User login
Medication-assisted treatment of opiate dependence is gaining favor
Experts have argued for decades about how best to manage opiate dependence, with practitioners generally subscribing to one of two strategies: either total abstinence or medication-assisted treatment (MAT).
Although MAT has proven efficacy, it has been slow to gain acceptance, and the gold standard of care since the 1930s has been abstinence-based treatment. Among elite institutional holdouts against MAT was the Hazelden Treatment Center, a leading treatment institution and publishing house that had been wedded to the abstinence model since it was founded in 1949.1 Now, Hazelden has gone on record as embracing MAT, raising the possibility that the two predominant treatment philosophies for opiate-dependent patients may no longer be at odds.
FROM ABSTINENCE TO METHADONE MAINTENANCE
The modern day abstinence-based movement in this country started in the decade before the founding of Hazelden. In 1935, the US government opened the first of two federal drug treatment centers, known as the United States Narcotic Farm, in Lexington, KY.2 The move by the government to get into the addiction treatment business largely stemmed from frustration over the growing problem of addiction at that time, coupled with a dearth of treatment options for addicts in the wake of the 1914 Harrison Narcotics Act.
The Narcotic Farm was an impressive facility—for all intents and purposes, a specialized prison—that initially housed 1,200 people. In addition to prisoners, it also accepted voluntary, nonprisoner patients. In many ways, it was ahead of its time. It offered a wide variety of services, including detoxification, group therapy, individual therapy, psychiatric and medical services, and vocational rehabilitation.2 Housed on the premises was the Addiction Research Center at Lexington, the first intramural research branch of the National Institute of Mental Health. After the “Blue Grass” mandatory commitment laws were passed in the 1940s, even the voluntary patients were ultimately committed for a 1-year sentence at Lexington. This facility, and its sister facility in Ft. Worth, TX, would have been the envy of any modern-day abstinence-based treatment center in terms of the services offered and the long lengths of stay.
The quality of the program, as evidenced by the impressive array of services and long stays, would lead one to expect that its treatment outcomes over nearly 40 years of operation were equally stellar. However, in terms of outcomes the Farm was an abysmal failure, as shown by numerous studies demonstrating relapse rates of more than 90% in the patients discharged from it.2,3
Similar frustrations at other abstinence-based treatment centers from the 1940s through the 1960s led Dr. Vincent Dole, the “father of methadone maintenance,” to conclude in 1971 that after detoxification from opiates, “human addicts almost always return to use of narcotics after they leave the hospital where they have been detoxified.”4 That realization inspired Dr. Dole and his wife and colleague Dr. Marie Nyswander to revisit the idea of medication-assisted treatment, an approach previously used by the morphine maintenance clinics of the early 1900s. This work led to the development of government-sanctioned methadone clinics across America and to the realization that long-term recovery was possible with medication, even without a lengthy hospital stay. For this revolutionary work on opiate addiction, Dr. Dole won the prestigious Lasker Award in 1988.
The major reason for the success of methadone was that, because of its pharmacokinetic profile, it could stabilize the patient through once-daily dosing without sedation or narcosis. As noted by Dr. Dole, once patients are on a stable dosing regimen, the obsessive preoccupation with drug use fades away.5
Despite its success, methadone maintenance had its share of detractors. It was fraught with controversy because it was viewed as a crutch, and those who were on it were often not considered by their abstinent peers as being in true recovery. The reasons for the negative attitudes toward MAT are unclear but may reflect antiquated beliefs that addiction may be indicative of a failure of morals or will, and that patients ought to be able to simply stop using.
Whatever the reason for the animosity surrounding MAT, it should be noted that an expert consensus panel convened by the Betty Ford Center in 2007 agreed that patients on MAT met their consensus definition of sobriety.6 The issue of what constitutes recovery remains a very complex and hotly debated topic that is beyond the scope of this paper and that has been discussed elsewhere.6,7
For more than 3 decades, methadone was the only medication available for MAT. Federal regulations limit the dispensing of methadone to licensed clinics, most of which are located in major metropolitan areas. Patients must go to the clinic every day to receive their dose of methadone—a major inconvenience, especially to those with transportation issues. Adding to the lack of appeal of methadone maintenance is that the clinics are typically located in the higher-crime areas of cities. Savvy drug dealers know the location of these clinics and often loiter on nearby street corners in an attempt to lure addicts away from recovery by flaunting their illicit drugs.
A final, very significant drawback of methadone is its safety profile. It is a full-agonist narcotic that can be fatal in overdose or in the induction phase, especially if taken with other drugs, such as benzodiazepines.
2003: BUPRENORPHINE-NALOXONE IS APPROVED
Such concerns led researchers to search for other medications to be used for MAT that could perhaps be prescribed in a typical outpatient physician practice. For many reasons, buprenorphine became the most promising candidate. In 2003, the US Food and Drug Administration approved the combination medication buprenorphine-naloxone (Sub-oxone) as only the second drug indicated for maintenance treatment of opioid dependence in the United States.
Buprenorphine differs from methadone in that it is a partial agonist at mu opiate receptors, and therefore has a “ceiling” or “plateau” effect in terms of dose-response and a much improved safety profile. Unlike methadone, buprenorphine can be prescribed in a doctor’s office and does not have to be dispensed at a government-approved clinic.
Unfortunately, buprenorphine-maintained patients seem to carry the same stigma in the recovery community as those maintained on methadone—that they are simply substituting one drug for another. Detractors usually fail to consider that, as with methadone, patients do not report getting “high” from taking buprenorphine. Patients will often state that when they first start taking it, they “feel something,” but after a few days of adjustment, they simply feel normal. They don’t feel high, they are no longer in withdrawal, their cravings are virtually eliminated, and their opiate receptors are effectively occupied and blocked, so there is no “high” in the event of a relapse.
What’s more, buprenorphine is not a medication that will help them deal with life’s stressors by “chemical coping.” Sober coping is a skill they must learn by actively participating in a solid 12-step-based recovery program and, in some cases, in psychotherapy. By removing the drug obsession, buprenorphine promotes and facilitates the important recovery goal of learning how to deal with life on life’s terms.
ADDICTION AS CHRONIC ILLNESS
Outcomes studies of addiction treatment have focused largely on rates of relapse after discharge from acute treatments such as residential rehabilitation, partial hospitalization, and intensive outpatient programs. With MAT, however, outcomes research has primarily looked at the duration of retention in treatment.
The change in focus between the two types of treatment coincides with a paradigm shift that views addiction as a chronic condition that requires ongoing care. Continued participation in prescribed care with demonstrated efficacy is considered to be the major indicator of success. Under the chronic illness model employed by MAT providers, if a patient reverted to briefly using a drug of abuse, this would be an issue to address in his ongoing treatment and would not necessarily indicate treatment failure as with the acute care model. Beyond retention rates, research has demonstrated that MAT with methadone results in reductions in rates of criminal activity, illicit drug use, acquisition of human immunodeficiency virus, and overall mortality.8–10
In outcomes studies, MAT has repeatedly shown better efficacy than abstinence-based approaches. During the first 5 years of its implementation, in 4,000 patients, methadone maintenance boasted 1-year retention rates exceeding 98%.11 Over the subsequent 3 years, with the number of patients approaching 35,000, the 1-year retention rates fell to around 60%—still far exceeding results of abstinence-based treatment and approximating the number cited in most modern studies.11
The retention rates in buprenorphine programs are similarly promising. Studies of 12 to 13 weeks duration have shown retention rates of 52% to 79%.12–15 Six-month studies have demonstrated retention rates of 43% to 100%.16–19 Another study showed that 38% of opiate-dependent patients remained in treatment with buprenorphine at 5 years.20 Surprisingly, most of the buprenorphine studies have been conducted in office-based practices, which are less structured than outpatient methadone programs.
MEDICATION-ASSISTED TREATMENT IS GAINING ACCEPTANCE
Data from decades of experience with MAT strongly support the conclusion that it is superior to abstinence-based approaches.
The importance of a patient staying in treatment cannot be overemphasized, as the consequence of failing in recovery may well be an early death. On average, heroin addicts lose about 18 years of life expectancy, and the mortality rate for injection users is roughly 2% per year.21 The mortality rate for heroin users is 6 to 20 times greater than for age-matched peers who are not drug users.22
As high as these numbers are, they are even higher for abusers of prescription narcotics. The annual death rate associated with opioid pain relievers (4.8 per 100,000) is nearly double that associated with illicit drugs (2.8 per 100,000).23
The recent and rather radical change in treatment philosophy by Hazelden came as a shock to some, a disappointment to others, and a welcome change to many who saw this as a move by one of the more respected treatment centers in the country to fall in line with the body of evidence that supports MAT for those suffering from opiate dependence. It remains a mystery why so many, if not most, addiction treatment centers in the United States cling to the abstinence-based philosophy despite the overwhelming data from decades of research and experience that show that abstinence does not work for the majority of opiate addicts.
Complete abstinence from opiate drugs of abuse and potentially addictive medications is a noble but perhaps unreachable goal for many sufferers. Hazelden’s announced acceptance of MAT gives credence to the value of recovery goals that are not entirely drug-free.
Dr. Dole was correct in stating that opiate addicts usually return to drugs if not provided with MAT. Treatment programs need to inform opiate-dependent patients that abstinence-based treatment offers only a 1 in 10 chance of success. Perhaps some patients, armed with the daunting statistics regarding abstinence, will be inspired to devote themselves wholeheartedly to their recovery in an effort to make it into that elite 10% group that achieves long-lasting recovery without the aid of medications. But for the other 90%, it is encouraging to hear that Hazelden, the model treatment center for most abstinence-based programs in this country, may now lead other abstinence-based centers to reconsider their treatment philosophies.
Historically, US doctors were not allowed by federal law to prescribe opiates for addiction treatment. With the passage of DATA 2000, buprenorphine (alone or in combination with naloxone) can be prescribed for addiction treatment only by providers who obtain a waiver from the US Drug Enforcement Administration (DEA). Any doctor can become qualified to prescribe buprenorphine or buprenorphinenaloxone after completing an 8-hour online training course (available at www.buppractice.com and at www.aaap.org/buprenorphine) and by obtaining a DATA 2000 waiver and a new prescribing number from the DEA. Doctors are initially limited to treating only 30 patients with buprenorphine-naloxone at any given time, but can apply for an extension to 100 patients after having had their waiver for 1 year.
As MAT continues to gain favor, demand will grow for more providers to obtain their waivers to prescribe buprenorphine and buprenorphine-naloxone. Historically, there have always been too few methadone clinics to meet the demand. One can hope that the growing number of waivered providers will greatly improve access to care by opiate addicts, no matter where they reside. Qualified prescribers of buprenorphine and buprenorphine-naloxone are limited by the federal restrictions on the numbers of patients they can treat. If the chronic disease of addiction is to be integrated into the continuing-care approach of modern medicine and managed alongside other chronic diseases, primary care providers who are not specialized in treating addiction will need to be become comfortable with maintaining patients on buprenorphine-naloxone.7 Presumably, such patients will have already been stabilized through participation in addiction treatment programs in their respective geographic areas. Primary care providers will need to develop relationships with local addictionologists and treatment programs so that they will be able to refer those in active addiction for induction and stabilization on MAT and will be able to refer those already stabilized on MAT back to such specialists when relapses occur.
We may finally be approaching a time when structured residential treatment and MAT are not mutually exclusive options for our patients. These treatment options must work together for optimal outcomes. Based on our experience with hundreds of patients at Cleveland Clinic’s Alcohol and Drug Recovery Center, we believe this change of treatment philosophy is long overdue. In clinical settings, patients do not fit cleanly into one treatment arm or another and often require a blended approach to effect long-lasting change. Hazelden’s shift of treatment philosophy is an indication that this research-supported viewpoint is gaining acceptance in the traditionally drug-free halls of addiction treatment programs.
- White WL. Slaying the Dragon. The History of Addiction Treatment and Recovery in America. Bloomington, IL: Chestnut Health Systems/Lighthouse Institute; 1998:124–125,201.
- Kosten TR, Gorelick DA. The Lexington narcotic farm. Am J Psychiatry 2002; 159:22.
- Hunt GH, Odoroff ME. Followup study of narcotic drug addicts after hospitalization. Public Health Rep 1962; 77:41–54.
- Dole VP. Narcotic addiction, physical dependence and relapse. N Engl J Med 1972; 286:988–992.
- Dole VP. Implications of methadone maintenance for theories of narcotic addiction. JAMA 1988; 260:3025–3029.
- Betty Ford Institute Consensus Panel. What is recovery? A working definition from the Betty Ford Institute. J Subst Abuse Treat 2007; 33:221–228.
- McLellan AT. Have we evaluated addiction treatment correctly? Implications from a chronic care perspective. Addiction 2002; 97:249–252.
- Grönbladh L, Ohlund LS, Gunne LM. Mortality in heroin addiction: impact of methadone treatment. Acta Psychiatr Scand 1990; 82:223–227.
- Ball JC, Lange WR, Myers CP, Friedman SR. Reducing the risk of AIDS through methadone maintenance treatment. J Health Soc Behav 1988; 29:214–226.
- Martin J, Zweben JE, Payte JT. Opioid maintenance treatment. In:Ries RK, Fiellin DA, Miller SC, Saitzeds R, editors. Principles of Addiction Medicine. 4th ed. Philadelphia, PA: Lippincottt Williams & Wilkins, 2009:671–688.
- Dole VP, Nyswander ME. Methadone maintenance treatment. A tenyear perspective. JAMA 1976; 235:2117–2119.
- Cunningham C, Giovanniello A, Sacajiu G, et al. Buprenorphine treatment in an urban community health center: what to expect. Fam Med 2008; 40:500–506.
- Fiellin DA, Pantalon MV, Pakes JP, O’Connor PG, Chawarski M, Schottenfeld RS. Treatment of heroin dependence with buprenorphine in primary care. Am J Drug Alcohol Abuse 2002; 28:231–241.
- Fudala PJ, Bridge TP, Herbert S, et al; Buprenorphine/Naloxone Collaborative Study Group. Office-based treatment of opiate addiction with a sublingual-tablet formulation of buprenorphine and naloxone. N Engl J Med 2003; 349:949–958.
- O’Connor PG, Oliveto AH, Shi JM, et al. A randomized trial of buprenorphine maintenance for heroin dependence in a primary care clinic for substance users versus a methadone clinic. Am J Med 1998; 105:100–105.
- Fiellin DA, Pantalon MV, Chawarski MC, et al. Counseling plus buprenorphine-naloxone maintenance therapy for opioid dependence. N Engl J Med 2006; 355:365–374.
- Moore BA, Fiellin DA, Barry DT, et al. Primary care office-based buprenorphine treatment: comparison of heroin and prescription opioid dependent patients. J Gen Intern Med 2007; 22:527–530.
- Mintzer IL, Eisenberg M, Terra M, MacVane C, Himmelstein DU, Woolhandler S. Treating opioid addiction with buprenorphine-naloxone in community-based primary care settings. Ann Fam Med 2007; 5:146–150.
- O’Connor PG, Oliveto AH, Shi JM, et al. A pilot study of primary-carebased buprenorphine maintenance for heroin dependence. Am J Drug Alcohol Abuse 1996; 22:523–531.
- Fiellin DA, Moore BA, Sullivan LE, et al. Long-term treatment with buprenorphine/naloxone in primary care: results at 2–5 years. Am J Addict 2008; 17:116–120.
- Smyth B, Hoffman V, Fan J, Hser YI. Years of potential life lost among heroin addicts 33 years after treatment. Prev Med 2007; 44:369–374.
- Sporer KA. Acute heroin overdose. Ann Intern Med 1999; 130:584–590.
- Centers for Disease Control and Prevention (CDC). Vital signs: overdoses of prescription opioid pain relievers—United States, 1999–2008. MMWR Morb Mortal Wkly Rep 2011; 60:1487–1492.
Experts have argued for decades about how best to manage opiate dependence, with practitioners generally subscribing to one of two strategies: either total abstinence or medication-assisted treatment (MAT).
Although MAT has proven efficacy, it has been slow to gain acceptance, and the gold standard of care since the 1930s has been abstinence-based treatment. Among elite institutional holdouts against MAT was the Hazelden Treatment Center, a leading treatment institution and publishing house that had been wedded to the abstinence model since it was founded in 1949.1 Now, Hazelden has gone on record as embracing MAT, raising the possibility that the two predominant treatment philosophies for opiate-dependent patients may no longer be at odds.
FROM ABSTINENCE TO METHADONE MAINTENANCE
The modern day abstinence-based movement in this country started in the decade before the founding of Hazelden. In 1935, the US government opened the first of two federal drug treatment centers, known as the United States Narcotic Farm, in Lexington, KY.2 The move by the government to get into the addiction treatment business largely stemmed from frustration over the growing problem of addiction at that time, coupled with a dearth of treatment options for addicts in the wake of the 1914 Harrison Narcotics Act.
The Narcotic Farm was an impressive facility—for all intents and purposes, a specialized prison—that initially housed 1,200 people. In addition to prisoners, it also accepted voluntary, nonprisoner patients. In many ways, it was ahead of its time. It offered a wide variety of services, including detoxification, group therapy, individual therapy, psychiatric and medical services, and vocational rehabilitation.2 Housed on the premises was the Addiction Research Center at Lexington, the first intramural research branch of the National Institute of Mental Health. After the “Blue Grass” mandatory commitment laws were passed in the 1940s, even the voluntary patients were ultimately committed for a 1-year sentence at Lexington. This facility, and its sister facility in Ft. Worth, TX, would have been the envy of any modern-day abstinence-based treatment center in terms of the services offered and the long lengths of stay.
The quality of the program, as evidenced by the impressive array of services and long stays, would lead one to expect that its treatment outcomes over nearly 40 years of operation were equally stellar. However, in terms of outcomes the Farm was an abysmal failure, as shown by numerous studies demonstrating relapse rates of more than 90% in the patients discharged from it.2,3
Similar frustrations at other abstinence-based treatment centers from the 1940s through the 1960s led Dr. Vincent Dole, the “father of methadone maintenance,” to conclude in 1971 that after detoxification from opiates, “human addicts almost always return to use of narcotics after they leave the hospital where they have been detoxified.”4 That realization inspired Dr. Dole and his wife and colleague Dr. Marie Nyswander to revisit the idea of medication-assisted treatment, an approach previously used by the morphine maintenance clinics of the early 1900s. This work led to the development of government-sanctioned methadone clinics across America and to the realization that long-term recovery was possible with medication, even without a lengthy hospital stay. For this revolutionary work on opiate addiction, Dr. Dole won the prestigious Lasker Award in 1988.
The major reason for the success of methadone was that, because of its pharmacokinetic profile, it could stabilize the patient through once-daily dosing without sedation or narcosis. As noted by Dr. Dole, once patients are on a stable dosing regimen, the obsessive preoccupation with drug use fades away.5
Despite its success, methadone maintenance had its share of detractors. It was fraught with controversy because it was viewed as a crutch, and those who were on it were often not considered by their abstinent peers as being in true recovery. The reasons for the negative attitudes toward MAT are unclear but may reflect antiquated beliefs that addiction may be indicative of a failure of morals or will, and that patients ought to be able to simply stop using.
Whatever the reason for the animosity surrounding MAT, it should be noted that an expert consensus panel convened by the Betty Ford Center in 2007 agreed that patients on MAT met their consensus definition of sobriety.6 The issue of what constitutes recovery remains a very complex and hotly debated topic that is beyond the scope of this paper and that has been discussed elsewhere.6,7
For more than 3 decades, methadone was the only medication available for MAT. Federal regulations limit the dispensing of methadone to licensed clinics, most of which are located in major metropolitan areas. Patients must go to the clinic every day to receive their dose of methadone—a major inconvenience, especially to those with transportation issues. Adding to the lack of appeal of methadone maintenance is that the clinics are typically located in the higher-crime areas of cities. Savvy drug dealers know the location of these clinics and often loiter on nearby street corners in an attempt to lure addicts away from recovery by flaunting their illicit drugs.
A final, very significant drawback of methadone is its safety profile. It is a full-agonist narcotic that can be fatal in overdose or in the induction phase, especially if taken with other drugs, such as benzodiazepines.
2003: BUPRENORPHINE-NALOXONE IS APPROVED
Such concerns led researchers to search for other medications to be used for MAT that could perhaps be prescribed in a typical outpatient physician practice. For many reasons, buprenorphine became the most promising candidate. In 2003, the US Food and Drug Administration approved the combination medication buprenorphine-naloxone (Sub-oxone) as only the second drug indicated for maintenance treatment of opioid dependence in the United States.
Buprenorphine differs from methadone in that it is a partial agonist at mu opiate receptors, and therefore has a “ceiling” or “plateau” effect in terms of dose-response and a much improved safety profile. Unlike methadone, buprenorphine can be prescribed in a doctor’s office and does not have to be dispensed at a government-approved clinic.
Unfortunately, buprenorphine-maintained patients seem to carry the same stigma in the recovery community as those maintained on methadone—that they are simply substituting one drug for another. Detractors usually fail to consider that, as with methadone, patients do not report getting “high” from taking buprenorphine. Patients will often state that when they first start taking it, they “feel something,” but after a few days of adjustment, they simply feel normal. They don’t feel high, they are no longer in withdrawal, their cravings are virtually eliminated, and their opiate receptors are effectively occupied and blocked, so there is no “high” in the event of a relapse.
What’s more, buprenorphine is not a medication that will help them deal with life’s stressors by “chemical coping.” Sober coping is a skill they must learn by actively participating in a solid 12-step-based recovery program and, in some cases, in psychotherapy. By removing the drug obsession, buprenorphine promotes and facilitates the important recovery goal of learning how to deal with life on life’s terms.
ADDICTION AS CHRONIC ILLNESS
Outcomes studies of addiction treatment have focused largely on rates of relapse after discharge from acute treatments such as residential rehabilitation, partial hospitalization, and intensive outpatient programs. With MAT, however, outcomes research has primarily looked at the duration of retention in treatment.
The change in focus between the two types of treatment coincides with a paradigm shift that views addiction as a chronic condition that requires ongoing care. Continued participation in prescribed care with demonstrated efficacy is considered to be the major indicator of success. Under the chronic illness model employed by MAT providers, if a patient reverted to briefly using a drug of abuse, this would be an issue to address in his ongoing treatment and would not necessarily indicate treatment failure as with the acute care model. Beyond retention rates, research has demonstrated that MAT with methadone results in reductions in rates of criminal activity, illicit drug use, acquisition of human immunodeficiency virus, and overall mortality.8–10
In outcomes studies, MAT has repeatedly shown better efficacy than abstinence-based approaches. During the first 5 years of its implementation, in 4,000 patients, methadone maintenance boasted 1-year retention rates exceeding 98%.11 Over the subsequent 3 years, with the number of patients approaching 35,000, the 1-year retention rates fell to around 60%—still far exceeding results of abstinence-based treatment and approximating the number cited in most modern studies.11
The retention rates in buprenorphine programs are similarly promising. Studies of 12 to 13 weeks duration have shown retention rates of 52% to 79%.12–15 Six-month studies have demonstrated retention rates of 43% to 100%.16–19 Another study showed that 38% of opiate-dependent patients remained in treatment with buprenorphine at 5 years.20 Surprisingly, most of the buprenorphine studies have been conducted in office-based practices, which are less structured than outpatient methadone programs.
MEDICATION-ASSISTED TREATMENT IS GAINING ACCEPTANCE
Data from decades of experience with MAT strongly support the conclusion that it is superior to abstinence-based approaches.
The importance of a patient staying in treatment cannot be overemphasized, as the consequence of failing in recovery may well be an early death. On average, heroin addicts lose about 18 years of life expectancy, and the mortality rate for injection users is roughly 2% per year.21 The mortality rate for heroin users is 6 to 20 times greater than for age-matched peers who are not drug users.22
As high as these numbers are, they are even higher for abusers of prescription narcotics. The annual death rate associated with opioid pain relievers (4.8 per 100,000) is nearly double that associated with illicit drugs (2.8 per 100,000).23
The recent and rather radical change in treatment philosophy by Hazelden came as a shock to some, a disappointment to others, and a welcome change to many who saw this as a move by one of the more respected treatment centers in the country to fall in line with the body of evidence that supports MAT for those suffering from opiate dependence. It remains a mystery why so many, if not most, addiction treatment centers in the United States cling to the abstinence-based philosophy despite the overwhelming data from decades of research and experience that show that abstinence does not work for the majority of opiate addicts.
Complete abstinence from opiate drugs of abuse and potentially addictive medications is a noble but perhaps unreachable goal for many sufferers. Hazelden’s announced acceptance of MAT gives credence to the value of recovery goals that are not entirely drug-free.
Dr. Dole was correct in stating that opiate addicts usually return to drugs if not provided with MAT. Treatment programs need to inform opiate-dependent patients that abstinence-based treatment offers only a 1 in 10 chance of success. Perhaps some patients, armed with the daunting statistics regarding abstinence, will be inspired to devote themselves wholeheartedly to their recovery in an effort to make it into that elite 10% group that achieves long-lasting recovery without the aid of medications. But for the other 90%, it is encouraging to hear that Hazelden, the model treatment center for most abstinence-based programs in this country, may now lead other abstinence-based centers to reconsider their treatment philosophies.
Historically, US doctors were not allowed by federal law to prescribe opiates for addiction treatment. With the passage of DATA 2000, buprenorphine (alone or in combination with naloxone) can be prescribed for addiction treatment only by providers who obtain a waiver from the US Drug Enforcement Administration (DEA). Any doctor can become qualified to prescribe buprenorphine or buprenorphinenaloxone after completing an 8-hour online training course (available at www.buppractice.com and at www.aaap.org/buprenorphine) and by obtaining a DATA 2000 waiver and a new prescribing number from the DEA. Doctors are initially limited to treating only 30 patients with buprenorphine-naloxone at any given time, but can apply for an extension to 100 patients after having had their waiver for 1 year.
As MAT continues to gain favor, demand will grow for more providers to obtain their waivers to prescribe buprenorphine and buprenorphine-naloxone. Historically, there have always been too few methadone clinics to meet the demand. One can hope that the growing number of waivered providers will greatly improve access to care by opiate addicts, no matter where they reside. Qualified prescribers of buprenorphine and buprenorphine-naloxone are limited by the federal restrictions on the numbers of patients they can treat. If the chronic disease of addiction is to be integrated into the continuing-care approach of modern medicine and managed alongside other chronic diseases, primary care providers who are not specialized in treating addiction will need to be become comfortable with maintaining patients on buprenorphine-naloxone.7 Presumably, such patients will have already been stabilized through participation in addiction treatment programs in their respective geographic areas. Primary care providers will need to develop relationships with local addictionologists and treatment programs so that they will be able to refer those in active addiction for induction and stabilization on MAT and will be able to refer those already stabilized on MAT back to such specialists when relapses occur.
We may finally be approaching a time when structured residential treatment and MAT are not mutually exclusive options for our patients. These treatment options must work together for optimal outcomes. Based on our experience with hundreds of patients at Cleveland Clinic’s Alcohol and Drug Recovery Center, we believe this change of treatment philosophy is long overdue. In clinical settings, patients do not fit cleanly into one treatment arm or another and often require a blended approach to effect long-lasting change. Hazelden’s shift of treatment philosophy is an indication that this research-supported viewpoint is gaining acceptance in the traditionally drug-free halls of addiction treatment programs.
Experts have argued for decades about how best to manage opiate dependence, with practitioners generally subscribing to one of two strategies: either total abstinence or medication-assisted treatment (MAT).
Although MAT has proven efficacy, it has been slow to gain acceptance, and the gold standard of care since the 1930s has been abstinence-based treatment. Among elite institutional holdouts against MAT was the Hazelden Treatment Center, a leading treatment institution and publishing house that had been wedded to the abstinence model since it was founded in 1949.1 Now, Hazelden has gone on record as embracing MAT, raising the possibility that the two predominant treatment philosophies for opiate-dependent patients may no longer be at odds.
FROM ABSTINENCE TO METHADONE MAINTENANCE
The modern day abstinence-based movement in this country started in the decade before the founding of Hazelden. In 1935, the US government opened the first of two federal drug treatment centers, known as the United States Narcotic Farm, in Lexington, KY.2 The move by the government to get into the addiction treatment business largely stemmed from frustration over the growing problem of addiction at that time, coupled with a dearth of treatment options for addicts in the wake of the 1914 Harrison Narcotics Act.
The Narcotic Farm was an impressive facility—for all intents and purposes, a specialized prison—that initially housed 1,200 people. In addition to prisoners, it also accepted voluntary, nonprisoner patients. In many ways, it was ahead of its time. It offered a wide variety of services, including detoxification, group therapy, individual therapy, psychiatric and medical services, and vocational rehabilitation.2 Housed on the premises was the Addiction Research Center at Lexington, the first intramural research branch of the National Institute of Mental Health. After the “Blue Grass” mandatory commitment laws were passed in the 1940s, even the voluntary patients were ultimately committed for a 1-year sentence at Lexington. This facility, and its sister facility in Ft. Worth, TX, would have been the envy of any modern-day abstinence-based treatment center in terms of the services offered and the long lengths of stay.
The quality of the program, as evidenced by the impressive array of services and long stays, would lead one to expect that its treatment outcomes over nearly 40 years of operation were equally stellar. However, in terms of outcomes the Farm was an abysmal failure, as shown by numerous studies demonstrating relapse rates of more than 90% in the patients discharged from it.2,3
Similar frustrations at other abstinence-based treatment centers from the 1940s through the 1960s led Dr. Vincent Dole, the “father of methadone maintenance,” to conclude in 1971 that after detoxification from opiates, “human addicts almost always return to use of narcotics after they leave the hospital where they have been detoxified.”4 That realization inspired Dr. Dole and his wife and colleague Dr. Marie Nyswander to revisit the idea of medication-assisted treatment, an approach previously used by the morphine maintenance clinics of the early 1900s. This work led to the development of government-sanctioned methadone clinics across America and to the realization that long-term recovery was possible with medication, even without a lengthy hospital stay. For this revolutionary work on opiate addiction, Dr. Dole won the prestigious Lasker Award in 1988.
The major reason for the success of methadone was that, because of its pharmacokinetic profile, it could stabilize the patient through once-daily dosing without sedation or narcosis. As noted by Dr. Dole, once patients are on a stable dosing regimen, the obsessive preoccupation with drug use fades away.5
Despite its success, methadone maintenance had its share of detractors. It was fraught with controversy because it was viewed as a crutch, and those who were on it were often not considered by their abstinent peers as being in true recovery. The reasons for the negative attitudes toward MAT are unclear but may reflect antiquated beliefs that addiction may be indicative of a failure of morals or will, and that patients ought to be able to simply stop using.
Whatever the reason for the animosity surrounding MAT, it should be noted that an expert consensus panel convened by the Betty Ford Center in 2007 agreed that patients on MAT met their consensus definition of sobriety.6 The issue of what constitutes recovery remains a very complex and hotly debated topic that is beyond the scope of this paper and that has been discussed elsewhere.6,7
For more than 3 decades, methadone was the only medication available for MAT. Federal regulations limit the dispensing of methadone to licensed clinics, most of which are located in major metropolitan areas. Patients must go to the clinic every day to receive their dose of methadone—a major inconvenience, especially to those with transportation issues. Adding to the lack of appeal of methadone maintenance is that the clinics are typically located in the higher-crime areas of cities. Savvy drug dealers know the location of these clinics and often loiter on nearby street corners in an attempt to lure addicts away from recovery by flaunting their illicit drugs.
A final, very significant drawback of methadone is its safety profile. It is a full-agonist narcotic that can be fatal in overdose or in the induction phase, especially if taken with other drugs, such as benzodiazepines.
2003: BUPRENORPHINE-NALOXONE IS APPROVED
Such concerns led researchers to search for other medications to be used for MAT that could perhaps be prescribed in a typical outpatient physician practice. For many reasons, buprenorphine became the most promising candidate. In 2003, the US Food and Drug Administration approved the combination medication buprenorphine-naloxone (Sub-oxone) as only the second drug indicated for maintenance treatment of opioid dependence in the United States.
Buprenorphine differs from methadone in that it is a partial agonist at mu opiate receptors, and therefore has a “ceiling” or “plateau” effect in terms of dose-response and a much improved safety profile. Unlike methadone, buprenorphine can be prescribed in a doctor’s office and does not have to be dispensed at a government-approved clinic.
Unfortunately, buprenorphine-maintained patients seem to carry the same stigma in the recovery community as those maintained on methadone—that they are simply substituting one drug for another. Detractors usually fail to consider that, as with methadone, patients do not report getting “high” from taking buprenorphine. Patients will often state that when they first start taking it, they “feel something,” but after a few days of adjustment, they simply feel normal. They don’t feel high, they are no longer in withdrawal, their cravings are virtually eliminated, and their opiate receptors are effectively occupied and blocked, so there is no “high” in the event of a relapse.
What’s more, buprenorphine is not a medication that will help them deal with life’s stressors by “chemical coping.” Sober coping is a skill they must learn by actively participating in a solid 12-step-based recovery program and, in some cases, in psychotherapy. By removing the drug obsession, buprenorphine promotes and facilitates the important recovery goal of learning how to deal with life on life’s terms.
ADDICTION AS CHRONIC ILLNESS
Outcomes studies of addiction treatment have focused largely on rates of relapse after discharge from acute treatments such as residential rehabilitation, partial hospitalization, and intensive outpatient programs. With MAT, however, outcomes research has primarily looked at the duration of retention in treatment.
The change in focus between the two types of treatment coincides with a paradigm shift that views addiction as a chronic condition that requires ongoing care. Continued participation in prescribed care with demonstrated efficacy is considered to be the major indicator of success. Under the chronic illness model employed by MAT providers, if a patient reverted to briefly using a drug of abuse, this would be an issue to address in his ongoing treatment and would not necessarily indicate treatment failure as with the acute care model. Beyond retention rates, research has demonstrated that MAT with methadone results in reductions in rates of criminal activity, illicit drug use, acquisition of human immunodeficiency virus, and overall mortality.8–10
In outcomes studies, MAT has repeatedly shown better efficacy than abstinence-based approaches. During the first 5 years of its implementation, in 4,000 patients, methadone maintenance boasted 1-year retention rates exceeding 98%.11 Over the subsequent 3 years, with the number of patients approaching 35,000, the 1-year retention rates fell to around 60%—still far exceeding results of abstinence-based treatment and approximating the number cited in most modern studies.11
The retention rates in buprenorphine programs are similarly promising. Studies of 12 to 13 weeks duration have shown retention rates of 52% to 79%.12–15 Six-month studies have demonstrated retention rates of 43% to 100%.16–19 Another study showed that 38% of opiate-dependent patients remained in treatment with buprenorphine at 5 years.20 Surprisingly, most of the buprenorphine studies have been conducted in office-based practices, which are less structured than outpatient methadone programs.
MEDICATION-ASSISTED TREATMENT IS GAINING ACCEPTANCE
Data from decades of experience with MAT strongly support the conclusion that it is superior to abstinence-based approaches.
The importance of a patient staying in treatment cannot be overemphasized, as the consequence of failing in recovery may well be an early death. On average, heroin addicts lose about 18 years of life expectancy, and the mortality rate for injection users is roughly 2% per year.21 The mortality rate for heroin users is 6 to 20 times greater than for age-matched peers who are not drug users.22
As high as these numbers are, they are even higher for abusers of prescription narcotics. The annual death rate associated with opioid pain relievers (4.8 per 100,000) is nearly double that associated with illicit drugs (2.8 per 100,000).23
The recent and rather radical change in treatment philosophy by Hazelden came as a shock to some, a disappointment to others, and a welcome change to many who saw this as a move by one of the more respected treatment centers in the country to fall in line with the body of evidence that supports MAT for those suffering from opiate dependence. It remains a mystery why so many, if not most, addiction treatment centers in the United States cling to the abstinence-based philosophy despite the overwhelming data from decades of research and experience that show that abstinence does not work for the majority of opiate addicts.
Complete abstinence from opiate drugs of abuse and potentially addictive medications is a noble but perhaps unreachable goal for many sufferers. Hazelden’s announced acceptance of MAT gives credence to the value of recovery goals that are not entirely drug-free.
Dr. Dole was correct in stating that opiate addicts usually return to drugs if not provided with MAT. Treatment programs need to inform opiate-dependent patients that abstinence-based treatment offers only a 1 in 10 chance of success. Perhaps some patients, armed with the daunting statistics regarding abstinence, will be inspired to devote themselves wholeheartedly to their recovery in an effort to make it into that elite 10% group that achieves long-lasting recovery without the aid of medications. But for the other 90%, it is encouraging to hear that Hazelden, the model treatment center for most abstinence-based programs in this country, may now lead other abstinence-based centers to reconsider their treatment philosophies.
Historically, US doctors were not allowed by federal law to prescribe opiates for addiction treatment. With the passage of DATA 2000, buprenorphine (alone or in combination with naloxone) can be prescribed for addiction treatment only by providers who obtain a waiver from the US Drug Enforcement Administration (DEA). Any doctor can become qualified to prescribe buprenorphine or buprenorphinenaloxone after completing an 8-hour online training course (available at www.buppractice.com and at www.aaap.org/buprenorphine) and by obtaining a DATA 2000 waiver and a new prescribing number from the DEA. Doctors are initially limited to treating only 30 patients with buprenorphine-naloxone at any given time, but can apply for an extension to 100 patients after having had their waiver for 1 year.
As MAT continues to gain favor, demand will grow for more providers to obtain their waivers to prescribe buprenorphine and buprenorphine-naloxone. Historically, there have always been too few methadone clinics to meet the demand. One can hope that the growing number of waivered providers will greatly improve access to care by opiate addicts, no matter where they reside. Qualified prescribers of buprenorphine and buprenorphine-naloxone are limited by the federal restrictions on the numbers of patients they can treat. If the chronic disease of addiction is to be integrated into the continuing-care approach of modern medicine and managed alongside other chronic diseases, primary care providers who are not specialized in treating addiction will need to be become comfortable with maintaining patients on buprenorphine-naloxone.7 Presumably, such patients will have already been stabilized through participation in addiction treatment programs in their respective geographic areas. Primary care providers will need to develop relationships with local addictionologists and treatment programs so that they will be able to refer those in active addiction for induction and stabilization on MAT and will be able to refer those already stabilized on MAT back to such specialists when relapses occur.
We may finally be approaching a time when structured residential treatment and MAT are not mutually exclusive options for our patients. These treatment options must work together for optimal outcomes. Based on our experience with hundreds of patients at Cleveland Clinic’s Alcohol and Drug Recovery Center, we believe this change of treatment philosophy is long overdue. In clinical settings, patients do not fit cleanly into one treatment arm or another and often require a blended approach to effect long-lasting change. Hazelden’s shift of treatment philosophy is an indication that this research-supported viewpoint is gaining acceptance in the traditionally drug-free halls of addiction treatment programs.
- White WL. Slaying the Dragon. The History of Addiction Treatment and Recovery in America. Bloomington, IL: Chestnut Health Systems/Lighthouse Institute; 1998:124–125,201.
- Kosten TR, Gorelick DA. The Lexington narcotic farm. Am J Psychiatry 2002; 159:22.
- Hunt GH, Odoroff ME. Followup study of narcotic drug addicts after hospitalization. Public Health Rep 1962; 77:41–54.
- Dole VP. Narcotic addiction, physical dependence and relapse. N Engl J Med 1972; 286:988–992.
- Dole VP. Implications of methadone maintenance for theories of narcotic addiction. JAMA 1988; 260:3025–3029.
- Betty Ford Institute Consensus Panel. What is recovery? A working definition from the Betty Ford Institute. J Subst Abuse Treat 2007; 33:221–228.
- McLellan AT. Have we evaluated addiction treatment correctly? Implications from a chronic care perspective. Addiction 2002; 97:249–252.
- Grönbladh L, Ohlund LS, Gunne LM. Mortality in heroin addiction: impact of methadone treatment. Acta Psychiatr Scand 1990; 82:223–227.
- Ball JC, Lange WR, Myers CP, Friedman SR. Reducing the risk of AIDS through methadone maintenance treatment. J Health Soc Behav 1988; 29:214–226.
- Martin J, Zweben JE, Payte JT. Opioid maintenance treatment. In:Ries RK, Fiellin DA, Miller SC, Saitzeds R, editors. Principles of Addiction Medicine. 4th ed. Philadelphia, PA: Lippincottt Williams & Wilkins, 2009:671–688.
- Dole VP, Nyswander ME. Methadone maintenance treatment. A tenyear perspective. JAMA 1976; 235:2117–2119.
- Cunningham C, Giovanniello A, Sacajiu G, et al. Buprenorphine treatment in an urban community health center: what to expect. Fam Med 2008; 40:500–506.
- Fiellin DA, Pantalon MV, Pakes JP, O’Connor PG, Chawarski M, Schottenfeld RS. Treatment of heroin dependence with buprenorphine in primary care. Am J Drug Alcohol Abuse 2002; 28:231–241.
- Fudala PJ, Bridge TP, Herbert S, et al; Buprenorphine/Naloxone Collaborative Study Group. Office-based treatment of opiate addiction with a sublingual-tablet formulation of buprenorphine and naloxone. N Engl J Med 2003; 349:949–958.
- O’Connor PG, Oliveto AH, Shi JM, et al. A randomized trial of buprenorphine maintenance for heroin dependence in a primary care clinic for substance users versus a methadone clinic. Am J Med 1998; 105:100–105.
- Fiellin DA, Pantalon MV, Chawarski MC, et al. Counseling plus buprenorphine-naloxone maintenance therapy for opioid dependence. N Engl J Med 2006; 355:365–374.
- Moore BA, Fiellin DA, Barry DT, et al. Primary care office-based buprenorphine treatment: comparison of heroin and prescription opioid dependent patients. J Gen Intern Med 2007; 22:527–530.
- Mintzer IL, Eisenberg M, Terra M, MacVane C, Himmelstein DU, Woolhandler S. Treating opioid addiction with buprenorphine-naloxone in community-based primary care settings. Ann Fam Med 2007; 5:146–150.
- O’Connor PG, Oliveto AH, Shi JM, et al. A pilot study of primary-carebased buprenorphine maintenance for heroin dependence. Am J Drug Alcohol Abuse 1996; 22:523–531.
- Fiellin DA, Moore BA, Sullivan LE, et al. Long-term treatment with buprenorphine/naloxone in primary care: results at 2–5 years. Am J Addict 2008; 17:116–120.
- Smyth B, Hoffman V, Fan J, Hser YI. Years of potential life lost among heroin addicts 33 years after treatment. Prev Med 2007; 44:369–374.
- Sporer KA. Acute heroin overdose. Ann Intern Med 1999; 130:584–590.
- Centers for Disease Control and Prevention (CDC). Vital signs: overdoses of prescription opioid pain relievers—United States, 1999–2008. MMWR Morb Mortal Wkly Rep 2011; 60:1487–1492.
- White WL. Slaying the Dragon. The History of Addiction Treatment and Recovery in America. Bloomington, IL: Chestnut Health Systems/Lighthouse Institute; 1998:124–125,201.
- Kosten TR, Gorelick DA. The Lexington narcotic farm. Am J Psychiatry 2002; 159:22.
- Hunt GH, Odoroff ME. Followup study of narcotic drug addicts after hospitalization. Public Health Rep 1962; 77:41–54.
- Dole VP. Narcotic addiction, physical dependence and relapse. N Engl J Med 1972; 286:988–992.
- Dole VP. Implications of methadone maintenance for theories of narcotic addiction. JAMA 1988; 260:3025–3029.
- Betty Ford Institute Consensus Panel. What is recovery? A working definition from the Betty Ford Institute. J Subst Abuse Treat 2007; 33:221–228.
- McLellan AT. Have we evaluated addiction treatment correctly? Implications from a chronic care perspective. Addiction 2002; 97:249–252.
- Grönbladh L, Ohlund LS, Gunne LM. Mortality in heroin addiction: impact of methadone treatment. Acta Psychiatr Scand 1990; 82:223–227.
- Ball JC, Lange WR, Myers CP, Friedman SR. Reducing the risk of AIDS through methadone maintenance treatment. J Health Soc Behav 1988; 29:214–226.
- Martin J, Zweben JE, Payte JT. Opioid maintenance treatment. In:Ries RK, Fiellin DA, Miller SC, Saitzeds R, editors. Principles of Addiction Medicine. 4th ed. Philadelphia, PA: Lippincottt Williams & Wilkins, 2009:671–688.
- Dole VP, Nyswander ME. Methadone maintenance treatment. A tenyear perspective. JAMA 1976; 235:2117–2119.
- Cunningham C, Giovanniello A, Sacajiu G, et al. Buprenorphine treatment in an urban community health center: what to expect. Fam Med 2008; 40:500–506.
- Fiellin DA, Pantalon MV, Pakes JP, O’Connor PG, Chawarski M, Schottenfeld RS. Treatment of heroin dependence with buprenorphine in primary care. Am J Drug Alcohol Abuse 2002; 28:231–241.
- Fudala PJ, Bridge TP, Herbert S, et al; Buprenorphine/Naloxone Collaborative Study Group. Office-based treatment of opiate addiction with a sublingual-tablet formulation of buprenorphine and naloxone. N Engl J Med 2003; 349:949–958.
- O’Connor PG, Oliveto AH, Shi JM, et al. A randomized trial of buprenorphine maintenance for heroin dependence in a primary care clinic for substance users versus a methadone clinic. Am J Med 1998; 105:100–105.
- Fiellin DA, Pantalon MV, Chawarski MC, et al. Counseling plus buprenorphine-naloxone maintenance therapy for opioid dependence. N Engl J Med 2006; 355:365–374.
- Moore BA, Fiellin DA, Barry DT, et al. Primary care office-based buprenorphine treatment: comparison of heroin and prescription opioid dependent patients. J Gen Intern Med 2007; 22:527–530.
- Mintzer IL, Eisenberg M, Terra M, MacVane C, Himmelstein DU, Woolhandler S. Treating opioid addiction with buprenorphine-naloxone in community-based primary care settings. Ann Fam Med 2007; 5:146–150.
- O’Connor PG, Oliveto AH, Shi JM, et al. A pilot study of primary-carebased buprenorphine maintenance for heroin dependence. Am J Drug Alcohol Abuse 1996; 22:523–531.
- Fiellin DA, Moore BA, Sullivan LE, et al. Long-term treatment with buprenorphine/naloxone in primary care: results at 2–5 years. Am J Addict 2008; 17:116–120.
- Smyth B, Hoffman V, Fan J, Hser YI. Years of potential life lost among heroin addicts 33 years after treatment. Prev Med 2007; 44:369–374.
- Sporer KA. Acute heroin overdose. Ann Intern Med 1999; 130:584–590.
- Centers for Disease Control and Prevention (CDC). Vital signs: overdoses of prescription opioid pain relievers—United States, 1999–2008. MMWR Morb Mortal Wkly Rep 2011; 60:1487–1492.
KEY POINTS
- Recidivism rates are high after detoxification without medication-assisted treatment.
- Whether staying in a maintenance program truly constitutes recovery continues to be debated, but patients on methadone or buprenorphine maintenance do not report getting “high”—they merely feel normal.
- Methadone is dispensed only in special clinics, whereas buprenorphine can be prescribed by a physician. Prescribing physicians must complete an 8-hour course online at www.buppractice.com or www.aaap.org/buprenorphine and obtain a waiver from the US Drug Enforcement Administration.
- With or without medication-assisted treatment, recovering addicts must learn the skill of sober coping by actively participating in a solid 12-step-based program and, in some cases, in psychotherapy.
Aspirin: Its risks, benefits, and optimal use in preventing cardiovascular events
A 57-year-old woman with no history of cardiovascular disease comes to the clinic for her annual evaluation. She does not have diabetes mellitus, but she does have hypertension and chronic osteoarthritis, currently treated with acetaminophen. Additionally, she admits to active tobacco use. Her systolic blood pressure is 130 mm Hg on therapy with hydrochlorothiazide. Her electrocardiogram demonstrates left ventricular hypertrophy. Her low-density lipoprotein (LDL) cholesterol level is 140 mg/dL, and her high-density lipoprotein (HDL) cholesterol level is 50 mg/dL. Should this patient be started on aspirin therapy?
Acetylsalicylic acid (aspirin) is an analgesic, antipyretic, and anti-inflammatory agent, but its more prominent use today is as an antithrombotic agent to treat or prevent cardiovascular events. Its antithrombotic properties are due to its effects on the enzyme cyclooxygenase. However, cyclooxygenase is also involved in regulation of the gastric mucosa, and so aspirin increases the risk of gastrointestinal bleeding.
Approximately 50 million people take aspirin on a daily basis to treat or prevent cardiovascular disease.1 Of these, at least half are taking more than 100 mg per day,2 reflecting the general belief that, for aspirin dosage, “more is better”—which is not true.
Additionally, recommendations about the use of aspirin were based on studies that included relatively few members of several important subgroups, such as people with diabetes without known cardiovascular disease, women, and the elderly, and thus may not reflect appropriate indications and dosages for these groups.
Here, we examine the literature, outline an individualized approach to aspirin therapy, and highlight areas for future study.
HISTORY OF ASPIRIN USE IN CARDIOVASCULAR DISEASE
- 1700s—Willow bark is used as an analgesic.
- 1897—Synthetic aspirin is developed as an antipyretic and anti-inflammatory agent.
- 1974—First landmark trial of aspirin for secondary prevention of myocardial infarction.3
- 1982—Nobel Prize awarded for discovery of aspirin mechanism.
- 1985—US Food and Drug Administration approves aspirin for the treatment and secondary prevention of acute myocardial infarction.
- 1998—The Second International Study of Infarct Survival (ISIS-2) finds that giving aspirin to patients with myocardial infarction within 24 hours of presentation leads to a significant reduction in vascular deaths.4
Ongoing uncertainties
Aspirin now carries a class I indication for all patients with suspected myocardial infarction. Since there are an estimated 600,000 new coronary events and 325,000 recurrent ischemic events per year in the United States,5 the need for aspirin will continue to remain great. It is also approved to prevent and treat stroke and in patients with unstable angina.

However, questions continue to emerge about aspirin’s dosing and appropriate use in specific populations. The initial prevention trials used a wide range of doses and, as mentioned, included few women, few people with diabetes, and few elderly people. The uncertainties are especially pertinent for patients without known vascular disease, in whom the absolute risk reduction is much less, making the assessment of bleeding risk particularly important. Furthermore, the absolute risk-to-benefit assessment may be different in certain populations.
Guidelines on the use of aspirin to prevent cardiovascular disease (Table 1)6–10 have evolved to take into account these possible disparities, and studies are taking place to further investigate aspirin use in these groups.
ASPIRIN AND GASTROINTESTINAL BLEEDING
Aspirin’s association with bleeding, particularly gastrointestinal bleeding, was recognized early as a use-limiting side effect. With or without aspirin, gastrointestinal bleeding is a common cause of morbidity and death, with an incidence of approximately 100 per 100,000 bleeding episodes in adults per year for upper gastrointestinal bleeding and 20 to 30 per 100,000 per year for lower gastrointestinal bleeding.11,12
The standard dosage (ie, 325 mg/day) is associated with a significantly higher risk of gastrointestinal bleeding (including fatal bleeds) than is 75 mg.13 However, even with lower doses, the risk of gastrointestinal bleeding is estimated to be twice as high as with no aspirin.14
And here is the irony: studies have shown that higher doses of aspirin offer no advantage in preventing thrombotic events compared with lower doses.15 For example, the Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent Events/Organization to Assess Strategies for Ischemic Stroke Syndromes study reported a higher rate of gastrointestinal bleeding with standard-dose aspirin therapy than with low-dose aspirin, with no additional cardiovascular benefit with the higher dose.16

Furthermore, several other risk factors increase the risk of gastrointestinal bleeding with aspirin use (Table 2). These risk factors are common in the general population but were not necessarily represented in participants in clinical trials. Thus, estimates of risk based on trial data most likely underestimate actual risk in the general population, and therefore, the individual patient’s risk of gastrointestinal bleeding, based on these and other factors, needs to be taken into consideration.
ASPIRIN IN PATIENTS WITH CORONARY ARTERY DISEASE
Randomized clinical trials have validated the benefits of aspirin in secondary prevention of cardiovascular events in patients who have had a myocardial infarction. Patients with coronary disease who withdraw from aspirin therapy or otherwise do not adhere to it have a risk of cardiovascular events three times higher than those who stay with it.17
Despite the strong data, however, several issues and questions remain about the use of aspirin for secondary prevention.
Bleeding risk must be considered, since gastrointestinal bleeding is associated with a higher risk of death and myocardial infarction in patients with cardiovascular disease.18 Many patients with coronary disease are on more than one antiplatelet or anticoagulant therapy for concomitant conditions such as atrial fibrillation or because they underwent a percutaneous intervention, which further increases the risk of bleeding.
This bleeding risk is reflected in changes in the most recent recommendations for aspirin dosing after percutaneous coronary intervention. Earlier guidelines advocated use of either 162 or 325 mg after the procedure. However, the most recent update (in 2011) now supports 81 mg for maintenance dosing after intervention.7
Patients with coronary disease but without prior myocardial infarction or intervention. Current guidelines recommend 75 to 162 mg of aspirin in all patients with coronary artery disease.6 However, this group is diverse and includes patients who have undergone percutaneous coronary intervention, patients with chronic stable angina, and patients with asymptomatic coronary artery disease found on imaging studies. The magnitude of benefit is not clear for those who have no symptoms or who have stable angina.
Most of the evidence supporting aspirin use in chronic angina came from a single trial in Sweden, in which 2,000 patients with chronic stable angina were given either 75 mg daily or placebo. Those who received aspirin had a 34% lower rate of myocardial infarction and sudden death.19
A substudy of the Physicians’ Health Study, with fewer patients, also noted a significant reduction in the rate of first myocardial infarction. The dose of aspirin in this study was 325 mg every other day.20
In the Women’s Health Initiative Observational Study, 70% of women with stable cardiovascular disease taking aspirin were taking 325 mg daily.21 This study demonstrated a significant reduction in the cardiovascular mortality rate, which supports current guidelines, and found no difference in outcomes with doses of 81 mg compared with 325 mg.21 This again corroborates that low-dose aspirin is preferential to standard-dose aspirin in women with cardiovascular disease.
These findings have not been validated in larger prospective trials. Thus, current guidelines for aspirin use may reflect extrapolation of aspirin benefit from higher-risk patients to lower-risk patients.
Nevertheless, although the debate continues, it has generally been accepted that in patients who are at high risk of vascular disease or who have had a myocardial infarction, the benefits of aspirin—a 20% relative reduction in vascular events22—clearly outweigh the risks.
ASPIRIN FOR PRIMARY PREVENTION
Assessing risk vs benefit is more complex when considering populations without known cardiovascular disease.
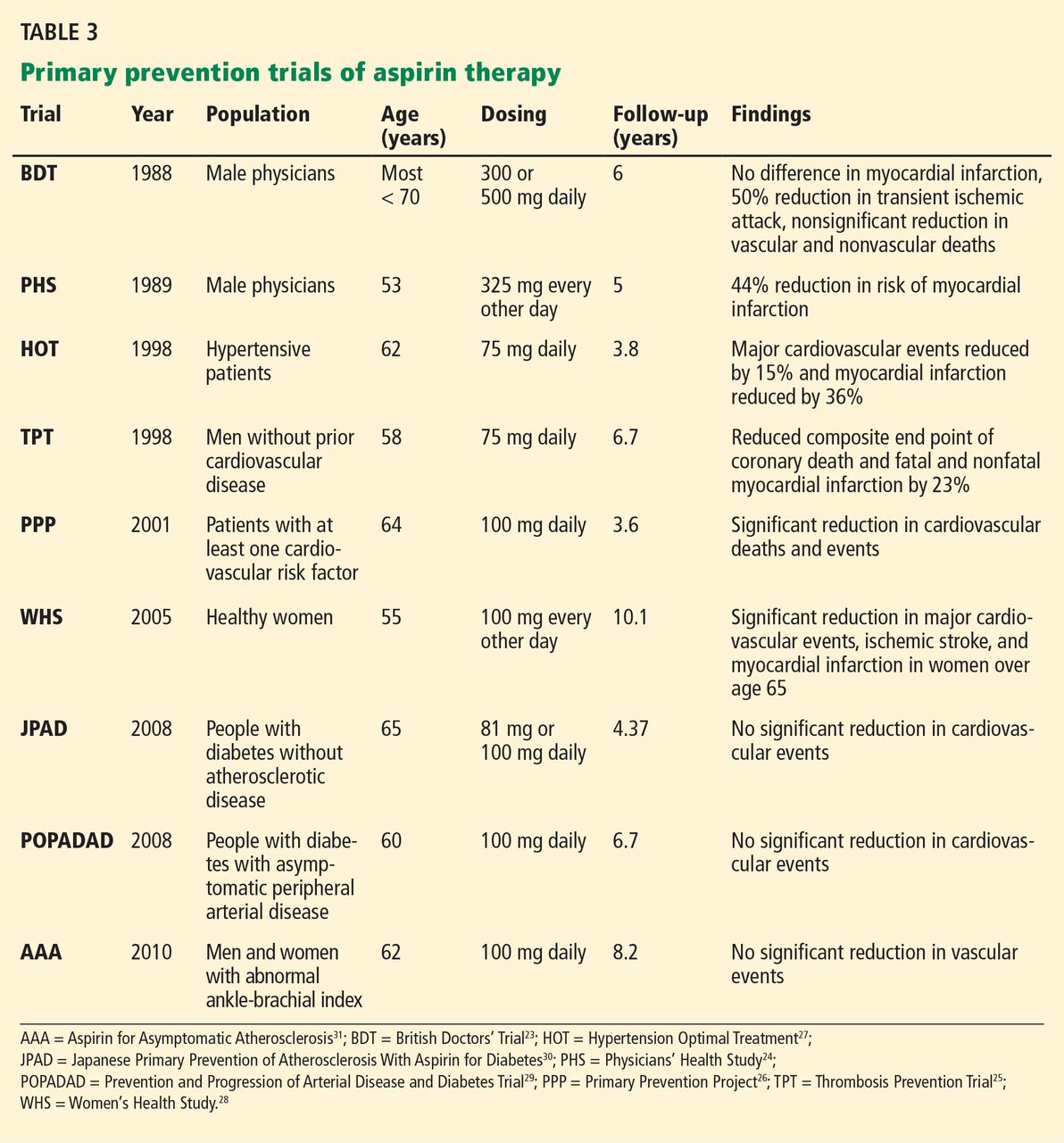
Only a few studies have specifically evaluated the use of aspirin for primary prevention (Table 3).23–31 The initial trials were in male physicians in the United Kingdom and the United States in the late 1980s and had somewhat conflicting results. A British study did not find a significant reduction in myocardial infarction,23 but the US Physician’s Health Study study did: the relative risk was 0.56 (95% confidence interval 0.45–0.70, P < .00001).24 The US study had more than four times the number of participants, used different dosing (325 mg every other day compared with 500 or 300 mg daily in the British study), and had a higher rate of compliance.
Several studies over the next decade demonstrated variable but significant reductions in cardiovascular events as well.25–27
A meta-analysis of primary prevention trials of aspirin was published in 2009.22 Although the relative risk reduction was similar in primary and secondary prevention, the absolute risk reduction in primary prevention was not nearly as great as in secondary prevention.
These findings are somewhat difficult to interpret, as the component trials included a wide spectrum of patients, ranging from healthy people with no symptoms and no known risk factors to those with limited risk factors. The trials were also performed over several decades during which primary prevention strategies were evolving. Additionally, most of the participants were middle-aged, nondiabetic men, so the results may not necessarily apply to people with diabetes, to women, or to the elderly. Thus, the pooled data in favor of aspirin for primary prevention may not be as broadly applicable to the general population as was once thought.
Aspirin for primary prevention in women
Guidelines for aspirin use in primary prevention were initially thought to be equally applicable to both sexes. However, concerns about the relatively low number of women participating in the studies and the possible mechanistic differences in aspirin efficacy in men vs women prompted further study.
A meta-analysis of randomized controlled trials found that aspirin was associated with a 12% relative reduction in the incidence of cardiovascular events in women and 14% in men. On the other hand, for stroke, the relative risk reduction was 17% in women, while men had no benefit.32
Most of the women in this meta-analysis were participants in the Women’s Health Study, and they were at low baseline risk.28 Although only about 10% of patients in this study were over age 65, this older group accounted for most of the benefit: these older women had a 26% risk reduction in major adverse cardiovascular events and 30% reduction in stroke.

