User login
A comment on a CME test question
To the Editor: Question 1 of the December 2013 CME test “Can an ARB be given to patients who have had angioedema on an ACE inhibitor?” presents the case of a 73-year-old woman with angioedema thought to be due to her taking enalapril; in addition, she takes hydrochlorothiazide. Her blood pressure is 118/72 mm Hg, and her heart rate is not specified. The question is what the next best step would be to manage her blood pressure medications. The “correct” answer is given as “substitute metoprolol for enalapril in her regimen.”
While this answer is the best choice given, I would take issue with it for two reasons. First, many elderly hypertension patients are overmedicated. With a blood pressure of 118/72 on two medications, it is entirely possible that she may not need to replace the enalapril with any other medication to maintain her pressure below the new JNC 8 threshold of 150/90 for the elderly, or even the 140/90 level specified in other guidelines.
I would recheck her pressure daily on her diuretic alone before adding back a second medication. If she does require a second blood pressure medication, JNC 8 (in agreement with other recent guidelines) recommends adding a calcium channel blocker. Beta-blockers are not recommended by any recent guidelines for first-line or second-line treatment of hypertension for elderly patients without special indications, such as tachyarrhythmias or history of myocardial infarction. No special indications for a beta-blocker were mentioned in this case. Indeed, elderly hypertensive patients often have slow-normal heart rates, or even mild resting bradycardia, which would make the addition of metoprolol contraindicated and potentially dangerous.
To the Editor: Question 1 of the December 2013 CME test “Can an ARB be given to patients who have had angioedema on an ACE inhibitor?” presents the case of a 73-year-old woman with angioedema thought to be due to her taking enalapril; in addition, she takes hydrochlorothiazide. Her blood pressure is 118/72 mm Hg, and her heart rate is not specified. The question is what the next best step would be to manage her blood pressure medications. The “correct” answer is given as “substitute metoprolol for enalapril in her regimen.”
While this answer is the best choice given, I would take issue with it for two reasons. First, many elderly hypertension patients are overmedicated. With a blood pressure of 118/72 on two medications, it is entirely possible that she may not need to replace the enalapril with any other medication to maintain her pressure below the new JNC 8 threshold of 150/90 for the elderly, or even the 140/90 level specified in other guidelines.
I would recheck her pressure daily on her diuretic alone before adding back a second medication. If she does require a second blood pressure medication, JNC 8 (in agreement with other recent guidelines) recommends adding a calcium channel blocker. Beta-blockers are not recommended by any recent guidelines for first-line or second-line treatment of hypertension for elderly patients without special indications, such as tachyarrhythmias or history of myocardial infarction. No special indications for a beta-blocker were mentioned in this case. Indeed, elderly hypertensive patients often have slow-normal heart rates, or even mild resting bradycardia, which would make the addition of metoprolol contraindicated and potentially dangerous.
To the Editor: Question 1 of the December 2013 CME test “Can an ARB be given to patients who have had angioedema on an ACE inhibitor?” presents the case of a 73-year-old woman with angioedema thought to be due to her taking enalapril; in addition, she takes hydrochlorothiazide. Her blood pressure is 118/72 mm Hg, and her heart rate is not specified. The question is what the next best step would be to manage her blood pressure medications. The “correct” answer is given as “substitute metoprolol for enalapril in her regimen.”
While this answer is the best choice given, I would take issue with it for two reasons. First, many elderly hypertension patients are overmedicated. With a blood pressure of 118/72 on two medications, it is entirely possible that she may not need to replace the enalapril with any other medication to maintain her pressure below the new JNC 8 threshold of 150/90 for the elderly, or even the 140/90 level specified in other guidelines.
I would recheck her pressure daily on her diuretic alone before adding back a second medication. If she does require a second blood pressure medication, JNC 8 (in agreement with other recent guidelines) recommends adding a calcium channel blocker. Beta-blockers are not recommended by any recent guidelines for first-line or second-line treatment of hypertension for elderly patients without special indications, such as tachyarrhythmias or history of myocardial infarction. No special indications for a beta-blocker were mentioned in this case. Indeed, elderly hypertensive patients often have slow-normal heart rates, or even mild resting bradycardia, which would make the addition of metoprolol contraindicated and potentially dangerous.
Prescribing for the pregnant patient
Primum non nocere: First, do no harm—a principle taught across the world to all medical students. It reminds the health care provider to consider the possible harm that any intervention might produce. Never is it more relevant in the mind of a clinician than when prescribing a medication for a pregnant woman. We are, after all, brought up in a society averse to medical risk.
When managing a pregnant patient, should the baby be the highest priority, whatever the mother may face? Or to take the extreme opposite position, should the mother be treated with the best possible options and the baby ignored?
And what about the views of the patient? There is a widespread cultural belief about the vulnerability of the mother and fetus during pregnancy. Therefore, when faced with the decision of whether to use a medication or not, what is the best recourse for the pregnant patient? Should she be the “good mother” and avoid all risk to the baby, or should she be the “responsible mother” who follows medical advice and takes treatment as recommended?
In truth, the path to safe management of a pregnant patient is rarely so dichotomous. In most cases, what is best for the mother is also best for the baby. However, caring for a pregnant or lactating woman can be challenging for clinicians facing insufficient information regarding medication safety, overestimation of the risk of medication by both the patient and the care provider, and increasing litigation costs.
This article provides key principles to guide clinicians caring for pregnant patients, as we find ourselves increasingly dependent on pharmacotherapy. It also includes sources of information clinicians can turn to when they need additional pregnancy safety data about a certain drug and when they want advice about conditions commonly seen in pregnancy and medications that can be justifiably used in those circumstances.
KEY CONCEPTS FOR PRESCRIBING IN PREGNANCY
The following concepts are key to prescribing for a pregnant patient:
No protective barrier exists between the maternal and fetal environments
The placenta contains a semipermeable membrane that selectively allows some substances to pass from the maternal to the fetal blood and excludes others. However, it is not really a “protective mechanism” when it comes to medications. Assume that the fetus will have exposure, at least to some degree.
In general, drugs that are lipophilic, of a low molecular weight, or not ionized at physiologic pH cross the placenta more efficiently than others. Heparin and insulin are notable exceptions to the rule that most drugs cross the placenta. They do not.
The gestational stage may determine the effect of a medication on the fetus
In animals and in humans, exposure of the embryo or fetus to a teratogen may produce a permanent abnormality of structure or function.
First-trimester exposures are most worrisome for structural malformations. However, fetal neurologic and behavioral development, fetal survival, and function of specific organs can be affected even after the first trimester. For example, while first-trimester exposure to angiotensin-converting enzyme inhibitors has been linked to a slight increase in congenital heart defects, exposure in the second or third trimester can result in fetal oligohydramnios, neonatal anuria, pulmonary hypoplasia, intrauterine growth restriction, and fetal death.
Physiologic changes of pregnancy affect the pharmacokinetics of medications
Pregnancy is associated with increased plasma volume, increased glomerular filtration rate, and dilutional hypoalbuminemia, which can all affect the bioavailability of medications. Absorption of oral agents also may be affected by slowed gastric motility in pregnancy.
Although these physiologic alterations do not routinely warrant a change in drug dosage, they may be important considerations when choosing an appropriate agent. For example, medications taken in multiple doses per day are more likely to have a sustained effect than once-daily medications, which would be rapidly cleared in a pregnant patient.
Sole reliance on the FDA pregnancy safety category may be inadequate

To help clinicians prescribe medications for pregnant women, the US Food and Drug Administration (FDA) assigns medications to one of five categories of risk (A, B, C, D, or X) (Table 1). Unfortunately, this classification system has several shortcomings:
- The categories are often seen as a grading system in which the risk increases from the lowest in category A to highest in category X, and the safety information in the accompanying narrative is not always appreciated by prescribers.
- Clinicians incorrectly assume that drugs in a particular category carry a similar risk. However, 65% to 70% of all medications are in category C. This category includes medications with adverse animal data or no animal data at all. In addition, adverse animal data may vary in severity from decreased fetal weight to major structural malformation and fetal loss, indicating a difference in expected risk.
- Most of the data on medication safety in pregnancy comes from animal studies, case reports, case series, case-control studies, or pregnancy registries, and each of these sources has significant limitations.
- The categories do not distinguish between supporting data from animal studies and human studies. For instance, a category-B drug may have animal studies that show no risk but no adequate human studies, or may have animal studies showing risk but human studies that do not.
Looking at the pregnancy risk classifications used in the United States (ie, the FDA system), Australia, and Sweden, researchers compared the classification of 236 drugs between the three systems and found that only one in four drugs was similarly classified into the same risk category. This discrepancy further brings into question the usefulness and reliability of these classifications.1
Finally, none of the classification systems tells us the potential harm from withholding a medication in pregnancy.
RESOURCES TO ASSESS MEDICATION SAFETY IN PREGNANCY
The FDA has proposed changes in the labeling of medications related to pregnancy and lactation.2 The proposed changes would eliminate the current categories and instead require a summary of the risks, the effects of the drug on the fetus, and clinical considerations for use during pregnancy. In addition, labeling would include a description of the medication’s effects on milk production, the amount of drug present in milk, and possible effects on the infant.
Until such changes are in place, what other resources can a busy clinician turn to for support?
The official drug labeling (or the package insert), also published in the Physicians’ Desk Reference, is one source of information, but it rarely provides up-to-date information about teratogenic risks in human pregnancies.
Several online databases review, summarize, and periodically update information from the peer-reviewed medical literature.3–7 The REPRORISK system4–7 maintained by Micromedex (Greenwood Village, CO) provides access to several databases that contain information about a wide range of individual medications: REPROTEXT, REPROTOX,5 Shepard’s Catalog of Teratogenic Agents,7 and the Teratogen Information System (TERIS).4 Online access and a smartphone “app” for these databases are available for a subscription fee. Summaries for individual medications can be ordered directly from TERIS, also for a fee. Several other resources are available in textbook format.8–10
In addition, health care providers can obtain information from or can refer pregnant and breastfeeding patients to a teratology information service for information and counseling about medication exposures. MotherToBaby,11 a service of the nonprofit Organization of Teratology Information Specialists, provides fact sheets, free phone consultation, risk assessment, and counseling by trained teratogen information specialists about environmental exposures, including prescription and over-the-counter medications and dietary and herbal supplements. Counselors from these services gather and synthesize information about exposures from the databases mentioned above, from the peer-reviewed medical literature, from drug manufacturers, and from other sources.

With the advent of electronic medical records and computerized provider order entry, clinical decision support systems hold promise as an additional resource for safe prescribing in pregnancy.
Fortunately, the list of teratogenic medications that are absolutely contraindicated in pregnancy remains small (Table 2).12,13
THE FOUR-QUESTION APPROACH TO CARING FOR THE PREGNANT PATIENT
Is the symptom self-limited or amenable to nonpharmacologic management?
It has been said that we live in a culture where every symptom warrants a pill. If this is true, there can be no better time for reevaluating this practice than during pregnancy.
Many of the medications most commonly used in pregnancy are for upper-respiratory-tract infections, headache, or psychological distress. Pregnancy is the ideal time to educate patients about the limited effectiveness of most cough-and-cold remedies and the inappropriateness of antibiotics for colds and viral bronchitis. It is also an ideal time for a trial of lifestyle modifications, relaxation, and biofeedback for a chronic headache problem. For cases of mild to moderate depression, it may be worth considering treatment with psychotherapy rather than medications.
Offering patients the option of no treatment or nonpharmacologic treatment for self-limited symptoms is an option worth considering.
How do the patient’s (and your) values and understanding affect the decision?
Is the patient willing to take medication? What are her beliefs with regard to her problem and how it should be managed in pregnancy?
Women and clinicians bring many worries and prejudices to the use of medications in pregnancy. The experiences of the patient and her family and friends may present huge obstacles to needed medication use in pregnancy. Misinformation in the media and from family members, friends, and other health care providers are other obstacles. The only way to deal with this issue is to ask your patient directly about her fears and concerns regarding each prescription written.
Clinicians also need to address fears or prejudices they themselves may have about medication safety in pregnancy. These may arise from a single bad experience in caring for a pregnant woman, discomfort with uncertainty, or a belief that pregnant women should avoid any and all risks of exposures, even when the mother’s condition warrants pharmacologic treatment.
Being informed, both scientifically and about one’s own biases or tendencies, is an essential foundation for rational prescribing in pregnancy.
Is the problem affected by pregnancy, and how?
Pregnancy can affect many medical conditions, and in different ways. Conditions such as asthma, migraine headache, and cardiac arrhythmia are exacerbated in pregnancy, placing the mother and fetus at increased risk of morbidity. Conditions such as Graves disease and hypertension may improve as pregnancy progresses, and medications often can be withdrawn as the patient progresses further along in gestation.
Understanding the effect of pregnancy on a particular problem may help the clinician to make an informed decision about medication use in pregnancy.
How does the problem affect pregnancy?
Considering the risk of untreated disease to the pregnancy may help in decision-making.
Many medical conditions can negatively affect the development of the fetus. A glaring example is diabetes mellitus, with poor glycemic control being linked to congenital malformations, spontaneous abortion, and fetal demise. Chronic conditions with periodic exacerbations such as asthma or epilepsy place the fetus at increased risk during a flare-up.
Therefore, for chronic conditions, continuing maintenance therapy is best. Preconception counseling in such cases is crucial, so that a drug with adequate safety data can be substituted before pregnancy. In this way, any risk to the mother or the embryo from exacerbation of disease as such adjustments are made is avoided.
For conditions arising de novo in pregnancy, the underlying principle remains the same. Is the risk of pharmacotherapy more than the risk of untreated disease? Invariably, the answer to this question supports medication use, and an educated provider will be able to choose a treatment that is justifiable in most circumstances.
CHOOSING A MEDICATION

Fetal well-being depends on maternal well-being. It therefore helps to think of medication use in pregnancy as “justified or not” rather than “safe or not.” Table 3 lists some conditions commonly seen in pregnancy, selected drugs of choice that can be safely used for treating those conditions, and alternates that may be justified in some circumstances.5,6,14–18
GOOD PRACTICES WHEN PRESCRIBING IN PREGNANCY
Prescribing in pregnancy will be most successful when both the patient and the prescribing physician consider the fetal benefit gained from optimizing maternal health. Good prescribing practices to ensure optimum therapeutic benefit when caring for a pregnant patient are to:
- Involve the patient in decision-making. Recognize her concerns, worries, and preferences regarding her illness and its treatment.
- Inform the patient of the risk of an untreated medical condition, weighed against the risk of medication.
- Choose medications with the most available safety data. Let the patient know what resources you have referred to in choosing the medication.
- It is advisable to perform a search each time a prescription is written for a pregnant or lactating woman.
- When possible, have the discussion in the preconception period.
- Consider the dynamic physiology of gestation. Choose the right drug for the right trimester.
- Discuss the plan with the patient and other providers.
- Define clear criteria for when to discontinue the treatment.
- Addis A, Sharabi S, Bonati M. Risk classification systems for drug use during pregnancy: are they a reliable source of information? Drug Saf 2000; 23:245–253.
- US Food and Drug Administration (FDA). Pregnancy and lactation labeling. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/Labeling/ucm093307.htm. Accessed April 4, 2014.
- Lagoy CT, Joshi N, Cragan JD, Rasmussen SA. Medication use during pregnancy and lactation: an urgent call for public health action. J Womens Health (Larchmt) 2005; 14:104–109.
- Clinical Teratology Website. University of Washington. http://depts.washington.edu/terisweb/teris/. Accessed April 4, 2014.
- REPROTOX, An Online Reproductive Toxicology Resource. Reproductive Toxicology Center. www.reprotox.org. Accessed April 4, 2014.
- REPRORISK. Micromedex, Inc. www.micromedex.com/products/reprorisk. Accessed April 4, 2014.
- Shepard TH. Catalog of teratogenic agents. 13th ed. Baltimore, MD: Johns Hopkins University Press; 2010.
- Briggs GG, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation: A reference guide to fetal and neonatal risk. Philadelphia, PA: Lippincott Williams & Wilkins; 2011.
- Koren G. Medication safety in pregnancy and breastfeeding. McGraw-Hill Professional Publishing; 2007.
- Friedman JM, Polifka JE. Teratogenic effects of drugs: A resource for clinicians (TERIS). Baltimore, MD: Johns Hopkins University Press; 2000.
- MotherToBaby. www.mothertobaby.org. Accessed April 4, 2014.
- Dunlop AL, Gardiner PM, Shellhaas CS, Menard MK, McDiarmid MA. The clinical content of preconception care: the use of medications and supplements among women of reproductive age. Am J Obstet Gynecol 2008; 199(suppl 2):S367–S372.
- Ciarkowski SL, Stalburg CM. Medication safety in obstetrics and gynecology. Clin Obstet Gynecol 2010; 53:482–499.
- Koren G, Pastuszak A, Ito S. Drugs in pregnancy. N Engl J Med 1998; 338:1128–1137.
- Lambert K, Holt RI. The use of insulin analogues in pregnancy. Diabetes Obes Metab 2013; 15:888–900.
- Chan WS, Anand S, Ginsberg JS. Anticoagulation of pregnant women with mechanical heart valves: a systematic review of the literature. Arch Intern Med 2000; 160:191–196.
- Nagler M, Haslauer M, Wuillemin WA. Fondaparinux—data on efficacy and safety in special situations. Thromb Res 2012; 129:407–417.
- Kweder SL, Powrie RO. Prescribing in pregnancy: a practical approach. In:Powrie RO, Greene M, Camann W, editors. De Swiet’s Medical disorders in Obstetric Practice. 5th ed. Hoboken, NJ: Wiley-Blackwell; 2010:633–640.
Primum non nocere: First, do no harm—a principle taught across the world to all medical students. It reminds the health care provider to consider the possible harm that any intervention might produce. Never is it more relevant in the mind of a clinician than when prescribing a medication for a pregnant woman. We are, after all, brought up in a society averse to medical risk.
When managing a pregnant patient, should the baby be the highest priority, whatever the mother may face? Or to take the extreme opposite position, should the mother be treated with the best possible options and the baby ignored?
And what about the views of the patient? There is a widespread cultural belief about the vulnerability of the mother and fetus during pregnancy. Therefore, when faced with the decision of whether to use a medication or not, what is the best recourse for the pregnant patient? Should she be the “good mother” and avoid all risk to the baby, or should she be the “responsible mother” who follows medical advice and takes treatment as recommended?
In truth, the path to safe management of a pregnant patient is rarely so dichotomous. In most cases, what is best for the mother is also best for the baby. However, caring for a pregnant or lactating woman can be challenging for clinicians facing insufficient information regarding medication safety, overestimation of the risk of medication by both the patient and the care provider, and increasing litigation costs.
This article provides key principles to guide clinicians caring for pregnant patients, as we find ourselves increasingly dependent on pharmacotherapy. It also includes sources of information clinicians can turn to when they need additional pregnancy safety data about a certain drug and when they want advice about conditions commonly seen in pregnancy and medications that can be justifiably used in those circumstances.
KEY CONCEPTS FOR PRESCRIBING IN PREGNANCY
The following concepts are key to prescribing for a pregnant patient:
No protective barrier exists between the maternal and fetal environments
The placenta contains a semipermeable membrane that selectively allows some substances to pass from the maternal to the fetal blood and excludes others. However, it is not really a “protective mechanism” when it comes to medications. Assume that the fetus will have exposure, at least to some degree.
In general, drugs that are lipophilic, of a low molecular weight, or not ionized at physiologic pH cross the placenta more efficiently than others. Heparin and insulin are notable exceptions to the rule that most drugs cross the placenta. They do not.
The gestational stage may determine the effect of a medication on the fetus
In animals and in humans, exposure of the embryo or fetus to a teratogen may produce a permanent abnormality of structure or function.
First-trimester exposures are most worrisome for structural malformations. However, fetal neurologic and behavioral development, fetal survival, and function of specific organs can be affected even after the first trimester. For example, while first-trimester exposure to angiotensin-converting enzyme inhibitors has been linked to a slight increase in congenital heart defects, exposure in the second or third trimester can result in fetal oligohydramnios, neonatal anuria, pulmonary hypoplasia, intrauterine growth restriction, and fetal death.
Physiologic changes of pregnancy affect the pharmacokinetics of medications
Pregnancy is associated with increased plasma volume, increased glomerular filtration rate, and dilutional hypoalbuminemia, which can all affect the bioavailability of medications. Absorption of oral agents also may be affected by slowed gastric motility in pregnancy.
Although these physiologic alterations do not routinely warrant a change in drug dosage, they may be important considerations when choosing an appropriate agent. For example, medications taken in multiple doses per day are more likely to have a sustained effect than once-daily medications, which would be rapidly cleared in a pregnant patient.
Sole reliance on the FDA pregnancy safety category may be inadequate
To help clinicians prescribe medications for pregnant women, the US Food and Drug Administration (FDA) assigns medications to one of five categories of risk (A, B, C, D, or X) (Table 1). Unfortunately, this classification system has several shortcomings:
- The categories are often seen as a grading system in which the risk increases from the lowest in category A to highest in category X, and the safety information in the accompanying narrative is not always appreciated by prescribers.
- Clinicians incorrectly assume that drugs in a particular category carry a similar risk. However, 65% to 70% of all medications are in category C. This category includes medications with adverse animal data or no animal data at all. In addition, adverse animal data may vary in severity from decreased fetal weight to major structural malformation and fetal loss, indicating a difference in expected risk.
- Most of the data on medication safety in pregnancy comes from animal studies, case reports, case series, case-control studies, or pregnancy registries, and each of these sources has significant limitations.
- The categories do not distinguish between supporting data from animal studies and human studies. For instance, a category-B drug may have animal studies that show no risk but no adequate human studies, or may have animal studies showing risk but human studies that do not.
Looking at the pregnancy risk classifications used in the United States (ie, the FDA system), Australia, and Sweden, researchers compared the classification of 236 drugs between the three systems and found that only one in four drugs was similarly classified into the same risk category. This discrepancy further brings into question the usefulness and reliability of these classifications.1
Finally, none of the classification systems tells us the potential harm from withholding a medication in pregnancy.
RESOURCES TO ASSESS MEDICATION SAFETY IN PREGNANCY
The FDA has proposed changes in the labeling of medications related to pregnancy and lactation.2 The proposed changes would eliminate the current categories and instead require a summary of the risks, the effects of the drug on the fetus, and clinical considerations for use during pregnancy. In addition, labeling would include a description of the medication’s effects on milk production, the amount of drug present in milk, and possible effects on the infant.
Until such changes are in place, what other resources can a busy clinician turn to for support?
The official drug labeling (or the package insert), also published in the Physicians’ Desk Reference, is one source of information, but it rarely provides up-to-date information about teratogenic risks in human pregnancies.
Several online databases review, summarize, and periodically update information from the peer-reviewed medical literature.3–7 The REPRORISK system4–7 maintained by Micromedex (Greenwood Village, CO) provides access to several databases that contain information about a wide range of individual medications: REPROTEXT, REPROTOX,5 Shepard’s Catalog of Teratogenic Agents,7 and the Teratogen Information System (TERIS).4 Online access and a smartphone “app” for these databases are available for a subscription fee. Summaries for individual medications can be ordered directly from TERIS, also for a fee. Several other resources are available in textbook format.8–10
In addition, health care providers can obtain information from or can refer pregnant and breastfeeding patients to a teratology information service for information and counseling about medication exposures. MotherToBaby,11 a service of the nonprofit Organization of Teratology Information Specialists, provides fact sheets, free phone consultation, risk assessment, and counseling by trained teratogen information specialists about environmental exposures, including prescription and over-the-counter medications and dietary and herbal supplements. Counselors from these services gather and synthesize information about exposures from the databases mentioned above, from the peer-reviewed medical literature, from drug manufacturers, and from other sources.
With the advent of electronic medical records and computerized provider order entry, clinical decision support systems hold promise as an additional resource for safe prescribing in pregnancy.
Fortunately, the list of teratogenic medications that are absolutely contraindicated in pregnancy remains small (Table 2).12,13
THE FOUR-QUESTION APPROACH TO CARING FOR THE PREGNANT PATIENT
Is the symptom self-limited or amenable to nonpharmacologic management?
It has been said that we live in a culture where every symptom warrants a pill. If this is true, there can be no better time for reevaluating this practice than during pregnancy.
Many of the medications most commonly used in pregnancy are for upper-respiratory-tract infections, headache, or psychological distress. Pregnancy is the ideal time to educate patients about the limited effectiveness of most cough-and-cold remedies and the inappropriateness of antibiotics for colds and viral bronchitis. It is also an ideal time for a trial of lifestyle modifications, relaxation, and biofeedback for a chronic headache problem. For cases of mild to moderate depression, it may be worth considering treatment with psychotherapy rather than medications.
Offering patients the option of no treatment or nonpharmacologic treatment for self-limited symptoms is an option worth considering.
How do the patient’s (and your) values and understanding affect the decision?
Is the patient willing to take medication? What are her beliefs with regard to her problem and how it should be managed in pregnancy?
Women and clinicians bring many worries and prejudices to the use of medications in pregnancy. The experiences of the patient and her family and friends may present huge obstacles to needed medication use in pregnancy. Misinformation in the media and from family members, friends, and other health care providers are other obstacles. The only way to deal with this issue is to ask your patient directly about her fears and concerns regarding each prescription written.
Clinicians also need to address fears or prejudices they themselves may have about medication safety in pregnancy. These may arise from a single bad experience in caring for a pregnant woman, discomfort with uncertainty, or a belief that pregnant women should avoid any and all risks of exposures, even when the mother’s condition warrants pharmacologic treatment.
Being informed, both scientifically and about one’s own biases or tendencies, is an essential foundation for rational prescribing in pregnancy.
Is the problem affected by pregnancy, and how?
Pregnancy can affect many medical conditions, and in different ways. Conditions such as asthma, migraine headache, and cardiac arrhythmia are exacerbated in pregnancy, placing the mother and fetus at increased risk of morbidity. Conditions such as Graves disease and hypertension may improve as pregnancy progresses, and medications often can be withdrawn as the patient progresses further along in gestation.
Understanding the effect of pregnancy on a particular problem may help the clinician to make an informed decision about medication use in pregnancy.
How does the problem affect pregnancy?
Considering the risk of untreated disease to the pregnancy may help in decision-making.
Many medical conditions can negatively affect the development of the fetus. A glaring example is diabetes mellitus, with poor glycemic control being linked to congenital malformations, spontaneous abortion, and fetal demise. Chronic conditions with periodic exacerbations such as asthma or epilepsy place the fetus at increased risk during a flare-up.
Therefore, for chronic conditions, continuing maintenance therapy is best. Preconception counseling in such cases is crucial, so that a drug with adequate safety data can be substituted before pregnancy. In this way, any risk to the mother or the embryo from exacerbation of disease as such adjustments are made is avoided.
For conditions arising de novo in pregnancy, the underlying principle remains the same. Is the risk of pharmacotherapy more than the risk of untreated disease? Invariably, the answer to this question supports medication use, and an educated provider will be able to choose a treatment that is justifiable in most circumstances.
CHOOSING A MEDICATION
Fetal well-being depends on maternal well-being. It therefore helps to think of medication use in pregnancy as “justified or not” rather than “safe or not.” Table 3 lists some conditions commonly seen in pregnancy, selected drugs of choice that can be safely used for treating those conditions, and alternates that may be justified in some circumstances.5,6,14–18
GOOD PRACTICES WHEN PRESCRIBING IN PREGNANCY
Prescribing in pregnancy will be most successful when both the patient and the prescribing physician consider the fetal benefit gained from optimizing maternal health. Good prescribing practices to ensure optimum therapeutic benefit when caring for a pregnant patient are to:
- Involve the patient in decision-making. Recognize her concerns, worries, and preferences regarding her illness and its treatment.
- Inform the patient of the risk of an untreated medical condition, weighed against the risk of medication.
- Choose medications with the most available safety data. Let the patient know what resources you have referred to in choosing the medication.
- It is advisable to perform a search each time a prescription is written for a pregnant or lactating woman.
- When possible, have the discussion in the preconception period.
- Consider the dynamic physiology of gestation. Choose the right drug for the right trimester.
- Discuss the plan with the patient and other providers.
- Define clear criteria for when to discontinue the treatment.
Primum non nocere: First, do no harm—a principle taught across the world to all medical students. It reminds the health care provider to consider the possible harm that any intervention might produce. Never is it more relevant in the mind of a clinician than when prescribing a medication for a pregnant woman. We are, after all, brought up in a society averse to medical risk.
When managing a pregnant patient, should the baby be the highest priority, whatever the mother may face? Or to take the extreme opposite position, should the mother be treated with the best possible options and the baby ignored?
And what about the views of the patient? There is a widespread cultural belief about the vulnerability of the mother and fetus during pregnancy. Therefore, when faced with the decision of whether to use a medication or not, what is the best recourse for the pregnant patient? Should she be the “good mother” and avoid all risk to the baby, or should she be the “responsible mother” who follows medical advice and takes treatment as recommended?
In truth, the path to safe management of a pregnant patient is rarely so dichotomous. In most cases, what is best for the mother is also best for the baby. However, caring for a pregnant or lactating woman can be challenging for clinicians facing insufficient information regarding medication safety, overestimation of the risk of medication by both the patient and the care provider, and increasing litigation costs.
This article provides key principles to guide clinicians caring for pregnant patients, as we find ourselves increasingly dependent on pharmacotherapy. It also includes sources of information clinicians can turn to when they need additional pregnancy safety data about a certain drug and when they want advice about conditions commonly seen in pregnancy and medications that can be justifiably used in those circumstances.
KEY CONCEPTS FOR PRESCRIBING IN PREGNANCY
The following concepts are key to prescribing for a pregnant patient:
No protective barrier exists between the maternal and fetal environments
The placenta contains a semipermeable membrane that selectively allows some substances to pass from the maternal to the fetal blood and excludes others. However, it is not really a “protective mechanism” when it comes to medications. Assume that the fetus will have exposure, at least to some degree.
In general, drugs that are lipophilic, of a low molecular weight, or not ionized at physiologic pH cross the placenta more efficiently than others. Heparin and insulin are notable exceptions to the rule that most drugs cross the placenta. They do not.
The gestational stage may determine the effect of a medication on the fetus
In animals and in humans, exposure of the embryo or fetus to a teratogen may produce a permanent abnormality of structure or function.
First-trimester exposures are most worrisome for structural malformations. However, fetal neurologic and behavioral development, fetal survival, and function of specific organs can be affected even after the first trimester. For example, while first-trimester exposure to angiotensin-converting enzyme inhibitors has been linked to a slight increase in congenital heart defects, exposure in the second or third trimester can result in fetal oligohydramnios, neonatal anuria, pulmonary hypoplasia, intrauterine growth restriction, and fetal death.
Physiologic changes of pregnancy affect the pharmacokinetics of medications
Pregnancy is associated with increased plasma volume, increased glomerular filtration rate, and dilutional hypoalbuminemia, which can all affect the bioavailability of medications. Absorption of oral agents also may be affected by slowed gastric motility in pregnancy.
Although these physiologic alterations do not routinely warrant a change in drug dosage, they may be important considerations when choosing an appropriate agent. For example, medications taken in multiple doses per day are more likely to have a sustained effect than once-daily medications, which would be rapidly cleared in a pregnant patient.
Sole reliance on the FDA pregnancy safety category may be inadequate
To help clinicians prescribe medications for pregnant women, the US Food and Drug Administration (FDA) assigns medications to one of five categories of risk (A, B, C, D, or X) (Table 1). Unfortunately, this classification system has several shortcomings:
- The categories are often seen as a grading system in which the risk increases from the lowest in category A to highest in category X, and the safety information in the accompanying narrative is not always appreciated by prescribers.
- Clinicians incorrectly assume that drugs in a particular category carry a similar risk. However, 65% to 70% of all medications are in category C. This category includes medications with adverse animal data or no animal data at all. In addition, adverse animal data may vary in severity from decreased fetal weight to major structural malformation and fetal loss, indicating a difference in expected risk.
- Most of the data on medication safety in pregnancy comes from animal studies, case reports, case series, case-control studies, or pregnancy registries, and each of these sources has significant limitations.
- The categories do not distinguish between supporting data from animal studies and human studies. For instance, a category-B drug may have animal studies that show no risk but no adequate human studies, or may have animal studies showing risk but human studies that do not.
Looking at the pregnancy risk classifications used in the United States (ie, the FDA system), Australia, and Sweden, researchers compared the classification of 236 drugs between the three systems and found that only one in four drugs was similarly classified into the same risk category. This discrepancy further brings into question the usefulness and reliability of these classifications.1
Finally, none of the classification systems tells us the potential harm from withholding a medication in pregnancy.
RESOURCES TO ASSESS MEDICATION SAFETY IN PREGNANCY
The FDA has proposed changes in the labeling of medications related to pregnancy and lactation.2 The proposed changes would eliminate the current categories and instead require a summary of the risks, the effects of the drug on the fetus, and clinical considerations for use during pregnancy. In addition, labeling would include a description of the medication’s effects on milk production, the amount of drug present in milk, and possible effects on the infant.
Until such changes are in place, what other resources can a busy clinician turn to for support?
The official drug labeling (or the package insert), also published in the Physicians’ Desk Reference, is one source of information, but it rarely provides up-to-date information about teratogenic risks in human pregnancies.
Several online databases review, summarize, and periodically update information from the peer-reviewed medical literature.3–7 The REPRORISK system4–7 maintained by Micromedex (Greenwood Village, CO) provides access to several databases that contain information about a wide range of individual medications: REPROTEXT, REPROTOX,5 Shepard’s Catalog of Teratogenic Agents,7 and the Teratogen Information System (TERIS).4 Online access and a smartphone “app” for these databases are available for a subscription fee. Summaries for individual medications can be ordered directly from TERIS, also for a fee. Several other resources are available in textbook format.8–10
In addition, health care providers can obtain information from or can refer pregnant and breastfeeding patients to a teratology information service for information and counseling about medication exposures. MotherToBaby,11 a service of the nonprofit Organization of Teratology Information Specialists, provides fact sheets, free phone consultation, risk assessment, and counseling by trained teratogen information specialists about environmental exposures, including prescription and over-the-counter medications and dietary and herbal supplements. Counselors from these services gather and synthesize information about exposures from the databases mentioned above, from the peer-reviewed medical literature, from drug manufacturers, and from other sources.
With the advent of electronic medical records and computerized provider order entry, clinical decision support systems hold promise as an additional resource for safe prescribing in pregnancy.
Fortunately, the list of teratogenic medications that are absolutely contraindicated in pregnancy remains small (Table 2).12,13
THE FOUR-QUESTION APPROACH TO CARING FOR THE PREGNANT PATIENT
Is the symptom self-limited or amenable to nonpharmacologic management?
It has been said that we live in a culture where every symptom warrants a pill. If this is true, there can be no better time for reevaluating this practice than during pregnancy.
Many of the medications most commonly used in pregnancy are for upper-respiratory-tract infections, headache, or psychological distress. Pregnancy is the ideal time to educate patients about the limited effectiveness of most cough-and-cold remedies and the inappropriateness of antibiotics for colds and viral bronchitis. It is also an ideal time for a trial of lifestyle modifications, relaxation, and biofeedback for a chronic headache problem. For cases of mild to moderate depression, it may be worth considering treatment with psychotherapy rather than medications.
Offering patients the option of no treatment or nonpharmacologic treatment for self-limited symptoms is an option worth considering.
How do the patient’s (and your) values and understanding affect the decision?
Is the patient willing to take medication? What are her beliefs with regard to her problem and how it should be managed in pregnancy?
Women and clinicians bring many worries and prejudices to the use of medications in pregnancy. The experiences of the patient and her family and friends may present huge obstacles to needed medication use in pregnancy. Misinformation in the media and from family members, friends, and other health care providers are other obstacles. The only way to deal with this issue is to ask your patient directly about her fears and concerns regarding each prescription written.
Clinicians also need to address fears or prejudices they themselves may have about medication safety in pregnancy. These may arise from a single bad experience in caring for a pregnant woman, discomfort with uncertainty, or a belief that pregnant women should avoid any and all risks of exposures, even when the mother’s condition warrants pharmacologic treatment.
Being informed, both scientifically and about one’s own biases or tendencies, is an essential foundation for rational prescribing in pregnancy.
Is the problem affected by pregnancy, and how?
Pregnancy can affect many medical conditions, and in different ways. Conditions such as asthma, migraine headache, and cardiac arrhythmia are exacerbated in pregnancy, placing the mother and fetus at increased risk of morbidity. Conditions such as Graves disease and hypertension may improve as pregnancy progresses, and medications often can be withdrawn as the patient progresses further along in gestation.
Understanding the effect of pregnancy on a particular problem may help the clinician to make an informed decision about medication use in pregnancy.
How does the problem affect pregnancy?
Considering the risk of untreated disease to the pregnancy may help in decision-making.
Many medical conditions can negatively affect the development of the fetus. A glaring example is diabetes mellitus, with poor glycemic control being linked to congenital malformations, spontaneous abortion, and fetal demise. Chronic conditions with periodic exacerbations such as asthma or epilepsy place the fetus at increased risk during a flare-up.
Therefore, for chronic conditions, continuing maintenance therapy is best. Preconception counseling in such cases is crucial, so that a drug with adequate safety data can be substituted before pregnancy. In this way, any risk to the mother or the embryo from exacerbation of disease as such adjustments are made is avoided.
For conditions arising de novo in pregnancy, the underlying principle remains the same. Is the risk of pharmacotherapy more than the risk of untreated disease? Invariably, the answer to this question supports medication use, and an educated provider will be able to choose a treatment that is justifiable in most circumstances.
CHOOSING A MEDICATION
Fetal well-being depends on maternal well-being. It therefore helps to think of medication use in pregnancy as “justified or not” rather than “safe or not.” Table 3 lists some conditions commonly seen in pregnancy, selected drugs of choice that can be safely used for treating those conditions, and alternates that may be justified in some circumstances.5,6,14–18
GOOD PRACTICES WHEN PRESCRIBING IN PREGNANCY
Prescribing in pregnancy will be most successful when both the patient and the prescribing physician consider the fetal benefit gained from optimizing maternal health. Good prescribing practices to ensure optimum therapeutic benefit when caring for a pregnant patient are to:
- Involve the patient in decision-making. Recognize her concerns, worries, and preferences regarding her illness and its treatment.
- Inform the patient of the risk of an untreated medical condition, weighed against the risk of medication.
- Choose medications with the most available safety data. Let the patient know what resources you have referred to in choosing the medication.
- It is advisable to perform a search each time a prescription is written for a pregnant or lactating woman.
- When possible, have the discussion in the preconception period.
- Consider the dynamic physiology of gestation. Choose the right drug for the right trimester.
- Discuss the plan with the patient and other providers.
- Define clear criteria for when to discontinue the treatment.
- Addis A, Sharabi S, Bonati M. Risk classification systems for drug use during pregnancy: are they a reliable source of information? Drug Saf 2000; 23:245–253.
- US Food and Drug Administration (FDA). Pregnancy and lactation labeling. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/Labeling/ucm093307.htm. Accessed April 4, 2014.
- Lagoy CT, Joshi N, Cragan JD, Rasmussen SA. Medication use during pregnancy and lactation: an urgent call for public health action. J Womens Health (Larchmt) 2005; 14:104–109.
- Clinical Teratology Website. University of Washington. http://depts.washington.edu/terisweb/teris/. Accessed April 4, 2014.
- REPROTOX, An Online Reproductive Toxicology Resource. Reproductive Toxicology Center. www.reprotox.org. Accessed April 4, 2014.
- REPRORISK. Micromedex, Inc. www.micromedex.com/products/reprorisk. Accessed April 4, 2014.
- Shepard TH. Catalog of teratogenic agents. 13th ed. Baltimore, MD: Johns Hopkins University Press; 2010.
- Briggs GG, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation: A reference guide to fetal and neonatal risk. Philadelphia, PA: Lippincott Williams & Wilkins; 2011.
- Koren G. Medication safety in pregnancy and breastfeeding. McGraw-Hill Professional Publishing; 2007.
- Friedman JM, Polifka JE. Teratogenic effects of drugs: A resource for clinicians (TERIS). Baltimore, MD: Johns Hopkins University Press; 2000.
- MotherToBaby. www.mothertobaby.org. Accessed April 4, 2014.
- Dunlop AL, Gardiner PM, Shellhaas CS, Menard MK, McDiarmid MA. The clinical content of preconception care: the use of medications and supplements among women of reproductive age. Am J Obstet Gynecol 2008; 199(suppl 2):S367–S372.
- Ciarkowski SL, Stalburg CM. Medication safety in obstetrics and gynecology. Clin Obstet Gynecol 2010; 53:482–499.
- Koren G, Pastuszak A, Ito S. Drugs in pregnancy. N Engl J Med 1998; 338:1128–1137.
- Lambert K, Holt RI. The use of insulin analogues in pregnancy. Diabetes Obes Metab 2013; 15:888–900.
- Chan WS, Anand S, Ginsberg JS. Anticoagulation of pregnant women with mechanical heart valves: a systematic review of the literature. Arch Intern Med 2000; 160:191–196.
- Nagler M, Haslauer M, Wuillemin WA. Fondaparinux—data on efficacy and safety in special situations. Thromb Res 2012; 129:407–417.
- Kweder SL, Powrie RO. Prescribing in pregnancy: a practical approach. In:Powrie RO, Greene M, Camann W, editors. De Swiet’s Medical disorders in Obstetric Practice. 5th ed. Hoboken, NJ: Wiley-Blackwell; 2010:633–640.
- Addis A, Sharabi S, Bonati M. Risk classification systems for drug use during pregnancy: are they a reliable source of information? Drug Saf 2000; 23:245–253.
- US Food and Drug Administration (FDA). Pregnancy and lactation labeling. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/Labeling/ucm093307.htm. Accessed April 4, 2014.
- Lagoy CT, Joshi N, Cragan JD, Rasmussen SA. Medication use during pregnancy and lactation: an urgent call for public health action. J Womens Health (Larchmt) 2005; 14:104–109.
- Clinical Teratology Website. University of Washington. http://depts.washington.edu/terisweb/teris/. Accessed April 4, 2014.
- REPROTOX, An Online Reproductive Toxicology Resource. Reproductive Toxicology Center. www.reprotox.org. Accessed April 4, 2014.
- REPRORISK. Micromedex, Inc. www.micromedex.com/products/reprorisk. Accessed April 4, 2014.
- Shepard TH. Catalog of teratogenic agents. 13th ed. Baltimore, MD: Johns Hopkins University Press; 2010.
- Briggs GG, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation: A reference guide to fetal and neonatal risk. Philadelphia, PA: Lippincott Williams & Wilkins; 2011.
- Koren G. Medication safety in pregnancy and breastfeeding. McGraw-Hill Professional Publishing; 2007.
- Friedman JM, Polifka JE. Teratogenic effects of drugs: A resource for clinicians (TERIS). Baltimore, MD: Johns Hopkins University Press; 2000.
- MotherToBaby. www.mothertobaby.org. Accessed April 4, 2014.
- Dunlop AL, Gardiner PM, Shellhaas CS, Menard MK, McDiarmid MA. The clinical content of preconception care: the use of medications and supplements among women of reproductive age. Am J Obstet Gynecol 2008; 199(suppl 2):S367–S372.
- Ciarkowski SL, Stalburg CM. Medication safety in obstetrics and gynecology. Clin Obstet Gynecol 2010; 53:482–499.
- Koren G, Pastuszak A, Ito S. Drugs in pregnancy. N Engl J Med 1998; 338:1128–1137.
- Lambert K, Holt RI. The use of insulin analogues in pregnancy. Diabetes Obes Metab 2013; 15:888–900.
- Chan WS, Anand S, Ginsberg JS. Anticoagulation of pregnant women with mechanical heart valves: a systematic review of the literature. Arch Intern Med 2000; 160:191–196.
- Nagler M, Haslauer M, Wuillemin WA. Fondaparinux—data on efficacy and safety in special situations. Thromb Res 2012; 129:407–417.
- Kweder SL, Powrie RO. Prescribing in pregnancy: a practical approach. In:Powrie RO, Greene M, Camann W, editors. De Swiet’s Medical disorders in Obstetric Practice. 5th ed. Hoboken, NJ: Wiley-Blackwell; 2010:633–640.
KEY POINTS
- There is no protective physiologic barrier between the maternal and fetal environments.
- The gestational stage may determine the effect of a medication on the fetus.
- The physiologic changes of pregnancy affect the pharmacokinetics of medications.
- Sole reliance on the US Food and Drug Administration’s pregnancy safety category may be inadequate.
- Key questions: Is the problem self-limited or amenable to nonpharmacologic management? How do the patient’s (and provider’s) presumptions affect decisions about this medication in pregnancy? How does pregnancy affect the problem, and how does the problem affect pregnancy?
Chronic obstructive pulmonary disease: An update for the primary physician
Chronic obstructive pulmonary disease (COPD) has seen several changes in its assessment and treatment in recent years, reflecting advances in our understanding of this common and serious disease.
This review updates busy practitioners on the major advances, including new assessment tools and new therapies.
COMMON AND INCREASING
COPD is the third leading cause of death in the United States, behind heart disease and cancer,1 and of the top five (the others being stroke and accidents), it is the only one that increased in incidence between 2007 and 2010.2 The 11th leading cause of disability-adjusted life years worldwide in 2002, COPD is projected to become the seventh by the year 2030.3
CHARACTERIZED BY OBSTRUCTION
COPD is characterized by persistent and progressive airflow obstruction associated with chronic airway inflammation in response to noxious particles and gases. Disease of the small airways (inflammation, mucus plugging, and fibrosis) and parenchymal destruction (emphysema) limit the flow of air.
COPD is diagnosed by spirometry—specifically, a ratio of forced expiratory volume in 1 second to forced vital capacity (FEV1/FVC) of less than 0.7 after a bronchodilator is given. The severity of airflow limitation is revealed by the FEV1 as a percent of the predicted value.
Cigarette smoking is the major cause of COPD, but the prevalence of COPD is 6.6% in people who have never smoked, and one-fourth of COPD patients in the United States have never smoked.4
GOLDEN GOALS: FEWER SYMPTOMS, LOWER RISK
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) periodically issues evidence-based statements on how to prevent and treat COPD.
In its 2013 update,5 GOLD suggested two goals: improving symptoms and reducing the risk of death, exacerbations, progression of disease, and treatment-related adverse effects. The latter goal—reducing risk—is relatively new.
Exacerbations are acute inflammatory events superimposed on chronic inflammation. The inflammation is often brought on by infection6 and increases the risk of death7 and the risk of a faster decline in lung function.8
Exacerbations may characterize a phenotype of COPD. The Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) analyzed the frequency of COPD exacerbations and associated factors in 2,138 patients with COPD over a period of 3 years.9 Although patients with more severe obstruction tended to have more exacerbations, some patients appeared susceptible to exacerbations irrespective of the severity of obstruction. The best predictor of exacerbations was a history of exacerbations.
HOW DO I ASSESS A PATIENT WITH COPD ON PRESENTATION?
Markers of airflow obstruction such as the FEV1 do not correlate strongly with exertional capacity and health status in patients with COPD.10,11
The BODE index (body mass index, obstruction, dyspnea score, and exercise oximetry) takes into account the multidimensional nature of COPD. It performs better than the FEV1 in predicting the risk of death.12 The propensity for exacerbations and comorbidities further modulates outcome.
Assessing symptoms
The modified British Medical Research Council (mMRC) dyspnea scale, based on work by Fletcher in 1952,13 has five grades, numbered 0 through 4:
- Grade 0—Breathless with strenuous exercise only
- Grade 1—Breathless when hurrying on level ground or walking up a slight hill
- Grade 2—Walks slower than people of the same age on level ground because of shortness of breath or has to stop when walking at own pace on level ground
- Grade 3—Stops for breath after walking about 100 yards or after a few minutes on level ground
- Grade 4—Too breathless to leave the house or breathless when dressing or undressing.
Grade 2 or higher separates symptomatic from asymptomatic COPD.
The COPD Assessment Test (CAT) (www.catestonline.org) is a proprietary questionnaire. Patients use a 6-point scale (numbered 0 though 5) to rate eight symptoms (cough, mucus production, chest tightness, shortness of breath on exertion, limitations in home activities, lack of confidence leaving the home, poor sleep, and lack of energy). A total score of 10 or higher is abnormal.
Four GOLD groups

The new GOLD guidelines (Table 1)5 define four groups of patients according to their severity of airflow obstruction, symptoms, and exacerbation history:
- Group A—fewer symptoms, low risk: Fewer symptoms (“less symptoms,” as worded in the guidelines) means a CAT score less than 10 or an mMRC grade less than 2; “low risk” means no more than one exacerbation per year and an FEV1 of at least 50%
- Group B—more symptoms, low risk: “More symptoms” means a CAT score of 10 or more or an mMRC grade of 2 or more
- Group C—fewer symptoms, high risk: “High risk” means two or more exacerbations per year or an FEV1 less than 50%
- Group D—more symptoms, high risk.
Thus, a patient with an FEV1 of 60% (moderate airflow limitation) who has had one exacerbation during the past year and a CAT score of 8 would be in group A. In contrast, a patient who has an FEV1 of 40% (severe airflow limitation), no history of exacerbations, and a CAT score of 20 would be in group D.
Updated GOLD guidelines suggest utilizing a stepwise approach to treatment, akin to asthma management guidelines, based on patient grouping.5
How accurate is the new GOLD system?
Although practical and suited for use in primary care, the new GOLD system is arbitrary and has not been thoroughly studied, and may therefore need refinement.
Lange et al14 compared the new GOLD system with the previous one in 6,628 patients with COPD. As anticipated, the new system was better at predicting exacerbations, as it incorporates a history of exacerbations in stratification. The presence of symptoms (as determined by an mMRC grade ≥ 2) was a marker of mortality risk that distinguished group A from group B, and group C from group D. Surprisingly, the rate of death was higher in group B (more symptoms, low risk) than in group C (fewer symptoms, high risk).
Notably, most patients in group C qualified for this group because of the severity of airflow obstruction, not because of a history of exacerbations. Therefore, patients whose symptoms are out of proportion to the severity of obstruction may be at higher risk of death, possibly because of comorbidities such as cardiovascular disease.15 Patients who qualified for groups C and D by having both a history of frequent exacerbations (≥ 2 per year) and symptoms rather than either one alone had a higher risk of death in 3 years.
Similarly, the symptom-assessment tool that is used—ie, the mMRC grade or the CAT score—also makes a difference.
The Health-Related Quality of Life in COPD in Europe Study16 retrospectively analyzed data from 1,817 patients to determine whether the cutoff points for symptoms as assessed by mMRC grade and CAT score were equivalent. Although the mMRC grade correlated well with overall health status, the cutoff mMRC grade of 2 or higher did not correspond to a CAT score of 10 or higher, classifying patients with health status impairment as asymptomatic (mean weighted kappa 0.626). The two tools agreed much better when the cutoff was set at an mMRC grade of 1 or higher (mean weighted kappa 0.792).16
Although assessment schemes continue to evolve as data accumulate, we believe the new system is a welcome initiative that reflects the changing notions of COPD.
Comorbidities matter
Another shift is the recognition that certain comorbidities increase the risk of death. In 1,664 patients with COPD who were followed for 51 months, 12 distinct comorbidities were associated with a higher risk of death after multivariate analysis.17
The COTE index (COPD-Specific Comorbidity Test) is based on these findings. It awards points as follows:
- 6 points for cancer of the lung, esophagus, pancreas, or breast, or for anxiety
- 2 points for all other cancers, liver cirrhosis, atrial fibrillation or flutter, diabetes with neuropathy, or pulmonary fibrosis
- 1 point for congestive heart failure, gastric or duodenal ulcer, or coronary artery disease.
A COTE index score of 4 or higher was associated with a risk of death 2.2 times higher in each quartile of the BODE index.
We strongly recommend being aware of comorbidities in COPD patients, particularly when symptoms are out of proportion to the severity of obstruction.
SHOULD I USE ANTIBIOTICS TO TREAT ALL COPD EXACERBATIONS?
Infections are thought to cause more than 80% of acute exacerbations of COPD.
Anthonisen et al,18 in a landmark trial, found broad-spectrum antibiotics to be most helpful if the patient had at least two of the three cardinal symptoms of COPD exacerbation (ie, shortness of breath, increase in sputum volume, and sputum purulence). Antibiotics decreased the rate of treatment failure and led to a more rapid clinical resolution of exacerbation. However, they did not help patients who had milder exacerbations.
Antibiotics may nevertheless have a role in ambulatory patients with mild to moderate COPD who present with exacerbations characterized by one or more cardinal symptoms.
Llor et al,19 in a multicenter randomized double-blind placebo-controlled trial in Spain, concluded that amoxicillin clavulanate (Augmentin) led to higher clinical cure rates and longer time to the next exacerbation in these patients. Most of the benefit was in patients with more symptoms, consistent with the results of the study by Anthonisen et al.18
There is also strong evidence to support the use of antibiotics in addition to systemic corticosteroids in hospitalized patients with acute exacerbations of COPD. A 7-day course of doxycycline (Vibramycin) added to a standard regimen of corticosteroids was associated with higher rates of clinical and microbiological cure on day 10 of the exacerbation.20 In a large retrospective cohort study in 84,621 hospitalized patients with COPD exacerbations, fewer of those who received antibiotics needed mechanical ventilation, died, or were readmitted.21 Although sicker patients received antibiotics more frequently, their mortality rate was lower than in those who did not receive antibiotics, who were presumably less sick.
A meta-analysis confirmed the salutary effect of antibiotics in inpatients and particularly those admitted to the intensive care unit.22 Mortality rates and hospital length of stay were not affected in patients who were not in intensive care.
Biomarkers such as procalcitonin might help reduce the unnecessary use of antibiotics. Stolz et al23 conducted a randomized controlled trial in which they based the decision to give antibiotics on a threshold procalcitonin level of at least 1 μg/L in hospitalized patients with COPD exacerbation. The rate of antibiotic use was reduced by more than 40% in the procalcitonin group without any difference in clinical outcomes, 6-month exacerbation rate, or rehospitalization compared with controls. Nonstandardized procalcitonin assays are a possible barrier to the widespread adoption of this threshold.
Comment. In general, we recommend antibiotics for hospitalized patients with COPD exacerbation and look forward to confirmatory data that support the use of biomarkers. For outpatients, we find the Anthonisen criteria useful for decision-making at the point of care.
ARE THERE ANY NEW INTERVENTIONS TO PREVENT COPD EXACERBATIONS?
Macrolides
Macrolides have a proven role in managing chronic suppurative respiratory diseases such as cystic fibrosis24 and diffuse panbronchiolitis.25 Since they are beneficial at lower doses than those used to treat infection, the mechanism may be anti-inflammatory rather than antimicrobial.
Albert et al26 assigned 1,142 patients who had had a COPD exacerbation within a year before enrollment or who were on home oxygen therapy to receive azithromycin (Zithromax) 250 mg daily or placebo.25 The azithromycin group had fewer acute exacerbations (hazard ratio 0.73, 95% CI 0.63–0.84, P < .001), and more patients in the azithromycin group achieved clinically significant improvements in quality of life, ie, a reduction in the St. George’s Respiratory Questionnaire (SGRQ) score of at least 4 points (43% vs 36%, P = .03). Adverse events that were more common in the azithromycin group were hearing loss (25% vs 20%) and macrolide-resistant strains in nasopharyngeal secretions (81% vs 41%). In subgroup analysis, the benefit in terms of reducing exacerbations was greater in patients over age 65, patients on home oxygen, and patients with moderate or severe obstruction compared with those with very severe obstruction.

