User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Cutaneous Complications Associated With Intraosseous Access Placement
Intraosseous (IO) access can afford a lifesaving means of vascular access in emergency settings, as it allows for the administration of large volumes of fluids, blood products, and medications at high flow rates directly into the highly vascularized osseous medullary cavity.1 Fortunately, the complication rate with this resuscitative effort is low, with many reports demonstrating complication rates of less than 1%.2 The most commonly reported complications include fluid extravasation, osteomyelitis, traumatic bone fracture, and epiphyseal plate damage.1-3 Although compartment syndrome and skin necrosis have been reported,4,5 there is no comprehensive list of sequelae resulting from fluid extravasation in the literature, and there are no known studies examining the incidence and types of cutaneous complications. In this study, we sought to evaluate the dermatologic impacts of this procedure.
Methods
We performed a retrospective chart review approved by the institutional review board at a large metropolitan level I trauma center in the Midwestern United States spanning 18 consecutive months to identify all patients who underwent IO line placement, either en route to or upon arrival at the trauma center. The electronic medical records of 113 patients (age range, 10 days–94 years) were identified using either an automated natural language look-up program with keywords including intraosseous access and IO or a Current Procedural Terminology code 36680. Data including patient age, reason for IO insertion, anatomic location of the IO, and complications secondary to IO line placement were recorded.
Results
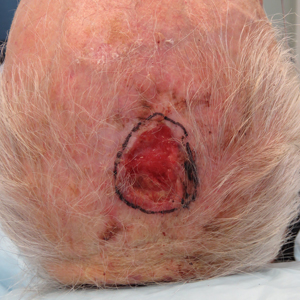
We identified an overall complication rate of 2.7% (3/113), with only 1 patient showing isolated cutaneous complications from IO line placement. The complications in the first 2 patients included compartment syndrome following IO line placement in the right tibia and needle breakage during IO line placement. The third patient, a 30-year-old heart transplant recipient, developed tense bullae on the left leg 5 days after a resuscitative effort required IO access through the bilateral tibiae. The patient had received vasopressors as well as 750 mL of normal saline through these access points. Two days after resuscitation, she developed an enlarg

At a scheduled 7-month dermatology follow-up, the wound bed appeared to be healing well with surrounding scarring with no residual bleeding or drainage (Figure 2) despite the patient reporting a protracted course of wound healing requiring debridement due to eschar formation and multiple follow-up appointments with the wound care service.

Comment
The most commonly reported complications with IO line placement result from fluid infiltration of the subcutaneous tissue secondary to catheter misplacement.1,3 Extravasated fluid may lead to tissue damage, compartment syndrome, and even tissue necrosis in some cases.1,4,5 Localized cellulitis and the formation of subcutaneous abscesses also have been reported, albeit rarely.3,5
In our retrospective cohort review, we identified an additional potential complication of IO line placement that has not been widely reported—development of large traumatic bullae. It is most likely that this patient’s IO catheter became dislodged, resulting in extravasation of fluids into the dermal and subcutaneous tissues.
Our findings support the previously noted complication rate of less than 1% following IO line placement, with an overall complication rate of 2.7% that included only 1 patient with a cutaneous complication.2 Given this low incidence, providers may not be used to recognizing such complications, leading to delayed or incorrect diagnosis of these entities. While there are certain conditions in which IO insertion is contraindicated, including severe bone diseases (eg, osteogenesis imperfecta, osteomyelitis), overlying cellulitis, and bone fracture, these conditions are rare and can be avoided in most cases by use of an alternative site for needle insertion.2 Due to the widespread utility of this tool and its few contraindications, its use in hospitalized patients is rapidly increasing, necessitating a need for quick recognition of potential complications.
From previous data on the incidence of traumatic blisters with underlying bone fractures, there are several identifiable risk factors that could be extended to patients at high risk for developing cutaneous IO complications secondary to the trauma associated with needle insertion,6 including wound-healing impairments in patients with fragile lymphatics, peripheral vascular disease, diabetes, or collagen vascular diseases (eg, lupus, rheumatoid arthritis, Sjögren syndrome). Patients with these conditions should be closely monitored for the development of bullae.6 While the patient we highlighted in our study did not have a history of such conditions, her history of cardiac disease, recent resuscitation attempts, and immunosuppression certainly could have contributed to suboptimal tissue agility and repair after IO line placement.
Conclusion
Intraosseous access is a safe, effective, and reliable option for vascular access in both pediatric and adult populations that is widely used in both prehospital (ie, paramedic administered) and hospital settings, including intensive care units, emergency departments, and any acute situation where rapid vascular access is necessary. This retrospective chart review examining the incidence and types of cutaneous complications associated with IO line placement at a level I trauma center revealed a total complication rate similar to those reported in previous studies and also highlighted a unique postprocedural cutaneous finding of traumatic bullae. Although no unified management recommendations currently exist, providers should consider this complication in the differential for hospitalized patients with large, atypical, asymmetric bullae in the absence of an alternative explanation for such skin findings.
- Day MW. Intraosseous devices for intravascular access in adult trauma patients. Crit Care Nurse. 2011;31:76-90. doi:10.4037/ccn2011615
- Petitpas F, Guenezan J, Vendeuvre T, et al. Use of intra-osseous access in adults: a systematic review. Crit Care. 2016;20:102. doi:10.1186/s13054-016-1277-6
- Desforges JF, Fiser DH. Intraosseous infusion. N Engl J Med. 1990;322:1579-1581. doi:10.1056/NEJM199005313222206
- Simmons CM, Johnson NE, Perkin RM, et al. Intraosseous extravasation complication reports. Ann Emerg Med. 1994;23:363-366. doi:10.1016/S0196-0644(94)70053-2
- Paxton JH. Intraosseous vascular access: a review. Trauma. 2012;14:195-232. doi:10.1177/1460408611430175
- Uebbing CM, Walsh M, Miller JB, et al. Fracture blisters. West J Emerg Med. 2011;12:131-133. doi:10.1016/S0190-9622(09)80152-7
Intraosseous (IO) access can afford a lifesaving means of vascular access in emergency settings, as it allows for the administration of large volumes of fluids, blood products, and medications at high flow rates directly into the highly vascularized osseous medullary cavity.1 Fortunately, the complication rate with this resuscitative effort is low, with many reports demonstrating complication rates of less than 1%.2 The most commonly reported complications include fluid extravasation, osteomyelitis, traumatic bone fracture, and epiphyseal plate damage.1-3 Although compartment syndrome and skin necrosis have been reported,4,5 there is no comprehensive list of sequelae resulting from fluid extravasation in the literature, and there are no known studies examining the incidence and types of cutaneous complications. In this study, we sought to evaluate the dermatologic impacts of this procedure.
Methods
We performed a retrospective chart review approved by the institutional review board at a large metropolitan level I trauma center in the Midwestern United States spanning 18 consecutive months to identify all patients who underwent IO line placement, either en route to or upon arrival at the trauma center. The electronic medical records of 113 patients (age range, 10 days–94 years) were identified using either an automated natural language look-up program with keywords including intraosseous access and IO or a Current Procedural Terminology code 36680. Data including patient age, reason for IO insertion, anatomic location of the IO, and complications secondary to IO line placement were recorded.
Results
We identified an overall complication rate of 2.7% (3/113), with only 1 patient showing isolated cutaneous complications from IO line placement. The complications in the first 2 patients included compartment syndrome following IO line placement in the right tibia and needle breakage during IO line placement. The third patient, a 30-year-old heart transplant recipient, developed tense bullae on the left leg 5 days after a resuscitative effort required IO access through the bilateral tibiae. The patient had received vasopressors as well as 750 mL of normal saline through these access points. Two days after resuscitation, she developed an enlarg
At a scheduled 7-month dermatology follow-up, the wound bed appeared to be healing well with surrounding scarring with no residual bleeding or drainage (Figure 2) despite the patient reporting a protracted course of wound healing requiring debridement due to eschar formation and multiple follow-up appointments with the wound care service.
Comment
The most commonly reported complications with IO line placement result from fluid infiltration of the subcutaneous tissue secondary to catheter misplacement.1,3 Extravasated fluid may lead to tissue damage, compartment syndrome, and even tissue necrosis in some cases.1,4,5 Localized cellulitis and the formation of subcutaneous abscesses also have been reported, albeit rarely.3,5
In our retrospective cohort review, we identified an additional potential complication of IO line placement that has not been widely reported—development of large traumatic bullae. It is most likely that this patient’s IO catheter became dislodged, resulting in extravasation of fluids into the dermal and subcutaneous tissues.
Our findings support the previously noted complication rate of less than 1% following IO line placement, with an overall complication rate of 2.7% that included only 1 patient with a cutaneous complication.2 Given this low incidence, providers may not be used to recognizing such complications, leading to delayed or incorrect diagnosis of these entities. While there are certain conditions in which IO insertion is contraindicated, including severe bone diseases (eg, osteogenesis imperfecta, osteomyelitis), overlying cellulitis, and bone fracture, these conditions are rare and can be avoided in most cases by use of an alternative site for needle insertion.2 Due to the widespread utility of this tool and its few contraindications, its use in hospitalized patients is rapidly increasing, necessitating a need for quick recognition of potential complications.
From previous data on the incidence of traumatic blisters with underlying bone fractures, there are several identifiable risk factors that could be extended to patients at high risk for developing cutaneous IO complications secondary to the trauma associated with needle insertion,6 including wound-healing impairments in patients with fragile lymphatics, peripheral vascular disease, diabetes, or collagen vascular diseases (eg, lupus, rheumatoid arthritis, Sjögren syndrome). Patients with these conditions should be closely monitored for the development of bullae.6 While the patient we highlighted in our study did not have a history of such conditions, her history of cardiac disease, recent resuscitation attempts, and immunosuppression certainly could have contributed to suboptimal tissue agility and repair after IO line placement.
Conclusion
Intraosseous access is a safe, effective, and reliable option for vascular access in both pediatric and adult populations that is widely used in both prehospital (ie, paramedic administered) and hospital settings, including intensive care units, emergency departments, and any acute situation where rapid vascular access is necessary. This retrospective chart review examining the incidence and types of cutaneous complications associated with IO line placement at a level I trauma center revealed a total complication rate similar to those reported in previous studies and also highlighted a unique postprocedural cutaneous finding of traumatic bullae. Although no unified management recommendations currently exist, providers should consider this complication in the differential for hospitalized patients with large, atypical, asymmetric bullae in the absence of an alternative explanation for such skin findings.
Intraosseous (IO) access can afford a lifesaving means of vascular access in emergency settings, as it allows for the administration of large volumes of fluids, blood products, and medications at high flow rates directly into the highly vascularized osseous medullary cavity.1 Fortunately, the complication rate with this resuscitative effort is low, with many reports demonstrating complication rates of less than 1%.2 The most commonly reported complications include fluid extravasation, osteomyelitis, traumatic bone fracture, and epiphyseal plate damage.1-3 Although compartment syndrome and skin necrosis have been reported,4,5 there is no comprehensive list of sequelae resulting from fluid extravasation in the literature, and there are no known studies examining the incidence and types of cutaneous complications. In this study, we sought to evaluate the dermatologic impacts of this procedure.
Methods
We performed a retrospective chart review approved by the institutional review board at a large metropolitan level I trauma center in the Midwestern United States spanning 18 consecutive months to identify all patients who underwent IO line placement, either en route to or upon arrival at the trauma center. The electronic medical records of 113 patients (age range, 10 days–94 years) were identified using either an automated natural language look-up program with keywords including intraosseous access and IO or a Current Procedural Terminology code 36680. Data including patient age, reason for IO insertion, anatomic location of the IO, and complications secondary to IO line placement were recorded.
Results
We identified an overall complication rate of 2.7% (3/113), with only 1 patient showing isolated cutaneous complications from IO line placement. The complications in the first 2 patients included compartment syndrome following IO line placement in the right tibia and needle breakage during IO line placement. The third patient, a 30-year-old heart transplant recipient, developed tense bullae on the left leg 5 days after a resuscitative effort required IO access through the bilateral tibiae. The patient had received vasopressors as well as 750 mL of normal saline through these access points. Two days after resuscitation, she developed an enlarg
At a scheduled 7-month dermatology follow-up, the wound bed appeared to be healing well with surrounding scarring with no residual bleeding or drainage (Figure 2) despite the patient reporting a protracted course of wound healing requiring debridement due to eschar formation and multiple follow-up appointments with the wound care service.
Comment
The most commonly reported complications with IO line placement result from fluid infiltration of the subcutaneous tissue secondary to catheter misplacement.1,3 Extravasated fluid may lead to tissue damage, compartment syndrome, and even tissue necrosis in some cases.1,4,5 Localized cellulitis and the formation of subcutaneous abscesses also have been reported, albeit rarely.3,5
In our retrospective cohort review, we identified an additional potential complication of IO line placement that has not been widely reported—development of large traumatic bullae. It is most likely that this patient’s IO catheter became dislodged, resulting in extravasation of fluids into the dermal and subcutaneous tissues.
Our findings support the previously noted complication rate of less than 1% following IO line placement, with an overall complication rate of 2.7% that included only 1 patient with a cutaneous complication.2 Given this low incidence, providers may not be used to recognizing such complications, leading to delayed or incorrect diagnosis of these entities. While there are certain conditions in which IO insertion is contraindicated, including severe bone diseases (eg, osteogenesis imperfecta, osteomyelitis), overlying cellulitis, and bone fracture, these conditions are rare and can be avoided in most cases by use of an alternative site for needle insertion.2 Due to the widespread utility of this tool and its few contraindications, its use in hospitalized patients is rapidly increasing, necessitating a need for quick recognition of potential complications.
From previous data on the incidence of traumatic blisters with underlying bone fractures, there are several identifiable risk factors that could be extended to patients at high risk for developing cutaneous IO complications secondary to the trauma associated with needle insertion,6 including wound-healing impairments in patients with fragile lymphatics, peripheral vascular disease, diabetes, or collagen vascular diseases (eg, lupus, rheumatoid arthritis, Sjögren syndrome). Patients with these conditions should be closely monitored for the development of bullae.6 While the patient we highlighted in our study did not have a history of such conditions, her history of cardiac disease, recent resuscitation attempts, and immunosuppression certainly could have contributed to suboptimal tissue agility and repair after IO line placement.
Conclusion
Intraosseous access is a safe, effective, and reliable option for vascular access in both pediatric and adult populations that is widely used in both prehospital (ie, paramedic administered) and hospital settings, including intensive care units, emergency departments, and any acute situation where rapid vascular access is necessary. This retrospective chart review examining the incidence and types of cutaneous complications associated with IO line placement at a level I trauma center revealed a total complication rate similar to those reported in previous studies and also highlighted a unique postprocedural cutaneous finding of traumatic bullae. Although no unified management recommendations currently exist, providers should consider this complication in the differential for hospitalized patients with large, atypical, asymmetric bullae in the absence of an alternative explanation for such skin findings.
- Day MW. Intraosseous devices for intravascular access in adult trauma patients. Crit Care Nurse. 2011;31:76-90. doi:10.4037/ccn2011615
- Petitpas F, Guenezan J, Vendeuvre T, et al. Use of intra-osseous access in adults: a systematic review. Crit Care. 2016;20:102. doi:10.1186/s13054-016-1277-6
- Desforges JF, Fiser DH. Intraosseous infusion. N Engl J Med. 1990;322:1579-1581. doi:10.1056/NEJM199005313222206
- Simmons CM, Johnson NE, Perkin RM, et al. Intraosseous extravasation complication reports. Ann Emerg Med. 1994;23:363-366. doi:10.1016/S0196-0644(94)70053-2
- Paxton JH. Intraosseous vascular access: a review. Trauma. 2012;14:195-232. doi:10.1177/1460408611430175
- Uebbing CM, Walsh M, Miller JB, et al. Fracture blisters. West J Emerg Med. 2011;12:131-133. doi:10.1016/S0190-9622(09)80152-7
- Day MW. Intraosseous devices for intravascular access in adult trauma patients. Crit Care Nurse. 2011;31:76-90. doi:10.4037/ccn2011615
- Petitpas F, Guenezan J, Vendeuvre T, et al. Use of intra-osseous access in adults: a systematic review. Crit Care. 2016;20:102. doi:10.1186/s13054-016-1277-6
- Desforges JF, Fiser DH. Intraosseous infusion. N Engl J Med. 1990;322:1579-1581. doi:10.1056/NEJM199005313222206
- Simmons CM, Johnson NE, Perkin RM, et al. Intraosseous extravasation complication reports. Ann Emerg Med. 1994;23:363-366. doi:10.1016/S0196-0644(94)70053-2
- Paxton JH. Intraosseous vascular access: a review. Trauma. 2012;14:195-232. doi:10.1177/1460408611430175
- Uebbing CM, Walsh M, Miller JB, et al. Fracture blisters. West J Emerg Med. 2011;12:131-133. doi:10.1016/S0190-9622(09)80152-7
Practice Points
- Intraosseous (IO) access provides rapid vascular access for the delivery of fluids, drugs, and blood products in emergent situations.
- Bullae are potential complications from IO line placement.
Progressive Axillary Hyperpigmentation
The Diagnosis: Dowling-Degos Disease
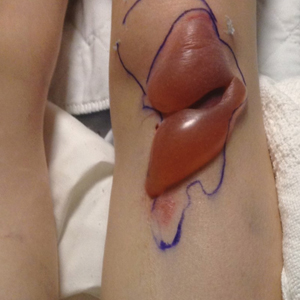
Histopathology demonstrated elongation of the epidermal rete ridges with increased basal pigmentation, suprapapillary epithelial thinning, dermal melanophages, and a mild lymphocytic infiltrate (Figure). Given the clinical and histologic findings, a diagnosis of Dowling-Degos disease (DDD) was made. The patient was counseled on the increased risk for her children developing DDD. Treatment with the erbium:YAG (Er:YAG) laser subsequently was initiated.

Dowling-Degos disease (also known as reticulate pigmented anomaly of the flexures) is an uncommon autosomal-dominant condition characterized by reticular hyperpigmentation involving the flexural and intertriginous sites. Classic DDD commonly is caused by lossof-function mutations in the keratin 5 gene, KRT51; however, DDD also may result from loss-of-function mutations in the protein O-fucosyltransferase 1, POFUT1, and protein O-glucosyltransferase 1, POGLUT1, genes.2
Rare cases of DDD associated with hidradenitis suppurativa are caused by mutations in the presenilin enhancer protein 2 gene, PSENEN.3
Of note, a missense mutation in KRT5 is implicated in epidermolysis bullosa simplex with mottled pigmentation. Onset of DDD typically occurs during the third to fourth decades of life. Reticulated hyperpigmented macules initially occur in the axillae and groin and progressively increase over time to involve the neck, inframammary folds, trunk, and flexural surfaces of the arms and thighs. Patients additionally may present with pitted perioral scars, comedolike lesions on the back and neck, epidermoid cysts, and hidradenitis suppurativa. Keratoacanthoma and squamous cell carcinoma rarely have been reported in association with classic DDD.4,5
Dowling-Degos disease usually is asymptomatic, though pruritus seldom may occur in the affected flexural areas. Histologically, the epidermal rete ridges are elongated in a filiform or antlerlike pattern with increased pigmentation of the basal layer and thinning of the suprapapillary epithelium. Dermal melanosis and a mild perivascular lymphohistiocytic infiltrate also are present with no increase in the number of melanocytes.6,7 Galli-Galli disease is a variant of DDD that shares similar clinical and histologic features of DDD but is distinguished from DDD by suprabasilar nondyskeratotic acantholysis on histology.8
Regarding other differential diagnoses for our patient, acanthosis nigricans may be distinguished clinically by the presence of velvety and/or verrucous plaques, commonly in the neck folds and axillae. Histologically, acanthosis nigricans is distinct from DDD and involves hyperkeratosis, acanthosis, and epidermal papillomatosis. Our patient had no history of diabetes mellitus or insulin resistance. Granular parakeratosis presents with hyperpigmented hyperkeratotic papules and plaques classically confined to the axillary region; however, the involvement of other intertriginous areas may occur. Histologically, granular parakeratosis demonstrates compact parakeratosis with small bluish keratohyalin granules within the stratum corneum. Confluent and reticulated papillomatosis presents with red-brown keratotic papules that initially appear in the intermammary region and spread laterally forming a reticulated pattern. Histology is similar to acanthosis nigricans and demonstrates hyperkeratosis, acanthosis, and papillomatosis. Inverse psoriasis presents with symmetric and sharply demarcated, erythematous, nonscaly plaques in the intertriginous areas. The plaques of inverse psoriasis may be pruritic and/or sore and occasionally may become macerated. Inverse psoriasis shares similar histologic findings compared to classic plaque psoriasis but may have less confluent parakeratosis.
Treatment of DDD essentially is reserved for cosmetic reasons. Topical hydroquinone, tretinoin, and corticosteroids have been used with limited to no success.5,9 Beneficial results after treatment with the Er:YAG laser have been reported.10
- Betz RC, Planko L, Eigelshoven S, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet. 2006;78:510-519.
- Basmanav FB, Oprisoreanu AM, Pasternack SM, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomaldominant Dowling-Degos disease. Am J Hum Genet. 2014;94:135-143.
- Pavlovsky M, Sarig O, Eskin-Schwartz M, et al. A phenotype combining hidradenitis suppurativa with Dowling-Degos disease caused by a founder mutation in PSENEN. Br J Dermatol. 2018;178:502-508.
- Ujihara M, Kamakura T, Ikeda M, et al. Dowling-Degos disease associated with squamous cell carcinomas on the dappled pigmentation. Br J Dermatol. 2002;147:568-571.
- Weber LA, Kantor GR, Bergfeld WF. Reticulate pigmented anomaly of the flexures (Dowling-Degos disease): a case report associated with hidradenitis suppurativa and squamous cell carcinoma. Cutis. 1990;45:446-450.
- Jones EW, Grice K. Reticulate pigmented anomaly of the flexures. Dowing Degos disease, a new genodermatosis. Arch Dermatol. 1978;114:1150-1157.
- Kim YC, Davis MD, Schanbacher CF, et al. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): a clinical and histopathologic study of 6 cases. J Am Acad Dermatol. 1999; 40:462-467.
- Reisenauer AK, Wordingham SV, York J, et al. Heterozygous frameshift mutation in keratin 5 in a family with Galli-Galli disease. Br J Dermatol. 2014;170:1362-1365.
- Oppolzer G, Schwarz T, Duschet P, et al. Dowling-Degos disease: unsuccessful therapeutic trial with retinoids [in German]. Hautarzt. 1987;38:615-618.
- Wenzel G, Petrow W, Tappe K, et al. Treatment of Dowling-Degos disease with Er:YAG-laser: results after 2.5 years. Dermatol Surg. 2003;29:1161-1162.
The Diagnosis: Dowling-Degos Disease
Histopathology demonstrated elongation of the epidermal rete ridges with increased basal pigmentation, suprapapillary epithelial thinning, dermal melanophages, and a mild lymphocytic infiltrate (Figure). Given the clinical and histologic findings, a diagnosis of Dowling-Degos disease (DDD) was made. The patient was counseled on the increased risk for her children developing DDD. Treatment with the erbium:YAG (Er:YAG) laser subsequently was initiated.
Dowling-Degos disease (also known as reticulate pigmented anomaly of the flexures) is an uncommon autosomal-dominant condition characterized by reticular hyperpigmentation involving the flexural and intertriginous sites. Classic DDD commonly is caused by lossof-function mutations in the keratin 5 gene, KRT51; however, DDD also may result from loss-of-function mutations in the protein O-fucosyltransferase 1, POFUT1, and protein O-glucosyltransferase 1, POGLUT1, genes.2
Rare cases of DDD associated with hidradenitis suppurativa are caused by mutations in the presenilin enhancer protein 2 gene, PSENEN.3
Of note, a missense mutation in KRT5 is implicated in epidermolysis bullosa simplex with mottled pigmentation. Onset of DDD typically occurs during the third to fourth decades of life. Reticulated hyperpigmented macules initially occur in the axillae and groin and progressively increase over time to involve the neck, inframammary folds, trunk, and flexural surfaces of the arms and thighs. Patients additionally may present with pitted perioral scars, comedolike lesions on the back and neck, epidermoid cysts, and hidradenitis suppurativa. Keratoacanthoma and squamous cell carcinoma rarely have been reported in association with classic DDD.4,5
Dowling-Degos disease usually is asymptomatic, though pruritus seldom may occur in the affected flexural areas. Histologically, the epidermal rete ridges are elongated in a filiform or antlerlike pattern with increased pigmentation of the basal layer and thinning of the suprapapillary epithelium. Dermal melanosis and a mild perivascular lymphohistiocytic infiltrate also are present with no increase in the number of melanocytes.6,7 Galli-Galli disease is a variant of DDD that shares similar clinical and histologic features of DDD but is distinguished from DDD by suprabasilar nondyskeratotic acantholysis on histology.8
Regarding other differential diagnoses for our patient, acanthosis nigricans may be distinguished clinically by the presence of velvety and/or verrucous plaques, commonly in the neck folds and axillae. Histologically, acanthosis nigricans is distinct from DDD and involves hyperkeratosis, acanthosis, and epidermal papillomatosis. Our patient had no history of diabetes mellitus or insulin resistance. Granular parakeratosis presents with hyperpigmented hyperkeratotic papules and plaques classically confined to the axillary region; however, the involvement of other intertriginous areas may occur. Histologically, granular parakeratosis demonstrates compact parakeratosis with small bluish keratohyalin granules within the stratum corneum. Confluent and reticulated papillomatosis presents with red-brown keratotic papules that initially appear in the intermammary region and spread laterally forming a reticulated pattern. Histology is similar to acanthosis nigricans and demonstrates hyperkeratosis, acanthosis, and papillomatosis. Inverse psoriasis presents with symmetric and sharply demarcated, erythematous, nonscaly plaques in the intertriginous areas. The plaques of inverse psoriasis may be pruritic and/or sore and occasionally may become macerated. Inverse psoriasis shares similar histologic findings compared to classic plaque psoriasis but may have less confluent parakeratosis.
Treatment of DDD essentially is reserved for cosmetic reasons. Topical hydroquinone, tretinoin, and corticosteroids have been used with limited to no success.5,9 Beneficial results after treatment with the Er:YAG laser have been reported.10
The Diagnosis: Dowling-Degos Disease
Histopathology demonstrated elongation of the epidermal rete ridges with increased basal pigmentation, suprapapillary epithelial thinning, dermal melanophages, and a mild lymphocytic infiltrate (Figure). Given the clinical and histologic findings, a diagnosis of Dowling-Degos disease (DDD) was made. The patient was counseled on the increased risk for her children developing DDD. Treatment with the erbium:YAG (Er:YAG) laser subsequently was initiated.
Dowling-Degos disease (also known as reticulate pigmented anomaly of the flexures) is an uncommon autosomal-dominant condition characterized by reticular hyperpigmentation involving the flexural and intertriginous sites. Classic DDD commonly is caused by lossof-function mutations in the keratin 5 gene, KRT51; however, DDD also may result from loss-of-function mutations in the protein O-fucosyltransferase 1, POFUT1, and protein O-glucosyltransferase 1, POGLUT1, genes.2
Rare cases of DDD associated with hidradenitis suppurativa are caused by mutations in the presenilin enhancer protein 2 gene, PSENEN.3
Of note, a missense mutation in KRT5 is implicated in epidermolysis bullosa simplex with mottled pigmentation. Onset of DDD typically occurs during the third to fourth decades of life. Reticulated hyperpigmented macules initially occur in the axillae and groin and progressively increase over time to involve the neck, inframammary folds, trunk, and flexural surfaces of the arms and thighs. Patients additionally may present with pitted perioral scars, comedolike lesions on the back and neck, epidermoid cysts, and hidradenitis suppurativa. Keratoacanthoma and squamous cell carcinoma rarely have been reported in association with classic DDD.4,5
Dowling-Degos disease usually is asymptomatic, though pruritus seldom may occur in the affected flexural areas. Histologically, the epidermal rete ridges are elongated in a filiform or antlerlike pattern with increased pigmentation of the basal layer and thinning of the suprapapillary epithelium. Dermal melanosis and a mild perivascular lymphohistiocytic infiltrate also are present with no increase in the number of melanocytes.6,7 Galli-Galli disease is a variant of DDD that shares similar clinical and histologic features of DDD but is distinguished from DDD by suprabasilar nondyskeratotic acantholysis on histology.8
Regarding other differential diagnoses for our patient, acanthosis nigricans may be distinguished clinically by the presence of velvety and/or verrucous plaques, commonly in the neck folds and axillae. Histologically, acanthosis nigricans is distinct from DDD and involves hyperkeratosis, acanthosis, and epidermal papillomatosis. Our patient had no history of diabetes mellitus or insulin resistance. Granular parakeratosis presents with hyperpigmented hyperkeratotic papules and plaques classically confined to the axillary region; however, the involvement of other intertriginous areas may occur. Histologically, granular parakeratosis demonstrates compact parakeratosis with small bluish keratohyalin granules within the stratum corneum. Confluent and reticulated papillomatosis presents with red-brown keratotic papules that initially appear in the intermammary region and spread laterally forming a reticulated pattern. Histology is similar to acanthosis nigricans and demonstrates hyperkeratosis, acanthosis, and papillomatosis. Inverse psoriasis presents with symmetric and sharply demarcated, erythematous, nonscaly plaques in the intertriginous areas. The plaques of inverse psoriasis may be pruritic and/or sore and occasionally may become macerated. Inverse psoriasis shares similar histologic findings compared to classic plaque psoriasis but may have less confluent parakeratosis.
Treatment of DDD essentially is reserved for cosmetic reasons. Topical hydroquinone, tretinoin, and corticosteroids have been used with limited to no success.5,9 Beneficial results after treatment with the Er:YAG laser have been reported.10
- Betz RC, Planko L, Eigelshoven S, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet. 2006;78:510-519.
- Basmanav FB, Oprisoreanu AM, Pasternack SM, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomaldominant Dowling-Degos disease. Am J Hum Genet. 2014;94:135-143.
- Pavlovsky M, Sarig O, Eskin-Schwartz M, et al. A phenotype combining hidradenitis suppurativa with Dowling-Degos disease caused by a founder mutation in PSENEN. Br J Dermatol. 2018;178:502-508.
- Ujihara M, Kamakura T, Ikeda M, et al. Dowling-Degos disease associated with squamous cell carcinomas on the dappled pigmentation. Br J Dermatol. 2002;147:568-571.
- Weber LA, Kantor GR, Bergfeld WF. Reticulate pigmented anomaly of the flexures (Dowling-Degos disease): a case report associated with hidradenitis suppurativa and squamous cell carcinoma. Cutis. 1990;45:446-450.
- Jones EW, Grice K. Reticulate pigmented anomaly of the flexures. Dowing Degos disease, a new genodermatosis. Arch Dermatol. 1978;114:1150-1157.
- Kim YC, Davis MD, Schanbacher CF, et al. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): a clinical and histopathologic study of 6 cases. J Am Acad Dermatol. 1999; 40:462-467.
- Reisenauer AK, Wordingham SV, York J, et al. Heterozygous frameshift mutation in keratin 5 in a family with Galli-Galli disease. Br J Dermatol. 2014;170:1362-1365.
- Oppolzer G, Schwarz T, Duschet P, et al. Dowling-Degos disease: unsuccessful therapeutic trial with retinoids [in German]. Hautarzt. 1987;38:615-618.
- Wenzel G, Petrow W, Tappe K, et al. Treatment of Dowling-Degos disease with Er:YAG-laser: results after 2.5 years. Dermatol Surg. 2003;29:1161-1162.
