescence, and enzyme-linked immunosorbent assay.
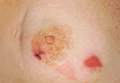
Clinical Pearl: The Squeeze Maneuver
Practice Gap
Warts may negatively impact a patient's quality of life, as they may cause not only discomfort and pain but also embarrassment and low self-esteem.1 Moreover, Ciconte et al1 demonstrated that study participants with warts on their feet were more likely to report physical discomfort than those with warts on their hands. Therefore, plantar warts should be diagnosed promptly to allow for proper treatment.
Warts may be identified by viewing the dilated capillaries that lie on their surface, which appear as small black dots to the naked eye.1 The formation of a plantar wart obliterates the normal plantar creases, thereby flattening the skin’s natural markings. However, a plantar wart may appear clinically similar to a callus and both lesions typically form in pressure point areas, warranting the use of a tool that aids in its diagnostic evaluation.1,2
Diagnostic Tools
Dermoscopy, a noninvasive tool that creates a microscopic visualization of lesions, is commonly used to distinguish dermatologic pathology if the clinical presentation overlaps with a similar lesion, such as a callus, corn, or plantar wart.1,3 However, there is another way of differentiating plantar warts from calluses using a simple 2-step clinical maneuver that we learned from Dr. Lewis Kaplan at the University of Miami.
Using the thumb or index finger, apply pressure at a perpendicular angle to the lesion on the sole of the patient’s foot, which will not create substantial discomfort or pain in a patient who has a plantar wart (Figure) but will be painful in a patient who has a callus due to the underlying bony spur. The next step involves applying pressure to the left and right sides of the lesion by squeezing toward the center with the thumb and index finger at a 45° angle. This maneuver will create substantial discomfort and pain in patients with plantar warts, thus helping to confirm the diagnosis.
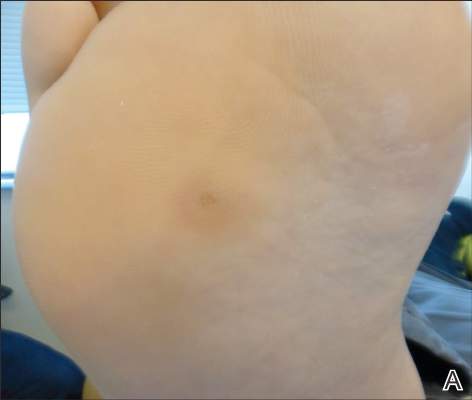
| 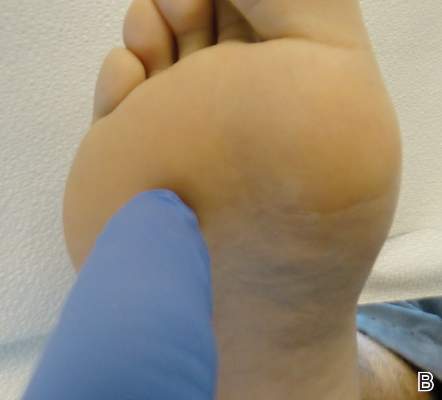
|
| A plantar wart before (A) and after undergoing the squeeze maneuver (B). The patient denied feelings of discomfort or pain. | |
Practice Implications
Rarely, a plantar wart can progress to form a verrucous carcinoma if left untreated.2 Thus, it is important to diagnose and treat plantar warts to avoid pain and potential complications. The technique discussed here, which we are coining as the “squeeze maneuver,” allows for easy diagnosis and negates the need for an expensive diagnostic tool.To submit a clinical pearl, contact our Editorial Office.
- Ciconte A, Campbell J, Tabrizi S, et al. Warts are not merely blemishes on the skin: a study on the morbidity associated with having viral cutaneous warts. Australas J Dermatol. 2003;44:169-173.
- Cardoso J, Calonje E. Cutaneous manifestations of human papillomaviruses: a review. Acta Dermatovenerol. 2011;20:145-154.
- Bae J, Kang H, Kim H, et al. Differential diagnosis of plantar wart from corn, callus and healed wart with the aid of dermoscopy. Br J Dermatol. 2009;160:220-222.
Practice Gap
Warts may negatively impact a patient's quality of life, as they may cause not only discomfort and pain but also embarrassment and low self-esteem.1 Moreover, Ciconte et al1 demonstrated that study participants with warts on their feet were more likely to report physical discomfort than those with warts on their hands. Therefore, plantar warts should be diagnosed promptly to allow for proper treatment.
Warts may be identified by viewing the dilated capillaries that lie on their surface, which appear as small black dots to the naked eye.1 The formation of a plantar wart obliterates the normal plantar creases, thereby flattening the skin’s natural markings. However, a plantar wart may appear clinically similar to a callus and both lesions typically form in pressure point areas, warranting the use of a tool that aids in its diagnostic evaluation.1,2
Diagnostic Tools
Dermoscopy, a noninvasive tool that creates a microscopic visualization of lesions, is commonly used to distinguish dermatologic pathology if the clinical presentation overlaps with a similar lesion, such as a callus, corn, or plantar wart.1,3 However, there is another way of differentiating plantar warts from calluses using a simple 2-step clinical maneuver that we learned from Dr. Lewis Kaplan at the University of Miami.
Using the thumb or index finger, apply pressure at a perpendicular angle to the lesion on the sole of the patient’s foot, which will not create substantial discomfort or pain in a patient who has a plantar wart (Figure) but will be painful in a patient who has a callus due to the underlying bony spur. The next step involves applying pressure to the left and right sides of the lesion by squeezing toward the center with the thumb and index finger at a 45° angle. This maneuver will create substantial discomfort and pain in patients with plantar warts, thus helping to confirm the diagnosis.
|
|
|
| A plantar wart before (A) and after undergoing the squeeze maneuver (B). The patient denied feelings of discomfort or pain. | |
Practice Implications
Rarely, a plantar wart can progress to form a verrucous carcinoma if left untreated.2 Thus, it is important to diagnose and treat plantar warts to avoid pain and potential complications. The technique discussed here, which we are coining as the “squeeze maneuver,” allows for easy diagnosis and negates the need for an expensive diagnostic tool.To submit a clinical pearl, contact our Editorial Office.
Practice Gap
Warts may negatively impact a patient's quality of life, as they may cause not only discomfort and pain but also embarrassment and low self-esteem.1 Moreover, Ciconte et al1 demonstrated that study participants with warts on their feet were more likely to report physical discomfort than those with warts on their hands. Therefore, plantar warts should be diagnosed promptly to allow for proper treatment.
Warts may be identified by viewing the dilated capillaries that lie on their surface, which appear as small black dots to the naked eye.1 The formation of a plantar wart obliterates the normal plantar creases, thereby flattening the skin’s natural markings. However, a plantar wart may appear clinically similar to a callus and both lesions typically form in pressure point areas, warranting the use of a tool that aids in its diagnostic evaluation.1,2
Diagnostic Tools
Dermoscopy, a noninvasive tool that creates a microscopic visualization of lesions, is commonly used to distinguish dermatologic pathology if the clinical presentation overlaps with a similar lesion, such as a callus, corn, or plantar wart.1,3 However, there is another way of differentiating plantar warts from calluses using a simple 2-step clinical maneuver that we learned from Dr. Lewis Kaplan at the University of Miami.
Using the thumb or index finger, apply pressure at a perpendicular angle to the lesion on the sole of the patient’s foot, which will not create substantial discomfort or pain in a patient who has a plantar wart (Figure) but will be painful in a patient who has a callus due to the underlying bony spur. The next step involves applying pressure to the left and right sides of the lesion by squeezing toward the center with the thumb and index finger at a 45° angle. This maneuver will create substantial discomfort and pain in patients with plantar warts, thus helping to confirm the diagnosis.
|
|
|
| A plantar wart before (A) and after undergoing the squeeze maneuver (B). The patient denied feelings of discomfort or pain. | |
Practice Implications
Rarely, a plantar wart can progress to form a verrucous carcinoma if left untreated.2 Thus, it is important to diagnose and treat plantar warts to avoid pain and potential complications. The technique discussed here, which we are coining as the “squeeze maneuver,” allows for easy diagnosis and negates the need for an expensive diagnostic tool.To submit a clinical pearl, contact our Editorial Office.
- Ciconte A, Campbell J, Tabrizi S, et al. Warts are not merely blemishes on the skin: a study on the morbidity associated with having viral cutaneous warts. Australas J Dermatol. 2003;44:169-173.
- Cardoso J, Calonje E. Cutaneous manifestations of human papillomaviruses: a review. Acta Dermatovenerol. 2011;20:145-154.
- Bae J, Kang H, Kim H, et al. Differential diagnosis of plantar wart from corn, callus and healed wart with the aid of dermoscopy. Br J Dermatol. 2009;160:220-222.