Comment. Macrolides are a valuable addition to the agents available for preventing COPD exacerbation (Table 2), but their role is still uncertain. Potential topics of research are whether these drugs have a role in patients already on preventive regimens, whether they would have a greater effect in distinct patient populations (eg, patients who have two or more exacerbations per year), and whether their broader use would lead to a change in the resident flora in the community.
Clinicians should exercise caution in the use of azithromycin in light of recent concern about associated cardiac morbidity and death. All patients should undergo electrocardiography to assess the QTc interval before starting treatment, as in the trial by Albert et al.26
Phosphodiesterase inhibitors

Roflumilast (Daliresp) is an oral phosphodiesterase 4 inhibitor approved for treating exacerbations and symptoms of chronic bronchitis in patients with severe COPD (Table 3). Phosphodiesterase 4, one of the 11 isoforms of the enzyme, is found in immune and inflammatory cells and promotes inflammatory responses. Roflumilast has anti-inflammatory properties but no acute bronchodilatory effect.27 Several phase 3 trials found the compound to have beneficial effects.
Calverley et al28 performed two placebo-controlled double-blind trials in outpatients with the clinical diagnosis of COPD who had chronic cough; increased sputum production; at least one recorded exacerbation requiring corticosteroids or hospitalization, or both; and an FEV1 of 50% or less. Patients were randomized to receive roflumilast 500 μg once a day (n = 1,537) or placebo (n = 1,554) for 1 year. The rate of moderate to severe exacerbations was 1.17 per year with roflumilast vs 1.37 with placebo (P < .0003). Adverse events were significantly more common with roflumilast and were related to the known side effects of the drug, namely, diarrhea, weight loss, decreased appetite, and nausea.
Fabbri et al29 performed two other placebo-controlled double-blind multicenter trials, studying the combinations of roflumilast with salmeterol (Serevent) and roflumilast with tiotropium (Spiriva) compared with placebo in 1,676 patients with COPD who had post-bronchodilator FEV1 values of 40% to 70% of predicted. The mean prebronchodilator FEV1 improved by 49 mL (P < .0001) in the salmeterol-plus-roflumilast trial and by 80 mL (P < .0001) in the tiotropium-plus-roflumilast trial compared with placebo. Fewer patients on roflumilast had exacerbations of any severity in both trials (risk ratio 0.82, P = .0419 and risk ratio 0.75, P = .0169, respectively).
No trial has yet addressed whether roflumilast is better than the combination of a long-acting muscarinic antagonist and a beta agonist, or whether roflumilast can be substituted for inhaled corticosteroids in a new triple-therapy combination. Clinicians should also be aware of psychiatric side effects of roflumilast, which include depression and, possibly, suicide.
ARE THERE ANY NEW BRONCHODILATORS FOR PATIENTS WITH COPD?
Long-acting muscarinic antagonists

Reversible airflow obstruction and mucus secretion are determined by the vagal cholinergic tone in patients with COPD.30 Antagonism of cholinergic (muscarinic) receptors results in bronchodilation and reduction in mucus production. Consequently, inhaled anticholinergic agents are the first-line therapy for COPD (Table 4).
Tiotropium bromide is a long-acting antimuscarinic approved in 2002 by the US Food and Drug Administration (FDA). The UPLIFT trial (Understanding Potential Long-Term Impacts on Function With Tiotropium)31 enrolled 5,993 patients with a mean FEV1 of 48% of predicted. Over a 4-year follow-up, significant improvements in mean FEV1 values (ranging from 87 mL to 103 mL before bronchodilation and 47 mL to 65 mL after bronchodilation, P < .001) in the tiotropium group were observed compared with placebo. The rate of the primary end point—the rate of decline in mean FEV1—was not different between tiotropium and placebo. However, there were important salutary effects in multiple clinical end points in the tiotropium group. Health-related quality of life as measured by the SGRQ improved in a clinically significant manner (> 4 points) in favor of tiotropium in a higher proportion of patients (45% vs 36%, P < .001). Tiotropium reduced the number of exacerbations per patient year (0.73 ± 0.02 vs 0.85 ± 0.02, RR = 0.86 (95% CI 0.81–0.91), P < .001) and the risk of respiratory failure (RR = 0.67, 95% CI 0.51–0.89). There were no significant differences in the risk of myocardial infarction, stroke, or pneumonia.
Aclidinium bromide (Tudorza Pressair) is a long-acting antimuscarinic recently approved by the FDA. Compared with tiotropium, it has a slightly faster onset of action and a considerably shorter half-life (29 hours vs 64 hours).32,33 Its dosage is 400 μg twice daily by inhalation. It provides sustained bronchodilation over 24 hours and may have a favorable side-effect profile, because it undergoes rapid hydrolysis in human plasma.34
ACCORD COPD I35 and ATTAIN,36 two phase 3 trials in patients with moderate-to severe COPD, found that twice-daily aclidinium was associated with statistically and clinically significant (> 100 mL) improvements in trough and peak FEV1 compared with placebo. Health status (assessed by SGRQ) and dyspnea (assessed by transitional dyspnea index) also improved significantly. However, improvements beyond minimum clinically significant thresholds were achieved only with 400 μg twice-daily dosing.
To date, no study has evaluated the impact of aclidinium on COPD exacerbation as a primary end point. Fewer moderate to severe exacerbations were reported in an earlier 52-week study of once-daily aclidinium (ACCLAIM COPD II) but not in ACCLAIM COPD I.37
Aclidinium may offer an advantage over tiotropium in patients who have nocturnal symptoms. Twice-daily aclidinium 400 μg was associated with superior FEV1 area-under-the-curve values compared with placebo and tiotropium, the difference mostly owing to improved nocturnal profile.38
Long-acting beta-2 agonists
Stimulation of airway beta-2 receptors relaxes smooth muscles and consequently dilates bronchioles via a cyclic adenosine monophosphate-dependent pathway.39
Short-acting beta-2 agonists such as albuterol and terbutaline have long been used as rescue medications for obstructive lung disease. Long-acting beta-2 agonists provide sustained bronchodilation and are therefore more efficacious as maintenance medications. Salmeterol, formoterol (Foradil), and arformoterol (Brovana) are long-acting beta-2 agonists in clinical use that are taken twice daily.
Clinical studies indicate that use of long-acting beta-2 agonists leads to significant improvements in FEV1,40–42 dynamic hyperinflation, exercise tolerance,43,44 and dyspnea.45,46 These drugs have also been associated with significant improvements in health-related quality of life and in the frequency of exacerbations.47–49
In patients with asthma, long-acting beta agonists may increase the risk of death.50 In contrast, in patients with COPD, they appear to offer a survival advantage when used in combination with inhaled corticosteroids,51 and some argue that this benefit is entirely from the long-acting beta agonist (a 17% reduction in mortality) rather than the inhaled corticosteroid (0% reduction in mortality).52

Indacaterol (Arcapta), approved in July 2011, is the first once-daily beta agonist or “ultra-long-acting” beta agonist (Table 5). Possibly because it has a high affinity for the lipid raft domain of the cell membrane where beta-2 receptors are coupled to second messengers,53 the drug has a 24-hour duration of action.
In patients with COPD, inhaled indacaterol 150 μg once daily improved airflow obstruction and health status as measured by SGRQ compared with salmeterol 50 μg twice daily and placebo.54 At the higher dose of 300 μg daily, the 52-week INVOLVE trial55 demonstrated early and more sustained improvement in FEV1 compared with placebo and formoterol. In this study, a lower exacerbation rate than with placebo was also noted. The drug has also shown equivalent bronchodilator efficacy at 150 μg and 300 μg daily dosing compared with tiotropium.56
The benefits of a longer-acting bronchodilator such as indacaterol are likely mediated by smoothing out airway bronchomotor tone over 24 hours without the dips seen with shorter-acting agents and by improvement of the FEV1 trough before the subsequent dose is due, aptly named “pharmacologic stenting.”57 Once-daily dosing should also foster better adherence. The safety profile appears excellent with no increase in cardiovascular or cerebrovascular events compared with placebo.58
The FDA approved the 75-μg daily dose instead of the higher doses used in the studies mentioned above. This decision was based on the observation that there appeared to be a flattened dose-response in patients with more severe COPD, with no further improvement in trough FEV1 at higher doses.59
DOES VITAMIN D SUPPLEMENTATION HAVE A ROLE IN COPD MANAGEMENT?
Vitamin D is vital for calcium and phosphate metabolism and bone health. Low vitamin D levels are associated with diminished leg strength and falls in the elderly.60 Osteoporosis, preventable with vitamin D and calcium supplementation, is linked to thoracic vertebral fracture and consequent reduced lung function.61,62

Patients with COPD are at higher risk of vitamin D deficiency, and more so if they also are obese, have advanced airflow obstruction, are depressed, or smoke.62 Therefore, there are sound reasons to look for vitamin D deficiency in patients with COPD and to treat it if the 25-hydroxyvitamin D level is less than 10 ng/ mL (Table 6).
Vitamin D may also have antimicrobial and immunomodulatory effects.63 Since COPD exacerbations are frequently caused by infection, it was hypothesized that vitamin D supplementation might reduce the rate of exacerbations.
In a study in 182 patients with moderate to very severe COPD and a history of recent exacerbations, high-dose vitamin D supplementation (100,000 IU) was given every 4 weeks for 1 year.64 There were no differences in the time to first exacerbation, in the rate of exacerbation, hospitalization, or death, or in quality of life between the placebo and intervention groups. However, subgroup analysis indicated that, in those with severe vitamin D deficiency at baseline, the exacerbation rate was reduced by more than 40%.
Comment. We recommend screening for vitamin D deficiency in patients with COPD. Supplementation is appropriate in those with low levels, but data indicate no role in those with normal levels.
WHAT ARE THE NONPHARMACOLOGIC APPROACHES TO COPD TREATMENT?
Noninvasive positive-pressure ventilation

Nocturnal noninvasive positive-pressure ventilation may be beneficial in patients with severe COPD, daytime hypercapnia, and nocturnal hypoventilation, particularly if higher inspiratory pressures are selected (Table 7).65,66
For instance, a randomized controlled trial of noninvasive positive-pressure ventilation plus long-term oxygen therapy compared with long-term oxygen therapy alone in hypercapnic COPD demonstrated a survival benefit in favor of ventilation (hazard ratio 0.6).67
In another randomized trial,68 settings that aimed to maximally reduce Paco2 (mean inspiratory positive airway pressure 29 cm H2O with a backup rate of 17.5/min) were compared with low-intensity positive airway pressure (mean inspiratory positive airway pressure 14 cm H2O, backup rate 8/min). The high inspiratory pressures increased the daily use of ventilation by 3.6 hours per day and improved exercise-related dyspnea, daytime Paco2, FEV1, vital capacity, and health-related quality of life66 without disrupting sleep quality.68
Caveats are that acclimation to the high pressures was achieved in the hospital, and the high pressures were associated with a significant increase in air leaks.66
Comments. Whether high-pressure noninvasive positive-pressure ventilation can be routinely implemented and adopted in the outpatient setting, and whether it is associated with a survival advantage remains to be determined. The advantages of noninvasive positive-pressure ventilation in the setting of hypercapnic COPD appear to augment those of pulmonary rehabilitation, with improved quality of life, gas exchange, and exercise tolerance, and a slower decline of lung function.69
Pulmonary rehabilitation

Pulmonary rehabilitation is a multidisciplinary approach to managing COPD (Table 8).
Patients participate in three to five supervised sessions per week, each lasting 3 to 4 hours, for 6 to 12 weeks. Less-frequent sessions may not be effective. For instance, in a randomized trial, exercising twice a week was not enough.70 Additionally, a program lasting longer than 12 weeks produced more sustained benefits than shorter programs.71
A key component is an exercise protocol centered on the lower extremities (walking, cycling, treadmill), with progressive exercise intensity to a target of about 60% to 80% of the maximal exercise tolerance,72 though more modest targets of about 50% can also be beneficial.73
Exercise should be tailored to the desired outcome. For instance, training of the upper arms may help with activities of daily living. In one study, unsupported (against gravity) arm training improved upper-extremity function more than supported arm training (by ergometer).74 Ventilatory muscle training is less common, as most randomized trials have not shown conclusive evidence of benefit. Current guidelines do not recommend routine inspiratory muscle training.71
Even though indices of pulmonary function do not improve after an exercise program, randomized trials have shown that pulmonary rehabilitation improves exercise capacity, dyspnea, and health-related quality of life; improves cost-effectiveness of health care utilization; and provides psychosocial benefits that often exceed those of other therapies. Although there is no significant evidence of whether pulmonary rehabilitation improves survival in patients with COPD,71 an observational study documented improvements in BODE scores as well as a reduction in respiratory mortality rates in patients undergoing pulmonary rehabilitation.75
A limitation of pulmonary rehabilitation is that endurance and psychological and cognitive function decline significantly if exercise is not maintained. However, the role of a maintenance program is uncertain, with long-term benefits considered modest.71
Lung-volume reduction surgery

