User login
Mitchel is a reporter for MDedge based in the Philadelphia area. He started with the company in 1992, when it was International Medical News Group (IMNG), and has since covered a range of medical specialties. Mitchel trained as a virologist at Roswell Park Memorial Institute in Buffalo, and then worked briefly as a researcher at Boston Children's Hospital before pivoting to journalism as a AAAS Mass Media Fellow in 1980. His first reporting job was with Science Digest magazine, and from the mid-1980s to early-1990s he was a reporter with Medical World News. @mitchelzoler
Pregnancy Does Not Preclude Safe DVT Treatment
MIAMI BEACH – Women who develop a deep vein thrombosis while pregnant can safely undergo removal of the clot without jeopardizing their pregnancy, based on a recent experience with 11 cases at one U.S. center.
"Not many vascular surgeons are willing to perform thrombectomy in pregnant women, and most physicians fear thrombolytic therapy in pregnant patients," Dr. Anthony J. Comerota said at ISET 2012, an international symposium on endovascular therapy.

But in reality, there is no evidence that thrombolysis poses any special risk during pregnancy, in part because the drugs that are usually used – urokinase, streptokinase, or tissue plasminogen activator (TPA) – do not cross the placenta, and therefore do not exert any fibrinolytic effect on a fetus, he said. In addition, the relatively recent introduction of pharmacomechanical methods for thrombolysis have "accelerated our enthusiasm and strengthened our confidence that this is an effective and safe strategy for treating extensive DVT [deep vein thrombosis] during pregnancy," said Dr. Comerota, a vascular surgeon and director of the Jobst Vascular Institute in Toledo, Ohio.
"Pregnancy need not be a major contraindication for successful management of extensive DVT. We need to minimize radiation, monitor the fetus, and control uterine contractions, but we should treat healthy, pregnant women [who have] a DVT the same way we treat healthy nonpregnant women" who have a DVT, he said in an interview. "The extraordinary concern for risk to the pregnancy [from clot removal] seems substantially overstated."
Because pregnancy produces a prothrombotic state, the risk that a healthy, pregnant woman has for DVT is about sixfold higher than her risk when not pregnant. Until now, most physicians deferred endovascular or surgical management of women who develop a DVT during pregnancy, and have relied exclusively on medical treatment with heparin or low-molecular-weight heparin, a strategy that is often ineffective.
"What pushed me to treat women earlier during their pregnancy was that I saw patients who did not receive therapy until after delivery, and by then the clot became more organized – a mix of collagen and scar inside the vein – -and caused severe postthrombotic disability," Dr. Comerota said.
Recently, he treated 11 pregnant women (10 with an iliofemoral thrombosis and 1 with the clot in her superior vena cava). One woman was in the first trimester, two were in the second, and eight were in the third trimester. Three patients underwent clot removal by open surgery, with the other eight treated by endovascular methods, including six who were treated with catheter-delivered TPA and one who received urokinase via catheter. Other endovascular tools that were used as needed included ultrasound clot treatment and rheolytic thrombectomy. In all cases, these treatments successfully removed the clot. One women developed hematuria and required a transfusion following her clot removal, and another patient developed a popliteal artery pseudoaneurysm. None of the patients had a recurrent DVT; two developed mild postthrombotic symptoms.
In one case, the fetus spontaneously aborted 5 days after the procedure secondary to antiphospholipid antibody syndrome, which had also triggered the thrombosis. The other 10 cases resulted in successful pregnancies and live deliveries, one surgically and the other nine vaginally. Three of the women had successful subsequent pregnancies, Dr. Comerota said.
Dr. Comerota said that he has been a speaker for and a consultant to Covidien, a company that makes some of the devices used in these procedures.
MIAMI BEACH – Women who develop a deep vein thrombosis while pregnant can safely undergo removal of the clot without jeopardizing their pregnancy, based on a recent experience with 11 cases at one U.S. center.
"Not many vascular surgeons are willing to perform thrombectomy in pregnant women, and most physicians fear thrombolytic therapy in pregnant patients," Dr. Anthony J. Comerota said at ISET 2012, an international symposium on endovascular therapy.
But in reality, there is no evidence that thrombolysis poses any special risk during pregnancy, in part because the drugs that are usually used – urokinase, streptokinase, or tissue plasminogen activator (TPA) – do not cross the placenta, and therefore do not exert any fibrinolytic effect on a fetus, he said. In addition, the relatively recent introduction of pharmacomechanical methods for thrombolysis have "accelerated our enthusiasm and strengthened our confidence that this is an effective and safe strategy for treating extensive DVT [deep vein thrombosis] during pregnancy," said Dr. Comerota, a vascular surgeon and director of the Jobst Vascular Institute in Toledo, Ohio.
"Pregnancy need not be a major contraindication for successful management of extensive DVT. We need to minimize radiation, monitor the fetus, and control uterine contractions, but we should treat healthy, pregnant women [who have] a DVT the same way we treat healthy nonpregnant women" who have a DVT, he said in an interview. "The extraordinary concern for risk to the pregnancy [from clot removal] seems substantially overstated."
Because pregnancy produces a prothrombotic state, the risk that a healthy, pregnant woman has for DVT is about sixfold higher than her risk when not pregnant. Until now, most physicians deferred endovascular or surgical management of women who develop a DVT during pregnancy, and have relied exclusively on medical treatment with heparin or low-molecular-weight heparin, a strategy that is often ineffective.
"What pushed me to treat women earlier during their pregnancy was that I saw patients who did not receive therapy until after delivery, and by then the clot became more organized – a mix of collagen and scar inside the vein – -and caused severe postthrombotic disability," Dr. Comerota said.
Recently, he treated 11 pregnant women (10 with an iliofemoral thrombosis and 1 with the clot in her superior vena cava). One woman was in the first trimester, two were in the second, and eight were in the third trimester. Three patients underwent clot removal by open surgery, with the other eight treated by endovascular methods, including six who were treated with catheter-delivered TPA and one who received urokinase via catheter. Other endovascular tools that were used as needed included ultrasound clot treatment and rheolytic thrombectomy. In all cases, these treatments successfully removed the clot. One women developed hematuria and required a transfusion following her clot removal, and another patient developed a popliteal artery pseudoaneurysm. None of the patients had a recurrent DVT; two developed mild postthrombotic symptoms.
In one case, the fetus spontaneously aborted 5 days after the procedure secondary to antiphospholipid antibody syndrome, which had also triggered the thrombosis. The other 10 cases resulted in successful pregnancies and live deliveries, one surgically and the other nine vaginally. Three of the women had successful subsequent pregnancies, Dr. Comerota said.
Dr. Comerota said that he has been a speaker for and a consultant to Covidien, a company that makes some of the devices used in these procedures.
MIAMI BEACH – Women who develop a deep vein thrombosis while pregnant can safely undergo removal of the clot without jeopardizing their pregnancy, based on a recent experience with 11 cases at one U.S. center.
"Not many vascular surgeons are willing to perform thrombectomy in pregnant women, and most physicians fear thrombolytic therapy in pregnant patients," Dr. Anthony J. Comerota said at ISET 2012, an international symposium on endovascular therapy.
But in reality, there is no evidence that thrombolysis poses any special risk during pregnancy, in part because the drugs that are usually used – urokinase, streptokinase, or tissue plasminogen activator (TPA) – do not cross the placenta, and therefore do not exert any fibrinolytic effect on a fetus, he said. In addition, the relatively recent introduction of pharmacomechanical methods for thrombolysis have "accelerated our enthusiasm and strengthened our confidence that this is an effective and safe strategy for treating extensive DVT [deep vein thrombosis] during pregnancy," said Dr. Comerota, a vascular surgeon and director of the Jobst Vascular Institute in Toledo, Ohio.
"Pregnancy need not be a major contraindication for successful management of extensive DVT. We need to minimize radiation, monitor the fetus, and control uterine contractions, but we should treat healthy, pregnant women [who have] a DVT the same way we treat healthy nonpregnant women" who have a DVT, he said in an interview. "The extraordinary concern for risk to the pregnancy [from clot removal] seems substantially overstated."
Because pregnancy produces a prothrombotic state, the risk that a healthy, pregnant woman has for DVT is about sixfold higher than her risk when not pregnant. Until now, most physicians deferred endovascular or surgical management of women who develop a DVT during pregnancy, and have relied exclusively on medical treatment with heparin or low-molecular-weight heparin, a strategy that is often ineffective.
"What pushed me to treat women earlier during their pregnancy was that I saw patients who did not receive therapy until after delivery, and by then the clot became more organized – a mix of collagen and scar inside the vein – -and caused severe postthrombotic disability," Dr. Comerota said.
Recently, he treated 11 pregnant women (10 with an iliofemoral thrombosis and 1 with the clot in her superior vena cava). One woman was in the first trimester, two were in the second, and eight were in the third trimester. Three patients underwent clot removal by open surgery, with the other eight treated by endovascular methods, including six who were treated with catheter-delivered TPA and one who received urokinase via catheter. Other endovascular tools that were used as needed included ultrasound clot treatment and rheolytic thrombectomy. In all cases, these treatments successfully removed the clot. One women developed hematuria and required a transfusion following her clot removal, and another patient developed a popliteal artery pseudoaneurysm. None of the patients had a recurrent DVT; two developed mild postthrombotic symptoms.
In one case, the fetus spontaneously aborted 5 days after the procedure secondary to antiphospholipid antibody syndrome, which had also triggered the thrombosis. The other 10 cases resulted in successful pregnancies and live deliveries, one surgically and the other nine vaginally. Three of the women had successful subsequent pregnancies, Dr. Comerota said.
Dr. Comerota said that he has been a speaker for and a consultant to Covidien, a company that makes some of the devices used in these procedures.
FROM ISET 2012, AN INTERNATIONAL SYMPOSIUM ON ENDOVASCULAR THERAPY
Major Finding: Treatment of 11 pregnant women with DVT resulted in 10 successful live births.
Data Source: A retrospective review of 11 cases managed at one U.S. center.
Disclosures: Dr. Comerota said that he has been a speaker for and a consultant to Covidien, a company that makes some of the devices used in these procedures.
Two Stents Show Peripheral Artery Efficacy
MIAMI BEACH – Two different self-expandable nitinol stents showed efficacy for treating peripheral arterial stenoses, according to results from two separate, pivotal trials. The Food and Drug Administration is reviewing data from both studies, with the agency’s decisions expected later this year, researchers said at ISET 2012, an international symposium on endovascular therapy.
The EverFlex stent underwent testing in femoropopliteal arteries in 287 patients with peripheral artery disease at 44 centers in the United States and Europe. The trial enrolled patients with Rutherford-Becker clinical categories of 2, 3, or 4, and at least a 50% stenosis. The average age of the patients enrolled was 68 years, two-thirds were men, 43% had diabetes, 88% had hypertension, 40% had Rutherford category 2 disease, and 56% had category 3 disease. The average lesion length was 9 cm, with 29% of patients having a lesion that was 20 cm long.
One of the study’s major goals was testing a single-stent strategy, and 95% of enrolled patients received a single stent, said Dr. Jon S. Matsumura, the trial’s principal investigator. Using single stents for long lesions may reduce the risk of stent fracture.

The primary efficacy end point of DURABILITY II (Safety and Effectiveness Study of EverFlex Stent to Treat Symptomatic Femoral-Popliteal Atherosclerosis) was primary patency in the treated vessel 1 year after stent placement. Study results from 226 patients with follow-up data showed primary patency in 77% after 1 year in an actuarial analysis. This level of success exceeded the 33% performance goal, set on the basis of literature controls using angioplasty interventions, said Dr. Matsumura, professor and chairman of the division of vascular surgery at the University of Wisconsin in Madison.
The study’s primary safety end point was the combined rate of major adverse effects after 30 days. The results showed no such events within that period, with no deaths, no amputations of treated limbs, and no instances of clinically driven target vessel revascularizations.
At 1 year after treatment, a single episode of stent fracture – a class V fracture in the stent’s transaxial spiral configuration – had occurred.
Also at 1 year, 84% of patients had at least a single-level improvement, compared with their baseline Rutherford clinical category, with 65% of patients improving by two or more categories. The researchers collected data on the baseline and 1-year follow-up treadmill walking tests in 29 patients, and 69% of these patients showed improvements in walking distance, with an average increase of about 420 feet per patient.
ORION (A Boston Scientific Trial of the Epic Nitinol Stent in the Treatment of Atherosclerotic Lesions in Iliac Arteries) enrolled 125 patients with a new or restenotic lesion in the common or external iliac artery, or in both, and with a Rutherford-Becker clinical category of 1, 2, 3, or 4. Lesions had to be no longer than 13 cm, with a reference vessel diameter of 5-11 mm. The study enrolled patients at 28 U.S. centers during May 2009–December 2010.
The study population had an average age of 61 years, two-thirds were men, a third of the patients had diabetes, and 76% had hypertension. The average lesion length was just over 3 cm, and the average vessel diameter was just under 8 mm. The operators in the study placed a total of 187 stents into 166 lesions in the enrolled patients.

This study’s primary end point was the combined rate of major adverse events after 9 months of follow-up. These events occurred in four of the 117 patients who were followed for 9 months, a 3.4% rate that consisted entirely of target vessel revascularizations; in three cases, these revascularizations occurred because of stent thrombosis episodes, said Dr. Daniel G. Clair, principal investigator for the study and chairman of vascular surgery at the Cleveland Clinic. The study patients had no other events that made up the major adverse event composite: no deaths within 30 days, no MIs during hospitalization, and no amputations of the index limb during 9 months of follow-up.
The 3.4% observed major adverse event rate ran significantly below the study’s prespecified performance goal of 17%. The 17% goal derived from an expected 8% rate based on the historical literature, plus an added margin of 9%. The actual rate "was much lower than predicted by the literature," based on how iliac artery stents performed in prior studies, Dr. Clair said.
The percentage of patients with a Rutherford-Becker clinical category of 2-4 dropped from 93% at baseline to 18% at 9 months. Average ankle brachial index increased from an average of 0.79 at baseline to 0.97 at 9 months. The primary patency rate at 9 months was 96%.
The results "support the safety and efficacy of the stent in iliac arteries," Dr. Clair concluded.
The DURABILITY II trial was sponsored by Covidien, the company developing the EverFlex stent. The ORION trial was sponsored by Boston Scientific, the company developing the Epic stent. Dr. Matsumura said that he has received research grants from Covidien and from Abbott, Cook, Endologix, and W.L. Gore. Dr. Clair said that he has been a consultant to Boston Scientific, and also to Covidien, Endologix, ev3, and Medtronic.
MIAMI BEACH – Two different self-expandable nitinol stents showed efficacy for treating peripheral arterial stenoses, according to results from two separate, pivotal trials. The Food and Drug Administration is reviewing data from both studies, with the agency’s decisions expected later this year, researchers said at ISET 2012, an international symposium on endovascular therapy.
The EverFlex stent underwent testing in femoropopliteal arteries in 287 patients with peripheral artery disease at 44 centers in the United States and Europe. The trial enrolled patients with Rutherford-Becker clinical categories of 2, 3, or 4, and at least a 50% stenosis. The average age of the patients enrolled was 68 years, two-thirds were men, 43% had diabetes, 88% had hypertension, 40% had Rutherford category 2 disease, and 56% had category 3 disease. The average lesion length was 9 cm, with 29% of patients having a lesion that was 20 cm long.
One of the study’s major goals was testing a single-stent strategy, and 95% of enrolled patients received a single stent, said Dr. Jon S. Matsumura, the trial’s principal investigator. Using single stents for long lesions may reduce the risk of stent fracture.
The primary efficacy end point of DURABILITY II (Safety and Effectiveness Study of EverFlex Stent to Treat Symptomatic Femoral-Popliteal Atherosclerosis) was primary patency in the treated vessel 1 year after stent placement. Study results from 226 patients with follow-up data showed primary patency in 77% after 1 year in an actuarial analysis. This level of success exceeded the 33% performance goal, set on the basis of literature controls using angioplasty interventions, said Dr. Matsumura, professor and chairman of the division of vascular surgery at the University of Wisconsin in Madison.
The study’s primary safety end point was the combined rate of major adverse effects after 30 days. The results showed no such events within that period, with no deaths, no amputations of treated limbs, and no instances of clinically driven target vessel revascularizations.
At 1 year after treatment, a single episode of stent fracture – a class V fracture in the stent’s transaxial spiral configuration – had occurred.
Also at 1 year, 84% of patients had at least a single-level improvement, compared with their baseline Rutherford clinical category, with 65% of patients improving by two or more categories. The researchers collected data on the baseline and 1-year follow-up treadmill walking tests in 29 patients, and 69% of these patients showed improvements in walking distance, with an average increase of about 420 feet per patient.
ORION (A Boston Scientific Trial of the Epic Nitinol Stent in the Treatment of Atherosclerotic Lesions in Iliac Arteries) enrolled 125 patients with a new or restenotic lesion in the common or external iliac artery, or in both, and with a Rutherford-Becker clinical category of 1, 2, 3, or 4. Lesions had to be no longer than 13 cm, with a reference vessel diameter of 5-11 mm. The study enrolled patients at 28 U.S. centers during May 2009–December 2010.
The study population had an average age of 61 years, two-thirds were men, a third of the patients had diabetes, and 76% had hypertension. The average lesion length was just over 3 cm, and the average vessel diameter was just under 8 mm. The operators in the study placed a total of 187 stents into 166 lesions in the enrolled patients.
This study’s primary end point was the combined rate of major adverse events after 9 months of follow-up. These events occurred in four of the 117 patients who were followed for 9 months, a 3.4% rate that consisted entirely of target vessel revascularizations; in three cases, these revascularizations occurred because of stent thrombosis episodes, said Dr. Daniel G. Clair, principal investigator for the study and chairman of vascular surgery at the Cleveland Clinic. The study patients had no other events that made up the major adverse event composite: no deaths within 30 days, no MIs during hospitalization, and no amputations of the index limb during 9 months of follow-up.
The 3.4% observed major adverse event rate ran significantly below the study’s prespecified performance goal of 17%. The 17% goal derived from an expected 8% rate based on the historical literature, plus an added margin of 9%. The actual rate "was much lower than predicted by the literature," based on how iliac artery stents performed in prior studies, Dr. Clair said.
The percentage of patients with a Rutherford-Becker clinical category of 2-4 dropped from 93% at baseline to 18% at 9 months. Average ankle brachial index increased from an average of 0.79 at baseline to 0.97 at 9 months. The primary patency rate at 9 months was 96%.
The results "support the safety and efficacy of the stent in iliac arteries," Dr. Clair concluded.
The DURABILITY II trial was sponsored by Covidien, the company developing the EverFlex stent. The ORION trial was sponsored by Boston Scientific, the company developing the Epic stent. Dr. Matsumura said that he has received research grants from Covidien and from Abbott, Cook, Endologix, and W.L. Gore. Dr. Clair said that he has been a consultant to Boston Scientific, and also to Covidien, Endologix, ev3, and Medtronic.
MIAMI BEACH – Two different self-expandable nitinol stents showed efficacy for treating peripheral arterial stenoses, according to results from two separate, pivotal trials. The Food and Drug Administration is reviewing data from both studies, with the agency’s decisions expected later this year, researchers said at ISET 2012, an international symposium on endovascular therapy.
The EverFlex stent underwent testing in femoropopliteal arteries in 287 patients with peripheral artery disease at 44 centers in the United States and Europe. The trial enrolled patients with Rutherford-Becker clinical categories of 2, 3, or 4, and at least a 50% stenosis. The average age of the patients enrolled was 68 years, two-thirds were men, 43% had diabetes, 88% had hypertension, 40% had Rutherford category 2 disease, and 56% had category 3 disease. The average lesion length was 9 cm, with 29% of patients having a lesion that was 20 cm long.
One of the study’s major goals was testing a single-stent strategy, and 95% of enrolled patients received a single stent, said Dr. Jon S. Matsumura, the trial’s principal investigator. Using single stents for long lesions may reduce the risk of stent fracture.
