User login
Advanced-stage calciphylaxis: Think before you punch
A 53-year-old woman presented with extensive, nonulcerated, painful plaques on both calves. She had long-standing diabetes mellitus and had recently started hemodialysis. She had no fever or trauma and did not appear to be in shock.
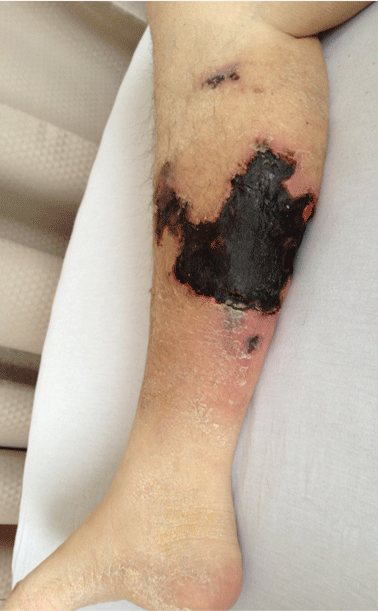
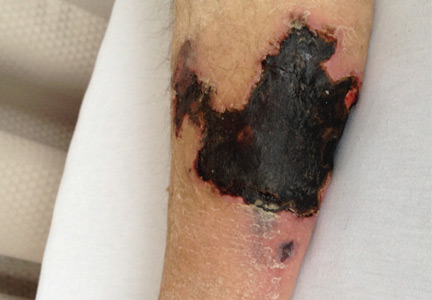
On physical examination, she had extensive, well-demarcated, nonulcerated, indurated dark eschar over the right calf (Figure 1). Her left calf had similar lesions that appeared as focal, discrete, nonulcerated, violaceous plaques, with associated tenderness. No significant erythema, edema, drainage, or fluctuance was noted.
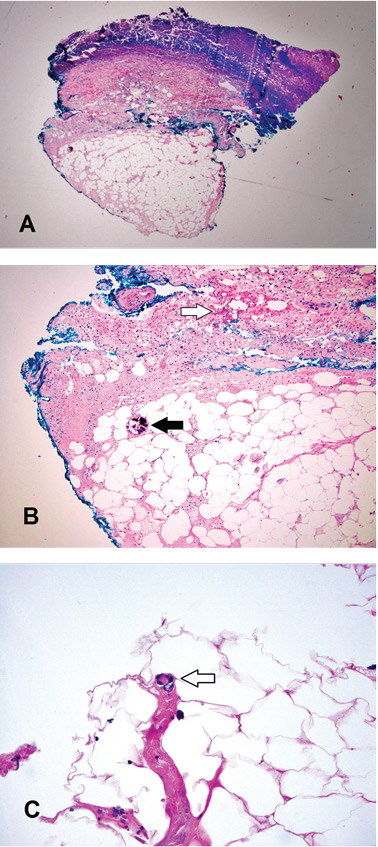
A broad-spectrum antibiotic was started empirically but was discontinued when routine blood testing and magnetic resonance imaging showed no evidence of infection. Histologic study of a full-thickness skin biopsy specimen (Figure 2) showed tissue necrosis, ulceration, and concentric calcification of small and medium-sized blood vessels, many with luminal thrombi, all of which together were diagnostic for calciphylaxis.
Treatment was started with cinacalcet, low-calcium dialysis baths, phosphate binders, and sodium thiosulfate. However, within a few days of the biopsy procedure, an infection developed at the biopsy site, and the patient developed sepsis and septic shock. She received broad-spectrum antibiotics and underwent extensive debridement with wound care. After a protracted hospital course, the infection resolved.
CALCIPHYLAXIS RISK FACTORS
Calciphylaxis, also referred to as calcific uremic arteriolopathy, is a rare and often fatal condition in patients with end-stage renal disease who are on hemodialysis (1% to 4% of dialysis patients).1–3 It is also seen in patients who have undergone renal transplant and in patients with chronic kidney disease who have a chronic inflammatory disease or who have been exposed to corticosteroids or warfarin. However, it can also occur in patients without chronic kidney disease or end-stage renal disease.
The term “calcific uremic arteriolopathy” is a misnomer, as this condition can occur in patients with normal renal function (nonuremic calciphylaxis). Also, despite what the term calciphylaxis implies, there is no systemic anaphylaxis.3–5
Documented risk factors include obesity; female sex; use of warfarin, corticosteroids, or vitamin D analogues; low serum albumin; hypercoagulable states; hyperparathyroidism; alcoholic liver disease; elevated calcium-phosphorus product; inflammation; connective tissue disease; and cancer.4–6
DIAGNOSTIC CLUES
There are no strict guidelines for the diagnosis of calciphylaxis, and the exact pathophysiology of calciphylaxis is not understood.1–4
Ulceration is considered the clinical hallmark, but there are increasing reports of patients presenting with nonulcerated plaques, as in our patient. The literature suggests a mortality rate of 33% at 6 months in these patients, but ulceration increases the risk of death to over 80%, and sepsis is the leading cause of death.7,8
Histologic features identified on full-thickness biopsy specimens are intravascular deposition of calcium in the media of the blood vessels, as well as fibrin thrombi formation, intimal proliferation, tissue necrosis, and resultant ischemia. However, as in our patient and as discussed below, the biopsy procedure can induce or exacerbate ulceration, increasing the risk of sepsis, and is thus controversial.7
In the early stages, lesions of calciphylaxis are focal and appear as erythema or livedo reticularis with or without subcutaneous plaques or ulcers. As the disease progresses, the ischemic changes coalesce to form denser violaceous, painful, plaquelike subcutaneous nodules with eschar. In the advanced stages, the eschar or ulceration involves an extensive area.
Diagnosis in the early stages is challenging because of the focal nature of involvement. The differential diagnosis includes potentially fatal conditions such as systemic vasculitis, nephrogenic systemic fibrosis, pyoderma gangrenosum, gangrene from peripheral arterial disease, cholesterol embolization, warfarin-induced necrosis, purpura fulminans, and oxalate vasculopathy.7
In the advanced stages, the diagnosis of calciphylaxis is clinically more evident, and the differential diagnosis usually narrows. Well-demarcated, necrotic, indurated lesions that are bilateral in a patient with end-stage renal disease without shock makes the diagnosis very likely.
The dangers of biopsy
As seen in our patient, biopsy for histologic confirmation of calciphylaxis can increase the risk of infection and sepsis.7 Also, the efficacy and clinical utility are uncertain because the quantity or depth of tissue obtained may not be enough for diagnosis. Deep incisional cutaneous biopsy is needed rather than punch biopsy to provide ample subcutaneous tissue for histologic study.3
Further, the biopsy procedure induces ulceration in the region of the incision, increasing the risk of infection and poor healing and escalating the risk of sepsis and death.7–9 Since extensive necrosis predisposes to a negative biopsy, a high clinical suspicion should drive early treatment of calciphylaxis.10 Noninvasive imaging studies such as plain radiography and bone scintigraphy can aid the diagnosis by detecting moderate to severe soft-tissue vascular calcification in these areas.7–11
DEBRIDEMENT IS CONTROVERSIAL
Conservative measures are the mainstay of care and include dietary alterations, noncalcium and nonaluminum phosphate binders, and low-calcium bath dialysis. There is mounting evidence for the use of calcimimetics and sodium thiosulfate.7,12–14
The role of wound debridement is controversial, as concomitant poor peripheral vascular perfusion can delay wound healing and, if ulceration ensues, there is a dramatic escalation of mortality risk. The decision for wound debridement is determined case by case, based on an assessment of the comorbidities, vascular perfusion, and status of the eschar.
Extensive wound debridement should be considered immediately after biopsy or with any signs of ulceration or infection—this in addition to meticulous wound care, which will promote healing and prevent serious complications secondary to infection.15
A TEAM APPROACH IMPROVES OUTCOMES
A multidisciplinary approach involving surgeons, nephrologists, dermatologists, dermatopathologists, wound or burn care team, nutrition team, pain management team, and infectious disease team is important to improve outcomes.7
Management mainly involves controlling pain; avoiding local trauma; treating and preventing infection; stopping causative agents such as warfarin and corticosteroids; intensive hemodialysis with an increase in both frequency and duration; intravenous sodium thiosulphate; non-calcium-phosphorus binders and cinacalcet in patients with elevated parathyroid hormone; and hyperbaric oxygen.12–14 There are also reports of success with oral etidronate and intravenous pamidronate.16,17
- Spanakis EK, Sellmeyer DE. Nonuremic calciphylaxis precipitated by teriparatide [rhPTH(1-34)] therapy in the setting of chronic warfarin and glucocorticoid treatment. Osteoporos Int 2014; 25:1411–1414.
- Brandenburg VM, Cozzolino M, Ketteler M. Calciphylaxis: a still unmet challenge. J Nephrol 2011; 24:142–148.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial 2002; 15:172–186.
- Rimtepathip P, Cohen D. A rare presentation of calciphylaxis in normal renal function. Int J Case Rep Images 2015; 6:366–369.
- Lonowski S, Martin S, Worswick S. Widespread calciphylaxis and normal renal function: no improvement with sodium thiosulfate. Dermatol Online J 2015; 21:13030/qt76845802.
- Zhou Q, Neubauer J, Kern JS, Grotz W, Walz G, Huber TB. Calciphylaxis. Lancet 2014; 383:1067.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis 2015; 66:133–146.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int 2002; 61:2210–2217.
- Hayashi M. Calciphylaxis: diagnosis and clinical features. Clin Exp Nephrol 2013; 17:498–503.
- Stavros K, Motiwala R, Zhou L, Sejdiu F, Shin S. Calciphylaxis in a dialysis patient diagnosed by muscle biopsy. J Clin Neuromuscul Dis 2014; 15:108–111.
- Bonchak JG, Park KK, Vethanayagamony T, Sheikh MM, Winterfield LS. Calciphylaxis: a case series and the role of radiology in diagnosis. Int J Dermatol 2015. [Epub ahead of print]
- Ross EA. Evolution of treatment strategies for calciphylaxis. Am J Nephrol 2011; 34:460–467.
- Cicone JS, Petronis JB, Embert CD, Spector DA. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am J Kidney Dis 2004; 43:1104–1108.
- Brandenburg VM, Kramann R, Specht P, Ketteler M. Calciphylaxis in CKD and beyond. Nephrol Dial Transplant 2012; 27:1314–1318.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage 2004; 50:64–66.
- Shiraishi N, Kitamura K, Miyoshi T, et al. Successful treatment of a patient with severe calcific uremic arteriolopathy (calciphylaxis) by etidronate disodium. Am J Kidney Dis 2006; 48:151–154.
- Hanafusa T, Yamaguchi Y, Tani M, Umegaki N, Nishimura Y, Katayama I. Intractable wounds caused by calcific uremic arteriolopathy treated with bisphosphonates. J Am Acad Dermatol 2007; 57:1021–1025.
A 53-year-old woman presented with extensive, nonulcerated, painful plaques on both calves. She had long-standing diabetes mellitus and had recently started hemodialysis. She had no fever or trauma and did not appear to be in shock.
On physical examination, she had extensive, well-demarcated, nonulcerated, indurated dark eschar over the right calf (Figure 1). Her left calf had similar lesions that appeared as focal, discrete, nonulcerated, violaceous plaques, with associated tenderness. No significant erythema, edema, drainage, or fluctuance was noted.
A broad-spectrum antibiotic was started empirically but was discontinued when routine blood testing and magnetic resonance imaging showed no evidence of infection. Histologic study of a full-thickness skin biopsy specimen (Figure 2) showed tissue necrosis, ulceration, and concentric calcification of small and medium-sized blood vessels, many with luminal thrombi, all of which together were diagnostic for calciphylaxis.
Treatment was started with cinacalcet, low-calcium dialysis baths, phosphate binders, and sodium thiosulfate. However, within a few days of the biopsy procedure, an infection developed at the biopsy site, and the patient developed sepsis and septic shock. She received broad-spectrum antibiotics and underwent extensive debridement with wound care. After a protracted hospital course, the infection resolved.
CALCIPHYLAXIS RISK FACTORS
Calciphylaxis, also referred to as calcific uremic arteriolopathy, is a rare and often fatal condition in patients with end-stage renal disease who are on hemodialysis (1% to 4% of dialysis patients).1–3 It is also seen in patients who have undergone renal transplant and in patients with chronic kidney disease who have a chronic inflammatory disease or who have been exposed to corticosteroids or warfarin. However, it can also occur in patients without chronic kidney disease or end-stage renal disease.
The term “calcific uremic arteriolopathy” is a misnomer, as this condition can occur in patients with normal renal function (nonuremic calciphylaxis). Also, despite what the term calciphylaxis implies, there is no systemic anaphylaxis.3–5
Documented risk factors include obesity; female sex; use of warfarin, corticosteroids, or vitamin D analogues; low serum albumin; hypercoagulable states; hyperparathyroidism; alcoholic liver disease; elevated calcium-phosphorus product; inflammation; connective tissue disease; and cancer.4–6
DIAGNOSTIC CLUES
There are no strict guidelines for the diagnosis of calciphylaxis, and the exact pathophysiology of calciphylaxis is not understood.1–4
Ulceration is considered the clinical hallmark, but there are increasing reports of patients presenting with nonulcerated plaques, as in our patient. The literature suggests a mortality rate of 33% at 6 months in these patients, but ulceration increases the risk of death to over 80%, and sepsis is the leading cause of death.7,8
Histologic features identified on full-thickness biopsy specimens are intravascular deposition of calcium in the media of the blood vessels, as well as fibrin thrombi formation, intimal proliferation, tissue necrosis, and resultant ischemia. However, as in our patient and as discussed below, the biopsy procedure can induce or exacerbate ulceration, increasing the risk of sepsis, and is thus controversial.7
In the early stages, lesions of calciphylaxis are focal and appear as erythema or livedo reticularis with or without subcutaneous plaques or ulcers. As the disease progresses, the ischemic changes coalesce to form denser violaceous, painful, plaquelike subcutaneous nodules with eschar. In the advanced stages, the eschar or ulceration involves an extensive area.
Diagnosis in the early stages is challenging because of the focal nature of involvement. The differential diagnosis includes potentially fatal conditions such as systemic vasculitis, nephrogenic systemic fibrosis, pyoderma gangrenosum, gangrene from peripheral arterial disease, cholesterol embolization, warfarin-induced necrosis, purpura fulminans, and oxalate vasculopathy.7
In the advanced stages, the diagnosis of calciphylaxis is clinically more evident, and the differential diagnosis usually narrows. Well-demarcated, necrotic, indurated lesions that are bilateral in a patient with end-stage renal disease without shock makes the diagnosis very likely.
The dangers of biopsy
As seen in our patient, biopsy for histologic confirmation of calciphylaxis can increase the risk of infection and sepsis.7 Also, the efficacy and clinical utility are uncertain because the quantity or depth of tissue obtained may not be enough for diagnosis. Deep incisional cutaneous biopsy is needed rather than punch biopsy to provide ample subcutaneous tissue for histologic study.3
Further, the biopsy procedure induces ulceration in the region of the incision, increasing the risk of infection and poor healing and escalating the risk of sepsis and death.7–9 Since extensive necrosis predisposes to a negative biopsy, a high clinical suspicion should drive early treatment of calciphylaxis.10 Noninvasive imaging studies such as plain radiography and bone scintigraphy can aid the diagnosis by detecting moderate to severe soft-tissue vascular calcification in these areas.7–11
DEBRIDEMENT IS CONTROVERSIAL
Conservative measures are the mainstay of care and include dietary alterations, noncalcium and nonaluminum phosphate binders, and low-calcium bath dialysis. There is mounting evidence for the use of calcimimetics and sodium thiosulfate.7,12–14
The role of wound debridement is controversial, as concomitant poor peripheral vascular perfusion can delay wound healing and, if ulceration ensues, there is a dramatic escalation of mortality risk. The decision for wound debridement is determined case by case, based on an assessment of the comorbidities, vascular perfusion, and status of the eschar.
Extensive wound debridement should be considered immediately after biopsy or with any signs of ulceration or infection—this in addition to meticulous wound care, which will promote healing and prevent serious complications secondary to infection.15
A TEAM APPROACH IMPROVES OUTCOMES
A multidisciplinary approach involving surgeons, nephrologists, dermatologists, dermatopathologists, wound or burn care team, nutrition team, pain management team, and infectious disease team is important to improve outcomes.7
Management mainly involves controlling pain; avoiding local trauma; treating and preventing infection; stopping causative agents such as warfarin and corticosteroids; intensive hemodialysis with an increase in both frequency and duration; intravenous sodium thiosulphate; non-calcium-phosphorus binders and cinacalcet in patients with elevated parathyroid hormone; and hyperbaric oxygen.12–14 There are also reports of success with oral etidronate and intravenous pamidronate.16,17
A 53-year-old woman presented with extensive, nonulcerated, painful plaques on both calves. She had long-standing diabetes mellitus and had recently started hemodialysis. She had no fever or trauma and did not appear to be in shock.
On physical examination, she had extensive, well-demarcated, nonulcerated, indurated dark eschar over the right calf (Figure 1). Her left calf had similar lesions that appeared as focal, discrete, nonulcerated, violaceous plaques, with associated tenderness. No significant erythema, edema, drainage, or fluctuance was noted.
A broad-spectrum antibiotic was started empirically but was discontinued when routine blood testing and magnetic resonance imaging showed no evidence of infection. Histologic study of a full-thickness skin biopsy specimen (Figure 2) showed tissue necrosis, ulceration, and concentric calcification of small and medium-sized blood vessels, many with luminal thrombi, all of which together were diagnostic for calciphylaxis.
Treatment was started with cinacalcet, low-calcium dialysis baths, phosphate binders, and sodium thiosulfate. However, within a few days of the biopsy procedure, an infection developed at the biopsy site, and the patient developed sepsis and septic shock. She received broad-spectrum antibiotics and underwent extensive debridement with wound care. After a protracted hospital course, the infection resolved.
CALCIPHYLAXIS RISK FACTORS
Calciphylaxis, also referred to as calcific uremic arteriolopathy, is a rare and often fatal condition in patients with end-stage renal disease who are on hemodialysis (1% to 4% of dialysis patients).1–3 It is also seen in patients who have undergone renal transplant and in patients with chronic kidney disease who have a chronic inflammatory disease or who have been exposed to corticosteroids or warfarin. However, it can also occur in patients without chronic kidney disease or end-stage renal disease.
The term “calcific uremic arteriolopathy” is a misnomer, as this condition can occur in patients with normal renal function (nonuremic calciphylaxis). Also, despite what the term calciphylaxis implies, there is no systemic anaphylaxis.3–5
Documented risk factors include obesity; female sex; use of warfarin, corticosteroids, or vitamin D analogues; low serum albumin; hypercoagulable states; hyperparathyroidism; alcoholic liver disease; elevated calcium-phosphorus product; inflammation; connective tissue disease; and cancer.4–6
DIAGNOSTIC CLUES
There are no strict guidelines for the diagnosis of calciphylaxis, and the exact pathophysiology of calciphylaxis is not understood.1–4
Ulceration is considered the clinical hallmark, but there are increasing reports of patients presenting with nonulcerated plaques, as in our patient. The literature suggests a mortality rate of 33% at 6 months in these patients, but ulceration increases the risk of death to over 80%, and sepsis is the leading cause of death.7,8
Histologic features identified on full-thickness biopsy specimens are intravascular deposition of calcium in the media of the blood vessels, as well as fibrin thrombi formation, intimal proliferation, tissue necrosis, and resultant ischemia. However, as in our patient and as discussed below, the biopsy procedure can induce or exacerbate ulceration, increasing the risk of sepsis, and is thus controversial.7
In the early stages, lesions of calciphylaxis are focal and appear as erythema or livedo reticularis with or without subcutaneous plaques or ulcers. As the disease progresses, the ischemic changes coalesce to form denser violaceous, painful, plaquelike subcutaneous nodules with eschar. In the advanced stages, the eschar or ulceration involves an extensive area.
Diagnosis in the early stages is challenging because of the focal nature of involvement. The differential diagnosis includes potentially fatal conditions such as systemic vasculitis, nephrogenic systemic fibrosis, pyoderma gangrenosum, gangrene from peripheral arterial disease, cholesterol embolization, warfarin-induced necrosis, purpura fulminans, and oxalate vasculopathy.7
In the advanced stages, the diagnosis of calciphylaxis is clinically more evident, and the differential diagnosis usually narrows. Well-demarcated, necrotic, indurated lesions that are bilateral in a patient with end-stage renal disease without shock makes the diagnosis very likely.
The dangers of biopsy
As seen in our patient, biopsy for histologic confirmation of calciphylaxis can increase the risk of infection and sepsis.7 Also, the efficacy and clinical utility are uncertain because the quantity or depth of tissue obtained may not be enough for diagnosis. Deep incisional cutaneous biopsy is needed rather than punch biopsy to provide ample subcutaneous tissue for histologic study.3
Further, the biopsy procedure induces ulceration in the region of the incision, increasing the risk of infection and poor healing and escalating the risk of sepsis and death.7–9 Since extensive necrosis predisposes to a negative biopsy, a high clinical suspicion should drive early treatment of calciphylaxis.10 Noninvasive imaging studies such as plain radiography and bone scintigraphy can aid the diagnosis by detecting moderate to severe soft-tissue vascular calcification in these areas.7–11
DEBRIDEMENT IS CONTROVERSIAL
Conservative measures are the mainstay of care and include dietary alterations, noncalcium and nonaluminum phosphate binders, and low-calcium bath dialysis. There is mounting evidence for the use of calcimimetics and sodium thiosulfate.7,12–14
The role of wound debridement is controversial, as concomitant poor peripheral vascular perfusion can delay wound healing and, if ulceration ensues, there is a dramatic escalation of mortality risk. The decision for wound debridement is determined case by case, based on an assessment of the comorbidities, vascular perfusion, and status of the eschar.
Extensive wound debridement should be considered immediately after biopsy or with any signs of ulceration or infection—this in addition to meticulous wound care, which will promote healing and prevent serious complications secondary to infection.15
A TEAM APPROACH IMPROVES OUTCOMES
A multidisciplinary approach involving surgeons, nephrologists, dermatologists, dermatopathologists, wound or burn care team, nutrition team, pain management team, and infectious disease team is important to improve outcomes.7
Management mainly involves controlling pain; avoiding local trauma; treating and preventing infection; stopping causative agents such as warfarin and corticosteroids; intensive hemodialysis with an increase in both frequency and duration; intravenous sodium thiosulphate; non-calcium-phosphorus binders and cinacalcet in patients with elevated parathyroid hormone; and hyperbaric oxygen.12–14 There are also reports of success with oral etidronate and intravenous pamidronate.16,17
- Spanakis EK, Sellmeyer DE. Nonuremic calciphylaxis precipitated by teriparatide [rhPTH(1-34)] therapy in the setting of chronic warfarin and glucocorticoid treatment. Osteoporos Int 2014; 25:1411–1414.
- Brandenburg VM, Cozzolino M, Ketteler M. Calciphylaxis: a still unmet challenge. J Nephrol 2011; 24:142–148.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial 2002; 15:172–186.
- Rimtepathip P, Cohen D. A rare presentation of calciphylaxis in normal renal function. Int J Case Rep Images 2015; 6:366–369.
- Lonowski S, Martin S, Worswick S. Widespread calciphylaxis and normal renal function: no improvement with sodium thiosulfate. Dermatol Online J 2015; 21:13030/qt76845802.
- Zhou Q, Neubauer J, Kern JS, Grotz W, Walz G, Huber TB. Calciphylaxis. Lancet 2014; 383:1067.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis 2015; 66:133–146.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int 2002; 61:2210–2217.
- Hayashi M. Calciphylaxis: diagnosis and clinical features. Clin Exp Nephrol 2013; 17:498–503.
- Stavros K, Motiwala R, Zhou L, Sejdiu F, Shin S. Calciphylaxis in a dialysis patient diagnosed by muscle biopsy. J Clin Neuromuscul Dis 2014; 15:108–111.
- Bonchak JG, Park KK, Vethanayagamony T, Sheikh MM, Winterfield LS. Calciphylaxis: a case series and the role of radiology in diagnosis. Int J Dermatol 2015. [Epub ahead of print]
- Ross EA. Evolution of treatment strategies for calciphylaxis. Am J Nephrol 2011; 34:460–467.
- Cicone JS, Petronis JB, Embert CD, Spector DA. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am J Kidney Dis 2004; 43:1104–1108.
- Brandenburg VM, Kramann R, Specht P, Ketteler M. Calciphylaxis in CKD and beyond. Nephrol Dial Transplant 2012; 27:1314–1318.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage 2004; 50:64–66.
- Shiraishi N, Kitamura K, Miyoshi T, et al. Successful treatment of a patient with severe calcific uremic arteriolopathy (calciphylaxis) by etidronate disodium. Am J Kidney Dis 2006; 48:151–154.
- Hanafusa T, Yamaguchi Y, Tani M, Umegaki N, Nishimura Y, Katayama I. Intractable wounds caused by calcific uremic arteriolopathy treated with bisphosphonates. J Am Acad Dermatol 2007; 57:1021–1025.
- Spanakis EK, Sellmeyer DE. Nonuremic calciphylaxis precipitated by teriparatide [rhPTH(1-34)] therapy in the setting of chronic warfarin and glucocorticoid treatment. Osteoporos Int 2014; 25:1411–1414.
- Brandenburg VM, Cozzolino M, Ketteler M. Calciphylaxis: a still unmet challenge. J Nephrol 2011; 24:142–148.
- Wilmer WA, Magro CM. Calciphylaxis: emerging concepts in prevention, diagnosis, and treatment. Semin Dial 2002; 15:172–186.
- Rimtepathip P, Cohen D. A rare presentation of calciphylaxis in normal renal function. Int J Case Rep Images 2015; 6:366–369.
- Lonowski S, Martin S, Worswick S. Widespread calciphylaxis and normal renal function: no improvement with sodium thiosulfate. Dermatol Online J 2015; 21:13030/qt76845802.
- Zhou Q, Neubauer J, Kern JS, Grotz W, Walz G, Huber TB. Calciphylaxis. Lancet 2014; 383:1067.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis 2015; 66:133–146.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int 2002; 61:2210–2217.
- Hayashi M. Calciphylaxis: diagnosis and clinical features. Clin Exp Nephrol 2013; 17:498–503.
- Stavros K, Motiwala R, Zhou L, Sejdiu F, Shin S. Calciphylaxis in a dialysis patient diagnosed by muscle biopsy. J Clin Neuromuscul Dis 2014; 15:108–111.
- Bonchak JG, Park KK, Vethanayagamony T, Sheikh MM, Winterfield LS. Calciphylaxis: a case series and the role of radiology in diagnosis. Int J Dermatol 2015. [Epub ahead of print]
- Ross EA. Evolution of treatment strategies for calciphylaxis. Am J Nephrol 2011; 34:460–467.
- Cicone JS, Petronis JB, Embert CD, Spector DA. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am J Kidney Dis 2004; 43:1104–1108.
- Brandenburg VM, Kramann R, Specht P, Ketteler M. Calciphylaxis in CKD and beyond. Nephrol Dial Transplant 2012; 27:1314–1318.
- Martin R. Mysterious calciphylaxis: wounds with eschar—to debride or not to debride? Ostomy Wound Manage 2004; 50:64–66.
- Shiraishi N, Kitamura K, Miyoshi T, et al. Successful treatment of a patient with severe calcific uremic arteriolopathy (calciphylaxis) by etidronate disodium. Am J Kidney Dis 2006; 48:151–154.
- Hanafusa T, Yamaguchi Y, Tani M, Umegaki N, Nishimura Y, Katayama I. Intractable wounds caused by calcific uremic arteriolopathy treated with bisphosphonates. J Am Acad Dermatol 2007; 57:1021–1025.
Renal failure in HCV cirrhosis
A 54-year-old man with a history of cirrhosis secondary to hepatitis C virus (HCV) infection has had a progressive decline in kidney function. He was diagnosed with hepatitis C 15 years ago; he tried interferon treatment, but this failed. He received a transjugular intrahepatic shunt 10 years ago after an episode of esophageal variceal bleeding. He has since been taking furosemide and spironolactone as maintenance treatment for ascites, and he has no other medical concerns such as hypertension or diabetes.

Two weeks ago, routine laboratory tests in the clinic showed that his serum creatinine level had increased from baseline. He was asked to stop his diuretics and increase his fluid intake. Nevertheless, his kidney function continued to decline (Table 1), and he was admitted to the hospital for further evaluation.
On admission, he appeared comfortable. He denied recent use of any medications, including nonsteroidal anti-inflammatory drugs, antibiotics, and diuretics, and he had no genitourinary symptoms. His temperature was normal, blood pressure 170/90 mm Hg, pulse rate 72 per minute, and respiratory rate 16. His skin and sclerae were not jaundiced; his abdomen was not tender, but it was grossly distended with ascites. He also had +3 pedal edema (on a scale of 4) extending to both knees. The rest of his physical examination was unremarkable. Results of further laboratory tests are shown in in Table 2.
Ultrasonography of the liver demonstrated cirrhosis with patent flow through the shunt, and ultrasonography of the kidneys showed that both were slightly enlarged with increased cortical echogenicity but no hydronephrosis or obstruction.
