User login
What is the difference between palliative care and hospice care?
Hospice care generally falls under the category of palliative care, despite being an older subspecialty. However, the two have different indications and goals and are often provided in different settings.
ORIGINS OF PALLIATIVE CARE
Prompted by what he perceived as neglect of dying patients in the acute care setting, Dr. Balfour Mount opened the first acute inpatient palliative care unit in Royal Victoria Hospital in Montréal, Québec, in 1976.1 His purpose was to provide a crisis-intervention service for patients who were actively dying, and this continues to be the main reason for consulting palliative care services in the hospital.
Palliative care has evolved since the 1970s and is now used in a variety of situations:
- A life-limiting illness in a patient who is not terminally ill
- A life-threatening illness in a patient who has symptoms but with the potential to recover
- A chronic illness such as heart failure or chronic obstructive pulmonary disease in a patient who is on disease-modifying therapy but has symptoms and will eventually succumb to the illness, but is expected to live longer than someone with advanced cancer.2
PALLIATIVE CARE IN CANCER PATIENTS
In patients with advanced cancer, palliative care is utilized earlier in the course of serious and life-limiting illness and is even involved in patient care when cure is the goal. Importantly, it now includes outpatient clinics to provide patients seamless care in conjunction with their oncologist’s care.3
Because palliative care focuses on the patient’s experience of the illness (sickness) rather than on disease itself (pathology), symptom management, psychosocial support, and assistance in decision-making are foremost. Initiating palliative care early in advanced cancer improves multiple outcomes and limits overly aggressive, ineffective therapies at the end of life (eg, late chemotherapy, late referral to hospice care, death in the intensive care unit), without hastening death. In fact, it may prolong life.3,4
Palliative care is indicated in a number of situations in oncology:
- Symptomatic presentations of cancer, even when curative treatments are available
- At the time of a sentinel event such as recurrence or unanticipated hospitalization
- When palliative radiation is needed
- When changes in chemotherapy are needed because of disease progression.
Also, cancer patients may develop symptoms that require a palliative procedure such as thoracentesis for pleural effusion, paracentesis for ascites, or surgery for a fracture or spinal cord compression. A palliative care consultation is also appropriate when patients change their goals of care (ie, palliation rather than cure), and when an oncologic crisis occurs and there is a need to offer support to the family and to clarify the goals of care.
PALLIATIVE CARE IN OTHER DISEASES
For patients with illnesses other than cancer, palliative care may be helpful when disease-modifying therapy becomes burdensome or ineffective, or when patients are symptomatic despite maximum therapy. Palliative care should also be considered when goals of care need to be explored, when a second opinion is needed on goals of care, or if the primary care provider and family are at odds.
WHEN A CONSULT IS INAPPROPRIATE
Palliative care consultation is inappropriate when used in lieu of an oncology consult in advanced cancer. Palliative care specialists are not experts in cancer care, whereas oncologists are familiar with rapid advancements in cancer care, including targeted agents that may offer benefit to patients with advanced cancer.
Palliative care consultation is also inappropriate if the patient does not want to see a palliative care specialist, or if the consult is used as a way to convince a patient to change advance directives or to choose not to be resuscitated. Also, cancer patients who are asymptomatic are unlikely to benefit from palliative care initially. The decision to consult palliative care should not depend on prognosis, and palliative care is more cost-effective when utilized early rather than as a crisis intervention near the end of life.3
THE PALLIATIVE CARE EVALUATION
The initial palliative care consultation usually involves an evaluation of the patient’s symptoms and concerns. Symptoms are targeted based on the patient’s priorities and on an assessment using validated questionnaires. A validated questionnaire is a better way to comprehensively gauge symptom burden than depending on patients to volunteer symptoms.5
As the relationship develops between patient, family, and palliative care specialist and as the disease takes its course, advance directives, prognosis, and end-of-life care goals can be addressed in follow-up consultations.3 Patients want to know about their prognosis, and they usually complete advance directives based on clinical circumstances rather than viewing them as an extension of patient autonomy, as originally intended.6
REIMBURSEMENT FOR PALLIATIVE CARE
Reimbursement for palliative care is similar to that for acute care and falls within the All Patient Refined Diagnosis-Related Group, or APR-DRG, system, and palliative care has its own V code for identification. Codes are used to designate disease, stage or location of metastases, disease complications, and symptoms, as well as for the discussion of goals of care.
WHAT PALLIATIVE CARE IS NOT
Palliative care has too often been tied to end-of-life care.7 The two often appear together in titles of reports in the literature. As a result, patients and physicians may be confused and, thus, reluctant to utilize palliative care services. To avoid the confusion, certain programs have included the term “supportive” oncology care in their title. This appears to facilitate palliative care referral, but may be misleading.8
WHAT IS HOSPICE CARE?
Hospice care is a service funded and capitated under Medicare part A and is largely provided as outpatient home care for those deemed terminally ill.9 An illness must be certified as terminal by two physicians. Medicare defines terminal illness as a life expectancy of 6 months or less if the illness runs its normal course.
The philosophy of hospice care is to provide comfort through intensive nurse management and home-based follow-up. In some cases, disease-modifying therapies are continued to control symptoms—eg, continuing angiotensin-converting enzyme inhibitors in heart failure patients. Hospice care is typically delivered at home, but it is also delivered in nursing homes, in hospital inpatient units, and at private or nonprofit hospice facilities.
Inpatient palliative care units are often mistaken for hospices. The purpose of hospice care is to provide quality of life and comfort and to avoid overly aggressive, expensive, and futile care at the end of life. The focus is on intensive, hands-on, personalized symptom care and family support at home. The goal is to provide a comfortable and dignified death among friends and family. The use of palliative radiation, transfusions, and antibiotics in hospice varies among hospice programs and is considered on a case-by-case basis.10
The Medicare per diem payment limits what hospices can afford, so they must be fiscally responsible. Hospice agencies are capitated and are responsible for providing medications and durable equipment necessary to treat symptoms related to the terminal illness. They also provide bereavement services for family members at no charge. Enrollment in hospice care can be revoked depending on circumstances and then reinstituted later as the goals of care change.
Care for nonterminal comorbid illnesses can be continued by a general practitioner or internist. This care is not covered under the Medicare hospice benefit, but it is covered under Medicare part B.
The patient and family can choose the hospice physician, who may be a family practitioner, internist, oncologist, or palliative care specialist, or may designate the hospice medical director as the hospice physician.
Criteria for hospice admission have been established for noncancer terminal illnesses and should be considered when practitioners decide to consult hospice.11–13
HOME-BASED PALLIATIVE CARE
Programs such as advanced illness management or home-based palliative care aim to improve the quality of care at home and prevent rehospitalization, particularly for patients with repeated hospitalizations.14 Home-based palliative care services are provided either by a clinician who makes home visits or by a certified home health care agency. Services are particularly useful for patients with serious illnesses who do not qualify for hospice services but are homebound. Consultations are obtained for ongoing supportive care at home, assessment for medication compliance, and disease monitoring at home. Consultations are scheduled at the time of hospital discharge.
Unlike hospice care, home-based palliative care does not include 24-hour on-call service. Comprehensive services (eg, home health aide, durable equipment, medications) are not provided as they are under hospice care: patients must qualify under Medicare stipulations for such services outside of hospice care. For example, home oxygen can only be supplied if the patient's oxygen saturation is less than 90%, while under the hospice benefit it is provided without regard to oxygen saturation and is based on symptom need. For home-based palliative care, patients must be largely homebound or unable to be seen regularly in the outpatient clinic. This type of care can be a bridge to hospice care for patients who feel they are not ready for hospice care at the time of discharge from acute care. Those who receive palliative care at home are less likely to be hospitalized at the end of life, are more likely to be transitioned to hospice at an appropriate time, and are more likely to have relief of symptoms.15
- Mount BM. The problem of caring for the dying in a general hospital; the palliative care unit as a possible solution. Can Med Assoc J 1976; 115:119–121.
- Higginson I. Palliative care: a review of past changes and future trends. J Public Health Med 1993; 15:3–8.
- Temel JS, Greer JA, Muzikansky A, et al. Early palliative care for patients with metastatic non-small-cell lung cancer. N Engl J Med 2010; 363:733–742.
- Zimmermann C, Riechelmann R, Krzyzanowska M, Rodin G, Tannock I. Effectiveness of specialized palliative care: a systematic review. JAMA 2008; 299:1698–1709.
- Homsi J, Walsh D, Rivera N, et al. Symptom evaluation in palliative medicine: patient report vs systematic assessment. Support Care Cancer 2006; 14:444–453.
- Tang ST, Liu TW, Lai MS, Liu LN, Chen CH, Koong SL. Congruence of knowledge, experiences, and p for disclosure of diagnosis and prognosis between terminally-ill cancer patients and their family caregivers in Taiwan. Cancer Invest 2006; 24:360–366.
- Bakitas M, Lyons KD, Hegel MT, Ahles T. Oncologists’ perspectives on concurrent palliative care in a National Cancer Institute-designated comprehensive cancer center. Palliat Support Care 2013; 11:415–423.
- Fadul N, Elsayem A, Palmer JL, et al. Supportive versus palliative care: what’s in a name: a survey of medical oncologists and midlevel providers at a comprehensive cancer center. Cancer 2009; 115:2013–2021.
- Rinaldo MJ. Medicare to cover hospice services. J Med Soc NJ 1982; 79:1015–1016.
- Enck RE. Palliative radiation therapy in hospice care. Am J Hosp Palliat Care 2002; 19:151–152.
- Luchins DJ, Hanrahan P, Murphy K. Criteria for enrolling dementia patients in hospice. J Am Geriatr Soc 1997; 45:1054–1059.
- Fox E, Landrum-McNiff K, Zhong Z, Dawson NV, Wu AW, Lynn J. Evaluation of prognostic criteria for determining hospice eligibility in patients with advanced lung, heart, or liver disease. JAMA 1999; 282:1638–1645.
- Stuart B. The NHO medical guidelines for non-cancer disease and local medical review policy: hospice access for patients with diseases other than cancer. Hosp J 1999; 14:139–154.
- McKinney M. Beyond hospice. New models of care focus on advanced illnesses. Mod Healthc 2013; 43:14–15.
- Gomes B, Calanzani N, Curiale V, McCrone P, Higginson IJ. Effectiveness and cost-effectiveness of home palliative care services for adults with advanced illness and their caregivers. Cochrane Database Syst Rev 2013; 6:CD007760.
Hospice care generally falls under the category of palliative care, despite being an older subspecialty. However, the two have different indications and goals and are often provided in different settings.
ORIGINS OF PALLIATIVE CARE
Prompted by what he perceived as neglect of dying patients in the acute care setting, Dr. Balfour Mount opened the first acute inpatient palliative care unit in Royal Victoria Hospital in Montréal, Québec, in 1976.1 His purpose was to provide a crisis-intervention service for patients who were actively dying, and this continues to be the main reason for consulting palliative care services in the hospital.
Palliative care has evolved since the 1970s and is now used in a variety of situations:
- A life-limiting illness in a patient who is not terminally ill
- A life-threatening illness in a patient who has symptoms but with the potential to recover
- A chronic illness such as heart failure or chronic obstructive pulmonary disease in a patient who is on disease-modifying therapy but has symptoms and will eventually succumb to the illness, but is expected to live longer than someone with advanced cancer.2
PALLIATIVE CARE IN CANCER PATIENTS
In patients with advanced cancer, palliative care is utilized earlier in the course of serious and life-limiting illness and is even involved in patient care when cure is the goal. Importantly, it now includes outpatient clinics to provide patients seamless care in conjunction with their oncologist’s care.3
Because palliative care focuses on the patient’s experience of the illness (sickness) rather than on disease itself (pathology), symptom management, psychosocial support, and assistance in decision-making are foremost. Initiating palliative care early in advanced cancer improves multiple outcomes and limits overly aggressive, ineffective therapies at the end of life (eg, late chemotherapy, late referral to hospice care, death in the intensive care unit), without hastening death. In fact, it may prolong life.3,4
Palliative care is indicated in a number of situations in oncology:
- Symptomatic presentations of cancer, even when curative treatments are available
- At the time of a sentinel event such as recurrence or unanticipated hospitalization
- When palliative radiation is needed
- When changes in chemotherapy are needed because of disease progression.
Also, cancer patients may develop symptoms that require a palliative procedure such as thoracentesis for pleural effusion, paracentesis for ascites, or surgery for a fracture or spinal cord compression. A palliative care consultation is also appropriate when patients change their goals of care (ie, palliation rather than cure), and when an oncologic crisis occurs and there is a need to offer support to the family and to clarify the goals of care.
PALLIATIVE CARE IN OTHER DISEASES
For patients with illnesses other than cancer, palliative care may be helpful when disease-modifying therapy becomes burdensome or ineffective, or when patients are symptomatic despite maximum therapy. Palliative care should also be considered when goals of care need to be explored, when a second opinion is needed on goals of care, or if the primary care provider and family are at odds.
WHEN A CONSULT IS INAPPROPRIATE
Palliative care consultation is inappropriate when used in lieu of an oncology consult in advanced cancer. Palliative care specialists are not experts in cancer care, whereas oncologists are familiar with rapid advancements in cancer care, including targeted agents that may offer benefit to patients with advanced cancer.
Palliative care consultation is also inappropriate if the patient does not want to see a palliative care specialist, or if the consult is used as a way to convince a patient to change advance directives or to choose not to be resuscitated. Also, cancer patients who are asymptomatic are unlikely to benefit from palliative care initially. The decision to consult palliative care should not depend on prognosis, and palliative care is more cost-effective when utilized early rather than as a crisis intervention near the end of life.3
THE PALLIATIVE CARE EVALUATION
The initial palliative care consultation usually involves an evaluation of the patient’s symptoms and concerns. Symptoms are targeted based on the patient’s priorities and on an assessment using validated questionnaires. A validated questionnaire is a better way to comprehensively gauge symptom burden than depending on patients to volunteer symptoms.5
As the relationship develops between patient, family, and palliative care specialist and as the disease takes its course, advance directives, prognosis, and end-of-life care goals can be addressed in follow-up consultations.3 Patients want to know about their prognosis, and they usually complete advance directives based on clinical circumstances rather than viewing them as an extension of patient autonomy, as originally intended.6
REIMBURSEMENT FOR PALLIATIVE CARE
Reimbursement for palliative care is similar to that for acute care and falls within the All Patient Refined Diagnosis-Related Group, or APR-DRG, system, and palliative care has its own V code for identification. Codes are used to designate disease, stage or location of metastases, disease complications, and symptoms, as well as for the discussion of goals of care.
WHAT PALLIATIVE CARE IS NOT
Palliative care has too often been tied to end-of-life care.7 The two often appear together in titles of reports in the literature. As a result, patients and physicians may be confused and, thus, reluctant to utilize palliative care services. To avoid the confusion, certain programs have included the term “supportive” oncology care in their title. This appears to facilitate palliative care referral, but may be misleading.8
WHAT IS HOSPICE CARE?
Hospice care is a service funded and capitated under Medicare part A and is largely provided as outpatient home care for those deemed terminally ill.9 An illness must be certified as terminal by two physicians. Medicare defines terminal illness as a life expectancy of 6 months or less if the illness runs its normal course.
The philosophy of hospice care is to provide comfort through intensive nurse management and home-based follow-up. In some cases, disease-modifying therapies are continued to control symptoms—eg, continuing angiotensin-converting enzyme inhibitors in heart failure patients. Hospice care is typically delivered at home, but it is also delivered in nursing homes, in hospital inpatient units, and at private or nonprofit hospice facilities.
Inpatient palliative care units are often mistaken for hospices. The purpose of hospice care is to provide quality of life and comfort and to avoid overly aggressive, expensive, and futile care at the end of life. The focus is on intensive, hands-on, personalized symptom care and family support at home. The goal is to provide a comfortable and dignified death among friends and family. The use of palliative radiation, transfusions, and antibiotics in hospice varies among hospice programs and is considered on a case-by-case basis.10
The Medicare per diem payment limits what hospices can afford, so they must be fiscally responsible. Hospice agencies are capitated and are responsible for providing medications and durable equipment necessary to treat symptoms related to the terminal illness. They also provide bereavement services for family members at no charge. Enrollment in hospice care can be revoked depending on circumstances and then reinstituted later as the goals of care change.
Care for nonterminal comorbid illnesses can be continued by a general practitioner or internist. This care is not covered under the Medicare hospice benefit, but it is covered under Medicare part B.
The patient and family can choose the hospice physician, who may be a family practitioner, internist, oncologist, or palliative care specialist, or may designate the hospice medical director as the hospice physician.
Criteria for hospice admission have been established for noncancer terminal illnesses and should be considered when practitioners decide to consult hospice.11–13
HOME-BASED PALLIATIVE CARE
Programs such as advanced illness management or home-based palliative care aim to improve the quality of care at home and prevent rehospitalization, particularly for patients with repeated hospitalizations.14 Home-based palliative care services are provided either by a clinician who makes home visits or by a certified home health care agency. Services are particularly useful for patients with serious illnesses who do not qualify for hospice services but are homebound. Consultations are obtained for ongoing supportive care at home, assessment for medication compliance, and disease monitoring at home. Consultations are scheduled at the time of hospital discharge.
Unlike hospice care, home-based palliative care does not include 24-hour on-call service. Comprehensive services (eg, home health aide, durable equipment, medications) are not provided as they are under hospice care: patients must qualify under Medicare stipulations for such services outside of hospice care. For example, home oxygen can only be supplied if the patient's oxygen saturation is less than 90%, while under the hospice benefit it is provided without regard to oxygen saturation and is based on symptom need. For home-based palliative care, patients must be largely homebound or unable to be seen regularly in the outpatient clinic. This type of care can be a bridge to hospice care for patients who feel they are not ready for hospice care at the time of discharge from acute care. Those who receive palliative care at home are less likely to be hospitalized at the end of life, are more likely to be transitioned to hospice at an appropriate time, and are more likely to have relief of symptoms.15
Hospice care generally falls under the category of palliative care, despite being an older subspecialty. However, the two have different indications and goals and are often provided in different settings.
ORIGINS OF PALLIATIVE CARE
Prompted by what he perceived as neglect of dying patients in the acute care setting, Dr. Balfour Mount opened the first acute inpatient palliative care unit in Royal Victoria Hospital in Montréal, Québec, in 1976.1 His purpose was to provide a crisis-intervention service for patients who were actively dying, and this continues to be the main reason for consulting palliative care services in the hospital.
Palliative care has evolved since the 1970s and is now used in a variety of situations:
- A life-limiting illness in a patient who is not terminally ill
- A life-threatening illness in a patient who has symptoms but with the potential to recover
- A chronic illness such as heart failure or chronic obstructive pulmonary disease in a patient who is on disease-modifying therapy but has symptoms and will eventually succumb to the illness, but is expected to live longer than someone with advanced cancer.2
PALLIATIVE CARE IN CANCER PATIENTS
In patients with advanced cancer, palliative care is utilized earlier in the course of serious and life-limiting illness and is even involved in patient care when cure is the goal. Importantly, it now includes outpatient clinics to provide patients seamless care in conjunction with their oncologist’s care.3
Because palliative care focuses on the patient’s experience of the illness (sickness) rather than on disease itself (pathology), symptom management, psychosocial support, and assistance in decision-making are foremost. Initiating palliative care early in advanced cancer improves multiple outcomes and limits overly aggressive, ineffective therapies at the end of life (eg, late chemotherapy, late referral to hospice care, death in the intensive care unit), without hastening death. In fact, it may prolong life.3,4
Palliative care is indicated in a number of situations in oncology:
- Symptomatic presentations of cancer, even when curative treatments are available
- At the time of a sentinel event such as recurrence or unanticipated hospitalization
- When palliative radiation is needed
- When changes in chemotherapy are needed because of disease progression.
Also, cancer patients may develop symptoms that require a palliative procedure such as thoracentesis for pleural effusion, paracentesis for ascites, or surgery for a fracture or spinal cord compression. A palliative care consultation is also appropriate when patients change their goals of care (ie, palliation rather than cure), and when an oncologic crisis occurs and there is a need to offer support to the family and to clarify the goals of care.
PALLIATIVE CARE IN OTHER DISEASES
For patients with illnesses other than cancer, palliative care may be helpful when disease-modifying therapy becomes burdensome or ineffective, or when patients are symptomatic despite maximum therapy. Palliative care should also be considered when goals of care need to be explored, when a second opinion is needed on goals of care, or if the primary care provider and family are at odds.
WHEN A CONSULT IS INAPPROPRIATE
Palliative care consultation is inappropriate when used in lieu of an oncology consult in advanced cancer. Palliative care specialists are not experts in cancer care, whereas oncologists are familiar with rapid advancements in cancer care, including targeted agents that may offer benefit to patients with advanced cancer.
Palliative care consultation is also inappropriate if the patient does not want to see a palliative care specialist, or if the consult is used as a way to convince a patient to change advance directives or to choose not to be resuscitated. Also, cancer patients who are asymptomatic are unlikely to benefit from palliative care initially. The decision to consult palliative care should not depend on prognosis, and palliative care is more cost-effective when utilized early rather than as a crisis intervention near the end of life.3
THE PALLIATIVE CARE EVALUATION
The initial palliative care consultation usually involves an evaluation of the patient’s symptoms and concerns. Symptoms are targeted based on the patient’s priorities and on an assessment using validated questionnaires. A validated questionnaire is a better way to comprehensively gauge symptom burden than depending on patients to volunteer symptoms.5
As the relationship develops between patient, family, and palliative care specialist and as the disease takes its course, advance directives, prognosis, and end-of-life care goals can be addressed in follow-up consultations.3 Patients want to know about their prognosis, and they usually complete advance directives based on clinical circumstances rather than viewing them as an extension of patient autonomy, as originally intended.6
REIMBURSEMENT FOR PALLIATIVE CARE
Reimbursement for palliative care is similar to that for acute care and falls within the All Patient Refined Diagnosis-Related Group, or APR-DRG, system, and palliative care has its own V code for identification. Codes are used to designate disease, stage or location of metastases, disease complications, and symptoms, as well as for the discussion of goals of care.
WHAT PALLIATIVE CARE IS NOT
Palliative care has too often been tied to end-of-life care.7 The two often appear together in titles of reports in the literature. As a result, patients and physicians may be confused and, thus, reluctant to utilize palliative care services. To avoid the confusion, certain programs have included the term “supportive” oncology care in their title. This appears to facilitate palliative care referral, but may be misleading.8
WHAT IS HOSPICE CARE?
Hospice care is a service funded and capitated under Medicare part A and is largely provided as outpatient home care for those deemed terminally ill.9 An illness must be certified as terminal by two physicians. Medicare defines terminal illness as a life expectancy of 6 months or less if the illness runs its normal course.
The philosophy of hospice care is to provide comfort through intensive nurse management and home-based follow-up. In some cases, disease-modifying therapies are continued to control symptoms—eg, continuing angiotensin-converting enzyme inhibitors in heart failure patients. Hospice care is typically delivered at home, but it is also delivered in nursing homes, in hospital inpatient units, and at private or nonprofit hospice facilities.
Inpatient palliative care units are often mistaken for hospices. The purpose of hospice care is to provide quality of life and comfort and to avoid overly aggressive, expensive, and futile care at the end of life. The focus is on intensive, hands-on, personalized symptom care and family support at home. The goal is to provide a comfortable and dignified death among friends and family. The use of palliative radiation, transfusions, and antibiotics in hospice varies among hospice programs and is considered on a case-by-case basis.10
The Medicare per diem payment limits what hospices can afford, so they must be fiscally responsible. Hospice agencies are capitated and are responsible for providing medications and durable equipment necessary to treat symptoms related to the terminal illness. They also provide bereavement services for family members at no charge. Enrollment in hospice care can be revoked depending on circumstances and then reinstituted later as the goals of care change.
Care for nonterminal comorbid illnesses can be continued by a general practitioner or internist. This care is not covered under the Medicare hospice benefit, but it is covered under Medicare part B.
The patient and family can choose the hospice physician, who may be a family practitioner, internist, oncologist, or palliative care specialist, or may designate the hospice medical director as the hospice physician.
Criteria for hospice admission have been established for noncancer terminal illnesses and should be considered when practitioners decide to consult hospice.11–13
HOME-BASED PALLIATIVE CARE
Programs such as advanced illness management or home-based palliative care aim to improve the quality of care at home and prevent rehospitalization, particularly for patients with repeated hospitalizations.14 Home-based palliative care services are provided either by a clinician who makes home visits or by a certified home health care agency. Services are particularly useful for patients with serious illnesses who do not qualify for hospice services but are homebound. Consultations are obtained for ongoing supportive care at home, assessment for medication compliance, and disease monitoring at home. Consultations are scheduled at the time of hospital discharge.
Unlike hospice care, home-based palliative care does not include 24-hour on-call service. Comprehensive services (eg, home health aide, durable equipment, medications) are not provided as they are under hospice care: patients must qualify under Medicare stipulations for such services outside of hospice care. For example, home oxygen can only be supplied if the patient's oxygen saturation is less than 90%, while under the hospice benefit it is provided without regard to oxygen saturation and is based on symptom need. For home-based palliative care, patients must be largely homebound or unable to be seen regularly in the outpatient clinic. This type of care can be a bridge to hospice care for patients who feel they are not ready for hospice care at the time of discharge from acute care. Those who receive palliative care at home are less likely to be hospitalized at the end of life, are more likely to be transitioned to hospice at an appropriate time, and are more likely to have relief of symptoms.15
- Mount BM. The problem of caring for the dying in a general hospital; the palliative care unit as a possible solution. Can Med Assoc J 1976; 115:119–121.
- Higginson I. Palliative care: a review of past changes and future trends. J Public Health Med 1993; 15:3–8.
- Temel JS, Greer JA, Muzikansky A, et al. Early palliative care for patients with metastatic non-small-cell lung cancer. N Engl J Med 2010; 363:733–742.
- Zimmermann C, Riechelmann R, Krzyzanowska M, Rodin G, Tannock I. Effectiveness of specialized palliative care: a systematic review. JAMA 2008; 299:1698–1709.
- Homsi J, Walsh D, Rivera N, et al. Symptom evaluation in palliative medicine: patient report vs systematic assessment. Support Care Cancer 2006; 14:444–453.
- Tang ST, Liu TW, Lai MS, Liu LN, Chen CH, Koong SL. Congruence of knowledge, experiences, and p for disclosure of diagnosis and prognosis between terminally-ill cancer patients and their family caregivers in Taiwan. Cancer Invest 2006; 24:360–366.
- Bakitas M, Lyons KD, Hegel MT, Ahles T. Oncologists’ perspectives on concurrent palliative care in a National Cancer Institute-designated comprehensive cancer center. Palliat Support Care 2013; 11:415–423.
- Fadul N, Elsayem A, Palmer JL, et al. Supportive versus palliative care: what’s in a name: a survey of medical oncologists and midlevel providers at a comprehensive cancer center. Cancer 2009; 115:2013–2021.
- Rinaldo MJ. Medicare to cover hospice services. J Med Soc NJ 1982; 79:1015–1016.
- Enck RE. Palliative radiation therapy in hospice care. Am J Hosp Palliat Care 2002; 19:151–152.
- Luchins DJ, Hanrahan P, Murphy K. Criteria for enrolling dementia patients in hospice. J Am Geriatr Soc 1997; 45:1054–1059.
- Fox E, Landrum-McNiff K, Zhong Z, Dawson NV, Wu AW, Lynn J. Evaluation of prognostic criteria for determining hospice eligibility in patients with advanced lung, heart, or liver disease. JAMA 1999; 282:1638–1645.
- Stuart B. The NHO medical guidelines for non-cancer disease and local medical review policy: hospice access for patients with diseases other than cancer. Hosp J 1999; 14:139–154.
- McKinney M. Beyond hospice. New models of care focus on advanced illnesses. Mod Healthc 2013; 43:14–15.
- Gomes B, Calanzani N, Curiale V, McCrone P, Higginson IJ. Effectiveness and cost-effectiveness of home palliative care services for adults with advanced illness and their caregivers. Cochrane Database Syst Rev 2013; 6:CD007760.
- Mount BM. The problem of caring for the dying in a general hospital; the palliative care unit as a possible solution. Can Med Assoc J 1976; 115:119–121.
- Higginson I. Palliative care: a review of past changes and future trends. J Public Health Med 1993; 15:3–8.
- Temel JS, Greer JA, Muzikansky A, et al. Early palliative care for patients with metastatic non-small-cell lung cancer. N Engl J Med 2010; 363:733–742.
- Zimmermann C, Riechelmann R, Krzyzanowska M, Rodin G, Tannock I. Effectiveness of specialized palliative care: a systematic review. JAMA 2008; 299:1698–1709.
- Homsi J, Walsh D, Rivera N, et al. Symptom evaluation in palliative medicine: patient report vs systematic assessment. Support Care Cancer 2006; 14:444–453.
- Tang ST, Liu TW, Lai MS, Liu LN, Chen CH, Koong SL. Congruence of knowledge, experiences, and p for disclosure of diagnosis and prognosis between terminally-ill cancer patients and their family caregivers in Taiwan. Cancer Invest 2006; 24:360–366.
- Bakitas M, Lyons KD, Hegel MT, Ahles T. Oncologists’ perspectives on concurrent palliative care in a National Cancer Institute-designated comprehensive cancer center. Palliat Support Care 2013; 11:415–423.
- Fadul N, Elsayem A, Palmer JL, et al. Supportive versus palliative care: what’s in a name: a survey of medical oncologists and midlevel providers at a comprehensive cancer center. Cancer 2009; 115:2013–2021.
- Rinaldo MJ. Medicare to cover hospice services. J Med Soc NJ 1982; 79:1015–1016.
- Enck RE. Palliative radiation therapy in hospice care. Am J Hosp Palliat Care 2002; 19:151–152.
- Luchins DJ, Hanrahan P, Murphy K. Criteria for enrolling dementia patients in hospice. J Am Geriatr Soc 1997; 45:1054–1059.
- Fox E, Landrum-McNiff K, Zhong Z, Dawson NV, Wu AW, Lynn J. Evaluation of prognostic criteria for determining hospice eligibility in patients with advanced lung, heart, or liver disease. JAMA 1999; 282:1638–1645.
