User login
JULIET: CAR T cells keep trucking against DLBCL
SAN DIEGO – Chimeric antigen receptor T-cell therapy with tisagenlecleucel (Kymriah) is associated with a high rate of durable responses in adults with relapsed or refractory diffuse large B-cell lymphoma, an updated analysis of the JULIET trial showed.

After a median follow-up of 19 months, two-thirds of adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who had early responses to chimeric antigen receptor (CAR) T-cell therapy with tisagenlecleucel remained in remission with no evidence of minimal residual disease, reported Richard Thomas Maziarz, MD, from the Oregon Health & Science Knight Cancer Institute in Portland, at the annual meeting of the American Society of Hematology.
“Since the previous report, no new deaths have been reported due to any cause other than patient disease progression. No treatment-related mortality was seen throughout the study, and there were three early deaths, all related to lymphoma that progressed,” he said in a briefing prior to presentation of the data in a scientific poster.
The updated study results were published simultaneously online in the New England Journal of Medicine.
JULIET then
In the phase 2, single-arm trial, investigators enrolled adults with DLBCL that had relapsed or was refractory after two or more prior lines of therapy and who were either ineligible for hematopoietic stem cell transplant or who experienced disease progression after transplant.
Interim results of the study were previously reported at the European Hematology Association Congress in 2017.
At that meeting, Gilles Salles, MD, PhD, from the University of Lyon (France), presented results of an analysis of available efficacy data on 51 patients with at least 3 months of follow-up. In this population, the best overall response rate was 59%. The 3-month overall response rate was 45%, consisting of 37% complete responses and 8% partial responses. Relapse-free survival at 6 months was 79% and all patients who had responses at 3 months continued to have responses at the time of data cutoff.
JULIET now
In the most recent analysis, completed after a median time from infusion to data cutoff of 14 months, the investigators reported on efficacy in 93 patients who received CAR T-cell infusions.
The best overall response rate, the primary endpoint, was 52%, comprising 40% complete responses and 12% partial responses. The response rates were consistent across all prognostic subgroups, including age, sex, previous response status, International Prognostic Index score at enrollment, prior therapy, molecular subtype, and other factors.
Estimated relapse-free survival 12 months after documentation of an initial response was 65%, and was 79% among patients who had complete responses.
The median duration of response had not been reached at the time of data cutoff; the median overall survival had not been reached for patients with a complete remission. Overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months.
Adverse events of special interest included grade 3 or 4 cytokine release syndrome (CRS) in 23% of patients, prolonged cytopenia in 34%, infections in 19%, neurologic events in 11%, febrile neutropenia in 15%, and tumor lysis syndrome in 2%.
There were no deaths attributable to CRS or to cerebral edema, a complication of CAR T-cell therapy that appears to be related to the costimulatory molecule used in various constructs.
“Patients with relapsed or refractory DLBCL who are not eligible for high-dose therapy and hematopoietic cell transplantation or for whom such therapy was not successful have very few treatment options. For these patients, tisagenlecleucel shows promise that will need to be confirmed through larger studies with longer follow-up,” the investigators wrote in the New England Journal of Medicine.
The JULIET Trial is supported by Novartis. Dr. Maziar reported personal fees from Incyte, Kite Therapeutics, and Athersys.
SOURCE: Maziarz RT et al. N Engl J Med. 2018 Dec 1. doi: 10.1056/NEJMoa1804980.
SAN DIEGO – Chimeric antigen receptor T-cell therapy with tisagenlecleucel (Kymriah) is associated with a high rate of durable responses in adults with relapsed or refractory diffuse large B-cell lymphoma, an updated analysis of the JULIET trial showed.
After a median follow-up of 19 months, two-thirds of adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who had early responses to chimeric antigen receptor (CAR) T-cell therapy with tisagenlecleucel remained in remission with no evidence of minimal residual disease, reported Richard Thomas Maziarz, MD, from the Oregon Health & Science Knight Cancer Institute in Portland, at the annual meeting of the American Society of Hematology.
“Since the previous report, no new deaths have been reported due to any cause other than patient disease progression. No treatment-related mortality was seen throughout the study, and there were three early deaths, all related to lymphoma that progressed,” he said in a briefing prior to presentation of the data in a scientific poster.
The updated study results were published simultaneously online in the New England Journal of Medicine.
JULIET then
In the phase 2, single-arm trial, investigators enrolled adults with DLBCL that had relapsed or was refractory after two or more prior lines of therapy and who were either ineligible for hematopoietic stem cell transplant or who experienced disease progression after transplant.
Interim results of the study were previously reported at the European Hematology Association Congress in 2017.
At that meeting, Gilles Salles, MD, PhD, from the University of Lyon (France), presented results of an analysis of available efficacy data on 51 patients with at least 3 months of follow-up. In this population, the best overall response rate was 59%. The 3-month overall response rate was 45%, consisting of 37% complete responses and 8% partial responses. Relapse-free survival at 6 months was 79% and all patients who had responses at 3 months continued to have responses at the time of data cutoff.
JULIET now
In the most recent analysis, completed after a median time from infusion to data cutoff of 14 months, the investigators reported on efficacy in 93 patients who received CAR T-cell infusions.
The best overall response rate, the primary endpoint, was 52%, comprising 40% complete responses and 12% partial responses. The response rates were consistent across all prognostic subgroups, including age, sex, previous response status, International Prognostic Index score at enrollment, prior therapy, molecular subtype, and other factors.
Estimated relapse-free survival 12 months after documentation of an initial response was 65%, and was 79% among patients who had complete responses.
The median duration of response had not been reached at the time of data cutoff; the median overall survival had not been reached for patients with a complete remission. Overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months.
Adverse events of special interest included grade 3 or 4 cytokine release syndrome (CRS) in 23% of patients, prolonged cytopenia in 34%, infections in 19%, neurologic events in 11%, febrile neutropenia in 15%, and tumor lysis syndrome in 2%.
There were no deaths attributable to CRS or to cerebral edema, a complication of CAR T-cell therapy that appears to be related to the costimulatory molecule used in various constructs.
“Patients with relapsed or refractory DLBCL who are not eligible for high-dose therapy and hematopoietic cell transplantation or for whom such therapy was not successful have very few treatment options. For these patients, tisagenlecleucel shows promise that will need to be confirmed through larger studies with longer follow-up,” the investigators wrote in the New England Journal of Medicine.
The JULIET Trial is supported by Novartis. Dr. Maziar reported personal fees from Incyte, Kite Therapeutics, and Athersys.
SOURCE: Maziarz RT et al. N Engl J Med. 2018 Dec 1. doi: 10.1056/NEJMoa1804980.
SAN DIEGO – Chimeric antigen receptor T-cell therapy with tisagenlecleucel (Kymriah) is associated with a high rate of durable responses in adults with relapsed or refractory diffuse large B-cell lymphoma, an updated analysis of the JULIET trial showed.
After a median follow-up of 19 months, two-thirds of adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who had early responses to chimeric antigen receptor (CAR) T-cell therapy with tisagenlecleucel remained in remission with no evidence of minimal residual disease, reported Richard Thomas Maziarz, MD, from the Oregon Health & Science Knight Cancer Institute in Portland, at the annual meeting of the American Society of Hematology.
“Since the previous report, no new deaths have been reported due to any cause other than patient disease progression. No treatment-related mortality was seen throughout the study, and there were three early deaths, all related to lymphoma that progressed,” he said in a briefing prior to presentation of the data in a scientific poster.
The updated study results were published simultaneously online in the New England Journal of Medicine.
JULIET then
In the phase 2, single-arm trial, investigators enrolled adults with DLBCL that had relapsed or was refractory after two or more prior lines of therapy and who were either ineligible for hematopoietic stem cell transplant or who experienced disease progression after transplant.
Interim results of the study were previously reported at the European Hematology Association Congress in 2017.
At that meeting, Gilles Salles, MD, PhD, from the University of Lyon (France), presented results of an analysis of available efficacy data on 51 patients with at least 3 months of follow-up. In this population, the best overall response rate was 59%. The 3-month overall response rate was 45%, consisting of 37% complete responses and 8% partial responses. Relapse-free survival at 6 months was 79% and all patients who had responses at 3 months continued to have responses at the time of data cutoff.
JULIET now
In the most recent analysis, completed after a median time from infusion to data cutoff of 14 months, the investigators reported on efficacy in 93 patients who received CAR T-cell infusions.
The best overall response rate, the primary endpoint, was 52%, comprising 40% complete responses and 12% partial responses. The response rates were consistent across all prognostic subgroups, including age, sex, previous response status, International Prognostic Index score at enrollment, prior therapy, molecular subtype, and other factors.
Estimated relapse-free survival 12 months after documentation of an initial response was 65%, and was 79% among patients who had complete responses.
The median duration of response had not been reached at the time of data cutoff; the median overall survival had not been reached for patients with a complete remission. Overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months.
Adverse events of special interest included grade 3 or 4 cytokine release syndrome (CRS) in 23% of patients, prolonged cytopenia in 34%, infections in 19%, neurologic events in 11%, febrile neutropenia in 15%, and tumor lysis syndrome in 2%.
There were no deaths attributable to CRS or to cerebral edema, a complication of CAR T-cell therapy that appears to be related to the costimulatory molecule used in various constructs.
“Patients with relapsed or refractory DLBCL who are not eligible for high-dose therapy and hematopoietic cell transplantation or for whom such therapy was not successful have very few treatment options. For these patients, tisagenlecleucel shows promise that will need to be confirmed through larger studies with longer follow-up,” the investigators wrote in the New England Journal of Medicine.