EXPLORING THE CAUSE OF RENAL FAILURE
1. Given this information, what is the likely cause of our patient’s renal failure?
- Volume depletion
- Acute tubular necrosis
- Hepatorenal syndrome
- HCV glomerulopathy
Renal failure is a common complication in cirrhosis and portends a higher risk of death.1 The differential diagnosis is broad, but a systematic approach incorporating data from the history, physical examination, and laboratory tests can help identify the cause and is essential in determining the prognosis and proper treatment.
Volume depletion
Volume depletion is a common cause of renal failure in cirrhotic patients. Common precipitants are excessive diuresis and gastrointestinal fluid loss from bleeding, vomiting, and diarrhea. Despite having ascites and edema, patients may have low fluid volume in the vascular space. Therefore, the first step in a patient with acute kidney injury is to withhold diuretics and give fluids. The renal failure usually rapidly reverses if the patient does not have renal parenchymal disease.2
Our patient did not present with any fluid losses, and his high blood pressure and normal heart rate did not suggest volume depletion. And most importantly, withholding his diuretics and giving fluids did not reverse his renal failure. Thus, volume depletion was an unlikely cause.
Acute tubular necrosis
The altered hemodynamics caused by cirrhosis predispose patients to acute tubular necrosis. Classically, this presents as muddy brown casts and renal tubular epithelial cells on urinalysis and as a fractional excretion of sodium greater than 2%.1 However, these microscopic findings lack sensitivity, and patients with cirrhosis may have marked sodium avidity and low urine sodium excretion despite tubular injury.3
This diagnosis must still be considered in patients with renal failure, especially after an insult such as hemorrhagic or septic shock or intake of nephrotoxins. However, because our patient did not have a history of any of these and because his renal failure had been progressing over weeks, acute tubular necrosis was considered unlikely.
Hepatorenal syndrome
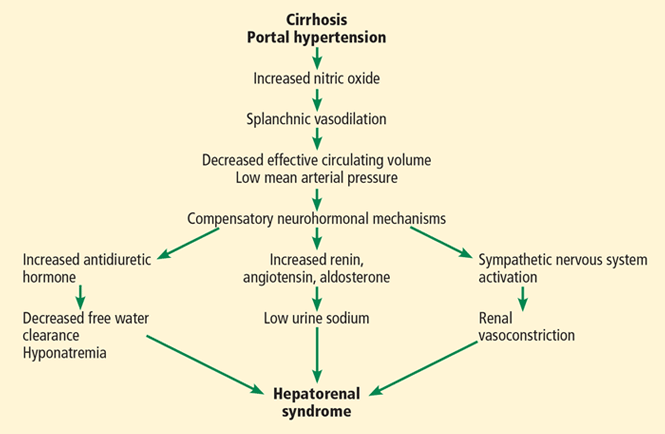
Hepatorenal syndrome is characterized by progressive renal failure in the absence of renal parenchymal disease. It is a functional disorder, ie, the decreased glomerular filtration rate results from renal vasoconstriction, which in turn is due to decreased systemic vascular resistance and increased compensatory activity of the renin-angiotensin-aldosterone axis and of antiduretic hormone release (Figure 1).
Hepatorenal syndrome often occurs in patients with advanced liver disease. These patients typically have a hyperdynamic circulation (systemic vasodilation, low blood pressure, and increased blood volume) with a low mean arterial pressure and increased renin and norepinephrine levels. Other frequent findings include hyponatremia, low urinary sodium excretion (< 2 mmol/day), and low free water clearance,4 all of which mark the high systemic levels of antidiuretic hormone and aldosterone.
Importantly, while hepatorenal syndrome is always considered in the differential diagnosis because of its unique prognosis and therapy, it remains a diagnosis of exclusion. The International Ascites Club5 has provided diagnostic criteria for hepatorenal syndrome:
- Cirrhosis and ascites
- Serum creatinine greater than 1.5 mg/dL
- Failure of serum creatinine to fall to less than 1.5 mg/dL after at least 48 hours of diuretic withdrawal and volume expansion with albumin (recommended dose 1 g/kg body weight per day up to a maximum of 100 g per day)
- Absence of shock
- No current or recent treatment with nephrotoxic drugs
- No signs of parenchymal kidney disease such as proteinuria (protein excretion > 500 mg/day), microhematuria (> 50 red blood cells per high-power field), or abnormalities on renal ultrasonography.
While these criteria are not perfect,6 they remind clinicians that there are other important causes of renal insufficiency in cirrhosis.
Clinically, our patient had no evidence of a hyperdynamic circulation and was instead hypertensive. He was eunatremic and did not have marked renal sodium avidity. His pyuria, proteinuria (his protein excretion was approximately 1.9 g/day as determined by urine spot protein-to-creatinine ratio), and results of ultrasonography also suggested underlying renal parenchymal disease. Therefore, hepatorenal syndrome was not the likely diagnosis.
HCV glomerulopathy
Intrinsic renal disease is likely, given our patient’s proteinuria, active urine sediment (ie, containing red blood cells, white blood cells, and protein), and abnormal findings on ultrasonography. In patients with HCV infection and no other cause of intrinsic kidney disease, immune complex deposition leading to glomerulonephritis is the most common pattern.7 Despite the intrinsic renal disease, fractional excretion of sodium may be less than 1% in glomerulonephritis. Hypertension in a patient such as ours with cirrhosis and renal insufficiency raises suspicion for glomerular disease, as hypertension is unlikely in advanced cirrhosis.8
Glomerulonephritis in patients with cirrhosis is often clinically silent and may be highly prevalent; some studies have shown glomerular involvement in 55% to 83% of patients with cirrhosis.9,10 This increases the risk of end-stage renal disease, and the Kidney Disease Improving Global Outcomes guideline recommends that HCV-infected patients be tested at least once a year for proteinuria, hematuria, and estimated glomerular filtration rate to detect possible HCV-associated kidney disease.11 According to current guidelines of the Infectious Diseases Society of America (IDSA) and American Association for the Study of Liver Diseases (AASLD) , detection of glomerulonephritis in HCV patients puts them in the highest priority class for treatment of HCV.12
HISTOLOGIC FINDINGS
Because of the high likelihood of glomerulopathy, our patient underwent renal biopsy.
2. What is the classic pathologic finding in HCV kidney disease?
- Focal segmental glomerulosclerosis
- Crescentic glomerulonephritis
- Membranoproliferative glomerulonephritis
- Membranous glomerulonephritis
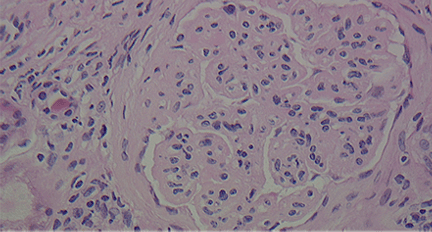
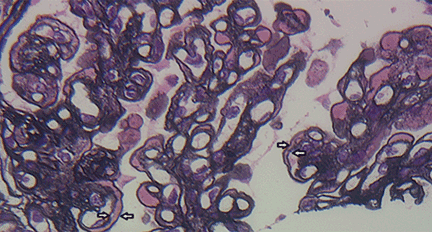
A number of pathologic patterns have been described in HCV kidney disease, including membranous glomerulonephritis, immunoglobulin A nephropathy, and focal segmental glomerulosclerosis. However, by far the most common pattern is type 1 membranoproliferative glomerulonephritis.13 (Types 2 and 3 are much less common, and we will not discuss them here.) In type 1, light microscopy shows increased mesangial cells and thickened capillary walls (lobular glomeruli), staining of the basement membrane reveals double contours (“tram tracking”) or splitting due to mesangial deposition, and immunofluorescence demonstrates immunoglobulin G and complement C3 deposition. All of these findings were seen in our patient (Figure 2, Figure 3).
Membranoproliferative glomerulonephritis in patients with HCV is most commonly associated with cryoglobulins, a mixture of monoclonal or polyclonal immunoglobulin (Ig) M that have antiglobulin (rheumatoid factor) activity and bind to polyclonal IgG. They reversibly precipitate at less than 37°C, (98.6°F), hence their name. Only 50% to 70% of patients with cryoglobulinemic membranoproliferative glomerulonephritis have detectable serum cryoglobulins; however, kidney biopsy may show globular accumulations of eosinophilic material and prominent hypercellularity due to infiltration of glomerular capillaries with mononuclear and polymorphonuclear leukocytes.
Noncryoglobulinemic membranoproliferative glomerulonephritis is also found in patients with HCV infection. Its histologic features are similar, but on biopsy, there is less prominent leukocytic infiltration and no eosinophilic material. Although the pathogenesis of glomerulonephritis in HCV infection is poorly understood, it is thought to result from deposition of circulating immune complexes of HCV, anti-HCV, and rheumatoid factor in the glomeruli.
3. What laboratory finding is often seen in membranoproliferative glomerulonephritis?
- Positive cytoplasmic antineutrophil cytoplasmic antibody
- serum complement Low levels
- Antiphospholipase A2 receptor antibodies
Cytoplasmic antineutrophil cytoplasmic antibody is seen in granulomatosis with polyangiitis, while antiphospholipid A2 receptor antibodies are seen in idiopathic membranous nephritis.
Low serum complement levels are frequently found in membranoproliferative glomerulonephritis. It is believed that immune complex deposition leads to glomerular damage through activation of the complement pathway and the subsequent influx of inflammatory cells, release of cytokines and proteases, and damage to capillary walls. When repair ensues, new mesangial matrix and basement membrane are deposited, leading to mesangial expansion and duplicated basement membrane.14
In cryoglobulinemic membranoproliferative glomerulonephritis, the complement C4 level is often much lower than C3, but in noncryoglobulinemic forms C3 is lower. A mnemonic to remember nephritic syndromes with low complement levels is “hy-PO-CO-MP-L-EM-ents”; PO for postinfectious, CO for cryoglobulins, MP for membranoproliferative glomerulonephritis, L for lupus, and EM for embolic.
BACK TO OUR PATIENT

In addition to kidney biopsy, we tested our patient for serum cryoglobulins, rheumatoid factor, and serum complements. Results from these tests (Table 3), in addition to the lack of cryoglobulins on his biopsy, led to the conclusion that he had noncryoglobulinemic membranoproliferative glomerulonephritis.
WHO SHOULD RECEIVE TREATMENT FOR HCV?
4. According to the current IDSA/AASLD guidelines, which of the following patients should not receive direct-acting antiviral therapy for HCV?
- Patients with HCV and only low-stage fibrosis
- Patients with decompensated cirrhosis
- Patients with a glomerular filtration rate less than 30 mL/minute
- None of the above—nearly all patients with HCV infection should receive treatment for it
While certain patients have compelling indications for HCV treatment, such as advanced fibrosis, severe extrahepatic manifestations of HCV (eg, glomerulonephritis, cryoglobulinemia), and posttransplant status, current guidelines recommend treatment for nearly all patients with HCV, including those with low-stage fibrosis.12
Patients with Child-Pugh grade B or C decompensated cirrhosis, even with hepatocellular carcinoma, may be considered for treatment. Multiple studies have demonstrated the efficacy and safety of direct-acting antiviral drugs in this patient population. In one randomized controlled trial,15 the combination of ledipasvir, sofosbuvir, and ribavirin resulted in high sustained virologic response rates at 12 weeks in patients infected with HCV genotype 1 or 4 with advanced liver disease, irrespective of transplant status (86% to 89% of patients were pretransplant). Sustained virologic response was associated with improvements in Model for End-Stage Liver Disease and Child-Pugh scores largely due to decreases in bilirubin and improvement in synthetic function (ie, albumin).
Similarly, even patients with a glomerular filtration rate less than 30 mL/min are candidates for treatment. Those with a glomerular filtration rate above 30 mL/min need no dosage adjustments for the most common regimens, while regimens are also available for those with a rate less than 30 mL/min. Although patients with low baseline renal function have a higher frequency of anemia (especially with ribavirin), worsening renal dysfunction, and more severe adverse events, treatment responses remain high and comparable to those without renal impairment.
The Hepatitis C Therapeutic Registry and Research Network (HCV-TARGET) is conducting an ongoing prospective study evaluating real-world use of direct-acting antiviral agents. The study has reported the safety and efficacy of sofosbuvir-containing regimens in patients with varying severities of kidney disease, including glomerular filtration rates less than 30 mL/min). The patients received different regimens that included sofosbuvir. The regimens were reportedly tolerated, and the rate of sustained viral response at 12 weeks remained high.16
The efficacy of direct-acting antiviral agents for HCV-associated glomerulonephritis remains to be studied but is promising. Earlier studies found that antiviral therapy based on interferon alfa with or without ribavirin can significantly decrease proteinuria and stabilize renal function.17–20 HCV RNA clearance has been found to best predict renal improvement.
OUR PATIENT’S COURSE
Unfortunately, our patient’s kidney function declined further over the next 3 months, and he is currently on dialysis awaiting simultaneous liver and kidney transplant.
- Ginès P, Schrier RW. Renal failure in cirrhosis. N Engl J Med 2009; 361:1279–1290.
- Mackelaite L, Alsauskas ZC, Ranganna K. Renal failure in patients with cirrhosis. Med Clin North Am 2009; 93:855–869.
- Wadei HM, Mai ML, Ahsan N, Gonwa TA. Hepatorenal syndrome: pathophysiology and management. Clin J Am Soc Nephrol 2006; 1:1066–1079.
- Gines A, Escorsell A, Gines P, et al. Incidence, predictive factors, and prognosis of the hepatorenal syndrome in cirrhosis with ascites. Gastroenterology 1993; 105:229–236.
- Salerno F, Gerbes A, Ginès P, Wong F, Arroyo V. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut 2007; 56:1310–1318.
- Watt K, Uhanova J, Minuk GY. Hepatorenal syndrome: diagnostic accuracy, clinical features, and outcome in a tertiary care center. Am J Gastroenterol 2002; 97:2046–2050.
- Graupera I, Cardenas A. Diagnostic approach to renal failure in cirrhosis. Clin Liver Dis 2013; 2:128–131.
- Dash SC, Bhowmik D. Glomerulopathy with liver disease: patterns and management. Saudi J Kidney Dis Transpl 2000; 11:414–420.
- Arase Y, Ikeda K, Murashima N, et al. Glomerulonephritis in autopsy cases with hepatitis C virus infection. Intern Med 1998; 37:836–840.
- McGuire BM, Julian BA, Bynon JS, et al. Brief communication: glomerulonephritis in patients with hepatitis C cirrhosis undergoing liver transplantation. Ann Intern Med 2006; 144:735–741.
- Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO clinical practice guidelines for the prevention, diagnosis, evaluation, and treatment of hepatitis C in chronic kidney disease. Kidney Int Suppl 2008; 109:S1–S99.
- American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America (IDSA). HCV guidance: recommendations for testing, managing, and treating hepatitis C. www.hcvguidelines.org/. Accessed July 10, 2016.
- Lai KN. Hepatitis-related renal disease. Future Virology 2011; 6:1361–1376.
- Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis—a new look at an old entity. N Engl J Med 2012; 366:1119–1131.
- Charlton M, Everson GT, Flamm SL, et al; SOLAR-1 Investigators. Ledipasvir and sofosbuvir plus ribavirin for treatment of HCV infection in patients with advanced liver disease. Gastroenterology 2015; 149:649–659.
- Saxena V, Koraishy FM, Sise ME, et al; HCV-TARGET. Safety and efficacy of sofosbuvir-containing regimens in hepatitis C-infected patients with impaired renal function. Liver Int 2016; 36:807–816.
- Feng B, Eknoyan G, Guo ZS, et al. Effect of interferon alpha-based antiviral therapy on hepatitis C virus-associated glomerulonephritis: a meta-analysis. Nephrol Dial Transplant 2012; 27:640–646.
- Bruchfeld A, Lindahl K, Ståhle L, Söderberg M, Schvarcz R. Interferon and ribavirin treatment in patients with hepatitis C-associated renal disease and renal insufficiency. Nephrol Dial Transplant 2003; 18:1573–1580.
- Rossi P, Bertani T, Baio P, et al. Hepatitis C virus-related cryoglobulinemic glomerulonephritis. Long-term remission after antiviral therapy. Kidney Int 2003; 63:2236–2241.
- Alric L, Plaisier E, Thebault S, et al. Influence of antiviral therapy in hepatitis C virus associated cryoglobulinemic MPGN. Am J Kidney Dis 2004; 43:617–623.
A 54-year-old man with a history of cirrhosis secondary to hepatitis C virus (HCV) infection has had a progressive decline in kidney function. He was diagnosed with hepatitis C 15 years ago; he tried interferon treatment, but this failed. He received a transjugular intrahepatic shunt 10 years ago after an episode of esophageal variceal bleeding. He has since been taking furosemide and spironolactone as maintenance treatment for ascites, and he has no other medical concerns such as hypertension or diabetes.
Two weeks ago, routine laboratory tests in the clinic showed that his serum creatinine level had increased from baseline. He was asked to stop his diuretics and increase his fluid intake. Nevertheless, his kidney function continued to decline (Table 1), and he was admitted to the hospital for further evaluation.
On admission, he appeared comfortable. He denied recent use of any medications, including nonsteroidal anti-inflammatory drugs, antibiotics, and diuretics, and he had no genitourinary symptoms. His temperature was normal, blood pressure 170/90 mm Hg, pulse rate 72 per minute, and respiratory rate 16. His skin and sclerae were not jaundiced; his abdomen was not tender, but it was grossly distended with ascites. He also had +3 pedal edema (on a scale of 4) extending to both knees. The rest of his physical examination was unremarkable. Results of further laboratory tests are shown in in Table 2.
Ultrasonography of the liver demonstrated cirrhosis with patent flow through the shunt, and ultrasonography of the kidneys showed that both were slightly enlarged with increased cortical echogenicity but no hydronephrosis or obstruction.
EXPLORING THE CAUSE OF RENAL FAILURE
1. Given this information, what is the likely cause of our patient’s renal failure?
- Volume depletion
- Acute tubular necrosis
- Hepatorenal syndrome
- HCV glomerulopathy
Renal failure is a common complication in cirrhosis and portends a higher risk of death.1 The differential diagnosis is broad, but a systematic approach incorporating data from the history, physical examination, and laboratory tests can help identify the cause and is essential in determining the prognosis and proper treatment.
Volume depletion
Volume depletion is a common cause of renal failure in cirrhotic patients. Common precipitants are excessive diuresis and gastrointestinal fluid loss from bleeding, vomiting, and diarrhea. Despite having ascites and edema, patients may have low fluid volume in the vascular space. Therefore, the first step in a patient with acute kidney injury is to withhold diuretics and give fluids. The renal failure usually rapidly reverses if the patient does not have renal parenchymal disease.2
Our patient did not present with any fluid losses, and his high blood pressure and normal heart rate did not suggest volume depletion. And most importantly, withholding his diuretics and giving fluids did not reverse his renal failure. Thus, volume depletion was an unlikely cause.
Acute tubular necrosis
The altered hemodynamics caused by cirrhosis predispose patients to acute tubular necrosis. Classically, this presents as muddy brown casts and renal tubular epithelial cells on urinalysis and as a fractional excretion of sodium greater than 2%.1 However, these microscopic findings lack sensitivity, and patients with cirrhosis may have marked sodium avidity and low urine sodium excretion despite tubular injury.3
This diagnosis must still be considered in patients with renal failure, especially after an insult such as hemorrhagic or septic shock or intake of nephrotoxins. However, because our patient did not have a history of any of these and because his renal failure had been progressing over weeks, acute tubular necrosis was considered unlikely.
Hepatorenal syndrome
Hepatorenal syndrome is characterized by progressive renal failure in the absence of renal parenchymal disease. It is a functional disorder, ie, the decreased glomerular filtration rate results from renal vasoconstriction, which in turn is due to decreased systemic vascular resistance and increased compensatory activity of the renin-angiotensin-aldosterone axis and of antiduretic hormone release (Figure 1).
Hepatorenal syndrome often occurs in patients with advanced liver disease. These patients typically have a hyperdynamic circulation (systemic vasodilation, low blood pressure, and increased blood volume) with a low mean arterial pressure and increased renin and norepinephrine levels. Other frequent findings include hyponatremia, low urinary sodium excretion (< 2 mmol/day), and low free water clearance,4 all of which mark the high systemic levels of antidiuretic hormone and aldosterone.
Importantly, while hepatorenal syndrome is always considered in the differential diagnosis because of its unique prognosis and therapy, it remains a diagnosis of exclusion. The International Ascites Club5 has provided diagnostic criteria for hepatorenal syndrome:
- Cirrhosis and ascites
- Serum creatinine greater than 1.5 mg/dL
- Failure of serum creatinine to fall to less than 1.5 mg/dL after at least 48 hours of diuretic withdrawal and volume expansion with albumin (recommended dose 1 g/kg body weight per day up to a maximum of 100 g per day)
- Absence of shock
- No current or recent treatment with nephrotoxic drugs
- No signs of parenchymal kidney disease such as proteinuria (protein excretion > 500 mg/day), microhematuria (> 50 red blood cells per high-power field), or abnormalities on renal ultrasonography.
While these criteria are not perfect,6 they remind clinicians that there are other important causes of renal insufficiency in cirrhosis.
Clinically, our patient had no evidence of a hyperdynamic circulation and was instead hypertensive. He was eunatremic and did not have marked renal sodium avidity. His pyuria, proteinuria (his protein excretion was approximately 1.9 g/day as determined by urine spot protein-to-creatinine ratio), and results of ultrasonography also suggested underlying renal parenchymal disease. Therefore, hepatorenal syndrome was not the likely diagnosis.
HCV glomerulopathy
Intrinsic renal disease is likely, given our patient’s proteinuria, active urine sediment (ie, containing red blood cells, white blood cells, and protein), and abnormal findings on ultrasonography. In patients with HCV infection and no other cause of intrinsic kidney disease, immune complex deposition leading to glomerulonephritis is the most common pattern.7 Despite the intrinsic renal disease, fractional excretion of sodium may be less than 1% in glomerulonephritis. Hypertension in a patient such as ours with cirrhosis and renal insufficiency raises suspicion for glomerular disease, as hypertension is unlikely in advanced cirrhosis.8
Glomerulonephritis in patients with cirrhosis is often clinically silent and may be highly prevalent; some studies have shown glomerular involvement in 55% to 83% of patients with cirrhosis.9,10 This increases the risk of end-stage renal disease, and the Kidney Disease Improving Global Outcomes guideline recommends that HCV-infected patients be tested at least once a year for proteinuria, hematuria, and estimated glomerular filtration rate to detect possible HCV-associated kidney disease.11 According to current guidelines of the Infectious Diseases Society of America (IDSA) and American Association for the Study of Liver Diseases (AASLD) , detection of glomerulonephritis in HCV patients puts them in the highest priority class for treatment of HCV.12
HISTOLOGIC FINDINGS
Because of the high likelihood of glomerulopathy, our patient underwent renal biopsy.
2. What is the classic pathologic finding in HCV kidney disease?
- Focal segmental glomerulosclerosis
- Crescentic glomerulonephritis
- Membranoproliferative glomerulonephritis
- Membranous glomerulonephritis
A number of pathologic patterns have been described in HCV kidney disease, including membranous glomerulonephritis, immunoglobulin A nephropathy, and focal segmental glomerulosclerosis. However, by far the most common pattern is type 1 membranoproliferative glomerulonephritis.13 (Types 2 and 3 are much less common, and we will not discuss them here.) In type 1, light microscopy shows increased mesangial cells and thickened capillary walls (lobular glomeruli), staining of the basement membrane reveals double contours (“tram tracking”) or splitting due to mesangial deposition, and immunofluorescence demonstrates immunoglobulin G and complement C3 deposition. All of these findings were seen in our patient (Figure 2, Figure 3).
Membranoproliferative glomerulonephritis in patients with HCV is most commonly associated with cryoglobulins, a mixture of monoclonal or polyclonal immunoglobulin (Ig) M that have antiglobulin (rheumatoid factor) activity and bind to polyclonal IgG. They reversibly precipitate at less than 37°C, (98.6°F), hence their name. Only 50% to 70% of patients with cryoglobulinemic membranoproliferative glomerulonephritis have detectable serum cryoglobulins; however, kidney biopsy may show globular accumulations of eosinophilic material and prominent hypercellularity due to infiltration of glomerular capillaries with mononuclear and polymorphonuclear leukocytes.
Noncryoglobulinemic membranoproliferative glomerulonephritis is also found in patients with HCV infection. Its histologic features are similar, but on biopsy, there is less prominent leukocytic infiltration and no eosinophilic material. Although the pathogenesis of glomerulonephritis in HCV infection is poorly understood, it is thought to result from deposition of circulating immune complexes of HCV, anti-HCV, and rheumatoid factor in the glomeruli.
3. What laboratory finding is often seen in membranoproliferative glomerulonephritis?
- Positive cytoplasmic antineutrophil cytoplasmic antibody
- serum complement Low levels
- Antiphospholipase A2 receptor antibodies
Cytoplasmic antineutrophil cytoplasmic antibody is seen in granulomatosis with polyangiitis, while antiphospholipid A2 receptor antibodies are seen in idiopathic membranous nephritis.
Low serum complement levels are frequently found in membranoproliferative glomerulonephritis. It is believed that immune complex deposition leads to glomerular damage through activation of the complement pathway and the subsequent influx of inflammatory cells, release of cytokines and proteases, and damage to capillary walls. When repair ensues, new mesangial matrix and basement membrane are deposited, leading to mesangial expansion and duplicated basement membrane.14
In cryoglobulinemic membranoproliferative glomerulonephritis, the complement C4 level is often much lower than C3, but in noncryoglobulinemic forms C3 is lower. A mnemonic to remember nephritic syndromes with low complement levels is “hy-PO-CO-MP-L-EM-ents”; PO for postinfectious, CO for cryoglobulins, MP for membranoproliferative glomerulonephritis, L for lupus, and EM for embolic.
BACK TO OUR PATIENT
In addition to kidney biopsy, we tested our patient for serum cryoglobulins, rheumatoid factor, and serum complements. Results from these tests (Table 3), in addition to the lack of cryoglobulins on his biopsy, led to the conclusion that he had noncryoglobulinemic membranoproliferative glomerulonephritis.
WHO SHOULD RECEIVE TREATMENT FOR HCV?
4. According to the current IDSA/AASLD guidelines, which of the following patients should not receive direct-acting antiviral therapy for HCV?
- Patients with HCV and only low-stage fibrosis
- Patients with decompensated cirrhosis
- Patients with a glomerular filtration rate less than 30 mL/minute
- None of the above—nearly all patients with HCV infection should receive treatment for it
While certain patients have compelling indications for HCV treatment, such as advanced fibrosis, severe extrahepatic manifestations of HCV (eg, glomerulonephritis, cryoglobulinemia), and posttransplant status, current guidelines recommend treatment for nearly all patients with HCV, including those with low-stage fibrosis.12
Patients with Child-Pugh grade B or C decompensated cirrhosis, even with hepatocellular carcinoma, may be considered for treatment. Multiple studies have demonstrated the efficacy and safety of direct-acting antiviral drugs in this patient population. In one randomized controlled trial,15 the combination of ledipasvir, sofosbuvir, and ribavirin resulted in high sustained virologic response rates at 12 weeks in patients infected with HCV genotype 1 or 4 with advanced liver disease, irrespective of transplant status (86% to 89% of patients were pretransplant). Sustained virologic response was associated with improvements in Model for End-Stage Liver Disease and Child-Pugh scores largely due to decreases in bilirubin and improvement in synthetic function (ie, albumin).
Similarly, even patients with a glomerular filtration rate less than 30 mL/min are candidates for treatment. Those with a glomerular filtration rate above 30 mL/min need no dosage adjustments for the most common regimens, while regimens are also available for those with a rate less than 30 mL/min. Although patients with low baseline renal function have a higher frequency of anemia (especially with ribavirin), worsening renal dysfunction, and more severe adverse events, treatment responses remain high and comparable to those without renal impairment.
The Hepatitis C Therapeutic Registry and Research Network (HCV-TARGET) is conducting an ongoing prospective study evaluating real-world use of direct-acting antiviral agents. The study has reported the safety and efficacy of sofosbuvir-containing regimens in patients with varying severities of kidney disease, including glomerular filtration rates less than 30 mL/min). The patients received different regimens that included sofosbuvir. The regimens were reportedly tolerated, and the rate of sustained viral response at 12 weeks remained high.16
The efficacy of direct-acting antiviral agents for HCV-associated glomerulonephritis remains to be studied but is promising. Earlier studies found that antiviral therapy based on interferon alfa with or without ribavirin can significantly decrease proteinuria and stabilize renal function.17–20 HCV RNA clearance has been found to best predict renal improvement.
OUR PATIENT’S COURSE
Unfortunately, our patient’s kidney function declined further over the next 3 months, and he is currently on dialysis awaiting simultaneous liver and kidney transplant.
A 54-year-old man with a history of cirrhosis secondary to hepatitis C virus (HCV) infection has had a progressive decline in kidney function. He was diagnosed with hepatitis C 15 years ago; he tried interferon treatment, but this failed. He received a transjugular intrahepatic shunt 10 years ago after an episode of esophageal variceal bleeding. He has since been taking furosemide and spironolactone as maintenance treatment for ascites, and he has no other medical concerns such as hypertension or diabetes.
Two weeks ago, routine laboratory tests in the clinic showed that his serum creatinine level had increased from baseline. He was asked to stop his diuretics and increase his fluid intake. Nevertheless, his kidney function continued to decline (Table 1), and he was admitted to the hospital for further evaluation.
