User login
Immunotherapy may benefit relapsed HSCT recipients
Photo from Business Wire
Results of a phase 1 study suggest that repeated doses of the immunotherapy drug ipilimumab is a feasible treatment option for patients with hematologic diseases who relapse after allogeneic hematopoietic stem cell transplant (HSCT).
Seven of the 28 patients studied responded to the treatment, but immune-mediated toxic effects and graft-vs-host disease (GVHD) occurred as well.
These results were published in NEJM.
Ipilimumab, which is already approved to treat unresectable or metastatic melanoma, works by blocking the immune checkpoint CTLA-4. Blockade of CTLA-4 has been shown to augment T-cell activation and proliferation.
“We believe [,in the case of relapse after HSCT,] the donor immune cells are present but can’t recognize the tumor cells because of inhibitory signals that disguise them,” said study author Matthew Davids, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
“By blocking the checkpoint, you allow the donor cells to see the cancer cells.”
Dr Davids and his colleagues tested this theory in 28 patients who had relapsed after allogeneic HSCT. The patients had acute myeloid leukemia (AML, n=12), Hodgkin lymphoma (n=7), non-Hodgkin lymphoma (n=4), myelodysplastic syndromes (MDS, n=2), multiple myeloma (n=1), myeloproliferative neoplasm (n=1), or acute lymphoblastic leukemia (n=1).
Patients had received a median of 3 prior treatment regimens, excluding HSCT (range, 1 to 14), and 20 patients (71%) had received treatment for relapse after transplant. Eight patients (29%) previously had grade 1/2 acute GVHD, and 16 (57%) previously had chronic GVHD.
The median time from transplant to initial treatment with ipilimumab was 675 days (range, 198 to 1830), and the median time from relapse to initial treatment with ipilimumab was 97 days (range, 0 to
1415).
Patients received induction therapy with ipilimumab at a dose of 3 mg/kg or 10 mg/kg every 3 weeks for a total of 4 doses. Those who had a clinical benefit received additional doses every 12 weeks for up to 60 weeks.
Safety
Five patients discontinued ipilimumab due to dose-limiting toxic effects. Four of these patients had GVHD, and 1 had severe immune-related adverse events.
Dose-limiting GVHD presented as chronic GVHD of the liver in 3 patients and acute GVHD of the gut in 1 patient.
Immune-related adverse events included death (n=1), pneumonitis (2 grade 2 events, 1 grade 4 event), colitis (1 grade 3 event), immune thrombocytopenia (1 grade 2 event), and diarrhea (1 grade 2 event).
Efficacy
There were no responses in patients who received ipilimumab at 3 mg/kg. Among the 22 patients who received ipilimumab at 10 mg/kg, 5 had a complete response, and 2 had a partial response.
Six other patients did not qualify as having responses but had a decrease in their tumor burden. Altogether, ipilimumab reduced tumor burden in 59% of patients.
The complete responses occurred in 4 patients with extramedullary AML and 1 patient with MDS developing into AML. Two of the AML patients remained in complete response at 12 and 15 months, and the patient with MDS remained in complete response at 16 months.
At a median follow-up of 15 months (range, 8 to 27), the median duration of response had not been reached. Responses were associated with in situ infiltration of cytotoxic CD8+ T cells, decreased activation of regulatory T cells, and expansion of subpopulations of effector T cells.
The 1-year overall survival rate was 49%.
The investigators said these encouraging results have set the stage for larger trials of checkpoint blockade in this patient population. Further research is planned to determine whether immunotherapy drugs could be given to high-risk patients to prevent relapse. ![]()
Photo from Business Wire
Results of a phase 1 study suggest that repeated doses of the immunotherapy drug ipilimumab is a feasible treatment option for patients with hematologic diseases who relapse after allogeneic hematopoietic stem cell transplant (HSCT).
Seven of the 28 patients studied responded to the treatment, but immune-mediated toxic effects and graft-vs-host disease (GVHD) occurred as well.
These results were published in NEJM.
Ipilimumab, which is already approved to treat unresectable or metastatic melanoma, works by blocking the immune checkpoint CTLA-4. Blockade of CTLA-4 has been shown to augment T-cell activation and proliferation.
“We believe [,in the case of relapse after HSCT,] the donor immune cells are present but can’t recognize the tumor cells because of inhibitory signals that disguise them,” said study author Matthew Davids, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
“By blocking the checkpoint, you allow the donor cells to see the cancer cells.”
Dr Davids and his colleagues tested this theory in 28 patients who had relapsed after allogeneic HSCT. The patients had acute myeloid leukemia (AML, n=12), Hodgkin lymphoma (n=7), non-Hodgkin lymphoma (n=4), myelodysplastic syndromes (MDS, n=2), multiple myeloma (n=1), myeloproliferative neoplasm (n=1), or acute lymphoblastic leukemia (n=1).
Patients had received a median of 3 prior treatment regimens, excluding HSCT (range, 1 to 14), and 20 patients (71%) had received treatment for relapse after transplant. Eight patients (29%) previously had grade 1/2 acute GVHD, and 16 (57%) previously had chronic GVHD.
The median time from transplant to initial treatment with ipilimumab was 675 days (range, 198 to 1830), and the median time from relapse to initial treatment with ipilimumab was 97 days (range, 0 to
1415).
Patients received induction therapy with ipilimumab at a dose of 3 mg/kg or 10 mg/kg every 3 weeks for a total of 4 doses. Those who had a clinical benefit received additional doses every 12 weeks for up to 60 weeks.
Safety
Five patients discontinued ipilimumab due to dose-limiting toxic effects. Four of these patients had GVHD, and 1 had severe immune-related adverse events.
Dose-limiting GVHD presented as chronic GVHD of the liver in 3 patients and acute GVHD of the gut in 1 patient.
Immune-related adverse events included death (n=1), pneumonitis (2 grade 2 events, 1 grade 4 event), colitis (1 grade 3 event), immune thrombocytopenia (1 grade 2 event), and diarrhea (1 grade 2 event).
Efficacy
There were no responses in patients who received ipilimumab at 3 mg/kg. Among the 22 patients who received ipilimumab at 10 mg/kg, 5 had a complete response, and 2 had a partial response.
Six other patients did not qualify as having responses but had a decrease in their tumor burden. Altogether, ipilimumab reduced tumor burden in 59% of patients.
The complete responses occurred in 4 patients with extramedullary AML and 1 patient with MDS developing into AML. Two of the AML patients remained in complete response at 12 and 15 months, and the patient with MDS remained in complete response at 16 months.
At a median follow-up of 15 months (range, 8 to 27), the median duration of response had not been reached. Responses were associated with in situ infiltration of cytotoxic CD8+ T cells, decreased activation of regulatory T cells, and expansion of subpopulations of effector T cells.
The 1-year overall survival rate was 49%.
The investigators said these encouraging results have set the stage for larger trials of checkpoint blockade in this patient population. Further research is planned to determine whether immunotherapy drugs could be given to high-risk patients to prevent relapse. ![]()
Photo from Business Wire
Results of a phase 1 study suggest that repeated doses of the immunotherapy drug ipilimumab is a feasible treatment option for patients with hematologic diseases who relapse after allogeneic hematopoietic stem cell transplant (HSCT).
Seven of the 28 patients studied responded to the treatment, but immune-mediated toxic effects and graft-vs-host disease (GVHD) occurred as well.
These results were published in NEJM.
Ipilimumab, which is already approved to treat unresectable or metastatic melanoma, works by blocking the immune checkpoint CTLA-4. Blockade of CTLA-4 has been shown to augment T-cell activation and proliferation.
“We believe [,in the case of relapse after HSCT,] the donor immune cells are present but can’t recognize the tumor cells because of inhibitory signals that disguise them,” said study author Matthew Davids, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
“By blocking the checkpoint, you allow the donor cells to see the cancer cells.”
Dr Davids and his colleagues tested this theory in 28 patients who had relapsed after allogeneic HSCT. The patients had acute myeloid leukemia (AML, n=12), Hodgkin lymphoma (n=7), non-Hodgkin lymphoma (n=4), myelodysplastic syndromes (MDS, n=2), multiple myeloma (n=1), myeloproliferative neoplasm (n=1), or acute lymphoblastic leukemia (n=1).
Patients had received a median of 3 prior treatment regimens, excluding HSCT (range, 1 to 14), and 20 patients (71%) had received treatment for relapse after transplant. Eight patients (29%) previously had grade 1/2 acute GVHD, and 16 (57%) previously had chronic GVHD.
The median time from transplant to initial treatment with ipilimumab was 675 days (range, 198 to 1830), and the median time from relapse to initial treatment with ipilimumab was 97 days (range, 0 to
1415).
Patients received induction therapy with ipilimumab at a dose of 3 mg/kg or 10 mg/kg every 3 weeks for a total of 4 doses. Those who had a clinical benefit received additional doses every 12 weeks for up to 60 weeks.
Safety
Five patients discontinued ipilimumab due to dose-limiting toxic effects. Four of these patients had GVHD, and 1 had severe immune-related adverse events.
Dose-limiting GVHD presented as chronic GVHD of the liver in 3 patients and acute GVHD of the gut in 1 patient.
Immune-related adverse events included death (n=1), pneumonitis (2 grade 2 events, 1 grade 4 event), colitis (1 grade 3 event), immune thrombocytopenia (1 grade 2 event), and diarrhea (1 grade 2 event).
Efficacy
There were no responses in patients who received ipilimumab at 3 mg/kg. Among the 22 patients who received ipilimumab at 10 mg/kg, 5 had a complete response, and 2 had a partial response.
Six other patients did not qualify as having responses but had a decrease in their tumor burden. Altogether, ipilimumab reduced tumor burden in 59% of patients.
The complete responses occurred in 4 patients with extramedullary AML and 1 patient with MDS developing into AML. Two of the AML patients remained in complete response at 12 and 15 months, and the patient with MDS remained in complete response at 16 months.
At a median follow-up of 15 months (range, 8 to 27), the median duration of response had not been reached. Responses were associated with in situ infiltration of cytotoxic CD8+ T cells, decreased activation of regulatory T cells, and expansion of subpopulations of effector T cells.
The 1-year overall survival rate was 49%.
The investigators said these encouraging results have set the stage for larger trials of checkpoint blockade in this patient population. Further research is planned to determine whether immunotherapy drugs could be given to high-risk patients to prevent relapse. ![]()
Ipilimumab may restore antitumor immunity after relapse from HSCT
Early data hint that immune checkpoint inhibitors may be able to restore antitumor activity in patients with hematologic malignancies that have relapsed after allogeneic transplant.
Among 22 patients with relapsed hematologic cancers following allogeneic hematopoietic stem cell transplantation (HSCT) in a phase I/Ib study, treatment with the anti-CTLA-4 antibody ipilimumab (Yervoy) at a dose of 10 mg/kg was associated with complete responses in five patients, partial responses in two, and decreased tumor burden in six, reported Matthew S. Davids, MD, of the Dana-Farber Cancer Institute in Boston, and his colleagues.

“CTLA-4 blockade was a feasible approach for the treatment of patients with relapsed hematologic cancer after transplantation. Complete remissions with some durability were observed, even in patients with refractory myeloid cancers,” they wrote (N Engl J Med. 2016 Jul 14. doi: 10.1056/NEJMoa1601202).
More than one-third of patients who undergo HSCT for hematologic malignancies such as lymphoma, multiple myeloma, or leukemia will experience a relapse, and most will die within a year of relapse despite salvage therapies or retransplantation, the authors noted.