- Betz RC, Planko L, Eigelshoven S, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet. 2006;78:510-519.
- Basmanav FB, Oprisoreanu AM, Pasternack SM, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomaldominant Dowling-Degos disease. Am J Hum Genet. 2014;94:135-143.
- Pavlovsky M, Sarig O, Eskin-Schwartz M, et al. A phenotype combining hidradenitis suppurativa with Dowling-Degos disease caused by a founder mutation in PSENEN. Br J Dermatol. 2018;178:502-508.
- Ujihara M, Kamakura T, Ikeda M, et al. Dowling-Degos disease associated with squamous cell carcinomas on the dappled pigmentation. Br J Dermatol. 2002;147:568-571.
- Weber LA, Kantor GR, Bergfeld WF. Reticulate pigmented anomaly of the flexures (Dowling-Degos disease): a case report associated with hidradenitis suppurativa and squamous cell carcinoma. Cutis. 1990;45:446-450.
- Jones EW, Grice K. Reticulate pigmented anomaly of the flexures. Dowing Degos disease, a new genodermatosis. Arch Dermatol. 1978;114:1150-1157.
- Kim YC, Davis MD, Schanbacher CF, et al. Dowling-Degos disease (reticulate pigmented anomaly of the flexures): a clinical and histopathologic study of 6 cases. J Am Acad Dermatol. 1999; 40:462-467.
- Reisenauer AK, Wordingham SV, York J, et al. Heterozygous frameshift mutation in keratin 5 in a family with Galli-Galli disease. Br J Dermatol. 2014;170:1362-1365.
- Oppolzer G, Schwarz T, Duschet P, et al. Dowling-Degos disease: unsuccessful therapeutic trial with retinoids [in German]. Hautarzt. 1987;38:615-618.
- Wenzel G, Petrow W, Tappe K, et al. Treatment of Dowling-Degos disease with Er:YAG-laser: results after 2.5 years. Dermatol Surg. 2003;29:1161-1162.

A 50-year-old Hispanic woman presented with asymptomatic, progressive, brown hyperpigmentation involving the axillae, neck, upper back, and inframammary areas of 5 years’ duration. She had no other notable medical history; family history was unremarkable. She had been treated with topical hydroquinone and tretinoin by an outside physician without improvement. Physical examination revealed reticulated hyperpigmented macules and patches involving the inverse regions of the neck, axillae, and inframammary regions. Additionally, acneform pitted scars involving the perioral region were seen. A 4.0-mm punch biopsy of the right axilla was performed.
Efficacy of Etanercept in the Treatment of Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis
Regarded as dermatologic emergencies, Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) represent a spectrum of blistering skin diseases that have a high mortality rate. Because of a misguided immune response to medications or infections, CD8+ T lymphocytes release proinflammatory cytokines, giving rise to the extensive epidermal destruction seen in SJS and TEN. The exact pathogenesis of SJS and TEN is still poorly defined, but studies have proposed that T cells mediate keratinocyte (KC) apoptosis through perforin and granzyme release and activation of the Fas/Fas ligand (FasL). Functioning as a transmembrane death receptor in the tumor necrosis factor (TNF) superfamily, Fas (CD95) activates Fas-associated death domain protein, caspases, and nucleases, resulting in organized cell destruction. Likewise, perforin and granzymes also have been shown to play a similar role in apoptosis via activation of caspases.1
Evidence for the role of TNF-α in SJS and TEN has been supported by findings of elevated levels of TNF-α within the blister fluid, serum, and KC cell surface. Additionally, TNF-α has been shown to upregulate inducible nitric oxide synthase in KCs, causing an accumulation of nitric oxide and subsequent FasL-mediated cell death.1-3 Notably, studies have demonstrated a relative lack of lymphocytes in the tissue of TEN patients despite the extensive destruction that is observed, thus emphasizing the importance of amplification and cell signaling via inflammatory mediators such as TNF-α.1 In this proposed model, T cells release IFN-γ, causing KCs to release TNF-α that subsequently promotes the upregulation of the aforementioned FasL.1 Tumor necrosis factor α also may promote increased MHC class I complex deposition on KC surfaces that may play a role in perforin and granzyme-mediated apoptosis of KCs.1
There is still debate on the standard of care for the treatment of SJS and TEN, attributed to the absence of randomized controlled trials and the rarity of the disease as well as the numerous conflicting studies evaluating potential treatments.1,4 Despite conflicting data to support their use, supportive care and intravenous immunoglobulin (IVIG) continue to be common treatments for SJS and TEN in hospitals worldwide. Elucidation of the role of TNF-α has prompted the use of infliximab and etanercept. In a case series of Italian patients with TEN (average SCORTEN, 3.6) treated with the TNF-α antagonist etanercept, no mortality was observed, which was well below the calculated expected mortality of 46.9%.2 Our retrospective study compared the use of a TNF antagonist to other therapies in the treatment of SJS/TEN. Our data suggest that etanercept is a lifesaving and disease-modifying therapy.
Methods
Twenty-two patients with SJS/TEN were included in this analysis. This included all patients who carried a clinical diagnosis of SJS/TEN with a confirmatory biopsy at our 2 university centers—University of California, Los Angeles, and Keck-LA County-Norris Hospital at the University of Southern California, Los Angeles—from 2013 to 2016. The diagnosis was rendered when a clinical diagnosis of SJS/TEN was given by a dermatologist and a confirmatory biopsy was performed. Every patient given a diagnosis of SJS/TEN at either university system from 2015 onward received an injection of etanercept given the positive results reported by Paradisi et al.2
The 9 patients who presented from 2013 to 2014 to our 2 hospital systems and were given a diagnosis of SJS/TEN received either IVIG or supportive care alone and had an average body surface area (BSA) affected of 23%. The 13 patients who presented from 2015 to 2016 were treated with etanercept in the form of a 50-mg subcutaneous injection given once to the right upper arm. Of this group, 4 patients received dual therapy with both IVIG and etanercept. In the etanercept-treated group (etanercept alone and etanercept plus IVIG), the average BSA affected was 30%. At the time of preliminary diagnosis, all patient medications were evaluated for a possible temporal relationship to the onset of rash and were discontinued if felt to be causative. The causative agent and treatment course for each patient is summarized in Table 1.
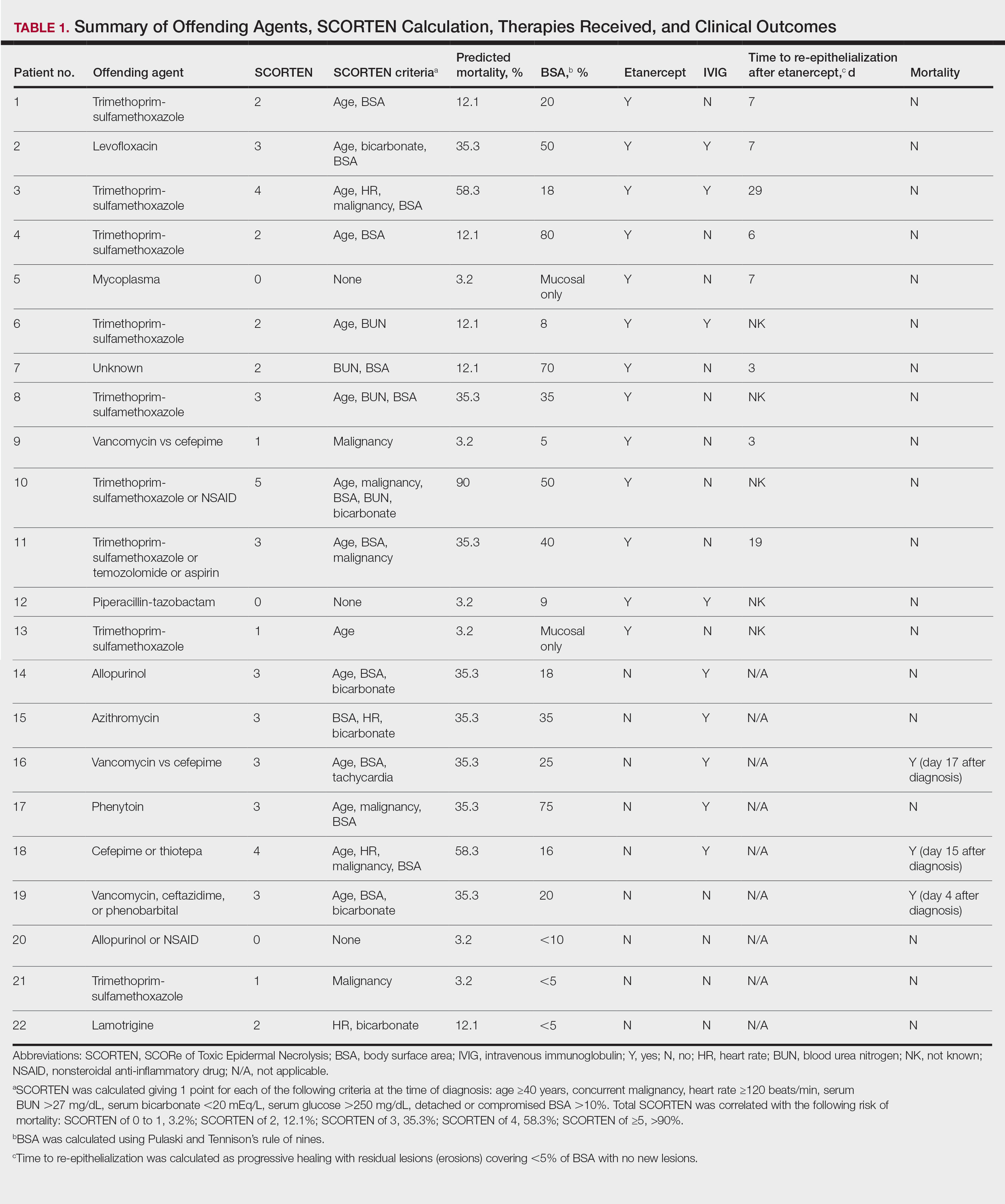
Patients were monitored daily in the hospital for improvement, and time to re-epithelialization was measured. Re-epithelialization was defined as progressive healing with residual lesions (erosions, ulcers, or bullae) covering no more than 5% BSA and was contingent on the patient having no new lesions within 24 hours.5 SCORe of Toxic Epidermal Necrosis (SCORTEN), a validated severity-of-illness score,6 was calculated by giving 1 point for each of the following criteria at the time of diagnosis: age ≥40 years, concurrent malignancy, heart rate ≥120 beats/min, serum blood urea nitrogen >27 mg/dL, serum bicarbonate <20 mEq/L, serum glucose >250 mg/dL, and detached or compromised BSA >10%. The total SCORTEN was correlated with the following risk of mortality as supported by prior validation studies: SCORTEN of 0 to 1, 3.2%; SCORTEN of 2, 12.1%; SCORTEN of 3, 35.3%; SCORTEN of 4, 58.3%; SCORTEN of ≥5, >90%.
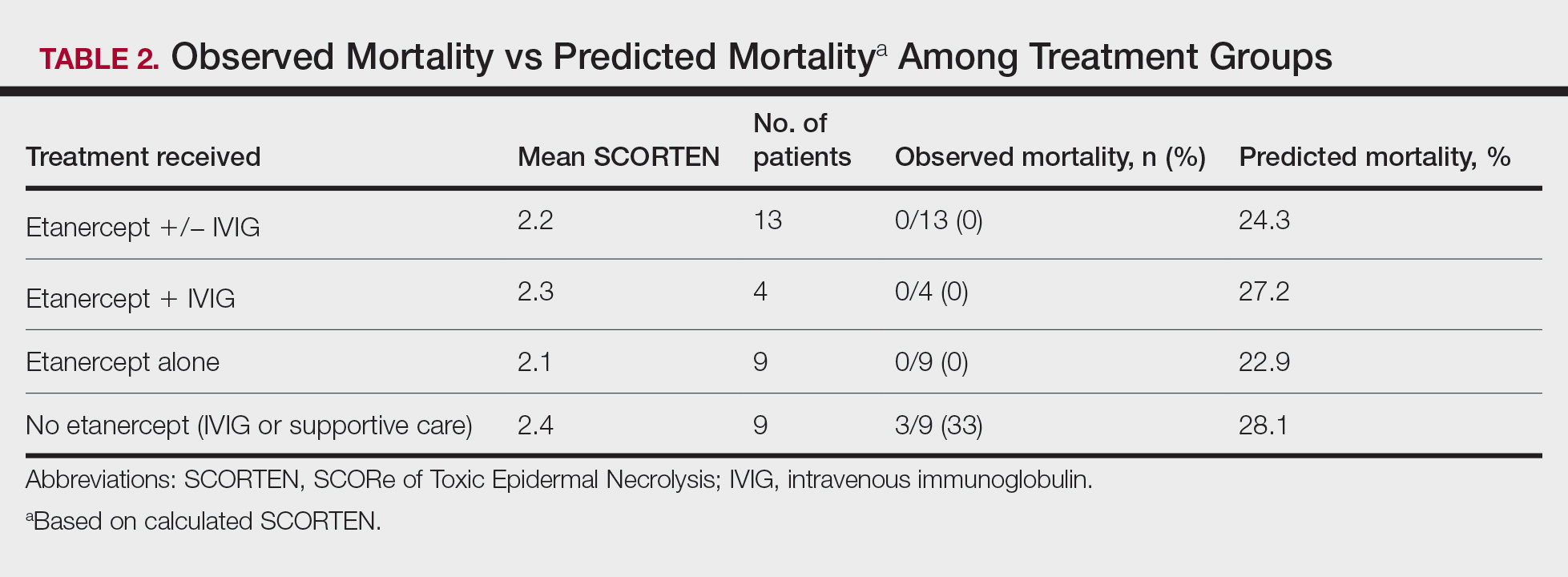
Results
A total of 13 patients received etanercept. The mean SCORTEN was 2.2. The observed mortality was 0%, which was markedly lower than the predicted mortality of 24.3% (as determined by linear interpolation). Of this cohort, 9 patients received etanercept alone (mean SCORTEN of 2.1, predicted mortality of 22.9%), whereas 4 patients received a combination of etanercept and IVIG (mean SCORTEN of 2.3, predicted mortality of 27.2%).
The 4 patients who received both etanercept and IVIG received dual therapy for varying reasons. In patient 2 (Table 1), the perceived severity of this case ultimately led to the decision to start IVIG in addition to etanercept, resulting in rapid recovery and discharge after only 1 week of hospitalization. Intravenous immunoglobulin also was given in patient 3 (SCORTEN of 4) and patient 6 (SCORTEN of 2) for progression of disease despite administration of etanercept, with subsequent cessation of progression after the addition of the second agent (IVIG). Patient 12 might have done well on etanercept monotherapy but was administered IVIG as a precautionary measure because of hospital treatment algorithms.
Nine patients did not receive etanercept. Of this group, 5 received IVIG and 4 were managed with supportive care alone. The average SCORTEN for this group was 2.4, only slightly higher than the group that received etanercept (Table 2). The mortality rate in this group was 33%, which was higher than the predicted mortality of 28.1%.
Re-epithelialization data were available for 8 patients who received etanercept. The average time to re-epithelialization for these patients was 8.9 days and ranged from 3 to 19 days. Of these patients, 2 received both IVIG and etanercept, with an average time to re-epithelialization of 13 days. For the 6 patients who received etanercept alone, the average time to re-epithelialization was 7.5 days. Re-epithelialization data were not available for any of the patients who received only IVIG or supportive care but to our recollection ranged from 14 to 21 days.
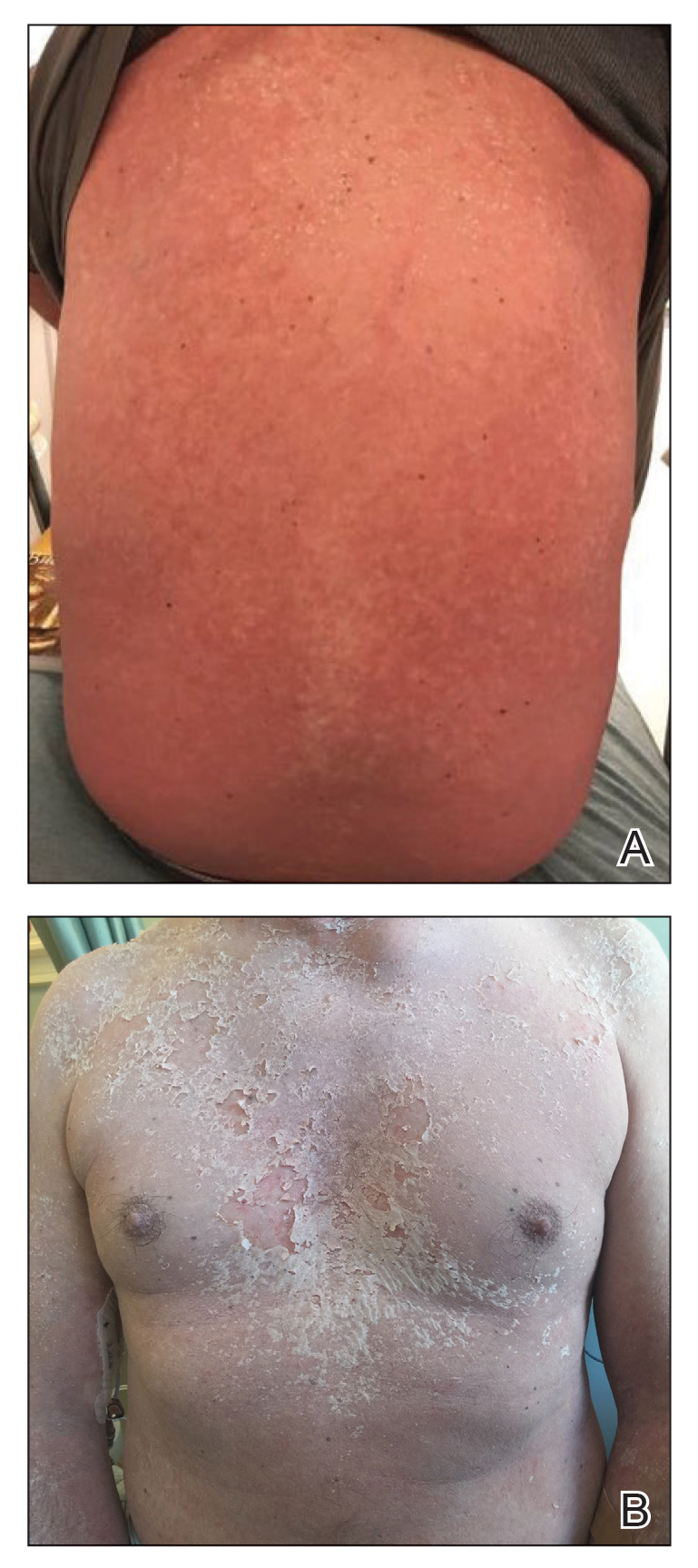
The clinical course of the 13 patients after the administration of a single dose of etanercept was remarkable, as there was complete absence of mortality and an increase in speed of recovery in most patients receiving this intervention (time to re-epithelialization, 3–19 days). We also observed another interesting trend from our patients treated with etanercept, which was the suggestion that treatment with etanercept may be less effective if IVIG and/or steroids are given prior to etanercept; likewise, treatment is more effective when etanercept is given quickly. For patients 1, 4, 5, 7, 9, and 11 (as shown in Table 1), no prior IVIG therapy or other immunosuppressive therapy had been given before etanercept was administered. In these 6 patients, the average time to re-epithelialization after etanercept administration was 7.5 days; average time to re-epithelialization, unfortunately, is not available for the patients who were not treated with etanercept. In addition, as shown in the Figure, it was noted in some patients that the depth of denudation was markedly more superficial than what would typically be clinically observed with TEN after administration of other immunomodulatory therapies such as IVIG or prednisone or with supportive care alone. In these 2 patients with superficial desquamation—patients 7 and 9—etanercept notably was given within 6 hours of onset of skin pain.
Comment
There is no definitive gold standard treatment of SJS, SJS/TEN overlap, or TEN. However, generally agreed upon management includes immediate discontinuation of the offending medication and supportive therapy with aggressive electrolyte replacement and wound care. Management in a burn unit or intensive care unit is recommended in severe cases. Contention over the efficacy of various medications in the treatment of SJS and TEN continues and largely is due to the rarity of SJS and TEN; studies are small and almost all lack randomization. Therapies that have been used include high-dose steroids, IVIG, plasmapheresis, cyclophosphamide, cyclosporine A, and TNF inhibitors (eg, etanercept, infliximab).1
Evidence for the use of anti–TNF-α antibodies has been limited thus far, with most of the literature focusing on infliximab and etanercept. Adalimumab, a fully humanized clonal antibody, has no reported cases in the dermatologic literature for use in patients with SJS/TEN. Two case reports of adalimumab paradoxically causing SJS have been documented. In both cases, adalimumab was stopped and patients responded to intravenous corticosteroids and infliximab.7,8 Similarly, thalidomide has not proven to be a promising anti–TNF-α agent for the treatment of SJS/TEN. In the only attempted randomized controlled trial for SJS and TEN, thalidomide appeared to increase mortality, eventuating in this trial being terminated prior to the planned end date.9Infliximab and etanercept have several case reports and a few case series highlighting potentially efficacious application of TNF-α inhibitors for the treatment of SJS/TEN.10-13 In 2002, Fischer et al10 reported the first case of TEN treated successfully with a single dose of infliximab 5 mg/kg. Kreft et al14 reported on etoricoxib-induced TEN that was treated with infliximab 5 mg/kg, which led to re-epithelialization within 5 weeks (notably a 5-week re-epithelialization time is not necessarily an improvement).
In 2005, Hunger et al3 demonstrated TNF-α’s release by KCs in the epidermis and by inflammatory cells in the dermis of a TEN patient. Twenty-four hours after the administration of infliximab 5 mg/kg in these patients, TNF-α was found to be below normal and epidermal detachment ceased.3 Wojtkietwicz et al13 demonstrated benefit following an infusion of infliximab 5 mg/kg in a patient whose disease continued to progress despite treatment with dexamethasone and 1.8 g/kg of IVIG.
Then 2 subsequent case series added further support for the efficacy of infliximab in the treatment of TEN. Patmanidis et al15 and Gaitanis et al16 reported similar results in 4 patients, each treated with infliximab 5 mg/kg immediately followed by initiation of high-dose IVIG (2 g/kg over 5 days). Zárate-Correa et al17 reported a 0% mortality rate and near-complete re-epithelialization after 5 to 14 days in 4 patients treated with a single 300-mg dose of infliximab.
However, the success of infliximab in the treatment of TEN has been countered by the pilot study by Paquet et al,18 which compared the efficacy of 150 mg/kg of N-acetylcysteine alone vs adding infliximab 5 mg/kg to treat 10 TEN patients. The study demonstrated no benefit at 48 hours in the group given infliximab, the time frame in which prior case reports touting infliximab’s benefit claimed the benefit was observed. Similarly, there was no effect on mortality for either treatment modality as assessed by illness auxiliary score.18
Evidence in support of the use of etanercept in the treatment of SJS/TEN is mounting, and some centers have begun to use it as the first-choice therapy for SJS/TEN. The first case was reported by Famularo et al,19 in which a patient with TEN was given 2 doses of etanercept 25 mg after failure to improve with prednisolone 1 mg/kg. The patient showed near-complete and rapid re-epithelization in 6 days before death due to disseminated intravascular coagulation 10 days after admission.19 Gubinelli et al20 and Sadighha21 independently reported cases of TEN and TEN/acute generalized exanthematous pustulosis overlap treated with a total of 50 mg of etanercept, demonstrating rapid cessation of lesion progression. Didona et al22 found similar benefit using etanercept 50 mg to treat TEN secondary to rituximab after failure to improve with prednisone and cyclophosphamide. Treatment of TEN with etanercept in an HIV-positive patient also has been reported. Lee et al23 described a patient who was administered 50-mg and 25-mg injections on days 3 and 5 of hospitalization, respectively, with re-epithelialization occurring by day 8. Finally, Owczarczyk-Saczonek et al24 reported a case of SJS in a patient with a 4-year history of etanercept and sulfasalazine treatment of rheumatoid arthritis; sulfasalazine was stopped, but this patient was continued on etanercept until resolution of skin and mucosal symptoms. However, it is important to consider the possibility of publication bias among these cases selected for their positive outcomes.
Perhaps the most compelling literature regarding the use of etanercept for TEN was described in a case series by Paradisi et al.2 This study included 10 patients with TEN, all of whom demonstrated complete re-epithelialization shortly after receiving etanercept 50 mg. Average SCORTEN was 3.6 with a range of 2 to 6. Eight patients in this study had severe comorbidities and all 10 patients survived, with a time to re-epithelialization ranging from 7 to 20 days.2 Additionally, a randomized controlled trial showed that 38 etanercept-treated patients had improved mortality (P=.266) and re-epithelialization time (P=.01) compared to patients treated with intravenous methylprednisolone.25Limitations to our study are similar to other reports of SJS/TEN and included the small number of cases and lack of randomization. Additionally, we do not have data available for all patients for time between onset of disease and treatment initiation. Because of these challenges, data presented in this case series is observational only. Additionally, the patients treated with etanercept alone had a slightly lower SCORTEN compared to the group that received IVIG or supportive care alone (2.1 and 2.4 respectively). However, the etanercept-only group actually had higher involvement of epidermal detachment (33%) compared to the non-etanercept group (23%).
Conclusion
Although treatment with etanercept lacks the support of a randomized controlled trial, similar to all other treatments currently used for SJS and TEN, preliminary reports highlight a benefit in disease progression and improvement in time to re-epithelialization. In particular, if etanercept 50 mg subcutaneously is given as monotherapy or is given early in the disease course (prior to other therapies being attempted and ideally within 6 hours of presentation), our data suggest an even greater trend toward improved mortality and decreased time to re-epithelialization. Additionally, our findings may suggest that in some patients, etanercept monotherapy is not an adequate intervention but the addition of IVIG may be helpful; however, the senior author (S.W.) notes anecdotally that in his experience with the patients treated at the University of California Los Angeles, the order of administration of combination therapies—etanercept followed by IVIG—was important in addition to the choice of therapy. These findings are promising enough to warrant a multicenter randomized controlled trial comparing the efficacy of etanercept to other more commonly used treatments for this spectrum of disease, including IVIG and/or cyclosporine. Based on the data presented in this case series, including the 13 patients who received etanercept and had a 0% mortality rate, etanercept may be viewed as a targeted therapeutic intervention for patients with SJS and TEN.
- Pereira FA, Mudgil AV, Rosmarin DM. Toxic epidermal necrolysis. J Am Acad Dermatol. 2007;56:181-200.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Hunger RE, Hunziker T, Buettiker U, et al. Rapid resolution of toxic epidermal necrolysis with anti-TNF-α treatment. J Allergy Clin Immunol. 2005;116:923-924.
- Worswick S, Cotliar J. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of treatment options. Dermatol Ther. 2011;24:207-218.
- Wallace AB. The exposure treatment of burns. Lancet Lond Engl. 1951;1:501-504.
- Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153.
- Mounach A, Rezqi A, Nouijai A, et al. Stevens-Johnson syndrome complicating adalimumab therapy in rheumatoid arthritis disease. Rheumatol Int. 2013;33:1351-1353.
- Salama M, Lawrance I-C. Stevens-Johnson syndrome complicating adalimumab therapy in Crohn’s disease. World J Gastroenterol. 2009;15:4449-4452.
- Wolkenstein P, Latarjet J, Roujeau JC, et al. Randomised comparison of thalidomide versus placebo in toxic epidermal necrolysis. Lancet Lond Engl. 1998;352:1586-1589.
- Fischer M, Fiedler E, Marsch WC, et al Antitumour necrosis factor-α antibodies (infliximab) in the treatment of a patient with toxic epidermal necrolysis. Br J Dermatol. 2002;146:707-709.
- Meiss F, Helmbold P, Meykadeh N, et al. Overlap of acute generalized exanthematous pustulosis and toxic epidermal necrolysis: response to antitumour necrosis factor-alpha antibody infliximab: report of three cases. J Eur Acad Dermatol Venereol. 2007;21:717-719.
- Al-Shouli S, Abouchala N, Bogusz MJ, et al. Toxic epidermal necrolysis associated with high intake of sildenafil and its response to infliximab. Acta Derm Venereol. 2005;85:534-535.
- Wojtkiewicz A, Wysocki M, Fortuna J, et al. Beneficial and rapid effect of infliximab on the course of toxic epidermal necrolysis. Acta Derm Venereol. 2008;88:420-421.
- Kreft B, Wohlrab J, Bramsiepe I, et al. Etoricoxib-induced toxic epidermal necrolysis: successful treatment with infliximab. J Dermatol. 2010;37:904-906.
- Patmanidis K, Sidiras A, Dolianitis K, et al. Combination of infliximab and high-dose intravenous immunoglobulin for toxic epidermal necrolysis: successful treatment of an elderly patient. Case Rep Dermatol Med. 2012;2012:915314.
- Gaitanis G, Spyridonos P, Patmanidis K, et al. Treatment of toxic epidermal necrolysis with the combination of infliximab and high-dose intravenous immunoglobulin. Dermatol Basel Switz. 2012;224:134-139.
- Zárate-Correa LC, Carrillo-Gómez DC, Ramírez-Escobar AF, et al. Toxic epidermal necrolysis successfully treated with infliximab. J Investig Allergol Clin Immunol. 2013;23:61-63.
- Paquet P, Jennes S, Rousseau AF, et al. Effect of N-acetylcysteine combined with infliximab on toxic epidermal necrolysis. a proof-of-concept study. Burns J Int Soc Burn Inj. 2014;40:1707-1712.
- Famularo G, Dona BD, Canzona F, et al. Etanercept for toxic epidermal necrolysis. Ann Pharmacother. 2007;41:1083-1084.
- Gubinelli E, Canzona F, Tonanzi T, et al. Toxic epidermal necrolysis successfully treated with etanercept. J Dermatol. 2009;36:150-153.
- Sadighha A. Etanercept in the treatment of a patient with acute generalized exanthematous pustulosis/toxic epidermal necrolysis: definition of a new model based on translational research. Int J Dermatol. 2009;48:913-914.
- Didona D, Paolino G, Garcovich S, et al. Successful use of etanercept in a case of toxic epidermal necrolysis induced by rituximab. J Eur Acad Dermatol Venereol. 2016;30:E83-E84.
- Lee Y-Y, Ko J-H, Wei C-H, et al. Use of etanercept to treat toxic epidermal necrolysis in a human immunodeficiency virus-positive patient. Dermatol Sin. 2013;31:78-81.
- Owczarczyk-Saczonek A, Zdanowska N, Znajewska-Pander A, et al. Stevens-Johnson syndrome in a patient with rheumatoid arthritis during long-term etanercept therapy. J Dermatol Case Rep. 2016;10:14-16.
- Wang CW, Yang LY, Chen CB, et al. Randomized, controlled trial of TNF-α antagonist in CTL mediated severe cutaneous adverse reactions. J Clin Invest. 2018;128:985-996.