- Ciconte A, Campbell J, Tabrizi S, et al. Warts are not merely blemishes on the skin: a study on the morbidity associated with having viral cutaneous warts. Australas J Dermatol. 2003;44:169-173.
- Cardoso J, Calonje E. Cutaneous manifestations of human papillomaviruses: a review. Acta Dermatovenerol. 2011;20:145-154.
- Bae J, Kang H, Kim H, et al. Differential diagnosis of plantar wart from corn, callus and healed wart with the aid of dermoscopy. Br J Dermatol. 2009;160:220-222.
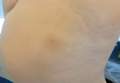
Diagnosing Porokeratosis of Mibelli Every Time: A Novel Biopsy Technique to Maximize Histopathologic Confirmation
Porokeratosis of Mibelli (PM) is a lesion characterized by a surrounding cornoid lamella with variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) in the center of the lesion that typically presents in infancy to early childhood.1 We report a case of PM in which a prior biopsy from the center of the lesion demonstrated papulosquamous dermatitis. We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM.
Case Report
A 3-year-old girl presented with an erythematous, hypopigmented, scaling plaque on the posterior aspect of the left ankle surrounded by a hard rim. The plaque was first noted at 12 months of age and had slowly enlarged as the patient grew. Six months prior, a biopsy from the center of the lesion performed at another facility demonstrated a papulosquamous dermatitis.
Physical examination revealed a lesion that was 4.2-cm long, 2.2-cm wide at the superior pole, and 3.5-cm wide at the inferior pole (Figure 1). A line was drawn with a skin marker perpendicular to the rim of the lesion (Figure 2A) and a 6-mm punch biopsy was performed, centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). The tissue was then bisected at the bedside along the skin marker line with a #15 blade (Figure 2C) and submitted in formalin for histologic processing. Histologic examination revealed an invagination of the epidermis producing a tier of parakeratotic cells with its apex pointed away from the center of the lesion. Dyskeratotic cells were noted at the base of the parakeratosis (Figure 3). Verrucous hyperplasia was present in the central portion of the specimen adjacent to the cornoid lamella. Based on these histopathologic findings, the correct diagnosis of PM was made.
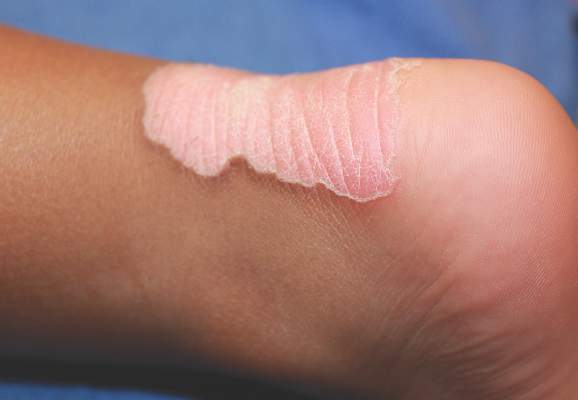
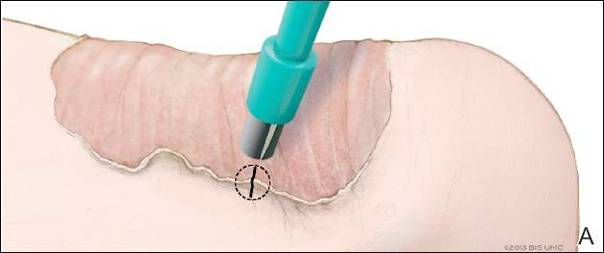
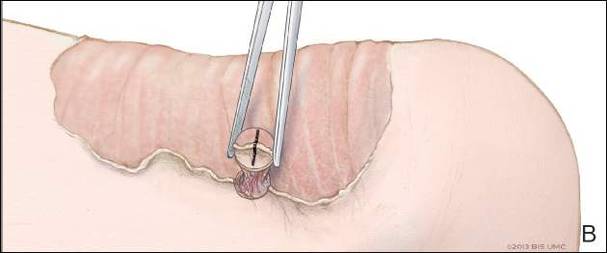
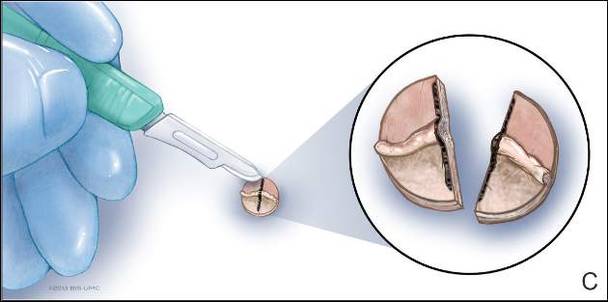
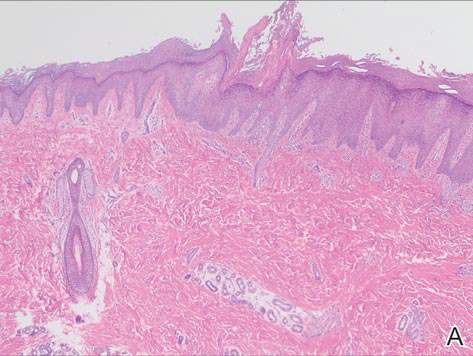
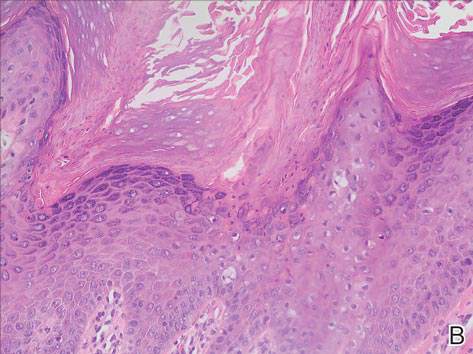
Comment
Porokeratosis of Mibelli is a rare condition that typically presents in infancy to early childhood.1 It may appear as small keratotic papules or larger plaques that reach several centimeters in diameter.2 There is a 7.5% risk for malignant transformation (eg, basal cell carcinoma, squamous cell carcinoma, Bowen disease).3 Variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) typically are present in the center of the lesion. In our case, a biopsy from the center of the plaque demonstrated verrucous hyperplasia. The incorrect diagnosis of PM as psoriasis also has been reported.4
We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM. First, draw a line perpendicular to the rim of the lesion to mark the biopsy site (Figure 2A). Second, perform a punch biopsy centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). Third, section the biopsied tissue with a #15 blade along the perpendicular line at the bedside (Figure 2C). The surgical pathology requisition should mention that the specimen has been transected and the cut edges should be placed down in the cassette, ensuring that the cornoid lamella will be present in cross-section on the slides.
If the punch biopsy specimen is not bisected, it can be difficult to orient it in the pathology laboratory, especially if the cornoid lamellae are not prominent. Furthermore, the technician processing the tissue may not be aware of the importance of sectioning the specimen perpendicular to the cornoid lamella. Following this procedure, diagnosis can be confirmed in virtually every case of PM.
- Richard G, Irvine A, Traupe H, et al. Ichthyosis and disorders of other conification. In: Schachner L, Hansen R, Krafchik B, et al, eds. Pediatric Dermatology. Philadelphia, PA: Elsevier Health Sciences; 2011:640-643.
- Pierson D, Bandel C, Ehrig, et al. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo J, Rapini R, et al, eds. Dermatology. 1st ed. Vol 2. Edinburgh, Scotland: Elsevier; 2003:1707-1709.
- Cort DF, Abdel-Aziz AH. Epithelioma arising in porokeratosis of Mibelli. Br J Plast Surg. 1972;25:318-328.
- De Simone C, Paradisi A, Massi G, et al. Giant verrucous porokeratosis of Mibelli mimicking psoriasis in a patient with psoriasis. J Am Acad Dermatol. 2007;57:665-668.
Porokeratosis of Mibelli (PM) is a lesion characterized by a surrounding cornoid lamella with variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) in the center of the lesion that typically presents in infancy to early childhood.1 We report a case of PM in which a prior biopsy from the center of the lesion demonstrated papulosquamous dermatitis. We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM.
Case Report
A 3-year-old girl presented with an erythematous, hypopigmented, scaling plaque on the posterior aspect of the left ankle surrounded by a hard rim. The plaque was first noted at 12 months of age and had slowly enlarged as the patient grew. Six months prior, a biopsy from the center of the lesion performed at another facility demonstrated a papulosquamous dermatitis.
Physical examination revealed a lesion that was 4.2-cm long, 2.2-cm wide at the superior pole, and 3.5-cm wide at the inferior pole (Figure 1). A line was drawn with a skin marker perpendicular to the rim of the lesion (Figure 2A) and a 6-mm punch biopsy was performed, centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). The tissue was then bisected at the bedside along the skin marker line with a #15 blade (Figure 2C) and submitted in formalin for histologic processing. Histologic examination revealed an invagination of the epidermis producing a tier of parakeratotic cells with its apex pointed away from the center of the lesion. Dyskeratotic cells were noted at the base of the parakeratosis (Figure 3). Verrucous hyperplasia was present in the central portion of the specimen adjacent to the cornoid lamella. Based on these histopathologic findings, the correct diagnosis of PM was made.