“Immune escape (i.e., tumor evasion of the donor immune system) contributes to relapse after allogeneic HSCT, and immune checkpoint inhibitory pathways probably play an important role,” they wrote.
Selective CTLA-4 blockade has been shown in mouse models to treat late relapse after transplantation by augmenting graft-versus-tumor response without apparent exacerbation of graft-versus-host disease (GVHD). To see whether the use of a CTLA-4 inhibitor could have the same effect in humans, the investigators instituted a single-group, open-label, dose-finding, safety and efficacy study of ipilimumab in 28 patients from six treatment sites.
The patients had all undergone allogeneic HSCT more than 3 months before the start of the study. The diagnoses included acute myeloid leukemia (AML) in 12 patients (including 3 with leukemia cutis and 1 with a myeloid sarcoma), Hodgkin lymphoma in 7, non-Hodgkin lymphoma in 4, myelodysplastic syndrome (MDS) in 2, and multiple myeloma, myeloproliferative neoplasm, and acute lymphoblastic leukemia in 1 patient each. Eight of the patients had previously had either grade I or II acute GVHD; 16 had had chronic GVHD.
Patients received induction therapy with ipilimumab at a dose of either 3 mg/kg (6 patients), or 10 mg/kg (22 patients) every 3 weeks for a total of 4 doses. Patients who experienced a clinical benefit from the drug could receive additional doses every 12 weeks for up to 60 weeks.
There were no clinical responses meeting study criteria in any of the patients who received the 3-mg/kg dose. Among the 22 who received the 10-mg/kg dose, however, the rate of complete responses was 23% (5 of 22), partial responses 9% (2 of 22), and decreased tumor burden 27% (6 of 22). The remaining nine patients experienced disease progression.
Four of the complete responses occurred in patients with extramedullary AML, and one occurred in a patient with MDS transforming into AML.
The safety analysis, which included all 28 patients evaluable for adverse events, showed four discontinuations due to dose-limiting chronic GVHD of the liver in the 3 patients, and acute GVHD of the gut in 1, and to severe immune-related events in one additional patient, leading to the patient’s death.
Other grade 3 or greater adverse events possibly related to ipilimumab included acute kidney injury (one patient) , corneal ulcer (one), thrombocytopenia (nine), neutropenia (three), anemia and pleural effusion (two).
The investigators point out that therapy to stimulate a graft-versus-tumor effect has the potential to promote or exacerbate GVHD, as occurred in four patients in the study. The GVHD in these patients was effectively managed with glucocorticoids, however.
The National Institutes of Health, Leukemia and Lymphoma Society, Pasquarello Tissue Bank, and Dana-Farber Cancer Institute supported the study. Dr. Davids disclosed grants from ASCO, the Pasquarello Tissue Bank, NIH, NCI, and Leukemia and Lymphoma society, and personal fees from several companies outside the study. Several coauthors disclosed relationships with various pharmaceutical companies, including Bristol-Myers Squibb, maker of ipilimumab.
Early data hint that immune checkpoint inhibitors may be able to restore antitumor activity in patients with hematologic malignancies that have relapsed after allogeneic transplant.
Among 22 patients with relapsed hematologic cancers following allogeneic hematopoietic stem cell transplantation (HSCT) in a phase I/Ib study, treatment with the anti-CTLA-4 antibody ipilimumab (Yervoy) at a dose of 10 mg/kg was associated with complete responses in five patients, partial responses in two, and decreased tumor burden in six, reported Matthew S. Davids, MD, of the Dana-Farber Cancer Institute in Boston, and his colleagues.
“CTLA-4 blockade was a feasible approach for the treatment of patients with relapsed hematologic cancer after transplantation. Complete remissions with some durability were observed, even in patients with refractory myeloid cancers,” they wrote (N Engl J Med. 2016 Jul 14. doi: 10.1056/NEJMoa1601202).
More than one-third of patients who undergo HSCT for hematologic malignancies such as lymphoma, multiple myeloma, or leukemia will experience a relapse, and most will die within a year of relapse despite salvage therapies or retransplantation, the authors noted.
“Immune escape (i.e., tumor evasion of the donor immune system) contributes to relapse after allogeneic HSCT, and immune checkpoint inhibitory pathways probably play an important role,” they wrote.
Selective CTLA-4 blockade has been shown in mouse models to treat late relapse after transplantation by augmenting graft-versus-tumor response without apparent exacerbation of graft-versus-host disease (GVHD). To see whether the use of a CTLA-4 inhibitor could have the same effect in humans, the investigators instituted a single-group, open-label, dose-finding, safety and efficacy study of ipilimumab in 28 patients from six treatment sites.
The patients had all undergone allogeneic HSCT more than 3 months before the start of the study. The diagnoses included acute myeloid leukemia (AML) in 12 patients (including 3 with leukemia cutis and 1 with a myeloid sarcoma), Hodgkin lymphoma in 7, non-Hodgkin lymphoma in 4, myelodysplastic syndrome (MDS) in 2, and multiple myeloma, myeloproliferative neoplasm, and acute lymphoblastic leukemia in 1 patient each. Eight of the patients had previously had either grade I or II acute GVHD; 16 had had chronic GVHD.
Patients received induction therapy with ipilimumab at a dose of either 3 mg/kg (6 patients), or 10 mg/kg (22 patients) every 3 weeks for a total of 4 doses. Patients who experienced a clinical benefit from the drug could receive additional doses every 12 weeks for up to 60 weeks.
There were no clinical responses meeting study criteria in any of the patients who received the 3-mg/kg dose. Among the 22 who received the 10-mg/kg dose, however, the rate of complete responses was 23% (5 of 22), partial responses 9% (2 of 22), and decreased tumor burden 27% (6 of 22). The remaining nine patients experienced disease progression.
Four of the complete responses occurred in patients with extramedullary AML, and one occurred in a patient with MDS transforming into AML.
The safety analysis, which included all 28 patients evaluable for adverse events, showed four discontinuations due to dose-limiting chronic GVHD of the liver in the 3 patients, and acute GVHD of the gut in 1, and to severe immune-related events in one additional patient, leading to the patient’s death.
Other grade 3 or greater adverse events possibly related to ipilimumab included acute kidney injury (one patient) , corneal ulcer (one), thrombocytopenia (nine), neutropenia (three), anemia and pleural effusion (two).
The investigators point out that therapy to stimulate a graft-versus-tumor effect has the potential to promote or exacerbate GVHD, as occurred in four patients in the study. The GVHD in these patients was effectively managed with glucocorticoids, however.
The National Institutes of Health, Leukemia and Lymphoma Society, Pasquarello Tissue Bank, and Dana-Farber Cancer Institute supported the study. Dr. Davids disclosed grants from ASCO, the Pasquarello Tissue Bank, NIH, NCI, and Leukemia and Lymphoma society, and personal fees from several companies outside the study. Several coauthors disclosed relationships with various pharmaceutical companies, including Bristol-Myers Squibb, maker of ipilimumab.
Early data hint that immune checkpoint inhibitors may be able to restore antitumor activity in patients with hematologic malignancies that have relapsed after allogeneic transplant.
Among 22 patients with relapsed hematologic cancers following allogeneic hematopoietic stem cell transplantation (HSCT) in a phase I/Ib study, treatment with the anti-CTLA-4 antibody ipilimumab (Yervoy) at a dose of 10 mg/kg was associated with complete responses in five patients, partial responses in two, and decreased tumor burden in six, reported Matthew S. Davids, MD, of the Dana-Farber Cancer Institute in Boston, and his colleagues.
“CTLA-4 blockade was a feasible approach for the treatment of patients with relapsed hematologic cancer after transplantation. Complete remissions with some durability were observed, even in patients with refractory myeloid cancers,” they wrote (N Engl J Med. 2016 Jul 14. doi: 10.1056/NEJMoa1601202).
More than one-third of patients who undergo HSCT for hematologic malignancies such as lymphoma, multiple myeloma, or leukemia will experience a relapse, and most will die within a year of relapse despite salvage therapies or retransplantation, the authors noted.
“Immune escape (i.e., tumor evasion of the donor immune system) contributes to relapse after allogeneic HSCT, and immune checkpoint inhibitory pathways probably play an important role,” they wrote.
Selective CTLA-4 blockade has been shown in mouse models to treat late relapse after transplantation by augmenting graft-versus-tumor response without apparent exacerbation of graft-versus-host disease (GVHD). To see whether the use of a CTLA-4 inhibitor could have the same effect in humans, the investigators instituted a single-group, open-label, dose-finding, safety and efficacy study of ipilimumab in 28 patients from six treatment sites.
The patients had all undergone allogeneic HSCT more than 3 months before the start of the study. The diagnoses included acute myeloid leukemia (AML) in 12 patients (including 3 with leukemia cutis and 1 with a myeloid sarcoma), Hodgkin lymphoma in 7, non-Hodgkin lymphoma in 4, myelodysplastic syndrome (MDS) in 2, and multiple myeloma, myeloproliferative neoplasm, and acute lymphoblastic leukemia in 1 patient each. Eight of the patients had previously had either grade I or II acute GVHD; 16 had had chronic GVHD.
Patients received induction therapy with ipilimumab at a dose of either 3 mg/kg (6 patients), or 10 mg/kg (22 patients) every 3 weeks for a total of 4 doses. Patients who experienced a clinical benefit from the drug could receive additional doses every 12 weeks for up to 60 weeks.
There were no clinical responses meeting study criteria in any of the patients who received the 3-mg/kg dose. Among the 22 who received the 10-mg/kg dose, however, the rate of complete responses was 23% (5 of 22), partial responses 9% (2 of 22), and decreased tumor burden 27% (6 of 22). The remaining nine patients experienced disease progression.
Four of the complete responses occurred in patients with extramedullary AML, and one occurred in a patient with MDS transforming into AML.
The safety analysis, which included all 28 patients evaluable for adverse events, showed four discontinuations due to dose-limiting chronic GVHD of the liver in the 3 patients, and acute GVHD of the gut in 1, and to severe immune-related events in one additional patient, leading to the patient’s death.
Other grade 3 or greater adverse events possibly related to ipilimumab included acute kidney injury (one patient) , corneal ulcer (one), thrombocytopenia (nine), neutropenia (three), anemia and pleural effusion (two).
The investigators point out that therapy to stimulate a graft-versus-tumor effect has the potential to promote or exacerbate GVHD, as occurred in four patients in the study. The GVHD in these patients was effectively managed with glucocorticoids, however.
The National Institutes of Health, Leukemia and Lymphoma Society, Pasquarello Tissue Bank, and Dana-Farber Cancer Institute supported the study. Dr. Davids disclosed grants from ASCO, the Pasquarello Tissue Bank, NIH, NCI, and Leukemia and Lymphoma society, and personal fees from several companies outside the study. Several coauthors disclosed relationships with various pharmaceutical companies, including Bristol-Myers Squibb, maker of ipilimumab.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Anti-CTLA-4 therapy may restore graft-versus-tumor effect in patients with hematologic malignancies relapsed after allogeneic transplantation.
Major finding: Five of 22 patients on a 10-mg/kg dose of ipilimumab had a complete response.
Data source: Phase I/Ib investigator-initiated study of 28 patients with hematologic malignancies relapsed after allogeneic hematopoietic stem cell transplantation.
Disclosures: The National Institutes of Health, Leukemia and Lymphoma Society, Pasquarello Tissue Bank, and Dana-Farber Cancer Institute supported the study. Dr. Davids disclosed grants from ASCO, the Pasquarello Tissue Bank, NIH, NCI, and Leukemia and Lymphoma society, and personal fees from several companies outside the study. Several coauthors disclosed relationships with various pharmaceutical companies, including Bristol-Myers Squibb, maker of ipilimumab.

