User login
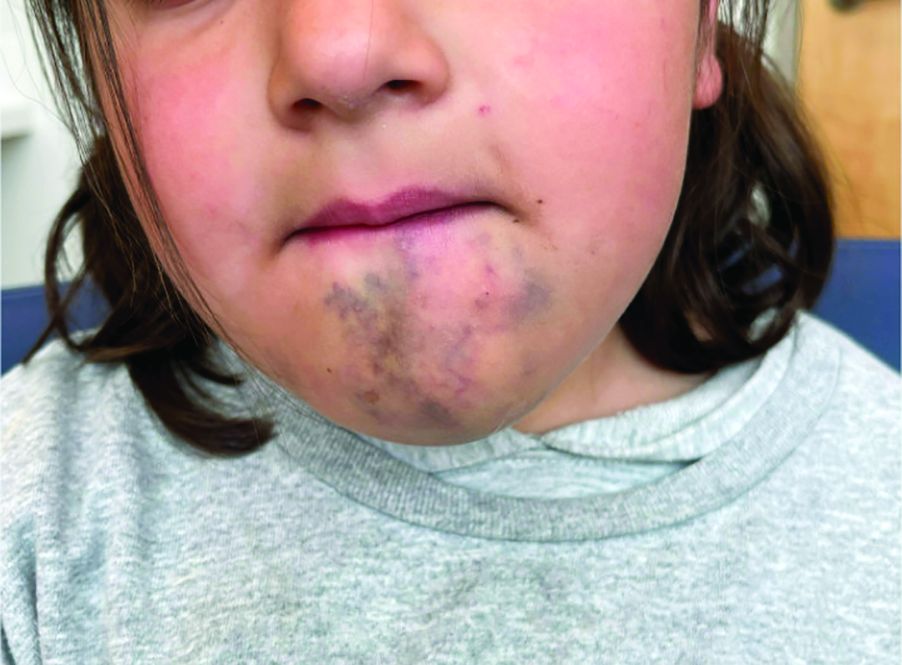
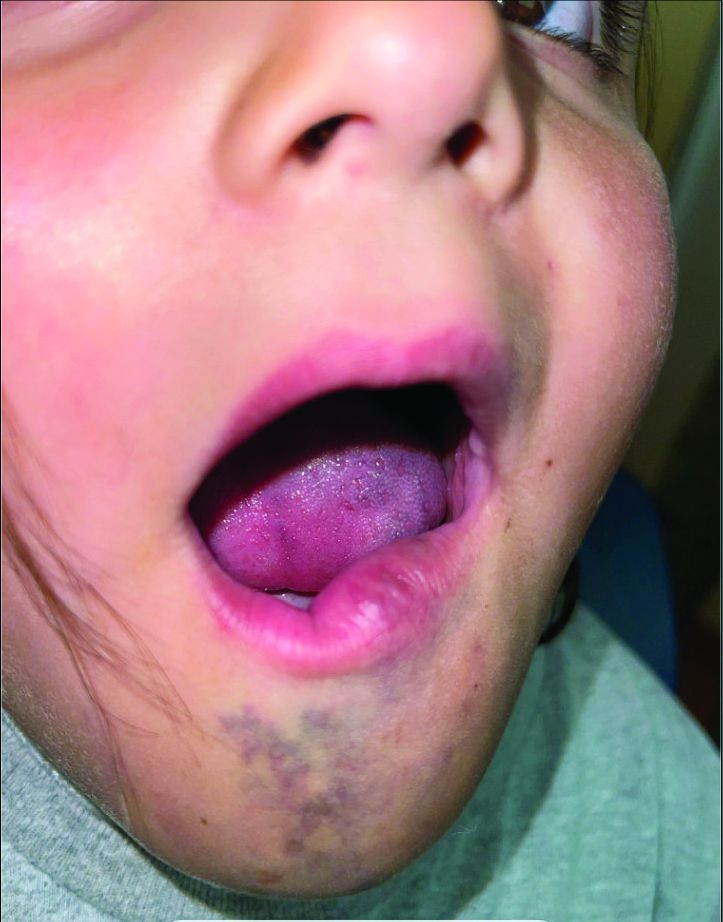
A 6-Year-Old Female Presents With a Bruise-Like Lesion on the Lip, Tongue, and Chin Area Present Since Birth
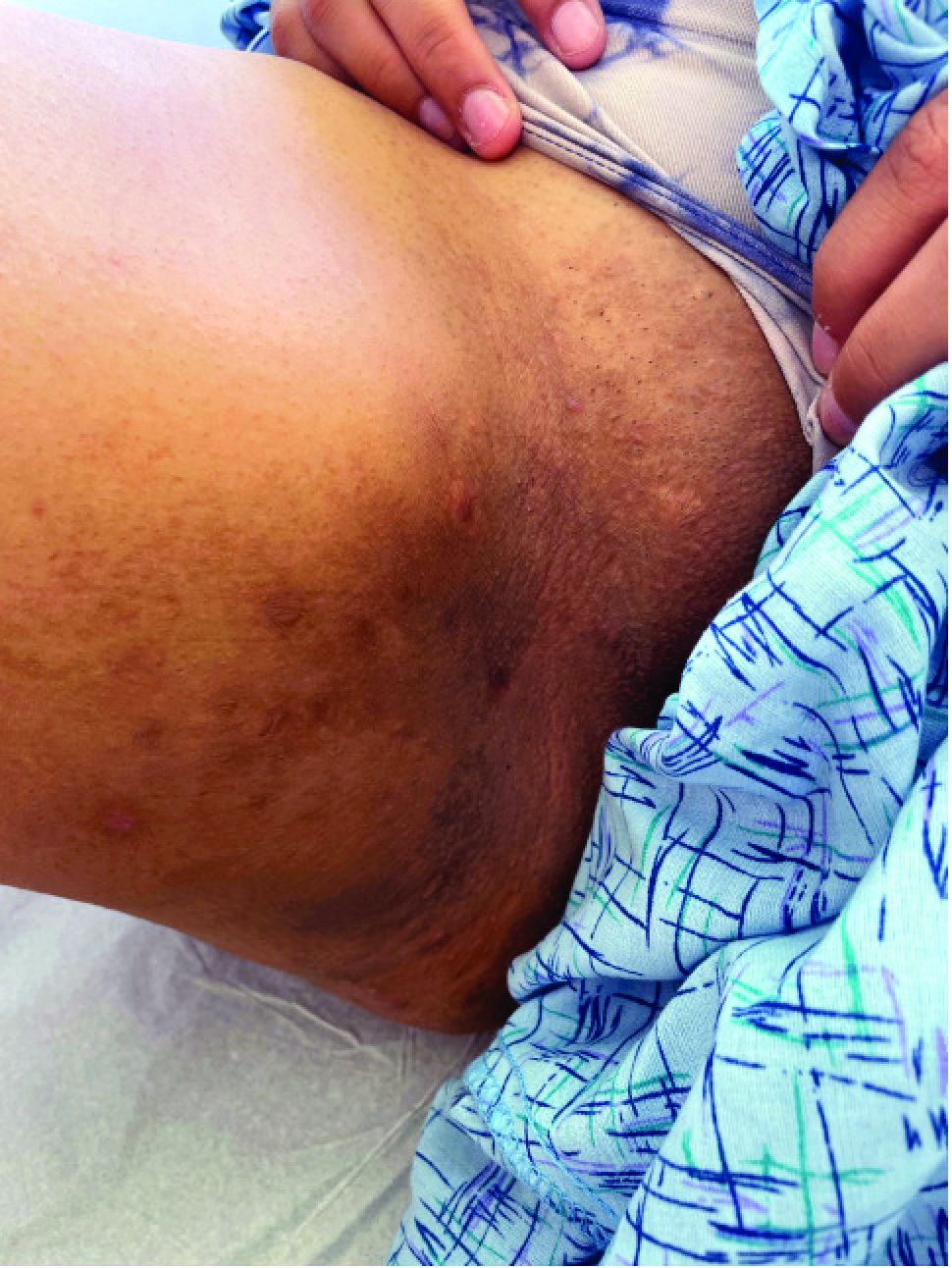
Diagnosis: Venous Malformation
Although present at birth, they are not always clinically evident early in life. They also tend to grow with the child without spontaneous regression, causing potential cosmetic concerns or complications from impingement on surrounding tissue.
Venous malformations appear with a bluish color appearing beneath the skin and can vary significantly in size and severity. Venous malformations are compressible and characterized by low to stagnant blood flow, which can spontaneously thrombose. Clinically, this may cause pain, swelling, skin changes, tissue and limb overgrowth, or functional impairment depending on location and size.
Venous malformations result from disorganized angiogenesis secondary to sporadic mutations in somatic cells. The most common implicated gene is TEK, a receptor tyrosine kinase. PIK3CA has also been involved. Both genes are involved in the PI3K/AKT/mTOR pathway, which regulates cell growth, proliferation, and angiogenesis. In venous endothelial cells, abnormal angiogenesis and vessel maturation may lead to venous malformation formation. Dysplastic vessels frequently separate from normal veins but may be contiguous with the deep venous system.
Diagnosis involves clinical history and physical examination. Imaging with ultrasound and magnetic resonance imaging (MRI) may be utilized. While ultrasound may be preferred for superficial venous malformations, MRI or MRI with MR angiography (MRA) is the preferred method for venous malformation assessment. Genetic testing may be appropriate for complex malformations, as classification of lesions by underlying mutation may allow targeted therapy.
This patient’s past MRI and MRA findings were consistent with a venous malformation.
Treatment
Venous malformations rarely regress spontaneously. Treatment is required if venous malformations are symptomatic, which may include pain, swelling, deformity, thrombosis, or interference with daily activities of living. Treatment plans require consideration of patient goals of care. The main categories of therapy are embolization/sclerotherapy, surgical resection, and molecular targeted therapy.

Sclerotherapy is a well-tolerated and efficacious first-line therapy. It can be used as either nonsurgical curative therapy or preoperative adjunct therapy to minimize blood loss before surgical resection. While surgical resection may cause scarring, multimodal approaches with sclerotherapy or laser therapy can decrease complications. Molecular therapies aim to reduce vascular proliferation and symptoms. Referral to hematology/oncology for evaluation and consideration of chemotherapeutic agents may be required. Sirolimus has been shown in mice models to inhibit an endothelial cell tyrosine kinase receptor that plays a role in venous malformation growth. Multiple studies have proved its efficacy in managing complicated vascular anomalies, including venous malformations. Alpelisib is an inhibitor of PI3KCA, which is part of the pathway that contributes to venous malformation formation. Dactolisib, a dual inhibitor of the PI3KA and mTOR pathways, and rebastinib, a TEK inhibitor, are being investigated.

Differential Diagnoses
The differential diagnosis includes dermal melanocytosis, nevus of Ota, hemangioma of infancy, and ashy dermatosis. In addition, venous malformations can be part of more complex vascular malformations.
Dermal melanocytosis, also known as Mongolian spots, are blue-gray patches of discoloration on the skin that appear at birth or shortly after. They result from the arrest of dermal melanocytes in the dermis during fetal life and tissue modeling. They are commonly observed in those of Asian or African descent with darker skin types. Most often, they are located in the lumbar or sacral-gluteal region. Unlike venous malformations, they are benign and do not involve vascular abnormalities. They typically fade over time.

Nevus of Ota is a benign congenital condition that presents with blue-gray or brown patches of pigmentation on the skin around the eyes, cheeks, and forehead. They are dermal melanocytes with a speckled instead of uniform appearance. Nevus of Ota primarily affects individuals of Asian descent and typically presents in the trigeminal nerve distribution region. Treatment can be done to minimize deformity, generally with pigmented laser surgery.
Hemangiomas of infancy are common benign tumors of infancy caused by endothelial cell proliferation. They are characterized by rapid growth followed by spontaneous involution within the first year of life and for several years. Hemangiomas can be superficial, deep, or mixed with features of both superficial and deep. Superficial hemangiomas present as raised, lobulated, and bright red while deep hemangiomas present as a bluish-hued nodule, plaque, or tumor. They are diagnosed clinically but skin biopsies and imaging can confirm the suspected diagnosis. While hemangiomas may self-resolve, complicated hemangiomas can be treated with topical timolol, oral propranolol, topical and intralesional corticosteroids, pulsed-dye laser, and surgical resection.
Ashy dermatosis is a term for asymptomatic, gray-blue or ashy patches distributed symmetrically on the trunk, head, neck, and upper extremities. It primarily affects individuals with darker skin types (Fitzpatrick III-V), and is more common in patients with Hispanic, Asian, or African backgrounds. The direct cause of ashy dermatosis is unknown but it is thought to be linked to drug ingestion, genetics, infection, and immune-mediated mechanisms. The general treatment includes topical corticosteroids, clofazimine, topical calcineurin inhibitors, oral dapsone, phototherapy, topical retinoids, or isotretinoin to reduce inflammation and pigmentation.
Danny Lee and Samuel Le serve as research fellows and Jolina Bui as research associate in the Pediatric Dermatology Division of the Department of Dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is Distinguished Professor of Dermatology and Pediatrics and Vice-Chair of the Department of Dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. The authors have no relevant financial disclosures.
Suggested Reading
Agarwal P, Patel BC. Nevus of Ota and Ito. [Updated 2023 Jul 10]. In: StatPearls [Internet]. StatPearls Publishing; 2024.
Dompmartin A et al. The VASCERN-VASCA Working Group Diagnostic and Management Pathways for Venous Malformations. J Vasc Anom (Phila). 2023 Mar 23;4(2):e064.
Dompmartin A et al. Venous malformation: Update on aetiopathogenesis, diagnosis and management. Phlebology. 2010 Oct;25(5):224-235.
Gupta D, Thappa DM. Mongolian spots. Indian J Dermatol Venereol Leprol. 2013 Jul-Aug;79(4):469-478.
Krowchuk DP et al. Clinical Practice Guideline for the Management of Infantile Hemangiomas. Pediatrics. 2019 Jan;143(1):e20183475.
Nguyen K, Khachemoune A. Ashy dermatosis: A review. Dermatol Online J. 2019 May 15;25(5):13030/qt44f462s8.
Patel ND, Chong AT et al. Venous Malformations. Semin Intervent Radiol. 2022 Dec 20;39(5):498-507.
Diagnosis: Venous Malformation
Although present at birth, they are not always clinically evident early in life. They also tend to grow with the child without spontaneous regression, causing potential cosmetic concerns or complications from impingement on surrounding tissue.
Venous malformations appear with a bluish color appearing beneath the skin and can vary significantly in size and severity. Venous malformations are compressible and characterized by low to stagnant blood flow, which can spontaneously thrombose. Clinically, this may cause pain, swelling, skin changes, tissue and limb overgrowth, or functional impairment depending on location and size.
Venous malformations result from disorganized angiogenesis secondary to sporadic mutations in somatic cells. The most common implicated gene is TEK, a receptor tyrosine kinase. PIK3CA has also been involved. Both genes are involved in the PI3K/AKT/mTOR pathway, which regulates cell growth, proliferation, and angiogenesis. In venous endothelial cells, abnormal angiogenesis and vessel maturation may lead to venous malformation formation. Dysplastic vessels frequently separate from normal veins but may be contiguous with the deep venous system.
Diagnosis involves clinical history and physical examination. Imaging with ultrasound and magnetic resonance imaging (MRI) may be utilized. While ultrasound may be preferred for superficial venous malformations, MRI or MRI with MR angiography (MRA) is the preferred method for venous malformation assessment. Genetic testing may be appropriate for complex malformations, as classification of lesions by underlying mutation may allow targeted therapy.
This patient’s past MRI and MRA findings were consistent with a venous malformation.
Treatment
Venous malformations rarely regress spontaneously. Treatment is required if venous malformations are symptomatic, which may include pain, swelling, deformity, thrombosis, or interference with daily activities of living. Treatment plans require consideration of patient goals of care. The main categories of therapy are embolization/sclerotherapy, surgical resection, and molecular targeted therapy.
Sclerotherapy is a well-tolerated and efficacious first-line therapy. It can be used as either nonsurgical curative therapy or preoperative adjunct therapy to minimize blood loss before surgical resection. While surgical resection may cause scarring, multimodal approaches with sclerotherapy or laser therapy can decrease complications. Molecular therapies aim to reduce vascular proliferation and symptoms. Referral to hematology/oncology for evaluation and consideration of chemotherapeutic agents may be required. Sirolimus has been shown in mice models to inhibit an endothelial cell tyrosine kinase receptor that plays a role in venous malformation growth. Multiple studies have proved its efficacy in managing complicated vascular anomalies, including venous malformations. Alpelisib is an inhibitor of PI3KCA, which is part of the pathway that contributes to venous malformation formation. Dactolisib, a dual inhibitor of the PI3KA and mTOR pathways, and rebastinib, a TEK inhibitor, are being investigated.
Differential Diagnoses
The differential diagnosis includes dermal melanocytosis, nevus of Ota, hemangioma of infancy, and ashy dermatosis. In addition, venous malformations can be part of more complex vascular malformations.
Dermal melanocytosis, also known as Mongolian spots, are blue-gray patches of discoloration on the skin that appear at birth or shortly after. They result from the arrest of dermal melanocytes in the dermis during fetal life and tissue modeling. They are commonly observed in those of Asian or African descent with darker skin types. Most often, they are located in the lumbar or sacral-gluteal region. Unlike venous malformations, they are benign and do not involve vascular abnormalities. They typically fade over time.
Nevus of Ota is a benign congenital condition that presents with blue-gray or brown patches of pigmentation on the skin around the eyes, cheeks, and forehead. They are dermal melanocytes with a speckled instead of uniform appearance. Nevus of Ota primarily affects individuals of Asian descent and typically presents in the trigeminal nerve distribution region. Treatment can be done to minimize deformity, generally with pigmented laser surgery.
Hemangiomas of infancy are common benign tumors of infancy caused by endothelial cell proliferation. They are characterized by rapid growth followed by spontaneous involution within the first year of life and for several years. Hemangiomas can be superficial, deep, or mixed with features of both superficial and deep. Superficial hemangiomas present as raised, lobulated, and bright red while deep hemangiomas present as a bluish-hued nodule, plaque, or tumor. They are diagnosed clinically but skin biopsies and imaging can confirm the suspected diagnosis. While hemangiomas may self-resolve, complicated hemangiomas can be treated with topical timolol, oral propranolol, topical and intralesional corticosteroids, pulsed-dye laser, and surgical resection.
Ashy dermatosis is a term for asymptomatic, gray-blue or ashy patches distributed symmetrically on the trunk, head, neck, and upper extremities. It primarily affects individuals with darker skin types (Fitzpatrick III-V), and is more common in patients with Hispanic, Asian, or African backgrounds. The direct cause of ashy dermatosis is unknown but it is thought to be linked to drug ingestion, genetics, infection, and immune-mediated mechanisms. The general treatment includes topical corticosteroids, clofazimine, topical calcineurin inhibitors, oral dapsone, phototherapy, topical retinoids, or isotretinoin to reduce inflammation and pigmentation.
Danny Lee and Samuel Le serve as research fellows and Jolina Bui as research associate in the Pediatric Dermatology Division of the Department of Dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is Distinguished Professor of Dermatology and Pediatrics and Vice-Chair of the Department of Dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. The authors have no relevant financial disclosures.
Suggested Reading
Agarwal P, Patel BC. Nevus of Ota and Ito. [Updated 2023 Jul 10]. In: StatPearls [Internet]. StatPearls Publishing; 2024.
Dompmartin A et al. The VASCERN-VASCA Working Group Diagnostic and Management Pathways for Venous Malformations. J Vasc Anom (Phila). 2023 Mar 23;4(2):e064.
Dompmartin A et al. Venous malformation: Update on aetiopathogenesis, diagnosis and management. Phlebology. 2010 Oct;25(5):224-235.
Gupta D, Thappa DM. Mongolian spots. Indian J Dermatol Venereol Leprol. 2013 Jul-Aug;79(4):469-478.
Krowchuk DP et al. Clinical Practice Guideline for the Management of Infantile Hemangiomas. Pediatrics. 2019 Jan;143(1):e20183475.
Nguyen K, Khachemoune A. Ashy dermatosis: A review. Dermatol Online J. 2019 May 15;25(5):13030/qt44f462s8.
Patel ND, Chong AT et al. Venous Malformations. Semin Intervent Radiol. 2022 Dec 20;39(5):498-507.