- Stuart B. The NHO medical guidelines for non-cancer disease and local medical review policy: hospice access for patients with diseases other than cancer. Hosp J 1999; 14:139–154.
- McKinney M. Beyond hospice. New models of care focus on advanced illnesses. Mod Healthc 2013; 43:14–15.
- Gomes B, Calanzani N, Curiale V, McCrone P, Higginson IJ. Effectiveness and cost-effectiveness of home palliative care services for adults with advanced illness and their caregivers. Cochrane Database Syst Rev 2013; 6:CD007760.
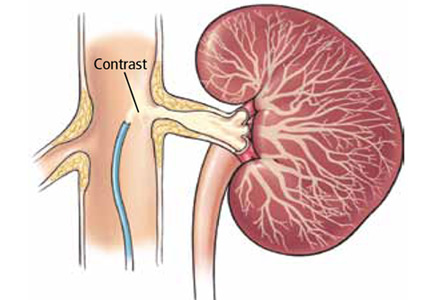
Does stenting of severe renal artery stenosis improve outomes compared with medical therapy alone?
No. In patients with severe atherosclerotic renal artery stenosis and hypertension or chronic kidney disease, renal artery stenting offers no additional benefit when added to comprehensive medical therapy.
In these patients, renal artery stenting in addition to antihypertensive drug therapy can improve blood pressure control modestly but has no significant effect on outcomes such as adverse cardiovascular events and death. And because renal artery stenting carries a risk of complications, medical management should continue to be the first-line therapy.
RENAL ARTERY STENOSIS
Renal artery stenosis is a common form of peripheral artery disease. Atherosclerosis is the most common cause, but it can also be caused by fibromuscular dysplasia or vasculitis (eg, Takayasu arteritis). It is most often unilateral, but bilateral disease has also been reported.
The prevalence of atherosclerotic renal vascular disease in the US Medicare population is 0.5%, and 5.5% in those with chronic kidney disease.1 Furthermore, renal artery stenosis is found in 6.8% of adults over age 65.2 The prevalence increases with age and is higher in patients with hyperlipidemia, peripheral arterial disease, and hypertension. The prevalence of renal artery stenosis in patients with atherosclerotic disease and renal dysfunction is as high as 50%.3
Patients with peripheral artery disease may be five times more likely to develop renal artery stenosis than people without peripheral artery disease.4 Significant stenosis can result in resistant arterial hypertension, renal insufficiency, left ventricular hypertrophy, and congestive heart failure.5
Nephropathy due to renal artery stenosis is complex and is caused by hypoperfusion and chronic microatheroembolism. Renal artery stenosis leads to oxidative stress, inflammation, fibrosis in the stenotic kidney, and, over time, loss of kidney function. Hypoperfusion also leads to activation of the renin-angiotensin-aldosterone system, which plays a role in development of left ventricular hypertrophy.5,6
Adequate blood pressure control, goal-directed lipid-lowering therapy, smoking cessation, and other preventive measures are the foundation of management.
RENAL ARTERY STENOSIS AND HYPERTENSION
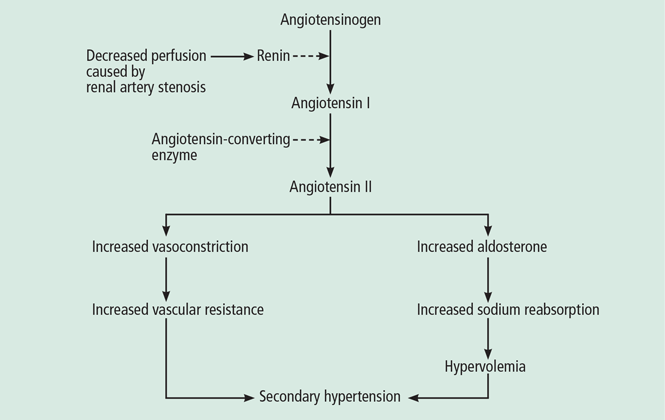
Renal artery stenosis is a cause of secondary hypertension. The stenosis decreases renal perfusion pressure, activating the release of renin and the production of angiotensin II, which in turn raises the blood pressure by two mechanisms (Figure 1): directly, by causing generalized vasoconstriction, and indirectly, by stimulating the release of aldosterone, which in turn increases the reabsorption of sodium and causes hypervolemia. These two mechanisms play a major role in renal vascular hypertension when renal artery stenosis is bilateral. In unilateral renal artery stenosis, pressure diuresis in the unaffected kidney compensates for the reabsorption of sodium in the affected kidney, keeping the blood pressure down. However, with time, the unaffected kidney will develop hypertensive nephropathy, and pressure diuresis will be lost.7,8 In addition, the activation of the renin-angiotensin-aldosterone system results in structural heart disease, such as left ventricular hypertrophy,5 and may shorten survival.
STENTING PLUS ANTIHYPERTENSIVE DRUG THERAPY
Because observational studies showed improvement in blood pressure control after endovascular stenting of atherosclerotic renal artery stenosis,9,10 this approach became a treatment option for uncontrolled hypertension in these patients. The 2005 joint guidelines of the American College of Cardiology and the American Heart Association11 considered percutaneous revascularization a reasonable option (level of evidence B) for patients who meet one of the following criteria:
- Hemodynamically significant stenosis and accelerated, resistant, or malignant hypertension, hypertension with an unexplained unilateral small kidney, or hypertension with intolerance to medication
- Renal artery stenosis and progressive chronic kidney disease with bilateral stenosis or stenosis in a solitary functioning kidney
- Hemodynamically significant stenosis and recurrent, unexplained congestive heart failure or sudden, unexplained pulmonary edema or unstable angina.11
However, no randomized study has shown a direct benefit of renal artery stenting on rates of cardiovascular events or renal function compared with drug therapy alone.
TRIALS OF STENTING VS MEDICAL THERAPY ALONE
Technical improvements have led to more widespread use of diagnostic and interventional endovascular tools for renal artery revascularization. Studies over the past 10 years examined the impact of stenting in patients with uncontrolled hypertension.
The STAR trial
In the Stent Placement and Blood Pressure and Lipid-lowering for the Prevention of Progression of Renal Dysfunction Caused by Atherosclerotic Ostial Stenosis of the Renal Artery (STAR) trial,9 patients with creatinine clearance less than 80 mL/min/1.73 m2, renal artery stenosis greater than 50%, and well-controlled blood pressure were randomized to either renal artery stenting plus medical therapy or medical therapy alone. The authors concluded that stenting had no effect on the progression of renal dysfunction but led to a small number of significant, procedure-related complications. The study was criticized for including patients with mild stenosis (< 50% stenosis) and for being underpowered for the primary end point.
The ASTRAL study
The Angioplasty and Stenting for Renal Artery Lesions (ASTRAL) study10 was a similar comparison with similar results, showing no benefit from stenting with respect to renal function, systolic blood pressure control, cardiovascular events, or death.
HERCULES
The Herculink Elite Cobalt Chromium Renal Stent Trial to Demonstrate Efficacy and Safety (HERCULES)12 was a prospective multicenter study of the effects of renal artery stenting in 202 patients with significant renal artery stenosis and uncontrolled hypertension. It showed a reduction in systolic blood pressure from baseline (P < .0001). However, follow-up was only 9 months, which was insufficient to show a significant effect on long-term cardiovascular and cerebrovascular outcomes.
The CORAL trial
The Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL) trial13 used more stringent definitions and longer follow-up. It randomized 947 patients to either stenting plus medical therapy or medical therapy alone. Patients had atherosclerotic renal artery stenosis, defined as stenosis of at least 80% or stenosis of 60% to 80% with a gradient of at least 20 mm Hg in the systolic pressure), and either systolic hypertension while taking two or more antihypertensive drugs or stage 3 or higher chronic kidney disease (glomerular filtration rate < 60 mL/min/1.73 m2 as calculated by the Modification of Diet in Renal Disease formula).
Participants were followed for 43 months to detect the occurrence of adverse cardiovascular and renal events. There was no significant difference in primary outcome between stenting plus drug therapy and drug therapy alone (35.1% and 35.8%, respectively; P = .58). However, stenting plus drug therapy was associated with modestly lower systolic pressures compared with drug therapy alone (−2.3 mm Hg, 95% confidence interval −4.4 to −0.2 mm Hg, P = .03).13 This study provided strong evidence that renal artery stenting offers no significant benefit to patients with moderately severe atherosclerotic renal artery stenosis, and that stenting may actually pose an unnecessary risk.
COMPLICATIONS OF RENAL ARTERY STENTING
Complications of renal artery stenting are a limiting factor compared with drug therapy alone, especially since the procedure offers no significant benefit in outcome. Procedural complication rates of 10% to 15% have been reported.9,10,12 The CORAL trial reported arterial dissection in 2.2%, branch-vessel occlusion in 1.2%, and distal embolization in 1.2% of patients undergoing stenting.13 Other reported complications have included stent misplacement requiring an additional stent, access-vessel damage, stent embolization, renal artery thrombosis or occlusion, and death.10,12
- Kalra PA, Guo H, Kausz AT, et al. Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int 2005; 68:293–301.
- Hansen KJ, Edwards MS, Craven TE, et al. Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg 2002; 36:443–451.
- Uzu T, Takeji M, Yamada N, et al. Prevalence and outcome of renal artery stenosis in atherosclerotic patients with renal dysfunction. Hypertens Res 2002; 25:537–542.
- Benjamin MM, Fazel P, Filardo G, Choi JW, Stoler RC. Prevalence of and risk factors of renal artery stenosis in patients with resistant hypertension. Am J Cardiol 2014; 113:687–690.
- Wu S, Polavarapu N, Stouffer GA. Left ventricular hypertrophy in patients with renal artery stenosis. Am J Med Sci 2006; 332:334–338.
- Lerman LO, Textor SC, Grande JP. Mechanisms of tissue injury in renal artery stenosis: ischemia and beyond. Prog Cardiovasc Dis 2009; 52:196–203.
- Black HR, Glickman MG, Schiff M Jr, Pingoud EG. Renovascular hypertension: pathophysiology, diagnosis, and treatment. Yale J Biol Med 1978; 51:635–654.
- Tobe SW, Burgess E, Lebel M. Atherosclerotic renovascular disease. Can J Cardiol 2006; 22:623–628.
- Bax L, Mali WP, Buskens E, et al; STAR Study Group. The benefit of stent placement and blood pressure and lipid-lowering for the prevention of progression of renal dysfunction caused by atherosclerotic ostial stenosis of the renal artery. The STAR-study: rationale and study design. J Nephrol 2003; 16:807–812.
- ASTRAL Investigators; Wheatley K, Ives N, Gray R, et al. Revascularization versus medical therapy for renal-artery stenosis. N Engl J Med 2009; 361:1953–1962.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al. ACC/AHA 2005 guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): executive summary. J Am Coll Cardiol 2006; 47:1239–1312.
No. In patients with severe atherosclerotic renal artery stenosis and hypertension or chronic kidney disease, renal artery stenting offers no additional benefit when added to comprehensive medical therapy.
In these patients, renal artery stenting in addition to antihypertensive drug therapy can improve blood pressure control modestly but has no significant effect on outcomes such as adverse cardiovascular events and death. And because renal artery stenting carries a risk of complications, medical management should continue to be the first-line therapy.
RENAL ARTERY STENOSIS
Renal artery stenosis is a common form of peripheral artery disease. Atherosclerosis is the most common cause, but it can also be caused by fibromuscular dysplasia or vasculitis (eg, Takayasu arteritis). It is most often unilateral, but bilateral disease has also been reported.
The prevalence of atherosclerotic renal vascular disease in the US Medicare population is 0.5%, and 5.5% in those with chronic kidney disease.1 Furthermore, renal artery stenosis is found in 6.8% of adults over age 65.2 The prevalence increases with age and is higher in patients with hyperlipidemia, peripheral arterial disease, and hypertension. The prevalence of renal artery stenosis in patients with atherosclerotic disease and renal dysfunction is as high as 50%.3
Patients with peripheral artery disease may be five times more likely to develop renal artery stenosis than people without peripheral artery disease.4 Significant stenosis can result in resistant arterial hypertension, renal insufficiency, left ventricular hypertrophy, and congestive heart failure.5
Nephropathy due to renal artery stenosis is complex and is caused by hypoperfusion and chronic microatheroembolism. Renal artery stenosis leads to oxidative stress, inflammation, fibrosis in the stenotic kidney, and, over time, loss of kidney function. Hypoperfusion also leads to activation of the renin-angiotensin-aldosterone system, which plays a role in development of left ventricular hypertrophy.5,6
Adequate blood pressure control, goal-directed lipid-lowering therapy, smoking cessation, and other preventive measures are the foundation of management.
RENAL ARTERY STENOSIS AND HYPERTENSION
Renal artery stenosis is a cause of secondary hypertension. The stenosis decreases renal perfusion pressure, activating the release of renin and the production of angiotensin II, which in turn raises the blood pressure by two mechanisms (Figure 1): directly, by causing generalized vasoconstriction, and indirectly, by stimulating the release of aldosterone, which in turn increases the reabsorption of sodium and causes hypervolemia. These two mechanisms play a major role in renal vascular hypertension when renal artery stenosis is bilateral. In unilateral renal artery stenosis, pressure diuresis in the unaffected kidney compensates for the reabsorption of sodium in the affected kidney, keeping the blood pressure down. However, with time, the unaffected kidney will develop hypertensive nephropathy, and pressure diuresis will be lost.7,8 In addition, the activation of the renin-angiotensin-aldosterone system results in structural heart disease, such as left ventricular hypertrophy,5 and may shorten survival.
STENTING PLUS ANTIHYPERTENSIVE DRUG THERAPY
Because observational studies showed improvement in blood pressure control after endovascular stenting of atherosclerotic renal artery stenosis,9,10 this approach became a treatment option for uncontrolled hypertension in these patients. The 2005 joint guidelines of the American College of Cardiology and the American Heart Association11 considered percutaneous revascularization a reasonable option (level of evidence B) for patients who meet one of the following criteria:
- Hemodynamically significant stenosis and accelerated, resistant, or malignant hypertension, hypertension with an unexplained unilateral small kidney, or hypertension with intolerance to medication
- Renal artery stenosis and progressive chronic kidney disease with bilateral stenosis or stenosis in a solitary functioning kidney
- Hemodynamically significant stenosis and recurrent, unexplained congestive heart failure or sudden, unexplained pulmonary edema or unstable angina.11
However, no randomized study has shown a direct benefit of renal artery stenting on rates of cardiovascular events or renal function compared with drug therapy alone.
TRIALS OF STENTING VS MEDICAL THERAPY ALONE
Technical improvements have led to more widespread use of diagnostic and interventional endovascular tools for renal artery revascularization. Studies over the past 10 years examined the impact of stenting in patients with uncontrolled hypertension.
The STAR trial
In the Stent Placement and Blood Pressure and Lipid-lowering for the Prevention of Progression of Renal Dysfunction Caused by Atherosclerotic Ostial Stenosis of the Renal Artery (STAR) trial,9 patients with creatinine clearance less than 80 mL/min/1.73 m2, renal artery stenosis greater than 50%, and well-controlled blood pressure were randomized to either renal artery stenting plus medical therapy or medical therapy alone. The authors concluded that stenting had no effect on the progression of renal dysfunction but led to a small number of significant, procedure-related complications. The study was criticized for including patients with mild stenosis (< 50% stenosis) and for being underpowered for the primary end point.
The ASTRAL study
The Angioplasty and Stenting for Renal Artery Lesions (ASTRAL) study10 was a similar comparison with similar results, showing no benefit from stenting with respect to renal function, systolic blood pressure control, cardiovascular events, or death.
HERCULES
The Herculink Elite Cobalt Chromium Renal Stent Trial to Demonstrate Efficacy and Safety (HERCULES)12 was a prospective multicenter study of the effects of renal artery stenting in 202 patients with significant renal artery stenosis and uncontrolled hypertension. It showed a reduction in systolic blood pressure from baseline (P < .0001). However, follow-up was only 9 months, which was insufficient to show a significant effect on long-term cardiovascular and cerebrovascular outcomes.
The CORAL trial
The Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL) trial13 used more stringent definitions and longer follow-up. It randomized 947 patients to either stenting plus medical therapy or medical therapy alone. Patients had atherosclerotic renal artery stenosis, defined as stenosis of at least 80% or stenosis of 60% to 80% with a gradient of at least 20 mm Hg in the systolic pressure), and either systolic hypertension while taking two or more antihypertensive drugs or stage 3 or higher chronic kidney disease (glomerular filtration rate < 60 mL/min/1.73 m2 as calculated by the Modification of Diet in Renal Disease formula).
Participants were followed for 43 months to detect the occurrence of adverse cardiovascular and renal events. There was no significant difference in primary outcome between stenting plus drug therapy and drug therapy alone (35.1% and 35.8%, respectively; P = .58). However, stenting plus drug therapy was associated with modestly lower systolic pressures compared with drug therapy alone (−2.3 mm Hg, 95% confidence interval −4.4 to −0.2 mm Hg, P = .03).13 This study provided strong evidence that renal artery stenting offers no significant benefit to patients with moderately severe atherosclerotic renal artery stenosis, and that stenting may actually pose an unnecessary risk.
COMPLICATIONS OF RENAL ARTERY STENTING
Complications of renal artery stenting are a limiting factor compared with drug therapy alone, especially since the procedure offers no significant benefit in outcome. Procedural complication rates of 10% to 15% have been reported.9,10,12 The CORAL trial reported arterial dissection in 2.2%, branch-vessel occlusion in 1.2%, and distal embolization in 1.2% of patients undergoing stenting.13 Other reported complications have included stent misplacement requiring an additional stent, access-vessel damage, stent embolization, renal artery thrombosis or occlusion, and death.10,12
No. In patients with severe atherosclerotic renal artery stenosis and hypertension or chronic kidney disease, renal artery stenting offers no additional benefit when added to comprehensive medical therapy.
In these patients, renal artery stenting in addition to antihypertensive drug therapy can improve blood pressure control modestly but has no significant effect on outcomes such as adverse cardiovascular events and death. And because renal artery stenting carries a risk of complications, medical management should continue to be the first-line therapy.
RENAL ARTERY STENOSIS
Renal artery stenosis is a common form of peripheral artery disease. Atherosclerosis is the most common cause, but it can also be caused by fibromuscular dysplasia or vasculitis (eg, Takayasu arteritis). It is most often unilateral, but bilateral disease has also been reported.
The prevalence of atherosclerotic renal vascular disease in the US Medicare population is 0.5%, and 5.5% in those with chronic kidney disease.1 Furthermore, renal artery stenosis is found in 6.8% of adults over age 65.2 The prevalence increases with age and is higher in patients with hyperlipidemia, peripheral arterial disease, and hypertension. The prevalence of renal artery stenosis in patients with atherosclerotic disease and renal dysfunction is as high as 50%.3
Patients with peripheral artery disease may be five times more likely to develop renal artery stenosis than people without peripheral artery disease.4 Significant stenosis can result in resistant arterial hypertension, renal insufficiency, left ventricular hypertrophy, and congestive heart failure.5
Nephropathy due to renal artery stenosis is complex and is caused by hypoperfusion and chronic microatheroembolism. Renal artery stenosis leads to oxidative stress, inflammation, fibrosis in the stenotic kidney, and, over time, loss of kidney function. Hypoperfusion also leads to activation of the renin-angiotensin-aldosterone system, which plays a role in development of left ventricular hypertrophy.5,6
Adequate blood pressure control, goal-directed lipid-lowering therapy, smoking cessation, and other preventive measures are the foundation of management.
RENAL ARTERY STENOSIS AND HYPERTENSION
Renal artery stenosis is a cause of secondary hypertension. The stenosis decreases renal perfusion pressure, activating the release of renin and the production of angiotensin II, which in turn raises the blood pressure by two mechanisms (Figure 1): directly, by causing generalized vasoconstriction, and indirectly, by stimulating the release of aldosterone, which in turn increases the reabsorption of sodium and causes hypervolemia. These two mechanisms play a major role in renal vascular hypertension when renal artery stenosis is bilateral. In unilateral renal artery stenosis, pressure diuresis in the unaffected kidney compensates for the reabsorption of sodium in the affected kidney, keeping the blood pressure down. However, with time, the unaffected kidney will develop hypertensive nephropathy, and pressure diuresis will be lost.7,8 In addition, the activation of the renin-angiotensin-aldosterone system results in structural heart disease, such as left ventricular hypertrophy,5 and may shorten survival.
STENTING PLUS ANTIHYPERTENSIVE DRUG THERAPY
Because observational studies showed improvement in blood pressure control after endovascular stenting of atherosclerotic renal artery stenosis,9,10 this approach became a treatment option for uncontrolled hypertension in these patients. The 2005 joint guidelines of the American College of Cardiology and the American Heart Association11 considered percutaneous revascularization a reasonable option (level of evidence B) for patients who meet one of the following criteria:
- Hemodynamically significant stenosis and accelerated, resistant, or malignant hypertension, hypertension with an unexplained unilateral small kidney, or hypertension with intolerance to medication
- Renal artery stenosis and progressive chronic kidney disease with bilateral stenosis or stenosis in a solitary functioning kidney
- Hemodynamically significant stenosis and recurrent, unexplained congestive heart failure or sudden, unexplained pulmonary edema or unstable angina.11
However, no randomized study has shown a direct benefit of renal artery stenting on rates of cardiovascular events or renal function compared with drug therapy alone.
TRIALS OF STENTING VS MEDICAL THERAPY ALONE
Technical improvements have led to more widespread use of diagnostic and interventional endovascular tools for renal artery revascularization. Studies over the past 10 years examined the impact of stenting in patients with uncontrolled hypertension.
The STAR trial
In the Stent Placement and Blood Pressure and Lipid-lowering for the Prevention of Progression of Renal Dysfunction Caused by Atherosclerotic Ostial Stenosis of the Renal Artery (STAR) trial,9 patients with creatinine clearance less than 80 mL/min/1.73 m2, renal artery stenosis greater than 50%, and well-controlled blood pressure were randomized to either renal artery stenting plus medical therapy or medical therapy alone. The authors concluded that stenting had no effect on the progression of renal dysfunction but led to a small number of significant, procedure-related complications. The study was criticized for including patients with mild stenosis (< 50% stenosis) and for being underpowered for the primary end point.
The ASTRAL study
The Angioplasty and Stenting for Renal Artery Lesions (ASTRAL) study10 was a similar comparison with similar results, showing no benefit from stenting with respect to renal function, systolic blood pressure control, cardiovascular events, or death.
HERCULES
The Herculink Elite Cobalt Chromium Renal Stent Trial to Demonstrate Efficacy and Safety (HERCULES)12 was a prospective multicenter study of the effects of renal artery stenting in 202 patients with significant renal artery stenosis and uncontrolled hypertension. It showed a reduction in systolic blood pressure from baseline (P < .0001). However, follow-up was only 9 months, which was insufficient to show a significant effect on long-term cardiovascular and cerebrovascular outcomes.
The CORAL trial
The Cardiovascular Outcomes in Renal Atherosclerotic Lesions (CORAL) trial13 used more stringent definitions and longer follow-up. It randomized 947 patients to either stenting plus medical therapy or medical therapy alone. Patients had atherosclerotic renal artery stenosis, defined as stenosis of at least 80% or stenosis of 60% to 80% with a gradient of at least 20 mm Hg in the systolic pressure), and either systolic hypertension while taking two or more antihypertensive drugs or stage 3 or higher chronic kidney disease (glomerular filtration rate < 60 mL/min/1.73 m2 as calculated by the Modification of Diet in Renal Disease formula).
Participants were followed for 43 months to detect the occurrence of adverse cardiovascular and renal events. There was no significant difference in primary outcome between stenting plus drug therapy and drug therapy alone (35.1% and 35.8%, respectively; P = .58). However, stenting plus drug therapy was associated with modestly lower systolic pressures compared with drug therapy alone (−2.3 mm Hg, 95% confidence interval −4.4 to −0.2 mm Hg, P = .03).13 This study provided strong evidence that renal artery stenting offers no significant benefit to patients with moderately severe atherosclerotic renal artery stenosis, and that stenting may actually pose an unnecessary risk.
COMPLICATIONS OF RENAL ARTERY STENTING
Complications of renal artery stenting are a limiting factor compared with drug therapy alone, especially since the procedure offers no significant benefit in outcome. Procedural complication rates of 10% to 15% have been reported.9,10,12 The CORAL trial reported arterial dissection in 2.2%, branch-vessel occlusion in 1.2%, and distal embolization in 1.2% of patients undergoing stenting.13 Other reported complications have included stent misplacement requiring an additional stent, access-vessel damage, stent embolization, renal artery thrombosis or occlusion, and death.10,12
- Kalra PA, Guo H, Kausz AT, et al. Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int 2005; 68:293–301.
- Hansen KJ, Edwards MS, Craven TE, et al. Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg 2002; 36:443–451.
- Uzu T, Takeji M, Yamada N, et al. Prevalence and outcome of renal artery stenosis in atherosclerotic patients with renal dysfunction. Hypertens Res 2002; 25:537–542.
- Benjamin MM, Fazel P, Filardo G, Choi JW, Stoler RC. Prevalence of and risk factors of renal artery stenosis in patients with resistant hypertension. Am J Cardiol 2014; 113:687–690.
- Wu S, Polavarapu N, Stouffer GA. Left ventricular hypertrophy in patients with renal artery stenosis. Am J Med Sci 2006; 332:334–338.
- Lerman LO, Textor SC, Grande JP. Mechanisms of tissue injury in renal artery stenosis: ischemia and beyond. Prog Cardiovasc Dis 2009; 52:196–203.
- Black HR, Glickman MG, Schiff M Jr, Pingoud EG. Renovascular hypertension: pathophysiology, diagnosis, and treatment. Yale J Biol Med 1978; 51:635–654.
- Tobe SW, Burgess E, Lebel M. Atherosclerotic renovascular disease. Can J Cardiol 2006; 22:623–628.
- Bax L, Mali WP, Buskens E, et al; STAR Study Group. The benefit of stent placement and blood pressure and lipid-lowering for the prevention of progression of renal dysfunction caused by atherosclerotic ostial stenosis of the renal artery. The STAR-study: rationale and study design. J Nephrol 2003; 16:807–812.
- ASTRAL Investigators; Wheatley K, Ives N, Gray R, et al. Revascularization versus medical therapy for renal-artery stenosis. N Engl J Med 2009; 361:1953–1962.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al. ACC/AHA 2005 guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): executive summary. J Am Coll Cardiol 2006; 47:1239–1312.
- Kalra PA, Guo H, Kausz AT, et al. Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int 2005; 68:293–301.
- Hansen KJ, Edwards MS, Craven TE, et al. Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg 2002; 36:443–451.
- Uzu T, Takeji M, Yamada N, et al. Prevalence and outcome of renal artery stenosis in atherosclerotic patients with renal dysfunction. Hypertens Res 2002; 25:537–542.
- Benjamin MM, Fazel P, Filardo G, Choi JW, Stoler RC. Prevalence of and risk factors of renal artery stenosis in patients with resistant hypertension. Am J Cardiol 2014; 113:687–690.
- Wu S, Polavarapu N, Stouffer GA. Left ventricular hypertrophy in patients with renal artery stenosis. Am J Med Sci 2006; 332:334–338.
- Lerman LO, Textor SC, Grande JP. Mechanisms of tissue injury in renal artery stenosis: ischemia and beyond. Prog Cardiovasc Dis 2009; 52:196–203.
- Black HR, Glickman MG, Schiff M Jr, Pingoud EG. Renovascular hypertension: pathophysiology, diagnosis, and treatment. Yale J Biol Med 1978; 51:635–654.
- Tobe SW, Burgess E, Lebel M. Atherosclerotic renovascular disease. Can J Cardiol 2006; 22:623–628.
- Bax L, Mali WP, Buskens E, et al; STAR Study Group. The benefit of stent placement and blood pressure and lipid-lowering for the prevention of progression of renal dysfunction caused by atherosclerotic ostial stenosis of the renal artery. The STAR-study: rationale and study design. J Nephrol 2003; 16:807–812.
- ASTRAL Investigators; Wheatley K, Ives N, Gray R, et al. Revascularization versus medical therapy for renal-artery stenosis. N Engl J Med 2009; 361:1953–1962.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al. ACC/AHA 2005 guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): executive summary. J Am Coll Cardiol 2006; 47:1239–1312.
Should thiopurine methyltransferase (TPMT) activity be determined before prescribing azathioprine, mercaptopurine, or thioguanine?
The thiopurines azathioprine, mercaptopurine, and thioguanine are prodrugs that are converted to active thioguanine nucleotide metabolites or methylated by thiopurine methyltransferase (TPMT) to compounds with less pharmacologic activity. In the absence of TPMT activity, patients are likely to have higher concentrations of thioguanine nucleotides, which can pose an increased risk of severe life-threatening myelosuppression. Determining TPMT activity, either directly by phenotyping or indirectly by determining the specific genetic allele (different alleles have different enzymatic activity), can help identify patients at greater risk of severe myelosuppression. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
THIOPURINES AND TPMT
Azathioprine, mercaptopurine, and thioguanine are used for treating autoimmune and inflammatory diseases1–3 and certain types of cancer such as leukemias and lymphomas.1,4–6 Typically, azathioprine is used to treat nonmalignant conditions, thioguanine is used to treat malignancies, and mercaptopurine can be used to treat both malignant and nonmalignant conditions.
Although the exact mechanism of action of these drugs has not been completely elucidated, the active thioguanine nucleotide metabolites are thought to be incorporated into the DNA of leukocytes, resulting in DNA damage that subsequently leads to cell death and myelosuppression.7–9
Variants of the TPMT gene may alter the activity of the TPMT enzyme, resulting in individual variability in thiopurine metabolism. Compared with people with normal (high) TPMT activity, those with intermediate or low TPMT activity metabolize the drugs more slowly, and are likely to have higher thioguanine nucleotide concentrations and therefore an increased risk of myelosuppression.