Three-drug regimen boosts progression-free survival in patients with relapsed CLL and adverse prognostic features
Progression-free survival was improved for poor prognosis patients with relapsed chronic lymphocytic leukemia when idelalisib was added to bendamustine and rituximab, based on the results of a randomized, double-blind, placebo-controlled phase III study.
The findings support the three-drug approach as an important treatment option for patients with relapsed CLL and adverse prognostic features, but the regimen also was associated with more grade 3 or greater adverse events, primarily neutropenia and opportunistic infections, that led to more study drug discontinuation, Jacqueline Claudia Barrientos, M.D., and her colleagues reported at the annual meeting of the American Society of Clinical Oncology.
The overall response rates were 68% in the three-drug group and 45% in the two-drug plus placebo group.
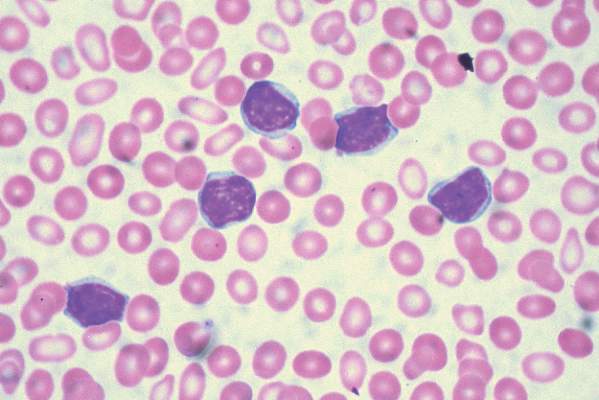
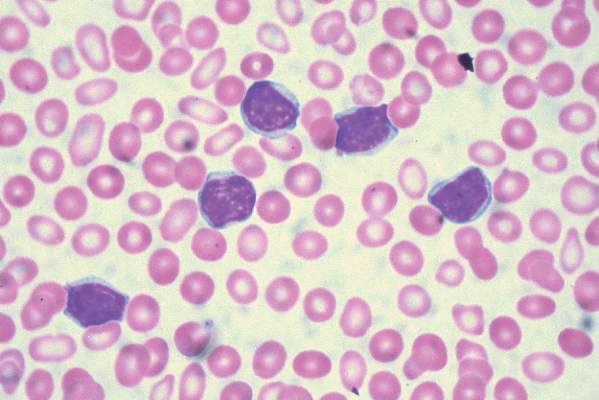
For the study, 416 patients were stratified based on whether they had 17p deletions or TP53 mutations, their IGHV mutation status, and whether they had refractory or relapsed disease. The 207 patients who received idelalisib, bendamustine, and rituximab had consistently better progression-free survival than the 209 patients who received placebo, bendamustine, and rituximab. The median progression-free survival was 23 months in the group that received idelalisib and 11 months in the group that received placebo, with an overall hazard ratio of 0.33 at a median follow-up of 12 months.
For those with 17p deletions or TP53 mutations, the median progression-free survival was 11 months with the three-drug regimen and 8 months for the two-drug plus placebo regimen. For those with neither of these abnormalities, progression-free survival was more than 24 months for patients given the three active drugs and 11 months for patients given two drugs plus placebo.
For those with the 11q deletion, median progression-free survival was 23 months with idelalisib, bendamustine, and rituximab and 9 months with placebo, bendamustine, and rituximab, said Dr. Barrientos of Hofstra University, Hempstead, N.Y.
For those with IGHV mutations, the median progression-free survival has not been reached in the idelalisib, bendamustine, and rituximab group and was 11 months in the placebo, bendamustine, and rituximab group.
Among patients with tumor burdens exceeding 5 cm, median progression-free survival was 23 months with idelalisib, bendamustine, and rituximab and about 10 months with placebo, bendamustine, and rituximab.
Grade 3 or greater adverse events affected 97% of patients given the three active drugs and 76% of patients given bendamustine and rituximab plus placebo. Adverse events resulted in drug dose reductions in 11% of those given the three active drugs and in 6% of those given bendamustine and rituximab plus placebo. The study drug was discontinued in 26% of those in the idelalisib, bendamustine, and rituximab group and in 13% of the placebo, bendamustine, and rituximab group. Death occurred in 10% of the study patients given idelalisib, bendamustine, and rituximab and in 7% of the patients given placebo, bendamustine, and rituximab.
To be eligible for the study (NCT01569295), patients needed to have previously treated recurrent CLL, have measurable lymphadenopathy, require therapy for CLL, and have experienced CLL progression for less than 36 months since the completion of their last prior therapy.
The treatment regimen consisted of 6 cycles every 28 days of bendamustine (70 mg/m2 on day 1 and day 2 of each cycle), rituximab (375 mg/m2 in cycle 1 and 500 mg/m2 in cycles 2-6), and idelalisib (150 mg twice daily) or placebo. Idelalisib or placebo was continued until an independent review committee confirmed disease progression, death, intolerable toxicity, or withdrawal of consent.
The study was sponsored by Gilead Sciences, the maker of idelalisib (Zydelig). Dr. Barrientos receives research funding from Gilead Sciences and Abbvie, and serves as a consultant or adviser to Celgene, Genentech, and Pharmacyclics.
On Twitter @maryjodales
Progression-free survival was improved for poor prognosis patients with relapsed chronic lymphocytic leukemia when idelalisib was added to bendamustine and rituximab, based on the results of a randomized, double-blind, placebo-controlled phase III study.
The findings support the three-drug approach as an important treatment option for patients with relapsed CLL and adverse prognostic features, but the regimen also was associated with more grade 3 or greater adverse events, primarily neutropenia and opportunistic infections, that led to more study drug discontinuation, Jacqueline Claudia Barrientos, M.D., and her colleagues reported at the annual meeting of the American Society of Clinical Oncology.
The overall response rates were 68% in the three-drug group and 45% in the two-drug plus placebo group.
For the study, 416 patients were stratified based on whether they had 17p deletions or TP53 mutations, their IGHV mutation status, and whether they had refractory or relapsed disease. The 207 patients who received idelalisib, bendamustine, and rituximab had consistently better progression-free survival than the 209 patients who received placebo, bendamustine, and rituximab. The median progression-free survival was 23 months in the group that received idelalisib and 11 months in the group that received placebo, with an overall hazard ratio of 0.33 at a median follow-up of 12 months.
For those with 17p deletions or TP53 mutations, the median progression-free survival was 11 months with the three-drug regimen and 8 months for the two-drug plus placebo regimen. For those with neither of these abnormalities, progression-free survival was more than 24 months for patients given the three active drugs and 11 months for patients given two drugs plus placebo.
For those with the 11q deletion, median progression-free survival was 23 months with idelalisib, bendamustine, and rituximab and 9 months with placebo, bendamustine, and rituximab, said Dr. Barrientos of Hofstra University, Hempstead, N.Y.
For those with IGHV mutations, the median progression-free survival has not been reached in the idelalisib, bendamustine, and rituximab group and was 11 months in the placebo, bendamustine, and rituximab group.
Among patients with tumor burdens exceeding 5 cm, median progression-free survival was 23 months with idelalisib, bendamustine, and rituximab and about 10 months with placebo, bendamustine, and rituximab.
Grade 3 or greater adverse events affected 97% of patients given the three active drugs and 76% of patients given bendamustine and rituximab plus placebo. Adverse events resulted in drug dose reductions in 11% of those given the three active drugs and in 6% of those given bendamustine and rituximab plus placebo. The study drug was discontinued in 26% of those in the idelalisib, bendamustine, and rituximab group and in 13% of the placebo, bendamustine, and rituximab group. Death occurred in 10% of the study patients given idelalisib, bendamustine, and rituximab and in 7% of the patients given placebo, bendamustine, and rituximab.
To be eligible for the study (NCT01569295), patients needed to have previously treated recurrent CLL, have measurable lymphadenopathy, require therapy for CLL, and have experienced CLL progression for less than 36 months since the completion of their last prior therapy.
The treatment regimen consisted of 6 cycles every 28 days of bendamustine (70 mg/m2 on day 1 and day 2 of each cycle), rituximab (375 mg/m2 in cycle 1 and 500 mg/m2 in cycles 2-6), and idelalisib (150 mg twice daily) or placebo. Idelalisib or placebo was continued until an independent review committee confirmed disease progression, death, intolerable toxicity, or withdrawal of consent.
The study was sponsored by Gilead Sciences, the maker of idelalisib (Zydelig). Dr. Barrientos receives research funding from Gilead Sciences and Abbvie, and serves as a consultant or adviser to Celgene, Genentech, and Pharmacyclics.
On Twitter @maryjodales
Progression-free survival was improved for poor prognosis patients with relapsed chronic lymphocytic leukemia when idelalisib was added to bendamustine and rituximab, based on the results of a randomized, double-blind, placebo-controlled phase III study.
The findings support the three-drug approach as an important treatment option for patients with relapsed CLL and adverse prognostic features, but the regimen also was associated with more grade 3 or greater adverse events, primarily neutropenia and opportunistic infections, that led to more study drug discontinuation, Jacqueline Claudia Barrientos, M.D., and her colleagues reported at the annual meeting of the American Society of Clinical Oncology.
The overall response rates were 68% in the three-drug group and 45% in the two-drug plus placebo group.
For the study, 416 patients were stratified based on whether they had 17p deletions or TP53 mutations, their IGHV mutation status, and whether they had refractory or relapsed disease. The 207 patients who received idelalisib, bendamustine, and rituximab had consistently better progression-free survival than the 209 patients who received placebo, bendamustine, and rituximab. The median progression-free survival was 23 months in the group that received idelalisib and 11 months in the group that received placebo, with an overall hazard ratio of 0.33 at a median follow-up of 12 months.
For those with 17p deletions or TP53 mutations, the median progression-free survival was 11 months with the three-drug regimen and 8 months for the two-drug plus placebo regimen. For those with neither of these abnormalities, progression-free survival was more than 24 months for patients given the three active drugs and 11 months for patients given two drugs plus placebo.
For those with the 11q deletion, median progression-free survival was 23 months with idelalisib, bendamustine, and rituximab and 9 months with placebo, bendamustine, and rituximab, said Dr. Barrientos of Hofstra University, Hempstead, N.Y.
For those with IGHV mutations, the median progression-free survival has not been reached in the idelalisib, bendamustine, and rituximab group and was 11 months in the placebo, bendamustine, and rituximab group.
Among patients with tumor burdens exceeding 5 cm, median progression-free survival was 23 months with idelalisib, bendamustine, and rituximab and about 10 months with placebo, bendamustine, and rituximab.
Grade 3 or greater adverse events affected 97% of patients given the three active drugs and 76% of patients given bendamustine and rituximab plus placebo. Adverse events resulted in drug dose reductions in 11% of those given the three active drugs and in 6% of those given bendamustine and rituximab plus placebo. The study drug was discontinued in 26% of those in the idelalisib, bendamustine, and rituximab group and in 13% of the placebo, bendamustine, and rituximab group. Death occurred in 10% of the study patients given idelalisib, bendamustine, and rituximab and in 7% of the patients given placebo, bendamustine, and rituximab.
To be eligible for the study (NCT01569295), patients needed to have previously treated recurrent CLL, have measurable lymphadenopathy, require therapy for CLL, and have experienced CLL progression for less than 36 months since the completion of their last prior therapy.