Lung-volume reduction consists of surgical wedge resections of emphysematous areas of the lung (Table 9).
The National Emphysema Treatment Trial76 randomized 1,218 patients to undergo either lung-volume reduction surgery or maximal medical therapy. Surgery improved survival, quality of life, and dyspnea in patients with upper-lobe emphysema and a low exercise capacity (corresponding to < 40 watts for men or < 25 watts for women in the maximal power achieved on cycle ergometry). While conferring no survival benefit in patients with upper-lobe-predominant emphysema and high exercise capacity, this surgery is likely to improve exercise capacity and quality of life in this subset of patients.
Importantly, the procedure is associated with a lower survival rate in patients with an FEV1 lower than 20%, homogeneous emphysema, a diffusing capacity of the lung for carbon monoxide lower than 20%, non-upper-lobe emphysema, or high baseline exercise capacity.
The proposed mechanisms of improvement of lung function include placing the diaphragm in a position with better mechanical advantage, reducing overall lung volume, better size-matching between the lungs and chest cavity, and restoring elastic recoil.76,77
Ongoing trials aim to replicate the success of lung-volume reduction using nonsurgical bronchoscopic techniques with one-way valves, coils, biologic sealants, thermal ablation, and airway stents.
- Miniño AM, Xu J, Kochanek KD. Deaths: preliminary data for 2008. National Vital Statistics Reports 2010; 59:1–52. http://www.cdc.gov/nchs/data/nvsr/nvsr59/nvsr59_02.pdf. Accessed April 10, 2014.
- Murphy SL, Xu J, Kochanek KD. Deaths: preliminary data from 2010. National Vital Statistics Reports 2012; 60:1–51. http://www.cdc.gov/nchs/data/nvsr/nvsr60/nvsr60_04.pdf. Accessed April 10, 2014.
- Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 2006; 3:e442.
- Behrendt CE. Mild and moderate-to-severe COPD in nonsmokers: distinct demographic profiles. Chest 2005; 128:1239–1244.
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). http://www.goldcopd.org/Guidelines/guidelines-resources.html. Accessed April 10, 2014.
- Papi A, Bellettato CM, Braccioni F, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med 2006; 173:1114–1121.
- Soler-Cataluña JJ, Martínez-García MA, Román Sánchez P, Salcedo E, Navarro M, Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax 2005; 60:925–931.
- Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002; 57:847–852.
- Hurst JR, Vestbo J, Anzueto A, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med 2010; 363:1128–1138.
- Cooper CB. The connection between chronic obstructive pulmonary disease symptoms and hyperinflation and its impact on exercise and function. Am J Med 2006; 119(suppl 1):21–31.
- Jones PW. Issues concerning health-related quality of life in COPD. Chest 1995; 107(suppl):187S–193S.
- Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med 2004; 350:1005–1012.
- Fletcher CM. The clinical diagnosis of pulmonary emphysema—an experimental study. J Royal Soc Med 1952; 45:577–584.
- Lange P, Marott JL, Vestbo J, et al. Prediction of the clinical course of chronic obstructive pulmonary disease, using the new GOLD classification: a study of the general population. Am J Respir Crit Care Med 2012; 186:975–981.
- Hurst J. Phenotype-based care in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:935–936.
- Jones PW, Adamek L, Nadeau G, Banik N. Comparisons of health status scores with MRC grades in COPD: implications for the GOLD 2011 classification. Eur Respir J 2013; 42:647–654.
- Divo M, Cote C, de Torres JP, et al; BODE Collaborative Group. Comorbidities and risk of mortality in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:155–161.
- Anthonisen NR, Manfreda J, Warren CP, Hershfield ES, Harding GK, Nelson NA. Antibiotic therapy in exacerbations of chronic obstructive pulmonary disease. Ann Intern Med 1987; 106:196–204.
- Llor C, Moragas A, Hernández S, Bayona C, Miravitlles M. Efficacy of antibiotic therapy for acute exacerbations of mild to moderate chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:716–723.
- Daniels JM, Snijders D, de Graaff CS, Vlaspolder F, Jansen HM, Boersma WG. Antibiotics in addition to systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010; 181:150–157.
- Rothberg MB, Pekow PS, Lahti M, Brody O, Skiest DJ, Lindenauer PK. Antibiotic therapy and treatment failure in patients hospitalized for acute exacerbations of chronic obstructive pulmonary disease. JAMA 2010; 303:2035–2042.
- Vollenweider DJ, Jarrett H, Steurer-Stey CA, Garcia-Aymerich J, Puhan MA. Antibiotics for exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2012; 12:CD010257.
- Stolz D, Christ-Crain M, Bingisser R, et al. Antibiotic treatment of exacerbations of COPD: a randomized, controlled trial comparing procalcitonin-guidance with standard therapy. Chest 2007; 131:9–19.
- Wolter J, Seeney S, Bell S, Bowler S, Masel P, McCormack J. Effect of long term treatment with azithromycin on disease parameters in cystic fibrosis: a randomised trial. Thorax 2002; 57:212–216.
- Kudoh S, Azuma A, Yamamoto M, Izumi T, Ando M. Improvement of survival in patients with diffuse panbronchiolitis treated with low-dose erythromycin. Am J Respir Crit Care Med 1998; 157:1829–1832.
- Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
- Gross NJ, Giembycz MA, Rennard SI. Treatment of chronic obstructive pulmonary disease with roflumilast, a new phosphodiesterase 4 inhibitor. COPD 2010; 7:141–153.
- Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ; M2-124 and M2-125 study groups. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 2009; 374:685–694.
- Fabbri LM, Calverley PM, Izquierdo-Alonso JL, et al; M2-127 and M2-128 study groups. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with long acting bronchodilators: two randomised clinical trials. Lancet 2009; 374:695–703.
- Gross NJ, Skorodin MS. Anticholinergic, antimuscarinic bronchodilators. Am Rev Respir Dis 1984; 129:856–870.
- Tashkin DP, Celli B, Senn S, et al; UPLIFT Study Investigators. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008; 359:1543–1554.
- Cazzola M. Aclidinium bromide, a novel long-acting muscarinic M3 antagonist for the treatment of COPD. Curr Opin Investig Drugs 2009; 10:482–490.
- Gavaldà A, Miralpeix M, Ramos I, et al. Characterization of aclidinium bromide, a novel inhaled muscarinic antagonist, with long duration of action and a favorable pharmacological profile. J Pharmacol Exp Ther 2009; 331:740–751.
- Alagha K, Bourdin A, Tummino C, Chanez P. An update on the efficacy and safety of aclidinium bromide in patients with COPD. Ther Adv Respir Dis 2011; 5:19–28.
- Kerwin EM, D’Urzo AD, Gelb AF, Lakkis H, Garcia Gil E, Caracta CF; ACCORD I study investigators. Efficacy and safety of a 12-week treatment with twice-daily aclidinium bromide in COPD patients (ACCORD COPD I). COPD 2012; 9:90–101.
- Jones PW, Singh D, Bateman ED, et al. Efficacy and safety of twice-daily aclidinium bromide in COPD patients: the ATTAIN study. Eur Respir J 2012; 40:830–836.
- Jones PW, Rennard SI, Agusti A, et al. Efficacy and safety of once-daily aclidinium in chronic obstructive pulmonary disease. Respir Res 2011; 12:55.
- Fuhr R, Magnussen H, Sarem K, et al. Efficacy of aclidinium bromide 400 μg twice daily compared with placebo and tiotropium in patients with moderate to severe COPD. Chest 2012; 141:745–752.
- Tashkin DP, Fabbri LM. Long-acting beta-agonists in the management of chronic obstructive pulmonary disease: current and future agents. Respir Res 2010; 11:149.
- Mahler DA, Donohue JF, Barbee RA, et al. Efficacy of salmeterol xinafoate in the treatment of COPD. Chest 1999; 115:957–965.
- Campbell M, Eliraz A, Johansson G, et al. Formoterol for maintenance and as-needed treatment of chronic obstructive pulmonary disease. Respir Med 2005; 99:1511–1520.
- Hanrahan JP, Hanania NA, Calhoun WJ, Sahn SA, Sciarappa K, Baumgartner RA. Effect of nebulized arformoterol on airway function in COPD: results from two randomized trials. COPD 2008; 5:25–34.
- Neder JA, Fuld JP, Overend T, et al. Effects of formoterol on exercise tolerance in severely disabled patients with COPD. Respir Med 2007; 101:2056–2064.
- O’Donnell DE, Voduc N, Fitzpatrick M, Webb KA. Effect of salmeterol on the ventilatory response to exercise in chronic obstructive pulmonary disease. Eur Respir J 2004; 24:86–94.
- Rennard SI, Anderson W, ZuWallack R, et al. Use of a long-acting inhaled beta 2-adrenergic agonist, salmeterol xinafoate, in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 163:1087–1092.
- Schultze-Werninghaus G. Multicenter 1-year trial on formoterol, a new long-acting beta 2-agonist, in chronic obstructive airway disease. Lung 1990; 168(suppl):83–89.
- Rodrigo GJ, Nannini LJ, Rodríguez-Roisin R. Safety of long-acting beta-agonists in stable COPD: a systematic review. Chest 2008; 133:1079–1087.
- Baker WL, Baker EL, Coleman CI. Pharmacologic treatments for chronic obstructive pulmonary disease: a mixed-treatment comparison meta-analysis. Pharmacotherapy 2009; 29:891–905.
- Rodrigo GJ, Castro-Rodriguez JA, Plaza V. Safety and efficacy of combined long-acting beta-agonists and inhaled corticosteroids vs long-acting beta-agonists monotherapy for stable COPD: a systematic review. Chest 2009; 136:1029–1038.
- Nelson HS, Weiss ST, Bleecker ER, Yancey SW, Dorinsky PM; SMART Study Group. The Salmeterol Multicenter Asthma Research Trial: a comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol. Chest 2006; 129:15–26.
- Calverley PM, Anderson JA, Celli B, et al; TORCH investigators. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007; 356:775–789.
- Suissa S, Ernst P, Vandemheen KL, Aaron SD. Methodological issues in therapeutic trials of COPD. Eur Respir J 2008; 31:927–933.
- Lombardi D, Cuenoud B, Krämer SD. Lipid membrane interactions of indacaterol and salmeterol: do they influence their pharmacological properties? Eur J Pharm Sci 2009; 38:533–547.
- Kornmann O, Dahl R, Centanni S, et al; INLIGHT-2 (Indacaterol Efficacy Evaluation Using 150-μg Doses with COPD Patients) study investigators. Once-daily indacaterol versus twice-daily salmeterol for COPD: a placebo-controlled comparison. Eur Respir J 2011; 37:273–279.
- Dahl R, Chung KF, Buhl R, et al; INVOLVE (INdacaterol: Value in COPD: Longer Term Validation of Efficacy and Safety) Study Investigators. Efficacy of a new once-daily long-acting inhaled beta2-agonist indacaterol versus twice-daily formoterol in COPD. Thorax 2010; 65:473–479.
- Donohue JF, Fogarty C, Lötvall J, et al; INHANCE Study Investigators. Once-daily bronchodilators for chronic obstructive pulmonary disease: indacaterol versus tiotropium. Am J Respir Crit Care Med 2010; 182:155–162.
- Beeh KM, Beier J. The short, the long and the “ultra-long”: why duration of bronchodilator action matters in chronic obstructive pulmonary disease. Adv Ther 2010; 27:150–159.
- Worth H, Chung KF, Felser JM, Hu H, Rueegg P. Cardio- and cerebrovascular safety of indacaterol vs formoterol, salmeterol, tiotropium and placebo in COPD. Respir Med 2011; 105:571–579.
- Chowdhury BA, Seymour SM, Michele TM, Durmowicz AG, Liu D, Rosebraugh CJ. The risks and benefits of indacaterol--the FDA’s review. N Engl J Med 2011; 365:2247–2249.
- Bischoff-Ferrari HA, Dawson-Hughes B, Willett WC, et al. Effect of vitamin D on falls: a meta-analysis. JAMA 2004; 291:1999–2006.
- Leech JA, Dulberg C, Kellie S, Pattee L, Gay J. Relationship of lung function to severity of osteoporosis in women. Am Rev Respir Dis 1990; 141:68–71.
- Persson LJ, Aanerud M, Hiemstra PS, Hardie JA, Bakke PS, Eagan TM. Chronic obstructive pulmonary disease is associated with low levels of vitamin D. PLoS One 2012; 7:e38934.
- Janssens W, Lehouck A, Carremans C, Bouillon R, Mathieu C, Decramer M. Vitamin D beyond bones in chronic obstructive pulmonary disease: time to act. Am J Respir Crit Care Med 2009; 179:630–636.
- Lehouck A, Mathieu C, Carremans C, et al. High doses of vitamin D to reduce exacerbations in chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med 2012; 156:105–114.
- Casanova C, Celli BR, Tost L, et al. Long-term controlled trial of nocturnal nasal positive pressure ventilation in patients with severe COPD. Chest 2000; 118:1582–1590.
- Dreher M, Storre JH, Schmoor C, Windisch W. High-intensity versus low-intensity non-invasive ventilation in patients with stable hypercapnic COPD: a randomised crossover trial. Thorax 2010; 65:303–308.
- McEvoy RD, Pierce RJ, Hillman D, et al; Australian trial of non-invasive Ventilation in Chronic Airflow Limitation (AVCAL) Study Group. Nocturnal non-invasive nasal ventilation in stable hypercapnic COPD: a randomised controlled trial. Thorax 2009; 64:561–566.
- Dreher M, Ekkernkamp E, Walterspacher S, et al. Noninvasive ventilation in COPD: impact of inspiratory pressure levels on sleep quality. Chest 2011; 140:939–945.
- Duiverman ML, Wempe JB, Bladder G, et al. Two-year home-based nocturnal noninvasive ventilation added to rehabilitation in chronic obstructive pulmonary disease patients: a randomized controlled trial. Respir Res 2011; 12:112.
- Ringbaek TJ, Broendum E, Hemmingsen L, et al. Rehabilitation of patients with chronic obstructive pulmonary disease. Exercise twice a week is not sufficient! Respir Med 2000; 94:150–154.
- Ries AL, Bauldoff GS, Carlin BW, et al. Pulmonary Rehabilitation: Joint ACCP/AACVPR Evidence-Based Clinical Practice Guidelines. Chest 2007; 131(suppl):4S–42S.
- Vallet G, Ahmaïdi S, Serres I, et al. Comparison of two training programmes in chronic airway limitation patients: standardized versus individualized protocols. Eur Respir J 1997; 10:114–122.
- Vogiatzis I, Williamson AF, Miles J, Taylor IK. Physiological response to moderate exercise workloads in a pulmonary rehabilitation program in patients with varying degrees of airflow obstruction. Chest 1999; 116:1200–1207.
- Martinez FJ, Vogel PD, Dupont DN, Stanopoulos I, Gray A, Beamis JF. Supported arm exercise vs unsupported arm exercise in the rehabilitation of patients with severe chronic airflow obstruction. Chest 1993; 103:1397–1402.
- Cote CG, Celli BR. Pulmonary rehabilitation and the BODE index in COPD. Eur Respir J 2005; 26:630–636.
- Fishman A, Martinez F, Naunheim K, et al; National Emphysema Treatment Trial Research Group. A randomized trial comparing lung-volume-reduction surgery with medical therapy for severe emphysema. N Engl J Med 2003; 348:2059–2073.
- Naunheim KS, Wood DE, Mohsenifar Z, et al; National Emphysema Treatment Trial Research Group. Long-term follow-up of patients receiving lung-volume-reduction surgery versus medical therapy for severe emphysema by the National Emphysema Treatment Trial Research Group. Ann Thorac Surg 2006; 82:431–443.
Chronic obstructive pulmonary disease (COPD) has seen several changes in its assessment and treatment in recent years, reflecting advances in our understanding of this common and serious disease.
This review updates busy practitioners on the major advances, including new assessment tools and new therapies.
COMMON AND INCREASING
COPD is the third leading cause of death in the United States, behind heart disease and cancer,1 and of the top five (the others being stroke and accidents), it is the only one that increased in incidence between 2007 and 2010.2 The 11th leading cause of disability-adjusted life years worldwide in 2002, COPD is projected to become the seventh by the year 2030.3
CHARACTERIZED BY OBSTRUCTION
COPD is characterized by persistent and progressive airflow obstruction associated with chronic airway inflammation in response to noxious particles and gases. Disease of the small airways (inflammation, mucus plugging, and fibrosis) and parenchymal destruction (emphysema) limit the flow of air.
COPD is diagnosed by spirometry—specifically, a ratio of forced expiratory volume in 1 second to forced vital capacity (FEV1/FVC) of less than 0.7 after a bronchodilator is given. The severity of airflow limitation is revealed by the FEV1 as a percent of the predicted value.
Cigarette smoking is the major cause of COPD, but the prevalence of COPD is 6.6% in people who have never smoked, and one-fourth of COPD patients in the United States have never smoked.4
GOLDEN GOALS: FEWER SYMPTOMS, LOWER RISK
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) periodically issues evidence-based statements on how to prevent and treat COPD.
In its 2013 update,5 GOLD suggested two goals: improving symptoms and reducing the risk of death, exacerbations, progression of disease, and treatment-related adverse effects. The latter goal—reducing risk—is relatively new.
Exacerbations are acute inflammatory events superimposed on chronic inflammation. The inflammation is often brought on by infection6 and increases the risk of death7 and the risk of a faster decline in lung function.8
Exacerbations may characterize a phenotype of COPD. The Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) analyzed the frequency of COPD exacerbations and associated factors in 2,138 patients with COPD over a period of 3 years.9 Although patients with more severe obstruction tended to have more exacerbations, some patients appeared susceptible to exacerbations irrespective of the severity of obstruction. The best predictor of exacerbations was a history of exacerbations.
HOW DO I ASSESS A PATIENT WITH COPD ON PRESENTATION?
Markers of airflow obstruction such as the FEV1 do not correlate strongly with exertional capacity and health status in patients with COPD.10,11
The BODE index (body mass index, obstruction, dyspnea score, and exercise oximetry) takes into account the multidimensional nature of COPD. It performs better than the FEV1 in predicting the risk of death.12 The propensity for exacerbations and comorbidities further modulates outcome.
Assessing symptoms
The modified British Medical Research Council (mMRC) dyspnea scale, based on work by Fletcher in 1952,13 has five grades, numbered 0 through 4:
- Grade 0—Breathless with strenuous exercise only
- Grade 1—Breathless when hurrying on level ground or walking up a slight hill
- Grade 2—Walks slower than people of the same age on level ground because of shortness of breath or has to stop when walking at own pace on level ground
- Grade 3—Stops for breath after walking about 100 yards or after a few minutes on level ground
- Grade 4—Too breathless to leave the house or breathless when dressing or undressing.
Grade 2 or higher separates symptomatic from asymptomatic COPD.
The COPD Assessment Test (CAT) (www.catestonline.org) is a proprietary questionnaire. Patients use a 6-point scale (numbered 0 though 5) to rate eight symptoms (cough, mucus production, chest tightness, shortness of breath on exertion, limitations in home activities, lack of confidence leaving the home, poor sleep, and lack of energy). A total score of 10 or higher is abnormal.
Four GOLD groups
The new GOLD guidelines (Table 1)5 define four groups of patients according to their severity of airflow obstruction, symptoms, and exacerbation history:
- Group A—fewer symptoms, low risk: Fewer symptoms (“less symptoms,” as worded in the guidelines) means a CAT score less than 10 or an mMRC grade less than 2; “low risk” means no more than one exacerbation per year and an FEV1 of at least 50%
- Group B—more symptoms, low risk: “More symptoms” means a CAT score of 10 or more or an mMRC grade of 2 or more
- Group C—fewer symptoms, high risk: “High risk” means two or more exacerbations per year or an FEV1 less than 50%
- Group D—more symptoms, high risk.
Thus, a patient with an FEV1 of 60% (moderate airflow limitation) who has had one exacerbation during the past year and a CAT score of 8 would be in group A. In contrast, a patient who has an FEV1 of 40% (severe airflow limitation), no history of exacerbations, and a CAT score of 20 would be in group D.
Updated GOLD guidelines suggest utilizing a stepwise approach to treatment, akin to asthma management guidelines, based on patient grouping.5
How accurate is the new GOLD system?
Although practical and suited for use in primary care, the new GOLD system is arbitrary and has not been thoroughly studied, and may therefore need refinement.
Lange et al14 compared the new GOLD system with the previous one in 6,628 patients with COPD. As anticipated, the new system was better at predicting exacerbations, as it incorporates a history of exacerbations in stratification. The presence of symptoms (as determined by an mMRC grade ≥ 2) was a marker of mortality risk that distinguished group A from group B, and group C from group D. Surprisingly, the rate of death was higher in group B (more symptoms, low risk) than in group C (fewer symptoms, high risk).
Notably, most patients in group C qualified for this group because of the severity of airflow obstruction, not because of a history of exacerbations. Therefore, patients whose symptoms are out of proportion to the severity of obstruction may be at higher risk of death, possibly because of comorbidities such as cardiovascular disease.15 Patients who qualified for groups C and D by having both a history of frequent exacerbations (≥ 2 per year) and symptoms rather than either one alone had a higher risk of death in 3 years.
Similarly, the symptom-assessment tool that is used—ie, the mMRC grade or the CAT score—also makes a difference.
The Health-Related Quality of Life in COPD in Europe Study16 retrospectively analyzed data from 1,817 patients to determine whether the cutoff points for symptoms as assessed by mMRC grade and CAT score were equivalent. Although the mMRC grade correlated well with overall health status, the cutoff mMRC grade of 2 or higher did not correspond to a CAT score of 10 or higher, classifying patients with health status impairment as asymptomatic (mean weighted kappa 0.626). The two tools agreed much better when the cutoff was set at an mMRC grade of 1 or higher (mean weighted kappa 0.792).16
Although assessment schemes continue to evolve as data accumulate, we believe the new system is a welcome initiative that reflects the changing notions of COPD.
Comorbidities matter
Another shift is the recognition that certain comorbidities increase the risk of death. In 1,664 patients with COPD who were followed for 51 months, 12 distinct comorbidities were associated with a higher risk of death after multivariate analysis.17
The COTE index (COPD-Specific Comorbidity Test) is based on these findings. It awards points as follows:
- 6 points for cancer of the lung, esophagus, pancreas, or breast, or for anxiety
- 2 points for all other cancers, liver cirrhosis, atrial fibrillation or flutter, diabetes with neuropathy, or pulmonary fibrosis
- 1 point for congestive heart failure, gastric or duodenal ulcer, or coronary artery disease.
A COTE index score of 4 or higher was associated with a risk of death 2.2 times higher in each quartile of the BODE index.
We strongly recommend being aware of comorbidities in COPD patients, particularly when symptoms are out of proportion to the severity of obstruction.
SHOULD I USE ANTIBIOTICS TO TREAT ALL COPD EXACERBATIONS?
Infections are thought to cause more than 80% of acute exacerbations of COPD.
Anthonisen et al,18 in a landmark trial, found broad-spectrum antibiotics to be most helpful if the patient had at least two of the three cardinal symptoms of COPD exacerbation (ie, shortness of breath, increase in sputum volume, and sputum purulence). Antibiotics decreased the rate of treatment failure and led to a more rapid clinical resolution of exacerbation. However, they did not help patients who had milder exacerbations.
Antibiotics may nevertheless have a role in ambulatory patients with mild to moderate COPD who present with exacerbations characterized by one or more cardinal symptoms.
Llor et al,19 in a multicenter randomized double-blind placebo-controlled trial in Spain, concluded that amoxicillin clavulanate (Augmentin) led to higher clinical cure rates and longer time to the next exacerbation in these patients. Most of the benefit was in patients with more symptoms, consistent with the results of the study by Anthonisen et al.18
There is also strong evidence to support the use of antibiotics in addition to systemic corticosteroids in hospitalized patients with acute exacerbations of COPD. A 7-day course of doxycycline (Vibramycin) added to a standard regimen of corticosteroids was associated with higher rates of clinical and microbiological cure on day 10 of the exacerbation.20 In a large retrospective cohort study in 84,621 hospitalized patients with COPD exacerbations, fewer of those who received antibiotics needed mechanical ventilation, died, or were readmitted.21 Although sicker patients received antibiotics more frequently, their mortality rate was lower than in those who did not receive antibiotics, who were presumably less sick.
A meta-analysis confirmed the salutary effect of antibiotics in inpatients and particularly those admitted to the intensive care unit.22 Mortality rates and hospital length of stay were not affected in patients who were not in intensive care.
Biomarkers such as procalcitonin might help reduce the unnecessary use of antibiotics. Stolz et al23 conducted a randomized controlled trial in which they based the decision to give antibiotics on a threshold procalcitonin level of at least 1 μg/L in hospitalized patients with COPD exacerbation. The rate of antibiotic use was reduced by more than 40% in the procalcitonin group without any difference in clinical outcomes, 6-month exacerbation rate, or rehospitalization compared with controls. Nonstandardized procalcitonin assays are a possible barrier to the widespread adoption of this threshold.
Comment. In general, we recommend antibiotics for hospitalized patients with COPD exacerbation and look forward to confirmatory data that support the use of biomarkers. For outpatients, we find the Anthonisen criteria useful for decision-making at the point of care.
ARE THERE ANY NEW INTERVENTIONS TO PREVENT COPD EXACERBATIONS?
Macrolides
Macrolides have a proven role in managing chronic suppurative respiratory diseases such as cystic fibrosis24 and diffuse panbronchiolitis.25 Since they are beneficial at lower doses than those used to treat infection, the mechanism may be anti-inflammatory rather than antimicrobial.
Albert et al26 assigned 1,142 patients who had had a COPD exacerbation within a year before enrollment or who were on home oxygen therapy to receive azithromycin (Zithromax) 250 mg daily or placebo.25 The azithromycin group had fewer acute exacerbations (hazard ratio 0.73, 95% CI 0.63–0.84, P < .001), and more patients in the azithromycin group achieved clinically significant improvements in quality of life, ie, a reduction in the St. George’s Respiratory Questionnaire (SGRQ) score of at least 4 points (43% vs 36%, P = .03). Adverse events that were more common in the azithromycin group were hearing loss (25% vs 20%) and macrolide-resistant strains in nasopharyngeal secretions (81% vs 41%). In subgroup analysis, the benefit in terms of reducing exacerbations was greater in patients over age 65, patients on home oxygen, and patients with moderate or severe obstruction compared with those with very severe obstruction.
Comment. Macrolides are a valuable addition to the agents available for preventing COPD exacerbation (Table 2), but their role is still uncertain. Potential topics of research are whether these drugs have a role in patients already on preventive regimens, whether they would have a greater effect in distinct patient populations (eg, patients who have two or more exacerbations per year), and whether their broader use would lead to a change in the resident flora in the community.
Clinicians should exercise caution in the use of azithromycin in light of recent concern about associated cardiac morbidity and death. All patients should undergo electrocardiography to assess the QTc interval before starting treatment, as in the trial by Albert et al.26
Phosphodiesterase inhibitors
Roflumilast (Daliresp) is an oral phosphodiesterase 4 inhibitor approved for treating exacerbations and symptoms of chronic bronchitis in patients with severe COPD (Table 3). Phosphodiesterase 4, one of the 11 isoforms of the enzyme, is found in immune and inflammatory cells and promotes inflammatory responses. Roflumilast has anti-inflammatory properties but no acute bronchodilatory effect.27 Several phase 3 trials found the compound to have beneficial effects.
Calverley et al28 performed two placebo-controlled double-blind trials in outpatients with the clinical diagnosis of COPD who had chronic cough; increased sputum production; at least one recorded exacerbation requiring corticosteroids or hospitalization, or both; and an FEV1 of 50% or less. Patients were randomized to receive roflumilast 500 μg once a day (n = 1,537) or placebo (n = 1,554) for 1 year. The rate of moderate to severe exacerbations was 1.17 per year with roflumilast vs 1.37 with placebo (P < .0003). Adverse events were significantly more common with roflumilast and were related to the known side effects of the drug, namely, diarrhea, weight loss, decreased appetite, and nausea.
Fabbri et al29 performed two other placebo-controlled double-blind multicenter trials, studying the combinations of roflumilast with salmeterol (Serevent) and roflumilast with tiotropium (Spiriva) compared with placebo in 1,676 patients with COPD who had post-bronchodilator FEV1 values of 40% to 70% of predicted. The mean prebronchodilator FEV1 improved by 49 mL (P < .0001) in the salmeterol-plus-roflumilast trial and by 80 mL (P < .0001) in the tiotropium-plus-roflumilast trial compared with placebo. Fewer patients on roflumilast had exacerbations of any severity in both trials (risk ratio 0.82, P = .0419 and risk ratio 0.75, P = .0169, respectively).
No trial has yet addressed whether roflumilast is better than the combination of a long-acting muscarinic antagonist and a beta agonist, or whether roflumilast can be substituted for inhaled corticosteroids in a new triple-therapy combination. Clinicians should also be aware of psychiatric side effects of roflumilast, which include depression and, possibly, suicide.
ARE THERE ANY NEW BRONCHODILATORS FOR PATIENTS WITH COPD?
Long-acting muscarinic antagonists
Reversible airflow obstruction and mucus secretion are determined by the vagal cholinergic tone in patients with COPD.30 Antagonism of cholinergic (muscarinic) receptors results in bronchodilation and reduction in mucus production. Consequently, inhaled anticholinergic agents are the first-line therapy for COPD (Table 4).
Tiotropium bromide is a long-acting antimuscarinic approved in 2002 by the US Food and Drug Administration (FDA). The UPLIFT trial (Understanding Potential Long-Term Impacts on Function With Tiotropium)31 enrolled 5,993 patients with a mean FEV1 of 48% of predicted. Over a 4-year follow-up, significant improvements in mean FEV1 values (ranging from 87 mL to 103 mL before bronchodilation and 47 mL to 65 mL after bronchodilation, P < .001) in the tiotropium group were observed compared with placebo. The rate of the primary end point—the rate of decline in mean FEV1—was not different between tiotropium and placebo. However, there were important salutary effects in multiple clinical end points in the tiotropium group. Health-related quality of life as measured by the SGRQ improved in a clinically significant manner (> 4 points) in favor of tiotropium in a higher proportion of patients (45% vs 36%, P < .001). Tiotropium reduced the number of exacerbations per patient year (0.73 ± 0.02 vs 0.85 ± 0.02, RR = 0.86 (95% CI 0.81–0.91), P < .001) and the risk of respiratory failure (RR = 0.67, 95% CI 0.51–0.89). There were no significant differences in the risk of myocardial infarction, stroke, or pneumonia.
Aclidinium bromide (Tudorza Pressair) is a long-acting antimuscarinic recently approved by the FDA. Compared with tiotropium, it has a slightly faster onset of action and a considerably shorter half-life (29 hours vs 64 hours).32,33 Its dosage is 400 μg twice daily by inhalation. It provides sustained bronchodilation over 24 hours and may have a favorable side-effect profile, because it undergoes rapid hydrolysis in human plasma.34
ACCORD COPD I35 and ATTAIN,36 two phase 3 trials in patients with moderate-to severe COPD, found that twice-daily aclidinium was associated with statistically and clinically significant (> 100 mL) improvements in trough and peak FEV1 compared with placebo. Health status (assessed by SGRQ) and dyspnea (assessed by transitional dyspnea index) also improved significantly. However, improvements beyond minimum clinically significant thresholds were achieved only with 400 μg twice-daily dosing.
To date, no study has evaluated the impact of aclidinium on COPD exacerbation as a primary end point. Fewer moderate to severe exacerbations were reported in an earlier 52-week study of once-daily aclidinium (ACCLAIM COPD II) but not in ACCLAIM COPD I.37
Aclidinium may offer an advantage over tiotropium in patients who have nocturnal symptoms. Twice-daily aclidinium 400 μg was associated with superior FEV1 area-under-the-curve values compared with placebo and tiotropium, the difference mostly owing to improved nocturnal profile.38
Long-acting beta-2 agonists
Stimulation of airway beta-2 receptors relaxes smooth muscles and consequently dilates bronchioles via a cyclic adenosine monophosphate-dependent pathway.39
Short-acting beta-2 agonists such as albuterol and terbutaline have long been used as rescue medications for obstructive lung disease. Long-acting beta-2 agonists provide sustained bronchodilation and are therefore more efficacious as maintenance medications. Salmeterol, formoterol (Foradil), and arformoterol (Brovana) are long-acting beta-2 agonists in clinical use that are taken twice daily.
Clinical studies indicate that use of long-acting beta-2 agonists leads to significant improvements in FEV1,40–42 dynamic hyperinflation, exercise tolerance,43,44 and dyspnea.45,46 These drugs have also been associated with significant improvements in health-related quality of life and in the frequency of exacerbations.47–49
In patients with asthma, long-acting beta agonists may increase the risk of death.50 In contrast, in patients with COPD, they appear to offer a survival advantage when used in combination with inhaled corticosteroids,51 and some argue that this benefit is entirely from the long-acting beta agonist (a 17% reduction in mortality) rather than the inhaled corticosteroid (0% reduction in mortality).52
Indacaterol (Arcapta), approved in July 2011, is the first once-daily beta agonist or “ultra-long-acting” beta agonist (Table 5). Possibly because it has a high affinity for the lipid raft domain of the cell membrane where beta-2 receptors are coupled to second messengers,53 the drug has a 24-hour duration of action.
In patients with COPD, inhaled indacaterol 150 μg once daily improved airflow obstruction and health status as measured by SGRQ compared with salmeterol 50 μg twice daily and placebo.54 At the higher dose of 300 μg daily, the 52-week INVOLVE trial55 demonstrated early and more sustained improvement in FEV1 compared with placebo and formoterol. In this study, a lower exacerbation rate than with placebo was also noted. The drug has also shown equivalent bronchodilator efficacy at 150 μg and 300 μg daily dosing compared with tiotropium.56
The benefits of a longer-acting bronchodilator such as indacaterol are likely mediated by smoothing out airway bronchomotor tone over 24 hours without the dips seen with shorter-acting agents and by improvement of the FEV1 trough before the subsequent dose is due, aptly named “pharmacologic stenting.”57 Once-daily dosing should also foster better adherence. The safety profile appears excellent with no increase in cardiovascular or cerebrovascular events compared with placebo.58
The FDA approved the 75-μg daily dose instead of the higher doses used in the studies mentioned above. This decision was based on the observation that there appeared to be a flattened dose-response in patients with more severe COPD, with no further improvement in trough FEV1 at higher doses.59
DOES VITAMIN D SUPPLEMENTATION HAVE A ROLE IN COPD MANAGEMENT?
Vitamin D is vital for calcium and phosphate metabolism and bone health. Low vitamin D levels are associated with diminished leg strength and falls in the elderly.60 Osteoporosis, preventable with vitamin D and calcium supplementation, is linked to thoracic vertebral fracture and consequent reduced lung function.61,62
Patients with COPD are at higher risk of vitamin D deficiency, and more so if they also are obese, have advanced airflow obstruction, are depressed, or smoke.62 Therefore, there are sound reasons to look for vitamin D deficiency in patients with COPD and to treat it if the 25-hydroxyvitamin D level is less than 10 ng/ mL (Table 6).
Vitamin D may also have antimicrobial and immunomodulatory effects.63 Since COPD exacerbations are frequently caused by infection, it was hypothesized that vitamin D supplementation might reduce the rate of exacerbations.
In a study in 182 patients with moderate to very severe COPD and a history of recent exacerbations, high-dose vitamin D supplementation (100,000 IU) was given every 4 weeks for 1 year.64 There were no differences in the time to first exacerbation, in the rate of exacerbation, hospitalization, or death, or in quality of life between the placebo and intervention groups. However, subgroup analysis indicated that, in those with severe vitamin D deficiency at baseline, the exacerbation rate was reduced by more than 40%.
Comment. We recommend screening for vitamin D deficiency in patients with COPD. Supplementation is appropriate in those with low levels, but data indicate no role in those with normal levels.
WHAT ARE THE NONPHARMACOLOGIC APPROACHES TO COPD TREATMENT?
Noninvasive positive-pressure ventilation
Nocturnal noninvasive positive-pressure ventilation may be beneficial in patients with severe COPD, daytime hypercapnia, and nocturnal hypoventilation, particularly if higher inspiratory pressures are selected (Table 7).65,66
For instance, a randomized controlled trial of noninvasive positive-pressure ventilation plus long-term oxygen therapy compared with long-term oxygen therapy alone in hypercapnic COPD demonstrated a survival benefit in favor of ventilation (hazard ratio 0.6).67
In another randomized trial,68 settings that aimed to maximally reduce Paco2 (mean inspiratory positive airway pressure 29 cm H2O with a backup rate of 17.5/min) were compared with low-intensity positive airway pressure (mean inspiratory positive airway pressure 14 cm H2O, backup rate 8/min). The high inspiratory pressures increased the daily use of ventilation by 3.6 hours per day and improved exercise-related dyspnea, daytime Paco2, FEV1, vital capacity, and health-related quality of life66 without disrupting sleep quality.68
Caveats are that acclimation to the high pressures was achieved in the hospital, and the high pressures were associated with a significant increase in air leaks.66
Comments. Whether high-pressure noninvasive positive-pressure ventilation can be routinely implemented and adopted in the outpatient setting, and whether it is associated with a survival advantage remains to be determined. The advantages of noninvasive positive-pressure ventilation in the setting of hypercapnic COPD appear to augment those of pulmonary rehabilitation, with improved quality of life, gas exchange, and exercise tolerance, and a slower decline of lung function.69
Pulmonary rehabilitation
Pulmonary rehabilitation is a multidisciplinary approach to managing COPD (Table 8).
Patients participate in three to five supervised sessions per week, each lasting 3 to 4 hours, for 6 to 12 weeks. Less-frequent sessions may not be effective. For instance, in a randomized trial, exercising twice a week was not enough.70 Additionally, a program lasting longer than 12 weeks produced more sustained benefits than shorter programs.71
A key component is an exercise protocol centered on the lower extremities (walking, cycling, treadmill), with progressive exercise intensity to a target of about 60% to 80% of the maximal exercise tolerance,72 though more modest targets of about 50% can also be beneficial.73
Exercise should be tailored to the desired outcome. For instance, training of the upper arms may help with activities of daily living. In one study, unsupported (against gravity) arm training improved upper-extremity function more than supported arm training (by ergometer).74 Ventilatory muscle training is less common, as most randomized trials have not shown conclusive evidence of benefit. Current guidelines do not recommend routine inspiratory muscle training.71
Even though indices of pulmonary function do not improve after an exercise program, randomized trials have shown that pulmonary rehabilitation improves exercise capacity, dyspnea, and health-related quality of life; improves cost-effectiveness of health care utilization; and provides psychosocial benefits that often exceed those of other therapies. Although there is no significant evidence of whether pulmonary rehabilitation improves survival in patients with COPD,71 an observational study documented improvements in BODE scores as well as a reduction in respiratory mortality rates in patients undergoing pulmonary rehabilitation.75
A limitation of pulmonary rehabilitation is that endurance and psychological and cognitive function decline significantly if exercise is not maintained. However, the role of a maintenance program is uncertain, with long-term benefits considered modest.71
Lung-volume reduction surgery
Lung-volume reduction consists of surgical wedge resections of emphysematous areas of the lung (Table 9).
The National Emphysema Treatment Trial76 randomized 1,218 patients to undergo either lung-volume reduction surgery or maximal medical therapy. Surgery improved survival, quality of life, and dyspnea in patients with upper-lobe emphysema and a low exercise capacity (corresponding to < 40 watts for men or < 25 watts for women in the maximal power achieved on cycle ergometry). While conferring no survival benefit in patients with upper-lobe-predominant emphysema and high exercise capacity, this surgery is likely to improve exercise capacity and quality of life in this subset of patients.
Importantly, the procedure is associated with a lower survival rate in patients with an FEV1 lower than 20%, homogeneous emphysema, a diffusing capacity of the lung for carbon monoxide lower than 20%, non-upper-lobe emphysema, or high baseline exercise capacity.
The proposed mechanisms of improvement of lung function include placing the diaphragm in a position with better mechanical advantage, reducing overall lung volume, better size-matching between the lungs and chest cavity, and restoring elastic recoil.76,77
Ongoing trials aim to replicate the success of lung-volume reduction using nonsurgical bronchoscopic techniques with one-way valves, coils, biologic sealants, thermal ablation, and airway stents.
Chronic obstructive pulmonary disease (COPD) has seen several changes in its assessment and treatment in recent years, reflecting advances in our understanding of this common and serious disease.
This review updates busy practitioners on the major advances, including new assessment tools and new therapies.
COMMON AND INCREASING
COPD is the third leading cause of death in the United States, behind heart disease and cancer,1 and of the top five (the others being stroke and accidents), it is the only one that increased in incidence between 2007 and 2010.2 The 11th leading cause of disability-adjusted life years worldwide in 2002, COPD is projected to become the seventh by the year 2030.3
CHARACTERIZED BY OBSTRUCTION
COPD is characterized by persistent and progressive airflow obstruction associated with chronic airway inflammation in response to noxious particles and gases. Disease of the small airways (inflammation, mucus plugging, and fibrosis) and parenchymal destruction (emphysema) limit the flow of air.
COPD is diagnosed by spirometry—specifically, a ratio of forced expiratory volume in 1 second to forced vital capacity (FEV1/FVC) of less than 0.7 after a bronchodilator is given. The severity of airflow limitation is revealed by the FEV1 as a percent of the predicted value.
Cigarette smoking is the major cause of COPD, but the prevalence of COPD is 6.6% in people who have never smoked, and one-fourth of COPD patients in the United States have never smoked.4
GOLDEN GOALS: FEWER SYMPTOMS, LOWER RISK
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) periodically issues evidence-based statements on how to prevent and treat COPD.
In its 2013 update,5 GOLD suggested two goals: improving symptoms and reducing the risk of death, exacerbations, progression of disease, and treatment-related adverse effects. The latter goal—reducing risk—is relatively new.
Exacerbations are acute inflammatory events superimposed on chronic inflammation. The inflammation is often brought on by infection6 and increases the risk of death7 and the risk of a faster decline in lung function.8
Exacerbations may characterize a phenotype of COPD. The Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) analyzed the frequency of COPD exacerbations and associated factors in 2,138 patients with COPD over a period of 3 years.9 Although patients with more severe obstruction tended to have more exacerbations, some patients appeared susceptible to exacerbations irrespective of the severity of obstruction. The best predictor of exacerbations was a history of exacerbations.
HOW DO I ASSESS A PATIENT WITH COPD ON PRESENTATION?
Markers of airflow obstruction such as the FEV1 do not correlate strongly with exertional capacity and health status in patients with COPD.10,11
The BODE index (body mass index, obstruction, dyspnea score, and exercise oximetry) takes into account the multidimensional nature of COPD. It performs better than the FEV1 in predicting the risk of death.12 The propensity for exacerbations and comorbidities further modulates outcome.
Assessing symptoms
The modified British Medical Research Council (mMRC) dyspnea scale, based on work by Fletcher in 1952,13 has five grades, numbered 0 through 4:
- Grade 0—Breathless with strenuous exercise only
- Grade 1—Breathless when hurrying on level ground or walking up a slight hill
- Grade 2—Walks slower than people of the same age on level ground because of shortness of breath or has to stop when walking at own pace on level ground
- Grade 3—Stops for breath after walking about 100 yards or after a few minutes on level ground
- Grade 4—Too breathless to leave the house or breathless when dressing or undressing.
Grade 2 or higher separates symptomatic from asymptomatic COPD.
The COPD Assessment Test (CAT) (www.catestonline.org) is a proprietary questionnaire. Patients use a 6-point scale (numbered 0 though 5) to rate eight symptoms (cough, mucus production, chest tightness, shortness of breath on exertion, limitations in home activities, lack of confidence leaving the home, poor sleep, and lack of energy). A total score of 10 or higher is abnormal.
Four GOLD groups
The new GOLD guidelines (Table 1)5 define four groups of patients according to their severity of airflow obstruction, symptoms, and exacerbation history:
- Group A—fewer symptoms, low risk: Fewer symptoms (“less symptoms,” as worded in the guidelines) means a CAT score less than 10 or an mMRC grade less than 2; “low risk” means no more than one exacerbation per year and an FEV1 of at least 50%
- Group B—more symptoms, low risk: “More symptoms” means a CAT score of 10 or more or an mMRC grade of 2 or more
- Group C—fewer symptoms, high risk: “High risk” means two or more exacerbations per year or an FEV1 less than 50%
- Group D—more symptoms, high risk.
Thus, a patient with an FEV1 of 60% (moderate airflow limitation) who has had one exacerbation during the past year and a CAT score of 8 would be in group A. In contrast, a patient who has an FEV1 of 40% (severe airflow limitation), no history of exacerbations, and a CAT score of 20 would be in group D.
Updated GOLD guidelines suggest utilizing a stepwise approach to treatment, akin to asthma management guidelines, based on patient grouping.5
How accurate is the new GOLD system?
Although practical and suited for use in primary care, the new GOLD system is arbitrary and has not been thoroughly studied, and may therefore need refinement.
Lange et al14 compared the new GOLD system with the previous one in 6,628 patients with COPD. As anticipated, the new system was better at predicting exacerbations, as it incorporates a history of exacerbations in stratification. The presence of symptoms (as determined by an mMRC grade ≥ 2) was a marker of mortality risk that distinguished group A from group B, and group C from group D. Surprisingly, the rate of death was higher in group B (more symptoms, low risk) than in group C (fewer symptoms, high risk).
Notably, most patients in group C qualified for this group because of the severity of airflow obstruction, not because of a history of exacerbations. Therefore, patients whose symptoms are out of proportion to the severity of obstruction may be at higher risk of death, possibly because of comorbidities such as cardiovascular disease.15 Patients who qualified for groups C and D by having both a history of frequent exacerbations (≥ 2 per year) and symptoms rather than either one alone had a higher risk of death in 3 years.
Similarly, the symptom-assessment tool that is used—ie, the mMRC grade or the CAT score—also makes a difference.
The Health-Related Quality of Life in COPD in Europe Study16 retrospectively analyzed data from 1,817 patients to determine whether the cutoff points for symptoms as assessed by mMRC grade and CAT score were equivalent. Although the mMRC grade correlated well with overall health status, the cutoff mMRC grade of 2 or higher did not correspond to a CAT score of 10 or higher, classifying patients with health status impairment as asymptomatic (mean weighted kappa 0.626). The two tools agreed much better when the cutoff was set at an mMRC grade of 1 or higher (mean weighted kappa 0.792).16
Although assessment schemes continue to evolve as data accumulate, we believe the new system is a welcome initiative that reflects the changing notions of COPD.
Comorbidities matter
Another shift is the recognition that certain comorbidities increase the risk of death. In 1,664 patients with COPD who were followed for 51 months, 12 distinct comorbidities were associated with a higher risk of death after multivariate analysis.17
The COTE index (COPD-Specific Comorbidity Test) is based on these findings. It awards points as follows:
- 6 points for cancer of the lung, esophagus, pancreas, or breast, or for anxiety
- 2 points for all other cancers, liver cirrhosis, atrial fibrillation or flutter, diabetes with neuropathy, or pulmonary fibrosis
- 1 point for congestive heart failure, gastric or duodenal ulcer, or coronary artery disease.
A COTE index score of 4 or higher was associated with a risk of death 2.2 times higher in each quartile of the BODE index.
We strongly recommend being aware of comorbidities in COPD patients, particularly when symptoms are out of proportion to the severity of obstruction.
SHOULD I USE ANTIBIOTICS TO TREAT ALL COPD EXACERBATIONS?
Infections are thought to cause more than 80% of acute exacerbations of COPD.
Anthonisen et al,18 in a landmark trial, found broad-spectrum antibiotics to be most helpful if the patient had at least two of the three cardinal symptoms of COPD exacerbation (ie, shortness of breath, increase in sputum volume, and sputum purulence). Antibiotics decreased the rate of treatment failure and led to a more rapid clinical resolution of exacerbation. However, they did not help patients who had milder exacerbations.
Antibiotics may nevertheless have a role in ambulatory patients with mild to moderate COPD who present with exacerbations characterized by one or more cardinal symptoms.
Llor et al,19 in a multicenter randomized double-blind placebo-controlled trial in Spain, concluded that amoxicillin clavulanate (Augmentin) led to higher clinical cure rates and longer time to the next exacerbation in these patients. Most of the benefit was in patients with more symptoms, consistent with the results of the study by Anthonisen et al.18
There is also strong evidence to support the use of antibiotics in addition to systemic corticosteroids in hospitalized patients with acute exacerbations of COPD. A 7-day course of doxycycline (Vibramycin) added to a standard regimen of corticosteroids was associated with higher rates of clinical and microbiological cure on day 10 of the exacerbation.20 In a large retrospective cohort study in 84,621 hospitalized patients with COPD exacerbations, fewer of those who received antibiotics needed mechanical ventilation, died, or were readmitted.21 Although sicker patients received antibiotics more frequently, their mortality rate was lower than in those who did not receive antibiotics, who were presumably less sick.
A meta-analysis confirmed the salutary effect of antibiotics in inpatients and particularly those admitted to the intensive care unit.22 Mortality rates and hospital length of stay were not affected in patients who were not in intensive care.
Biomarkers such as procalcitonin might help reduce the unnecessary use of antibiotics. Stolz et al23 conducted a randomized controlled trial in which they based the decision to give antibiotics on a threshold procalcitonin level of at least 1 μg/L in hospitalized patients with COPD exacerbation. The rate of antibiotic use was reduced by more than 40% in the procalcitonin group without any difference in clinical outcomes, 6-month exacerbation rate, or rehospitalization compared with controls. Nonstandardized procalcitonin assays are a possible barrier to the widespread adoption of this threshold.
Comment. In general, we recommend antibiotics for hospitalized patients with COPD exacerbation and look forward to confirmatory data that support the use of biomarkers. For outpatients, we find the Anthonisen criteria useful for decision-making at the point of care.
ARE THERE ANY NEW INTERVENTIONS TO PREVENT COPD EXACERBATIONS?
Macrolides
Macrolides have a proven role in managing chronic suppurative respiratory diseases such as cystic fibrosis24 and diffuse panbronchiolitis.25 Since they are beneficial at lower doses than those used to treat infection, the mechanism may be anti-inflammatory rather than antimicrobial.
Albert et al26 assigned 1,142 patients who had had a COPD exacerbation within a year before enrollment or who were on home oxygen therapy to receive azithromycin (Zithromax) 250 mg daily or placebo.25 The azithromycin group had fewer acute exacerbations (hazard ratio 0.73, 95% CI 0.63–0.84, P < .001), and more patients in the azithromycin group achieved clinically significant improvements in quality of life, ie, a reduction in the St. George’s Respiratory Questionnaire (SGRQ) score of at least 4 points (43% vs 36%, P = .03). Adverse events that were more common in the azithromycin group were hearing loss (25% vs 20%) and macrolide-resistant strains in nasopharyngeal secretions (81% vs 41%). In subgroup analysis, the benefit in terms of reducing exacerbations was greater in patients over age 65, patients on home oxygen, and patients with moderate or severe obstruction compared with those with very severe obstruction.
Comment. Macrolides are a valuable addition to the agents available for preventing COPD exacerbation (Table 2), but their role is still uncertain. Potential topics of research are whether these drugs have a role in patients already on preventive regimens, whether they would have a greater effect in distinct patient populations (eg, patients who have two or more exacerbations per year), and whether their broader use would lead to a change in the resident flora in the community.
Clinicians should exercise caution in the use of azithromycin in light of recent concern about associated cardiac morbidity and death. All patients should undergo electrocardiography to assess the QTc interval before starting treatment, as in the trial by Albert et al.26
Phosphodiesterase inhibitors
Roflumilast (Daliresp) is an oral phosphodiesterase 4 inhibitor approved for treating exacerbations and symptoms of chronic bronchitis in patients with severe COPD (Table 3). Phosphodiesterase 4, one of the 11 isoforms of the enzyme, is found in immune and inflammatory cells and promotes inflammatory responses. Roflumilast has anti-inflammatory properties but no acute bronchodilatory effect.27 Several phase 3 trials found the compound to have beneficial effects.
Calverley et al28 performed two placebo-controlled double-blind trials in outpatients with the clinical diagnosis of COPD who had chronic cough; increased sputum production; at least one recorded exacerbation requiring corticosteroids or hospitalization, or both; and an FEV1 of 50% or less. Patients were randomized to receive roflumilast 500 μg once a day (n = 1,537) or placebo (n = 1,554) for 1 year. The rate of moderate to severe exacerbations was 1.17 per year with roflumilast vs 1.37 with placebo (P < .0003). Adverse events were significantly more common with roflumilast and were related to the known side effects of the drug, namely, diarrhea, weight loss, decreased appetite, and nausea.
