User login
Can we reduce the risk of readmission for a patient with an exacerbation of COPD?
We think so. Some strategies to reduce readmission rates, such as coordinating care and managing comorbidities, apply to chronic diseases in general, while others are disease-specific. To reduce the need for hospital readmission for chronic obstructive pulmonary disease (COPD), coordinated efforts involving both inpatient and outpatient care are necessary. This can be achieved by using a checklist before discharge (Table 1) and by implementing outpatient COPD programs that continue patient education and provide rapid access to medical support if needed.

There is room for improvement. COPD is common and expensive, with high rates of hospital readmission,1 and up to 70% of the money we spend on it goes for hospital care.2 No wonder then that the Centers for Medicare and Medicaid Services has now expanded its Readmissions Reduction Program to include acute COPD exacerbations.3 Yet in a retrospective study, Yip et al4 found that fewer than half of patients hospitalized with acute exacerbation of COPD received appropriate vaccinations, counseling on smoking cessation, and long-acting inhalers—all of which are on our checklist.4
The following interventions have been demonstrated to be useful in reducing COPD hospital admissions and the risk of death.
SMOKING CESSATION
Cigarette smoking is the most common and easily identifiable risk factor for COPD exacerbation.5
Au et al5 found that quitting smoking reduces the risk of COPD exacerbation (adjusted hazard ratio 0.78, 95% confidence interval [CI] 0.75–0.87), and the risk keeps decreasing the longer the patient stays off tobacco.5
Whether counseling hospitalized patients on smoking cessation reduces the COPD readmission rate has not been well studied. However, a meta-analysis of nine randomized controlled trials, two of which were done in the hospital, revealed higher abstinence rates in COPD patients who received extensive counseling on smoking cessation.7 For these reasons, hospitalized COPD patients who smoke should be strongly encouraged to quit.6
PNEUMOCOCCAL AND INFLUENZA VACCINATIONS
In a large retrospective study,8 pneumococcal vaccination was associated with a significantly lower risk of hospitalization for pneumonia in patients with chronic lung disease, including those with COPD (relative risk [RR] 0.57, 95% CI 0.38–0.84). The benefit was even greater with pneumococcal and influenza vaccinations during the influenza season (RR 0.28, 95% CI 0.14–0.58).
Randomized controlled trials indicate that influenza vaccination may reduce the rate of COPD exacerbations, especially in epidemic years when the proportion of exacerbations due to influenza is higher.9
Wongsurakiat et al10 found a significant reduction in the incidence of influenza-related acute respiratory illness in COPD patients in a well-designed randomized, placebo-controlled trial (RR 0.24, P = .005).10
Similarly, in another randomized controlled trial, pneumococcal vaccination was effective in preventing community-acquired pneumonia in COPD patients under age 65 and in those with severe airflow obstruction, although no statistically significant differences were found among other groups of patients with COPD.11
Therefore, influenza and pneumococcal vaccinations are recommended by major COPD guidelines, such as GOLD (Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease).6
INHALERS
Inhaler therapy is recommended based on COPD severity according to GOLD classification, and appropriate inhaler therapy with proper inhaler technique reduces the number of COPD exacerbations and hospitalizations.6
Long-acting beta-agonists and anticholinergics reduce the risk of COPD exacerbation and hospitalization and so are preferred over short-acting formulations except for patients in GOLD group A, ie, those who have few symptoms and are at low risk of exacerbations.6
Long-term treatment with inhaled corticosteroids with long-acting bronchodilators is recommended for patients at high risk of exacerbations (ie, those with two or more exacerbations in the previous year or a forced expiratory volume in 1 second [FEV1] less than 50% of predicted).6
OXYGEN THERAPY
Two older randomized controlled trials, the Nocturnal Oxygen Therapy Trial and the Medical Research Council study, reviewed by Stoller et al,12 provided clear evidence that oxygen therapy reduces the death rate and improves quality of life in COPD patients who have chronic resting hypoxemia (room air Pao2 ≤ 55 mm Hg, or ≤ 59 mm Hg with signs of right-sided heart strain or polycythemia).
PULMONARY REHABILITATION
Pulmonary rehabilitation likely reduces hospital admissions by improving exercise capacity.13 A systematic review of six trials in 230 patients found that respiratory rehabilitation after an acute COPD exacerbation reduced the risk of COPD hospital admission (RR 0.26, 95% CI 0.12–0.54) and the risk of death (RR 0.45, 95% CI 0.22–0.91).13
OTHER INTERVENTIONS
Home noninvasive ventilator support reduced hospital and intensive care unit readmissions in select patients recurrently hospitalized for acidotic exacerbations of COPD in one small study.14
Long-term antibiotic therapy. Although there is evidence that azithromycin, taken daily for 1 year, decreases the frequency of COPD exacerbations,15 concern persists that this approach promotes antibiotic resistance, and the GOLD guidelines do not recommend routinely using antibiotics in patients with clinically stable COPD.6
Roflumilast. According to the GOLD guidelines, the phosphodiesterase-4 inhibitor roflumilast (Daliresp) may be useful in reducing exacerbations in patients who have an FEV1 less than 50% of predicted, chronic bronchitis, and frequent exacerbations.6
Referral. Patients who have severe recurrent COPD exacerbations despite appropriate therapy will likely benefit from referral to a pulmonary specialist for other options such as theophylline, lung-reduction surgery, and lung transplantation.
PATIENT EDUCATION AND OUTPATIENT COPD PROGRAMS
There is growing evidence that outpatient programs that provide education and medical support significantly reduce the rate of hospitalizations for COPD.16–18 Patient education includes symptom monitoring, early recognition of an exacerbation, appropriate use of inhalers and nebulizers, and advice on smoking cessation.16
On the other hand, a Veterans Administration randomized controlled trial was stopped early because of a higher rate of death in the group that underwent a comprehensive care-management program of COPD education, an action plan for identification and treatment of exacerbations, and scheduled proactive telephone calls for case management.19
Further study is needed to investigate the cost-effectiveness and safety of COPD management programs and whether to adopt such programs on a systematic level.
In conclusion, COPD patients require a comprehensive approach based on studied interventions. This may be achieved through systematic methods that allow each patient to benefit from all possible interventions appropriate for him or her. Hospitalization of COPD patients provides an excellent opportunity to implement this comprehensive approach.
- Westert GP, Lagoe RJ, Keskimäki I, Leyland A, Murphy M. An international study of hospital readmissions and related utilization in Europe and the USA. Health Policy 2002; 61:269–278.
- Halpern MT, Stanford RH, Borker R. The burden of COPD in the USA: results from the Confronting COPD survey. Respir Med 2003; 97(suppl C):S81–S89.
- Centers for Medicare and Medicaid Services. Readmissions reduction program. www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/AcuteInpatientPPS/Readmissions-Reduction-Program.html. Accessed August 9, 2014.
- Yip NH, Yuen G, Lazar EJ, et al. Analysis of hospitalizations for COPD exacerbation: opportunities for improving care. COPD 2010; 7:85–92.
- Au DH, Bryson CL, Chien JW, et al. The effects of smoking cessation on the risk of chronic obstructive pulmonary disease exacerbations. J Gen Intern Med 2009; 24:457–463.
- Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2013; 187:347–365.
- Thabane MCOPD Working Group. Smoking cessation for patients with chronic obstructive pulmonary disease (COPD): an evidence-based analysis. Ont Health Technol Assess Ser 2012; 12:1–50.
- Nichol KL, Baken L, Wuorenma J, Nelson A. The health and economic benefits associated with pneumococcal vaccination of elderly persons with chronic lung disease. Arch Intern Med 1999; 159:2437–2442.
- Poole PJ, Chacko E, Wood-Baker RW, Cates CJ. Influenza vaccine for patients with chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2006; 1:CD002733.
- Wongsurakiat P, Maranetra KN, Wasi C, Kositanont U, Dejsomritrutai W, Charoenratanakul S. Acute respiratory illness in patients with COPD and the effectiveness of influenza vaccination: a randomized controlled study. Chest 2004; 125:2011–2020.
- Alfageme I, Vazquez R, Reyes N, et al. Clinical efficacy of anti-pneumococcal vaccination in patients with COPD. Thorax 2006; 61:189–195.
- Stoller JK, Panos RJ, Krachman S, Doherty DE, Make B; Long-term Oxygen Treatment Trial Research Group. Oxygen therapy for patients with COPD: current evidence and the long-term oxygen treatment trial. Chest 2010; 138:179–187.
- Puhan MA, Scharplatz M, Troosters T, Steurer J. Respiratory rehabilitation after acute exacerbation of COPD may reduce risk for readmission and mortality—a systematic review. Respir Res 2005; 6:54.
- Tuggey JM, Plant PK, Elliott MW. Domiciliary non-invasive ventilation for recurrent acidotic exacerbations of COPD: an economic analysis. Thorax 2003; 58:867–871.
- Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
- Lawlor M, Kealy S, Agnew M, et al. Early discharge care with ongoing follow-up support may reduce hospital readmissions in COPD. Int J Chron Obstruct Pulmon Dis 2009; 4:55–60.
- Gadoury MA, Schwartzman K, Rouleau M, et al; Chronic Obstructive Pulmonary Disease axis of the Respiratory Health Network, Fonds de la Recherche en Santé du Québec (FRSQ). Self-management reduces both short- and long-term hospitalisation in COPD. Eur Respir J 2005; 26:853–857.
- Rice KL, Dewan N, Bloomfield HE, et al. Disease management program for chronic obstructive pulmonary disease: a randomized controlled trial. Am J Respir Crit Care Med 2010; 182:890–896.
- Fan VS, Gaziano JM, Lew R, et al. A comprehensive care management program to prevent chronic obstructive pulmonary disease hospitalizations: a randomized, controlled trial. Ann Intern Med 2012; 156:673–683.
- COPD Working Group. Noninvasive positive pressure ventilation for chronic respiratory failure patients with stable chronic obstructive pulmonary disease (COPD): an evidence-based analysis. Ont Health Technol Assess Ser 2012; 12( 9):1–51.
We think so. Some strategies to reduce readmission rates, such as coordinating care and managing comorbidities, apply to chronic diseases in general, while others are disease-specific. To reduce the need for hospital readmission for chronic obstructive pulmonary disease (COPD), coordinated efforts involving both inpatient and outpatient care are necessary. This can be achieved by using a checklist before discharge (Table 1) and by implementing outpatient COPD programs that continue patient education and provide rapid access to medical support if needed.
There is room for improvement. COPD is common and expensive, with high rates of hospital readmission,1 and up to 70% of the money we spend on it goes for hospital care.2 No wonder then that the Centers for Medicare and Medicaid Services has now expanded its Readmissions Reduction Program to include acute COPD exacerbations.3 Yet in a retrospective study, Yip et al4 found that fewer than half of patients hospitalized with acute exacerbation of COPD received appropriate vaccinations, counseling on smoking cessation, and long-acting inhalers—all of which are on our checklist.4
The following interventions have been demonstrated to be useful in reducing COPD hospital admissions and the risk of death.
SMOKING CESSATION
Cigarette smoking is the most common and easily identifiable risk factor for COPD exacerbation.5
Au et al5 found that quitting smoking reduces the risk of COPD exacerbation (adjusted hazard ratio 0.78, 95% confidence interval [CI] 0.75–0.87), and the risk keeps decreasing the longer the patient stays off tobacco.5
Whether counseling hospitalized patients on smoking cessation reduces the COPD readmission rate has not been well studied. However, a meta-analysis of nine randomized controlled trials, two of which were done in the hospital, revealed higher abstinence rates in COPD patients who received extensive counseling on smoking cessation.7 For these reasons, hospitalized COPD patients who smoke should be strongly encouraged to quit.6
PNEUMOCOCCAL AND INFLUENZA VACCINATIONS
In a large retrospective study,8 pneumococcal vaccination was associated with a significantly lower risk of hospitalization for pneumonia in patients with chronic lung disease, including those with COPD (relative risk [RR] 0.57, 95% CI 0.38–0.84). The benefit was even greater with pneumococcal and influenza vaccinations during the influenza season (RR 0.28, 95% CI 0.14–0.58).
Randomized controlled trials indicate that influenza vaccination may reduce the rate of COPD exacerbations, especially in epidemic years when the proportion of exacerbations due to influenza is higher.9
Wongsurakiat et al10 found a significant reduction in the incidence of influenza-related acute respiratory illness in COPD patients in a well-designed randomized, placebo-controlled trial (RR 0.24, P = .005).10
Similarly, in another randomized controlled trial, pneumococcal vaccination was effective in preventing community-acquired pneumonia in COPD patients under age 65 and in those with severe airflow obstruction, although no statistically significant differences were found among other groups of patients with COPD.11
Therefore, influenza and pneumococcal vaccinations are recommended by major COPD guidelines, such as GOLD (Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease).6
INHALERS
Inhaler therapy is recommended based on COPD severity according to GOLD classification, and appropriate inhaler therapy with proper inhaler technique reduces the number of COPD exacerbations and hospitalizations.6
Long-acting beta-agonists and anticholinergics reduce the risk of COPD exacerbation and hospitalization and so are preferred over short-acting formulations except for patients in GOLD group A, ie, those who have few symptoms and are at low risk of exacerbations.6
Long-term treatment with inhaled corticosteroids with long-acting bronchodilators is recommended for patients at high risk of exacerbations (ie, those with two or more exacerbations in the previous year or a forced expiratory volume in 1 second [FEV1] less than 50% of predicted).6
OXYGEN THERAPY
Two older randomized controlled trials, the Nocturnal Oxygen Therapy Trial and the Medical Research Council study, reviewed by Stoller et al,12 provided clear evidence that oxygen therapy reduces the death rate and improves quality of life in COPD patients who have chronic resting hypoxemia (room air Pao2 ≤ 55 mm Hg, or ≤ 59 mm Hg with signs of right-sided heart strain or polycythemia).
PULMONARY REHABILITATION
Pulmonary rehabilitation likely reduces hospital admissions by improving exercise capacity.13 A systematic review of six trials in 230 patients found that respiratory rehabilitation after an acute COPD exacerbation reduced the risk of COPD hospital admission (RR 0.26, 95% CI 0.12–0.54) and the risk of death (RR 0.45, 95% CI 0.22–0.91).13
OTHER INTERVENTIONS
Home noninvasive ventilator support reduced hospital and intensive care unit readmissions in select patients recurrently hospitalized for acidotic exacerbations of COPD in one small study.14
Long-term antibiotic therapy. Although there is evidence that azithromycin, taken daily for 1 year, decreases the frequency of COPD exacerbations,15 concern persists that this approach promotes antibiotic resistance, and the GOLD guidelines do not recommend routinely using antibiotics in patients with clinically stable COPD.6
Roflumilast. According to the GOLD guidelines, the phosphodiesterase-4 inhibitor roflumilast (Daliresp) may be useful in reducing exacerbations in patients who have an FEV1 less than 50% of predicted, chronic bronchitis, and frequent exacerbations.6
Referral. Patients who have severe recurrent COPD exacerbations despite appropriate therapy will likely benefit from referral to a pulmonary specialist for other options such as theophylline, lung-reduction surgery, and lung transplantation.
PATIENT EDUCATION AND OUTPATIENT COPD PROGRAMS
There is growing evidence that outpatient programs that provide education and medical support significantly reduce the rate of hospitalizations for COPD.16–18 Patient education includes symptom monitoring, early recognition of an exacerbation, appropriate use of inhalers and nebulizers, and advice on smoking cessation.16
On the other hand, a Veterans Administration randomized controlled trial was stopped early because of a higher rate of death in the group that underwent a comprehensive care-management program of COPD education, an action plan for identification and treatment of exacerbations, and scheduled proactive telephone calls for case management.19
Further study is needed to investigate the cost-effectiveness and safety of COPD management programs and whether to adopt such programs on a systematic level.
In conclusion, COPD patients require a comprehensive approach based on studied interventions. This may be achieved through systematic methods that allow each patient to benefit from all possible interventions appropriate for him or her. Hospitalization of COPD patients provides an excellent opportunity to implement this comprehensive approach.
We think so. Some strategies to reduce readmission rates, such as coordinating care and managing comorbidities, apply to chronic diseases in general, while others are disease-specific. To reduce the need for hospital readmission for chronic obstructive pulmonary disease (COPD), coordinated efforts involving both inpatient and outpatient care are necessary. This can be achieved by using a checklist before discharge (Table 1) and by implementing outpatient COPD programs that continue patient education and provide rapid access to medical support if needed.
There is room for improvement. COPD is common and expensive, with high rates of hospital readmission,1 and up to 70% of the money we spend on it goes for hospital care.2 No wonder then that the Centers for Medicare and Medicaid Services has now expanded its Readmissions Reduction Program to include acute COPD exacerbations.3 Yet in a retrospective study, Yip et al4 found that fewer than half of patients hospitalized with acute exacerbation of COPD received appropriate vaccinations, counseling on smoking cessation, and long-acting inhalers—all of which are on our checklist.4
The following interventions have been demonstrated to be useful in reducing COPD hospital admissions and the risk of death.
SMOKING CESSATION
Cigarette smoking is the most common and easily identifiable risk factor for COPD exacerbation.5
Au et al5 found that quitting smoking reduces the risk of COPD exacerbation (adjusted hazard ratio 0.78, 95% confidence interval [CI] 0.75–0.87), and the risk keeps decreasing the longer the patient stays off tobacco.5
Whether counseling hospitalized patients on smoking cessation reduces the COPD readmission rate has not been well studied. However, a meta-analysis of nine randomized controlled trials, two of which were done in the hospital, revealed higher abstinence rates in COPD patients who received extensive counseling on smoking cessation.7 For these reasons, hospitalized COPD patients who smoke should be strongly encouraged to quit.6
PNEUMOCOCCAL AND INFLUENZA VACCINATIONS
In a large retrospective study,8 pneumococcal vaccination was associated with a significantly lower risk of hospitalization for pneumonia in patients with chronic lung disease, including those with COPD (relative risk [RR] 0.57, 95% CI 0.38–0.84). The benefit was even greater with pneumococcal and influenza vaccinations during the influenza season (RR 0.28, 95% CI 0.14–0.58).
Randomized controlled trials indicate that influenza vaccination may reduce the rate of COPD exacerbations, especially in epidemic years when the proportion of exacerbations due to influenza is higher.9
Wongsurakiat et al10 found a significant reduction in the incidence of influenza-related acute respiratory illness in COPD patients in a well-designed randomized, placebo-controlled trial (RR 0.24, P = .005).10
Similarly, in another randomized controlled trial, pneumococcal vaccination was effective in preventing community-acquired pneumonia in COPD patients under age 65 and in those with severe airflow obstruction, although no statistically significant differences were found among other groups of patients with COPD.11
Therefore, influenza and pneumococcal vaccinations are recommended by major COPD guidelines, such as GOLD (Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease).6
INHALERS
Inhaler therapy is recommended based on COPD severity according to GOLD classification, and appropriate inhaler therapy with proper inhaler technique reduces the number of COPD exacerbations and hospitalizations.6
Long-acting beta-agonists and anticholinergics reduce the risk of COPD exacerbation and hospitalization and so are preferred over short-acting formulations except for patients in GOLD group A, ie, those who have few symptoms and are at low risk of exacerbations.6
Long-term treatment with inhaled corticosteroids with long-acting bronchodilators is recommended for patients at high risk of exacerbations (ie, those with two or more exacerbations in the previous year or a forced expiratory volume in 1 second [FEV1] less than 50% of predicted).6
OXYGEN THERAPY
Two older randomized controlled trials, the Nocturnal Oxygen Therapy Trial and the Medical Research Council study, reviewed by Stoller et al,12 provided clear evidence that oxygen therapy reduces the death rate and improves quality of life in COPD patients who have chronic resting hypoxemia (room air Pao2 ≤ 55 mm Hg, or ≤ 59 mm Hg with signs of right-sided heart strain or polycythemia).
PULMONARY REHABILITATION
Pulmonary rehabilitation likely reduces hospital admissions by improving exercise capacity.13 A systematic review of six trials in 230 patients found that respiratory rehabilitation after an acute COPD exacerbation reduced the risk of COPD hospital admission (RR 0.26, 95% CI 0.12–0.54) and the risk of death (RR 0.45, 95% CI 0.22–0.91).13
OTHER INTERVENTIONS
Home noninvasive ventilator support reduced hospital and intensive care unit readmissions in select patients recurrently hospitalized for acidotic exacerbations of COPD in one small study.14
Long-term antibiotic therapy. Although there is evidence that azithromycin, taken daily for 1 year, decreases the frequency of COPD exacerbations,15 concern persists that this approach promotes antibiotic resistance, and the GOLD guidelines do not recommend routinely using antibiotics in patients with clinically stable COPD.6
Roflumilast. According to the GOLD guidelines, the phosphodiesterase-4 inhibitor roflumilast (Daliresp) may be useful in reducing exacerbations in patients who have an FEV1 less than 50% of predicted, chronic bronchitis, and frequent exacerbations.6
Referral. Patients who have severe recurrent COPD exacerbations despite appropriate therapy will likely benefit from referral to a pulmonary specialist for other options such as theophylline, lung-reduction surgery, and lung transplantation.
PATIENT EDUCATION AND OUTPATIENT COPD PROGRAMS
There is growing evidence that outpatient programs that provide education and medical support significantly reduce the rate of hospitalizations for COPD.16–18 Patient education includes symptom monitoring, early recognition of an exacerbation, appropriate use of inhalers and nebulizers, and advice on smoking cessation.16
On the other hand, a Veterans Administration randomized controlled trial was stopped early because of a higher rate of death in the group that underwent a comprehensive care-management program of COPD education, an action plan for identification and treatment of exacerbations, and scheduled proactive telephone calls for case management.19
Further study is needed to investigate the cost-effectiveness and safety of COPD management programs and whether to adopt such programs on a systematic level.
In conclusion, COPD patients require a comprehensive approach based on studied interventions. This may be achieved through systematic methods that allow each patient to benefit from all possible interventions appropriate for him or her. Hospitalization of COPD patients provides an excellent opportunity to implement this comprehensive approach.
- Westert GP, Lagoe RJ, Keskimäki I, Leyland A, Murphy M. An international study of hospital readmissions and related utilization in Europe and the USA. Health Policy 2002; 61:269–278.
- Halpern MT, Stanford RH, Borker R. The burden of COPD in the USA: results from the Confronting COPD survey. Respir Med 2003; 97(suppl C):S81–S89.
- Centers for Medicare and Medicaid Services. Readmissions reduction program. www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/AcuteInpatientPPS/Readmissions-Reduction-Program.html. Accessed August 9, 2014.
- Yip NH, Yuen G, Lazar EJ, et al. Analysis of hospitalizations for COPD exacerbation: opportunities for improving care. COPD 2010; 7:85–92.
- Au DH, Bryson CL, Chien JW, et al. The effects of smoking cessation on the risk of chronic obstructive pulmonary disease exacerbations. J Gen Intern Med 2009; 24:457–463.
- Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2013; 187:347–365.
- Thabane MCOPD Working Group. Smoking cessation for patients with chronic obstructive pulmonary disease (COPD): an evidence-based analysis. Ont Health Technol Assess Ser 2012; 12:1–50.
- Nichol KL, Baken L, Wuorenma J, Nelson A. The health and economic benefits associated with pneumococcal vaccination of elderly persons with chronic lung disease. Arch Intern Med 1999; 159:2437–2442.
- Poole PJ, Chacko E, Wood-Baker RW, Cates CJ. Influenza vaccine for patients with chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2006; 1:CD002733.
- Wongsurakiat P, Maranetra KN, Wasi C, Kositanont U, Dejsomritrutai W, Charoenratanakul S. Acute respiratory illness in patients with COPD and the effectiveness of influenza vaccination: a randomized controlled study. Chest 2004; 125:2011–2020.
- Alfageme I, Vazquez R, Reyes N, et al. Clinical efficacy of anti-pneumococcal vaccination in patients with COPD. Thorax 2006; 61:189–195.
- Stoller JK, Panos RJ, Krachman S, Doherty DE, Make B; Long-term Oxygen Treatment Trial Research Group. Oxygen therapy for patients with COPD: current evidence and the long-term oxygen treatment trial. Chest 2010; 138:179–187.
- Puhan MA, Scharplatz M, Troosters T, Steurer J. Respiratory rehabilitation after acute exacerbation of COPD may reduce risk for readmission and mortality—a systematic review. Respir Res 2005; 6:54.
- Tuggey JM, Plant PK, Elliott MW. Domiciliary non-invasive ventilation for recurrent acidotic exacerbations of COPD: an economic analysis. Thorax 2003; 58:867–871.
- Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
- Lawlor M, Kealy S, Agnew M, et al. Early discharge care with ongoing follow-up support may reduce hospital readmissions in COPD. Int J Chron Obstruct Pulmon Dis 2009; 4:55–60.
- Gadoury MA, Schwartzman K, Rouleau M, et al; Chronic Obstructive Pulmonary Disease axis of the Respiratory Health Network, Fonds de la Recherche en Santé du Québec (FRSQ). Self-management reduces both short- and long-term hospitalisation in COPD. Eur Respir J 2005; 26:853–857.
- Rice KL, Dewan N, Bloomfield HE, et al. Disease management program for chronic obstructive pulmonary disease: a randomized controlled trial. Am J Respir Crit Care Med 2010; 182:890–896.
- Fan VS, Gaziano JM, Lew R, et al. A comprehensive care management program to prevent chronic obstructive pulmonary disease hospitalizations: a randomized, controlled trial. Ann Intern Med 2012; 156:673–683.
- COPD Working Group. Noninvasive positive pressure ventilation for chronic respiratory failure patients with stable chronic obstructive pulmonary disease (COPD): an evidence-based analysis. Ont Health Technol Assess Ser 2012; 12( 9):1–51.
- Westert GP, Lagoe RJ, Keskimäki I, Leyland A, Murphy M. An international study of hospital readmissions and related utilization in Europe and the USA. Health Policy 2002; 61:269–278.
- Halpern MT, Stanford RH, Borker R. The burden of COPD in the USA: results from the Confronting COPD survey. Respir Med 2003; 97(suppl C):S81–S89.
- Centers for Medicare and Medicaid Services. Readmissions reduction program. www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/AcuteInpatientPPS/Readmissions-Reduction-Program.html. Accessed August 9, 2014.
- Yip NH, Yuen G, Lazar EJ, et al. Analysis of hospitalizations for COPD exacerbation: opportunities for improving care. COPD 2010; 7:85–92.
- Au DH, Bryson CL, Chien JW, et al. The effects of smoking cessation on the risk of chronic obstructive pulmonary disease exacerbations. J Gen Intern Med 2009; 24:457–463.
- Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2013; 187:347–365.
- Thabane MCOPD Working Group. Smoking cessation for patients with chronic obstructive pulmonary disease (COPD): an evidence-based analysis. Ont Health Technol Assess Ser 2012; 12:1–50.
- Nichol KL, Baken L, Wuorenma J, Nelson A. The health and economic benefits associated with pneumococcal vaccination of elderly persons with chronic lung disease. Arch Intern Med 1999; 159:2437–2442.
- Poole PJ, Chacko E, Wood-Baker RW, Cates CJ. Influenza vaccine for patients with chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2006; 1:CD002733.
- Wongsurakiat P, Maranetra KN, Wasi C, Kositanont U, Dejsomritrutai W, Charoenratanakul S. Acute respiratory illness in patients with COPD and the effectiveness of influenza vaccination: a randomized controlled study. Chest 2004; 125:2011–2020.
- Alfageme I, Vazquez R, Reyes N, et al. Clinical efficacy of anti-pneumococcal vaccination in patients with COPD. Thorax 2006; 61:189–195.
- Stoller JK, Panos RJ, Krachman S, Doherty DE, Make B; Long-term Oxygen Treatment Trial Research Group. Oxygen therapy for patients with COPD: current evidence and the long-term oxygen treatment trial. Chest 2010; 138:179–187.
- Puhan MA, Scharplatz M, Troosters T, Steurer J. Respiratory rehabilitation after acute exacerbation of COPD may reduce risk for readmission and mortality—a systematic review. Respir Res 2005; 6:54.
- Tuggey JM, Plant PK, Elliott MW. Domiciliary non-invasive ventilation for recurrent acidotic exacerbations of COPD: an economic analysis. Thorax 2003; 58:867–871.
- Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
- Lawlor M, Kealy S, Agnew M, et al. Early discharge care with ongoing follow-up support may reduce hospital readmissions in COPD. Int J Chron Obstruct Pulmon Dis 2009; 4:55–60.
- Gadoury MA, Schwartzman K, Rouleau M, et al; Chronic Obstructive Pulmonary Disease axis of the Respiratory Health Network, Fonds de la Recherche en Santé du Québec (FRSQ). Self-management reduces both short- and long-term hospitalisation in COPD. Eur Respir J 2005; 26:853–857.
- Rice KL, Dewan N, Bloomfield HE, et al. Disease management program for chronic obstructive pulmonary disease: a randomized controlled trial. Am J Respir Crit Care Med 2010; 182:890–896.
- Fan VS, Gaziano JM, Lew R, et al. A comprehensive care management program to prevent chronic obstructive pulmonary disease hospitalizations: a randomized, controlled trial. Ann Intern Med 2012; 156:673–683.
- COPD Working Group. Noninvasive positive pressure ventilation for chronic respiratory failure patients with stable chronic obstructive pulmonary disease (COPD): an evidence-based analysis. Ont Health Technol Assess Ser 2012; 12( 9):1–51.
Do patients who received only two doses of hepatitis B vaccine need a booster?
The Advisory Committee on Immunization Practices (ACIP) currently recommends that people who have not completed the three-dose vaccination series against hepatitis B virus (HBV) should receive the missed doses: ie, the three-dose regimen does not need to be restarted.1 However, evidence suggests that a two-dose regimen may provide adequate seroprotection for healthy young adults.
As the three-dose regimen has been shown to protect 90% to 100% of adults,2 it has gained widespread acceptance and is now standard clinical practice.2 However, deviating from the three-dose regimen may not leave healthy young adults vulnerable to HBV infection.
RECOMMENDED DOSES AND SCHEDULES
Widespread use of the three-dose regimen for HBV stemmed from the first clinical evaluation of the recombinant vaccine, in which three 10-μg doses were given at 0, 1, and 6 months to healthy, low-risk adult volunteers.3 This regimen was shown to provide seroprotection in over 95% of adolescents and 90% of healthy adults.2

Currently, three HBV vaccines for adults are approved in the United States: Recombivax HB, Engerix-B, and Twinrix (Table 1). While Recombivax has a seroprotection rate of 89% in healthy adults over age 40, it has higher seroprotection rates in younger people: eg, two doses of Recombivax given 4 to 6 months apart provide seroprotection to 99% of children aged 11 to 15.4 On the other hand, patients on hemodialysis require three 40-μg doses of Recombivax or four 40-μg doses of Engerix-B.
Evidence for a two-dose regimen
Since the development of the recombinant HBV vaccine used today, studies have shown that a two-dose regimen offers seroprotection comparable with, if not better than, the three-dose regimen in adolescents and healthy young adults. Marsano et al5 found that with a two-dose regimen, 96% to 99% of young adults attained seroprotection, with immune memory persisting for up to 2 years.5 Moreover, Cassidy et al6 randomized adolescents to a two-dose or a three-dose regimen and found the two regimens to be equally effective in conferring immunogenicity and immunologic memory.6
Other studies in adolescents have confirmed these findings and offered new evidence in support of the two-dose regimen.7,8 For example, studies found that the two-dose regimen conferred seroprotection at even lower doses than previously studied, and that it conferred immune memory lasting at least 5 years.6,7
However, because these studies were conducted in adolescents and healthy young adults, the findings may not hold true for other populations. Studies suggest that the three-dose regimen is best for those over age 40. Moreover, it is advisable to adhere to a three-dose regimen when treating people at high risk of contracting HBV, such as health care workers; people with chronic liver disease, diabetes mellitus, or end-stage renal disease on hemodialysis; people who have multiple sex partners; and men who have sex with men.
The impact of long intervals between doses
Although the aforementioned studies focused on a two-dose regimen with a 6-month interval, longer intervals between doses do not impair seroprotection and in some cases may even prove beneficial. Heron et al9 demonstrated that a two-dose regimen with a 12-month interval induces seroprotection as effectively as a standard three-dose or two-dose regimen with a 6-month interval.9 Moreover, studies of the impact of deviating from a three-dose regimen found that intervals of longer than 1 year did not impede seroprotection. Not only may seroprotection be attained with intervals of 5 to 10 years before the final dose, but final antibody levels tend to increase with increasing time between doses.10
Nevertheless, even though an extended interval between doses may prove beneficial after the final dose is received, delaying doses may leave patients unprotected. Indeed, alternative three-dose and even four-dose schedules with shorter intervals between doses exist for certain high-risk populations, such as those recently exposed to HBV and travelers to areas of high prevalence. Therefore, intentionally extending intervals between doses may be inappropriate.
SEROPROTECTION AND PROTECTION AGAINST INFECTION
Legitimate concerns exist about the final antibody level attained with a two-dose regimen, which is typically lower than that attained with a three-dose regimen. As HBV antibody levels decline with time, lower final antibody levels theoretically increase the risk of losing seroprotection. Study of vaccine efficacy has defined seroprotection as antibody levels greater than or equal to 10 mIU/mL.11 Yet evidence suggests that even when antibody levels drop below this level, the risk of symptomatic HBV infection does not increase. Evidence also suggests that immune memory outlasts the presence of seroprotective antibody levels, indicating that true protection against significant infection does not necessarily correlate with, and may even exceed, seroprotection.2 This may relate to HBV’s long incubation period, which allows memory cells time to generate an effective immune response.10 For example, Floreani et al12 showed that even though 15% of adults lost seroprotective antibody levels 10 years after vaccination, none demonstrated hepatitis B antigen reactivity or seroconversion.