The primary efficacy end point of DURABILITY II (Safety and Effectiveness Study of EverFlex Stent to Treat Symptomatic Femoral-Popliteal Atherosclerosis) was primary patency in the treated vessel 1 year after stent placement. Study results from 226 patients with follow-up data showed primary patency in 77% after 1 year in an actuarial analysis. This level of success exceeded the 33% performance goal, set on the basis of literature controls using angioplasty interventions, said Dr. Matsumura, professor and chairman of the division of vascular surgery at the University of Wisconsin in Madison.
The study’s primary safety end point was the combined rate of major adverse effects after 30 days. The results showed no such events within that period, with no deaths, no amputations of treated limbs, and no instances of clinically driven target vessel revascularizations.
At 1 year after treatment, a single episode of stent fracture – a class V fracture in the stent’s transaxial spiral configuration – had occurred.
Also at 1 year, 84% of patients had at least a single-level improvement, compared with their baseline Rutherford clinical category, with 65% of patients improving by two or more categories. The researchers collected data on the baseline and 1-year follow-up treadmill walking tests in 29 patients, and 69% of these patients showed improvements in walking distance, with an average increase of about 420 feet per patient.
ORION (A Boston Scientific Trial of the Epic Nitinol Stent in the Treatment of Atherosclerotic Lesions in Iliac Arteries) enrolled 125 patients with a new or restenotic lesion in the common or external iliac artery, or in both, and with a Rutherford-Becker clinical category of 1, 2, 3, or 4. Lesions had to be no longer than 13 cm, with a reference vessel diameter of 5-11 mm. The study enrolled patients at 28 U.S. centers during May 2009–December 2010.
The study population had an average age of 61 years, two-thirds were men, a third of the patients had diabetes, and 76% had hypertension. The average lesion length was just over 3 cm, and the average vessel diameter was just under 8 mm. The operators in the study placed a total of 187 stents into 166 lesions in the enrolled patients.
This study’s primary end point was the combined rate of major adverse events after 9 months of follow-up. These events occurred in four of the 117 patients who were followed for 9 months, a 3.4% rate that consisted entirely of target vessel revascularizations; in three cases, these revascularizations occurred because of stent thrombosis episodes, said Dr. Daniel G. Clair, principal investigator for the study and chairman of vascular surgery at the Cleveland Clinic. The study patients had no other events that made up the major adverse event composite: no deaths within 30 days, no MIs during hospitalization, and no amputations of the index limb during 9 months of follow-up.
The 3.4% observed major adverse event rate ran significantly below the study’s prespecified performance goal of 17%. The 17% goal derived from an expected 8% rate based on the historical literature, plus an added margin of 9%. The actual rate "was much lower than predicted by the literature," based on how iliac artery stents performed in prior studies, Dr. Clair said.
The percentage of patients with a Rutherford-Becker clinical category of 2-4 dropped from 93% at baseline to 18% at 9 months. Average ankle brachial index increased from an average of 0.79 at baseline to 0.97 at 9 months. The primary patency rate at 9 months was 96%.
The results "support the safety and efficacy of the stent in iliac arteries," Dr. Clair concluded.
The DURABILITY II trial was sponsored by Covidien, the company developing the EverFlex stent. The ORION trial was sponsored by Boston Scientific, the company developing the Epic stent. Dr. Matsumura said that he has received research grants from Covidien and from Abbott, Cook, Endologix, and W.L. Gore. Dr. Clair said that he has been a consultant to Boston Scientific, and also to Covidien, Endologix, ev3, and Medtronic.
FROM ISET 2012, AN INTERNATIONAL SYMPOSIUM ON ENDOVASCULAR THERAPY
Major Finding: In femoropopliteal arteries, the EverFlex stent produced a 77% actuarial primary patency rate after 1 year. In iliac arteries, the Epic stent produced a 3.4% major adverse event rate after 9 months.
Data Source: The DURABILITY II trial enrolled 287 patients in a single-arm study at 44 U.S. and European centers. The ORION study enrolled 125 patients in a single-arm study at 28 U.S. centers.
Disclosures: The DURABILITY II trial was sponsored by Covidien, the company developing the EverFlex stent. The ORION trial was sponsored by Boston Scientific, the company developing the Epic stent. Dr. Matsumura said that he has received research grants from Covidien and from Abbott, Cook, Endologix, and W.L. Gore. Dr. Clair said that he has been a consultant to Boston Scientific, and also to Covidien, Endologix, ev3, and Medtronic.
Aortic Regurgitation After TAVR Poses Threat
MIAMI BEACH – Now that transcatheter aortic valve replacement is available for routine U.S. use, interventionalists have focused on making the procedure better and safer, such as cutting the incidence of significant regurgitation through the replacement aortic valve.
Toward that end, researchers have developed a way to quickly and objectively assess the risk for aortic valve regurgitation based on the measurement of aortic blood pressures immediately after transcatheter valve replacement (TAVR). This new measure, the "aortic regurgitation index," showed significant correlation with periprocedural aortic regurgitation as well as with patients’ 1-year survival following their procedure, Dr. Eberhard Grube said at ISET 2012, an international symposium on endovascular therapy.

Until now, "there has been no way to quantify aortic regurgitation; it’s subjective," said Dr. Grube, professor and chief of cardiology and angiology at the Helios Heart Centre in Siegburg, Germany. The aortic regurgitation index (ARI) allows physicians "to quantify aortic regurgitation periprocedurally by objectively looking at the patient’s hemodynamics, regardless of the subjective evaluation of aortic insufficiency by angiography or by echo," he said. "We can see the ARI and know what we need to do for postdeployment treatment."
Dr. Grube and his associates set the ARI as equal to the patient’s diastolic aortic pressure minus the left ventricular end diastolic pressure, then divided by the aortic systolic pressure, and multiplied by 100.
For example, a patient with mild periprocedural aortic regurgitation might have an aortic diastolic pressure of 60 mm Hg, a left ventricular end diastolic pressure of 15 mm Hg, and an aortic systolic pressure of 150 mm Hg, which would produce an ARI of 30, showing that the patient had a low risk for dying during the subsequent year. In contrast, a patient with moderate or severe periprocedural aortic regurgitation could have an aortic diastolic pressure of 40 mm Hg, a left ventricular end diastolic pressure of 20 mm Hg, and an aortic systolic pressure of 120 mm Hg, which would produce an ARI of 16.7, flagging a higher mortality risk during the following year.
His group assessed the prognostic ability of the ARI in a series of 146 patients who underwent TAVR. The group included 124 patients with no or mild aortic regurgitation following TAVR and 22 who showed moderate or severe regurgitation. During 1-year follow-up, the 96 patients with a periprocedural ARI of 25 or greater had an 83% survival rate; the 50 patients with a periprocedural ARI of less than 25 had a survival rate of 54%, a statistically significant difference. The analysis also showed a significant correlation between the ARI and the severity of aortic regurgitation. Among patients with no discernible regurgitation, the average ARI was about 30, in those with mild regurgitation the average ARI was about 25, in patients with moderate regurgitation the average was about 18, and in patients with severe regurgitation the ARI averaged about 10.
One recently identified key to limiting aortic regurgitation following TAVR is proper valve sizing relative to the patient’s aortic annulus. "Appropriate valve sizing is likely the most important factor that will influence both the degree of perivalvular regurgitation and pacemaker need after TAVR," said Dr. Jeffrey J. Popma in a separate talk at the meeting.
Until recently, many operators used transthoracic echocardiography for annulus sizing, but recently published evidence showed that imaging by CT or by MRI provide superior information, said Dr. Popma, director of interventional cardiology at Beth Israel Deaconess Medical Center in Boston. He cited a British report published last November that compared transthoracic echo, CT, and MRI in 202 patients who underwent TAVR, which found both CT and MRI superior to echo for annulus measurement prior to TAVR (J. Am. Coll. Cardiol. 2011;58:2165-73).
Dr. Grube said that he has financial relationships with Direct Flow, Claret, Biosensors, Medtronic, Mitralign, Boston Scientific, Cordis, Abbott Vascular, and InSeal. Dr. Popma said that he has financial relationships with Abbott, Boston Scientific, ev3 (Covidien), Medtronic, and Cordis.
MIAMI BEACH – Now that transcatheter aortic valve replacement is available for routine U.S. use, interventionalists have focused on making the procedure better and safer, such as cutting the incidence of significant regurgitation through the replacement aortic valve.
Toward that end, researchers have developed a way to quickly and objectively assess the risk for aortic valve regurgitation based on the measurement of aortic blood pressures immediately after transcatheter valve replacement (TAVR). This new measure, the "aortic regurgitation index," showed significant correlation with periprocedural aortic regurgitation as well as with patients’ 1-year survival following their procedure, Dr. Eberhard Grube said at ISET 2012, an international symposium on endovascular therapy.
Until now, "there has been no way to quantify aortic regurgitation; it’s subjective," said Dr. Grube, professor and chief of cardiology and angiology at the Helios Heart Centre in Siegburg, Germany. The aortic regurgitation index (ARI) allows physicians "to quantify aortic regurgitation periprocedurally by objectively looking at the patient’s hemodynamics, regardless of the subjective evaluation of aortic insufficiency by angiography or by echo," he said. "We can see the ARI and know what we need to do for postdeployment treatment."
Dr. Grube and his associates set the ARI as equal to the patient’s diastolic aortic pressure minus the left ventricular end diastolic pressure, then divided by the aortic systolic pressure, and multiplied by 100.
For example, a patient with mild periprocedural aortic regurgitation might have an aortic diastolic pressure of 60 mm Hg, a left ventricular end diastolic pressure of 15 mm Hg, and an aortic systolic pressure of 150 mm Hg, which would produce an ARI of 30, showing that the patient had a low risk for dying during the subsequent year. In contrast, a patient with moderate or severe periprocedural aortic regurgitation could have an aortic diastolic pressure of 40 mm Hg, a left ventricular end diastolic pressure of 20 mm Hg, and an aortic systolic pressure of 120 mm Hg, which would produce an ARI of 16.7, flagging a higher mortality risk during the following year.
His group assessed the prognostic ability of the ARI in a series of 146 patients who underwent TAVR. The group included 124 patients with no or mild aortic regurgitation following TAVR and 22 who showed moderate or severe regurgitation. During 1-year follow-up, the 96 patients with a periprocedural ARI of 25 or greater had an 83% survival rate; the 50 patients with a periprocedural ARI of less than 25 had a survival rate of 54%, a statistically significant difference. The analysis also showed a significant correlation between the ARI and the severity of aortic regurgitation. Among patients with no discernible regurgitation, the average ARI was about 30, in those with mild regurgitation the average ARI was about 25, in patients with moderate regurgitation the average was about 18, and in patients with severe regurgitation the ARI averaged about 10.
One recently identified key to limiting aortic regurgitation following TAVR is proper valve sizing relative to the patient’s aortic annulus. "Appropriate valve sizing is likely the most important factor that will influence both the degree of perivalvular regurgitation and pacemaker need after TAVR," said Dr. Jeffrey J. Popma in a separate talk at the meeting.
Until recently, many operators used transthoracic echocardiography for annulus sizing, but recently published evidence showed that imaging by CT or by MRI provide superior information, said Dr. Popma, director of interventional cardiology at Beth Israel Deaconess Medical Center in Boston. He cited a British report published last November that compared transthoracic echo, CT, and MRI in 202 patients who underwent TAVR, which found both CT and MRI superior to echo for annulus measurement prior to TAVR (J. Am. Coll. Cardiol. 2011;58:2165-73).
Dr. Grube said that he has financial relationships with Direct Flow, Claret, Biosensors, Medtronic, Mitralign, Boston Scientific, Cordis, Abbott Vascular, and InSeal. Dr. Popma said that he has financial relationships with Abbott, Boston Scientific, ev3 (Covidien), Medtronic, and Cordis.
MIAMI BEACH – Now that transcatheter aortic valve replacement is available for routine U.S. use, interventionalists have focused on making the procedure better and safer, such as cutting the incidence of significant regurgitation through the replacement aortic valve.
Toward that end, researchers have developed a way to quickly and objectively assess the risk for aortic valve regurgitation based on the measurement of aortic blood pressures immediately after transcatheter valve replacement (TAVR). This new measure, the "aortic regurgitation index," showed significant correlation with periprocedural aortic regurgitation as well as with patients’ 1-year survival following their procedure, Dr. Eberhard Grube said at ISET 2012, an international symposium on endovascular therapy.
Until now, "there has been no way to quantify aortic regurgitation; it’s subjective," said Dr. Grube, professor and chief of cardiology and angiology at the Helios Heart Centre in Siegburg, Germany. The aortic regurgitation index (ARI) allows physicians "to quantify aortic regurgitation periprocedurally by objectively looking at the patient’s hemodynamics, regardless of the subjective evaluation of aortic insufficiency by angiography or by echo," he said. "We can see the ARI and know what we need to do for postdeployment treatment."
Dr. Grube and his associates set the ARI as equal to the patient’s diastolic aortic pressure minus the left ventricular end diastolic pressure, then divided by the aortic systolic pressure, and multiplied by 100.
For example, a patient with mild periprocedural aortic regurgitation might have an aortic diastolic pressure of 60 mm Hg, a left ventricular end diastolic pressure of 15 mm Hg, and an aortic systolic pressure of 150 mm Hg, which would produce an ARI of 30, showing that the patient had a low risk for dying during the subsequent year. In contrast, a patient with moderate or severe periprocedural aortic regurgitation could have an aortic diastolic pressure of 40 mm Hg, a left ventricular end diastolic pressure of 20 mm Hg, and an aortic systolic pressure of 120 mm Hg, which would produce an ARI of 16.7, flagging a higher mortality risk during the following year.
His group assessed the prognostic ability of the ARI in a series of 146 patients who underwent TAVR. The group included 124 patients with no or mild aortic regurgitation following TAVR and 22 who showed moderate or severe regurgitation. During 1-year follow-up, the 96 patients with a periprocedural ARI of 25 or greater had an 83% survival rate; the 50 patients with a periprocedural ARI of less than 25 had a survival rate of 54%, a statistically significant difference. The analysis also showed a significant correlation between the ARI and the severity of aortic regurgitation. Among patients with no discernible regurgitation, the average ARI was about 30, in those with mild regurgitation the average ARI was about 25, in patients with moderate regurgitation the average was about 18, and in patients with severe regurgitation the ARI averaged about 10.
One recently identified key to limiting aortic regurgitation following TAVR is proper valve sizing relative to the patient’s aortic annulus. "Appropriate valve sizing is likely the most important factor that will influence both the degree of perivalvular regurgitation and pacemaker need after TAVR," said Dr. Jeffrey J. Popma in a separate talk at the meeting.
Until recently, many operators used transthoracic echocardiography for annulus sizing, but recently published evidence showed that imaging by CT or by MRI provide superior information, said Dr. Popma, director of interventional cardiology at Beth Israel Deaconess Medical Center in Boston. He cited a British report published last November that compared transthoracic echo, CT, and MRI in 202 patients who underwent TAVR, which found both CT and MRI superior to echo for annulus measurement prior to TAVR (J. Am. Coll. Cardiol. 2011;58:2165-73).
Dr. Grube said that he has financial relationships with Direct Flow, Claret, Biosensors, Medtronic, Mitralign, Boston Scientific, Cordis, Abbott Vascular, and InSeal. Dr. Popma said that he has financial relationships with Abbott, Boston Scientific, ev3 (Covidien), Medtronic, and Cordis.
FROM ISET 2012, AN INTERNATIONAL SYMPOSIUM ON ENDOVASCULAR THERAPY
Major Finding: An aortic regurgitation index of 25 or higher was linked with a 1-year survival post TAVR of 83%, compared with 54% among patients with an index of less than 25.
Data Source: One-year follow-up of 146 patients who underwent transcatheter aortic valve replacement.
Disclosures: Dr. Grube said that he has financial relationships with Direct Flow, Claret, Biosensors, Medtronic, Mitralign, Boston Scientific, Cordis, Abbott Vascular, and InSeal. Dr. Popma said that he has financial relationships with Abbott, Boston Scientific, ev3 (Covidien), Medtronic, and Cordis.
Excitement builds for renal denervation in hypertension
MIAMI BEACH – The U.S. pivotal trial assessing renal denervation for blood pressure reduction began in October and a report on its final results won’t appear until next year, but excitement based on earlier study findings and the European clinical experience is already running high for this new approach to treating drug-resistant hypertension.
"Resistant hypertension is common, and is becoming more common despite a huge armamentarium of pharmacologic agents. The early literature [on renal denervation] is very impressive" for safety and efficacy, and the approach is plausibly "based on physiology," Dr. Michael R. Jaff said at ISET 2012, an international symposium on endovascular therapy. Moreover, on top of the expected antihypertensive effect of renal denervation, recent results from a pilot, randomized controlled study with 50 hypertensive patients suggested that the intervention could also help normalize glucose metabolism and reverse type 2 diabetes in some patients.

Renal denervation "could arguably be the most exciting advance in interventional vascular medicine," said Dr. Jaff, medical director of the vascular center at Massachusetts General Hospital in Boston. "People’s imaginations are running wild about the potential implications of this type of therapy for a whole host of patients. In the near term I’m incredibly bullish [about this treatment] and in the far term I’m even more enthusiastic."
In addition to the clinical efficacy that renal denervation so far has shown, it has the pluses of being relatively simple and easy to do, easy to learn, and fairly quick; the procedure involves modestly priced equipment and appears very safe as well, noted Dr. Horst Sievert, an interventional cardiologist who collaborated on one of the efficacy trials and now routinely performs renal denervations at the cardiovascular center of Saint Katharinen Hospital in Frankfurt, Germany.
The Symplicity Renal Denervation System, developed by Ardian, which is now owned by Medtronic, has been available for routine use in Europe and Australia since 2010. The U.S. pivotal trial, Symplicity HTN-3 (Renal Denervation in Patients With Uncontrolled Hypertension), plans to enroll 530 patients at 60 U.S. centers and should be completed by March of next year.
The procedure, which requires just a flexible catheter and radiofrequency generator, delivers four to eight low-power treatments along the length of both main renal arteries using a 6F sheath, targeting the renal nerves in the adventitia of the renal arteries and producing reduced sympathetic activity and renin release. According to Dr. Sievert, the typical procedure takes 45-60 minutes, and is successful in significantly reducing systolic and diastolic pressure in 75%-80% of drug-refractory patients. The worldwide case experience so far has led to few complications, no reported thrombus formations, no detrimental effects on renal blood flow or function, and no reported cases of severe hypotension following treatment, he said.
"I believe that renal denervation will become as important as PCI [percutaneous coronary intervention] or PTA [percutaneous transluminal angioplasty]," said Dr. Sievert, director of the cardiovascular center and also on the faculty of the University of Frankfurt.
"The risks are so few and the complication rate so low that it’s not worth figuring out which patients will respond and who won’t. I would just do the treatment and see if the patient responds," Dr. Sievert said at the meeting. No explanation exists as to why some patients don’t respond, and no marker exists to distinguish patients who will respond and those who won’t.
The largest systematic assessment of the antihypertensive impact of renal denervation came in a randomized trial, Symplicity HTN-2, with 106 patients, which was sponsored by Ardian and published in 2010. All patients had an entry systolic pressure of at least 160 mm Hg (or at least 150 mm Hg if they also had type 2 diabetes) despite current treatment with at least three antihypertensive medications. At 6 months after treatment, the 52 patients treated with denervation had an average blood pressure reduction of 32/12 mm Hg, compared with an average systolic pressure rise of 1 mm Hg and no average diastolic change in the 54 control patients (Lancet 2010;376:1903-9).
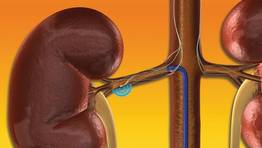
In the denervation group, 84% of patients had at least a 10-mm Hg drop in their systolic pressure 6 months after treatment compared with 35% of the control patients having this response, and 39% of the denervation patients had their systolic pressure fall below 140 mm Hg at 6 months compared with 6% of control patients. A subsequent report on a total of 153 patients treated with renal denervation documented that the blood pressure reductions seen at 6 months persisted out to 2 years (Hypertension 2011;57:911-7).