Thus, for women, aspirin seems to become effective for primary prevention at an older age than in men, and the guidelines have been changed accordingly (Figure 1).
More women should be taking aspirin than actually are. For example, Rivera et al33 found that only 41% of eligible women were receiving aspirin for primary prevention and 48% of eligible women were receiving it for secondary prevention.
People with diabetes
People with diabetes without overt cardiovascular disease are at higher risk of cardiovascular events than age- and sex-matched controls.34 On the other hand, people with diabetes may be more prone to aspirin resistance and may not derive as much cardiovascular benefit from aspirin.
Early primary prevention studies included few people with diabetes. Subsequent meta-analyses of trials that used a wide range of aspirin doses found a relative risk reduction of 9%, which was not statistically significant.9,35,36
But there is some evidence that people with diabetes, with37 and without22 coronary disease, may be at higher inherent risk of bleeding than people without diabetes. Although aspirin may not necessarily increase the risk of bleeding in diabetic patients, recent data suggest no benefit in terms of a reduction in vascular events.38
The balance of risk vs benefit for aspirin in this special population is not clear, although some argue that these patients should be treated somewhere on the spectrum of risk between primary and secondary prevention.
The US Preventive Services Task Force did not differentiate between people with or without diabetes in its 2009 guidelines for aspirin for primary prevention.8 However, the debate is reflected in a change in 2010 American College of Cardiology/American Diabetes Association guidelines regarding aspirin use in people with diabetes without known cardiovascular disease.39 As opposed to earlier recommendations from these organizations in favor of aspirin for all people with diabetes regardless of 10-year risk, current recommendations advise low-dose aspirin (81–162 mg) for diabetic patients without known vascular disease who have a greater than 10% risk of a cardiovascular event and are not at increased risk of bleeding.
These changes were based on the findings of two trials: the Prevention and Progression of Arterial Disease and Diabetes Trial (POPADAD) and the Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes (JPAD) study. These did not show a statistically significant benefit in prevention of cardiovascular events with aspirin.29,30
After the new guidelines came out, a meta-analysis further bolstered its recommendations. 40 In seven randomized clinical trials in 11,000 patients, the relative risk reduction was 9% with aspirin, which did not reach statistical significance.
Statins may dilute the benefit of aspirin
The use of statins has been increasing, and this trend may have played a role in the marginal benefit of aspirin therapy in these recent studies. In the Japanese trial, approximately 25% of the patients were known to be using a statin; the percentage of statin use was not reported specifically in POPADAD, but both of these studies were published in 2008, when the proportion of diabetic patients taking a statin would be expected to be higher than in earlier primary prevention trials, which were performed primarily in the 1990s. Thus, the beneficial effects of statins may have somewhat diluted the risk reduction attributable to aspirin.
Trials under way in patients with diabetes
The evolving and somewhat conflicting guidelines highlight the need for further study in patients with diabetes. To address this area, two trials are in progress: the Aspirin and Simvastatin Combination for Cardiovascular Events Prevention Trial in Diabetes (ACCEPT-D) and A Study of Cardiovascular Events in Diabetes (ASCEND).41,42
ACCEPT-D is testing low-dose aspirin (100 mg daily) in diabetic patients who are also on simvastatin. This study also includes prespecified subgroups stratified by sex, age, and baseline lipid levels.
The ASCEND trial will use the same aspirin dose as ACCEPT-D, with a target enrollment of 10,000 patients with diabetes without known vascular disease.
More frequent dosing for people with diabetes?
Although not supported by current guidelines, recent work has suggested that people with diabetes may need more-frequent dosing of aspirin.43 This topic warrants further investigation.
Aspirin as primary prevention in elderly patients
The incidence of cardiovascular events increases with age37—but so does the incidence of gastrointestinal bleeding.44 Upper gastrointestinal bleeding is especially worrisome in the elderly, in whom the estimated case-fatality rate is high.12 Assessment of risk and benefit is particularly important in patients over age 65 without known coronary disease.
Uncertainty about aspirin use in this population is reflected in the most recent US Preventive Services Task Force guidelines, which do not advocate either for or against regular aspirin use for primary prevention in those over the age of 80.
Data on this topic from clinical trials are limited. The Antithrombotic Trialists’ Collaboration (2009) found that although age is associated with a risk of major coronary events similar to that of other traditional risk factors such as diabetes, hypertension, and tobacco use, older age is also associated with the highest risk of major extracranial bleeding.22
Because of the lack of data in this population, several studies are currently under way. The Aspirin in Reducing Events in the Elderly (ASPREE) trial is studying 100 mg daily in nondiabetic patients without known cardiovascular disease who are age 70 and older.45 An additional trial will study patients age 60 to 85 with concurrent diagnoses of hypertension, hyperlipidemia, or diabetes and will test the same aspirin dose as in ASPREE.46 These trials should provide further insight into the safety and efficacy of aspirin for primary prevention in the elderly.
FUTURE DIRECTIONS
Aspirin remains a cornerstone of therapy in patients with cardiovascular disease and in secondary prevention of adverse cardiovascular events, but its role in primary prevention remains under scrutiny. Recommendations have evolved to reflect emerging data in special populations, and an algorithm based on Framingham risk assessment in men for myocardial infarction and ischemic stroke assessment in women for assessing appropriateness of aspirin therapy based on currently available guidelines is presented in Figure 1.6,8,47–49 Targeted studies have advanced our understanding of aspirin use in women, and future studies in people with diabetes and in the elderly should provide further insight into the role of aspirin for primary prevention in these specific groups as well.
Additionally, the range of doses used in clinical studies has propagated the general misperception that higher doses of aspirin are more efficacious. Future studies should continue to use lower doses of aspirin to minimize bleeding risk with an added focus on re-examining its net benefit in the modern era of increasing statin use, which may reduce the absolute risk reduction attributable to aspirin.
One particular area of debate is whether enteric coating can result in functional aspirin resistance. Grosser et al50 found that sustained aspirin resistance was rare, and “pseudoresistance” was related to the use of a single enteric-coated aspirin instead of immediate-release aspirin in people who had not been taking aspirin up to then. This complements an earlier study, which found that enteric-coated aspirin had an appropriate effect when given for 7 days.51 Therefore, for patients who have not been taking aspirin, the first dose should always be immediate-release, not enteric coated.
SHOULD OUR PATIENT RECEIVE ASPIRIN?
The patient we described at the beginning of this article has several risk factors—hypertension, dyslipidemia, left ventricular hypertrophy, and smoking—but no known cardiovascular disease as yet. Her risk of an adverse cardiovascular event appears moderate. However, her 10-year risk of stroke by the Framingham risk calculation is 10%, which would qualify her for aspirin for primary prevention. Of particular note is that the significance of left ventricular hypertrophy as a risk factor for stroke in women is higher than in men and in our case accounts for half of this patient’s risk.
We should explain to the patient that the anticipated benefits of aspirin for stroke prevention outweigh bleeding risks, and thus aspirin therapy would be recommended. However, with her elevated LDL-cholesterol, she may benefit from a statin, which could lessen the relative risk reduction from additional aspirin use.
- Chan FK, Graham DY. Review article: prevention of non-steroidal anti-inflammatory drug gastrointestinal complications—review and recommendations based on risk assessment. Aliment Pharmacol Ther 2004; 19:1051–1061.
- Peters RJ, Mehta SR, Fox KA, et al; Clopidogrel in Unstable angina to prevent Recurrent Events (CURE) Trial Investigators. Effects of aspirin dose when used alone or in combination with clopidogrel in patients with acute coronary syndromes: observations from the Clopidogrel in Unstable angina to prevent Recurrent Events (CURE) study. Circulation 2003; 108:1682–1687.
- Elwood PC, Cochrane AL, Burr ML, et al. A randomized controlled trial of acetyl salicylic acid in the secondary prevention of mortality from myocardial infarction. Br Med J 1974; 1:436–440.
- Baigent C, Collins R, Appleby P, Parish S, Sleight P, Peto R. ISIS-2: 10 year survival among patients with suspected acute myocardial infarction in randomised comparison of intravenous streptokinase, oral aspirin, both, or neither. The ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. BMJ 1998; 316:1337–1343.
- Roger VL, Go AS, Lloyd-Jones DM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2011 update: a report from the American Heart Association. Circulation 2011; 123:e18–e209.
- Smith SC, Benjamin EJ, Bonow RO, et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation endorsed by the World Heart Federation and the Preventive Cardiovascular Nurses Association. J Am Coll Cardiol 2011; 58:2432–2446.
- Levine GN, Bates ER, Blankenship JC, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; Society for Cardiovascular Angiography and Interventions. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol 2011; 58:e44–e122.
- US Preventive Services Task Force. Aspirin for the prevention of cardiovascular disease: US Preventive Services Task Force recommendation statement. Ann Intern Med 2009; 150:396–404.
- Pignone M, Alberts MJ, Colwell JA, et al. Aspirin for primary prevention of cardiovascular events in people with diabetes: a position statement of the American Diabetes Association, a scientific statement of the American Heart Association, and an expert consensus document of the American College of Cardiology Foundation. Circulation 2010; 121:2694–2701.
- Mosca L, Benjamin EJ, Berra K, et al. Effectiveness-based guidelines for the prevention of cardiovascular disease in women—2011 update: a guideline from the American Heart Association. Circulation 2011; 123:1243–1262.
- Strate LL. Lower GI bleeding: epidemiology and diagnosis. Gastroenterol Clin North Am 2005; 34:643–664.
- Rockall TA, Logan RF, Devlin HB, Northfield TC. Incidence of and mortality from acute upper gastrointestinal haemorrhage in the United Kingdom. Steering Committee and members of the National Audit of Acute Upper Gastrointestinal Haemorrhage. BMJ 1995; 311:222–226.
- Campbell CL, Smyth S, Montalescot G, Steinhubl SR. Aspirin dose for the prevention of cardiovascular disease: a systematic review. JAMA 2007; 297:2018–2024.
- Weil J, Colin-Jones D, Langman M, et al. Prophylactic aspirin and risk of peptic ulcer bleeding. BMJ 1995; 310:827–830.
- Reilly IA, FitzGerald GA. Inhibition of thromboxane formation in vivo and ex vivo: implications for therapy with platelet inhibitory drugs. Blood 1987; 69:180–186.
- CURRENT-OASIS 7 Investigators; Mehta SR, Bassand JP, Chrolavicius S, et al. Dose comparisons of clopidogrel and aspirin in acute coronary syndromes. N Engl J Med 2010; 363:930–942.
- Biondi-Zoccai GG, Lotrionte M, Agostoni P, et al. A systematic review and meta-analysis on the hazards of discontinuing or not adhering to aspirin among 50,279 patients at risk for coronary artery disease. Eur Heart J 2006; 27:2667–2674.
- Berger PB, Bhatt DL, Fuster V, et al; CHARISMA Investigators. Bleeding complications with dual antiplatelet therapy among patients with stable vascular disease or risk factors for vascular disease: results from the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial. Circulation 2010; 121:2575–2583.
- Juul-Möller S, Edvardsson N, Jahnmatz B, Rosén A, Sørensen S, Omblus R. Double-blind trial of aspirin in primary prevention of myocardial infarction in patients with stable chronic angina pectoris. The Swedish Angina Pectoris Aspirin Trial (SAPAT) Group. Lancet 1992; 340:1421–1425.
- Ridker PM, Manson JE, Gaziano JM, Buring JE, Hennekens CH. Low-dose aspirin therapy for chronic stable angina. A randomized, placebo-controlled clinical trial. Ann Intern Med 1991; 114:835–839.
- Berger JS, Brown DL, Burke GL, et al. Aspirin use, dose, and clinical outcomes in postmenopausal women with stable cardiovascular disease: the Women’s Health Initiative Observational Study. Circ Cardiovasc Qual Outcomes 2009; 2:78–87.
- Antithrombotic Trialists’ (ATT) Collaboration; Baigent C, Blackwell L, Collins R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet 2009; 373:1849–1860.
- Peto R, Gray R, Collins R, et al. Randomised trial of prophylactic daily aspirin in British male doctors. Br Med J (Clin Res Ed) 1988; 296:313–316.
- Final report on the aspirin component of the ongoing Physicians’ Health Study. Steering Committee of the Physicians’ Health Study Research Group. N Engl J Med 1989; 321:129–135.
- Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. The Medical Research Council’s General Practice Research Framework. Lancet 1998; 351:233–241.
- de Gaetano GCollaborative Group of the Primary Prevention Project. Low-dose aspirin and vitamin E in people at cardiovascular risk: a randomised trial in general practice. Collaborative Group of the Primary Prevention Project. Lancet 2001; 357:89–95.
- Hansson L, Zanchetti A, Carruthers SG, et al. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. HOT Study Group. Lancet 1998; 351:1755–1762.
- Ridker PM, Cook NR, Lee IM, et al. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med 2005; 352:1293–1304.
- Belch J, MacCuish A, Campbell I, et al; Prevention of Progression of Arterial Disease and Diabetes Study Group; Diabetes Registry Group; Royal College of Physicians Edinburgh. The prevention of progression of arterial disease and diabetes (POPADAD) trial: factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ 2008; 337:a1840.
- Ogawa H, Nakayama M, Morimoto T, et al; Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes (JPAD) Trial Investigators. Low-dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: a randomized controlled trial. JAMA 2008; 300:2134–2141.
- Fowkes FG, Price JF, Stewart MC, et al; Aspirin for Asymptomatic Atherosclerosis Trialists. Aspirin for prevention of cardiovascular events in a general population screened for a low ankle brachial index: a randomized controlled trial. JAMA 2010; 303:841–848.
- Berger JS, Roncaglioni MC, Avanzini F, Pangrazzi I, Tognoni G, Brown DL. Aspirin for the primary prevention of cardiovascular events in women and men: a sex-specific meta-analysis of randomized controlled trials. JAMA 2006; 295:306–313.
- Rivera CM, Song J, Copeland L, Buirge C, Ory M, McNeal CJ. Underuse of aspirin for primary and secondary prevention of cardiovascular disease events in women. J Womens Health (Larchmt) 2012; 21:379–387.
- Wilson R, Gazzala J, House J. Aspirin in primary and secondary prevention in elderly adults revisited. South Med J 2012; 105:82–86.
- De Berardis G, Sacco M, Strippoli GF, et al. Aspirin for primary prevention of cardiovascular events in people with diabetes: meta-analysis of randomised controlled trials. BMJ 2009; 339:b4531.
- Zhang C, Sun A, Zhang P, et al. Aspirin for primary prevention of cardiovascular events in patients with diabetes: a meta-analysis. Diabetes Res Clin Pract 2010; 87:211–218.
- Moukarbel GV, Signorovitch JE, Pfeffer MA, et al. Gastrointestinal bleeding in high risk survivors of myocardial infarction: the VALIANT Trial. Eur Heart J 2009; 30:2226–2232.
- De Berardis G, Lucisano G, D’Ettorre A, et al. Association of aspirin use with major bleeding in patients with and without diabetes. JAMA 2012; 307:2286–2294.
- Pignone M, Alberts MJ, Colwell JA, et al; American Diabetes Association; American Heart Association; American College of Cardiology Foundation. Aspirin for primary prevention of cardiovascular events in people with diabetes. J Am Coll Cardiol 2010; 55:2878–2886.
- Butalia S, Leung AA, Ghali WA, Rabi DM. Aspirin effect on the incidence of major adverse cardiovascular events in patients with diabetes mellitus: a systematic review and meta-analysis. Cardiovasc Diabetol 2011; 10:25.
- De Berardis G, Sacco M, Evangelista V, et al; ACCEPT-D Study Group. Aspirin and Simvastatin Combination for Cardiovascular Events Prevention Trial in Diabetes (ACCEPT-D): design of a randomized study of the efficacy of low-dose aspirin in the prevention of cardiovascular events in subjects with diabetes mellitus treated with statins. Trials 2007; 8:21.
- British Heart Foundation. ASCEND: A Study of Cardiovascular Events in Diabetes. http://www.ctsu.ox.ac.uk/ascend. Accessed April 1, 2013.
- Rocca B, Santilli F, Pitocco D, et al. The recovery of platelet cyclooxygenase activity explains interindividual variability in responsiveness to low-dose aspirin in patients with and without diabetes. J Thromb Haemost 2012; 10:1220–1230.
- Hernández-Díaz S, Garcia Rodriguez LA. Cardioprotective aspirin users and their excess risk for upper gastrointestinal complications. BMC Med 2006; 4:22.
- Nelson MR, Reid CM, Ames DA, et al. Feasibility of conducting a primary prevention trial of low-dose aspirin for major adverse cardiovascular events in older people in Australia: results from the ASPirin in Reducing Events in the Elderly (ASPREE) pilot study. Med J Aust 2008; 189:105–109.
- Teramoto T, Shimada K, Uchiyama S, et al. Rationale, design, and baseline data of the Japanese Primary Prevention Project (JPPP)—a randomized, open-label, controlled trial of aspirin versus no aspirin in patients with multiple risk factors for vascular events. Am Heart J 2010; 159:361–369.e4.
- Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology; American Heart Association; Canadian Cardiovascular Society. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction—executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to revise the 1999 guidelines for the management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:671–719.
- Brott TG, Halperin JL, Abbara S, et al. 2011 ASA/ACCF/AHA/AANN/AANS/ACR/ASNR/CNS/SAIP/SCAI/SIR/SNIS/SVM/SVS Guideline on the Management of Patients With Extracranial Carotid and Vertebral Artery Disease A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, and the American Stroke Association, American Association of Neuroscience Nurses, American Association of Neurological Surgeons, American College of Radiology, American Society of Neuroradiology, Congress of Neurological Surgeons, Society of Atherosclerosis Imaging and Prevention, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of NeuroInterventional Surgery, Society for Vascular Medicine, and Society for Vascular Surgery Developed in Collaboration With the American Academy of Neurology and Society of Cardiovascular Computed Tomography. J Am Coll Cardiol 2011; 57:e16–e94.
- Wright RS, Anderson JL, Adams CD, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2011 ACCF/AHA focused update incorporated into the ACC/AHA 2007 Guidelines for the Management of Patients with Unstable Angina/Non-ST-Elevation Myocardial Infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines developed in collaboration with the American Academy of Family Physicians, Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons. J Am Coll Cardiol 2011; 57:e215–e367.
- Grosser T, Fries S, Lawson JA, Kapoor SC, Grant GR, Fitzgerald GA. Drug resistance and pseudoresistance: An unintended consequence of enteric coating aspirin. Circulation 2012; Epub ahead of print.
- Karha J, Rajagopal V, Kottke-Marchant K, Bhatt DL. Lack of effect of enteric coating on aspirin-induced inhibition of platelet aggregation in healthy volunteers. Am Heart J 2006; 151:976.e7–e11.
A 57-year-old woman with no history of cardiovascular disease comes to the clinic for her annual evaluation. She does not have diabetes mellitus, but she does have hypertension and chronic osteoarthritis, currently treated with acetaminophen. Additionally, she admits to active tobacco use. Her systolic blood pressure is 130 mm Hg on therapy with hydrochlorothiazide. Her electrocardiogram demonstrates left ventricular hypertrophy. Her low-density lipoprotein (LDL) cholesterol level is 140 mg/dL, and her high-density lipoprotein (HDL) cholesterol level is 50 mg/dL. Should this patient be started on aspirin therapy?
Acetylsalicylic acid (aspirin) is an analgesic, antipyretic, and anti-inflammatory agent, but its more prominent use today is as an antithrombotic agent to treat or prevent cardiovascular events. Its antithrombotic properties are due to its effects on the enzyme cyclooxygenase. However, cyclooxygenase is also involved in regulation of the gastric mucosa, and so aspirin increases the risk of gastrointestinal bleeding.
Approximately 50 million people take aspirin on a daily basis to treat or prevent cardiovascular disease.1 Of these, at least half are taking more than 100 mg per day,2 reflecting the general belief that, for aspirin dosage, “more is better”—which is not true.
Additionally, recommendations about the use of aspirin were based on studies that included relatively few members of several important subgroups, such as people with diabetes without known cardiovascular disease, women, and the elderly, and thus may not reflect appropriate indications and dosages for these groups.
Here, we examine the literature, outline an individualized approach to aspirin therapy, and highlight areas for future study.
HISTORY OF ASPIRIN USE IN CARDIOVASCULAR DISEASE
- 1700s—Willow bark is used as an analgesic.
- 1897—Synthetic aspirin is developed as an antipyretic and anti-inflammatory agent.
- 1974—First landmark trial of aspirin for secondary prevention of myocardial infarction.3
- 1982—Nobel Prize awarded for discovery of aspirin mechanism.
- 1985—US Food and Drug Administration approves aspirin for the treatment and secondary prevention of acute myocardial infarction.
- 1998—The Second International Study of Infarct Survival (ISIS-2) finds that giving aspirin to patients with myocardial infarction within 24 hours of presentation leads to a significant reduction in vascular deaths.4
Ongoing uncertainties
Aspirin now carries a class I indication for all patients with suspected myocardial infarction. Since there are an estimated 600,000 new coronary events and 325,000 recurrent ischemic events per year in the United States,5 the need for aspirin will continue to remain great. It is also approved to prevent and treat stroke and in patients with unstable angina.
However, questions continue to emerge about aspirin’s dosing and appropriate use in specific populations. The initial prevention trials used a wide range of doses and, as mentioned, included few women, few people with diabetes, and few elderly people. The uncertainties are especially pertinent for patients without known vascular disease, in whom the absolute risk reduction is much less, making the assessment of bleeding risk particularly important. Furthermore, the absolute risk-to-benefit assessment may be different in certain populations.
Guidelines on the use of aspirin to prevent cardiovascular disease (Table 1)6–10 have evolved to take into account these possible disparities, and studies are taking place to further investigate aspirin use in these groups.
ASPIRIN AND GASTROINTESTINAL BLEEDING
Aspirin’s association with bleeding, particularly gastrointestinal bleeding, was recognized early as a use-limiting side effect. With or without aspirin, gastrointestinal bleeding is a common cause of morbidity and death, with an incidence of approximately 100 per 100,000 bleeding episodes in adults per year for upper gastrointestinal bleeding and 20 to 30 per 100,000 per year for lower gastrointestinal bleeding.11,12
The standard dosage (ie, 325 mg/day) is associated with a significantly higher risk of gastrointestinal bleeding (including fatal bleeds) than is 75 mg.13 However, even with lower doses, the risk of gastrointestinal bleeding is estimated to be twice as high as with no aspirin.14
And here is the irony: studies have shown that higher doses of aspirin offer no advantage in preventing thrombotic events compared with lower doses.15 For example, the Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent Events/Organization to Assess Strategies for Ischemic Stroke Syndromes study reported a higher rate of gastrointestinal bleeding with standard-dose aspirin therapy than with low-dose aspirin, with no additional cardiovascular benefit with the higher dose.16
Furthermore, several other risk factors increase the risk of gastrointestinal bleeding with aspirin use (Table 2). These risk factors are common in the general population but were not necessarily represented in participants in clinical trials. Thus, estimates of risk based on trial data most likely underestimate actual risk in the general population, and therefore, the individual patient’s risk of gastrointestinal bleeding, based on these and other factors, needs to be taken into consideration.
ASPIRIN IN PATIENTS WITH CORONARY ARTERY DISEASE
Randomized clinical trials have validated the benefits of aspirin in secondary prevention of cardiovascular events in patients who have had a myocardial infarction. Patients with coronary disease who withdraw from aspirin therapy or otherwise do not adhere to it have a risk of cardiovascular events three times higher than those who stay with it.17
Despite the strong data, however, several issues and questions remain about the use of aspirin for secondary prevention.
Bleeding risk must be considered, since gastrointestinal bleeding is associated with a higher risk of death and myocardial infarction in patients with cardiovascular disease.18 Many patients with coronary disease are on more than one antiplatelet or anticoagulant therapy for concomitant conditions such as atrial fibrillation or because they underwent a percutaneous intervention, which further increases the risk of bleeding.
This bleeding risk is reflected in changes in the most recent recommendations for aspirin dosing after percutaneous coronary intervention. Earlier guidelines advocated use of either 162 or 325 mg after the procedure. However, the most recent update (in 2011) now supports 81 mg for maintenance dosing after intervention.7
Patients with coronary disease but without prior myocardial infarction or intervention. Current guidelines recommend 75 to 162 mg of aspirin in all patients with coronary artery disease.6 However, this group is diverse and includes patients who have undergone percutaneous coronary intervention, patients with chronic stable angina, and patients with asymptomatic coronary artery disease found on imaging studies. The magnitude of benefit is not clear for those who have no symptoms or who have stable angina.
Most of the evidence supporting aspirin use in chronic angina came from a single trial in Sweden, in which 2,000 patients with chronic stable angina were given either 75 mg daily or placebo. Those who received aspirin had a 34% lower rate of myocardial infarction and sudden death.19
A substudy of the Physicians’ Health Study, with fewer patients, also noted a significant reduction in the rate of first myocardial infarction. The dose of aspirin in this study was 325 mg every other day.20
In the Women’s Health Initiative Observational Study, 70% of women with stable cardiovascular disease taking aspirin were taking 325 mg daily.21 This study demonstrated a significant reduction in the cardiovascular mortality rate, which supports current guidelines, and found no difference in outcomes with doses of 81 mg compared with 325 mg.21 This again corroborates that low-dose aspirin is preferential to standard-dose aspirin in women with cardiovascular disease.
These findings have not been validated in larger prospective trials. Thus, current guidelines for aspirin use may reflect extrapolation of aspirin benefit from higher-risk patients to lower-risk patients.
Nevertheless, although the debate continues, it has generally been accepted that in patients who are at high risk of vascular disease or who have had a myocardial infarction, the benefits of aspirin—a 20% relative reduction in vascular events22—clearly outweigh the risks.
ASPIRIN FOR PRIMARY PREVENTION
Assessing risk vs benefit is more complex when considering populations without known cardiovascular disease.
Only a few studies have specifically evaluated the use of aspirin for primary prevention (Table 3).23–31 The initial trials were in male physicians in the United Kingdom and the United States in the late 1980s and had somewhat conflicting results. A British study did not find a significant reduction in myocardial infarction,23 but the US Physician’s Health Study study did: the relative risk was 0.56 (95% confidence interval 0.45–0.70, P < .00001).24 The US study had more than four times the number of participants, used different dosing (325 mg every other day compared with 500 or 300 mg daily in the British study), and had a higher rate of compliance.
Several studies over the next decade demonstrated variable but significant reductions in cardiovascular events as well.25–27
A meta-analysis of primary prevention trials of aspirin was published in 2009.22 Although the relative risk reduction was similar in primary and secondary prevention, the absolute risk reduction in primary prevention was not nearly as great as in secondary prevention.
These findings are somewhat difficult to interpret, as the component trials included a wide spectrum of patients, ranging from healthy people with no symptoms and no known risk factors to those with limited risk factors. The trials were also performed over several decades during which primary prevention strategies were evolving. Additionally, most of the participants were middle-aged, nondiabetic men, so the results may not necessarily apply to people with diabetes, to women, or to the elderly. Thus, the pooled data in favor of aspirin for primary prevention may not be as broadly applicable to the general population as was once thought.
Aspirin for primary prevention in women
Guidelines for aspirin use in primary prevention were initially thought to be equally applicable to both sexes. However, concerns about the relatively low number of women participating in the studies and the possible mechanistic differences in aspirin efficacy in men vs women prompted further study.
A meta-analysis of randomized controlled trials found that aspirin was associated with a 12% relative reduction in the incidence of cardiovascular events in women and 14% in men. On the other hand, for stroke, the relative risk reduction was 17% in women, while men had no benefit.32
Most of the women in this meta-analysis were participants in the Women’s Health Study, and they were at low baseline risk.28 Although only about 10% of patients in this study were over age 65, this older group accounted for most of the benefit: these older women had a 26% risk reduction in major adverse cardiovascular events and 30% reduction in stroke.
Thus, for women, aspirin seems to become effective for primary prevention at an older age than in men, and the guidelines have been changed accordingly (Figure 1).
More women should be taking aspirin than actually are. For example, Rivera et al33 found that only 41% of eligible women were receiving aspirin for primary prevention and 48% of eligible women were receiving it for secondary prevention.
People with diabetes
People with diabetes without overt cardiovascular disease are at higher risk of cardiovascular events than age- and sex-matched controls.34 On the other hand, people with diabetes may be more prone to aspirin resistance and may not derive as much cardiovascular benefit from aspirin.
Early primary prevention studies included few people with diabetes. Subsequent meta-analyses of trials that used a wide range of aspirin doses found a relative risk reduction of 9%, which was not statistically significant.9,35,36
But there is some evidence that people with diabetes, with37 and without22 coronary disease, may be at higher inherent risk of bleeding than people without diabetes. Although aspirin may not necessarily increase the risk of bleeding in diabetic patients, recent data suggest no benefit in terms of a reduction in vascular events.38
The balance of risk vs benefit for aspirin in this special population is not clear, although some argue that these patients should be treated somewhere on the spectrum of risk between primary and secondary prevention.
The US Preventive Services Task Force did not differentiate between people with or without diabetes in its 2009 guidelines for aspirin for primary prevention.8 However, the debate is reflected in a change in 2010 American College of Cardiology/American Diabetes Association guidelines regarding aspirin use in people with diabetes without known cardiovascular disease.39 As opposed to earlier recommendations from these organizations in favor of aspirin for all people with diabetes regardless of 10-year risk, current recommendations advise low-dose aspirin (81–162 mg) for diabetic patients without known vascular disease who have a greater than 10% risk of a cardiovascular event and are not at increased risk of bleeding.
These changes were based on the findings of two trials: the Prevention and Progression of Arterial Disease and Diabetes Trial (POPADAD) and the Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes (JPAD) study. These did not show a statistically significant benefit in prevention of cardiovascular events with aspirin.29,30
After the new guidelines came out, a meta-analysis further bolstered its recommendations. 40 In seven randomized clinical trials in 11,000 patients, the relative risk reduction was 9% with aspirin, which did not reach statistical significance.
Statins may dilute the benefit of aspirin
The use of statins has been increasing, and this trend may have played a role in the marginal benefit of aspirin therapy in these recent studies. In the Japanese trial, approximately 25% of the patients were known to be using a statin; the percentage of statin use was not reported specifically in POPADAD, but both of these studies were published in 2008, when the proportion of diabetic patients taking a statin would be expected to be higher than in earlier primary prevention trials, which were performed primarily in the 1990s. Thus, the beneficial effects of statins may have somewhat diluted the risk reduction attributable to aspirin.
Trials under way in patients with diabetes
The evolving and somewhat conflicting guidelines highlight the need for further study in patients with diabetes. To address this area, two trials are in progress: the Aspirin and Simvastatin Combination for Cardiovascular Events Prevention Trial in Diabetes (ACCEPT-D) and A Study of Cardiovascular Events in Diabetes (ASCEND).41,42
ACCEPT-D is testing low-dose aspirin (100 mg daily) in diabetic patients who are also on simvastatin. This study also includes prespecified subgroups stratified by sex, age, and baseline lipid levels.
The ASCEND trial will use the same aspirin dose as ACCEPT-D, with a target enrollment of 10,000 patients with diabetes without known vascular disease.
More frequent dosing for people with diabetes?
Although not supported by current guidelines, recent work has suggested that people with diabetes may need more-frequent dosing of aspirin.43 This topic warrants further investigation.
Aspirin as primary prevention in elderly patients
The incidence of cardiovascular events increases with age37—but so does the incidence of gastrointestinal bleeding.44 Upper gastrointestinal bleeding is especially worrisome in the elderly, in whom the estimated case-fatality rate is high.12 Assessment of risk and benefit is particularly important in patients over age 65 without known coronary disease.
Uncertainty about aspirin use in this population is reflected in the most recent US Preventive Services Task Force guidelines, which do not advocate either for or against regular aspirin use for primary prevention in those over the age of 80.
Data on this topic from clinical trials are limited. The Antithrombotic Trialists’ Collaboration (2009) found that although age is associated with a risk of major coronary events similar to that of other traditional risk factors such as diabetes, hypertension, and tobacco use, older age is also associated with the highest risk of major extracranial bleeding.22
Because of the lack of data in this population, several studies are currently under way. The Aspirin in Reducing Events in the Elderly (ASPREE) trial is studying 100 mg daily in nondiabetic patients without known cardiovascular disease who are age 70 and older.45 An additional trial will study patients age 60 to 85 with concurrent diagnoses of hypertension, hyperlipidemia, or diabetes and will test the same aspirin dose as in ASPREE.46 These trials should provide further insight into the safety and efficacy of aspirin for primary prevention in the elderly.
FUTURE DIRECTIONS
Aspirin remains a cornerstone of therapy in patients with cardiovascular disease and in secondary prevention of adverse cardiovascular events, but its role in primary prevention remains under scrutiny. Recommendations have evolved to reflect emerging data in special populations, and an algorithm based on Framingham risk assessment in men for myocardial infarction and ischemic stroke assessment in women for assessing appropriateness of aspirin therapy based on currently available guidelines is presented in Figure 1.6,8,47–49 Targeted studies have advanced our understanding of aspirin use in women, and future studies in people with diabetes and in the elderly should provide further insight into the role of aspirin for primary prevention in these specific groups as well.
Additionally, the range of doses used in clinical studies has propagated the general misperception that higher doses of aspirin are more efficacious. Future studies should continue to use lower doses of aspirin to minimize bleeding risk with an added focus on re-examining its net benefit in the modern era of increasing statin use, which may reduce the absolute risk reduction attributable to aspirin.
One particular area of debate is whether enteric coating can result in functional aspirin resistance. Grosser et al50 found that sustained aspirin resistance was rare, and “pseudoresistance” was related to the use of a single enteric-coated aspirin instead of immediate-release aspirin in people who had not been taking aspirin up to then. This complements an earlier study, which found that enteric-coated aspirin had an appropriate effect when given for 7 days.51 Therefore, for patients who have not been taking aspirin, the first dose should always be immediate-release, not enteric coated.
SHOULD OUR PATIENT RECEIVE ASPIRIN?
The patient we described at the beginning of this article has several risk factors—hypertension, dyslipidemia, left ventricular hypertrophy, and smoking—but no known cardiovascular disease as yet. Her risk of an adverse cardiovascular event appears moderate. However, her 10-year risk of stroke by the Framingham risk calculation is 10%, which would qualify her for aspirin for primary prevention. Of particular note is that the significance of left ventricular hypertrophy as a risk factor for stroke in women is higher than in men and in our case accounts for half of this patient’s risk.
We should explain to the patient that the anticipated benefits of aspirin for stroke prevention outweigh bleeding risks, and thus aspirin therapy would be recommended. However, with her elevated LDL-cholesterol, she may benefit from a statin, which could lessen the relative risk reduction from additional aspirin use.
A 57-year-old woman with no history of cardiovascular disease comes to the clinic for her annual evaluation. She does not have diabetes mellitus, but she does have hypertension and chronic osteoarthritis, currently treated with acetaminophen. Additionally, she admits to active tobacco use. Her systolic blood pressure is 130 mm Hg on therapy with hydrochlorothiazide. Her electrocardiogram demonstrates left ventricular hypertrophy. Her low-density lipoprotein (LDL) cholesterol level is 140 mg/dL, and her high-density lipoprotein (HDL) cholesterol level is 50 mg/dL. Should this patient be started on aspirin therapy?
Acetylsalicylic acid (aspirin) is an analgesic, antipyretic, and anti-inflammatory agent, but its more prominent use today is as an antithrombotic agent to treat or prevent cardiovascular events. Its antithrombotic properties are due to its effects on the enzyme cyclooxygenase. However, cyclooxygenase is also involved in regulation of the gastric mucosa, and so aspirin increases the risk of gastrointestinal bleeding.
Approximately 50 million people take aspirin on a daily basis to treat or prevent cardiovascular disease.1 Of these, at least half are taking more than 100 mg per day,2 reflecting the general belief that, for aspirin dosage, “more is better”—which is not true.
Additionally, recommendations about the use of aspirin were based on studies that included relatively few members of several important subgroups, such as people with diabetes without known cardiovascular disease, women, and the elderly, and thus may not reflect appropriate indications and dosages for these groups.
Here, we examine the literature, outline an individualized approach to aspirin therapy, and highlight areas for future study.
HISTORY OF ASPIRIN USE IN CARDIOVASCULAR DISEASE
- 1700s—Willow bark is used as an analgesic.
- 1897—Synthetic aspirin is developed as an antipyretic and anti-inflammatory agent.
- 1974—First landmark trial of aspirin for secondary prevention of myocardial infarction.3
- 1982—Nobel Prize awarded for discovery of aspirin mechanism.
- 1985—US Food and Drug Administration approves aspirin for the treatment and secondary prevention of acute myocardial infarction.
- 1998—The Second International Study of Infarct Survival (ISIS-2) finds that giving aspirin to patients with myocardial infarction within 24 hours of presentation leads to a significant reduction in vascular deaths.4
Ongoing uncertainties
Aspirin now carries a class I indication for all patients with suspected myocardial infarction. Since there are an estimated 600,000 new coronary events and 325,000 recurrent ischemic events per year in the United States,5 the need for aspirin will continue to remain great. It is also approved to prevent and treat stroke and in patients with unstable angina.
However, questions continue to emerge about aspirin’s dosing and appropriate use in specific populations. The initial prevention trials used a wide range of doses and, as mentioned, included few women, few people with diabetes, and few elderly people. The uncertainties are especially pertinent for patients without known vascular disease, in whom the absolute risk reduction is much less, making the assessment of bleeding risk particularly important. Furthermore, the absolute risk-to-benefit assessment may be different in certain populations.
Guidelines on the use of aspirin to prevent cardiovascular disease (Table 1)6–10 have evolved to take into account these possible disparities, and studies are taking place to further investigate aspirin use in these groups.
ASPIRIN AND GASTROINTESTINAL BLEEDING
Aspirin’s association with bleeding, particularly gastrointestinal bleeding, was recognized early as a use-limiting side effect. With or without aspirin, gastrointestinal bleeding is a common cause of morbidity and death, with an incidence of approximately 100 per 100,000 bleeding episodes in adults per year for upper gastrointestinal bleeding and 20 to 30 per 100,000 per year for lower gastrointestinal bleeding.11,12
The standard dosage (ie, 325 mg/day) is associated with a significantly higher risk of gastrointestinal bleeding (including fatal bleeds) than is 75 mg.13 However, even with lower doses, the risk of gastrointestinal bleeding is estimated to be twice as high as with no aspirin.14
And here is the irony: studies have shown that higher doses of aspirin offer no advantage in preventing thrombotic events compared with lower doses.15 For example, the Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent Events/Organization to Assess Strategies for Ischemic Stroke Syndromes study reported a higher rate of gastrointestinal bleeding with standard-dose aspirin therapy than with low-dose aspirin, with no additional cardiovascular benefit with the higher dose.16
Furthermore, several other risk factors increase the risk of gastrointestinal bleeding with aspirin use (Table 2). These risk factors are common in the general population but were not necessarily represented in participants in clinical trials. Thus, estimates of risk based on trial data most likely underestimate actual risk in the general population, and therefore, the individual patient’s risk of gastrointestinal bleeding, based on these and other factors, needs to be taken into consideration.
ASPIRIN IN PATIENTS WITH CORONARY ARTERY DISEASE
Randomized clinical trials have validated the benefits of aspirin in secondary prevention of cardiovascular events in patients who have had a myocardial infarction. Patients with coronary disease who withdraw from aspirin therapy or otherwise do not adhere to it have a risk of cardiovascular events three times higher than those who stay with it.17
Despite the strong data, however, several issues and questions remain about the use of aspirin for secondary prevention.
Bleeding risk must be considered, since gastrointestinal bleeding is associated with a higher risk of death and myocardial infarction in patients with cardiovascular disease.18 Many patients with coronary disease are on more than one antiplatelet or anticoagulant therapy for concomitant conditions such as atrial fibrillation or because they underwent a percutaneous intervention, which further increases the risk of bleeding.
This bleeding risk is reflected in changes in the most recent recommendations for aspirin dosing after percutaneous coronary intervention. Earlier guidelines advocated use of either 162 or 325 mg after the procedure. However, the most recent update (in 2011) now supports 81 mg for maintenance dosing after intervention.7
Patients with coronary disease but without prior myocardial infarction or intervention. Current guidelines recommend 75 to 162 mg of aspirin in all patients with coronary artery disease.6 However, this group is diverse and includes patients who have undergone percutaneous coronary intervention, patients with chronic stable angina, and patients with asymptomatic coronary artery disease found on imaging studies. The magnitude of benefit is not clear for those who have no symptoms or who have stable angina.
Most of the evidence supporting aspirin use in chronic angina came from a single trial in Sweden, in which 2,000 patients with chronic stable angina were given either 75 mg daily or placebo. Those who received aspirin had a 34% lower rate of myocardial infarction and sudden death.19
A substudy of the Physicians’ Health Study, with fewer patients, also noted a significant reduction in the rate of first myocardial infarction. The dose of aspirin in this study was 325 mg every other day.20
In the Women’s Health Initiative Observational Study, 70% of women with stable cardiovascular disease taking aspirin were taking 325 mg daily.21 This study demonstrated a significant reduction in the cardiovascular mortality rate, which supports current guidelines, and found no difference in outcomes with doses of 81 mg compared with 325 mg.21 This again corroborates that low-dose aspirin is preferential to standard-dose aspirin in women with cardiovascular disease.
These findings have not been validated in larger prospective trials. Thus, current guidelines for aspirin use may reflect extrapolation of aspirin benefit from higher-risk patients to lower-risk patients.
Nevertheless, although the debate continues, it has generally been accepted that in patients who are at high risk of vascular disease or who have had a myocardial infarction, the benefits of aspirin—a 20% relative reduction in vascular events22—clearly outweigh the risks.
ASPIRIN FOR PRIMARY PREVENTION
Assessing risk vs benefit is more complex when considering populations without known cardiovascular disease.
Only a few studies have specifically evaluated the use of aspirin for primary prevention (Table 3).23–31 The initial trials were in male physicians in the United Kingdom and the United States in the late 1980s and had somewhat conflicting results. A British study did not find a significant reduction in myocardial infarction,23 but the US Physician’s Health Study study did: the relative risk was 0.56 (95% confidence interval 0.45–0.70, P < .00001).24 The US study had more than four times the number of participants, used different dosing (325 mg every other day compared with 500 or 300 mg daily in the British study), and had a higher rate of compliance.
Several studies over the next decade demonstrated variable but significant reductions in cardiovascular events as well.25–27
A meta-analysis of primary prevention trials of aspirin was published in 2009.22 Although the relative risk reduction was similar in primary and secondary prevention, the absolute risk reduction in primary prevention was not nearly as great as in secondary prevention.
These findings are somewhat difficult to interpret, as the component trials included a wide spectrum of patients, ranging from healthy people with no symptoms and no known risk factors to those with limited risk factors. The trials were also performed over several decades during which primary prevention strategies were evolving. Additionally, most of the participants were middle-aged, nondiabetic men, so the results may not necessarily apply to people with diabetes, to women, or to the elderly. Thus, the pooled data in favor of aspirin for primary prevention may not be as broadly applicable to the general population as was once thought.
Aspirin for primary prevention in women
Guidelines for aspirin use in primary prevention were initially thought to be equally applicable to both sexes. However, concerns about the relatively low number of women participating in the studies and the possible mechanistic differences in aspirin efficacy in men vs women prompted further study.
A meta-analysis of randomized controlled trials found that aspirin was associated with a 12% relative reduction in the incidence of cardiovascular events in women and 14% in men. On the other hand, for stroke, the relative risk reduction was 17% in women, while men had no benefit.32
Most of the women in this meta-analysis were participants in the Women’s Health Study, and they were at low baseline risk.28 Although only about 10% of patients in this study were over age 65, this older group accounted for most of the benefit: these older women had a 26% risk reduction in major adverse cardiovascular events and 30% reduction in stroke.
Thus, for women, aspirin seems to become effective for primary prevention at an older age than in men, and the guidelines have been changed accordingly (Figure 1).
More women should be taking aspirin than actually are. For example, Rivera et al33 found that only 41% of eligible women were receiving aspirin for primary prevention and 48% of eligible women were receiving it for secondary prevention.
People with diabetes
People with diabetes without overt cardiovascular disease are at higher risk of cardiovascular events than age- and sex-matched controls.34 On the other hand, people with diabetes may be more prone to aspirin resistance and may not derive as much cardiovascular benefit from aspirin.
Early primary prevention studies included few people with diabetes. Subsequent meta-analyses of trials that used a wide range of aspirin doses found a relative risk reduction of 9%, which was not statistically significant.9,35,36
But there is some evidence that people with diabetes, with37 and without22 coronary disease, may be at higher inherent risk of bleeding than people without diabetes. Although aspirin may not necessarily increase the risk of bleeding in diabetic patients, recent data suggest no benefit in terms of a reduction in vascular events.38
The balance of risk vs benefit for aspirin in this special population is not clear, although some argue that these patients should be treated somewhere on the spectrum of risk between primary and secondary prevention.
The US Preventive Services Task Force did not differentiate between people with or without diabetes in its 2009 guidelines for aspirin for primary prevention.8 However, the debate is reflected in a change in 2010 American College of Cardiology/American Diabetes Association guidelines regarding aspirin use in people with diabetes without known cardiovascular disease.39 As opposed to earlier recommendations from these organizations in favor of aspirin for all people with diabetes regardless of 10-year risk, current recommendations advise low-dose aspirin (81–162 mg) for diabetic patients without known vascular disease who have a greater than 10% risk of a cardiovascular event and are not at increased risk of bleeding.
These changes were based on the findings of two trials: the Prevention and Progression of Arterial Disease and Diabetes Trial (POPADAD) and the Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes (JPAD) study. These did not show a statistically significant benefit in prevention of cardiovascular events with aspirin.29,30
After the new guidelines came out, a meta-analysis further bolstered its recommendations. 40 In seven randomized clinical trials in 11,000 patients, the relative risk reduction was 9% with aspirin, which did not reach statistical significance.
Statins may dilute the benefit of aspirin
The use of statins has been increasing, and this trend may have played a role in the marginal benefit of aspirin therapy in these recent studies. In the Japanese trial, approximately 25% of the patients were known to be using a statin; the percentage of statin use was not reported specifically in POPADAD, but both of these studies were published in 2008, when the proportion of diabetic patients taking a statin would be expected to be higher than in earlier primary prevention trials, which were performed primarily in the 1990s. Thus, the beneficial effects of statins may have somewhat diluted the risk reduction attributable to aspirin.
Trials under way in patients with diabetes
The evolving and somewhat conflicting guidelines highlight the need for further study in patients with diabetes. To address this area, two trials are in progress: the Aspirin and Simvastatin Combination for Cardiovascular Events Prevention Trial in Diabetes (ACCEPT-D) and A Study of Cardiovascular Events in Diabetes (ASCEND).41,42
ACCEPT-D is testing low-dose aspirin (100 mg daily) in diabetic patients who are also on simvastatin. This study also includes prespecified subgroups stratified by sex, age, and baseline lipid levels.
The ASCEND trial will use the same aspirin dose as ACCEPT-D, with a target enrollment of 10,000 patients with diabetes without known vascular disease.
More frequent dosing for people with diabetes?
Although not supported by current guidelines, recent work has suggested that people with diabetes may need more-frequent dosing of aspirin.43 This topic warrants further investigation.
Aspirin as primary prevention in elderly patients
The incidence of cardiovascular events increases with age37—but so does the incidence of gastrointestinal bleeding.44 Upper gastrointestinal bleeding is especially worrisome in the elderly, in whom the estimated case-fatality rate is high.12 Assessment of risk and benefit is particularly important in patients over age 65 without known coronary disease.
Uncertainty about aspirin use in this population is reflected in the most recent US Preventive Services Task Force guidelines, which do not advocate either for or against regular aspirin use for primary prevention in those over the age of 80.
Data on this topic from clinical trials are limited. The Antithrombotic Trialists’ Collaboration (2009) found that although age is associated with a risk of major coronary events similar to that of other traditional risk factors such as diabetes, hypertension, and tobacco use, older age is also associated with the highest risk of major extracranial bleeding.22
Because of the lack of data in this population, several studies are currently under way. The Aspirin in Reducing Events in the Elderly (ASPREE) trial is studying 100 mg daily in nondiabetic patients without known cardiovascular disease who are age 70 and older.45 An additional trial will study patients age 60 to 85 with concurrent diagnoses of hypertension, hyperlipidemia, or diabetes and will test the same aspirin dose as in ASPREE.46 These trials should provide further insight into the safety and efficacy of aspirin for primary prevention in the elderly.
FUTURE DIRECTIONS
Aspirin remains a cornerstone of therapy in patients with cardiovascular disease and in secondary prevention of adverse cardiovascular events, but its role in primary prevention remains under scrutiny. Recommendations have evolved to reflect emerging data in special populations, and an algorithm based on Framingham risk assessment in men for myocardial infarction and ischemic stroke assessment in women for assessing appropriateness of aspirin therapy based on currently available guidelines is presented in Figure 1.6,8,47–49 Targeted studies have advanced our understanding of aspirin use in women, and future studies in people with diabetes and in the elderly should provide further insight into the role of aspirin for primary prevention in these specific groups as well.
Additionally, the range of doses used in clinical studies has propagated the general misperception that higher doses of aspirin are more efficacious. Future studies should continue to use lower doses of aspirin to minimize bleeding risk with an added focus on re-examining its net benefit in the modern era of increasing statin use, which may reduce the absolute risk reduction attributable to aspirin.
One particular area of debate is whether enteric coating can result in functional aspirin resistance. Grosser et al50 found that sustained aspirin resistance was rare, and “pseudoresistance” was related to the use of a single enteric-coated aspirin instead of immediate-release aspirin in people who had not been taking aspirin up to then. This complements an earlier study, which found that enteric-coated aspirin had an appropriate effect when given for 7 days.51 Therefore, for patients who have not been taking aspirin, the first dose should always be immediate-release, not enteric coated.
SHOULD OUR PATIENT RECEIVE ASPIRIN?
The patient we described at the beginning of this article has several risk factors—hypertension, dyslipidemia, left ventricular hypertrophy, and smoking—but no known cardiovascular disease as yet. Her risk of an adverse cardiovascular event appears moderate. However, her 10-year risk of stroke by the Framingham risk calculation is 10%, which would qualify her for aspirin for primary prevention. Of particular note is that the significance of left ventricular hypertrophy as a risk factor for stroke in women is higher than in men and in our case accounts for half of this patient’s risk.
We should explain to the patient that the anticipated benefits of aspirin for stroke prevention outweigh bleeding risks, and thus aspirin therapy would be recommended. However, with her elevated LDL-cholesterol, she may benefit from a statin, which could lessen the relative risk reduction from additional aspirin use.
- Chan FK, Graham DY. Review article: prevention of non-steroidal anti-inflammatory drug gastrointestinal complications—review and recommendations based on risk assessment. Aliment Pharmacol Ther 2004; 19:1051–1061.
- Peters RJ, Mehta SR, Fox KA, et al; Clopidogrel in Unstable angina to prevent Recurrent Events (CURE) Trial Investigators. Effects of aspirin dose when used alone or in combination with clopidogrel in patients with acute coronary syndromes: observations from the Clopidogrel in Unstable angina to prevent Recurrent Events (CURE) study. Circulation 2003; 108:1682–1687.
- Elwood PC, Cochrane AL, Burr ML, et al. A randomized controlled trial of acetyl salicylic acid in the secondary prevention of mortality from myocardial infarction. Br Med J 1974; 1:436–440.
- Baigent C, Collins R, Appleby P, Parish S, Sleight P, Peto R. ISIS-2: 10 year survival among patients with suspected acute myocardial infarction in randomised comparison of intravenous streptokinase, oral aspirin, both, or neither. The ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. BMJ 1998; 316:1337–1343.
- Roger VL, Go AS, Lloyd-Jones DM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2011 update: a report from the American Heart Association. Circulation 2011; 123:e18–e209.
- Smith SC, Benjamin EJ, Bonow RO, et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation endorsed by the World Heart Federation and the Preventive Cardiovascular Nurses Association. J Am Coll Cardiol 2011; 58:2432–2446.
- Levine GN, Bates ER, Blankenship JC, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; Society for Cardiovascular Angiography and Interventions. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol 2011; 58:e44–e122.
- US Preventive Services Task Force. Aspirin for the prevention of cardiovascular disease: US Preventive Services Task Force recommendation statement. Ann Intern Med 2009; 150:396–404.
- Pignone M, Alberts MJ, Colwell JA, et al. Aspirin for primary prevention of cardiovascular events in people with diabetes: a position statement of the American Diabetes Association, a scientific statement of the American Heart Association, and an expert consensus document of the American College of Cardiology Foundation. Circulation 2010; 121:2694–2701.
- Mosca L, Benjamin EJ, Berra K, et al. Effectiveness-based guidelines for the prevention of cardiovascular disease in women—2011 update: a guideline from the American Heart Association. Circulation 2011; 123:1243–1262.
- Strate LL. Lower GI bleeding: epidemiology and diagnosis. Gastroenterol Clin North Am 2005; 34:643–664.
- Rockall TA, Logan RF, Devlin HB, Northfield TC. Incidence of and mortality from acute upper gastrointestinal haemorrhage in the United Kingdom. Steering Committee and members of the National Audit of Acute Upper Gastrointestinal Haemorrhage. BMJ 1995; 311:222–226.
- Campbell CL, Smyth S, Montalescot G, Steinhubl SR. Aspirin dose for the prevention of cardiovascular disease: a systematic review. JAMA 2007; 297:2018–2024.
- Weil J, Colin-Jones D, Langman M, et al. Prophylactic aspirin and risk of peptic ulcer bleeding. BMJ 1995; 310:827–830.
- Reilly IA, FitzGerald GA. Inhibition of thromboxane formation in vivo and ex vivo: implications for therapy with platelet inhibitory drugs. Blood 1987; 69:180–186.
- CURRENT-OASIS 7 Investigators; Mehta SR, Bassand JP, Chrolavicius S, et al. Dose comparisons of clopidogrel and aspirin in acute coronary syndromes. N Engl J Med 2010; 363:930–942.
- Biondi-Zoccai GG, Lotrionte M, Agostoni P, et al. A systematic review and meta-analysis on the hazards of discontinuing or not adhering to aspirin among 50,279 patients at risk for coronary artery disease. Eur Heart J 2006; 27:2667–2674.
- Berger PB, Bhatt DL, Fuster V, et al; CHARISMA Investigators. Bleeding complications with dual antiplatelet therapy among patients with stable vascular disease or risk factors for vascular disease: results from the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial. Circulation 2010; 121:2575–2583.
- Juul-Möller S, Edvardsson N, Jahnmatz B, Rosén A, Sørensen S, Omblus R. Double-blind trial of aspirin in primary prevention of myocardial infarction in patients with stable chronic angina pectoris. The Swedish Angina Pectoris Aspirin Trial (SAPAT) Group. Lancet 1992; 340:1421–1425.
- Ridker PM, Manson JE, Gaziano JM, Buring JE, Hennekens CH. Low-dose aspirin therapy for chronic stable angina. A randomized, placebo-controlled clinical trial. Ann Intern Med 1991; 114:835–839.
- Berger JS, Brown DL, Burke GL, et al. Aspirin use, dose, and clinical outcomes in postmenopausal women with stable cardiovascular disease: the Women’s Health Initiative Observational Study. Circ Cardiovasc Qual Outcomes 2009; 2:78–87.
- Antithrombotic Trialists’ (ATT) Collaboration; Baigent C, Blackwell L, Collins R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet 2009; 373:1849–1860.
- Peto R, Gray R, Collins R, et al. Randomised trial of prophylactic daily aspirin in British male doctors. Br Med J (Clin Res Ed) 1988; 296:313–316.
- Final report on the aspirin component of the ongoing Physicians’ Health Study. Steering Committee of the Physicians’ Health Study Research Group. N Engl J Med 1989; 321:129–135.
- Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. The Medical Research Council’s General Practice Research Framework. Lancet 1998; 351:233–241.
- de Gaetano GCollaborative Group of the Primary Prevention Project. Low-dose aspirin and vitamin E in people at cardiovascular risk: a randomised trial in general practice. Collaborative Group of the Primary Prevention Project. Lancet 2001; 357:89–95.
- Hansson L, Zanchetti A, Carruthers SG, et al. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. HOT Study Group. Lancet 1998; 351:1755–1762.
- Ridker PM, Cook NR, Lee IM, et al. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med 2005; 352:1293–1304.
- Belch J, MacCuish A, Campbell I, et al; Prevention of Progression of Arterial Disease and Diabetes Study Group; Diabetes Registry Group; Royal College of Physicians Edinburgh. The prevention of progression of arterial disease and diabetes (POPADAD) trial: factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ 2008; 337:a1840.
- Ogawa H, Nakayama M, Morimoto T, et al; Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes (JPAD) Trial Investigators. Low-dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: a randomized controlled trial. JAMA 2008; 300:2134–2141.
- Fowkes FG, Price JF, Stewart MC, et al; Aspirin for Asymptomatic Atherosclerosis Trialists. Aspirin for prevention of cardiovascular events in a general population screened for a low ankle brachial index: a randomized controlled trial. JAMA 2010; 303:841–848.
- Berger JS, Roncaglioni MC, Avanzini F, Pangrazzi I, Tognoni G, Brown DL. Aspirin for the primary prevention of cardiovascular events in women and men: a sex-specific meta-analysis of randomized controlled trials. JAMA 2006; 295:306–313.
- Rivera CM, Song J, Copeland L, Buirge C, Ory M, McNeal CJ. Underuse of aspirin for primary and secondary prevention of cardiovascular disease events in women. J Womens Health (Larchmt) 2012; 21:379–387.
- Wilson R, Gazzala J, House J. Aspirin in primary and secondary prevention in elderly adults revisited. South Med J 2012; 105:82–86.
- De Berardis G, Sacco M, Strippoli GF, et al. Aspirin for primary prevention of cardiovascular events in people with diabetes: meta-analysis of randomised controlled trials. BMJ 2009; 339:b4531.
- Zhang C, Sun A, Zhang P, et al. Aspirin for primary prevention of cardiovascular events in patients with diabetes: a meta-analysis. Diabetes Res Clin Pract 2010; 87:211–218.
- Moukarbel GV, Signorovitch JE, Pfeffer MA, et al. Gastrointestinal bleeding in high risk survivors of myocardial infarction: the VALIANT Trial. Eur Heart J 2009; 30:2226–2232.
- De Berardis G, Lucisano G, D’Ettorre A, et al. Association of aspirin use with major bleeding in patients with and without diabetes. JAMA 2012; 307:2286–2294.
- Pignone M, Alberts MJ, Colwell JA, et al; American Diabetes Association; American Heart Association; American College of Cardiology Foundation. Aspirin for primary prevention of cardiovascular events in people with diabetes. J Am Coll Cardiol 2010; 55:2878–2886.
- Butalia S, Leung AA, Ghali WA, Rabi DM. Aspirin effect on the incidence of major adverse cardiovascular events in patients with diabetes mellitus: a systematic review and meta-analysis. Cardiovasc Diabetol 2011; 10:25.
- De Berardis G, Sacco M, Evangelista V, et al; ACCEPT-D Study Group. Aspirin and Simvastatin Combination for Cardiovascular Events Prevention Trial in Diabetes (ACCEPT-D): design of a randomized study of the efficacy of low-dose aspirin in the prevention of cardiovascular events in subjects with diabetes mellitus treated with statins. Trials 2007; 8:21.
- British Heart Foundation. ASCEND: A Study of Cardiovascular Events in Diabetes. http://www.ctsu.ox.ac.uk/ascend. Accessed April 1, 2013.
- Rocca B, Santilli F, Pitocco D, et al. The recovery of platelet cyclooxygenase activity explains interindividual variability in responsiveness to low-dose aspirin in patients with and without diabetes. J Thromb Haemost 2012; 10:1220–1230.
- Hernández-Díaz S, Garcia Rodriguez LA. Cardioprotective aspirin users and their excess risk for upper gastrointestinal complications. BMC Med 2006; 4:22.
- Nelson MR, Reid CM, Ames DA, et al. Feasibility of conducting a primary prevention trial of low-dose aspirin for major adverse cardiovascular events in older people in Australia: results from the ASPirin in Reducing Events in the Elderly (ASPREE) pilot study. Med J Aust 2008; 189:105–109.
- Teramoto T, Shimada K, Uchiyama S, et al. Rationale, design, and baseline data of the Japanese Primary Prevention Project (JPPP)—a randomized, open-label, controlled trial of aspirin versus no aspirin in patients with multiple risk factors for vascular events. Am Heart J 2010; 159:361–369.e4.
- Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology; American Heart Association; Canadian Cardiovascular Society. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction—executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to revise the 1999 guidelines for the management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:671–719.
- Brott TG, Halperin JL, Abbara S, et al. 2011 ASA/ACCF/AHA/AANN/AANS/ACR/ASNR/CNS/SAIP/SCAI/SIR/SNIS/SVM/SVS Guideline on the Management of Patients With Extracranial Carotid and Vertebral Artery Disease A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, and the American Stroke Association, American Association of Neuroscience Nurses, American Association of Neurological Surgeons, American College of Radiology, American Society of Neuroradiology, Congress of Neurological Surgeons, Society of Atherosclerosis Imaging and Prevention, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of NeuroInterventional Surgery, Society for Vascular Medicine, and Society for Vascular Surgery Developed in Collaboration With the American Academy of Neurology and Society of Cardiovascular Computed Tomography. J Am Coll Cardiol 2011; 57:e16–e94.
- Wright RS, Anderson JL, Adams CD, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2011 ACCF/AHA focused update incorporated into the ACC/AHA 2007 Guidelines for the Management of Patients with Unstable Angina/Non-ST-Elevation Myocardial Infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines developed in collaboration with the American Academy of Family Physicians, Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons. J Am Coll Cardiol 2011; 57:e215–e367.
- Grosser T, Fries S, Lawson JA, Kapoor SC, Grant GR, Fitzgerald GA. Drug resistance and pseudoresistance: An unintended consequence of enteric coating aspirin. Circulation 2012; Epub ahead of print.
- Karha J, Rajagopal V, Kottke-Marchant K, Bhatt DL. Lack of effect of enteric coating on aspirin-induced inhibition of platelet aggregation in healthy volunteers. Am Heart J 2006; 151:976.e7–e11.
- Chan FK, Graham DY. Review article: prevention of non-steroidal anti-inflammatory drug gastrointestinal complications—review and recommendations based on risk assessment. Aliment Pharmacol Ther 2004; 19:1051–1061.
- Peters RJ, Mehta SR, Fox KA, et al; Clopidogrel in Unstable angina to prevent Recurrent Events (CURE) Trial Investigators. Effects of aspirin dose when used alone or in combination with clopidogrel in patients with acute coronary syndromes: observations from the Clopidogrel in Unstable angina to prevent Recurrent Events (CURE) study. Circulation 2003; 108:1682–1687.
- Elwood PC, Cochrane AL, Burr ML, et al. A randomized controlled trial of acetyl salicylic acid in the secondary prevention of mortality from myocardial infarction. Br Med J 1974; 1:436–440.
- Baigent C, Collins R, Appleby P, Parish S, Sleight P, Peto R. ISIS-2: 10 year survival among patients with suspected acute myocardial infarction in randomised comparison of intravenous streptokinase, oral aspirin, both, or neither. The ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. BMJ 1998; 316:1337–1343.
- Roger VL, Go AS, Lloyd-Jones DM, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2011 update: a report from the American Heart Association. Circulation 2011; 123:e18–e209.
- Smith SC, Benjamin EJ, Bonow RO, et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation endorsed by the World Heart Federation and the Preventive Cardiovascular Nurses Association. J Am Coll Cardiol 2011; 58:2432–2446.
- Levine GN, Bates ER, Blankenship JC, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; Society for Cardiovascular Angiography and Interventions. 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol 2011; 58:e44–e122.
- US Preventive Services Task Force. Aspirin for the prevention of cardiovascular disease: US Preventive Services Task Force recommendation statement. Ann Intern Med 2009; 150:396–404.
- Pignone M, Alberts MJ, Colwell JA, et al. Aspirin for primary prevention of cardiovascular events in people with diabetes: a position statement of the American Diabetes Association, a scientific statement of the American Heart Association, and an expert consensus document of the American College of Cardiology Foundation. Circulation 2010; 121:2694–2701.
- Mosca L, Benjamin EJ, Berra K, et al. Effectiveness-based guidelines for the prevention of cardiovascular disease in women—2011 update: a guideline from the American Heart Association. Circulation 2011; 123:1243–1262.
- Strate LL. Lower GI bleeding: epidemiology and diagnosis. Gastroenterol Clin North Am 2005; 34:643–664.
- Rockall TA, Logan RF, Devlin HB, Northfield TC. Incidence of and mortality from acute upper gastrointestinal haemorrhage in the United Kingdom. Steering Committee and members of the National Audit of Acute Upper Gastrointestinal Haemorrhage. BMJ 1995; 311:222–226.
- Campbell CL, Smyth S, Montalescot G, Steinhubl SR. Aspirin dose for the prevention of cardiovascular disease: a systematic review. JAMA 2007; 297:2018–2024.
- Weil J, Colin-Jones D, Langman M, et al. Prophylactic aspirin and risk of peptic ulcer bleeding. BMJ 1995; 310:827–830.
- Reilly IA, FitzGerald GA. Inhibition of thromboxane formation in vivo and ex vivo: implications for therapy with platelet inhibitory drugs. Blood 1987; 69:180–186.
- CURRENT-OASIS 7 Investigators; Mehta SR, Bassand JP, Chrolavicius S, et al. Dose comparisons of clopidogrel and aspirin in acute coronary syndromes. N Engl J Med 2010; 363:930–942.
- Biondi-Zoccai GG, Lotrionte M, Agostoni P, et al. A systematic review and meta-analysis on the hazards of discontinuing or not adhering to aspirin among 50,279 patients at risk for coronary artery disease. Eur Heart J 2006; 27:2667–2674.
- Berger PB, Bhatt DL, Fuster V, et al; CHARISMA Investigators. Bleeding complications with dual antiplatelet therapy among patients with stable vascular disease or risk factors for vascular disease: results from the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial. Circulation 2010; 121:2575–2583.
- Juul-Möller S, Edvardsson N, Jahnmatz B, Rosén A, Sørensen S, Omblus R. Double-blind trial of aspirin in primary prevention of myocardial infarction in patients with stable chronic angina pectoris. The Swedish Angina Pectoris Aspirin Trial (SAPAT) Group. Lancet 1992; 340:1421–1425.
- Ridker PM, Manson JE, Gaziano JM, Buring JE, Hennekens CH. Low-dose aspirin therapy for chronic stable angina. A randomized, placebo-controlled clinical trial. Ann Intern Med 1991; 114:835–839.
- Berger JS, Brown DL, Burke GL, et al. Aspirin use, dose, and clinical outcomes in postmenopausal women with stable cardiovascular disease: the Women’s Health Initiative Observational Study. Circ Cardiovasc Qual Outcomes 2009; 2:78–87.
- Antithrombotic Trialists’ (ATT) Collaboration; Baigent C, Blackwell L, Collins R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet 2009; 373:1849–1860.
- Peto R, Gray R, Collins R, et al. Randomised trial of prophylactic daily aspirin in British male doctors. Br Med J (Clin Res Ed) 1988; 296:313–316.
- Final report on the aspirin component of the ongoing Physicians’ Health Study. Steering Committee of the Physicians’ Health Study Research Group. N Engl J Med 1989; 321:129–135.
- Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. The Medical Research Council’s General Practice Research Framework. Lancet 1998; 351:233–241.
- de Gaetano GCollaborative Group of the Primary Prevention Project. Low-dose aspirin and vitamin E in people at cardiovascular risk: a randomised trial in general practice. Collaborative Group of the Primary Prevention Project. Lancet 2001; 357:89–95.
- Hansson L, Zanchetti A, Carruthers SG, et al. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. HOT Study Group. Lancet 1998; 351:1755–1762.
- Ridker PM, Cook NR, Lee IM, et al. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med 2005; 352:1293–1304.
- Belch J, MacCuish A, Campbell I, et al; Prevention of Progression of Arterial Disease and Diabetes Study Group; Diabetes Registry Group; Royal College of Physicians Edinburgh. The prevention of progression of arterial disease and diabetes (POPADAD) trial: factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ 2008; 337:a1840.
- Ogawa H, Nakayama M, Morimoto T, et al; Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes (JPAD) Trial Investigators. Low-dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: a randomized controlled trial. JAMA 2008; 300:2134–2141.
- Fowkes FG, Price JF, Stewart MC, et al; Aspirin for Asymptomatic Atherosclerosis Trialists. Aspirin for prevention of cardiovascular events in a general population screened for a low ankle brachial index: a randomized controlled trial. JAMA 2010; 303:841–848.
- Berger JS, Roncaglioni MC, Avanzini F, Pangrazzi I, Tognoni G, Brown DL. Aspirin for the primary prevention of cardiovascular events in women and men: a sex-specific meta-analysis of randomized controlled trials. JAMA 2006; 295:306–313.
- Rivera CM, Song J, Copeland L, Buirge C, Ory M, McNeal CJ. Underuse of aspirin for primary and secondary prevention of cardiovascular disease events in women. J Womens Health (Larchmt) 2012; 21:379–387.
- Wilson R, Gazzala J, House J. Aspirin in primary and secondary prevention in elderly adults revisited. South Med J 2012; 105:82–86.
- De Berardis G, Sacco M, Strippoli GF, et al. Aspirin for primary prevention of cardiovascular events in people with diabetes: meta-analysis of randomised controlled trials. BMJ 2009; 339:b4531.
- Zhang C, Sun A, Zhang P, et al. Aspirin for primary prevention of cardiovascular events in patients with diabetes: a meta-analysis. Diabetes Res Clin Pract 2010; 87:211–218.
- Moukarbel GV, Signorovitch JE, Pfeffer MA, et al. Gastrointestinal bleeding in high risk survivors of myocardial infarction: the VALIANT Trial. Eur Heart J 2009; 30:2226–2232.
- De Berardis G, Lucisano G, D’Ettorre A, et al. Association of aspirin use with major bleeding in patients with and without diabetes. JAMA 2012; 307:2286–2294.
- Pignone M, Alberts MJ, Colwell JA, et al; American Diabetes Association; American Heart Association; American College of Cardiology Foundation. Aspirin for primary prevention of cardiovascular events in people with diabetes. J Am Coll Cardiol 2010; 55:2878–2886.
- Butalia S, Leung AA, Ghali WA, Rabi DM. Aspirin effect on the incidence of major adverse cardiovascular events in patients with diabetes mellitus: a systematic review and meta-analysis. Cardiovasc Diabetol 2011; 10:25.
- De Berardis G, Sacco M, Evangelista V, et al; ACCEPT-D Study Group. Aspirin and Simvastatin Combination for Cardiovascular Events Prevention Trial in Diabetes (ACCEPT-D): design of a randomized study of the efficacy of low-dose aspirin in the prevention of cardiovascular events in subjects with diabetes mellitus treated with statins. Trials 2007; 8:21.
- British Heart Foundation. ASCEND: A Study of Cardiovascular Events in Diabetes. http://www.ctsu.ox.ac.uk/ascend. Accessed April 1, 2013.
- Rocca B, Santilli F, Pitocco D, et al. The recovery of platelet cyclooxygenase activity explains interindividual variability in responsiveness to low-dose aspirin in patients with and without diabetes. J Thromb Haemost 2012; 10:1220–1230.
- Hernández-Díaz S, Garcia Rodriguez LA. Cardioprotective aspirin users and their excess risk for upper gastrointestinal complications. BMC Med 2006; 4:22.
- Nelson MR, Reid CM, Ames DA, et al. Feasibility of conducting a primary prevention trial of low-dose aspirin for major adverse cardiovascular events in older people in Australia: results from the ASPirin in Reducing Events in the Elderly (ASPREE) pilot study. Med J Aust 2008; 189:105–109.
- Teramoto T, Shimada K, Uchiyama S, et al. Rationale, design, and baseline data of the Japanese Primary Prevention Project (JPPP)—a randomized, open-label, controlled trial of aspirin versus no aspirin in patients with multiple risk factors for vascular events. Am Heart J 2010; 159:361–369.e4.
- Antman EM, Anbe DT, Armstrong PW, et al; American College of Cardiology; American Heart Association; Canadian Cardiovascular Society. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction—executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to revise the 1999 guidelines for the management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:671–719.
- Brott TG, Halperin JL, Abbara S, et al. 2011 ASA/ACCF/AHA/AANN/AANS/ACR/ASNR/CNS/SAIP/SCAI/SIR/SNIS/SVM/SVS Guideline on the Management of Patients With Extracranial Carotid and Vertebral Artery Disease A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, and the American Stroke Association, American Association of Neuroscience Nurses, American Association of Neurological Surgeons, American College of Radiology, American Society of Neuroradiology, Congress of Neurological Surgeons, Society of Atherosclerosis Imaging and Prevention, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of NeuroInterventional Surgery, Society for Vascular Medicine, and Society for Vascular Surgery Developed in Collaboration With the American Academy of Neurology and Society of Cardiovascular Computed Tomography. J Am Coll Cardiol 2011; 57:e16–e94.
- Wright RS, Anderson JL, Adams CD, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2011 ACCF/AHA focused update incorporated into the ACC/AHA 2007 Guidelines for the Management of Patients with Unstable Angina/Non-ST-Elevation Myocardial Infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines developed in collaboration with the American Academy of Family Physicians, Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons. J Am Coll Cardiol 2011; 57:e215–e367.
- Grosser T, Fries S, Lawson JA, Kapoor SC, Grant GR, Fitzgerald GA. Drug resistance and pseudoresistance: An unintended consequence of enteric coating aspirin. Circulation 2012; Epub ahead of print.
- Karha J, Rajagopal V, Kottke-Marchant K, Bhatt DL. Lack of effect of enteric coating on aspirin-induced inhibition of platelet aggregation in healthy volunteers. Am Heart J 2006; 151:976.e7–e11.
KEY POINTS
- Aspirin is as beneficial in low doses (eg, 81 mg daily) as it is in standard doses (325 mg) and poses less risk of gastrointestinal bleeding, although the bleeding risk is still twice as high as without aspirin.
- Since the absolute reduction in heart attacks and strokes is less in primary prevention than in secondary prevention, the risk of bleeding may for some groups outweigh the benefit, and the decision to use aspirin must be more individualized.
- Whether to prescribe aspirin for primary prevention depends on the combination of the individual patient’s sex, age, and 10-year risk of myocardial infarction (in men) or of stroke (in women).
Resistance of man and bug

Why individual clinicians make specific decisions usually can be sorted out. But our behavior as a group is more difficult to understand and, even when there are pressing and convincing reasons to change, behavior is difficult to alter.
In this issue, Drs. Federico Perez and David Van Duin discuss the emergence of carbapenem-resistant bacteria, dubbed “superbugs” by the media. Antibiotic resistance is not new; it was reported in Staphylococcus species within several years of the introduction of penicillin.1 However, it has been increasing in prevalence and molecular complexity after years of relatively promiscuous antibiotic use. As the percentage of inpatients with immunosuppression and frailty increases in this environment of known antibiotic resistance, initial empiric antibiotic choices will by necessity include drugs likely to further promote development of resistant strains. But why do physicians still prescribe antibiotics for uncomplicated upper respiratory tract infections and asymptomatic bacteriuria, despite numerous studies and guidelines suggesting this practice has little benefit? Is it because patients expect a prescription in return for their copayment? Is it the path of least resistance? Or do physicians not accept the data showing that it is unnecessary?
Dr. Gerald Appel discusses diabetic nephropathy, an area that involves resistance of another kind, ie, the apparent resistance of physicians and patients to achieving evidence-based treatment targets. We hold controlled trials as the Holy Grail of evidence-based medicine, yet we seem to have an aversion to following guidelines based on trial-derived evidence. (I do not refer here to blind guideline adherence, ignoring individual patient characteristics.)
So how can physicians’ behavior be altered and our resistance to change be reduced? Experiments are under way, such as paying physicians based on their performance, linking patients’ insurance rates to achieving selected outcomes, and linking physician practice self-review to certification. Perhaps naively, I continue to believe that the most effective impetus to changing personal practice is the dissemination of data from high-quality trials (tempered by our accumulated experience and keeping our eyes wide open), coupled with our desire to do the best for our patients.
- Barber M. Coagulase-positive staphylococci resistant to penicillin. J Pathol Bacteriol 1947; 59:373–384.
Why individual clinicians make specific decisions usually can be sorted out. But our behavior as a group is more difficult to understand and, even when there are pressing and convincing reasons to change, behavior is difficult to alter.
In this issue, Drs. Federico Perez and David Van Duin discuss the emergence of carbapenem-resistant bacteria, dubbed “superbugs” by the media. Antibiotic resistance is not new; it was reported in Staphylococcus species within several years of the introduction of penicillin.1 However, it has been increasing in prevalence and molecular complexity after years of relatively promiscuous antibiotic use. As the percentage of inpatients with immunosuppression and frailty increases in this environment of known antibiotic resistance, initial empiric antibiotic choices will by necessity include drugs likely to further promote development of resistant strains. But why do physicians still prescribe antibiotics for uncomplicated upper respiratory tract infections and asymptomatic bacteriuria, despite numerous studies and guidelines suggesting this practice has little benefit? Is it because patients expect a prescription in return for their copayment? Is it the path of least resistance? Or do physicians not accept the data showing that it is unnecessary?
Dr. Gerald Appel discusses diabetic nephropathy, an area that involves resistance of another kind, ie, the apparent resistance of physicians and patients to achieving evidence-based treatment targets. We hold controlled trials as the Holy Grail of evidence-based medicine, yet we seem to have an aversion to following guidelines based on trial-derived evidence. (I do not refer here to blind guideline adherence, ignoring individual patient characteristics.)
So how can physicians’ behavior be altered and our resistance to change be reduced? Experiments are under way, such as paying physicians based on their performance, linking patients’ insurance rates to achieving selected outcomes, and linking physician practice self-review to certification. Perhaps naively, I continue to believe that the most effective impetus to changing personal practice is the dissemination of data from high-quality trials (tempered by our accumulated experience and keeping our eyes wide open), coupled with our desire to do the best for our patients.
Why individual clinicians make specific decisions usually can be sorted out. But our behavior as a group is more difficult to understand and, even when there are pressing and convincing reasons to change, behavior is difficult to alter.
In this issue, Drs. Federico Perez and David Van Duin discuss the emergence of carbapenem-resistant bacteria, dubbed “superbugs” by the media. Antibiotic resistance is not new; it was reported in Staphylococcus species within several years of the introduction of penicillin.1 However, it has been increasing in prevalence and molecular complexity after years of relatively promiscuous antibiotic use. As the percentage of inpatients with immunosuppression and frailty increases in this environment of known antibiotic resistance, initial empiric antibiotic choices will by necessity include drugs likely to further promote development of resistant strains. But why do physicians still prescribe antibiotics for uncomplicated upper respiratory tract infections and asymptomatic bacteriuria, despite numerous studies and guidelines suggesting this practice has little benefit? Is it because patients expect a prescription in return for their copayment? Is it the path of least resistance? Or do physicians not accept the data showing that it is unnecessary?
Dr. Gerald Appel discusses diabetic nephropathy, an area that involves resistance of another kind, ie, the apparent resistance of physicians and patients to achieving evidence-based treatment targets. We hold controlled trials as the Holy Grail of evidence-based medicine, yet we seem to have an aversion to following guidelines based on trial-derived evidence. (I do not refer here to blind guideline adherence, ignoring individual patient characteristics.)
So how can physicians’ behavior be altered and our resistance to change be reduced? Experiments are under way, such as paying physicians based on their performance, linking patients’ insurance rates to achieving selected outcomes, and linking physician practice self-review to certification. Perhaps naively, I continue to believe that the most effective impetus to changing personal practice is the dissemination of data from high-quality trials (tempered by our accumulated experience and keeping our eyes wide open), coupled with our desire to do the best for our patients.
- Barber M. Coagulase-positive staphylococci resistant to penicillin. J Pathol Bacteriol 1947; 59:373–384.
- Barber M. Coagulase-positive staphylococci resistant to penicillin. J Pathol Bacteriol 1947; 59:373–384.
Detecting and controlling diabetic nephropathy: What do we know?
Diabetes is on the rise, and so is diabetic nephropathy. In view of this epidemic, physicians should consider strategies to detect and control kidney disease in their diabetic patients.
This article will focus on kidney disease in adult-onset type 2 diabetes. Although it has different pathogenetic mechanisms than type 1 diabetes, the clinical course of the two conditions is very similar in terms of the prevalence of proteinuria after diagnosis, the progression to renal failure after the onset of proteinuria, and treatment options.1
DIABETES AND DIABETIC KIDNEY DISEASE ARE ON THE RISE
The incidence of diabetes increases with age, and with the aging of the baby boomers, its prevalence is growing dramatically. The 2005– 2008 National Health and Nutrition Examination Survey estimated the prevalence as 3.7% in adults age 20 to 44, 13.7% at age 45 to 64, and 26.9% in people age 65 and older. The obesity epidemic is also contributing to the increase in diabetes in all age groups.
Diabetic kidney disease has increased in the United States from about 4 million cases 20 years ago to about 7 million in 2005–2008.2 Diabetes is the major cause of end-stage renal disease in the developed world, accounting for 40% to 50% of cases. Other major causes are hypertension (27%) and glomerulonephritis (13%).3
Physicians in nearly every field of medicine now care for patients with diabetic nephropathy. The classic presentation—a patient who has impaired vision, fluid retention with edema, and hypertension—is commonly seen in dialysis units and ophthalmology and cardiovascular clinics.
CLINICAL PROGRESSION
Early in the course of diabetic nephropathy, blood pressure is normal and microalbuminuria is not evident, but many patients have a high glomerular filtration rate (GFR), indicating temporarily “enhanced” renal function or hyperfiltration. The next stage is characterized by microalbuminuria, correlating with glomerular mesangial expansion: the GFR falls back into the normal range and blood pressure starts to increase. Finally, macroalbuminuria occurs, accompanied by rising blood pressure and a declining GFR, correlating with the histologic appearance of glomerulosclerosis and Kimmelstiel-Wilson nodules.4
Hypertension develops in 5% of patients by 10 years after type 1 diabetes is diagnosed, 33% by 20 years, and 70% by 40 years. In contrast, 40% of patients with type 2 diabetes have high blood pressure at diagnosis.
Unfortunately, in most cases, this progression is a one-way street, so it is critical to intervene to try to slow the progression early in the course of the disease process.
SCREENING FOR DIABETIC NEPHROPATHY
Nephropathy screening guidelines for patients with diabetes are provided in Table 1.5

Blood pressure should be monitored at each office visit (Table 1). The goal for adults with diabetes should be to reduce blood pressure to 130/80 mm Hg. Reduction beyond this level may be associated with an increased mortality rate.6 Very high blood pressure (> 180 mm Hg systolic) should be lowered slowly. Lowering blood pressure delays the progression from microalbuminuria (30–299 mg/day or 20–199 μg/min) to macroalbuminuria (> 300 mg/day or > 200 μg/min) and slows the progression to renal failure.
Urinary albumin. Proteinuria takes 5 to 10 years to develop after the onset of diabetes. Because it is possible for patients with type 2 diabetes to have had the disease for some time before being diagnosed, urinary albumin screening should be performed at diagnosis and annually thereafter. Patients with type 1 are usually diagnosed with diabetes at or near onset of disease; therefore, annual screening for urinary albumin can begin 5 years after diagnosis.5
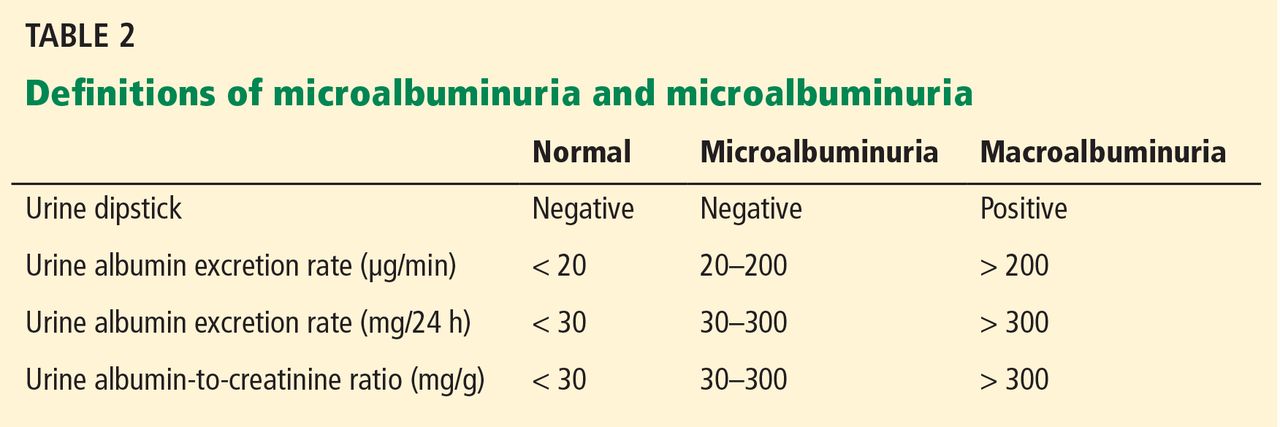
Proteinuria can be measured in different ways (Table 2). The basic screening test for clinical proteinuria is the urine dipstick, which is very sensitive to albumin and relatively insensitive to other proteins. “Trace-positive” results are common in healthy people, so proteinuria is not confirmed unless a patient has repeatedly positive results.
Microalbuminuria is important to measure, especially if it helps determine therapy. It is not detectable by the urinary dipstick, but can be measured in the following ways:
- Measurement of the albumin-creatinine ratio in a random spot collection
- 24-hour collection (creatinine should simultaneously be measured and creatinine clearance calculated)
- Timed collection (4 hours or overnight).
The first method is preferred, and any positive test result must be confirmed by repeat analyses of urinary albumin before a patient is diagnosed with microalbuminuria.
Occasionally a patient presenting with proteinuria but normal blood sugar and hemoglobin A1c will have a biopsy that reveals morphologic changes of classic diabetic nephropathy. Most such patients have a history of hyperglycemia, indicating that they actually have been diabetic.
Proteinuria—the best marker of disease progression
Proteinuria is the strongest predictor of renal outcomes. The Reduction in End Points in Noninsulin-Dependent Diabetes Mellitus With the Angiotensin II Antagonist Losartan (RENAAL) study was a randomized, placebo-controlled trial in more than 1,500 patients with type 2 diabetes to test the effects of losartan on renal outcome. Those with high albuminuria (> 3.0 g albumin/g creatinine) at baseline were five times more likely to reach a renal end point and were eight times more likely to have progression to end-stage renal disease than patients with low albuminuria (< 1.5 g/g).7 The degree of albuminuria after 6 months of treatment showed similar predictive trends, indicating that monitoring and treating proteinuria are extremely important goals.
STRATEGY 1 TO LIMIT RENAL INJURY: REDUCE BLOOD PRESSURE
Blood pressure control improves renal and cardiovascular function.
As early as 1983, Parving et al,8 in a study of only 10 insulin-dependent diabetic patients, showed strong evidence that early aggressive antihypertensive treatment improved the course of diabetic nephropathy. During the mean pretreatment period of 29 months, the GFR decreased significantly and the urinary albumin excretion rate and arterial blood pressure rose significantly. During the mean 39-month period of antihypertensive treatment with metoprolol, hydralazine, and furosemide or a thiazide, mean arterial blood pressure fell from 144/97 to 128/84 mm Hg and urinary albumin excretion from 977 to 433 μg/ min. The rate of decline in GFR slowed from 0.91 mL/min/month before treatment to 0.39 mL/min/month during treatment.
The Action in Diabetes and Vascular Disease: Preterax and Diamicron MR Controlled Evaluation (ADVANCE) trial9 enrolled more than 11,000 patients internationally with type 2 diabetes at high risk for cardiovascular events. In addition to standard therapy, blood pressure was intensively controlled in one group with a combination of the angiotensin-converting enzyme (ACE) inhibitor perindopril and the diuretic indapamide. The intensive-therapy group achieved blood pressures less than 140/80 mm Hg and had a mean reduction of systolic blood pressure of 5.6 mm Hg and diastolic blood pressure of 2.2 mm Hg vs controls. Despite these apparently modest reductions, the intensively controlled group had a significant 9% reduction of the primary outcome of combined macrovascular events (cardiovascular death, myocardial infarction, and stroke) and microvascular events (new or worsening nephropathy, or retinopathy).10
A meta-analysis of studies of patients with type 2 diabetes found reduced nephropathy with systolic blood pressure control to less than 130 mm Hg.11
The United Kingdom Prospective Diabetes Study (UKPDS) is a series of studies of diabetes. The original study in 1998 enrolled 5,102 patients with newly diagnosed type 2 diabetes.12 The more than 1,000 patients with hypertension were randomized to either tight blood pressure control or regular care. The intensive treatment group had a mean blood pressure reduction of 9 mm Hg systolic and 3 mm Hg diastolic, along with major reductions in all diabetes end points, diabetes deaths, microvascular disease, and stroke over a median follow-up of 8.4 years.
Continuous blood pressure control is critical
Tight blood pressure control must be maintained to have continued benefit. During the 10 years following the UKPDS, no attempts were made to maintain the previously assigned therapies. A follow-up study13 of 884 UKPDS patients found that blood pressures were the same again between the two groups 2 years after the trial was stopped, and no beneficial legacy effect from previous blood pressure control was evident on end points.
Control below 120 mm Hg systolic not needed
Blood pressure control slows kidney disease and prevents major macrovascular disease, but there is no evidence that lowering systolic blood pressure below 120 mm Hg provides additional benefit. In the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial,14 more than 10,000 patients with type 2 diabetes and existing cardiovascular disease or additional cardiovascular risk factors were randomized to a goal of systolic blood pressure less than 120 mm Hg or less than 140 mm Hg (actual mean systolic pressures were 119 vs 134 mm Hg, respectively). Over nearly 5 years, there was no difference in cardiovascular events or deaths between the two groups.15