POSTVACCINATION TESTING AND ADDITIONAL DOSES
At times, it may be wise to measure antibody levels after the final dose to confirm seroprotection. Seroprotection should be documented when knowledge of the patient’s immune status will affect subsequent management. As recommended by the US Centers for Disease Control and Prevention, health care workers, hemodialysis patients, immunocompromised patients, and sexual partners of patients with chronic HBV infection should undergo antibody testing 1 to 2 months after the completion of a three-dose vaccination regimen. Hemodialysis patients require annual confirmation of seroprotection and should receive booster doses of HBV vaccine if necessary.
Postvaccination testing (quantitative HBV surface antibody testing) costs about the same as a single dose of HBV vaccine. Therefore, if postvaccination testing is considered because of missed vaccine doses, it may be more cost-efficient to simply administer the missed dose.
- Department of Health and Human Services. Appendix A Immunization Management Issues. http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5516a2.htm. Accessed April 6, 2014.
- Leuridan E, Van Damme P. Hepatitis B and the need for a booster dose. Clin Infect Dis 2011; 53:68–75.
- Scolnick EM, McLean AA, West DJ, McAleer WJ, Miller WJ, Buynak EB. Clinical evaluation in healthy adults of a hepatitis B vaccine made by recombinant DNA. JAMA 1984; 251:2812–2815.
- Merck and Co, Inc. 1998. Recombivax HB. http://www.merck.com/product/usa/pi_circulars/r/recombivax_hb/re-combivax_pi.pdf. Accessed April 7, 2014.
- Marsano LS, West DJ, Chan I, et al. A two-dose hepatitis B vaccine regimen: proof of priming and memory responses in young adults. Vaccine 1998; 16:624–629.
- Cassidy WM, Watson B, Ioli VA, Williams K, Bird S, West DJ. A randomized trial of alternative two- and three-dose hepatitis B vaccination regimens in adolescents: antibody responses, safety, and immunologic memory. Pediatrics 2001; 107:626–631.
- Van Damme P, Moiseeva A, Marichev I, et al. Five years follow-up following two or three doses of a hepatitis B vaccine in adolescents aged 11–15 years: a randomised controlled study. BMC Infect Dis 2010; 10:357.
- Heron L, Selnikova O, Moiseieva A, et al. Immunogenicity, reactogenicity and safety of two-dose versus three-dose (standard care) hepatitis B immunisation of healthy adolescents aged 11–15 years: a randomised controlled trial. Vaccine 2007; 25:2817–2822.
- Heron LG, Chant KG, Jalaludin BB. A novel hepatitis B vaccination regimen for adolescents: two doses 12 months apart. Vaccine 2002; 20:3472–3476.
- Jackson Y, Chappuis F, Mezger N, Kanappa K, Loutan L. High immunogenicity of delayed third dose of hepatitis B vaccine in travellers. Vaccine 2007; 25:3482–3484.
- Jack AD, Hall AJ, Maine N, Mendy M, Whittle HC. What level of hepatitis B antibody is protective? J Infect Dis 1999; 179:489–492.
- Floreani A, Baldo V, Cristofoletti M, et al. Long-term persistence of anti-HBs after vaccination against HBV: an 18 year experience in health care workers. Vaccine 2004; 22:607–610.
The Advisory Committee on Immunization Practices (ACIP) currently recommends that people who have not completed the three-dose vaccination series against hepatitis B virus (HBV) should receive the missed doses: ie, the three-dose regimen does not need to be restarted.1 However, evidence suggests that a two-dose regimen may provide adequate seroprotection for healthy young adults.
As the three-dose regimen has been shown to protect 90% to 100% of adults,2 it has gained widespread acceptance and is now standard clinical practice.2 However, deviating from the three-dose regimen may not leave healthy young adults vulnerable to HBV infection.
RECOMMENDED DOSES AND SCHEDULES
Widespread use of the three-dose regimen for HBV stemmed from the first clinical evaluation of the recombinant vaccine, in which three 10-μg doses were given at 0, 1, and 6 months to healthy, low-risk adult volunteers.3 This regimen was shown to provide seroprotection in over 95% of adolescents and 90% of healthy adults.2
Currently, three HBV vaccines for adults are approved in the United States: Recombivax HB, Engerix-B, and Twinrix (Table 1). While Recombivax has a seroprotection rate of 89% in healthy adults over age 40, it has higher seroprotection rates in younger people: eg, two doses of Recombivax given 4 to 6 months apart provide seroprotection to 99% of children aged 11 to 15.4 On the other hand, patients on hemodialysis require three 40-μg doses of Recombivax or four 40-μg doses of Engerix-B.
Evidence for a two-dose regimen
Since the development of the recombinant HBV vaccine used today, studies have shown that a two-dose regimen offers seroprotection comparable with, if not better than, the three-dose regimen in adolescents and healthy young adults. Marsano et al5 found that with a two-dose regimen, 96% to 99% of young adults attained seroprotection, with immune memory persisting for up to 2 years.5 Moreover, Cassidy et al6 randomized adolescents to a two-dose or a three-dose regimen and found the two regimens to be equally effective in conferring immunogenicity and immunologic memory.6
Other studies in adolescents have confirmed these findings and offered new evidence in support of the two-dose regimen.7,8 For example, studies found that the two-dose regimen conferred seroprotection at even lower doses than previously studied, and that it conferred immune memory lasting at least 5 years.6,7
However, because these studies were conducted in adolescents and healthy young adults, the findings may not hold true for other populations. Studies suggest that the three-dose regimen is best for those over age 40. Moreover, it is advisable to adhere to a three-dose regimen when treating people at high risk of contracting HBV, such as health care workers; people with chronic liver disease, diabetes mellitus, or end-stage renal disease on hemodialysis; people who have multiple sex partners; and men who have sex with men.
The impact of long intervals between doses
Although the aforementioned studies focused on a two-dose regimen with a 6-month interval, longer intervals between doses do not impair seroprotection and in some cases may even prove beneficial. Heron et al9 demonstrated that a two-dose regimen with a 12-month interval induces seroprotection as effectively as a standard three-dose or two-dose regimen with a 6-month interval.9 Moreover, studies of the impact of deviating from a three-dose regimen found that intervals of longer than 1 year did not impede seroprotection. Not only may seroprotection be attained with intervals of 5 to 10 years before the final dose, but final antibody levels tend to increase with increasing time between doses.10
Nevertheless, even though an extended interval between doses may prove beneficial after the final dose is received, delaying doses may leave patients unprotected. Indeed, alternative three-dose and even four-dose schedules with shorter intervals between doses exist for certain high-risk populations, such as those recently exposed to HBV and travelers to areas of high prevalence. Therefore, intentionally extending intervals between doses may be inappropriate.
SEROPROTECTION AND PROTECTION AGAINST INFECTION
Legitimate concerns exist about the final antibody level attained with a two-dose regimen, which is typically lower than that attained with a three-dose regimen. As HBV antibody levels decline with time, lower final antibody levels theoretically increase the risk of losing seroprotection. Study of vaccine efficacy has defined seroprotection as antibody levels greater than or equal to 10 mIU/mL.11 Yet evidence suggests that even when antibody levels drop below this level, the risk of symptomatic HBV infection does not increase. Evidence also suggests that immune memory outlasts the presence of seroprotective antibody levels, indicating that true protection against significant infection does not necessarily correlate with, and may even exceed, seroprotection.2 This may relate to HBV’s long incubation period, which allows memory cells time to generate an effective immune response.10 For example, Floreani et al12 showed that even though 15% of adults lost seroprotective antibody levels 10 years after vaccination, none demonstrated hepatitis B antigen reactivity or seroconversion.
POSTVACCINATION TESTING AND ADDITIONAL DOSES
At times, it may be wise to measure antibody levels after the final dose to confirm seroprotection. Seroprotection should be documented when knowledge of the patient’s immune status will affect subsequent management. As recommended by the US Centers for Disease Control and Prevention, health care workers, hemodialysis patients, immunocompromised patients, and sexual partners of patients with chronic HBV infection should undergo antibody testing 1 to 2 months after the completion of a three-dose vaccination regimen. Hemodialysis patients require annual confirmation of seroprotection and should receive booster doses of HBV vaccine if necessary.
Postvaccination testing (quantitative HBV surface antibody testing) costs about the same as a single dose of HBV vaccine. Therefore, if postvaccination testing is considered because of missed vaccine doses, it may be more cost-efficient to simply administer the missed dose.
The Advisory Committee on Immunization Practices (ACIP) currently recommends that people who have not completed the three-dose vaccination series against hepatitis B virus (HBV) should receive the missed doses: ie, the three-dose regimen does not need to be restarted.1 However, evidence suggests that a two-dose regimen may provide adequate seroprotection for healthy young adults.
As the three-dose regimen has been shown to protect 90% to 100% of adults,2 it has gained widespread acceptance and is now standard clinical practice.2 However, deviating from the three-dose regimen may not leave healthy young adults vulnerable to HBV infection.
RECOMMENDED DOSES AND SCHEDULES
Widespread use of the three-dose regimen for HBV stemmed from the first clinical evaluation of the recombinant vaccine, in which three 10-μg doses were given at 0, 1, and 6 months to healthy, low-risk adult volunteers.3 This regimen was shown to provide seroprotection in over 95% of adolescents and 90% of healthy adults.2
Currently, three HBV vaccines for adults are approved in the United States: Recombivax HB, Engerix-B, and Twinrix (Table 1). While Recombivax has a seroprotection rate of 89% in healthy adults over age 40, it has higher seroprotection rates in younger people: eg, two doses of Recombivax given 4 to 6 months apart provide seroprotection to 99% of children aged 11 to 15.4 On the other hand, patients on hemodialysis require three 40-μg doses of Recombivax or four 40-μg doses of Engerix-B.
Evidence for a two-dose regimen
Since the development of the recombinant HBV vaccine used today, studies have shown that a two-dose regimen offers seroprotection comparable with, if not better than, the three-dose regimen in adolescents and healthy young adults. Marsano et al5 found that with a two-dose regimen, 96% to 99% of young adults attained seroprotection, with immune memory persisting for up to 2 years.5 Moreover, Cassidy et al6 randomized adolescents to a two-dose or a three-dose regimen and found the two regimens to be equally effective in conferring immunogenicity and immunologic memory.6
Other studies in adolescents have confirmed these findings and offered new evidence in support of the two-dose regimen.7,8 For example, studies found that the two-dose regimen conferred seroprotection at even lower doses than previously studied, and that it conferred immune memory lasting at least 5 years.6,7
However, because these studies were conducted in adolescents and healthy young adults, the findings may not hold true for other populations. Studies suggest that the three-dose regimen is best for those over age 40. Moreover, it is advisable to adhere to a three-dose regimen when treating people at high risk of contracting HBV, such as health care workers; people with chronic liver disease, diabetes mellitus, or end-stage renal disease on hemodialysis; people who have multiple sex partners; and men who have sex with men.
The impact of long intervals between doses
Although the aforementioned studies focused on a two-dose regimen with a 6-month interval, longer intervals between doses do not impair seroprotection and in some cases may even prove beneficial. Heron et al9 demonstrated that a two-dose regimen with a 12-month interval induces seroprotection as effectively as a standard three-dose or two-dose regimen with a 6-month interval.9 Moreover, studies of the impact of deviating from a three-dose regimen found that intervals of longer than 1 year did not impede seroprotection. Not only may seroprotection be attained with intervals of 5 to 10 years before the final dose, but final antibody levels tend to increase with increasing time between doses.10
Nevertheless, even though an extended interval between doses may prove beneficial after the final dose is received, delaying doses may leave patients unprotected. Indeed, alternative three-dose and even four-dose schedules with shorter intervals between doses exist for certain high-risk populations, such as those recently exposed to HBV and travelers to areas of high prevalence. Therefore, intentionally extending intervals between doses may be inappropriate.
SEROPROTECTION AND PROTECTION AGAINST INFECTION
Legitimate concerns exist about the final antibody level attained with a two-dose regimen, which is typically lower than that attained with a three-dose regimen. As HBV antibody levels decline with time, lower final antibody levels theoretically increase the risk of losing seroprotection. Study of vaccine efficacy has defined seroprotection as antibody levels greater than or equal to 10 mIU/mL.11 Yet evidence suggests that even when antibody levels drop below this level, the risk of symptomatic HBV infection does not increase. Evidence also suggests that immune memory outlasts the presence of seroprotective antibody levels, indicating that true protection against significant infection does not necessarily correlate with, and may even exceed, seroprotection.2 This may relate to HBV’s long incubation period, which allows memory cells time to generate an effective immune response.10 For example, Floreani et al12 showed that even though 15% of adults lost seroprotective antibody levels 10 years after vaccination, none demonstrated hepatitis B antigen reactivity or seroconversion.
POSTVACCINATION TESTING AND ADDITIONAL DOSES
At times, it may be wise to measure antibody levels after the final dose to confirm seroprotection. Seroprotection should be documented when knowledge of the patient’s immune status will affect subsequent management. As recommended by the US Centers for Disease Control and Prevention, health care workers, hemodialysis patients, immunocompromised patients, and sexual partners of patients with chronic HBV infection should undergo antibody testing 1 to 2 months after the completion of a three-dose vaccination regimen. Hemodialysis patients require annual confirmation of seroprotection and should receive booster doses of HBV vaccine if necessary.
Postvaccination testing (quantitative HBV surface antibody testing) costs about the same as a single dose of HBV vaccine. Therefore, if postvaccination testing is considered because of missed vaccine doses, it may be more cost-efficient to simply administer the missed dose.
- Department of Health and Human Services. Appendix A Immunization Management Issues. http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5516a2.htm. Accessed April 6, 2014.
- Leuridan E, Van Damme P. Hepatitis B and the need for a booster dose. Clin Infect Dis 2011; 53:68–75.
- Scolnick EM, McLean AA, West DJ, McAleer WJ, Miller WJ, Buynak EB. Clinical evaluation in healthy adults of a hepatitis B vaccine made by recombinant DNA. JAMA 1984; 251:2812–2815.
- Merck and Co, Inc. 1998. Recombivax HB. http://www.merck.com/product/usa/pi_circulars/r/recombivax_hb/re-combivax_pi.pdf. Accessed April 7, 2014.
- Marsano LS, West DJ, Chan I, et al. A two-dose hepatitis B vaccine regimen: proof of priming and memory responses in young adults. Vaccine 1998; 16:624–629.
- Cassidy WM, Watson B, Ioli VA, Williams K, Bird S, West DJ. A randomized trial of alternative two- and three-dose hepatitis B vaccination regimens in adolescents: antibody responses, safety, and immunologic memory. Pediatrics 2001; 107:626–631.
- Van Damme P, Moiseeva A, Marichev I, et al. Five years follow-up following two or three doses of a hepatitis B vaccine in adolescents aged 11–15 years: a randomised controlled study. BMC Infect Dis 2010; 10:357.
- Heron L, Selnikova O, Moiseieva A, et al. Immunogenicity, reactogenicity and safety of two-dose versus three-dose (standard care) hepatitis B immunisation of healthy adolescents aged 11–15 years: a randomised controlled trial. Vaccine 2007; 25:2817–2822.
- Heron LG, Chant KG, Jalaludin BB. A novel hepatitis B vaccination regimen for adolescents: two doses 12 months apart. Vaccine 2002; 20:3472–3476.
- Jackson Y, Chappuis F, Mezger N, Kanappa K, Loutan L. High immunogenicity of delayed third dose of hepatitis B vaccine in travellers. Vaccine 2007; 25:3482–3484.
- Jack AD, Hall AJ, Maine N, Mendy M, Whittle HC. What level of hepatitis B antibody is protective? J Infect Dis 1999; 179:489–492.
- Floreani A, Baldo V, Cristofoletti M, et al. Long-term persistence of anti-HBs after vaccination against HBV: an 18 year experience in health care workers. Vaccine 2004; 22:607–610.
- Department of Health and Human Services. Appendix A Immunization Management Issues. http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5516a2.htm. Accessed April 6, 2014.
- Leuridan E, Van Damme P. Hepatitis B and the need for a booster dose. Clin Infect Dis 2011; 53:68–75.
- Scolnick EM, McLean AA, West DJ, McAleer WJ, Miller WJ, Buynak EB. Clinical evaluation in healthy adults of a hepatitis B vaccine made by recombinant DNA. JAMA 1984; 251:2812–2815.
- Merck and Co, Inc. 1998. Recombivax HB. http://www.merck.com/product/usa/pi_circulars/r/recombivax_hb/re-combivax_pi.pdf. Accessed April 7, 2014.
- Marsano LS, West DJ, Chan I, et al. A two-dose hepatitis B vaccine regimen: proof of priming and memory responses in young adults. Vaccine 1998; 16:624–629.
- Cassidy WM, Watson B, Ioli VA, Williams K, Bird S, West DJ. A randomized trial of alternative two- and three-dose hepatitis B vaccination regimens in adolescents: antibody responses, safety, and immunologic memory. Pediatrics 2001; 107:626–631.
- Van Damme P, Moiseeva A, Marichev I, et al. Five years follow-up following two or three doses of a hepatitis B vaccine in adolescents aged 11–15 years: a randomised controlled study. BMC Infect Dis 2010; 10:357.
- Heron L, Selnikova O, Moiseieva A, et al. Immunogenicity, reactogenicity and safety of two-dose versus three-dose (standard care) hepatitis B immunisation of healthy adolescents aged 11–15 years: a randomised controlled trial. Vaccine 2007; 25:2817–2822.
- Heron LG, Chant KG, Jalaludin BB. A novel hepatitis B vaccination regimen for adolescents: two doses 12 months apart. Vaccine 2002; 20:3472–3476.
- Jackson Y, Chappuis F, Mezger N, Kanappa K, Loutan L. High immunogenicity of delayed third dose of hepatitis B vaccine in travellers. Vaccine 2007; 25:3482–3484.
- Jack AD, Hall AJ, Maine N, Mendy M, Whittle HC. What level of hepatitis B antibody is protective? J Infect Dis 1999; 179:489–492.
- Floreani A, Baldo V, Cristofoletti M, et al. Long-term persistence of anti-HBs after vaccination against HBV: an 18 year experience in health care workers. Vaccine 2004; 22:607–610.
Is hemoglobin A1c an accurate measure of glycemic control in all diabetic patients?
No. Hemoglobin A1c has been validated as a predictor of diabetes-related complications and is a standard measure of the adequacy of glucose control. But sometimes we need to regard its values with suspicion, especially when they are not concordant with the patient’s self-monitored blood glucose levels.
UNIVERSALLY USED
Measuring glycated hemoglobin has become an essential tool for detecting impaired glucose tolerance (when levels are between 5.7% and 6.5%), for diagnosing diabetes mellitus (when levels are ≥ 6.5%), and for following the adequacy of control in established disease. The results reflect glycemic control over the preceding 2 to 3 months and possibly indicate the risk of complications, particularly microvascular disease in the long term.
The significance of hemoglobin A1c was further accentuated with the results of the DETECT-2 project,1 which showed that the risk of diabetic retinopathy is insignificant with levels lower than 6% and rises substantially when it is greater than 6.5%.
However, because the biochemical hallmark of diabetes is hyperglycemia (and not the glycation of proteins), concerns have been raised about the universal validity of hemoglobin A1c in all diabetic patients, especially when it is used to monitor glucose control in the long term.2
FACTORS THAT AFFECT THE GLYCATED HEMOGLOBIN LEVEL
Altered glycation
Although the hemoglobin A1c value correlates well with the mean blood glucose level over the previous months, it is affected more by the most recent glucose levels than by earlier levels, and it is especially affected by the most recent peak in blood glucose.3 It is estimated that approximately 50% of the hemoglobin A1c level is determined by the plasma glucose level during the preceding 1-month period.3
Other factors that affect levels of glycated hemoglobin independently of the average glucose level during the previous months include genetic predisposition (some people are “rapid glycators”), labile glycation (ie, transient glycation of hemoglobin when exposed to very high concentrations of glucose), and the 2,3-diphosphoglycerate concentration and pH of the blood.2
Hemoglobin factors
Age of red blood cells. Red blood cells last about 120 days, and the mean age of all red blood cells in circulation ranges from 38 to 60 days (50 on average). Turnover is dictated by a number of factors, including ethnicity, which in turn significantly affect hemoglobin A1c values.
Race and ethnicity. African American, Asian, and Hispanic patients may have higher hemoglobin A1c values than white people who have the same blood glucose levels. In one study of racial and ethnic differences in mean plasma glucose, levels were higher by 0.37% in African American patients, 0.27% in Hispanics, and 0.33% in Asians than in white patients, and the differences were statistically significant.4 However, there is no clear evidence that these differences are associated with differences in the incidence of microvascular disease.5
Effects due to heritable factors could vary among ethnic groups. Racial differences in hemoglobin A1c may be ascribed to the degree of glycation, caused by multiple factors, and to socioeconomic status. Interestingly, many of the interracial differences in conditions that affect erythrocyte turnover would in theory lead to a lower hemoglobin A1c in nonwhites, which is not the case.6
Pregnancy. The mechanisms of hemoglobin A1c discrepancy in pregnancy are not clear. It has been demonstrated that pregnant women may have lower hemoglobin A1c levels than nonpregnant women.7–9 Hemodilution and increased cell turnover have been postulated to account for the decrease, although a mechanism has not been described. Interestingly, conflicting data have been reported regarding hemoglobin A1c in the last trimester of pregnancy (increase, decrease, or no change). Iron deficiency has been presumed to cause the increase of hemoglobin A1c in the last trimester.10
Moreover, hemoglobin A1c may reflect glucose levels during a shorter time because of increased turnover of red blood cells that occurs during this state. Erythropoietin and erythrocyte production are increased during normal pregnancy while hemoglobin and hematocrit continuously dilute into the third trimester. In normal pregnancy, the red blood cell life span is decreased due to “emergency hemopoiesis” in response to these elevated erythropoietin levels.
Anemia. Hemolytic anemia, acute bleeding, and iron-deficiency anemia all influence glycated hemoglobin levels. The formation of reticulocytes whose hemoglobin lacks glycosylation may lead to falsely low hemoglobin A1c values. Interestingly, iron deficiency by itself has been observed to cause elevation of hemoglobin A1c through unclear mechanisms11; however, iron replacement may lead to reticulocytosis. Alternatively, asplenic patients may have deceptively higher hemoglobin A1c values because of the increased life span of their red blood cells.12
Hemoglobinopathy. Hemoglobin F may cause overestimation of hemoglobin A1c levels, whereas hemoglobin S and hemoglobin C may cause underestimation. Of note, these effects are method-specific, and newer immunoassay techniques are relatively robust even in the presence of common hemoglobin variants. Clinicians should be aware of their institution’s laboratory method for measuring glycated hemoglobin.13
Comorbidities
Chronic illnesses can cause fluctuation in hemoglobin A1c and make it unreliable. Uremia, severe hypertriglyceridemia, severe hyperbilirubinemia, chronic alcoholism, chronic salicylate use, chronic opioid use, and lead poisoning all can falsely increase hemoglobin A1c levels.
Vitamin and mineral deficiencies (eg, deficiencies of vitamin B12 and iron) can reduce red blood cell turnover and therefore falsely elevate hemoglobin A1c levels. Conversely, medical replacement of these deficiencies could lead to higher red blood cell turnover and reduced hemoglobin A1c levels.
Blood transfusions. Recent reports suggest that red blood cell transfusions reduce the hemoglobin A1c concentration in diabetic patients. This effect was most pronounced in patients who received large transfusion volumes or who had a high hemoglobin A1c level before the transfusion.14
Renal failure. Patients with renal failure have higher levels of carbamylated hemoglobin, which is reported to interfere with measurement and interpretation of hemoglobin A1c. Moreover, there is concern that hemoglobin A1c values may be falsely low in these patients because of shortened erythrocyte survival. Other factors that influence hemoglobin A1c and cause the measured levels to be misleadingly low in renal failure patients include use of recombinant human erythropoietin, the uremic environment, and blood transfusions.15
It has been suggested that glycated albumin may be a better marker for assessing glycemic control in patients with severe chronic kidney disease.16
Medications and supplements that affect hemoglobin
Drugs that may cause hemolysis could lower hemoglobin A1c levels. Examples are dapsone, ribavirin, and sulfonamides. Other drugs can change the structure of hemoglobin. For example, hydroxyurea alters hemoglobin A into hemoglobin F, thus lowering the hemoglobin A1c level. Chronic opiate use has been reported to increase hemoglobin A1c levels through mechanisms yet unclear.
Aspirin, vitamin C, and vitamin E have been postulated to interfere with hemoglobin A1c measurement assays, although studies have not been consistent in demonstrating these effects.
Labile diabetes
In some patients with diabetes, blood glucose levels are labile and oscillate between states of hypoglycemia and hyperglycemia, despite optimal hemoglobin A1c levels.17 In these patients, the average blood glucose level may very well correlate appropriately with the glycated hemoglobin level, but the degree of control would not be acceptable. Fasting hyperglycemia or postprandial hyperglycemia, or both, especially in the setting of significant glycemic variability over the month before testing, may not be captured by the hemoglobin A1c measurement. These glycemic excursions may be important, as data suggest that this variability may independently worsen microvascular complications in diabetic patients.18
ALTERNATIVES TO MEASURING THE GLYCATED HEMOGLOBIN
When hemoglobin A1c levels are suspected to be inaccurate, other tests of the adequacy of glycemic control can be used.19
Continuous glucose monitoring is the gold standard and precisely shows the degree of glycemic variability, usually over 5 days. It is often used when hypoglycemia and wide fluctuations in within-day and day-to-day glucose levels are suspected. In addition, we believe that continuous monitoring could be used to confirm the validity of hemoglobin A1c testing. In a clinical setting in which the level does not seem to match the fingerstick blood glucose readings, it can be a useful tool to assess the range and variation in glycemic control.
This method, however, is not practical in all diabetic patients, and it certainly does not have the same long-term predictive prognostic value. Yet it may still have a role in validating measures of long-term glycemic control (eg, hemoglobin A1c). There is evidence that using continuous glucose monitoring periodically can improve glycemic control, lower hemoglobin A1c levels, and lead to fewer hypoglycemic events.20 As discussed earlier, patients who have labile glycemic excursions and higher risk of microvascular complications can still have “normal” hemoglobin A1c levels; in this scenario, the use of continuous glucose monitoring can lead to lower risk and better control.
1,5-anhydroglucitol and fructosamine are circulating biomarkers that reflect short-term glucose control, ie, over 2 to 3 weeks. The higher the average blood glucose level, the lower the 1,5-anhydroglucitol level, since higher glucose levels competitively inhibit renal reabsorption of this molecule. However, its utility is limited in renal failure, liver disease, and pregnancy.
Fructosamines are nonenzymatically glycated proteins. As markers, they are reliable in renal disease but are unreliable in hypoproteinemic states such as liver disease, nephrosis, and lipemia. This group of proteins represents all of serum-stable glycated proteins; they are strongly influenced by the concentration of serum proteins, as well as by coexisting low-molecular-weight substances in the plasma.
Glycated albumin is superior to glycated hemoglobin in reflecting glycemic control, as it has a faster metabolic turnover than hemoglobin and is not affected by hemoglobin-opathies. Unlike fructosamines, it is not influenced by the serum albumin concentration. Moreover, it may be superior to the hemoglobin A1c in patients who have postprandial hypoglycemia.21
Interestingly, recent cross-sectional analyses suggest that fructosamines and glycated albumin are at least as strongly associated with microvascular complications as the hemoglobin A1c is.22
BE ALERT TO FACTORS THAT AFFECT GLYCATED HEMOGLOBIN
Hemoglobin A1c reflects exposure of red blood cells to glucose. Multiple factors—pathologic, physiologic, and environmental—can influence the glycation process, red blood cell turnover, and the hemoglobin structure in ways that can decrease the reliability of the hemoglobin A1c measurement.
Clinicians should be vigilant for the various clinical situations in which hemoglobin A1c is hard to interpret, and they should be familiar with alternative tests (eg, continuous glucose monitoring, 1,5-anhydroglucitol, fructosamines) that can be used to monitor adequate glycemic control in these patients.
- Colaguiri S, Lee CM, Wong TY, Balkau B, Shaw JE, Borch-Johnsen K; DETECT-2 Collaboration Writing Group. Glycemic thresholds for diabetes-specific retinopathy: implications for diagnostic criteria for diabetes. Diabetes Care 2011; 34:145–150.
- Bonora E, Tuomilehto J. The pros and cons of diagnosing diabetes with A1C. Diabetes Care 2011; 34(suppl 2):S184–S190.
- Rohlfing CL, Wiedmeyer HM, Little RR, England JD, Tennill A, Goldstein DE. Defining the relationship between plasma glucose and HbA(1c): analysis of glucose profiles and HbA(1c) in the Diabetes Control and Complications Trial. Diabetes Care 2002; 25:275–278.
- Herman WH, Dungan KM, Wolffenbuttel BH, et al. Racial and ethnic differences in mean plasma glucose, hemoglobin A1c, and 1,5-anhydroglucitol in over 2000 patients with type 2 diabetes. J Clin Endocrinol Metab 2009; 94:1689–1694.
- Selvin E, Steffes MW, Zhu H, et al. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N Engl J Med 2010; 362:800–811.
- Tahara Y, Shima K. The response of GHb to stepwise plasma glucose change over time in diabetic patients. Diabetes Care 1993; 16:1313–1314.
- Radder JK, van Roosmalen J. HbA1c in healthy, pregnant women. Neth J Med 2005; 63:256–259.
- Mosca A, Paleari R, Dalfra MG, et al. Reference intervals for hemoglobin A1c in pregnant women: data from an Italian multicenter study. Clin Chem 2006; 52:1138–1143.
- Nielsen LR, Ekbom P, Damm P, et al. HbA1c levels are significantly lower in early and late pregnancy. Diabetes Care 2004; 27:1200–1201.
- Makris K, Spanou L. Is there a relationship between mean blood glucose and glycated hemoglobin? J Diabetes Sci Technol 2011; 5:1572–1583.
- Tarim O, Kucukerdogan A, Gunay U, Eralp O, Ercan I. Effects of iron deficiency anemia on hemoglobin A1c in type 1 diabetes mellitus. Pediatr Int 1999; 41:357–362.
- Panzer S, Kronik G, Lechner K, Bettelheim P, Neumann E, Dudczak R. Glycosylated hemoglobins (GHb): an index of red cell survival. Blood 1982; 59:1348–1350.
- National Glycohemoglobin Standardization Program. HbA1c assay interferences. www.ngsp.org/interf.asp. Accessed December 27, 2013.
- Spencer DH, Grossman BJ, Scott MG. Red cell transfusion decreases hemoglobin A1c in patients with diabetes. Clin Chem 2011; 57:344–346.
- Little RR, Rohlfing CL, Tennill AL, et al. Measurement of Hba(1C) in patients with chronic renal failure. Clin Chim Acta 2013; 418:73–76.
- Vos FE, Schollum JB, Walker RJ. Glycated albumin is the preferred marker for assessing glycaemic control in advanced chronic kidney disease. NDT Plus 2011; 4:368–375.
- Kilpatrick ES, Rigby AS, Goode K, Atkin SL. Relating mean blood glucose and glucose variability to the risk of multiple episodes of hypoglycaemia in type 1 diabetes. Diabetologia 2007; 50:2553–2561.
- Monnier L, Mas E, Ginet C, et al. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006; 295:1681–1687.
- Radin MS. Pitfalls in hemoglobin A1c measurement: when results may be misleading. J Gen Intern Med 2013; Sep 4 [epub ahead of print]. http://link.springer.com/article/10.1007%2Fs11606-013-2595-x/fulltext.html. Accessed January 29, 2014.
- Leinung M, Nardacci E, Patel N, Bettadahalli S, Paika K, Thompson S. Benefits of short-term professional continuous glucose monitoring in clinical practice. Diabetes Technol Ther 2013; 15:744–747.
- Koga M, Kasayama S. Clinical impact of glycated albumin as another glycemic control marker. Endocr J 2010; 57:751–762.
- Selvin E, Francis LM, Ballantyne CM, et al. Nontraditional markers of glycemia: associations with microvascular conditions. Diabetes Care 2011; 34:960–967.
No. Hemoglobin A1c has been validated as a predictor of diabetes-related complications and is a standard measure of the adequacy of glucose control. But sometimes we need to regard its values with suspicion, especially when they are not concordant with the patient’s self-monitored blood glucose levels.
UNIVERSALLY USED
Measuring glycated hemoglobin has become an essential tool for detecting impaired glucose tolerance (when levels are between 5.7% and 6.5%), for diagnosing diabetes mellitus (when levels are ≥ 6.5%), and for following the adequacy of control in established disease. The results reflect glycemic control over the preceding 2 to 3 months and possibly indicate the risk of complications, particularly microvascular disease in the long term.
The significance of hemoglobin A1c was further accentuated with the results of the DETECT-2 project,1 which showed that the risk of diabetic retinopathy is insignificant with levels lower than 6% and rises substantially when it is greater than 6.5%.
However, because the biochemical hallmark of diabetes is hyperglycemia (and not the glycation of proteins), concerns have been raised about the universal validity of hemoglobin A1c in all diabetic patients, especially when it is used to monitor glucose control in the long term.2
FACTORS THAT AFFECT THE GLYCATED HEMOGLOBIN LEVEL
Altered glycation
Although the hemoglobin A1c value correlates well with the mean blood glucose level over the previous months, it is affected more by the most recent glucose levels than by earlier levels, and it is especially affected by the most recent peak in blood glucose.3 It is estimated that approximately 50% of the hemoglobin A1c level is determined by the plasma glucose level during the preceding 1-month period.3
Other factors that affect levels of glycated hemoglobin independently of the average glucose level during the previous months include genetic predisposition (some people are “rapid glycators”), labile glycation (ie, transient glycation of hemoglobin when exposed to very high concentrations of glucose), and the 2,3-diphosphoglycerate concentration and pH of the blood.2
Hemoglobin factors
Age of red blood cells. Red blood cells last about 120 days, and the mean age of all red blood cells in circulation ranges from 38 to 60 days (50 on average). Turnover is dictated by a number of factors, including ethnicity, which in turn significantly affect hemoglobin A1c values.