Striking as this effect was, perhaps even more notable was a report last year from a separate pilot study that randomized 50 patients who met the same definition of refractory hypertension. In addition to showing similar blood pressure results in the denervated patients, the researchers on this study also looked at several measures of glucose metabolism. At 3 months after intervention, the 37 denervated patients had an average 9% cut in their fasting blood glucose level and an average 12% drop in their average fasting blood insulin levels, both statistically significant changes from baseline, compared with no significant change in the 13 control patients. The percent of patients with normal glucose tolerance rose by 11% in the denervated group, and dropped by 7% in the control group. The authors of the report concluded that renal sympathetic activity plays a role in insulin resistance (Circulation 2011;123:1940-6).
Future studies will likely examine the possible impact of renal denervation on heart failure and on obstructive sleep apnea, noted Dr. Jaff.
Dr. Jaff said that he has financial relationships with several cardiovascular device companies including Medtronic Hansen Medical and Medtronic Vascular. Dr. Sievert said that he has financial relationships with several cardiovascular device and pharmaceutical companies including Ardian and Medtronic.
MIAMI BEACH – The U.S. pivotal trial assessing renal denervation for blood pressure reduction began in October and a report on its final results won’t appear until next year, but excitement based on earlier study findings and the European clinical experience is already running high for this new approach to treating drug-resistant hypertension.
"Resistant hypertension is common, and is becoming more common despite a huge armamentarium of pharmacologic agents. The early literature [on renal denervation] is very impressive" for safety and efficacy, and the approach is plausibly "based on physiology," Dr. Michael R. Jaff said at ISET 2012, an international symposium on endovascular therapy. Moreover, on top of the expected antihypertensive effect of renal denervation, recent results from a pilot, randomized controlled study with 50 hypertensive patients suggested that the intervention could also help normalize glucose metabolism and reverse type 2 diabetes in some patients.
Renal denervation "could arguably be the most exciting advance in interventional vascular medicine," said Dr. Jaff, medical director of the vascular center at Massachusetts General Hospital in Boston. "People’s imaginations are running wild about the potential implications of this type of therapy for a whole host of patients. In the near term I’m incredibly bullish [about this treatment] and in the far term I’m even more enthusiastic."
In addition to the clinical efficacy that renal denervation so far has shown, it has the pluses of being relatively simple and easy to do, easy to learn, and fairly quick; the procedure involves modestly priced equipment and appears very safe as well, noted Dr. Horst Sievert, an interventional cardiologist who collaborated on one of the efficacy trials and now routinely performs renal denervations at the cardiovascular center of Saint Katharinen Hospital in Frankfurt, Germany.
The Symplicity Renal Denervation System, developed by Ardian, which is now owned by Medtronic, has been available for routine use in Europe and Australia since 2010. The U.S. pivotal trial, Symplicity HTN-3 (Renal Denervation in Patients With Uncontrolled Hypertension), plans to enroll 530 patients at 60 U.S. centers and should be completed by March of next year.
The procedure, which requires just a flexible catheter and radiofrequency generator, delivers four to eight low-power treatments along the length of both main renal arteries using a 6F sheath, targeting the renal nerves in the adventitia of the renal arteries and producing reduced sympathetic activity and renin release. According to Dr. Sievert, the typical procedure takes 45-60 minutes, and is successful in significantly reducing systolic and diastolic pressure in 75%-80% of drug-refractory patients. The worldwide case experience so far has led to few complications, no reported thrombus formations, no detrimental effects on renal blood flow or function, and no reported cases of severe hypotension following treatment, he said.
"I believe that renal denervation will become as important as PCI [percutaneous coronary intervention] or PTA [percutaneous transluminal angioplasty]," said Dr. Sievert, director of the cardiovascular center and also on the faculty of the University of Frankfurt.
"The risks are so few and the complication rate so low that it’s not worth figuring out which patients will respond and who won’t. I would just do the treatment and see if the patient responds," Dr. Sievert said at the meeting. No explanation exists as to why some patients don’t respond, and no marker exists to distinguish patients who will respond and those who won’t.
The largest systematic assessment of the antihypertensive impact of renal denervation came in a randomized trial, Symplicity HTN-2, with 106 patients, which was sponsored by Ardian and published in 2010. All patients had an entry systolic pressure of at least 160 mm Hg (or at least 150 mm Hg if they also had type 2 diabetes) despite current treatment with at least three antihypertensive medications. At 6 months after treatment, the 52 patients treated with denervation had an average blood pressure reduction of 32/12 mm Hg, compared with an average systolic pressure rise of 1 mm Hg and no average diastolic change in the 54 control patients (Lancet 2010;376:1903-9).
In the denervation group, 84% of patients had at least a 10-mm Hg drop in their systolic pressure 6 months after treatment compared with 35% of the control patients having this response, and 39% of the denervation patients had their systolic pressure fall below 140 mm Hg at 6 months compared with 6% of control patients. A subsequent report on a total of 153 patients treated with renal denervation documented that the blood pressure reductions seen at 6 months persisted out to 2 years (Hypertension 2011;57:911-7).
Striking as this effect was, perhaps even more notable was a report last year from a separate pilot study that randomized 50 patients who met the same definition of refractory hypertension. In addition to showing similar blood pressure results in the denervated patients, the researchers on this study also looked at several measures of glucose metabolism. At 3 months after intervention, the 37 denervated patients had an average 9% cut in their fasting blood glucose level and an average 12% drop in their average fasting blood insulin levels, both statistically significant changes from baseline, compared with no significant change in the 13 control patients. The percent of patients with normal glucose tolerance rose by 11% in the denervated group, and dropped by 7% in the control group. The authors of the report concluded that renal sympathetic activity plays a role in insulin resistance (Circulation 2011;123:1940-6).
Future studies will likely examine the possible impact of renal denervation on heart failure and on obstructive sleep apnea, noted Dr. Jaff.
Dr. Jaff said that he has financial relationships with several cardiovascular device companies including Medtronic Hansen Medical and Medtronic Vascular. Dr. Sievert said that he has financial relationships with several cardiovascular device and pharmaceutical companies including Ardian and Medtronic.
MIAMI BEACH – The U.S. pivotal trial assessing renal denervation for blood pressure reduction began in October and a report on its final results won’t appear until next year, but excitement based on earlier study findings and the European clinical experience is already running high for this new approach to treating drug-resistant hypertension.
"Resistant hypertension is common, and is becoming more common despite a huge armamentarium of pharmacologic agents. The early literature [on renal denervation] is very impressive" for safety and efficacy, and the approach is plausibly "based on physiology," Dr. Michael R. Jaff said at ISET 2012, an international symposium on endovascular therapy. Moreover, on top of the expected antihypertensive effect of renal denervation, recent results from a pilot, randomized controlled study with 50 hypertensive patients suggested that the intervention could also help normalize glucose metabolism and reverse type 2 diabetes in some patients.
Renal denervation "could arguably be the most exciting advance in interventional vascular medicine," said Dr. Jaff, medical director of the vascular center at Massachusetts General Hospital in Boston. "People’s imaginations are running wild about the potential implications of this type of therapy for a whole host of patients. In the near term I’m incredibly bullish [about this treatment] and in the far term I’m even more enthusiastic."
In addition to the clinical efficacy that renal denervation so far has shown, it has the pluses of being relatively simple and easy to do, easy to learn, and fairly quick; the procedure involves modestly priced equipment and appears very safe as well, noted Dr. Horst Sievert, an interventional cardiologist who collaborated on one of the efficacy trials and now routinely performs renal denervations at the cardiovascular center of Saint Katharinen Hospital in Frankfurt, Germany.
The Symplicity Renal Denervation System, developed by Ardian, which is now owned by Medtronic, has been available for routine use in Europe and Australia since 2010. The U.S. pivotal trial, Symplicity HTN-3 (Renal Denervation in Patients With Uncontrolled Hypertension), plans to enroll 530 patients at 60 U.S. centers and should be completed by March of next year.
The procedure, which requires just a flexible catheter and radiofrequency generator, delivers four to eight low-power treatments along the length of both main renal arteries using a 6F sheath, targeting the renal nerves in the adventitia of the renal arteries and producing reduced sympathetic activity and renin release. According to Dr. Sievert, the typical procedure takes 45-60 minutes, and is successful in significantly reducing systolic and diastolic pressure in 75%-80% of drug-refractory patients. The worldwide case experience so far has led to few complications, no reported thrombus formations, no detrimental effects on renal blood flow or function, and no reported cases of severe hypotension following treatment, he said.
"I believe that renal denervation will become as important as PCI [percutaneous coronary intervention] or PTA [percutaneous transluminal angioplasty]," said Dr. Sievert, director of the cardiovascular center and also on the faculty of the University of Frankfurt.
"The risks are so few and the complication rate so low that it’s not worth figuring out which patients will respond and who won’t. I would just do the treatment and see if the patient responds," Dr. Sievert said at the meeting. No explanation exists as to why some patients don’t respond, and no marker exists to distinguish patients who will respond and those who won’t.
The largest systematic assessment of the antihypertensive impact of renal denervation came in a randomized trial, Symplicity HTN-2, with 106 patients, which was sponsored by Ardian and published in 2010. All patients had an entry systolic pressure of at least 160 mm Hg (or at least 150 mm Hg if they also had type 2 diabetes) despite current treatment with at least three antihypertensive medications. At 6 months after treatment, the 52 patients treated with denervation had an average blood pressure reduction of 32/12 mm Hg, compared with an average systolic pressure rise of 1 mm Hg and no average diastolic change in the 54 control patients (Lancet 2010;376:1903-9).
In the denervation group, 84% of patients had at least a 10-mm Hg drop in their systolic pressure 6 months after treatment compared with 35% of the control patients having this response, and 39% of the denervation patients had their systolic pressure fall below 140 mm Hg at 6 months compared with 6% of control patients. A subsequent report on a total of 153 patients treated with renal denervation documented that the blood pressure reductions seen at 6 months persisted out to 2 years (Hypertension 2011;57:911-7).
Striking as this effect was, perhaps even more notable was a report last year from a separate pilot study that randomized 50 patients who met the same definition of refractory hypertension. In addition to showing similar blood pressure results in the denervated patients, the researchers on this study also looked at several measures of glucose metabolism. At 3 months after intervention, the 37 denervated patients had an average 9% cut in their fasting blood glucose level and an average 12% drop in their average fasting blood insulin levels, both statistically significant changes from baseline, compared with no significant change in the 13 control patients. The percent of patients with normal glucose tolerance rose by 11% in the denervated group, and dropped by 7% in the control group. The authors of the report concluded that renal sympathetic activity plays a role in insulin resistance (Circulation 2011;123:1940-6).
Future studies will likely examine the possible impact of renal denervation on heart failure and on obstructive sleep apnea, noted Dr. Jaff.
Dr. Jaff said that he has financial relationships with several cardiovascular device companies including Medtronic Hansen Medical and Medtronic Vascular. Dr. Sievert said that he has financial relationships with several cardiovascular device and pharmaceutical companies including Ardian and Medtronic.
EXPERT ANALYSIS FROM ISET 2012, AN INTERNATIONAL SYMPOSIUM ON ENDOVASCULAR THERAPY
First U.S. Cell-Based Flu Vaccine Plant Awaits Pandemic
With the official dedication Dec. 12 of America’s first facility for making influenza vaccine in cell culture, the United States took a small but important step toward improved protection against the next flu pandemic, experts said.
But some question just how many weeks this new facility will shave off of the timeline for moving from a candidate pandemic vaccine strain to getting the vaccine into the arms of the American public. And by relying on an inherently old-fashioned approach to flu protection with limited effectiveness, some see this development as merely refining an archaic vaccine that is overdue for replacement by a newer and better method of immunoprotection against pandemic influenza.

"It’s a small, incremental step in preparedness, but the current hemagglutinin-antigen based vaccine really is inadequate for providing the kind of immunologic protection we need, and so anything you do to speed up the amount of [this type of] vaccine is only a marginal improvement in protecting against influenza," commented Michael T. Osterholm, Ph.D., professor in the school of public health and director of the Center for Infectious Disease Research and Policy at the University of Minnesota, Minneapolis.
"Just because you can make more of this [vaccine] faster, what does that mean? It’s better than nothing, absolutely, but that doesn’t mean it will be the answer for a very serious pandemic," Dr. Osterholm said in an interview.
"Having a new facility of substantial production capacity in the United States is a comfort," said Dr. William Schaffner, professor of medicine and chairman of preventive medicine at Vanderbilt University, Nashville, Tenn. "When you’re creating a pandemic vaccine, every week counts," and the cell culture–based technology that the new plant employs "is supposed to be somewhat faster" than conventional, egg-based production of flu vaccine, he noted. By not relying on chicken-based egg production that might conceivably be vulnerable to an avian flu pandemic, the new facility also sidesteps that potential danger.
"Having this facility in the United States is a significant step, as is having more vaccine," he said.
The new vaccine plant, in Holly Springs, N.C., culminates a 7-year project by Novartis working on contract with the Health and Human Services department, which funded 49% of the construction costs. The new plant brings cell-based flu vaccine production into the United States, and eventually onto the U.S. vaccine market for the first time, and also serves to beef up U.S.-based flu vaccine production of all types.
HHS officials first envisioned the new facility in 2004, when the American flu vaccine supply became threatened by many previous manufacturers going out of business, and when the supply from British-based Chiron temporarily went off line, said Robin Robinson, Ph.D., director of the HSS Biomedical Advanced Research and Development Authority. The facility uses cultured canine kidney cells to grow influenza vaccine.

The Holly Springs plant first became operational in late 2009, and now has produced several commercial-scale lots that have undergone testing in Phase III trials, Dr. Robinson said in an interview. The facility is positioned to immediately start making as much as 50 million doses of pandemic flu vaccine using cell-culture production, which could receive emergency-use FDA licensing and be available to the American public.
In the absence of a pandemic, the facility will primarily focus on making seasonal influenza vaccine (as well as certain other vaccines), but with much less urgency. The seasonal vaccine will undergo less speedy FDA licensing and will likely not appear on the U.S. market until 2013 or 2014, he said. Currently, U.S. residents received roughly 130 million doses of seasonal flu vaccine annually.
For pandemics, a major goal of cell-based flu vaccine production is to have "more vaccine available sooner," Dr. Robinson said. "The goal is 4 months" once a candidate vaccine strain is isolated during a new pandemic, but a more realistic expectation is that facility will send out significant amounts of vaccine after "4-5 months," he said. That compares with the 5 months it took to have the first egg-based vaccine appear during the H1N1 pandemic of 2009, he noted.

"If a pandemic happened now, and if everything went right, we’d cut the time to vaccine by about 1 month. That would have a huge impact. We hope it would eliminate the confusion" and the rationing that characterized the first weeks of pandemic vaccine availability in 2009, Dr. Robinson said.
Even saving just a couple weeks or so could make a difference, some experts said. "If the H1N1 vaccine had been available in quantity 1 month earlier, it would have made a difference in containing the [2009] pandemic," said Dr. W. Paul Glezen, a professor of molecular virology and microbiology at Baylor College of Medicine, Houston. "Using a tissue culture substrate to grow influenza viruses provides flexibility to vaccine production not available before using embryonated eggs. It should be safer as well, with less danger of contamination. Holly Springs is the beginning, and a good start. Most of the other manufacturers are also developing facilities for growing vaccine in tissue culture. The evidence suggests that all influenza vaccine should be produced in mammalian cells because adapting human influenza virus to grow in eggs alters important antigens that may result in vaccine that is less effective. The shorter production time also means the decision for [seasonal] vaccine constituents can be delayed to allow more time to assess the emergence of new variants to include in the vaccine."
"One month less" time to produce pandemic vaccine "is probably the best-case scenario. We might not get a full month, but it is supposed to be somewhat faster" than egg-based production, said Dr. Schaffner. "It could get the program going quicker, it could give the public a measure of confidence that the response is speedy and more optimistic. In 2009, it was perceived that the government over-promised and under-delivered. If we can start getting vaccine into the pipeline quicker, the public will think [the public health response] is on the ball."
But the reality may be more sobering. A Novartis cell-culture plant in Europe made H1N1 pandemic vaccine in 2009 that did not arrive any quicker than egg-based vaccine, Dr. Osterholm noted. Even if the 2009 European vaccine had glitches that could be avoided in America when the next flu pandemic strikes, what difference might a few weeks make for public health? "It means that vaccine would arrive at about the peak of the second [infection] wave, which is still really late," he said. "Even if you shaved a month off in 2009, it still would have had a limited impact. We need to do our best to have vaccine before the second wave."
Dr. Robinson agreed that even faster would be better. Next on the agenda for the Biomedical Advanced Research and Development Authority and HHS is to finish plans to make pandemic flu vaccine using recombinant technology. Dr. Robinson said this could produce vaccine within about 12 weeks, and that this capability is roughly 2 years away.
But recombinant vaccine may not be ideal either. Investigational recombinant vaccines have had effectiveness levels comparable to the 60% seen in trivalent inactivated vaccines. The answer, Dr. Osterholm said, lies in better understanding the immunity produced by natural flu infection and using that as a model to develop new vaccine antigens that reach much higher levels of effectiveness.
"Much of what we know about flu immunity is for hemagglutinin, and that’s not much," he said. "In 2009, there was clear evidence that 65- to 70-year-olds had residual protection to H1N1 from when that virus last circulated in the 1950s. These elderly people were entering immunosenescence, but they still had good protection. Yet we can’t get good protection from year to year with hemagglutinin-based vaccines. That shows something else going on immunologically that we should think about. Several possible target antigens have been identified, but so far they are not going anywhere because there has not been sufficient investment.
"Novel antigens are the ultimate flu vaccine goal. Is the Holly Springs plant a good thing for the short term? Sure, it’s an incremental increase in preparedness. But it’s not a major change in preparedness," Dr. Osterholm said.
Dr. Osterholm and Dr. Robinson said that they had no disclosures. Dr. Schaffner said that he has been a consultant to GlaxoSmithKline, Dynavax, Novartis, and Pfizer, and that he has served on data safety and monitoring boards for Sanofi-Pasteur and Merck. Dr. Glezen said that he has received a research grant from MedImmune.
With the official dedication Dec. 12 of America’s first facility for making influenza vaccine in cell culture, the United States took a small but important step toward improved protection against the next flu pandemic, experts said.
But some question just how many weeks this new facility will shave off of the timeline for moving from a candidate pandemic vaccine strain to getting the vaccine into the arms of the American public. And by relying on an inherently old-fashioned approach to flu protection with limited effectiveness, some see this development as merely refining an archaic vaccine that is overdue for replacement by a newer and better method of immunoprotection against pandemic influenza.
"It’s a small, incremental step in preparedness, but the current hemagglutinin-antigen based vaccine really is inadequate for providing the kind of immunologic protection we need, and so anything you do to speed up the amount of [this type of] vaccine is only a marginal improvement in protecting against influenza," commented Michael T. Osterholm, Ph.D., professor in the school of public health and director of the Center for Infectious Disease Research and Policy at the University of Minnesota, Minneapolis.
"Just because you can make more of this [vaccine] faster, what does that mean? It’s better than nothing, absolutely, but that doesn’t mean it will be the answer for a very serious pandemic," Dr. Osterholm said in an interview.
"Having a new facility of substantial production capacity in the United States is a comfort," said Dr. William Schaffner, professor of medicine and chairman of preventive medicine at Vanderbilt University, Nashville, Tenn. "When you’re creating a pandemic vaccine, every week counts," and the cell culture–based technology that the new plant employs "is supposed to be somewhat faster" than conventional, egg-based production of flu vaccine, he noted. By not relying on chicken-based egg production that might conceivably be vulnerable to an avian flu pandemic, the new facility also sidesteps that potential danger.
"Having this facility in the United States is a significant step, as is having more vaccine," he said.