One of the earliest correlations between TPMT activity and thiopurine-induced myelosuppression was described in a pediatric patient with acute lymphocytic leukemia.10 After being prescribed a conventional mercaptopurine dosage (75 mg/m2 daily), the patient developed severe myelosuppression and was observed to have a thioguanine nucleotide metabolite concentration seven times the observed population median. TPMT phenotyping demonstrated that the patient had low TPMT activity. Reducing the mercaptopurine dose by approximately 90% resulted in normalization of thioguanine nucleotide metabolite concentrations, and the myelosuppression subsequently resolved.
Approximately 10% of the population has intermediate TPMT activity and 0.3% has low or absent TPMT activity, though these percentages vary depending on ancestry.1 Research has demonstrated that approximately 30% to 60% of those with intermediate TPMT activity cannot tolerate a full thiopurine dose (eg, azathioprine 2–3 mg/kg/day or mercaptopurine 1.5 mg/kg/day).1 Almost all patients with low TPMT activity will develop life-threating myelosuppression if prescribed a full thiopurine dose.1
SHOULD TPMT ACTIVITY BE DETERMINED FOR EVERY PATIENT PRESCRIBED A THIOPURINE?
Although determining TPMT activity in thiopurine-naïve patients will assist clinicians in selecting a thiopurine starting dose or in deciding if an alternative agent is warranted, there are instances when a clinician may elect to not perform a TPMT genotype or phenotype test. For example, determining TPMT activity is not recommended for patients who previously tolerated thiopurine therapy at full steady-state doses.
The required starting dose of a thiopurine can influence the decision on whether or not to test for TPMT activity. TPMT genotyping or phenotyping may be of most benefit for patients requiring immediate full doses of a thiopurine.11 Ideally, TPMT activity should be determined before prescribing immediate full doses of a thiopurine. This could be achieved by preemptively ordering a TPMT test in patients likely to require immunosuppression—for example, in patients diagnosed with inflammatory or autoimmune diseases. If therapy cannot be delayed and TPMT activity is unknown, ordering a TPMT test at the time of prescribing a full thiopurine dose is still of benefit. Depending on the clinical laboratory utilized for testing, TPMT phenotype results are usually reported in 3 to 5 days, and TPMT genotype results are usually reported in 5 to 7 days. Because most patients will not reach steady-state concentrations for 2 to 6 weeks, clinicians could initiate immediate full doses of a thiopurine and modify therapy based on TPMT test results before accumulation of thioguanine nucleotide metabolites occurs. Caution should be used with this approach, particularly in situations where the clinical laboratory may not return results in a timely manner.
For patients who are candidates for an initial low dose of a thiopurine, clinicians may choose to slowly titrate doses based on response and tolerability instead of determining TPMT activity.11 Depending on the starting dose and how slowly titration occurs, initiating a thiopurine at a low dose and titrating based on response can be a feasible approach for patients with intermediate TPMT activity. Because drastic thiopurine dose reductions of approximately 10-fold are required for patients with low TPMT activity, which is a much smaller dosage than most clinicians will initially prescribe, the starting dosage will likely not be low enough to prevent myelosuppression in patients with low TPMT activity.1,10
Determining TPMT activity can help clinicians establish an appropriate titration schedule. Patients with normal TPMT activity will usually reach thiopurine steady-state concentrations in 2 weeks, and the dosage can be titrated based on response.1 Alterations in TPMT activity influence the pharmacokinetic parameters of thiopurines, and the time to reach steady-state is extended to 4 or 6 weeks for those with intermediate or low TPMT activity.1 Increasing the thiopurine dosage before reaching steady state can lead to the prescribing of doses that will not be tolerated, resulting in myelosuppression.
Factors to consider when deciding if TPMT activity should be assessed include the disease state being treated and corresponding starting dose, the need for immediate full doses, and previous documented tolerance of thiopurines at steady-state doses. As with many aspects of medicine that have multiple options, coupled with an increase in patient access to healthcare information, the decision to test for TPMT activity may include shared decision-making between patients and providers. Although TPMT genotyping or phenotyping can help identify those at greatest risk of severe myelosuppression, such assays do not replace routine monitoring for myelosuppression, hepatotoxicity, or pancreatitis that may be caused by thiopurines.
WHAT TESTS ARE AVAILABLE TO DETERMINE TPMT ACTIVITY?
Patients with intermediate or low TPMT activity can be identified by either genotyping or phenotyping. There are considerations, though, that clinicians should be aware of before selecting a particular test.
TPMT genotyping
![]()
Four TPMT alleles, TPMT*2, *3A, *3B, and *3C, account for over 90% of inactivating polymorphisms.12 Therefore, most reference laboratories only analyze for those genetic variants. Based on the reported test result, a predicted phenotype (eg, normal, intermediate, or low TPMT activity) can be assigned. Table 1 lists the predicted phenotypes for select genotyping results.
TPMT phenotyping
Phenotyping quantitates TPMT enzyme activity in erythrocytes, and based on the result, patients are classified as having normal, intermediate, or low TPMT activity. Because internal standards and other testing conditions may differ between reference laboratories, test results must be interpreted in the context of the laboratory that performed the assay.
Which test is right for my patient?
In most cases, either the genotype or the phenotype test provides sufficient information to guide thiopurine therapy. There are certain circumstances, though, in which the genotype or phenotype test is less informative.
TPMT genotyping, when performed using a blood specimen, is not recommended in those with a history of allogeneic bone marrow transplantation, as the result would reflect the donor’s genotype, not the patient’s. In such instances, monitoring of white blood cell counts and thiopurine metabolites may be more beneficial.
TPMT phenotyping may be inaccurate if performed within 30 to 90 days of an erythrocyte transfusion, as the test result may be influenced by donor erythrocytes. If a patient is receiving erythrocyte transfusions, TPMT genotyping is preferable to phenotyping.
Test cost may also be a consideration when determining if the genotype or phenotype test is best for your patient. Costs vary by laboratory, but phenotyping is generally less expensive than genotyping. The cost of genotyping, though, continues to decrease.13 The approximate commercial cost is $200 for phenotyping and $450 for genotyping, but laboratory fees may be substantially higher. Several insurance plans, including Medicare, cover TPMT testing, but reimbursement and copayments vary, depending on the patient’s specific plan.
There are conflicting data as to whether determining TPMT status is11,14–18 or is not19 cost-effective. Multiple studies suggest that the cost of genotyping a sufficient number of patients to identify a single individual at high risk of myelosuppression is cheaper than the costs associated with treating an adverse event. Additional cost-benefit studies are needed, particularly studies that consider how bundled payments and outcomes-based reimbursement influence cost-effectiveness.
MODIFYING THIOPURINE THERAPY BASED ON TPMT ACTIVITY
![]()
There is a strong correlation between TPMT activity and tolerated thiopurine doses, with those having intermediate or low TPMT activity requiring lower doses.10,20–23 Adjusting mercaptopurine doses based on TPMT activity to prevent hematopoietic toxicity has been successfully demonstrated in pediatric patients with acute lymphoblastic leukemia.24 Furthermore, reducing initial thiopurine doses to avoid myelosuppression and titrating based on response has been shown to not compromise outcomes.1,25,26 The Clinical Pharmacogenetic Implementation Consortium (CPIC) has developed an evidence-based guideline on how to adjust thiopurine doses based on TPMT activity,1 summarized in Table 2. These dosing recommendations are classified as “strong.”
Patients with normal TPMT activity should be prescribed the usual thiopurine starting dose as indicated by disease-specific guidelines.
For those with intermediate TPMT activity, the CPIC guideline recommends reducing the initial targeted full dose of azathioprine and mercaptopurine by 30% to 70% and reducing the targeted full dose of thioguanine by 30% to 50%. The percentage of dose reduction depends on the targeted full dose. Siegel and Sands27 suggested that for those who are diagnosed with inflammatory bowel disease and have intermediate TPMT activity, azathioprine should be initiated at a low dose and titrated to 1.25 mg/kg and mercaptopurine should be initiated at a low dose and titrated to 0.75 mg/kg. Based on these titration goals, if the targeted full dose for mercaptopurine is 1 mg/kg, then a dose reduction of approximately 30% would be more appropriate. If the targeted full dose is 1.5 mg/kg, a dose reduction of approximately 50% would be more appropriate. Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 2 to 4 weeks to reach steady state before dose titration.
For those with low TPMT activity, alternative therapy should be considered for nonmalignant conditions because of the risk of severe myelosuppression. For malignancy, or if a thiopurine is warranted for a nonmalignant condition, consider a 90% dose reduction and give the drug three times per week instead of daily. For example, acute lymphoblastic leukemia patients with low TPMT activity can be started on mercaptopurine 10 mg/m2 three times per week instead of the usual starting dose.10 Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 4 to 6 weeks to reach steady state before dose titration.
RECOMMENDATIONS
Individuals with intermediate or low TPMT activity have an increased risk of myelosuppression. Because of the elevated risk for morbidity and death, especially for patients with low TPMT activity, multiple guidelines and regulatory agencies recommend TPMT genotyping or phenotyping if a thiopurine is prescribed.25,28–32 Although additional cost-benefit analysis studies are needed, evidence suggests testing for TPMT activity may be cheaper than the costs associated with treating myelosuppression.
In view of treatment guidelines, the recommendations of regulatory agencies, cost-benefit analyses, and the availability of gene-based dosing recommendations, we consider the benefits of testing for TPMT activity to greatly outweigh any associated risks. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Ansari A, Arenas M, Greenfield SM, et al. Prospective evaluation of the pharmacogenetics of azathioprine in the treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2008; 28:973–983.
- Beswick L, Friedman AB, Sparrow MP. The role of thiopurine metabolite monitoring in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol 2014; 8:383–392.
- Gervasini G, Vagace JM. Impact of genetic polymorphisms on chemotherapy toxicity in childhood acute lymphoblastic leukemia. Front Genet 2012; 3:249.
- Levinsen M, Rotevatn EØ, Rosthøj S, et al; Nordic Society of Paediatric Haematology, Oncology. Pharmacogenetically based dosing of thiopurines in childhood acute lymphoblastic leukemia: influence on cure rates and risk of second cancer. Pediatr Blood Cancer 2014; 61:797–802.
- Adam de Beaumais T, Jacqz-Aigrain E. Pharmacogenetic determinants of mercaptopurine disposition in children with acute lymphoblastic leukemia. Eur J Clin Pharmacol 2012; 68:1233–1242.
- Derijks LJ, Gilissen LP, Hooymans PM, Hommes DW. Review article: thiopurines in inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:715–729.
- Fairchild CR, Maybaum J, Kennedy KA. Concurrent unilateral chromatid damage and DNA strand breakage in response to 6-thioguanine treatment. Biochem Pharmacol 1986; 35:3533–3541.
- Karran P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br Med Bull 2006; 79–80:153–170.
- Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr 1991; 119:985–989.
- Gardiner SJ, Gearry RB, Barclay ML, Begg EJ. Two cases of thiopurine methyltransferase (TPMT) deficiency—a lucky save and a near miss with azathioprine. Br J Clin Pharmacol 2006; 62:473–476.
- Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther 2013; 93:324–325.
- Altman RB. Pharmacogenomics: “noninferiority” is sufficient for initial implementation. Clin Pharmacol Ther 2011; 89:348–350.
- van den Akker-van Marle ME, Gurwitz D, Detmar SB, et al. Cost-effectiveness of pharmacogenomics in clinical practice: a case study of thiopurine methyltransferase genotyping in acute lymphoblastic leukemia in Europe. Pharmacogenomics 2006; 7:783–792.
- Clunie GP, Lennard L. Relevance of thiopurine methyltransferase status in rheumatology patients receiving azathioprine. Rheumatology (Oxford) 2004; 43:13–18.
- Dubinsky MC, Reyes E, Ofman J, Chiou CF, Wade S, Sandborn WJ. A cost-effectiveness analysis of alternative disease management strategies in patients with Crohn’s disease treated with azathioprine or 6-mercaptopurine. Am J Gastroenterol 2005; 100:2239–2247.
- Winter J, Walker A, Shapiro D, Gaffney D, Spooner RJ, Mills PR. Cost-effectiveness of thiopurine methyltransferase genotype screening in patients about to commence azathioprine therapy for treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20:593–599.
- Marra CA, Esdaile JM, Anis AH. Practical pharmacogenetics: the cost effectiveness of screening for thiopurine s-methyltransferase polymorphisms in patients with rheumatological conditions treated with azathioprine. J Rheumatol 2002; 29:2507–2512.
- Donnan JR, Ungar WJ, Mathews M, Hancock-Howard RL, Rahman P. A cost effectiveness analysis of thiopurine methyltransferase testing for guiding 6-mercaptopurine dosing in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 2011; 57:231–239.
- Lennard L, Gibson BE, Nicole T, Lilleyman JS. Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch Dis Child 1993; 69:577–579.
- Hindorf U, Lindqvist M, Hildebrand H, Fagerberg U, Almer S. Adverse events leading to modification of therapy in a large cohort of patients with inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:331–342.
- Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 1999; 91:2001–2008.
- Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 1999; 93:2817–2823.
- Pui CH, Pei D, Sandlund JT, et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 2010; 24:371–382.
- Ford LT, Berg JD. Thiopurine S-methyltransferase (TPMT) assessment prior to starting thiopurine drug treatment; a pharmacogenomic test whose time has come. J Clin Pathol 2010; 63:288–295.
- Schmiegelow K, Forestier E, Hellebostad M, et al; Nordic Society of Paediatric Haematology and Oncology. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 2010; 24:345–354.
- Siegel CA, Sands BE. Review article: practical management of inflammatory bowel disease patients taking immunomodulators. Aliment Pharmacol Ther 2005; 22:1–16.
- Mayberry JF, Lobo A, Ford AC, Thomas A. NICE clinical guideline (CG152): the management of Crohn’s disease in adults, children and young people. Aliment Pharmacol Ther 2013; 37:195–203
- Mowat C, Cole A, Windsor A, et al; IBD Section of the British Society of Gastroenterology. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011; 60:571–607.
- Turner D, Levine A, Escher JC, et al; European Crohn’s and Colitis Organization; European Society for Paediatric Gastroenterology, Hepatology, and Nutrition. Management of pediatric ulcerative colitis: joint ECCO and ESPGHAN evidence-based consensus guidelines. J Pediatr Gastroenterol Nutr 2012; 55:340–361.
- Bernstein CN, Fried M, Krabshuis JH, et al. World Gastroenterology Organization Practice Guidelines for the diagnosis and management of IBD in 2010. Inflamm Bowel Dis 2010; 16:112–124.
- Becquemont L, Alfirevic A, Amstutz U, et al. Practical recommendations for pharmacogenomics-based prescription: 2010 ESF-UB Conference on Pharmacogenetics and Pharmacogenomics. Pharmacogenomics 2011; 12:113–124.
The thiopurines azathioprine, mercaptopurine, and thioguanine are prodrugs that are converted to active thioguanine nucleotide metabolites or methylated by thiopurine methyltransferase (TPMT) to compounds with less pharmacologic activity. In the absence of TPMT activity, patients are likely to have higher concentrations of thioguanine nucleotides, which can pose an increased risk of severe life-threatening myelosuppression. Determining TPMT activity, either directly by phenotyping or indirectly by determining the specific genetic allele (different alleles have different enzymatic activity), can help identify patients at greater risk of severe myelosuppression. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
THIOPURINES AND TPMT
Azathioprine, mercaptopurine, and thioguanine are used for treating autoimmune and inflammatory diseases1–3 and certain types of cancer such as leukemias and lymphomas.1,4–6 Typically, azathioprine is used to treat nonmalignant conditions, thioguanine is used to treat malignancies, and mercaptopurine can be used to treat both malignant and nonmalignant conditions.
Although the exact mechanism of action of these drugs has not been completely elucidated, the active thioguanine nucleotide metabolites are thought to be incorporated into the DNA of leukocytes, resulting in DNA damage that subsequently leads to cell death and myelosuppression.7–9
Variants of the TPMT gene may alter the activity of the TPMT enzyme, resulting in individual variability in thiopurine metabolism. Compared with people with normal (high) TPMT activity, those with intermediate or low TPMT activity metabolize the drugs more slowly, and are likely to have higher thioguanine nucleotide concentrations and therefore an increased risk of myelosuppression.
One of the earliest correlations between TPMT activity and thiopurine-induced myelosuppression was described in a pediatric patient with acute lymphocytic leukemia.10 After being prescribed a conventional mercaptopurine dosage (75 mg/m2 daily), the patient developed severe myelosuppression and was observed to have a thioguanine nucleotide metabolite concentration seven times the observed population median. TPMT phenotyping demonstrated that the patient had low TPMT activity. Reducing the mercaptopurine dose by approximately 90% resulted in normalization of thioguanine nucleotide metabolite concentrations, and the myelosuppression subsequently resolved.
Approximately 10% of the population has intermediate TPMT activity and 0.3% has low or absent TPMT activity, though these percentages vary depending on ancestry.1 Research has demonstrated that approximately 30% to 60% of those with intermediate TPMT activity cannot tolerate a full thiopurine dose (eg, azathioprine 2–3 mg/kg/day or mercaptopurine 1.5 mg/kg/day).1 Almost all patients with low TPMT activity will develop life-threating myelosuppression if prescribed a full thiopurine dose.1
SHOULD TPMT ACTIVITY BE DETERMINED FOR EVERY PATIENT PRESCRIBED A THIOPURINE?
Although determining TPMT activity in thiopurine-naïve patients will assist clinicians in selecting a thiopurine starting dose or in deciding if an alternative agent is warranted, there are instances when a clinician may elect to not perform a TPMT genotype or phenotype test. For example, determining TPMT activity is not recommended for patients who previously tolerated thiopurine therapy at full steady-state doses.
The required starting dose of a thiopurine can influence the decision on whether or not to test for TPMT activity. TPMT genotyping or phenotyping may be of most benefit for patients requiring immediate full doses of a thiopurine.11 Ideally, TPMT activity should be determined before prescribing immediate full doses of a thiopurine. This could be achieved by preemptively ordering a TPMT test in patients likely to require immunosuppression—for example, in patients diagnosed with inflammatory or autoimmune diseases. If therapy cannot be delayed and TPMT activity is unknown, ordering a TPMT test at the time of prescribing a full thiopurine dose is still of benefit. Depending on the clinical laboratory utilized for testing, TPMT phenotype results are usually reported in 3 to 5 days, and TPMT genotype results are usually reported in 5 to 7 days. Because most patients will not reach steady-state concentrations for 2 to 6 weeks, clinicians could initiate immediate full doses of a thiopurine and modify therapy based on TPMT test results before accumulation of thioguanine nucleotide metabolites occurs. Caution should be used with this approach, particularly in situations where the clinical laboratory may not return results in a timely manner.
For patients who are candidates for an initial low dose of a thiopurine, clinicians may choose to slowly titrate doses based on response and tolerability instead of determining TPMT activity.11 Depending on the starting dose and how slowly titration occurs, initiating a thiopurine at a low dose and titrating based on response can be a feasible approach for patients with intermediate TPMT activity. Because drastic thiopurine dose reductions of approximately 10-fold are required for patients with low TPMT activity, which is a much smaller dosage than most clinicians will initially prescribe, the starting dosage will likely not be low enough to prevent myelosuppression in patients with low TPMT activity.1,10
Determining TPMT activity can help clinicians establish an appropriate titration schedule. Patients with normal TPMT activity will usually reach thiopurine steady-state concentrations in 2 weeks, and the dosage can be titrated based on response.1 Alterations in TPMT activity influence the pharmacokinetic parameters of thiopurines, and the time to reach steady-state is extended to 4 or 6 weeks for those with intermediate or low TPMT activity.1 Increasing the thiopurine dosage before reaching steady state can lead to the prescribing of doses that will not be tolerated, resulting in myelosuppression.
Factors to consider when deciding if TPMT activity should be assessed include the disease state being treated and corresponding starting dose, the need for immediate full doses, and previous documented tolerance of thiopurines at steady-state doses. As with many aspects of medicine that have multiple options, coupled with an increase in patient access to healthcare information, the decision to test for TPMT activity may include shared decision-making between patients and providers. Although TPMT genotyping or phenotyping can help identify those at greatest risk of severe myelosuppression, such assays do not replace routine monitoring for myelosuppression, hepatotoxicity, or pancreatitis that may be caused by thiopurines.
WHAT TESTS ARE AVAILABLE TO DETERMINE TPMT ACTIVITY?
Patients with intermediate or low TPMT activity can be identified by either genotyping or phenotyping. There are considerations, though, that clinicians should be aware of before selecting a particular test.
TPMT genotyping
![]()
Four TPMT alleles, TPMT*2, *3A, *3B, and *3C, account for over 90% of inactivating polymorphisms.12 Therefore, most reference laboratories only analyze for those genetic variants. Based on the reported test result, a predicted phenotype (eg, normal, intermediate, or low TPMT activity) can be assigned. Table 1 lists the predicted phenotypes for select genotyping results.
TPMT phenotyping
Phenotyping quantitates TPMT enzyme activity in erythrocytes, and based on the result, patients are classified as having normal, intermediate, or low TPMT activity. Because internal standards and other testing conditions may differ between reference laboratories, test results must be interpreted in the context of the laboratory that performed the assay.
Which test is right for my patient?
In most cases, either the genotype or the phenotype test provides sufficient information to guide thiopurine therapy. There are certain circumstances, though, in which the genotype or phenotype test is less informative.
TPMT genotyping, when performed using a blood specimen, is not recommended in those with a history of allogeneic bone marrow transplantation, as the result would reflect the donor’s genotype, not the patient’s. In such instances, monitoring of white blood cell counts and thiopurine metabolites may be more beneficial.
TPMT phenotyping may be inaccurate if performed within 30 to 90 days of an erythrocyte transfusion, as the test result may be influenced by donor erythrocytes. If a patient is receiving erythrocyte transfusions, TPMT genotyping is preferable to phenotyping.
Test cost may also be a consideration when determining if the genotype or phenotype test is best for your patient. Costs vary by laboratory, but phenotyping is generally less expensive than genotyping. The cost of genotyping, though, continues to decrease.13 The approximate commercial cost is $200 for phenotyping and $450 for genotyping, but laboratory fees may be substantially higher. Several insurance plans, including Medicare, cover TPMT testing, but reimbursement and copayments vary, depending on the patient’s specific plan.
There are conflicting data as to whether determining TPMT status is11,14–18 or is not19 cost-effective. Multiple studies suggest that the cost of genotyping a sufficient number of patients to identify a single individual at high risk of myelosuppression is cheaper than the costs associated with treating an adverse event. Additional cost-benefit studies are needed, particularly studies that consider how bundled payments and outcomes-based reimbursement influence cost-effectiveness.
MODIFYING THIOPURINE THERAPY BASED ON TPMT ACTIVITY
![]()
There is a strong correlation between TPMT activity and tolerated thiopurine doses, with those having intermediate or low TPMT activity requiring lower doses.10,20–23 Adjusting mercaptopurine doses based on TPMT activity to prevent hematopoietic toxicity has been successfully demonstrated in pediatric patients with acute lymphoblastic leukemia.24 Furthermore, reducing initial thiopurine doses to avoid myelosuppression and titrating based on response has been shown to not compromise outcomes.1,25,26 The Clinical Pharmacogenetic Implementation Consortium (CPIC) has developed an evidence-based guideline on how to adjust thiopurine doses based on TPMT activity,1 summarized in Table 2. These dosing recommendations are classified as “strong.”
Patients with normal TPMT activity should be prescribed the usual thiopurine starting dose as indicated by disease-specific guidelines.
For those with intermediate TPMT activity, the CPIC guideline recommends reducing the initial targeted full dose of azathioprine and mercaptopurine by 30% to 70% and reducing the targeted full dose of thioguanine by 30% to 50%. The percentage of dose reduction depends on the targeted full dose. Siegel and Sands27 suggested that for those who are diagnosed with inflammatory bowel disease and have intermediate TPMT activity, azathioprine should be initiated at a low dose and titrated to 1.25 mg/kg and mercaptopurine should be initiated at a low dose and titrated to 0.75 mg/kg. Based on these titration goals, if the targeted full dose for mercaptopurine is 1 mg/kg, then a dose reduction of approximately 30% would be more appropriate. If the targeted full dose is 1.5 mg/kg, a dose reduction of approximately 50% would be more appropriate. Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 2 to 4 weeks to reach steady state before dose titration.
For those with low TPMT activity, alternative therapy should be considered for nonmalignant conditions because of the risk of severe myelosuppression. For malignancy, or if a thiopurine is warranted for a nonmalignant condition, consider a 90% dose reduction and give the drug three times per week instead of daily. For example, acute lymphoblastic leukemia patients with low TPMT activity can be started on mercaptopurine 10 mg/m2 three times per week instead of the usual starting dose.10 Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 4 to 6 weeks to reach steady state before dose titration.
RECOMMENDATIONS
Individuals with intermediate or low TPMT activity have an increased risk of myelosuppression. Because of the elevated risk for morbidity and death, especially for patients with low TPMT activity, multiple guidelines and regulatory agencies recommend TPMT genotyping or phenotyping if a thiopurine is prescribed.25,28–32 Although additional cost-benefit analysis studies are needed, evidence suggests testing for TPMT activity may be cheaper than the costs associated with treating myelosuppression.
In view of treatment guidelines, the recommendations of regulatory agencies, cost-benefit analyses, and the availability of gene-based dosing recommendations, we consider the benefits of testing for TPMT activity to greatly outweigh any associated risks. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
The thiopurines azathioprine, mercaptopurine, and thioguanine are prodrugs that are converted to active thioguanine nucleotide metabolites or methylated by thiopurine methyltransferase (TPMT) to compounds with less pharmacologic activity. In the absence of TPMT activity, patients are likely to have higher concentrations of thioguanine nucleotides, which can pose an increased risk of severe life-threatening myelosuppression. Determining TPMT activity, either directly by phenotyping or indirectly by determining the specific genetic allele (different alleles have different enzymatic activity), can help identify patients at greater risk of severe myelosuppression. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
THIOPURINES AND TPMT
Azathioprine, mercaptopurine, and thioguanine are used for treating autoimmune and inflammatory diseases1–3 and certain types of cancer such as leukemias and lymphomas.1,4–6 Typically, azathioprine is used to treat nonmalignant conditions, thioguanine is used to treat malignancies, and mercaptopurine can be used to treat both malignant and nonmalignant conditions.
Although the exact mechanism of action of these drugs has not been completely elucidated, the active thioguanine nucleotide metabolites are thought to be incorporated into the DNA of leukocytes, resulting in DNA damage that subsequently leads to cell death and myelosuppression.7–9
Variants of the TPMT gene may alter the activity of the TPMT enzyme, resulting in individual variability in thiopurine metabolism. Compared with people with normal (high) TPMT activity, those with intermediate or low TPMT activity metabolize the drugs more slowly, and are likely to have higher thioguanine nucleotide concentrations and therefore an increased risk of myelosuppression.
One of the earliest correlations between TPMT activity and thiopurine-induced myelosuppression was described in a pediatric patient with acute lymphocytic leukemia.10 After being prescribed a conventional mercaptopurine dosage (75 mg/m2 daily), the patient developed severe myelosuppression and was observed to have a thioguanine nucleotide metabolite concentration seven times the observed population median. TPMT phenotyping demonstrated that the patient had low TPMT activity. Reducing the mercaptopurine dose by approximately 90% resulted in normalization of thioguanine nucleotide metabolite concentrations, and the myelosuppression subsequently resolved.
Approximately 10% of the population has intermediate TPMT activity and 0.3% has low or absent TPMT activity, though these percentages vary depending on ancestry.1 Research has demonstrated that approximately 30% to 60% of those with intermediate TPMT activity cannot tolerate a full thiopurine dose (eg, azathioprine 2–3 mg/kg/day or mercaptopurine 1.5 mg/kg/day).1 Almost all patients with low TPMT activity will develop life-threating myelosuppression if prescribed a full thiopurine dose.1
SHOULD TPMT ACTIVITY BE DETERMINED FOR EVERY PATIENT PRESCRIBED A THIOPURINE?
Although determining TPMT activity in thiopurine-naïve patients will assist clinicians in selecting a thiopurine starting dose or in deciding if an alternative agent is warranted, there are instances when a clinician may elect to not perform a TPMT genotype or phenotype test. For example, determining TPMT activity is not recommended for patients who previously tolerated thiopurine therapy at full steady-state doses.
The required starting dose of a thiopurine can influence the decision on whether or not to test for TPMT activity. TPMT genotyping or phenotyping may be of most benefit for patients requiring immediate full doses of a thiopurine.11 Ideally, TPMT activity should be determined before prescribing immediate full doses of a thiopurine. This could be achieved by preemptively ordering a TPMT test in patients likely to require immunosuppression—for example, in patients diagnosed with inflammatory or autoimmune diseases. If therapy cannot be delayed and TPMT activity is unknown, ordering a TPMT test at the time of prescribing a full thiopurine dose is still of benefit. Depending on the clinical laboratory utilized for testing, TPMT phenotype results are usually reported in 3 to 5 days, and TPMT genotype results are usually reported in 5 to 7 days. Because most patients will not reach steady-state concentrations for 2 to 6 weeks, clinicians could initiate immediate full doses of a thiopurine and modify therapy based on TPMT test results before accumulation of thioguanine nucleotide metabolites occurs. Caution should be used with this approach, particularly in situations where the clinical laboratory may not return results in a timely manner.
For patients who are candidates for an initial low dose of a thiopurine, clinicians may choose to slowly titrate doses based on response and tolerability instead of determining TPMT activity.11 Depending on the starting dose and how slowly titration occurs, initiating a thiopurine at a low dose and titrating based on response can be a feasible approach for patients with intermediate TPMT activity. Because drastic thiopurine dose reductions of approximately 10-fold are required for patients with low TPMT activity, which is a much smaller dosage than most clinicians will initially prescribe, the starting dosage will likely not be low enough to prevent myelosuppression in patients with low TPMT activity.1,10
Determining TPMT activity can help clinicians establish an appropriate titration schedule. Patients with normal TPMT activity will usually reach thiopurine steady-state concentrations in 2 weeks, and the dosage can be titrated based on response.1 Alterations in TPMT activity influence the pharmacokinetic parameters of thiopurines, and the time to reach steady-state is extended to 4 or 6 weeks for those with intermediate or low TPMT activity.1 Increasing the thiopurine dosage before reaching steady state can lead to the prescribing of doses that will not be tolerated, resulting in myelosuppression.