Race and ethnicity. African American, Asian, and Hispanic patients may have higher hemoglobin A1c values than white people who have the same blood glucose levels. In one study of racial and ethnic differences in mean plasma glucose, levels were higher by 0.37% in African American patients, 0.27% in Hispanics, and 0.33% in Asians than in white patients, and the differences were statistically significant.4 However, there is no clear evidence that these differences are associated with differences in the incidence of microvascular disease.5
Effects due to heritable factors could vary among ethnic groups. Racial differences in hemoglobin A1c may be ascribed to the degree of glycation, caused by multiple factors, and to socioeconomic status. Interestingly, many of the interracial differences in conditions that affect erythrocyte turnover would in theory lead to a lower hemoglobin A1c in nonwhites, which is not the case.6
Pregnancy. The mechanisms of hemoglobin A1c discrepancy in pregnancy are not clear. It has been demonstrated that pregnant women may have lower hemoglobin A1c levels than nonpregnant women.7–9 Hemodilution and increased cell turnover have been postulated to account for the decrease, although a mechanism has not been described. Interestingly, conflicting data have been reported regarding hemoglobin A1c in the last trimester of pregnancy (increase, decrease, or no change). Iron deficiency has been presumed to cause the increase of hemoglobin A1c in the last trimester.10
Moreover, hemoglobin A1c may reflect glucose levels during a shorter time because of increased turnover of red blood cells that occurs during this state. Erythropoietin and erythrocyte production are increased during normal pregnancy while hemoglobin and hematocrit continuously dilute into the third trimester. In normal pregnancy, the red blood cell life span is decreased due to “emergency hemopoiesis” in response to these elevated erythropoietin levels.
Anemia. Hemolytic anemia, acute bleeding, and iron-deficiency anemia all influence glycated hemoglobin levels. The formation of reticulocytes whose hemoglobin lacks glycosylation may lead to falsely low hemoglobin A1c values. Interestingly, iron deficiency by itself has been observed to cause elevation of hemoglobin A1c through unclear mechanisms11; however, iron replacement may lead to reticulocytosis. Alternatively, asplenic patients may have deceptively higher hemoglobin A1c values because of the increased life span of their red blood cells.12
Hemoglobinopathy. Hemoglobin F may cause overestimation of hemoglobin A1c levels, whereas hemoglobin S and hemoglobin C may cause underestimation. Of note, these effects are method-specific, and newer immunoassay techniques are relatively robust even in the presence of common hemoglobin variants. Clinicians should be aware of their institution’s laboratory method for measuring glycated hemoglobin.13
Comorbidities
Chronic illnesses can cause fluctuation in hemoglobin A1c and make it unreliable. Uremia, severe hypertriglyceridemia, severe hyperbilirubinemia, chronic alcoholism, chronic salicylate use, chronic opioid use, and lead poisoning all can falsely increase hemoglobin A1c levels.
Vitamin and mineral deficiencies (eg, deficiencies of vitamin B12 and iron) can reduce red blood cell turnover and therefore falsely elevate hemoglobin A1c levels. Conversely, medical replacement of these deficiencies could lead to higher red blood cell turnover and reduced hemoglobin A1c levels.
Blood transfusions. Recent reports suggest that red blood cell transfusions reduce the hemoglobin A1c concentration in diabetic patients. This effect was most pronounced in patients who received large transfusion volumes or who had a high hemoglobin A1c level before the transfusion.14
Renal failure. Patients with renal failure have higher levels of carbamylated hemoglobin, which is reported to interfere with measurement and interpretation of hemoglobin A1c. Moreover, there is concern that hemoglobin A1c values may be falsely low in these patients because of shortened erythrocyte survival. Other factors that influence hemoglobin A1c and cause the measured levels to be misleadingly low in renal failure patients include use of recombinant human erythropoietin, the uremic environment, and blood transfusions.15
It has been suggested that glycated albumin may be a better marker for assessing glycemic control in patients with severe chronic kidney disease.16
Medications and supplements that affect hemoglobin
Drugs that may cause hemolysis could lower hemoglobin A1c levels. Examples are dapsone, ribavirin, and sulfonamides. Other drugs can change the structure of hemoglobin. For example, hydroxyurea alters hemoglobin A into hemoglobin F, thus lowering the hemoglobin A1c level. Chronic opiate use has been reported to increase hemoglobin A1c levels through mechanisms yet unclear.
Aspirin, vitamin C, and vitamin E have been postulated to interfere with hemoglobin A1c measurement assays, although studies have not been consistent in demonstrating these effects.
Labile diabetes
In some patients with diabetes, blood glucose levels are labile and oscillate between states of hypoglycemia and hyperglycemia, despite optimal hemoglobin A1c levels.17 In these patients, the average blood glucose level may very well correlate appropriately with the glycated hemoglobin level, but the degree of control would not be acceptable. Fasting hyperglycemia or postprandial hyperglycemia, or both, especially in the setting of significant glycemic variability over the month before testing, may not be captured by the hemoglobin A1c measurement. These glycemic excursions may be important, as data suggest that this variability may independently worsen microvascular complications in diabetic patients.18
ALTERNATIVES TO MEASURING THE GLYCATED HEMOGLOBIN
When hemoglobin A1c levels are suspected to be inaccurate, other tests of the adequacy of glycemic control can be used.19
Continuous glucose monitoring is the gold standard and precisely shows the degree of glycemic variability, usually over 5 days. It is often used when hypoglycemia and wide fluctuations in within-day and day-to-day glucose levels are suspected. In addition, we believe that continuous monitoring could be used to confirm the validity of hemoglobin A1c testing. In a clinical setting in which the level does not seem to match the fingerstick blood glucose readings, it can be a useful tool to assess the range and variation in glycemic control.
This method, however, is not practical in all diabetic patients, and it certainly does not have the same long-term predictive prognostic value. Yet it may still have a role in validating measures of long-term glycemic control (eg, hemoglobin A1c). There is evidence that using continuous glucose monitoring periodically can improve glycemic control, lower hemoglobin A1c levels, and lead to fewer hypoglycemic events.20 As discussed earlier, patients who have labile glycemic excursions and higher risk of microvascular complications can still have “normal” hemoglobin A1c levels; in this scenario, the use of continuous glucose monitoring can lead to lower risk and better control.
1,5-anhydroglucitol and fructosamine are circulating biomarkers that reflect short-term glucose control, ie, over 2 to 3 weeks. The higher the average blood glucose level, the lower the 1,5-anhydroglucitol level, since higher glucose levels competitively inhibit renal reabsorption of this molecule. However, its utility is limited in renal failure, liver disease, and pregnancy.
Fructosamines are nonenzymatically glycated proteins. As markers, they are reliable in renal disease but are unreliable in hypoproteinemic states such as liver disease, nephrosis, and lipemia. This group of proteins represents all of serum-stable glycated proteins; they are strongly influenced by the concentration of serum proteins, as well as by coexisting low-molecular-weight substances in the plasma.
Glycated albumin is superior to glycated hemoglobin in reflecting glycemic control, as it has a faster metabolic turnover than hemoglobin and is not affected by hemoglobin-opathies. Unlike fructosamines, it is not influenced by the serum albumin concentration. Moreover, it may be superior to the hemoglobin A1c in patients who have postprandial hypoglycemia.21
Interestingly, recent cross-sectional analyses suggest that fructosamines and glycated albumin are at least as strongly associated with microvascular complications as the hemoglobin A1c is.22
BE ALERT TO FACTORS THAT AFFECT GLYCATED HEMOGLOBIN
Hemoglobin A1c reflects exposure of red blood cells to glucose. Multiple factors—pathologic, physiologic, and environmental—can influence the glycation process, red blood cell turnover, and the hemoglobin structure in ways that can decrease the reliability of the hemoglobin A1c measurement.
Clinicians should be vigilant for the various clinical situations in which hemoglobin A1c is hard to interpret, and they should be familiar with alternative tests (eg, continuous glucose monitoring, 1,5-anhydroglucitol, fructosamines) that can be used to monitor adequate glycemic control in these patients.
No. Hemoglobin A1c has been validated as a predictor of diabetes-related complications and is a standard measure of the adequacy of glucose control. But sometimes we need to regard its values with suspicion, especially when they are not concordant with the patient’s self-monitored blood glucose levels.
UNIVERSALLY USED
Measuring glycated hemoglobin has become an essential tool for detecting impaired glucose tolerance (when levels are between 5.7% and 6.5%), for diagnosing diabetes mellitus (when levels are ≥ 6.5%), and for following the adequacy of control in established disease. The results reflect glycemic control over the preceding 2 to 3 months and possibly indicate the risk of complications, particularly microvascular disease in the long term.
The significance of hemoglobin A1c was further accentuated with the results of the DETECT-2 project,1 which showed that the risk of diabetic retinopathy is insignificant with levels lower than 6% and rises substantially when it is greater than 6.5%.
However, because the biochemical hallmark of diabetes is hyperglycemia (and not the glycation of proteins), concerns have been raised about the universal validity of hemoglobin A1c in all diabetic patients, especially when it is used to monitor glucose control in the long term.2
FACTORS THAT AFFECT THE GLYCATED HEMOGLOBIN LEVEL
Altered glycation
Although the hemoglobin A1c value correlates well with the mean blood glucose level over the previous months, it is affected more by the most recent glucose levels than by earlier levels, and it is especially affected by the most recent peak in blood glucose.3 It is estimated that approximately 50% of the hemoglobin A1c level is determined by the plasma glucose level during the preceding 1-month period.3
Other factors that affect levels of glycated hemoglobin independently of the average glucose level during the previous months include genetic predisposition (some people are “rapid glycators”), labile glycation (ie, transient glycation of hemoglobin when exposed to very high concentrations of glucose), and the 2,3-diphosphoglycerate concentration and pH of the blood.2
Hemoglobin factors
Age of red blood cells. Red blood cells last about 120 days, and the mean age of all red blood cells in circulation ranges from 38 to 60 days (50 on average). Turnover is dictated by a number of factors, including ethnicity, which in turn significantly affect hemoglobin A1c values.
Race and ethnicity. African American, Asian, and Hispanic patients may have higher hemoglobin A1c values than white people who have the same blood glucose levels. In one study of racial and ethnic differences in mean plasma glucose, levels were higher by 0.37% in African American patients, 0.27% in Hispanics, and 0.33% in Asians than in white patients, and the differences were statistically significant.4 However, there is no clear evidence that these differences are associated with differences in the incidence of microvascular disease.5
Effects due to heritable factors could vary among ethnic groups. Racial differences in hemoglobin A1c may be ascribed to the degree of glycation, caused by multiple factors, and to socioeconomic status. Interestingly, many of the interracial differences in conditions that affect erythrocyte turnover would in theory lead to a lower hemoglobin A1c in nonwhites, which is not the case.6
Pregnancy. The mechanisms of hemoglobin A1c discrepancy in pregnancy are not clear. It has been demonstrated that pregnant women may have lower hemoglobin A1c levels than nonpregnant women.7–9 Hemodilution and increased cell turnover have been postulated to account for the decrease, although a mechanism has not been described. Interestingly, conflicting data have been reported regarding hemoglobin A1c in the last trimester of pregnancy (increase, decrease, or no change). Iron deficiency has been presumed to cause the increase of hemoglobin A1c in the last trimester.10
Moreover, hemoglobin A1c may reflect glucose levels during a shorter time because of increased turnover of red blood cells that occurs during this state. Erythropoietin and erythrocyte production are increased during normal pregnancy while hemoglobin and hematocrit continuously dilute into the third trimester. In normal pregnancy, the red blood cell life span is decreased due to “emergency hemopoiesis” in response to these elevated erythropoietin levels.
Anemia. Hemolytic anemia, acute bleeding, and iron-deficiency anemia all influence glycated hemoglobin levels. The formation of reticulocytes whose hemoglobin lacks glycosylation may lead to falsely low hemoglobin A1c values. Interestingly, iron deficiency by itself has been observed to cause elevation of hemoglobin A1c through unclear mechanisms11; however, iron replacement may lead to reticulocytosis. Alternatively, asplenic patients may have deceptively higher hemoglobin A1c values because of the increased life span of their red blood cells.12
Hemoglobinopathy. Hemoglobin F may cause overestimation of hemoglobin A1c levels, whereas hemoglobin S and hemoglobin C may cause underestimation. Of note, these effects are method-specific, and newer immunoassay techniques are relatively robust even in the presence of common hemoglobin variants. Clinicians should be aware of their institution’s laboratory method for measuring glycated hemoglobin.13
Comorbidities
Chronic illnesses can cause fluctuation in hemoglobin A1c and make it unreliable. Uremia, severe hypertriglyceridemia, severe hyperbilirubinemia, chronic alcoholism, chronic salicylate use, chronic opioid use, and lead poisoning all can falsely increase hemoglobin A1c levels.
Vitamin and mineral deficiencies (eg, deficiencies of vitamin B12 and iron) can reduce red blood cell turnover and therefore falsely elevate hemoglobin A1c levels. Conversely, medical replacement of these deficiencies could lead to higher red blood cell turnover and reduced hemoglobin A1c levels.
Blood transfusions. Recent reports suggest that red blood cell transfusions reduce the hemoglobin A1c concentration in diabetic patients. This effect was most pronounced in patients who received large transfusion volumes or who had a high hemoglobin A1c level before the transfusion.14
Renal failure. Patients with renal failure have higher levels of carbamylated hemoglobin, which is reported to interfere with measurement and interpretation of hemoglobin A1c. Moreover, there is concern that hemoglobin A1c values may be falsely low in these patients because of shortened erythrocyte survival. Other factors that influence hemoglobin A1c and cause the measured levels to be misleadingly low in renal failure patients include use of recombinant human erythropoietin, the uremic environment, and blood transfusions.15
It has been suggested that glycated albumin may be a better marker for assessing glycemic control in patients with severe chronic kidney disease.16
Medications and supplements that affect hemoglobin
Drugs that may cause hemolysis could lower hemoglobin A1c levels. Examples are dapsone, ribavirin, and sulfonamides. Other drugs can change the structure of hemoglobin. For example, hydroxyurea alters hemoglobin A into hemoglobin F, thus lowering the hemoglobin A1c level. Chronic opiate use has been reported to increase hemoglobin A1c levels through mechanisms yet unclear.
Aspirin, vitamin C, and vitamin E have been postulated to interfere with hemoglobin A1c measurement assays, although studies have not been consistent in demonstrating these effects.
Labile diabetes
In some patients with diabetes, blood glucose levels are labile and oscillate between states of hypoglycemia and hyperglycemia, despite optimal hemoglobin A1c levels.17 In these patients, the average blood glucose level may very well correlate appropriately with the glycated hemoglobin level, but the degree of control would not be acceptable. Fasting hyperglycemia or postprandial hyperglycemia, or both, especially in the setting of significant glycemic variability over the month before testing, may not be captured by the hemoglobin A1c measurement. These glycemic excursions may be important, as data suggest that this variability may independently worsen microvascular complications in diabetic patients.18
ALTERNATIVES TO MEASURING THE GLYCATED HEMOGLOBIN
When hemoglobin A1c levels are suspected to be inaccurate, other tests of the adequacy of glycemic control can be used.19
Continuous glucose monitoring is the gold standard and precisely shows the degree of glycemic variability, usually over 5 days. It is often used when hypoglycemia and wide fluctuations in within-day and day-to-day glucose levels are suspected. In addition, we believe that continuous monitoring could be used to confirm the validity of hemoglobin A1c testing. In a clinical setting in which the level does not seem to match the fingerstick blood glucose readings, it can be a useful tool to assess the range and variation in glycemic control.
This method, however, is not practical in all diabetic patients, and it certainly does not have the same long-term predictive prognostic value. Yet it may still have a role in validating measures of long-term glycemic control (eg, hemoglobin A1c). There is evidence that using continuous glucose monitoring periodically can improve glycemic control, lower hemoglobin A1c levels, and lead to fewer hypoglycemic events.20 As discussed earlier, patients who have labile glycemic excursions and higher risk of microvascular complications can still have “normal” hemoglobin A1c levels; in this scenario, the use of continuous glucose monitoring can lead to lower risk and better control.
1,5-anhydroglucitol and fructosamine are circulating biomarkers that reflect short-term glucose control, ie, over 2 to 3 weeks. The higher the average blood glucose level, the lower the 1,5-anhydroglucitol level, since higher glucose levels competitively inhibit renal reabsorption of this molecule. However, its utility is limited in renal failure, liver disease, and pregnancy.
Fructosamines are nonenzymatically glycated proteins. As markers, they are reliable in renal disease but are unreliable in hypoproteinemic states such as liver disease, nephrosis, and lipemia. This group of proteins represents all of serum-stable glycated proteins; they are strongly influenced by the concentration of serum proteins, as well as by coexisting low-molecular-weight substances in the plasma.
Glycated albumin is superior to glycated hemoglobin in reflecting glycemic control, as it has a faster metabolic turnover than hemoglobin and is not affected by hemoglobin-opathies. Unlike fructosamines, it is not influenced by the serum albumin concentration. Moreover, it may be superior to the hemoglobin A1c in patients who have postprandial hypoglycemia.21
Interestingly, recent cross-sectional analyses suggest that fructosamines and glycated albumin are at least as strongly associated with microvascular complications as the hemoglobin A1c is.22
BE ALERT TO FACTORS THAT AFFECT GLYCATED HEMOGLOBIN
Hemoglobin A1c reflects exposure of red blood cells to glucose. Multiple factors—pathologic, physiologic, and environmental—can influence the glycation process, red blood cell turnover, and the hemoglobin structure in ways that can decrease the reliability of the hemoglobin A1c measurement.
Clinicians should be vigilant for the various clinical situations in which hemoglobin A1c is hard to interpret, and they should be familiar with alternative tests (eg, continuous glucose monitoring, 1,5-anhydroglucitol, fructosamines) that can be used to monitor adequate glycemic control in these patients.
- Colaguiri S, Lee CM, Wong TY, Balkau B, Shaw JE, Borch-Johnsen K; DETECT-2 Collaboration Writing Group. Glycemic thresholds for diabetes-specific retinopathy: implications for diagnostic criteria for diabetes. Diabetes Care 2011; 34:145–150.
- Bonora E, Tuomilehto J. The pros and cons of diagnosing diabetes with A1C. Diabetes Care 2011; 34(suppl 2):S184–S190.
- Rohlfing CL, Wiedmeyer HM, Little RR, England JD, Tennill A, Goldstein DE. Defining the relationship between plasma glucose and HbA(1c): analysis of glucose profiles and HbA(1c) in the Diabetes Control and Complications Trial. Diabetes Care 2002; 25:275–278.
- Herman WH, Dungan KM, Wolffenbuttel BH, et al. Racial and ethnic differences in mean plasma glucose, hemoglobin A1c, and 1,5-anhydroglucitol in over 2000 patients with type 2 diabetes. J Clin Endocrinol Metab 2009; 94:1689–1694.
- Selvin E, Steffes MW, Zhu H, et al. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N Engl J Med 2010; 362:800–811.
- Tahara Y, Shima K. The response of GHb to stepwise plasma glucose change over time in diabetic patients. Diabetes Care 1993; 16:1313–1314.
- Radder JK, van Roosmalen J. HbA1c in healthy, pregnant women. Neth J Med 2005; 63:256–259.
- Mosca A, Paleari R, Dalfra MG, et al. Reference intervals for hemoglobin A1c in pregnant women: data from an Italian multicenter study. Clin Chem 2006; 52:1138–1143.
- Nielsen LR, Ekbom P, Damm P, et al. HbA1c levels are significantly lower in early and late pregnancy. Diabetes Care 2004; 27:1200–1201.
- Makris K, Spanou L. Is there a relationship between mean blood glucose and glycated hemoglobin? J Diabetes Sci Technol 2011; 5:1572–1583.
- Tarim O, Kucukerdogan A, Gunay U, Eralp O, Ercan I. Effects of iron deficiency anemia on hemoglobin A1c in type 1 diabetes mellitus. Pediatr Int 1999; 41:357–362.
- Panzer S, Kronik G, Lechner K, Bettelheim P, Neumann E, Dudczak R. Glycosylated hemoglobins (GHb): an index of red cell survival. Blood 1982; 59:1348–1350.
- National Glycohemoglobin Standardization Program. HbA1c assay interferences. www.ngsp.org/interf.asp. Accessed December 27, 2013.
- Spencer DH, Grossman BJ, Scott MG. Red cell transfusion decreases hemoglobin A1c in patients with diabetes. Clin Chem 2011; 57:344–346.
- Little RR, Rohlfing CL, Tennill AL, et al. Measurement of Hba(1C) in patients with chronic renal failure. Clin Chim Acta 2013; 418:73–76.
- Vos FE, Schollum JB, Walker RJ. Glycated albumin is the preferred marker for assessing glycaemic control in advanced chronic kidney disease. NDT Plus 2011; 4:368–375.
- Kilpatrick ES, Rigby AS, Goode K, Atkin SL. Relating mean blood glucose and glucose variability to the risk of multiple episodes of hypoglycaemia in type 1 diabetes. Diabetologia 2007; 50:2553–2561.
- Monnier L, Mas E, Ginet C, et al. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006; 295:1681–1687.
- Radin MS. Pitfalls in hemoglobin A1c measurement: when results may be misleading. J Gen Intern Med 2013; Sep 4 [epub ahead of print]. http://link.springer.com/article/10.1007%2Fs11606-013-2595-x/fulltext.html. Accessed January 29, 2014.
- Leinung M, Nardacci E, Patel N, Bettadahalli S, Paika K, Thompson S. Benefits of short-term professional continuous glucose monitoring in clinical practice. Diabetes Technol Ther 2013; 15:744–747.
- Koga M, Kasayama S. Clinical impact of glycated albumin as another glycemic control marker. Endocr J 2010; 57:751–762.
- Selvin E, Francis LM, Ballantyne CM, et al. Nontraditional markers of glycemia: associations with microvascular conditions. Diabetes Care 2011; 34:960–967.
- Colaguiri S, Lee CM, Wong TY, Balkau B, Shaw JE, Borch-Johnsen K; DETECT-2 Collaboration Writing Group. Glycemic thresholds for diabetes-specific retinopathy: implications for diagnostic criteria for diabetes. Diabetes Care 2011; 34:145–150.
- Bonora E, Tuomilehto J. The pros and cons of diagnosing diabetes with A1C. Diabetes Care 2011; 34(suppl 2):S184–S190.
- Rohlfing CL, Wiedmeyer HM, Little RR, England JD, Tennill A, Goldstein DE. Defining the relationship between plasma glucose and HbA(1c): analysis of glucose profiles and HbA(1c) in the Diabetes Control and Complications Trial. Diabetes Care 2002; 25:275–278.
- Herman WH, Dungan KM, Wolffenbuttel BH, et al. Racial and ethnic differences in mean plasma glucose, hemoglobin A1c, and 1,5-anhydroglucitol in over 2000 patients with type 2 diabetes. J Clin Endocrinol Metab 2009; 94:1689–1694.
- Selvin E, Steffes MW, Zhu H, et al. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N Engl J Med 2010; 362:800–811.
- Tahara Y, Shima K. The response of GHb to stepwise plasma glucose change over time in diabetic patients. Diabetes Care 1993; 16:1313–1314.
- Radder JK, van Roosmalen J. HbA1c in healthy, pregnant women. Neth J Med 2005; 63:256–259.
- Mosca A, Paleari R, Dalfra MG, et al. Reference intervals for hemoglobin A1c in pregnant women: data from an Italian multicenter study. Clin Chem 2006; 52:1138–1143.
- Nielsen LR, Ekbom P, Damm P, et al. HbA1c levels are significantly lower in early and late pregnancy. Diabetes Care 2004; 27:1200–1201.
- Makris K, Spanou L. Is there a relationship between mean blood glucose and glycated hemoglobin? J Diabetes Sci Technol 2011; 5:1572–1583.
- Tarim O, Kucukerdogan A, Gunay U, Eralp O, Ercan I. Effects of iron deficiency anemia on hemoglobin A1c in type 1 diabetes mellitus. Pediatr Int 1999; 41:357–362.
- Panzer S, Kronik G, Lechner K, Bettelheim P, Neumann E, Dudczak R. Glycosylated hemoglobins (GHb): an index of red cell survival. Blood 1982; 59:1348–1350.
- National Glycohemoglobin Standardization Program. HbA1c assay interferences. www.ngsp.org/interf.asp. Accessed December 27, 2013.
- Spencer DH, Grossman BJ, Scott MG. Red cell transfusion decreases hemoglobin A1c in patients with diabetes. Clin Chem 2011; 57:344–346.
- Little RR, Rohlfing CL, Tennill AL, et al. Measurement of Hba(1C) in patients with chronic renal failure. Clin Chim Acta 2013; 418:73–76.
- Vos FE, Schollum JB, Walker RJ. Glycated albumin is the preferred marker for assessing glycaemic control in advanced chronic kidney disease. NDT Plus 2011; 4:368–375.
- Kilpatrick ES, Rigby AS, Goode K, Atkin SL. Relating mean blood glucose and glucose variability to the risk of multiple episodes of hypoglycaemia in type 1 diabetes. Diabetologia 2007; 50:2553–2561.
- Monnier L, Mas E, Ginet C, et al. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006; 295:1681–1687.
- Radin MS. Pitfalls in hemoglobin A1c measurement: when results may be misleading. J Gen Intern Med 2013; Sep 4 [epub ahead of print]. http://link.springer.com/article/10.1007%2Fs11606-013-2595-x/fulltext.html. Accessed January 29, 2014.
- Leinung M, Nardacci E, Patel N, Bettadahalli S, Paika K, Thompson S. Benefits of short-term professional continuous glucose monitoring in clinical practice. Diabetes Technol Ther 2013; 15:744–747.
- Koga M, Kasayama S. Clinical impact of glycated albumin as another glycemic control marker. Endocr J 2010; 57:751–762.
- Selvin E, Francis LM, Ballantyne CM, et al. Nontraditional markers of glycemia: associations with microvascular conditions. Diabetes Care 2011; 34:960–967.
Should patients with gout avoid thiazides for hypertension?
The decision should be individualized, taking into consideration the degree to which the thiazide increases the serum urate level, whether this increase can be managed without overly complicating the patient’s hypouricemic therapy, and, most importantly, what effect switching to another drug will have on the control of the patient’s hypertension. No study has directly addressed this issue.
My practice in most patients, for reasons I explain below, is to use a thiazide if it helps to control the blood pressure and to adjust the dose of the hypouricemic therapy as needed to reduce the serum urate to the desired level.
THIAZIDES REMAIN IMPORTANT IN ANTIHYPERTENSIVE THERAPY
Many patients with gout also have hypertension, perhaps due in part to the same hyperuricemia that caused their gouty arthritis. It is well documented that thiazide diuretics can raise the serum urate level.1 In some studies2 (but not all3), patients using thiazides had a higher incidence of gouty arthritis. Thus, it is reasonable to ask if we should avoid thiazides in patients with coexistent gout and hypertension.
Many hypertensive patients fail to reach their target blood pressures (although with the “looser” recommendations in the latest guidelines,4 we may appear to be doing a better job). The reasons for failing to reach target pressures are complex and many: physicians may simply not be aggressive enough in pursuing a target blood pressure; patients cannot tolerate the drugs or cannot afford the drugs; and many patients need two or more antihypertensive drugs to achieve adequate control. Thiazides are cheap and effective5 and work synergistically with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers.6

Thus, in many patients, avoiding or discontinuing a thiazide may inhibit our ability to control their hypertension, which is a key contributor to cardiovascular events and chronic kidney injury in patients with gout. Since other diuretics (eg, loop diuretics, which can lower blood pressure but often require split doses) also raise the serum urate level, switching to one of them will not eliminate concern over hyperuricemia.
Thiazides and serum urate
Thiazides slightly increase the serum urate level and in a dose-dependent manner. At the doses commonly used in treating hypertension (12.5 or 25 mg once a day), hydrochlorothiazide increases the serum urate level by 0.8 mg/dL or less in patients with normal renal function, as shown in a number of older hypertension treatment trials and in a recent prospective study.1 The effect of chlorthalidone is similar.
In patients with chronic gout treated with a xanthine oxidase inhibitor (allopurinol or febuxostat) to lower the serum urate to the American College of Rheumatology’s recommended target level7 of less than 6.0 mg/dL (or < 5 mg/dL in the British Rheumatology guidelines), this small elevation in serum urate is unlikely to negate the clinical efficacy of these drugs when dosing is optimized. Small studies have demonstrated a clinically insignificant pharmacodynamic interaction between thiazides and xanthine oxidase inhibitors.8,9 When I add a thiazide to a patient’s regimen, I do not usually need to increase the dose of allopurinol significantly to keep the serum urate level below the desired target.
Switch antihypertensive therapy
Occasionally, in a patient with chronic gout and mild hypertension who has a serum urate level marginally above the estimated precipitation threshold of 6.7 mg/dL, it is reasonable to simply switch the thiazide to another antihypertensive, such as losartan. Losartan is a weak uricosuric and can lower the serum urate level slightly, possibly making the addition of another hypouricemic agent unnecessary, while still controlling the blood pressure with a single pill. This decision must be individualized, taking into consideration the efficacy and cost of the alternative antihypertensive drug, as well as the potential but as yet unproven cardiovascular and renal benefits of lowering the serum urate with a more potent hypouricemic to a degree not likely to be attained with losartan alone.
Continue thiazide, adjust gout therapy
Discontinuing a thiazide or switching to another antihypertensive drug may increase the cost and decrease the efficacy of hypertensive therapy. Continuing thiazide therapy and, if necessary, adjusting hypouricemic therapy will not worsen the control of the serum urate level or gouty arthritis, and in most patients will not complicate the management of gout.
ASPIRIN AND HYPERURICEMIA
In answer to a separate but related question, aspirin in low doses for cardioprotection (81 mg daily) also need not be stopped in patients with hyperuricemia or gout in an effort to better control the serum urate level. Low-dose aspirin increases the serum urate level by about 0.3 mg/dL. Since patients with gout have a higher risk of having cardiovascular disease, metabolic syndrome, and chronic kidney disease, many will benefit from low-dose aspirin therapy.
- McAdams DeMarco MA, Maynard JW, Baer AN, et al. Diuretic use, increased serum urate levels, and risk of incident gout in a population-based study of adults with hypertension: the Atherosclerosis Risk in Communities cohort study. Arthritis Rheum 2012; 64:121–129.
- Choi HK, Soriano LC, Zhang Y, Rodríguez LA. Antihypertensive drugs and risk of incident gout among patients with hypertension: population based case-control study. BMJ 2012; 344:d8190.
- Hueskes BA, Roovers EA, Mantel-Teeuwisse AK, Janssens HJ, van de Lisdonk EH, Janssen M. Use of diuretics and the risk of gouty arthritis: a systematic review. Semin Arthritis Rheum 2012; 41:879–889.
- James PA, Oparil S, Carter BL, et al. 2014 Evidence-based guideline for the management of high blood pressure in adults. Report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2013; doi: 10.1001/jama2013.284427
- Fuchs FD. Diuretics: still essential drugs for the management of hypertension. Expert Rev Cardiovasc Ther 2009; 7:591–598.
- Sood N, Reinhart KM, Baker WL. Combination therapy for the management of hypertension: a review of the evidence. Am J Health Syst Pharm 2010; 67:885–894.
- Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken) 2012; 64:1431–1446.
- Löffler W, Landthaler R, de Vries JX, et al. Interaction of allopurinol and hydrochlorothiazide during prolonged oral administration of both drugs in normal subjects. I. Uric acid kinetics. Clin Investig 1994; 72:1071–1075.
- Grabowski B, Khosravan R, Wu JT, Vernillet L, Lademacher C. Effect of hydrochlorothiazide on the pharmacokinetics and pharmacodynamics of febuxostat, a non-purine selective inhibitor of xanthine oxidase. Br J Clin Pharmacol 2010; 70:57–64.
The decision should be individualized, taking into consideration the degree to which the thiazide increases the serum urate level, whether this increase can be managed without overly complicating the patient’s hypouricemic therapy, and, most importantly, what effect switching to another drug will have on the control of the patient’s hypertension. No study has directly addressed this issue.
My practice in most patients, for reasons I explain below, is to use a thiazide if it helps to control the blood pressure and to adjust the dose of the hypouricemic therapy as needed to reduce the serum urate to the desired level.
THIAZIDES REMAIN IMPORTANT IN ANTIHYPERTENSIVE THERAPY
Many patients with gout also have hypertension, perhaps due in part to the same hyperuricemia that caused their gouty arthritis. It is well documented that thiazide diuretics can raise the serum urate level.1 In some studies2 (but not all3), patients using thiazides had a higher incidence of gouty arthritis. Thus, it is reasonable to ask if we should avoid thiazides in patients with coexistent gout and hypertension.
Many hypertensive patients fail to reach their target blood pressures (although with the “looser” recommendations in the latest guidelines,4 we may appear to be doing a better job). The reasons for failing to reach target pressures are complex and many: physicians may simply not be aggressive enough in pursuing a target blood pressure; patients cannot tolerate the drugs or cannot afford the drugs; and many patients need two or more antihypertensive drugs to achieve adequate control. Thiazides are cheap and effective5 and work synergistically with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers.6
Thus, in many patients, avoiding or discontinuing a thiazide may inhibit our ability to control their hypertension, which is a key contributor to cardiovascular events and chronic kidney injury in patients with gout. Since other diuretics (eg, loop diuretics, which can lower blood pressure but often require split doses) also raise the serum urate level, switching to one of them will not eliminate concern over hyperuricemia.