Comment
Porokeratosis of Mibelli is a rare condition that typically presents in infancy to early childhood.1 It may appear as small keratotic papules or larger plaques that reach several centimeters in diameter.2 There is a 7.5% risk for malignant transformation (eg, basal cell carcinoma, squamous cell carcinoma, Bowen disease).3 Variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) typically are present in the center of the lesion. In our case, a biopsy from the center of the plaque demonstrated verrucous hyperplasia. The incorrect diagnosis of PM as psoriasis also has been reported.4
We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM. First, draw a line perpendicular to the rim of the lesion to mark the biopsy site (Figure 2A). Second, perform a punch biopsy centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). Third, section the biopsied tissue with a #15 blade along the perpendicular line at the bedside (Figure 2C). The surgical pathology requisition should mention that the specimen has been transected and the cut edges should be placed down in the cassette, ensuring that the cornoid lamella will be present in cross-section on the slides.
If the punch biopsy specimen is not bisected, it can be difficult to orient it in the pathology laboratory, especially if the cornoid lamellae are not prominent. Furthermore, the technician processing the tissue may not be aware of the importance of sectioning the specimen perpendicular to the cornoid lamella. Following this procedure, diagnosis can be confirmed in virtually every case of PM.
Porokeratosis of Mibelli (PM) is a lesion characterized by a surrounding cornoid lamella with variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) in the center of the lesion that typically presents in infancy to early childhood.1 We report a case of PM in which a prior biopsy from the center of the lesion demonstrated papulosquamous dermatitis. We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM.
Case Report
A 3-year-old girl presented with an erythematous, hypopigmented, scaling plaque on the posterior aspect of the left ankle surrounded by a hard rim. The plaque was first noted at 12 months of age and had slowly enlarged as the patient grew. Six months prior, a biopsy from the center of the lesion performed at another facility demonstrated a papulosquamous dermatitis.
Physical examination revealed a lesion that was 4.2-cm long, 2.2-cm wide at the superior pole, and 3.5-cm wide at the inferior pole (Figure 1). A line was drawn with a skin marker perpendicular to the rim of the lesion (Figure 2A) and a 6-mm punch biopsy was performed, centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). The tissue was then bisected at the bedside along the skin marker line with a #15 blade (Figure 2C) and submitted in formalin for histologic processing. Histologic examination revealed an invagination of the epidermis producing a tier of parakeratotic cells with its apex pointed away from the center of the lesion. Dyskeratotic cells were noted at the base of the parakeratosis (Figure 3). Verrucous hyperplasia was present in the central portion of the specimen adjacent to the cornoid lamella. Based on these histopathologic findings, the correct diagnosis of PM was made.
Comment
Porokeratosis of Mibelli is a rare condition that typically presents in infancy to early childhood.1 It may appear as small keratotic papules or larger plaques that reach several centimeters in diameter.2 There is a 7.5% risk for malignant transformation (eg, basal cell carcinoma, squamous cell carcinoma, Bowen disease).3 Variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) typically are present in the center of the lesion. In our case, a biopsy from the center of the plaque demonstrated verrucous hyperplasia. The incorrect diagnosis of PM as psoriasis also has been reported.4
We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM. First, draw a line perpendicular to the rim of the lesion to mark the biopsy site (Figure 2A). Second, perform a punch biopsy centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). Third, section the biopsied tissue with a #15 blade along the perpendicular line at the bedside (Figure 2C). The surgical pathology requisition should mention that the specimen has been transected and the cut edges should be placed down in the cassette, ensuring that the cornoid lamella will be present in cross-section on the slides.
If the punch biopsy specimen is not bisected, it can be difficult to orient it in the pathology laboratory, especially if the cornoid lamellae are not prominent. Furthermore, the technician processing the tissue may not be aware of the importance of sectioning the specimen perpendicular to the cornoid lamella. Following this procedure, diagnosis can be confirmed in virtually every case of PM.
- Richard G, Irvine A, Traupe H, et al. Ichthyosis and disorders of other conification. In: Schachner L, Hansen R, Krafchik B, et al, eds. Pediatric Dermatology. Philadelphia, PA: Elsevier Health Sciences; 2011:640-643.
- Pierson D, Bandel C, Ehrig, et al. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo J, Rapini R, et al, eds. Dermatology. 1st ed. Vol 2. Edinburgh, Scotland: Elsevier; 2003:1707-1709.
- Cort DF, Abdel-Aziz AH. Epithelioma arising in porokeratosis of Mibelli. Br J Plast Surg. 1972;25:318-328.
- De Simone C, Paradisi A, Massi G, et al. Giant verrucous porokeratosis of Mibelli mimicking psoriasis in a patient with psoriasis. J Am Acad Dermatol. 2007;57:665-668.
- Richard G, Irvine A, Traupe H, et al. Ichthyosis and disorders of other conification. In: Schachner L, Hansen R, Krafchik B, et al, eds. Pediatric Dermatology. Philadelphia, PA: Elsevier Health Sciences; 2011:640-643.
- Pierson D, Bandel C, Ehrig, et al. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo J, Rapini R, et al, eds. Dermatology. 1st ed. Vol 2. Edinburgh, Scotland: Elsevier; 2003:1707-1709.
- Cort DF, Abdel-Aziz AH. Epithelioma arising in porokeratosis of Mibelli. Br J Plast Surg. 1972;25:318-328.
- De Simone C, Paradisi A, Massi G, et al. Giant verrucous porokeratosis of Mibelli mimicking psoriasis in a patient with psoriasis. J Am Acad Dermatol. 2007;57:665-668.
Practice Points
- A biopsy from the center of a plaque of porokeratosis will produce nonspecific findings.
- Bisecting the punch specimen at the bedside along a line drawn perpendicular to the cornoid lamella guarantees proper orientation of the specimen.
Brown Macule on the Waist
The best diagnosis is:
a. granular cell tumor
b. intradermal nevus
c. Langerhans cell disease
d. mastocytosis
e. multicentric reticulohistiocytosis
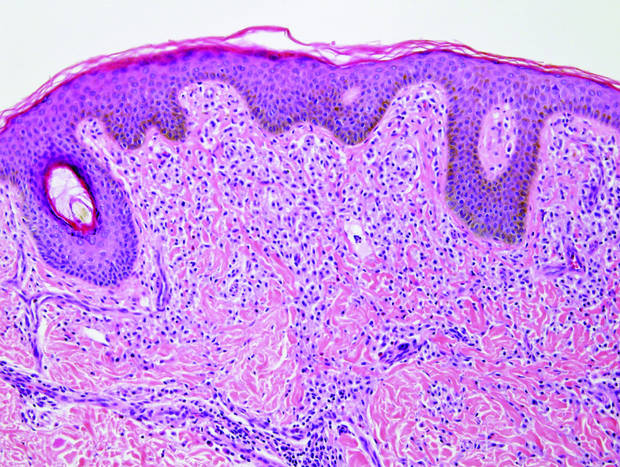
|
| Monomorphic cell infiltrate in the upper dermis (H&E, original magnification ×100). |
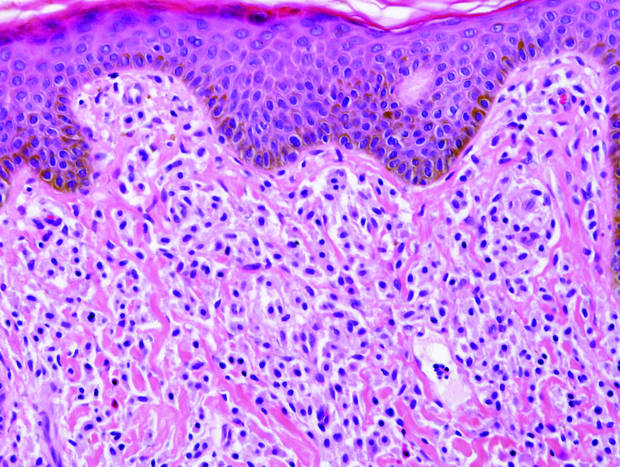
|
| A closer view reveals cuboidal or spindle cells with basal hyperpigmentation (H&E, original magnification ×200). |
Continue to the next page for the diagnosis >>
Mastocytosis
Mastocytosis is a clonal proliferation of mast cells in the skin and various systems of the body including the bone marrow, liver, lymph nodes, and gastrointestinal tract.1,2 Mast cell proliferation is closely associated with germline and acquired activating KIT mutations.3-5 Adult-onset mastocytosis is likely to involve several organs, whereas pediatric mastocytosis usually affects only the skin and is self-limiting. Patients with profound mast cell infiltration in the skin or other organs are likely to have attacks of flushing, palpitation, or diarrhea resulting from the degranulation of mast cells and release of histamine.6,7 In a majority of patients with advanced systemic mastocytosis, mast cells are positive for the Ki-1 antigen (CD30), whereas in most patients with indolent systemic mastocytosis, only a few mast cells are positive for CD30.8 Recently, CD30 was reported as a new drug target in patients with CD30+ advanced systemic mastocytosis.9 Because the skin frequently is involved and easily accessible in comparison with other organs, skin biopsy often is performed to establish a diagnosis of mastocytosis. Cutaneous mastocytosis comprises urticaria pigmentosa, solitary mastocytoma, diffuse cutaneous mastocytosis, and telangiectasia macularis eruptiva perstans; approximately 80% of all cases have urticaria pigmentosa.10-12 In cutaneous mastocytosis, skin biopsy typically shows monomorphous mast cell infiltrate mostly in the upper third of the dermis. The density of mast cells varies according to the clinical variant. For example, a lesion of telangiectasia macularis eruptiva perstans has only a perivascular mast cell infiltrate, whereas a solitary mastocytoma has sheets of mast cells in the dermis, sometimes extending into the subcutis. A skin biopsy of the brown macule on the waist showed a number of cuboidal or spindle mast cells in the upper dermis with occasional eosinophils. These mast cells are monomorphous, and no mitotic figures, necrotic cells, or atypical cells are seen. Mast cells have metachromatic granules in the cytoplasm, which can be seen with toluidine blue or Giemsa stain. CD117 (c-kit) also is positive. Mast cells in urticaria pigmentosa easily may be mistaken for nevus cells. Hyperpigmentation of the basal layer, a characteristic feature seen in urticaria pigmentosa, also may erroneously suggest a diagnosis of a melanocytic nevus.