Diagnosis: Venous Malformation
Although present at birth, they are not always clinically evident early in life. They also tend to grow with the child without spontaneous regression, causing potential cosmetic concerns or complications from impingement on surrounding tissue.
Venous malformations appear with a bluish color appearing beneath the skin and can vary significantly in size and severity. Venous malformations are compressible and characterized by low to stagnant blood flow, which can spontaneously thrombose. Clinically, this may cause pain, swelling, skin changes, tissue and limb overgrowth, or functional impairment depending on location and size.
Venous malformations result from disorganized angiogenesis secondary to sporadic mutations in somatic cells. The most common implicated gene is TEK, a receptor tyrosine kinase. PIK3CA has also been involved. Both genes are involved in the PI3K/AKT/mTOR pathway, which regulates cell growth, proliferation, and angiogenesis. In venous endothelial cells, abnormal angiogenesis and vessel maturation may lead to venous malformation formation. Dysplastic vessels frequently separate from normal veins but may be contiguous with the deep venous system.
Diagnosis involves clinical history and physical examination. Imaging with ultrasound and magnetic resonance imaging (MRI) may be utilized. While ultrasound may be preferred for superficial venous malformations, MRI or MRI with MR angiography (MRA) is the preferred method for venous malformation assessment. Genetic testing may be appropriate for complex malformations, as classification of lesions by underlying mutation may allow targeted therapy.
This patient’s past MRI and MRA findings were consistent with a venous malformation.
Treatment
Venous malformations rarely regress spontaneously. Treatment is required if venous malformations are symptomatic, which may include pain, swelling, deformity, thrombosis, or interference with daily activities of living. Treatment plans require consideration of patient goals of care. The main categories of therapy are embolization/sclerotherapy, surgical resection, and molecular targeted therapy.
Sclerotherapy is a well-tolerated and efficacious first-line therapy. It can be used as either nonsurgical curative therapy or preoperative adjunct therapy to minimize blood loss before surgical resection. While surgical resection may cause scarring, multimodal approaches with sclerotherapy or laser therapy can decrease complications. Molecular therapies aim to reduce vascular proliferation and symptoms. Referral to hematology/oncology for evaluation and consideration of chemotherapeutic agents may be required. Sirolimus has been shown in mice models to inhibit an endothelial cell tyrosine kinase receptor that plays a role in venous malformation growth. Multiple studies have proved its efficacy in managing complicated vascular anomalies, including venous malformations. Alpelisib is an inhibitor of PI3KCA, which is part of the pathway that contributes to venous malformation formation. Dactolisib, a dual inhibitor of the PI3KA and mTOR pathways, and rebastinib, a TEK inhibitor, are being investigated.
Differential Diagnoses
The differential diagnosis includes dermal melanocytosis, nevus of Ota, hemangioma of infancy, and ashy dermatosis. In addition, venous malformations can be part of more complex vascular malformations.
Dermal melanocytosis, also known as Mongolian spots, are blue-gray patches of discoloration on the skin that appear at birth or shortly after. They result from the arrest of dermal melanocytes in the dermis during fetal life and tissue modeling. They are commonly observed in those of Asian or African descent with darker skin types. Most often, they are located in the lumbar or sacral-gluteal region. Unlike venous malformations, they are benign and do not involve vascular abnormalities. They typically fade over time.
Nevus of Ota is a benign congenital condition that presents with blue-gray or brown patches of pigmentation on the skin around the eyes, cheeks, and forehead. They are dermal melanocytes with a speckled instead of uniform appearance. Nevus of Ota primarily affects individuals of Asian descent and typically presents in the trigeminal nerve distribution region. Treatment can be done to minimize deformity, generally with pigmented laser surgery.
Hemangiomas of infancy are common benign tumors of infancy caused by endothelial cell proliferation. They are characterized by rapid growth followed by spontaneous involution within the first year of life and for several years. Hemangiomas can be superficial, deep, or mixed with features of both superficial and deep. Superficial hemangiomas present as raised, lobulated, and bright red while deep hemangiomas present as a bluish-hued nodule, plaque, or tumor. They are diagnosed clinically but skin biopsies and imaging can confirm the suspected diagnosis. While hemangiomas may self-resolve, complicated hemangiomas can be treated with topical timolol, oral propranolol, topical and intralesional corticosteroids, pulsed-dye laser, and surgical resection.
Ashy dermatosis is a term for asymptomatic, gray-blue or ashy patches distributed symmetrically on the trunk, head, neck, and upper extremities. It primarily affects individuals with darker skin types (Fitzpatrick III-V), and is more common in patients with Hispanic, Asian, or African backgrounds. The direct cause of ashy dermatosis is unknown but it is thought to be linked to drug ingestion, genetics, infection, and immune-mediated mechanisms. The general treatment includes topical corticosteroids, clofazimine, topical calcineurin inhibitors, oral dapsone, phototherapy, topical retinoids, or isotretinoin to reduce inflammation and pigmentation.
Danny Lee and Samuel Le serve as research fellows and Jolina Bui as research associate in the Pediatric Dermatology Division of the Department of Dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is Distinguished Professor of Dermatology and Pediatrics and Vice-Chair of the Department of Dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. The authors have no relevant financial disclosures.
Suggested Reading
Agarwal P, Patel BC. Nevus of Ota and Ito. [Updated 2023 Jul 10]. In: StatPearls [Internet]. StatPearls Publishing; 2024.
Dompmartin A et al. The VASCERN-VASCA Working Group Diagnostic and Management Pathways for Venous Malformations. J Vasc Anom (Phila). 2023 Mar 23;4(2):e064.
Dompmartin A et al. Venous malformation: Update on aetiopathogenesis, diagnosis and management. Phlebology. 2010 Oct;25(5):224-235.
Gupta D, Thappa DM. Mongolian spots. Indian J Dermatol Venereol Leprol. 2013 Jul-Aug;79(4):469-478.
Krowchuk DP et al. Clinical Practice Guideline for the Management of Infantile Hemangiomas. Pediatrics. 2019 Jan;143(1):e20183475.
Nguyen K, Khachemoune A. Ashy dermatosis: A review. Dermatol Online J. 2019 May 15;25(5):13030/qt44f462s8.
Patel ND, Chong AT et al. Venous Malformations. Semin Intervent Radiol. 2022 Dec 20;39(5):498-507.
A 6-year-old girl presents with a bruise-like lesion on the lip, tongue, and chin area present since birth. The family states that her tongue has been increasing in size and is painful. On physical exam, she presents with left lower mucosal lip fullness and an overlying violaceous hue extending into the oral mucosa and onto the left tongue. The left portion of the dorsal tongue displays an increased thickness and bluish discoloration and there is a pink, smooth papule on the left anterolateral tongue.
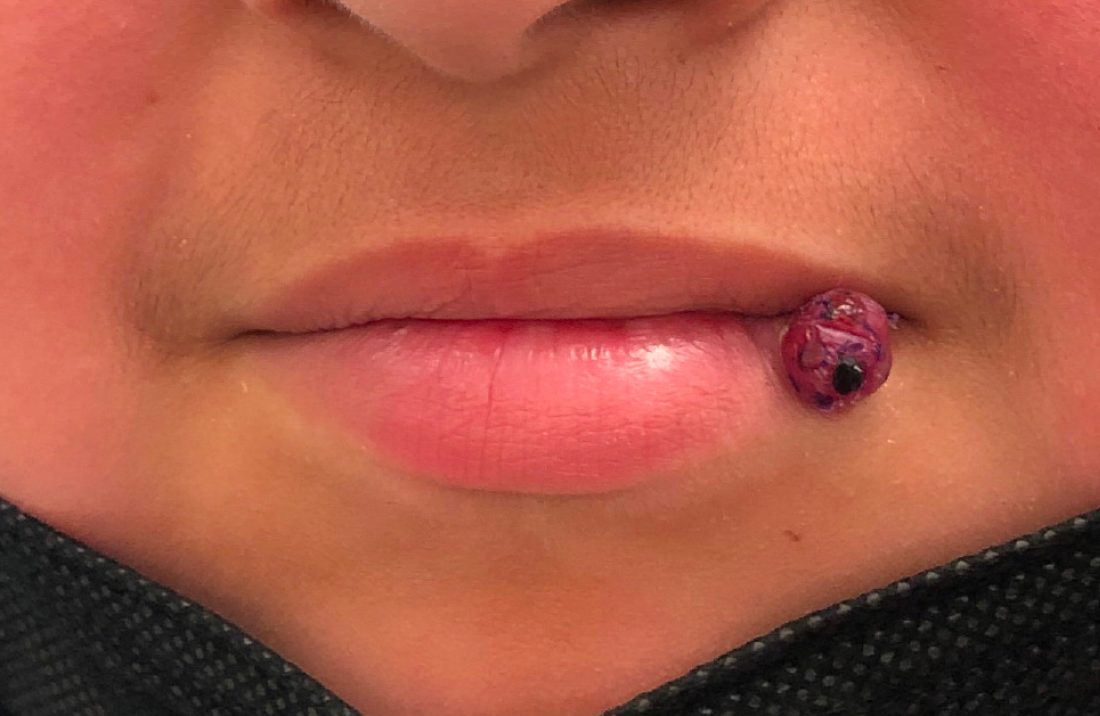
A 10-year-old with a red bump on her lower lip
The patient’s history and examination are consistent with a diagnosis of pyogenic granuloma. Specifically, the history of rapid growth, friable nature, associated bleeding, and hemorrhagic crusting point to pyogenic granuloma as the most likely diagnosis.
Pyogenic granuloma is an acquired benign vascular growth of the skin or mucous membranes.1 It most frequently occurs in children and young adults and most commonly affects the skin of the head, trunk, and extremities.2 Common mucosal sites include the gingiva, lips, and tongue.2 The etiology of pyogenic granuloma is unknown, though it is thought to be a process akin to the overgrowth of granulation tissue.3,4 Expression of angiogenic factors and subsequent vascular hyperplasia are also implicated as key players in the pathogenesis of pyogenic granuloma.1,4 In addition, several associated factors and inciting triggers have been proposed including trauma, infections, and hormonal fluctuations.3-5 However, the majority of patients do not report predisposing factors or a history of prior trauma at the site.3,6

Clinically, pyogenic granuloma usually presents as a painless, erythematous, dome-shaped friable papule or nodule that easily bleeds and may ulcerate. It typically undergoes a period of growth over weeks to months followed by stabilization. Occasionally, pyogenic granulomas will spontaneously involute, though most do not.7 Pyogenic granuloma may occur within an existing capillary malformation, such as a port wine stain, spontaneously or as a sequela of laser treatment.8,9 Diagnosis of pyogenic granuloma can typically be made clinically on the basis of history and exam. Dermoscopic evaluation of pyogenic granuloma will reveal a homogeneous papule with a surrounding white-brown collarette, and potentially white intersecting lines.10 Histopathologic evaluation may be necessary to differentiate lesions from conditions that may mimic pyogenic granuloma.
What’s on the differential?
The differential diagnosis for pyogenic granuloma consists of Spitz nevus, cherry hemangioma, amelanotic melanoma, and glomus tumor.
Spitz nevus
Spitz nevus (spindle and epithelial cell nevus) is a benign melanocytic lesion that classically appears as a sharply circumscribed, smooth, dome-shaped, pink-red, or brown papule or plaque. There is typically a history of rapid growth over several months followed by stabilization. It usually presents in childhood or adolescence and is most commonly located on the face and extremities. While there are similarities in the appearance of Spitz nevi and pyogenic granuloma, Spitz nevi are not usually friable nor associated with bleeding as in our patient. Furthermore, on dermoscopy, Spitz nevus typically exhibits a starburst pattern with regularly distributed dotted vessels, or a peripheral globular pattern with reticular depigmentation. The definitive diagnosis of Spitz nevi relies on histopathologic evaluation, which is critical for discriminating Spitz nevi from melanoma.
Cherry hemangioma
Cherry angiomas are the most common type of acquired benign vascular proliferation. They present as small, bright red or violaceous macules or papules. However, they typically appear in early to midadulthood and increase in number with age. The age of our patient and solitary presentation of the lesion make this diagnosis unlikely. In addition, cherry angiomas are not usually associated with bleeding. It is important to note that, depending on the age of the patient, pyogenic granuloma may also be confused with infantile hemangioma. Infantile hemangiomas may become bright red papules, nodules, or plaques that appear in early infancy. They characteristically involute, which does not typically happen with pyogenic granuloma.
Amelanotic melanoma
Amelanotic melanoma is an uncommon variant of melanoma with little to no pigmentation. It may appear as a skin-colored to light-brown, pink, or red macule, papule, or nodule. The lesion may be asymmetric with irregular and well-defined borders. The variable and uncharacteristic appearance of this melanoma variant makes it diagnostically challenging and it is often confused with benign lesions including pyogenic granuloma. Dermoscopy can help distinguish amelanotic melanoma from other benign conditions, and will reveal areas of pink to white, polymorphous vessels and crystalline structures. However, ultimately biopsy and histopathological evaluation is necessary for accurate diagnosis.
Glomus tumor
Glomus tumors are rare, benign neoplasms originating from cells of the glomus body that presents as a red-purple, vascular papule or nodule. They are usually found in areas rich in glomus bodies, such as the subungual regions, fingertips, palms, wrists, and forearms. Glomus tumors are typically associated with tenderness, paroxysmal pain, and cold sensitivity. They do not bleed or ulcerate. While pyogenic granuloma may be confused for glomus tumor when present on the fingers or extremities, the location of the lesion in our patient is not consistent with a diagnosis of glomus tumor.
Management and disease course
Management with procedural or topical interventions is usually pursued for pyogenic granuloma because of frequent bleeding and ulceration of lesions. The most common approach is simple excision by a scoop or shave technique, with or without curettage and most commonly with electrocautery of the base. Other options include full-thickness excision, destruction with laser therapy, cryotherapy, or topical treatments (for example, timolol).11 Lesion recurrence can occur with both surgical and nonsurgical management.11 Regardless of management technique, it is useful to obtain histopathologic evaluation of tissue for accurate diagnosis.
Our patient underwent surgical destruction of her lower-lip lesion with shave excision followed by electrocautery. The surgical specimen was sent for pathology, which confirmed the diagnosis of pyogenic granuloma. The patient experienced no complications from the procedure and did not have recurrence of the lesion.
Ms. Sui is a research associate in the department of dermatology, division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Ms. Sui nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Lin RL and Janniger CK. Cutis. 2004 Oct;74(4):229-33.
2. Harris MN et al. J Am Acad Dermatol. 2000 Jun;42(6):1012-6.
3. Pagliai KA and Cohen BA. Pediatr Dermatol. 2004 Jan-Feb;21(1):10-3.
4. Kamal R et al. J Oral Maxillofac Pathol. 2012 Jan;16(1):79-82.
5. Requena L and Sangueza OP. J Am Acad Dermatol. 1997 Dec;37(6):887-919.
6. Patrice SJ et al. Pediatr Dermatol. 1991 Dec;8(4):267-76.
7. Luba MC et al. Am Fam Physician. 2003 Feb 15;67(4):729-38.
8. Swerlick RA and Cooper PH. J Am Acad Dermatol. 1983 May;8(5):627-30.
9. Sheehan DJ and Lesher JL Jr. Cutis. 2004 Mar;73(3):175-80.
10. Zaballos P et al. Br J Dermatol. 2006 Jun;154(6):1108-11.
11. Lee J et al. J Plast Reconstr Aesthet Surg. 2011 Sep;64(9):1216-20. .
The patient’s history and examination are consistent with a diagnosis of pyogenic granuloma. Specifically, the history of rapid growth, friable nature, associated bleeding, and hemorrhagic crusting point to pyogenic granuloma as the most likely diagnosis.
Pyogenic granuloma is an acquired benign vascular growth of the skin or mucous membranes.1 It most frequently occurs in children and young adults and most commonly affects the skin of the head, trunk, and extremities.2 Common mucosal sites include the gingiva, lips, and tongue.2 The etiology of pyogenic granuloma is unknown, though it is thought to be a process akin to the overgrowth of granulation tissue.3,4 Expression of angiogenic factors and subsequent vascular hyperplasia are also implicated as key players in the pathogenesis of pyogenic granuloma.1,4 In addition, several associated factors and inciting triggers have been proposed including trauma, infections, and hormonal fluctuations.3-5 However, the majority of patients do not report predisposing factors or a history of prior trauma at the site.3,6
Clinically, pyogenic granuloma usually presents as a painless, erythematous, dome-shaped friable papule or nodule that easily bleeds and may ulcerate. It typically undergoes a period of growth over weeks to months followed by stabilization. Occasionally, pyogenic granulomas will spontaneously involute, though most do not.7 Pyogenic granuloma may occur within an existing capillary malformation, such as a port wine stain, spontaneously or as a sequela of laser treatment.8,9 Diagnosis of pyogenic granuloma can typically be made clinically on the basis of history and exam. Dermoscopic evaluation of pyogenic granuloma will reveal a homogeneous papule with a surrounding white-brown collarette, and potentially white intersecting lines.10 Histopathologic evaluation may be necessary to differentiate lesions from conditions that may mimic pyogenic granuloma.
What’s on the differential?
The differential diagnosis for pyogenic granuloma consists of Spitz nevus, cherry hemangioma, amelanotic melanoma, and glomus tumor.
Spitz nevus
Spitz nevus (spindle and epithelial cell nevus) is a benign melanocytic lesion that classically appears as a sharply circumscribed, smooth, dome-shaped, pink-red, or brown papule or plaque. There is typically a history of rapid growth over several months followed by stabilization. It usually presents in childhood or adolescence and is most commonly located on the face and extremities. While there are similarities in the appearance of Spitz nevi and pyogenic granuloma, Spitz nevi are not usually friable nor associated with bleeding as in our patient. Furthermore, on dermoscopy, Spitz nevus typically exhibits a starburst pattern with regularly distributed dotted vessels, or a peripheral globular pattern with reticular depigmentation. The definitive diagnosis of Spitz nevi relies on histopathologic evaluation, which is critical for discriminating Spitz nevi from melanoma.
Cherry hemangioma
Cherry angiomas are the most common type of acquired benign vascular proliferation. They present as small, bright red or violaceous macules or papules. However, they typically appear in early to midadulthood and increase in number with age. The age of our patient and solitary presentation of the lesion make this diagnosis unlikely. In addition, cherry angiomas are not usually associated with bleeding. It is important to note that, depending on the age of the patient, pyogenic granuloma may also be confused with infantile hemangioma. Infantile hemangiomas may become bright red papules, nodules, or plaques that appear in early infancy. They characteristically involute, which does not typically happen with pyogenic granuloma.
Amelanotic melanoma
Amelanotic melanoma is an uncommon variant of melanoma with little to no pigmentation. It may appear as a skin-colored to light-brown, pink, or red macule, papule, or nodule. The lesion may be asymmetric with irregular and well-defined borders. The variable and uncharacteristic appearance of this melanoma variant makes it diagnostically challenging and it is often confused with benign lesions including pyogenic granuloma. Dermoscopy can help distinguish amelanotic melanoma from other benign conditions, and will reveal areas of pink to white, polymorphous vessels and crystalline structures. However, ultimately biopsy and histopathological evaluation is necessary for accurate diagnosis.
Glomus tumor
Glomus tumors are rare, benign neoplasms originating from cells of the glomus body that presents as a red-purple, vascular papule or nodule. They are usually found in areas rich in glomus bodies, such as the subungual regions, fingertips, palms, wrists, and forearms. Glomus tumors are typically associated with tenderness, paroxysmal pain, and cold sensitivity. They do not bleed or ulcerate. While pyogenic granuloma may be confused for glomus tumor when present on the fingers or extremities, the location of the lesion in our patient is not consistent with a diagnosis of glomus tumor.
Management and disease course
Management with procedural or topical interventions is usually pursued for pyogenic granuloma because of frequent bleeding and ulceration of lesions. The most common approach is simple excision by a scoop or shave technique, with or without curettage and most commonly with electrocautery of the base. Other options include full-thickness excision, destruction with laser therapy, cryotherapy, or topical treatments (for example, timolol).11 Lesion recurrence can occur with both surgical and nonsurgical management.11 Regardless of management technique, it is useful to obtain histopathologic evaluation of tissue for accurate diagnosis.
Our patient underwent surgical destruction of her lower-lip lesion with shave excision followed by electrocautery. The surgical specimen was sent for pathology, which confirmed the diagnosis of pyogenic granuloma. The patient experienced no complications from the procedure and did not have recurrence of the lesion.