On admission, he appeared comfortable. He denied recent use of any medications, including nonsteroidal anti-inflammatory drugs, antibiotics, and diuretics, and he had no genitourinary symptoms. His temperature was normal, blood pressure 170/90 mm Hg, pulse rate 72 per minute, and respiratory rate 16. His skin and sclerae were not jaundiced; his abdomen was not tender, but it was grossly distended with ascites. He also had +3 pedal edema (on a scale of 4) extending to both knees. The rest of his physical examination was unremarkable. Results of further laboratory tests are shown in in Table 2.
Ultrasonography of the liver demonstrated cirrhosis with patent flow through the shunt, and ultrasonography of the kidneys showed that both were slightly enlarged with increased cortical echogenicity but no hydronephrosis or obstruction.
EXPLORING THE CAUSE OF RENAL FAILURE
1. Given this information, what is the likely cause of our patient’s renal failure?
- Volume depletion
- Acute tubular necrosis
- Hepatorenal syndrome
- HCV glomerulopathy
Renal failure is a common complication in cirrhosis and portends a higher risk of death.1 The differential diagnosis is broad, but a systematic approach incorporating data from the history, physical examination, and laboratory tests can help identify the cause and is essential in determining the prognosis and proper treatment.
Volume depletion
Volume depletion is a common cause of renal failure in cirrhotic patients. Common precipitants are excessive diuresis and gastrointestinal fluid loss from bleeding, vomiting, and diarrhea. Despite having ascites and edema, patients may have low fluid volume in the vascular space. Therefore, the first step in a patient with acute kidney injury is to withhold diuretics and give fluids. The renal failure usually rapidly reverses if the patient does not have renal parenchymal disease.2
Our patient did not present with any fluid losses, and his high blood pressure and normal heart rate did not suggest volume depletion. And most importantly, withholding his diuretics and giving fluids did not reverse his renal failure. Thus, volume depletion was an unlikely cause.
Acute tubular necrosis
The altered hemodynamics caused by cirrhosis predispose patients to acute tubular necrosis. Classically, this presents as muddy brown casts and renal tubular epithelial cells on urinalysis and as a fractional excretion of sodium greater than 2%.1 However, these microscopic findings lack sensitivity, and patients with cirrhosis may have marked sodium avidity and low urine sodium excretion despite tubular injury.3
This diagnosis must still be considered in patients with renal failure, especially after an insult such as hemorrhagic or septic shock or intake of nephrotoxins. However, because our patient did not have a history of any of these and because his renal failure had been progressing over weeks, acute tubular necrosis was considered unlikely.
Hepatorenal syndrome
Hepatorenal syndrome is characterized by progressive renal failure in the absence of renal parenchymal disease. It is a functional disorder, ie, the decreased glomerular filtration rate results from renal vasoconstriction, which in turn is due to decreased systemic vascular resistance and increased compensatory activity of the renin-angiotensin-aldosterone axis and of antiduretic hormone release (Figure 1).
Hepatorenal syndrome often occurs in patients with advanced liver disease. These patients typically have a hyperdynamic circulation (systemic vasodilation, low blood pressure, and increased blood volume) with a low mean arterial pressure and increased renin and norepinephrine levels. Other frequent findings include hyponatremia, low urinary sodium excretion (< 2 mmol/day), and low free water clearance,4 all of which mark the high systemic levels of antidiuretic hormone and aldosterone.
Importantly, while hepatorenal syndrome is always considered in the differential diagnosis because of its unique prognosis and therapy, it remains a diagnosis of exclusion. The International Ascites Club5 has provided diagnostic criteria for hepatorenal syndrome:
- Cirrhosis and ascites
- Serum creatinine greater than 1.5 mg/dL
- Failure of serum creatinine to fall to less than 1.5 mg/dL after at least 48 hours of diuretic withdrawal and volume expansion with albumin (recommended dose 1 g/kg body weight per day up to a maximum of 100 g per day)
- Absence of shock
- No current or recent treatment with nephrotoxic drugs
- No signs of parenchymal kidney disease such as proteinuria (protein excretion > 500 mg/day), microhematuria (> 50 red blood cells per high-power field), or abnormalities on renal ultrasonography.
While these criteria are not perfect,6 they remind clinicians that there are other important causes of renal insufficiency in cirrhosis.
Clinically, our patient had no evidence of a hyperdynamic circulation and was instead hypertensive. He was eunatremic and did not have marked renal sodium avidity. His pyuria, proteinuria (his protein excretion was approximately 1.9 g/day as determined by urine spot protein-to-creatinine ratio), and results of ultrasonography also suggested underlying renal parenchymal disease. Therefore, hepatorenal syndrome was not the likely diagnosis.
HCV glomerulopathy
Intrinsic renal disease is likely, given our patient’s proteinuria, active urine sediment (ie, containing red blood cells, white blood cells, and protein), and abnormal findings on ultrasonography. In patients with HCV infection and no other cause of intrinsic kidney disease, immune complex deposition leading to glomerulonephritis is the most common pattern.7 Despite the intrinsic renal disease, fractional excretion of sodium may be less than 1% in glomerulonephritis. Hypertension in a patient such as ours with cirrhosis and renal insufficiency raises suspicion for glomerular disease, as hypertension is unlikely in advanced cirrhosis.8
Glomerulonephritis in patients with cirrhosis is often clinically silent and may be highly prevalent; some studies have shown glomerular involvement in 55% to 83% of patients with cirrhosis.9,10 This increases the risk of end-stage renal disease, and the Kidney Disease Improving Global Outcomes guideline recommends that HCV-infected patients be tested at least once a year for proteinuria, hematuria, and estimated glomerular filtration rate to detect possible HCV-associated kidney disease.11 According to current guidelines of the Infectious Diseases Society of America (IDSA) and American Association for the Study of Liver Diseases (AASLD) , detection of glomerulonephritis in HCV patients puts them in the highest priority class for treatment of HCV.12
HISTOLOGIC FINDINGS
Because of the high likelihood of glomerulopathy, our patient underwent renal biopsy.
2. What is the classic pathologic finding in HCV kidney disease?
- Focal segmental glomerulosclerosis
- Crescentic glomerulonephritis
- Membranoproliferative glomerulonephritis
- Membranous glomerulonephritis
A number of pathologic patterns have been described in HCV kidney disease, including membranous glomerulonephritis, immunoglobulin A nephropathy, and focal segmental glomerulosclerosis. However, by far the most common pattern is type 1 membranoproliferative glomerulonephritis.13 (Types 2 and 3 are much less common, and we will not discuss them here.) In type 1, light microscopy shows increased mesangial cells and thickened capillary walls (lobular glomeruli), staining of the basement membrane reveals double contours (“tram tracking”) or splitting due to mesangial deposition, and immunofluorescence demonstrates immunoglobulin G and complement C3 deposition. All of these findings were seen in our patient (Figure 2, Figure 3).
Membranoproliferative glomerulonephritis in patients with HCV is most commonly associated with cryoglobulins, a mixture of monoclonal or polyclonal immunoglobulin (Ig) M that have antiglobulin (rheumatoid factor) activity and bind to polyclonal IgG. They reversibly precipitate at less than 37°C, (98.6°F), hence their name. Only 50% to 70% of patients with cryoglobulinemic membranoproliferative glomerulonephritis have detectable serum cryoglobulins; however, kidney biopsy may show globular accumulations of eosinophilic material and prominent hypercellularity due to infiltration of glomerular capillaries with mononuclear and polymorphonuclear leukocytes.
Noncryoglobulinemic membranoproliferative glomerulonephritis is also found in patients with HCV infection. Its histologic features are similar, but on biopsy, there is less prominent leukocytic infiltration and no eosinophilic material. Although the pathogenesis of glomerulonephritis in HCV infection is poorly understood, it is thought to result from deposition of circulating immune complexes of HCV, anti-HCV, and rheumatoid factor in the glomeruli.
3. What laboratory finding is often seen in membranoproliferative glomerulonephritis?
- Positive cytoplasmic antineutrophil cytoplasmic antibody
- serum complement Low levels
- Antiphospholipase A2 receptor antibodies
Cytoplasmic antineutrophil cytoplasmic antibody is seen in granulomatosis with polyangiitis, while antiphospholipid A2 receptor antibodies are seen in idiopathic membranous nephritis.
Low serum complement levels are frequently found in membranoproliferative glomerulonephritis. It is believed that immune complex deposition leads to glomerular damage through activation of the complement pathway and the subsequent influx of inflammatory cells, release of cytokines and proteases, and damage to capillary walls. When repair ensues, new mesangial matrix and basement membrane are deposited, leading to mesangial expansion and duplicated basement membrane.14
In cryoglobulinemic membranoproliferative glomerulonephritis, the complement C4 level is often much lower than C3, but in noncryoglobulinemic forms C3 is lower. A mnemonic to remember nephritic syndromes with low complement levels is “hy-PO-CO-MP-L-EM-ents”; PO for postinfectious, CO for cryoglobulins, MP for membranoproliferative glomerulonephritis, L for lupus, and EM for embolic.
BACK TO OUR PATIENT
In addition to kidney biopsy, we tested our patient for serum cryoglobulins, rheumatoid factor, and serum complements. Results from these tests (Table 3), in addition to the lack of cryoglobulins on his biopsy, led to the conclusion that he had noncryoglobulinemic membranoproliferative glomerulonephritis.
WHO SHOULD RECEIVE TREATMENT FOR HCV?
4. According to the current IDSA/AASLD guidelines, which of the following patients should not receive direct-acting antiviral therapy for HCV?
- Patients with HCV and only low-stage fibrosis
- Patients with decompensated cirrhosis
- Patients with a glomerular filtration rate less than 30 mL/minute
- None of the above—nearly all patients with HCV infection should receive treatment for it
While certain patients have compelling indications for HCV treatment, such as advanced fibrosis, severe extrahepatic manifestations of HCV (eg, glomerulonephritis, cryoglobulinemia), and posttransplant status, current guidelines recommend treatment for nearly all patients with HCV, including those with low-stage fibrosis.12
Patients with Child-Pugh grade B or C decompensated cirrhosis, even with hepatocellular carcinoma, may be considered for treatment. Multiple studies have demonstrated the efficacy and safety of direct-acting antiviral drugs in this patient population. In one randomized controlled trial,15 the combination of ledipasvir, sofosbuvir, and ribavirin resulted in high sustained virologic response rates at 12 weeks in patients infected with HCV genotype 1 or 4 with advanced liver disease, irrespective of transplant status (86% to 89% of patients were pretransplant). Sustained virologic response was associated with improvements in Model for End-Stage Liver Disease and Child-Pugh scores largely due to decreases in bilirubin and improvement in synthetic function (ie, albumin).
Similarly, even patients with a glomerular filtration rate less than 30 mL/min are candidates for treatment. Those with a glomerular filtration rate above 30 mL/min need no dosage adjustments for the most common regimens, while regimens are also available for those with a rate less than 30 mL/min. Although patients with low baseline renal function have a higher frequency of anemia (especially with ribavirin), worsening renal dysfunction, and more severe adverse events, treatment responses remain high and comparable to those without renal impairment.
The Hepatitis C Therapeutic Registry and Research Network (HCV-TARGET) is conducting an ongoing prospective study evaluating real-world use of direct-acting antiviral agents. The study has reported the safety and efficacy of sofosbuvir-containing regimens in patients with varying severities of kidney disease, including glomerular filtration rates less than 30 mL/min). The patients received different regimens that included sofosbuvir. The regimens were reportedly tolerated, and the rate of sustained viral response at 12 weeks remained high.16
The efficacy of direct-acting antiviral agents for HCV-associated glomerulonephritis remains to be studied but is promising. Earlier studies found that antiviral therapy based on interferon alfa with or without ribavirin can significantly decrease proteinuria and stabilize renal function.17–20 HCV RNA clearance has been found to best predict renal improvement.
OUR PATIENT’S COURSE
Unfortunately, our patient’s kidney function declined further over the next 3 months, and he is currently on dialysis awaiting simultaneous liver and kidney transplant.
- Ginès P, Schrier RW. Renal failure in cirrhosis. N Engl J Med 2009; 361:1279–1290.
- Mackelaite L, Alsauskas ZC, Ranganna K. Renal failure in patients with cirrhosis. Med Clin North Am 2009; 93:855–869.
- Wadei HM, Mai ML, Ahsan N, Gonwa TA. Hepatorenal syndrome: pathophysiology and management. Clin J Am Soc Nephrol 2006; 1:1066–1079.
- Gines A, Escorsell A, Gines P, et al. Incidence, predictive factors, and prognosis of the hepatorenal syndrome in cirrhosis with ascites. Gastroenterology 1993; 105:229–236.
- Salerno F, Gerbes A, Ginès P, Wong F, Arroyo V. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut 2007; 56:1310–1318.
- Watt K, Uhanova J, Minuk GY. Hepatorenal syndrome: diagnostic accuracy, clinical features, and outcome in a tertiary care center. Am J Gastroenterol 2002; 97:2046–2050.
- Graupera I, Cardenas A. Diagnostic approach to renal failure in cirrhosis. Clin Liver Dis 2013; 2:128–131.
- Dash SC, Bhowmik D. Glomerulopathy with liver disease: patterns and management. Saudi J Kidney Dis Transpl 2000; 11:414–420.
- Arase Y, Ikeda K, Murashima N, et al. Glomerulonephritis in autopsy cases with hepatitis C virus infection. Intern Med 1998; 37:836–840.
- McGuire BM, Julian BA, Bynon JS, et al. Brief communication: glomerulonephritis in patients with hepatitis C cirrhosis undergoing liver transplantation. Ann Intern Med 2006; 144:735–741.
- Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO clinical practice guidelines for the prevention, diagnosis, evaluation, and treatment of hepatitis C in chronic kidney disease. Kidney Int Suppl 2008; 109:S1–S99.
- American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America (IDSA). HCV guidance: recommendations for testing, managing, and treating hepatitis C. www.hcvguidelines.org/. Accessed July 10, 2016.
- Lai KN. Hepatitis-related renal disease. Future Virology 2011; 6:1361–1376.
- Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis—a new look at an old entity. N Engl J Med 2012; 366:1119–1131.
- Charlton M, Everson GT, Flamm SL, et al; SOLAR-1 Investigators. Ledipasvir and sofosbuvir plus ribavirin for treatment of HCV infection in patients with advanced liver disease. Gastroenterology 2015; 149:649–659.
- Saxena V, Koraishy FM, Sise ME, et al; HCV-TARGET. Safety and efficacy of sofosbuvir-containing regimens in hepatitis C-infected patients with impaired renal function. Liver Int 2016; 36:807–816.
- Feng B, Eknoyan G, Guo ZS, et al. Effect of interferon alpha-based antiviral therapy on hepatitis C virus-associated glomerulonephritis: a meta-analysis. Nephrol Dial Transplant 2012; 27:640–646.
- Bruchfeld A, Lindahl K, Ståhle L, Söderberg M, Schvarcz R. Interferon and ribavirin treatment in patients with hepatitis C-associated renal disease and renal insufficiency. Nephrol Dial Transplant 2003; 18:1573–1580.
- Rossi P, Bertani T, Baio P, et al. Hepatitis C virus-related cryoglobulinemic glomerulonephritis. Long-term remission after antiviral therapy. Kidney Int 2003; 63:2236–2241.
- Alric L, Plaisier E, Thebault S, et al. Influence of antiviral therapy in hepatitis C virus associated cryoglobulinemic MPGN. Am J Kidney Dis 2004; 43:617–623.
- Ginès P, Schrier RW. Renal failure in cirrhosis. N Engl J Med 2009; 361:1279–1290.
- Mackelaite L, Alsauskas ZC, Ranganna K. Renal failure in patients with cirrhosis. Med Clin North Am 2009; 93:855–869.
- Wadei HM, Mai ML, Ahsan N, Gonwa TA. Hepatorenal syndrome: pathophysiology and management. Clin J Am Soc Nephrol 2006; 1:1066–1079.
- Gines A, Escorsell A, Gines P, et al. Incidence, predictive factors, and prognosis of the hepatorenal syndrome in cirrhosis with ascites. Gastroenterology 1993; 105:229–236.
- Salerno F, Gerbes A, Ginès P, Wong F, Arroyo V. Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Gut 2007; 56:1310–1318.
- Watt K, Uhanova J, Minuk GY. Hepatorenal syndrome: diagnostic accuracy, clinical features, and outcome in a tertiary care center. Am J Gastroenterol 2002; 97:2046–2050.
- Graupera I, Cardenas A. Diagnostic approach to renal failure in cirrhosis. Clin Liver Dis 2013; 2:128–131.
- Dash SC, Bhowmik D. Glomerulopathy with liver disease: patterns and management. Saudi J Kidney Dis Transpl 2000; 11:414–420.
- Arase Y, Ikeda K, Murashima N, et al. Glomerulonephritis in autopsy cases with hepatitis C virus infection. Intern Med 1998; 37:836–840.
- McGuire BM, Julian BA, Bynon JS, et al. Brief communication: glomerulonephritis in patients with hepatitis C cirrhosis undergoing liver transplantation. Ann Intern Med 2006; 144:735–741.
- Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO clinical practice guidelines for the prevention, diagnosis, evaluation, and treatment of hepatitis C in chronic kidney disease. Kidney Int Suppl 2008; 109:S1–S99.
- American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America (IDSA). HCV guidance: recommendations for testing, managing, and treating hepatitis C. www.hcvguidelines.org/. Accessed July 10, 2016.
- Lai KN. Hepatitis-related renal disease. Future Virology 2011; 6:1361–1376.
- Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis—a new look at an old entity. N Engl J Med 2012; 366:1119–1131.
- Charlton M, Everson GT, Flamm SL, et al; SOLAR-1 Investigators. Ledipasvir and sofosbuvir plus ribavirin for treatment of HCV infection in patients with advanced liver disease. Gastroenterology 2015; 149:649–659.
- Saxena V, Koraishy FM, Sise ME, et al; HCV-TARGET. Safety and efficacy of sofosbuvir-containing regimens in hepatitis C-infected patients with impaired renal function. Liver Int 2016; 36:807–816.
- Feng B, Eknoyan G, Guo ZS, et al. Effect of interferon alpha-based antiviral therapy on hepatitis C virus-associated glomerulonephritis: a meta-analysis. Nephrol Dial Transplant 2012; 27:640–646.
- Bruchfeld A, Lindahl K, Ståhle L, Söderberg M, Schvarcz R. Interferon and ribavirin treatment in patients with hepatitis C-associated renal disease and renal insufficiency. Nephrol Dial Transplant 2003; 18:1573–1580.
- Rossi P, Bertani T, Baio P, et al. Hepatitis C virus-related cryoglobulinemic glomerulonephritis. Long-term remission after antiviral therapy. Kidney Int 2003; 63:2236–2241.
- Alric L, Plaisier E, Thebault S, et al. Influence of antiviral therapy in hepatitis C virus associated cryoglobulinemic MPGN. Am J Kidney Dis 2004; 43:617–623.
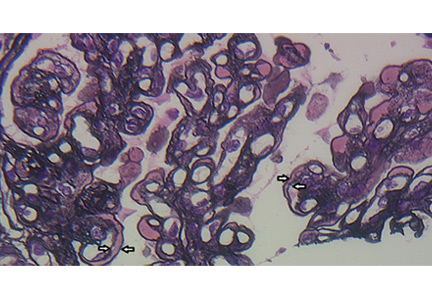
Trust the thyroid thermostat

Primary hypothyroidism is common. Most patients acquire it when the thyroid gland is damaged by autoimmune inflammation. It is readily and reliably treated with the orally administered synthetic hormone levothyroxine, or less reliably with thyroid gland extracts. Absorption of either medication is significantly influenced by food, so patients need to pay attention to the timing of ingestion. But occasional blood testing can be used to easily monitor the sufficiency of replacement therapy.
The predominant active thyroid hormone is triiodothyronine (T3), most of which is converted from thyroxine (T4) by deiodination outside of the thyroid gland. Circulating thyroid-binding globulins tie up significant amounts of these hormones in the blood, and this protein binding is affected by a number of factors. Free T3 and T4—not bound—are the substances that exert physiologic effects on target organs and also give feedback information to the pituitary gland which, completing the loop, releases thyroid-stimulating hormone (TSH) and ultimately controls the healthy thyroid gland’s production and release of its hormones. Hence, the total circulating thyroid hormone levels are not as biologically relevant as the free T3 and T4 levels. Even in the absence of a functioning thyroid gland, the TSH level reliably reflects the bioactivity of circulating thyroid hormones so long as the pituitary gland is functioning normally.
Routine tracking of the biologic effects of thyroid hormone, such as the metabolic rate, is unreasonable, and other biologic effects such as the cholesterol level are influenced by so many factors in addition to T3 as to be unreliable indicators of thyroid hormone levels. Assuming the patient’s hypothalamic-pituitary axis is normal, the most reasonable and reliable way to track the biologic effect of thyroid hormone is to follow the TSH level. Although the exact relationship between free thyroid hormone and TSH levels is slightly different between patients with normal thyroid glands and those with damaged glands receiving replacement therapy, TSH measurement is an excellent indicator of the level that the brain wants thyroid function to be. Other than in specific nonhomeostatic circumstances, the pituitary gland is usually a superb thermostat for thyroid hormone activity.
In this issue of the Journal, Dr. Christian Nasr discusses the rationale for routinely using TSH measurement alone to direct exogenous thyroid replacement, explaining why it is cost-effective and clinically appropriate.
While following T3 and T4 may occasionally be useful in a few patients, the wealth of clinical data does not support this practice. As a routine practice it is certainly financially wasteful, and may lead to inappropriate clinical decisions.
Why, then, do some physicians persist in regularly following T3 and T4 levels in addition to TSH? There is no single answer. Although some patients may feel “better” if they take a little more rather than a little less levothyroxine, whether this benefit outweighs the metabolic price in the long run is not at all clear. Plus, in the published experience with treating subclinical hypothyroidism,1 patients did not generally feel better or experience desired weight loss when they received slightly more exogenous thyroid hormone. Somewhat analogously, if the TSH level is already normal, increasing thyroid replacement to attain a free T3 or T4 level in the high-normal range is unlikely to improve clinical outcomes in a meaningful way and may well be detrimental in the long term.
Despite a lot of chatter on Internet blogs regarding the multiple benefits of selective T3 replacement and higher T3 levels, akin to testosterone supplementation above what the (normal functioning) hypothalamic-pituitary axis has determined to be biologically appropriate, there is limited clinical evidence to support this practice. When replacing the output of a diseased or absent thyroid gland, it is reasonable clinical practice to trust the pituitary readings of the thyroid thermostat.
- Rugge JB, Bougatsos C, Chou R. Screening and treatment of thyroid dysfunction: an evidence review for the U.S. Preventive Services Task Force. Ann Intern Med 2015; 162:35–45.
Primary hypothyroidism is common. Most patients acquire it when the thyroid gland is damaged by autoimmune inflammation. It is readily and reliably treated with the orally administered synthetic hormone levothyroxine, or less reliably with thyroid gland extracts. Absorption of either medication is significantly influenced by food, so patients need to pay attention to the timing of ingestion. But occasional blood testing can be used to easily monitor the sufficiency of replacement therapy.
The predominant active thyroid hormone is triiodothyronine (T3), most of which is converted from thyroxine (T4) by deiodination outside of the thyroid gland. Circulating thyroid-binding globulins tie up significant amounts of these hormones in the blood, and this protein binding is affected by a number of factors. Free T3 and T4—not bound—are the substances that exert physiologic effects on target organs and also give feedback information to the pituitary gland which, completing the loop, releases thyroid-stimulating hormone (TSH) and ultimately controls the healthy thyroid gland’s production and release of its hormones. Hence, the total circulating thyroid hormone levels are not as biologically relevant as the free T3 and T4 levels. Even in the absence of a functioning thyroid gland, the TSH level reliably reflects the bioactivity of circulating thyroid hormones so long as the pituitary gland is functioning normally.
Routine tracking of the biologic effects of thyroid hormone, such as the metabolic rate, is unreasonable, and other biologic effects such as the cholesterol level are influenced by so many factors in addition to T3 as to be unreliable indicators of thyroid hormone levels. Assuming the patient’s hypothalamic-pituitary axis is normal, the most reasonable and reliable way to track the biologic effect of thyroid hormone is to follow the TSH level. Although the exact relationship between free thyroid hormone and TSH levels is slightly different between patients with normal thyroid glands and those with damaged glands receiving replacement therapy, TSH measurement is an excellent indicator of the level that the brain wants thyroid function to be. Other than in specific nonhomeostatic circumstances, the pituitary gland is usually a superb thermostat for thyroid hormone activity.
In this issue of the Journal, Dr. Christian Nasr discusses the rationale for routinely using TSH measurement alone to direct exogenous thyroid replacement, explaining why it is cost-effective and clinically appropriate.
While following T3 and T4 may occasionally be useful in a few patients, the wealth of clinical data does not support this practice. As a routine practice it is certainly financially wasteful, and may lead to inappropriate clinical decisions.
Why, then, do some physicians persist in regularly following T3 and T4 levels in addition to TSH? There is no single answer. Although some patients may feel “better” if they take a little more rather than a little less levothyroxine, whether this benefit outweighs the metabolic price in the long run is not at all clear. Plus, in the published experience with treating subclinical hypothyroidism,1 patients did not generally feel better or experience desired weight loss when they received slightly more exogenous thyroid hormone. Somewhat analogously, if the TSH level is already normal, increasing thyroid replacement to attain a free T3 or T4 level in the high-normal range is unlikely to improve clinical outcomes in a meaningful way and may well be detrimental in the long term.
Despite a lot of chatter on Internet blogs regarding the multiple benefits of selective T3 replacement and higher T3 levels, akin to testosterone supplementation above what the (normal functioning) hypothalamic-pituitary axis has determined to be biologically appropriate, there is limited clinical evidence to support this practice. When replacing the output of a diseased or absent thyroid gland, it is reasonable clinical practice to trust the pituitary readings of the thyroid thermostat.
Primary hypothyroidism is common. Most patients acquire it when the thyroid gland is damaged by autoimmune inflammation. It is readily and reliably treated with the orally administered synthetic hormone levothyroxine, or less reliably with thyroid gland extracts. Absorption of either medication is significantly influenced by food, so patients need to pay attention to the timing of ingestion. But occasional blood testing can be used to easily monitor the sufficiency of replacement therapy.
The predominant active thyroid hormone is triiodothyronine (T3), most of which is converted from thyroxine (T4) by deiodination outside of the thyroid gland. Circulating thyroid-binding globulins tie up significant amounts of these hormones in the blood, and this protein binding is affected by a number of factors. Free T3 and T4—not bound—are the substances that exert physiologic effects on target organs and also give feedback information to the pituitary gland which, completing the loop, releases thyroid-stimulating hormone (TSH) and ultimately controls the healthy thyroid gland’s production and release of its hormones. Hence, the total circulating thyroid hormone levels are not as biologically relevant as the free T3 and T4 levels. Even in the absence of a functioning thyroid gland, the TSH level reliably reflects the bioactivity of circulating thyroid hormones so long as the pituitary gland is functioning normally.
Routine tracking of the biologic effects of thyroid hormone, such as the metabolic rate, is unreasonable, and other biologic effects such as the cholesterol level are influenced by so many factors in addition to T3 as to be unreliable indicators of thyroid hormone levels. Assuming the patient’s hypothalamic-pituitary axis is normal, the most reasonable and reliable way to track the biologic effect of thyroid hormone is to follow the TSH level. Although the exact relationship between free thyroid hormone and TSH levels is slightly different between patients with normal thyroid glands and those with damaged glands receiving replacement therapy, TSH measurement is an excellent indicator of the level that the brain wants thyroid function to be. Other than in specific nonhomeostatic circumstances, the pituitary gland is usually a superb thermostat for thyroid hormone activity.
In this issue of the Journal, Dr. Christian Nasr discusses the rationale for routinely using TSH measurement alone to direct exogenous thyroid replacement, explaining why it is cost-effective and clinically appropriate.
While following T3 and T4 may occasionally be useful in a few patients, the wealth of clinical data does not support this practice. As a routine practice it is certainly financially wasteful, and may lead to inappropriate clinical decisions.
Why, then, do some physicians persist in regularly following T3 and T4 levels in addition to TSH? There is no single answer. Although some patients may feel “better” if they take a little more rather than a little less levothyroxine, whether this benefit outweighs the metabolic price in the long run is not at all clear. Plus, in the published experience with treating subclinical hypothyroidism,1 patients did not generally feel better or experience desired weight loss when they received slightly more exogenous thyroid hormone. Somewhat analogously, if the TSH level is already normal, increasing thyroid replacement to attain a free T3 or T4 level in the high-normal range is unlikely to improve clinical outcomes in a meaningful way and may well be detrimental in the long term.
Despite a lot of chatter on Internet blogs regarding the multiple benefits of selective T3 replacement and higher T3 levels, akin to testosterone supplementation above what the (normal functioning) hypothalamic-pituitary axis has determined to be biologically appropriate, there is limited clinical evidence to support this practice. When replacing the output of a diseased or absent thyroid gland, it is reasonable clinical practice to trust the pituitary readings of the thyroid thermostat.
- Rugge JB, Bougatsos C, Chou R. Screening and treatment of thyroid dysfunction: an evidence review for the U.S. Preventive Services Task Force. Ann Intern Med 2015; 162:35–45.
- Rugge JB, Bougatsos C, Chou R. Screening and treatment of thyroid dysfunction: an evidence review for the U.S. Preventive Services Task Force. Ann Intern Med 2015; 162:35–45.