Fabbri et al29 performed two other placebo-controlled double-blind multicenter trials, studying the combinations of roflumilast with salmeterol (Serevent) and roflumilast with tiotropium (Spiriva) compared with placebo in 1,676 patients with COPD who had post-bronchodilator FEV1 values of 40% to 70% of predicted. The mean prebronchodilator FEV1 improved by 49 mL (P < .0001) in the salmeterol-plus-roflumilast trial and by 80 mL (P < .0001) in the tiotropium-plus-roflumilast trial compared with placebo. Fewer patients on roflumilast had exacerbations of any severity in both trials (risk ratio 0.82, P = .0419 and risk ratio 0.75, P = .0169, respectively).
No trial has yet addressed whether roflumilast is better than the combination of a long-acting muscarinic antagonist and a beta agonist, or whether roflumilast can be substituted for inhaled corticosteroids in a new triple-therapy combination. Clinicians should also be aware of psychiatric side effects of roflumilast, which include depression and, possibly, suicide.
ARE THERE ANY NEW BRONCHODILATORS FOR PATIENTS WITH COPD?
Long-acting muscarinic antagonists
Reversible airflow obstruction and mucus secretion are determined by the vagal cholinergic tone in patients with COPD.30 Antagonism of cholinergic (muscarinic) receptors results in bronchodilation and reduction in mucus production. Consequently, inhaled anticholinergic agents are the first-line therapy for COPD (Table 4).
Tiotropium bromide is a long-acting antimuscarinic approved in 2002 by the US Food and Drug Administration (FDA). The UPLIFT trial (Understanding Potential Long-Term Impacts on Function With Tiotropium)31 enrolled 5,993 patients with a mean FEV1 of 48% of predicted. Over a 4-year follow-up, significant improvements in mean FEV1 values (ranging from 87 mL to 103 mL before bronchodilation and 47 mL to 65 mL after bronchodilation, P < .001) in the tiotropium group were observed compared with placebo. The rate of the primary end point—the rate of decline in mean FEV1—was not different between tiotropium and placebo. However, there were important salutary effects in multiple clinical end points in the tiotropium group. Health-related quality of life as measured by the SGRQ improved in a clinically significant manner (> 4 points) in favor of tiotropium in a higher proportion of patients (45% vs 36%, P < .001). Tiotropium reduced the number of exacerbations per patient year (0.73 ± 0.02 vs 0.85 ± 0.02, RR = 0.86 (95% CI 0.81–0.91), P < .001) and the risk of respiratory failure (RR = 0.67, 95% CI 0.51–0.89). There were no significant differences in the risk of myocardial infarction, stroke, or pneumonia.
Aclidinium bromide (Tudorza Pressair) is a long-acting antimuscarinic recently approved by the FDA. Compared with tiotropium, it has a slightly faster onset of action and a considerably shorter half-life (29 hours vs 64 hours).32,33 Its dosage is 400 μg twice daily by inhalation. It provides sustained bronchodilation over 24 hours and may have a favorable side-effect profile, because it undergoes rapid hydrolysis in human plasma.34
ACCORD COPD I35 and ATTAIN,36 two phase 3 trials in patients with moderate-to severe COPD, found that twice-daily aclidinium was associated with statistically and clinically significant (> 100 mL) improvements in trough and peak FEV1 compared with placebo. Health status (assessed by SGRQ) and dyspnea (assessed by transitional dyspnea index) also improved significantly. However, improvements beyond minimum clinically significant thresholds were achieved only with 400 μg twice-daily dosing.
To date, no study has evaluated the impact of aclidinium on COPD exacerbation as a primary end point. Fewer moderate to severe exacerbations were reported in an earlier 52-week study of once-daily aclidinium (ACCLAIM COPD II) but not in ACCLAIM COPD I.37
Aclidinium may offer an advantage over tiotropium in patients who have nocturnal symptoms. Twice-daily aclidinium 400 μg was associated with superior FEV1 area-under-the-curve values compared with placebo and tiotropium, the difference mostly owing to improved nocturnal profile.38
Long-acting beta-2 agonists
Stimulation of airway beta-2 receptors relaxes smooth muscles and consequently dilates bronchioles via a cyclic adenosine monophosphate-dependent pathway.39
Short-acting beta-2 agonists such as albuterol and terbutaline have long been used as rescue medications for obstructive lung disease. Long-acting beta-2 agonists provide sustained bronchodilation and are therefore more efficacious as maintenance medications. Salmeterol, formoterol (Foradil), and arformoterol (Brovana) are long-acting beta-2 agonists in clinical use that are taken twice daily.
Clinical studies indicate that use of long-acting beta-2 agonists leads to significant improvements in FEV1,40–42 dynamic hyperinflation, exercise tolerance,43,44 and dyspnea.45,46 These drugs have also been associated with significant improvements in health-related quality of life and in the frequency of exacerbations.47–49
In patients with asthma, long-acting beta agonists may increase the risk of death.50 In contrast, in patients with COPD, they appear to offer a survival advantage when used in combination with inhaled corticosteroids,51 and some argue that this benefit is entirely from the long-acting beta agonist (a 17% reduction in mortality) rather than the inhaled corticosteroid (0% reduction in mortality).52
Indacaterol (Arcapta), approved in July 2011, is the first once-daily beta agonist or “ultra-long-acting” beta agonist (Table 5). Possibly because it has a high affinity for the lipid raft domain of the cell membrane where beta-2 receptors are coupled to second messengers,53 the drug has a 24-hour duration of action.
In patients with COPD, inhaled indacaterol 150 μg once daily improved airflow obstruction and health status as measured by SGRQ compared with salmeterol 50 μg twice daily and placebo.54 At the higher dose of 300 μg daily, the 52-week INVOLVE trial55 demonstrated early and more sustained improvement in FEV1 compared with placebo and formoterol. In this study, a lower exacerbation rate than with placebo was also noted. The drug has also shown equivalent bronchodilator efficacy at 150 μg and 300 μg daily dosing compared with tiotropium.56
The benefits of a longer-acting bronchodilator such as indacaterol are likely mediated by smoothing out airway bronchomotor tone over 24 hours without the dips seen with shorter-acting agents and by improvement of the FEV1 trough before the subsequent dose is due, aptly named “pharmacologic stenting.”57 Once-daily dosing should also foster better adherence. The safety profile appears excellent with no increase in cardiovascular or cerebrovascular events compared with placebo.58
The FDA approved the 75-μg daily dose instead of the higher doses used in the studies mentioned above. This decision was based on the observation that there appeared to be a flattened dose-response in patients with more severe COPD, with no further improvement in trough FEV1 at higher doses.59
DOES VITAMIN D SUPPLEMENTATION HAVE A ROLE IN COPD MANAGEMENT?
Vitamin D is vital for calcium and phosphate metabolism and bone health. Low vitamin D levels are associated with diminished leg strength and falls in the elderly.60 Osteoporosis, preventable with vitamin D and calcium supplementation, is linked to thoracic vertebral fracture and consequent reduced lung function.61,62
Patients with COPD are at higher risk of vitamin D deficiency, and more so if they also are obese, have advanced airflow obstruction, are depressed, or smoke.62 Therefore, there are sound reasons to look for vitamin D deficiency in patients with COPD and to treat it if the 25-hydroxyvitamin D level is less than 10 ng/ mL (Table 6).
Vitamin D may also have antimicrobial and immunomodulatory effects.63 Since COPD exacerbations are frequently caused by infection, it was hypothesized that vitamin D supplementation might reduce the rate of exacerbations.
In a study in 182 patients with moderate to very severe COPD and a history of recent exacerbations, high-dose vitamin D supplementation (100,000 IU) was given every 4 weeks for 1 year.64 There were no differences in the time to first exacerbation, in the rate of exacerbation, hospitalization, or death, or in quality of life between the placebo and intervention groups. However, subgroup analysis indicated that, in those with severe vitamin D deficiency at baseline, the exacerbation rate was reduced by more than 40%.
Comment. We recommend screening for vitamin D deficiency in patients with COPD. Supplementation is appropriate in those with low levels, but data indicate no role in those with normal levels.
WHAT ARE THE NONPHARMACOLOGIC APPROACHES TO COPD TREATMENT?
Noninvasive positive-pressure ventilation
Nocturnal noninvasive positive-pressure ventilation may be beneficial in patients with severe COPD, daytime hypercapnia, and nocturnal hypoventilation, particularly if higher inspiratory pressures are selected (Table 7).65,66
For instance, a randomized controlled trial of noninvasive positive-pressure ventilation plus long-term oxygen therapy compared with long-term oxygen therapy alone in hypercapnic COPD demonstrated a survival benefit in favor of ventilation (hazard ratio 0.6).67
In another randomized trial,68 settings that aimed to maximally reduce Paco2 (mean inspiratory positive airway pressure 29 cm H2O with a backup rate of 17.5/min) were compared with low-intensity positive airway pressure (mean inspiratory positive airway pressure 14 cm H2O, backup rate 8/min). The high inspiratory pressures increased the daily use of ventilation by 3.6 hours per day and improved exercise-related dyspnea, daytime Paco2, FEV1, vital capacity, and health-related quality of life66 without disrupting sleep quality.68
Caveats are that acclimation to the high pressures was achieved in the hospital, and the high pressures were associated with a significant increase in air leaks.66
Comments. Whether high-pressure noninvasive positive-pressure ventilation can be routinely implemented and adopted in the outpatient setting, and whether it is associated with a survival advantage remains to be determined. The advantages of noninvasive positive-pressure ventilation in the setting of hypercapnic COPD appear to augment those of pulmonary rehabilitation, with improved quality of life, gas exchange, and exercise tolerance, and a slower decline of lung function.69
Pulmonary rehabilitation
Pulmonary rehabilitation is a multidisciplinary approach to managing COPD (Table 8).
Patients participate in three to five supervised sessions per week, each lasting 3 to 4 hours, for 6 to 12 weeks. Less-frequent sessions may not be effective. For instance, in a randomized trial, exercising twice a week was not enough.70 Additionally, a program lasting longer than 12 weeks produced more sustained benefits than shorter programs.71
A key component is an exercise protocol centered on the lower extremities (walking, cycling, treadmill), with progressive exercise intensity to a target of about 60% to 80% of the maximal exercise tolerance,72 though more modest targets of about 50% can also be beneficial.73
Exercise should be tailored to the desired outcome. For instance, training of the upper arms may help with activities of daily living. In one study, unsupported (against gravity) arm training improved upper-extremity function more than supported arm training (by ergometer).74 Ventilatory muscle training is less common, as most randomized trials have not shown conclusive evidence of benefit. Current guidelines do not recommend routine inspiratory muscle training.71
Even though indices of pulmonary function do not improve after an exercise program, randomized trials have shown that pulmonary rehabilitation improves exercise capacity, dyspnea, and health-related quality of life; improves cost-effectiveness of health care utilization; and provides psychosocial benefits that often exceed those of other therapies. Although there is no significant evidence of whether pulmonary rehabilitation improves survival in patients with COPD,71 an observational study documented improvements in BODE scores as well as a reduction in respiratory mortality rates in patients undergoing pulmonary rehabilitation.75
A limitation of pulmonary rehabilitation is that endurance and psychological and cognitive function decline significantly if exercise is not maintained. However, the role of a maintenance program is uncertain, with long-term benefits considered modest.71
Lung-volume reduction surgery
Lung-volume reduction consists of surgical wedge resections of emphysematous areas of the lung (Table 9).
The National Emphysema Treatment Trial76 randomized 1,218 patients to undergo either lung-volume reduction surgery or maximal medical therapy. Surgery improved survival, quality of life, and dyspnea in patients with upper-lobe emphysema and a low exercise capacity (corresponding to < 40 watts for men or < 25 watts for women in the maximal power achieved on cycle ergometry). While conferring no survival benefit in patients with upper-lobe-predominant emphysema and high exercise capacity, this surgery is likely to improve exercise capacity and quality of life in this subset of patients.
Importantly, the procedure is associated with a lower survival rate in patients with an FEV1 lower than 20%, homogeneous emphysema, a diffusing capacity of the lung for carbon monoxide lower than 20%, non-upper-lobe emphysema, or high baseline exercise capacity.
The proposed mechanisms of improvement of lung function include placing the diaphragm in a position with better mechanical advantage, reducing overall lung volume, better size-matching between the lungs and chest cavity, and restoring elastic recoil.76,77
Ongoing trials aim to replicate the success of lung-volume reduction using nonsurgical bronchoscopic techniques with one-way valves, coils, biologic sealants, thermal ablation, and airway stents.
- Miniño AM, Xu J, Kochanek KD. Deaths: preliminary data for 2008. National Vital Statistics Reports 2010; 59:1–52. http://www.cdc.gov/nchs/data/nvsr/nvsr59/nvsr59_02.pdf. Accessed April 10, 2014.
- Murphy SL, Xu J, Kochanek KD. Deaths: preliminary data from 2010. National Vital Statistics Reports 2012; 60:1–51. http://www.cdc.gov/nchs/data/nvsr/nvsr60/nvsr60_04.pdf. Accessed April 10, 2014.
- Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 2006; 3:e442.
- Behrendt CE. Mild and moderate-to-severe COPD in nonsmokers: distinct demographic profiles. Chest 2005; 128:1239–1244.
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). http://www.goldcopd.org/Guidelines/guidelines-resources.html. Accessed April 10, 2014.
- Papi A, Bellettato CM, Braccioni F, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med 2006; 173:1114–1121.
- Soler-Cataluña JJ, Martínez-García MA, Román Sánchez P, Salcedo E, Navarro M, Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax 2005; 60:925–931.
- Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002; 57:847–852.
- Hurst JR, Vestbo J, Anzueto A, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med 2010; 363:1128–1138.
- Cooper CB. The connection between chronic obstructive pulmonary disease symptoms and hyperinflation and its impact on exercise and function. Am J Med 2006; 119(suppl 1):21–31.
- Jones PW. Issues concerning health-related quality of life in COPD. Chest 1995; 107(suppl):187S–193S.
- Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med 2004; 350:1005–1012.
- Fletcher CM. The clinical diagnosis of pulmonary emphysema—an experimental study. J Royal Soc Med 1952; 45:577–584.
- Lange P, Marott JL, Vestbo J, et al. Prediction of the clinical course of chronic obstructive pulmonary disease, using the new GOLD classification: a study of the general population. Am J Respir Crit Care Med 2012; 186:975–981.
- Hurst J. Phenotype-based care in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:935–936.
- Jones PW, Adamek L, Nadeau G, Banik N. Comparisons of health status scores with MRC grades in COPD: implications for the GOLD 2011 classification. Eur Respir J 2013; 42:647–654.
- Divo M, Cote C, de Torres JP, et al; BODE Collaborative Group. Comorbidities and risk of mortality in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:155–161.
- Anthonisen NR, Manfreda J, Warren CP, Hershfield ES, Harding GK, Nelson NA. Antibiotic therapy in exacerbations of chronic obstructive pulmonary disease. Ann Intern Med 1987; 106:196–204.
- Llor C, Moragas A, Hernández S, Bayona C, Miravitlles M. Efficacy of antibiotic therapy for acute exacerbations of mild to moderate chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:716–723.
- Daniels JM, Snijders D, de Graaff CS, Vlaspolder F, Jansen HM, Boersma WG. Antibiotics in addition to systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010; 181:150–157.
- Rothberg MB, Pekow PS, Lahti M, Brody O, Skiest DJ, Lindenauer PK. Antibiotic therapy and treatment failure in patients hospitalized for acute exacerbations of chronic obstructive pulmonary disease. JAMA 2010; 303:2035–2042.
- Vollenweider DJ, Jarrett H, Steurer-Stey CA, Garcia-Aymerich J, Puhan MA. Antibiotics for exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2012; 12:CD010257.
- Stolz D, Christ-Crain M, Bingisser R, et al. Antibiotic treatment of exacerbations of COPD: a randomized, controlled trial comparing procalcitonin-guidance with standard therapy. Chest 2007; 131:9–19.
- Wolter J, Seeney S, Bell S, Bowler S, Masel P, McCormack J. Effect of long term treatment with azithromycin on disease parameters in cystic fibrosis: a randomised trial. Thorax 2002; 57:212–216.
- Kudoh S, Azuma A, Yamamoto M, Izumi T, Ando M. Improvement of survival in patients with diffuse panbronchiolitis treated with low-dose erythromycin. Am J Respir Crit Care Med 1998; 157:1829–1832.
- Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
- Gross NJ, Giembycz MA, Rennard SI. Treatment of chronic obstructive pulmonary disease with roflumilast, a new phosphodiesterase 4 inhibitor. COPD 2010; 7:141–153.
- Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ; M2-124 and M2-125 study groups. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 2009; 374:685–694.
- Fabbri LM, Calverley PM, Izquierdo-Alonso JL, et al; M2-127 and M2-128 study groups. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with long acting bronchodilators: two randomised clinical trials. Lancet 2009; 374:695–703.
- Gross NJ, Skorodin MS. Anticholinergic, antimuscarinic bronchodilators. Am Rev Respir Dis 1984; 129:856–870.
- Tashkin DP, Celli B, Senn S, et al; UPLIFT Study Investigators. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008; 359:1543–1554.
- Cazzola M. Aclidinium bromide, a novel long-acting muscarinic M3 antagonist for the treatment of COPD. Curr Opin Investig Drugs 2009; 10:482–490.
- Gavaldà A, Miralpeix M, Ramos I, et al. Characterization of aclidinium bromide, a novel inhaled muscarinic antagonist, with long duration of action and a favorable pharmacological profile. J Pharmacol Exp Ther 2009; 331:740–751.
- Alagha K, Bourdin A, Tummino C, Chanez P. An update on the efficacy and safety of aclidinium bromide in patients with COPD. Ther Adv Respir Dis 2011; 5:19–28.
- Kerwin EM, D’Urzo AD, Gelb AF, Lakkis H, Garcia Gil E, Caracta CF; ACCORD I study investigators. Efficacy and safety of a 12-week treatment with twice-daily aclidinium bromide in COPD patients (ACCORD COPD I). COPD 2012; 9:90–101.
- Jones PW, Singh D, Bateman ED, et al. Efficacy and safety of twice-daily aclidinium bromide in COPD patients: the ATTAIN study. Eur Respir J 2012; 40:830–836.
- Jones PW, Rennard SI, Agusti A, et al. Efficacy and safety of once-daily aclidinium in chronic obstructive pulmonary disease. Respir Res 2011; 12:55.
- Fuhr R, Magnussen H, Sarem K, et al. Efficacy of aclidinium bromide 400 μg twice daily compared with placebo and tiotropium in patients with moderate to severe COPD. Chest 2012; 141:745–752.
- Tashkin DP, Fabbri LM. Long-acting beta-agonists in the management of chronic obstructive pulmonary disease: current and future agents. Respir Res 2010; 11:149.
- Mahler DA, Donohue JF, Barbee RA, et al. Efficacy of salmeterol xinafoate in the treatment of COPD. Chest 1999; 115:957–965.
- Campbell M, Eliraz A, Johansson G, et al. Formoterol for maintenance and as-needed treatment of chronic obstructive pulmonary disease. Respir Med 2005; 99:1511–1520.
- Hanrahan JP, Hanania NA, Calhoun WJ, Sahn SA, Sciarappa K, Baumgartner RA. Effect of nebulized arformoterol on airway function in COPD: results from two randomized trials. COPD 2008; 5:25–34.
- Neder JA, Fuld JP, Overend T, et al. Effects of formoterol on exercise tolerance in severely disabled patients with COPD. Respir Med 2007; 101:2056–2064.
- O’Donnell DE, Voduc N, Fitzpatrick M, Webb KA. Effect of salmeterol on the ventilatory response to exercise in chronic obstructive pulmonary disease. Eur Respir J 2004; 24:86–94.
- Rennard SI, Anderson W, ZuWallack R, et al. Use of a long-acting inhaled beta 2-adrenergic agonist, salmeterol xinafoate, in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 163:1087–1092.
- Schultze-Werninghaus G. Multicenter 1-year trial on formoterol, a new long-acting beta 2-agonist, in chronic obstructive airway disease. Lung 1990; 168(suppl):83–89.
- Rodrigo GJ, Nannini LJ, Rodríguez-Roisin R. Safety of long-acting beta-agonists in stable COPD: a systematic review. Chest 2008; 133:1079–1087.
- Baker WL, Baker EL, Coleman CI. Pharmacologic treatments for chronic obstructive pulmonary disease: a mixed-treatment comparison meta-analysis. Pharmacotherapy 2009; 29:891–905.
- Rodrigo GJ, Castro-Rodriguez JA, Plaza V. Safety and efficacy of combined long-acting beta-agonists and inhaled corticosteroids vs long-acting beta-agonists monotherapy for stable COPD: a systematic review. Chest 2009; 136:1029–1038.
- Nelson HS, Weiss ST, Bleecker ER, Yancey SW, Dorinsky PM; SMART Study Group. The Salmeterol Multicenter Asthma Research Trial: a comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol. Chest 2006; 129:15–26.
- Calverley PM, Anderson JA, Celli B, et al; TORCH investigators. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007; 356:775–789.
- Suissa S, Ernst P, Vandemheen KL, Aaron SD. Methodological issues in therapeutic trials of COPD. Eur Respir J 2008; 31:927–933.
- Lombardi D, Cuenoud B, Krämer SD. Lipid membrane interactions of indacaterol and salmeterol: do they influence their pharmacological properties? Eur J Pharm Sci 2009; 38:533–547.
- Kornmann O, Dahl R, Centanni S, et al; INLIGHT-2 (Indacaterol Efficacy Evaluation Using 150-μg Doses with COPD Patients) study investigators. Once-daily indacaterol versus twice-daily salmeterol for COPD: a placebo-controlled comparison. Eur Respir J 2011; 37:273–279.
- Dahl R, Chung KF, Buhl R, et al; INVOLVE (INdacaterol: Value in COPD: Longer Term Validation of Efficacy and Safety) Study Investigators. Efficacy of a new once-daily long-acting inhaled beta2-agonist indacaterol versus twice-daily formoterol in COPD. Thorax 2010; 65:473–479.
- Donohue JF, Fogarty C, Lötvall J, et al; INHANCE Study Investigators. Once-daily bronchodilators for chronic obstructive pulmonary disease: indacaterol versus tiotropium. Am J Respir Crit Care Med 2010; 182:155–162.
- Beeh KM, Beier J. The short, the long and the “ultra-long”: why duration of bronchodilator action matters in chronic obstructive pulmonary disease. Adv Ther 2010; 27:150–159.
- Worth H, Chung KF, Felser JM, Hu H, Rueegg P. Cardio- and cerebrovascular safety of indacaterol vs formoterol, salmeterol, tiotropium and placebo in COPD. Respir Med 2011; 105:571–579.
- Chowdhury BA, Seymour SM, Michele TM, Durmowicz AG, Liu D, Rosebraugh CJ. The risks and benefits of indacaterol--the FDA’s review. N Engl J Med 2011; 365:2247–2249.
- Bischoff-Ferrari HA, Dawson-Hughes B, Willett WC, et al. Effect of vitamin D on falls: a meta-analysis. JAMA 2004; 291:1999–2006.
- Leech JA, Dulberg C, Kellie S, Pattee L, Gay J. Relationship of lung function to severity of osteoporosis in women. Am Rev Respir Dis 1990; 141:68–71.
- Persson LJ, Aanerud M, Hiemstra PS, Hardie JA, Bakke PS, Eagan TM. Chronic obstructive pulmonary disease is associated with low levels of vitamin D. PLoS One 2012; 7:e38934.
- Janssens W, Lehouck A, Carremans C, Bouillon R, Mathieu C, Decramer M. Vitamin D beyond bones in chronic obstructive pulmonary disease: time to act. Am J Respir Crit Care Med 2009; 179:630–636.
- Lehouck A, Mathieu C, Carremans C, et al. High doses of vitamin D to reduce exacerbations in chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med 2012; 156:105–114.
- Casanova C, Celli BR, Tost L, et al. Long-term controlled trial of nocturnal nasal positive pressure ventilation in patients with severe COPD. Chest 2000; 118:1582–1590.
- Dreher M, Storre JH, Schmoor C, Windisch W. High-intensity versus low-intensity non-invasive ventilation in patients with stable hypercapnic COPD: a randomised crossover trial. Thorax 2010; 65:303–308.
- McEvoy RD, Pierce RJ, Hillman D, et al; Australian trial of non-invasive Ventilation in Chronic Airflow Limitation (AVCAL) Study Group. Nocturnal non-invasive nasal ventilation in stable hypercapnic COPD: a randomised controlled trial. Thorax 2009; 64:561–566.
- Dreher M, Ekkernkamp E, Walterspacher S, et al. Noninvasive ventilation in COPD: impact of inspiratory pressure levels on sleep quality. Chest 2011; 140:939–945.
- Duiverman ML, Wempe JB, Bladder G, et al. Two-year home-based nocturnal noninvasive ventilation added to rehabilitation in chronic obstructive pulmonary disease patients: a randomized controlled trial. Respir Res 2011; 12:112.
- Ringbaek TJ, Broendum E, Hemmingsen L, et al. Rehabilitation of patients with chronic obstructive pulmonary disease. Exercise twice a week is not sufficient! Respir Med 2000; 94:150–154.
- Ries AL, Bauldoff GS, Carlin BW, et al. Pulmonary Rehabilitation: Joint ACCP/AACVPR Evidence-Based Clinical Practice Guidelines. Chest 2007; 131(suppl):4S–42S.
- Vallet G, Ahmaïdi S, Serres I, et al. Comparison of two training programmes in chronic airway limitation patients: standardized versus individualized protocols. Eur Respir J 1997; 10:114–122.
- Vogiatzis I, Williamson AF, Miles J, Taylor IK. Physiological response to moderate exercise workloads in a pulmonary rehabilitation program in patients with varying degrees of airflow obstruction. Chest 1999; 116:1200–1207.
- Martinez FJ, Vogel PD, Dupont DN, Stanopoulos I, Gray A, Beamis JF. Supported arm exercise vs unsupported arm exercise in the rehabilitation of patients with severe chronic airflow obstruction. Chest 1993; 103:1397–1402.
- Cote CG, Celli BR. Pulmonary rehabilitation and the BODE index in COPD. Eur Respir J 2005; 26:630–636.
- Fishman A, Martinez F, Naunheim K, et al; National Emphysema Treatment Trial Research Group. A randomized trial comparing lung-volume-reduction surgery with medical therapy for severe emphysema. N Engl J Med 2003; 348:2059–2073.
- Naunheim KS, Wood DE, Mohsenifar Z, et al; National Emphysema Treatment Trial Research Group. Long-term follow-up of patients receiving lung-volume-reduction surgery versus medical therapy for severe emphysema by the National Emphysema Treatment Trial Research Group. Ann Thorac Surg 2006; 82:431–443.
- Miniño AM, Xu J, Kochanek KD. Deaths: preliminary data for 2008. National Vital Statistics Reports 2010; 59:1–52. http://www.cdc.gov/nchs/data/nvsr/nvsr59/nvsr59_02.pdf. Accessed April 10, 2014.
- Murphy SL, Xu J, Kochanek KD. Deaths: preliminary data from 2010. National Vital Statistics Reports 2012; 60:1–51. http://www.cdc.gov/nchs/data/nvsr/nvsr60/nvsr60_04.pdf. Accessed April 10, 2014.
- Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 2006; 3:e442.
- Behrendt CE. Mild and moderate-to-severe COPD in nonsmokers: distinct demographic profiles. Chest 2005; 128:1239–1244.
- Global Initiative for Chronic Obstructive Lung Disease (GOLD). http://www.goldcopd.org/Guidelines/guidelines-resources.html. Accessed April 10, 2014.
- Papi A, Bellettato CM, Braccioni F, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med 2006; 173:1114–1121.
- Soler-Cataluña JJ, Martínez-García MA, Román Sánchez P, Salcedo E, Navarro M, Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax 2005; 60:925–931.
- Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002; 57:847–852.
- Hurst JR, Vestbo J, Anzueto A, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) Investigators. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med 2010; 363:1128–1138.
- Cooper CB. The connection between chronic obstructive pulmonary disease symptoms and hyperinflation and its impact on exercise and function. Am J Med 2006; 119(suppl 1):21–31.
- Jones PW. Issues concerning health-related quality of life in COPD. Chest 1995; 107(suppl):187S–193S.
- Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med 2004; 350:1005–1012.
- Fletcher CM. The clinical diagnosis of pulmonary emphysema—an experimental study. J Royal Soc Med 1952; 45:577–584.
- Lange P, Marott JL, Vestbo J, et al. Prediction of the clinical course of chronic obstructive pulmonary disease, using the new GOLD classification: a study of the general population. Am J Respir Crit Care Med 2012; 186:975–981.
- Hurst J. Phenotype-based care in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:935–936.
- Jones PW, Adamek L, Nadeau G, Banik N. Comparisons of health status scores with MRC grades in COPD: implications for the GOLD 2011 classification. Eur Respir J 2013; 42:647–654.
- Divo M, Cote C, de Torres JP, et al; BODE Collaborative Group. Comorbidities and risk of mortality in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:155–161.
- Anthonisen NR, Manfreda J, Warren CP, Hershfield ES, Harding GK, Nelson NA. Antibiotic therapy in exacerbations of chronic obstructive pulmonary disease. Ann Intern Med 1987; 106:196–204.
- Llor C, Moragas A, Hernández S, Bayona C, Miravitlles M. Efficacy of antibiotic therapy for acute exacerbations of mild to moderate chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2012; 186:716–723.
- Daniels JM, Snijders D, de Graaff CS, Vlaspolder F, Jansen HM, Boersma WG. Antibiotics in addition to systemic corticosteroids for acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010; 181:150–157.
- Rothberg MB, Pekow PS, Lahti M, Brody O, Skiest DJ, Lindenauer PK. Antibiotic therapy and treatment failure in patients hospitalized for acute exacerbations of chronic obstructive pulmonary disease. JAMA 2010; 303:2035–2042.
- Vollenweider DJ, Jarrett H, Steurer-Stey CA, Garcia-Aymerich J, Puhan MA. Antibiotics for exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2012; 12:CD010257.
- Stolz D, Christ-Crain M, Bingisser R, et al. Antibiotic treatment of exacerbations of COPD: a randomized, controlled trial comparing procalcitonin-guidance with standard therapy. Chest 2007; 131:9–19.
- Wolter J, Seeney S, Bell S, Bowler S, Masel P, McCormack J. Effect of long term treatment with azithromycin on disease parameters in cystic fibrosis: a randomised trial. Thorax 2002; 57:212–216.
- Kudoh S, Azuma A, Yamamoto M, Izumi T, Ando M. Improvement of survival in patients with diffuse panbronchiolitis treated with low-dose erythromycin. Am J Respir Crit Care Med 1998; 157:1829–1832.
- Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
- Gross NJ, Giembycz MA, Rennard SI. Treatment of chronic obstructive pulmonary disease with roflumilast, a new phosphodiesterase 4 inhibitor. COPD 2010; 7:141–153.
- Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ; M2-124 and M2-125 study groups. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 2009; 374:685–694.
- Fabbri LM, Calverley PM, Izquierdo-Alonso JL, et al; M2-127 and M2-128 study groups. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with long acting bronchodilators: two randomised clinical trials. Lancet 2009; 374:695–703.
- Gross NJ, Skorodin MS. Anticholinergic, antimuscarinic bronchodilators. Am Rev Respir Dis 1984; 129:856–870.
- Tashkin DP, Celli B, Senn S, et al; UPLIFT Study Investigators. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008; 359:1543–1554.
- Cazzola M. Aclidinium bromide, a novel long-acting muscarinic M3 antagonist for the treatment of COPD. Curr Opin Investig Drugs 2009; 10:482–490.
- Gavaldà A, Miralpeix M, Ramos I, et al. Characterization of aclidinium bromide, a novel inhaled muscarinic antagonist, with long duration of action and a favorable pharmacological profile. J Pharmacol Exp Ther 2009; 331:740–751.
- Alagha K, Bourdin A, Tummino C, Chanez P. An update on the efficacy and safety of aclidinium bromide in patients with COPD. Ther Adv Respir Dis 2011; 5:19–28.
- Kerwin EM, D’Urzo AD, Gelb AF, Lakkis H, Garcia Gil E, Caracta CF; ACCORD I study investigators. Efficacy and safety of a 12-week treatment with twice-daily aclidinium bromide in COPD patients (ACCORD COPD I). COPD 2012; 9:90–101.
- Jones PW, Singh D, Bateman ED, et al. Efficacy and safety of twice-daily aclidinium bromide in COPD patients: the ATTAIN study. Eur Respir J 2012; 40:830–836.
- Jones PW, Rennard SI, Agusti A, et al. Efficacy and safety of once-daily aclidinium in chronic obstructive pulmonary disease. Respir Res 2011; 12:55.
- Fuhr R, Magnussen H, Sarem K, et al. Efficacy of aclidinium bromide 400 μg twice daily compared with placebo and tiotropium in patients with moderate to severe COPD. Chest 2012; 141:745–752.
- Tashkin DP, Fabbri LM. Long-acting beta-agonists in the management of chronic obstructive pulmonary disease: current and future agents. Respir Res 2010; 11:149.
- Mahler DA, Donohue JF, Barbee RA, et al. Efficacy of salmeterol xinafoate in the treatment of COPD. Chest 1999; 115:957–965.
- Campbell M, Eliraz A, Johansson G, et al. Formoterol for maintenance and as-needed treatment of chronic obstructive pulmonary disease. Respir Med 2005; 99:1511–1520.
- Hanrahan JP, Hanania NA, Calhoun WJ, Sahn SA, Sciarappa K, Baumgartner RA. Effect of nebulized arformoterol on airway function in COPD: results from two randomized trials. COPD 2008; 5:25–34.
- Neder JA, Fuld JP, Overend T, et al. Effects of formoterol on exercise tolerance in severely disabled patients with COPD. Respir Med 2007; 101:2056–2064.
- O’Donnell DE, Voduc N, Fitzpatrick M, Webb KA. Effect of salmeterol on the ventilatory response to exercise in chronic obstructive pulmonary disease. Eur Respir J 2004; 24:86–94.
- Rennard SI, Anderson W, ZuWallack R, et al. Use of a long-acting inhaled beta 2-adrenergic agonist, salmeterol xinafoate, in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 163:1087–1092.
- Schultze-Werninghaus G. Multicenter 1-year trial on formoterol, a new long-acting beta 2-agonist, in chronic obstructive airway disease. Lung 1990; 168(suppl):83–89.
- Rodrigo GJ, Nannini LJ, Rodríguez-Roisin R. Safety of long-acting beta-agonists in stable COPD: a systematic review. Chest 2008; 133:1079–1087.
- Baker WL, Baker EL, Coleman CI. Pharmacologic treatments for chronic obstructive pulmonary disease: a mixed-treatment comparison meta-analysis. Pharmacotherapy 2009; 29:891–905.
- Rodrigo GJ, Castro-Rodriguez JA, Plaza V. Safety and efficacy of combined long-acting beta-agonists and inhaled corticosteroids vs long-acting beta-agonists monotherapy for stable COPD: a systematic review. Chest 2009; 136:1029–1038.
- Nelson HS, Weiss ST, Bleecker ER, Yancey SW, Dorinsky PM; SMART Study Group. The Salmeterol Multicenter Asthma Research Trial: a comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol. Chest 2006; 129:15–26.
- Calverley PM, Anderson JA, Celli B, et al; TORCH investigators. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007; 356:775–789.
- Suissa S, Ernst P, Vandemheen KL, Aaron SD. Methodological issues in therapeutic trials of COPD. Eur Respir J 2008; 31:927–933.
- Lombardi D, Cuenoud B, Krämer SD. Lipid membrane interactions of indacaterol and salmeterol: do they influence their pharmacological properties? Eur J Pharm Sci 2009; 38:533–547.
- Kornmann O, Dahl R, Centanni S, et al; INLIGHT-2 (Indacaterol Efficacy Evaluation Using 150-μg Doses with COPD Patients) study investigators. Once-daily indacaterol versus twice-daily salmeterol for COPD: a placebo-controlled comparison. Eur Respir J 2011; 37:273–279.
- Dahl R, Chung KF, Buhl R, et al; INVOLVE (INdacaterol: Value in COPD: Longer Term Validation of Efficacy and Safety) Study Investigators. Efficacy of a new once-daily long-acting inhaled beta2-agonist indacaterol versus twice-daily formoterol in COPD. Thorax 2010; 65:473–479.
- Donohue JF, Fogarty C, Lötvall J, et al; INHANCE Study Investigators. Once-daily bronchodilators for chronic obstructive pulmonary disease: indacaterol versus tiotropium. Am J Respir Crit Care Med 2010; 182:155–162.
- Beeh KM, Beier J. The short, the long and the “ultra-long”: why duration of bronchodilator action matters in chronic obstructive pulmonary disease. Adv Ther 2010; 27:150–159.
- Worth H, Chung KF, Felser JM, Hu H, Rueegg P. Cardio- and cerebrovascular safety of indacaterol vs formoterol, salmeterol, tiotropium and placebo in COPD. Respir Med 2011; 105:571–579.
- Chowdhury BA, Seymour SM, Michele TM, Durmowicz AG, Liu D, Rosebraugh CJ. The risks and benefits of indacaterol--the FDA’s review. N Engl J Med 2011; 365:2247–2249.
- Bischoff-Ferrari HA, Dawson-Hughes B, Willett WC, et al. Effect of vitamin D on falls: a meta-analysis. JAMA 2004; 291:1999–2006.
- Leech JA, Dulberg C, Kellie S, Pattee L, Gay J. Relationship of lung function to severity of osteoporosis in women. Am Rev Respir Dis 1990; 141:68–71.
- Persson LJ, Aanerud M, Hiemstra PS, Hardie JA, Bakke PS, Eagan TM. Chronic obstructive pulmonary disease is associated with low levels of vitamin D. PLoS One 2012; 7:e38934.
- Janssens W, Lehouck A, Carremans C, Bouillon R, Mathieu C, Decramer M. Vitamin D beyond bones in chronic obstructive pulmonary disease: time to act. Am J Respir Crit Care Med 2009; 179:630–636.
- Lehouck A, Mathieu C, Carremans C, et al. High doses of vitamin D to reduce exacerbations in chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med 2012; 156:105–114.
- Casanova C, Celli BR, Tost L, et al. Long-term controlled trial of nocturnal nasal positive pressure ventilation in patients with severe COPD. Chest 2000; 118:1582–1590.
- Dreher M, Storre JH, Schmoor C, Windisch W. High-intensity versus low-intensity non-invasive ventilation in patients with stable hypercapnic COPD: a randomised crossover trial. Thorax 2010; 65:303–308.
- McEvoy RD, Pierce RJ, Hillman D, et al; Australian trial of non-invasive Ventilation in Chronic Airflow Limitation (AVCAL) Study Group. Nocturnal non-invasive nasal ventilation in stable hypercapnic COPD: a randomised controlled trial. Thorax 2009; 64:561–566.
- Dreher M, Ekkernkamp E, Walterspacher S, et al. Noninvasive ventilation in COPD: impact of inspiratory pressure levels on sleep quality. Chest 2011; 140:939–945.
- Duiverman ML, Wempe JB, Bladder G, et al. Two-year home-based nocturnal noninvasive ventilation added to rehabilitation in chronic obstructive pulmonary disease patients: a randomized controlled trial. Respir Res 2011; 12:112.
- Ringbaek TJ, Broendum E, Hemmingsen L, et al. Rehabilitation of patients with chronic obstructive pulmonary disease. Exercise twice a week is not sufficient! Respir Med 2000; 94:150–154.
- Ries AL, Bauldoff GS, Carlin BW, et al. Pulmonary Rehabilitation: Joint ACCP/AACVPR Evidence-Based Clinical Practice Guidelines. Chest 2007; 131(suppl):4S–42S.
- Vallet G, Ahmaïdi S, Serres I, et al. Comparison of two training programmes in chronic airway limitation patients: standardized versus individualized protocols. Eur Respir J 1997; 10:114–122.
- Vogiatzis I, Williamson AF, Miles J, Taylor IK. Physiological response to moderate exercise workloads in a pulmonary rehabilitation program in patients with varying degrees of airflow obstruction. Chest 1999; 116:1200–1207.
- Martinez FJ, Vogel PD, Dupont DN, Stanopoulos I, Gray A, Beamis JF. Supported arm exercise vs unsupported arm exercise in the rehabilitation of patients with severe chronic airflow obstruction. Chest 1993; 103:1397–1402.
- Cote CG, Celli BR. Pulmonary rehabilitation and the BODE index in COPD. Eur Respir J 2005; 26:630–636.
- Fishman A, Martinez F, Naunheim K, et al; National Emphysema Treatment Trial Research Group. A randomized trial comparing lung-volume-reduction surgery with medical therapy for severe emphysema. N Engl J Med 2003; 348:2059–2073.
- Naunheim KS, Wood DE, Mohsenifar Z, et al; National Emphysema Treatment Trial Research Group. Long-term follow-up of patients receiving lung-volume-reduction surgery versus medical therapy for severe emphysema by the National Emphysema Treatment Trial Research Group. Ann Thorac Surg 2006; 82:431–443.
KEY POINTS
- A new COPD classification scheme is based on severity, symptoms, and exacerbations.
- Azithromycin 250 mg daily prevents exacerbations of COPD in those at high risk.
- Long-acting muscarinic antagonists such as aclidinium and tiotropium are first-line therapy.
- Relatively new options include roflumilast, an oral phosphodiesterase inhibitor, and indacaterol, an ultra-long-acting beta agonist that is taken once daily.
- Nondrug interventions include pulmonary rehabilitation, vitamin D supplementation, noninvasive positive-pressure ventilation, and lung-volume reduction surgery.
Syncope during a pharmacologic nuclear stress test
A 60-year-old woman was referred for pharmacologic nuclear stress testing before treatment for breast cancer. She had hypertension, diabetes mellitus, coronary artery disease, and a remote history of stroke, and she was taking clonidine (Catapres), labetalol (Normodyne, Trandate), furosemide (Lasix), hydralazine, valsartan (Diovan), insulin, and the aspirin-dipyridamole combination Aggrenox. Her vital signs and electrocardiogram before the stress test were normal.
The stress test was started with a standard protocol of adenosine (Adenoscan) infused intravenously over 4 minutes. For the first 2 minutes, she was stable and had no symptoms, but then sinus pauses and second-degree atrioventricular block type 2 developed, after which her heart stopped beating (Figure 1). The infusion was immediately stopped, but she became unresponsive and remained pulseless.
Cardiopulmonary resuscitation was started, aminophylline 100 mg was given intravenously, and she regained a pulse and blood pressure within a few minutes. She was then transferred to the emergency room, where she returned to her baseline clinical and neurologic status without symptoms.
AN UNRECOGNIZED DRUG INTERACTION
Asystole occurred in this patient because of the interaction of intravenous adenosine with the dipyridamole in the medication Aggrenox. Although adenosine, given during pharmacologic stress testing, is known to interact with various medications, the potential for this interaction may be overlooked if the culprit is present in a combination drug. Aggrenox is commonly given for secondary stroke prevention and should be discontinued before pharmacologic nuclear stress testing.
Pharmacologic stress testing involves two commonly used stress agents, adenosine and regadenoson (Lexiscan), which cause coronary vasodilation through their action on A2A receptors in the heart. Coronary vasodilation results in flow heterogeneity in the region of a stenotic artery, which can be detected with nuclear perfusion agents. In addition, adenosine has a short-lived effect on the A1 receptors that block atrioventricular conduction.1
Dipyridamole (Persantine) is contraindicated when either adenosine or regadenoson is used. Dipyridamole enhances the effect of exogenous and endogenous adenosine by inhibiting its uptake by cardiac cells, thus enhancing the action of these coronary vasodilators.2 Atrioventricular block is common during adenosine stress testing but is transient because adenosine has a short half-life (< 10 seconds), and complete heart block or asystole, as seen in this patient, is rare. Giving intravenous adenosine or regadenoson to patients on dipyridamole may have a marked effect on adenosine receptors, so that profound bradycardia and even asystole leading to cardiac collapse may occur. No data are available on the specific interaction of dipyridamole and regadenoson.
Even though the pharmacodynamics of the interaction between dipyridamole and adenosine are known,3 few reports are available detailing serious adverse events. The contraindication to pharmacologic stress testing in patients taking dipyridamole is noted in the American Society of Nuclear Cardiology Guidelines for stress protocols,4 which advise discontinuing dipyridamole-containing drugs at least 48 hours before the use of adenosine or regadenoson. Similarly, the American Heart Association guidelines5 for the management of supraventricular tachycardia recommend an initial dose of 3 mg of adenosine rather than 6 mg in patients who have been taking dipyridamole.
The dose of aminophylline for reversing the adverse effects of adenosine or regadenoson is 50 to 250 mg intravenously over 30 to 60 seconds. But since these adverse effects are short-lived once the infusion is stopped, aminophylline is usually given only if the adverse effects are severe, as in this patient.
Pharmacologic nuclear stress testing with adenosine receptor agonists (eg, adenosine or regadenoson) is contraindicated in patients taking dipyridamole or the combination pill Aggrenox because of the potential for profound bradyarrhythmias or asystole.
- Zoghbi GJ, Iskandrian AE. Selective adenosine agonists and myocardial perfusion imaging. J Nucl Cardiol 2012; 19:126–141.
- Lerman BB, Wesley RC, Belardinelli L. Electrophysiologic effects of dipyridamole on atrioventricular nodal conduction and supraventricular tachycardia. Role of endogenous adenosine. Circulation 1989; 80:1536–1543.
- Biaggioni I, Onrot J, Hollister AS, Robertson D. Cardiovascular effects of adenosine infusion in man and their modulation by dipyridamole. Life Sci 1986; 39:2229–2236.
- Henzlova MJ, Cerqueira MD, Mahmarian JJ, Yao SS; Quality Assurance Committee of the American Society of Nuclear Cardiology. Stress protocols and tracers. J Nucl Cardiol 2006; 13:e80–e90.
- ECC Committee, Subcommittees and Task Forces of the American Heart Association. 2005 American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Part 7.3: management of symptomatic bradycardia and tachycardia. Circulation 2005; 112(suppl 24):IV67–IV77.
A 60-year-old woman was referred for pharmacologic nuclear stress testing before treatment for breast cancer. She had hypertension, diabetes mellitus, coronary artery disease, and a remote history of stroke, and she was taking clonidine (Catapres), labetalol (Normodyne, Trandate), furosemide (Lasix), hydralazine, valsartan (Diovan), insulin, and the aspirin-dipyridamole combination Aggrenox. Her vital signs and electrocardiogram before the stress test were normal.
The stress test was started with a standard protocol of adenosine (Adenoscan) infused intravenously over 4 minutes. For the first 2 minutes, she was stable and had no symptoms, but then sinus pauses and second-degree atrioventricular block type 2 developed, after which her heart stopped beating (Figure 1). The infusion was immediately stopped, but she became unresponsive and remained pulseless.
Cardiopulmonary resuscitation was started, aminophylline 100 mg was given intravenously, and she regained a pulse and blood pressure within a few minutes. She was then transferred to the emergency room, where she returned to her baseline clinical and neurologic status without symptoms.
AN UNRECOGNIZED DRUG INTERACTION
Asystole occurred in this patient because of the interaction of intravenous adenosine with the dipyridamole in the medication Aggrenox. Although adenosine, given during pharmacologic stress testing, is known to interact with various medications, the potential for this interaction may be overlooked if the culprit is present in a combination drug. Aggrenox is commonly given for secondary stroke prevention and should be discontinued before pharmacologic nuclear stress testing.
Pharmacologic stress testing involves two commonly used stress agents, adenosine and regadenoson (Lexiscan), which cause coronary vasodilation through their action on A2A receptors in the heart. Coronary vasodilation results in flow heterogeneity in the region of a stenotic artery, which can be detected with nuclear perfusion agents. In addition, adenosine has a short-lived effect on the A1 receptors that block atrioventricular conduction.1
Dipyridamole (Persantine) is contraindicated when either adenosine or regadenoson is used. Dipyridamole enhances the effect of exogenous and endogenous adenosine by inhibiting its uptake by cardiac cells, thus enhancing the action of these coronary vasodilators.2 Atrioventricular block is common during adenosine stress testing but is transient because adenosine has a short half-life (< 10 seconds), and complete heart block or asystole, as seen in this patient, is rare. Giving intravenous adenosine or regadenoson to patients on dipyridamole may have a marked effect on adenosine receptors, so that profound bradycardia and even asystole leading to cardiac collapse may occur. No data are available on the specific interaction of dipyridamole and regadenoson.
Even though the pharmacodynamics of the interaction between dipyridamole and adenosine are known,3 few reports are available detailing serious adverse events. The contraindication to pharmacologic stress testing in patients taking dipyridamole is noted in the American Society of Nuclear Cardiology Guidelines for stress protocols,4 which advise discontinuing dipyridamole-containing drugs at least 48 hours before the use of adenosine or regadenoson. Similarly, the American Heart Association guidelines5 for the management of supraventricular tachycardia recommend an initial dose of 3 mg of adenosine rather than 6 mg in patients who have been taking dipyridamole.
The dose of aminophylline for reversing the adverse effects of adenosine or regadenoson is 50 to 250 mg intravenously over 30 to 60 seconds. But since these adverse effects are short-lived once the infusion is stopped, aminophylline is usually given only if the adverse effects are severe, as in this patient.
Pharmacologic nuclear stress testing with adenosine receptor agonists (eg, adenosine or regadenoson) is contraindicated in patients taking dipyridamole or the combination pill Aggrenox because of the potential for profound bradyarrhythmias or asystole.
A 60-year-old woman was referred for pharmacologic nuclear stress testing before treatment for breast cancer. She had hypertension, diabetes mellitus, coronary artery disease, and a remote history of stroke, and she was taking clonidine (Catapres), labetalol (Normodyne, Trandate), furosemide (Lasix), hydralazine, valsartan (Diovan), insulin, and the aspirin-dipyridamole combination Aggrenox. Her vital signs and electrocardiogram before the stress test were normal.
The stress test was started with a standard protocol of adenosine (Adenoscan) infused intravenously over 4 minutes. For the first 2 minutes, she was stable and had no symptoms, but then sinus pauses and second-degree atrioventricular block type 2 developed, after which her heart stopped beating (Figure 1). The infusion was immediately stopped, but she became unresponsive and remained pulseless.
Cardiopulmonary resuscitation was started, aminophylline 100 mg was given intravenously, and she regained a pulse and blood pressure within a few minutes. She was then transferred to the emergency room, where she returned to her baseline clinical and neurologic status without symptoms.
AN UNRECOGNIZED DRUG INTERACTION
Asystole occurred in this patient because of the interaction of intravenous adenosine with the dipyridamole in the medication Aggrenox. Although adenosine, given during pharmacologic stress testing, is known to interact with various medications, the potential for this interaction may be overlooked if the culprit is present in a combination drug. Aggrenox is commonly given for secondary stroke prevention and should be discontinued before pharmacologic nuclear stress testing.
Pharmacologic stress testing involves two commonly used stress agents, adenosine and regadenoson (Lexiscan), which cause coronary vasodilation through their action on A2A receptors in the heart. Coronary vasodilation results in flow heterogeneity in the region of a stenotic artery, which can be detected with nuclear perfusion agents. In addition, adenosine has a short-lived effect on the A1 receptors that block atrioventricular conduction.1
Dipyridamole (Persantine) is contraindicated when either adenosine or regadenoson is used. Dipyridamole enhances the effect of exogenous and endogenous adenosine by inhibiting its uptake by cardiac cells, thus enhancing the action of these coronary vasodilators.2 Atrioventricular block is common during adenosine stress testing but is transient because adenosine has a short half-life (< 10 seconds), and complete heart block or asystole, as seen in this patient, is rare. Giving intravenous adenosine or regadenoson to patients on dipyridamole may have a marked effect on adenosine receptors, so that profound bradycardia and even asystole leading to cardiac collapse may occur. No data are available on the specific interaction of dipyridamole and regadenoson.
Even though the pharmacodynamics of the interaction between dipyridamole and adenosine are known,3 few reports are available detailing serious adverse events. The contraindication to pharmacologic stress testing in patients taking dipyridamole is noted in the American Society of Nuclear Cardiology Guidelines for stress protocols,4 which advise discontinuing dipyridamole-containing drugs at least 48 hours before the use of adenosine or regadenoson. Similarly, the American Heart Association guidelines5 for the management of supraventricular tachycardia recommend an initial dose of 3 mg of adenosine rather than 6 mg in patients who have been taking dipyridamole.
The dose of aminophylline for reversing the adverse effects of adenosine or regadenoson is 50 to 250 mg intravenously over 30 to 60 seconds. But since these adverse effects are short-lived once the infusion is stopped, aminophylline is usually given only if the adverse effects are severe, as in this patient.
Pharmacologic nuclear stress testing with adenosine receptor agonists (eg, adenosine or regadenoson) is contraindicated in patients taking dipyridamole or the combination pill Aggrenox because of the potential for profound bradyarrhythmias or asystole.
- Zoghbi GJ, Iskandrian AE. Selective adenosine agonists and myocardial perfusion imaging. J Nucl Cardiol 2012; 19:126–141.
- Lerman BB, Wesley RC, Belardinelli L. Electrophysiologic effects of dipyridamole on atrioventricular nodal conduction and supraventricular tachycardia. Role of endogenous adenosine. Circulation 1989; 80:1536–1543.
- Biaggioni I, Onrot J, Hollister AS, Robertson D. Cardiovascular effects of adenosine infusion in man and their modulation by dipyridamole. Life Sci 1986; 39:2229–2236.
- Henzlova MJ, Cerqueira MD, Mahmarian JJ, Yao SS; Quality Assurance Committee of the American Society of Nuclear Cardiology. Stress protocols and tracers. J Nucl Cardiol 2006; 13:e80–e90.
- ECC Committee, Subcommittees and Task Forces of the American Heart Association. 2005 American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Part 7.3: management of symptomatic bradycardia and tachycardia. Circulation 2005; 112(suppl 24):IV67–IV77.
- Zoghbi GJ, Iskandrian AE. Selective adenosine agonists and myocardial perfusion imaging. J Nucl Cardiol 2012; 19:126–141.
- Lerman BB, Wesley RC, Belardinelli L. Electrophysiologic effects of dipyridamole on atrioventricular nodal conduction and supraventricular tachycardia. Role of endogenous adenosine. Circulation 1989; 80:1536–1543.
- Biaggioni I, Onrot J, Hollister AS, Robertson D. Cardiovascular effects of adenosine infusion in man and their modulation by dipyridamole. Life Sci 1986; 39:2229–2236.
- Henzlova MJ, Cerqueira MD, Mahmarian JJ, Yao SS; Quality Assurance Committee of the American Society of Nuclear Cardiology. Stress protocols and tracers. J Nucl Cardiol 2006; 13:e80–e90.
- ECC Committee, Subcommittees and Task Forces of the American Heart Association. 2005 American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Part 7.3: management of symptomatic bradycardia and tachycardia. Circulation 2005; 112(suppl 24):IV67–IV77.
Managing acute coronary syndromes: Decades of progress
Most decisions for managing acute coronary syndromes can be based on ample data from large randomized trials with hard clinical end points, so there is little reason to provide care that is not evidence-based.
This article reviews some of the trials that provide guidance on diagnosing and managing acute coronary syndromes, including the timing of reperfusion and adjunctive therapies in different situations.
MOST ACUTE CORONARY SYNDROMES ARE NON-ST-ELEVATION CONDITIONS
Acute coronary syndromes range from unstable angina and non-ST-elevation myocardial infarction (NSTEMI) to ST-elevation MI (STEMI), reflecting a continuum of severity of coronary stenosis. The degree of coronary occlusion may ultimately determine whether a patient has unstable angina or MI with or without ST elevation.1
The substrate for all of these is vulnerable plaque. Angiographic studies have indicated that in many cases medium-size plaques (30%–40% stenosis) are more likely to rupture than larger, more obstructive ones. Moderate plaques may be vulnerable because they are less mature, with a large lipid core and a thin cap prone to rupture or erode, exposing the thrombogenic subendothelial components.2