The JULIET Trial is supported by Novartis. Dr. Maziar reported personal fees from Incyte, Kite Therapeutics, and Athersys.
SOURCE: Maziarz RT et al. N Engl J Med. 2018 Dec 1. doi: 10.1056/NEJMoa1804980.
REPORTING FROM ASH 2018
Key clinical point: Chimeric antigen receptor T-cell therapy produced durable responses in patients with heavily pretreated diffuse large B-cell lymphoma.
Major finding: The best overall response rate, the primary endpoint, was 52%, comprising 40% complete responses and 12% partial responses.
Study details: A single-arm, open-label study of tisagenlecleucel in adults with relapsed or refractory diffuse large B-cell lymphoma.
Disclosures: The JULIET trial is supported by Novartis. Dr. Maziarz reported personal fees from Incyte, Kite Therapeutics, and Athersys.
Source: Maziarz RT et al. N Engl J Med. 2018 Dec 1. doi: 10.1056/NEJMoa1804980.
New PCNSL guidelines emphasize importance of patient fitness
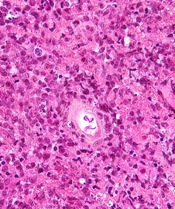
New guidelines on the diagnosis and management of patients with primary central nervous system lymphoma (PCNSL) emphasize prompt diagnosis, aggressive treatment whenever possible, and multidisciplinary team support.
A unique aspect for hematologic cancers, the guidelines note, is that appropriate treatment for PCNSL requires input from neurology specialists.
And the guidelines recommend methotrexate-based treatment only be administered at centers experienced in delivering intensive chemotherapy.
Christopher P. Fox, MD, of the Nottingham University Hospitals NHS Trust in Nottingham, U.K., and his colleagues on behalf of the British Society for Haematology published the guidelines in BJH.
The authors incorporated findings from studies published since the society’s last comprehensive PCNSL guidelines were issued more than a decade ago.
The new guidelines provide recommendations for diagnosis and imaging, primary treatment of PCNSL, consolidation chemotherapy, follow-up, management of relapsed/refractory disease, and neuropsychological assessments.
Highlights include:
- People with suspected PCNSL must receive quick and coordinated attention from a multidisciplinary team of neurologists, hematologist-oncologists, and ocular specialists
- Histological diagnoses in addition to imaging findings should be performed
- Corticosteroids should be avoided or discontinued before biopsy, as even a short course of steroids can impede diagnosis
- Aggressive induction treatment should be chosen based on the patient’s fitness
- Patients should be offered entry into clinical trials whenever possible
- Universal screening for eye involvement should be conducted.
Primary treatment
Dr. Fox and his colleagues say definitive treatment for PCNSL—induction of remission followed by consolidation—should start within 2 weeks of diagnosis, and a treatment regimen should be chosen according to a patient’s physiological fitness, not age.
The fittest patients, who have better organ function and fewer comorbidities, should be eligible for intensive combination immuno-chemotherapy incorporating high-dose methotrexate (HD-MTX)—optimally, four cycles of HD-MTX, cytarabine, thiotepa, and rituximab.
Those deemed unfit for this regimen should be offered induction treatment with HD-MTX, rituximab, and procarbazine, the guidelines say.
If patients cannot tolerate HD-MTX, oral chemotherapy, whole-brain radiotherapy (WBRT), or corticosteroids may be used.
The authors do not recommend intrathecal chemotherapy alongside systemic CNS-directed therapy.
Response should be assessed with contrast-enhanced magnetic resonance imaging (MRI) routinely after every two cycles of HD-MTX-based therapy and at the end of remission induction.
Consolidation chemotherapy
Consolidation therapy should be initiated after induction for all patients with non-progressive disease. High-dose thiotepa-based chemotherapy with autologous stem cell transplant (ASCT) is the recommended first-line option for consolidation.
Patients ineligible for high-dose therapy followed by ASCT who have residual disease after induction therapy should be considered for WBRT. This is also the case for patients with residual disease after thiotepa-based ASCT.
However, Dr. Fox and his colleagues say WBRT consolidation is “contentious” for patients in complete response after HD-MTX regimens but ineligible for ASCT. The authors suggest carefully balancing potential improvement in progression-free survival against risks of neurocognitive toxicity.
Response to consolidation, again measured with contrast-enhanced MRI, should be carried out between 1 and 2 months after therapy is completed, and patients should be referred for neuropsychological testing to assess cognitive function.
Patients with relapsed or refractory disease should be approached with maximum urgency—the guidelines offer an algorithm for retreatment options—and offered clinical trial entry wherever possible.
Some coauthors, including the lead author, disclosed receiving fees from pharmaceutical manufacturers Adienne and/or F. Hoffman-La Roche.
New guidelines on the diagnosis and management of patients with primary central nervous system lymphoma (PCNSL) emphasize prompt diagnosis, aggressive treatment whenever possible, and multidisciplinary team support.
A unique aspect for hematologic cancers, the guidelines note, is that appropriate treatment for PCNSL requires input from neurology specialists.
And the guidelines recommend methotrexate-based treatment only be administered at centers experienced in delivering intensive chemotherapy.
Christopher P. Fox, MD, of the Nottingham University Hospitals NHS Trust in Nottingham, U.K., and his colleagues on behalf of the British Society for Haematology published the guidelines in BJH.
The authors incorporated findings from studies published since the society’s last comprehensive PCNSL guidelines were issued more than a decade ago.
The new guidelines provide recommendations for diagnosis and imaging, primary treatment of PCNSL, consolidation chemotherapy, follow-up, management of relapsed/refractory disease, and neuropsychological assessments.
Highlights include:
- People with suspected PCNSL must receive quick and coordinated attention from a multidisciplinary team of neurologists, hematologist-oncologists, and ocular specialists
- Histological diagnoses in addition to imaging findings should be performed
- Corticosteroids should be avoided or discontinued before biopsy, as even a short course of steroids can impede diagnosis
- Aggressive induction treatment should be chosen based on the patient’s fitness
- Patients should be offered entry into clinical trials whenever possible
- Universal screening for eye involvement should be conducted.
Primary treatment
Dr. Fox and his colleagues say definitive treatment for PCNSL—induction of remission followed by consolidation—should start within 2 weeks of diagnosis, and a treatment regimen should be chosen according to a patient’s physiological fitness, not age.
The fittest patients, who have better organ function and fewer comorbidities, should be eligible for intensive combination immuno-chemotherapy incorporating high-dose methotrexate (HD-MTX)—optimally, four cycles of HD-MTX, cytarabine, thiotepa, and rituximab.
Those deemed unfit for this regimen should be offered induction treatment with HD-MTX, rituximab, and procarbazine, the guidelines say.
If patients cannot tolerate HD-MTX, oral chemotherapy, whole-brain radiotherapy (WBRT), or corticosteroids may be used.
The authors do not recommend intrathecal chemotherapy alongside systemic CNS-directed therapy.
Response should be assessed with contrast-enhanced magnetic resonance imaging (MRI) routinely after every two cycles of HD-MTX-based therapy and at the end of remission induction.
Consolidation chemotherapy
Consolidation therapy should be initiated after induction for all patients with non-progressive disease. High-dose thiotepa-based chemotherapy with autologous stem cell transplant (ASCT) is the recommended first-line option for consolidation.
Patients ineligible for high-dose therapy followed by ASCT who have residual disease after induction therapy should be considered for WBRT. This is also the case for patients with residual disease after thiotepa-based ASCT.
However, Dr. Fox and his colleagues say WBRT consolidation is “contentious” for patients in complete response after HD-MTX regimens but ineligible for ASCT. The authors suggest carefully balancing potential improvement in progression-free survival against risks of neurocognitive toxicity.
Response to consolidation, again measured with contrast-enhanced MRI, should be carried out between 1 and 2 months after therapy is completed, and patients should be referred for neuropsychological testing to assess cognitive function.
Patients with relapsed or refractory disease should be approached with maximum urgency—the guidelines offer an algorithm for retreatment options—and offered clinical trial entry wherever possible.
Some coauthors, including the lead author, disclosed receiving fees from pharmaceutical manufacturers Adienne and/or F. Hoffman-La Roche.
New guidelines on the diagnosis and management of patients with primary central nervous system lymphoma (PCNSL) emphasize prompt diagnosis, aggressive treatment whenever possible, and multidisciplinary team support.
A unique aspect for hematologic cancers, the guidelines note, is that appropriate treatment for PCNSL requires input from neurology specialists.
And the guidelines recommend methotrexate-based treatment only be administered at centers experienced in delivering intensive chemotherapy.
Christopher P. Fox, MD, of the Nottingham University Hospitals NHS Trust in Nottingham, U.K., and his colleagues on behalf of the British Society for Haematology published the guidelines in BJH.
The authors incorporated findings from studies published since the society’s last comprehensive PCNSL guidelines were issued more than a decade ago.
The new guidelines provide recommendations for diagnosis and imaging, primary treatment of PCNSL, consolidation chemotherapy, follow-up, management of relapsed/refractory disease, and neuropsychological assessments.
Highlights include:
- People with suspected PCNSL must receive quick and coordinated attention from a multidisciplinary team of neurologists, hematologist-oncologists, and ocular specialists
- Histological diagnoses in addition to imaging findings should be performed
- Corticosteroids should be avoided or discontinued before biopsy, as even a short course of steroids can impede diagnosis
- Aggressive induction treatment should be chosen based on the patient’s fitness
- Patients should be offered entry into clinical trials whenever possible
- Universal screening for eye involvement should be conducted.