Is a serum TSH measurement sufficient to monitor the treatment of primary hypothyroidism?
A 28-year-old woman returns for follow-up of her hypothyroidism. She was diagnosed 4 years ago when she presented with fatigue, “foggy” thinking, poor concentration, cold intolerance, and constipation. Her thyroid-stimulating hormone (TSH) level at that time was elevated at 15 mIU/L (reference range 0.4–4). She was started on 50 µg of levothyroxine daily, which helped her symptoms, but she continued to complain of tiredness and the inability to lose weight. She has been on 100 µg of levothyroxine daily since her last visit 1 year ago.
On examination, she has a small, diffuse, and firm goiter; she has no Cushing-like features, visual field abnormalities, or signs of hypothyroidism.
Her TSH level today is 1.2 mIU/L. Based on this, you recommend no change in her daily levothyroxine dose. She expresses dissatisfaction that you had ordered only a TSH, and she asks you to order thyroxine (T4) and triiodothyronine (T3) measurements because she read on the Internet that those were needed to determine the appropriateness of the levothyroxine dose.
Should T4 or T3 be routinely measured when adjusting thyroid replacement therapy?
IN PRIMARY HYPOTHYROIDISM, TSH IS ENOUGH
In a patient with primary hypothyroidism and no suspicion of pituitary abnormality, a serum TSH is sufficient for monitoring thyroid status and adjusting the dose of thyroid hormone.
Hypothyroidism is one of the most common endocrine disorders, affecting about 4% of the adult US population.1 In areas of iodine sufficiency, primary hypothyroidism is due predominantly to Hashimoto thyroiditis.
The role of the lack of thyroid hormone in the pathogenesis of myxedema was recognized in the late 19th century through the observation of a “cretinoid” state occurring in middle-aged women, associated with atrophy of the thyroid gland and a similar severe state noted after total thyroidectomy.2
In 1891, George R. Murray was able to “cure” myxedema in a patient by injecting sheep thyroid extract subcutaneously. Thyroid extracts continued to be the only treatment for hypothyroidism until 1950, when levothyroxine was introduced and later became the main treatment. Around that time, T3 was discovered and was described as being the physiologically active thyroid hormone. Later, it was noted that 80% to 90% of circulating T3 is generated through peripheral deiodination of T4, the latter being considered a prohormone.2
The pituitary-thyroid axis is regulated through negative feedback. At concentrations of free T4 below normal, plasma TSH rises rapidly with small decrements in T4 levels.3 The opposite phenomenon occurs with free T4 concentrations above normal. Since T4 has a long disappearance half-time—about 7 days—a normal TSH tends to stay relatively stable in the same individual.4 The relationship between TSH and T4 was long thought to be inverse log-linear, but Hadlow et al5 found that it is complex and nonlinear and differs by age and sex. TSH and T4 concentrations have narrower within-individual variability than inter-individual variability. Although environmental factors may affect this hypothalamic-pituitary set-point, there is evidence that heritability is a major determinant of individual variability.6
GUIDELINES AND CHOOSING WISELY
In 2014, the American Thyroid Association published comprehensive, evidence-based guidelines for the treatment of hypothyroidism.7 The guidelines state that the goal of thyroid hormone replacement is to achieve clinical and biochemical euthyroidism.7 TSH continues to be the most reliable marker of adequacy of thyroid hormone replacement in primary hypothyroidism. The guidelines recommend aiming for a TSH in the normal range (generally 0.4–4 mIU/L).
Most studies of the risks associated with hypothyroidism or thyrotoxicosis have looked at TSH levels. Significantly increased risk of cardiovascular mortality and morbidity is seen in individuals with TSH levels higher than 10 mIU/L.8 On the other hand, excess thyroid hormone leading to a TSH level lower than 0.1 mIU/L has been associated with an increased risk of atrial fibrillation in older persons and osteoporosis in postmenopausal women.
The classic symptoms and signs of hypothyroidism correlate with biochemical hypothyroidism and usually improve with the restoration of euthyroidism. Some of these symptoms, however, lack sensitivity and specificity, especially with modest degrees of hypothyroidism.7 A randomized controlled trial showed that patients were unable to detect any difference in symptoms when the levothyroxine dose was changed by about 20%.9
HARMS ASSOCIATED WITH ORDERING T4 AND T3
Other than the financial burden to the patient and society, there is no major morbidity caused by obtaining T4 or T3 levels, or both. However, knowing the T4 or T3 level does not help with management beyond the information offered by the TSH value. Hypothyroid patients treated with levothyroxine to maintain a normal TSH generally have higher free T4 levels and lower free T3 levels than euthyroid patients with similar TSH values.10 Therefore, reacting to a high T4 level or a low T3 level in a treated hypothyroid patient with a normal TSH may lead to inappropriate dose adjustment. On the other hand, increasing the dose of thyroid hormone in a patient with a low TSH whose T3 level is low-normal may lead to morbidity.
SPECIAL SCENARIO: PITUITARY COMPROMISE
We assume that the patient described above has primary hypothyroidism and that her pituitary-thyroid axis is intact. Primary hypothyroidism is diagnosed by a high TSH along with a low or low-normal T4. In this typical case, TSH can be used to guide therapy without the need for other tests.
However, when there is pituitary compromise (hypopituitarism, congenital central hypothyroidism), the TSH will not be reliable to monitor the adequacy of thyroid hormone replacement therapy. The aim of levothyroxine management in these patients is to maintain a free T4 concentration in the upper half of the normal range. If the free T3 concentration is followed and is found to be elevated, the dose of levothyroxine should be reduced.11
CLINICAL BOTTOM LINE
Since our patient’s dose of levothyroxine has been stable and her TSH is not elevated, measuring serum levels of T4 and T3 would not contribute to her management. For such a patient, if the TSH were less than 3 mIU/L, increasing the dose would be unlikely to offer clinical benefit.
On the other hand, if her TSH was higher than 4 mIU/L, then it would be legitimate to tweak the dose upward and reassess her thyroid state clinically and biochemically 6 to 8 weeks later. One would need to be careful not to induce thyrotoxicosis through such an intervention because of the potential morbidity.
The TSH level is typically monitored every 6 to 12 months when the patient is clinically stable. It should be measured sooner in circumstances that include the following:
- Symptoms of hypothyroidism or thyrotoxicosis
- Starting a new medication known to affect thyroid hormone levels
- Significant weight change
- Hospitalization
- Pregnancy.
- Aoki Y, Belin RM, Clickner R, Jeffries R, Phillips L, Mahaffey KR. Serum TSH and total T4 in the United States population and their association with participant characteristics: National Health and Nutrition Examination Survey (NHANES 1999–2002). Thyroid 2007; 17:1211–1223.
- Kopp PE. Commentary on: guidelines for the treatment of hypothyroidism. Thyroid 2014; 24:1667–1669.
- Reichlin S, Utiger RD. Regulation of the pituitary-thyroid axis in man: relationship of TSH concentration to concentration of free and total thyroxine in plasma. J Clin Endocrinol Metab 1967; 27:251–255.
- Azukizawa M, Pekary AE, Hershman JM, Parker DC. Plasma thyrotropin, thyroxine, and triiodothyronine relationships in man. J Clin Endocrinol Metab 1976; 43:533–542.
- Hadlow NC, Rothacker KM, Wardrop R, Brown SJ, Mun Lim E, Walsh JP. The relationship between TSH and free T4 in a large population is complex and nonlinear and differs by age and sex. J Clin Endocrinol Metab 2013; 98:2936–2943.
- Clark PM, Holder RL, Haque SM, Hobbs FDR, Roberts LM, Franklyn JA. The relationship between serum TSH and free T4 in older people. J Clin Pathol 2012; 65:463–465.
- Jonklaas J, Bianco AC, Bauer AJ, et al; American Thyroid Association Task Force on Thyroid Hormone Replacement. Guidelines for the treatment of hypothyroidism. Thyroid 2014; 24:1670–1751.
- Rodondi N, den Elzen WP, Bauer DC, et al; Thyroid Studies Collaboration. Subclinical hypothyroidism and the risk of coronary heart disease and mortality. JAMA 2010; 304:1365–1374.
- Walsh JP, Ward LC, Burke V, et al. Small changes in thyroxine dosage do not produce measurable changes in hypothyroid symptoms, well-being, or quality of life: results of a double-blind, randomized clinical trial. J Clin Endocrinol Metab 2006; 91:2624–2630.
- Woeber KA. Levothyroxine therapy and serum free thyroxine and free triiodothyronine concentrations. J Endocrinol Invest 2002; 25:106–109.
- Grunenwald S, Caron P. Central hypothyroidism in adults: better understanding for better care. Pituitary 2015; 18:169–175.
A 28-year-old woman returns for follow-up of her hypothyroidism. She was diagnosed 4 years ago when she presented with fatigue, “foggy” thinking, poor concentration, cold intolerance, and constipation. Her thyroid-stimulating hormone (TSH) level at that time was elevated at 15 mIU/L (reference range 0.4–4). She was started on 50 µg of levothyroxine daily, which helped her symptoms, but she continued to complain of tiredness and the inability to lose weight. She has been on 100 µg of levothyroxine daily since her last visit 1 year ago.
On examination, she has a small, diffuse, and firm goiter; she has no Cushing-like features, visual field abnormalities, or signs of hypothyroidism.
Her TSH level today is 1.2 mIU/L. Based on this, you recommend no change in her daily levothyroxine dose. She expresses dissatisfaction that you had ordered only a TSH, and she asks you to order thyroxine (T4) and triiodothyronine (T3) measurements because she read on the Internet that those were needed to determine the appropriateness of the levothyroxine dose.
Should T4 or T3 be routinely measured when adjusting thyroid replacement therapy?
IN PRIMARY HYPOTHYROIDISM, TSH IS ENOUGH
In a patient with primary hypothyroidism and no suspicion of pituitary abnormality, a serum TSH is sufficient for monitoring thyroid status and adjusting the dose of thyroid hormone.
Hypothyroidism is one of the most common endocrine disorders, affecting about 4% of the adult US population.1 In areas of iodine sufficiency, primary hypothyroidism is due predominantly to Hashimoto thyroiditis.
The role of the lack of thyroid hormone in the pathogenesis of myxedema was recognized in the late 19th century through the observation of a “cretinoid” state occurring in middle-aged women, associated with atrophy of the thyroid gland and a similar severe state noted after total thyroidectomy.2
In 1891, George R. Murray was able to “cure” myxedema in a patient by injecting sheep thyroid extract subcutaneously. Thyroid extracts continued to be the only treatment for hypothyroidism until 1950, when levothyroxine was introduced and later became the main treatment. Around that time, T3 was discovered and was described as being the physiologically active thyroid hormone. Later, it was noted that 80% to 90% of circulating T3 is generated through peripheral deiodination of T4, the latter being considered a prohormone.2
The pituitary-thyroid axis is regulated through negative feedback. At concentrations of free T4 below normal, plasma TSH rises rapidly with small decrements in T4 levels.3 The opposite phenomenon occurs with free T4 concentrations above normal. Since T4 has a long disappearance half-time—about 7 days—a normal TSH tends to stay relatively stable in the same individual.4 The relationship between TSH and T4 was long thought to be inverse log-linear, but Hadlow et al5 found that it is complex and nonlinear and differs by age and sex. TSH and T4 concentrations have narrower within-individual variability than inter-individual variability. Although environmental factors may affect this hypothalamic-pituitary set-point, there is evidence that heritability is a major determinant of individual variability.6
GUIDELINES AND CHOOSING WISELY
In 2014, the American Thyroid Association published comprehensive, evidence-based guidelines for the treatment of hypothyroidism.7 The guidelines state that the goal of thyroid hormone replacement is to achieve clinical and biochemical euthyroidism.7 TSH continues to be the most reliable marker of adequacy of thyroid hormone replacement in primary hypothyroidism. The guidelines recommend aiming for a TSH in the normal range (generally 0.4–4 mIU/L).
Most studies of the risks associated with hypothyroidism or thyrotoxicosis have looked at TSH levels. Significantly increased risk of cardiovascular mortality and morbidity is seen in individuals with TSH levels higher than 10 mIU/L.8 On the other hand, excess thyroid hormone leading to a TSH level lower than 0.1 mIU/L has been associated with an increased risk of atrial fibrillation in older persons and osteoporosis in postmenopausal women.
The classic symptoms and signs of hypothyroidism correlate with biochemical hypothyroidism and usually improve with the restoration of euthyroidism. Some of these symptoms, however, lack sensitivity and specificity, especially with modest degrees of hypothyroidism.7 A randomized controlled trial showed that patients were unable to detect any difference in symptoms when the levothyroxine dose was changed by about 20%.9
HARMS ASSOCIATED WITH ORDERING T4 AND T3
Other than the financial burden to the patient and society, there is no major morbidity caused by obtaining T4 or T3 levels, or both. However, knowing the T4 or T3 level does not help with management beyond the information offered by the TSH value. Hypothyroid patients treated with levothyroxine to maintain a normal TSH generally have higher free T4 levels and lower free T3 levels than euthyroid patients with similar TSH values.10 Therefore, reacting to a high T4 level or a low T3 level in a treated hypothyroid patient with a normal TSH may lead to inappropriate dose adjustment. On the other hand, increasing the dose of thyroid hormone in a patient with a low TSH whose T3 level is low-normal may lead to morbidity.
SPECIAL SCENARIO: PITUITARY COMPROMISE
We assume that the patient described above has primary hypothyroidism and that her pituitary-thyroid axis is intact. Primary hypothyroidism is diagnosed by a high TSH along with a low or low-normal T4. In this typical case, TSH can be used to guide therapy without the need for other tests.
However, when there is pituitary compromise (hypopituitarism, congenital central hypothyroidism), the TSH will not be reliable to monitor the adequacy of thyroid hormone replacement therapy. The aim of levothyroxine management in these patients is to maintain a free T4 concentration in the upper half of the normal range. If the free T3 concentration is followed and is found to be elevated, the dose of levothyroxine should be reduced.11
CLINICAL BOTTOM LINE
Since our patient’s dose of levothyroxine has been stable and her TSH is not elevated, measuring serum levels of T4 and T3 would not contribute to her management. For such a patient, if the TSH were less than 3 mIU/L, increasing the dose would be unlikely to offer clinical benefit.
On the other hand, if her TSH was higher than 4 mIU/L, then it would be legitimate to tweak the dose upward and reassess her thyroid state clinically and biochemically 6 to 8 weeks later. One would need to be careful not to induce thyrotoxicosis through such an intervention because of the potential morbidity.
The TSH level is typically monitored every 6 to 12 months when the patient is clinically stable. It should be measured sooner in circumstances that include the following:
- Symptoms of hypothyroidism or thyrotoxicosis
- Starting a new medication known to affect thyroid hormone levels
- Significant weight change
- Hospitalization
- Pregnancy.
A 28-year-old woman returns for follow-up of her hypothyroidism. She was diagnosed 4 years ago when she presented with fatigue, “foggy” thinking, poor concentration, cold intolerance, and constipation. Her thyroid-stimulating hormone (TSH) level at that time was elevated at 15 mIU/L (reference range 0.4–4). She was started on 50 µg of levothyroxine daily, which helped her symptoms, but she continued to complain of tiredness and the inability to lose weight. She has been on 100 µg of levothyroxine daily since her last visit 1 year ago.
On examination, she has a small, diffuse, and firm goiter; she has no Cushing-like features, visual field abnormalities, or signs of hypothyroidism.
Her TSH level today is 1.2 mIU/L. Based on this, you recommend no change in her daily levothyroxine dose. She expresses dissatisfaction that you had ordered only a TSH, and she asks you to order thyroxine (T4) and triiodothyronine (T3) measurements because she read on the Internet that those were needed to determine the appropriateness of the levothyroxine dose.
Should T4 or T3 be routinely measured when adjusting thyroid replacement therapy?
IN PRIMARY HYPOTHYROIDISM, TSH IS ENOUGH
In a patient with primary hypothyroidism and no suspicion of pituitary abnormality, a serum TSH is sufficient for monitoring thyroid status and adjusting the dose of thyroid hormone.
Hypothyroidism is one of the most common endocrine disorders, affecting about 4% of the adult US population.1 In areas of iodine sufficiency, primary hypothyroidism is due predominantly to Hashimoto thyroiditis.
The role of the lack of thyroid hormone in the pathogenesis of myxedema was recognized in the late 19th century through the observation of a “cretinoid” state occurring in middle-aged women, associated with atrophy of the thyroid gland and a similar severe state noted after total thyroidectomy.2
In 1891, George R. Murray was able to “cure” myxedema in a patient by injecting sheep thyroid extract subcutaneously. Thyroid extracts continued to be the only treatment for hypothyroidism until 1950, when levothyroxine was introduced and later became the main treatment. Around that time, T3 was discovered and was described as being the physiologically active thyroid hormone. Later, it was noted that 80% to 90% of circulating T3 is generated through peripheral deiodination of T4, the latter being considered a prohormone.2
The pituitary-thyroid axis is regulated through negative feedback. At concentrations of free T4 below normal, plasma TSH rises rapidly with small decrements in T4 levels.3 The opposite phenomenon occurs with free T4 concentrations above normal. Since T4 has a long disappearance half-time—about 7 days—a normal TSH tends to stay relatively stable in the same individual.4 The relationship between TSH and T4 was long thought to be inverse log-linear, but Hadlow et al5 found that it is complex and nonlinear and differs by age and sex. TSH and T4 concentrations have narrower within-individual variability than inter-individual variability. Although environmental factors may affect this hypothalamic-pituitary set-point, there is evidence that heritability is a major determinant of individual variability.6
GUIDELINES AND CHOOSING WISELY
In 2014, the American Thyroid Association published comprehensive, evidence-based guidelines for the treatment of hypothyroidism.7 The guidelines state that the goal of thyroid hormone replacement is to achieve clinical and biochemical euthyroidism.7 TSH continues to be the most reliable marker of adequacy of thyroid hormone replacement in primary hypothyroidism. The guidelines recommend aiming for a TSH in the normal range (generally 0.4–4 mIU/L).
Most studies of the risks associated with hypothyroidism or thyrotoxicosis have looked at TSH levels. Significantly increased risk of cardiovascular mortality and morbidity is seen in individuals with TSH levels higher than 10 mIU/L.8 On the other hand, excess thyroid hormone leading to a TSH level lower than 0.1 mIU/L has been associated with an increased risk of atrial fibrillation in older persons and osteoporosis in postmenopausal women.
The classic symptoms and signs of hypothyroidism correlate with biochemical hypothyroidism and usually improve with the restoration of euthyroidism. Some of these symptoms, however, lack sensitivity and specificity, especially with modest degrees of hypothyroidism.7 A randomized controlled trial showed that patients were unable to detect any difference in symptoms when the levothyroxine dose was changed by about 20%.9
HARMS ASSOCIATED WITH ORDERING T4 AND T3
Other than the financial burden to the patient and society, there is no major morbidity caused by obtaining T4 or T3 levels, or both. However, knowing the T4 or T3 level does not help with management beyond the information offered by the TSH value. Hypothyroid patients treated with levothyroxine to maintain a normal TSH generally have higher free T4 levels and lower free T3 levels than euthyroid patients with similar TSH values.10 Therefore, reacting to a high T4 level or a low T3 level in a treated hypothyroid patient with a normal TSH may lead to inappropriate dose adjustment. On the other hand, increasing the dose of thyroid hormone in a patient with a low TSH whose T3 level is low-normal may lead to morbidity.
SPECIAL SCENARIO: PITUITARY COMPROMISE
We assume that the patient described above has primary hypothyroidism and that her pituitary-thyroid axis is intact. Primary hypothyroidism is diagnosed by a high TSH along with a low or low-normal T4. In this typical case, TSH can be used to guide therapy without the need for other tests.
However, when there is pituitary compromise (hypopituitarism, congenital central hypothyroidism), the TSH will not be reliable to monitor the adequacy of thyroid hormone replacement therapy. The aim of levothyroxine management in these patients is to maintain a free T4 concentration in the upper half of the normal range. If the free T3 concentration is followed and is found to be elevated, the dose of levothyroxine should be reduced.11
CLINICAL BOTTOM LINE
Since our patient’s dose of levothyroxine has been stable and her TSH is not elevated, measuring serum levels of T4 and T3 would not contribute to her management. For such a patient, if the TSH were less than 3 mIU/L, increasing the dose would be unlikely to offer clinical benefit.
On the other hand, if her TSH was higher than 4 mIU/L, then it would be legitimate to tweak the dose upward and reassess her thyroid state clinically and biochemically 6 to 8 weeks later. One would need to be careful not to induce thyrotoxicosis through such an intervention because of the potential morbidity.
The TSH level is typically monitored every 6 to 12 months when the patient is clinically stable. It should be measured sooner in circumstances that include the following:
- Symptoms of hypothyroidism or thyrotoxicosis
- Starting a new medication known to affect thyroid hormone levels
- Significant weight change
- Hospitalization
- Pregnancy.
- Aoki Y, Belin RM, Clickner R, Jeffries R, Phillips L, Mahaffey KR. Serum TSH and total T4 in the United States population and their association with participant characteristics: National Health and Nutrition Examination Survey (NHANES 1999–2002). Thyroid 2007; 17:1211–1223.
- Kopp PE. Commentary on: guidelines for the treatment of hypothyroidism. Thyroid 2014; 24:1667–1669.
- Reichlin S, Utiger RD. Regulation of the pituitary-thyroid axis in man: relationship of TSH concentration to concentration of free and total thyroxine in plasma. J Clin Endocrinol Metab 1967; 27:251–255.
- Azukizawa M, Pekary AE, Hershman JM, Parker DC. Plasma thyrotropin, thyroxine, and triiodothyronine relationships in man. J Clin Endocrinol Metab 1976; 43:533–542.
- Hadlow NC, Rothacker KM, Wardrop R, Brown SJ, Mun Lim E, Walsh JP. The relationship between TSH and free T4 in a large population is complex and nonlinear and differs by age and sex. J Clin Endocrinol Metab 2013; 98:2936–2943.
- Clark PM, Holder RL, Haque SM, Hobbs FDR, Roberts LM, Franklyn JA. The relationship between serum TSH and free T4 in older people. J Clin Pathol 2012; 65:463–465.
- Jonklaas J, Bianco AC, Bauer AJ, et al; American Thyroid Association Task Force on Thyroid Hormone Replacement. Guidelines for the treatment of hypothyroidism. Thyroid 2014; 24:1670–1751.
- Rodondi N, den Elzen WP, Bauer DC, et al; Thyroid Studies Collaboration. Subclinical hypothyroidism and the risk of coronary heart disease and mortality. JAMA 2010; 304:1365–1374.
- Walsh JP, Ward LC, Burke V, et al. Small changes in thyroxine dosage do not produce measurable changes in hypothyroid symptoms, well-being, or quality of life: results of a double-blind, randomized clinical trial. J Clin Endocrinol Metab 2006; 91:2624–2630.
- Woeber KA. Levothyroxine therapy and serum free thyroxine and free triiodothyronine concentrations. J Endocrinol Invest 2002; 25:106–109.
- Grunenwald S, Caron P. Central hypothyroidism in adults: better understanding for better care. Pituitary 2015; 18:169–175.
- Aoki Y, Belin RM, Clickner R, Jeffries R, Phillips L, Mahaffey KR. Serum TSH and total T4 in the United States population and their association with participant characteristics: National Health and Nutrition Examination Survey (NHANES 1999–2002). Thyroid 2007; 17:1211–1223.
- Kopp PE. Commentary on: guidelines for the treatment of hypothyroidism. Thyroid 2014; 24:1667–1669.
- Reichlin S, Utiger RD. Regulation of the pituitary-thyroid axis in man: relationship of TSH concentration to concentration of free and total thyroxine in plasma. J Clin Endocrinol Metab 1967; 27:251–255.
- Azukizawa M, Pekary AE, Hershman JM, Parker DC. Plasma thyrotropin, thyroxine, and triiodothyronine relationships in man. J Clin Endocrinol Metab 1976; 43:533–542.
- Hadlow NC, Rothacker KM, Wardrop R, Brown SJ, Mun Lim E, Walsh JP. The relationship between TSH and free T4 in a large population is complex and nonlinear and differs by age and sex. J Clin Endocrinol Metab 2013; 98:2936–2943.
- Clark PM, Holder RL, Haque SM, Hobbs FDR, Roberts LM, Franklyn JA. The relationship between serum TSH and free T4 in older people. J Clin Pathol 2012; 65:463–465.
- Jonklaas J, Bianco AC, Bauer AJ, et al; American Thyroid Association Task Force on Thyroid Hormone Replacement. Guidelines for the treatment of hypothyroidism. Thyroid 2014; 24:1670–1751.
- Rodondi N, den Elzen WP, Bauer DC, et al; Thyroid Studies Collaboration. Subclinical hypothyroidism and the risk of coronary heart disease and mortality. JAMA 2010; 304:1365–1374.
- Walsh JP, Ward LC, Burke V, et al. Small changes in thyroxine dosage do not produce measurable changes in hypothyroid symptoms, well-being, or quality of life: results of a double-blind, randomized clinical trial. J Clin Endocrinol Metab 2006; 91:2624–2630.
- Woeber KA. Levothyroxine therapy and serum free thyroxine and free triiodothyronine concentrations. J Endocrinol Invest 2002; 25:106–109.
- Grunenwald S, Caron P. Central hypothyroidism in adults: better understanding for better care. Pituitary 2015; 18:169–175.
Your patient has chronic leukemia: Now what?
The advent of targeted therapies has dramatically changed the management of chronic leukemia. Chemotherapy—highly toxic, nonspecific drugs that can be dangerous to patients and providers and result in only modest success—is gradually being replaced by biologic targeting of malignancy. Scientists are rapidly identifying extracellular and intracellular targets on tumor cells and are developing and testing promising new therapies aimed at these targets. Survival of cancer patients has become so common that clinicians outside the specialties of hematology and oncology are now caring for them.
This article describes new biologic therapies for chronic myelogenous leukemia (CML) and chronic lymphocytic leukemia (CLL), along with the diagnosis of these diseases and management of survivors in the primary care setting.
CHRONIC MYELOGENOUS LEUKEMIA
A seemingly healthy person needs laboratory blood work, perhaps for an insurance physical examination or for a preoperative workup. Or a patient comes to the emergency department with a sore throat and routine blood tests are ordered. Their laboratory values:
- White blood cell count 250 × 109/L (reference range 3–11)
- Neutrophils 70% (40%–70%)
- Blasts 1% (0)
- Metacytes and myelocytes 5% (0)
- Bands 5% (0)
- Lymphocytes 10% (22%–40%)
- Monocytes 5% (0–7%)
- Basophils 3% (0–1%)
- Eosinophils 1% (0–4%)
- Hemoglobin 12.1 g/dL (11.5–15.5 in women, 13.0–17.0 in men)
- Platelet count 525 × 109/L (150–400).
Leukocytosis and a ‘left shift’
Although this scenario often raises concern for acute leukemia, a careful look shows evidence of a chronic myeloproliferative disorder instead. Specifically, this patient’s laboratory values show a “left shift”—an increase in immature neutrophils, ie, blasts, myelocytes, and bands.
This picture is characteristic of CML, an uncommon leukemia with about 4,500 new cases annually in the United States. Patients can present at any age, but the disease occurs more often in older people, with a median age of 66.1
The presentation is usually subtle: about half of cases are detected by routine laboratory testing, which typically reveals a left-shifted leukocytosis with basophilia and a few blasts. Mild anemia is common. The platelet count is elevated in 30% to 50% of patients at diagnosis. Bone marrow aspirate shows significant myeloid hyperplasia without dysplasia, and sometimes shows mild fibrosis.
Philadelphia chromosome is diagnostic
A definitive diagnosis is made by demonstration of an abnormally short chromosome 22. Described in 1960 by Peter Nowell of the University of Pennsylvania and David Hugerford of the Institute for Cancer Research,2 this abnormality, called the Philadelphia chromosome, was the first specific genetic abnormality associated with a human cancer. Later, researchers used banding techniques to find that the Philadelphia chromosome results from a reciprocal translocation of genetic material between the BCR gene on chromosome 22 and the ABL1 gene on chromosome 9, t(9:22).3,4 The resulting chimeric gene, called BCR-ABL, codes for an oncogenic protein, a tyrosine kinase with constitutive activity.
The Philadelphia chromosome is present in 95% of patients with CML and can be found in all myeloid cell lineages, including erythrocytes, granulocytes, monocytes, and megakaryocytes as well as some cells of lymphocytic lineage, indicating that malignant transformation to CML takes place at the stem cell level.
The mutation causes several problems: the abnormal tyrosine kinase increases cell proliferation, inhibits apoptosis, and alters adhesion molecules in the stroma of the bone marrow, allowing immature cells to leak into the bloodstream. Most important, the mutation increases genomic instability so that additional mutations are likelier to occur over time, making it inevitable that, without treatment, the disease will progress to a fatal blast crisis within an average of 5 years of diagnosis.
CML has three clinical phases
Untreated, CML progresses through three distinct phases: chronic, accelerated, and blast crisis, defined by abnormalities in the blood smear and bone marrow (Table 1).5,6 Most patients (85%) are diagnosed during the chronic phase. The accelerated and blastic phases resemble acute leukemia.
