User login
Step-down etanercept fails to control ankylosing spondylitis
LIVERPOOL, ENGLAND – Reducing the once-weekly maintenance dose of etanercept from 50 mg to 25 mg was associated with worsening control of ankylosing spondylitis in an open-label, multicenter, pilot study.
Results of the ANSWER (Ankylosing Spondylitis with Etanercept Regimens) trial showed that halving the dose of the biologic almost halved the percentage of patients maintaining disease control at 6 months, from 92% to 52% of patients.
Although this was a small study, involving just 47 patients who were randomized to continue etanercept at a weekly dose of 50 mg (n = 24) or to drop down to 25 mg (n = 23) after competing 6 months’ treatment, the results suggest that other approaches are needed to see if the long-term dose of the drug can be reduced, commented study investigator Dr. Karl Gaffney of Norfolk and Norwich University Hospital NHS Foundation Trust, Norwich, England.
"A larger study is required to identify patients suitable for step-down," Dr. Gaffney said at the British Society for Rheumatology annual conference.
"We all know that anti-TNF [tumor necrosis factor] therapy is firmly established in clinical practice for patients with ankylosing spondylitis and this treatment has transformed the quality of life for many of our patients," Dr. Gaffney observed.
"The concept of dose reduction is very appealing from an economic perspective, but also in terms of the cumulative exposure and the risk of adverse effects," he added.
"In patients with rheumatoid arthritis, it’s been shown that lower doses may maintain the clinical response," Dr. Gaffney explained, citing the PRESERVE study published last year (Lancet 2013;381:918-29). However, there are limited data about whether reducing the dose of etanercept could also be successful in ankylosing spondylitis (AS) patients. This is why the ANSWER study was performed.
Patients with active AS who were not responding to conventional therapies and had not yet been treated with a biologic drug were consecutively recruited at two hospitals in England between September 2010 and September 2012. For inclusion, patients had to have sustained spinal disease and a Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score of four or higher. The mean age of patients in the trial was 46.7 years, and the mean BASDAI at baseline was 6.8.
All recruited patients were treated with 50 mg etanercept, once weekly, for 6 months, and those with a "sufficient" clinical response were randomized to either continue this dose for a further 6 months or step down to 25 mg for continued maintenance treatment. A sufficient clinical response was defined as a 50% reduction in BASDAI or a fall of two or more units plus reduction in axial pain of 2 cm or more.
Three months into the maintenance period, patients who had been randomized to the step-down arm started to experience loss of disease control (60.9% vs. 91.7% of 50 mg–treated patients), with the gap widening by 6 months postrandomization and 39% fewer patients maintaining disease control (P = .003).
"There’s no doubt that the 50-mg group did better," Dr. Gaffney said. There were also statistically significant differences favoring the higher dose in a number of secondary outcome measures, which included other measurements of AS disease activity and quality of life. There were no significant differences in terms of adverse events between the two groups.
"However, 52% of the 25-mg arm did maintain their BASDAI response. So theoretically, there may be a subset of patients in whom we can offer this therapeutic option," Dr. Gaffney said. He noted that even in this small group of patients with short-term follow-up that the potential cost savings of being able to reduce the dose would be significant, at around £100,000 (about U.S. $170,000).
"We all have patients who do very well [on etanercept], and I think it would be very nice to be able to say, from 6 months, who we can attempt to step down," commented Dr. Nicola Goodson of University Hospital Aintree, Liverpool. Dr. Goodson chaired the session where the study findings were reported.
Although the ANSWER investigators did look for predictors of response, none were found to be significant. Patients continue to be monitored and the investigators may look at this again. Dr. Gaffney noted that all patients who had their 50-mg dose reinstated regained their clinical response.
He also highlighted during discussion: "A number of patients in the 50-mg group asked to be moved down to 25 mg at the end of the study because they wanted to explore the option. So I think this may well have some long-term consequences, especially in a disease area where we know that the treatment is more symptom modifying than radiographically modifying."
The study was funded by Pfizer. Dr. Gaffney has received research support and honoraria from Pfizer, Merck Sharp & Dohme, AbbVie, and UCB.
LIVERPOOL, ENGLAND – Reducing the once-weekly maintenance dose of etanercept from 50 mg to 25 mg was associated with worsening control of ankylosing spondylitis in an open-label, multicenter, pilot study.
Results of the ANSWER (Ankylosing Spondylitis with Etanercept Regimens) trial showed that halving the dose of the biologic almost halved the percentage of patients maintaining disease control at 6 months, from 92% to 52% of patients.
Although this was a small study, involving just 47 patients who were randomized to continue etanercept at a weekly dose of 50 mg (n = 24) or to drop down to 25 mg (n = 23) after competing 6 months’ treatment, the results suggest that other approaches are needed to see if the long-term dose of the drug can be reduced, commented study investigator Dr. Karl Gaffney of Norfolk and Norwich University Hospital NHS Foundation Trust, Norwich, England.
"A larger study is required to identify patients suitable for step-down," Dr. Gaffney said at the British Society for Rheumatology annual conference.
"We all know that anti-TNF [tumor necrosis factor] therapy is firmly established in clinical practice for patients with ankylosing spondylitis and this treatment has transformed the quality of life for many of our patients," Dr. Gaffney observed.
"The concept of dose reduction is very appealing from an economic perspective, but also in terms of the cumulative exposure and the risk of adverse effects," he added.
"In patients with rheumatoid arthritis, it’s been shown that lower doses may maintain the clinical response," Dr. Gaffney explained, citing the PRESERVE study published last year (Lancet 2013;381:918-29). However, there are limited data about whether reducing the dose of etanercept could also be successful in ankylosing spondylitis (AS) patients. This is why the ANSWER study was performed.
Patients with active AS who were not responding to conventional therapies and had not yet been treated with a biologic drug were consecutively recruited at two hospitals in England between September 2010 and September 2012. For inclusion, patients had to have sustained spinal disease and a Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score of four or higher. The mean age of patients in the trial was 46.7 years, and the mean BASDAI at baseline was 6.8.
All recruited patients were treated with 50 mg etanercept, once weekly, for 6 months, and those with a "sufficient" clinical response were randomized to either continue this dose for a further 6 months or step down to 25 mg for continued maintenance treatment. A sufficient clinical response was defined as a 50% reduction in BASDAI or a fall of two or more units plus reduction in axial pain of 2 cm or more.
Three months into the maintenance period, patients who had been randomized to the step-down arm started to experience loss of disease control (60.9% vs. 91.7% of 50 mg–treated patients), with the gap widening by 6 months postrandomization and 39% fewer patients maintaining disease control (P = .003).
"There’s no doubt that the 50-mg group did better," Dr. Gaffney said. There were also statistically significant differences favoring the higher dose in a number of secondary outcome measures, which included other measurements of AS disease activity and quality of life. There were no significant differences in terms of adverse events between the two groups.
"However, 52% of the 25-mg arm did maintain their BASDAI response. So theoretically, there may be a subset of patients in whom we can offer this therapeutic option," Dr. Gaffney said. He noted that even in this small group of patients with short-term follow-up that the potential cost savings of being able to reduce the dose would be significant, at around £100,000 (about U.S. $170,000).
"We all have patients who do very well [on etanercept], and I think it would be very nice to be able to say, from 6 months, who we can attempt to step down," commented Dr. Nicola Goodson of University Hospital Aintree, Liverpool. Dr. Goodson chaired the session where the study findings were reported.
Although the ANSWER investigators did look for predictors of response, none were found to be significant. Patients continue to be monitored and the investigators may look at this again. Dr. Gaffney noted that all patients who had their 50-mg dose reinstated regained their clinical response.
He also highlighted during discussion: "A number of patients in the 50-mg group asked to be moved down to 25 mg at the end of the study because they wanted to explore the option. So I think this may well have some long-term consequences, especially in a disease area where we know that the treatment is more symptom modifying than radiographically modifying."
The study was funded by Pfizer. Dr. Gaffney has received research support and honoraria from Pfizer, Merck Sharp & Dohme, AbbVie, and UCB.
LIVERPOOL, ENGLAND – Reducing the once-weekly maintenance dose of etanercept from 50 mg to 25 mg was associated with worsening control of ankylosing spondylitis in an open-label, multicenter, pilot study.
Results of the ANSWER (Ankylosing Spondylitis with Etanercept Regimens) trial showed that halving the dose of the biologic almost halved the percentage of patients maintaining disease control at 6 months, from 92% to 52% of patients.
Although this was a small study, involving just 47 patients who were randomized to continue etanercept at a weekly dose of 50 mg (n = 24) or to drop down to 25 mg (n = 23) after competing 6 months’ treatment, the results suggest that other approaches are needed to see if the long-term dose of the drug can be reduced, commented study investigator Dr. Karl Gaffney of Norfolk and Norwich University Hospital NHS Foundation Trust, Norwich, England.
"A larger study is required to identify patients suitable for step-down," Dr. Gaffney said at the British Society for Rheumatology annual conference.
"We all know that anti-TNF [tumor necrosis factor] therapy is firmly established in clinical practice for patients with ankylosing spondylitis and this treatment has transformed the quality of life for many of our patients," Dr. Gaffney observed.
"The concept of dose reduction is very appealing from an economic perspective, but also in terms of the cumulative exposure and the risk of adverse effects," he added.
"In patients with rheumatoid arthritis, it’s been shown that lower doses may maintain the clinical response," Dr. Gaffney explained, citing the PRESERVE study published last year (Lancet 2013;381:918-29). However, there are limited data about whether reducing the dose of etanercept could also be successful in ankylosing spondylitis (AS) patients. This is why the ANSWER study was performed.
Patients with active AS who were not responding to conventional therapies and had not yet been treated with a biologic drug were consecutively recruited at two hospitals in England between September 2010 and September 2012. For inclusion, patients had to have sustained spinal disease and a Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score of four or higher. The mean age of patients in the trial was 46.7 years, and the mean BASDAI at baseline was 6.8.
All recruited patients were treated with 50 mg etanercept, once weekly, for 6 months, and those with a "sufficient" clinical response were randomized to either continue this dose for a further 6 months or step down to 25 mg for continued maintenance treatment. A sufficient clinical response was defined as a 50% reduction in BASDAI or a fall of two or more units plus reduction in axial pain of 2 cm or more.
Three months into the maintenance period, patients who had been randomized to the step-down arm started to experience loss of disease control (60.9% vs. 91.7% of 50 mg–treated patients), with the gap widening by 6 months postrandomization and 39% fewer patients maintaining disease control (P = .003).
"There’s no doubt that the 50-mg group did better," Dr. Gaffney said. There were also statistically significant differences favoring the higher dose in a number of secondary outcome measures, which included other measurements of AS disease activity and quality of life. There were no significant differences in terms of adverse events between the two groups.
"However, 52% of the 25-mg arm did maintain their BASDAI response. So theoretically, there may be a subset of patients in whom we can offer this therapeutic option," Dr. Gaffney said. He noted that even in this small group of patients with short-term follow-up that the potential cost savings of being able to reduce the dose would be significant, at around £100,000 (about U.S. $170,000).
"We all have patients who do very well [on etanercept], and I think it would be very nice to be able to say, from 6 months, who we can attempt to step down," commented Dr. Nicola Goodson of University Hospital Aintree, Liverpool. Dr. Goodson chaired the session where the study findings were reported.
Although the ANSWER investigators did look for predictors of response, none were found to be significant. Patients continue to be monitored and the investigators may look at this again. Dr. Gaffney noted that all patients who had their 50-mg dose reinstated regained their clinical response.
He also highlighted during discussion: "A number of patients in the 50-mg group asked to be moved down to 25 mg at the end of the study because they wanted to explore the option. So I think this may well have some long-term consequences, especially in a disease area where we know that the treatment is more symptom modifying than radiographically modifying."
The study was funded by Pfizer. Dr. Gaffney has received research support and honoraria from Pfizer, Merck Sharp & Dohme, AbbVie, and UCB.
AT RHEUMATOLOGY 2014
Key clinical point: All the patients who did not maintain disease control after halving their dose of etanercept were able to regain clinical response.
Major finding: At 6 months, 52% vs. 92% of the 25 mg– and 50 mg–treated patients maintained a clinical response (P = .003).
Data source: An open-label, multicenter, noninferiority, randomized, pilot study of 47 patients with ankylosing spondylitis treated with weekly etanercept.
Disclosures: The study was funded by Pfizer. Dr. Gaffney has received research support and honoraria from Pfizer, Merck Sharp & Dohme, AbbVie, and UCB.
FDA approves PDE-4 inhibitor for treating psoriatic arthritis
Apremilast, an oral phosphodiesterase-4 inhibitor, has been approved for treating adults with active psoriatic arthritis, based on the results of three studies of 1,493 patients, the Food and Drug Administration announced on March 21.
As a postmarketing requirement, the manufacturer will evaluate the effects of exposure to treatment in pregnant women with a pregnancy registry, according to an FDA statement.
In the three studies, the signs and symptoms of psoriatic arthritis improved among patients treated with apremilast, compared with those on placebo. Diarrhea, nausea, and headache were the most common adverse events associated with treatment. Depression was reported more frequently among those treated with apremilast in the studies, according to the FDA.
Results of the phase III studies were reported in 2013 at the American College of Rheumatology annual meeting and at the annual European Congress of Rheumatology.