Granular cell tumors predominantly affect the oral cavity, but the skin also can be involved. It comprises a fascicular infiltrate of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (Figure 1).13 Cell membranes are not always distinct. Although the nuclei usually are small and centrally located, irregular and plump nuclei with distinct nucleoli also may be seen. The overlying epidermis tends to be hyperplastic. Granular cell tumor is considered a group of lesions of varying histogenesis. Cases in which tumors originated from a neural crest–derived peripheral nerve–related cell as well as a Schwann cell have been reported.14,15 The origin of granular cell tumors is controversial.
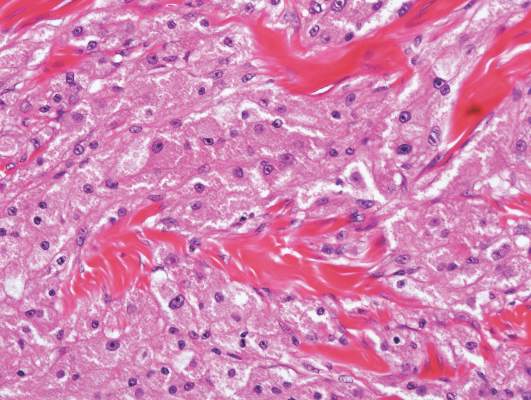
|
| Figure 1. Granular cell tumor showing fascicles of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (H&E, original magnification ×200). |

|
| Figure 2. Intradermal nevus showing nests with melanin in the uppermost area of the lesion and neurotized nevus cells in the lower part (H&E, original magnification ×100). Pseudovascular spaces are seen on the right side. |
Intradermal nevus usually has nests and cords of nevus cells in the upper dermis. The uppermost melanocytes often contain a moderate amount of melanin, whereas nevus cells in the mid and lower dermis usually do not contain melanin (Figure 2). Shrinkage during tissue processing maycause clefts between nevus cells, resulting in pseudovascular spaces.16 The deeper dermis may have a neuroid appearance with spindle-shaped cells and Meissner corpuscle–like structures.17
Although Langerhans cell disease was formerly known as Langerhans cell histiocytosis and subdivided into several clinical subtypes, including Letterer-Siwe disease, Hand-Schüller-Christian disease, and eosinophilic granuloma, these clinical subtypes commonly overlapped. Langerhans cell disease is now used as a terminology that encompasses all subtypes.18,19 Langerhans cell disease is characterized by a proliferation of Langerhans cells with a variable mixture of other inflammatory cells. The constituent cells are large and ovoid with a distinct folded or lobulated, often kidney-shaped nucleus.20 Langerhans cells usually infiltrate the upper dermis and occasionally the epidermis (Figure 3). CD1a, HLA-DR, S-100 protein, and langerin are positive in Langerhans cells.21

|
| Figure 3. Langerhans cell disease showing an infiltrate of large and ovoid Langerhans cells with a distinct folded or lobulated, often kidney-shaped nucleus in the upper dermis and epidermis (H&E, original magnification ×200). |

|
| Figure 4. Multicentric reticulohistiocytosis showing a mixture of mononuclear and multinucleate histiocytes with abundant eosinophilic and finely granular cytoplasm (H&E, original magnification ×200). |
Multicentric reticulohistiocytosis is characterized by a combination of papulonodular cutaneous lesions and severe arthropathy.22 An irregular mixture of mononuclear and multinucleate histiocytes showing abundant eosinophilic and finely granular cytoplasm, often with a ground-glass appearance, is seen along with lymphocytic infiltration (Figure 4).23 A few giant cells may be seen in early lesions; older lesions more commonly have giant cells and fibrosis.
1. Arock M, Valent P. Pathogenesis, classification and treatment of mastocytosis: state of the art in 2010 and future perspectives. Expert Rev Hematol. 2010;3:497-516.
2. Pardanani A. Systemic mastocytosis in adults: 2013 update on diagnosis, risk stratification, and management. Am J Hematol. 2013;88:612-624.
3. Orfao A, Garcia-Montero AC, Sanchez L, et al. Recent advances in the understanding of mastocytosis: the role of KIT mutations. Br J Haematol. 2007;138:12-30.
4. Yanagihori H, Oyama N, Nakamura K, et al. c-KIT mutations in patients with childhood-onset mastocytosis and genotype-phenotype correlation. J Mol Diagn. 2005;7:252-257.
5. Bodemer C, Hermine O, Palmérini F, et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol. 2010;130:804-815.
6. Kettelhut BV, Metcalfe DD. Pediatric mastocytosis. Ann Allergy. 1994;73:197-202; quiz 202-207.
7. Longley J, Duffy TP, Kohn S. The mast cell and mast cell disease. J Am Acad Dermatol. 1995;32:545-561; quiz 562-564.
8. Sotlar K, Cerny-Reiterer S, Petat-Dutter K, et al. Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol. 2011;24:585-595.
9. Blatt K, Cerny-Reiterer S, Schwaab J, et al. Identification of the Ki-1 antigen (CD30) as a novel therapeutic target in systemic mastocytosis [published online October 20, 2015]. Blood. 2015;126:2832-2841.
10. Kiszewski AE, Duran-Mckinster C, Orozco-Covarrubias L, et al. Cutaneous mastocytosis in children: a clinical analysis of 71 cases. J Eur Acad Dermatol Venereol. 2004;18:285-290.
11. Akoglu G, Erkin G, Cakir B, et al. Cutaneous mastocytosis: demographic aspects and clinical features of 55 patients. J Eur Acad Dermatol Venereol. 2006;20:969-973.
12. Sarkany RP, Monk BE, Handfield-Jones SE. Telangiectasia macularis eruptiva perstans: a case report and review of the literature. Clin Exp Dermatol. 1998;23:38-39.
13. Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
14. Buley ID, Gatter KC, Kelly PM, et al. Granular cell tumours revisited. an immunohistological and ultrastructural study. Histopathology. 1988;12:263-274.
15. Penneys NS, Adachi K, Ziegels-Weissman J, et al. Granular cell tumors of the skin contain myelin basic protein. Arch Pathol Lab Med. 1983;107:302-303.
16. Modlin RL, Gottlieb B, Taylor C, et al. Identification of cells lining pseudovascular spaces of benign pigmented nevi. Am J Dermatopathol. 1984;(suppl 6):25-29.
17. Fullen DR, Reed JA, Finnerty B, et al. S100A6 preferentially labels type C nevus cells and nevic corpuscles: additional support for Schwannian differentiation of intradermal nevi. J Cutan Pathol. 2001;28:393-399.
18. Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
19. Weedon D. Cutaneous infiltrates—non-lymphoid. In: Weedon D, ed. Weedon’s Skin Pathology. 3rd ed. Amsterdam, Netherlands: Elsevier; 2010:937-970.
20. Harrist TJ, Bhan AK, Murphy GF, et al. Histiocytosis-X: in situ characterization of cutaneous infiltrates with monoclonal antibodies. Am J Clin Pathol. 1983;79:294-300.