Thiazides and serum urate
Thiazides slightly increase the serum urate level and in a dose-dependent manner. At the doses commonly used in treating hypertension (12.5 or 25 mg once a day), hydrochlorothiazide increases the serum urate level by 0.8 mg/dL or less in patients with normal renal function, as shown in a number of older hypertension treatment trials and in a recent prospective study.1 The effect of chlorthalidone is similar.
In patients with chronic gout treated with a xanthine oxidase inhibitor (allopurinol or febuxostat) to lower the serum urate to the American College of Rheumatology’s recommended target level7 of less than 6.0 mg/dL (or < 5 mg/dL in the British Rheumatology guidelines), this small elevation in serum urate is unlikely to negate the clinical efficacy of these drugs when dosing is optimized. Small studies have demonstrated a clinically insignificant pharmacodynamic interaction between thiazides and xanthine oxidase inhibitors.8,9 When I add a thiazide to a patient’s regimen, I do not usually need to increase the dose of allopurinol significantly to keep the serum urate level below the desired target.
Switch antihypertensive therapy
Occasionally, in a patient with chronic gout and mild hypertension who has a serum urate level marginally above the estimated precipitation threshold of 6.7 mg/dL, it is reasonable to simply switch the thiazide to another antihypertensive, such as losartan. Losartan is a weak uricosuric and can lower the serum urate level slightly, possibly making the addition of another hypouricemic agent unnecessary, while still controlling the blood pressure with a single pill. This decision must be individualized, taking into consideration the efficacy and cost of the alternative antihypertensive drug, as well as the potential but as yet unproven cardiovascular and renal benefits of lowering the serum urate with a more potent hypouricemic to a degree not likely to be attained with losartan alone.
Continue thiazide, adjust gout therapy
Discontinuing a thiazide or switching to another antihypertensive drug may increase the cost and decrease the efficacy of hypertensive therapy. Continuing thiazide therapy and, if necessary, adjusting hypouricemic therapy will not worsen the control of the serum urate level or gouty arthritis, and in most patients will not complicate the management of gout.
ASPIRIN AND HYPERURICEMIA
In answer to a separate but related question, aspirin in low doses for cardioprotection (81 mg daily) also need not be stopped in patients with hyperuricemia or gout in an effort to better control the serum urate level. Low-dose aspirin increases the serum urate level by about 0.3 mg/dL. Since patients with gout have a higher risk of having cardiovascular disease, metabolic syndrome, and chronic kidney disease, many will benefit from low-dose aspirin therapy.
The decision should be individualized, taking into consideration the degree to which the thiazide increases the serum urate level, whether this increase can be managed without overly complicating the patient’s hypouricemic therapy, and, most importantly, what effect switching to another drug will have on the control of the patient’s hypertension. No study has directly addressed this issue.
My practice in most patients, for reasons I explain below, is to use a thiazide if it helps to control the blood pressure and to adjust the dose of the hypouricemic therapy as needed to reduce the serum urate to the desired level.
THIAZIDES REMAIN IMPORTANT IN ANTIHYPERTENSIVE THERAPY
Many patients with gout also have hypertension, perhaps due in part to the same hyperuricemia that caused their gouty arthritis. It is well documented that thiazide diuretics can raise the serum urate level.1 In some studies2 (but not all3), patients using thiazides had a higher incidence of gouty arthritis. Thus, it is reasonable to ask if we should avoid thiazides in patients with coexistent gout and hypertension.
Many hypertensive patients fail to reach their target blood pressures (although with the “looser” recommendations in the latest guidelines,4 we may appear to be doing a better job). The reasons for failing to reach target pressures are complex and many: physicians may simply not be aggressive enough in pursuing a target blood pressure; patients cannot tolerate the drugs or cannot afford the drugs; and many patients need two or more antihypertensive drugs to achieve adequate control. Thiazides are cheap and effective5 and work synergistically with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers.6
Thus, in many patients, avoiding or discontinuing a thiazide may inhibit our ability to control their hypertension, which is a key contributor to cardiovascular events and chronic kidney injury in patients with gout. Since other diuretics (eg, loop diuretics, which can lower blood pressure but often require split doses) also raise the serum urate level, switching to one of them will not eliminate concern over hyperuricemia.
Thiazides and serum urate
Thiazides slightly increase the serum urate level and in a dose-dependent manner. At the doses commonly used in treating hypertension (12.5 or 25 mg once a day), hydrochlorothiazide increases the serum urate level by 0.8 mg/dL or less in patients with normal renal function, as shown in a number of older hypertension treatment trials and in a recent prospective study.1 The effect of chlorthalidone is similar.
In patients with chronic gout treated with a xanthine oxidase inhibitor (allopurinol or febuxostat) to lower the serum urate to the American College of Rheumatology’s recommended target level7 of less than 6.0 mg/dL (or < 5 mg/dL in the British Rheumatology guidelines), this small elevation in serum urate is unlikely to negate the clinical efficacy of these drugs when dosing is optimized. Small studies have demonstrated a clinically insignificant pharmacodynamic interaction between thiazides and xanthine oxidase inhibitors.8,9 When I add a thiazide to a patient’s regimen, I do not usually need to increase the dose of allopurinol significantly to keep the serum urate level below the desired target.
Switch antihypertensive therapy
Occasionally, in a patient with chronic gout and mild hypertension who has a serum urate level marginally above the estimated precipitation threshold of 6.7 mg/dL, it is reasonable to simply switch the thiazide to another antihypertensive, such as losartan. Losartan is a weak uricosuric and can lower the serum urate level slightly, possibly making the addition of another hypouricemic agent unnecessary, while still controlling the blood pressure with a single pill. This decision must be individualized, taking into consideration the efficacy and cost of the alternative antihypertensive drug, as well as the potential but as yet unproven cardiovascular and renal benefits of lowering the serum urate with a more potent hypouricemic to a degree not likely to be attained with losartan alone.
Continue thiazide, adjust gout therapy
Discontinuing a thiazide or switching to another antihypertensive drug may increase the cost and decrease the efficacy of hypertensive therapy. Continuing thiazide therapy and, if necessary, adjusting hypouricemic therapy will not worsen the control of the serum urate level or gouty arthritis, and in most patients will not complicate the management of gout.
ASPIRIN AND HYPERURICEMIA
In answer to a separate but related question, aspirin in low doses for cardioprotection (81 mg daily) also need not be stopped in patients with hyperuricemia or gout in an effort to better control the serum urate level. Low-dose aspirin increases the serum urate level by about 0.3 mg/dL. Since patients with gout have a higher risk of having cardiovascular disease, metabolic syndrome, and chronic kidney disease, many will benefit from low-dose aspirin therapy.
- McAdams DeMarco MA, Maynard JW, Baer AN, et al. Diuretic use, increased serum urate levels, and risk of incident gout in a population-based study of adults with hypertension: the Atherosclerosis Risk in Communities cohort study. Arthritis Rheum 2012; 64:121–129.
- Choi HK, Soriano LC, Zhang Y, Rodríguez LA. Antihypertensive drugs and risk of incident gout among patients with hypertension: population based case-control study. BMJ 2012; 344:d8190.
- Hueskes BA, Roovers EA, Mantel-Teeuwisse AK, Janssens HJ, van de Lisdonk EH, Janssen M. Use of diuretics and the risk of gouty arthritis: a systematic review. Semin Arthritis Rheum 2012; 41:879–889.
- James PA, Oparil S, Carter BL, et al. 2014 Evidence-based guideline for the management of high blood pressure in adults. Report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2013; doi: 10.1001/jama2013.284427
- Fuchs FD. Diuretics: still essential drugs for the management of hypertension. Expert Rev Cardiovasc Ther 2009; 7:591–598.
- Sood N, Reinhart KM, Baker WL. Combination therapy for the management of hypertension: a review of the evidence. Am J Health Syst Pharm 2010; 67:885–894.
- Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken) 2012; 64:1431–1446.
- Löffler W, Landthaler R, de Vries JX, et al. Interaction of allopurinol and hydrochlorothiazide during prolonged oral administration of both drugs in normal subjects. I. Uric acid kinetics. Clin Investig 1994; 72:1071–1075.
- Grabowski B, Khosravan R, Wu JT, Vernillet L, Lademacher C. Effect of hydrochlorothiazide on the pharmacokinetics and pharmacodynamics of febuxostat, a non-purine selective inhibitor of xanthine oxidase. Br J Clin Pharmacol 2010; 70:57–64.
- McAdams DeMarco MA, Maynard JW, Baer AN, et al. Diuretic use, increased serum urate levels, and risk of incident gout in a population-based study of adults with hypertension: the Atherosclerosis Risk in Communities cohort study. Arthritis Rheum 2012; 64:121–129.
- Choi HK, Soriano LC, Zhang Y, Rodríguez LA. Antihypertensive drugs and risk of incident gout among patients with hypertension: population based case-control study. BMJ 2012; 344:d8190.
- Hueskes BA, Roovers EA, Mantel-Teeuwisse AK, Janssens HJ, van de Lisdonk EH, Janssen M. Use of diuretics and the risk of gouty arthritis: a systematic review. Semin Arthritis Rheum 2012; 41:879–889.
- James PA, Oparil S, Carter BL, et al. 2014 Evidence-based guideline for the management of high blood pressure in adults. Report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2013; doi: 10.1001/jama2013.284427
- Fuchs FD. Diuretics: still essential drugs for the management of hypertension. Expert Rev Cardiovasc Ther 2009; 7:591–598.
- Sood N, Reinhart KM, Baker WL. Combination therapy for the management of hypertension: a review of the evidence. Am J Health Syst Pharm 2010; 67:885–894.
- Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken) 2012; 64:1431–1446.
- Löffler W, Landthaler R, de Vries JX, et al. Interaction of allopurinol and hydrochlorothiazide during prolonged oral administration of both drugs in normal subjects. I. Uric acid kinetics. Clin Investig 1994; 72:1071–1075.
- Grabowski B, Khosravan R, Wu JT, Vernillet L, Lademacher C. Effect of hydrochlorothiazide on the pharmacokinetics and pharmacodynamics of febuxostat, a non-purine selective inhibitor of xanthine oxidase. Br J Clin Pharmacol 2010; 70:57–64.
Do all hospitalized patients need stress ulcer prophylaxis?
No. Based on current evidence and guidelines, routine acid-suppressive therapy to prevent stress ulcers has no benefit in hospitalized patients outside the critical-care setting. Only critically ill patients who meet specific criteria, as described in the guidelines of the American Society of Health System Pharmacists, should receive acid-suppressive therapy.
Unfortunately, routine stress ulcer prophylaxis is common in US hospitals, unnecessarily putting patients at risk of complications and adding costs.
STRESS ULCER AND CRITICAL ILLNESS
Stress ulcers—ulcerations of the upper part of the gastrointestinal (GI) mucosa in the setting of acute disease—usually involve the fundus and body of the stomach. The stomach is lined with a glycoprotein mucous layer rich in bicarbonates, forming a physiologic barrier to protect the gastric wall from acid insult by neutralizing hydrogen ions. Disruption of this protective layer can occur in critically ill patients (eg, those with shock or sepsis) through overproduction of uremic toxins, increased reflux of bile salts, compromised blood flow, and increased stomach acidity through gastrin stimulation of parietal cells.
More than 75% of patients with major burns or cranial trauma develop endoscopic mucosal abnormalities within 72 hours of injury.1 In critically ill patients, the risk of ulcer-related overt bleeding is estimated to be 5% to 25%. Furthermore, 1% to 5% of stress ulcers can be deep enough to erode into the submucosa, causing clinically significant GI bleeding, defined as bleeding complicated by hemodynamic compromise or a drop in hemoglobin that requires a blood transfusion.2 In contrast, in inpatients who are not critically ill, the risk of overt bleeding from stress ulcers is less than 1%.3
ADDRESSING RISK
A multicenter prospective cohort study of 2,252 intensive care patients2 reported two main risk factors for significant bleeding caused by stress ulcers: mechanical ventilation for more than 48 hours and coagulopathy, defined as a platelet count below 50 × 109/L, an international normalized ratio greater than 1.5, or a partial thromboplastin time more than twice the control value.4 In hemodynamically stable patients receiving anticoagulation in a general medical or surgical ward, the risk of GI bleeding was low, and acid suppression failed to lower the rate of stress ulcer occurrence.3
Other risk factors include severe sepsis, shock, liver failure, kidney failure, burns over 35% of the total body surface, organ transplantation, cranial trauma, spinal cord trauma, history of peptic ulcer disease, and history of upper GI bleeding.3,5,6 Steroid therapy is not considered a risk factor for stress ulcers unless it is used in the presence of another risk factor such as use of aspirin or nonsteroidal antiinflammatory drugs (NSAIDs).2
INDICATIONS FOR PROPHYLAXIS
Prophylaxis with a proton pump inhibitor (PPI) is indicated in specific conditions—ie, peptic ulcer disease, gastroesophageal reflux disease, chronic NSAID therapy, and Zollinger-Ellison syndrome—and to eradicate Helicobacter pylori infection.7 But in the United States, stress ulcer prophylaxis is overused in general-care floors despite the lack of supporting evidence.
The American Society of Health System Pharmacists guidelines recommend it in the intensive care unit for patients with any of the following: coagulopathy, prolonged mechanical ventilation (more than 48 hours), GI ulcer or bleeding within the past year, sepsis, a stay longer than 1 week in the intensive care unit, occult GI bleeding for 6 or more days, and steroid therapy with more than 250 mg of hydrocortisone daily.8 Hemodynamically stable patients admitted to general-care floors should not receive stress ulcer prophylaxis, as it only negligibly decreases the rate of GI bleeding, from 0.33% to 0.22%.9
WHY ROUTINE ULCER PROPHYLAXIS IS NOT FOR ALL HOSPITALIZED PATIENTS
Although stress ulcer prophylaxis is often considered benign, its lack of proven benefit, additional cost, and risk of adverse effects, including interactions with foods and other drugs, preclude using it routinely for all hospitalized patients.10,11 Chronic use of PPIs has been associated with complications, as discussed below.
Infection
Acid suppression may impair the destruction of ingested microorganisms, resulting in overgrowth of bacteria.12 Overuse of PPIs may increase the risk of several infections:
- Diarrhea due to Clostridium difficile12
- Community-acquired pneumonia, from increased microaspiration of overgrown microorganisms into the lung.12
- Spontaneous bacterial peritonitis in patients with cirrhosis,13 although the mechanism is not clear. (Small-bowel bacterial overgrowth is the hypothesized cause.)
Bone fracture
PPIs lower gastric acidity, and this can inhibit intestinal calcium absorption. Furthermore, PPIs may directly inhibit bone resorption by osteoclasts.14
Reduction in clopidogrel efficacy
PPIs may reduce the efficacy of clopidogrel as a result of competitive inhibition of cytochrome CYP2C19, which is necessary to metabolize clopidogrel to its active forms. Therefore, concomitant use of clopidogrel with omeprazole, esomeprazole, or other CYP2C19 inhibitors is not recommended.15
Nutritional deficiencies
The overgrown microorganisms consume cobalamin in the stomach, resulting in vitamin B12 deficiency. Acid-suppressive therapy can also reduce the absorption of magnesium and iron.12
Unnecessary cost
Heidelbaugh and Inadomi16 reviewed the non-evidence-based use of stress ulcer prophylaxis in patients admitted to a large university hospital and estimated that it entailed a cost to the hospital of $111,791 over the course of a year.
WHICH ULCER PROPHYLAXIS SHOULD BE USED IN CRITICALLY ILL PATIENTS?
Studies have shown histamine-2 blockers to be superior to antacids and sucralfate in preventing stress ulcer and GI bleeding,8,15 but no study has compared PPIs with sucralfate and antacids.
When indicated, an oral PPI is preferred over an oral histamine-2 blocker for GI prophylaxis.17 This practice is considered cost-effective and is associated with lower rates of stress ulcer and GI bleeding. In intubated patients, however, an intravenous histamine-2 blocker is preferable to an intravenous PPI.3,8,11 Interestingly, no difference was reported between PPIs and histamine-2 blockers in terms of mortality rate or reduction in the incidence of nosocomial pneumonia.17
OUR RECOMMENDATION
Only critically ill patients who meet the specific criteria described here should receive stress ulcer prophylaxis. More effort is needed to educate residents, medical staff, and pharmacists about current guidelines. Computerized ordering templates and reminders to discontinue prophylaxis at discharge or step-down may decrease overall use, reduce costs, and limit potential side effects.18
- DePriest JL. Stress ulcer prophylaxis. Do critically ill patients need it? Postgrad Med 1995; 98:159–168.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- Qadeer MA, Richter JE, Brotman DJ. Hospital-acquired gastrointestinal bleeding outside the critical care unit: risk factors, role of acid suppression, and endoscopy findings. J Hosp Med 2006; 1:13–20.
- Shuman RB, Schuster DP, Zuckerman GR. Prophylactic therapy for stress ulcer bleeding: a reappraisal. Ann Intern Med 1987; 106:562–567.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Cook DJ, Reeve BK, Guyatt GH, et al. Stress ulcer prophylaxis in critically ill patients. Resolving discordant meta-analyses. JAMA 1996; 275:308–314.
- Kahrilas PJ, Shaheen NJ, Vaezi MF, et al; American Gastroenterological Association. American Gastroenterological Association Medical Position Statement on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1383–1391.e1–1391.e5.
- Barkun AN, Bardou M, Pham CQ, Martel M. Proton pump inhibitors vs histamine 2 receptor antagonists for stress-related mucosal bleeding prophylaxis in critically ill patients: a meta-analysis. Am J Gastroenterol 2012; 107:507–520.
- Herzig SJ, Vaughn BP, Howell MD, Ngo LH, Marcantonio ER. Acid-suppressive medication use and the risk for nosocomial gastrointestinal tract bleeding. Arch Intern Med 2011; 171:991–997.
- Cook DJ. Stress ulcer prophylaxis: gastrointestinal bleeding and nosocomial pneumonia. Best evidence synthesis. Scand J Gastroenterol Suppl 1995; 210:48–52.
- Messori A, Trippoli S, Vaiani M, Gorini M, Corrado A. Bleeding and pneumonia in intensive care patients given ranitidine and sucralfate for prevention of stress ulcer: meta-analysis of randomised controlled trials. BMJ 2000; 321:1103–1106.
- Heidelbaugh JJ, Kim AH, Chang R, Walker PC. Overutilization of proton-pump inhibitors: what the clinician needs to know. Therap Adv Gastroenterol 2012; 5:219–232.
- Deshpande A, Pasupuleti V, Thota P, et al. Acid-suppressive therapy is associated with spontaneous bacterial peritonitis in cirrhotic patients: a meta-analysis. J Gastroenterol Hepatol 2013; 28:235–242.
- Farina C, Gagliardi S. Selective inhibition of osteoclast vacuolar H(+)- ATPase. Curr Pharm Des 2002; 8:2033–2048.
- ASHP Therapeutic Guidelines on Stress Ulcer Prophylaxis. ASHP Commission on Therapeutics and approved by the ASHP Board of Directors on November 14, 1998. Am J Health Syst Pharm 1999; 56:347–379.
- Heidelbaugh JJ, Inadomi JM. Magnitude and economic impact of inappropriate use of stress ulcer prophylaxis in non-ICU hospitalized patients. Am J Gastroenterol 2006; 101:2200–2205.
- Alhazzani W, Alenezi F, Jaeschke RZ, Moayyedi P, Cook DJ. Proton pump inhibitors versus histamine 2 receptor antagonists for stress ulcer prophylaxis in critically ill patients: a systematic review and meta-analysis. Crit Care Med 2013; 41:693–705.
- Liberman JD, Whelan CT. Brief report: reducing inappropriate usage of stress ulcer prophylaxis among internal medicine residents. A practice-based educational intervention. J Gen Intern Med 2006; 21:498–500.
No. Based on current evidence and guidelines, routine acid-suppressive therapy to prevent stress ulcers has no benefit in hospitalized patients outside the critical-care setting. Only critically ill patients who meet specific criteria, as described in the guidelines of the American Society of Health System Pharmacists, should receive acid-suppressive therapy.
Unfortunately, routine stress ulcer prophylaxis is common in US hospitals, unnecessarily putting patients at risk of complications and adding costs.
STRESS ULCER AND CRITICAL ILLNESS
Stress ulcers—ulcerations of the upper part of the gastrointestinal (GI) mucosa in the setting of acute disease—usually involve the fundus and body of the stomach. The stomach is lined with a glycoprotein mucous layer rich in bicarbonates, forming a physiologic barrier to protect the gastric wall from acid insult by neutralizing hydrogen ions. Disruption of this protective layer can occur in critically ill patients (eg, those with shock or sepsis) through overproduction of uremic toxins, increased reflux of bile salts, compromised blood flow, and increased stomach acidity through gastrin stimulation of parietal cells.
More than 75% of patients with major burns or cranial trauma develop endoscopic mucosal abnormalities within 72 hours of injury.1 In critically ill patients, the risk of ulcer-related overt bleeding is estimated to be 5% to 25%. Furthermore, 1% to 5% of stress ulcers can be deep enough to erode into the submucosa, causing clinically significant GI bleeding, defined as bleeding complicated by hemodynamic compromise or a drop in hemoglobin that requires a blood transfusion.2 In contrast, in inpatients who are not critically ill, the risk of overt bleeding from stress ulcers is less than 1%.3
ADDRESSING RISK
A multicenter prospective cohort study of 2,252 intensive care patients2 reported two main risk factors for significant bleeding caused by stress ulcers: mechanical ventilation for more than 48 hours and coagulopathy, defined as a platelet count below 50 × 109/L, an international normalized ratio greater than 1.5, or a partial thromboplastin time more than twice the control value.4 In hemodynamically stable patients receiving anticoagulation in a general medical or surgical ward, the risk of GI bleeding was low, and acid suppression failed to lower the rate of stress ulcer occurrence.3
Other risk factors include severe sepsis, shock, liver failure, kidney failure, burns over 35% of the total body surface, organ transplantation, cranial trauma, spinal cord trauma, history of peptic ulcer disease, and history of upper GI bleeding.3,5,6 Steroid therapy is not considered a risk factor for stress ulcers unless it is used in the presence of another risk factor such as use of aspirin or nonsteroidal antiinflammatory drugs (NSAIDs).2
INDICATIONS FOR PROPHYLAXIS
Prophylaxis with a proton pump inhibitor (PPI) is indicated in specific conditions—ie, peptic ulcer disease, gastroesophageal reflux disease, chronic NSAID therapy, and Zollinger-Ellison syndrome—and to eradicate Helicobacter pylori infection.7 But in the United States, stress ulcer prophylaxis is overused in general-care floors despite the lack of supporting evidence.
The American Society of Health System Pharmacists guidelines recommend it in the intensive care unit for patients with any of the following: coagulopathy, prolonged mechanical ventilation (more than 48 hours), GI ulcer or bleeding within the past year, sepsis, a stay longer than 1 week in the intensive care unit, occult GI bleeding for 6 or more days, and steroid therapy with more than 250 mg of hydrocortisone daily.8 Hemodynamically stable patients admitted to general-care floors should not receive stress ulcer prophylaxis, as it only negligibly decreases the rate of GI bleeding, from 0.33% to 0.22%.9
WHY ROUTINE ULCER PROPHYLAXIS IS NOT FOR ALL HOSPITALIZED PATIENTS
Although stress ulcer prophylaxis is often considered benign, its lack of proven benefit, additional cost, and risk of adverse effects, including interactions with foods and other drugs, preclude using it routinely for all hospitalized patients.10,11 Chronic use of PPIs has been associated with complications, as discussed below.
Infection
Acid suppression may impair the destruction of ingested microorganisms, resulting in overgrowth of bacteria.12 Overuse of PPIs may increase the risk of several infections:
- Diarrhea due to Clostridium difficile12
- Community-acquired pneumonia, from increased microaspiration of overgrown microorganisms into the lung.12
- Spontaneous bacterial peritonitis in patients with cirrhosis,13 although the mechanism is not clear. (Small-bowel bacterial overgrowth is the hypothesized cause.)
Bone fracture
PPIs lower gastric acidity, and this can inhibit intestinal calcium absorption. Furthermore, PPIs may directly inhibit bone resorption by osteoclasts.14
Reduction in clopidogrel efficacy
PPIs may reduce the efficacy of clopidogrel as a result of competitive inhibition of cytochrome CYP2C19, which is necessary to metabolize clopidogrel to its active forms. Therefore, concomitant use of clopidogrel with omeprazole, esomeprazole, or other CYP2C19 inhibitors is not recommended.15
Nutritional deficiencies
The overgrown microorganisms consume cobalamin in the stomach, resulting in vitamin B12 deficiency. Acid-suppressive therapy can also reduce the absorption of magnesium and iron.12
Unnecessary cost
Heidelbaugh and Inadomi16 reviewed the non-evidence-based use of stress ulcer prophylaxis in patients admitted to a large university hospital and estimated that it entailed a cost to the hospital of $111,791 over the course of a year.
WHICH ULCER PROPHYLAXIS SHOULD BE USED IN CRITICALLY ILL PATIENTS?
Studies have shown histamine-2 blockers to be superior to antacids and sucralfate in preventing stress ulcer and GI bleeding,8,15 but no study has compared PPIs with sucralfate and antacids.
When indicated, an oral PPI is preferred over an oral histamine-2 blocker for GI prophylaxis.17 This practice is considered cost-effective and is associated with lower rates of stress ulcer and GI bleeding. In intubated patients, however, an intravenous histamine-2 blocker is preferable to an intravenous PPI.3,8,11 Interestingly, no difference was reported between PPIs and histamine-2 blockers in terms of mortality rate or reduction in the incidence of nosocomial pneumonia.17
OUR RECOMMENDATION
Only critically ill patients who meet the specific criteria described here should receive stress ulcer prophylaxis. More effort is needed to educate residents, medical staff, and pharmacists about current guidelines. Computerized ordering templates and reminders to discontinue prophylaxis at discharge or step-down may decrease overall use, reduce costs, and limit potential side effects.18
No. Based on current evidence and guidelines, routine acid-suppressive therapy to prevent stress ulcers has no benefit in hospitalized patients outside the critical-care setting. Only critically ill patients who meet specific criteria, as described in the guidelines of the American Society of Health System Pharmacists, should receive acid-suppressive therapy.
Unfortunately, routine stress ulcer prophylaxis is common in US hospitals, unnecessarily putting patients at risk of complications and adding costs.
STRESS ULCER AND CRITICAL ILLNESS
Stress ulcers—ulcerations of the upper part of the gastrointestinal (GI) mucosa in the setting of acute disease—usually involve the fundus and body of the stomach. The stomach is lined with a glycoprotein mucous layer rich in bicarbonates, forming a physiologic barrier to protect the gastric wall from acid insult by neutralizing hydrogen ions. Disruption of this protective layer can occur in critically ill patients (eg, those with shock or sepsis) through overproduction of uremic toxins, increased reflux of bile salts, compromised blood flow, and increased stomach acidity through gastrin stimulation of parietal cells.
More than 75% of patients with major burns or cranial trauma develop endoscopic mucosal abnormalities within 72 hours of injury.1 In critically ill patients, the risk of ulcer-related overt bleeding is estimated to be 5% to 25%. Furthermore, 1% to 5% of stress ulcers can be deep enough to erode into the submucosa, causing clinically significant GI bleeding, defined as bleeding complicated by hemodynamic compromise or a drop in hemoglobin that requires a blood transfusion.2 In contrast, in inpatients who are not critically ill, the risk of overt bleeding from stress ulcers is less than 1%.3
ADDRESSING RISK
A multicenter prospective cohort study of 2,252 intensive care patients2 reported two main risk factors for significant bleeding caused by stress ulcers: mechanical ventilation for more than 48 hours and coagulopathy, defined as a platelet count below 50 × 109/L, an international normalized ratio greater than 1.5, or a partial thromboplastin time more than twice the control value.4 In hemodynamically stable patients receiving anticoagulation in a general medical or surgical ward, the risk of GI bleeding was low, and acid suppression failed to lower the rate of stress ulcer occurrence.3
Other risk factors include severe sepsis, shock, liver failure, kidney failure, burns over 35% of the total body surface, organ transplantation, cranial trauma, spinal cord trauma, history of peptic ulcer disease, and history of upper GI bleeding.3,5,6 Steroid therapy is not considered a risk factor for stress ulcers unless it is used in the presence of another risk factor such as use of aspirin or nonsteroidal antiinflammatory drugs (NSAIDs).2
INDICATIONS FOR PROPHYLAXIS
Prophylaxis with a proton pump inhibitor (PPI) is indicated in specific conditions—ie, peptic ulcer disease, gastroesophageal reflux disease, chronic NSAID therapy, and Zollinger-Ellison syndrome—and to eradicate Helicobacter pylori infection.7 But in the United States, stress ulcer prophylaxis is overused in general-care floors despite the lack of supporting evidence.
The American Society of Health System Pharmacists guidelines recommend it in the intensive care unit for patients with any of the following: coagulopathy, prolonged mechanical ventilation (more than 48 hours), GI ulcer or bleeding within the past year, sepsis, a stay longer than 1 week in the intensive care unit, occult GI bleeding for 6 or more days, and steroid therapy with more than 250 mg of hydrocortisone daily.8 Hemodynamically stable patients admitted to general-care floors should not receive stress ulcer prophylaxis, as it only negligibly decreases the rate of GI bleeding, from 0.33% to 0.22%.9
WHY ROUTINE ULCER PROPHYLAXIS IS NOT FOR ALL HOSPITALIZED PATIENTS
Although stress ulcer prophylaxis is often considered benign, its lack of proven benefit, additional cost, and risk of adverse effects, including interactions with foods and other drugs, preclude using it routinely for all hospitalized patients.10,11 Chronic use of PPIs has been associated with complications, as discussed below.
Infection
Acid suppression may impair the destruction of ingested microorganisms, resulting in overgrowth of bacteria.12 Overuse of PPIs may increase the risk of several infections:
- Diarrhea due to Clostridium difficile12
- Community-acquired pneumonia, from increased microaspiration of overgrown microorganisms into the lung.12
- Spontaneous bacterial peritonitis in patients with cirrhosis,13 although the mechanism is not clear. (Small-bowel bacterial overgrowth is the hypothesized cause.)
Bone fracture
PPIs lower gastric acidity, and this can inhibit intestinal calcium absorption. Furthermore, PPIs may directly inhibit bone resorption by osteoclasts.14
Reduction in clopidogrel efficacy
PPIs may reduce the efficacy of clopidogrel as a result of competitive inhibition of cytochrome CYP2C19, which is necessary to metabolize clopidogrel to its active forms. Therefore, concomitant use of clopidogrel with omeprazole, esomeprazole, or other CYP2C19 inhibitors is not recommended.15
Nutritional deficiencies
The overgrown microorganisms consume cobalamin in the stomach, resulting in vitamin B12 deficiency. Acid-suppressive therapy can also reduce the absorption of magnesium and iron.12
Unnecessary cost
Heidelbaugh and Inadomi16 reviewed the non-evidence-based use of stress ulcer prophylaxis in patients admitted to a large university hospital and estimated that it entailed a cost to the hospital of $111,791 over the course of a year.
WHICH ULCER PROPHYLAXIS SHOULD BE USED IN CRITICALLY ILL PATIENTS?
Studies have shown histamine-2 blockers to be superior to antacids and sucralfate in preventing stress ulcer and GI bleeding,8,15 but no study has compared PPIs with sucralfate and antacids.
When indicated, an oral PPI is preferred over an oral histamine-2 blocker for GI prophylaxis.17 This practice is considered cost-effective and is associated with lower rates of stress ulcer and GI bleeding. In intubated patients, however, an intravenous histamine-2 blocker is preferable to an intravenous PPI.3,8,11 Interestingly, no difference was reported between PPIs and histamine-2 blockers in terms of mortality rate or reduction in the incidence of nosocomial pneumonia.17
OUR RECOMMENDATION
Only critically ill patients who meet the specific criteria described here should receive stress ulcer prophylaxis. More effort is needed to educate residents, medical staff, and pharmacists about current guidelines. Computerized ordering templates and reminders to discontinue prophylaxis at discharge or step-down may decrease overall use, reduce costs, and limit potential side effects.18
- DePriest JL. Stress ulcer prophylaxis. Do critically ill patients need it? Postgrad Med 1995; 98:159–168.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- Qadeer MA, Richter JE, Brotman DJ. Hospital-acquired gastrointestinal bleeding outside the critical care unit: risk factors, role of acid suppression, and endoscopy findings. J Hosp Med 2006; 1:13–20.
- Shuman RB, Schuster DP, Zuckerman GR. Prophylactic therapy for stress ulcer bleeding: a reappraisal. Ann Intern Med 1987; 106:562–567.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Cook DJ, Reeve BK, Guyatt GH, et al. Stress ulcer prophylaxis in critically ill patients. Resolving discordant meta-analyses. JAMA 1996; 275:308–314.
- Kahrilas PJ, Shaheen NJ, Vaezi MF, et al; American Gastroenterological Association. American Gastroenterological Association Medical Position Statement on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1383–1391.e1–1391.e5.
- Barkun AN, Bardou M, Pham CQ, Martel M. Proton pump inhibitors vs histamine 2 receptor antagonists for stress-related mucosal bleeding prophylaxis in critically ill patients: a meta-analysis. Am J Gastroenterol 2012; 107:507–520.
- Herzig SJ, Vaughn BP, Howell MD, Ngo LH, Marcantonio ER. Acid-suppressive medication use and the risk for nosocomial gastrointestinal tract bleeding. Arch Intern Med 2011; 171:991–997.