During treatment, health care professionals are advised to regularly monitor the weight of patients, and "if unexplained or clinically significant weight loss occurs, the weight loss should be evaluated and discontinuation of treatment should be considered," the statement said. Celgene Corporation will market apremilast under the brand name Otezla.
Apremilast, an oral phosphodiesterase-4 inhibitor, has been approved for treating adults with active psoriatic arthritis, based on the results of three studies of 1,493 patients, the Food and Drug Administration announced on March 21.
As a postmarketing requirement, the manufacturer will evaluate the effects of exposure to treatment in pregnant women with a pregnancy registry, according to an FDA statement.
In the three studies, the signs and symptoms of psoriatic arthritis improved among patients treated with apremilast, compared with those on placebo. Diarrhea, nausea, and headache were the most common adverse events associated with treatment. Depression was reported more frequently among those treated with apremilast in the studies, according to the FDA.
Results of the phase III studies were reported in 2013 at the American College of Rheumatology annual meeting and at the annual European Congress of Rheumatology.
During treatment, health care professionals are advised to regularly monitor the weight of patients, and "if unexplained or clinically significant weight loss occurs, the weight loss should be evaluated and discontinuation of treatment should be considered," the statement said. Celgene Corporation will market apremilast under the brand name Otezla.
Apremilast, an oral phosphodiesterase-4 inhibitor, has been approved for treating adults with active psoriatic arthritis, based on the results of three studies of 1,493 patients, the Food and Drug Administration announced on March 21.
As a postmarketing requirement, the manufacturer will evaluate the effects of exposure to treatment in pregnant women with a pregnancy registry, according to an FDA statement.
In the three studies, the signs and symptoms of psoriatic arthritis improved among patients treated with apremilast, compared with those on placebo. Diarrhea, nausea, and headache were the most common adverse events associated with treatment. Depression was reported more frequently among those treated with apremilast in the studies, according to the FDA.
Results of the phase III studies were reported in 2013 at the American College of Rheumatology annual meeting and at the annual European Congress of Rheumatology.
During treatment, health care professionals are advised to regularly monitor the weight of patients, and "if unexplained or clinically significant weight loss occurs, the weight loss should be evaluated and discontinuation of treatment should be considered," the statement said. Celgene Corporation will market apremilast under the brand name Otezla.
Gut-joint connection promising in psoriatic arthritis
SNOWMASS, COLO. – Mounting circumstantial evidence points to perturbation of bacterial communities in the gut and skin as important environmental triggers for psoriasis and psoriatic arthritis.
A distinctive pattern of alterations in the skin microbiota, termed bacterial "cutaneotypes," has recently been documented in lesional and uninvolved skin of psoriasis patients. Similarly, psoriatic arthritis patients show decreased diversity of their intestinal bacterial community in a pattern similar to patients with inflammatory bowel disease, Dr. Jose U. Scher said at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.

The current working hypothesis of disease pathogenesis is that, in individuals genetically predisposed to psoriasis or psoriatic arthritis, this microbial dysbiosis at the cutaneous and gut levels provides an environmental trigger for overt expression of clinical disease.
"This dysbiosis is potentially relevant as a diagnostic and/or therapeutic target in psoriasis and psoriatic arthritis. For example, it may eventually become possible to assess the gut microbiota to predict which psoriasis patients will later develop psoriatic arthritis. And reconstituting the gut flora may turn out to have therapeutic benefit. But much more work is needed," explained Dr. Scher, director of the Microbiome Center for Rheumatology and Autoimmunity at New York University.
About 25%-30% of psoriasis patients develop inflammatory psoriatic arthritis, most often roughly 7 years after onset of their skin disease. Genetics clearly plays a role, as shown in a classic Danish twin registry study with more than 21,000 subjects. Fifty-five percent of the siblings of monozygotic twins with psoriatic arthritis had skin psoriasis, but only 10% of the siblings had psoriatic arthritis, as did 3.8% of siblings of dizygotic twins with psoriatic arthritis. The lesser concordance rate seen for psoriatic arthritis hinted at the importance of environmental factors in disease genesis (Ann. Rheum. Dis. 2008;67:1417-21).
Subclinical gut inflammation is common in psoriatic arthritis. In one early study, histologic evidence of mild or moderate gut mucosal inflammation was detected in 45% of a group of psoriatic arthritis patients, compared with 15% of patients with rheumatoid arthritis and 0% of controls (Scand. J. Rheumatol. 1997;26:92-8).
Also, psoriasis patients are at a roughly 3.5-fold increased risk of developing Crohn’s disease. Among patients with established psoriatic arthritis, this risk climbs to 6.5-fold greater than in nonpsoriatic controls (Ann. Rheum. Dis. 2013;72:1200-5).
In a soon-to-be-published study, Dr. Scher and his coworkers have taken the field a step further, employing high-throughput gene sequencing to analyze the gut bacterial communities of psoriatic arthritis patients and controls. These were all new-onset psoriatic arthritis patients who had never been exposed to systemic corticosteroids, biologic agents, or conventional disease-modifying antirheumatic drugs.
Compared with the stool samples of healthy controls, several major bacterial species were underrepresented or absent in the gut microbiota of psoriatic arthritis patients. These included Akkermansia, the most common mucolytic bacterium in healthy subjects. Intriguingly, Akkermansia counts are decreased 15-fold in Crohn’s disease and 92-fold in ulcerative colitis, according to the rheumatologist.
Other bacterial species markedly less abundant in the psoriatic arthritis patients’ gut flora were Ruminoccocus, Alistipes, and Roseburia. Like Akkermansia, these are mucin-degrading bacteria that promote a healthy gut environment, and they, too, are reduced in inflammatory bowel disease, Dr. Scher said.
He theorized that these disruptions of the bacterial ecosystem might arise from a course of antibiotics, a change in diet, or other insults. The result is activation of dendritic cells to produce interleukin-23, which triggers a proinflammatory cascade including tumor necrosis factor-alpha, interleukin-22, and antimicrobial peptides.
As shown by other investigators (Mucosal Immunol. 2013;6:666-77), these proinflammatory cytokines inhibit RANK ligand, which is the critical factor for differentiation of microfold cells in the gut. These microfold cells, or M cells, are specialized epithelial cells that transport antigens across the gut epithelium and play an important role in maintenance of an efficient immune response. It’s plausible that, when these M cells are defective, the resultant loss of tolerance and chronic inflammatory state can result in psoriatic arthritis, he explained.
Dr. Scher’s colleagues at New York University have used high-throughput gene sequencing to analyze the cutaneous microbiota of lesional and nonlesional skin in psoriasis patients, as well as skin samples from the same sites in healthy controls. The impetus for this study was a hypothesis that psoriasis might represent an inappropriate cutaneous immune response directed against offending bacteria in the skin microbiota.
Sure enough, the bacterial community present in psoriatic lesions displayed a markedly decreased diversity, compared with controls. This decreased diversity also was present, albeit to a lesser extent, in the psoriasis patients’ nonlesional skin. Both the lesional and nonlesional skin of psoriasis patients contained an increased abundance of Corynebacterium, Staphylococcus, and Streptococcus, compared with controls.
In addition, the skin microbiota could be classified into one of two characteristic patterns, which the investigators termed "cutaneotypes." Cutaneotype 1, which predominated in the skin of normal controls, contained an abundance of Proteobacteria. In contrast, cutaneotype 2, which was 3.5-fold more prevalent in psoriatic lesions than in controls, was enriched in Firmicutes and Actinobacteria. The psoriatic patients’ nonlesional skin contained a balance of cutaneotypes 1 and 2 (Microbiome 2013 Dec. 23 [doi:10.1186/2049-2618-1-31]).
Dr. Scher said that the next step in his own research is to learn whether the gut microbiota of psoriasis patients differs from that of psoriatic arthritis patients.
"That’s to me the most important piece of data. I think there’s a lot of work to do, and we’re doing it now, in prospective fashion. We’re following patients with psoriasis, and for those who later convert to psoriatic arthritis we’ll want to know if there’s alteration of their bacterial immunity, both in the gut and the skin," he explained.
Dr. Scher reported having no financial conflicts.
SNOWMASS, COLO. – Mounting circumstantial evidence points to perturbation of bacterial communities in the gut and skin as important environmental triggers for psoriasis and psoriatic arthritis.
A distinctive pattern of alterations in the skin microbiota, termed bacterial "cutaneotypes," has recently been documented in lesional and uninvolved skin of psoriasis patients. Similarly, psoriatic arthritis patients show decreased diversity of their intestinal bacterial community in a pattern similar to patients with inflammatory bowel disease, Dr. Jose U. Scher said at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
The current working hypothesis of disease pathogenesis is that, in individuals genetically predisposed to psoriasis or psoriatic arthritis, this microbial dysbiosis at the cutaneous and gut levels provides an environmental trigger for overt expression of clinical disease.
"This dysbiosis is potentially relevant as a diagnostic and/or therapeutic target in psoriasis and psoriatic arthritis. For example, it may eventually become possible to assess the gut microbiota to predict which psoriasis patients will later develop psoriatic arthritis. And reconstituting the gut flora may turn out to have therapeutic benefit. But much more work is needed," explained Dr. Scher, director of the Microbiome Center for Rheumatology and Autoimmunity at New York University.
About 25%-30% of psoriasis patients develop inflammatory psoriatic arthritis, most often roughly 7 years after onset of their skin disease. Genetics clearly plays a role, as shown in a classic Danish twin registry study with more than 21,000 subjects. Fifty-five percent of the siblings of monozygotic twins with psoriatic arthritis had skin psoriasis, but only 10% of the siblings had psoriatic arthritis, as did 3.8% of siblings of dizygotic twins with psoriatic arthritis. The lesser concordance rate seen for psoriatic arthritis hinted at the importance of environmental factors in disease genesis (Ann. Rheum. Dis. 2008;67:1417-21).
Subclinical gut inflammation is common in psoriatic arthritis. In one early study, histologic evidence of mild or moderate gut mucosal inflammation was detected in 45% of a group of psoriatic arthritis patients, compared with 15% of patients with rheumatoid arthritis and 0% of controls (Scand. J. Rheumatol. 1997;26:92-8).
Also, psoriasis patients are at a roughly 3.5-fold increased risk of developing Crohn’s disease. Among patients with established psoriatic arthritis, this risk climbs to 6.5-fold greater than in nonpsoriatic controls (Ann. Rheum. Dis. 2013;72:1200-5).
In a soon-to-be-published study, Dr. Scher and his coworkers have taken the field a step further, employing high-throughput gene sequencing to analyze the gut bacterial communities of psoriatic arthritis patients and controls. These were all new-onset psoriatic arthritis patients who had never been exposed to systemic corticosteroids, biologic agents, or conventional disease-modifying antirheumatic drugs.
Compared with the stool samples of healthy controls, several major bacterial species were underrepresented or absent in the gut microbiota of psoriatic arthritis patients. These included Akkermansia, the most common mucolytic bacterium in healthy subjects. Intriguingly, Akkermansia counts are decreased 15-fold in Crohn’s disease and 92-fold in ulcerative colitis, according to the rheumatologist.
Other bacterial species markedly less abundant in the psoriatic arthritis patients’ gut flora were Ruminoccocus, Alistipes, and Roseburia. Like Akkermansia, these are mucin-degrading bacteria that promote a healthy gut environment, and they, too, are reduced in inflammatory bowel disease, Dr. Scher said.
He theorized that these disruptions of the bacterial ecosystem might arise from a course of antibiotics, a change in diet, or other insults. The result is activation of dendritic cells to produce interleukin-23, which triggers a proinflammatory cascade including tumor necrosis factor-alpha, interleukin-22, and antimicrobial peptides.
As shown by other investigators (Mucosal Immunol. 2013;6:666-77), these proinflammatory cytokines inhibit RANK ligand, which is the critical factor for differentiation of microfold cells in the gut. These microfold cells, or M cells, are specialized epithelial cells that transport antigens across the gut epithelium and play an important role in maintenance of an efficient immune response. It’s plausible that, when these M cells are defective, the resultant loss of tolerance and chronic inflammatory state can result in psoriatic arthritis, he explained.
Dr. Scher’s colleagues at New York University have used high-throughput gene sequencing to analyze the cutaneous microbiota of lesional and nonlesional skin in psoriasis patients, as well as skin samples from the same sites in healthy controls. The impetus for this study was a hypothesis that psoriasis might represent an inappropriate cutaneous immune response directed against offending bacteria in the skin microbiota.
Sure enough, the bacterial community present in psoriatic lesions displayed a markedly decreased diversity, compared with controls. This decreased diversity also was present, albeit to a lesser extent, in the psoriasis patients’ nonlesional skin. Both the lesional and nonlesional skin of psoriasis patients contained an increased abundance of Corynebacterium, Staphylococcus, and Streptococcus, compared with controls.
In addition, the skin microbiota could be classified into one of two characteristic patterns, which the investigators termed "cutaneotypes." Cutaneotype 1, which predominated in the skin of normal controls, contained an abundance of Proteobacteria. In contrast, cutaneotype 2, which was 3.5-fold more prevalent in psoriatic lesions than in controls, was enriched in Firmicutes and Actinobacteria. The psoriatic patients’ nonlesional skin contained a balance of cutaneotypes 1 and 2 (Microbiome 2013 Dec. 23 [doi:10.1186/2049-2618-1-31]).
Dr. Scher said that the next step in his own research is to learn whether the gut microbiota of psoriasis patients differs from that of psoriatic arthritis patients.