Ms. Sui is a research associate in the department of dermatology, division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Ms. Sui nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Lin RL and Janniger CK. Cutis. 2004 Oct;74(4):229-33.
2. Harris MN et al. J Am Acad Dermatol. 2000 Jun;42(6):1012-6.
3. Pagliai KA and Cohen BA. Pediatr Dermatol. 2004 Jan-Feb;21(1):10-3.
4. Kamal R et al. J Oral Maxillofac Pathol. 2012 Jan;16(1):79-82.
5. Requena L and Sangueza OP. J Am Acad Dermatol. 1997 Dec;37(6):887-919.
6. Patrice SJ et al. Pediatr Dermatol. 1991 Dec;8(4):267-76.
7. Luba MC et al. Am Fam Physician. 2003 Feb 15;67(4):729-38.
8. Swerlick RA and Cooper PH. J Am Acad Dermatol. 1983 May;8(5):627-30.
9. Sheehan DJ and Lesher JL Jr. Cutis. 2004 Mar;73(3):175-80.
10. Zaballos P et al. Br J Dermatol. 2006 Jun;154(6):1108-11.
11. Lee J et al. J Plast Reconstr Aesthet Surg. 2011 Sep;64(9):1216-20. .
The patient’s history and examination are consistent with a diagnosis of pyogenic granuloma. Specifically, the history of rapid growth, friable nature, associated bleeding, and hemorrhagic crusting point to pyogenic granuloma as the most likely diagnosis.
Pyogenic granuloma is an acquired benign vascular growth of the skin or mucous membranes.1 It most frequently occurs in children and young adults and most commonly affects the skin of the head, trunk, and extremities.2 Common mucosal sites include the gingiva, lips, and tongue.2 The etiology of pyogenic granuloma is unknown, though it is thought to be a process akin to the overgrowth of granulation tissue.3,4 Expression of angiogenic factors and subsequent vascular hyperplasia are also implicated as key players in the pathogenesis of pyogenic granuloma.1,4 In addition, several associated factors and inciting triggers have been proposed including trauma, infections, and hormonal fluctuations.3-5 However, the majority of patients do not report predisposing factors or a history of prior trauma at the site.3,6
Clinically, pyogenic granuloma usually presents as a painless, erythematous, dome-shaped friable papule or nodule that easily bleeds and may ulcerate. It typically undergoes a period of growth over weeks to months followed by stabilization. Occasionally, pyogenic granulomas will spontaneously involute, though most do not.7 Pyogenic granuloma may occur within an existing capillary malformation, such as a port wine stain, spontaneously or as a sequela of laser treatment.8,9 Diagnosis of pyogenic granuloma can typically be made clinically on the basis of history and exam. Dermoscopic evaluation of pyogenic granuloma will reveal a homogeneous papule with a surrounding white-brown collarette, and potentially white intersecting lines.10 Histopathologic evaluation may be necessary to differentiate lesions from conditions that may mimic pyogenic granuloma.
What’s on the differential?
The differential diagnosis for pyogenic granuloma consists of Spitz nevus, cherry hemangioma, amelanotic melanoma, and glomus tumor.
Spitz nevus
Spitz nevus (spindle and epithelial cell nevus) is a benign melanocytic lesion that classically appears as a sharply circumscribed, smooth, dome-shaped, pink-red, or brown papule or plaque. There is typically a history of rapid growth over several months followed by stabilization. It usually presents in childhood or adolescence and is most commonly located on the face and extremities. While there are similarities in the appearance of Spitz nevi and pyogenic granuloma, Spitz nevi are not usually friable nor associated with bleeding as in our patient. Furthermore, on dermoscopy, Spitz nevus typically exhibits a starburst pattern with regularly distributed dotted vessels, or a peripheral globular pattern with reticular depigmentation. The definitive diagnosis of Spitz nevi relies on histopathologic evaluation, which is critical for discriminating Spitz nevi from melanoma.
Cherry hemangioma
Cherry angiomas are the most common type of acquired benign vascular proliferation. They present as small, bright red or violaceous macules or papules. However, they typically appear in early to midadulthood and increase in number with age. The age of our patient and solitary presentation of the lesion make this diagnosis unlikely. In addition, cherry angiomas are not usually associated with bleeding. It is important to note that, depending on the age of the patient, pyogenic granuloma may also be confused with infantile hemangioma. Infantile hemangiomas may become bright red papules, nodules, or plaques that appear in early infancy. They characteristically involute, which does not typically happen with pyogenic granuloma.
Amelanotic melanoma
Amelanotic melanoma is an uncommon variant of melanoma with little to no pigmentation. It may appear as a skin-colored to light-brown, pink, or red macule, papule, or nodule. The lesion may be asymmetric with irregular and well-defined borders. The variable and uncharacteristic appearance of this melanoma variant makes it diagnostically challenging and it is often confused with benign lesions including pyogenic granuloma. Dermoscopy can help distinguish amelanotic melanoma from other benign conditions, and will reveal areas of pink to white, polymorphous vessels and crystalline structures. However, ultimately biopsy and histopathological evaluation is necessary for accurate diagnosis.
Glomus tumor
Glomus tumors are rare, benign neoplasms originating from cells of the glomus body that presents as a red-purple, vascular papule or nodule. They are usually found in areas rich in glomus bodies, such as the subungual regions, fingertips, palms, wrists, and forearms. Glomus tumors are typically associated with tenderness, paroxysmal pain, and cold sensitivity. They do not bleed or ulcerate. While pyogenic granuloma may be confused for glomus tumor when present on the fingers or extremities, the location of the lesion in our patient is not consistent with a diagnosis of glomus tumor.
Management and disease course
Management with procedural or topical interventions is usually pursued for pyogenic granuloma because of frequent bleeding and ulceration of lesions. The most common approach is simple excision by a scoop or shave technique, with or without curettage and most commonly with electrocautery of the base. Other options include full-thickness excision, destruction with laser therapy, cryotherapy, or topical treatments (for example, timolol).11 Lesion recurrence can occur with both surgical and nonsurgical management.11 Regardless of management technique, it is useful to obtain histopathologic evaluation of tissue for accurate diagnosis.
Our patient underwent surgical destruction of her lower-lip lesion with shave excision followed by electrocautery. The surgical specimen was sent for pathology, which confirmed the diagnosis of pyogenic granuloma. The patient experienced no complications from the procedure and did not have recurrence of the lesion.
Ms. Sui is a research associate in the department of dermatology, division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Ms. Sui nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Lin RL and Janniger CK. Cutis. 2004 Oct;74(4):229-33.
2. Harris MN et al. J Am Acad Dermatol. 2000 Jun;42(6):1012-6.
3. Pagliai KA and Cohen BA. Pediatr Dermatol. 2004 Jan-Feb;21(1):10-3.
4. Kamal R et al. J Oral Maxillofac Pathol. 2012 Jan;16(1):79-82.
5. Requena L and Sangueza OP. J Am Acad Dermatol. 1997 Dec;37(6):887-919.
6. Patrice SJ et al. Pediatr Dermatol. 1991 Dec;8(4):267-76.
7. Luba MC et al. Am Fam Physician. 2003 Feb 15;67(4):729-38.
8. Swerlick RA and Cooper PH. J Am Acad Dermatol. 1983 May;8(5):627-30.
9. Sheehan DJ and Lesher JL Jr. Cutis. 2004 Mar;73(3):175-80.
10. Zaballos P et al. Br J Dermatol. 2006 Jun;154(6):1108-11.
11. Lee J et al. J Plast Reconstr Aesthet Surg. 2011 Sep;64(9):1216-20. .
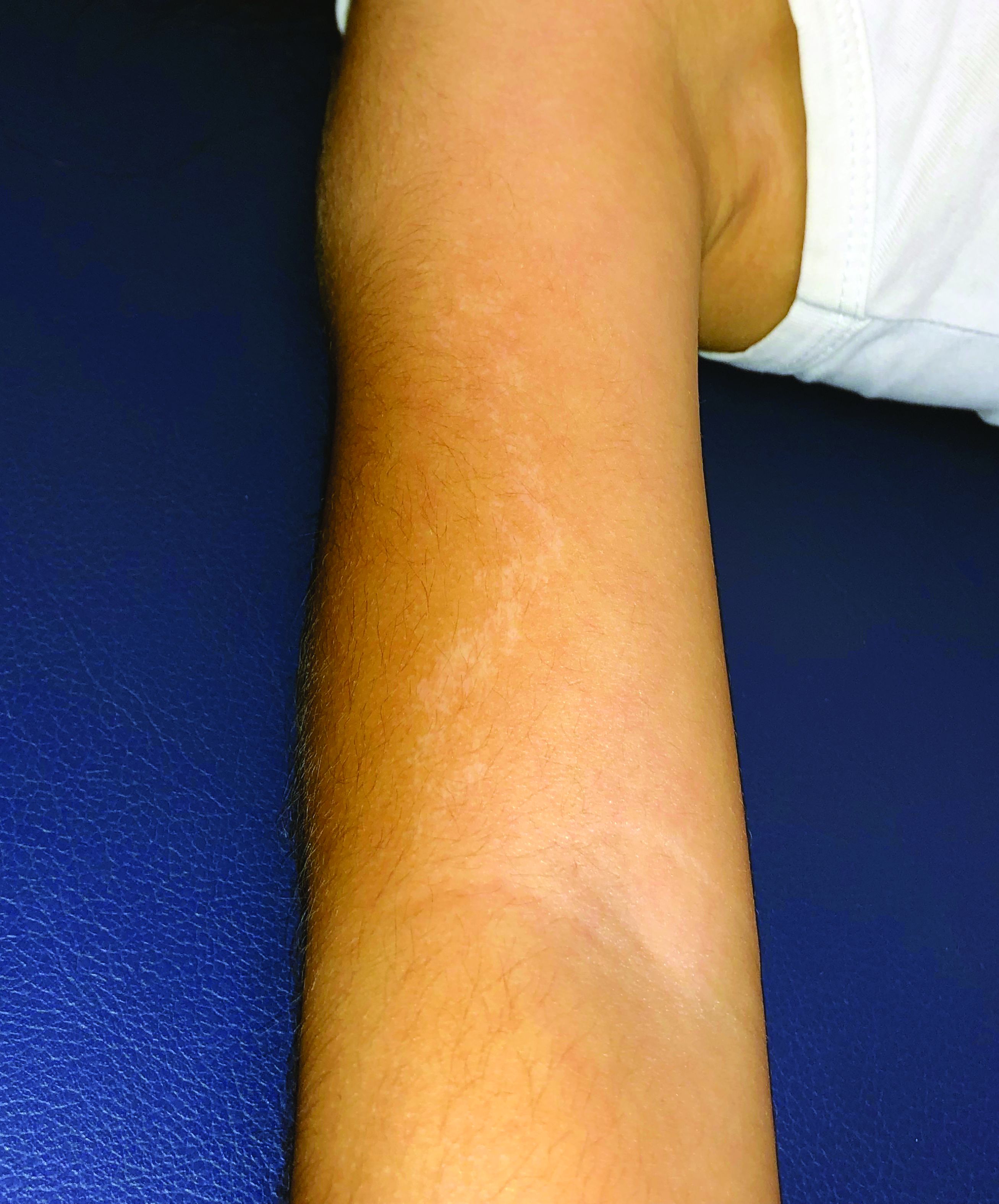
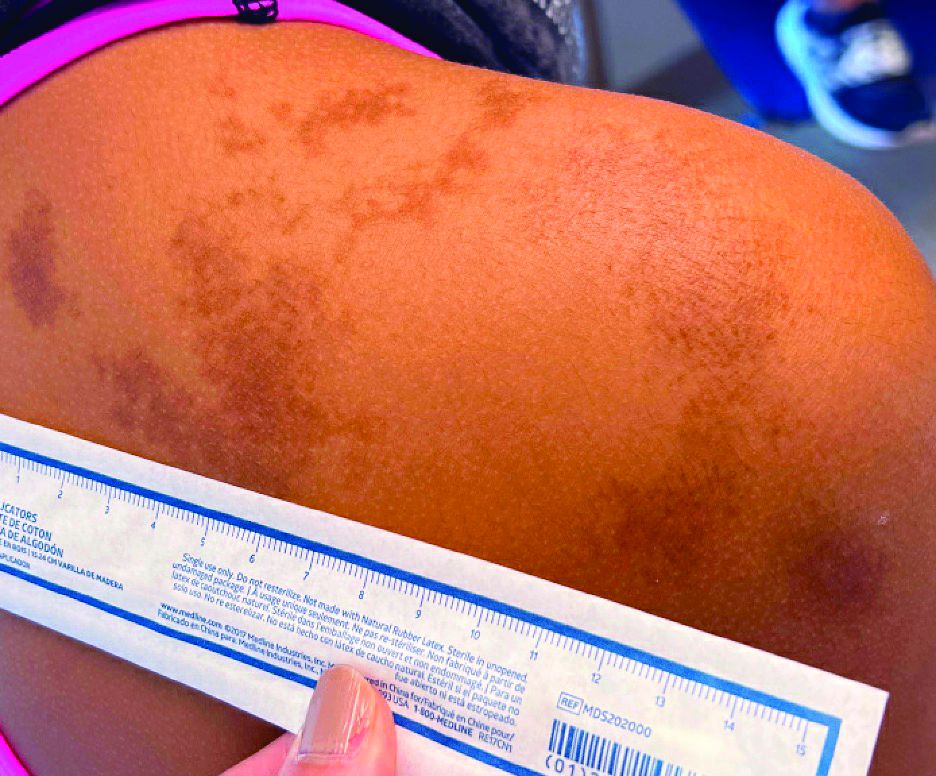
An 11-year-old female presented with skin discoloration on her back
Becker’s nevus
The history and physical exam are most consistent with Becker’s nevus, also known as Becker’s melanosis. This is a benign cutaneous hamartoma, usually found in males, characterized by a large, irregularly shaped brown patch, often with hypertrichosis.1 Becker’s nevus can be congenital but is more commonly noticed in late childhood or early adolescence, with thickening, increased pigmentation, and hair growth. Becker’s nevus is considered an overgrowth of epidermal pigment cells and hair follicles and is thought to be attributable to postzygotic mutations (with ACTB mutations most reported).1 It is often located unilaterally on the upper trunk but is occasionally present elsewhere on the body. Acne may occasionally develop within the nevus, which is believed to be triggered by puberty-associated androgens.1 The lesion tends to persist indefinitely but has no propensity for malignant transformation.

Becker’s nevus is generally an isolated skin lesion without other anomalies. However, in rare instances, it may be associated with ipsilateral breast hypoplasia or hypoplastic defects of the muscle, skin, or skeleton, which is known as Becker’s nevus syndrome.2 Treatment is not medically warranted for an isolated Becker’s nevus but may be pursued for cosmetic reasons. Although treatment is generally discouraged because of variable success, laser hair removal and laser therapy may be pursued to address the hypertrichosis and hyperpigmentation, respectively.
What is on the differential?
A café-au-lait macule (CALM) is a light- to dark-brown, oval lesion that commonly presents at birth or in early childhood. CALMs vary widely in size from less than 1.5 cm to more than 20 cm in diameter. They are asymptomatic and grow in proportion to the individual over time.3 Becker’s nevus can be distinguished from CALMs by the development of hypertrichosis, typical location and course, and other skin changes within the nevus.
Postinflammatory hyperpigmentation (PIH) is characterized by asymptomatic, darkened macules or patches that are brown to blue-gray in color. It is one of the most common causes of hyperpigmentation, particularly in skin of color, and can take months to years to resolve. PIH is caused by increased melanin production in response to a cutaneous inflammatory process, such as a drug reaction, allergy, mechanical or thermal injury, infection, phototoxicity, or an underlying skin condition.3 Our patient’s history with the lack of an inciting inflammatory process is more consistent with Becker’s nevus.
Erythema ab igne is a cutaneous reaction to heat that presents as a hyperpigmented patch with a reticular or mottled configuration and superficial venular telangiectasia. The lesion is initially erythematous and progresses to a pale pink to purplish dark-brown color.4 Causes include long-term use of a heating pad, laptop, electric blanket, or a hot water bottle. The absence of prolonged heat exposure in our patient’s history does not favor erythema ab igne.
Pigmentary mosaicism is characterized by a distinctive pattern of hyperpigmentation that follows the lines of ectodermal embryologic development, known as the lines of Blaschko.5 This condition is also known as linear and whorled nevoid hypermelanosis because of its streaky or swirl-like pattern. Pigmentary mosaicism can be present at birth or appear within the first few weeks of life. It is caused by genetic heterogeneity in neuroectodermal cells, which results in skin with areas of varying colors. Pigmentary mosaicism is unlikely in this case as our patient’s lesion does not follow the lines of Blaschko.
Ms. Laborada is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital. Ms. Laborada and Dr. Eichenfield have no relevant financial disclosures.
References
1. Atzmony L et al. J Cutan Pathol. 2020;47(8):681-5.
2. Danarti R et al. J Am Acad Dermatol. 2004;51(6):965-9.
3. Paller A and Mancini AJ. “Hurwitz Clinical Pediatric Dermatology: A textbook of skin disorders of childhood and adolescence” 4th ed. Philadelphia: Elsevier Saunders, 2011.
4. Patel DP. JAMA Dermatol. 2017;153(7):685.
5. Kromann AB et al. Orphanet J Rare Dis. 2018;13(1):39.
Becker’s nevus
The history and physical exam are most consistent with Becker’s nevus, also known as Becker’s melanosis. This is a benign cutaneous hamartoma, usually found in males, characterized by a large, irregularly shaped brown patch, often with hypertrichosis.1 Becker’s nevus can be congenital but is more commonly noticed in late childhood or early adolescence, with thickening, increased pigmentation, and hair growth. Becker’s nevus is considered an overgrowth of epidermal pigment cells and hair follicles and is thought to be attributable to postzygotic mutations (with ACTB mutations most reported).1 It is often located unilaterally on the upper trunk but is occasionally present elsewhere on the body. Acne may occasionally develop within the nevus, which is believed to be triggered by puberty-associated androgens.1 The lesion tends to persist indefinitely but has no propensity for malignant transformation.
Becker’s nevus is generally an isolated skin lesion without other anomalies. However, in rare instances, it may be associated with ipsilateral breast hypoplasia or hypoplastic defects of the muscle, skin, or skeleton, which is known as Becker’s nevus syndrome.2 Treatment is not medically warranted for an isolated Becker’s nevus but may be pursued for cosmetic reasons. Although treatment is generally discouraged because of variable success, laser hair removal and laser therapy may be pursued to address the hypertrichosis and hyperpigmentation, respectively.
What is on the differential?
A café-au-lait macule (CALM) is a light- to dark-brown, oval lesion that commonly presents at birth or in early childhood. CALMs vary widely in size from less than 1.5 cm to more than 20 cm in diameter. They are asymptomatic and grow in proportion to the individual over time.3 Becker’s nevus can be distinguished from CALMs by the development of hypertrichosis, typical location and course, and other skin changes within the nevus.
Postinflammatory hyperpigmentation (PIH) is characterized by asymptomatic, darkened macules or patches that are brown to blue-gray in color. It is one of the most common causes of hyperpigmentation, particularly in skin of color, and can take months to years to resolve. PIH is caused by increased melanin production in response to a cutaneous inflammatory process, such as a drug reaction, allergy, mechanical or thermal injury, infection, phototoxicity, or an underlying skin condition.3 Our patient’s history with the lack of an inciting inflammatory process is more consistent with Becker’s nevus.
Erythema ab igne is a cutaneous reaction to heat that presents as a hyperpigmented patch with a reticular or mottled configuration and superficial venular telangiectasia. The lesion is initially erythematous and progresses to a pale pink to purplish dark-brown color.4 Causes include long-term use of a heating pad, laptop, electric blanket, or a hot water bottle. The absence of prolonged heat exposure in our patient’s history does not favor erythema ab igne.
Pigmentary mosaicism is characterized by a distinctive pattern of hyperpigmentation that follows the lines of ectodermal embryologic development, known as the lines of Blaschko.5 This condition is also known as linear and whorled nevoid hypermelanosis because of its streaky or swirl-like pattern. Pigmentary mosaicism can be present at birth or appear within the first few weeks of life. It is caused by genetic heterogeneity in neuroectodermal cells, which results in skin with areas of varying colors. Pigmentary mosaicism is unlikely in this case as our patient’s lesion does not follow the lines of Blaschko.