Because the vulnerability of a coronary plaque may not correlate with the severity of stenosis before the plaque ruptures, stress tests and symptoms may not predict the risk of MI. The key role of thrombosis in the pathogenesis also highlights the importance of antithrombotic therapy in the acute phases of acute coronary syndromes, which can significantly reduce mortality and morbidity rates.
Perhaps because of the widespread use of aspirin and statins, most patients who currently present with an acute coronary syndrome have either unstable angina or NSTEMI: of about 1.57 million hospital admissions in 2004 for acute coronary syndromes, for example, only 330,000 (21%) were for STEMI.3
DIAGNOSING ACUTE CORONARY SYNDROME
Symptoms may not be classic
The classic symptoms of acute coronary syndromes are intense, oppressive chest pressure radiating to the left arm, but nearly any discomfort “between the nose and navel” (eg, including the jaw, arm, and epigastric and abdominal areas) may be an acute coronary syndrome. Associated symptoms may include chest heaviness or burning, radiation to the jaw, neck, shoulder, back, or arms, and dyspnea.
Particularly in older, female, postoperative, or diabetic patients, the presentation may be atypical or “silent,” including nausea or vomiting; breathlessness; sweating; arrhythmias; or light-headedness. Especially in these groups, symptoms may be mild or subtle, and acute coronary syndrome may manifest only as “not feeling well.”
The differential diagnosis of acute coronary syndromes is broad. Most important to immediately consider are pulmonary embolism and aortic dissection, as they are life-threatening and are treated differently from acute coronary syndromes. Otherwise, it is best to err on the side of caution and treat for an acute coronary syndrome until it is proven otherwise.
Electrocardiography is critical
Electrocardiography (ECG) gives valuable information about the location, extent, and prognosis of infarction, and it is critically important for distinguishing STEMI from NSTEMI, with ST elevation classically diagnostic of complete coronary occlusion. Q waves can occur early and do not necessarily signify completed infarction, as traditionally thought. ST depression or T inversion indicates that total coronary occlusion is unlikely unless they are in a pattern of circumflex infarct associated with an enlarging R wave in lead V1. An ST elevation in RV4 indicates right ventricular infarction.
The appearance on ECG may evolve over time, so a patient with atypical symptoms and a nonspecific electrocardiogram should be observed for 24 hours or until more specific criteria develop.
Biomarkers in NSTEMI
In MI, cardiac troponin levels begin to rise about 3 hours after the onset of chest pain, and elevations can last for up to 14 days. Levels can also be mildly elevated chronically in patients with renal dysfunction, so positive biomarker tests in that population should be interpreted cautiously.
For STEMI, the opportunity to reperfuse is lost if one waits for cardiac biomarkers to become elevated. But for NSTEMI, they are highly sensitive and specific for identifying patients at high risk and determining who should be treated aggressively. Patients who are biomarker-negative have a better prognosis than patients with identical symptoms and electrocardiograms who are biomarker-positive.
MI is currently defined as a rise in any biomarker (usually troponin) above the 99th percentile for a reference population, with at least one of the following:
- Ischemic symptoms
- New ST/T changes or left bundle branch block
- Pathologic Q waves
- Loss of myocardium or abnormal wall motion seen by imaging
- Intracoronary thrombus.
REPERFUSION FOR ACUTE STEMI
Because acute coronary syndromes have a common pathophysiology, for the most part, lessons from clinical trials in one syndrome are relevant to the others. However, important differences exist regarding the need for immediate reperfusion in STEMI, since in most cases these patients have total rather than partial occlusion.
Fibrinolysis has limitations
The standard of management for STEMI is immediate reperfusion. The goal is to interrupt the wave front of myocardial necrosis, salvage threatened myocardium, and ultimately improve survival.
Five placebo-controlled trials showed a 30% reduction in the death rate in patients who received fibrinolytic therapy within 6 to 12 hours of presentation.4
Patients with ST elevation or with new bundle branch block benefit most from fibrinolytic therapy. Those with ST depression, T inversion, or nonspecific changes on ECG do not benefit; they probably do not have complete coronary occlusion, so the prothrombotic or platelet-activating effects of fibrinolytic therapy may make them worse.5 Further, fibrinolytic therapy poses the risk of intracranial hemorrhage, which, although rare (occurring in up to 1% of cases depending on the drug regimen), is a devastating complication.
In general, absolute contraindications to fibrinolysis include intracranial abnormalities, hemorrhage, and head trauma. An important relative contraindication is uncontrolled blood pressure (> 180/110 mm Hg at any point during hospitalization, including during the immediate presentation). Studies show that even if blood pressure can be controlled, the risk of intracranial hemorrhage is substantially higher, although the risk may not outweigh the benefit of reperfusion, particularly for large infarctions when percutaneous coronary intervention (PCI) is not available as an alternative to fibrinolysis.
Prompt PCI is preferable to fibrinolysis
If PCI is available on site, there is nearly no role for fibrinolytic therapy. PCI is better than fibrinolytic therapy in terms of the degree of reperfusion, reocclusion, MI recurrence, and mortality rate, and it poses little or no risk of intracranial hemorrhage.6
For either fibrinolytic therapy or percutaneous therapy, “time is muscle”: the longer the ischemic time, the higher the mortality rate (relative risk = 1.075 for every 30 minutes of delay, P = .041).7
At centers that do not have PCI on site, studies (mainly from Europe) have shown that it is better to transport the patient for PCI than to give immediate fibrinolytic therapy.7,8 But because the centers studied tended to have short transport times (usually 40 minutes or less), it is uncertain whether the results are applicable throughout the United States.
The delay between symptom onset and presentation is also relevant. Reperfusion within the first 1 to 2 hours after the onset of symptoms provides the greatest degree of myocardial salvage and of reduction in the risk of death; the extent of benefit thereafter is substantially less. As a result, patients who present very early after symptom onset have the most to lose if their reperfusion is delayed by even a few more hours, whereas patients who have already experienced several hours of pain are affected less by additional delay.9 Thus, patients presenting within the “golden” 1 or 2 hours after symptoms begin should be considered for fibrinolytic therapy if transfer for PCI cannot be done expeditiously. It is important for hospitals without PCI available on site to have a system in place for rapid transport of patients when needed.
Guidelines advise that patients with STEMI should undergo PCI rather than receive fibrinolytic therapy as long as PCI is available within 90 minutes of first medical contact. Otherwise, fibrinolysis should be started within 30 minutes.10 For patients who present several hours after symptom onset, PCI may still be preferable even if the transport time is somewhat longer.
PCI after fibrinolytic therapy
In prior decades, PCI immediately after fibrinolytic therapy was associated with an increased risk of bleeding complications and reinfarction. That has changed with improvements in equipment and antithrombotic therapy.
Two large trials conclusively found that routinely transferring high-risk patients for PCI immediately after receiving fibrinolytic therapy (combined half-dose reteplase [Retavase] and abciximab [ReoPro]11 or full-dose tenecteplase [TNKase]12) resulted in much lower rates of ischemic end points without an increase in bleeding complications compared with transferring patients only for rescue PCI after fibrinolytic therapy.
Routine transfer is now the standard of care for high-risk patients after fibrinolytic therapy and probably is best for all patients after an MI.
MANAGING NSTEMI AND UNSTABLE ANGINA
For patients with NSTEMI, immediate reperfusion is usually not required, although initial triage for “early invasive” vs “initial conservative” management must be done early in the hospital course. Randomized trials have evaluated these two approaches, with most studies in the contemporary era reporting improved outcomes with an early invasive approach.
The TACTICS trial,13 the most important of these, enrolled more than 2,200 patients with unstable angina or NSTEMI and randomized them to an early invasive strategy or a conservative strategy. Overall, results were better with the early invasive strategy.
The ICTUS trial.14 Although several studies showed that an early invasive approach was better, the most recent study using the most modern practices—the ICTUS trial—did not find that it reduced death rates. Most patients eventually underwent angiography and revascularization, but not early on. However, all studies showed that rates of recurrent unstable angina and hospitalization were reduced by an early invasive approach, so revascularization does have a role in stabilizing the patient. But in situations of aggressive medical management with antithrombotic and other therapies, an early conservative approach may be an appropriate alternative for many patients.15
The selection of an invasive vs a conservative approach should include a consideration of risk, which can be estimated using a number of criteria, including the Thrombolysis in Myocardial Infarction (TIMI) or the GRACE risk score. When risk was stratified using the TIMI risk score,16 in the TACTICS trial, the higher the risk score, the more likely patients were to benefit from early revascularization.
When an invasive approach is chosen, it does not appear necessary to take patients to catheterization immediately (within 2–24 hours) compared with later during the hospital course.
The TIMACS trial,17 with more than 3,000 patients, tested the benefits of very early vs later revascularization for patients with NSTEMI and unstable angina. Early intervention did not significantly improve outcomes for the primary composite end point of death, MI, and stroke in the overall population enrolled in the trial, but when the secondary end point of refractory ischemia was added in, early intervention was found to be beneficial overall. Moreover, when stratified by risk, high-risk patients significantly benefited from early intervention for the primary end point.
Guidelines for NSTEMI and unstable angina continue to prefer an early invasive strategy, particularly for high-risk patients, although a conservative strategy is considered acceptable if patients receive intensive evidence-based medical therapy and remain clinically stable.18
ANTITHROMBOTIC THERAPIES
Once a revascularization strategy has been chosen, adjunctive therapies should be considered. The most important are the antithrombotic therapies.
Many drugs target platelet activity. Most important are the thromboxane inhibitor aspirin, the adenosine diphosphate (ADP) receptor antagonists clopidogrel (Plavix), prasugrel (Effient), and ticagrelor (Brilinta), and the glycoprotein (GP) IIb/IIIa antagonists abciximab and eptifibatide (Integrilin). Others, such as thrombin receptor antagonists, are under investigation.19
Aspirin for secondary prevention
Evidence is unequivocal for the benefit of aspirin therapy in patients with established or suspected vascular disease.
The ISIS-2 trial20 compared 35-day mortality rates in 16,000 patients with STEMI who were given aspirin, streptokinase, combined streptokinase and aspirin, or placebo. Mortality rates were reduced by aspirin compared with placebo by an extent similar to that achieved with streptokinase, with a further reduction when aspirin and streptokinase were given together.
Therefore, patients with STEMI should be given aspirin daily indefinitely unless they have true aspirin allergy. The dose is 165 to 325 mg initially and 75 to 162 mg daily thereafter.
For NSTEMI and even for secondary prevention in less-acute situations, a number of smaller trials also provide clear evidence of benefit from aspirin therapy.
The CURRENT-OASIS 7 trial21 showed that low maintenance dosages of aspirin (75–100 mg per day) resulted in the same incidence of ischemic end points (cardiovascular death, MI, or stroke) as higher dosages. Although rates of major bleeding events did not differ, a higher rate of gastrointestinal bleeding was evident at just 30 days in patients taking the higher doses. This large trial clearly established that there is no advantage to daily aspirin doses of more than 100 mg.
DUAL ANTIPLATELET THERAPY IS STANDARD
Standard practice now is to use aspirin plus another antiplatelet agent that acts by inhibiting either the ADP receptor (for which there is the most evidence) or the GP IIb/IIIa receptor (which is becoming less used). Dual therapy should begin early in patients with acute coronary syndrome.
Clopidogrel: Well studied with aspirin
The most commonly used ADP antagonist is clopidogrel, a thienopyridine. Much evidence exists for its benefit.
The CURE trial22 randomized more than 12,000 patients with NSTEMI or unstable angina to aspirin plus either clopidogrel or placebo. The incidence of the combined end point of MI, stroke, and cardiovascular death was 20% lower in the clopidogrel group than in the placebo group over 12 months of follow-up. The benefit of clopidogrel began to occur within the first 24 hours after randomization, with a 33% relative risk reduction in the combined end point of cardiovascular death, MI, stroke, and severe ischemia, demonstrating the importance of starting this agent early in the hospital course.
COMMIT23 found a benefit in adding clopidogrel to aspirin in patients with acute STEMI. Although it was only a 30-day trial, significant risk reduction was found in the dual-therapy group for combined death, stroke, or reinfarction. The results of this brief trial were less definitive, but the pathophysiology was similar to non-ST-elevation acute coronary syndromes, so it is reasonable to extrapolate the long-term findings to this setting.
The CURRENT-OASIS 7 trial21 randomized more than 25,000 patients to either clopidogrel in a double dosage (600 mg load, 150 mg/day for 6 days, then 75 mg/day) or standard dosage (300 mg load, 75 mg/day thereafter). Although no overall benefit was found for the higher dosage, a subgroup of more than 17,000 patients who underwent PCI after randomization had a lower risk of developing stent thrombosis. On the other hand, higher doses of clopidogrel caused more major bleeding events.
Ticagrelor and prasugrel: New alternatives to clopidogrel
The principal limitation of clopidogrel is its metabolism. It is a prodrug, ie, it is not active as taken and must be converted to its active state by cytochrome P450 enzymes in the liver. Patients who bear certain polymorphisms in the genes for these enzymes or who are taking other medications that affect this enzymatic pathway may derive less platelet inhibition from the drug, leading to considerable patient-to-patient variability in the degree of antiplatelet effect.
Alternatives to clopidogrel have been developed that inhibit platelets more intensely, are activated more rapidly, and have less interpatient variability. Available now are ticagrelor and prasugrel.24 Like clopidogrel, prasugrel is absorbed as an inactive prodrug, but it is efficiently metabolized by esterases to an active form, and then by a simpler step within the liver to its fully active metabolite.25 Ticagrelor is active as absorbed.26
Pharmacodynamically, the two drugs perform almost identically and much faster than clopidogrel, with equilibrium platelet inhibition reached in less than 1 hour. The degree of platelet inhibition is also more—sometimes twice as much—with the new drugs compared with clopidogrel, and the effect is much more consistent between patients.
Both clopidogrel and prasugrel permanently inhibit the platelet ADP receptor, and 3 to 7 days are therefore required for their antiplatelet effects to completely wear off. In contrast, ticagrelor is a reversible inhibitor and its effects wear off more rapidly. Despite achieving a much higher level of platelet inhibition than clopidogrel, ticagrelor’s activity falls below that of clopidogrel’s by 48 hours of discontinuing the drugs.
Trial of prasugrel vs clopidogrel
The TRITON-TIMI 38 trial27 enrolled more than 13,000 patients with acute coronary syndromes, randomized to receive, either prasugrel or clopidogrel, in addition to aspirin. The patients were all undergoing PCI, so the findings do not apply to patients treated medically with an early conservative approach. The study drug was given only after the decision was made to perform PCI in patients with non-ST-elevation acute coronary syndrome (but given immediately for patients with STEMI, because nearly all those patients undergo PCI).
Prasugrel was clearly beneficial, with a significant 20% lower rate of the combined end point of cardiovascular death, MI, and stroke at 15 months. However, bleeding risk was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% confidence interval 1.02–1.68, P = .03). Looking at individual end points, the advantages of prasugrel were primarily in reducing rates of stent thrombosis and nonfatal MI. Death rates with the two drugs were equivalent, possibly because of the higher risk of bleeding with prasugrel. Bleeding in the prasugrel group was particularly increased in patients who underwent bypass surgery; more patients also needed transfusion.
Subgroup analysis showed that patients with a history of stroke or transient ischemic attack had higher rates of ischemic and bleeding events with prasugrel than with clopidogrel, leading to these being labeled as absolute contraindications to prasugrel. Patients over age 75 or who weighed less than 60 kg experienced excess bleeding risk that closely matched the reduction in ischemic event rates and thus did not have a net benefit with prasugrel.
Trial of ticagrelor vs clopidogrel
The PLATO trial28 included 18,000 patients, of whom 65% underwent revascularization and 35% were treated medically. The drug—clopidogrel or ticagrelor—was given in addition to aspirin at randomization (within 24 hours of symptom onset); this more closely follows clinical practice, in which dual antiplatelet therapy is started as soon as possible. This difference makes the PLATO study more relevant to practice for patients with non-ST-elevation acute coronary syndrome. Also, because they gave the drugs to all patients regardless of whether they were to undergo PCI, this study likely had a higher-risk population, which may be refected in the higher mortality rate at 30 days (5.9% in the clopidogrel group in the PLATO study vs 3.2% in the clopidogrel group in the TRITON study).
Another important difference between the trials testing prasugrel and ticagrelor is that patients who had already received a thienopyridine were excluded from the prasugrel trial but not from the ticagrelor trial. Nearly half the patients in the ticagrelor group were already taking clopidogrel. The clinical implication is that for patients who arrive from another facility and already have been given clopidogrel, it is safe to give ticagrelor. There is limited information about whether that is also true for prasugrel, although there is no known reason why the safety of adding prasugrel to clopidogrel should be different from that of ticagrelor.
The rate of ischemic events was 20% lower in the ticagrelor group than in the clopidogrel group, importantly including reductions in the incidence of death, MI, and stent thrombosis. There was no increase with ticagrelor compared with clopidogrel in bleeding associated with coronary artery bypass graft surgery, likely because of the more rapid washout of the ticagrelor effect, or in the need for blood transfusions. However, the rate of bleeding unrelated to coronary artery bypass was about 20% higher with ticagrelor.
In summary, more intense platelet inhibition reduces the risk of ischemic events, but, particularly for the irreversible inhibitor prasugrel, at the cost of a higher risk of bleeding. In general, the net benefit of these agents in preventing the irreversible complications of MI and (in the case of ticagrelor) death favor the use of the more intense ADP inhibitors in appropriate patients. Ticagrelor is indicated in patients with acute coronary syndromes undergoing invasive or conservative management; prasugrel is indicated in patients undergoing PCI, but contraindicated in patients with a previous stroke or transient ischemic event. Neither drug is indicated in patients undergoing elective PCI outside the setting of acute coronary syndromes, although these agents may be appropriate in patients with intolerance or allergy to clopidogrel.
Glycoprotein IIb/IIIa antagonists for select cases only
GP IIb/IIIa antagonists such as abciximab were previously used more commonly than they are today. Now, with routine pretreatment using thienopyridines, their role in acute coronary syndromes is less clear. They still play a role when routine dual antiplatelet therapy is not used, when prasugrel or ticagrelor is not used, and when heparin rather than an alternative antithrombin agent is used.
A meta-analysis29 of 3,755 patients showed a clear reduction in ischemic complications with abciximab as an adjunct to primary PCI for STEMI in patients treated with heparin.
Kastrati et al30 found that patients with non-ST-elevation acute coronary syndromes benefited from abciximab at the time of PCI with heparin, even though they had been routinely pretreated with clopidogrel. However, benefits were seen only in high-risk patients who had presented with elevated troponins.
On the other hand, the role of GP IIb/IIIa blockade for “upstream” medical management in patients with acute coronary syndromes has been eroded by several studies.
The ACUITY trial31 randomized more than 9,000 patients to receive either routine treatment with a GP IIb/IIIa inhibitor before angiography or deferred selective use in the catheterization laboratory only for patients undergoing PCI. No significant differences were found in rates of MI and death.
The Early ACS trial32 compared early routine eptifibatide vs delayed, provisional eptifibatide in 9,492 patients with acute coronary syndromes without ST elevation and who were assigned to an invasive strategy. The early-eptifibatide group received two boluses and an infusion of eptifibatide before angiography; the others received a placebo infusion, with provisional eptifibatide after angiography if the patient underwent PCI and was deemed at high risk. No significant difference in rates of death or MI were noted, and the early-eptifibatide group had significantly higher rates of bleeding and need for transfusion.
The FINESSE trial33 also discredited “facilitating” PCI by giving GP IIb/IIIa antagonists in patients with STEMI before arrival in the catheterization laboratory, with no benefit to giving abciximab ahead of time vs in the catheterization laboratory, and with an increased risk of bleeding complications.
These studies have helped narrow the use of GP IIb/IIIa inhibitors to the catheterization laboratory in conjunction with heparin anticoagulation (as compared with bivalirudin [Angiomax]; see below) and only in select or high-risk cases. These drugs are indicated in the medical phase of management only if patients cannot be stabilized by aspirin or ADP inhibition.
NEWER ANTITHROMBOTICS: ADVANTAGES UNCLEAR
The complex coagulation cascade has a number of components, but only a few are targeted by drugs that are approved and recommended: fondaparinux (Arixtra) and oral factor Xa inhibitors affect the prothrombinase complex (including factor X); bivalirudin and oral factor IIa inhibitors affect thrombin; and heparin and the low-molecular-weight heparins inhibit both targets.
Low-molecular-weight heparins
The SYNERGY trial34 randomized nearly 10,000 patients with non-ST-elevation acute coronary syndromes at high risk for ischemic cardiac complications managed with an invasive approach to either the low-molecular-weight heparin enoxaparin (Lovenox) or intravenous unfractionated heparin immediately after enrollment. Most patients underwent catheterization and revascularization. No clinical advantage was found for enoxaparin, and bleeding complications were increased.
The EXTRACT-TIMI 25 trial35 randomized more than 20,000 patients with STEMI who were about to undergo fibrinolysis to receive either enoxaparin throughout hospitalization (average of 8 days) or unfractionated heparin for at least 48 hours. The enoxaparin group had a lower rate of recurrent MI, but it was unclear if the difference was in part attributable to the longer therapy time. The enoxaparin group also had more bleeding.
Fondaparinux
The OASIS-5 trial36,37 compared enoxaparin and fondaparinux, an exclusive factor Xa inhibitor, in more than 20,000 patients with unstable angina or NSTEMI. Fondaparinux was associated with a lower risk of death and reinfarction as well as fewer bleeding events. However, the benefits were almost exclusively in patients treated medically. In those undergoing PCI within the first 8 days, no benefit was found, although there was still a significant reduction in major bleeding events. Catheter thrombosis was also increased in patients taking fondaparinux, but only in those who did not receive adequate unfractionated heparin treatment before PCI.
Bivalirudin superior at time of catheterization
The most significant advance in antithrombotic therapy for patients with acute coronary syndromes is bivalirudin. This drug has a clear role only in the catheterization laboratory, where patients can be switched to it from heparin, low-molecular-weight heparin, or fondaparinux.
Three trials38–40 evaluated the drug in a total of more than 20,000 patients receiving invasive management of coronary artery disease undergoing PCI for elective indications, NSTEMI, or STEMI.
Results were remarkably similar across the three trials. Patients who were treated with bivalirudin alone had the same rate of ischemic end points at 30 days as those receiving heparin plus a GP IIb/IIIa inhibitor, but bivalirudin was associated with a consistent and significant 40% to 50% lower bleeding risk. For the highest-risk patients, those with STEMI, the bivalirudin group also had a significantly lower risk of death at 1 year.41
OTHER DRUGS: EARLY TREATMENT NO LONGER ROUTINE
Most data for the use of therapies aside from antithrombotics are from studies of patients with STEMI, but findings can logically be extrapolated to those with non-ST-elevation acute coronary syndromes.
Beta-blockers: Cardiogenic shock a risk
For beta-blockers, many historical trials were done in stable coronary disease, but there are no large trials in the setting of NSTEMI or unstable angina, and only recently have there been large trials for STEMI. Before the availability of recent evidence, standard practice was to treat STEMI routinely with intravenous metoprolol (Lopressor) and then oral metoprolol.
When large studies were finally conducted, the results were sobering.
COMMIT.42 Nearly 46,000 patients with suspected acute MI were randomized to receive either metoprolol (up to 15 mg intravenously, then 200 mg by mouth daily until discharge or for up to 4 weeks in the hospital) or placebo. Surprisingly, although rates of reinfarction and ventricular fibrillation were lower with metoprolol, a higher risk of cardiogenic shock with early beta-blockade offset these benefits and the net mortality rate was not reduced. This study led to a reduction in the early use of beta-blockers in patients with STEMI.
The standard of care has now shifted from beta-blockers in everyone as early as possible after MI to being more cautious in patients with contraindications, including signs of heart failure or a low-output state, or even in those of advanced age or with borderline low blood pressure or a high heart rate. Patients who present late and therefore may have a larger infarct are also at higher risk.
Although the goal should be to ultimately discharge patients on beta-blocker therapy after an MI, there should be no rush to start one early.
Carvedilol now preferred after STEMI
The CAPRICORN trial43 randomized nearly 2,000 patients following MI with left ventricular dysfunction (an ejection fraction of 40% or below) to either placebo or the beta-blocker carvedilol (Coreg). Patients taking the drug had a clear reduction in rates of death and reinfarction, leading to this drug becoming the beta-blocker of choice in patients with ventricular dysfunction after STEMI.
Angiotensin-converting enzyme inhibitors: Early risk of cardiogenic shock
The use of angiotensin-converting enzyme (ACE) inhibitors after MI is also supported by several studies.44 Two very large studies, one of nearly 60,000 patients and one of nearly 20,000, showed a clear reduction in the mortality rate in those who received an ACE inhibitor. Most of the benefit was in patients with an ejection fraction of less than 40%. On the basis of these trials, ACE inhibitors are indicated for all patients for the first 30 days after MI and then indefinitely for those with left ventricular dysfunction. However, the trial in which an ACE inhibitor was given intravenously early on had to be stopped prematurely because of worse outcomes owing to cardiogenic shock.
These studies highlight again that for patients who are unstable in the first few days of an acute coronary syndrome, it is best to wait until their condition stabilizes and to start these therapies before hospital discharge.
Intensive statin therapy
In the last 20 years, unequivocal evidence has emerged to support the beneficial role of statins for secondary prevention in patients with established coronary artery disease. More-recent trials have also shown that intensive statin therapy (a high dose of a potent statin) improves outcomes better than lower doses.
The PROVE-IT TIMI 22 trial45 randomized patients after an acute coronary syndrome to receive either standard therapy (pravastatin [Pravachol] 40 mg) or intensive therapy (atorvastatin [Lipitor] 80 mg). The intensive-therapy group had a significantly lower rate of major cardiovascular events, and the difference persisted and grew over 30 months of follow-up.
A number of studies confirmed this and broadened the patient population to those with unstable or stable coronary disease. Regardless of the risk profile, the effects were consistent and showed that high-dose statins were better in preventing coronary death and MI.46
Guidelines are evolving toward recommendation of highest doses of statins independently of the target level of low-density lipoprotein cholesterol.
- Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines; Canadian Cardiovascular Society. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction. Circulation 2004; 110:e82–e292. Erratum in: Circulation 2005; 111:2013–2014.
- Davies MJ. The pathophysiology of acute coronary syndromes. Heart 2000; 83:361–366.
- Rosamond W, Flegal K, Friday G, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics–2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2007; 115:e69–e171.
- Granger CB, Califf RM, Topol EJ. Thrombolytic therapy for acute myocardial infarction. A review. Drugs 1992; 44:293–325.
- Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Lancet 1994; 343:311–322.
- Keely EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet 2003; 361:13–20
- De Luca G, Suryapranata H, Ottervanger JP, Antman EM. Time delay to treatment and mortality in primary angioplasty for acute myocardial infarction: every minute of delay counts. Circulation 2004; 109:1223–1225.
- Dalby M, Bouzamondo A, Lechat P, Montalescot G. Transfer for primary angioplasty versus immediate thrombolysis in acute myocardial infarction: a meta-analysis. Circulation 2003; 108:1809–1814.
- Gersh BJ, Stone GW, White HD, Holmes DR Jr. Pharmacological facilitation of primary percutaneous coronary intervention for acute myocardial infarction: is the slope of the curve the shape of the future? JAMA 2005; 293:979–986.
- Antman EM, Hand M, Armstron PW, et al; Canadian Cardiovascular Society; American Academy of Family Physicians; American College of Cardiology; American Heart Association. 2007 focused update of the ACC/AHA 2004 guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2008; 51:210–247.
- Di Mario C, Dudek D, Piscione F, et al; CARESS-in-AMI (Combined Abciximab Reteplase Stent Study in Acute Myocardial Infarction) Investigators. Immediate angioplasty versus standard therapy with rescue angioplasty after thrombolysis in the Combined Abciximab REteplase Stent Study in Acute Myocardial Infarction (CARESS-in-AMI): an open, prospective, randomised, multicentre trial. Lancet 2008; 371:559–568.
- Cantor WJ, Fitchett D, Borgundvaag B, et al; TRANSFER-AMI Trial Investigators. Routine early angioplasty after fibrinolysis for acute myocardial infarction. N Engl J Med 2009; 360:2705–2718.
- Cannon CP, Weintraub WS, Demopoulos LA, et al; TACTICS (Treat Angina With Aggrastat and Determine Cost of Therapy With an Invasive or Conservative Strategy)–Thrombolysis in Myocardial Infarction 18 Investigators. Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med 2001; 344:1879–1887.
- Damman P, Hirsch A, Windhausen F, Tijssen JG, de Winter RJ; ICTUS Investigators. 5-year clinical outcomes in the ICTUS (Invasive versus Conservative Treatment in Unstable coronary Syndromes) trial a randomized comparison of an early invasive versus selective invasive management in patients with non-ST-segment elevation acute coronary syndrome. J Am Coll Cardiol 2010; 55:858–864.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- Antman EM, Cohen M, Bernink PJ, et al. The TIMI risk score for unstable angina/non-ST elevation MI: a method for prognostication and therapeutic decision making. JAMA 2000; 284:835–842.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- Yousef O, Bhatt DL. The evolution of antiplatelet therapy in cardiovascular disease. Nat Rev Cardiol 2011; 8:547–559.
- ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- CURRENT-OASIS 7 Investigators; Mehta SR, Bassand JP, Chrolavicius S, et al. Dose comparisons of clopidogrel and aspirin in acute coronary syndromes. N Engl J Med 2010; 363:930–942.
- Yusuf S, Mehta SR, Zhao F, et al; Clopidogrel in Unstable angina to prevent Recurrent Events Trial Investigators. Early and late effects of clopidogrel in patients with acute coronary syndromes. Circulation 2003; 107:966–972.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Schömig A. Ticagrelor—is there need for a new player in the antiplatelet-therapy field? N Engl J Med 2009; 361:1108–1111.
- Wiviott SD, Antman EM, Braunwald E. Prasugrel. Circulation 2010; 122:394–403.
- Gurbel PA, Bliden KP, Butler K, et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: the ONSET/OFFSET study. Circulation 2009; 120:2577–2585.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- de Queiroz Fernandes Araujo JO, Veloso HH, Braga De Paiva JM, Fiho MW, Vincenzo De Paola AA. Efficacy and safety of abciximab on acute myocardial infarction treated with percutaneous coronary interventions: a meta-analysis of randomized, controlled trials. Am Heart J 2004; 148:937–943.
- Kastrati A, Mehilli J, Neuman FJ, et al; Intracoronary Stenting and Antithrombotic: Regimen Rapid Early Action for Coronary Treatment 2 (ISAR-REACT 2) Trial Investigators. Abciximab in patients with acute coronary syndromes undergoing percutaneous coronary intervention after clopidogrel pretreatment: the ISAR-REACT 2 randomized trial. JAMA 2006; 295:1531–1538.
- Stone GW, Bertrand ME, Moses JW, et al; ACUITY Investigators. Routine upstream initiation vs deferred selective use of glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: the ACUITY Timing trial. JAMA 2007; 297:591–602.
- Giugliano RP, White JA, Bode C, et al; Early ACS Investigators. Early vs delayed, provisional eptifibatide in acute coronary syndromes. N Engl J Med 2009; 360:2176–2190.
- Ellis SG, Tendera M, de Belder MA, et al; FINESSE Investigators. Facilitated PCI in patients with ST-elevation myocardial infarction. N Engl J Med 2008; 358:2205–2217.
- Fergusson JJ, Califf RM, Antman EM, et al; SYNERGY Trial Investigators. Enoxaparin vs unfractionated heparin in high-risk patients with non-ST-segment elevation acute coronary syndromes managed with an intended early invasive strategy: primary results of the SYNERGY randomized trial. JAMA 2004; 292:45–54.
- Antman EM, Morrow DA, McCabe CH; EXTRACT-TIMI 25 Investigators. Enoxaparin versus unfractionated heparin with fibrinolysis for ST-elevation myocardial infarction. N Engl J Med 2006; 354:1477–1488.
- The Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Mehta SR, Granger CB, Eikelboom JW, et al. Efficacy and safety of fondaparinux versus enoxaparin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: results from the OASIS-5 trial. J Am Coll Cardiol 2007; 50:1742–1751.
- Lincoff AM, Bittl JA, Harrington RA, et al; REPLACE-2 Investigators. Bivalirudin and provisional glycoprotein IIb/IIIa blockade compared with heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary intervention: REPLACE-2 randomized trial. JAMA 2003; 289:853–863.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Stone GW, Witzenbichler B, Guagliumi G, et al; HORIZONS-AMI Trial Investigators. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med 2007; 358:2218–2230.
- Mehran R, Lansky AJ, Witzenbichler B, et al; HORIZONS-AMI Trial Investigators. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet 2009; 374:1149–1159.
- Chen ZM, Pan HC, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) Collaborative Group. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1622–1632.
- Dargie JH. Effect of carvedilol on outcome after myocardial infarction in patients with left-ventricular dysfunction: the CAPRICORN randomised trial. Lancet 2001; 357:1385–1390.
- Hennekens CH, Albert CM, Godfried SL, Gaziano JM, Buring JE. Adjunctive drug therapy of acute myocardial infarction—evidence from clinical trials. N Engl J Med 1996; 335:1660–1667.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Cannon CP, Steinberg BA, Murphy SA, Mega JL, Braunwald E. Meta-analysis of cardiovascular outcomes trials comparing intensive versus moderate statin therapy. J Am Coll Cardiol 2006; 48:438–445.
Most decisions for managing acute coronary syndromes can be based on ample data from large randomized trials with hard clinical end points, so there is little reason to provide care that is not evidence-based.
This article reviews some of the trials that provide guidance on diagnosing and managing acute coronary syndromes, including the timing of reperfusion and adjunctive therapies in different situations.
MOST ACUTE CORONARY SYNDROMES ARE NON-ST-ELEVATION CONDITIONS
Acute coronary syndromes range from unstable angina and non-ST-elevation myocardial infarction (NSTEMI) to ST-elevation MI (STEMI), reflecting a continuum of severity of coronary stenosis. The degree of coronary occlusion may ultimately determine whether a patient has unstable angina or MI with or without ST elevation.1
The substrate for all of these is vulnerable plaque. Angiographic studies have indicated that in many cases medium-size plaques (30%–40% stenosis) are more likely to rupture than larger, more obstructive ones. Moderate plaques may be vulnerable because they are less mature, with a large lipid core and a thin cap prone to rupture or erode, exposing the thrombogenic subendothelial components.2
Because the vulnerability of a coronary plaque may not correlate with the severity of stenosis before the plaque ruptures, stress tests and symptoms may not predict the risk of MI. The key role of thrombosis in the pathogenesis also highlights the importance of antithrombotic therapy in the acute phases of acute coronary syndromes, which can significantly reduce mortality and morbidity rates.
Perhaps because of the widespread use of aspirin and statins, most patients who currently present with an acute coronary syndrome have either unstable angina or NSTEMI: of about 1.57 million hospital admissions in 2004 for acute coronary syndromes, for example, only 330,000 (21%) were for STEMI.3
DIAGNOSING ACUTE CORONARY SYNDROME
Symptoms may not be classic
The classic symptoms of acute coronary syndromes are intense, oppressive chest pressure radiating to the left arm, but nearly any discomfort “between the nose and navel” (eg, including the jaw, arm, and epigastric and abdominal areas) may be an acute coronary syndrome. Associated symptoms may include chest heaviness or burning, radiation to the jaw, neck, shoulder, back, or arms, and dyspnea.
Particularly in older, female, postoperative, or diabetic patients, the presentation may be atypical or “silent,” including nausea or vomiting; breathlessness; sweating; arrhythmias; or light-headedness. Especially in these groups, symptoms may be mild or subtle, and acute coronary syndrome may manifest only as “not feeling well.”
The differential diagnosis of acute coronary syndromes is broad. Most important to immediately consider are pulmonary embolism and aortic dissection, as they are life-threatening and are treated differently from acute coronary syndromes. Otherwise, it is best to err on the side of caution and treat for an acute coronary syndrome until it is proven otherwise.
Electrocardiography is critical
Electrocardiography (ECG) gives valuable information about the location, extent, and prognosis of infarction, and it is critically important for distinguishing STEMI from NSTEMI, with ST elevation classically diagnostic of complete coronary occlusion. Q waves can occur early and do not necessarily signify completed infarction, as traditionally thought. ST depression or T inversion indicates that total coronary occlusion is unlikely unless they are in a pattern of circumflex infarct associated with an enlarging R wave in lead V1. An ST elevation in RV4 indicates right ventricular infarction.
The appearance on ECG may evolve over time, so a patient with atypical symptoms and a nonspecific electrocardiogram should be observed for 24 hours or until more specific criteria develop.
Biomarkers in NSTEMI
In MI, cardiac troponin levels begin to rise about 3 hours after the onset of chest pain, and elevations can last for up to 14 days. Levels can also be mildly elevated chronically in patients with renal dysfunction, so positive biomarker tests in that population should be interpreted cautiously.
For STEMI, the opportunity to reperfuse is lost if one waits for cardiac biomarkers to become elevated. But for NSTEMI, they are highly sensitive and specific for identifying patients at high risk and determining who should be treated aggressively. Patients who are biomarker-negative have a better prognosis than patients with identical symptoms and electrocardiograms who are biomarker-positive.
MI is currently defined as a rise in any biomarker (usually troponin) above the 99th percentile for a reference population, with at least one of the following:
- Ischemic symptoms
- New ST/T changes or left bundle branch block
- Pathologic Q waves
- Loss of myocardium or abnormal wall motion seen by imaging
- Intracoronary thrombus.
REPERFUSION FOR ACUTE STEMI
Because acute coronary syndromes have a common pathophysiology, for the most part, lessons from clinical trials in one syndrome are relevant to the others. However, important differences exist regarding the need for immediate reperfusion in STEMI, since in most cases these patients have total rather than partial occlusion.
Fibrinolysis has limitations
The standard of management for STEMI is immediate reperfusion. The goal is to interrupt the wave front of myocardial necrosis, salvage threatened myocardium, and ultimately improve survival.
Five placebo-controlled trials showed a 30% reduction in the death rate in patients who received fibrinolytic therapy within 6 to 12 hours of presentation.4
Patients with ST elevation or with new bundle branch block benefit most from fibrinolytic therapy. Those with ST depression, T inversion, or nonspecific changes on ECG do not benefit; they probably do not have complete coronary occlusion, so the prothrombotic or platelet-activating effects of fibrinolytic therapy may make them worse.5 Further, fibrinolytic therapy poses the risk of intracranial hemorrhage, which, although rare (occurring in up to 1% of cases depending on the drug regimen), is a devastating complication.
In general, absolute contraindications to fibrinolysis include intracranial abnormalities, hemorrhage, and head trauma. An important relative contraindication is uncontrolled blood pressure (> 180/110 mm Hg at any point during hospitalization, including during the immediate presentation). Studies show that even if blood pressure can be controlled, the risk of intracranial hemorrhage is substantially higher, although the risk may not outweigh the benefit of reperfusion, particularly for large infarctions when percutaneous coronary intervention (PCI) is not available as an alternative to fibrinolysis.
Prompt PCI is preferable to fibrinolysis
If PCI is available on site, there is nearly no role for fibrinolytic therapy. PCI is better than fibrinolytic therapy in terms of the degree of reperfusion, reocclusion, MI recurrence, and mortality rate, and it poses little or no risk of intracranial hemorrhage.6
For either fibrinolytic therapy or percutaneous therapy, “time is muscle”: the longer the ischemic time, the higher the mortality rate (relative risk = 1.075 for every 30 minutes of delay, P = .041).7
At centers that do not have PCI on site, studies (mainly from Europe) have shown that it is better to transport the patient for PCI than to give immediate fibrinolytic therapy.7,8 But because the centers studied tended to have short transport times (usually 40 minutes or less), it is uncertain whether the results are applicable throughout the United States.
The delay between symptom onset and presentation is also relevant. Reperfusion within the first 1 to 2 hours after the onset of symptoms provides the greatest degree of myocardial salvage and of reduction in the risk of death; the extent of benefit thereafter is substantially less. As a result, patients who present very early after symptom onset have the most to lose if their reperfusion is delayed by even a few more hours, whereas patients who have already experienced several hours of pain are affected less by additional delay.9 Thus, patients presenting within the “golden” 1 or 2 hours after symptoms begin should be considered for fibrinolytic therapy if transfer for PCI cannot be done expeditiously. It is important for hospitals without PCI available on site to have a system in place for rapid transport of patients when needed.
Guidelines advise that patients with STEMI should undergo PCI rather than receive fibrinolytic therapy as long as PCI is available within 90 minutes of first medical contact. Otherwise, fibrinolysis should be started within 30 minutes.10 For patients who present several hours after symptom onset, PCI may still be preferable even if the transport time is somewhat longer.
PCI after fibrinolytic therapy
In prior decades, PCI immediately after fibrinolytic therapy was associated with an increased risk of bleeding complications and reinfarction. That has changed with improvements in equipment and antithrombotic therapy.
Two large trials conclusively found that routinely transferring high-risk patients for PCI immediately after receiving fibrinolytic therapy (combined half-dose reteplase [Retavase] and abciximab [ReoPro]11 or full-dose tenecteplase [TNKase]12) resulted in much lower rates of ischemic end points without an increase in bleeding complications compared with transferring patients only for rescue PCI after fibrinolytic therapy.
Routine transfer is now the standard of care for high-risk patients after fibrinolytic therapy and probably is best for all patients after an MI.
MANAGING NSTEMI AND UNSTABLE ANGINA
For patients with NSTEMI, immediate reperfusion is usually not required, although initial triage for “early invasive” vs “initial conservative” management must be done early in the hospital course. Randomized trials have evaluated these two approaches, with most studies in the contemporary era reporting improved outcomes with an early invasive approach.
The TACTICS trial,13 the most important of these, enrolled more than 2,200 patients with unstable angina or NSTEMI and randomized them to an early invasive strategy or a conservative strategy. Overall, results were better with the early invasive strategy.
The ICTUS trial.14 Although several studies showed that an early invasive approach was better, the most recent study using the most modern practices—the ICTUS trial—did not find that it reduced death rates. Most patients eventually underwent angiography and revascularization, but not early on. However, all studies showed that rates of recurrent unstable angina and hospitalization were reduced by an early invasive approach, so revascularization does have a role in stabilizing the patient. But in situations of aggressive medical management with antithrombotic and other therapies, an early conservative approach may be an appropriate alternative for many patients.15
The selection of an invasive vs a conservative approach should include a consideration of risk, which can be estimated using a number of criteria, including the Thrombolysis in Myocardial Infarction (TIMI) or the GRACE risk score. When risk was stratified using the TIMI risk score,16 in the TACTICS trial, the higher the risk score, the more likely patients were to benefit from early revascularization.
When an invasive approach is chosen, it does not appear necessary to take patients to catheterization immediately (within 2–24 hours) compared with later during the hospital course.
The TIMACS trial,17 with more than 3,000 patients, tested the benefits of very early vs later revascularization for patients with NSTEMI and unstable angina. Early intervention did not significantly improve outcomes for the primary composite end point of death, MI, and stroke in the overall population enrolled in the trial, but when the secondary end point of refractory ischemia was added in, early intervention was found to be beneficial overall. Moreover, when stratified by risk, high-risk patients significantly benefited from early intervention for the primary end point.
Guidelines for NSTEMI and unstable angina continue to prefer an early invasive strategy, particularly for high-risk patients, although a conservative strategy is considered acceptable if patients receive intensive evidence-based medical therapy and remain clinically stable.18
ANTITHROMBOTIC THERAPIES
Once a revascularization strategy has been chosen, adjunctive therapies should be considered. The most important are the antithrombotic therapies.
Many drugs target platelet activity. Most important are the thromboxane inhibitor aspirin, the adenosine diphosphate (ADP) receptor antagonists clopidogrel (Plavix), prasugrel (Effient), and ticagrelor (Brilinta), and the glycoprotein (GP) IIb/IIIa antagonists abciximab and eptifibatide (Integrilin). Others, such as thrombin receptor antagonists, are under investigation.19
Aspirin for secondary prevention
Evidence is unequivocal for the benefit of aspirin therapy in patients with established or suspected vascular disease.
The ISIS-2 trial20 compared 35-day mortality rates in 16,000 patients with STEMI who were given aspirin, streptokinase, combined streptokinase and aspirin, or placebo. Mortality rates were reduced by aspirin compared with placebo by an extent similar to that achieved with streptokinase, with a further reduction when aspirin and streptokinase were given together.
Therefore, patients with STEMI should be given aspirin daily indefinitely unless they have true aspirin allergy. The dose is 165 to 325 mg initially and 75 to 162 mg daily thereafter.
For NSTEMI and even for secondary prevention in less-acute situations, a number of smaller trials also provide clear evidence of benefit from aspirin therapy.
The CURRENT-OASIS 7 trial21 showed that low maintenance dosages of aspirin (75–100 mg per day) resulted in the same incidence of ischemic end points (cardiovascular death, MI, or stroke) as higher dosages. Although rates of major bleeding events did not differ, a higher rate of gastrointestinal bleeding was evident at just 30 days in patients taking the higher doses. This large trial clearly established that there is no advantage to daily aspirin doses of more than 100 mg.
DUAL ANTIPLATELET THERAPY IS STANDARD
Standard practice now is to use aspirin plus another antiplatelet agent that acts by inhibiting either the ADP receptor (for which there is the most evidence) or the GP IIb/IIIa receptor (which is becoming less used). Dual therapy should begin early in patients with acute coronary syndrome.
Clopidogrel: Well studied with aspirin
The most commonly used ADP antagonist is clopidogrel, a thienopyridine. Much evidence exists for its benefit.
The CURE trial22 randomized more than 12,000 patients with NSTEMI or unstable angina to aspirin plus either clopidogrel or placebo. The incidence of the combined end point of MI, stroke, and cardiovascular death was 20% lower in the clopidogrel group than in the placebo group over 12 months of follow-up. The benefit of clopidogrel began to occur within the first 24 hours after randomization, with a 33% relative risk reduction in the combined end point of cardiovascular death, MI, stroke, and severe ischemia, demonstrating the importance of starting this agent early in the hospital course.
COMMIT23 found a benefit in adding clopidogrel to aspirin in patients with acute STEMI. Although it was only a 30-day trial, significant risk reduction was found in the dual-therapy group for combined death, stroke, or reinfarction. The results of this brief trial were less definitive, but the pathophysiology was similar to non-ST-elevation acute coronary syndromes, so it is reasonable to extrapolate the long-term findings to this setting.
The CURRENT-OASIS 7 trial21 randomized more than 25,000 patients to either clopidogrel in a double dosage (600 mg load, 150 mg/day for 6 days, then 75 mg/day) or standard dosage (300 mg load, 75 mg/day thereafter). Although no overall benefit was found for the higher dosage, a subgroup of more than 17,000 patients who underwent PCI after randomization had a lower risk of developing stent thrombosis. On the other hand, higher doses of clopidogrel caused more major bleeding events.
Ticagrelor and prasugrel: New alternatives to clopidogrel
The principal limitation of clopidogrel is its metabolism. It is a prodrug, ie, it is not active as taken and must be converted to its active state by cytochrome P450 enzymes in the liver. Patients who bear certain polymorphisms in the genes for these enzymes or who are taking other medications that affect this enzymatic pathway may derive less platelet inhibition from the drug, leading to considerable patient-to-patient variability in the degree of antiplatelet effect.
Alternatives to clopidogrel have been developed that inhibit platelets more intensely, are activated more rapidly, and have less interpatient variability. Available now are ticagrelor and prasugrel.24 Like clopidogrel, prasugrel is absorbed as an inactive prodrug, but it is efficiently metabolized by esterases to an active form, and then by a simpler step within the liver to its fully active metabolite.25 Ticagrelor is active as absorbed.26
Pharmacodynamically, the two drugs perform almost identically and much faster than clopidogrel, with equilibrium platelet inhibition reached in less than 1 hour. The degree of platelet inhibition is also more—sometimes twice as much—with the new drugs compared with clopidogrel, and the effect is much more consistent between patients.
Both clopidogrel and prasugrel permanently inhibit the platelet ADP receptor, and 3 to 7 days are therefore required for their antiplatelet effects to completely wear off. In contrast, ticagrelor is a reversible inhibitor and its effects wear off more rapidly. Despite achieving a much higher level of platelet inhibition than clopidogrel, ticagrelor’s activity falls below that of clopidogrel’s by 48 hours of discontinuing the drugs.
Trial of prasugrel vs clopidogrel
The TRITON-TIMI 38 trial27 enrolled more than 13,000 patients with acute coronary syndromes, randomized to receive, either prasugrel or clopidogrel, in addition to aspirin. The patients were all undergoing PCI, so the findings do not apply to patients treated medically with an early conservative approach. The study drug was given only after the decision was made to perform PCI in patients with non-ST-elevation acute coronary syndrome (but given immediately for patients with STEMI, because nearly all those patients undergo PCI).
Prasugrel was clearly beneficial, with a significant 20% lower rate of the combined end point of cardiovascular death, MI, and stroke at 15 months. However, bleeding risk was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% confidence interval 1.02–1.68, P = .03). Looking at individual end points, the advantages of prasugrel were primarily in reducing rates of stent thrombosis and nonfatal MI. Death rates with the two drugs were equivalent, possibly because of the higher risk of bleeding with prasugrel. Bleeding in the prasugrel group was particularly increased in patients who underwent bypass surgery; more patients also needed transfusion.
Subgroup analysis showed that patients with a history of stroke or transient ischemic attack had higher rates of ischemic and bleeding events with prasugrel than with clopidogrel, leading to these being labeled as absolute contraindications to prasugrel. Patients over age 75 or who weighed less than 60 kg experienced excess bleeding risk that closely matched the reduction in ischemic event rates and thus did not have a net benefit with prasugrel.
Trial of ticagrelor vs clopidogrel
The PLATO trial28 included 18,000 patients, of whom 65% underwent revascularization and 35% were treated medically. The drug—clopidogrel or ticagrelor—was given in addition to aspirin at randomization (within 24 hours of symptom onset); this more closely follows clinical practice, in which dual antiplatelet therapy is started as soon as possible. This difference makes the PLATO study more relevant to practice for patients with non-ST-elevation acute coronary syndrome. Also, because they gave the drugs to all patients regardless of whether they were to undergo PCI, this study likely had a higher-risk population, which may be refected in the higher mortality rate at 30 days (5.9% in the clopidogrel group in the PLATO study vs 3.2% in the clopidogrel group in the TRITON study).
Another important difference between the trials testing prasugrel and ticagrelor is that patients who had already received a thienopyridine were excluded from the prasugrel trial but not from the ticagrelor trial. Nearly half the patients in the ticagrelor group were already taking clopidogrel. The clinical implication is that for patients who arrive from another facility and already have been given clopidogrel, it is safe to give ticagrelor. There is limited information about whether that is also true for prasugrel, although there is no known reason why the safety of adding prasugrel to clopidogrel should be different from that of ticagrelor.
The rate of ischemic events was 20% lower in the ticagrelor group than in the clopidogrel group, importantly including reductions in the incidence of death, MI, and stent thrombosis. There was no increase with ticagrelor compared with clopidogrel in bleeding associated with coronary artery bypass graft surgery, likely because of the more rapid washout of the ticagrelor effect, or in the need for blood transfusions. However, the rate of bleeding unrelated to coronary artery bypass was about 20% higher with ticagrelor.
In summary, more intense platelet inhibition reduces the risk of ischemic events, but, particularly for the irreversible inhibitor prasugrel, at the cost of a higher risk of bleeding. In general, the net benefit of these agents in preventing the irreversible complications of MI and (in the case of ticagrelor) death favor the use of the more intense ADP inhibitors in appropriate patients. Ticagrelor is indicated in patients with acute coronary syndromes undergoing invasive or conservative management; prasugrel is indicated in patients undergoing PCI, but contraindicated in patients with a previous stroke or transient ischemic event. Neither drug is indicated in patients undergoing elective PCI outside the setting of acute coronary syndromes, although these agents may be appropriate in patients with intolerance or allergy to clopidogrel.
Glycoprotein IIb/IIIa antagonists for select cases only
GP IIb/IIIa antagonists such as abciximab were previously used more commonly than they are today. Now, with routine pretreatment using thienopyridines, their role in acute coronary syndromes is less clear. They still play a role when routine dual antiplatelet therapy is not used, when prasugrel or ticagrelor is not used, and when heparin rather than an alternative antithrombin agent is used.
A meta-analysis29 of 3,755 patients showed a clear reduction in ischemic complications with abciximab as an adjunct to primary PCI for STEMI in patients treated with heparin.
Kastrati et al30 found that patients with non-ST-elevation acute coronary syndromes benefited from abciximab at the time of PCI with heparin, even though they had been routinely pretreated with clopidogrel. However, benefits were seen only in high-risk patients who had presented with elevated troponins.
On the other hand, the role of GP IIb/IIIa blockade for “upstream” medical management in patients with acute coronary syndromes has been eroded by several studies.
The ACUITY trial31 randomized more than 9,000 patients to receive either routine treatment with a GP IIb/IIIa inhibitor before angiography or deferred selective use in the catheterization laboratory only for patients undergoing PCI. No significant differences were found in rates of MI and death.
The Early ACS trial32 compared early routine eptifibatide vs delayed, provisional eptifibatide in 9,492 patients with acute coronary syndromes without ST elevation and who were assigned to an invasive strategy. The early-eptifibatide group received two boluses and an infusion of eptifibatide before angiography; the others received a placebo infusion, with provisional eptifibatide after angiography if the patient underwent PCI and was deemed at high risk. No significant difference in rates of death or MI were noted, and the early-eptifibatide group had significantly higher rates of bleeding and need for transfusion.
The FINESSE trial33 also discredited “facilitating” PCI by giving GP IIb/IIIa antagonists in patients with STEMI before arrival in the catheterization laboratory, with no benefit to giving abciximab ahead of time vs in the catheterization laboratory, and with an increased risk of bleeding complications.
These studies have helped narrow the use of GP IIb/IIIa inhibitors to the catheterization laboratory in conjunction with heparin anticoagulation (as compared with bivalirudin [Angiomax]; see below) and only in select or high-risk cases. These drugs are indicated in the medical phase of management only if patients cannot be stabilized by aspirin or ADP inhibition.
NEWER ANTITHROMBOTICS: ADVANTAGES UNCLEAR
The complex coagulation cascade has a number of components, but only a few are targeted by drugs that are approved and recommended: fondaparinux (Arixtra) and oral factor Xa inhibitors affect the prothrombinase complex (including factor X); bivalirudin and oral factor IIa inhibitors affect thrombin; and heparin and the low-molecular-weight heparins inhibit both targets.
Low-molecular-weight heparins
The SYNERGY trial34 randomized nearly 10,000 patients with non-ST-elevation acute coronary syndromes at high risk for ischemic cardiac complications managed with an invasive approach to either the low-molecular-weight heparin enoxaparin (Lovenox) or intravenous unfractionated heparin immediately after enrollment. Most patients underwent catheterization and revascularization. No clinical advantage was found for enoxaparin, and bleeding complications were increased.
The EXTRACT-TIMI 25 trial35 randomized more than 20,000 patients with STEMI who were about to undergo fibrinolysis to receive either enoxaparin throughout hospitalization (average of 8 days) or unfractionated heparin for at least 48 hours. The enoxaparin group had a lower rate of recurrent MI, but it was unclear if the difference was in part attributable to the longer therapy time. The enoxaparin group also had more bleeding.
Fondaparinux
The OASIS-5 trial36,37 compared enoxaparin and fondaparinux, an exclusive factor Xa inhibitor, in more than 20,000 patients with unstable angina or NSTEMI. Fondaparinux was associated with a lower risk of death and reinfarction as well as fewer bleeding events. However, the benefits were almost exclusively in patients treated medically. In those undergoing PCI within the first 8 days, no benefit was found, although there was still a significant reduction in major bleeding events. Catheter thrombosis was also increased in patients taking fondaparinux, but only in those who did not receive adequate unfractionated heparin treatment before PCI.
Bivalirudin superior at time of catheterization
The most significant advance in antithrombotic therapy for patients with acute coronary syndromes is bivalirudin. This drug has a clear role only in the catheterization laboratory, where patients can be switched to it from heparin, low-molecular-weight heparin, or fondaparinux.
Three trials38–40 evaluated the drug in a total of more than 20,000 patients receiving invasive management of coronary artery disease undergoing PCI for elective indications, NSTEMI, or STEMI.
Results were remarkably similar across the three trials. Patients who were treated with bivalirudin alone had the same rate of ischemic end points at 30 days as those receiving heparin plus a GP IIb/IIIa inhibitor, but bivalirudin was associated with a consistent and significant 40% to 50% lower bleeding risk. For the highest-risk patients, those with STEMI, the bivalirudin group also had a significantly lower risk of death at 1 year.41
OTHER DRUGS: EARLY TREATMENT NO LONGER ROUTINE
Most data for the use of therapies aside from antithrombotics are from studies of patients with STEMI, but findings can logically be extrapolated to those with non-ST-elevation acute coronary syndromes.
Beta-blockers: Cardiogenic shock a risk
For beta-blockers, many historical trials were done in stable coronary disease, but there are no large trials in the setting of NSTEMI or unstable angina, and only recently have there been large trials for STEMI. Before the availability of recent evidence, standard practice was to treat STEMI routinely with intravenous metoprolol (Lopressor) and then oral metoprolol.
When large studies were finally conducted, the results were sobering.
COMMIT.42 Nearly 46,000 patients with suspected acute MI were randomized to receive either metoprolol (up to 15 mg intravenously, then 200 mg by mouth daily until discharge or for up to 4 weeks in the hospital) or placebo. Surprisingly, although rates of reinfarction and ventricular fibrillation were lower with metoprolol, a higher risk of cardiogenic shock with early beta-blockade offset these benefits and the net mortality rate was not reduced. This study led to a reduction in the early use of beta-blockers in patients with STEMI.
The standard of care has now shifted from beta-blockers in everyone as early as possible after MI to being more cautious in patients with contraindications, including signs of heart failure or a low-output state, or even in those of advanced age or with borderline low blood pressure or a high heart rate. Patients who present late and therefore may have a larger infarct are also at higher risk.
Although the goal should be to ultimately discharge patients on beta-blocker therapy after an MI, there should be no rush to start one early.
Carvedilol now preferred after STEMI
The CAPRICORN trial43 randomized nearly 2,000 patients following MI with left ventricular dysfunction (an ejection fraction of 40% or below) to either placebo or the beta-blocker carvedilol (Coreg). Patients taking the drug had a clear reduction in rates of death and reinfarction, leading to this drug becoming the beta-blocker of choice in patients with ventricular dysfunction after STEMI.
Angiotensin-converting enzyme inhibitors: Early risk of cardiogenic shock
The use of angiotensin-converting enzyme (ACE) inhibitors after MI is also supported by several studies.44 Two very large studies, one of nearly 60,000 patients and one of nearly 20,000, showed a clear reduction in the mortality rate in those who received an ACE inhibitor. Most of the benefit was in patients with an ejection fraction of less than 40%. On the basis of these trials, ACE inhibitors are indicated for all patients for the first 30 days after MI and then indefinitely for those with left ventricular dysfunction. However, the trial in which an ACE inhibitor was given intravenously early on had to be stopped prematurely because of worse outcomes owing to cardiogenic shock.
These studies highlight again that for patients who are unstable in the first few days of an acute coronary syndrome, it is best to wait until their condition stabilizes and to start these therapies before hospital discharge.
Intensive statin therapy
In the last 20 years, unequivocal evidence has emerged to support the beneficial role of statins for secondary prevention in patients with established coronary artery disease. More-recent trials have also shown that intensive statin therapy (a high dose of a potent statin) improves outcomes better than lower doses.
The PROVE-IT TIMI 22 trial45 randomized patients after an acute coronary syndrome to receive either standard therapy (pravastatin [Pravachol] 40 mg) or intensive therapy (atorvastatin [Lipitor] 80 mg). The intensive-therapy group had a significantly lower rate of major cardiovascular events, and the difference persisted and grew over 30 months of follow-up.
A number of studies confirmed this and broadened the patient population to those with unstable or stable coronary disease. Regardless of the risk profile, the effects were consistent and showed that high-dose statins were better in preventing coronary death and MI.46
Guidelines are evolving toward recommendation of highest doses of statins independently of the target level of low-density lipoprotein cholesterol.
Most decisions for managing acute coronary syndromes can be based on ample data from large randomized trials with hard clinical end points, so there is little reason to provide care that is not evidence-based.
This article reviews some of the trials that provide guidance on diagnosing and managing acute coronary syndromes, including the timing of reperfusion and adjunctive therapies in different situations.
MOST ACUTE CORONARY SYNDROMES ARE NON-ST-ELEVATION CONDITIONS
Acute coronary syndromes range from unstable angina and non-ST-elevation myocardial infarction (NSTEMI) to ST-elevation MI (STEMI), reflecting a continuum of severity of coronary stenosis. The degree of coronary occlusion may ultimately determine whether a patient has unstable angina or MI with or without ST elevation.1
The substrate for all of these is vulnerable plaque. Angiographic studies have indicated that in many cases medium-size plaques (30%–40% stenosis) are more likely to rupture than larger, more obstructive ones. Moderate plaques may be vulnerable because they are less mature, with a large lipid core and a thin cap prone to rupture or erode, exposing the thrombogenic subendothelial components.2
Because the vulnerability of a coronary plaque may not correlate with the severity of stenosis before the plaque ruptures, stress tests and symptoms may not predict the risk of MI. The key role of thrombosis in the pathogenesis also highlights the importance of antithrombotic therapy in the acute phases of acute coronary syndromes, which can significantly reduce mortality and morbidity rates.
Perhaps because of the widespread use of aspirin and statins, most patients who currently present with an acute coronary syndrome have either unstable angina or NSTEMI: of about 1.57 million hospital admissions in 2004 for acute coronary syndromes, for example, only 330,000 (21%) were for STEMI.3
DIAGNOSING ACUTE CORONARY SYNDROME
Symptoms may not be classic
The classic symptoms of acute coronary syndromes are intense, oppressive chest pressure radiating to the left arm, but nearly any discomfort “between the nose and navel” (eg, including the jaw, arm, and epigastric and abdominal areas) may be an acute coronary syndrome. Associated symptoms may include chest heaviness or burning, radiation to the jaw, neck, shoulder, back, or arms, and dyspnea.
Particularly in older, female, postoperative, or diabetic patients, the presentation may be atypical or “silent,” including nausea or vomiting; breathlessness; sweating; arrhythmias; or light-headedness. Especially in these groups, symptoms may be mild or subtle, and acute coronary syndrome may manifest only as “not feeling well.”
The differential diagnosis of acute coronary syndromes is broad. Most important to immediately consider are pulmonary embolism and aortic dissection, as they are life-threatening and are treated differently from acute coronary syndromes. Otherwise, it is best to err on the side of caution and treat for an acute coronary syndrome until it is proven otherwise.
Electrocardiography is critical
Electrocardiography (ECG) gives valuable information about the location, extent, and prognosis of infarction, and it is critically important for distinguishing STEMI from NSTEMI, with ST elevation classically diagnostic of complete coronary occlusion. Q waves can occur early and do not necessarily signify completed infarction, as traditionally thought. ST depression or T inversion indicates that total coronary occlusion is unlikely unless they are in a pattern of circumflex infarct associated with an enlarging R wave in lead V1. An ST elevation in RV4 indicates right ventricular infarction.
The appearance on ECG may evolve over time, so a patient with atypical symptoms and a nonspecific electrocardiogram should be observed for 24 hours or until more specific criteria develop.
Biomarkers in NSTEMI
In MI, cardiac troponin levels begin to rise about 3 hours after the onset of chest pain, and elevations can last for up to 14 days. Levels can also be mildly elevated chronically in patients with renal dysfunction, so positive biomarker tests in that population should be interpreted cautiously.
For STEMI, the opportunity to reperfuse is lost if one waits for cardiac biomarkers to become elevated. But for NSTEMI, they are highly sensitive and specific for identifying patients at high risk and determining who should be treated aggressively. Patients who are biomarker-negative have a better prognosis than patients with identical symptoms and electrocardiograms who are biomarker-positive.
MI is currently defined as a rise in any biomarker (usually troponin) above the 99th percentile for a reference population, with at least one of the following:
- Ischemic symptoms
- New ST/T changes or left bundle branch block
- Pathologic Q waves
- Loss of myocardium or abnormal wall motion seen by imaging
- Intracoronary thrombus.
REPERFUSION FOR ACUTE STEMI
Because acute coronary syndromes have a common pathophysiology, for the most part, lessons from clinical trials in one syndrome are relevant to the others. However, important differences exist regarding the need for immediate reperfusion in STEMI, since in most cases these patients have total rather than partial occlusion.
Fibrinolysis has limitations
The standard of management for STEMI is immediate reperfusion. The goal is to interrupt the wave front of myocardial necrosis, salvage threatened myocardium, and ultimately improve survival.
Five placebo-controlled trials showed a 30% reduction in the death rate in patients who received fibrinolytic therapy within 6 to 12 hours of presentation.4
Patients with ST elevation or with new bundle branch block benefit most from fibrinolytic therapy. Those with ST depression, T inversion, or nonspecific changes on ECG do not benefit; they probably do not have complete coronary occlusion, so the prothrombotic or platelet-activating effects of fibrinolytic therapy may make them worse.5 Further, fibrinolytic therapy poses the risk of intracranial hemorrhage, which, although rare (occurring in up to 1% of cases depending on the drug regimen), is a devastating complication.
In general, absolute contraindications to fibrinolysis include intracranial abnormalities, hemorrhage, and head trauma. An important relative contraindication is uncontrolled blood pressure (> 180/110 mm Hg at any point during hospitalization, including during the immediate presentation). Studies show that even if blood pressure can be controlled, the risk of intracranial hemorrhage is substantially higher, although the risk may not outweigh the benefit of reperfusion, particularly for large infarctions when percutaneous coronary intervention (PCI) is not available as an alternative to fibrinolysis.
Prompt PCI is preferable to fibrinolysis
If PCI is available on site, there is nearly no role for fibrinolytic therapy. PCI is better than fibrinolytic therapy in terms of the degree of reperfusion, reocclusion, MI recurrence, and mortality rate, and it poses little or no risk of intracranial hemorrhage.6
For either fibrinolytic therapy or percutaneous therapy, “time is muscle”: the longer the ischemic time, the higher the mortality rate (relative risk = 1.075 for every 30 minutes of delay, P = .041).7
At centers that do not have PCI on site, studies (mainly from Europe) have shown that it is better to transport the patient for PCI than to give immediate fibrinolytic therapy.7,8 But because the centers studied tended to have short transport times (usually 40 minutes or less), it is uncertain whether the results are applicable throughout the United States.
The delay between symptom onset and presentation is also relevant. Reperfusion within the first 1 to 2 hours after the onset of symptoms provides the greatest degree of myocardial salvage and of reduction in the risk of death; the extent of benefit thereafter is substantially less. As a result, patients who present very early after symptom onset have the most to lose if their reperfusion is delayed by even a few more hours, whereas patients who have already experienced several hours of pain are affected less by additional delay.9 Thus, patients presenting within the “golden” 1 or 2 hours after symptoms begin should be considered for fibrinolytic therapy if transfer for PCI cannot be done expeditiously. It is important for hospitals without PCI available on site to have a system in place for rapid transport of patients when needed.
Guidelines advise that patients with STEMI should undergo PCI rather than receive fibrinolytic therapy as long as PCI is available within 90 minutes of first medical contact. Otherwise, fibrinolysis should be started within 30 minutes.10 For patients who present several hours after symptom onset, PCI may still be preferable even if the transport time is somewhat longer.
PCI after fibrinolytic therapy
In prior decades, PCI immediately after fibrinolytic therapy was associated with an increased risk of bleeding complications and reinfarction. That has changed with improvements in equipment and antithrombotic therapy.
Two large trials conclusively found that routinely transferring high-risk patients for PCI immediately after receiving fibrinolytic therapy (combined half-dose reteplase [Retavase] and abciximab [ReoPro]11 or full-dose tenecteplase [TNKase]12) resulted in much lower rates of ischemic end points without an increase in bleeding complications compared with transferring patients only for rescue PCI after fibrinolytic therapy.
Routine transfer is now the standard of care for high-risk patients after fibrinolytic therapy and probably is best for all patients after an MI.
MANAGING NSTEMI AND UNSTABLE ANGINA
For patients with NSTEMI, immediate reperfusion is usually not required, although initial triage for “early invasive” vs “initial conservative” management must be done early in the hospital course. Randomized trials have evaluated these two approaches, with most studies in the contemporary era reporting improved outcomes with an early invasive approach.
The TACTICS trial,13 the most important of these, enrolled more than 2,200 patients with unstable angina or NSTEMI and randomized them to an early invasive strategy or a conservative strategy. Overall, results were better with the early invasive strategy.
The ICTUS trial.14 Although several studies showed that an early invasive approach was better, the most recent study using the most modern practices—the ICTUS trial—did not find that it reduced death rates. Most patients eventually underwent angiography and revascularization, but not early on. However, all studies showed that rates of recurrent unstable angina and hospitalization were reduced by an early invasive approach, so revascularization does have a role in stabilizing the patient. But in situations of aggressive medical management with antithrombotic and other therapies, an early conservative approach may be an appropriate alternative for many patients.15
The selection of an invasive vs a conservative approach should include a consideration of risk, which can be estimated using a number of criteria, including the Thrombolysis in Myocardial Infarction (TIMI) or the GRACE risk score. When risk was stratified using the TIMI risk score,16 in the TACTICS trial, the higher the risk score, the more likely patients were to benefit from early revascularization.
When an invasive approach is chosen, it does not appear necessary to take patients to catheterization immediately (within 2–24 hours) compared with later during the hospital course.
The TIMACS trial,17 with more than 3,000 patients, tested the benefits of very early vs later revascularization for patients with NSTEMI and unstable angina. Early intervention did not significantly improve outcomes for the primary composite end point of death, MI, and stroke in the overall population enrolled in the trial, but when the secondary end point of refractory ischemia was added in, early intervention was found to be beneficial overall. Moreover, when stratified by risk, high-risk patients significantly benefited from early intervention for the primary end point.
Guidelines for NSTEMI and unstable angina continue to prefer an early invasive strategy, particularly for high-risk patients, although a conservative strategy is considered acceptable if patients receive intensive evidence-based medical therapy and remain clinically stable.18
ANTITHROMBOTIC THERAPIES
Once a revascularization strategy has been chosen, adjunctive therapies should be considered. The most important are the antithrombotic therapies.
Many drugs target platelet activity. Most important are the thromboxane inhibitor aspirin, the adenosine diphosphate (ADP) receptor antagonists clopidogrel (Plavix), prasugrel (Effient), and ticagrelor (Brilinta), and the glycoprotein (GP) IIb/IIIa antagonists abciximab and eptifibatide (Integrilin). Others, such as thrombin receptor antagonists, are under investigation.19
Aspirin for secondary prevention
Evidence is unequivocal for the benefit of aspirin therapy in patients with established or suspected vascular disease.
The ISIS-2 trial20 compared 35-day mortality rates in 16,000 patients with STEMI who were given aspirin, streptokinase, combined streptokinase and aspirin, or placebo. Mortality rates were reduced by aspirin compared with placebo by an extent similar to that achieved with streptokinase, with a further reduction when aspirin and streptokinase were given together.
Therefore, patients with STEMI should be given aspirin daily indefinitely unless they have true aspirin allergy. The dose is 165 to 325 mg initially and 75 to 162 mg daily thereafter.
For NSTEMI and even for secondary prevention in less-acute situations, a number of smaller trials also provide clear evidence of benefit from aspirin therapy.
The CURRENT-OASIS 7 trial21 showed that low maintenance dosages of aspirin (75–100 mg per day) resulted in the same incidence of ischemic end points (cardiovascular death, MI, or stroke) as higher dosages. Although rates of major bleeding events did not differ, a higher rate of gastrointestinal bleeding was evident at just 30 days in patients taking the higher doses. This large trial clearly established that there is no advantage to daily aspirin doses of more than 100 mg.
DUAL ANTIPLATELET THERAPY IS STANDARD
Standard practice now is to use aspirin plus another antiplatelet agent that acts by inhibiting either the ADP receptor (for which there is the most evidence) or the GP IIb/IIIa receptor (which is becoming less used). Dual therapy should begin early in patients with acute coronary syndrome.
Clopidogrel: Well studied with aspirin
The most commonly used ADP antagonist is clopidogrel, a thienopyridine. Much evidence exists for its benefit.
The CURE trial22 randomized more than 12,000 patients with NSTEMI or unstable angina to aspirin plus either clopidogrel or placebo. The incidence of the combined end point of MI, stroke, and cardiovascular death was 20% lower in the clopidogrel group than in the placebo group over 12 months of follow-up. The benefit of clopidogrel began to occur within the first 24 hours after randomization, with a 33% relative risk reduction in the combined end point of cardiovascular death, MI, stroke, and severe ischemia, demonstrating the importance of starting this agent early in the hospital course.
COMMIT23 found a benefit in adding clopidogrel to aspirin in patients with acute STEMI. Although it was only a 30-day trial, significant risk reduction was found in the dual-therapy group for combined death, stroke, or reinfarction. The results of this brief trial were less definitive, but the pathophysiology was similar to non-ST-elevation acute coronary syndromes, so it is reasonable to extrapolate the long-term findings to this setting.
The CURRENT-OASIS 7 trial21 randomized more than 25,000 patients to either clopidogrel in a double dosage (600 mg load, 150 mg/day for 6 days, then 75 mg/day) or standard dosage (300 mg load, 75 mg/day thereafter). Although no overall benefit was found for the higher dosage, a subgroup of more than 17,000 patients who underwent PCI after randomization had a lower risk of developing stent thrombosis. On the other hand, higher doses of clopidogrel caused more major bleeding events.
Ticagrelor and prasugrel: New alternatives to clopidogrel
The principal limitation of clopidogrel is its metabolism. It is a prodrug, ie, it is not active as taken and must be converted to its active state by cytochrome P450 enzymes in the liver. Patients who bear certain polymorphisms in the genes for these enzymes or who are taking other medications that affect this enzymatic pathway may derive less platelet inhibition from the drug, leading to considerable patient-to-patient variability in the degree of antiplatelet effect.
Alternatives to clopidogrel have been developed that inhibit platelets more intensely, are activated more rapidly, and have less interpatient variability. Available now are ticagrelor and prasugrel.24 Like clopidogrel, prasugrel is absorbed as an inactive prodrug, but it is efficiently metabolized by esterases to an active form, and then by a simpler step within the liver to its fully active metabolite.25 Ticagrelor is active as absorbed.26
Pharmacodynamically, the two drugs perform almost identically and much faster than clopidogrel, with equilibrium platelet inhibition reached in less than 1 hour. The degree of platelet inhibition is also more—sometimes twice as much—with the new drugs compared with clopidogrel, and the effect is much more consistent between patients.
Both clopidogrel and prasugrel permanently inhibit the platelet ADP receptor, and 3 to 7 days are therefore required for their antiplatelet effects to completely wear off. In contrast, ticagrelor is a reversible inhibitor and its effects wear off more rapidly. Despite achieving a much higher level of platelet inhibition than clopidogrel, ticagrelor’s activity falls below that of clopidogrel’s by 48 hours of discontinuing the drugs.
Trial of prasugrel vs clopidogrel
The TRITON-TIMI 38 trial27 enrolled more than 13,000 patients with acute coronary syndromes, randomized to receive, either prasugrel or clopidogrel, in addition to aspirin. The patients were all undergoing PCI, so the findings do not apply to patients treated medically with an early conservative approach. The study drug was given only after the decision was made to perform PCI in patients with non-ST-elevation acute coronary syndrome (but given immediately for patients with STEMI, because nearly all those patients undergo PCI).
Prasugrel was clearly beneficial, with a significant 20% lower rate of the combined end point of cardiovascular death, MI, and stroke at 15 months. However, bleeding risk was higher with prasugrel (2.4% vs 1.8%, hazard ratio 1.32, 95% confidence interval 1.02–1.68, P = .03). Looking at individual end points, the advantages of prasugrel were primarily in reducing rates of stent thrombosis and nonfatal MI. Death rates with the two drugs were equivalent, possibly because of the higher risk of bleeding with prasugrel. Bleeding in the prasugrel group was particularly increased in patients who underwent bypass surgery; more patients also needed transfusion.
Subgroup analysis showed that patients with a history of stroke or transient ischemic attack had higher rates of ischemic and bleeding events with prasugrel than with clopidogrel, leading to these being labeled as absolute contraindications to prasugrel. Patients over age 75 or who weighed less than 60 kg experienced excess bleeding risk that closely matched the reduction in ischemic event rates and thus did not have a net benefit with prasugrel.
Trial of ticagrelor vs clopidogrel
The PLATO trial28 included 18,000 patients, of whom 65% underwent revascularization and 35% were treated medically. The drug—clopidogrel or ticagrelor—was given in addition to aspirin at randomization (within 24 hours of symptom onset); this more closely follows clinical practice, in which dual antiplatelet therapy is started as soon as possible. This difference makes the PLATO study more relevant to practice for patients with non-ST-elevation acute coronary syndrome. Also, because they gave the drugs to all patients regardless of whether they were to undergo PCI, this study likely had a higher-risk population, which may be refected in the higher mortality rate at 30 days (5.9% in the clopidogrel group in the PLATO study vs 3.2% in the clopidogrel group in the TRITON study).
Another important difference between the trials testing prasugrel and ticagrelor is that patients who had already received a thienopyridine were excluded from the prasugrel trial but not from the ticagrelor trial. Nearly half the patients in the ticagrelor group were already taking clopidogrel. The clinical implication is that for patients who arrive from another facility and already have been given clopidogrel, it is safe to give ticagrelor. There is limited information about whether that is also true for prasugrel, although there is no known reason why the safety of adding prasugrel to clopidogrel should be different from that of ticagrelor.
The rate of ischemic events was 20% lower in the ticagrelor group than in the clopidogrel group, importantly including reductions in the incidence of death, MI, and stent thrombosis. There was no increase with ticagrelor compared with clopidogrel in bleeding associated with coronary artery bypass graft surgery, likely because of the more rapid washout of the ticagrelor effect, or in the need for blood transfusions. However, the rate of bleeding unrelated to coronary artery bypass was about 20% higher with ticagrelor.
In summary, more intense platelet inhibition reduces the risk of ischemic events, but, particularly for the irreversible inhibitor prasugrel, at the cost of a higher risk of bleeding. In general, the net benefit of these agents in preventing the irreversible complications of MI and (in the case of ticagrelor) death favor the use of the more intense ADP inhibitors in appropriate patients. Ticagrelor is indicated in patients with acute coronary syndromes undergoing invasive or conservative management; prasugrel is indicated in patients undergoing PCI, but contraindicated in patients with a previous stroke or transient ischemic event. Neither drug is indicated in patients undergoing elective PCI outside the setting of acute coronary syndromes, although these agents may be appropriate in patients with intolerance or allergy to clopidogrel.
Glycoprotein IIb/IIIa antagonists for select cases only
GP IIb/IIIa antagonists such as abciximab were previously used more commonly than they are today. Now, with routine pretreatment using thienopyridines, their role in acute coronary syndromes is less clear. They still play a role when routine dual antiplatelet therapy is not used, when prasugrel or ticagrelor is not used, and when heparin rather than an alternative antithrombin agent is used.
A meta-analysis29 of 3,755 patients showed a clear reduction in ischemic complications with abciximab as an adjunct to primary PCI for STEMI in patients treated with heparin.
Kastrati et al30 found that patients with non-ST-elevation acute coronary syndromes benefited from abciximab at the time of PCI with heparin, even though they had been routinely pretreated with clopidogrel. However, benefits were seen only in high-risk patients who had presented with elevated troponins.
On the other hand, the role of GP IIb/IIIa blockade for “upstream” medical management in patients with acute coronary syndromes has been eroded by several studies.
The ACUITY trial31 randomized more than 9,000 patients to receive either routine treatment with a GP IIb/IIIa inhibitor before angiography or deferred selective use in the catheterization laboratory only for patients undergoing PCI. No significant differences were found in rates of MI and death.
The Early ACS trial32 compared early routine eptifibatide vs delayed, provisional eptifibatide in 9,492 patients with acute coronary syndromes without ST elevation and who were assigned to an invasive strategy. The early-eptifibatide group received two boluses and an infusion of eptifibatide before angiography; the others received a placebo infusion, with provisional eptifibatide after angiography if the patient underwent PCI and was deemed at high risk. No significant difference in rates of death or MI were noted, and the early-eptifibatide group had significantly higher rates of bleeding and need for transfusion.
The FINESSE trial33 also discredited “facilitating” PCI by giving GP IIb/IIIa antagonists in patients with STEMI before arrival in the catheterization laboratory, with no benefit to giving abciximab ahead of time vs in the catheterization laboratory, and with an increased risk of bleeding complications.
These studies have helped narrow the use of GP IIb/IIIa inhibitors to the catheterization laboratory in conjunction with heparin anticoagulation (as compared with bivalirudin [Angiomax]; see below) and only in select or high-risk cases. These drugs are indicated in the medical phase of management only if patients cannot be stabilized by aspirin or ADP inhibition.
NEWER ANTITHROMBOTICS: ADVANTAGES UNCLEAR
The complex coagulation cascade has a number of components, but only a few are targeted by drugs that are approved and recommended: fondaparinux (Arixtra) and oral factor Xa inhibitors affect the prothrombinase complex (including factor X); bivalirudin and oral factor IIa inhibitors affect thrombin; and heparin and the low-molecular-weight heparins inhibit both targets.
Low-molecular-weight heparins
The SYNERGY trial34 randomized nearly 10,000 patients with non-ST-elevation acute coronary syndromes at high risk for ischemic cardiac complications managed with an invasive approach to either the low-molecular-weight heparin enoxaparin (Lovenox) or intravenous unfractionated heparin immediately after enrollment. Most patients underwent catheterization and revascularization. No clinical advantage was found for enoxaparin, and bleeding complications were increased.
The EXTRACT-TIMI 25 trial35 randomized more than 20,000 patients with STEMI who were about to undergo fibrinolysis to receive either enoxaparin throughout hospitalization (average of 8 days) or unfractionated heparin for at least 48 hours. The enoxaparin group had a lower rate of recurrent MI, but it was unclear if the difference was in part attributable to the longer therapy time. The enoxaparin group also had more bleeding.
Fondaparinux
The OASIS-5 trial36,37 compared enoxaparin and fondaparinux, an exclusive factor Xa inhibitor, in more than 20,000 patients with unstable angina or NSTEMI. Fondaparinux was associated with a lower risk of death and reinfarction as well as fewer bleeding events. However, the benefits were almost exclusively in patients treated medically. In those undergoing PCI within the first 8 days, no benefit was found, although there was still a significant reduction in major bleeding events. Catheter thrombosis was also increased in patients taking fondaparinux, but only in those who did not receive adequate unfractionated heparin treatment before PCI.
Bivalirudin superior at time of catheterization
The most significant advance in antithrombotic therapy for patients with acute coronary syndromes is bivalirudin. This drug has a clear role only in the catheterization laboratory, where patients can be switched to it from heparin, low-molecular-weight heparin, or fondaparinux.
Three trials38–40 evaluated the drug in a total of more than 20,000 patients receiving invasive management of coronary artery disease undergoing PCI for elective indications, NSTEMI, or STEMI.
Results were remarkably similar across the three trials. Patients who were treated with bivalirudin alone had the same rate of ischemic end points at 30 days as those receiving heparin plus a GP IIb/IIIa inhibitor, but bivalirudin was associated with a consistent and significant 40% to 50% lower bleeding risk. For the highest-risk patients, those with STEMI, the bivalirudin group also had a significantly lower risk of death at 1 year.41
OTHER DRUGS: EARLY TREATMENT NO LONGER ROUTINE
Most data for the use of therapies aside from antithrombotics are from studies of patients with STEMI, but findings can logically be extrapolated to those with non-ST-elevation acute coronary syndromes.
Beta-blockers: Cardiogenic shock a risk
For beta-blockers, many historical trials were done in stable coronary disease, but there are no large trials in the setting of NSTEMI or unstable angina, and only recently have there been large trials for STEMI. Before the availability of recent evidence, standard practice was to treat STEMI routinely with intravenous metoprolol (Lopressor) and then oral metoprolol.
When large studies were finally conducted, the results were sobering.
COMMIT.42 Nearly 46,000 patients with suspected acute MI were randomized to receive either metoprolol (up to 15 mg intravenously, then 200 mg by mouth daily until discharge or for up to 4 weeks in the hospital) or placebo. Surprisingly, although rates of reinfarction and ventricular fibrillation were lower with metoprolol, a higher risk of cardiogenic shock with early beta-blockade offset these benefits and the net mortality rate was not reduced. This study led to a reduction in the early use of beta-blockers in patients with STEMI.
The standard of care has now shifted from beta-blockers in everyone as early as possible after MI to being more cautious in patients with contraindications, including signs of heart failure or a low-output state, or even in those of advanced age or with borderline low blood pressure or a high heart rate. Patients who present late and therefore may have a larger infarct are also at higher risk.
Although the goal should be to ultimately discharge patients on beta-blocker therapy after an MI, there should be no rush to start one early.
Carvedilol now preferred after STEMI
The CAPRICORN trial43 randomized nearly 2,000 patients following MI with left ventricular dysfunction (an ejection fraction of 40% or below) to either placebo or the beta-blocker carvedilol (Coreg). Patients taking the drug had a clear reduction in rates of death and reinfarction, leading to this drug becoming the beta-blocker of choice in patients with ventricular dysfunction after STEMI.
Angiotensin-converting enzyme inhibitors: Early risk of cardiogenic shock
The use of angiotensin-converting enzyme (ACE) inhibitors after MI is also supported by several studies.44 Two very large studies, one of nearly 60,000 patients and one of nearly 20,000, showed a clear reduction in the mortality rate in those who received an ACE inhibitor. Most of the benefit was in patients with an ejection fraction of less than 40%. On the basis of these trials, ACE inhibitors are indicated for all patients for the first 30 days after MI and then indefinitely for those with left ventricular dysfunction. However, the trial in which an ACE inhibitor was given intravenously early on had to be stopped prematurely because of worse outcomes owing to cardiogenic shock.
These studies highlight again that for patients who are unstable in the first few days of an acute coronary syndrome, it is best to wait until their condition stabilizes and to start these therapies before hospital discharge.
Intensive statin therapy
In the last 20 years, unequivocal evidence has emerged to support the beneficial role of statins for secondary prevention in patients with established coronary artery disease. More-recent trials have also shown that intensive statin therapy (a high dose of a potent statin) improves outcomes better than lower doses.
The PROVE-IT TIMI 22 trial45 randomized patients after an acute coronary syndrome to receive either standard therapy (pravastatin [Pravachol] 40 mg) or intensive therapy (atorvastatin [Lipitor] 80 mg). The intensive-therapy group had a significantly lower rate of major cardiovascular events, and the difference persisted and grew over 30 months of follow-up.
A number of studies confirmed this and broadened the patient population to those with unstable or stable coronary disease. Regardless of the risk profile, the effects were consistent and showed that high-dose statins were better in preventing coronary death and MI.46
Guidelines are evolving toward recommendation of highest doses of statins independently of the target level of low-density lipoprotein cholesterol.
- Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines; Canadian Cardiovascular Society. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction. Circulation 2004; 110:e82–e292. Erratum in: Circulation 2005; 111:2013–2014.
- Davies MJ. The pathophysiology of acute coronary syndromes. Heart 2000; 83:361–366.
- Rosamond W, Flegal K, Friday G, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics–2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2007; 115:e69–e171.
- Granger CB, Califf RM, Topol EJ. Thrombolytic therapy for acute myocardial infarction. A review. Drugs 1992; 44:293–325.
- Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Lancet 1994; 343:311–322.
- Keely EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet 2003; 361:13–20
- De Luca G, Suryapranata H, Ottervanger JP, Antman EM. Time delay to treatment and mortality in primary angioplasty for acute myocardial infarction: every minute of delay counts. Circulation 2004; 109:1223–1225.
- Dalby M, Bouzamondo A, Lechat P, Montalescot G. Transfer for primary angioplasty versus immediate thrombolysis in acute myocardial infarction: a meta-analysis. Circulation 2003; 108:1809–1814.
- Gersh BJ, Stone GW, White HD, Holmes DR Jr. Pharmacological facilitation of primary percutaneous coronary intervention for acute myocardial infarction: is the slope of the curve the shape of the future? JAMA 2005; 293:979–986.
- Antman EM, Hand M, Armstron PW, et al; Canadian Cardiovascular Society; American Academy of Family Physicians; American College of Cardiology; American Heart Association. 2007 focused update of the ACC/AHA 2004 guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2008; 51:210–247.
- Di Mario C, Dudek D, Piscione F, et al; CARESS-in-AMI (Combined Abciximab Reteplase Stent Study in Acute Myocardial Infarction) Investigators. Immediate angioplasty versus standard therapy with rescue angioplasty after thrombolysis in the Combined Abciximab REteplase Stent Study in Acute Myocardial Infarction (CARESS-in-AMI): an open, prospective, randomised, multicentre trial. Lancet 2008; 371:559–568.
- Cantor WJ, Fitchett D, Borgundvaag B, et al; TRANSFER-AMI Trial Investigators. Routine early angioplasty after fibrinolysis for acute myocardial infarction. N Engl J Med 2009; 360:2705–2718.
- Cannon CP, Weintraub WS, Demopoulos LA, et al; TACTICS (Treat Angina With Aggrastat and Determine Cost of Therapy With an Invasive or Conservative Strategy)–Thrombolysis in Myocardial Infarction 18 Investigators. Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med 2001; 344:1879–1887.
- Damman P, Hirsch A, Windhausen F, Tijssen JG, de Winter RJ; ICTUS Investigators. 5-year clinical outcomes in the ICTUS (Invasive versus Conservative Treatment in Unstable coronary Syndromes) trial a randomized comparison of an early invasive versus selective invasive management in patients with non-ST-segment elevation acute coronary syndrome. J Am Coll Cardiol 2010; 55:858–864.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- Antman EM, Cohen M, Bernink PJ, et al. The TIMI risk score for unstable angina/non-ST elevation MI: a method for prognostication and therapeutic decision making. JAMA 2000; 284:835–842.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- Yousef O, Bhatt DL. The evolution of antiplatelet therapy in cardiovascular disease. Nat Rev Cardiol 2011; 8:547–559.
- ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- CURRENT-OASIS 7 Investigators; Mehta SR, Bassand JP, Chrolavicius S, et al. Dose comparisons of clopidogrel and aspirin in acute coronary syndromes. N Engl J Med 2010; 363:930–942.
- Yusuf S, Mehta SR, Zhao F, et al; Clopidogrel in Unstable angina to prevent Recurrent Events Trial Investigators. Early and late effects of clopidogrel in patients with acute coronary syndromes. Circulation 2003; 107:966–972.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Schömig A. Ticagrelor—is there need for a new player in the antiplatelet-therapy field? N Engl J Med 2009; 361:1108–1111.
- Wiviott SD, Antman EM, Braunwald E. Prasugrel. Circulation 2010; 122:394–403.
- Gurbel PA, Bliden KP, Butler K, et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: the ONSET/OFFSET study. Circulation 2009; 120:2577–2585.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- de Queiroz Fernandes Araujo JO, Veloso HH, Braga De Paiva JM, Fiho MW, Vincenzo De Paola AA. Efficacy and safety of abciximab on acute myocardial infarction treated with percutaneous coronary interventions: a meta-analysis of randomized, controlled trials. Am Heart J 2004; 148:937–943.
- Kastrati A, Mehilli J, Neuman FJ, et al; Intracoronary Stenting and Antithrombotic: Regimen Rapid Early Action for Coronary Treatment 2 (ISAR-REACT 2) Trial Investigators. Abciximab in patients with acute coronary syndromes undergoing percutaneous coronary intervention after clopidogrel pretreatment: the ISAR-REACT 2 randomized trial. JAMA 2006; 295:1531–1538.
- Stone GW, Bertrand ME, Moses JW, et al; ACUITY Investigators. Routine upstream initiation vs deferred selective use of glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: the ACUITY Timing trial. JAMA 2007; 297:591–602.
- Giugliano RP, White JA, Bode C, et al; Early ACS Investigators. Early vs delayed, provisional eptifibatide in acute coronary syndromes. N Engl J Med 2009; 360:2176–2190.
- Ellis SG, Tendera M, de Belder MA, et al; FINESSE Investigators. Facilitated PCI in patients with ST-elevation myocardial infarction. N Engl J Med 2008; 358:2205–2217.
- Fergusson JJ, Califf RM, Antman EM, et al; SYNERGY Trial Investigators. Enoxaparin vs unfractionated heparin in high-risk patients with non-ST-segment elevation acute coronary syndromes managed with an intended early invasive strategy: primary results of the SYNERGY randomized trial. JAMA 2004; 292:45–54.
- Antman EM, Morrow DA, McCabe CH; EXTRACT-TIMI 25 Investigators. Enoxaparin versus unfractionated heparin with fibrinolysis for ST-elevation myocardial infarction. N Engl J Med 2006; 354:1477–1488.
- The Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Mehta SR, Granger CB, Eikelboom JW, et al. Efficacy and safety of fondaparinux versus enoxaparin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: results from the OASIS-5 trial. J Am Coll Cardiol 2007; 50:1742–1751.
- Lincoff AM, Bittl JA, Harrington RA, et al; REPLACE-2 Investigators. Bivalirudin and provisional glycoprotein IIb/IIIa blockade compared with heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary intervention: REPLACE-2 randomized trial. JAMA 2003; 289:853–863.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Stone GW, Witzenbichler B, Guagliumi G, et al; HORIZONS-AMI Trial Investigators. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med 2007; 358:2218–2230.
- Mehran R, Lansky AJ, Witzenbichler B, et al; HORIZONS-AMI Trial Investigators. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet 2009; 374:1149–1159.
- Chen ZM, Pan HC, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) Collaborative Group. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1622–1632.
- Dargie JH. Effect of carvedilol on outcome after myocardial infarction in patients with left-ventricular dysfunction: the CAPRICORN randomised trial. Lancet 2001; 357:1385–1390.
- Hennekens CH, Albert CM, Godfried SL, Gaziano JM, Buring JE. Adjunctive drug therapy of acute myocardial infarction—evidence from clinical trials. N Engl J Med 1996; 335:1660–1667.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Cannon CP, Steinberg BA, Murphy SA, Mega JL, Braunwald E. Meta-analysis of cardiovascular outcomes trials comparing intensive versus moderate statin therapy. J Am Coll Cardiol 2006; 48:438–445.
- Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology; American Heart Association Task Force on Practice Guidelines; Canadian Cardiovascular Society. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction. Circulation 2004; 110:e82–e292. Erratum in: Circulation 2005; 111:2013–2014.
- Davies MJ. The pathophysiology of acute coronary syndromes. Heart 2000; 83:361–366.
- Rosamond W, Flegal K, Friday G, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics–2007 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2007; 115:e69–e171.
- Granger CB, Califf RM, Topol EJ. Thrombolytic therapy for acute myocardial infarction. A review. Drugs 1992; 44:293–325.
- Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Lancet 1994; 343:311–322.
- Keely EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet 2003; 361:13–20
- De Luca G, Suryapranata H, Ottervanger JP, Antman EM. Time delay to treatment and mortality in primary angioplasty for acute myocardial infarction: every minute of delay counts. Circulation 2004; 109:1223–1225.
- Dalby M, Bouzamondo A, Lechat P, Montalescot G. Transfer for primary angioplasty versus immediate thrombolysis in acute myocardial infarction: a meta-analysis. Circulation 2003; 108:1809–1814.
- Gersh BJ, Stone GW, White HD, Holmes DR Jr. Pharmacological facilitation of primary percutaneous coronary intervention for acute myocardial infarction: is the slope of the curve the shape of the future? JAMA 2005; 293:979–986.
- Antman EM, Hand M, Armstron PW, et al; Canadian Cardiovascular Society; American Academy of Family Physicians; American College of Cardiology; American Heart Association. 2007 focused update of the ACC/AHA 2004 guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2008; 51:210–247.
- Di Mario C, Dudek D, Piscione F, et al; CARESS-in-AMI (Combined Abciximab Reteplase Stent Study in Acute Myocardial Infarction) Investigators. Immediate angioplasty versus standard therapy with rescue angioplasty after thrombolysis in the Combined Abciximab REteplase Stent Study in Acute Myocardial Infarction (CARESS-in-AMI): an open, prospective, randomised, multicentre trial. Lancet 2008; 371:559–568.
- Cantor WJ, Fitchett D, Borgundvaag B, et al; TRANSFER-AMI Trial Investigators. Routine early angioplasty after fibrinolysis for acute myocardial infarction. N Engl J Med 2009; 360:2705–2718.
- Cannon CP, Weintraub WS, Demopoulos LA, et al; TACTICS (Treat Angina With Aggrastat and Determine Cost of Therapy With an Invasive or Conservative Strategy)–Thrombolysis in Myocardial Infarction 18 Investigators. Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med 2001; 344:1879–1887.
- Damman P, Hirsch A, Windhausen F, Tijssen JG, de Winter RJ; ICTUS Investigators. 5-year clinical outcomes in the ICTUS (Invasive versus Conservative Treatment in Unstable coronary Syndromes) trial a randomized comparison of an early invasive versus selective invasive management in patients with non-ST-segment elevation acute coronary syndrome. J Am Coll Cardiol 2010; 55:858–864.
- Bavry AA, Kumbhani DJ, Rassi AN, Bhatt DL, Askari AT. Benefit of early invasive therapy in acute coronary syndromes: a meta-analysis of contemporary randomized clinical trials. J Am Coll Cardiol 2006; 48:1319–1325.
- Antman EM, Cohen M, Bernink PJ, et al. The TIMI risk score for unstable angina/non-ST elevation MI: a method for prognostication and therapeutic decision making. JAMA 2000; 284:835–842.
- Mehta SR, Granger CB, Boden WE, et al; TIMACS Investigators. Early versus delayed invasive intervention in acute coronary syndromes. N Engl J Med 2009; 360:2165–2175.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- Yousef O, Bhatt DL. The evolution of antiplatelet therapy in cardiovascular disease. Nat Rev Cardiol 2011; 8:547–559.
- ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988; 2:349–360.
- CURRENT-OASIS 7 Investigators; Mehta SR, Bassand JP, Chrolavicius S, et al. Dose comparisons of clopidogrel and aspirin in acute coronary syndromes. N Engl J Med 2010; 363:930–942.
- Yusuf S, Mehta SR, Zhao F, et al; Clopidogrel in Unstable angina to prevent Recurrent Events Trial Investigators. Early and late effects of clopidogrel in patients with acute coronary syndromes. Circulation 2003; 107:966–972.
- Chen ZM, Jiang LX, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) collaborative group. Addition of clopidogrel to aspirin in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
- Schömig A. Ticagrelor—is there need for a new player in the antiplatelet-therapy field? N Engl J Med 2009; 361:1108–1111.
- Wiviott SD, Antman EM, Braunwald E. Prasugrel. Circulation 2010; 122:394–403.
- Gurbel PA, Bliden KP, Butler K, et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: the ONSET/OFFSET study. Circulation 2009; 120:2577–2585.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Wallentin L, Becker RC, Budaj A, et al; PLATO Investigators. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2009; 361:1045–1057.
- de Queiroz Fernandes Araujo JO, Veloso HH, Braga De Paiva JM, Fiho MW, Vincenzo De Paola AA. Efficacy and safety of abciximab on acute myocardial infarction treated with percutaneous coronary interventions: a meta-analysis of randomized, controlled trials. Am Heart J 2004; 148:937–943.
- Kastrati A, Mehilli J, Neuman FJ, et al; Intracoronary Stenting and Antithrombotic: Regimen Rapid Early Action for Coronary Treatment 2 (ISAR-REACT 2) Trial Investigators. Abciximab in patients with acute coronary syndromes undergoing percutaneous coronary intervention after clopidogrel pretreatment: the ISAR-REACT 2 randomized trial. JAMA 2006; 295:1531–1538.
- Stone GW, Bertrand ME, Moses JW, et al; ACUITY Investigators. Routine upstream initiation vs deferred selective use of glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: the ACUITY Timing trial. JAMA 2007; 297:591–602.
- Giugliano RP, White JA, Bode C, et al; Early ACS Investigators. Early vs delayed, provisional eptifibatide in acute coronary syndromes. N Engl J Med 2009; 360:2176–2190.
- Ellis SG, Tendera M, de Belder MA, et al; FINESSE Investigators. Facilitated PCI in patients with ST-elevation myocardial infarction. N Engl J Med 2008; 358:2205–2217.
- Fergusson JJ, Califf RM, Antman EM, et al; SYNERGY Trial Investigators. Enoxaparin vs unfractionated heparin in high-risk patients with non-ST-segment elevation acute coronary syndromes managed with an intended early invasive strategy: primary results of the SYNERGY randomized trial. JAMA 2004; 292:45–54.
- Antman EM, Morrow DA, McCabe CH; EXTRACT-TIMI 25 Investigators. Enoxaparin versus unfractionated heparin with fibrinolysis for ST-elevation myocardial infarction. N Engl J Med 2006; 354:1477–1488.
- The Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Mehta SR, Granger CB, Eikelboom JW, et al. Efficacy and safety of fondaparinux versus enoxaparin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: results from the OASIS-5 trial. J Am Coll Cardiol 2007; 50:1742–1751.
- Lincoff AM, Bittl JA, Harrington RA, et al; REPLACE-2 Investigators. Bivalirudin and provisional glycoprotein IIb/IIIa blockade compared with heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary intervention: REPLACE-2 randomized trial. JAMA 2003; 289:853–863.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Stone GW, Witzenbichler B, Guagliumi G, et al; HORIZONS-AMI Trial Investigators. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med 2007; 358:2218–2230.
- Mehran R, Lansky AJ, Witzenbichler B, et al; HORIZONS-AMI Trial Investigators. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet 2009; 374:1149–1159.
- Chen ZM, Pan HC, Chen YP, et al; COMMIT (Clopidogrel and Metoprolol in Myocardial Infarction Trial) Collaborative Group. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet 2005; 366:1622–1632.
- Dargie JH. Effect of carvedilol on outcome after myocardial infarction in patients with left-ventricular dysfunction: the CAPRICORN randomised trial. Lancet 2001; 357:1385–1390.
- Hennekens CH, Albert CM, Godfried SL, Gaziano JM, Buring JE. Adjunctive drug therapy of acute myocardial infarction—evidence from clinical trials. N Engl J Med 1996; 335:1660–1667.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Cannon CP, Steinberg BA, Murphy SA, Mega JL, Braunwald E. Meta-analysis of cardiovascular outcomes trials comparing intensive versus moderate statin therapy. J Am Coll Cardiol 2006; 48:438–445.
KEY POINTS
- For acute ST-elevation myocardial infarction, primary percutaneous coronary intervention is preferred over fibrinolytic therapy if it is available within 90 minutes of first medical contact.
- For non-ST-elevation acute coronary syndromes, either an early invasive or conservative strategy is recommended depending on patient risk and whether intensive medical therapy is available and appropriate.
- Daily aspirin therapy is indicated for all patients with acute coronary syndromes unless they have a true aspirin allergy.
- Adenosine diphosphate receptor inhibitors—clopidogrel, prasugrel, and ticagrelor—reduce ischemic events but increase bleeding risk and should be used only for patients with no history of stroke or transient ischemic attack.
Hepatitis C virus: Here comes all-oral treatment
In late 2013, the US Food and Drug Administration (FDA) approved sofosbuvir and simeprevir, the newest direct-acting antiviral agents for treating chronic hepatitis C virus (HCV) infection. Multiple clinical trials have demonstrated dramatically improved treatment outcomes with these agents, opening the door to all-oral regimens or interferon-free regimens as the future standard of care for HCV.
In this article, we discuss the results of the trials that established the efficacy and safety of sofosbuvir and simeprevir and led to their FDA approval. We also summarize the importance of these agents and evaluate other direct-acting antivirals currently in the pipeline for HCV treatment.
HCV IS A RISING PROBLEM
Chronic HCV infection is a major clinical and public health problem, with the estimated number of people infected exceeding 170 million worldwide, including 3.2 million in the United States.1 It is a leading cause of cirrhosis, and its complications include hepatocellular carcinoma and liver failure. Cirrhosis due to HCV remains the leading indication for liver transplantation in the United States, accounting for nearly 40% of liver transplants in adults.2