Chronic phase management
Therapies over the years have included arsenic (Fowler solution), splenic radiotherapy, busulfan, hydroxyurea, cytarabine, and interferon. All had some palliative success, but usually did not suppress leukemic progression.7
In contrast, patients undergoing allogeneic bone marrow transplant had a 5-year survival rate of 60% to 80% during the chronic phase of CML, 40% to 60% during the accelerated phase, and 10% to 20% during a blast crisis.8 Long-term survival confirmed the ability of transplant to cure CML, and bone marrow transplant with matched donors was the standard of care for younger patients until the end of the 20th century.
Tyrosine kinase inhibition
A new paradigm in treatment began with the development of imatinib, a tyrosine kinase inhibitor that directly interferes with the product of the chimeric BCR-ABL gene.9
Patients treated with imatinib during the chronic phase of CML have survival rates similar to those of people without the disease, and they usually do not progress to the accelerated and blast phases. As a result of this success, the number of transplants for CML has fallen precipitously.
Other tyrosine kinase inhibitors (dasatinib, nilotinib) that have since been developed have shown even better results in achieving remission and preventing progression. Improved survival is more difficult to demonstrate because the control groups in studies receive imatinib and have 10-year survival rates of about 90%.10–12
With the tyrosine kinase inhibitors, CML can be regarded as functionally cured.13 Patients take these drugs for life and usually experience a relapse if they stop. Patients with CML are now more likely to die of a comorbidity than of CML.
Choose therapy by tolerability
Which tyrosine kinase inhibitor to use depends more on the side-effect profile of the drug than on its efficacy. Nilotinib should be avoided in patients with vascular disease, and dasatinib avoided in patients with pulmonary disease. Each drug may be associated with some degree of nausea, diarrhea, cramps, rash, and edema.10–12
CML is not an immunosuppressive disease, nor are the drugs used to treat it. Patients with CML have an intact immune system. Therefore, precautions taken for patients with acute leukemia or lymphoid malignancy are not required for patients with CML.
Managing survivors
Since imatinib was introduced in 2000, the US Food and Drug Administration (FDA) has approved approximately 20 tyrosine kinase inhibitors for various cancers. These drugs are improving survival rates so well that patients with cancer are increasingly being seen by their primary care doctors for their medical problems.
Some problems have emerged that are consequences of this successful therapy. Angiogenesis inhibitors such as bevacizumab affect vascular endothelial growth factors, which injure endothelial cells. These effects may result in high blood pressure and arterial occlusive disease. Algorithms have been proposed for managing cardiovascular complications for patients taking tyrosine kinase inhibitors.14 Further, cardiovascular risk factors such as hyperlipidemia, diabetes, and obesity must be aggressively managed in patients taking tyrosine kinase inhibitors.
Vascular effects, rashes, and drug interactions may best be managed by primary care physicians, cardiologists, and nephrologists, who deal with such problems regularly.
CHRONIC LYMPHOCYTIC LEUKEMIA
A patient undergoes routine laboratory blood work in the emergency department or clinic, with these results:
- White blood cell count 250 × 109/L
- Neutrophils 1%
- Lymphocytes 99%
- Hemoglobin 12.1 g/dL
- Platelet count 160 × 109/L.
Like patients with CML, those with CLL usually present with no symptoms. The complete blood cell count reveals numerous white blood cells and lymphocytosis. Patients may have painless lymphadenopathy, anemia, and thrombocytopenia, but they do not typically have fever, sweats, or weight loss.
The disease is characterized by clonal proliferation and accumulation of mature-appearing neoplastic B lymphocytes in the blood, bone marrow, lymph nodes, and spleen. The peripheral blood smear shows “smudge cells,” indicating fragile lymphocytes.
The median age at diagnosis is about 70, with fewer than 15% of newly diagnosed patients under age 50.
CLL is the most common leukemia in the Western world, accounting for about 30% of cases of leukemia in adults. It is rare in Asians, probably because of genetic differences.
Monoclonal B-cell lymphocytosis precedes CLL
Monoclonal B-cell lymphocytosis is related to CLL and always precedes it. It is a common condition, detectable in up to 5% of older adults. The differential count shows a less severe lymphocytosis than in CLL.
Because monoclonal B-cell lymphocytosis does not always convert to leukemia, it is important for insurance coverage purposes not to diagnose it as a leukemia. Treatment-free survival of patients diagnosed with monoclonal B-cell lymphocytosis is 87% at 5 years.15,16
Diagnosing CLL
Lymphocytosis can indicate other low-grade lymphoproliferative diseases and malignancies, so further evaluation is critical. To diagnose CLL, the B-cell count by flow cytometry (not the absolute lymphocyte count from the complete blood cell count) must be at least 5 × 109/L. Below that threshold, monoclonal B-cell lymphocytosis is diagnosed unless lymphadenopathy is present, indicating small lymphocytic lymphoma. Unlike in benign lymphoproliferations, CLL lymphocytes coexpress the B-cell marker CD19 and the T-cell marker CD5.17 Bone marrow examination is rarely needed for the diagnosis of CLL.
Two types of CLL can be defined, depending on whether the B cells carry V genes that are mutated or unmutated. B cells expressing ZAP-70 and CD38 tend to carry the unmutated gene, which is associated with a worse prognosis.18 Regardless of which type a patient has, treatments and the indications for treatment are the same.
Increasing immune dysfunction

CLL is staged according to effects on lymph tissue and hematopoiesis. The Rai system for clinical staging of CLL has been used since 1975 with little alteration (Table 2).19
CLL is often an indolent lymphoproliferative malignancy and does not always progress to a fatal end stage. Therefore, treatment may be deferred, with a watch-and-wait approach until symptoms develop or the disease progresses. Approximately half of patients never require treatment.20 Progression involves increasing bone marrow impairment with greater susceptibility to infection (due to intrinsic features of CLL and its therapy) and hypogammaglobulinemia in advanced disease.21,22 Systemic infection is the cause of death for most patients.
Because CLL is a disease of the immune system, the development of autoantibodies is a cardinal feature. Autoimmune complications are almost exclusively limited to blood and can include hemolytic anemia, pure red cell aplasia, immune-mediated thrombocytopenia, and granulocytopenia. Other autoimmune diseases, such as rheumatoid arthritis, thyroiditis, and Addison disease, are uncommon.23,24
Other complications may occur in patients who have been treated with chemotherapy, and these are usually fatal. The Richter transformation (to an aggressive lymphoma) occurs in about 15%. Other less common complications include prolymphocytoid transformation and secondary malignancies, particularly carcinomas of the lung and gastrointestinal tract and acute (myeloid) leukemia.25
Survival rates in CLL have improved substantially over the past decades,26–28 with significant gains following the introduction of antibiotics and, to a lesser extent, transfusions. Median survival is generally between 6 and 9 years, but many patients live for years without requiring therapy.
Chemotherapy: The mainstay of treatment
When to begin therapy remains one of the most challenging issues of patient management. Unlike in CML, there is no advantage to starting at diagnosis when most patients are asymptomatic.29

In 1996, the National Cancer Institute issued guidelines for starting treatment, which were updated in 2008 with very little change (Table 3).30 In general, the onset of symptoms and evidence of impaired marrow function, including an abnormal hemoglobin level and platelet count, are indications. The white blood cell count continuously increases during the disease course but is not usually an important factor for initiating treatment.
The therapeutic goal for most patients who require treatment has historically been palliation of symptoms. Therapy must be individualized to a patient’s age and clinical status, with a heavier reliance on chemotherapeutic agents for patients who can tolerate it and on immunotherapy for others. General strategies are as follows:
- “Go-Go” patients—young, fit, with few comorbidities, good renal function—are the minority. Recommendation: combination chemotherapy with fludarabine, cyclophosphamide, and rituximab (FCR).
- “Slo-Go” patients are reasonably fit and can tolerate chemotherapy but not FCR. Recommendation: combination therapy with either bendamustine and rituximab or chlorambucil and rituximab (for less fit patients). Recent evidence indicates ibrutinib may be useful for such patients.31
- “No-Go” patients are frail with short life expectancy. Recommendation: rituximab or observation (see below)
All CLL treatments are potentially toxic. Chemotherapy damages DNA and often causes blood cell counts to fall. Immunosuppression worsens with almost any treatment, involving a substantial risk of secondary malignancy. Although survival improves with therapy, relapse is universal.
Targeting CLL pathways
The new paradigm for cancer therapy is to identify a cellular pathway that drives oncogenesis or proliferation and interfere with it. The B-cell receptor pathway is enormously complex with numerous complex factors, making it difficult to discern the critical mutation that drives the proliferation of lymphocytes.
Bruton tyrosine kinase (Btk) is one factor that is critical for CLL proliferation. Patients with congenitally mutated or dysfunctional Btk have lymphopenia and agammaglobulinemia, making it a promising target for patients with B-cell disorders. Other experimental therapies are based on other such identified factors.
In 2014, the FDA approved two drugs for CLL—ibrutinib, a Btk inhibitor, and idelalisib, an inhibitor of phosphoinositide 3-kinase—after they were shown in clinical trials to dramatically improve outcomes in patients with relapsed CLL.32,33 Trials with these drugs are ongoing. These drugs also inhibit tyrosine kinase and so have vascular side effects in addition to their own idiosyncratic effects.
Ibrutinib has anticoagulant effects and should be stopped before surgery. It also can cause or exacerbate atrial fibrillation, making management of CLL difficult. It is associated with hypogammaglobulinemia, often requiring ongoing immunoglobulin replacement.
Idelalisib tends to cause systemic autoimmune phenomena such as pneumonitis and colitis.
Using T cells as therapy
It has long been observed that patients who undergo bone marrow transplant for leukemia have lower relapse rates if the transplant is allogeneic rather than from a twin. Further, if T cells are removed from the donor graft, graft-vs-host disease may be prevented but the risk of relapses increases. Finally, the presence of graft-vs-host disease tends to reduce the risk of relapse.34 Therefore, T cells clearly are key ingredients for success in the setting of bone marrow transplant. In fact, merely providing T cells for a relapse after allogeneic transplant can induce remission. However, because donor T cells are not targeted, acute and chronic graft-vs-host disease often can ensue.
‘Designer’ monoclonal antibodies
The B lymphocyte has multiple potential targets for new therapies for CLL as well as other cancers involving B cells. CD20 was identified on the surface of B cells in 1988 and is the target protein of the monoclonal antibody drug rituximab. Monoclonal antibodies can be modified to target other surface antigens, to link radioisotopes to deliver radiation therapy, and to deliver drugs that would otherwise be too toxic to be given systemically.35 Monoclonal antibodies can also be modified to enhance function.
Antibodies alone, however, must often rely on the host T cells for cytotoxicity and they are often compromised by either the underlying disease or treatment. Adapting the targeting function of antibodies to enhance or genetically alter T cells to recognize cancer-specific antigens is now being explored for leukemias.36
In 2014, the FDA approved blinatumomab for the treatment of relapsed or refractory acute lymphoblastic leukemia. This biopharmaceutical agent recruits T cells with one antibody-like moiety and targets the CD19 receptor of B cells with another. Given as a single intravenous treatment without chemotherapy, it has an almost 50% response rate, and those who respond tend to stay in remission. Other similar drugs are being developed, and using them earlier in treatment and for other B-cell leukemias is being explored.
New B-cell targeted therapy with CAR-Ts
Newer treatments are being developed based on chimeric antigen receptor T (CAR-T) cells. These engineered T cells express an anti-CD19 moiety that targets B cells, but also activate upon binding to them.37 CAR-T technology is being refined and shows great promise for cancer treatment.
Multiple clinical trials are currently under way in which the investigators collect autologous T cells by leukopheresis from a patient with a relapsed or refractory B-cell malignancy, transduce the T cells with retroviral vectors into anti-CD19 CAR-T cells, and then reinfuse them into the patient following modest chemotherapy.38
Study results from a small number of patients with relapsing or refractory CLL showed that some patients achieved long-term, progression-free survival.39 The most success with this therapy, however, has been in acute lymphoblastic leukemia.40 Possibly, this treatment could be applied to other lymphoid malignancies that also express CD19.
More advances
CAR-T cell therapy has drawbacks. The cells attack only the target antigen, which currently limits their use mostly to hematologic malignancies. In addition, autologous T cells are not robust. Also, the use of allogeneic T cells is restricted by their major histocompatibility complex, and the cells will be rejected by the recipient if not matched.
An attempt to overcome some of these drawbacks is to develop T cells redirected for universal cytokine killing. CAR-T cells are modified with a gene that causes them to excrete interleukin 12, which attracts macrophages and natural killer cells to the environment to better fight the tumor.41
Other modifications include editing out certain genes including the major histocompatibility complex, which avoids the problem of rejection. Another modification is to insert a “suicide gene” that allows the engineered T cells to be killed with an antidote if they do not work as planned.
Such gene-editing techniques hold great promise for curing cancers without chemotherapy in the not so distant future.
- National Cancer Institute Surveillance, Epidemiology, and End Results Program. SEER Stat Fact Sheets: Chronic Myeloid Leukemia. http://seer.cancer.gov/statfacts/html/cmyl.html. Accessed July 1, 2016.
- Nowell PC, Hungerford DA. A minute chromosome in human chronic granulocytic leukemia. Science 1960; 132:1497.
- Melo JV. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood 1996; 88:2375–2384.
- Pasternak G, Hochhaus A, Schultheis B, Hehlmann R. Chronic myelogenous leukemia: molecular and cellular aspects. J Cancer Res Clin Oncol 1998; 124:643–660.
- Faderl S, Kantarjian HM, Talpaz M. Chronic myelogenous leukemia: update on biology and treatment. Oncology (Williston Park) 1999; 13:169–184.
- Sawyers CL. Chronic myeloid leukemia. N Engl J Med 1999; 340:1330–1340.
- Hehlmann R, Heimpel H, Hasford J, et al. Randomized comparison of interferon-alpha with busulfan and hydroxyurea in chronic myelogenous leukemia. The German CML Study Group. Blood 1994; 84:4064–4077.
- Radich JP, Olavarria E, Apperley JF. Allogeneic hematopoietic stem cell transplantation for chronic myeloid leukemia. Hematol Oncol Clin North Am 2004; 18:685–702.
- Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood 2008; 112:4808–4817.
- O’Brien SG, Guilhot F, Larson RA, et al; IRIS Investigators. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2003; 348:994-1004.
- Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2010; 362:2260–2270.
- Saglio G, Kim DW, Issaragrisil S, et al; ENESTnd Investigators. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med 2010; 362:2251–2259.
- Pfirrmann M, Baccarani M, Saussele S, et al. Prognosis of long-term survival considering disease-specific death in patients with chronic myeloid leukemia. Leukemia 2016; 30:48-56.
- Li W, Croce K, Steensma DP, McDermott DF, Ben-Yehuda O, Moslehi J. Vascular and metabolic implications of novel targeted cancer therapies: focus on kinase inhibitors. J Am Coll Cardiol 2015; 66:1160–1178.
- Rawstron AC, Bennett F, Hillmen P. The biological and clinical relationship between CD5+23+ monoclonal B-cell lymphocytosis and chronic lymphocytic leukaemia. Br J Haematol 2007; 139:724–729.
- Rawstron AC, Bennett FL, O’Connor SJ, et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med 2008; 359:575–583.
- Hallek M, Cheson BD, Catovsky D, et al; International Workshop on Chronic Lymphocytic Leukemia. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008; 111:5446–5456.
- Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med 2005; 352:804–815.
- Rai KR, Sawitsky A, Cronkite EP, Chanana AD, Levy RN, Pasternack BS. Clinical staging of chronic lymphocytic leukemia. Blood 1975; 46:219–234.
- Dierlamm J, Michaux L, Criel A, Wlodarska I, Van den Berghe H, Hossfeld DK. Genetic abnormalities in chronic lymphocytic leukemia and their clinical and prognostic implications. Cancer Genet Cytogenet 1997; 94:27–35.
- Rozman C, Montserrat E. Chronic lymphocytic leukemia. N Engl J Med 1995; 333:1052–1057. Erratum in: N Engl J Med 1995; 333:1515.
- Jemal A, Thomas A, Murray T, Thun M. Cancer statistics, 2002. CA Cancer J Clin 2002; 52:23-47. Errata in: CA Cancer J Clin 2002; 52:119. CA Cancer J Clin 2002; 52:181–182.
- Caligaris-Cappio F, Hamblin TJ. B-cell chronic lymphocytic leukemia: a bird of a different feather. J Clin Oncol 1999; 17:399–408.
- Keating MJ. Chronic lymphocytic leukemia. Semin Oncol 1999; 26(suppl 14):107–114.
- Kalil N, Cheson BD. Management of chronic lymphocytic leukaemia. Drugs Aging 2000; 16:9–27.
- Minot GR, Buckman TE, Isaacs R. Chronic myelogenous leukemia: age incidence, duration, and benefit derived from irradiation. JAMA 1924; 82:1489–1494.
- Reinhard EH, Neely CL, Samples DM. Radioactive phosphorus in the treatment of chronic leukemias: long-term results over a period of 15 years. Cancer 1959; 50:942–958.
- Diehl LF, Karnell LH, Menck HR. The American College of Surgeons Commission on Cancer and the American Cancer Society. The National Cancer Data Base report on age, gender, treatment, and outcomes of patients with chronic lymphocytic leukemia. Cancer 1999; 86:2684–2692.
- Chemotherapeutic options in chronic lymphocytic leukemia: a meta-analysis of the randomized trials. CLL Trialists’ Collaborative Group. J Natl Cancer Inst 1999; 91:861–868.
- Cheson BD, Bennett JM, Grever M, et al. National Cancer Institute-sponsored working group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood 1996; 87:4990–4997.
- Burger JA, Tedeschi A, Barr PM, et al; RESONATE-2 Investigators. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med 2015; 373:2425–2437.
- Byrd JC, Brown JR, O’Brien S, et al; RESONATE Investigators. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med 2014; 371:213–223.
- Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med 2014; 370:997–1007.
- Horowitz MM, Gale RP, Sondel PM, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood 1990; 75:555–562.
- Weiner GJ. Building better monoclonal antibody-based therapeutics. Nat Rev Cancer 2015; 15:361–370.
- Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer 2013; 13:525–541.
- Urba WJ, Longo DL. Redirecting T cells. N Engl J Med 2011; 365:754–757.
- Klebanoff CA, Yamamoto TN, Restifo NP. Immunotherapy: treatment of aggressive lymphomas with anti-CD19 CAR T cells. Nat Rev Clin Oncol 2014; 11:685-686.
- Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 2015; 7:303ra139.
- Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015; 385:517–528.
- Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev 2014; 257:83–90.
The advent of targeted therapies has dramatically changed the management of chronic leukemia. Chemotherapy—highly toxic, nonspecific drugs that can be dangerous to patients and providers and result in only modest success—is gradually being replaced by biologic targeting of malignancy. Scientists are rapidly identifying extracellular and intracellular targets on tumor cells and are developing and testing promising new therapies aimed at these targets. Survival of cancer patients has become so common that clinicians outside the specialties of hematology and oncology are now caring for them.
This article describes new biologic therapies for chronic myelogenous leukemia (CML) and chronic lymphocytic leukemia (CLL), along with the diagnosis of these diseases and management of survivors in the primary care setting.
CHRONIC MYELOGENOUS LEUKEMIA
A seemingly healthy person needs laboratory blood work, perhaps for an insurance physical examination or for a preoperative workup. Or a patient comes to the emergency department with a sore throat and routine blood tests are ordered. Their laboratory values:
- White blood cell count 250 × 109/L (reference range 3–11)
- Neutrophils 70% (40%–70%)
- Blasts 1% (0)
- Metacytes and myelocytes 5% (0)
- Bands 5% (0)
- Lymphocytes 10% (22%–40%)
- Monocytes 5% (0–7%)
- Basophils 3% (0–1%)
- Eosinophils 1% (0–4%)
- Hemoglobin 12.1 g/dL (11.5–15.5 in women, 13.0–17.0 in men)
- Platelet count 525 × 109/L (150–400).
Leukocytosis and a ‘left shift’
Although this scenario often raises concern for acute leukemia, a careful look shows evidence of a chronic myeloproliferative disorder instead. Specifically, this patient’s laboratory values show a “left shift”—an increase in immature neutrophils, ie, blasts, myelocytes, and bands.
This picture is characteristic of CML, an uncommon leukemia with about 4,500 new cases annually in the United States. Patients can present at any age, but the disease occurs more often in older people, with a median age of 66.1
The presentation is usually subtle: about half of cases are detected by routine laboratory testing, which typically reveals a left-shifted leukocytosis with basophilia and a few blasts. Mild anemia is common. The platelet count is elevated in 30% to 50% of patients at diagnosis. Bone marrow aspirate shows significant myeloid hyperplasia without dysplasia, and sometimes shows mild fibrosis.
Philadelphia chromosome is diagnostic
A definitive diagnosis is made by demonstration of an abnormally short chromosome 22. Described in 1960 by Peter Nowell of the University of Pennsylvania and David Hugerford of the Institute for Cancer Research,2 this abnormality, called the Philadelphia chromosome, was the first specific genetic abnormality associated with a human cancer. Later, researchers used banding techniques to find that the Philadelphia chromosome results from a reciprocal translocation of genetic material between the BCR gene on chromosome 22 and the ABL1 gene on chromosome 9, t(9:22).3,4 The resulting chimeric gene, called BCR-ABL, codes for an oncogenic protein, a tyrosine kinase with constitutive activity.
The Philadelphia chromosome is present in 95% of patients with CML and can be found in all myeloid cell lineages, including erythrocytes, granulocytes, monocytes, and megakaryocytes as well as some cells of lymphocytic lineage, indicating that malignant transformation to CML takes place at the stem cell level.
The mutation causes several problems: the abnormal tyrosine kinase increases cell proliferation, inhibits apoptosis, and alters adhesion molecules in the stroma of the bone marrow, allowing immature cells to leak into the bloodstream. Most important, the mutation increases genomic instability so that additional mutations are likelier to occur over time, making it inevitable that, without treatment, the disease will progress to a fatal blast crisis within an average of 5 years of diagnosis.
CML has three clinical phases
Untreated, CML progresses through three distinct phases: chronic, accelerated, and blast crisis, defined by abnormalities in the blood smear and bone marrow (Table 1).5,6 Most patients (85%) are diagnosed during the chronic phase. The accelerated and blastic phases resemble acute leukemia.
Chronic phase management
Therapies over the years have included arsenic (Fowler solution), splenic radiotherapy, busulfan, hydroxyurea, cytarabine, and interferon. All had some palliative success, but usually did not suppress leukemic progression.7
In contrast, patients undergoing allogeneic bone marrow transplant had a 5-year survival rate of 60% to 80% during the chronic phase of CML, 40% to 60% during the accelerated phase, and 10% to 20% during a blast crisis.8 Long-term survival confirmed the ability of transplant to cure CML, and bone marrow transplant with matched donors was the standard of care for younger patients until the end of the 20th century.
Tyrosine kinase inhibition
A new paradigm in treatment began with the development of imatinib, a tyrosine kinase inhibitor that directly interferes with the product of the chimeric BCR-ABL gene.9
Patients treated with imatinib during the chronic phase of CML have survival rates similar to those of people without the disease, and they usually do not progress to the accelerated and blast phases. As a result of this success, the number of transplants for CML has fallen precipitously.
Other tyrosine kinase inhibitors (dasatinib, nilotinib) that have since been developed have shown even better results in achieving remission and preventing progression. Improved survival is more difficult to demonstrate because the control groups in studies receive imatinib and have 10-year survival rates of about 90%.10–12
With the tyrosine kinase inhibitors, CML can be regarded as functionally cured.13 Patients take these drugs for life and usually experience a relapse if they stop. Patients with CML are now more likely to die of a comorbidity than of CML.
Choose therapy by tolerability
Which tyrosine kinase inhibitor to use depends more on the side-effect profile of the drug than on its efficacy. Nilotinib should be avoided in patients with vascular disease, and dasatinib avoided in patients with pulmonary disease. Each drug may be associated with some degree of nausea, diarrhea, cramps, rash, and edema.10–12
CML is not an immunosuppressive disease, nor are the drugs used to treat it. Patients with CML have an intact immune system. Therefore, precautions taken for patients with acute leukemia or lymphoid malignancy are not required for patients with CML.
Managing survivors
Since imatinib was introduced in 2000, the US Food and Drug Administration (FDA) has approved approximately 20 tyrosine kinase inhibitors for various cancers. These drugs are improving survival rates so well that patients with cancer are increasingly being seen by their primary care doctors for their medical problems.
Some problems have emerged that are consequences of this successful therapy. Angiogenesis inhibitors such as bevacizumab affect vascular endothelial growth factors, which injure endothelial cells. These effects may result in high blood pressure and arterial occlusive disease. Algorithms have been proposed for managing cardiovascular complications for patients taking tyrosine kinase inhibitors.14 Further, cardiovascular risk factors such as hyperlipidemia, diabetes, and obesity must be aggressively managed in patients taking tyrosine kinase inhibitors.
Vascular effects, rashes, and drug interactions may best be managed by primary care physicians, cardiologists, and nephrologists, who deal with such problems regularly.
CHRONIC LYMPHOCYTIC LEUKEMIA
A patient undergoes routine laboratory blood work in the emergency department or clinic, with these results:
- White blood cell count 250 × 109/L
- Neutrophils 1%
- Lymphocytes 99%
- Hemoglobin 12.1 g/dL
- Platelet count 160 × 109/L.
Like patients with CML, those with CLL usually present with no symptoms. The complete blood cell count reveals numerous white blood cells and lymphocytosis. Patients may have painless lymphadenopathy, anemia, and thrombocytopenia, but they do not typically have fever, sweats, or weight loss.
The disease is characterized by clonal proliferation and accumulation of mature-appearing neoplastic B lymphocytes in the blood, bone marrow, lymph nodes, and spleen. The peripheral blood smear shows “smudge cells,” indicating fragile lymphocytes.
The median age at diagnosis is about 70, with fewer than 15% of newly diagnosed patients under age 50.
CLL is the most common leukemia in the Western world, accounting for about 30% of cases of leukemia in adults. It is rare in Asians, probably because of genetic differences.
Monoclonal B-cell lymphocytosis precedes CLL
Monoclonal B-cell lymphocytosis is related to CLL and always precedes it. It is a common condition, detectable in up to 5% of older adults. The differential count shows a less severe lymphocytosis than in CLL.
Because monoclonal B-cell lymphocytosis does not always convert to leukemia, it is important for insurance coverage purposes not to diagnose it as a leukemia. Treatment-free survival of patients diagnosed with monoclonal B-cell lymphocytosis is 87% at 5 years.15,16
Diagnosing CLL
Lymphocytosis can indicate other low-grade lymphoproliferative diseases and malignancies, so further evaluation is critical. To diagnose CLL, the B-cell count by flow cytometry (not the absolute lymphocyte count from the complete blood cell count) must be at least 5 × 109/L. Below that threshold, monoclonal B-cell lymphocytosis is diagnosed unless lymphadenopathy is present, indicating small lymphocytic lymphoma. Unlike in benign lymphoproliferations, CLL lymphocytes coexpress the B-cell marker CD19 and the T-cell marker CD5.17 Bone marrow examination is rarely needed for the diagnosis of CLL.
Two types of CLL can be defined, depending on whether the B cells carry V genes that are mutated or unmutated. B cells expressing ZAP-70 and CD38 tend to carry the unmutated gene, which is associated with a worse prognosis.18 Regardless of which type a patient has, treatments and the indications for treatment are the same.
Increasing immune dysfunction
CLL is staged according to effects on lymph tissue and hematopoiesis. The Rai system for clinical staging of CLL has been used since 1975 with little alteration (Table 2).19
CLL is often an indolent lymphoproliferative malignancy and does not always progress to a fatal end stage. Therefore, treatment may be deferred, with a watch-and-wait approach until symptoms develop or the disease progresses. Approximately half of patients never require treatment.20 Progression involves increasing bone marrow impairment with greater susceptibility to infection (due to intrinsic features of CLL and its therapy) and hypogammaglobulinemia in advanced disease.21,22 Systemic infection is the cause of death for most patients.
Because CLL is a disease of the immune system, the development of autoantibodies is a cardinal feature. Autoimmune complications are almost exclusively limited to blood and can include hemolytic anemia, pure red cell aplasia, immune-mediated thrombocytopenia, and granulocytopenia. Other autoimmune diseases, such as rheumatoid arthritis, thyroiditis, and Addison disease, are uncommon.23,24
Other complications may occur in patients who have been treated with chemotherapy, and these are usually fatal. The Richter transformation (to an aggressive lymphoma) occurs in about 15%. Other less common complications include prolymphocytoid transformation and secondary malignancies, particularly carcinomas of the lung and gastrointestinal tract and acute (myeloid) leukemia.25
Survival rates in CLL have improved substantially over the past decades,26–28 with significant gains following the introduction of antibiotics and, to a lesser extent, transfusions. Median survival is generally between 6 and 9 years, but many patients live for years without requiring therapy.
Chemotherapy: The mainstay of treatment
When to begin therapy remains one of the most challenging issues of patient management. Unlike in CML, there is no advantage to starting at diagnosis when most patients are asymptomatic.29
In 1996, the National Cancer Institute issued guidelines for starting treatment, which were updated in 2008 with very little change (Table 3).30 In general, the onset of symptoms and evidence of impaired marrow function, including an abnormal hemoglobin level and platelet count, are indications. The white blood cell count continuously increases during the disease course but is not usually an important factor for initiating treatment.