Ms. Laborada is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital. Ms. Laborada and Dr. Eichenfield have no relevant financial disclosures.
References
1. Atzmony L et al. J Cutan Pathol. 2020;47(8):681-5.
2. Danarti R et al. J Am Acad Dermatol. 2004;51(6):965-9.
3. Paller A and Mancini AJ. “Hurwitz Clinical Pediatric Dermatology: A textbook of skin disorders of childhood and adolescence” 4th ed. Philadelphia: Elsevier Saunders, 2011.
4. Patel DP. JAMA Dermatol. 2017;153(7):685.
5. Kromann AB et al. Orphanet J Rare Dis. 2018;13(1):39.
Becker’s nevus
The history and physical exam are most consistent with Becker’s nevus, also known as Becker’s melanosis. This is a benign cutaneous hamartoma, usually found in males, characterized by a large, irregularly shaped brown patch, often with hypertrichosis.1 Becker’s nevus can be congenital but is more commonly noticed in late childhood or early adolescence, with thickening, increased pigmentation, and hair growth. Becker’s nevus is considered an overgrowth of epidermal pigment cells and hair follicles and is thought to be attributable to postzygotic mutations (with ACTB mutations most reported).1 It is often located unilaterally on the upper trunk but is occasionally present elsewhere on the body. Acne may occasionally develop within the nevus, which is believed to be triggered by puberty-associated androgens.1 The lesion tends to persist indefinitely but has no propensity for malignant transformation.
Becker’s nevus is generally an isolated skin lesion without other anomalies. However, in rare instances, it may be associated with ipsilateral breast hypoplasia or hypoplastic defects of the muscle, skin, or skeleton, which is known as Becker’s nevus syndrome.2 Treatment is not medically warranted for an isolated Becker’s nevus but may be pursued for cosmetic reasons. Although treatment is generally discouraged because of variable success, laser hair removal and laser therapy may be pursued to address the hypertrichosis and hyperpigmentation, respectively.
What is on the differential?
A café-au-lait macule (CALM) is a light- to dark-brown, oval lesion that commonly presents at birth or in early childhood. CALMs vary widely in size from less than 1.5 cm to more than 20 cm in diameter. They are asymptomatic and grow in proportion to the individual over time.3 Becker’s nevus can be distinguished from CALMs by the development of hypertrichosis, typical location and course, and other skin changes within the nevus.
Postinflammatory hyperpigmentation (PIH) is characterized by asymptomatic, darkened macules or patches that are brown to blue-gray in color. It is one of the most common causes of hyperpigmentation, particularly in skin of color, and can take months to years to resolve. PIH is caused by increased melanin production in response to a cutaneous inflammatory process, such as a drug reaction, allergy, mechanical or thermal injury, infection, phototoxicity, or an underlying skin condition.3 Our patient’s history with the lack of an inciting inflammatory process is more consistent with Becker’s nevus.
Erythema ab igne is a cutaneous reaction to heat that presents as a hyperpigmented patch with a reticular or mottled configuration and superficial venular telangiectasia. The lesion is initially erythematous and progresses to a pale pink to purplish dark-brown color.4 Causes include long-term use of a heating pad, laptop, electric blanket, or a hot water bottle. The absence of prolonged heat exposure in our patient’s history does not favor erythema ab igne.
Pigmentary mosaicism is characterized by a distinctive pattern of hyperpigmentation that follows the lines of ectodermal embryologic development, known as the lines of Blaschko.5 This condition is also known as linear and whorled nevoid hypermelanosis because of its streaky or swirl-like pattern. Pigmentary mosaicism can be present at birth or appear within the first few weeks of life. It is caused by genetic heterogeneity in neuroectodermal cells, which results in skin with areas of varying colors. Pigmentary mosaicism is unlikely in this case as our patient’s lesion does not follow the lines of Blaschko.
Ms. Laborada is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital. Ms. Laborada and Dr. Eichenfield have no relevant financial disclosures.
References
1. Atzmony L et al. J Cutan Pathol. 2020;47(8):681-5.
2. Danarti R et al. J Am Acad Dermatol. 2004;51(6):965-9.
3. Paller A and Mancini AJ. “Hurwitz Clinical Pediatric Dermatology: A textbook of skin disorders of childhood and adolescence” 4th ed. Philadelphia: Elsevier Saunders, 2011.
4. Patel DP. JAMA Dermatol. 2017;153(7):685.
5. Kromann AB et al. Orphanet J Rare Dis. 2018;13(1):39.
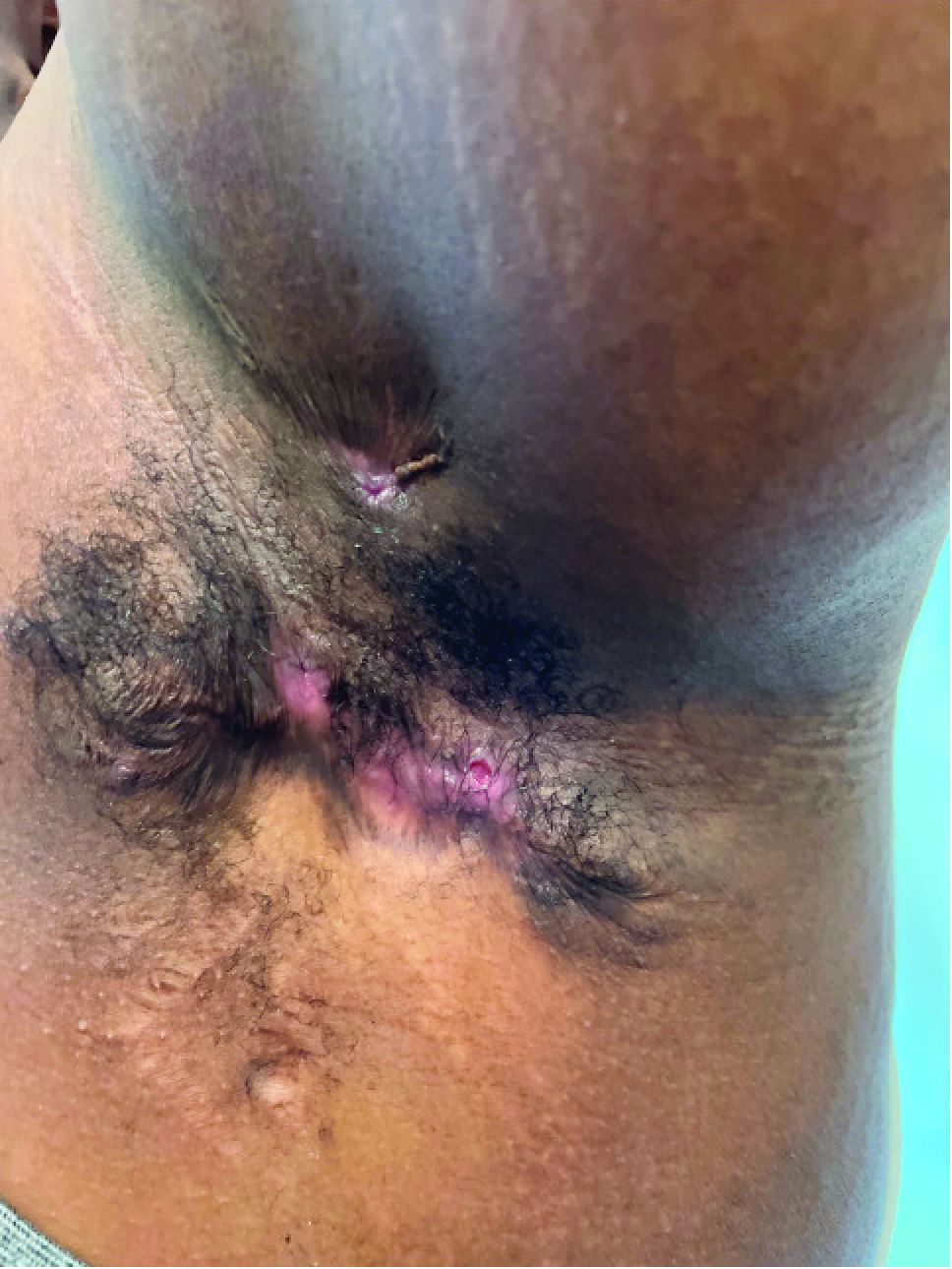
What is the diagnosis?
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that is becoming more recognized in children. It has a variable presentation, most commonly presenting as painful, recurrent cysts, abscesses, nodules, and/or pustules in classic locations with associated scarring and sinus tract formation.
The majority of patients present with bilateral lesions found most commonly in the axillae and inguinal folds.1 There are myriad other potential sites of involvement including the inframammary folds, inner thighs, buttocks, and groin.1 Diagnosis is made based on history and physical exam. There is a standard severity classification scheme called the Hurley score, which stratifies disease severity based on the presence of sinus tracts and extent of disease.1 HS is associated with comorbid conditions such as obesity, overweight, acne, and inflammatory bowel and joint disease.2 This painful, persistent condition is well documented to have a negative impact on quality of life in adult patients, and similar impairment has been found in pediatric patients.3,4

HS may be increasing in pediatric and adolescent patients, with recent studies showing onset coinciding most commonly with the onset of puberty.1,2 There is often a period of several years between symptom onset and diagnosis.1 A recent editorial highlighted the disparities that exist in HS, with disease more common in Black children and limited information about disease prevalence in Hispanic children.5
What’s the treatment plan?
HS is a difficult disease to treat, with few patients achieving remission and a significant proportion of patients with treatment-refractory disease.1 There are limited studies of HS treatment in pediatric patients. Topical and systemic antibiotic therapy are mainstays of HS treatment, with tetracyclines and a combination of clindamycin plus rifampin commonly used in adults and children alike. Topical therapies including topical antibiotics and antibacterial solutions are frequently used as adjunctive therapy.6 Adalimumab, a tumor necrosis factor receptor blocker, has been Food and Drug Administration approved for HS for ages 12 and up and is currently the only FDA-approved medication for HS in pediatric patients. Our patient was started on 100 mg doxycycline twice daily, with short-dose topical corticosteroids for symptom management of the most inflamed lesions.
What’s on the differential?
Acne conglobata
Acne conglobata is an uncommon, severe variant of acne vulgaris which arise in patients with a history of acne vulgaris and presents with comedones, cysts, abscesses, and scarring with possible drainage of pus. Lesions can present diffusely on the face, back, and body, including in the axillae, groin, and buttocks, and as such can be confused with HS.7
However, in contrast with HS, patients with acne conglobata will also develop disease in non–apocrine gland–bearing skin. This patient’s lack of preceding acne and restriction of lesions to the axillae, inguinal folds, and buttocks makes acne conglobata less likely.
Epidermal inclusion cyst
Epidermal inclusion cyst (EIC) is a common cutaneous cyst, presenting as a well-circumscribed nodule(s) with a central punctum. If not excised, lesions can sometimes become infected and painful.8 In contrast with HS, EIC presents only uncommonly as multiple lesions arising in different areas, and spontaneous drainage is uncommon. Our patient’s development of multiple draining lesions makes this diagnosis unlikely.
Furunculosis
Furunculosis is a common bacterial infection of the skin, presenting with inflammatory nodules or pustules centered around the hair follicle. Lesions may commonly present at sites of skin trauma and are found most frequently on the extremities.9 Though furunculosis lesions may drain pus and can coalesce to form larger “carbuncles,” our patient’s presence of significant scarring and lack of extremity involvement makes HS more likely.
Recurrent MRSA abscesses
Methicillin-resistant Staphylococcus aureus skin and soft-tissue infections are not uncommon in the pediatric population, with presentation of infection ranging from cellulitis to fluid-containing abscesses.10 Recurrent abscesses may be seen in MRSA infection, however in this patient the presence of draining, scarring lesions in multiple locations typical for HS over time is more consistent with a diagnosis of HS.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Appiah is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Appiah have no relevant financial disclosures.
References
1. Liy-Wong C et al. JAMA Dermatol. 2021;157(4):385-91.
2. Choi E et al. J Am Acad Dermatol. 2022;86(1):140-7.
3. Machado MO et al. JAMA Dermatol. 2019;155(8):939-45.
4. McAndrew R et al. J Am Acad Dermatol. 2021;84(3):829-30.
5. Kirby JS and Zaenglein AL. JAMA Dermatol. 2021;157(4):379-80.
6. Alikhan A et al. J Am Acad Dermatol. 2019;81(1):91-101.
7. Greydanus DE et al. Dis Mon. 2021;67(4):101103.
8. Weir CB, St. Hilaire NJ. Epidermal Inclusion Cyst, in “StatPearls.” Treasure Island, Fla: StatPearls Publishing, 2021.
9. Atanaskova N and Tomecki KJ. Dermatol Clin. 2010;28(3):479-87.
10. Papastefan ST et al. J Surg Res. 2019;242:70-7.
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that is becoming more recognized in children. It has a variable presentation, most commonly presenting as painful, recurrent cysts, abscesses, nodules, and/or pustules in classic locations with associated scarring and sinus tract formation.
The majority of patients present with bilateral lesions found most commonly in the axillae and inguinal folds.1 There are myriad other potential sites of involvement including the inframammary folds, inner thighs, buttocks, and groin.1 Diagnosis is made based on history and physical exam. There is a standard severity classification scheme called the Hurley score, which stratifies disease severity based on the presence of sinus tracts and extent of disease.1 HS is associated with comorbid conditions such as obesity, overweight, acne, and inflammatory bowel and joint disease.2 This painful, persistent condition is well documented to have a negative impact on quality of life in adult patients, and similar impairment has been found in pediatric patients.3,4
HS may be increasing in pediatric and adolescent patients, with recent studies showing onset coinciding most commonly with the onset of puberty.1,2 There is often a period of several years between symptom onset and diagnosis.1 A recent editorial highlighted the disparities that exist in HS, with disease more common in Black children and limited information about disease prevalence in Hispanic children.5
What’s the treatment plan?
HS is a difficult disease to treat, with few patients achieving remission and a significant proportion of patients with treatment-refractory disease.1 There are limited studies of HS treatment in pediatric patients. Topical and systemic antibiotic therapy are mainstays of HS treatment, with tetracyclines and a combination of clindamycin plus rifampin commonly used in adults and children alike. Topical therapies including topical antibiotics and antibacterial solutions are frequently used as adjunctive therapy.6 Adalimumab, a tumor necrosis factor receptor blocker, has been Food and Drug Administration approved for HS for ages 12 and up and is currently the only FDA-approved medication for HS in pediatric patients. Our patient was started on 100 mg doxycycline twice daily, with short-dose topical corticosteroids for symptom management of the most inflamed lesions.
What’s on the differential?
Acne conglobata
Acne conglobata is an uncommon, severe variant of acne vulgaris which arise in patients with a history of acne vulgaris and presents with comedones, cysts, abscesses, and scarring with possible drainage of pus. Lesions can present diffusely on the face, back, and body, including in the axillae, groin, and buttocks, and as such can be confused with HS.7
However, in contrast with HS, patients with acne conglobata will also develop disease in non–apocrine gland–bearing skin. This patient’s lack of preceding acne and restriction of lesions to the axillae, inguinal folds, and buttocks makes acne conglobata less likely.
Epidermal inclusion cyst
Epidermal inclusion cyst (EIC) is a common cutaneous cyst, presenting as a well-circumscribed nodule(s) with a central punctum. If not excised, lesions can sometimes become infected and painful.8 In contrast with HS, EIC presents only uncommonly as multiple lesions arising in different areas, and spontaneous drainage is uncommon. Our patient’s development of multiple draining lesions makes this diagnosis unlikely.
Furunculosis
Furunculosis is a common bacterial infection of the skin, presenting with inflammatory nodules or pustules centered around the hair follicle. Lesions may commonly present at sites of skin trauma and are found most frequently on the extremities.9 Though furunculosis lesions may drain pus and can coalesce to form larger “carbuncles,” our patient’s presence of significant scarring and lack of extremity involvement makes HS more likely.
Recurrent MRSA abscesses
Methicillin-resistant Staphylococcus aureus skin and soft-tissue infections are not uncommon in the pediatric population, with presentation of infection ranging from cellulitis to fluid-containing abscesses.10 Recurrent abscesses may be seen in MRSA infection, however in this patient the presence of draining, scarring lesions in multiple locations typical for HS over time is more consistent with a diagnosis of HS.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Appiah is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Appiah have no relevant financial disclosures.
References
1. Liy-Wong C et al. JAMA Dermatol. 2021;157(4):385-91.
2. Choi E et al. J Am Acad Dermatol. 2022;86(1):140-7.
3. Machado MO et al. JAMA Dermatol. 2019;155(8):939-45.
4. McAndrew R et al. J Am Acad Dermatol. 2021;84(3):829-30.
5. Kirby JS and Zaenglein AL. JAMA Dermatol. 2021;157(4):379-80.
6. Alikhan A et al. J Am Acad Dermatol. 2019;81(1):91-101.
7. Greydanus DE et al. Dis Mon. 2021;67(4):101103.
8. Weir CB, St. Hilaire NJ. Epidermal Inclusion Cyst, in “StatPearls.” Treasure Island, Fla: StatPearls Publishing, 2021.
9. Atanaskova N and Tomecki KJ. Dermatol Clin. 2010;28(3):479-87.
10. Papastefan ST et al. J Surg Res. 2019;242:70-7.
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that is becoming more recognized in children. It has a variable presentation, most commonly presenting as painful, recurrent cysts, abscesses, nodules, and/or pustules in classic locations with associated scarring and sinus tract formation.
The majority of patients present with bilateral lesions found most commonly in the axillae and inguinal folds.1 There are myriad other potential sites of involvement including the inframammary folds, inner thighs, buttocks, and groin.1 Diagnosis is made based on history and physical exam. There is a standard severity classification scheme called the Hurley score, which stratifies disease severity based on the presence of sinus tracts and extent of disease.1 HS is associated with comorbid conditions such as obesity, overweight, acne, and inflammatory bowel and joint disease.2 This painful, persistent condition is well documented to have a negative impact on quality of life in adult patients, and similar impairment has been found in pediatric patients.3,4
HS may be increasing in pediatric and adolescent patients, with recent studies showing onset coinciding most commonly with the onset of puberty.1,2 There is often a period of several years between symptom onset and diagnosis.1 A recent editorial highlighted the disparities that exist in HS, with disease more common in Black children and limited information about disease prevalence in Hispanic children.5
What’s the treatment plan?
HS is a difficult disease to treat, with few patients achieving remission and a significant proportion of patients with treatment-refractory disease.1 There are limited studies of HS treatment in pediatric patients. Topical and systemic antibiotic therapy are mainstays of HS treatment, with tetracyclines and a combination of clindamycin plus rifampin commonly used in adults and children alike. Topical therapies including topical antibiotics and antibacterial solutions are frequently used as adjunctive therapy.6 Adalimumab, a tumor necrosis factor receptor blocker, has been Food and Drug Administration approved for HS for ages 12 and up and is currently the only FDA-approved medication for HS in pediatric patients. Our patient was started on 100 mg doxycycline twice daily, with short-dose topical corticosteroids for symptom management of the most inflamed lesions.
What’s on the differential?
Acne conglobata
Acne conglobata is an uncommon, severe variant of acne vulgaris which arise in patients with a history of acne vulgaris and presents with comedones, cysts, abscesses, and scarring with possible drainage of pus. Lesions can present diffusely on the face, back, and body, including in the axillae, groin, and buttocks, and as such can be confused with HS.7
However, in contrast with HS, patients with acne conglobata will also develop disease in non–apocrine gland–bearing skin. This patient’s lack of preceding acne and restriction of lesions to the axillae, inguinal folds, and buttocks makes acne conglobata less likely.
Epidermal inclusion cyst
Epidermal inclusion cyst (EIC) is a common cutaneous cyst, presenting as a well-circumscribed nodule(s) with a central punctum. If not excised, lesions can sometimes become infected and painful.8 In contrast with HS, EIC presents only uncommonly as multiple lesions arising in different areas, and spontaneous drainage is uncommon. Our patient’s development of multiple draining lesions makes this diagnosis unlikely.
Furunculosis
Furunculosis is a common bacterial infection of the skin, presenting with inflammatory nodules or pustules centered around the hair follicle. Lesions may commonly present at sites of skin trauma and are found most frequently on the extremities.9 Though furunculosis lesions may drain pus and can coalesce to form larger “carbuncles,” our patient’s presence of significant scarring and lack of extremity involvement makes HS more likely.