Primary treatment
Dr. Fox and his colleagues say definitive treatment for PCNSL—induction of remission followed by consolidation—should start within 2 weeks of diagnosis, and a treatment regimen should be chosen according to a patient’s physiological fitness, not age.
The fittest patients, who have better organ function and fewer comorbidities, should be eligible for intensive combination immuno-chemotherapy incorporating high-dose methotrexate (HD-MTX)—optimally, four cycles of HD-MTX, cytarabine, thiotepa, and rituximab.
Those deemed unfit for this regimen should be offered induction treatment with HD-MTX, rituximab, and procarbazine, the guidelines say.
If patients cannot tolerate HD-MTX, oral chemotherapy, whole-brain radiotherapy (WBRT), or corticosteroids may be used.
The authors do not recommend intrathecal chemotherapy alongside systemic CNS-directed therapy.
Response should be assessed with contrast-enhanced magnetic resonance imaging (MRI) routinely after every two cycles of HD-MTX-based therapy and at the end of remission induction.
Consolidation chemotherapy
Consolidation therapy should be initiated after induction for all patients with non-progressive disease. High-dose thiotepa-based chemotherapy with autologous stem cell transplant (ASCT) is the recommended first-line option for consolidation.
Patients ineligible for high-dose therapy followed by ASCT who have residual disease after induction therapy should be considered for WBRT. This is also the case for patients with residual disease after thiotepa-based ASCT.
However, Dr. Fox and his colleagues say WBRT consolidation is “contentious” for patients in complete response after HD-MTX regimens but ineligible for ASCT. The authors suggest carefully balancing potential improvement in progression-free survival against risks of neurocognitive toxicity.
Response to consolidation, again measured with contrast-enhanced MRI, should be carried out between 1 and 2 months after therapy is completed, and patients should be referred for neuropsychological testing to assess cognitive function.
Patients with relapsed or refractory disease should be approached with maximum urgency—the guidelines offer an algorithm for retreatment options—and offered clinical trial entry wherever possible.
Some coauthors, including the lead author, disclosed receiving fees from pharmaceutical manufacturers Adienne and/or F. Hoffman-La Roche.
FLYER: Four cycles of R-CHOP as good as six in low-risk DLBCL
SAN DIEGO – A shortened regimen of four cycles of rituximab plus CHOP chemotherapy was noninferior in efficacy to the standard six cycles of R-CHOP in patients aged under age 60 years with favorable-risk diffuse large B-cell lymphoma (DLBCL), and the truncated regimen was associated with about a one-third reduction in nonhematologic adverse events, investigators in the FLYER trial reported.

Among 588 evaluable patients aged younger than 60 years with favorable-prognosis diffuse DLBCL, there were no significant differences in either progression-free survival (PFS), event-free survival, or overall survival (OS) between patients who were randomly assigned to therapy with four cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone), compared with patients assigned to six cycles, reported Viola Poeschel, MD, of Saarland University in Homburg, Germany.
“Six cycles of R-CHOP led to a higher toxicity with respect to leukocytopenia and anemia, both of any grades and also of grades 3 to 4, compared to four cycles of R-CHOP,” she said in a briefing at the annual meeting of the American Society of Hematology.
For younger patients with favorable-prognosis DLBCL – defined as an age-adjusted International Prognostic Index score of 0 and low tumor burden (less than 7.5 cm) – four cycles of R-CHOP can be a new standard of care, Dr. Poeschel said.
The investigators were prompted to look at the question of a shorter R-CHOP regimen by the results of the MInT trial, in which a subpopulation of favorable-prognosis DLBCL had a 3-year PFS rate of 89% (Lancet Oncol. 2006 May;7[5]379-91). The FLYER trial was designed as a noninferiority study to see whether in a similar group of patients reducing the number of R-CHOP cycles could maintain efficacy while reducing toxicity.
At a median follow-up of 66 months, the PFS rate, the primary endpoint, was 94% in the R-CHOP 6 group, compared with 96% for R-CHOP 4.
“As the lower limit of the 95% confidence interval of our experimental arm was 94%, it is shown that it is definitely noninferior to the standard arm, six cycles of R-CHOP,” Dr. Poeschel said.
Similarly, the rate of 3-year OS was 98% in the six-cycle group, compared with 99% in the four-cycle group, and the survival curves were virtually superimposable out to more than 10 years of follow-up.
R-CHOP 6 was associated with more frequent hematologic adverse events, compared with R-CHOP 4, with leukopenia of any grade occurring in 237 versus 171 patients, respectively, and grade 3 or 4 events occurring in 110 versus 80 patients, respectively.
Any grade anemia occurred in 172 patients assigned to six cycles versus 107 assigned to four cycles. Rates of grade 3-4 anemia and thrombocytopenia of any grade or of grade 3-4 were similar between the groups.
Nonhematologic adverse events of any grade or of grade 3 or 4 that were more frequent with R-CHOP 6 versus R-CHOP 4 included all events considered together, paresthesias, nausea, infection, vomiting, and mucositis.
As noted before, the total number of nonhematologic adverse events was reduced by about one-third.
“We are certainly always looking for ways to make treatments easier for our patients to reduce adverse effects, and certainly for this subgroup of patients it appears that we can make their treatment shorter and have less burden but equivalent efficacy,” commented David Steensma, MD, from the Dana-Farber Cancer Institute/Harvard Cancer Center in Boston, who moderated the briefing.
Dr. Steensma and Dr. Poeschel both cautioned that the results of the study pertain only to those patients with DLBCL who are younger and have favorable-prognosis disease.
“We can’t extend it to other subtypes of large cell lymphoma, but that’s always a laudable goal, so I think this will immediately influence clinical practice,” Dr. Steensma said.
The study was sponsored by the German High-Grade Non-Hodgkin’s Lymphoma Study Group. Dr. Poeschel reporteed travel grants from Roche and Amgen. Dr. Steensma reported no disclosures relevant to the study.
SOURCE: Poeschel V et al. ASH 2018, Abstract 781.
SAN DIEGO – A shortened regimen of four cycles of rituximab plus CHOP chemotherapy was noninferior in efficacy to the standard six cycles of R-CHOP in patients aged under age 60 years with favorable-risk diffuse large B-cell lymphoma (DLBCL), and the truncated regimen was associated with about a one-third reduction in nonhematologic adverse events, investigators in the FLYER trial reported.
Among 588 evaluable patients aged younger than 60 years with favorable-prognosis diffuse DLBCL, there were no significant differences in either progression-free survival (PFS), event-free survival, or overall survival (OS) between patients who were randomly assigned to therapy with four cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone), compared with patients assigned to six cycles, reported Viola Poeschel, MD, of Saarland University in Homburg, Germany.
“Six cycles of R-CHOP led to a higher toxicity with respect to leukocytopenia and anemia, both of any grades and also of grades 3 to 4, compared to four cycles of R-CHOP,” she said in a briefing at the annual meeting of the American Society of Hematology.
For younger patients with favorable-prognosis DLBCL – defined as an age-adjusted International Prognostic Index score of 0 and low tumor burden (less than 7.5 cm) – four cycles of R-CHOP can be a new standard of care, Dr. Poeschel said.
The investigators were prompted to look at the question of a shorter R-CHOP regimen by the results of the MInT trial, in which a subpopulation of favorable-prognosis DLBCL had a 3-year PFS rate of 89% (Lancet Oncol. 2006 May;7[5]379-91). The FLYER trial was designed as a noninferiority study to see whether in a similar group of patients reducing the number of R-CHOP cycles could maintain efficacy while reducing toxicity.
At a median follow-up of 66 months, the PFS rate, the primary endpoint, was 94% in the R-CHOP 6 group, compared with 96% for R-CHOP 4.
“As the lower limit of the 95% confidence interval of our experimental arm was 94%, it is shown that it is definitely noninferior to the standard arm, six cycles of R-CHOP,” Dr. Poeschel said.
Similarly, the rate of 3-year OS was 98% in the six-cycle group, compared with 99% in the four-cycle group, and the survival curves were virtually superimposable out to more than 10 years of follow-up.
R-CHOP 6 was associated with more frequent hematologic adverse events, compared with R-CHOP 4, with leukopenia of any grade occurring in 237 versus 171 patients, respectively, and grade 3 or 4 events occurring in 110 versus 80 patients, respectively.
Any grade anemia occurred in 172 patients assigned to six cycles versus 107 assigned to four cycles. Rates of grade 3-4 anemia and thrombocytopenia of any grade or of grade 3-4 were similar between the groups.
Nonhematologic adverse events of any grade or of grade 3 or 4 that were more frequent with R-CHOP 6 versus R-CHOP 4 included all events considered together, paresthesias, nausea, infection, vomiting, and mucositis.
As noted before, the total number of nonhematologic adverse events was reduced by about one-third.
“We are certainly always looking for ways to make treatments easier for our patients to reduce adverse effects, and certainly for this subgroup of patients it appears that we can make their treatment shorter and have less burden but equivalent efficacy,” commented David Steensma, MD, from the Dana-Farber Cancer Institute/Harvard Cancer Center in Boston, who moderated the briefing.
Dr. Steensma and Dr. Poeschel both cautioned that the results of the study pertain only to those patients with DLBCL who are younger and have favorable-prognosis disease.
“We can’t extend it to other subtypes of large cell lymphoma, but that’s always a laudable goal, so I think this will immediately influence clinical practice,” Dr. Steensma said.
The study was sponsored by the German High-Grade Non-Hodgkin’s Lymphoma Study Group. Dr. Poeschel reporteed travel grants from Roche and Amgen. Dr. Steensma reported no disclosures relevant to the study.