Since 1997, six international organizations have revised their recommended blood pressure goals in diabetes mellitus and renal diseases. Randomized clinical trials and observational studies have demonstrated the importance of blood pressure control to the level of 125/75 to 140/80 mm Hg. The National Kidney Foundation, the American Diabetes Association, and the Canadian Hypertension Society have developed consensus guidelines for blood pressure control to less than 130/80 mm Hg.16–21 Table 3 summarizes blood pressure goals for patients with diabetes.
STRATEGY 2: CONTROL BLOOD SUGAR
Recommendations for blood sugar goals are more controversial.
The Diabetes Control and Complications Trial22 provided early evidence that tight blood sugar control slows the development of microalbuminuria and macroalbuminuria. The study randomized more than 1,400 patients with type 1 diabetes to either standard therapy (1 or 2 daily insulin injections) or intensive therapy (an external insulin pump or 3 or more insulin injections guided by frequent blood glucose monitoring) to keep blood glucose levels close to normal. About half the patients had mild retinopathy at baseline and the others had no retinopathy. After 6.5 years, intensive therapy was found to significantly delay the onset and slow the progression of diabetic retinopathy and nephropathy.
The Kumamoto Study23 randomized 110 patients with type 2 diabetes and either no retinopathy (primary prevention cohort) or simple retinopathy (secondary prevention cohort) to receive either multiple insulin injections or conventional insulin therapy over 8 years. Intensive therapy led to lower rates of retinopathy (7.7% vs 32% in primary prevention and 19% vs 44% in secondary prevention) and progressive nephropathy (7% vs 28% in primary prevention at 6 years and 11% vs 32% in secondary prevention).
In addition to studying the effects of blood pressure control, the UKPDS also studied the effects of intensive blood glucose control.24,25 Nearly 4,000 patients with newly diagnosed type 2 diabetes were randomized to intensive treatment with a sulfonylurea or insulin, or to conventional treatment with diet. Over 10 years, the mean hemoglobin A1c was reduced to 7.0% in the intensive group and 7.9% in the conventional group. The risk of any diabetes-related end point was 12% lower in the intensive group, 10% lower for diabetes-related death, and 6% lower for all-cause mortality. There was also a 25% reduction in microvascular disease (retinopathy and nephropathy). However, the intensive group had more hypoglycemic episodes than the conventional group and a tendency to some increase in macrovascular events. A legacy effect was evident: patients who had intensive treatment had less microvascular disease progression years after stopping therapy.
Tight glycemic control reduces nephropathy, but does it increase cardiovascular risk?
Earlier trials provided strong evidence that blood glucose control prevents or slows retinopathy and nephropathy. The critical question is, “At what expense?” Although diabetes is the most common cause of kidney failure in the United States, most people with diabetes do not die of kidney failure, but of cardiovascular disease. Two recent large trials had different results regarding glycemic control below hemoglobin A1c of 7.0% and macrovascular risk, creating a controversy about what recommendations are best.
The ADVANCE trial, enrolling 11,140 patients with type 2 diabetes, was largely conducted in Australia and used the sulfonylurea glipizide for glycemic control. Compared with the group that received standard therapy (n=5,569), the intensive-treatment group (n=5,571) achieved mean hemoglobin A1c levels of 6.5% compared with 7.3% in the standard group, and had less nephropathy, less microalbuminuria, less doubling of creatinine, and a lower rate of end-stage renal disease (4% vs 5% in the standard therapy group). No difference between the two groups was found in retinopathy. Rates of all-cause mortality did not differ between the groups.9
The ACCORD trial had more than 10,000 subjects with type 2 diabetes and took place mostly in the United States. Using mainly rosiglitazone for intensive therapy, the intensive group achieved hemoglobin A1c levels of 6.4% vs 7.5% in the standard-therapy group. The trial was stopped early, at 3.7 years, because of a higher risk of death and cardiovascular events in the group with intensive glycemic control. However, the intensive-therapy group did have a significant decrease in microvascular renal outcomes and a reduction in the progression of retinopathy.14,26
In summary, tighter glycemic control improves microvascular complications—both retinopathy and nephropathy—in patients with type 2 diabetes. The benefit of intensive therapy on macrovascular complications (stroke, myocardial infarction) in long-standing diabetes has not been convincingly demonstrated in randomized trials. The UKPDS suggested that maintaining a hemoglobin A1c of 7% in patients newly diagnosed with type 2 diabetes confers long-term cardiovascular benefits. The target hemoglobin A1c for type 2 diabetes should be tailored to the patient: 7% is a reasonable goal for most patients, but the goal should be higher for the elderly and frail. Reducing the risk of cardiovascular death is still best done by controlling blood pressure, reducing lipids, quitting smoking, and losing weight.
STRATEGY 3: INHIBIT THE RENIN-ANGIOTENSIN-ALDOSTERONE AXIS
Components of the renin-angiotensin-aldo-sterone system are present not only in the circulation but also in many tissues, including the heart, brain, kidney, blood vessels, and adrenal glands. The role of renin-angiotensin-aldosterone system blockers in treating and preventing diabetic nephropathy has become controversial in recent years with findings from new studies.
The renin-angiotensin-aldosterone system is important in the development or maintenance of high blood pressure and the resultant damage to the brain, heart, and kidney. Drug development has focused on inhibiting steps in the biochemical pathway. ACE inhibitors block the formation of angiotensin II—the most biologically potent angiotensin peptide—and are among the most commonly used drugs to treat hypertension and concomitant conditions, such as renal insufficiency, proteinuria, and heart failure. Angiotensin receptor blockers (ARBs) interact with the angiotensin AT1 receptor and block most of its actions. They are approved by the US Food and Drug Administration (FDA) for the treatment of hypertension, and they help prevent left ventricular hypertrophy and mesangial sclerosis. Large studies have shown that ACE inhibitors and ARBs offer similar cardiovascular benefit.
The glomerulus has the only capillary bed with a blood supply that drains into an efferent arteriole instead of a venule, providing high resistance to aid filtration. Efferent arterioles are rich in AT1 receptors. In the presence of angiotensin II they constrict, increasing pressure in the glomerulus, which can lead to proteinuria and glomerulosclerosis. ACE inhibitors and ARBs relax the efferent arteriole, allowing increased blood flow through the glomerulus. This reduction in intraglomerular pressure is associated with less proteinuria and less glomerulosclerosis.
Diabetes promotes renal disease in many ways. Glucose and advanced glycation end products can lead to increased blood flow and increased pressure in the glomerulus. Through a variety of pathways, hyperglycemia, acting on angiotensin II, leads to NF-kapa beta production, profibrotic cytokines, increased matrix, and eventual fibrosis. ACE inhibitors and ARBs counteract many of these.
ACE inhibitors and ARBs slow nephropathy progression beyond blood pressure control
Several major clinical trials27–32 examined the effects of either ACE inhibitors or ARBs in slowing the progression of diabetic nephropathy and have had consistently positive results.
The Collaborative Study Group30 was a 3-year randomized trial in 419 patients with type 1 diabetes, using the ACE inhibitor captopril vs placebo. Captopril was associated with less decline in kidney function and a 50% reduction in the risk of the combined end points of death, dialysis, and transplantation that was independent of the small difference in blood pressures between the two groups.
The Irbesartan Diabetic Nephropathy Trial (IDNT)31 studied the effect of the ARB irbesartan vs the calcium channel blocker amlodipine vs placebo over 2.6 years in 1,715 patients with type 2 diabetes. Irbesartan was found to be significantly more effective in protecting against the progression of nephropathy, independent of reduction in blood pressure.
The RENAAL trial,32 published in 2001, was a 3-year, randomized, double-blind study comparing the ARB losartan at increasing dosages with placebo (both taken in addition to conventional antihypertensive treatment) in 1,513 patients with type 2 diabetes and nephropathy. The blood pressure goal was 140/90 mm Hg in both groups, but the losartan group had a lower rate of doubling of serum creatinine, end-stage renal disease, and combined end-stage renal disease or death.
‘Aldosterone escape’ motivates the search for new therapies
An important reason for developing more ways to block the renin-angiotensin-aldosterone system is because of “aldosterone escape,” the phenomenon of angiotensin II or aldosterone returning to pretreatment levels despite continued ACE inhibition.
Biollaz et al,33 in a 1982 study of 19 patients with hypertension, showed that despite reducing blood pressure and keeping the blood level of ACE very low with twice-daily enalapril 20 mg, blood and urine levels of angiotensin II steadily rose back to baseline levels within a few months.
A growing body of evidence suggests that despite effective inhibition of angiotensin II activity, non-ACE synthetic pathways still permit angiotensin II generation via serine proteases such as chymase, cathepsin G, and tissue plasminogen activator.
Thus, efforts have been made to block the renin-angiotensin system in other places. In addition to ACE inhibitors and ARBs, two aldosterone receptor antagonists are available, spironolactone and eplerenone, both used to treat heart failure. A direct renin inhibitor, aliskiren, is also available.
Combination therapy—less proteinuria, but…
A number of studies have shown that combination treatment with agents having different targets in the renin-angiotensin-aldosterone system leads to larger reductions in albuminuria than does single-agent therapy.
Mogensen et al34 studied the effect of the ACE inhibitor lisinopril (20 mg per day) plus the ARB candesartan (16 mg per day) in subjects with microalbuminuria, hypertension, and type 2 diabetes. Combined treatment was more effective in reducing proteinuria.
Epstein et al35 studied the effects of the ACE inhibitor enalapril (20 mg/day) combined with either of two doses of the selective aldosterone receptor antagonist eplerenone (50 or 100 mg/day) or placebo. Both eplerenone dosages, when added to the enalapril treatment, significantly reduced albuminuria from baseline as early as week 4 (P < .001), but placebo treatment added to the enalapril did not result in any significant decrease in urinary albumin excretion. Systolic blood pressure decreased significantly in all treatment groups and by about the same amount.
The Aliskiren Combined With Losartan in Type 2 Diabetes and Nephropathy (AVOID) trial36 randomized more than 600 patients with type 2 diabetes and nephropathy to aliskiren (a renin inhibitor) or placebo added to the ARB losartan. Again, combination treatment was more renoprotective, independent of blood pressure lowering.
Worse outcomes with combination therapy?
More recent studies have indicated that although combination therapy reduces proteinuria to a greater extent than monotherapy, overall it worsens major renal and cardiovascular outcomes. The multicenter Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET)37 randomized more than 25,000 patients age 55 and older with established atherosclerotic vascular disease or with diabetes and end-organ damage to receive either the ARB telmisartan 80 mg daily, the ACE inhibitor ramipril 10 mg daily, or both. Mean follow-up was 56 months. The combination-treatment group had higher rates of death and renal disease than the single-therapy groups (which did not differ from one another).
Why the combination therapy had poorer outcomes is under debate. Patients may get sudden drops in blood pressure that are not detected with only periodic monitoring. Renal failure was mostly acute rather than chronic, and the estimated GFR declined more in the combined therapy group than in the single-therapy groups.
The Aliskiren Trial in Type 2 Diabetes Using Cardiovascular and Renal Disease Endpoints (ALTITUDE) was designed to test the effect of the direct renin inhibitor aliskiren or placebo, both arms combined with either an ACE inhibitor or an ARB in patients with type 2 diabetes at high risk for cardiovascular and renal events. The trial was terminated early because of more strokes and deaths in the combination therapy arms. The results led the FDA to issue black box warnings against using aliskiren with these other classes of agents, and all studies testing similar combinations have been stopped. (In one study that was stopped and has not yet been published, 100 patients with proteinuria were treated with either aliskiren, the ARB losartan, or both, to evaluate the effects of aldosterone escape. Results showed no differences: about one-third of each group had this phenomenon.)
My personal recommendation is as follows: for younger patients with proteinuria, at lower risk for cardiovascular events and with disease due not to diabetes but to immunoglobulin A nephropathy or another proteinuric kidney disease, treat with both an ACE inhibitor and ARB. But the combination should not be used for patients at high risk of cardiovascular disease, which includes almost all patients with diabetes.
If more aggressive renin-angiotensin system blockade is needed against diabetic nephropathy, adding a diuretic increases the impact of blocking the renin-angiotensin-aldosterone system on both proteinuria and progression of renal disease. The aldosterone blocker spironolactone 25 mg can be added if potassium levels are carefully monitored.
ACE inhibitor plus calcium channel blocker is safer than ACE inhibitor plus diuretic
The Avoiding Cardiovascular Events Through Combination Therapy in Patients Living With Systolic Hypertension (ACCOMPLISH) trial38 randomized more than 11,000 high-risk patients with hypertension to receive an ACE inhibitor (benazepril) plus either a calcium channel blocker (amlodipine) or thiazide diuretic (hydrochlorothiazide). Blood pressures were identical between the two groups, but the trial was terminated early, at 36 months, because of a higher risk of the combined end point of cardiovascular death, myocardial infarction, stroke, and other major cardiac events in the ACE inhibitor-thiazide group.
Although some experts believe this study is definitive and indicates that high blood pressure should never be treated with an ACE inhibitor-thiazide combination, I believe that caution is needed in interpreting these findings. This regimen should be avoided in older patients with diabetes at high risk for cardiovascular disease, but otherwise, getting blood pressure under control is critical, and this combination can be used if it works and the patient is tolerating it well.
In summary, the choice of blood pressure-lowering medications is based on reducing cardiovascular events and slowing the progression of kidney disease. Either an ACE inhibitor or an ARB is the first choice for patients with diabetes, hypertension, and any degree of proteinuria. Many experts recommend beginning one of these agents even if proteinuria is not present. However, the combination of an ACE inhibitor and ARB should not be used in diabetic patients, especially if they have cardiovascular disease, until further data clarify the results of the ONTARGET and ALTITUDE trials.
STRATEGY 4: METABOLIC MANIPULATION WITH NOVEL AGENTS
Several new agents have recently been studied for the treatment of diabetic nephropathy, including aminoguanidine, which reduces levels of advanced glycation end-products, and sulodexide, which blocks basement membrane permeability. Neither agent has been shown to be safe and effective in diabetic nephropathy. The newest agent is bardoxolone methyl. It induces the Keap1–Nrf2 pathway, which up-regulates cytoprotective factors, suppressing inflammatory and other cytokines that are major mediators of progression of chronic kidney disease.39
Pergola et al,40 in a phase 2, double-blind trial, randomized 227 adults with diabetic kidney disease and a low estimated GFR (20–45 mL/min/1.73 m2) to receive placebo or bardoxolone 25, 75, or 150 mg daily. Drug treatment was associated with improvement in the estimated GFR, a finding that persisted throughout the 52 weeks of treatment. Surprisingly, proteinuria did not decrease with drug treatment.
As of this writing, a large multicenter controlled randomized trial has been halted because of concerns by the data safety monitoring board, which found increased rates of death and fluid retention with the drug. A number of recent trials have shown a beneficial effect of sodium bicarbonate therapy in patients with late-stage chronic kidney disease. They have shown slowing of the progression of GFR decline in a number of renal diseases, including diabetes.
- Ritz E, Orth SR. Nephropathy in patients with type 2 diabetes mellitus. N Engl J Med 1999; 341:1127–1133.
- de Boer IH, Rue TC, Hall YN, Heagerty PJ, Weiss NS, Himmelfarb J. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA 2011; 305:2532–2539.
- United States Renal Data System (USRDS) 2000 Annual Data Report. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases – Division of Kidney, Urologic and Hematologic Diseases. USRDS Coordinating Center operated by the Minneapolis Medical Research Foundation. www.usrds.org
- Macisaac RJ, Jerums G. Diabetic kidney disease with and without albuminuria. Curr Opin Nephrol Hypertens 2011; 20:246–257.
- Molitch ME, DeFronzo RA, Franz MJ, et al; American Diabetes Association. Nephropathy in diabetes. Diabetes Care 2004; 27(suppl 1):S79–S83.
- Vamos EP, Harris M, Millett C, et al. Association of systolic and diastolic blood pressure and all cause mortality in people with newly diagnosed type 2 diabetes: retrospective cohort study. BMJ 2012; 345:e5567.
- de Zeeuw D, Remuzzi G, Parving HH, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int 2004; 65:2309–2320.
- Parving HH, Andersen AR, Smidt UM, Svendsen PA. Early aggressive antihypertensive treatment reduces rate of decline in kidney function in diabetic nephropathy. Lancet 1983; 1:1175–1179.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): a randomised controlled trial. Lancet 2007; 370:829–840.
- Bangalore S, Kumar S, Lobach I, Messerli FH. Blood pressure targets in subjects with type 2 diabetes mellitus/impaired fasting glucose: observations from traditional and bayesian random-effects meta-analyses of randomized trials. Circulation 2011; 123:2799–2810.
- UK Prospective Diabetes Study Group. Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. BMJ 1998; 317:703–713.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577–1589.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- American Diabetes Association. Standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S11–S63. (Erratum in: Diabetes Care 2012; 35:660.)
- Bakris GL, Williams M, Dworkin L, et al. Preserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertension and Diabetes Executive Committees Working Group. Am J Kidney Dis 2000; 36:646–661.
- Ramsay L, Williams B, Johnston G, et al. Guidelines for management of hypertension: report of the third working party of the British Hypertension Society. J Hum Hypertens 1999; 13:569–592.
- Feldman RD, Campbell N, Larochelle P, et al. 1999 Canadian recommendations for the management of hypertension. Task Force for the Development of the 1999 Canadian Recommendations for the Management of Hypertension. CMAJ 1999; 161(suppl):12:S1–S17.
- Chalmers J, MacMahon S, Mancia G, et al. 1999 World Health Organization-International Society of Hypertension Guidelines for the management of hypertension. Guidelines Sub-committee of the World Health Organization. Clin Exp Hypertens 1999; 21:1009–1060.
- The seventh report of the Joint National Committee on Prevention, Detection Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 1993; 329:977–986.
- Shichiri M, Kishikawa H, Ohkubo Y, Wake N. Long-term results of the Kumamoto Study on optimal diabetes control in type 2 diabetic patients. Diabetes Care 2000; 23(suppl 2):B21–B29.
- Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998; 352:837–853. Erratum in: Lancet 1999; 354:602.
- Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. UK Prospective Diabetes Study Group. BMJ 1998; 317:703–713. Erratum in: BMJ 1999; 318:29.
- Ismail-Beigi F, Craven T, Banerji MA, et al; ACCORD trial group. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: an analysis of the ACCORD randomised trial. Lancet 2010; 376:419–430. Erratum in: Lancet 2010; 376:1466.
- Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Heart Outcomes Prevention Evaluation Study Investigators. Lancet 2000: 355:253–259. Erratum in: Lancet2000; 356:860.
- Parving HH, Lehnert H, Bröchner-Mortensen J, et al; Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria Study Group. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med 2001; 345:870–878.
- Viberti G, Wheeldon NM; MicroAlbuminuria Reduction With VALsartan (MARVAL) Study Investigators. Microalbuminuria reduction with valsartan in patients with type 2 diabetes mellitus: a blood pressure-independent effect. Circulation 2002; 106:672–678.
- Lewis EJ, Hunsicker LG, Bain R P, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 1993; 329:1456–1462.
- Lewis EJ, Hunsicker LG, Clarke WR, et al; Collaborative Study Group. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345:851–860.
- Brenner BM, Cooper ME, de Zeeuw D, et al; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345:861–869.
- Biollaz J, Brunner HR, Gavras I, Waeber B, Gavras H. Antihypertensive therapy with MK 421: angiotensin II--renin relationships to evaluate efficacy of converting enzyme blockade. J Cardiovasc Pharmacol 1982; 4:966–972.
- Mogensen CE, Neldam S, Tikkanen I, et al. Randomised controlled trial of dual blockade of renin-angiotensin system in patients with hypertension, microalbuminuria, and non-insulin dependent diabetes: the candesartan and lisinopril microalbuminuria (CALM) study. BMJ 2000; 321:1440–1444.
- Epstein M, Williams GH, Weinberger M, et al. Selective aldosterone blockade with eplerenone reduces albuminuria in patients with type 2 diabetes. Clin J Am Soc Nephrol 2006; 1:940–951.
- Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK; AVOID Study Investigators. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med 2008; 358:2433–2446.
- Mann JF, Schmieder RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Jamerson K, Weber MA, Bakris GL, et al; ACCOMPLISH Trial Investigators. Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N Engl J Med 2008; 359:2417–2428.
- Kim HJ, Vaziri ND. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am J Physiol Renal Physiol 2010; 298:F662–F671.
- Pergola PE, Raskin P, Toto RD, et al; BEAM Study Investigators Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 2011; 365:327–336.
Diabetes is on the rise, and so is diabetic nephropathy. In view of this epidemic, physicians should consider strategies to detect and control kidney disease in their diabetic patients.
This article will focus on kidney disease in adult-onset type 2 diabetes. Although it has different pathogenetic mechanisms than type 1 diabetes, the clinical course of the two conditions is very similar in terms of the prevalence of proteinuria after diagnosis, the progression to renal failure after the onset of proteinuria, and treatment options.1
DIABETES AND DIABETIC KIDNEY DISEASE ARE ON THE RISE
The incidence of diabetes increases with age, and with the aging of the baby boomers, its prevalence is growing dramatically. The 2005– 2008 National Health and Nutrition Examination Survey estimated the prevalence as 3.7% in adults age 20 to 44, 13.7% at age 45 to 64, and 26.9% in people age 65 and older. The obesity epidemic is also contributing to the increase in diabetes in all age groups.
Diabetic kidney disease has increased in the United States from about 4 million cases 20 years ago to about 7 million in 2005–2008.2 Diabetes is the major cause of end-stage renal disease in the developed world, accounting for 40% to 50% of cases. Other major causes are hypertension (27%) and glomerulonephritis (13%).3
Physicians in nearly every field of medicine now care for patients with diabetic nephropathy. The classic presentation—a patient who has impaired vision, fluid retention with edema, and hypertension—is commonly seen in dialysis units and ophthalmology and cardiovascular clinics.
CLINICAL PROGRESSION
Early in the course of diabetic nephropathy, blood pressure is normal and microalbuminuria is not evident, but many patients have a high glomerular filtration rate (GFR), indicating temporarily “enhanced” renal function or hyperfiltration. The next stage is characterized by microalbuminuria, correlating with glomerular mesangial expansion: the GFR falls back into the normal range and blood pressure starts to increase. Finally, macroalbuminuria occurs, accompanied by rising blood pressure and a declining GFR, correlating with the histologic appearance of glomerulosclerosis and Kimmelstiel-Wilson nodules.4
Hypertension develops in 5% of patients by 10 years after type 1 diabetes is diagnosed, 33% by 20 years, and 70% by 40 years. In contrast, 40% of patients with type 2 diabetes have high blood pressure at diagnosis.
Unfortunately, in most cases, this progression is a one-way street, so it is critical to intervene to try to slow the progression early in the course of the disease process.
SCREENING FOR DIABETIC NEPHROPATHY
Nephropathy screening guidelines for patients with diabetes are provided in Table 1.5
Blood pressure should be monitored at each office visit (Table 1). The goal for adults with diabetes should be to reduce blood pressure to 130/80 mm Hg. Reduction beyond this level may be associated with an increased mortality rate.6 Very high blood pressure (> 180 mm Hg systolic) should be lowered slowly. Lowering blood pressure delays the progression from microalbuminuria (30–299 mg/day or 20–199 μg/min) to macroalbuminuria (> 300 mg/day or > 200 μg/min) and slows the progression to renal failure.
Urinary albumin. Proteinuria takes 5 to 10 years to develop after the onset of diabetes. Because it is possible for patients with type 2 diabetes to have had the disease for some time before being diagnosed, urinary albumin screening should be performed at diagnosis and annually thereafter. Patients with type 1 are usually diagnosed with diabetes at or near onset of disease; therefore, annual screening for urinary albumin can begin 5 years after diagnosis.5
Proteinuria can be measured in different ways (Table 2). The basic screening test for clinical proteinuria is the urine dipstick, which is very sensitive to albumin and relatively insensitive to other proteins. “Trace-positive” results are common in healthy people, so proteinuria is not confirmed unless a patient has repeatedly positive results.
Microalbuminuria is important to measure, especially if it helps determine therapy. It is not detectable by the urinary dipstick, but can be measured in the following ways:
- Measurement of the albumin-creatinine ratio in a random spot collection
- 24-hour collection (creatinine should simultaneously be measured and creatinine clearance calculated)
- Timed collection (4 hours or overnight).
The first method is preferred, and any positive test result must be confirmed by repeat analyses of urinary albumin before a patient is diagnosed with microalbuminuria.
Occasionally a patient presenting with proteinuria but normal blood sugar and hemoglobin A1c will have a biopsy that reveals morphologic changes of classic diabetic nephropathy. Most such patients have a history of hyperglycemia, indicating that they actually have been diabetic.
Proteinuria—the best marker of disease progression
Proteinuria is the strongest predictor of renal outcomes. The Reduction in End Points in Noninsulin-Dependent Diabetes Mellitus With the Angiotensin II Antagonist Losartan (RENAAL) study was a randomized, placebo-controlled trial in more than 1,500 patients with type 2 diabetes to test the effects of losartan on renal outcome. Those with high albuminuria (> 3.0 g albumin/g creatinine) at baseline were five times more likely to reach a renal end point and were eight times more likely to have progression to end-stage renal disease than patients with low albuminuria (< 1.5 g/g).7 The degree of albuminuria after 6 months of treatment showed similar predictive trends, indicating that monitoring and treating proteinuria are extremely important goals.
STRATEGY 1 TO LIMIT RENAL INJURY: REDUCE BLOOD PRESSURE
Blood pressure control improves renal and cardiovascular function.
As early as 1983, Parving et al,8 in a study of only 10 insulin-dependent diabetic patients, showed strong evidence that early aggressive antihypertensive treatment improved the course of diabetic nephropathy. During the mean pretreatment period of 29 months, the GFR decreased significantly and the urinary albumin excretion rate and arterial blood pressure rose significantly. During the mean 39-month period of antihypertensive treatment with metoprolol, hydralazine, and furosemide or a thiazide, mean arterial blood pressure fell from 144/97 to 128/84 mm Hg and urinary albumin excretion from 977 to 433 μg/ min. The rate of decline in GFR slowed from 0.91 mL/min/month before treatment to 0.39 mL/min/month during treatment.
The Action in Diabetes and Vascular Disease: Preterax and Diamicron MR Controlled Evaluation (ADVANCE) trial9 enrolled more than 11,000 patients internationally with type 2 diabetes at high risk for cardiovascular events. In addition to standard therapy, blood pressure was intensively controlled in one group with a combination of the angiotensin-converting enzyme (ACE) inhibitor perindopril and the diuretic indapamide. The intensive-therapy group achieved blood pressures less than 140/80 mm Hg and had a mean reduction of systolic blood pressure of 5.6 mm Hg and diastolic blood pressure of 2.2 mm Hg vs controls. Despite these apparently modest reductions, the intensively controlled group had a significant 9% reduction of the primary outcome of combined macrovascular events (cardiovascular death, myocardial infarction, and stroke) and microvascular events (new or worsening nephropathy, or retinopathy).10
A meta-analysis of studies of patients with type 2 diabetes found reduced nephropathy with systolic blood pressure control to less than 130 mm Hg.11
The United Kingdom Prospective Diabetes Study (UKPDS) is a series of studies of diabetes. The original study in 1998 enrolled 5,102 patients with newly diagnosed type 2 diabetes.12 The more than 1,000 patients with hypertension were randomized to either tight blood pressure control or regular care. The intensive treatment group had a mean blood pressure reduction of 9 mm Hg systolic and 3 mm Hg diastolic, along with major reductions in all diabetes end points, diabetes deaths, microvascular disease, and stroke over a median follow-up of 8.4 years.
Continuous blood pressure control is critical
Tight blood pressure control must be maintained to have continued benefit. During the 10 years following the UKPDS, no attempts were made to maintain the previously assigned therapies. A follow-up study13 of 884 UKPDS patients found that blood pressures were the same again between the two groups 2 years after the trial was stopped, and no beneficial legacy effect from previous blood pressure control was evident on end points.
Control below 120 mm Hg systolic not needed
Blood pressure control slows kidney disease and prevents major macrovascular disease, but there is no evidence that lowering systolic blood pressure below 120 mm Hg provides additional benefit. In the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial,14 more than 10,000 patients with type 2 diabetes and existing cardiovascular disease or additional cardiovascular risk factors were randomized to a goal of systolic blood pressure less than 120 mm Hg or less than 140 mm Hg (actual mean systolic pressures were 119 vs 134 mm Hg, respectively). Over nearly 5 years, there was no difference in cardiovascular events or deaths between the two groups.15
Since 1997, six international organizations have revised their recommended blood pressure goals in diabetes mellitus and renal diseases. Randomized clinical trials and observational studies have demonstrated the importance of blood pressure control to the level of 125/75 to 140/80 mm Hg. The National Kidney Foundation, the American Diabetes Association, and the Canadian Hypertension Society have developed consensus guidelines for blood pressure control to less than 130/80 mm Hg.16–21 Table 3 summarizes blood pressure goals for patients with diabetes.
STRATEGY 2: CONTROL BLOOD SUGAR
Recommendations for blood sugar goals are more controversial.
The Diabetes Control and Complications Trial22 provided early evidence that tight blood sugar control slows the development of microalbuminuria and macroalbuminuria. The study randomized more than 1,400 patients with type 1 diabetes to either standard therapy (1 or 2 daily insulin injections) or intensive therapy (an external insulin pump or 3 or more insulin injections guided by frequent blood glucose monitoring) to keep blood glucose levels close to normal. About half the patients had mild retinopathy at baseline and the others had no retinopathy. After 6.5 years, intensive therapy was found to significantly delay the onset and slow the progression of diabetic retinopathy and nephropathy.
The Kumamoto Study23 randomized 110 patients with type 2 diabetes and either no retinopathy (primary prevention cohort) or simple retinopathy (secondary prevention cohort) to receive either multiple insulin injections or conventional insulin therapy over 8 years. Intensive therapy led to lower rates of retinopathy (7.7% vs 32% in primary prevention and 19% vs 44% in secondary prevention) and progressive nephropathy (7% vs 28% in primary prevention at 6 years and 11% vs 32% in secondary prevention).
In addition to studying the effects of blood pressure control, the UKPDS also studied the effects of intensive blood glucose control.24,25 Nearly 4,000 patients with newly diagnosed type 2 diabetes were randomized to intensive treatment with a sulfonylurea or insulin, or to conventional treatment with diet. Over 10 years, the mean hemoglobin A1c was reduced to 7.0% in the intensive group and 7.9% in the conventional group. The risk of any diabetes-related end point was 12% lower in the intensive group, 10% lower for diabetes-related death, and 6% lower for all-cause mortality. There was also a 25% reduction in microvascular disease (retinopathy and nephropathy). However, the intensive group had more hypoglycemic episodes than the conventional group and a tendency to some increase in macrovascular events. A legacy effect was evident: patients who had intensive treatment had less microvascular disease progression years after stopping therapy.
Tight glycemic control reduces nephropathy, but does it increase cardiovascular risk?
Earlier trials provided strong evidence that blood glucose control prevents or slows retinopathy and nephropathy. The critical question is, “At what expense?” Although diabetes is the most common cause of kidney failure in the United States, most people with diabetes do not die of kidney failure, but of cardiovascular disease. Two recent large trials had different results regarding glycemic control below hemoglobin A1c of 7.0% and macrovascular risk, creating a controversy about what recommendations are best.
The ADVANCE trial, enrolling 11,140 patients with type 2 diabetes, was largely conducted in Australia and used the sulfonylurea glipizide for glycemic control. Compared with the group that received standard therapy (n=5,569), the intensive-treatment group (n=5,571) achieved mean hemoglobin A1c levels of 6.5% compared with 7.3% in the standard group, and had less nephropathy, less microalbuminuria, less doubling of creatinine, and a lower rate of end-stage renal disease (4% vs 5% in the standard therapy group). No difference between the two groups was found in retinopathy. Rates of all-cause mortality did not differ between the groups.9
The ACCORD trial had more than 10,000 subjects with type 2 diabetes and took place mostly in the United States. Using mainly rosiglitazone for intensive therapy, the intensive group achieved hemoglobin A1c levels of 6.4% vs 7.5% in the standard-therapy group. The trial was stopped early, at 3.7 years, because of a higher risk of death and cardiovascular events in the group with intensive glycemic control. However, the intensive-therapy group did have a significant decrease in microvascular renal outcomes and a reduction in the progression of retinopathy.14,26
In summary, tighter glycemic control improves microvascular complications—both retinopathy and nephropathy—in patients with type 2 diabetes. The benefit of intensive therapy on macrovascular complications (stroke, myocardial infarction) in long-standing diabetes has not been convincingly demonstrated in randomized trials. The UKPDS suggested that maintaining a hemoglobin A1c of 7% in patients newly diagnosed with type 2 diabetes confers long-term cardiovascular benefits. The target hemoglobin A1c for type 2 diabetes should be tailored to the patient: 7% is a reasonable goal for most patients, but the goal should be higher for the elderly and frail. Reducing the risk of cardiovascular death is still best done by controlling blood pressure, reducing lipids, quitting smoking, and losing weight.
STRATEGY 3: INHIBIT THE RENIN-ANGIOTENSIN-ALDOSTERONE AXIS
Components of the renin-angiotensin-aldo-sterone system are present not only in the circulation but also in many tissues, including the heart, brain, kidney, blood vessels, and adrenal glands. The role of renin-angiotensin-aldosterone system blockers in treating and preventing diabetic nephropathy has become controversial in recent years with findings from new studies.
The renin-angiotensin-aldosterone system is important in the development or maintenance of high blood pressure and the resultant damage to the brain, heart, and kidney. Drug development has focused on inhibiting steps in the biochemical pathway. ACE inhibitors block the formation of angiotensin II—the most biologically potent angiotensin peptide—and are among the most commonly used drugs to treat hypertension and concomitant conditions, such as renal insufficiency, proteinuria, and heart failure. Angiotensin receptor blockers (ARBs) interact with the angiotensin AT1 receptor and block most of its actions. They are approved by the US Food and Drug Administration (FDA) for the treatment of hypertension, and they help prevent left ventricular hypertrophy and mesangial sclerosis. Large studies have shown that ACE inhibitors and ARBs offer similar cardiovascular benefit.
The glomerulus has the only capillary bed with a blood supply that drains into an efferent arteriole instead of a venule, providing high resistance to aid filtration. Efferent arterioles are rich in AT1 receptors. In the presence of angiotensin II they constrict, increasing pressure in the glomerulus, which can lead to proteinuria and glomerulosclerosis. ACE inhibitors and ARBs relax the efferent arteriole, allowing increased blood flow through the glomerulus. This reduction in intraglomerular pressure is associated with less proteinuria and less glomerulosclerosis.
Diabetes promotes renal disease in many ways. Glucose and advanced glycation end products can lead to increased blood flow and increased pressure in the glomerulus. Through a variety of pathways, hyperglycemia, acting on angiotensin II, leads to NF-kapa beta production, profibrotic cytokines, increased matrix, and eventual fibrosis. ACE inhibitors and ARBs counteract many of these.
ACE inhibitors and ARBs slow nephropathy progression beyond blood pressure control
Several major clinical trials27–32 examined the effects of either ACE inhibitors or ARBs in slowing the progression of diabetic nephropathy and have had consistently positive results.
The Collaborative Study Group30 was a 3-year randomized trial in 419 patients with type 1 diabetes, using the ACE inhibitor captopril vs placebo. Captopril was associated with less decline in kidney function and a 50% reduction in the risk of the combined end points of death, dialysis, and transplantation that was independent of the small difference in blood pressures between the two groups.
The Irbesartan Diabetic Nephropathy Trial (IDNT)31 studied the effect of the ARB irbesartan vs the calcium channel blocker amlodipine vs placebo over 2.6 years in 1,715 patients with type 2 diabetes. Irbesartan was found to be significantly more effective in protecting against the progression of nephropathy, independent of reduction in blood pressure.
The RENAAL trial,32 published in 2001, was a 3-year, randomized, double-blind study comparing the ARB losartan at increasing dosages with placebo (both taken in addition to conventional antihypertensive treatment) in 1,513 patients with type 2 diabetes and nephropathy. The blood pressure goal was 140/90 mm Hg in both groups, but the losartan group had a lower rate of doubling of serum creatinine, end-stage renal disease, and combined end-stage renal disease or death.
‘Aldosterone escape’ motivates the search for new therapies
An important reason for developing more ways to block the renin-angiotensin-aldosterone system is because of “aldosterone escape,” the phenomenon of angiotensin II or aldosterone returning to pretreatment levels despite continued ACE inhibition.
Biollaz et al,33 in a 1982 study of 19 patients with hypertension, showed that despite reducing blood pressure and keeping the blood level of ACE very low with twice-daily enalapril 20 mg, blood and urine levels of angiotensin II steadily rose back to baseline levels within a few months.
A growing body of evidence suggests that despite effective inhibition of angiotensin II activity, non-ACE synthetic pathways still permit angiotensin II generation via serine proteases such as chymase, cathepsin G, and tissue plasminogen activator.
Thus, efforts have been made to block the renin-angiotensin system in other places. In addition to ACE inhibitors and ARBs, two aldosterone receptor antagonists are available, spironolactone and eplerenone, both used to treat heart failure. A direct renin inhibitor, aliskiren, is also available.
Combination therapy—less proteinuria, but…
A number of studies have shown that combination treatment with agents having different targets in the renin-angiotensin-aldosterone system leads to larger reductions in albuminuria than does single-agent therapy.
Mogensen et al34 studied the effect of the ACE inhibitor lisinopril (20 mg per day) plus the ARB candesartan (16 mg per day) in subjects with microalbuminuria, hypertension, and type 2 diabetes. Combined treatment was more effective in reducing proteinuria.
Epstein et al35 studied the effects of the ACE inhibitor enalapril (20 mg/day) combined with either of two doses of the selective aldosterone receptor antagonist eplerenone (50 or 100 mg/day) or placebo. Both eplerenone dosages, when added to the enalapril treatment, significantly reduced albuminuria from baseline as early as week 4 (P < .001), but placebo treatment added to the enalapril did not result in any significant decrease in urinary albumin excretion. Systolic blood pressure decreased significantly in all treatment groups and by about the same amount.
The Aliskiren Combined With Losartan in Type 2 Diabetes and Nephropathy (AVOID) trial36 randomized more than 600 patients with type 2 diabetes and nephropathy to aliskiren (a renin inhibitor) or placebo added to the ARB losartan. Again, combination treatment was more renoprotective, independent of blood pressure lowering.
Worse outcomes with combination therapy?
More recent studies have indicated that although combination therapy reduces proteinuria to a greater extent than monotherapy, overall it worsens major renal and cardiovascular outcomes. The multicenter Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET)37 randomized more than 25,000 patients age 55 and older with established atherosclerotic vascular disease or with diabetes and end-organ damage to receive either the ARB telmisartan 80 mg daily, the ACE inhibitor ramipril 10 mg daily, or both. Mean follow-up was 56 months. The combination-treatment group had higher rates of death and renal disease than the single-therapy groups (which did not differ from one another).
Why the combination therapy had poorer outcomes is under debate. Patients may get sudden drops in blood pressure that are not detected with only periodic monitoring. Renal failure was mostly acute rather than chronic, and the estimated GFR declined more in the combined therapy group than in the single-therapy groups.
The Aliskiren Trial in Type 2 Diabetes Using Cardiovascular and Renal Disease Endpoints (ALTITUDE) was designed to test the effect of the direct renin inhibitor aliskiren or placebo, both arms combined with either an ACE inhibitor or an ARB in patients with type 2 diabetes at high risk for cardiovascular and renal events. The trial was terminated early because of more strokes and deaths in the combination therapy arms. The results led the FDA to issue black box warnings against using aliskiren with these other classes of agents, and all studies testing similar combinations have been stopped. (In one study that was stopped and has not yet been published, 100 patients with proteinuria were treated with either aliskiren, the ARB losartan, or both, to evaluate the effects of aldosterone escape. Results showed no differences: about one-third of each group had this phenomenon.)
My personal recommendation is as follows: for younger patients with proteinuria, at lower risk for cardiovascular events and with disease due not to diabetes but to immunoglobulin A nephropathy or another proteinuric kidney disease, treat with both an ACE inhibitor and ARB. But the combination should not be used for patients at high risk of cardiovascular disease, which includes almost all patients with diabetes.
If more aggressive renin-angiotensin system blockade is needed against diabetic nephropathy, adding a diuretic increases the impact of blocking the renin-angiotensin-aldosterone system on both proteinuria and progression of renal disease. The aldosterone blocker spironolactone 25 mg can be added if potassium levels are carefully monitored.
ACE inhibitor plus calcium channel blocker is safer than ACE inhibitor plus diuretic
The Avoiding Cardiovascular Events Through Combination Therapy in Patients Living With Systolic Hypertension (ACCOMPLISH) trial38 randomized more than 11,000 high-risk patients with hypertension to receive an ACE inhibitor (benazepril) plus either a calcium channel blocker (amlodipine) or thiazide diuretic (hydrochlorothiazide). Blood pressures were identical between the two groups, but the trial was terminated early, at 36 months, because of a higher risk of the combined end point of cardiovascular death, myocardial infarction, stroke, and other major cardiac events in the ACE inhibitor-thiazide group.
Although some experts believe this study is definitive and indicates that high blood pressure should never be treated with an ACE inhibitor-thiazide combination, I believe that caution is needed in interpreting these findings. This regimen should be avoided in older patients with diabetes at high risk for cardiovascular disease, but otherwise, getting blood pressure under control is critical, and this combination can be used if it works and the patient is tolerating it well.
In summary, the choice of blood pressure-lowering medications is based on reducing cardiovascular events and slowing the progression of kidney disease. Either an ACE inhibitor or an ARB is the first choice for patients with diabetes, hypertension, and any degree of proteinuria. Many experts recommend beginning one of these agents even if proteinuria is not present. However, the combination of an ACE inhibitor and ARB should not be used in diabetic patients, especially if they have cardiovascular disease, until further data clarify the results of the ONTARGET and ALTITUDE trials.
STRATEGY 4: METABOLIC MANIPULATION WITH NOVEL AGENTS
Several new agents have recently been studied for the treatment of diabetic nephropathy, including aminoguanidine, which reduces levels of advanced glycation end-products, and sulodexide, which blocks basement membrane permeability. Neither agent has been shown to be safe and effective in diabetic nephropathy. The newest agent is bardoxolone methyl. It induces the Keap1–Nrf2 pathway, which up-regulates cytoprotective factors, suppressing inflammatory and other cytokines that are major mediators of progression of chronic kidney disease.39
Pergola et al,40 in a phase 2, double-blind trial, randomized 227 adults with diabetic kidney disease and a low estimated GFR (20–45 mL/min/1.73 m2) to receive placebo or bardoxolone 25, 75, or 150 mg daily. Drug treatment was associated with improvement in the estimated GFR, a finding that persisted throughout the 52 weeks of treatment. Surprisingly, proteinuria did not decrease with drug treatment.
As of this writing, a large multicenter controlled randomized trial has been halted because of concerns by the data safety monitoring board, which found increased rates of death and fluid retention with the drug. A number of recent trials have shown a beneficial effect of sodium bicarbonate therapy in patients with late-stage chronic kidney disease. They have shown slowing of the progression of GFR decline in a number of renal diseases, including diabetes.
Diabetes is on the rise, and so is diabetic nephropathy. In view of this epidemic, physicians should consider strategies to detect and control kidney disease in their diabetic patients.
This article will focus on kidney disease in adult-onset type 2 diabetes. Although it has different pathogenetic mechanisms than type 1 diabetes, the clinical course of the two conditions is very similar in terms of the prevalence of proteinuria after diagnosis, the progression to renal failure after the onset of proteinuria, and treatment options.1
DIABETES AND DIABETIC KIDNEY DISEASE ARE ON THE RISE
The incidence of diabetes increases with age, and with the aging of the baby boomers, its prevalence is growing dramatically. The 2005– 2008 National Health and Nutrition Examination Survey estimated the prevalence as 3.7% in adults age 20 to 44, 13.7% at age 45 to 64, and 26.9% in people age 65 and older. The obesity epidemic is also contributing to the increase in diabetes in all age groups.
Diabetic kidney disease has increased in the United States from about 4 million cases 20 years ago to about 7 million in 2005–2008.2 Diabetes is the major cause of end-stage renal disease in the developed world, accounting for 40% to 50% of cases. Other major causes are hypertension (27%) and glomerulonephritis (13%).3
Physicians in nearly every field of medicine now care for patients with diabetic nephropathy. The classic presentation—a patient who has impaired vision, fluid retention with edema, and hypertension—is commonly seen in dialysis units and ophthalmology and cardiovascular clinics.
CLINICAL PROGRESSION
Early in the course of diabetic nephropathy, blood pressure is normal and microalbuminuria is not evident, but many patients have a high glomerular filtration rate (GFR), indicating temporarily “enhanced” renal function or hyperfiltration. The next stage is characterized by microalbuminuria, correlating with glomerular mesangial expansion: the GFR falls back into the normal range and blood pressure starts to increase. Finally, macroalbuminuria occurs, accompanied by rising blood pressure and a declining GFR, correlating with the histologic appearance of glomerulosclerosis and Kimmelstiel-Wilson nodules.4
Hypertension develops in 5% of patients by 10 years after type 1 diabetes is diagnosed, 33% by 20 years, and 70% by 40 years. In contrast, 40% of patients with type 2 diabetes have high blood pressure at diagnosis.
Unfortunately, in most cases, this progression is a one-way street, so it is critical to intervene to try to slow the progression early in the course of the disease process.
SCREENING FOR DIABETIC NEPHROPATHY
Nephropathy screening guidelines for patients with diabetes are provided in Table 1.5
Blood pressure should be monitored at each office visit (Table 1). The goal for adults with diabetes should be to reduce blood pressure to 130/80 mm Hg. Reduction beyond this level may be associated with an increased mortality rate.6 Very high blood pressure (> 180 mm Hg systolic) should be lowered slowly. Lowering blood pressure delays the progression from microalbuminuria (30–299 mg/day or 20–199 μg/min) to macroalbuminuria (> 300 mg/day or > 200 μg/min) and slows the progression to renal failure.
Urinary albumin. Proteinuria takes 5 to 10 years to develop after the onset of diabetes. Because it is possible for patients with type 2 diabetes to have had the disease for some time before being diagnosed, urinary albumin screening should be performed at diagnosis and annually thereafter. Patients with type 1 are usually diagnosed with diabetes at or near onset of disease; therefore, annual screening for urinary albumin can begin 5 years after diagnosis.5
Proteinuria can be measured in different ways (Table 2). The basic screening test for clinical proteinuria is the urine dipstick, which is very sensitive to albumin and relatively insensitive to other proteins. “Trace-positive” results are common in healthy people, so proteinuria is not confirmed unless a patient has repeatedly positive results.
Microalbuminuria is important to measure, especially if it helps determine therapy. It is not detectable by the urinary dipstick, but can be measured in the following ways:
- Measurement of the albumin-creatinine ratio in a random spot collection
- 24-hour collection (creatinine should simultaneously be measured and creatinine clearance calculated)
- Timed collection (4 hours or overnight).
The first method is preferred, and any positive test result must be confirmed by repeat analyses of urinary albumin before a patient is diagnosed with microalbuminuria.
Occasionally a patient presenting with proteinuria but normal blood sugar and hemoglobin A1c will have a biopsy that reveals morphologic changes of classic diabetic nephropathy. Most such patients have a history of hyperglycemia, indicating that they actually have been diabetic.
Proteinuria—the best marker of disease progression
Proteinuria is the strongest predictor of renal outcomes. The Reduction in End Points in Noninsulin-Dependent Diabetes Mellitus With the Angiotensin II Antagonist Losartan (RENAAL) study was a randomized, placebo-controlled trial in more than 1,500 patients with type 2 diabetes to test the effects of losartan on renal outcome. Those with high albuminuria (> 3.0 g albumin/g creatinine) at baseline were five times more likely to reach a renal end point and were eight times more likely to have progression to end-stage renal disease than patients with low albuminuria (< 1.5 g/g).7 The degree of albuminuria after 6 months of treatment showed similar predictive trends, indicating that monitoring and treating proteinuria are extremely important goals.
STRATEGY 1 TO LIMIT RENAL INJURY: REDUCE BLOOD PRESSURE
Blood pressure control improves renal and cardiovascular function.
As early as 1983, Parving et al,8 in a study of only 10 insulin-dependent diabetic patients, showed strong evidence that early aggressive antihypertensive treatment improved the course of diabetic nephropathy. During the mean pretreatment period of 29 months, the GFR decreased significantly and the urinary albumin excretion rate and arterial blood pressure rose significantly. During the mean 39-month period of antihypertensive treatment with metoprolol, hydralazine, and furosemide or a thiazide, mean arterial blood pressure fell from 144/97 to 128/84 mm Hg and urinary albumin excretion from 977 to 433 μg/ min. The rate of decline in GFR slowed from 0.91 mL/min/month before treatment to 0.39 mL/min/month during treatment.
The Action in Diabetes and Vascular Disease: Preterax and Diamicron MR Controlled Evaluation (ADVANCE) trial9 enrolled more than 11,000 patients internationally with type 2 diabetes at high risk for cardiovascular events. In addition to standard therapy, blood pressure was intensively controlled in one group with a combination of the angiotensin-converting enzyme (ACE) inhibitor perindopril and the diuretic indapamide. The intensive-therapy group achieved blood pressures less than 140/80 mm Hg and had a mean reduction of systolic blood pressure of 5.6 mm Hg and diastolic blood pressure of 2.2 mm Hg vs controls. Despite these apparently modest reductions, the intensively controlled group had a significant 9% reduction of the primary outcome of combined macrovascular events (cardiovascular death, myocardial infarction, and stroke) and microvascular events (new or worsening nephropathy, or retinopathy).10
A meta-analysis of studies of patients with type 2 diabetes found reduced nephropathy with systolic blood pressure control to less than 130 mm Hg.11
The United Kingdom Prospective Diabetes Study (UKPDS) is a series of studies of diabetes. The original study in 1998 enrolled 5,102 patients with newly diagnosed type 2 diabetes.12 The more than 1,000 patients with hypertension were randomized to either tight blood pressure control or regular care. The intensive treatment group had a mean blood pressure reduction of 9 mm Hg systolic and 3 mm Hg diastolic, along with major reductions in all diabetes end points, diabetes deaths, microvascular disease, and stroke over a median follow-up of 8.4 years.
Continuous blood pressure control is critical
Tight blood pressure control must be maintained to have continued benefit. During the 10 years following the UKPDS, no attempts were made to maintain the previously assigned therapies. A follow-up study13 of 884 UKPDS patients found that blood pressures were the same again between the two groups 2 years after the trial was stopped, and no beneficial legacy effect from previous blood pressure control was evident on end points.
Control below 120 mm Hg systolic not needed
Blood pressure control slows kidney disease and prevents major macrovascular disease, but there is no evidence that lowering systolic blood pressure below 120 mm Hg provides additional benefit. In the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial,14 more than 10,000 patients with type 2 diabetes and existing cardiovascular disease or additional cardiovascular risk factors were randomized to a goal of systolic blood pressure less than 120 mm Hg or less than 140 mm Hg (actual mean systolic pressures were 119 vs 134 mm Hg, respectively). Over nearly 5 years, there was no difference in cardiovascular events or deaths between the two groups.15
Since 1997, six international organizations have revised their recommended blood pressure goals in diabetes mellitus and renal diseases. Randomized clinical trials and observational studies have demonstrated the importance of blood pressure control to the level of 125/75 to 140/80 mm Hg. The National Kidney Foundation, the American Diabetes Association, and the Canadian Hypertension Society have developed consensus guidelines for blood pressure control to less than 130/80 mm Hg.16–21 Table 3 summarizes blood pressure goals for patients with diabetes.
STRATEGY 2: CONTROL BLOOD SUGAR
Recommendations for blood sugar goals are more controversial.
The Diabetes Control and Complications Trial22 provided early evidence that tight blood sugar control slows the development of microalbuminuria and macroalbuminuria. The study randomized more than 1,400 patients with type 1 diabetes to either standard therapy (1 or 2 daily insulin injections) or intensive therapy (an external insulin pump or 3 or more insulin injections guided by frequent blood glucose monitoring) to keep blood glucose levels close to normal. About half the patients had mild retinopathy at baseline and the others had no retinopathy. After 6.5 years, intensive therapy was found to significantly delay the onset and slow the progression of diabetic retinopathy and nephropathy.
The Kumamoto Study23 randomized 110 patients with type 2 diabetes and either no retinopathy (primary prevention cohort) or simple retinopathy (secondary prevention cohort) to receive either multiple insulin injections or conventional insulin therapy over 8 years. Intensive therapy led to lower rates of retinopathy (7.7% vs 32% in primary prevention and 19% vs 44% in secondary prevention) and progressive nephropathy (7% vs 28% in primary prevention at 6 years and 11% vs 32% in secondary prevention).
In addition to studying the effects of blood pressure control, the UKPDS also studied the effects of intensive blood glucose control.24,25 Nearly 4,000 patients with newly diagnosed type 2 diabetes were randomized to intensive treatment with a sulfonylurea or insulin, or to conventional treatment with diet. Over 10 years, the mean hemoglobin A1c was reduced to 7.0% in the intensive group and 7.9% in the conventional group. The risk of any diabetes-related end point was 12% lower in the intensive group, 10% lower for diabetes-related death, and 6% lower for all-cause mortality. There was also a 25% reduction in microvascular disease (retinopathy and nephropathy). However, the intensive group had more hypoglycemic episodes than the conventional group and a tendency to some increase in macrovascular events. A legacy effect was evident: patients who had intensive treatment had less microvascular disease progression years after stopping therapy.
Tight glycemic control reduces nephropathy, but does it increase cardiovascular risk?
Earlier trials provided strong evidence that blood glucose control prevents or slows retinopathy and nephropathy. The critical question is, “At what expense?” Although diabetes is the most common cause of kidney failure in the United States, most people with diabetes do not die of kidney failure, but of cardiovascular disease. Two recent large trials had different results regarding glycemic control below hemoglobin A1c of 7.0% and macrovascular risk, creating a controversy about what recommendations are best.
The ADVANCE trial, enrolling 11,140 patients with type 2 diabetes, was largely conducted in Australia and used the sulfonylurea glipizide for glycemic control. Compared with the group that received standard therapy (n=5,569), the intensive-treatment group (n=5,571) achieved mean hemoglobin A1c levels of 6.5% compared with 7.3% in the standard group, and had less nephropathy, less microalbuminuria, less doubling of creatinine, and a lower rate of end-stage renal disease (4% vs 5% in the standard therapy group). No difference between the two groups was found in retinopathy. Rates of all-cause mortality did not differ between the groups.9
The ACCORD trial had more than 10,000 subjects with type 2 diabetes and took place mostly in the United States. Using mainly rosiglitazone for intensive therapy, the intensive group achieved hemoglobin A1c levels of 6.4% vs 7.5% in the standard-therapy group. The trial was stopped early, at 3.7 years, because of a higher risk of death and cardiovascular events in the group with intensive glycemic control. However, the intensive-therapy group did have a significant decrease in microvascular renal outcomes and a reduction in the progression of retinopathy.14,26
In summary, tighter glycemic control improves microvascular complications—both retinopathy and nephropathy—in patients with type 2 diabetes. The benefit of intensive therapy on macrovascular complications (stroke, myocardial infarction) in long-standing diabetes has not been convincingly demonstrated in randomized trials. The UKPDS suggested that maintaining a hemoglobin A1c of 7% in patients newly diagnosed with type 2 diabetes confers long-term cardiovascular benefits. The target hemoglobin A1c for type 2 diabetes should be tailored to the patient: 7% is a reasonable goal for most patients, but the goal should be higher for the elderly and frail. Reducing the risk of cardiovascular death is still best done by controlling blood pressure, reducing lipids, quitting smoking, and losing weight.
STRATEGY 3: INHIBIT THE RENIN-ANGIOTENSIN-ALDOSTERONE AXIS
Components of the renin-angiotensin-aldo-sterone system are present not only in the circulation but also in many tissues, including the heart, brain, kidney, blood vessels, and adrenal glands. The role of renin-angiotensin-aldosterone system blockers in treating and preventing diabetic nephropathy has become controversial in recent years with findings from new studies.
The renin-angiotensin-aldosterone system is important in the development or maintenance of high blood pressure and the resultant damage to the brain, heart, and kidney. Drug development has focused on inhibiting steps in the biochemical pathway. ACE inhibitors block the formation of angiotensin II—the most biologically potent angiotensin peptide—and are among the most commonly used drugs to treat hypertension and concomitant conditions, such as renal insufficiency, proteinuria, and heart failure. Angiotensin receptor blockers (ARBs) interact with the angiotensin AT1 receptor and block most of its actions. They are approved by the US Food and Drug Administration (FDA) for the treatment of hypertension, and they help prevent left ventricular hypertrophy and mesangial sclerosis. Large studies have shown that ACE inhibitors and ARBs offer similar cardiovascular benefit.
The glomerulus has the only capillary bed with a blood supply that drains into an efferent arteriole instead of a venule, providing high resistance to aid filtration. Efferent arterioles are rich in AT1 receptors. In the presence of angiotensin II they constrict, increasing pressure in the glomerulus, which can lead to proteinuria and glomerulosclerosis. ACE inhibitors and ARBs relax the efferent arteriole, allowing increased blood flow through the glomerulus. This reduction in intraglomerular pressure is associated with less proteinuria and less glomerulosclerosis.
Diabetes promotes renal disease in many ways. Glucose and advanced glycation end products can lead to increased blood flow and increased pressure in the glomerulus. Through a variety of pathways, hyperglycemia, acting on angiotensin II, leads to NF-kapa beta production, profibrotic cytokines, increased matrix, and eventual fibrosis. ACE inhibitors and ARBs counteract many of these.
ACE inhibitors and ARBs slow nephropathy progression beyond blood pressure control
Several major clinical trials27–32 examined the effects of either ACE inhibitors or ARBs in slowing the progression of diabetic nephropathy and have had consistently positive results.
The Collaborative Study Group30 was a 3-year randomized trial in 419 patients with type 1 diabetes, using the ACE inhibitor captopril vs placebo. Captopril was associated with less decline in kidney function and a 50% reduction in the risk of the combined end points of death, dialysis, and transplantation that was independent of the small difference in blood pressures between the two groups.
The Irbesartan Diabetic Nephropathy Trial (IDNT)31 studied the effect of the ARB irbesartan vs the calcium channel blocker amlodipine vs placebo over 2.6 years in 1,715 patients with type 2 diabetes. Irbesartan was found to be significantly more effective in protecting against the progression of nephropathy, independent of reduction in blood pressure.
The RENAAL trial,32 published in 2001, was a 3-year, randomized, double-blind study comparing the ARB losartan at increasing dosages with placebo (both taken in addition to conventional antihypertensive treatment) in 1,513 patients with type 2 diabetes and nephropathy. The blood pressure goal was 140/90 mm Hg in both groups, but the losartan group had a lower rate of doubling of serum creatinine, end-stage renal disease, and combined end-stage renal disease or death.
‘Aldosterone escape’ motivates the search for new therapies
An important reason for developing more ways to block the renin-angiotensin-aldosterone system is because of “aldosterone escape,” the phenomenon of angiotensin II or aldosterone returning to pretreatment levels despite continued ACE inhibition.
Biollaz et al,33 in a 1982 study of 19 patients with hypertension, showed that despite reducing blood pressure and keeping the blood level of ACE very low with twice-daily enalapril 20 mg, blood and urine levels of angiotensin II steadily rose back to baseline levels within a few months.
A growing body of evidence suggests that despite effective inhibition of angiotensin II activity, non-ACE synthetic pathways still permit angiotensin II generation via serine proteases such as chymase, cathepsin G, and tissue plasminogen activator.
Thus, efforts have been made to block the renin-angiotensin system in other places. In addition to ACE inhibitors and ARBs, two aldosterone receptor antagonists are available, spironolactone and eplerenone, both used to treat heart failure. A direct renin inhibitor, aliskiren, is also available.
Combination therapy—less proteinuria, but…
A number of studies have shown that combination treatment with agents having different targets in the renin-angiotensin-aldosterone system leads to larger reductions in albuminuria than does single-agent therapy.
Mogensen et al34 studied the effect of the ACE inhibitor lisinopril (20 mg per day) plus the ARB candesartan (16 mg per day) in subjects with microalbuminuria, hypertension, and type 2 diabetes. Combined treatment was more effective in reducing proteinuria.
Epstein et al35 studied the effects of the ACE inhibitor enalapril (20 mg/day) combined with either of two doses of the selective aldosterone receptor antagonist eplerenone (50 or 100 mg/day) or placebo. Both eplerenone dosages, when added to the enalapril treatment, significantly reduced albuminuria from baseline as early as week 4 (P < .001), but placebo treatment added to the enalapril did not result in any significant decrease in urinary albumin excretion. Systolic blood pressure decreased significantly in all treatment groups and by about the same amount.
The Aliskiren Combined With Losartan in Type 2 Diabetes and Nephropathy (AVOID) trial36 randomized more than 600 patients with type 2 diabetes and nephropathy to aliskiren (a renin inhibitor) or placebo added to the ARB losartan. Again, combination treatment was more renoprotective, independent of blood pressure lowering.
Worse outcomes with combination therapy?
More recent studies have indicated that although combination therapy reduces proteinuria to a greater extent than monotherapy, overall it worsens major renal and cardiovascular outcomes. The multicenter Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET)37 randomized more than 25,000 patients age 55 and older with established atherosclerotic vascular disease or with diabetes and end-organ damage to receive either the ARB telmisartan 80 mg daily, the ACE inhibitor ramipril 10 mg daily, or both. Mean follow-up was 56 months. The combination-treatment group had higher rates of death and renal disease than the single-therapy groups (which did not differ from one another).
Why the combination therapy had poorer outcomes is under debate. Patients may get sudden drops in blood pressure that are not detected with only periodic monitoring. Renal failure was mostly acute rather than chronic, and the estimated GFR declined more in the combined therapy group than in the single-therapy groups.
The Aliskiren Trial in Type 2 Diabetes Using Cardiovascular and Renal Disease Endpoints (ALTITUDE) was designed to test the effect of the direct renin inhibitor aliskiren or placebo, both arms combined with either an ACE inhibitor or an ARB in patients with type 2 diabetes at high risk for cardiovascular and renal events. The trial was terminated early because of more strokes and deaths in the combination therapy arms. The results led the FDA to issue black box warnings against using aliskiren with these other classes of agents, and all studies testing similar combinations have been stopped. (In one study that was stopped and has not yet been published, 100 patients with proteinuria were treated with either aliskiren, the ARB losartan, or both, to evaluate the effects of aldosterone escape. Results showed no differences: about one-third of each group had this phenomenon.)
My personal recommendation is as follows: for younger patients with proteinuria, at lower risk for cardiovascular events and with disease due not to diabetes but to immunoglobulin A nephropathy or another proteinuric kidney disease, treat with both an ACE inhibitor and ARB. But the combination should not be used for patients at high risk of cardiovascular disease, which includes almost all patients with diabetes.
If more aggressive renin-angiotensin system blockade is needed against diabetic nephropathy, adding a diuretic increases the impact of blocking the renin-angiotensin-aldosterone system on both proteinuria and progression of renal disease. The aldosterone blocker spironolactone 25 mg can be added if potassium levels are carefully monitored.
ACE inhibitor plus calcium channel blocker is safer than ACE inhibitor plus diuretic
The Avoiding Cardiovascular Events Through Combination Therapy in Patients Living With Systolic Hypertension (ACCOMPLISH) trial38 randomized more than 11,000 high-risk patients with hypertension to receive an ACE inhibitor (benazepril) plus either a calcium channel blocker (amlodipine) or thiazide diuretic (hydrochlorothiazide). Blood pressures were identical between the two groups, but the trial was terminated early, at 36 months, because of a higher risk of the combined end point of cardiovascular death, myocardial infarction, stroke, and other major cardiac events in the ACE inhibitor-thiazide group.
Although some experts believe this study is definitive and indicates that high blood pressure should never be treated with an ACE inhibitor-thiazide combination, I believe that caution is needed in interpreting these findings. This regimen should be avoided in older patients with diabetes at high risk for cardiovascular disease, but otherwise, getting blood pressure under control is critical, and this combination can be used if it works and the patient is tolerating it well.
In summary, the choice of blood pressure-lowering medications is based on reducing cardiovascular events and slowing the progression of kidney disease. Either an ACE inhibitor or an ARB is the first choice for patients with diabetes, hypertension, and any degree of proteinuria. Many experts recommend beginning one of these agents even if proteinuria is not present. However, the combination of an ACE inhibitor and ARB should not be used in diabetic patients, especially if they have cardiovascular disease, until further data clarify the results of the ONTARGET and ALTITUDE trials.
STRATEGY 4: METABOLIC MANIPULATION WITH NOVEL AGENTS
Several new agents have recently been studied for the treatment of diabetic nephropathy, including aminoguanidine, which reduces levels of advanced glycation end-products, and sulodexide, which blocks basement membrane permeability. Neither agent has been shown to be safe and effective in diabetic nephropathy. The newest agent is bardoxolone methyl. It induces the Keap1–Nrf2 pathway, which up-regulates cytoprotective factors, suppressing inflammatory and other cytokines that are major mediators of progression of chronic kidney disease.39
Pergola et al,40 in a phase 2, double-blind trial, randomized 227 adults with diabetic kidney disease and a low estimated GFR (20–45 mL/min/1.73 m2) to receive placebo or bardoxolone 25, 75, or 150 mg daily. Drug treatment was associated with improvement in the estimated GFR, a finding that persisted throughout the 52 weeks of treatment. Surprisingly, proteinuria did not decrease with drug treatment.
As of this writing, a large multicenter controlled randomized trial has been halted because of concerns by the data safety monitoring board, which found increased rates of death and fluid retention with the drug. A number of recent trials have shown a beneficial effect of sodium bicarbonate therapy in patients with late-stage chronic kidney disease. They have shown slowing of the progression of GFR decline in a number of renal diseases, including diabetes.
- Ritz E, Orth SR. Nephropathy in patients with type 2 diabetes mellitus. N Engl J Med 1999; 341:1127–1133.
- de Boer IH, Rue TC, Hall YN, Heagerty PJ, Weiss NS, Himmelfarb J. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA 2011; 305:2532–2539.
- United States Renal Data System (USRDS) 2000 Annual Data Report. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases – Division of Kidney, Urologic and Hematologic Diseases. USRDS Coordinating Center operated by the Minneapolis Medical Research Foundation. www.usrds.org
- Macisaac RJ, Jerums G. Diabetic kidney disease with and without albuminuria. Curr Opin Nephrol Hypertens 2011; 20:246–257.
- Molitch ME, DeFronzo RA, Franz MJ, et al; American Diabetes Association. Nephropathy in diabetes. Diabetes Care 2004; 27(suppl 1):S79–S83.
- Vamos EP, Harris M, Millett C, et al. Association of systolic and diastolic blood pressure and all cause mortality in people with newly diagnosed type 2 diabetes: retrospective cohort study. BMJ 2012; 345:e5567.
- de Zeeuw D, Remuzzi G, Parving HH, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int 2004; 65:2309–2320.
- Parving HH, Andersen AR, Smidt UM, Svendsen PA. Early aggressive antihypertensive treatment reduces rate of decline in kidney function in diabetic nephropathy. Lancet 1983; 1:1175–1179.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): a randomised controlled trial. Lancet 2007; 370:829–840.
- Bangalore S, Kumar S, Lobach I, Messerli FH. Blood pressure targets in subjects with type 2 diabetes mellitus/impaired fasting glucose: observations from traditional and bayesian random-effects meta-analyses of randomized trials. Circulation 2011; 123:2799–2810.
- UK Prospective Diabetes Study Group. Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. BMJ 1998; 317:703–713.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577–1589.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- American Diabetes Association. Standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S11–S63. (Erratum in: Diabetes Care 2012; 35:660.)
- Bakris GL, Williams M, Dworkin L, et al. Preserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertension and Diabetes Executive Committees Working Group. Am J Kidney Dis 2000; 36:646–661.
- Ramsay L, Williams B, Johnston G, et al. Guidelines for management of hypertension: report of the third working party of the British Hypertension Society. J Hum Hypertens 1999; 13:569–592.
- Feldman RD, Campbell N, Larochelle P, et al. 1999 Canadian recommendations for the management of hypertension. Task Force for the Development of the 1999 Canadian Recommendations for the Management of Hypertension. CMAJ 1999; 161(suppl):12:S1–S17.
- Chalmers J, MacMahon S, Mancia G, et al. 1999 World Health Organization-International Society of Hypertension Guidelines for the management of hypertension. Guidelines Sub-committee of the World Health Organization. Clin Exp Hypertens 1999; 21:1009–1060.
- The seventh report of the Joint National Committee on Prevention, Detection Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 1993; 329:977–986.
- Shichiri M, Kishikawa H, Ohkubo Y, Wake N. Long-term results of the Kumamoto Study on optimal diabetes control in type 2 diabetic patients. Diabetes Care 2000; 23(suppl 2):B21–B29.
- Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998; 352:837–853. Erratum in: Lancet 1999; 354:602.
- Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. UK Prospective Diabetes Study Group. BMJ 1998; 317:703–713. Erratum in: BMJ 1999; 318:29.
- Ismail-Beigi F, Craven T, Banerji MA, et al; ACCORD trial group. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: an analysis of the ACCORD randomised trial. Lancet 2010; 376:419–430. Erratum in: Lancet 2010; 376:1466.
- Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Heart Outcomes Prevention Evaluation Study Investigators. Lancet 2000: 355:253–259. Erratum in: Lancet2000; 356:860.
- Parving HH, Lehnert H, Bröchner-Mortensen J, et al; Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria Study Group. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med 2001; 345:870–878.
- Viberti G, Wheeldon NM; MicroAlbuminuria Reduction With VALsartan (MARVAL) Study Investigators. Microalbuminuria reduction with valsartan in patients with type 2 diabetes mellitus: a blood pressure-independent effect. Circulation 2002; 106:672–678.
- Lewis EJ, Hunsicker LG, Bain R P, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 1993; 329:1456–1462.
- Lewis EJ, Hunsicker LG, Clarke WR, et al; Collaborative Study Group. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345:851–860.
- Brenner BM, Cooper ME, de Zeeuw D, et al; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345:861–869.
- Biollaz J, Brunner HR, Gavras I, Waeber B, Gavras H. Antihypertensive therapy with MK 421: angiotensin II--renin relationships to evaluate efficacy of converting enzyme blockade. J Cardiovasc Pharmacol 1982; 4:966–972.
- Mogensen CE, Neldam S, Tikkanen I, et al. Randomised controlled trial of dual blockade of renin-angiotensin system in patients with hypertension, microalbuminuria, and non-insulin dependent diabetes: the candesartan and lisinopril microalbuminuria (CALM) study. BMJ 2000; 321:1440–1444.
- Epstein M, Williams GH, Weinberger M, et al. Selective aldosterone blockade with eplerenone reduces albuminuria in patients with type 2 diabetes. Clin J Am Soc Nephrol 2006; 1:940–951.
- Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK; AVOID Study Investigators. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med 2008; 358:2433–2446.
- Mann JF, Schmieder RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Jamerson K, Weber MA, Bakris GL, et al; ACCOMPLISH Trial Investigators. Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N Engl J Med 2008; 359:2417–2428.
- Kim HJ, Vaziri ND. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am J Physiol Renal Physiol 2010; 298:F662–F671.
- Pergola PE, Raskin P, Toto RD, et al; BEAM Study Investigators Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 2011; 365:327–336.
- Ritz E, Orth SR. Nephropathy in patients with type 2 diabetes mellitus. N Engl J Med 1999; 341:1127–1133.
- de Boer IH, Rue TC, Hall YN, Heagerty PJ, Weiss NS, Himmelfarb J. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA 2011; 305:2532–2539.
- United States Renal Data System (USRDS) 2000 Annual Data Report. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases – Division of Kidney, Urologic and Hematologic Diseases. USRDS Coordinating Center operated by the Minneapolis Medical Research Foundation. www.usrds.org
- Macisaac RJ, Jerums G. Diabetic kidney disease with and without albuminuria. Curr Opin Nephrol Hypertens 2011; 20:246–257.
- Molitch ME, DeFronzo RA, Franz MJ, et al; American Diabetes Association. Nephropathy in diabetes. Diabetes Care 2004; 27(suppl 1):S79–S83.
- Vamos EP, Harris M, Millett C, et al. Association of systolic and diastolic blood pressure and all cause mortality in people with newly diagnosed type 2 diabetes: retrospective cohort study. BMJ 2012; 345:e5567.
- de Zeeuw D, Remuzzi G, Parving HH, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int 2004; 65:2309–2320.
- Parving HH, Andersen AR, Smidt UM, Svendsen PA. Early aggressive antihypertensive treatment reduces rate of decline in kidney function in diabetic nephropathy. Lancet 1983; 1:1175–1179.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): a randomised controlled trial. Lancet 2007; 370:829–840.
- Bangalore S, Kumar S, Lobach I, Messerli FH. Blood pressure targets in subjects with type 2 diabetes mellitus/impaired fasting glucose: observations from traditional and bayesian random-effects meta-analyses of randomized trials. Circulation 2011; 123:2799–2810.
- UK Prospective Diabetes Study Group. Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. BMJ 1998; 317:703–713.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577–1589.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- American Diabetes Association. Standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S11–S63. (Erratum in: Diabetes Care 2012; 35:660.)
- Bakris GL, Williams M, Dworkin L, et al. Preserving renal function in adults with hypertension and diabetes: a consensus approach. National Kidney Foundation Hypertension and Diabetes Executive Committees Working Group. Am J Kidney Dis 2000; 36:646–661.
- Ramsay L, Williams B, Johnston G, et al. Guidelines for management of hypertension: report of the third working party of the British Hypertension Society. J Hum Hypertens 1999; 13:569–592.
- Feldman RD, Campbell N, Larochelle P, et al. 1999 Canadian recommendations for the management of hypertension. Task Force for the Development of the 1999 Canadian Recommendations for the Management of Hypertension. CMAJ 1999; 161(suppl):12:S1–S17.
- Chalmers J, MacMahon S, Mancia G, et al. 1999 World Health Organization-International Society of Hypertension Guidelines for the management of hypertension. Guidelines Sub-committee of the World Health Organization. Clin Exp Hypertens 1999; 21:1009–1060.
- The seventh report of the Joint National Committee on Prevention, Detection Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 1993; 329:977–986.
- Shichiri M, Kishikawa H, Ohkubo Y, Wake N. Long-term results of the Kumamoto Study on optimal diabetes control in type 2 diabetic patients. Diabetes Care 2000; 23(suppl 2):B21–B29.
- Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998; 352:837–853. Erratum in: Lancet 1999; 354:602.
- Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. UK Prospective Diabetes Study Group. BMJ 1998; 317:703–713. Erratum in: BMJ 1999; 318:29.
- Ismail-Beigi F, Craven T, Banerji MA, et al; ACCORD trial group. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: an analysis of the ACCORD randomised trial. Lancet 2010; 376:419–430. Erratum in: Lancet 2010; 376:1466.
- Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Heart Outcomes Prevention Evaluation Study Investigators. Lancet 2000: 355:253–259. Erratum in: Lancet2000; 356:860.
- Parving HH, Lehnert H, Bröchner-Mortensen J, et al; Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria Study Group. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med 2001; 345:870–878.
- Viberti G, Wheeldon NM; MicroAlbuminuria Reduction With VALsartan (MARVAL) Study Investigators. Microalbuminuria reduction with valsartan in patients with type 2 diabetes mellitus: a blood pressure-independent effect. Circulation 2002; 106:672–678.
- Lewis EJ, Hunsicker LG, Bain R P, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 1993; 329:1456–1462.
- Lewis EJ, Hunsicker LG, Clarke WR, et al; Collaborative Study Group. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345:851–860.
- Brenner BM, Cooper ME, de Zeeuw D, et al; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345:861–869.
- Biollaz J, Brunner HR, Gavras I, Waeber B, Gavras H. Antihypertensive therapy with MK 421: angiotensin II--renin relationships to evaluate efficacy of converting enzyme blockade. J Cardiovasc Pharmacol 1982; 4:966–972.
- Mogensen CE, Neldam S, Tikkanen I, et al. Randomised controlled trial of dual blockade of renin-angiotensin system in patients with hypertension, microalbuminuria, and non-insulin dependent diabetes: the candesartan and lisinopril microalbuminuria (CALM) study. BMJ 2000; 321:1440–1444.
- Epstein M, Williams GH, Weinberger M, et al. Selective aldosterone blockade with eplerenone reduces albuminuria in patients with type 2 diabetes. Clin J Am Soc Nephrol 2006; 1:940–951.
- Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK; AVOID Study Investigators. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med 2008; 358:2433–2446.
- Mann JF, Schmieder RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Jamerson K, Weber MA, Bakris GL, et al; ACCOMPLISH Trial Investigators. Benazepril plus amlodipine or hydrochlorothiazide for hypertension in high-risk patients. N Engl J Med 2008; 359:2417–2428.
- Kim HJ, Vaziri ND. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am J Physiol Renal Physiol 2010; 298:F662–F671.
- Pergola PE, Raskin P, Toto RD, et al; BEAM Study Investigators Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 2011; 365:327–336.
KEY POINTS
- The progression from no proteinuria to microalbuminuria to clinical proteinuria parallels glomerular changes of thickening of the basement membrane, mesangial expansion, and the development of Kimmelstiel-Wilson nodules and sclerosis.
- Blood pressure control to 130/80 mm Hg slows microvascular and macrovascular disease, but the goal should not be lower in older patients with diabetes.
- Glycemic control slows microvascular disease: the goal for most patients for hemoglobin A1c is 7.0%. Tighter control may increase cardiovascular risk.
- Either an angiotensin-converting enzyme inhibitor or an angiotensin receptor blocker is the first-line treatment for diabetic nephropathy; combining the two is no longer recommended.
- If more aggressive treatment is needed, a diuretic or spironolactone (with potassium monitoring) can be added.
- The role of sodium bicarbonate and new agents such as blockers of transcription factors is still emerging.
Carbapenem-resistant Enterobacteriaceae: A menace to our most vulnerable patients
The past 10 years have brought a formidable challenge to the clinical arena, as carbapenems, until now the most reliable antibiotics against Klebsiella species, Escherichia coli, and other Enterobacteriaceae, are becoming increasingly ineffective.
Infections caused by carbapenem-resistant Enterobacteriaceae (CRE) pose a serious threat to hospitalized patients. Moreover, CRE often demonstrate resistance to many other classes of antibiotics, thus limiting our therapeutic options. Furthermore, few new antibiotics are in line to replace carbapenems. This public health crisis demands redefined and refocused efforts in the diagnosis, treatment, and control of infections in hospitalized patients.
Here, we present an overview of CRE and discuss avenues to escape a new era of untreatable infections.
INCREASED USE OF CARBAPENEMS AND EMERGENCE OF RESISTANCE
Developed in the 1980s, carbapenems are derivatives of thyanamycin. Imipenem and meropenem, the first members of the class, had a broad spectrum of antimicrobial activity that included coverage of Pseudomonas aeruginosa, adequately positioning them for the treatment of nosocomial infections. Back then, nearly all Enterobacteriaceae were susceptible to carbapenems.1

In the 1990s, Enterobacteriaceae started to develop resistance to cephalosporins—till then, the first-line antibiotics for these organisms—by acquiring extended-spectrum betalactamases, which inactivate those agents. Consequently, the use of cephalosporins had to be restricted, while carbapenems, which remained impervious to these enzymes, had to be used more.2 In pivotal international studies in the treatment of infections caused by strains of K pneumoniae that produced these inactivating enzymes, outcomes were better with carbapenems than with cephalosporins and fluoroquinolones.3,4
Ertapenem, a carbapenem without antipseudomonal activity and highly bound to protein, was released in 2001. Its prolonged half-life permitted once-daily dosing, which positioned it as an option for treating infections in community dwellers.5 Doripenem is the newest member of the class of carbapenems, and its spectrum of activity is similar to that of imipenem and meropenem and includes P aeruginosa.6 The use of carbapenems, measured in a representative sample of 35 university hospitals in the United States, increased by 59% between 2002 and 2006.7
In the early 2000s, carbapenem resistance in K pneumoniae and other Enterobacteriaceae was rare in North America. But then, after initial outbreaks occurred in hospitals in the Northeast (especially New York City), CRE began to spread throughout the United States. By 2009–2010, the National Healthcare Safety Network from the Centers for Disease Control and Prevention (CDC) revealed that 12.8% of K pneumoniae isolates associated with bloodstream infections were resistant to carbapenems.8
In March 2013, the CDC disclosed that 3.9% of short-stay acute-care hospitals and 17.8% of long-term acute-care hospitals reported at least one CRE health care-associated infection in 2012. CRE had extended to 42 states, and the proportion of Enterobacteriaceae that are CRE had increased fourfold over the past 10 years.9
Coinciding with the increased use of carbapenems, multiple factors and modifiers likely contributed to the dramatic increase in CRE. These include use of other antibiotics in humans and animals, their relative penetration and selective effect on the gut microbiota, case-mix and infection control practices in different health care settings, and travel patterns.
POWERFUL ENZYMES THAT TRAVEL FAR
Bacterial acquisition of carbapenemases, enzymes that inactivate carbapenems, is crucial to the emergence of CRE. The enzyme in the sentinel carbapenem-resistant K pneumoniae isolate found in 1996 in North Carolina was designated K pneumoniae carbapenemase (KPC-1). This mechanism also conferred resistance to all cephalosporins, aztreonam, and beta-lactamase inhibitors such as clavulanic acid and tazobactam.10
KPC-2 (later determined to be identical to KPC-1) was found in K pneumoniae from Baltimore, and KPC-3 caused an early outbreak in New York City.11,12 To date, 12 additional variants of blaKPC, the gene encoding for the KPC enzyme, have been described.13
The genes encoding carbapenemases are usually found on plasmids or other common mobile genetic elements.14 These genetic elements allow the organism to acquire genes conferring resistance to other classes of antimicrobials, such as aminoglycoside-modifying enzymes and fluoroquinolone-resistance determinants, and beta-lactamases.15,16 The result is that CRE isolates are increasingly multidrug-resistant (ie, resistant to three or more classes of antimicrobials), extensively drug-resistant (ie, resistant to all but one or two classes), or pandrug-resistant (ie, resistant to all available classes of antibiotics).17 Thus, up to 98% of KPC-producing K pneumoniae are resistant to trimethoprim-sulfamethoxazole, 90% are resistant to fluoroquinolones, and 60% are resistant to gentamicin or amikacin.15
The mobility of these genetic elements has also allowed for dispersion into diverse Enterobacteriaceae such as E coli, Klebsiella oxytoca, Enterobacter, Serratia, and Salmonella species. Furthermore, KPC has been described in non-Enterobacteriaceae such as Acinetobacter baumannii and P aeruginosa.
Extending globally, KPC is now endemic in the Mediterranean basin, including Israel, Greece, and Italy; in South America, especially Colombia, Argentina, and Brazil; and in China.18 Most interesting is the intercontinental transfer of these strains: it has been documented that the index patient with KPC-producing K pneumoniae in Medellin, Colombia, came from Israel to undergo liver transplantation.19 Likewise, KPC-producing K pneumoniae in France and Israel could be linked epidemiologically and genetically to the predominant US strain.20,21
Even more explosive has been the surge of another carbapenemase, the Ambler Class B New Delhi metallo-beta-lactamase, or NDM-1. Initially reported in a urinary isolate of K pneumoniae from a Swedish patient who had been hospitalized in New Delhi in 2008, NDM-1 was soon found throughout India, in Pakistan, and in the United Kingdom.22 Interestingly, several of the UK patients with NDM-1-harboring bacteria had received organ transplants in the Indian subcontinent. Reports from elsewhere in Europe, Australia, and Africa followed suit, usually with a connection to the Indian subcontinent epicenter. In contrast, several other cases in Europe were traced to the Balkans, where there appears to be another focus of NDM-1.23
Penetration of NDM-1 into North America has begun, with cases and outbreaks reported in several US and Canadian regions, and in a military medical facility in Afghanistan. In several of these instances, there has been a documented link with travel and hospitalizations overseas.24–27 However, no such link with travel could be established in a recent outbreak in Ontario.27

In addition, resistance to carbapenems may result from other enzymes (Table 1), or from combinations of changes in outer membrane porins and the production of extended spectrum beta-lactamases or other cephalosporinases.28
DEADLY IMPACT ON THE MOST VULNERABLE
Regardless of the resistance pattern, Enterobacteriaceae are an important cause of health care-associated infections, including urinary and bloodstream infections in patients with indwelling catheters, pneumonia (often in association with mechanical ventilation), and, less frequently, infections of skin and soft tissues and the central nervous system.29–31
Several studies have examined the clinical characteristics and outcomes of patients with CRE infections. Those typically affected are elderly and debilitated and have multiple comorbidities, including diabetes mellitus and immunosuppression. They are heavily exposed to health care with frequent antecedent hospitalizations and invasive procedures. Furthermore, they are often severely ill and require intensive care. Patients infected with carbapenem-resistant K pneumoniae, compared with those with carbapenem-susceptible strains, are more likely to have undergone organ or stem cell transplantation or mechanical ventilation, and to have had a longer hospital stay before infection.
They also experience a high mortality rate, which ranges from 30% in patients with nonbacteremic infections to 72% in series of patients with liver transplants or bloodstream infections.32–37
More recently, CRE has been reported in other vulnerable populations, such as children with critical illness or cancer and in burn patients.38–40
Elderly and critically ill patients with bacteremia originating from a high-risk source (eg, pneumonia) typically face the most adverse outcomes. With increasing drug resistance, inadequate initial antimicrobial therapy is more commonly seen and may account for some of these poor outcomes.37,41
LONG-TERM CARE FACILITIES IN THE EYE OF THE STORM
A growing body of evidence suggests that long-term care facilities play a crucial role in the spread of CRE.
In an investigation into carbapenem-resistant A baumanii and K pneumoniae in a hospital system,36 75% of patients with carbapenem-resistant K pneumoniae were admitted from long-term care facilities, and only 1 of 13 patients was discharged home.
In a series of patients with carbapenem-resistant K pneumoniae bloodstream infections, 42% survived their index hospital stay. Of these patients, only 32% were discharged home, and readmissions were very common.32
Admission from a long-term care facility or transfer from another hospital is significantly associated with carbapenem resistance in patients with Enterobacteriaceae.42 Similarly, in Israel, a large reservoir of CRE was found in postacute care facilities.43
It is clear that long-term care residents are at increased risk of colonization and infection with CRE. However, further studies are needed to evaluate whether this simply refects an overlap in risk factors, or whether significant patient-to-patient transmission occurs in these settings.
INFECTION CONTROL TAKES CENTER STAGE
It is important to note that risk factors for CRE match those of various nosocomial infections, including other resistant gram-negative bacilli, methicillin-resistant Staphylococcus aureus, vancomycin-resistant enterococci, Candida species, and Clostridium difficile; in fact, CRE often coexist with other multidrug-resistant organisms.44,45
Common risk factors include residence in a long-term care facility, an intensive care unit stay, use of lines and catheters, and antibiotic exposure. This commonality of risk factors implies that systematic infection-prevention measures will have an impact on the prevalence and incidence rates of multidrug-resistant organism infections across the board, CRE included. It should be emphasized that strict compliance with hand hygiene is still the foundation of any infection-prevention strategy.
Infection prevention and the control of transmission of CRE in long-term care facilities pose unique challenges. Guidelines from the Society for Healthcare Epidemiology and the Association for Professionals in Infection Control recommend the use of contact precautions for patients with multidrug-resistant organisms, including CRE, who are ill and totally dependent on health care workers for activities of daily living or whose secretions or drainage cannot be contained. These same guidelines advise against attempting to eradicate multidrug-resistant organism colonization status.46
In acute care facilities, Best Infection Control Practices from the CDC and the Healthcare Infection Control Practices Advisory Committee encourage mechanisms for the rapid recognition and reporting of CRE cases to infection prevention personnel so that contact precautions can be implemented. Furthermore, facilities without CRE cases should carry out periodic laboratory reviews to identify cases, and patients exposed to CRE cases should be screened with surveillance cultures.47
Outbreaks of CRE may require extraordinary infection control measures. An approach combining point-prevalence surveillance of colonization, detection of environmental and common-equipment contamination, with the implementation of a bundle consisting of chlorhexidine baths, cohorting of colonized patients and health care personnel, increased environmental cleaning, and staff education may be effective in controlling outbreaks of CRE.48
Nevertheless, control of CRE may prove exceptionally difficult. A recent high-profile outbreak of carbapenem-resistant K pneumoniae at the National Institutes of Health Clinical Center in Maryland caused infections in 18 patients, 11 of whom died.49 Of note, carbapenem-resistant K pneumoniae was detected in this outbreak in both respiratory equipment and sink drains. The outbreak was ultimately contained by detection through surveillance cultures and by strict cohorting of colonized patients, which minimized common medical equipment and personnel between affected patients and other patients in the hospital. Additionally, rooms were sanitized with hydrogen peroxide vapor, and sinks and drains where carbapenem-resistant K pneumoniae was detected were removed.
CHALLENGES IN THE MICROBIOLOGY LABORATORY
Adequate treatment and control of CRE infections is predicated upon their accurate and prompt diagnosis from patient samples in the clinical microbiology laboratory.50
Traditional and current culture-based methods take several days to provide that information, delaying effective antibiotic therapy and permitting the transmission of undetected CRE. Furthermore, interpretative criteria of minimal inhibitory concentrations (MICs) of carbapenems recently required readjustment, as many KPC-producing strains of K pneumoniae had MICs below the previous breakpoint of resistance. In the past, this contributed to instances of “silent” dissemination of KPC-producing K pneumoniae.51
In contrast, using the new lower breakpoints of resistance for carbapenems without using a phenotypic test such as the modified Hodge test or the carbapenem-EDTA combination tests will result in a lack of differentiation between various mechanisms of carbapenem resistance.28,52,53 This may be clinically relevant, as the clinical response to carbapenem therapy may vary depending on the mechanism of resistance.
GENERAL PRINCIPLES APPLY
In treating patients infected with CRE, clinicians need to strictly observe general principles of infectious disease management to ensure the best possible outcomes. These include:
Timely and accurate diagnosis, as discussed above.
Source control, which should include drainage of any infected collections, and removal of lines, devices, and urinary catheters.
Distinguishing between infection and colonization. CRE are often encountered as urinary isolates, and the distinction between asymptomatic bacteriuria and urinary tract infection may be extremely difficult, especially in residents of long-term care facilities with chronic indwelling catheters, who are thegroup at highest risk of CRE colonization and infection. Urinalysis may be helpful in the absence of pyuria, as this rules out an infection; however, it must be emphasized that the presence of pyuria is not a helpful feature, as pyuria is common in both asymptomatic bacteriuria and urinary tract infection.54 Symptoms should be carefully evaluated in every patient with bacteriuria, and urinary tract infection should be a diagnosis of exclusion in patients with functional symptoms such as confusion or falls.
Selection of the most appropriate antibiotic regimen. While the emphasis is often on the antibiotic regimen, the above elements should not be neglected.
A DWINDLING THERAPEUTIC ARSENAL
Clinicians treating CRE infections are left with only a few antibiotic options. These options are generally limited by a lack of clinical data on efficacy, as well as by concerns about toxicity. These “drugs of last resort” include polymyxins (such as colistin), aminoglycosides, tigecycline, and fosfomycin. The role of carbapenem therapy, potentially in combination regimens, in a high-dose prolonged infusion, or even “double carbapenem therapy” remains to be determined.37,55,56
Colistin
Colistin is one of the first-line agents for treating CRE infections. First introduced in the 1950s, its use was mostly abandoned in favor of aminoglycosides. A proportion of the data on safety and efficacy of colistin, therefore, is based on older, less rigorous studies.
Neurotoxicity and nephrotoxicity are the two main concerns with colistin, and while the incidence of these adverse events does appear to be lower with modern preparations, it is still substantial.57 Dosing issues have not been completely clarified either, especially in relation to renal clearance and in patients on renal replacement therapy.58,59 Unfortunately, there have been reports of outbreaks of CRE displaying resistance to colistin.60
Tigecycline
Tigecycline is a newer antibiotic of the glycylcycline class. Like colistin, it has no oral preparation for systemic infections.
The main side effect of tigecycline is nausea.61 Other reported issues include pancreatitis and extreme alkaline phosphatase elevations.
The efficacy of tigecycline has come into question in view of meta-analyses of clinical trials, some of which have shown higher mortality rates in patients treated with tigecycline than with comparator agents.62–65 Based on these data, the US Food and Drug Administration issued a warning in 2010 regarding the increased mortality risk. Although these meta-analyses did not include patients with CRE for whom available comparators would have been ineffective, it is an important safety signal.
The efficacy of tigecycline is further limited by increasing in vitro resistance in CRE. Serum and urinary levels of tigecycline are low, and most experts discourage the use of tigecycline as monotherapy for blood stream or urinary tract infections.
Aminoglycosides
CRE display variable in vitro susceptibility to different aminoglycosides. If the organism is susceptible, aminoglycosides may be very useful in the treatment of CRE infections, especially urinary tract infectons. In a study of carbapenem-resistant K pneumoniae urinary tract infections, patients who were treated with polymyxins or tigecycline were significantly less likely to have clearance of their urine as compared with patients treated with aminoglycosides.66
Ototoxicity and nephrotoxicity are demonstrated adverse effects of aminoglycosides. Close monitoring of serum levels, interval audiology examinations at baseline and during therapy, and the use of extended-interval dosing may help to decrease the incidence of these toxicities.
Fosfomycin
Fosfomycin is only available as an oral formulation in the United States, although intravenous administration has been used in other countries. It is exclusively used to treat urinary tract infections.
CRE often retain susceptibility to fosfomycin, and clearance of urine in cystitis may be attempted with this agent to avoid the need for intravenous treatment.29,67
Combination therapy, other topics to be explored
Recent observational reports from Greece, Italy, and the United States describe higher survival rates in patients with CRE infections treated with a combination regimen rather than monotherapy with colistin or tigecycline. This is despite reliable activity of colistin and tigecycline, and often in regimens containing carbapenems. Clinical experiments are needed to clarify the value of combination regimens that include carbapenems for the treatment of CRE infections.
Similarly, the role of carbapenems given as a high-dose prolonged infusion or as double carbapenem therapy needs to be explored further.37,55,56,68
Also to be determined is the optimal duration of treatment. To date, there is no evidence that increasing the duration of treatment beyond that recommended for infections with more susceptible bacteria results in improved outcomes. Therefore, commonly used durations include 1 week for complicated urinary tract infections, 2 weeks for bacteremia (from the first day with negative blood cultures and source control), and 8 to 14 days for pneumonia.
A SERIOUS THREAT
The emergence of CRE is a serious threat to the safety of patients in our health care system. CRE are highly successful nosocomial pathogens selected by the use of antibiotics, which burden patients debilitated by advanced age, comorbidities, and medical interventions. Infections with CRE result in poor outcomes, and available treatments of last resort such as tigecycline and colistin are of unclear efficacy and safety.