The clinical impact of HCV will only continue to escalate, and in parallel, so will the cost to society. Models suggest that HCV-related deaths will double between 2010 and 2019, and considering only direct medical costs, the projected financial burden of treating HCV-related disease during this interval is estimated at between $6.5 and $13.6 billion.3
AN RNA VIRUS WITH SIX GENOTYPES

HCV, first identified in 1989, is an enveloped, single-stranded RNA flavivirus of the Hepacivirus genus measuring 50 to 60 nm in diameter.4 There are six viral genotypes, with genotype 1 being the most common in the United States and traditionally the most difficult to treat.
Once inside the host cell, the virus releases its RNA strand, which is translated into a single polyprotein of about 3,000 amino acids. This large molecule is then cleaved by proteases into several domains: three structural proteins (C, E1, and E2), a small protein called p7, and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (Figure 1).5 These nonstructural proteins enable the virus to replicate.
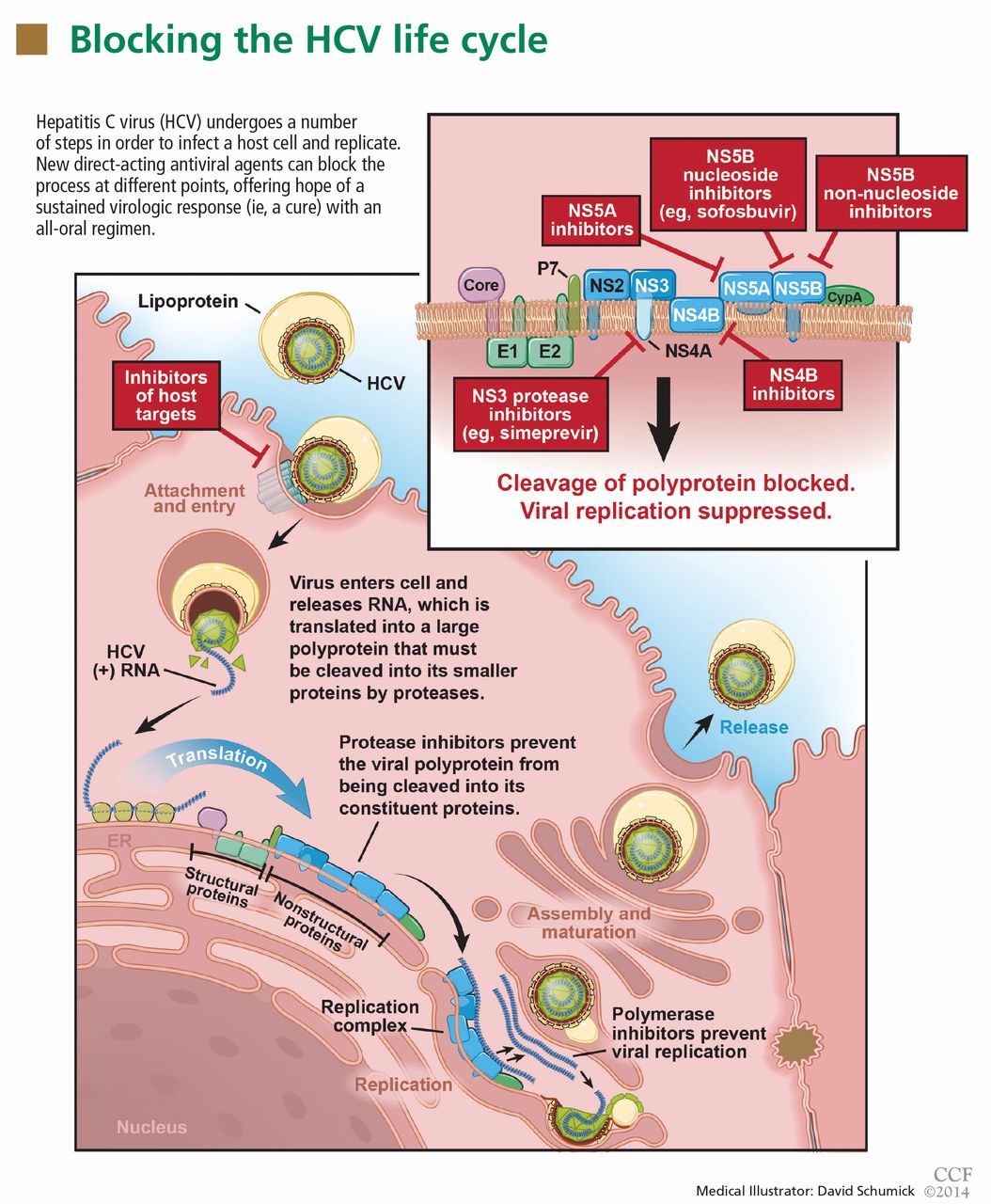
GOAL OF TREATING HCV: A SUSTAINED VIROLOGIC RESPONSE
The aim of HCV treatment is to achieve a sustained virologic response, defined as having no detectable viral RNA after completion of antiviral therapy. This is associated with substantially better clinical outcomes, lower rates of liver-related morbidity and all-cause mortality, and stabilization of or even improvement in liver histology.6,7 This end point has traditionally been assessed at 6 months after the end of therapy, but recent data suggest the rates at 12 weeks are essentially equivalent.

Table 1 summarizes the patterns of virologic response in treating HCV infection.
Interferon plus ribavirin: The standard of care for many years
HCV treatment has evolved over the past 20 years. Before 2011, the standard of care was a combination of interferon alfa-polyethylene glycol (peg-interferon), given as a weekly injection, and oral ribavirin. Neither drug has specific antiviral activity, and when they are used together they result in a sustained virologic response in fewer than 50% of patients with HCV genotype 1 and, at best, in 70% to 80% of patients with other genotypes.8
Nearly all patients receiving interferon experience side effects, which can be serious. Fatigue and flu-like symptoms are common, and the drug can also cause psychiatric symptoms (including depression or psychosis), weight loss, seizures, peripheral neuropathy, and bone marrow suppression. Ribavirin causes hemolysis and skin complications and is teratogenic.9
An important bit of information to know when using interferon is the patient’s IL28B genotype. This refers to a single-nucleotide polymorphism (C or T) on chromosome 19q13 (rs12979860) upstream of the IL28B gene encoding for interferon lambda-3. It is strongly associated with responsiveness to interferon: patients with the IL28B CC genotype have a much better chance of a sustained virologic response with interferon than do patients with CT or TT.
Boceprevir and telaprevir: First-generation protease inhibitors
In May 2011, the FDA approved the NS3/4A protease inhibitors boceprevir and telaprevir for treating HCV genotype 1, marking the beginning of the era of direct-acting antiviral agents.10 When these drugs are used in combination with peg-interferon alfa and ribavirin, up to 75% of patients with HCV genotype 1 who have had no previous treatment achieve a sustained virologic response.
But despite greatly improving the response rate, these first-generation protease inhibitors have substantial limitations. Twenty-five percent of patients with HCV genotype 1 who have received no previous treatment and 71% of patients who did not respond to previous treatment will not achieve a sustained virologic response with these agents.11 Further, they are effective only against HCV genotype 1, being highly specific for the amino acid target sequence of the NS3 region.
Also, they must be used in combination with interferon alfa and ribavirin because the virus needs to mutate only a little—a few amino-acid substitutions—to gain resistance to them.12 Therefore, patients are still exposed to interferon and ribavirin, with their toxicity. In addition, dysgeusia is seen with boceprevir, rash with telaprevir, and anemia with both.13,14
Finally, serious drug-drug interactions prompted the FDA to impose warnings for the use of these agents with other medications that interact with CYP3A4, the principal enzyme responsible for their metabolism. Thus, these significant adverse effects dampen the enthusiasm of patients contemplating a long course of treatment with these agents.
The need to improve the rate of sustained virologic response, shorten the duration of treatment, avoid serious side effects, improve efficacy in treating patients infected with genotypes other than 1, and, importantly, eliminate the need for interferon alfa and its serious adverse effects have driven the development of new direct-acting antiviral agents, including the two newly FDA-approved drugs, sofosbuvir and simeprevir.
SOFOSBUVIR: A POLYMERASE INHIBITOR
Sofosbuvir is a uridine nucleotide analogue that selectively inhibits the HCV NS5B RNA-dependent RNA polymerase (Figure 1). It targets the highly conserved nucleotide-binding pocket of this enzyme and functions as a chain terminator.15 While the protease inhibitors are genotype-dependent, inhibition of the highly conserved viral polymerase has an impact that spans genotypes.
Early clinical trials of sofosbuvir

Sofosbuvir has been tested in combination with interferon alfa and ribavirin, as well as in interferon-free regimens (Table 2).16–20
Rodriguez-Torres et al,15
- 56% with sofosbuvir 100 mg, peg-interferon, and ribavirin
- 83% with sofosbuvir 200 mg, peg-interferon, and ribavirin
- 80% with sofosbuvir 400 mg, peg-interferon, and ribavirin
- 43% with peg-interferon and ribavirin alone.
The ATOMIC trial16 tested the efficacy and safety of sofosbuvir in combination with peg-interferon and ribavirin in patients with HCV genotype 1, 4, or 6, without cirrhosis, who had not received any previous treatment. Patients with HCV genotype 1 were randomized to three treatments:
- Sofosbuvir 400 mg orally once daily plus peg-interferon and ribavirin for 12 weeks
- The same regimen, but for 24 weeks
- Sofosbuvir plus peg-interferon and ribavirin for 12 weeks, followed by 12 weeks of either sofosbuvir monotherapy or sofosbuvir plus ribavirin.
The rates of sustained virologic response were very high and were not significantly different among the three groups: 89%, 89%, and 87%, respectively. Patients who were able to complete a full course of therapy achieved even higher rates of sustained virologic response, ranging from 96% to 98%. The likelihood of response was not adversely affected by the usual markers of a poorer prognosis, such as a high viral load (≥ 800,000 IU/mL) or a non-CC IL28B genotype. Although patients with cirrhosis (another predictor of no response) were excluded from this study, the presence of bridging fibrosis did not seem to affect the rate of sustained virologic response. The results in patients with genotypes other than 1 were very encouraging, but the small number of patients enrolled precluded drawing firm conclusions in this group.
Important implications of the ATOMIC trial include the following:
There is no benefit in prolonging treatment with sofosbuvir beyond 12 weeks, since adverse events increased without any improvement in the rate of sustained virologic response.
There is a very low likelihood of developing viral resistance or mutation when using sofosbuvir.
There is no role for response-guided therapy, a concept used with protease inhibitor-based regimens in which patients who have complete clearance of the virus within the first 4 weeks of treatment (a rapid virologic response) and remain clear through 12 weeks of treatment (an extended rapid viral response) can be treated for a shorter duration without decreasing the likelihood of a sustained virologic response.
Lawitz et al17 conducted a randomized double-blind phase 2 trial to evaluate the effect of sofosbuvir dosing on response in noncirrhotic, previously untreated patients with HCV genotype 1, 2, or 3. Patients with HCV genotype 1 were randomized to one of three treatment groups in a 2:2:1 ratio: sofosbuvir 200 mg orally once daily, sofosbuvir 400 mg orally once daily, or placebo, all for 12 weeks in combination with peg-interferon (180 μg weekly) and ribavirin in a dosage based on weight. Depending on the viral response, patients continued peg-interferon and ribavirin for an additional 12 weeks if they achieved an extended rapid viral response, or 36 weeks if they did not achieve an extended rapid virologic response, and in all patients who received placebo. Patients with HCV genotype 2 or 3 were given sofosbuvir 400 mg once daily in combination with interferon and ribavirin for 12 weeks.
As in the ATOMIC trial, all patients treated with sofosbuvir had a very rapid reduction in viral load: 98% of patients with genotype 1 developed a rapid virologic response, and therefore almost all were eligible for the shorter treatment course of 24 weeks.17 The latter finding again suggested that response-guided treatment is not relevant with sofosbuvir-based regimens.
Very high rates of sustained virologic response were seen: 90% in patients with genotype 1 treated with sofosbuvir 200 mg, 91% in those with genotype 1 treated with 400 mg, and 92% in those with genotype 2 or 3. Although 6% of patients in the 200-mg group had virologic breakthrough after completing sofosbuvir treatment, no virologic breakthrough was observed in the 400-mg group, suggesting that the 400-mg dose might suppress the virus more effectively.17
The ELECTRON trial18 was a phase 2 study designed to evaluate the efficacy and safety of sofosbuvir and ribavirin in interferon-sparing and interferon-free regimens in patients with HCV genotype 1, 2, or 3 infection. Sofosbuvir was tested with peg-interferon and ribavirin, with ribavirin alone, and as monotherapy in previously untreated patients with genotype 2 or 3. A small number of patients with genotype 1 who were previously untreated and who were previously nonresponders were also treated with sofosbuvir and ribavirin.
All patients had a rapid virologic response, and viral suppression was sustained through the end of treatment. All patients with genotype 2 or 3 treated with double therapy (sofosbuvir and ribavirin) or triple therapy (sofosbuvir, peg-interferon, and ribavirin) achieved a sustained virologic response, compared with only 60% of patients treated with sofosbuvir monotherapy. The monotherapy group had an equal number of relapsers among those with genotype 2 or 3. Of the genotype 1 patients treated with sofosbuvir and ribavirin, 84% of those previously untreated developed a sustained virologic response, whereas only 10% of the previous nonresponders did.
Phase 3 clinical trials of sofosbuvir
The NEUTRINO trial19 studied the efficacy and safety of sofosbuvir in previously untreated patients with HCV genotype 1, 4, 5, or 6. In this phase 3 open-label study, all patients received sofosbuvir plus peg-interferon and weight-based ribavirin therapy for 12 weeks. Of the patients enrolled, 89% had genotype 1, while 9% had genotype 4 and 2% had genotype 5 or 6. Overall, 17% of the patients had cirrhosis.
The viral load rapidly decreased in all patients treated with sofosbuvir irrespective of the HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Ninety-nine percent of patients with genotype 1, 4, 5, or 6 achieved a rapid virologic response, and 90% achieved a sustained virologic response at 12 weeks after completion of treatment with sofosbuvir and ribavirin. Patients with cirrhosis had a slightly lower rate of sustained virologic response (80%, compared with 92% in patients without cirrhosis). Also, patients with non-CC IL28B genotypes had a lower rate of sustained virologic response (87% in non-CC allele vs 98% in patients with the favorable CC allele).
The FISSION trial19 recruited previously untreated patients with genotype 2 or 3 and randomized them to therapy with either sofosbuvir plus ribavirin in a weight-based dose for 12 weeks, or 24 weeks of interferon and ribavirin. In this study, 20% of patients in each treatment group had cirrhosis.
As in the NEUTRINO trial, the viral load rapidly decreased in all patients treated with sofosbuvir irrespective of HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Here, 100% of patients with genotype 2 or 3 who were treated with sofosbuvir and ribavirin achieved a rapid virologic response. Differences in outcome emerged based on genotype: 97% of those with genotype 2 and 56% of those with genotype 3 achieved a sustained virologic response. The overall rate was 67%, which was not different from patients treated with peg-interferon and ribavirin. In the subgroup of patients with cirrhosis, 47% of those treated with sofosbuvir and ribavirin achieved a sustained virologic response, vs 38% of those who received peg-interferon plus ribavirin.
In both the NEUTRINO and FISSION trials, few patients discontinued treatment, with higher rates of most adverse events occurring in patients treated with peg-interferon and ribavirin.
POSITRON,20 a phase 3 clinical trial, tested sofosbuvir in patients with HCV genotype 2 or 3 who were ineligible for peg-interferon, unwilling to take peg-interferon, or unable to tolerate peg-interferon (mainly because of clinically significant psychiatric disorders). Patients were randomized to two treatment groups for 12 weeks: sofosbuvir plus ribavirin, or placebo. About 50% of patients had HCV genotype 3, and 16% had cirrhosis.
The overall rate of sustained virologic response at 12 weeks after treatment was 78% in the sofosbuvir-and-ribavirin group (93% in genotype 2 patients and 61% in genotype 3 patients). Again, cirrhosis was associated with a lower rate of sustained virologic response (61% of patients with cirrhosis achieved a sustained virologic response vs 81% of patients without cirrhosis). None of the sofosbuvir-treated patients had virologic failure while on treatment.
FUSION,20 another phase 3 trial, evaluated sofosbuvir in patients infected with HCV genotype 2 or 3 for whom interferon-based treatment had failed. They were randomized to either 12 weeks or 16 weeks of sofosbuvir and weight-based ribavirin treatment. About 60% of patients had HCV genotype 3, and 34% had cirrhosis.
The overall sustained virologic response rate was 50% in the patients treated for 12 weeks and 73% in those treated for 16 weeks: specifically, 86% of patients with genotype 2 achieved a sustained virologic response at 12 weeks and 94% at 16 weeks, whereas in those with genotype 3 the rates were 30% at 12 weeks and 62% at 16 weeks.
Cirrhosis was again a predictor of lack of response to sofosbuvir. In the group treated for 12 weeks, 31% of those with cirrhosis achieved a sustained virologic response compared with 61% in those without cirrhosis. In the group treated for 16 weeks, 61% of those with cirrhosis achieved a sustained virologic response compared with 76% in those without cirrhosis.
In both the POSITRON and FUSION trials, relapse accounted for all treatment failures, and no virologic resistance was detected in patients who did not have a sustained virologic response. The investigators concluded that 12 weeks of treatment with sofosbuvir and ribavirin can be effective for HCV genotype 2 infection, but extending the treatment to 16 weeks may be beneficial for genotype 3. This may be especially important in patients with cirrhosis or those who did not have a response to peg-interferon-based treatment.
VALENCE,21 an ongoing phase 3 trial in Europe, is assessing the safety and efficacy of sofosbuvir 400 mg once daily and weight-based ribavirin in patients with HCV genotype 2 or 3. Eighty-five percent of the trial participants have received previous treatment, and 21% have cirrhosis. Patients were originally randomized in a 4:1 ratio to receive sofosbuvir plus ribavirin for 12 weeks or matching placebo, but as a result of emerging data suggesting that patients with genotype 3 would benefit from more than 12 weeks of treatment, the study was subsequently amended to extend treatment to 24 weeks for patients with genotype 3.
Overall rates of sustained virologic response were 93% in patients with genotype 2 and 85% in patients with genotype 3. In previously treated patients with genotype 2 who were treated for 12 weeks, the rates of sustained virologic response were 91% in those without cirrhosis vs 88% in those with cirrhosis. In previously treated patients with genotype 3, the rates in those treated for 24 weeks were 87% in patients without cirrhosis vs 60% with cirrhosis. The safety profile was consistent with that of ribavirin.
Side effects of sofosbuvir
In clinical trials, side effects occurred most often when sofosbuvir was combined with interferon and ribavirin and were consistent with the known side effects of the latter two agents. The most frequently reported side effects included fatigue, insomnia, nausea, rash, anemia, headache, and arthralgia, with most of these adverse events rated by treating clinicians as being mild in severity.15,20
In the ATOMIC trial, the most common events leading to drug discontinuation were anemia and neutropenia, both associated with interferon and ribavirin. Patients receiving sofosbuvir monotherapy after 12 weeks of triple therapy showed rapid improvement in hemoglobin levels and neutrophil counts, indicating that hematologic abnormalities attributed solely to sofosbuvir are minimal. In the FISSION trial, the incidence of adverse events was consistently lower in those receiving sofosbuvir-ribavirin than in patients receiving interferon-ribavirin without sofosbuvir.19
In the POSITRON trial, discontinuation of sofosbuvir because of adverse events was uncommon, and there were no differences in the incidence of adverse events and laboratory abnormalities between patients with and without cirrhosis when they received sofosbuvir and ribavirin.20
Sofosbuvir dosage and indications

Sofosbuvir is approved in an oral dose of 400 mg once daily in combination with ribavirin for patients infected with HCV genotype 2 or 3 and in combination with ribavirin and interferon alfa in patients infected with HCV genotype 1 or 4 (Table 3). It could be considered for HCV genotype 1 in combination with ribavirin alone for 24 weeks in patients who are ineligible for interferon.
Sofosbuvir is also recommended in combination with ribavirin in HCV-infected patients with hepatocellular carcinoma who are awaiting liver transplantation, for up to 48 weeks or until they receive a transplant, to prevent posttransplant reinfection with HCV.
Sofosbuvir is expensive
A course of therapy is expected to cost about $84,000, which is significantly more than the cost of previous triple therapy (peg-interferon, ribavirin, and either boceprevir or telaprevir).22 This high cost will undoubtedly lead to less widespread use in developing countries, and potentially even in the United States. As newer direct-acting antiviral agents become available, the price will likely come down, enhancing access to these drugs.
SIMEPREVIR: A SECOND-GENERATION PROTEASE INHIBITOR
Telaprevir and boceprevir are NS3/A4 protease inhibitors that belong to the alfa-ketoamid derivative class. Simeprevir belongs to the macrocyclic class and has a different way of binding to the target enzyme.23 Like sofosbuvir, simeprevir was recently approved by the FDA for the treatment of HCV genotype 1.
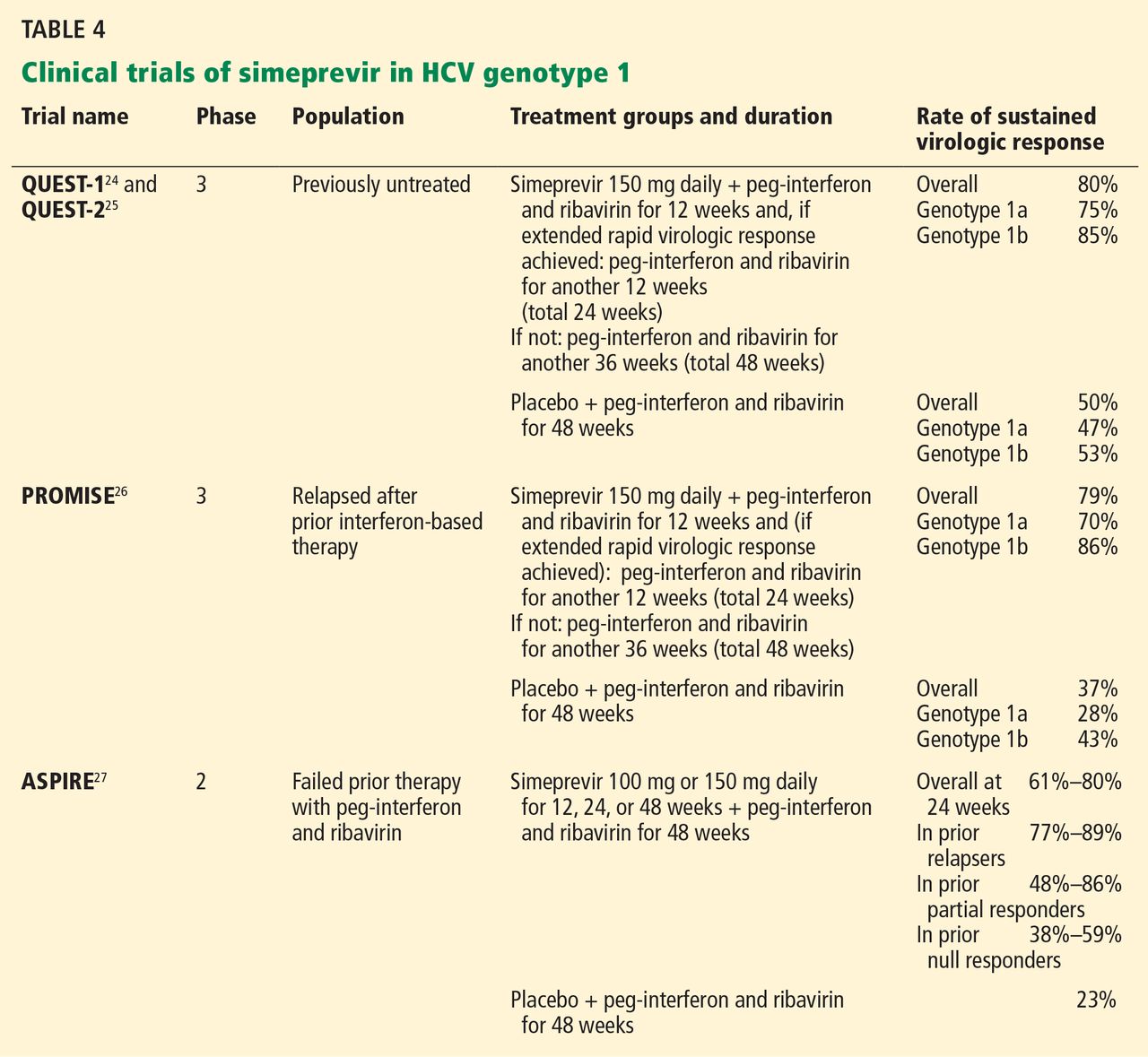
The therapeutic efficacy of simeprevir has been tested in several clinical trials (Table 4), including QUEST-124 and QUEST-225 (in previously untreated patients), PROMISE26 (in prior relapsers), and ASPIRE27 (in prior partial and null responders). Results from these trials showed high overall rates of sustained virologic response with triple therapy (ie, simeprevir combined with peg-interferon and ribavirin). It was generally well tolerated, and most adverse events reported during 12 weeks of treatment were of mild to moderate severity.
In QUEST-1 and QUEST-2, both double-blind phase 3 clinical trials, previously untreated patients infected with HCV genotype 1 were randomized in a 2:1 ratio to receive either simeprevir 150 mg daily or placebo for 12 weeks; both groups also received peg-interferon and ribavirin. Patients then received peg-interferon and ribavirin alone for 12 or 36 weeks in the simeprevir group (based on response) and for 36 weeks in the placebo group.
The overall rate of sustained virologic response at 12 weeks was 80% in the simeprevir group (75% in those with genotype 1a and 85% in those with genotype 1b) vs 50% in the placebo group (receiving peg-interferon and ribavirin alone).24,25
PROMISE,26 another double-blind randomized phase 3 clinical trial, evaluated simeprevir in patients with HCV genotype 1 who relapsed after previous interferon-based therapy. It had a similar design to QUEST-1 and QUEST-2, and 15% of all patients had cirrhosis.
The overall sustained virologic response rate at 12 weeks after treatment was 79% in the simeprevir group (70% in patients with genotype 1a and 86% in those with genotype 1b) vs 37% in the placebo group. Rates were similar in patients with absent to moderate fibrosis (82%), advanced fibrosis (73%), or cirrhosis (74%).
ASPIRE.27 Simeprevir efficacy in patients with HCV genotype 1 for whom previous therapy with peg-interferon and ribavirin had failed was tested in ASPIRE, a double-blind randomized phase 2 clinical trial. Patients were randomized to receive simeprevir (either 100 mg or 150 mg daily) for 12, 24, or 48 weeks in combination with 48 weeks of peg-interferon and ribavirin, or placebo plus peg-interferon and ribavirin for 48 weeks.
The primary end point was the rate of sustained virologic response at 24 weeks. Overall, rates were 61% to 80% for the simeprevir treatment groups compared with 23% with placebo, regardless of prior response to peg-interferon and ribavirin. By subgroup, rates were:
- 77% to 89% with simeprevir vs 37% with placebo in prior relapsers
- 48% to 86% with simeprevir vs 9% with placebo in prior partial responders
- 38% to 59% with placebo vs 19% for prior nonresponders.
The best rates of sustained viral response at 24 weeks were in the groups that received simeprevir 150 mg daily: 85% in prior relapsers, 75% in prior partial responders, and 51% in prior nonresponders.
Simeprevir vs other direct-acting antiviral drugs
Advantages of simeprevir over the earlier protease inhibitors include once-daily dosing, a lower rate of adverse events (the most common being fatigue, headache, rash, photosensitivity, and pruritus), a lower likelihood of discontinuation because of adverse events, and fewer drug-drug interactions (since it is a weak inhibitor of the CYP3A4 enzyme).
Unlike sofosbuvir, simeprevir was FDA-approved only for HCV genotype 1 and in combination with interferon alfa and ribavirin. Compared with sofosbuvir, the treatment duration with simeprevir regimens is longer overall (interferon alfa and ribavirin are given for 12 weeks in sofosbuvir-based regimens vs 24 to 48 weeks with simeprevir). As with sofosbuvir, the estimated cost of simeprevir is high, about $66,000 for a 12-week course.
Simeprevir dosage and indications
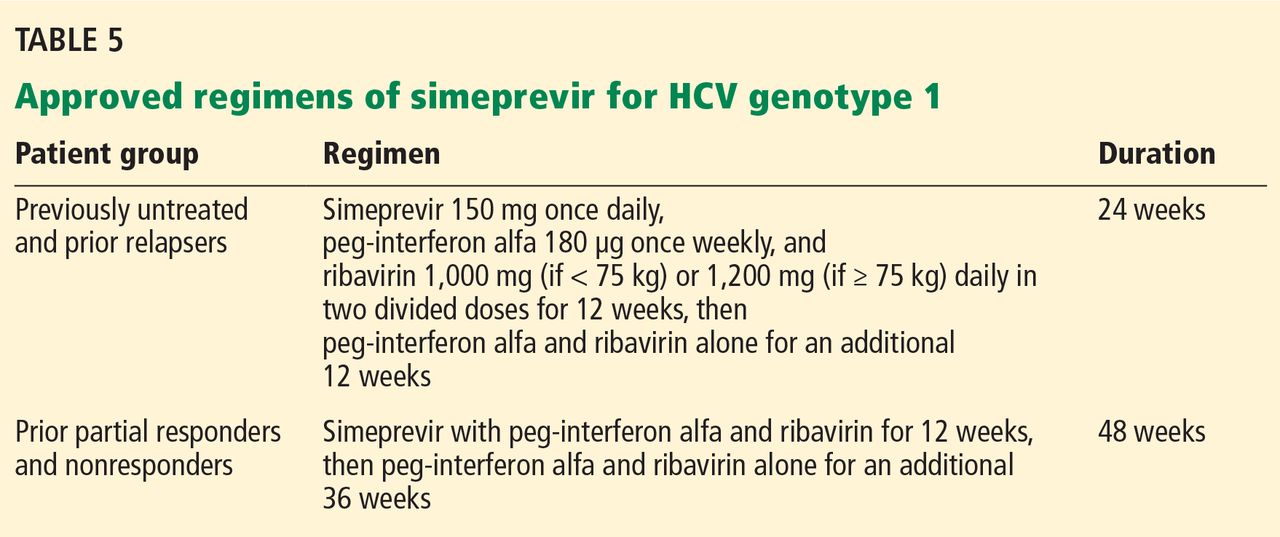
Simeprevir was approved at an oral dose of 150 mg once daily in combination with ribavirin and interferon alfa in patients with HCV genotype 1 (Table 5).
The approved regimens for simeprevir are fixed in total duration based on the patient’s treatment history. Specifically, all patients receive the drug in combination with peg-interferon and ribavirin for 12 weeks. Then, previously untreated patients and prior relapsers continue to receive peg-interferon and ribavirin alone for another 12 weeks, and those with a partial or null response continue with these drugs for another 36 weeks.
Patients infected with HCV genotype 1a should be screened for the NS3 Q80K polymorphism at baseline, as it has been associated with substantially reduced response to simeprevir.
Sofosbuvir and simeprevir in combination
The COSMOS trial.28 Given their differences in mechanism of action, sofosbuvir and simeprevir are being tested in combination. The COSMOS trial is an ongoing phase 2 randomized open-label study investigating the efficacy and safety of simeprevir and sofosbuvir in combination with and without ribavirin in patients with HCV genotype 1, including nonresponders and those with cirrhosis. Early results are promising, with very high rates of sustained virologic response with the sofosbuvir-simeprevir combination (93% to 100%) and indicate that the addition of ribavirin might not be needed to achieve sustained virologic response in this patient population.
THE FUTURE
The emergence of all-oral regimens for HCV treatment with increasingly sophisticated agents such as sofosbuvir and simeprevir will dramatically alter the management of HCV patients. In view of the improvement in sustained virologic response rates with these treatments, and since most HCV-infected persons have no symptoms, the US Centers for Disease Control and Prevention29 recently recommended one-time testing of the cohort in which the prevalence of HCV infection is highest: all persons born between 1945 and 1965. This undoubtedly will increase the detection of this infection—and the number of new patients expecting treatment.

Future drugs promise further improvements (Table 6).30–35 Advances in knowledge of the HCV molecular structure have led to the development of numerous direct-acting antiviral agents with very specific viral targets. A second wave of protease inhibitors and of nucleoside and nonnucleoside polymerase inhibitors will soon be available. Inhibitors of NS5A (a protein important in the assembly of the viral replication complex) such as daclatasvir and ledipasvir, are currently in phase 3 clinical trials. Other viral proteins involved in assembly of the virus, including the core protein and p7, are being explored as drug targets. In addition, inhibiting host targets such as cyclophilin A and miR122 has gained traction recently, with specific agents currently in phase 2 and 3 clinical trials.
Factors that previously were major determinants of response to treatment, such as IL28B genotype, viral load, race, age, extent of fibrosis, and genotype 1 subtypes, will become much less important with the introduction of highly potent direct-acting antiviral agents.
Many all-oral combinations are being evaluated in clinical trials. For example, the open-label, phase 2 LONESTAR trial tested the utility of combining sofosbuvir and ledipasvir (an NS5A inhibitor) with and without ribavirin for 8 or 12 weeks in previously untreated patients with HCV genotype 1, and for 12 weeks in patients with HCV genotype 1 who did not achieve a sustained virologic response after receiving a protease inhibitor-based regimen (half of whom had compensated cirrhosis).36 Sustained virologic response rates were very high (95% to 100%) in both previously treated and previously untreated patients, including those with cirrhosis. Similar rates were achieved by the 8-week and 12-week groups in noncirrhotic patients who had not been previously treated for HCV. The typical hematologic abnormalities associated with interferon were not observed except for mild anemia in patients who received ribavirin. These results suggest that the combination of sofosbuvir and ledipasvir could offer a very effective, short, all-oral treatment for patients with HCV genotype 1, including those with cirrhosis, who up to now have been difficult to treat.
Challenges remaining