- Cook DJ. Stress ulcer prophylaxis: gastrointestinal bleeding and nosocomial pneumonia. Best evidence synthesis. Scand J Gastroenterol Suppl 1995; 210:48–52.
- Messori A, Trippoli S, Vaiani M, Gorini M, Corrado A. Bleeding and pneumonia in intensive care patients given ranitidine and sucralfate for prevention of stress ulcer: meta-analysis of randomised controlled trials. BMJ 2000; 321:1103–1106.
- Heidelbaugh JJ, Kim AH, Chang R, Walker PC. Overutilization of proton-pump inhibitors: what the clinician needs to know. Therap Adv Gastroenterol 2012; 5:219–232.
- Deshpande A, Pasupuleti V, Thota P, et al. Acid-suppressive therapy is associated with spontaneous bacterial peritonitis in cirrhotic patients: a meta-analysis. J Gastroenterol Hepatol 2013; 28:235–242.
- Farina C, Gagliardi S. Selective inhibition of osteoclast vacuolar H(+)- ATPase. Curr Pharm Des 2002; 8:2033–2048.
- ASHP Therapeutic Guidelines on Stress Ulcer Prophylaxis. ASHP Commission on Therapeutics and approved by the ASHP Board of Directors on November 14, 1998. Am J Health Syst Pharm 1999; 56:347–379.
- Heidelbaugh JJ, Inadomi JM. Magnitude and economic impact of inappropriate use of stress ulcer prophylaxis in non-ICU hospitalized patients. Am J Gastroenterol 2006; 101:2200–2205.
- Alhazzani W, Alenezi F, Jaeschke RZ, Moayyedi P, Cook DJ. Proton pump inhibitors versus histamine 2 receptor antagonists for stress ulcer prophylaxis in critically ill patients: a systematic review and meta-analysis. Crit Care Med 2013; 41:693–705.
- Liberman JD, Whelan CT. Brief report: reducing inappropriate usage of stress ulcer prophylaxis among internal medicine residents. A practice-based educational intervention. J Gen Intern Med 2006; 21:498–500.
- DePriest JL. Stress ulcer prophylaxis. Do critically ill patients need it? Postgrad Med 1995; 98:159–168.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- Qadeer MA, Richter JE, Brotman DJ. Hospital-acquired gastrointestinal bleeding outside the critical care unit: risk factors, role of acid suppression, and endoscopy findings. J Hosp Med 2006; 1:13–20.
- Shuman RB, Schuster DP, Zuckerman GR. Prophylactic therapy for stress ulcer bleeding: a reappraisal. Ann Intern Med 1987; 106:562–567.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Cook DJ, Reeve BK, Guyatt GH, et al. Stress ulcer prophylaxis in critically ill patients. Resolving discordant meta-analyses. JAMA 1996; 275:308–314.
- Kahrilas PJ, Shaheen NJ, Vaezi MF, et al; American Gastroenterological Association. American Gastroenterological Association Medical Position Statement on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1383–1391.e1–1391.e5.
- Barkun AN, Bardou M, Pham CQ, Martel M. Proton pump inhibitors vs histamine 2 receptor antagonists for stress-related mucosal bleeding prophylaxis in critically ill patients: a meta-analysis. Am J Gastroenterol 2012; 107:507–520.
- Herzig SJ, Vaughn BP, Howell MD, Ngo LH, Marcantonio ER. Acid-suppressive medication use and the risk for nosocomial gastrointestinal tract bleeding. Arch Intern Med 2011; 171:991–997.
- Cook DJ. Stress ulcer prophylaxis: gastrointestinal bleeding and nosocomial pneumonia. Best evidence synthesis. Scand J Gastroenterol Suppl 1995; 210:48–52.
- Messori A, Trippoli S, Vaiani M, Gorini M, Corrado A. Bleeding and pneumonia in intensive care patients given ranitidine and sucralfate for prevention of stress ulcer: meta-analysis of randomised controlled trials. BMJ 2000; 321:1103–1106.
- Heidelbaugh JJ, Kim AH, Chang R, Walker PC. Overutilization of proton-pump inhibitors: what the clinician needs to know. Therap Adv Gastroenterol 2012; 5:219–232.
- Deshpande A, Pasupuleti V, Thota P, et al. Acid-suppressive therapy is associated with spontaneous bacterial peritonitis in cirrhotic patients: a meta-analysis. J Gastroenterol Hepatol 2013; 28:235–242.
- Farina C, Gagliardi S. Selective inhibition of osteoclast vacuolar H(+)- ATPase. Curr Pharm Des 2002; 8:2033–2048.
- ASHP Therapeutic Guidelines on Stress Ulcer Prophylaxis. ASHP Commission on Therapeutics and approved by the ASHP Board of Directors on November 14, 1998. Am J Health Syst Pharm 1999; 56:347–379.
- Heidelbaugh JJ, Inadomi JM. Magnitude and economic impact of inappropriate use of stress ulcer prophylaxis in non-ICU hospitalized patients. Am J Gastroenterol 2006; 101:2200–2205.
- Alhazzani W, Alenezi F, Jaeschke RZ, Moayyedi P, Cook DJ. Proton pump inhibitors versus histamine 2 receptor antagonists for stress ulcer prophylaxis in critically ill patients: a systematic review and meta-analysis. Crit Care Med 2013; 41:693–705.
- Liberman JD, Whelan CT. Brief report: reducing inappropriate usage of stress ulcer prophylaxis among internal medicine residents. A practice-based educational intervention. J Gen Intern Med 2006; 21:498–500.
Can an ARB be given to patients who have had angioedema on an ACE inhibitor?
Current evidence suggests no absolute contraindication to angiotensin receptor blockers (ARBs) in patients who have had angioedema attributable to an angiotensin-converting enzyme (ACE) inhibitor. However, since ARBs can also cause angioedema, they should be prescribed with extreme caution after a thorough risk-benefit analysis and after educating the patient to watch for signs of angioedema while taking the drug.
A GROWING PROBLEM
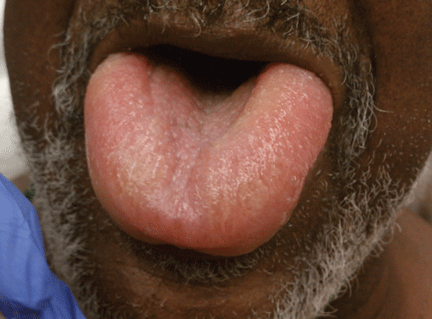
Angioedema is a potentially life-threatening swelling of the skin and subcutaneous tissues, often affecting the lips and tongue (Figure 1), and in some cases interfering with breathing and requiring tracheostomy.1 The incidence rate of angioedema in patients taking ACE inhibitors ranges from 0.1% to 0.7%.2–4 Although this rate may seem low, the widespread and growing use of ACE inhibitors and ARBs in patients with diabetes, diabetic nephropathy, and congestive heart failure5 makes angioedema fairly common in clinical practice.
ACE inhibitor-induced angioedema most commonly occurs within days of initiating therapy, but it also may occur weeks, months, or even years after the start of treatment.1 Patients who are over age 65, black, or female are at higher risk, as are renal transplant recipients taking mTOR inhibitors such as sirolimus. Diabetes appears to be associated with a lower risk.4,6,7 This adverse reaction to ACE inhibitors is thought to be a class side effect, and the future use of this class of drugs would be contraindicated.8,9
ACE inhibitors cause angioedema by direct interference with the degradation of bradykinin, thereby increasing bradykinin levels and potentiating its biologic effect, leading to increased vascular permeability, inflammation, and activation of nociceptors.8
EVIDENCE TO SUPPORT THE USE OF ARBs
ACE inhibitors and ARBs both block the renin-angiotensin-aldosterone pathway and confer similar advantages in patients with congestive heart failure, renal failure, and diabetes. But since ARBs directly inhibit the angiotensin receptor and do not interfere with bradykinin degradation, how they cause angioedema is unclear, and clinicians have questioned whether these agents might be used safely in patients who have had angioedema on an ACE inhibitor.
In a large meta-analysis of randomized clinical trials, Makani et al2 investigated the risk of angioedema with ARB use in 35,479 patients and compared this with other commonly used antihypertensive drugs. The weighted incidence of angioedema was 0.30% with an ACE inhibitor, 0.11% with an ARB, and 0.07% with placebo.2 In seven trials included in this study that compared ARBs with placebo, there was no significant difference in the risk of angioedema. Even in such a large study, the event rate was small, making definite conclusions difficult.
In a retrospective observational study of 4 million patients by Toh et al,3 patients on beta-blockers were used as a reference, and propensity scoring was used to estimate the hazard ratio of angioedema separately for drugs targeting the renin-angiotensin-aldosterone system, including ACE inhibitors and ARBs. The risk of angioedema, as measured by the cumulative incidence and incidence rate, was highest for ACE inhibitors and was similar between ARBs and beta-blockers. The risk of serious angioedema was three times higher with ACE inhibitors than with beta-blockers, and there was no higher risk of serious angioedema with ARBs than with beta-blockers.3
Looking specifically at the use of ARBs in patients who developed angioedema on an ACE inhibitor, Haymore et al10 performed a meta-analysis evaluating only three studies that showed the estimated risk of angioedema with an ARB was between 3.5% and 9.4% in patients with a history of ACE inhibitor-induced angioedema. Later, when the results of the Telmisartan Randomised Assessment Study in ACE Intolerant Subjects With Cardiovascular Disease trial11 were published, the previous meta-analysis was updated12: the risk of angioedema with an ARB was only 2.5% (95% confidence interval 0%–6.6%), and there was no statistically significant difference in the odds (odds ratio 1.1; 95% confidence interval 0.07–17) of angioedema between ARBs and placebo.10,12 Again, these results should be interpreted with caution, as only two patients in the ARB (telmisartan) group and three patients in the placebo group developed angioedema.
In another review, Beavers et al13 advised that the prescribing practitioner should carefully perform a risk-benefit analysis before substituting an ARB in patients with ACE inhibitor-induced angioedema. They concluded that an ARB could be considered in patients who are likely to have a large clinical benefit from an ARB, such as those with heart failure. They also suggested that angioedema related to ARBs was less severe and occurred earlier than with that linked to ACE inhibitors.
No large clinical trial has yet been specifically designed to address the use of ARBs in patients with a history of ACE inhibitor-induced angioedema. The package insert for the ARB losartan mentions that the risk of this adverse reaction might be higher in patients who have had angioedema on an ACE inhibitor. However, the issue of recurrent angioedema is not further addressed for this or other commonly used ARBs.
GENERAL RECOMMENDATIONS
The mechanisms of ARB-induced angioedema are yet unknown. However, studies have shown that the incidence of angioedema while on an ARB is low and is probably comparable to that of placebo.2,3,12–14 And since ARBs share many of the cardiac and renal protective effects of ACE inhibitors, ARBs may be beneficial for patients who discontinue an ACE inhibitor because of adverse effects including angioedema.9,15,16 Based on the discussion above, there is no clear evidence to suggest that ARBs are contraindicated in such patients, especially if there is a compelling indication for an ARB.
The National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) guidelines on hypertension in chronic kidney disease recommend caution when substituting an ARB for an ACE inhibitor after angioedema.15 The joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA) for the diagnosis and management of heart failure in adults advise “extreme caution.”9,16
The risks and benefits of ARB therapy in this setting should be analyzed by the prescribing physician and discussed with the patient. The patient should be closely monitored for the recurrence of angioedema and should be given a clear plan of action should symptoms recur.
OUR ADVICE
In patients with ACE inhibitor-induced angioedema, we recommend the following:
- Determine that the patient truly has one of the evidence-based, compelling indications for an ARB. Carefully weigh the risks and benefits for the individual patient, and discuss the risk of angioedema based on age, race, sex, and medical history, and the availability of immediate medical care should angioedema occur.
- If there is an evidence-based indication for an ARB that outweighs the risk of angioedema, an ARB may be started with caution.
- Specifically discuss with the patient the possibility of recurrence of angioedema while on an ARB, and provide instructions on how to proceed if this should occur.
- Kaplan AP, Greaves MW. Angioedema. J Am Acad Dermatol 2005; 53:373–388.
- Makani H, Messerli FH, Romero J, et al. Meta-analysis of randomized trials of angioedema as an adverse event of renin-angiotensin system inhibitors. Am J Cardiol 2012; 110:383–391.
- Toh S, Reichman ME, Houstoun M, et al. Comparative risk for angioedema associated with the use of drugs that target the renin-angiotensin-aldosterone system. Arch Intern Med 2012; 172:1582–1589.
- Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med 2005; 165:1637–1642.
- Taylor AA, Siragy H, Nesbitt S. Angiotensin receptor blockers: pharmacology, efficacy, and safety. J Clin Hypertens (Greenwich) 2011; 13:677–686.
- Duerr M, Glander P, Diekmann F, Dragun D, Neumayer HH, Budde K. Increased incidence of angioedema with ACE inhibitors in combination with mTOR inhibitors in kidney transplant recipients. Clin J Am Soc Nephrol 2010; 5:703–708.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am 2006; 26:725–737.
- Inomata N. Recent advances in drug-induced angioedema. Allergol Int 2012; 61:545–557.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology Foundation; American Heart Association. 2009 Focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the International Society for Heart and Lung Transplantation. J Am Coll Cardiol 2009; 53:e1–e90.
- Haymore BR, Yoon J, Mikita CP, Klote MM, DeZee KJ. Risk of angioedema with angiotensin receptor blockers in patients with prior angioedema associated with angiotensin-converting enzyme inhibitors: a meta-analysis. Ann Allergy Asthma Immunol 2008; 101:495–499.
- Telmisartan Randomised Assessment Study in ACE Intolerant Subjects with Cardiovascular Disease (TRANSCEND) Investigators. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: a randomised controlled trial. Lancet 2008; 372:1174–1183.
- Haymore BR, DeZee KJ. Use of angiotensin receptor blockers after angioedema with an angiotensin-converting enzyme inhibitor. Ann Allergy Asthma Immunol 2009; 103:83–84.
- Beavers CJ, Dunn SP, Macaulay TE. The role of angiotensin receptor blockers in patients with angiotensin-converting enzyme inhibitor-induced angioedema. Ann Pharmacother 2011; 45:520–524.
- Caldeira D, David C, Sampaio C. Tolerability of angiotensin-receptor blockers in patients with intolerance to angiotensin-converting enzyme inhibitors: a systematic review and meta-analysis. Am J Cardiovasc Drugs 2012; 12:263–277.
- Kidney Disease Outcomes Quality Initiative (K/DOQI). K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004; 43(suppl 1):S1–S290.
- Smith SC, Benjamin EJ, Bonow RO, et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation endorsed by the World Heart Federation and the Preventive Cardiovascular Nurses Association. J Am Coll Cardiol 2011; 58:2432–2446.
Current evidence suggests no absolute contraindication to angiotensin receptor blockers (ARBs) in patients who have had angioedema attributable to an angiotensin-converting enzyme (ACE) inhibitor. However, since ARBs can also cause angioedema, they should be prescribed with extreme caution after a thorough risk-benefit analysis and after educating the patient to watch for signs of angioedema while taking the drug.
A GROWING PROBLEM
Angioedema is a potentially life-threatening swelling of the skin and subcutaneous tissues, often affecting the lips and tongue (Figure 1), and in some cases interfering with breathing and requiring tracheostomy.1 The incidence rate of angioedema in patients taking ACE inhibitors ranges from 0.1% to 0.7%.2–4 Although this rate may seem low, the widespread and growing use of ACE inhibitors and ARBs in patients with diabetes, diabetic nephropathy, and congestive heart failure5 makes angioedema fairly common in clinical practice.
ACE inhibitor-induced angioedema most commonly occurs within days of initiating therapy, but it also may occur weeks, months, or even years after the start of treatment.1 Patients who are over age 65, black, or female are at higher risk, as are renal transplant recipients taking mTOR inhibitors such as sirolimus. Diabetes appears to be associated with a lower risk.4,6,7 This adverse reaction to ACE inhibitors is thought to be a class side effect, and the future use of this class of drugs would be contraindicated.8,9
ACE inhibitors cause angioedema by direct interference with the degradation of bradykinin, thereby increasing bradykinin levels and potentiating its biologic effect, leading to increased vascular permeability, inflammation, and activation of nociceptors.8
EVIDENCE TO SUPPORT THE USE OF ARBs
ACE inhibitors and ARBs both block the renin-angiotensin-aldosterone pathway and confer similar advantages in patients with congestive heart failure, renal failure, and diabetes. But since ARBs directly inhibit the angiotensin receptor and do not interfere with bradykinin degradation, how they cause angioedema is unclear, and clinicians have questioned whether these agents might be used safely in patients who have had angioedema on an ACE inhibitor.
In a large meta-analysis of randomized clinical trials, Makani et al2 investigated the risk of angioedema with ARB use in 35,479 patients and compared this with other commonly used antihypertensive drugs. The weighted incidence of angioedema was 0.30% with an ACE inhibitor, 0.11% with an ARB, and 0.07% with placebo.2 In seven trials included in this study that compared ARBs with placebo, there was no significant difference in the risk of angioedema. Even in such a large study, the event rate was small, making definite conclusions difficult.
In a retrospective observational study of 4 million patients by Toh et al,3 patients on beta-blockers were used as a reference, and propensity scoring was used to estimate the hazard ratio of angioedema separately for drugs targeting the renin-angiotensin-aldosterone system, including ACE inhibitors and ARBs. The risk of angioedema, as measured by the cumulative incidence and incidence rate, was highest for ACE inhibitors and was similar between ARBs and beta-blockers. The risk of serious angioedema was three times higher with ACE inhibitors than with beta-blockers, and there was no higher risk of serious angioedema with ARBs than with beta-blockers.3
Looking specifically at the use of ARBs in patients who developed angioedema on an ACE inhibitor, Haymore et al10 performed a meta-analysis evaluating only three studies that showed the estimated risk of angioedema with an ARB was between 3.5% and 9.4% in patients with a history of ACE inhibitor-induced angioedema. Later, when the results of the Telmisartan Randomised Assessment Study in ACE Intolerant Subjects With Cardiovascular Disease trial11 were published, the previous meta-analysis was updated12: the risk of angioedema with an ARB was only 2.5% (95% confidence interval 0%–6.6%), and there was no statistically significant difference in the odds (odds ratio 1.1; 95% confidence interval 0.07–17) of angioedema between ARBs and placebo.10,12 Again, these results should be interpreted with caution, as only two patients in the ARB (telmisartan) group and three patients in the placebo group developed angioedema.
In another review, Beavers et al13 advised that the prescribing practitioner should carefully perform a risk-benefit analysis before substituting an ARB in patients with ACE inhibitor-induced angioedema. They concluded that an ARB could be considered in patients who are likely to have a large clinical benefit from an ARB, such as those with heart failure. They also suggested that angioedema related to ARBs was less severe and occurred earlier than with that linked to ACE inhibitors.
No large clinical trial has yet been specifically designed to address the use of ARBs in patients with a history of ACE inhibitor-induced angioedema. The package insert for the ARB losartan mentions that the risk of this adverse reaction might be higher in patients who have had angioedema on an ACE inhibitor. However, the issue of recurrent angioedema is not further addressed for this or other commonly used ARBs.
GENERAL RECOMMENDATIONS
The mechanisms of ARB-induced angioedema are yet unknown. However, studies have shown that the incidence of angioedema while on an ARB is low and is probably comparable to that of placebo.2,3,12–14 And since ARBs share many of the cardiac and renal protective effects of ACE inhibitors, ARBs may be beneficial for patients who discontinue an ACE inhibitor because of adverse effects including angioedema.9,15,16 Based on the discussion above, there is no clear evidence to suggest that ARBs are contraindicated in such patients, especially if there is a compelling indication for an ARB.
The National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) guidelines on hypertension in chronic kidney disease recommend caution when substituting an ARB for an ACE inhibitor after angioedema.15 The joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA) for the diagnosis and management of heart failure in adults advise “extreme caution.”9,16
The risks and benefits of ARB therapy in this setting should be analyzed by the prescribing physician and discussed with the patient. The patient should be closely monitored for the recurrence of angioedema and should be given a clear plan of action should symptoms recur.
OUR ADVICE
In patients with ACE inhibitor-induced angioedema, we recommend the following:
- Determine that the patient truly has one of the evidence-based, compelling indications for an ARB. Carefully weigh the risks and benefits for the individual patient, and discuss the risk of angioedema based on age, race, sex, and medical history, and the availability of immediate medical care should angioedema occur.
- If there is an evidence-based indication for an ARB that outweighs the risk of angioedema, an ARB may be started with caution.
- Specifically discuss with the patient the possibility of recurrence of angioedema while on an ARB, and provide instructions on how to proceed if this should occur.
Current evidence suggests no absolute contraindication to angiotensin receptor blockers (ARBs) in patients who have had angioedema attributable to an angiotensin-converting enzyme (ACE) inhibitor. However, since ARBs can also cause angioedema, they should be prescribed with extreme caution after a thorough risk-benefit analysis and after educating the patient to watch for signs of angioedema while taking the drug.
A GROWING PROBLEM
Angioedema is a potentially life-threatening swelling of the skin and subcutaneous tissues, often affecting the lips and tongue (Figure 1), and in some cases interfering with breathing and requiring tracheostomy.1 The incidence rate of angioedema in patients taking ACE inhibitors ranges from 0.1% to 0.7%.2–4 Although this rate may seem low, the widespread and growing use of ACE inhibitors and ARBs in patients with diabetes, diabetic nephropathy, and congestive heart failure5 makes angioedema fairly common in clinical practice.
ACE inhibitor-induced angioedema most commonly occurs within days of initiating therapy, but it also may occur weeks, months, or even years after the start of treatment.1 Patients who are over age 65, black, or female are at higher risk, as are renal transplant recipients taking mTOR inhibitors such as sirolimus. Diabetes appears to be associated with a lower risk.4,6,7 This adverse reaction to ACE inhibitors is thought to be a class side effect, and the future use of this class of drugs would be contraindicated.8,9
ACE inhibitors cause angioedema by direct interference with the degradation of bradykinin, thereby increasing bradykinin levels and potentiating its biologic effect, leading to increased vascular permeability, inflammation, and activation of nociceptors.8
EVIDENCE TO SUPPORT THE USE OF ARBs
ACE inhibitors and ARBs both block the renin-angiotensin-aldosterone pathway and confer similar advantages in patients with congestive heart failure, renal failure, and diabetes. But since ARBs directly inhibit the angiotensin receptor and do not interfere with bradykinin degradation, how they cause angioedema is unclear, and clinicians have questioned whether these agents might be used safely in patients who have had angioedema on an ACE inhibitor.
In a large meta-analysis of randomized clinical trials, Makani et al2 investigated the risk of angioedema with ARB use in 35,479 patients and compared this with other commonly used antihypertensive drugs. The weighted incidence of angioedema was 0.30% with an ACE inhibitor, 0.11% with an ARB, and 0.07% with placebo.2 In seven trials included in this study that compared ARBs with placebo, there was no significant difference in the risk of angioedema. Even in such a large study, the event rate was small, making definite conclusions difficult.
In a retrospective observational study of 4 million patients by Toh et al,3 patients on beta-blockers were used as a reference, and propensity scoring was used to estimate the hazard ratio of angioedema separately for drugs targeting the renin-angiotensin-aldosterone system, including ACE inhibitors and ARBs. The risk of angioedema, as measured by the cumulative incidence and incidence rate, was highest for ACE inhibitors and was similar between ARBs and beta-blockers. The risk of serious angioedema was three times higher with ACE inhibitors than with beta-blockers, and there was no higher risk of serious angioedema with ARBs than with beta-blockers.3
Looking specifically at the use of ARBs in patients who developed angioedema on an ACE inhibitor, Haymore et al10 performed a meta-analysis evaluating only three studies that showed the estimated risk of angioedema with an ARB was between 3.5% and 9.4% in patients with a history of ACE inhibitor-induced angioedema. Later, when the results of the Telmisartan Randomised Assessment Study in ACE Intolerant Subjects With Cardiovascular Disease trial11 were published, the previous meta-analysis was updated12: the risk of angioedema with an ARB was only 2.5% (95% confidence interval 0%–6.6%), and there was no statistically significant difference in the odds (odds ratio 1.1; 95% confidence interval 0.07–17) of angioedema between ARBs and placebo.10,12 Again, these results should be interpreted with caution, as only two patients in the ARB (telmisartan) group and three patients in the placebo group developed angioedema.
In another review, Beavers et al13 advised that the prescribing practitioner should carefully perform a risk-benefit analysis before substituting an ARB in patients with ACE inhibitor-induced angioedema. They concluded that an ARB could be considered in patients who are likely to have a large clinical benefit from an ARB, such as those with heart failure. They also suggested that angioedema related to ARBs was less severe and occurred earlier than with that linked to ACE inhibitors.
No large clinical trial has yet been specifically designed to address the use of ARBs in patients with a history of ACE inhibitor-induced angioedema. The package insert for the ARB losartan mentions that the risk of this adverse reaction might be higher in patients who have had angioedema on an ACE inhibitor. However, the issue of recurrent angioedema is not further addressed for this or other commonly used ARBs.
GENERAL RECOMMENDATIONS
The mechanisms of ARB-induced angioedema are yet unknown. However, studies have shown that the incidence of angioedema while on an ARB is low and is probably comparable to that of placebo.2,3,12–14 And since ARBs share many of the cardiac and renal protective effects of ACE inhibitors, ARBs may be beneficial for patients who discontinue an ACE inhibitor because of adverse effects including angioedema.9,15,16 Based on the discussion above, there is no clear evidence to suggest that ARBs are contraindicated in such patients, especially if there is a compelling indication for an ARB.
The National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) guidelines on hypertension in chronic kidney disease recommend caution when substituting an ARB for an ACE inhibitor after angioedema.15 The joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA) for the diagnosis and management of heart failure in adults advise “extreme caution.”9,16
The risks and benefits of ARB therapy in this setting should be analyzed by the prescribing physician and discussed with the patient. The patient should be closely monitored for the recurrence of angioedema and should be given a clear plan of action should symptoms recur.
OUR ADVICE
In patients with ACE inhibitor-induced angioedema, we recommend the following:
- Determine that the patient truly has one of the evidence-based, compelling indications for an ARB. Carefully weigh the risks and benefits for the individual patient, and discuss the risk of angioedema based on age, race, sex, and medical history, and the availability of immediate medical care should angioedema occur.
- If there is an evidence-based indication for an ARB that outweighs the risk of angioedema, an ARB may be started with caution.
- Specifically discuss with the patient the possibility of recurrence of angioedema while on an ARB, and provide instructions on how to proceed if this should occur.
- Kaplan AP, Greaves MW. Angioedema. J Am Acad Dermatol 2005; 53:373–388.
- Makani H, Messerli FH, Romero J, et al. Meta-analysis of randomized trials of angioedema as an adverse event of renin-angiotensin system inhibitors. Am J Cardiol 2012; 110:383–391.
- Toh S, Reichman ME, Houstoun M, et al. Comparative risk for angioedema associated with the use of drugs that target the renin-angiotensin-aldosterone system. Arch Intern Med 2012; 172:1582–1589.
- Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med 2005; 165:1637–1642.
- Taylor AA, Siragy H, Nesbitt S. Angiotensin receptor blockers: pharmacology, efficacy, and safety. J Clin Hypertens (Greenwich) 2011; 13:677–686.
- Duerr M, Glander P, Diekmann F, Dragun D, Neumayer HH, Budde K. Increased incidence of angioedema with ACE inhibitors in combination with mTOR inhibitors in kidney transplant recipients. Clin J Am Soc Nephrol 2010; 5:703–708.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am 2006; 26:725–737.
- Inomata N. Recent advances in drug-induced angioedema. Allergol Int 2012; 61:545–557.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology Foundation; American Heart Association. 2009 Focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the International Society for Heart and Lung Transplantation. J Am Coll Cardiol 2009; 53:e1–e90.
- Haymore BR, Yoon J, Mikita CP, Klote MM, DeZee KJ. Risk of angioedema with angiotensin receptor blockers in patients with prior angioedema associated with angiotensin-converting enzyme inhibitors: a meta-analysis. Ann Allergy Asthma Immunol 2008; 101:495–499.
- Telmisartan Randomised Assessment Study in ACE Intolerant Subjects with Cardiovascular Disease (TRANSCEND) Investigators. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: a randomised controlled trial. Lancet 2008; 372:1174–1183.
- Haymore BR, DeZee KJ. Use of angiotensin receptor blockers after angioedema with an angiotensin-converting enzyme inhibitor. Ann Allergy Asthma Immunol 2009; 103:83–84.
- Beavers CJ, Dunn SP, Macaulay TE. The role of angiotensin receptor blockers in patients with angiotensin-converting enzyme inhibitor-induced angioedema. Ann Pharmacother 2011; 45:520–524.
- Caldeira D, David C, Sampaio C. Tolerability of angiotensin-receptor blockers in patients with intolerance to angiotensin-converting enzyme inhibitors: a systematic review and meta-analysis. Am J Cardiovasc Drugs 2012; 12:263–277.
- Kidney Disease Outcomes Quality Initiative (K/DOQI). K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004; 43(suppl 1):S1–S290.
- Smith SC, Benjamin EJ, Bonow RO, et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation endorsed by the World Heart Federation and the Preventive Cardiovascular Nurses Association. J Am Coll Cardiol 2011; 58:2432–2446.
- Kaplan AP, Greaves MW. Angioedema. J Am Acad Dermatol 2005; 53:373–388.
- Makani H, Messerli FH, Romero J, et al. Meta-analysis of randomized trials of angioedema as an adverse event of renin-angiotensin system inhibitors. Am J Cardiol 2012; 110:383–391.
- Toh S, Reichman ME, Houstoun M, et al. Comparative risk for angioedema associated with the use of drugs that target the renin-angiotensin-aldosterone system. Arch Intern Med 2012; 172:1582–1589.
- Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med 2005; 165:1637–1642.
- Taylor AA, Siragy H, Nesbitt S. Angiotensin receptor blockers: pharmacology, efficacy, and safety. J Clin Hypertens (Greenwich) 2011; 13:677–686.
- Duerr M, Glander P, Diekmann F, Dragun D, Neumayer HH, Budde K. Increased incidence of angioedema with ACE inhibitors in combination with mTOR inhibitors in kidney transplant recipients. Clin J Am Soc Nephrol 2010; 5:703–708.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am 2006; 26:725–737.
- Inomata N. Recent advances in drug-induced angioedema. Allergol Int 2012; 61:545–557.
- Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology Foundation; American Heart Association. 2009 Focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the International Society for Heart and Lung Transplantation. J Am Coll Cardiol 2009; 53:e1–e90.
- Haymore BR, Yoon J, Mikita CP, Klote MM, DeZee KJ. Risk of angioedema with angiotensin receptor blockers in patients with prior angioedema associated with angiotensin-converting enzyme inhibitors: a meta-analysis. Ann Allergy Asthma Immunol 2008; 101:495–499.
- Telmisartan Randomised Assessment Study in ACE Intolerant Subjects with Cardiovascular Disease (TRANSCEND) Investigators. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: a randomised controlled trial. Lancet 2008; 372:1174–1183.
- Haymore BR, DeZee KJ. Use of angiotensin receptor blockers after angioedema with an angiotensin-converting enzyme inhibitor. Ann Allergy Asthma Immunol 2009; 103:83–84.
- Beavers CJ, Dunn SP, Macaulay TE. The role of angiotensin receptor blockers in patients with angiotensin-converting enzyme inhibitor-induced angioedema. Ann Pharmacother 2011; 45:520–524.
- Caldeira D, David C, Sampaio C. Tolerability of angiotensin-receptor blockers in patients with intolerance to angiotensin-converting enzyme inhibitors: a systematic review and meta-analysis. Am J Cardiovasc Drugs 2012; 12:263–277.
- Kidney Disease Outcomes Quality Initiative (K/DOQI). K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004; 43(suppl 1):S1–S290.
- Smith SC, Benjamin EJ, Bonow RO, et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation endorsed by the World Heart Federation and the Preventive Cardiovascular Nurses Association. J Am Coll Cardiol 2011; 58:2432–2446.
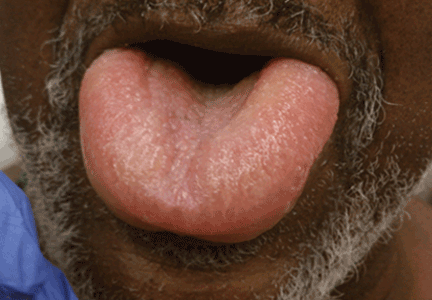
How should we manage insulin therapy before surgery?
Continuing at least part of the basal insulin is the reasonable, physiologic approach to controlling glucose levels before surgery in patients with diabetes. The process involves three basic steps:
- Ascertaining the type of diabetes
- Adjusting the basal insulin dosage
- Stopping the prandial insulin.
The steps are the same whether the surgery is major or minor. These recommendations are based on general principles of insulin action, data from large databases of surgical inpatients, and expert clinical experience translated into standardized protocols.1,2
WHY CONTINUE THE INSULIN?
Stopping or decreasing insulin because of a fear of hypoglycemia is not appropriate, as the resulting hyperglycemia can lead to delayed wound healing, wound infection, fluid and electrolyte shifts, diabetic ketoacidosis, and hyperosmolar states.
Insulin inhibits both gluconeogenesis and conversion of glycogen to glucose, processes that occur regardless of food intake. It also inhibits degradation of fats to fatty acids and of fatty acids to ketones. This is why inadequate insulin dosing can lead to uncontrolled hyperglycemia and even ketoacidosis, and thus why long-acting insulin is needed in a fasting state.
STEP 1: ASCERTAIN THE TYPE OF DIABETES
Does the patient have type 1 or type 2 diabetes, and does that even matter?