Control of CRE transmission is hindered by the transit of patients through long-term care facilities, and detection of CRE is difficult because of the myriad mechanisms involved and the imperfect methods currently available. Clinicians are concerned and frustrated, especially given the paucity of antibiotics in development to address the therapeutic dilemma posed by CRE. The challenge of CRE and other multidrug-resistant organisms requires the concerted response of professionals in various disciplines, including pharmacists, microbiologists, infection control practitioners, and infectious disease clinicians (Table 2).
Control of transmission by infection prevention strategies and by antimicrobial stewardship is going to be crucial in the years to come, not only for limiting the spread of CRE, but also for preventing the next multidrug-resistant “superbug” from emerging. However, the current reality is that health care providers will be faced with increased numbers of patients infected with CRE.
Prospective studies into transmission, molecular characteristics, and, most of all, treatment regimens are urgently needed. In addition, the development of new antimicrobials and nontraditional antimicrobial methods should have international priority.
- Papp-Wallace KM, Endimiani A, Taracila MA, Bonomo RA. Carbapenems: past, present, and future. Antimicrob Agents Chemother 2011; 55:4943–4960.
- Rahal JJ, Urban C, Horn D, et al. Class restriction of cephalosporin use to control total cephalosporin resistance in nosocomial Klebsiella. JAMA 1998; 280:1233–1237.
- Paterson DL, Ko WC, Von Gottberg A, et al. International prospective study of Klebsiella pneumoniae bacteremia: implications of extended-spectrum beta-lactamase production in nosocomial Infections. Ann Intern Med 2004; 140:26–32.
- Endimiani A, Luzzaro F, Perilli M, et al. Bacteremia due to Klebsiella pneumoniae isolates producing the TEM-52 extended-spectrum beta-lactamase: treatment outcome of patients receiving imipenem or ciprofoxacin. Clin Infect Dis 2004; 38:243–251.
- Livermore DM, Sefton AM, Scott GM. Properties and potential of ertapenem. J Antimicrob Chemother 2003; 52:331–344.
- Bazan JA, Martin SI, Kaye KM. Newer beta-lactam antibiotics: doripenem, ceftobiprole, ceftaroline, and cefepime. Infect Dis Clin North Am 2009; 23:983–996, ix.
- Pakyz AL, MacDougall C, Oinonen M, Polk RE. Trends in antibacterial use in US academic health centers: 2002 to 2006. Arch Intern Med 2008; 168:2254–2260.
- Sievert DM, Ricks P, Edwards JR, et al. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect Control Hosp Epidemiol 2013; 34:1–14.
- Centers for Disease Control and Prevention. Vital signs: carbapenem-resistant Enterobacteriaceae. MMWR 2013; 62:165–170.
- Yigit H, Queenan AM, Anderson GJ, et al. Novel carbapenem-hydrolyzing beta-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob Agents Chemother 2001; 45:1151–1161.
- Smith Moland E, Hanson ND, Herrera VL, et al. Plasmid-mediated, carbapenem-hydrolysing beta-lactamase, KPC-2, in Klebsiella pneumoniae isolates. J Antimicrob Chemother 2003; 51:711–714.
- Woodford N, Tierno PM, Young K, et al. Outbreak of Klebsiella pneumoniae producing a new carbapenem-hydrolyzing class A beta-lactamase, KPC-3, in a New York medical center. Antimicrob Agents Chemother 2004; 48:4793–4799.
- Lehey Clinic. OXA-type β-Lactamases. http://www.lahey.org/Studies/other.asp#table1. Accessed March 11, 2013.
- Mathers AJ, Cox HL, Kitchel B, et al. Molecular dissection of an outbreak of carbapenem-resistant Enterobacteriaceae reveals intergenus KPC carbapenemase transmission through a promiscuous plasmid. MBio 2011; 2 6:e00204–11.
- Endimiani A, Hujer AM, Perez F, et al. Characterization of blaKPC-containing Klebsiella pneumoniae isolates detected in different institutions in the Eastern USA. J Antimicrob Chemother 2009; 63:427–437.
- Endimiani A, Carias LL, Hujer AM, et al. Presence of plasmid-mediated quinolone resistance in Klebsiella pneumoniae isolates possessing blaKPC in the United States. Antimicro Agents Chemother 2008; 52:2680–2682.
- Magiorakos A P, Srinivasan A, Carey RB, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect 2012; 18:268–281.
- Tzouvelekis LS, Markogiannakis A, Psichogiou M, Tassios PT, Daikos GL. Carbapenemases in Klebsiella pneumoniae and other Enterobacteriaceae: an evolving crisis of global dimensions. Clin Microbiol Rev 2012; 25:682–707.
- Lopez JA, Correa A, Navon-Venezia S, et al. Intercontinental spread from Israel to Colombia of a KPC-3-producing Klebsiella pneumoniae strain. Clin Microbiol Infect 2011; 17:52–56.
- Naas T, Nordmann P, Vedel G, Poyart C. Plasmid-mediated carbapenem-hydrolyzing beta-lactamase KPC in a Klebsiella pneumoniae isolate from France. Antimicrob Agents Chemother 2005; 49:4423–4424.
- Navon-Venezia S, Leavitt A, Schwaber MJ, et al. First report on a hyperepidemic clone of KPC-3-producing Klebsiella pneumoniae in Israel genetically related to a strain causing outbreaks in the United States. Antimicrob Agents Chemother 2009; 53:818–820.
- Yong D, Toleman MA, Giske CG, et al. Characterization of a new metallo-beta-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob Agents Chemother 2009; 53:5046–5054.
- Livermore DM, Walsh TR, Toleman M, Woodford N. Balkan NDM-1: escape or transplant? Lancet Infect Dis 2011; 11:164.
- Centers for Disease Control and Prevention. Carbapenem-resistant enterobacteriaceae containing New Delhi metallo-beta-lactamase in two patients - Rhode Island, March 2012. MMWR Morb Mortal Wkly Rep 2012Jun 22; 61:446–448.
- Centers for Disease Control and Prevention. Detection of Enterobacteriaceae isolates carrying metallo-beta-lactamase—United States, 2010. MMWR Morb Mortal Wkly Rep 2010; 59:750.
- McGann P, Hang J, Clifford RJ, et al. Complete sequence of a novel 178-kilobase plasmid carrying bla(NDM-1) in a Providencia stuartii strain isolated in Afghanistan. Antimicrob Agents Chemother 2012; 56:1673–1679.
- Borgia S, Lastovetska O, Richardson D, et al. Outbreak of carbapenem-resistant Enterobacteriaceae containing blaNDM-1, Ontario, Canada. Clin Infect Dis 2012; 55:e109–e117.
- Endimiani A, Perez F, Bajaksouzian S, et al. Evaluation of updated interpretative criteria for categorizing Klebsiella pneumoniae with reduced carbapenem susceptibility. J Clinic Microbiol 2010; 48:4417–4425.
- Neuner EA, Sekeres J, Hall GS, van Duin D. Experience with fosfomycin for treatment of urinary tract infections due to multidrug-resistant organisms. Antimicrob Agents Chemother 2012; 56:5744–5748.
- Neuner EA, Yeh JY, Hall GS, et al. Treatment and outcomes in carbapenem-resistant Klebsiella pneumoniae bloodstream infections. Diagnostic Microbiol Infect Dis 2011; 69:357–362.
- van Duin D, Kaye KS, Neuner EA, Bonomo RA. Carbapenem-resistant Enterobacteriaceae: a review of treatment and outcomes. Diagnostic Microbiol Infect Dis 2013; 75:115–120.
- Neuner EA, Yeh J-Y, Hall GS, et al. Treatment and outcomes in carbapenem-resistant Klebsiella pneumoniae bloodstream infections. Diagn Microbiol Infect Dis 2011; 69:357–362.
- Patel G, Huprikar S, Factor SH, Jenkins SG, Calfee DP. Outcomes of carbapenem-resistant Klebsiella pneumoniae infection and the impact of antimicrobial and adjunctive therapies. Infect Control Hosp Epidemiol 2008; 29:1099–1106.
- Borer A, Saidel-Odes L, Riesenberg K, et al. Attributable mortality rate for carbapenem-resistant Klebsiella pneumoniae bacteremia. Infect Control Hosp Epidemiol 2009; 30:972–976.
- Marchaim D, Chopra T, Perez F, et al. Outcomes and genetic relatedness of carbapenem-resistant Enterobacteriaceae at Detroit medical center. Infect Control Hosp Epidemiol 2011; 32:861–871.
- Perez F, Endimiani A, Ray AJ, et al. Carbapenem-resistant Acinetobacter baumannii and Klebsiella pneumoniae across a hospital system: impact of post-acute care facilities on dissemination. J Antimicrob Chemother 2010; 65:1807–1818.
- Tumbarello M, Viale P, Viscoli C, et al. Predictors of mortality in bloodstream infections caused by Klebsiella pneumoniae carbapenemase-producing K. pneumoniae: importance of combination therapy. Clin Infect Dis 2012; 55:943–950.
- Little ML, Qin X, Zerr DM, Weissman SJ. Molecular diversity in mechanisms of carbapenem resistance in paediatric Enterobacteriaceae. Int J Antimicrob Agents 2012; 39:52–57.
- Logan LK. Carbapenem-resistant Enterobacteriaceae: an emerging problem in children. Clin Infect Dis 2012; 55:852–859.
- Rastegar Lari A, Azimi L, Rahbar M, Fallah F, Alaghehbandan R. Phenotypic detection of Klebsiella pneumoniae carbapenemase among burns patients: first report from Iran. Burns 2013; 39:174–176.
- Zarkotou O, Pournaras S, Tselioti P, et al. Predictors of mortality in patients with bloodstream infections caused by KPC-producing Klebsiella pneumoniae and impact of appropriate antimicrobial treatment. Clin Microbiol Infect 2011; 17:1798–1803.
- Hyle EP, Ferraro MJ, Silver M, Lee H, Hooper DC. Ertapenem-resistant Enterobacteriaceae: risk factors for acquisition and outcomes. Infect Control Hosp Epidemiol 2010; 31:1242–1249.
- Ben-David D, Masarwa S, Navon-Venezia S, et al. Carbapenem-resistant Klebsiella pneumoniae in post-acute-care facilities in Israel. Infect Control Hosp Epidemiol 2011; 32:845–853.
- Safdar N, Maki DG. The commonality of risk factors for nosocomial colonization and infection with antimicrobial-resistant Staphylococcus aureus, enterococcus, gram-negative bacilli, Clostridium difficile, and Candida. Ann Intern Med 2002; 136:834–844.
- Marchaim D, Perez F, Lee J, et al. “Swimming in resistance”: co-colonization with carbapenem-resistant Enterobacteriaceae and Acinetobacter baumannii or Pseudomonas aeruginosa.” Am J Infect Control 2012; 40:830–835.
- Smith PW, Bennett G, Bradley S, et al. SHEA/APIC Guideline: Infection prevention and control in the long-term care facility. Am J Infect Control 2008; 36:504–535.
- Centers for Disease Control and Prevention. Guidance for control of infections with carbapenem-resistant or carbapenemase-producing Enterobacteriaceae in acute care facilities. MMWR 2009; 58:256–260.
- Munoz-Price LS, De La Cuesta C, Adams S, et al. Successful eradication of a monoclonal strain of Klebsiella pneumoniae during a K. pneumoniae carbapenemase-producing K. pneumoniae outbreak in a surgical intensive care unit in Miami, Florida. Infect Control Hosp Epidemiol 2010; 31:1074–1077.
- Snitkin ES, Zelazny AM, Thomas PJ, et al. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with wholegenome sequencing. Sci Transl Med 2012; 4:148ra16.
- Srinivasan A, Patel JB. Klebsiella pneumoniae carbapenemase-producing organisms: an ounce of prevention really is worth a pound of cure. Infect Control Hosp Epidemiol 2008; 29:1107–1109.
- Viau RA, Hujer AM, Marshall SH, et al. “Silent” dissemination of Klebsiella pneumoniae isolates bearing K pneumoniae carbapenemase in a long-term care facility for children and young adults in Northeast Ohio”. Clin Infect Dis 2012; 54:1314–1321.
- Galani I, Rekatsina PD, Hatzaki D, Plachouras D, Souli M, Giamarellou H. Evaluation of different laboratory tests for the detection of metallo-beta-lactamase production in Enterobacteriaceae. J Antimicrob Chemother 2008; 61:548–553.
- Anderson KF, Lonsway DR, Rasheed JK, et al. Evaluation of methods to identify the Klebsiella pneumoniae carbapenemase in Enterobacteriaceae. J Clin Microbiol 2007; 45:2723–2725.
- Nicolle LE, Bradley S, Colgan R, Rice JC, Schaeffer A, Hooton TM. Infectious Diseases Society of America guidelines for the diagnosis and treatment of asymptomatic bacteriuria in adults. Clin Infect Dis 2005; 40:643–654.
- Daikos GL, Markogiannakis A. Carbapenemase-producing Klebsiella pneumoniae: (when) might we still consider treating with carbapenems? Clin Microbiol Infect 2011; 17:1135–1141.
- Bulik CC, Nicolau DP. Double-carbapenem therapy for carbapenemase-producing Klebsiella pneumoniae. Antimicrob Agents Chemother 2011; 55:3002–3004.
- Pogue JM, Lee J, Marchaim D, et al. Incidence of and risk factors for colistin-associated nephrotoxicity in a large academic health system. Clin Infect Dis 2011; 53:879–884.
- Garonzik SM, Li J, Thamlikitkul V, et al. Population pharmacokinetics of colistin methanesulfonate and formed colistin in critically ill patients from a multicenter study provide dosing suggestions for various categories of patients. Antimicrob Agents Chemother 2011; 55:3284–3294.
- Dalfno L, Puntillo F, Mosca A, et al. High-dose, extended-interval colistin administration in critically ill patients: is this the right dosing strategy? A preliminary study. Clin Infect Dis 2012; 54:1720–1726.
- Marchaim D, Chopra T, Pogue JM, et al. Outbreak of colistin-resistant, carbapenem-resistant Klebsiella pneumoniae in metropolitan Detroit, Michigan. Antimicrob Agents Chemother 2011; 55:593–599.
- Bonilla MF, Avery RK, Rehm SJ, Neuner EA, Isada CM, van Duin D. Extreme alkaline phosphatase elevation associated with tigecycline. J Antimicrob Chemother 2011; 66:952–953.
- Prasad P, Sun J, Danner RL, Natanson C. Excess deaths associated with tigecycline after approval based on noninferiority trials. Clin Infect Dis 2012; 54:1699–1709.
- Tasina E, Haidich AB, Kokkali S, Arvanitidou M. Efficacy and safety of tigecycline for the treatment of infectious diseases: a meta-analysis. Lancet Infect Dis 2011; 11:834–844.
- Cai Y, Wang R, Liang B, Bai N, Liu Y. Systematic review and meta-analysis of the effectiveness and safety of tigecycline for treatment of infectious disease. Antimicrob Agents Chemother 2011; 55:1162–1172.
- Yahav D, Lador A, Paul M, Leibovici L. Efficacy and safety of tigecycline: a systematic review and meta-analysis. J Antimicrob Chemother 2011; 66:1963–1971.
- Satlin MJ, Kubin CJ, Blumenthal JS, et al. Comparative effectiveness of aminoglycosides, polymyxin B, and tigecycline for clearance of carbapenem-resistant Klebsiella pneumoniae from urine. Antimicrob Agents Chemother 2011; 55:5893–5899.
- Endimiani A, Patel G, Hujer KM, et al. In vitro activity of fosfomycin against blaKPC-containing Klebsiella pneumoniae isolates, including those nonsusceptible to tigecycline and/or colistin. Antimicrob Agents Chemother 2010; 54:526–529.
- Qureshi ZA, Paterson DL, Potoski BA, et al. Treatment outcome of bacteremia due to KPC-producing Klebsiella pneumoniae: superiority of combination antimicrobial regimens. Antimicrob Agents Chemother 2012; 56:2108–2113.
The past 10 years have brought a formidable challenge to the clinical arena, as carbapenems, until now the most reliable antibiotics against Klebsiella species, Escherichia coli, and other Enterobacteriaceae, are becoming increasingly ineffective.
Infections caused by carbapenem-resistant Enterobacteriaceae (CRE) pose a serious threat to hospitalized patients. Moreover, CRE often demonstrate resistance to many other classes of antibiotics, thus limiting our therapeutic options. Furthermore, few new antibiotics are in line to replace carbapenems. This public health crisis demands redefined and refocused efforts in the diagnosis, treatment, and control of infections in hospitalized patients.
Here, we present an overview of CRE and discuss avenues to escape a new era of untreatable infections.
INCREASED USE OF CARBAPENEMS AND EMERGENCE OF RESISTANCE
Developed in the 1980s, carbapenems are derivatives of thyanamycin. Imipenem and meropenem, the first members of the class, had a broad spectrum of antimicrobial activity that included coverage of Pseudomonas aeruginosa, adequately positioning them for the treatment of nosocomial infections. Back then, nearly all Enterobacteriaceae were susceptible to carbapenems.1
In the 1990s, Enterobacteriaceae started to develop resistance to cephalosporins—till then, the first-line antibiotics for these organisms—by acquiring extended-spectrum betalactamases, which inactivate those agents. Consequently, the use of cephalosporins had to be restricted, while carbapenems, which remained impervious to these enzymes, had to be used more.2 In pivotal international studies in the treatment of infections caused by strains of K pneumoniae that produced these inactivating enzymes, outcomes were better with carbapenems than with cephalosporins and fluoroquinolones.3,4
Ertapenem, a carbapenem without antipseudomonal activity and highly bound to protein, was released in 2001. Its prolonged half-life permitted once-daily dosing, which positioned it as an option for treating infections in community dwellers.5 Doripenem is the newest member of the class of carbapenems, and its spectrum of activity is similar to that of imipenem and meropenem and includes P aeruginosa.6 The use of carbapenems, measured in a representative sample of 35 university hospitals in the United States, increased by 59% between 2002 and 2006.7
In the early 2000s, carbapenem resistance in K pneumoniae and other Enterobacteriaceae was rare in North America. But then, after initial outbreaks occurred in hospitals in the Northeast (especially New York City), CRE began to spread throughout the United States. By 2009–2010, the National Healthcare Safety Network from the Centers for Disease Control and Prevention (CDC) revealed that 12.8% of K pneumoniae isolates associated with bloodstream infections were resistant to carbapenems.8
In March 2013, the CDC disclosed that 3.9% of short-stay acute-care hospitals and 17.8% of long-term acute-care hospitals reported at least one CRE health care-associated infection in 2012. CRE had extended to 42 states, and the proportion of Enterobacteriaceae that are CRE had increased fourfold over the past 10 years.9
Coinciding with the increased use of carbapenems, multiple factors and modifiers likely contributed to the dramatic increase in CRE. These include use of other antibiotics in humans and animals, their relative penetration and selective effect on the gut microbiota, case-mix and infection control practices in different health care settings, and travel patterns.
POWERFUL ENZYMES THAT TRAVEL FAR
Bacterial acquisition of carbapenemases, enzymes that inactivate carbapenems, is crucial to the emergence of CRE. The enzyme in the sentinel carbapenem-resistant K pneumoniae isolate found in 1996 in North Carolina was designated K pneumoniae carbapenemase (KPC-1). This mechanism also conferred resistance to all cephalosporins, aztreonam, and beta-lactamase inhibitors such as clavulanic acid and tazobactam.10
KPC-2 (later determined to be identical to KPC-1) was found in K pneumoniae from Baltimore, and KPC-3 caused an early outbreak in New York City.11,12 To date, 12 additional variants of blaKPC, the gene encoding for the KPC enzyme, have been described.13
The genes encoding carbapenemases are usually found on plasmids or other common mobile genetic elements.14 These genetic elements allow the organism to acquire genes conferring resistance to other classes of antimicrobials, such as aminoglycoside-modifying enzymes and fluoroquinolone-resistance determinants, and beta-lactamases.15,16 The result is that CRE isolates are increasingly multidrug-resistant (ie, resistant to three or more classes of antimicrobials), extensively drug-resistant (ie, resistant to all but one or two classes), or pandrug-resistant (ie, resistant to all available classes of antibiotics).17 Thus, up to 98% of KPC-producing K pneumoniae are resistant to trimethoprim-sulfamethoxazole, 90% are resistant to fluoroquinolones, and 60% are resistant to gentamicin or amikacin.15
The mobility of these genetic elements has also allowed for dispersion into diverse Enterobacteriaceae such as E coli, Klebsiella oxytoca, Enterobacter, Serratia, and Salmonella species. Furthermore, KPC has been described in non-Enterobacteriaceae such as Acinetobacter baumannii and P aeruginosa.
Extending globally, KPC is now endemic in the Mediterranean basin, including Israel, Greece, and Italy; in South America, especially Colombia, Argentina, and Brazil; and in China.18 Most interesting is the intercontinental transfer of these strains: it has been documented that the index patient with KPC-producing K pneumoniae in Medellin, Colombia, came from Israel to undergo liver transplantation.19 Likewise, KPC-producing K pneumoniae in France and Israel could be linked epidemiologically and genetically to the predominant US strain.20,21
Even more explosive has been the surge of another carbapenemase, the Ambler Class B New Delhi metallo-beta-lactamase, or NDM-1. Initially reported in a urinary isolate of K pneumoniae from a Swedish patient who had been hospitalized in New Delhi in 2008, NDM-1 was soon found throughout India, in Pakistan, and in the United Kingdom.22 Interestingly, several of the UK patients with NDM-1-harboring bacteria had received organ transplants in the Indian subcontinent. Reports from elsewhere in Europe, Australia, and Africa followed suit, usually with a connection to the Indian subcontinent epicenter. In contrast, several other cases in Europe were traced to the Balkans, where there appears to be another focus of NDM-1.23
Penetration of NDM-1 into North America has begun, with cases and outbreaks reported in several US and Canadian regions, and in a military medical facility in Afghanistan. In several of these instances, there has been a documented link with travel and hospitalizations overseas.24–27 However, no such link with travel could be established in a recent outbreak in Ontario.27
In addition, resistance to carbapenems may result from other enzymes (Table 1), or from combinations of changes in outer membrane porins and the production of extended spectrum beta-lactamases or other cephalosporinases.28
DEADLY IMPACT ON THE MOST VULNERABLE
Regardless of the resistance pattern, Enterobacteriaceae are an important cause of health care-associated infections, including urinary and bloodstream infections in patients with indwelling catheters, pneumonia (often in association with mechanical ventilation), and, less frequently, infections of skin and soft tissues and the central nervous system.29–31
Several studies have examined the clinical characteristics and outcomes of patients with CRE infections. Those typically affected are elderly and debilitated and have multiple comorbidities, including diabetes mellitus and immunosuppression. They are heavily exposed to health care with frequent antecedent hospitalizations and invasive procedures. Furthermore, they are often severely ill and require intensive care. Patients infected with carbapenem-resistant K pneumoniae, compared with those with carbapenem-susceptible strains, are more likely to have undergone organ or stem cell transplantation or mechanical ventilation, and to have had a longer hospital stay before infection.
They also experience a high mortality rate, which ranges from 30% in patients with nonbacteremic infections to 72% in series of patients with liver transplants or bloodstream infections.32–37
More recently, CRE has been reported in other vulnerable populations, such as children with critical illness or cancer and in burn patients.38–40
Elderly and critically ill patients with bacteremia originating from a high-risk source (eg, pneumonia) typically face the most adverse outcomes. With increasing drug resistance, inadequate initial antimicrobial therapy is more commonly seen and may account for some of these poor outcomes.37,41
LONG-TERM CARE FACILITIES IN THE EYE OF THE STORM
A growing body of evidence suggests that long-term care facilities play a crucial role in the spread of CRE.
In an investigation into carbapenem-resistant A baumanii and K pneumoniae in a hospital system,36 75% of patients with carbapenem-resistant K pneumoniae were admitted from long-term care facilities, and only 1 of 13 patients was discharged home.
In a series of patients with carbapenem-resistant K pneumoniae bloodstream infections, 42% survived their index hospital stay. Of these patients, only 32% were discharged home, and readmissions were very common.32
Admission from a long-term care facility or transfer from another hospital is significantly associated with carbapenem resistance in patients with Enterobacteriaceae.42 Similarly, in Israel, a large reservoir of CRE was found in postacute care facilities.43
It is clear that long-term care residents are at increased risk of colonization and infection with CRE. However, further studies are needed to evaluate whether this simply refects an overlap in risk factors, or whether significant patient-to-patient transmission occurs in these settings.
INFECTION CONTROL TAKES CENTER STAGE
It is important to note that risk factors for CRE match those of various nosocomial infections, including other resistant gram-negative bacilli, methicillin-resistant Staphylococcus aureus, vancomycin-resistant enterococci, Candida species, and Clostridium difficile; in fact, CRE often coexist with other multidrug-resistant organisms.44,45
Common risk factors include residence in a long-term care facility, an intensive care unit stay, use of lines and catheters, and antibiotic exposure. This commonality of risk factors implies that systematic infection-prevention measures will have an impact on the prevalence and incidence rates of multidrug-resistant organism infections across the board, CRE included. It should be emphasized that strict compliance with hand hygiene is still the foundation of any infection-prevention strategy.
Infection prevention and the control of transmission of CRE in long-term care facilities pose unique challenges. Guidelines from the Society for Healthcare Epidemiology and the Association for Professionals in Infection Control recommend the use of contact precautions for patients with multidrug-resistant organisms, including CRE, who are ill and totally dependent on health care workers for activities of daily living or whose secretions or drainage cannot be contained. These same guidelines advise against attempting to eradicate multidrug-resistant organism colonization status.46
In acute care facilities, Best Infection Control Practices from the CDC and the Healthcare Infection Control Practices Advisory Committee encourage mechanisms for the rapid recognition and reporting of CRE cases to infection prevention personnel so that contact precautions can be implemented. Furthermore, facilities without CRE cases should carry out periodic laboratory reviews to identify cases, and patients exposed to CRE cases should be screened with surveillance cultures.47
Outbreaks of CRE may require extraordinary infection control measures. An approach combining point-prevalence surveillance of colonization, detection of environmental and common-equipment contamination, with the implementation of a bundle consisting of chlorhexidine baths, cohorting of colonized patients and health care personnel, increased environmental cleaning, and staff education may be effective in controlling outbreaks of CRE.48
Nevertheless, control of CRE may prove exceptionally difficult. A recent high-profile outbreak of carbapenem-resistant K pneumoniae at the National Institutes of Health Clinical Center in Maryland caused infections in 18 patients, 11 of whom died.49 Of note, carbapenem-resistant K pneumoniae was detected in this outbreak in both respiratory equipment and sink drains. The outbreak was ultimately contained by detection through surveillance cultures and by strict cohorting of colonized patients, which minimized common medical equipment and personnel between affected patients and other patients in the hospital. Additionally, rooms were sanitized with hydrogen peroxide vapor, and sinks and drains where carbapenem-resistant K pneumoniae was detected were removed.
CHALLENGES IN THE MICROBIOLOGY LABORATORY
Adequate treatment and control of CRE infections is predicated upon their accurate and prompt diagnosis from patient samples in the clinical microbiology laboratory.50
Traditional and current culture-based methods take several days to provide that information, delaying effective antibiotic therapy and permitting the transmission of undetected CRE. Furthermore, interpretative criteria of minimal inhibitory concentrations (MICs) of carbapenems recently required readjustment, as many KPC-producing strains of K pneumoniae had MICs below the previous breakpoint of resistance. In the past, this contributed to instances of “silent” dissemination of KPC-producing K pneumoniae.51
In contrast, using the new lower breakpoints of resistance for carbapenems without using a phenotypic test such as the modified Hodge test or the carbapenem-EDTA combination tests will result in a lack of differentiation between various mechanisms of carbapenem resistance.28,52,53 This may be clinically relevant, as the clinical response to carbapenem therapy may vary depending on the mechanism of resistance.
GENERAL PRINCIPLES APPLY
In treating patients infected with CRE, clinicians need to strictly observe general principles of infectious disease management to ensure the best possible outcomes. These include:
Timely and accurate diagnosis, as discussed above.
Source control, which should include drainage of any infected collections, and removal of lines, devices, and urinary catheters.
Distinguishing between infection and colonization. CRE are often encountered as urinary isolates, and the distinction between asymptomatic bacteriuria and urinary tract infection may be extremely difficult, especially in residents of long-term care facilities with chronic indwelling catheters, who are thegroup at highest risk of CRE colonization and infection. Urinalysis may be helpful in the absence of pyuria, as this rules out an infection; however, it must be emphasized that the presence of pyuria is not a helpful feature, as pyuria is common in both asymptomatic bacteriuria and urinary tract infection.54 Symptoms should be carefully evaluated in every patient with bacteriuria, and urinary tract infection should be a diagnosis of exclusion in patients with functional symptoms such as confusion or falls.
Selection of the most appropriate antibiotic regimen. While the emphasis is often on the antibiotic regimen, the above elements should not be neglected.
A DWINDLING THERAPEUTIC ARSENAL
Clinicians treating CRE infections are left with only a few antibiotic options. These options are generally limited by a lack of clinical data on efficacy, as well as by concerns about toxicity. These “drugs of last resort” include polymyxins (such as colistin), aminoglycosides, tigecycline, and fosfomycin. The role of carbapenem therapy, potentially in combination regimens, in a high-dose prolonged infusion, or even “double carbapenem therapy” remains to be determined.37,55,56
Colistin
Colistin is one of the first-line agents for treating CRE infections. First introduced in the 1950s, its use was mostly abandoned in favor of aminoglycosides. A proportion of the data on safety and efficacy of colistin, therefore, is based on older, less rigorous studies.
Neurotoxicity and nephrotoxicity are the two main concerns with colistin, and while the incidence of these adverse events does appear to be lower with modern preparations, it is still substantial.57 Dosing issues have not been completely clarified either, especially in relation to renal clearance and in patients on renal replacement therapy.58,59 Unfortunately, there have been reports of outbreaks of CRE displaying resistance to colistin.60
Tigecycline
Tigecycline is a newer antibiotic of the glycylcycline class. Like colistin, it has no oral preparation for systemic infections.
The main side effect of tigecycline is nausea.61 Other reported issues include pancreatitis and extreme alkaline phosphatase elevations.
The efficacy of tigecycline has come into question in view of meta-analyses of clinical trials, some of which have shown higher mortality rates in patients treated with tigecycline than with comparator agents.62–65 Based on these data, the US Food and Drug Administration issued a warning in 2010 regarding the increased mortality risk. Although these meta-analyses did not include patients with CRE for whom available comparators would have been ineffective, it is an important safety signal.
The efficacy of tigecycline is further limited by increasing in vitro resistance in CRE. Serum and urinary levels of tigecycline are low, and most experts discourage the use of tigecycline as monotherapy for blood stream or urinary tract infections.
Aminoglycosides
CRE display variable in vitro susceptibility to different aminoglycosides. If the organism is susceptible, aminoglycosides may be very useful in the treatment of CRE infections, especially urinary tract infectons. In a study of carbapenem-resistant K pneumoniae urinary tract infections, patients who were treated with polymyxins or tigecycline were significantly less likely to have clearance of their urine as compared with patients treated with aminoglycosides.66
Ototoxicity and nephrotoxicity are demonstrated adverse effects of aminoglycosides. Close monitoring of serum levels, interval audiology examinations at baseline and during therapy, and the use of extended-interval dosing may help to decrease the incidence of these toxicities.
Fosfomycin
Fosfomycin is only available as an oral formulation in the United States, although intravenous administration has been used in other countries. It is exclusively used to treat urinary tract infections.
CRE often retain susceptibility to fosfomycin, and clearance of urine in cystitis may be attempted with this agent to avoid the need for intravenous treatment.29,67
Combination therapy, other topics to be explored
Recent observational reports from Greece, Italy, and the United States describe higher survival rates in patients with CRE infections treated with a combination regimen rather than monotherapy with colistin or tigecycline. This is despite reliable activity of colistin and tigecycline, and often in regimens containing carbapenems. Clinical experiments are needed to clarify the value of combination regimens that include carbapenems for the treatment of CRE infections.
Similarly, the role of carbapenems given as a high-dose prolonged infusion or as double carbapenem therapy needs to be explored further.37,55,56,68
Also to be determined is the optimal duration of treatment. To date, there is no evidence that increasing the duration of treatment beyond that recommended for infections with more susceptible bacteria results in improved outcomes. Therefore, commonly used durations include 1 week for complicated urinary tract infections, 2 weeks for bacteremia (from the first day with negative blood cultures and source control), and 8 to 14 days for pneumonia.
A SERIOUS THREAT
The emergence of CRE is a serious threat to the safety of patients in our health care system. CRE are highly successful nosocomial pathogens selected by the use of antibiotics, which burden patients debilitated by advanced age, comorbidities, and medical interventions. Infections with CRE result in poor outcomes, and available treatments of last resort such as tigecycline and colistin are of unclear efficacy and safety.
Control of CRE transmission is hindered by the transit of patients through long-term care facilities, and detection of CRE is difficult because of the myriad mechanisms involved and the imperfect methods currently available. Clinicians are concerned and frustrated, especially given the paucity of antibiotics in development to address the therapeutic dilemma posed by CRE. The challenge of CRE and other multidrug-resistant organisms requires the concerted response of professionals in various disciplines, including pharmacists, microbiologists, infection control practitioners, and infectious disease clinicians (Table 2).
Control of transmission by infection prevention strategies and by antimicrobial stewardship is going to be crucial in the years to come, not only for limiting the spread of CRE, but also for preventing the next multidrug-resistant “superbug” from emerging. However, the current reality is that health care providers will be faced with increased numbers of patients infected with CRE.
Prospective studies into transmission, molecular characteristics, and, most of all, treatment regimens are urgently needed. In addition, the development of new antimicrobials and nontraditional antimicrobial methods should have international priority.
The past 10 years have brought a formidable challenge to the clinical arena, as carbapenems, until now the most reliable antibiotics against Klebsiella species, Escherichia coli, and other Enterobacteriaceae, are becoming increasingly ineffective.
Infections caused by carbapenem-resistant Enterobacteriaceae (CRE) pose a serious threat to hospitalized patients. Moreover, CRE often demonstrate resistance to many other classes of antibiotics, thus limiting our therapeutic options. Furthermore, few new antibiotics are in line to replace carbapenems. This public health crisis demands redefined and refocused efforts in the diagnosis, treatment, and control of infections in hospitalized patients.
Here, we present an overview of CRE and discuss avenues to escape a new era of untreatable infections.
INCREASED USE OF CARBAPENEMS AND EMERGENCE OF RESISTANCE
Developed in the 1980s, carbapenems are derivatives of thyanamycin. Imipenem and meropenem, the first members of the class, had a broad spectrum of antimicrobial activity that included coverage of Pseudomonas aeruginosa, adequately positioning them for the treatment of nosocomial infections. Back then, nearly all Enterobacteriaceae were susceptible to carbapenems.1
In the 1990s, Enterobacteriaceae started to develop resistance to cephalosporins—till then, the first-line antibiotics for these organisms—by acquiring extended-spectrum betalactamases, which inactivate those agents. Consequently, the use of cephalosporins had to be restricted, while carbapenems, which remained impervious to these enzymes, had to be used more.2 In pivotal international studies in the treatment of infections caused by strains of K pneumoniae that produced these inactivating enzymes, outcomes were better with carbapenems than with cephalosporins and fluoroquinolones.3,4
Ertapenem, a carbapenem without antipseudomonal activity and highly bound to protein, was released in 2001. Its prolonged half-life permitted once-daily dosing, which positioned it as an option for treating infections in community dwellers.5 Doripenem is the newest member of the class of carbapenems, and its spectrum of activity is similar to that of imipenem and meropenem and includes P aeruginosa.6 The use of carbapenems, measured in a representative sample of 35 university hospitals in the United States, increased by 59% between 2002 and 2006.7
In the early 2000s, carbapenem resistance in K pneumoniae and other Enterobacteriaceae was rare in North America. But then, after initial outbreaks occurred in hospitals in the Northeast (especially New York City), CRE began to spread throughout the United States. By 2009–2010, the National Healthcare Safety Network from the Centers for Disease Control and Prevention (CDC) revealed that 12.8% of K pneumoniae isolates associated with bloodstream infections were resistant to carbapenems.8
In March 2013, the CDC disclosed that 3.9% of short-stay acute-care hospitals and 17.8% of long-term acute-care hospitals reported at least one CRE health care-associated infection in 2012. CRE had extended to 42 states, and the proportion of Enterobacteriaceae that are CRE had increased fourfold over the past 10 years.9
Coinciding with the increased use of carbapenems, multiple factors and modifiers likely contributed to the dramatic increase in CRE. These include use of other antibiotics in humans and animals, their relative penetration and selective effect on the gut microbiota, case-mix and infection control practices in different health care settings, and travel patterns.
POWERFUL ENZYMES THAT TRAVEL FAR
Bacterial acquisition of carbapenemases, enzymes that inactivate carbapenems, is crucial to the emergence of CRE. The enzyme in the sentinel carbapenem-resistant K pneumoniae isolate found in 1996 in North Carolina was designated K pneumoniae carbapenemase (KPC-1). This mechanism also conferred resistance to all cephalosporins, aztreonam, and beta-lactamase inhibitors such as clavulanic acid and tazobactam.10
KPC-2 (later determined to be identical to KPC-1) was found in K pneumoniae from Baltimore, and KPC-3 caused an early outbreak in New York City.11,12 To date, 12 additional variants of blaKPC, the gene encoding for the KPC enzyme, have been described.13
The genes encoding carbapenemases are usually found on plasmids or other common mobile genetic elements.14 These genetic elements allow the organism to acquire genes conferring resistance to other classes of antimicrobials, such as aminoglycoside-modifying enzymes and fluoroquinolone-resistance determinants, and beta-lactamases.15,16 The result is that CRE isolates are increasingly multidrug-resistant (ie, resistant to three or more classes of antimicrobials), extensively drug-resistant (ie, resistant to all but one or two classes), or pandrug-resistant (ie, resistant to all available classes of antibiotics).17 Thus, up to 98% of KPC-producing K pneumoniae are resistant to trimethoprim-sulfamethoxazole, 90% are resistant to fluoroquinolones, and 60% are resistant to gentamicin or amikacin.15
The mobility of these genetic elements has also allowed for dispersion into diverse Enterobacteriaceae such as E coli, Klebsiella oxytoca, Enterobacter, Serratia, and Salmonella species. Furthermore, KPC has been described in non-Enterobacteriaceae such as Acinetobacter baumannii and P aeruginosa.
Extending globally, KPC is now endemic in the Mediterranean basin, including Israel, Greece, and Italy; in South America, especially Colombia, Argentina, and Brazil; and in China.18 Most interesting is the intercontinental transfer of these strains: it has been documented that the index patient with KPC-producing K pneumoniae in Medellin, Colombia, came from Israel to undergo liver transplantation.19 Likewise, KPC-producing K pneumoniae in France and Israel could be linked epidemiologically and genetically to the predominant US strain.20,21
Even more explosive has been the surge of another carbapenemase, the Ambler Class B New Delhi metallo-beta-lactamase, or NDM-1. Initially reported in a urinary isolate of K pneumoniae from a Swedish patient who had been hospitalized in New Delhi in 2008, NDM-1 was soon found throughout India, in Pakistan, and in the United Kingdom.22 Interestingly, several of the UK patients with NDM-1-harboring bacteria had received organ transplants in the Indian subcontinent. Reports from elsewhere in Europe, Australia, and Africa followed suit, usually with a connection to the Indian subcontinent epicenter. In contrast, several other cases in Europe were traced to the Balkans, where there appears to be another focus of NDM-1.23
Penetration of NDM-1 into North America has begun, with cases and outbreaks reported in several US and Canadian regions, and in a military medical facility in Afghanistan. In several of these instances, there has been a documented link with travel and hospitalizations overseas.24–27 However, no such link with travel could be established in a recent outbreak in Ontario.27
In addition, resistance to carbapenems may result from other enzymes (Table 1), or from combinations of changes in outer membrane porins and the production of extended spectrum beta-lactamases or other cephalosporinases.28
DEADLY IMPACT ON THE MOST VULNERABLE
Regardless of the resistance pattern, Enterobacteriaceae are an important cause of health care-associated infections, including urinary and bloodstream infections in patients with indwelling catheters, pneumonia (often in association with mechanical ventilation), and, less frequently, infections of skin and soft tissues and the central nervous system.29–31
Several studies have examined the clinical characteristics and outcomes of patients with CRE infections. Those typically affected are elderly and debilitated and have multiple comorbidities, including diabetes mellitus and immunosuppression. They are heavily exposed to health care with frequent antecedent hospitalizations and invasive procedures. Furthermore, they are often severely ill and require intensive care. Patients infected with carbapenem-resistant K pneumoniae, compared with those with carbapenem-susceptible strains, are more likely to have undergone organ or stem cell transplantation or mechanical ventilation, and to have had a longer hospital stay before infection.
They also experience a high mortality rate, which ranges from 30% in patients with nonbacteremic infections to 72% in series of patients with liver transplants or bloodstream infections.32–37
More recently, CRE has been reported in other vulnerable populations, such as children with critical illness or cancer and in burn patients.38–40
Elderly and critically ill patients with bacteremia originating from a high-risk source (eg, pneumonia) typically face the most adverse outcomes. With increasing drug resistance, inadequate initial antimicrobial therapy is more commonly seen and may account for some of these poor outcomes.37,41
LONG-TERM CARE FACILITIES IN THE EYE OF THE STORM
A growing body of evidence suggests that long-term care facilities play a crucial role in the spread of CRE.
In an investigation into carbapenem-resistant A baumanii and K pneumoniae in a hospital system,36 75% of patients with carbapenem-resistant K pneumoniae were admitted from long-term care facilities, and only 1 of 13 patients was discharged home.
In a series of patients with carbapenem-resistant K pneumoniae bloodstream infections, 42% survived their index hospital stay. Of these patients, only 32% were discharged home, and readmissions were very common.32
Admission from a long-term care facility or transfer from another hospital is significantly associated with carbapenem resistance in patients with Enterobacteriaceae.42 Similarly, in Israel, a large reservoir of CRE was found in postacute care facilities.43
It is clear that long-term care residents are at increased risk of colonization and infection with CRE. However, further studies are needed to evaluate whether this simply refects an overlap in risk factors, or whether significant patient-to-patient transmission occurs in these settings.
INFECTION CONTROL TAKES CENTER STAGE
It is important to note that risk factors for CRE match those of various nosocomial infections, including other resistant gram-negative bacilli, methicillin-resistant Staphylococcus aureus, vancomycin-resistant enterococci, Candida species, and Clostridium difficile; in fact, CRE often coexist with other multidrug-resistant organisms.44,45
Common risk factors include residence in a long-term care facility, an intensive care unit stay, use of lines and catheters, and antibiotic exposure. This commonality of risk factors implies that systematic infection-prevention measures will have an impact on the prevalence and incidence rates of multidrug-resistant organism infections across the board, CRE included. It should be emphasized that strict compliance with hand hygiene is still the foundation of any infection-prevention strategy.
Infection prevention and the control of transmission of CRE in long-term care facilities pose unique challenges. Guidelines from the Society for Healthcare Epidemiology and the Association for Professionals in Infection Control recommend the use of contact precautions for patients with multidrug-resistant organisms, including CRE, who are ill and totally dependent on health care workers for activities of daily living or whose secretions or drainage cannot be contained. These same guidelines advise against attempting to eradicate multidrug-resistant organism colonization status.46
In acute care facilities, Best Infection Control Practices from the CDC and the Healthcare Infection Control Practices Advisory Committee encourage mechanisms for the rapid recognition and reporting of CRE cases to infection prevention personnel so that contact precautions can be implemented. Furthermore, facilities without CRE cases should carry out periodic laboratory reviews to identify cases, and patients exposed to CRE cases should be screened with surveillance cultures.47
Outbreaks of CRE may require extraordinary infection control measures. An approach combining point-prevalence surveillance of colonization, detection of environmental and common-equipment contamination, with the implementation of a bundle consisting of chlorhexidine baths, cohorting of colonized patients and health care personnel, increased environmental cleaning, and staff education may be effective in controlling outbreaks of CRE.48
Nevertheless, control of CRE may prove exceptionally difficult. A recent high-profile outbreak of carbapenem-resistant K pneumoniae at the National Institutes of Health Clinical Center in Maryland caused infections in 18 patients, 11 of whom died.49 Of note, carbapenem-resistant K pneumoniae was detected in this outbreak in both respiratory equipment and sink drains. The outbreak was ultimately contained by detection through surveillance cultures and by strict cohorting of colonized patients, which minimized common medical equipment and personnel between affected patients and other patients in the hospital. Additionally, rooms were sanitized with hydrogen peroxide vapor, and sinks and drains where carbapenem-resistant K pneumoniae was detected were removed.
CHALLENGES IN THE MICROBIOLOGY LABORATORY
Adequate treatment and control of CRE infections is predicated upon their accurate and prompt diagnosis from patient samples in the clinical microbiology laboratory.50
Traditional and current culture-based methods take several days to provide that information, delaying effective antibiotic therapy and permitting the transmission of undetected CRE. Furthermore, interpretative criteria of minimal inhibitory concentrations (MICs) of carbapenems recently required readjustment, as many KPC-producing strains of K pneumoniae had MICs below the previous breakpoint of resistance. In the past, this contributed to instances of “silent” dissemination of KPC-producing K pneumoniae.51
In contrast, using the new lower breakpoints of resistance for carbapenems without using a phenotypic test such as the modified Hodge test or the carbapenem-EDTA combination tests will result in a lack of differentiation between various mechanisms of carbapenem resistance.28,52,53 This may be clinically relevant, as the clinical response to carbapenem therapy may vary depending on the mechanism of resistance.
GENERAL PRINCIPLES APPLY
In treating patients infected with CRE, clinicians need to strictly observe general principles of infectious disease management to ensure the best possible outcomes. These include:
Timely and accurate diagnosis, as discussed above.
Source control, which should include drainage of any infected collections, and removal of lines, devices, and urinary catheters.
Distinguishing between infection and colonization. CRE are often encountered as urinary isolates, and the distinction between asymptomatic bacteriuria and urinary tract infection may be extremely difficult, especially in residents of long-term care facilities with chronic indwelling catheters, who are thegroup at highest risk of CRE colonization and infection. Urinalysis may be helpful in the absence of pyuria, as this rules out an infection; however, it must be emphasized that the presence of pyuria is not a helpful feature, as pyuria is common in both asymptomatic bacteriuria and urinary tract infection.54 Symptoms should be carefully evaluated in every patient with bacteriuria, and urinary tract infection should be a diagnosis of exclusion in patients with functional symptoms such as confusion or falls.
Selection of the most appropriate antibiotic regimen. While the emphasis is often on the antibiotic regimen, the above elements should not be neglected.
A DWINDLING THERAPEUTIC ARSENAL
Clinicians treating CRE infections are left with only a few antibiotic options. These options are generally limited by a lack of clinical data on efficacy, as well as by concerns about toxicity. These “drugs of last resort” include polymyxins (such as colistin), aminoglycosides, tigecycline, and fosfomycin. The role of carbapenem therapy, potentially in combination regimens, in a high-dose prolonged infusion, or even “double carbapenem therapy” remains to be determined.37,55,56
Colistin
Colistin is one of the first-line agents for treating CRE infections. First introduced in the 1950s, its use was mostly abandoned in favor of aminoglycosides. A proportion of the data on safety and efficacy of colistin, therefore, is based on older, less rigorous studies.
Neurotoxicity and nephrotoxicity are the two main concerns with colistin, and while the incidence of these adverse events does appear to be lower with modern preparations, it is still substantial.57 Dosing issues have not been completely clarified either, especially in relation to renal clearance and in patients on renal replacement therapy.58,59 Unfortunately, there have been reports of outbreaks of CRE displaying resistance to colistin.60
Tigecycline
Tigecycline is a newer antibiotic of the glycylcycline class. Like colistin, it has no oral preparation for systemic infections.
The main side effect of tigecycline is nausea.61 Other reported issues include pancreatitis and extreme alkaline phosphatase elevations.
The efficacy of tigecycline has come into question in view of meta-analyses of clinical trials, some of which have shown higher mortality rates in patients treated with tigecycline than with comparator agents.62–65 Based on these data, the US Food and Drug Administration issued a warning in 2010 regarding the increased mortality risk. Although these meta-analyses did not include patients with CRE for whom available comparators would have been ineffective, it is an important safety signal.
The efficacy of tigecycline is further limited by increasing in vitro resistance in CRE. Serum and urinary levels of tigecycline are low, and most experts discourage the use of tigecycline as monotherapy for blood stream or urinary tract infections.
Aminoglycosides
CRE display variable in vitro susceptibility to different aminoglycosides. If the organism is susceptible, aminoglycosides may be very useful in the treatment of CRE infections, especially urinary tract infectons. In a study of carbapenem-resistant K pneumoniae urinary tract infections, patients who were treated with polymyxins or tigecycline were significantly less likely to have clearance of their urine as compared with patients treated with aminoglycosides.66
Ototoxicity and nephrotoxicity are demonstrated adverse effects of aminoglycosides. Close monitoring of serum levels, interval audiology examinations at baseline and during therapy, and the use of extended-interval dosing may help to decrease the incidence of these toxicities.
Fosfomycin
Fosfomycin is only available as an oral formulation in the United States, although intravenous administration has been used in other countries. It is exclusively used to treat urinary tract infections.
CRE often retain susceptibility to fosfomycin, and clearance of urine in cystitis may be attempted with this agent to avoid the need for intravenous treatment.29,67
Combination therapy, other topics to be explored
Recent observational reports from Greece, Italy, and the United States describe higher survival rates in patients with CRE infections treated with a combination regimen rather than monotherapy with colistin or tigecycline. This is despite reliable activity of colistin and tigecycline, and often in regimens containing carbapenems. Clinical experiments are needed to clarify the value of combination regimens that include carbapenems for the treatment of CRE infections.
Similarly, the role of carbapenems given as a high-dose prolonged infusion or as double carbapenem therapy needs to be explored further.37,55,56,68
Also to be determined is the optimal duration of treatment. To date, there is no evidence that increasing the duration of treatment beyond that recommended for infections with more susceptible bacteria results in improved outcomes. Therefore, commonly used durations include 1 week for complicated urinary tract infections, 2 weeks for bacteremia (from the first day with negative blood cultures and source control), and 8 to 14 days for pneumonia.
A SERIOUS THREAT
The emergence of CRE is a serious threat to the safety of patients in our health care system. CRE are highly successful nosocomial pathogens selected by the use of antibiotics, which burden patients debilitated by advanced age, comorbidities, and medical interventions. Infections with CRE result in poor outcomes, and available treatments of last resort such as tigecycline and colistin are of unclear efficacy and safety.
Control of CRE transmission is hindered by the transit of patients through long-term care facilities, and detection of CRE is difficult because of the myriad mechanisms involved and the imperfect methods currently available. Clinicians are concerned and frustrated, especially given the paucity of antibiotics in development to address the therapeutic dilemma posed by CRE. The challenge of CRE and other multidrug-resistant organisms requires the concerted response of professionals in various disciplines, including pharmacists, microbiologists, infection control practitioners, and infectious disease clinicians (Table 2).
Control of transmission by infection prevention strategies and by antimicrobial stewardship is going to be crucial in the years to come, not only for limiting the spread of CRE, but also for preventing the next multidrug-resistant “superbug” from emerging. However, the current reality is that health care providers will be faced with increased numbers of patients infected with CRE.
Prospective studies into transmission, molecular characteristics, and, most of all, treatment regimens are urgently needed. In addition, the development of new antimicrobials and nontraditional antimicrobial methods should have international priority.
- Papp-Wallace KM, Endimiani A, Taracila MA, Bonomo RA. Carbapenems: past, present, and future. Antimicrob Agents Chemother 2011; 55:4943–4960.
- Rahal JJ, Urban C, Horn D, et al. Class restriction of cephalosporin use to control total cephalosporin resistance in nosocomial Klebsiella. JAMA 1998; 280:1233–1237.
- Paterson DL, Ko WC, Von Gottberg A, et al. International prospective study of Klebsiella pneumoniae bacteremia: implications of extended-spectrum beta-lactamase production in nosocomial Infections. Ann Intern Med 2004; 140:26–32.
- Endimiani A, Luzzaro F, Perilli M, et al. Bacteremia due to Klebsiella pneumoniae isolates producing the TEM-52 extended-spectrum beta-lactamase: treatment outcome of patients receiving imipenem or ciprofoxacin. Clin Infect Dis 2004; 38:243–251.
- Livermore DM, Sefton AM, Scott GM. Properties and potential of ertapenem. J Antimicrob Chemother 2003; 52:331–344.
- Bazan JA, Martin SI, Kaye KM. Newer beta-lactam antibiotics: doripenem, ceftobiprole, ceftaroline, and cefepime. Infect Dis Clin North Am 2009; 23:983–996, ix.
- Pakyz AL, MacDougall C, Oinonen M, Polk RE. Trends in antibacterial use in US academic health centers: 2002 to 2006. Arch Intern Med 2008; 168:2254–2260.
- Sievert DM, Ricks P, Edwards JR, et al. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect Control Hosp Epidemiol 2013; 34:1–14.
- Centers for Disease Control and Prevention. Vital signs: carbapenem-resistant Enterobacteriaceae. MMWR 2013; 62:165–170.
- Yigit H, Queenan AM, Anderson GJ, et al. Novel carbapenem-hydrolyzing beta-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob Agents Chemother 2001; 45:1151–1161.
- Smith Moland E, Hanson ND, Herrera VL, et al. Plasmid-mediated, carbapenem-hydrolysing beta-lactamase, KPC-2, in Klebsiella pneumoniae isolates. J Antimicrob Chemother 2003; 51:711–714.
- Woodford N, Tierno PM, Young K, et al. Outbreak of Klebsiella pneumoniae producing a new carbapenem-hydrolyzing class A beta-lactamase, KPC-3, in a New York medical center. Antimicrob Agents Chemother 2004; 48:4793–4799.
- Lehey Clinic. OXA-type β-Lactamases. http://www.lahey.org/Studies/other.asp#table1. Accessed March 11, 2013.
- Mathers AJ, Cox HL, Kitchel B, et al. Molecular dissection of an outbreak of carbapenem-resistant Enterobacteriaceae reveals intergenus KPC carbapenemase transmission through a promiscuous plasmid. MBio 2011; 2 6:e00204–11.
- Endimiani A, Hujer AM, Perez F, et al. Characterization of blaKPC-containing Klebsiella pneumoniae isolates detected in different institutions in the Eastern USA. J Antimicrob Chemother 2009; 63:427–437.
- Endimiani A, Carias LL, Hujer AM, et al. Presence of plasmid-mediated quinolone resistance in Klebsiella pneumoniae isolates possessing blaKPC in the United States. Antimicro Agents Chemother 2008; 52:2680–2682.
- Magiorakos A P, Srinivasan A, Carey RB, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect 2012; 18:268–281.
- Tzouvelekis LS, Markogiannakis A, Psichogiou M, Tassios PT, Daikos GL. Carbapenemases in Klebsiella pneumoniae and other Enterobacteriaceae: an evolving crisis of global dimensions. Clin Microbiol Rev 2012; 25:682–707.
- Lopez JA, Correa A, Navon-Venezia S, et al. Intercontinental spread from Israel to Colombia of a KPC-3-producing Klebsiella pneumoniae strain. Clin Microbiol Infect 2011; 17:52–56.
- Naas T, Nordmann P, Vedel G, Poyart C. Plasmid-mediated carbapenem-hydrolyzing beta-lactamase KPC in a Klebsiella pneumoniae isolate from France. Antimicrob Agents Chemother 2005; 49:4423–4424.
- Navon-Venezia S, Leavitt A, Schwaber MJ, et al. First report on a hyperepidemic clone of KPC-3-producing Klebsiella pneumoniae in Israel genetically related to a strain causing outbreaks in the United States. Antimicrob Agents Chemother 2009; 53:818–820.
- Yong D, Toleman MA, Giske CG, et al. Characterization of a new metallo-beta-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob Agents Chemother 2009; 53:5046–5054.
- Livermore DM, Walsh TR, Toleman M, Woodford N. Balkan NDM-1: escape or transplant? Lancet Infect Dis 2011; 11:164.
- Centers for Disease Control and Prevention. Carbapenem-resistant enterobacteriaceae containing New Delhi metallo-beta-lactamase in two patients - Rhode Island, March 2012. MMWR Morb Mortal Wkly Rep 2012Jun 22; 61:446–448.
- Centers for Disease Control and Prevention. Detection of Enterobacteriaceae isolates carrying metallo-beta-lactamase—United States, 2010. MMWR Morb Mortal Wkly Rep 2010; 59:750.
- McGann P, Hang J, Clifford RJ, et al. Complete sequence of a novel 178-kilobase plasmid carrying bla(NDM-1) in a Providencia stuartii strain isolated in Afghanistan. Antimicrob Agents Chemother 2012; 56:1673–1679.
- Borgia S, Lastovetska O, Richardson D, et al. Outbreak of carbapenem-resistant Enterobacteriaceae containing blaNDM-1, Ontario, Canada. Clin Infect Dis 2012; 55:e109–e117.
- Endimiani A, Perez F, Bajaksouzian S, et al. Evaluation of updated interpretative criteria for categorizing Klebsiella pneumoniae with reduced carbapenem susceptibility. J Clinic Microbiol 2010; 48:4417–4425.
- Neuner EA, Sekeres J, Hall GS, van Duin D. Experience with fosfomycin for treatment of urinary tract infections due to multidrug-resistant organisms. Antimicrob Agents Chemother 2012; 56:5744–5748.
- Neuner EA, Yeh JY, Hall GS, et al. Treatment and outcomes in carbapenem-resistant Klebsiella pneumoniae bloodstream infections. Diagnostic Microbiol Infect Dis 2011; 69:357–362.
- van Duin D, Kaye KS, Neuner EA, Bonomo RA. Carbapenem-resistant Enterobacteriaceae: a review of treatment and outcomes. Diagnostic Microbiol Infect Dis 2013; 75:115–120.
- Neuner EA, Yeh J-Y, Hall GS, et al. Treatment and outcomes in carbapenem-resistant Klebsiella pneumoniae bloodstream infections. Diagn Microbiol Infect Dis 2011; 69:357–362.
- Patel G, Huprikar S, Factor SH, Jenkins SG, Calfee DP. Outcomes of carbapenem-resistant Klebsiella pneumoniae infection and the impact of antimicrobial and adjunctive therapies. Infect Control Hosp Epidemiol 2008; 29:1099–1106.
- Borer A, Saidel-Odes L, Riesenberg K, et al. Attributable mortality rate for carbapenem-resistant Klebsiella pneumoniae bacteremia. Infect Control Hosp Epidemiol 2009; 30:972–976.
- Marchaim D, Chopra T, Perez F, et al. Outcomes and genetic relatedness of carbapenem-resistant Enterobacteriaceae at Detroit medical center. Infect Control Hosp Epidemiol 2011; 32:861–871.
- Perez F, Endimiani A, Ray AJ, et al. Carbapenem-resistant Acinetobacter baumannii and Klebsiella pneumoniae across a hospital system: impact of post-acute care facilities on dissemination. J Antimicrob Chemother 2010; 65:1807–1818.
- Tumbarello M, Viale P, Viscoli C, et al. Predictors of mortality in bloodstream infections caused by Klebsiella pneumoniae carbapenemase-producing K. pneumoniae: importance of combination therapy. Clin Infect Dis 2012; 55:943–950.
- Little ML, Qin X, Zerr DM, Weissman SJ. Molecular diversity in mechanisms of carbapenem resistance in paediatric Enterobacteriaceae. Int J Antimicrob Agents 2012; 39:52–57.
- Logan LK. Carbapenem-resistant Enterobacteriaceae: an emerging problem in children. Clin Infect Dis 2012; 55:852–859.
- Rastegar Lari A, Azimi L, Rahbar M, Fallah F, Alaghehbandan R. Phenotypic detection of Klebsiella pneumoniae carbapenemase among burns patients: first report from Iran. Burns 2013; 39:174–176.
- Zarkotou O, Pournaras S, Tselioti P, et al. Predictors of mortality in patients with bloodstream infections caused by KPC-producing Klebsiella pneumoniae and impact of appropriate antimicrobial treatment. Clin Microbiol Infect 2011; 17:1798–1803.
- Hyle EP, Ferraro MJ, Silver M, Lee H, Hooper DC. Ertapenem-resistant Enterobacteriaceae: risk factors for acquisition and outcomes. Infect Control Hosp Epidemiol 2010; 31:1242–1249.
- Ben-David D, Masarwa S, Navon-Venezia S, et al. Carbapenem-resistant Klebsiella pneumoniae in post-acute-care facilities in Israel. Infect Control Hosp Epidemiol 2011; 32:845–853.
- Safdar N, Maki DG. The commonality of risk factors for nosocomial colonization and infection with antimicrobial-resistant Staphylococcus aureus, enterococcus, gram-negative bacilli, Clostridium difficile, and Candida. Ann Intern Med 2002; 136:834–844.
- Marchaim D, Perez F, Lee J, et al. “Swimming in resistance”: co-colonization with carbapenem-resistant Enterobacteriaceae and Acinetobacter baumannii or Pseudomonas aeruginosa.” Am J Infect Control 2012; 40:830–835.
- Smith PW, Bennett G, Bradley S, et al. SHEA/APIC Guideline: Infection prevention and control in the long-term care facility. Am J Infect Control 2008; 36:504–535.
- Centers for Disease Control and Prevention. Guidance for control of infections with carbapenem-resistant or carbapenemase-producing Enterobacteriaceae in acute care facilities. MMWR 2009; 58:256–260.
- Munoz-Price LS, De La Cuesta C, Adams S, et al. Successful eradication of a monoclonal strain of Klebsiella pneumoniae during a K. pneumoniae carbapenemase-producing K. pneumoniae outbreak in a surgical intensive care unit in Miami, Florida. Infect Control Hosp Epidemiol 2010; 31:1074–1077.
- Snitkin ES, Zelazny AM, Thomas PJ, et al. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with wholegenome sequencing. Sci Transl Med 2012; 4:148ra16.
- Srinivasan A, Patel JB. Klebsiella pneumoniae carbapenemase-producing organisms: an ounce of prevention really is worth a pound of cure. Infect Control Hosp Epidemiol 2008; 29:1107–1109.
- Viau RA, Hujer AM, Marshall SH, et al. “Silent” dissemination of Klebsiella pneumoniae isolates bearing K pneumoniae carbapenemase in a long-term care facility for children and young adults in Northeast Ohio”. Clin Infect Dis 2012; 54:1314–1321.
- Galani I, Rekatsina PD, Hatzaki D, Plachouras D, Souli M, Giamarellou H. Evaluation of different laboratory tests for the detection of metallo-beta-lactamase production in Enterobacteriaceae. J Antimicrob Chemother 2008; 61:548–553.
- Anderson KF, Lonsway DR, Rasheed JK, et al. Evaluation of methods to identify the Klebsiella pneumoniae carbapenemase in Enterobacteriaceae. J Clin Microbiol 2007; 45:2723–2725.
- Nicolle LE, Bradley S, Colgan R, Rice JC, Schaeffer A, Hooton TM. Infectious Diseases Society of America guidelines for the diagnosis and treatment of asymptomatic bacteriuria in adults. Clin Infect Dis 2005; 40:643–654.
- Daikos GL, Markogiannakis A. Carbapenemase-producing Klebsiella pneumoniae: (when) might we still consider treating with carbapenems? Clin Microbiol Infect 2011; 17:1135–1141.
- Bulik CC, Nicolau DP. Double-carbapenem therapy for carbapenemase-producing Klebsiella pneumoniae. Antimicrob Agents Chemother 2011; 55:3002–3004.
- Pogue JM, Lee J, Marchaim D, et al. Incidence of and risk factors for colistin-associated nephrotoxicity in a large academic health system. Clin Infect Dis 2011; 53:879–884.
- Garonzik SM, Li J, Thamlikitkul V, et al. Population pharmacokinetics of colistin methanesulfonate and formed colistin in critically ill patients from a multicenter study provide dosing suggestions for various categories of patients. Antimicrob Agents Chemother 2011; 55:3284–3294.
- Dalfno L, Puntillo F, Mosca A, et al. High-dose, extended-interval colistin administration in critically ill patients: is this the right dosing strategy? A preliminary study. Clin Infect Dis 2012; 54:1720–1726.
- Marchaim D, Chopra T, Pogue JM, et al. Outbreak of colistin-resistant, carbapenem-resistant Klebsiella pneumoniae in metropolitan Detroit, Michigan. Antimicrob Agents Chemother 2011; 55:593–599.
- Bonilla MF, Avery RK, Rehm SJ, Neuner EA, Isada CM, van Duin D. Extreme alkaline phosphatase elevation associated with tigecycline. J Antimicrob Chemother 2011; 66:952–953.
- Prasad P, Sun J, Danner RL, Natanson C. Excess deaths associated with tigecycline after approval based on noninferiority trials. Clin Infect Dis 2012; 54:1699–1709.
- Tasina E, Haidich AB, Kokkali S, Arvanitidou M. Efficacy and safety of tigecycline for the treatment of infectious diseases: a meta-analysis. Lancet Infect Dis 2011; 11:834–844.
- Cai Y, Wang R, Liang B, Bai N, Liu Y. Systematic review and meta-analysis of the effectiveness and safety of tigecycline for treatment of infectious disease. Antimicrob Agents Chemother 2011; 55:1162–1172.
- Yahav D, Lador A, Paul M, Leibovici L. Efficacy and safety of tigecycline: a systematic review and meta-analysis. J Antimicrob Chemother 2011; 66:1963–1971.
- Satlin MJ, Kubin CJ, Blumenthal JS, et al. Comparative effectiveness of aminoglycosides, polymyxin B, and tigecycline for clearance of carbapenem-resistant Klebsiella pneumoniae from urine. Antimicrob Agents Chemother 2011; 55:5893–5899.
- Endimiani A, Patel G, Hujer KM, et al. In vitro activity of fosfomycin against blaKPC-containing Klebsiella pneumoniae isolates, including those nonsusceptible to tigecycline and/or colistin. Antimicrob Agents Chemother 2010; 54:526–529.
- Qureshi ZA, Paterson DL, Potoski BA, et al. Treatment outcome of bacteremia due to KPC-producing Klebsiella pneumoniae: superiority of combination antimicrobial regimens. Antimicrob Agents Chemother 2012; 56:2108–2113.
- Papp-Wallace KM, Endimiani A, Taracila MA, Bonomo RA. Carbapenems: past, present, and future. Antimicrob Agents Chemother 2011; 55:4943–4960.
- Rahal JJ, Urban C, Horn D, et al. Class restriction of cephalosporin use to control total cephalosporin resistance in nosocomial Klebsiella. JAMA 1998; 280:1233–1237.
- Paterson DL, Ko WC, Von Gottberg A, et al. International prospective study of Klebsiella pneumoniae bacteremia: implications of extended-spectrum beta-lactamase production in nosocomial Infections. Ann Intern Med 2004; 140:26–32.
- Endimiani A, Luzzaro F, Perilli M, et al. Bacteremia due to Klebsiella pneumoniae isolates producing the TEM-52 extended-spectrum beta-lactamase: treatment outcome of patients receiving imipenem or ciprofoxacin. Clin Infect Dis 2004; 38:243–251.
- Livermore DM, Sefton AM, Scott GM. Properties and potential of ertapenem. J Antimicrob Chemother 2003; 52:331–344.
- Bazan JA, Martin SI, Kaye KM. Newer beta-lactam antibiotics: doripenem, ceftobiprole, ceftaroline, and cefepime. Infect Dis Clin North Am 2009; 23:983–996, ix.
- Pakyz AL, MacDougall C, Oinonen M, Polk RE. Trends in antibacterial use in US academic health centers: 2002 to 2006. Arch Intern Med 2008; 168:2254–2260.
- Sievert DM, Ricks P, Edwards JR, et al. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect Control Hosp Epidemiol 2013; 34:1–14.
- Centers for Disease Control and Prevention. Vital signs: carbapenem-resistant Enterobacteriaceae. MMWR 2013; 62:165–170.
- Yigit H, Queenan AM, Anderson GJ, et al. Novel carbapenem-hydrolyzing beta-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob Agents Chemother 2001; 45:1151–1161.
- Smith Moland E, Hanson ND, Herrera VL, et al. Plasmid-mediated, carbapenem-hydrolysing beta-lactamase, KPC-2, in Klebsiella pneumoniae isolates. J Antimicrob Chemother 2003; 51:711–714.
- Woodford N, Tierno PM, Young K, et al. Outbreak of Klebsiella pneumoniae producing a new carbapenem-hydrolyzing class A beta-lactamase, KPC-3, in a New York medical center. Antimicrob Agents Chemother 2004; 48:4793–4799.
- Lehey Clinic. OXA-type β-Lactamases. http://www.lahey.org/Studies/other.asp#table1. Accessed March 11, 2013.
- Mathers AJ, Cox HL, Kitchel B, et al. Molecular dissection of an outbreak of carbapenem-resistant Enterobacteriaceae reveals intergenus KPC carbapenemase transmission through a promiscuous plasmid. MBio 2011; 2 6:e00204–11.
- Endimiani A, Hujer AM, Perez F, et al. Characterization of blaKPC-containing Klebsiella pneumoniae isolates detected in different institutions in the Eastern USA. J Antimicrob Chemother 2009; 63:427–437.
- Endimiani A, Carias LL, Hujer AM, et al. Presence of plasmid-mediated quinolone resistance in Klebsiella pneumoniae isolates possessing blaKPC in the United States. Antimicro Agents Chemother 2008; 52:2680–2682.
- Magiorakos A P, Srinivasan A, Carey RB, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect 2012; 18:268–281.
- Tzouvelekis LS, Markogiannakis A, Psichogiou M, Tassios PT, Daikos GL. Carbapenemases in Klebsiella pneumoniae and other Enterobacteriaceae: an evolving crisis of global dimensions. Clin Microbiol Rev 2012; 25:682–707.
- Lopez JA, Correa A, Navon-Venezia S, et al. Intercontinental spread from Israel to Colombia of a KPC-3-producing Klebsiella pneumoniae strain. Clin Microbiol Infect 2011; 17:52–56.
- Naas T, Nordmann P, Vedel G, Poyart C. Plasmid-mediated carbapenem-hydrolyzing beta-lactamase KPC in a Klebsiella pneumoniae isolate from France. Antimicrob Agents Chemother 2005; 49:4423–4424.
- Navon-Venezia S, Leavitt A, Schwaber MJ, et al. First report on a hyperepidemic clone of KPC-3-producing Klebsiella pneumoniae in Israel genetically related to a strain causing outbreaks in the United States. Antimicrob Agents Chemother 2009; 53:818–820.
- Yong D, Toleman MA, Giske CG, et al. Characterization of a new metallo-beta-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob Agents Chemother 2009; 53:5046–5054.
- Livermore DM, Walsh TR, Toleman M, Woodford N. Balkan NDM-1: escape or transplant? Lancet Infect Dis 2011; 11:164.
- Centers for Disease Control and Prevention. Carbapenem-resistant enterobacteriaceae containing New Delhi metallo-beta-lactamase in two patients - Rhode Island, March 2012. MMWR Morb Mortal Wkly Rep 2012Jun 22; 61:446–448.
- Centers for Disease Control and Prevention. Detection of Enterobacteriaceae isolates carrying metallo-beta-lactamase—United States, 2010. MMWR Morb Mortal Wkly Rep 2010; 59:750.
- McGann P, Hang J, Clifford RJ, et al. Complete sequence of a novel 178-kilobase plasmid carrying bla(NDM-1) in a Providencia stuartii strain isolated in Afghanistan. Antimicrob Agents Chemother 2012; 56:1673–1679.
- Borgia S, Lastovetska O, Richardson D, et al. Outbreak of carbapenem-resistant Enterobacteriaceae containing blaNDM-1, Ontario, Canada. Clin Infect Dis 2012; 55:e109–e117.
- Endimiani A, Perez F, Bajaksouzian S, et al. Evaluation of updated interpretative criteria for categorizing Klebsiella pneumoniae with reduced carbapenem susceptibility. J Clinic Microbiol 2010; 48:4417–4425.
- Neuner EA, Sekeres J, Hall GS, van Duin D. Experience with fosfomycin for treatment of urinary tract infections due to multidrug-resistant organisms. Antimicrob Agents Chemother 2012; 56:5744–5748.
- Neuner EA, Yeh JY, Hall GS, et al. Treatment and outcomes in carbapenem-resistant Klebsiella pneumoniae bloodstream infections. Diagnostic Microbiol Infect Dis 2011; 69:357–362.
- van Duin D, Kaye KS, Neuner EA, Bonomo RA. Carbapenem-resistant Enterobacteriaceae: a review of treatment and outcomes. Diagnostic Microbiol Infect Dis 2013; 75:115–120.
- Neuner EA, Yeh J-Y, Hall GS, et al. Treatment and outcomes in carbapenem-resistant Klebsiella pneumoniae bloodstream infections. Diagn Microbiol Infect Dis 2011; 69:357–362.
- Patel G, Huprikar S, Factor SH, Jenkins SG, Calfee DP. Outcomes of carbapenem-resistant Klebsiella pneumoniae infection and the impact of antimicrobial and adjunctive therapies. Infect Control Hosp Epidemiol 2008; 29:1099–1106.
- Borer A, Saidel-Odes L, Riesenberg K, et al. Attributable mortality rate for carbapenem-resistant Klebsiella pneumoniae bacteremia. Infect Control Hosp Epidemiol 2009; 30:972–976.
- Marchaim D, Chopra T, Perez F, et al. Outcomes and genetic relatedness of carbapenem-resistant Enterobacteriaceae at Detroit medical center. Infect Control Hosp Epidemiol 2011; 32:861–871.
- Perez F, Endimiani A, Ray AJ, et al. Carbapenem-resistant Acinetobacter baumannii and Klebsiella pneumoniae across a hospital system: impact of post-acute care facilities on dissemination. J Antimicrob Chemother 2010; 65:1807–1818.
- Tumbarello M, Viale P, Viscoli C, et al. Predictors of mortality in bloodstream infections caused by Klebsiella pneumoniae carbapenemase-producing K. pneumoniae: importance of combination therapy. Clin Infect Dis 2012; 55:943–950.
- Little ML, Qin X, Zerr DM, Weissman SJ. Molecular diversity in mechanisms of carbapenem resistance in paediatric Enterobacteriaceae. Int J Antimicrob Agents 2012; 39:52–57.
- Logan LK. Carbapenem-resistant Enterobacteriaceae: an emerging problem in children. Clin Infect Dis 2012; 55:852–859.
- Rastegar Lari A, Azimi L, Rahbar M, Fallah F, Alaghehbandan R. Phenotypic detection of Klebsiella pneumoniae carbapenemase among burns patients: first report from Iran. Burns 2013; 39:174–176.
- Zarkotou O, Pournaras S, Tselioti P, et al. Predictors of mortality in patients with bloodstream infections caused by KPC-producing Klebsiella pneumoniae and impact of appropriate antimicrobial treatment. Clin Microbiol Infect 2011; 17:1798–1803.
- Hyle EP, Ferraro MJ, Silver M, Lee H, Hooper DC. Ertapenem-resistant Enterobacteriaceae: risk factors for acquisition and outcomes. Infect Control Hosp Epidemiol 2010; 31:1242–1249.
- Ben-David D, Masarwa S, Navon-Venezia S, et al. Carbapenem-resistant Klebsiella pneumoniae in post-acute-care facilities in Israel. Infect Control Hosp Epidemiol 2011; 32:845–853.
- Safdar N, Maki DG. The commonality of risk factors for nosocomial colonization and infection with antimicrobial-resistant Staphylococcus aureus, enterococcus, gram-negative bacilli, Clostridium difficile, and Candida. Ann Intern Med 2002; 136:834–844.
- Marchaim D, Perez F, Lee J, et al. “Swimming in resistance”: co-colonization with carbapenem-resistant Enterobacteriaceae and Acinetobacter baumannii or Pseudomonas aeruginosa.” Am J Infect Control 2012; 40:830–835.
- Smith PW, Bennett G, Bradley S, et al. SHEA/APIC Guideline: Infection prevention and control in the long-term care facility. Am J Infect Control 2008; 36:504–535.
- Centers for Disease Control and Prevention. Guidance for control of infections with carbapenem-resistant or carbapenemase-producing Enterobacteriaceae in acute care facilities. MMWR 2009; 58:256–260.
- Munoz-Price LS, De La Cuesta C, Adams S, et al. Successful eradication of a monoclonal strain of Klebsiella pneumoniae during a K. pneumoniae carbapenemase-producing K. pneumoniae outbreak in a surgical intensive care unit in Miami, Florida. Infect Control Hosp Epidemiol 2010; 31:1074–1077.
- Snitkin ES, Zelazny AM, Thomas PJ, et al. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with wholegenome sequencing. Sci Transl Med 2012; 4:148ra16.
- Srinivasan A, Patel JB. Klebsiella pneumoniae carbapenemase-producing organisms: an ounce of prevention really is worth a pound of cure. Infect Control Hosp Epidemiol 2008; 29:1107–1109.
- Viau RA, Hujer AM, Marshall SH, et al. “Silent” dissemination of Klebsiella pneumoniae isolates bearing K pneumoniae carbapenemase in a long-term care facility for children and young adults in Northeast Ohio”. Clin Infect Dis 2012; 54:1314–1321.
- Galani I, Rekatsina PD, Hatzaki D, Plachouras D, Souli M, Giamarellou H. Evaluation of different laboratory tests for the detection of metallo-beta-lactamase production in Enterobacteriaceae. J Antimicrob Chemother 2008; 61:548–553.
- Anderson KF, Lonsway DR, Rasheed JK, et al. Evaluation of methods to identify the Klebsiella pneumoniae carbapenemase in Enterobacteriaceae. J Clin Microbiol 2007; 45:2723–2725.
- Nicolle LE, Bradley S, Colgan R, Rice JC, Schaeffer A, Hooton TM. Infectious Diseases Society of America guidelines for the diagnosis and treatment of asymptomatic bacteriuria in adults. Clin Infect Dis 2005; 40:643–654.
- Daikos GL, Markogiannakis A. Carbapenemase-producing Klebsiella pneumoniae: (when) might we still consider treating with carbapenems? Clin Microbiol Infect 2011; 17:1135–1141.
- Bulik CC, Nicolau DP. Double-carbapenem therapy for carbapenemase-producing Klebsiella pneumoniae. Antimicrob Agents Chemother 2011; 55:3002–3004.
- Pogue JM, Lee J, Marchaim D, et al. Incidence of and risk factors for colistin-associated nephrotoxicity in a large academic health system. Clin Infect Dis 2011; 53:879–884.
- Garonzik SM, Li J, Thamlikitkul V, et al. Population pharmacokinetics of colistin methanesulfonate and formed colistin in critically ill patients from a multicenter study provide dosing suggestions for various categories of patients. Antimicrob Agents Chemother 2011; 55:3284–3294.
- Dalfno L, Puntillo F, Mosca A, et al. High-dose, extended-interval colistin administration in critically ill patients: is this the right dosing strategy? A preliminary study. Clin Infect Dis 2012; 54:1720–1726.
- Marchaim D, Chopra T, Pogue JM, et al. Outbreak of colistin-resistant, carbapenem-resistant Klebsiella pneumoniae in metropolitan Detroit, Michigan. Antimicrob Agents Chemother 2011; 55:593–599.
- Bonilla MF, Avery RK, Rehm SJ, Neuner EA, Isada CM, van Duin D. Extreme alkaline phosphatase elevation associated with tigecycline. J Antimicrob Chemother 2011; 66:952–953.
- Prasad P, Sun J, Danner RL, Natanson C. Excess deaths associated with tigecycline after approval based on noninferiority trials. Clin Infect Dis 2012; 54:1699–1709.
- Tasina E, Haidich AB, Kokkali S, Arvanitidou M. Efficacy and safety of tigecycline for the treatment of infectious diseases: a meta-analysis. Lancet Infect Dis 2011; 11:834–844.
- Cai Y, Wang R, Liang B, Bai N, Liu Y. Systematic review and meta-analysis of the effectiveness and safety of tigecycline for treatment of infectious disease. Antimicrob Agents Chemother 2011; 55:1162–1172.
- Yahav D, Lador A, Paul M, Leibovici L. Efficacy and safety of tigecycline: a systematic review and meta-analysis. J Antimicrob Chemother 2011; 66:1963–1971.
- Satlin MJ, Kubin CJ, Blumenthal JS, et al. Comparative effectiveness of aminoglycosides, polymyxin B, and tigecycline for clearance of carbapenem-resistant Klebsiella pneumoniae from urine. Antimicrob Agents Chemother 2011; 55:5893–5899.
- Endimiani A, Patel G, Hujer KM, et al. In vitro activity of fosfomycin against blaKPC-containing Klebsiella pneumoniae isolates, including those nonsusceptible to tigecycline and/or colistin. Antimicrob Agents Chemother 2010; 54:526–529.
- Qureshi ZA, Paterson DL, Potoski BA, et al. Treatment outcome of bacteremia due to KPC-producing Klebsiella pneumoniae: superiority of combination antimicrobial regimens. Antimicrob Agents Chemother 2012; 56:2108–2113.
KEY POINTS
- The utility of carbapenems is being undermined by the emergence of resistance in Enterobacteriaceae and other bacteria.
- The clinical impact of CRE falls on elderly patients exposed to these organisms in hospitals and long-term care facilities. In this vulnerable group, invasive infections with CRE exact a high death rate.
- Long-term care facilities play an important role in the transmission dynamics of CRE.
- Tigecycline and colistin are treatments of last resort against infections caused by CRE. Their use in combination with other agents, especially carbapenems, may improve outcomes and needs to be explored further.
- Early detection of CRE in the microbiology laboratory is key to guiding infection control and treatment decisions and supporting surveillance efforts.
Recent recommendations on steroid-induced osteoporosis: More targeted, but more complicated
Whenever a patient begins treatment with a glucocorticoid drug, we need to think about bone loss.
The American College of Rheumatology (ACR) issued recommendations for preventing and treating glucocorticoid-induced osteoporosis in 2010.1 Compared with its previous guidelines,2 the new ones are more tailored and nuanced but may be more difficult for physicians to follow. The guidelines call for assessing fracture risk using the computer-based Fracture Risk Assessment Tool, or FRAX (www/shef.ac.uk/FRAX), developed by the World Health Organization (WHO). For those without a computer or ready access to the Web, an application of FRAX is available for download on smartphones.
In this article, my purpose is to review the new recommendations and to offer my perspective, which does not necessarily reflect the opinions of the ACR.
DESPITE EVIDENCE, MANY PATIENTS RECEIVE NO INTERVENTION
Use of glucocorticoids is the most common cause of secondary osteoporosis. During the first 6 to 12 months of use, these drugs can cause a rapid loss of bone mass due to increased bone resorption; with continued use, they cause a slower but steady decline in bone mass due to reduced bone formation.3 Epidemiologic studies have found that the risk of fractures increases with dose, starting with doses as low as 2.5 mg per day of prednisone or its equivalent.4
Numerous clinical trials have evaluated the effect of bisphosphonates and teriparatide (Forteo) on bone mass and fracture risk in patients on glucocorticoid therapy. The bisphosphonates alendronate (Fosamax) and risedronate (Actonel) have both been shown to increase bone mass and reduce vertebral fracture risk in glucocorticoid recipients.5–8 Zoledronic acid (Reclast), a parenteral bisphosphonate given in one annual dose, was shown to increase bone mass more than oral risedronate taken daily,9 and teriparatide, a formulation of parathyroid hormone, was better than alendronate.10
However, despite the known risk of fractures with glucocorticoid use and the demonstrated efficacy of available agents in preventing bone loss and fracture, many patients do not receive any intervention.11,12
WHAT HAS HAPPENED SINCE 2001?
In the interval since 2001, several guidelines for managing glucocorticoid-induced osteoporosis have been published in other countries.13–17 Broadly speaking, they recommend starting preventive drug therapy for patients at risk of fracture at the same time glucocorticoid drugs are started if the patient is expected to take glucocorticoids for more than 3 to 6 months in doses higher than 5 to 7.5 mg of prednisone or its equivalent daily.
Recommendations for patients who have been on glucocorticoids for longer than 3 to 6 months at initial evaluation have been based largely on T scores derived from dual-energy x-ray absorptiometry (DXA). Thresholds for initiating therapy have varied: the ACR in 2001 recommended preventive treatment if the T score is lower than −1.0, whereas British guidelines said −1.5 and Dutch guidelines said −2.5.
In the United States, since 2001 when the ACR published its last guidelines,2 zoledronic acid and teriparatide have been approved for use in glucocorticoid-induced osteoporosis. In addition, guideline-development methodology has evolved and now is more scientifically rigorous. Finally, a risk-assessment tool has been developed that enables a more tailored approach (see below).
FRAX (www.shef.ac.uk/FRAX)
FRAX is a tool developed by the WHO to calculate the risk of fracture. If you go to the FRAX Web site and enter the required clinical information (race, age, sex, weight, height, previous fracture, family history of a fractured hip in a parent, current smoking, use of glucocorticoids, rheumatoid arthritis, secondary osteoporosis, consumption of three or more units of alcohol per day, and bone mineral density of the femoral neck), it will tell you the patient’s 10-year absolute (not relative) risk of major osteoporotic fracture and of hip fracture.
Since FRAX was unveiled in 2008, calculation of absolute fracture risk has become the standard method for making treatment decisions in patients with low bone mass who have not yet received any fracture-preventing treatment.18 The use of clinical risk factors in FRAX increases its ability to predict risk over and above the use of bone density by itself. And glucocorticoids are one of the clinical risk factors in FRAX.
But in which patients is treatment with a bisphosphonate or teriparatide cost-effective?
Thresholds for cost-effectiveness have been developed on the basis of economic assumptions that are country-specific. In the United States, the National Osteoporosis Foundation recommends drug therapy if the 10-year absolute risk of a major osteoporotic fracture of the hip, spine (clinical, not radiographic), wrist, or humerus is greater than 20% or if the risk of a hip fracture is greater than 3%.19
At equivalent bone densities, women taking glucocorticoids are at considerably higher risk of fracture than nonusers.20 For example, consider a 65-year-old white woman, weight 59 kg, height 163 cm, no previous fractures, no parent with a fractured hip, no current smoking, no rheumatoid arthritis, no secondary osteoporosis, no excessive alcohol use, and a T score of −2.2 in the femoral neck. (Try this on the FRAX Web site.) If she does not use glucocorticoids, her 10-year risk of hip fracture is 2.0%; using glucocorticoids increases the risk to 3.6%. This is higher than the 3% National Osteoporosis Foundation guideline; thus, treatment would be recommended.
Also using FRAX, a 55-year-old white woman with a T score of −1.8 and on glucocorticoid therapy has a 67% higher risk of major osteoporotic fracture and an 80% higher risk of hip fracture.
For a third example, a white woman age 60, weight 70 kg, height 168 cm, negative for all the other risk factors but with a T score of −2.1 and on glucocorticoids has a calculated 10-year fracture risk of 2.1%, which is below the National Osteoporosis Foundation treatment threshold. However, most clinicians would probably recommend treatment for her, depending on the anticipated dose and duration of glucocorticoid therapy.
A caveat. In FRAX, glucocorticoid therapy is a categorical variable—a yes-or-no question—and yes is defined as having ever used a glucocorticoid in a dose greater than 5 mg for more than 3 months. Therefore, according to FRAX, a patient who took 5 mg of prednisone for 3 months 5 years ago has the same fracture risk as a patient on 60 mg of prednisone after a diagnosis of temporal arteritis. For this reason, the FRAX tool is likely to underestimate fracture risk, especially in patients currently taking glucocorticoids and those on higher doses of these drugs.
Kanis et al used the General Practice Research Database to adjust the fracture risk for glucocorticoid use in FRAX.21 At doses higher than 7.5 mg, the fracture risk had to be revised upward by 10% to 25% depending on the fracture site (hip vs any major osteoporotic fracture) and age (greater at age 40 than at age 90).
The underestimation of fracture risk led the ACR Expert Advisory Panel to create risk strata for major osteoporotic fractures, ie, low (< 10% risk per 10 years), medium (10%–20%), and high (> 20%) and uses these cut points to make treatment recommendations.
HOW THE 2010 GUIDELINES WERE DEVELOPED
Whereas the 2001 recommendations were based on a more informal consensus approach, the 2010 recommendations use a more scientifically rigorous methodology for guideline development, the Research and Development/University of California at Los Angeles (RAND/UCLA) Appropriateness Method. The RAND/UCLA method combines the best available scientific evidence with expert opinion to develop practice guidelines.
In drawing up the 2010 recommendations the ACR used three panels of experts. The Core Executive Panel conducted a systematic review of controlled clinical trials of therapies currently approved for treating glucocorticoid-induced osteoporosis in the United States, Canada, or the European Union. They found 53 articles meeting their inclusion criteria; an evidence report was produced that informed the development of the recommendations. This evidence report and guideline development process is available at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658. The Expert Advisory Panel framed the recommendations, and the Task Force Panel voted on them. The Core Executive Panel and Expert Advisory Panel constructed 48 patient-specific clinical scenarios using four variables: sex, age, race/ethnicity, and femoral neck T scores.
The members of the Task Force Panel were asked to use the evidence report and their expert judgment to vote on and rate the appropriateness of using a specific therapy in the context of each scenario on a 9-point Likert scale (1 = appropriate; 9 = not appropriate). Agreement occurred when 7 or more of the 10 panel members rated a scenario 1, 2, or 3. Disagreements were defined as 3 or more of the 10 members rating the scenario between 4 and 9 while the other members rated it lower.
Disagreements in voting were discussed in an attempt to achieve consensus, and a second vote was conducted which determined the final recommendations. If disagreement remained after the vote, no recommendation was made.
No attempt was made to assign priority of one drug over another when multiple drugs were deemed appropriate, although the final recommendations did differentiate drugs based on patient categories.
START WITH COUNSELING, ASSESSMENT

For patients starting or already on glucocorticoid therapy that is expected to last at least 3 months, the first step is to counsel them on lifestyle modifications (Table 1) and to assess their risk factors (Figure 1). Recommendations for monitoring patients receiving glucocorticoid therapy for at least 3 months are presented in Table 2.
These recommendations are based on literature review, and the strength of evidence is graded:
- Grade A—derived from multiple randomized controlled trials or a meta-analysis
- Grade B—derived from a single randomized controlled trial or nonrandomized study
- Grade C—derived from consensus, expert opinion, or case series.
This system is the same one used by the American College of Cardiology and is based on clinical trial data.22

Recommendations for calcium intake and vitamin D supplementation were graded A; all other recommendations were graded C (Tables 1 and 2). It is important to note that practices that receive a grade of C may still be accepted as standard of care, such as fall assessment and smoking cessation.
FOR POSTMENOPAUSAL WOMEN AND FOR MEN AGE 50 AND OLDER
FRAX low-risk group