"That’s to me the most important piece of data. I think there’s a lot of work to do, and we’re doing it now, in prospective fashion. We’re following patients with psoriasis, and for those who later convert to psoriatic arthritis we’ll want to know if there’s alteration of their bacterial immunity, both in the gut and the skin," he explained.
Dr. Scher reported having no financial conflicts.
SNOWMASS, COLO. – Mounting circumstantial evidence points to perturbation of bacterial communities in the gut and skin as important environmental triggers for psoriasis and psoriatic arthritis.
A distinctive pattern of alterations in the skin microbiota, termed bacterial "cutaneotypes," has recently been documented in lesional and uninvolved skin of psoriasis patients. Similarly, psoriatic arthritis patients show decreased diversity of their intestinal bacterial community in a pattern similar to patients with inflammatory bowel disease, Dr. Jose U. Scher said at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
The current working hypothesis of disease pathogenesis is that, in individuals genetically predisposed to psoriasis or psoriatic arthritis, this microbial dysbiosis at the cutaneous and gut levels provides an environmental trigger for overt expression of clinical disease.
"This dysbiosis is potentially relevant as a diagnostic and/or therapeutic target in psoriasis and psoriatic arthritis. For example, it may eventually become possible to assess the gut microbiota to predict which psoriasis patients will later develop psoriatic arthritis. And reconstituting the gut flora may turn out to have therapeutic benefit. But much more work is needed," explained Dr. Scher, director of the Microbiome Center for Rheumatology and Autoimmunity at New York University.
About 25%-30% of psoriasis patients develop inflammatory psoriatic arthritis, most often roughly 7 years after onset of their skin disease. Genetics clearly plays a role, as shown in a classic Danish twin registry study with more than 21,000 subjects. Fifty-five percent of the siblings of monozygotic twins with psoriatic arthritis had skin psoriasis, but only 10% of the siblings had psoriatic arthritis, as did 3.8% of siblings of dizygotic twins with psoriatic arthritis. The lesser concordance rate seen for psoriatic arthritis hinted at the importance of environmental factors in disease genesis (Ann. Rheum. Dis. 2008;67:1417-21).
Subclinical gut inflammation is common in psoriatic arthritis. In one early study, histologic evidence of mild or moderate gut mucosal inflammation was detected in 45% of a group of psoriatic arthritis patients, compared with 15% of patients with rheumatoid arthritis and 0% of controls (Scand. J. Rheumatol. 1997;26:92-8).
Also, psoriasis patients are at a roughly 3.5-fold increased risk of developing Crohn’s disease. Among patients with established psoriatic arthritis, this risk climbs to 6.5-fold greater than in nonpsoriatic controls (Ann. Rheum. Dis. 2013;72:1200-5).
In a soon-to-be-published study, Dr. Scher and his coworkers have taken the field a step further, employing high-throughput gene sequencing to analyze the gut bacterial communities of psoriatic arthritis patients and controls. These were all new-onset psoriatic arthritis patients who had never been exposed to systemic corticosteroids, biologic agents, or conventional disease-modifying antirheumatic drugs.
Compared with the stool samples of healthy controls, several major bacterial species were underrepresented or absent in the gut microbiota of psoriatic arthritis patients. These included Akkermansia, the most common mucolytic bacterium in healthy subjects. Intriguingly, Akkermansia counts are decreased 15-fold in Crohn’s disease and 92-fold in ulcerative colitis, according to the rheumatologist.
Other bacterial species markedly less abundant in the psoriatic arthritis patients’ gut flora were Ruminoccocus, Alistipes, and Roseburia. Like Akkermansia, these are mucin-degrading bacteria that promote a healthy gut environment, and they, too, are reduced in inflammatory bowel disease, Dr. Scher said.
He theorized that these disruptions of the bacterial ecosystem might arise from a course of antibiotics, a change in diet, or other insults. The result is activation of dendritic cells to produce interleukin-23, which triggers a proinflammatory cascade including tumor necrosis factor-alpha, interleukin-22, and antimicrobial peptides.
As shown by other investigators (Mucosal Immunol. 2013;6:666-77), these proinflammatory cytokines inhibit RANK ligand, which is the critical factor for differentiation of microfold cells in the gut. These microfold cells, or M cells, are specialized epithelial cells that transport antigens across the gut epithelium and play an important role in maintenance of an efficient immune response. It’s plausible that, when these M cells are defective, the resultant loss of tolerance and chronic inflammatory state can result in psoriatic arthritis, he explained.
Dr. Scher’s colleagues at New York University have used high-throughput gene sequencing to analyze the cutaneous microbiota of lesional and nonlesional skin in psoriasis patients, as well as skin samples from the same sites in healthy controls. The impetus for this study was a hypothesis that psoriasis might represent an inappropriate cutaneous immune response directed against offending bacteria in the skin microbiota.
Sure enough, the bacterial community present in psoriatic lesions displayed a markedly decreased diversity, compared with controls. This decreased diversity also was present, albeit to a lesser extent, in the psoriasis patients’ nonlesional skin. Both the lesional and nonlesional skin of psoriasis patients contained an increased abundance of Corynebacterium, Staphylococcus, and Streptococcus, compared with controls.
In addition, the skin microbiota could be classified into one of two characteristic patterns, which the investigators termed "cutaneotypes." Cutaneotype 1, which predominated in the skin of normal controls, contained an abundance of Proteobacteria. In contrast, cutaneotype 2, which was 3.5-fold more prevalent in psoriatic lesions than in controls, was enriched in Firmicutes and Actinobacteria. The psoriatic patients’ nonlesional skin contained a balance of cutaneotypes 1 and 2 (Microbiome 2013 Dec. 23 [doi:10.1186/2049-2618-1-31]).
Dr. Scher said that the next step in his own research is to learn whether the gut microbiota of psoriasis patients differs from that of psoriatic arthritis patients.
"That’s to me the most important piece of data. I think there’s a lot of work to do, and we’re doing it now, in prospective fashion. We’re following patients with psoriasis, and for those who later convert to psoriatic arthritis we’ll want to know if there’s alteration of their bacterial immunity, both in the gut and the skin," he explained.
Dr. Scher reported having no financial conflicts.
EXPERT ANALYSIS FROM THE WINTER RHEUMATOLOGY SYMPOSIUM

Shift in approach needed for managing inflammatory arthritis in pregnancy
SNOWMASS, COLO. – The common strategy of managing inflammatory arthritis during pregnancy by stopping all prepregnancy medications and prescribing prednisone is counterproductive, Dr. Megan E.B. Clowse said at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
"This sort of safe approach is actually not the safest or best approach. We all need to shift our attitude and get comfortable with at least a couple of other medications in order to help our patients have better outcomes," asserted Dr. Clowse, director of the Duke Autoimmunity in Pregnancy (DAP) Registry at Duke University, Durham, N.C.

It’s often true that rheumatoid arthritis (RA) tends to get better in pregnancy. Nonetheless, pregnancy may not be enough to control disease activity in many women, as illustrated by a nationwide Dutch study in which half of RA patients had moderate to severe disease activity by their third trimester (Arthritis Rheum. 2008;59:1241-8).
"In my experience, the patients who were on tumor necrosis factor inhibitors prepregnancy do not do as well when they come off them. So pregnancy might be good for RA disease activity, but it’s definitely not as good as a tumor necrosis factor inhibitor," according to the rheumatologist.
Roughly one-quarter of the more than 230 prospectively studied pregnancies in the DAP Registry have been in women with RA or other inflammatory arthritides. The preterm birth rate in RA patients in DAP, as in other RA registries, is higher than in the general population. Notably, all of the DAP patients with RA who had preterm birth had moderate to severe disease activity in the first and second trimesters, compared with just a single RA patient with a term delivery.
"Allowing disease to flare may actually harm the pregnancy," Dr. Clowse commented.
Her two top recommendations for nonbiologic treatment of inflammatory arthritis during pregnancy are hydroxychloroquine and sulfasalazine. Hydroxychloroquine is effective for mild arthritis, and it’s rated by the Food and Drug Administration as Pregnancy Category C, with no reports of human toxicity. Sulfasalazine has a good safety profile in pregnancy – it is Pregnancy Category B – and has good efficacy for peripheral arthritis.
"These are two very important medications that are much better – and definitely much safer – options than prednisone. These are just great options, and I try to encourage patients to take them. It’s definitely better than letting patients flare while off methotrexate before they deliver," she continued.
"TNF [tumor necrosis factor] inhibitors are, I think, another reasonable option, particularly in the population with moderate to severe disease activity. If any of these medications help you avoid prednisone, then I think you come out ahead," Dr. Clowse said.
Reassuring data regarding the safety of anti-TNF biologics in pregnancy comes from several sources. A systematic literature review by Dr. Evelyne Vinet and her coworkers at McGill University, Montreal, found no evidence of excess risk of adverse pregnancy or fetal outcomes in women exposed to etanercept (Enbrel), infliximab (Remicade), or adalimumab (Humira) during pregnancy (Arthritis Rheum. 2009;61:587-92). A report from the Organization of Teratology Information Specialists (OTIS) Registry, presented at the 2012 annual meeting of the American College of Rheumatology, found no significant difference between RA patients exposed to adalimumab in pregnancy and RA controls in rates of live birth, miscarriage, stillbirth, congenital abnormalities, or serious infections in the first year of life.
Moreover, DAP Registry data support the notion that for some patients, anti-TNF biologics during pregnancy may be a good option. In Dr. Clowse’s series of 29 RA patients, disease activity during the first trimester neatly delineated three distinct groups. Eleven patients who had a 28-joint Disease Activity Score using C-reactive protein (DAS28-CRP) averaging 1.5 in the first trimester required neither TNF inhibitors nor prednisone during their pregnancy, and they had no abnormal outcomes. Seven patients with significant first-trimester disease activity as defined by an average DAS28-CRP score of 3.4 went back on their TNF inhibitor during pregnancy, and their disease essentially went into remission, with only one preterm birth. In contrast, 11 other patients with significant first-trimester disease activity went on prednisone because they rejected TNF inhibitor therapy; their disease activity remained substantial, and 6 of the 11 had a preterm birth and/or preeclampsia.
"All of the preeclampsia occurred in the prednisone group. They also had a lot of disease activity postpartum," Dr. Clowse observed.
Infliximab and adalimumab freely pass across the placenta; etanercept and certolizumab pegol (Cimzia) do not. Regardless, no infant exposed in utero to a TNF inhibitor should receive the rotavirus vaccine during the first 5 months of life. That’s the only live-virus vaccine given during infancy in the United States; all the other vaccines can safely be given according to schedule.
Dr. Clowse said there is essentially no published literature regarding psoriatic arthritis in pregnancy. The DAP registry experience includes seven pregnancies in women with psoriatic arthritis. There were no pregnancy losses, one preterm birth, and two cases of preeclampsia.
"What I see is dramatically increased disease activity when women with psoriatic arthritis stop their TNF inhibitor. The worst psoriasis I’ve ever seen is in pregnant patients; they are really miserable off their TNF inhibitors," she said.
There is also very little in the literature about axial spondyloarthritis in pregnancy. The DAP registry’s 10 cases constitute one of the largest series. There were no pregnancy losses, no preeclampsia, and one preterm birth. Eight of the women were delivered by C-section.
"That reflects my personal bias, which is that the position required for vaginal delivery is pretty horrible for sacroiliac joint pain and leads to significant flares postpartum. That’s particularly true if the patient has an epidural and doesn’t know how hard she’s pushing on those SI joints," according to the rheumatologist.
Dr. Clowse reported serving as a consultant to UCB, which markets certolizumab pegol.
SNOWMASS, COLO. – The common strategy of managing inflammatory arthritis during pregnancy by stopping all prepregnancy medications and prescribing prednisone is counterproductive, Dr. Megan E.B. Clowse said at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
"This sort of safe approach is actually not the safest or best approach. We all need to shift our attitude and get comfortable with at least a couple of other medications in order to help our patients have better outcomes," asserted Dr. Clowse, director of the Duke Autoimmunity in Pregnancy (DAP) Registry at Duke University, Durham, N.C.
It’s often true that rheumatoid arthritis (RA) tends to get better in pregnancy. Nonetheless, pregnancy may not be enough to control disease activity in many women, as illustrated by a nationwide Dutch study in which half of RA patients had moderate to severe disease activity by their third trimester (Arthritis Rheum. 2008;59:1241-8).
"In my experience, the patients who were on tumor necrosis factor inhibitors prepregnancy do not do as well when they come off them. So pregnancy might be good for RA disease activity, but it’s definitely not as good as a tumor necrosis factor inhibitor," according to the rheumatologist.
Roughly one-quarter of the more than 230 prospectively studied pregnancies in the DAP Registry have been in women with RA or other inflammatory arthritides. The preterm birth rate in RA patients in DAP, as in other RA registries, is higher than in the general population. Notably, all of the DAP patients with RA who had preterm birth had moderate to severe disease activity in the first and second trimesters, compared with just a single RA patient with a term delivery.
"Allowing disease to flare may actually harm the pregnancy," Dr. Clowse commented.
Her two top recommendations for nonbiologic treatment of inflammatory arthritis during pregnancy are hydroxychloroquine and sulfasalazine. Hydroxychloroquine is effective for mild arthritis, and it’s rated by the Food and Drug Administration as Pregnancy Category C, with no reports of human toxicity. Sulfasalazine has a good safety profile in pregnancy – it is Pregnancy Category B – and has good efficacy for peripheral arthritis.