21. Lau SK, Chu PG, Weiss LM. Immunohistochemical expression of langerin in Langerhans cell histiocytosis and non-Langerhans cell histiocytic disorders. Am J Surg Pathol. 2008;32:615-619.
22. Lesher JL Jr, Allen BS. Multicentric reticulohistiocytosis. J Am Acad Dermatol. 1984;11:713-723.
23. Heathcote JG, Guenther LC, Wallace AC. Multicentric reticulohistiocytosis: a report of a case and a review of the pathology. Pathology. 1985;17:601-608.
The best diagnosis is:
a. granular cell tumor
b. intradermal nevus
c. Langerhans cell disease
d. mastocytosis
e. multicentric reticulohistiocytosis
|
|
| Monomorphic cell infiltrate in the upper dermis (H&E, original magnification ×100). |
|
|
| A closer view reveals cuboidal or spindle cells with basal hyperpigmentation (H&E, original magnification ×200). |
Continue to the next page for the diagnosis >>
Mastocytosis
Mastocytosis is a clonal proliferation of mast cells in the skin and various systems of the body including the bone marrow, liver, lymph nodes, and gastrointestinal tract.1,2 Mast cell proliferation is closely associated with germline and acquired activating KIT mutations.3-5 Adult-onset mastocytosis is likely to involve several organs, whereas pediatric mastocytosis usually affects only the skin and is self-limiting. Patients with profound mast cell infiltration in the skin or other organs are likely to have attacks of flushing, palpitation, or diarrhea resulting from the degranulation of mast cells and release of histamine.6,7 In a majority of patients with advanced systemic mastocytosis, mast cells are positive for the Ki-1 antigen (CD30), whereas in most patients with indolent systemic mastocytosis, only a few mast cells are positive for CD30.8 Recently, CD30 was reported as a new drug target in patients with CD30+ advanced systemic mastocytosis.9 Because the skin frequently is involved and easily accessible in comparison with other organs, skin biopsy often is performed to establish a diagnosis of mastocytosis. Cutaneous mastocytosis comprises urticaria pigmentosa, solitary mastocytoma, diffuse cutaneous mastocytosis, and telangiectasia macularis eruptiva perstans; approximately 80% of all cases have urticaria pigmentosa.10-12 In cutaneous mastocytosis, skin biopsy typically shows monomorphous mast cell infiltrate mostly in the upper third of the dermis. The density of mast cells varies according to the clinical variant. For example, a lesion of telangiectasia macularis eruptiva perstans has only a perivascular mast cell infiltrate, whereas a solitary mastocytoma has sheets of mast cells in the dermis, sometimes extending into the subcutis. A skin biopsy of the brown macule on the waist showed a number of cuboidal or spindle mast cells in the upper dermis with occasional eosinophils. These mast cells are monomorphous, and no mitotic figures, necrotic cells, or atypical cells are seen. Mast cells have metachromatic granules in the cytoplasm, which can be seen with toluidine blue or Giemsa stain. CD117 (c-kit) also is positive. Mast cells in urticaria pigmentosa easily may be mistaken for nevus cells. Hyperpigmentation of the basal layer, a characteristic feature seen in urticaria pigmentosa, also may erroneously suggest a diagnosis of a melanocytic nevus.
Granular cell tumors predominantly affect the oral cavity, but the skin also can be involved. It comprises a fascicular infiltrate of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (Figure 1).13 Cell membranes are not always distinct. Although the nuclei usually are small and centrally located, irregular and plump nuclei with distinct nucleoli also may be seen. The overlying epidermis tends to be hyperplastic. Granular cell tumor is considered a group of lesions of varying histogenesis. Cases in which tumors originated from a neural crest–derived peripheral nerve–related cell as well as a Schwann cell have been reported.14,15 The origin of granular cell tumors is controversial.
|
|
| Figure 1. Granular cell tumor showing fascicles of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (H&E, original magnification ×200). |
|
|
| Figure 2. Intradermal nevus showing nests with melanin in the uppermost area of the lesion and neurotized nevus cells in the lower part (H&E, original magnification ×100). Pseudovascular spaces are seen on the right side. |
Intradermal nevus usually has nests and cords of nevus cells in the upper dermis. The uppermost melanocytes often contain a moderate amount of melanin, whereas nevus cells in the mid and lower dermis usually do not contain melanin (Figure 2). Shrinkage during tissue processing maycause clefts between nevus cells, resulting in pseudovascular spaces.16 The deeper dermis may have a neuroid appearance with spindle-shaped cells and Meissner corpuscle–like structures.17
Although Langerhans cell disease was formerly known as Langerhans cell histiocytosis and subdivided into several clinical subtypes, including Letterer-Siwe disease, Hand-Schüller-Christian disease, and eosinophilic granuloma, these clinical subtypes commonly overlapped. Langerhans cell disease is now used as a terminology that encompasses all subtypes.18,19 Langerhans cell disease is characterized by a proliferation of Langerhans cells with a variable mixture of other inflammatory cells. The constituent cells are large and ovoid with a distinct folded or lobulated, often kidney-shaped nucleus.20 Langerhans cells usually infiltrate the upper dermis and occasionally the epidermis (Figure 3). CD1a, HLA-DR, S-100 protein, and langerin are positive in Langerhans cells.21
|
|
| Figure 3. Langerhans cell disease showing an infiltrate of large and ovoid Langerhans cells with a distinct folded or lobulated, often kidney-shaped nucleus in the upper dermis and epidermis (H&E, original magnification ×200). |
|
|
| Figure 4. Multicentric reticulohistiocytosis showing a mixture of mononuclear and multinucleate histiocytes with abundant eosinophilic and finely granular cytoplasm (H&E, original magnification ×200). |
Multicentric reticulohistiocytosis is characterized by a combination of papulonodular cutaneous lesions and severe arthropathy.22 An irregular mixture of mononuclear and multinucleate histiocytes showing abundant eosinophilic and finely granular cytoplasm, often with a ground-glass appearance, is seen along with lymphocytic infiltration (Figure 4).23 A few giant cells may be seen in early lesions; older lesions more commonly have giant cells and fibrosis.
The best diagnosis is:
a. granular cell tumor
b. intradermal nevus
c. Langerhans cell disease
d. mastocytosis
e. multicentric reticulohistiocytosis
|
|
| Monomorphic cell infiltrate in the upper dermis (H&E, original magnification ×100). |
|
|
| A closer view reveals cuboidal or spindle cells with basal hyperpigmentation (H&E, original magnification ×200). |
Continue to the next page for the diagnosis >>
Mastocytosis
Mastocytosis is a clonal proliferation of mast cells in the skin and various systems of the body including the bone marrow, liver, lymph nodes, and gastrointestinal tract.1,2 Mast cell proliferation is closely associated with germline and acquired activating KIT mutations.3-5 Adult-onset mastocytosis is likely to involve several organs, whereas pediatric mastocytosis usually affects only the skin and is self-limiting. Patients with profound mast cell infiltration in the skin or other organs are likely to have attacks of flushing, palpitation, or diarrhea resulting from the degranulation of mast cells and release of histamine.6,7 In a majority of patients with advanced systemic mastocytosis, mast cells are positive for the Ki-1 antigen (CD30), whereas in most patients with indolent systemic mastocytosis, only a few mast cells are positive for CD30.8 Recently, CD30 was reported as a new drug target in patients with CD30+ advanced systemic mastocytosis.9 Because the skin frequently is involved and easily accessible in comparison with other organs, skin biopsy often is performed to establish a diagnosis of mastocytosis. Cutaneous mastocytosis comprises urticaria pigmentosa, solitary mastocytoma, diffuse cutaneous mastocytosis, and telangiectasia macularis eruptiva perstans; approximately 80% of all cases have urticaria pigmentosa.10-12 In cutaneous mastocytosis, skin biopsy typically shows monomorphous mast cell infiltrate mostly in the upper third of the dermis. The density of mast cells varies according to the clinical variant. For example, a lesion of telangiectasia macularis eruptiva perstans has only a perivascular mast cell infiltrate, whereas a solitary mastocytoma has sheets of mast cells in the dermis, sometimes extending into the subcutis. A skin biopsy of the brown macule on the waist showed a number of cuboidal or spindle mast cells in the upper dermis with occasional eosinophils. These mast cells are monomorphous, and no mitotic figures, necrotic cells, or atypical cells are seen. Mast cells have metachromatic granules in the cytoplasm, which can be seen with toluidine blue or Giemsa stain. CD117 (c-kit) also is positive. Mast cells in urticaria pigmentosa easily may be mistaken for nevus cells. Hyperpigmentation of the basal layer, a characteristic feature seen in urticaria pigmentosa, also may erroneously suggest a diagnosis of a melanocytic nevus.