The type of diabetes should not matter, since ideally the insulin should be dosed the same for both types. However, the consequences of inappropriate insulin management may be different.
Usually, the type of diabetes can be ascertained by the history. If the patient was diagnosed at age 40 or later and was on oral medication for years before insulin was started, then he or she most likely has type 2. If the patient was younger than 40 at the time of diagnosis, was lean, and was started on insulin within a year of diagnosis, then he or she likely has type 1.
If this information is not available or is unreliable and the patient has been on insulin for many years, we recommend viewing the patient as being insulinopenic, ie, not producing enough insulin endogenously and thus requiring insulin at all times.
Though checking for antibody markers of type 1 diabetes might give a more definitive answer, it is not practical before surgery.
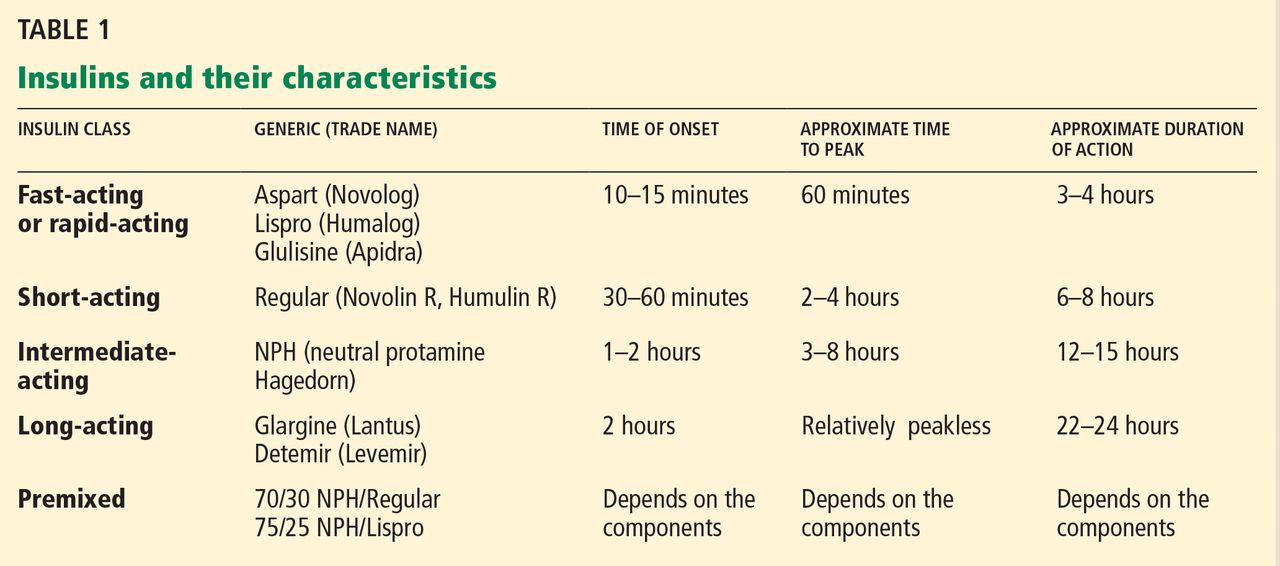
In the setting of surgical stress, withholding the basal insulin preoperatively and just giving a small dose of fast-acting (see Table 1 for the different classes of insulin) or shortacting insulin as part of a sliding scale (ie, insulin given only when the blood glucose reaches a certain high level) can send a patient with type 1 diabetes into diabetic ketoacidosis by the end of the day. This is less likely to occur in a patient with type 2 diabetes with some endogenous insulin secretion.
STEP 2: ADJUST THE BASAL INSULIN
Basal insulin is the insulin that the healthy person’s body produces when fasting. For a diabetic patient already on insulin, basal insulin is insulin injected to prevent ketogenesis, glycogenolysis, and gluconeogenesis in the fasting state.
If the basal insulin is long-acting
Long-acting insulins have a relatively peakless profile and, when properly dosed, should not result in hypoglycemia when a patient is fasting.
Preoperatively, the patient should take it as close as possible to the usual time of injection. This could be at home either at bedtime the night before surgery or the morning of surgery. If there is concern for hypoglycemia, the injection can be given when the patient is at the hospital.
- If the patient does not tend to have hypoglycemic episodes and the total daily basal insulin dose is roughly the same as the total daily mealtime (prandial) dose (eg, 50% basal, 50% prandial ratio), the full dose of basal insulin can be given.3
Example: If the patient is on insulin glargine 30 U at bedtime and insulin lispro 10 U with each meal and does not have hypoglycemic episodes, then insulin glargine 30 U should be taken at bedtime.
- If the patient has hypoglycemic episodes at home, then the basal insulin can be reduced by 25%.3
Example: If the patient is on insulin glargine 30 U at bedtime and insulin glulisine 10 U with each meal (appropriate proportion of doses, similar to the example above) but has hypoglycemic episodes at home on this regimen, then only 22 U of insulin glargine should be taken at bedtime.
- If the patient’s regimen has disproportionately more basal insulin than mealtime insulin, then the total daily doses can be added and half can be given as the basal insulin.
Example: If the patient is on insulin detemir 30 U every morning at 6 am and insulin aspart 6 U with each meal and has no hypoglycemic episodes, then 24 U of insulin detemir should be taken in the morning (ie, half of the total of 30 + 6 + 6 + 6).
- In the less common scenario of diabetes managed only with basal insulin (no other diabetes injections or oral agents), then half of the dose can be given.
- If the patient is on twice-daily long-acting basal insulin, then both the dose the night before surgery and the dose the morning of surgery should be adjusted.
If the basal insulin is intermediate-acting
The intermediate-acting insulin neutral protamine Hagedorn (NPH) is usually given twice a day because of its profile (Table 1).
- On the night before surgery, the full dose of NPH insulin should be taken, unless the patient will now skip a nighttime meal because of taking nothing by mouth, in which case the dose can be decreased by 25%.1
- On the morning of surgery, since the patient will be skipping breakfast and probably also lunch, the dose should be reduced by 50%.3,4
Special situation: Premixed insulins
Premixed insulins (70/30, 75/25) are a combination of intermediate-acting insulin and either fast-acting or short-acting insulin. In other words, they are combinations of basal and prandial insulin. Their use is thus not ideal in the preoperative period. There are two options in these situations.
One option is to switch to a regimen that includes long-acting insulin. If the patient is admitted for surgery, then the hospital staff can change the insulin regimen to long-acting basal insulin. A quick formula for conversion is to add all the premixed insulin doses and give half as basal insulin on the morning of surgery, similar to the scenario above for the patient with long-acting basal insulin that was out of proportion to the prandial insulin injections.
For example, if the usual regimen is insulin 70/30 NPH/Regular, 60 U with breakfast, 30 U with dinner, then the patient can take 45 U of insulin glargine (which is half of 60 + 30) in the morning or evening before surgery.
Another option is to adjust the dose of pre-mixed insulin. Sometimes it is not feasible or economical to change the patient’s premixed insulin just before surgery. In these situations, the patient can take half of the morning dose, followed by dextrose-containing intravenous fluids and blood glucose checks.
We recommend preoperatively giving at least part of the patient’s previous basal insulin, regardless of the type of diabetes, the type of surgery, or the fasting period.
STEP 3: STOP THE PRANDIAL INSULIN
Prandial insulin—given before each meal to cover the carbohydrates to be consumed—should be stopped the morning of surgery.3,4
WHAT ABOUT SLIDING SCALE INSULIN?
Using a sliding scale alone has no known benefit. Although it can be a quick fix to correct a high glucose level, it should be added to the basal insulin and not used as the sole insulin therapy. If a sliding scale is used, fast-acting insulin (aspart, glulisine, lispro) is preferred over regular insulin because of the more rapid onset and shorter duration of action.
Patients already using a supplemental insulin scale can apply it to correct a blood glucose above 200 mg/dL on the morning of surgery.
MAINTENANCE FLUIDS
As long as glucose levels are not very elevated (ie, > 200 mg/dL), after 12 hours on a nothing-by-mouth regimen, provide dextrose in the IV fluid to prevent hypoglycemia (eg, the patient received long-acting insulin and the glucose levels are running low) or to prevent starvation ketosis, which may result in ketones in the blood or urine. We recommend 5% dextrose in half-normal (0.45%) saline at 50 to 75 mL/hour as maintenance fluid; the infusion rate should be lower if fluid overload is a concern.
POSTOPERATIVE INSULIN MANAGEMENT
Once patients are discharged and can go back to their previous routine, they can restart their usual insulin regimen the same evening. The prandial insulin will be resumed when the regular diet is reintroduced, and the doses will be adjusted according to food intake.
- Joshi GP, Chung F, Vann MA, et al; Society for Ambulatory Anesthesia. Society for Ambulatory Anesthesia consensus statement on perioperative blood glucose management in diabetic patients undergoing ambulatory surgery. Anesth Analg 2010; 111:1378–1387.
- DiNardo M, Donihi AC, Forte P, Gieraltowski L, Korytkowski M. Standardized glycemic management and perioperative glycemic outcomes in patients with diabetes mellitus who undergo same-day surgery. Endocr Pract 2011; 17:404–411.
- Vann MA. Perioperative management of ambulatory surgical patients with diabetes mellitus. Curr Opin Anaesthesiol 2009; 22:718–724.
- Meneghini LF. Perioperative management of diabetes: translating evidence into practice. Cleve Clin J Med 2009; 76(suppl 4):S53–S59.
Continuing at least part of the basal insulin is the reasonable, physiologic approach to controlling glucose levels before surgery in patients with diabetes. The process involves three basic steps:
- Ascertaining the type of diabetes
- Adjusting the basal insulin dosage
- Stopping the prandial insulin.
The steps are the same whether the surgery is major or minor. These recommendations are based on general principles of insulin action, data from large databases of surgical inpatients, and expert clinical experience translated into standardized protocols.1,2
WHY CONTINUE THE INSULIN?
Stopping or decreasing insulin because of a fear of hypoglycemia is not appropriate, as the resulting hyperglycemia can lead to delayed wound healing, wound infection, fluid and electrolyte shifts, diabetic ketoacidosis, and hyperosmolar states.
Insulin inhibits both gluconeogenesis and conversion of glycogen to glucose, processes that occur regardless of food intake. It also inhibits degradation of fats to fatty acids and of fatty acids to ketones. This is why inadequate insulin dosing can lead to uncontrolled hyperglycemia and even ketoacidosis, and thus why long-acting insulin is needed in a fasting state.
STEP 1: ASCERTAIN THE TYPE OF DIABETES
Does the patient have type 1 or type 2 diabetes, and does that even matter?
The type of diabetes should not matter, since ideally the insulin should be dosed the same for both types. However, the consequences of inappropriate insulin management may be different.
Usually, the type of diabetes can be ascertained by the history. If the patient was diagnosed at age 40 or later and was on oral medication for years before insulin was started, then he or she most likely has type 2. If the patient was younger than 40 at the time of diagnosis, was lean, and was started on insulin within a year of diagnosis, then he or she likely has type 1.
If this information is not available or is unreliable and the patient has been on insulin for many years, we recommend viewing the patient as being insulinopenic, ie, not producing enough insulin endogenously and thus requiring insulin at all times.
Though checking for antibody markers of type 1 diabetes might give a more definitive answer, it is not practical before surgery.
In the setting of surgical stress, withholding the basal insulin preoperatively and just giving a small dose of fast-acting (see Table 1 for the different classes of insulin) or shortacting insulin as part of a sliding scale (ie, insulin given only when the blood glucose reaches a certain high level) can send a patient with type 1 diabetes into diabetic ketoacidosis by the end of the day. This is less likely to occur in a patient with type 2 diabetes with some endogenous insulin secretion.
STEP 2: ADJUST THE BASAL INSULIN
Basal insulin is the insulin that the healthy person’s body produces when fasting. For a diabetic patient already on insulin, basal insulin is insulin injected to prevent ketogenesis, glycogenolysis, and gluconeogenesis in the fasting state.
If the basal insulin is long-acting
Long-acting insulins have a relatively peakless profile and, when properly dosed, should not result in hypoglycemia when a patient is fasting.
Preoperatively, the patient should take it as close as possible to the usual time of injection. This could be at home either at bedtime the night before surgery or the morning of surgery. If there is concern for hypoglycemia, the injection can be given when the patient is at the hospital.
- If the patient does not tend to have hypoglycemic episodes and the total daily basal insulin dose is roughly the same as the total daily mealtime (prandial) dose (eg, 50% basal, 50% prandial ratio), the full dose of basal insulin can be given.3
Example: If the patient is on insulin glargine 30 U at bedtime and insulin lispro 10 U with each meal and does not have hypoglycemic episodes, then insulin glargine 30 U should be taken at bedtime.
- If the patient has hypoglycemic episodes at home, then the basal insulin can be reduced by 25%.3
Example: If the patient is on insulin glargine 30 U at bedtime and insulin glulisine 10 U with each meal (appropriate proportion of doses, similar to the example above) but has hypoglycemic episodes at home on this regimen, then only 22 U of insulin glargine should be taken at bedtime.
- If the patient’s regimen has disproportionately more basal insulin than mealtime insulin, then the total daily doses can be added and half can be given as the basal insulin.
Example: If the patient is on insulin detemir 30 U every morning at 6 am and insulin aspart 6 U with each meal and has no hypoglycemic episodes, then 24 U of insulin detemir should be taken in the morning (ie, half of the total of 30 + 6 + 6 + 6).
- In the less common scenario of diabetes managed only with basal insulin (no other diabetes injections or oral agents), then half of the dose can be given.
- If the patient is on twice-daily long-acting basal insulin, then both the dose the night before surgery and the dose the morning of surgery should be adjusted.
If the basal insulin is intermediate-acting
The intermediate-acting insulin neutral protamine Hagedorn (NPH) is usually given twice a day because of its profile (Table 1).
- On the night before surgery, the full dose of NPH insulin should be taken, unless the patient will now skip a nighttime meal because of taking nothing by mouth, in which case the dose can be decreased by 25%.1
- On the morning of surgery, since the patient will be skipping breakfast and probably also lunch, the dose should be reduced by 50%.3,4
Special situation: Premixed insulins
Premixed insulins (70/30, 75/25) are a combination of intermediate-acting insulin and either fast-acting or short-acting insulin. In other words, they are combinations of basal and prandial insulin. Their use is thus not ideal in the preoperative period. There are two options in these situations.
One option is to switch to a regimen that includes long-acting insulin. If the patient is admitted for surgery, then the hospital staff can change the insulin regimen to long-acting basal insulin. A quick formula for conversion is to add all the premixed insulin doses and give half as basal insulin on the morning of surgery, similar to the scenario above for the patient with long-acting basal insulin that was out of proportion to the prandial insulin injections.
For example, if the usual regimen is insulin 70/30 NPH/Regular, 60 U with breakfast, 30 U with dinner, then the patient can take 45 U of insulin glargine (which is half of 60 + 30) in the morning or evening before surgery.
Another option is to adjust the dose of pre-mixed insulin. Sometimes it is not feasible or economical to change the patient’s premixed insulin just before surgery. In these situations, the patient can take half of the morning dose, followed by dextrose-containing intravenous fluids and blood glucose checks.
We recommend preoperatively giving at least part of the patient’s previous basal insulin, regardless of the type of diabetes, the type of surgery, or the fasting period.
STEP 3: STOP THE PRANDIAL INSULIN
Prandial insulin—given before each meal to cover the carbohydrates to be consumed—should be stopped the morning of surgery.3,4
WHAT ABOUT SLIDING SCALE INSULIN?
Using a sliding scale alone has no known benefit. Although it can be a quick fix to correct a high glucose level, it should be added to the basal insulin and not used as the sole insulin therapy. If a sliding scale is used, fast-acting insulin (aspart, glulisine, lispro) is preferred over regular insulin because of the more rapid onset and shorter duration of action.
Patients already using a supplemental insulin scale can apply it to correct a blood glucose above 200 mg/dL on the morning of surgery.
MAINTENANCE FLUIDS
As long as glucose levels are not very elevated (ie, > 200 mg/dL), after 12 hours on a nothing-by-mouth regimen, provide dextrose in the IV fluid to prevent hypoglycemia (eg, the patient received long-acting insulin and the glucose levels are running low) or to prevent starvation ketosis, which may result in ketones in the blood or urine. We recommend 5% dextrose in half-normal (0.45%) saline at 50 to 75 mL/hour as maintenance fluid; the infusion rate should be lower if fluid overload is a concern.
POSTOPERATIVE INSULIN MANAGEMENT
Once patients are discharged and can go back to their previous routine, they can restart their usual insulin regimen the same evening. The prandial insulin will be resumed when the regular diet is reintroduced, and the doses will be adjusted according to food intake.
Continuing at least part of the basal insulin is the reasonable, physiologic approach to controlling glucose levels before surgery in patients with diabetes. The process involves three basic steps:
- Ascertaining the type of diabetes
- Adjusting the basal insulin dosage
- Stopping the prandial insulin.
The steps are the same whether the surgery is major or minor. These recommendations are based on general principles of insulin action, data from large databases of surgical inpatients, and expert clinical experience translated into standardized protocols.1,2
WHY CONTINUE THE INSULIN?
Stopping or decreasing insulin because of a fear of hypoglycemia is not appropriate, as the resulting hyperglycemia can lead to delayed wound healing, wound infection, fluid and electrolyte shifts, diabetic ketoacidosis, and hyperosmolar states.
Insulin inhibits both gluconeogenesis and conversion of glycogen to glucose, processes that occur regardless of food intake. It also inhibits degradation of fats to fatty acids and of fatty acids to ketones. This is why inadequate insulin dosing can lead to uncontrolled hyperglycemia and even ketoacidosis, and thus why long-acting insulin is needed in a fasting state.
STEP 1: ASCERTAIN THE TYPE OF DIABETES
Does the patient have type 1 or type 2 diabetes, and does that even matter?
The type of diabetes should not matter, since ideally the insulin should be dosed the same for both types. However, the consequences of inappropriate insulin management may be different.
Usually, the type of diabetes can be ascertained by the history. If the patient was diagnosed at age 40 or later and was on oral medication for years before insulin was started, then he or she most likely has type 2. If the patient was younger than 40 at the time of diagnosis, was lean, and was started on insulin within a year of diagnosis, then he or she likely has type 1.
If this information is not available or is unreliable and the patient has been on insulin for many years, we recommend viewing the patient as being insulinopenic, ie, not producing enough insulin endogenously and thus requiring insulin at all times.
Though checking for antibody markers of type 1 diabetes might give a more definitive answer, it is not practical before surgery.
In the setting of surgical stress, withholding the basal insulin preoperatively and just giving a small dose of fast-acting (see Table 1 for the different classes of insulin) or shortacting insulin as part of a sliding scale (ie, insulin given only when the blood glucose reaches a certain high level) can send a patient with type 1 diabetes into diabetic ketoacidosis by the end of the day. This is less likely to occur in a patient with type 2 diabetes with some endogenous insulin secretion.
STEP 2: ADJUST THE BASAL INSULIN
Basal insulin is the insulin that the healthy person’s body produces when fasting. For a diabetic patient already on insulin, basal insulin is insulin injected to prevent ketogenesis, glycogenolysis, and gluconeogenesis in the fasting state.
If the basal insulin is long-acting
Long-acting insulins have a relatively peakless profile and, when properly dosed, should not result in hypoglycemia when a patient is fasting.
Preoperatively, the patient should take it as close as possible to the usual time of injection. This could be at home either at bedtime the night before surgery or the morning of surgery. If there is concern for hypoglycemia, the injection can be given when the patient is at the hospital.
- If the patient does not tend to have hypoglycemic episodes and the total daily basal insulin dose is roughly the same as the total daily mealtime (prandial) dose (eg, 50% basal, 50% prandial ratio), the full dose of basal insulin can be given.3
Example: If the patient is on insulin glargine 30 U at bedtime and insulin lispro 10 U with each meal and does not have hypoglycemic episodes, then insulin glargine 30 U should be taken at bedtime.
- If the patient has hypoglycemic episodes at home, then the basal insulin can be reduced by 25%.3
Example: If the patient is on insulin glargine 30 U at bedtime and insulin glulisine 10 U with each meal (appropriate proportion of doses, similar to the example above) but has hypoglycemic episodes at home on this regimen, then only 22 U of insulin glargine should be taken at bedtime.
- If the patient’s regimen has disproportionately more basal insulin than mealtime insulin, then the total daily doses can be added and half can be given as the basal insulin.
Example: If the patient is on insulin detemir 30 U every morning at 6 am and insulin aspart 6 U with each meal and has no hypoglycemic episodes, then 24 U of insulin detemir should be taken in the morning (ie, half of the total of 30 + 6 + 6 + 6).
- In the less common scenario of diabetes managed only with basal insulin (no other diabetes injections or oral agents), then half of the dose can be given.
- If the patient is on twice-daily long-acting basal insulin, then both the dose the night before surgery and the dose the morning of surgery should be adjusted.
If the basal insulin is intermediate-acting
The intermediate-acting insulin neutral protamine Hagedorn (NPH) is usually given twice a day because of its profile (Table 1).
- On the night before surgery, the full dose of NPH insulin should be taken, unless the patient will now skip a nighttime meal because of taking nothing by mouth, in which case the dose can be decreased by 25%.1
- On the morning of surgery, since the patient will be skipping breakfast and probably also lunch, the dose should be reduced by 50%.3,4
Special situation: Premixed insulins
Premixed insulins (70/30, 75/25) are a combination of intermediate-acting insulin and either fast-acting or short-acting insulin. In other words, they are combinations of basal and prandial insulin. Their use is thus not ideal in the preoperative period. There are two options in these situations.
One option is to switch to a regimen that includes long-acting insulin. If the patient is admitted for surgery, then the hospital staff can change the insulin regimen to long-acting basal insulin. A quick formula for conversion is to add all the premixed insulin doses and give half as basal insulin on the morning of surgery, similar to the scenario above for the patient with long-acting basal insulin that was out of proportion to the prandial insulin injections.
For example, if the usual regimen is insulin 70/30 NPH/Regular, 60 U with breakfast, 30 U with dinner, then the patient can take 45 U of insulin glargine (which is half of 60 + 30) in the morning or evening before surgery.
Another option is to adjust the dose of pre-mixed insulin. Sometimes it is not feasible or economical to change the patient’s premixed insulin just before surgery. In these situations, the patient can take half of the morning dose, followed by dextrose-containing intravenous fluids and blood glucose checks.
We recommend preoperatively giving at least part of the patient’s previous basal insulin, regardless of the type of diabetes, the type of surgery, or the fasting period.
STEP 3: STOP THE PRANDIAL INSULIN
Prandial insulin—given before each meal to cover the carbohydrates to be consumed—should be stopped the morning of surgery.3,4
WHAT ABOUT SLIDING SCALE INSULIN?
Using a sliding scale alone has no known benefit. Although it can be a quick fix to correct a high glucose level, it should be added to the basal insulin and not used as the sole insulin therapy. If a sliding scale is used, fast-acting insulin (aspart, glulisine, lispro) is preferred over regular insulin because of the more rapid onset and shorter duration of action.
Patients already using a supplemental insulin scale can apply it to correct a blood glucose above 200 mg/dL on the morning of surgery.
MAINTENANCE FLUIDS
As long as glucose levels are not very elevated (ie, > 200 mg/dL), after 12 hours on a nothing-by-mouth regimen, provide dextrose in the IV fluid to prevent hypoglycemia (eg, the patient received long-acting insulin and the glucose levels are running low) or to prevent starvation ketosis, which may result in ketones in the blood or urine. We recommend 5% dextrose in half-normal (0.45%) saline at 50 to 75 mL/hour as maintenance fluid; the infusion rate should be lower if fluid overload is a concern.
POSTOPERATIVE INSULIN MANAGEMENT
Once patients are discharged and can go back to their previous routine, they can restart their usual insulin regimen the same evening. The prandial insulin will be resumed when the regular diet is reintroduced, and the doses will be adjusted according to food intake.
- Joshi GP, Chung F, Vann MA, et al; Society for Ambulatory Anesthesia. Society for Ambulatory Anesthesia consensus statement on perioperative blood glucose management in diabetic patients undergoing ambulatory surgery. Anesth Analg 2010; 111:1378–1387.
- DiNardo M, Donihi AC, Forte P, Gieraltowski L, Korytkowski M. Standardized glycemic management and perioperative glycemic outcomes in patients with diabetes mellitus who undergo same-day surgery. Endocr Pract 2011; 17:404–411.
- Vann MA. Perioperative management of ambulatory surgical patients with diabetes mellitus. Curr Opin Anaesthesiol 2009; 22:718–724.
- Meneghini LF. Perioperative management of diabetes: translating evidence into practice. Cleve Clin J Med 2009; 76(suppl 4):S53–S59.
- Joshi GP, Chung F, Vann MA, et al; Society for Ambulatory Anesthesia. Society for Ambulatory Anesthesia consensus statement on perioperative blood glucose management in diabetic patients undergoing ambulatory surgery. Anesth Analg 2010; 111:1378–1387.
- DiNardo M, Donihi AC, Forte P, Gieraltowski L, Korytkowski M. Standardized glycemic management and perioperative glycemic outcomes in patients with diabetes mellitus who undergo same-day surgery. Endocr Pract 2011; 17:404–411.
- Vann MA. Perioperative management of ambulatory surgical patients with diabetes mellitus. Curr Opin Anaesthesiol 2009; 22:718–724.
- Meneghini LF. Perioperative management of diabetes: translating evidence into practice. Cleve Clin J Med 2009; 76(suppl 4):S53–S59.
Is anticoagulation appropriate for all patients with portal vein thrombosis?
No. in general, the decision to treat portal vein thrombosis with anticoagulant drugs is complex and depends on whether the thrombosis is acute or chronic, and whether the cause is a local factor, cirrhosis of the liver, or a systemic condition (Table 1). A “one-size-fits-all” approach should be avoided (Figure 1).
ACUTE PORTAL VEIN THROMBOSIS WITHOUT CIRRHOSIS

No randomized controlled trial has yet evaluated anticoagulation in acute portal vein thrombosis. But a prospective study published in 2010 showed that the portal vein and its left or right branch were patent in 39% of anticoagulated patients (vs 13% initially), the splenic vein in 80% (vs 57% initially), and the superior mesenteric vein in 73% (vs 42% initially).1 Further, there appears to be a 20% reduction in the overall mortality rate associated with anticoagulation for acute portal vein thrombosis in retrospective studies.2
In the absence of contraindications, anticoagulation with heparin or low-molecular-weight heparin is recommended, with complete bridging to oral anticoagulation with a vitamin K antagonist. Anticoagulation should be continued for at least 3 months, and indefinitely in patients with permanent hypercoaguable risk factors.3
CHRONIC PORTAL VEIN THROMBOSIS WITHOUT CIRRHOSIS
All patients with chronic portal vein thrombosis should undergo esophagogastroduodenoscopy to evaluate for varices. Patients with large varices should be treated orally with a nonselective beta-adrenergic blocker or endoscopically. Though no prospective study has validated this practice, a retrospective analysis showed a decreased risk of first or recurrent bleeding.4
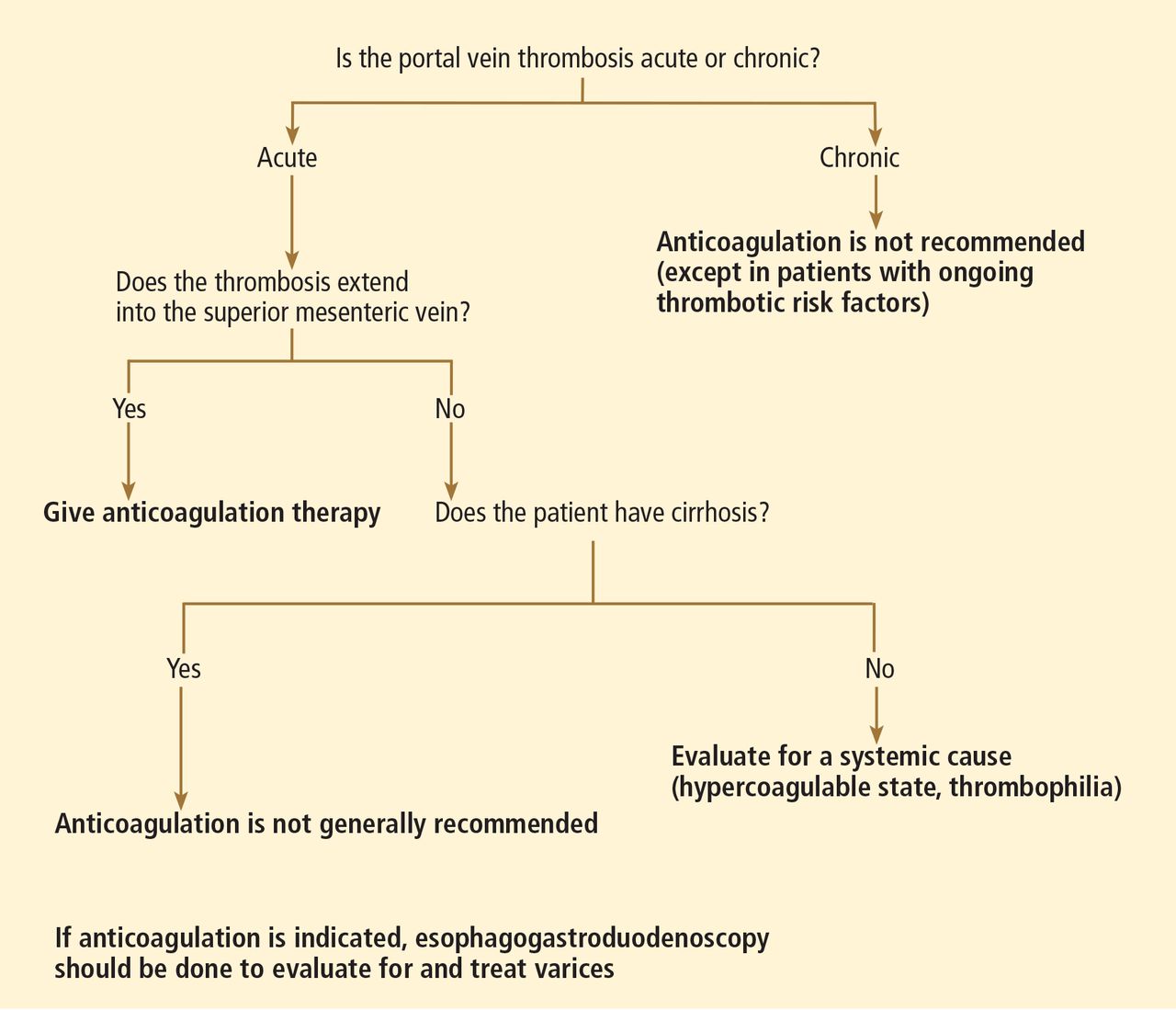
In 2007, a retrospective study showed a lower rate of death in patients with portomesenteric venous thrombosis treated with an oral vitamin K antagonist.5 Patients with chronic portal vein thrombosis with ongoing thrombotic risk factors should be treated with long-term anticoagulation after screening for varices, and if varices are present, primary prophylaxis should be started.3 With this approach, less than 5% of patients died from classic complications of portal vein thrombosis at 5 years of follow-up.4
ACUTE OR CHRONIC PORTAL VEIN THROMBOSIS WITH CIRRHOSIS
Portal vein thrombosis is common in patients with underlying cirrhosis. The risk in patients with cirrhosis significantly increases as liver function worsens. In patients with well-compensated cirrhosis, the risk is less than 1% vs 8% to 25% in those with advanced cirrhosis.6
In patients awaiting liver transplantation, a large retrospective study7 showed that the rate of partial or complete recanalization of the splanchnic veins was significantly higher in those who received anticoagulation (8 of 19) than in those who did not (0 of 10, P = .002). The rate of survival was significantly lower in those who had complete thrombotic obstruction of the portal vein at the time of surgery (P = .04). However, there was no difference in survival rates between those with partial obstruction who received anticoagulation and those with a patent portal vein.7
A later retrospective study8 showed no significant benefit in the rate of transplantation-free survival or survival after liver transplantation in patients with or without chronic portal vein thrombosis.8
Unfortunately, we have no data from prospective controlled trials and only limited data from retrospective studies to make a strong recommendation for or against anticoagulation in either acute and chronic portal vein thrombosis associated with cirrhosis. As such, each case must be evaluated on an individual basis in association with expert consultation.
In our experience, the risk of bleeding in patients with liver cirrhosis is substantial because of the decreased synthesis of coagulation factors and the presence of varices, whereas the efficacy and the benefits of recanalizing the portal vein in asymptomatic patients with liver cirrhosis and portal vein thrombosis are unknown. Therefore, unless the thrombosis extends into the mesenteric vein, thus posing a risk of mesenteric ischemia, we do not generally recommend anticoagulation in asymptomatic portal vein thrombosis in patients with cirrhosis.
- Plessier A, Darwish-Murad S, Hernandez-Guerra M, et al; European Network for Vascular Disorders of the Liver (EN-Vie). Acute portal vein thrombosis unrelated to cirrhosis: a prospective multicenter follow-up study. Hepatology 2010; 51:210–218.
- Kumar S, Sarr MG, Kamath PS. Mesenteric venous thrombosis. N Engl J Med 2001; 345:1683–1688.
- de Franchis R. Evolving consensus in portal hypertension. Report of the Baveno IV consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol 2005; 43:167–176.
- Condat B, Pessione F, Hillaire S, et al. Current outcome of portal vein thrombosis in adults: risk and benefit of anticoagulant therapy. Gastroenterology 2001; 120:490–497.