The treatment regimen consisted of 6 cycles every 28 days of bendamustine (70 mg/m2 on day 1 and day 2 of each cycle), rituximab (375 mg/m2 in cycle 1 and 500 mg/m2 in cycles 2-6), and idelalisib (150 mg twice daily) or placebo. Idelalisib or placebo was continued until an independent review committee confirmed disease progression, death, intolerable toxicity, or withdrawal of consent.
The study was sponsored by Gilead Sciences, the maker of idelalisib (Zydelig). Dr. Barrientos receives research funding from Gilead Sciences and Abbvie, and serves as a consultant or adviser to Celgene, Genentech, and Pharmacyclics.
On Twitter @maryjodales
FROM 2016 ASCO ANNUAL MEETING
Key clinical point: Progression-free survival was improved for poor prognosis patients with relapsed chronic lymphocytic leukemia when idelalisib was added to bendamustine, and rituximab.
Major finding: The median progression-free survival was 23 months in the group that received idelalisib and 11 months in the group that received placebo, with an overall hazard ratio of 0.33 at a median follow-up of 12 months.
Data source: A randomized, double-blind, placebo-controlled phase III study of 416 patients stratified on the basis of 17p deletions or TP53 mutations, IGHV mutation status, and whether they had refractory or relapsed disease.
Disclosures: The study was sponsored by Gilead Sciences, the maker of idelalisib (Zydelig). Dr. Barrientos receives research funding from Gilead Sciences and Abbvie, and serves as a consultant or adviser to Celgene, Genentech, and Pharmacyclics.
ATRA-arsenic ‘new standard of care’ for APL
The combination of all-trans-retinoic acid (ATRA) and arsenic trioxide continues to show advantages over ATRA and chemotherapy as first-line therapy for patients with low- or intermediate-risk acute promyelocytic leukemia (APL), investigators report.
The final analysis of theAPL046 study, a noninferiority trial of ATRA plus arsenic trioxide (ATRA-ATO) vs. ATRA and standard chemotherapy, showed that event-free survival (EFS) and overall survival (OS) were significantly better among patients assigned to ATRA-ATO. Patients in this group also had a significantly lower cumulative incidence of relapse, reported Francesco Lo-Coco, MD, of University Tor Vergata in Rome and colleagues.
“Our results support the use of ATRA-ATO in patients with newly diagnosed APL and point to this strategy as the new standard of care for low- or intermediate-risk patients. Studies exploring the role of ATRA-ATO are warranted in other APL subsets including high-risk, pediatric, and elderly patients,” they wrote (J Clin Oncol. 2016 July 11. doi: 10.1200/JCO.2016.67.1982).
In the trial, 276 patients with newly diagnosed APL were randomly assigned to receive either ATRA-ATO or ATRA plus chemotherapy with idarubicin, mercaptopurine, and methotrexate.
A total of 263 patients were evaluable for response to induction, including 127 assigned to ATRA-ATO and 136 assigned to ATRA-chemotherapy. Following induction, all patients assigned to ATRA-ATO and 127 assigned to chemotherapy (97%) achieved a complete remission (CR).
After a median follow-up of 40.6 months, the rate of EFS, the primary outcome, was 97.3% for ATRA-ATO vs. 80% for ATRA-chemotherapy (P less than .001). The cumulative incidences of relapse were 1.9% and 13.9%, respectively (P = .0013), and overall survival rates at 50 months were 99.2% vs. 92.6% (P = . 0073).
Hematologic toxicities were more frequent among patients assigned to chemotherapy, while liver function abnormalities occurred more often among those assigned to ATO.
One patient assigned to ATRA-ATO died while in CR, from bronchopneumonia caused by the H1N1 (influenza) virus. Five patients assigned to chemotherapy died in CR, from hemorrhagic shock, pulmonary embolism, bronchopneumonia (two patients) and secondary myelodysplastic syndrome.
Of the 17 patients who experienced relapses during follow-up, 2 assigned to ATRA-ATO had relapses at 22 and 27 months. The remaining 15 relapses were among patients assigned to ATRA-chemotherapy, occurring at a median of 14 months.
The authors noted that the superior EFS and cumulative incidence of relapse with ATRA-ATO emerged only after longer follow-up, indicating that the advantage of ATRA-ATO over ATRA-chemotherapy increases over time and that “the inclusion of ATO in the treatment of low- or intermediate-risk APL not only reduces mortality and hematologic toxicity, but also results in improved and sustained antileukemic activity.”
The study was supported by the Gruppo Italiano Malattie Ematologiche dell’Adulto Foundation; the Associazione Italiana Contro le Leucemie, Linfomi e Mieloma, Associazione Italiana per la Ricerca sul Cancro, and the German Federal Ministry of Education and Research. Dr. Lo-Coco disclosed honoraria, consulting, and speakers’ bureau activities with Teva Pharmaceutical, Lundbeck, Novartis, Baxalta, and Pfizer. Several other coauthors disclosed similar relationships.
The combination of all-trans-retinoic acid (ATRA) and arsenic trioxide continues to show advantages over ATRA and chemotherapy as first-line therapy for patients with low- or intermediate-risk acute promyelocytic leukemia (APL), investigators report.
The final analysis of theAPL046 study, a noninferiority trial of ATRA plus arsenic trioxide (ATRA-ATO) vs. ATRA and standard chemotherapy, showed that event-free survival (EFS) and overall survival (OS) were significantly better among patients assigned to ATRA-ATO. Patients in this group also had a significantly lower cumulative incidence of relapse, reported Francesco Lo-Coco, MD, of University Tor Vergata in Rome and colleagues.
“Our results support the use of ATRA-ATO in patients with newly diagnosed APL and point to this strategy as the new standard of care for low- or intermediate-risk patients. Studies exploring the role of ATRA-ATO are warranted in other APL subsets including high-risk, pediatric, and elderly patients,” they wrote (J Clin Oncol. 2016 July 11. doi: 10.1200/JCO.2016.67.1982).
In the trial, 276 patients with newly diagnosed APL were randomly assigned to receive either ATRA-ATO or ATRA plus chemotherapy with idarubicin, mercaptopurine, and methotrexate.
A total of 263 patients were evaluable for response to induction, including 127 assigned to ATRA-ATO and 136 assigned to ATRA-chemotherapy. Following induction, all patients assigned to ATRA-ATO and 127 assigned to chemotherapy (97%) achieved a complete remission (CR).
After a median follow-up of 40.6 months, the rate of EFS, the primary outcome, was 97.3% for ATRA-ATO vs. 80% for ATRA-chemotherapy (P less than .001). The cumulative incidences of relapse were 1.9% and 13.9%, respectively (P = .0013), and overall survival rates at 50 months were 99.2% vs. 92.6% (P = . 0073).
Hematologic toxicities were more frequent among patients assigned to chemotherapy, while liver function abnormalities occurred more often among those assigned to ATO.
One patient assigned to ATRA-ATO died while in CR, from bronchopneumonia caused by the H1N1 (influenza) virus. Five patients assigned to chemotherapy died in CR, from hemorrhagic shock, pulmonary embolism, bronchopneumonia (two patients) and secondary myelodysplastic syndrome.
Of the 17 patients who experienced relapses during follow-up, 2 assigned to ATRA-ATO had relapses at 22 and 27 months. The remaining 15 relapses were among patients assigned to ATRA-chemotherapy, occurring at a median of 14 months.
The authors noted that the superior EFS and cumulative incidence of relapse with ATRA-ATO emerged only after longer follow-up, indicating that the advantage of ATRA-ATO over ATRA-chemotherapy increases over time and that “the inclusion of ATO in the treatment of low- or intermediate-risk APL not only reduces mortality and hematologic toxicity, but also results in improved and sustained antileukemic activity.”
The study was supported by the Gruppo Italiano Malattie Ematologiche dell’Adulto Foundation; the Associazione Italiana Contro le Leucemie, Linfomi e Mieloma, Associazione Italiana per la Ricerca sul Cancro, and the German Federal Ministry of Education and Research. Dr. Lo-Coco disclosed honoraria, consulting, and speakers’ bureau activities with Teva Pharmaceutical, Lundbeck, Novartis, Baxalta, and Pfizer. Several other coauthors disclosed similar relationships.
The combination of all-trans-retinoic acid (ATRA) and arsenic trioxide continues to show advantages over ATRA and chemotherapy as first-line therapy for patients with low- or intermediate-risk acute promyelocytic leukemia (APL), investigators report.
The final analysis of theAPL046 study, a noninferiority trial of ATRA plus arsenic trioxide (ATRA-ATO) vs. ATRA and standard chemotherapy, showed that event-free survival (EFS) and overall survival (OS) were significantly better among patients assigned to ATRA-ATO. Patients in this group also had a significantly lower cumulative incidence of relapse, reported Francesco Lo-Coco, MD, of University Tor Vergata in Rome and colleagues.
“Our results support the use of ATRA-ATO in patients with newly diagnosed APL and point to this strategy as the new standard of care for low- or intermediate-risk patients. Studies exploring the role of ATRA-ATO are warranted in other APL subsets including high-risk, pediatric, and elderly patients,” they wrote (J Clin Oncol. 2016 July 11. doi: 10.1200/JCO.2016.67.1982).
In the trial, 276 patients with newly diagnosed APL were randomly assigned to receive either ATRA-ATO or ATRA plus chemotherapy with idarubicin, mercaptopurine, and methotrexate.
A total of 263 patients were evaluable for response to induction, including 127 assigned to ATRA-ATO and 136 assigned to ATRA-chemotherapy. Following induction, all patients assigned to ATRA-ATO and 127 assigned to chemotherapy (97%) achieved a complete remission (CR).
After a median follow-up of 40.6 months, the rate of EFS, the primary outcome, was 97.3% for ATRA-ATO vs. 80% for ATRA-chemotherapy (P less than .001). The cumulative incidences of relapse were 1.9% and 13.9%, respectively (P = .0013), and overall survival rates at 50 months were 99.2% vs. 92.6% (P = . 0073).
Hematologic toxicities were more frequent among patients assigned to chemotherapy, while liver function abnormalities occurred more often among those assigned to ATO.
One patient assigned to ATRA-ATO died while in CR, from bronchopneumonia caused by the H1N1 (influenza) virus. Five patients assigned to chemotherapy died in CR, from hemorrhagic shock, pulmonary embolism, bronchopneumonia (two patients) and secondary myelodysplastic syndrome.
Of the 17 patients who experienced relapses during follow-up, 2 assigned to ATRA-ATO had relapses at 22 and 27 months. The remaining 15 relapses were among patients assigned to ATRA-chemotherapy, occurring at a median of 14 months.
The authors noted that the superior EFS and cumulative incidence of relapse with ATRA-ATO emerged only after longer follow-up, indicating that the advantage of ATRA-ATO over ATRA-chemotherapy increases over time and that “the inclusion of ATO in the treatment of low- or intermediate-risk APL not only reduces mortality and hematologic toxicity, but also results in improved and sustained antileukemic activity.”
The study was supported by the Gruppo Italiano Malattie Ematologiche dell’Adulto Foundation; the Associazione Italiana Contro le Leucemie, Linfomi e Mieloma, Associazione Italiana per la Ricerca sul Cancro, and the German Federal Ministry of Education and Research. Dr. Lo-Coco disclosed honoraria, consulting, and speakers’ bureau activities with Teva Pharmaceutical, Lundbeck, Novartis, Baxalta, and Pfizer. Several other coauthors disclosed similar relationships.
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: ATRA-ATO was superior to ATRA-chemotherapy as first-line therapy for acute promyelocytic leukemia.