Recall that “low risk” based on the new ACR guidelines means that the 10-year absolute risk of a major osteoporotic fracture, as calculated with FRAX, is less than 10%.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is less than 7.5 mg/day, no pharmacologic treatment is recommended.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. These are the most straightforward of the recommendations. All three bisphosphonates are recommended as treatment options if the glucocorticoid dose is at least 7.5 mg/day and the duration at least 3 months. Ibandronate (Boniva) was not included because it has no data from clinical trials.
FRAX medium-risk group
“Medium risk” means that the 10-year absolute fracture risk of major osteoporotic fractures is 10% to 20%.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is less than 7.5 mg/day, alendronate or risedronate is recommended.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. Treatment is recommended at all glucocorticoid doses for patients in the medium-risk category if the duration of glucocorticoid treatment is at least 3 months, with one difference: zoledronic acid is recommended only if the glucocorticoid dose is 7.5 mg/day or higher. This inconsistency persisted after a second round of voting by the Task Force Panel.
FRAX high-risk group
In this group, the 10-year risk of major osteoporotic fractures is higher than 20%.
- If the glucocorticoid dose is less than 5 mg/day for up to 1 month, alendronate, risedronate, or zoledronic acid is recommended.
- If the dose is 5 mg/day or more for up to 1 month, or any dose for more than 1 month, alendronate, risedronate, zoledronic acid or teriparatide is recommended.
Comment. Based on current National Osteoporosis Foundation guidelines, all patients with a 10-year risk greater than 20% are recommended for treatment for any duration and dose of glucocorticoid use. However, teriparatide is recommended only if the duration of glucocorticoid therapy is more than 1 month.
FOR PREMENOPAUSAL WOMEN AND FOR MEN YOUNGER THAN AGE 50
Use of FRAX is not appropriate in premenopausal women or in men younger than 50 years.
Younger patients with no prevalent fracture
For men younger than 50 and premenopausal women who have not had a previous fracture, data were considered inadequate to make a recommendation, and no votes were taken.
Prevalent fracture in premenopausal women of nonchildbearing potential
In premenopausal women of nonchildbearing potential who have had a fracture:
- If the glucocorticoid duration is 1 to 3 months and the dose is 5 mg/day or higher, alendronate or risedronate is recommended.
- If the duration is 1 to 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended
- If the duration is more than 3 months, alendronate, risedronate, zoledronic acid, or teriparatide is recommended.
Comment. Treatment is recommended with any of the four medications in patients with a fracture and treated with glucocorticoids for more than 3 months. For shorter-duration glucocorticoid use (1–3 months) at 5 mg/day or higher, only alendronate and risedronate are recommended. If the dose is 7.5 mg/day or higher, any bisphosphonate is recommended. Zoledronic acid was consistently differentiated by the expert panel on the basis of dose and duration of glucocorticoid use, in view of its 1-year duration of effect after one dose.
Prevalent fracture in women of childbearing potential
- If the glucocorticoid duration is 1 to 3 months, there was no consensus (ie, voting disagreements could not be resolved).
- If the glucocorticoid duration is more than 3 months and the dose is 7.5 mg/day or more, alendronate, risedronate, or teriparatide is recommended.
- If the glucocorticoid duration is more than 3 months and the dose is less than 7.5 mg/day, there was no consensus.
Comment. Childbearing potential creates further complexities because of concern about fetal toxicity with bisphosphonates. For short-term glucocorticoid therapy at any dose and for therapy longer than 3 months at less than 7.5 mg, no consensus could be reached. For therapy longer than 3 months and with 7.5 mg/day or higher, treatment is recommended but not with zoledronic acid, based on the long half-life of the drug and concern for fetal toxicity.
Additional risk stratification
The panel recommended that if the following were present, a shift to a higher fracture risk category should be considered (low to medium, or medium to high):
- High daily dose of glucocorticoid
- High cumulative glucocorticoid dose
- Declining bone mineral density on serial DXA.
These are known risk factors that increase fracture risk but would not affect fracture risk in the FRAX model.
WHAT IS NEW IN THE 2010 RECOMMENDATIONS?
Recommendations for counseling now include fall risk assessment, height measurement, 25-hydroxyvitamin D measurement, and evaluation of patients for prevalent and incident fractures using vertebral fracture assessment by DXA or radiographic imaging of the spine.
Recommended drugs now include teriparatide and zoledronic acid, while estrogen and testosterone are no longer recommended as therapies for glucocorticoid-induced osteoporosis. Ibandronate is not included, since there have been no randomized controlled trials of this bisphosphonate in glucocorticoid-induced osteoporosis.
Recommendations for treatment in 2001 were based on T scores alone, while the 2010 recommendations use an assessment of absolute fracture risk based on FRAX for postmenopausal women and for men age 50 and older.
A clinician’s guide that summarizes the ACR recommendations is available at www.rheumatology.org/practice/clinical/guidelines/.
RECOMMENDATIONS DO NOT REPLACE CLINICAL JUDGMENT
Although the 2010 recommendations were more rigorous in their development process than those of 2001, they have limitations and they should not replace clinical judgment. Rather, they are intended to provide an evidence-based approach to guide clinicians in making treatment choices in patients on glucocorticoid therapy.
CONSIDERING ABSOLUTE FRACTURE RISK IN TREATMENT DECISIONS
The 2001 ACR guidelines recommended fracture-preventing treatment in all patients starting glucocorticoid therapy at more than 5 mg/day if the planned duration of treatment was at least 3 months, and in patients on long-term glucocorticoid therapy if the T score was less than −1.0. While these guidelines were simple and easy to use, they were not specific enough to provide useful guidance in specific scenarios.
A model of absolute fracture risk was not available in 2001. A 55-year old white woman with a T score of −1.1 who smoked, who had been using 5 mg of prednisone for the last 12 months, and who had stable bone mass on serial DXA scans would have been recommended for treatment based on the 2001 recommendations. If this patient’s FRAX-calculated 10-year absolute risk of a major osteoporotic fracture is less than 10%, that would be well below the National Osteoporosis Foundation’s cost-effective treatment threshold of 20%. The new guidelines suggest no treatment is needed, since the risk category is low and the dose is less than 7.5 mg. However, if on serial DXA this patient had a significant decline in bone mass, the guidelines suggest shifting the patient to a higher risk category, ie, from low to medium risk, which would result in a recommendation in favor of treatment.
The 2010 recommendations are not as simple to use as those from 2001. They encourage using FRAX to calculate fracture risk; thus, knowledge of the strengths and limitations of FRAX is required. Access to the internet in the examination room or use of the FRAX tool on a smartphone as well as willingness to spend a minute to calculate fracture risk are needed. For those who cannot or choose not to use the FRAX tool, the ACR publication provides tables for patient risk assessment based on age and T score. However, the tables would have to be readily available in the clinic, which may not be practical.
The 2010 recommendation provide a more nuanced approach to treatment in patients on glucocorticoid therapy and are likely to change treatment decisions based on their use, just as FRAX has altered treatment decisions in patients with primary osteoporosis.23
FRAX has limitations
FRAX underestimates the effect of glucocorticoids on fracture risk because steroid use is a yes-or-no question and its weight represents the average risk in a population that has ever used steroids, most of whom were using doses between 2.5 and 7.5 mg.
The WHO recognized this limitation and suggested an upward adjustment of risk for patients on 7.5 mg or more, ranging from 10% to 25%.21 For patients on high doses of steroids, this adjustment is still likely to result in underestimation of fracture risk and undertreatment of glucocorticoid-treated patients.
The 2010 recommendations adjust for this limitation, recommending treatment in the low-risk and medium-risk categories if the glucocorticoid dose is 7.5 mg or higher. If a patient is using high daily doses of steroids or has a declining bone density, the 2010 recommendations suggest increasing the risk category from low to medium or medium to high.
FRAX risk factors are dichotomous (yes/no) and are not adjusted for dose effects such as multiple fractures (vs a single fracture), heavy smoking (vs light smoking), heavy alcohol use (6 units per day vs 3 units), or severe rheumatoid arthritis (vs mild disease). Family history of osteoporosis in the FRAX is limited to parents with a hip fracture—vertebral fractures in a family member do not count.
Since FRAX uses the bone mineral density in the hip, it underestimates fracture risk in patients with low spine density but normal hip density. It may also underestimate fracture risk in patients with declining bone mass; the 2010 recommendations suggest the clinician should increase the risk category in this situation.
LIMITATIONS OF THE GUIDELINES
The 2010 recommendations do not include several important groups in which steroids are used, including transplant recipients, children, and patients on inhaled corticosteroids. The panel thought that there were insufficient data to make recommendations for these populations, as well as for premenopausal women and men younger than 50 years who did not have a prevalent fracture. The absence of a recommendation in these situations should not be considered a recommendation for no treatment; it is an acknowledgment of a lack of evidence, a lack of consensus among experts, and the need for additional clinical trials.
For premenopausal women and men under age 50 with a fracture, the recommendations are complicated and not intuitive. Zoledronic acid is not recommended for women of non-childbearing potential with a glucocorticoid duration of 1 to 3 months unless the steroid dose is at least 7.5 mg. This recommendation was based on panel voting and consensus that giving zoledronic acid, a medication with a 1-year duration of effect, in a patient on steroids for only 1 to 3 months was not warranted.
Teriparatide was recommended only if glucocorticoids are used for at least 3 months, although anyone who already has a fracture might be considered at high enough risk to warrant anabolic therapy regardless of steroid use or duration.
Zoledronic acid was excluded in women of childbearing potential, based on panel voting and consensus that drugs given in smaller amounts over 1 year might be less harmful to a fetus than one with a longer half-life given in a larger bolus once a year.
The panel could reach no consensus on women of childbearing potential with a prevalent fracture who were using less than 7.5 mg/day of glucocorticoids. A lack of consensus was the result of insufficient data to make evidence-based decisions and a disagreement among experts on the correct treatment.
The guidelines do not address the duration of treatment with bisphosphonates, a topic of importance because of concern for the potential long-term side effects of these medications.
THE BOTTOM LINE
The 2010 recommendations add a degree of complexity, with different medications recommended on the basis of glucocorticoid dose and duration as well as patient age, menopausal status, and childbearing potential. Guideline developers and clinicians face a difficult trade-off: easy-to-follow guidelines or more targeted guidelines that are more complex and therefore more difficult to use than previous guidelines.
This criticism is reasonable. The complexity is a result of insufficient evidence from clinical trials to make more exact and user-friendly recommendations, and also a result of the RAND/UCLA methodology. In cases that lack sufficient evidence on which to make a decision, the guideline development uses voting among experts in an attempt to develop consensus. This often results in complexity, lack of consensus, or inconsistencies.
The guidelines are straightforward for postmenopausal women and men age 50 and older on at least 7.5 mg prednisone for more than 3 months.
Since there is substantial evidence that many patients on glucocorticoid therapy go untreated, the risk of fracture in this population would be substantially reduced if clinicians would adhere to the recommendations.
- Grossman JM, Gordon R, Ranganath VK, et al; American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken) 2010; 62:1515–1526.
- Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Arthritis Rheum 2001; 44:1496–1503.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Oral corticosteroids and fracture risk: relationship to daily and cumulative doses. Rheumatology (Oxford) 2000; 39:1383–1389.
- Saag KG, Emkey R, Schnitzer TJ, et al. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-Induced Osteoporosis Intervention Study Group. N Engl J Med 1998; 339:292–299.
- Cohen S, Levy RM, Keller M, et al. Risedronate therapy prevents corticosteroid-induced bone loss: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum 1999; 42:2309–2318.
- Reid DM, Hughes RA, Laan RF, et al. Efficacy and safety of daily risedronate in the treatment of corticosteroid-induced osteoporosis in men and women: a randomized trial. European Corticosteroid-Induced Osteoporosis Treatment Study. J Bone Miner Res 2000; 15:1006–1013.
- Wallach S, Cohen S, Reid DM, et al. Effects of risedronate treatment on bone density and vertebral fracture in patients on corticosteroid therapy. Calcif Tissue Int 2000; 67:277–285.
- Reid DM, Devogelaer JP, Saag K, et al; HORIZON investigators. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 2009; 373:1253–1263.
- Saag KG, Shane E, Boonen S, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 2007; 357:2028–2039.
- Curtis JR, Westfall AO, Allison JJ, et al. Longitudinal patterns in the prevention of osteoporosis in glucocorticoid-treated patients. Arthritis Rheum 2005; 52:2485–2494.
- Feldstein AC, Elmer PJ, Nichols GA, Herson M. Practice patterns in patients at risk for glucocorticoid-induced osteoporosis. Osteoporos Int 2005; 16:2168–2174.
- Brown JP, Josse RG; Scientific Advisory Council of the Osteoporosis Society of Canada. 2002 clinical practice guidelines for the diagnosis and management of osteoporosis in Canada. CMAJ 2002; 167(suppl 10):S1–S34.
- Devogelaer JP, Goemaere S, Boonen S, et al. Evidence-based guidelines for the prevention and treatment of glucocorticoid-induced osteoporosis: a consensus document of the Belgian Bone Club. Osteoporos Int 2006; 17:8–19.
- Gourlay M, Franceschini N, Sheyn Y. Prevention and treatment strategies for glucocorticoid-induced osteoporotic fractures. Clin Rheumatol 2007; 26:144–153.
- Nawata H, Soen S, Takayanagi R, et al; Subcommittee to Study Diagnostic Criteria for Glucocorticoid-Induced Osteoporosis. Guidelines on the management and treatment of glucocorticoid-induced osteoporosis of the Japanese Society for Bone and Mineral Research (2004). J Bone Miner Metab 2005; 23:105–109.
- Geusens PP, Lems WF, Verhaar HJ, et al. Review and evaluation of the Dutch guidelines for osteoporosis. J Eval Clin Pract 2006; 12:539–548.
- Kanis JA, Johnell O, Oden A, Johansson H, McCloskey E. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int 2008; 19:385–389.
- National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Washington, DC, National Osteoporosis Foundation, 2010. http://nof.org/files/nof/public/content/file/344/upload/159.pdf. Accessed December 31, 2012.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Guidance for the adjustment of FRAX according to the dose of glucocorticoids. Osteoporos Int 2011; 22:809–816.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation 2005; 112:e154–e235.
- Dawson-Hughes B, Tosteson AN, Melton LJ, et al; National Osteoporosis Foundation Guide Committee. Implications of absolute fracture risk assessment for osteoporosis practice guidelines in the USA. Osteoporos Int 2008; 19:449–458.
Whenever a patient begins treatment with a glucocorticoid drug, we need to think about bone loss.
The American College of Rheumatology (ACR) issued recommendations for preventing and treating glucocorticoid-induced osteoporosis in 2010.1 Compared with its previous guidelines,2 the new ones are more tailored and nuanced but may be more difficult for physicians to follow. The guidelines call for assessing fracture risk using the computer-based Fracture Risk Assessment Tool, or FRAX (www/shef.ac.uk/FRAX), developed by the World Health Organization (WHO). For those without a computer or ready access to the Web, an application of FRAX is available for download on smartphones.
In this article, my purpose is to review the new recommendations and to offer my perspective, which does not necessarily reflect the opinions of the ACR.
DESPITE EVIDENCE, MANY PATIENTS RECEIVE NO INTERVENTION
Use of glucocorticoids is the most common cause of secondary osteoporosis. During the first 6 to 12 months of use, these drugs can cause a rapid loss of bone mass due to increased bone resorption; with continued use, they cause a slower but steady decline in bone mass due to reduced bone formation.3 Epidemiologic studies have found that the risk of fractures increases with dose, starting with doses as low as 2.5 mg per day of prednisone or its equivalent.4
Numerous clinical trials have evaluated the effect of bisphosphonates and teriparatide (Forteo) on bone mass and fracture risk in patients on glucocorticoid therapy. The bisphosphonates alendronate (Fosamax) and risedronate (Actonel) have both been shown to increase bone mass and reduce vertebral fracture risk in glucocorticoid recipients.5–8 Zoledronic acid (Reclast), a parenteral bisphosphonate given in one annual dose, was shown to increase bone mass more than oral risedronate taken daily,9 and teriparatide, a formulation of parathyroid hormone, was better than alendronate.10
However, despite the known risk of fractures with glucocorticoid use and the demonstrated efficacy of available agents in preventing bone loss and fracture, many patients do not receive any intervention.11,12
WHAT HAS HAPPENED SINCE 2001?
In the interval since 2001, several guidelines for managing glucocorticoid-induced osteoporosis have been published in other countries.13–17 Broadly speaking, they recommend starting preventive drug therapy for patients at risk of fracture at the same time glucocorticoid drugs are started if the patient is expected to take glucocorticoids for more than 3 to 6 months in doses higher than 5 to 7.5 mg of prednisone or its equivalent daily.
Recommendations for patients who have been on glucocorticoids for longer than 3 to 6 months at initial evaluation have been based largely on T scores derived from dual-energy x-ray absorptiometry (DXA). Thresholds for initiating therapy have varied: the ACR in 2001 recommended preventive treatment if the T score is lower than −1.0, whereas British guidelines said −1.5 and Dutch guidelines said −2.5.
In the United States, since 2001 when the ACR published its last guidelines,2 zoledronic acid and teriparatide have been approved for use in glucocorticoid-induced osteoporosis. In addition, guideline-development methodology has evolved and now is more scientifically rigorous. Finally, a risk-assessment tool has been developed that enables a more tailored approach (see below).
FRAX (www.shef.ac.uk/FRAX)
FRAX is a tool developed by the WHO to calculate the risk of fracture. If you go to the FRAX Web site and enter the required clinical information (race, age, sex, weight, height, previous fracture, family history of a fractured hip in a parent, current smoking, use of glucocorticoids, rheumatoid arthritis, secondary osteoporosis, consumption of three or more units of alcohol per day, and bone mineral density of the femoral neck), it will tell you the patient’s 10-year absolute (not relative) risk of major osteoporotic fracture and of hip fracture.
Since FRAX was unveiled in 2008, calculation of absolute fracture risk has become the standard method for making treatment decisions in patients with low bone mass who have not yet received any fracture-preventing treatment.18 The use of clinical risk factors in FRAX increases its ability to predict risk over and above the use of bone density by itself. And glucocorticoids are one of the clinical risk factors in FRAX.
But in which patients is treatment with a bisphosphonate or teriparatide cost-effective?
Thresholds for cost-effectiveness have been developed on the basis of economic assumptions that are country-specific. In the United States, the National Osteoporosis Foundation recommends drug therapy if the 10-year absolute risk of a major osteoporotic fracture of the hip, spine (clinical, not radiographic), wrist, or humerus is greater than 20% or if the risk of a hip fracture is greater than 3%.19
At equivalent bone densities, women taking glucocorticoids are at considerably higher risk of fracture than nonusers.20 For example, consider a 65-year-old white woman, weight 59 kg, height 163 cm, no previous fractures, no parent with a fractured hip, no current smoking, no rheumatoid arthritis, no secondary osteoporosis, no excessive alcohol use, and a T score of −2.2 in the femoral neck. (Try this on the FRAX Web site.) If she does not use glucocorticoids, her 10-year risk of hip fracture is 2.0%; using glucocorticoids increases the risk to 3.6%. This is higher than the 3% National Osteoporosis Foundation guideline; thus, treatment would be recommended.
Also using FRAX, a 55-year-old white woman with a T score of −1.8 and on glucocorticoid therapy has a 67% higher risk of major osteoporotic fracture and an 80% higher risk of hip fracture.
For a third example, a white woman age 60, weight 70 kg, height 168 cm, negative for all the other risk factors but with a T score of −2.1 and on glucocorticoids has a calculated 10-year fracture risk of 2.1%, which is below the National Osteoporosis Foundation treatment threshold. However, most clinicians would probably recommend treatment for her, depending on the anticipated dose and duration of glucocorticoid therapy.
A caveat. In FRAX, glucocorticoid therapy is a categorical variable—a yes-or-no question—and yes is defined as having ever used a glucocorticoid in a dose greater than 5 mg for more than 3 months. Therefore, according to FRAX, a patient who took 5 mg of prednisone for 3 months 5 years ago has the same fracture risk as a patient on 60 mg of prednisone after a diagnosis of temporal arteritis. For this reason, the FRAX tool is likely to underestimate fracture risk, especially in patients currently taking glucocorticoids and those on higher doses of these drugs.
Kanis et al used the General Practice Research Database to adjust the fracture risk for glucocorticoid use in FRAX.21 At doses higher than 7.5 mg, the fracture risk had to be revised upward by 10% to 25% depending on the fracture site (hip vs any major osteoporotic fracture) and age (greater at age 40 than at age 90).
The underestimation of fracture risk led the ACR Expert Advisory Panel to create risk strata for major osteoporotic fractures, ie, low (< 10% risk per 10 years), medium (10%–20%), and high (> 20%) and uses these cut points to make treatment recommendations.
HOW THE 2010 GUIDELINES WERE DEVELOPED
Whereas the 2001 recommendations were based on a more informal consensus approach, the 2010 recommendations use a more scientifically rigorous methodology for guideline development, the Research and Development/University of California at Los Angeles (RAND/UCLA) Appropriateness Method. The RAND/UCLA method combines the best available scientific evidence with expert opinion to develop practice guidelines.
In drawing up the 2010 recommendations the ACR used three panels of experts. The Core Executive Panel conducted a systematic review of controlled clinical trials of therapies currently approved for treating glucocorticoid-induced osteoporosis in the United States, Canada, or the European Union. They found 53 articles meeting their inclusion criteria; an evidence report was produced that informed the development of the recommendations. This evidence report and guideline development process is available at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658. The Expert Advisory Panel framed the recommendations, and the Task Force Panel voted on them. The Core Executive Panel and Expert Advisory Panel constructed 48 patient-specific clinical scenarios using four variables: sex, age, race/ethnicity, and femoral neck T scores.
The members of the Task Force Panel were asked to use the evidence report and their expert judgment to vote on and rate the appropriateness of using a specific therapy in the context of each scenario on a 9-point Likert scale (1 = appropriate; 9 = not appropriate). Agreement occurred when 7 or more of the 10 panel members rated a scenario 1, 2, or 3. Disagreements were defined as 3 or more of the 10 members rating the scenario between 4 and 9 while the other members rated it lower.
Disagreements in voting were discussed in an attempt to achieve consensus, and a second vote was conducted which determined the final recommendations. If disagreement remained after the vote, no recommendation was made.
No attempt was made to assign priority of one drug over another when multiple drugs were deemed appropriate, although the final recommendations did differentiate drugs based on patient categories.
START WITH COUNSELING, ASSESSMENT
For patients starting or already on glucocorticoid therapy that is expected to last at least 3 months, the first step is to counsel them on lifestyle modifications (Table 1) and to assess their risk factors (Figure 1). Recommendations for monitoring patients receiving glucocorticoid therapy for at least 3 months are presented in Table 2.
These recommendations are based on literature review, and the strength of evidence is graded:
- Grade A—derived from multiple randomized controlled trials or a meta-analysis
- Grade B—derived from a single randomized controlled trial or nonrandomized study
- Grade C—derived from consensus, expert opinion, or case series.
This system is the same one used by the American College of Cardiology and is based on clinical trial data.22
Recommendations for calcium intake and vitamin D supplementation were graded A; all other recommendations were graded C (Tables 1 and 2). It is important to note that practices that receive a grade of C may still be accepted as standard of care, such as fall assessment and smoking cessation.
FOR POSTMENOPAUSAL WOMEN AND FOR MEN AGE 50 AND OLDER
FRAX low-risk group
Recall that “low risk” based on the new ACR guidelines means that the 10-year absolute risk of a major osteoporotic fracture, as calculated with FRAX, is less than 10%.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is less than 7.5 mg/day, no pharmacologic treatment is recommended.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. These are the most straightforward of the recommendations. All three bisphosphonates are recommended as treatment options if the glucocorticoid dose is at least 7.5 mg/day and the duration at least 3 months. Ibandronate (Boniva) was not included because it has no data from clinical trials.
FRAX medium-risk group
“Medium risk” means that the 10-year absolute fracture risk of major osteoporotic fractures is 10% to 20%.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is less than 7.5 mg/day, alendronate or risedronate is recommended.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. Treatment is recommended at all glucocorticoid doses for patients in the medium-risk category if the duration of glucocorticoid treatment is at least 3 months, with one difference: zoledronic acid is recommended only if the glucocorticoid dose is 7.5 mg/day or higher. This inconsistency persisted after a second round of voting by the Task Force Panel.
FRAX high-risk group
In this group, the 10-year risk of major osteoporotic fractures is higher than 20%.
- If the glucocorticoid dose is less than 5 mg/day for up to 1 month, alendronate, risedronate, or zoledronic acid is recommended.
- If the dose is 5 mg/day or more for up to 1 month, or any dose for more than 1 month, alendronate, risedronate, zoledronic acid or teriparatide is recommended.
Comment. Based on current National Osteoporosis Foundation guidelines, all patients with a 10-year risk greater than 20% are recommended for treatment for any duration and dose of glucocorticoid use. However, teriparatide is recommended only if the duration of glucocorticoid therapy is more than 1 month.
FOR PREMENOPAUSAL WOMEN AND FOR MEN YOUNGER THAN AGE 50
Use of FRAX is not appropriate in premenopausal women or in men younger than 50 years.
Younger patients with no prevalent fracture
For men younger than 50 and premenopausal women who have not had a previous fracture, data were considered inadequate to make a recommendation, and no votes were taken.
Prevalent fracture in premenopausal women of nonchildbearing potential
In premenopausal women of nonchildbearing potential who have had a fracture:
- If the glucocorticoid duration is 1 to 3 months and the dose is 5 mg/day or higher, alendronate or risedronate is recommended.
- If the duration is 1 to 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended
- If the duration is more than 3 months, alendronate, risedronate, zoledronic acid, or teriparatide is recommended.
Comment. Treatment is recommended with any of the four medications in patients with a fracture and treated with glucocorticoids for more than 3 months. For shorter-duration glucocorticoid use (1–3 months) at 5 mg/day or higher, only alendronate and risedronate are recommended. If the dose is 7.5 mg/day or higher, any bisphosphonate is recommended. Zoledronic acid was consistently differentiated by the expert panel on the basis of dose and duration of glucocorticoid use, in view of its 1-year duration of effect after one dose.
Prevalent fracture in women of childbearing potential
- If the glucocorticoid duration is 1 to 3 months, there was no consensus (ie, voting disagreements could not be resolved).
- If the glucocorticoid duration is more than 3 months and the dose is 7.5 mg/day or more, alendronate, risedronate, or teriparatide is recommended.
- If the glucocorticoid duration is more than 3 months and the dose is less than 7.5 mg/day, there was no consensus.
Comment. Childbearing potential creates further complexities because of concern about fetal toxicity with bisphosphonates. For short-term glucocorticoid therapy at any dose and for therapy longer than 3 months at less than 7.5 mg, no consensus could be reached. For therapy longer than 3 months and with 7.5 mg/day or higher, treatment is recommended but not with zoledronic acid, based on the long half-life of the drug and concern for fetal toxicity.
Additional risk stratification
The panel recommended that if the following were present, a shift to a higher fracture risk category should be considered (low to medium, or medium to high):
- High daily dose of glucocorticoid
- High cumulative glucocorticoid dose
- Declining bone mineral density on serial DXA.
These are known risk factors that increase fracture risk but would not affect fracture risk in the FRAX model.
WHAT IS NEW IN THE 2010 RECOMMENDATIONS?
Recommendations for counseling now include fall risk assessment, height measurement, 25-hydroxyvitamin D measurement, and evaluation of patients for prevalent and incident fractures using vertebral fracture assessment by DXA or radiographic imaging of the spine.
Recommended drugs now include teriparatide and zoledronic acid, while estrogen and testosterone are no longer recommended as therapies for glucocorticoid-induced osteoporosis. Ibandronate is not included, since there have been no randomized controlled trials of this bisphosphonate in glucocorticoid-induced osteoporosis.
Recommendations for treatment in 2001 were based on T scores alone, while the 2010 recommendations use an assessment of absolute fracture risk based on FRAX for postmenopausal women and for men age 50 and older.
A clinician’s guide that summarizes the ACR recommendations is available at www.rheumatology.org/practice/clinical/guidelines/.
RECOMMENDATIONS DO NOT REPLACE CLINICAL JUDGMENT
Although the 2010 recommendations were more rigorous in their development process than those of 2001, they have limitations and they should not replace clinical judgment. Rather, they are intended to provide an evidence-based approach to guide clinicians in making treatment choices in patients on glucocorticoid therapy.
CONSIDERING ABSOLUTE FRACTURE RISK IN TREATMENT DECISIONS
The 2001 ACR guidelines recommended fracture-preventing treatment in all patients starting glucocorticoid therapy at more than 5 mg/day if the planned duration of treatment was at least 3 months, and in patients on long-term glucocorticoid therapy if the T score was less than −1.0. While these guidelines were simple and easy to use, they were not specific enough to provide useful guidance in specific scenarios.
A model of absolute fracture risk was not available in 2001. A 55-year old white woman with a T score of −1.1 who smoked, who had been using 5 mg of prednisone for the last 12 months, and who had stable bone mass on serial DXA scans would have been recommended for treatment based on the 2001 recommendations. If this patient’s FRAX-calculated 10-year absolute risk of a major osteoporotic fracture is less than 10%, that would be well below the National Osteoporosis Foundation’s cost-effective treatment threshold of 20%. The new guidelines suggest no treatment is needed, since the risk category is low and the dose is less than 7.5 mg. However, if on serial DXA this patient had a significant decline in bone mass, the guidelines suggest shifting the patient to a higher risk category, ie, from low to medium risk, which would result in a recommendation in favor of treatment.
The 2010 recommendations are not as simple to use as those from 2001. They encourage using FRAX to calculate fracture risk; thus, knowledge of the strengths and limitations of FRAX is required. Access to the internet in the examination room or use of the FRAX tool on a smartphone as well as willingness to spend a minute to calculate fracture risk are needed. For those who cannot or choose not to use the FRAX tool, the ACR publication provides tables for patient risk assessment based on age and T score. However, the tables would have to be readily available in the clinic, which may not be practical.
The 2010 recommendation provide a more nuanced approach to treatment in patients on glucocorticoid therapy and are likely to change treatment decisions based on their use, just as FRAX has altered treatment decisions in patients with primary osteoporosis.23
FRAX has limitations
FRAX underestimates the effect of glucocorticoids on fracture risk because steroid use is a yes-or-no question and its weight represents the average risk in a population that has ever used steroids, most of whom were using doses between 2.5 and 7.5 mg.
The WHO recognized this limitation and suggested an upward adjustment of risk for patients on 7.5 mg or more, ranging from 10% to 25%.21 For patients on high doses of steroids, this adjustment is still likely to result in underestimation of fracture risk and undertreatment of glucocorticoid-treated patients.
The 2010 recommendations adjust for this limitation, recommending treatment in the low-risk and medium-risk categories if the glucocorticoid dose is 7.5 mg or higher. If a patient is using high daily doses of steroids or has a declining bone density, the 2010 recommendations suggest increasing the risk category from low to medium or medium to high.
FRAX risk factors are dichotomous (yes/no) and are not adjusted for dose effects such as multiple fractures (vs a single fracture), heavy smoking (vs light smoking), heavy alcohol use (6 units per day vs 3 units), or severe rheumatoid arthritis (vs mild disease). Family history of osteoporosis in the FRAX is limited to parents with a hip fracture—vertebral fractures in a family member do not count.
Since FRAX uses the bone mineral density in the hip, it underestimates fracture risk in patients with low spine density but normal hip density. It may also underestimate fracture risk in patients with declining bone mass; the 2010 recommendations suggest the clinician should increase the risk category in this situation.
LIMITATIONS OF THE GUIDELINES
The 2010 recommendations do not include several important groups in which steroids are used, including transplant recipients, children, and patients on inhaled corticosteroids. The panel thought that there were insufficient data to make recommendations for these populations, as well as for premenopausal women and men younger than 50 years who did not have a prevalent fracture. The absence of a recommendation in these situations should not be considered a recommendation for no treatment; it is an acknowledgment of a lack of evidence, a lack of consensus among experts, and the need for additional clinical trials.
For premenopausal women and men under age 50 with a fracture, the recommendations are complicated and not intuitive. Zoledronic acid is not recommended for women of non-childbearing potential with a glucocorticoid duration of 1 to 3 months unless the steroid dose is at least 7.5 mg. This recommendation was based on panel voting and consensus that giving zoledronic acid, a medication with a 1-year duration of effect, in a patient on steroids for only 1 to 3 months was not warranted.
Teriparatide was recommended only if glucocorticoids are used for at least 3 months, although anyone who already has a fracture might be considered at high enough risk to warrant anabolic therapy regardless of steroid use or duration.
Zoledronic acid was excluded in women of childbearing potential, based on panel voting and consensus that drugs given in smaller amounts over 1 year might be less harmful to a fetus than one with a longer half-life given in a larger bolus once a year.
The panel could reach no consensus on women of childbearing potential with a prevalent fracture who were using less than 7.5 mg/day of glucocorticoids. A lack of consensus was the result of insufficient data to make evidence-based decisions and a disagreement among experts on the correct treatment.
The guidelines do not address the duration of treatment with bisphosphonates, a topic of importance because of concern for the potential long-term side effects of these medications.
THE BOTTOM LINE
The 2010 recommendations add a degree of complexity, with different medications recommended on the basis of glucocorticoid dose and duration as well as patient age, menopausal status, and childbearing potential. Guideline developers and clinicians face a difficult trade-off: easy-to-follow guidelines or more targeted guidelines that are more complex and therefore more difficult to use than previous guidelines.
This criticism is reasonable. The complexity is a result of insufficient evidence from clinical trials to make more exact and user-friendly recommendations, and also a result of the RAND/UCLA methodology. In cases that lack sufficient evidence on which to make a decision, the guideline development uses voting among experts in an attempt to develop consensus. This often results in complexity, lack of consensus, or inconsistencies.
The guidelines are straightforward for postmenopausal women and men age 50 and older on at least 7.5 mg prednisone for more than 3 months.
Since there is substantial evidence that many patients on glucocorticoid therapy go untreated, the risk of fracture in this population would be substantially reduced if clinicians would adhere to the recommendations.
Whenever a patient begins treatment with a glucocorticoid drug, we need to think about bone loss.
The American College of Rheumatology (ACR) issued recommendations for preventing and treating glucocorticoid-induced osteoporosis in 2010.1 Compared with its previous guidelines,2 the new ones are more tailored and nuanced but may be more difficult for physicians to follow. The guidelines call for assessing fracture risk using the computer-based Fracture Risk Assessment Tool, or FRAX (www/shef.ac.uk/FRAX), developed by the World Health Organization (WHO). For those without a computer or ready access to the Web, an application of FRAX is available for download on smartphones.
In this article, my purpose is to review the new recommendations and to offer my perspective, which does not necessarily reflect the opinions of the ACR.
DESPITE EVIDENCE, MANY PATIENTS RECEIVE NO INTERVENTION
Use of glucocorticoids is the most common cause of secondary osteoporosis. During the first 6 to 12 months of use, these drugs can cause a rapid loss of bone mass due to increased bone resorption; with continued use, they cause a slower but steady decline in bone mass due to reduced bone formation.3 Epidemiologic studies have found that the risk of fractures increases with dose, starting with doses as low as 2.5 mg per day of prednisone or its equivalent.4
Numerous clinical trials have evaluated the effect of bisphosphonates and teriparatide (Forteo) on bone mass and fracture risk in patients on glucocorticoid therapy. The bisphosphonates alendronate (Fosamax) and risedronate (Actonel) have both been shown to increase bone mass and reduce vertebral fracture risk in glucocorticoid recipients.5–8 Zoledronic acid (Reclast), a parenteral bisphosphonate given in one annual dose, was shown to increase bone mass more than oral risedronate taken daily,9 and teriparatide, a formulation of parathyroid hormone, was better than alendronate.10
However, despite the known risk of fractures with glucocorticoid use and the demonstrated efficacy of available agents in preventing bone loss and fracture, many patients do not receive any intervention.11,12
WHAT HAS HAPPENED SINCE 2001?
In the interval since 2001, several guidelines for managing glucocorticoid-induced osteoporosis have been published in other countries.13–17 Broadly speaking, they recommend starting preventive drug therapy for patients at risk of fracture at the same time glucocorticoid drugs are started if the patient is expected to take glucocorticoids for more than 3 to 6 months in doses higher than 5 to 7.5 mg of prednisone or its equivalent daily.
Recommendations for patients who have been on glucocorticoids for longer than 3 to 6 months at initial evaluation have been based largely on T scores derived from dual-energy x-ray absorptiometry (DXA). Thresholds for initiating therapy have varied: the ACR in 2001 recommended preventive treatment if the T score is lower than −1.0, whereas British guidelines said −1.5 and Dutch guidelines said −2.5.
In the United States, since 2001 when the ACR published its last guidelines,2 zoledronic acid and teriparatide have been approved for use in glucocorticoid-induced osteoporosis. In addition, guideline-development methodology has evolved and now is more scientifically rigorous. Finally, a risk-assessment tool has been developed that enables a more tailored approach (see below).
FRAX (www.shef.ac.uk/FRAX)
FRAX is a tool developed by the WHO to calculate the risk of fracture. If you go to the FRAX Web site and enter the required clinical information (race, age, sex, weight, height, previous fracture, family history of a fractured hip in a parent, current smoking, use of glucocorticoids, rheumatoid arthritis, secondary osteoporosis, consumption of three or more units of alcohol per day, and bone mineral density of the femoral neck), it will tell you the patient’s 10-year absolute (not relative) risk of major osteoporotic fracture and of hip fracture.
Since FRAX was unveiled in 2008, calculation of absolute fracture risk has become the standard method for making treatment decisions in patients with low bone mass who have not yet received any fracture-preventing treatment.18 The use of clinical risk factors in FRAX increases its ability to predict risk over and above the use of bone density by itself. And glucocorticoids are one of the clinical risk factors in FRAX.
But in which patients is treatment with a bisphosphonate or teriparatide cost-effective?
Thresholds for cost-effectiveness have been developed on the basis of economic assumptions that are country-specific. In the United States, the National Osteoporosis Foundation recommends drug therapy if the 10-year absolute risk of a major osteoporotic fracture of the hip, spine (clinical, not radiographic), wrist, or humerus is greater than 20% or if the risk of a hip fracture is greater than 3%.19
At equivalent bone densities, women taking glucocorticoids are at considerably higher risk of fracture than nonusers.20 For example, consider a 65-year-old white woman, weight 59 kg, height 163 cm, no previous fractures, no parent with a fractured hip, no current smoking, no rheumatoid arthritis, no secondary osteoporosis, no excessive alcohol use, and a T score of −2.2 in the femoral neck. (Try this on the FRAX Web site.) If she does not use glucocorticoids, her 10-year risk of hip fracture is 2.0%; using glucocorticoids increases the risk to 3.6%. This is higher than the 3% National Osteoporosis Foundation guideline; thus, treatment would be recommended.
Also using FRAX, a 55-year-old white woman with a T score of −1.8 and on glucocorticoid therapy has a 67% higher risk of major osteoporotic fracture and an 80% higher risk of hip fracture.
For a third example, a white woman age 60, weight 70 kg, height 168 cm, negative for all the other risk factors but with a T score of −2.1 and on glucocorticoids has a calculated 10-year fracture risk of 2.1%, which is below the National Osteoporosis Foundation treatment threshold. However, most clinicians would probably recommend treatment for her, depending on the anticipated dose and duration of glucocorticoid therapy.
A caveat. In FRAX, glucocorticoid therapy is a categorical variable—a yes-or-no question—and yes is defined as having ever used a glucocorticoid in a dose greater than 5 mg for more than 3 months. Therefore, according to FRAX, a patient who took 5 mg of prednisone for 3 months 5 years ago has the same fracture risk as a patient on 60 mg of prednisone after a diagnosis of temporal arteritis. For this reason, the FRAX tool is likely to underestimate fracture risk, especially in patients currently taking glucocorticoids and those on higher doses of these drugs.
Kanis et al used the General Practice Research Database to adjust the fracture risk for glucocorticoid use in FRAX.21 At doses higher than 7.5 mg, the fracture risk had to be revised upward by 10% to 25% depending on the fracture site (hip vs any major osteoporotic fracture) and age (greater at age 40 than at age 90).
The underestimation of fracture risk led the ACR Expert Advisory Panel to create risk strata for major osteoporotic fractures, ie, low (< 10% risk per 10 years), medium (10%–20%), and high (> 20%) and uses these cut points to make treatment recommendations.
HOW THE 2010 GUIDELINES WERE DEVELOPED
Whereas the 2001 recommendations were based on a more informal consensus approach, the 2010 recommendations use a more scientifically rigorous methodology for guideline development, the Research and Development/University of California at Los Angeles (RAND/UCLA) Appropriateness Method. The RAND/UCLA method combines the best available scientific evidence with expert opinion to develop practice guidelines.
In drawing up the 2010 recommendations the ACR used three panels of experts. The Core Executive Panel conducted a systematic review of controlled clinical trials of therapies currently approved for treating glucocorticoid-induced osteoporosis in the United States, Canada, or the European Union. They found 53 articles meeting their inclusion criteria; an evidence report was produced that informed the development of the recommendations. This evidence report and guideline development process is available at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658. The Expert Advisory Panel framed the recommendations, and the Task Force Panel voted on them. The Core Executive Panel and Expert Advisory Panel constructed 48 patient-specific clinical scenarios using four variables: sex, age, race/ethnicity, and femoral neck T scores.
The members of the Task Force Panel were asked to use the evidence report and their expert judgment to vote on and rate the appropriateness of using a specific therapy in the context of each scenario on a 9-point Likert scale (1 = appropriate; 9 = not appropriate). Agreement occurred when 7 or more of the 10 panel members rated a scenario 1, 2, or 3. Disagreements were defined as 3 or more of the 10 members rating the scenario between 4 and 9 while the other members rated it lower.
Disagreements in voting were discussed in an attempt to achieve consensus, and a second vote was conducted which determined the final recommendations. If disagreement remained after the vote, no recommendation was made.
No attempt was made to assign priority of one drug over another when multiple drugs were deemed appropriate, although the final recommendations did differentiate drugs based on patient categories.
START WITH COUNSELING, ASSESSMENT
For patients starting or already on glucocorticoid therapy that is expected to last at least 3 months, the first step is to counsel them on lifestyle modifications (Table 1) and to assess their risk factors (Figure 1). Recommendations for monitoring patients receiving glucocorticoid therapy for at least 3 months are presented in Table 2.
These recommendations are based on literature review, and the strength of evidence is graded:
- Grade A—derived from multiple randomized controlled trials or a meta-analysis
- Grade B—derived from a single randomized controlled trial or nonrandomized study
- Grade C—derived from consensus, expert opinion, or case series.
This system is the same one used by the American College of Cardiology and is based on clinical trial data.22
Recommendations for calcium intake and vitamin D supplementation were graded A; all other recommendations were graded C (Tables 1 and 2). It is important to note that practices that receive a grade of C may still be accepted as standard of care, such as fall assessment and smoking cessation.
FOR POSTMENOPAUSAL WOMEN AND FOR MEN AGE 50 AND OLDER
FRAX low-risk group
Recall that “low risk” based on the new ACR guidelines means that the 10-year absolute risk of a major osteoporotic fracture, as calculated with FRAX, is less than 10%.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is less than 7.5 mg/day, no pharmacologic treatment is recommended.
- If glucocorticoid use is expected to last or has already lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. These are the most straightforward of the recommendations. All three bisphosphonates are recommended as treatment options if the glucocorticoid dose is at least 7.5 mg/day and the duration at least 3 months. Ibandronate (Boniva) was not included because it has no data from clinical trials.
FRAX medium-risk group
“Medium risk” means that the 10-year absolute fracture risk of major osteoporotic fractures is 10% to 20%.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is less than 7.5 mg/day, alendronate or risedronate is recommended.
- If glucocorticoid use is anticipated to last or has lasted at least 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended.
Comment. Treatment is recommended at all glucocorticoid doses for patients in the medium-risk category if the duration of glucocorticoid treatment is at least 3 months, with one difference: zoledronic acid is recommended only if the glucocorticoid dose is 7.5 mg/day or higher. This inconsistency persisted after a second round of voting by the Task Force Panel.
FRAX high-risk group
In this group, the 10-year risk of major osteoporotic fractures is higher than 20%.
- If the glucocorticoid dose is less than 5 mg/day for up to 1 month, alendronate, risedronate, or zoledronic acid is recommended.
- If the dose is 5 mg/day or more for up to 1 month, or any dose for more than 1 month, alendronate, risedronate, zoledronic acid or teriparatide is recommended.
Comment. Based on current National Osteoporosis Foundation guidelines, all patients with a 10-year risk greater than 20% are recommended for treatment for any duration and dose of glucocorticoid use. However, teriparatide is recommended only if the duration of glucocorticoid therapy is more than 1 month.
FOR PREMENOPAUSAL WOMEN AND FOR MEN YOUNGER THAN AGE 50
Use of FRAX is not appropriate in premenopausal women or in men younger than 50 years.
Younger patients with no prevalent fracture
For men younger than 50 and premenopausal women who have not had a previous fracture, data were considered inadequate to make a recommendation, and no votes were taken.
Prevalent fracture in premenopausal women of nonchildbearing potential
In premenopausal women of nonchildbearing potential who have had a fracture:
- If the glucocorticoid duration is 1 to 3 months and the dose is 5 mg/day or higher, alendronate or risedronate is recommended.
- If the duration is 1 to 3 months and the dose is 7.5 mg/day or higher, alendronate, risedronate, or zoledronic acid is recommended
- If the duration is more than 3 months, alendronate, risedronate, zoledronic acid, or teriparatide is recommended.
Comment. Treatment is recommended with any of the four medications in patients with a fracture and treated with glucocorticoids for more than 3 months. For shorter-duration glucocorticoid use (1–3 months) at 5 mg/day or higher, only alendronate and risedronate are recommended. If the dose is 7.5 mg/day or higher, any bisphosphonate is recommended. Zoledronic acid was consistently differentiated by the expert panel on the basis of dose and duration of glucocorticoid use, in view of its 1-year duration of effect after one dose.
Prevalent fracture in women of childbearing potential
- If the glucocorticoid duration is 1 to 3 months, there was no consensus (ie, voting disagreements could not be resolved).
- If the glucocorticoid duration is more than 3 months and the dose is 7.5 mg/day or more, alendronate, risedronate, or teriparatide is recommended.
- If the glucocorticoid duration is more than 3 months and the dose is less than 7.5 mg/day, there was no consensus.
Comment. Childbearing potential creates further complexities because of concern about fetal toxicity with bisphosphonates. For short-term glucocorticoid therapy at any dose and for therapy longer than 3 months at less than 7.5 mg, no consensus could be reached. For therapy longer than 3 months and with 7.5 mg/day or higher, treatment is recommended but not with zoledronic acid, based on the long half-life of the drug and concern for fetal toxicity.
Additional risk stratification
The panel recommended that if the following were present, a shift to a higher fracture risk category should be considered (low to medium, or medium to high):
- High daily dose of glucocorticoid
- High cumulative glucocorticoid dose
- Declining bone mineral density on serial DXA.
These are known risk factors that increase fracture risk but would not affect fracture risk in the FRAX model.
WHAT IS NEW IN THE 2010 RECOMMENDATIONS?
Recommendations for counseling now include fall risk assessment, height measurement, 25-hydroxyvitamin D measurement, and evaluation of patients for prevalent and incident fractures using vertebral fracture assessment by DXA or radiographic imaging of the spine.
Recommended drugs now include teriparatide and zoledronic acid, while estrogen and testosterone are no longer recommended as therapies for glucocorticoid-induced osteoporosis. Ibandronate is not included, since there have been no randomized controlled trials of this bisphosphonate in glucocorticoid-induced osteoporosis.
Recommendations for treatment in 2001 were based on T scores alone, while the 2010 recommendations use an assessment of absolute fracture risk based on FRAX for postmenopausal women and for men age 50 and older.
A clinician’s guide that summarizes the ACR recommendations is available at www.rheumatology.org/practice/clinical/guidelines/.
RECOMMENDATIONS DO NOT REPLACE CLINICAL JUDGMENT
Although the 2010 recommendations were more rigorous in their development process than those of 2001, they have limitations and they should not replace clinical judgment. Rather, they are intended to provide an evidence-based approach to guide clinicians in making treatment choices in patients on glucocorticoid therapy.
CONSIDERING ABSOLUTE FRACTURE RISK IN TREATMENT DECISIONS
The 2001 ACR guidelines recommended fracture-preventing treatment in all patients starting glucocorticoid therapy at more than 5 mg/day if the planned duration of treatment was at least 3 months, and in patients on long-term glucocorticoid therapy if the T score was less than −1.0. While these guidelines were simple and easy to use, they were not specific enough to provide useful guidance in specific scenarios.
A model of absolute fracture risk was not available in 2001. A 55-year old white woman with a T score of −1.1 who smoked, who had been using 5 mg of prednisone for the last 12 months, and who had stable bone mass on serial DXA scans would have been recommended for treatment based on the 2001 recommendations. If this patient’s FRAX-calculated 10-year absolute risk of a major osteoporotic fracture is less than 10%, that would be well below the National Osteoporosis Foundation’s cost-effective treatment threshold of 20%. The new guidelines suggest no treatment is needed, since the risk category is low and the dose is less than 7.5 mg. However, if on serial DXA this patient had a significant decline in bone mass, the guidelines suggest shifting the patient to a higher risk category, ie, from low to medium risk, which would result in a recommendation in favor of treatment.
The 2010 recommendations are not as simple to use as those from 2001. They encourage using FRAX to calculate fracture risk; thus, knowledge of the strengths and limitations of FRAX is required. Access to the internet in the examination room or use of the FRAX tool on a smartphone as well as willingness to spend a minute to calculate fracture risk are needed. For those who cannot or choose not to use the FRAX tool, the ACR publication provides tables for patient risk assessment based on age and T score. However, the tables would have to be readily available in the clinic, which may not be practical.
The 2010 recommendation provide a more nuanced approach to treatment in patients on glucocorticoid therapy and are likely to change treatment decisions based on their use, just as FRAX has altered treatment decisions in patients with primary osteoporosis.23
FRAX has limitations
FRAX underestimates the effect of glucocorticoids on fracture risk because steroid use is a yes-or-no question and its weight represents the average risk in a population that has ever used steroids, most of whom were using doses between 2.5 and 7.5 mg.
The WHO recognized this limitation and suggested an upward adjustment of risk for patients on 7.5 mg or more, ranging from 10% to 25%.21 For patients on high doses of steroids, this adjustment is still likely to result in underestimation of fracture risk and undertreatment of glucocorticoid-treated patients.
The 2010 recommendations adjust for this limitation, recommending treatment in the low-risk and medium-risk categories if the glucocorticoid dose is 7.5 mg or higher. If a patient is using high daily doses of steroids or has a declining bone density, the 2010 recommendations suggest increasing the risk category from low to medium or medium to high.
FRAX risk factors are dichotomous (yes/no) and are not adjusted for dose effects such as multiple fractures (vs a single fracture), heavy smoking (vs light smoking), heavy alcohol use (6 units per day vs 3 units), or severe rheumatoid arthritis (vs mild disease). Family history of osteoporosis in the FRAX is limited to parents with a hip fracture—vertebral fractures in a family member do not count.
Since FRAX uses the bone mineral density in the hip, it underestimates fracture risk in patients with low spine density but normal hip density. It may also underestimate fracture risk in patients with declining bone mass; the 2010 recommendations suggest the clinician should increase the risk category in this situation.
LIMITATIONS OF THE GUIDELINES
The 2010 recommendations do not include several important groups in which steroids are used, including transplant recipients, children, and patients on inhaled corticosteroids. The panel thought that there were insufficient data to make recommendations for these populations, as well as for premenopausal women and men younger than 50 years who did not have a prevalent fracture. The absence of a recommendation in these situations should not be considered a recommendation for no treatment; it is an acknowledgment of a lack of evidence, a lack of consensus among experts, and the need for additional clinical trials.
For premenopausal women and men under age 50 with a fracture, the recommendations are complicated and not intuitive. Zoledronic acid is not recommended for women of non-childbearing potential with a glucocorticoid duration of 1 to 3 months unless the steroid dose is at least 7.5 mg. This recommendation was based on panel voting and consensus that giving zoledronic acid, a medication with a 1-year duration of effect, in a patient on steroids for only 1 to 3 months was not warranted.
Teriparatide was recommended only if glucocorticoids are used for at least 3 months, although anyone who already has a fracture might be considered at high enough risk to warrant anabolic therapy regardless of steroid use or duration.
Zoledronic acid was excluded in women of childbearing potential, based on panel voting and consensus that drugs given in smaller amounts over 1 year might be less harmful to a fetus than one with a longer half-life given in a larger bolus once a year.
The panel could reach no consensus on women of childbearing potential with a prevalent fracture who were using less than 7.5 mg/day of glucocorticoids. A lack of consensus was the result of insufficient data to make evidence-based decisions and a disagreement among experts on the correct treatment.
The guidelines do not address the duration of treatment with bisphosphonates, a topic of importance because of concern for the potential long-term side effects of these medications.
THE BOTTOM LINE
The 2010 recommendations add a degree of complexity, with different medications recommended on the basis of glucocorticoid dose and duration as well as patient age, menopausal status, and childbearing potential. Guideline developers and clinicians face a difficult trade-off: easy-to-follow guidelines or more targeted guidelines that are more complex and therefore more difficult to use than previous guidelines.
This criticism is reasonable. The complexity is a result of insufficient evidence from clinical trials to make more exact and user-friendly recommendations, and also a result of the RAND/UCLA methodology. In cases that lack sufficient evidence on which to make a decision, the guideline development uses voting among experts in an attempt to develop consensus. This often results in complexity, lack of consensus, or inconsistencies.
The guidelines are straightforward for postmenopausal women and men age 50 and older on at least 7.5 mg prednisone for more than 3 months.
Since there is substantial evidence that many patients on glucocorticoid therapy go untreated, the risk of fracture in this population would be substantially reduced if clinicians would adhere to the recommendations.
- Grossman JM, Gordon R, Ranganath VK, et al; American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken) 2010; 62:1515–1526.
- Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Arthritis Rheum 2001; 44:1496–1503.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Oral corticosteroids and fracture risk: relationship to daily and cumulative doses. Rheumatology (Oxford) 2000; 39:1383–1389.
- Saag KG, Emkey R, Schnitzer TJ, et al. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-Induced Osteoporosis Intervention Study Group. N Engl J Med 1998; 339:292–299.
- Cohen S, Levy RM, Keller M, et al. Risedronate therapy prevents corticosteroid-induced bone loss: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum 1999; 42:2309–2318.
- Reid DM, Hughes RA, Laan RF, et al. Efficacy and safety of daily risedronate in the treatment of corticosteroid-induced osteoporosis in men and women: a randomized trial. European Corticosteroid-Induced Osteoporosis Treatment Study. J Bone Miner Res 2000; 15:1006–1013.
- Wallach S, Cohen S, Reid DM, et al. Effects of risedronate treatment on bone density and vertebral fracture in patients on corticosteroid therapy. Calcif Tissue Int 2000; 67:277–285.
- Reid DM, Devogelaer JP, Saag K, et al; HORIZON investigators. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 2009; 373:1253–1263.
- Saag KG, Shane E, Boonen S, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 2007; 357:2028–2039.
- Curtis JR, Westfall AO, Allison JJ, et al. Longitudinal patterns in the prevention of osteoporosis in glucocorticoid-treated patients. Arthritis Rheum 2005; 52:2485–2494.
- Feldstein AC, Elmer PJ, Nichols GA, Herson M. Practice patterns in patients at risk for glucocorticoid-induced osteoporosis. Osteoporos Int 2005; 16:2168–2174.
- Brown JP, Josse RG; Scientific Advisory Council of the Osteoporosis Society of Canada. 2002 clinical practice guidelines for the diagnosis and management of osteoporosis in Canada. CMAJ 2002; 167(suppl 10):S1–S34.
- Devogelaer JP, Goemaere S, Boonen S, et al. Evidence-based guidelines for the prevention and treatment of glucocorticoid-induced osteoporosis: a consensus document of the Belgian Bone Club. Osteoporos Int 2006; 17:8–19.
- Gourlay M, Franceschini N, Sheyn Y. Prevention and treatment strategies for glucocorticoid-induced osteoporotic fractures. Clin Rheumatol 2007; 26:144–153.
- Nawata H, Soen S, Takayanagi R, et al; Subcommittee to Study Diagnostic Criteria for Glucocorticoid-Induced Osteoporosis. Guidelines on the management and treatment of glucocorticoid-induced osteoporosis of the Japanese Society for Bone and Mineral Research (2004). J Bone Miner Metab 2005; 23:105–109.
- Geusens PP, Lems WF, Verhaar HJ, et al. Review and evaluation of the Dutch guidelines for osteoporosis. J Eval Clin Pract 2006; 12:539–548.
- Kanis JA, Johnell O, Oden A, Johansson H, McCloskey E. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int 2008; 19:385–389.
- National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Washington, DC, National Osteoporosis Foundation, 2010. http://nof.org/files/nof/public/content/file/344/upload/159.pdf. Accessed December 31, 2012.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Guidance for the adjustment of FRAX according to the dose of glucocorticoids. Osteoporos Int 2011; 22:809–816.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation 2005; 112:e154–e235.
- Dawson-Hughes B, Tosteson AN, Melton LJ, et al; National Osteoporosis Foundation Guide Committee. Implications of absolute fracture risk assessment for osteoporosis practice guidelines in the USA. Osteoporos Int 2008; 19:449–458.
- Grossman JM, Gordon R, Ranganath VK, et al; American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken) 2010; 62:1515–1526.
- Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Arthritis Rheum 2001; 44:1496–1503.
- Compston J. Management of glucocorticoid-induced osteoporosis. Nat Rev Rheumatol 2010; 6:82–88.
- van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Oral corticosteroids and fracture risk: relationship to daily and cumulative doses. Rheumatology (Oxford) 2000; 39:1383–1389.
- Saag KG, Emkey R, Schnitzer TJ, et al. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-Induced Osteoporosis Intervention Study Group. N Engl J Med 1998; 339:292–299.
- Cohen S, Levy RM, Keller M, et al. Risedronate therapy prevents corticosteroid-induced bone loss: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum 1999; 42:2309–2318.
- Reid DM, Hughes RA, Laan RF, et al. Efficacy and safety of daily risedronate in the treatment of corticosteroid-induced osteoporosis in men and women: a randomized trial. European Corticosteroid-Induced Osteoporosis Treatment Study. J Bone Miner Res 2000; 15:1006–1013.
- Wallach S, Cohen S, Reid DM, et al. Effects of risedronate treatment on bone density and vertebral fracture in patients on corticosteroid therapy. Calcif Tissue Int 2000; 67:277–285.
- Reid DM, Devogelaer JP, Saag K, et al; HORIZON investigators. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 2009; 373:1253–1263.
- Saag KG, Shane E, Boonen S, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 2007; 357:2028–2039.
- Curtis JR, Westfall AO, Allison JJ, et al. Longitudinal patterns in the prevention of osteoporosis in glucocorticoid-treated patients. Arthritis Rheum 2005; 52:2485–2494.
- Feldstein AC, Elmer PJ, Nichols GA, Herson M. Practice patterns in patients at risk for glucocorticoid-induced osteoporosis. Osteoporos Int 2005; 16:2168–2174.
- Brown JP, Josse RG; Scientific Advisory Council of the Osteoporosis Society of Canada. 2002 clinical practice guidelines for the diagnosis and management of osteoporosis in Canada. CMAJ 2002; 167(suppl 10):S1–S34.
- Devogelaer JP, Goemaere S, Boonen S, et al. Evidence-based guidelines for the prevention and treatment of glucocorticoid-induced osteoporosis: a consensus document of the Belgian Bone Club. Osteoporos Int 2006; 17:8–19.
- Gourlay M, Franceschini N, Sheyn Y. Prevention and treatment strategies for glucocorticoid-induced osteoporotic fractures. Clin Rheumatol 2007; 26:144–153.
- Nawata H, Soen S, Takayanagi R, et al; Subcommittee to Study Diagnostic Criteria for Glucocorticoid-Induced Osteoporosis. Guidelines on the management and treatment of glucocorticoid-induced osteoporosis of the Japanese Society for Bone and Mineral Research (2004). J Bone Miner Metab 2005; 23:105–109.
- Geusens PP, Lems WF, Verhaar HJ, et al. Review and evaluation of the Dutch guidelines for osteoporosis. J Eval Clin Pract 2006; 12:539–548.
- Kanis JA, Johnell O, Oden A, Johansson H, McCloskey E. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int 2008; 19:385–389.
- National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. Washington, DC, National Osteoporosis Foundation, 2010. http://nof.org/files/nof/public/content/file/344/upload/159.pdf. Accessed December 31, 2012.
- Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum 2003; 48:3224–3229.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Guidance for the adjustment of FRAX according to the dose of glucocorticoids. Osteoporos Int 2011; 22:809–816.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation 2005; 112:e154–e235.
- Dawson-Hughes B, Tosteson AN, Melton LJ, et al; National Osteoporosis Foundation Guide Committee. Implications of absolute fracture risk assessment for osteoporosis practice guidelines in the USA. Osteoporos Int 2008; 19:449–458.
KEY POINTS
- The risk of fracture should be assessed at the start of glucocorticoid therapy.
- Factors that affect the decision to prescribe osteoporosis drugs include the patient’s risk of fractures as assessed with FRAX (www.shef.ac.uk/FRAX), the dose of glucocorticoid, and the projected duration of treatment.
- Since FRAX treats glucocorticoid use simply as a yes-or-no question, it likely underestimates the fracture risk in current users and at high doses. The estimate of risk should be adjusted upward in these situations.
Statins and diabetes risk: Fact, fiction, and clinical implications
On february 28, 2012, the US Food and Drug Administration (FDA) updated its labeling requirements for statins. In addition to revising its recommendations for monitoring liver function and its alerts about reports of memory loss, the FDA also warned of the possibility of new-onset diabetes mellitus and worse glycemic control in patients taking statin drugs.1
This change stoked an ongoing debate about the risk of diabetes with statin use and the implications of such an effect. To understand the clinical consequences of this alert and its effect on treatment decisions, we need to consider the degree to which statins lower the risk of cardiovascular disease in patients at high risk (including diabetic patients), the magnitude of the risk of developing new diabetes while on statin therapy, and the ratio of risk to benefit in treated populations.
This review will discuss the evidence for this possible adverse effect and the implications for clinical practice.
DO STATINS CAUSE DIABETES?
Individual controlled trials dating back more than a decade have had conflicting results about new diabetes and poorer diabetic control in patients taking statins.
The West of Scotland Coronary Prevention Study (WOSCOPS)2 suggested that the incidence of diabetes was 30% lower in patients taking pravastatin (Pravachol) 40 mg/day than with placebo. However, this was not observed with atorvastatin (Lipitor) 10 mg/day in the Anglo-Scandinavian Cardiac Outcomes Trial–Lipid-Lowering Arm (ASCOT-LLA)3 in hypertensive patients or in the Collaborative Atorvastatin Diabetes Study (CARDS)4 in diabetic patients,4 nor was it noted with simvastatin (Zocor) 40 mg/day in the Heart Protection Study (HPS).5
The Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER),6 using the more potent agent rosuvastatin (Crestor) 20 mg/day in patients with elevated levels of C-reactive protein (CRP), was stopped early when an interim analysis found a 44% lower incidence of the primary end point. However, the trial also reported a 26% higher incidence of diabetes in follow-up of less than 2 years.
In the Prospective Study of Pravastatin in the Elderly at Risk (PROSPER),7 with a mean age at entry of 75, there was a 32% higher incidence of diabetes with pravastatin therapy.7
Results of meta-analyses
Several meta-analyses have addressed these differences.
Rajpathak et al8 performed a meta-analysis, published in 2009, of six trials—WOSCOPS,2 ASCOT-LLA,3 JUPITER,6 HPS,5 the Long-term Intervention With Pravastatin in Ischaemic Disease (LIPID) study,9 and the Controlled Rosuvastatin Multinational Study in Heart Failure (CORONA),10 with a total of 57,593 patients. They calculated that the incidence of diabetes was 13% higher (an absolute difference of 0.5%) in statin recipients, which was statistically significant. In their initial analysis, the authors excluded WOSCOPS, describing it as hypothesis-generating. The relative increase in risk was less—6%—and was not statistically significant when WOSCOPS was included.
Sattar et al,11 in a larger meta-analysis published in 2010, included 91,140 participants in 13 major statin trials conducted between 1994 and 2009; each trial had more than 1,000 patients and more than 1 year of follow-up.2,3,5–7,9,10,12–17 New diabetes was defined as physician reporting of new diabetes, new diabetic medication use, or a fasting glucose greater than 7 mmol/L (126 mg/dL).
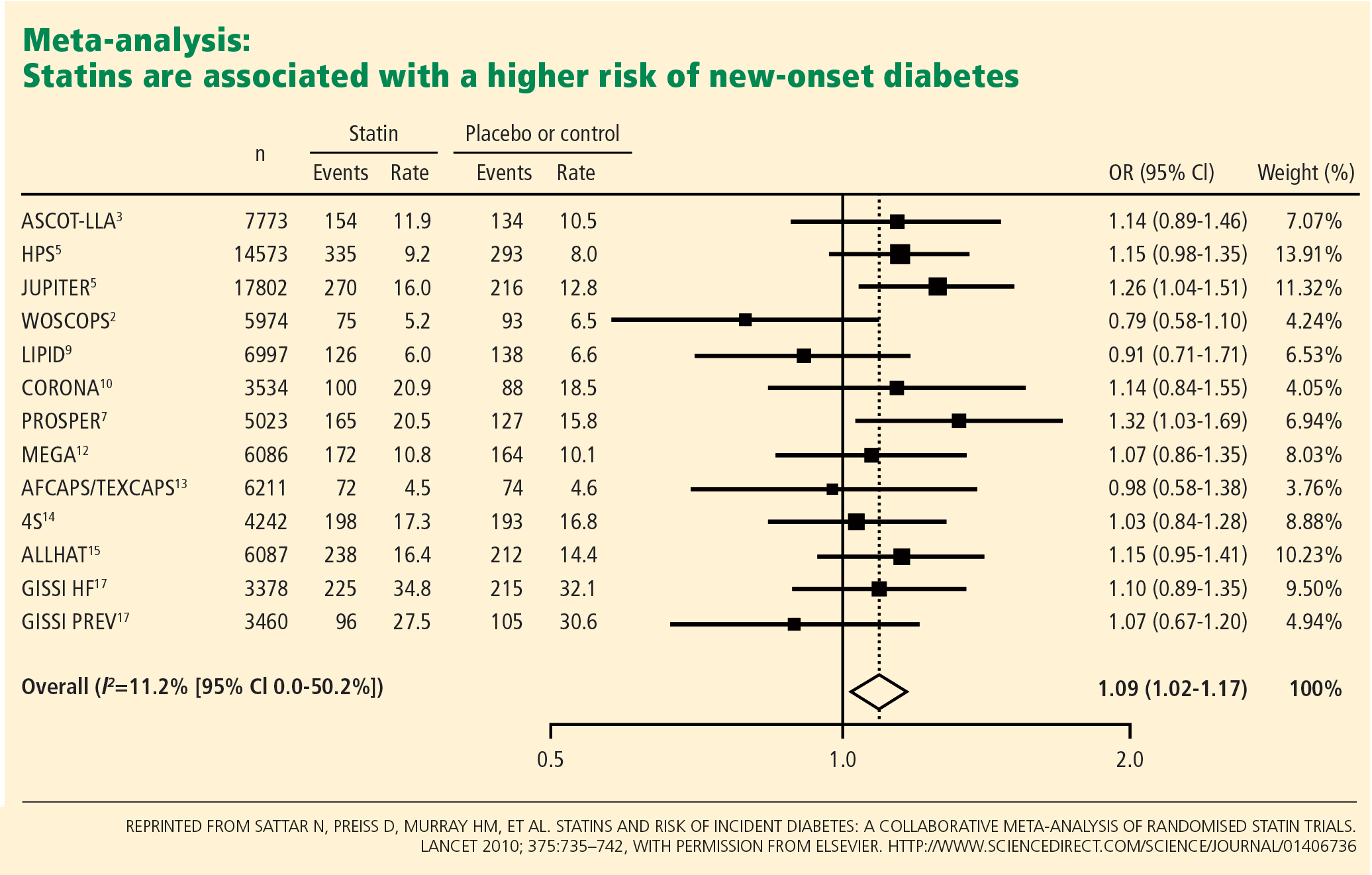
New diabetes occurred in 2,226 (4.89%) of the statin recipients and in 2,052 (4.5%) of the placebo recipients, an absolute difference of 0.39%, or 9% more (odds ratio [OR] 1.09; 95% confidence interval [CI] 1.02–1.17) (Figure 1).
The incidence of diabetes varied substantially among the 13 trials, with only JUPITER6 and PROSPER7 finding statistically significant increases in rates (26% and 32%, respectively). Of the other 11 trials, 4 had nonsignificant trends toward lower incidence,2,9,13,17 while the 7 others had nonsignificant trends toward higher incidence.
Does the specific statin make a difference?
Questions have been raised as to whether the type of statin used, the intensity of therapy, or the population studied contributed to these differences. Various studies suggest that factors such as using hydrophilic vs lipophilic statins (hydrophilic statins include pravastatin and rosuvastatin; lipophilic statins include atorvastatin, lovastatin, and simvastatin), the dose, the extent of lowering of low-density lipoprotein cholesterol (LDL-C), and the age or clinical characteristics of the population studied may influence this relationship.18–20
Yamakawa et al18 examined the effect of atorvastatin 10 mg/day, pravastatin 10 mg/day, and pitavastatin (Livalo) 2 mg/day on glycemic control over 3 months in a retrospective analysis. Random blood glucose and hemoglobin A1c levels were increased in the atorvastatin group but not in the other two.18
A prospective comparison of atorvastatin 20 mg vs pitavastatin 4 mg in patients with type 2 diabetes, presented at the American College of Cardiology’s 2011 annual meeting, reported a significant increase in fasting glucose levels with atorvastatin, particularly in women, but not with pitavastatin.19
In the Compare the Effect of Rosuvastatin With Atorvastatin on Apo B/Apo A-1 Ratio in Patients With Type 2 Diabetes Mellitus and Dyslipidaemia (CORALL) study,20 both high-dose rosuvastatin (40 mg) and high-dose atorvastatin (80 mg) were associated with significant increases in hemoglobin A1c, although the mean fasting glucose levels were not significantly different at 18 weeks of therapy.
A meta-analysis by Sattar et al11 did not find a clear difference between lipophilic statins (OR 1.10 vs placebo) and hydrophilic statins (OR 1.08). In analysis by statin type, the combined rosuvastatin trials were statistically significant in favor of a higher diabetes risk (OR 1.18, 95% CI 1.04–1.44). Nonsignificant trends were noted for atorvastatin trials (OR 1.14) and simvastatin trials (OR 1.11) and less so for pravastatin (OR 1.03); the OR for lovastatin was 0.98. This may suggest that there is a stronger effect with more potent statins or with greater lowering of LDL-C.
Meta-regression analysis in this study demonstrated that diabetes risk with statins was higher in older patients but was not influenced by body mass index or by the extent that LDL-C was lowered.
Statin dose as a risk factor
Intensive-dose statin therapy has been shown to reduce cardiovascular risk more than low-dose or moderate-dose therapy, thus supporting more aggressive treatment of LDL-C in higher-risk patients. However, some controlled studies comparing more-potent with less-potent statin regimens suggest that there may also be a higher risk of incident diabetes at higher doses.21–24
In a post hoc analysis of the Pravastatin or Atorvastatin Evaluation and Infection Therapy– Thrombolysis in Myocardial Infarction 22 (PROVE-IT TIMI 22) trial,21 patients who had experienced an acute coronary syndrome had a greater increase in hemoglobin A1c if treated with atorvastatin 80 mg/day than with pravastatin 40 mg/day.
Waters et al23 reported a higher risk of new diabetes with atorvastatin 80 mg than with placebo and a trend toward a higher risk with atorvastatin 80 mg than with atorvastatin 10 mg or simvastatin 20 mg.
In contrast, a review by Yousef et al24 of the data from the Enhanced Feedback for Effective Cardiac Treatment (EFFECT) study did not find a higher diabetes risk with more intensive statin therapy based on the magnitude of LDL-C reduction. A propensity-matched examination of deaths, recurrent acute ischemic events, or new diabetes in patients previously hospitalized with myocardial infarction found no differences in these end points each year out to 5 years. The risk of diabetes was in fact lower (but the difference was not statistically significant) in the high-dose groups out to 5 years. The risk of myocardial infarction or death was numerically different in the high-dose groups, but the difference was not statistically significant.
Preiss et al25 in 2011 performed a meta-analysis of the impact of intensity of statin therapy on diabetes risk. They examined data from 32,752 participants without diabetes at baseline in five randomized controlled trials with more than 1,000 participants and more than 1 year of follow-up, comparing high-dose therapy against moderate-dose statin therapy.21,22,26–28 New diabetes was considered present if there was an adverse event report of diabetes, if glucose-lowering drugs were started, or if two fasting plasma glucose measurements were higher than 7 mmol/L (126 mg/dL).
Diabetes developed in 1,449 (8.8%) of the intensive-therapy group and 1,300 (8.0%) of the moderate-therapy group (OR 1.12, 95% CI 1.04–1.22). In contrast, incident cardiovascular disease occurred in 3,134 (19.1%) of the intensive-therapy group and 3,550 (21.7%) of the moderate-therapy group (OR 0.84, 95% CI 0.75–0.94). Therefore, there was an 0.8% absolute increase in diabetes cases on high-dose statins and a 2.6% absolute reduction in adverse cardiovascular events.
CAUTION IN INTERPRETING THESE DATA
There are many reasons for caution in interpreting these studies.
The trials were not designed to look for diabetes
The data supporting the relationship between statin therapy and higher risk of diabetes are primarily from observational studies. These studies were not prospectively designed to address this question, and we therefore need to view this as association and not as causation.
The definition of diabetes varied between trials, and new-onset diabetes was often not rigorously screened for. In many trials the outcome of diabetes was at least partially based on nonstandardized, nonadjudicated physician reporting.
Consequently, if statins reduce the risk of diabetes, the results from WOSCOPS may overstate the reduction, since this study used a non-standard definition of incident diabetes (fasting plasma glucose > 126 mg/dL plus a > 36 mg/dL increase from baseline). When Sattar et al11 reanalyzed WOSCOPS data using a more standard definition, they found a smaller effect.
On the other hand, nonstandardized physician reporting may overstate an adverse effect. Sattar et al11 also found that when fasting plasma glucose levels alone were used as the definition for diabetes, the overall risk was attenuated and was no longer statistically significant (OR 1.07, 95% CI 0.97–1.17).
Perhaps statin therapy uncovers diabetes only in people at risk of diabetes
Perhaps statin therapy uncovers diabetes only in people at higher baseline risk of developing diabetes. Therefore, this adverse effect may be restricted to certain groups and not applicable to the general population.
In JUPITER, one of the two trials in which, on independent analysis, statin use was associated with new diabetes, 77% of patients in the rosuvastatin group who developed diabetes had impaired fasting glucose at entry and therefore were at higher risk of developing diabetes.6
Possibly, the relationship is driven by preexisting metabolic syndrome or other risk factors for diabetes. In the two studies that reported a statistically significantly higher incidence of new diabetes, more than 40% of patients in JUPITER met the criteria for metabolic syndrome, and metabolic syndrome, which increases in prevalence with age, was likely more prominent in the elderly population in PROSPER.
Waters et al23 grouped patients according to whether they had risk factors for diabetes (impaired fasting glucose, obesity, elevated triglycerides, and hypertension) and found that those who had none or one of these risk factors had no difference in the rate of new-onset diabetes with either moderate or intensive statin therapy, but the risk was pronounced in those who had three or four risk factors.
Ridker et al29 reanalyzed the JUPITER data from patients who did not have cardiovascular disease at baseline. Overall, for every 54 new cases of diabetes in follow-up, 134 cardiovascular events or deaths were prevented. In subgroup analysis, those who had one or more risk factors for diabetes at baseline (metabolic syndrome, impaired fasting glucose, obesity, or hemoglobin A1c > 6%) had a 39% reduction in the primary end point and a 28% increase in new diabetes. Those who had none of these risk factors had a 52% lower rate of cardiovascular events but no increase in diabetes.
Other confounding factors
Bias and confounding factors are difficult to control for in studies without prospectively defined, recognized, and analyzed outcomes.
Although it may be a bit of a stretch, residual confounding factors such as myalgia side effects while on statins may reduce exercise in the statin-treatment groups. Perhaps a change to a healthier lifestyle after cardiovascular events may be more common in placebo groups. Improved survival with statins may allow more people at risk of diabetes to live longer and present with the diagnosis.30
POSSIBLE EXPLANATIONS, BUT NO UNIFYING MECHANISM
If mechanisms could be identified to explain the association between statins and diabetes, this would strengthen the argument that it is a cause-and-effect relationship. Many explanations have been proposed as to how statins may influence glucose metabolism and insulin sensitivity.31–34 These are possible explanations based on other observations.
In theory, statins may improve insulin sensitivity via their anti-inflammatory effect, since inflammatory markers and proinflammatory cytokines have been linked with insulin resistance. However, other effects of statins may adversely affect glycemic control.
In vivo analysis has shown that some but not all statins increase insulin levels and decrease insulin sensitivity in a dose-dependent fashion. Some statins decrease adiponectin and may worsen glycemic control through loss of adiponectin’s proposed protective anti-proliferative and antiangiogenic properties. In vitro studies and animal studies have demonstrated a decrease in expression of insulin-responsive glucose transporter 4 (GLUT4) with atorvastatin, and an increase in GLUT1. It has been hypothesized that reduction in isoprenoid biosynthesis or decreased insulin signaling may explain these effects and that changes in glucose transport in adipocytes may cause insulin resistance. Other studies suggest that dysregulation of cellular cholesterol may attenuate beta-cell function. Impaired biosynthesis of ubiquinones may result in delayed production of adenosine triphosphate and consequently diminish insulin release.
But different effects have been reported for atorvastatin, simvastatin, and pravastatin, arguing against a unifying explanation or, alternatively, suggesting that differences in lipophilicity and potency among statins are important. Hydrophilic statins may be less likely to be taken up by extrahepatic cells such as pancreatic cells and adipocytes, possibly lessening these effects. However, the strong association between rosuvastatin (which is hydrophilic) and new diabetes would not support this hypothesis.
Despite these speculations, lack of conformity in response to different statins and discrepancies in the clinical outcomes noted in trials fail to clearly identify a common causative mechanism.
OTHER COMMON THERAPIES MAY INFLUENCE GLYCEMIC CONTROL
Statins are not the first drugs for reducing cardiovascular risk that have been shown to affect glucose levels during treatment.
Niacin
Niacin has been known to increase glucose levels but has long been used as a treatment for dyslipidemia despite this caution. Reduced glycemic control during niacin treatment in diabetic patients does not seem to alter the beneficial effects of treatment.35–37
In a post hoc analysis of the Coronary Drug Project (CDP), in patient subgroups defined by baseline fasting plasma glucose and compared with placebo, niacin reduced the 6-year risk of recurrent myocardial infarction and the combined end point of coronary heart disease death or nonfatal myocardial infarction similarly (interactive P value nonsignificant) across all levels of baseline fasting plasma glucose, including levels of 126 mg/dL or higher at study entry.36
In another post hoc analysis of CDP patient subgroups defined by the change in glycemic status from baseline to 1 year, niacin reduced the 6-year risk of the same end points similarly (interactive P value nonsignificant) across all levels of change in fasting plasma glucose from baseline to year 1, whether baseline fasting plasma glucose levels decreased, stayed the same, or increased to 10 mg/dL or higher on niacin therapy.36
Therefore, the beneficial effect of niacin of reducing the rate of recurrent nonfatal myocardial infarction and coronary heart disease events was not significantly diminished when impaired fasting glucose or diabetes was present when therapy was started or by on-therapy increases from baseline fasting plasma glucose.
In addition, on-therapy changes in glycemic control may be dose-related and minimized by surveillance and therapy adjustments. The Assessment of Diabetes Control and Evaluation of the Efficacy of Niaspan Trial (ADVENT)38 found that changes in glycemic control were minimal as measured by fasting glucose and hemoglobin A1c; were associated with a higher niacin dose (1.5 g/day vs 1 g/day); and, when present, were successfully managed by adjusting the diabetes treatment regimen.
Antihypertensive drugs
Diuretics as well as beta-blockers have been reported to increase the incidence of diabetes in patients with hypertension.15,38–40
A retrospective longitudinal cohort study40 in 2009 examined the development of new-onset diabetes (defined as a new ICD-9 code for diabetes or initiation of diabetes treatment) in 24,688 treated hypertensive patients without diabetes at baseline; 4,385 (17.8%) of the patients developed diabetes. After adjusting for sex and age, the risk of new diabetes was significant in users of diuretics (OR 1.10), beta-blockers (OR 1.12), and calcium channel blockers (OR 1.10) compared with users of angiotensin-converting enzyme inhibitors, (OR 0.92), angiotensin receptor blockers (OR 0.90), or alpha-blockers (OR 0.88).
However, the increase in blood glucose does not seem to attenuate the beneficial effects of reducing cardiovascular events. In the Antihypertensive and Lipid-lowering Treatment to Prevent Heart Attack Trial (ALLHAT),15 a long-term follow-up of those developing new-onset diabetes while taking chlorthalidone (Hygroton) found no difference in the risk of death from cardiovascular disease or from any cause (hazard ratio = 1.04).15
CLINICAL IMPLICATIONS
Balancing the benefits and risks of statins
It is important to examine how the 0.4% increase in absolute risk of new-onset diabetes as calculated in meta-analyses compares with the benefits of statin treatment in terms of cardiovascular risk reduction.
Using data from the Cholesterol Treatment Trialists (CTT) meta-analysis of statin trials in 71,370 participants, Sattar et al11 estimated that statin treatment is associated with 5.4 fewer deaths from coronary heart disease and cases of nonfatal myocardial infarction per 255 patients treated over 4 years for each 1-mmol/L (39 mg/dL) reduction in LDL-C compared with controls. The benefit would be even greater if stroke, revascularization, and hospitalization are included as end points. This benefit is contrasted with the risk of developing 1 additional case of diabetes for every 255 patients treated with statins over the same period.
Preiss et al25 calculated that there were 2 more cases of diabetes per 1,000 patient-years in patients receiving intensive doses than in those receiving moderate doses (18.9 vs 16.9), corresponding to 1 additional case of diabetes for every 498 patients treated per year. However, there were 6.5 fewer first major cardiovascular events per 1,000 patient-years (44.5 vs 51.0), corresponding to a number needed to treat per year to prevent 1 cardiovascular event of 155. Most of the benefit was due to fewer revascularizations, followed by nonfatal myocardial infarctions. The 12% increase in new diabetes with high-dose therapy contrasted with a 16% reduction in new cardiovascular disease combined events (OR 0.84, 95% CI 0.75–0.94).
As previously noted, in the JUPITER trial, the benefits of preventing cardiovascular events with statin therapy outweighed the risk of new diabetes in people both with and without baseline risk factors for diabetes.29 Similar to the observations with niacin and some antihypertensive drugs, the increase in blood glucose with statins does not appear to reduce the benefits of cardiovascular risk reduction in these patients at moderate to high risk, even when used at high doses.
People with diabetes need aggressive lipid-lowering—with statins
Diabetes is a coronary heart disease risk equivalent and is associated with high risk of cardiovascular events.41–46 Overall, the risk for these adverse events is two to four times greater in people with diabetes than without. Atherosclerosis-related events account for approximately 65% to 75% of all deaths in people with diabetes, and 75% of these events are coronary. Lipid abnormalities are strongly correlated with the risk of cardiovascular disease in people with diabetes, and aggressive treatment of risk factors, particularly lipid abnormalities, has been shown to reduce this risk.47–49 And data from multiple clinical trials support the use of statins to lower LDL-C as the first-line therapy for dyslipidemia in people with diabetes, just as it is in the general population.3–7,9,13,23,50–61
Analyses of diabetic subgroups encompassing 18,000 to 20,000 patients in the large statin trials have clearly demonstrated the benefits of statin therapy. A recent metaanalysis of 10 placebo-controlled trials that included approximately 16,000 patients with diabetes and 54,000 without diabetes demonstrated a 30% reduction in coronary heart disease, a 19% reduction in strokes, and a 12% reduction in mortality.54 Furthermore, in another meta-analysis of 14 trials, a similar 22% reduction in coronary heart disease was noted in people with diabetes whether or not they had a history of cardiovascular disease.55
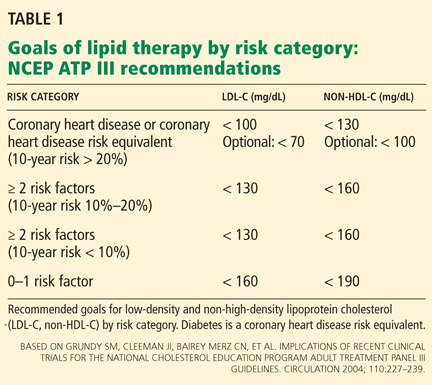
Therefore, aggressive treatment of lipid abnormalities with statins as primary treatment has generally been adopted as a standard of care in diabetic patients, particularly those with clinical cardiovascular disease or one or more risk factors. The Adult Treatment Panel III guidelines recommend a minimum LDL-C goal of less than 100 mg/dL and a goal of less than 70 mg/dL as an option for patients with diabetes (Table 1).41,62 Similar recommendations have been issued by the American Diabetes Association together with the American College of Cardiology (Table 2),30 the American Diabetes Association by itself,63 and the American Academy of Pediatrics.6
Is new-onset diabetes as dangerous as established diabetes?
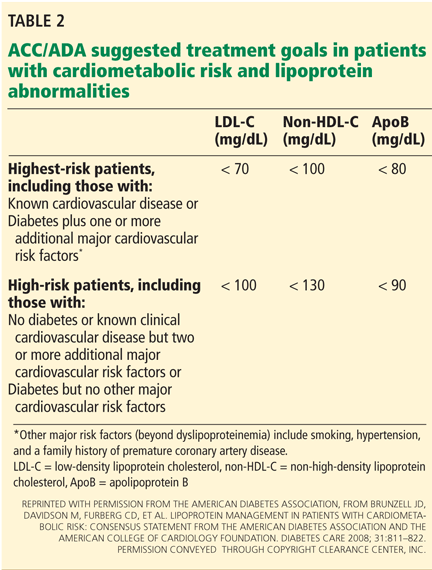
In studies to date, there did not appear to be more events in those who developed new-onset diabetes.
Waters et al,24 evaluating three trials of high-dose atorvastatin therapy, found that major cardiovascular events occurred in 11.3% of those with new-onset diabetes, 10.8% of those without new-onset diabetes (HR 1.02, 95% CI 0.77–1.35), and 17.5% of those who had diabetes at baseline.
Therefore, it may not be appropriate to extrapolate the glucose changes seen on statin therapy to an equivalent increase in adverse cardiovascular events as seen in other diabetic patients. The beneficial reduction in cardiovascular events does not appear to be diminished in those developing diabetes. It is not clear that the increase in glucose on statins has the same implications of a new diagnosis of diabetes. Does this elevation in glucose represent true diabetes or some downstream effect? For example, thiazide diuretics have been known to increase blood glucose levels, but the levels drop when these drugs are discontinued, even after many years of treatment.
On the other hand, it is possible that follow-up of 5 years or less in clinical trials has not allowed sufficient time to examine the influence of the increase in new-onset diabetes on future cardiovascular events. In addition, because of the widespread use of statins across a broad range of cardiovascular risk, even if the effect is small in absolute terms, the potential adverse effects are magnified, particularly in a low-risk population in which the cardiovascular benefits are smaller.
The association is real, but questions remain
In view of the evidence, it is difficult to refute that an association exits between statin use and new-onset diabetes, at least in some subgroups. The dose response noted in some studies further reinforces the conclusion that the association is real. However, many questions remain unanswered regarding mechanism of effect, whether there are differences depending on the particular statin or dose used, or differential effects in the populations treated (such as patients with metabolic syndrome or the elderly).
Until the contradictory observations can be resolved and plausible mechanisms of action elucidated, causality cannot be established. From a clinical standpoint there is no current evidence suggesting that the elevations in blood glucose seen while on lipid-lowering or blood-pressure-lowering therapy are associated with an increased risk of cardiovascular events or that they attenuate the beneficial effects of the therapy.
Statins should continue to be used in patients at high risk
Until further studies are done, statins should continue to be used, after assessing the risks and the benefits.
Primary prevention patients at moderate to high risk and secondary prevention patients stand to gain from statin therapy, and it should not be denied or doses reduced on the basis of concerns about the development of new-onset diabetes. The recognized modest risk of developing diabetes does not appear to blunt the cardioprotective effects of statin therapy in these moderate-to high-risk groups.
Rather than stop statins in patients at risk of diabetes such as the elderly or those with prediabetes, insulin resistance, or metabolic syndrome who are on therapy for appropriate reasons, it is reasonable to continue these drugs, monitoring glucose more closely and emphasizing the importance of weight reduction, diet, and aerobic exercise for preventing diabetes. The Diabetes Prevention Program Research Group, for example, reduced the incidence of diabetes by 58% over 2.8 years of follow-up with intensive lifestyle interventions (a low-calorie, low-fat diet plus moderate physical activity 150 minutes per week) vs usual care in at-risk populations.65
Should statins be used more cautiously in patients at lower risk?
The most recent Cholesterol Treatment Trialists meta-analysis of 27 randomized clinical trials (22 placebo-controlled, 134,537 people; 5 high-dose vs low-dose, 39,612 people) reported that reducing LDL-C with statins lowered cardiovascular risk even in low-risk patients.66 Overall, there were 21% fewer major cardiovascular events (coronary heart disease, stroke, or coronary revascularization) for every 1-mmol/L reduction in LDL-C.
The proportional reduction in events was at least as large in the two lowest-risk groups (estimated 5-year risk of < 5% and 5% to < 10%, 53,152 people) as in the higher-risk groups. This was reflected mainly in fewer nonfatal myocardial infarctions and coronary revascularizations. In these groups, the absolute reduction in risk for each 1-mmol/L reduction in LDL-C was 11 per 1,000 patients over 5 years. Even in this low-risk population, the reduction in cardiovascular risk seems to compare favorably with the small estimated increase risk of diabetes.
However, even in the lowest-risk group studied, the average baseline LDL-C level was greater than 130 mg/dL.
Therefore, in groups in which the benefits of statins on cardiovascular risk reduction are less robust (eg, low-risk primary prevention groups without significant elevations in LDLC, particularly the elderly), it would not be difficult to justify the case for more cautious use of statin therapy. If statins are used in these low-risk groups, restricting their use to those with at least moderate LDL-C elevation, using less aggressive LDL-C-lowering targets, and regular monitoring of fasting glucose seem reasonable until further information is available.
- US Food and Drug Administration. Statin drugs—drug safety communication: class labeling change. February 28, 2012. http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm293670.htm.
- Freeman DJ, Norrie J, Sattar N, et al. Pravastatin and the development of diabetes mellitus: evidence for a protective treatment effect in the West of Scotland Coronary Prevention Study. Circulation 2001; 103:357–362.
- Sever PS, Dahlof B, Poulter NR, et al; ASCOT investigators. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial–Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet 2003; 361:1149–1158.
- Colhoun HM, Betteridge DJ, Durrington PN, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet 2004; 364:685–696.
- Collins R, Armitage J, Parish S, Sleigh P, Peto R; for the Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet 2003; 361:2005–2016.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Shepherd J, Blauw GJ, Murphy MB, et al; PROSPER Study Group. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002; 360:1623–1630.
- Rajpathak SN, Kumbhani DJ, Crandall J, Barzilai N, Alderman M, Ridker PM. Statin therapy and risk of developing type 2 diabetes: a meta-analysis. Diabetes Care 2009; 32:1924–1929.
- Keech A, Colquhoun D, Best J, et al. Secondary prevention of cardiovascular events with long-term pravastatin in patients with diabetes or impaired fasting glucose—results from the LIPID trial. Diabetes Care 2003; 26:2713–2721.
- Kjekshus J, Apetrei E, Barrios V, et al. Rosuvastatin in older patients with systolic heart failure. N Engl J Med 2007; 357:2248–2261.
- Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 2010; 375:735–742.
- Nakamura H, Arakawa K, Itakura H, et al. Primary prevention of cardiovascular disease with pravastatin in Japan (MEGA Study): a prospective randomised controlled trial. Lancet 2006; 368:1155–1163.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Scandinavian Simvastatin Survival Study study group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- Barzilay JI, Davis BR, Pressel SL, et al; ALLHAT Collaborative Research Group. Long-term effects of incident diabetes mellitus on cardiovascular outcomes in people treated for hypertension: the ALLHAT Diabetes Extension Study. Circ Cardiovasc Qual Outcomes 2012; 5:153–162.
- Tavazzi L, Maggioni AP, Marchioli R, et al. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet 2008; 372:1231–1239.
- GISSI Prevenzione Investigators (Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico). Results of the low-dose (20 mg) pravastatin GISSI Prevenzione trial in 4271 patients with recent myocardial infarction: do stopped trials contribute to overall knowledge? Ital Heart J 2000; 1:810–820.
- Yamakawa T, Takano T, Tanaka S, Kadonosono K, Terauchi Y. Influence of pitavastatin on glucose tolerance in patients with type 2 diabetes mellitus. J Atheroscler Thromb 2008; 15:269–275.
- Kryzhanovski V, Gumprecht J, Zhu B, Yu CY, Hounslow N, Sponseller CA. Atorvastatin but not pitavastatin significantly increases fasting plasma glucose in patients with type 2 diabetes and combined dyslipidemia (abstract). J Am Coll Cardiol 2011; 57:E575.
- Simsek S, Schalkwijk CG, Wolffenbuttel BH. Effects of rosuvastatin and atorvastatin on glycaemic control in type 2 diabetes—the CORALL study. Diabet Med 2012; 29:628–631.
- Sabatine MS, Morrow DA, Giugliano RP, et al. Implications of upstream glycoprotein IIb/IIIa inhibition and coronary artery stenting in the invasive management of unstable angina/non-ST-elevation myocardial infarction: a comparison of the Thrombolysis In Myocardial Infarction (TIMI) IIIB trial and the Treat angina with Aggrastat and determine Cost of Therapy with Invasive or Conservative Strategy (TACTICS)-TIMI 18 trial. Circulation 2004; 110(suppl III):834–880.
- Shepherd J, Barter P, Carmena R, et al. Effect of lowering LDL cholesterol substantially below currently recommended levels in patients with coronary heart disease and diabetes: the Treating to New Targets (TNT) study. Diabetes Care 2006; 29:1220–1226.
- Waters DD, Ho JE, DeMicco DA, et al. Predictors of new-onset diabetes in patients treated with atorvastatin: results from 3 large randomized clinical trials. J Am Coll Cardiol 2011; 57:1535–1545.
- Yousef A, Tu JV, Wang J, Donovan L, Ko DT. The association of intensive statin therapy on long-term risks of cardiovascular events and diabetes following acute myocardial infarction (abstract). Circulation 2012; 125:e859.
- Preiss D, Seshasai SR, Welsh P, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a metaanalysis. JAMA 2011; 305:2556–2564.
- de Lemos JA, Blazing MA, Wiviott SD, et al; A to Z Investigators. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- Pedersen TR, Faegeman O, Kastelein JJ, et al; Incremental Decrease in End Points Through Aggressive Lipid Lowering (IDEAL) Study Group. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Armitage J, Bowman L, Wallendszus K, et al; Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet 2010; 37:1658–1669.
- Ridker PM, Pradhan A, MacFadyen JG, Libby P, Glynn RJ. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: an analysis from the JUPITER trial. Lancet 2012; 380:565–571.
- Brunzell JD, Davidson M, Furberg CD, et al; American Diabetes Association; American College of Cardiology Foundation. Lipoprotein management in patients with cardiometabolic risk: consensus statement from the American Diabetes Association and the American College of Cardiology Foundation. Diabetes Care 2008; 31:811–822.
- Koh KK, Quon MJ, Han SH, Lee Y, Kim SJ, Shin EK. Atorvastatin causes insulin resistance and increases ambient glycemia in hypercholesterolemic patients. J Am Coll Cardiol 2010; 55:1209–1216.
- Koh KK, Quon MJ, Han SH, et al. Differential metabolic effects of pravastatin and simvastatin in hypercholesterolemic patients. Atherosclerosis 2009; 204:483–490.
- Nakata M, Nagasaka S, Kusaka I, Matsuoka H, Ishibashi S, Yada T. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): implications in glycaemic control. Diabetologia 2006; 49:1881–1892.
- Yada T, Nakata M, Shiraishi T, Kakei M. Inhibition by simvastatin, but not pravastatin, of glucose-induced cytosolic Ca2+ signalling and insulin secretion due to blockade of L-type Ca2+ channels in rat islet beta-cells. Br J Pharmacol 1999; 126:1205–1213.
- Guyton JR, Fazio S, Adewale AJ, et al. Effect of extended-release niacin on new-onset diabetes among hyperlipidemic patients treated with ezetimibe/simvastatin in a randomized controlled trial. Diabetes Care 2012; 35:857–860.
- Canner PL, Furberg CD, Terrin ML, McGovern ME. Benefits of niacin by glycemic status in patients with healed myocardial infarction (from the Coronary Drug Project). Am J Cardiol 2005; 95:254–257.
- Grundy SM, Vega GL, McGovern ME, et al; Diabetes Multicenter Research Group. Efficacy, safety, and tolerability of once-daily niacin for the treatment of dyslipidemia associated with type 2 diabetes: results of the Assessment of Diabetes Control and Evaluation of the Efficacy of Niaspan Trial. Arch Intern Med 2002; 162:1568–1576.
- Gupta AK, Dahlof B, Dobson J, Sever PS, Wedel H, Poulter NRAnglo-Scandinavian Cardiac Outcomes Trial Investigators. Determinants of new-onset diabetes among 19,257 hypertensive patients randomized in the Anglo-Scandinavian Cardiac Outcomes Trial—Blood Pressure Lowering Arm and the relative influence of antihypertensive medication. Diabetes Care 2008; 31:982–988.
- Elliott WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet 2007; 369:201–207.
- Jong JP, Chang MH, Tien L, et al. Antihypertensive drugs and new-onset diabetes: a retrospective longitudinal cohort study. Cardiovasc Ther 2009; 27:159–163.
- Third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Norhammar A, Tenerz A, Nilsson G, et al. Glucose metabolism in patients with acute myocardial infarction and no previous diagnosis of diabetes mellitus: a prospective study Lancet 2002; 359:2140–2144.
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Sprafka JM, Burke GL, Folsom AR, McGovbern PG, Hahn LP. Trends in prevalence of diabetes mellitus in patients with myocardial infarction and effect of diabetes on survival. The Minnesota Heart Survey. Diabetes Care 1991; 14:537–543.
- Geiss LS, Herman WH, Smith PJ. Mortality in non-insulin-dependent diabetes. In:Harris MI, Cowie CC, Stern MP, et al, editors. Diabetes in America. 2nd ed. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, 1995:233–257.
- Stamler J, Vaccaro O, Neaton JD, Wentworth D. Diabetes, other risk factors, and 12-yr cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial. Diabetes Care 1993; 16:434–444.
- Turner RC, Millns H, Neil HA, et al. Risk factors for coronary artery disease in non-insulin dependent diabetes mellitus: United Kingdom Prospective Diabetes Study (UKPDS: 23). BMJ 1998; 316:823–828.
- Gaede P, Vedel P, Larsen N, Jensen GV, Parving HH, Pedersen O. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Engl J Med 2003; 348:383–393.
- Gaede P, Pederson O. Intensive integrated therapy of type 2 diabetes: implications for long-term prognosis. Diabetes 2004; 53:S39–S47.
- Goldberg RB, Mellies MJ, Sacks FM, et al. Cardiovascular events and their reduction with pravastatin in diabetic and glucose-intolerant myocardial infarction survivors with average cholesterol levels: subgroup analyses in the cholesterol and recurrent events (CARE) trial. The Care Investigators. Circulation 1998; 98:2513–2519.
- Pyðrälä K, Pedersen TR, Kjekshus J, Faergeman O, Olsson AG, Thorgeirsson G. Cholesterol lowering with simvastatin improves prognosis of diabetic patients with coronary heart disease. A subgroup analysis of the Scandinavian Simvastatin Survival Study (4S). Diabetes Care 1997; 20:614–620.
- Vijan S, Hayward RA; American College of Physicians. Pharmacologic lipid-lowering therapy in type 2 diabetes mellitus: background paper for the American College of Physicians. Ann Intern Med 2004; 140:650–658.
- Baigent C, Keech A, Kearney PM, et al; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278. Errata in Lancet 2008; 371:2084, Lancet 2005; 366:1358.
- Brugts JJ, Yetgin T, Hoeks SE, et al. The benefits of statins in people without established cardiovascular disease but with cardiovascular risk factors: meta-analysis of randomised controlled trials. BMJ 2009; 338:b2376.
- Kearney PM, Blackwell L, Collins R, Keech A, Simes J, Baigent C; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet 2008; 371:117–125.
- Nissen SE, Nicholls SJ, Sipahi I, et al; ASTEROID Investigators. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes et al. N Engl J Med 2004; 350:1495–1504.
- LaRosa JC, Grundy SM, Waters DD, et al; Treating to New Targets (TNT) Investigators. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Baigent C, Blackwell L, Emberson J, et al; Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670–1681.
- LIPID Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med 1998; 339:1349–1357.
- Grundy SM, Cleeman JI, Bairey Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- American Diabetes Association. Executive summary: standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S5–S10.
- Daniels SR, Greer FR; Committee on Nutrition. Lipid screening and cardiovascular health in childhood. Pediatrics 2008; 122:198–208.
- Knowler WC, Barrett-Connor E, Fowler SE, et al; Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346:393–403.
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Mihaylova B, Emberson J, Blackwell L, et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet 2012; 380:581–590.
On february 28, 2012, the US Food and Drug Administration (FDA) updated its labeling requirements for statins. In addition to revising its recommendations for monitoring liver function and its alerts about reports of memory loss, the FDA also warned of the possibility of new-onset diabetes mellitus and worse glycemic control in patients taking statin drugs.1
This change stoked an ongoing debate about the risk of diabetes with statin use and the implications of such an effect. To understand the clinical consequences of this alert and its effect on treatment decisions, we need to consider the degree to which statins lower the risk of cardiovascular disease in patients at high risk (including diabetic patients), the magnitude of the risk of developing new diabetes while on statin therapy, and the ratio of risk to benefit in treated populations.
This review will discuss the evidence for this possible adverse effect and the implications for clinical practice.
DO STATINS CAUSE DIABETES?
Individual controlled trials dating back more than a decade have had conflicting results about new diabetes and poorer diabetic control in patients taking statins.
The West of Scotland Coronary Prevention Study (WOSCOPS)2 suggested that the incidence of diabetes was 30% lower in patients taking pravastatin (Pravachol) 40 mg/day than with placebo. However, this was not observed with atorvastatin (Lipitor) 10 mg/day in the Anglo-Scandinavian Cardiac Outcomes Trial–Lipid-Lowering Arm (ASCOT-LLA)3 in hypertensive patients or in the Collaborative Atorvastatin Diabetes Study (CARDS)4 in diabetic patients,4 nor was it noted with simvastatin (Zocor) 40 mg/day in the Heart Protection Study (HPS).5
The Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER),6 using the more potent agent rosuvastatin (Crestor) 20 mg/day in patients with elevated levels of C-reactive protein (CRP), was stopped early when an interim analysis found a 44% lower incidence of the primary end point. However, the trial also reported a 26% higher incidence of diabetes in follow-up of less than 2 years.
In the Prospective Study of Pravastatin in the Elderly at Risk (PROSPER),7 with a mean age at entry of 75, there was a 32% higher incidence of diabetes with pravastatin therapy.7
Results of meta-analyses
Several meta-analyses have addressed these differences.
Rajpathak et al8 performed a meta-analysis, published in 2009, of six trials—WOSCOPS,2 ASCOT-LLA,3 JUPITER,6 HPS,5 the Long-term Intervention With Pravastatin in Ischaemic Disease (LIPID) study,9 and the Controlled Rosuvastatin Multinational Study in Heart Failure (CORONA),10 with a total of 57,593 patients. They calculated that the incidence of diabetes was 13% higher (an absolute difference of 0.5%) in statin recipients, which was statistically significant. In their initial analysis, the authors excluded WOSCOPS, describing it as hypothesis-generating. The relative increase in risk was less—6%—and was not statistically significant when WOSCOPS was included.
Sattar et al,11 in a larger meta-analysis published in 2010, included 91,140 participants in 13 major statin trials conducted between 1994 and 2009; each trial had more than 1,000 patients and more than 1 year of follow-up.2,3,5–7,9,10,12–17 New diabetes was defined as physician reporting of new diabetes, new diabetic medication use, or a fasting glucose greater than 7 mmol/L (126 mg/dL).
New diabetes occurred in 2,226 (4.89%) of the statin recipients and in 2,052 (4.5%) of the placebo recipients, an absolute difference of 0.39%, or 9% more (odds ratio [OR] 1.09; 95% confidence interval [CI] 1.02–1.17) (Figure 1).
The incidence of diabetes varied substantially among the 13 trials, with only JUPITER6 and PROSPER7 finding statistically significant increases in rates (26% and 32%, respectively). Of the other 11 trials, 4 had nonsignificant trends toward lower incidence,2,9,13,17 while the 7 others had nonsignificant trends toward higher incidence.
Does the specific statin make a difference?
Questions have been raised as to whether the type of statin used, the intensity of therapy, or the population studied contributed to these differences. Various studies suggest that factors such as using hydrophilic vs lipophilic statins (hydrophilic statins include pravastatin and rosuvastatin; lipophilic statins include atorvastatin, lovastatin, and simvastatin), the dose, the extent of lowering of low-density lipoprotein cholesterol (LDL-C), and the age or clinical characteristics of the population studied may influence this relationship.18–20
Yamakawa et al18 examined the effect of atorvastatin 10 mg/day, pravastatin 10 mg/day, and pitavastatin (Livalo) 2 mg/day on glycemic control over 3 months in a retrospective analysis. Random blood glucose and hemoglobin A1c levels were increased in the atorvastatin group but not in the other two.18
A prospective comparison of atorvastatin 20 mg vs pitavastatin 4 mg in patients with type 2 diabetes, presented at the American College of Cardiology’s 2011 annual meeting, reported a significant increase in fasting glucose levels with atorvastatin, particularly in women, but not with pitavastatin.19
In the Compare the Effect of Rosuvastatin With Atorvastatin on Apo B/Apo A-1 Ratio in Patients With Type 2 Diabetes Mellitus and Dyslipidaemia (CORALL) study,20 both high-dose rosuvastatin (40 mg) and high-dose atorvastatin (80 mg) were associated with significant increases in hemoglobin A1c, although the mean fasting glucose levels were not significantly different at 18 weeks of therapy.
A meta-analysis by Sattar et al11 did not find a clear difference between lipophilic statins (OR 1.10 vs placebo) and hydrophilic statins (OR 1.08). In analysis by statin type, the combined rosuvastatin trials were statistically significant in favor of a higher diabetes risk (OR 1.18, 95% CI 1.04–1.44). Nonsignificant trends were noted for atorvastatin trials (OR 1.14) and simvastatin trials (OR 1.11) and less so for pravastatin (OR 1.03); the OR for lovastatin was 0.98. This may suggest that there is a stronger effect with more potent statins or with greater lowering of LDL-C.
Meta-regression analysis in this study demonstrated that diabetes risk with statins was higher in older patients but was not influenced by body mass index or by the extent that LDL-C was lowered.
Statin dose as a risk factor
Intensive-dose statin therapy has been shown to reduce cardiovascular risk more than low-dose or moderate-dose therapy, thus supporting more aggressive treatment of LDL-C in higher-risk patients. However, some controlled studies comparing more-potent with less-potent statin regimens suggest that there may also be a higher risk of incident diabetes at higher doses.21–24
In a post hoc analysis of the Pravastatin or Atorvastatin Evaluation and Infection Therapy– Thrombolysis in Myocardial Infarction 22 (PROVE-IT TIMI 22) trial,21 patients who had experienced an acute coronary syndrome had a greater increase in hemoglobin A1c if treated with atorvastatin 80 mg/day than with pravastatin 40 mg/day.
Waters et al23 reported a higher risk of new diabetes with atorvastatin 80 mg than with placebo and a trend toward a higher risk with atorvastatin 80 mg than with atorvastatin 10 mg or simvastatin 20 mg.
In contrast, a review by Yousef et al24 of the data from the Enhanced Feedback for Effective Cardiac Treatment (EFFECT) study did not find a higher diabetes risk with more intensive statin therapy based on the magnitude of LDL-C reduction. A propensity-matched examination of deaths, recurrent acute ischemic events, or new diabetes in patients previously hospitalized with myocardial infarction found no differences in these end points each year out to 5 years. The risk of diabetes was in fact lower (but the difference was not statistically significant) in the high-dose groups out to 5 years. The risk of myocardial infarction or death was numerically different in the high-dose groups, but the difference was not statistically significant.
Preiss et al25 in 2011 performed a meta-analysis of the impact of intensity of statin therapy on diabetes risk. They examined data from 32,752 participants without diabetes at baseline in five randomized controlled trials with more than 1,000 participants and more than 1 year of follow-up, comparing high-dose therapy against moderate-dose statin therapy.21,22,26–28 New diabetes was considered present if there was an adverse event report of diabetes, if glucose-lowering drugs were started, or if two fasting plasma glucose measurements were higher than 7 mmol/L (126 mg/dL).
Diabetes developed in 1,449 (8.8%) of the intensive-therapy group and 1,300 (8.0%) of the moderate-therapy group (OR 1.12, 95% CI 1.04–1.22). In contrast, incident cardiovascular disease occurred in 3,134 (19.1%) of the intensive-therapy group and 3,550 (21.7%) of the moderate-therapy group (OR 0.84, 95% CI 0.75–0.94). Therefore, there was an 0.8% absolute increase in diabetes cases on high-dose statins and a 2.6% absolute reduction in adverse cardiovascular events.
CAUTION IN INTERPRETING THESE DATA
There are many reasons for caution in interpreting these studies.
The trials were not designed to look for diabetes
The data supporting the relationship between statin therapy and higher risk of diabetes are primarily from observational studies. These studies were not prospectively designed to address this question, and we therefore need to view this as association and not as causation.
The definition of diabetes varied between trials, and new-onset diabetes was often not rigorously screened for. In many trials the outcome of diabetes was at least partially based on nonstandardized, nonadjudicated physician reporting.
Consequently, if statins reduce the risk of diabetes, the results from WOSCOPS may overstate the reduction, since this study used a non-standard definition of incident diabetes (fasting plasma glucose > 126 mg/dL plus a > 36 mg/dL increase from baseline). When Sattar et al11 reanalyzed WOSCOPS data using a more standard definition, they found a smaller effect.
On the other hand, nonstandardized physician reporting may overstate an adverse effect. Sattar et al11 also found that when fasting plasma glucose levels alone were used as the definition for diabetes, the overall risk was attenuated and was no longer statistically significant (OR 1.07, 95% CI 0.97–1.17).
Perhaps statin therapy uncovers diabetes only in people at risk of diabetes
Perhaps statin therapy uncovers diabetes only in people at higher baseline risk of developing diabetes. Therefore, this adverse effect may be restricted to certain groups and not applicable to the general population.
In JUPITER, one of the two trials in which, on independent analysis, statin use was associated with new diabetes, 77% of patients in the rosuvastatin group who developed diabetes had impaired fasting glucose at entry and therefore were at higher risk of developing diabetes.6
Possibly, the relationship is driven by preexisting metabolic syndrome or other risk factors for diabetes. In the two studies that reported a statistically significantly higher incidence of new diabetes, more than 40% of patients in JUPITER met the criteria for metabolic syndrome, and metabolic syndrome, which increases in prevalence with age, was likely more prominent in the elderly population in PROSPER.
Waters et al23 grouped patients according to whether they had risk factors for diabetes (impaired fasting glucose, obesity, elevated triglycerides, and hypertension) and found that those who had none or one of these risk factors had no difference in the rate of new-onset diabetes with either moderate or intensive statin therapy, but the risk was pronounced in those who had three or four risk factors.
Ridker et al29 reanalyzed the JUPITER data from patients who did not have cardiovascular disease at baseline. Overall, for every 54 new cases of diabetes in follow-up, 134 cardiovascular events or deaths were prevented. In subgroup analysis, those who had one or more risk factors for diabetes at baseline (metabolic syndrome, impaired fasting glucose, obesity, or hemoglobin A1c > 6%) had a 39% reduction in the primary end point and a 28% increase in new diabetes. Those who had none of these risk factors had a 52% lower rate of cardiovascular events but no increase in diabetes.
Other confounding factors
Bias and confounding factors are difficult to control for in studies without prospectively defined, recognized, and analyzed outcomes.
Although it may be a bit of a stretch, residual confounding factors such as myalgia side effects while on statins may reduce exercise in the statin-treatment groups. Perhaps a change to a healthier lifestyle after cardiovascular events may be more common in placebo groups. Improved survival with statins may allow more people at risk of diabetes to live longer and present with the diagnosis.30
POSSIBLE EXPLANATIONS, BUT NO UNIFYING MECHANISM
If mechanisms could be identified to explain the association between statins and diabetes, this would strengthen the argument that it is a cause-and-effect relationship. Many explanations have been proposed as to how statins may influence glucose metabolism and insulin sensitivity.31–34 These are possible explanations based on other observations.
In theory, statins may improve insulin sensitivity via their anti-inflammatory effect, since inflammatory markers and proinflammatory cytokines have been linked with insulin resistance. However, other effects of statins may adversely affect glycemic control.
In vivo analysis has shown that some but not all statins increase insulin levels and decrease insulin sensitivity in a dose-dependent fashion. Some statins decrease adiponectin and may worsen glycemic control through loss of adiponectin’s proposed protective anti-proliferative and antiangiogenic properties. In vitro studies and animal studies have demonstrated a decrease in expression of insulin-responsive glucose transporter 4 (GLUT4) with atorvastatin, and an increase in GLUT1. It has been hypothesized that reduction in isoprenoid biosynthesis or decreased insulin signaling may explain these effects and that changes in glucose transport in adipocytes may cause insulin resistance. Other studies suggest that dysregulation of cellular cholesterol may attenuate beta-cell function. Impaired biosynthesis of ubiquinones may result in delayed production of adenosine triphosphate and consequently diminish insulin release.
But different effects have been reported for atorvastatin, simvastatin, and pravastatin, arguing against a unifying explanation or, alternatively, suggesting that differences in lipophilicity and potency among statins are important. Hydrophilic statins may be less likely to be taken up by extrahepatic cells such as pancreatic cells and adipocytes, possibly lessening these effects. However, the strong association between rosuvastatin (which is hydrophilic) and new diabetes would not support this hypothesis.
Despite these speculations, lack of conformity in response to different statins and discrepancies in the clinical outcomes noted in trials fail to clearly identify a common causative mechanism.
OTHER COMMON THERAPIES MAY INFLUENCE GLYCEMIC CONTROL
Statins are not the first drugs for reducing cardiovascular risk that have been shown to affect glucose levels during treatment.
Niacin
Niacin has been known to increase glucose levels but has long been used as a treatment for dyslipidemia despite this caution. Reduced glycemic control during niacin treatment in diabetic patients does not seem to alter the beneficial effects of treatment.35–37
In a post hoc analysis of the Coronary Drug Project (CDP), in patient subgroups defined by baseline fasting plasma glucose and compared with placebo, niacin reduced the 6-year risk of recurrent myocardial infarction and the combined end point of coronary heart disease death or nonfatal myocardial infarction similarly (interactive P value nonsignificant) across all levels of baseline fasting plasma glucose, including levels of 126 mg/dL or higher at study entry.36
In another post hoc analysis of CDP patient subgroups defined by the change in glycemic status from baseline to 1 year, niacin reduced the 6-year risk of the same end points similarly (interactive P value nonsignificant) across all levels of change in fasting plasma glucose from baseline to year 1, whether baseline fasting plasma glucose levels decreased, stayed the same, or increased to 10 mg/dL or higher on niacin therapy.36
Therefore, the beneficial effect of niacin of reducing the rate of recurrent nonfatal myocardial infarction and coronary heart disease events was not significantly diminished when impaired fasting glucose or diabetes was present when therapy was started or by on-therapy increases from baseline fasting plasma glucose.
In addition, on-therapy changes in glycemic control may be dose-related and minimized by surveillance and therapy adjustments. The Assessment of Diabetes Control and Evaluation of the Efficacy of Niaspan Trial (ADVENT)38 found that changes in glycemic control were minimal as measured by fasting glucose and hemoglobin A1c; were associated with a higher niacin dose (1.5 g/day vs 1 g/day); and, when present, were successfully managed by adjusting the diabetes treatment regimen.
Antihypertensive drugs
Diuretics as well as beta-blockers have been reported to increase the incidence of diabetes in patients with hypertension.15,38–40
A retrospective longitudinal cohort study40 in 2009 examined the development of new-onset diabetes (defined as a new ICD-9 code for diabetes or initiation of diabetes treatment) in 24,688 treated hypertensive patients without diabetes at baseline; 4,385 (17.8%) of the patients developed diabetes. After adjusting for sex and age, the risk of new diabetes was significant in users of diuretics (OR 1.10), beta-blockers (OR 1.12), and calcium channel blockers (OR 1.10) compared with users of angiotensin-converting enzyme inhibitors, (OR 0.92), angiotensin receptor blockers (OR 0.90), or alpha-blockers (OR 0.88).
However, the increase in blood glucose does not seem to attenuate the beneficial effects of reducing cardiovascular events. In the Antihypertensive and Lipid-lowering Treatment to Prevent Heart Attack Trial (ALLHAT),15 a long-term follow-up of those developing new-onset diabetes while taking chlorthalidone (Hygroton) found no difference in the risk of death from cardiovascular disease or from any cause (hazard ratio = 1.04).15
CLINICAL IMPLICATIONS
Balancing the benefits and risks of statins
It is important to examine how the 0.4% increase in absolute risk of new-onset diabetes as calculated in meta-analyses compares with the benefits of statin treatment in terms of cardiovascular risk reduction.
Using data from the Cholesterol Treatment Trialists (CTT) meta-analysis of statin trials in 71,370 participants, Sattar et al11 estimated that statin treatment is associated with 5.4 fewer deaths from coronary heart disease and cases of nonfatal myocardial infarction per 255 patients treated over 4 years for each 1-mmol/L (39 mg/dL) reduction in LDL-C compared with controls. The benefit would be even greater if stroke, revascularization, and hospitalization are included as end points. This benefit is contrasted with the risk of developing 1 additional case of diabetes for every 255 patients treated with statins over the same period.
Preiss et al25 calculated that there were 2 more cases of diabetes per 1,000 patient-years in patients receiving intensive doses than in those receiving moderate doses (18.9 vs 16.9), corresponding to 1 additional case of diabetes for every 498 patients treated per year. However, there were 6.5 fewer first major cardiovascular events per 1,000 patient-years (44.5 vs 51.0), corresponding to a number needed to treat per year to prevent 1 cardiovascular event of 155. Most of the benefit was due to fewer revascularizations, followed by nonfatal myocardial infarctions. The 12% increase in new diabetes with high-dose therapy contrasted with a 16% reduction in new cardiovascular disease combined events (OR 0.84, 95% CI 0.75–0.94).
As previously noted, in the JUPITER trial, the benefits of preventing cardiovascular events with statin therapy outweighed the risk of new diabetes in people both with and without baseline risk factors for diabetes.29 Similar to the observations with niacin and some antihypertensive drugs, the increase in blood glucose with statins does not appear to reduce the benefits of cardiovascular risk reduction in these patients at moderate to high risk, even when used at high doses.
People with diabetes need aggressive lipid-lowering—with statins
Diabetes is a coronary heart disease risk equivalent and is associated with high risk of cardiovascular events.41–46 Overall, the risk for these adverse events is two to four times greater in people with diabetes than without. Atherosclerosis-related events account for approximately 65% to 75% of all deaths in people with diabetes, and 75% of these events are coronary. Lipid abnormalities are strongly correlated with the risk of cardiovascular disease in people with diabetes, and aggressive treatment of risk factors, particularly lipid abnormalities, has been shown to reduce this risk.47–49 And data from multiple clinical trials support the use of statins to lower LDL-C as the first-line therapy for dyslipidemia in people with diabetes, just as it is in the general population.3–7,9,13,23,50–61
Analyses of diabetic subgroups encompassing 18,000 to 20,000 patients in the large statin trials have clearly demonstrated the benefits of statin therapy. A recent metaanalysis of 10 placebo-controlled trials that included approximately 16,000 patients with diabetes and 54,000 without diabetes demonstrated a 30% reduction in coronary heart disease, a 19% reduction in strokes, and a 12% reduction in mortality.54 Furthermore, in another meta-analysis of 14 trials, a similar 22% reduction in coronary heart disease was noted in people with diabetes whether or not they had a history of cardiovascular disease.55
Therefore, aggressive treatment of lipid abnormalities with statins as primary treatment has generally been adopted as a standard of care in diabetic patients, particularly those with clinical cardiovascular disease or one or more risk factors. The Adult Treatment Panel III guidelines recommend a minimum LDL-C goal of less than 100 mg/dL and a goal of less than 70 mg/dL as an option for patients with diabetes (Table 1).41,62 Similar recommendations have been issued by the American Diabetes Association together with the American College of Cardiology (Table 2),30 the American Diabetes Association by itself,63 and the American Academy of Pediatrics.6
Is new-onset diabetes as dangerous as established diabetes?
In studies to date, there did not appear to be more events in those who developed new-onset diabetes.
Waters et al,24 evaluating three trials of high-dose atorvastatin therapy, found that major cardiovascular events occurred in 11.3% of those with new-onset diabetes, 10.8% of those without new-onset diabetes (HR 1.02, 95% CI 0.77–1.35), and 17.5% of those who had diabetes at baseline.
Therefore, it may not be appropriate to extrapolate the glucose changes seen on statin therapy to an equivalent increase in adverse cardiovascular events as seen in other diabetic patients. The beneficial reduction in cardiovascular events does not appear to be diminished in those developing diabetes. It is not clear that the increase in glucose on statins has the same implications of a new diagnosis of diabetes. Does this elevation in glucose represent true diabetes or some downstream effect? For example, thiazide diuretics have been known to increase blood glucose levels, but the levels drop when these drugs are discontinued, even after many years of treatment.
On the other hand, it is possible that follow-up of 5 years or less in clinical trials has not allowed sufficient time to examine the influence of the increase in new-onset diabetes on future cardiovascular events. In addition, because of the widespread use of statins across a broad range of cardiovascular risk, even if the effect is small in absolute terms, the potential adverse effects are magnified, particularly in a low-risk population in which the cardiovascular benefits are smaller.
The association is real, but questions remain
In view of the evidence, it is difficult to refute that an association exits between statin use and new-onset diabetes, at least in some subgroups. The dose response noted in some studies further reinforces the conclusion that the association is real. However, many questions remain unanswered regarding mechanism of effect, whether there are differences depending on the particular statin or dose used, or differential effects in the populations treated (such as patients with metabolic syndrome or the elderly).
Until the contradictory observations can be resolved and plausible mechanisms of action elucidated, causality cannot be established. From a clinical standpoint there is no current evidence suggesting that the elevations in blood glucose seen while on lipid-lowering or blood-pressure-lowering therapy are associated with an increased risk of cardiovascular events or that they attenuate the beneficial effects of the therapy.
Statins should continue to be used in patients at high risk
Until further studies are done, statins should continue to be used, after assessing the risks and the benefits.
Primary prevention patients at moderate to high risk and secondary prevention patients stand to gain from statin therapy, and it should not be denied or doses reduced on the basis of concerns about the development of new-onset diabetes. The recognized modest risk of developing diabetes does not appear to blunt the cardioprotective effects of statin therapy in these moderate-to high-risk groups.
Rather than stop statins in patients at risk of diabetes such as the elderly or those with prediabetes, insulin resistance, or metabolic syndrome who are on therapy for appropriate reasons, it is reasonable to continue these drugs, monitoring glucose more closely and emphasizing the importance of weight reduction, diet, and aerobic exercise for preventing diabetes. The Diabetes Prevention Program Research Group, for example, reduced the incidence of diabetes by 58% over 2.8 years of follow-up with intensive lifestyle interventions (a low-calorie, low-fat diet plus moderate physical activity 150 minutes per week) vs usual care in at-risk populations.65
Should statins be used more cautiously in patients at lower risk?
The most recent Cholesterol Treatment Trialists meta-analysis of 27 randomized clinical trials (22 placebo-controlled, 134,537 people; 5 high-dose vs low-dose, 39,612 people) reported that reducing LDL-C with statins lowered cardiovascular risk even in low-risk patients.66 Overall, there were 21% fewer major cardiovascular events (coronary heart disease, stroke, or coronary revascularization) for every 1-mmol/L reduction in LDL-C.
The proportional reduction in events was at least as large in the two lowest-risk groups (estimated 5-year risk of < 5% and 5% to < 10%, 53,152 people) as in the higher-risk groups. This was reflected mainly in fewer nonfatal myocardial infarctions and coronary revascularizations. In these groups, the absolute reduction in risk for each 1-mmol/L reduction in LDL-C was 11 per 1,000 patients over 5 years. Even in this low-risk population, the reduction in cardiovascular risk seems to compare favorably with the small estimated increase risk of diabetes.
However, even in the lowest-risk group studied, the average baseline LDL-C level was greater than 130 mg/dL.
Therefore, in groups in which the benefits of statins on cardiovascular risk reduction are less robust (eg, low-risk primary prevention groups without significant elevations in LDLC, particularly the elderly), it would not be difficult to justify the case for more cautious use of statin therapy. If statins are used in these low-risk groups, restricting their use to those with at least moderate LDL-C elevation, using less aggressive LDL-C-lowering targets, and regular monitoring of fasting glucose seem reasonable until further information is available.
On february 28, 2012, the US Food and Drug Administration (FDA) updated its labeling requirements for statins. In addition to revising its recommendations for monitoring liver function and its alerts about reports of memory loss, the FDA also warned of the possibility of new-onset diabetes mellitus and worse glycemic control in patients taking statin drugs.1
This change stoked an ongoing debate about the risk of diabetes with statin use and the implications of such an effect. To understand the clinical consequences of this alert and its effect on treatment decisions, we need to consider the degree to which statins lower the risk of cardiovascular disease in patients at high risk (including diabetic patients), the magnitude of the risk of developing new diabetes while on statin therapy, and the ratio of risk to benefit in treated populations.
This review will discuss the evidence for this possible adverse effect and the implications for clinical practice.
DO STATINS CAUSE DIABETES?
Individual controlled trials dating back more than a decade have had conflicting results about new diabetes and poorer diabetic control in patients taking statins.
The West of Scotland Coronary Prevention Study (WOSCOPS)2 suggested that the incidence of diabetes was 30% lower in patients taking pravastatin (Pravachol) 40 mg/day than with placebo. However, this was not observed with atorvastatin (Lipitor) 10 mg/day in the Anglo-Scandinavian Cardiac Outcomes Trial–Lipid-Lowering Arm (ASCOT-LLA)3 in hypertensive patients or in the Collaborative Atorvastatin Diabetes Study (CARDS)4 in diabetic patients,4 nor was it noted with simvastatin (Zocor) 40 mg/day in the Heart Protection Study (HPS).5
The Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER),6 using the more potent agent rosuvastatin (Crestor) 20 mg/day in patients with elevated levels of C-reactive protein (CRP), was stopped early when an interim analysis found a 44% lower incidence of the primary end point. However, the trial also reported a 26% higher incidence of diabetes in follow-up of less than 2 years.
In the Prospective Study of Pravastatin in the Elderly at Risk (PROSPER),7 with a mean age at entry of 75, there was a 32% higher incidence of diabetes with pravastatin therapy.7
Results of meta-analyses
Several meta-analyses have addressed these differences.
Rajpathak et al8 performed a meta-analysis, published in 2009, of six trials—WOSCOPS,2 ASCOT-LLA,3 JUPITER,6 HPS,5 the Long-term Intervention With Pravastatin in Ischaemic Disease (LIPID) study,9 and the Controlled Rosuvastatin Multinational Study in Heart Failure (CORONA),10 with a total of 57,593 patients. They calculated that the incidence of diabetes was 13% higher (an absolute difference of 0.5%) in statin recipients, which was statistically significant. In their initial analysis, the authors excluded WOSCOPS, describing it as hypothesis-generating. The relative increase in risk was less—6%—and was not statistically significant when WOSCOPS was included.
Sattar et al,11 in a larger meta-analysis published in 2010, included 91,140 participants in 13 major statin trials conducted between 1994 and 2009; each trial had more than 1,000 patients and more than 1 year of follow-up.2,3,5–7,9,10,12–17 New diabetes was defined as physician reporting of new diabetes, new diabetic medication use, or a fasting glucose greater than 7 mmol/L (126 mg/dL).
New diabetes occurred in 2,226 (4.89%) of the statin recipients and in 2,052 (4.5%) of the placebo recipients, an absolute difference of 0.39%, or 9% more (odds ratio [OR] 1.09; 95% confidence interval [CI] 1.02–1.17) (Figure 1).
The incidence of diabetes varied substantially among the 13 trials, with only JUPITER6 and PROSPER7 finding statistically significant increases in rates (26% and 32%, respectively). Of the other 11 trials, 4 had nonsignificant trends toward lower incidence,2,9,13,17 while the 7 others had nonsignificant trends toward higher incidence.
Does the specific statin make a difference?
Questions have been raised as to whether the type of statin used, the intensity of therapy, or the population studied contributed to these differences. Various studies suggest that factors such as using hydrophilic vs lipophilic statins (hydrophilic statins include pravastatin and rosuvastatin; lipophilic statins include atorvastatin, lovastatin, and simvastatin), the dose, the extent of lowering of low-density lipoprotein cholesterol (LDL-C), and the age or clinical characteristics of the population studied may influence this relationship.18–20
Yamakawa et al18 examined the effect of atorvastatin 10 mg/day, pravastatin 10 mg/day, and pitavastatin (Livalo) 2 mg/day on glycemic control over 3 months in a retrospective analysis. Random blood glucose and hemoglobin A1c levels were increased in the atorvastatin group but not in the other two.18
A prospective comparison of atorvastatin 20 mg vs pitavastatin 4 mg in patients with type 2 diabetes, presented at the American College of Cardiology’s 2011 annual meeting, reported a significant increase in fasting glucose levels with atorvastatin, particularly in women, but not with pitavastatin.19
In the Compare the Effect of Rosuvastatin With Atorvastatin on Apo B/Apo A-1 Ratio in Patients With Type 2 Diabetes Mellitus and Dyslipidaemia (CORALL) study,20 both high-dose rosuvastatin (40 mg) and high-dose atorvastatin (80 mg) were associated with significant increases in hemoglobin A1c, although the mean fasting glucose levels were not significantly different at 18 weeks of therapy.
A meta-analysis by Sattar et al11 did not find a clear difference between lipophilic statins (OR 1.10 vs placebo) and hydrophilic statins (OR 1.08). In analysis by statin type, the combined rosuvastatin trials were statistically significant in favor of a higher diabetes risk (OR 1.18, 95% CI 1.04–1.44). Nonsignificant trends were noted for atorvastatin trials (OR 1.14) and simvastatin trials (OR 1.11) and less so for pravastatin (OR 1.03); the OR for lovastatin was 0.98. This may suggest that there is a stronger effect with more potent statins or with greater lowering of LDL-C.
Meta-regression analysis in this study demonstrated that diabetes risk with statins was higher in older patients but was not influenced by body mass index or by the extent that LDL-C was lowered.
Statin dose as a risk factor
Intensive-dose statin therapy has been shown to reduce cardiovascular risk more than low-dose or moderate-dose therapy, thus supporting more aggressive treatment of LDL-C in higher-risk patients. However, some controlled studies comparing more-potent with less-potent statin regimens suggest that there may also be a higher risk of incident diabetes at higher doses.21–24
In a post hoc analysis of the Pravastatin or Atorvastatin Evaluation and Infection Therapy– Thrombolysis in Myocardial Infarction 22 (PROVE-IT TIMI 22) trial,21 patients who had experienced an acute coronary syndrome had a greater increase in hemoglobin A1c if treated with atorvastatin 80 mg/day than with pravastatin 40 mg/day.
Waters et al23 reported a higher risk of new diabetes with atorvastatin 80 mg than with placebo and a trend toward a higher risk with atorvastatin 80 mg than with atorvastatin 10 mg or simvastatin 20 mg.
In contrast, a review by Yousef et al24 of the data from the Enhanced Feedback for Effective Cardiac Treatment (EFFECT) study did not find a higher diabetes risk with more intensive statin therapy based on the magnitude of LDL-C reduction. A propensity-matched examination of deaths, recurrent acute ischemic events, or new diabetes in patients previously hospitalized with myocardial infarction found no differences in these end points each year out to 5 years. The risk of diabetes was in fact lower (but the difference was not statistically significant) in the high-dose groups out to 5 years. The risk of myocardial infarction or death was numerically different in the high-dose groups, but the difference was not statistically significant.
Preiss et al25 in 2011 performed a meta-analysis of the impact of intensity of statin therapy on diabetes risk. They examined data from 32,752 participants without diabetes at baseline in five randomized controlled trials with more than 1,000 participants and more than 1 year of follow-up, comparing high-dose therapy against moderate-dose statin therapy.21,22,26–28 New diabetes was considered present if there was an adverse event report of diabetes, if glucose-lowering drugs were started, or if two fasting plasma glucose measurements were higher than 7 mmol/L (126 mg/dL).
Diabetes developed in 1,449 (8.8%) of the intensive-therapy group and 1,300 (8.0%) of the moderate-therapy group (OR 1.12, 95% CI 1.04–1.22). In contrast, incident cardiovascular disease occurred in 3,134 (19.1%) of the intensive-therapy group and 3,550 (21.7%) of the moderate-therapy group (OR 0.84, 95% CI 0.75–0.94). Therefore, there was an 0.8% absolute increase in diabetes cases on high-dose statins and a 2.6% absolute reduction in adverse cardiovascular events.
CAUTION IN INTERPRETING THESE DATA
There are many reasons for caution in interpreting these studies.
The trials were not designed to look for diabetes
The data supporting the relationship between statin therapy and higher risk of diabetes are primarily from observational studies. These studies were not prospectively designed to address this question, and we therefore need to view this as association and not as causation.
The definition of diabetes varied between trials, and new-onset diabetes was often not rigorously screened for. In many trials the outcome of diabetes was at least partially based on nonstandardized, nonadjudicated physician reporting.
Consequently, if statins reduce the risk of diabetes, the results from WOSCOPS may overstate the reduction, since this study used a non-standard definition of incident diabetes (fasting plasma glucose > 126 mg/dL plus a > 36 mg/dL increase from baseline). When Sattar et al11 reanalyzed WOSCOPS data using a more standard definition, they found a smaller effect.
On the other hand, nonstandardized physician reporting may overstate an adverse effect. Sattar et al11 also found that when fasting plasma glucose levels alone were used as the definition for diabetes, the overall risk was attenuated and was no longer statistically significant (OR 1.07, 95% CI 0.97–1.17).
Perhaps statin therapy uncovers diabetes only in people at risk of diabetes
Perhaps statin therapy uncovers diabetes only in people at higher baseline risk of developing diabetes. Therefore, this adverse effect may be restricted to certain groups and not applicable to the general population.
In JUPITER, one of the two trials in which, on independent analysis, statin use was associated with new diabetes, 77% of patients in the rosuvastatin group who developed diabetes had impaired fasting glucose at entry and therefore were at higher risk of developing diabetes.6
Possibly, the relationship is driven by preexisting metabolic syndrome or other risk factors for diabetes. In the two studies that reported a statistically significantly higher incidence of new diabetes, more than 40% of patients in JUPITER met the criteria for metabolic syndrome, and metabolic syndrome, which increases in prevalence with age, was likely more prominent in the elderly population in PROSPER.
Waters et al23 grouped patients according to whether they had risk factors for diabetes (impaired fasting glucose, obesity, elevated triglycerides, and hypertension) and found that those who had none or one of these risk factors had no difference in the rate of new-onset diabetes with either moderate or intensive statin therapy, but the risk was pronounced in those who had three or four risk factors.
Ridker et al29 reanalyzed the JUPITER data from patients who did not have cardiovascular disease at baseline. Overall, for every 54 new cases of diabetes in follow-up, 134 cardiovascular events or deaths were prevented. In subgroup analysis, those who had one or more risk factors for diabetes at baseline (metabolic syndrome, impaired fasting glucose, obesity, or hemoglobin A1c > 6%) had a 39% reduction in the primary end point and a 28% increase in new diabetes. Those who had none of these risk factors had a 52% lower rate of cardiovascular events but no increase in diabetes.
Other confounding factors
Bias and confounding factors are difficult to control for in studies without prospectively defined, recognized, and analyzed outcomes.
Although it may be a bit of a stretch, residual confounding factors such as myalgia side effects while on statins may reduce exercise in the statin-treatment groups. Perhaps a change to a healthier lifestyle after cardiovascular events may be more common in placebo groups. Improved survival with statins may allow more people at risk of diabetes to live longer and present with the diagnosis.30
POSSIBLE EXPLANATIONS, BUT NO UNIFYING MECHANISM
If mechanisms could be identified to explain the association between statins and diabetes, this would strengthen the argument that it is a cause-and-effect relationship. Many explanations have been proposed as to how statins may influence glucose metabolism and insulin sensitivity.31–34 These are possible explanations based on other observations.
In theory, statins may improve insulin sensitivity via their anti-inflammatory effect, since inflammatory markers and proinflammatory cytokines have been linked with insulin resistance. However, other effects of statins may adversely affect glycemic control.
In vivo analysis has shown that some but not all statins increase insulin levels and decrease insulin sensitivity in a dose-dependent fashion. Some statins decrease adiponectin and may worsen glycemic control through loss of adiponectin’s proposed protective anti-proliferative and antiangiogenic properties. In vitro studies and animal studies have demonstrated a decrease in expression of insulin-responsive glucose transporter 4 (GLUT4) with atorvastatin, and an increase in GLUT1. It has been hypothesized that reduction in isoprenoid biosynthesis or decreased insulin signaling may explain these effects and that changes in glucose transport in adipocytes may cause insulin resistance. Other studies suggest that dysregulation of cellular cholesterol may attenuate beta-cell function. Impaired biosynthesis of ubiquinones may result in delayed production of adenosine triphosphate and consequently diminish insulin release.
But different effects have been reported for atorvastatin, simvastatin, and pravastatin, arguing against a unifying explanation or, alternatively, suggesting that differences in lipophilicity and potency among statins are important. Hydrophilic statins may be less likely to be taken up by extrahepatic cells such as pancreatic cells and adipocytes, possibly lessening these effects. However, the strong association between rosuvastatin (which is hydrophilic) and new diabetes would not support this hypothesis.
Despite these speculations, lack of conformity in response to different statins and discrepancies in the clinical outcomes noted in trials fail to clearly identify a common causative mechanism.
OTHER COMMON THERAPIES MAY INFLUENCE GLYCEMIC CONTROL
Statins are not the first drugs for reducing cardiovascular risk that have been shown to affect glucose levels during treatment.
Niacin
Niacin has been known to increase glucose levels but has long been used as a treatment for dyslipidemia despite this caution. Reduced glycemic control during niacin treatment in diabetic patients does not seem to alter the beneficial effects of treatment.35–37
In a post hoc analysis of the Coronary Drug Project (CDP), in patient subgroups defined by baseline fasting plasma glucose and compared with placebo, niacin reduced the 6-year risk of recurrent myocardial infarction and the combined end point of coronary heart disease death or nonfatal myocardial infarction similarly (interactive P value nonsignificant) across all levels of baseline fasting plasma glucose, including levels of 126 mg/dL or higher at study entry.36
In another post hoc analysis of CDP patient subgroups defined by the change in glycemic status from baseline to 1 year, niacin reduced the 6-year risk of the same end points similarly (interactive P value nonsignificant) across all levels of change in fasting plasma glucose from baseline to year 1, whether baseline fasting plasma glucose levels decreased, stayed the same, or increased to 10 mg/dL or higher on niacin therapy.36
Therefore, the beneficial effect of niacin of reducing the rate of recurrent nonfatal myocardial infarction and coronary heart disease events was not significantly diminished when impaired fasting glucose or diabetes was present when therapy was started or by on-therapy increases from baseline fasting plasma glucose.
In addition, on-therapy changes in glycemic control may be dose-related and minimized by surveillance and therapy adjustments. The Assessment of Diabetes Control and Evaluation of the Efficacy of Niaspan Trial (ADVENT)38 found that changes in glycemic control were minimal as measured by fasting glucose and hemoglobin A1c; were associated with a higher niacin dose (1.5 g/day vs 1 g/day); and, when present, were successfully managed by adjusting the diabetes treatment regimen.
Antihypertensive drugs
Diuretics as well as beta-blockers have been reported to increase the incidence of diabetes in patients with hypertension.15,38–40
A retrospective longitudinal cohort study40 in 2009 examined the development of new-onset diabetes (defined as a new ICD-9 code for diabetes or initiation of diabetes treatment) in 24,688 treated hypertensive patients without diabetes at baseline; 4,385 (17.8%) of the patients developed diabetes. After adjusting for sex and age, the risk of new diabetes was significant in users of diuretics (OR 1.10), beta-blockers (OR 1.12), and calcium channel blockers (OR 1.10) compared with users of angiotensin-converting enzyme inhibitors, (OR 0.92), angiotensin receptor blockers (OR 0.90), or alpha-blockers (OR 0.88).
However, the increase in blood glucose does not seem to attenuate the beneficial effects of reducing cardiovascular events. In the Antihypertensive and Lipid-lowering Treatment to Prevent Heart Attack Trial (ALLHAT),15 a long-term follow-up of those developing new-onset diabetes while taking chlorthalidone (Hygroton) found no difference in the risk of death from cardiovascular disease or from any cause (hazard ratio = 1.04).15
CLINICAL IMPLICATIONS
Balancing the benefits and risks of statins
It is important to examine how the 0.4% increase in absolute risk of new-onset diabetes as calculated in meta-analyses compares with the benefits of statin treatment in terms of cardiovascular risk reduction.
Using data from the Cholesterol Treatment Trialists (CTT) meta-analysis of statin trials in 71,370 participants, Sattar et al11 estimated that statin treatment is associated with 5.4 fewer deaths from coronary heart disease and cases of nonfatal myocardial infarction per 255 patients treated over 4 years for each 1-mmol/L (39 mg/dL) reduction in LDL-C compared with controls. The benefit would be even greater if stroke, revascularization, and hospitalization are included as end points. This benefit is contrasted with the risk of developing 1 additional case of diabetes for every 255 patients treated with statins over the same period.
Preiss et al25 calculated that there were 2 more cases of diabetes per 1,000 patient-years in patients receiving intensive doses than in those receiving moderate doses (18.9 vs 16.9), corresponding to 1 additional case of diabetes for every 498 patients treated per year. However, there were 6.5 fewer first major cardiovascular events per 1,000 patient-years (44.5 vs 51.0), corresponding to a number needed to treat per year to prevent 1 cardiovascular event of 155. Most of the benefit was due to fewer revascularizations, followed by nonfatal myocardial infarctions. The 12% increase in new diabetes with high-dose therapy contrasted with a 16% reduction in new cardiovascular disease combined events (OR 0.84, 95% CI 0.75–0.94).
As previously noted, in the JUPITER trial, the benefits of preventing cardiovascular events with statin therapy outweighed the risk of new diabetes in people both with and without baseline risk factors for diabetes.29 Similar to the observations with niacin and some antihypertensive drugs, the increase in blood glucose with statins does not appear to reduce the benefits of cardiovascular risk reduction in these patients at moderate to high risk, even when used at high doses.
People with diabetes need aggressive lipid-lowering—with statins
Diabetes is a coronary heart disease risk equivalent and is associated with high risk of cardiovascular events.41–46 Overall, the risk for these adverse events is two to four times greater in people with diabetes than without. Atherosclerosis-related events account for approximately 65% to 75% of all deaths in people with diabetes, and 75% of these events are coronary. Lipid abnormalities are strongly correlated with the risk of cardiovascular disease in people with diabetes, and aggressive treatment of risk factors, particularly lipid abnormalities, has been shown to reduce this risk.47–49 And data from multiple clinical trials support the use of statins to lower LDL-C as the first-line therapy for dyslipidemia in people with diabetes, just as it is in the general population.3–7,9,13,23,50–61
Analyses of diabetic subgroups encompassing 18,000 to 20,000 patients in the large statin trials have clearly demonstrated the benefits of statin therapy. A recent metaanalysis of 10 placebo-controlled trials that included approximately 16,000 patients with diabetes and 54,000 without diabetes demonstrated a 30% reduction in coronary heart disease, a 19% reduction in strokes, and a 12% reduction in mortality.54 Furthermore, in another meta-analysis of 14 trials, a similar 22% reduction in coronary heart disease was noted in people with diabetes whether or not they had a history of cardiovascular disease.55
Therefore, aggressive treatment of lipid abnormalities with statins as primary treatment has generally been adopted as a standard of care in diabetic patients, particularly those with clinical cardiovascular disease or one or more risk factors. The Adult Treatment Panel III guidelines recommend a minimum LDL-C goal of less than 100 mg/dL and a goal of less than 70 mg/dL as an option for patients with diabetes (Table 1).41,62 Similar recommendations have been issued by the American Diabetes Association together with the American College of Cardiology (Table 2),30 the American Diabetes Association by itself,63 and the American Academy of Pediatrics.6
Is new-onset diabetes as dangerous as established diabetes?
In studies to date, there did not appear to be more events in those who developed new-onset diabetes.
Waters et al,24 evaluating three trials of high-dose atorvastatin therapy, found that major cardiovascular events occurred in 11.3% of those with new-onset diabetes, 10.8% of those without new-onset diabetes (HR 1.02, 95% CI 0.77–1.35), and 17.5% of those who had diabetes at baseline.
Therefore, it may not be appropriate to extrapolate the glucose changes seen on statin therapy to an equivalent increase in adverse cardiovascular events as seen in other diabetic patients. The beneficial reduction in cardiovascular events does not appear to be diminished in those developing diabetes. It is not clear that the increase in glucose on statins has the same implications of a new diagnosis of diabetes. Does this elevation in glucose represent true diabetes or some downstream effect? For example, thiazide diuretics have been known to increase blood glucose levels, but the levels drop when these drugs are discontinued, even after many years of treatment.
On the other hand, it is possible that follow-up of 5 years or less in clinical trials has not allowed sufficient time to examine the influence of the increase in new-onset diabetes on future cardiovascular events. In addition, because of the widespread use of statins across a broad range of cardiovascular risk, even if the effect is small in absolute terms, the potential adverse effects are magnified, particularly in a low-risk population in which the cardiovascular benefits are smaller.
The association is real, but questions remain
In view of the evidence, it is difficult to refute that an association exits between statin use and new-onset diabetes, at least in some subgroups. The dose response noted in some studies further reinforces the conclusion that the association is real. However, many questions remain unanswered regarding mechanism of effect, whether there are differences depending on the particular statin or dose used, or differential effects in the populations treated (such as patients with metabolic syndrome or the elderly).
Until the contradictory observations can be resolved and plausible mechanisms of action elucidated, causality cannot be established. From a clinical standpoint there is no current evidence suggesting that the elevations in blood glucose seen while on lipid-lowering or blood-pressure-lowering therapy are associated with an increased risk of cardiovascular events or that they attenuate the beneficial effects of the therapy.
Statins should continue to be used in patients at high risk
Until further studies are done, statins should continue to be used, after assessing the risks and the benefits.
Primary prevention patients at moderate to high risk and secondary prevention patients stand to gain from statin therapy, and it should not be denied or doses reduced on the basis of concerns about the development of new-onset diabetes. The recognized modest risk of developing diabetes does not appear to blunt the cardioprotective effects of statin therapy in these moderate-to high-risk groups.
Rather than stop statins in patients at risk of diabetes such as the elderly or those with prediabetes, insulin resistance, or metabolic syndrome who are on therapy for appropriate reasons, it is reasonable to continue these drugs, monitoring glucose more closely and emphasizing the importance of weight reduction, diet, and aerobic exercise for preventing diabetes. The Diabetes Prevention Program Research Group, for example, reduced the incidence of diabetes by 58% over 2.8 years of follow-up with intensive lifestyle interventions (a low-calorie, low-fat diet plus moderate physical activity 150 minutes per week) vs usual care in at-risk populations.65
Should statins be used more cautiously in patients at lower risk?
The most recent Cholesterol Treatment Trialists meta-analysis of 27 randomized clinical trials (22 placebo-controlled, 134,537 people; 5 high-dose vs low-dose, 39,612 people) reported that reducing LDL-C with statins lowered cardiovascular risk even in low-risk patients.66 Overall, there were 21% fewer major cardiovascular events (coronary heart disease, stroke, or coronary revascularization) for every 1-mmol/L reduction in LDL-C.
The proportional reduction in events was at least as large in the two lowest-risk groups (estimated 5-year risk of < 5% and 5% to < 10%, 53,152 people) as in the higher-risk groups. This was reflected mainly in fewer nonfatal myocardial infarctions and coronary revascularizations. In these groups, the absolute reduction in risk for each 1-mmol/L reduction in LDL-C was 11 per 1,000 patients over 5 years. Even in this low-risk population, the reduction in cardiovascular risk seems to compare favorably with the small estimated increase risk of diabetes.
However, even in the lowest-risk group studied, the average baseline LDL-C level was greater than 130 mg/dL.
Therefore, in groups in which the benefits of statins on cardiovascular risk reduction are less robust (eg, low-risk primary prevention groups without significant elevations in LDLC, particularly the elderly), it would not be difficult to justify the case for more cautious use of statin therapy. If statins are used in these low-risk groups, restricting their use to those with at least moderate LDL-C elevation, using less aggressive LDL-C-lowering targets, and regular monitoring of fasting glucose seem reasonable until further information is available.
- US Food and Drug Administration. Statin drugs—drug safety communication: class labeling change. February 28, 2012. http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm293670.htm.
- Freeman DJ, Norrie J, Sattar N, et al. Pravastatin and the development of diabetes mellitus: evidence for a protective treatment effect in the West of Scotland Coronary Prevention Study. Circulation 2001; 103:357–362.
- Sever PS, Dahlof B, Poulter NR, et al; ASCOT investigators. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial–Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet 2003; 361:1149–1158.
- Colhoun HM, Betteridge DJ, Durrington PN, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet 2004; 364:685–696.
- Collins R, Armitage J, Parish S, Sleigh P, Peto R; for the Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet 2003; 361:2005–2016.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Shepherd J, Blauw GJ, Murphy MB, et al; PROSPER Study Group. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002; 360:1623–1630.
- Rajpathak SN, Kumbhani DJ, Crandall J, Barzilai N, Alderman M, Ridker PM. Statin therapy and risk of developing type 2 diabetes: a meta-analysis. Diabetes Care 2009; 32:1924–1929.
- Keech A, Colquhoun D, Best J, et al. Secondary prevention of cardiovascular events with long-term pravastatin in patients with diabetes or impaired fasting glucose—results from the LIPID trial. Diabetes Care 2003; 26:2713–2721.
- Kjekshus J, Apetrei E, Barrios V, et al. Rosuvastatin in older patients with systolic heart failure. N Engl J Med 2007; 357:2248–2261.
- Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 2010; 375:735–742.
- Nakamura H, Arakawa K, Itakura H, et al. Primary prevention of cardiovascular disease with pravastatin in Japan (MEGA Study): a prospective randomised controlled trial. Lancet 2006; 368:1155–1163.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Scandinavian Simvastatin Survival Study study group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- Barzilay JI, Davis BR, Pressel SL, et al; ALLHAT Collaborative Research Group. Long-term effects of incident diabetes mellitus on cardiovascular outcomes in people treated for hypertension: the ALLHAT Diabetes Extension Study. Circ Cardiovasc Qual Outcomes 2012; 5:153–162.
- Tavazzi L, Maggioni AP, Marchioli R, et al. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet 2008; 372:1231–1239.
- GISSI Prevenzione Investigators (Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico). Results of the low-dose (20 mg) pravastatin GISSI Prevenzione trial in 4271 patients with recent myocardial infarction: do stopped trials contribute to overall knowledge? Ital Heart J 2000; 1:810–820.
- Yamakawa T, Takano T, Tanaka S, Kadonosono K, Terauchi Y. Influence of pitavastatin on glucose tolerance in patients with type 2 diabetes mellitus. J Atheroscler Thromb 2008; 15:269–275.
- Kryzhanovski V, Gumprecht J, Zhu B, Yu CY, Hounslow N, Sponseller CA. Atorvastatin but not pitavastatin significantly increases fasting plasma glucose in patients with type 2 diabetes and combined dyslipidemia (abstract). J Am Coll Cardiol 2011; 57:E575.
- Simsek S, Schalkwijk CG, Wolffenbuttel BH. Effects of rosuvastatin and atorvastatin on glycaemic control in type 2 diabetes—the CORALL study. Diabet Med 2012; 29:628–631.
- Sabatine MS, Morrow DA, Giugliano RP, et al. Implications of upstream glycoprotein IIb/IIIa inhibition and coronary artery stenting in the invasive management of unstable angina/non-ST-elevation myocardial infarction: a comparison of the Thrombolysis In Myocardial Infarction (TIMI) IIIB trial and the Treat angina with Aggrastat and determine Cost of Therapy with Invasive or Conservative Strategy (TACTICS)-TIMI 18 trial. Circulation 2004; 110(suppl III):834–880.
- Shepherd J, Barter P, Carmena R, et al. Effect of lowering LDL cholesterol substantially below currently recommended levels in patients with coronary heart disease and diabetes: the Treating to New Targets (TNT) study. Diabetes Care 2006; 29:1220–1226.
- Waters DD, Ho JE, DeMicco DA, et al. Predictors of new-onset diabetes in patients treated with atorvastatin: results from 3 large randomized clinical trials. J Am Coll Cardiol 2011; 57:1535–1545.
- Yousef A, Tu JV, Wang J, Donovan L, Ko DT. The association of intensive statin therapy on long-term risks of cardiovascular events and diabetes following acute myocardial infarction (abstract). Circulation 2012; 125:e859.
- Preiss D, Seshasai SR, Welsh P, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a metaanalysis. JAMA 2011; 305:2556–2564.
- de Lemos JA, Blazing MA, Wiviott SD, et al; A to Z Investigators. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- Pedersen TR, Faegeman O, Kastelein JJ, et al; Incremental Decrease in End Points Through Aggressive Lipid Lowering (IDEAL) Study Group. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Armitage J, Bowman L, Wallendszus K, et al; Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet 2010; 37:1658–1669.
- Ridker PM, Pradhan A, MacFadyen JG, Libby P, Glynn RJ. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: an analysis from the JUPITER trial. Lancet 2012; 380:565–571.
- Brunzell JD, Davidson M, Furberg CD, et al; American Diabetes Association; American College of Cardiology Foundation. Lipoprotein management in patients with cardiometabolic risk: consensus statement from the American Diabetes Association and the American College of Cardiology Foundation. Diabetes Care 2008; 31:811–822.
- Koh KK, Quon MJ, Han SH, Lee Y, Kim SJ, Shin EK. Atorvastatin causes insulin resistance and increases ambient glycemia in hypercholesterolemic patients. J Am Coll Cardiol 2010; 55:1209–1216.
- Koh KK, Quon MJ, Han SH, et al. Differential metabolic effects of pravastatin and simvastatin in hypercholesterolemic patients. Atherosclerosis 2009; 204:483–490.
- Nakata M, Nagasaka S, Kusaka I, Matsuoka H, Ishibashi S, Yada T. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): implications in glycaemic control. Diabetologia 2006; 49:1881–1892.
- Yada T, Nakata M, Shiraishi T, Kakei M. Inhibition by simvastatin, but not pravastatin, of glucose-induced cytosolic Ca2+ signalling and insulin secretion due to blockade of L-type Ca2+ channels in rat islet beta-cells. Br J Pharmacol 1999; 126:1205–1213.
- Guyton JR, Fazio S, Adewale AJ, et al. Effect of extended-release niacin on new-onset diabetes among hyperlipidemic patients treated with ezetimibe/simvastatin in a randomized controlled trial. Diabetes Care 2012; 35:857–860.
- Canner PL, Furberg CD, Terrin ML, McGovern ME. Benefits of niacin by glycemic status in patients with healed myocardial infarction (from the Coronary Drug Project). Am J Cardiol 2005; 95:254–257.
- Grundy SM, Vega GL, McGovern ME, et al; Diabetes Multicenter Research Group. Efficacy, safety, and tolerability of once-daily niacin for the treatment of dyslipidemia associated with type 2 diabetes: results of the Assessment of Diabetes Control and Evaluation of the Efficacy of Niaspan Trial. Arch Intern Med 2002; 162:1568–1576.
- Gupta AK, Dahlof B, Dobson J, Sever PS, Wedel H, Poulter NRAnglo-Scandinavian Cardiac Outcomes Trial Investigators. Determinants of new-onset diabetes among 19,257 hypertensive patients randomized in the Anglo-Scandinavian Cardiac Outcomes Trial—Blood Pressure Lowering Arm and the relative influence of antihypertensive medication. Diabetes Care 2008; 31:982–988.
- Elliott WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet 2007; 369:201–207.
- Jong JP, Chang MH, Tien L, et al. Antihypertensive drugs and new-onset diabetes: a retrospective longitudinal cohort study. Cardiovasc Ther 2009; 27:159–163.
- Third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Norhammar A, Tenerz A, Nilsson G, et al. Glucose metabolism in patients with acute myocardial infarction and no previous diagnosis of diabetes mellitus: a prospective study Lancet 2002; 359:2140–2144.
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Sprafka JM, Burke GL, Folsom AR, McGovbern PG, Hahn LP. Trends in prevalence of diabetes mellitus in patients with myocardial infarction and effect of diabetes on survival. The Minnesota Heart Survey. Diabetes Care 1991; 14:537–543.
- Geiss LS, Herman WH, Smith PJ. Mortality in non-insulin-dependent diabetes. In:Harris MI, Cowie CC, Stern MP, et al, editors. Diabetes in America. 2nd ed. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, 1995:233–257.
- Stamler J, Vaccaro O, Neaton JD, Wentworth D. Diabetes, other risk factors, and 12-yr cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial. Diabetes Care 1993; 16:434–444.
- Turner RC, Millns H, Neil HA, et al. Risk factors for coronary artery disease in non-insulin dependent diabetes mellitus: United Kingdom Prospective Diabetes Study (UKPDS: 23). BMJ 1998; 316:823–828.
- Gaede P, Vedel P, Larsen N, Jensen GV, Parving HH, Pedersen O. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Engl J Med 2003; 348:383–393.
- Gaede P, Pederson O. Intensive integrated therapy of type 2 diabetes: implications for long-term prognosis. Diabetes 2004; 53:S39–S47.
- Goldberg RB, Mellies MJ, Sacks FM, et al. Cardiovascular events and their reduction with pravastatin in diabetic and glucose-intolerant myocardial infarction survivors with average cholesterol levels: subgroup analyses in the cholesterol and recurrent events (CARE) trial. The Care Investigators. Circulation 1998; 98:2513–2519.
- Pyðrälä K, Pedersen TR, Kjekshus J, Faergeman O, Olsson AG, Thorgeirsson G. Cholesterol lowering with simvastatin improves prognosis of diabetic patients with coronary heart disease. A subgroup analysis of the Scandinavian Simvastatin Survival Study (4S). Diabetes Care 1997; 20:614–620.
- Vijan S, Hayward RA; American College of Physicians. Pharmacologic lipid-lowering therapy in type 2 diabetes mellitus: background paper for the American College of Physicians. Ann Intern Med 2004; 140:650–658.
- Baigent C, Keech A, Kearney PM, et al; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278. Errata in Lancet 2008; 371:2084, Lancet 2005; 366:1358.
- Brugts JJ, Yetgin T, Hoeks SE, et al. The benefits of statins in people without established cardiovascular disease but with cardiovascular risk factors: meta-analysis of randomised controlled trials. BMJ 2009; 338:b2376.
- Kearney PM, Blackwell L, Collins R, Keech A, Simes J, Baigent C; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet 2008; 371:117–125.
- Nissen SE, Nicholls SJ, Sipahi I, et al; ASTEROID Investigators. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes et al. N Engl J Med 2004; 350:1495–1504.
- LaRosa JC, Grundy SM, Waters DD, et al; Treating to New Targets (TNT) Investigators. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Baigent C, Blackwell L, Emberson J, et al; Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670–1681.
- LIPID Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med 1998; 339:1349–1357.
- Grundy SM, Cleeman JI, Bairey Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- American Diabetes Association. Executive summary: standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S5–S10.
- Daniels SR, Greer FR; Committee on Nutrition. Lipid screening and cardiovascular health in childhood. Pediatrics 2008; 122:198–208.
- Knowler WC, Barrett-Connor E, Fowler SE, et al; Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346:393–403.
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Mihaylova B, Emberson J, Blackwell L, et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet 2012; 380:581–590.
- US Food and Drug Administration. Statin drugs—drug safety communication: class labeling change. February 28, 2012. http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm293670.htm.
- Freeman DJ, Norrie J, Sattar N, et al. Pravastatin and the development of diabetes mellitus: evidence for a protective treatment effect in the West of Scotland Coronary Prevention Study. Circulation 2001; 103:357–362.
- Sever PS, Dahlof B, Poulter NR, et al; ASCOT investigators. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial–Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet 2003; 361:1149–1158.
- Colhoun HM, Betteridge DJ, Durrington PN, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet 2004; 364:685–696.
- Collins R, Armitage J, Parish S, Sleigh P, Peto R; for the Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet 2003; 361:2005–2016.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Shepherd J, Blauw GJ, Murphy MB, et al; PROSPER Study Group. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002; 360:1623–1630.
- Rajpathak SN, Kumbhani DJ, Crandall J, Barzilai N, Alderman M, Ridker PM. Statin therapy and risk of developing type 2 diabetes: a meta-analysis. Diabetes Care 2009; 32:1924–1929.
- Keech A, Colquhoun D, Best J, et al. Secondary prevention of cardiovascular events with long-term pravastatin in patients with diabetes or impaired fasting glucose—results from the LIPID trial. Diabetes Care 2003; 26:2713–2721.
- Kjekshus J, Apetrei E, Barrios V, et al. Rosuvastatin in older patients with systolic heart failure. N Engl J Med 2007; 357:2248–2261.
- Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 2010; 375:735–742.
- Nakamura H, Arakawa K, Itakura H, et al. Primary prevention of cardiovascular disease with pravastatin in Japan (MEGA Study): a prospective randomised controlled trial. Lancet 2006; 368:1155–1163.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Scandinavian Simvastatin Survival Study study group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- Barzilay JI, Davis BR, Pressel SL, et al; ALLHAT Collaborative Research Group. Long-term effects of incident diabetes mellitus on cardiovascular outcomes in people treated for hypertension: the ALLHAT Diabetes Extension Study. Circ Cardiovasc Qual Outcomes 2012; 5:153–162.
- Tavazzi L, Maggioni AP, Marchioli R, et al. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet 2008; 372:1231–1239.
- GISSI Prevenzione Investigators (Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico). Results of the low-dose (20 mg) pravastatin GISSI Prevenzione trial in 4271 patients with recent myocardial infarction: do stopped trials contribute to overall knowledge? Ital Heart J 2000; 1:810–820.
- Yamakawa T, Takano T, Tanaka S, Kadonosono K, Terauchi Y. Influence of pitavastatin on glucose tolerance in patients with type 2 diabetes mellitus. J Atheroscler Thromb 2008; 15:269–275.
- Kryzhanovski V, Gumprecht J, Zhu B, Yu CY, Hounslow N, Sponseller CA. Atorvastatin but not pitavastatin significantly increases fasting plasma glucose in patients with type 2 diabetes and combined dyslipidemia (abstract). J Am Coll Cardiol 2011; 57:E575.
- Simsek S, Schalkwijk CG, Wolffenbuttel BH. Effects of rosuvastatin and atorvastatin on glycaemic control in type 2 diabetes—the CORALL study. Diabet Med 2012; 29:628–631.
- Sabatine MS, Morrow DA, Giugliano RP, et al. Implications of upstream glycoprotein IIb/IIIa inhibition and coronary artery stenting in the invasive management of unstable angina/non-ST-elevation myocardial infarction: a comparison of the Thrombolysis In Myocardial Infarction (TIMI) IIIB trial and the Treat angina with Aggrastat and determine Cost of Therapy with Invasive or Conservative Strategy (TACTICS)-TIMI 18 trial. Circulation 2004; 110(suppl III):834–880.
- Shepherd J, Barter P, Carmena R, et al. Effect of lowering LDL cholesterol substantially below currently recommended levels in patients with coronary heart disease and diabetes: the Treating to New Targets (TNT) study. Diabetes Care 2006; 29:1220–1226.
- Waters DD, Ho JE, DeMicco DA, et al. Predictors of new-onset diabetes in patients treated with atorvastatin: results from 3 large randomized clinical trials. J Am Coll Cardiol 2011; 57:1535–1545.
- Yousef A, Tu JV, Wang J, Donovan L, Ko DT. The association of intensive statin therapy on long-term risks of cardiovascular events and diabetes following acute myocardial infarction (abstract). Circulation 2012; 125:e859.
- Preiss D, Seshasai SR, Welsh P, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a metaanalysis. JAMA 2011; 305:2556–2564.
- de Lemos JA, Blazing MA, Wiviott SD, et al; A to Z Investigators. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- Pedersen TR, Faegeman O, Kastelein JJ, et al; Incremental Decrease in End Points Through Aggressive Lipid Lowering (IDEAL) Study Group. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Armitage J, Bowman L, Wallendszus K, et al; Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet 2010; 37:1658–1669.
- Ridker PM, Pradhan A, MacFadyen JG, Libby P, Glynn RJ. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: an analysis from the JUPITER trial. Lancet 2012; 380:565–571.
- Brunzell JD, Davidson M, Furberg CD, et al; American Diabetes Association; American College of Cardiology Foundation. Lipoprotein management in patients with cardiometabolic risk: consensus statement from the American Diabetes Association and the American College of Cardiology Foundation. Diabetes Care 2008; 31:811–822.
- Koh KK, Quon MJ, Han SH, Lee Y, Kim SJ, Shin EK. Atorvastatin causes insulin resistance and increases ambient glycemia in hypercholesterolemic patients. J Am Coll Cardiol 2010; 55:1209–1216.
- Koh KK, Quon MJ, Han SH, et al. Differential metabolic effects of pravastatin and simvastatin in hypercholesterolemic patients. Atherosclerosis 2009; 204:483–490.
- Nakata M, Nagasaka S, Kusaka I, Matsuoka H, Ishibashi S, Yada T. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): implications in glycaemic control. Diabetologia 2006; 49:1881–1892.
- Yada T, Nakata M, Shiraishi T, Kakei M. Inhibition by simvastatin, but not pravastatin, of glucose-induced cytosolic Ca2+ signalling and insulin secretion due to blockade of L-type Ca2+ channels in rat islet beta-cells. Br J Pharmacol 1999; 126:1205–1213.
- Guyton JR, Fazio S, Adewale AJ, et al. Effect of extended-release niacin on new-onset diabetes among hyperlipidemic patients treated with ezetimibe/simvastatin in a randomized controlled trial. Diabetes Care 2012; 35:857–860.
- Canner PL, Furberg CD, Terrin ML, McGovern ME. Benefits of niacin by glycemic status in patients with healed myocardial infarction (from the Coronary Drug Project). Am J Cardiol 2005; 95:254–257.
- Grundy SM, Vega GL, McGovern ME, et al; Diabetes Multicenter Research Group. Efficacy, safety, and tolerability of once-daily niacin for the treatment of dyslipidemia associated with type 2 diabetes: results of the Assessment of Diabetes Control and Evaluation of the Efficacy of Niaspan Trial. Arch Intern Med 2002; 162:1568–1576.
- Gupta AK, Dahlof B, Dobson J, Sever PS, Wedel H, Poulter NRAnglo-Scandinavian Cardiac Outcomes Trial Investigators. Determinants of new-onset diabetes among 19,257 hypertensive patients randomized in the Anglo-Scandinavian Cardiac Outcomes Trial—Blood Pressure Lowering Arm and the relative influence of antihypertensive medication. Diabetes Care 2008; 31:982–988.
- Elliott WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet 2007; 369:201–207.
- Jong JP, Chang MH, Tien L, et al. Antihypertensive drugs and new-onset diabetes: a retrospective longitudinal cohort study. Cardiovasc Ther 2009; 27:159–163.
- Third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Norhammar A, Tenerz A, Nilsson G, et al. Glucose metabolism in patients with acute myocardial infarction and no previous diagnosis of diabetes mellitus: a prospective study Lancet 2002; 359:2140–2144.
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Sprafka JM, Burke GL, Folsom AR, McGovbern PG, Hahn LP. Trends in prevalence of diabetes mellitus in patients with myocardial infarction and effect of diabetes on survival. The Minnesota Heart Survey. Diabetes Care 1991; 14:537–543.
- Geiss LS, Herman WH, Smith PJ. Mortality in non-insulin-dependent diabetes. In:Harris MI, Cowie CC, Stern MP, et al, editors. Diabetes in America. 2nd ed. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, 1995:233–257.
- Stamler J, Vaccaro O, Neaton JD, Wentworth D. Diabetes, other risk factors, and 12-yr cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial. Diabetes Care 1993; 16:434–444.
- Turner RC, Millns H, Neil HA, et al. Risk factors for coronary artery disease in non-insulin dependent diabetes mellitus: United Kingdom Prospective Diabetes Study (UKPDS: 23). BMJ 1998; 316:823–828.
- Gaede P, Vedel P, Larsen N, Jensen GV, Parving HH, Pedersen O. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Engl J Med 2003; 348:383–393.
- Gaede P, Pederson O. Intensive integrated therapy of type 2 diabetes: implications for long-term prognosis. Diabetes 2004; 53:S39–S47.
- Goldberg RB, Mellies MJ, Sacks FM, et al. Cardiovascular events and their reduction with pravastatin in diabetic and glucose-intolerant myocardial infarction survivors with average cholesterol levels: subgroup analyses in the cholesterol and recurrent events (CARE) trial. The Care Investigators. Circulation 1998; 98:2513–2519.
- Pyðrälä K, Pedersen TR, Kjekshus J, Faergeman O, Olsson AG, Thorgeirsson G. Cholesterol lowering with simvastatin improves prognosis of diabetic patients with coronary heart disease. A subgroup analysis of the Scandinavian Simvastatin Survival Study (4S). Diabetes Care 1997; 20:614–620.
- Vijan S, Hayward RA; American College of Physicians. Pharmacologic lipid-lowering therapy in type 2 diabetes mellitus: background paper for the American College of Physicians. Ann Intern Med 2004; 140:650–658.
- Baigent C, Keech A, Kearney PM, et al; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278. Errata in Lancet 2008; 371:2084, Lancet 2005; 366:1358.
- Brugts JJ, Yetgin T, Hoeks SE, et al. The benefits of statins in people without established cardiovascular disease but with cardiovascular risk factors: meta-analysis of randomised controlled trials. BMJ 2009; 338:b2376.
- Kearney PM, Blackwell L, Collins R, Keech A, Simes J, Baigent C; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet 2008; 371:117–125.
- Nissen SE, Nicholls SJ, Sipahi I, et al; ASTEROID Investigators. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes et al. N Engl J Med 2004; 350:1495–1504.
- LaRosa JC, Grundy SM, Waters DD, et al; Treating to New Targets (TNT) Investigators. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360:7–22.
- Baigent C, Blackwell L, Emberson J, et al; Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670–1681.
- LIPID Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med 1998; 339:1349–1357.
- Grundy SM, Cleeman JI, Bairey Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- American Diabetes Association. Executive summary: standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S5–S10.
- Daniels SR, Greer FR; Committee on Nutrition. Lipid screening and cardiovascular health in childhood. Pediatrics 2008; 122:198–208.
- Knowler WC, Barrett-Connor E, Fowler SE, et al; Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346:393–403.
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Mihaylova B, Emberson J, Blackwell L, et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet 2012; 380:581–590.
KEY POINTS
- The evidence from individual clinical trials is mixed, but meta-analyses indicate that statin therapy is associated with approximately a 9% higher risk of diabetes (an absolute difference of about 0.4%).
- We need to interpret this information cautiously. Many potentially confounding factors are involved, and rigorous prospective trials are needed to examine this issue.
- The benefit of preventing serious cardiovascular events seems to outweigh the higher risks of diabetes and poorer glycemic control, and we should continue to give statins to patients at moderate to high risk, including those with diabetes, with vigilance for these side effects.
Male hypogonadism: More than just a low testosterone
Editor’s note: This article on the differential diagnosis of hypogonadism in men is the first of two articles. The second, to be published next month, focuses on the appropriate use of testosterone therapy.
A 54-year-old man is referred for evaluation of low testosterone. He had seen his primary care physician for complaints of diminished libido and erectile dysfunction for the past year and worsening fatigue over the past few years. He has not been formally diagnosed with any medical condition. His serum testosterone level is 180 ng/dL (reference range 249–836 ng/dL).
On physical examination, he is obese (body mass index 31 kg/m2) with a normal-appearing male body habitus, no gynecomastia, and normal testicles and prostate gland.
How should this patient be evaluated?
LOW TESTOSTERONE HAS MANY CAUSES
Male hypogonadism, ie, failure of the testes to produce adequate amounts of androgen or sperm, has become a common clinical finding, particularly in the older population. This is more likely the result of an increase in awareness and detection of the disorder by physicians rather than a true increase in prevalence.
The finding of a low serum testosterone value needs to be confirmed and thoroughly evaluated before starting treatment. It is important to determine whether the cause is a primary (hypergonadotropic) testicular disorder or secondary to a hypothalamic-pituitary process (hypogonadotropic or normogonadotropic).
THE HYPOTHALAMIC-PITUITARY-GONADAL AXIS
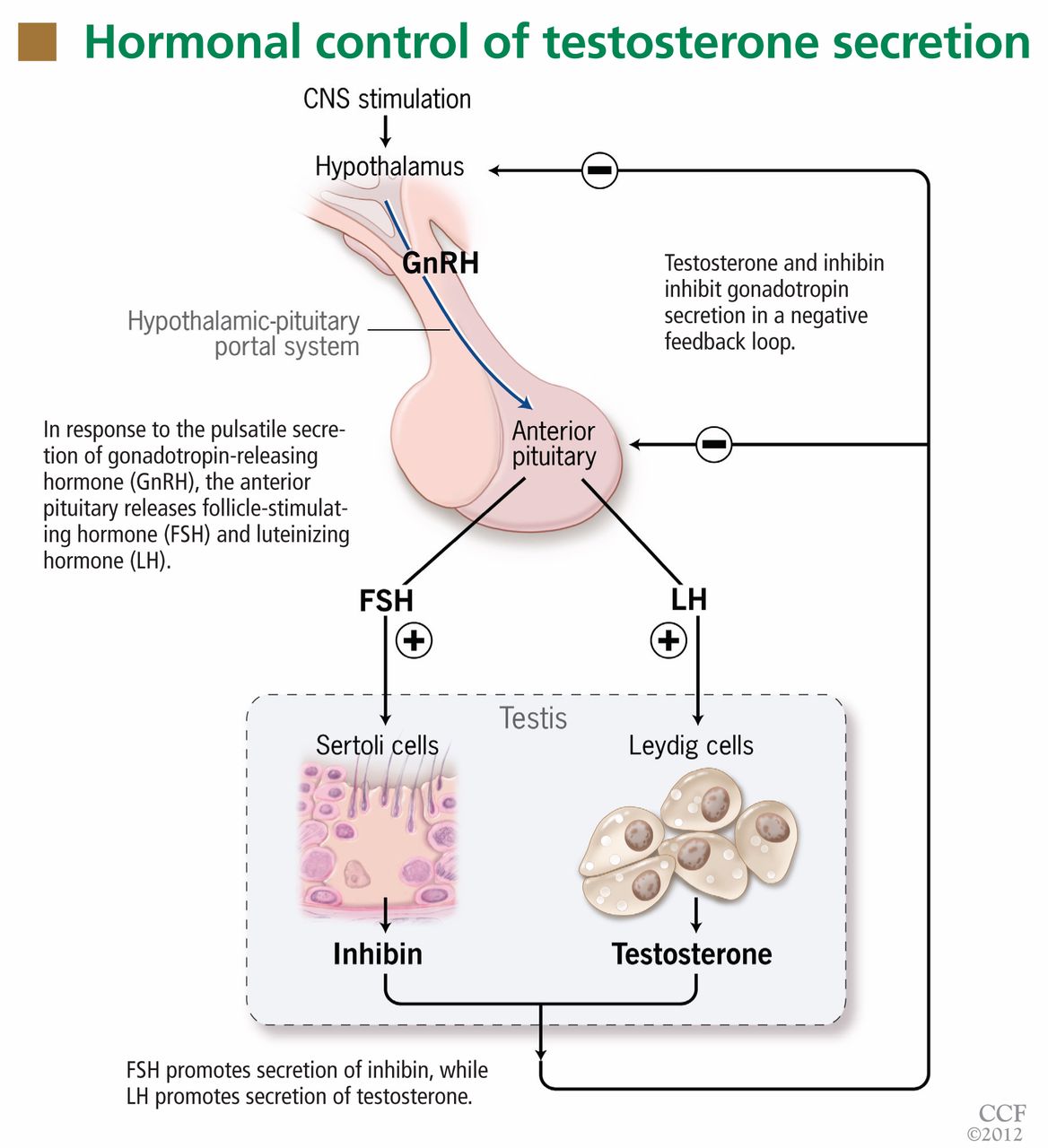
Testosterone production is under the control of luteinizing hormone (LH), whereas sperm production is under the control of follicle-stimulating hormone (FSH) (Figure 1). Both of these pituitary hormones are regulated by the pulsatile secretion of hypothalamic gonadotropin-releasing hormone (GnRH).
Testosterone (produced by Leydig cells) and inhibin B (produced by Sertoli cells within the seminiferous tubules) result in negative feedback inhibition of gonadotropin (LH and FSH) secretion. Testosterone and estradiol (produced by aromatization of testosterone) act at both pituitary and hypothalamic sites and are the principal regulators of LH secretion.1,2 Inhibin B is the major regulator of FSH secretion in men,3 but steroid feedback also occurs.2,4
TO FOLLOW UP A LOW TESTOSTERONE, CONFIRM THE VALUE NEAR 8 am
If a testosterone value is found to be low, it is important to determine the time that the sample was obtained. Serum testosterone levels follow a diurnal rhythm, at least in younger men, with values near 8 am being, on average, 30% higher than the trough levels later in the day.5–7 The timing of the diurnal variation may be different in night-shift workers, who may require assessment at a more appropriate time of the day (ie, upon awakening).
Another factor affecting testosterone levels is the patient’s health status at the time of testing. Values obtained in the hospital during an acute illness should be repeated once the event has resolved, as testosterone values decrease considerably in this setting.8 Even in outpatients, particularly in men over age 60, one must be sure that the low testosterone level was not obtained during a period of decompensation of one of the many comorbidities seen in these patients, such as coronary artery disease, congestive heart failure, or uncontrolled diabetes.
If an 8 am testosterone value is low, it is reasonable to obtain at least one confirmatory testosterone level on another day, near 8 am, in the next few weeks, when the patient is in good health. Confirming the testosterone level is important, particularly since commercially available testosterone assays are not well standardized and some are frankly unreliable.9,10 A repeat confirmatory level should always be performed by a reliable reference laboratory. If the testosterone level is still low, further evaluation is warranted.
TOTAL TESTOSTERONE VS BIOAVAILABLE TESTOSTERONE VS FREE TESTOSTERONE
Of the total circulating testosterone, 60% is bound to sex hormone-binding globulin (SHBG), 38% is bound to albumin, and only 2% is free. All of these fractions can be measured to assess for testosterone deficiency.
Free testosterone is the biologically active form of this hormone and, thus, the free testosterone level is considered to be a better representation of the true testosterone status. However, some clinicians believe that bioavailable testosterone (testosterone loosely bound to albumin + free testosterone) is a better reflection of the true level of the active hormone than the level of free testosterone alone.
There are situations in which the total testosterone level is low but bioavailable or free testosterone levels are normal. The level of total testosterone is affected by alterations in the levels of SHBG and albumin. A reduction in the level of SHBG can result in low total serum testosterone levels in patients with obesity or type 2 diabetes (states of insulin resistance), and also in cachexia, malnutrition, advanced cirrhosis, acromegaly, hypothyroidism, and nephrotic syndrome. SHBG can also be low in patients taking glucocorticoids, progestins, or androgenic steroids.11 In these settings, checking the level of free testosterone (the active hormone), bioavailable testosterone, or both, by a reliable reference laboratory, may be more appropriate.9,10
But regardless of which measurement is chosen, all testosterone levels—especially bioavailable and free testosterone values—should be interpreted with caution if they are not measured at a reliable reference laboratory.9,10 Interested readers may wish to see the US Centers for Disease Control and Prevention (CDC) Hormone Standardization Program Web site (www.cdc.gov/labstandards/hs.html) for more details, including a list of CDC-certified laboratories.
CLINICAL FEATURES OF LOW TESTOSTERONE
A history of erectile dysfunction, decreased libido, and fatigue may be seen in patients with low testosterone. However, one must realize that these symptoms—as well as others reported by men with low testosterone, such as depression, difficulty concentrating, irritability, and insomnia—are nonspecific and may be related to other medical conditions.12
Likewise, physical findings such as muscle weakness, reduced body hair, and altered fat distribution (abdominal obesity) are seen in men with low testosterone, but also in those with a number of other medical conditions.
Additional features suggest specific disorders, eg, anosmia in Kallmann syndrome; eunuchoid body habitus, gynecomastia, and small testes in Klinefelter syndrome.
Men with low testosterone may have low bone mineral density or anemia, or both.
Careful examination of the breasts for gynecomastia and the testes for size, consistency, and masses (testicular tumors) helps in formulating a differential diagnosis and in appropriately directing subsequent laboratory evaluation and diagnostic imaging.
LOW TESTOSTERONE: PRIMARY VS SECONDARY
A history of testicular trauma, systemic chemotherapy, or mumps orchitis should direct the physician’s attention to a testicular etiology. On the other hand, darkened or tanned skin (suggesting hemochromatosis), galactorrhea (suggesting hyperprolactinemia), or visual field deficits (suggesting a sellar mass) should direct the physician’s attention toward a pituitary-hypothalamic process.