The success of sofosbuvir and simeprevir paves the way for interferon-free regimens.37 For a long time, the treatment of HCV infection required close monitoring of patients while managing the side effects of interferon, but the current and emerging direct-acting antiviral agents will soon change this practice. Given the synergistic effects of combination therapy—targeting the virus at multiple locations, decreasing the likelihood of drug resistance, and improving efficacy—combination regimens seem to be the optimal solution to the HCV epidemic. Lower risk of side effects and shorter treatment duration will definitely improve the acceptance of any new regimen. New agents that act against conserved viral targets, thereby yielding activity across multiple genotypes, will be advantageous as well. Table 7 compares the rates of sustained virologic response of the different currently approved HCV treatment regimens.
Clinical challenges remain, including the management of special patient populations for whom data are still limited. These include patients with cirrhosis, chronic kidney disease, renal failure, and concurrent infection with human immunodeficiency virus, and patients who have undergone solid organ transplantation. Clinical trials are under way to evaluate the treatment options for these patients, who will likely need to wait for the emergence of additional agents before dramatic improvement in sustained virologic response rates may be expected.38
As the treatment of HCV becomes simpler, safer, and more effective, primary care physicians will increasingly be expected to manage it. Difficult-to-treat patients, including the special populations above, will require specialist management and individualized treatment regimens, at least until better therapies are available. The high projected cost of the new agents may limit access, at least initially. However, the dramatic improvement in sustained virologic response rates and all that that implies in terms of decreased risk of advanced liver disease and its complications will undoubtedly make these therapies cost-effective.39
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the united states. Clin Infect Dis 2012; 55(suppl 1):S10–S15.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first international liver transplantation society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transplant 2003; 9:S1–S9.
- Wong JB, McQuillan GM, McHutchison JG, Poynard T. Estimating future hepatitis C morbidity, mortality, and costs in the United States. Am J Public Health 2000; 90:1562–1569.
- Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007; 132:1979–1998.
- Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol 2000; 81:1631–1648.
- Singal AG, Volk ML, Jensen D, Di Bisceglie AM, Schoenfeld PS. A sustained viral response is associated with reduced liver-related morbidity and mortality in patients with hepatitis C virus. Clin Gastroenterol Hepatol 2010; 8:280–288,288.e1.
- Camma C, Di Bona D, Schepis F, et al. Effect of peginterferon alfa-2a on liver histology in chronic hepatitis C: a meta-analysis of individual patient data. Hepatology 2004; 39:333–342.
- Paeshuyse J, Dallmeier K, Neyts J. Ribavirin for the treatment of chronic hepatitis C virus infection: a review of the proposed mechanisms of action. Curr Opin Virol 2011; 1:590–598.
- Thomas E, Ghany MG, Liang TJ. The application and mechanism of action of ribavirin in therapy of hepatitis C. Antivir Chem Chemother 2012; 23:1–12.
- Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB; American Association for Study of Liver Diseases. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54:1433–1444.
- Soriano V, Vispo E, Poveda E, Labarga P, Barreiro P. Treatment failure with new hepatitis C drugs. Expert Opin Pharmacother 2012; 13:313–323.
- Asselah T, Marcellin P. Interferon free therapy with direct acting antivirals for HCV. Liver Int 2013; 33(suppl 1):93–104.
- Manns MP, McCone J, Davis MN, et al. Overall safety profile of boceprevir plus peginterferon alfa-2b and ribavirin in patients with chronic hepatitis C genotype 1: a combined analysis of 3 phase 2/3 clinical trials. Liver Int 2013; Aug 2. doi: 10.1111/liv.12300. [Epub ahead of print]
- Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Rodriguez-Torres M, Lawitz E, Kowdley KV, et al. Sofosbuvir (GS-7977) plus peginterferon/ribavirin in treatment-naive patients with HCV genotype 1: a randomized, 28-day, dose-ranging trial. J Hepatol 2013; 58:663–668.
- Kowdley KV, Lawitz E, Crespo I, et al. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet 2013; 381:2100–2107.
- Lawitz E, Lalezari JP, Hassanein T, et al. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect Dis 2013; 13:401–408.
- Gane EJ, Stedman CA, Hyland RH, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 2013; 368:34–44.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Jacobson IM, Gordon SC, Kowdley KV, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med 2013; 368:1867–1877.
- Zeuzem S, Dusheiko G, Salupere R, et al. Sofosbuvir + ribavirin for 12 or 24 weeks for patients with HCV genotype 2 or 3: the VALENCE trial [abstract no.1085]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- You DM, Pockros PJ. Simeprevir for the treatment of chronic hepatitis C. Expert Opin Pharmacother 2013; 14:2581–2589.
- Jacobson IM, Dore GJ, Foster G, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naive patients: results from Quest-1, a phase III trial [abstract no. 1425]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, Netherlands.
- Manns M, Marcellin P, Poordad FP, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naïve patients: results from QUEST-2, a phase III trial [abstract no. 1413]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, The Netherlands.
- Lawitz E, Forns X, Zeuzem S, et al. Simeprevir (TMC435) with peginterferon/ribavirin for treatment of chronic HCV genotype 1 infection in patients who relapsed after previous interferon-based therapy: results from promise, a phase III trial [abstract no. 869b]. Digestive Disease Week; May 18–21, 2013; Orlando, FL.
- Zeuzem S, Berg T, Gane E, et al. Simeprevir increases rate of sustained virologic response among treatment-experienced patients with HCV genotype-1 infection: a phase IIb trial. Gastroenterology epub Oct 31, 2013.
- Jacobson IM, Ghalib RM, Rodriguez-Torres M, et al. SVR results of a once-daily regimen of simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in cirrhotic and non-cirrhotic HCV genotype 1 treatment-naive and prior null responder patients: the COSMOS study [abstract LB-3]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Smith BD, Morgan RL, Beckett GA, et al. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945–1965. MMWR Recomm Rep 2012; 61( RR-4):1–32.
- Sulkowski MS, Kang M, Matining R, et al. Safety and antiviral activity of the HCV entry inhibitor ITX5061 in treatment-naive HCV-infected adults: a randomized, double-blind, phase 1b study. J Infect Dis 2013 Oct 9. [Epub ahead of print]
- Pawlotsky JM. NS5A inhibitors in the treatment of hepatitis C. J Hepatol 2013; 59:375–382.
- Yu M, Corsa AC, Xu S, et al. In vitro efficacy of approved and experimental antivirals against novel genotype 3 hepatitis C virus subgenomic replicons. Antiviral Res 2013; 100:439–445.
- Aghemo A, De Francesco R. New horizons in hepatitis C antiviral therapy with direct-acting antivirals. Hepatology 2013; 58:428–438.
- Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med 2013; 368:1907–1917.
- Flisiak R, Jaroszewicz J, Parfieniuk-Kowerda A. Emerging treatments for hepatitis C. Expert Opin Emerg Drugs 2013; 18:461–475.
- Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet 2013 Nov 1. doi: 10.1016/S0140-6736(13)62121-2 [Epub ahead of print]
- Drenth JP. HCV treatment—no more room for interferonologists? N Engl J Med 2013; 368:1931–1932.
- Casey LC, Lee WM. Hepatitis C virus therapy update 2013. Curr Opin Gastroenterol 2013; 29:243–249.
- Afdhal NH, Zeuzem S, Schooley RT, et al. The new paradigm of hepatitis C therapy: integration of oral therapies into best practices. J Viral Hepatol 2013; 20:745–760.
In late 2013, the US Food and Drug Administration (FDA) approved sofosbuvir and simeprevir, the newest direct-acting antiviral agents for treating chronic hepatitis C virus (HCV) infection. Multiple clinical trials have demonstrated dramatically improved treatment outcomes with these agents, opening the door to all-oral regimens or interferon-free regimens as the future standard of care for HCV.
In this article, we discuss the results of the trials that established the efficacy and safety of sofosbuvir and simeprevir and led to their FDA approval. We also summarize the importance of these agents and evaluate other direct-acting antivirals currently in the pipeline for HCV treatment.
HCV IS A RISING PROBLEM
Chronic HCV infection is a major clinical and public health problem, with the estimated number of people infected exceeding 170 million worldwide, including 3.2 million in the United States.1 It is a leading cause of cirrhosis, and its complications include hepatocellular carcinoma and liver failure. Cirrhosis due to HCV remains the leading indication for liver transplantation in the United States, accounting for nearly 40% of liver transplants in adults.2
The clinical impact of HCV will only continue to escalate, and in parallel, so will the cost to society. Models suggest that HCV-related deaths will double between 2010 and 2019, and considering only direct medical costs, the projected financial burden of treating HCV-related disease during this interval is estimated at between $6.5 and $13.6 billion.3
AN RNA VIRUS WITH SIX GENOTYPES
HCV, first identified in 1989, is an enveloped, single-stranded RNA flavivirus of the Hepacivirus genus measuring 50 to 60 nm in diameter.4 There are six viral genotypes, with genotype 1 being the most common in the United States and traditionally the most difficult to treat.
Once inside the host cell, the virus releases its RNA strand, which is translated into a single polyprotein of about 3,000 amino acids. This large molecule is then cleaved by proteases into several domains: three structural proteins (C, E1, and E2), a small protein called p7, and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (Figure 1).5 These nonstructural proteins enable the virus to replicate.
GOAL OF TREATING HCV: A SUSTAINED VIROLOGIC RESPONSE
The aim of HCV treatment is to achieve a sustained virologic response, defined as having no detectable viral RNA after completion of antiviral therapy. This is associated with substantially better clinical outcomes, lower rates of liver-related morbidity and all-cause mortality, and stabilization of or even improvement in liver histology.6,7 This end point has traditionally been assessed at 6 months after the end of therapy, but recent data suggest the rates at 12 weeks are essentially equivalent.
Table 1 summarizes the patterns of virologic response in treating HCV infection.
Interferon plus ribavirin: The standard of care for many years
HCV treatment has evolved over the past 20 years. Before 2011, the standard of care was a combination of interferon alfa-polyethylene glycol (peg-interferon), given as a weekly injection, and oral ribavirin. Neither drug has specific antiviral activity, and when they are used together they result in a sustained virologic response in fewer than 50% of patients with HCV genotype 1 and, at best, in 70% to 80% of patients with other genotypes.8
Nearly all patients receiving interferon experience side effects, which can be serious. Fatigue and flu-like symptoms are common, and the drug can also cause psychiatric symptoms (including depression or psychosis), weight loss, seizures, peripheral neuropathy, and bone marrow suppression. Ribavirin causes hemolysis and skin complications and is teratogenic.9
An important bit of information to know when using interferon is the patient’s IL28B genotype. This refers to a single-nucleotide polymorphism (C or T) on chromosome 19q13 (rs12979860) upstream of the IL28B gene encoding for interferon lambda-3. It is strongly associated with responsiveness to interferon: patients with the IL28B CC genotype have a much better chance of a sustained virologic response with interferon than do patients with CT or TT.
Boceprevir and telaprevir: First-generation protease inhibitors
In May 2011, the FDA approved the NS3/4A protease inhibitors boceprevir and telaprevir for treating HCV genotype 1, marking the beginning of the era of direct-acting antiviral agents.10 When these drugs are used in combination with peg-interferon alfa and ribavirin, up to 75% of patients with HCV genotype 1 who have had no previous treatment achieve a sustained virologic response.
But despite greatly improving the response rate, these first-generation protease inhibitors have substantial limitations. Twenty-five percent of patients with HCV genotype 1 who have received no previous treatment and 71% of patients who did not respond to previous treatment will not achieve a sustained virologic response with these agents.11 Further, they are effective only against HCV genotype 1, being highly specific for the amino acid target sequence of the NS3 region.
Also, they must be used in combination with interferon alfa and ribavirin because the virus needs to mutate only a little—a few amino-acid substitutions—to gain resistance to them.12 Therefore, patients are still exposed to interferon and ribavirin, with their toxicity. In addition, dysgeusia is seen with boceprevir, rash with telaprevir, and anemia with both.13,14
Finally, serious drug-drug interactions prompted the FDA to impose warnings for the use of these agents with other medications that interact with CYP3A4, the principal enzyme responsible for their metabolism. Thus, these significant adverse effects dampen the enthusiasm of patients contemplating a long course of treatment with these agents.
The need to improve the rate of sustained virologic response, shorten the duration of treatment, avoid serious side effects, improve efficacy in treating patients infected with genotypes other than 1, and, importantly, eliminate the need for interferon alfa and its serious adverse effects have driven the development of new direct-acting antiviral agents, including the two newly FDA-approved drugs, sofosbuvir and simeprevir.
SOFOSBUVIR: A POLYMERASE INHIBITOR
Sofosbuvir is a uridine nucleotide analogue that selectively inhibits the HCV NS5B RNA-dependent RNA polymerase (Figure 1). It targets the highly conserved nucleotide-binding pocket of this enzyme and functions as a chain terminator.15 While the protease inhibitors are genotype-dependent, inhibition of the highly conserved viral polymerase has an impact that spans genotypes.
Early clinical trials of sofosbuvir
Sofosbuvir has been tested in combination with interferon alfa and ribavirin, as well as in interferon-free regimens (Table 2).16–20
Rodriguez-Torres et al,15
- 56% with sofosbuvir 100 mg, peg-interferon, and ribavirin
- 83% with sofosbuvir 200 mg, peg-interferon, and ribavirin
- 80% with sofosbuvir 400 mg, peg-interferon, and ribavirin
- 43% with peg-interferon and ribavirin alone.
The ATOMIC trial16 tested the efficacy and safety of sofosbuvir in combination with peg-interferon and ribavirin in patients with HCV genotype 1, 4, or 6, without cirrhosis, who had not received any previous treatment. Patients with HCV genotype 1 were randomized to three treatments:
- Sofosbuvir 400 mg orally once daily plus peg-interferon and ribavirin for 12 weeks
- The same regimen, but for 24 weeks
- Sofosbuvir plus peg-interferon and ribavirin for 12 weeks, followed by 12 weeks of either sofosbuvir monotherapy or sofosbuvir plus ribavirin.
The rates of sustained virologic response were very high and were not significantly different among the three groups: 89%, 89%, and 87%, respectively. Patients who were able to complete a full course of therapy achieved even higher rates of sustained virologic response, ranging from 96% to 98%. The likelihood of response was not adversely affected by the usual markers of a poorer prognosis, such as a high viral load (≥ 800,000 IU/mL) or a non-CC IL28B genotype. Although patients with cirrhosis (another predictor of no response) were excluded from this study, the presence of bridging fibrosis did not seem to affect the rate of sustained virologic response. The results in patients with genotypes other than 1 were very encouraging, but the small number of patients enrolled precluded drawing firm conclusions in this group.
Important implications of the ATOMIC trial include the following:
There is no benefit in prolonging treatment with sofosbuvir beyond 12 weeks, since adverse events increased without any improvement in the rate of sustained virologic response.
There is a very low likelihood of developing viral resistance or mutation when using sofosbuvir.
There is no role for response-guided therapy, a concept used with protease inhibitor-based regimens in which patients who have complete clearance of the virus within the first 4 weeks of treatment (a rapid virologic response) and remain clear through 12 weeks of treatment (an extended rapid viral response) can be treated for a shorter duration without decreasing the likelihood of a sustained virologic response.
Lawitz et al17 conducted a randomized double-blind phase 2 trial to evaluate the effect of sofosbuvir dosing on response in noncirrhotic, previously untreated patients with HCV genotype 1, 2, or 3. Patients with HCV genotype 1 were randomized to one of three treatment groups in a 2:2:1 ratio: sofosbuvir 200 mg orally once daily, sofosbuvir 400 mg orally once daily, or placebo, all for 12 weeks in combination with peg-interferon (180 μg weekly) and ribavirin in a dosage based on weight. Depending on the viral response, patients continued peg-interferon and ribavirin for an additional 12 weeks if they achieved an extended rapid viral response, or 36 weeks if they did not achieve an extended rapid virologic response, and in all patients who received placebo. Patients with HCV genotype 2 or 3 were given sofosbuvir 400 mg once daily in combination with interferon and ribavirin for 12 weeks.
As in the ATOMIC trial, all patients treated with sofosbuvir had a very rapid reduction in viral load: 98% of patients with genotype 1 developed a rapid virologic response, and therefore almost all were eligible for the shorter treatment course of 24 weeks.17 The latter finding again suggested that response-guided treatment is not relevant with sofosbuvir-based regimens.
Very high rates of sustained virologic response were seen: 90% in patients with genotype 1 treated with sofosbuvir 200 mg, 91% in those with genotype 1 treated with 400 mg, and 92% in those with genotype 2 or 3. Although 6% of patients in the 200-mg group had virologic breakthrough after completing sofosbuvir treatment, no virologic breakthrough was observed in the 400-mg group, suggesting that the 400-mg dose might suppress the virus more effectively.17
The ELECTRON trial18 was a phase 2 study designed to evaluate the efficacy and safety of sofosbuvir and ribavirin in interferon-sparing and interferon-free regimens in patients with HCV genotype 1, 2, or 3 infection. Sofosbuvir was tested with peg-interferon and ribavirin, with ribavirin alone, and as monotherapy in previously untreated patients with genotype 2 or 3. A small number of patients with genotype 1 who were previously untreated and who were previously nonresponders were also treated with sofosbuvir and ribavirin.
All patients had a rapid virologic response, and viral suppression was sustained through the end of treatment. All patients with genotype 2 or 3 treated with double therapy (sofosbuvir and ribavirin) or triple therapy (sofosbuvir, peg-interferon, and ribavirin) achieved a sustained virologic response, compared with only 60% of patients treated with sofosbuvir monotherapy. The monotherapy group had an equal number of relapsers among those with genotype 2 or 3. Of the genotype 1 patients treated with sofosbuvir and ribavirin, 84% of those previously untreated developed a sustained virologic response, whereas only 10% of the previous nonresponders did.
Phase 3 clinical trials of sofosbuvir
The NEUTRINO trial19 studied the efficacy and safety of sofosbuvir in previously untreated patients with HCV genotype 1, 4, 5, or 6. In this phase 3 open-label study, all patients received sofosbuvir plus peg-interferon and weight-based ribavirin therapy for 12 weeks. Of the patients enrolled, 89% had genotype 1, while 9% had genotype 4 and 2% had genotype 5 or 6. Overall, 17% of the patients had cirrhosis.
The viral load rapidly decreased in all patients treated with sofosbuvir irrespective of the HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Ninety-nine percent of patients with genotype 1, 4, 5, or 6 achieved a rapid virologic response, and 90% achieved a sustained virologic response at 12 weeks after completion of treatment with sofosbuvir and ribavirin. Patients with cirrhosis had a slightly lower rate of sustained virologic response (80%, compared with 92% in patients without cirrhosis). Also, patients with non-CC IL28B genotypes had a lower rate of sustained virologic response (87% in non-CC allele vs 98% in patients with the favorable CC allele).
The FISSION trial19 recruited previously untreated patients with genotype 2 or 3 and randomized them to therapy with either sofosbuvir plus ribavirin in a weight-based dose for 12 weeks, or 24 weeks of interferon and ribavirin. In this study, 20% of patients in each treatment group had cirrhosis.
As in the NEUTRINO trial, the viral load rapidly decreased in all patients treated with sofosbuvir irrespective of HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Here, 100% of patients with genotype 2 or 3 who were treated with sofosbuvir and ribavirin achieved a rapid virologic response. Differences in outcome emerged based on genotype: 97% of those with genotype 2 and 56% of those with genotype 3 achieved a sustained virologic response. The overall rate was 67%, which was not different from patients treated with peg-interferon and ribavirin. In the subgroup of patients with cirrhosis, 47% of those treated with sofosbuvir and ribavirin achieved a sustained virologic response, vs 38% of those who received peg-interferon plus ribavirin.
In both the NEUTRINO and FISSION trials, few patients discontinued treatment, with higher rates of most adverse events occurring in patients treated with peg-interferon and ribavirin.
POSITRON,20 a phase 3 clinical trial, tested sofosbuvir in patients with HCV genotype 2 or 3 who were ineligible for peg-interferon, unwilling to take peg-interferon, or unable to tolerate peg-interferon (mainly because of clinically significant psychiatric disorders). Patients were randomized to two treatment groups for 12 weeks: sofosbuvir plus ribavirin, or placebo. About 50% of patients had HCV genotype 3, and 16% had cirrhosis.
The overall rate of sustained virologic response at 12 weeks after treatment was 78% in the sofosbuvir-and-ribavirin group (93% in genotype 2 patients and 61% in genotype 3 patients). Again, cirrhosis was associated with a lower rate of sustained virologic response (61% of patients with cirrhosis achieved a sustained virologic response vs 81% of patients without cirrhosis). None of the sofosbuvir-treated patients had virologic failure while on treatment.
FUSION,20 another phase 3 trial, evaluated sofosbuvir in patients infected with HCV genotype 2 or 3 for whom interferon-based treatment had failed. They were randomized to either 12 weeks or 16 weeks of sofosbuvir and weight-based ribavirin treatment. About 60% of patients had HCV genotype 3, and 34% had cirrhosis.
The overall sustained virologic response rate was 50% in the patients treated for 12 weeks and 73% in those treated for 16 weeks: specifically, 86% of patients with genotype 2 achieved a sustained virologic response at 12 weeks and 94% at 16 weeks, whereas in those with genotype 3 the rates were 30% at 12 weeks and 62% at 16 weeks.
Cirrhosis was again a predictor of lack of response to sofosbuvir. In the group treated for 12 weeks, 31% of those with cirrhosis achieved a sustained virologic response compared with 61% in those without cirrhosis. In the group treated for 16 weeks, 61% of those with cirrhosis achieved a sustained virologic response compared with 76% in those without cirrhosis.
In both the POSITRON and FUSION trials, relapse accounted for all treatment failures, and no virologic resistance was detected in patients who did not have a sustained virologic response. The investigators concluded that 12 weeks of treatment with sofosbuvir and ribavirin can be effective for HCV genotype 2 infection, but extending the treatment to 16 weeks may be beneficial for genotype 3. This may be especially important in patients with cirrhosis or those who did not have a response to peg-interferon-based treatment.
VALENCE,21 an ongoing phase 3 trial in Europe, is assessing the safety and efficacy of sofosbuvir 400 mg once daily and weight-based ribavirin in patients with HCV genotype 2 or 3. Eighty-five percent of the trial participants have received previous treatment, and 21% have cirrhosis. Patients were originally randomized in a 4:1 ratio to receive sofosbuvir plus ribavirin for 12 weeks or matching placebo, but as a result of emerging data suggesting that patients with genotype 3 would benefit from more than 12 weeks of treatment, the study was subsequently amended to extend treatment to 24 weeks for patients with genotype 3.
Overall rates of sustained virologic response were 93% in patients with genotype 2 and 85% in patients with genotype 3. In previously treated patients with genotype 2 who were treated for 12 weeks, the rates of sustained virologic response were 91% in those without cirrhosis vs 88% in those with cirrhosis. In previously treated patients with genotype 3, the rates in those treated for 24 weeks were 87% in patients without cirrhosis vs 60% with cirrhosis. The safety profile was consistent with that of ribavirin.
Side effects of sofosbuvir
In clinical trials, side effects occurred most often when sofosbuvir was combined with interferon and ribavirin and were consistent with the known side effects of the latter two agents. The most frequently reported side effects included fatigue, insomnia, nausea, rash, anemia, headache, and arthralgia, with most of these adverse events rated by treating clinicians as being mild in severity.15,20
In the ATOMIC trial, the most common events leading to drug discontinuation were anemia and neutropenia, both associated with interferon and ribavirin. Patients receiving sofosbuvir monotherapy after 12 weeks of triple therapy showed rapid improvement in hemoglobin levels and neutrophil counts, indicating that hematologic abnormalities attributed solely to sofosbuvir are minimal. In the FISSION trial, the incidence of adverse events was consistently lower in those receiving sofosbuvir-ribavirin than in patients receiving interferon-ribavirin without sofosbuvir.19
In the POSITRON trial, discontinuation of sofosbuvir because of adverse events was uncommon, and there were no differences in the incidence of adverse events and laboratory abnormalities between patients with and without cirrhosis when they received sofosbuvir and ribavirin.20
Sofosbuvir dosage and indications
Sofosbuvir is approved in an oral dose of 400 mg once daily in combination with ribavirin for patients infected with HCV genotype 2 or 3 and in combination with ribavirin and interferon alfa in patients infected with HCV genotype 1 or 4 (Table 3). It could be considered for HCV genotype 1 in combination with ribavirin alone for 24 weeks in patients who are ineligible for interferon.
Sofosbuvir is also recommended in combination with ribavirin in HCV-infected patients with hepatocellular carcinoma who are awaiting liver transplantation, for up to 48 weeks or until they receive a transplant, to prevent posttransplant reinfection with HCV.
Sofosbuvir is expensive
A course of therapy is expected to cost about $84,000, which is significantly more than the cost of previous triple therapy (peg-interferon, ribavirin, and either boceprevir or telaprevir).22 This high cost will undoubtedly lead to less widespread use in developing countries, and potentially even in the United States. As newer direct-acting antiviral agents become available, the price will likely come down, enhancing access to these drugs.
SIMEPREVIR: A SECOND-GENERATION PROTEASE INHIBITOR
Telaprevir and boceprevir are NS3/A4 protease inhibitors that belong to the alfa-ketoamid derivative class. Simeprevir belongs to the macrocyclic class and has a different way of binding to the target enzyme.23 Like sofosbuvir, simeprevir was recently approved by the FDA for the treatment of HCV genotype 1.
The therapeutic efficacy of simeprevir has been tested in several clinical trials (Table 4), including QUEST-124 and QUEST-225 (in previously untreated patients), PROMISE26 (in prior relapsers), and ASPIRE27 (in prior partial and null responders). Results from these trials showed high overall rates of sustained virologic response with triple therapy (ie, simeprevir combined with peg-interferon and ribavirin). It was generally well tolerated, and most adverse events reported during 12 weeks of treatment were of mild to moderate severity.
In QUEST-1 and QUEST-2, both double-blind phase 3 clinical trials, previously untreated patients infected with HCV genotype 1 were randomized in a 2:1 ratio to receive either simeprevir 150 mg daily or placebo for 12 weeks; both groups also received peg-interferon and ribavirin. Patients then received peg-interferon and ribavirin alone for 12 or 36 weeks in the simeprevir group (based on response) and for 36 weeks in the placebo group.
The overall rate of sustained virologic response at 12 weeks was 80% in the simeprevir group (75% in those with genotype 1a and 85% in those with genotype 1b) vs 50% in the placebo group (receiving peg-interferon and ribavirin alone).24,25
PROMISE,26 another double-blind randomized phase 3 clinical trial, evaluated simeprevir in patients with HCV genotype 1 who relapsed after previous interferon-based therapy. It had a similar design to QUEST-1 and QUEST-2, and 15% of all patients had cirrhosis.
The overall sustained virologic response rate at 12 weeks after treatment was 79% in the simeprevir group (70% in patients with genotype 1a and 86% in those with genotype 1b) vs 37% in the placebo group. Rates were similar in patients with absent to moderate fibrosis (82%), advanced fibrosis (73%), or cirrhosis (74%).
ASPIRE.27 Simeprevir efficacy in patients with HCV genotype 1 for whom previous therapy with peg-interferon and ribavirin had failed was tested in ASPIRE, a double-blind randomized phase 2 clinical trial. Patients were randomized to receive simeprevir (either 100 mg or 150 mg daily) for 12, 24, or 48 weeks in combination with 48 weeks of peg-interferon and ribavirin, or placebo plus peg-interferon and ribavirin for 48 weeks.
The primary end point was the rate of sustained virologic response at 24 weeks. Overall, rates were 61% to 80% for the simeprevir treatment groups compared with 23% with placebo, regardless of prior response to peg-interferon and ribavirin. By subgroup, rates were:
- 77% to 89% with simeprevir vs 37% with placebo in prior relapsers
- 48% to 86% with simeprevir vs 9% with placebo in prior partial responders
- 38% to 59% with placebo vs 19% for prior nonresponders.
The best rates of sustained viral response at 24 weeks were in the groups that received simeprevir 150 mg daily: 85% in prior relapsers, 75% in prior partial responders, and 51% in prior nonresponders.
Simeprevir vs other direct-acting antiviral drugs
Advantages of simeprevir over the earlier protease inhibitors include once-daily dosing, a lower rate of adverse events (the most common being fatigue, headache, rash, photosensitivity, and pruritus), a lower likelihood of discontinuation because of adverse events, and fewer drug-drug interactions (since it is a weak inhibitor of the CYP3A4 enzyme).
Unlike sofosbuvir, simeprevir was FDA-approved only for HCV genotype 1 and in combination with interferon alfa and ribavirin. Compared with sofosbuvir, the treatment duration with simeprevir regimens is longer overall (interferon alfa and ribavirin are given for 12 weeks in sofosbuvir-based regimens vs 24 to 48 weeks with simeprevir). As with sofosbuvir, the estimated cost of simeprevir is high, about $66,000 for a 12-week course.
Simeprevir dosage and indications
Simeprevir was approved at an oral dose of 150 mg once daily in combination with ribavirin and interferon alfa in patients with HCV genotype 1 (Table 5).
The approved regimens for simeprevir are fixed in total duration based on the patient’s treatment history. Specifically, all patients receive the drug in combination with peg-interferon and ribavirin for 12 weeks. Then, previously untreated patients and prior relapsers continue to receive peg-interferon and ribavirin alone for another 12 weeks, and those with a partial or null response continue with these drugs for another 36 weeks.
Patients infected with HCV genotype 1a should be screened for the NS3 Q80K polymorphism at baseline, as it has been associated with substantially reduced response to simeprevir.
Sofosbuvir and simeprevir in combination
The COSMOS trial.28 Given their differences in mechanism of action, sofosbuvir and simeprevir are being tested in combination. The COSMOS trial is an ongoing phase 2 randomized open-label study investigating the efficacy and safety of simeprevir and sofosbuvir in combination with and without ribavirin in patients with HCV genotype 1, including nonresponders and those with cirrhosis. Early results are promising, with very high rates of sustained virologic response with the sofosbuvir-simeprevir combination (93% to 100%) and indicate that the addition of ribavirin might not be needed to achieve sustained virologic response in this patient population.
THE FUTURE
The emergence of all-oral regimens for HCV treatment with increasingly sophisticated agents such as sofosbuvir and simeprevir will dramatically alter the management of HCV patients. In view of the improvement in sustained virologic response rates with these treatments, and since most HCV-infected persons have no symptoms, the US Centers for Disease Control and Prevention29 recently recommended one-time testing of the cohort in which the prevalence of HCV infection is highest: all persons born between 1945 and 1965. This undoubtedly will increase the detection of this infection—and the number of new patients expecting treatment.
Future drugs promise further improvements (Table 6).30–35 Advances in knowledge of the HCV molecular structure have led to the development of numerous direct-acting antiviral agents with very specific viral targets. A second wave of protease inhibitors and of nucleoside and nonnucleoside polymerase inhibitors will soon be available. Inhibitors of NS5A (a protein important in the assembly of the viral replication complex) such as daclatasvir and ledipasvir, are currently in phase 3 clinical trials. Other viral proteins involved in assembly of the virus, including the core protein and p7, are being explored as drug targets. In addition, inhibiting host targets such as cyclophilin A and miR122 has gained traction recently, with specific agents currently in phase 2 and 3 clinical trials.
Factors that previously were major determinants of response to treatment, such as IL28B genotype, viral load, race, age, extent of fibrosis, and genotype 1 subtypes, will become much less important with the introduction of highly potent direct-acting antiviral agents.
Many all-oral combinations are being evaluated in clinical trials. For example, the open-label, phase 2 LONESTAR trial tested the utility of combining sofosbuvir and ledipasvir (an NS5A inhibitor) with and without ribavirin for 8 or 12 weeks in previously untreated patients with HCV genotype 1, and for 12 weeks in patients with HCV genotype 1 who did not achieve a sustained virologic response after receiving a protease inhibitor-based regimen (half of whom had compensated cirrhosis).36 Sustained virologic response rates were very high (95% to 100%) in both previously treated and previously untreated patients, including those with cirrhosis. Similar rates were achieved by the 8-week and 12-week groups in noncirrhotic patients who had not been previously treated for HCV. The typical hematologic abnormalities associated with interferon were not observed except for mild anemia in patients who received ribavirin. These results suggest that the combination of sofosbuvir and ledipasvir could offer a very effective, short, all-oral treatment for patients with HCV genotype 1, including those with cirrhosis, who up to now have been difficult to treat.
Challenges remaining
The success of sofosbuvir and simeprevir paves the way for interferon-free regimens.37 For a long time, the treatment of HCV infection required close monitoring of patients while managing the side effects of interferon, but the current and emerging direct-acting antiviral agents will soon change this practice. Given the synergistic effects of combination therapy—targeting the virus at multiple locations, decreasing the likelihood of drug resistance, and improving efficacy—combination regimens seem to be the optimal solution to the HCV epidemic. Lower risk of side effects and shorter treatment duration will definitely improve the acceptance of any new regimen. New agents that act against conserved viral targets, thereby yielding activity across multiple genotypes, will be advantageous as well. Table 7 compares the rates of sustained virologic response of the different currently approved HCV treatment regimens.
Clinical challenges remain, including the management of special patient populations for whom data are still limited. These include patients with cirrhosis, chronic kidney disease, renal failure, and concurrent infection with human immunodeficiency virus, and patients who have undergone solid organ transplantation. Clinical trials are under way to evaluate the treatment options for these patients, who will likely need to wait for the emergence of additional agents before dramatic improvement in sustained virologic response rates may be expected.38
As the treatment of HCV becomes simpler, safer, and more effective, primary care physicians will increasingly be expected to manage it. Difficult-to-treat patients, including the special populations above, will require specialist management and individualized treatment regimens, at least until better therapies are available. The high projected cost of the new agents may limit access, at least initially. However, the dramatic improvement in sustained virologic response rates and all that that implies in terms of decreased risk of advanced liver disease and its complications will undoubtedly make these therapies cost-effective.39
In late 2013, the US Food and Drug Administration (FDA) approved sofosbuvir and simeprevir, the newest direct-acting antiviral agents for treating chronic hepatitis C virus (HCV) infection. Multiple clinical trials have demonstrated dramatically improved treatment outcomes with these agents, opening the door to all-oral regimens or interferon-free regimens as the future standard of care for HCV.
In this article, we discuss the results of the trials that established the efficacy and safety of sofosbuvir and simeprevir and led to their FDA approval. We also summarize the importance of these agents and evaluate other direct-acting antivirals currently in the pipeline for HCV treatment.
HCV IS A RISING PROBLEM
Chronic HCV infection is a major clinical and public health problem, with the estimated number of people infected exceeding 170 million worldwide, including 3.2 million in the United States.1 It is a leading cause of cirrhosis, and its complications include hepatocellular carcinoma and liver failure. Cirrhosis due to HCV remains the leading indication for liver transplantation in the United States, accounting for nearly 40% of liver transplants in adults.2
The clinical impact of HCV will only continue to escalate, and in parallel, so will the cost to society. Models suggest that HCV-related deaths will double between 2010 and 2019, and considering only direct medical costs, the projected financial burden of treating HCV-related disease during this interval is estimated at between $6.5 and $13.6 billion.3
AN RNA VIRUS WITH SIX GENOTYPES
HCV, first identified in 1989, is an enveloped, single-stranded RNA flavivirus of the Hepacivirus genus measuring 50 to 60 nm in diameter.4 There are six viral genotypes, with genotype 1 being the most common in the United States and traditionally the most difficult to treat.
Once inside the host cell, the virus releases its RNA strand, which is translated into a single polyprotein of about 3,000 amino acids. This large molecule is then cleaved by proteases into several domains: three structural proteins (C, E1, and E2), a small protein called p7, and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (Figure 1).5 These nonstructural proteins enable the virus to replicate.
GOAL OF TREATING HCV: A SUSTAINED VIROLOGIC RESPONSE
The aim of HCV treatment is to achieve a sustained virologic response, defined as having no detectable viral RNA after completion of antiviral therapy. This is associated with substantially better clinical outcomes, lower rates of liver-related morbidity and all-cause mortality, and stabilization of or even improvement in liver histology.6,7 This end point has traditionally been assessed at 6 months after the end of therapy, but recent data suggest the rates at 12 weeks are essentially equivalent.
Table 1 summarizes the patterns of virologic response in treating HCV infection.
Interferon plus ribavirin: The standard of care for many years
HCV treatment has evolved over the past 20 years. Before 2011, the standard of care was a combination of interferon alfa-polyethylene glycol (peg-interferon), given as a weekly injection, and oral ribavirin. Neither drug has specific antiviral activity, and when they are used together they result in a sustained virologic response in fewer than 50% of patients with HCV genotype 1 and, at best, in 70% to 80% of patients with other genotypes.8
Nearly all patients receiving interferon experience side effects, which can be serious. Fatigue and flu-like symptoms are common, and the drug can also cause psychiatric symptoms (including depression or psychosis), weight loss, seizures, peripheral neuropathy, and bone marrow suppression. Ribavirin causes hemolysis and skin complications and is teratogenic.9
An important bit of information to know when using interferon is the patient’s IL28B genotype. This refers to a single-nucleotide polymorphism (C or T) on chromosome 19q13 (rs12979860) upstream of the IL28B gene encoding for interferon lambda-3. It is strongly associated with responsiveness to interferon: patients with the IL28B CC genotype have a much better chance of a sustained virologic response with interferon than do patients with CT or TT.
Boceprevir and telaprevir: First-generation protease inhibitors
In May 2011, the FDA approved the NS3/4A protease inhibitors boceprevir and telaprevir for treating HCV genotype 1, marking the beginning of the era of direct-acting antiviral agents.10 When these drugs are used in combination with peg-interferon alfa and ribavirin, up to 75% of patients with HCV genotype 1 who have had no previous treatment achieve a sustained virologic response.
But despite greatly improving the response rate, these first-generation protease inhibitors have substantial limitations. Twenty-five percent of patients with HCV genotype 1 who have received no previous treatment and 71% of patients who did not respond to previous treatment will not achieve a sustained virologic response with these agents.11 Further, they are effective only against HCV genotype 1, being highly specific for the amino acid target sequence of the NS3 region.
Also, they must be used in combination with interferon alfa and ribavirin because the virus needs to mutate only a little—a few amino-acid substitutions—to gain resistance to them.12 Therefore, patients are still exposed to interferon and ribavirin, with their toxicity. In addition, dysgeusia is seen with boceprevir, rash with telaprevir, and anemia with both.13,14
Finally, serious drug-drug interactions prompted the FDA to impose warnings for the use of these agents with other medications that interact with CYP3A4, the principal enzyme responsible for their metabolism. Thus, these significant adverse effects dampen the enthusiasm of patients contemplating a long course of treatment with these agents.
The need to improve the rate of sustained virologic response, shorten the duration of treatment, avoid serious side effects, improve efficacy in treating patients infected with genotypes other than 1, and, importantly, eliminate the need for interferon alfa and its serious adverse effects have driven the development of new direct-acting antiviral agents, including the two newly FDA-approved drugs, sofosbuvir and simeprevir.
SOFOSBUVIR: A POLYMERASE INHIBITOR
Sofosbuvir is a uridine nucleotide analogue that selectively inhibits the HCV NS5B RNA-dependent RNA polymerase (Figure 1). It targets the highly conserved nucleotide-binding pocket of this enzyme and functions as a chain terminator.15 While the protease inhibitors are genotype-dependent, inhibition of the highly conserved viral polymerase has an impact that spans genotypes.
Early clinical trials of sofosbuvir
Sofosbuvir has been tested in combination with interferon alfa and ribavirin, as well as in interferon-free regimens (Table 2).16–20
Rodriguez-Torres et al,15
- 56% with sofosbuvir 100 mg, peg-interferon, and ribavirin
- 83% with sofosbuvir 200 mg, peg-interferon, and ribavirin
- 80% with sofosbuvir 400 mg, peg-interferon, and ribavirin
- 43% with peg-interferon and ribavirin alone.
The ATOMIC trial16 tested the efficacy and safety of sofosbuvir in combination with peg-interferon and ribavirin in patients with HCV genotype 1, 4, or 6, without cirrhosis, who had not received any previous treatment. Patients with HCV genotype 1 were randomized to three treatments:
- Sofosbuvir 400 mg orally once daily plus peg-interferon and ribavirin for 12 weeks
- The same regimen, but for 24 weeks
- Sofosbuvir plus peg-interferon and ribavirin for 12 weeks, followed by 12 weeks of either sofosbuvir monotherapy or sofosbuvir plus ribavirin.
The rates of sustained virologic response were very high and were not significantly different among the three groups: 89%, 89%, and 87%, respectively. Patients who were able to complete a full course of therapy achieved even higher rates of sustained virologic response, ranging from 96% to 98%. The likelihood of response was not adversely affected by the usual markers of a poorer prognosis, such as a high viral load (≥ 800,000 IU/mL) or a non-CC IL28B genotype. Although patients with cirrhosis (another predictor of no response) were excluded from this study, the presence of bridging fibrosis did not seem to affect the rate of sustained virologic response. The results in patients with genotypes other than 1 were very encouraging, but the small number of patients enrolled precluded drawing firm conclusions in this group.
Important implications of the ATOMIC trial include the following:
There is no benefit in prolonging treatment with sofosbuvir beyond 12 weeks, since adverse events increased without any improvement in the rate of sustained virologic response.
There is a very low likelihood of developing viral resistance or mutation when using sofosbuvir.
There is no role for response-guided therapy, a concept used with protease inhibitor-based regimens in which patients who have complete clearance of the virus within the first 4 weeks of treatment (a rapid virologic response) and remain clear through 12 weeks of treatment (an extended rapid viral response) can be treated for a shorter duration without decreasing the likelihood of a sustained virologic response.
Lawitz et al17 conducted a randomized double-blind phase 2 trial to evaluate the effect of sofosbuvir dosing on response in noncirrhotic, previously untreated patients with HCV genotype 1, 2, or 3. Patients with HCV genotype 1 were randomized to one of three treatment groups in a 2:2:1 ratio: sofosbuvir 200 mg orally once daily, sofosbuvir 400 mg orally once daily, or placebo, all for 12 weeks in combination with peg-interferon (180 μg weekly) and ribavirin in a dosage based on weight. Depending on the viral response, patients continued peg-interferon and ribavirin for an additional 12 weeks if they achieved an extended rapid viral response, or 36 weeks if they did not achieve an extended rapid virologic response, and in all patients who received placebo. Patients with HCV genotype 2 or 3 were given sofosbuvir 400 mg once daily in combination with interferon and ribavirin for 12 weeks.
As in the ATOMIC trial, all patients treated with sofosbuvir had a very rapid reduction in viral load: 98% of patients with genotype 1 developed a rapid virologic response, and therefore almost all were eligible for the shorter treatment course of 24 weeks.17 The latter finding again suggested that response-guided treatment is not relevant with sofosbuvir-based regimens.
Very high rates of sustained virologic response were seen: 90% in patients with genotype 1 treated with sofosbuvir 200 mg, 91% in those with genotype 1 treated with 400 mg, and 92% in those with genotype 2 or 3. Although 6% of patients in the 200-mg group had virologic breakthrough after completing sofosbuvir treatment, no virologic breakthrough was observed in the 400-mg group, suggesting that the 400-mg dose might suppress the virus more effectively.17
The ELECTRON trial18 was a phase 2 study designed to evaluate the efficacy and safety of sofosbuvir and ribavirin in interferon-sparing and interferon-free regimens in patients with HCV genotype 1, 2, or 3 infection. Sofosbuvir was tested with peg-interferon and ribavirin, with ribavirin alone, and as monotherapy in previously untreated patients with genotype 2 or 3. A small number of patients with genotype 1 who were previously untreated and who were previously nonresponders were also treated with sofosbuvir and ribavirin.
All patients had a rapid virologic response, and viral suppression was sustained through the end of treatment. All patients with genotype 2 or 3 treated with double therapy (sofosbuvir and ribavirin) or triple therapy (sofosbuvir, peg-interferon, and ribavirin) achieved a sustained virologic response, compared with only 60% of patients treated with sofosbuvir monotherapy. The monotherapy group had an equal number of relapsers among those with genotype 2 or 3. Of the genotype 1 patients treated with sofosbuvir and ribavirin, 84% of those previously untreated developed a sustained virologic response, whereas only 10% of the previous nonresponders did.
Phase 3 clinical trials of sofosbuvir
The NEUTRINO trial19 studied the efficacy and safety of sofosbuvir in previously untreated patients with HCV genotype 1, 4, 5, or 6. In this phase 3 open-label study, all patients received sofosbuvir plus peg-interferon and weight-based ribavirin therapy for 12 weeks. Of the patients enrolled, 89% had genotype 1, while 9% had genotype 4 and 2% had genotype 5 or 6. Overall, 17% of the patients had cirrhosis.
The viral load rapidly decreased in all patients treated with sofosbuvir irrespective of the HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Ninety-nine percent of patients with genotype 1, 4, 5, or 6 achieved a rapid virologic response, and 90% achieved a sustained virologic response at 12 weeks after completion of treatment with sofosbuvir and ribavirin. Patients with cirrhosis had a slightly lower rate of sustained virologic response (80%, compared with 92% in patients without cirrhosis). Also, patients with non-CC IL28B genotypes had a lower rate of sustained virologic response (87% in non-CC allele vs 98% in patients with the favorable CC allele).
The FISSION trial19 recruited previously untreated patients with genotype 2 or 3 and randomized them to therapy with either sofosbuvir plus ribavirin in a weight-based dose for 12 weeks, or 24 weeks of interferon and ribavirin. In this study, 20% of patients in each treatment group had cirrhosis.
As in the NEUTRINO trial, the viral load rapidly decreased in all patients treated with sofosbuvir irrespective of HCV genotype, IL28B status, race, or the presence or absence of cirrhosis. Here, 100% of patients with genotype 2 or 3 who were treated with sofosbuvir and ribavirin achieved a rapid virologic response. Differences in outcome emerged based on genotype: 97% of those with genotype 2 and 56% of those with genotype 3 achieved a sustained virologic response. The overall rate was 67%, which was not different from patients treated with peg-interferon and ribavirin. In the subgroup of patients with cirrhosis, 47% of those treated with sofosbuvir and ribavirin achieved a sustained virologic response, vs 38% of those who received peg-interferon plus ribavirin.
In both the NEUTRINO and FISSION trials, few patients discontinued treatment, with higher rates of most adverse events occurring in patients treated with peg-interferon and ribavirin.
POSITRON,20 a phase 3 clinical trial, tested sofosbuvir in patients with HCV genotype 2 or 3 who were ineligible for peg-interferon, unwilling to take peg-interferon, or unable to tolerate peg-interferon (mainly because of clinically significant psychiatric disorders). Patients were randomized to two treatment groups for 12 weeks: sofosbuvir plus ribavirin, or placebo. About 50% of patients had HCV genotype 3, and 16% had cirrhosis.
The overall rate of sustained virologic response at 12 weeks after treatment was 78% in the sofosbuvir-and-ribavirin group (93% in genotype 2 patients and 61% in genotype 3 patients). Again, cirrhosis was associated with a lower rate of sustained virologic response (61% of patients with cirrhosis achieved a sustained virologic response vs 81% of patients without cirrhosis). None of the sofosbuvir-treated patients had virologic failure while on treatment.
FUSION,20 another phase 3 trial, evaluated sofosbuvir in patients infected with HCV genotype 2 or 3 for whom interferon-based treatment had failed. They were randomized to either 12 weeks or 16 weeks of sofosbuvir and weight-based ribavirin treatment. About 60% of patients had HCV genotype 3, and 34% had cirrhosis.
The overall sustained virologic response rate was 50% in the patients treated for 12 weeks and 73% in those treated for 16 weeks: specifically, 86% of patients with genotype 2 achieved a sustained virologic response at 12 weeks and 94% at 16 weeks, whereas in those with genotype 3 the rates were 30% at 12 weeks and 62% at 16 weeks.
Cirrhosis was again a predictor of lack of response to sofosbuvir. In the group treated for 12 weeks, 31% of those with cirrhosis achieved a sustained virologic response compared with 61% in those without cirrhosis. In the group treated for 16 weeks, 61% of those with cirrhosis achieved a sustained virologic response compared with 76% in those without cirrhosis.
In both the POSITRON and FUSION trials, relapse accounted for all treatment failures, and no virologic resistance was detected in patients who did not have a sustained virologic response. The investigators concluded that 12 weeks of treatment with sofosbuvir and ribavirin can be effective for HCV genotype 2 infection, but extending the treatment to 16 weeks may be beneficial for genotype 3. This may be especially important in patients with cirrhosis or those who did not have a response to peg-interferon-based treatment.
VALENCE,21 an ongoing phase 3 trial in Europe, is assessing the safety and efficacy of sofosbuvir 400 mg once daily and weight-based ribavirin in patients with HCV genotype 2 or 3. Eighty-five percent of the trial participants have received previous treatment, and 21% have cirrhosis. Patients were originally randomized in a 4:1 ratio to receive sofosbuvir plus ribavirin for 12 weeks or matching placebo, but as a result of emerging data suggesting that patients with genotype 3 would benefit from more than 12 weeks of treatment, the study was subsequently amended to extend treatment to 24 weeks for patients with genotype 3.
Overall rates of sustained virologic response were 93% in patients with genotype 2 and 85% in patients with genotype 3. In previously treated patients with genotype 2 who were treated for 12 weeks, the rates of sustained virologic response were 91% in those without cirrhosis vs 88% in those with cirrhosis. In previously treated patients with genotype 3, the rates in those treated for 24 weeks were 87% in patients without cirrhosis vs 60% with cirrhosis. The safety profile was consistent with that of ribavirin.
Side effects of sofosbuvir
In clinical trials, side effects occurred most often when sofosbuvir was combined with interferon and ribavirin and were consistent with the known side effects of the latter two agents. The most frequently reported side effects included fatigue, insomnia, nausea, rash, anemia, headache, and arthralgia, with most of these adverse events rated by treating clinicians as being mild in severity.15,20
In the ATOMIC trial, the most common events leading to drug discontinuation were anemia and neutropenia, both associated with interferon and ribavirin. Patients receiving sofosbuvir monotherapy after 12 weeks of triple therapy showed rapid improvement in hemoglobin levels and neutrophil counts, indicating that hematologic abnormalities attributed solely to sofosbuvir are minimal. In the FISSION trial, the incidence of adverse events was consistently lower in those receiving sofosbuvir-ribavirin than in patients receiving interferon-ribavirin without sofosbuvir.19
In the POSITRON trial, discontinuation of sofosbuvir because of adverse events was uncommon, and there were no differences in the incidence of adverse events and laboratory abnormalities between patients with and without cirrhosis when they received sofosbuvir and ribavirin.20
Sofosbuvir dosage and indications
Sofosbuvir is approved in an oral dose of 400 mg once daily in combination with ribavirin for patients infected with HCV genotype 2 or 3 and in combination with ribavirin and interferon alfa in patients infected with HCV genotype 1 or 4 (Table 3). It could be considered for HCV genotype 1 in combination with ribavirin alone for 24 weeks in patients who are ineligible for interferon.
Sofosbuvir is also recommended in combination with ribavirin in HCV-infected patients with hepatocellular carcinoma who are awaiting liver transplantation, for up to 48 weeks or until they receive a transplant, to prevent posttransplant reinfection with HCV.
Sofosbuvir is expensive
A course of therapy is expected to cost about $84,000, which is significantly more than the cost of previous triple therapy (peg-interferon, ribavirin, and either boceprevir or telaprevir).22 This high cost will undoubtedly lead to less widespread use in developing countries, and potentially even in the United States. As newer direct-acting antiviral agents become available, the price will likely come down, enhancing access to these drugs.
SIMEPREVIR: A SECOND-GENERATION PROTEASE INHIBITOR
Telaprevir and boceprevir are NS3/A4 protease inhibitors that belong to the alfa-ketoamid derivative class. Simeprevir belongs to the macrocyclic class and has a different way of binding to the target enzyme.23 Like sofosbuvir, simeprevir was recently approved by the FDA for the treatment of HCV genotype 1.
The therapeutic efficacy of simeprevir has been tested in several clinical trials (Table 4), including QUEST-124 and QUEST-225 (in previously untreated patients), PROMISE26 (in prior relapsers), and ASPIRE27 (in prior partial and null responders). Results from these trials showed high overall rates of sustained virologic response with triple therapy (ie, simeprevir combined with peg-interferon and ribavirin). It was generally well tolerated, and most adverse events reported during 12 weeks of treatment were of mild to moderate severity.
In QUEST-1 and QUEST-2, both double-blind phase 3 clinical trials, previously untreated patients infected with HCV genotype 1 were randomized in a 2:1 ratio to receive either simeprevir 150 mg daily or placebo for 12 weeks; both groups also received peg-interferon and ribavirin. Patients then received peg-interferon and ribavirin alone for 12 or 36 weeks in the simeprevir group (based on response) and for 36 weeks in the placebo group.
The overall rate of sustained virologic response at 12 weeks was 80% in the simeprevir group (75% in those with genotype 1a and 85% in those with genotype 1b) vs 50% in the placebo group (receiving peg-interferon and ribavirin alone).24,25
PROMISE,26 another double-blind randomized phase 3 clinical trial, evaluated simeprevir in patients with HCV genotype 1 who relapsed after previous interferon-based therapy. It had a similar design to QUEST-1 and QUEST-2, and 15% of all patients had cirrhosis.
The overall sustained virologic response rate at 12 weeks after treatment was 79% in the simeprevir group (70% in patients with genotype 1a and 86% in those with genotype 1b) vs 37% in the placebo group. Rates were similar in patients with absent to moderate fibrosis (82%), advanced fibrosis (73%), or cirrhosis (74%).
ASPIRE.27 Simeprevir efficacy in patients with HCV genotype 1 for whom previous therapy with peg-interferon and ribavirin had failed was tested in ASPIRE, a double-blind randomized phase 2 clinical trial. Patients were randomized to receive simeprevir (either 100 mg or 150 mg daily) for 12, 24, or 48 weeks in combination with 48 weeks of peg-interferon and ribavirin, or placebo plus peg-interferon and ribavirin for 48 weeks.
The primary end point was the rate of sustained virologic response at 24 weeks. Overall, rates were 61% to 80% for the simeprevir treatment groups compared with 23% with placebo, regardless of prior response to peg-interferon and ribavirin. By subgroup, rates were:
- 77% to 89% with simeprevir vs 37% with placebo in prior relapsers
- 48% to 86% with simeprevir vs 9% with placebo in prior partial responders
- 38% to 59% with placebo vs 19% for prior nonresponders.
The best rates of sustained viral response at 24 weeks were in the groups that received simeprevir 150 mg daily: 85% in prior relapsers, 75% in prior partial responders, and 51% in prior nonresponders.
Simeprevir vs other direct-acting antiviral drugs
Advantages of simeprevir over the earlier protease inhibitors include once-daily dosing, a lower rate of adverse events (the most common being fatigue, headache, rash, photosensitivity, and pruritus), a lower likelihood of discontinuation because of adverse events, and fewer drug-drug interactions (since it is a weak inhibitor of the CYP3A4 enzyme).
Unlike sofosbuvir, simeprevir was FDA-approved only for HCV genotype 1 and in combination with interferon alfa and ribavirin. Compared with sofosbuvir, the treatment duration with simeprevir regimens is longer overall (interferon alfa and ribavirin are given for 12 weeks in sofosbuvir-based regimens vs 24 to 48 weeks with simeprevir). As with sofosbuvir, the estimated cost of simeprevir is high, about $66,000 for a 12-week course.
Simeprevir dosage and indications
Simeprevir was approved at an oral dose of 150 mg once daily in combination with ribavirin and interferon alfa in patients with HCV genotype 1 (Table 5).
The approved regimens for simeprevir are fixed in total duration based on the patient’s treatment history. Specifically, all patients receive the drug in combination with peg-interferon and ribavirin for 12 weeks. Then, previously untreated patients and prior relapsers continue to receive peg-interferon and ribavirin alone for another 12 weeks, and those with a partial or null response continue with these drugs for another 36 weeks.
Patients infected with HCV genotype 1a should be screened for the NS3 Q80K polymorphism at baseline, as it has been associated with substantially reduced response to simeprevir.
Sofosbuvir and simeprevir in combination
The COSMOS trial.28 Given their differences in mechanism of action, sofosbuvir and simeprevir are being tested in combination. The COSMOS trial is an ongoing phase 2 randomized open-label study investigating the efficacy and safety of simeprevir and sofosbuvir in combination with and without ribavirin in patients with HCV genotype 1, including nonresponders and those with cirrhosis. Early results are promising, with very high rates of sustained virologic response with the sofosbuvir-simeprevir combination (93% to 100%) and indicate that the addition of ribavirin might not be needed to achieve sustained virologic response in this patient population.
THE FUTURE
The emergence of all-oral regimens for HCV treatment with increasingly sophisticated agents such as sofosbuvir and simeprevir will dramatically alter the management of HCV patients. In view of the improvement in sustained virologic response rates with these treatments, and since most HCV-infected persons have no symptoms, the US Centers for Disease Control and Prevention29 recently recommended one-time testing of the cohort in which the prevalence of HCV infection is highest: all persons born between 1945 and 1965. This undoubtedly will increase the detection of this infection—and the number of new patients expecting treatment.
Future drugs promise further improvements (Table 6).30–35 Advances in knowledge of the HCV molecular structure have led to the development of numerous direct-acting antiviral agents with very specific viral targets. A second wave of protease inhibitors and of nucleoside and nonnucleoside polymerase inhibitors will soon be available. Inhibitors of NS5A (a protein important in the assembly of the viral replication complex) such as daclatasvir and ledipasvir, are currently in phase 3 clinical trials. Other viral proteins involved in assembly of the virus, including the core protein and p7, are being explored as drug targets. In addition, inhibiting host targets such as cyclophilin A and miR122 has gained traction recently, with specific agents currently in phase 2 and 3 clinical trials.
Factors that previously were major determinants of response to treatment, such as IL28B genotype, viral load, race, age, extent of fibrosis, and genotype 1 subtypes, will become much less important with the introduction of highly potent direct-acting antiviral agents.
Many all-oral combinations are being evaluated in clinical trials. For example, the open-label, phase 2 LONESTAR trial tested the utility of combining sofosbuvir and ledipasvir (an NS5A inhibitor) with and without ribavirin for 8 or 12 weeks in previously untreated patients with HCV genotype 1, and for 12 weeks in patients with HCV genotype 1 who did not achieve a sustained virologic response after receiving a protease inhibitor-based regimen (half of whom had compensated cirrhosis).36 Sustained virologic response rates were very high (95% to 100%) in both previously treated and previously untreated patients, including those with cirrhosis. Similar rates were achieved by the 8-week and 12-week groups in noncirrhotic patients who had not been previously treated for HCV. The typical hematologic abnormalities associated with interferon were not observed except for mild anemia in patients who received ribavirin. These results suggest that the combination of sofosbuvir and ledipasvir could offer a very effective, short, all-oral treatment for patients with HCV genotype 1, including those with cirrhosis, who up to now have been difficult to treat.
Challenges remaining
The success of sofosbuvir and simeprevir paves the way for interferon-free regimens.37 For a long time, the treatment of HCV infection required close monitoring of patients while managing the side effects of interferon, but the current and emerging direct-acting antiviral agents will soon change this practice. Given the synergistic effects of combination therapy—targeting the virus at multiple locations, decreasing the likelihood of drug resistance, and improving efficacy—combination regimens seem to be the optimal solution to the HCV epidemic. Lower risk of side effects and shorter treatment duration will definitely improve the acceptance of any new regimen. New agents that act against conserved viral targets, thereby yielding activity across multiple genotypes, will be advantageous as well. Table 7 compares the rates of sustained virologic response of the different currently approved HCV treatment regimens.