Granular cell tumors predominantly affect the oral cavity, but the skin also can be involved. It comprises a fascicular infiltrate of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (Figure 1).13 Cell membranes are not always distinct. Although the nuclei usually are small and centrally located, irregular and plump nuclei with distinct nucleoli also may be seen. The overlying epidermis tends to be hyperplastic. Granular cell tumor is considered a group of lesions of varying histogenesis. Cases in which tumors originated from a neural crest–derived peripheral nerve–related cell as well as a Schwann cell have been reported.14,15 The origin of granular cell tumors is controversial.
|
|
| Figure 1. Granular cell tumor showing fascicles of large and polygonal cells with characteristic eosinophilic granular cytoplasm in the dermis (H&E, original magnification ×200). |
|
|
| Figure 2. Intradermal nevus showing nests with melanin in the uppermost area of the lesion and neurotized nevus cells in the lower part (H&E, original magnification ×100). Pseudovascular spaces are seen on the right side. |
Intradermal nevus usually has nests and cords of nevus cells in the upper dermis. The uppermost melanocytes often contain a moderate amount of melanin, whereas nevus cells in the mid and lower dermis usually do not contain melanin (Figure 2). Shrinkage during tissue processing maycause clefts between nevus cells, resulting in pseudovascular spaces.16 The deeper dermis may have a neuroid appearance with spindle-shaped cells and Meissner corpuscle–like structures.17
Although Langerhans cell disease was formerly known as Langerhans cell histiocytosis and subdivided into several clinical subtypes, including Letterer-Siwe disease, Hand-Schüller-Christian disease, and eosinophilic granuloma, these clinical subtypes commonly overlapped. Langerhans cell disease is now used as a terminology that encompasses all subtypes.18,19 Langerhans cell disease is characterized by a proliferation of Langerhans cells with a variable mixture of other inflammatory cells. The constituent cells are large and ovoid with a distinct folded or lobulated, often kidney-shaped nucleus.20 Langerhans cells usually infiltrate the upper dermis and occasionally the epidermis (Figure 3). CD1a, HLA-DR, S-100 protein, and langerin are positive in Langerhans cells.21
|
|
| Figure 3. Langerhans cell disease showing an infiltrate of large and ovoid Langerhans cells with a distinct folded or lobulated, often kidney-shaped nucleus in the upper dermis and epidermis (H&E, original magnification ×200). |
|
|
| Figure 4. Multicentric reticulohistiocytosis showing a mixture of mononuclear and multinucleate histiocytes with abundant eosinophilic and finely granular cytoplasm (H&E, original magnification ×200). |
Multicentric reticulohistiocytosis is characterized by a combination of papulonodular cutaneous lesions and severe arthropathy.22 An irregular mixture of mononuclear and multinucleate histiocytes showing abundant eosinophilic and finely granular cytoplasm, often with a ground-glass appearance, is seen along with lymphocytic infiltration (Figure 4).23 A few giant cells may be seen in early lesions; older lesions more commonly have giant cells and fibrosis.
1. Arock M, Valent P. Pathogenesis, classification and treatment of mastocytosis: state of the art in 2010 and future perspectives. Expert Rev Hematol. 2010;3:497-516.
2. Pardanani A. Systemic mastocytosis in adults: 2013 update on diagnosis, risk stratification, and management. Am J Hematol. 2013;88:612-624.
3. Orfao A, Garcia-Montero AC, Sanchez L, et al. Recent advances in the understanding of mastocytosis: the role of KIT mutations. Br J Haematol. 2007;138:12-30.
4. Yanagihori H, Oyama N, Nakamura K, et al. c-KIT mutations in patients with childhood-onset mastocytosis and genotype-phenotype correlation. J Mol Diagn. 2005;7:252-257.
5. Bodemer C, Hermine O, Palmérini F, et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol. 2010;130:804-815.
6. Kettelhut BV, Metcalfe DD. Pediatric mastocytosis. Ann Allergy. 1994;73:197-202; quiz 202-207.
7. Longley J, Duffy TP, Kohn S. The mast cell and mast cell disease. J Am Acad Dermatol. 1995;32:545-561; quiz 562-564.
8. Sotlar K, Cerny-Reiterer S, Petat-Dutter K, et al. Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol. 2011;24:585-595.
9. Blatt K, Cerny-Reiterer S, Schwaab J, et al. Identification of the Ki-1 antigen (CD30) as a novel therapeutic target in systemic mastocytosis [published online October 20, 2015]. Blood. 2015;126:2832-2841.
10. Kiszewski AE, Duran-Mckinster C, Orozco-Covarrubias L, et al. Cutaneous mastocytosis in children: a clinical analysis of 71 cases. J Eur Acad Dermatol Venereol. 2004;18:285-290.
11. Akoglu G, Erkin G, Cakir B, et al. Cutaneous mastocytosis: demographic aspects and clinical features of 55 patients. J Eur Acad Dermatol Venereol. 2006;20:969-973.
12. Sarkany RP, Monk BE, Handfield-Jones SE. Telangiectasia macularis eruptiva perstans: a case report and review of the literature. Clin Exp Dermatol. 1998;23:38-39.
13. Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
14. Buley ID, Gatter KC, Kelly PM, et al. Granular cell tumours revisited. an immunohistological and ultrastructural study. Histopathology. 1988;12:263-274.
15. Penneys NS, Adachi K, Ziegels-Weissman J, et al. Granular cell tumors of the skin contain myelin basic protein. Arch Pathol Lab Med. 1983;107:302-303.
16. Modlin RL, Gottlieb B, Taylor C, et al. Identification of cells lining pseudovascular spaces of benign pigmented nevi. Am J Dermatopathol. 1984;(suppl 6):25-29.
17. Fullen DR, Reed JA, Finnerty B, et al. S100A6 preferentially labels type C nevus cells and nevic corpuscles: additional support for Schwannian differentiation of intradermal nevi. J Cutan Pathol. 2001;28:393-399.
18. Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
19. Weedon D. Cutaneous infiltrates—non-lymphoid. In: Weedon D, ed. Weedon’s Skin Pathology. 3rd ed. Amsterdam, Netherlands: Elsevier; 2010:937-970.
20. Harrist TJ, Bhan AK, Murphy GF, et al. Histiocytosis-X: in situ characterization of cutaneous infiltrates with monoclonal antibodies. Am J Clin Pathol. 1983;79:294-300.
21. Lau SK, Chu PG, Weiss LM. Immunohistochemical expression of langerin in Langerhans cell histiocytosis and non-Langerhans cell histiocytic disorders. Am J Surg Pathol. 2008;32:615-619.
22. Lesher JL Jr, Allen BS. Multicentric reticulohistiocytosis. J Am Acad Dermatol. 1984;11:713-723.
23. Heathcote JG, Guenther LC, Wallace AC. Multicentric reticulohistiocytosis: a report of a case and a review of the pathology. Pathology. 1985;17:601-608.
1. Arock M, Valent P. Pathogenesis, classification and treatment of mastocytosis: state of the art in 2010 and future perspectives. Expert Rev Hematol. 2010;3:497-516.
2. Pardanani A. Systemic mastocytosis in adults: 2013 update on diagnosis, risk stratification, and management. Am J Hematol. 2013;88:612-624.
3. Orfao A, Garcia-Montero AC, Sanchez L, et al. Recent advances in the understanding of mastocytosis: the role of KIT mutations. Br J Haematol. 2007;138:12-30.
4. Yanagihori H, Oyama N, Nakamura K, et al. c-KIT mutations in patients with childhood-onset mastocytosis and genotype-phenotype correlation. J Mol Diagn. 2005;7:252-257.
5. Bodemer C, Hermine O, Palmérini F, et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol. 2010;130:804-815.
6. Kettelhut BV, Metcalfe DD. Pediatric mastocytosis. Ann Allergy. 1994;73:197-202; quiz 202-207.
7. Longley J, Duffy TP, Kohn S. The mast cell and mast cell disease. J Am Acad Dermatol. 1995;32:545-561; quiz 562-564.
8. Sotlar K, Cerny-Reiterer S, Petat-Dutter K, et al. Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol. 2011;24:585-595.
9. Blatt K, Cerny-Reiterer S, Schwaab J, et al. Identification of the Ki-1 antigen (CD30) as a novel therapeutic target in systemic mastocytosis [published online October 20, 2015]. Blood. 2015;126:2832-2841.
10. Kiszewski AE, Duran-Mckinster C, Orozco-Covarrubias L, et al. Cutaneous mastocytosis in children: a clinical analysis of 71 cases. J Eur Acad Dermatol Venereol. 2004;18:285-290.
11. Akoglu G, Erkin G, Cakir B, et al. Cutaneous mastocytosis: demographic aspects and clinical features of 55 patients. J Eur Acad Dermatol Venereol. 2006;20:969-973.
12. Sarkany RP, Monk BE, Handfield-Jones SE. Telangiectasia macularis eruptiva perstans: a case report and review of the literature. Clin Exp Dermatol. 1998;23:38-39.