Major finding: Event-free survival after a median 40.6 months was 97.3% for ATRA-ATO vs. 80% for ATRA-chemotherapy
Data source: Randomized controlled trial comparing ATRA-ATO with ATRA-chemotherapy in 263 patients with newly diagnosed APL.
Disclosures: The study was supported by the Gruppo Italiano Malattie Ematologiche dell’Adulto Foundation; the Associazione Italiana Contro le Leucemie, Linfomi e Mieloma, Associazione Italiana per la Ricerca sul Cancro, and the German Federal Ministry of Education and Research. Dr. Lo-Coco disclosed honoraria, consulting, and speakers’ bureau activities with Teva Pharmaceutical, Lundbeck, Novartis, Baxalta, and Pfizer. Several other co-authors disclosed similar relationships.
Method reveals cells of origin in AML

in the bone marrow
Whole-genome profiling of open chromatin is a reliable way to identify the cells of origin in acute myeloid leukemia (AML), according to research published in Nature Communications.
“Knowing the cell of origin of cancer cells can provide insight into tumor subtypes and possibly diagnostic and therapeutic benefit,” said study author Jennifer Trowbridge, PhD, of the Jackson Laboratory for Mammalian Genetics in Bar Harbor, Maine.
“But existing methods to identify cell of origin from bulk tumor cell samples have been unsuccessful.”
Dr Trowbridge and her colleagues hypothesized that analyzing open chromatin in bulk tumor cells might provide a better method for identifying cancer cells of origin because of the cell-type specificity of chromatin structure.
The researchers worked with a mouse model of AML driven by expression of MLL-AF9, a fusion oncogene formed by a chromosome translocation between chromosomes 9 and 11.
The team began with 5 distinct, normal cell types found in the bone marrow in both mice and humans: long-term hematopoietic stem cells (HSCs), short-term HSCs, multipotent progenitors, common myeloid progenitors, and granulocyte macrophage progenitors.
The AML that developed from these different cells of origin had different penetrance and aggressiveness when engrafted in mice. The stem cell-derived lines proved the most aggressive and the committed progenitor lines the least aggressive.
These patterns were also reflected in the frequency of leukemia-initiating cells in each cell line, with HSCs having the highest frequency and committed progenitors having the lowest.
The researchers then set out to profile the open chromatin in these distinct AML samples and compare them to open chromatin patterns in normal cells using computational models.
The team identified open chromatin signatures and gene expression patterns in AML samples that may allow stem-cell-derived AML to be distinguished from progenitor-cell-of-origin AML.
These results support findings in human data suggesting the stage of a progenitor cell when it transforms to leukemia impacts clinical progression, with earlier-stage cell-of-origin cancers being more aggressive.
The researchers noted that, with further study of open chromatin in normal human stem and progenitor cell types as well as AML patient cohorts, this profiling approach could reveal precise regions with prognostic significance based on cell of origin; in other words, a valuable cancer biomarker. ![]()
in the bone marrow
Whole-genome profiling of open chromatin is a reliable way to identify the cells of origin in acute myeloid leukemia (AML), according to research published in Nature Communications.
“Knowing the cell of origin of cancer cells can provide insight into tumor subtypes and possibly diagnostic and therapeutic benefit,” said study author Jennifer Trowbridge, PhD, of the Jackson Laboratory for Mammalian Genetics in Bar Harbor, Maine.
“But existing methods to identify cell of origin from bulk tumor cell samples have been unsuccessful.”
Dr Trowbridge and her colleagues hypothesized that analyzing open chromatin in bulk tumor cells might provide a better method for identifying cancer cells of origin because of the cell-type specificity of chromatin structure.
The researchers worked with a mouse model of AML driven by expression of MLL-AF9, a fusion oncogene formed by a chromosome translocation between chromosomes 9 and 11.
The team began with 5 distinct, normal cell types found in the bone marrow in both mice and humans: long-term hematopoietic stem cells (HSCs), short-term HSCs, multipotent progenitors, common myeloid progenitors, and granulocyte macrophage progenitors.
The AML that developed from these different cells of origin had different penetrance and aggressiveness when engrafted in mice. The stem cell-derived lines proved the most aggressive and the committed progenitor lines the least aggressive.
These patterns were also reflected in the frequency of leukemia-initiating cells in each cell line, with HSCs having the highest frequency and committed progenitors having the lowest.
The researchers then set out to profile the open chromatin in these distinct AML samples and compare them to open chromatin patterns in normal cells using computational models.
The team identified open chromatin signatures and gene expression patterns in AML samples that may allow stem-cell-derived AML to be distinguished from progenitor-cell-of-origin AML.
These results support findings in human data suggesting the stage of a progenitor cell when it transforms to leukemia impacts clinical progression, with earlier-stage cell-of-origin cancers being more aggressive.
The researchers noted that, with further study of open chromatin in normal human stem and progenitor cell types as well as AML patient cohorts, this profiling approach could reveal precise regions with prognostic significance based on cell of origin; in other words, a valuable cancer biomarker. ![]()
in the bone marrow
Whole-genome profiling of open chromatin is a reliable way to identify the cells of origin in acute myeloid leukemia (AML), according to research published in Nature Communications.
“Knowing the cell of origin of cancer cells can provide insight into tumor subtypes and possibly diagnostic and therapeutic benefit,” said study author Jennifer Trowbridge, PhD, of the Jackson Laboratory for Mammalian Genetics in Bar Harbor, Maine.
“But existing methods to identify cell of origin from bulk tumor cell samples have been unsuccessful.”
Dr Trowbridge and her colleagues hypothesized that analyzing open chromatin in bulk tumor cells might provide a better method for identifying cancer cells of origin because of the cell-type specificity of chromatin structure.
The researchers worked with a mouse model of AML driven by expression of MLL-AF9, a fusion oncogene formed by a chromosome translocation between chromosomes 9 and 11.
The team began with 5 distinct, normal cell types found in the bone marrow in both mice and humans: long-term hematopoietic stem cells (HSCs), short-term HSCs, multipotent progenitors, common myeloid progenitors, and granulocyte macrophage progenitors.
The AML that developed from these different cells of origin had different penetrance and aggressiveness when engrafted in mice. The stem cell-derived lines proved the most aggressive and the committed progenitor lines the least aggressive.
These patterns were also reflected in the frequency of leukemia-initiating cells in each cell line, with HSCs having the highest frequency and committed progenitors having the lowest.
The researchers then set out to profile the open chromatin in these distinct AML samples and compare them to open chromatin patterns in normal cells using computational models.
The team identified open chromatin signatures and gene expression patterns in AML samples that may allow stem-cell-derived AML to be distinguished from progenitor-cell-of-origin AML.
These results support findings in human data suggesting the stage of a progenitor cell when it transforms to leukemia impacts clinical progression, with earlier-stage cell-of-origin cancers being more aggressive.
The researchers noted that, with further study of open chromatin in normal human stem and progenitor cell types as well as AML patient cohorts, this profiling approach could reveal precise regions with prognostic significance based on cell of origin; in other words, a valuable cancer biomarker. ![]()
Cord blood cell infusions reduce cGVHD incidence
Repeat infusions of mesenchymal stromal stem cells appear to inhibit the development of chronic graft-versus-host disease (cGVHD) in patients who have undergone an allogeneic stem cell transplant.
The 2-year cumulative incidence of cGVHD among those randomized to receive repeated infusions of umbilical cord–derived mesenchymal stromal cells (MSCs) was half that of controls treated with a saline placebo, based on results from a randomized phase II, double blind trial in 124 patients with hematologic malignancies who underwent an HLA-haploidentical allogeneic hematopoietic stem cell transplantation (HSCT).
“Our goal was to minimize the incidence of cGVHD, reduce the severity of cGVHD, and demonstrate the safety of MSC infusions. We performed repeated infusions of MSCs once a month for a total of four rounds for each patient. Over the median 47-month posttransplantation period, the incidence of cGVHD was lower in the MSCs group than in the non-MSCs control group,” Lei Gao, MD, of the Third Military Medical University in Chongqing, China, and colleagues wrote in the Journal of Clinical Oncology (2016 Jul 11. doi: 10.1200/JCO.2015.65.3642).
Although cGVHD is associated with a reduced risk of leukemia relapse, it is still the leading cause of nonrelapse deaths after HSCT. The incidence of cGVHD is higher among recipients of HLA-haploidentical HSCT, in which the donor and recipient have identical HLA alleles on only one copy of chromosome, than among HLA-matched recipients, who have identical alleles on both copies.
The researchers randomly assigned 124 patients who had undergo HLA-haploidentical HSCT to receive either placebo or MSCs at a dose of 3 x 107 cells/100 mL per month for four cycles beginning 4 months after HSCT
Of the 124 randomized patients, 12 discontinued the study due to cGVHD or disease progression.
The 2-year cumulative incidence of cGVHD among patients treated with MSCs was 27%, compared with 49% for placebo-treated controls (P = .021). Seven patients in the control group but none in the MSC-treated group developed typical lung cGVHD (P = .047).
The investigators also observed increases in memory B lymphocytes and regulatory T cells, and in the ratio of type 1 to type 2 T-helper cells, as well as a decrease in natural killer cells.
The finding that the MSC infusions increased the number of regulatory T cells while decreasing the incidence of cGVHD suggests that regulatory T cells play an inhibitory role, the investigators said.
The study was supported by the Chinese Academy of Sciences. Chinese National Natural Science Foundation, and other Chinese government grants. The authors reported having no relationships to disclose.
On the basis of the Gao et al. study, future investigations of cGVHD prophylaxis using MSCs should be explored. Approximately 90% of their population was younger than age 40, however, and it will be interesting to observe if this strategy is effective in older adults. The permutations and combinations for using different cell sources for deriving the MSCs, and in the context of different neoplastic disease, type and stage, conditioning regimen intensity, GVHD prophylaxis, graft and donor source, among other variables, are daunting. Nonetheless, the results of their trial encourage us to further explore this approach.
Hillard M. Lazarus, MD, is with Case Western Reserve University, Cleveland. Steven Z. Pavletic, MD, is with the National Institutes of Health, Bethesda, Md. Their comments were taken from an accompanying editorial (J Clin Oncol. 2016 Jul 11. doi: 10.1200/JCO.2016.67.7344).
On the basis of the Gao et al. study, future investigations of cGVHD prophylaxis using MSCs should be explored. Approximately 90% of their population was younger than age 40, however, and it will be interesting to observe if this strategy is effective in older adults. The permutations and combinations for using different cell sources for deriving the MSCs, and in the context of different neoplastic disease, type and stage, conditioning regimen intensity, GVHD prophylaxis, graft and donor source, among other variables, are daunting. Nonetheless, the results of their trial encourage us to further explore this approach.
Hillard M. Lazarus, MD, is with Case Western Reserve University, Cleveland. Steven Z. Pavletic, MD, is with the National Institutes of Health, Bethesda, Md. Their comments were taken from an accompanying editorial (J Clin Oncol. 2016 Jul 11. doi: 10.1200/JCO.2016.67.7344).
On the basis of the Gao et al. study, future investigations of cGVHD prophylaxis using MSCs should be explored. Approximately 90% of their population was younger than age 40, however, and it will be interesting to observe if this strategy is effective in older adults. The permutations and combinations for using different cell sources for deriving the MSCs, and in the context of different neoplastic disease, type and stage, conditioning regimen intensity, GVHD prophylaxis, graft and donor source, among other variables, are daunting. Nonetheless, the results of their trial encourage us to further explore this approach.
Hillard M. Lazarus, MD, is with Case Western Reserve University, Cleveland. Steven Z. Pavletic, MD, is with the National Institutes of Health, Bethesda, Md. Their comments were taken from an accompanying editorial (J Clin Oncol. 2016 Jul 11. doi: 10.1200/JCO.2016.67.7344).