- Orr DW, Harrison PM, Devlin J, et al. Chronic mesenteric venous thrombosis: evaluation and determinants of survival during long-term follow-up. Clin Gastroenterol Hepatol 2007; 5:80–86.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Francoz C, Belghiti J, Vilgrain V, et al. Splanchnic vein thrombosis in candidates for liver transplantation: usefulness of screening and anticoagulation. Gut 2005; 54:691–697.
- John BV, Konjeti VR, Aggarwal A, et al. The impact of portal vein thrombosis (PVT) on cirrhotics awaiting liver transplantation (abstract). Hepatology 2010; 52(suppl1):888A–889A.
No. in general, the decision to treat portal vein thrombosis with anticoagulant drugs is complex and depends on whether the thrombosis is acute or chronic, and whether the cause is a local factor, cirrhosis of the liver, or a systemic condition (Table 1). A “one-size-fits-all” approach should be avoided (Figure 1).
ACUTE PORTAL VEIN THROMBOSIS WITHOUT CIRRHOSIS
No randomized controlled trial has yet evaluated anticoagulation in acute portal vein thrombosis. But a prospective study published in 2010 showed that the portal vein and its left or right branch were patent in 39% of anticoagulated patients (vs 13% initially), the splenic vein in 80% (vs 57% initially), and the superior mesenteric vein in 73% (vs 42% initially).1 Further, there appears to be a 20% reduction in the overall mortality rate associated with anticoagulation for acute portal vein thrombosis in retrospective studies.2
In the absence of contraindications, anticoagulation with heparin or low-molecular-weight heparin is recommended, with complete bridging to oral anticoagulation with a vitamin K antagonist. Anticoagulation should be continued for at least 3 months, and indefinitely in patients with permanent hypercoaguable risk factors.3
CHRONIC PORTAL VEIN THROMBOSIS WITHOUT CIRRHOSIS
All patients with chronic portal vein thrombosis should undergo esophagogastroduodenoscopy to evaluate for varices. Patients with large varices should be treated orally with a nonselective beta-adrenergic blocker or endoscopically. Though no prospective study has validated this practice, a retrospective analysis showed a decreased risk of first or recurrent bleeding.4
In 2007, a retrospective study showed a lower rate of death in patients with portomesenteric venous thrombosis treated with an oral vitamin K antagonist.5 Patients with chronic portal vein thrombosis with ongoing thrombotic risk factors should be treated with long-term anticoagulation after screening for varices, and if varices are present, primary prophylaxis should be started.3 With this approach, less than 5% of patients died from classic complications of portal vein thrombosis at 5 years of follow-up.4
ACUTE OR CHRONIC PORTAL VEIN THROMBOSIS WITH CIRRHOSIS
Portal vein thrombosis is common in patients with underlying cirrhosis. The risk in patients with cirrhosis significantly increases as liver function worsens. In patients with well-compensated cirrhosis, the risk is less than 1% vs 8% to 25% in those with advanced cirrhosis.6
In patients awaiting liver transplantation, a large retrospective study7 showed that the rate of partial or complete recanalization of the splanchnic veins was significantly higher in those who received anticoagulation (8 of 19) than in those who did not (0 of 10, P = .002). The rate of survival was significantly lower in those who had complete thrombotic obstruction of the portal vein at the time of surgery (P = .04). However, there was no difference in survival rates between those with partial obstruction who received anticoagulation and those with a patent portal vein.7
A later retrospective study8 showed no significant benefit in the rate of transplantation-free survival or survival after liver transplantation in patients with or without chronic portal vein thrombosis.8
Unfortunately, we have no data from prospective controlled trials and only limited data from retrospective studies to make a strong recommendation for or against anticoagulation in either acute and chronic portal vein thrombosis associated with cirrhosis. As such, each case must be evaluated on an individual basis in association with expert consultation.
In our experience, the risk of bleeding in patients with liver cirrhosis is substantial because of the decreased synthesis of coagulation factors and the presence of varices, whereas the efficacy and the benefits of recanalizing the portal vein in asymptomatic patients with liver cirrhosis and portal vein thrombosis are unknown. Therefore, unless the thrombosis extends into the mesenteric vein, thus posing a risk of mesenteric ischemia, we do not generally recommend anticoagulation in asymptomatic portal vein thrombosis in patients with cirrhosis.
No. in general, the decision to treat portal vein thrombosis with anticoagulant drugs is complex and depends on whether the thrombosis is acute or chronic, and whether the cause is a local factor, cirrhosis of the liver, or a systemic condition (Table 1). A “one-size-fits-all” approach should be avoided (Figure 1).
ACUTE PORTAL VEIN THROMBOSIS WITHOUT CIRRHOSIS
No randomized controlled trial has yet evaluated anticoagulation in acute portal vein thrombosis. But a prospective study published in 2010 showed that the portal vein and its left or right branch were patent in 39% of anticoagulated patients (vs 13% initially), the splenic vein in 80% (vs 57% initially), and the superior mesenteric vein in 73% (vs 42% initially).1 Further, there appears to be a 20% reduction in the overall mortality rate associated with anticoagulation for acute portal vein thrombosis in retrospective studies.2
In the absence of contraindications, anticoagulation with heparin or low-molecular-weight heparin is recommended, with complete bridging to oral anticoagulation with a vitamin K antagonist. Anticoagulation should be continued for at least 3 months, and indefinitely in patients with permanent hypercoaguable risk factors.3
CHRONIC PORTAL VEIN THROMBOSIS WITHOUT CIRRHOSIS
All patients with chronic portal vein thrombosis should undergo esophagogastroduodenoscopy to evaluate for varices. Patients with large varices should be treated orally with a nonselective beta-adrenergic blocker or endoscopically. Though no prospective study has validated this practice, a retrospective analysis showed a decreased risk of first or recurrent bleeding.4
In 2007, a retrospective study showed a lower rate of death in patients with portomesenteric venous thrombosis treated with an oral vitamin K antagonist.5 Patients with chronic portal vein thrombosis with ongoing thrombotic risk factors should be treated with long-term anticoagulation after screening for varices, and if varices are present, primary prophylaxis should be started.3 With this approach, less than 5% of patients died from classic complications of portal vein thrombosis at 5 years of follow-up.4
ACUTE OR CHRONIC PORTAL VEIN THROMBOSIS WITH CIRRHOSIS
Portal vein thrombosis is common in patients with underlying cirrhosis. The risk in patients with cirrhosis significantly increases as liver function worsens. In patients with well-compensated cirrhosis, the risk is less than 1% vs 8% to 25% in those with advanced cirrhosis.6
In patients awaiting liver transplantation, a large retrospective study7 showed that the rate of partial or complete recanalization of the splanchnic veins was significantly higher in those who received anticoagulation (8 of 19) than in those who did not (0 of 10, P = .002). The rate of survival was significantly lower in those who had complete thrombotic obstruction of the portal vein at the time of surgery (P = .04). However, there was no difference in survival rates between those with partial obstruction who received anticoagulation and those with a patent portal vein.7
A later retrospective study8 showed no significant benefit in the rate of transplantation-free survival or survival after liver transplantation in patients with or without chronic portal vein thrombosis.8
Unfortunately, we have no data from prospective controlled trials and only limited data from retrospective studies to make a strong recommendation for or against anticoagulation in either acute and chronic portal vein thrombosis associated with cirrhosis. As such, each case must be evaluated on an individual basis in association with expert consultation.
In our experience, the risk of bleeding in patients with liver cirrhosis is substantial because of the decreased synthesis of coagulation factors and the presence of varices, whereas the efficacy and the benefits of recanalizing the portal vein in asymptomatic patients with liver cirrhosis and portal vein thrombosis are unknown. Therefore, unless the thrombosis extends into the mesenteric vein, thus posing a risk of mesenteric ischemia, we do not generally recommend anticoagulation in asymptomatic portal vein thrombosis in patients with cirrhosis.
- Plessier A, Darwish-Murad S, Hernandez-Guerra M, et al; European Network for Vascular Disorders of the Liver (EN-Vie). Acute portal vein thrombosis unrelated to cirrhosis: a prospective multicenter follow-up study. Hepatology 2010; 51:210–218.
- Kumar S, Sarr MG, Kamath PS. Mesenteric venous thrombosis. N Engl J Med 2001; 345:1683–1688.
- de Franchis R. Evolving consensus in portal hypertension. Report of the Baveno IV consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol 2005; 43:167–176.
- Condat B, Pessione F, Hillaire S, et al. Current outcome of portal vein thrombosis in adults: risk and benefit of anticoagulant therapy. Gastroenterology 2001; 120:490–497.
- Orr DW, Harrison PM, Devlin J, et al. Chronic mesenteric venous thrombosis: evaluation and determinants of survival during long-term follow-up. Clin Gastroenterol Hepatol 2007; 5:80–86.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Francoz C, Belghiti J, Vilgrain V, et al. Splanchnic vein thrombosis in candidates for liver transplantation: usefulness of screening and anticoagulation. Gut 2005; 54:691–697.
- John BV, Konjeti VR, Aggarwal A, et al. The impact of portal vein thrombosis (PVT) on cirrhotics awaiting liver transplantation (abstract). Hepatology 2010; 52(suppl1):888A–889A.
- Plessier A, Darwish-Murad S, Hernandez-Guerra M, et al; European Network for Vascular Disorders of the Liver (EN-Vie). Acute portal vein thrombosis unrelated to cirrhosis: a prospective multicenter follow-up study. Hepatology 2010; 51:210–218.
- Kumar S, Sarr MG, Kamath PS. Mesenteric venous thrombosis. N Engl J Med 2001; 345:1683–1688.
- de Franchis R. Evolving consensus in portal hypertension. Report of the Baveno IV consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol 2005; 43:167–176.
- Condat B, Pessione F, Hillaire S, et al. Current outcome of portal vein thrombosis in adults: risk and benefit of anticoagulant therapy. Gastroenterology 2001; 120:490–497.
- Orr DW, Harrison PM, Devlin J, et al. Chronic mesenteric venous thrombosis: evaluation and determinants of survival during long-term follow-up. Clin Gastroenterol Hepatol 2007; 5:80–86.
- DeLeve LD, Valla DC, Garcia-Tsao G; American Association for the Study of Liver Diseases. Vascular disorders of the liver. Hepatology 2009; 49:1729–1764.
- Francoz C, Belghiti J, Vilgrain V, et al. Splanchnic vein thrombosis in candidates for liver transplantation: usefulness of screening and anticoagulation. Gut 2005; 54:691–697.
- John BV, Konjeti VR, Aggarwal A, et al. The impact of portal vein thrombosis (PVT) on cirrhotics awaiting liver transplantation (abstract). Hepatology 2010; 52(suppl1):888A–889A.
Which lower-extremity DVTs should be removed early?
Early thrombus removal for lower-extremity deep venous thrombosis (DVT) is at present only modestly supported by evidence and so remains controversial. It is largely aimed at preventing postthrombotic syndrome.
The decision to pursue early thrombus removal demands weighing the patient’s risk of postthrombotic syndrome against the risks and costs associated with thrombolysis and thrombectomy, such as bleeding complications. In the final analysis, this remains a subjective decision.
With these caveats in mind, the best candidate for early thrombus removal is a young patient with iliofemoral DVT with symptoms lasting fewer than 14 days.
POSTTHROMBOTIC SYNDROME IS COMMON
Anticoagulation with heparin and warfarin is the mainstay of DVT therapy. Indeed, the safety of this therapy and its effectiveness in reducing thrombus propagation and DVT recurrence are well established. Neither heparin nor warfarin, however, actively reduces the thrombus burden. Rather, both prevent the clot from propagating while it is, hopefully, gradually reabsorbed through endogenous mechanisms.
Up to 50% of DVT patients develop postthrombotic syndrome. A variety of mechanisms are involved, including persistent obstructive thrombosis and valvular injury.1 But much remains unknown about the etiology, and some patients develop the condition in the absence of abnormalities on objective testing.
Symptoms of postthrombotic syndrome can range from mild heaviness, edema, erythema, and cramping in the affected limb to debilitating pain with classic signs of venous hypertension (eg, venous ectasia and ulcers). It accounts for significant health care costs and has a detrimental effect on quality of life.1 Thus, there has been interest in early thrombus removal as initial therapy for DVT.
THROMBUS REMOVAL
Venous clots can be removed with open surgery or, more typically, with percutaneous catheter-based thrombolysis and thrombectomy devices that use high-velocity saline jets, ultrasonic energy, or wire oscillation to mechanically fragment the venous clot. All of these mechanisms help with drug delivery and pose a minimal risk of pulmonary embolism.
Evidence is weak
Patients with DVT of the iliac venous system or common femoral vein are at highest risk of postthrombotic syndrome. Therefore, the Society for Vascular Surgery and the American Venous Forum have issued a grade 2C (ie, weak) recommendation in favor of early thrombus removal in patients with a first-time episode of iliofemoral DVT with fewer than 14 days of symptoms.2 Moreover, patients must have a low risk of bleeding complications, be ambulatory, and have reasonable life expectancy.
The recommendation is buttressed by a Cochrane meta-analysis that included 101 patients.3 It concluded that there was a significant decrement in the development of postthrombotic syndrome with thrombolysis (but without mechanical thrombectomy) compared with standard therapy: the rate was 48% (29/61) with thrombolysis, and 65% (26/40) with standard therapy.3
More recently, the Catheter-Directed Thrombolysis Versus Standard Treatment for Acute Iliofemoral Deep Vein Thrombosis (CaVenT) study, a randomized prospective trial in 189 patients, demonstrated a lower rate of postthrombotic syndrome at 24 months and increased iliofemoral patency at 6 months with catheter-directed thrombolysis with alteplase (41.1% and 65.9%) vs anticoagulation with heparin and warfarin alone (55.6% and 47.4%).4
The Acute Venous Thrombosis: Thrombus Removal With Adjunctive Catheter-directed Thrombolysis (ATTRACT) trial is an ongoing prospective randomized multicenter trial of the effect of thrombolysis on postthrombotic syndrome that also hopes to clarify the relative benefits of different methods of pharmacomechanical clot removal.
While CaVenT has not been criticized extensively in the literature, other studies supporting early intervention for iliofemoral venous thrombosis generally have been noted to have a number of shortcomings, including a lack of randomization, and consequent bias, and the use of surrogate end points instead of a direct assessment of postthrombotic syndrome.
Reflecting the weakness of the evidence, the American College of Chest Physicians has issued a grade 2C recommendation against catheter-directed thrombolysis and against thrombectomy in favor of anticoagulant therapy.5
A subjective, case-by-case decision
The decision on standard vs interventional therapy must be made case by case. For example, thrombus removal may be more appropriate for a physically active young patient who is more likely to be impaired by postthrombotic syndrome, whereas standard warfarin therapy may be preferable for a sedentary patient. We are also more inclined to offer thrombus removal to patients who have worse symptoms.
Complicating the issue, many patients present with a mix of variables that support and oppose intervention—eg, a moderately active elderly patient with an unclear life expectancy and a history of gastrointestinal bleeding. At present, there is no way to quantitatively evaluate the risks and rewards of thrombus removal, and the final decision is essentially subjective.
Additional facts warranting consideration include the possibility that thrombolysis may require several days of therapy with daily venography for evaluation. Monitoring in the intensive care unit is normally required during the period of thrombolysis. Patients should be apprised of these elements of therapy beforehand; obviously, those who are unwilling to comply are not candidates.
Not a substitute for anticoagulation
It is important to recognize that thrombus removal is not a substitute for standard heparin-warfarin anticoagulation, which must also be prescribed.5 Thus, patients who cannot tolerate standard post-DVT anticoagulation should not undergo thrombus removal. Furthermore, the current evidence supports the use of standard anticoagulation over early thrombus removal of DVTs that are more distal in the lower extremity, such as those in the popliteal vein.5
PHLEGMASIA CERULEA DOLENS IS A SPECIAL CASE
Phlegmasia cerulea dolens—acute venous outflow obstruction associated with edema, cyanosis, and pain that in the worst cases may lead to shock, limb loss, and death—constitutes a special case. Although we lack robust supporting evidence, phlegmasia is a commonly accepted indication for early thrombus removal as a means of limb salvage.2,6
- Kahn SR. The post thrombotic syndrome. Thromb Res 2011; 127 (suppl 3):S89–S92.
- Meissner MH, Gloviczki P, Comerota AJ, et al; Society for Vascular Surgery; American Venous Forum. Early thrombus removal strategies for acute deep venous thrombosis: clinical practice guidelines of the Society for Vascular Surgery and the American Venous Forum. J Vasc Surg 2012; 55:1449–1462.
- Watson LI, Armon MP. Thrombolysis for acute deep vein thrombosis. Cochrane Database Syst Rev 2004; 4:CD002783.
- Enden T, Haig Y, Kløw NE, et al; CaVenT Study Group. Long-term outcome after additional catheter-directed thrombolysis versus standard treatment for acute iliofemoral deep vein thrombosis (the CaVenT study): a randomised controlled trial. Lancet 2012; 379:31–38.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141 (suppl 2):e419S–e494S.
- Patterson BO, Hinchliffe R, Loftus IM, Thompson MM, Holt PJ. Indications for catheter-directed thrombolysis in the management of acute proximal deep venous thrombosis. Arterioscler Thromb Vasc Biol 2010; 30:669–674.
Early thrombus removal for lower-extremity deep venous thrombosis (DVT) is at present only modestly supported by evidence and so remains controversial. It is largely aimed at preventing postthrombotic syndrome.
The decision to pursue early thrombus removal demands weighing the patient’s risk of postthrombotic syndrome against the risks and costs associated with thrombolysis and thrombectomy, such as bleeding complications. In the final analysis, this remains a subjective decision.
With these caveats in mind, the best candidate for early thrombus removal is a young patient with iliofemoral DVT with symptoms lasting fewer than 14 days.
POSTTHROMBOTIC SYNDROME IS COMMON
Anticoagulation with heparin and warfarin is the mainstay of DVT therapy. Indeed, the safety of this therapy and its effectiveness in reducing thrombus propagation and DVT recurrence are well established. Neither heparin nor warfarin, however, actively reduces the thrombus burden. Rather, both prevent the clot from propagating while it is, hopefully, gradually reabsorbed through endogenous mechanisms.
Up to 50% of DVT patients develop postthrombotic syndrome. A variety of mechanisms are involved, including persistent obstructive thrombosis and valvular injury.1 But much remains unknown about the etiology, and some patients develop the condition in the absence of abnormalities on objective testing.
Symptoms of postthrombotic syndrome can range from mild heaviness, edema, erythema, and cramping in the affected limb to debilitating pain with classic signs of venous hypertension (eg, venous ectasia and ulcers). It accounts for significant health care costs and has a detrimental effect on quality of life.1 Thus, there has been interest in early thrombus removal as initial therapy for DVT.
THROMBUS REMOVAL
Venous clots can be removed with open surgery or, more typically, with percutaneous catheter-based thrombolysis and thrombectomy devices that use high-velocity saline jets, ultrasonic energy, or wire oscillation to mechanically fragment the venous clot. All of these mechanisms help with drug delivery and pose a minimal risk of pulmonary embolism.
Evidence is weak
Patients with DVT of the iliac venous system or common femoral vein are at highest risk of postthrombotic syndrome. Therefore, the Society for Vascular Surgery and the American Venous Forum have issued a grade 2C (ie, weak) recommendation in favor of early thrombus removal in patients with a first-time episode of iliofemoral DVT with fewer than 14 days of symptoms.2 Moreover, patients must have a low risk of bleeding complications, be ambulatory, and have reasonable life expectancy.
The recommendation is buttressed by a Cochrane meta-analysis that included 101 patients.3 It concluded that there was a significant decrement in the development of postthrombotic syndrome with thrombolysis (but without mechanical thrombectomy) compared with standard therapy: the rate was 48% (29/61) with thrombolysis, and 65% (26/40) with standard therapy.3
More recently, the Catheter-Directed Thrombolysis Versus Standard Treatment for Acute Iliofemoral Deep Vein Thrombosis (CaVenT) study, a randomized prospective trial in 189 patients, demonstrated a lower rate of postthrombotic syndrome at 24 months and increased iliofemoral patency at 6 months with catheter-directed thrombolysis with alteplase (41.1% and 65.9%) vs anticoagulation with heparin and warfarin alone (55.6% and 47.4%).4
The Acute Venous Thrombosis: Thrombus Removal With Adjunctive Catheter-directed Thrombolysis (ATTRACT) trial is an ongoing prospective randomized multicenter trial of the effect of thrombolysis on postthrombotic syndrome that also hopes to clarify the relative benefits of different methods of pharmacomechanical clot removal.
While CaVenT has not been criticized extensively in the literature, other studies supporting early intervention for iliofemoral venous thrombosis generally have been noted to have a number of shortcomings, including a lack of randomization, and consequent bias, and the use of surrogate end points instead of a direct assessment of postthrombotic syndrome.
Reflecting the weakness of the evidence, the American College of Chest Physicians has issued a grade 2C recommendation against catheter-directed thrombolysis and against thrombectomy in favor of anticoagulant therapy.5
A subjective, case-by-case decision
The decision on standard vs interventional therapy must be made case by case. For example, thrombus removal may be more appropriate for a physically active young patient who is more likely to be impaired by postthrombotic syndrome, whereas standard warfarin therapy may be preferable for a sedentary patient. We are also more inclined to offer thrombus removal to patients who have worse symptoms.
Complicating the issue, many patients present with a mix of variables that support and oppose intervention—eg, a moderately active elderly patient with an unclear life expectancy and a history of gastrointestinal bleeding. At present, there is no way to quantitatively evaluate the risks and rewards of thrombus removal, and the final decision is essentially subjective.
Additional facts warranting consideration include the possibility that thrombolysis may require several days of therapy with daily venography for evaluation. Monitoring in the intensive care unit is normally required during the period of thrombolysis. Patients should be apprised of these elements of therapy beforehand; obviously, those who are unwilling to comply are not candidates.
Not a substitute for anticoagulation
It is important to recognize that thrombus removal is not a substitute for standard heparin-warfarin anticoagulation, which must also be prescribed.5 Thus, patients who cannot tolerate standard post-DVT anticoagulation should not undergo thrombus removal. Furthermore, the current evidence supports the use of standard anticoagulation over early thrombus removal of DVTs that are more distal in the lower extremity, such as those in the popliteal vein.5
PHLEGMASIA CERULEA DOLENS IS A SPECIAL CASE
Phlegmasia cerulea dolens—acute venous outflow obstruction associated with edema, cyanosis, and pain that in the worst cases may lead to shock, limb loss, and death—constitutes a special case. Although we lack robust supporting evidence, phlegmasia is a commonly accepted indication for early thrombus removal as a means of limb salvage.2,6
Early thrombus removal for lower-extremity deep venous thrombosis (DVT) is at present only modestly supported by evidence and so remains controversial. It is largely aimed at preventing postthrombotic syndrome.
The decision to pursue early thrombus removal demands weighing the patient’s risk of postthrombotic syndrome against the risks and costs associated with thrombolysis and thrombectomy, such as bleeding complications. In the final analysis, this remains a subjective decision.
With these caveats in mind, the best candidate for early thrombus removal is a young patient with iliofemoral DVT with symptoms lasting fewer than 14 days.
POSTTHROMBOTIC SYNDROME IS COMMON
Anticoagulation with heparin and warfarin is the mainstay of DVT therapy. Indeed, the safety of this therapy and its effectiveness in reducing thrombus propagation and DVT recurrence are well established. Neither heparin nor warfarin, however, actively reduces the thrombus burden. Rather, both prevent the clot from propagating while it is, hopefully, gradually reabsorbed through endogenous mechanisms.
Up to 50% of DVT patients develop postthrombotic syndrome. A variety of mechanisms are involved, including persistent obstructive thrombosis and valvular injury.1 But much remains unknown about the etiology, and some patients develop the condition in the absence of abnormalities on objective testing.
Symptoms of postthrombotic syndrome can range from mild heaviness, edema, erythema, and cramping in the affected limb to debilitating pain with classic signs of venous hypertension (eg, venous ectasia and ulcers). It accounts for significant health care costs and has a detrimental effect on quality of life.1 Thus, there has been interest in early thrombus removal as initial therapy for DVT.
THROMBUS REMOVAL
Venous clots can be removed with open surgery or, more typically, with percutaneous catheter-based thrombolysis and thrombectomy devices that use high-velocity saline jets, ultrasonic energy, or wire oscillation to mechanically fragment the venous clot. All of these mechanisms help with drug delivery and pose a minimal risk of pulmonary embolism.
Evidence is weak
Patients with DVT of the iliac venous system or common femoral vein are at highest risk of postthrombotic syndrome. Therefore, the Society for Vascular Surgery and the American Venous Forum have issued a grade 2C (ie, weak) recommendation in favor of early thrombus removal in patients with a first-time episode of iliofemoral DVT with fewer than 14 days of symptoms.2 Moreover, patients must have a low risk of bleeding complications, be ambulatory, and have reasonable life expectancy.
The recommendation is buttressed by a Cochrane meta-analysis that included 101 patients.3 It concluded that there was a significant decrement in the development of postthrombotic syndrome with thrombolysis (but without mechanical thrombectomy) compared with standard therapy: the rate was 48% (29/61) with thrombolysis, and 65% (26/40) with standard therapy.3
More recently, the Catheter-Directed Thrombolysis Versus Standard Treatment for Acute Iliofemoral Deep Vein Thrombosis (CaVenT) study, a randomized prospective trial in 189 patients, demonstrated a lower rate of postthrombotic syndrome at 24 months and increased iliofemoral patency at 6 months with catheter-directed thrombolysis with alteplase (41.1% and 65.9%) vs anticoagulation with heparin and warfarin alone (55.6% and 47.4%).4
The Acute Venous Thrombosis: Thrombus Removal With Adjunctive Catheter-directed Thrombolysis (ATTRACT) trial is an ongoing prospective randomized multicenter trial of the effect of thrombolysis on postthrombotic syndrome that also hopes to clarify the relative benefits of different methods of pharmacomechanical clot removal.
While CaVenT has not been criticized extensively in the literature, other studies supporting early intervention for iliofemoral venous thrombosis generally have been noted to have a number of shortcomings, including a lack of randomization, and consequent bias, and the use of surrogate end points instead of a direct assessment of postthrombotic syndrome.
Reflecting the weakness of the evidence, the American College of Chest Physicians has issued a grade 2C recommendation against catheter-directed thrombolysis and against thrombectomy in favor of anticoagulant therapy.5
A subjective, case-by-case decision
The decision on standard vs interventional therapy must be made case by case. For example, thrombus removal may be more appropriate for a physically active young patient who is more likely to be impaired by postthrombotic syndrome, whereas standard warfarin therapy may be preferable for a sedentary patient. We are also more inclined to offer thrombus removal to patients who have worse symptoms.
Complicating the issue, many patients present with a mix of variables that support and oppose intervention—eg, a moderately active elderly patient with an unclear life expectancy and a history of gastrointestinal bleeding. At present, there is no way to quantitatively evaluate the risks and rewards of thrombus removal, and the final decision is essentially subjective.
Additional facts warranting consideration include the possibility that thrombolysis may require several days of therapy with daily venography for evaluation. Monitoring in the intensive care unit is normally required during the period of thrombolysis. Patients should be apprised of these elements of therapy beforehand; obviously, those who are unwilling to comply are not candidates.
Not a substitute for anticoagulation
It is important to recognize that thrombus removal is not a substitute for standard heparin-warfarin anticoagulation, which must also be prescribed.5 Thus, patients who cannot tolerate standard post-DVT anticoagulation should not undergo thrombus removal. Furthermore, the current evidence supports the use of standard anticoagulation over early thrombus removal of DVTs that are more distal in the lower extremity, such as those in the popliteal vein.5
PHLEGMASIA CERULEA DOLENS IS A SPECIAL CASE
Phlegmasia cerulea dolens—acute venous outflow obstruction associated with edema, cyanosis, and pain that in the worst cases may lead to shock, limb loss, and death—constitutes a special case. Although we lack robust supporting evidence, phlegmasia is a commonly accepted indication for early thrombus removal as a means of limb salvage.2,6
- Kahn SR. The post thrombotic syndrome. Thromb Res 2011; 127 (suppl 3):S89–S92.
- Meissner MH, Gloviczki P, Comerota AJ, et al; Society for Vascular Surgery; American Venous Forum. Early thrombus removal strategies for acute deep venous thrombosis: clinical practice guidelines of the Society for Vascular Surgery and the American Venous Forum. J Vasc Surg 2012; 55:1449–1462.
- Watson LI, Armon MP. Thrombolysis for acute deep vein thrombosis. Cochrane Database Syst Rev 2004; 4:CD002783.
- Enden T, Haig Y, Kløw NE, et al; CaVenT Study Group. Long-term outcome after additional catheter-directed thrombolysis versus standard treatment for acute iliofemoral deep vein thrombosis (the CaVenT study): a randomised controlled trial. Lancet 2012; 379:31–38.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141 (suppl 2):e419S–e494S.
- Patterson BO, Hinchliffe R, Loftus IM, Thompson MM, Holt PJ. Indications for catheter-directed thrombolysis in the management of acute proximal deep venous thrombosis. Arterioscler Thromb Vasc Biol 2010; 30:669–674.
- Kahn SR. The post thrombotic syndrome. Thromb Res 2011; 127 (suppl 3):S89–S92.
- Meissner MH, Gloviczki P, Comerota AJ, et al; Society for Vascular Surgery; American Venous Forum. Early thrombus removal strategies for acute deep venous thrombosis: clinical practice guidelines of the Society for Vascular Surgery and the American Venous Forum. J Vasc Surg 2012; 55:1449–1462.
- Watson LI, Armon MP. Thrombolysis for acute deep vein thrombosis. Cochrane Database Syst Rev 2004; 4:CD002783.
- Enden T, Haig Y, Kløw NE, et al; CaVenT Study Group. Long-term outcome after additional catheter-directed thrombolysis versus standard treatment for acute iliofemoral deep vein thrombosis (the CaVenT study): a randomised controlled trial. Lancet 2012; 379:31–38.
- Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141 (suppl 2):e419S–e494S.
- Patterson BO, Hinchliffe R, Loftus IM, Thompson MM, Holt PJ. Indications for catheter-directed thrombolysis in the management of acute proximal deep venous thrombosis. Arterioscler Thromb Vasc Biol 2010; 30:669–674.
Which patients may benefit from coronary artery calcification scoring?
Although we still have no evidence from randomized trials that patients have better outcomes if we measure the calcification in their coronary arteries, a growing body of evidence shows that we can estimate risk more accurately than with a risk model score alone if we also score coronary artery calcification in asymptomatic patients, especially those at intermediate risk.
Current guidelines1 recommend using the Framingham Risk Score or a similar tool to estimate coronary risk in asymptomatic patients, but these tools have only modest accuracy. Calcification scoring is accurate, inexpensive, quick, widely available, low-risk, and does not appear to increase medical costs afterward. In addition to improving risk stratification, it may also encourage patients to adhere better to drug therapy and lifestyle modification.
HOW IS CORONARY ARTERY CALCIFICATION MEASURED?
Calcification of the coronary arteries is synonymous with atherosclerosis. It can easily be detected with computed tomography without contrast (Figure 1), and the amount can be quantified with a scoring system such as the volumetric score or the Agatston score. The latter, which is more commonly used, is based on the product of the area of the calcium deposits and the x-ray attenuation in Hounsfield units.
Scores can be roughly categorized (with some overlap owing to data from different studies) as:
- Low risk: 0 Agatston units (AU)
- Average risk: 1–112 AU
- Moderate risk: 100–400 AU
- High risk: 400–999 AU
- Very high risk: 1,000 AU.2
The actual test takes only a few seconds, and the patient can usually be out the door in 15 minutes or less. It does not require iodinated contrast and the radiation dose is minimal, usually less than 1 mSv, equivalent to fewer than 10 chest radiographs.3
The cost is typically between $200 and $500. The test is usually not covered by health insurance, but this differs by insurer and by state; for example, coverage is mandated in Texas, and the test is covered by United Healthcare.
WHAT IS THE EVIDENCE IN FAVOR OF CALCIFICATION SCORING?
Cohort studies with long-term follow-up show that calcification scoring has robust prognostic ability. A pooled analysis of several of these studies2 showed that a higher score strongly correlated with a higher risk of cardiac events over 3 to 5 years. Compared with the risk in people with a score of 0, the risk was twice as high in those with a score of 1 to 112, four times as high with a score of 100 to 400, seven times as high with a score of 400 to 499, and 10 times as high with a score greater than 1,000.2
A cohort study of more than 25,000 patients had similar conclusions about the magnitude of risk associated with coronary calcification.4 It also found that the 10-year risk of death was 0.6% in patients with a score of 0, 3.4% with a score of 101 to 399, 5.3% with a score of 400 to 699, 6.1% with a score of 700 to 999, and 12.2% with a score greater than 1,000.
Although progression of coronary artery calcification may predict the risk of death from any cause,5 the clinical utility of serial measurements is not yet apparent, especially since statin therapy—our front-line treatment for coronary disease—has not been shown to slow the progression of calcification.
Improving the accuracy of risk prediction
If a patient’s 10-year coronary risk is intermediate (10% to 20%), calcification scoring can reclassify the risk as low or high in about 50% of cases and can improve the accuracy of risk prediction.6–8
For example, Elias-Smale et al6 evaluated the effect of calcification scoring in 2,028 asymptomatic patients, with median follow-up of 9.2 years and 135 coronary events observed. Adding the calcification score to the Framingham model significantly improved risk classification, with a net reclassification improvement (NRI) of 0.14 (P < .01). (NRI is a measure of discriminatory performance for a diagnostic test; higher is better.9) Reclassification was most robust in those at intermediate risk, 52% of whom were reclassified, with 30% reclassified to low risk and 22% reclassified to high risk.