The therapeutic goal for most patients who require treatment has historically been palliation of symptoms. Therapy must be individualized to a patient’s age and clinical status, with a heavier reliance on chemotherapeutic agents for patients who can tolerate it and on immunotherapy for others. General strategies are as follows:
- “Go-Go” patients—young, fit, with few comorbidities, good renal function—are the minority. Recommendation: combination chemotherapy with fludarabine, cyclophosphamide, and rituximab (FCR).
- “Slo-Go” patients are reasonably fit and can tolerate chemotherapy but not FCR. Recommendation: combination therapy with either bendamustine and rituximab or chlorambucil and rituximab (for less fit patients). Recent evidence indicates ibrutinib may be useful for such patients.31
- “No-Go” patients are frail with short life expectancy. Recommendation: rituximab or observation (see below)
All CLL treatments are potentially toxic. Chemotherapy damages DNA and often causes blood cell counts to fall. Immunosuppression worsens with almost any treatment, involving a substantial risk of secondary malignancy. Although survival improves with therapy, relapse is universal.
Targeting CLL pathways
The new paradigm for cancer therapy is to identify a cellular pathway that drives oncogenesis or proliferation and interfere with it. The B-cell receptor pathway is enormously complex with numerous complex factors, making it difficult to discern the critical mutation that drives the proliferation of lymphocytes.
Bruton tyrosine kinase (Btk) is one factor that is critical for CLL proliferation. Patients with congenitally mutated or dysfunctional Btk have lymphopenia and agammaglobulinemia, making it a promising target for patients with B-cell disorders. Other experimental therapies are based on other such identified factors.
In 2014, the FDA approved two drugs for CLL—ibrutinib, a Btk inhibitor, and idelalisib, an inhibitor of phosphoinositide 3-kinase—after they were shown in clinical trials to dramatically improve outcomes in patients with relapsed CLL.32,33 Trials with these drugs are ongoing. These drugs also inhibit tyrosine kinase and so have vascular side effects in addition to their own idiosyncratic effects.
Ibrutinib has anticoagulant effects and should be stopped before surgery. It also can cause or exacerbate atrial fibrillation, making management of CLL difficult. It is associated with hypogammaglobulinemia, often requiring ongoing immunoglobulin replacement.
Idelalisib tends to cause systemic autoimmune phenomena such as pneumonitis and colitis.
Using T cells as therapy
It has long been observed that patients who undergo bone marrow transplant for leukemia have lower relapse rates if the transplant is allogeneic rather than from a twin. Further, if T cells are removed from the donor graft, graft-vs-host disease may be prevented but the risk of relapses increases. Finally, the presence of graft-vs-host disease tends to reduce the risk of relapse.34 Therefore, T cells clearly are key ingredients for success in the setting of bone marrow transplant. In fact, merely providing T cells for a relapse after allogeneic transplant can induce remission. However, because donor T cells are not targeted, acute and chronic graft-vs-host disease often can ensue.
‘Designer’ monoclonal antibodies
The B lymphocyte has multiple potential targets for new therapies for CLL as well as other cancers involving B cells. CD20 was identified on the surface of B cells in 1988 and is the target protein of the monoclonal antibody drug rituximab. Monoclonal antibodies can be modified to target other surface antigens, to link radioisotopes to deliver radiation therapy, and to deliver drugs that would otherwise be too toxic to be given systemically.35 Monoclonal antibodies can also be modified to enhance function.
Antibodies alone, however, must often rely on the host T cells for cytotoxicity and they are often compromised by either the underlying disease or treatment. Adapting the targeting function of antibodies to enhance or genetically alter T cells to recognize cancer-specific antigens is now being explored for leukemias.36
In 2014, the FDA approved blinatumomab for the treatment of relapsed or refractory acute lymphoblastic leukemia. This biopharmaceutical agent recruits T cells with one antibody-like moiety and targets the CD19 receptor of B cells with another. Given as a single intravenous treatment without chemotherapy, it has an almost 50% response rate, and those who respond tend to stay in remission. Other similar drugs are being developed, and using them earlier in treatment and for other B-cell leukemias is being explored.
New B-cell targeted therapy with CAR-Ts
Newer treatments are being developed based on chimeric antigen receptor T (CAR-T) cells. These engineered T cells express an anti-CD19 moiety that targets B cells, but also activate upon binding to them.37 CAR-T technology is being refined and shows great promise for cancer treatment.
Multiple clinical trials are currently under way in which the investigators collect autologous T cells by leukopheresis from a patient with a relapsed or refractory B-cell malignancy, transduce the T cells with retroviral vectors into anti-CD19 CAR-T cells, and then reinfuse them into the patient following modest chemotherapy.38
Study results from a small number of patients with relapsing or refractory CLL showed that some patients achieved long-term, progression-free survival.39 The most success with this therapy, however, has been in acute lymphoblastic leukemia.40 Possibly, this treatment could be applied to other lymphoid malignancies that also express CD19.
More advances
CAR-T cell therapy has drawbacks. The cells attack only the target antigen, which currently limits their use mostly to hematologic malignancies. In addition, autologous T cells are not robust. Also, the use of allogeneic T cells is restricted by their major histocompatibility complex, and the cells will be rejected by the recipient if not matched.
An attempt to overcome some of these drawbacks is to develop T cells redirected for universal cytokine killing. CAR-T cells are modified with a gene that causes them to excrete interleukin 12, which attracts macrophages and natural killer cells to the environment to better fight the tumor.41
Other modifications include editing out certain genes including the major histocompatibility complex, which avoids the problem of rejection. Another modification is to insert a “suicide gene” that allows the engineered T cells to be killed with an antidote if they do not work as planned.
Such gene-editing techniques hold great promise for curing cancers without chemotherapy in the not so distant future.
The advent of targeted therapies has dramatically changed the management of chronic leukemia. Chemotherapy—highly toxic, nonspecific drugs that can be dangerous to patients and providers and result in only modest success—is gradually being replaced by biologic targeting of malignancy. Scientists are rapidly identifying extracellular and intracellular targets on tumor cells and are developing and testing promising new therapies aimed at these targets. Survival of cancer patients has become so common that clinicians outside the specialties of hematology and oncology are now caring for them.
This article describes new biologic therapies for chronic myelogenous leukemia (CML) and chronic lymphocytic leukemia (CLL), along with the diagnosis of these diseases and management of survivors in the primary care setting.
CHRONIC MYELOGENOUS LEUKEMIA
A seemingly healthy person needs laboratory blood work, perhaps for an insurance physical examination or for a preoperative workup. Or a patient comes to the emergency department with a sore throat and routine blood tests are ordered. Their laboratory values:
- White blood cell count 250 × 109/L (reference range 3–11)
- Neutrophils 70% (40%–70%)
- Blasts 1% (0)
- Metacytes and myelocytes 5% (0)
- Bands 5% (0)
- Lymphocytes 10% (22%–40%)
- Monocytes 5% (0–7%)
- Basophils 3% (0–1%)
- Eosinophils 1% (0–4%)
- Hemoglobin 12.1 g/dL (11.5–15.5 in women, 13.0–17.0 in men)
- Platelet count 525 × 109/L (150–400).
Leukocytosis and a ‘left shift’
Although this scenario often raises concern for acute leukemia, a careful look shows evidence of a chronic myeloproliferative disorder instead. Specifically, this patient’s laboratory values show a “left shift”—an increase in immature neutrophils, ie, blasts, myelocytes, and bands.
This picture is characteristic of CML, an uncommon leukemia with about 4,500 new cases annually in the United States. Patients can present at any age, but the disease occurs more often in older people, with a median age of 66.1
The presentation is usually subtle: about half of cases are detected by routine laboratory testing, which typically reveals a left-shifted leukocytosis with basophilia and a few blasts. Mild anemia is common. The platelet count is elevated in 30% to 50% of patients at diagnosis. Bone marrow aspirate shows significant myeloid hyperplasia without dysplasia, and sometimes shows mild fibrosis.
Philadelphia chromosome is diagnostic
A definitive diagnosis is made by demonstration of an abnormally short chromosome 22. Described in 1960 by Peter Nowell of the University of Pennsylvania and David Hugerford of the Institute for Cancer Research,2 this abnormality, called the Philadelphia chromosome, was the first specific genetic abnormality associated with a human cancer. Later, researchers used banding techniques to find that the Philadelphia chromosome results from a reciprocal translocation of genetic material between the BCR gene on chromosome 22 and the ABL1 gene on chromosome 9, t(9:22).3,4 The resulting chimeric gene, called BCR-ABL, codes for an oncogenic protein, a tyrosine kinase with constitutive activity.
The Philadelphia chromosome is present in 95% of patients with CML and can be found in all myeloid cell lineages, including erythrocytes, granulocytes, monocytes, and megakaryocytes as well as some cells of lymphocytic lineage, indicating that malignant transformation to CML takes place at the stem cell level.
The mutation causes several problems: the abnormal tyrosine kinase increases cell proliferation, inhibits apoptosis, and alters adhesion molecules in the stroma of the bone marrow, allowing immature cells to leak into the bloodstream. Most important, the mutation increases genomic instability so that additional mutations are likelier to occur over time, making it inevitable that, without treatment, the disease will progress to a fatal blast crisis within an average of 5 years of diagnosis.
CML has three clinical phases
Untreated, CML progresses through three distinct phases: chronic, accelerated, and blast crisis, defined by abnormalities in the blood smear and bone marrow (Table 1).5,6 Most patients (85%) are diagnosed during the chronic phase. The accelerated and blastic phases resemble acute leukemia.
Chronic phase management
Therapies over the years have included arsenic (Fowler solution), splenic radiotherapy, busulfan, hydroxyurea, cytarabine, and interferon. All had some palliative success, but usually did not suppress leukemic progression.7
In contrast, patients undergoing allogeneic bone marrow transplant had a 5-year survival rate of 60% to 80% during the chronic phase of CML, 40% to 60% during the accelerated phase, and 10% to 20% during a blast crisis.8 Long-term survival confirmed the ability of transplant to cure CML, and bone marrow transplant with matched donors was the standard of care for younger patients until the end of the 20th century.
Tyrosine kinase inhibition
A new paradigm in treatment began with the development of imatinib, a tyrosine kinase inhibitor that directly interferes with the product of the chimeric BCR-ABL gene.9
Patients treated with imatinib during the chronic phase of CML have survival rates similar to those of people without the disease, and they usually do not progress to the accelerated and blast phases. As a result of this success, the number of transplants for CML has fallen precipitously.
Other tyrosine kinase inhibitors (dasatinib, nilotinib) that have since been developed have shown even better results in achieving remission and preventing progression. Improved survival is more difficult to demonstrate because the control groups in studies receive imatinib and have 10-year survival rates of about 90%.10–12
With the tyrosine kinase inhibitors, CML can be regarded as functionally cured.13 Patients take these drugs for life and usually experience a relapse if they stop. Patients with CML are now more likely to die of a comorbidity than of CML.
Choose therapy by tolerability
Which tyrosine kinase inhibitor to use depends more on the side-effect profile of the drug than on its efficacy. Nilotinib should be avoided in patients with vascular disease, and dasatinib avoided in patients with pulmonary disease. Each drug may be associated with some degree of nausea, diarrhea, cramps, rash, and edema.10–12
CML is not an immunosuppressive disease, nor are the drugs used to treat it. Patients with CML have an intact immune system. Therefore, precautions taken for patients with acute leukemia or lymphoid malignancy are not required for patients with CML.
Managing survivors
Since imatinib was introduced in 2000, the US Food and Drug Administration (FDA) has approved approximately 20 tyrosine kinase inhibitors for various cancers. These drugs are improving survival rates so well that patients with cancer are increasingly being seen by their primary care doctors for their medical problems.
Some problems have emerged that are consequences of this successful therapy. Angiogenesis inhibitors such as bevacizumab affect vascular endothelial growth factors, which injure endothelial cells. These effects may result in high blood pressure and arterial occlusive disease. Algorithms have been proposed for managing cardiovascular complications for patients taking tyrosine kinase inhibitors.14 Further, cardiovascular risk factors such as hyperlipidemia, diabetes, and obesity must be aggressively managed in patients taking tyrosine kinase inhibitors.
Vascular effects, rashes, and drug interactions may best be managed by primary care physicians, cardiologists, and nephrologists, who deal with such problems regularly.
CHRONIC LYMPHOCYTIC LEUKEMIA
A patient undergoes routine laboratory blood work in the emergency department or clinic, with these results:
- White blood cell count 250 × 109/L
- Neutrophils 1%
- Lymphocytes 99%
- Hemoglobin 12.1 g/dL
- Platelet count 160 × 109/L.
Like patients with CML, those with CLL usually present with no symptoms. The complete blood cell count reveals numerous white blood cells and lymphocytosis. Patients may have painless lymphadenopathy, anemia, and thrombocytopenia, but they do not typically have fever, sweats, or weight loss.
The disease is characterized by clonal proliferation and accumulation of mature-appearing neoplastic B lymphocytes in the blood, bone marrow, lymph nodes, and spleen. The peripheral blood smear shows “smudge cells,” indicating fragile lymphocytes.
The median age at diagnosis is about 70, with fewer than 15% of newly diagnosed patients under age 50.
CLL is the most common leukemia in the Western world, accounting for about 30% of cases of leukemia in adults. It is rare in Asians, probably because of genetic differences.
Monoclonal B-cell lymphocytosis precedes CLL
Monoclonal B-cell lymphocytosis is related to CLL and always precedes it. It is a common condition, detectable in up to 5% of older adults. The differential count shows a less severe lymphocytosis than in CLL.
Because monoclonal B-cell lymphocytosis does not always convert to leukemia, it is important for insurance coverage purposes not to diagnose it as a leukemia. Treatment-free survival of patients diagnosed with monoclonal B-cell lymphocytosis is 87% at 5 years.15,16
Diagnosing CLL
Lymphocytosis can indicate other low-grade lymphoproliferative diseases and malignancies, so further evaluation is critical. To diagnose CLL, the B-cell count by flow cytometry (not the absolute lymphocyte count from the complete blood cell count) must be at least 5 × 109/L. Below that threshold, monoclonal B-cell lymphocytosis is diagnosed unless lymphadenopathy is present, indicating small lymphocytic lymphoma. Unlike in benign lymphoproliferations, CLL lymphocytes coexpress the B-cell marker CD19 and the T-cell marker CD5.17 Bone marrow examination is rarely needed for the diagnosis of CLL.
Two types of CLL can be defined, depending on whether the B cells carry V genes that are mutated or unmutated. B cells expressing ZAP-70 and CD38 tend to carry the unmutated gene, which is associated with a worse prognosis.18 Regardless of which type a patient has, treatments and the indications for treatment are the same.
Increasing immune dysfunction
CLL is staged according to effects on lymph tissue and hematopoiesis. The Rai system for clinical staging of CLL has been used since 1975 with little alteration (Table 2).19
CLL is often an indolent lymphoproliferative malignancy and does not always progress to a fatal end stage. Therefore, treatment may be deferred, with a watch-and-wait approach until symptoms develop or the disease progresses. Approximately half of patients never require treatment.20 Progression involves increasing bone marrow impairment with greater susceptibility to infection (due to intrinsic features of CLL and its therapy) and hypogammaglobulinemia in advanced disease.21,22 Systemic infection is the cause of death for most patients.
Because CLL is a disease of the immune system, the development of autoantibodies is a cardinal feature. Autoimmune complications are almost exclusively limited to blood and can include hemolytic anemia, pure red cell aplasia, immune-mediated thrombocytopenia, and granulocytopenia. Other autoimmune diseases, such as rheumatoid arthritis, thyroiditis, and Addison disease, are uncommon.23,24
Other complications may occur in patients who have been treated with chemotherapy, and these are usually fatal. The Richter transformation (to an aggressive lymphoma) occurs in about 15%. Other less common complications include prolymphocytoid transformation and secondary malignancies, particularly carcinomas of the lung and gastrointestinal tract and acute (myeloid) leukemia.25
Survival rates in CLL have improved substantially over the past decades,26–28 with significant gains following the introduction of antibiotics and, to a lesser extent, transfusions. Median survival is generally between 6 and 9 years, but many patients live for years without requiring therapy.
Chemotherapy: The mainstay of treatment
When to begin therapy remains one of the most challenging issues of patient management. Unlike in CML, there is no advantage to starting at diagnosis when most patients are asymptomatic.29
In 1996, the National Cancer Institute issued guidelines for starting treatment, which were updated in 2008 with very little change (Table 3).30 In general, the onset of symptoms and evidence of impaired marrow function, including an abnormal hemoglobin level and platelet count, are indications. The white blood cell count continuously increases during the disease course but is not usually an important factor for initiating treatment.
The therapeutic goal for most patients who require treatment has historically been palliation of symptoms. Therapy must be individualized to a patient’s age and clinical status, with a heavier reliance on chemotherapeutic agents for patients who can tolerate it and on immunotherapy for others. General strategies are as follows:
- “Go-Go” patients—young, fit, with few comorbidities, good renal function—are the minority. Recommendation: combination chemotherapy with fludarabine, cyclophosphamide, and rituximab (FCR).
- “Slo-Go” patients are reasonably fit and can tolerate chemotherapy but not FCR. Recommendation: combination therapy with either bendamustine and rituximab or chlorambucil and rituximab (for less fit patients). Recent evidence indicates ibrutinib may be useful for such patients.31
- “No-Go” patients are frail with short life expectancy. Recommendation: rituximab or observation (see below)
All CLL treatments are potentially toxic. Chemotherapy damages DNA and often causes blood cell counts to fall. Immunosuppression worsens with almost any treatment, involving a substantial risk of secondary malignancy. Although survival improves with therapy, relapse is universal.
Targeting CLL pathways
The new paradigm for cancer therapy is to identify a cellular pathway that drives oncogenesis or proliferation and interfere with it. The B-cell receptor pathway is enormously complex with numerous complex factors, making it difficult to discern the critical mutation that drives the proliferation of lymphocytes.
Bruton tyrosine kinase (Btk) is one factor that is critical for CLL proliferation. Patients with congenitally mutated or dysfunctional Btk have lymphopenia and agammaglobulinemia, making it a promising target for patients with B-cell disorders. Other experimental therapies are based on other such identified factors.
In 2014, the FDA approved two drugs for CLL—ibrutinib, a Btk inhibitor, and idelalisib, an inhibitor of phosphoinositide 3-kinase—after they were shown in clinical trials to dramatically improve outcomes in patients with relapsed CLL.32,33 Trials with these drugs are ongoing. These drugs also inhibit tyrosine kinase and so have vascular side effects in addition to their own idiosyncratic effects.
Ibrutinib has anticoagulant effects and should be stopped before surgery. It also can cause or exacerbate atrial fibrillation, making management of CLL difficult. It is associated with hypogammaglobulinemia, often requiring ongoing immunoglobulin replacement.
Idelalisib tends to cause systemic autoimmune phenomena such as pneumonitis and colitis.
Using T cells as therapy
It has long been observed that patients who undergo bone marrow transplant for leukemia have lower relapse rates if the transplant is allogeneic rather than from a twin. Further, if T cells are removed from the donor graft, graft-vs-host disease may be prevented but the risk of relapses increases. Finally, the presence of graft-vs-host disease tends to reduce the risk of relapse.34 Therefore, T cells clearly are key ingredients for success in the setting of bone marrow transplant. In fact, merely providing T cells for a relapse after allogeneic transplant can induce remission. However, because donor T cells are not targeted, acute and chronic graft-vs-host disease often can ensue.
‘Designer’ monoclonal antibodies
The B lymphocyte has multiple potential targets for new therapies for CLL as well as other cancers involving B cells. CD20 was identified on the surface of B cells in 1988 and is the target protein of the monoclonal antibody drug rituximab. Monoclonal antibodies can be modified to target other surface antigens, to link radioisotopes to deliver radiation therapy, and to deliver drugs that would otherwise be too toxic to be given systemically.35 Monoclonal antibodies can also be modified to enhance function.
Antibodies alone, however, must often rely on the host T cells for cytotoxicity and they are often compromised by either the underlying disease or treatment. Adapting the targeting function of antibodies to enhance or genetically alter T cells to recognize cancer-specific antigens is now being explored for leukemias.36
In 2014, the FDA approved blinatumomab for the treatment of relapsed or refractory acute lymphoblastic leukemia. This biopharmaceutical agent recruits T cells with one antibody-like moiety and targets the CD19 receptor of B cells with another. Given as a single intravenous treatment without chemotherapy, it has an almost 50% response rate, and those who respond tend to stay in remission. Other similar drugs are being developed, and using them earlier in treatment and for other B-cell leukemias is being explored.
New B-cell targeted therapy with CAR-Ts
Newer treatments are being developed based on chimeric antigen receptor T (CAR-T) cells. These engineered T cells express an anti-CD19 moiety that targets B cells, but also activate upon binding to them.37 CAR-T technology is being refined and shows great promise for cancer treatment.
Multiple clinical trials are currently under way in which the investigators collect autologous T cells by leukopheresis from a patient with a relapsed or refractory B-cell malignancy, transduce the T cells with retroviral vectors into anti-CD19 CAR-T cells, and then reinfuse them into the patient following modest chemotherapy.38
Study results from a small number of patients with relapsing or refractory CLL showed that some patients achieved long-term, progression-free survival.39 The most success with this therapy, however, has been in acute lymphoblastic leukemia.40 Possibly, this treatment could be applied to other lymphoid malignancies that also express CD19.
More advances
CAR-T cell therapy has drawbacks. The cells attack only the target antigen, which currently limits their use mostly to hematologic malignancies. In addition, autologous T cells are not robust. Also, the use of allogeneic T cells is restricted by their major histocompatibility complex, and the cells will be rejected by the recipient if not matched.
An attempt to overcome some of these drawbacks is to develop T cells redirected for universal cytokine killing. CAR-T cells are modified with a gene that causes them to excrete interleukin 12, which attracts macrophages and natural killer cells to the environment to better fight the tumor.41
Other modifications include editing out certain genes including the major histocompatibility complex, which avoids the problem of rejection. Another modification is to insert a “suicide gene” that allows the engineered T cells to be killed with an antidote if they do not work as planned.
Such gene-editing techniques hold great promise for curing cancers without chemotherapy in the not so distant future.
- National Cancer Institute Surveillance, Epidemiology, and End Results Program. SEER Stat Fact Sheets: Chronic Myeloid Leukemia. http://seer.cancer.gov/statfacts/html/cmyl.html. Accessed July 1, 2016.
- Nowell PC, Hungerford DA. A minute chromosome in human chronic granulocytic leukemia. Science 1960; 132:1497.
- Melo JV. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood 1996; 88:2375–2384.
- Pasternak G, Hochhaus A, Schultheis B, Hehlmann R. Chronic myelogenous leukemia: molecular and cellular aspects. J Cancer Res Clin Oncol 1998; 124:643–660.
- Faderl S, Kantarjian HM, Talpaz M. Chronic myelogenous leukemia: update on biology and treatment. Oncology (Williston Park) 1999; 13:169–184.
- Sawyers CL. Chronic myeloid leukemia. N Engl J Med 1999; 340:1330–1340.
- Hehlmann R, Heimpel H, Hasford J, et al. Randomized comparison of interferon-alpha with busulfan and hydroxyurea in chronic myelogenous leukemia. The German CML Study Group. Blood 1994; 84:4064–4077.
- Radich JP, Olavarria E, Apperley JF. Allogeneic hematopoietic stem cell transplantation for chronic myeloid leukemia. Hematol Oncol Clin North Am 2004; 18:685–702.
- Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood 2008; 112:4808–4817.
- O’Brien SG, Guilhot F, Larson RA, et al; IRIS Investigators. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2003; 348:994-1004.
- Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2010; 362:2260–2270.
- Saglio G, Kim DW, Issaragrisil S, et al; ENESTnd Investigators. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med 2010; 362:2251–2259.
- Pfirrmann M, Baccarani M, Saussele S, et al. Prognosis of long-term survival considering disease-specific death in patients with chronic myeloid leukemia. Leukemia 2016; 30:48-56.
- Li W, Croce K, Steensma DP, McDermott DF, Ben-Yehuda O, Moslehi J. Vascular and metabolic implications of novel targeted cancer therapies: focus on kinase inhibitors. J Am Coll Cardiol 2015; 66:1160–1178.
- Rawstron AC, Bennett F, Hillmen P. The biological and clinical relationship between CD5+23+ monoclonal B-cell lymphocytosis and chronic lymphocytic leukaemia. Br J Haematol 2007; 139:724–729.
- Rawstron AC, Bennett FL, O’Connor SJ, et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med 2008; 359:575–583.
- Hallek M, Cheson BD, Catovsky D, et al; International Workshop on Chronic Lymphocytic Leukemia. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008; 111:5446–5456.
- Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med 2005; 352:804–815.
- Rai KR, Sawitsky A, Cronkite EP, Chanana AD, Levy RN, Pasternack BS. Clinical staging of chronic lymphocytic leukemia. Blood 1975; 46:219–234.
- Dierlamm J, Michaux L, Criel A, Wlodarska I, Van den Berghe H, Hossfeld DK. Genetic abnormalities in chronic lymphocytic leukemia and their clinical and prognostic implications. Cancer Genet Cytogenet 1997; 94:27–35.
- Rozman C, Montserrat E. Chronic lymphocytic leukemia. N Engl J Med 1995; 333:1052–1057. Erratum in: N Engl J Med 1995; 333:1515.
- Jemal A, Thomas A, Murray T, Thun M. Cancer statistics, 2002. CA Cancer J Clin 2002; 52:23-47. Errata in: CA Cancer J Clin 2002; 52:119. CA Cancer J Clin 2002; 52:181–182.
- Caligaris-Cappio F, Hamblin TJ. B-cell chronic lymphocytic leukemia: a bird of a different feather. J Clin Oncol 1999; 17:399–408.
- Keating MJ. Chronic lymphocytic leukemia. Semin Oncol 1999; 26(suppl 14):107–114.
- Kalil N, Cheson BD. Management of chronic lymphocytic leukaemia. Drugs Aging 2000; 16:9–27.
- Minot GR, Buckman TE, Isaacs R. Chronic myelogenous leukemia: age incidence, duration, and benefit derived from irradiation. JAMA 1924; 82:1489–1494.
- Reinhard EH, Neely CL, Samples DM. Radioactive phosphorus in the treatment of chronic leukemias: long-term results over a period of 15 years. Cancer 1959; 50:942–958.
- Diehl LF, Karnell LH, Menck HR. The American College of Surgeons Commission on Cancer and the American Cancer Society. The National Cancer Data Base report on age, gender, treatment, and outcomes of patients with chronic lymphocytic leukemia. Cancer 1999; 86:2684–2692.
- Chemotherapeutic options in chronic lymphocytic leukemia: a meta-analysis of the randomized trials. CLL Trialists’ Collaborative Group. J Natl Cancer Inst 1999; 91:861–868.
- Cheson BD, Bennett JM, Grever M, et al. National Cancer Institute-sponsored working group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood 1996; 87:4990–4997.
- Burger JA, Tedeschi A, Barr PM, et al; RESONATE-2 Investigators. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med 2015; 373:2425–2437.
- Byrd JC, Brown JR, O’Brien S, et al; RESONATE Investigators. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med 2014; 371:213–223.
- Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med 2014; 370:997–1007.
- Horowitz MM, Gale RP, Sondel PM, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood 1990; 75:555–562.
- Weiner GJ. Building better monoclonal antibody-based therapeutics. Nat Rev Cancer 2015; 15:361–370.
- Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer 2013; 13:525–541.
- Urba WJ, Longo DL. Redirecting T cells. N Engl J Med 2011; 365:754–757.
- Klebanoff CA, Yamamoto TN, Restifo NP. Immunotherapy: treatment of aggressive lymphomas with anti-CD19 CAR T cells. Nat Rev Clin Oncol 2014; 11:685-686.
- Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 2015; 7:303ra139.
- Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015; 385:517–528.
- Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev 2014; 257:83–90.
- National Cancer Institute Surveillance, Epidemiology, and End Results Program. SEER Stat Fact Sheets: Chronic Myeloid Leukemia. http://seer.cancer.gov/statfacts/html/cmyl.html. Accessed July 1, 2016.
- Nowell PC, Hungerford DA. A minute chromosome in human chronic granulocytic leukemia. Science 1960; 132:1497.
- Melo JV. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood 1996; 88:2375–2384.
- Pasternak G, Hochhaus A, Schultheis B, Hehlmann R. Chronic myelogenous leukemia: molecular and cellular aspects. J Cancer Res Clin Oncol 1998; 124:643–660.
- Faderl S, Kantarjian HM, Talpaz M. Chronic myelogenous leukemia: update on biology and treatment. Oncology (Williston Park) 1999; 13:169–184.
- Sawyers CL. Chronic myeloid leukemia. N Engl J Med 1999; 340:1330–1340.
- Hehlmann R, Heimpel H, Hasford J, et al. Randomized comparison of interferon-alpha with busulfan and hydroxyurea in chronic myelogenous leukemia. The German CML Study Group. Blood 1994; 84:4064–4077.
- Radich JP, Olavarria E, Apperley JF. Allogeneic hematopoietic stem cell transplantation for chronic myeloid leukemia. Hematol Oncol Clin North Am 2004; 18:685–702.
- Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood 2008; 112:4808–4817.
- O’Brien SG, Guilhot F, Larson RA, et al; IRIS Investigators. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2003; 348:994-1004.
- Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2010; 362:2260–2270.
- Saglio G, Kim DW, Issaragrisil S, et al; ENESTnd Investigators. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med 2010; 362:2251–2259.