The new vaccine plant, in Holly Springs, N.C., culminates a 7-year project by Novartis working on contract with the Health and Human Services department, which funded 49% of the construction costs. The new plant brings cell-based flu vaccine production into the United States, and eventually onto the U.S. vaccine market for the first time, and also serves to beef up U.S.-based flu vaccine production of all types.
HHS officials first envisioned the new facility in 2004, when the American flu vaccine supply became threatened by many previous manufacturers going out of business, and when the supply from British-based Chiron temporarily went off line, said Robin Robinson, Ph.D., director of the HSS Biomedical Advanced Research and Development Authority. The facility uses cultured canine kidney cells to grow influenza vaccine.
The Holly Springs plant first became operational in late 2009, and now has produced several commercial-scale lots that have undergone testing in Phase III trials, Dr. Robinson said in an interview. The facility is positioned to immediately start making as much as 50 million doses of pandemic flu vaccine using cell-culture production, which could receive emergency-use FDA licensing and be available to the American public.
In the absence of a pandemic, the facility will primarily focus on making seasonal influenza vaccine (as well as certain other vaccines), but with much less urgency. The seasonal vaccine will undergo less speedy FDA licensing and will likely not appear on the U.S. market until 2013 or 2014, he said. Currently, U.S. residents received roughly 130 million doses of seasonal flu vaccine annually.
For pandemics, a major goal of cell-based flu vaccine production is to have "more vaccine available sooner," Dr. Robinson said. "The goal is 4 months" once a candidate vaccine strain is isolated during a new pandemic, but a more realistic expectation is that facility will send out significant amounts of vaccine after "4-5 months," he said. That compares with the 5 months it took to have the first egg-based vaccine appear during the H1N1 pandemic of 2009, he noted.
"If a pandemic happened now, and if everything went right, we’d cut the time to vaccine by about 1 month. That would have a huge impact. We hope it would eliminate the confusion" and the rationing that characterized the first weeks of pandemic vaccine availability in 2009, Dr. Robinson said.
Even saving just a couple weeks or so could make a difference, some experts said. "If the H1N1 vaccine had been available in quantity 1 month earlier, it would have made a difference in containing the [2009] pandemic," said Dr. W. Paul Glezen, a professor of molecular virology and microbiology at Baylor College of Medicine, Houston. "Using a tissue culture substrate to grow influenza viruses provides flexibility to vaccine production not available before using embryonated eggs. It should be safer as well, with less danger of contamination. Holly Springs is the beginning, and a good start. Most of the other manufacturers are also developing facilities for growing vaccine in tissue culture. The evidence suggests that all influenza vaccine should be produced in mammalian cells because adapting human influenza virus to grow in eggs alters important antigens that may result in vaccine that is less effective. The shorter production time also means the decision for [seasonal] vaccine constituents can be delayed to allow more time to assess the emergence of new variants to include in the vaccine."
"One month less" time to produce pandemic vaccine "is probably the best-case scenario. We might not get a full month, but it is supposed to be somewhat faster" than egg-based production, said Dr. Schaffner. "It could get the program going quicker, it could give the public a measure of confidence that the response is speedy and more optimistic. In 2009, it was perceived that the government over-promised and under-delivered. If we can start getting vaccine into the pipeline quicker, the public will think [the public health response] is on the ball."
But the reality may be more sobering. A Novartis cell-culture plant in Europe made H1N1 pandemic vaccine in 2009 that did not arrive any quicker than egg-based vaccine, Dr. Osterholm noted. Even if the 2009 European vaccine had glitches that could be avoided in America when the next flu pandemic strikes, what difference might a few weeks make for public health? "It means that vaccine would arrive at about the peak of the second [infection] wave, which is still really late," he said. "Even if you shaved a month off in 2009, it still would have had a limited impact. We need to do our best to have vaccine before the second wave."
Dr. Robinson agreed that even faster would be better. Next on the agenda for the Biomedical Advanced Research and Development Authority and HHS is to finish plans to make pandemic flu vaccine using recombinant technology. Dr. Robinson said this could produce vaccine within about 12 weeks, and that this capability is roughly 2 years away.
But recombinant vaccine may not be ideal either. Investigational recombinant vaccines have had effectiveness levels comparable to the 60% seen in trivalent inactivated vaccines. The answer, Dr. Osterholm said, lies in better understanding the immunity produced by natural flu infection and using that as a model to develop new vaccine antigens that reach much higher levels of effectiveness.
"Much of what we know about flu immunity is for hemagglutinin, and that’s not much," he said. "In 2009, there was clear evidence that 65- to 70-year-olds had residual protection to H1N1 from when that virus last circulated in the 1950s. These elderly people were entering immunosenescence, but they still had good protection. Yet we can’t get good protection from year to year with hemagglutinin-based vaccines. That shows something else going on immunologically that we should think about. Several possible target antigens have been identified, but so far they are not going anywhere because there has not been sufficient investment.
"Novel antigens are the ultimate flu vaccine goal. Is the Holly Springs plant a good thing for the short term? Sure, it’s an incremental increase in preparedness. But it’s not a major change in preparedness," Dr. Osterholm said.
Dr. Osterholm and Dr. Robinson said that they had no disclosures. Dr. Schaffner said that he has been a consultant to GlaxoSmithKline, Dynavax, Novartis, and Pfizer, and that he has served on data safety and monitoring boards for Sanofi-Pasteur and Merck. Dr. Glezen said that he has received a research grant from MedImmune.
With the official dedication Dec. 12 of America’s first facility for making influenza vaccine in cell culture, the United States took a small but important step toward improved protection against the next flu pandemic, experts said.
But some question just how many weeks this new facility will shave off of the timeline for moving from a candidate pandemic vaccine strain to getting the vaccine into the arms of the American public. And by relying on an inherently old-fashioned approach to flu protection with limited effectiveness, some see this development as merely refining an archaic vaccine that is overdue for replacement by a newer and better method of immunoprotection against pandemic influenza.
"It’s a small, incremental step in preparedness, but the current hemagglutinin-antigen based vaccine really is inadequate for providing the kind of immunologic protection we need, and so anything you do to speed up the amount of [this type of] vaccine is only a marginal improvement in protecting against influenza," commented Michael T. Osterholm, Ph.D., professor in the school of public health and director of the Center for Infectious Disease Research and Policy at the University of Minnesota, Minneapolis.
"Just because you can make more of this [vaccine] faster, what does that mean? It’s better than nothing, absolutely, but that doesn’t mean it will be the answer for a very serious pandemic," Dr. Osterholm said in an interview.
"Having a new facility of substantial production capacity in the United States is a comfort," said Dr. William Schaffner, professor of medicine and chairman of preventive medicine at Vanderbilt University, Nashville, Tenn. "When you’re creating a pandemic vaccine, every week counts," and the cell culture–based technology that the new plant employs "is supposed to be somewhat faster" than conventional, egg-based production of flu vaccine, he noted. By not relying on chicken-based egg production that might conceivably be vulnerable to an avian flu pandemic, the new facility also sidesteps that potential danger.
"Having this facility in the United States is a significant step, as is having more vaccine," he said.
The new vaccine plant, in Holly Springs, N.C., culminates a 7-year project by Novartis working on contract with the Health and Human Services department, which funded 49% of the construction costs. The new plant brings cell-based flu vaccine production into the United States, and eventually onto the U.S. vaccine market for the first time, and also serves to beef up U.S.-based flu vaccine production of all types.
HHS officials first envisioned the new facility in 2004, when the American flu vaccine supply became threatened by many previous manufacturers going out of business, and when the supply from British-based Chiron temporarily went off line, said Robin Robinson, Ph.D., director of the HSS Biomedical Advanced Research and Development Authority. The facility uses cultured canine kidney cells to grow influenza vaccine.
The Holly Springs plant first became operational in late 2009, and now has produced several commercial-scale lots that have undergone testing in Phase III trials, Dr. Robinson said in an interview. The facility is positioned to immediately start making as much as 50 million doses of pandemic flu vaccine using cell-culture production, which could receive emergency-use FDA licensing and be available to the American public.
In the absence of a pandemic, the facility will primarily focus on making seasonal influenza vaccine (as well as certain other vaccines), but with much less urgency. The seasonal vaccine will undergo less speedy FDA licensing and will likely not appear on the U.S. market until 2013 or 2014, he said. Currently, U.S. residents received roughly 130 million doses of seasonal flu vaccine annually.
For pandemics, a major goal of cell-based flu vaccine production is to have "more vaccine available sooner," Dr. Robinson said. "The goal is 4 months" once a candidate vaccine strain is isolated during a new pandemic, but a more realistic expectation is that facility will send out significant amounts of vaccine after "4-5 months," he said. That compares with the 5 months it took to have the first egg-based vaccine appear during the H1N1 pandemic of 2009, he noted.
"If a pandemic happened now, and if everything went right, we’d cut the time to vaccine by about 1 month. That would have a huge impact. We hope it would eliminate the confusion" and the rationing that characterized the first weeks of pandemic vaccine availability in 2009, Dr. Robinson said.
Even saving just a couple weeks or so could make a difference, some experts said. "If the H1N1 vaccine had been available in quantity 1 month earlier, it would have made a difference in containing the [2009] pandemic," said Dr. W. Paul Glezen, a professor of molecular virology and microbiology at Baylor College of Medicine, Houston. "Using a tissue culture substrate to grow influenza viruses provides flexibility to vaccine production not available before using embryonated eggs. It should be safer as well, with less danger of contamination. Holly Springs is the beginning, and a good start. Most of the other manufacturers are also developing facilities for growing vaccine in tissue culture. The evidence suggests that all influenza vaccine should be produced in mammalian cells because adapting human influenza virus to grow in eggs alters important antigens that may result in vaccine that is less effective. The shorter production time also means the decision for [seasonal] vaccine constituents can be delayed to allow more time to assess the emergence of new variants to include in the vaccine."
"One month less" time to produce pandemic vaccine "is probably the best-case scenario. We might not get a full month, but it is supposed to be somewhat faster" than egg-based production, said Dr. Schaffner. "It could get the program going quicker, it could give the public a measure of confidence that the response is speedy and more optimistic. In 2009, it was perceived that the government over-promised and under-delivered. If we can start getting vaccine into the pipeline quicker, the public will think [the public health response] is on the ball."
But the reality may be more sobering. A Novartis cell-culture plant in Europe made H1N1 pandemic vaccine in 2009 that did not arrive any quicker than egg-based vaccine, Dr. Osterholm noted. Even if the 2009 European vaccine had glitches that could be avoided in America when the next flu pandemic strikes, what difference might a few weeks make for public health? "It means that vaccine would arrive at about the peak of the second [infection] wave, which is still really late," he said. "Even if you shaved a month off in 2009, it still would have had a limited impact. We need to do our best to have vaccine before the second wave."
Dr. Robinson agreed that even faster would be better. Next on the agenda for the Biomedical Advanced Research and Development Authority and HHS is to finish plans to make pandemic flu vaccine using recombinant technology. Dr. Robinson said this could produce vaccine within about 12 weeks, and that this capability is roughly 2 years away.
But recombinant vaccine may not be ideal either. Investigational recombinant vaccines have had effectiveness levels comparable to the 60% seen in trivalent inactivated vaccines. The answer, Dr. Osterholm said, lies in better understanding the immunity produced by natural flu infection and using that as a model to develop new vaccine antigens that reach much higher levels of effectiveness.
"Much of what we know about flu immunity is for hemagglutinin, and that’s not much," he said. "In 2009, there was clear evidence that 65- to 70-year-olds had residual protection to H1N1 from when that virus last circulated in the 1950s. These elderly people were entering immunosenescence, but they still had good protection. Yet we can’t get good protection from year to year with hemagglutinin-based vaccines. That shows something else going on immunologically that we should think about. Several possible target antigens have been identified, but so far they are not going anywhere because there has not been sufficient investment.
"Novel antigens are the ultimate flu vaccine goal. Is the Holly Springs plant a good thing for the short term? Sure, it’s an incremental increase in preparedness. But it’s not a major change in preparedness," Dr. Osterholm said.
Dr. Osterholm and Dr. Robinson said that they had no disclosures. Dr. Schaffner said that he has been a consultant to GlaxoSmithKline, Dynavax, Novartis, and Pfizer, and that he has served on data safety and monitoring boards for Sanofi-Pasteur and Merck. Dr. Glezen said that he has received a research grant from MedImmune.
Remission Entrenched as RA Management Goal
Remission has become an accepted goal in the management of rheumatoid arthritis, but its definition remains in flux.
About a year ago, a panel assembled jointly by the American College of Rheumatology (ACR) and the European League Against Rheumatism (EULAR) published two provisional definitions of remission in rheumatoid arthritis (RA) for clinical trials: a Simplified Disease Activity Index (SDAI) score of 3.3 or less; or no more than 1 tender and 1 swollen on a 28-joint examination, a C reactive protein (CRP) level of 1 mg/dL or less, and a patient global assessment score of 1 or less on a 0-10 scale (Arthritis Rheum. 2011;63:573-86; Ann. Rheum. Dis. 2011;70:404-13).
The ACR/EULAR panel explicitly said that their definitions of remission were intended for use only in clinical trials for the time being, and the group did not yet endorse their use in routine clinical practice, in part because the definitions had not yet been tested in that setting, and in part because in clinical practice data on acute phase reactants, such as CRP, "are frequently not immediately available."
Dr. David T. Felson, professor of medicine and epidemiology at Boston University as well as first author on the ACR/EULAR remission paper, noted in an interview that "We studied data from clinical trials to develop these remission criteria, and trial patients are not generalizable to those in practice, nor are their assessments as comprehensive."
But even with the new definitions not formally designed for routine practice, their designation by a combined ACR and EULAR panel appears to have helped solidify remission as the benchmark goal for management of most RA patients, capping a decade-long trend. It’s already well accepted that remission is achievable in "at least half" of patients with new-onset RA, noted Dr. James R. O’Dell and Dr. Ted R. Mikuls in an editorial that accompanied the publication of the provisional definition (Arthritis Rheum. 2011;63:587-9). The remission rate in patients with long-standing RA is much lower, more on the order of perhaps 10%, Dr. O’Dell said in an interview.
Noted Dr. Daniel E. Furst: "When I started working on rheumatoid arthritis, we used the word remission with the hope that some day it would be possible. Because of advances in treatment, over the last 10 years it has become possible, and consequently it is totally appropriate that we aim for remission."
The provisional definitions that the panel set for clinical trials serve as "a reasonable set of criteria" for routine practice, said Dr. Furst, who is Carl M. Pearson professor in rheumatology at the University of California, Los Angeles and a member of the ACR/EULAR panel.
"I do this all the time. It requires physicians to routinely quantify patient responses, which is not common right now, but it will become more common. The need for a lab test [measurement of CRP] as part of the definition makes it a little more difficult to use because it usually takes some time to get the blood-test result. What I do is a CDAI [Clinical Disease Activity Index, the sum of tender and swollen joint counts, and physician and patient global assessments] at the same time that I’m obtaining the other results that take time. That’s more practical to do in everyday practice," he said in an interview.
"Right now, my associates and I generally use a DAS28, but we’re rethinking that," in part prompted by the new remission definitions, said Dr. O’Dell, who is Larson professor of medicine and chief of rheumatology at the University of Nebraska in Omaha. "DAS28 is an imperfect measure. I can have a patient with a very low [ESR] of 2 [mm/hr] and their DAS28 will look pretty good until they have three or four swollen joints. But the flip side is I can have a patient who is doing terrific, with no swollen joints, and their [ESR] is 25. Since the ARC/EULAR definition, we have thought about whether we should do more CDAI or SDAI. [ESR] and CRP give information in a different way than what we get from joints, but they often aren’t available in real time.
"The ACR Quality Measures Committee will issue a white paper in late spring on the disease activity measures that it thinks are feasible and that clinicians can use, including the CADI, SDAI, DAS28, RAPID3 [Routine Assessment of Patient Index Data 3], and PAS [Patient Activity Score]. It’s far more important that physicians measure a patient’s disease activity with some scale than which scale you use," he said in an interview. "If you don’t want to do 28-joint counts, then do a RAPID3. The ACR/EULAR panel set its remission criteria for trials, but for routine practice there should be more flexibility" for physicians to use the scale that best fits their approach to practice, Dr. O’Dell said. "The RAPID3 is much easier to do and is available in real time, and is very good. It’s not quite as good as some others, but if that’s what you use, you’ll do fine."
Assessing the ACR/EULAR Criteria’s Performance
In addition to not having been derived from patients in routine practice, the ACR/EULAR definitions of remission may also show variability from physician to physician, and unstable reproducibility within individual patients.
Last November, researchers from the Arthritis and Rheumatology Clinics of Kansas in Wichita and their collaborators published an analysis that applied the ACR/EULAR provisional remission definition to patients in two cohorts: 1,341 patients from the Department of Veteran’s Affairs, and 1,153 patients in a cohort assembled at the Arthritis and Rheumatology Clinics of Kansas. The two ACR/EULAR criteria identified about 5%-10% of patients as being in remission at any one time, depending on the cohort and specific definition applied, they reported (Arthritis Rheum. 2011;63:3204-15).
However, the probability of a specific patient being in remission at two or more visits ranged from 2%-5%. The analysis also showed "substantial evidence to indicate that inter-rater reliability is poor with respect to the examination of tender and swollen joints." The authors estimated that the probability of remission using the ACR/EULAR definitions in these two cohorts could vary by twofold depending solely on the physician examiner and independent of disease activity.
The researchers concluded that "problems with reliability and agreement limit the usefulness of these criteria in the individual patient." But several rheumatologists interviewed noted that variability among physicians in scoring swollen and tender joint counts is expected. The finding simply underscores that the best way to serially monitor joint status in a patient is for the same physician to do it every time. They also commented that the low rates of remission seen in this study highlight just how challenging it is to maintain RA patients at a very low level of disease activity.
"These criteria were developed for clinical trials, and I agree that they’re reasonable for trials," said Dr. Frederick Wolfe, the senior author on this study and a member of the ACR/EULAR definition panel. "Until now, there were lots of different ideas of what was remission, which limited its use. We wanted to make a standard for remission [in trials], and did a pretty good job. But in the clinic, physicians know more than they can get from questionnaires," he said in an interview.
For example, Dr. Wolfe cited a study he and his associates ran in which patients were asked what influenced their global self rating. "We found that some patients who all physicians would agree were in remission gave themselves poor scores because they had pain in other regions, mostly back pain, or because of fatigue, or other things" not related to RA. "In clinical practice, we’d ask patients to ignore that, or to explain their high global self-assessment score, so in the clinic you can decide whether patients are in remission," said Dr. Wolfe, director of the National Databank for Rheumatic Diseases in Wichita, Kan.
For measuring RA activity in routine practice, Dr. Wolfe said that he favors the CDAI "as an overall measure of disease activity." He also endorsed patient-centered measures of pain, such as the Health Assessment Questionnaire (HAQ), global self-assessment, RAPID3, and the PAS.
A more positive assessment of the ACR/EULAR definitions in clinical practice came from a recent study that applied them to a cohort of 535 RA patients at Brigham and Women’s Hospital in Boston followed for 2 years by serial joint radiographs. The authors examined the ability of the definitions to predict good radiographic and functional outcomes over time in these clinical-practice patients. They found that 30 patients (6%) of the 535 met the four-part ACR/EULAR definition of remission at baseline, while 26 (5%) met the CDAI definition, 37 (7%) met a SDAI definition of remission, and 106 (20%) met a DAS28-CRP definition. The best of these measures for predicting freedom from radiographic joint progression during 2 years of follow-up was the CDAI, with a positive likelihood ratio of 2.8, followed closely by the four-part ACR/EULAR definition, with a positive likelihood ratio of 2.7. The ratio for SDAI was 2.1, and for DAS28-CRP 1.5 (Ann. Rheum. Dis. 2011 Oct. 12 [doi:10.1136/ard.2011.154625]).