13. Lack EE, Worsham GF, Callihan MD, et al. Granular cell tumor: a clinicopathologic study of 110 patients. J Surg Oncol. 1980;13:301-316.
14. Buley ID, Gatter KC, Kelly PM, et al. Granular cell tumours revisited. an immunohistological and ultrastructural study. Histopathology. 1988;12:263-274.
15. Penneys NS, Adachi K, Ziegels-Weissman J, et al. Granular cell tumors of the skin contain myelin basic protein. Arch Pathol Lab Med. 1983;107:302-303.
16. Modlin RL, Gottlieb B, Taylor C, et al. Identification of cells lining pseudovascular spaces of benign pigmented nevi. Am J Dermatopathol. 1984;(suppl 6):25-29.
17. Fullen DR, Reed JA, Finnerty B, et al. S100A6 preferentially labels type C nevus cells and nevic corpuscles: additional support for Schwannian differentiation of intradermal nevi. J Cutan Pathol. 2001;28:393-399.
18. Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
19. Weedon D. Cutaneous infiltrates—non-lymphoid. In: Weedon D, ed. Weedon’s Skin Pathology. 3rd ed. Amsterdam, Netherlands: Elsevier; 2010:937-970.
20. Harrist TJ, Bhan AK, Murphy GF, et al. Histiocytosis-X: in situ characterization of cutaneous infiltrates with monoclonal antibodies. Am J Clin Pathol. 1983;79:294-300.
21. Lau SK, Chu PG, Weiss LM. Immunohistochemical expression of langerin in Langerhans cell histiocytosis and non-Langerhans cell histiocytic disorders. Am J Surg Pathol. 2008;32:615-619.
22. Lesher JL Jr, Allen BS. Multicentric reticulohistiocytosis. J Am Acad Dermatol. 1984;11:713-723.
23. Heathcote JG, Guenther LC, Wallace AC. Multicentric reticulohistiocytosis: a report of a case and a review of the pathology. Pathology. 1985;17:601-608.
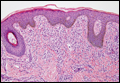
Multiple Superficial White Nodules on the Bilateral Helical Rims
The Diagnosis: Bilateral Auricular Tophaceous Gout
Histopathologic evaluation with hematoxylin and eosin staining demonstrated clusters of abundant granular amorphous material within the subcutaneous tissue (Figure 1). The overlying epidermis and dermis were unremarkable. The granular amorphous material demonstrated numerous monosodium urate crystals under polarized light (Figure 2). At a return visit following the biopsy results, the patient reported a history of a single episode of monoarticular gouty arthritis involving the right hallux approximately 6 months after the onset of the skin lesions. With the added clinical history and the biopsy results, his serum uric acid level was obtained and was found to be elevated at 9.2 mg/dL (reference range, 3.5–8 mg/dL).
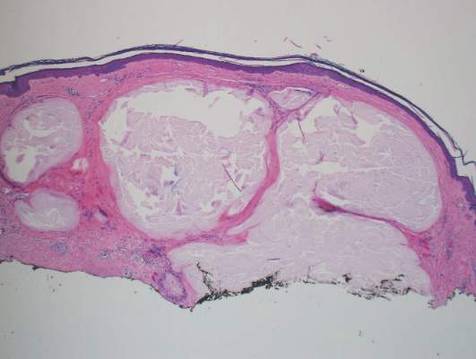
|
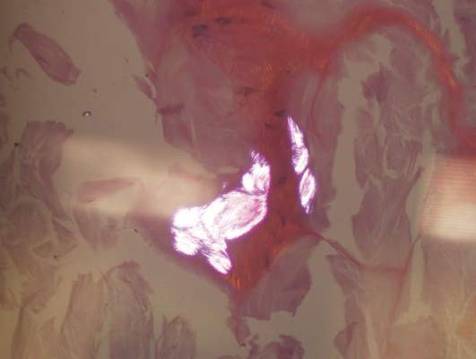
|
In our patient, the clinical differential diagnosis included calcium deposits, weathering nodules, and tophaceous gout. The differential diagnosis of auricular lesions is broad, and benign lesions may mimic cancerous entities such as basal cell carcinoma and squamous cell carcinoma.1 Therefore a detailed history, thorough physical examination, and tissue sampling are key to establishing the correct diagnosis. Our patient’s history of monoarticular gouty arthritis was only elucidated after a diagnosis of bilateral auricular tophaceous gout was made based on the biopsy results.
Subcutaneous tophi represent a chronic state of hyperuricemia and tend to manifest after long-standing polyarthritis and repeated acute gout attacks.2-5 These lesions develop in approximately 50% of gout patients and usually occur an average of 11.6 years after the onset of disease.2 There is a subset of individuals that are at higher risk for developing tophi, including elderly and female patients, diuretic and chronic nonsteroidal anti-inflammatory drug users, patients with a history of cyclosporine therapy, and patients with underlying chronic renal insufficiency.2,6,7 The most commonly affected tissues are those with poor blood supply and lower temperatures, such as the ear helix and first metacarpal joint.4 The auricle is the most common site of tophi on the head and neck. Tophi of the helices are generally asymptomatic and nontender; however, tophi can become large, inflamed, and ulcerated, causing pressure and discomfort.2 Combination treatment with dietary modification and antihyperuricemic therapy (eg, allopurinol) has been shown to reduce the size of lesions and prevent future tophi formation. However, these results may take months, warranting excision of large and symptomatic lesions.4,8
Our case is unusual in that the onset of the auricular lesions predated the articular gout by 6 months. Gouty tophi as the initial presentation of hyperuricemia is rare; however, tophi formation without concomitant arthritis has been reported.2,3,7,9 Wernick et al7 described 6 patients presenting with tophi before the onset of inflammatory arthritis that they attributed to changes in active inflammation by age (eg, elderly patients were more commonly immunosuppressed), chronic illnesses, and anti-inflammatory medications (eg, nonsteroidal anti-inflammatory drugs). Another possible explanation for this atypical presentation is misdiagnosis caused by other forms of arthritis (eg, rheumatoid arthritis, osteoarthritis) masking acute gout episodes. It also has been reported that monosodium urate crystals can be found in synovial fluid with no inflammation and therefore no symptoms.7
Tophi, although rare, may be the sole clinical manifestation of underlying gouty disease. It is important to be aware of this atypical presentation to prevent misdiagnosis and provide appropriate treatment.
- Dompmartin A. Nodules of the external ear [in French]. Ann Dermatol Venereol. 1999;126:261-266.
- Griffin G, Munns J, Fullen D, et al. Auricular tophi as the initial presentation of gout. Otolaryngol Head Neck Surg. 2009;141:153-154.
- Koley S, Salodkar A, Choudhary S, et al. Tophi as first manifestation of gout. Indian J Dermatol Venerol. 2010;76:393-393-396.
- Moriwaki Y. Tophaceous gout [in Japanese]. Nihon Rinsho. 2008;66:711-716.
- Eggebeen AT. Gout: an update. Am Fam Physician. 2007;76:801-808.
- Hollingworth P, Scott JT, Burry HC. Nonarticular gout: hyperuricemia and tophus formation without gouty arthritis. Arthritis Rheum. 1983;26:98-101.
- Wernick R, Winkler C, Campbell S. Tophi as the initial manifestation of gout. report of six cases and review of the literature. Arch Intern Med. 1992;152:873-876.
- Caldas CA, Fuller R. Excellent response to the clinical treatment of tophaceous gout. Clin Rheumatol. 2009;26:1553-1555.
- Iglesias A, Londono JC, Saaibi DL, et al. Gout nodulosis: widespread subcutaneous deposits without gout. Arthritis Care Res. 1996;9:74-77.
The Diagnosis: Bilateral Auricular Tophaceous Gout
Histopathologic evaluation with hematoxylin and eosin staining demonstrated clusters of abundant granular amorphous material within the subcutaneous tissue (Figure 1). The overlying epidermis and dermis were unremarkable. The granular amorphous material demonstrated numerous monosodium urate crystals under polarized light (Figure 2). At a return visit following the biopsy results, the patient reported a history of a single episode of monoarticular gouty arthritis involving the right hallux approximately 6 months after the onset of the skin lesions. With the added clinical history and the biopsy results, his serum uric acid level was obtained and was found to be elevated at 9.2 mg/dL (reference range, 3.5–8 mg/dL).
|
|
|
|
In our patient, the clinical differential diagnosis included calcium deposits, weathering nodules, and tophaceous gout. The differential diagnosis of auricular lesions is broad, and benign lesions may mimic cancerous entities such as basal cell carcinoma and squamous cell carcinoma.1 Therefore a detailed history, thorough physical examination, and tissue sampling are key to establishing the correct diagnosis. Our patient’s history of monoarticular gouty arthritis was only elucidated after a diagnosis of bilateral auricular tophaceous gout was made based on the biopsy results.