Once the low testosterone value has been confirmed at least one time near 8 am, one should obtain LH and FSH values to help direct further evaluation in deciphering the etiology (Figure 2). Elevated (hypergonadotropic) values indicate a testicular disorder (primary hypogonadism), whereas low (hypogonadotropic) or normal (normogonadotropic) values point to a pituitary-hypothalamic process (secondary hypogonadism). It should be emphasized that, in the setting of a low testosterone level, LH and FSH values within the normal range are “inappropriately normal” so that further investigation is required.
This evaluation should also include serum prolactin, thyroid-stimulating hormone (TSH, also known as thyrotropin), free thyroxine (T4), and ferritin levels, the latter because hemochromatosis (iron overload) can cause both primary and secondary hypogonadism. If at any time in the evaluation the laboratory results suggest secondary hypogonadism, a full assessment of pituitary function should be undertaken.
Semen analysis is usually reserved for patients presenting with the primary complaint of infertility.
PRIMARY HYPOGONADISM
The patient should be carefully questioned about the age at which his problems began, about pubertal development, and about fertility. Causes of primary hypogonadism include:
- Karyotype abnormalities—Klinefelter syndrome (47, XXY syndrome) is the most common
- Toxin exposure, chemotherapy
- Congenital defects—anorchia, cryptorchidism13
- Orchitis (mumps, autoimmune)
- Testicular trauma or infarction
- Hemochromatosis
- Medications that inhibit androgen biosynthesis, eg, ketoconazole (Nizoral)14
- Increase in temperature of the testicular environment (due to varicocele or a large panniculus).
SECONDARY HYPOGONADISM
Causes of secondary hypogonadism include the following:
Congenital disorders
These disorders are usually diagnosed in childhood or adolescence, often after the patient is brought to the physician because of short stature or pubertal delay.
- Kallmann syndrome (anosmia and GnRH deficiency)15
- GnRH receptor mutation and deficiency16
- Genetic mutations associated with pituitary hormone deficiencies, eg, PROP-1 mutation.17
Acquired disorders that suppress gonadotrophs
Drugs. Long-term therapy with common medications such as opioids or corticosteroids can result in secondary hypogonadism.18–20 Others are GnRH analogues such as leuprolide (Lupron), which are used in treating advanced prostate cancer. The hypogonadism is usually transient and resolves after stopping the offending agent.
Obesity and related conditions such as obstructive sleep apnea, insulin resistance, and type 2 diabetes mellitus are associated with low testosterone levels.21 Treatment should be directed at these underlying conditions and should include lifestyle measures such as weight loss and exercise, rather than simple prescribing of testosterone supplementation, as these efforts may provide multiple health benefits in addition to raising testosterone levels.22
Insulin resistance. In the setting of obesity, the total testosterone level may be low but the bioavailable and free testosterone (active hormone) levels may be normal. This is due to the effect of hyperinsulinemia on the liver, which results in a reduction in SHBG production.23 Low levels of both total and free testosterone can be seen in morbid obesity,24 but the cause remains unclear.
Type 2 diabetes mellitus. Testosterone levels have been reported to be lower in obese men who have diabetes than in those with obesity alone.24 This decrement, comparable in magnitude to that seen with other chronic diseases, suggests that low testosterone may simply be a marker of poor health.22,25,26
Sleep apnea. Disturbances in the sleep cycle, regardless of the underlying cause, can result in decreases in serum testosterone levels. Often, correcting the underlying sleep disturbance can result in a normalization of serum testosterone levels.27,28 A caveat about testosterone therapy: a thorough evaluation for sleep apnea should be undertaken in patients at high risk, since testosterone replacement therapy can adversely affect ventilatory drive and induce or worsen obstructive sleep apnea.29
Aging. Most reports have shown an agerelated decline in both total and free serum testosterone levels (commonly referred to as “andropause”), particularly in men over 60 years of age. There also appears to be a loss of circadian rhythm,30 although not all reports agree.6 It appears that factors such as functional status and overall health may play a more important role in the pathophysiology of hypogonadism in men of advanced age than age alone.
Hemochromatosis. Iron overload, regardless of the cause, can result in hypogonadism via deposition of iron in the hypothalamus, pituitary, or testes. Hereditary hemochromatosis is a common autosomal recessive disease characterized by increased iron absorption. Although both primary and secondary hypogonadism can occur with long-standing iron overload, the latter is much more common.31 Some cases of hypogonadism have been reported to reverse with iron depletion therapy.32
Hyperprolactinemia. Recognized causes of hyperprolactinemia in men include medications (dopamine antagonists, antipsychotics, metoclopramide [Reglan]), pituitary adenomas (microadenomas < 10 mm, macroadenomas ≥ 10 mm), lactotroph hyperfunction (stalk compression interrupting or reducing the tonic suppression of prolactin secretion by dopamine), hypothyroidism, stress, chronic renal failure, cirrhosis, chest wall injury (trauma), and active herpes zoster. The ensuing hypogonadism may be due to the compressive effect of a sellar mass or the direct effect of the prolactin elevation alone, since prolactin disrupts the pulsatile release of GnRH from the hypothalamus,33 required for normal LH and FSH secretion.
Estrogen excess can be either exogenous (from exposure to estrogen-containing contraceptives and creams) or endogenous (from testicular34,35 or very rare adrenal36 estrogen-secreting tumors). Of note, some cases of testicular neoplasms may be detectable only with ultrasonography. Computed tomography may be performed if an adrenal lesion is suspected.
Anabolic steroid abuse. Exposure to anabolic steroids, deliberately or inadvertently, can result in secondary hypogonadism and testicular atrophy, both of which may persist for years after stopping the anabolic agents. If you suspect anabolic steroid abuse, a urine anabolic steroid screen can be obtained.
Anorexia nervosa is far less common in men than in women.37,38 Elements in the history that suggest this disorder include excessive exercise and a low body mass index. Chronic malnutrition (cachexia), regardless of the cause, can result in secondary hypogonadism.
Acute illness (gonadotroph sick syndrome). Hypogonadism is a relatively common finding in any critical illness (analogous to euthyroid sick syndrome with respect to the hypothalamic-pituitary-thyroid axis).8 Testosterone levels are invariably low, so that assessment of testosterone status is not recommended in this setting. The low testosterone phase is usually transient and resolves with resolution or improvement of the underlying medical condition, such as sepsis or myocardial infarction.
HIV. Human immunodeficiency virus (HIV) infection can result in primary or secondary hypogonadism. It can occur with active HIV infection, in patients in whom control of viral replication has been achieved with highly active antiretroviral therapy, and even in patients who have normalized CD4+ cell counts.39 Hypogonadism in HIV patients is multifactorial and may be related to weight loss, opportunistic infections of the pituitary-hypothalamus or testes, or medications such as opioids (licit or illicit), ganciclovir (Cytovene), ketoconazole, the appetite stimulant megestrol (Megace), or cyclophosphamide (Cytoxan). Testosterone replacement therapy does not adversely affect the HIV disease process and in fact may help to avoid complications.
Chronic medical conditions such as cirrhosis, renal failure, and rheumatoid arthritis commonly result in hypogonadism, the pathogenesis of which may involve dysfunction at all levels of the hypothalamic-pituitary-go-nadal axis.40–45 Hypogonadism in the setting of chronic disease is multifactorial, being due not only to the metabolic disturbances seen with these illnesses (uremia in renal failure, elevated circulating estrogens in liver cirrhosis), but also to recurrent acute illness and hospitalization for infection in these immuno-compromised hosts, either from the underlying medical condition or as a result of medications (corticosteroids).
Alcohol abuse. Alcohol can have adverse effects at all levels of the hypothalamic-pituitary-gonadal axis, resulting in low serum testosterone and reduced spermatogenesis.46
Severe chronic primary hypothyroidism, manifested by an extreme elevation of serum thyroid-stimulating hormone (TSH), can result in hypopituitarism. Pituitary function usually recovers with restoration of euthyroidism.47,48
Pubertal delay. Depending on the age of presentation, differentiating pubertal delay from permanent hypogonadotropic hypogonadism can be challenging.
Acquired disorders that damage gonadotrophs
- Sellar mass or cyst—pituitary adenoma, craniopharyngioma, Rathke cleft cyst, meningioma
- Infiltrative lesion—lymphocytic hypophysitis, Langerhans cell histiocytosis, hemochromatosis, sarcoidosis, infection
- Metastatic lesion
- Trauma (head injury)
- Radiation exposure
- Surgery
- Stalk severance
- Pituitary apoplexy.
See Table 1 for a summary of the causes of male hypogonadism.
WHEN IS MRI INDICATED IN EVALUATING SECONDARY HYPOGONADISM?