SOURCE: Poeschel V et al. ASH 2018, Abstract 781.
SAN DIEGO – A shortened regimen of four cycles of rituximab plus CHOP chemotherapy was noninferior in efficacy to the standard six cycles of R-CHOP in patients aged under age 60 years with favorable-risk diffuse large B-cell lymphoma (DLBCL), and the truncated regimen was associated with about a one-third reduction in nonhematologic adverse events, investigators in the FLYER trial reported.
Among 588 evaluable patients aged younger than 60 years with favorable-prognosis diffuse DLBCL, there were no significant differences in either progression-free survival (PFS), event-free survival, or overall survival (OS) between patients who were randomly assigned to therapy with four cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone), compared with patients assigned to six cycles, reported Viola Poeschel, MD, of Saarland University in Homburg, Germany.
“Six cycles of R-CHOP led to a higher toxicity with respect to leukocytopenia and anemia, both of any grades and also of grades 3 to 4, compared to four cycles of R-CHOP,” she said in a briefing at the annual meeting of the American Society of Hematology.
For younger patients with favorable-prognosis DLBCL – defined as an age-adjusted International Prognostic Index score of 0 and low tumor burden (less than 7.5 cm) – four cycles of R-CHOP can be a new standard of care, Dr. Poeschel said.
The investigators were prompted to look at the question of a shorter R-CHOP regimen by the results of the MInT trial, in which a subpopulation of favorable-prognosis DLBCL had a 3-year PFS rate of 89% (Lancet Oncol. 2006 May;7[5]379-91). The FLYER trial was designed as a noninferiority study to see whether in a similar group of patients reducing the number of R-CHOP cycles could maintain efficacy while reducing toxicity.
At a median follow-up of 66 months, the PFS rate, the primary endpoint, was 94% in the R-CHOP 6 group, compared with 96% for R-CHOP 4.
“As the lower limit of the 95% confidence interval of our experimental arm was 94%, it is shown that it is definitely noninferior to the standard arm, six cycles of R-CHOP,” Dr. Poeschel said.
Similarly, the rate of 3-year OS was 98% in the six-cycle group, compared with 99% in the four-cycle group, and the survival curves were virtually superimposable out to more than 10 years of follow-up.
R-CHOP 6 was associated with more frequent hematologic adverse events, compared with R-CHOP 4, with leukopenia of any grade occurring in 237 versus 171 patients, respectively, and grade 3 or 4 events occurring in 110 versus 80 patients, respectively.
Any grade anemia occurred in 172 patients assigned to six cycles versus 107 assigned to four cycles. Rates of grade 3-4 anemia and thrombocytopenia of any grade or of grade 3-4 were similar between the groups.
Nonhematologic adverse events of any grade or of grade 3 or 4 that were more frequent with R-CHOP 6 versus R-CHOP 4 included all events considered together, paresthesias, nausea, infection, vomiting, and mucositis.
As noted before, the total number of nonhematologic adverse events was reduced by about one-third.
“We are certainly always looking for ways to make treatments easier for our patients to reduce adverse effects, and certainly for this subgroup of patients it appears that we can make their treatment shorter and have less burden but equivalent efficacy,” commented David Steensma, MD, from the Dana-Farber Cancer Institute/Harvard Cancer Center in Boston, who moderated the briefing.
Dr. Steensma and Dr. Poeschel both cautioned that the results of the study pertain only to those patients with DLBCL who are younger and have favorable-prognosis disease.
“We can’t extend it to other subtypes of large cell lymphoma, but that’s always a laudable goal, so I think this will immediately influence clinical practice,” Dr. Steensma said.
The study was sponsored by the German High-Grade Non-Hodgkin’s Lymphoma Study Group. Dr. Poeschel reporteed travel grants from Roche and Amgen. Dr. Steensma reported no disclosures relevant to the study.
SOURCE: Poeschel V et al. ASH 2018, Abstract 781.
REPORTING FROM ASH 2018
Key clinical point: Four cycles of R-CHOP was noninferior to six cycles in younger patients with favorable-prognosis diffuse large B-cell lymphoma.
Major finding: R-CHOP 4 was noninferior to R-CHOP 6 for the primary progression-free survival endpoint.
Study details: A randomized trial in 588 patients with favorable-prognosis diffuse large B-cell lymphoma.
Disclosures: The study was sponsored by the German High-Grade Non-Hodgkin’s Lymphoma Study Group. Dr. Poeschel reporteed travel grants from Roche and Amgen. Dr. Steensma reported no disclosures relevant to the study.
Source: Poeschel V et al. ASH 2018, Abstract 781.
FLYER: R-CHOP 4 safer, as effective for low-risk DLBCL patients under 60
SAN DIEGO – Patients aged younger than 60 years with favorable-prognosis diffuse large B-cell lymphoma who were randomly assigned to therapy with four cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) had progression-free, event-free, and overall survival rates comparable with those of patients assigned to six cycles, investigators in the FLYER trial reported.
The four-cycle regimen was associated with a marked reduction in adverse events, with an overall drop in nonhematologic malignancies of approximately one-third compared with the six-cycle regimen.
For younger patients with favorable-prognosis DLBCL – defined as an age-adjusted International Prognostic Index score of 0 and low tumor burden (less than 7.5 cm) – four cycles of R-CHOP can be a new standard of care.
In this video interview at the annual meeting of the American Society of Hematology, Viola Poeschel, MD, of Saarland University in Homburg, Germany, describes the patient population who may benefit from shorter duration therapy.
SAN DIEGO – Patients aged younger than 60 years with favorable-prognosis diffuse large B-cell lymphoma who were randomly assigned to therapy with four cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) had progression-free, event-free, and overall survival rates comparable with those of patients assigned to six cycles, investigators in the FLYER trial reported.
The four-cycle regimen was associated with a marked reduction in adverse events, with an overall drop in nonhematologic malignancies of approximately one-third compared with the six-cycle regimen.
For younger patients with favorable-prognosis DLBCL – defined as an age-adjusted International Prognostic Index score of 0 and low tumor burden (less than 7.5 cm) – four cycles of R-CHOP can be a new standard of care.
In this video interview at the annual meeting of the American Society of Hematology, Viola Poeschel, MD, of Saarland University in Homburg, Germany, describes the patient population who may benefit from shorter duration therapy.
SAN DIEGO – Patients aged younger than 60 years with favorable-prognosis diffuse large B-cell lymphoma who were randomly assigned to therapy with four cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) had progression-free, event-free, and overall survival rates comparable with those of patients assigned to six cycles, investigators in the FLYER trial reported.
The four-cycle regimen was associated with a marked reduction in adverse events, with an overall drop in nonhematologic malignancies of approximately one-third compared with the six-cycle regimen.
For younger patients with favorable-prognosis DLBCL – defined as an age-adjusted International Prognostic Index score of 0 and low tumor burden (less than 7.5 cm) – four cycles of R-CHOP can be a new standard of care.
In this video interview at the annual meeting of the American Society of Hematology, Viola Poeschel, MD, of Saarland University in Homburg, Germany, describes the patient population who may benefit from shorter duration therapy.
REPORTING FROM ASH 2018

New data further support curability of myeloma
finds a retrospective cohort study of the International Myeloma Working Group. That figure may be even higher today because more than 90% of patients in the study – the largest yet to look at outcome predictors in this population – were treated in the era before novel therapies became available.
Investigators led by Saad Z. Usmani, MD, director/chief of plasma cell disorders and director of clinical research (hematologic malignancies) at the Levine Cancer Institute/Atrium Health in Charlotte, N.C., studied 7,291 patients with newly diagnosed multiple myeloma who were up to 75 years old and eligible for high-dose melphalan and autologous stem cell transplant. The patients were treated in clinical trials in 10 countries.
Compared with counterparts who did not achieve complete response 1 year after diagnosis, patients who did had better median progression-free survival (3.3 vs. 2.6 years; P less than .0001) and median overall survival (8.5 vs. 6.3 years; P less than .0001), according to study results report in Blood Cancer Journal.
The investigators next performed multivariate analyses to assess clinical variables at diagnosis associated with 10-year survival as compared with 2-year death.
Results here indicated that patients were less likely to be alive at 10 years if they were older than 65 years (odds ratio for death, 1.87; P = .002); had an immunoglobulin A isotype (OR, 1.53; P = .004); had a low albumin level, defined as less than 3.5 g/dL (OR, 1.36; P = .023); had an elevated beta2-microglobulin level, defined as at least 3.5 mg/dL (OR, 1.86; P less than .001); had a higher serum creatinine level, defined as at least 2 mg/dL (OR, 1.77; P = .005); had a lower hemoglobin level, defined as less than 10 g/dL (OR, 1.55; P = .003); or had a lower platelet count, defined as less than 150,000/μL (OR, 2.26; P less than .001).
Cytogenetic abnormalities did not independently predict long-term survival, but these abnormalities were obtained only by conventional band karyotyping and were not available for some patients.
Overall, the cohort had a relative survival of about 0.9 when compared with the matched general population. With follow-up out to about 20 years, the cure fraction (proportion achieving or exceeding expected survival when compared with the matched general population) was 14.3%.
Identification of early complete response as a predictor of long-term survival “underscores the importance of depth of response as we explore novel regimens for newly diagnosed [multiple myeloma] along with [minimal residual disease] endpoints,” Dr. Usmani and his colleagues wrote while acknowledging that the patients studied were a selected group eligible for transplant and treated on trials.