Regarded as dermatologic emergencies, Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) represent a spectrum of blistering skin diseases that have a high mortality rate. Because of a misguided immune response to medications or infections, CD8+ T lymphocytes release proinflammatory cytokines, giving rise to the extensive epidermal destruction seen in SJS and TEN. The exact pathogenesis of SJS and TEN is still poorly defined, but studies have proposed that T cells mediate keratinocyte (KC) apoptosis through perforin and granzyme release and activation of the Fas/Fas ligand (FasL). Functioning as a transmembrane death receptor in the tumor necrosis factor (TNF) superfamily, Fas (CD95) activates Fas-associated death domain protein, caspases, and nucleases, resulting in organized cell destruction. Likewise, perforin and granzymes also have been shown to play a similar role in apoptosis via activation of caspases.1
Evidence for the role of TNF-α in SJS and TEN has been supported by findings of elevated levels of TNF-α within the blister fluid, serum, and KC cell surface. Additionally, TNF-α has been shown to upregulate inducible nitric oxide synthase in KCs, causing an accumulation of nitric oxide and subsequent FasL-mediated cell death.1-3 Notably, studies have demonstrated a relative lack of lymphocytes in the tissue of TEN patients despite the extensive destruction that is observed, thus emphasizing the importance of amplification and cell signaling via inflammatory mediators such as TNF-α.1 In this proposed model, T cells release IFN-γ, causing KCs to release TNF-α that subsequently promotes the upregulation of the aforementioned FasL.1 Tumor necrosis factor α also may promote increased MHC class I complex deposition on KC surfaces that may play a role in perforin and granzyme-mediated apoptosis of KCs.1
There is still debate on the standard of care for the treatment of SJS and TEN, attributed to the absence of randomized controlled trials and the rarity of the disease as well as the numerous conflicting studies evaluating potential treatments.1,4 Despite conflicting data to support their use, supportive care and intravenous immunoglobulin (IVIG) continue to be common treatments for SJS and TEN in hospitals worldwide. Elucidation of the role of TNF-α has prompted the use of infliximab and etanercept. In a case series of Italian patients with TEN (average SCORTEN, 3.6) treated with the TNF-α antagonist etanercept, no mortality was observed, which was well below the calculated expected mortality of 46.9%.2 Our retrospective study compared the use of a TNF antagonist to other therapies in the treatment of SJS/TEN. Our data suggest that etanercept is a lifesaving and disease-modifying therapy.
Methods
Twenty-two patients with SJS/TEN were included in this analysis. This included all patients who carried a clinical diagnosis of SJS/TEN with a confirmatory biopsy at our 2 university centers—University of California, Los Angeles, and Keck-LA County-Norris Hospital at the University of Southern California, Los Angeles—from 2013 to 2016. The diagnosis was rendered when a clinical diagnosis of SJS/TEN was given by a dermatologist and a confirmatory biopsy was performed. Every patient given a diagnosis of SJS/TEN at either university system from 2015 onward received an injection of etanercept given the positive results reported by Paradisi et al.2
The 9 patients who presented from 2013 to 2014 to our 2 hospital systems and were given a diagnosis of SJS/TEN received either IVIG or supportive care alone and had an average body surface area (BSA) affected of 23%. The 13 patients who presented from 2015 to 2016 were treated with etanercept in the form of a 50-mg subcutaneous injection given once to the right upper arm. Of this group, 4 patients received dual therapy with both IVIG and etanercept. In the etanercept-treated group (etanercept alone and etanercept plus IVIG), the average BSA affected was 30%. At the time of preliminary diagnosis, all patient medications were evaluated for a possible temporal relationship to the onset of rash and were discontinued if felt to be causative. The causative agent and treatment course for each patient is summarized in Table 1.
Patients were monitored daily in the hospital for improvement, and time to re-epithelialization was measured. Re-epithelialization was defined as progressive healing with residual lesions (erosions, ulcers, or bullae) covering no more than 5% BSA and was contingent on the patient having no new lesions within 24 hours.5 SCORe of Toxic Epidermal Necrosis (SCORTEN), a validated severity-of-illness score,6 was calculated by giving 1 point for each of the following criteria at the time of diagnosis: age ≥40 years, concurrent malignancy, heart rate ≥120 beats/min, serum blood urea nitrogen >27 mg/dL, serum bicarbonate <20 mEq/L, serum glucose >250 mg/dL, and detached or compromised BSA >10%. The total SCORTEN was correlated with the following risk of mortality as supported by prior validation studies: SCORTEN of 0 to 1, 3.2%; SCORTEN of 2, 12.1%; SCORTEN of 3, 35.3%; SCORTEN of 4, 58.3%; SCORTEN of ≥5, >90%.
Results
A total of 13 patients received etanercept. The mean SCORTEN was 2.2. The observed mortality was 0%, which was markedly lower than the predicted mortality of 24.3% (as determined by linear interpolation). Of this cohort, 9 patients received etanercept alone (mean SCORTEN of 2.1, predicted mortality of 22.9%), whereas 4 patients received a combination of etanercept and IVIG (mean SCORTEN of 2.3, predicted mortality of 27.2%).
The 4 patients who received both etanercept and IVIG received dual therapy for varying reasons. In patient 2 (Table 1), the perceived severity of this case ultimately led to the decision to start IVIG in addition to etanercept, resulting in rapid recovery and discharge after only 1 week of hospitalization. Intravenous immunoglobulin also was given in patient 3 (SCORTEN of 4) and patient 6 (SCORTEN of 2) for progression of disease despite administration of etanercept, with subsequent cessation of progression after the addition of the second agent (IVIG). Patient 12 might have done well on etanercept monotherapy but was administered IVIG as a precautionary measure because of hospital treatment algorithms.
Nine patients did not receive etanercept. Of this group, 5 received IVIG and 4 were managed with supportive care alone. The average SCORTEN for this group was 2.4, only slightly higher than the group that received etanercept (Table 2). The mortality rate in this group was 33%, which was higher than the predicted mortality of 28.1%.
Re-epithelialization data were available for 8 patients who received etanercept. The average time to re-epithelialization for these patients was 8.9 days and ranged from 3 to 19 days. Of these patients, 2 received both IVIG and etanercept, with an average time to re-epithelialization of 13 days. For the 6 patients who received etanercept alone, the average time to re-epithelialization was 7.5 days. Re-epithelialization data were not available for any of the patients who received only IVIG or supportive care but to our recollection ranged from 14 to 21 days.
The clinical course of the 13 patients after the administration of a single dose of etanercept was remarkable, as there was complete absence of mortality and an increase in speed of recovery in most patients receiving this intervention (time to re-epithelialization, 3–19 days). We also observed another interesting trend from our patients treated with etanercept, which was the suggestion that treatment with etanercept may be less effective if IVIG and/or steroids are given prior to etanercept; likewise, treatment is more effective when etanercept is given quickly. For patients 1, 4, 5, 7, 9, and 11 (as shown in Table 1), no prior IVIG therapy or other immunosuppressive therapy had been given before etanercept was administered. In these 6 patients, the average time to re-epithelialization after etanercept administration was 7.5 days; average time to re-epithelialization, unfortunately, is not available for the patients who were not treated with etanercept. In addition, as shown in the Figure, it was noted in some patients that the depth of denudation was markedly more superficial than what would typically be clinically observed with TEN after administration of other immunomodulatory therapies such as IVIG or prednisone or with supportive care alone. In these 2 patients with superficial desquamation—patients 7 and 9—etanercept notably was given within 6 hours of onset of skin pain.
Comment
There is no definitive gold standard treatment of SJS, SJS/TEN overlap, or TEN. However, generally agreed upon management includes immediate discontinuation of the offending medication and supportive therapy with aggressive electrolyte replacement and wound care. Management in a burn unit or intensive care unit is recommended in severe cases. Contention over the efficacy of various medications in the treatment of SJS and TEN continues and largely is due to the rarity of SJS and TEN; studies are small and almost all lack randomization. Therapies that have been used include high-dose steroids, IVIG, plasmapheresis, cyclophosphamide, cyclosporine A, and TNF inhibitors (eg, etanercept, infliximab).1
Evidence for the use of anti–TNF-α antibodies has been limited thus far, with most of the literature focusing on infliximab and etanercept. Adalimumab, a fully humanized clonal antibody, has no reported cases in the dermatologic literature for use in patients with SJS/TEN. Two case reports of adalimumab paradoxically causing SJS have been documented. In both cases, adalimumab was stopped and patients responded to intravenous corticosteroids and infliximab.7,8 Similarly, thalidomide has not proven to be a promising anti–TNF-α agent for the treatment of SJS/TEN. In the only attempted randomized controlled trial for SJS and TEN, thalidomide appeared to increase mortality, eventuating in this trial being terminated prior to the planned end date.9Infliximab and etanercept have several case reports and a few case series highlighting potentially efficacious application of TNF-α inhibitors for the treatment of SJS/TEN.10-13 In 2002, Fischer et al10 reported the first case of TEN treated successfully with a single dose of infliximab 5 mg/kg. Kreft et al14 reported on etoricoxib-induced TEN that was treated with infliximab 5 mg/kg, which led to re-epithelialization within 5 weeks (notably a 5-week re-epithelialization time is not necessarily an improvement).
In 2005, Hunger et al3 demonstrated TNF-α’s release by KCs in the epidermis and by inflammatory cells in the dermis of a TEN patient. Twenty-four hours after the administration of infliximab 5 mg/kg in these patients, TNF-α was found to be below normal and epidermal detachment ceased.3 Wojtkietwicz et al13 demonstrated benefit following an infusion of infliximab 5 mg/kg in a patient whose disease continued to progress despite treatment with dexamethasone and 1.8 g/kg of IVIG.
Then 2 subsequent case series added further support for the efficacy of infliximab in the treatment of TEN. Patmanidis et al15 and Gaitanis et al16 reported similar results in 4 patients, each treated with infliximab 5 mg/kg immediately followed by initiation of high-dose IVIG (2 g/kg over 5 days). Zárate-Correa et al17 reported a 0% mortality rate and near-complete re-epithelialization after 5 to 14 days in 4 patients treated with a single 300-mg dose of infliximab.
However, the success of infliximab in the treatment of TEN has been countered by the pilot study by Paquet et al,18 which compared the efficacy of 150 mg/kg of N-acetylcysteine alone vs adding infliximab 5 mg/kg to treat 10 TEN patients. The study demonstrated no benefit at 48 hours in the group given infliximab, the time frame in which prior case reports touting infliximab’s benefit claimed the benefit was observed. Similarly, there was no effect on mortality for either treatment modality as assessed by illness auxiliary score.18
Evidence in support of the use of etanercept in the treatment of SJS/TEN is mounting, and some centers have begun to use it as the first-choice therapy for SJS/TEN. The first case was reported by Famularo et al,19 in which a patient with TEN was given 2 doses of etanercept 25 mg after failure to improve with prednisolone 1 mg/kg. The patient showed near-complete and rapid re-epithelization in 6 days before death due to disseminated intravascular coagulation 10 days after admission.19 Gubinelli et al20 and Sadighha21 independently reported cases of TEN and TEN/acute generalized exanthematous pustulosis overlap treated with a total of 50 mg of etanercept, demonstrating rapid cessation of lesion progression. Didona et al22 found similar benefit using etanercept 50 mg to treat TEN secondary to rituximab after failure to improve with prednisone and cyclophosphamide. Treatment of TEN with etanercept in an HIV-positive patient also has been reported. Lee et al23 described a patient who was administered 50-mg and 25-mg injections on days 3 and 5 of hospitalization, respectively, with re-epithelialization occurring by day 8. Finally, Owczarczyk-Saczonek et al24 reported a case of SJS in a patient with a 4-year history of etanercept and sulfasalazine treatment of rheumatoid arthritis; sulfasalazine was stopped, but this patient was continued on etanercept until resolution of skin and mucosal symptoms. However, it is important to consider the possibility of publication bias among these cases selected for their positive outcomes.
Perhaps the most compelling literature regarding the use of etanercept for TEN was described in a case series by Paradisi et al.2 This study included 10 patients with TEN, all of whom demonstrated complete re-epithelialization shortly after receiving etanercept 50 mg. Average SCORTEN was 3.6 with a range of 2 to 6. Eight patients in this study had severe comorbidities and all 10 patients survived, with a time to re-epithelialization ranging from 7 to 20 days.2 Additionally, a randomized controlled trial showed that 38 etanercept-treated patients had improved mortality (P=.266) and re-epithelialization time (P=.01) compared to patients treated with intravenous methylprednisolone.25Limitations to our study are similar to other reports of SJS/TEN and included the small number of cases and lack of randomization. Additionally, we do not have data available for all patients for time between onset of disease and treatment initiation. Because of these challenges, data presented in this case series is observational only. Additionally, the patients treated with etanercept alone had a slightly lower SCORTEN compared to the group that received IVIG or supportive care alone (2.1 and 2.4 respectively). However, the etanercept-only group actually had higher involvement of epidermal detachment (33%) compared to the non-etanercept group (23%).
Conclusion
Although treatment with etanercept lacks the support of a randomized controlled trial, similar to all other treatments currently used for SJS and TEN, preliminary reports highlight a benefit in disease progression and improvement in time to re-epithelialization. In particular, if etanercept 50 mg subcutaneously is given as monotherapy or is given early in the disease course (prior to other therapies being attempted and ideally within 6 hours of presentation), our data suggest an even greater trend toward improved mortality and decreased time to re-epithelialization. Additionally, our findings may suggest that in some patients, etanercept monotherapy is not an adequate intervention but the addition of IVIG may be helpful; however, the senior author (S.W.) notes anecdotally that in his experience with the patients treated at the University of California Los Angeles, the order of administration of combination therapies—etanercept followed by IVIG—was important in addition to the choice of therapy. These findings are promising enough to warrant a multicenter randomized controlled trial comparing the efficacy of etanercept to other more commonly used treatments for this spectrum of disease, including IVIG and/or cyclosporine. Based on the data presented in this case series, including the 13 patients who received etanercept and had a 0% mortality rate, etanercept may be viewed as a targeted therapeutic intervention for patients with SJS and TEN.
Regarded as dermatologic emergencies, Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) represent a spectrum of blistering skin diseases that have a high mortality rate. Because of a misguided immune response to medications or infections, CD8+ T lymphocytes release proinflammatory cytokines, giving rise to the extensive epidermal destruction seen in SJS and TEN. The exact pathogenesis of SJS and TEN is still poorly defined, but studies have proposed that T cells mediate keratinocyte (KC) apoptosis through perforin and granzyme release and activation of the Fas/Fas ligand (FasL). Functioning as a transmembrane death receptor in the tumor necrosis factor (TNF) superfamily, Fas (CD95) activates Fas-associated death domain protein, caspases, and nucleases, resulting in organized cell destruction. Likewise, perforin and granzymes also have been shown to play a similar role in apoptosis via activation of caspases.1
Evidence for the role of TNF-α in SJS and TEN has been supported by findings of elevated levels of TNF-α within the blister fluid, serum, and KC cell surface. Additionally, TNF-α has been shown to upregulate inducible nitric oxide synthase in KCs, causing an accumulation of nitric oxide and subsequent FasL-mediated cell death.1-3 Notably, studies have demonstrated a relative lack of lymphocytes in the tissue of TEN patients despite the extensive destruction that is observed, thus emphasizing the importance of amplification and cell signaling via inflammatory mediators such as TNF-α.1 In this proposed model, T cells release IFN-γ, causing KCs to release TNF-α that subsequently promotes the upregulation of the aforementioned FasL.1 Tumor necrosis factor α also may promote increased MHC class I complex deposition on KC surfaces that may play a role in perforin and granzyme-mediated apoptosis of KCs.1
There is still debate on the standard of care for the treatment of SJS and TEN, attributed to the absence of randomized controlled trials and the rarity of the disease as well as the numerous conflicting studies evaluating potential treatments.1,4 Despite conflicting data to support their use, supportive care and intravenous immunoglobulin (IVIG) continue to be common treatments for SJS and TEN in hospitals worldwide. Elucidation of the role of TNF-α has prompted the use of infliximab and etanercept. In a case series of Italian patients with TEN (average SCORTEN, 3.6) treated with the TNF-α antagonist etanercept, no mortality was observed, which was well below the calculated expected mortality of 46.9%.2 Our retrospective study compared the use of a TNF antagonist to other therapies in the treatment of SJS/TEN. Our data suggest that etanercept is a lifesaving and disease-modifying therapy.
Methods
Twenty-two patients with SJS/TEN were included in this analysis. This included all patients who carried a clinical diagnosis of SJS/TEN with a confirmatory biopsy at our 2 university centers—University of California, Los Angeles, and Keck-LA County-Norris Hospital at the University of Southern California, Los Angeles—from 2013 to 2016. The diagnosis was rendered when a clinical diagnosis of SJS/TEN was given by a dermatologist and a confirmatory biopsy was performed. Every patient given a diagnosis of SJS/TEN at either university system from 2015 onward received an injection of etanercept given the positive results reported by Paradisi et al.2
The 9 patients who presented from 2013 to 2014 to our 2 hospital systems and were given a diagnosis of SJS/TEN received either IVIG or supportive care alone and had an average body surface area (BSA) affected of 23%. The 13 patients who presented from 2015 to 2016 were treated with etanercept in the form of a 50-mg subcutaneous injection given once to the right upper arm. Of this group, 4 patients received dual therapy with both IVIG and etanercept. In the etanercept-treated group (etanercept alone and etanercept plus IVIG), the average BSA affected was 30%. At the time of preliminary diagnosis, all patient medications were evaluated for a possible temporal relationship to the onset of rash and were discontinued if felt to be causative. The causative agent and treatment course for each patient is summarized in Table 1.
Patients were monitored daily in the hospital for improvement, and time to re-epithelialization was measured. Re-epithelialization was defined as progressive healing with residual lesions (erosions, ulcers, or bullae) covering no more than 5% BSA and was contingent on the patient having no new lesions within 24 hours.5 SCORe of Toxic Epidermal Necrosis (SCORTEN), a validated severity-of-illness score,6 was calculated by giving 1 point for each of the following criteria at the time of diagnosis: age ≥40 years, concurrent malignancy, heart rate ≥120 beats/min, serum blood urea nitrogen >27 mg/dL, serum bicarbonate <20 mEq/L, serum glucose >250 mg/dL, and detached or compromised BSA >10%. The total SCORTEN was correlated with the following risk of mortality as supported by prior validation studies: SCORTEN of 0 to 1, 3.2%; SCORTEN of 2, 12.1%; SCORTEN of 3, 35.3%; SCORTEN of 4, 58.3%; SCORTEN of ≥5, >90%.
Results
A total of 13 patients received etanercept. The mean SCORTEN was 2.2. The observed mortality was 0%, which was markedly lower than the predicted mortality of 24.3% (as determined by linear interpolation). Of this cohort, 9 patients received etanercept alone (mean SCORTEN of 2.1, predicted mortality of 22.9%), whereas 4 patients received a combination of etanercept and IVIG (mean SCORTEN of 2.3, predicted mortality of 27.2%).
The 4 patients who received both etanercept and IVIG received dual therapy for varying reasons. In patient 2 (Table 1), the perceived severity of this case ultimately led to the decision to start IVIG in addition to etanercept, resulting in rapid recovery and discharge after only 1 week of hospitalization. Intravenous immunoglobulin also was given in patient 3 (SCORTEN of 4) and patient 6 (SCORTEN of 2) for progression of disease despite administration of etanercept, with subsequent cessation of progression after the addition of the second agent (IVIG). Patient 12 might have done well on etanercept monotherapy but was administered IVIG as a precautionary measure because of hospital treatment algorithms.
Nine patients did not receive etanercept. Of this group, 5 received IVIG and 4 were managed with supportive care alone. The average SCORTEN for this group was 2.4, only slightly higher than the group that received etanercept (Table 2). The mortality rate in this group was 33%, which was higher than the predicted mortality of 28.1%.
Re-epithelialization data were available for 8 patients who received etanercept. The average time to re-epithelialization for these patients was 8.9 days and ranged from 3 to 19 days. Of these patients, 2 received both IVIG and etanercept, with an average time to re-epithelialization of 13 days. For the 6 patients who received etanercept alone, the average time to re-epithelialization was 7.5 days. Re-epithelialization data were not available for any of the patients who received only IVIG or supportive care but to our recollection ranged from 14 to 21 days.
The clinical course of the 13 patients after the administration of a single dose of etanercept was remarkable, as there was complete absence of mortality and an increase in speed of recovery in most patients receiving this intervention (time to re-epithelialization, 3–19 days). We also observed another interesting trend from our patients treated with etanercept, which was the suggestion that treatment with etanercept may be less effective if IVIG and/or steroids are given prior to etanercept; likewise, treatment is more effective when etanercept is given quickly. For patients 1, 4, 5, 7, 9, and 11 (as shown in Table 1), no prior IVIG therapy or other immunosuppressive therapy had been given before etanercept was administered. In these 6 patients, the average time to re-epithelialization after etanercept administration was 7.5 days; average time to re-epithelialization, unfortunately, is not available for the patients who were not treated with etanercept. In addition, as shown in the Figure, it was noted in some patients that the depth of denudation was markedly more superficial than what would typically be clinically observed with TEN after administration of other immunomodulatory therapies such as IVIG or prednisone or with supportive care alone. In these 2 patients with superficial desquamation—patients 7 and 9—etanercept notably was given within 6 hours of onset of skin pain.
Comment
There is no definitive gold standard treatment of SJS, SJS/TEN overlap, or TEN. However, generally agreed upon management includes immediate discontinuation of the offending medication and supportive therapy with aggressive electrolyte replacement and wound care. Management in a burn unit or intensive care unit is recommended in severe cases. Contention over the efficacy of various medications in the treatment of SJS and TEN continues and largely is due to the rarity of SJS and TEN; studies are small and almost all lack randomization. Therapies that have been used include high-dose steroids, IVIG, plasmapheresis, cyclophosphamide, cyclosporine A, and TNF inhibitors (eg, etanercept, infliximab).1
Evidence for the use of anti–TNF-α antibodies has been limited thus far, with most of the literature focusing on infliximab and etanercept. Adalimumab, a fully humanized clonal antibody, has no reported cases in the dermatologic literature for use in patients with SJS/TEN. Two case reports of adalimumab paradoxically causing SJS have been documented. In both cases, adalimumab was stopped and patients responded to intravenous corticosteroids and infliximab.7,8 Similarly, thalidomide has not proven to be a promising anti–TNF-α agent for the treatment of SJS/TEN. In the only attempted randomized controlled trial for SJS and TEN, thalidomide appeared to increase mortality, eventuating in this trial being terminated prior to the planned end date.9Infliximab and etanercept have several case reports and a few case series highlighting potentially efficacious application of TNF-α inhibitors for the treatment of SJS/TEN.10-13 In 2002, Fischer et al10 reported the first case of TEN treated successfully with a single dose of infliximab 5 mg/kg. Kreft et al14 reported on etoricoxib-induced TEN that was treated with infliximab 5 mg/kg, which led to re-epithelialization within 5 weeks (notably a 5-week re-epithelialization time is not necessarily an improvement).
In 2005, Hunger et al3 demonstrated TNF-α’s release by KCs in the epidermis and by inflammatory cells in the dermis of a TEN patient. Twenty-four hours after the administration of infliximab 5 mg/kg in these patients, TNF-α was found to be below normal and epidermal detachment ceased.3 Wojtkietwicz et al13 demonstrated benefit following an infusion of infliximab 5 mg/kg in a patient whose disease continued to progress despite treatment with dexamethasone and 1.8 g/kg of IVIG.
Then 2 subsequent case series added further support for the efficacy of infliximab in the treatment of TEN. Patmanidis et al15 and Gaitanis et al16 reported similar results in 4 patients, each treated with infliximab 5 mg/kg immediately followed by initiation of high-dose IVIG (2 g/kg over 5 days). Zárate-Correa et al17 reported a 0% mortality rate and near-complete re-epithelialization after 5 to 14 days in 4 patients treated with a single 300-mg dose of infliximab.
However, the success of infliximab in the treatment of TEN has been countered by the pilot study by Paquet et al,18 which compared the efficacy of 150 mg/kg of N-acetylcysteine alone vs adding infliximab 5 mg/kg to treat 10 TEN patients. The study demonstrated no benefit at 48 hours in the group given infliximab, the time frame in which prior case reports touting infliximab’s benefit claimed the benefit was observed. Similarly, there was no effect on mortality for either treatment modality as assessed by illness auxiliary score.18
Evidence in support of the use of etanercept in the treatment of SJS/TEN is mounting, and some centers have begun to use it as the first-choice therapy for SJS/TEN. The first case was reported by Famularo et al,19 in which a patient with TEN was given 2 doses of etanercept 25 mg after failure to improve with prednisolone 1 mg/kg. The patient showed near-complete and rapid re-epithelization in 6 days before death due to disseminated intravascular coagulation 10 days after admission.19 Gubinelli et al20 and Sadighha21 independently reported cases of TEN and TEN/acute generalized exanthematous pustulosis overlap treated with a total of 50 mg of etanercept, demonstrating rapid cessation of lesion progression. Didona et al22 found similar benefit using etanercept 50 mg to treat TEN secondary to rituximab after failure to improve with prednisone and cyclophosphamide. Treatment of TEN with etanercept in an HIV-positive patient also has been reported. Lee et al23 described a patient who was administered 50-mg and 25-mg injections on days 3 and 5 of hospitalization, respectively, with re-epithelialization occurring by day 8. Finally, Owczarczyk-Saczonek et al24 reported a case of SJS in a patient with a 4-year history of etanercept and sulfasalazine treatment of rheumatoid arthritis; sulfasalazine was stopped, but this patient was continued on etanercept until resolution of skin and mucosal symptoms. However, it is important to consider the possibility of publication bias among these cases selected for their positive outcomes.
Perhaps the most compelling literature regarding the use of etanercept for TEN was described in a case series by Paradisi et al.2 This study included 10 patients with TEN, all of whom demonstrated complete re-epithelialization shortly after receiving etanercept 50 mg. Average SCORTEN was 3.6 with a range of 2 to 6. Eight patients in this study had severe comorbidities and all 10 patients survived, with a time to re-epithelialization ranging from 7 to 20 days.2 Additionally, a randomized controlled trial showed that 38 etanercept-treated patients had improved mortality (P=.266) and re-epithelialization time (P=.01) compared to patients treated with intravenous methylprednisolone.25Limitations to our study are similar to other reports of SJS/TEN and included the small number of cases and lack of randomization. Additionally, we do not have data available for all patients for time between onset of disease and treatment initiation. Because of these challenges, data presented in this case series is observational only. Additionally, the patients treated with etanercept alone had a slightly lower SCORTEN compared to the group that received IVIG or supportive care alone (2.1 and 2.4 respectively). However, the etanercept-only group actually had higher involvement of epidermal detachment (33%) compared to the non-etanercept group (23%).
Conclusion
Although treatment with etanercept lacks the support of a randomized controlled trial, similar to all other treatments currently used for SJS and TEN, preliminary reports highlight a benefit in disease progression and improvement in time to re-epithelialization. In particular, if etanercept 50 mg subcutaneously is given as monotherapy or is given early in the disease course (prior to other therapies being attempted and ideally within 6 hours of presentation), our data suggest an even greater trend toward improved mortality and decreased time to re-epithelialization. Additionally, our findings may suggest that in some patients, etanercept monotherapy is not an adequate intervention but the addition of IVIG may be helpful; however, the senior author (S.W.) notes anecdotally that in his experience with the patients treated at the University of California Los Angeles, the order of administration of combination therapies—etanercept followed by IVIG—was important in addition to the choice of therapy. These findings are promising enough to warrant a multicenter randomized controlled trial comparing the efficacy of etanercept to other more commonly used treatments for this spectrum of disease, including IVIG and/or cyclosporine. Based on the data presented in this case series, including the 13 patients who received etanercept and had a 0% mortality rate, etanercept may be viewed as a targeted therapeutic intervention for patients with SJS and TEN.
- Pereira FA, Mudgil AV, Rosmarin DM. Toxic epidermal necrolysis. J Am Acad Dermatol. 2007;56:181-200.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Hunger RE, Hunziker T, Buettiker U, et al. Rapid resolution of toxic epidermal necrolysis with anti-TNF-α treatment. J Allergy Clin Immunol. 2005;116:923-924.
- Worswick S, Cotliar J. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of treatment options. Dermatol Ther. 2011;24:207-218.
- Wallace AB. The exposure treatment of burns. Lancet Lond Engl. 1951;1:501-504.
- Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153.
- Mounach A, Rezqi A, Nouijai A, et al. Stevens-Johnson syndrome complicating adalimumab therapy in rheumatoid arthritis disease. Rheumatol Int. 2013;33:1351-1353.
- Salama M, Lawrance I-C. Stevens-Johnson syndrome complicating adalimumab therapy in Crohn’s disease. World J Gastroenterol. 2009;15:4449-4452.
- Wolkenstein P, Latarjet J, Roujeau JC, et al. Randomised comparison of thalidomide versus placebo in toxic epidermal necrolysis. Lancet Lond Engl. 1998;352:1586-1589.
- Fischer M, Fiedler E, Marsch WC, et al Antitumour necrosis factor-α antibodies (infliximab) in the treatment of a patient with toxic epidermal necrolysis. Br J Dermatol. 2002;146:707-709.
- Meiss F, Helmbold P, Meykadeh N, et al. Overlap of acute generalized exanthematous pustulosis and toxic epidermal necrolysis: response to antitumour necrosis factor-alpha antibody infliximab: report of three cases. J Eur Acad Dermatol Venereol. 2007;21:717-719.
- Al-Shouli S, Abouchala N, Bogusz MJ, et al. Toxic epidermal necrolysis associated with high intake of sildenafil and its response to infliximab. Acta Derm Venereol. 2005;85:534-535.
- Wojtkiewicz A, Wysocki M, Fortuna J, et al. Beneficial and rapid effect of infliximab on the course of toxic epidermal necrolysis. Acta Derm Venereol. 2008;88:420-421.
- Kreft B, Wohlrab J, Bramsiepe I, et al. Etoricoxib-induced toxic epidermal necrolysis: successful treatment with infliximab. J Dermatol. 2010;37:904-906.
- Patmanidis K, Sidiras A, Dolianitis K, et al. Combination of infliximab and high-dose intravenous immunoglobulin for toxic epidermal necrolysis: successful treatment of an elderly patient. Case Rep Dermatol Med. 2012;2012:915314.
- Gaitanis G, Spyridonos P, Patmanidis K, et al. Treatment of toxic epidermal necrolysis with the combination of infliximab and high-dose intravenous immunoglobulin. Dermatol Basel Switz. 2012;224:134-139.
- Zárate-Correa LC, Carrillo-Gómez DC, Ramírez-Escobar AF, et al. Toxic epidermal necrolysis successfully treated with infliximab. J Investig Allergol Clin Immunol. 2013;23:61-63.
- Paquet P, Jennes S, Rousseau AF, et al. Effect of N-acetylcysteine combined with infliximab on toxic epidermal necrolysis. a proof-of-concept study. Burns J Int Soc Burn Inj. 2014;40:1707-1712.
- Famularo G, Dona BD, Canzona F, et al. Etanercept for toxic epidermal necrolysis. Ann Pharmacother. 2007;41:1083-1084.
- Gubinelli E, Canzona F, Tonanzi T, et al. Toxic epidermal necrolysis successfully treated with etanercept. J Dermatol. 2009;36:150-153.
- Sadighha A. Etanercept in the treatment of a patient with acute generalized exanthematous pustulosis/toxic epidermal necrolysis: definition of a new model based on translational research. Int J Dermatol. 2009;48:913-914.
- Didona D, Paolino G, Garcovich S, et al. Successful use of etanercept in a case of toxic epidermal necrolysis induced by rituximab. J Eur Acad Dermatol Venereol. 2016;30:E83-E84.