"These are two very important medications that are much better – and definitely much safer – options than prednisone. These are just great options, and I try to encourage patients to take them. It’s definitely better than letting patients flare while off methotrexate before they deliver," she continued.
"TNF [tumor necrosis factor] inhibitors are, I think, another reasonable option, particularly in the population with moderate to severe disease activity. If any of these medications help you avoid prednisone, then I think you come out ahead," Dr. Clowse said.
Reassuring data regarding the safety of anti-TNF biologics in pregnancy comes from several sources. A systematic literature review by Dr. Evelyne Vinet and her coworkers at McGill University, Montreal, found no evidence of excess risk of adverse pregnancy or fetal outcomes in women exposed to etanercept (Enbrel), infliximab (Remicade), or adalimumab (Humira) during pregnancy (Arthritis Rheum. 2009;61:587-92). A report from the Organization of Teratology Information Specialists (OTIS) Registry, presented at the 2012 annual meeting of the American College of Rheumatology, found no significant difference between RA patients exposed to adalimumab in pregnancy and RA controls in rates of live birth, miscarriage, stillbirth, congenital abnormalities, or serious infections in the first year of life.
Moreover, DAP Registry data support the notion that for some patients, anti-TNF biologics during pregnancy may be a good option. In Dr. Clowse’s series of 29 RA patients, disease activity during the first trimester neatly delineated three distinct groups. Eleven patients who had a 28-joint Disease Activity Score using C-reactive protein (DAS28-CRP) averaging 1.5 in the first trimester required neither TNF inhibitors nor prednisone during their pregnancy, and they had no abnormal outcomes. Seven patients with significant first-trimester disease activity as defined by an average DAS28-CRP score of 3.4 went back on their TNF inhibitor during pregnancy, and their disease essentially went into remission, with only one preterm birth. In contrast, 11 other patients with significant first-trimester disease activity went on prednisone because they rejected TNF inhibitor therapy; their disease activity remained substantial, and 6 of the 11 had a preterm birth and/or preeclampsia.
"All of the preeclampsia occurred in the prednisone group. They also had a lot of disease activity postpartum," Dr. Clowse observed.
Infliximab and adalimumab freely pass across the placenta; etanercept and certolizumab pegol (Cimzia) do not. Regardless, no infant exposed in utero to a TNF inhibitor should receive the rotavirus vaccine during the first 5 months of life. That’s the only live-virus vaccine given during infancy in the United States; all the other vaccines can safely be given according to schedule.
Dr. Clowse said there is essentially no published literature regarding psoriatic arthritis in pregnancy. The DAP registry experience includes seven pregnancies in women with psoriatic arthritis. There were no pregnancy losses, one preterm birth, and two cases of preeclampsia.
"What I see is dramatically increased disease activity when women with psoriatic arthritis stop their TNF inhibitor. The worst psoriasis I’ve ever seen is in pregnant patients; they are really miserable off their TNF inhibitors," she said.
There is also very little in the literature about axial spondyloarthritis in pregnancy. The DAP registry’s 10 cases constitute one of the largest series. There were no pregnancy losses, no preeclampsia, and one preterm birth. Eight of the women were delivered by C-section.
"That reflects my personal bias, which is that the position required for vaginal delivery is pretty horrible for sacroiliac joint pain and leads to significant flares postpartum. That’s particularly true if the patient has an epidural and doesn’t know how hard she’s pushing on those SI joints," according to the rheumatologist.
Dr. Clowse reported serving as a consultant to UCB, which markets certolizumab pegol.
SNOWMASS, COLO. – The common strategy of managing inflammatory arthritis during pregnancy by stopping all prepregnancy medications and prescribing prednisone is counterproductive, Dr. Megan E.B. Clowse said at the Winter Rheumatology Symposium sponsored by the American College of Rheumatology.
"This sort of safe approach is actually not the safest or best approach. We all need to shift our attitude and get comfortable with at least a couple of other medications in order to help our patients have better outcomes," asserted Dr. Clowse, director of the Duke Autoimmunity in Pregnancy (DAP) Registry at Duke University, Durham, N.C.
It’s often true that rheumatoid arthritis (RA) tends to get better in pregnancy. Nonetheless, pregnancy may not be enough to control disease activity in many women, as illustrated by a nationwide Dutch study in which half of RA patients had moderate to severe disease activity by their third trimester (Arthritis Rheum. 2008;59:1241-8).
"In my experience, the patients who were on tumor necrosis factor inhibitors prepregnancy do not do as well when they come off them. So pregnancy might be good for RA disease activity, but it’s definitely not as good as a tumor necrosis factor inhibitor," according to the rheumatologist.
Roughly one-quarter of the more than 230 prospectively studied pregnancies in the DAP Registry have been in women with RA or other inflammatory arthritides. The preterm birth rate in RA patients in DAP, as in other RA registries, is higher than in the general population. Notably, all of the DAP patients with RA who had preterm birth had moderate to severe disease activity in the first and second trimesters, compared with just a single RA patient with a term delivery.
"Allowing disease to flare may actually harm the pregnancy," Dr. Clowse commented.
Her two top recommendations for nonbiologic treatment of inflammatory arthritis during pregnancy are hydroxychloroquine and sulfasalazine. Hydroxychloroquine is effective for mild arthritis, and it’s rated by the Food and Drug Administration as Pregnancy Category C, with no reports of human toxicity. Sulfasalazine has a good safety profile in pregnancy – it is Pregnancy Category B – and has good efficacy for peripheral arthritis.
"These are two very important medications that are much better – and definitely much safer – options than prednisone. These are just great options, and I try to encourage patients to take them. It’s definitely better than letting patients flare while off methotrexate before they deliver," she continued.
"TNF [tumor necrosis factor] inhibitors are, I think, another reasonable option, particularly in the population with moderate to severe disease activity. If any of these medications help you avoid prednisone, then I think you come out ahead," Dr. Clowse said.
Reassuring data regarding the safety of anti-TNF biologics in pregnancy comes from several sources. A systematic literature review by Dr. Evelyne Vinet and her coworkers at McGill University, Montreal, found no evidence of excess risk of adverse pregnancy or fetal outcomes in women exposed to etanercept (Enbrel), infliximab (Remicade), or adalimumab (Humira) during pregnancy (Arthritis Rheum. 2009;61:587-92). A report from the Organization of Teratology Information Specialists (OTIS) Registry, presented at the 2012 annual meeting of the American College of Rheumatology, found no significant difference between RA patients exposed to adalimumab in pregnancy and RA controls in rates of live birth, miscarriage, stillbirth, congenital abnormalities, or serious infections in the first year of life.
Moreover, DAP Registry data support the notion that for some patients, anti-TNF biologics during pregnancy may be a good option. In Dr. Clowse’s series of 29 RA patients, disease activity during the first trimester neatly delineated three distinct groups. Eleven patients who had a 28-joint Disease Activity Score using C-reactive protein (DAS28-CRP) averaging 1.5 in the first trimester required neither TNF inhibitors nor prednisone during their pregnancy, and they had no abnormal outcomes. Seven patients with significant first-trimester disease activity as defined by an average DAS28-CRP score of 3.4 went back on their TNF inhibitor during pregnancy, and their disease essentially went into remission, with only one preterm birth. In contrast, 11 other patients with significant first-trimester disease activity went on prednisone because they rejected TNF inhibitor therapy; their disease activity remained substantial, and 6 of the 11 had a preterm birth and/or preeclampsia.
"All of the preeclampsia occurred in the prednisone group. They also had a lot of disease activity postpartum," Dr. Clowse observed.
Infliximab and adalimumab freely pass across the placenta; etanercept and certolizumab pegol (Cimzia) do not. Regardless, no infant exposed in utero to a TNF inhibitor should receive the rotavirus vaccine during the first 5 months of life. That’s the only live-virus vaccine given during infancy in the United States; all the other vaccines can safely be given according to schedule.
Dr. Clowse said there is essentially no published literature regarding psoriatic arthritis in pregnancy. The DAP registry experience includes seven pregnancies in women with psoriatic arthritis. There were no pregnancy losses, one preterm birth, and two cases of preeclampsia.
"What I see is dramatically increased disease activity when women with psoriatic arthritis stop their TNF inhibitor. The worst psoriasis I’ve ever seen is in pregnant patients; they are really miserable off their TNF inhibitors," she said.
There is also very little in the literature about axial spondyloarthritis in pregnancy. The DAP registry’s 10 cases constitute one of the largest series. There were no pregnancy losses, no preeclampsia, and one preterm birth. Eight of the women were delivered by C-section.
"That reflects my personal bias, which is that the position required for vaginal delivery is pretty horrible for sacroiliac joint pain and leads to significant flares postpartum. That’s particularly true if the patient has an epidural and doesn’t know how hard she’s pushing on those SI joints," according to the rheumatologist.
Dr. Clowse reported serving as a consultant to UCB, which markets certolizumab pegol.
EXPERT ANALYSIS FROM THE WINTER RHEUMATOLOGY SYMPOSIUM

Apremilast’s positive study results made it the talk of ACR 2013
SAN DIEGO – The novel oral phosphodiesterase-4 inhibitor apremilast cut an impressively wide swath through the annual meeting of the American College of Rheumatology on the strength of positive results in three separate pivotal phase III trials for psoriatic arthritis and a favorable phase II study in Behçet’s syndrome.
Dr. Alvin F. Wells reported that apremilast resulted in clinically meaningful improvement in psoriatic arthritis symptoms, physical function, and associated skin psoriasis at 16 weeks in the PALACE 4 trial. Moreover, the improvement remained durable through 52 weeks in the phase III clinical trial.
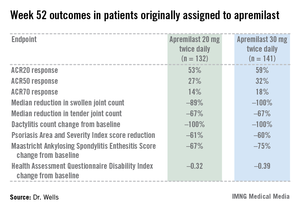
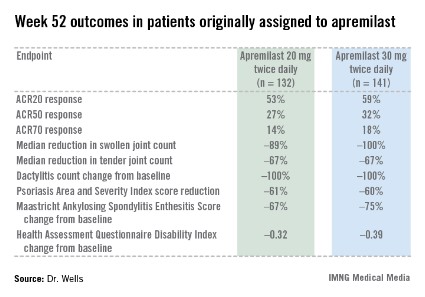
PALACE 4 compared apremilast against placebo as a first-line treatment in 527 patients with psoriatic arthritis not previously treated with a disease-modifying antirheumatic drug. This was a population with active disease: a mean baseline 11 swollen and 20 tender joints; a Health Assessment Questionnaire Disability Index score averaging 1.07; and a 50% prevalence of dactylitis, with a mean severity score of 2.0. Sixty-five percent of patients had enthesitis, with a median Maastricht Ankylosing Spondylitis Enthesitis Score (MASES) of 3.0. Participants had a mean 16-year history of psoriasis and a 3.4-year duration of psoriatic arthritis.
The primary study endpoint was attainment of an American College of Rheumatology 20% improvement (ACR20) response at 16 weeks. This was achieved in 29.2% of patients randomized to apremilast at 20 mg twice daily, 32.3% on 30 mg twice daily, and 16.9% on placebo. At week 16, patients on placebo were re-randomized to apremilast at 20 mg or 30 mg twice weekly. By week 52, an ACR20 response was achieved by 53% of patients in the apremilast 20 mg twice-daily group and 59% of those on the higher dose, according to Dr. Wells, director of the Rheumatology and Immunotherapy Center in Franklin, Wisc.
Apremilast also scored well on numerous secondary endpoints (see chart). For example, the swollen joint count decreased by a median of 89% over the course of 52 weeks in patients on apremilast at 20 mg twice daily.
"The way I like to think of that result is at least 50% of patients had an 89% improvement in their swollen joint count," he explained.
During the first 16 weeks of PALACE 4, there were fewer serious adverse events in apremilast-treated patients than with placebo-treated patients. The most frequent side effects of apremilast were nausea, diarrhea, and headache, affecting 16%, 12%, and 8%, respectively, of patients in the higher-dose arm. These adverse events were almost exclusively mild or moderate, they began within the first 2 weeks of treatment, and they resolved in 4 weeks despite continued treatment. Less than 2% of apremilast-treated patients dropped out of the study due to nausea or diarrhea through week 52.
PALACE 4 differed from the PALACE 2 and 3 trials, also presented at the ACR meeting, in that PALACE 2 and 3 involved only patients with psoriatic arthritis previously treated with biologic agents or other disease-modifying antirheumatic drugs. All three studies had the same design, and the results in terms of both efficacy and safety were consistent across the full clinical trial program. No clinically meaningful changes in laboratory values were seen in any of the PALACE trials, suggesting ongoing lab monitoring may not be necessary.
Also at the ACR annual meeting, Dr. Gülen Hatemi of Istanbul University, Turkey, in a reprise of her report several weeks earlier at the annual congress of the European Academy of Dermatology and Venereology, presented a phase II study showing apremilast to be highly effective in treating the oral ulcers that are the cardinal feature of Behçet’s syndrome.
Celgene filed for approval of apremilast for psoriatic arthritis with the Food and Drug Administration earlier this year and anticipates a decision in March 2014. The company also plans to apply to the European regulatory agency for the same indication before year’s end, and to petition the FDA for approval in psoriasis within the same time frame. A 500-patient phase III study of apremilast in ankylosing spondylitis, known as POSTURE, is well underway.