Factors to consider when deciding if TPMT activity should be assessed include the disease state being treated and corresponding starting dose, the need for immediate full doses, and previous documented tolerance of thiopurines at steady-state doses. As with many aspects of medicine that have multiple options, coupled with an increase in patient access to healthcare information, the decision to test for TPMT activity may include shared decision-making between patients and providers. Although TPMT genotyping or phenotyping can help identify those at greatest risk of severe myelosuppression, such assays do not replace routine monitoring for myelosuppression, hepatotoxicity, or pancreatitis that may be caused by thiopurines.
WHAT TESTS ARE AVAILABLE TO DETERMINE TPMT ACTIVITY?
Patients with intermediate or low TPMT activity can be identified by either genotyping or phenotyping. There are considerations, though, that clinicians should be aware of before selecting a particular test.
TPMT genotyping
![]()
Four TPMT alleles, TPMT*2, *3A, *3B, and *3C, account for over 90% of inactivating polymorphisms.12 Therefore, most reference laboratories only analyze for those genetic variants. Based on the reported test result, a predicted phenotype (eg, normal, intermediate, or low TPMT activity) can be assigned. Table 1 lists the predicted phenotypes for select genotyping results.
TPMT phenotyping
Phenotyping quantitates TPMT enzyme activity in erythrocytes, and based on the result, patients are classified as having normal, intermediate, or low TPMT activity. Because internal standards and other testing conditions may differ between reference laboratories, test results must be interpreted in the context of the laboratory that performed the assay.
Which test is right for my patient?
In most cases, either the genotype or the phenotype test provides sufficient information to guide thiopurine therapy. There are certain circumstances, though, in which the genotype or phenotype test is less informative.
TPMT genotyping, when performed using a blood specimen, is not recommended in those with a history of allogeneic bone marrow transplantation, as the result would reflect the donor’s genotype, not the patient’s. In such instances, monitoring of white blood cell counts and thiopurine metabolites may be more beneficial.
TPMT phenotyping may be inaccurate if performed within 30 to 90 days of an erythrocyte transfusion, as the test result may be influenced by donor erythrocytes. If a patient is receiving erythrocyte transfusions, TPMT genotyping is preferable to phenotyping.
Test cost may also be a consideration when determining if the genotype or phenotype test is best for your patient. Costs vary by laboratory, but phenotyping is generally less expensive than genotyping. The cost of genotyping, though, continues to decrease.13 The approximate commercial cost is $200 for phenotyping and $450 for genotyping, but laboratory fees may be substantially higher. Several insurance plans, including Medicare, cover TPMT testing, but reimbursement and copayments vary, depending on the patient’s specific plan.
There are conflicting data as to whether determining TPMT status is11,14–18 or is not19 cost-effective. Multiple studies suggest that the cost of genotyping a sufficient number of patients to identify a single individual at high risk of myelosuppression is cheaper than the costs associated with treating an adverse event. Additional cost-benefit studies are needed, particularly studies that consider how bundled payments and outcomes-based reimbursement influence cost-effectiveness.
MODIFYING THIOPURINE THERAPY BASED ON TPMT ACTIVITY
![]()
There is a strong correlation between TPMT activity and tolerated thiopurine doses, with those having intermediate or low TPMT activity requiring lower doses.10,20–23 Adjusting mercaptopurine doses based on TPMT activity to prevent hematopoietic toxicity has been successfully demonstrated in pediatric patients with acute lymphoblastic leukemia.24 Furthermore, reducing initial thiopurine doses to avoid myelosuppression and titrating based on response has been shown to not compromise outcomes.1,25,26 The Clinical Pharmacogenetic Implementation Consortium (CPIC) has developed an evidence-based guideline on how to adjust thiopurine doses based on TPMT activity,1 summarized in Table 2. These dosing recommendations are classified as “strong.”
Patients with normal TPMT activity should be prescribed the usual thiopurine starting dose as indicated by disease-specific guidelines.
For those with intermediate TPMT activity, the CPIC guideline recommends reducing the initial targeted full dose of azathioprine and mercaptopurine by 30% to 70% and reducing the targeted full dose of thioguanine by 30% to 50%. The percentage of dose reduction depends on the targeted full dose. Siegel and Sands27 suggested that for those who are diagnosed with inflammatory bowel disease and have intermediate TPMT activity, azathioprine should be initiated at a low dose and titrated to 1.25 mg/kg and mercaptopurine should be initiated at a low dose and titrated to 0.75 mg/kg. Based on these titration goals, if the targeted full dose for mercaptopurine is 1 mg/kg, then a dose reduction of approximately 30% would be more appropriate. If the targeted full dose is 1.5 mg/kg, a dose reduction of approximately 50% would be more appropriate. Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 2 to 4 weeks to reach steady state before dose titration.
For those with low TPMT activity, alternative therapy should be considered for nonmalignant conditions because of the risk of severe myelosuppression. For malignancy, or if a thiopurine is warranted for a nonmalignant condition, consider a 90% dose reduction and give the drug three times per week instead of daily. For example, acute lymphoblastic leukemia patients with low TPMT activity can be started on mercaptopurine 10 mg/m2 three times per week instead of the usual starting dose.10 Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 4 to 6 weeks to reach steady state before dose titration.
RECOMMENDATIONS
Individuals with intermediate or low TPMT activity have an increased risk of myelosuppression. Because of the elevated risk for morbidity and death, especially for patients with low TPMT activity, multiple guidelines and regulatory agencies recommend TPMT genotyping or phenotyping if a thiopurine is prescribed.25,28–32 Although additional cost-benefit analysis studies are needed, evidence suggests testing for TPMT activity may be cheaper than the costs associated with treating myelosuppression.
In view of treatment guidelines, the recommendations of regulatory agencies, cost-benefit analyses, and the availability of gene-based dosing recommendations, we consider the benefits of testing for TPMT activity to greatly outweigh any associated risks. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Ansari A, Arenas M, Greenfield SM, et al. Prospective evaluation of the pharmacogenetics of azathioprine in the treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2008; 28:973–983.
- Beswick L, Friedman AB, Sparrow MP. The role of thiopurine metabolite monitoring in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol 2014; 8:383–392.
- Gervasini G, Vagace JM. Impact of genetic polymorphisms on chemotherapy toxicity in childhood acute lymphoblastic leukemia. Front Genet 2012; 3:249.
- Levinsen M, Rotevatn EØ, Rosthøj S, et al; Nordic Society of Paediatric Haematology, Oncology. Pharmacogenetically based dosing of thiopurines in childhood acute lymphoblastic leukemia: influence on cure rates and risk of second cancer. Pediatr Blood Cancer 2014; 61:797–802.
- Adam de Beaumais T, Jacqz-Aigrain E. Pharmacogenetic determinants of mercaptopurine disposition in children with acute lymphoblastic leukemia. Eur J Clin Pharmacol 2012; 68:1233–1242.
- Derijks LJ, Gilissen LP, Hooymans PM, Hommes DW. Review article: thiopurines in inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:715–729.
- Fairchild CR, Maybaum J, Kennedy KA. Concurrent unilateral chromatid damage and DNA strand breakage in response to 6-thioguanine treatment. Biochem Pharmacol 1986; 35:3533–3541.
- Karran P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br Med Bull 2006; 79–80:153–170.
- Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr 1991; 119:985–989.
- Gardiner SJ, Gearry RB, Barclay ML, Begg EJ. Two cases of thiopurine methyltransferase (TPMT) deficiency—a lucky save and a near miss with azathioprine. Br J Clin Pharmacol 2006; 62:473–476.
- Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther 2013; 93:324–325.
- Altman RB. Pharmacogenomics: “noninferiority” is sufficient for initial implementation. Clin Pharmacol Ther 2011; 89:348–350.
- van den Akker-van Marle ME, Gurwitz D, Detmar SB, et al. Cost-effectiveness of pharmacogenomics in clinical practice: a case study of thiopurine methyltransferase genotyping in acute lymphoblastic leukemia in Europe. Pharmacogenomics 2006; 7:783–792.
- Clunie GP, Lennard L. Relevance of thiopurine methyltransferase status in rheumatology patients receiving azathioprine. Rheumatology (Oxford) 2004; 43:13–18.
- Dubinsky MC, Reyes E, Ofman J, Chiou CF, Wade S, Sandborn WJ. A cost-effectiveness analysis of alternative disease management strategies in patients with Crohn’s disease treated with azathioprine or 6-mercaptopurine. Am J Gastroenterol 2005; 100:2239–2247.
- Winter J, Walker A, Shapiro D, Gaffney D, Spooner RJ, Mills PR. Cost-effectiveness of thiopurine methyltransferase genotype screening in patients about to commence azathioprine therapy for treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20:593–599.
- Marra CA, Esdaile JM, Anis AH. Practical pharmacogenetics: the cost effectiveness of screening for thiopurine s-methyltransferase polymorphisms in patients with rheumatological conditions treated with azathioprine. J Rheumatol 2002; 29:2507–2512.
- Donnan JR, Ungar WJ, Mathews M, Hancock-Howard RL, Rahman P. A cost effectiveness analysis of thiopurine methyltransferase testing for guiding 6-mercaptopurine dosing in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 2011; 57:231–239.
- Lennard L, Gibson BE, Nicole T, Lilleyman JS. Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch Dis Child 1993; 69:577–579.
- Hindorf U, Lindqvist M, Hildebrand H, Fagerberg U, Almer S. Adverse events leading to modification of therapy in a large cohort of patients with inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:331–342.
- Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 1999; 91:2001–2008.
- Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 1999; 93:2817–2823.
- Pui CH, Pei D, Sandlund JT, et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 2010; 24:371–382.
- Ford LT, Berg JD. Thiopurine S-methyltransferase (TPMT) assessment prior to starting thiopurine drug treatment; a pharmacogenomic test whose time has come. J Clin Pathol 2010; 63:288–295.
- Schmiegelow K, Forestier E, Hellebostad M, et al; Nordic Society of Paediatric Haematology and Oncology. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 2010; 24:345–354.
- Siegel CA, Sands BE. Review article: practical management of inflammatory bowel disease patients taking immunomodulators. Aliment Pharmacol Ther 2005; 22:1–16.
- Mayberry JF, Lobo A, Ford AC, Thomas A. NICE clinical guideline (CG152): the management of Crohn’s disease in adults, children and young people. Aliment Pharmacol Ther 2013; 37:195–203
- Mowat C, Cole A, Windsor A, et al; IBD Section of the British Society of Gastroenterology. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011; 60:571–607.
- Turner D, Levine A, Escher JC, et al; European Crohn’s and Colitis Organization; European Society for Paediatric Gastroenterology, Hepatology, and Nutrition. Management of pediatric ulcerative colitis: joint ECCO and ESPGHAN evidence-based consensus guidelines. J Pediatr Gastroenterol Nutr 2012; 55:340–361.
- Bernstein CN, Fried M, Krabshuis JH, et al. World Gastroenterology Organization Practice Guidelines for the diagnosis and management of IBD in 2010. Inflamm Bowel Dis 2010; 16:112–124.
- Becquemont L, Alfirevic A, Amstutz U, et al. Practical recommendations for pharmacogenomics-based prescription: 2010 ESF-UB Conference on Pharmacogenetics and Pharmacogenomics. Pharmacogenomics 2011; 12:113–124.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Ansari A, Arenas M, Greenfield SM, et al. Prospective evaluation of the pharmacogenetics of azathioprine in the treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2008; 28:973–983.
- Beswick L, Friedman AB, Sparrow MP. The role of thiopurine metabolite monitoring in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol 2014; 8:383–392.
- Gervasini G, Vagace JM. Impact of genetic polymorphisms on chemotherapy toxicity in childhood acute lymphoblastic leukemia. Front Genet 2012; 3:249.
- Levinsen M, Rotevatn EØ, Rosthøj S, et al; Nordic Society of Paediatric Haematology, Oncology. Pharmacogenetically based dosing of thiopurines in childhood acute lymphoblastic leukemia: influence on cure rates and risk of second cancer. Pediatr Blood Cancer 2014; 61:797–802.
- Adam de Beaumais T, Jacqz-Aigrain E. Pharmacogenetic determinants of mercaptopurine disposition in children with acute lymphoblastic leukemia. Eur J Clin Pharmacol 2012; 68:1233–1242.
- Derijks LJ, Gilissen LP, Hooymans PM, Hommes DW. Review article: thiopurines in inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:715–729.
- Fairchild CR, Maybaum J, Kennedy KA. Concurrent unilateral chromatid damage and DNA strand breakage in response to 6-thioguanine treatment. Biochem Pharmacol 1986; 35:3533–3541.
- Karran P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br Med Bull 2006; 79–80:153–170.
- Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr 1991; 119:985–989.
- Gardiner SJ, Gearry RB, Barclay ML, Begg EJ. Two cases of thiopurine methyltransferase (TPMT) deficiency—a lucky save and a near miss with azathioprine. Br J Clin Pharmacol 2006; 62:473–476.
- Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther 2013; 93:324–325.
- Altman RB. Pharmacogenomics: “noninferiority” is sufficient for initial implementation. Clin Pharmacol Ther 2011; 89:348–350.
- van den Akker-van Marle ME, Gurwitz D, Detmar SB, et al. Cost-effectiveness of pharmacogenomics in clinical practice: a case study of thiopurine methyltransferase genotyping in acute lymphoblastic leukemia in Europe. Pharmacogenomics 2006; 7:783–792.
- Clunie GP, Lennard L. Relevance of thiopurine methyltransferase status in rheumatology patients receiving azathioprine. Rheumatology (Oxford) 2004; 43:13–18.
- Dubinsky MC, Reyes E, Ofman J, Chiou CF, Wade S, Sandborn WJ. A cost-effectiveness analysis of alternative disease management strategies in patients with Crohn’s disease treated with azathioprine or 6-mercaptopurine. Am J Gastroenterol 2005; 100:2239–2247.
- Winter J, Walker A, Shapiro D, Gaffney D, Spooner RJ, Mills PR. Cost-effectiveness of thiopurine methyltransferase genotype screening in patients about to commence azathioprine therapy for treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20:593–599.
- Marra CA, Esdaile JM, Anis AH. Practical pharmacogenetics: the cost effectiveness of screening for thiopurine s-methyltransferase polymorphisms in patients with rheumatological conditions treated with azathioprine. J Rheumatol 2002; 29:2507–2512.
- Donnan JR, Ungar WJ, Mathews M, Hancock-Howard RL, Rahman P. A cost effectiveness analysis of thiopurine methyltransferase testing for guiding 6-mercaptopurine dosing in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 2011; 57:231–239.
- Lennard L, Gibson BE, Nicole T, Lilleyman JS. Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch Dis Child 1993; 69:577–579.
- Hindorf U, Lindqvist M, Hildebrand H, Fagerberg U, Almer S. Adverse events leading to modification of therapy in a large cohort of patients with inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:331–342.
- Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 1999; 91:2001–2008.
- Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 1999; 93:2817–2823.
- Pui CH, Pei D, Sandlund JT, et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 2010; 24:371–382.
- Ford LT, Berg JD. Thiopurine S-methyltransferase (TPMT) assessment prior to starting thiopurine drug treatment; a pharmacogenomic test whose time has come. J Clin Pathol 2010; 63:288–295.
- Schmiegelow K, Forestier E, Hellebostad M, et al; Nordic Society of Paediatric Haematology and Oncology. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 2010; 24:345–354.
- Siegel CA, Sands BE. Review article: practical management of inflammatory bowel disease patients taking immunomodulators. Aliment Pharmacol Ther 2005; 22:1–16.
- Mayberry JF, Lobo A, Ford AC, Thomas A. NICE clinical guideline (CG152): the management of Crohn’s disease in adults, children and young people. Aliment Pharmacol Ther 2013; 37:195–203
- Mowat C, Cole A, Windsor A, et al; IBD Section of the British Society of Gastroenterology. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011; 60:571–607.
- Turner D, Levine A, Escher JC, et al; European Crohn’s and Colitis Organization; European Society for Paediatric Gastroenterology, Hepatology, and Nutrition. Management of pediatric ulcerative colitis: joint ECCO and ESPGHAN evidence-based consensus guidelines. J Pediatr Gastroenterol Nutr 2012; 55:340–361.
- Bernstein CN, Fried M, Krabshuis JH, et al. World Gastroenterology Organization Practice Guidelines for the diagnosis and management of IBD in 2010. Inflamm Bowel Dis 2010; 16:112–124.
- Becquemont L, Alfirevic A, Amstutz U, et al. Practical recommendations for pharmacogenomics-based prescription: 2010 ESF-UB Conference on Pharmacogenetics and Pharmacogenomics. Pharmacogenomics 2011; 12:113–124.
When does pericarditis merit a workup for autoimmune or inflammatory disease?
Pericarditis is in most cases a one-time disease simply treated with anti-inflammatory drugs. It requires no extensive workup for systemic inflammatory or autoimmune disease. Further evaluation is required for patients who have recurrent pericarditis resistant to conventional therapy or pericarditis with manifestations of systemic disease.
ACUTE PERICARDITIS
Pericardial disease has different presentations: acute, recurrent, constrictive, effusive-constrictive, and pericardial effusion with or without tamponade. Acute pericarditis is the most common of these and can affect people of all ages. The typical acute manifestations are chest pain (usually pleuritic), a pericardial friction rub, and widespread ST-segment elevation on the electrocardiogram.1,2 The chest pain tends to be sharp and long-lasting; it radiates to the trapezius ridge and increases during respiration or body movements.
Acute pericarditis usually responds to an anti-inflammatory drug such as colchicine 0.6 mg/day for 3 months, a nonsteroidal anti-inflammatory drug such as ibuprofen 600 mg three times a day for 10 days, and in advanced resistant cases, an oral corticosteroid.3,4
Most often, pericarditis is either idiopathic or occurs after a respiratory viral illness. Much less common causes include bacterial infection, postpericardiotomy syndrome, myocardial infarction, primary or metastatic tumors, trauma, radiation, and uremia. However, pericarditis can also be part of the presentation of systemic inflammatory and autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus; hereditary periodic fever syndromes such as familial Mediterranean fever; and systemic-onset juvenile idiopathic arthritis.1,5
In acute pericarditis, a complex workup is usually not justified, since the results will have limited usefulness in the clinical management of the patient. It is most often diagnosed by the presenting symptoms, auscultation, electrocardiography, echocardiography, and chest radiography, and by additional basic tests that include a complete blood cell count, complete metabolic profile, erythrocyte sedimentation rate, and C-reactive protein level. However, if pericarditis does not respond to anti-inflammatory treatment and if an autoimmune or infectious disease is suspected, further evaluation may include antinuclear antibody testing and testing for human immunodeficiency virus and tuberculosis. If the diagnosis of acute pericarditis remains uncertain, cardiac magnetic resonance imaging (MRI) may be useful.
RECURRENT PERICARDITIS
Although acute pericarditis most often has a benign course and responds well to anti-inflammatory drugs, 20% to 30% of patients who have a first attack of acute pericarditis have a recurrence, and up to 50% of patients who have one recurrence will have another.3,4
Disease activity can be followed with serial testing of inflammatory markers—eg, erythrocyte sedimentation rate and C-reactive protein level. Echocardiography, cardiac computed tomography, and cardiac MRI can characterize active inflammation, edema, pericardial thickness, and pericardial effusion.6–8
Recurrent pericarditis is often resistant to standard therapy and requires corticosteroids in high doses, which paradoxically can increase the risk of recurrence. Therefore, further workup for underlying autoimmune disease, systemic inflammatory disease, or infection is necessary. More potent immunosuppressive therapy may be required, not only in pericarditis associated with systemic autoimmune or inflammatory conditions, but even in idiopathic recurrent pericarditis, either to control symptoms or to mitigate the effects of corticosteroids.
SYSTEMIC INFLAMMATION
The true prevalence of pericardial disease in most systemic inflammatory and autoimmune diseases is difficult to determine from current data. But advances in serologic testing and imaging techniques have shown cardiac involvement in a number of inflammatory diseases.9
In one study, a serologic autoimmune workup in patients with acute pericarditis found that 2% had collagen vascular disease.9 Pericardial involvement is likely in systemic lupus erythematosus,10 and a postmortem study of patients with systemic sclerosis found that 72% had pericarditis.11 Mixed connective tissue disease has been associated with pericarditis in 29% of cases and 56% in autopsy studies.12,13 Pericarditis may be the initial manifestation of vasculitis—eg, Takayasu arteritis or granulomatosis with polyangiitis (formerly known as Wegener granulomatosis).
Other diseases with pericardial involvement include Still disease, Sjögren syndrome, sarcoidosis, and inflammatory bowel disease. Symptomatic pericarditis occurs in about 25% of patients with Sjögren syndrome and asymptomatic pericardial involvement in more than half. Autopsy studies reported pericardial involvement in up to 80% of patients with systemic lupus erythematosus. Cardiac tamponade occurs in fewer than 2%, and constrictive pericarditis is extremely rare.5,9–11
RECOMMENDATIONS
Patients with a first episode of pericarditis should be treated with an anti-inflammatory medication, with no comprehensive testing for autoimmune disease. An evaluation for autoimmune and infectious disease should be carried out in patients with fever (temperature > 38°C; 100.4°F), recurrent pericarditis, recurrent large pericardial effusion or tamponade, or night sweats despite conventional medical therapy. Signs of systemic disease such as renal failure, elevated liver enzymes, or skin rash merit further evaluation.
Prospective studies using appropriate serologic testing and imaging are needed to determine the correlation between myopericardial involvement and inflammatory diseases because of increased morbidity and mortality in several of these diseases.
- Troughton RW, Asher CR, Klein AL. Pericarditis. Lancet 2004; 363:717–727.
- Alraies MC, Klein AL. Should we still use electrocardiography to diagnose pericardial disease? Cleve Clin J Med 2013; 80:97–100.
- Imazio M, Brucato A, Cemin R, et al; ICAP Investigators. A randomized trial of colchicine for acute pericarditis. N Engl J Med 2013; 369:1522–1528.
- Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation 2007; 115:2739–2744.
- Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol 1995; 75:378–382.
- Verhaert D, Gabriel RS, Johnston D, Lytle BW, Desai MY, Klein AL. The role of multimodality imaging in the management of pericardial disease. Circ Cardiovasc Imaging 2010; 3:333–343.
- Klein AL, Abbara S, Agler DA, et al. American Society of Echocardiography clinical recommendations for multimodality cardiovascular imaging of patients with pericardial disease: endorsed by the Society for Cardiovascular Magnetic Resonance and Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr 2013; 26:965–1012.e15.
- Yingchoncharoen T, Alraies MC, Kwon DH, Rodriguez ER, Tan CD, Klein AL. Emerging role of multimodality imaging in management of inflammatory pericardial diseases. Expert Rev Cardiovasc Ther 2013; 11:1211–1225.
- Knockaert DC. Cardiac involvement in systemic inflammatory diseases. Eur Heart J 2007; 28:1797–1804.
- Doria A, Iaccarino L, Sarzi-Puttini P, Atzeni F, Turriel M, Petri M. Cardiac involvement in systemic lupus erythematosus. Lupus 2005; 14:683–686.
- Byers RJ, Marshall DA, Freemont AJ. Pericardial involvement in systemic sclerosis. Ann Rheum Dis 1997; 56:393–394.
- Kasukawa R. Mixed connective tissue disease. Intern Med 1999; 38:386–393.
- Bezerra MC, Saraiva F Jr, Carvalho JF, Caleiro MT, Goncalves CR, Borba EF. Cardiac tamponade due to massive pericardial effusion in mixed connective tissue disease: reversal with steroid therapy. Lupus 2004; 13:618–620.
Pericarditis is in most cases a one-time disease simply treated with anti-inflammatory drugs. It requires no extensive workup for systemic inflammatory or autoimmune disease. Further evaluation is required for patients who have recurrent pericarditis resistant to conventional therapy or pericarditis with manifestations of systemic disease.
ACUTE PERICARDITIS
Pericardial disease has different presentations: acute, recurrent, constrictive, effusive-constrictive, and pericardial effusion with or without tamponade. Acute pericarditis is the most common of these and can affect people of all ages. The typical acute manifestations are chest pain (usually pleuritic), a pericardial friction rub, and widespread ST-segment elevation on the electrocardiogram.1,2 The chest pain tends to be sharp and long-lasting; it radiates to the trapezius ridge and increases during respiration or body movements.
Acute pericarditis usually responds to an anti-inflammatory drug such as colchicine 0.6 mg/day for 3 months, a nonsteroidal anti-inflammatory drug such as ibuprofen 600 mg three times a day for 10 days, and in advanced resistant cases, an oral corticosteroid.3,4
Most often, pericarditis is either idiopathic or occurs after a respiratory viral illness. Much less common causes include bacterial infection, postpericardiotomy syndrome, myocardial infarction, primary or metastatic tumors, trauma, radiation, and uremia. However, pericarditis can also be part of the presentation of systemic inflammatory and autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus; hereditary periodic fever syndromes such as familial Mediterranean fever; and systemic-onset juvenile idiopathic arthritis.1,5
In acute pericarditis, a complex workup is usually not justified, since the results will have limited usefulness in the clinical management of the patient. It is most often diagnosed by the presenting symptoms, auscultation, electrocardiography, echocardiography, and chest radiography, and by additional basic tests that include a complete blood cell count, complete metabolic profile, erythrocyte sedimentation rate, and C-reactive protein level. However, if pericarditis does not respond to anti-inflammatory treatment and if an autoimmune or infectious disease is suspected, further evaluation may include antinuclear antibody testing and testing for human immunodeficiency virus and tuberculosis. If the diagnosis of acute pericarditis remains uncertain, cardiac magnetic resonance imaging (MRI) may be useful.
RECURRENT PERICARDITIS
Although acute pericarditis most often has a benign course and responds well to anti-inflammatory drugs, 20% to 30% of patients who have a first attack of acute pericarditis have a recurrence, and up to 50% of patients who have one recurrence will have another.3,4
Disease activity can be followed with serial testing of inflammatory markers—eg, erythrocyte sedimentation rate and C-reactive protein level. Echocardiography, cardiac computed tomography, and cardiac MRI can characterize active inflammation, edema, pericardial thickness, and pericardial effusion.6–8
Recurrent pericarditis is often resistant to standard therapy and requires corticosteroids in high doses, which paradoxically can increase the risk of recurrence. Therefore, further workup for underlying autoimmune disease, systemic inflammatory disease, or infection is necessary. More potent immunosuppressive therapy may be required, not only in pericarditis associated with systemic autoimmune or inflammatory conditions, but even in idiopathic recurrent pericarditis, either to control symptoms or to mitigate the effects of corticosteroids.
SYSTEMIC INFLAMMATION
The true prevalence of pericardial disease in most systemic inflammatory and autoimmune diseases is difficult to determine from current data. But advances in serologic testing and imaging techniques have shown cardiac involvement in a number of inflammatory diseases.9
In one study, a serologic autoimmune workup in patients with acute pericarditis found that 2% had collagen vascular disease.9 Pericardial involvement is likely in systemic lupus erythematosus,10 and a postmortem study of patients with systemic sclerosis found that 72% had pericarditis.11 Mixed connective tissue disease has been associated with pericarditis in 29% of cases and 56% in autopsy studies.12,13 Pericarditis may be the initial manifestation of vasculitis—eg, Takayasu arteritis or granulomatosis with polyangiitis (formerly known as Wegener granulomatosis).
Other diseases with pericardial involvement include Still disease, Sjögren syndrome, sarcoidosis, and inflammatory bowel disease. Symptomatic pericarditis occurs in about 25% of patients with Sjögren syndrome and asymptomatic pericardial involvement in more than half. Autopsy studies reported pericardial involvement in up to 80% of patients with systemic lupus erythematosus. Cardiac tamponade occurs in fewer than 2%, and constrictive pericarditis is extremely rare.5,9–11
RECOMMENDATIONS
Patients with a first episode of pericarditis should be treated with an anti-inflammatory medication, with no comprehensive testing for autoimmune disease. An evaluation for autoimmune and infectious disease should be carried out in patients with fever (temperature > 38°C; 100.4°F), recurrent pericarditis, recurrent large pericardial effusion or tamponade, or night sweats despite conventional medical therapy. Signs of systemic disease such as renal failure, elevated liver enzymes, or skin rash merit further evaluation.
Prospective studies using appropriate serologic testing and imaging are needed to determine the correlation between myopericardial involvement and inflammatory diseases because of increased morbidity and mortality in several of these diseases.
Pericarditis is in most cases a one-time disease simply treated with anti-inflammatory drugs. It requires no extensive workup for systemic inflammatory or autoimmune disease. Further evaluation is required for patients who have recurrent pericarditis resistant to conventional therapy or pericarditis with manifestations of systemic disease.
ACUTE PERICARDITIS
Pericardial disease has different presentations: acute, recurrent, constrictive, effusive-constrictive, and pericardial effusion with or without tamponade. Acute pericarditis is the most common of these and can affect people of all ages. The typical acute manifestations are chest pain (usually pleuritic), a pericardial friction rub, and widespread ST-segment elevation on the electrocardiogram.1,2 The chest pain tends to be sharp and long-lasting; it radiates to the trapezius ridge and increases during respiration or body movements.
Acute pericarditis usually responds to an anti-inflammatory drug such as colchicine 0.6 mg/day for 3 months, a nonsteroidal anti-inflammatory drug such as ibuprofen 600 mg three times a day for 10 days, and in advanced resistant cases, an oral corticosteroid.3,4
Most often, pericarditis is either idiopathic or occurs after a respiratory viral illness. Much less common causes include bacterial infection, postpericardiotomy syndrome, myocardial infarction, primary or metastatic tumors, trauma, radiation, and uremia. However, pericarditis can also be part of the presentation of systemic inflammatory and autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus; hereditary periodic fever syndromes such as familial Mediterranean fever; and systemic-onset juvenile idiopathic arthritis.1,5
In acute pericarditis, a complex workup is usually not justified, since the results will have limited usefulness in the clinical management of the patient. It is most often diagnosed by the presenting symptoms, auscultation, electrocardiography, echocardiography, and chest radiography, and by additional basic tests that include a complete blood cell count, complete metabolic profile, erythrocyte sedimentation rate, and C-reactive protein level. However, if pericarditis does not respond to anti-inflammatory treatment and if an autoimmune or infectious disease is suspected, further evaluation may include antinuclear antibody testing and testing for human immunodeficiency virus and tuberculosis. If the diagnosis of acute pericarditis remains uncertain, cardiac magnetic resonance imaging (MRI) may be useful.