- Lee Y-Y, Ko J-H, Wei C-H, et al. Use of etanercept to treat toxic epidermal necrolysis in a human immunodeficiency virus-positive patient. Dermatol Sin. 2013;31:78-81.
- Owczarczyk-Saczonek A, Zdanowska N, Znajewska-Pander A, et al. Stevens-Johnson syndrome in a patient with rheumatoid arthritis during long-term etanercept therapy. J Dermatol Case Rep. 2016;10:14-16.
- Wang CW, Yang LY, Chen CB, et al. Randomized, controlled trial of TNF-α antagonist in CTL mediated severe cutaneous adverse reactions. J Clin Invest. 2018;128:985-996.
- Pereira FA, Mudgil AV, Rosmarin DM. Toxic epidermal necrolysis. J Am Acad Dermatol. 2007;56:181-200.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Hunger RE, Hunziker T, Buettiker U, et al. Rapid resolution of toxic epidermal necrolysis with anti-TNF-α treatment. J Allergy Clin Immunol. 2005;116:923-924.
- Worswick S, Cotliar J. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of treatment options. Dermatol Ther. 2011;24:207-218.
- Wallace AB. The exposure treatment of burns. Lancet Lond Engl. 1951;1:501-504.
- Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153.
- Mounach A, Rezqi A, Nouijai A, et al. Stevens-Johnson syndrome complicating adalimumab therapy in rheumatoid arthritis disease. Rheumatol Int. 2013;33:1351-1353.
- Salama M, Lawrance I-C. Stevens-Johnson syndrome complicating adalimumab therapy in Crohn’s disease. World J Gastroenterol. 2009;15:4449-4452.
- Wolkenstein P, Latarjet J, Roujeau JC, et al. Randomised comparison of thalidomide versus placebo in toxic epidermal necrolysis. Lancet Lond Engl. 1998;352:1586-1589.
- Fischer M, Fiedler E, Marsch WC, et al Antitumour necrosis factor-α antibodies (infliximab) in the treatment of a patient with toxic epidermal necrolysis. Br J Dermatol. 2002;146:707-709.
- Meiss F, Helmbold P, Meykadeh N, et al. Overlap of acute generalized exanthematous pustulosis and toxic epidermal necrolysis: response to antitumour necrosis factor-alpha antibody infliximab: report of three cases. J Eur Acad Dermatol Venereol. 2007;21:717-719.
- Al-Shouli S, Abouchala N, Bogusz MJ, et al. Toxic epidermal necrolysis associated with high intake of sildenafil and its response to infliximab. Acta Derm Venereol. 2005;85:534-535.
- Wojtkiewicz A, Wysocki M, Fortuna J, et al. Beneficial and rapid effect of infliximab on the course of toxic epidermal necrolysis. Acta Derm Venereol. 2008;88:420-421.
- Kreft B, Wohlrab J, Bramsiepe I, et al. Etoricoxib-induced toxic epidermal necrolysis: successful treatment with infliximab. J Dermatol. 2010;37:904-906.
- Patmanidis K, Sidiras A, Dolianitis K, et al. Combination of infliximab and high-dose intravenous immunoglobulin for toxic epidermal necrolysis: successful treatment of an elderly patient. Case Rep Dermatol Med. 2012;2012:915314.
- Gaitanis G, Spyridonos P, Patmanidis K, et al. Treatment of toxic epidermal necrolysis with the combination of infliximab and high-dose intravenous immunoglobulin. Dermatol Basel Switz. 2012;224:134-139.
- Zárate-Correa LC, Carrillo-Gómez DC, Ramírez-Escobar AF, et al. Toxic epidermal necrolysis successfully treated with infliximab. J Investig Allergol Clin Immunol. 2013;23:61-63.
- Paquet P, Jennes S, Rousseau AF, et al. Effect of N-acetylcysteine combined with infliximab on toxic epidermal necrolysis. a proof-of-concept study. Burns J Int Soc Burn Inj. 2014;40:1707-1712.
- Famularo G, Dona BD, Canzona F, et al. Etanercept for toxic epidermal necrolysis. Ann Pharmacother. 2007;41:1083-1084.
- Gubinelli E, Canzona F, Tonanzi T, et al. Toxic epidermal necrolysis successfully treated with etanercept. J Dermatol. 2009;36:150-153.
- Sadighha A. Etanercept in the treatment of a patient with acute generalized exanthematous pustulosis/toxic epidermal necrolysis: definition of a new model based on translational research. Int J Dermatol. 2009;48:913-914.
- Didona D, Paolino G, Garcovich S, et al. Successful use of etanercept in a case of toxic epidermal necrolysis induced by rituximab. J Eur Acad Dermatol Venereol. 2016;30:E83-E84.
- Lee Y-Y, Ko J-H, Wei C-H, et al. Use of etanercept to treat toxic epidermal necrolysis in a human immunodeficiency virus-positive patient. Dermatol Sin. 2013;31:78-81.
- Owczarczyk-Saczonek A, Zdanowska N, Znajewska-Pander A, et al. Stevens-Johnson syndrome in a patient with rheumatoid arthritis during long-term etanercept therapy. J Dermatol Case Rep. 2016;10:14-16.
- Wang CW, Yang LY, Chen CB, et al. Randomized, controlled trial of TNF-α antagonist in CTL mediated severe cutaneous adverse reactions. J Clin Invest. 2018;128:985-996.
Practice Points
- Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are life-threatening dermatologic emergencies without a universally accepted treatment.
- Results of this study support the use of single-dose subcutaneous etanercept 50 mg as a potentially lifesaving therapy for patients with SJS/TEN.
Dermatopathology Etiquette 101
The Accreditation Council for Graduate Medical Education has established core competencies to serve as a foundation for the training received in a dermatology residency program.1 Although programs are required to have the same concentrations—patient care, medical knowledge, practice-based learning and improvement, interpersonal and communication skills, professionalism, and systems-based practice—no specific guidelines are in place regarding how each of these competencies should be reached within a training period.2 Instead, it remains the responsibility of each program to formulate an individualized curriculum to facilitate proficiency in the multiple areas encompassed by a residency.
In many dermatology residency programs, dermatopathology is a substantial component of educational objectives and the curriculum.1 Residents may spend as much as 25% of their training on dermatopathology. However, there is great variability among programs in methods of teaching dermatopathology. When Hinshaw3 surveyed 52 of 109 dermatology residency programs, they identified differences in dermatopathology teaching that included, but was not limited to, utilization of problem-based learning (in 40.4% of programs), integration of journal reviews (53.8%), and computer-based learning (19.2%). In addition, differences were identified in the recommended primary textbook and the makeup of faculty who taught dermatopathology.3
Although residency programs vary in their methods of teaching this important component of dermatology, most use a multiheaded microscope in some capacity for didactics or sign-out. For most trainees, the dermatopathology laboratory is a new environment compared to the clinical space that medical students and residents become accustomed to throughout their education, thus creating a knowledge gap for trainees on proper dermatopathology etiquette and universal guidelines.
With medical students, residents, and fellows in mind, we have prepared a basic “dermatopathology etiquette” reference for trainees. Just as there are universal rules in the operating room for surgery (eg, sterile technique), we want to establish a code of conduct at the microscope. We hope that these 10 tips will, first, be useful to those who are unsure how to approach their first experience with dermatopathology and, second, serve as a guideline to aid development of appropriate communication skills and functioning within this novel setting. This list also can serve as a resource for dermatopathology attendings to provide to rotating residents and students.
1. New to pathology? It’s okay to ask. Do not hesitate to ask upper-year residents, fellows, and attendings for instructions on such matters as how to adjust your eyepiece to get the best resolution.
2. If a slide drops on the floor, do not move! Your first instinct might be to move your chair to look for the dropped slide, but you might roll over it and break it.
3. When the attending is looking through the scope, you look through the scope. Dermatopathology is a visual exercise. Getting in your “optic mileage” is best done under the guidance of an experienced dermatopathologist.
4. Rules regarding food and drink at the microscope vary by pathologist. It’s best to ask what each attending prefers. Safe advice is to avoid foods that make noise, such as chewing gum and chips, and food that has a strong odor, such as microwaved leftovers.
5. Limit use of a laptop, cell phone, and smartwatch. If you think that using any of these is necessary, it generally is best to announce that you are looking up something related to the case and then share your findings (but not the most recent post on your Facebook News Feed).
6. If you notice that something needs correcting on the report, speak up! We are all human; we all make typos. Do not hesitate to mention this as soon as possible, especially before the case is signed out. You will likely be thanked by your attending because it is harder to rectify once the report has been signed out.
7. Small talk often is welcome during large excisions. This is a great time to ask what others are doing next weekend or what happened in clinic earlier that day, or just to tell a good (clean) joke that is making the rounds. Conversely, if the case is complex, it often is best to wait until it is completed before asking questions.
8. When participating in a roundtable diagnosis, you are welcome to directly state the diagnosis for bread-and-butter cases, such as basal cell carcinomas and seborrheic keratoses. It is appropriate to be more descriptive and methodical in more complex cases. When evaluating a rash, give the general inflammatory pattern first. For example, is it spongiotic? Psoriasiform? Interface? Or a mixed pattern?
9. Extra points for identifying special sites! These include mucosal, genital, and acral sites. You might even get bonus points if you can determine something about the patient (child or adult) based on the pathologic features, such as variation in collagen patterns.
10. Whenever you are in doubt, just describe what you see. You can use the traditional top-down approach or start with stating the most evident finding, then proceed to a top-down description. If it is a neoplasm, describe the overall architecture; then, what you see at a cellular level will get you some points as well.
We acknowledge that this list of 10 tips is not comprehensive and might vary by attending and each institution’s distinctive training format. We are hopeful, however, that these 10 points of etiquette can serve as a guideline.
- Hinshaw M, Hsu P, Lee L-Y, et al. The current state of dermatopathology education: a survey of the Association of Professors of Dermatology. J Cutan Pathol. 2009;36:620-628. doi:10.1111/j.1600-0560.2008.01128.x
- Hinshaw MA, Stratman EJ. Core competencies in dermatopathology. J Cutan Pathol. 2006;33:160-165. doi:10.1111/j.0303-6987.2006.00442.x
- Hinshaw MA. Dermatopathology education: an update. Dermatol Clin. 2012;30:815-826. doi:10.1016/j.det.2012.06.003
The Accreditation Council for Graduate Medical Education has established core competencies to serve as a foundation for the training received in a dermatology residency program.1 Although programs are required to have the same concentrations—patient care, medical knowledge, practice-based learning and improvement, interpersonal and communication skills, professionalism, and systems-based practice—no specific guidelines are in place regarding how each of these competencies should be reached within a training period.2 Instead, it remains the responsibility of each program to formulate an individualized curriculum to facilitate proficiency in the multiple areas encompassed by a residency.
In many dermatology residency programs, dermatopathology is a substantial component of educational objectives and the curriculum.1 Residents may spend as much as 25% of their training on dermatopathology. However, there is great variability among programs in methods of teaching dermatopathology. When Hinshaw3 surveyed 52 of 109 dermatology residency programs, they identified differences in dermatopathology teaching that included, but was not limited to, utilization of problem-based learning (in 40.4% of programs), integration of journal reviews (53.8%), and computer-based learning (19.2%). In addition, differences were identified in the recommended primary textbook and the makeup of faculty who taught dermatopathology.3
Although residency programs vary in their methods of teaching this important component of dermatology, most use a multiheaded microscope in some capacity for didactics or sign-out. For most trainees, the dermatopathology laboratory is a new environment compared to the clinical space that medical students and residents become accustomed to throughout their education, thus creating a knowledge gap for trainees on proper dermatopathology etiquette and universal guidelines.
With medical students, residents, and fellows in mind, we have prepared a basic “dermatopathology etiquette” reference for trainees. Just as there are universal rules in the operating room for surgery (eg, sterile technique), we want to establish a code of conduct at the microscope. We hope that these 10 tips will, first, be useful to those who are unsure how to approach their first experience with dermatopathology and, second, serve as a guideline to aid development of appropriate communication skills and functioning within this novel setting. This list also can serve as a resource for dermatopathology attendings to provide to rotating residents and students.
1. New to pathology? It’s okay to ask. Do not hesitate to ask upper-year residents, fellows, and attendings for instructions on such matters as how to adjust your eyepiece to get the best resolution.
2. If a slide drops on the floor, do not move! Your first instinct might be to move your chair to look for the dropped slide, but you might roll over it and break it.
3. When the attending is looking through the scope, you look through the scope. Dermatopathology is a visual exercise. Getting in your “optic mileage” is best done under the guidance of an experienced dermatopathologist.
4. Rules regarding food and drink at the microscope vary by pathologist. It’s best to ask what each attending prefers. Safe advice is to avoid foods that make noise, such as chewing gum and chips, and food that has a strong odor, such as microwaved leftovers.
5. Limit use of a laptop, cell phone, and smartwatch. If you think that using any of these is necessary, it generally is best to announce that you are looking up something related to the case and then share your findings (but not the most recent post on your Facebook News Feed).
6. If you notice that something needs correcting on the report, speak up! We are all human; we all make typos. Do not hesitate to mention this as soon as possible, especially before the case is signed out. You will likely be thanked by your attending because it is harder to rectify once the report has been signed out.
7. Small talk often is welcome during large excisions. This is a great time to ask what others are doing next weekend or what happened in clinic earlier that day, or just to tell a good (clean) joke that is making the rounds. Conversely, if the case is complex, it often is best to wait until it is completed before asking questions.
8. When participating in a roundtable diagnosis, you are welcome to directly state the diagnosis for bread-and-butter cases, such as basal cell carcinomas and seborrheic keratoses. It is appropriate to be more descriptive and methodical in more complex cases. When evaluating a rash, give the general inflammatory pattern first. For example, is it spongiotic? Psoriasiform? Interface? Or a mixed pattern?
9. Extra points for identifying special sites! These include mucosal, genital, and acral sites. You might even get bonus points if you can determine something about the patient (child or adult) based on the pathologic features, such as variation in collagen patterns.
10. Whenever you are in doubt, just describe what you see. You can use the traditional top-down approach or start with stating the most evident finding, then proceed to a top-down description. If it is a neoplasm, describe the overall architecture; then, what you see at a cellular level will get you some points as well.
We acknowledge that this list of 10 tips is not comprehensive and might vary by attending and each institution’s distinctive training format. We are hopeful, however, that these 10 points of etiquette can serve as a guideline.
The Accreditation Council for Graduate Medical Education has established core competencies to serve as a foundation for the training received in a dermatology residency program.1 Although programs are required to have the same concentrations—patient care, medical knowledge, practice-based learning and improvement, interpersonal and communication skills, professionalism, and systems-based practice—no specific guidelines are in place regarding how each of these competencies should be reached within a training period.2 Instead, it remains the responsibility of each program to formulate an individualized curriculum to facilitate proficiency in the multiple areas encompassed by a residency.
In many dermatology residency programs, dermatopathology is a substantial component of educational objectives and the curriculum.1 Residents may spend as much as 25% of their training on dermatopathology. However, there is great variability among programs in methods of teaching dermatopathology. When Hinshaw3 surveyed 52 of 109 dermatology residency programs, they identified differences in dermatopathology teaching that included, but was not limited to, utilization of problem-based learning (in 40.4% of programs), integration of journal reviews (53.8%), and computer-based learning (19.2%). In addition, differences were identified in the recommended primary textbook and the makeup of faculty who taught dermatopathology.3
Although residency programs vary in their methods of teaching this important component of dermatology, most use a multiheaded microscope in some capacity for didactics or sign-out. For most trainees, the dermatopathology laboratory is a new environment compared to the clinical space that medical students and residents become accustomed to throughout their education, thus creating a knowledge gap for trainees on proper dermatopathology etiquette and universal guidelines.
With medical students, residents, and fellows in mind, we have prepared a basic “dermatopathology etiquette” reference for trainees. Just as there are universal rules in the operating room for surgery (eg, sterile technique), we want to establish a code of conduct at the microscope. We hope that these 10 tips will, first, be useful to those who are unsure how to approach their first experience with dermatopathology and, second, serve as a guideline to aid development of appropriate communication skills and functioning within this novel setting. This list also can serve as a resource for dermatopathology attendings to provide to rotating residents and students.
1. New to pathology? It’s okay to ask. Do not hesitate to ask upper-year residents, fellows, and attendings for instructions on such matters as how to adjust your eyepiece to get the best resolution.
2. If a slide drops on the floor, do not move! Your first instinct might be to move your chair to look for the dropped slide, but you might roll over it and break it.
3. When the attending is looking through the scope, you look through the scope. Dermatopathology is a visual exercise. Getting in your “optic mileage” is best done under the guidance of an experienced dermatopathologist.
4. Rules regarding food and drink at the microscope vary by pathologist. It’s best to ask what each attending prefers. Safe advice is to avoid foods that make noise, such as chewing gum and chips, and food that has a strong odor, such as microwaved leftovers.
5. Limit use of a laptop, cell phone, and smartwatch. If you think that using any of these is necessary, it generally is best to announce that you are looking up something related to the case and then share your findings (but not the most recent post on your Facebook News Feed).
6. If you notice that something needs correcting on the report, speak up! We are all human; we all make typos. Do not hesitate to mention this as soon as possible, especially before the case is signed out. You will likely be thanked by your attending because it is harder to rectify once the report has been signed out.
7. Small talk often is welcome during large excisions. This is a great time to ask what others are doing next weekend or what happened in clinic earlier that day, or just to tell a good (clean) joke that is making the rounds. Conversely, if the case is complex, it often is best to wait until it is completed before asking questions.
8. When participating in a roundtable diagnosis, you are welcome to directly state the diagnosis for bread-and-butter cases, such as basal cell carcinomas and seborrheic keratoses. It is appropriate to be more descriptive and methodical in more complex cases. When evaluating a rash, give the general inflammatory pattern first. For example, is it spongiotic? Psoriasiform? Interface? Or a mixed pattern?
9. Extra points for identifying special sites! These include mucosal, genital, and acral sites. You might even get bonus points if you can determine something about the patient (child or adult) based on the pathologic features, such as variation in collagen patterns.
10. Whenever you are in doubt, just describe what you see. You can use the traditional top-down approach or start with stating the most evident finding, then proceed to a top-down description. If it is a neoplasm, describe the overall architecture; then, what you see at a cellular level will get you some points as well.
We acknowledge that this list of 10 tips is not comprehensive and might vary by attending and each institution’s distinctive training format. We are hopeful, however, that these 10 points of etiquette can serve as a guideline.
- Hinshaw M, Hsu P, Lee L-Y, et al. The current state of dermatopathology education: a survey of the Association of Professors of Dermatology. J Cutan Pathol. 2009;36:620-628. doi:10.1111/j.1600-0560.2008.01128.x
- Hinshaw MA, Stratman EJ. Core competencies in dermatopathology. J Cutan Pathol. 2006;33:160-165. doi:10.1111/j.0303-6987.2006.00442.x
- Hinshaw MA. Dermatopathology education: an update. Dermatol Clin. 2012;30:815-826. doi:10.1016/j.det.2012.06.003
- Hinshaw M, Hsu P, Lee L-Y, et al. The current state of dermatopathology education: a survey of the Association of Professors of Dermatology. J Cutan Pathol. 2009;36:620-628. doi:10.1111/j.1600-0560.2008.01128.x
- Hinshaw MA, Stratman EJ. Core competencies in dermatopathology. J Cutan Pathol. 2006;33:160-165. doi:10.1111/j.0303-6987.2006.00442.x
- Hinshaw MA. Dermatopathology education: an update. Dermatol Clin. 2012;30:815-826. doi:10.1016/j.det.2012.06.003
Wiping Away Cellulitis: A Case of Factitious Disorder
To the Editor:
Patients with psychocutaneous disorders present unique challenges to physicians. We illustrate the critical role that dermoscopy may play to illuminate exogenous skin pathology.
A 50-year-old woman with a reported medical history of systemic lupus erythematosus, chronic pain, and nonhealing leg ulcers presented to the emergency department with severe pain of the left lower leg and redness that was concerning for cellulitis. She sought treatment at an outside hospital for cellulitis 2 weeks prior but left against medical advice. Symptomatic review revealed chest pain, shortness of breath, nausea, vomiting, and diarrhea. The primary team started her on intravenous clindamycin and vancomycin for the presumed infection and scheduled narcotic medications due to concerns of intractable pain in the left leg. The dermatology department was consulted after failure to improve with 1 week of systemic antibiotics.
Physical examination revealed a geometric, atrophic, purple plaque on the left anterior shin from a prior leg ulcer as well as a diffuse red-pink patch extending from the knee to the ankle. Notably, the cellulitis spared the left posterior calf resting against the sheet and had a sharp line of demarcation at the distal shin. The leg was cool to the touch while the patient was distractible. She later reported that the leg was extremely tender to palpation. Dermoscopy revealed linear red pigments within skin furrows that accentuated skin lines (Figure). These findings raised suspicions of an external manipulation. The skin was wiped with an alcohol pad that removed a shimmering pink substance consistent in appearance to a cosmetic product. The skin beneath the cellulitis appeared normal.
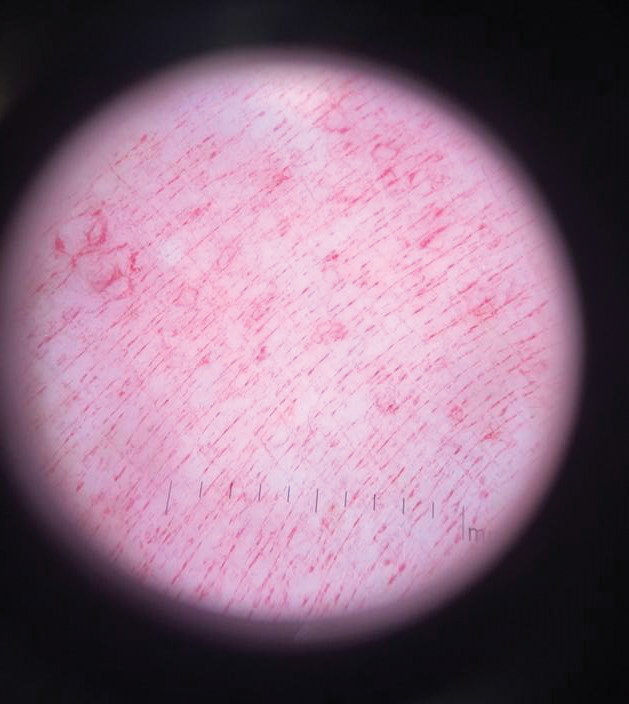
On further review of the patient’s medical record, it was noted that she was admitted several months ago for ulcers of the left leg. She had been to multiple hospitals and had numerous rounds of antibiotics. Biopsy of an ulcer revealed dermal fibrosis consistent with scarring. Aerobic bacteria, atypical mycobacteria, and fungal cultures were all negative. The physicians suspected a self-induced etiology consistent with dermatitis artefacta. The patient emphasized multiple psychosocial stressors as well as having frequent lupus flares despite repeated negative workup. Given the exaggerated symptoms and unnecessary hospital visits, she was given the diagnosis of factitious disorder (malingering or Munchausen syndrome). After extensive discussion, the patient was amenable to outpatient mental health counseling.
Dermoscopy is not a standard method to diagnose cellulitis of the skin; however, when patients present with an atypical response to appropriate care, the presumed diagnosis must be challenged. This patient had dramatized symptoms, false medical history, and numerous hospitalizations that were suspicious for factitious disorder.1 Furthermore, the physical examination was inconsistent with the classic course of cellulitis. In this case, dermoscopy had advantages over biopsies because it was noninvasive, gave immediate feedback, and provided a macroscopic view of the morphology. Via dermoscopy, we had an objective lens to distinguish cellulitis from cosmetic product and to obtain the correct diagnosis.
- Harth W, Taube KM, Gieler U. Facticious disorders in dermatology. J Dtsch Dermatol Ges. 2010;8:361-372.
To the Editor:
Patients with psychocutaneous disorders present unique challenges to physicians. We illustrate the critical role that dermoscopy may play to illuminate exogenous skin pathology.
A 50-year-old woman with a reported medical history of systemic lupus erythematosus, chronic pain, and nonhealing leg ulcers presented to the emergency department with severe pain of the left lower leg and redness that was concerning for cellulitis. She sought treatment at an outside hospital for cellulitis 2 weeks prior but left against medical advice. Symptomatic review revealed chest pain, shortness of breath, nausea, vomiting, and diarrhea. The primary team started her on intravenous clindamycin and vancomycin for the presumed infection and scheduled narcotic medications due to concerns of intractable pain in the left leg. The dermatology department was consulted after failure to improve with 1 week of systemic antibiotics.
Physical examination revealed a geometric, atrophic, purple plaque on the left anterior shin from a prior leg ulcer as well as a diffuse red-pink patch extending from the knee to the ankle. Notably, the cellulitis spared the left posterior calf resting against the sheet and had a sharp line of demarcation at the distal shin. The leg was cool to the touch while the patient was distractible. She later reported that the leg was extremely tender to palpation. Dermoscopy revealed linear red pigments within skin furrows that accentuated skin lines (Figure). These findings raised suspicions of an external manipulation. The skin was wiped with an alcohol pad that removed a shimmering pink substance consistent in appearance to a cosmetic product. The skin beneath the cellulitis appeared normal.
On further review of the patient’s medical record, it was noted that she was admitted several months ago for ulcers of the left leg. She had been to multiple hospitals and had numerous rounds of antibiotics. Biopsy of an ulcer revealed dermal fibrosis consistent with scarring. Aerobic bacteria, atypical mycobacteria, and fungal cultures were all negative. The physicians suspected a self-induced etiology consistent with dermatitis artefacta. The patient emphasized multiple psychosocial stressors as well as having frequent lupus flares despite repeated negative workup. Given the exaggerated symptoms and unnecessary hospital visits, she was given the diagnosis of factitious disorder (malingering or Munchausen syndrome). After extensive discussion, the patient was amenable to outpatient mental health counseling.
Dermoscopy is not a standard method to diagnose cellulitis of the skin; however, when patients present with an atypical response to appropriate care, the presumed diagnosis must be challenged. This patient had dramatized symptoms, false medical history, and numerous hospitalizations that were suspicious for factitious disorder.1 Furthermore, the physical examination was inconsistent with the classic course of cellulitis. In this case, dermoscopy had advantages over biopsies because it was noninvasive, gave immediate feedback, and provided a macroscopic view of the morphology. Via dermoscopy, we had an objective lens to distinguish cellulitis from cosmetic product and to obtain the correct diagnosis.
To the Editor:
Patients with psychocutaneous disorders present unique challenges to physicians. We illustrate the critical role that dermoscopy may play to illuminate exogenous skin pathology.
A 50-year-old woman with a reported medical history of systemic lupus erythematosus, chronic pain, and nonhealing leg ulcers presented to the emergency department with severe pain of the left lower leg and redness that was concerning for cellulitis. She sought treatment at an outside hospital for cellulitis 2 weeks prior but left against medical advice. Symptomatic review revealed chest pain, shortness of breath, nausea, vomiting, and diarrhea. The primary team started her on intravenous clindamycin and vancomycin for the presumed infection and scheduled narcotic medications due to concerns of intractable pain in the left leg. The dermatology department was consulted after failure to improve with 1 week of systemic antibiotics.
Physical examination revealed a geometric, atrophic, purple plaque on the left anterior shin from a prior leg ulcer as well as a diffuse red-pink patch extending from the knee to the ankle. Notably, the cellulitis spared the left posterior calf resting against the sheet and had a sharp line of demarcation at the distal shin. The leg was cool to the touch while the patient was distractible. She later reported that the leg was extremely tender to palpation. Dermoscopy revealed linear red pigments within skin furrows that accentuated skin lines (Figure). These findings raised suspicions of an external manipulation. The skin was wiped with an alcohol pad that removed a shimmering pink substance consistent in appearance to a cosmetic product. The skin beneath the cellulitis appeared normal.
On further review of the patient’s medical record, it was noted that she was admitted several months ago for ulcers of the left leg. She had been to multiple hospitals and had numerous rounds of antibiotics. Biopsy of an ulcer revealed dermal fibrosis consistent with scarring. Aerobic bacteria, atypical mycobacteria, and fungal cultures were all negative. The physicians suspected a self-induced etiology consistent with dermatitis artefacta. The patient emphasized multiple psychosocial stressors as well as having frequent lupus flares despite repeated negative workup. Given the exaggerated symptoms and unnecessary hospital visits, she was given the diagnosis of factitious disorder (malingering or Munchausen syndrome). After extensive discussion, the patient was amenable to outpatient mental health counseling.
Dermoscopy is not a standard method to diagnose cellulitis of the skin; however, when patients present with an atypical response to appropriate care, the presumed diagnosis must be challenged. This patient had dramatized symptoms, false medical history, and numerous hospitalizations that were suspicious for factitious disorder.1 Furthermore, the physical examination was inconsistent with the classic course of cellulitis. In this case, dermoscopy had advantages over biopsies because it was noninvasive, gave immediate feedback, and provided a macroscopic view of the morphology. Via dermoscopy, we had an objective lens to distinguish cellulitis from cosmetic product and to obtain the correct diagnosis.
- Harth W, Taube KM, Gieler U. Facticious disorders in dermatology. J Dtsch Dermatol Ges. 2010;8:361-372.
- Harth W, Taube KM, Gieler U. Facticious disorders in dermatology. J Dtsch Dermatol Ges. 2010;8:361-372.
Practice Points
- Consider exogenous factors or alternative diagnoses when a patient does not respond to appropriate care.
- Although dermoscopy is not used to diagnose cellulitis, it could be helpful in distinguishing cosmetic products used in dermatitis artefacta.
Pigmented Basal Cell Carcinoma With Annular Leukoderma
To the Editor:
Annular leukoderma, or the halo phenomenon, is a circular reaction of hypopigmentation that most commonly is observed alongside congenital nevi, acquired melanocytic nevi, blue nevi, Spitz nevi, vitiligo, and rarely melanoma.1 There is limited literature on the mechanism of the halo phenomenon. Most of the literature proposes a T cell–mediated immune response to antigens, which causes not only surrounding pigment loss but also heralds the regression of central lesions.2 Others have suggested a vascular mechanism, with blood shunted away from the lesions.3 Because guidelines discourage biopsy of typical halo nevi, it becomes important to evaluate lesions for worrisome features such as ulceration or asymmetry, especially in older patients. We present a case of a pigmented basal cell carcinoma (BCC) that exhibited the halo phenomenon. Four other cases have been described in the literature.3-6
A 53-year-old man presented for evaluation of an asymptomatic lesion on the left side of the abdomen of approximately 8 months’ duration. He had no personal or family history of skin cancer. Physical examination revealed a central 1-cm, pink, verrucous papule surrounded by a 2×1.2-cm, depigmented, circular patch on the left side of the inferior abdomen (Figure 1). Upon questioning, the patient produced cell phone photographs of the trunk from 3 years prior, which did not show any lesions present. Full-body skin examination did not reveal any other concerning pigmented lesions. Excisional biopsy was performed due to concern for amelanotic melanoma, and histopathology revealed a superficial and pigmented BCC (Figure 2). Immunohistochemistry with Melan-A was negative for atypical melanocytes, with no uptake in the leukoderma areas.