PALACE 4 was sponsored by Celgene. Dr. Wells reported receiving a research grant from the company.
SAN DIEGO – The novel oral phosphodiesterase-4 inhibitor apremilast cut an impressively wide swath through the annual meeting of the American College of Rheumatology on the strength of positive results in three separate pivotal phase III trials for psoriatic arthritis and a favorable phase II study in Behçet’s syndrome.
Dr. Alvin F. Wells reported that apremilast resulted in clinically meaningful improvement in psoriatic arthritis symptoms, physical function, and associated skin psoriasis at 16 weeks in the PALACE 4 trial. Moreover, the improvement remained durable through 52 weeks in the phase III clinical trial.
PALACE 4 compared apremilast against placebo as a first-line treatment in 527 patients with psoriatic arthritis not previously treated with a disease-modifying antirheumatic drug. This was a population with active disease: a mean baseline 11 swollen and 20 tender joints; a Health Assessment Questionnaire Disability Index score averaging 1.07; and a 50% prevalence of dactylitis, with a mean severity score of 2.0. Sixty-five percent of patients had enthesitis, with a median Maastricht Ankylosing Spondylitis Enthesitis Score (MASES) of 3.0. Participants had a mean 16-year history of psoriasis and a 3.4-year duration of psoriatic arthritis.
The primary study endpoint was attainment of an American College of Rheumatology 20% improvement (ACR20) response at 16 weeks. This was achieved in 29.2% of patients randomized to apremilast at 20 mg twice daily, 32.3% on 30 mg twice daily, and 16.9% on placebo. At week 16, patients on placebo were re-randomized to apremilast at 20 mg or 30 mg twice weekly. By week 52, an ACR20 response was achieved by 53% of patients in the apremilast 20 mg twice-daily group and 59% of those on the higher dose, according to Dr. Wells, director of the Rheumatology and Immunotherapy Center in Franklin, Wisc.
Apremilast also scored well on numerous secondary endpoints (see chart). For example, the swollen joint count decreased by a median of 89% over the course of 52 weeks in patients on apremilast at 20 mg twice daily.
"The way I like to think of that result is at least 50% of patients had an 89% improvement in their swollen joint count," he explained.
During the first 16 weeks of PALACE 4, there were fewer serious adverse events in apremilast-treated patients than with placebo-treated patients. The most frequent side effects of apremilast were nausea, diarrhea, and headache, affecting 16%, 12%, and 8%, respectively, of patients in the higher-dose arm. These adverse events were almost exclusively mild or moderate, they began within the first 2 weeks of treatment, and they resolved in 4 weeks despite continued treatment. Less than 2% of apremilast-treated patients dropped out of the study due to nausea or diarrhea through week 52.
PALACE 4 differed from the PALACE 2 and 3 trials, also presented at the ACR meeting, in that PALACE 2 and 3 involved only patients with psoriatic arthritis previously treated with biologic agents or other disease-modifying antirheumatic drugs. All three studies had the same design, and the results in terms of both efficacy and safety were consistent across the full clinical trial program. No clinically meaningful changes in laboratory values were seen in any of the PALACE trials, suggesting ongoing lab monitoring may not be necessary.
Also at the ACR annual meeting, Dr. Gülen Hatemi of Istanbul University, Turkey, in a reprise of her report several weeks earlier at the annual congress of the European Academy of Dermatology and Venereology, presented a phase II study showing apremilast to be highly effective in treating the oral ulcers that are the cardinal feature of Behçet’s syndrome.
Celgene filed for approval of apremilast for psoriatic arthritis with the Food and Drug Administration earlier this year and anticipates a decision in March 2014. The company also plans to apply to the European regulatory agency for the same indication before year’s end, and to petition the FDA for approval in psoriasis within the same time frame. A 500-patient phase III study of apremilast in ankylosing spondylitis, known as POSTURE, is well underway.
PALACE 4 was sponsored by Celgene. Dr. Wells reported receiving a research grant from the company.
SAN DIEGO – The novel oral phosphodiesterase-4 inhibitor apremilast cut an impressively wide swath through the annual meeting of the American College of Rheumatology on the strength of positive results in three separate pivotal phase III trials for psoriatic arthritis and a favorable phase II study in Behçet’s syndrome.
Dr. Alvin F. Wells reported that apremilast resulted in clinically meaningful improvement in psoriatic arthritis symptoms, physical function, and associated skin psoriasis at 16 weeks in the PALACE 4 trial. Moreover, the improvement remained durable through 52 weeks in the phase III clinical trial.
PALACE 4 compared apremilast against placebo as a first-line treatment in 527 patients with psoriatic arthritis not previously treated with a disease-modifying antirheumatic drug. This was a population with active disease: a mean baseline 11 swollen and 20 tender joints; a Health Assessment Questionnaire Disability Index score averaging 1.07; and a 50% prevalence of dactylitis, with a mean severity score of 2.0. Sixty-five percent of patients had enthesitis, with a median Maastricht Ankylosing Spondylitis Enthesitis Score (MASES) of 3.0. Participants had a mean 16-year history of psoriasis and a 3.4-year duration of psoriatic arthritis.
The primary study endpoint was attainment of an American College of Rheumatology 20% improvement (ACR20) response at 16 weeks. This was achieved in 29.2% of patients randomized to apremilast at 20 mg twice daily, 32.3% on 30 mg twice daily, and 16.9% on placebo. At week 16, patients on placebo were re-randomized to apremilast at 20 mg or 30 mg twice weekly. By week 52, an ACR20 response was achieved by 53% of patients in the apremilast 20 mg twice-daily group and 59% of those on the higher dose, according to Dr. Wells, director of the Rheumatology and Immunotherapy Center in Franklin, Wisc.
Apremilast also scored well on numerous secondary endpoints (see chart). For example, the swollen joint count decreased by a median of 89% over the course of 52 weeks in patients on apremilast at 20 mg twice daily.
"The way I like to think of that result is at least 50% of patients had an 89% improvement in their swollen joint count," he explained.
During the first 16 weeks of PALACE 4, there were fewer serious adverse events in apremilast-treated patients than with placebo-treated patients. The most frequent side effects of apremilast were nausea, diarrhea, and headache, affecting 16%, 12%, and 8%, respectively, of patients in the higher-dose arm. These adverse events were almost exclusively mild or moderate, they began within the first 2 weeks of treatment, and they resolved in 4 weeks despite continued treatment. Less than 2% of apremilast-treated patients dropped out of the study due to nausea or diarrhea through week 52.
PALACE 4 differed from the PALACE 2 and 3 trials, also presented at the ACR meeting, in that PALACE 2 and 3 involved only patients with psoriatic arthritis previously treated with biologic agents or other disease-modifying antirheumatic drugs. All three studies had the same design, and the results in terms of both efficacy and safety were consistent across the full clinical trial program. No clinically meaningful changes in laboratory values were seen in any of the PALACE trials, suggesting ongoing lab monitoring may not be necessary.
Also at the ACR annual meeting, Dr. Gülen Hatemi of Istanbul University, Turkey, in a reprise of her report several weeks earlier at the annual congress of the European Academy of Dermatology and Venereology, presented a phase II study showing apremilast to be highly effective in treating the oral ulcers that are the cardinal feature of Behçet’s syndrome.
Celgene filed for approval of apremilast for psoriatic arthritis with the Food and Drug Administration earlier this year and anticipates a decision in March 2014. The company also plans to apply to the European regulatory agency for the same indication before year’s end, and to petition the FDA for approval in psoriasis within the same time frame. A 500-patient phase III study of apremilast in ankylosing spondylitis, known as POSTURE, is well underway.
PALACE 4 was sponsored by Celgene. Dr. Wells reported receiving a research grant from the company.
AT THE ACR ANNUAL MEETING
Major finding: After 16 weeks of treatment, the investigational drug apremilast achieved an ACR20 response of 29.2% at 20 mg twice daily as first-line therapy in patients with psoriatic arthritis and 32.3% at 30 mg twice daily, whereas placebo-treated controls achieved a rate of 16.9%.
Data source: The PALACE 4 study was a pivotal phase III, randomized, placebo-controlled trial including 527 patients with active psoriatic arthritis.
Disclosures: PALACE 4 was sponsored by Celgene. Dr. Wells reported receiving a research grant from the company.
Treat-to-target approach for psoriatic arthritis found beneficial
SAN DIEGO – Compared with standard care, intensive management of psoriatic arthritis using a treat-to-target approach significantly improved joint and skin outcomes for patients newly diagnosed with the disease, results from a multicenter, randomized controlled trial showed.
"Treating to target works in this disease, and it’s going to result in better long-term outcomes," lead investigator Dr. Philip Helliwell said during a press briefing at the annual meeting of the American College of Rheumatology.

Dr. Helliwell and his associates at eight centers in the United Kingdom randomized 206 patients with early psoriatic arthritis to standard care or intensive management, and followed them for 48 weeks. Patients in the standard care group were treated by a rheumatologist with no set protocol and no limitations, while those in the intensive management group followed a strict treatment protocol with escalation of therapy if minimal disease activity criteria were not met.
Patients in the intensive management group were started on methotrexate with rapid escalation to a dose of *25 mg/week after 6 weeks if they tolerated the drug. If they did not meet minimal disease criteria after 12 weeks, they received a more powerful combination of disease-modifying antirheumatic drugs (DMARDs).
After another 12 weeks, patients in the intensive management group were given anti–tumor necrosis factor therapy if they had three or more tender joints. If they had fewer than three tender or swollen joints but did not meet the minimal disease activity criteria, they were given methotrexate and an alternative DMARD. Patients in the standard care group were treated with DMARDs, but with no set time limits for drug therapy escalation or measurements to reach.
The primary outcome measures were the proportion of patients in both groups who achieved ACR20, ACR50, and ACR70 criteria for disease activity, which represent disease improvement of 20%, 50%, and 70%, respectively. Dr. Helliwell reported that compared with the standard care group, a higher proportion of patients in the intensive management group achieved ACR20 (62% vs. 45%, respectively), ACR50 (51% vs. 25%), and ACR70 (38% vs. 17%). A higher proportion of patients in the intensive management group also achieved a Psoriasis Area and Severity Index 75 compared with their counterparts in the standard care group (59% vs. 33%).
Research in psoriatic arthritis has "lagged behind that of rheumatoid arthritis for years in terms of pathogenesis and treatment paradigms," noted Dr. Helliwell, a senior lecturer in rheumatology at the Institute of Rheumatic and Musculoskeletal Medicine at the University of Leeds (England). "I think the study we’ve reported brings psoriatic arthritis right up to date alongside RA."
He said that up to one-third of people with psoriasis will develop psoriatic arthritis, "so it’s not an insignificant arthritis. It’s often not recognized. One survey we did of people with psoriasis in the community in the U.K. found that up to half of the people with psoriatic arthritis didn’t know they had it. They’d seen their doctor for various reasons and had been told they’d had another form of arthritis, or they were fobbed off with other diagnoses."
The researchers have yet to perform a cost analysis comparing the two treatment groups.
The study was funded by Arthritis Research UK and Pfizer. Dr. Helliwell disclosed that he has received consulting fees from Pfizer.
*Correction 11/11/13: A previous version of this story misstated the methotrexate dosage used in the study. This version has been updated to reflect the correct dosage.
SAN DIEGO – Compared with standard care, intensive management of psoriatic arthritis using a treat-to-target approach significantly improved joint and skin outcomes for patients newly diagnosed with the disease, results from a multicenter, randomized controlled trial showed.
"Treating to target works in this disease, and it’s going to result in better long-term outcomes," lead investigator Dr. Philip Helliwell said during a press briefing at the annual meeting of the American College of Rheumatology.
Dr. Helliwell and his associates at eight centers in the United Kingdom randomized 206 patients with early psoriatic arthritis to standard care or intensive management, and followed them for 48 weeks. Patients in the standard care group were treated by a rheumatologist with no set protocol and no limitations, while those in the intensive management group followed a strict treatment protocol with escalation of therapy if minimal disease activity criteria were not met.
Patients in the intensive management group were started on methotrexate with rapid escalation to a dose of *25 mg/week after 6 weeks if they tolerated the drug. If they did not meet minimal disease criteria after 12 weeks, they received a more powerful combination of disease-modifying antirheumatic drugs (DMARDs).
After another 12 weeks, patients in the intensive management group were given anti–tumor necrosis factor therapy if they had three or more tender joints. If they had fewer than three tender or swollen joints but did not meet the minimal disease activity criteria, they were given methotrexate and an alternative DMARD. Patients in the standard care group were treated with DMARDs, but with no set time limits for drug therapy escalation or measurements to reach.
The primary outcome measures were the proportion of patients in both groups who achieved ACR20, ACR50, and ACR70 criteria for disease activity, which represent disease improvement of 20%, 50%, and 70%, respectively. Dr. Helliwell reported that compared with the standard care group, a higher proportion of patients in the intensive management group achieved ACR20 (62% vs. 45%, respectively), ACR50 (51% vs. 25%), and ACR70 (38% vs. 17%). A higher proportion of patients in the intensive management group also achieved a Psoriasis Area and Severity Index 75 compared with their counterparts in the standard care group (59% vs. 33%).
Research in psoriatic arthritis has "lagged behind that of rheumatoid arthritis for years in terms of pathogenesis and treatment paradigms," noted Dr. Helliwell, a senior lecturer in rheumatology at the Institute of Rheumatic and Musculoskeletal Medicine at the University of Leeds (England). "I think the study we’ve reported brings psoriatic arthritis right up to date alongside RA."