"Our findings strengthen the applicability of the ACR/EULAR criteria in clinical practice," the researchers concluded, and also noted that "the findings might have been different in a patient population of early RA patients." The cohort they examined had been diagnosed with RA for a median of 11 years, with 75% diagnosed for 4 years or longer.
The authors also said "a significant proportion of patients who do not fulfill the remission criteria still experience a good outcome of their disease, especially for radiographic joint damage. ... A byproduct of more stringent remission criteria as a treatment target will be more patients in low disease activity with good disease outcome, potentially risking overtreatment."
Several other assessments of the remission criteria in other data sets are on the way, Dr. O’Dell said. "I’ve heard of them, and people are talking about them," and these reports will likely appear in the literature over the next year. Advance word is that the remission criteria "stood up very well," Dr. O’Dell added.
Is Remission the Right Goal?
The last comment from the Brigham and Women’s group raises the important question of whether remission is the best target for most RA patients or is low disease activity enough.
"About 10%-20% of patients in trials reach remission according to the new definitions. Newly diagnosed patients tend to have disease that is more responsive to treatment, so that the proportion achieving remission is likely to be higher, but probably not reaching 40%," said Dr. Felson. "In trials, regimens will target" the new ARC/EULAR definition, "but I’m less sure about this as a target for patients [in routine practice]. It may be difficult for many patients to reach this threshold, even with optimal current treatment."
For the time being, in patients with an established RA diagnosis using MRI or ultrasound to pick up inflammation that is not clinically apparent "is not practical," said Dr. O’Dell. One area where joint imaging may be especially helpful is in patients who are doing well on treatment and can be considered for tapering down treatment. Studies are now looking at whether ultrasound or MRI can identify patients who are good candidates for dose reductions, Dr. O’Dell said.
"Remission, whether you define it with the new definition or an older one, is necessary for the long-term health of patients," said Dr. Furst. "The goal is preventing x-ray damage, and allowing patients to have optimal long-term function and quality of life. That’s what remission criteria are all about: They give physicians a measuring stick for telling how well a patient is doing" with respect to these long-term treatment goals. "There is always a balance between achieving these goals and making sure the patient is not harmed" by aggressive treatment. The ACR/EULAR criteria "emphasize the need to quantify patient response, and in that sense they are slowly changing practice," Dr. Furst said.
"There is no doubt that there is increasing movement toward treating to a target, but most rheumatologists don’t believe that remission is the right goal for every patient." said Dr. O’Dell. "We’d love to have remission, but sometimes it’s more problematic than it’s worth. What we don’t know is the long-term difference between a patient who is barely in remission compared with one who is close but not in remission. How far should we push it? If the patient is not having trouble with treatment, then we should definitely push it. If a patient is on 20 mg/week of methotrexate and is nearly there, then trying 25 mg of methotrexate/week is clearly the right thing to do in most patients. But if the patient is not at remission but also is not always tolerating 20 mg/week, if they don’t feel good the day after their dose, what’s the right answer? We don’t know; it’s clinical judgment."
Dr. Felson, Dr. O’Dell and Dr. Wolfe said they had no disclosures. Dr. Furst said that he has financial relationships with Abbott, Actelion, Amgen, Biogen Idec, Bristol-Myers Squibb, Centocor, Genentech, Gilead, GlaxoSmithKline, Merck, Nitec, Novartis, Roche, UCB, Wyeth, and Xoma.
Remission has become an accepted goal in the management of rheumatoid arthritis, but its definition remains in flux.
About a year ago, a panel assembled jointly by the American College of Rheumatology (ACR) and the European League Against Rheumatism (EULAR) published two provisional definitions of remission in rheumatoid arthritis (RA) for clinical trials: a Simplified Disease Activity Index (SDAI) score of 3.3 or less; or no more than 1 tender and 1 swollen on a 28-joint examination, a C reactive protein (CRP) level of 1 mg/dL or less, and a patient global assessment score of 1 or less on a 0-10 scale (Arthritis Rheum. 2011;63:573-86; Ann. Rheum. Dis. 2011;70:404-13).
The ACR/EULAR panel explicitly said that their definitions of remission were intended for use only in clinical trials for the time being, and the group did not yet endorse their use in routine clinical practice, in part because the definitions had not yet been tested in that setting, and in part because in clinical practice data on acute phase reactants, such as CRP, "are frequently not immediately available."
Dr. David T. Felson, professor of medicine and epidemiology at Boston University as well as first author on the ACR/EULAR remission paper, noted in an interview that "We studied data from clinical trials to develop these remission criteria, and trial patients are not generalizable to those in practice, nor are their assessments as comprehensive."
But even with the new definitions not formally designed for routine practice, their designation by a combined ACR and EULAR panel appears to have helped solidify remission as the benchmark goal for management of most RA patients, capping a decade-long trend. It’s already well accepted that remission is achievable in "at least half" of patients with new-onset RA, noted Dr. James R. O’Dell and Dr. Ted R. Mikuls in an editorial that accompanied the publication of the provisional definition (Arthritis Rheum. 2011;63:587-9). The remission rate in patients with long-standing RA is much lower, more on the order of perhaps 10%, Dr. O’Dell said in an interview.
Noted Dr. Daniel E. Furst: "When I started working on rheumatoid arthritis, we used the word remission with the hope that some day it would be possible. Because of advances in treatment, over the last 10 years it has become possible, and consequently it is totally appropriate that we aim for remission."
The provisional definitions that the panel set for clinical trials serve as "a reasonable set of criteria" for routine practice, said Dr. Furst, who is Carl M. Pearson professor in rheumatology at the University of California, Los Angeles and a member of the ACR/EULAR panel.
"I do this all the time. It requires physicians to routinely quantify patient responses, which is not common right now, but it will become more common. The need for a lab test [measurement of CRP] as part of the definition makes it a little more difficult to use because it usually takes some time to get the blood-test result. What I do is a CDAI [Clinical Disease Activity Index, the sum of tender and swollen joint counts, and physician and patient global assessments] at the same time that I’m obtaining the other results that take time. That’s more practical to do in everyday practice," he said in an interview.
"Right now, my associates and I generally use a DAS28, but we’re rethinking that," in part prompted by the new remission definitions, said Dr. O’Dell, who is Larson professor of medicine and chief of rheumatology at the University of Nebraska in Omaha. "DAS28 is an imperfect measure. I can have a patient with a very low [ESR] of 2 [mm/hr] and their DAS28 will look pretty good until they have three or four swollen joints. But the flip side is I can have a patient who is doing terrific, with no swollen joints, and their [ESR] is 25. Since the ARC/EULAR definition, we have thought about whether we should do more CDAI or SDAI. [ESR] and CRP give information in a different way than what we get from joints, but they often aren’t available in real time.
"The ACR Quality Measures Committee will issue a white paper in late spring on the disease activity measures that it thinks are feasible and that clinicians can use, including the CADI, SDAI, DAS28, RAPID3 [Routine Assessment of Patient Index Data 3], and PAS [Patient Activity Score]. It’s far more important that physicians measure a patient’s disease activity with some scale than which scale you use," he said in an interview. "If you don’t want to do 28-joint counts, then do a RAPID3. The ACR/EULAR panel set its remission criteria for trials, but for routine practice there should be more flexibility" for physicians to use the scale that best fits their approach to practice, Dr. O’Dell said. "The RAPID3 is much easier to do and is available in real time, and is very good. It’s not quite as good as some others, but if that’s what you use, you’ll do fine."
Assessing the ACR/EULAR Criteria’s Performance
In addition to not having been derived from patients in routine practice, the ACR/EULAR definitions of remission may also show variability from physician to physician, and unstable reproducibility within individual patients.
Last November, researchers from the Arthritis and Rheumatology Clinics of Kansas in Wichita and their collaborators published an analysis that applied the ACR/EULAR provisional remission definition to patients in two cohorts: 1,341 patients from the Department of Veteran’s Affairs, and 1,153 patients in a cohort assembled at the Arthritis and Rheumatology Clinics of Kansas. The two ACR/EULAR criteria identified about 5%-10% of patients as being in remission at any one time, depending on the cohort and specific definition applied, they reported (Arthritis Rheum. 2011;63:3204-15).
However, the probability of a specific patient being in remission at two or more visits ranged from 2%-5%. The analysis also showed "substantial evidence to indicate that inter-rater reliability is poor with respect to the examination of tender and swollen joints." The authors estimated that the probability of remission using the ACR/EULAR definitions in these two cohorts could vary by twofold depending solely on the physician examiner and independent of disease activity.
The researchers concluded that "problems with reliability and agreement limit the usefulness of these criteria in the individual patient." But several rheumatologists interviewed noted that variability among physicians in scoring swollen and tender joint counts is expected. The finding simply underscores that the best way to serially monitor joint status in a patient is for the same physician to do it every time. They also commented that the low rates of remission seen in this study highlight just how challenging it is to maintain RA patients at a very low level of disease activity.
"These criteria were developed for clinical trials, and I agree that they’re reasonable for trials," said Dr. Frederick Wolfe, the senior author on this study and a member of the ACR/EULAR definition panel. "Until now, there were lots of different ideas of what was remission, which limited its use. We wanted to make a standard for remission [in trials], and did a pretty good job. But in the clinic, physicians know more than they can get from questionnaires," he said in an interview.
For example, Dr. Wolfe cited a study he and his associates ran in which patients were asked what influenced their global self rating. "We found that some patients who all physicians would agree were in remission gave themselves poor scores because they had pain in other regions, mostly back pain, or because of fatigue, or other things" not related to RA. "In clinical practice, we’d ask patients to ignore that, or to explain their high global self-assessment score, so in the clinic you can decide whether patients are in remission," said Dr. Wolfe, director of the National Databank for Rheumatic Diseases in Wichita, Kan.
For measuring RA activity in routine practice, Dr. Wolfe said that he favors the CDAI "as an overall measure of disease activity." He also endorsed patient-centered measures of pain, such as the Health Assessment Questionnaire (HAQ), global self-assessment, RAPID3, and the PAS.
A more positive assessment of the ACR/EULAR definitions in clinical practice came from a recent study that applied them to a cohort of 535 RA patients at Brigham and Women’s Hospital in Boston followed for 2 years by serial joint radiographs. The authors examined the ability of the definitions to predict good radiographic and functional outcomes over time in these clinical-practice patients. They found that 30 patients (6%) of the 535 met the four-part ACR/EULAR definition of remission at baseline, while 26 (5%) met the CDAI definition, 37 (7%) met a SDAI definition of remission, and 106 (20%) met a DAS28-CRP definition. The best of these measures for predicting freedom from radiographic joint progression during 2 years of follow-up was the CDAI, with a positive likelihood ratio of 2.8, followed closely by the four-part ACR/EULAR definition, with a positive likelihood ratio of 2.7. The ratio for SDAI was 2.1, and for DAS28-CRP 1.5 (Ann. Rheum. Dis. 2011 Oct. 12 [doi:10.1136/ard.2011.154625]).
"Our findings strengthen the applicability of the ACR/EULAR criteria in clinical practice," the researchers concluded, and also noted that "the findings might have been different in a patient population of early RA patients." The cohort they examined had been diagnosed with RA for a median of 11 years, with 75% diagnosed for 4 years or longer.
The authors also said "a significant proportion of patients who do not fulfill the remission criteria still experience a good outcome of their disease, especially for radiographic joint damage. ... A byproduct of more stringent remission criteria as a treatment target will be more patients in low disease activity with good disease outcome, potentially risking overtreatment."
Several other assessments of the remission criteria in other data sets are on the way, Dr. O’Dell said. "I’ve heard of them, and people are talking about them," and these reports will likely appear in the literature over the next year. Advance word is that the remission criteria "stood up very well," Dr. O’Dell added.
Is Remission the Right Goal?
The last comment from the Brigham and Women’s group raises the important question of whether remission is the best target for most RA patients or is low disease activity enough.
"About 10%-20% of patients in trials reach remission according to the new definitions. Newly diagnosed patients tend to have disease that is more responsive to treatment, so that the proportion achieving remission is likely to be higher, but probably not reaching 40%," said Dr. Felson. "In trials, regimens will target" the new ARC/EULAR definition, "but I’m less sure about this as a target for patients [in routine practice]. It may be difficult for many patients to reach this threshold, even with optimal current treatment."
For the time being, in patients with an established RA diagnosis using MRI or ultrasound to pick up inflammation that is not clinically apparent "is not practical," said Dr. O’Dell. One area where joint imaging may be especially helpful is in patients who are doing well on treatment and can be considered for tapering down treatment. Studies are now looking at whether ultrasound or MRI can identify patients who are good candidates for dose reductions, Dr. O’Dell said.
"Remission, whether you define it with the new definition or an older one, is necessary for the long-term health of patients," said Dr. Furst. "The goal is preventing x-ray damage, and allowing patients to have optimal long-term function and quality of life. That’s what remission criteria are all about: They give physicians a measuring stick for telling how well a patient is doing" with respect to these long-term treatment goals. "There is always a balance between achieving these goals and making sure the patient is not harmed" by aggressive treatment. The ACR/EULAR criteria "emphasize the need to quantify patient response, and in that sense they are slowly changing practice," Dr. Furst said.
"There is no doubt that there is increasing movement toward treating to a target, but most rheumatologists don’t believe that remission is the right goal for every patient." said Dr. O’Dell. "We’d love to have remission, but sometimes it’s more problematic than it’s worth. What we don’t know is the long-term difference between a patient who is barely in remission compared with one who is close but not in remission. How far should we push it? If the patient is not having trouble with treatment, then we should definitely push it. If a patient is on 20 mg/week of methotrexate and is nearly there, then trying 25 mg of methotrexate/week is clearly the right thing to do in most patients. But if the patient is not at remission but also is not always tolerating 20 mg/week, if they don’t feel good the day after their dose, what’s the right answer? We don’t know; it’s clinical judgment."
Dr. Felson, Dr. O’Dell and Dr. Wolfe said they had no disclosures. Dr. Furst said that he has financial relationships with Abbott, Actelion, Amgen, Biogen Idec, Bristol-Myers Squibb, Centocor, Genentech, Gilead, GlaxoSmithKline, Merck, Nitec, Novartis, Roche, UCB, Wyeth, and Xoma.
Remission has become an accepted goal in the management of rheumatoid arthritis, but its definition remains in flux.
About a year ago, a panel assembled jointly by the American College of Rheumatology (ACR) and the European League Against Rheumatism (EULAR) published two provisional definitions of remission in rheumatoid arthritis (RA) for clinical trials: a Simplified Disease Activity Index (SDAI) score of 3.3 or less; or no more than 1 tender and 1 swollen on a 28-joint examination, a C reactive protein (CRP) level of 1 mg/dL or less, and a patient global assessment score of 1 or less on a 0-10 scale (Arthritis Rheum. 2011;63:573-86; Ann. Rheum. Dis. 2011;70:404-13).
The ACR/EULAR panel explicitly said that their definitions of remission were intended for use only in clinical trials for the time being, and the group did not yet endorse their use in routine clinical practice, in part because the definitions had not yet been tested in that setting, and in part because in clinical practice data on acute phase reactants, such as CRP, "are frequently not immediately available."
Dr. David T. Felson, professor of medicine and epidemiology at Boston University as well as first author on the ACR/EULAR remission paper, noted in an interview that "We studied data from clinical trials to develop these remission criteria, and trial patients are not generalizable to those in practice, nor are their assessments as comprehensive."
But even with the new definitions not formally designed for routine practice, their designation by a combined ACR and EULAR panel appears to have helped solidify remission as the benchmark goal for management of most RA patients, capping a decade-long trend. It’s already well accepted that remission is achievable in "at least half" of patients with new-onset RA, noted Dr. James R. O’Dell and Dr. Ted R. Mikuls in an editorial that accompanied the publication of the provisional definition (Arthritis Rheum. 2011;63:587-9). The remission rate in patients with long-standing RA is much lower, more on the order of perhaps 10%, Dr. O’Dell said in an interview.
Noted Dr. Daniel E. Furst: "When I started working on rheumatoid arthritis, we used the word remission with the hope that some day it would be possible. Because of advances in treatment, over the last 10 years it has become possible, and consequently it is totally appropriate that we aim for remission."
The provisional definitions that the panel set for clinical trials serve as "a reasonable set of criteria" for routine practice, said Dr. Furst, who is Carl M. Pearson professor in rheumatology at the University of California, Los Angeles and a member of the ACR/EULAR panel.
"I do this all the time. It requires physicians to routinely quantify patient responses, which is not common right now, but it will become more common. The need for a lab test [measurement of CRP] as part of the definition makes it a little more difficult to use because it usually takes some time to get the blood-test result. What I do is a CDAI [Clinical Disease Activity Index, the sum of tender and swollen joint counts, and physician and patient global assessments] at the same time that I’m obtaining the other results that take time. That’s more practical to do in everyday practice," he said in an interview.
"Right now, my associates and I generally use a DAS28, but we’re rethinking that," in part prompted by the new remission definitions, said Dr. O’Dell, who is Larson professor of medicine and chief of rheumatology at the University of Nebraska in Omaha. "DAS28 is an imperfect measure. I can have a patient with a very low [ESR] of 2 [mm/hr] and their DAS28 will look pretty good until they have three or four swollen joints. But the flip side is I can have a patient who is doing terrific, with no swollen joints, and their [ESR] is 25. Since the ARC/EULAR definition, we have thought about whether we should do more CDAI or SDAI. [ESR] and CRP give information in a different way than what we get from joints, but they often aren’t available in real time.
"The ACR Quality Measures Committee will issue a white paper in late spring on the disease activity measures that it thinks are feasible and that clinicians can use, including the CADI, SDAI, DAS28, RAPID3 [Routine Assessment of Patient Index Data 3], and PAS [Patient Activity Score]. It’s far more important that physicians measure a patient’s disease activity with some scale than which scale you use," he said in an interview. "If you don’t want to do 28-joint counts, then do a RAPID3. The ACR/EULAR panel set its remission criteria for trials, but for routine practice there should be more flexibility" for physicians to use the scale that best fits their approach to practice, Dr. O’Dell said. "The RAPID3 is much easier to do and is available in real time, and is very good. It’s not quite as good as some others, but if that’s what you use, you’ll do fine."
Assessing the ACR/EULAR Criteria’s Performance
In addition to not having been derived from patients in routine practice, the ACR/EULAR definitions of remission may also show variability from physician to physician, and unstable reproducibility within individual patients.
Last November, researchers from the Arthritis and Rheumatology Clinics of Kansas in Wichita and their collaborators published an analysis that applied the ACR/EULAR provisional remission definition to patients in two cohorts: 1,341 patients from the Department of Veteran’s Affairs, and 1,153 patients in a cohort assembled at the Arthritis and Rheumatology Clinics of Kansas. The two ACR/EULAR criteria identified about 5%-10% of patients as being in remission at any one time, depending on the cohort and specific definition applied, they reported (Arthritis Rheum. 2011;63:3204-15).
However, the probability of a specific patient being in remission at two or more visits ranged from 2%-5%. The analysis also showed "substantial evidence to indicate that inter-rater reliability is poor with respect to the examination of tender and swollen joints." The authors estimated that the probability of remission using the ACR/EULAR definitions in these two cohorts could vary by twofold depending solely on the physician examiner and independent of disease activity.
The researchers concluded that "problems with reliability and agreement limit the usefulness of these criteria in the individual patient." But several rheumatologists interviewed noted that variability among physicians in scoring swollen and tender joint counts is expected. The finding simply underscores that the best way to serially monitor joint status in a patient is for the same physician to do it every time. They also commented that the low rates of remission seen in this study highlight just how challenging it is to maintain RA patients at a very low level of disease activity.
"These criteria were developed for clinical trials, and I agree that they’re reasonable for trials," said Dr. Frederick Wolfe, the senior author on this study and a member of the ACR/EULAR definition panel. "Until now, there were lots of different ideas of what was remission, which limited its use. We wanted to make a standard for remission [in trials], and did a pretty good job. But in the clinic, physicians know more than they can get from questionnaires," he said in an interview.