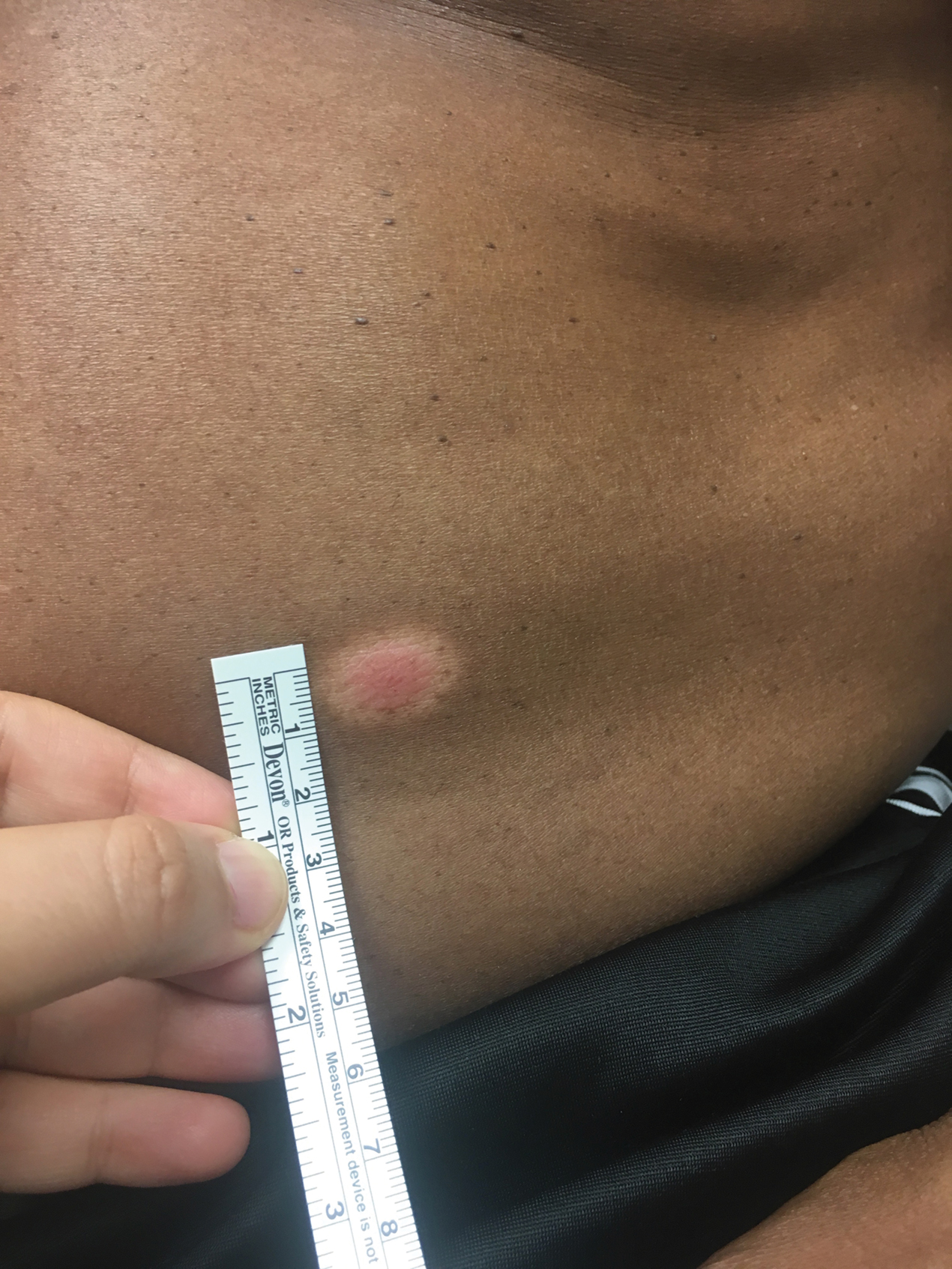
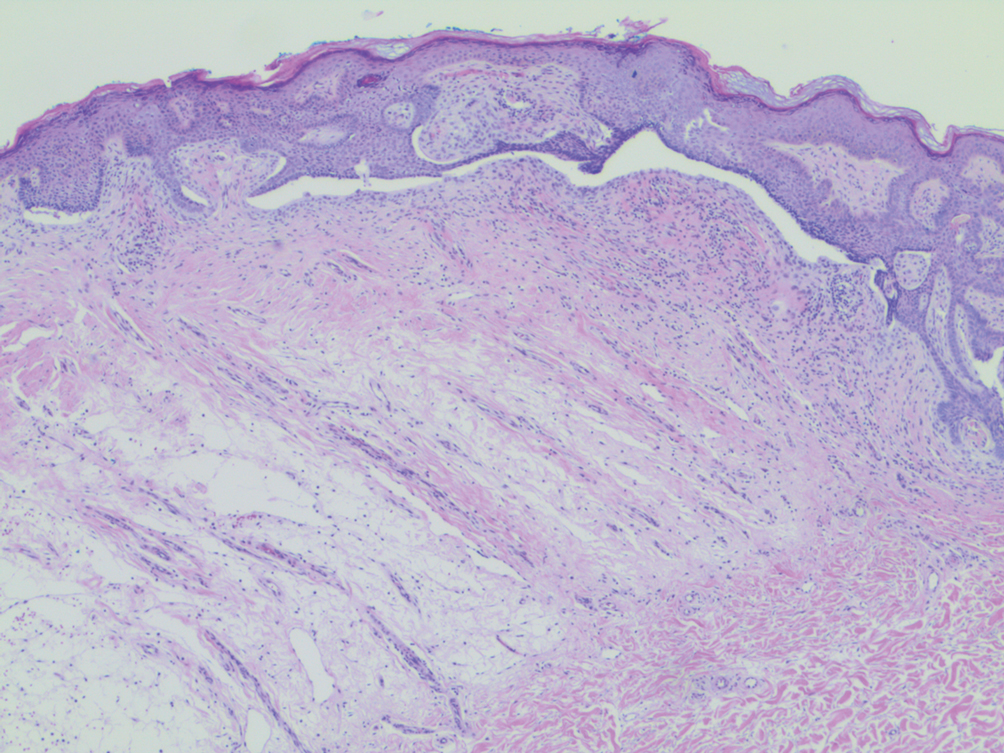
The clinical presentation initially was concerning for amelanotic melanoma. All melanoma subtypes may appear as hypomelanotic lesions, though these most commonly are observed in the desmoplastic or nodular subtypes. Amelanotic melanomas may present as well-defined red or pink macules, plaques, or nodules, with some tumors presenting with light brown pigmentation.7
The differential diagnosis for lesions with the halo phenomenon is large. In adults, the halo phenomenon may be concerning for malignant or regressing melanoma. As an immunogenic tumor, melanoma’s immunogenic melanocytes may incite a cell-mediated immune response to antigens common to neoplastic and normal melanocytes, which can clinically manifest not only as local annular leukoderma but also as distant vitiligo or halo nevi.7 The halo phenomenon more commonly is associated with benign processes such as vitiligo and halo nevi in children. In most children, halo nevi occur as an isolated phenomenon but still warrant a complete skin examination for melanoma and vitiligo. The presence of halo nevi has been associated with distant vitiligo—possibly through shared immunologic mechanisms—especially if patients present with the Koebner phenomenon, multiple halo nevi, or a family history of vitiligo.8 A prospective study also found that the presence of halo nevi was an independent risk factor for the progression of segmental vitiligo to mixed vitiligo.9 Hormones also may play a role in the leukoderma acquisitum centrifugum, or halo, nevi. Halo nevi most commonly affect adolescents and pregnant women. It has been postulated that congenital nevi may be unique in their response to altered estrogen levels, increasing the rate not only of halo nevi but also of melanoma in pregnant women.10
Our patient’s final histologic diagnosis was pigmented BCC, which comprises only 6% of all BCCs.3 The proposed mechanism is that melanocytes colonize the tumor in the surrounding stroma and produce excess melanin. Basal cell carcinoma with halo phenomenon is a rare presentation. As in our case, 2 prior BCC reports also involved patients older than 50 years,3,5 with the 2 other cases describing women in their late twenties and early thirties.4,6 Additionally, 2 of 4 reports described patients with a history of multiple BCCs.3,5
In summary, the seemingly benign halo phenomenon may accompany malignant processes such as nonmelanoma skin cancer. Careful consideration of lesion time course and atypia is imperative for proper clinical suspicion in such cases.
- Mooney MA, Barr RJ, Buxton MG. Halo nevus or halo phenomenon? a study of 142 cases. J Cutan Pathol. 1995;22:342-348.
- Zeff RA, Freitag A, Grin CM, et al. The immune response in halo nevi. J Am Acad Dermatol. 1997;37:620-624.
- Johnson DB, Ceilley RI. Basal cell carcinoma with annular leukoderma mimicking leukoderma acquisitum centrifugum. Arch Dermatol. 1980;116:352-353.
- Basak PY, Meric G, Ciris M. Basal cell carcinoma with halo phenomenon in a young female: significance of dermatoscopy in early diagnosis. Indian J Dermatol. 2015;60:214.
- Pembroke AC, Liddell K. Basal cell epithelioma with a hypopigmented halo. Arch Dermatol. 1981;117:317.
- Rustemeyer J, Günther L, Deichert L. A rare association: basal cell carcinoma in a vitiliginous macula. Oral Maxillofac Surg. 2011;15:175-177.
- Naveh HP, Rao UN, Butterfield LH. Melanoma‐associated leukoderma—immunology in black and white? Pigment Cell Melanoma Res. 2013;26:796-804.
- Zhou H, Wu L-C, Chen M-K, et al. Factors associated with development of vitiligo in patients with halo nevus. Chinese Med J. 2017;130:2703.
- Ezzedine K, Diallo A, Léauté‐Labrèze C, et al. Halo naevi and leukotrichia are strong predictors of the passage to mixed vitiligo in a subgroup of segmental vitiligo. Br J Dermatol. 2012;166:539-544.
- Nading MA, Nanney LB, Ellis DL. Pregnancy and estrogen receptor β expression in a large congenital nevus. Arch Dermatol. 2009;145:691-694.
To the Editor:
Annular leukoderma, or the halo phenomenon, is a circular reaction of hypopigmentation that most commonly is observed alongside congenital nevi, acquired melanocytic nevi, blue nevi, Spitz nevi, vitiligo, and rarely melanoma.1 There is limited literature on the mechanism of the halo phenomenon. Most of the literature proposes a T cell–mediated immune response to antigens, which causes not only surrounding pigment loss but also heralds the regression of central lesions.2 Others have suggested a vascular mechanism, with blood shunted away from the lesions.3 Because guidelines discourage biopsy of typical halo nevi, it becomes important to evaluate lesions for worrisome features such as ulceration or asymmetry, especially in older patients. We present a case of a pigmented basal cell carcinoma (BCC) that exhibited the halo phenomenon. Four other cases have been described in the literature.3-6
A 53-year-old man presented for evaluation of an asymptomatic lesion on the left side of the abdomen of approximately 8 months’ duration. He had no personal or family history of skin cancer. Physical examination revealed a central 1-cm, pink, verrucous papule surrounded by a 2×1.2-cm, depigmented, circular patch on the left side of the inferior abdomen (Figure 1). Upon questioning, the patient produced cell phone photographs of the trunk from 3 years prior, which did not show any lesions present. Full-body skin examination did not reveal any other concerning pigmented lesions. Excisional biopsy was performed due to concern for amelanotic melanoma, and histopathology revealed a superficial and pigmented BCC (Figure 2). Immunohistochemistry with Melan-A was negative for atypical melanocytes, with no uptake in the leukoderma areas.
The clinical presentation initially was concerning for amelanotic melanoma. All melanoma subtypes may appear as hypomelanotic lesions, though these most commonly are observed in the desmoplastic or nodular subtypes. Amelanotic melanomas may present as well-defined red or pink macules, plaques, or nodules, with some tumors presenting with light brown pigmentation.7
The differential diagnosis for lesions with the halo phenomenon is large. In adults, the halo phenomenon may be concerning for malignant or regressing melanoma. As an immunogenic tumor, melanoma’s immunogenic melanocytes may incite a cell-mediated immune response to antigens common to neoplastic and normal melanocytes, which can clinically manifest not only as local annular leukoderma but also as distant vitiligo or halo nevi.7 The halo phenomenon more commonly is associated with benign processes such as vitiligo and halo nevi in children. In most children, halo nevi occur as an isolated phenomenon but still warrant a complete skin examination for melanoma and vitiligo. The presence of halo nevi has been associated with distant vitiligo—possibly through shared immunologic mechanisms—especially if patients present with the Koebner phenomenon, multiple halo nevi, or a family history of vitiligo.8 A prospective study also found that the presence of halo nevi was an independent risk factor for the progression of segmental vitiligo to mixed vitiligo.9 Hormones also may play a role in the leukoderma acquisitum centrifugum, or halo, nevi. Halo nevi most commonly affect adolescents and pregnant women. It has been postulated that congenital nevi may be unique in their response to altered estrogen levels, increasing the rate not only of halo nevi but also of melanoma in pregnant women.10
Our patient’s final histologic diagnosis was pigmented BCC, which comprises only 6% of all BCCs.3 The proposed mechanism is that melanocytes colonize the tumor in the surrounding stroma and produce excess melanin. Basal cell carcinoma with halo phenomenon is a rare presentation. As in our case, 2 prior BCC reports also involved patients older than 50 years,3,5 with the 2 other cases describing women in their late twenties and early thirties.4,6 Additionally, 2 of 4 reports described patients with a history of multiple BCCs.3,5
In summary, the seemingly benign halo phenomenon may accompany malignant processes such as nonmelanoma skin cancer. Careful consideration of lesion time course and atypia is imperative for proper clinical suspicion in such cases.
To the Editor:
Annular leukoderma, or the halo phenomenon, is a circular reaction of hypopigmentation that most commonly is observed alongside congenital nevi, acquired melanocytic nevi, blue nevi, Spitz nevi, vitiligo, and rarely melanoma.1 There is limited literature on the mechanism of the halo phenomenon. Most of the literature proposes a T cell–mediated immune response to antigens, which causes not only surrounding pigment loss but also heralds the regression of central lesions.2 Others have suggested a vascular mechanism, with blood shunted away from the lesions.3 Because guidelines discourage biopsy of typical halo nevi, it becomes important to evaluate lesions for worrisome features such as ulceration or asymmetry, especially in older patients. We present a case of a pigmented basal cell carcinoma (BCC) that exhibited the halo phenomenon. Four other cases have been described in the literature.3-6
A 53-year-old man presented for evaluation of an asymptomatic lesion on the left side of the abdomen of approximately 8 months’ duration. He had no personal or family history of skin cancer. Physical examination revealed a central 1-cm, pink, verrucous papule surrounded by a 2×1.2-cm, depigmented, circular patch on the left side of the inferior abdomen (Figure 1). Upon questioning, the patient produced cell phone photographs of the trunk from 3 years prior, which did not show any lesions present. Full-body skin examination did not reveal any other concerning pigmented lesions. Excisional biopsy was performed due to concern for amelanotic melanoma, and histopathology revealed a superficial and pigmented BCC (Figure 2). Immunohistochemistry with Melan-A was negative for atypical melanocytes, with no uptake in the leukoderma areas.
The clinical presentation initially was concerning for amelanotic melanoma. All melanoma subtypes may appear as hypomelanotic lesions, though these most commonly are observed in the desmoplastic or nodular subtypes. Amelanotic melanomas may present as well-defined red or pink macules, plaques, or nodules, with some tumors presenting with light brown pigmentation.7
The differential diagnosis for lesions with the halo phenomenon is large. In adults, the halo phenomenon may be concerning for malignant or regressing melanoma. As an immunogenic tumor, melanoma’s immunogenic melanocytes may incite a cell-mediated immune response to antigens common to neoplastic and normal melanocytes, which can clinically manifest not only as local annular leukoderma but also as distant vitiligo or halo nevi.7 The halo phenomenon more commonly is associated with benign processes such as vitiligo and halo nevi in children. In most children, halo nevi occur as an isolated phenomenon but still warrant a complete skin examination for melanoma and vitiligo. The presence of halo nevi has been associated with distant vitiligo—possibly through shared immunologic mechanisms—especially if patients present with the Koebner phenomenon, multiple halo nevi, or a family history of vitiligo.8 A prospective study also found that the presence of halo nevi was an independent risk factor for the progression of segmental vitiligo to mixed vitiligo.9 Hormones also may play a role in the leukoderma acquisitum centrifugum, or halo, nevi. Halo nevi most commonly affect adolescents and pregnant women. It has been postulated that congenital nevi may be unique in their response to altered estrogen levels, increasing the rate not only of halo nevi but also of melanoma in pregnant women.10
Our patient’s final histologic diagnosis was pigmented BCC, which comprises only 6% of all BCCs.3 The proposed mechanism is that melanocytes colonize the tumor in the surrounding stroma and produce excess melanin. Basal cell carcinoma with halo phenomenon is a rare presentation. As in our case, 2 prior BCC reports also involved patients older than 50 years,3,5 with the 2 other cases describing women in their late twenties and early thirties.4,6 Additionally, 2 of 4 reports described patients with a history of multiple BCCs.3,5
In summary, the seemingly benign halo phenomenon may accompany malignant processes such as nonmelanoma skin cancer. Careful consideration of lesion time course and atypia is imperative for proper clinical suspicion in such cases.
- Mooney MA, Barr RJ, Buxton MG. Halo nevus or halo phenomenon? a study of 142 cases. J Cutan Pathol. 1995;22:342-348.
- Zeff RA, Freitag A, Grin CM, et al. The immune response in halo nevi. J Am Acad Dermatol. 1997;37:620-624.
- Johnson DB, Ceilley RI. Basal cell carcinoma with annular leukoderma mimicking leukoderma acquisitum centrifugum. Arch Dermatol. 1980;116:352-353.
- Basak PY, Meric G, Ciris M. Basal cell carcinoma with halo phenomenon in a young female: significance of dermatoscopy in early diagnosis. Indian J Dermatol. 2015;60:214.
- Pembroke AC, Liddell K. Basal cell epithelioma with a hypopigmented halo. Arch Dermatol. 1981;117:317.
- Rustemeyer J, Günther L, Deichert L. A rare association: basal cell carcinoma in a vitiliginous macula. Oral Maxillofac Surg. 2011;15:175-177.
- Naveh HP, Rao UN, Butterfield LH. Melanoma‐associated leukoderma—immunology in black and white? Pigment Cell Melanoma Res. 2013;26:796-804.
- Zhou H, Wu L-C, Chen M-K, et al. Factors associated with development of vitiligo in patients with halo nevus. Chinese Med J. 2017;130:2703.
- Ezzedine K, Diallo A, Léauté‐Labrèze C, et al. Halo naevi and leukotrichia are strong predictors of the passage to mixed vitiligo in a subgroup of segmental vitiligo. Br J Dermatol. 2012;166:539-544.
- Nading MA, Nanney LB, Ellis DL. Pregnancy and estrogen receptor β expression in a large congenital nevus. Arch Dermatol. 2009;145:691-694.
- Mooney MA, Barr RJ, Buxton MG. Halo nevus or halo phenomenon? a study of 142 cases. J Cutan Pathol. 1995;22:342-348.
- Zeff RA, Freitag A, Grin CM, et al. The immune response in halo nevi. J Am Acad Dermatol. 1997;37:620-624.
- Johnson DB, Ceilley RI. Basal cell carcinoma with annular leukoderma mimicking leukoderma acquisitum centrifugum. Arch Dermatol. 1980;116:352-353.
- Basak PY, Meric G, Ciris M. Basal cell carcinoma with halo phenomenon in a young female: significance of dermatoscopy in early diagnosis. Indian J Dermatol. 2015;60:214.
- Pembroke AC, Liddell K. Basal cell epithelioma with a hypopigmented halo. Arch Dermatol. 1981;117:317.
- Rustemeyer J, Günther L, Deichert L. A rare association: basal cell carcinoma in a vitiliginous macula. Oral Maxillofac Surg. 2011;15:175-177.
- Naveh HP, Rao UN, Butterfield LH. Melanoma‐associated leukoderma—immunology in black and white? Pigment Cell Melanoma Res. 2013;26:796-804.
- Zhou H, Wu L-C, Chen M-K, et al. Factors associated with development of vitiligo in patients with halo nevus. Chinese Med J. 2017;130:2703.
- Ezzedine K, Diallo A, Léauté‐Labrèze C, et al. Halo naevi and leukotrichia are strong predictors of the passage to mixed vitiligo in a subgroup of segmental vitiligo. Br J Dermatol. 2012;166:539-544.
- Nading MA, Nanney LB, Ellis DL. Pregnancy and estrogen receptor β expression in a large congenital nevus. Arch Dermatol. 2009;145:691-694.
Practice Points
- Annular leukoderma, or the halo phenomenon, is a circular reaction of hypopigmentation that more commonly is associated with benign processes such as halo nevi.
- The halo phenomenon may accompany malignant processes, such as nonmelanoma skin cancer. Careful consideration of lesion time course and atypia is imperative for proper clinical suspicion in such cases.
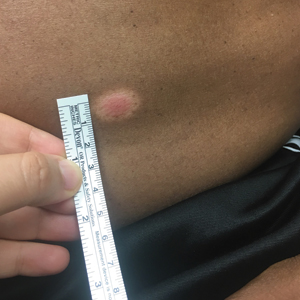
Dermatoethics for Dermatology Residents
As dermatology residents, we have a lot on our plates. With so many diagnoses to learn and treatments to understand, the sheer volume of knowledge we are expected to be familiar with sometimes can be overwhelming. The thought of adding yet another thing to the list of many things we already need to know—least of all a topic such as dermatoethics—may be unappealing. This article will discuss the importance of ethics training in dermatology residency as well as provide helpful resources for how this training can be achieved.
Professionalism as a Core Competency
The Accreditation Council for Graduate Medical Education (ACGME) considers professionalism as 1 of its 6 core competencies.1 These competencies provide a conceptual framework detailing the domains physicians should be proficient in before they can enter autonomous practice. When it comes to professionalism, residents are expected to demonstrate compassion, integrity, and respect for others; honesty with patients; respect for patient confidentiality and autonomy; appropriate relationships with patients; accountability to patients, society, and the profession; and a sensitivity and responsiveness to diverse patient population.1
The ACGME milestones are intended to assess resident development within the 6 competencies with more specific parameters for evaluation.2 Those pertaining to professionalism evaluate a resident’s ability to demonstrate professional behavior, an understanding of ethical principles, accountability, and conscientiousness, as well as self-awareness and the ability to seek help for personal or professional well-being. The crux of the kinds of activities that constitute acquisition of these professional skills are specialty specific. The ACGME ultimately believes that having a working knowledge of professionalism and ethical principles prepares residents for practicing medicine in the real world. Because of these requirements, residency programs are expected to provide resources for residents to explore ethical problems faced by dermatologists.
Beyond “Passing” Residency
The reality is that learning about medical ethics and practicing professional behavior is not just about ticking boxes to get ACGME accreditation or to “pass” residency. The data suggest that having a strong foundation in these principles is good for overall personal well-being, job satisfaction, and patient care. Studies have shown that unprofessional behavior in medical school is correlated to disciplinary action by state licensing boards against practicing physicians.3,4 In fact, a study found that in one cohort of physicians (N=68), 95% of disciplinary actions were for lapses in professionalism, which included activities such as sexual misconduct and inappropriate prescribing.4 Behaving appropriately protects your license to practice medicine.
Thinking through these problematic ethical scenarios also goes beyond coming up with the right answer. Exploring ethical conundrums is thought to develop analytical skills that can help one navigate future tricky situations that can be morally distressing and can lead to burnout. Introspection and self-awareness coupled with these skills ideally will help physicians think through sensitive and difficult situations with the courage to hold true to their convictions and ultimately uphold the professionalism of the specialty.5
Self-awareness has the additional bonus of empowering physicians to acknowledge personal and professional limitations with the goal of seeking help when it is needed before it is too late. It comes as no surprise that how we feel as physicians directly impacts how we treat our patients. One study found that depressed residents were more than 6 times more likely to make medication errors compared to nondepressed colleagues.6 Regularly taking stock of our professional and personal reserves can go a long way to improving overall well-being.
Resources for Dermatoethics Training
The best starting point for developing a robust dermatoethics curriculum is the material provided by the American Board of Dermatology, which is available online.7 An ad hoc subcommittee of the American Board of Dermatology composed of experts in dermatoethics and resident education reviewed relevant ethics literature and identified 6 core domains considered fundamental to dermatology resident education in ethics and professionalism.8 This team also provided a thorough list of relevant background readings for each topic. To cover pertinent material, the subcommittee recommended a 60-minute teaching session every other month with the intent of covering all the material over a 3-year period. If your program directors are not aware of this great resource and you feel your own ethics training may be lacking, bringing this up as a template might be helpful. A detailed description of an innovative dermatoethics curriculum organized at the Department of Dermatology at the Warren Alpert Medical School of Brown University (Providence, Rhode Island) in 2001 also may serve as a guide for programs hoping to design their own approach.5
For those interested in self-study, there is an excellent text dedicated to dermatoethics, which is aptly entitled Dermatoethics: Contemporary Ethics and Professionalism in Dermatology.9 This book offers superb case-based discussions on a wide range of ethical quandaries that dermatologists may face, ranging from unsolicited dermatologic advice (eg, Is it wrong to tell the person next to you in the grocery store that they might have a melanoma?) to research and publication ethics. This text provides a toolkit for handling tough situations in the clinic and beyond. The Journal of the American Academy of Dermatology publishes an Ethics Journal Club for which contributors can submit real-life practical ethical dilemmas, and the journal solicits a resolution or response from a dermatoethicist.
Additionally, a pilot curriculum project out of the University of Utah (Salt Lake City, Utah), of which I am a team member, currently is designing and testing several dermatoethics PowerPoint modules with the intention of making this material widely available through medical education portals.
The Hidden Curriculum
A formal curriculum can only provide so much when it comes to ethics training. In truth, much of what we learn as ethically minded dermatologists comes from our day-to-day practice.10 Paying attention to the more informal curriculum that we are immersed in during routine as well as unusual encounters also is important for achieving milestones. Teaching moments for thinking through ethical dilemmas abound, and this approach easily can be incorporated into routine workflow.11 Next time you encounter an ethical situation that gives you pause (eg, Can I biopsy an intubated patient without getting appropriate consent?), talk it through with your supervisor. Gems of autonomous practice often can be mined from these off-the-cuff conversations.
Can Professionalism Be Taught?
Finally, it is worth mentioning that while the number of resources available to dermatology residents for honing their ethics skills is increasing, ways of measuring the impact of this additional training in vivo are not.12 There are no good tools available to determine how ethics training influences resident behaviors. Similarly, there is no good evidence for what constitutes the most effective method for teaching medical ethics to trainees. It is a growing field with lots of room for more robust research. For now, the overall goal of a dermatoethics curriculum is to provide a mix of curriculum opportunities, ranging from formal lectures and readings to more informal conversations, with the hope of providing residents a toolbox for dealing with ethical dilemmas and a working knowledge of professionalism.
Final Thoughts
There are several resources available for dermatology programs to provide quality dermatoethics training to their residents. These can be mixed and matched to create a tailored formal curriculum alongside the more informal ethics training that happens in the clinic and on the wards. Providing this education is about more than just fulfilling accreditation requirements. Understanding ethical principles and how they can be applied to navigate sensitive situations is ultimately good for both professional and personal well-being.
- Accreditation Council for Graduate Medical Education. ACGME common program requirements (residency). ACGME website. Accessed June 10, 2021. https://www.acgme.org/Portals/0/PFAssets/ProgramRequirements/CPRResidency2020.pdf
- Edgar L, McLean S, Hogan SO, et al. The milestones guidebook. Accreditation Council for Graduate Medical Education website. Accessed June 10, 2021. acgme.org/portals/0/MilestonesGuidebook.pdf
- Papadakis MA, Teherani A, Banach MA, et al. Disciplinary action by medical boards and prior behavior in medical school. N Engl J Med. 2005;353:2673-2682.
- Papadakis MA, Hodgson CS, Teherani A, et al. Unprofessional behavior in medical school is associated with subsequent disciplinary action by a state medical board. Acad Med. 2004;79:244-249.
- Bercovitch L, Long TP. Dermatoethics: a curriculum in bioethics and professionalism for dermatology residents at Brown Medical School. J Am Acad Dermatol. 2007;56:679-682.
- Fahrenkopf AM, Sectish TC, Barger LK, et al. Rates of medication errors among depressed and burnt out residents: prospective cohort study. BMJ. 2008;336:488-491.
- Recommended topics for 3-year dermatoethics curricular cycle. American Board of Dermatology website. Accessed June 10, 2021. https://www.abderm.org/residents-and-fellows/dermatoethics.aspx
- Stoff BK, Grant-Kels JM, Brodell RT, et al. Introducing a curriculum in ethics and professionalism for dermatology residencies. J Am Acad Dermatol. 2018;78:1032-1034.
- Bercovitch L, Perlis C, Stoff BK, et al, eds. Dermatoethics: Contemporary Ethics and Professionalism in Dermatology. 2nd ed. Springer International Publishing; 2021.
- Hafferty FW, Franks R. The hidden curriculum, ethics teaching, and the structure of medical education. Acad Med. 1994;69:861-871.
- Aldrich N, Mostow E. Incorporating teaching dermatoethics in a busy outpatient clinic. J Am Acad Dermatol. 2011;65:423-424.
- de la Garza S, Phuoc V, Throneberry S, et al. Teaching medical ethics in graduate and undergraduate medical education: a systematic review of effectiveness. Acad Psychiatry. 2017;41:520-525.
As dermatology residents, we have a lot on our plates. With so many diagnoses to learn and treatments to understand, the sheer volume of knowledge we are expected to be familiar with sometimes can be overwhelming. The thought of adding yet another thing to the list of many things we already need to know—least of all a topic such as dermatoethics—may be unappealing. This article will discuss the importance of ethics training in dermatology residency as well as provide helpful resources for how this training can be achieved.
Professionalism as a Core Competency
The Accreditation Council for Graduate Medical Education (ACGME) considers professionalism as 1 of its 6 core competencies.1 These competencies provide a conceptual framework detailing the domains physicians should be proficient in before they can enter autonomous practice. When it comes to professionalism, residents are expected to demonstrate compassion, integrity, and respect for others; honesty with patients; respect for patient confidentiality and autonomy; appropriate relationships with patients; accountability to patients, society, and the profession; and a sensitivity and responsiveness to diverse patient population.1
The ACGME milestones are intended to assess resident development within the 6 competencies with more specific parameters for evaluation.2 Those pertaining to professionalism evaluate a resident’s ability to demonstrate professional behavior, an understanding of ethical principles, accountability, and conscientiousness, as well as self-awareness and the ability to seek help for personal or professional well-being. The crux of the kinds of activities that constitute acquisition of these professional skills are specialty specific. The ACGME ultimately believes that having a working knowledge of professionalism and ethical principles prepares residents for practicing medicine in the real world. Because of these requirements, residency programs are expected to provide resources for residents to explore ethical problems faced by dermatologists.
Beyond “Passing” Residency
The reality is that learning about medical ethics and practicing professional behavior is not just about ticking boxes to get ACGME accreditation or to “pass” residency. The data suggest that having a strong foundation in these principles is good for overall personal well-being, job satisfaction, and patient care. Studies have shown that unprofessional behavior in medical school is correlated to disciplinary action by state licensing boards against practicing physicians.3,4 In fact, a study found that in one cohort of physicians (N=68), 95% of disciplinary actions were for lapses in professionalism, which included activities such as sexual misconduct and inappropriate prescribing.4 Behaving appropriately protects your license to practice medicine.