The yield of pituitary-hypothalamic imaging in older men with secondary hypogonadism is fairly low in the absence of other pituitary hormone abnormalities and deficiencies. There are limited data regarding appropriate criteria for performing hypothalamic-pituitary imaging studies. However, a patient who has multiple anterior pituitary abnormalities on laboratory evaluation should undergo dedicated hypothalamic-pituitary magnetic resonance imaging (MRI).
The Endocrine Society Clinical Practice Guidelines11 recommend that MRI be performed to exclude a pituitary or hypothalamic tumor or infiltrative disease if the patient has severe secondary hypogonadism (serum testosterone < 150 ng/dL), panhypopituitarism, persistent hyperprolactinemia, or symptoms or signs of tumor mass effect such as headache, visual impairment, or a visual field defect.
WHO SHOULD UNDERGO ASSESSMENT OF TESTOSTERONE STATUS?
Screening for androgen deficiency in the asymptomatic general population is not recommended.11 The nonspecific nature of many of the signs and symptoms of androgen deficiency makes it difficult to give concrete recommendations as to who should have testosterone levels measured. Clinicians should consider testing if there is evidence of certain clinical disorders that are associated with low testosterone levels (see earlier discussion on the specific causes of primary and secondary hypogonadism).
When a male patient complains of erectile dysfunction, the investigation should include an assessment of serum testosterone. However, if a man who has a constellation of nonspecific symptoms asks for his testosterone level to be assessed (which is common, given the aggressive marketing of testosterone replacement by the pharmaceutical industry), we would recommend a basic evaluation that includes a comprehensive metabolic panel, complete blood count, and TSH level. Further testing should be determined by the history and physical examination. If no obvious explanation has been found for the patient’s symptoms at that point, assessment of serum testosterone may be warranted. More often than not the patient’s weight and limited physical activity are the driving forces behind the nonspecific symptoms, and counseling a patient on a life-style change can provide much benefit if the patient follows through with the physician’s recommendations.
Men whom we believe should not undergo assessment for testosterone deficiency are those who are acutely ill and hospitalized and those who are severely obese and are complaining of fatigue. Testosterone levels should be assessed only after the acute illness has resolved and, in a severely obese patient with fatigue, only after a thorough evaluation for sleep apnea has been undertaken.
TREAT THE UNDERLYING CAUSE, IF ONE CAN BE FOUND
If the evaluation of low testosterone leads to the diagnosis of a clear underlying condition that is amenable to treatment, such as prolactin elevation or sleep apnea, then treatment should be directed at the underlying cause, with subsequent monitoring of the patient’s symptoms and response in serum testosterone levels. In general, the use of dopamine agonist therapy in the management of hyperprolactinemia and, in cases of panhypopituitarism, of replacement therapy with levothyroxine (Synthroid), hydrocortisone, and possibly growth hormone and desmopressin (DDAVP), fall best under the purview of an endocrinologist. A caveat: serum TSH cannot be used to monitor levothyroxine replacement therapy in cases of secondary hypothyroidism. The clinical picture and serum free T4 and free T3 levels are used instead.
In the absence of a correctable (or immediately correctable) cause, testosterone supplementation can be initiated on an individualized basis in select patients who have clinical signs and symptoms of androgen deficiency if the benefits of treatment appear to outweigh the potential risks, and only after a thorough discussion with the patient.11 The Endocrine Society recommends against offering testosterone therapy to all older men with low testosterone.11
INFERTILITY
In men presenting with low serum testosterone, semen analysis is not routine. It is usually reserved for patients presenting with the primary complaint of infertility.
If an endocrine disorder such as prolactin elevation or hypothyroidism is the suspected cause of infertility, the patient should be referred to an endocrinologist for further evaluation and management. Treatment of male infertility should be directed at the underlying cause, but often requires exogenous human chorionic gonadotropin, FSH, GnRH (via a pulsatile pump), and possibly sperm harvesting from the testis with subsequent in vitro fertilization with intracytoplasmic sperm injection. It is critical that the partner be included in the evaluation of infertility.
These patients should be referred to a urologic or fertility center specializing in the diagnosis and treatment of infertility. For further information regarding male infertility, patients can be directed to www.fertilitylifelines.com.
CASE CONCLUDED
The patient’s low serum testosterone was confirmed on subsequent measurements at 8 am, with levels of 128 and 182 ng/dL (reference range 249–836). Other laboratory values:
- LH 1.4 mIU/mL (reference range 1.2–8.6)
- FSH 2.7 mIU/mL (1.3–9.9 mIU/mL)
(Both of these values are inappropriately normal in the setting of the low testosterone.)
- TSH 248 μIU/mL (0.4–5.5)
- Prolactin 24.6 ng/mL (1.6–18.8).
The patient was started on levothyroxine replacement therapy and after 3 months was noted to be euthyroid (TSH 1.8 μIU/mL) and to have a normal serum prolactin level. Testosterone levels (8 am) at this time were 350 ng/dL and 420 ng/dL.
Therefore, the cause of this patient’s hypogonadism was severe hypothyroidism and associated mild hyperprolactinemia. This case shows that a thorough evaluation is warranted before initiating testosterone therapy.
- Pitteloud N, Dwyer AA, DeCruz S, et al. Inhibition of luteinizing hormone secretion by testosterone in men requires aromatization for its pituitary but not its hypo-thalamic effects: evidence from the tandem study of normal and gonadotropin-releasing hormone-deficient men. J Clin Endocrinol Metab 2008; 93:784–791.
- Hayes FJ, DeCruz S, Seminara SB, Boepple PA, Crowley WF. Differential regulation of gonadotropin secretion by testosterone in the human male: absence of a negative feedback effect of testosterone on follicle-stimulating hormone secretion. J Clin Endocrinol Metab 2001; 86:53–58.
- Hayes FJ, Pitteloud N, DeCruz S, Crowley WF, Boepple PA. Importance of inhibin B in the regulation of FSH secretion in the human male. J Clin Endocrinol Metab 2001; 86:5541–5546.
- Pitteloud N, Dwyer AA, DeCruz S, et al. The relative role of gonadal sex steroids and gonadotropin-releasing hormone pulse frequency in the regulation of follicle-stimulating hormone secretion in men. J Clin Endocrinol Metab 2008; 93:2686–2692.
- Cooke RR, McIntosh JE, McIntosh RP. Circadian variation in serum free and non-SHBG-bound testosterone in normal men: measurements, and simulation using a mass action model. Clin Endocrinol (Oxf) 1993; 39:163–171.
- Diver MJ, Imtiaz KE, Ahmad AM, Vora JP, Fraser WD. Diurnal rhythms of serum total, free and bioavailable testosterone and of SHBG in middle-aged men compared with those in young men. Clin Endocrinol (Oxf) 2003; 58:710–717.
- Clair P, Claustrat B, Jordan D, Dechaud H, Sassolas G. Daily variations of plasma sex hormone-binding globulin binding capacity, testosterone and luteinizing hormone concentrations in healthy rested adult males. Horm Res 1985; 21:220–223.
- Woolf PD, Hamill RW, McDonald JV, Lee LA, Kelly M. Transient hypogonadotropic hypogonadism caused by critical illness. J Clin Endocrinol Metab 1985; 60:444–450.
- Rosner W, Auchus RJ, Azziz R, Sluss PM, Raff H. Position statement: utility, limitations, and pitfalls in measuring testosterone: an Endocrine Society position statement. J Clin Endocrinol Metab 2007; 92:405–413.
- Rosner W, Vesper H, et al; Endocrine Society; American Association for Clinical Chemistry; American Association of Clinical Endocrinologists; et al. Toward excellence in testosterone testing: a consensus statement. J Clin Endocrinol Metab 2010; 95:4542–4548.
- Bhasin S, Cunningham GR, Hayes FJ, et al; Task Force, Endocrine Society. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010; 95:2536–2559.
- Wu FC, Tajar A, Beynon JM, et al; EMAS Group. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med 2010; 363:123–135.
- Farrer JH, Sikka SC, Xie HW, Constantinide D, Rajfer J. Impaired testosterone biosynthesis in cryptorchidism. Fertil Steril 1985; 44:125–132.
- Sikka SC, Swerdloff RS, Rajfer J. In vitro inhibition of testosterone biosynthesis by ketoconazole. Endocrinology 1985; 116:1920–1925.
- Pallais JC, Au M, Pitteloud N, Seminara S, Crowley WF Jr. Kallmann syndrome. In:Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, eds. GeneReviews™ (Internet). Seattle, WA: University of Washington; 1993.
- Chevrier L, Guimiot F, de Roux N. GnRH receptor mutations in isolated gonadotropic deficiency. Mol Cell Endocrinol 2011; 346:21–28.
- Romero CJ, Pine-Twaddell E, Radovick S. Novel mutations associated with combined pituitary hormone deficiency. J Mol Endocrinol 2011; 46:R93–R102.
- Colameco S, Coren JS, Ciervo CA. Continuous opioid treatment for chronic noncancer pain: a time for moderation in prescribing. Postgrad Med 2009; 121:61–66.
- Fraser LA, Morrison D, Morley-Forster P, et al. Oral opioids for chronic non-cancer pain: higher prevalence of hypogonadism in men than in women. Exp Clin Endocrinol Diabetes 2009; 117:38–43.
- Morrison D, Capewell S, Reynolds SP, et al. Testosterone levels during systemic and inhaled corticosteroid therapy. Respir Med 1994; 88:659–663.
- Mah PM, Wittert GA. Obesity and testicular function. Mol Cell Endocrinol 2010; 316:180–186.
- Grossmann M. Low testosterone in men with type 2 diabetes: significance and treatment. J Clin Endocrinol Metab 2011; 96:2341–2353.
- Gascón F, Valle M, Martos R, et al. Sex hormone-binding globulin as a marker for hyperinsulinemia and/or insulin resistance in obese children. Eur J Endocrinol 2000; 143:85–89.
- Dhindsa S, Miller MG, McWhirter CL, et al. Testosterone concentrations in diabetic and nondiabetic obese men. Diabetes Care 2010; 33:1186–1192.
- Grossmann M, Gianatti EJ, Zajac JD. Testosterone and type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 2010; 17:247–256.
- Andersson B, Mårin P, Lissner L, Vermeulen A, Björntorp P. Testosterone concentrations in women and men with NIDDM. Diabetes Care 1994; 17:405–411.
- Santamaria JD, Prior JC, Fleetham JA. Reversible reproductive dysfunction in men with obstructive sleep apnoea. Clin Endocrinol (Oxf) 1988; 28:461–470.
- Grunstein RR, Handelsman DJ, Lawrence SJ, Blackwell C, Caterson ID, Sullivan CE. Neuroendocrine dysfunction in sleep apnea: reversal by continuous positive airways pressure therapy. J Clin Endocrinol Metab 1989; 68:352–358.
- Matsumoto AM, Sandblom RE, Schoene RB, et al. Testosterone replacement in hypogonadal men: effects on obstructive sleep apnoea, respiratory drives, and sleep. Clin Endocrinol (Oxf) 1985; 22:713–721.
- Bremner WJ, Vitiello MV, Prinz PN. Loss of circadian rhythmicity in blood testosterone levels with aging in normal men. J Clin Endocrinol Metab 1983; 56:1278–1281.
- McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab 2005; 90:2451–2455.
- Kelly TM, Edwards CQ, Meikle AW, Kushner JP. Hypogonadism in hemochromatosis: reversal with iron depletion. Ann Intern Med 1984; 101:629–632.
- Milenkovic L, D’Angelo G, Kelly PA, Weiner RI. Inhibition of gonadotropin hormone-releasing hormone release by prolactin from GT1 neuronal cell lines through prolactin receptors. Proc Natl Acad Sci U S A 1994; 91:1244–1247.
- Valensi P, Coussieu C, Kemeny JL, Attali JR, Amouroux J, Sebaoun J. Endocrine investigations in two cases of feminizing Leydig cell tumour. Acta Endocrinol (Copenh) 1987; 115:365–372.
- Young S, Gooneratne S, Straus FH, Zeller WP, Bulun SE, Rosenthal IM. Feminizing Sertoli cell tumors in boys with Peutz-Jeghers syndrome. Am J Surg Pathol 1995; 19:50–58.
- Zayed A, Stock JL, Liepman MK, Wollin M, Longcope C. Feminization as a result of both peripheral conversion of androgens and direct estrogen production from an adrenocortical carcinoma. J Endocrinol Invest 1994; 17:275–278.
- Russ MJ, Ackerman SH, Barakat R, Levy B. Hypogonadotropic hypogonadism and delayed puberty in a man with anorexia nervosa. Psychosomatics 1986; 27:737–739.
- Rigotti NA, Neer RM, Jameson L. Osteopenia and bone fractures in a man with anorexia nervosa and hypogonadism. JAMA 1986; 256:385–388.
- Cohan GR. HIV-associated hypogonadism. AIDS Read 2006; 16:341–345,348,352–354.
- Handelsman DJ, Strasser S, McDonald JA, Conway AJ, McCaughan GW. Hypothalamic-pituitary-testicular function in end-stage nonalcoholic liver disease before and after liver transplantation. Clin Endocrinol (Oxf) 1995; 43:331–337.
- Lim VS, Fang VS. Gonadal dysfunction in uremic men. A study of the hypothalamo-pituitary-testicular axis before and after renal transplantation. Am J Med 1975; 58:655–662.
- Handelsman DJ, Dong Q. Hypothalamo-pituitary gonadal axis in chronic renal failure. Endocrinol Metab Clin North Am 1993; 22:145–161.
- Handelsman DJ, Spaliviero JA, Turtle JR. Hypothalamic-pituitary function in experimental uremic hypogonadism. Endocrinology 1985; 117:1984–1995.
- Tengstrand B, Carlström K, Hafström I. Bioavailable testosterone in men with rheumatoid arthritis-high frequency of hypogonadism. Rheumatology (Oxford) 2002; 41:285–289.
- Tengstrand B, Carlström K, Hafström I. Gonadal hormones in men with rheumatoid arthritis--from onset through 2 years. J Rheumatol 2009; 36:887–892.
- Emanuele MA, Emanuele NV. Alcohol’s effects on male reproduction. Alcohol Health Res World 1998; 22:195–201.
- Meikle AW. The interrelationships between thyroid dysfunction and hypogonadism in men and boys. Thyroid 2004; 14( suppl 1):S17–S25.
- Vagenakis AG, Dole K, Braverman LE. Pituitary enlargement, pituitary failure, and primary hypothyroidism. Ann Intern Med 1976; 85:195–198.
Editor’s note: This article on the differential diagnosis of hypogonadism in men is the first of two articles. The second, to be published next month, focuses on the appropriate use of testosterone therapy.
A 54-year-old man is referred for evaluation of low testosterone. He had seen his primary care physician for complaints of diminished libido and erectile dysfunction for the past year and worsening fatigue over the past few years. He has not been formally diagnosed with any medical condition. His serum testosterone level is 180 ng/dL (reference range 249–836 ng/dL).
On physical examination, he is obese (body mass index 31 kg/m2) with a normal-appearing male body habitus, no gynecomastia, and normal testicles and prostate gland.
How should this patient be evaluated?
LOW TESTOSTERONE HAS MANY CAUSES
Male hypogonadism, ie, failure of the testes to produce adequate amounts of androgen or sperm, has become a common clinical finding, particularly in the older population. This is more likely the result of an increase in awareness and detection of the disorder by physicians rather than a true increase in prevalence.
The finding of a low serum testosterone value needs to be confirmed and thoroughly evaluated before starting treatment. It is important to determine whether the cause is a primary (hypergonadotropic) testicular disorder or secondary to a hypothalamic-pituitary process (hypogonadotropic or normogonadotropic).
THE HYPOTHALAMIC-PITUITARY-GONADAL AXIS
Testosterone production is under the control of luteinizing hormone (LH), whereas sperm production is under the control of follicle-stimulating hormone (FSH) (Figure 1). Both of these pituitary hormones are regulated by the pulsatile secretion of hypothalamic gonadotropin-releasing hormone (GnRH).
Testosterone (produced by Leydig cells) and inhibin B (produced by Sertoli cells within the seminiferous tubules) result in negative feedback inhibition of gonadotropin (LH and FSH) secretion. Testosterone and estradiol (produced by aromatization of testosterone) act at both pituitary and hypothalamic sites and are the principal regulators of LH secretion.1,2 Inhibin B is the major regulator of FSH secretion in men,3 but steroid feedback also occurs.2,4
TO FOLLOW UP A LOW TESTOSTERONE, CONFIRM THE VALUE NEAR 8 am
If a testosterone value is found to be low, it is important to determine the time that the sample was obtained. Serum testosterone levels follow a diurnal rhythm, at least in younger men, with values near 8 am being, on average, 30% higher than the trough levels later in the day.5–7 The timing of the diurnal variation may be different in night-shift workers, who may require assessment at a more appropriate time of the day (ie, upon awakening).
Another factor affecting testosterone levels is the patient’s health status at the time of testing. Values obtained in the hospital during an acute illness should be repeated once the event has resolved, as testosterone values decrease considerably in this setting.8 Even in outpatients, particularly in men over age 60, one must be sure that the low testosterone level was not obtained during a period of decompensation of one of the many comorbidities seen in these patients, such as coronary artery disease, congestive heart failure, or uncontrolled diabetes.
If an 8 am testosterone value is low, it is reasonable to obtain at least one confirmatory testosterone level on another day, near 8 am, in the next few weeks, when the patient is in good health. Confirming the testosterone level is important, particularly since commercially available testosterone assays are not well standardized and some are frankly unreliable.9,10 A repeat confirmatory level should always be performed by a reliable reference laboratory. If the testosterone level is still low, further evaluation is warranted.
TOTAL TESTOSTERONE VS BIOAVAILABLE TESTOSTERONE VS FREE TESTOSTERONE
Of the total circulating testosterone, 60% is bound to sex hormone-binding globulin (SHBG), 38% is bound to albumin, and only 2% is free. All of these fractions can be measured to assess for testosterone deficiency.
Free testosterone is the biologically active form of this hormone and, thus, the free testosterone level is considered to be a better representation of the true testosterone status. However, some clinicians believe that bioavailable testosterone (testosterone loosely bound to albumin + free testosterone) is a better reflection of the true level of the active hormone than the level of free testosterone alone.
There are situations in which the total testosterone level is low but bioavailable or free testosterone levels are normal. The level of total testosterone is affected by alterations in the levels of SHBG and albumin. A reduction in the level of SHBG can result in low total serum testosterone levels in patients with obesity or type 2 diabetes (states of insulin resistance), and also in cachexia, malnutrition, advanced cirrhosis, acromegaly, hypothyroidism, and nephrotic syndrome. SHBG can also be low in patients taking glucocorticoids, progestins, or androgenic steroids.11 In these settings, checking the level of free testosterone (the active hormone), bioavailable testosterone, or both, by a reliable reference laboratory, may be more appropriate.9,10
But regardless of which measurement is chosen, all testosterone levels—especially bioavailable and free testosterone values—should be interpreted with caution if they are not measured at a reliable reference laboratory.9,10 Interested readers may wish to see the US Centers for Disease Control and Prevention (CDC) Hormone Standardization Program Web site (www.cdc.gov/labstandards/hs.html) for more details, including a list of CDC-certified laboratories.
CLINICAL FEATURES OF LOW TESTOSTERONE
A history of erectile dysfunction, decreased libido, and fatigue may be seen in patients with low testosterone. However, one must realize that these symptoms—as well as others reported by men with low testosterone, such as depression, difficulty concentrating, irritability, and insomnia—are nonspecific and may be related to other medical conditions.12
Likewise, physical findings such as muscle weakness, reduced body hair, and altered fat distribution (abdominal obesity) are seen in men with low testosterone, but also in those with a number of other medical conditions.
Additional features suggest specific disorders, eg, anosmia in Kallmann syndrome; eunuchoid body habitus, gynecomastia, and small testes in Klinefelter syndrome.
Men with low testosterone may have low bone mineral density or anemia, or both.
Careful examination of the breasts for gynecomastia and the testes for size, consistency, and masses (testicular tumors) helps in formulating a differential diagnosis and in appropriately directing subsequent laboratory evaluation and diagnostic imaging.
LOW TESTOSTERONE: PRIMARY VS SECONDARY
A history of testicular trauma, systemic chemotherapy, or mumps orchitis should direct the physician’s attention to a testicular etiology. On the other hand, darkened or tanned skin (suggesting hemochromatosis), galactorrhea (suggesting hyperprolactinemia), or visual field deficits (suggesting a sellar mass) should direct the physician’s attention toward a pituitary-hypothalamic process.
Once the low testosterone value has been confirmed at least one time near 8 am, one should obtain LH and FSH values to help direct further evaluation in deciphering the etiology (Figure 2). Elevated (hypergonadotropic) values indicate a testicular disorder (primary hypogonadism), whereas low (hypogonadotropic) or normal (normogonadotropic) values point to a pituitary-hypothalamic process (secondary hypogonadism). It should be emphasized that, in the setting of a low testosterone level, LH and FSH values within the normal range are “inappropriately normal” so that further investigation is required.
This evaluation should also include serum prolactin, thyroid-stimulating hormone (TSH, also known as thyrotropin), free thyroxine (T4), and ferritin levels, the latter because hemochromatosis (iron overload) can cause both primary and secondary hypogonadism. If at any time in the evaluation the laboratory results suggest secondary hypogonadism, a full assessment of pituitary function should be undertaken.
Semen analysis is usually reserved for patients presenting with the primary complaint of infertility.
PRIMARY HYPOGONADISM
The patient should be carefully questioned about the age at which his problems began, about pubertal development, and about fertility. Causes of primary hypogonadism include:
- Karyotype abnormalities—Klinefelter syndrome (47, XXY syndrome) is the most common
- Toxin exposure, chemotherapy
- Congenital defects—anorchia, cryptorchidism13
- Orchitis (mumps, autoimmune)
- Testicular trauma or infarction
- Hemochromatosis
- Medications that inhibit androgen biosynthesis, eg, ketoconazole (Nizoral)14
- Increase in temperature of the testicular environment (due to varicocele or a large panniculus).
SECONDARY HYPOGONADISM
Causes of secondary hypogonadism include the following:
Congenital disorders
These disorders are usually diagnosed in childhood or adolescence, often after the patient is brought to the physician because of short stature or pubertal delay.
- Kallmann syndrome (anosmia and GnRH deficiency)15
- GnRH receptor mutation and deficiency16
- Genetic mutations associated with pituitary hormone deficiencies, eg, PROP-1 mutation.17
Acquired disorders that suppress gonadotrophs
Drugs. Long-term therapy with common medications such as opioids or corticosteroids can result in secondary hypogonadism.18–20 Others are GnRH analogues such as leuprolide (Lupron), which are used in treating advanced prostate cancer. The hypogonadism is usually transient and resolves after stopping the offending agent.
Obesity and related conditions such as obstructive sleep apnea, insulin resistance, and type 2 diabetes mellitus are associated with low testosterone levels.21 Treatment should be directed at these underlying conditions and should include lifestyle measures such as weight loss and exercise, rather than simple prescribing of testosterone supplementation, as these efforts may provide multiple health benefits in addition to raising testosterone levels.22
Insulin resistance. In the setting of obesity, the total testosterone level may be low but the bioavailable and free testosterone (active hormone) levels may be normal. This is due to the effect of hyperinsulinemia on the liver, which results in a reduction in SHBG production.23 Low levels of both total and free testosterone can be seen in morbid obesity,24 but the cause remains unclear.
Type 2 diabetes mellitus. Testosterone levels have been reported to be lower in obese men who have diabetes than in those with obesity alone.24 This decrement, comparable in magnitude to that seen with other chronic diseases, suggests that low testosterone may simply be a marker of poor health.22,25,26
Sleep apnea. Disturbances in the sleep cycle, regardless of the underlying cause, can result in decreases in serum testosterone levels. Often, correcting the underlying sleep disturbance can result in a normalization of serum testosterone levels.27,28 A caveat about testosterone therapy: a thorough evaluation for sleep apnea should be undertaken in patients at high risk, since testosterone replacement therapy can adversely affect ventilatory drive and induce or worsen obstructive sleep apnea.29
Aging. Most reports have shown an agerelated decline in both total and free serum testosterone levels (commonly referred to as “andropause”), particularly in men over 60 years of age. There also appears to be a loss of circadian rhythm,30 although not all reports agree.6 It appears that factors such as functional status and overall health may play a more important role in the pathophysiology of hypogonadism in men of advanced age than age alone.
Hemochromatosis. Iron overload, regardless of the cause, can result in hypogonadism via deposition of iron in the hypothalamus, pituitary, or testes. Hereditary hemochromatosis is a common autosomal recessive disease characterized by increased iron absorption. Although both primary and secondary hypogonadism can occur with long-standing iron overload, the latter is much more common.31 Some cases of hypogonadism have been reported to reverse with iron depletion therapy.32
Hyperprolactinemia. Recognized causes of hyperprolactinemia in men include medications (dopamine antagonists, antipsychotics, metoclopramide [Reglan]), pituitary adenomas (microadenomas < 10 mm, macroadenomas ≥ 10 mm), lactotroph hyperfunction (stalk compression interrupting or reducing the tonic suppression of prolactin secretion by dopamine), hypothyroidism, stress, chronic renal failure, cirrhosis, chest wall injury (trauma), and active herpes zoster. The ensuing hypogonadism may be due to the compressive effect of a sellar mass or the direct effect of the prolactin elevation alone, since prolactin disrupts the pulsatile release of GnRH from the hypothalamus,33 required for normal LH and FSH secretion.
Estrogen excess can be either exogenous (from exposure to estrogen-containing contraceptives and creams) or endogenous (from testicular34,35 or very rare adrenal36 estrogen-secreting tumors). Of note, some cases of testicular neoplasms may be detectable only with ultrasonography. Computed tomography may be performed if an adrenal lesion is suspected.
Anabolic steroid abuse. Exposure to anabolic steroids, deliberately or inadvertently, can result in secondary hypogonadism and testicular atrophy, both of which may persist for years after stopping the anabolic agents. If you suspect anabolic steroid abuse, a urine anabolic steroid screen can be obtained.
Anorexia nervosa is far less common in men than in women.37,38 Elements in the history that suggest this disorder include excessive exercise and a low body mass index. Chronic malnutrition (cachexia), regardless of the cause, can result in secondary hypogonadism.
Acute illness (gonadotroph sick syndrome). Hypogonadism is a relatively common finding in any critical illness (analogous to euthyroid sick syndrome with respect to the hypothalamic-pituitary-thyroid axis).8 Testosterone levels are invariably low, so that assessment of testosterone status is not recommended in this setting. The low testosterone phase is usually transient and resolves with resolution or improvement of the underlying medical condition, such as sepsis or myocardial infarction.
HIV. Human immunodeficiency virus (HIV) infection can result in primary or secondary hypogonadism. It can occur with active HIV infection, in patients in whom control of viral replication has been achieved with highly active antiretroviral therapy, and even in patients who have normalized CD4+ cell counts.39 Hypogonadism in HIV patients is multifactorial and may be related to weight loss, opportunistic infections of the pituitary-hypothalamus or testes, or medications such as opioids (licit or illicit), ganciclovir (Cytovene), ketoconazole, the appetite stimulant megestrol (Megace), or cyclophosphamide (Cytoxan). Testosterone replacement therapy does not adversely affect the HIV disease process and in fact may help to avoid complications.
Chronic medical conditions such as cirrhosis, renal failure, and rheumatoid arthritis commonly result in hypogonadism, the pathogenesis of which may involve dysfunction at all levels of the hypothalamic-pituitary-go-nadal axis.40–45 Hypogonadism in the setting of chronic disease is multifactorial, being due not only to the metabolic disturbances seen with these illnesses (uremia in renal failure, elevated circulating estrogens in liver cirrhosis), but also to recurrent acute illness and hospitalization for infection in these immuno-compromised hosts, either from the underlying medical condition or as a result of medications (corticosteroids).
Alcohol abuse. Alcohol can have adverse effects at all levels of the hypothalamic-pituitary-gonadal axis, resulting in low serum testosterone and reduced spermatogenesis.46
Severe chronic primary hypothyroidism, manifested by an extreme elevation of serum thyroid-stimulating hormone (TSH), can result in hypopituitarism. Pituitary function usually recovers with restoration of euthyroidism.47,48
Pubertal delay. Depending on the age of presentation, differentiating pubertal delay from permanent hypogonadotropic hypogonadism can be challenging.
Acquired disorders that damage gonadotrophs
- Sellar mass or cyst—pituitary adenoma, craniopharyngioma, Rathke cleft cyst, meningioma
- Infiltrative lesion—lymphocytic hypophysitis, Langerhans cell histiocytosis, hemochromatosis, sarcoidosis, infection
- Metastatic lesion
- Trauma (head injury)
- Radiation exposure
- Surgery
- Stalk severance
- Pituitary apoplexy.
See Table 1 for a summary of the causes of male hypogonadism.
WHEN IS MRI INDICATED IN EVALUATING SECONDARY HYPOGONADISM?
The yield of pituitary-hypothalamic imaging in older men with secondary hypogonadism is fairly low in the absence of other pituitary hormone abnormalities and deficiencies. There are limited data regarding appropriate criteria for performing hypothalamic-pituitary imaging studies. However, a patient who has multiple anterior pituitary abnormalities on laboratory evaluation should undergo dedicated hypothalamic-pituitary magnetic resonance imaging (MRI).
The Endocrine Society Clinical Practice Guidelines11 recommend that MRI be performed to exclude a pituitary or hypothalamic tumor or infiltrative disease if the patient has severe secondary hypogonadism (serum testosterone < 150 ng/dL), panhypopituitarism, persistent hyperprolactinemia, or symptoms or signs of tumor mass effect such as headache, visual impairment, or a visual field defect.
WHO SHOULD UNDERGO ASSESSMENT OF TESTOSTERONE STATUS?
Screening for androgen deficiency in the asymptomatic general population is not recommended.11 The nonspecific nature of many of the signs and symptoms of androgen deficiency makes it difficult to give concrete recommendations as to who should have testosterone levels measured. Clinicians should consider testing if there is evidence of certain clinical disorders that are associated with low testosterone levels (see earlier discussion on the specific causes of primary and secondary hypogonadism).
When a male patient complains of erectile dysfunction, the investigation should include an assessment of serum testosterone. However, if a man who has a constellation of nonspecific symptoms asks for his testosterone level to be assessed (which is common, given the aggressive marketing of testosterone replacement by the pharmaceutical industry), we would recommend a basic evaluation that includes a comprehensive metabolic panel, complete blood count, and TSH level. Further testing should be determined by the history and physical examination. If no obvious explanation has been found for the patient’s symptoms at that point, assessment of serum testosterone may be warranted. More often than not the patient’s weight and limited physical activity are the driving forces behind the nonspecific symptoms, and counseling a patient on a life-style change can provide much benefit if the patient follows through with the physician’s recommendations.
Men whom we believe should not undergo assessment for testosterone deficiency are those who are acutely ill and hospitalized and those who are severely obese and are complaining of fatigue. Testosterone levels should be assessed only after the acute illness has resolved and, in a severely obese patient with fatigue, only after a thorough evaluation for sleep apnea has been undertaken.
TREAT THE UNDERLYING CAUSE, IF ONE CAN BE FOUND
If the evaluation of low testosterone leads to the diagnosis of a clear underlying condition that is amenable to treatment, such as prolactin elevation or sleep apnea, then treatment should be directed at the underlying cause, with subsequent monitoring of the patient’s symptoms and response in serum testosterone levels. In general, the use of dopamine agonist therapy in the management of hyperprolactinemia and, in cases of panhypopituitarism, of replacement therapy with levothyroxine (Synthroid), hydrocortisone, and possibly growth hormone and desmopressin (DDAVP), fall best under the purview of an endocrinologist. A caveat: serum TSH cannot be used to monitor levothyroxine replacement therapy in cases of secondary hypothyroidism. The clinical picture and serum free T4 and free T3 levels are used instead.
In the absence of a correctable (or immediately correctable) cause, testosterone supplementation can be initiated on an individualized basis in select patients who have clinical signs and symptoms of androgen deficiency if the benefits of treatment appear to outweigh the potential risks, and only after a thorough discussion with the patient.11 The Endocrine Society recommends against offering testosterone therapy to all older men with low testosterone.11
INFERTILITY
In men presenting with low serum testosterone, semen analysis is not routine. It is usually reserved for patients presenting with the primary complaint of infertility.
If an endocrine disorder such as prolactin elevation or hypothyroidism is the suspected cause of infertility, the patient should be referred to an endocrinologist for further evaluation and management. Treatment of male infertility should be directed at the underlying cause, but often requires exogenous human chorionic gonadotropin, FSH, GnRH (via a pulsatile pump), and possibly sperm harvesting from the testis with subsequent in vitro fertilization with intracytoplasmic sperm injection. It is critical that the partner be included in the evaluation of infertility.
These patients should be referred to a urologic or fertility center specializing in the diagnosis and treatment of infertility. For further information regarding male infertility, patients can be directed to www.fertilitylifelines.com.
CASE CONCLUDED
The patient’s low serum testosterone was confirmed on subsequent measurements at 8 am, with levels of 128 and 182 ng/dL (reference range 249–836). Other laboratory values:
- LH 1.4 mIU/mL (reference range 1.2–8.6)
- FSH 2.7 mIU/mL (1.3–9.9 mIU/mL)
(Both of these values are inappropriately normal in the setting of the low testosterone.)
- TSH 248 μIU/mL (0.4–5.5)
- Prolactin 24.6 ng/mL (1.6–18.8).
The patient was started on levothyroxine replacement therapy and after 3 months was noted to be euthyroid (TSH 1.8 μIU/mL) and to have a normal serum prolactin level. Testosterone levels (8 am) at this time were 350 ng/dL and 420 ng/dL.
Therefore, the cause of this patient’s hypogonadism was severe hypothyroidism and associated mild hyperprolactinemia. This case shows that a thorough evaluation is warranted before initiating testosterone therapy.
Editor’s note: This article on the differential diagnosis of hypogonadism in men is the first of two articles. The second, to be published next month, focuses on the appropriate use of testosterone therapy.
A 54-year-old man is referred for evaluation of low testosterone. He had seen his primary care physician for complaints of diminished libido and erectile dysfunction for the past year and worsening fatigue over the past few years. He has not been formally diagnosed with any medical condition. His serum testosterone level is 180 ng/dL (reference range 249–836 ng/dL).
On physical examination, he is obese (body mass index 31 kg/m2) with a normal-appearing male body habitus, no gynecomastia, and normal testicles and prostate gland.
How should this patient be evaluated?
LOW TESTOSTERONE HAS MANY CAUSES
Male hypogonadism, ie, failure of the testes to produce adequate amounts of androgen or sperm, has become a common clinical finding, particularly in the older population. This is more likely the result of an increase in awareness and detection of the disorder by physicians rather than a true increase in prevalence.
The finding of a low serum testosterone value needs to be confirmed and thoroughly evaluated before starting treatment. It is important to determine whether the cause is a primary (hypergonadotropic) testicular disorder or secondary to a hypothalamic-pituitary process (hypogonadotropic or normogonadotropic).
THE HYPOTHALAMIC-PITUITARY-GONADAL AXIS
Testosterone production is under the control of luteinizing hormone (LH), whereas sperm production is under the control of follicle-stimulating hormone (FSH) (Figure 1). Both of these pituitary hormones are regulated by the pulsatile secretion of hypothalamic gonadotropin-releasing hormone (GnRH).
Testosterone (produced by Leydig cells) and inhibin B (produced by Sertoli cells within the seminiferous tubules) result in negative feedback inhibition of gonadotropin (LH and FSH) secretion. Testosterone and estradiol (produced by aromatization of testosterone) act at both pituitary and hypothalamic sites and are the principal regulators of LH secretion.1,2 Inhibin B is the major regulator of FSH secretion in men,3 but steroid feedback also occurs.2,4
TO FOLLOW UP A LOW TESTOSTERONE, CONFIRM THE VALUE NEAR 8 am
If a testosterone value is found to be low, it is important to determine the time that the sample was obtained. Serum testosterone levels follow a diurnal rhythm, at least in younger men, with values near 8 am being, on average, 30% higher than the trough levels later in the day.5–7 The timing of the diurnal variation may be different in night-shift workers, who may require assessment at a more appropriate time of the day (ie, upon awakening).
Another factor affecting testosterone levels is the patient’s health status at the time of testing. Values obtained in the hospital during an acute illness should be repeated once the event has resolved, as testosterone values decrease considerably in this setting.8 Even in outpatients, particularly in men over age 60, one must be sure that the low testosterone level was not obtained during a period of decompensation of one of the many comorbidities seen in these patients, such as coronary artery disease, congestive heart failure, or uncontrolled diabetes.
If an 8 am testosterone value is low, it is reasonable to obtain at least one confirmatory testosterone level on another day, near 8 am, in the next few weeks, when the patient is in good health. Confirming the testosterone level is important, particularly since commercially available testosterone assays are not well standardized and some are frankly unreliable.9,10 A repeat confirmatory level should always be performed by a reliable reference laboratory. If the testosterone level is still low, further evaluation is warranted.
TOTAL TESTOSTERONE VS BIOAVAILABLE TESTOSTERONE VS FREE TESTOSTERONE
Of the total circulating testosterone, 60% is bound to sex hormone-binding globulin (SHBG), 38% is bound to albumin, and only 2% is free. All of these fractions can be measured to assess for testosterone deficiency.
Free testosterone is the biologically active form of this hormone and, thus, the free testosterone level is considered to be a better representation of the true testosterone status. However, some clinicians believe that bioavailable testosterone (testosterone loosely bound to albumin + free testosterone) is a better reflection of the true level of the active hormone than the level of free testosterone alone.
There are situations in which the total testosterone level is low but bioavailable or free testosterone levels are normal. The level of total testosterone is affected by alterations in the levels of SHBG and albumin. A reduction in the level of SHBG can result in low total serum testosterone levels in patients with obesity or type 2 diabetes (states of insulin resistance), and also in cachexia, malnutrition, advanced cirrhosis, acromegaly, hypothyroidism, and nephrotic syndrome. SHBG can also be low in patients taking glucocorticoids, progestins, or androgenic steroids.11 In these settings, checking the level of free testosterone (the active hormone), bioavailable testosterone, or both, by a reliable reference laboratory, may be more appropriate.9,10
But regardless of which measurement is chosen, all testosterone levels—especially bioavailable and free testosterone values—should be interpreted with caution if they are not measured at a reliable reference laboratory.9,10 Interested readers may wish to see the US Centers for Disease Control and Prevention (CDC) Hormone Standardization Program Web site (www.cdc.gov/labstandards/hs.html) for more details, including a list of CDC-certified laboratories.
CLINICAL FEATURES OF LOW TESTOSTERONE
A history of erectile dysfunction, decreased libido, and fatigue may be seen in patients with low testosterone. However, one must realize that these symptoms—as well as others reported by men with low testosterone, such as depression, difficulty concentrating, irritability, and insomnia—are nonspecific and may be related to other medical conditions.12
Likewise, physical findings such as muscle weakness, reduced body hair, and altered fat distribution (abdominal obesity) are seen in men with low testosterone, but also in those with a number of other medical conditions.
Additional features suggest specific disorders, eg, anosmia in Kallmann syndrome; eunuchoid body habitus, gynecomastia, and small testes in Klinefelter syndrome.
Men with low testosterone may have low bone mineral density or anemia, or both.
Careful examination of the breasts for gynecomastia and the testes for size, consistency, and masses (testicular tumors) helps in formulating a differential diagnosis and in appropriately directing subsequent laboratory evaluation and diagnostic imaging.
LOW TESTOSTERONE: PRIMARY VS SECONDARY
A history of testicular trauma, systemic chemotherapy, or mumps orchitis should direct the physician’s attention to a testicular etiology. On the other hand, darkened or tanned skin (suggesting hemochromatosis), galactorrhea (suggesting hyperprolactinemia), or visual field deficits (suggesting a sellar mass) should direct the physician’s attention toward a pituitary-hypothalamic process.
Once the low testosterone value has been confirmed at least one time near 8 am, one should obtain LH and FSH values to help direct further evaluation in deciphering the etiology (Figure 2). Elevated (hypergonadotropic) values indicate a testicular disorder (primary hypogonadism), whereas low (hypogonadotropic) or normal (normogonadotropic) values point to a pituitary-hypothalamic process (secondary hypogonadism). It should be emphasized that, in the setting of a low testosterone level, LH and FSH values within the normal range are “inappropriately normal” so that further investigation is required.
This evaluation should also include serum prolactin, thyroid-stimulating hormone (TSH, also known as thyrotropin), free thyroxine (T4), and ferritin levels, the latter because hemochromatosis (iron overload) can cause both primary and secondary hypogonadism. If at any time in the evaluation the laboratory results suggest secondary hypogonadism, a full assessment of pituitary function should be undertaken.
Semen analysis is usually reserved for patients presenting with the primary complaint of infertility.
PRIMARY HYPOGONADISM
The patient should be carefully questioned about the age at which his problems began, about pubertal development, and about fertility. Causes of primary hypogonadism include:
- Karyotype abnormalities—Klinefelter syndrome (47, XXY syndrome) is the most common
- Toxin exposure, chemotherapy
- Congenital defects—anorchia, cryptorchidism13
- Orchitis (mumps, autoimmune)
- Testicular trauma or infarction
- Hemochromatosis
- Medications that inhibit androgen biosynthesis, eg, ketoconazole (Nizoral)14
- Increase in temperature of the testicular environment (due to varicocele or a large panniculus).
SECONDARY HYPOGONADISM
Causes of secondary hypogonadism include the following:
Congenital disorders
These disorders are usually diagnosed in childhood or adolescence, often after the patient is brought to the physician because of short stature or pubertal delay.
- Kallmann syndrome (anosmia and GnRH deficiency)15
- GnRH receptor mutation and deficiency16
- Genetic mutations associated with pituitary hormone deficiencies, eg, PROP-1 mutation.17
Acquired disorders that suppress gonadotrophs
Drugs. Long-term therapy with common medications such as opioids or corticosteroids can result in secondary hypogonadism.18–20 Others are GnRH analogues such as leuprolide (Lupron), which are used in treating advanced prostate cancer. The hypogonadism is usually transient and resolves after stopping the offending agent.
Obesity and related conditions such as obstructive sleep apnea, insulin resistance, and type 2 diabetes mellitus are associated with low testosterone levels.21 Treatment should be directed at these underlying conditions and should include lifestyle measures such as weight loss and exercise, rather than simple prescribing of testosterone supplementation, as these efforts may provide multiple health benefits in addition to raising testosterone levels.22
Insulin resistance. In the setting of obesity, the total testosterone level may be low but the bioavailable and free testosterone (active hormone) levels may be normal. This is due to the effect of hyperinsulinemia on the liver, which results in a reduction in SHBG production.23 Low levels of both total and free testosterone can be seen in morbid obesity,24 but the cause remains unclear.
Type 2 diabetes mellitus. Testosterone levels have been reported to be lower in obese men who have diabetes than in those with obesity alone.24 This decrement, comparable in magnitude to that seen with other chronic diseases, suggests that low testosterone may simply be a marker of poor health.22,25,26
Sleep apnea. Disturbances in the sleep cycle, regardless of the underlying cause, can result in decreases in serum testosterone levels. Often, correcting the underlying sleep disturbance can result in a normalization of serum testosterone levels.27,28 A caveat about testosterone therapy: a thorough evaluation for sleep apnea should be undertaken in patients at high risk, since testosterone replacement therapy can adversely affect ventilatory drive and induce or worsen obstructive sleep apnea.29
Aging. Most reports have shown an agerelated decline in both total and free serum testosterone levels (commonly referred to as “andropause”), particularly in men over 60 years of age. There also appears to be a loss of circadian rhythm,30 although not all reports agree.6 It appears that factors such as functional status and overall health may play a more important role in the pathophysiology of hypogonadism in men of advanced age than age alone.
Hemochromatosis. Iron overload, regardless of the cause, can result in hypogonadism via deposition of iron in the hypothalamus, pituitary, or testes. Hereditary hemochromatosis is a common autosomal recessive disease characterized by increased iron absorption. Although both primary and secondary hypogonadism can occur with long-standing iron overload, the latter is much more common.31 Some cases of hypogonadism have been reported to reverse with iron depletion therapy.32
Hyperprolactinemia. Recognized causes of hyperprolactinemia in men include medications (dopamine antagonists, antipsychotics, metoclopramide [Reglan]), pituitary adenomas (microadenomas < 10 mm, macroadenomas ≥ 10 mm), lactotroph hyperfunction (stalk compression interrupting or reducing the tonic suppression of prolactin secretion by dopamine), hypothyroidism, stress, chronic renal failure, cirrhosis, chest wall injury (trauma), and active herpes zoster. The ensuing hypogonadism may be due to the compressive effect of a sellar mass or the direct effect of the prolactin elevation alone, since prolactin disrupts the pulsatile release of GnRH from the hypothalamus,33 required for normal LH and FSH secretion.
Estrogen excess can be either exogenous (from exposure to estrogen-containing contraceptives and creams) or endogenous (from testicular34,35 or very rare adrenal36 estrogen-secreting tumors). Of note, some cases of testicular neoplasms may be detectable only with ultrasonography. Computed tomography may be performed if an adrenal lesion is suspected.
Anabolic steroid abuse. Exposure to anabolic steroids, deliberately or inadvertently, can result in secondary hypogonadism and testicular atrophy, both of which may persist for years after stopping the anabolic agents. If you suspect anabolic steroid abuse, a urine anabolic steroid screen can be obtained.
Anorexia nervosa is far less common in men than in women.37,38 Elements in the history that suggest this disorder include excessive exercise and a low body mass index. Chronic malnutrition (cachexia), regardless of the cause, can result in secondary hypogonadism.
Acute illness (gonadotroph sick syndrome). Hypogonadism is a relatively common finding in any critical illness (analogous to euthyroid sick syndrome with respect to the hypothalamic-pituitary-thyroid axis).8 Testosterone levels are invariably low, so that assessment of testosterone status is not recommended in this setting. The low testosterone phase is usually transient and resolves with resolution or improvement of the underlying medical condition, such as sepsis or myocardial infarction.
HIV. Human immunodeficiency virus (HIV) infection can result in primary or secondary hypogonadism. It can occur with active HIV infection, in patients in whom control of viral replication has been achieved with highly active antiretroviral therapy, and even in patients who have normalized CD4+ cell counts.39 Hypogonadism in HIV patients is multifactorial and may be related to weight loss, opportunistic infections of the pituitary-hypothalamus or testes, or medications such as opioids (licit or illicit), ganciclovir (Cytovene), ketoconazole, the appetite stimulant megestrol (Megace), or cyclophosphamide (Cytoxan). Testosterone replacement therapy does not adversely affect the HIV disease process and in fact may help to avoid complications.
Chronic medical conditions such as cirrhosis, renal failure, and rheumatoid arthritis commonly result in hypogonadism, the pathogenesis of which may involve dysfunction at all levels of the hypothalamic-pituitary-go-nadal axis.40–45 Hypogonadism in the setting of chronic disease is multifactorial, being due not only to the metabolic disturbances seen with these illnesses (uremia in renal failure, elevated circulating estrogens in liver cirrhosis), but also to recurrent acute illness and hospitalization for infection in these immuno-compromised hosts, either from the underlying medical condition or as a result of medications (corticosteroids).
Alcohol abuse. Alcohol can have adverse effects at all levels of the hypothalamic-pituitary-gonadal axis, resulting in low serum testosterone and reduced spermatogenesis.46
Severe chronic primary hypothyroidism, manifested by an extreme elevation of serum thyroid-stimulating hormone (TSH), can result in hypopituitarism. Pituitary function usually recovers with restoration of euthyroidism.47,48
Pubertal delay. Depending on the age of presentation, differentiating pubertal delay from permanent hypogonadotropic hypogonadism can be challenging.
Acquired disorders that damage gonadotrophs
- Sellar mass or cyst—pituitary adenoma, craniopharyngioma, Rathke cleft cyst, meningioma
- Infiltrative lesion—lymphocytic hypophysitis, Langerhans cell histiocytosis, hemochromatosis, sarcoidosis, infection
- Metastatic lesion
- Trauma (head injury)
- Radiation exposure
- Surgery
- Stalk severance
- Pituitary apoplexy.
See Table 1 for a summary of the causes of male hypogonadism.
WHEN IS MRI INDICATED IN EVALUATING SECONDARY HYPOGONADISM?
The yield of pituitary-hypothalamic imaging in older men with secondary hypogonadism is fairly low in the absence of other pituitary hormone abnormalities and deficiencies. There are limited data regarding appropriate criteria for performing hypothalamic-pituitary imaging studies. However, a patient who has multiple anterior pituitary abnormalities on laboratory evaluation should undergo dedicated hypothalamic-pituitary magnetic resonance imaging (MRI).
The Endocrine Society Clinical Practice Guidelines11 recommend that MRI be performed to exclude a pituitary or hypothalamic tumor or infiltrative disease if the patient has severe secondary hypogonadism (serum testosterone < 150 ng/dL), panhypopituitarism, persistent hyperprolactinemia, or symptoms or signs of tumor mass effect such as headache, visual impairment, or a visual field defect.
WHO SHOULD UNDERGO ASSESSMENT OF TESTOSTERONE STATUS?
Screening for androgen deficiency in the asymptomatic general population is not recommended.11 The nonspecific nature of many of the signs and symptoms of androgen deficiency makes it difficult to give concrete recommendations as to who should have testosterone levels measured. Clinicians should consider testing if there is evidence of certain clinical disorders that are associated with low testosterone levels (see earlier discussion on the specific causes of primary and secondary hypogonadism).
When a male patient complains of erectile dysfunction, the investigation should include an assessment of serum testosterone. However, if a man who has a constellation of nonspecific symptoms asks for his testosterone level to be assessed (which is common, given the aggressive marketing of testosterone replacement by the pharmaceutical industry), we would recommend a basic evaluation that includes a comprehensive metabolic panel, complete blood count, and TSH level. Further testing should be determined by the history and physical examination. If no obvious explanation has been found for the patient’s symptoms at that point, assessment of serum testosterone may be warranted. More often than not the patient’s weight and limited physical activity are the driving forces behind the nonspecific symptoms, and counseling a patient on a life-style change can provide much benefit if the patient follows through with the physician’s recommendations.
Men whom we believe should not undergo assessment for testosterone deficiency are those who are acutely ill and hospitalized and those who are severely obese and are complaining of fatigue. Testosterone levels should be assessed only after the acute illness has resolved and, in a severely obese patient with fatigue, only after a thorough evaluation for sleep apnea has been undertaken.
TREAT THE UNDERLYING CAUSE, IF ONE CAN BE FOUND
If the evaluation of low testosterone leads to the diagnosis of a clear underlying condition that is amenable to treatment, such as prolactin elevation or sleep apnea, then treatment should be directed at the underlying cause, with subsequent monitoring of the patient’s symptoms and response in serum testosterone levels. In general, the use of dopamine agonist therapy in the management of hyperprolactinemia and, in cases of panhypopituitarism, of replacement therapy with levothyroxine (Synthroid), hydrocortisone, and possibly growth hormone and desmopressin (DDAVP), fall best under the purview of an endocrinologist. A caveat: serum TSH cannot be used to monitor levothyroxine replacement therapy in cases of secondary hypothyroidism. The clinical picture and serum free T4 and free T3 levels are used instead.
In the absence of a correctable (or immediately correctable) cause, testosterone supplementation can be initiated on an individualized basis in select patients who have clinical signs and symptoms of androgen deficiency if the benefits of treatment appear to outweigh the potential risks, and only after a thorough discussion with the patient.11 The Endocrine Society recommends against offering testosterone therapy to all older men with low testosterone.11
INFERTILITY
In men presenting with low serum testosterone, semen analysis is not routine. It is usually reserved for patients presenting with the primary complaint of infertility.
If an endocrine disorder such as prolactin elevation or hypothyroidism is the suspected cause of infertility, the patient should be referred to an endocrinologist for further evaluation and management. Treatment of male infertility should be directed at the underlying cause, but often requires exogenous human chorionic gonadotropin, FSH, GnRH (via a pulsatile pump), and possibly sperm harvesting from the testis with subsequent in vitro fertilization with intracytoplasmic sperm injection. It is critical that the partner be included in the evaluation of infertility.
These patients should be referred to a urologic or fertility center specializing in the diagnosis and treatment of infertility. For further information regarding male infertility, patients can be directed to www.fertilitylifelines.com.
CASE CONCLUDED
The patient’s low serum testosterone was confirmed on subsequent measurements at 8 am, with levels of 128 and 182 ng/dL (reference range 249–836). Other laboratory values:
- LH 1.4 mIU/mL (reference range 1.2–8.6)
- FSH 2.7 mIU/mL (1.3–9.9 mIU/mL)
(Both of these values are inappropriately normal in the setting of the low testosterone.)
- TSH 248 μIU/mL (0.4–5.5)
- Prolactin 24.6 ng/mL (1.6–18.8).
The patient was started on levothyroxine replacement therapy and after 3 months was noted to be euthyroid (TSH 1.8 μIU/mL) and to have a normal serum prolactin level. Testosterone levels (8 am) at this time were 350 ng/dL and 420 ng/dL.
Therefore, the cause of this patient’s hypogonadism was severe hypothyroidism and associated mild hyperprolactinemia. This case shows that a thorough evaluation is warranted before initiating testosterone therapy.
- Pitteloud N, Dwyer AA, DeCruz S, et al. Inhibition of luteinizing hormone secretion by testosterone in men requires aromatization for its pituitary but not its hypo-thalamic effects: evidence from the tandem study of normal and gonadotropin-releasing hormone-deficient men. J Clin Endocrinol Metab 2008; 93:784–791.
- Hayes FJ, DeCruz S, Seminara SB, Boepple PA, Crowley WF. Differential regulation of gonadotropin secretion by testosterone in the human male: absence of a negative feedback effect of testosterone on follicle-stimulating hormone secretion. J Clin Endocrinol Metab 2001; 86:53–58.
- Hayes FJ, Pitteloud N, DeCruz S, Crowley WF, Boepple PA. Importance of inhibin B in the regulation of FSH secretion in the human male. J Clin Endocrinol Metab 2001; 86:5541–5546.
- Pitteloud N, Dwyer AA, DeCruz S, et al. The relative role of gonadal sex steroids and gonadotropin-releasing hormone pulse frequency in the regulation of follicle-stimulating hormone secretion in men. J Clin Endocrinol Metab 2008; 93:2686–2692.
- Cooke RR, McIntosh JE, McIntosh RP. Circadian variation in serum free and non-SHBG-bound testosterone in normal men: measurements, and simulation using a mass action model. Clin Endocrinol (Oxf) 1993; 39:163–171.
- Diver MJ, Imtiaz KE, Ahmad AM, Vora JP, Fraser WD. Diurnal rhythms of serum total, free and bioavailable testosterone and of SHBG in middle-aged men compared with those in young men. Clin Endocrinol (Oxf) 2003; 58:710–717.
- Clair P, Claustrat B, Jordan D, Dechaud H, Sassolas G. Daily variations of plasma sex hormone-binding globulin binding capacity, testosterone and luteinizing hormone concentrations in healthy rested adult males. Horm Res 1985; 21:220–223.
- Woolf PD, Hamill RW, McDonald JV, Lee LA, Kelly M. Transient hypogonadotropic hypogonadism caused by critical illness. J Clin Endocrinol Metab 1985; 60:444–450.
- Rosner W, Auchus RJ, Azziz R, Sluss PM, Raff H. Position statement: utility, limitations, and pitfalls in measuring testosterone: an Endocrine Society position statement. J Clin Endocrinol Metab 2007; 92:405–413.
- Rosner W, Vesper H, et al; Endocrine Society; American Association for Clinical Chemistry; American Association of Clinical Endocrinologists; et al. Toward excellence in testosterone testing: a consensus statement. J Clin Endocrinol Metab 2010; 95:4542–4548.
- Bhasin S, Cunningham GR, Hayes FJ, et al; Task Force, Endocrine Society. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010; 95:2536–2559.
- Wu FC, Tajar A, Beynon JM, et al; EMAS Group. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med 2010; 363:123–135.
- Farrer JH, Sikka SC, Xie HW, Constantinide D, Rajfer J. Impaired testosterone biosynthesis in cryptorchidism. Fertil Steril 1985; 44:125–132.
- Sikka SC, Swerdloff RS, Rajfer J. In vitro inhibition of testosterone biosynthesis by ketoconazole. Endocrinology 1985; 116:1920–1925.
- Pallais JC, Au M, Pitteloud N, Seminara S, Crowley WF Jr. Kallmann syndrome. In:Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, eds. GeneReviews™ (Internet). Seattle, WA: University of Washington; 1993.
- Chevrier L, Guimiot F, de Roux N. GnRH receptor mutations in isolated gonadotropic deficiency. Mol Cell Endocrinol 2011; 346:21–28.
- Romero CJ, Pine-Twaddell E, Radovick S. Novel mutations associated with combined pituitary hormone deficiency. J Mol Endocrinol 2011; 46:R93–R102.
- Colameco S, Coren JS, Ciervo CA. Continuous opioid treatment for chronic noncancer pain: a time for moderation in prescribing. Postgrad Med 2009; 121:61–66.
- Fraser LA, Morrison D, Morley-Forster P, et al. Oral opioids for chronic non-cancer pain: higher prevalence of hypogonadism in men than in women. Exp Clin Endocrinol Diabetes 2009; 117:38–43.
- Morrison D, Capewell S, Reynolds SP, et al. Testosterone levels during systemic and inhaled corticosteroid therapy. Respir Med 1994; 88:659–663.
- Mah PM, Wittert GA. Obesity and testicular function. Mol Cell Endocrinol 2010; 316:180–186.
- Grossmann M. Low testosterone in men with type 2 diabetes: significance and treatment. J Clin Endocrinol Metab 2011; 96:2341–2353.
- Gascón F, Valle M, Martos R, et al. Sex hormone-binding globulin as a marker for hyperinsulinemia and/or insulin resistance in obese children. Eur J Endocrinol 2000; 143:85–89.
- Dhindsa S, Miller MG, McWhirter CL, et al. Testosterone concentrations in diabetic and nondiabetic obese men. Diabetes Care 2010; 33:1186–1192.
- Grossmann M, Gianatti EJ, Zajac JD. Testosterone and type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 2010; 17:247–256.
- Andersson B, Mårin P, Lissner L, Vermeulen A, Björntorp P. Testosterone concentrations in women and men with NIDDM. Diabetes Care 1994; 17:405–411.
- Santamaria JD, Prior JC, Fleetham JA. Reversible reproductive dysfunction in men with obstructive sleep apnoea. Clin Endocrinol (Oxf) 1988; 28:461–470.
- Grunstein RR, Handelsman DJ, Lawrence SJ, Blackwell C, Caterson ID, Sullivan CE. Neuroendocrine dysfunction in sleep apnea: reversal by continuous positive airways pressure therapy. J Clin Endocrinol Metab 1989; 68:352–358.
- Matsumoto AM, Sandblom RE, Schoene RB, et al. Testosterone replacement in hypogonadal men: effects on obstructive sleep apnoea, respiratory drives, and sleep. Clin Endocrinol (Oxf) 1985; 22:713–721.
- Bremner WJ, Vitiello MV, Prinz PN. Loss of circadian rhythmicity in blood testosterone levels with aging in normal men. J Clin Endocrinol Metab 1983; 56:1278–1281.
- McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab 2005; 90:2451–2455.
- Kelly TM, Edwards CQ, Meikle AW, Kushner JP. Hypogonadism in hemochromatosis: reversal with iron depletion. Ann Intern Med 1984; 101:629–632.
- Milenkovic L, D’Angelo G, Kelly PA, Weiner RI. Inhibition of gonadotropin hormone-releasing hormone release by prolactin from GT1 neuronal cell lines through prolactin receptors. Proc Natl Acad Sci U S A 1994; 91:1244–1247.
- Valensi P, Coussieu C, Kemeny JL, Attali JR, Amouroux J, Sebaoun J. Endocrine investigations in two cases of feminizing Leydig cell tumour. Acta Endocrinol (Copenh) 1987; 115:365–372.
- Young S, Gooneratne S, Straus FH, Zeller WP, Bulun SE, Rosenthal IM. Feminizing Sertoli cell tumors in boys with Peutz-Jeghers syndrome. Am J Surg Pathol 1995; 19:50–58.
- Zayed A, Stock JL, Liepman MK, Wollin M, Longcope C. Feminization as a result of both peripheral conversion of androgens and direct estrogen production from an adrenocortical carcinoma. J Endocrinol Invest 1994; 17:275–278.
- Russ MJ, Ackerman SH, Barakat R, Levy B. Hypogonadotropic hypogonadism and delayed puberty in a man with anorexia nervosa. Psychosomatics 1986; 27:737–739.
- Rigotti NA, Neer RM, Jameson L. Osteopenia and bone fractures in a man with anorexia nervosa and hypogonadism. JAMA 1986; 256:385–388.
- Cohan GR. HIV-associated hypogonadism. AIDS Read 2006; 16:341–345,348,352–354.
- Handelsman DJ, Strasser S, McDonald JA, Conway AJ, McCaughan GW. Hypothalamic-pituitary-testicular function in end-stage nonalcoholic liver disease before and after liver transplantation. Clin Endocrinol (Oxf) 1995; 43:331–337.
- Lim VS, Fang VS. Gonadal dysfunction in uremic men. A study of the hypothalamo-pituitary-testicular axis before and after renal transplantation. Am J Med 1975; 58:655–662.
- Handelsman DJ, Dong Q. Hypothalamo-pituitary gonadal axis in chronic renal failure. Endocrinol Metab Clin North Am 1993; 22:145–161.
- Handelsman DJ, Spaliviero JA, Turtle JR. Hypothalamic-pituitary function in experimental uremic hypogonadism. Endocrinology 1985; 117:1984–1995.
- Tengstrand B, Carlström K, Hafström I. Bioavailable testosterone in men with rheumatoid arthritis-high frequency of hypogonadism. Rheumatology (Oxford) 2002; 41:285–289.
- Tengstrand B, Carlström K, Hafström I. Gonadal hormones in men with rheumatoid arthritis--from onset through 2 years. J Rheumatol 2009; 36:887–892.
- Emanuele MA, Emanuele NV. Alcohol’s effects on male reproduction. Alcohol Health Res World 1998; 22:195–201.
- Meikle AW. The interrelationships between thyroid dysfunction and hypogonadism in men and boys. Thyroid 2004; 14( suppl 1):S17–S25.
- Vagenakis AG, Dole K, Braverman LE. Pituitary enlargement, pituitary failure, and primary hypothyroidism. Ann Intern Med 1976; 85:195–198.
- Pitteloud N, Dwyer AA, DeCruz S, et al. Inhibition of luteinizing hormone secretion by testosterone in men requires aromatization for its pituitary but not its hypo-thalamic effects: evidence from the tandem study of normal and gonadotropin-releasing hormone-deficient men. J Clin Endocrinol Metab 2008; 93:784–791.
- Hayes FJ, DeCruz S, Seminara SB, Boepple PA, Crowley WF. Differential regulation of gonadotropin secretion by testosterone in the human male: absence of a negative feedback effect of testosterone on follicle-stimulating hormone secretion. J Clin Endocrinol Metab 2001; 86:53–58.
- Hayes FJ, Pitteloud N, DeCruz S, Crowley WF, Boepple PA. Importance of inhibin B in the regulation of FSH secretion in the human male. J Clin Endocrinol Metab 2001; 86:5541–5546.
- Pitteloud N, Dwyer AA, DeCruz S, et al. The relative role of gonadal sex steroids and gonadotropin-releasing hormone pulse frequency in the regulation of follicle-stimulating hormone secretion in men. J Clin Endocrinol Metab 2008; 93:2686–2692.
- Cooke RR, McIntosh JE, McIntosh RP. Circadian variation in serum free and non-SHBG-bound testosterone in normal men: measurements, and simulation using a mass action model. Clin Endocrinol (Oxf) 1993; 39:163–171.
- Diver MJ, Imtiaz KE, Ahmad AM, Vora JP, Fraser WD. Diurnal rhythms of serum total, free and bioavailable testosterone and of SHBG in middle-aged men compared with those in young men. Clin Endocrinol (Oxf) 2003; 58:710–717.
- Clair P, Claustrat B, Jordan D, Dechaud H, Sassolas G. Daily variations of plasma sex hormone-binding globulin binding capacity, testosterone and luteinizing hormone concentrations in healthy rested adult males. Horm Res 1985; 21:220–223.
- Woolf PD, Hamill RW, McDonald JV, Lee LA, Kelly M. Transient hypogonadotropic hypogonadism caused by critical illness. J Clin Endocrinol Metab 1985; 60:444–450.
- Rosner W, Auchus RJ, Azziz R, Sluss PM, Raff H. Position statement: utility, limitations, and pitfalls in measuring testosterone: an Endocrine Society position statement. J Clin Endocrinol Metab 2007; 92:405–413.
- Rosner W, Vesper H, et al; Endocrine Society; American Association for Clinical Chemistry; American Association of Clinical Endocrinologists; et al. Toward excellence in testosterone testing: a consensus statement. J Clin Endocrinol Metab 2010; 95:4542–4548.
- Bhasin S, Cunningham GR, Hayes FJ, et al; Task Force, Endocrine Society. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010; 95:2536–2559.
- Wu FC, Tajar A, Beynon JM, et al; EMAS Group. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med 2010; 363:123–135.
- Farrer JH, Sikka SC, Xie HW, Constantinide D, Rajfer J. Impaired testosterone biosynthesis in cryptorchidism. Fertil Steril 1985; 44:125–132.
- Sikka SC, Swerdloff RS, Rajfer J. In vitro inhibition of testosterone biosynthesis by ketoconazole. Endocrinology 1985; 116:1920–1925.
- Pallais JC, Au M, Pitteloud N, Seminara S, Crowley WF Jr. Kallmann syndrome. In:Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, eds. GeneReviews™ (Internet). Seattle, WA: University of Washington; 1993.
- Chevrier L, Guimiot F, de Roux N. GnRH receptor mutations in isolated gonadotropic deficiency. Mol Cell Endocrinol 2011; 346:21–28.
- Romero CJ, Pine-Twaddell E, Radovick S. Novel mutations associated with combined pituitary hormone deficiency. J Mol Endocrinol 2011; 46:R93–R102.
- Colameco S, Coren JS, Ciervo CA. Continuous opioid treatment for chronic noncancer pain: a time for moderation in prescribing. Postgrad Med 2009; 121:61–66.
- Fraser LA, Morrison D, Morley-Forster P, et al. Oral opioids for chronic non-cancer pain: higher prevalence of hypogonadism in men than in women. Exp Clin Endocrinol Diabetes 2009; 117:38–43.
- Morrison D, Capewell S, Reynolds SP, et al. Testosterone levels during systemic and inhaled corticosteroid therapy. Respir Med 1994; 88:659–663.
- Mah PM, Wittert GA. Obesity and testicular function. Mol Cell Endocrinol 2010; 316:180–186.
- Grossmann M. Low testosterone in men with type 2 diabetes: significance and treatment. J Clin Endocrinol Metab 2011; 96:2341–2353.
- Gascón F, Valle M, Martos R, et al. Sex hormone-binding globulin as a marker for hyperinsulinemia and/or insulin resistance in obese children. Eur J Endocrinol 2000; 143:85–89.
- Dhindsa S, Miller MG, McWhirter CL, et al. Testosterone concentrations in diabetic and nondiabetic obese men. Diabetes Care 2010; 33:1186–1192.
- Grossmann M, Gianatti EJ, Zajac JD. Testosterone and type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 2010; 17:247–256.
- Andersson B, Mårin P, Lissner L, Vermeulen A, Björntorp P. Testosterone concentrations in women and men with NIDDM. Diabetes Care 1994; 17:405–411.
- Santamaria JD, Prior JC, Fleetham JA. Reversible reproductive dysfunction in men with obstructive sleep apnoea. Clin Endocrinol (Oxf) 1988; 28:461–470.
- Grunstein RR, Handelsman DJ, Lawrence SJ, Blackwell C, Caterson ID, Sullivan CE. Neuroendocrine dysfunction in sleep apnea: reversal by continuous positive airways pressure therapy. J Clin Endocrinol Metab 1989; 68:352–358.
- Matsumoto AM, Sandblom RE, Schoene RB, et al. Testosterone replacement in hypogonadal men: effects on obstructive sleep apnoea, respiratory drives, and sleep. Clin Endocrinol (Oxf) 1985; 22:713–721.
- Bremner WJ, Vitiello MV, Prinz PN. Loss of circadian rhythmicity in blood testosterone levels with aging in normal men. J Clin Endocrinol Metab 1983; 56:1278–1281.
- McDermott JH, Walsh CH. Hypogonadism in hereditary hemochromatosis. J Clin Endocrinol Metab 2005; 90:2451–2455.
- Kelly TM, Edwards CQ, Meikle AW, Kushner JP. Hypogonadism in hemochromatosis: reversal with iron depletion. Ann Intern Med 1984; 101:629–632.
- Milenkovic L, D’Angelo G, Kelly PA, Weiner RI. Inhibition of gonadotropin hormone-releasing hormone release by prolactin from GT1 neuronal cell lines through prolactin receptors. Proc Natl Acad Sci U S A 1994; 91:1244–1247.
- Valensi P, Coussieu C, Kemeny JL, Attali JR, Amouroux J, Sebaoun J. Endocrine investigations in two cases of feminizing Leydig cell tumour. Acta Endocrinol (Copenh) 1987; 115:365–372.
- Young S, Gooneratne S, Straus FH, Zeller WP, Bulun SE, Rosenthal IM. Feminizing Sertoli cell tumors in boys with Peutz-Jeghers syndrome. Am J Surg Pathol 1995; 19:50–58.
- Zayed A, Stock JL, Liepman MK, Wollin M, Longcope C. Feminization as a result of both peripheral conversion of androgens and direct estrogen production from an adrenocortical carcinoma. J Endocrinol Invest 1994; 17:275–278.
- Russ MJ, Ackerman SH, Barakat R, Levy B. Hypogonadotropic hypogonadism and delayed puberty in a man with anorexia nervosa. Psychosomatics 1986; 27:737–739.
- Rigotti NA, Neer RM, Jameson L. Osteopenia and bone fractures in a man with anorexia nervosa and hypogonadism. JAMA 1986; 256:385–388.
- Cohan GR. HIV-associated hypogonadism. AIDS Read 2006; 16:341–345,348,352–354.
- Handelsman DJ, Strasser S, McDonald JA, Conway AJ, McCaughan GW. Hypothalamic-pituitary-testicular function in end-stage nonalcoholic liver disease before and after liver transplantation. Clin Endocrinol (Oxf) 1995; 43:331–337.
- Lim VS, Fang VS. Gonadal dysfunction in uremic men. A study of the hypothalamo-pituitary-testicular axis before and after renal transplantation. Am J Med 1975; 58:655–662.
- Handelsman DJ, Dong Q. Hypothalamo-pituitary gonadal axis in chronic renal failure. Endocrinol Metab Clin North Am 1993; 22:145–161.
- Handelsman DJ, Spaliviero JA, Turtle JR. Hypothalamic-pituitary function in experimental uremic hypogonadism. Endocrinology 1985; 117:1984–1995.
- Tengstrand B, Carlström K, Hafström I. Bioavailable testosterone in men with rheumatoid arthritis-high frequency of hypogonadism. Rheumatology (Oxford) 2002; 41:285–289.
- Tengstrand B, Carlström K, Hafström I. Gonadal hormones in men with rheumatoid arthritis--from onset through 2 years. J Rheumatol 2009; 36:887–892.
- Emanuele MA, Emanuele NV. Alcohol’s effects on male reproduction. Alcohol Health Res World 1998; 22:195–201.
- Meikle AW. The interrelationships between thyroid dysfunction and hypogonadism in men and boys. Thyroid 2004; 14( suppl 1):S17–S25.
- Vagenakis AG, Dole K, Braverman LE. Pituitary enlargement, pituitary failure, and primary hypothyroidism. Ann Intern Med 1976; 85:195–198.
KEY POINTS
- Blood samples for testosterone measurements should be drawn near 8 am.
- A low serum testosterone value should always be confirmed by a reliable reference laboratory.
- The definition of a low testosterone level varies from laboratory to laboratory. In general, values less than 200 or 250 ng/dL are considered low, and values between 250 and 350 ng/dL may be considered borderline low.
- If testosterone is low, determine if the cause is primary (testicular) or secondary (hypothalamic-pituitary).
- Acute illness and treatment with opioids, anabolic steroids, or corticosteroids can result in transient hypogonadism.
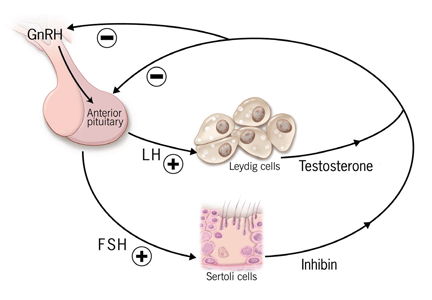
The role of aldosterone receptor antagonists in the management of heart failure: An update
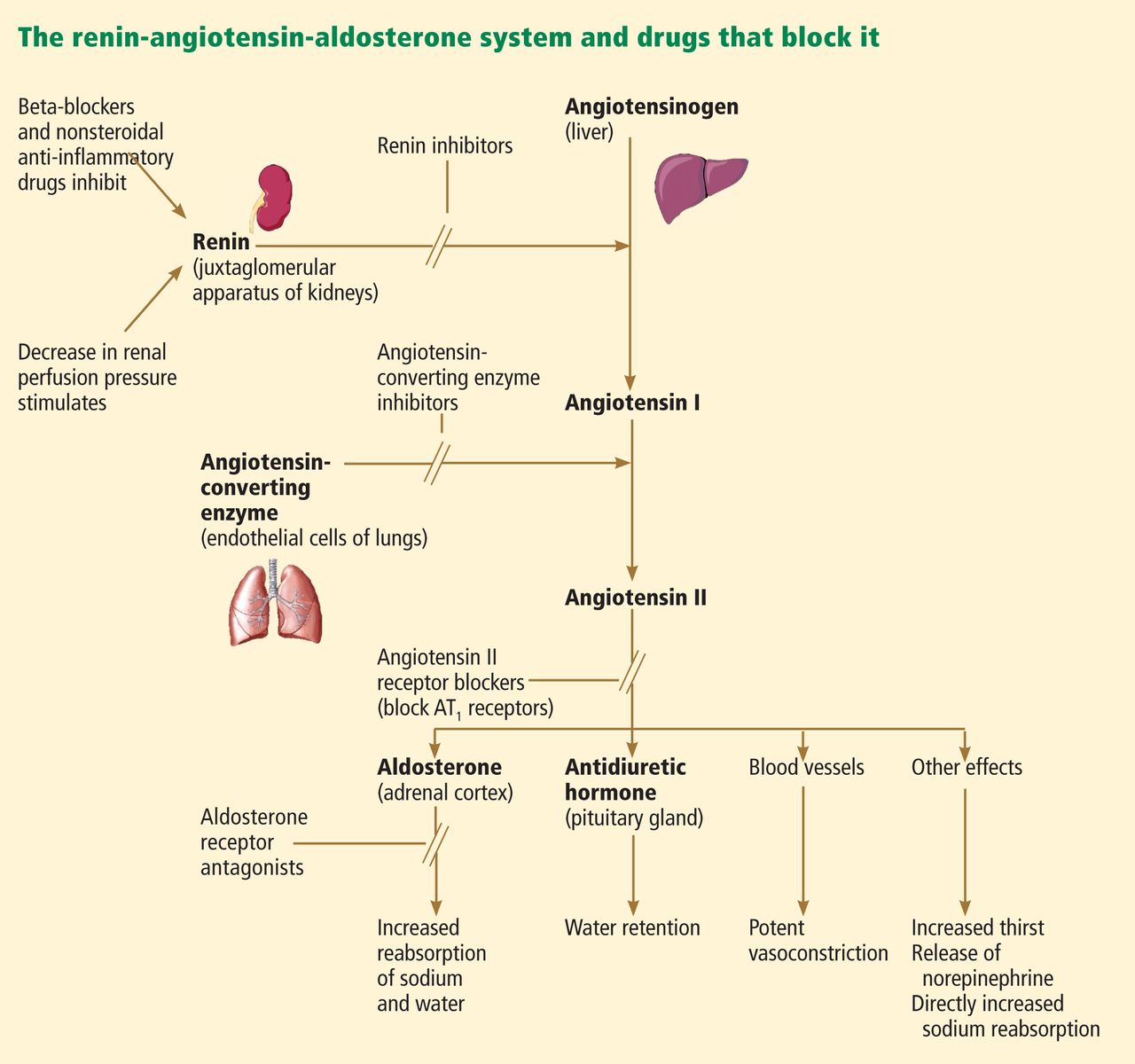
Over the past 30 years, the focus of treating heart failure has shifted from managing symptoms to prolonging lives. When the neurohormonal hypothesis (ie, the concept that neurohormonal dysregulation and not merely hemodynamic changes are responsible for the onset and progression of heart failure) was introduced, it brought a dramatic change that included new classes of drugs that interfere with the renin-angiotensin-aldosterone system, ie, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and, most recently, aldosterone receptor antagonists (ARAs) (Figure 1).
Evidence supporting the use of the ARAs spironolactone (Aldactone) and eplerenone (Inspra) in heart failure has been growing, as has evidence of their usefulness in treating diabetes and chronic renal disease. Still, these drugs must be used cautiously, as they can cause hyperkalemia.
This paper will review the clinical use of ARAs in symptomatic systolic heart failure, their side effects, the findings and implications of recent trials, and controversies in this area, notably whether there is any evidence favoring the use of one drug over another.
ALDOSTERONE IN HEART FAILURE
Aldosterone, a hormone secreted by the zona glomerulosa of the adrenal gland, was first isolated by Simpson and Tait more than half a century ago.1 Later, it was found to promote reabsorption of sodium and excretion of potassium in the kidneys and hence was categorized as a mineralocorticoid hormone.
Release of aldosterone is stimulated by decreased renal perfusion via angiotensin II, hyperkalemia, and possibly adrenocorticotropic hormone.2 Aldosterone exerts its effects by binding to mineralocorticoid receptors in renal epithelial cells.
Aldosterone has several deleterious effects on the failing heart, primarily sodium and fluid retention, but also endothelial dysfunction, left ventricular hypertrophy, and myocardial fibrosis.2,3 Plasma aldosterone levels can be markedly elevated in patients with heart failure, likely due to activation of the renin-angiotensin-aldosterone system. Elevated aldosterone and angiotensin II levels have been associated with higher mortality rates.4
ALDOSTERONE ‘ESCAPE’ BLUNTS THE EFFECT OF ACE INHIBITORS AND ARBs
ACE inhibitors and ARBs have become standards of care for patients with systolic heart failure, and for many years, it was believed that these drugs suppressed aldosterone levels sufficiently. But elevated aldosterone levels have been noted in up to 38% of patients on chronic ACE inhibitor therapy.5 In one study, patients on dual blockade, ie, on both an ACE inhibitor and an ARB, had significantly lower aldosterone levels at 17 weeks of therapy, but not at 43 weeks.6 This phenomenon is known as “aldosterone escape.”
Several mechanisms might explain this phenomenon. Angiotensin II, a potent inducer of aldosterone, is “reactivated” during long-term ACE inhibitor therapy. Interestingly, patients progress toward aldosterone escape regardless of whether the ACE inhibitor dose is low or high.7 There is evidence that some aldosterone is produced by endothelial cells and vascular smooth muscle in the heart and blood vessels,8 but ACE inhibitors and ARBs suppress only the aldosterone secreted by the adrenal glands.
Regardless of the mechanism, aldosterone escape can blunt the effects of ACE inhibitors and ARBs, reducing their favorable effects on the risk of death in heart failure patients. This is the rationale for also using ARAs.
ARAs IN HEART FAILURE
Aldosterone acts by regulating gene expression after binding to mineralocorticoid receptors. These receptors are found not only in epithelial tissue in the kidneys and glands, but also in nonepithelial tissues such as cardiomyocytes, vessel walls, and the hippocampus of the brain.9 The nonepithelial effects were first demonstrated 2 decades ago by Brilla et al,10 who noted that chronically elevated aldosterone levels in rats promoted cardiac fibroblast growth, collagen accumulation, and, hence, ventricular remodeling.
The hypertensive effect of aldosterone may also be mediated through mineralocorticoid receptors in the brain. Gomez-Sanchez et al11 found that infusing aldosterone into the cerebral ventricles caused significant hypertension. A selective mineralocorticoid antagonist inhibited this effect when infused into the cerebral ventricles but not when given systemically.
In 1959, Cella and Kagawa created spironolactone, a nonselective ARA, by combining elements of progesterone for its antimineralocorticoid effect and elements of digitoxin for its cardiotonic effect.12 Although spironolactone is very effective in treating hypertension and heart failure, its use is limited by progestational and antiandrogenic side effects. This led, in 1987, to the invention by de Gasparo et al of a newer molecule, a selective ARA now called eplerenone.13 Although eplerenone may be somewhat less potent than spironolactone in blocking mineralocorticoid receptors, no significant difference in efficacy has been noted in randomized clinical trials, and its antiandrogenic action is negligible.12
Although these drugs target aldosterone receptors, newer drugs may target different aspects of mineralocorticoid activities, and thus the term “mineralocorticoid receptor antagonist” has been proposed.
TRIALS OF ARAs IN HEART FAILURE
An online data supplement that accompanies this paper at provides a detailed comparison of the three major trials of ARAs in patients with heart failure.
The Randomized Aldactone Evaluation Study (RALES)
The first major clinical trial of an ARA was the Randomized Aldactone Evaluation Study (RALES),14 a randomized, double-blind, controlled comparison of spironolactone and placebo.
The 1,663 patients in the trial all had severe heart failure (New York Heart Association class [NYHA] III and ambulatory class IV symptoms) and a left ventricular ejection fraction of 35% or less. Most were on an ACE inhibitor, a loop diuretic, and digoxin, but only 10% of patients in both groups were on a beta-blocker. Patients with chronic renal failure (serum creatinine > 2.5 mg/dL) or hyperkalemia (potassium > 5.0 mmol/L) were excluded.
RALES was halted early when an interim analysis at a mean follow-up of 24 months showed that significantly fewer patients were dying in the spironolactone group; their all-cause mortality rate was 30% lower (relative risk [RR] 0.70, 95% confidence interval [CI] 0.60–0.82, P < .001), and their cardiac mortality rate was 31% lower (RR 0.69, 95% CI 0.58–0.82, P < .001). This was concordant with a lower risk of both sudden cardiac death and death from progressive heart failure. The risk of hospitalization for cardiac causes was also 30% lower for patients in the spironolactone group, who also experienced significant symptom improvement.
Gynecomastia and breast pain occurred in about 10% of patients in the spironolactone group, and adverse effects leading to study drug discontinuation occurred in 2%.14
The Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS)
The next landmark trial of an ARA was the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS).15 A total of 6,632 patients were randomized to receive eplerenone or placebo in this multicenter, double-blind trial. To be enrolled, patients had to have acute myocardial infarction, a left ventricular ejection fraction of 40% or less, and either clinical signs of heart failure 3 to 14 days after the infarction or a history of diabetes mellitus. Patients were excluded if they had chronic kidney disease (defined as a serum creatinine > 2.5 mg/dL or an estimated glomerular filtration rate < 30 mL/min/1.73 m2) or hyperkalemia (a serum potassium > 5.0 mmol/L). All the patients received optimal medical therapy and reperfusion therapy, if warranted.
This event-driven trial was stopped when 1,012 deaths had occurred. During a mean follow-up of 16 months, there was a 15% lower rate of all-cause mortality in the eplerenone group (RR 0.85, 95% CI 0.75–0.96, P = .008) and a 13% lower rate of cardiovascular mortality (RR 0.83, 95% CI 0.72–0.94, P = .005). The reduction in the cardiovascular mortality rate was attributed to a 21% reduction in the rate of sudden cardiac deaths. The rate of heart failure hospitalization was also lower in the eplerenone group.
Serious hyperkalemia occurred significantly more frequently in the eplerenone group (5.5% vs 3.9%, P = .002), but similar rates of gynecomastia were observed. The incidence of hyperkalemia was higher in patients with a creatinine clearance less than 50 mL/min.
Further analyses revealed a 31% lower rate of all-cause mortality (95% CI 0.54–0.89, P = .004) and a 32% lower rate of cardiovascular mortality (95% CI 0.53–0.88, P = .003) at 30 days after randomization in the eplerenone group.16 Importantly, 25% of all deaths in the EPHESUS study during the 16-month follow-up period occurred in the first 30 days after randomization. The Kaplan-Meier survival curves showed separation as early as 5 days after randomization. Hence, the 30-day mortality results from EPHESUS further indicated that starting eplerenone early may be particularly beneficial.
The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF)
After RALES and EPHESUS, a gap remained in our knowledge, ie, how to use ARAs in patients with mild heart failure, who account for most cases. This led to the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure) trial, which expanded the indications for ARAs to patients with chronic systolic heart failure with mild symptoms.17
In this double-blind trial, 2,737 patients with NYHA class II heart failure with a left ventricular ejection fraction of 35% or less were randomized to receive oral eplerenone 25 mg or placebo once daily. All patients were already on a beta-blocker; they were also all on an ACE inhibitor, an ARB, or both at the recommended or maximal tolerated dose. Patients with a glomerular filtration rate between 30 and 49 mL/min were started on alternate-day dosing, and those with glomerular filtration rates below 30 mL/min were excluded.
To ensure that the event rate was high enough to give this trial sufficient power:
- Only patients age 55 years or older were included
- Patients with a left ventricular ejection fraction greater than 30% were enrolled only if the QRS duration was greater than 130 ms (only 3.5% of patients in both groups were enrolled based on this criterion)
- Patients either had to have been hospitalized for cardiovascular reasons in the 6 months before randomization or had to have elevated natriuretic peptides (B-type natriuretic peptide [BNP] level > 250 pg/mL or N-terminal pro-BNP > 500 pg/mL in men and > 750 pg/mL in women).
The study was stopped early at a median follow-up of 21 months after an interim analysis showed a significantly lower rate of the primary composite end point (death from a cardiovascular cause or hospitalization for heart failure) in the eplerenone group: 18.3% vs 25.9% (hazard ratio [HR] 0.63, 95% CI 0.54– 0.74, P < .001). The rates of all-cause mortality were 12.5% vs 15.5% (HR 0.76, 95% CI 0.62–0.93, P = .008), and the rates of cardiovascular mortality were 10.8% vs 13.5% (HR 0.76, 95% CI 0.61–0.94, P = .01). Kaplan-Meier curves for all-cause mortality showed significant separation only after 1 year, which was not the case in EPHESUS and RALES. But the curves for hospitalization separated within a few weeks after randomization.
The incidence of hyperkalemia (serum potassium level > 5.5 mmol/L) was significantly higher in the eplerenone group (11.8% vs 7.2%, P < .001), but there was no statistically significant difference between groups when potassium levels above 6 mmol/L were considered (2.5% vs 1.9%, P = .29). This is despite one-third of patients having an estimated glomerular filtration rate less than 60 mL/min/1.73 m2. Breast symptoms were very rare, occurring in 1% or fewer patients in both groups. The discontinuation rate of the study drug was similar in both groups.
HOW DO ARAs PREVENT DEATH?
Multiple studies show that spironolactone and eplerenone lower blood pressure in a dose-related manner.18 These drugs reduce fluid volume and pulmonary congestion, which could have been the primary mechanism for the reduction in heart failure hospitalizations in the EMPHASIS-HF trial. But other mechanisms might explain the reduction in cardiovascular mortality rates in the trials summarized above.
Transcardiac extraction of aldosterone was increased in a study of patients with heart failure. 19 The transcardiac gradient of plasma aldosterone correlated with levels of procollagen III N-terminal propeptide, a biochemical marker of myocardial fibrosis. This suggests that aldosterone could be a stimulant of myocardial fibrosis. Spironolactone inhibited the transcardiac extraction of aldosterone in the same study.19
In another study,20 spironolactone significantly suppressed elevation of procollagen III N-terminal propeptide after myocardial infarction. It was also demonstrated that spironolactone prevented left ventricular remodeling after infarction, even in patients receiving an ACE inhibitor. Similar results, ie, decreased left ventricular myocardial fibrosis and remodeling, were noted in another trial in which eplerenone was added to an ARB.21
Myocardial fibrosis is a known substrate for ventricular arrhythmias. In a randomized study in 35 patients, spironolactone decreased the incidence of ventricular arrhythmias.22 This finding correlates with the decreased incidence of sudden cardiac death in the RALES and EPHESUS trials.
ADVERSE EFFECTS OF ARAs
Hyperkalemia, hyperkalemia, hyperkalemia
Potassium excretion is physiologically regulated by the serum aldosterone concentration and by the delivery of sodium to the distal nephron. Aldosterone increases potassium excretion. As a result of decreased renal perfusion that occurs with heart failure, sodium is intensely reabsorbed in the proximal tubule, and very little sodium reaches the distal nephron. When aldosterone receptors are blocked by ARAs, the risk of hyperkalemia increases.23