He said that up to one-third of people with psoriasis will develop psoriatic arthritis, "so it’s not an insignificant arthritis. It’s often not recognized. One survey we did of people with psoriasis in the community in the U.K. found that up to half of the people with psoriatic arthritis didn’t know they had it. They’d seen their doctor for various reasons and had been told they’d had another form of arthritis, or they were fobbed off with other diagnoses."
The researchers have yet to perform a cost analysis comparing the two treatment groups.
The study was funded by Arthritis Research UK and Pfizer. Dr. Helliwell disclosed that he has received consulting fees from Pfizer.
*Correction 11/11/13: A previous version of this story misstated the methotrexate dosage used in the study. This version has been updated to reflect the correct dosage.
SAN DIEGO – Compared with standard care, intensive management of psoriatic arthritis using a treat-to-target approach significantly improved joint and skin outcomes for patients newly diagnosed with the disease, results from a multicenter, randomized controlled trial showed.
"Treating to target works in this disease, and it’s going to result in better long-term outcomes," lead investigator Dr. Philip Helliwell said during a press briefing at the annual meeting of the American College of Rheumatology.
Dr. Helliwell and his associates at eight centers in the United Kingdom randomized 206 patients with early psoriatic arthritis to standard care or intensive management, and followed them for 48 weeks. Patients in the standard care group were treated by a rheumatologist with no set protocol and no limitations, while those in the intensive management group followed a strict treatment protocol with escalation of therapy if minimal disease activity criteria were not met.
Patients in the intensive management group were started on methotrexate with rapid escalation to a dose of *25 mg/week after 6 weeks if they tolerated the drug. If they did not meet minimal disease criteria after 12 weeks, they received a more powerful combination of disease-modifying antirheumatic drugs (DMARDs).
After another 12 weeks, patients in the intensive management group were given anti–tumor necrosis factor therapy if they had three or more tender joints. If they had fewer than three tender or swollen joints but did not meet the minimal disease activity criteria, they were given methotrexate and an alternative DMARD. Patients in the standard care group were treated with DMARDs, but with no set time limits for drug therapy escalation or measurements to reach.
The primary outcome measures were the proportion of patients in both groups who achieved ACR20, ACR50, and ACR70 criteria for disease activity, which represent disease improvement of 20%, 50%, and 70%, respectively. Dr. Helliwell reported that compared with the standard care group, a higher proportion of patients in the intensive management group achieved ACR20 (62% vs. 45%, respectively), ACR50 (51% vs. 25%), and ACR70 (38% vs. 17%). A higher proportion of patients in the intensive management group also achieved a Psoriasis Area and Severity Index 75 compared with their counterparts in the standard care group (59% vs. 33%).
Research in psoriatic arthritis has "lagged behind that of rheumatoid arthritis for years in terms of pathogenesis and treatment paradigms," noted Dr. Helliwell, a senior lecturer in rheumatology at the Institute of Rheumatic and Musculoskeletal Medicine at the University of Leeds (England). "I think the study we’ve reported brings psoriatic arthritis right up to date alongside RA."
He said that up to one-third of people with psoriasis will develop psoriatic arthritis, "so it’s not an insignificant arthritis. It’s often not recognized. One survey we did of people with psoriasis in the community in the U.K. found that up to half of the people with psoriatic arthritis didn’t know they had it. They’d seen their doctor for various reasons and had been told they’d had another form of arthritis, or they were fobbed off with other diagnoses."
The researchers have yet to perform a cost analysis comparing the two treatment groups.
The study was funded by Arthritis Research UK and Pfizer. Dr. Helliwell disclosed that he has received consulting fees from Pfizer.
*Correction 11/11/13: A previous version of this story misstated the methotrexate dosage used in the study. This version has been updated to reflect the correct dosage.
AT THE ACR ANNUAL MEETING
Major finding: At 48 weeks, a higher proportion of psoriatic arthritis patients in the intensive management group achieved ACR70, compared with those in the standard care group (38% vs. 17%, respectively).
Data source: A multicenter trial of 206 patients with early psoriatic arthritis who were randomized to standard care or intensive management and followed for 48 weeks.
Disclosures: The study was funded by Arthritis Research UK and Pfizer. Dr. Helliwell disclosed that he has received consulting fees from Pfizer.

Smoking has damaging impact in ankylosing spondylitis
SAN DIEGO – Smoking increases the effect of inflammation on x-ray damage in people with ankylosing spondylitis by more than fivefold, results from a Dutch study demonstrated. In addition, the effect is amplified by more than 13-fold in male smokers, compared with females who don’t smoke.
Those are the key findings from OASIS (Outcome in AS International Study), which followed ankylosing spondylitis patients for up to 12 years with biannual clinical assessments, including radiographs. Lead investigator Dr. Sofia Ramiro of the Academic Medical Center at the University of Amsterdam discussed the study during a press briefing at the annual meeting of the American College of Rheumatology.

Two reviewers independently scored the x-rays of 127 patients with ankylosing spondylitis according to the Modified Stoke Ankylosing Spondylitis Supine Score (mSASSS). Dr. Ramiro and her associates evaluated the relationship between the AS Disease Activity Score and x-ray damage over a period of 12 years by assessing the effect of inflammation at one point in time on the progression of damage 2 years later.
The mean age of the 127 patients was 41 years and 71% were male. Their mean symptom duration was 18 years, 82% were human leukocyte antigen B27 (HLA-B27) positive, and 40% were smokers.
Overall, smokers had a 5.5-fold higher effect of inflammation on x-ray damage, compared with nonsmokers, and male smokers had a 13.4-fold higher effect of inflammation on damage, compared with female nonsmokers. In addition, smokers with a short symptom duration – defined as those with fewer than 18 years of symptoms – had an 8.1-fold higher effect of inflammation on damage, compared with nonsmokers with long symptom duration.
"Smoking cessation, especially among young males, seems to be an important take-home message," Dr. Ramiro commented. "It will probably lead to less radiographic damage and to better long-term outcomes."
The press briefing’s moderator, Dr. Christie M. Bartels of the division of rheumatology at the University of Wisconsin in Madison, characterized the findings as "additional compelling evidence to say that smoking cessation would be an important process measure to look at in the rheumatology clinic, so that we can help our patients achieve their optimal outcomes."
As to the possible mechanism of action behind the relationship, Dr. Ramiro speculated that "it’s not really smoking itself that’s contributing to the worse outcomes. Smoking is increasing the impact of inflammation on radiographic damage. It’s inflammation that causes the damage."
In an interview, she acknowledged certain limitations of the study, including the fact that the sample size "is not large enough to split the data into several relevant subgroups. For instance, the number of females is rather small, as is the number of HLA-B27–negative males and females."
Dr. Ramiro said that she had no relevant financial conflicts to disclose.
SAN DIEGO – Smoking increases the effect of inflammation on x-ray damage in people with ankylosing spondylitis by more than fivefold, results from a Dutch study demonstrated. In addition, the effect is amplified by more than 13-fold in male smokers, compared with females who don’t smoke.
Those are the key findings from OASIS (Outcome in AS International Study), which followed ankylosing spondylitis patients for up to 12 years with biannual clinical assessments, including radiographs. Lead investigator Dr. Sofia Ramiro of the Academic Medical Center at the University of Amsterdam discussed the study during a press briefing at the annual meeting of the American College of Rheumatology.
Two reviewers independently scored the x-rays of 127 patients with ankylosing spondylitis according to the Modified Stoke Ankylosing Spondylitis Supine Score (mSASSS). Dr. Ramiro and her associates evaluated the relationship between the AS Disease Activity Score and x-ray damage over a period of 12 years by assessing the effect of inflammation at one point in time on the progression of damage 2 years later.
The mean age of the 127 patients was 41 years and 71% were male. Their mean symptom duration was 18 years, 82% were human leukocyte antigen B27 (HLA-B27) positive, and 40% were smokers.
Overall, smokers had a 5.5-fold higher effect of inflammation on x-ray damage, compared with nonsmokers, and male smokers had a 13.4-fold higher effect of inflammation on damage, compared with female nonsmokers. In addition, smokers with a short symptom duration – defined as those with fewer than 18 years of symptoms – had an 8.1-fold higher effect of inflammation on damage, compared with nonsmokers with long symptom duration.
"Smoking cessation, especially among young males, seems to be an important take-home message," Dr. Ramiro commented. "It will probably lead to less radiographic damage and to better long-term outcomes."
The press briefing’s moderator, Dr. Christie M. Bartels of the division of rheumatology at the University of Wisconsin in Madison, characterized the findings as "additional compelling evidence to say that smoking cessation would be an important process measure to look at in the rheumatology clinic, so that we can help our patients achieve their optimal outcomes."
As to the possible mechanism of action behind the relationship, Dr. Ramiro speculated that "it’s not really smoking itself that’s contributing to the worse outcomes. Smoking is increasing the impact of inflammation on radiographic damage. It’s inflammation that causes the damage."
In an interview, she acknowledged certain limitations of the study, including the fact that the sample size "is not large enough to split the data into several relevant subgroups. For instance, the number of females is rather small, as is the number of HLA-B27–negative males and females."
Dr. Ramiro said that she had no relevant financial conflicts to disclose.
SAN DIEGO – Smoking increases the effect of inflammation on x-ray damage in people with ankylosing spondylitis by more than fivefold, results from a Dutch study demonstrated. In addition, the effect is amplified by more than 13-fold in male smokers, compared with females who don’t smoke.
Those are the key findings from OASIS (Outcome in AS International Study), which followed ankylosing spondylitis patients for up to 12 years with biannual clinical assessments, including radiographs. Lead investigator Dr. Sofia Ramiro of the Academic Medical Center at the University of Amsterdam discussed the study during a press briefing at the annual meeting of the American College of Rheumatology.
Two reviewers independently scored the x-rays of 127 patients with ankylosing spondylitis according to the Modified Stoke Ankylosing Spondylitis Supine Score (mSASSS). Dr. Ramiro and her associates evaluated the relationship between the AS Disease Activity Score and x-ray damage over a period of 12 years by assessing the effect of inflammation at one point in time on the progression of damage 2 years later.
The mean age of the 127 patients was 41 years and 71% were male. Their mean symptom duration was 18 years, 82% were human leukocyte antigen B27 (HLA-B27) positive, and 40% were smokers.
Overall, smokers had a 5.5-fold higher effect of inflammation on x-ray damage, compared with nonsmokers, and male smokers had a 13.4-fold higher effect of inflammation on damage, compared with female nonsmokers. In addition, smokers with a short symptom duration – defined as those with fewer than 18 years of symptoms – had an 8.1-fold higher effect of inflammation on damage, compared with nonsmokers with long symptom duration.
"Smoking cessation, especially among young males, seems to be an important take-home message," Dr. Ramiro commented. "It will probably lead to less radiographic damage and to better long-term outcomes."
The press briefing’s moderator, Dr. Christie M. Bartels of the division of rheumatology at the University of Wisconsin in Madison, characterized the findings as "additional compelling evidence to say that smoking cessation would be an important process measure to look at in the rheumatology clinic, so that we can help our patients achieve their optimal outcomes."
As to the possible mechanism of action behind the relationship, Dr. Ramiro speculated that "it’s not really smoking itself that’s contributing to the worse outcomes. Smoking is increasing the impact of inflammation on radiographic damage. It’s inflammation that causes the damage."
In an interview, she acknowledged certain limitations of the study, including the fact that the sample size "is not large enough to split the data into several relevant subgroups. For instance, the number of females is rather small, as is the number of HLA-B27–negative males and females."
Dr. Ramiro said that she had no relevant financial conflicts to disclose.
AT THE ACR ANNUAL MEETING
Major finding: Smokers had a 5.5-fold higher effect of inflammation on x-ray damage, compared with nonsmokers, and male smokers had a 13.4-fold higher effect of inflammation on damage, compared with female nonsmokers.
Data source: An assessment of 127 patients enrolled in the Outcome in AS International Study (OASIS), which followed ankylosing spondylitis patients for up to 12 years with biannual clinical assessments, including radiographs.
Disclosures: Dr. Ramiro said that she had no relevant financial conflicts to disclose.

Ustekinumab approved for psoriatic arthritis
Ustekinumab, a human interleukin-12 and -23 antagonist, has been approved for the treatment of adults with active psoriatic arthritis, the manufacturer has announced.
The approved indication is for use alone or in combination with methotrexate, in the form of a 45-mg subcutaneous injection at weeks 0 and 4, and then every 12 weeks. For patients with coexistent moderate-to-severe plaque psoriasis who weigh more than 220 pounds, the recommended dose is a 90-mg subcutaneous injection at weeks 0 and 4, and then every 12 weeks.
Ustekinumab, approved for treating psoriasis in 2009, is marketed as Stelara by Janssen Biotech, which announced the approval in a press release on Sept. 23. Ustekinumab is the first treatment targeting the cytokines interleukin-12 and interleukin-23, according to the statement.
Approval was based on the results of two phase III multicenter, randomized double-blind studies comparing ustekinumab to placebo in 927 patients with active psoriatic arthritis, with at least five tender and five swollen joints and a C-reactive protein (CRP) level of at least 0.3 mg/dL, despite previous treatment with conventional therapy. The primary endpoint was the ACR 20 response at 24 weeks.