Thinking through these problematic ethical scenarios also goes beyond coming up with the right answer. Exploring ethical conundrums is thought to develop analytical skills that can help one navigate future tricky situations that can be morally distressing and can lead to burnout. Introspection and self-awareness coupled with these skills ideally will help physicians think through sensitive and difficult situations with the courage to hold true to their convictions and ultimately uphold the professionalism of the specialty.5
Self-awareness has the additional bonus of empowering physicians to acknowledge personal and professional limitations with the goal of seeking help when it is needed before it is too late. It comes as no surprise that how we feel as physicians directly impacts how we treat our patients. One study found that depressed residents were more than 6 times more likely to make medication errors compared to nondepressed colleagues.6 Regularly taking stock of our professional and personal reserves can go a long way to improving overall well-being.
Resources for Dermatoethics Training
The best starting point for developing a robust dermatoethics curriculum is the material provided by the American Board of Dermatology, which is available online.7 An ad hoc subcommittee of the American Board of Dermatology composed of experts in dermatoethics and resident education reviewed relevant ethics literature and identified 6 core domains considered fundamental to dermatology resident education in ethics and professionalism.8 This team also provided a thorough list of relevant background readings for each topic. To cover pertinent material, the subcommittee recommended a 60-minute teaching session every other month with the intent of covering all the material over a 3-year period. If your program directors are not aware of this great resource and you feel your own ethics training may be lacking, bringing this up as a template might be helpful. A detailed description of an innovative dermatoethics curriculum organized at the Department of Dermatology at the Warren Alpert Medical School of Brown University (Providence, Rhode Island) in 2001 also may serve as a guide for programs hoping to design their own approach.5
For those interested in self-study, there is an excellent text dedicated to dermatoethics, which is aptly entitled Dermatoethics: Contemporary Ethics and Professionalism in Dermatology.9 This book offers superb case-based discussions on a wide range of ethical quandaries that dermatologists may face, ranging from unsolicited dermatologic advice (eg, Is it wrong to tell the person next to you in the grocery store that they might have a melanoma?) to research and publication ethics. This text provides a toolkit for handling tough situations in the clinic and beyond. The Journal of the American Academy of Dermatology publishes an Ethics Journal Club for which contributors can submit real-life practical ethical dilemmas, and the journal solicits a resolution or response from a dermatoethicist.
Additionally, a pilot curriculum project out of the University of Utah (Salt Lake City, Utah), of which I am a team member, currently is designing and testing several dermatoethics PowerPoint modules with the intention of making this material widely available through medical education portals.
The Hidden Curriculum
A formal curriculum can only provide so much when it comes to ethics training. In truth, much of what we learn as ethically minded dermatologists comes from our day-to-day practice.10 Paying attention to the more informal curriculum that we are immersed in during routine as well as unusual encounters also is important for achieving milestones. Teaching moments for thinking through ethical dilemmas abound, and this approach easily can be incorporated into routine workflow.11 Next time you encounter an ethical situation that gives you pause (eg, Can I biopsy an intubated patient without getting appropriate consent?), talk it through with your supervisor. Gems of autonomous practice often can be mined from these off-the-cuff conversations.
Can Professionalism Be Taught?
Finally, it is worth mentioning that while the number of resources available to dermatology residents for honing their ethics skills is increasing, ways of measuring the impact of this additional training in vivo are not.12 There are no good tools available to determine how ethics training influences resident behaviors. Similarly, there is no good evidence for what constitutes the most effective method for teaching medical ethics to trainees. It is a growing field with lots of room for more robust research. For now, the overall goal of a dermatoethics curriculum is to provide a mix of curriculum opportunities, ranging from formal lectures and readings to more informal conversations, with the hope of providing residents a toolbox for dealing with ethical dilemmas and a working knowledge of professionalism.
Final Thoughts
There are several resources available for dermatology programs to provide quality dermatoethics training to their residents. These can be mixed and matched to create a tailored formal curriculum alongside the more informal ethics training that happens in the clinic and on the wards. Providing this education is about more than just fulfilling accreditation requirements. Understanding ethical principles and how they can be applied to navigate sensitive situations is ultimately good for both professional and personal well-being.
As dermatology residents, we have a lot on our plates. With so many diagnoses to learn and treatments to understand, the sheer volume of knowledge we are expected to be familiar with sometimes can be overwhelming. The thought of adding yet another thing to the list of many things we already need to know—least of all a topic such as dermatoethics—may be unappealing. This article will discuss the importance of ethics training in dermatology residency as well as provide helpful resources for how this training can be achieved.
Professionalism as a Core Competency
The Accreditation Council for Graduate Medical Education (ACGME) considers professionalism as 1 of its 6 core competencies.1 These competencies provide a conceptual framework detailing the domains physicians should be proficient in before they can enter autonomous practice. When it comes to professionalism, residents are expected to demonstrate compassion, integrity, and respect for others; honesty with patients; respect for patient confidentiality and autonomy; appropriate relationships with patients; accountability to patients, society, and the profession; and a sensitivity and responsiveness to diverse patient population.1
The ACGME milestones are intended to assess resident development within the 6 competencies with more specific parameters for evaluation.2 Those pertaining to professionalism evaluate a resident’s ability to demonstrate professional behavior, an understanding of ethical principles, accountability, and conscientiousness, as well as self-awareness and the ability to seek help for personal or professional well-being. The crux of the kinds of activities that constitute acquisition of these professional skills are specialty specific. The ACGME ultimately believes that having a working knowledge of professionalism and ethical principles prepares residents for practicing medicine in the real world. Because of these requirements, residency programs are expected to provide resources for residents to explore ethical problems faced by dermatologists.
Beyond “Passing” Residency
The reality is that learning about medical ethics and practicing professional behavior is not just about ticking boxes to get ACGME accreditation or to “pass” residency. The data suggest that having a strong foundation in these principles is good for overall personal well-being, job satisfaction, and patient care. Studies have shown that unprofessional behavior in medical school is correlated to disciplinary action by state licensing boards against practicing physicians.3,4 In fact, a study found that in one cohort of physicians (N=68), 95% of disciplinary actions were for lapses in professionalism, which included activities such as sexual misconduct and inappropriate prescribing.4 Behaving appropriately protects your license to practice medicine.
Thinking through these problematic ethical scenarios also goes beyond coming up with the right answer. Exploring ethical conundrums is thought to develop analytical skills that can help one navigate future tricky situations that can be morally distressing and can lead to burnout. Introspection and self-awareness coupled with these skills ideally will help physicians think through sensitive and difficult situations with the courage to hold true to their convictions and ultimately uphold the professionalism of the specialty.5
Self-awareness has the additional bonus of empowering physicians to acknowledge personal and professional limitations with the goal of seeking help when it is needed before it is too late. It comes as no surprise that how we feel as physicians directly impacts how we treat our patients. One study found that depressed residents were more than 6 times more likely to make medication errors compared to nondepressed colleagues.6 Regularly taking stock of our professional and personal reserves can go a long way to improving overall well-being.
Resources for Dermatoethics Training
The best starting point for developing a robust dermatoethics curriculum is the material provided by the American Board of Dermatology, which is available online.7 An ad hoc subcommittee of the American Board of Dermatology composed of experts in dermatoethics and resident education reviewed relevant ethics literature and identified 6 core domains considered fundamental to dermatology resident education in ethics and professionalism.8 This team also provided a thorough list of relevant background readings for each topic. To cover pertinent material, the subcommittee recommended a 60-minute teaching session every other month with the intent of covering all the material over a 3-year period. If your program directors are not aware of this great resource and you feel your own ethics training may be lacking, bringing this up as a template might be helpful. A detailed description of an innovative dermatoethics curriculum organized at the Department of Dermatology at the Warren Alpert Medical School of Brown University (Providence, Rhode Island) in 2001 also may serve as a guide for programs hoping to design their own approach.5
For those interested in self-study, there is an excellent text dedicated to dermatoethics, which is aptly entitled Dermatoethics: Contemporary Ethics and Professionalism in Dermatology.9 This book offers superb case-based discussions on a wide range of ethical quandaries that dermatologists may face, ranging from unsolicited dermatologic advice (eg, Is it wrong to tell the person next to you in the grocery store that they might have a melanoma?) to research and publication ethics. This text provides a toolkit for handling tough situations in the clinic and beyond. The Journal of the American Academy of Dermatology publishes an Ethics Journal Club for which contributors can submit real-life practical ethical dilemmas, and the journal solicits a resolution or response from a dermatoethicist.
Additionally, a pilot curriculum project out of the University of Utah (Salt Lake City, Utah), of which I am a team member, currently is designing and testing several dermatoethics PowerPoint modules with the intention of making this material widely available through medical education portals.
The Hidden Curriculum
A formal curriculum can only provide so much when it comes to ethics training. In truth, much of what we learn as ethically minded dermatologists comes from our day-to-day practice.10 Paying attention to the more informal curriculum that we are immersed in during routine as well as unusual encounters also is important for achieving milestones. Teaching moments for thinking through ethical dilemmas abound, and this approach easily can be incorporated into routine workflow.11 Next time you encounter an ethical situation that gives you pause (eg, Can I biopsy an intubated patient without getting appropriate consent?), talk it through with your supervisor. Gems of autonomous practice often can be mined from these off-the-cuff conversations.
Can Professionalism Be Taught?
Finally, it is worth mentioning that while the number of resources available to dermatology residents for honing their ethics skills is increasing, ways of measuring the impact of this additional training in vivo are not.12 There are no good tools available to determine how ethics training influences resident behaviors. Similarly, there is no good evidence for what constitutes the most effective method for teaching medical ethics to trainees. It is a growing field with lots of room for more robust research. For now, the overall goal of a dermatoethics curriculum is to provide a mix of curriculum opportunities, ranging from formal lectures and readings to more informal conversations, with the hope of providing residents a toolbox for dealing with ethical dilemmas and a working knowledge of professionalism.
Final Thoughts
There are several resources available for dermatology programs to provide quality dermatoethics training to their residents. These can be mixed and matched to create a tailored formal curriculum alongside the more informal ethics training that happens in the clinic and on the wards. Providing this education is about more than just fulfilling accreditation requirements. Understanding ethical principles and how they can be applied to navigate sensitive situations is ultimately good for both professional and personal well-being.
- Accreditation Council for Graduate Medical Education. ACGME common program requirements (residency). ACGME website. Accessed June 10, 2021. https://www.acgme.org/Portals/0/PFAssets/ProgramRequirements/CPRResidency2020.pdf
- Edgar L, McLean S, Hogan SO, et al. The milestones guidebook. Accreditation Council for Graduate Medical Education website. Accessed June 10, 2021. acgme.org/portals/0/MilestonesGuidebook.pdf
- Papadakis MA, Teherani A, Banach MA, et al. Disciplinary action by medical boards and prior behavior in medical school. N Engl J Med. 2005;353:2673-2682.
- Papadakis MA, Hodgson CS, Teherani A, et al. Unprofessional behavior in medical school is associated with subsequent disciplinary action by a state medical board. Acad Med. 2004;79:244-249.
- Bercovitch L, Long TP. Dermatoethics: a curriculum in bioethics and professionalism for dermatology residents at Brown Medical School. J Am Acad Dermatol. 2007;56:679-682.
- Fahrenkopf AM, Sectish TC, Barger LK, et al. Rates of medication errors among depressed and burnt out residents: prospective cohort study. BMJ. 2008;336:488-491.
- Recommended topics for 3-year dermatoethics curricular cycle. American Board of Dermatology website. Accessed June 10, 2021. https://www.abderm.org/residents-and-fellows/dermatoethics.aspx
- Stoff BK, Grant-Kels JM, Brodell RT, et al. Introducing a curriculum in ethics and professionalism for dermatology residencies. J Am Acad Dermatol. 2018;78:1032-1034.
- Bercovitch L, Perlis C, Stoff BK, et al, eds. Dermatoethics: Contemporary Ethics and Professionalism in Dermatology. 2nd ed. Springer International Publishing; 2021.
- Hafferty FW, Franks R. The hidden curriculum, ethics teaching, and the structure of medical education. Acad Med. 1994;69:861-871.
- Aldrich N, Mostow E. Incorporating teaching dermatoethics in a busy outpatient clinic. J Am Acad Dermatol. 2011;65:423-424.
- de la Garza S, Phuoc V, Throneberry S, et al. Teaching medical ethics in graduate and undergraduate medical education: a systematic review of effectiveness. Acad Psychiatry. 2017;41:520-525.
- Accreditation Council for Graduate Medical Education. ACGME common program requirements (residency). ACGME website. Accessed June 10, 2021. https://www.acgme.org/Portals/0/PFAssets/ProgramRequirements/CPRResidency2020.pdf
- Edgar L, McLean S, Hogan SO, et al. The milestones guidebook. Accreditation Council for Graduate Medical Education website. Accessed June 10, 2021. acgme.org/portals/0/MilestonesGuidebook.pdf
- Papadakis MA, Teherani A, Banach MA, et al. Disciplinary action by medical boards and prior behavior in medical school. N Engl J Med. 2005;353:2673-2682.
- Papadakis MA, Hodgson CS, Teherani A, et al. Unprofessional behavior in medical school is associated with subsequent disciplinary action by a state medical board. Acad Med. 2004;79:244-249.
- Bercovitch L, Long TP. Dermatoethics: a curriculum in bioethics and professionalism for dermatology residents at Brown Medical School. J Am Acad Dermatol. 2007;56:679-682.
- Fahrenkopf AM, Sectish TC, Barger LK, et al. Rates of medication errors among depressed and burnt out residents: prospective cohort study. BMJ. 2008;336:488-491.
- Recommended topics for 3-year dermatoethics curricular cycle. American Board of Dermatology website. Accessed June 10, 2021. https://www.abderm.org/residents-and-fellows/dermatoethics.aspx
- Stoff BK, Grant-Kels JM, Brodell RT, et al. Introducing a curriculum in ethics and professionalism for dermatology residencies. J Am Acad Dermatol. 2018;78:1032-1034.
- Bercovitch L, Perlis C, Stoff BK, et al, eds. Dermatoethics: Contemporary Ethics and Professionalism in Dermatology. 2nd ed. Springer International Publishing; 2021.
- Hafferty FW, Franks R. The hidden curriculum, ethics teaching, and the structure of medical education. Acad Med. 1994;69:861-871.
- Aldrich N, Mostow E. Incorporating teaching dermatoethics in a busy outpatient clinic. J Am Acad Dermatol. 2011;65:423-424.
- de la Garza S, Phuoc V, Throneberry S, et al. Teaching medical ethics in graduate and undergraduate medical education: a systematic review of effectiveness. Acad Psychiatry. 2017;41:520-525.
Resident Pearls
- Professionalism is one of the 6 core competencies used by the Accreditation Council for Graduate Medical Education (ACGME) to evaluate physician preparedness for autonomous practice. Dermatology residency programs are expected to provide resources for achieving this competency.
- Several resources for exploring ethical issues in dermatology are available and can be utilized to create a formal curriculum alongside the more tacit learning that takes place in daily practice.
- Learning about ethical principles and their application can ultimately help practicing physicians avoid disciplinary action and improve overall well-being.

Hard Nodular Plaque on the Scalp
The Diagnosis: Platelike Osteoma Cutis
Histopathologic examination revealed extensive cutaneous ossification in the dermis and subcutis with dermal fibrosis and minimal surrounding inflammation (Figure 1). There was no evidence of infection or neoplasm. Further evaluation did not demonstrate any additional physical dysmorphia, and there were no imbalances of calcium-phosphate metabolism or abnormalities in parathyroid hormone or thyroid hormone function. A diagnosis of platelike osteoma cutis (PLOC) was favored. Computed tomography of the head showed material at the posterior skull of similar density to the adjacent calvarial skull and centered within the dermis, consistent with osteoma cutis (Figure 2).
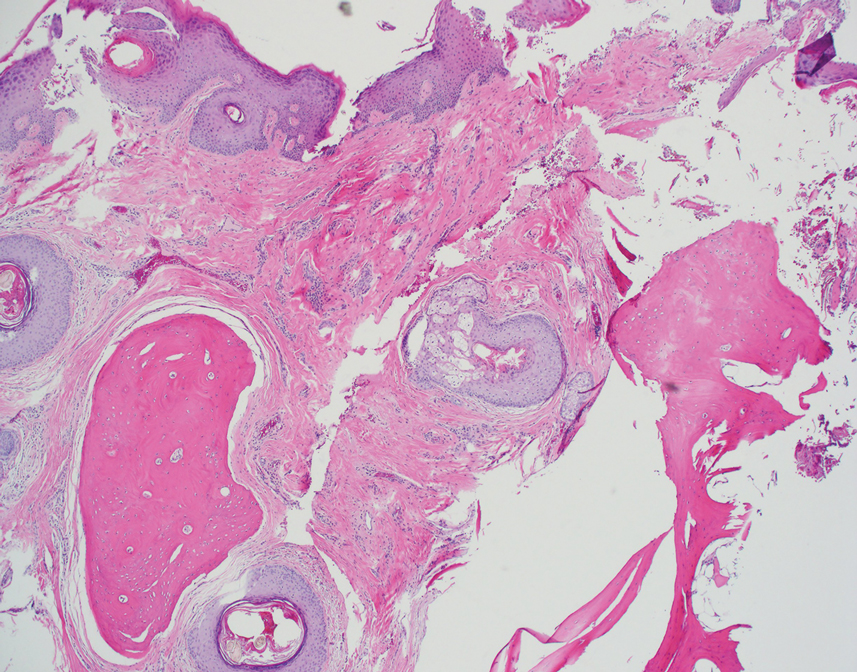
Osteoma cutis describes the formation of bone within the skin. It occurs when hydroxyapatite crystals in a proteinaceous matrix are deposited within the skin, ultimately leading to the formation of bone ultrastructure. Ossification of the skin most often occurs secondary to trauma, inflammation, or neoplasm; however, it rarely may be a primary event.1,2
Platelike osteoma cutis is a rare form of primary cutaneous ossification in which bone forms within the skin in a platelike manner. It most frequently affects the scalp but also has been observed on the trunk and extremities.1 A driving metabolic or endocrine abnormality typically is not identified.2
Platelike osteoma cutis can occur as an isolated finding or as a feature of Albright hereditary osteodystrophy (AHO) or progressive osseous heteroplasia (POH). In addition to cutaneous ossification, AHO involves short stature, endocrinopathy, obesity, shortened fourth and fifth metacarpals, and mental retardation. Progressive osseous heteroplasia is characterized by progressive ossification of the skin and deeper tissues such as muscle and fascia, leading to severe movement restriction; it is believed to be a localized nonprogressive variant of POH.3,4 Mutations in the guanine nucleotide binding protein, alpha stimulating activity polypeptide 1 gene, GNAS1, a key regulatory gene involved in AHO and POH, have been found in several cases of PLOC.3 Our patient lacked any dysmorphic features or laboratory abnormalities suggestive of AHO or POH. Moreover, testing of the tissue and blood for the GNAS1 mutation was negative. Treatment of PLOC often is difficult. Our patient underwent a trial of ablative fractional laser resurfacing, which failed to lead to perceivable improvement.
The differential diagnoses include a kerion, dissecting cellulitis of the scalp, folliculitis decalvans, and acne keloidalis nuchae. A kerion is a manifestation of tinea capitis characterized by an inflammatory plaque, often with pain or tenderness. Kerions most frequently occur in children aged 5 to 10 years.5 Failure to treat a kerion may result in scarring alopecia. Treatment consists of oral antifungals.
Dissecting cellulitis of the scalp is thought to occur secondary to follicular occlusion. It is characterized by boggy suppurative nodules primarily on the posterior and vertex scalp. Patchy hair loss is present and typically progresses to cicatricial alopecia. Histology characteristically shows areas of dense, predominantly neutrophilic, perifollicular dermal infiltrates.6
Folliculitis decalvans is a primary neutrophilic cicatricial alopecia that primarily occurs in adults. Patients with folliculitis decalvans tend to have multiple pustules on the periphery of confluent areas of scarring alopecia. It is theorized that an immune response to staphylococcal superantigens contributes to this disease process.7
The clinical findings of acne keloidalis nuchae include inflammatory pustules and papules with keloidlike plaques on the posterior neck and scalp. It occurs predominantly in teenaged and adult males of African ancestry.8 Treatment is aimed at reducing inflammation and preventing exacerbating factors. Severe disease courses may lead to scarring alopecia.
- Sanmartín O, Alegre V, Martinez-Aparicio A, et al. Congenital platelike osteoma cutis: case report and review of the literature. Pediatr Dermatol. 1993;10:182-186.
- Talsania N, Jolliffe V, O’Toole EA, et al. Platelike osteoma cutis. J Am Acad Dermatol. 2009;64:613-615.
- Yeh GL, Mathur S, Wivel A, et al. GNAS1 mutation and Cbfa1 misexpression in a child with severe congenital platelike osteoma cutis. J Bone Miner Res. 2000;15:2063-2073.
- Hernandez-Martin A, Perez-Mies B, Torrelo A. Congenital plate-like osteoma cutis in an infant. Pediatr Dermatol. 2009;26:479-481.
- Zaraa I, Hawilo A, Aounallah A, et al. Inflammatory tinea capitis: a 12-year study and a review of the literature. Mycoses. 2013;56:110-116.
- Scheinfeld N. Dissecting cellulitis (perifolliculitis capitis abscedens et suffodiens): a comprehensive review focusing on new treatments and findings of the last decade with commentary comparing the therapies and causes of dissecting cellulitis to hidradenitis suppurativa. Dermatol Online J. 2014;20:22692.
- Ross EK, Tan E, Shapiro J. Update on primary cicatricial alopecias. J Am Acad Dermatol. 2005;53:1-37.
- Knable AL Jr, Hanke CW, Gonin R. Prevalence of acne keloidalis nuchae in football players. J Am Acad Dermatol. 1997;37:570-574.
The Diagnosis: Platelike Osteoma Cutis
Histopathologic examination revealed extensive cutaneous ossification in the dermis and subcutis with dermal fibrosis and minimal surrounding inflammation (Figure 1). There was no evidence of infection or neoplasm. Further evaluation did not demonstrate any additional physical dysmorphia, and there were no imbalances of calcium-phosphate metabolism or abnormalities in parathyroid hormone or thyroid hormone function. A diagnosis of platelike osteoma cutis (PLOC) was favored. Computed tomography of the head showed material at the posterior skull of similar density to the adjacent calvarial skull and centered within the dermis, consistent with osteoma cutis (Figure 2).
Osteoma cutis describes the formation of bone within the skin. It occurs when hydroxyapatite crystals in a proteinaceous matrix are deposited within the skin, ultimately leading to the formation of bone ultrastructure. Ossification of the skin most often occurs secondary to trauma, inflammation, or neoplasm; however, it rarely may be a primary event.1,2
Platelike osteoma cutis is a rare form of primary cutaneous ossification in which bone forms within the skin in a platelike manner. It most frequently affects the scalp but also has been observed on the trunk and extremities.1 A driving metabolic or endocrine abnormality typically is not identified.2
Platelike osteoma cutis can occur as an isolated finding or as a feature of Albright hereditary osteodystrophy (AHO) or progressive osseous heteroplasia (POH). In addition to cutaneous ossification, AHO involves short stature, endocrinopathy, obesity, shortened fourth and fifth metacarpals, and mental retardation. Progressive osseous heteroplasia is characterized by progressive ossification of the skin and deeper tissues such as muscle and fascia, leading to severe movement restriction; it is believed to be a localized nonprogressive variant of POH.3,4 Mutations in the guanine nucleotide binding protein, alpha stimulating activity polypeptide 1 gene, GNAS1, a key regulatory gene involved in AHO and POH, have been found in several cases of PLOC.3 Our patient lacked any dysmorphic features or laboratory abnormalities suggestive of AHO or POH. Moreover, testing of the tissue and blood for the GNAS1 mutation was negative. Treatment of PLOC often is difficult. Our patient underwent a trial of ablative fractional laser resurfacing, which failed to lead to perceivable improvement.
The differential diagnoses include a kerion, dissecting cellulitis of the scalp, folliculitis decalvans, and acne keloidalis nuchae. A kerion is a manifestation of tinea capitis characterized by an inflammatory plaque, often with pain or tenderness. Kerions most frequently occur in children aged 5 to 10 years.5 Failure to treat a kerion may result in scarring alopecia. Treatment consists of oral antifungals.
Dissecting cellulitis of the scalp is thought to occur secondary to follicular occlusion. It is characterized by boggy suppurative nodules primarily on the posterior and vertex scalp. Patchy hair loss is present and typically progresses to cicatricial alopecia. Histology characteristically shows areas of dense, predominantly neutrophilic, perifollicular dermal infiltrates.6
Folliculitis decalvans is a primary neutrophilic cicatricial alopecia that primarily occurs in adults. Patients with folliculitis decalvans tend to have multiple pustules on the periphery of confluent areas of scarring alopecia. It is theorized that an immune response to staphylococcal superantigens contributes to this disease process.7
The clinical findings of acne keloidalis nuchae include inflammatory pustules and papules with keloidlike plaques on the posterior neck and scalp. It occurs predominantly in teenaged and adult males of African ancestry.8 Treatment is aimed at reducing inflammation and preventing exacerbating factors. Severe disease courses may lead to scarring alopecia.
The Diagnosis: Platelike Osteoma Cutis
Histopathologic examination revealed extensive cutaneous ossification in the dermis and subcutis with dermal fibrosis and minimal surrounding inflammation (Figure 1). There was no evidence of infection or neoplasm. Further evaluation did not demonstrate any additional physical dysmorphia, and there were no imbalances of calcium-phosphate metabolism or abnormalities in parathyroid hormone or thyroid hormone function. A diagnosis of platelike osteoma cutis (PLOC) was favored. Computed tomography of the head showed material at the posterior skull of similar density to the adjacent calvarial skull and centered within the dermis, consistent with osteoma cutis (Figure 2).
Osteoma cutis describes the formation of bone within the skin. It occurs when hydroxyapatite crystals in a proteinaceous matrix are deposited within the skin, ultimately leading to the formation of bone ultrastructure. Ossification of the skin most often occurs secondary to trauma, inflammation, or neoplasm; however, it rarely may be a primary event.1,2
Platelike osteoma cutis is a rare form of primary cutaneous ossification in which bone forms within the skin in a platelike manner. It most frequently affects the scalp but also has been observed on the trunk and extremities.1 A driving metabolic or endocrine abnormality typically is not identified.2
Platelike osteoma cutis can occur as an isolated finding or as a feature of Albright hereditary osteodystrophy (AHO) or progressive osseous heteroplasia (POH). In addition to cutaneous ossification, AHO involves short stature, endocrinopathy, obesity, shortened fourth and fifth metacarpals, and mental retardation. Progressive osseous heteroplasia is characterized by progressive ossification of the skin and deeper tissues such as muscle and fascia, leading to severe movement restriction; it is believed to be a localized nonprogressive variant of POH.3,4 Mutations in the guanine nucleotide binding protein, alpha stimulating activity polypeptide 1 gene, GNAS1, a key regulatory gene involved in AHO and POH, have been found in several cases of PLOC.3 Our patient lacked any dysmorphic features or laboratory abnormalities suggestive of AHO or POH. Moreover, testing of the tissue and blood for the GNAS1 mutation was negative. Treatment of PLOC often is difficult. Our patient underwent a trial of ablative fractional laser resurfacing, which failed to lead to perceivable improvement.
The differential diagnoses include a kerion, dissecting cellulitis of the scalp, folliculitis decalvans, and acne keloidalis nuchae. A kerion is a manifestation of tinea capitis characterized by an inflammatory plaque, often with pain or tenderness. Kerions most frequently occur in children aged 5 to 10 years.5 Failure to treat a kerion may result in scarring alopecia. Treatment consists of oral antifungals.
Dissecting cellulitis of the scalp is thought to occur secondary to follicular occlusion. It is characterized by boggy suppurative nodules primarily on the posterior and vertex scalp. Patchy hair loss is present and typically progresses to cicatricial alopecia. Histology characteristically shows areas of dense, predominantly neutrophilic, perifollicular dermal infiltrates.6
Folliculitis decalvans is a primary neutrophilic cicatricial alopecia that primarily occurs in adults. Patients with folliculitis decalvans tend to have multiple pustules on the periphery of confluent areas of scarring alopecia. It is theorized that an immune response to staphylococcal superantigens contributes to this disease process.7
The clinical findings of acne keloidalis nuchae include inflammatory pustules and papules with keloidlike plaques on the posterior neck and scalp. It occurs predominantly in teenaged and adult males of African ancestry.8 Treatment is aimed at reducing inflammation and preventing exacerbating factors. Severe disease courses may lead to scarring alopecia.
- Sanmartín O, Alegre V, Martinez-Aparicio A, et al. Congenital platelike osteoma cutis: case report and review of the literature. Pediatr Dermatol. 1993;10:182-186.
- Talsania N, Jolliffe V, O’Toole EA, et al. Platelike osteoma cutis. J Am Acad Dermatol. 2009;64:613-615.
- Yeh GL, Mathur S, Wivel A, et al. GNAS1 mutation and Cbfa1 misexpression in a child with severe congenital platelike osteoma cutis. J Bone Miner Res. 2000;15:2063-2073.
- Hernandez-Martin A, Perez-Mies B, Torrelo A. Congenital plate-like osteoma cutis in an infant. Pediatr Dermatol. 2009;26:479-481.
- Zaraa I, Hawilo A, Aounallah A, et al. Inflammatory tinea capitis: a 12-year study and a review of the literature. Mycoses. 2013;56:110-116.
- Scheinfeld N. Dissecting cellulitis (perifolliculitis capitis abscedens et suffodiens): a comprehensive review focusing on new treatments and findings of the last decade with commentary comparing the therapies and causes of dissecting cellulitis to hidradenitis suppurativa. Dermatol Online J. 2014;20:22692.
- Ross EK, Tan E, Shapiro J. Update on primary cicatricial alopecias. J Am Acad Dermatol. 2005;53:1-37.
- Knable AL Jr, Hanke CW, Gonin R. Prevalence of acne keloidalis nuchae in football players. J Am Acad Dermatol. 1997;37:570-574.
- Sanmartín O, Alegre V, Martinez-Aparicio A, et al. Congenital platelike osteoma cutis: case report and review of the literature. Pediatr Dermatol. 1993;10:182-186.
- Talsania N, Jolliffe V, O’Toole EA, et al. Platelike osteoma cutis. J Am Acad Dermatol. 2009;64:613-615.
- Yeh GL, Mathur S, Wivel A, et al. GNAS1 mutation and Cbfa1 misexpression in a child with severe congenital platelike osteoma cutis. J Bone Miner Res. 2000;15:2063-2073.
- Hernandez-Martin A, Perez-Mies B, Torrelo A. Congenital plate-like osteoma cutis in an infant. Pediatr Dermatol. 2009;26:479-481.
- Zaraa I, Hawilo A, Aounallah A, et al. Inflammatory tinea capitis: a 12-year study and a review of the literature. Mycoses. 2013;56:110-116.
- Scheinfeld N. Dissecting cellulitis (perifolliculitis capitis abscedens et suffodiens): a comprehensive review focusing on new treatments and findings of the last decade with commentary comparing the therapies and causes of dissecting cellulitis to hidradenitis suppurativa. Dermatol Online J. 2014;20:22692.
- Ross EK, Tan E, Shapiro J. Update on primary cicatricial alopecias. J Am Acad Dermatol. 2005;53:1-37.
- Knable AL Jr, Hanke CW, Gonin R. Prevalence of acne keloidalis nuchae in football players. J Am Acad Dermatol. 1997;37:570-574.