Recurrent MRSA abscesses
Methicillin-resistant Staphylococcus aureus skin and soft-tissue infections are not uncommon in the pediatric population, with presentation of infection ranging from cellulitis to fluid-containing abscesses.10 Recurrent abscesses may be seen in MRSA infection, however in this patient the presence of draining, scarring lesions in multiple locations typical for HS over time is more consistent with a diagnosis of HS.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Appiah is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Appiah have no relevant financial disclosures.
References
1. Liy-Wong C et al. JAMA Dermatol. 2021;157(4):385-91.
2. Choi E et al. J Am Acad Dermatol. 2022;86(1):140-7.
3. Machado MO et al. JAMA Dermatol. 2019;155(8):939-45.
4. McAndrew R et al. J Am Acad Dermatol. 2021;84(3):829-30.
5. Kirby JS and Zaenglein AL. JAMA Dermatol. 2021;157(4):379-80.
6. Alikhan A et al. J Am Acad Dermatol. 2019;81(1):91-101.
7. Greydanus DE et al. Dis Mon. 2021;67(4):101103.
8. Weir CB, St. Hilaire NJ. Epidermal Inclusion Cyst, in “StatPearls.” Treasure Island, Fla: StatPearls Publishing, 2021.
9. Atanaskova N and Tomecki KJ. Dermatol Clin. 2010;28(3):479-87.
10. Papastefan ST et al. J Surg Res. 2019;242:70-7.
A female toddler presents with an itchy yellow nodule
Juvenile xanthogranuloma (JXG) is a benign disorder presenting as firm, yellow-red skin papules or nodules, usually in infancy or early childhood. It derives its name based on its yellowish color and the histologic finding of lipid-filled histiocytes. In fact, it is a form of non-Langerhans’ cell histiocytosis. It most commonly presents on the head, neck, and trunk, but can arise anywhere on the body as demonstrated by this case. While often pink to reddish early on, the characteristic yellow or orange, brown appearance over time is common, occasionally with overlying telangiectasia, and ranging in size from 1 mm to 2 cm. While typically asymptomatic, it is possible for lesions to itch. JXG is usually self-limiting, and spontaneously resolves over several years. On dermoscopy (with polarized light), it has a characteristic “setting sun” appearance because of its central yellow area surrounded by a reddish periphery.
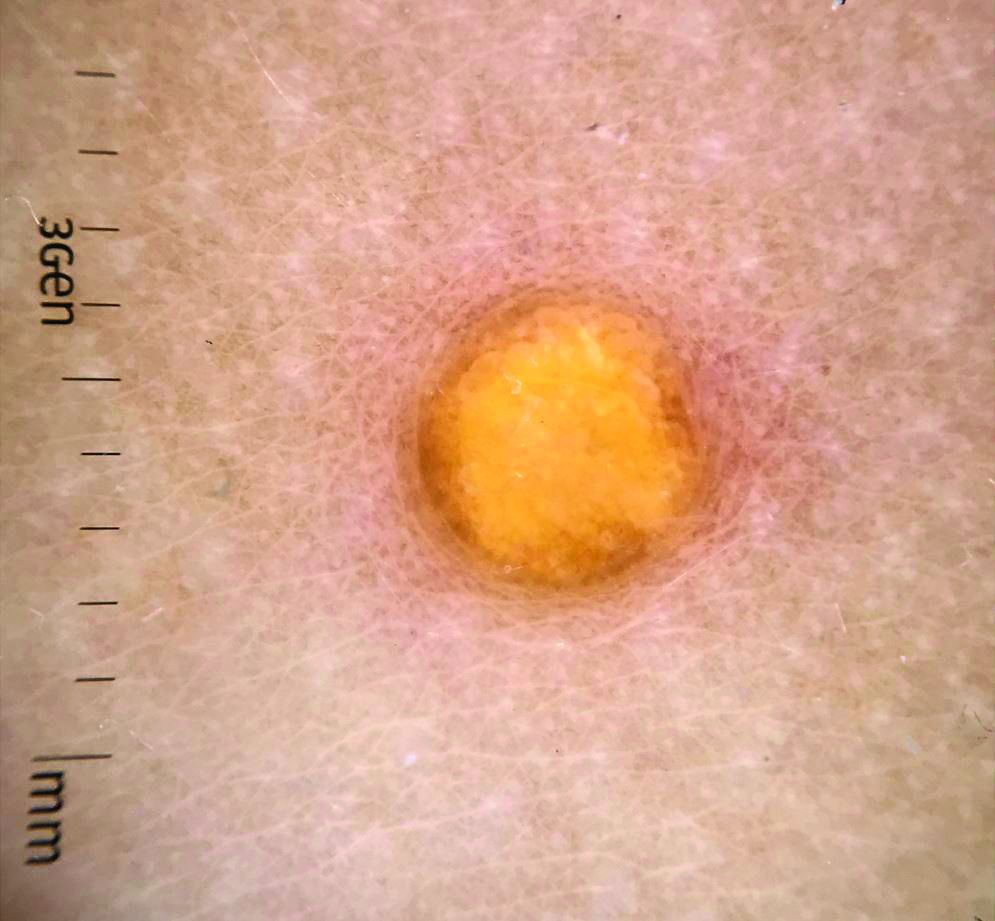
JXGs have been associated with neurofibromatosis-1 and a “triple association” of NF-1, JXG, and juvenile myelomonocytic leukemia (JMML) has been debated. Many cases are diagnosed on clinical grounds without histologic confirmation, so while the absolute incidence is unknown, they are not uncommon.
What is on the differential?
Spitz nevus is a melanocytic lesion which typically presents as a sharply circumscribed, dome-shaped, pink-red or brown papule or nodule, and is composed of large epithelioid and/or spindled cells. These nevi can present with a spectrum of morphology and biologic activity; commonly with benign melanocytic proliferations and a symmetric appearance or, rarely, with atypical tumors or lesions, characterized as Spitzoid melanomas. The yellowish color of JXG is distinct from the appearance of Spitz tumors.
Molluscum contagiosum is a common pox viral infection seen in children that presents with round, flat-topped firm papules on the skin and distinctive whitish centers with or without umbilication. Like JXG, molluscum contagiosum papules may grow over time and cause pruritus. However, this diagnosis is less likely given the absence of other lesions on the skin, lack of known contacts with similar lesions, and yellowish color without a more typical appearance of molluscum.
Dermatofibromas occur in people of all ages, although more commonly between the ages of 20 and 40 and in those with a history of trauma at the lesion. Like JXGs, dermatofibromas tend to be firm, solitary papules or nodules. They usually are hyperpigmented, and classically “dimple when pinched” as they are fixed to the subcutaneous tissue. However, this patient’s age, lack of trauma, and the lesion morphology are not consistent with dermatofibromas.

Like XJGs, mastocytomas commonly present in the first 2 years of life with maculopapular or nodular lesions that itch. However, the history of new-onset itch in recent months as the lesion grew larger and the yellow color on dermoscopy are more consistent with JXG.
Eruptive xanthomas typically appear suddenly as multiple erythematous yellow, dome-shaped papules on the extensor surfaces of the extremities, buttocks, and hands. They are usually present with hypertriglyceridemia and are very rare in young children. The presence of a solitary lesion in a 6-month-old patient without a history of lipid abnormalities favors the diagnosis of XJG.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Kleinman is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Kleinman have no relevant financial disclosures.
References
Hernandez-Martin A et al. J Am Acad Dermatol. 1997 Mar;36(3 Pt 1):355-67.
Prendiville J. Lumps, bumps and hamartomas in “Neonatal and Infant Dermatology,” 3rd ed. (Philadelphia: Elsevier, 2015).
Püttgen KB. Juvenile xanthogranuloma. UpToDate, 2021.
Schaffer JV. Am J Clin Dermatol. 2021 Mar;22(2):205-20.
Juvenile xanthogranuloma (JXG) is a benign disorder presenting as firm, yellow-red skin papules or nodules, usually in infancy or early childhood. It derives its name based on its yellowish color and the histologic finding of lipid-filled histiocytes. In fact, it is a form of non-Langerhans’ cell histiocytosis. It most commonly presents on the head, neck, and trunk, but can arise anywhere on the body as demonstrated by this case. While often pink to reddish early on, the characteristic yellow or orange, brown appearance over time is common, occasionally with overlying telangiectasia, and ranging in size from 1 mm to 2 cm. While typically asymptomatic, it is possible for lesions to itch. JXG is usually self-limiting, and spontaneously resolves over several years. On dermoscopy (with polarized light), it has a characteristic “setting sun” appearance because of its central yellow area surrounded by a reddish periphery.
JXGs have been associated with neurofibromatosis-1 and a “triple association” of NF-1, JXG, and juvenile myelomonocytic leukemia (JMML) has been debated. Many cases are diagnosed on clinical grounds without histologic confirmation, so while the absolute incidence is unknown, they are not uncommon.
What is on the differential?
Spitz nevus is a melanocytic lesion which typically presents as a sharply circumscribed, dome-shaped, pink-red or brown papule or nodule, and is composed of large epithelioid and/or spindled cells. These nevi can present with a spectrum of morphology and biologic activity; commonly with benign melanocytic proliferations and a symmetric appearance or, rarely, with atypical tumors or lesions, characterized as Spitzoid melanomas. The yellowish color of JXG is distinct from the appearance of Spitz tumors.
Molluscum contagiosum is a common pox viral infection seen in children that presents with round, flat-topped firm papules on the skin and distinctive whitish centers with or without umbilication. Like JXG, molluscum contagiosum papules may grow over time and cause pruritus. However, this diagnosis is less likely given the absence of other lesions on the skin, lack of known contacts with similar lesions, and yellowish color without a more typical appearance of molluscum.
Dermatofibromas occur in people of all ages, although more commonly between the ages of 20 and 40 and in those with a history of trauma at the lesion. Like JXGs, dermatofibromas tend to be firm, solitary papules or nodules. They usually are hyperpigmented, and classically “dimple when pinched” as they are fixed to the subcutaneous tissue. However, this patient’s age, lack of trauma, and the lesion morphology are not consistent with dermatofibromas.
Like XJGs, mastocytomas commonly present in the first 2 years of life with maculopapular or nodular lesions that itch. However, the history of new-onset itch in recent months as the lesion grew larger and the yellow color on dermoscopy are more consistent with JXG.
Eruptive xanthomas typically appear suddenly as multiple erythematous yellow, dome-shaped papules on the extensor surfaces of the extremities, buttocks, and hands. They are usually present with hypertriglyceridemia and are very rare in young children. The presence of a solitary lesion in a 6-month-old patient without a history of lipid abnormalities favors the diagnosis of XJG.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Kleinman is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Kleinman have no relevant financial disclosures.
References
Hernandez-Martin A et al. J Am Acad Dermatol. 1997 Mar;36(3 Pt 1):355-67.
Prendiville J. Lumps, bumps and hamartomas in “Neonatal and Infant Dermatology,” 3rd ed. (Philadelphia: Elsevier, 2015).
Püttgen KB. Juvenile xanthogranuloma. UpToDate, 2021.
Schaffer JV. Am J Clin Dermatol. 2021 Mar;22(2):205-20.
Juvenile xanthogranuloma (JXG) is a benign disorder presenting as firm, yellow-red skin papules or nodules, usually in infancy or early childhood. It derives its name based on its yellowish color and the histologic finding of lipid-filled histiocytes. In fact, it is a form of non-Langerhans’ cell histiocytosis. It most commonly presents on the head, neck, and trunk, but can arise anywhere on the body as demonstrated by this case. While often pink to reddish early on, the characteristic yellow or orange, brown appearance over time is common, occasionally with overlying telangiectasia, and ranging in size from 1 mm to 2 cm. While typically asymptomatic, it is possible for lesions to itch. JXG is usually self-limiting, and spontaneously resolves over several years. On dermoscopy (with polarized light), it has a characteristic “setting sun” appearance because of its central yellow area surrounded by a reddish periphery.
JXGs have been associated with neurofibromatosis-1 and a “triple association” of NF-1, JXG, and juvenile myelomonocytic leukemia (JMML) has been debated. Many cases are diagnosed on clinical grounds without histologic confirmation, so while the absolute incidence is unknown, they are not uncommon.
What is on the differential?
Spitz nevus is a melanocytic lesion which typically presents as a sharply circumscribed, dome-shaped, pink-red or brown papule or nodule, and is composed of large epithelioid and/or spindled cells. These nevi can present with a spectrum of morphology and biologic activity; commonly with benign melanocytic proliferations and a symmetric appearance or, rarely, with atypical tumors or lesions, characterized as Spitzoid melanomas. The yellowish color of JXG is distinct from the appearance of Spitz tumors.
Molluscum contagiosum is a common pox viral infection seen in children that presents with round, flat-topped firm papules on the skin and distinctive whitish centers with or without umbilication. Like JXG, molluscum contagiosum papules may grow over time and cause pruritus. However, this diagnosis is less likely given the absence of other lesions on the skin, lack of known contacts with similar lesions, and yellowish color without a more typical appearance of molluscum.
Dermatofibromas occur in people of all ages, although more commonly between the ages of 20 and 40 and in those with a history of trauma at the lesion. Like JXGs, dermatofibromas tend to be firm, solitary papules or nodules. They usually are hyperpigmented, and classically “dimple when pinched” as they are fixed to the subcutaneous tissue. However, this patient’s age, lack of trauma, and the lesion morphology are not consistent with dermatofibromas.
Like XJGs, mastocytomas commonly present in the first 2 years of life with maculopapular or nodular lesions that itch. However, the history of new-onset itch in recent months as the lesion grew larger and the yellow color on dermoscopy are more consistent with JXG.
Eruptive xanthomas typically appear suddenly as multiple erythematous yellow, dome-shaped papules on the extensor surfaces of the extremities, buttocks, and hands. They are usually present with hypertriglyceridemia and are very rare in young children. The presence of a solitary lesion in a 6-month-old patient without a history of lipid abnormalities favors the diagnosis of XJG.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Kleinman is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Kleinman have no relevant financial disclosures.
References
Hernandez-Martin A et al. J Am Acad Dermatol. 1997 Mar;36(3 Pt 1):355-67.
Prendiville J. Lumps, bumps and hamartomas in “Neonatal and Infant Dermatology,” 3rd ed. (Philadelphia: Elsevier, 2015).
Püttgen KB. Juvenile xanthogranuloma. UpToDate, 2021.
Schaffer JV. Am J Clin Dermatol. 2021 Mar;22(2):205-20.
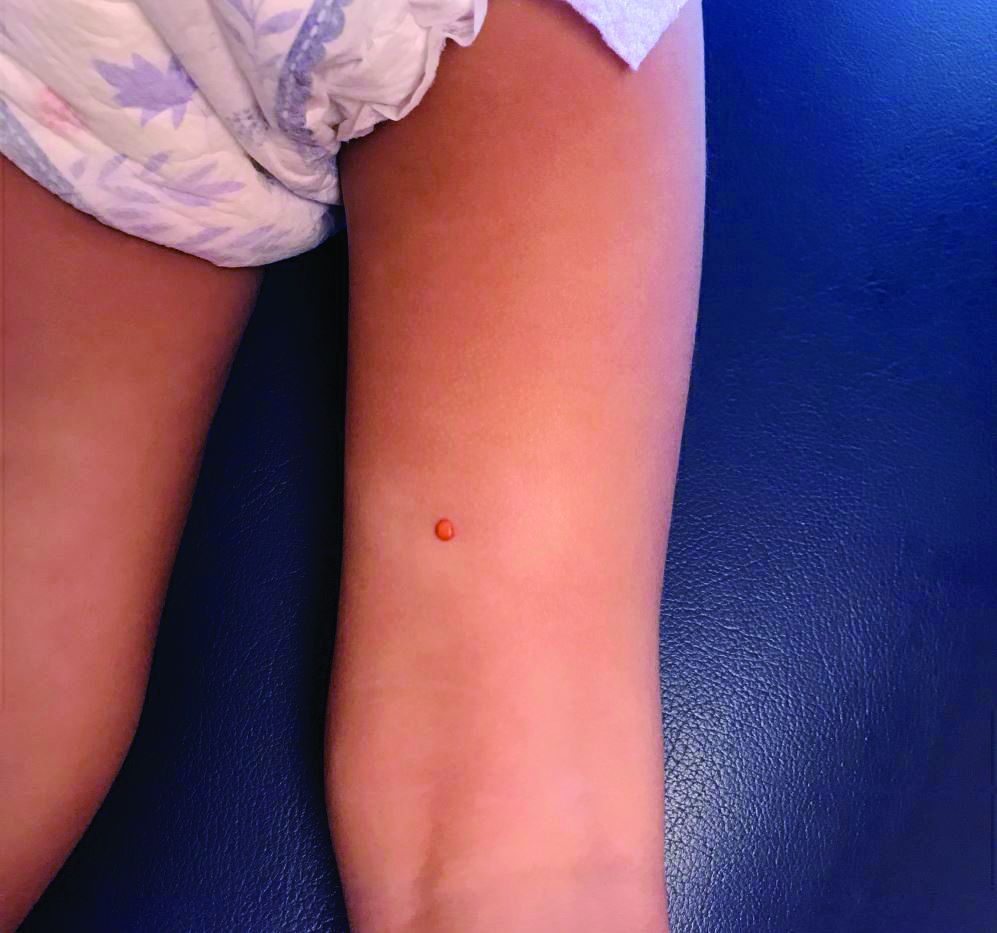
An infant girl presents with a growing pink-red leg nodule
The history of a brownish to pink patch with color change and rapid growth within the first year combined with the exam findings, are suggestive of a tufted angioma, though the findings presented may be nonspecific.
A tufted angioma is a rare vascular tumor of infancy or early childhood, that is present at birth in approximately half of cases. It may initially present as a faint pink to brown plaque, but develops as a firm, red to violaceous nodule or plaque, usually with “lumpiness” or nodularity.1-3 Lesions usually are infiltrative with indistinct borders. They are named for their histologic appearance, with lobules of capillaries which appear as “tufts” in the dermis and subdermis with “cannonball” appearance, and are considered to be on a spectrum with another vascular tumor called kaposiform hemangioendothelioma (KHE).4 These vascular tumors can trigger Kasabach-Merritt syndrome, a disease process in which vascular tumors trap platelets and clotting factors, resulting in a life-threatening thrombocytopenia and consumptive coagulopathy with a high risk of bleeding and high-output heart failure.5
What’s the differential diagnosis?
The differential diagnosis of tufted angioma includes other potentially large vascular lesions including infantile hemangioma, congenital hemangioma, port-wine birth marks (capillary malformations), hemangioendotheliomas, and rhabdomyosarcomas.

Infantile hemangiomas (IH) are common vascular tumors of infancy seen in 4%-5% of infants that are characterized by a growth and involution phase. Classically, lesions can be absent or minimally evident at birth, becoming noticeable within the first months of life with a rapid growth phase and typical progression to bright red papules, nodules, or plaques. Deeper hemangiomas may appear more skin colored on the surface with a bluish coloration underneath. They are usually more discreet, with relatively defined borders. Diagnosis is typically clinical and many IHs self-resolve, albeit with residual findings including skin atrophy, scarring, and telangiectasia. Observation or topical timolol are first-line treatment options for more superficial lesions while systemic propranolol is the treatment of choice for deeper IHs or those resulting in possible airway or vision compromise.
Congenital hemangiomas (CH) are another type of vascular growth characterized by a solitary erythematous to violaceous plaque or nodule present at birth with overlying telangiectasia. CHs can be subdivided into categories including rapidly involuting (RICH), partially involuting (PICH), and noninvoluting (NICH). Diagnosis is usually clinical and, depending on the subtype, treatment can involve watchful waiting (for RICHs) or more active intervention such as pulse dye laser or surgical resection (for PICHs or NICHs). The growing nature of this patient’s mass makes a diagnosis of CH unlikely.
Port-wine birth mark, also known as nevus flammeus, is a vascular malformation that appears at birth as a nonpalpable irregular erythematous to violaceous macular plaque. Port-wine stains may be isolated birthmarks, or associated with Sturge-Weber syndrome, complex vascular malformations, or soft-tissue overgrowth. Klippel-Trenauny syndrome (KTS) describes capillary-venous malformations with limb overgrowth, with or without lymphatic malformations, and many are associated with somatic mutations in the PIK3CA gene. While KTS could be considered in this patient, the nodular appearance with lumpy texture and rapid growth makes a vascular tumor more likely.