RECURRENT PERICARDITIS
Although acute pericarditis most often has a benign course and responds well to anti-inflammatory drugs, 20% to 30% of patients who have a first attack of acute pericarditis have a recurrence, and up to 50% of patients who have one recurrence will have another.3,4
Disease activity can be followed with serial testing of inflammatory markers—eg, erythrocyte sedimentation rate and C-reactive protein level. Echocardiography, cardiac computed tomography, and cardiac MRI can characterize active inflammation, edema, pericardial thickness, and pericardial effusion.6–8
Recurrent pericarditis is often resistant to standard therapy and requires corticosteroids in high doses, which paradoxically can increase the risk of recurrence. Therefore, further workup for underlying autoimmune disease, systemic inflammatory disease, or infection is necessary. More potent immunosuppressive therapy may be required, not only in pericarditis associated with systemic autoimmune or inflammatory conditions, but even in idiopathic recurrent pericarditis, either to control symptoms or to mitigate the effects of corticosteroids.
SYSTEMIC INFLAMMATION
The true prevalence of pericardial disease in most systemic inflammatory and autoimmune diseases is difficult to determine from current data. But advances in serologic testing and imaging techniques have shown cardiac involvement in a number of inflammatory diseases.9
In one study, a serologic autoimmune workup in patients with acute pericarditis found that 2% had collagen vascular disease.9 Pericardial involvement is likely in systemic lupus erythematosus,10 and a postmortem study of patients with systemic sclerosis found that 72% had pericarditis.11 Mixed connective tissue disease has been associated with pericarditis in 29% of cases and 56% in autopsy studies.12,13 Pericarditis may be the initial manifestation of vasculitis—eg, Takayasu arteritis or granulomatosis with polyangiitis (formerly known as Wegener granulomatosis).
Other diseases with pericardial involvement include Still disease, Sjögren syndrome, sarcoidosis, and inflammatory bowel disease. Symptomatic pericarditis occurs in about 25% of patients with Sjögren syndrome and asymptomatic pericardial involvement in more than half. Autopsy studies reported pericardial involvement in up to 80% of patients with systemic lupus erythematosus. Cardiac tamponade occurs in fewer than 2%, and constrictive pericarditis is extremely rare.5,9–11
RECOMMENDATIONS
Patients with a first episode of pericarditis should be treated with an anti-inflammatory medication, with no comprehensive testing for autoimmune disease. An evaluation for autoimmune and infectious disease should be carried out in patients with fever (temperature > 38°C; 100.4°F), recurrent pericarditis, recurrent large pericardial effusion or tamponade, or night sweats despite conventional medical therapy. Signs of systemic disease such as renal failure, elevated liver enzymes, or skin rash merit further evaluation.
Prospective studies using appropriate serologic testing and imaging are needed to determine the correlation between myopericardial involvement and inflammatory diseases because of increased morbidity and mortality in several of these diseases.
- Troughton RW, Asher CR, Klein AL. Pericarditis. Lancet 2004; 363:717–727.
- Alraies MC, Klein AL. Should we still use electrocardiography to diagnose pericardial disease? Cleve Clin J Med 2013; 80:97–100.
- Imazio M, Brucato A, Cemin R, et al; ICAP Investigators. A randomized trial of colchicine for acute pericarditis. N Engl J Med 2013; 369:1522–1528.
- Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation 2007; 115:2739–2744.
- Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol 1995; 75:378–382.
- Verhaert D, Gabriel RS, Johnston D, Lytle BW, Desai MY, Klein AL. The role of multimodality imaging in the management of pericardial disease. Circ Cardiovasc Imaging 2010; 3:333–343.
- Klein AL, Abbara S, Agler DA, et al. American Society of Echocardiography clinical recommendations for multimodality cardiovascular imaging of patients with pericardial disease: endorsed by the Society for Cardiovascular Magnetic Resonance and Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr 2013; 26:965–1012.e15.
- Yingchoncharoen T, Alraies MC, Kwon DH, Rodriguez ER, Tan CD, Klein AL. Emerging role of multimodality imaging in management of inflammatory pericardial diseases. Expert Rev Cardiovasc Ther 2013; 11:1211–1225.
- Knockaert DC. Cardiac involvement in systemic inflammatory diseases. Eur Heart J 2007; 28:1797–1804.
- Doria A, Iaccarino L, Sarzi-Puttini P, Atzeni F, Turriel M, Petri M. Cardiac involvement in systemic lupus erythematosus. Lupus 2005; 14:683–686.
- Byers RJ, Marshall DA, Freemont AJ. Pericardial involvement in systemic sclerosis. Ann Rheum Dis 1997; 56:393–394.
- Kasukawa R. Mixed connective tissue disease. Intern Med 1999; 38:386–393.
- Bezerra MC, Saraiva F Jr, Carvalho JF, Caleiro MT, Goncalves CR, Borba EF. Cardiac tamponade due to massive pericardial effusion in mixed connective tissue disease: reversal with steroid therapy. Lupus 2004; 13:618–620.
- Troughton RW, Asher CR, Klein AL. Pericarditis. Lancet 2004; 363:717–727.
- Alraies MC, Klein AL. Should we still use electrocardiography to diagnose pericardial disease? Cleve Clin J Med 2013; 80:97–100.
- Imazio M, Brucato A, Cemin R, et al; ICAP Investigators. A randomized trial of colchicine for acute pericarditis. N Engl J Med 2013; 369:1522–1528.
- Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation 2007; 115:2739–2744.
- Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol 1995; 75:378–382.
- Verhaert D, Gabriel RS, Johnston D, Lytle BW, Desai MY, Klein AL. The role of multimodality imaging in the management of pericardial disease. Circ Cardiovasc Imaging 2010; 3:333–343.
- Klein AL, Abbara S, Agler DA, et al. American Society of Echocardiography clinical recommendations for multimodality cardiovascular imaging of patients with pericardial disease: endorsed by the Society for Cardiovascular Magnetic Resonance and Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr 2013; 26:965–1012.e15.
- Yingchoncharoen T, Alraies MC, Kwon DH, Rodriguez ER, Tan CD, Klein AL. Emerging role of multimodality imaging in management of inflammatory pericardial diseases. Expert Rev Cardiovasc Ther 2013; 11:1211–1225.
- Knockaert DC. Cardiac involvement in systemic inflammatory diseases. Eur Heart J 2007; 28:1797–1804.
- Doria A, Iaccarino L, Sarzi-Puttini P, Atzeni F, Turriel M, Petri M. Cardiac involvement in systemic lupus erythematosus. Lupus 2005; 14:683–686.
- Byers RJ, Marshall DA, Freemont AJ. Pericardial involvement in systemic sclerosis. Ann Rheum Dis 1997; 56:393–394.
- Kasukawa R. Mixed connective tissue disease. Intern Med 1999; 38:386–393.
- Bezerra MC, Saraiva F Jr, Carvalho JF, Caleiro MT, Goncalves CR, Borba EF. Cardiac tamponade due to massive pericardial effusion in mixed connective tissue disease: reversal with steroid therapy. Lupus 2004; 13:618–620.
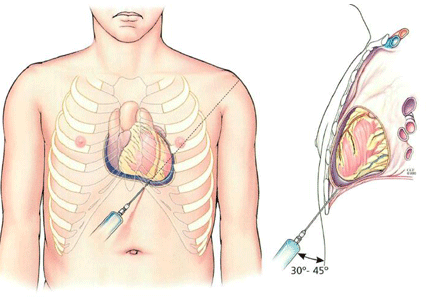
Can the test for human papillomavirus DNA be used as the stand-alone, first-line screening test for cervical cancer?
Yes. Growing evidence demonstrates that the human papillomavirus (HPV) DNA test is more sensitive than the Papanicolaou (Pap) test, with a better negative predictive value—ie, women who have negative test results can be more certain that they are truly free of cervical cancer.1–3
On April 24, 2014, the US Food and Drug Administration (FDA) approved the Cobas HPV test developed by Roche for use as the first-line screening test for cervical cancer in women age 25 and older.4 The approval follows the unanimous recommendation from an independent panel of experts, the Microbiology Devices Panel of the FDA’s Medical Devices Advisory Committee, on March 12, 2014.
PAP-HPV COTESTING IS EFFECTIVE BUT NOT PERFECT
Based on conclusive evidence of a direct link between HPV infection (specifically, infection with certain high-risk HPV genotypes) and almost all cases of invasive cervical cancer,5,6 the American Cancer Society (ACS), American Society for Colposcopy and Cervical Pathology (ASCCP), American Society for Clinical Pathology (ASCP), US Preventive Services Task Force (USPSTF), and American Congress of Obstetricians and Gynecologists (ACOG) issued a consensus recommendation for Pap-HPV cotesting as the preferred screening strategy starting at age 30 and continuing through age 65.7–9
Compared with Pap testing alone, cotesting offers improved detection of cervical intraepithelial neoplasia grade 2 or worse (CIN2+) and the ability to safely extend the screening interval to every 5 years in women who have negative results on both tests. It is an effective screening strategy and remains the standard of care today.
However, this strategy is not perfect and presents several problems for clinicians. The results of the two tests often conflict—the results of the Pap test might be positive while those of the HPV test are negative, or vice versa. Integrating the results of cotesting into triaging can be confusing and complicated. In addition, performing two tests on all women increases the cost of care. And furthermore, the cotesting strategy increases the number of women who require immediate or short-term follow-up,1,2,10–12 such as colposcopy, which is unnecessary for many.
THE HPV TEST DETECTS 14 HIGH-RISK GENOTYPES
The FDA-approved HPV test detects 14 high-risk genotypes. The results for 12 of these are pooled and reported collectively as either positive or negative, while the other two—HPV 16 and HPV 18—are reported separately. (HPV 16 and HPV 18 are the highest-risk genotypes, and together they account for more than two-thirds of cases of invasive cervical cancer.)
ADVANTAGES OF HPV-ONLY TESTING: FINDINGS FROM THE ATHENA TRIAL
The FDA’s decision to approve the Cobas HPV test for use by itself for screening was based on the landmark ATHENA (Addressing the Need for Advanced HPV Diagnostics) trial.13 ATHENA, the largest prospective study of cervical cancer screening performed in the United States to date, enrolled 47,208 women at 61 sites in 23 states. The study revealed the following findings:
- The HPV DNA test had higher sensitivity for detecting CIN3+ (37% higher than the Pap test) and equivalent specificity.
- The HPV test’s positive predictive value was nearly twice as high (12.25% vs 6.47%), and it had a higher negative predictive value (99.58% vs 99.41%) in detecting CIN3+ than with the Pap test.
- HPV testing by itself performed better than Pap-HPV cotesting, with positive predictive values of 12.25% vs 11.04% and negative predictive values of 99.58% vs 99.52% (data presented to the FDA Medical Devices Advisory Committee, Microbiology Panel. March 12, 2014. FDA Executive Summary).
For women whose results were negative for HPV 16 and 18 but positive for the 12-genotype pooled panel, the sample was automatically submitted for cytologic (Pap) testing. Reserving Pap testing for samples in this category improved the specificity of the test and resulted in fewer colposcopy referrals. The ATHENA researchers found that 11.4% of the participants who tested positive for either HPV 16 or 18 had CIN2+.13 Other large cohort studies14,15 also showed that the short-term risk of developing CIN3+ reached 10% over 1 to 5 years in women who tested positive for HPV 16 or 18.
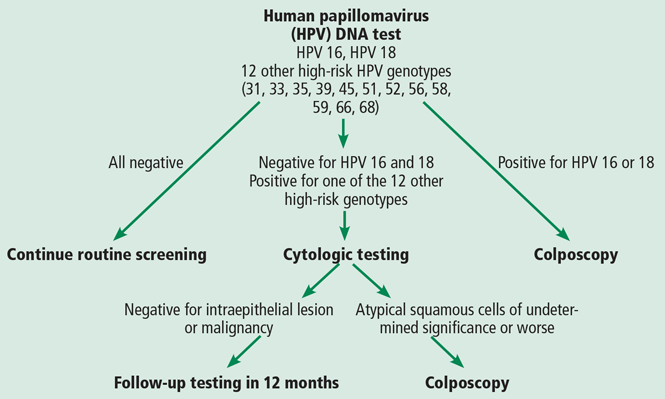
The proposed algorithm for screening (Figure 1) takes advantage of the superior sensitivity of the HPV test, the built-in risk stratification of HPV 16 and 18 genotyping, and the excellent specificity of the Pap test in triaging women whose results are positive for high-risk HPV genotypes other than HPV 16 and 18. Thus, women who have a negative HPV test result can be assured of remaining disease-free for 3 years. The algorithm also identifies women who are at highest risk, ie, those who test positive for HPV 16 or 18. In contrast, the current cotesting approach uses the Qiagen Hybrid Capture HPV testing system, which is a panel of 13 high-risk genotypes, but, if the result is positive, it does not tell you which one the patient has. Furthermore, the new algorithm provides efficient triage, using the Pap test, for women who test positive for the 12 other high-risk HPV genotypes.
Data from large clinical trials other than ATHENA are limited.
FDA APPROVAL DOES NOT CHANGE THE GUIDELINES—YET
The cervical cancer screening guidelines are developed by several organizations other than the FDA. The current guidelines issued by the ACS, ASCCP, ASCP, USPSTF, and ACOG in 2012 call for Pap testing every 3 years in women younger than 30 and Pap-HPV cotesting every 5 years in women ages 30 to 65.7–9 However, FDA approval of the new indication of the HPV DNA test as a stand-alone first-line screening test is an important milestone. It heralds the shifting of the practice paradigm from morphologically based Pap testing to molecular testing in cervical cancer screening.
The ACS and ASCCP have announced that they are reviewing the evidence and may issue updated guidelines for clinicians in the near future.16,17 We anticipate that other organizations may take similar steps. As primary care physicians, we need to stay tuned and follow the most up-to-date evidence-based practice guidelines to provide the best care for our patients.
- Katki HA, Kinney WK, Fetterman B, et al. Cervical cancer risk for women undergoing concurrent testing for human papillomavirus and cervical cytology: a population-based study in routine clinical practice. Lancet Oncol 2011; 12:663–672.
- Ronco G, Giorgi-Rossi P, Carozzi F, et al; New Technologies for Cervical Cancer screening (NTCC) Working Group. Efficacy of human papillomavirus testing for the detection of invasive cervical cancers and cervical intraepithelial neoplasia: a randomized controlled trial. Lancet Oncol 2010; 11:249–257.
- Dillner J, Rebolj M, Birembaut P, et al; Joint European Cohort Study. Long term predictive values of cytology and human papillomavirus testing in cervical cancer screening: joint European cohort study. BMJ 2008; 337:a1754.
- US Food and Drug Administration. FDA approves first human papillomavirus test for primary cervical cancer screening. www.fda.gov/newsevents/newsroom/pressannouncements/ucm394773.htm. Accessed March 3, 2015.
- Muñoz N, Castellsagué X, de González AB, Gissmann L. Chapter 1: HPV in the etiology of human cancer. Vaccine 2006; 24(suppl 3):S3/1–S3/10.
- Walboomers JM, Jacobs MV, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 1999; 189:12–19.
- Saslow D, Solomon D, Lawson HW, et al; American Cancer Society; American Society for Colposcopy and Cervical Pathology; American Society for Clinical Pathology. American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer. Am J Clin Pathol 2012; 137:516–542.
- Moyer VA; US Preventive Services Task Force. Screening for cervical cancer: US Preventive Services Task Force recommendation statement. Ann Intern Med 2012; 156:880–891.
- Committee on Practice Bulletins—Gynecology. ACOG practice bulletin number 131: screening for cervical cancer. Obstet Gynecol 2012; 120:1222–1238.
- Castle PE, Stoler MH, Wright TC Jr, Sharma A, Wright TL, Behrens CM. Performance of carcinogenic human papillomavirus (HPV) testing and HPV16 or HPV18 genotyping for cervical cancer screening of women aged 25 years and older: a subanalysis of the ATHENA study. Lancet Oncol 2011; 12:880–890.
- Kitchener HC, Almonte M, Thomson C, et al. HPV testing in combination with liquid-based cytology in primary cervical screening (ARTISTIC): a randomized controlled trial. Lancet Oncol 2009; 10:672–682.
- Naucler P, Ryd W, Tornberg S, et al. Efficacy of HPV DNA testing with cytology triage and/or repeat HPV DNA testing in primary cervical cancer screening. J Natl Cancer Inst 2009; 101:88–99.
- Wright TC Jr, Stoler MH, Sharma A, Zhang G, Behrens C, Wright TL; ATHENA (Addressing The Need for Advanced HPV Diagnostics) Study Group. Evaluation of HPV-16 and HPV-18 genotyping for the triage of women with high-risk HPV+ cytology-negative results. Am J Clin Pathol 2011; 136:578–586.
- Kjaer SK, Frederiksen K, Munk C, Iftner T. Long-term absolute risk of cervical intraepithelial neoplasia grade 3 or worse following human papillomavirus infection: role of persistence. J Natl Cancer Inst 2010; 102:1478–1488.
- Khan MJ, Castle PE, Lorincz AT, et al. The elevated 10-year risk of cervical precancer and cancer in women with human papillomavirus (HPV) type 16 or 18 and the possible utility of type-specific HPV testing in clinical practice. J Natl Cancer Inst 2005; 97:1072–1079.
- American Cancer Society. FDA approves HPV test as first line screening for cervical cancer. www.cancer.org/cancer/news/fda-approves-hpv-test-as-first-line-screening-for-cervical-cancer. Accessed March 3, 2015.
- American Society for Colposcopy and Cervical Pathology. Medical societies recommend consideration of primary HPV testing for cervical cancer screening. www.asccp.org/About-ASCCP/News-Announcements. Accessed March 3, 2015.
Yes. Growing evidence demonstrates that the human papillomavirus (HPV) DNA test is more sensitive than the Papanicolaou (Pap) test, with a better negative predictive value—ie, women who have negative test results can be more certain that they are truly free of cervical cancer.1–3
On April 24, 2014, the US Food and Drug Administration (FDA) approved the Cobas HPV test developed by Roche for use as the first-line screening test for cervical cancer in women age 25 and older.4 The approval follows the unanimous recommendation from an independent panel of experts, the Microbiology Devices Panel of the FDA’s Medical Devices Advisory Committee, on March 12, 2014.
PAP-HPV COTESTING IS EFFECTIVE BUT NOT PERFECT
Based on conclusive evidence of a direct link between HPV infection (specifically, infection with certain high-risk HPV genotypes) and almost all cases of invasive cervical cancer,5,6 the American Cancer Society (ACS), American Society for Colposcopy and Cervical Pathology (ASCCP), American Society for Clinical Pathology (ASCP), US Preventive Services Task Force (USPSTF), and American Congress of Obstetricians and Gynecologists (ACOG) issued a consensus recommendation for Pap-HPV cotesting as the preferred screening strategy starting at age 30 and continuing through age 65.7–9
Compared with Pap testing alone, cotesting offers improved detection of cervical intraepithelial neoplasia grade 2 or worse (CIN2+) and the ability to safely extend the screening interval to every 5 years in women who have negative results on both tests. It is an effective screening strategy and remains the standard of care today.
However, this strategy is not perfect and presents several problems for clinicians. The results of the two tests often conflict—the results of the Pap test might be positive while those of the HPV test are negative, or vice versa. Integrating the results of cotesting into triaging can be confusing and complicated. In addition, performing two tests on all women increases the cost of care. And furthermore, the cotesting strategy increases the number of women who require immediate or short-term follow-up,1,2,10–12 such as colposcopy, which is unnecessary for many.
THE HPV TEST DETECTS 14 HIGH-RISK GENOTYPES
The FDA-approved HPV test detects 14 high-risk genotypes. The results for 12 of these are pooled and reported collectively as either positive or negative, while the other two—HPV 16 and HPV 18—are reported separately. (HPV 16 and HPV 18 are the highest-risk genotypes, and together they account for more than two-thirds of cases of invasive cervical cancer.)
ADVANTAGES OF HPV-ONLY TESTING: FINDINGS FROM THE ATHENA TRIAL
The FDA’s decision to approve the Cobas HPV test for use by itself for screening was based on the landmark ATHENA (Addressing the Need for Advanced HPV Diagnostics) trial.13 ATHENA, the largest prospective study of cervical cancer screening performed in the United States to date, enrolled 47,208 women at 61 sites in 23 states. The study revealed the following findings:
- The HPV DNA test had higher sensitivity for detecting CIN3+ (37% higher than the Pap test) and equivalent specificity.
- The HPV test’s positive predictive value was nearly twice as high (12.25% vs 6.47%), and it had a higher negative predictive value (99.58% vs 99.41%) in detecting CIN3+ than with the Pap test.
- HPV testing by itself performed better than Pap-HPV cotesting, with positive predictive values of 12.25% vs 11.04% and negative predictive values of 99.58% vs 99.52% (data presented to the FDA Medical Devices Advisory Committee, Microbiology Panel. March 12, 2014. FDA Executive Summary).
For women whose results were negative for HPV 16 and 18 but positive for the 12-genotype pooled panel, the sample was automatically submitted for cytologic (Pap) testing. Reserving Pap testing for samples in this category improved the specificity of the test and resulted in fewer colposcopy referrals. The ATHENA researchers found that 11.4% of the participants who tested positive for either HPV 16 or 18 had CIN2+.13 Other large cohort studies14,15 also showed that the short-term risk of developing CIN3+ reached 10% over 1 to 5 years in women who tested positive for HPV 16 or 18.
The proposed algorithm for screening (Figure 1) takes advantage of the superior sensitivity of the HPV test, the built-in risk stratification of HPV 16 and 18 genotyping, and the excellent specificity of the Pap test in triaging women whose results are positive for high-risk HPV genotypes other than HPV 16 and 18. Thus, women who have a negative HPV test result can be assured of remaining disease-free for 3 years. The algorithm also identifies women who are at highest risk, ie, those who test positive for HPV 16 or 18. In contrast, the current cotesting approach uses the Qiagen Hybrid Capture HPV testing system, which is a panel of 13 high-risk genotypes, but, if the result is positive, it does not tell you which one the patient has. Furthermore, the new algorithm provides efficient triage, using the Pap test, for women who test positive for the 12 other high-risk HPV genotypes.
Data from large clinical trials other than ATHENA are limited.
FDA APPROVAL DOES NOT CHANGE THE GUIDELINES—YET
The cervical cancer screening guidelines are developed by several organizations other than the FDA. The current guidelines issued by the ACS, ASCCP, ASCP, USPSTF, and ACOG in 2012 call for Pap testing every 3 years in women younger than 30 and Pap-HPV cotesting every 5 years in women ages 30 to 65.7–9 However, FDA approval of the new indication of the HPV DNA test as a stand-alone first-line screening test is an important milestone. It heralds the shifting of the practice paradigm from morphologically based Pap testing to molecular testing in cervical cancer screening.
The ACS and ASCCP have announced that they are reviewing the evidence and may issue updated guidelines for clinicians in the near future.16,17 We anticipate that other organizations may take similar steps. As primary care physicians, we need to stay tuned and follow the most up-to-date evidence-based practice guidelines to provide the best care for our patients.
Yes. Growing evidence demonstrates that the human papillomavirus (HPV) DNA test is more sensitive than the Papanicolaou (Pap) test, with a better negative predictive value—ie, women who have negative test results can be more certain that they are truly free of cervical cancer.1–3
On April 24, 2014, the US Food and Drug Administration (FDA) approved the Cobas HPV test developed by Roche for use as the first-line screening test for cervical cancer in women age 25 and older.4 The approval follows the unanimous recommendation from an independent panel of experts, the Microbiology Devices Panel of the FDA’s Medical Devices Advisory Committee, on March 12, 2014.
PAP-HPV COTESTING IS EFFECTIVE BUT NOT PERFECT
Based on conclusive evidence of a direct link between HPV infection (specifically, infection with certain high-risk HPV genotypes) and almost all cases of invasive cervical cancer,5,6 the American Cancer Society (ACS), American Society for Colposcopy and Cervical Pathology (ASCCP), American Society for Clinical Pathology (ASCP), US Preventive Services Task Force (USPSTF), and American Congress of Obstetricians and Gynecologists (ACOG) issued a consensus recommendation for Pap-HPV cotesting as the preferred screening strategy starting at age 30 and continuing through age 65.7–9
Compared with Pap testing alone, cotesting offers improved detection of cervical intraepithelial neoplasia grade 2 or worse (CIN2+) and the ability to safely extend the screening interval to every 5 years in women who have negative results on both tests. It is an effective screening strategy and remains the standard of care today.
However, this strategy is not perfect and presents several problems for clinicians. The results of the two tests often conflict—the results of the Pap test might be positive while those of the HPV test are negative, or vice versa. Integrating the results of cotesting into triaging can be confusing and complicated. In addition, performing two tests on all women increases the cost of care. And furthermore, the cotesting strategy increases the number of women who require immediate or short-term follow-up,1,2,10–12 such as colposcopy, which is unnecessary for many.
THE HPV TEST DETECTS 14 HIGH-RISK GENOTYPES
The FDA-approved HPV test detects 14 high-risk genotypes. The results for 12 of these are pooled and reported collectively as either positive or negative, while the other two—HPV 16 and HPV 18—are reported separately. (HPV 16 and HPV 18 are the highest-risk genotypes, and together they account for more than two-thirds of cases of invasive cervical cancer.)
ADVANTAGES OF HPV-ONLY TESTING: FINDINGS FROM THE ATHENA TRIAL
The FDA’s decision to approve the Cobas HPV test for use by itself for screening was based on the landmark ATHENA (Addressing the Need for Advanced HPV Diagnostics) trial.13 ATHENA, the largest prospective study of cervical cancer screening performed in the United States to date, enrolled 47,208 women at 61 sites in 23 states. The study revealed the following findings:
- The HPV DNA test had higher sensitivity for detecting CIN3+ (37% higher than the Pap test) and equivalent specificity.
- The HPV test’s positive predictive value was nearly twice as high (12.25% vs 6.47%), and it had a higher negative predictive value (99.58% vs 99.41%) in detecting CIN3+ than with the Pap test.
- HPV testing by itself performed better than Pap-HPV cotesting, with positive predictive values of 12.25% vs 11.04% and negative predictive values of 99.58% vs 99.52% (data presented to the FDA Medical Devices Advisory Committee, Microbiology Panel. March 12, 2014. FDA Executive Summary).
For women whose results were negative for HPV 16 and 18 but positive for the 12-genotype pooled panel, the sample was automatically submitted for cytologic (Pap) testing. Reserving Pap testing for samples in this category improved the specificity of the test and resulted in fewer colposcopy referrals. The ATHENA researchers found that 11.4% of the participants who tested positive for either HPV 16 or 18 had CIN2+.13 Other large cohort studies14,15 also showed that the short-term risk of developing CIN3+ reached 10% over 1 to 5 years in women who tested positive for HPV 16 or 18.
The proposed algorithm for screening (Figure 1) takes advantage of the superior sensitivity of the HPV test, the built-in risk stratification of HPV 16 and 18 genotyping, and the excellent specificity of the Pap test in triaging women whose results are positive for high-risk HPV genotypes other than HPV 16 and 18. Thus, women who have a negative HPV test result can be assured of remaining disease-free for 3 years. The algorithm also identifies women who are at highest risk, ie, those who test positive for HPV 16 or 18. In contrast, the current cotesting approach uses the Qiagen Hybrid Capture HPV testing system, which is a panel of 13 high-risk genotypes, but, if the result is positive, it does not tell you which one the patient has. Furthermore, the new algorithm provides efficient triage, using the Pap test, for women who test positive for the 12 other high-risk HPV genotypes.
Data from large clinical trials other than ATHENA are limited.
FDA APPROVAL DOES NOT CHANGE THE GUIDELINES—YET
The cervical cancer screening guidelines are developed by several organizations other than the FDA. The current guidelines issued by the ACS, ASCCP, ASCP, USPSTF, and ACOG in 2012 call for Pap testing every 3 years in women younger than 30 and Pap-HPV cotesting every 5 years in women ages 30 to 65.7–9 However, FDA approval of the new indication of the HPV DNA test as a stand-alone first-line screening test is an important milestone. It heralds the shifting of the practice paradigm from morphologically based Pap testing to molecular testing in cervical cancer screening.
The ACS and ASCCP have announced that they are reviewing the evidence and may issue updated guidelines for clinicians in the near future.16,17 We anticipate that other organizations may take similar steps. As primary care physicians, we need to stay tuned and follow the most up-to-date evidence-based practice guidelines to provide the best care for our patients.
- Katki HA, Kinney WK, Fetterman B, et al. Cervical cancer risk for women undergoing concurrent testing for human papillomavirus and cervical cytology: a population-based study in routine clinical practice. Lancet Oncol 2011; 12:663–672.
- Ronco G, Giorgi-Rossi P, Carozzi F, et al; New Technologies for Cervical Cancer screening (NTCC) Working Group. Efficacy of human papillomavirus testing for the detection of invasive cervical cancers and cervical intraepithelial neoplasia: a randomized controlled trial. Lancet Oncol 2010; 11:249–257.
- Dillner J, Rebolj M, Birembaut P, et al; Joint European Cohort Study. Long term predictive values of cytology and human papillomavirus testing in cervical cancer screening: joint European cohort study. BMJ 2008; 337:a1754.
- US Food and Drug Administration. FDA approves first human papillomavirus test for primary cervical cancer screening. www.fda.gov/newsevents/newsroom/pressannouncements/ucm394773.htm. Accessed March 3, 2015.