For example, Dr. Wolfe cited a study he and his associates ran in which patients were asked what influenced their global self rating. "We found that some patients who all physicians would agree were in remission gave themselves poor scores because they had pain in other regions, mostly back pain, or because of fatigue, or other things" not related to RA. "In clinical practice, we’d ask patients to ignore that, or to explain their high global self-assessment score, so in the clinic you can decide whether patients are in remission," said Dr. Wolfe, director of the National Databank for Rheumatic Diseases in Wichita, Kan.
For measuring RA activity in routine practice, Dr. Wolfe said that he favors the CDAI "as an overall measure of disease activity." He also endorsed patient-centered measures of pain, such as the Health Assessment Questionnaire (HAQ), global self-assessment, RAPID3, and the PAS.
A more positive assessment of the ACR/EULAR definitions in clinical practice came from a recent study that applied them to a cohort of 535 RA patients at Brigham and Women’s Hospital in Boston followed for 2 years by serial joint radiographs. The authors examined the ability of the definitions to predict good radiographic and functional outcomes over time in these clinical-practice patients. They found that 30 patients (6%) of the 535 met the four-part ACR/EULAR definition of remission at baseline, while 26 (5%) met the CDAI definition, 37 (7%) met a SDAI definition of remission, and 106 (20%) met a DAS28-CRP definition. The best of these measures for predicting freedom from radiographic joint progression during 2 years of follow-up was the CDAI, with a positive likelihood ratio of 2.8, followed closely by the four-part ACR/EULAR definition, with a positive likelihood ratio of 2.7. The ratio for SDAI was 2.1, and for DAS28-CRP 1.5 (Ann. Rheum. Dis. 2011 Oct. 12 [doi:10.1136/ard.2011.154625]).
"Our findings strengthen the applicability of the ACR/EULAR criteria in clinical practice," the researchers concluded, and also noted that "the findings might have been different in a patient population of early RA patients." The cohort they examined had been diagnosed with RA for a median of 11 years, with 75% diagnosed for 4 years or longer.
The authors also said "a significant proportion of patients who do not fulfill the remission criteria still experience a good outcome of their disease, especially for radiographic joint damage. ... A byproduct of more stringent remission criteria as a treatment target will be more patients in low disease activity with good disease outcome, potentially risking overtreatment."
Several other assessments of the remission criteria in other data sets are on the way, Dr. O’Dell said. "I’ve heard of them, and people are talking about them," and these reports will likely appear in the literature over the next year. Advance word is that the remission criteria "stood up very well," Dr. O’Dell added.
Is Remission the Right Goal?
The last comment from the Brigham and Women’s group raises the important question of whether remission is the best target for most RA patients or is low disease activity enough.
"About 10%-20% of patients in trials reach remission according to the new definitions. Newly diagnosed patients tend to have disease that is more responsive to treatment, so that the proportion achieving remission is likely to be higher, but probably not reaching 40%," said Dr. Felson. "In trials, regimens will target" the new ARC/EULAR definition, "but I’m less sure about this as a target for patients [in routine practice]. It may be difficult for many patients to reach this threshold, even with optimal current treatment."
For the time being, in patients with an established RA diagnosis using MRI or ultrasound to pick up inflammation that is not clinically apparent "is not practical," said Dr. O’Dell. One area where joint imaging may be especially helpful is in patients who are doing well on treatment and can be considered for tapering down treatment. Studies are now looking at whether ultrasound or MRI can identify patients who are good candidates for dose reductions, Dr. O’Dell said.
"Remission, whether you define it with the new definition or an older one, is necessary for the long-term health of patients," said Dr. Furst. "The goal is preventing x-ray damage, and allowing patients to have optimal long-term function and quality of life. That’s what remission criteria are all about: They give physicians a measuring stick for telling how well a patient is doing" with respect to these long-term treatment goals. "There is always a balance between achieving these goals and making sure the patient is not harmed" by aggressive treatment. The ACR/EULAR criteria "emphasize the need to quantify patient response, and in that sense they are slowly changing practice," Dr. Furst said.
"There is no doubt that there is increasing movement toward treating to a target, but most rheumatologists don’t believe that remission is the right goal for every patient." said Dr. O’Dell. "We’d love to have remission, but sometimes it’s more problematic than it’s worth. What we don’t know is the long-term difference between a patient who is barely in remission compared with one who is close but not in remission. How far should we push it? If the patient is not having trouble with treatment, then we should definitely push it. If a patient is on 20 mg/week of methotrexate and is nearly there, then trying 25 mg of methotrexate/week is clearly the right thing to do in most patients. But if the patient is not at remission but also is not always tolerating 20 mg/week, if they don’t feel good the day after their dose, what’s the right answer? We don’t know; it’s clinical judgment."
Dr. Felson, Dr. O’Dell and Dr. Wolfe said they had no disclosures. Dr. Furst said that he has financial relationships with Abbott, Actelion, Amgen, Biogen Idec, Bristol-Myers Squibb, Centocor, Genentech, Gilead, GlaxoSmithKline, Merck, Nitec, Novartis, Roche, UCB, Wyeth, and Xoma.
First U.S. Fibromuscular Dysplasia Registry Yields New Clues
MIAMI BEACH – The mystery shrouding fibromuscular dysplasia, a clinically important and surprisingly prevalent vascular disease of unknown etiology, began to lift with initial findings from 339 patients enrolled in the first U.S. registry for the disease.
Baseline observations from the U.S. Registry for Fibromuscular Dysplasia (FMD), which enrolled its first patient in 2008 and currently has women as 91% of enrolled patients, show that the disease first occurs across a broad swath of age groups, with an age at first diagnosis ranging from 5 to 83 years old. In addition to the distinctive "string of beads" arterial fibrosis appearance that defines the disease and is apparent on imaging, and which usually occurs in the renal or carotid artery, or both, 19% of patients also had a dissection in an artery somewhere in their body (14% with a dissection in a carotid artery) and 17% had an arterial aneurysm somewhere (5% with a renal artery aneurysm and 4% with a carotid aneurysm).
"A patient can have normal blood pressure and normal-appearing carotid and renal arteries but have terrible headaches and FMD. Why? We don’t know."
These dissection and aneurysm prevalence rates had not previously been appreciated to run so high in FMD patients. "It suggests a diffuse arteriopathy that can present in several different ways," Dr. Jeffrey W. Olin said at ISET 2012, an international symposium on endovascular therapy.
Another notable finding was that, besides hypertension, which was the most common presenting manifestation of FMD (seen in 66% of the registry patients at initial diagnosis), other common presenting symptoms included significant, often migraine-like headache in 53%, pulsatile tinnitus (a whooshing sound patients hear) in 30%, and dizziness in 28%. The high prevalence of headache in FMD had been "first reported 30 years ago, but sort of got lost," Dr. Olin said in an interview.
"A patient can have normal blood pressure and normal-appearing carotid and renal arteries but have terrible headaches and FMD. Why? We don’t know. We have no idea what causes the headaches," said Dr. Olin, a cardiologist who is professor of medicine and director of vascular medicine at Mount Sinai Medical Center in New York. Four percent of the registry patients had no symptoms on presentation; physicians found their FMD incidentally during imaging examinations for other reasons.

Other flags to trigger suspicion of FMD include hypertension that begins before age 35 (although it can also start later in life), treatment-resistant hypertension, epigastric bruit and hypertension, renal infarction, cervical bruit in a patient less than 60 years old, transient ischemic attack or stroke in a patient less than 60 years old, or an aortic aneurysm in a patient less than 60 years old.
Perhaps the most surprising new finding so far from the registry is evidence for a strong family history of cardiovascular disease, with 81% of first- or second-degree relatives of FMD patients having hypertension, 59% with hyperlipidemia, 53% with a history of a stroke, 22% with an aneurysm, and 21% with a history of sudden death. The prevalence of a first- or second-degree relative also having a diagnosis of FMD was 7%, not much higher than the currently estimated 4% prevalence of FMD in the general population.
Dr. Olin and his associates from the registry hypothesize that patients who develop FMD have an as-yet unidentified genetic predisposition that interacts with an environmental trigger. His hope is that, by continuing to expand the registry and by receiving substantially more research support than FMD now gets, a more concerted research effort can address the genetic questions raised by the family-history findings.
Current treatment of FMD is symptom driven and usually focuses on trying to resolve patients’ hypertension, which is often treatment resistant.

"FMD is potentially treatable for hypertension," with endovascular treatment of affected renal arteries the standard intervention for patients with resistant hypertension, Dr. Robert A. Lookstein said in a separate talk at the meeting. Hypertension cure rates from balloon angioplasty, however, are "surprisingly low" – 45% in one meta-analysis – probably because of suboptimal interventional treatment, said Dr. Lookstein, chief of the division of interventional radiology at Mount Sinai.
Assessing a patient’s renal arteries before and during treatment using intravascular ultrasound (IVUS) and a pressure wire provides the key to successful resolution of hypertension by renal-artery angioplasty in FMD patients, he said.
These two techniques, especially pressure-wire measurements, allow the operator to assess the patient’s renal arteries before and during treatment to determine whether the intervention is having a meaningful effect. "You can’t tell [whether the angioplasty produced benefit] by the angiography appearance alone," he cautioned. "You need to be compulsive with IVUS and measuring pressure gradients to determine where to start treatment and when to stop."
The U. S. Registry for FMD began with seven U.S. centers enrolling patients and now involves 10 centers and has enrolled more than 500 patients. The first 339 enrollees included 44% who were patients at the Cleveland Clinic and 20% who were patients at Mount Sinai.
The average age of the enrolled patients at first symptom appearance was 47 years old; first diagnosis did not occur until an average of more than 4 years later, at an average age of 52 years old.
Vascular bed involvement among the first registry patients included one or both renal arteries in 69%, at least one extracranial carotid in 62%, vertebral arteries in 19%, and mesenteric arteries in 12%. At the time of enrollment, 7% of patients had a history of coronary artery disease and 10% had a history of stroke. The arterial fibrosis characteristic of FMD notably appears in portions of the renal and carotid arteries where atherosclerotic disease usually does not occur – in the distal renal artery and in the distal carotid, "higher than physicians usually look when they do carotid ultrasound" examinations, Dr. Olin said.
When he finds carotid fibrosis, Dr. Olin starts those patients on a daily aspirin, although the benefit of this strategy hasn’t been proven. If carotid lesions are substantial, treatment by angioplasty is possible; carotid stenting is not used on FMD patients, nor is endarterectomy, as there is no atherosclerotic plaque to remove, he said.
Dr. Olin said that he has been a consultant to Merck, and that he chairs the advisory board of the Fibromuscular Dysplasia Society of America. Dr. Lookstein said that he has been a consultant to Medrad and Cordis.
MIAMI BEACH – The mystery shrouding fibromuscular dysplasia, a clinically important and surprisingly prevalent vascular disease of unknown etiology, began to lift with initial findings from 339 patients enrolled in the first U.S. registry for the disease.
Baseline observations from the U.S. Registry for Fibromuscular Dysplasia (FMD), which enrolled its first patient in 2008 and currently has women as 91% of enrolled patients, show that the disease first occurs across a broad swath of age groups, with an age at first diagnosis ranging from 5 to 83 years old. In addition to the distinctive "string of beads" arterial fibrosis appearance that defines the disease and is apparent on imaging, and which usually occurs in the renal or carotid artery, or both, 19% of patients also had a dissection in an artery somewhere in their body (14% with a dissection in a carotid artery) and 17% had an arterial aneurysm somewhere (5% with a renal artery aneurysm and 4% with a carotid aneurysm).
"A patient can have normal blood pressure and normal-appearing carotid and renal arteries but have terrible headaches and FMD. Why? We don’t know."
These dissection and aneurysm prevalence rates had not previously been appreciated to run so high in FMD patients. "It suggests a diffuse arteriopathy that can present in several different ways," Dr. Jeffrey W. Olin said at ISET 2012, an international symposium on endovascular therapy.
Another notable finding was that, besides hypertension, which was the most common presenting manifestation of FMD (seen in 66% of the registry patients at initial diagnosis), other common presenting symptoms included significant, often migraine-like headache in 53%, pulsatile tinnitus (a whooshing sound patients hear) in 30%, and dizziness in 28%. The high prevalence of headache in FMD had been "first reported 30 years ago, but sort of got lost," Dr. Olin said in an interview.
"A patient can have normal blood pressure and normal-appearing carotid and renal arteries but have terrible headaches and FMD. Why? We don’t know. We have no idea what causes the headaches," said Dr. Olin, a cardiologist who is professor of medicine and director of vascular medicine at Mount Sinai Medical Center in New York. Four percent of the registry patients had no symptoms on presentation; physicians found their FMD incidentally during imaging examinations for other reasons.
Other flags to trigger suspicion of FMD include hypertension that begins before age 35 (although it can also start later in life), treatment-resistant hypertension, epigastric bruit and hypertension, renal infarction, cervical bruit in a patient less than 60 years old, transient ischemic attack or stroke in a patient less than 60 years old, or an aortic aneurysm in a patient less than 60 years old.
Perhaps the most surprising new finding so far from the registry is evidence for a strong family history of cardiovascular disease, with 81% of first- or second-degree relatives of FMD patients having hypertension, 59% with hyperlipidemia, 53% with a history of a stroke, 22% with an aneurysm, and 21% with a history of sudden death. The prevalence of a first- or second-degree relative also having a diagnosis of FMD was 7%, not much higher than the currently estimated 4% prevalence of FMD in the general population.
Dr. Olin and his associates from the registry hypothesize that patients who develop FMD have an as-yet unidentified genetic predisposition that interacts with an environmental trigger. His hope is that, by continuing to expand the registry and by receiving substantially more research support than FMD now gets, a more concerted research effort can address the genetic questions raised by the family-history findings.
Current treatment of FMD is symptom driven and usually focuses on trying to resolve patients’ hypertension, which is often treatment resistant.
"FMD is potentially treatable for hypertension," with endovascular treatment of affected renal arteries the standard intervention for patients with resistant hypertension, Dr. Robert A. Lookstein said in a separate talk at the meeting. Hypertension cure rates from balloon angioplasty, however, are "surprisingly low" – 45% in one meta-analysis – probably because of suboptimal interventional treatment, said Dr. Lookstein, chief of the division of interventional radiology at Mount Sinai.
Assessing a patient’s renal arteries before and during treatment using intravascular ultrasound (IVUS) and a pressure wire provides the key to successful resolution of hypertension by renal-artery angioplasty in FMD patients, he said.
These two techniques, especially pressure-wire measurements, allow the operator to assess the patient’s renal arteries before and during treatment to determine whether the intervention is having a meaningful effect. "You can’t tell [whether the angioplasty produced benefit] by the angiography appearance alone," he cautioned. "You need to be compulsive with IVUS and measuring pressure gradients to determine where to start treatment and when to stop."
The U. S. Registry for FMD began with seven U.S. centers enrolling patients and now involves 10 centers and has enrolled more than 500 patients. The first 339 enrollees included 44% who were patients at the Cleveland Clinic and 20% who were patients at Mount Sinai.
The average age of the enrolled patients at first symptom appearance was 47 years old; first diagnosis did not occur until an average of more than 4 years later, at an average age of 52 years old.
Vascular bed involvement among the first registry patients included one or both renal arteries in 69%, at least one extracranial carotid in 62%, vertebral arteries in 19%, and mesenteric arteries in 12%. At the time of enrollment, 7% of patients had a history of coronary artery disease and 10% had a history of stroke. The arterial fibrosis characteristic of FMD notably appears in portions of the renal and carotid arteries where atherosclerotic disease usually does not occur – in the distal renal artery and in the distal carotid, "higher than physicians usually look when they do carotid ultrasound" examinations, Dr. Olin said.
When he finds carotid fibrosis, Dr. Olin starts those patients on a daily aspirin, although the benefit of this strategy hasn’t been proven. If carotid lesions are substantial, treatment by angioplasty is possible; carotid stenting is not used on FMD patients, nor is endarterectomy, as there is no atherosclerotic plaque to remove, he said.
Dr. Olin said that he has been a consultant to Merck, and that he chairs the advisory board of the Fibromuscular Dysplasia Society of America. Dr. Lookstein said that he has been a consultant to Medrad and Cordis.
MIAMI BEACH – The mystery shrouding fibromuscular dysplasia, a clinically important and surprisingly prevalent vascular disease of unknown etiology, began to lift with initial findings from 339 patients enrolled in the first U.S. registry for the disease.
Baseline observations from the U.S. Registry for Fibromuscular Dysplasia (FMD), which enrolled its first patient in 2008 and currently has women as 91% of enrolled patients, show that the disease first occurs across a broad swath of age groups, with an age at first diagnosis ranging from 5 to 83 years old. In addition to the distinctive "string of beads" arterial fibrosis appearance that defines the disease and is apparent on imaging, and which usually occurs in the renal or carotid artery, or both, 19% of patients also had a dissection in an artery somewhere in their body (14% with a dissection in a carotid artery) and 17% had an arterial aneurysm somewhere (5% with a renal artery aneurysm and 4% with a carotid aneurysm).
"A patient can have normal blood pressure and normal-appearing carotid and renal arteries but have terrible headaches and FMD. Why? We don’t know."
These dissection and aneurysm prevalence rates had not previously been appreciated to run so high in FMD patients. "It suggests a diffuse arteriopathy that can present in several different ways," Dr. Jeffrey W. Olin said at ISET 2012, an international symposium on endovascular therapy.
Another notable finding was that, besides hypertension, which was the most common presenting manifestation of FMD (seen in 66% of the registry patients at initial diagnosis), other common presenting symptoms included significant, often migraine-like headache in 53%, pulsatile tinnitus (a whooshing sound patients hear) in 30%, and dizziness in 28%. The high prevalence of headache in FMD had been "first reported 30 years ago, but sort of got lost," Dr. Olin said in an interview.
"A patient can have normal blood pressure and normal-appearing carotid and renal arteries but have terrible headaches and FMD. Why? We don’t know. We have no idea what causes the headaches," said Dr. Olin, a cardiologist who is professor of medicine and director of vascular medicine at Mount Sinai Medical Center in New York. Four percent of the registry patients had no symptoms on presentation; physicians found their FMD incidentally during imaging examinations for other reasons.
Other flags to trigger suspicion of FMD include hypertension that begins before age 35 (although it can also start later in life), treatment-resistant hypertension, epigastric bruit and hypertension, renal infarction, cervical bruit in a patient less than 60 years old, transient ischemic attack or stroke in a patient less than 60 years old, or an aortic aneurysm in a patient less than 60 years old.
Perhaps the most surprising new finding so far from the registry is evidence for a strong family history of cardiovascular disease, with 81% of first- or second-degree relatives of FMD patients having hypertension, 59% with hyperlipidemia, 53% with a history of a stroke, 22% with an aneurysm, and 21% with a history of sudden death. The prevalence of a first- or second-degree relative also having a diagnosis of FMD was 7%, not much higher than the currently estimated 4% prevalence of FMD in the general population.
Dr. Olin and his associates from the registry hypothesize that patients who develop FMD have an as-yet unidentified genetic predisposition that interacts with an environmental trigger. His hope is that, by continuing to expand the registry and by receiving substantially more research support than FMD now gets, a more concerted research effort can address the genetic questions raised by the family-history findings.
Current treatment of FMD is symptom driven and usually focuses on trying to resolve patients’ hypertension, which is often treatment resistant.
"FMD is potentially treatable for hypertension," with endovascular treatment of affected renal arteries the standard intervention for patients with resistant hypertension, Dr. Robert A. Lookstein said in a separate talk at the meeting. Hypertension cure rates from balloon angioplasty, however, are "surprisingly low" – 45% in one meta-analysis – probably because of suboptimal interventional treatment, said Dr. Lookstein, chief of the division of interventional radiology at Mount Sinai.
Assessing a patient’s renal arteries before and during treatment using intravascular ultrasound (IVUS) and a pressure wire provides the key to successful resolution of hypertension by renal-artery angioplasty in FMD patients, he said.