A 35-year-old man presented to the dermatology clinic with a slow-growing plaque on the scalp of 10 years’ duration. The lesion was mildly pruritic and was never associated with any pain or discharge. He denied antecedent trauma or infection. A hard, erythematous, nodular, alopecic plaque with punctate hyperkeratosis on the left posterior temporal and parietal scalp was noted on physical examination. The lesion was slightly tender to palpation.
Argyria From a Topical Home Remedy
To the Editor:
Argyria is a rare disease caused by chronic exposure to products with high silver content (eg, oral ingestion, inhalation, percutaneous absorption). With time, the blood levels of silver surpass the body’s renal and hepatic excretory capacities that lead to silver granules being deposited in the skin and internal organs, including the liver, spleen, adrenal glands, and bone marrow.1 The cutaneous deposition results in a blue or blue-gray pigmentation of the skin, mucous membranes, and nails. Intervals of exposure that span from 8 months to 5 years prior to symptom onset have been described in the literature.2 The discoloration that results often is permanent, with no established way of effectively removing silver deposits from the tissue.3
A 22-year-old autistic man, who was completely dependent on his mother’s care, presented to the emergency department with a primary concern of abdominal pain. The mother reported that he was indicating abdominal pain by motioning to his stomach for the last 5 days. The mother also reported he did not have a bowel movement during this time, and she noticed his hands were shaking. Prior to presentation, the mother had given him 2 enemas and had him on a 3-day strict liquid fast consisting of water, lemon juice, cayenne pepper, honey, and orange juice. Notably, the mother had a strong history of using naturopathic remedies for treatment of her son’s ailments.
On admission, the patient was stable. There was a 2-point decrease in the patient’s body mass index over the last month. Initial serum electrolytes were highly abnormal with a serum sodium level of 124 mEq/L (reference range, 135–145 mEq/L), blood urea nitrogen of 3 mg/dL (reference range, 7–20 mg/dL), creatinine of 0.77 mg/dL (reference range, 0.74–1.35 mg/dL), and lactic acid of 2.1 mEq/L (reference range, 0.5–1 mEq/L). Serum osmolality was 272 mOsm/kg (reference range, 275–295 mOsm/kg). Urine osmolality was 114 mOsm/kg (reference range, 500–850 mOsm/kg) with a low-normal urine sodium level of 41 mmol/24 hr (reference range, 40–220 mmol/24 hr). Abnormalities were felt to be secondary to malnutrition from the strict liquid diet (blood urea nitrogen and creatinine ratio of 3:1 suggestive of notable protein calorie malnutrition). The patient was given 1 L of normal saline in the emergency department, with further fluids held so as not to increase serum sodium level too rapidly. A regular diet was started.
Physical examination revealed dry mucosal membranes but otherwise was unremarkable. Active bowel sounds were noted, as well as a soft, nontender, and nondistended abdomen; however, when examining the patient’s hands for reported shaking, a distinct abnormality of the nails was noticed. The patient had slate blue discoloration of the lunula, along with hyperpigmented violaceous discoloration of the proximal nail bed on all 10 fingernails (Figure 1). No abnormalities were seen on the toenails. The mother had a distinct bluish gray discoloration of the face as well as similar nail findings (Figure 2), strongly suggestive of colloidal silver use. An urgent serum silver level was ordered on the patient as well as a heavy metal panel. The mother was found applying numerous “natural remedies” to the patient’s skin while in the hospital, including a liquid spray and lotion, both in unmarked bottles. At that time, the mother was informed that no external supplements should be applied to her son. The serum silver level was elevated substantially at 94.3 ng/mL (reference range, <1.0 ng/mL). When the mother was confronted, she initially denied use of silver but later admitted to notable silver content in the cream she was applying to her son’s skin. The mother reported that she read online that colloidal silver had been historically used to cure numerous ailments and she was ordering products from an online company. She was counseled on the dangers of both topical application and ingestion of silver, and all supplements were removed from the home.
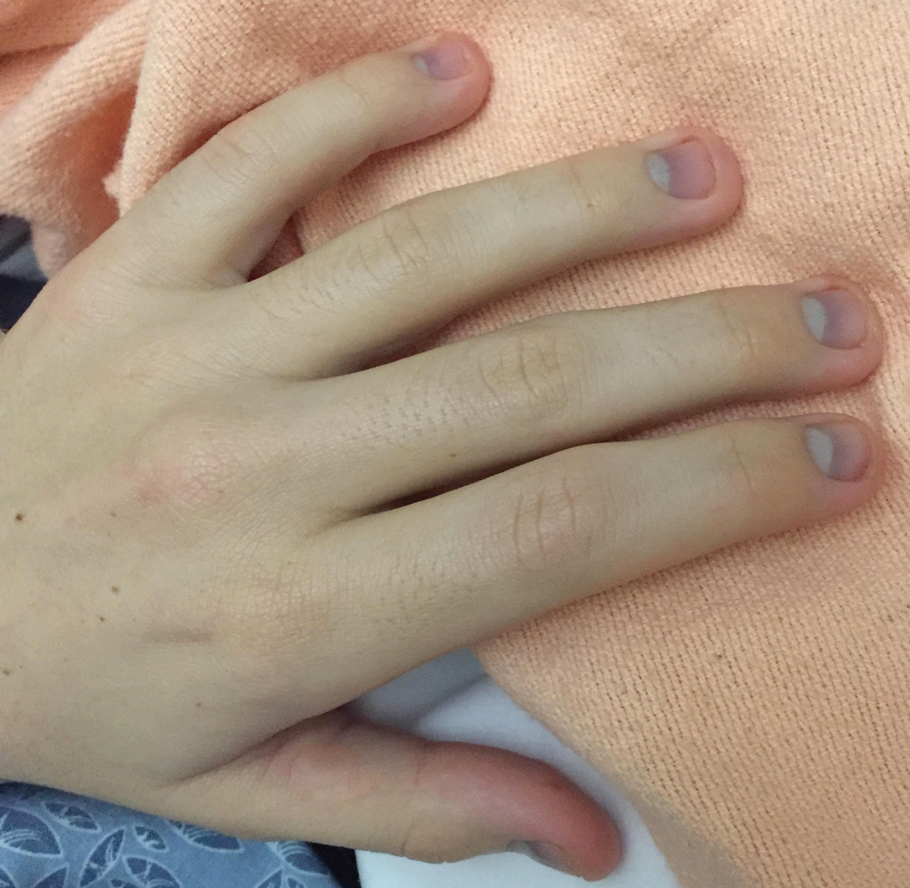
Argyria is a rare condition caused by chronic exposure to silver and is characterized by a blue-gray pigmentation in the skin and appendages, mucous membranes, and internal organs.4 Clinically, argyria is classified as generalized or localized. Generalized argyria results from ingestion or inhalation of silver compounds, where granules deposit preferentially in sun-exposed areas of skin as well as internal organs, with the highest concentration in the liver, spleen, and adrenal glands; discoloration often is permanent.5 On the contrary, localized argyria results from direct external contact with silver and granules deposited in the hands, eyes, and mucosa.5 Although the exact mechanism of penetration from topical silver remains unknown, it is thought to enter via the eccrine sweat ducts, as histopathology reveals silver granules found in highest concentration surrounding sweat glands in the dermis.6
Initial differential diagnoses for altered nail pigmentation include drug-induced causes, systemic diseases, cyanosis, and exposure to metals.7 The most commonly indicated medications resulting in blue nail pigment changes include antimalarials, minocycline, zidovudine, and phenothiazine. Systemic diseases that may cause blue nail color change include Wilson disease, hemochromatosis, Addison disease, methemoglobinemia, and alkaptonuria.7 Metals include gold, mercury, arsenic, bismuth, lead, and silver.4 After a thorough review of the patient’s medications and lack of support for any underlying disease process, contact with metals, particularly silver, was ranked highly on our differential list. In support of this theory, the mother’s bluish gray facial skin led to high clinical suspicion that she was ingesting colloidal silver and also was exposing her son to silver.
Treatment of argyria is challenging but first and foremost involves discontinuation of the source of chronic silver exposure. Unfortunately, the discoloration of generalized argyria often is permanent. Sunscreen can be used to help prevent any further darkening of pigment. The pigment in localized argyria has been reported to slowly fade with time, and there also have been reports of successful treatment using a low-fluence Q-switched 1064-nm Nd:YAG laser.8
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Lencastre A, Lobo M, João A. Argyria—case report. An Bras Dermatol. 2013;88:413-416.
- Park S-W, Kim J-H, Shin H-T, et al. An effective modality for argyria treatment: Q-switched 1,064-nm Nd:YAG laser. Ann Dermatol. 2013;25:511-512.
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Garcias-Ladaria J, Hernandez-Bel P, Torregrosa-Calatayud JL, et al. Localized cutaneous argyria: a report of 2 cases. Actas Dermosifiliogr. 2013;104:253-254.
- Kapur N, Landon G, Yu RC. Localized argyria in an antique restorer. Br J Dermatol. 2001;144:191-192.
- Kubba A, Kubba R, Batrani M, Pal T. Argyria an unrecognized cause of cutaneous pigmentation in Indian patients: a case series and review of the literature. Indian J Dermatol Venereol Leprol. 2013;79:805-811.
- Han TY, Chang HS, Lee HK, et al. Successful treatment of argyria using a low-fluence Q-switched 1064-nm Nd:YAG laser. Int J Dermatol. 2011;50:751-753.
To the Editor:
Argyria is a rare disease caused by chronic exposure to products with high silver content (eg, oral ingestion, inhalation, percutaneous absorption). With time, the blood levels of silver surpass the body’s renal and hepatic excretory capacities that lead to silver granules being deposited in the skin and internal organs, including the liver, spleen, adrenal glands, and bone marrow.1 The cutaneous deposition results in a blue or blue-gray pigmentation of the skin, mucous membranes, and nails. Intervals of exposure that span from 8 months to 5 years prior to symptom onset have been described in the literature.2 The discoloration that results often is permanent, with no established way of effectively removing silver deposits from the tissue.3
A 22-year-old autistic man, who was completely dependent on his mother’s care, presented to the emergency department with a primary concern of abdominal pain. The mother reported that he was indicating abdominal pain by motioning to his stomach for the last 5 days. The mother also reported he did not have a bowel movement during this time, and she noticed his hands were shaking. Prior to presentation, the mother had given him 2 enemas and had him on a 3-day strict liquid fast consisting of water, lemon juice, cayenne pepper, honey, and orange juice. Notably, the mother had a strong history of using naturopathic remedies for treatment of her son’s ailments.
On admission, the patient was stable. There was a 2-point decrease in the patient’s body mass index over the last month. Initial serum electrolytes were highly abnormal with a serum sodium level of 124 mEq/L (reference range, 135–145 mEq/L), blood urea nitrogen of 3 mg/dL (reference range, 7–20 mg/dL), creatinine of 0.77 mg/dL (reference range, 0.74–1.35 mg/dL), and lactic acid of 2.1 mEq/L (reference range, 0.5–1 mEq/L). Serum osmolality was 272 mOsm/kg (reference range, 275–295 mOsm/kg). Urine osmolality was 114 mOsm/kg (reference range, 500–850 mOsm/kg) with a low-normal urine sodium level of 41 mmol/24 hr (reference range, 40–220 mmol/24 hr). Abnormalities were felt to be secondary to malnutrition from the strict liquid diet (blood urea nitrogen and creatinine ratio of 3:1 suggestive of notable protein calorie malnutrition). The patient was given 1 L of normal saline in the emergency department, with further fluids held so as not to increase serum sodium level too rapidly. A regular diet was started.
Physical examination revealed dry mucosal membranes but otherwise was unremarkable. Active bowel sounds were noted, as well as a soft, nontender, and nondistended abdomen; however, when examining the patient’s hands for reported shaking, a distinct abnormality of the nails was noticed. The patient had slate blue discoloration of the lunula, along with hyperpigmented violaceous discoloration of the proximal nail bed on all 10 fingernails (Figure 1). No abnormalities were seen on the toenails. The mother had a distinct bluish gray discoloration of the face as well as similar nail findings (Figure 2), strongly suggestive of colloidal silver use. An urgent serum silver level was ordered on the patient as well as a heavy metal panel. The mother was found applying numerous “natural remedies” to the patient’s skin while in the hospital, including a liquid spray and lotion, both in unmarked bottles. At that time, the mother was informed that no external supplements should be applied to her son. The serum silver level was elevated substantially at 94.3 ng/mL (reference range, <1.0 ng/mL). When the mother was confronted, she initially denied use of silver but later admitted to notable silver content in the cream she was applying to her son’s skin. The mother reported that she read online that colloidal silver had been historically used to cure numerous ailments and she was ordering products from an online company. She was counseled on the dangers of both topical application and ingestion of silver, and all supplements were removed from the home.
Argyria is a rare condition caused by chronic exposure to silver and is characterized by a blue-gray pigmentation in the skin and appendages, mucous membranes, and internal organs.4 Clinically, argyria is classified as generalized or localized. Generalized argyria results from ingestion or inhalation of silver compounds, where granules deposit preferentially in sun-exposed areas of skin as well as internal organs, with the highest concentration in the liver, spleen, and adrenal glands; discoloration often is permanent.5 On the contrary, localized argyria results from direct external contact with silver and granules deposited in the hands, eyes, and mucosa.5 Although the exact mechanism of penetration from topical silver remains unknown, it is thought to enter via the eccrine sweat ducts, as histopathology reveals silver granules found in highest concentration surrounding sweat glands in the dermis.6
Initial differential diagnoses for altered nail pigmentation include drug-induced causes, systemic diseases, cyanosis, and exposure to metals.7 The most commonly indicated medications resulting in blue nail pigment changes include antimalarials, minocycline, zidovudine, and phenothiazine. Systemic diseases that may cause blue nail color change include Wilson disease, hemochromatosis, Addison disease, methemoglobinemia, and alkaptonuria.7 Metals include gold, mercury, arsenic, bismuth, lead, and silver.4 After a thorough review of the patient’s medications and lack of support for any underlying disease process, contact with metals, particularly silver, was ranked highly on our differential list. In support of this theory, the mother’s bluish gray facial skin led to high clinical suspicion that she was ingesting colloidal silver and also was exposing her son to silver.
Treatment of argyria is challenging but first and foremost involves discontinuation of the source of chronic silver exposure. Unfortunately, the discoloration of generalized argyria often is permanent. Sunscreen can be used to help prevent any further darkening of pigment. The pigment in localized argyria has been reported to slowly fade with time, and there also have been reports of successful treatment using a low-fluence Q-switched 1064-nm Nd:YAG laser.8
To the Editor:
Argyria is a rare disease caused by chronic exposure to products with high silver content (eg, oral ingestion, inhalation, percutaneous absorption). With time, the blood levels of silver surpass the body’s renal and hepatic excretory capacities that lead to silver granules being deposited in the skin and internal organs, including the liver, spleen, adrenal glands, and bone marrow.1 The cutaneous deposition results in a blue or blue-gray pigmentation of the skin, mucous membranes, and nails. Intervals of exposure that span from 8 months to 5 years prior to symptom onset have been described in the literature.2 The discoloration that results often is permanent, with no established way of effectively removing silver deposits from the tissue.3
A 22-year-old autistic man, who was completely dependent on his mother’s care, presented to the emergency department with a primary concern of abdominal pain. The mother reported that he was indicating abdominal pain by motioning to his stomach for the last 5 days. The mother also reported he did not have a bowel movement during this time, and she noticed his hands were shaking. Prior to presentation, the mother had given him 2 enemas and had him on a 3-day strict liquid fast consisting of water, lemon juice, cayenne pepper, honey, and orange juice. Notably, the mother had a strong history of using naturopathic remedies for treatment of her son’s ailments.
On admission, the patient was stable. There was a 2-point decrease in the patient’s body mass index over the last month. Initial serum electrolytes were highly abnormal with a serum sodium level of 124 mEq/L (reference range, 135–145 mEq/L), blood urea nitrogen of 3 mg/dL (reference range, 7–20 mg/dL), creatinine of 0.77 mg/dL (reference range, 0.74–1.35 mg/dL), and lactic acid of 2.1 mEq/L (reference range, 0.5–1 mEq/L). Serum osmolality was 272 mOsm/kg (reference range, 275–295 mOsm/kg). Urine osmolality was 114 mOsm/kg (reference range, 500–850 mOsm/kg) with a low-normal urine sodium level of 41 mmol/24 hr (reference range, 40–220 mmol/24 hr). Abnormalities were felt to be secondary to malnutrition from the strict liquid diet (blood urea nitrogen and creatinine ratio of 3:1 suggestive of notable protein calorie malnutrition). The patient was given 1 L of normal saline in the emergency department, with further fluids held so as not to increase serum sodium level too rapidly. A regular diet was started.
Physical examination revealed dry mucosal membranes but otherwise was unremarkable. Active bowel sounds were noted, as well as a soft, nontender, and nondistended abdomen; however, when examining the patient’s hands for reported shaking, a distinct abnormality of the nails was noticed. The patient had slate blue discoloration of the lunula, along with hyperpigmented violaceous discoloration of the proximal nail bed on all 10 fingernails (Figure 1). No abnormalities were seen on the toenails. The mother had a distinct bluish gray discoloration of the face as well as similar nail findings (Figure 2), strongly suggestive of colloidal silver use. An urgent serum silver level was ordered on the patient as well as a heavy metal panel. The mother was found applying numerous “natural remedies” to the patient’s skin while in the hospital, including a liquid spray and lotion, both in unmarked bottles. At that time, the mother was informed that no external supplements should be applied to her son. The serum silver level was elevated substantially at 94.3 ng/mL (reference range, <1.0 ng/mL). When the mother was confronted, she initially denied use of silver but later admitted to notable silver content in the cream she was applying to her son’s skin. The mother reported that she read online that colloidal silver had been historically used to cure numerous ailments and she was ordering products from an online company. She was counseled on the dangers of both topical application and ingestion of silver, and all supplements were removed from the home.
Argyria is a rare condition caused by chronic exposure to silver and is characterized by a blue-gray pigmentation in the skin and appendages, mucous membranes, and internal organs.4 Clinically, argyria is classified as generalized or localized. Generalized argyria results from ingestion or inhalation of silver compounds, where granules deposit preferentially in sun-exposed areas of skin as well as internal organs, with the highest concentration in the liver, spleen, and adrenal glands; discoloration often is permanent.5 On the contrary, localized argyria results from direct external contact with silver and granules deposited in the hands, eyes, and mucosa.5 Although the exact mechanism of penetration from topical silver remains unknown, it is thought to enter via the eccrine sweat ducts, as histopathology reveals silver granules found in highest concentration surrounding sweat glands in the dermis.6
Initial differential diagnoses for altered nail pigmentation include drug-induced causes, systemic diseases, cyanosis, and exposure to metals.7 The most commonly indicated medications resulting in blue nail pigment changes include antimalarials, minocycline, zidovudine, and phenothiazine. Systemic diseases that may cause blue nail color change include Wilson disease, hemochromatosis, Addison disease, methemoglobinemia, and alkaptonuria.7 Metals include gold, mercury, arsenic, bismuth, lead, and silver.4 After a thorough review of the patient’s medications and lack of support for any underlying disease process, contact with metals, particularly silver, was ranked highly on our differential list. In support of this theory, the mother’s bluish gray facial skin led to high clinical suspicion that she was ingesting colloidal silver and also was exposing her son to silver.
Treatment of argyria is challenging but first and foremost involves discontinuation of the source of chronic silver exposure. Unfortunately, the discoloration of generalized argyria often is permanent. Sunscreen can be used to help prevent any further darkening of pigment. The pigment in localized argyria has been reported to slowly fade with time, and there also have been reports of successful treatment using a low-fluence Q-switched 1064-nm Nd:YAG laser.8
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Lencastre A, Lobo M, João A. Argyria—case report. An Bras Dermatol. 2013;88:413-416.
- Park S-W, Kim J-H, Shin H-T, et al. An effective modality for argyria treatment: Q-switched 1,064-nm Nd:YAG laser. Ann Dermatol. 2013;25:511-512.
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Garcias-Ladaria J, Hernandez-Bel P, Torregrosa-Calatayud JL, et al. Localized cutaneous argyria: a report of 2 cases. Actas Dermosifiliogr. 2013;104:253-254.
- Kapur N, Landon G, Yu RC. Localized argyria in an antique restorer. Br J Dermatol. 2001;144:191-192.
- Kubba A, Kubba R, Batrani M, Pal T. Argyria an unrecognized cause of cutaneous pigmentation in Indian patients: a case series and review of the literature. Indian J Dermatol Venereol Leprol. 2013;79:805-811.
- Han TY, Chang HS, Lee HK, et al. Successful treatment of argyria using a low-fluence Q-switched 1064-nm Nd:YAG laser. Int J Dermatol. 2011;50:751-753.
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Lencastre A, Lobo M, João A. Argyria—case report. An Bras Dermatol. 2013;88:413-416.
- Park S-W, Kim J-H, Shin H-T, et al. An effective modality for argyria treatment: Q-switched 1,064-nm Nd:YAG laser. Ann Dermatol. 2013;25:511-512.
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Garcias-Ladaria J, Hernandez-Bel P, Torregrosa-Calatayud JL, et al. Localized cutaneous argyria: a report of 2 cases. Actas Dermosifiliogr. 2013;104:253-254.
- Kapur N, Landon G, Yu RC. Localized argyria in an antique restorer. Br J Dermatol. 2001;144:191-192.
- Kubba A, Kubba R, Batrani M, Pal T. Argyria an unrecognized cause of cutaneous pigmentation in Indian patients: a case series and review of the literature. Indian J Dermatol Venereol Leprol. 2013;79:805-811.
- Han TY, Chang HS, Lee HK, et al. Successful treatment of argyria using a low-fluence Q-switched 1064-nm Nd:YAG laser. Int J Dermatol. 2011;50:751-753.
Practice Points
- Argyria results from chronic exposure to products with a high silver content and may result in abnormalities of the skin and internal organs.
- Examination of the fingernails can provide important clues to underlying systemic conditions or external exposures.
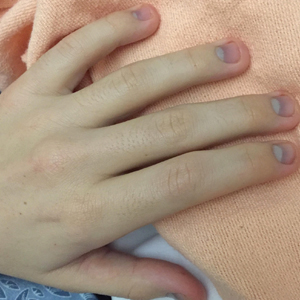
Squamoid Eccrine Ductal Carcinoma
Squamoid eccrine ductal carcinoma (SEDC) is an aggressive underrecognized cutaneous malignancy of unknown etiology.1 It is most likely to occur in sun-exposed areas of the body, most commonly the head and neck. Risk factors include male sex, increased age, and chronic immunosuppression.1-4 Current reports suggest that SEDC is likely a high-grade subtype of squamous cell carcinoma (SCC) with a high risk for local recurrence (25%) and metastasis (13%).1,3,5,6 There are as few as 56 cases of SEDC reported in the literature; however, the number of cases may be closer to 100 due to SEDC being classified as either adenosquamous carcinoma of the skin or ductal eccrine carcinoma with squamous differentiation.1
Clinically, SEDC mimics keratinocyte carcinomas. Histologically, SEDC is biphasic, with a superficial portion resembling well-differentiated SCC and a deeply invasive portion having infiltrative irregular cords with ductal differentiation. Perineural invasion (PNI) frequently is present. Multiple connections to the overlying epidermis also can be seen, serving as a subtle clue to the diagnosis on broad superficial specimens.1-3 Due to superficial sampling, approximately 50% of reported cases are misdiagnosed as SCC during the initial biopsy.4 The diagnosis of SEDC often is made during complete excision when deeper tissue is sampled. Establishing an accurate diagnosis is important given the more aggressive nature of SEDC compared with SCC and its proclivity for PNI.1,3,6 The purpose of this review is to increase awareness of this underrecognized entity and describe the histologic findings that help distinguish SEDC from SCC.
Patient Chart Review
We reviewed chart notes as well as frozen and formalin-fixed paraffin-embedded tissue sections from all 5 patients diagnosed with SEDC at a single institution between November 2018 and May 2020. The mean age of patients was 81 years, and 4 were male. Four of the patients presented for MMS with a preoperative diagnosis of SCC per the original biopsy results. Only 1 patient had a preoperative diagnosis of SEDC. The details of each case are recorded in the Table. All tumors were greater than 2 cm in diameter on initial presentation, were located on the head, and clinically resembled keratinocyte carcinoma with either a nodular or plaquelike appearance (Figure 1).
Intraoperative histologic examination of the excised tissue revealed a biphasic pattern consisting of superficial SCC features overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation in all 5 patients (Figures 2–4). Immunohistochemical staining with cytokeratin AE1/AE3 revealed thin strands of carcinoma in the mid to deeper dermis with squamous differentiation and eccrine ductal differentiation (Figure 5), thus confirming the diagnosis in all 5 patients.
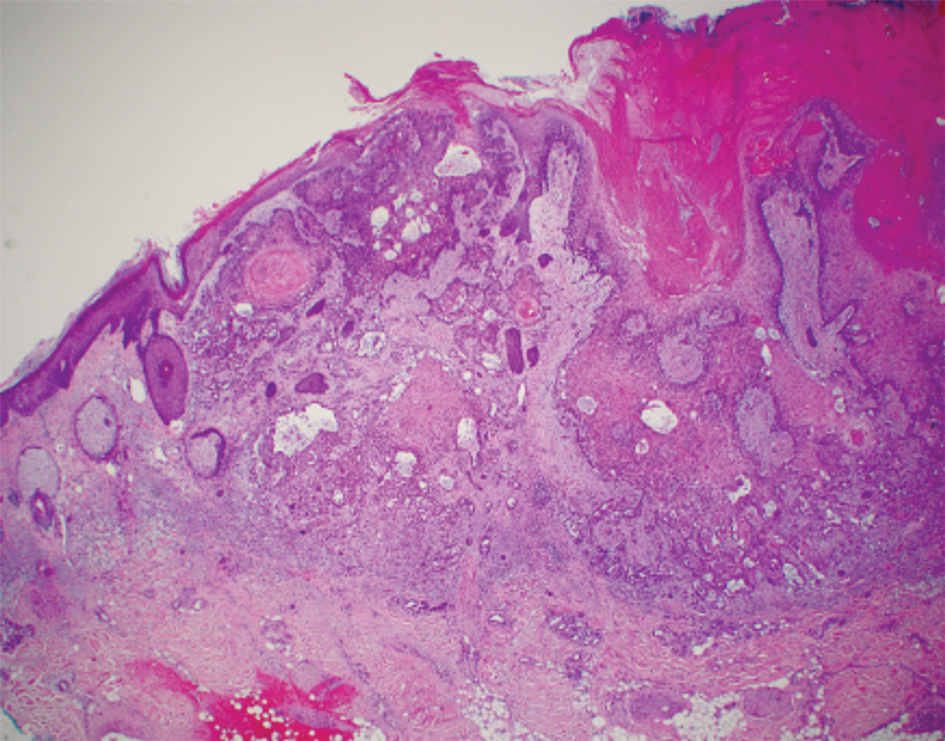


The median depth of tumor invasion was 4.1 mm (range, 2.2–5.45 mm). Ulceration was seen in 3 of the patients, and PNI of large-caliber nerves was observed in all 5 patients. A connection with the overlying epidermis was present in all 5 patients. All 5 patients required more than 1 Mohs stage for complete tumor clearance (Table).
In 4 of the patients, nodal imaging performed at the time of diagnosis revealed no evidence of metastasis. Two patients received adjuvant radiation therapy, and none demonstrated evidence of recurrence. The mean follow-up time was 11 months (range, 6.5–18 months) for the 4 cases with available follow-up data (Table).
Literature Review
A PubMed review of the literature using the search term squamoid eccrine ductal carcinoma resulted in 28 articles, 19 of which were included in the review based on inclusion criteria (original articles available in English, in full text, and pertained to SEDC). Our review yielded 56 cases of SEDC.1-19 The mean age of patients with SEDC was 72 years. The number of male and female cases was 52% (29/56) and 48% (27/56), respectively. The most common location of SEDC was on the head or neck (71% [40/56]), followed by the extremities (19% [11/56]). Immunosuppression was noted in 9% (5/56) of cases. Wide local excision was the most commonly employed treatment modality (91% [51/56]), with MMS being used in 4 patients (7%). Adjuvant radiation was reported in 5% (3/56) of cases. Perineural invasion was reported in 34% (19/56) of cases. Recurrence was seen in 23% (13/56) of cases, with a mean time to recurrence of 10.4 months. Metastasis to regional lymph nodes was observed in 13% (7/56) of cases, with 7% (4/56) of those cases having distant metastases.
Comment
Squamoid eccrine ductal carcinoma was successfully treated with MMS in all 5 of the patients we reviewed. Recognition of a distinct biphasic pattern consisting of squamous differentiation superficially with epidermal connection overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation should lead to consideration of this diagnosis. A thorough inspection for PNI also should be performed, as this finding was present in all of 5 cases and in 34% of reported cases in our literature review.
The differential diagnosis for SEDC includes SCC, metastatic adenocarcinoma with squamoid features, and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma (MAC), and porocarcinoma with squamous differentiation. The combination of histologic features with the immunoexpression profile of carcinoembryonic antigen (CEA), epithelial membrane antigen (EMA), cytokeratin (CK) 5/6, and p63 can effectively exclude the other entities in the differential and confirm the diagnosis of SEDC.1,3,4 While the diagnosis of SEDC relies on the specific histologic features of multiple surface attachments and superficial squamoid changes with deep ductular elements, immunohistochemistry can nonetheless be adjunctive in difficult cases. Positive immunohistochemical staining for CEA and EMA can help to highlight and delineate true glandular elements, whereas CK5/6 highlights the overall contour of the tumor, displaying more clearly the multiple epidermal attachments and the subtle infiltrative nature of the deeper components of invasive cords and ducts. In addition, the combination of CK5/6 and p63 positivity supports the primary cutaneous nature of the lesion rather than metastatic adenocarcinoma.13,20 Other markers of eccrine secretory coils, such as CK7, CAM5.2, and S100, also are sometimes used for confirmation, some of which can aid in distinction from noneccrine sweat gland differentiation, as CK7 and CAM5.2 are negative in both luminal and basal cells of the dermal duct while being positive within the secretory coil, and S100 protein is expressed within eccrine secretory coil but negative within the apocrine sweat glands.2,4,21
The clinical findings from our chart review corroborated those reported in the literature. The mean age of SEDC in the 5 patients we reviewed was 81 years, and all cases presented on the head, consistent with the findings observed in the literature. Although 4 of our cases were male, there may not be a difference in risk based on sex as previously thought.1 Our literature review revealed an almost equivalent percentage of male and female cases, with 52% being male.