Recent therapeutic advances “have reignited the debate on possible functional curability of a subset MM patients,” they noted. “[T]here are perhaps more effective drugs and drug classes in the clinician’s armamentarium than [were] available for MM patients being treated in the 1990s or even early 2000s. This may mean that the depth of response after induction therapy may continue to improve over time, potentially further improving the PFS/OS of [the] biologic subset who previously achieved [partial response] yet had good long-term survival.”
Dr. Usmani disclosed that he is a consultant for AbbVie, Amgen, BMS, Celgene, Janssen, Takeda, Sanofi, and SkylineDx; receives speaker’s fees for Amgen, Celgene, Janssen, and Takeda; and receives research funding from Amgen, Array Biopharma, BMS, Celgene, Janssen, Pharmacyclics, Sanofi, and Takeda.
SOURCE: Usmani SZ et al. Blood Cancer J. 2018 Nov 23;8(12):123..
finds a retrospective cohort study of the International Myeloma Working Group. That figure may be even higher today because more than 90% of patients in the study – the largest yet to look at outcome predictors in this population – were treated in the era before novel therapies became available.
Investigators led by Saad Z. Usmani, MD, director/chief of plasma cell disorders and director of clinical research (hematologic malignancies) at the Levine Cancer Institute/Atrium Health in Charlotte, N.C., studied 7,291 patients with newly diagnosed multiple myeloma who were up to 75 years old and eligible for high-dose melphalan and autologous stem cell transplant. The patients were treated in clinical trials in 10 countries.
Compared with counterparts who did not achieve complete response 1 year after diagnosis, patients who did had better median progression-free survival (3.3 vs. 2.6 years; P less than .0001) and median overall survival (8.5 vs. 6.3 years; P less than .0001), according to study results report in Blood Cancer Journal.
The investigators next performed multivariate analyses to assess clinical variables at diagnosis associated with 10-year survival as compared with 2-year death.
Results here indicated that patients were less likely to be alive at 10 years if they were older than 65 years (odds ratio for death, 1.87; P = .002); had an immunoglobulin A isotype (OR, 1.53; P = .004); had a low albumin level, defined as less than 3.5 g/dL (OR, 1.36; P = .023); had an elevated beta2-microglobulin level, defined as at least 3.5 mg/dL (OR, 1.86; P less than .001); had a higher serum creatinine level, defined as at least 2 mg/dL (OR, 1.77; P = .005); had a lower hemoglobin level, defined as less than 10 g/dL (OR, 1.55; P = .003); or had a lower platelet count, defined as less than 150,000/μL (OR, 2.26; P less than .001).
Cytogenetic abnormalities did not independently predict long-term survival, but these abnormalities were obtained only by conventional band karyotyping and were not available for some patients.
Overall, the cohort had a relative survival of about 0.9 when compared with the matched general population. With follow-up out to about 20 years, the cure fraction (proportion achieving or exceeding expected survival when compared with the matched general population) was 14.3%.
Identification of early complete response as a predictor of long-term survival “underscores the importance of depth of response as we explore novel regimens for newly diagnosed [multiple myeloma] along with [minimal residual disease] endpoints,” Dr. Usmani and his colleagues wrote while acknowledging that the patients studied were a selected group eligible for transplant and treated on trials.
Recent therapeutic advances “have reignited the debate on possible functional curability of a subset MM patients,” they noted. “[T]here are perhaps more effective drugs and drug classes in the clinician’s armamentarium than [were] available for MM patients being treated in the 1990s or even early 2000s. This may mean that the depth of response after induction therapy may continue to improve over time, potentially further improving the PFS/OS of [the] biologic subset who previously achieved [partial response] yet had good long-term survival.”
Dr. Usmani disclosed that he is a consultant for AbbVie, Amgen, BMS, Celgene, Janssen, Takeda, Sanofi, and SkylineDx; receives speaker’s fees for Amgen, Celgene, Janssen, and Takeda; and receives research funding from Amgen, Array Biopharma, BMS, Celgene, Janssen, Pharmacyclics, Sanofi, and Takeda.
SOURCE: Usmani SZ et al. Blood Cancer J. 2018 Nov 23;8(12):123..
finds a retrospective cohort study of the International Myeloma Working Group. That figure may be even higher today because more than 90% of patients in the study – the largest yet to look at outcome predictors in this population – were treated in the era before novel therapies became available.
Investigators led by Saad Z. Usmani, MD, director/chief of plasma cell disorders and director of clinical research (hematologic malignancies) at the Levine Cancer Institute/Atrium Health in Charlotte, N.C., studied 7,291 patients with newly diagnosed multiple myeloma who were up to 75 years old and eligible for high-dose melphalan and autologous stem cell transplant. The patients were treated in clinical trials in 10 countries.
Compared with counterparts who did not achieve complete response 1 year after diagnosis, patients who did had better median progression-free survival (3.3 vs. 2.6 years; P less than .0001) and median overall survival (8.5 vs. 6.3 years; P less than .0001), according to study results report in Blood Cancer Journal.
The investigators next performed multivariate analyses to assess clinical variables at diagnosis associated with 10-year survival as compared with 2-year death.
Results here indicated that patients were less likely to be alive at 10 years if they were older than 65 years (odds ratio for death, 1.87; P = .002); had an immunoglobulin A isotype (OR, 1.53; P = .004); had a low albumin level, defined as less than 3.5 g/dL (OR, 1.36; P = .023); had an elevated beta2-microglobulin level, defined as at least 3.5 mg/dL (OR, 1.86; P less than .001); had a higher serum creatinine level, defined as at least 2 mg/dL (OR, 1.77; P = .005); had a lower hemoglobin level, defined as less than 10 g/dL (OR, 1.55; P = .003); or had a lower platelet count, defined as less than 150,000/μL (OR, 2.26; P less than .001).
Cytogenetic abnormalities did not independently predict long-term survival, but these abnormalities were obtained only by conventional band karyotyping and were not available for some patients.
Overall, the cohort had a relative survival of about 0.9 when compared with the matched general population. With follow-up out to about 20 years, the cure fraction (proportion achieving or exceeding expected survival when compared with the matched general population) was 14.3%.
Identification of early complete response as a predictor of long-term survival “underscores the importance of depth of response as we explore novel regimens for newly diagnosed [multiple myeloma] along with [minimal residual disease] endpoints,” Dr. Usmani and his colleagues wrote while acknowledging that the patients studied were a selected group eligible for transplant and treated on trials.
Recent therapeutic advances “have reignited the debate on possible functional curability of a subset MM patients,” they noted. “[T]here are perhaps more effective drugs and drug classes in the clinician’s armamentarium than [were] available for MM patients being treated in the 1990s or even early 2000s. This may mean that the depth of response after induction therapy may continue to improve over time, potentially further improving the PFS/OS of [the] biologic subset who previously achieved [partial response] yet had good long-term survival.”
Dr. Usmani disclosed that he is a consultant for AbbVie, Amgen, BMS, Celgene, Janssen, Takeda, Sanofi, and SkylineDx; receives speaker’s fees for Amgen, Celgene, Janssen, and Takeda; and receives research funding from Amgen, Array Biopharma, BMS, Celgene, Janssen, Pharmacyclics, Sanofi, and Takeda.
SOURCE: Usmani SZ et al. Blood Cancer J. 2018 Nov 23;8(12):123..
FROM BLOOD CANCER JOURNAL
Key clinical point: Some patients with newly diagnosed multiple myeloma eligible for transplant are likely now being cured.
Major finding: The cure fraction (proportion of patients achieving or exceeding expected survival compared with the matched general population) was 14.3%.
Study details: An international retrospective cohort study of 7,291 patients with newly diagnosed multiple myeloma eligible for high-dose melphalan and autologous stem cell transplant who were treated in clinical trials.
Disclosures: Dr. Usmani disclosed that he is a consultant for AbbVie, Amgen, BMS, Celgene, Janssen, Takeda, Sanofi, and SkylineDx; receives speaker’s fees for Amgen, Celgene, Janssen, and Takeda; and receives research funding from Amgen, Array Biopharma, BMS, Celgene, Janssen, Pharmacyclics, Sanofi, and Takeda.
Source: Usmani SZ et al. Blood Cancer J. 2018 Nov 23;8(12):123.
FDA approves biosimilar rituximab for NHL

The U.S. Food and Drug Administration (FDA) has approved a biosimilar rituximab product for the treatment of non-Hodgkin lymphoma (NHL).
Celltrion’s Truxima (rituximab-abbs) is a biosimilar of Genentech’s Rituxan and the first biosimilar approved in the United States to treat NHL.
Truxima (formerly CT-P10) is approved to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy.
Specifically, Truxima is approved as a single agent to treat relapsed or refractory, low grade or follicular, CD20-positive, B-cell NHL.
Truxima is approved in combination with first-line chemotherapy to treat previously untreated follicular, CD20-positive, B-cell NHL.
Truxima is approved as single-agent maintenance therapy in patients with follicular, CD20-positive, B-cell NHL who achieve a complete or partial response to a rituximab product in combination with chemotherapy.
And Truxima is approved as a single agent to treat non-progressing, low-grade, CD20-positive, B-cell NHL after first-line treatment with cyclophosphamide, vincristine, and prednisone.
The label for Truxima contains a boxed warning detailing the risk of fatal infusion reactions, severe skin and mouth reactions (some with fatal outcomes), hepatitis B virus reactivation that may cause serious liver problems (including liver failure and death), and progressive multifocal leukoencephalopathy.
The FDA said its approval of Truxima is “based on a review of evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic data, clinical immunogenicity data, and other clinical data that demonstrates Truxima is biosimilar to Rituxan.”