These two techniques, especially pressure-wire measurements, allow the operator to assess the patient’s renal arteries before and during treatment to determine whether the intervention is having a meaningful effect. "You can’t tell [whether the angioplasty produced benefit] by the angiography appearance alone," he cautioned. "You need to be compulsive with IVUS and measuring pressure gradients to determine where to start treatment and when to stop."
The U. S. Registry for FMD began with seven U.S. centers enrolling patients and now involves 10 centers and has enrolled more than 500 patients. The first 339 enrollees included 44% who were patients at the Cleveland Clinic and 20% who were patients at Mount Sinai.
The average age of the enrolled patients at first symptom appearance was 47 years old; first diagnosis did not occur until an average of more than 4 years later, at an average age of 52 years old.
Vascular bed involvement among the first registry patients included one or both renal arteries in 69%, at least one extracranial carotid in 62%, vertebral arteries in 19%, and mesenteric arteries in 12%. At the time of enrollment, 7% of patients had a history of coronary artery disease and 10% had a history of stroke. The arterial fibrosis characteristic of FMD notably appears in portions of the renal and carotid arteries where atherosclerotic disease usually does not occur – in the distal renal artery and in the distal carotid, "higher than physicians usually look when they do carotid ultrasound" examinations, Dr. Olin said.
When he finds carotid fibrosis, Dr. Olin starts those patients on a daily aspirin, although the benefit of this strategy hasn’t been proven. If carotid lesions are substantial, treatment by angioplasty is possible; carotid stenting is not used on FMD patients, nor is endarterectomy, as there is no atherosclerotic plaque to remove, he said.
Dr. Olin said that he has been a consultant to Merck, and that he chairs the advisory board of the Fibromuscular Dysplasia Society of America. Dr. Lookstein said that he has been a consultant to Medrad and Cordis.
FROM ISET 2012, AN INTERNATIONAL SYMPOSIUM ON ENDOVASCULAR THERAPY
Major Finding: The most common presenting symptoms in patients with fibromuscular dysplasia were hypertension in 66%, headache in 53%, pulsatile tinnitus in 30%, and dizziness in 28%.
Data Source: Baseline findings from the first 339 patients enrolled in the U.S. Registry for Fibromuscular Dysplasia.
Disclosures: Dr. Olin said that he has been a consultant to Merck, and that he chairs the advisory board of the Fibromuscular Dysplasia Society of America. Dr. Lookstein said that he has been a consultant to Medrad and Cordis.
Local, Regional Anesthesia Surpass General for AAA EVAR
MIAMI BEACH – Local or regional anesthesia is a better option than is general anesthesia for patients undergoing endovascular repair of an abdominal aortic aneurysm, based on findings from a registry with nearly 1,200 patients.
Both local anesthesia and regional anesthesia each surpassed general anesthesia in two periprocedural measures: significantly reducing procedure time, and significantly reducing postoperative hospitalization, Dr. Rutger A. Stokmans said at ISET 2012, an international symposium on endovascular therapy.

In addition, both local and regional anesthesia led to trends in reduced rates of major adverse events during the 30 days following surgery, although these differences did not reach statistical significance. All three anesthesia types linked with similar rates of both technical and clinical success of the aneurysm repairs. Regional anesthesia also led to a significantly lower rate of ICU admission, compared with both general and local anesthesia; local anesthesia showed no significant difference for this measure, compared with general anesthesia.
Based on these findings, local or regional anesthesia should be preferred when performing endovascular aneurysm repair (EVAR), whereas general anesthesia should usually be avoided, said Dr. Stokmans, a vascular surgeon at Catharina Hospital in Eindhoven, the Netherlands.
These findings support the most recent anesthesia recommendations of the European Society for Vascular Surgery, which in 2011 guidelines for managing abdominal aortic aneurysms (AAA) cited local anesthesia as preferred for EVAR, with regional or general anesthesia reserved for patients with contraindications for local anesthesia, he said (Euro. J. Vasc. Endovasc. Surg .2011;41[suppl. 1]:S1-S58). The most recent guidelines for AAA management from the Society for Vascular Surgery suggested using local or regional anesthesia over general anesthesia, he added (J. Vasc. Surg. 2009[suppl.]:50:S2-S49).
But despite these recommendations, the most commonly used anesthesia type worldwide for EVAR repair of AAA has been general anesthesia, followed by regional anesthesia, with local treatment used least often, according to the registry data reported by Dr. Stokmans. Among the 1,199 patients enrolled in ENGAGE (Endurant Stent Graft Natural Selection Global Postmarketing Registry) during March 2009 to December 2010 in 30 countries on five continents, 749 (62%) underwent their EVAR with general anesthesia, 325 (27%) with regional, and 125 (10%) with local anesthesia. (Percentages do not add up to 100% because of rounding.)
The registry data also showed striking regional variations in anesthesia use, with general anesthesia used on about 90% of patients in Canada, Australia, and New Zealand, and on about 70% of patients in Scandinavian countries and the United Kingdom. But in Central Europe, regional anesthesia – used on nearly 70% of EVAR patients – dominated. The only region favoring local anesthesia was South America (Argentina, Columbia, and Uruguay), where about 50% of patients received local, but more than 40% received general anesthesia, he said. The registry contained no U.S. patients, although the Endurant AAA stent graft system is marketed in the United States.
The average age of the EVAR patients in the registry was about 73 years. Those patients who underwent general anesthesia were significantly older, by an average of about 18 months, compared with those who received local or regional anesthesia.
The proportions of patients undergoing general, regional, or local anesthesia were similar in the subgroups of patients with American Society of Anesthesiologists (ASA) physical status scores of 1, 2, or 3. However, among the highest-risk patients included in the study – those with an ASA score of 4 – a significantly greater proportion of patients received general anesthesia. The multivariate models used in the analysis, therefore, were adjusted for age and for ASA score. About 42% of patients were in ASA class 2, and another 42% in class 3.
All patients were hemodynamically stable at the time of their enrollment. The maximum AAA diameter of registry patients was 6 cm, and about 88% of patients had an AAA diameter greater than 5 cm.
Average procedure times were 106 minutes in the general anesthesia patients, 95 minutes in the regional patients, and 81 minutes in the local anesthesia patients – statistically significant differences among the three groups.
The average postoperative hospitalization was 5.2 days in the general anesthesia patients, 4.3 days in the regional patients, and 3.6 days in the local anesthesia patients, differences that were statistically significant among each of the three groups.
The rate of technical surgical success was 98% in all three subgroups, and the rate of clinical success reached 97%-98% in all three groups.
The rates of major adverse events during the 30 days following surgery were 5.1% in the general anesthesia patients, 3.2% in the local patients, and 2.2% in the regional anesthesia patients. None of these differences reached statistical significance. Major adverse events included death, MI, stroke, renal failure, blood loss greater than 1 L, and bowel ischemia. No patients developed paraplegia or respiratory failure.
Postoperative ICU admission occurred for 27% of the regional anesthesia patients, 35% of the general patients, and 42% of the local anesthesia patients. The rate among the regional patients was significantly less than in the other two groups, but the difference in rates between the general and local anesthesia patients did not reach statistical significance.
The ENGAGE registry was organized and sponsored by Medtronic, which markets the Endurant stent. Dr. Stokmans and his associates received an unrestricted research grant from Medtronic. Dr. Stokmans said that he had no other disclosures.
MIAMI BEACH – Local or regional anesthesia is a better option than is general anesthesia for patients undergoing endovascular repair of an abdominal aortic aneurysm, based on findings from a registry with nearly 1,200 patients.
Both local anesthesia and regional anesthesia each surpassed general anesthesia in two periprocedural measures: significantly reducing procedure time, and significantly reducing postoperative hospitalization, Dr. Rutger A. Stokmans said at ISET 2012, an international symposium on endovascular therapy.
In addition, both local and regional anesthesia led to trends in reduced rates of major adverse events during the 30 days following surgery, although these differences did not reach statistical significance. All three anesthesia types linked with similar rates of both technical and clinical success of the aneurysm repairs. Regional anesthesia also led to a significantly lower rate of ICU admission, compared with both general and local anesthesia; local anesthesia showed no significant difference for this measure, compared with general anesthesia.
Based on these findings, local or regional anesthesia should be preferred when performing endovascular aneurysm repair (EVAR), whereas general anesthesia should usually be avoided, said Dr. Stokmans, a vascular surgeon at Catharina Hospital in Eindhoven, the Netherlands.
These findings support the most recent anesthesia recommendations of the European Society for Vascular Surgery, which in 2011 guidelines for managing abdominal aortic aneurysms (AAA) cited local anesthesia as preferred for EVAR, with regional or general anesthesia reserved for patients with contraindications for local anesthesia, he said (Euro. J. Vasc. Endovasc. Surg .2011;41[suppl. 1]:S1-S58). The most recent guidelines for AAA management from the Society for Vascular Surgery suggested using local or regional anesthesia over general anesthesia, he added (J. Vasc. Surg. 2009[suppl.]:50:S2-S49).
But despite these recommendations, the most commonly used anesthesia type worldwide for EVAR repair of AAA has been general anesthesia, followed by regional anesthesia, with local treatment used least often, according to the registry data reported by Dr. Stokmans. Among the 1,199 patients enrolled in ENGAGE (Endurant Stent Graft Natural Selection Global Postmarketing Registry) during March 2009 to December 2010 in 30 countries on five continents, 749 (62%) underwent their EVAR with general anesthesia, 325 (27%) with regional, and 125 (10%) with local anesthesia. (Percentages do not add up to 100% because of rounding.)
The registry data also showed striking regional variations in anesthesia use, with general anesthesia used on about 90% of patients in Canada, Australia, and New Zealand, and on about 70% of patients in Scandinavian countries and the United Kingdom. But in Central Europe, regional anesthesia – used on nearly 70% of EVAR patients – dominated. The only region favoring local anesthesia was South America (Argentina, Columbia, and Uruguay), where about 50% of patients received local, but more than 40% received general anesthesia, he said. The registry contained no U.S. patients, although the Endurant AAA stent graft system is marketed in the United States.
The average age of the EVAR patients in the registry was about 73 years. Those patients who underwent general anesthesia were significantly older, by an average of about 18 months, compared with those who received local or regional anesthesia.
The proportions of patients undergoing general, regional, or local anesthesia were similar in the subgroups of patients with American Society of Anesthesiologists (ASA) physical status scores of 1, 2, or 3. However, among the highest-risk patients included in the study – those with an ASA score of 4 – a significantly greater proportion of patients received general anesthesia. The multivariate models used in the analysis, therefore, were adjusted for age and for ASA score. About 42% of patients were in ASA class 2, and another 42% in class 3.
All patients were hemodynamically stable at the time of their enrollment. The maximum AAA diameter of registry patients was 6 cm, and about 88% of patients had an AAA diameter greater than 5 cm.
Average procedure times were 106 minutes in the general anesthesia patients, 95 minutes in the regional patients, and 81 minutes in the local anesthesia patients – statistically significant differences among the three groups.
The average postoperative hospitalization was 5.2 days in the general anesthesia patients, 4.3 days in the regional patients, and 3.6 days in the local anesthesia patients, differences that were statistically significant among each of the three groups.
The rate of technical surgical success was 98% in all three subgroups, and the rate of clinical success reached 97%-98% in all three groups.
The rates of major adverse events during the 30 days following surgery were 5.1% in the general anesthesia patients, 3.2% in the local patients, and 2.2% in the regional anesthesia patients. None of these differences reached statistical significance. Major adverse events included death, MI, stroke, renal failure, blood loss greater than 1 L, and bowel ischemia. No patients developed paraplegia or respiratory failure.
Postoperative ICU admission occurred for 27% of the regional anesthesia patients, 35% of the general patients, and 42% of the local anesthesia patients. The rate among the regional patients was significantly less than in the other two groups, but the difference in rates between the general and local anesthesia patients did not reach statistical significance.
The ENGAGE registry was organized and sponsored by Medtronic, which markets the Endurant stent. Dr. Stokmans and his associates received an unrestricted research grant from Medtronic. Dr. Stokmans said that he had no other disclosures.
MIAMI BEACH – Local or regional anesthesia is a better option than is general anesthesia for patients undergoing endovascular repair of an abdominal aortic aneurysm, based on findings from a registry with nearly 1,200 patients.
Both local anesthesia and regional anesthesia each surpassed general anesthesia in two periprocedural measures: significantly reducing procedure time, and significantly reducing postoperative hospitalization, Dr. Rutger A. Stokmans said at ISET 2012, an international symposium on endovascular therapy.
In addition, both local and regional anesthesia led to trends in reduced rates of major adverse events during the 30 days following surgery, although these differences did not reach statistical significance. All three anesthesia types linked with similar rates of both technical and clinical success of the aneurysm repairs. Regional anesthesia also led to a significantly lower rate of ICU admission, compared with both general and local anesthesia; local anesthesia showed no significant difference for this measure, compared with general anesthesia.
Based on these findings, local or regional anesthesia should be preferred when performing endovascular aneurysm repair (EVAR), whereas general anesthesia should usually be avoided, said Dr. Stokmans, a vascular surgeon at Catharina Hospital in Eindhoven, the Netherlands.
These findings support the most recent anesthesia recommendations of the European Society for Vascular Surgery, which in 2011 guidelines for managing abdominal aortic aneurysms (AAA) cited local anesthesia as preferred for EVAR, with regional or general anesthesia reserved for patients with contraindications for local anesthesia, he said (Euro. J. Vasc. Endovasc. Surg .2011;41[suppl. 1]:S1-S58). The most recent guidelines for AAA management from the Society for Vascular Surgery suggested using local or regional anesthesia over general anesthesia, he added (J. Vasc. Surg. 2009[suppl.]:50:S2-S49).
But despite these recommendations, the most commonly used anesthesia type worldwide for EVAR repair of AAA has been general anesthesia, followed by regional anesthesia, with local treatment used least often, according to the registry data reported by Dr. Stokmans. Among the 1,199 patients enrolled in ENGAGE (Endurant Stent Graft Natural Selection Global Postmarketing Registry) during March 2009 to December 2010 in 30 countries on five continents, 749 (62%) underwent their EVAR with general anesthesia, 325 (27%) with regional, and 125 (10%) with local anesthesia. (Percentages do not add up to 100% because of rounding.)
The registry data also showed striking regional variations in anesthesia use, with general anesthesia used on about 90% of patients in Canada, Australia, and New Zealand, and on about 70% of patients in Scandinavian countries and the United Kingdom. But in Central Europe, regional anesthesia – used on nearly 70% of EVAR patients – dominated. The only region favoring local anesthesia was South America (Argentina, Columbia, and Uruguay), where about 50% of patients received local, but more than 40% received general anesthesia, he said. The registry contained no U.S. patients, although the Endurant AAA stent graft system is marketed in the United States.
The average age of the EVAR patients in the registry was about 73 years. Those patients who underwent general anesthesia were significantly older, by an average of about 18 months, compared with those who received local or regional anesthesia.
The proportions of patients undergoing general, regional, or local anesthesia were similar in the subgroups of patients with American Society of Anesthesiologists (ASA) physical status scores of 1, 2, or 3. However, among the highest-risk patients included in the study – those with an ASA score of 4 – a significantly greater proportion of patients received general anesthesia. The multivariate models used in the analysis, therefore, were adjusted for age and for ASA score. About 42% of patients were in ASA class 2, and another 42% in class 3.
All patients were hemodynamically stable at the time of their enrollment. The maximum AAA diameter of registry patients was 6 cm, and about 88% of patients had an AAA diameter greater than 5 cm.
Average procedure times were 106 minutes in the general anesthesia patients, 95 minutes in the regional patients, and 81 minutes in the local anesthesia patients – statistically significant differences among the three groups.
The average postoperative hospitalization was 5.2 days in the general anesthesia patients, 4.3 days in the regional patients, and 3.6 days in the local anesthesia patients, differences that were statistically significant among each of the three groups.
The rate of technical surgical success was 98% in all three subgroups, and the rate of clinical success reached 97%-98% in all three groups.
The rates of major adverse events during the 30 days following surgery were 5.1% in the general anesthesia patients, 3.2% in the local patients, and 2.2% in the regional anesthesia patients. None of these differences reached statistical significance. Major adverse events included death, MI, stroke, renal failure, blood loss greater than 1 L, and bowel ischemia. No patients developed paraplegia or respiratory failure.
Postoperative ICU admission occurred for 27% of the regional anesthesia patients, 35% of the general patients, and 42% of the local anesthesia patients. The rate among the regional patients was significantly less than in the other two groups, but the difference in rates between the general and local anesthesia patients did not reach statistical significance.
The ENGAGE registry was organized and sponsored by Medtronic, which markets the Endurant stent. Dr. Stokmans and his associates received an unrestricted research grant from Medtronic. Dr. Stokmans said that he had no other disclosures.
FROM ISET 2012, AN INTERNATIONAL SYMPOSIUM ON ENDOVASCULAR THERAPY
Major Finding: Postoperative hospitalization averaged 5.2 days in EVAR patients treated with general anesthesia, 4.3 days with regional anesthesia, and 3.6 days with local anesthesia.
Data Source: ENGAGE, an international registry of 1,199 patients who underwent EVAR for an abdominal aortic aneurysm during 2009 or 2010.
Disclosures: The ENGAGE registry was organized and sponsored by Medtronic, which markets the Endurant EVAR stent. Dr. Stokmans and his associates received an unrestricted research grant from Medtronic. Dr. Stokmans said that he had no other disclosures.
Noninvasive Scan Has Promise For Lung Cancer Genotyping
AMSTERDAM – An experimental combination of PET scanning and a positron-emitting form of erlotinib appeared to work as a noninvasive way of identifying patients with advanced non–small cell lung cancer tumors that have the right genotype to receive erlotinib therapy.
"[11C]erlotinib PET shows promise as a noninvasive, in vivo means of selecting patients who may benefit from thymidine kinase inhibitor therapy," Dr. Idris Bahce said, reporting on a pilot study of 10 patients. Erlotinib (Tarceva) is from the thymidine kinase inhibitor drug class.
In his study, uptake of 11C-labeled erlotinib was significantly linked to the patients’ having an activating mutation in their epidermal growth factor receptor (EGFR) gene, specifically an exon 19 deletion.
Patients positive for erlotinib uptake on the PET scan also showed a tendency for better clinical responses to a therapeutic erlotinib regimen, reported Dr. Bahce, a pulmonologist at VU University, Amsterdam, during the World Conference on Lung Cancer.
Until now, the only way to identify advanced non–small cell lung cancer (NSCLC) tumors that are candidates for treatment with a tyrosine kinase inhibitor has been to biopsy the tumor and run an in vitro genetic analysis on the tumor cells. That can be challenging in some patients, such as when the tumor is not easy to biopsy, a limited amount of tissue is available, or the tumor is genetically heterogeneous. To get a reliable result from biopsy and testing, at least 30% of the specimen must contain malignant cells, Dr. Bahce said at the conference, sponsored by the International Association for the Study of Lung Cancer.
"It is a very early study, but ... it’s important because personalized treatment [for cancer] has gone to the next level, where we use new agents and match them to the right patients by doing biopsies," commented Dr. Roy S. Herbst, chief of medical oncology at the Yale Cancer Center in New Haven. "The PET method also allows physicians to assess the volume of cancer carrying the EGFR mutation following treatment, a way to track treatment efficacy," said Dr. Herbst in an interview.
"Instead of getting tissue at one point in time, you can image more frequently. It’s a way to track the course of treatment noninvasively," and in real time, he said.
He also predicted that the [11C]erlotinib PET test will become commercialized, although currently Dr. Bahce’s studies do not have any commercial funding.
"This is a proof of concept study," commented Dr. Luis Paz-Ares, chief of medical oncology at University Hospital Virgin del Rocio in Seville, Spain. "We need to define the positive predictive value and the negative predictive value" of the test, he added. The long-term future of a test like this may also be limited because future testing will probably need to look at multiple biomarkers, Dr. Paz-Ares said.