Repeat infusions of mesenchymal stromal stem cells appear to inhibit the development of chronic graft-versus-host disease (cGVHD) in patients who have undergone an allogeneic stem cell transplant.
The 2-year cumulative incidence of cGVHD among those randomized to receive repeated infusions of umbilical cord–derived mesenchymal stromal cells (MSCs) was half that of controls treated with a saline placebo, based on results from a randomized phase II, double blind trial in 124 patients with hematologic malignancies who underwent an HLA-haploidentical allogeneic hematopoietic stem cell transplantation (HSCT).
“Our goal was to minimize the incidence of cGVHD, reduce the severity of cGVHD, and demonstrate the safety of MSC infusions. We performed repeated infusions of MSCs once a month for a total of four rounds for each patient. Over the median 47-month posttransplantation period, the incidence of cGVHD was lower in the MSCs group than in the non-MSCs control group,” Lei Gao, MD, of the Third Military Medical University in Chongqing, China, and colleagues wrote in the Journal of Clinical Oncology (2016 Jul 11. doi: 10.1200/JCO.2015.65.3642).
Although cGVHD is associated with a reduced risk of leukemia relapse, it is still the leading cause of nonrelapse deaths after HSCT. The incidence of cGVHD is higher among recipients of HLA-haploidentical HSCT, in which the donor and recipient have identical HLA alleles on only one copy of chromosome, than among HLA-matched recipients, who have identical alleles on both copies.
The researchers randomly assigned 124 patients who had undergo HLA-haploidentical HSCT to receive either placebo or MSCs at a dose of 3 x 107 cells/100 mL per month for four cycles beginning 4 months after HSCT
Of the 124 randomized patients, 12 discontinued the study due to cGVHD or disease progression.
The 2-year cumulative incidence of cGVHD among patients treated with MSCs was 27%, compared with 49% for placebo-treated controls (P = .021). Seven patients in the control group but none in the MSC-treated group developed typical lung cGVHD (P = .047).
The investigators also observed increases in memory B lymphocytes and regulatory T cells, and in the ratio of type 1 to type 2 T-helper cells, as well as a decrease in natural killer cells.
The finding that the MSC infusions increased the number of regulatory T cells while decreasing the incidence of cGVHD suggests that regulatory T cells play an inhibitory role, the investigators said.
The study was supported by the Chinese Academy of Sciences. Chinese National Natural Science Foundation, and other Chinese government grants. The authors reported having no relationships to disclose.
Repeat infusions of mesenchymal stromal stem cells appear to inhibit the development of chronic graft-versus-host disease (cGVHD) in patients who have undergone an allogeneic stem cell transplant.
The 2-year cumulative incidence of cGVHD among those randomized to receive repeated infusions of umbilical cord–derived mesenchymal stromal cells (MSCs) was half that of controls treated with a saline placebo, based on results from a randomized phase II, double blind trial in 124 patients with hematologic malignancies who underwent an HLA-haploidentical allogeneic hematopoietic stem cell transplantation (HSCT).
“Our goal was to minimize the incidence of cGVHD, reduce the severity of cGVHD, and demonstrate the safety of MSC infusions. We performed repeated infusions of MSCs once a month for a total of four rounds for each patient. Over the median 47-month posttransplantation period, the incidence of cGVHD was lower in the MSCs group than in the non-MSCs control group,” Lei Gao, MD, of the Third Military Medical University in Chongqing, China, and colleagues wrote in the Journal of Clinical Oncology (2016 Jul 11. doi: 10.1200/JCO.2015.65.3642).
Although cGVHD is associated with a reduced risk of leukemia relapse, it is still the leading cause of nonrelapse deaths after HSCT. The incidence of cGVHD is higher among recipients of HLA-haploidentical HSCT, in which the donor and recipient have identical HLA alleles on only one copy of chromosome, than among HLA-matched recipients, who have identical alleles on both copies.
The researchers randomly assigned 124 patients who had undergo HLA-haploidentical HSCT to receive either placebo or MSCs at a dose of 3 x 107 cells/100 mL per month for four cycles beginning 4 months after HSCT
Of the 124 randomized patients, 12 discontinued the study due to cGVHD or disease progression.
The 2-year cumulative incidence of cGVHD among patients treated with MSCs was 27%, compared with 49% for placebo-treated controls (P = .021). Seven patients in the control group but none in the MSC-treated group developed typical lung cGVHD (P = .047).
The investigators also observed increases in memory B lymphocytes and regulatory T cells, and in the ratio of type 1 to type 2 T-helper cells, as well as a decrease in natural killer cells.
The finding that the MSC infusions increased the number of regulatory T cells while decreasing the incidence of cGVHD suggests that regulatory T cells play an inhibitory role, the investigators said.
The study was supported by the Chinese Academy of Sciences. Chinese National Natural Science Foundation, and other Chinese government grants. The authors reported having no relationships to disclose.
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Mesenchymal stromal cell infusions may reduce risk of chronic graft-versus-host disease following allogeneic stem cell transplants.
Major finding: The 2-year cumulative incidence of cGVHD among patients treated with MSCs was 27.4%, compared with 49% for placebo-treated controls.
Data source: Randomized, double-blind, controlled trial in 124 patients following HSCT for hematologic malignancies.
Disclosures: The study was supported by the Chinese Academy of Sciences. Chinese National Natural Science Foundation, and other Chinese government grants. The authors reported having no relationships to disclose.
Lack of hematology-specific measures not barrier to quality end of life care
Standard oncology measures are acceptable indicators of good-quality end of life care for hematologic malignancies, investigators report.
Six out of eight standard end of life (EOL) care measures were deemed acceptable (defined as greater than 55% agreement) based on 349 returned surveys from hematologic oncologists. Hospice enrollment more than 7 days before death, no chemotherapy less than 14 days before death, and no CPR and intubation within the last 30 days of life were acceptable measures to more than 75% of those surveyed.
No more than one hospitalization within 30 days of death and no more than one emergency room visit within 30 days of death did not meet the threshold for acceptance by the oncologists.
“These data suggest that the absence of hematology-specific measures is unlikely to be a major barrier to quality care and that more resources should be directed toward addressing barriers to compliance with established EOL quality measures than toward creating new ones,” Oreofe O. Odejide, MD, of the Dana-Farber Cancer Institute, Boston, and her associates reported (J Clin Oncol. 2016 July. doi: 10.1200/JCO.2016.67.8177).
The top barriers to end of life care were unrealistic patient expectations (97.3%), clinician concern about diminishing patient and family hope (71.3%), and unrealistic clinician expectations (59.0%), according to investigators.
Surveyed oncologists also reported that increasing access to palliative care services (93.7%), increasing access to inpatient hospice facilities (92.2%), and allowing hospice enrollment with disease-directed treatment (90.1%) would be the most helpful interventions to improve end of life care.
The Lymphoma Research Foundation, the Conquer Cancer Foundation, and investigators’ respective institutions funded this research. Dr. Odejide and four of her associates had no disclosures to report. Two investigators reported serving in advisory roles, holding patents, or having stock or ownership interest in UnitedHealthcare, UpToDate, or Recap Information Systems.
On Twitter @jessnicolecraig
Standard oncology measures are acceptable indicators of good-quality end of life care for hematologic malignancies, investigators report.
Six out of eight standard end of life (EOL) care measures were deemed acceptable (defined as greater than 55% agreement) based on 349 returned surveys from hematologic oncologists. Hospice enrollment more than 7 days before death, no chemotherapy less than 14 days before death, and no CPR and intubation within the last 30 days of life were acceptable measures to more than 75% of those surveyed.
No more than one hospitalization within 30 days of death and no more than one emergency room visit within 30 days of death did not meet the threshold for acceptance by the oncologists.
“These data suggest that the absence of hematology-specific measures is unlikely to be a major barrier to quality care and that more resources should be directed toward addressing barriers to compliance with established EOL quality measures than toward creating new ones,” Oreofe O. Odejide, MD, of the Dana-Farber Cancer Institute, Boston, and her associates reported (J Clin Oncol. 2016 July. doi: 10.1200/JCO.2016.67.8177).
The top barriers to end of life care were unrealistic patient expectations (97.3%), clinician concern about diminishing patient and family hope (71.3%), and unrealistic clinician expectations (59.0%), according to investigators.
Surveyed oncologists also reported that increasing access to palliative care services (93.7%), increasing access to inpatient hospice facilities (92.2%), and allowing hospice enrollment with disease-directed treatment (90.1%) would be the most helpful interventions to improve end of life care.
The Lymphoma Research Foundation, the Conquer Cancer Foundation, and investigators’ respective institutions funded this research. Dr. Odejide and four of her associates had no disclosures to report. Two investigators reported serving in advisory roles, holding patents, or having stock or ownership interest in UnitedHealthcare, UpToDate, or Recap Information Systems.
On Twitter @jessnicolecraig
Standard oncology measures are acceptable indicators of good-quality end of life care for hematologic malignancies, investigators report.
Six out of eight standard end of life (EOL) care measures were deemed acceptable (defined as greater than 55% agreement) based on 349 returned surveys from hematologic oncologists. Hospice enrollment more than 7 days before death, no chemotherapy less than 14 days before death, and no CPR and intubation within the last 30 days of life were acceptable measures to more than 75% of those surveyed.
No more than one hospitalization within 30 days of death and no more than one emergency room visit within 30 days of death did not meet the threshold for acceptance by the oncologists.
“These data suggest that the absence of hematology-specific measures is unlikely to be a major barrier to quality care and that more resources should be directed toward addressing barriers to compliance with established EOL quality measures than toward creating new ones,” Oreofe O. Odejide, MD, of the Dana-Farber Cancer Institute, Boston, and her associates reported (J Clin Oncol. 2016 July. doi: 10.1200/JCO.2016.67.8177).
The top barriers to end of life care were unrealistic patient expectations (97.3%), clinician concern about diminishing patient and family hope (71.3%), and unrealistic clinician expectations (59.0%), according to investigators.
Surveyed oncologists also reported that increasing access to palliative care services (93.7%), increasing access to inpatient hospice facilities (92.2%), and allowing hospice enrollment with disease-directed treatment (90.1%) would be the most helpful interventions to improve end of life care.
The Lymphoma Research Foundation, the Conquer Cancer Foundation, and investigators’ respective institutions funded this research. Dr. Odejide and four of her associates had no disclosures to report. Two investigators reported serving in advisory roles, holding patents, or having stock or ownership interest in UnitedHealthcare, UpToDate, or Recap Information Systems.
On Twitter @jessnicolecraig
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Standard measures are acceptable indicators of good quality end of life care for patients with hematologic malignancies.
Major finding: All but two standard measures developed for solid tumors were found to be acceptable (defined as greater than 55% agreement) indicators of good quality end of life care for hematologic malignancies.
Data source: A national, pilot-tested survey of 349 hematologic oncologists.
Disclosures: In addition to internal funding, the Lymphoma Research Foundation and the Conquer Cancer Foundation funded this research. Dr. Odejide and four of her associates had no disclosures to report. Two investigators reported serving in advisory roles, holding patents, or having stock or ownership interest in UnitedHealthcare, UpToDate, or Recap Information Systems.
New insights into infant leukemia
Photo by Matthias Zepper
Researchers may have identified the cells responsible for MLL-AF4+ infant B-cell acute lymphoblastic leukemia, according to a paper published in Cell Reports.
The team analyzed mouse embryos and discovered a “window of opportunity” during which a pre-leukemic state can take hold.