A phase 3 trial recently published in The Lancet Haematology suggested that Truxima is equivalent to the reference product in patients with low-tumor-burden follicular lymphoma.
For more details on Truxima, see the prescribing information.
The U.S. Food and Drug Administration (FDA) has approved a biosimilar rituximab product for the treatment of non-Hodgkin lymphoma (NHL).
Celltrion’s Truxima (rituximab-abbs) is a biosimilar of Genentech’s Rituxan and the first biosimilar approved in the United States to treat NHL.
Truxima (formerly CT-P10) is approved to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy.
Specifically, Truxima is approved as a single agent to treat relapsed or refractory, low grade or follicular, CD20-positive, B-cell NHL.
Truxima is approved in combination with first-line chemotherapy to treat previously untreated follicular, CD20-positive, B-cell NHL.
Truxima is approved as single-agent maintenance therapy in patients with follicular, CD20-positive, B-cell NHL who achieve a complete or partial response to a rituximab product in combination with chemotherapy.
And Truxima is approved as a single agent to treat non-progressing, low-grade, CD20-positive, B-cell NHL after first-line treatment with cyclophosphamide, vincristine, and prednisone.
The label for Truxima contains a boxed warning detailing the risk of fatal infusion reactions, severe skin and mouth reactions (some with fatal outcomes), hepatitis B virus reactivation that may cause serious liver problems (including liver failure and death), and progressive multifocal leukoencephalopathy.
The FDA said its approval of Truxima is “based on a review of evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic data, clinical immunogenicity data, and other clinical data that demonstrates Truxima is biosimilar to Rituxan.”
A phase 3 trial recently published in The Lancet Haematology suggested that Truxima is equivalent to the reference product in patients with low-tumor-burden follicular lymphoma.
For more details on Truxima, see the prescribing information.
The U.S. Food and Drug Administration (FDA) has approved a biosimilar rituximab product for the treatment of non-Hodgkin lymphoma (NHL).
Celltrion’s Truxima (rituximab-abbs) is a biosimilar of Genentech’s Rituxan and the first biosimilar approved in the United States to treat NHL.
Truxima (formerly CT-P10) is approved to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy.
Specifically, Truxima is approved as a single agent to treat relapsed or refractory, low grade or follicular, CD20-positive, B-cell NHL.
Truxima is approved in combination with first-line chemotherapy to treat previously untreated follicular, CD20-positive, B-cell NHL.
Truxima is approved as single-agent maintenance therapy in patients with follicular, CD20-positive, B-cell NHL who achieve a complete or partial response to a rituximab product in combination with chemotherapy.
And Truxima is approved as a single agent to treat non-progressing, low-grade, CD20-positive, B-cell NHL after first-line treatment with cyclophosphamide, vincristine, and prednisone.
The label for Truxima contains a boxed warning detailing the risk of fatal infusion reactions, severe skin and mouth reactions (some with fatal outcomes), hepatitis B virus reactivation that may cause serious liver problems (including liver failure and death), and progressive multifocal leukoencephalopathy.
The FDA said its approval of Truxima is “based on a review of evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic data, clinical immunogenicity data, and other clinical data that demonstrates Truxima is biosimilar to Rituxan.”
A phase 3 trial recently published in The Lancet Haematology suggested that Truxima is equivalent to the reference product in patients with low-tumor-burden follicular lymphoma.
For more details on Truxima, see the prescribing information.
FDA approves rituximab biosimilar for lymphoma
(NHL).

Celltrion’s Truxima (rituximab-abbs) is a biosimilar of Genentech’s Rituxan (rituximab) and the first biosimilar approved in the United States to treat NHL.
Truxima (formerly CT-P10) is approved to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy. Truxima is approved as a single agent to treat relapsed or refractory, low grade or follicular, CD20-positive, B-cell NHL. Truxima is approved in combination with first-line chemotherapy to treat previously untreated follicular, CD20-positive, B-cell NHL.
Truxima is approved as single-agent maintenance therapy in patients with follicular, CD20-positive, B-cell NHL who achieve a complete or partial response to a rituximab product in combination with chemotherapy. Truxima also is approved as a single agent to treat nonprogressing, low-grade, CD20-positive, B-cell NHL after first-line treatment with cyclophosphamide, vincristine, and prednisone.The label for Truxima contains a boxed warning detailing the risk of fatal infusion reactions, severe skin and mouth reactions (some with fatal outcomes), hepatitis B virus reactivation that may cause serious liver problems (including liver failure and death), and progressive multifocal leukoencephalopathy.
The FDA said its approval of Truxima is “based on a review of evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic data, clinical immunogenicity data, and other clinical data that demonstrates Truxima is biosimilar to Rituxan.”
Findings from a phase 3 trial suggested that Truxima is equivalent to the reference product in patients with low-tumor-burden follicular lymphoma (Lancet Haematol. 2018 Nov;5[11]:e543-53).
(NHL).
Celltrion’s Truxima (rituximab-abbs) is a biosimilar of Genentech’s Rituxan (rituximab) and the first biosimilar approved in the United States to treat NHL.
Truxima (formerly CT-P10) is approved to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy. Truxima is approved as a single agent to treat relapsed or refractory, low grade or follicular, CD20-positive, B-cell NHL. Truxima is approved in combination with first-line chemotherapy to treat previously untreated follicular, CD20-positive, B-cell NHL.
Truxima is approved as single-agent maintenance therapy in patients with follicular, CD20-positive, B-cell NHL who achieve a complete or partial response to a rituximab product in combination with chemotherapy. Truxima also is approved as a single agent to treat nonprogressing, low-grade, CD20-positive, B-cell NHL after first-line treatment with cyclophosphamide, vincristine, and prednisone.The label for Truxima contains a boxed warning detailing the risk of fatal infusion reactions, severe skin and mouth reactions (some with fatal outcomes), hepatitis B virus reactivation that may cause serious liver problems (including liver failure and death), and progressive multifocal leukoencephalopathy.
The FDA said its approval of Truxima is “based on a review of evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic data, clinical immunogenicity data, and other clinical data that demonstrates Truxima is biosimilar to Rituxan.”
Findings from a phase 3 trial suggested that Truxima is equivalent to the reference product in patients with low-tumor-burden follicular lymphoma (Lancet Haematol. 2018 Nov;5[11]:e543-53).
(NHL).
Celltrion’s Truxima (rituximab-abbs) is a biosimilar of Genentech’s Rituxan (rituximab) and the first biosimilar approved in the United States to treat NHL.
Truxima (formerly CT-P10) is approved to treat adults with CD20-positive, B-cell NHL, either as a single agent or in combination with chemotherapy. Truxima is approved as a single agent to treat relapsed or refractory, low grade or follicular, CD20-positive, B-cell NHL. Truxima is approved in combination with first-line chemotherapy to treat previously untreated follicular, CD20-positive, B-cell NHL.
Truxima is approved as single-agent maintenance therapy in patients with follicular, CD20-positive, B-cell NHL who achieve a complete or partial response to a rituximab product in combination with chemotherapy. Truxima also is approved as a single agent to treat nonprogressing, low-grade, CD20-positive, B-cell NHL after first-line treatment with cyclophosphamide, vincristine, and prednisone.The label for Truxima contains a boxed warning detailing the risk of fatal infusion reactions, severe skin and mouth reactions (some with fatal outcomes), hepatitis B virus reactivation that may cause serious liver problems (including liver failure and death), and progressive multifocal leukoencephalopathy.
The FDA said its approval of Truxima is “based on a review of evidence that included extensive structural and functional characterization, animal study data, human pharmacokinetic data, clinical immunogenicity data, and other clinical data that demonstrates Truxima is biosimilar to Rituxan.”
Findings from a phase 3 trial suggested that Truxima is equivalent to the reference product in patients with low-tumor-burden follicular lymphoma (Lancet Haematol. 2018 Nov;5[11]:e543-53).
Cortactin expression aids in CLL-MCL differential
The presence or absence in tumor cells of cortactin, a cytoskeleton-remodeling adapter protein, may be a marker to help pathologists distinguish between chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL), investigators suggest.
A study of cortactin expression in tumor samples from patients with B-cell CLL, MCL, and other hematologic malignancies showed that while cortactin was present in 14 of 17 CLL samples, it was not expressed on any of 16 MCL samples, reported Marco Pizzi, MD, PhD, from the University of Padova (Italy) and his colleagues.
“In particular, cortactin may contribute to the differential diagnosis between CLL and MCL, two neoplasms with similar histological features but very different clinical outcome. Further studies are needed to clarify the molecular mechanisms of deranged cortactin expression in MCL and CLL and to investigate any possible relationship between cortactin status and the biological features of these lymphomas,” they wrote in Human Pathology.
Overexpression of cortactin has been reported in several solid tumors, and increased expression of CTTN, the gene encoding for cortactin, has been associated with aggressive, poor prognosis disease, the investigators noted.
To characterize cortactin expression in lymphoid and hematopoietic cells and detect potential associations between cortactin and virulence of hematologic malignancies, the investigators performed immunohistochemical analysis on samples from 131 patients treated at their center. The samples included 17 cases of CLL, 16 of MCL, 25 of follicular lymphoma (FL), 30 of marginal zone lymphoma (MZL), 10 of hairy cell leukemia, three of splenic diffuse red pulp small B-cell lymphomas (SDRPBL), and 30 of diffuse large B-cell lymphoma (DLBCL).