- Muñoz N, Castellsagué X, de González AB, Gissmann L. Chapter 1: HPV in the etiology of human cancer. Vaccine 2006; 24(suppl 3):S3/1–S3/10.
- Walboomers JM, Jacobs MV, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 1999; 189:12–19.
- Saslow D, Solomon D, Lawson HW, et al; American Cancer Society; American Society for Colposcopy and Cervical Pathology; American Society for Clinical Pathology. American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer. Am J Clin Pathol 2012; 137:516–542.
- Moyer VA; US Preventive Services Task Force. Screening for cervical cancer: US Preventive Services Task Force recommendation statement. Ann Intern Med 2012; 156:880–891.
- Committee on Practice Bulletins—Gynecology. ACOG practice bulletin number 131: screening for cervical cancer. Obstet Gynecol 2012; 120:1222–1238.
- Castle PE, Stoler MH, Wright TC Jr, Sharma A, Wright TL, Behrens CM. Performance of carcinogenic human papillomavirus (HPV) testing and HPV16 or HPV18 genotyping for cervical cancer screening of women aged 25 years and older: a subanalysis of the ATHENA study. Lancet Oncol 2011; 12:880–890.
- Kitchener HC, Almonte M, Thomson C, et al. HPV testing in combination with liquid-based cytology in primary cervical screening (ARTISTIC): a randomized controlled trial. Lancet Oncol 2009; 10:672–682.
- Naucler P, Ryd W, Tornberg S, et al. Efficacy of HPV DNA testing with cytology triage and/or repeat HPV DNA testing in primary cervical cancer screening. J Natl Cancer Inst 2009; 101:88–99.
- Wright TC Jr, Stoler MH, Sharma A, Zhang G, Behrens C, Wright TL; ATHENA (Addressing The Need for Advanced HPV Diagnostics) Study Group. Evaluation of HPV-16 and HPV-18 genotyping for the triage of women with high-risk HPV+ cytology-negative results. Am J Clin Pathol 2011; 136:578–586.
- Kjaer SK, Frederiksen K, Munk C, Iftner T. Long-term absolute risk of cervical intraepithelial neoplasia grade 3 or worse following human papillomavirus infection: role of persistence. J Natl Cancer Inst 2010; 102:1478–1488.
- Khan MJ, Castle PE, Lorincz AT, et al. The elevated 10-year risk of cervical precancer and cancer in women with human papillomavirus (HPV) type 16 or 18 and the possible utility of type-specific HPV testing in clinical practice. J Natl Cancer Inst 2005; 97:1072–1079.
- American Cancer Society. FDA approves HPV test as first line screening for cervical cancer. www.cancer.org/cancer/news/fda-approves-hpv-test-as-first-line-screening-for-cervical-cancer. Accessed March 3, 2015.
- American Society for Colposcopy and Cervical Pathology. Medical societies recommend consideration of primary HPV testing for cervical cancer screening. www.asccp.org/About-ASCCP/News-Announcements. Accessed March 3, 2015.
- Katki HA, Kinney WK, Fetterman B, et al. Cervical cancer risk for women undergoing concurrent testing for human papillomavirus and cervical cytology: a population-based study in routine clinical practice. Lancet Oncol 2011; 12:663–672.
- Ronco G, Giorgi-Rossi P, Carozzi F, et al; New Technologies for Cervical Cancer screening (NTCC) Working Group. Efficacy of human papillomavirus testing for the detection of invasive cervical cancers and cervical intraepithelial neoplasia: a randomized controlled trial. Lancet Oncol 2010; 11:249–257.
- Dillner J, Rebolj M, Birembaut P, et al; Joint European Cohort Study. Long term predictive values of cytology and human papillomavirus testing in cervical cancer screening: joint European cohort study. BMJ 2008; 337:a1754.
- US Food and Drug Administration. FDA approves first human papillomavirus test for primary cervical cancer screening. www.fda.gov/newsevents/newsroom/pressannouncements/ucm394773.htm. Accessed March 3, 2015.
- Muñoz N, Castellsagué X, de González AB, Gissmann L. Chapter 1: HPV in the etiology of human cancer. Vaccine 2006; 24(suppl 3):S3/1–S3/10.
- Walboomers JM, Jacobs MV, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 1999; 189:12–19.
- Saslow D, Solomon D, Lawson HW, et al; American Cancer Society; American Society for Colposcopy and Cervical Pathology; American Society for Clinical Pathology. American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer. Am J Clin Pathol 2012; 137:516–542.
- Moyer VA; US Preventive Services Task Force. Screening for cervical cancer: US Preventive Services Task Force recommendation statement. Ann Intern Med 2012; 156:880–891.
- Committee on Practice Bulletins—Gynecology. ACOG practice bulletin number 131: screening for cervical cancer. Obstet Gynecol 2012; 120:1222–1238.
- Castle PE, Stoler MH, Wright TC Jr, Sharma A, Wright TL, Behrens CM. Performance of carcinogenic human papillomavirus (HPV) testing and HPV16 or HPV18 genotyping for cervical cancer screening of women aged 25 years and older: a subanalysis of the ATHENA study. Lancet Oncol 2011; 12:880–890.
- Kitchener HC, Almonte M, Thomson C, et al. HPV testing in combination with liquid-based cytology in primary cervical screening (ARTISTIC): a randomized controlled trial. Lancet Oncol 2009; 10:672–682.
- Naucler P, Ryd W, Tornberg S, et al. Efficacy of HPV DNA testing with cytology triage and/or repeat HPV DNA testing in primary cervical cancer screening. J Natl Cancer Inst 2009; 101:88–99.
- Wright TC Jr, Stoler MH, Sharma A, Zhang G, Behrens C, Wright TL; ATHENA (Addressing The Need for Advanced HPV Diagnostics) Study Group. Evaluation of HPV-16 and HPV-18 genotyping for the triage of women with high-risk HPV+ cytology-negative results. Am J Clin Pathol 2011; 136:578–586.
- Kjaer SK, Frederiksen K, Munk C, Iftner T. Long-term absolute risk of cervical intraepithelial neoplasia grade 3 or worse following human papillomavirus infection: role of persistence. J Natl Cancer Inst 2010; 102:1478–1488.
- Khan MJ, Castle PE, Lorincz AT, et al. The elevated 10-year risk of cervical precancer and cancer in women with human papillomavirus (HPV) type 16 or 18 and the possible utility of type-specific HPV testing in clinical practice. J Natl Cancer Inst 2005; 97:1072–1079.
- American Cancer Society. FDA approves HPV test as first line screening for cervical cancer. www.cancer.org/cancer/news/fda-approves-hpv-test-as-first-line-screening-for-cervical-cancer. Accessed March 3, 2015.
- American Society for Colposcopy and Cervical Pathology. Medical societies recommend consideration of primary HPV testing for cervical cancer screening. www.asccp.org/About-ASCCP/News-Announcements. Accessed March 3, 2015.
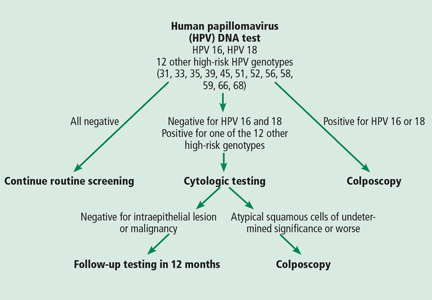
Should we be concerned about thyroid cancer in patients taking glucagon-like peptide 1 receptor agonists?
The question is complicated, as there are different types of thyroid cancer, and a causal relationship is hard to prove.
Glucagon-like peptide 1 (GLP-1) receptor agonists can be safely used in all patients with thyroid cancers that are derived from the thyroid follicular epithelium (papillary and follicular thyroid cancer). However, they are currently contraindicated in patients with medullary thyroid cancer and in patients with multiple endocrine neoplasia 2 (MEN-2), which is not a form of thyroid cancer but is relevant to our discussion. We probably should be cautious about using them in patients with familial thyroid cancer and those with a genetic predisposition for papillary or follicular thyroid cancer.
GLP-1 DRUGS ARE WIDELY USED
The glucagon-like peptide 1 (GLP-1) receptor agonists are widely used to treat type 2 diabetes mellitus. The currently available drugs of this class—exenatide (Byetta), liraglutide (Victoza), albiglutide (Tanzeum), dulaglutide (Trulicity), and extended-release exenatide (Bydureon)—are popular because they lower glucose levels, pose a low risk of hypoglycemia, can induce weight loss,1 and, in the case of extended-release exenatide and albiglutide, are given once weekly. They are currently recommended as add-on therapy to metformin. These drugs mimic the action of GLP-1, an endogenous hormone released by the intestine in response to food. They bind to receptors on beta cells, stimulating insulin production.1
FOUR TYPES OF THYROID CANCER
There are four types of thyroid cancer: medullary (a contraindication to GLP-1 agonists), papillary, follicular, and anaplastic.
Medullary thyroid cancer is extremely rare in humans, with 976 cases diagnosed from 1992 to 2006 in the United States, compared with 36,583 cases of papillary and 4,560 cases of follicular cancer. Anaplastic cancer is also rare (556 cases).2 The highest incidence rates of medullary thyroid cancer are in people of Hispanic descent (0.21 per 100,000 woman-years and 0.18 per 100,000 man-years).2
EXPERIMENTAL EVIDENCE
Pancreatic beta cells are not the only cells in the body that can express GLP-1 receptors. Notably, the parafollicular cells (also called C cells) of the thyroid, which secrete calcitonin and which are the cells involved in medullary thyroid cancer, also sometimes express these receptors if cancer develops.
In experiments in mice and rats, the incidence of thyroid C-cell tumors was higher in animals given GLP-1 analogues. Liraglutide, exenatide, taspoglutide, and lixisenatide potently activated GLP-1 receptors in thyroid C cells, increasing calcitonin gene expression and stimulating calcitonin release in a dose-dependent manner.3 Moreover, sustained activation of these receptors caused C-cell hyperplasia and resulted in medullary thyroid cancer. However medullary thyroid cancer also occurred in rodents receiving placebo.
C cells in monkeys and humans express fewer GLP-1 receptors than those in rodents; in fact, healthy human C cells do not express them at all.3,4 In rats with C-cell hyperplasia or medullary thyroid cancer, GLP-1 receptors are present in 100% of cases (and in increased density), compared with 27% of human medullary thyroid cancers.4
In addition to medullary thyroid cancer, various other human tumors have been shown to express GLP-1 receptors.5 Based on limited data, KÖrner et al5 found that these receptors are also present in various other human tumors, eg:
- Pheochromocytoma (60%)
- Paraganglioma (28%)
- Meningioma (35%)
- Astrocytoma (25%)
- Glioblastoma (9%)
- Ependymoma (16%)
- Medulloblastoma (25%)
- Nephroblastoma (22%)
- Neuroblastoma (18%)
- Ovarian adenocarcinoma (16%)
- Prostate carcinoma (5%).
Madsen et al6 reported that liraglutide binding to the GLP-1 receptor on murine thyroid C cells led to C-cell hyperplasia. However, prolonged administration of liraglutide at very high doses did not produce C-cell proliferation in monkeys.3
Gier et al7 looked at GLP-1 receptor expression in normal human C cells, hyperplastic C cells, and medullary thyroid cancer cells, as well as in papillary thyroid cancer cells, which do not arise from C cells. They demonstrated concurrent calcitonin and GLP-1 receptor immunostaining, not only in those with C-cell hyperplasia (9 of 9 cases) and medullary thyroid cancer (11 of 12 cases), but also in 3 (18%) of 17 patients with papillary thyroid cancer and 5 (33%) of 15 with normal thyroid follicular cells. However, the choice of polyclonal antibodies and radioligands used and concerns about methodology have led investigators to interpret these results cautiously.8–10
STUDIES OF GLP-1 AGONISTS IN HUMANS
Several prospective clinical studies showed no increase in calcitonin levels during therapy with GLP-1 receptor agonists in patients with type 2 diabetes.3,11 Long-term use of liraglutide in high doses (up to 3 mg per day) did not lead to elevations in serum calcitonin levels.11
In a retrospective Adverse Event Reporting System database review, the incidence rate of thyroid cancer in patients treated with exenatide was higher—with an odds ratio of 4.7 (30 events)—than with a panel of control drugs (3 events).12 However, this study did not differentiate between types of thyroid cancer, and the inherent limitations of retrospective databases complicate its interpretation. Such a high odds ratio would imply a significant increase in the incidence of medullary thyroid cancer, but this does not seem to be true.
Alves et al13 performed a meta-analysis of randomized controlled trials and long-term observational studies. None of the studies evaluating exenatide reported cases of thyroid cancer, whereas five of the studies evaluating liraglutide did. In total, nine patients treated with liraglutide were diagnosed with thyroid cancer, compared with one patient on glimepiride. The odds ratio for thyroid cancer occurrence associated with liraglutide treatment was 1.54, but that was not statistically significant (95% confidence interval 0.40–6.02, P = .53, I2 = 0%).
These studies are hypothesis-generating and do not prove that GLP-1 receptor agonists cause medullary thyroid cancer. Given the extremely low incidence of medullary thyroid cancer, to prove or disprove a causal relationship would require an enormous number of patients, who would need to be followed for several years.
OFFICIAL RECOMMENDATIONS
Considerable differences in the biology of the rodent vs human thyroid GLP-1 receptor systems have led regulatory authorities to conclude that the risk for development of medullary thyroid cancer with GLP-1 therapy in humans is difficult to quantify, but low.14 Consequently, the US Food and Drug Administration recommends neither monitoring of calcitonin levels nor ultrasound imaging as a screening tool in patients taking GLP-1 agonists.14
BENEFITS OUTWEIGH RISKS
At present, the benefits of using GLP-1 receptor agonists to treat type 2 diabetes mellitus outweigh the risks, and there seems to be little reason to withhold this effective therapy except in patients who have a personal or family history of medullary thyroid cancer or MEN-2. Until the effects of GLP-1 agonists are systematically studied in follicular-cell-derived thyroid cancer, we also recommend caution when considering their use in patients with familial thyroid cancer and those with a genetic predisposition for papillary and follicular thyroid cancer—eg, patients with familial adenomatous polyposis, phosphate and tensin homolog hamartoma tumor syndrome, Carney complex type 1, Werner syndrome, or familial papillary thyroid cancer.
Methodologically superior studies and careful long-term monitoring of patients treated with GLP-1 agonists are required to clarify the risk vs benefit of these therapies.
- Samson SL, Garber A. GLP-1R agonist therapy for diabetes: benefits and potential risks. Curr Opin Endocrinol Diabetes Obes 2013; 20:87–97.
- Aschebrook-Kilfoy B, Ward MH, Sabra MM, Devesa SS. Thyroid cancer incidence patterns in the United States by histologic type, 1992–2006. Thyroid 2011; 21:125–134.
- Bjerre Knudsen L, Madsen LW, Andersen S, et al. Glucagon-like peptide-1 receptor agonists activate rodent thyroid C-cells causing calcitonin release and C-cell proliferation. Endocrinology 2010; 151:1473–1486.
- Waser B, Beetschen K, Pellegata NS, Reubi JC. Incretin receptors in non-neoplastic and neoplastic thyroid C cells in rodents and humans: relevance for incretin-based diabetes therapy. Neuroendocrinology 2011; 94:291–301.
- Körner M, Stöckli M, Waser B, Reubi JC. GLP-1 receptor expression in human tumors and human normal tissues: potential for in vivo targeting. J Nucl Med 2007; 48:736–743.
- Madsen LW, Knauf JA, Gotfredsen C, et al. GLP-1 receptor agonists and the thyroid: C-cell effects in mice are mediated via the GLP-1 receptor and not associated with RET activation. Endocrinology 2012; 153:1538–1547.
- Gier B, Butler PC, Lai CK, Kirakossian D, DeNicola MM, Yeh MW. Glucagon like peptide-1 receptor expression in the human thyroid gland. J Clin Endocrinol Metab 2012; 97:121–131.
- Drucker DJ, Sherman SI, Bergenstal RM, Buse JB. The safety of incretin-based therapies—review of the scientific evidence. J Clin Endocrinol Metab 2011; 96:2027–2031.
- Gagel RF. Activation of G-protein-coupled receptors and thyroid malignant tumors: the jury is still out. Endocr Pract 2011; 17:957–959.
- Nauck MA. A critical analysis of the clinical use of incretin-based therapies: the benefits by far outweigh the potential risks. Diabetes Care 2013; 36:2126–2132.
- Hegedüs L, Moses AC, Zdravkovic M, Le Thi T, Daniels GH. GLP-1 and calcitonin concentration in humans: lack of evidence of calcitonin release from sequential screening in over 5000 subjects with type 2 diabetes or nondiabetic obese subjects treated with the human GLP-1 analog, liraglutide. J Clin Endocrinol Metab 2011; 96:853–860.
- Elashoff M, Matveyenko AV, Gier B, Elashoff R, Butler PC. Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology 2011; 141:150–156.
- Alves C, Batel-Marques F, Macedo AF. A meta-analysis of serious adverse events reported with exenatide and liraglutide: acute pancreatitis and cancer. Diabetes Res Clin Pract 2012; 98:271–284.
- Parks M, Rosebraugh C. Weighing risks and benefits of liraglutide—the FDA’s review of a new antidiabetic therapy. N Engl J Med 2010; 362:774–777.
The question is complicated, as there are different types of thyroid cancer, and a causal relationship is hard to prove.
Glucagon-like peptide 1 (GLP-1) receptor agonists can be safely used in all patients with thyroid cancers that are derived from the thyroid follicular epithelium (papillary and follicular thyroid cancer). However, they are currently contraindicated in patients with medullary thyroid cancer and in patients with multiple endocrine neoplasia 2 (MEN-2), which is not a form of thyroid cancer but is relevant to our discussion. We probably should be cautious about using them in patients with familial thyroid cancer and those with a genetic predisposition for papillary or follicular thyroid cancer.
GLP-1 DRUGS ARE WIDELY USED
The glucagon-like peptide 1 (GLP-1) receptor agonists are widely used to treat type 2 diabetes mellitus. The currently available drugs of this class—exenatide (Byetta), liraglutide (Victoza), albiglutide (Tanzeum), dulaglutide (Trulicity), and extended-release exenatide (Bydureon)—are popular because they lower glucose levels, pose a low risk of hypoglycemia, can induce weight loss,1 and, in the case of extended-release exenatide and albiglutide, are given once weekly. They are currently recommended as add-on therapy to metformin. These drugs mimic the action of GLP-1, an endogenous hormone released by the intestine in response to food. They bind to receptors on beta cells, stimulating insulin production.1
FOUR TYPES OF THYROID CANCER
There are four types of thyroid cancer: medullary (a contraindication to GLP-1 agonists), papillary, follicular, and anaplastic.
Medullary thyroid cancer is extremely rare in humans, with 976 cases diagnosed from 1992 to 2006 in the United States, compared with 36,583 cases of papillary and 4,560 cases of follicular cancer. Anaplastic cancer is also rare (556 cases).2 The highest incidence rates of medullary thyroid cancer are in people of Hispanic descent (0.21 per 100,000 woman-years and 0.18 per 100,000 man-years).2
EXPERIMENTAL EVIDENCE
Pancreatic beta cells are not the only cells in the body that can express GLP-1 receptors. Notably, the parafollicular cells (also called C cells) of the thyroid, which secrete calcitonin and which are the cells involved in medullary thyroid cancer, also sometimes express these receptors if cancer develops.
In experiments in mice and rats, the incidence of thyroid C-cell tumors was higher in animals given GLP-1 analogues. Liraglutide, exenatide, taspoglutide, and lixisenatide potently activated GLP-1 receptors in thyroid C cells, increasing calcitonin gene expression and stimulating calcitonin release in a dose-dependent manner.3 Moreover, sustained activation of these receptors caused C-cell hyperplasia and resulted in medullary thyroid cancer. However medullary thyroid cancer also occurred in rodents receiving placebo.
C cells in monkeys and humans express fewer GLP-1 receptors than those in rodents; in fact, healthy human C cells do not express them at all.3,4 In rats with C-cell hyperplasia or medullary thyroid cancer, GLP-1 receptors are present in 100% of cases (and in increased density), compared with 27% of human medullary thyroid cancers.4
In addition to medullary thyroid cancer, various other human tumors have been shown to express GLP-1 receptors.5 Based on limited data, KÖrner et al5 found that these receptors are also present in various other human tumors, eg:
- Pheochromocytoma (60%)
- Paraganglioma (28%)
- Meningioma (35%)
- Astrocytoma (25%)
- Glioblastoma (9%)
- Ependymoma (16%)
- Medulloblastoma (25%)
- Nephroblastoma (22%)
- Neuroblastoma (18%)
- Ovarian adenocarcinoma (16%)
- Prostate carcinoma (5%).
Madsen et al6 reported that liraglutide binding to the GLP-1 receptor on murine thyroid C cells led to C-cell hyperplasia. However, prolonged administration of liraglutide at very high doses did not produce C-cell proliferation in monkeys.3
Gier et al7 looked at GLP-1 receptor expression in normal human C cells, hyperplastic C cells, and medullary thyroid cancer cells, as well as in papillary thyroid cancer cells, which do not arise from C cells. They demonstrated concurrent calcitonin and GLP-1 receptor immunostaining, not only in those with C-cell hyperplasia (9 of 9 cases) and medullary thyroid cancer (11 of 12 cases), but also in 3 (18%) of 17 patients with papillary thyroid cancer and 5 (33%) of 15 with normal thyroid follicular cells. However, the choice of polyclonal antibodies and radioligands used and concerns about methodology have led investigators to interpret these results cautiously.8–10
STUDIES OF GLP-1 AGONISTS IN HUMANS
Several prospective clinical studies showed no increase in calcitonin levels during therapy with GLP-1 receptor agonists in patients with type 2 diabetes.3,11 Long-term use of liraglutide in high doses (up to 3 mg per day) did not lead to elevations in serum calcitonin levels.11
In a retrospective Adverse Event Reporting System database review, the incidence rate of thyroid cancer in patients treated with exenatide was higher—with an odds ratio of 4.7 (30 events)—than with a panel of control drugs (3 events).12 However, this study did not differentiate between types of thyroid cancer, and the inherent limitations of retrospective databases complicate its interpretation. Such a high odds ratio would imply a significant increase in the incidence of medullary thyroid cancer, but this does not seem to be true.
Alves et al13 performed a meta-analysis of randomized controlled trials and long-term observational studies. None of the studies evaluating exenatide reported cases of thyroid cancer, whereas five of the studies evaluating liraglutide did. In total, nine patients treated with liraglutide were diagnosed with thyroid cancer, compared with one patient on glimepiride. The odds ratio for thyroid cancer occurrence associated with liraglutide treatment was 1.54, but that was not statistically significant (95% confidence interval 0.40–6.02, P = .53, I2 = 0%).
These studies are hypothesis-generating and do not prove that GLP-1 receptor agonists cause medullary thyroid cancer. Given the extremely low incidence of medullary thyroid cancer, to prove or disprove a causal relationship would require an enormous number of patients, who would need to be followed for several years.
OFFICIAL RECOMMENDATIONS
Considerable differences in the biology of the rodent vs human thyroid GLP-1 receptor systems have led regulatory authorities to conclude that the risk for development of medullary thyroid cancer with GLP-1 therapy in humans is difficult to quantify, but low.14 Consequently, the US Food and Drug Administration recommends neither monitoring of calcitonin levels nor ultrasound imaging as a screening tool in patients taking GLP-1 agonists.14
BENEFITS OUTWEIGH RISKS
At present, the benefits of using GLP-1 receptor agonists to treat type 2 diabetes mellitus outweigh the risks, and there seems to be little reason to withhold this effective therapy except in patients who have a personal or family history of medullary thyroid cancer or MEN-2. Until the effects of GLP-1 agonists are systematically studied in follicular-cell-derived thyroid cancer, we also recommend caution when considering their use in patients with familial thyroid cancer and those with a genetic predisposition for papillary and follicular thyroid cancer—eg, patients with familial adenomatous polyposis, phosphate and tensin homolog hamartoma tumor syndrome, Carney complex type 1, Werner syndrome, or familial papillary thyroid cancer.
Methodologically superior studies and careful long-term monitoring of patients treated with GLP-1 agonists are required to clarify the risk vs benefit of these therapies.
The question is complicated, as there are different types of thyroid cancer, and a causal relationship is hard to prove.
Glucagon-like peptide 1 (GLP-1) receptor agonists can be safely used in all patients with thyroid cancers that are derived from the thyroid follicular epithelium (papillary and follicular thyroid cancer). However, they are currently contraindicated in patients with medullary thyroid cancer and in patients with multiple endocrine neoplasia 2 (MEN-2), which is not a form of thyroid cancer but is relevant to our discussion. We probably should be cautious about using them in patients with familial thyroid cancer and those with a genetic predisposition for papillary or follicular thyroid cancer.
GLP-1 DRUGS ARE WIDELY USED
The glucagon-like peptide 1 (GLP-1) receptor agonists are widely used to treat type 2 diabetes mellitus. The currently available drugs of this class—exenatide (Byetta), liraglutide (Victoza), albiglutide (Tanzeum), dulaglutide (Trulicity), and extended-release exenatide (Bydureon)—are popular because they lower glucose levels, pose a low risk of hypoglycemia, can induce weight loss,1 and, in the case of extended-release exenatide and albiglutide, are given once weekly. They are currently recommended as add-on therapy to metformin. These drugs mimic the action of GLP-1, an endogenous hormone released by the intestine in response to food. They bind to receptors on beta cells, stimulating insulin production.1
FOUR TYPES OF THYROID CANCER
There are four types of thyroid cancer: medullary (a contraindication to GLP-1 agonists), papillary, follicular, and anaplastic.
Medullary thyroid cancer is extremely rare in humans, with 976 cases diagnosed from 1992 to 2006 in the United States, compared with 36,583 cases of papillary and 4,560 cases of follicular cancer. Anaplastic cancer is also rare (556 cases).2 The highest incidence rates of medullary thyroid cancer are in people of Hispanic descent (0.21 per 100,000 woman-years and 0.18 per 100,000 man-years).2
EXPERIMENTAL EVIDENCE
Pancreatic beta cells are not the only cells in the body that can express GLP-1 receptors. Notably, the parafollicular cells (also called C cells) of the thyroid, which secrete calcitonin and which are the cells involved in medullary thyroid cancer, also sometimes express these receptors if cancer develops.
In experiments in mice and rats, the incidence of thyroid C-cell tumors was higher in animals given GLP-1 analogues. Liraglutide, exenatide, taspoglutide, and lixisenatide potently activated GLP-1 receptors in thyroid C cells, increasing calcitonin gene expression and stimulating calcitonin release in a dose-dependent manner.3 Moreover, sustained activation of these receptors caused C-cell hyperplasia and resulted in medullary thyroid cancer. However medullary thyroid cancer also occurred in rodents receiving placebo.
C cells in monkeys and humans express fewer GLP-1 receptors than those in rodents; in fact, healthy human C cells do not express them at all.3,4 In rats with C-cell hyperplasia or medullary thyroid cancer, GLP-1 receptors are present in 100% of cases (and in increased density), compared with 27% of human medullary thyroid cancers.4
In addition to medullary thyroid cancer, various other human tumors have been shown to express GLP-1 receptors.5 Based on limited data, KÖrner et al5 found that these receptors are also present in various other human tumors, eg:
- Pheochromocytoma (60%)
- Paraganglioma (28%)
- Meningioma (35%)
- Astrocytoma (25%)
- Glioblastoma (9%)
- Ependymoma (16%)
- Medulloblastoma (25%)
- Nephroblastoma (22%)
- Neuroblastoma (18%)
- Ovarian adenocarcinoma (16%)
- Prostate carcinoma (5%).
Madsen et al6 reported that liraglutide binding to the GLP-1 receptor on murine thyroid C cells led to C-cell hyperplasia. However, prolonged administration of liraglutide at very high doses did not produce C-cell proliferation in monkeys.3
Gier et al7 looked at GLP-1 receptor expression in normal human C cells, hyperplastic C cells, and medullary thyroid cancer cells, as well as in papillary thyroid cancer cells, which do not arise from C cells. They demonstrated concurrent calcitonin and GLP-1 receptor immunostaining, not only in those with C-cell hyperplasia (9 of 9 cases) and medullary thyroid cancer (11 of 12 cases), but also in 3 (18%) of 17 patients with papillary thyroid cancer and 5 (33%) of 15 with normal thyroid follicular cells. However, the choice of polyclonal antibodies and radioligands used and concerns about methodology have led investigators to interpret these results cautiously.8–10
STUDIES OF GLP-1 AGONISTS IN HUMANS
Several prospective clinical studies showed no increase in calcitonin levels during therapy with GLP-1 receptor agonists in patients with type 2 diabetes.3,11 Long-term use of liraglutide in high doses (up to 3 mg per day) did not lead to elevations in serum calcitonin levels.11
In a retrospective Adverse Event Reporting System database review, the incidence rate of thyroid cancer in patients treated with exenatide was higher—with an odds ratio of 4.7 (30 events)—than with a panel of control drugs (3 events).12 However, this study did not differentiate between types of thyroid cancer, and the inherent limitations of retrospective databases complicate its interpretation. Such a high odds ratio would imply a significant increase in the incidence of medullary thyroid cancer, but this does not seem to be true.
Alves et al13 performed a meta-analysis of randomized controlled trials and long-term observational studies. None of the studies evaluating exenatide reported cases of thyroid cancer, whereas five of the studies evaluating liraglutide did. In total, nine patients treated with liraglutide were diagnosed with thyroid cancer, compared with one patient on glimepiride. The odds ratio for thyroid cancer occurrence associated with liraglutide treatment was 1.54, but that was not statistically significant (95% confidence interval 0.40–6.02, P = .53, I2 = 0%).
These studies are hypothesis-generating and do not prove that GLP-1 receptor agonists cause medullary thyroid cancer. Given the extremely low incidence of medullary thyroid cancer, to prove or disprove a causal relationship would require an enormous number of patients, who would need to be followed for several years.