They also found evidence to suggest this pre-leukemic state is driven by lymphoid-primed multipotent progenitor cells.
“One of the most common and aggressive types of infant blood cancer is associated with the MLL-AF4 fusion gene, which arises during pregnancy,” said study author Katrin Ottersbach, PhD, of the University of Edinburgh in the UK.
“One of the main impediments to improving the survival rates in infants is the lack of knowledge on where and when during development this mutation arises and how it affects the developing blood system of the baby.”
To gain some insight, Dr Ottersbach and her colleagues bred mice where one parent carries an inactive form of the MLL-AF4 fusion gene and the other parent expresses a gene for an enzyme that activates the fusion gene.
The embryos from this pairing entered a pre-leukemic state between days 12 and 14. The researchers found that MLL-AF4 imparted enhanced B lymphoid potential and increased repopulation and self-renewal capacity during this time.
Further investigation suggested that lymphoid-primed multipotent progenitor cells were the major contributors to the enhanced B lymphoid output and may therefore be the cells of origin.
The researchers noted that the mice did not actually develop infant leukemia, so more research is needed to identify the events required for progression to MLL-AF4+ infant B-cell acute lymphoblastic leukemia.
“Our findings reveal the first changes that take place in blood development caused by the MLL-AF4 mutation during a pre-cancerous state,” Dr Ottersbach said. “This has increased our knowledge on how this aggressive disease develops and will help identify early signs of disease and points for therapeutic intervention.” ![]()
Photo by Matthias Zepper
Researchers may have identified the cells responsible for MLL-AF4+ infant B-cell acute lymphoblastic leukemia, according to a paper published in Cell Reports.
The team analyzed mouse embryos and discovered a “window of opportunity” during which a pre-leukemic state can take hold.
They also found evidence to suggest this pre-leukemic state is driven by lymphoid-primed multipotent progenitor cells.
“One of the most common and aggressive types of infant blood cancer is associated with the MLL-AF4 fusion gene, which arises during pregnancy,” said study author Katrin Ottersbach, PhD, of the University of Edinburgh in the UK.
“One of the main impediments to improving the survival rates in infants is the lack of knowledge on where and when during development this mutation arises and how it affects the developing blood system of the baby.”
To gain some insight, Dr Ottersbach and her colleagues bred mice where one parent carries an inactive form of the MLL-AF4 fusion gene and the other parent expresses a gene for an enzyme that activates the fusion gene.
The embryos from this pairing entered a pre-leukemic state between days 12 and 14. The researchers found that MLL-AF4 imparted enhanced B lymphoid potential and increased repopulation and self-renewal capacity during this time.
Further investigation suggested that lymphoid-primed multipotent progenitor cells were the major contributors to the enhanced B lymphoid output and may therefore be the cells of origin.
The researchers noted that the mice did not actually develop infant leukemia, so more research is needed to identify the events required for progression to MLL-AF4+ infant B-cell acute lymphoblastic leukemia.
“Our findings reveal the first changes that take place in blood development caused by the MLL-AF4 mutation during a pre-cancerous state,” Dr Ottersbach said. “This has increased our knowledge on how this aggressive disease develops and will help identify early signs of disease and points for therapeutic intervention.” ![]()
Photo by Matthias Zepper
Researchers may have identified the cells responsible for MLL-AF4+ infant B-cell acute lymphoblastic leukemia, according to a paper published in Cell Reports.
The team analyzed mouse embryos and discovered a “window of opportunity” during which a pre-leukemic state can take hold.
They also found evidence to suggest this pre-leukemic state is driven by lymphoid-primed multipotent progenitor cells.
“One of the most common and aggressive types of infant blood cancer is associated with the MLL-AF4 fusion gene, which arises during pregnancy,” said study author Katrin Ottersbach, PhD, of the University of Edinburgh in the UK.
“One of the main impediments to improving the survival rates in infants is the lack of knowledge on where and when during development this mutation arises and how it affects the developing blood system of the baby.”
To gain some insight, Dr Ottersbach and her colleagues bred mice where one parent carries an inactive form of the MLL-AF4 fusion gene and the other parent expresses a gene for an enzyme that activates the fusion gene.
The embryos from this pairing entered a pre-leukemic state between days 12 and 14. The researchers found that MLL-AF4 imparted enhanced B lymphoid potential and increased repopulation and self-renewal capacity during this time.
Further investigation suggested that lymphoid-primed multipotent progenitor cells were the major contributors to the enhanced B lymphoid output and may therefore be the cells of origin.
The researchers noted that the mice did not actually develop infant leukemia, so more research is needed to identify the events required for progression to MLL-AF4+ infant B-cell acute lymphoblastic leukemia.
“Our findings reveal the first changes that take place in blood development caused by the MLL-AF4 mutation during a pre-cancerous state,” Dr Ottersbach said. “This has increased our knowledge on how this aggressive disease develops and will help identify early signs of disease and points for therapeutic intervention.” ![]()
Drug’s benefits outweigh risks, PRAC says

Photo courtesy of
Gilead Sciences, Inc.
The European Medicines Agency’s Pharmacovigilance Risk Assessment Committee (PRAC) has completed its review of the PI3Kδ inhibitor idelalisib (Zyedelig) and concluded that the drug’s benefits outweigh its risks in the treatment of chronic lymphocytic leukemia (CLL) and follicular lymphoma.
However, the PRAC also confirmed that the drug increases the risk of serious infections, including Pneumocystis jirovecii pneumonia.
And the committee updated its previous recommendations to manage this risk.
The PRAC’s recommendations will now be sent to the Committee for Medicinal Products for Human Use, which will adopt the EMA’s final opinion. The final stage of the review procedure is the adoption by the European Commission of a legally binding decision applicable in all member states of the European Union (EU).
About idelalisib
In the EU, idelalisib is approved for use in combination with rituximab to treat adults with CLL who have received at least 1 prior therapy or as first-line treatment in the presence of 17p deletion or TP53 mutation in CLL patients unsuitable for chemo-immunotherapy.
Idelalisib is also approved as monotherapy for adults with follicular lymphoma that is refractory to 2 prior lines of treatment.
About the review
The PRAC’s review of idelalisib began after a higher rate of serious adverse events, including deaths, was seen in 3 clinical trials evaluating the addition of idelalisib to standard therapy in first-line CLL and relapsed indolent non-Hodgkin lymphoma (NHL).
Most of the deaths were related to infections such as Pneumocystis jirovecii pneumonia and cytomegalovirus infection. Other excess deaths were related mainly to respiratory events.
The NHL studies (NCT01732926 and NCT01732913) included patients with disease characteristics different from those covered by the currently approved indications for idelalisib and investigated combinations of drugs that are not currently approved in the EU—idelalisib plus rituximab for NHL and idelalisib plus bendamustine and rituximab for NHL.
The CLL trial (NCT01980888) involved patients who had not received previous treatment, some of whom had the 17p deletion or TP53 mutation. However, the trial also investigated a combination of drugs not currently approved in the EU—idelalisib plus bendamustine and rituximab.
PRAC’s recommendations
The PRAC noted that, although the aforementioned trials did not all use idelalisib as currently authorized, the risk of serious infection is considered relevant to the authorized use.
Therefore, the PRAC recommends that all patients treated with idelalisib receive antibiotics to prevent Pneumocystis jirovecii pneumonia during treatment and for up to 2 to 6 months after treatment has stopped.
Patients should also be monitored for infection and have regular blood tests for white cell counts because low counts can increase their risk of infection.
Furthermore, idelalisib should not be started in patients with a generalized infection.
At the beginning of its review, the PRAC had said idelalisib should not be started in patients with previously untreated CLL and 17p deletion or TP53 mutation.
Now, the PRAC has concluded that idelalisib can be initiated in these patients, provided they cannot take any alternative treatment and that the recommended measures to prevent infection are followed. ![]()
Photo courtesy of
Gilead Sciences, Inc.
The European Medicines Agency’s Pharmacovigilance Risk Assessment Committee (PRAC) has completed its review of the PI3Kδ inhibitor idelalisib (Zyedelig) and concluded that the drug’s benefits outweigh its risks in the treatment of chronic lymphocytic leukemia (CLL) and follicular lymphoma.
However, the PRAC also confirmed that the drug increases the risk of serious infections, including Pneumocystis jirovecii pneumonia.
And the committee updated its previous recommendations to manage this risk.
The PRAC’s recommendations will now be sent to the Committee for Medicinal Products for Human Use, which will adopt the EMA’s final opinion. The final stage of the review procedure is the adoption by the European Commission of a legally binding decision applicable in all member states of the European Union (EU).
About idelalisib
In the EU, idelalisib is approved for use in combination with rituximab to treat adults with CLL who have received at least 1 prior therapy or as first-line treatment in the presence of 17p deletion or TP53 mutation in CLL patients unsuitable for chemo-immunotherapy.
Idelalisib is also approved as monotherapy for adults with follicular lymphoma that is refractory to 2 prior lines of treatment.
About the review
The PRAC’s review of idelalisib began after a higher rate of serious adverse events, including deaths, was seen in 3 clinical trials evaluating the addition of idelalisib to standard therapy in first-line CLL and relapsed indolent non-Hodgkin lymphoma (NHL).
Most of the deaths were related to infections such as Pneumocystis jirovecii pneumonia and cytomegalovirus infection. Other excess deaths were related mainly to respiratory events.
The NHL studies (NCT01732926 and NCT01732913) included patients with disease characteristics different from those covered by the currently approved indications for idelalisib and investigated combinations of drugs that are not currently approved in the EU—idelalisib plus rituximab for NHL and idelalisib plus bendamustine and rituximab for NHL.
The CLL trial (NCT01980888) involved patients who had not received previous treatment, some of whom had the 17p deletion or TP53 mutation. However, the trial also investigated a combination of drugs not currently approved in the EU—idelalisib plus bendamustine and rituximab.
PRAC’s recommendations
The PRAC noted that, although the aforementioned trials did not all use idelalisib as currently authorized, the risk of serious infection is considered relevant to the authorized use.
Therefore, the PRAC recommends that all patients treated with idelalisib receive antibiotics to prevent Pneumocystis jirovecii pneumonia during treatment and for up to 2 to 6 months after treatment has stopped.
Patients should also be monitored for infection and have regular blood tests for white cell counts because low counts can increase their risk of infection.
Furthermore, idelalisib should not be started in patients with a generalized infection.
At the beginning of its review, the PRAC had said idelalisib should not be started in patients with previously untreated CLL and 17p deletion or TP53 mutation.
Now, the PRAC has concluded that idelalisib can be initiated in these patients, provided they cannot take any alternative treatment and that the recommended measures to prevent infection are followed. ![]()
Photo courtesy of
Gilead Sciences, Inc.
The European Medicines Agency’s Pharmacovigilance Risk Assessment Committee (PRAC) has completed its review of the PI3Kδ inhibitor idelalisib (Zyedelig) and concluded that the drug’s benefits outweigh its risks in the treatment of chronic lymphocytic leukemia (CLL) and follicular lymphoma.
However, the PRAC also confirmed that the drug increases the risk of serious infections, including Pneumocystis jirovecii pneumonia.
And the committee updated its previous recommendations to manage this risk.
The PRAC’s recommendations will now be sent to the Committee for Medicinal Products for Human Use, which will adopt the EMA’s final opinion. The final stage of the review procedure is the adoption by the European Commission of a legally binding decision applicable in all member states of the European Union (EU).