They found that cortactin was expressed in 14 of the 17 CLL samples, all 10 of the HCL samples, and 22 of the 30 DLBCL samples. In contrast, there was no cortactin expression detected in any of either 16 MCL or three SDRPBL samples. The researchers found that 13 of 30 MZL samples had low-level staining. In FL, cortactin was expressed in 2 of the samples but in the remaining 23 cases the researchers found only scattered cortactin-positive lymphoid elements of non–B-cell lineage.
The investigators also found that cortactin expression in CLL correlated with other CLL-specific markers, and found that expression of two or more of the markers had 89.1% sensitivity, 100% specificity, a 100% positive predictive value, and 90.5% negative predictive value for a diagnosis of CLL.
In addition, they saw that the immunohistochemical results were similar to those for CTTN gene expression assessed by in silico analysis.
The investigators noted that CLL and MCL are challenging to differentiate from one another because of morphologic similarities and partially overlapping immunophenotypes.
“In this context, cortactin expression would strongly sustain a diagnosis of CLL over MCL, particularly in association with other CLL markers (i.e., LEF1 and CD200),” they wrote.
The study was internally supported. The authors declared no conflicts of interest.
SOURCE: Pizzi M et al. Hum Pathol. 2018 Nov 17. doi: 10.1016/j.humpath.2018.10.038.
The presence or absence in tumor cells of cortactin, a cytoskeleton-remodeling adapter protein, may be a marker to help pathologists distinguish between chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL), investigators suggest.
A study of cortactin expression in tumor samples from patients with B-cell CLL, MCL, and other hematologic malignancies showed that while cortactin was present in 14 of 17 CLL samples, it was not expressed on any of 16 MCL samples, reported Marco Pizzi, MD, PhD, from the University of Padova (Italy) and his colleagues.
“In particular, cortactin may contribute to the differential diagnosis between CLL and MCL, two neoplasms with similar histological features but very different clinical outcome. Further studies are needed to clarify the molecular mechanisms of deranged cortactin expression in MCL and CLL and to investigate any possible relationship between cortactin status and the biological features of these lymphomas,” they wrote in Human Pathology.
Overexpression of cortactin has been reported in several solid tumors, and increased expression of CTTN, the gene encoding for cortactin, has been associated with aggressive, poor prognosis disease, the investigators noted.
To characterize cortactin expression in lymphoid and hematopoietic cells and detect potential associations between cortactin and virulence of hematologic malignancies, the investigators performed immunohistochemical analysis on samples from 131 patients treated at their center. The samples included 17 cases of CLL, 16 of MCL, 25 of follicular lymphoma (FL), 30 of marginal zone lymphoma (MZL), 10 of hairy cell leukemia, three of splenic diffuse red pulp small B-cell lymphomas (SDRPBL), and 30 of diffuse large B-cell lymphoma (DLBCL).
They found that cortactin was expressed in 14 of the 17 CLL samples, all 10 of the HCL samples, and 22 of the 30 DLBCL samples. In contrast, there was no cortactin expression detected in any of either 16 MCL or three SDRPBL samples. The researchers found that 13 of 30 MZL samples had low-level staining. In FL, cortactin was expressed in 2 of the samples but in the remaining 23 cases the researchers found only scattered cortactin-positive lymphoid elements of non–B-cell lineage.
The investigators also found that cortactin expression in CLL correlated with other CLL-specific markers, and found that expression of two or more of the markers had 89.1% sensitivity, 100% specificity, a 100% positive predictive value, and 90.5% negative predictive value for a diagnosis of CLL.
In addition, they saw that the immunohistochemical results were similar to those for CTTN gene expression assessed by in silico analysis.
The investigators noted that CLL and MCL are challenging to differentiate from one another because of morphologic similarities and partially overlapping immunophenotypes.
“In this context, cortactin expression would strongly sustain a diagnosis of CLL over MCL, particularly in association with other CLL markers (i.e., LEF1 and CD200),” they wrote.
The study was internally supported. The authors declared no conflicts of interest.
SOURCE: Pizzi M et al. Hum Pathol. 2018 Nov 17. doi: 10.1016/j.humpath.2018.10.038.
The presence or absence in tumor cells of cortactin, a cytoskeleton-remodeling adapter protein, may be a marker to help pathologists distinguish between chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL), investigators suggest.
A study of cortactin expression in tumor samples from patients with B-cell CLL, MCL, and other hematologic malignancies showed that while cortactin was present in 14 of 17 CLL samples, it was not expressed on any of 16 MCL samples, reported Marco Pizzi, MD, PhD, from the University of Padova (Italy) and his colleagues.
“In particular, cortactin may contribute to the differential diagnosis between CLL and MCL, two neoplasms with similar histological features but very different clinical outcome. Further studies are needed to clarify the molecular mechanisms of deranged cortactin expression in MCL and CLL and to investigate any possible relationship between cortactin status and the biological features of these lymphomas,” they wrote in Human Pathology.
Overexpression of cortactin has been reported in several solid tumors, and increased expression of CTTN, the gene encoding for cortactin, has been associated with aggressive, poor prognosis disease, the investigators noted.
To characterize cortactin expression in lymphoid and hematopoietic cells and detect potential associations between cortactin and virulence of hematologic malignancies, the investigators performed immunohistochemical analysis on samples from 131 patients treated at their center. The samples included 17 cases of CLL, 16 of MCL, 25 of follicular lymphoma (FL), 30 of marginal zone lymphoma (MZL), 10 of hairy cell leukemia, three of splenic diffuse red pulp small B-cell lymphomas (SDRPBL), and 30 of diffuse large B-cell lymphoma (DLBCL).
They found that cortactin was expressed in 14 of the 17 CLL samples, all 10 of the HCL samples, and 22 of the 30 DLBCL samples. In contrast, there was no cortactin expression detected in any of either 16 MCL or three SDRPBL samples. The researchers found that 13 of 30 MZL samples had low-level staining. In FL, cortactin was expressed in 2 of the samples but in the remaining 23 cases the researchers found only scattered cortactin-positive lymphoid elements of non–B-cell lineage.
The investigators also found that cortactin expression in CLL correlated with other CLL-specific markers, and found that expression of two or more of the markers had 89.1% sensitivity, 100% specificity, a 100% positive predictive value, and 90.5% negative predictive value for a diagnosis of CLL.
In addition, they saw that the immunohistochemical results were similar to those for CTTN gene expression assessed by in silico analysis.
The investigators noted that CLL and MCL are challenging to differentiate from one another because of morphologic similarities and partially overlapping immunophenotypes.
“In this context, cortactin expression would strongly sustain a diagnosis of CLL over MCL, particularly in association with other CLL markers (i.e., LEF1 and CD200),” they wrote.
The study was internally supported. The authors declared no conflicts of interest.
SOURCE: Pizzi M et al. Hum Pathol. 2018 Nov 17. doi: 10.1016/j.humpath.2018.10.038.
FROM HUMAN PATHOLOGY
Key clinical point:
Major finding: Cortactin was expressed on 14 of 17 CLL samples vs. none of 16 MCL samples.
Study details: Immunohistochemistry analysis of samples from 131 patients with B-cell lineage non-Hodgkin lymphomas.
Disclosures: The study was internally supported. The authors reported having no conflicts of interest.
Source: Pizzi M et al. Hum Pathol. 2018 Nov 17. doi: 10.1016/j.humpath.2018.10.038.
EC approves mogamulizumab for MF, SS

The European Commission (EC) has granted marketing authorization for mogamulizumab (Poteligeo), a humanized monoclonal antibody directed against CCR4.
This means mogamulizumab is approved for use in adults with mycosis fungoides (MF) or Sézary syndrome (SS) who have received at least one prior systemic therapy.
This approval is valid in all countries of the European Union as well as Norway, Iceland, and Liechtenstein.
Kyowa Kirin plans to launch mogamulizumab in various European markets starting next year.
The EC’s decision to approve mogamulizumab was supported by the phase 3 MAVORIC trial. Results from this trial were published in The Lancet Oncology in August.
MAVORIC was a comparison of mogamulizumab and vorinostat in 372 adults with MF or SS who had received at least one prior systemic therapy.
Mogamulizumab provided a significant improvement in progression-free survival (PFS), the study’s primary endpoint.
According to investigators, the median PFS was 7.7 months with mogamulizumab and 3.1 months with vorinostat (hazard ratio=0.53, P<0.0001).
According to an independent review committee, the median PFS was 6.7 months and 3.8 months, respectively (hazard ratio=0.64, P<0.0007).
There was a significant improvement in overall response rate (ORR) with mogamulizumab.
According to independent review, the global ORR was 23% (43/186) in the mogamulizumab arm and 4% (7/186) in the vorinostat arm (risk ratio=19.4, P<0.0001).
According to investigators, the global ORR was 28% (52/186) and 5% (9/186), respectively (risk ratio=23.1, P<0.0001).
For patients with MF, the investigator-assessed ORR was 21% (22/105) with mogamulizumab and 7% (7/99) with vorinostat.
For SS patients, the investigator-assessed ORR was 37% (30/81) and 2% (2/87), respectively.
Grade 3 adverse events (AEs) in the mogamulizumab arm included drug eruptions (n=8), hypertension (n=8), pneumonia (n=6), fatigue (n=3), cellulitis (n=3), infusion-related reactions (n=3), sepsis (n=2), decreased appetite (n=2), AST increase (n=2), weight decrease (n=1), pyrexia (n=1), constipation (n=1), nausea (n=1), and diarrhea (n=1).
Grade 4 AEs with mogamulizumab were cellulitis (n=1) and pneumonia (n=1). Grade 5 AEs included pneumonia (n=1) and sepsis (n=1).
The European Commission (EC) has granted marketing authorization for mogamulizumab (Poteligeo), a humanized monoclonal antibody directed against CCR4.