In the first trial, PSUMMIT 1, 42% of those on the 45-mg dose and 50% of those on the 90-mg dose achieved an ACR 20 at 24 weeks, vs. 23% of those on placebo. In addition, 57% of those on the 45-mg dose and 62% of those on the 90-mg dose achieved a PASI (Psoriasis Area and Severity Index) 75 response, vs. 11% of those on placebo.
In the second study, PSUMMIT II, 44% of those on a 45-mg dose and 44% of those on a 90-mg dose met the primary endpoint, vs 20% of those on placebo. Also, 51% of those on the 45-mg dose and 56% of those on the 90-mg dose achieved a PASI 75 response, vs. 5% of those on placebo.
The results of the PSUMMIT 1 study were published online in the Lancet on June 13 (doi:10.1016/S0140-6736[13]60594-2).
The warnings and precautions section of the ustekinumab label includes information about the risk of serious infections and other risks associated with treatment.
The updated label is available here.
Ustekinumab, a human interleukin-12 and -23 antagonist, has been approved for the treatment of adults with active psoriatic arthritis, the manufacturer has announced.
The approved indication is for use alone or in combination with methotrexate, in the form of a 45-mg subcutaneous injection at weeks 0 and 4, and then every 12 weeks. For patients with coexistent moderate-to-severe plaque psoriasis who weigh more than 220 pounds, the recommended dose is a 90-mg subcutaneous injection at weeks 0 and 4, and then every 12 weeks.
Ustekinumab, approved for treating psoriasis in 2009, is marketed as Stelara by Janssen Biotech, which announced the approval in a press release on Sept. 23. Ustekinumab is the first treatment targeting the cytokines interleukin-12 and interleukin-23, according to the statement.
Approval was based on the results of two phase III multicenter, randomized double-blind studies comparing ustekinumab to placebo in 927 patients with active psoriatic arthritis, with at least five tender and five swollen joints and a C-reactive protein (CRP) level of at least 0.3 mg/dL, despite previous treatment with conventional therapy. The primary endpoint was the ACR 20 response at 24 weeks.
In the first trial, PSUMMIT 1, 42% of those on the 45-mg dose and 50% of those on the 90-mg dose achieved an ACR 20 at 24 weeks, vs. 23% of those on placebo. In addition, 57% of those on the 45-mg dose and 62% of those on the 90-mg dose achieved a PASI (Psoriasis Area and Severity Index) 75 response, vs. 11% of those on placebo.
In the second study, PSUMMIT II, 44% of those on a 45-mg dose and 44% of those on a 90-mg dose met the primary endpoint, vs 20% of those on placebo. Also, 51% of those on the 45-mg dose and 56% of those on the 90-mg dose achieved a PASI 75 response, vs. 5% of those on placebo.
The results of the PSUMMIT 1 study were published online in the Lancet on June 13 (doi:10.1016/S0140-6736[13]60594-2).
The warnings and precautions section of the ustekinumab label includes information about the risk of serious infections and other risks associated with treatment.
The updated label is available here.
Ustekinumab, a human interleukin-12 and -23 antagonist, has been approved for the treatment of adults with active psoriatic arthritis, the manufacturer has announced.
The approved indication is for use alone or in combination with methotrexate, in the form of a 45-mg subcutaneous injection at weeks 0 and 4, and then every 12 weeks. For patients with coexistent moderate-to-severe plaque psoriasis who weigh more than 220 pounds, the recommended dose is a 90-mg subcutaneous injection at weeks 0 and 4, and then every 12 weeks.
Ustekinumab, approved for treating psoriasis in 2009, is marketed as Stelara by Janssen Biotech, which announced the approval in a press release on Sept. 23. Ustekinumab is the first treatment targeting the cytokines interleukin-12 and interleukin-23, according to the statement.
Approval was based on the results of two phase III multicenter, randomized double-blind studies comparing ustekinumab to placebo in 927 patients with active psoriatic arthritis, with at least five tender and five swollen joints and a C-reactive protein (CRP) level of at least 0.3 mg/dL, despite previous treatment with conventional therapy. The primary endpoint was the ACR 20 response at 24 weeks.
In the first trial, PSUMMIT 1, 42% of those on the 45-mg dose and 50% of those on the 90-mg dose achieved an ACR 20 at 24 weeks, vs. 23% of those on placebo. In addition, 57% of those on the 45-mg dose and 62% of those on the 90-mg dose achieved a PASI (Psoriasis Area and Severity Index) 75 response, vs. 11% of those on placebo.
In the second study, PSUMMIT II, 44% of those on a 45-mg dose and 44% of those on a 90-mg dose met the primary endpoint, vs 20% of those on placebo. Also, 51% of those on the 45-mg dose and 56% of those on the 90-mg dose achieved a PASI 75 response, vs. 5% of those on placebo.
The results of the PSUMMIT 1 study were published online in the Lancet on June 13 (doi:10.1016/S0140-6736[13]60594-2).
The warnings and precautions section of the ustekinumab label includes information about the risk of serious infections and other risks associated with treatment.
The updated label is available here.
Proposed axial spondyloarthritis indication for certolizumab divides FDA panel
SILVER SPRING, MD. – A Food and Drug Administration advisory panel split its vote on whether to recommend the approval of certolizumab as a treatment for axial spondyloarthritis, based on a study of 325 patients with ankylosing spondylitis or nonradiographic axial spondyloarthritis.
At a July 23 meeting, the FDA’s Arthritis Advisory Committee voted 7-6, with one abstention, to recommend approval of the tumor necrosis factor (TNF) blocker for the manufacturer’s proposed indication: treatment of "active axial spondyloarthritis, including patients with ankylosing spondylitis."
Certolizumab, a TNF blocker, was approved in 2008 for treating Crohn’s disease and in 2009 for treating rheumatoid arthritis (RA). It is marketed as Cimzia by UCB and is administered by subcutaneous injection every 4 weeks for Crohn’s disease and every other week or every 4 weeks for RA, as maintenance therapy. Currently, four other TNF inhibitors are approved for ankylosing spondylitis (AS) but none are approved for treating nonradiographic axial spondyloarthritis (nr-axSpA).
The pivotal study compared two dosing regimens of certolizumab against placebo in patients with active axial spondyloarthritis who had an inadequate response to NSAIDs or a contraindication to NSAIDs (178 patients with AS, and 147 patients with nr-axSpA). The dosing regimens were the same as those currently approved for rheumatoid arthritis: a loading dose of 400 mg in weeks 0, 2, and 4, followed by either 200 mg every 2 weeks (Q2W) or 400 mg every 4 weeks (Q4W). Requirements for enrollment included chronic back pain for at least 3 months; onset before age 45 years; and imaging criteria that included sacroiliitis (on MRI or X-ray) with at least one spondyloarthritis feature, such as inflammation, back pain, arthritis, uveitis, or dactylitis.
At 12 weeks, 58% of patients on the Q2W regimen and 64% on the Q4W regimen had achieved an ASAS (Assessment of SpondyloArthritis international Society) 20 response, the primary endpoint, compared with 38% of those on placebo, both of which were statistically significant differences. The differences remained significant at 24 weeks. ASAS20 responses in the two subpopulations, those with AS and those with nr-axSpA, also favored those treated with certolizumab.
Treatment with certolizumab also resulted in significant improvements in function and spinal mobility, with similar responses at both dosage regimens, compared with placebo, as well as improvements in spinal and sacroiliac joint inflammation, productivity, and health-related quality of life, according to UCB.
The rates of serious adverse events were similar in the certolizumab-treated and placebo groups, the safety profile of certolizumab was consistent with the known safety profile of TNF inhibitors, and no new safety signals were identified. The rate of serious infections, a known risk, was higher among those on certolizumab.
The main issue raised by the FDA was whether the data from this study were adequate to support the indication, especially for patients with nr-axSpA, which would be a new indication for an approved treatment, according to Dr. Janet Maynard, acting clinical team leader in the FDA’s Division of Pulmonary, Allergy, and Rheumatology Products. But the FDA’s analysis of the data did not suggest that the efficacy results were driven by any subgroup, and the efficacy data for AS "appear reasonable," she said at the meeting.
The panel had no substantial safety concerns – voting 13-1 that the safety profile of certolizumab was adequate to support approval of the new indication – and agreed there was evidence of efficacy. Those voting against approval, however, thought that the indication was too broad and had concerns that included the small number of patients for a new indication (nr-axSpA), the lack of a clear definition of active disease in the indication, and the possibility that primary care physicians would prescribe the biologic drug for patients with low back pain.
Panelists supporting approval said that adequate prescribing information would result in appropriate use for patients with active disease in clinical practice.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, a panelist might be given a waiver, but not at this meeting.
Certolizumab, a TNF blocker, Crohn’s disease, rheumatoid arthritis (RA), Cimzia, UCB, subcutaneous injection, Dr. Janet Maynard, FDA’s Division of Pulmonary, Allergy, and Rheumatology Products,
SILVER SPRING, MD. – A Food and Drug Administration advisory panel split its vote on whether to recommend the approval of certolizumab as a treatment for axial spondyloarthritis, based on a study of 325 patients with ankylosing spondylitis or nonradiographic axial spondyloarthritis.
At a July 23 meeting, the FDA’s Arthritis Advisory Committee voted 7-6, with one abstention, to recommend approval of the tumor necrosis factor (TNF) blocker for the manufacturer’s proposed indication: treatment of "active axial spondyloarthritis, including patients with ankylosing spondylitis."
Certolizumab, a TNF blocker, was approved in 2008 for treating Crohn’s disease and in 2009 for treating rheumatoid arthritis (RA). It is marketed as Cimzia by UCB and is administered by subcutaneous injection every 4 weeks for Crohn’s disease and every other week or every 4 weeks for RA, as maintenance therapy. Currently, four other TNF inhibitors are approved for ankylosing spondylitis (AS) but none are approved for treating nonradiographic axial spondyloarthritis (nr-axSpA).
The pivotal study compared two dosing regimens of certolizumab against placebo in patients with active axial spondyloarthritis who had an inadequate response to NSAIDs or a contraindication to NSAIDs (178 patients with AS, and 147 patients with nr-axSpA). The dosing regimens were the same as those currently approved for rheumatoid arthritis: a loading dose of 400 mg in weeks 0, 2, and 4, followed by either 200 mg every 2 weeks (Q2W) or 400 mg every 4 weeks (Q4W). Requirements for enrollment included chronic back pain for at least 3 months; onset before age 45 years; and imaging criteria that included sacroiliitis (on MRI or X-ray) with at least one spondyloarthritis feature, such as inflammation, back pain, arthritis, uveitis, or dactylitis.
At 12 weeks, 58% of patients on the Q2W regimen and 64% on the Q4W regimen had achieved an ASAS (Assessment of SpondyloArthritis international Society) 20 response, the primary endpoint, compared with 38% of those on placebo, both of which were statistically significant differences. The differences remained significant at 24 weeks. ASAS20 responses in the two subpopulations, those with AS and those with nr-axSpA, also favored those treated with certolizumab.
Treatment with certolizumab also resulted in significant improvements in function and spinal mobility, with similar responses at both dosage regimens, compared with placebo, as well as improvements in spinal and sacroiliac joint inflammation, productivity, and health-related quality of life, according to UCB.
The rates of serious adverse events were similar in the certolizumab-treated and placebo groups, the safety profile of certolizumab was consistent with the known safety profile of TNF inhibitors, and no new safety signals were identified. The rate of serious infections, a known risk, was higher among those on certolizumab.
The main issue raised by the FDA was whether the data from this study were adequate to support the indication, especially for patients with nr-axSpA, which would be a new indication for an approved treatment, according to Dr. Janet Maynard, acting clinical team leader in the FDA’s Division of Pulmonary, Allergy, and Rheumatology Products. But the FDA’s analysis of the data did not suggest that the efficacy results were driven by any subgroup, and the efficacy data for AS "appear reasonable," she said at the meeting.
The panel had no substantial safety concerns – voting 13-1 that the safety profile of certolizumab was adequate to support approval of the new indication – and agreed there was evidence of efficacy. Those voting against approval, however, thought that the indication was too broad and had concerns that included the small number of patients for a new indication (nr-axSpA), the lack of a clear definition of active disease in the indication, and the possibility that primary care physicians would prescribe the biologic drug for patients with low back pain.
Panelists supporting approval said that adequate prescribing information would result in appropriate use for patients with active disease in clinical practice.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, a panelist might be given a waiver, but not at this meeting.
SILVER SPRING, MD. – A Food and Drug Administration advisory panel split its vote on whether to recommend the approval of certolizumab as a treatment for axial spondyloarthritis, based on a study of 325 patients with ankylosing spondylitis or nonradiographic axial spondyloarthritis.
At a July 23 meeting, the FDA’s Arthritis Advisory Committee voted 7-6, with one abstention, to recommend approval of the tumor necrosis factor (TNF) blocker for the manufacturer’s proposed indication: treatment of "active axial spondyloarthritis, including patients with ankylosing spondylitis."
Certolizumab, a TNF blocker, was approved in 2008 for treating Crohn’s disease and in 2009 for treating rheumatoid arthritis (RA). It is marketed as Cimzia by UCB and is administered by subcutaneous injection every 4 weeks for Crohn’s disease and every other week or every 4 weeks for RA, as maintenance therapy. Currently, four other TNF inhibitors are approved for ankylosing spondylitis (AS) but none are approved for treating nonradiographic axial spondyloarthritis (nr-axSpA).