- Pfirrmann M, Baccarani M, Saussele S, et al. Prognosis of long-term survival considering disease-specific death in patients with chronic myeloid leukemia. Leukemia 2016; 30:48-56.
- Li W, Croce K, Steensma DP, McDermott DF, Ben-Yehuda O, Moslehi J. Vascular and metabolic implications of novel targeted cancer therapies: focus on kinase inhibitors. J Am Coll Cardiol 2015; 66:1160–1178.
- Rawstron AC, Bennett F, Hillmen P. The biological and clinical relationship between CD5+23+ monoclonal B-cell lymphocytosis and chronic lymphocytic leukaemia. Br J Haematol 2007; 139:724–729.
- Rawstron AC, Bennett FL, O’Connor SJ, et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med 2008; 359:575–583.
- Hallek M, Cheson BD, Catovsky D, et al; International Workshop on Chronic Lymphocytic Leukemia. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 2008; 111:5446–5456.
- Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med 2005; 352:804–815.
- Rai KR, Sawitsky A, Cronkite EP, Chanana AD, Levy RN, Pasternack BS. Clinical staging of chronic lymphocytic leukemia. Blood 1975; 46:219–234.
- Dierlamm J, Michaux L, Criel A, Wlodarska I, Van den Berghe H, Hossfeld DK. Genetic abnormalities in chronic lymphocytic leukemia and their clinical and prognostic implications. Cancer Genet Cytogenet 1997; 94:27–35.
- Rozman C, Montserrat E. Chronic lymphocytic leukemia. N Engl J Med 1995; 333:1052–1057. Erratum in: N Engl J Med 1995; 333:1515.
- Jemal A, Thomas A, Murray T, Thun M. Cancer statistics, 2002. CA Cancer J Clin 2002; 52:23-47. Errata in: CA Cancer J Clin 2002; 52:119. CA Cancer J Clin 2002; 52:181–182.
- Caligaris-Cappio F, Hamblin TJ. B-cell chronic lymphocytic leukemia: a bird of a different feather. J Clin Oncol 1999; 17:399–408.
- Keating MJ. Chronic lymphocytic leukemia. Semin Oncol 1999; 26(suppl 14):107–114.
- Kalil N, Cheson BD. Management of chronic lymphocytic leukaemia. Drugs Aging 2000; 16:9–27.
- Minot GR, Buckman TE, Isaacs R. Chronic myelogenous leukemia: age incidence, duration, and benefit derived from irradiation. JAMA 1924; 82:1489–1494.
- Reinhard EH, Neely CL, Samples DM. Radioactive phosphorus in the treatment of chronic leukemias: long-term results over a period of 15 years. Cancer 1959; 50:942–958.
- Diehl LF, Karnell LH, Menck HR. The American College of Surgeons Commission on Cancer and the American Cancer Society. The National Cancer Data Base report on age, gender, treatment, and outcomes of patients with chronic lymphocytic leukemia. Cancer 1999; 86:2684–2692.
- Chemotherapeutic options in chronic lymphocytic leukemia: a meta-analysis of the randomized trials. CLL Trialists’ Collaborative Group. J Natl Cancer Inst 1999; 91:861–868.
- Cheson BD, Bennett JM, Grever M, et al. National Cancer Institute-sponsored working group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood 1996; 87:4990–4997.
- Burger JA, Tedeschi A, Barr PM, et al; RESONATE-2 Investigators. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med 2015; 373:2425–2437.
- Byrd JC, Brown JR, O’Brien S, et al; RESONATE Investigators. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med 2014; 371:213–223.
- Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med 2014; 370:997–1007.
- Horowitz MM, Gale RP, Sondel PM, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood 1990; 75:555–562.
- Weiner GJ. Building better monoclonal antibody-based therapeutics. Nat Rev Cancer 2015; 15:361–370.
- Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer 2013; 13:525–541.
- Urba WJ, Longo DL. Redirecting T cells. N Engl J Med 2011; 365:754–757.
- Klebanoff CA, Yamamoto TN, Restifo NP. Immunotherapy: treatment of aggressive lymphomas with anti-CD19 CAR T cells. Nat Rev Clin Oncol 2014; 11:685-686.
- Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 2015; 7:303ra139.
- Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015; 385:517–528.
- Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev 2014; 257:83–90.
KEY POINTS
- Chronic myelogenous leukemia (CML) can now be functionally cured with tyrosine kinase inhibitors, which interfere with the product of the oncogene causing the disease.
- Patients diagnosed with CML should begin therapy immediately even if they have no symptoms.
- Tyrosine kinase inhibitors have side effects that increase cardiovascular risk.
- Chronic lymphocytic leukemia (CLL) is an immunologic disease involving clonal proliferation of B cells. Chemotherapy for CLL should begin only when symptoms or indicators of impaired marrow function reach a certain threshold.
- New treatments for CLL increase the risk of atrial fibrillation and autoimmunity.
- Experimental B-cell–targeted therapies have demonstrated encouraging results even when chemotherapy fails in CLL and other B-cell cancers.

Support Our Veterans and PA History!
If you are a veteran PA or a PA on active duty in the uniformed services, you may want to take advantage of the PA History Society’s plans to upgrade the Veteran Memorial Garden at the Eugene A. Stead, Jr, Center for Physician Assistants in Durham, North Carolina, into a “place of remembrance.”

The Society is selling 9 x 9-in engraved brick pavers for $100 each. For those interested in purchasing more than one paver, they are offering a sliding scale: 1 for $100, 2 for $175, 3 for $250, 4 for $325, and 5 for $400. The engraved paver will include the appropriate uniformed service logo and 3 lines for name, branch, and years of service. The pavers will be embedded in the wheelchair accessible walkway and in the patio area surrounding a life-size bronze combat medic statue— the centerpiece of the garden.

This is a chance to honor yourself and other PA colleagues who have served or are currently serving their country. Construction and landscaping is to begin in October 2016, with a dedication ceremony scheduled for April 2017.
Order now via the Society’s website at http://pahx.org/ to make sure that your paver is displayed prominently in the garden.
Reginald Carter, PhD, PA
Historian Emeritus
PA History Society
If you are a veteran PA or a PA on active duty in the uniformed services, you may want to take advantage of the PA History Society’s plans to upgrade the Veteran Memorial Garden at the Eugene A. Stead, Jr, Center for Physician Assistants in Durham, North Carolina, into a “place of remembrance.”
The Society is selling 9 x 9-in engraved brick pavers for $100 each. For those interested in purchasing more than one paver, they are offering a sliding scale: 1 for $100, 2 for $175, 3 for $250, 4 for $325, and 5 for $400. The engraved paver will include the appropriate uniformed service logo and 3 lines for name, branch, and years of service. The pavers will be embedded in the wheelchair accessible walkway and in the patio area surrounding a life-size bronze combat medic statue— the centerpiece of the garden.
This is a chance to honor yourself and other PA colleagues who have served or are currently serving their country. Construction and landscaping is to begin in October 2016, with a dedication ceremony scheduled for April 2017.
Order now via the Society’s website at http://pahx.org/ to make sure that your paver is displayed prominently in the garden.
Reginald Carter, PhD, PA
Historian Emeritus
PA History Society
If you are a veteran PA or a PA on active duty in the uniformed services, you may want to take advantage of the PA History Society’s plans to upgrade the Veteran Memorial Garden at the Eugene A. Stead, Jr, Center for Physician Assistants in Durham, North Carolina, into a “place of remembrance.”
The Society is selling 9 x 9-in engraved brick pavers for $100 each. For those interested in purchasing more than one paver, they are offering a sliding scale: 1 for $100, 2 for $175, 3 for $250, 4 for $325, and 5 for $400. The engraved paver will include the appropriate uniformed service logo and 3 lines for name, branch, and years of service. The pavers will be embedded in the wheelchair accessible walkway and in the patio area surrounding a life-size bronze combat medic statue— the centerpiece of the garden.
This is a chance to honor yourself and other PA colleagues who have served or are currently serving their country. Construction and landscaping is to begin in October 2016, with a dedication ceremony scheduled for April 2017.
Order now via the Society’s website at http://pahx.org/ to make sure that your paver is displayed prominently in the garden.
Reginald Carter, PhD, PA
Historian Emeritus
PA History Society

Who benefits most from immediate HIV therapy?
DURBAN, SOUTH AFRICA – Immediate initiation of antiretroviral therapy in asymptomatic treatment-naive HIV-infected adults with a CD4+ cell count greater than 500/mL brings considerably more bang for the buck in selected patient subgroups, according to a secondary analysis from the landmark START trial.
Four subgroups in START stood out as having larger absolute risk reductions and lower numbers-needed-to-treat with a strategy of immediate treatment: patients above age 50, those with a baseline Framingham Risk Score in excess of 10%, individuals whose plasma HIV RNA level exceeds 50,000 copies/mL, and patients with a CD4:CD8 ratio below 0.5, Dr. Jean-Michel Molina reported at the 21st International AIDS Conference.
“These patients might be prioritized for immediate access to ART,” observed Dr. Molina, professor of infectious diseases at the University of Paris-Diderot and head of the infectious diseases department at Saint-Louis Hospital, also in Paris.
The START (Strategic Timing of AntiRetroviral Treatment) study was a major clinical trial conducted in 35 countries. Investigators randomized 4,685 treatment-naive HIV-infected men and women with CD4+ cell counts in the normal range to immediate antiretroviral therapy or to deferral of treatment until their CD4+ cell count dropped to 350 cells/mL. After 3 years of prospective follow-up, the immediate-treatment strategy was associated with a 47% reduction in risk for the primary endpoint, a composite of AIDS, major cardiovascular or other non-AIDS events, and death. The number needed to treat immediately for 1 year in order to prevent one major event was 128 (N Engl J Med. 2015;373:795-807).
The START findings prompted a revision in World Health Organization guidelines, which now recommend universal antiretroviral treatment (ART) in patients with HIV infection regardless of their CD4+ cell count.
But some patients are reluctant to go on lifetime ART, particularly since they still feel normal while in the initial phases of HIV infection. In such cases, these new subgroup data may tip the balance in decision-making. Moreover, the new START findings should help physicians and policy makers in prioritizing access to immediate ART in settings where it isn’t universally available, according to Dr. Molina.
In the prespecified subgroup analysis, patients aged 50 and up at enrollment had a 2.9% incidence of the primary composite endpoint at 3 years if randomized to immediate ART and an 11.7% rate if they were assigned to deferred ART. The number of 50-plus-year-olds needed to treat (NNT) immediately for 1 year in order to prevent one additional case of AIDS, a major non-AIDS event, or death was just 45, compared to NNTs of 151 and 206 in patients aged 30-49 and younger than 30, respectively. Patients aged 50 and older accounted for nearly 12% of the overall study population.
For the roughly 28% of START participants whose baseline CD4:CD8 ratio was less than 0.5, the NNT for immediate rather than deferred therapy was 60, substantially more favorable than the NNTs of 214 in patients with a baseline ratio of 0.5-0.8 and 248 in patients with a CD4:CD8 ratio greater than 0.8. The incidence of the primary endpoint at 3 years of follow-up in patients with a CD4:CD8 ratio of less than 0.5 was 0.5% in the immediate ART group and 6.3% with deferred therapy.
Similarly, patients with a baseline 10-year Framingham Risk Score (FRS) of 10% or higher had an NNT of 69, compared with NNTs of 111 in subjects with an FRS of 1%-9.9% and 276 in those with an FRS of less than 1%. Patients with an FRS of 10% or more had a 2.4% incidence of the primary endpoint at 3 years if assigned to immediate ART and a 10.1% rate with deferred therapy. Patients with an FRS of 10% or more comprised only 9.6% of the study population, Dr. Molina continued.
Patients with a heavy baseline viral load as evidenced by a plasma HIV RNA level of at least 50,000 copies/mL accounted for roughly 22% of the total study sample. Their 3-year rate of the primary outcome was 2.1% with immediate ART and 6.9% with deferred treatment. The NNT was 67, compared to an NNT of 122 in patients with 3,000-49,999 copies/mL and 992 in the one-quarter of START participants with a baseline plasma HIV RNA level of less than 3,000 copies/mL.
Variables that weren’t related to the magnitude of absolute risk reduction and NNT in the START subgroup analysis were race, gender, geographic region, baseline CD4 cell count, and whether an individual resided in a high- or lower-income country.
Several audience members rose to assert that the CD4:CD8 ratio and viral load might very well be redundant predictors measuring the same thing, since they are typically tightly correlated. Dr. Molina replied that the START investigators are planning to conduct a multivariate analysis of the data in the near future, which should provide a definitive answer.
Another audience member expressed surprise at what struck him as a low cardiovascular event rate in the START study, given that HIV infection is known to be associated with accelerated atherosclerosis. Dr. Molina said the explanation for the low number of cardiovascular events lies in the fact that cardiovascular risk is so heavily age-dependent, and START participants were relatively young, with a median age of 36 years.
The START trial was carried out by the International Network for Strategic Initiatives in Global HIV Trials (INSIGHT) with funding provided mainly by the National Institutes of Health. Dr. Molina reported having no financial conflicts of interest.
DURBAN, SOUTH AFRICA – Immediate initiation of antiretroviral therapy in asymptomatic treatment-naive HIV-infected adults with a CD4+ cell count greater than 500/mL brings considerably more bang for the buck in selected patient subgroups, according to a secondary analysis from the landmark START trial.
Four subgroups in START stood out as having larger absolute risk reductions and lower numbers-needed-to-treat with a strategy of immediate treatment: patients above age 50, those with a baseline Framingham Risk Score in excess of 10%, individuals whose plasma HIV RNA level exceeds 50,000 copies/mL, and patients with a CD4:CD8 ratio below 0.5, Dr. Jean-Michel Molina reported at the 21st International AIDS Conference.
“These patients might be prioritized for immediate access to ART,” observed Dr. Molina, professor of infectious diseases at the University of Paris-Diderot and head of the infectious diseases department at Saint-Louis Hospital, also in Paris.
The START (Strategic Timing of AntiRetroviral Treatment) study was a major clinical trial conducted in 35 countries. Investigators randomized 4,685 treatment-naive HIV-infected men and women with CD4+ cell counts in the normal range to immediate antiretroviral therapy or to deferral of treatment until their CD4+ cell count dropped to 350 cells/mL. After 3 years of prospective follow-up, the immediate-treatment strategy was associated with a 47% reduction in risk for the primary endpoint, a composite of AIDS, major cardiovascular or other non-AIDS events, and death. The number needed to treat immediately for 1 year in order to prevent one major event was 128 (N Engl J Med. 2015;373:795-807).
The START findings prompted a revision in World Health Organization guidelines, which now recommend universal antiretroviral treatment (ART) in patients with HIV infection regardless of their CD4+ cell count.
But some patients are reluctant to go on lifetime ART, particularly since they still feel normal while in the initial phases of HIV infection. In such cases, these new subgroup data may tip the balance in decision-making. Moreover, the new START findings should help physicians and policy makers in prioritizing access to immediate ART in settings where it isn’t universally available, according to Dr. Molina.
In the prespecified subgroup analysis, patients aged 50 and up at enrollment had a 2.9% incidence of the primary composite endpoint at 3 years if randomized to immediate ART and an 11.7% rate if they were assigned to deferred ART. The number of 50-plus-year-olds needed to treat (NNT) immediately for 1 year in order to prevent one additional case of AIDS, a major non-AIDS event, or death was just 45, compared to NNTs of 151 and 206 in patients aged 30-49 and younger than 30, respectively. Patients aged 50 and older accounted for nearly 12% of the overall study population.
For the roughly 28% of START participants whose baseline CD4:CD8 ratio was less than 0.5, the NNT for immediate rather than deferred therapy was 60, substantially more favorable than the NNTs of 214 in patients with a baseline ratio of 0.5-0.8 and 248 in patients with a CD4:CD8 ratio greater than 0.8. The incidence of the primary endpoint at 3 years of follow-up in patients with a CD4:CD8 ratio of less than 0.5 was 0.5% in the immediate ART group and 6.3% with deferred therapy.
Similarly, patients with a baseline 10-year Framingham Risk Score (FRS) of 10% or higher had an NNT of 69, compared with NNTs of 111 in subjects with an FRS of 1%-9.9% and 276 in those with an FRS of less than 1%. Patients with an FRS of 10% or more had a 2.4% incidence of the primary endpoint at 3 years if assigned to immediate ART and a 10.1% rate with deferred therapy. Patients with an FRS of 10% or more comprised only 9.6% of the study population, Dr. Molina continued.
Patients with a heavy baseline viral load as evidenced by a plasma HIV RNA level of at least 50,000 copies/mL accounted for roughly 22% of the total study sample. Their 3-year rate of the primary outcome was 2.1% with immediate ART and 6.9% with deferred treatment. The NNT was 67, compared to an NNT of 122 in patients with 3,000-49,999 copies/mL and 992 in the one-quarter of START participants with a baseline plasma HIV RNA level of less than 3,000 copies/mL.
Variables that weren’t related to the magnitude of absolute risk reduction and NNT in the START subgroup analysis were race, gender, geographic region, baseline CD4 cell count, and whether an individual resided in a high- or lower-income country.
Several audience members rose to assert that the CD4:CD8 ratio and viral load might very well be redundant predictors measuring the same thing, since they are typically tightly correlated. Dr. Molina replied that the START investigators are planning to conduct a multivariate analysis of the data in the near future, which should provide a definitive answer.
Another audience member expressed surprise at what struck him as a low cardiovascular event rate in the START study, given that HIV infection is known to be associated with accelerated atherosclerosis. Dr. Molina said the explanation for the low number of cardiovascular events lies in the fact that cardiovascular risk is so heavily age-dependent, and START participants were relatively young, with a median age of 36 years.
The START trial was carried out by the International Network for Strategic Initiatives in Global HIV Trials (INSIGHT) with funding provided mainly by the National Institutes of Health. Dr. Molina reported having no financial conflicts of interest.
DURBAN, SOUTH AFRICA – Immediate initiation of antiretroviral therapy in asymptomatic treatment-naive HIV-infected adults with a CD4+ cell count greater than 500/mL brings considerably more bang for the buck in selected patient subgroups, according to a secondary analysis from the landmark START trial.
Four subgroups in START stood out as having larger absolute risk reductions and lower numbers-needed-to-treat with a strategy of immediate treatment: patients above age 50, those with a baseline Framingham Risk Score in excess of 10%, individuals whose plasma HIV RNA level exceeds 50,000 copies/mL, and patients with a CD4:CD8 ratio below 0.5, Dr. Jean-Michel Molina reported at the 21st International AIDS Conference.
“These patients might be prioritized for immediate access to ART,” observed Dr. Molina, professor of infectious diseases at the University of Paris-Diderot and head of the infectious diseases department at Saint-Louis Hospital, also in Paris.
The START (Strategic Timing of AntiRetroviral Treatment) study was a major clinical trial conducted in 35 countries. Investigators randomized 4,685 treatment-naive HIV-infected men and women with CD4+ cell counts in the normal range to immediate antiretroviral therapy or to deferral of treatment until their CD4+ cell count dropped to 350 cells/mL. After 3 years of prospective follow-up, the immediate-treatment strategy was associated with a 47% reduction in risk for the primary endpoint, a composite of AIDS, major cardiovascular or other non-AIDS events, and death. The number needed to treat immediately for 1 year in order to prevent one major event was 128 (N Engl J Med. 2015;373:795-807).
The START findings prompted a revision in World Health Organization guidelines, which now recommend universal antiretroviral treatment (ART) in patients with HIV infection regardless of their CD4+ cell count.
But some patients are reluctant to go on lifetime ART, particularly since they still feel normal while in the initial phases of HIV infection. In such cases, these new subgroup data may tip the balance in decision-making. Moreover, the new START findings should help physicians and policy makers in prioritizing access to immediate ART in settings where it isn’t universally available, according to Dr. Molina.
In the prespecified subgroup analysis, patients aged 50 and up at enrollment had a 2.9% incidence of the primary composite endpoint at 3 years if randomized to immediate ART and an 11.7% rate if they were assigned to deferred ART. The number of 50-plus-year-olds needed to treat (NNT) immediately for 1 year in order to prevent one additional case of AIDS, a major non-AIDS event, or death was just 45, compared to NNTs of 151 and 206 in patients aged 30-49 and younger than 30, respectively. Patients aged 50 and older accounted for nearly 12% of the overall study population.
For the roughly 28% of START participants whose baseline CD4:CD8 ratio was less than 0.5, the NNT for immediate rather than deferred therapy was 60, substantially more favorable than the NNTs of 214 in patients with a baseline ratio of 0.5-0.8 and 248 in patients with a CD4:CD8 ratio greater than 0.8. The incidence of the primary endpoint at 3 years of follow-up in patients with a CD4:CD8 ratio of less than 0.5 was 0.5% in the immediate ART group and 6.3% with deferred therapy.
Similarly, patients with a baseline 10-year Framingham Risk Score (FRS) of 10% or higher had an NNT of 69, compared with NNTs of 111 in subjects with an FRS of 1%-9.9% and 276 in those with an FRS of less than 1%. Patients with an FRS of 10% or more had a 2.4% incidence of the primary endpoint at 3 years if assigned to immediate ART and a 10.1% rate with deferred therapy. Patients with an FRS of 10% or more comprised only 9.6% of the study population, Dr. Molina continued.
Patients with a heavy baseline viral load as evidenced by a plasma HIV RNA level of at least 50,000 copies/mL accounted for roughly 22% of the total study sample. Their 3-year rate of the primary outcome was 2.1% with immediate ART and 6.9% with deferred treatment. The NNT was 67, compared to an NNT of 122 in patients with 3,000-49,999 copies/mL and 992 in the one-quarter of START participants with a baseline plasma HIV RNA level of less than 3,000 copies/mL.
Variables that weren’t related to the magnitude of absolute risk reduction and NNT in the START subgroup analysis were race, gender, geographic region, baseline CD4 cell count, and whether an individual resided in a high- or lower-income country.
Several audience members rose to assert that the CD4:CD8 ratio and viral load might very well be redundant predictors measuring the same thing, since they are typically tightly correlated. Dr. Molina replied that the START investigators are planning to conduct a multivariate analysis of the data in the near future, which should provide a definitive answer.
Another audience member expressed surprise at what struck him as a low cardiovascular event rate in the START study, given that HIV infection is known to be associated with accelerated atherosclerosis. Dr. Molina said the explanation for the low number of cardiovascular events lies in the fact that cardiovascular risk is so heavily age-dependent, and START participants were relatively young, with a median age of 36 years.
The START trial was carried out by the International Network for Strategic Initiatives in Global HIV Trials (INSIGHT) with funding provided mainly by the National Institutes of Health. Dr. Molina reported having no financial conflicts of interest.
AT AIDS 2016
Key clinical point: Four specific subgroups of asymptomatic HIV-infected adults who obtain the most clinical benefit from immediate rather than deferred antiretroviral therapy have been identified.
Major finding: The number of asymptomatic HIV-infected adults needed to treat immediately with antiretroviral therapy for 1 year instead of deferring treatment in order to avoid one case of AIDS or serious non-AIDS illness is 45 in patients aged 50 and older, compared with NNTs of 151 in 30- to 49-year-olds and 206 in patients younger than age 30.
Data source: This was a prespecified subgroup analysis of the landmark START trial, in which 4,685 treatment-naive asymptomatic HIV-infected adults with more than 500 CD4+ cells/mL were randomized to immediate or deferred antiretroviral therapy.
Disclosures: The START trial was funded chiefly by the National Institutes of Health. The presenter reported having no financial conflicts of interest.
Prophylaxis key to preventing medication overuse migraine
LAKE BUENA VISTA, FLA. – The etiology of intractable daily headaches is broad and includes life-threatening diagnoses. But a large proportion of those headaches results from overuse of therapies for migraine, making them in some cases an iatrogenic and avoidable complication, according to a headache specialist.
After ruling out serious systemic diseases, one of the first questions to ask patients with chronic daily headache is whether they have a history of migraine and, if this history is positive, how often they have been taking medications to abort symptoms, reported Wendy L. Wright, MS, a headache specialist and family nurse practitioner in private practice in Amherst, N.H.

“Use of any medicine for the treatment of migraine more than 2 or 3 times per week can result in medication overuse headache,” according to Ms. Wright, who maintained that medication overuse headache is “almost always transformed migraine.”
Medication overuse headaches do not stem from prescription drugs only, Ms. Wright said at the meeting, which was held by the American Pain Society and Global Academy for Medical Education. Global Academy and this organization are owned by the same company. She cited data indicating that acetaminophen is implicated in almost half of overuse headaches, but most patients are taking this drug or others in combinations. One study found that at the time that overuse headache developed, the average number of daily doses of headache drugs, including different types of drugs, was 5.2, Ms. Wright said.
Controlling medication overuse headaches is challenging and often requires several steps, she said. Overuse of butalbital, for example, requires tapering.
“You do not want to cold turkey individuals who have been taking high doses of butalbiltal because they can actually have a seizure,” Ms. Wright cautioned.
A more prudent strategy outlined by Ms. Wright involves a slow taper of the medication that the patient has been overusing while simultaneously uptitrating prophylactic therapies, such as beta blockers, divalproex, or topiramate. For butalbital, specifically, Ms. Wright recommended reducing the dose by about 10% per week with complete withdrawal in 2 to 3 months. For treatment of migraine, abortive medications should be used that have a different mechanism of action from the one implicated in the overuse complication.
“Here is one of my strategies: 0.5 mg to 1.0 mg per day of prednisone along with a [proton pump inhibitor],” Ms. Wright reported. “I taper the prednisone over 21 days, but at the same time I am pulling away their abortive medications.”
As migraine transforms from medication overuse into chronic daily headache, the presentation often shifts from its rapid attack-like onset into a less severe presentation, often losing the aura for those who had aura previously, Ms. Wright said. For migraine patients who develop chronic daily headache, other etiologies, such as meningitis or a tumor, must be considered. However, suspicion of an overuse syndrome should intensify for patients who report taking drugs like triptans 10 or more days per month or analgesics such as acetaminophen or nonsteroidal anti-inflammatory medications 15 days or more per days per month.
In some cases, patients take it upon themselves to increase the frequency of drugs they use to control migraine. This is particularly common for nonprescription agents, such as acetaminophen, that patients consider to be benign. However, many patients come to her specialty clinic from another provider who increased the frequency of abortive medications without understanding or considering the overuse phenomenon. Patients should be educated about the risks of medication overuse, but clinicians can avoid overuse by increasing their focus on prophylaxis.
Prophylaxis is particularly useful in patients with known triggers or a consistent pattern of migraine, such as migraine related to the menstrual cycle, Ms. Wright said. She referred to joint guidelines from the American Headache Society and the American Academy of Neurology (AHS/AAN) that have outlined available prophylactic therapies grouped by level of supporting evidence (Headache. 2012 Jun;52[6]:930-45).
Medication overuse headache is such a well-recognized phenomenon that it has been given its own ICD-10 code for reimbursement, but Ms. Wright said. In addition to prophylactic therapies recommended by AHS/AAN, she recommended pursuing adjunctive nonpharmacologic strategies for migraine prevention. Acupuncture is one such option. In addition, patients must be educated about the risks.
Ms. Wright has financial relationships with Merck, Pfizer, and Takeda.
LAKE BUENA VISTA, FLA. – The etiology of intractable daily headaches is broad and includes life-threatening diagnoses. But a large proportion of those headaches results from overuse of therapies for migraine, making them in some cases an iatrogenic and avoidable complication, according to a headache specialist.
After ruling out serious systemic diseases, one of the first questions to ask patients with chronic daily headache is whether they have a history of migraine and, if this history is positive, how often they have been taking medications to abort symptoms, reported Wendy L. Wright, MS, a headache specialist and family nurse practitioner in private practice in Amherst, N.H.
“Use of any medicine for the treatment of migraine more than 2 or 3 times per week can result in medication overuse headache,” according to Ms. Wright, who maintained that medication overuse headache is “almost always transformed migraine.”
Medication overuse headaches do not stem from prescription drugs only, Ms. Wright said at the meeting, which was held by the American Pain Society and Global Academy for Medical Education. Global Academy and this organization are owned by the same company. She cited data indicating that acetaminophen is implicated in almost half of overuse headaches, but most patients are taking this drug or others in combinations. One study found that at the time that overuse headache developed, the average number of daily doses of headache drugs, including different types of drugs, was 5.2, Ms. Wright said.
Controlling medication overuse headaches is challenging and often requires several steps, she said. Overuse of butalbital, for example, requires tapering.
“You do not want to cold turkey individuals who have been taking high doses of butalbiltal because they can actually have a seizure,” Ms. Wright cautioned.
A more prudent strategy outlined by Ms. Wright involves a slow taper of the medication that the patient has been overusing while simultaneously uptitrating prophylactic therapies, such as beta blockers, divalproex, or topiramate. For butalbital, specifically, Ms. Wright recommended reducing the dose by about 10% per week with complete withdrawal in 2 to 3 months. For treatment of migraine, abortive medications should be used that have a different mechanism of action from the one implicated in the overuse complication.
“Here is one of my strategies: 0.5 mg to 1.0 mg per day of prednisone along with a [proton pump inhibitor],” Ms. Wright reported. “I taper the prednisone over 21 days, but at the same time I am pulling away their abortive medications.”