This means mogamulizumab is approved for use in adults with mycosis fungoides (MF) or Sézary syndrome (SS) who have received at least one prior systemic therapy.
This approval is valid in all countries of the European Union as well as Norway, Iceland, and Liechtenstein.
Kyowa Kirin plans to launch mogamulizumab in various European markets starting next year.
The EC’s decision to approve mogamulizumab was supported by the phase 3 MAVORIC trial. Results from this trial were published in The Lancet Oncology in August.
MAVORIC was a comparison of mogamulizumab and vorinostat in 372 adults with MF or SS who had received at least one prior systemic therapy.
Mogamulizumab provided a significant improvement in progression-free survival (PFS), the study’s primary endpoint.
According to investigators, the median PFS was 7.7 months with mogamulizumab and 3.1 months with vorinostat (hazard ratio=0.53, P<0.0001).
According to an independent review committee, the median PFS was 6.7 months and 3.8 months, respectively (hazard ratio=0.64, P<0.0007).
There was a significant improvement in overall response rate (ORR) with mogamulizumab.
According to independent review, the global ORR was 23% (43/186) in the mogamulizumab arm and 4% (7/186) in the vorinostat arm (risk ratio=19.4, P<0.0001).
According to investigators, the global ORR was 28% (52/186) and 5% (9/186), respectively (risk ratio=23.1, P<0.0001).
For patients with MF, the investigator-assessed ORR was 21% (22/105) with mogamulizumab and 7% (7/99) with vorinostat.
For SS patients, the investigator-assessed ORR was 37% (30/81) and 2% (2/87), respectively.
Grade 3 adverse events (AEs) in the mogamulizumab arm included drug eruptions (n=8), hypertension (n=8), pneumonia (n=6), fatigue (n=3), cellulitis (n=3), infusion-related reactions (n=3), sepsis (n=2), decreased appetite (n=2), AST increase (n=2), weight decrease (n=1), pyrexia (n=1), constipation (n=1), nausea (n=1), and diarrhea (n=1).
Grade 4 AEs with mogamulizumab were cellulitis (n=1) and pneumonia (n=1). Grade 5 AEs included pneumonia (n=1) and sepsis (n=1).
The European Commission (EC) has granted marketing authorization for mogamulizumab (Poteligeo), a humanized monoclonal antibody directed against CCR4.
This means mogamulizumab is approved for use in adults with mycosis fungoides (MF) or Sézary syndrome (SS) who have received at least one prior systemic therapy.
This approval is valid in all countries of the European Union as well as Norway, Iceland, and Liechtenstein.
Kyowa Kirin plans to launch mogamulizumab in various European markets starting next year.
The EC’s decision to approve mogamulizumab was supported by the phase 3 MAVORIC trial. Results from this trial were published in The Lancet Oncology in August.
MAVORIC was a comparison of mogamulizumab and vorinostat in 372 adults with MF or SS who had received at least one prior systemic therapy.
Mogamulizumab provided a significant improvement in progression-free survival (PFS), the study’s primary endpoint.
According to investigators, the median PFS was 7.7 months with mogamulizumab and 3.1 months with vorinostat (hazard ratio=0.53, P<0.0001).
According to an independent review committee, the median PFS was 6.7 months and 3.8 months, respectively (hazard ratio=0.64, P<0.0007).
There was a significant improvement in overall response rate (ORR) with mogamulizumab.
According to independent review, the global ORR was 23% (43/186) in the mogamulizumab arm and 4% (7/186) in the vorinostat arm (risk ratio=19.4, P<0.0001).
According to investigators, the global ORR was 28% (52/186) and 5% (9/186), respectively (risk ratio=23.1, P<0.0001).
For patients with MF, the investigator-assessed ORR was 21% (22/105) with mogamulizumab and 7% (7/99) with vorinostat.
For SS patients, the investigator-assessed ORR was 37% (30/81) and 2% (2/87), respectively.
Grade 3 adverse events (AEs) in the mogamulizumab arm included drug eruptions (n=8), hypertension (n=8), pneumonia (n=6), fatigue (n=3), cellulitis (n=3), infusion-related reactions (n=3), sepsis (n=2), decreased appetite (n=2), AST increase (n=2), weight decrease (n=1), pyrexia (n=1), constipation (n=1), nausea (n=1), and diarrhea (n=1).
Grade 4 AEs with mogamulizumab were cellulitis (n=1) and pneumonia (n=1). Grade 5 AEs included pneumonia (n=1) and sepsis (n=1).
Pegfilgrastim biosimilar approved by EC

The European Commission (EC) has granted marketing authorization for Sandoz’s pegfilgrastim product Ziextenzo®, a biosimilar of Amgen’s Neulasta.
Ziextenzo is approved for the same use as the reference medicine—to reduce the duration of neutropenia and the incidence of febrile neutropenia in adults receiving cytotoxic chemotherapy for malignancies except chronic myeloid leukemia and myelodysplastic syndromes.
The approval is valid in all countries of the European Union as well as Norway, Iceland, and Liechtenstein.
The EC’s approval was based on research suggesting Ziextenzo is comparable to Neulasta in terms of safety, efficacy, pharmacokinetics, and pharmacodynamics.1,2,3,4
1. Blackwell K. et al. Pooled analysis of two randomized, double-blind trials comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Ann Oncol 28, 2272-2277 (2017).
2. Nakov R. et al. Proposed biosimilar pegfilgrastim LA-EP2006 shows similarity in pharmacokinetics and pharmacodynamics to reference pegfilgrastim in healthy subjects. 2017 San Antonio Breast Cancer Symposium, abstract P3-14-10.
3. Blackwell K. et al. A Comparison of Proposed Biosimilar LA-EP2006 and Reference Pegfilgrastim for the Prevention of Neutropenia in Patients With Early-Stage Breast Cancer Receiving Myelosuppressive Adjuvant or Neoadjuvant Chemotherapy: Pegfilgrastim Randomized Oncology (Supportive Care) Trial to Evaluate Comparative Treatment (PROTECT-2), a Phase III, Randomized, Double-Blind Trial. Oncologist 21, 789-794 (2016).
4. Harbeck N. et al. Randomized, double-blind study comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Future Oncol 12, 1359-1367 (2016).
The European Commission (EC) has granted marketing authorization for Sandoz’s pegfilgrastim product Ziextenzo®, a biosimilar of Amgen’s Neulasta.
Ziextenzo is approved for the same use as the reference medicine—to reduce the duration of neutropenia and the incidence of febrile neutropenia in adults receiving cytotoxic chemotherapy for malignancies except chronic myeloid leukemia and myelodysplastic syndromes.
The approval is valid in all countries of the European Union as well as Norway, Iceland, and Liechtenstein.
The EC’s approval was based on research suggesting Ziextenzo is comparable to Neulasta in terms of safety, efficacy, pharmacokinetics, and pharmacodynamics.1,2,3,4
1. Blackwell K. et al. Pooled analysis of two randomized, double-blind trials comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Ann Oncol 28, 2272-2277 (2017).
2. Nakov R. et al. Proposed biosimilar pegfilgrastim LA-EP2006 shows similarity in pharmacokinetics and pharmacodynamics to reference pegfilgrastim in healthy subjects. 2017 San Antonio Breast Cancer Symposium, abstract P3-14-10.
3. Blackwell K. et al. A Comparison of Proposed Biosimilar LA-EP2006 and Reference Pegfilgrastim for the Prevention of Neutropenia in Patients With Early-Stage Breast Cancer Receiving Myelosuppressive Adjuvant or Neoadjuvant Chemotherapy: Pegfilgrastim Randomized Oncology (Supportive Care) Trial to Evaluate Comparative Treatment (PROTECT-2), a Phase III, Randomized, Double-Blind Trial. Oncologist 21, 789-794 (2016).
4. Harbeck N. et al. Randomized, double-blind study comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Future Oncol 12, 1359-1367 (2016).
The European Commission (EC) has granted marketing authorization for Sandoz’s pegfilgrastim product Ziextenzo®, a biosimilar of Amgen’s Neulasta.
Ziextenzo is approved for the same use as the reference medicine—to reduce the duration of neutropenia and the incidence of febrile neutropenia in adults receiving cytotoxic chemotherapy for malignancies except chronic myeloid leukemia and myelodysplastic syndromes.
The approval is valid in all countries of the European Union as well as Norway, Iceland, and Liechtenstein.
The EC’s approval was based on research suggesting Ziextenzo is comparable to Neulasta in terms of safety, efficacy, pharmacokinetics, and pharmacodynamics.1,2,3,4
1. Blackwell K. et al. Pooled analysis of two randomized, double-blind trials comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Ann Oncol 28, 2272-2277 (2017).
2. Nakov R. et al. Proposed biosimilar pegfilgrastim LA-EP2006 shows similarity in pharmacokinetics and pharmacodynamics to reference pegfilgrastim in healthy subjects. 2017 San Antonio Breast Cancer Symposium, abstract P3-14-10.
3. Blackwell K. et al. A Comparison of Proposed Biosimilar LA-EP2006 and Reference Pegfilgrastim for the Prevention of Neutropenia in Patients With Early-Stage Breast Cancer Receiving Myelosuppressive Adjuvant or Neoadjuvant Chemotherapy: Pegfilgrastim Randomized Oncology (Supportive Care) Trial to Evaluate Comparative Treatment (PROTECT-2), a Phase III, Randomized, Double-Blind Trial. Oncologist 21, 789-794 (2016).
4. Harbeck N. et al. Randomized, double-blind study comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Future Oncol 12, 1359-1367 (2016).