Rhabdomyosarcoma is a malignancy of skeletal muscle lineage and the most common soft tissue tumor in pediatrics. Cutaneous rhabdomyosarcomas present as erythematous nodules, markedly firm, often “fixed” to deep tissue. A rapidly growing atypical, firm tumor of infancy should raise the consideration of rhabdomyosarcoma and imaging and biopsy are appropriate for evaluation.
What should the evaluation and management of this patient be?
Initial workup should include a complete blood count with platelet count as well as coagulation studies including D-dimer, fibrinogen, prothrombin time, and activated partial thromboplastin time, to assess for any thrombocytopenia or coagulopathy.6 Ultrasound and/or MRI may also be performed to determine lesion extent. While typical MRI findings might be suggestive of a tufted angioma or hemangioendothelioma, biopsy for histologic examination is usually the approach to diagnosis, which will demonstrate stereotypic round lobules of capillaries in a “tufted” distribution.2,7 Biopsy may be performed by a surgeon or dermatologist but bleeding at time of biopsy needs to be considered before moving forward with the procedure.
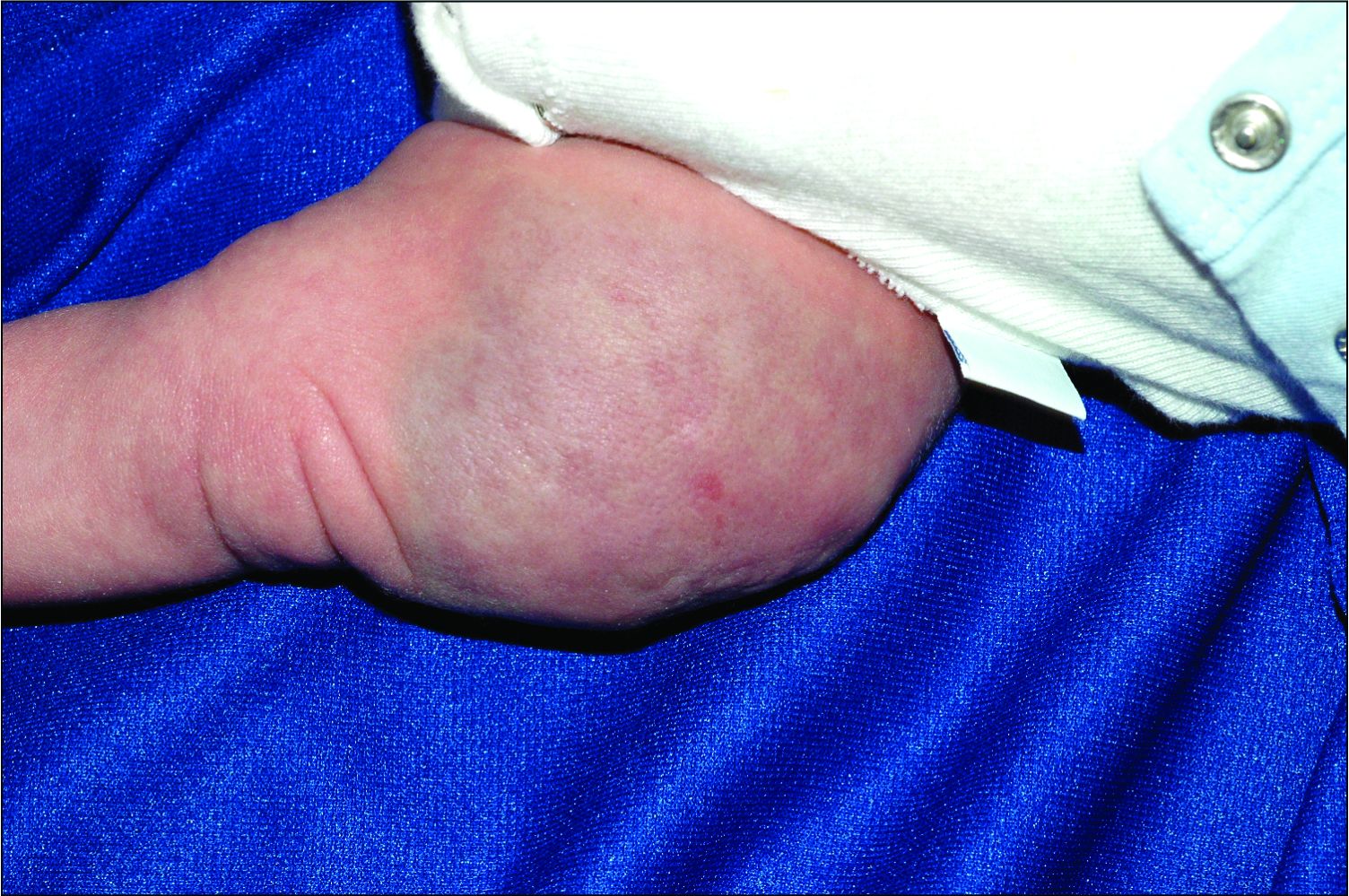
Tufted angiomas of early life may regress spontaneously, though lesions with symptoms, with functional significance, or associated with KHE may require therapy. Surgical excision is one option, but it may be difficult to execute given that these lesions often have poorly defined margins.1 Other treatment choices include but are not limited to aspirin, systemic corticosteroids, vincristine, interferon-alpha, embolization, and sirolimus.8 No specific expert-directed consensus guidelines exist for these lesions, and suspicion of this lesion should prompt urgent referral to a pediatric dermatologist. Concern for Kasabach-Merritt syndrome should trigger immediate referral for rapid evaluation and management.
Complete blood count with platelet count and coagulation studies were normal in our patient. This infant underwent biopsy to confirm the diagnosis of tufted angioma and MRI to determine lesion extent. The lesion slowly involuted spontaneously without recurrence.
Mr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is MS4 at the University of Rochester, N.Y. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Herron MD et al. Pediatr Dermatol. 2002;19(5):394-401.
2. Jones EW and Orkin M. J Am Acad Dermatol. 1989;20(2 Pt 1):214-25.
3. Wong SN and Tay YK. Pediatr Dermatol. 2002;19(5):388-93.
4. Croteau SE and Gupta D. Semin Cutan Med Surg. 2016;35(3):147-52.
5. Kelly M. Pediatr Clin North Am. 2010;57(5):1085-9.
6. Osio A et al. Arch Dermatol. 2010;146(7):758-63.
7. Padilla RS et al. Am J Dermatopathol. 1987;9(4):292-300.
8. Liu XH et al. Int J Cancer. 2016;139(7):1658-66.
The history of a brownish to pink patch with color change and rapid growth within the first year combined with the exam findings, are suggestive of a tufted angioma, though the findings presented may be nonspecific.
A tufted angioma is a rare vascular tumor of infancy or early childhood, that is present at birth in approximately half of cases. It may initially present as a faint pink to brown plaque, but develops as a firm, red to violaceous nodule or plaque, usually with “lumpiness” or nodularity.1-3 Lesions usually are infiltrative with indistinct borders. They are named for their histologic appearance, with lobules of capillaries which appear as “tufts” in the dermis and subdermis with “cannonball” appearance, and are considered to be on a spectrum with another vascular tumor called kaposiform hemangioendothelioma (KHE).4 These vascular tumors can trigger Kasabach-Merritt syndrome, a disease process in which vascular tumors trap platelets and clotting factors, resulting in a life-threatening thrombocytopenia and consumptive coagulopathy with a high risk of bleeding and high-output heart failure.5
What’s the differential diagnosis?
The differential diagnosis of tufted angioma includes other potentially large vascular lesions including infantile hemangioma, congenital hemangioma, port-wine birth marks (capillary malformations), hemangioendotheliomas, and rhabdomyosarcomas.
Infantile hemangiomas (IH) are common vascular tumors of infancy seen in 4%-5% of infants that are characterized by a growth and involution phase. Classically, lesions can be absent or minimally evident at birth, becoming noticeable within the first months of life with a rapid growth phase and typical progression to bright red papules, nodules, or plaques. Deeper hemangiomas may appear more skin colored on the surface with a bluish coloration underneath. They are usually more discreet, with relatively defined borders. Diagnosis is typically clinical and many IHs self-resolve, albeit with residual findings including skin atrophy, scarring, and telangiectasia. Observation or topical timolol are first-line treatment options for more superficial lesions while systemic propranolol is the treatment of choice for deeper IHs or those resulting in possible airway or vision compromise.
Congenital hemangiomas (CH) are another type of vascular growth characterized by a solitary erythematous to violaceous plaque or nodule present at birth with overlying telangiectasia. CHs can be subdivided into categories including rapidly involuting (RICH), partially involuting (PICH), and noninvoluting (NICH). Diagnosis is usually clinical and, depending on the subtype, treatment can involve watchful waiting (for RICHs) or more active intervention such as pulse dye laser or surgical resection (for PICHs or NICHs). The growing nature of this patient’s mass makes a diagnosis of CH unlikely.
Port-wine birth mark, also known as nevus flammeus, is a vascular malformation that appears at birth as a nonpalpable irregular erythematous to violaceous macular plaque. Port-wine stains may be isolated birthmarks, or associated with Sturge-Weber syndrome, complex vascular malformations, or soft-tissue overgrowth. Klippel-Trenauny syndrome (KTS) describes capillary-venous malformations with limb overgrowth, with or without lymphatic malformations, and many are associated with somatic mutations in the PIK3CA gene. While KTS could be considered in this patient, the nodular appearance with lumpy texture and rapid growth makes a vascular tumor more likely.
Rhabdomyosarcoma is a malignancy of skeletal muscle lineage and the most common soft tissue tumor in pediatrics. Cutaneous rhabdomyosarcomas present as erythematous nodules, markedly firm, often “fixed” to deep tissue. A rapidly growing atypical, firm tumor of infancy should raise the consideration of rhabdomyosarcoma and imaging and biopsy are appropriate for evaluation.
What should the evaluation and management of this patient be?
Initial workup should include a complete blood count with platelet count as well as coagulation studies including D-dimer, fibrinogen, prothrombin time, and activated partial thromboplastin time, to assess for any thrombocytopenia or coagulopathy.6 Ultrasound and/or MRI may also be performed to determine lesion extent. While typical MRI findings might be suggestive of a tufted angioma or hemangioendothelioma, biopsy for histologic examination is usually the approach to diagnosis, which will demonstrate stereotypic round lobules of capillaries in a “tufted” distribution.2,7 Biopsy may be performed by a surgeon or dermatologist but bleeding at time of biopsy needs to be considered before moving forward with the procedure.
Tufted angiomas of early life may regress spontaneously, though lesions with symptoms, with functional significance, or associated with KHE may require therapy. Surgical excision is one option, but it may be difficult to execute given that these lesions often have poorly defined margins.1 Other treatment choices include but are not limited to aspirin, systemic corticosteroids, vincristine, interferon-alpha, embolization, and sirolimus.8 No specific expert-directed consensus guidelines exist for these lesions, and suspicion of this lesion should prompt urgent referral to a pediatric dermatologist. Concern for Kasabach-Merritt syndrome should trigger immediate referral for rapid evaluation and management.
Complete blood count with platelet count and coagulation studies were normal in our patient. This infant underwent biopsy to confirm the diagnosis of tufted angioma and MRI to determine lesion extent. The lesion slowly involuted spontaneously without recurrence.
Mr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is MS4 at the University of Rochester, N.Y. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Herron MD et al. Pediatr Dermatol. 2002;19(5):394-401.
2. Jones EW and Orkin M. J Am Acad Dermatol. 1989;20(2 Pt 1):214-25.
3. Wong SN and Tay YK. Pediatr Dermatol. 2002;19(5):388-93.
4. Croteau SE and Gupta D. Semin Cutan Med Surg. 2016;35(3):147-52.
5. Kelly M. Pediatr Clin North Am. 2010;57(5):1085-9.
6. Osio A et al. Arch Dermatol. 2010;146(7):758-63.
7. Padilla RS et al. Am J Dermatopathol. 1987;9(4):292-300.
8. Liu XH et al. Int J Cancer. 2016;139(7):1658-66.
The history of a brownish to pink patch with color change and rapid growth within the first year combined with the exam findings, are suggestive of a tufted angioma, though the findings presented may be nonspecific.
A tufted angioma is a rare vascular tumor of infancy or early childhood, that is present at birth in approximately half of cases. It may initially present as a faint pink to brown plaque, but develops as a firm, red to violaceous nodule or plaque, usually with “lumpiness” or nodularity.1-3 Lesions usually are infiltrative with indistinct borders. They are named for their histologic appearance, with lobules of capillaries which appear as “tufts” in the dermis and subdermis with “cannonball” appearance, and are considered to be on a spectrum with another vascular tumor called kaposiform hemangioendothelioma (KHE).4 These vascular tumors can trigger Kasabach-Merritt syndrome, a disease process in which vascular tumors trap platelets and clotting factors, resulting in a life-threatening thrombocytopenia and consumptive coagulopathy with a high risk of bleeding and high-output heart failure.5
What’s the differential diagnosis?
The differential diagnosis of tufted angioma includes other potentially large vascular lesions including infantile hemangioma, congenital hemangioma, port-wine birth marks (capillary malformations), hemangioendotheliomas, and rhabdomyosarcomas.
Infantile hemangiomas (IH) are common vascular tumors of infancy seen in 4%-5% of infants that are characterized by a growth and involution phase. Classically, lesions can be absent or minimally evident at birth, becoming noticeable within the first months of life with a rapid growth phase and typical progression to bright red papules, nodules, or plaques. Deeper hemangiomas may appear more skin colored on the surface with a bluish coloration underneath. They are usually more discreet, with relatively defined borders. Diagnosis is typically clinical and many IHs self-resolve, albeit with residual findings including skin atrophy, scarring, and telangiectasia. Observation or topical timolol are first-line treatment options for more superficial lesions while systemic propranolol is the treatment of choice for deeper IHs or those resulting in possible airway or vision compromise.
Congenital hemangiomas (CH) are another type of vascular growth characterized by a solitary erythematous to violaceous plaque or nodule present at birth with overlying telangiectasia. CHs can be subdivided into categories including rapidly involuting (RICH), partially involuting (PICH), and noninvoluting (NICH). Diagnosis is usually clinical and, depending on the subtype, treatment can involve watchful waiting (for RICHs) or more active intervention such as pulse dye laser or surgical resection (for PICHs or NICHs). The growing nature of this patient’s mass makes a diagnosis of CH unlikely.
Port-wine birth mark, also known as nevus flammeus, is a vascular malformation that appears at birth as a nonpalpable irregular erythematous to violaceous macular plaque. Port-wine stains may be isolated birthmarks, or associated with Sturge-Weber syndrome, complex vascular malformations, or soft-tissue overgrowth. Klippel-Trenauny syndrome (KTS) describes capillary-venous malformations with limb overgrowth, with or without lymphatic malformations, and many are associated with somatic mutations in the PIK3CA gene. While KTS could be considered in this patient, the nodular appearance with lumpy texture and rapid growth makes a vascular tumor more likely.
Rhabdomyosarcoma is a malignancy of skeletal muscle lineage and the most common soft tissue tumor in pediatrics. Cutaneous rhabdomyosarcomas present as erythematous nodules, markedly firm, often “fixed” to deep tissue. A rapidly growing atypical, firm tumor of infancy should raise the consideration of rhabdomyosarcoma and imaging and biopsy are appropriate for evaluation.
What should the evaluation and management of this patient be?
Initial workup should include a complete blood count with platelet count as well as coagulation studies including D-dimer, fibrinogen, prothrombin time, and activated partial thromboplastin time, to assess for any thrombocytopenia or coagulopathy.6 Ultrasound and/or MRI may also be performed to determine lesion extent. While typical MRI findings might be suggestive of a tufted angioma or hemangioendothelioma, biopsy for histologic examination is usually the approach to diagnosis, which will demonstrate stereotypic round lobules of capillaries in a “tufted” distribution.2,7 Biopsy may be performed by a surgeon or dermatologist but bleeding at time of biopsy needs to be considered before moving forward with the procedure.
Tufted angiomas of early life may regress spontaneously, though lesions with symptoms, with functional significance, or associated with KHE may require therapy. Surgical excision is one option, but it may be difficult to execute given that these lesions often have poorly defined margins.1 Other treatment choices include but are not limited to aspirin, systemic corticosteroids, vincristine, interferon-alpha, embolization, and sirolimus.8 No specific expert-directed consensus guidelines exist for these lesions, and suspicion of this lesion should prompt urgent referral to a pediatric dermatologist. Concern for Kasabach-Merritt syndrome should trigger immediate referral for rapid evaluation and management.
Complete blood count with platelet count and coagulation studies were normal in our patient. This infant underwent biopsy to confirm the diagnosis of tufted angioma and MRI to determine lesion extent. The lesion slowly involuted spontaneously without recurrence.
Mr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is MS4 at the University of Rochester, N.Y. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Herron MD et al. Pediatr Dermatol. 2002;19(5):394-401.
2. Jones EW and Orkin M. J Am Acad Dermatol. 1989;20(2 Pt 1):214-25.
3. Wong SN and Tay YK. Pediatr Dermatol. 2002;19(5):388-93.
4. Croteau SE and Gupta D. Semin Cutan Med Surg. 2016;35(3):147-52.
5. Kelly M. Pediatr Clin North Am. 2010;57(5):1085-9.
6. Osio A et al. Arch Dermatol. 2010;146(7):758-63.
7. Padilla RS et al. Am J Dermatopathol. 1987;9(4):292-300.
8. Liu XH et al. Int J Cancer. 2016;139(7):1658-66.
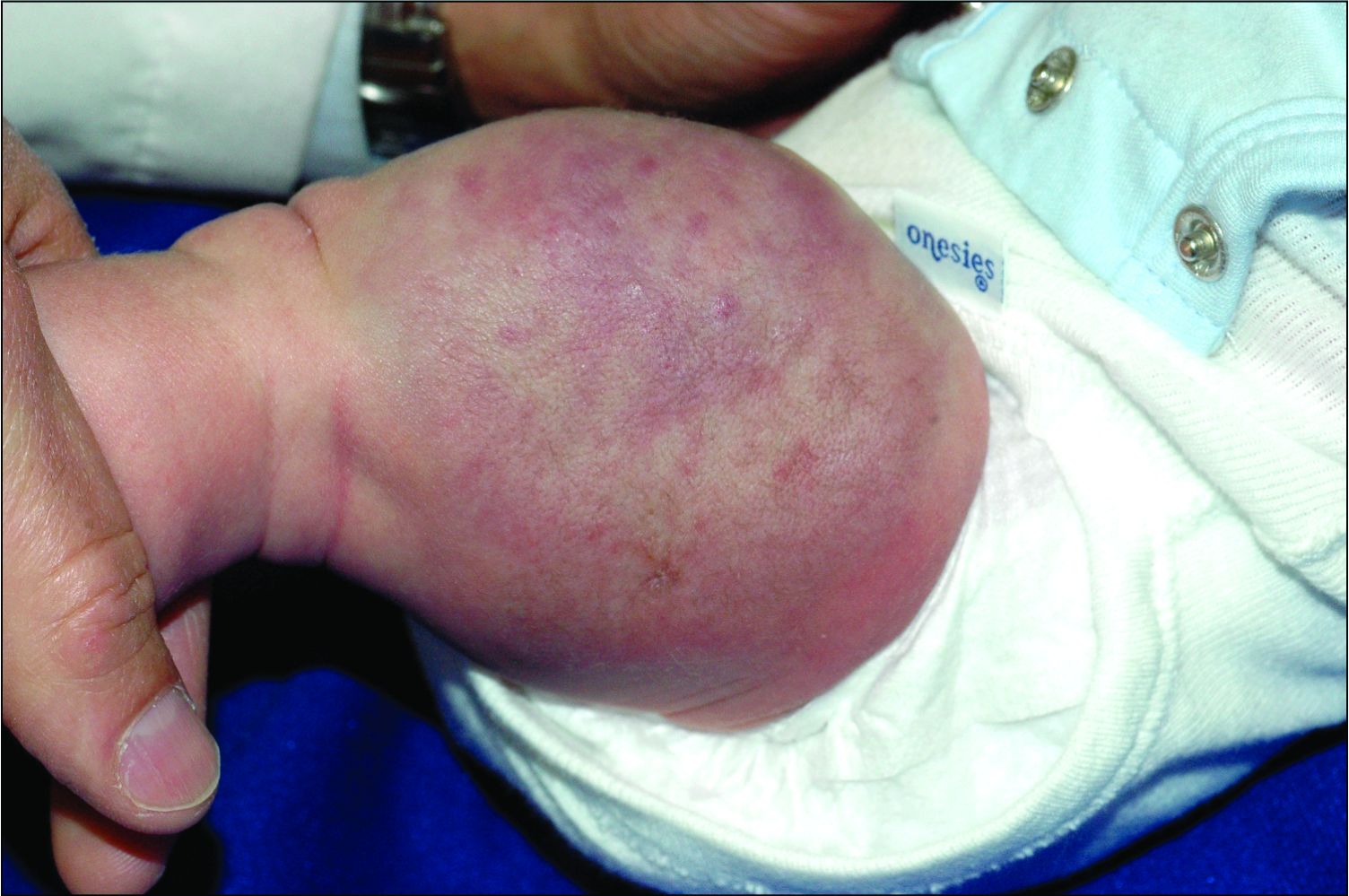
On physical exam, you see an infant with a mass of the left lower extremity. Close examination reveals an approximately 7 cm x 8 cm poorly defined mass with overlying central erythematous to violaceous color of the left anterior upper leg with a lumpy texture. The lesion is moderately firm and mildly tender on palpation.