OFFICIAL RECOMMENDATIONS
Considerable differences in the biology of the rodent vs human thyroid GLP-1 receptor systems have led regulatory authorities to conclude that the risk for development of medullary thyroid cancer with GLP-1 therapy in humans is difficult to quantify, but low.14 Consequently, the US Food and Drug Administration recommends neither monitoring of calcitonin levels nor ultrasound imaging as a screening tool in patients taking GLP-1 agonists.14
BENEFITS OUTWEIGH RISKS
At present, the benefits of using GLP-1 receptor agonists to treat type 2 diabetes mellitus outweigh the risks, and there seems to be little reason to withhold this effective therapy except in patients who have a personal or family history of medullary thyroid cancer or MEN-2. Until the effects of GLP-1 agonists are systematically studied in follicular-cell-derived thyroid cancer, we also recommend caution when considering their use in patients with familial thyroid cancer and those with a genetic predisposition for papillary and follicular thyroid cancer—eg, patients with familial adenomatous polyposis, phosphate and tensin homolog hamartoma tumor syndrome, Carney complex type 1, Werner syndrome, or familial papillary thyroid cancer.
Methodologically superior studies and careful long-term monitoring of patients treated with GLP-1 agonists are required to clarify the risk vs benefit of these therapies.
- Samson SL, Garber A. GLP-1R agonist therapy for diabetes: benefits and potential risks. Curr Opin Endocrinol Diabetes Obes 2013; 20:87–97.
- Aschebrook-Kilfoy B, Ward MH, Sabra MM, Devesa SS. Thyroid cancer incidence patterns in the United States by histologic type, 1992–2006. Thyroid 2011; 21:125–134.
- Bjerre Knudsen L, Madsen LW, Andersen S, et al. Glucagon-like peptide-1 receptor agonists activate rodent thyroid C-cells causing calcitonin release and C-cell proliferation. Endocrinology 2010; 151:1473–1486.
- Waser B, Beetschen K, Pellegata NS, Reubi JC. Incretin receptors in non-neoplastic and neoplastic thyroid C cells in rodents and humans: relevance for incretin-based diabetes therapy. Neuroendocrinology 2011; 94:291–301.
- Körner M, Stöckli M, Waser B, Reubi JC. GLP-1 receptor expression in human tumors and human normal tissues: potential for in vivo targeting. J Nucl Med 2007; 48:736–743.
- Madsen LW, Knauf JA, Gotfredsen C, et al. GLP-1 receptor agonists and the thyroid: C-cell effects in mice are mediated via the GLP-1 receptor and not associated with RET activation. Endocrinology 2012; 153:1538–1547.
- Gier B, Butler PC, Lai CK, Kirakossian D, DeNicola MM, Yeh MW. Glucagon like peptide-1 receptor expression in the human thyroid gland. J Clin Endocrinol Metab 2012; 97:121–131.
- Drucker DJ, Sherman SI, Bergenstal RM, Buse JB. The safety of incretin-based therapies—review of the scientific evidence. J Clin Endocrinol Metab 2011; 96:2027–2031.
- Gagel RF. Activation of G-protein-coupled receptors and thyroid malignant tumors: the jury is still out. Endocr Pract 2011; 17:957–959.
- Nauck MA. A critical analysis of the clinical use of incretin-based therapies: the benefits by far outweigh the potential risks. Diabetes Care 2013; 36:2126–2132.
- Hegedüs L, Moses AC, Zdravkovic M, Le Thi T, Daniels GH. GLP-1 and calcitonin concentration in humans: lack of evidence of calcitonin release from sequential screening in over 5000 subjects with type 2 diabetes or nondiabetic obese subjects treated with the human GLP-1 analog, liraglutide. J Clin Endocrinol Metab 2011; 96:853–860.
- Elashoff M, Matveyenko AV, Gier B, Elashoff R, Butler PC. Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology 2011; 141:150–156.
- Alves C, Batel-Marques F, Macedo AF. A meta-analysis of serious adverse events reported with exenatide and liraglutide: acute pancreatitis and cancer. Diabetes Res Clin Pract 2012; 98:271–284.
- Parks M, Rosebraugh C. Weighing risks and benefits of liraglutide—the FDA’s review of a new antidiabetic therapy. N Engl J Med 2010; 362:774–777.
- Samson SL, Garber A. GLP-1R agonist therapy for diabetes: benefits and potential risks. Curr Opin Endocrinol Diabetes Obes 2013; 20:87–97.
- Aschebrook-Kilfoy B, Ward MH, Sabra MM, Devesa SS. Thyroid cancer incidence patterns in the United States by histologic type, 1992–2006. Thyroid 2011; 21:125–134.
- Bjerre Knudsen L, Madsen LW, Andersen S, et al. Glucagon-like peptide-1 receptor agonists activate rodent thyroid C-cells causing calcitonin release and C-cell proliferation. Endocrinology 2010; 151:1473–1486.
- Waser B, Beetschen K, Pellegata NS, Reubi JC. Incretin receptors in non-neoplastic and neoplastic thyroid C cells in rodents and humans: relevance for incretin-based diabetes therapy. Neuroendocrinology 2011; 94:291–301.
- Körner M, Stöckli M, Waser B, Reubi JC. GLP-1 receptor expression in human tumors and human normal tissues: potential for in vivo targeting. J Nucl Med 2007; 48:736–743.
- Madsen LW, Knauf JA, Gotfredsen C, et al. GLP-1 receptor agonists and the thyroid: C-cell effects in mice are mediated via the GLP-1 receptor and not associated with RET activation. Endocrinology 2012; 153:1538–1547.
- Gier B, Butler PC, Lai CK, Kirakossian D, DeNicola MM, Yeh MW. Glucagon like peptide-1 receptor expression in the human thyroid gland. J Clin Endocrinol Metab 2012; 97:121–131.
- Drucker DJ, Sherman SI, Bergenstal RM, Buse JB. The safety of incretin-based therapies—review of the scientific evidence. J Clin Endocrinol Metab 2011; 96:2027–2031.
- Gagel RF. Activation of G-protein-coupled receptors and thyroid malignant tumors: the jury is still out. Endocr Pract 2011; 17:957–959.
- Nauck MA. A critical analysis of the clinical use of incretin-based therapies: the benefits by far outweigh the potential risks. Diabetes Care 2013; 36:2126–2132.
- Hegedüs L, Moses AC, Zdravkovic M, Le Thi T, Daniels GH. GLP-1 and calcitonin concentration in humans: lack of evidence of calcitonin release from sequential screening in over 5000 subjects with type 2 diabetes or nondiabetic obese subjects treated with the human GLP-1 analog, liraglutide. J Clin Endocrinol Metab 2011; 96:853–860.
- Elashoff M, Matveyenko AV, Gier B, Elashoff R, Butler PC. Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology 2011; 141:150–156.
- Alves C, Batel-Marques F, Macedo AF. A meta-analysis of serious adverse events reported with exenatide and liraglutide: acute pancreatitis and cancer. Diabetes Res Clin Pract 2012; 98:271–284.
- Parks M, Rosebraugh C. Weighing risks and benefits of liraglutide—the FDA’s review of a new antidiabetic therapy. N Engl J Med 2010; 362:774–777.
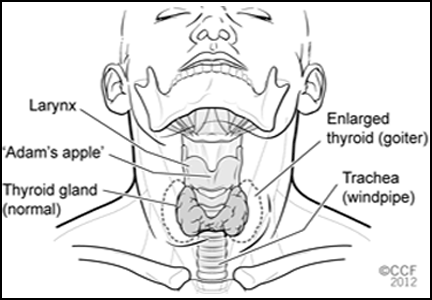
Should patients stop taking aspirin for primary prevention?
In view of current evidence, we do not recommend routinely using aspirin for primary prevention of cardiovascular disease, even in patients with diabetes mellitus. The decision must be individualized on the basis of the patient’s risks of cardiovascular disease and bleeding, especially the risk of serious bleeding events such as gastrointestinal and intracranial hemorrhage.
For example, patients with a family history of myocardial infarction at an early age and patients who smoke or have multiple cardiovascular risk factors may be most likely to benefit, whereas those with risk factors for gastrointestinal bleeding such as dyspepsia or ulcer would not be good candidates. Of note, current recommendations are mixed and confusing and will need to be reevaluated as new trial data become available.
TRIALS THAT SET THE STAGE FOR CURRENT PRACTICE
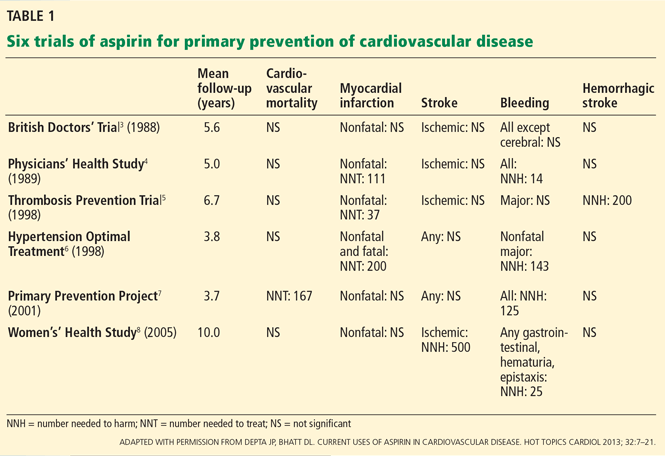
Routine use of aspirin for primary prevention of cardiovascular disease remains controversial.1,2 Aspirin’s safety and efficacy for this indication was studied in six major trials (Table 1).3–8 In the late 1980s, the first two primary prevention trials of aspirin enrolled healthy male physicians who had minimal cardiovascular risk factors3,4:
The British Doctors’ Trial3 observed no significant differences between aspirin (300–500 mg/day) and no aspirin in the rates of the primary end point of cardiovascular death or in the individual secondary end points of nonfatal myocardial infarction, nonfatal stroke, or bleeding.3
The Physicians’ Health Study4 found no differences in the rates of cardiovascular mortality or ischemic stroke between aspirin (325 mg every other day) and placebo. The rate of nonfatal myocardial infarction was significantly lower with aspirin than with placebo, but with a higher risk of bleeding. Relative risks and 95% confidence intervals with aspirin vs placebo:
- Nonfatal myocardial infarction
0.59 (0.47–0.74), P < .00001 - Bleeding
1.32 (1.25–1.40), P < .00001 - Blood transfusions
1.71 (1.09–2.69), P = .02 - Hemorrhagic stroke
2.14 (0.96–4.77), P = .06.
A subgroup analysis revealed that the benefit of aspirin for myocardial infarction in the Physicians’ Health Study was predominantly in those age 50 and older.4 This finding established the common clinical practice of routinely using aspirin for primary prevention in men age 50 and older.1
Later, aspirin for primary prevention was studied in four trials,5–8 three of which enrolled patients at higher cardiovascular risk5–7:
The Thrombosis Prevention Trial5 was conducted in men in the highest quintile of cardiovascular risk. The aspirin dosage was 75 mg/day.
The Hypertension Optimal Treatment6 trial included men and women ages 50 to 80 with hypertension. Aspirin dosage: 75 mg/day.
The Primary Prevention Project7 involved men and women age 50 and older with at least one risk factor for cardiovascular disease.1,5–7 The aspirin dosage was 100 mg/day.
In these trials (Table 1), aspirin significantly lowered the rate of ischemic events compared with placebo or control: nonfatal myocardial infarction in the Thrombosis Prevention Trial; myocardial infarction and major adverse cardiac event (ie, cardiovascular death, myocardial infarction, or stroke) in the Hypertension Optimal Treatment trial; and cardiovascular mortality and major cardiovascular events (cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, angina pectoris, transient ischemic attack, peripheral artery disease, or revascularization procedures) in the Primary Prevention Project. However, aspirin’s benefit in each trial was largely offset by a higher rate of various bleeding end points.5–7
The Women’s Health Study
A subgroup analysis of the Hypertension Optimal Treatment trial suggested that sex may influence the efficacy of aspirin—specifically, aspirin did not prevent nonfatal myocardial infarction in women.9 Given the paucity of female participants in the previous primary prevention trials, the Women’s Health Study8 was designed to determine the efficacy and safety of aspirin (100 mg every other day) in women age 45 and older with very few cardiovascular risk factors.8
Aspirin did not significantly reduce the rate of the primary end point of cardiovascular death, myocardial infarction, or stroke, though a significant effect was observed in the subgroup of women age 65 and older. Although overall the Women’s Health Study found no benefit in the rate of myocardial infarction, there was a significant reduction in the rate of ischemic stroke (which needs to be interpreted cautiously in an overall neutral trial) and a nonsignificant increase in the rate of hemorrhagic stroke. As in other trials, rates of bleeding, including gastrointestinal bleeding, were higher with aspirin.
A meta-analysis of six trials of aspirin for primary prevention
In 2009, the Antithrombotic Trialists’ Collaboration10 published a meta-analysis of six trials of aspirin for primary prevention. In this analysis, aspirin did not reduce the rate of cardiovascular death, but it did reduce the yearly risk of:
- Death from coronary heart disease or nonfatal myocardial infarction
(0.28% vs 0.34%, P < .0001) - Nonfatal myocardial infarction
(0.18% vs 0.23%, P < .0001) - Ischemic stroke
(0.11% vs 0.12%, P = .05).10
Despite aspirin’s apparent efficacy, the absolute yearly risk for major extracranial bleeding and hemorrhagic stroke was also significantly increased with aspirin use by 0.3% and 0.1%, respectively. The efficacy of aspirin for preventing all serious vascular events (vascular death, myocardial infarction, or stroke) was similar in men and women.10 The authors concluded that the net benefit of aspirin did not outweigh the increased risks of bleeding.
WHAT ABOUT PATIENTS WITH DIABETES?
When considering whether to prescribe aspirin for primary prevention, the individual patient’s risks of cardiovascular disease and bleeding must be carefully assessed. Those at highest risk of cardiovascular disease and at low risk of bleeding may still benefit, but current evidence does not clearly support this strategy.
For example, diabetes mellitus has traditionally been considered a coronary heart disease equivalent, and aspirin was routinely prescribed as “secondary prevention.”11 In the six trials of aspirin for primary prevention, the prevalence of diabetic patients ranged from 1% to 17%, the efficacy of aspirin in this subgroup was inconsistent among the trials, and aspirin did not confer a net clinical benefit according to the 2009 Antithrombotic Trialists’ Collaboration meta-analysis.1,3–8,10
Additionally, two trials of aspirin for primary prevention in diabetes12,13 failed to demonstrate significant efficacy for aspirin compared with no aspirin, either in Japanese patients with type 2 diabetes and no history of cardiovascular disease12 or in patients with asymptomatic peripheral artery disease.13
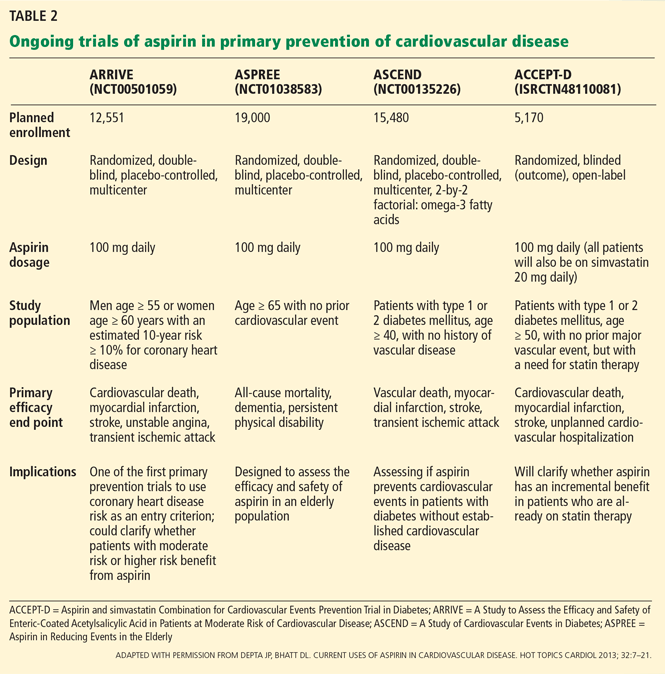
Thus, the current evidence for aspirin for primary prevention in diabetes does not demonstrate a net clinical benefit, but ongoing trials (Table 2) may provide evidence for the use of aspirin in this important subgroup.
An important finding from the 2009 Antithrombotic Trialists’ Collaboration was that traditional risk factors for cardiovascular disease also increase the risk of major bleeding, thus making it difficult to determine who will receive the maximum net clinical benefit.10 Additionally, many of the aspirin primary prevention trials predated the widespread use of statins and the current lower prevalence of smoking, which may further limit the generalizability of the positive signals seen in earlier trials.
THE DATA ARE MIXED, BUT ONE MESSAGE IS CLEAR

Based on the current available evidence, the US Food and Drug Administration recently issued a Consumer Update that does not support aspirin for primary prevention and warns patients about the risk of serious bleeding complications.14 Moreover, current guidelines and consensus panels (Table 3) for aspirin in primary prevention differ from one another,15–21 making it challenging for clinicians to determine which patients would benefit. One message is clear in the most current clinical guidelines, namely, that routine use of aspirin for primary prevention is not recommended.15–21 Several ongoing trials may resolve this important clinical dilemma.
- Depta JP, Bhatt DL. Current uses of aspirin in cardiovascular disease. Hot Topics Cardiol 2013; 32:7–21.
- Nemerovski CW, Salinitri FD, Morbitzer KA, Moser LR. Aspirin for primary prevention of cardiovascular disease events. Pharmacotherapy 2012; 32:1020–1035.
- Peto R, Gray R, Collins R, et al. Randomised trial of prophylactic daily aspirin in British male doctors. Br Med J (Clin Res Ed) 1988; 296:313–316.
- Final report on the aspirin component of the ongoing Physicians’ Health Study. Steering Committee of the Physicians’ Health Study Research Group. N Engl J Med 1989; 321:129–135.
- Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. The Medical Research Council’s General Practice Research Framework. Lancet 1998; 351:233–241.
- Hansson L, Zanchetti A, Carruthers SG, et al. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. HOT Study Group. Lancet 1998; 351:1755–1762.
- de Gaetano G; Collaborative Group of the Primary Prevention Project. Low-dose aspirin and vitamin E in people at cardiovascular risk: a randomised trial in general practice. Collaborative Group of the Primary Prevention Project. Lancet 2001; 357:89–95.
- Ridker PM, Cook NR, Lee IM, et al. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med 2005; 352:1293–1304.
- Kjeldsen SE, Kolloch RE, Leonetti G, et al. Influence of gender and age on preventing cardiovascular disease by antihypertensive treatment and acetylsalicylic acid. The HOT study. Hypertension Optimal Treatment. J Hypertens 2000; 18:629–642.
- Antithrombotic Trialists’ (ATT) Collaboration; Baigent C, Blackwell L, Collins R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet 2009; 373:1849–1860.
- Buse JB, Ginsberg HN, Bakris GL, et al; American Heart Association; American Diabetes Association. Primary prevention of cardiovascular diseases in people with diabetes mellitus: a scientific statement from the American Heart Association and the American Diabetes Association. Circulation 2007; 115:114–126.
- Ogawa H, Nakayama M, Morimoto T, et al; Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes (JPAD) Trial Investigators. Low-dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: a randomized controlled trial. JAMA 2008; 300:2134–2141.
- Belch J, MacCuish A, Campbell I, et al; Prevention of Progression of Arterial Disease and Diabetes Study Group; Diabetes Registry Group; Royal College of Physicians Edinburgh. The prevention of progression of arterial disease and diabetes (POPADAD) trial: factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ 2008; 337:a1840.
- US Food and Drug Administration (FDA). Use of aspirin for primary prevention of heart attack and stroke. http://www.fda.gov/drugs/resourcesforyou/consumers/ucm390574.htm. Accessed January 9, 2015.
- Vandvik PO, Lincoff AM, Gore JM, et al; American College of Chest Physicians. Primary and secondary prevention of cardiovascular disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e637S–e668S.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Pearson TA, Blair SN, Daniels SR, et al. AHA Guidelines for Primary Prevention of Cardiovascular Disease and Stroke: 2002 Update: Consensus Panel Guide to Comprehensive Risk Reduction for Adult Patients Without Coronary or Other Atherosclerotic Vascular Diseases. American Heart Association Science Advisory and Coordinating Committee. Circulation 2002; 106:388–391.
- Mosca L, Benjamin EJ, Berra K, et al. Effectiveness-based Guidelines for the Prevention of Cardiovascular Disease in Women—2011 Update: a Guideline from the American Heart Association. Circulation 2011; 123:1243–1262.
- Bell AD, Roussin A, Cartier R, et al; Canadian Cardiovascular Society. The use of antiplatelet therapy in the outpatient setting: Canadian Cardiovascular Society Guidelines. Can J Cardiol 2011; 27(suppl A):S1–S59.
- Perk J, De Backer G, Gohlke H, et al; European Association for Cardiovascular Prevention & Rehabilitation (EACPR); ESC Committee for Practice Guidelines (CPG). European guidelines on cardiovascular disease prevention in clinical practice (version 2012). The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Eur Heart J 2012; 33:1635–1701.
- US Preventive Services Task Force. Aspirin for the prevention of cardiovascular disease: US Preventive Services Task Force recommendation statement. Ann Intern Med 2009; 150:396–404.
In view of current evidence, we do not recommend routinely using aspirin for primary prevention of cardiovascular disease, even in patients with diabetes mellitus. The decision must be individualized on the basis of the patient’s risks of cardiovascular disease and bleeding, especially the risk of serious bleeding events such as gastrointestinal and intracranial hemorrhage.
For example, patients with a family history of myocardial infarction at an early age and patients who smoke or have multiple cardiovascular risk factors may be most likely to benefit, whereas those with risk factors for gastrointestinal bleeding such as dyspepsia or ulcer would not be good candidates. Of note, current recommendations are mixed and confusing and will need to be reevaluated as new trial data become available.
TRIALS THAT SET THE STAGE FOR CURRENT PRACTICE
Routine use of aspirin for primary prevention of cardiovascular disease remains controversial.1,2 Aspirin’s safety and efficacy for this indication was studied in six major trials (Table 1).3–8 In the late 1980s, the first two primary prevention trials of aspirin enrolled healthy male physicians who had minimal cardiovascular risk factors3,4:
The British Doctors’ Trial3 observed no significant differences between aspirin (300–500 mg/day) and no aspirin in the rates of the primary end point of cardiovascular death or in the individual secondary end points of nonfatal myocardial infarction, nonfatal stroke, or bleeding.3
The Physicians’ Health Study4 found no differences in the rates of cardiovascular mortality or ischemic stroke between aspirin (325 mg every other day) and placebo. The rate of nonfatal myocardial infarction was significantly lower with aspirin than with placebo, but with a higher risk of bleeding. Relative risks and 95% confidence intervals with aspirin vs placebo:
- Nonfatal myocardial infarction
0.59 (0.47–0.74), P < .00001 - Bleeding
1.32 (1.25–1.40), P < .00001 - Blood transfusions
1.71 (1.09–2.69), P = .02 - Hemorrhagic stroke
2.14 (0.96–4.77), P = .06.
A subgroup analysis revealed that the benefit of aspirin for myocardial infarction in the Physicians’ Health Study was predominantly in those age 50 and older.4 This finding established the common clinical practice of routinely using aspirin for primary prevention in men age 50 and older.1
Later, aspirin for primary prevention was studied in four trials,5–8 three of which enrolled patients at higher cardiovascular risk5–7:
The Thrombosis Prevention Trial5 was conducted in men in the highest quintile of cardiovascular risk. The aspirin dosage was 75 mg/day.
The Hypertension Optimal Treatment6 trial included men and women ages 50 to 80 with hypertension. Aspirin dosage: 75 mg/day.
The Primary Prevention Project7 involved men and women age 50 and older with at least one risk factor for cardiovascular disease.1,5–7 The aspirin dosage was 100 mg/day.
In these trials (Table 1), aspirin significantly lowered the rate of ischemic events compared with placebo or control: nonfatal myocardial infarction in the Thrombosis Prevention Trial; myocardial infarction and major adverse cardiac event (ie, cardiovascular death, myocardial infarction, or stroke) in the Hypertension Optimal Treatment trial; and cardiovascular mortality and major cardiovascular events (cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, angina pectoris, transient ischemic attack, peripheral artery disease, or revascularization procedures) in the Primary Prevention Project. However, aspirin’s benefit in each trial was largely offset by a higher rate of various bleeding end points.5–7
The Women’s Health Study
A subgroup analysis of the Hypertension Optimal Treatment trial suggested that sex may influence the efficacy of aspirin—specifically, aspirin did not prevent nonfatal myocardial infarction in women.9 Given the paucity of female participants in the previous primary prevention trials, the Women’s Health Study8 was designed to determine the efficacy and safety of aspirin (100 mg every other day) in women age 45 and older with very few cardiovascular risk factors.8
Aspirin did not significantly reduce the rate of the primary end point of cardiovascular death, myocardial infarction, or stroke, though a significant effect was observed in the subgroup of women age 65 and older. Although overall the Women’s Health Study found no benefit in the rate of myocardial infarction, there was a significant reduction in the rate of ischemic stroke (which needs to be interpreted cautiously in an overall neutral trial) and a nonsignificant increase in the rate of hemorrhagic stroke. As in other trials, rates of bleeding, including gastrointestinal bleeding, were higher with aspirin.
A meta-analysis of six trials of aspirin for primary prevention
In 2009, the Antithrombotic Trialists’ Collaboration10 published a meta-analysis of six trials of aspirin for primary prevention. In this analysis, aspirin did not reduce the rate of cardiovascular death, but it did reduce the yearly risk of:
- Death from coronary heart disease or nonfatal myocardial infarction
(0.28% vs 0.34%, P < .0001) - Nonfatal myocardial infarction
(0.18% vs 0.23%, P < .0001) - Ischemic stroke
(0.11% vs 0.12%, P = .05).10
Despite aspirin’s apparent efficacy, the absolute yearly risk for major extracranial bleeding and hemorrhagic stroke was also significantly increased with aspirin use by 0.3% and 0.1%, respectively. The efficacy of aspirin for preventing all serious vascular events (vascular death, myocardial infarction, or stroke) was similar in men and women.10 The authors concluded that the net benefit of aspirin did not outweigh the increased risks of bleeding.
WHAT ABOUT PATIENTS WITH DIABETES?
When considering whether to prescribe aspirin for primary prevention, the individual patient’s risks of cardiovascular disease and bleeding must be carefully assessed. Those at highest risk of cardiovascular disease and at low risk of bleeding may still benefit, but current evidence does not clearly support this strategy.
For example, diabetes mellitus has traditionally been considered a coronary heart disease equivalent, and aspirin was routinely prescribed as “secondary prevention.”11 In the six trials of aspirin for primary prevention, the prevalence of diabetic patients ranged from 1% to 17%, the efficacy of aspirin in this subgroup was inconsistent among the trials, and aspirin did not confer a net clinical benefit according to the 2009 Antithrombotic Trialists’ Collaboration meta-analysis.1,3–8,10
Additionally, two trials of aspirin for primary prevention in diabetes12,13 failed to demonstrate significant efficacy for aspirin compared with no aspirin, either in Japanese patients with type 2 diabetes and no history of cardiovascular disease12 or in patients with asymptomatic peripheral artery disease.13
Thus, the current evidence for aspirin for primary prevention in diabetes does not demonstrate a net clinical benefit, but ongoing trials (Table 2) may provide evidence for the use of aspirin in this important subgroup.
An important finding from the 2009 Antithrombotic Trialists’ Collaboration was that traditional risk factors for cardiovascular disease also increase the risk of major bleeding, thus making it difficult to determine who will receive the maximum net clinical benefit.10 Additionally, many of the aspirin primary prevention trials predated the widespread use of statins and the current lower prevalence of smoking, which may further limit the generalizability of the positive signals seen in earlier trials.
THE DATA ARE MIXED, BUT ONE MESSAGE IS CLEAR
Based on the current available evidence, the US Food and Drug Administration recently issued a Consumer Update that does not support aspirin for primary prevention and warns patients about the risk of serious bleeding complications.14 Moreover, current guidelines and consensus panels (Table 3) for aspirin in primary prevention differ from one another,15–21 making it challenging for clinicians to determine which patients would benefit. One message is clear in the most current clinical guidelines, namely, that routine use of aspirin for primary prevention is not recommended.15–21 Several ongoing trials may resolve this important clinical dilemma.
In view of current evidence, we do not recommend routinely using aspirin for primary prevention of cardiovascular disease, even in patients with diabetes mellitus. The decision must be individualized on the basis of the patient’s risks of cardiovascular disease and bleeding, especially the risk of serious bleeding events such as gastrointestinal and intracranial hemorrhage.
For example, patients with a family history of myocardial infarction at an early age and patients who smoke or have multiple cardiovascular risk factors may be most likely to benefit, whereas those with risk factors for gastrointestinal bleeding such as dyspepsia or ulcer would not be good candidates. Of note, current recommendations are mixed and confusing and will need to be reevaluated as new trial data become available.
TRIALS THAT SET THE STAGE FOR CURRENT PRACTICE
Routine use of aspirin for primary prevention of cardiovascular disease remains controversial.1,2 Aspirin’s safety and efficacy for this indication was studied in six major trials (Table 1).3–8 In the late 1980s, the first two primary prevention trials of aspirin enrolled healthy male physicians who had minimal cardiovascular risk factors3,4:
The British Doctors’ Trial3 observed no significant differences between aspirin (300–500 mg/day) and no aspirin in the rates of the primary end point of cardiovascular death or in the individual secondary end points of nonfatal myocardial infarction, nonfatal stroke, or bleeding.3
The Physicians’ Health Study4 found no differences in the rates of cardiovascular mortality or ischemic stroke between aspirin (325 mg every other day) and placebo. The rate of nonfatal myocardial infarction was significantly lower with aspirin than with placebo, but with a higher risk of bleeding. Relative risks and 95% confidence intervals with aspirin vs placebo:
- Nonfatal myocardial infarction
0.59 (0.47–0.74), P < .00001 - Bleeding
1.32 (1.25–1.40), P < .00001 - Blood transfusions
1.71 (1.09–2.69), P = .02 - Hemorrhagic stroke
2.14 (0.96–4.77), P = .06.