Erbel et al7 reported data from the Heinz Nixdorf Recall study, which used calcification scoring to estimate the NRI in 4,129 patients followed for 5 years. During this time there were 93 coronary deaths and non-fatal myocardial infarctions. The addition of the calcification score to the Framingham risk model resulted in an NRI of 0.21 (P = .0002) for patients with a risk of 6% to 20% and 0.31 (P < .0001) for those with a risk of 10% to 20%. Erbel et al also estimated the C statistic (area under the receiver operating characteristic curve; the maximum value is 1.0 and the higher the value the better) for the addition of the calcification score to the Framingham risk model and to the Adult Treatment Panel (ATP) III algorithm. They reported a significant increase of 0.681 to 0.749 with the Framingham model and 0.653 to 0.755 with the ATP III algorithm.
Polonsky et al8 studied a cohort of 5,878 participants from the Multi-Ethnic Study of Atherosclerosis (MESA) and estimated the event risk using a model based on Framingham risk characteristics. When the calcification score was added to the prediction model, 26% of the sample was reclassified to a new risk category. In intermediate-risk patients, 292 (16%) were reclassified as high risk, and 712 (39%) were reclassified as low risk, achieving an NRI of 0.55 (95% confidence interval 0.41 to 0.69; P < .001). In addition, the C statistic for the prediction of cardiovascular events was 0.76 for the model based on Framingham risk characteristics and increased to 0.81 (P < .001) with the addition of calcification scoring.
Improving adherence and care
Knowing that a patient has a higher calcification score, physicians are more likely to prescribe lipid-lowering and antihypertensive drugs (Table 1),10–12 and patients with a higher score are also more often adherent to recommendations regarding diet and exercise.13
Rozanski et al,14 in a randomized controlled trial, showed that measuring coronary artery calcification did not increase downstream medical spending. A modest improvement in systolic blood pressure (P = .02), serum low-density lipoprotein level (P = .04), and waist circumference (P = .01) was observed in patients who had their calcification measured. Patients with the highest scores had the greatest improvement in coronary risk factors, including blood pressure, cholesterol, weight, and regular exercise.
On the other hand, other analyses have suggested that imaging tests are not effective for motivating behavioral changes. This topic deserves more research.15
Less utility in symptomatic disease
Coronary artery calcification scoring has less clinical utility in patients who already have coronary symptoms. Villines et al16 described a cohort of 10,037 patients with coronary symptoms who underwent calcification scoring and computed tomographic coronary angiography and found that stenosis of greater than 50% was present in 3.5% of those who had a score of 0 and in 29% of those with a score higher than 0. Therefore, a score of 0 does not rule out obstructive coronary heart disease if the patient has symptoms. Conversely, these patients may still have coronary artery calcification even if perfusion stress imaging is normal,17,18 and calcification scoring may have a role in the evaluation of equivocal stress tests.19
CALCIFICATION SCORING GUIDELINES
In their most recent (2010) joint guidelines for assessing risk of coronary heart disease in asymptomatic patients,20 the American College of Cardiology and the American Heart Association say coronary artery calcification scoring:
- Is recommended for asymptomatic patients at intermediate 10-year risk (10% to 20%) of coronary heart disease (class IIa recommendation, level of evidence B)
- May be acceptable for asymptomatic patients at low to intermediate risk (6% to 10%) (class IIb recommendation)
- Is discouraged for those at low risk (< 6%) (class III recommendation).
The most recent (2010) criteria for the appropriate use of cardiac computed tomography21 provide similar recommendations. Specifically, coronary artery calcification scoring with noncontrast computed tomography was rated as appropriate for patients at intermediate risk (10% to 20%) of coronary heart disease and for the specific subset of patients who are at low risk (6% to 10%) but who have a family history of premature coronary heart disease.
These recommendations are based on multiple lines of evidence that calcification scoring is a robust risk-predictor, can enhance risk estimates beyond traditional scoring strategies, and may—in theory—improve outcomes.
CALCIFICATION SCORING’S LIMITATIONS
The images used for measuring coronary calcification do predict risk of cardiovascular events, but they are not adequate to assess the severity of coronary stenosis. Further, calcification scoring often leads to incidental findings, which can cause anxiety and possibly lead to more imaging, entailing more radiation exposure and expense. And as noted, there are no randomized trial data demonstrating a reduction in cardiovascular events with the use of calcification scoring.
- Redberg RF, Benjamin EJ, Bittner V, et al. ACCF/AHA 2009 performance measures for primary prevention of cardiovascular disease in adults. J Am Coll Cardiol 2009; 54:1364–1405.
- Greenland P, Bonow RO, Brundage BH, et al. ACCF/AHA 2007 clinical expert consensus document on coronary artery calcium scoring by computed tomography in global cardiovascular risk assessment and in evaluation of patients with chest pain. J Am Coll Cardiol 2007; 49:378–402.
- Winchester DE, Wymer DC, Shifrin RY, Kraft SM, Hill JA. Responsible use of computed tomography in the evaluation of coronary artery disease and chest pain. Mayo Clin Proc 2010; 85:358–364.
- Budoff MJ, Shaw LJ, Liu ST, et al. Long-term prognosis associated with coronary calcification: observations from a registry of 25,253 patients. J Am Coll Cardiol 2007; 49:1860–1870.
- Budoff MJ, Hokanson JE, Nasir K, et al. Progression of coronary artery calcium predicts all-cause mortality. JACC Cardiovasc Imaging 2010; 3:1229–1236.
- Elias-Smale SE, Proença RV, Koller MT, et al. Coronary calcium score improves classification of coronary heart disease risk in the elderly: The Rotterdam study. J Am Coll Cardiol 2010; 56:1407–1414.
- Erbel R, Möhlenkamp S, Moebus S, et al; Heinz Nixdorf Recall Study Investigative Group. Coronary risk stratification, discrimination, and reclassification improvement based on quantification of subclinical coronary atherosclerosis: the Heinz Nixdorf Recall study. J Am Coll Cardiol 2010; 56:1397–1406.
- Polonsky TS, McClelland RL, Jorgensen NW, et al. Coronary artery calcium score and risk classification for coronary heart disease prediction. JAMA 2010; 303:1610–1616.
- Pencina MJ, Agostino RB, Agostino RB, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Statist Med 2008; 27:157–172.
- Kalia NK, Miller LG, Nasir K, Blumenthal RS, Agrawal N, Budoff MJ. Visualizing coronary calcium is associated with improvements in adherence to statin therapy. Atherosclerosis 2006; 185:394–399.
- Nasir K, McClelland RL, Blumenthal RS, et al. Coronary artery calcium in relation to initiation and continuation of cardiovascular preventive medications: the Multi-Ethnic Study of Atherosclerosis (MESA). Circ Cardiovasc Qual Outcomes 2010; 3:228–235.
- Taylor AJ, Bindeman J, Feuerstein I, et al. Community-based provision of statin and aspirin after the detection of coronary artery calcium within a community-based screening cohort. J Am Coll Cardiol 2008; 51:1337–1341.
- Orakzai RH, Nasir K, Orakzai SH, et al. Effect of patient visualization of coronary calcium by electron beam computed tomography on changes in beneficial lifestyle behaviors. Am J Cardiol 2008; 101:999–1002.
- Rozanski A, Gransar H, Shaw LJ, et al. Impact of coronary artery calcium scanning on coronary risk factors and downstream testing the EISNER (Early Identification of Subclinical Atherosclerosis by Noninvasive Imaging Research) prospective randomized trial. J Am Coll Cardiol 2011; 57:1622–1632.
- Hackam DG, Shojania KG, Spence JD, et al. Influence of noninvasive cardiovascular imaging in primary prevention: systematic review and meta-analysis of randomized trials. Arch Intern Med 2011; 171:977–982.
- Villines TC, Hulten EA, Shaw LJ, et al; CONFIRM Registry Investigators. Prevalence and severity of coronary artery disease and adverse events among symptomatic patients with coronary artery calcification scores of zero undergoing coronary computed tomography angiography. J Am Coll Cardiol 2011; 58:2533–2540.
- Schenker MP, Dorbala S, Hong EC, et al. Interrelation of coronary calcification, myocardial ischemia, and outcomes in patients with intermediate likelihood of coronary artery disease: a combined positron emission tomography/computed tomography study. Circulation 2008; 117:1693–1700.
- Bybee KA, Lee J, Markiewicz R, et al. Diagnostic and clinical benefit of combined coronary calcium and perfusion assessment in patients undergoing PET/CT myocardial perfusion stress imaging. J Nucl Cardiol 2010; 17:188–196.
- Schmermund A, Baumgart D, Sack S, et al. Assessment of coronary calcification by electron-beam computed tomography in symptomatic patients with normal, abnormal or equivocal exercise stress test. Eur Heart J 2000; 21:1674–1682.
- Greenland P, Alpert JS, Beller GA, et al. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults. J Am Coll Cardiol 2010; 56:e50–e103.
- Taylor AJ, Cerqueira M, Hodgson JM, et al. ACCF/SCCT/ACR/AHA/ASE/ASNC/NASCI/SCAI/SCMR 2010 appropriate use criteria for cardiac computed tomography. J Am Coll Cardiol 2010; 56:1864–1894.
Although we still have no evidence from randomized trials that patients have better outcomes if we measure the calcification in their coronary arteries, a growing body of evidence shows that we can estimate risk more accurately than with a risk model score alone if we also score coronary artery calcification in asymptomatic patients, especially those at intermediate risk.
Current guidelines1 recommend using the Framingham Risk Score or a similar tool to estimate coronary risk in asymptomatic patients, but these tools have only modest accuracy. Calcification scoring is accurate, inexpensive, quick, widely available, low-risk, and does not appear to increase medical costs afterward. In addition to improving risk stratification, it may also encourage patients to adhere better to drug therapy and lifestyle modification.
HOW IS CORONARY ARTERY CALCIFICATION MEASURED?
Calcification of the coronary arteries is synonymous with atherosclerosis. It can easily be detected with computed tomography without contrast (Figure 1), and the amount can be quantified with a scoring system such as the volumetric score or the Agatston score. The latter, which is more commonly used, is based on the product of the area of the calcium deposits and the x-ray attenuation in Hounsfield units.
Scores can be roughly categorized (with some overlap owing to data from different studies) as:
- Low risk: 0 Agatston units (AU)
- Average risk: 1–112 AU
- Moderate risk: 100–400 AU
- High risk: 400–999 AU
- Very high risk: 1,000 AU.2
The actual test takes only a few seconds, and the patient can usually be out the door in 15 minutes or less. It does not require iodinated contrast and the radiation dose is minimal, usually less than 1 mSv, equivalent to fewer than 10 chest radiographs.3
The cost is typically between $200 and $500. The test is usually not covered by health insurance, but this differs by insurer and by state; for example, coverage is mandated in Texas, and the test is covered by United Healthcare.
WHAT IS THE EVIDENCE IN FAVOR OF CALCIFICATION SCORING?
Cohort studies with long-term follow-up show that calcification scoring has robust prognostic ability. A pooled analysis of several of these studies2 showed that a higher score strongly correlated with a higher risk of cardiac events over 3 to 5 years. Compared with the risk in people with a score of 0, the risk was twice as high in those with a score of 1 to 112, four times as high with a score of 100 to 400, seven times as high with a score of 400 to 499, and 10 times as high with a score greater than 1,000.2
A cohort study of more than 25,000 patients had similar conclusions about the magnitude of risk associated with coronary calcification.4 It also found that the 10-year risk of death was 0.6% in patients with a score of 0, 3.4% with a score of 101 to 399, 5.3% with a score of 400 to 699, 6.1% with a score of 700 to 999, and 12.2% with a score greater than 1,000.
Although progression of coronary artery calcification may predict the risk of death from any cause,5 the clinical utility of serial measurements is not yet apparent, especially since statin therapy—our front-line treatment for coronary disease—has not been shown to slow the progression of calcification.
Improving the accuracy of risk prediction
If a patient’s 10-year coronary risk is intermediate (10% to 20%), calcification scoring can reclassify the risk as low or high in about 50% of cases and can improve the accuracy of risk prediction.6–8
For example, Elias-Smale et al6 evaluated the effect of calcification scoring in 2,028 asymptomatic patients, with median follow-up of 9.2 years and 135 coronary events observed. Adding the calcification score to the Framingham model significantly improved risk classification, with a net reclassification improvement (NRI) of 0.14 (P < .01). (NRI is a measure of discriminatory performance for a diagnostic test; higher is better.9) Reclassification was most robust in those at intermediate risk, 52% of whom were reclassified, with 30% reclassified to low risk and 22% reclassified to high risk.
Erbel et al7 reported data from the Heinz Nixdorf Recall study, which used calcification scoring to estimate the NRI in 4,129 patients followed for 5 years. During this time there were 93 coronary deaths and non-fatal myocardial infarctions. The addition of the calcification score to the Framingham risk model resulted in an NRI of 0.21 (P = .0002) for patients with a risk of 6% to 20% and 0.31 (P < .0001) for those with a risk of 10% to 20%. Erbel et al also estimated the C statistic (area under the receiver operating characteristic curve; the maximum value is 1.0 and the higher the value the better) for the addition of the calcification score to the Framingham risk model and to the Adult Treatment Panel (ATP) III algorithm. They reported a significant increase of 0.681 to 0.749 with the Framingham model and 0.653 to 0.755 with the ATP III algorithm.
Polonsky et al8 studied a cohort of 5,878 participants from the Multi-Ethnic Study of Atherosclerosis (MESA) and estimated the event risk using a model based on Framingham risk characteristics. When the calcification score was added to the prediction model, 26% of the sample was reclassified to a new risk category. In intermediate-risk patients, 292 (16%) were reclassified as high risk, and 712 (39%) were reclassified as low risk, achieving an NRI of 0.55 (95% confidence interval 0.41 to 0.69; P < .001). In addition, the C statistic for the prediction of cardiovascular events was 0.76 for the model based on Framingham risk characteristics and increased to 0.81 (P < .001) with the addition of calcification scoring.
Improving adherence and care
Knowing that a patient has a higher calcification score, physicians are more likely to prescribe lipid-lowering and antihypertensive drugs (Table 1),10–12 and patients with a higher score are also more often adherent to recommendations regarding diet and exercise.13
Rozanski et al,14 in a randomized controlled trial, showed that measuring coronary artery calcification did not increase downstream medical spending. A modest improvement in systolic blood pressure (P = .02), serum low-density lipoprotein level (P = .04), and waist circumference (P = .01) was observed in patients who had their calcification measured. Patients with the highest scores had the greatest improvement in coronary risk factors, including blood pressure, cholesterol, weight, and regular exercise.
On the other hand, other analyses have suggested that imaging tests are not effective for motivating behavioral changes. This topic deserves more research.15
Less utility in symptomatic disease
Coronary artery calcification scoring has less clinical utility in patients who already have coronary symptoms. Villines et al16 described a cohort of 10,037 patients with coronary symptoms who underwent calcification scoring and computed tomographic coronary angiography and found that stenosis of greater than 50% was present in 3.5% of those who had a score of 0 and in 29% of those with a score higher than 0. Therefore, a score of 0 does not rule out obstructive coronary heart disease if the patient has symptoms. Conversely, these patients may still have coronary artery calcification even if perfusion stress imaging is normal,17,18 and calcification scoring may have a role in the evaluation of equivocal stress tests.19
CALCIFICATION SCORING GUIDELINES
In their most recent (2010) joint guidelines for assessing risk of coronary heart disease in asymptomatic patients,20 the American College of Cardiology and the American Heart Association say coronary artery calcification scoring:
- Is recommended for asymptomatic patients at intermediate 10-year risk (10% to 20%) of coronary heart disease (class IIa recommendation, level of evidence B)
- May be acceptable for asymptomatic patients at low to intermediate risk (6% to 10%) (class IIb recommendation)
- Is discouraged for those at low risk (< 6%) (class III recommendation).
The most recent (2010) criteria for the appropriate use of cardiac computed tomography21 provide similar recommendations. Specifically, coronary artery calcification scoring with noncontrast computed tomography was rated as appropriate for patients at intermediate risk (10% to 20%) of coronary heart disease and for the specific subset of patients who are at low risk (6% to 10%) but who have a family history of premature coronary heart disease.
These recommendations are based on multiple lines of evidence that calcification scoring is a robust risk-predictor, can enhance risk estimates beyond traditional scoring strategies, and may—in theory—improve outcomes.
CALCIFICATION SCORING’S LIMITATIONS
The images used for measuring coronary calcification do predict risk of cardiovascular events, but they are not adequate to assess the severity of coronary stenosis. Further, calcification scoring often leads to incidental findings, which can cause anxiety and possibly lead to more imaging, entailing more radiation exposure and expense. And as noted, there are no randomized trial data demonstrating a reduction in cardiovascular events with the use of calcification scoring.
Although we still have no evidence from randomized trials that patients have better outcomes if we measure the calcification in their coronary arteries, a growing body of evidence shows that we can estimate risk more accurately than with a risk model score alone if we also score coronary artery calcification in asymptomatic patients, especially those at intermediate risk.
Current guidelines1 recommend using the Framingham Risk Score or a similar tool to estimate coronary risk in asymptomatic patients, but these tools have only modest accuracy. Calcification scoring is accurate, inexpensive, quick, widely available, low-risk, and does not appear to increase medical costs afterward. In addition to improving risk stratification, it may also encourage patients to adhere better to drug therapy and lifestyle modification.
HOW IS CORONARY ARTERY CALCIFICATION MEASURED?
Calcification of the coronary arteries is synonymous with atherosclerosis. It can easily be detected with computed tomography without contrast (Figure 1), and the amount can be quantified with a scoring system such as the volumetric score or the Agatston score. The latter, which is more commonly used, is based on the product of the area of the calcium deposits and the x-ray attenuation in Hounsfield units.
Scores can be roughly categorized (with some overlap owing to data from different studies) as:
- Low risk: 0 Agatston units (AU)
- Average risk: 1–112 AU
- Moderate risk: 100–400 AU
- High risk: 400–999 AU
- Very high risk: 1,000 AU.2
The actual test takes only a few seconds, and the patient can usually be out the door in 15 minutes or less. It does not require iodinated contrast and the radiation dose is minimal, usually less than 1 mSv, equivalent to fewer than 10 chest radiographs.3
The cost is typically between $200 and $500. The test is usually not covered by health insurance, but this differs by insurer and by state; for example, coverage is mandated in Texas, and the test is covered by United Healthcare.
WHAT IS THE EVIDENCE IN FAVOR OF CALCIFICATION SCORING?
Cohort studies with long-term follow-up show that calcification scoring has robust prognostic ability. A pooled analysis of several of these studies2 showed that a higher score strongly correlated with a higher risk of cardiac events over 3 to 5 years. Compared with the risk in people with a score of 0, the risk was twice as high in those with a score of 1 to 112, four times as high with a score of 100 to 400, seven times as high with a score of 400 to 499, and 10 times as high with a score greater than 1,000.2
A cohort study of more than 25,000 patients had similar conclusions about the magnitude of risk associated with coronary calcification.4 It also found that the 10-year risk of death was 0.6% in patients with a score of 0, 3.4% with a score of 101 to 399, 5.3% with a score of 400 to 699, 6.1% with a score of 700 to 999, and 12.2% with a score greater than 1,000.
Although progression of coronary artery calcification may predict the risk of death from any cause,5 the clinical utility of serial measurements is not yet apparent, especially since statin therapy—our front-line treatment for coronary disease—has not been shown to slow the progression of calcification.
Improving the accuracy of risk prediction
If a patient’s 10-year coronary risk is intermediate (10% to 20%), calcification scoring can reclassify the risk as low or high in about 50% of cases and can improve the accuracy of risk prediction.6–8
For example, Elias-Smale et al6 evaluated the effect of calcification scoring in 2,028 asymptomatic patients, with median follow-up of 9.2 years and 135 coronary events observed. Adding the calcification score to the Framingham model significantly improved risk classification, with a net reclassification improvement (NRI) of 0.14 (P < .01). (NRI is a measure of discriminatory performance for a diagnostic test; higher is better.9) Reclassification was most robust in those at intermediate risk, 52% of whom were reclassified, with 30% reclassified to low risk and 22% reclassified to high risk.
Erbel et al7 reported data from the Heinz Nixdorf Recall study, which used calcification scoring to estimate the NRI in 4,129 patients followed for 5 years. During this time there were 93 coronary deaths and non-fatal myocardial infarctions. The addition of the calcification score to the Framingham risk model resulted in an NRI of 0.21 (P = .0002) for patients with a risk of 6% to 20% and 0.31 (P < .0001) for those with a risk of 10% to 20%. Erbel et al also estimated the C statistic (area under the receiver operating characteristic curve; the maximum value is 1.0 and the higher the value the better) for the addition of the calcification score to the Framingham risk model and to the Adult Treatment Panel (ATP) III algorithm. They reported a significant increase of 0.681 to 0.749 with the Framingham model and 0.653 to 0.755 with the ATP III algorithm.
Polonsky et al8 studied a cohort of 5,878 participants from the Multi-Ethnic Study of Atherosclerosis (MESA) and estimated the event risk using a model based on Framingham risk characteristics. When the calcification score was added to the prediction model, 26% of the sample was reclassified to a new risk category. In intermediate-risk patients, 292 (16%) were reclassified as high risk, and 712 (39%) were reclassified as low risk, achieving an NRI of 0.55 (95% confidence interval 0.41 to 0.69; P < .001). In addition, the C statistic for the prediction of cardiovascular events was 0.76 for the model based on Framingham risk characteristics and increased to 0.81 (P < .001) with the addition of calcification scoring.
Improving adherence and care
Knowing that a patient has a higher calcification score, physicians are more likely to prescribe lipid-lowering and antihypertensive drugs (Table 1),10–12 and patients with a higher score are also more often adherent to recommendations regarding diet and exercise.13
Rozanski et al,14 in a randomized controlled trial, showed that measuring coronary artery calcification did not increase downstream medical spending. A modest improvement in systolic blood pressure (P = .02), serum low-density lipoprotein level (P = .04), and waist circumference (P = .01) was observed in patients who had their calcification measured. Patients with the highest scores had the greatest improvement in coronary risk factors, including blood pressure, cholesterol, weight, and regular exercise.
On the other hand, other analyses have suggested that imaging tests are not effective for motivating behavioral changes. This topic deserves more research.15
Less utility in symptomatic disease
Coronary artery calcification scoring has less clinical utility in patients who already have coronary symptoms. Villines et al16 described a cohort of 10,037 patients with coronary symptoms who underwent calcification scoring and computed tomographic coronary angiography and found that stenosis of greater than 50% was present in 3.5% of those who had a score of 0 and in 29% of those with a score higher than 0. Therefore, a score of 0 does not rule out obstructive coronary heart disease if the patient has symptoms. Conversely, these patients may still have coronary artery calcification even if perfusion stress imaging is normal,17,18 and calcification scoring may have a role in the evaluation of equivocal stress tests.19
CALCIFICATION SCORING GUIDELINES
In their most recent (2010) joint guidelines for assessing risk of coronary heart disease in asymptomatic patients,20 the American College of Cardiology and the American Heart Association say coronary artery calcification scoring:
- Is recommended for asymptomatic patients at intermediate 10-year risk (10% to 20%) of coronary heart disease (class IIa recommendation, level of evidence B)
- May be acceptable for asymptomatic patients at low to intermediate risk (6% to 10%) (class IIb recommendation)
- Is discouraged for those at low risk (< 6%) (class III recommendation).
The most recent (2010) criteria for the appropriate use of cardiac computed tomography21 provide similar recommendations. Specifically, coronary artery calcification scoring with noncontrast computed tomography was rated as appropriate for patients at intermediate risk (10% to 20%) of coronary heart disease and for the specific subset of patients who are at low risk (6% to 10%) but who have a family history of premature coronary heart disease.
These recommendations are based on multiple lines of evidence that calcification scoring is a robust risk-predictor, can enhance risk estimates beyond traditional scoring strategies, and may—in theory—improve outcomes.
CALCIFICATION SCORING’S LIMITATIONS
The images used for measuring coronary calcification do predict risk of cardiovascular events, but they are not adequate to assess the severity of coronary stenosis. Further, calcification scoring often leads to incidental findings, which can cause anxiety and possibly lead to more imaging, entailing more radiation exposure and expense. And as noted, there are no randomized trial data demonstrating a reduction in cardiovascular events with the use of calcification scoring.
- Redberg RF, Benjamin EJ, Bittner V, et al. ACCF/AHA 2009 performance measures for primary prevention of cardiovascular disease in adults. J Am Coll Cardiol 2009; 54:1364–1405.
- Greenland P, Bonow RO, Brundage BH, et al. ACCF/AHA 2007 clinical expert consensus document on coronary artery calcium scoring by computed tomography in global cardiovascular risk assessment and in evaluation of patients with chest pain. J Am Coll Cardiol 2007; 49:378–402.
- Winchester DE, Wymer DC, Shifrin RY, Kraft SM, Hill JA. Responsible use of computed tomography in the evaluation of coronary artery disease and chest pain. Mayo Clin Proc 2010; 85:358–364.
- Budoff MJ, Shaw LJ, Liu ST, et al. Long-term prognosis associated with coronary calcification: observations from a registry of 25,253 patients. J Am Coll Cardiol 2007; 49:1860–1870.
- Budoff MJ, Hokanson JE, Nasir K, et al. Progression of coronary artery calcium predicts all-cause mortality. JACC Cardiovasc Imaging 2010; 3:1229–1236.
- Elias-Smale SE, Proença RV, Koller MT, et al. Coronary calcium score improves classification of coronary heart disease risk in the elderly: The Rotterdam study. J Am Coll Cardiol 2010; 56:1407–1414.
- Erbel R, Möhlenkamp S, Moebus S, et al; Heinz Nixdorf Recall Study Investigative Group. Coronary risk stratification, discrimination, and reclassification improvement based on quantification of subclinical coronary atherosclerosis: the Heinz Nixdorf Recall study. J Am Coll Cardiol 2010; 56:1397–1406.
- Polonsky TS, McClelland RL, Jorgensen NW, et al. Coronary artery calcium score and risk classification for coronary heart disease prediction. JAMA 2010; 303:1610–1616.
- Pencina MJ, Agostino RB, Agostino RB, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Statist Med 2008; 27:157–172.
- Kalia NK, Miller LG, Nasir K, Blumenthal RS, Agrawal N, Budoff MJ. Visualizing coronary calcium is associated with improvements in adherence to statin therapy. Atherosclerosis 2006; 185:394–399.
- Nasir K, McClelland RL, Blumenthal RS, et al. Coronary artery calcium in relation to initiation and continuation of cardiovascular preventive medications: the Multi-Ethnic Study of Atherosclerosis (MESA). Circ Cardiovasc Qual Outcomes 2010; 3:228–235.
- Taylor AJ, Bindeman J, Feuerstein I, et al. Community-based provision of statin and aspirin after the detection of coronary artery calcium within a community-based screening cohort. J Am Coll Cardiol 2008; 51:1337–1341.
- Orakzai RH, Nasir K, Orakzai SH, et al. Effect of patient visualization of coronary calcium by electron beam computed tomography on changes in beneficial lifestyle behaviors. Am J Cardiol 2008; 101:999–1002.
- Rozanski A, Gransar H, Shaw LJ, et al. Impact of coronary artery calcium scanning on coronary risk factors and downstream testing the EISNER (Early Identification of Subclinical Atherosclerosis by Noninvasive Imaging Research) prospective randomized trial. J Am Coll Cardiol 2011; 57:1622–1632.
- Hackam DG, Shojania KG, Spence JD, et al. Influence of noninvasive cardiovascular imaging in primary prevention: systematic review and meta-analysis of randomized trials. Arch Intern Med 2011; 171:977–982.
- Villines TC, Hulten EA, Shaw LJ, et al; CONFIRM Registry Investigators. Prevalence and severity of coronary artery disease and adverse events among symptomatic patients with coronary artery calcification scores of zero undergoing coronary computed tomography angiography. J Am Coll Cardiol 2011; 58:2533–2540.
- Schenker MP, Dorbala S, Hong EC, et al. Interrelation of coronary calcification, myocardial ischemia, and outcomes in patients with intermediate likelihood of coronary artery disease: a combined positron emission tomography/computed tomography study. Circulation 2008; 117:1693–1700.
- Bybee KA, Lee J, Markiewicz R, et al. Diagnostic and clinical benefit of combined coronary calcium and perfusion assessment in patients undergoing PET/CT myocardial perfusion stress imaging. J Nucl Cardiol 2010; 17:188–196.
- Schmermund A, Baumgart D, Sack S, et al. Assessment of coronary calcification by electron-beam computed tomography in symptomatic patients with normal, abnormal or equivocal exercise stress test. Eur Heart J 2000; 21:1674–1682.
- Greenland P, Alpert JS, Beller GA, et al. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults. J Am Coll Cardiol 2010; 56:e50–e103.
- Taylor AJ, Cerqueira M, Hodgson JM, et al. ACCF/SCCT/ACR/AHA/ASE/ASNC/NASCI/SCAI/SCMR 2010 appropriate use criteria for cardiac computed tomography. J Am Coll Cardiol 2010; 56:1864–1894.
- Redberg RF, Benjamin EJ, Bittner V, et al. ACCF/AHA 2009 performance measures for primary prevention of cardiovascular disease in adults. J Am Coll Cardiol 2009; 54:1364–1405.
- Greenland P, Bonow RO, Brundage BH, et al. ACCF/AHA 2007 clinical expert consensus document on coronary artery calcium scoring by computed tomography in global cardiovascular risk assessment and in evaluation of patients with chest pain. J Am Coll Cardiol 2007; 49:378–402.
- Winchester DE, Wymer DC, Shifrin RY, Kraft SM, Hill JA. Responsible use of computed tomography in the evaluation of coronary artery disease and chest pain. Mayo Clin Proc 2010; 85:358–364.
- Budoff MJ, Shaw LJ, Liu ST, et al. Long-term prognosis associated with coronary calcification: observations from a registry of 25,253 patients. J Am Coll Cardiol 2007; 49:1860–1870.
- Budoff MJ, Hokanson JE, Nasir K, et al. Progression of coronary artery calcium predicts all-cause mortality. JACC Cardiovasc Imaging 2010; 3:1229–1236.
- Elias-Smale SE, Proença RV, Koller MT, et al. Coronary calcium score improves classification of coronary heart disease risk in the elderly: The Rotterdam study. J Am Coll Cardiol 2010; 56:1407–1414.
- Erbel R, Möhlenkamp S, Moebus S, et al; Heinz Nixdorf Recall Study Investigative Group. Coronary risk stratification, discrimination, and reclassification improvement based on quantification of subclinical coronary atherosclerosis: the Heinz Nixdorf Recall study. J Am Coll Cardiol 2010; 56:1397–1406.
- Polonsky TS, McClelland RL, Jorgensen NW, et al. Coronary artery calcium score and risk classification for coronary heart disease prediction. JAMA 2010; 303:1610–1616.
- Pencina MJ, Agostino RB, Agostino RB, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Statist Med 2008; 27:157–172.
- Kalia NK, Miller LG, Nasir K, Blumenthal RS, Agrawal N, Budoff MJ. Visualizing coronary calcium is associated with improvements in adherence to statin therapy. Atherosclerosis 2006; 185:394–399.
- Nasir K, McClelland RL, Blumenthal RS, et al. Coronary artery calcium in relation to initiation and continuation of cardiovascular preventive medications: the Multi-Ethnic Study of Atherosclerosis (MESA). Circ Cardiovasc Qual Outcomes 2010; 3:228–235.
- Taylor AJ, Bindeman J, Feuerstein I, et al. Community-based provision of statin and aspirin after the detection of coronary artery calcium within a community-based screening cohort. J Am Coll Cardiol 2008; 51:1337–1341.
- Orakzai RH, Nasir K, Orakzai SH, et al. Effect of patient visualization of coronary calcium by electron beam computed tomography on changes in beneficial lifestyle behaviors. Am J Cardiol 2008; 101:999–1002.
- Rozanski A, Gransar H, Shaw LJ, et al. Impact of coronary artery calcium scanning on coronary risk factors and downstream testing the EISNER (Early Identification of Subclinical Atherosclerosis by Noninvasive Imaging Research) prospective randomized trial. J Am Coll Cardiol 2011; 57:1622–1632.
- Hackam DG, Shojania KG, Spence JD, et al. Influence of noninvasive cardiovascular imaging in primary prevention: systematic review and meta-analysis of randomized trials. Arch Intern Med 2011; 171:977–982.
- Villines TC, Hulten EA, Shaw LJ, et al; CONFIRM Registry Investigators. Prevalence and severity of coronary artery disease and adverse events among symptomatic patients with coronary artery calcification scores of zero undergoing coronary computed tomography angiography. J Am Coll Cardiol 2011; 58:2533–2540.
- Schenker MP, Dorbala S, Hong EC, et al. Interrelation of coronary calcification, myocardial ischemia, and outcomes in patients with intermediate likelihood of coronary artery disease: a combined positron emission tomography/computed tomography study. Circulation 2008; 117:1693–1700.
- Bybee KA, Lee J, Markiewicz R, et al. Diagnostic and clinical benefit of combined coronary calcium and perfusion assessment in patients undergoing PET/CT myocardial perfusion stress imaging. J Nucl Cardiol 2010; 17:188–196.
- Schmermund A, Baumgart D, Sack S, et al. Assessment of coronary calcification by electron-beam computed tomography in symptomatic patients with normal, abnormal or equivocal exercise stress test. Eur Heart J 2000; 21:1674–1682.
- Greenland P, Alpert JS, Beller GA, et al. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults. J Am Coll Cardiol 2010; 56:e50–e103.
- Taylor AJ, Cerqueira M, Hodgson JM, et al. ACCF/SCCT/ACR/AHA/ASE/ASNC/NASCI/SCAI/SCMR 2010 appropriate use criteria for cardiac computed tomography. J Am Coll Cardiol 2010; 56:1864–1894.