A young girl presents with ‘itchy, rashy’ hands
Given the presence of erythema, lichenification, fissuring, and scale of the hands over the course of more than 3 months with the absence of nail findings is most consistent with a diagnosis of chronic hand eczema.
Chronic hand eczema (CHE) is an inflammatory dermatitis of the hands or wrists that persists for longer than 3 months or recurs twice or more in a 12-month timespan.1,2 Hand eczema can be a manifestation of atopic dermatitis, allergic contact dermatitis, or irritant contact dermatitis. Its multifactorial pathogenesis includes epidermal injury and disturbed epidermal barrier function from exogenous factors such as irritants or contact allergens, as well as endogenous factors including atopic dermatitis.3 In pediatrics, it often presents after an acute phase of hand dermatitis with chronic pruritus, erythema, and dry skin with scale.4 Examination findings vary widely with erythema, vesicles, scale, fissures, crusting, hyperkeratosis, and/or lichenification.3,5 Diagnosis is often achieved with careful history, asking about potential exposures that may induce lesions, and physical exam of the entire skin, including the feet. Based upon clinical history or persistent dermatitis, allergic contact dermatitis patch testing should be considered.2
What’s the treatment plan?
Given that CHE is an inflammatory disease process, the goal of treatment is to reduce inflammation and allow for skin barrier repair. Unfortunately, only one study has investigated therapeutics for pediatric CHE,6 with the remainder of the literature based on adult CHE. Current CHE guidelines recommend avoidance of allergens, irritants, or other triggers of the disease as well as liberal and regular use of emollients. Because of the relative thickness of hand skin, higher-potency topical corticosteroids are often used as first-line therapy, with lower-strength topical steroids, calcineurin inhibitors, or crisaborole used as maintenance therapy. Other treatment options include phototherapy, and rarely, systemic therapies are utilized for atopic dermatitis.
What’s the differential diagnosis?
The differential diagnosis of CHE includes other scaling or hyperkeratotic skin conditions including psoriasis and tinea manuum. Other skin conditions that localize to extremities including scabies and hand-foot-and-mouth disease are discussed below.
Psoriasis can present on the hands with erythematous, well-demarcated, silver scaling plaques. However, additional plaques may be found on the elbows, knees, scalp, umbilicus, and sacrum. Nails can demonstrate pitting, oil drops, splinter hemorrhages, or onycholysis. First-line treatment includes a combination of topical steroids, topical vitamin D analogues, and keratolytics.
Tinea mannum is a dermatophyte infection of the skin of the hands. Typically, only one hand is affected with concomitant bilateral tinea pedis. It results in a white scaly plaque with dorsal hand involvement demonstrating an annular appearance, elevated edge, and central clearing. KOH prep will demonstrate septate hyphae, and cultures will grow dermatophyte colonies. Treatment includes topical antifungals or systemic antifungals for recalcitrant disease.
Scabies presents with short linear hypopigmented lesions with a black dot on one end as well as erythematous pruritic papules. These appear on the interdigital web spaces, wrists, axilla, buttocks, and genital region. Skin scraping prep with mineral oil can show mites and eggs. All individuals in an affected household should be treated with either topical permethrin or oral ivermectin to avoid reinfection or parasitic spread. All contacted linens must be cleaned with hot water and dried on high heat.
Hand-foot-and-mouth disease, classically caused by coxsackievirus, is an acute viral illness that results in an eruption of erythematous macules, papules, and vesicles on the ventral hands, soles of the feet, and oral mucosa. Diagnosis is achieved clinically and treatment is symptomatic as the lesions are self-limited.
Our patient underwent patch testing but did not return positive to any allergens. She was started on potent topical corticosteroids, educated on trigger avoidance, and gradually achieved good disease control.
Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
Michael Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is a 4th year medical student at the University of Rochester (N.Y.). Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital.
References
1. Diepgen TL et al. Br J Dermatol. 2009;160(2):353-8.
2. Diepgen TL et al. J Dtsch Dermatol Ges. 2015;13(1):e1-22.
3. Agner T and Elsner P. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:4-12.
4. Mortz CG et al. Br J Dermatol. 2001;144(3):523-32.
5. Silvestre Salvador JF et al. Actas Dermosifiliogr. 2020;111(1):26-40.
6. Luchsinger I et al. J Eur Acad Dermatol Venereol. 2020;34(5):1037-42.
7. English J et al. Clin Exp Dermatol. 2009;34(7):761-9.
8. Elsner P and Agner T. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:13-21.
Given the presence of erythema, lichenification, fissuring, and scale of the hands over the course of more than 3 months with the absence of nail findings is most consistent with a diagnosis of chronic hand eczema.
Chronic hand eczema (CHE) is an inflammatory dermatitis of the hands or wrists that persists for longer than 3 months or recurs twice or more in a 12-month timespan.1,2 Hand eczema can be a manifestation of atopic dermatitis, allergic contact dermatitis, or irritant contact dermatitis. Its multifactorial pathogenesis includes epidermal injury and disturbed epidermal barrier function from exogenous factors such as irritants or contact allergens, as well as endogenous factors including atopic dermatitis.3 In pediatrics, it often presents after an acute phase of hand dermatitis with chronic pruritus, erythema, and dry skin with scale.4 Examination findings vary widely with erythema, vesicles, scale, fissures, crusting, hyperkeratosis, and/or lichenification.3,5 Diagnosis is often achieved with careful history, asking about potential exposures that may induce lesions, and physical exam of the entire skin, including the feet. Based upon clinical history or persistent dermatitis, allergic contact dermatitis patch testing should be considered.2
What’s the treatment plan?
Given that CHE is an inflammatory disease process, the goal of treatment is to reduce inflammation and allow for skin barrier repair. Unfortunately, only one study has investigated therapeutics for pediatric CHE,6 with the remainder of the literature based on adult CHE. Current CHE guidelines recommend avoidance of allergens, irritants, or other triggers of the disease as well as liberal and regular use of emollients. Because of the relative thickness of hand skin, higher-potency topical corticosteroids are often used as first-line therapy, with lower-strength topical steroids, calcineurin inhibitors, or crisaborole used as maintenance therapy. Other treatment options include phototherapy, and rarely, systemic therapies are utilized for atopic dermatitis.
What’s the differential diagnosis?
The differential diagnosis of CHE includes other scaling or hyperkeratotic skin conditions including psoriasis and tinea manuum. Other skin conditions that localize to extremities including scabies and hand-foot-and-mouth disease are discussed below.
Psoriasis can present on the hands with erythematous, well-demarcated, silver scaling plaques. However, additional plaques may be found on the elbows, knees, scalp, umbilicus, and sacrum. Nails can demonstrate pitting, oil drops, splinter hemorrhages, or onycholysis. First-line treatment includes a combination of topical steroids, topical vitamin D analogues, and keratolytics.
Tinea mannum is a dermatophyte infection of the skin of the hands. Typically, only one hand is affected with concomitant bilateral tinea pedis. It results in a white scaly plaque with dorsal hand involvement demonstrating an annular appearance, elevated edge, and central clearing. KOH prep will demonstrate septate hyphae, and cultures will grow dermatophyte colonies. Treatment includes topical antifungals or systemic antifungals for recalcitrant disease.
Scabies presents with short linear hypopigmented lesions with a black dot on one end as well as erythematous pruritic papules. These appear on the interdigital web spaces, wrists, axilla, buttocks, and genital region. Skin scraping prep with mineral oil can show mites and eggs. All individuals in an affected household should be treated with either topical permethrin or oral ivermectin to avoid reinfection or parasitic spread. All contacted linens must be cleaned with hot water and dried on high heat.
Hand-foot-and-mouth disease, classically caused by coxsackievirus, is an acute viral illness that results in an eruption of erythematous macules, papules, and vesicles on the ventral hands, soles of the feet, and oral mucosa. Diagnosis is achieved clinically and treatment is symptomatic as the lesions are self-limited.
Our patient underwent patch testing but did not return positive to any allergens. She was started on potent topical corticosteroids, educated on trigger avoidance, and gradually achieved good disease control.
Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
Michael Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is a 4th year medical student at the University of Rochester (N.Y.). Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital.
References
1. Diepgen TL et al. Br J Dermatol. 2009;160(2):353-8.
2. Diepgen TL et al. J Dtsch Dermatol Ges. 2015;13(1):e1-22.
3. Agner T and Elsner P. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:4-12.
4. Mortz CG et al. Br J Dermatol. 2001;144(3):523-32.
5. Silvestre Salvador JF et al. Actas Dermosifiliogr. 2020;111(1):26-40.
6. Luchsinger I et al. J Eur Acad Dermatol Venereol. 2020;34(5):1037-42.
7. English J et al. Clin Exp Dermatol. 2009;34(7):761-9.
8. Elsner P and Agner T. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:13-21.
Given the presence of erythema, lichenification, fissuring, and scale of the hands over the course of more than 3 months with the absence of nail findings is most consistent with a diagnosis of chronic hand eczema.
Chronic hand eczema (CHE) is an inflammatory dermatitis of the hands or wrists that persists for longer than 3 months or recurs twice or more in a 12-month timespan.1,2 Hand eczema can be a manifestation of atopic dermatitis, allergic contact dermatitis, or irritant contact dermatitis. Its multifactorial pathogenesis includes epidermal injury and disturbed epidermal barrier function from exogenous factors such as irritants or contact allergens, as well as endogenous factors including atopic dermatitis.3 In pediatrics, it often presents after an acute phase of hand dermatitis with chronic pruritus, erythema, and dry skin with scale.4 Examination findings vary widely with erythema, vesicles, scale, fissures, crusting, hyperkeratosis, and/or lichenification.3,5 Diagnosis is often achieved with careful history, asking about potential exposures that may induce lesions, and physical exam of the entire skin, including the feet. Based upon clinical history or persistent dermatitis, allergic contact dermatitis patch testing should be considered.2
What’s the treatment plan?
Given that CHE is an inflammatory disease process, the goal of treatment is to reduce inflammation and allow for skin barrier repair. Unfortunately, only one study has investigated therapeutics for pediatric CHE,6 with the remainder of the literature based on adult CHE. Current CHE guidelines recommend avoidance of allergens, irritants, or other triggers of the disease as well as liberal and regular use of emollients. Because of the relative thickness of hand skin, higher-potency topical corticosteroids are often used as first-line therapy, with lower-strength topical steroids, calcineurin inhibitors, or crisaborole used as maintenance therapy. Other treatment options include phototherapy, and rarely, systemic therapies are utilized for atopic dermatitis.
What’s the differential diagnosis?
The differential diagnosis of CHE includes other scaling or hyperkeratotic skin conditions including psoriasis and tinea manuum. Other skin conditions that localize to extremities including scabies and hand-foot-and-mouth disease are discussed below.
Psoriasis can present on the hands with erythematous, well-demarcated, silver scaling plaques. However, additional plaques may be found on the elbows, knees, scalp, umbilicus, and sacrum. Nails can demonstrate pitting, oil drops, splinter hemorrhages, or onycholysis. First-line treatment includes a combination of topical steroids, topical vitamin D analogues, and keratolytics.
Tinea mannum is a dermatophyte infection of the skin of the hands. Typically, only one hand is affected with concomitant bilateral tinea pedis. It results in a white scaly plaque with dorsal hand involvement demonstrating an annular appearance, elevated edge, and central clearing. KOH prep will demonstrate septate hyphae, and cultures will grow dermatophyte colonies. Treatment includes topical antifungals or systemic antifungals for recalcitrant disease.
Scabies presents with short linear hypopigmented lesions with a black dot on one end as well as erythematous pruritic papules. These appear on the interdigital web spaces, wrists, axilla, buttocks, and genital region. Skin scraping prep with mineral oil can show mites and eggs. All individuals in an affected household should be treated with either topical permethrin or oral ivermectin to avoid reinfection or parasitic spread. All contacted linens must be cleaned with hot water and dried on high heat.
Hand-foot-and-mouth disease, classically caused by coxsackievirus, is an acute viral illness that results in an eruption of erythematous macules, papules, and vesicles on the ventral hands, soles of the feet, and oral mucosa. Diagnosis is achieved clinically and treatment is symptomatic as the lesions are self-limited.
Our patient underwent patch testing but did not return positive to any allergens. She was started on potent topical corticosteroids, educated on trigger avoidance, and gradually achieved good disease control.
Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
Michael Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is a 4th year medical student at the University of Rochester (N.Y.). Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital.
References
1. Diepgen TL et al. Br J Dermatol. 2009;160(2):353-8.
2. Diepgen TL et al. J Dtsch Dermatol Ges. 2015;13(1):e1-22.
3. Agner T and Elsner P. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:4-12.
4. Mortz CG et al. Br J Dermatol. 2001;144(3):523-32.
5. Silvestre Salvador JF et al. Actas Dermosifiliogr. 2020;111(1):26-40.
6. Luchsinger I et al. J Eur Acad Dermatol Venereol. 2020;34(5):1037-42.
7. English J et al. Clin Exp Dermatol. 2009;34(7):761-9.
8. Elsner P and Agner T. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:13-21.
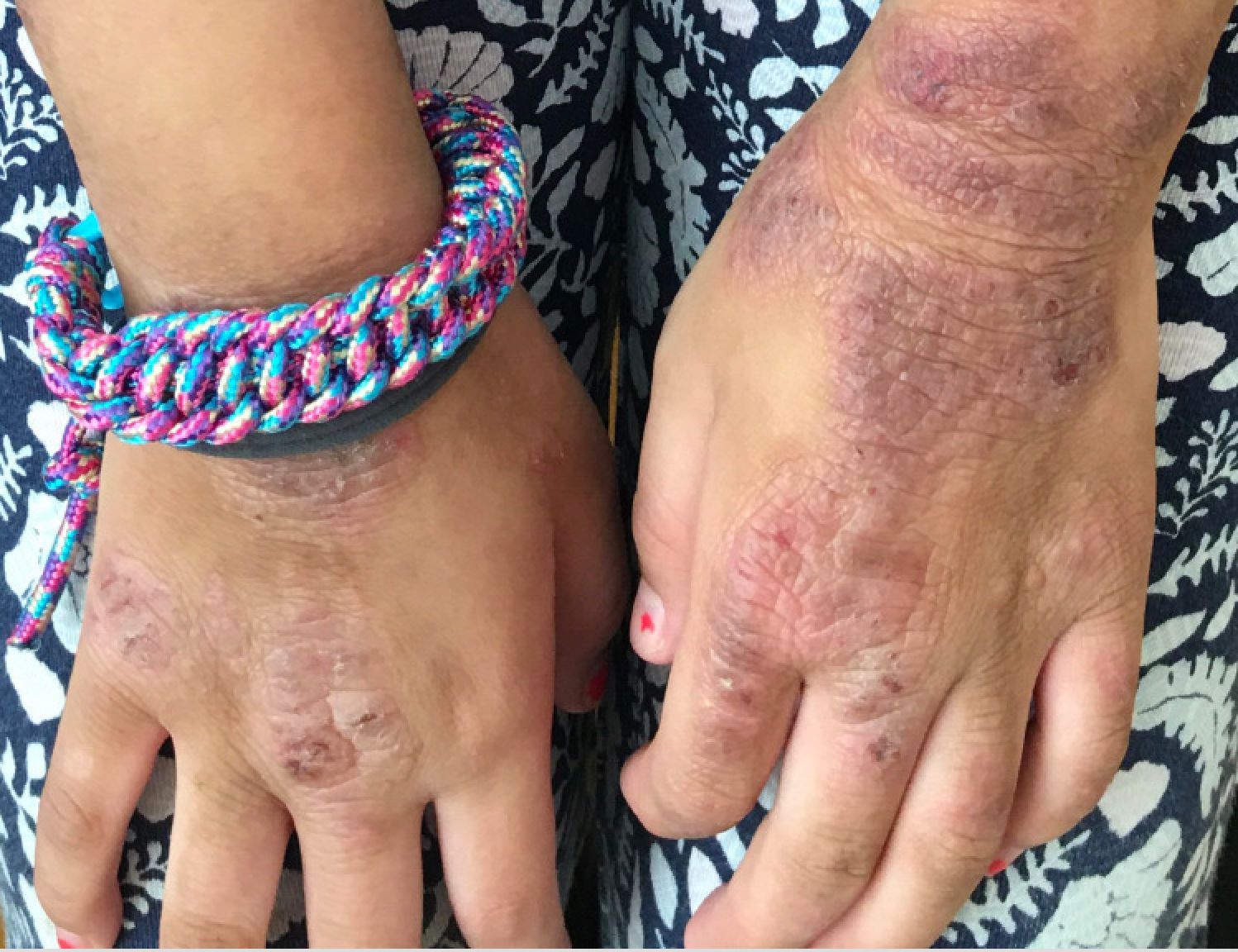
Examination findings of the bilateral hands and wrists demonstrate plaques of erythema, lichenification, and scale of the dorsal surfaces of the hands and digits. Closer inspection reveals fissuring and erythematous crust of the affected skin but normal nails. The rest of the skin exam is unremarkable.
An 11-year-old female with a 3-year history of alopecia
Given the longstanding scarring alopecia, with negative fungal cultures and with perifollicular erythema and scaling, this diagnosis is most consistent with lichen planopilaris.
Lichen planopilaris (LPP) is considered one of the primary scarring alopecias, a group of diseases characterized by inflammation and subsequent irreversible hair loss.1 LPP specifically is believed to be caused by dysfunction of cell-mediated immunity, resulting in T lymphocytes attacking follicular hair stem cells.2 It typically presents with hair loss, pruritus, scaling, burning pain, and tenderness of the scalp when active,1,3 with exam showing perifollicular scale and erythema on the borders of the patches of alopecia.4,5 Over time, scarring of the scalp develops with loss of follicular ostia.1 Definitive diagnosis typically requires punch biopsy of the affected scalp, as such can determine the presence or absence of inflammation in affected areas of the scalp.1
What’s the treatment plan?
Given that LPP is an autoimmune inflammatory disease process, the goal of treatment is to calm down the inflammation of the scalp to prevent further progression of a patient’s hair loss. This is typically achieved with superpotent topical corticosteroids, such as clobetasol applied directly to the scalp, and/or intralesional corticosteroids, such as triamcinolone acetonide suspension injected directly to the affected scalp.3,6,7 Other treatment options include systemic agents, such as hydroxychloroquine, methotrexate, mycophenolate mofetil, pioglitazone, and doxycycline.3,6 Hair loss is not reversible as loss of follicular ostia and hair stem cells results in permanent scarring.1 Management often requires a referral to dermatology for aggressive treatment to prevent further hair loss.
What’s the differential diagnosis?
The differential diagnosis of lichen planopilaris includes other scarring alopecias, including central centrifugal cicatricial alopecia, discoid lupus erythematosus, folliculitis decalvans. While nonscarring, alopecia areata, trichotillomania, and telogen effluvium are discussed below as well.
Central centrifugal cicatricial alopecia is very rare in pediatrics, and is a type of asymptomatic scarring alopecia that begins at the vertex of the scalp, spreading centrifugally and resulting in shiny plaque development. Treatment involves reduction of hair grooming as well as topical and intralesional steroids.