As migraine transforms from medication overuse into chronic daily headache, the presentation often shifts from its rapid attack-like onset into a less severe presentation, often losing the aura for those who had aura previously, Ms. Wright said. For migraine patients who develop chronic daily headache, other etiologies, such as meningitis or a tumor, must be considered. However, suspicion of an overuse syndrome should intensify for patients who report taking drugs like triptans 10 or more days per month or analgesics such as acetaminophen or nonsteroidal anti-inflammatory medications 15 days or more per days per month.
In some cases, patients take it upon themselves to increase the frequency of drugs they use to control migraine. This is particularly common for nonprescription agents, such as acetaminophen, that patients consider to be benign. However, many patients come to her specialty clinic from another provider who increased the frequency of abortive medications without understanding or considering the overuse phenomenon. Patients should be educated about the risks of medication overuse, but clinicians can avoid overuse by increasing their focus on prophylaxis.
Prophylaxis is particularly useful in patients with known triggers or a consistent pattern of migraine, such as migraine related to the menstrual cycle, Ms. Wright said. She referred to joint guidelines from the American Headache Society and the American Academy of Neurology (AHS/AAN) that have outlined available prophylactic therapies grouped by level of supporting evidence (Headache. 2012 Jun;52[6]:930-45).
Medication overuse headache is such a well-recognized phenomenon that it has been given its own ICD-10 code for reimbursement, but Ms. Wright said. In addition to prophylactic therapies recommended by AHS/AAN, she recommended pursuing adjunctive nonpharmacologic strategies for migraine prevention. Acupuncture is one such option. In addition, patients must be educated about the risks.
Ms. Wright has financial relationships with Merck, Pfizer, and Takeda.
LAKE BUENA VISTA, FLA. – The etiology of intractable daily headaches is broad and includes life-threatening diagnoses. But a large proportion of those headaches results from overuse of therapies for migraine, making them in some cases an iatrogenic and avoidable complication, according to a headache specialist.
After ruling out serious systemic diseases, one of the first questions to ask patients with chronic daily headache is whether they have a history of migraine and, if this history is positive, how often they have been taking medications to abort symptoms, reported Wendy L. Wright, MS, a headache specialist and family nurse practitioner in private practice in Amherst, N.H.
“Use of any medicine for the treatment of migraine more than 2 or 3 times per week can result in medication overuse headache,” according to Ms. Wright, who maintained that medication overuse headache is “almost always transformed migraine.”
Medication overuse headaches do not stem from prescription drugs only, Ms. Wright said at the meeting, which was held by the American Pain Society and Global Academy for Medical Education. Global Academy and this organization are owned by the same company. She cited data indicating that acetaminophen is implicated in almost half of overuse headaches, but most patients are taking this drug or others in combinations. One study found that at the time that overuse headache developed, the average number of daily doses of headache drugs, including different types of drugs, was 5.2, Ms. Wright said.
Controlling medication overuse headaches is challenging and often requires several steps, she said. Overuse of butalbital, for example, requires tapering.
“You do not want to cold turkey individuals who have been taking high doses of butalbiltal because they can actually have a seizure,” Ms. Wright cautioned.
A more prudent strategy outlined by Ms. Wright involves a slow taper of the medication that the patient has been overusing while simultaneously uptitrating prophylactic therapies, such as beta blockers, divalproex, or topiramate. For butalbital, specifically, Ms. Wright recommended reducing the dose by about 10% per week with complete withdrawal in 2 to 3 months. For treatment of migraine, abortive medications should be used that have a different mechanism of action from the one implicated in the overuse complication.
“Here is one of my strategies: 0.5 mg to 1.0 mg per day of prednisone along with a [proton pump inhibitor],” Ms. Wright reported. “I taper the prednisone over 21 days, but at the same time I am pulling away their abortive medications.”
As migraine transforms from medication overuse into chronic daily headache, the presentation often shifts from its rapid attack-like onset into a less severe presentation, often losing the aura for those who had aura previously, Ms. Wright said. For migraine patients who develop chronic daily headache, other etiologies, such as meningitis or a tumor, must be considered. However, suspicion of an overuse syndrome should intensify for patients who report taking drugs like triptans 10 or more days per month or analgesics such as acetaminophen or nonsteroidal anti-inflammatory medications 15 days or more per days per month.
In some cases, patients take it upon themselves to increase the frequency of drugs they use to control migraine. This is particularly common for nonprescription agents, such as acetaminophen, that patients consider to be benign. However, many patients come to her specialty clinic from another provider who increased the frequency of abortive medications without understanding or considering the overuse phenomenon. Patients should be educated about the risks of medication overuse, but clinicians can avoid overuse by increasing their focus on prophylaxis.
Prophylaxis is particularly useful in patients with known triggers or a consistent pattern of migraine, such as migraine related to the menstrual cycle, Ms. Wright said. She referred to joint guidelines from the American Headache Society and the American Academy of Neurology (AHS/AAN) that have outlined available prophylactic therapies grouped by level of supporting evidence (Headache. 2012 Jun;52[6]:930-45).
Medication overuse headache is such a well-recognized phenomenon that it has been given its own ICD-10 code for reimbursement, but Ms. Wright said. In addition to prophylactic therapies recommended by AHS/AAN, she recommended pursuing adjunctive nonpharmacologic strategies for migraine prevention. Acupuncture is one such option. In addition, patients must be educated about the risks.
Ms. Wright has financial relationships with Merck, Pfizer, and Takeda.
EXPERT ANALYSIS FROM PAIN CARE FOR PRIMARY CARE

New SHM Members – August 2016
J. Nicholson, Alabama
N. Tangutur, MD, Alabama
E. Ali, MD, Arizona
B. Cabrera, MD, Arizona
J. Castrolondono, MD, Arizona
T. Djurisic, MD, Arizona
R. Ernst, MD, Arizona
A. M. Mendez, Arizona
B. Mozaffari, DO, Arizona
A. Sharma, MD, Arizona
R. Soni, MD, Arizona
G. Neaville, MD, Arkansas
D. Sidhu, PA-C, British Columbia
S. Sidhu, EMBA, British Columbia
G. Bean, MD, MPH, MBA, FAAP, California
K. Bechler, MD, California
K. Chauhan, MPH, MD, California
N. Dave, MD, California
G. Dhanoa, California
A. Fisher, California
Y. Youssef, MD, California
S. De La Garza, MD, Colorado
V. Gundu, MD, Colorado
B. McCoy, DO, Colorado
J. Costanzo-Brown, FNP, Delaware
G. Siu, MD, Delaware
Y. Tal, MD, Delaware
J. Browning, NP-BC, DCNP, Florida
A. Chamseddin, MD, Florida
J. Florindez, MD, Florida
I. Gadalla, PA-C, Florida
G. Guess, Florida
M. E. Huckestein, ANP, Florida
D. Keerty, Florida
M. Mayo, DO, Florida
H. Nasser, MD, Florida
S. Rothstein, MSc, Florida
L. Succari, MD, Florida
S. Zimmer, MD, Florida
C. Ezigbo, Georgia
A. Mann, MD, Georgia
D. Wilmoth, Georgia
A. M. Sanchez Varela, MD, Guam
S. Cline, PhD, MBA, RN, Idaho
K. Abe, PA-C, Illinois
S. Chaudhry-Khan, MD, Illinois
K. Gallagher, Illinois
S. Kuhns, RN, Illinois
S. Pulimi, MD, Illinois
A. Urbonas, MD, Illinois
J. Chounramany, Iowa
B. Daniel, MD, Iowa
S. Joy, ARNP, Iowa
E. Kuperman, MD, Iowa
E. Shinozaki, MD, Iowa
S. Velur, MBBS, Iowa
L. Amos, MD, Kansas
L. Olson, MD, Kansas
M. Schultz, ANP, Kansas
M. Sharma, MD, Kansas
C. Castellanos, Kentucky
B. Mauldin, Louisiana
A. Thompson Soileau, MD, Louisiana
K. Hartman, MD, Maine
K. Carr, MD, Maryland
M. J. Dales, Maryland
K. Jansen, MPAS, PA-C, Maryland
A. Jubon, PA-C, Maryland
O. Schwartz, MD, Maryland
J. Withey, MD, Maryland
J. Louloudes, PA-C, Massachusetts
A. Susheelo, Massachusetts
T. Vu, Massachusetts
K. Bhatti, PA-C, Manitoba
R. Akram, Michigan
S. Federico, APRN-BC, Michigan
A. Mohammed, MBBS, Michigan
Q. Salamah, MD, Michigan
M. Schmuker, DO, Michigan
J. Dressen, Minnesota
G. Larson, MHA, Minnesota
T. Starkey, MD, Minnesota
V. Adike, MBBS, Mississippi
A. Collins, MD, Mississippi
J. Foreman, AGNP, Mississippi
J. Grady, MD, Mississippi
K. Heintzelman, DO, Mississippi
M. Moon, MD, Mississippi
A. Pamarthy, MD, Mississippi
J. Shores, Mississippi
J. Halsey, MD, MA, Missouri
M. Hendrix, MD, Missouri
U. Inampudi, MD, Missouri
C. Paris, APRN, FNP, Missouri
D. Payton, MD, Missouri
N. Crump, MD, Nebraska
T. Langenhan, MD, Nebraska
S. Garrett, MD, Nevada
C. Szot, MD, New Hampshire
D. Abbasi, MD, MBBS, New Jersey
S. M. Abel, ACNP, New Jersey
K. Alban, New Jersey
R. Amarini, New Jersey
J. Bauer, New Jersey
M. Branca, New Jersey
A. Hamarich, DO, New Jersey
S. Jaleel, MD, New Jersey
J. Knight, New Jersey
C. Lucchese, New Jersey
J. Peterson, New Jersey
M. Sohaib, MD, New Jersey
A. Azhar, MD, New York
B. Changlai, MD, New York
D. Gerling, New York
Y. Jin, New York
E. Palermo, ACNP, New York
V. Phillips, FNP, New York
P. Shi, DO, New York
B. Wertheimer, MD, New York
M. Yarowsky, MD, New York
C. Yates, MD, New York
J. Adams, MD, North Carolina
S. Akkaladevi, North Carolina
M. Arapian, MD, North Carolina
J. Cunningham, MD, North Carolina
K. Desronvil, ACNP, North Carolina
M. Dittmer, PA-C, North Carolina
Z. Edinger, ANP, North Carolina
T. Elswick, PA-C, North Carolina
D. Goble, MD, North Carolina
O. Jeelani, MD, MBBS, North Carolina
S. Lateef, North Carolina
G. Shalhoub, MD, North Carolina
J. Townsend, North Carolina
T. Turbett, North Carolina
K. Broderick-Forsgren, MD, Ohio
D. Foote, ACNP, Ohio
R. Muriithi, MBchB, Ohio
K. Patel, MD, Ohio
P. Veeramreddy, MBBS, Ohio
Y. Chen, MD, Oklahoma
M. Langmacher, BS, MD, Oklahoma
R. Mourh, MD, Oklahoma
M. Lukban, MD, Oregon
B. Ongole, MD, Oregon
J. Brunner, BS, MBA, Pennsylvania
M. Butala, Pennsylvania
B. Y. Chen, Pennsylvania
Z. Chen, MD, Pennsylvania
R. House, CRNP, Pennsylvania
M. Mar Fan, MD, Pennsylvania
E. McCamant, Pennsylvania
O. Okonkwo, MD, FACP, Pennsylvania
A. Savini, PA-C, Pennsylvania
A. Singh, MD, Pennsylvania
B. Smith, Pennsylvania
A. Tarique, MD, Pennsylvania
A. Whitsel, CRNP, Pennsylvania
P. Woods, MD, Pennsylvania
R. Ball, MHA, South Carolina
D. Burns, South Carolina
A. Kachalia, MD, South Carolina
L. Teague, South Carolina
D. Kindelspire, South Dakota
J. Bynum, Tennessee
A. Davidson, APRN-BC, Tennessee
P. Goleb, Tennessee
P. McCain, FNP, Tennessee
S. Patel, MD, Tennessee
A. Proffitt, ACNP, ANP, APRN, MSN, Tennessee
J. Tompkins, MD, Tennessee
F. Cardona, MD, Texas
N. Civunigunta, Texas
S. Khan, Texas
M. Mann, Texas
J. Muntz, MD, Texas
L. Swift, Texas
M. Abbott, FACHE, MBA, PharmD, Virginia
M. Alfaris, MD, Virginia
H. Aros, MD, Virginia
S. Naidu, MD, Virginia
M. Shaub, Virginia
O. Adeyeri, West Virginia
F. Farahmand, MD, West Virginia
J. Guinto, ARNP, West Virginia
S. Shiveley, MD, West Virginia
J. Singh, MBBch, West Virginia
G. Johnson, DO, Wisconsin
V. McFadden, MD, PhD, Wisconsin
S. Alam, Dhaka, Bangladesh
A. Fathala, MD, Saudi Arabia
J. Nicholson, Alabama
N. Tangutur, MD, Alabama
E. Ali, MD, Arizona
B. Cabrera, MD, Arizona
J. Castrolondono, MD, Arizona
T. Djurisic, MD, Arizona
R. Ernst, MD, Arizona
A. M. Mendez, Arizona
B. Mozaffari, DO, Arizona
A. Sharma, MD, Arizona
R. Soni, MD, Arizona
G. Neaville, MD, Arkansas
D. Sidhu, PA-C, British Columbia
S. Sidhu, EMBA, British Columbia
G. Bean, MD, MPH, MBA, FAAP, California
K. Bechler, MD, California
K. Chauhan, MPH, MD, California
N. Dave, MD, California
G. Dhanoa, California
A. Fisher, California
Y. Youssef, MD, California
S. De La Garza, MD, Colorado
V. Gundu, MD, Colorado
B. McCoy, DO, Colorado
J. Costanzo-Brown, FNP, Delaware
G. Siu, MD, Delaware
Y. Tal, MD, Delaware
J. Browning, NP-BC, DCNP, Florida
A. Chamseddin, MD, Florida
J. Florindez, MD, Florida
I. Gadalla, PA-C, Florida
G. Guess, Florida
M. E. Huckestein, ANP, Florida
D. Keerty, Florida
M. Mayo, DO, Florida
H. Nasser, MD, Florida
S. Rothstein, MSc, Florida
L. Succari, MD, Florida
S. Zimmer, MD, Florida
C. Ezigbo, Georgia
A. Mann, MD, Georgia
D. Wilmoth, Georgia
A. M. Sanchez Varela, MD, Guam
S. Cline, PhD, MBA, RN, Idaho
K. Abe, PA-C, Illinois
S. Chaudhry-Khan, MD, Illinois
K. Gallagher, Illinois
S. Kuhns, RN, Illinois
S. Pulimi, MD, Illinois
A. Urbonas, MD, Illinois
J. Chounramany, Iowa
B. Daniel, MD, Iowa
S. Joy, ARNP, Iowa
E. Kuperman, MD, Iowa
E. Shinozaki, MD, Iowa
S. Velur, MBBS, Iowa
L. Amos, MD, Kansas
L. Olson, MD, Kansas
M. Schultz, ANP, Kansas
M. Sharma, MD, Kansas
C. Castellanos, Kentucky
B. Mauldin, Louisiana
A. Thompson Soileau, MD, Louisiana
K. Hartman, MD, Maine
K. Carr, MD, Maryland
M. J. Dales, Maryland
K. Jansen, MPAS, PA-C, Maryland
A. Jubon, PA-C, Maryland
O. Schwartz, MD, Maryland
J. Withey, MD, Maryland
J. Louloudes, PA-C, Massachusetts
A. Susheelo, Massachusetts
T. Vu, Massachusetts
K. Bhatti, PA-C, Manitoba
R. Akram, Michigan
S. Federico, APRN-BC, Michigan
A. Mohammed, MBBS, Michigan
Q. Salamah, MD, Michigan
M. Schmuker, DO, Michigan
J. Dressen, Minnesota
G. Larson, MHA, Minnesota
T. Starkey, MD, Minnesota
V. Adike, MBBS, Mississippi
A. Collins, MD, Mississippi
J. Foreman, AGNP, Mississippi
J. Grady, MD, Mississippi
K. Heintzelman, DO, Mississippi
M. Moon, MD, Mississippi
A. Pamarthy, MD, Mississippi
J. Shores, Mississippi
J. Halsey, MD, MA, Missouri
M. Hendrix, MD, Missouri
U. Inampudi, MD, Missouri
C. Paris, APRN, FNP, Missouri
D. Payton, MD, Missouri
N. Crump, MD, Nebraska
T. Langenhan, MD, Nebraska
S. Garrett, MD, Nevada
C. Szot, MD, New Hampshire
D. Abbasi, MD, MBBS, New Jersey
S. M. Abel, ACNP, New Jersey
K. Alban, New Jersey
R. Amarini, New Jersey
J. Bauer, New Jersey
M. Branca, New Jersey
A. Hamarich, DO, New Jersey
S. Jaleel, MD, New Jersey
J. Knight, New Jersey
C. Lucchese, New Jersey
J. Peterson, New Jersey
M. Sohaib, MD, New Jersey
A. Azhar, MD, New York
B. Changlai, MD, New York
D. Gerling, New York
Y. Jin, New York
E. Palermo, ACNP, New York
V. Phillips, FNP, New York
P. Shi, DO, New York
B. Wertheimer, MD, New York
M. Yarowsky, MD, New York
C. Yates, MD, New York
J. Adams, MD, North Carolina
S. Akkaladevi, North Carolina
M. Arapian, MD, North Carolina
J. Cunningham, MD, North Carolina
K. Desronvil, ACNP, North Carolina
M. Dittmer, PA-C, North Carolina
Z. Edinger, ANP, North Carolina
T. Elswick, PA-C, North Carolina
D. Goble, MD, North Carolina
O. Jeelani, MD, MBBS, North Carolina
S. Lateef, North Carolina
G. Shalhoub, MD, North Carolina
J. Townsend, North Carolina
T. Turbett, North Carolina
K. Broderick-Forsgren, MD, Ohio
D. Foote, ACNP, Ohio
R. Muriithi, MBchB, Ohio
K. Patel, MD, Ohio
P. Veeramreddy, MBBS, Ohio
Y. Chen, MD, Oklahoma
M. Langmacher, BS, MD, Oklahoma
R. Mourh, MD, Oklahoma
M. Lukban, MD, Oregon
B. Ongole, MD, Oregon
J. Brunner, BS, MBA, Pennsylvania
M. Butala, Pennsylvania
B. Y. Chen, Pennsylvania
Z. Chen, MD, Pennsylvania
R. House, CRNP, Pennsylvania
M. Mar Fan, MD, Pennsylvania
E. McCamant, Pennsylvania
O. Okonkwo, MD, FACP, Pennsylvania
A. Savini, PA-C, Pennsylvania
A. Singh, MD, Pennsylvania
B. Smith, Pennsylvania
A. Tarique, MD, Pennsylvania
A. Whitsel, CRNP, Pennsylvania
P. Woods, MD, Pennsylvania
R. Ball, MHA, South Carolina
D. Burns, South Carolina
A. Kachalia, MD, South Carolina
L. Teague, South Carolina
D. Kindelspire, South Dakota
J. Bynum, Tennessee
A. Davidson, APRN-BC, Tennessee
P. Goleb, Tennessee
P. McCain, FNP, Tennessee
S. Patel, MD, Tennessee
A. Proffitt, ACNP, ANP, APRN, MSN, Tennessee
J. Tompkins, MD, Tennessee
F. Cardona, MD, Texas
N. Civunigunta, Texas
S. Khan, Texas
M. Mann, Texas
J. Muntz, MD, Texas
L. Swift, Texas
M. Abbott, FACHE, MBA, PharmD, Virginia
M. Alfaris, MD, Virginia
H. Aros, MD, Virginia
S. Naidu, MD, Virginia
M. Shaub, Virginia
O. Adeyeri, West Virginia
F. Farahmand, MD, West Virginia
J. Guinto, ARNP, West Virginia
S. Shiveley, MD, West Virginia
J. Singh, MBBch, West Virginia
G. Johnson, DO, Wisconsin
V. McFadden, MD, PhD, Wisconsin
S. Alam, Dhaka, Bangladesh
A. Fathala, MD, Saudi Arabia
J. Nicholson, Alabama
N. Tangutur, MD, Alabama
E. Ali, MD, Arizona
B. Cabrera, MD, Arizona
J. Castrolondono, MD, Arizona
T. Djurisic, MD, Arizona
R. Ernst, MD, Arizona
A. M. Mendez, Arizona
B. Mozaffari, DO, Arizona
A. Sharma, MD, Arizona
R. Soni, MD, Arizona
G. Neaville, MD, Arkansas
D. Sidhu, PA-C, British Columbia
S. Sidhu, EMBA, British Columbia
G. Bean, MD, MPH, MBA, FAAP, California
K. Bechler, MD, California
K. Chauhan, MPH, MD, California
N. Dave, MD, California
G. Dhanoa, California
A. Fisher, California
Y. Youssef, MD, California
S. De La Garza, MD, Colorado
V. Gundu, MD, Colorado
B. McCoy, DO, Colorado
J. Costanzo-Brown, FNP, Delaware
G. Siu, MD, Delaware
Y. Tal, MD, Delaware
J. Browning, NP-BC, DCNP, Florida
A. Chamseddin, MD, Florida
J. Florindez, MD, Florida
I. Gadalla, PA-C, Florida
G. Guess, Florida
M. E. Huckestein, ANP, Florida
D. Keerty, Florida
M. Mayo, DO, Florida
H. Nasser, MD, Florida
S. Rothstein, MSc, Florida
L. Succari, MD, Florida
S. Zimmer, MD, Florida
C. Ezigbo, Georgia
A. Mann, MD, Georgia
D. Wilmoth, Georgia
A. M. Sanchez Varela, MD, Guam
S. Cline, PhD, MBA, RN, Idaho
K. Abe, PA-C, Illinois
S. Chaudhry-Khan, MD, Illinois
K. Gallagher, Illinois
S. Kuhns, RN, Illinois
S. Pulimi, MD, Illinois
A. Urbonas, MD, Illinois
J. Chounramany, Iowa
B. Daniel, MD, Iowa
S. Joy, ARNP, Iowa
E. Kuperman, MD, Iowa
E. Shinozaki, MD, Iowa
S. Velur, MBBS, Iowa
L. Amos, MD, Kansas
L. Olson, MD, Kansas
M. Schultz, ANP, Kansas
M. Sharma, MD, Kansas
C. Castellanos, Kentucky
B. Mauldin, Louisiana
A. Thompson Soileau, MD, Louisiana
K. Hartman, MD, Maine
K. Carr, MD, Maryland
M. J. Dales, Maryland
K. Jansen, MPAS, PA-C, Maryland
A. Jubon, PA-C, Maryland
O. Schwartz, MD, Maryland
J. Withey, MD, Maryland
J. Louloudes, PA-C, Massachusetts
A. Susheelo, Massachusetts
T. Vu, Massachusetts
K. Bhatti, PA-C, Manitoba
R. Akram, Michigan
S. Federico, APRN-BC, Michigan
A. Mohammed, MBBS, Michigan
Q. Salamah, MD, Michigan
M. Schmuker, DO, Michigan
J. Dressen, Minnesota
G. Larson, MHA, Minnesota
T. Starkey, MD, Minnesota
V. Adike, MBBS, Mississippi
A. Collins, MD, Mississippi
J. Foreman, AGNP, Mississippi
J. Grady, MD, Mississippi
K. Heintzelman, DO, Mississippi
M. Moon, MD, Mississippi
A. Pamarthy, MD, Mississippi
J. Shores, Mississippi
J. Halsey, MD, MA, Missouri
M. Hendrix, MD, Missouri
U. Inampudi, MD, Missouri
C. Paris, APRN, FNP, Missouri
D. Payton, MD, Missouri
N. Crump, MD, Nebraska
T. Langenhan, MD, Nebraska
S. Garrett, MD, Nevada
C. Szot, MD, New Hampshire
D. Abbasi, MD, MBBS, New Jersey
S. M. Abel, ACNP, New Jersey
K. Alban, New Jersey
R. Amarini, New Jersey
J. Bauer, New Jersey
M. Branca, New Jersey
A. Hamarich, DO, New Jersey
S. Jaleel, MD, New Jersey
J. Knight, New Jersey
C. Lucchese, New Jersey
J. Peterson, New Jersey
M. Sohaib, MD, New Jersey
A. Azhar, MD, New York
B. Changlai, MD, New York
D. Gerling, New York
Y. Jin, New York
E. Palermo, ACNP, New York
V. Phillips, FNP, New York
P. Shi, DO, New York
B. Wertheimer, MD, New York
M. Yarowsky, MD, New York
C. Yates, MD, New York
J. Adams, MD, North Carolina
S. Akkaladevi, North Carolina
M. Arapian, MD, North Carolina
J. Cunningham, MD, North Carolina
K. Desronvil, ACNP, North Carolina
M. Dittmer, PA-C, North Carolina
Z. Edinger, ANP, North Carolina
T. Elswick, PA-C, North Carolina
D. Goble, MD, North Carolina
O. Jeelani, MD, MBBS, North Carolina
S. Lateef, North Carolina
G. Shalhoub, MD, North Carolina
J. Townsend, North Carolina
T. Turbett, North Carolina
K. Broderick-Forsgren, MD, Ohio
D. Foote, ACNP, Ohio
R. Muriithi, MBchB, Ohio
K. Patel, MD, Ohio
P. Veeramreddy, MBBS, Ohio
Y. Chen, MD, Oklahoma
M. Langmacher, BS, MD, Oklahoma
R. Mourh, MD, Oklahoma
M. Lukban, MD, Oregon
B. Ongole, MD, Oregon
J. Brunner, BS, MBA, Pennsylvania
M. Butala, Pennsylvania
B. Y. Chen, Pennsylvania
Z. Chen, MD, Pennsylvania
R. House, CRNP, Pennsylvania
M. Mar Fan, MD, Pennsylvania
E. McCamant, Pennsylvania
O. Okonkwo, MD, FACP, Pennsylvania
A. Savini, PA-C, Pennsylvania
A. Singh, MD, Pennsylvania
B. Smith, Pennsylvania
A. Tarique, MD, Pennsylvania
A. Whitsel, CRNP, Pennsylvania
P. Woods, MD, Pennsylvania
R. Ball, MHA, South Carolina
D. Burns, South Carolina
A. Kachalia, MD, South Carolina
L. Teague, South Carolina
D. Kindelspire, South Dakota
J. Bynum, Tennessee
A. Davidson, APRN-BC, Tennessee
P. Goleb, Tennessee
P. McCain, FNP, Tennessee
S. Patel, MD, Tennessee
A. Proffitt, ACNP, ANP, APRN, MSN, Tennessee
J. Tompkins, MD, Tennessee
F. Cardona, MD, Texas
N. Civunigunta, Texas
S. Khan, Texas
M. Mann, Texas
J. Muntz, MD, Texas
L. Swift, Texas
M. Abbott, FACHE, MBA, PharmD, Virginia
M. Alfaris, MD, Virginia
H. Aros, MD, Virginia
S. Naidu, MD, Virginia
M. Shaub, Virginia
O. Adeyeri, West Virginia
F. Farahmand, MD, West Virginia
J. Guinto, ARNP, West Virginia
S. Shiveley, MD, West Virginia
J. Singh, MBBch, West Virginia
G. Johnson, DO, Wisconsin
V. McFadden, MD, PhD, Wisconsin
S. Alam, Dhaka, Bangladesh
A. Fathala, MD, Saudi Arabia
Register for Academic Hospitalist Academy
Don’t miss the eighth annual Academic Hospitalist Academy (AHA), Sept. 12–15, at the scenic Lakeway Resort and Spa in Austin, Texas. You will experience an energizing, interactive learning environment featuring didactics, small-group exercises, and skill-building breakout sessions. Each full day of learning is facilitated by leading clinician-educators, hospitalist-researchers, and clinical administrators in a 1-to-10 faculty-to-student ratio.
AHA’s principal goals are to:
- Develop junior academic hospitalists as the premier teachers and educational leaders at their institutions
- Help academic hospitalists develop scholarly work and increase scholarly output
- Enhance awareness of the value of quality improvement and patient safety work
- Support academic promotion of all attendees
Register now at www.academichospitalist.org.
Don’t miss the eighth annual Academic Hospitalist Academy (AHA), Sept. 12–15, at the scenic Lakeway Resort and Spa in Austin, Texas. You will experience an energizing, interactive learning environment featuring didactics, small-group exercises, and skill-building breakout sessions. Each full day of learning is facilitated by leading clinician-educators, hospitalist-researchers, and clinical administrators in a 1-to-10 faculty-to-student ratio.
AHA’s principal goals are to:
- Develop junior academic hospitalists as the premier teachers and educational leaders at their institutions
- Help academic hospitalists develop scholarly work and increase scholarly output
- Enhance awareness of the value of quality improvement and patient safety work
- Support academic promotion of all attendees
Register now at www.academichospitalist.org.
Don’t miss the eighth annual Academic Hospitalist Academy (AHA), Sept. 12–15, at the scenic Lakeway Resort and Spa in Austin, Texas. You will experience an energizing, interactive learning environment featuring didactics, small-group exercises, and skill-building breakout sessions. Each full day of learning is facilitated by leading clinician-educators, hospitalist-researchers, and clinical administrators in a 1-to-10 faculty-to-student ratio.
AHA’s principal goals are to:
- Develop junior academic hospitalists as the premier teachers and educational leaders at their institutions
- Help academic hospitalists develop scholarly work and increase scholarly output
- Enhance awareness of the value of quality improvement and patient safety work
- Support academic promotion of all attendees
Register now at www.academichospitalist.org.