The pivotal study compared two dosing regimens of certolizumab against placebo in patients with active axial spondyloarthritis who had an inadequate response to NSAIDs or a contraindication to NSAIDs (178 patients with AS, and 147 patients with nr-axSpA). The dosing regimens were the same as those currently approved for rheumatoid arthritis: a loading dose of 400 mg in weeks 0, 2, and 4, followed by either 200 mg every 2 weeks (Q2W) or 400 mg every 4 weeks (Q4W). Requirements for enrollment included chronic back pain for at least 3 months; onset before age 45 years; and imaging criteria that included sacroiliitis (on MRI or X-ray) with at least one spondyloarthritis feature, such as inflammation, back pain, arthritis, uveitis, or dactylitis.
At 12 weeks, 58% of patients on the Q2W regimen and 64% on the Q4W regimen had achieved an ASAS (Assessment of SpondyloArthritis international Society) 20 response, the primary endpoint, compared with 38% of those on placebo, both of which were statistically significant differences. The differences remained significant at 24 weeks. ASAS20 responses in the two subpopulations, those with AS and those with nr-axSpA, also favored those treated with certolizumab.
Treatment with certolizumab also resulted in significant improvements in function and spinal mobility, with similar responses at both dosage regimens, compared with placebo, as well as improvements in spinal and sacroiliac joint inflammation, productivity, and health-related quality of life, according to UCB.
The rates of serious adverse events were similar in the certolizumab-treated and placebo groups, the safety profile of certolizumab was consistent with the known safety profile of TNF inhibitors, and no new safety signals were identified. The rate of serious infections, a known risk, was higher among those on certolizumab.
The main issue raised by the FDA was whether the data from this study were adequate to support the indication, especially for patients with nr-axSpA, which would be a new indication for an approved treatment, according to Dr. Janet Maynard, acting clinical team leader in the FDA’s Division of Pulmonary, Allergy, and Rheumatology Products. But the FDA’s analysis of the data did not suggest that the efficacy results were driven by any subgroup, and the efficacy data for AS "appear reasonable," she said at the meeting.
The panel had no substantial safety concerns – voting 13-1 that the safety profile of certolizumab was adequate to support approval of the new indication – and agreed there was evidence of efficacy. Those voting against approval, however, thought that the indication was too broad and had concerns that included the small number of patients for a new indication (nr-axSpA), the lack of a clear definition of active disease in the indication, and the possibility that primary care physicians would prescribe the biologic drug for patients with low back pain.
Panelists supporting approval said that adequate prescribing information would result in appropriate use for patients with active disease in clinical practice.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, a panelist might be given a waiver, but not at this meeting.
Certolizumab, a TNF blocker, Crohn’s disease, rheumatoid arthritis (RA), Cimzia, UCB, subcutaneous injection, Dr. Janet Maynard, FDA’s Division of Pulmonary, Allergy, and Rheumatology Products,
Certolizumab, a TNF blocker, Crohn’s disease, rheumatoid arthritis (RA), Cimzia, UCB, subcutaneous injection, Dr. Janet Maynard, FDA’s Division of Pulmonary, Allergy, and Rheumatology Products,
AT THE FDA ADVISORY COMMITTEE MEETING
FDA panel votes against new AS indication for TNF-blocker
SILVER SPRING, MD. – Citing uncertainties about the efficacy data in a pivotal trial, the majority of a Food and Drug Administration advisory panel recommended against expanding the approval of adalimumab to include patients with axial spondyloarthritis who do not have radiographic evidence of ankylosing spondylitis.
At a July 23 meeting, the FDA’s Arthritis Advisory Committee voted 12-1, with 1 abstention, against recommending approval of adalimumab for the indication proposed by the manufacturer, AbbVie Inc.: reducing signs and symptoms in adults with active nonradiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation (elevated C-reactive protein or MRI findings) and who have had an inadequate response to, or are intolerant of nonsteroidal anti-inflammatory drugs.
Adalimumab is a monoclonal antibody against tumor necrosis factor (TNF) that was initially approved by the FDA in December 2002 as a treatment for rheumatoid arthritis in adults and was subsequently approved in 2006 for reducing the signs and symptoms of active ankylosing spondylitis (AS) in adults. It is also approved for psoriatic arthritis, juvenile idiopathic arthritis, Crohn’s disease, ulcerative colitis, and plaque psoriasis. It is administered by subcutaneous injection every other week.
Axial spondyloarthritis (axial SpA) includes AS and nr-axSpA; there are no FDA-approved treatments for people with nr-axSpA.
The majority of the panel agreed that no new safety signals were seen in the pivotal study and that the safety profile of adalimumab would not be expected to be different in patients with nr-axSpA and those with RA and other approved indications. But the panel voted that the manufacturer’s data did not provide substantial evidence that the drug resulted in a "clinically meaningful beneficial effect" for these patients, the efficacy criteria needed for approval.
The study compared adalimumab to placebo in 192 patients with active nr-axSpA, who had an inadequate response to NSAIDs, could not tolerate NSAIDs, or had a contraindication to NSAIDs. Patients were able to continue treatment with DMARDs, including sulfasalazine and methotrexate. The primary endpoint used to evaluate clinical responses was the ASAS (Assessment of SpondyloArthritis International Society) 40 response, which is defined as an improvement of at least 40% and absolute improvement of at least two units from baseline in at least three of four domains with no deterioration in the remaining domain (pain, function, morning stiffness, and Patient’s Global Assessment of Disease Activity).
At 12 weeks, 36% of those on adalimumab at a dose of 40 mg every other week had achieved an ASAS40 response, compared with 15% of those on placebo, a statistically significant difference. About 20% of those on adalimumab were in clinical remission at week 12, and efficacy was maintained or improved further among those who continued adalimumab in the open-label period through 68 weeks, according to the company. Adverse events were similar to those associated with the use of adalimumab in patients with AS, psoriatic arthritis, and rheumatoid arthritis.
But based on patient x-rays read at a central facility, a substantial proportion of patients in the trial had radiographic evidence of AS, an approved indication for adalimumab, which the FDA reviewer said may have affected the efficacy results. The FDA’s analysis of ASAS40 responses identified a markedly greater beneficial effect among those who had AS and had positive findings on x-rays, compared with those who had negative x-ray results and had nr-axSpA. In addition, the estimated effect of treatment in the nr-axSpA patients was described as "modest" by the FDA.
Panelists said that there is an unmet need for treatments in patients with this group, and encouraged the company to conduct another, better-designed study.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, a panelist might be given a waiver, but not at this meeting.
In July 2012, adalimumab was approved in the European Union for nr-axSpA.
SILVER SPRING, MD. – Citing uncertainties about the efficacy data in a pivotal trial, the majority of a Food and Drug Administration advisory panel recommended against expanding the approval of adalimumab to include patients with axial spondyloarthritis who do not have radiographic evidence of ankylosing spondylitis.
At a July 23 meeting, the FDA’s Arthritis Advisory Committee voted 12-1, with 1 abstention, against recommending approval of adalimumab for the indication proposed by the manufacturer, AbbVie Inc.: reducing signs and symptoms in adults with active nonradiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation (elevated C-reactive protein or MRI findings) and who have had an inadequate response to, or are intolerant of nonsteroidal anti-inflammatory drugs.
Adalimumab is a monoclonal antibody against tumor necrosis factor (TNF) that was initially approved by the FDA in December 2002 as a treatment for rheumatoid arthritis in adults and was subsequently approved in 2006 for reducing the signs and symptoms of active ankylosing spondylitis (AS) in adults. It is also approved for psoriatic arthritis, juvenile idiopathic arthritis, Crohn’s disease, ulcerative colitis, and plaque psoriasis. It is administered by subcutaneous injection every other week.
Axial spondyloarthritis (axial SpA) includes AS and nr-axSpA; there are no FDA-approved treatments for people with nr-axSpA.
The majority of the panel agreed that no new safety signals were seen in the pivotal study and that the safety profile of adalimumab would not be expected to be different in patients with nr-axSpA and those with RA and other approved indications. But the panel voted that the manufacturer’s data did not provide substantial evidence that the drug resulted in a "clinically meaningful beneficial effect" for these patients, the efficacy criteria needed for approval.
The study compared adalimumab to placebo in 192 patients with active nr-axSpA, who had an inadequate response to NSAIDs, could not tolerate NSAIDs, or had a contraindication to NSAIDs. Patients were able to continue treatment with DMARDs, including sulfasalazine and methotrexate. The primary endpoint used to evaluate clinical responses was the ASAS (Assessment of SpondyloArthritis International Society) 40 response, which is defined as an improvement of at least 40% and absolute improvement of at least two units from baseline in at least three of four domains with no deterioration in the remaining domain (pain, function, morning stiffness, and Patient’s Global Assessment of Disease Activity).
At 12 weeks, 36% of those on adalimumab at a dose of 40 mg every other week had achieved an ASAS40 response, compared with 15% of those on placebo, a statistically significant difference. About 20% of those on adalimumab were in clinical remission at week 12, and efficacy was maintained or improved further among those who continued adalimumab in the open-label period through 68 weeks, according to the company. Adverse events were similar to those associated with the use of adalimumab in patients with AS, psoriatic arthritis, and rheumatoid arthritis.
But based on patient x-rays read at a central facility, a substantial proportion of patients in the trial had radiographic evidence of AS, an approved indication for adalimumab, which the FDA reviewer said may have affected the efficacy results. The FDA’s analysis of ASAS40 responses identified a markedly greater beneficial effect among those who had AS and had positive findings on x-rays, compared with those who had negative x-ray results and had nr-axSpA. In addition, the estimated effect of treatment in the nr-axSpA patients was described as "modest" by the FDA.
Panelists said that there is an unmet need for treatments in patients with this group, and encouraged the company to conduct another, better-designed study.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, a panelist might be given a waiver, but not at this meeting.
In July 2012, adalimumab was approved in the European Union for nr-axSpA.
SILVER SPRING, MD. – Citing uncertainties about the efficacy data in a pivotal trial, the majority of a Food and Drug Administration advisory panel recommended against expanding the approval of adalimumab to include patients with axial spondyloarthritis who do not have radiographic evidence of ankylosing spondylitis.
At a July 23 meeting, the FDA’s Arthritis Advisory Committee voted 12-1, with 1 abstention, against recommending approval of adalimumab for the indication proposed by the manufacturer, AbbVie Inc.: reducing signs and symptoms in adults with active nonradiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation (elevated C-reactive protein or MRI findings) and who have had an inadequate response to, or are intolerant of nonsteroidal anti-inflammatory drugs.
Adalimumab is a monoclonal antibody against tumor necrosis factor (TNF) that was initially approved by the FDA in December 2002 as a treatment for rheumatoid arthritis in adults and was subsequently approved in 2006 for reducing the signs and symptoms of active ankylosing spondylitis (AS) in adults. It is also approved for psoriatic arthritis, juvenile idiopathic arthritis, Crohn’s disease, ulcerative colitis, and plaque psoriasis. It is administered by subcutaneous injection every other week.
Axial spondyloarthritis (axial SpA) includes AS and nr-axSpA; there are no FDA-approved treatments for people with nr-axSpA.
The majority of the panel agreed that no new safety signals were seen in the pivotal study and that the safety profile of adalimumab would not be expected to be different in patients with nr-axSpA and those with RA and other approved indications. But the panel voted that the manufacturer’s data did not provide substantial evidence that the drug resulted in a "clinically meaningful beneficial effect" for these patients, the efficacy criteria needed for approval.
The study compared adalimumab to placebo in 192 patients with active nr-axSpA, who had an inadequate response to NSAIDs, could not tolerate NSAIDs, or had a contraindication to NSAIDs. Patients were able to continue treatment with DMARDs, including sulfasalazine and methotrexate. The primary endpoint used to evaluate clinical responses was the ASAS (Assessment of SpondyloArthritis International Society) 40 response, which is defined as an improvement of at least 40% and absolute improvement of at least two units from baseline in at least three of four domains with no deterioration in the remaining domain (pain, function, morning stiffness, and Patient’s Global Assessment of Disease Activity).
At 12 weeks, 36% of those on adalimumab at a dose of 40 mg every other week had achieved an ASAS40 response, compared with 15% of those on placebo, a statistically significant difference. About 20% of those on adalimumab were in clinical remission at week 12, and efficacy was maintained or improved further among those who continued adalimumab in the open-label period through 68 weeks, according to the company. Adverse events were similar to those associated with the use of adalimumab in patients with AS, psoriatic arthritis, and rheumatoid arthritis.
But based on patient x-rays read at a central facility, a substantial proportion of patients in the trial had radiographic evidence of AS, an approved indication for adalimumab, which the FDA reviewer said may have affected the efficacy results. The FDA’s analysis of ASAS40 responses identified a markedly greater beneficial effect among those who had AS and had positive findings on x-rays, compared with those who had negative x-ray results and had nr-axSpA. In addition, the estimated effect of treatment in the nr-axSpA patients was described as "modest" by the FDA.
Panelists said that there is an unmet need for treatments in patients with this group, and encouraged the company to conduct another, better-designed study.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting; occasionally, a panelist might be given a waiver, but not at this meeting.
In July 2012, adalimumab was approved in the European Union for nr-axSpA.
AT AN FDA ADVISORY COMMITTEE MEETING