Other electrolyte abnormalities associated with ARAs are hyponatremia and hyperchloremic metabolic acidosis (Table 1). There could be a reversible decline in the glomerular filtration rate as well.24 Of note, most patients with chronic systolic heart failure in the RALES and EMPHASIS-HF trials were already receiving a diuretic; thus, the adverse effect profile of ARAs in otherwise euvolemic (or even hypovolemic) patients is not well appreciated.
Failure to closely monitor electrolyte levels increases the risk of hyperkalemia and renal failure, so there is a need for regular follow-up visits for patients taking an ARA.25 This was made clear when a population-based analysis from Canada compared the rates of hyperkalemia-related hospitalization and death before and after the RALES trial was published. The prescription rate for spironolactone increased threefold, but the rate of hyperkalemia-related hospitalization increased fourfold and the rate of death increased sixfold.26
Although caution is recommended when starting a patient on an ARA, a recent trial conducted in 167 cardiology practices noted that ARAs were the most underused drugs for heart failure. In this study, an ARA was prescribed to only 35% of eligible patients. The prescription rate was not significantly higher even in dedicated heart failure clinics.27 Possible reasons suggested by the authors were drug side effects, the need for closer monitoring of laboratory values, and a lack of knowledge.
A population-based analysis from the United Kingdom found a significant increase over time in spironolactone prescriptions after the release of the RALES trial results, but there was no increase in the rate of serious hyperkalemia (serum potassium > 6 mmol/L) or hyperkalemia-related hospitalization.28 The authors suggested that careful monitoring could prevent hyperkalemia-related complications. They also observed that 75% of patients who had spironolactone-associated hyperkalemia were over 65 years old. Hence, we recommend closer monitoring when starting an elderly patient on an ARA.
Breast, gastrointestinal symptoms
The nonselective ARA spironolactone is associated with antiandrogenic side effects. In a smaller study in patients with resistant hypertension, Nishizaka et al noted that low-dose spironolactone (up to 50 mg/day) was associated with breast tenderness in about 10%.29 Breast symptoms with spironolactone are dose-related, and the incidence can be as high as 50% when the drug is used in dosages of 150 mg/day or higher.30
In one population-based case-control study, spironolactone was associated with a 2.7 times higher risk of gastrointestinal side effects (bleeding or ulcer).31
ARAs IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
The concept of diastolic heart failure or “heart failure with preserved ejection fraction” has been growing. A significant proportion of patients with a diagnosis of heart failure have preserved left ventricular ejection fraction (≥ 50%) and diastolic dysfunction.
Despite multiple trials, no treatment has been shown to lower the mortality rate in heart failure with preserved ejection fraction.32,33 A recently published randomized controlled trial in 44 patients with this condition showed reduction in serum biochemical markers of collagen turnover and improvement in diastolic function with ARAs, but there was no difference in exercise capacity.34 A larger double-blind randomized control trial, Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF), is under way to evaluate the effects of ARAs on exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction.35
In January 2012, the Trial of Aldosterone Antagonist Therapy in Adults With Preserved Ejection Fraction Congestive Heart Failure (TOPCAT) completed enrollment of 3,445 patients to study the effect of ARAs in reducing the composite end point of cardiovascular mortality, aborted cardiac arrest, and heart failure hospitalization. Long-term follow-up of this event-driven study is currently under way.
ARAs IN DIABETES MELLITUS AND CHRONIC KIDNEY DISEASE
Under physiologic conditions, the serum aldosterone level is regulated by volume status through the renin-angiotensin system. But in patients with chronic kidney disease, the serum aldosterone level could be elevated without renin-angiotensin system stimulation.36
High aldosterone levels were associated with proteinuria and glomerulosclerosis in rats.37 In a study in 83 patients, aldosterone receptor blockade was shown to decrease proteinuria and possibly to retard the progression of chronic kidney disease. In this trial, baseline serum aldosterone levels correlated with proteinuria.38 Animal studies suggest that adipocyte-derived factors may stimulate aldosterone, which may be relevant in patients who have both chronic kidney disease and metabolic syndrome.39
The impact of ARAs in patients with diabetes mellitus is often overlooked. In EPHESUS, diabetes mellitus was an inclusion criterion even in the absence of heart failure signs and symptoms in the postinfarction setting of impaired left ventricular ejection fraction.15
In patients with diabetic nephropathy, there is growing evidence that ARAs can decrease proteinuria, even if the serum aldosterone level is normal. For example, in a study in 20 patients with diabetic nephropathy, spironolactone reduced proteinuria by 32%. This reduction was independent of serum aldosterone levels.40
In diabetic rats, hyperglycemia was noted to cause podocyte injury through mineralocorticoid receptor-mediated production of reactive oxygen species, independently of serum aldosterone levels. Spironolactone decreased the production of reactive oxygen species, thereby potentially reducing proteinuria.41
RECOMMENDATIONS ARE BEING REVISED
The most recent joint guidelines of the American Heart Association and the American College of Cardiology for the management of heart failure42 were published in 2009, which was before the EMPHASIS-HF results. An update is expected soon. In the 2009 version, ARAs received a class I recommendation for patients with moderately severe to severe symptoms, decreased ejection fraction, normal renal function, and normal potassium levels. The guidelines also said that the risks of ARAs may outweigh their benefits if regular monitoring is not possible.
The recommended starting dosage is 12.5 mg/day of spironolactone or 25 mg/day of eplerenone; the dose can be doubled, if tolerated.
Close monitoring is recommended, ie, measuring serum potassium and renal function 3 and 7 days after starting therapy and then monthly for the first 3 months. Closer monitoring is needed if an ACE inhibitor or an ARB is added later. In elderly patients, the glomerular filtration rate is preferred over the serum creatinine level, and ARA therapy is not advisable if the glomerular filtration rate is less than 30 mL/min/1.73 m2.
Avoid concomitant use of the following:
- Potassium supplements (unless persistent hypokalemia is present)
- Nonsteroidal anti-inflammatory drugs
- An ACE inhibitor and an ARB in combination
- A high dose of an ACE inhibitor or ARB.
Conditions that can lead to dehydration (eg, diarrhea, excessive use of diuretics) or acute illness should warrant reduction (or even withholding) of ARAs. When to discontinue ARA therapy is not well described, nor is the safety of starting ARAs in the hospital. However, it is clear that many patients who are potentially eligible for ARAs are not prescribed them.43
The guidelines are currently being revised, and will likely incorporate the new data from EMPHASIS-HF to extend to a broader population. The benefits of ARAs can be met only if the risks are minimized.
WHICH ARA IS BETTER?
The pharmacologic differences between the two ARAs have been described earlier, and guidelines have advocated evidence-based use of ARAs for their respective indications. There have been no large-scale, head-to-head comparisons of spironolactone and eplerenone in the heart failure population, and in clinical practice the drugs are prescribed interchangeably in most patients.
A double-blind randomized controlled trial in 141 patients with hypertension and primary hyperaldosteronism found that spironolactone lowered diastolic blood pressure more, but it also caused antiandrogenic effects more often.44
There is some evidence to suggest that eplerenone has a better metabolic profile than spironolactone. The data came from a small randomized controlled trial in 107 stable outpatients with mild heart failure.45 Patients who were prescribed spironolactone had a higher cortisol level and hemoglobin A1c level 4 months after starting treatment. This effect was not seen in patients who were on eplerenone. However, these findings need to be confirmed in larger trials.
While the differences between the two drugs remain to be determined, the most important differences in clinical practice are selectivity for receptors (and hence their antiandrogenic side effects) and price. Even though it is available as a generic drug, eplerenone still costs at least three times more than spironolactone for the same dosage and indication.
- Simpson SA, Tait JF, Bush IE. Secretion of a salt-retaining hormone by the mammalian adrenal cortex. Lancet 1952; 2:226–228.
- Struthers AD, MacDonald TM. Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc Res 2004; 61:663–670.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the “aldosterone receptor blockade in diastolic heart failure” trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 1990; 82:1730–1736.
- MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart 1999; 82:57–61.
- McKelvie RS, Yusuf S, Pericak D, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation 1999; 100:1056–1064.
- Tang WH, Vagelos RH, Yee YG, et al. Neurohormonal and clinical responses to high- versus low-dose enalapril therapy in chronic heart failure. J Am Coll Cardiol 2002; 39:70–78.
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001; 345:1689–1697.
- Funder JW. The role of aldosterone and mineralocorticoid receptors in cardiovascular disease. Am J Cardiovasc Drugs 2007; 7:151–157.
- Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 1990; 67:1355–1364.
- Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol 1992; 262:E96–E99.
- Garthwaite SM, McMahon EG. The evolution of aldosterone antagonists. Mol Cell Endocrinol 2004; 217:27–31.
- de Gasparo M, Joss U, Ramjoué HP, et al. Three new epoxy-spirolactone derivatives: characterization in vivo and in vitro. J Pharmacol Exp Ther 1987; 240:650–656.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am J Hypertens 2002; 15:709–716.
- Tsutamoto T, Wada A, Maeda K, et al. Spironolactone inhibits the transcardiac extraction of aldosterone in patients with congestive heart failure. J Am Coll Cardiol 2000; 36:838–844.
- Hayashi M, Tsutamoto T, Wada A, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents postinfarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003; 107:2559–2565.
- Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additive amelioration of left ventricular remodeling and molecular alterations by combined aldosterone and angiotensin receptor blockade after myocardial infarction. Cardiovasc Res 2005; 67:97–105.
- Ramires FJ, Mansur A, Coelho O, et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idiopathic dilated or to ischemic cardiomyopathy. Am J Cardiol 2000; 85:1207–1211.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the reninangiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Sica DA. The risks and benefits of therapy with aldosterone receptor antagonist therapy. Curr Drug Saf 2007; 2:71–77.
- Shah KB, Rao K, Sawyer R, Gottlieb SS. The adequacy of laboratory monitoring in patients treated with spironolactone for congestive heart failure. J Am Coll Cardiol 2005; 46:845–849.
- Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004; 351:543–551.
- Albert NM, Fonarow GC, Yancy CW, et al. Influence of dedicated heart failure clinics on delivery of recommended therapies in outpatient cardiology practices: findings from the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Am Heart J 2010; 159:238–244.
- Wei L, Struthers AD, Fahey T, Watson AD, Macdonald TM. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ 2010; 340:c1768.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Jeunemaitre X, Chatellier G, Kreft-Jais C, et al. Efficacy and tolerance of spironolactone in essential hypertension. Am J Cardiol 1987; 60:820–825.
- Verhamme K, Mosis G, Dieleman J, Stricker B, Sturkenboom M. Spironolactone and risk of upper gastrointestinal events: population based case-control study. BMJ 2006; 333:330.
- Massie BM, Carson PE, McMurray JJ, et al; I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 2008; 359:2456–2467.
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362:777–781.
- Deswal A, Richardson P, Bozkurt B, Mann DL. Results of the Randomized Aldosterone Antagonism in Heart Failure With Preserved Ejection Fraction Trial (RAAM-PEF). J Card Fail 2011; 17:634–642.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the ‘aldosterone receptor blockade in diastolic heart failure’ trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Hené RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int 1982; 21:98–101.
- Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 1996; 98:1063–1068.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int 2006; 70:2116–2123.
- Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol 2006; 17:3438–3446.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int 2006; 70:536–542.
- Toyonaga J, Tsuruya K, Ikeda H, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant 2011; 26:2475–2484.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- Albert NM, Yancy CW, Liang L, et al. Use of aldosterone antagonists in heart failure. JAMA 2009; 302:1658–1665.
- Parthasarathy HK, Ménard J, White WB, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 2011; 29:980–990.
- Yamaji M, Tsutamoto T, Kawahara C, et al. Effect of eplerenone versus spironolactone on cortisol and hemoglobin A1(c) levels in patients with chronic heart failure. Am Heart J 2010; 160:915–921.
Over the past 30 years, the focus of treating heart failure has shifted from managing symptoms to prolonging lives. When the neurohormonal hypothesis (ie, the concept that neurohormonal dysregulation and not merely hemodynamic changes are responsible for the onset and progression of heart failure) was introduced, it brought a dramatic change that included new classes of drugs that interfere with the renin-angiotensin-aldosterone system, ie, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and, most recently, aldosterone receptor antagonists (ARAs) (Figure 1).
Evidence supporting the use of the ARAs spironolactone (Aldactone) and eplerenone (Inspra) in heart failure has been growing, as has evidence of their usefulness in treating diabetes and chronic renal disease. Still, these drugs must be used cautiously, as they can cause hyperkalemia.
This paper will review the clinical use of ARAs in symptomatic systolic heart failure, their side effects, the findings and implications of recent trials, and controversies in this area, notably whether there is any evidence favoring the use of one drug over another.
ALDOSTERONE IN HEART FAILURE
Aldosterone, a hormone secreted by the zona glomerulosa of the adrenal gland, was first isolated by Simpson and Tait more than half a century ago.1 Later, it was found to promote reabsorption of sodium and excretion of potassium in the kidneys and hence was categorized as a mineralocorticoid hormone.
Release of aldosterone is stimulated by decreased renal perfusion via angiotensin II, hyperkalemia, and possibly adrenocorticotropic hormone.2 Aldosterone exerts its effects by binding to mineralocorticoid receptors in renal epithelial cells.
Aldosterone has several deleterious effects on the failing heart, primarily sodium and fluid retention, but also endothelial dysfunction, left ventricular hypertrophy, and myocardial fibrosis.2,3 Plasma aldosterone levels can be markedly elevated in patients with heart failure, likely due to activation of the renin-angiotensin-aldosterone system. Elevated aldosterone and angiotensin II levels have been associated with higher mortality rates.4
ALDOSTERONE ‘ESCAPE’ BLUNTS THE EFFECT OF ACE INHIBITORS AND ARBs
ACE inhibitors and ARBs have become standards of care for patients with systolic heart failure, and for many years, it was believed that these drugs suppressed aldosterone levels sufficiently. But elevated aldosterone levels have been noted in up to 38% of patients on chronic ACE inhibitor therapy.5 In one study, patients on dual blockade, ie, on both an ACE inhibitor and an ARB, had significantly lower aldosterone levels at 17 weeks of therapy, but not at 43 weeks.6 This phenomenon is known as “aldosterone escape.”
Several mechanisms might explain this phenomenon. Angiotensin II, a potent inducer of aldosterone, is “reactivated” during long-term ACE inhibitor therapy. Interestingly, patients progress toward aldosterone escape regardless of whether the ACE inhibitor dose is low or high.7 There is evidence that some aldosterone is produced by endothelial cells and vascular smooth muscle in the heart and blood vessels,8 but ACE inhibitors and ARBs suppress only the aldosterone secreted by the adrenal glands.
Regardless of the mechanism, aldosterone escape can blunt the effects of ACE inhibitors and ARBs, reducing their favorable effects on the risk of death in heart failure patients. This is the rationale for also using ARAs.
ARAs IN HEART FAILURE
Aldosterone acts by regulating gene expression after binding to mineralocorticoid receptors. These receptors are found not only in epithelial tissue in the kidneys and glands, but also in nonepithelial tissues such as cardiomyocytes, vessel walls, and the hippocampus of the brain.9 The nonepithelial effects were first demonstrated 2 decades ago by Brilla et al,10 who noted that chronically elevated aldosterone levels in rats promoted cardiac fibroblast growth, collagen accumulation, and, hence, ventricular remodeling.
The hypertensive effect of aldosterone may also be mediated through mineralocorticoid receptors in the brain. Gomez-Sanchez et al11 found that infusing aldosterone into the cerebral ventricles caused significant hypertension. A selective mineralocorticoid antagonist inhibited this effect when infused into the cerebral ventricles but not when given systemically.
In 1959, Cella and Kagawa created spironolactone, a nonselective ARA, by combining elements of progesterone for its antimineralocorticoid effect and elements of digitoxin for its cardiotonic effect.12 Although spironolactone is very effective in treating hypertension and heart failure, its use is limited by progestational and antiandrogenic side effects. This led, in 1987, to the invention by de Gasparo et al of a newer molecule, a selective ARA now called eplerenone.13 Although eplerenone may be somewhat less potent than spironolactone in blocking mineralocorticoid receptors, no significant difference in efficacy has been noted in randomized clinical trials, and its antiandrogenic action is negligible.12
Although these drugs target aldosterone receptors, newer drugs may target different aspects of mineralocorticoid activities, and thus the term “mineralocorticoid receptor antagonist” has been proposed.
TRIALS OF ARAs IN HEART FAILURE
An online data supplement that accompanies this paper at provides a detailed comparison of the three major trials of ARAs in patients with heart failure.
The Randomized Aldactone Evaluation Study (RALES)
The first major clinical trial of an ARA was the Randomized Aldactone Evaluation Study (RALES),14 a randomized, double-blind, controlled comparison of spironolactone and placebo.
The 1,663 patients in the trial all had severe heart failure (New York Heart Association class [NYHA] III and ambulatory class IV symptoms) and a left ventricular ejection fraction of 35% or less. Most were on an ACE inhibitor, a loop diuretic, and digoxin, but only 10% of patients in both groups were on a beta-blocker. Patients with chronic renal failure (serum creatinine > 2.5 mg/dL) or hyperkalemia (potassium > 5.0 mmol/L) were excluded.
RALES was halted early when an interim analysis at a mean follow-up of 24 months showed that significantly fewer patients were dying in the spironolactone group; their all-cause mortality rate was 30% lower (relative risk [RR] 0.70, 95% confidence interval [CI] 0.60–0.82, P < .001), and their cardiac mortality rate was 31% lower (RR 0.69, 95% CI 0.58–0.82, P < .001). This was concordant with a lower risk of both sudden cardiac death and death from progressive heart failure. The risk of hospitalization for cardiac causes was also 30% lower for patients in the spironolactone group, who also experienced significant symptom improvement.
Gynecomastia and breast pain occurred in about 10% of patients in the spironolactone group, and adverse effects leading to study drug discontinuation occurred in 2%.14
The Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS)
The next landmark trial of an ARA was the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS).15 A total of 6,632 patients were randomized to receive eplerenone or placebo in this multicenter, double-blind trial. To be enrolled, patients had to have acute myocardial infarction, a left ventricular ejection fraction of 40% or less, and either clinical signs of heart failure 3 to 14 days after the infarction or a history of diabetes mellitus. Patients were excluded if they had chronic kidney disease (defined as a serum creatinine > 2.5 mg/dL or an estimated glomerular filtration rate < 30 mL/min/1.73 m2) or hyperkalemia (a serum potassium > 5.0 mmol/L). All the patients received optimal medical therapy and reperfusion therapy, if warranted.
This event-driven trial was stopped when 1,012 deaths had occurred. During a mean follow-up of 16 months, there was a 15% lower rate of all-cause mortality in the eplerenone group (RR 0.85, 95% CI 0.75–0.96, P = .008) and a 13% lower rate of cardiovascular mortality (RR 0.83, 95% CI 0.72–0.94, P = .005). The reduction in the cardiovascular mortality rate was attributed to a 21% reduction in the rate of sudden cardiac deaths. The rate of heart failure hospitalization was also lower in the eplerenone group.
Serious hyperkalemia occurred significantly more frequently in the eplerenone group (5.5% vs 3.9%, P = .002), but similar rates of gynecomastia were observed. The incidence of hyperkalemia was higher in patients with a creatinine clearance less than 50 mL/min.
Further analyses revealed a 31% lower rate of all-cause mortality (95% CI 0.54–0.89, P = .004) and a 32% lower rate of cardiovascular mortality (95% CI 0.53–0.88, P = .003) at 30 days after randomization in the eplerenone group.16 Importantly, 25% of all deaths in the EPHESUS study during the 16-month follow-up period occurred in the first 30 days after randomization. The Kaplan-Meier survival curves showed separation as early as 5 days after randomization. Hence, the 30-day mortality results from EPHESUS further indicated that starting eplerenone early may be particularly beneficial.
The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF)
After RALES and EPHESUS, a gap remained in our knowledge, ie, how to use ARAs in patients with mild heart failure, who account for most cases. This led to the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure) trial, which expanded the indications for ARAs to patients with chronic systolic heart failure with mild symptoms.17
In this double-blind trial, 2,737 patients with NYHA class II heart failure with a left ventricular ejection fraction of 35% or less were randomized to receive oral eplerenone 25 mg or placebo once daily. All patients were already on a beta-blocker; they were also all on an ACE inhibitor, an ARB, or both at the recommended or maximal tolerated dose. Patients with a glomerular filtration rate between 30 and 49 mL/min were started on alternate-day dosing, and those with glomerular filtration rates below 30 mL/min were excluded.
To ensure that the event rate was high enough to give this trial sufficient power:
- Only patients age 55 years or older were included
- Patients with a left ventricular ejection fraction greater than 30% were enrolled only if the QRS duration was greater than 130 ms (only 3.5% of patients in both groups were enrolled based on this criterion)
- Patients either had to have been hospitalized for cardiovascular reasons in the 6 months before randomization or had to have elevated natriuretic peptides (B-type natriuretic peptide [BNP] level > 250 pg/mL or N-terminal pro-BNP > 500 pg/mL in men and > 750 pg/mL in women).
The study was stopped early at a median follow-up of 21 months after an interim analysis showed a significantly lower rate of the primary composite end point (death from a cardiovascular cause or hospitalization for heart failure) in the eplerenone group: 18.3% vs 25.9% (hazard ratio [HR] 0.63, 95% CI 0.54– 0.74, P < .001). The rates of all-cause mortality were 12.5% vs 15.5% (HR 0.76, 95% CI 0.62–0.93, P = .008), and the rates of cardiovascular mortality were 10.8% vs 13.5% (HR 0.76, 95% CI 0.61–0.94, P = .01). Kaplan-Meier curves for all-cause mortality showed significant separation only after 1 year, which was not the case in EPHESUS and RALES. But the curves for hospitalization separated within a few weeks after randomization.
The incidence of hyperkalemia (serum potassium level > 5.5 mmol/L) was significantly higher in the eplerenone group (11.8% vs 7.2%, P < .001), but there was no statistically significant difference between groups when potassium levels above 6 mmol/L were considered (2.5% vs 1.9%, P = .29). This is despite one-third of patients having an estimated glomerular filtration rate less than 60 mL/min/1.73 m2. Breast symptoms were very rare, occurring in 1% or fewer patients in both groups. The discontinuation rate of the study drug was similar in both groups.
HOW DO ARAs PREVENT DEATH?
Multiple studies show that spironolactone and eplerenone lower blood pressure in a dose-related manner.18 These drugs reduce fluid volume and pulmonary congestion, which could have been the primary mechanism for the reduction in heart failure hospitalizations in the EMPHASIS-HF trial. But other mechanisms might explain the reduction in cardiovascular mortality rates in the trials summarized above.
Transcardiac extraction of aldosterone was increased in a study of patients with heart failure. 19 The transcardiac gradient of plasma aldosterone correlated with levels of procollagen III N-terminal propeptide, a biochemical marker of myocardial fibrosis. This suggests that aldosterone could be a stimulant of myocardial fibrosis. Spironolactone inhibited the transcardiac extraction of aldosterone in the same study.19
In another study,20 spironolactone significantly suppressed elevation of procollagen III N-terminal propeptide after myocardial infarction. It was also demonstrated that spironolactone prevented left ventricular remodeling after infarction, even in patients receiving an ACE inhibitor. Similar results, ie, decreased left ventricular myocardial fibrosis and remodeling, were noted in another trial in which eplerenone was added to an ARB.21
Myocardial fibrosis is a known substrate for ventricular arrhythmias. In a randomized study in 35 patients, spironolactone decreased the incidence of ventricular arrhythmias.22 This finding correlates with the decreased incidence of sudden cardiac death in the RALES and EPHESUS trials.
ADVERSE EFFECTS OF ARAs
Hyperkalemia, hyperkalemia, hyperkalemia
Potassium excretion is physiologically regulated by the serum aldosterone concentration and by the delivery of sodium to the distal nephron. Aldosterone increases potassium excretion. As a result of decreased renal perfusion that occurs with heart failure, sodium is intensely reabsorbed in the proximal tubule, and very little sodium reaches the distal nephron. When aldosterone receptors are blocked by ARAs, the risk of hyperkalemia increases.23
Other electrolyte abnormalities associated with ARAs are hyponatremia and hyperchloremic metabolic acidosis (Table 1). There could be a reversible decline in the glomerular filtration rate as well.24 Of note, most patients with chronic systolic heart failure in the RALES and EMPHASIS-HF trials were already receiving a diuretic; thus, the adverse effect profile of ARAs in otherwise euvolemic (or even hypovolemic) patients is not well appreciated.
Failure to closely monitor electrolyte levels increases the risk of hyperkalemia and renal failure, so there is a need for regular follow-up visits for patients taking an ARA.25 This was made clear when a population-based analysis from Canada compared the rates of hyperkalemia-related hospitalization and death before and after the RALES trial was published. The prescription rate for spironolactone increased threefold, but the rate of hyperkalemia-related hospitalization increased fourfold and the rate of death increased sixfold.26
Although caution is recommended when starting a patient on an ARA, a recent trial conducted in 167 cardiology practices noted that ARAs were the most underused drugs for heart failure. In this study, an ARA was prescribed to only 35% of eligible patients. The prescription rate was not significantly higher even in dedicated heart failure clinics.27 Possible reasons suggested by the authors were drug side effects, the need for closer monitoring of laboratory values, and a lack of knowledge.
A population-based analysis from the United Kingdom found a significant increase over time in spironolactone prescriptions after the release of the RALES trial results, but there was no increase in the rate of serious hyperkalemia (serum potassium > 6 mmol/L) or hyperkalemia-related hospitalization.28 The authors suggested that careful monitoring could prevent hyperkalemia-related complications. They also observed that 75% of patients who had spironolactone-associated hyperkalemia were over 65 years old. Hence, we recommend closer monitoring when starting an elderly patient on an ARA.
Breast, gastrointestinal symptoms
The nonselective ARA spironolactone is associated with antiandrogenic side effects. In a smaller study in patients with resistant hypertension, Nishizaka et al noted that low-dose spironolactone (up to 50 mg/day) was associated with breast tenderness in about 10%.29 Breast symptoms with spironolactone are dose-related, and the incidence can be as high as 50% when the drug is used in dosages of 150 mg/day or higher.30
In one population-based case-control study, spironolactone was associated with a 2.7 times higher risk of gastrointestinal side effects (bleeding or ulcer).31
ARAs IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
The concept of diastolic heart failure or “heart failure with preserved ejection fraction” has been growing. A significant proportion of patients with a diagnosis of heart failure have preserved left ventricular ejection fraction (≥ 50%) and diastolic dysfunction.
Despite multiple trials, no treatment has been shown to lower the mortality rate in heart failure with preserved ejection fraction.32,33 A recently published randomized controlled trial in 44 patients with this condition showed reduction in serum biochemical markers of collagen turnover and improvement in diastolic function with ARAs, but there was no difference in exercise capacity.34 A larger double-blind randomized control trial, Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF), is under way to evaluate the effects of ARAs on exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction.35
In January 2012, the Trial of Aldosterone Antagonist Therapy in Adults With Preserved Ejection Fraction Congestive Heart Failure (TOPCAT) completed enrollment of 3,445 patients to study the effect of ARAs in reducing the composite end point of cardiovascular mortality, aborted cardiac arrest, and heart failure hospitalization. Long-term follow-up of this event-driven study is currently under way.
ARAs IN DIABETES MELLITUS AND CHRONIC KIDNEY DISEASE
Under physiologic conditions, the serum aldosterone level is regulated by volume status through the renin-angiotensin system. But in patients with chronic kidney disease, the serum aldosterone level could be elevated without renin-angiotensin system stimulation.36
High aldosterone levels were associated with proteinuria and glomerulosclerosis in rats.37 In a study in 83 patients, aldosterone receptor blockade was shown to decrease proteinuria and possibly to retard the progression of chronic kidney disease. In this trial, baseline serum aldosterone levels correlated with proteinuria.38 Animal studies suggest that adipocyte-derived factors may stimulate aldosterone, which may be relevant in patients who have both chronic kidney disease and metabolic syndrome.39
The impact of ARAs in patients with diabetes mellitus is often overlooked. In EPHESUS, diabetes mellitus was an inclusion criterion even in the absence of heart failure signs and symptoms in the postinfarction setting of impaired left ventricular ejection fraction.15
In patients with diabetic nephropathy, there is growing evidence that ARAs can decrease proteinuria, even if the serum aldosterone level is normal. For example, in a study in 20 patients with diabetic nephropathy, spironolactone reduced proteinuria by 32%. This reduction was independent of serum aldosterone levels.40
In diabetic rats, hyperglycemia was noted to cause podocyte injury through mineralocorticoid receptor-mediated production of reactive oxygen species, independently of serum aldosterone levels. Spironolactone decreased the production of reactive oxygen species, thereby potentially reducing proteinuria.41
RECOMMENDATIONS ARE BEING REVISED
The most recent joint guidelines of the American Heart Association and the American College of Cardiology for the management of heart failure42 were published in 2009, which was before the EMPHASIS-HF results. An update is expected soon. In the 2009 version, ARAs received a class I recommendation for patients with moderately severe to severe symptoms, decreased ejection fraction, normal renal function, and normal potassium levels. The guidelines also said that the risks of ARAs may outweigh their benefits if regular monitoring is not possible.
The recommended starting dosage is 12.5 mg/day of spironolactone or 25 mg/day of eplerenone; the dose can be doubled, if tolerated.
Close monitoring is recommended, ie, measuring serum potassium and renal function 3 and 7 days after starting therapy and then monthly for the first 3 months. Closer monitoring is needed if an ACE inhibitor or an ARB is added later. In elderly patients, the glomerular filtration rate is preferred over the serum creatinine level, and ARA therapy is not advisable if the glomerular filtration rate is less than 30 mL/min/1.73 m2.
Avoid concomitant use of the following:
- Potassium supplements (unless persistent hypokalemia is present)
- Nonsteroidal anti-inflammatory drugs
- An ACE inhibitor and an ARB in combination
- A high dose of an ACE inhibitor or ARB.
Conditions that can lead to dehydration (eg, diarrhea, excessive use of diuretics) or acute illness should warrant reduction (or even withholding) of ARAs. When to discontinue ARA therapy is not well described, nor is the safety of starting ARAs in the hospital. However, it is clear that many patients who are potentially eligible for ARAs are not prescribed them.43
The guidelines are currently being revised, and will likely incorporate the new data from EMPHASIS-HF to extend to a broader population. The benefits of ARAs can be met only if the risks are minimized.
WHICH ARA IS BETTER?
The pharmacologic differences between the two ARAs have been described earlier, and guidelines have advocated evidence-based use of ARAs for their respective indications. There have been no large-scale, head-to-head comparisons of spironolactone and eplerenone in the heart failure population, and in clinical practice the drugs are prescribed interchangeably in most patients.
A double-blind randomized controlled trial in 141 patients with hypertension and primary hyperaldosteronism found that spironolactone lowered diastolic blood pressure more, but it also caused antiandrogenic effects more often.44
There is some evidence to suggest that eplerenone has a better metabolic profile than spironolactone. The data came from a small randomized controlled trial in 107 stable outpatients with mild heart failure.45 Patients who were prescribed spironolactone had a higher cortisol level and hemoglobin A1c level 4 months after starting treatment. This effect was not seen in patients who were on eplerenone. However, these findings need to be confirmed in larger trials.
While the differences between the two drugs remain to be determined, the most important differences in clinical practice are selectivity for receptors (and hence their antiandrogenic side effects) and price. Even though it is available as a generic drug, eplerenone still costs at least three times more than spironolactone for the same dosage and indication.
Over the past 30 years, the focus of treating heart failure has shifted from managing symptoms to prolonging lives. When the neurohormonal hypothesis (ie, the concept that neurohormonal dysregulation and not merely hemodynamic changes are responsible for the onset and progression of heart failure) was introduced, it brought a dramatic change that included new classes of drugs that interfere with the renin-angiotensin-aldosterone system, ie, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and, most recently, aldosterone receptor antagonists (ARAs) (Figure 1).
Evidence supporting the use of the ARAs spironolactone (Aldactone) and eplerenone (Inspra) in heart failure has been growing, as has evidence of their usefulness in treating diabetes and chronic renal disease. Still, these drugs must be used cautiously, as they can cause hyperkalemia.
This paper will review the clinical use of ARAs in symptomatic systolic heart failure, their side effects, the findings and implications of recent trials, and controversies in this area, notably whether there is any evidence favoring the use of one drug over another.
ALDOSTERONE IN HEART FAILURE
Aldosterone, a hormone secreted by the zona glomerulosa of the adrenal gland, was first isolated by Simpson and Tait more than half a century ago.1 Later, it was found to promote reabsorption of sodium and excretion of potassium in the kidneys and hence was categorized as a mineralocorticoid hormone.
Release of aldosterone is stimulated by decreased renal perfusion via angiotensin II, hyperkalemia, and possibly adrenocorticotropic hormone.2 Aldosterone exerts its effects by binding to mineralocorticoid receptors in renal epithelial cells.
Aldosterone has several deleterious effects on the failing heart, primarily sodium and fluid retention, but also endothelial dysfunction, left ventricular hypertrophy, and myocardial fibrosis.2,3 Plasma aldosterone levels can be markedly elevated in patients with heart failure, likely due to activation of the renin-angiotensin-aldosterone system. Elevated aldosterone and angiotensin II levels have been associated with higher mortality rates.4
ALDOSTERONE ‘ESCAPE’ BLUNTS THE EFFECT OF ACE INHIBITORS AND ARBs
ACE inhibitors and ARBs have become standards of care for patients with systolic heart failure, and for many years, it was believed that these drugs suppressed aldosterone levels sufficiently. But elevated aldosterone levels have been noted in up to 38% of patients on chronic ACE inhibitor therapy.5 In one study, patients on dual blockade, ie, on both an ACE inhibitor and an ARB, had significantly lower aldosterone levels at 17 weeks of therapy, but not at 43 weeks.6 This phenomenon is known as “aldosterone escape.”
Several mechanisms might explain this phenomenon. Angiotensin II, a potent inducer of aldosterone, is “reactivated” during long-term ACE inhibitor therapy. Interestingly, patients progress toward aldosterone escape regardless of whether the ACE inhibitor dose is low or high.7 There is evidence that some aldosterone is produced by endothelial cells and vascular smooth muscle in the heart and blood vessels,8 but ACE inhibitors and ARBs suppress only the aldosterone secreted by the adrenal glands.
Regardless of the mechanism, aldosterone escape can blunt the effects of ACE inhibitors and ARBs, reducing their favorable effects on the risk of death in heart failure patients. This is the rationale for also using ARAs.
ARAs IN HEART FAILURE
Aldosterone acts by regulating gene expression after binding to mineralocorticoid receptors. These receptors are found not only in epithelial tissue in the kidneys and glands, but also in nonepithelial tissues such as cardiomyocytes, vessel walls, and the hippocampus of the brain.9 The nonepithelial effects were first demonstrated 2 decades ago by Brilla et al,10 who noted that chronically elevated aldosterone levels in rats promoted cardiac fibroblast growth, collagen accumulation, and, hence, ventricular remodeling.
The hypertensive effect of aldosterone may also be mediated through mineralocorticoid receptors in the brain. Gomez-Sanchez et al11 found that infusing aldosterone into the cerebral ventricles caused significant hypertension. A selective mineralocorticoid antagonist inhibited this effect when infused into the cerebral ventricles but not when given systemically.
In 1959, Cella and Kagawa created spironolactone, a nonselective ARA, by combining elements of progesterone for its antimineralocorticoid effect and elements of digitoxin for its cardiotonic effect.12 Although spironolactone is very effective in treating hypertension and heart failure, its use is limited by progestational and antiandrogenic side effects. This led, in 1987, to the invention by de Gasparo et al of a newer molecule, a selective ARA now called eplerenone.13 Although eplerenone may be somewhat less potent than spironolactone in blocking mineralocorticoid receptors, no significant difference in efficacy has been noted in randomized clinical trials, and its antiandrogenic action is negligible.12
Although these drugs target aldosterone receptors, newer drugs may target different aspects of mineralocorticoid activities, and thus the term “mineralocorticoid receptor antagonist” has been proposed.
TRIALS OF ARAs IN HEART FAILURE
An online data supplement that accompanies this paper at provides a detailed comparison of the three major trials of ARAs in patients with heart failure.
The Randomized Aldactone Evaluation Study (RALES)
The first major clinical trial of an ARA was the Randomized Aldactone Evaluation Study (RALES),14 a randomized, double-blind, controlled comparison of spironolactone and placebo.
The 1,663 patients in the trial all had severe heart failure (New York Heart Association class [NYHA] III and ambulatory class IV symptoms) and a left ventricular ejection fraction of 35% or less. Most were on an ACE inhibitor, a loop diuretic, and digoxin, but only 10% of patients in both groups were on a beta-blocker. Patients with chronic renal failure (serum creatinine > 2.5 mg/dL) or hyperkalemia (potassium > 5.0 mmol/L) were excluded.
RALES was halted early when an interim analysis at a mean follow-up of 24 months showed that significantly fewer patients were dying in the spironolactone group; their all-cause mortality rate was 30% lower (relative risk [RR] 0.70, 95% confidence interval [CI] 0.60–0.82, P < .001), and their cardiac mortality rate was 31% lower (RR 0.69, 95% CI 0.58–0.82, P < .001). This was concordant with a lower risk of both sudden cardiac death and death from progressive heart failure. The risk of hospitalization for cardiac causes was also 30% lower for patients in the spironolactone group, who also experienced significant symptom improvement.
Gynecomastia and breast pain occurred in about 10% of patients in the spironolactone group, and adverse effects leading to study drug discontinuation occurred in 2%.14
The Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS)
The next landmark trial of an ARA was the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS).15 A total of 6,632 patients were randomized to receive eplerenone or placebo in this multicenter, double-blind trial. To be enrolled, patients had to have acute myocardial infarction, a left ventricular ejection fraction of 40% or less, and either clinical signs of heart failure 3 to 14 days after the infarction or a history of diabetes mellitus. Patients were excluded if they had chronic kidney disease (defined as a serum creatinine > 2.5 mg/dL or an estimated glomerular filtration rate < 30 mL/min/1.73 m2) or hyperkalemia (a serum potassium > 5.0 mmol/L). All the patients received optimal medical therapy and reperfusion therapy, if warranted.
This event-driven trial was stopped when 1,012 deaths had occurred. During a mean follow-up of 16 months, there was a 15% lower rate of all-cause mortality in the eplerenone group (RR 0.85, 95% CI 0.75–0.96, P = .008) and a 13% lower rate of cardiovascular mortality (RR 0.83, 95% CI 0.72–0.94, P = .005). The reduction in the cardiovascular mortality rate was attributed to a 21% reduction in the rate of sudden cardiac deaths. The rate of heart failure hospitalization was also lower in the eplerenone group.
Serious hyperkalemia occurred significantly more frequently in the eplerenone group (5.5% vs 3.9%, P = .002), but similar rates of gynecomastia were observed. The incidence of hyperkalemia was higher in patients with a creatinine clearance less than 50 mL/min.
Further analyses revealed a 31% lower rate of all-cause mortality (95% CI 0.54–0.89, P = .004) and a 32% lower rate of cardiovascular mortality (95% CI 0.53–0.88, P = .003) at 30 days after randomization in the eplerenone group.16 Importantly, 25% of all deaths in the EPHESUS study during the 16-month follow-up period occurred in the first 30 days after randomization. The Kaplan-Meier survival curves showed separation as early as 5 days after randomization. Hence, the 30-day mortality results from EPHESUS further indicated that starting eplerenone early may be particularly beneficial.
The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF)
After RALES and EPHESUS, a gap remained in our knowledge, ie, how to use ARAs in patients with mild heart failure, who account for most cases. This led to the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure) trial, which expanded the indications for ARAs to patients with chronic systolic heart failure with mild symptoms.17
In this double-blind trial, 2,737 patients with NYHA class II heart failure with a left ventricular ejection fraction of 35% or less were randomized to receive oral eplerenone 25 mg or placebo once daily. All patients were already on a beta-blocker; they were also all on an ACE inhibitor, an ARB, or both at the recommended or maximal tolerated dose. Patients with a glomerular filtration rate between 30 and 49 mL/min were started on alternate-day dosing, and those with glomerular filtration rates below 30 mL/min were excluded.
To ensure that the event rate was high enough to give this trial sufficient power:
- Only patients age 55 years or older were included
- Patients with a left ventricular ejection fraction greater than 30% were enrolled only if the QRS duration was greater than 130 ms (only 3.5% of patients in both groups were enrolled based on this criterion)
- Patients either had to have been hospitalized for cardiovascular reasons in the 6 months before randomization or had to have elevated natriuretic peptides (B-type natriuretic peptide [BNP] level > 250 pg/mL or N-terminal pro-BNP > 500 pg/mL in men and > 750 pg/mL in women).
The study was stopped early at a median follow-up of 21 months after an interim analysis showed a significantly lower rate of the primary composite end point (death from a cardiovascular cause or hospitalization for heart failure) in the eplerenone group: 18.3% vs 25.9% (hazard ratio [HR] 0.63, 95% CI 0.54– 0.74, P < .001). The rates of all-cause mortality were 12.5% vs 15.5% (HR 0.76, 95% CI 0.62–0.93, P = .008), and the rates of cardiovascular mortality were 10.8% vs 13.5% (HR 0.76, 95% CI 0.61–0.94, P = .01). Kaplan-Meier curves for all-cause mortality showed significant separation only after 1 year, which was not the case in EPHESUS and RALES. But the curves for hospitalization separated within a few weeks after randomization.
The incidence of hyperkalemia (serum potassium level > 5.5 mmol/L) was significantly higher in the eplerenone group (11.8% vs 7.2%, P < .001), but there was no statistically significant difference between groups when potassium levels above 6 mmol/L were considered (2.5% vs 1.9%, P = .29). This is despite one-third of patients having an estimated glomerular filtration rate less than 60 mL/min/1.73 m2. Breast symptoms were very rare, occurring in 1% or fewer patients in both groups. The discontinuation rate of the study drug was similar in both groups.
HOW DO ARAs PREVENT DEATH?
Multiple studies show that spironolactone and eplerenone lower blood pressure in a dose-related manner.18 These drugs reduce fluid volume and pulmonary congestion, which could have been the primary mechanism for the reduction in heart failure hospitalizations in the EMPHASIS-HF trial. But other mechanisms might explain the reduction in cardiovascular mortality rates in the trials summarized above.
Transcardiac extraction of aldosterone was increased in a study of patients with heart failure. 19 The transcardiac gradient of plasma aldosterone correlated with levels of procollagen III N-terminal propeptide, a biochemical marker of myocardial fibrosis. This suggests that aldosterone could be a stimulant of myocardial fibrosis. Spironolactone inhibited the transcardiac extraction of aldosterone in the same study.19
In another study,20 spironolactone significantly suppressed elevation of procollagen III N-terminal propeptide after myocardial infarction. It was also demonstrated that spironolactone prevented left ventricular remodeling after infarction, even in patients receiving an ACE inhibitor. Similar results, ie, decreased left ventricular myocardial fibrosis and remodeling, were noted in another trial in which eplerenone was added to an ARB.21
Myocardial fibrosis is a known substrate for ventricular arrhythmias. In a randomized study in 35 patients, spironolactone decreased the incidence of ventricular arrhythmias.22 This finding correlates with the decreased incidence of sudden cardiac death in the RALES and EPHESUS trials.
ADVERSE EFFECTS OF ARAs
Hyperkalemia, hyperkalemia, hyperkalemia
Potassium excretion is physiologically regulated by the serum aldosterone concentration and by the delivery of sodium to the distal nephron. Aldosterone increases potassium excretion. As a result of decreased renal perfusion that occurs with heart failure, sodium is intensely reabsorbed in the proximal tubule, and very little sodium reaches the distal nephron. When aldosterone receptors are blocked by ARAs, the risk of hyperkalemia increases.23
Other electrolyte abnormalities associated with ARAs are hyponatremia and hyperchloremic metabolic acidosis (Table 1). There could be a reversible decline in the glomerular filtration rate as well.24 Of note, most patients with chronic systolic heart failure in the RALES and EMPHASIS-HF trials were already receiving a diuretic; thus, the adverse effect profile of ARAs in otherwise euvolemic (or even hypovolemic) patients is not well appreciated.
Failure to closely monitor electrolyte levels increases the risk of hyperkalemia and renal failure, so there is a need for regular follow-up visits for patients taking an ARA.25 This was made clear when a population-based analysis from Canada compared the rates of hyperkalemia-related hospitalization and death before and after the RALES trial was published. The prescription rate for spironolactone increased threefold, but the rate of hyperkalemia-related hospitalization increased fourfold and the rate of death increased sixfold.26
Although caution is recommended when starting a patient on an ARA, a recent trial conducted in 167 cardiology practices noted that ARAs were the most underused drugs for heart failure. In this study, an ARA was prescribed to only 35% of eligible patients. The prescription rate was not significantly higher even in dedicated heart failure clinics.27 Possible reasons suggested by the authors were drug side effects, the need for closer monitoring of laboratory values, and a lack of knowledge.
A population-based analysis from the United Kingdom found a significant increase over time in spironolactone prescriptions after the release of the RALES trial results, but there was no increase in the rate of serious hyperkalemia (serum potassium > 6 mmol/L) or hyperkalemia-related hospitalization.28 The authors suggested that careful monitoring could prevent hyperkalemia-related complications. They also observed that 75% of patients who had spironolactone-associated hyperkalemia were over 65 years old. Hence, we recommend closer monitoring when starting an elderly patient on an ARA.
Breast, gastrointestinal symptoms
The nonselective ARA spironolactone is associated with antiandrogenic side effects. In a smaller study in patients with resistant hypertension, Nishizaka et al noted that low-dose spironolactone (up to 50 mg/day) was associated with breast tenderness in about 10%.29 Breast symptoms with spironolactone are dose-related, and the incidence can be as high as 50% when the drug is used in dosages of 150 mg/day or higher.30
In one population-based case-control study, spironolactone was associated with a 2.7 times higher risk of gastrointestinal side effects (bleeding or ulcer).31
ARAs IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
The concept of diastolic heart failure or “heart failure with preserved ejection fraction” has been growing. A significant proportion of patients with a diagnosis of heart failure have preserved left ventricular ejection fraction (≥ 50%) and diastolic dysfunction.
Despite multiple trials, no treatment has been shown to lower the mortality rate in heart failure with preserved ejection fraction.32,33 A recently published randomized controlled trial in 44 patients with this condition showed reduction in serum biochemical markers of collagen turnover and improvement in diastolic function with ARAs, but there was no difference in exercise capacity.34 A larger double-blind randomized control trial, Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF), is under way to evaluate the effects of ARAs on exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction.35
In January 2012, the Trial of Aldosterone Antagonist Therapy in Adults With Preserved Ejection Fraction Congestive Heart Failure (TOPCAT) completed enrollment of 3,445 patients to study the effect of ARAs in reducing the composite end point of cardiovascular mortality, aborted cardiac arrest, and heart failure hospitalization. Long-term follow-up of this event-driven study is currently under way.
ARAs IN DIABETES MELLITUS AND CHRONIC KIDNEY DISEASE
Under physiologic conditions, the serum aldosterone level is regulated by volume status through the renin-angiotensin system. But in patients with chronic kidney disease, the serum aldosterone level could be elevated without renin-angiotensin system stimulation.36
High aldosterone levels were associated with proteinuria and glomerulosclerosis in rats.37 In a study in 83 patients, aldosterone receptor blockade was shown to decrease proteinuria and possibly to retard the progression of chronic kidney disease. In this trial, baseline serum aldosterone levels correlated with proteinuria.38 Animal studies suggest that adipocyte-derived factors may stimulate aldosterone, which may be relevant in patients who have both chronic kidney disease and metabolic syndrome.39
The impact of ARAs in patients with diabetes mellitus is often overlooked. In EPHESUS, diabetes mellitus was an inclusion criterion even in the absence of heart failure signs and symptoms in the postinfarction setting of impaired left ventricular ejection fraction.15
In patients with diabetic nephropathy, there is growing evidence that ARAs can decrease proteinuria, even if the serum aldosterone level is normal. For example, in a study in 20 patients with diabetic nephropathy, spironolactone reduced proteinuria by 32%. This reduction was independent of serum aldosterone levels.40
In diabetic rats, hyperglycemia was noted to cause podocyte injury through mineralocorticoid receptor-mediated production of reactive oxygen species, independently of serum aldosterone levels. Spironolactone decreased the production of reactive oxygen species, thereby potentially reducing proteinuria.41
RECOMMENDATIONS ARE BEING REVISED
The most recent joint guidelines of the American Heart Association and the American College of Cardiology for the management of heart failure42 were published in 2009, which was before the EMPHASIS-HF results. An update is expected soon. In the 2009 version, ARAs received a class I recommendation for patients with moderately severe to severe symptoms, decreased ejection fraction, normal renal function, and normal potassium levels. The guidelines also said that the risks of ARAs may outweigh their benefits if regular monitoring is not possible.
The recommended starting dosage is 12.5 mg/day of spironolactone or 25 mg/day of eplerenone; the dose can be doubled, if tolerated.
Close monitoring is recommended, ie, measuring serum potassium and renal function 3 and 7 days after starting therapy and then monthly for the first 3 months. Closer monitoring is needed if an ACE inhibitor or an ARB is added later. In elderly patients, the glomerular filtration rate is preferred over the serum creatinine level, and ARA therapy is not advisable if the glomerular filtration rate is less than 30 mL/min/1.73 m2.
Avoid concomitant use of the following:
- Potassium supplements (unless persistent hypokalemia is present)
- Nonsteroidal anti-inflammatory drugs
- An ACE inhibitor and an ARB in combination
- A high dose of an ACE inhibitor or ARB.
Conditions that can lead to dehydration (eg, diarrhea, excessive use of diuretics) or acute illness should warrant reduction (or even withholding) of ARAs. When to discontinue ARA therapy is not well described, nor is the safety of starting ARAs in the hospital. However, it is clear that many patients who are potentially eligible for ARAs are not prescribed them.43
The guidelines are currently being revised, and will likely incorporate the new data from EMPHASIS-HF to extend to a broader population. The benefits of ARAs can be met only if the risks are minimized.
WHICH ARA IS BETTER?
The pharmacologic differences between the two ARAs have been described earlier, and guidelines have advocated evidence-based use of ARAs for their respective indications. There have been no large-scale, head-to-head comparisons of spironolactone and eplerenone in the heart failure population, and in clinical practice the drugs are prescribed interchangeably in most patients.
A double-blind randomized controlled trial in 141 patients with hypertension and primary hyperaldosteronism found that spironolactone lowered diastolic blood pressure more, but it also caused antiandrogenic effects more often.44
There is some evidence to suggest that eplerenone has a better metabolic profile than spironolactone. The data came from a small randomized controlled trial in 107 stable outpatients with mild heart failure.45 Patients who were prescribed spironolactone had a higher cortisol level and hemoglobin A1c level 4 months after starting treatment. This effect was not seen in patients who were on eplerenone. However, these findings need to be confirmed in larger trials.
While the differences between the two drugs remain to be determined, the most important differences in clinical practice are selectivity for receptors (and hence their antiandrogenic side effects) and price. Even though it is available as a generic drug, eplerenone still costs at least three times more than spironolactone for the same dosage and indication.
- Simpson SA, Tait JF, Bush IE. Secretion of a salt-retaining hormone by the mammalian adrenal cortex. Lancet 1952; 2:226–228.
- Struthers AD, MacDonald TM. Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc Res 2004; 61:663–670.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the “aldosterone receptor blockade in diastolic heart failure” trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 1990; 82:1730–1736.
- MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart 1999; 82:57–61.
- McKelvie RS, Yusuf S, Pericak D, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation 1999; 100:1056–1064.
- Tang WH, Vagelos RH, Yee YG, et al. Neurohormonal and clinical responses to high- versus low-dose enalapril therapy in chronic heart failure. J Am Coll Cardiol 2002; 39:70–78.
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001; 345:1689–1697.
- Funder JW. The role of aldosterone and mineralocorticoid receptors in cardiovascular disease. Am J Cardiovasc Drugs 2007; 7:151–157.
- Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 1990; 67:1355–1364.
- Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol 1992; 262:E96–E99.
- Garthwaite SM, McMahon EG. The evolution of aldosterone antagonists. Mol Cell Endocrinol 2004; 217:27–31.
- de Gasparo M, Joss U, Ramjoué HP, et al. Three new epoxy-spirolactone derivatives: characterization in vivo and in vitro. J Pharmacol Exp Ther 1987; 240:650–656.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am J Hypertens 2002; 15:709–716.
- Tsutamoto T, Wada A, Maeda K, et al. Spironolactone inhibits the transcardiac extraction of aldosterone in patients with congestive heart failure. J Am Coll Cardiol 2000; 36:838–844.
- Hayashi M, Tsutamoto T, Wada A, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents postinfarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003; 107:2559–2565.
- Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additive amelioration of left ventricular remodeling and molecular alterations by combined aldosterone and angiotensin receptor blockade after myocardial infarction. Cardiovasc Res 2005; 67:97–105.
- Ramires FJ, Mansur A, Coelho O, et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idiopathic dilated or to ischemic cardiomyopathy. Am J Cardiol 2000; 85:1207–1211.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the reninangiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Sica DA. The risks and benefits of therapy with aldosterone receptor antagonist therapy. Curr Drug Saf 2007; 2:71–77.
- Shah KB, Rao K, Sawyer R, Gottlieb SS. The adequacy of laboratory monitoring in patients treated with spironolactone for congestive heart failure. J Am Coll Cardiol 2005; 46:845–849.
- Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004; 351:543–551.
- Albert NM, Fonarow GC, Yancy CW, et al. Influence of dedicated heart failure clinics on delivery of recommended therapies in outpatient cardiology practices: findings from the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Am Heart J 2010; 159:238–244.
- Wei L, Struthers AD, Fahey T, Watson AD, Macdonald TM. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ 2010; 340:c1768.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Jeunemaitre X, Chatellier G, Kreft-Jais C, et al. Efficacy and tolerance of spironolactone in essential hypertension. Am J Cardiol 1987; 60:820–825.
- Verhamme K, Mosis G, Dieleman J, Stricker B, Sturkenboom M. Spironolactone and risk of upper gastrointestinal events: population based case-control study. BMJ 2006; 333:330.
- Massie BM, Carson PE, McMurray JJ, et al; I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 2008; 359:2456–2467.
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362:777–781.
- Deswal A, Richardson P, Bozkurt B, Mann DL. Results of the Randomized Aldosterone Antagonism in Heart Failure With Preserved Ejection Fraction Trial (RAAM-PEF). J Card Fail 2011; 17:634–642.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the ‘aldosterone receptor blockade in diastolic heart failure’ trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Hené RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int 1982; 21:98–101.
- Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 1996; 98:1063–1068.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int 2006; 70:2116–2123.
- Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol 2006; 17:3438–3446.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int 2006; 70:536–542.
- Toyonaga J, Tsuruya K, Ikeda H, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant 2011; 26:2475–2484.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- Albert NM, Yancy CW, Liang L, et al. Use of aldosterone antagonists in heart failure. JAMA 2009; 302:1658–1665.
- Parthasarathy HK, Ménard J, White WB, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 2011; 29:980–990.
- Yamaji M, Tsutamoto T, Kawahara C, et al. Effect of eplerenone versus spironolactone on cortisol and hemoglobin A1(c) levels in patients with chronic heart failure. Am Heart J 2010; 160:915–921.
- Simpson SA, Tait JF, Bush IE. Secretion of a salt-retaining hormone by the mammalian adrenal cortex. Lancet 1952; 2:226–228.
- Struthers AD, MacDonald TM. Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc Res 2004; 61:663–670.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the “aldosterone receptor blockade in diastolic heart failure” trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 1990; 82:1730–1736.
- MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart 1999; 82:57–61.
- McKelvie RS, Yusuf S, Pericak D, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation 1999; 100:1056–1064.
- Tang WH, Vagelos RH, Yee YG, et al. Neurohormonal and clinical responses to high- versus low-dose enalapril therapy in chronic heart failure. J Am Coll Cardiol 2002; 39:70–78.
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001; 345:1689–1697.
- Funder JW. The role of aldosterone and mineralocorticoid receptors in cardiovascular disease. Am J Cardiovasc Drugs 2007; 7:151–157.
- Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 1990; 67:1355–1364.
- Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol 1992; 262:E96–E99.
- Garthwaite SM, McMahon EG. The evolution of aldosterone antagonists. Mol Cell Endocrinol 2004; 217:27–31.
- de Gasparo M, Joss U, Ramjoué HP, et al. Three new epoxy-spirolactone derivatives: characterization in vivo and in vitro. J Pharmacol Exp Ther 1987; 240:650–656.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am J Hypertens 2002; 15:709–716.
- Tsutamoto T, Wada A, Maeda K, et al. Spironolactone inhibits the transcardiac extraction of aldosterone in patients with congestive heart failure. J Am Coll Cardiol 2000; 36:838–844.
- Hayashi M, Tsutamoto T, Wada A, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents postinfarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003; 107:2559–2565.
- Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additive amelioration of left ventricular remodeling and molecular alterations by combined aldosterone and angiotensin receptor blockade after myocardial infarction. Cardiovasc Res 2005; 67:97–105.
- Ramires FJ, Mansur A, Coelho O, et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idiopathic dilated or to ischemic cardiomyopathy. Am J Cardiol 2000; 85:1207–1211.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the reninangiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Sica DA. The risks and benefits of therapy with aldosterone receptor antagonist therapy. Curr Drug Saf 2007; 2:71–77.
- Shah KB, Rao K, Sawyer R, Gottlieb SS. The adequacy of laboratory monitoring in patients treated with spironolactone for congestive heart failure. J Am Coll Cardiol 2005; 46:845–849.
- Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004; 351:543–551.
- Albert NM, Fonarow GC, Yancy CW, et al. Influence of dedicated heart failure clinics on delivery of recommended therapies in outpatient cardiology practices: findings from the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Am Heart J 2010; 159:238–244.
- Wei L, Struthers AD, Fahey T, Watson AD, Macdonald TM. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ 2010; 340:c1768.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Jeunemaitre X, Chatellier G, Kreft-Jais C, et al. Efficacy and tolerance of spironolactone in essential hypertension. Am J Cardiol 1987; 60:820–825.
- Verhamme K, Mosis G, Dieleman J, Stricker B, Sturkenboom M. Spironolactone and risk of upper gastrointestinal events: population based case-control study. BMJ 2006; 333:330.
- Massie BM, Carson PE, McMurray JJ, et al; I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 2008; 359:2456–2467.
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362:777–781.
- Deswal A, Richardson P, Bozkurt B, Mann DL. Results of the Randomized Aldosterone Antagonism in Heart Failure With Preserved Ejection Fraction Trial (RAAM-PEF). J Card Fail 2011; 17:634–642.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the ‘aldosterone receptor blockade in diastolic heart failure’ trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Hené RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int 1982; 21:98–101.
- Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 1996; 98:1063–1068.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int 2006; 70:2116–2123.
- Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol 2006; 17:3438–3446.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int 2006; 70:536–542.
- Toyonaga J, Tsuruya K, Ikeda H, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant 2011; 26:2475–2484.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- Albert NM, Yancy CW, Liang L, et al. Use of aldosterone antagonists in heart failure. JAMA 2009; 302:1658–1665.
- Parthasarathy HK, Ménard J, White WB, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 2011; 29:980–990.
- Yamaji M, Tsutamoto T, Kawahara C, et al. Effect of eplerenone versus spironolactone on cortisol and hemoglobin A1(c) levels in patients with chronic heart failure. Am Heart J 2010; 160:915–921.
KEY POINTS
- Although caution is advised in starting ARAs, these drugs are commonly underused in heart failure.
- Aldosterone “escape” can blunt the effects of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. This is the rationale for also using ARAs.
- The major trials of ARAs in heart failure to date have been the Randomized Aldactone Evaluation Study (RALES), the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS), and the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF).
- Close monitoring is essential when starting an ARA, as severe hyperkalemia and renal insufficiency can occur.
A woman with a swollen uvula
A 39-year-old woman on patient-controlled analgesia with morphine after cesarean delivery suddenly developed shortness of breath. On examination, the uvula was notably edematous, pale, and translucent, with no sign of erythema (Figure 1). The previous night, when the morphine was started, she had mild pruritus, which responded to treatment with oral diphenhydramine (Benadryl).
Given the extent of the uvular edema, emergency intubation was performed, and epinephrine and corticosteroids were given.
Q: Which is the most likely diagnosis at this point?
- Hereditary angioedema
- Infection causing epiglottitis masquerading as uvular swelling
- Opioid-induced uvular hydrops
- Myxedematous infiltration due to hypothyroidism
A: Opioid-induced uvular hydrops is the most likely diagnosis in this case, although it is rare. The most common side effect of opioids is constipation; others include lethargy, delirium, and sedation.
Hereditary angioedema is unlikely in this patient, as it usually presents in childhood or adolescence and there is usually a family history. Also, her cesarean delivery was done under regional anesthesia, which requires no oral or uvular manipulation and so cannot cause uvular swelling. In the absence of pain, fever, or signs and symptoms of pharyngitis, infection was unlikely. And she had no history of hypothyroidism.
UVULAR HYDROPS
This condition may be caused by opioid-induced direct degranulation of mast cells and basophils, inciting an inflammatory response.
Differential diagnoses include:
- Hereditary angioedema
- Effects of other drugs (angiotensin-converting enzyme inhibitors, cocaine, non-steroidal anti-inflammatory drugs)
- Infection (Haemophilus influenzae, Streptococcus pneumoniae)
- Trauma (intubation during surgery)
- Myxedematous infiltration due to hypothyroidism
- Granulomatous infiltration due to sarcoidosis.
When the patient’s condition has been stabilized, several outpatient tests may help narrow the differential diagnosis:
- Serum complement levels, of which the most reliable and cost-effective screening test for hereditary angioedema is a serum C4 level
- A serum thyroid-stimulating hormone level to rule out hypothyroidism
- Skin and lymph node biopsy (if skin lesions or lymphadenopathy is present), and chest radiograph to rule out sarcoidosis
- A urine drug screen and a skin-prick test for opioids (even though a negative skin test does not exclude opiate sensitivity).
OUR PATIENT’S COURSE
Our patient was discharged and underwent further outpatient evaluation. At discharge, she was advised to avoid opioids in the future.
SUGGESTED READING
Grigoriadou S, Longhurst HJ. Clinical Immunology Review Series: An approach to the patient with angio-oedema. Clin Exp Immunol 2009; 155:367–377.
Marx JA, Hockberger RS, Walls RM. Urticaria and angioedema. In:Marx JA, Hockberger RS, Walls RM, et aleditors. Rosen’s Emergency Medicine. 7th ed. Philadelphia, PA: Mosby/Elsevier, 2010.
Morgan BP. Hereditary angioedema—therapies old and new. N Engl J Med 2010; 363:581–583.
Neustein SM. Acute uvular edema after regional anesthesia. J Clin Anesth 2007; 19:365–366.
A 39-year-old woman on patient-controlled analgesia with morphine after cesarean delivery suddenly developed shortness of breath. On examination, the uvula was notably edematous, pale, and translucent, with no sign of erythema (Figure 1). The previous night, when the morphine was started, she had mild pruritus, which responded to treatment with oral diphenhydramine (Benadryl).
Given the extent of the uvular edema, emergency intubation was performed, and epinephrine and corticosteroids were given.
Q: Which is the most likely diagnosis at this point?
- Hereditary angioedema
- Infection causing epiglottitis masquerading as uvular swelling
- Opioid-induced uvular hydrops
- Myxedematous infiltration due to hypothyroidism
A: Opioid-induced uvular hydrops is the most likely diagnosis in this case, although it is rare. The most common side effect of opioids is constipation; others include lethargy, delirium, and sedation.
Hereditary angioedema is unlikely in this patient, as it usually presents in childhood or adolescence and there is usually a family history. Also, her cesarean delivery was done under regional anesthesia, which requires no oral or uvular manipulation and so cannot cause uvular swelling. In the absence of pain, fever, or signs and symptoms of pharyngitis, infection was unlikely. And she had no history of hypothyroidism.
UVULAR HYDROPS
This condition may be caused by opioid-induced direct degranulation of mast cells and basophils, inciting an inflammatory response.
Differential diagnoses include:
- Hereditary angioedema
- Effects of other drugs (angiotensin-converting enzyme inhibitors, cocaine, non-steroidal anti-inflammatory drugs)
- Infection (Haemophilus influenzae, Streptococcus pneumoniae)
- Trauma (intubation during surgery)
- Myxedematous infiltration due to hypothyroidism
- Granulomatous infiltration due to sarcoidosis.
When the patient’s condition has been stabilized, several outpatient tests may help narrow the differential diagnosis:
- Serum complement levels, of which the most reliable and cost-effective screening test for hereditary angioedema is a serum C4 level
- A serum thyroid-stimulating hormone level to rule out hypothyroidism
- Skin and lymph node biopsy (if skin lesions or lymphadenopathy is present), and chest radiograph to rule out sarcoidosis
- A urine drug screen and a skin-prick test for opioids (even though a negative skin test does not exclude opiate sensitivity).
OUR PATIENT’S COURSE
Our patient was discharged and underwent further outpatient evaluation. At discharge, she was advised to avoid opioids in the future.
A 39-year-old woman on patient-controlled analgesia with morphine after cesarean delivery suddenly developed shortness of breath. On examination, the uvula was notably edematous, pale, and translucent, with no sign of erythema (Figure 1). The previous night, when the morphine was started, she had mild pruritus, which responded to treatment with oral diphenhydramine (Benadryl).
Given the extent of the uvular edema, emergency intubation was performed, and epinephrine and corticosteroids were given.
Q: Which is the most likely diagnosis at this point?
- Hereditary angioedema
- Infection causing epiglottitis masquerading as uvular swelling
- Opioid-induced uvular hydrops
- Myxedematous infiltration due to hypothyroidism
A: Opioid-induced uvular hydrops is the most likely diagnosis in this case, although it is rare. The most common side effect of opioids is constipation; others include lethargy, delirium, and sedation.
Hereditary angioedema is unlikely in this patient, as it usually presents in childhood or adolescence and there is usually a family history. Also, her cesarean delivery was done under regional anesthesia, which requires no oral or uvular manipulation and so cannot cause uvular swelling. In the absence of pain, fever, or signs and symptoms of pharyngitis, infection was unlikely. And she had no history of hypothyroidism.
UVULAR HYDROPS
This condition may be caused by opioid-induced direct degranulation of mast cells and basophils, inciting an inflammatory response.
Differential diagnoses include:
- Hereditary angioedema
- Effects of other drugs (angiotensin-converting enzyme inhibitors, cocaine, non-steroidal anti-inflammatory drugs)
- Infection (Haemophilus influenzae, Streptococcus pneumoniae)
- Trauma (intubation during surgery)
- Myxedematous infiltration due to hypothyroidism
- Granulomatous infiltration due to sarcoidosis.
When the patient’s condition has been stabilized, several outpatient tests may help narrow the differential diagnosis:
- Serum complement levels, of which the most reliable and cost-effective screening test for hereditary angioedema is a serum C4 level
- A serum thyroid-stimulating hormone level to rule out hypothyroidism
- Skin and lymph node biopsy (if skin lesions or lymphadenopathy is present), and chest radiograph to rule out sarcoidosis
- A urine drug screen and a skin-prick test for opioids (even though a negative skin test does not exclude opiate sensitivity).
OUR PATIENT’S COURSE
Our patient was discharged and underwent further outpatient evaluation. At discharge, she was advised to avoid opioids in the future.
SUGGESTED READING
Grigoriadou S, Longhurst HJ. Clinical Immunology Review Series: An approach to the patient with angio-oedema. Clin Exp Immunol 2009; 155:367–377.
Marx JA, Hockberger RS, Walls RM. Urticaria and angioedema. In:Marx JA, Hockberger RS, Walls RM, et aleditors. Rosen’s Emergency Medicine. 7th ed. Philadelphia, PA: Mosby/Elsevier, 2010.
Morgan BP. Hereditary angioedema—therapies old and new. N Engl J Med 2010; 363:581–583.
Neustein SM. Acute uvular edema after regional anesthesia. J Clin Anesth 2007; 19:365–366.
SUGGESTED READING
Grigoriadou S, Longhurst HJ. Clinical Immunology Review Series: An approach to the patient with angio-oedema. Clin Exp Immunol 2009; 155:367–377.
Marx JA, Hockberger RS, Walls RM. Urticaria and angioedema. In:Marx JA, Hockberger RS, Walls RM, et aleditors. Rosen’s Emergency Medicine. 7th ed. Philadelphia, PA: Mosby/Elsevier, 2010.
Morgan BP. Hereditary angioedema—therapies old and new. N Engl J Med 2010; 363:581–583.
Neustein SM. Acute uvular edema after regional anesthesia. J Clin Anesth 2007; 19:365–366.