Clinical challenges remain, including the management of special patient populations for whom data are still limited. These include patients with cirrhosis, chronic kidney disease, renal failure, and concurrent infection with human immunodeficiency virus, and patients who have undergone solid organ transplantation. Clinical trials are under way to evaluate the treatment options for these patients, who will likely need to wait for the emergence of additional agents before dramatic improvement in sustained virologic response rates may be expected.38
As the treatment of HCV becomes simpler, safer, and more effective, primary care physicians will increasingly be expected to manage it. Difficult-to-treat patients, including the special populations above, will require specialist management and individualized treatment regimens, at least until better therapies are available. The high projected cost of the new agents may limit access, at least initially. However, the dramatic improvement in sustained virologic response rates and all that that implies in terms of decreased risk of advanced liver disease and its complications will undoubtedly make these therapies cost-effective.39
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the united states. Clin Infect Dis 2012; 55(suppl 1):S10–S15.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first international liver transplantation society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transplant 2003; 9:S1–S9.
- Wong JB, McQuillan GM, McHutchison JG, Poynard T. Estimating future hepatitis C morbidity, mortality, and costs in the United States. Am J Public Health 2000; 90:1562–1569.
- Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007; 132:1979–1998.
- Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol 2000; 81:1631–1648.
- Singal AG, Volk ML, Jensen D, Di Bisceglie AM, Schoenfeld PS. A sustained viral response is associated with reduced liver-related morbidity and mortality in patients with hepatitis C virus. Clin Gastroenterol Hepatol 2010; 8:280–288,288.e1.
- Camma C, Di Bona D, Schepis F, et al. Effect of peginterferon alfa-2a on liver histology in chronic hepatitis C: a meta-analysis of individual patient data. Hepatology 2004; 39:333–342.
- Paeshuyse J, Dallmeier K, Neyts J. Ribavirin for the treatment of chronic hepatitis C virus infection: a review of the proposed mechanisms of action. Curr Opin Virol 2011; 1:590–598.
- Thomas E, Ghany MG, Liang TJ. The application and mechanism of action of ribavirin in therapy of hepatitis C. Antivir Chem Chemother 2012; 23:1–12.
- Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB; American Association for Study of Liver Diseases. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54:1433–1444.
- Soriano V, Vispo E, Poveda E, Labarga P, Barreiro P. Treatment failure with new hepatitis C drugs. Expert Opin Pharmacother 2012; 13:313–323.
- Asselah T, Marcellin P. Interferon free therapy with direct acting antivirals for HCV. Liver Int 2013; 33(suppl 1):93–104.
- Manns MP, McCone J, Davis MN, et al. Overall safety profile of boceprevir plus peginterferon alfa-2b and ribavirin in patients with chronic hepatitis C genotype 1: a combined analysis of 3 phase 2/3 clinical trials. Liver Int 2013; Aug 2. doi: 10.1111/liv.12300. [Epub ahead of print]
- Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Rodriguez-Torres M, Lawitz E, Kowdley KV, et al. Sofosbuvir (GS-7977) plus peginterferon/ribavirin in treatment-naive patients with HCV genotype 1: a randomized, 28-day, dose-ranging trial. J Hepatol 2013; 58:663–668.
- Kowdley KV, Lawitz E, Crespo I, et al. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet 2013; 381:2100–2107.
- Lawitz E, Lalezari JP, Hassanein T, et al. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect Dis 2013; 13:401–408.
- Gane EJ, Stedman CA, Hyland RH, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 2013; 368:34–44.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Jacobson IM, Gordon SC, Kowdley KV, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med 2013; 368:1867–1877.
- Zeuzem S, Dusheiko G, Salupere R, et al. Sofosbuvir + ribavirin for 12 or 24 weeks for patients with HCV genotype 2 or 3: the VALENCE trial [abstract no.1085]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- You DM, Pockros PJ. Simeprevir for the treatment of chronic hepatitis C. Expert Opin Pharmacother 2013; 14:2581–2589.
- Jacobson IM, Dore GJ, Foster G, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naive patients: results from Quest-1, a phase III trial [abstract no. 1425]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, Netherlands.
- Manns M, Marcellin P, Poordad FP, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naïve patients: results from QUEST-2, a phase III trial [abstract no. 1413]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, The Netherlands.
- Lawitz E, Forns X, Zeuzem S, et al. Simeprevir (TMC435) with peginterferon/ribavirin for treatment of chronic HCV genotype 1 infection in patients who relapsed after previous interferon-based therapy: results from promise, a phase III trial [abstract no. 869b]. Digestive Disease Week; May 18–21, 2013; Orlando, FL.
- Zeuzem S, Berg T, Gane E, et al. Simeprevir increases rate of sustained virologic response among treatment-experienced patients with HCV genotype-1 infection: a phase IIb trial. Gastroenterology epub Oct 31, 2013.
- Jacobson IM, Ghalib RM, Rodriguez-Torres M, et al. SVR results of a once-daily regimen of simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in cirrhotic and non-cirrhotic HCV genotype 1 treatment-naive and prior null responder patients: the COSMOS study [abstract LB-3]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Smith BD, Morgan RL, Beckett GA, et al. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945–1965. MMWR Recomm Rep 2012; 61( RR-4):1–32.
- Sulkowski MS, Kang M, Matining R, et al. Safety and antiviral activity of the HCV entry inhibitor ITX5061 in treatment-naive HCV-infected adults: a randomized, double-blind, phase 1b study. J Infect Dis 2013 Oct 9. [Epub ahead of print]
- Pawlotsky JM. NS5A inhibitors in the treatment of hepatitis C. J Hepatol 2013; 59:375–382.
- Yu M, Corsa AC, Xu S, et al. In vitro efficacy of approved and experimental antivirals against novel genotype 3 hepatitis C virus subgenomic replicons. Antiviral Res 2013; 100:439–445.
- Aghemo A, De Francesco R. New horizons in hepatitis C antiviral therapy with direct-acting antivirals. Hepatology 2013; 58:428–438.
- Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med 2013; 368:1907–1917.
- Flisiak R, Jaroszewicz J, Parfieniuk-Kowerda A. Emerging treatments for hepatitis C. Expert Opin Emerg Drugs 2013; 18:461–475.
- Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet 2013 Nov 1. doi: 10.1016/S0140-6736(13)62121-2 [Epub ahead of print]
- Drenth JP. HCV treatment—no more room for interferonologists? N Engl J Med 2013; 368:1931–1932.
- Casey LC, Lee WM. Hepatitis C virus therapy update 2013. Curr Opin Gastroenterol 2013; 29:243–249.
- Afdhal NH, Zeuzem S, Schooley RT, et al. The new paradigm of hepatitis C therapy: integration of oral therapies into best practices. J Viral Hepatol 2013; 20:745–760.
- Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the united states. Clin Infect Dis 2012; 55(suppl 1):S10–S15.
- Wiesner RH, Sorrell M, Villamil F; International Liver Transplantation Society Expert Panel. Report of the first international liver transplantation society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transplant 2003; 9:S1–S9.
- Wong JB, McQuillan GM, McHutchison JG, Poynard T. Estimating future hepatitis C morbidity, mortality, and costs in the United States. Am J Public Health 2000; 90:1562–1569.
- Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007; 132:1979–1998.
- Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol 2000; 81:1631–1648.
- Singal AG, Volk ML, Jensen D, Di Bisceglie AM, Schoenfeld PS. A sustained viral response is associated with reduced liver-related morbidity and mortality in patients with hepatitis C virus. Clin Gastroenterol Hepatol 2010; 8:280–288,288.e1.
- Camma C, Di Bona D, Schepis F, et al. Effect of peginterferon alfa-2a on liver histology in chronic hepatitis C: a meta-analysis of individual patient data. Hepatology 2004; 39:333–342.
- Paeshuyse J, Dallmeier K, Neyts J. Ribavirin for the treatment of chronic hepatitis C virus infection: a review of the proposed mechanisms of action. Curr Opin Virol 2011; 1:590–598.
- Thomas E, Ghany MG, Liang TJ. The application and mechanism of action of ribavirin in therapy of hepatitis C. Antivir Chem Chemother 2012; 23:1–12.
- Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB; American Association for Study of Liver Diseases. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54:1433–1444.
- Soriano V, Vispo E, Poveda E, Labarga P, Barreiro P. Treatment failure with new hepatitis C drugs. Expert Opin Pharmacother 2012; 13:313–323.
- Asselah T, Marcellin P. Interferon free therapy with direct acting antivirals for HCV. Liver Int 2013; 33(suppl 1):93–104.
- Manns MP, McCone J, Davis MN, et al. Overall safety profile of boceprevir plus peginterferon alfa-2b and ribavirin in patients with chronic hepatitis C genotype 1: a combined analysis of 3 phase 2/3 clinical trials. Liver Int 2013; Aug 2. doi: 10.1111/liv.12300. [Epub ahead of print]
- Jacobson IM, McHutchison JG, Dusheiko G, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Rodriguez-Torres M, Lawitz E, Kowdley KV, et al. Sofosbuvir (GS-7977) plus peginterferon/ribavirin in treatment-naive patients with HCV genotype 1: a randomized, 28-day, dose-ranging trial. J Hepatol 2013; 58:663–668.
- Kowdley KV, Lawitz E, Crespo I, et al. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet 2013; 381:2100–2107.
- Lawitz E, Lalezari JP, Hassanein T, et al. Sofosbuvir in combination with peginterferon alfa-2a and ribavirin for non-cirrhotic, treatment-naive patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double-blind, phase 2 trial. Lancet Infect Dis 2013; 13:401–408.
- Gane EJ, Stedman CA, Hyland RH, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 2013; 368:34–44.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Jacobson IM, Gordon SC, Kowdley KV, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med 2013; 368:1867–1877.
- Zeuzem S, Dusheiko G, Salupere R, et al. Sofosbuvir + ribavirin for 12 or 24 weeks for patients with HCV genotype 2 or 3: the VALENCE trial [abstract no.1085]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- You DM, Pockros PJ. Simeprevir for the treatment of chronic hepatitis C. Expert Opin Pharmacother 2013; 14:2581–2589.
- Jacobson IM, Dore GJ, Foster G, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naive patients: results from Quest-1, a phase III trial [abstract no. 1425]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, Netherlands.
- Manns M, Marcellin P, Poordad FP, et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naïve patients: results from QUEST-2, a phase III trial [abstract no. 1413]. Annual Meeting of the European Association for the Study of the Liver; April 24–28, 2013; Amsterdam, The Netherlands.
- Lawitz E, Forns X, Zeuzem S, et al. Simeprevir (TMC435) with peginterferon/ribavirin for treatment of chronic HCV genotype 1 infection in patients who relapsed after previous interferon-based therapy: results from promise, a phase III trial [abstract no. 869b]. Digestive Disease Week; May 18–21, 2013; Orlando, FL.
- Zeuzem S, Berg T, Gane E, et al. Simeprevir increases rate of sustained virologic response among treatment-experienced patients with HCV genotype-1 infection: a phase IIb trial. Gastroenterology epub Oct 31, 2013.
- Jacobson IM, Ghalib RM, Rodriguez-Torres M, et al. SVR results of a once-daily regimen of simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in cirrhotic and non-cirrhotic HCV genotype 1 treatment-naive and prior null responder patients: the COSMOS study [abstract LB-3]. 64th Annual Meeting of the American Association for the Study of Liver Diseases; November 1–5, 2013; Washington, DC.
- Smith BD, Morgan RL, Beckett GA, et al. Recommendations for the identification of chronic hepatitis C virus infection among persons born during 1945–1965. MMWR Recomm Rep 2012; 61( RR-4):1–32.
- Sulkowski MS, Kang M, Matining R, et al. Safety and antiviral activity of the HCV entry inhibitor ITX5061 in treatment-naive HCV-infected adults: a randomized, double-blind, phase 1b study. J Infect Dis 2013 Oct 9. [Epub ahead of print]
- Pawlotsky JM. NS5A inhibitors in the treatment of hepatitis C. J Hepatol 2013; 59:375–382.
- Yu M, Corsa AC, Xu S, et al. In vitro efficacy of approved and experimental antivirals against novel genotype 3 hepatitis C virus subgenomic replicons. Antiviral Res 2013; 100:439–445.
- Aghemo A, De Francesco R. New horizons in hepatitis C antiviral therapy with direct-acting antivirals. Hepatology 2013; 58:428–438.
- Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med 2013; 368:1907–1917.
- Flisiak R, Jaroszewicz J, Parfieniuk-Kowerda A. Emerging treatments for hepatitis C. Expert Opin Emerg Drugs 2013; 18:461–475.
- Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet 2013 Nov 1. doi: 10.1016/S0140-6736(13)62121-2 [Epub ahead of print]
- Drenth JP. HCV treatment—no more room for interferonologists? N Engl J Med 2013; 368:1931–1932.
- Casey LC, Lee WM. Hepatitis C virus therapy update 2013. Curr Opin Gastroenterol 2013; 29:243–249.
- Afdhal NH, Zeuzem S, Schooley RT, et al. The new paradigm of hepatitis C therapy: integration of oral therapies into best practices. J Viral Hepatol 2013; 20:745–760.
KEY POINTS
- In clinical trials of treatment for chronic HCV infection, regimens that included a direct-acting antiviral agent were more effective than ones that did not.
- Sofosbuvir is approved in an oral dose of 400 mg once daily in combination with ribavirin for patients infected with HCV genotype 2 or 3, and in combination with ribavirin and interferon in patients infected with HCV genotype 1 or 4. It is also recommended in combination with ribavirin in HCV-infected patients with hepatocellular carcinoma who are awaiting liver transplantation.
- Simeprevir is approved in an oral dose of 150 mg once daily in combination with ribavirin and interferon for patients with HCV genotype 1.
- The new drugs are expensive, a potential barrier for many patients. As more direct-acting antiviral agents become available, their cost will likely decrease.
- Combinations of direct-acting antiviral agents of different classes may prove even more effective and could eliminate the need for interferon entirely.
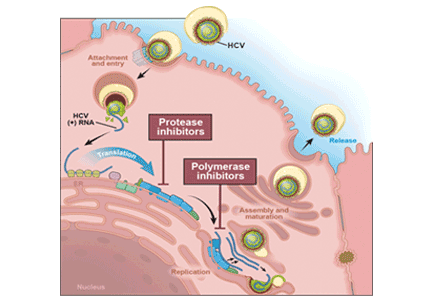
When are effective medications just too expensive?
The era of all-oral agents for hepatitis C virus infection has begun. Previous treatments for this disease included pegylated interferon and ribavirin, which had limited effectiveness and side effects severe enough to reduce adherence and quality of life. Recent trials have documented the effectiveness of the new direct-acting antiviral agents.1 These new drugs work better and offer the promise of an all-oral treatment regimen that avoids pegylated interferon.
But they cost a lot. Prices of more than $50,000 are estimated for a 2-to-3-month course of treatment.2 These new medications reflect the kind of societal advances that justify a long-term investment in basic and clinical research. But do we value advances at any cost?
DOES COST MATTER?
Leaving aside the question of whether these particular drugs are too expensive, the general question remains whether effective therapies can ever be so expensive that we should not use them.
Does cost matter? Well, we all know that it does. We pay attention to cost in our individual purchasing and in how we think about business and government spending. And yet, while everyone agrees that we shouldn’t pay for care that provides no benefit, many of us stop at just that line, and think or act as if we can’t put a price on those elements of health care that offer some potential to save lives. It’s a comfortable position, because in going after pure waste we feel like fiscally temperate guardians of societal resources without feeling responsible for heart-rending choices about overspending on things that do work. Yet that spending threatens societal resources just as much as useless therapies.
In the end, though, it is an illogical position. The illogic is easy to understand once you walk it through: if you are unwilling to put a price on life, then you are saying that there is no price too high for any potential health benefit, no matter how small. That means you commit all your resources to health and you go bankrupt.
So, implicitly or explicitly (our society does so implicitly—and inconsistently, at that), you have to put a maximum price on life. But at that point, you are (again, implicitly) saying that when there are treatments that cost more, you shouldn’t buy them.3 Admittedly, it doesn’t sound good, and in health care, which touches us so intimately, it doesn’t feel good either.
SHOULD PHYSICIANS CARE ABOUT COST?
Many of us were taught in medical school that it isn’t the doctor’s job to think about cost. Physicians are to be clinical advocates for their patients without consideration of cost—but that can’t be right, and it isn’t right.
First, even if physicians are patient advocates first, they ought to consider cost when the patient is paying. The rise in the use of high-deductible health insurance plans has expanded the financial risk that individual patients face in their own health care decisions. Physicians may be unprepared to help patients with those decisions, but it seems like a service they ought to provide.
Second, the line between cost to the individual and cost to society is blurred at best. Our societal health care spending is nothing more than the aggregation of our individual health care spending. Even if we don’t want physicians to focus on cost when with an individual patient at the bedside or at the examination table, don’t we want societal cost to be at least in their peripheral vision?
Many obstacles impede this view. Even if physicians can keep societal costs in their peripheral vision, they certainly can’t see to the edges of the broad canvas that all of health care represents, and they have no easy decision rules for how to turn what vision they have into a decision for a particular patient.
A variety of stakeholders have succeeded in turning what might have been seen as socially responsible thinking into a dirty word. The same politicians who use the term “stewardship” when they are in favor of considering societal implications call it “rationing” when they feel the other way. As a result, some of our most important institutions—eg, Medicare—are prohibited from considering price. Commercial insurers, still smarting from the managed-care backlash of the 1990s, have limited ability to effectively manage costs while maintaining quality. In some sense, this vacuum creates an opportunity for physician leadership.
COST-EFFECTIVENESS ANALYSIS AND ITS LIMITATIONS
Cost-effectiveness analysis, which represents the health care value of a therapy as the ratio of its financial cost to its benefit (eg, cost per quality-adjusted life-year), offers a disciplined approach to these conflicts between individual good and social good.4
The long-term costs of hepatitis C are substantial and include multiple diagnostic tests, hospitalization, surgery, and death. A major treatment for both liver failure and hepatocellular cancer is liver transplantation, which can entail hundreds of thousands of dollars in cost for the surgery and ongoing care. Preventing just one transplant can provide enormous savings, in addition to freeing up cadaveric organs for another patient. A careful cost-effectiveness analysis could tell us whether the new direct-acting antiviral agents are worth their cost.
These analyses are appealing because they are formal and disciplined, but it turns out that they are far from value-free. Their methodology is complicated and is sensitive to subjective modeling assumptions whose implications are often not straightforward, are hard to report in the compact methods sections of manuscripts, and are harder still to interpret by most readers of these articles.
Further, these models focus exclusively on economic efficiency, so even the most carefully constructed cost-effectiveness analyses need to be tempered by a sense of social equity not captured in these models. For example, an emphasis on increasing quality-adjusted life-years will naturally lead to policy decisions that favor groups that have more life-years remaining. That may sound fine if we are comfortable with the idea that, in general, we should target our resources toward younger people rather than older people. But the same thinking means we should target our resources away from men (who don’t live as long as women) or away from members of racial minority groups (who don’t live as long as whites).
Finally, although some throw about numbers like $50,000 to $100,000 per quality-adjusted life-year as a guide, the price thresholds revealed by our current practices and policies are inconsistent. Hemodialysis is funded through Medicare by a federal mandate, but more cost-effective vaccines and preventive care are not covered to the same degree. Cost-effectiveness analyses are essential to establish a quantitative sense about the efficient use of resources, but they need to be interpreted alongside other considerations we also value. Cost-effectiveness analyses don’t take us all the way to the decision line by themselves.
WHY ARE NEW DRUGS SO EXPENSIVE?
The high cost of the new direct-acting antivirals for just months of therapy seems excessive on its face. Even though most patients will not pay these costs directly, they are borne by society through higher taxes or premiums for commercial insurance, which are paid out-of-pocket by those who purchase individual insurance, or substitute for wages in employment-based health insurance.
We know that the actual cost to manufacture these drugs is significantly less than the prices charged by pharmaceutical companies5 and that the government subsidizes both the research and the reimbursement for certain therapies. However, the companies need to cover the long-term costs of research and development not only for these drugs but for other drugs that did not make it through the pipeline but might have.6
There are at least two sides to this economy. First, the more we are willing to pay for successful drugs that go to market, the more the developers of those drugs will be willing to invest in finding new ones. If we were to pay less for individual successes, we would in the end have fewer trials and fewer overall successes.
Second, pharmaceutical companies hire economists to do their own cost-effectiveness calculations. One reason it should be no surprise that new drugs often arrive on the market at prices that are pretty close to commonly accepted thresholds for cost-effectiveness is that this is partly how they were priced in the first place. Pharmaceutical companies naturally want to price their products as high as they can. Since there is a limit to what people are willing to pay for the benefit they get in return, determining that limit and setting the price at that point helps firms extract as much of the surplus as possible.
AN OPPORTUNITY FOR LEADERSHIP
A disciplined analysis of the costs and benefits of new drug therapies is critical to any medical policy decision, rather than cost alone. There will always be a point where new treatments are too expensive—a point not based on absolute cost, but on cost relative to what is gained over and above the next best alternative.7 However, we should acknowledge that these analyses are based on estimates that may change over time, that they require modeling assumptions that are often subjective and opaque, and that the interpretation and implementation of these policies within their social context is just as important as the analysis of their economic efficiency.
As challenging as these decisions are, they offer an opportunity for leadership from medicine. Some organizations have already taken a stance on eliminating waste—through their participation in the Choosing Wisely initiative led by the American Board of Internal Medicine8 or through stands against the use of drugs and procedures that offer no benefit over cheaper alternatives.9 As these decisions get harder and as we aim to reduce not just zero-value care, but also low-value care, physicians have an enormous amount to contribute.
- Dugum M, O’Shea R. Hepatitis C virus: here comes alloral treatment. Cleve Clin J Med 2014; 81:159–172.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- Asch DA. Basic lessons in resource allocation: sharing, setting limits, and being fair. Pharos Alpha Omega Alpha Honor Med Soc 1995; 58:33–34.
- Weinstein MC, Stason WB. Foundations of cost-effectiveness analysis for health and medical practices. N Engl J Med 1977; 296:716–721.
- Hill A, Khoo S, Fortunak J, Simmons B, Ford N. Minimum costs for producing hepatitis C direct acting antivirals, for use in large-scale treatment access programs in developing countries. Clin Infect Dis 2014; Jan 6 [Epub ahead of print].
- Adams CP, Brantner VV. Estimating the cost of new drug development: is it really 802 million dollars? Health Aff (Millwood) 2006; 25:420–428.
- Eisenberg JM. Clinical economics. A guide to the economic analysis of clinical practices. JAMA 1989; 262:2879–2886.
- Cassel CK, Guest JA. Choosing wisely: helping physicians and patients make smart decisions about their care. JAMA 2012; 307:1801–1802.
- Bach PB, Saltz LB, Wittes RE. In cancer care, cost matters. New York Times. October 15, 2012:A25.
The era of all-oral agents for hepatitis C virus infection has begun. Previous treatments for this disease included pegylated interferon and ribavirin, which had limited effectiveness and side effects severe enough to reduce adherence and quality of life. Recent trials have documented the effectiveness of the new direct-acting antiviral agents.1 These new drugs work better and offer the promise of an all-oral treatment regimen that avoids pegylated interferon.
But they cost a lot. Prices of more than $50,000 are estimated for a 2-to-3-month course of treatment.2 These new medications reflect the kind of societal advances that justify a long-term investment in basic and clinical research. But do we value advances at any cost?
DOES COST MATTER?
Leaving aside the question of whether these particular drugs are too expensive, the general question remains whether effective therapies can ever be so expensive that we should not use them.
Does cost matter? Well, we all know that it does. We pay attention to cost in our individual purchasing and in how we think about business and government spending. And yet, while everyone agrees that we shouldn’t pay for care that provides no benefit, many of us stop at just that line, and think or act as if we can’t put a price on those elements of health care that offer some potential to save lives. It’s a comfortable position, because in going after pure waste we feel like fiscally temperate guardians of societal resources without feeling responsible for heart-rending choices about overspending on things that do work. Yet that spending threatens societal resources just as much as useless therapies.
In the end, though, it is an illogical position. The illogic is easy to understand once you walk it through: if you are unwilling to put a price on life, then you are saying that there is no price too high for any potential health benefit, no matter how small. That means you commit all your resources to health and you go bankrupt.
So, implicitly or explicitly (our society does so implicitly—and inconsistently, at that), you have to put a maximum price on life. But at that point, you are (again, implicitly) saying that when there are treatments that cost more, you shouldn’t buy them.3 Admittedly, it doesn’t sound good, and in health care, which touches us so intimately, it doesn’t feel good either.
SHOULD PHYSICIANS CARE ABOUT COST?
Many of us were taught in medical school that it isn’t the doctor’s job to think about cost. Physicians are to be clinical advocates for their patients without consideration of cost—but that can’t be right, and it isn’t right.
First, even if physicians are patient advocates first, they ought to consider cost when the patient is paying. The rise in the use of high-deductible health insurance plans has expanded the financial risk that individual patients face in their own health care decisions. Physicians may be unprepared to help patients with those decisions, but it seems like a service they ought to provide.
Second, the line between cost to the individual and cost to society is blurred at best. Our societal health care spending is nothing more than the aggregation of our individual health care spending. Even if we don’t want physicians to focus on cost when with an individual patient at the bedside or at the examination table, don’t we want societal cost to be at least in their peripheral vision?
Many obstacles impede this view. Even if physicians can keep societal costs in their peripheral vision, they certainly can’t see to the edges of the broad canvas that all of health care represents, and they have no easy decision rules for how to turn what vision they have into a decision for a particular patient.
A variety of stakeholders have succeeded in turning what might have been seen as socially responsible thinking into a dirty word. The same politicians who use the term “stewardship” when they are in favor of considering societal implications call it “rationing” when they feel the other way. As a result, some of our most important institutions—eg, Medicare—are prohibited from considering price. Commercial insurers, still smarting from the managed-care backlash of the 1990s, have limited ability to effectively manage costs while maintaining quality. In some sense, this vacuum creates an opportunity for physician leadership.
COST-EFFECTIVENESS ANALYSIS AND ITS LIMITATIONS
Cost-effectiveness analysis, which represents the health care value of a therapy as the ratio of its financial cost to its benefit (eg, cost per quality-adjusted life-year), offers a disciplined approach to these conflicts between individual good and social good.4
The long-term costs of hepatitis C are substantial and include multiple diagnostic tests, hospitalization, surgery, and death. A major treatment for both liver failure and hepatocellular cancer is liver transplantation, which can entail hundreds of thousands of dollars in cost for the surgery and ongoing care. Preventing just one transplant can provide enormous savings, in addition to freeing up cadaveric organs for another patient. A careful cost-effectiveness analysis could tell us whether the new direct-acting antiviral agents are worth their cost.
These analyses are appealing because they are formal and disciplined, but it turns out that they are far from value-free. Their methodology is complicated and is sensitive to subjective modeling assumptions whose implications are often not straightforward, are hard to report in the compact methods sections of manuscripts, and are harder still to interpret by most readers of these articles.
Further, these models focus exclusively on economic efficiency, so even the most carefully constructed cost-effectiveness analyses need to be tempered by a sense of social equity not captured in these models. For example, an emphasis on increasing quality-adjusted life-years will naturally lead to policy decisions that favor groups that have more life-years remaining. That may sound fine if we are comfortable with the idea that, in general, we should target our resources toward younger people rather than older people. But the same thinking means we should target our resources away from men (who don’t live as long as women) or away from members of racial minority groups (who don’t live as long as whites).
Finally, although some throw about numbers like $50,000 to $100,000 per quality-adjusted life-year as a guide, the price thresholds revealed by our current practices and policies are inconsistent. Hemodialysis is funded through Medicare by a federal mandate, but more cost-effective vaccines and preventive care are not covered to the same degree. Cost-effectiveness analyses are essential to establish a quantitative sense about the efficient use of resources, but they need to be interpreted alongside other considerations we also value. Cost-effectiveness analyses don’t take us all the way to the decision line by themselves.
WHY ARE NEW DRUGS SO EXPENSIVE?
The high cost of the new direct-acting antivirals for just months of therapy seems excessive on its face. Even though most patients will not pay these costs directly, they are borne by society through higher taxes or premiums for commercial insurance, which are paid out-of-pocket by those who purchase individual insurance, or substitute for wages in employment-based health insurance.
We know that the actual cost to manufacture these drugs is significantly less than the prices charged by pharmaceutical companies5 and that the government subsidizes both the research and the reimbursement for certain therapies. However, the companies need to cover the long-term costs of research and development not only for these drugs but for other drugs that did not make it through the pipeline but might have.6
There are at least two sides to this economy. First, the more we are willing to pay for successful drugs that go to market, the more the developers of those drugs will be willing to invest in finding new ones. If we were to pay less for individual successes, we would in the end have fewer trials and fewer overall successes.
Second, pharmaceutical companies hire economists to do their own cost-effectiveness calculations. One reason it should be no surprise that new drugs often arrive on the market at prices that are pretty close to commonly accepted thresholds for cost-effectiveness is that this is partly how they were priced in the first place. Pharmaceutical companies naturally want to price their products as high as they can. Since there is a limit to what people are willing to pay for the benefit they get in return, determining that limit and setting the price at that point helps firms extract as much of the surplus as possible.
AN OPPORTUNITY FOR LEADERSHIP
A disciplined analysis of the costs and benefits of new drug therapies is critical to any medical policy decision, rather than cost alone. There will always be a point where new treatments are too expensive—a point not based on absolute cost, but on cost relative to what is gained over and above the next best alternative.7 However, we should acknowledge that these analyses are based on estimates that may change over time, that they require modeling assumptions that are often subjective and opaque, and that the interpretation and implementation of these policies within their social context is just as important as the analysis of their economic efficiency.
As challenging as these decisions are, they offer an opportunity for leadership from medicine. Some organizations have already taken a stance on eliminating waste—through their participation in the Choosing Wisely initiative led by the American Board of Internal Medicine8 or through stands against the use of drugs and procedures that offer no benefit over cheaper alternatives.9 As these decisions get harder and as we aim to reduce not just zero-value care, but also low-value care, physicians have an enormous amount to contribute.
The era of all-oral agents for hepatitis C virus infection has begun. Previous treatments for this disease included pegylated interferon and ribavirin, which had limited effectiveness and side effects severe enough to reduce adherence and quality of life. Recent trials have documented the effectiveness of the new direct-acting antiviral agents.1 These new drugs work better and offer the promise of an all-oral treatment regimen that avoids pegylated interferon.
But they cost a lot. Prices of more than $50,000 are estimated for a 2-to-3-month course of treatment.2 These new medications reflect the kind of societal advances that justify a long-term investment in basic and clinical research. But do we value advances at any cost?
DOES COST MATTER?
Leaving aside the question of whether these particular drugs are too expensive, the general question remains whether effective therapies can ever be so expensive that we should not use them.
Does cost matter? Well, we all know that it does. We pay attention to cost in our individual purchasing and in how we think about business and government spending. And yet, while everyone agrees that we shouldn’t pay for care that provides no benefit, many of us stop at just that line, and think or act as if we can’t put a price on those elements of health care that offer some potential to save lives. It’s a comfortable position, because in going after pure waste we feel like fiscally temperate guardians of societal resources without feeling responsible for heart-rending choices about overspending on things that do work. Yet that spending threatens societal resources just as much as useless therapies.
In the end, though, it is an illogical position. The illogic is easy to understand once you walk it through: if you are unwilling to put a price on life, then you are saying that there is no price too high for any potential health benefit, no matter how small. That means you commit all your resources to health and you go bankrupt.
So, implicitly or explicitly (our society does so implicitly—and inconsistently, at that), you have to put a maximum price on life. But at that point, you are (again, implicitly) saying that when there are treatments that cost more, you shouldn’t buy them.3 Admittedly, it doesn’t sound good, and in health care, which touches us so intimately, it doesn’t feel good either.
SHOULD PHYSICIANS CARE ABOUT COST?
Many of us were taught in medical school that it isn’t the doctor’s job to think about cost. Physicians are to be clinical advocates for their patients without consideration of cost—but that can’t be right, and it isn’t right.
First, even if physicians are patient advocates first, they ought to consider cost when the patient is paying. The rise in the use of high-deductible health insurance plans has expanded the financial risk that individual patients face in their own health care decisions. Physicians may be unprepared to help patients with those decisions, but it seems like a service they ought to provide.
Second, the line between cost to the individual and cost to society is blurred at best. Our societal health care spending is nothing more than the aggregation of our individual health care spending. Even if we don’t want physicians to focus on cost when with an individual patient at the bedside or at the examination table, don’t we want societal cost to be at least in their peripheral vision?
Many obstacles impede this view. Even if physicians can keep societal costs in their peripheral vision, they certainly can’t see to the edges of the broad canvas that all of health care represents, and they have no easy decision rules for how to turn what vision they have into a decision for a particular patient.
A variety of stakeholders have succeeded in turning what might have been seen as socially responsible thinking into a dirty word. The same politicians who use the term “stewardship” when they are in favor of considering societal implications call it “rationing” when they feel the other way. As a result, some of our most important institutions—eg, Medicare—are prohibited from considering price. Commercial insurers, still smarting from the managed-care backlash of the 1990s, have limited ability to effectively manage costs while maintaining quality. In some sense, this vacuum creates an opportunity for physician leadership.
COST-EFFECTIVENESS ANALYSIS AND ITS LIMITATIONS
Cost-effectiveness analysis, which represents the health care value of a therapy as the ratio of its financial cost to its benefit (eg, cost per quality-adjusted life-year), offers a disciplined approach to these conflicts between individual good and social good.4
The long-term costs of hepatitis C are substantial and include multiple diagnostic tests, hospitalization, surgery, and death. A major treatment for both liver failure and hepatocellular cancer is liver transplantation, which can entail hundreds of thousands of dollars in cost for the surgery and ongoing care. Preventing just one transplant can provide enormous savings, in addition to freeing up cadaveric organs for another patient. A careful cost-effectiveness analysis could tell us whether the new direct-acting antiviral agents are worth their cost.
These analyses are appealing because they are formal and disciplined, but it turns out that they are far from value-free. Their methodology is complicated and is sensitive to subjective modeling assumptions whose implications are often not straightforward, are hard to report in the compact methods sections of manuscripts, and are harder still to interpret by most readers of these articles.
Further, these models focus exclusively on economic efficiency, so even the most carefully constructed cost-effectiveness analyses need to be tempered by a sense of social equity not captured in these models. For example, an emphasis on increasing quality-adjusted life-years will naturally lead to policy decisions that favor groups that have more life-years remaining. That may sound fine if we are comfortable with the idea that, in general, we should target our resources toward younger people rather than older people. But the same thinking means we should target our resources away from men (who don’t live as long as women) or away from members of racial minority groups (who don’t live as long as whites).
Finally, although some throw about numbers like $50,000 to $100,000 per quality-adjusted life-year as a guide, the price thresholds revealed by our current practices and policies are inconsistent. Hemodialysis is funded through Medicare by a federal mandate, but more cost-effective vaccines and preventive care are not covered to the same degree. Cost-effectiveness analyses are essential to establish a quantitative sense about the efficient use of resources, but they need to be interpreted alongside other considerations we also value. Cost-effectiveness analyses don’t take us all the way to the decision line by themselves.
WHY ARE NEW DRUGS SO EXPENSIVE?
The high cost of the new direct-acting antivirals for just months of therapy seems excessive on its face. Even though most patients will not pay these costs directly, they are borne by society through higher taxes or premiums for commercial insurance, which are paid out-of-pocket by those who purchase individual insurance, or substitute for wages in employment-based health insurance.
We know that the actual cost to manufacture these drugs is significantly less than the prices charged by pharmaceutical companies5 and that the government subsidizes both the research and the reimbursement for certain therapies. However, the companies need to cover the long-term costs of research and development not only for these drugs but for other drugs that did not make it through the pipeline but might have.6
There are at least two sides to this economy. First, the more we are willing to pay for successful drugs that go to market, the more the developers of those drugs will be willing to invest in finding new ones. If we were to pay less for individual successes, we would in the end have fewer trials and fewer overall successes.
Second, pharmaceutical companies hire economists to do their own cost-effectiveness calculations. One reason it should be no surprise that new drugs often arrive on the market at prices that are pretty close to commonly accepted thresholds for cost-effectiveness is that this is partly how they were priced in the first place. Pharmaceutical companies naturally want to price their products as high as they can. Since there is a limit to what people are willing to pay for the benefit they get in return, determining that limit and setting the price at that point helps firms extract as much of the surplus as possible.
AN OPPORTUNITY FOR LEADERSHIP
A disciplined analysis of the costs and benefits of new drug therapies is critical to any medical policy decision, rather than cost alone. There will always be a point where new treatments are too expensive—a point not based on absolute cost, but on cost relative to what is gained over and above the next best alternative.7 However, we should acknowledge that these analyses are based on estimates that may change over time, that they require modeling assumptions that are often subjective and opaque, and that the interpretation and implementation of these policies within their social context is just as important as the analysis of their economic efficiency.
As challenging as these decisions are, they offer an opportunity for leadership from medicine. Some organizations have already taken a stance on eliminating waste—through their participation in the Choosing Wisely initiative led by the American Board of Internal Medicine8 or through stands against the use of drugs and procedures that offer no benefit over cheaper alternatives.9 As these decisions get harder and as we aim to reduce not just zero-value care, but also low-value care, physicians have an enormous amount to contribute.
- Dugum M, O’Shea R. Hepatitis C virus: here comes alloral treatment. Cleve Clin J Med 2014; 81:159–172.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- Asch DA. Basic lessons in resource allocation: sharing, setting limits, and being fair. Pharos Alpha Omega Alpha Honor Med Soc 1995; 58:33–34.
- Weinstein MC, Stason WB. Foundations of cost-effectiveness analysis for health and medical practices. N Engl J Med 1977; 296:716–721.
- Hill A, Khoo S, Fortunak J, Simmons B, Ford N. Minimum costs for producing hepatitis C direct acting antivirals, for use in large-scale treatment access programs in developing countries. Clin Infect Dis 2014; Jan 6 [Epub ahead of print].
- Adams CP, Brantner VV. Estimating the cost of new drug development: is it really 802 million dollars? Health Aff (Millwood) 2006; 25:420–428.
- Eisenberg JM. Clinical economics. A guide to the economic analysis of clinical practices. JAMA 1989; 262:2879–2886.
- Cassel CK, Guest JA. Choosing wisely: helping physicians and patients make smart decisions about their care. JAMA 2012; 307:1801–1802.
- Bach PB, Saltz LB, Wittes RE. In cancer care, cost matters. New York Times. October 15, 2012:A25.
- Dugum M, O’Shea R. Hepatitis C virus: here comes alloral treatment. Cleve Clin J Med 2014; 81:159–172.
- Soriano V, Vispo E, de Mendoza C, et al. Hepatitis C therapy with HCV NS5B polymerase inhibitors. Expert Opin Pharmacother 2013; 14:1161–1170.
- Asch DA. Basic lessons in resource allocation: sharing, setting limits, and being fair. Pharos Alpha Omega Alpha Honor Med Soc 1995; 58:33–34.
- Weinstein MC, Stason WB. Foundations of cost-effectiveness analysis for health and medical practices. N Engl J Med 1977; 296:716–721.
- Hill A, Khoo S, Fortunak J, Simmons B, Ford N. Minimum costs for producing hepatitis C direct acting antivirals, for use in large-scale treatment access programs in developing countries. Clin Infect Dis 2014; Jan 6 [Epub ahead of print].
- Adams CP, Brantner VV. Estimating the cost of new drug development: is it really 802 million dollars? Health Aff (Millwood) 2006; 25:420–428.
- Eisenberg JM. Clinical economics. A guide to the economic analysis of clinical practices. JAMA 1989; 262:2879–2886.
- Cassel CK, Guest JA. Choosing wisely: helping physicians and patients make smart decisions about their care. JAMA 2012; 307:1801–1802.
- Bach PB, Saltz LB, Wittes RE. In cancer care, cost matters. New York Times. October 15, 2012:A25.
Canagliflozin
To the Editor: We found Dr. Vouyiouklis’s article about the recently approved sodium-glucose cotransport 2 (SGLT) inhibitor canagliflozin very useful. However, we strongly believe there are some issues that should be addressed.
In discussing the canagliflozin trials, Dr. Vouyiouklis did not mention a phase III randomized, double-blind, double-arm study, in which canagliflozin (100 and 300 mg) in addition to metformin was compared with placebo and sitagliptin (100 mg) in patients with type 2 diabetes.1 This study recruited 1,284 participants in 22 countries. At week 52, hemoglobin A1c levels had declined by 0.73% in the sitagliptin group, 0.73% in the canagliflozin 100 mg group, and 0.88% in the canagliflozin 300 mg group. Thus, canagliflozin 100 mg demonstrated noninferiority, and canagliflozin 300 mg demonstrated superiority. In addition, as previously described by other trials, a significant statistical reduction was observed in weight and blood pressure with modest elevations in LDL cholesterol and the incidence of mycotic urinary infections.
Current guidelines and recommendations give a wide variety of therapeutic options as the second step if lifestyle interventions and metformin fail to achieve glycemic control.2 The best combination regimen is still debated and, because of their excellent side-effect profile, dipeptidyl peptidase-4 inhibitors (gliptins) are one of the most used therapeutic classes. We believe this study adds important evidence that could help with decision-making in routine clinical practice.
Also, canagliflozin’s favorable effects on weight and blood pressure inevitably lead to the question, Are the weight loss and decreased systolic blood pressure due to osmotic diuresis or to lean or body fat weight loss? The mechanism of action of SGLT2 inhibitors, per se, favors osmotic diuresis, and several trials have demonstrated this same effect, as well as postural dizziness and orthostatic hypotension.3,4 Until now, the exact cause of this weight loss has not been elucidated, and no trial has demonstrated with precision a reduction in lean or fat body weight as a direct effect of SGLT2 inhibitors. This, in addition to LDL elevation, could have important clinical implications, as diuretic osmosis will subsequently activate the renin-angiotensin-aldosterone system. This might initially blunt this blood pressure reduction and promote parasympathetic inhibition, sympathetic activation, and myocardial and vascular fibrosis that can potentially lead in the long term to adverse cardiovascular outcomes.5
- Lavalle-González FJ, Januszewicz A, Davidson J, et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomized trial. Diabetologia 2013; 56:2582–2592.
- Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012; 35:1364–1379.
- Stenlöf K, Cefalu WT, Kim KA, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab 2013; 15:372–382.
- INVOKANA (canagliflozin) tablets, for oral use. Prescribing Information. Janssen Pharmaceuticals, Inc. www.janssenpharmaceuticalsinc.com/assets/invokana_prescribing_info.pdf. Accessed January 12, 2014.
- MacFadyen RJ, Barr CS, Struthers AD. Aldosterone blockade reduces vascular collagen turnover, improves heart rate variability and reduces early morning rise in heart rate in heart failure patients. Cardiovasc Res 1997; 35:30–34.
To the Editor: We found Dr. Vouyiouklis’s article about the recently approved sodium-glucose cotransport 2 (SGLT) inhibitor canagliflozin very useful. However, we strongly believe there are some issues that should be addressed.
In discussing the canagliflozin trials, Dr. Vouyiouklis did not mention a phase III randomized, double-blind, double-arm study, in which canagliflozin (100 and 300 mg) in addition to metformin was compared with placebo and sitagliptin (100 mg) in patients with type 2 diabetes.1 This study recruited 1,284 participants in 22 countries. At week 52, hemoglobin A1c levels had declined by 0.73% in the sitagliptin group, 0.73% in the canagliflozin 100 mg group, and 0.88% in the canagliflozin 300 mg group. Thus, canagliflozin 100 mg demonstrated noninferiority, and canagliflozin 300 mg demonstrated superiority. In addition, as previously described by other trials, a significant statistical reduction was observed in weight and blood pressure with modest elevations in LDL cholesterol and the incidence of mycotic urinary infections.
Current guidelines and recommendations give a wide variety of therapeutic options as the second step if lifestyle interventions and metformin fail to achieve glycemic control.2 The best combination regimen is still debated and, because of their excellent side-effect profile, dipeptidyl peptidase-4 inhibitors (gliptins) are one of the most used therapeutic classes. We believe this study adds important evidence that could help with decision-making in routine clinical practice.
Also, canagliflozin’s favorable effects on weight and blood pressure inevitably lead to the question, Are the weight loss and decreased systolic blood pressure due to osmotic diuresis or to lean or body fat weight loss? The mechanism of action of SGLT2 inhibitors, per se, favors osmotic diuresis, and several trials have demonstrated this same effect, as well as postural dizziness and orthostatic hypotension.3,4 Until now, the exact cause of this weight loss has not been elucidated, and no trial has demonstrated with precision a reduction in lean or fat body weight as a direct effect of SGLT2 inhibitors. This, in addition to LDL elevation, could have important clinical implications, as diuretic osmosis will subsequently activate the renin-angiotensin-aldosterone system. This might initially blunt this blood pressure reduction and promote parasympathetic inhibition, sympathetic activation, and myocardial and vascular fibrosis that can potentially lead in the long term to adverse cardiovascular outcomes.5
To the Editor: We found Dr. Vouyiouklis’s article about the recently approved sodium-glucose cotransport 2 (SGLT) inhibitor canagliflozin very useful. However, we strongly believe there are some issues that should be addressed.
In discussing the canagliflozin trials, Dr. Vouyiouklis did not mention a phase III randomized, double-blind, double-arm study, in which canagliflozin (100 and 300 mg) in addition to metformin was compared with placebo and sitagliptin (100 mg) in patients with type 2 diabetes.1 This study recruited 1,284 participants in 22 countries. At week 52, hemoglobin A1c levels had declined by 0.73% in the sitagliptin group, 0.73% in the canagliflozin 100 mg group, and 0.88% in the canagliflozin 300 mg group. Thus, canagliflozin 100 mg demonstrated noninferiority, and canagliflozin 300 mg demonstrated superiority. In addition, as previously described by other trials, a significant statistical reduction was observed in weight and blood pressure with modest elevations in LDL cholesterol and the incidence of mycotic urinary infections.
Current guidelines and recommendations give a wide variety of therapeutic options as the second step if lifestyle interventions and metformin fail to achieve glycemic control.2 The best combination regimen is still debated and, because of their excellent side-effect profile, dipeptidyl peptidase-4 inhibitors (gliptins) are one of the most used therapeutic classes. We believe this study adds important evidence that could help with decision-making in routine clinical practice.
Also, canagliflozin’s favorable effects on weight and blood pressure inevitably lead to the question, Are the weight loss and decreased systolic blood pressure due to osmotic diuresis or to lean or body fat weight loss? The mechanism of action of SGLT2 inhibitors, per se, favors osmotic diuresis, and several trials have demonstrated this same effect, as well as postural dizziness and orthostatic hypotension.3,4 Until now, the exact cause of this weight loss has not been elucidated, and no trial has demonstrated with precision a reduction in lean or fat body weight as a direct effect of SGLT2 inhibitors. This, in addition to LDL elevation, could have important clinical implications, as diuretic osmosis will subsequently activate the renin-angiotensin-aldosterone system. This might initially blunt this blood pressure reduction and promote parasympathetic inhibition, sympathetic activation, and myocardial and vascular fibrosis that can potentially lead in the long term to adverse cardiovascular outcomes.5
- Lavalle-González FJ, Januszewicz A, Davidson J, et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomized trial. Diabetologia 2013; 56:2582–2592.
- Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012; 35:1364–1379.
- Stenlöf K, Cefalu WT, Kim KA, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab 2013; 15:372–382.
- INVOKANA (canagliflozin) tablets, for oral use. Prescribing Information. Janssen Pharmaceuticals, Inc. www.janssenpharmaceuticalsinc.com/assets/invokana_prescribing_info.pdf. Accessed January 12, 2014.
- MacFadyen RJ, Barr CS, Struthers AD. Aldosterone blockade reduces vascular collagen turnover, improves heart rate variability and reduces early morning rise in heart rate in heart failure patients. Cardiovasc Res 1997; 35:30–34.
- Lavalle-González FJ, Januszewicz A, Davidson J, et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomized trial. Diabetologia 2013; 56:2582–2592.
- Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012; 35:1364–1379.
- Stenlöf K, Cefalu WT, Kim KA, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab 2013; 15:372–382.
- INVOKANA (canagliflozin) tablets, for oral use. Prescribing Information. Janssen Pharmaceuticals, Inc. www.janssenpharmaceuticalsinc.com/assets/invokana_prescribing_info.pdf. Accessed January 12, 2014.
- MacFadyen RJ, Barr CS, Struthers AD. Aldosterone blockade reduces vascular collagen turnover, improves heart rate variability and reduces early morning rise in heart rate in heart failure patients. Cardiovasc Res 1997; 35:30–34.
Canagliflozin
To the Editor: In a recent CCJM review of canagliflozin,1 this novel antihyperglycemic medication was noted to be associated with a dose-dependent increase in low-density lipoprotein (LDL) cholesterol, with an increase in LDL of 8.3 mg/dL (0.215 mmol/L) seen with the 300-mg/day dose of canagliflozin.
The Cholesterol Treatment Trialists’ (CTT) meta-analysis2 showed a significant 21% proportional reduction in major vascular events per 1.0 mmol/L reduction in LDL cholesterol in people with diabetes treated with statins over an average of 4.3 years. If we assume that raising LDL cholesterol by 1.0 mmol/L has the opposite effect, then patients taking 300 mg per day of canagliflozin would be expected to suffer an increase in major vascular events of about 4.5% over 4.3 years. Put another way, for every 22 diabetic patients treated with canagliflozin over 4.3 years, one additional major vascular event would be expected on the basis of the associated increase in LDL cholesterol.
The CTT data also showed a significant 9% decrease in all-cause mortality for every 1.0 mmol/L decrease in LDL cholesterol. Again, assuming that raising LDL has the opposite effect of lowering it, then we should expect an additional death for each 52 diabetic patients treated with 300 mg/day of canagliflozin per day for 4.3 years.
The hypotensive side effect of canagliflozin might tend to mitigate some of the above adverse effects, as might its antihyperglycemic effect. Still, it would seem prudent to use this novel agent only as a second- or third-line choice, particularly in diabetic patients who have already suffered a major vascular event.
- Vouyiouklis M. Canagliflozin: improving diabetes by making urine sweet. Cleve Clin J Med 2013; 80:683–687.
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Kearney PM, Blackwell L, Collins R, et al. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet 2008; 371:117–125.
To the Editor: In a recent CCJM review of canagliflozin,1 this novel antihyperglycemic medication was noted to be associated with a dose-dependent increase in low-density lipoprotein (LDL) cholesterol, with an increase in LDL of 8.3 mg/dL (0.215 mmol/L) seen with the 300-mg/day dose of canagliflozin.
The Cholesterol Treatment Trialists’ (CTT) meta-analysis2 showed a significant 21% proportional reduction in major vascular events per 1.0 mmol/L reduction in LDL cholesterol in people with diabetes treated with statins over an average of 4.3 years. If we assume that raising LDL cholesterol by 1.0 mmol/L has the opposite effect, then patients taking 300 mg per day of canagliflozin would be expected to suffer an increase in major vascular events of about 4.5% over 4.3 years. Put another way, for every 22 diabetic patients treated with canagliflozin over 4.3 years, one additional major vascular event would be expected on the basis of the associated increase in LDL cholesterol.
The CTT data also showed a significant 9% decrease in all-cause mortality for every 1.0 mmol/L decrease in LDL cholesterol. Again, assuming that raising LDL has the opposite effect of lowering it, then we should expect an additional death for each 52 diabetic patients treated with 300 mg/day of canagliflozin per day for 4.3 years.
The hypotensive side effect of canagliflozin might tend to mitigate some of the above adverse effects, as might its antihyperglycemic effect. Still, it would seem prudent to use this novel agent only as a second- or third-line choice, particularly in diabetic patients who have already suffered a major vascular event.
To the Editor: In a recent CCJM review of canagliflozin,1 this novel antihyperglycemic medication was noted to be associated with a dose-dependent increase in low-density lipoprotein (LDL) cholesterol, with an increase in LDL of 8.3 mg/dL (0.215 mmol/L) seen with the 300-mg/day dose of canagliflozin.
The Cholesterol Treatment Trialists’ (CTT) meta-analysis2 showed a significant 21% proportional reduction in major vascular events per 1.0 mmol/L reduction in LDL cholesterol in people with diabetes treated with statins over an average of 4.3 years. If we assume that raising LDL cholesterol by 1.0 mmol/L has the opposite effect, then patients taking 300 mg per day of canagliflozin would be expected to suffer an increase in major vascular events of about 4.5% over 4.3 years. Put another way, for every 22 diabetic patients treated with canagliflozin over 4.3 years, one additional major vascular event would be expected on the basis of the associated increase in LDL cholesterol.
The CTT data also showed a significant 9% decrease in all-cause mortality for every 1.0 mmol/L decrease in LDL cholesterol. Again, assuming that raising LDL has the opposite effect of lowering it, then we should expect an additional death for each 52 diabetic patients treated with 300 mg/day of canagliflozin per day for 4.3 years.
The hypotensive side effect of canagliflozin might tend to mitigate some of the above adverse effects, as might its antihyperglycemic effect. Still, it would seem prudent to use this novel agent only as a second- or third-line choice, particularly in diabetic patients who have already suffered a major vascular event.
- Vouyiouklis M. Canagliflozin: improving diabetes by making urine sweet. Cleve Clin J Med 2013; 80:683–687.
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Kearney PM, Blackwell L, Collins R, et al. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet 2008; 371:117–125.
- Vouyiouklis M. Canagliflozin: improving diabetes by making urine sweet. Cleve Clin J Med 2013; 80:683–687.
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Kearney PM, Blackwell L, Collins R, et al. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet 2008; 371:117–125.
In reply: Canagliflozin
In Reply: I would like to thank these readers very much for their response and comments.
Additional data provided from the study conducted by Lavalle-González et al evaluating the efficacy and safety of canagliflozin (100-mg and 300-mg doses) vs placebo and sitagliptin in patients with type 2 diabetes showed similar findings in weight and blood pressure reduction with slight LDL elevation with the studies mentioned in my article.1 At 52 weeks, as noted, canagliflozin 100 mg demonstrated noninferiority, and canagliflozin 300 mg showed a statistically significant superiority to sitagliptin in lowering hemoglobin A1c (a change of −0.73% with canagliflozin 100 mg, −0.88% with canagliflozin 300 mg, and −0.73% with sitagliptin), which may be considered in treatment decisions along with the other possible effects of this drug.1
The decision to use canagliflozin as second-or third-line therapy should be individualized after considering all of the patient’s risk factors as well as the potential benefit vs side effectsof this drug. Metformin remains my first-line choice in the management of type 2 diabetes. In my clinical practice, thus far, I have not used canagliflozin in patients with known coronary disease or a history of cardiovascular events. I have ensured that the LDL is certainly below goal before starting any patient on this drug, and I have followed the LDL closely, without hesitating to increase the statin drug to keep the LDL below goal. I agree that the slight increase of LDL is of concern, and certainly long-term studies are necessary to see whether there will be any increase in cardiovascular events from the use of canagliflozin.
- Lavalle-González FJ, Januszewicz A, Davidson J, et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomized trial. Diabetologia 2013; 56:2582–2592.
In Reply: I would like to thank these readers very much for their response and comments.
Additional data provided from the study conducted by Lavalle-González et al evaluating the efficacy and safety of canagliflozin (100-mg and 300-mg doses) vs placebo and sitagliptin in patients with type 2 diabetes showed similar findings in weight and blood pressure reduction with slight LDL elevation with the studies mentioned in my article.1 At 52 weeks, as noted, canagliflozin 100 mg demonstrated noninferiority, and canagliflozin 300 mg showed a statistically significant superiority to sitagliptin in lowering hemoglobin A1c (a change of −0.73% with canagliflozin 100 mg, −0.88% with canagliflozin 300 mg, and −0.73% with sitagliptin), which may be considered in treatment decisions along with the other possible effects of this drug.1
The decision to use canagliflozin as second-or third-line therapy should be individualized after considering all of the patient’s risk factors as well as the potential benefit vs side effectsof this drug. Metformin remains my first-line choice in the management of type 2 diabetes. In my clinical practice, thus far, I have not used canagliflozin in patients with known coronary disease or a history of cardiovascular events. I have ensured that the LDL is certainly below goal before starting any patient on this drug, and I have followed the LDL closely, without hesitating to increase the statin drug to keep the LDL below goal. I agree that the slight increase of LDL is of concern, and certainly long-term studies are necessary to see whether there will be any increase in cardiovascular events from the use of canagliflozin.
In Reply: I would like to thank these readers very much for their response and comments.
Additional data provided from the study conducted by Lavalle-González et al evaluating the efficacy and safety of canagliflozin (100-mg and 300-mg doses) vs placebo and sitagliptin in patients with type 2 diabetes showed similar findings in weight and blood pressure reduction with slight LDL elevation with the studies mentioned in my article.1 At 52 weeks, as noted, canagliflozin 100 mg demonstrated noninferiority, and canagliflozin 300 mg showed a statistically significant superiority to sitagliptin in lowering hemoglobin A1c (a change of −0.73% with canagliflozin 100 mg, −0.88% with canagliflozin 300 mg, and −0.73% with sitagliptin), which may be considered in treatment decisions along with the other possible effects of this drug.1
The decision to use canagliflozin as second-or third-line therapy should be individualized after considering all of the patient’s risk factors as well as the potential benefit vs side effectsof this drug. Metformin remains my first-line choice in the management of type 2 diabetes. In my clinical practice, thus far, I have not used canagliflozin in patients with known coronary disease or a history of cardiovascular events. I have ensured that the LDL is certainly below goal before starting any patient on this drug, and I have followed the LDL closely, without hesitating to increase the statin drug to keep the LDL below goal. I agree that the slight increase of LDL is of concern, and certainly long-term studies are necessary to see whether there will be any increase in cardiovascular events from the use of canagliflozin.
- Lavalle-González FJ, Januszewicz A, Davidson J, et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomized trial. Diabetologia 2013; 56:2582–2592.
- Lavalle-González FJ, Januszewicz A, Davidson J, et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomized trial. Diabetologia 2013; 56:2582–2592.