Immunosuppression has been associated with an increased risk for SEDC. Our literature review revealed that approximately 9% (5/56) of cases occurred in immunosuppressed individuals. Two of these reported cases were in the setting of underlying chronic lymphocytic leukemia, 2 in individuals with a history of organ transplant, and 1 treated with azathioprine for myasthenia gravis.2,4,10,12,13 Our chart review supported this correlation, as all 5 patients had a medical history potentially consistent with being in an immunocompromised state (Table). Notably, patient 5 represents a unique case of SEDC occurring in the setting of HIV. The patient had HIV for 33 years, with his most recent CD4+ count of 794 mm3 and HIV-1 RNA load of 35 copies/mL. Given that HIV-positive individuals may have more than a 2-fold increased risk of SCC, a greater degree of suspicion for SEDC should be maintained for these patients.22,23
The etiology of SEDC is controversial but is thought to be either an SCC arising from eccrine glands or a variant of eccrine carcinoma with extensive squamoid differentiation.4,6,13,14,17,24 While SEDC certainly appears to share the proclivity for PNI with the malignant eccrine tumor MAC, it is simultaneously quite distinct, demonstrating nuclear pleomorphism and mitotic activity, both of which are lacking in the bland nature of MACs.12,25
The exact prevalence of SEDC is difficult to ascertain because of its frequent misdiagnosis and variable nomenclature used within the literature. Most reported cases of SEDC are mistakenly diagnosed as SCC on the initial shave or punch biopsy because of superficial sampling. This also was the case in 4 of the patients we reviewed. In addition, there are reported cases of SEDC that were referred to by the investigators as cutaneous adenosquamous carcinoma (cASC), among other descriptors, such as ductal eccrine carcinoma with squamous differentiation, adnexal carcinoma with squamous and ductal differentiation, and syringoid eccrine carcinoma.26-32 While the World Health Organization classifies SEDC as a distinct variant of cASC, which is a rare variant of SCC in itself, the 2 can be differentiated. Despite the similar clinical and histologic features shared between cASC and SEDC, the neoplastic aggregates in SEDC exhibit ductal differentiation containing lumina positive for CEA and EMA.4 Overall, we favor the term squamoid eccrine ductal carcinoma, as there has recently been more uniformity for the designation of this disease entity as such.
It is unclear whether the high incidence of local recurrence (23% [13/56]) of SEDC reported in the literature is related to the treatment modality employed (ie, wide local excision) or due to the innate aggressiveness of SEDC.1,3,5 The literature has shown that MMS has lower recurrence rates than other treatments at 5-year follow-up for SCC (3.1%–5%) and eccrine carcinomas (0%–5%).33,34 Although studies assessing tumor behavior or comparing treatment modalities are limited because of the rarity and underrecognition of SEDC, MMS has been used several times for SEDC with only 1 recurrence reported.4,13,17,24 Given that all 5 of the patients we reviewed required more than 1 Mohs stage for complete tumor clearance and none demonstrated evidence of recurrence or metastasis (Table), we recommend MMS as the treatment of choice for SEDC.
Conclusion
Squamoid eccrine ductal carcinoma is a rare but likely underdiagnosed cutaneous tumor of uncertain etiology. Because of its propensity for recurrence and metastasis, excision of SEDC with complete circumferential peripheral and deep margin assessment with close follow-up is recommended.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Jacob J, Kugelman L. Squamoid eccrine ductal carcinoma. Cutis. 2018;101:378-380, 385.
- Yim S, Lee YH, Chae SW, et al. Squamoid eccrine ductal carcinoma of the ear helix. Clin Case Rep. 2019;7:1409-1411.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Jung YH, Jo HJ, Kang MS. Squamoid eccrine ductal carcinoma of the scalp. Korean J Pathol. 2012;46:278-281.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;91:799-802.
- Phan K, Kim L, Lim P, et al. A case report of temple squamoid eccrine ductal carcinoma: a diagnostic challenge beneath the tip of the iceberg. Dermatol Ther. 2020;33:E13213.
- McKissack SS, Wohltmann W, Dalton SR, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Lobo-Jardim MM, Souza BdCE, Kakizaki P, et al. Dermoscopy of squamoid eccrine ductal carcinoma: an aid for early diagnosis. An Bras Dermatol. 2018;93:893-895.
- Chan H, Howard V, Moir D, et al. Squamoid eccrine ductal carcinoma of the scalp. Aust J Dermatol. 2016;57:E117-E119.
- Wang B, Jarell AD, Bingham JL, et al. PET/CT imaging of squamoid eccrine ductal carcinoma. Clin Nucl Med. 2015;40:322-324.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma). J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. J Clin Aesthet Dermatol. 2013;6:33-36.
- Pusiol T, Morichetti D, Zorzi MG, et al. Squamoid eccrine ductal carcinoma: inappropriate diagnosis. Dermatol Surg. 2011;37:1819-1820.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Wasserman DI, Sack J, Gonzalez-Serva A, et al. Sentinel lymph node biopsy for a squamoid eccrine carcinoma with lymphatic invasion. Dermatol Surg. 2007;33:1126-1129.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Herrero J, Monteagudo C, Jorda E, et al. Squamoid eccrine ductal carcinoma. Histopathology. 1998;32:478-480.
- Wong TY, Suster S, Mihm MC. Squamoid eccrine ductal carcinoma. Histopathology. 1997;30:288-293.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Dabbs DJ. Diagnostic Immunohistochemistry: Theranostic and Genomic Applications. 4th ed. Elsevier/Saunders; 2014.
- Silverberg MJ, Leyden W, Warton EM, et al. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J Natl Cancer Inst. 2013;105:350-360.
- Asgari MM, Ray GT, Quesenberry CP Jr, et al. Association of multiple primary skin cancers with human immunodeficiency virus infection, CD4 count, and viral load. JAMA Dermatol. 2017;153:892-896.
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207.
- Kazakov DV. Cutaneous Adnexal Tumors. Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2012.
- Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin- and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
- Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
- Ko CJ, Leffell DJ, McNiff JM. Adenosquamous carcinoma: a report of nine cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
- Patel V, Squires SM, Liu DY, et al. Cutaneous adenosquamous carcinoma: a rare neoplasm with biphasic differentiation. Cutis. 2014;94:231-233.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
- Azorín D, López-Ríos F, Ballestín C, et al. Primary cutaneous adenosquamous carcinoma: a case report and review of the literature. J Cutan Pathol. 2001;28:542-545.
- Wildemore JK, Lee JB, Humphreys TR. Mohs surgery for malignant eccrine neoplasms. Dermatol Surg. 2004;30(12 pt 2):1574-1579.
- Garcia-Zuazaga J, Olbricht SM. Cutaneous squamous cell carcinoma. Adv Dermatol. 2008;24:33-57.
Squamoid eccrine ductal carcinoma (SEDC) is an aggressive underrecognized cutaneous malignancy of unknown etiology.1 It is most likely to occur in sun-exposed areas of the body, most commonly the head and neck. Risk factors include male sex, increased age, and chronic immunosuppression.1-4 Current reports suggest that SEDC is likely a high-grade subtype of squamous cell carcinoma (SCC) with a high risk for local recurrence (25%) and metastasis (13%).1,3,5,6 There are as few as 56 cases of SEDC reported in the literature; however, the number of cases may be closer to 100 due to SEDC being classified as either adenosquamous carcinoma of the skin or ductal eccrine carcinoma with squamous differentiation.1
Clinically, SEDC mimics keratinocyte carcinomas. Histologically, SEDC is biphasic, with a superficial portion resembling well-differentiated SCC and a deeply invasive portion having infiltrative irregular cords with ductal differentiation. Perineural invasion (PNI) frequently is present. Multiple connections to the overlying epidermis also can be seen, serving as a subtle clue to the diagnosis on broad superficial specimens.1-3 Due to superficial sampling, approximately 50% of reported cases are misdiagnosed as SCC during the initial biopsy.4 The diagnosis of SEDC often is made during complete excision when deeper tissue is sampled. Establishing an accurate diagnosis is important given the more aggressive nature of SEDC compared with SCC and its proclivity for PNI.1,3,6 The purpose of this review is to increase awareness of this underrecognized entity and describe the histologic findings that help distinguish SEDC from SCC.
Patient Chart Review
We reviewed chart notes as well as frozen and formalin-fixed paraffin-embedded tissue sections from all 5 patients diagnosed with SEDC at a single institution between November 2018 and May 2020. The mean age of patients was 81 years, and 4 were male. Four of the patients presented for MMS with a preoperative diagnosis of SCC per the original biopsy results. Only 1 patient had a preoperative diagnosis of SEDC. The details of each case are recorded in the Table. All tumors were greater than 2 cm in diameter on initial presentation, were located on the head, and clinically resembled keratinocyte carcinoma with either a nodular or plaquelike appearance (Figure 1).
Intraoperative histologic examination of the excised tissue revealed a biphasic pattern consisting of superficial SCC features overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation in all 5 patients (Figures 2–4). Immunohistochemical staining with cytokeratin AE1/AE3 revealed thin strands of carcinoma in the mid to deeper dermis with squamous differentiation and eccrine ductal differentiation (Figure 5), thus confirming the diagnosis in all 5 patients.
The median depth of tumor invasion was 4.1 mm (range, 2.2–5.45 mm). Ulceration was seen in 3 of the patients, and PNI of large-caliber nerves was observed in all 5 patients. A connection with the overlying epidermis was present in all 5 patients. All 5 patients required more than 1 Mohs stage for complete tumor clearance (Table).
In 4 of the patients, nodal imaging performed at the time of diagnosis revealed no evidence of metastasis. Two patients received adjuvant radiation therapy, and none demonstrated evidence of recurrence. The mean follow-up time was 11 months (range, 6.5–18 months) for the 4 cases with available follow-up data (Table).
Literature Review
A PubMed review of the literature using the search term squamoid eccrine ductal carcinoma resulted in 28 articles, 19 of which were included in the review based on inclusion criteria (original articles available in English, in full text, and pertained to SEDC). Our review yielded 56 cases of SEDC.1-19 The mean age of patients with SEDC was 72 years. The number of male and female cases was 52% (29/56) and 48% (27/56), respectively. The most common location of SEDC was on the head or neck (71% [40/56]), followed by the extremities (19% [11/56]). Immunosuppression was noted in 9% (5/56) of cases. Wide local excision was the most commonly employed treatment modality (91% [51/56]), with MMS being used in 4 patients (7%). Adjuvant radiation was reported in 5% (3/56) of cases. Perineural invasion was reported in 34% (19/56) of cases. Recurrence was seen in 23% (13/56) of cases, with a mean time to recurrence of 10.4 months. Metastasis to regional lymph nodes was observed in 13% (7/56) of cases, with 7% (4/56) of those cases having distant metastases.
Comment
Squamoid eccrine ductal carcinoma was successfully treated with MMS in all 5 of the patients we reviewed. Recognition of a distinct biphasic pattern consisting of squamous differentiation superficially with epidermal connection overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation should lead to consideration of this diagnosis. A thorough inspection for PNI also should be performed, as this finding was present in all of 5 cases and in 34% of reported cases in our literature review.
The differential diagnosis for SEDC includes SCC, metastatic adenocarcinoma with squamoid features, and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma (MAC), and porocarcinoma with squamous differentiation. The combination of histologic features with the immunoexpression profile of carcinoembryonic antigen (CEA), epithelial membrane antigen (EMA), cytokeratin (CK) 5/6, and p63 can effectively exclude the other entities in the differential and confirm the diagnosis of SEDC.1,3,4 While the diagnosis of SEDC relies on the specific histologic features of multiple surface attachments and superficial squamoid changes with deep ductular elements, immunohistochemistry can nonetheless be adjunctive in difficult cases. Positive immunohistochemical staining for CEA and EMA can help to highlight and delineate true glandular elements, whereas CK5/6 highlights the overall contour of the tumor, displaying more clearly the multiple epidermal attachments and the subtle infiltrative nature of the deeper components of invasive cords and ducts. In addition, the combination of CK5/6 and p63 positivity supports the primary cutaneous nature of the lesion rather than metastatic adenocarcinoma.13,20 Other markers of eccrine secretory coils, such as CK7, CAM5.2, and S100, also are sometimes used for confirmation, some of which can aid in distinction from noneccrine sweat gland differentiation, as CK7 and CAM5.2 are negative in both luminal and basal cells of the dermal duct while being positive within the secretory coil, and S100 protein is expressed within eccrine secretory coil but negative within the apocrine sweat glands.2,4,21
The clinical findings from our chart review corroborated those reported in the literature. The mean age of SEDC in the 5 patients we reviewed was 81 years, and all cases presented on the head, consistent with the findings observed in the literature. Although 4 of our cases were male, there may not be a difference in risk based on sex as previously thought.1 Our literature review revealed an almost equivalent percentage of male and female cases, with 52% being male.
Immunosuppression has been associated with an increased risk for SEDC. Our literature review revealed that approximately 9% (5/56) of cases occurred in immunosuppressed individuals. Two of these reported cases were in the setting of underlying chronic lymphocytic leukemia, 2 in individuals with a history of organ transplant, and 1 treated with azathioprine for myasthenia gravis.2,4,10,12,13 Our chart review supported this correlation, as all 5 patients had a medical history potentially consistent with being in an immunocompromised state (Table). Notably, patient 5 represents a unique case of SEDC occurring in the setting of HIV. The patient had HIV for 33 years, with his most recent CD4+ count of 794 mm3 and HIV-1 RNA load of 35 copies/mL. Given that HIV-positive individuals may have more than a 2-fold increased risk of SCC, a greater degree of suspicion for SEDC should be maintained for these patients.22,23
The etiology of SEDC is controversial but is thought to be either an SCC arising from eccrine glands or a variant of eccrine carcinoma with extensive squamoid differentiation.4,6,13,14,17,24 While SEDC certainly appears to share the proclivity for PNI with the malignant eccrine tumor MAC, it is simultaneously quite distinct, demonstrating nuclear pleomorphism and mitotic activity, both of which are lacking in the bland nature of MACs.12,25
The exact prevalence of SEDC is difficult to ascertain because of its frequent misdiagnosis and variable nomenclature used within the literature. Most reported cases of SEDC are mistakenly diagnosed as SCC on the initial shave or punch biopsy because of superficial sampling. This also was the case in 4 of the patients we reviewed. In addition, there are reported cases of SEDC that were referred to by the investigators as cutaneous adenosquamous carcinoma (cASC), among other descriptors, such as ductal eccrine carcinoma with squamous differentiation, adnexal carcinoma with squamous and ductal differentiation, and syringoid eccrine carcinoma.26-32 While the World Health Organization classifies SEDC as a distinct variant of cASC, which is a rare variant of SCC in itself, the 2 can be differentiated. Despite the similar clinical and histologic features shared between cASC and SEDC, the neoplastic aggregates in SEDC exhibit ductal differentiation containing lumina positive for CEA and EMA.4 Overall, we favor the term squamoid eccrine ductal carcinoma, as there has recently been more uniformity for the designation of this disease entity as such.
It is unclear whether the high incidence of local recurrence (23% [13/56]) of SEDC reported in the literature is related to the treatment modality employed (ie, wide local excision) or due to the innate aggressiveness of SEDC.1,3,5 The literature has shown that MMS has lower recurrence rates than other treatments at 5-year follow-up for SCC (3.1%–5%) and eccrine carcinomas (0%–5%).33,34 Although studies assessing tumor behavior or comparing treatment modalities are limited because of the rarity and underrecognition of SEDC, MMS has been used several times for SEDC with only 1 recurrence reported.4,13,17,24 Given that all 5 of the patients we reviewed required more than 1 Mohs stage for complete tumor clearance and none demonstrated evidence of recurrence or metastasis (Table), we recommend MMS as the treatment of choice for SEDC.
Conclusion
Squamoid eccrine ductal carcinoma is a rare but likely underdiagnosed cutaneous tumor of uncertain etiology. Because of its propensity for recurrence and metastasis, excision of SEDC with complete circumferential peripheral and deep margin assessment with close follow-up is recommended.
Squamoid eccrine ductal carcinoma (SEDC) is an aggressive underrecognized cutaneous malignancy of unknown etiology.1 It is most likely to occur in sun-exposed areas of the body, most commonly the head and neck. Risk factors include male sex, increased age, and chronic immunosuppression.1-4 Current reports suggest that SEDC is likely a high-grade subtype of squamous cell carcinoma (SCC) with a high risk for local recurrence (25%) and metastasis (13%).1,3,5,6 There are as few as 56 cases of SEDC reported in the literature; however, the number of cases may be closer to 100 due to SEDC being classified as either adenosquamous carcinoma of the skin or ductal eccrine carcinoma with squamous differentiation.1
Clinically, SEDC mimics keratinocyte carcinomas. Histologically, SEDC is biphasic, with a superficial portion resembling well-differentiated SCC and a deeply invasive portion having infiltrative irregular cords with ductal differentiation. Perineural invasion (PNI) frequently is present. Multiple connections to the overlying epidermis also can be seen, serving as a subtle clue to the diagnosis on broad superficial specimens.1-3 Due to superficial sampling, approximately 50% of reported cases are misdiagnosed as SCC during the initial biopsy.4 The diagnosis of SEDC often is made during complete excision when deeper tissue is sampled. Establishing an accurate diagnosis is important given the more aggressive nature of SEDC compared with SCC and its proclivity for PNI.1,3,6 The purpose of this review is to increase awareness of this underrecognized entity and describe the histologic findings that help distinguish SEDC from SCC.
Patient Chart Review
We reviewed chart notes as well as frozen and formalin-fixed paraffin-embedded tissue sections from all 5 patients diagnosed with SEDC at a single institution between November 2018 and May 2020. The mean age of patients was 81 years, and 4 were male. Four of the patients presented for MMS with a preoperative diagnosis of SCC per the original biopsy results. Only 1 patient had a preoperative diagnosis of SEDC. The details of each case are recorded in the Table. All tumors were greater than 2 cm in diameter on initial presentation, were located on the head, and clinically resembled keratinocyte carcinoma with either a nodular or plaquelike appearance (Figure 1).
Intraoperative histologic examination of the excised tissue revealed a biphasic pattern consisting of superficial SCC features overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation in all 5 patients (Figures 2–4). Immunohistochemical staining with cytokeratin AE1/AE3 revealed thin strands of carcinoma in the mid to deeper dermis with squamous differentiation and eccrine ductal differentiation (Figure 5), thus confirming the diagnosis in all 5 patients.
The median depth of tumor invasion was 4.1 mm (range, 2.2–5.45 mm). Ulceration was seen in 3 of the patients, and PNI of large-caliber nerves was observed in all 5 patients. A connection with the overlying epidermis was present in all 5 patients. All 5 patients required more than 1 Mohs stage for complete tumor clearance (Table).
In 4 of the patients, nodal imaging performed at the time of diagnosis revealed no evidence of metastasis. Two patients received adjuvant radiation therapy, and none demonstrated evidence of recurrence. The mean follow-up time was 11 months (range, 6.5–18 months) for the 4 cases with available follow-up data (Table).
Literature Review
A PubMed review of the literature using the search term squamoid eccrine ductal carcinoma resulted in 28 articles, 19 of which were included in the review based on inclusion criteria (original articles available in English, in full text, and pertained to SEDC). Our review yielded 56 cases of SEDC.1-19 The mean age of patients with SEDC was 72 years. The number of male and female cases was 52% (29/56) and 48% (27/56), respectively. The most common location of SEDC was on the head or neck (71% [40/56]), followed by the extremities (19% [11/56]). Immunosuppression was noted in 9% (5/56) of cases. Wide local excision was the most commonly employed treatment modality (91% [51/56]), with MMS being used in 4 patients (7%). Adjuvant radiation was reported in 5% (3/56) of cases. Perineural invasion was reported in 34% (19/56) of cases. Recurrence was seen in 23% (13/56) of cases, with a mean time to recurrence of 10.4 months. Metastasis to regional lymph nodes was observed in 13% (7/56) of cases, with 7% (4/56) of those cases having distant metastases.
Comment
Squamoid eccrine ductal carcinoma was successfully treated with MMS in all 5 of the patients we reviewed. Recognition of a distinct biphasic pattern consisting of squamous differentiation superficially with epidermal connection overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation should lead to consideration of this diagnosis. A thorough inspection for PNI also should be performed, as this finding was present in all of 5 cases and in 34% of reported cases in our literature review.
The differential diagnosis for SEDC includes SCC, metastatic adenocarcinoma with squamoid features, and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma (MAC), and porocarcinoma with squamous differentiation. The combination of histologic features with the immunoexpression profile of carcinoembryonic antigen (CEA), epithelial membrane antigen (EMA), cytokeratin (CK) 5/6, and p63 can effectively exclude the other entities in the differential and confirm the diagnosis of SEDC.1,3,4 While the diagnosis of SEDC relies on the specific histologic features of multiple surface attachments and superficial squamoid changes with deep ductular elements, immunohistochemistry can nonetheless be adjunctive in difficult cases. Positive immunohistochemical staining for CEA and EMA can help to highlight and delineate true glandular elements, whereas CK5/6 highlights the overall contour of the tumor, displaying more clearly the multiple epidermal attachments and the subtle infiltrative nature of the deeper components of invasive cords and ducts. In addition, the combination of CK5/6 and p63 positivity supports the primary cutaneous nature of the lesion rather than metastatic adenocarcinoma.13,20 Other markers of eccrine secretory coils, such as CK7, CAM5.2, and S100, also are sometimes used for confirmation, some of which can aid in distinction from noneccrine sweat gland differentiation, as CK7 and CAM5.2 are negative in both luminal and basal cells of the dermal duct while being positive within the secretory coil, and S100 protein is expressed within eccrine secretory coil but negative within the apocrine sweat glands.2,4,21
The clinical findings from our chart review corroborated those reported in the literature. The mean age of SEDC in the 5 patients we reviewed was 81 years, and all cases presented on the head, consistent with the findings observed in the literature. Although 4 of our cases were male, there may not be a difference in risk based on sex as previously thought.1 Our literature review revealed an almost equivalent percentage of male and female cases, with 52% being male.
Immunosuppression has been associated with an increased risk for SEDC. Our literature review revealed that approximately 9% (5/56) of cases occurred in immunosuppressed individuals. Two of these reported cases were in the setting of underlying chronic lymphocytic leukemia, 2 in individuals with a history of organ transplant, and 1 treated with azathioprine for myasthenia gravis.2,4,10,12,13 Our chart review supported this correlation, as all 5 patients had a medical history potentially consistent with being in an immunocompromised state (Table). Notably, patient 5 represents a unique case of SEDC occurring in the setting of HIV. The patient had HIV for 33 years, with his most recent CD4+ count of 794 mm3 and HIV-1 RNA load of 35 copies/mL. Given that HIV-positive individuals may have more than a 2-fold increased risk of SCC, a greater degree of suspicion for SEDC should be maintained for these patients.22,23
The etiology of SEDC is controversial but is thought to be either an SCC arising from eccrine glands or a variant of eccrine carcinoma with extensive squamoid differentiation.4,6,13,14,17,24 While SEDC certainly appears to share the proclivity for PNI with the malignant eccrine tumor MAC, it is simultaneously quite distinct, demonstrating nuclear pleomorphism and mitotic activity, both of which are lacking in the bland nature of MACs.12,25
The exact prevalence of SEDC is difficult to ascertain because of its frequent misdiagnosis and variable nomenclature used within the literature. Most reported cases of SEDC are mistakenly diagnosed as SCC on the initial shave or punch biopsy because of superficial sampling. This also was the case in 4 of the patients we reviewed. In addition, there are reported cases of SEDC that were referred to by the investigators as cutaneous adenosquamous carcinoma (cASC), among other descriptors, such as ductal eccrine carcinoma with squamous differentiation, adnexal carcinoma with squamous and ductal differentiation, and syringoid eccrine carcinoma.26-32 While the World Health Organization classifies SEDC as a distinct variant of cASC, which is a rare variant of SCC in itself, the 2 can be differentiated. Despite the similar clinical and histologic features shared between cASC and SEDC, the neoplastic aggregates in SEDC exhibit ductal differentiation containing lumina positive for CEA and EMA.4 Overall, we favor the term squamoid eccrine ductal carcinoma, as there has recently been more uniformity for the designation of this disease entity as such.
It is unclear whether the high incidence of local recurrence (23% [13/56]) of SEDC reported in the literature is related to the treatment modality employed (ie, wide local excision) or due to the innate aggressiveness of SEDC.1,3,5 The literature has shown that MMS has lower recurrence rates than other treatments at 5-year follow-up for SCC (3.1%–5%) and eccrine carcinomas (0%–5%).33,34 Although studies assessing tumor behavior or comparing treatment modalities are limited because of the rarity and underrecognition of SEDC, MMS has been used several times for SEDC with only 1 recurrence reported.4,13,17,24 Given that all 5 of the patients we reviewed required more than 1 Mohs stage for complete tumor clearance and none demonstrated evidence of recurrence or metastasis (Table), we recommend MMS as the treatment of choice for SEDC.
Conclusion
Squamoid eccrine ductal carcinoma is a rare but likely underdiagnosed cutaneous tumor of uncertain etiology. Because of its propensity for recurrence and metastasis, excision of SEDC with complete circumferential peripheral and deep margin assessment with close follow-up is recommended.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Jacob J, Kugelman L. Squamoid eccrine ductal carcinoma. Cutis. 2018;101:378-380, 385.
- Yim S, Lee YH, Chae SW, et al. Squamoid eccrine ductal carcinoma of the ear helix. Clin Case Rep. 2019;7:1409-1411.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Jung YH, Jo HJ, Kang MS. Squamoid eccrine ductal carcinoma of the scalp. Korean J Pathol. 2012;46:278-281.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;91:799-802.
- Phan K, Kim L, Lim P, et al. A case report of temple squamoid eccrine ductal carcinoma: a diagnostic challenge beneath the tip of the iceberg. Dermatol Ther. 2020;33:E13213.
- McKissack SS, Wohltmann W, Dalton SR, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Lobo-Jardim MM, Souza BdCE, Kakizaki P, et al. Dermoscopy of squamoid eccrine ductal carcinoma: an aid for early diagnosis. An Bras Dermatol. 2018;93:893-895.
- Chan H, Howard V, Moir D, et al. Squamoid eccrine ductal carcinoma of the scalp. Aust J Dermatol. 2016;57:E117-E119.
- Wang B, Jarell AD, Bingham JL, et al. PET/CT imaging of squamoid eccrine ductal carcinoma. Clin Nucl Med. 2015;40:322-324.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma). J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. J Clin Aesthet Dermatol. 2013;6:33-36.
- Pusiol T, Morichetti D, Zorzi MG, et al. Squamoid eccrine ductal carcinoma: inappropriate diagnosis. Dermatol Surg. 2011;37:1819-1820.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Wasserman DI, Sack J, Gonzalez-Serva A, et al. Sentinel lymph node biopsy for a squamoid eccrine carcinoma with lymphatic invasion. Dermatol Surg. 2007;33:1126-1129.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Herrero J, Monteagudo C, Jorda E, et al. Squamoid eccrine ductal carcinoma. Histopathology. 1998;32:478-480.
- Wong TY, Suster S, Mihm MC. Squamoid eccrine ductal carcinoma. Histopathology. 1997;30:288-293.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Dabbs DJ. Diagnostic Immunohistochemistry: Theranostic and Genomic Applications. 4th ed. Elsevier/Saunders; 2014.
- Silverberg MJ, Leyden W, Warton EM, et al. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J Natl Cancer Inst. 2013;105:350-360.
- Asgari MM, Ray GT, Quesenberry CP Jr, et al. Association of multiple primary skin cancers with human immunodeficiency virus infection, CD4 count, and viral load. JAMA Dermatol. 2017;153:892-896.
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207.
- Kazakov DV. Cutaneous Adnexal Tumors. Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2012.
- Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin- and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
- Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
- Ko CJ, Leffell DJ, McNiff JM. Adenosquamous carcinoma: a report of nine cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
- Patel V, Squires SM, Liu DY, et al. Cutaneous adenosquamous carcinoma: a rare neoplasm with biphasic differentiation. Cutis. 2014;94:231-233.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
- Azorín D, López-Ríos F, Ballestín C, et al. Primary cutaneous adenosquamous carcinoma: a case report and review of the literature. J Cutan Pathol. 2001;28:542-545.
- Wildemore JK, Lee JB, Humphreys TR. Mohs surgery for malignant eccrine neoplasms. Dermatol Surg. 2004;30(12 pt 2):1574-1579.
- Garcia-Zuazaga J, Olbricht SM. Cutaneous squamous cell carcinoma. Adv Dermatol. 2008;24:33-57.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Jacob J, Kugelman L. Squamoid eccrine ductal carcinoma. Cutis. 2018;101:378-380, 385.
- Yim S, Lee YH, Chae SW, et al. Squamoid eccrine ductal carcinoma of the ear helix. Clin Case Rep. 2019;7:1409-1411.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Jung YH, Jo HJ, Kang MS. Squamoid eccrine ductal carcinoma of the scalp. Korean J Pathol. 2012;46:278-281.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;91:799-802.
- Phan K, Kim L, Lim P, et al. A case report of temple squamoid eccrine ductal carcinoma: a diagnostic challenge beneath the tip of the iceberg. Dermatol Ther. 2020;33:E13213.
- McKissack SS, Wohltmann W, Dalton SR, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Lobo-Jardim MM, Souza BdCE, Kakizaki P, et al. Dermoscopy of squamoid eccrine ductal carcinoma: an aid for early diagnosis. An Bras Dermatol. 2018;93:893-895.
- Chan H, Howard V, Moir D, et al. Squamoid eccrine ductal carcinoma of the scalp. Aust J Dermatol. 2016;57:E117-E119.
- Wang B, Jarell AD, Bingham JL, et al. PET/CT imaging of squamoid eccrine ductal carcinoma. Clin Nucl Med. 2015;40:322-324.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma). J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. J Clin Aesthet Dermatol. 2013;6:33-36.
- Pusiol T, Morichetti D, Zorzi MG, et al. Squamoid eccrine ductal carcinoma: inappropriate diagnosis. Dermatol Surg. 2011;37:1819-1820.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Wasserman DI, Sack J, Gonzalez-Serva A, et al. Sentinel lymph node biopsy for a squamoid eccrine carcinoma with lymphatic invasion. Dermatol Surg. 2007;33:1126-1129.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Herrero J, Monteagudo C, Jorda E, et al. Squamoid eccrine ductal carcinoma. Histopathology. 1998;32:478-480.
- Wong TY, Suster S, Mihm MC. Squamoid eccrine ductal carcinoma. Histopathology. 1997;30:288-293.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Dabbs DJ. Diagnostic Immunohistochemistry: Theranostic and Genomic Applications. 4th ed. Elsevier/Saunders; 2014.
- Silverberg MJ, Leyden W, Warton EM, et al. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J Natl Cancer Inst. 2013;105:350-360.
- Asgari MM, Ray GT, Quesenberry CP Jr, et al. Association of multiple primary skin cancers with human immunodeficiency virus infection, CD4 count, and viral load. JAMA Dermatol. 2017;153:892-896.
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207.
- Kazakov DV. Cutaneous Adnexal Tumors. Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2012.
- Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin- and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
- Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
- Ko CJ, Leffell DJ, McNiff JM. Adenosquamous carcinoma: a report of nine cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
- Patel V, Squires SM, Liu DY, et al. Cutaneous adenosquamous carcinoma: a rare neoplasm with biphasic differentiation. Cutis. 2014;94:231-233.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
- Azorín D, López-Ríos F, Ballestín C, et al. Primary cutaneous adenosquamous carcinoma: a case report and review of the literature. J Cutan Pathol. 2001;28:542-545.
- Wildemore JK, Lee JB, Humphreys TR. Mohs surgery for malignant eccrine neoplasms. Dermatol Surg. 2004;30(12 pt 2):1574-1579.
- Garcia-Zuazaga J, Olbricht SM. Cutaneous squamous cell carcinoma. Adv Dermatol. 2008;24:33-57.
PRACTICE POINTS
- Squamoid eccrine ductal carcinoma is an aggressive underrecognized cutaneous malignancy that often is misdiagnosed as squamous cell carcinoma (SCC) during initial biopsy.
- Squamoid eccrine ductal carcinoma has a biphasic histologic appearance with a superficial portion resembling well-differentiated SCC and a deeply invasive portion comprised of infiltrative irregular cords with ductal differentiation.
- Excision with complete circumferential peripheral and deep margin assessment with close follow-up is recommended for these patients because of the high risk for recurrence and metastasis.