About idelalisib
In the EU, idelalisib is approved for use in combination with rituximab to treat adults with CLL who have received at least 1 prior therapy or as first-line treatment in the presence of 17p deletion or TP53 mutation in CLL patients unsuitable for chemo-immunotherapy.
Idelalisib is also approved as monotherapy for adults with follicular lymphoma that is refractory to 2 prior lines of treatment.
About the review
The PRAC’s review of idelalisib began after a higher rate of serious adverse events, including deaths, was seen in 3 clinical trials evaluating the addition of idelalisib to standard therapy in first-line CLL and relapsed indolent non-Hodgkin lymphoma (NHL).
Most of the deaths were related to infections such as Pneumocystis jirovecii pneumonia and cytomegalovirus infection. Other excess deaths were related mainly to respiratory events.
The NHL studies (NCT01732926 and NCT01732913) included patients with disease characteristics different from those covered by the currently approved indications for idelalisib and investigated combinations of drugs that are not currently approved in the EU—idelalisib plus rituximab for NHL and idelalisib plus bendamustine and rituximab for NHL.
The CLL trial (NCT01980888) involved patients who had not received previous treatment, some of whom had the 17p deletion or TP53 mutation. However, the trial also investigated a combination of drugs not currently approved in the EU—idelalisib plus bendamustine and rituximab.
PRAC’s recommendations
The PRAC noted that, although the aforementioned trials did not all use idelalisib as currently authorized, the risk of serious infection is considered relevant to the authorized use.
Therefore, the PRAC recommends that all patients treated with idelalisib receive antibiotics to prevent Pneumocystis jirovecii pneumonia during treatment and for up to 2 to 6 months after treatment has stopped.
Patients should also be monitored for infection and have regular blood tests for white cell counts because low counts can increase their risk of infection.
Furthermore, idelalisib should not be started in patients with a generalized infection.
At the beginning of its review, the PRAC had said idelalisib should not be started in patients with previously untreated CLL and 17p deletion or TP53 mutation.
Now, the PRAC has concluded that idelalisib can be initiated in these patients, provided they cannot take any alternative treatment and that the recommended measures to prevent infection are followed. ![]()
Deaths prompt clinical hold for JCAR015 trial

Image from UNSW
Update: The hold on this trial has been lifted. Click here for additional details.
A trial of the chimeric antigen receptor (CAR) T-cell therapy JCAR015 has been placed on clinical hold following 3 patient deaths.
The trial, known as ROCKET, is a phase 2 study of adults with relapsed or refractory B-cell acute lymphoblastic leukemia.
The US Food and Drug Administration (FDA) placed a hold on the trial after 3 patients died of cerebral edema.
All 3 patients had received conditioning with fludarabine, and Juno Therapeutics, the company developing JCAR015, believes this may have caused the patients’ deaths.
Patients enrolled on the ROCKET trial previously received conditioning with cyclophosphamide alone, but investigators decided to add fludarabine in hopes of increasing efficacy.
The addition of fludarabine to conditioning had been shown to increase the efficacy of 2 of Juno’s other CAR T-cell therapies, JCAR014 and JCAR017, in phase 1/2 trials.
“However, since adding fludarabine to the preconditioning on the ROCKET trial, we have seen an increase in the incidence of severe neurotoxicity, which has, unfortunately, included 2 patient deaths that occurred last week from cerebral edema that appeared to be treatment-related,” Hans Bishop, Juno’s president and chief executive officer, said in a conference call.
“After the first of these 2 deaths, we immediately paused the trial for an internal review and review with our Data Safety Monitoring Board [DSMB] and the FDA. There was also 1 previous death from cerebral edema on the trial in May. After a review of that event, we, along with the FDA and our DSMB, concluded there were confounding factors, and a change in our plans at the time was not warranted.”
After the more recent deaths, Juno investigated several factors that could have contributed, including the conditioning regimen, patient characteristics, toxicity management, product characteristics, and cell dose.
“Although more than 1 factor may have contributed, based on our review of the data available . . . , we believe the addition of fludarabine, when combined with JCAR015, is the most likely and the most appropriately modifiable factor,” Bishop said.
“Indeed, with cy[clophosphamide] alone, which we have used in the greatest number of patients treated in the ROCKET trial to date, there have not been any treatment-related deaths, and the incidence of severe neurotoxicity is within the range of what we expected in light of the Memorial Sloan-Kettering experience [phase 1 trial of JCAR015].”
Therefore, Juno has proposed continuing the ROCKET trial using conditioning with cyclophosphamide alone.
In response to this request, the FDA has requested that Juno submit:
- A revised patient informed consent form
- A revised investigator brochure
- A revised trial protocol
- A copy of a presentation the company made to the FDA.
The FDA said it will expedite the review of these documents and expects to complete the review within 30 days of receiving them.
If the clinical hold on the ROCKET trial is lifted, Juno plans to continue the trial. However, the hold will likely impact the company’s goal of gaining FDA approval for JCAR015 in 2017.
Juno’s trials and plans for its other CD19-directed CAR-T cell product candidates are not affected by the clinical hold placed on ROCKET.
ROCKET is not the first trial of JCAR015 to be placed on hold. The phase 1 trial of the therapy was placed on clinical hold in 2014, after 2 patients died of cytokine release syndrome.
That hold was lifted following changes to enrollment criteria and dosing. Results from this trial were presented at ASCO 2015 and ASCO 2016. ![]()
Image from UNSW
Update: The hold on this trial has been lifted. Click here for additional details.
A trial of the chimeric antigen receptor (CAR) T-cell therapy JCAR015 has been placed on clinical hold following 3 patient deaths.
The trial, known as ROCKET, is a phase 2 study of adults with relapsed or refractory B-cell acute lymphoblastic leukemia.
The US Food and Drug Administration (FDA) placed a hold on the trial after 3 patients died of cerebral edema.
All 3 patients had received conditioning with fludarabine, and Juno Therapeutics, the company developing JCAR015, believes this may have caused the patients’ deaths.
Patients enrolled on the ROCKET trial previously received conditioning with cyclophosphamide alone, but investigators decided to add fludarabine in hopes of increasing efficacy.
The addition of fludarabine to conditioning had been shown to increase the efficacy of 2 of Juno’s other CAR T-cell therapies, JCAR014 and JCAR017, in phase 1/2 trials.
“However, since adding fludarabine to the preconditioning on the ROCKET trial, we have seen an increase in the incidence of severe neurotoxicity, which has, unfortunately, included 2 patient deaths that occurred last week from cerebral edema that appeared to be treatment-related,” Hans Bishop, Juno’s president and chief executive officer, said in a conference call.
“After the first of these 2 deaths, we immediately paused the trial for an internal review and review with our Data Safety Monitoring Board [DSMB] and the FDA. There was also 1 previous death from cerebral edema on the trial in May. After a review of that event, we, along with the FDA and our DSMB, concluded there were confounding factors, and a change in our plans at the time was not warranted.”
After the more recent deaths, Juno investigated several factors that could have contributed, including the conditioning regimen, patient characteristics, toxicity management, product characteristics, and cell dose.
“Although more than 1 factor may have contributed, based on our review of the data available . . . , we believe the addition of fludarabine, when combined with JCAR015, is the most likely and the most appropriately modifiable factor,” Bishop said.
“Indeed, with cy[clophosphamide] alone, which we have used in the greatest number of patients treated in the ROCKET trial to date, there have not been any treatment-related deaths, and the incidence of severe neurotoxicity is within the range of what we expected in light of the Memorial Sloan-Kettering experience [phase 1 trial of JCAR015].”
Therefore, Juno has proposed continuing the ROCKET trial using conditioning with cyclophosphamide alone.
In response to this request, the FDA has requested that Juno submit:
- A revised patient informed consent form
- A revised investigator brochure
- A revised trial protocol
- A copy of a presentation the company made to the FDA.
The FDA said it will expedite the review of these documents and expects to complete the review within 30 days of receiving them.
If the clinical hold on the ROCKET trial is lifted, Juno plans to continue the trial. However, the hold will likely impact the company’s goal of gaining FDA approval for JCAR015 in 2017.
Juno’s trials and plans for its other CD19-directed CAR-T cell product candidates are not affected by the clinical hold placed on ROCKET.
ROCKET is not the first trial of JCAR015 to be placed on hold. The phase 1 trial of the therapy was placed on clinical hold in 2014, after 2 patients died of cytokine release syndrome.
That hold was lifted following changes to enrollment criteria and dosing. Results from this trial were presented at ASCO 2015 and ASCO 2016. ![]()
Image from UNSW
Update: The hold on this trial has been lifted. Click here for additional details.
A trial of the chimeric antigen receptor (CAR) T-cell therapy JCAR015 has been placed on clinical hold following 3 patient deaths.
The trial, known as ROCKET, is a phase 2 study of adults with relapsed or refractory B-cell acute lymphoblastic leukemia.
The US Food and Drug Administration (FDA) placed a hold on the trial after 3 patients died of cerebral edema.
All 3 patients had received conditioning with fludarabine, and Juno Therapeutics, the company developing JCAR015, believes this may have caused the patients’ deaths.
Patients enrolled on the ROCKET trial previously received conditioning with cyclophosphamide alone, but investigators decided to add fludarabine in hopes of increasing efficacy.
The addition of fludarabine to conditioning had been shown to increase the efficacy of 2 of Juno’s other CAR T-cell therapies, JCAR014 and JCAR017, in phase 1/2 trials.
“However, since adding fludarabine to the preconditioning on the ROCKET trial, we have seen an increase in the incidence of severe neurotoxicity, which has, unfortunately, included 2 patient deaths that occurred last week from cerebral edema that appeared to be treatment-related,” Hans Bishop, Juno’s president and chief executive officer, said in a conference call.
“After the first of these 2 deaths, we immediately paused the trial for an internal review and review with our Data Safety Monitoring Board [DSMB] and the FDA. There was also 1 previous death from cerebral edema on the trial in May. After a review of that event, we, along with the FDA and our DSMB, concluded there were confounding factors, and a change in our plans at the time was not warranted.”
After the more recent deaths, Juno investigated several factors that could have contributed, including the conditioning regimen, patient characteristics, toxicity management, product characteristics, and cell dose.
“Although more than 1 factor may have contributed, based on our review of the data available . . . , we believe the addition of fludarabine, when combined with JCAR015, is the most likely and the most appropriately modifiable factor,” Bishop said.
“Indeed, with cy[clophosphamide] alone, which we have used in the greatest number of patients treated in the ROCKET trial to date, there have not been any treatment-related deaths, and the incidence of severe neurotoxicity is within the range of what we expected in light of the Memorial Sloan-Kettering experience [phase 1 trial of JCAR015].”
Therefore, Juno has proposed continuing the ROCKET trial using conditioning with cyclophosphamide alone.
In response to this request, the FDA has requested that Juno submit:
- A revised patient informed consent form
- A revised investigator brochure
- A revised trial protocol
- A copy of a presentation the company made to the FDA.
The FDA said it will expedite the review of these documents and expects to complete the review within 30 days of receiving them.
If the clinical hold on the ROCKET trial is lifted, Juno plans to continue the trial. However, the hold will likely impact the company’s goal of gaining FDA approval for JCAR015 in 2017.
Juno’s trials and plans for its other CD19-directed CAR-T cell product candidates are not affected by the clinical hold placed on ROCKET.
ROCKET is not the first trial of JCAR015 to be placed on hold. The phase 1 trial of the therapy was placed on clinical hold in 2014, after 2 patients died of cytokine release syndrome.
That hold was lifted following changes to enrollment criteria and dosing. Results from this trial were presented at ASCO 2015 and ASCO 2016. ![]()