Discoid lupus erythematosus presents as scaling erythematous plaques on the face and scalp that result in skin pigment changes and atrophy over time. Scalp involvement results in scarring alopecia. Treatment includes the use of high-potency topical corticosteroids, topical calcineurin inhibitors, and hydroxychloroquine.
Folliculitis decalvans is another form of scarring alopecia believed to be caused by an inflammatory response to Staphylococcus aureus in the scalp, resulting in the formation of scarring of the scalp and perifollicular pustules. Treatment is topical antibiotics and intralesional steroids.
Alopecia areata is a form of nonscarring alopecia resulting in small round patches of partially reversible hair loss characterized by the pathognomonic finding of so-called exclamation point hairs that are broader distally and taper toward the scalp on physical exam. Considered an autoimmune disorder, it varies greatly in extent and course. While focal hair loss is the hallmark of this disease, usually hair follicles are present.
Trichotillosis, also known as trichotillomania (hair pulling), results in alopecia with irregular borders and broken hairs of different lengths secondary to the urge to remove or pull one’s own hair, resulting in nonscarring alopecia. It may be associated with stress or anxiety, obsessive-compulsive disorders, or other repetitive body-altering behaviors. Treatments include reassurance and education as it can be self-limited in some, behavior modification, or systemic therapy including tricyclic antidepressants or SSRIs.
Our patient underwent scalp punch biopsy to confirm the diagnosis and was started on potent topical corticosteroids with good disease control.
Dr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university, and he is chief of pediatric and adolescent dermatology at the hospital. Neither of the doctors had any relevant financial disclosures. Email them at pdnews@mdedge.com.
References
1. J Am Acad Dermatol. 2005 Jul. doi: 10.1016/j.jaad.2004.06.015.
2. J Pathol. 2013 Oct. doi: 10.1002/path.4233.
3. Pediatr Dermatol. 2015 Sep-Oct. doi: 10.1111/pde.12624.
4. J Am Acad Dermatol. 2004 Jan. doi: 10.1016/j.jaad.2003.04.001.
5. J Am Acad Dermatol. 1992 Dec. doi: 10.1016/0190-9622(92)70290-v.
6. Clin Cosmet Investig Dermatol. 2018 Feb 27. doi: 10.2147/CCID.S137870.
7. Semin Cutan Med Surg. 2009 Mar. doi: 10.1016/j.sder.2008.12.006.
Given the longstanding scarring alopecia, with negative fungal cultures and with perifollicular erythema and scaling, this diagnosis is most consistent with lichen planopilaris.
Lichen planopilaris (LPP) is considered one of the primary scarring alopecias, a group of diseases characterized by inflammation and subsequent irreversible hair loss.1 LPP specifically is believed to be caused by dysfunction of cell-mediated immunity, resulting in T lymphocytes attacking follicular hair stem cells.2 It typically presents with hair loss, pruritus, scaling, burning pain, and tenderness of the scalp when active,1,3 with exam showing perifollicular scale and erythema on the borders of the patches of alopecia.4,5 Over time, scarring of the scalp develops with loss of follicular ostia.1 Definitive diagnosis typically requires punch biopsy of the affected scalp, as such can determine the presence or absence of inflammation in affected areas of the scalp.1
What’s the treatment plan?
Given that LPP is an autoimmune inflammatory disease process, the goal of treatment is to calm down the inflammation of the scalp to prevent further progression of a patient’s hair loss. This is typically achieved with superpotent topical corticosteroids, such as clobetasol applied directly to the scalp, and/or intralesional corticosteroids, such as triamcinolone acetonide suspension injected directly to the affected scalp.3,6,7 Other treatment options include systemic agents, such as hydroxychloroquine, methotrexate, mycophenolate mofetil, pioglitazone, and doxycycline.3,6 Hair loss is not reversible as loss of follicular ostia and hair stem cells results in permanent scarring.1 Management often requires a referral to dermatology for aggressive treatment to prevent further hair loss.
What’s the differential diagnosis?
The differential diagnosis of lichen planopilaris includes other scarring alopecias, including central centrifugal cicatricial alopecia, discoid lupus erythematosus, folliculitis decalvans. While nonscarring, alopecia areata, trichotillomania, and telogen effluvium are discussed below as well.
Central centrifugal cicatricial alopecia is very rare in pediatrics, and is a type of asymptomatic scarring alopecia that begins at the vertex of the scalp, spreading centrifugally and resulting in shiny plaque development. Treatment involves reduction of hair grooming as well as topical and intralesional steroids.
Discoid lupus erythematosus presents as scaling erythematous plaques on the face and scalp that result in skin pigment changes and atrophy over time. Scalp involvement results in scarring alopecia. Treatment includes the use of high-potency topical corticosteroids, topical calcineurin inhibitors, and hydroxychloroquine.
Folliculitis decalvans is another form of scarring alopecia believed to be caused by an inflammatory response to Staphylococcus aureus in the scalp, resulting in the formation of scarring of the scalp and perifollicular pustules. Treatment is topical antibiotics and intralesional steroids.
Alopecia areata is a form of nonscarring alopecia resulting in small round patches of partially reversible hair loss characterized by the pathognomonic finding of so-called exclamation point hairs that are broader distally and taper toward the scalp on physical exam. Considered an autoimmune disorder, it varies greatly in extent and course. While focal hair loss is the hallmark of this disease, usually hair follicles are present.
Trichotillosis, also known as trichotillomania (hair pulling), results in alopecia with irregular borders and broken hairs of different lengths secondary to the urge to remove or pull one’s own hair, resulting in nonscarring alopecia. It may be associated with stress or anxiety, obsessive-compulsive disorders, or other repetitive body-altering behaviors. Treatments include reassurance and education as it can be self-limited in some, behavior modification, or systemic therapy including tricyclic antidepressants or SSRIs.
Our patient underwent scalp punch biopsy to confirm the diagnosis and was started on potent topical corticosteroids with good disease control.
Dr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university, and he is chief of pediatric and adolescent dermatology at the hospital. Neither of the doctors had any relevant financial disclosures. Email them at pdnews@mdedge.com.
References
1. J Am Acad Dermatol. 2005 Jul. doi: 10.1016/j.jaad.2004.06.015.
2. J Pathol. 2013 Oct. doi: 10.1002/path.4233.
3. Pediatr Dermatol. 2015 Sep-Oct. doi: 10.1111/pde.12624.
4. J Am Acad Dermatol. 2004 Jan. doi: 10.1016/j.jaad.2003.04.001.
5. J Am Acad Dermatol. 1992 Dec. doi: 10.1016/0190-9622(92)70290-v.
6. Clin Cosmet Investig Dermatol. 2018 Feb 27. doi: 10.2147/CCID.S137870.
7. Semin Cutan Med Surg. 2009 Mar. doi: 10.1016/j.sder.2008.12.006.
Given the longstanding scarring alopecia, with negative fungal cultures and with perifollicular erythema and scaling, this diagnosis is most consistent with lichen planopilaris.
Lichen planopilaris (LPP) is considered one of the primary scarring alopecias, a group of diseases characterized by inflammation and subsequent irreversible hair loss.1 LPP specifically is believed to be caused by dysfunction of cell-mediated immunity, resulting in T lymphocytes attacking follicular hair stem cells.2 It typically presents with hair loss, pruritus, scaling, burning pain, and tenderness of the scalp when active,1,3 with exam showing perifollicular scale and erythema on the borders of the patches of alopecia.4,5 Over time, scarring of the scalp develops with loss of follicular ostia.1 Definitive diagnosis typically requires punch biopsy of the affected scalp, as such can determine the presence or absence of inflammation in affected areas of the scalp.1
What’s the treatment plan?
Given that LPP is an autoimmune inflammatory disease process, the goal of treatment is to calm down the inflammation of the scalp to prevent further progression of a patient’s hair loss. This is typically achieved with superpotent topical corticosteroids, such as clobetasol applied directly to the scalp, and/or intralesional corticosteroids, such as triamcinolone acetonide suspension injected directly to the affected scalp.3,6,7 Other treatment options include systemic agents, such as hydroxychloroquine, methotrexate, mycophenolate mofetil, pioglitazone, and doxycycline.3,6 Hair loss is not reversible as loss of follicular ostia and hair stem cells results in permanent scarring.1 Management often requires a referral to dermatology for aggressive treatment to prevent further hair loss.
What’s the differential diagnosis?
The differential diagnosis of lichen planopilaris includes other scarring alopecias, including central centrifugal cicatricial alopecia, discoid lupus erythematosus, folliculitis decalvans. While nonscarring, alopecia areata, trichotillomania, and telogen effluvium are discussed below as well.
Central centrifugal cicatricial alopecia is very rare in pediatrics, and is a type of asymptomatic scarring alopecia that begins at the vertex of the scalp, spreading centrifugally and resulting in shiny plaque development. Treatment involves reduction of hair grooming as well as topical and intralesional steroids.
Discoid lupus erythematosus presents as scaling erythematous plaques on the face and scalp that result in skin pigment changes and atrophy over time. Scalp involvement results in scarring alopecia. Treatment includes the use of high-potency topical corticosteroids, topical calcineurin inhibitors, and hydroxychloroquine.
Folliculitis decalvans is another form of scarring alopecia believed to be caused by an inflammatory response to Staphylococcus aureus in the scalp, resulting in the formation of scarring of the scalp and perifollicular pustules. Treatment is topical antibiotics and intralesional steroids.
Alopecia areata is a form of nonscarring alopecia resulting in small round patches of partially reversible hair loss characterized by the pathognomonic finding of so-called exclamation point hairs that are broader distally and taper toward the scalp on physical exam. Considered an autoimmune disorder, it varies greatly in extent and course. While focal hair loss is the hallmark of this disease, usually hair follicles are present.
Trichotillosis, also known as trichotillomania (hair pulling), results in alopecia with irregular borders and broken hairs of different lengths secondary to the urge to remove or pull one’s own hair, resulting in nonscarring alopecia. It may be associated with stress or anxiety, obsessive-compulsive disorders, or other repetitive body-altering behaviors. Treatments include reassurance and education as it can be self-limited in some, behavior modification, or systemic therapy including tricyclic antidepressants or SSRIs.
Our patient underwent scalp punch biopsy to confirm the diagnosis and was started on potent topical corticosteroids with good disease control.
Dr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university, and he is chief of pediatric and adolescent dermatology at the hospital. Neither of the doctors had any relevant financial disclosures. Email them at pdnews@mdedge.com.
References
1. J Am Acad Dermatol. 2005 Jul. doi: 10.1016/j.jaad.2004.06.015.
2. J Pathol. 2013 Oct. doi: 10.1002/path.4233.
3. Pediatr Dermatol. 2015 Sep-Oct. doi: 10.1111/pde.12624.
4. J Am Acad Dermatol. 2004 Jan. doi: 10.1016/j.jaad.2003.04.001.
5. J Am Acad Dermatol. 1992 Dec. doi: 10.1016/0190-9622(92)70290-v.
6. Clin Cosmet Investig Dermatol. 2018 Feb 27. doi: 10.2147/CCID.S137870.
7. Semin Cutan Med Surg. 2009 Mar. doi: 10.1016/j.sder.2008.12.006.
An 11-year-old female is seen in clinic with a 3-year history of alopecia. The patient recently immigrated to the United States from Afghanistan. Prior to immigrating, she was evaluated for "scarring alopecia" and had been treated with oral and topical steroids as well as oral and topical antifungals. When active, she had itching and tenderness. She is not actively losing any hair at this time, but she has not regrown any of her hair. The patient has no family members with alopecia. She reports some burning pain and itching of her scalp, and denies any muscle pain or weakness or sun sensitivity.
On physical exam, you see 50% loss of hair on the superior scalp with preservation of the anterior hair line. Patches of hair can be seen throughout, with segments of smooth-skinned alopecia, without pustules. There is a loss of the follicle pattern in scarred areas, and magnification or "dermoscopy" shows perifollicular erythema and scaling at the border of the affected scalp. Labs are all within normal limits. Bacterial and fungal cultures of the scalp do not grow organisms.
A 5-year-old boy with a papular rash on his arm
Lichen striatus (LS) is a common benign skin condition that presents in children between the ages of 5 and 15 years.1 The rash is typically unilateral and most frequently on the extremities, although it may appear on the face, trunk, or buttocks. The lesions start as pink or skin-colored asymptomatic papules in a linear orientation following the lines of Blaschko. There may be residual postinflammatory hypo- or hyperpigmentation which often improves within a few years.

Of note, there are subsets of lichen striatus: Hypopigmented lichen striatus with minimal papules has been termed “lichen striatus albus.” Nail lichen striatus may present as onycholysis or fissuring of nails, present as an isolated finding, or more commonly in association with concurrent affected skin. Nail lichen striatus typically resolves on its own, however there are case reports of improvement with intralesional steroids.2
There is no established etiology for LS. Autoimmune disease, viruses, immunizations, medications, and hypersensitivity reactions have been associated with triggering LS in various case reports, although strength of the associations is low. Children have been reported to have LS following scarlet fever and Candida vulvitis.3 Diagnosis usually is clinical, although biopsy may be helpful for histopathologic confirmation. No work-up for associated infections or conditions is warranted.
The differential for linear papular lesions includes inflammatory linear verrucous epidermal nevus (ILVEN), blaschkitis, or linear morphea. ILVEN is a hamartoma that usually is congenital or presents in early childhood; presents with linear or whorled, hyperkeratotic papules and plaque in similar linear “line of Blaschko” patterns; and represents cutaneous mosaicism. It is often difficult to differentiate between lichen striatus and ILVEN, however lichen striatus is not congenital, and is a self-limited condition. Under dermoscopy (polarized light systems) findings of LS more frequently demonstrate gray granular pigmentation. ILVEN is more frequently associated with cerebriform pattern.4 Blaschkitis is a term for a blaschkoid inflammation of the skin that presents with more eczematous findings and histology of spongiosis, unlike the lichenoid findings of LS. It is typically accompanied by noticeable pruritus and broader bands of involved area, and has older age of onset than LS. Linear morphea is a deeper inflammatory process of the dermis or subcutaneous fat, presenting with sclerotic skin, and typically has associated atrophy.
Treatment need not be pursued for lichen striatus because it is a benign condition. The lesions typically self-resolve without any residual scarring. If patients have associated pruritus then low- to midpotency topical steroids can be used for symptomatic relief.
Dr. Kaushik is with the division of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego, and Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Dr. Kaushik or Dr. Eichenfield. Email them at pdnews@mdedge.com.
References
1. Gupta D, Mathes E. Lichen Striatus. (Levy ML ed.) 2019: UpToDate.
2. Dermatol Ther. 2018 Nov;31(6):e12713.
3. Int J Dermatol. 2018 Sep;57(9):1118-9.
4. J Dermatol. 2017 Dec;44(12):e355-6.
Lichen striatus (LS) is a common benign skin condition that presents in children between the ages of 5 and 15 years.1 The rash is typically unilateral and most frequently on the extremities, although it may appear on the face, trunk, or buttocks. The lesions start as pink or skin-colored asymptomatic papules in a linear orientation following the lines of Blaschko. There may be residual postinflammatory hypo- or hyperpigmentation which often improves within a few years.
Of note, there are subsets of lichen striatus: Hypopigmented lichen striatus with minimal papules has been termed “lichen striatus albus.” Nail lichen striatus may present as onycholysis or fissuring of nails, present as an isolated finding, or more commonly in association with concurrent affected skin. Nail lichen striatus typically resolves on its own, however there are case reports of improvement with intralesional steroids.2
There is no established etiology for LS. Autoimmune disease, viruses, immunizations, medications, and hypersensitivity reactions have been associated with triggering LS in various case reports, although strength of the associations is low. Children have been reported to have LS following scarlet fever and Candida vulvitis.3 Diagnosis usually is clinical, although biopsy may be helpful for histopathologic confirmation. No work-up for associated infections or conditions is warranted.
The differential for linear papular lesions includes inflammatory linear verrucous epidermal nevus (ILVEN), blaschkitis, or linear morphea. ILVEN is a hamartoma that usually is congenital or presents in early childhood; presents with linear or whorled, hyperkeratotic papules and plaque in similar linear “line of Blaschko” patterns; and represents cutaneous mosaicism. It is often difficult to differentiate between lichen striatus and ILVEN, however lichen striatus is not congenital, and is a self-limited condition. Under dermoscopy (polarized light systems) findings of LS more frequently demonstrate gray granular pigmentation. ILVEN is more frequently associated with cerebriform pattern.4 Blaschkitis is a term for a blaschkoid inflammation of the skin that presents with more eczematous findings and histology of spongiosis, unlike the lichenoid findings of LS. It is typically accompanied by noticeable pruritus and broader bands of involved area, and has older age of onset than LS. Linear morphea is a deeper inflammatory process of the dermis or subcutaneous fat, presenting with sclerotic skin, and typically has associated atrophy.
Treatment need not be pursued for lichen striatus because it is a benign condition. The lesions typically self-resolve without any residual scarring. If patients have associated pruritus then low- to midpotency topical steroids can be used for symptomatic relief.
Dr. Kaushik is with the division of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego, and Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Dr. Kaushik or Dr. Eichenfield. Email them at pdnews@mdedge.com.
References
1. Gupta D, Mathes E. Lichen Striatus. (Levy ML ed.) 2019: UpToDate.
2. Dermatol Ther. 2018 Nov;31(6):e12713.
3. Int J Dermatol. 2018 Sep;57(9):1118-9.
4. J Dermatol. 2017 Dec;44(12):e355-6.
Lichen striatus (LS) is a common benign skin condition that presents in children between the ages of 5 and 15 years.1 The rash is typically unilateral and most frequently on the extremities, although it may appear on the face, trunk, or buttocks. The lesions start as pink or skin-colored asymptomatic papules in a linear orientation following the lines of Blaschko. There may be residual postinflammatory hypo- or hyperpigmentation which often improves within a few years.
Of note, there are subsets of lichen striatus: Hypopigmented lichen striatus with minimal papules has been termed “lichen striatus albus.” Nail lichen striatus may present as onycholysis or fissuring of nails, present as an isolated finding, or more commonly in association with concurrent affected skin. Nail lichen striatus typically resolves on its own, however there are case reports of improvement with intralesional steroids.2
There is no established etiology for LS. Autoimmune disease, viruses, immunizations, medications, and hypersensitivity reactions have been associated with triggering LS in various case reports, although strength of the associations is low. Children have been reported to have LS following scarlet fever and Candida vulvitis.3 Diagnosis usually is clinical, although biopsy may be helpful for histopathologic confirmation. No work-up for associated infections or conditions is warranted.
The differential for linear papular lesions includes inflammatory linear verrucous epidermal nevus (ILVEN), blaschkitis, or linear morphea. ILVEN is a hamartoma that usually is congenital or presents in early childhood; presents with linear or whorled, hyperkeratotic papules and plaque in similar linear “line of Blaschko” patterns; and represents cutaneous mosaicism. It is often difficult to differentiate between lichen striatus and ILVEN, however lichen striatus is not congenital, and is a self-limited condition. Under dermoscopy (polarized light systems) findings of LS more frequently demonstrate gray granular pigmentation. ILVEN is more frequently associated with cerebriform pattern.4 Blaschkitis is a term for a blaschkoid inflammation of the skin that presents with more eczematous findings and histology of spongiosis, unlike the lichenoid findings of LS. It is typically accompanied by noticeable pruritus and broader bands of involved area, and has older age of onset than LS. Linear morphea is a deeper inflammatory process of the dermis or subcutaneous fat, presenting with sclerotic skin, and typically has associated atrophy.
Treatment need not be pursued for lichen striatus because it is a benign condition. The lesions typically self-resolve without any residual scarring. If patients have associated pruritus then low- to midpotency topical steroids can be used for symptomatic relief.
Dr. Kaushik is with the division of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego, and Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital-San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. There are no conflicts of interest or financial disclosures for Dr. Kaushik or Dr. Eichenfield. Email them at pdnews@mdedge.com.
References
1. Gupta D, Mathes E. Lichen Striatus. (Levy ML ed.) 2019: UpToDate.
2. Dermatol Ther. 2018 Nov;31(6):e12713.
3. Int J Dermatol. 2018 Sep;57(9):1118-9.
4. J Dermatol. 2017 Dec;44(12):e355-6.