The study enrolled five patients with advanced NSCLC who had exon 19 deletion EGFR mutations, and five advanced NSCLC patients with wild-type EGFR genes. Each patient underwent a pair of [11C]erlotinib PET scans, each preceded by a [15O]water PET scan to assess blood perfusion of the tumors. A 4-hour interval separated the two sets of scans.
The scan results showed that the volume of distribution of the tagged erlotinib in the patients with EGFR mutations ran about 50% higher than in the patients with wild-type tumors, a difference that was significant (P = .03). Clinically, two of the five wild-type patients had nonetheless received erlotinib treatment prior to testing, and neither patient responded, with both showing progressive disease.
Three of the five patients with an EGFR mutation began receiving erlotinib treatment after testing and responded. In one of these patients, the tumor remained in check for 13 months. In a second patient, the tumor began to progress after 17 months of no progression on treatment. In the third patient, the tumor began to progress again after about 4 weeks of no progression on erlotinib treatment, Dr. Bahce said. A fourth patient went on erlotinib treatment before testing, and did not respond and continued to have progressive disease.
The two patient subgroups showed no difference in blood perfusion into the tumors or in EGFR expression in cell membranes.
Dr. Bahce said he had no disclosures.☐
AMSTERDAM – An experimental combination of PET scanning and a positron-emitting form of erlotinib appeared to work as a noninvasive way of identifying patients with advanced non–small cell lung cancer tumors that have the right genotype to receive erlotinib therapy.
"[11C]erlotinib PET shows promise as a noninvasive, in vivo means of selecting patients who may benefit from thymidine kinase inhibitor therapy," Dr. Idris Bahce said, reporting on a pilot study of 10 patients. Erlotinib (Tarceva) is from the thymidine kinase inhibitor drug class.
In his study, uptake of 11C-labeled erlotinib was significantly linked to the patients’ having an activating mutation in their epidermal growth factor receptor (EGFR) gene, specifically an exon 19 deletion.
Patients positive for erlotinib uptake on the PET scan also showed a tendency for better clinical responses to a therapeutic erlotinib regimen, reported Dr. Bahce, a pulmonologist at VU University, Amsterdam, during the World Conference on Lung Cancer.
Until now, the only way to identify advanced non–small cell lung cancer (NSCLC) tumors that are candidates for treatment with a tyrosine kinase inhibitor has been to biopsy the tumor and run an in vitro genetic analysis on the tumor cells. That can be challenging in some patients, such as when the tumor is not easy to biopsy, a limited amount of tissue is available, or the tumor is genetically heterogeneous. To get a reliable result from biopsy and testing, at least 30% of the specimen must contain malignant cells, Dr. Bahce said at the conference, sponsored by the International Association for the Study of Lung Cancer.
"It is a very early study, but ... it’s important because personalized treatment [for cancer] has gone to the next level, where we use new agents and match them to the right patients by doing biopsies," commented Dr. Roy S. Herbst, chief of medical oncology at the Yale Cancer Center in New Haven. "The PET method also allows physicians to assess the volume of cancer carrying the EGFR mutation following treatment, a way to track treatment efficacy," said Dr. Herbst in an interview.
"Instead of getting tissue at one point in time, you can image more frequently. It’s a way to track the course of treatment noninvasively," and in real time, he said.
He also predicted that the [11C]erlotinib PET test will become commercialized, although currently Dr. Bahce’s studies do not have any commercial funding.
"This is a proof of concept study," commented Dr. Luis Paz-Ares, chief of medical oncology at University Hospital Virgin del Rocio in Seville, Spain. "We need to define the positive predictive value and the negative predictive value" of the test, he added. The long-term future of a test like this may also be limited because future testing will probably need to look at multiple biomarkers, Dr. Paz-Ares said.
The study enrolled five patients with advanced NSCLC who had exon 19 deletion EGFR mutations, and five advanced NSCLC patients with wild-type EGFR genes. Each patient underwent a pair of [11C]erlotinib PET scans, each preceded by a [15O]water PET scan to assess blood perfusion of the tumors. A 4-hour interval separated the two sets of scans.
The scan results showed that the volume of distribution of the tagged erlotinib in the patients with EGFR mutations ran about 50% higher than in the patients with wild-type tumors, a difference that was significant (P = .03). Clinically, two of the five wild-type patients had nonetheless received erlotinib treatment prior to testing, and neither patient responded, with both showing progressive disease.
Three of the five patients with an EGFR mutation began receiving erlotinib treatment after testing and responded. In one of these patients, the tumor remained in check for 13 months. In a second patient, the tumor began to progress after 17 months of no progression on treatment. In the third patient, the tumor began to progress again after about 4 weeks of no progression on erlotinib treatment, Dr. Bahce said. A fourth patient went on erlotinib treatment before testing, and did not respond and continued to have progressive disease.
The two patient subgroups showed no difference in blood perfusion into the tumors or in EGFR expression in cell membranes.
Dr. Bahce said he had no disclosures.☐
AMSTERDAM – An experimental combination of PET scanning and a positron-emitting form of erlotinib appeared to work as a noninvasive way of identifying patients with advanced non–small cell lung cancer tumors that have the right genotype to receive erlotinib therapy.
"[11C]erlotinib PET shows promise as a noninvasive, in vivo means of selecting patients who may benefit from thymidine kinase inhibitor therapy," Dr. Idris Bahce said, reporting on a pilot study of 10 patients. Erlotinib (Tarceva) is from the thymidine kinase inhibitor drug class.
In his study, uptake of 11C-labeled erlotinib was significantly linked to the patients’ having an activating mutation in their epidermal growth factor receptor (EGFR) gene, specifically an exon 19 deletion.
Patients positive for erlotinib uptake on the PET scan also showed a tendency for better clinical responses to a therapeutic erlotinib regimen, reported Dr. Bahce, a pulmonologist at VU University, Amsterdam, during the World Conference on Lung Cancer.
Until now, the only way to identify advanced non–small cell lung cancer (NSCLC) tumors that are candidates for treatment with a tyrosine kinase inhibitor has been to biopsy the tumor and run an in vitro genetic analysis on the tumor cells. That can be challenging in some patients, such as when the tumor is not easy to biopsy, a limited amount of tissue is available, or the tumor is genetically heterogeneous. To get a reliable result from biopsy and testing, at least 30% of the specimen must contain malignant cells, Dr. Bahce said at the conference, sponsored by the International Association for the Study of Lung Cancer.
"It is a very early study, but ... it’s important because personalized treatment [for cancer] has gone to the next level, where we use new agents and match them to the right patients by doing biopsies," commented Dr. Roy S. Herbst, chief of medical oncology at the Yale Cancer Center in New Haven. "The PET method also allows physicians to assess the volume of cancer carrying the EGFR mutation following treatment, a way to track treatment efficacy," said Dr. Herbst in an interview.
"Instead of getting tissue at one point in time, you can image more frequently. It’s a way to track the course of treatment noninvasively," and in real time, he said.
He also predicted that the [11C]erlotinib PET test will become commercialized, although currently Dr. Bahce’s studies do not have any commercial funding.
"This is a proof of concept study," commented Dr. Luis Paz-Ares, chief of medical oncology at University Hospital Virgin del Rocio in Seville, Spain. "We need to define the positive predictive value and the negative predictive value" of the test, he added. The long-term future of a test like this may also be limited because future testing will probably need to look at multiple biomarkers, Dr. Paz-Ares said.
The study enrolled five patients with advanced NSCLC who had exon 19 deletion EGFR mutations, and five advanced NSCLC patients with wild-type EGFR genes. Each patient underwent a pair of [11C]erlotinib PET scans, each preceded by a [15O]water PET scan to assess blood perfusion of the tumors. A 4-hour interval separated the two sets of scans.
The scan results showed that the volume of distribution of the tagged erlotinib in the patients with EGFR mutations ran about 50% higher than in the patients with wild-type tumors, a difference that was significant (P = .03). Clinically, two of the five wild-type patients had nonetheless received erlotinib treatment prior to testing, and neither patient responded, with both showing progressive disease.
Three of the five patients with an EGFR mutation began receiving erlotinib treatment after testing and responded. In one of these patients, the tumor remained in check for 13 months. In a second patient, the tumor began to progress after 17 months of no progression on treatment. In the third patient, the tumor began to progress again after about 4 weeks of no progression on erlotinib treatment, Dr. Bahce said. A fourth patient went on erlotinib treatment before testing, and did not respond and continued to have progressive disease.
The two patient subgroups showed no difference in blood perfusion into the tumors or in EGFR expression in cell membranes.
Dr. Bahce said he had no disclosures.☐
Major Finding: Advanced non–small cell lung cancer tumors with an epidermal growth factor receptor (EGFR)–activating mutation bound significantly more radiolabeled erlotinib than did tumors with wild-type EGFR genes (P = .03).
Data Source: A pilot study in 10 patients.
Disclosures: Dr. Bahce said he had no disclosures.
Noninvasive Scan Has Promise For Lung Cancer Genotyping
AMSTERDAM – An experimental combination of PET scanning and a positron-emitting form of erlotinib appeared to work as a noninvasive way of identifying patients with advanced non–small cell lung cancer tumors that have the right genotype to receive erlotinib therapy.
"[11C]erlotinib PET shows promise as a noninvasive, in vivo means of selecting patients who may benefit from thymidine kinase inhibitor therapy," Dr. Idris Bahce said, reporting on a pilot study of 10 patients. Erlotinib (Tarceva) is from the thymidine kinase inhibitor drug class.
In his study, uptake of 11C-labeled erlotinib was significantly linked to the patients’ having an activating mutation in their epidermal growth factor receptor (EGFR) gene, specifically an exon 19 deletion.
Patients positive for erlotinib uptake on the PET scan also showed a tendency for better clinical responses to a therapeutic erlotinib regimen, reported Dr. Bahce, a pulmonologist at VU University, Amsterdam, during the World Conference on Lung Cancer.
Until now, the only way to identify advanced non–small cell lung cancer (NSCLC) tumors that are candidates for treatment with a tyrosine kinase inhibitor has been to biopsy the tumor and run an in vitro genetic analysis on the tumor cells. That can be challenging in some patients, such as when the tumor is not easy to biopsy, a limited amount of tissue is available, or the tumor is genetically heterogeneous. To get a reliable result from biopsy and testing, at least 30% of the specimen must contain malignant cells, Dr. Bahce said at the conference, sponsored by the International Association for the Study of Lung Cancer.
"It is a very early study, but ... it’s important because personalized treatment [for cancer] has gone to the next level, where we use new agents and match them to the right patients by doing biopsies," commented Dr. Roy S. Herbst, chief of medical oncology at the Yale Cancer Center in New Haven. "The PET method also allows physicians to assess the volume of cancer carrying the EGFR mutation following treatment, a way to track treatment efficacy," said Dr. Herbst in an interview.
"Instead of getting tissue at one point in time, you can image more frequently. It’s a way to track the course of treatment noninvasively," and in real time, he said.
He also predicted that the [11C]erlotinib PET test will become commercialized, although currently Dr. Bahce’s studies do not have any commercial funding.
"This is a proof of concept study," commented Dr. Luis Paz-Ares, chief of medical oncology at University Hospital Virgin del Rocio in Seville, Spain. "We need to define the positive predictive value and the negative predictive value" of the test, he added. The long-term future of a test like this may also be limited because future testing will probably need to look at multiple biomarkers, Dr. Paz-Ares said.
The study enrolled five patients with advanced NSCLC who had exon 19 deletion EGFR mutations, and five advanced NSCLC patients with wild-type EGFR genes. Each patient underwent a pair of [11C]erlotinib PET scans, each preceded by a [15O]water PET scan to assess blood perfusion of the tumors. A 4-hour interval separated the two sets of scans.
The scan results showed that the volume of distribution of the tagged erlotinib in the patients with EGFR mutations ran about 50% higher than in the patients with wild-type tumors, a difference that was significant (P = .03). Clinically, two of the five wild-type patients had nonetheless received erlotinib treatment prior to testing, and neither patient responded, with both showing progressive disease.
Three of the five patients with an EGFR mutation began receiving erlotinib treatment after testing and responded. In one of these patients, the tumor remained in check for 13 months. In a second patient, the tumor began to progress after 17 months of no progression on treatment. In the third patient, the tumor began to progress again after about 4 weeks of no progression on erlotinib treatment, Dr. Bahce said. A fourth patient went on erlotinib treatment before testing, and did not respond and continued to have progressive disease.
The two patient subgroups showed no difference in blood perfusion into the tumors or in EGFR expression in cell membranes.
Dr. Bahce said he had no disclosures.☐
AMSTERDAM – An experimental combination of PET scanning and a positron-emitting form of erlotinib appeared to work as a noninvasive way of identifying patients with advanced non–small cell lung cancer tumors that have the right genotype to receive erlotinib therapy.
"[11C]erlotinib PET shows promise as a noninvasive, in vivo means of selecting patients who may benefit from thymidine kinase inhibitor therapy," Dr. Idris Bahce said, reporting on a pilot study of 10 patients. Erlotinib (Tarceva) is from the thymidine kinase inhibitor drug class.
In his study, uptake of 11C-labeled erlotinib was significantly linked to the patients’ having an activating mutation in their epidermal growth factor receptor (EGFR) gene, specifically an exon 19 deletion.
Patients positive for erlotinib uptake on the PET scan also showed a tendency for better clinical responses to a therapeutic erlotinib regimen, reported Dr. Bahce, a pulmonologist at VU University, Amsterdam, during the World Conference on Lung Cancer.
Until now, the only way to identify advanced non–small cell lung cancer (NSCLC) tumors that are candidates for treatment with a tyrosine kinase inhibitor has been to biopsy the tumor and run an in vitro genetic analysis on the tumor cells. That can be challenging in some patients, such as when the tumor is not easy to biopsy, a limited amount of tissue is available, or the tumor is genetically heterogeneous. To get a reliable result from biopsy and testing, at least 30% of the specimen must contain malignant cells, Dr. Bahce said at the conference, sponsored by the International Association for the Study of Lung Cancer.
"It is a very early study, but ... it’s important because personalized treatment [for cancer] has gone to the next level, where we use new agents and match them to the right patients by doing biopsies," commented Dr. Roy S. Herbst, chief of medical oncology at the Yale Cancer Center in New Haven. "The PET method also allows physicians to assess the volume of cancer carrying the EGFR mutation following treatment, a way to track treatment efficacy," said Dr. Herbst in an interview.
"Instead of getting tissue at one point in time, you can image more frequently. It’s a way to track the course of treatment noninvasively," and in real time, he said.
He also predicted that the [11C]erlotinib PET test will become commercialized, although currently Dr. Bahce’s studies do not have any commercial funding.
"This is a proof of concept study," commented Dr. Luis Paz-Ares, chief of medical oncology at University Hospital Virgin del Rocio in Seville, Spain. "We need to define the positive predictive value and the negative predictive value" of the test, he added. The long-term future of a test like this may also be limited because future testing will probably need to look at multiple biomarkers, Dr. Paz-Ares said.
The study enrolled five patients with advanced NSCLC who had exon 19 deletion EGFR mutations, and five advanced NSCLC patients with wild-type EGFR genes. Each patient underwent a pair of [11C]erlotinib PET scans, each preceded by a [15O]water PET scan to assess blood perfusion of the tumors. A 4-hour interval separated the two sets of scans.
The scan results showed that the volume of distribution of the tagged erlotinib in the patients with EGFR mutations ran about 50% higher than in the patients with wild-type tumors, a difference that was significant (P = .03). Clinically, two of the five wild-type patients had nonetheless received erlotinib treatment prior to testing, and neither patient responded, with both showing progressive disease.
Three of the five patients with an EGFR mutation began receiving erlotinib treatment after testing and responded. In one of these patients, the tumor remained in check for 13 months. In a second patient, the tumor began to progress after 17 months of no progression on treatment. In the third patient, the tumor began to progress again after about 4 weeks of no progression on erlotinib treatment, Dr. Bahce said. A fourth patient went on erlotinib treatment before testing, and did not respond and continued to have progressive disease.
The two patient subgroups showed no difference in blood perfusion into the tumors or in EGFR expression in cell membranes.
Dr. Bahce said he had no disclosures.☐
AMSTERDAM – An experimental combination of PET scanning and a positron-emitting form of erlotinib appeared to work as a noninvasive way of identifying patients with advanced non–small cell lung cancer tumors that have the right genotype to receive erlotinib therapy.
"[11C]erlotinib PET shows promise as a noninvasive, in vivo means of selecting patients who may benefit from thymidine kinase inhibitor therapy," Dr. Idris Bahce said, reporting on a pilot study of 10 patients. Erlotinib (Tarceva) is from the thymidine kinase inhibitor drug class.
In his study, uptake of 11C-labeled erlotinib was significantly linked to the patients’ having an activating mutation in their epidermal growth factor receptor (EGFR) gene, specifically an exon 19 deletion.
Patients positive for erlotinib uptake on the PET scan also showed a tendency for better clinical responses to a therapeutic erlotinib regimen, reported Dr. Bahce, a pulmonologist at VU University, Amsterdam, during the World Conference on Lung Cancer.
Until now, the only way to identify advanced non–small cell lung cancer (NSCLC) tumors that are candidates for treatment with a tyrosine kinase inhibitor has been to biopsy the tumor and run an in vitro genetic analysis on the tumor cells. That can be challenging in some patients, such as when the tumor is not easy to biopsy, a limited amount of tissue is available, or the tumor is genetically heterogeneous. To get a reliable result from biopsy and testing, at least 30% of the specimen must contain malignant cells, Dr. Bahce said at the conference, sponsored by the International Association for the Study of Lung Cancer.
"It is a very early study, but ... it’s important because personalized treatment [for cancer] has gone to the next level, where we use new agents and match them to the right patients by doing biopsies," commented Dr. Roy S. Herbst, chief of medical oncology at the Yale Cancer Center in New Haven. "The PET method also allows physicians to assess the volume of cancer carrying the EGFR mutation following treatment, a way to track treatment efficacy," said Dr. Herbst in an interview.
"Instead of getting tissue at one point in time, you can image more frequently. It’s a way to track the course of treatment noninvasively," and in real time, he said.
He also predicted that the [11C]erlotinib PET test will become commercialized, although currently Dr. Bahce’s studies do not have any commercial funding.
"This is a proof of concept study," commented Dr. Luis Paz-Ares, chief of medical oncology at University Hospital Virgin del Rocio in Seville, Spain. "We need to define the positive predictive value and the negative predictive value" of the test, he added. The long-term future of a test like this may also be limited because future testing will probably need to look at multiple biomarkers, Dr. Paz-Ares said.
The study enrolled five patients with advanced NSCLC who had exon 19 deletion EGFR mutations, and five advanced NSCLC patients with wild-type EGFR genes. Each patient underwent a pair of [11C]erlotinib PET scans, each preceded by a [15O]water PET scan to assess blood perfusion of the tumors. A 4-hour interval separated the two sets of scans.
The scan results showed that the volume of distribution of the tagged erlotinib in the patients with EGFR mutations ran about 50% higher than in the patients with wild-type tumors, a difference that was significant (P = .03). Clinically, two of the five wild-type patients had nonetheless received erlotinib treatment prior to testing, and neither patient responded, with both showing progressive disease.
Three of the five patients with an EGFR mutation began receiving erlotinib treatment after testing and responded. In one of these patients, the tumor remained in check for 13 months. In a second patient, the tumor began to progress after 17 months of no progression on treatment. In the third patient, the tumor began to progress again after about 4 weeks of no progression on erlotinib treatment, Dr. Bahce said. A fourth patient went on erlotinib treatment before testing, and did not respond and continued to have progressive disease.
The two patient subgroups showed no difference in blood perfusion into the tumors or in EGFR expression in cell membranes.
Dr. Bahce said he had no disclosures.☐
Major Finding: Advanced non–small cell lung cancer tumors with an epidermal growth factor receptor (EGFR)–activating mutation bound significantly more radiolabeled erlotinib than did tumors with wild-type EGFR genes (P = .03).
Data Source: A pilot study in 10 patients.
Disclosures: Dr. Bahce said he had no disclosures.