Subcutaneous tophi represent a chronic state of hyperuricemia and tend to manifest after long-standing polyarthritis and repeated acute gout attacks.2-5 These lesions develop in approximately 50% of gout patients and usually occur an average of 11.6 years after the onset of disease.2 There is a subset of individuals that are at higher risk for developing tophi, including elderly and female patients, diuretic and chronic nonsteroidal anti-inflammatory drug users, patients with a history of cyclosporine therapy, and patients with underlying chronic renal insufficiency.2,6,7 The most commonly affected tissues are those with poor blood supply and lower temperatures, such as the ear helix and first metacarpal joint.4 The auricle is the most common site of tophi on the head and neck. Tophi of the helices are generally asymptomatic and nontender; however, tophi can become large, inflamed, and ulcerated, causing pressure and discomfort.2 Combination treatment with dietary modification and antihyperuricemic therapy (eg, allopurinol) has been shown to reduce the size of lesions and prevent future tophi formation. However, these results may take months, warranting excision of large and symptomatic lesions.4,8
Our case is unusual in that the onset of the auricular lesions predated the articular gout by 6 months. Gouty tophi as the initial presentation of hyperuricemia is rare; however, tophi formation without concomitant arthritis has been reported.2,3,7,9 Wernick et al7 described 6 patients presenting with tophi before the onset of inflammatory arthritis that they attributed to changes in active inflammation by age (eg, elderly patients were more commonly immunosuppressed), chronic illnesses, and anti-inflammatory medications (eg, nonsteroidal anti-inflammatory drugs). Another possible explanation for this atypical presentation is misdiagnosis caused by other forms of arthritis (eg, rheumatoid arthritis, osteoarthritis) masking acute gout episodes. It also has been reported that monosodium urate crystals can be found in synovial fluid with no inflammation and therefore no symptoms.7
Tophi, although rare, may be the sole clinical manifestation of underlying gouty disease. It is important to be aware of this atypical presentation to prevent misdiagnosis and provide appropriate treatment.
The Diagnosis: Bilateral Auricular Tophaceous Gout
Histopathologic evaluation with hematoxylin and eosin staining demonstrated clusters of abundant granular amorphous material within the subcutaneous tissue (Figure 1). The overlying epidermis and dermis were unremarkable. The granular amorphous material demonstrated numerous monosodium urate crystals under polarized light (Figure 2). At a return visit following the biopsy results, the patient reported a history of a single episode of monoarticular gouty arthritis involving the right hallux approximately 6 months after the onset of the skin lesions. With the added clinical history and the biopsy results, his serum uric acid level was obtained and was found to be elevated at 9.2 mg/dL (reference range, 3.5–8 mg/dL).
|
|
|
|
In our patient, the clinical differential diagnosis included calcium deposits, weathering nodules, and tophaceous gout. The differential diagnosis of auricular lesions is broad, and benign lesions may mimic cancerous entities such as basal cell carcinoma and squamous cell carcinoma.1 Therefore a detailed history, thorough physical examination, and tissue sampling are key to establishing the correct diagnosis. Our patient’s history of monoarticular gouty arthritis was only elucidated after a diagnosis of bilateral auricular tophaceous gout was made based on the biopsy results.
Subcutaneous tophi represent a chronic state of hyperuricemia and tend to manifest after long-standing polyarthritis and repeated acute gout attacks.2-5 These lesions develop in approximately 50% of gout patients and usually occur an average of 11.6 years after the onset of disease.2 There is a subset of individuals that are at higher risk for developing tophi, including elderly and female patients, diuretic and chronic nonsteroidal anti-inflammatory drug users, patients with a history of cyclosporine therapy, and patients with underlying chronic renal insufficiency.2,6,7 The most commonly affected tissues are those with poor blood supply and lower temperatures, such as the ear helix and first metacarpal joint.4 The auricle is the most common site of tophi on the head and neck. Tophi of the helices are generally asymptomatic and nontender; however, tophi can become large, inflamed, and ulcerated, causing pressure and discomfort.2 Combination treatment with dietary modification and antihyperuricemic therapy (eg, allopurinol) has been shown to reduce the size of lesions and prevent future tophi formation. However, these results may take months, warranting excision of large and symptomatic lesions.4,8
Our case is unusual in that the onset of the auricular lesions predated the articular gout by 6 months. Gouty tophi as the initial presentation of hyperuricemia is rare; however, tophi formation without concomitant arthritis has been reported.2,3,7,9 Wernick et al7 described 6 patients presenting with tophi before the onset of inflammatory arthritis that they attributed to changes in active inflammation by age (eg, elderly patients were more commonly immunosuppressed), chronic illnesses, and anti-inflammatory medications (eg, nonsteroidal anti-inflammatory drugs). Another possible explanation for this atypical presentation is misdiagnosis caused by other forms of arthritis (eg, rheumatoid arthritis, osteoarthritis) masking acute gout episodes. It also has been reported that monosodium urate crystals can be found in synovial fluid with no inflammation and therefore no symptoms.7
Tophi, although rare, may be the sole clinical manifestation of underlying gouty disease. It is important to be aware of this atypical presentation to prevent misdiagnosis and provide appropriate treatment.
- Dompmartin A. Nodules of the external ear [in French]. Ann Dermatol Venereol. 1999;126:261-266.
- Griffin G, Munns J, Fullen D, et al. Auricular tophi as the initial presentation of gout. Otolaryngol Head Neck Surg. 2009;141:153-154.
- Koley S, Salodkar A, Choudhary S, et al. Tophi as first manifestation of gout. Indian J Dermatol Venerol. 2010;76:393-393-396.
- Moriwaki Y. Tophaceous gout [in Japanese]. Nihon Rinsho. 2008;66:711-716.
- Eggebeen AT. Gout: an update. Am Fam Physician. 2007;76:801-808.
- Hollingworth P, Scott JT, Burry HC. Nonarticular gout: hyperuricemia and tophus formation without gouty arthritis. Arthritis Rheum. 1983;26:98-101.
- Wernick R, Winkler C, Campbell S. Tophi as the initial manifestation of gout. report of six cases and review of the literature. Arch Intern Med. 1992;152:873-876.
- Caldas CA, Fuller R. Excellent response to the clinical treatment of tophaceous gout. Clin Rheumatol. 2009;26:1553-1555.
- Iglesias A, Londono JC, Saaibi DL, et al. Gout nodulosis: widespread subcutaneous deposits without gout. Arthritis Care Res. 1996;9:74-77.
- Dompmartin A. Nodules of the external ear [in French]. Ann Dermatol Venereol. 1999;126:261-266.
- Griffin G, Munns J, Fullen D, et al. Auricular tophi as the initial presentation of gout. Otolaryngol Head Neck Surg. 2009;141:153-154.
- Koley S, Salodkar A, Choudhary S, et al. Tophi as first manifestation of gout. Indian J Dermatol Venerol. 2010;76:393-393-396.
- Moriwaki Y. Tophaceous gout [in Japanese]. Nihon Rinsho. 2008;66:711-716.
- Eggebeen AT. Gout: an update. Am Fam Physician. 2007;76:801-808.
- Hollingworth P, Scott JT, Burry HC. Nonarticular gout: hyperuricemia and tophus formation without gouty arthritis. Arthritis Rheum. 1983;26:98-101.
- Wernick R, Winkler C, Campbell S. Tophi as the initial manifestation of gout. report of six cases and review of the literature. Arch Intern Med. 1992;152:873-876.
- Caldas CA, Fuller R. Excellent response to the clinical treatment of tophaceous gout. Clin Rheumatol. 2009;26:1553-1555.
- Iglesias A, Londono JC, Saaibi DL, et al. Gout nodulosis: widespread subcutaneous deposits without gout. Arthritis Care Res. 1996;9:74-77.
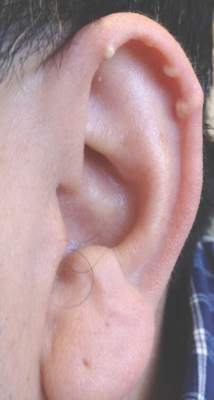
A 40-year-old man presented for evaluation of multiple small nodules on the bilateral auricles primarily involving the helices of 1 year’s duration. The lesions were nontender with no associated bleeding, burning, or pruritus. He denied any trauma to these sites and denied any systemic symptoms including fever, chills, joint pain, or weight loss. His medical history was remarkable for type 2 diabetes mellitus. He had no history of similar skin lesions or renal disease and denied any alcohol intake. He also denied taking any over-the-counter or prescription medications. Physical examination revealed several 1- to 4-mm superficial white dermal nodules located on the bilateral helical rims. The lesions were firm and well circumscribed and the surrounding skin showed mild erythema. Shave biopsies of the nodules were performed.