A subgroup analysis revealed that the benefit of aspirin for myocardial infarction in the Physicians’ Health Study was predominantly in those age 50 and older.4 This finding established the common clinical practice of routinely using aspirin for primary prevention in men age 50 and older.1
Later, aspirin for primary prevention was studied in four trials,5–8 three of which enrolled patients at higher cardiovascular risk5–7:
The Thrombosis Prevention Trial5 was conducted in men in the highest quintile of cardiovascular risk. The aspirin dosage was 75 mg/day.
The Hypertension Optimal Treatment6 trial included men and women ages 50 to 80 with hypertension. Aspirin dosage: 75 mg/day.
The Primary Prevention Project7 involved men and women age 50 and older with at least one risk factor for cardiovascular disease.1,5–7 The aspirin dosage was 100 mg/day.
In these trials (Table 1), aspirin significantly lowered the rate of ischemic events compared with placebo or control: nonfatal myocardial infarction in the Thrombosis Prevention Trial; myocardial infarction and major adverse cardiac event (ie, cardiovascular death, myocardial infarction, or stroke) in the Hypertension Optimal Treatment trial; and cardiovascular mortality and major cardiovascular events (cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, angina pectoris, transient ischemic attack, peripheral artery disease, or revascularization procedures) in the Primary Prevention Project. However, aspirin’s benefit in each trial was largely offset by a higher rate of various bleeding end points.5–7
The Women’s Health Study
A subgroup analysis of the Hypertension Optimal Treatment trial suggested that sex may influence the efficacy of aspirin—specifically, aspirin did not prevent nonfatal myocardial infarction in women.9 Given the paucity of female participants in the previous primary prevention trials, the Women’s Health Study8 was designed to determine the efficacy and safety of aspirin (100 mg every other day) in women age 45 and older with very few cardiovascular risk factors.8
Aspirin did not significantly reduce the rate of the primary end point of cardiovascular death, myocardial infarction, or stroke, though a significant effect was observed in the subgroup of women age 65 and older. Although overall the Women’s Health Study found no benefit in the rate of myocardial infarction, there was a significant reduction in the rate of ischemic stroke (which needs to be interpreted cautiously in an overall neutral trial) and a nonsignificant increase in the rate of hemorrhagic stroke. As in other trials, rates of bleeding, including gastrointestinal bleeding, were higher with aspirin.
A meta-analysis of six trials of aspirin for primary prevention
In 2009, the Antithrombotic Trialists’ Collaboration10 published a meta-analysis of six trials of aspirin for primary prevention. In this analysis, aspirin did not reduce the rate of cardiovascular death, but it did reduce the yearly risk of:
- Death from coronary heart disease or nonfatal myocardial infarction
(0.28% vs 0.34%, P < .0001) - Nonfatal myocardial infarction
(0.18% vs 0.23%, P < .0001) - Ischemic stroke
(0.11% vs 0.12%, P = .05).10
Despite aspirin’s apparent efficacy, the absolute yearly risk for major extracranial bleeding and hemorrhagic stroke was also significantly increased with aspirin use by 0.3% and 0.1%, respectively. The efficacy of aspirin for preventing all serious vascular events (vascular death, myocardial infarction, or stroke) was similar in men and women.10 The authors concluded that the net benefit of aspirin did not outweigh the increased risks of bleeding.
WHAT ABOUT PATIENTS WITH DIABETES?
When considering whether to prescribe aspirin for primary prevention, the individual patient’s risks of cardiovascular disease and bleeding must be carefully assessed. Those at highest risk of cardiovascular disease and at low risk of bleeding may still benefit, but current evidence does not clearly support this strategy.
For example, diabetes mellitus has traditionally been considered a coronary heart disease equivalent, and aspirin was routinely prescribed as “secondary prevention.”11 In the six trials of aspirin for primary prevention, the prevalence of diabetic patients ranged from 1% to 17%, the efficacy of aspirin in this subgroup was inconsistent among the trials, and aspirin did not confer a net clinical benefit according to the 2009 Antithrombotic Trialists’ Collaboration meta-analysis.1,3–8,10
Additionally, two trials of aspirin for primary prevention in diabetes12,13 failed to demonstrate significant efficacy for aspirin compared with no aspirin, either in Japanese patients with type 2 diabetes and no history of cardiovascular disease12 or in patients with asymptomatic peripheral artery disease.13
Thus, the current evidence for aspirin for primary prevention in diabetes does not demonstrate a net clinical benefit, but ongoing trials (Table 2) may provide evidence for the use of aspirin in this important subgroup.
An important finding from the 2009 Antithrombotic Trialists’ Collaboration was that traditional risk factors for cardiovascular disease also increase the risk of major bleeding, thus making it difficult to determine who will receive the maximum net clinical benefit.10 Additionally, many of the aspirin primary prevention trials predated the widespread use of statins and the current lower prevalence of smoking, which may further limit the generalizability of the positive signals seen in earlier trials.
THE DATA ARE MIXED, BUT ONE MESSAGE IS CLEAR
Based on the current available evidence, the US Food and Drug Administration recently issued a Consumer Update that does not support aspirin for primary prevention and warns patients about the risk of serious bleeding complications.14 Moreover, current guidelines and consensus panels (Table 3) for aspirin in primary prevention differ from one another,15–21 making it challenging for clinicians to determine which patients would benefit. One message is clear in the most current clinical guidelines, namely, that routine use of aspirin for primary prevention is not recommended.15–21 Several ongoing trials may resolve this important clinical dilemma.
- Depta JP, Bhatt DL. Current uses of aspirin in cardiovascular disease. Hot Topics Cardiol 2013; 32:7–21.
- Nemerovski CW, Salinitri FD, Morbitzer KA, Moser LR. Aspirin for primary prevention of cardiovascular disease events. Pharmacotherapy 2012; 32:1020–1035.
- Peto R, Gray R, Collins R, et al. Randomised trial of prophylactic daily aspirin in British male doctors. Br Med J (Clin Res Ed) 1988; 296:313–316.
- Final report on the aspirin component of the ongoing Physicians’ Health Study. Steering Committee of the Physicians’ Health Study Research Group. N Engl J Med 1989; 321:129–135.
- Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. The Medical Research Council’s General Practice Research Framework. Lancet 1998; 351:233–241.
- Hansson L, Zanchetti A, Carruthers SG, et al. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. HOT Study Group. Lancet 1998; 351:1755–1762.
- de Gaetano G; Collaborative Group of the Primary Prevention Project. Low-dose aspirin and vitamin E in people at cardiovascular risk: a randomised trial in general practice. Collaborative Group of the Primary Prevention Project. Lancet 2001; 357:89–95.
- Ridker PM, Cook NR, Lee IM, et al. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med 2005; 352:1293–1304.
- Kjeldsen SE, Kolloch RE, Leonetti G, et al. Influence of gender and age on preventing cardiovascular disease by antihypertensive treatment and acetylsalicylic acid. The HOT study. Hypertension Optimal Treatment. J Hypertens 2000; 18:629–642.
- Antithrombotic Trialists’ (ATT) Collaboration; Baigent C, Blackwell L, Collins R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet 2009; 373:1849–1860.
- Buse JB, Ginsberg HN, Bakris GL, et al; American Heart Association; American Diabetes Association. Primary prevention of cardiovascular diseases in people with diabetes mellitus: a scientific statement from the American Heart Association and the American Diabetes Association. Circulation 2007; 115:114–126.
- Ogawa H, Nakayama M, Morimoto T, et al; Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes (JPAD) Trial Investigators. Low-dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: a randomized controlled trial. JAMA 2008; 300:2134–2141.
- Belch J, MacCuish A, Campbell I, et al; Prevention of Progression of Arterial Disease and Diabetes Study Group; Diabetes Registry Group; Royal College of Physicians Edinburgh. The prevention of progression of arterial disease and diabetes (POPADAD) trial: factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ 2008; 337:a1840.
- US Food and Drug Administration (FDA). Use of aspirin for primary prevention of heart attack and stroke. http://www.fda.gov/drugs/resourcesforyou/consumers/ucm390574.htm. Accessed January 9, 2015.
- Vandvik PO, Lincoff AM, Gore JM, et al; American College of Chest Physicians. Primary and secondary prevention of cardiovascular disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e637S–e668S.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Pearson TA, Blair SN, Daniels SR, et al. AHA Guidelines for Primary Prevention of Cardiovascular Disease and Stroke: 2002 Update: Consensus Panel Guide to Comprehensive Risk Reduction for Adult Patients Without Coronary or Other Atherosclerotic Vascular Diseases. American Heart Association Science Advisory and Coordinating Committee. Circulation 2002; 106:388–391.
- Mosca L, Benjamin EJ, Berra K, et al. Effectiveness-based Guidelines for the Prevention of Cardiovascular Disease in Women—2011 Update: a Guideline from the American Heart Association. Circulation 2011; 123:1243–1262.
- Bell AD, Roussin A, Cartier R, et al; Canadian Cardiovascular Society. The use of antiplatelet therapy in the outpatient setting: Canadian Cardiovascular Society Guidelines. Can J Cardiol 2011; 27(suppl A):S1–S59.
- Perk J, De Backer G, Gohlke H, et al; European Association for Cardiovascular Prevention & Rehabilitation (EACPR); ESC Committee for Practice Guidelines (CPG). European guidelines on cardiovascular disease prevention in clinical practice (version 2012). The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Eur Heart J 2012; 33:1635–1701.
- US Preventive Services Task Force. Aspirin for the prevention of cardiovascular disease: US Preventive Services Task Force recommendation statement. Ann Intern Med 2009; 150:396–404.
- Depta JP, Bhatt DL. Current uses of aspirin in cardiovascular disease. Hot Topics Cardiol 2013; 32:7–21.
- Nemerovski CW, Salinitri FD, Morbitzer KA, Moser LR. Aspirin for primary prevention of cardiovascular disease events. Pharmacotherapy 2012; 32:1020–1035.
- Peto R, Gray R, Collins R, et al. Randomised trial of prophylactic daily aspirin in British male doctors. Br Med J (Clin Res Ed) 1988; 296:313–316.
- Final report on the aspirin component of the ongoing Physicians’ Health Study. Steering Committee of the Physicians’ Health Study Research Group. N Engl J Med 1989; 321:129–135.
- Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. The Medical Research Council’s General Practice Research Framework. Lancet 1998; 351:233–241.
- Hansson L, Zanchetti A, Carruthers SG, et al. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. HOT Study Group. Lancet 1998; 351:1755–1762.
- de Gaetano G; Collaborative Group of the Primary Prevention Project. Low-dose aspirin and vitamin E in people at cardiovascular risk: a randomised trial in general practice. Collaborative Group of the Primary Prevention Project. Lancet 2001; 357:89–95.
- Ridker PM, Cook NR, Lee IM, et al. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med 2005; 352:1293–1304.
- Kjeldsen SE, Kolloch RE, Leonetti G, et al. Influence of gender and age on preventing cardiovascular disease by antihypertensive treatment and acetylsalicylic acid. The HOT study. Hypertension Optimal Treatment. J Hypertens 2000; 18:629–642.
- Antithrombotic Trialists’ (ATT) Collaboration; Baigent C, Blackwell L, Collins R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet 2009; 373:1849–1860.
- Buse JB, Ginsberg HN, Bakris GL, et al; American Heart Association; American Diabetes Association. Primary prevention of cardiovascular diseases in people with diabetes mellitus: a scientific statement from the American Heart Association and the American Diabetes Association. Circulation 2007; 115:114–126.
- Ogawa H, Nakayama M, Morimoto T, et al; Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes (JPAD) Trial Investigators. Low-dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: a randomized controlled trial. JAMA 2008; 300:2134–2141.
- Belch J, MacCuish A, Campbell I, et al; Prevention of Progression of Arterial Disease and Diabetes Study Group; Diabetes Registry Group; Royal College of Physicians Edinburgh. The prevention of progression of arterial disease and diabetes (POPADAD) trial: factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. BMJ 2008; 337:a1840.
- US Food and Drug Administration (FDA). Use of aspirin for primary prevention of heart attack and stroke. http://www.fda.gov/drugs/resourcesforyou/consumers/ucm390574.htm. Accessed January 9, 2015.
- Vandvik PO, Lincoff AM, Gore JM, et al; American College of Chest Physicians. Primary and secondary prevention of cardiovascular disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e637S–e668S.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Pearson TA, Blair SN, Daniels SR, et al. AHA Guidelines for Primary Prevention of Cardiovascular Disease and Stroke: 2002 Update: Consensus Panel Guide to Comprehensive Risk Reduction for Adult Patients Without Coronary or Other Atherosclerotic Vascular Diseases. American Heart Association Science Advisory and Coordinating Committee. Circulation 2002; 106:388–391.
- Mosca L, Benjamin EJ, Berra K, et al. Effectiveness-based Guidelines for the Prevention of Cardiovascular Disease in Women—2011 Update: a Guideline from the American Heart Association. Circulation 2011; 123:1243–1262.
- Bell AD, Roussin A, Cartier R, et al; Canadian Cardiovascular Society. The use of antiplatelet therapy in the outpatient setting: Canadian Cardiovascular Society Guidelines. Can J Cardiol 2011; 27(suppl A):S1–S59.
- Perk J, De Backer G, Gohlke H, et al; European Association for Cardiovascular Prevention & Rehabilitation (EACPR); ESC Committee for Practice Guidelines (CPG). European guidelines on cardiovascular disease prevention in clinical practice (version 2012). The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Eur Heart J 2012; 33:1635–1701.
- US Preventive Services Task Force. Aspirin for the prevention of cardiovascular disease: US Preventive Services Task Force recommendation statement. Ann Intern Med 2009; 150:396–404.

When should I discuss driving with my older patients?
Most older drivers are safe drivers and are less likely than younger people to drive recklessly, at high speeds, or under the influence of alcohol.1 However, motor vehicle injuries are the second leading cause of injury-related deaths among older adults. Very old adults (80 years and over) have higher rates of fatality and injury in motor vehicle crashes per million miles driven than any other age group except for teenagers.1 Therefore, consider safety screening of all very old drivers plus any older adult with certain high-risk medical conditions, including the following.
NEUROCOGNITIVE DISORDERS
Drivers with Alzheimer disease—the most common type of major neurocognitive disorder (dementia) in older adults in the United States—are at high risk for adverse driving events due to impaired memory, attentiveness, problem-solving skills, multitasking, orientation, judgment, and reaction speed. Even in amnesic mild cognitive impairment—a mild neurocognitive disorder without functional decline—driving skills such as lane control may be impaired.2
Frontotemporal dementia, a less common cause of dementia in older adults, is associated with profound impairments in reasoning, task flexibility, planning, and execution. Persons with frontotemporal dementia are more likely to speed, run stop signs, and suffer more off-road crashes and collisions.3
The diagnosis of dementia, however, is less predictive of driving risk than the stage of dementia. The American Academy of Neurology recommends that health care providers clinically “stage” all demented individuals using a validated tool at diagnosis and periodically afterwards. The Clinical Dementia Rating (CDR) scale is appropriate for staging dementia in the office. The CDR has also been shown to identify people with dementia who are at an increased risk of unsafe driving, with strong evidence (level of evidence A) relating dementia stage to driving risk.4 The CDR assigns a score of 1 for mild dementia (function impaired in at least one complex activity); 2 for moderate dementia (function impaired in at least one basic activity); and 3 for severe dementia. Individuals with a CDR score of 2 or higher are considered to be at very high risk if still driving. These persons should be encouraged to surrender their driving privileges.4 Even with mild dementia (CDR score of 1), as few as 41% of drivers may drive safely.4 Most persons with mild cognitive impairment (CDR score of 0.5) are safe drivers.

Patients often have poor insight into their driving safety. However, a caregiver’s rating of driving skills as marginal or unsafe is useful in identifying unsafe drivers (level of evidence B) and can be considered a red flag.4 Predictors with less support in the literature (level of evidence C) include recent traffic citations, motor vehicle accidents, and self-reported situational avoidance, such as limiting driving to familiar roadways. Additional predictors include Mini-Mental State Examination scores of 24 or less, and/or the emergence of an aggressive or impulsive personality (Table 1). A driver evaluation is helpful when there is mild cognitive impairment or mild dementia with at least one red flag.
Clinicians who are not comfortable with staging dementia as mild, moderate, or severe may consider referring to a neurologist or geriatrician.
There is no evidence to support or refute the benefit of interventional strategies such as driver rehabilitation for drivers with dementia.
PARKINSON DISEASE
Individuals with mild motor disability from Parkinson disease may be fit drivers. As the disease progresses, drivers with Parkinson disease may make more errors than healthy elders in visual scanning, signaling, vehicle positioning, and velocity regulation (eg, traveling so slowly that it may be unsafe).5 Clinicians can consider referring a patient with Parkinson disease for a baseline driving evaluation upon diagnosis, and then every 1 to 2 years for reassessment. Alternate transportation should be arranged as the disease progresses.
EPISODIC INCAPACITATION
Approximately 1% to 3% of all motor vehicle accidents are due to sudden incapacitation of an otherwise safe driver.
Syncope. Neurally mediated (vasovagal) syncope accounts for 30% to 35% of syncopal episodes while driving.6 Cardiac arrhythmias are the next most common cause and include bradyarrhythmias (7%), supraventricular tachyarrhythmias (2%–15%), and ventricular tachyarrhythmias (5%–17%). Because neurocardiogenic syncope often recurs, consider restricting driving for those with recurrent or severe neurocardiogenic syncopal episodes until symptoms are controlled.
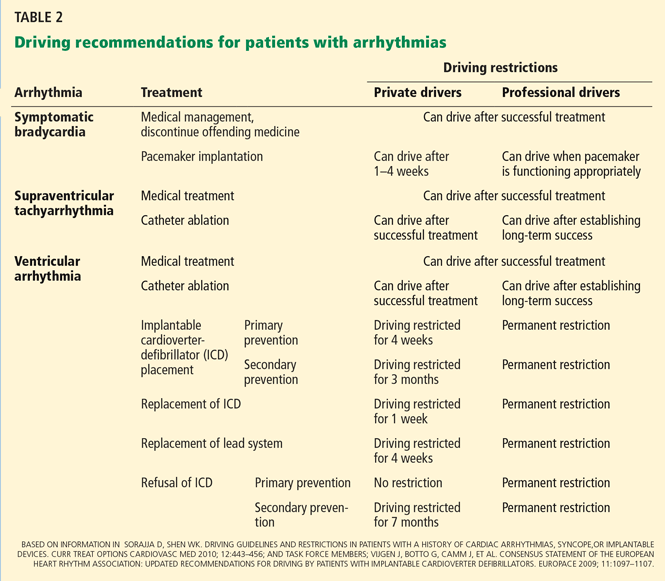
Arrhythmias. Driving recommendations for various arrhythmias7,8 are listed in Table 2.
Many patients who have an implantable cardioverter-defibrillator (ICD) device experience an unexpected shock. For individuals with a history of ventricular tachycardia or fibrillation, the 5-year actuarial incidence of appropriate ICD shocks ranges between 55% and 70%. However, data indicate that 90% to 100% of drivers who received ICD discharges while driving continued to drive without causing motor vehicle accidents.9,10
Seizures. States differ in their rules for reporting drivers who have epilepsy or breakthrough seizures. Physicians should refer to their state regulations when counseling these patients.
POLYPHARMACY
Polypharmacy is common in older adults. Many take psychoactive drugs that can impair tracking, alertness, coordination, and reaction time. With the “Roadwise Rx” tool (www.roadwiserx.com), health care providers and patients can enter the names of medicines to check if they affect driving ability. Nonproprietary on-line tools such as “START” (Screening Tool to Alert doctors to Right Treatment) and “STOPP” (Screening Tool of Older Persons’ Potentially Inappropriate Prescriptions) can be used to prune medication lists.
DRIVING EVALUATION
America is a nation of highways overflowing with cars. Cars provide transportation but also reflect wealth and personality, particularly for men. Practically, the ability to drive a car allows older men and women to socialize in the community, shop for essentials, and take care of themselves without being a burden. Driving cessation can cause social isolation and depressive symptoms and can strain caregiver resources.
It is therefore understandable for health care providers to feel reluctant or uncomfortable counseling older adults to give up their driving privileges. A health care provider who identifies driving safety concerns can refer a patient to a geriatrician for further risk assessment or to a certified driver rehabilitation specialist (CDRS) for a driving evaluation. A CDRS will also offer the patient and caregiver information on local resources for transportation alternatives. A list of local CDRSs can be found on the Association for Driver Rehabilitation Specialists website (www.aded.net). Many hospitals have occupational therapists who are CDRSs.
The evaluation typically involves an assessment of the driver’s knowledge of traffic signs and laws, a cognitive assessment, possibly a simulation, and finally an on-road driving evaluation if deemed appropriate. Medicare coverage depends on diagnosis and the state carrier.
- Williams AF. Teenage drivers: patterns of risk. J Safety Res 2003; 34:5–15.
- Griffith HR, Okonkwo OC, Stewart CC, et al. Lower hippocampal volume predicts decrements in lane control among drivers with amnestic mild cognitive impairment. J Geriatr Psychiatry Neurol 2013; 26:259–266.
- de Simone V, Kaplan L, Patronas N, Wassermann EM, Grafman J. Driving abilities in frontotemporal dementia patients. Dement Geriatr Cogn Disord 2007; 23:1–7.
- Iverson DJ, Gronseth GS, Reger MA, Classen S, Dubinsky RM, Rizzo M; Quality Standards Subcomittee of the American Academy of Neurology. Practice parameter update: evaluation and management of driving risk in dementia: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2010; 74:1316–1324.
- Classen S, Brumback B, Monahan M, et al. Driving errors in Parkinson’s disease: moving closer to predicting on-road outcomes. Am J Occup Ther 2014; 68:77–85.
- Blitzer ML, Saliba BC, Ghantous AE, Marieb MA, Schoenfeld MH. Causes of impaired consciousness while driving a motorized vehicle. Am J Cardiol 2003; 91:1373–1374.
- Sorajja D, Shen WK. Driving guidelines and restrictions in patients with a history of cardiac arrhythmias, syncope,or implantable devices. Curr Treat Options Cardiovasc Med 2010; 12:443–456.
- Task force members; Vijgen J, Botto G, Camm J, et al. Consensus statement of the European Heart Rhythm Association: updated recommendations for driving by patients with implantable cardioverter defibrillators. Europace 2009; 11:1097–1107.
- Conti JB, Woodard DA, Tucker KJ, Bryant B, King LC, Curtis AB. Modification of patient driving behavior after implantation of a cardioverter defibrillator. Pacing Clin Electrophysiol 1997; 20:2200–2204.
- Lerecouvreux M, Aït Saïd M, Paziaud O, et al. Automobile driving and implantable defibrillators. Arch Mal Coeur Vaiss 2005; 98:288–293. Article in French.
Most older drivers are safe drivers and are less likely than younger people to drive recklessly, at high speeds, or under the influence of alcohol.1 However, motor vehicle injuries are the second leading cause of injury-related deaths among older adults. Very old adults (80 years and over) have higher rates of fatality and injury in motor vehicle crashes per million miles driven than any other age group except for teenagers.1 Therefore, consider safety screening of all very old drivers plus any older adult with certain high-risk medical conditions, including the following.
NEUROCOGNITIVE DISORDERS
Drivers with Alzheimer disease—the most common type of major neurocognitive disorder (dementia) in older adults in the United States—are at high risk for adverse driving events due to impaired memory, attentiveness, problem-solving skills, multitasking, orientation, judgment, and reaction speed. Even in amnesic mild cognitive impairment—a mild neurocognitive disorder without functional decline—driving skills such as lane control may be impaired.2
Frontotemporal dementia, a less common cause of dementia in older adults, is associated with profound impairments in reasoning, task flexibility, planning, and execution. Persons with frontotemporal dementia are more likely to speed, run stop signs, and suffer more off-road crashes and collisions.3
The diagnosis of dementia, however, is less predictive of driving risk than the stage of dementia. The American Academy of Neurology recommends that health care providers clinically “stage” all demented individuals using a validated tool at diagnosis and periodically afterwards. The Clinical Dementia Rating (CDR) scale is appropriate for staging dementia in the office. The CDR has also been shown to identify people with dementia who are at an increased risk of unsafe driving, with strong evidence (level of evidence A) relating dementia stage to driving risk.4 The CDR assigns a score of 1 for mild dementia (function impaired in at least one complex activity); 2 for moderate dementia (function impaired in at least one basic activity); and 3 for severe dementia. Individuals with a CDR score of 2 or higher are considered to be at very high risk if still driving. These persons should be encouraged to surrender their driving privileges.4 Even with mild dementia (CDR score of 1), as few as 41% of drivers may drive safely.4 Most persons with mild cognitive impairment (CDR score of 0.5) are safe drivers.
Patients often have poor insight into their driving safety. However, a caregiver’s rating of driving skills as marginal or unsafe is useful in identifying unsafe drivers (level of evidence B) and can be considered a red flag.4 Predictors with less support in the literature (level of evidence C) include recent traffic citations, motor vehicle accidents, and self-reported situational avoidance, such as limiting driving to familiar roadways. Additional predictors include Mini-Mental State Examination scores of 24 or less, and/or the emergence of an aggressive or impulsive personality (Table 1). A driver evaluation is helpful when there is mild cognitive impairment or mild dementia with at least one red flag.
Clinicians who are not comfortable with staging dementia as mild, moderate, or severe may consider referring to a neurologist or geriatrician.
There is no evidence to support or refute the benefit of interventional strategies such as driver rehabilitation for drivers with dementia.
PARKINSON DISEASE
Individuals with mild motor disability from Parkinson disease may be fit drivers. As the disease progresses, drivers with Parkinson disease may make more errors than healthy elders in visual scanning, signaling, vehicle positioning, and velocity regulation (eg, traveling so slowly that it may be unsafe).5 Clinicians can consider referring a patient with Parkinson disease for a baseline driving evaluation upon diagnosis, and then every 1 to 2 years for reassessment. Alternate transportation should be arranged as the disease progresses.
EPISODIC INCAPACITATION
Approximately 1% to 3% of all motor vehicle accidents are due to sudden incapacitation of an otherwise safe driver.
Syncope. Neurally mediated (vasovagal) syncope accounts for 30% to 35% of syncopal episodes while driving.6 Cardiac arrhythmias are the next most common cause and include bradyarrhythmias (7%), supraventricular tachyarrhythmias (2%–15%), and ventricular tachyarrhythmias (5%–17%). Because neurocardiogenic syncope often recurs, consider restricting driving for those with recurrent or severe neurocardiogenic syncopal episodes until symptoms are controlled.
Arrhythmias. Driving recommendations for various arrhythmias7,8 are listed in Table 2.
Many patients who have an implantable cardioverter-defibrillator (ICD) device experience an unexpected shock. For individuals with a history of ventricular tachycardia or fibrillation, the 5-year actuarial incidence of appropriate ICD shocks ranges between 55% and 70%. However, data indicate that 90% to 100% of drivers who received ICD discharges while driving continued to drive without causing motor vehicle accidents.9,10
Seizures. States differ in their rules for reporting drivers who have epilepsy or breakthrough seizures. Physicians should refer to their state regulations when counseling these patients.
POLYPHARMACY
Polypharmacy is common in older adults. Many take psychoactive drugs that can impair tracking, alertness, coordination, and reaction time. With the “Roadwise Rx” tool (www.roadwiserx.com), health care providers and patients can enter the names of medicines to check if they affect driving ability. Nonproprietary on-line tools such as “START” (Screening Tool to Alert doctors to Right Treatment) and “STOPP” (Screening Tool of Older Persons’ Potentially Inappropriate Prescriptions) can be used to prune medication lists.
DRIVING EVALUATION
America is a nation of highways overflowing with cars. Cars provide transportation but also reflect wealth and personality, particularly for men. Practically, the ability to drive a car allows older men and women to socialize in the community, shop for essentials, and take care of themselves without being a burden. Driving cessation can cause social isolation and depressive symptoms and can strain caregiver resources.
It is therefore understandable for health care providers to feel reluctant or uncomfortable counseling older adults to give up their driving privileges. A health care provider who identifies driving safety concerns can refer a patient to a geriatrician for further risk assessment or to a certified driver rehabilitation specialist (CDRS) for a driving evaluation. A CDRS will also offer the patient and caregiver information on local resources for transportation alternatives. A list of local CDRSs can be found on the Association for Driver Rehabilitation Specialists website (www.aded.net). Many hospitals have occupational therapists who are CDRSs.
The evaluation typically involves an assessment of the driver’s knowledge of traffic signs and laws, a cognitive assessment, possibly a simulation, and finally an on-road driving evaluation if deemed appropriate. Medicare coverage depends on diagnosis and the state carrier.
Most older drivers are safe drivers and are less likely than younger people to drive recklessly, at high speeds, or under the influence of alcohol.1 However, motor vehicle injuries are the second leading cause of injury-related deaths among older adults. Very old adults (80 years and over) have higher rates of fatality and injury in motor vehicle crashes per million miles driven than any other age group except for teenagers.1 Therefore, consider safety screening of all very old drivers plus any older adult with certain high-risk medical conditions, including the following.
NEUROCOGNITIVE DISORDERS
Drivers with Alzheimer disease—the most common type of major neurocognitive disorder (dementia) in older adults in the United States—are at high risk for adverse driving events due to impaired memory, attentiveness, problem-solving skills, multitasking, orientation, judgment, and reaction speed. Even in amnesic mild cognitive impairment—a mild neurocognitive disorder without functional decline—driving skills such as lane control may be impaired.2
Frontotemporal dementia, a less common cause of dementia in older adults, is associated with profound impairments in reasoning, task flexibility, planning, and execution. Persons with frontotemporal dementia are more likely to speed, run stop signs, and suffer more off-road crashes and collisions.3
The diagnosis of dementia, however, is less predictive of driving risk than the stage of dementia. The American Academy of Neurology recommends that health care providers clinically “stage” all demented individuals using a validated tool at diagnosis and periodically afterwards. The Clinical Dementia Rating (CDR) scale is appropriate for staging dementia in the office. The CDR has also been shown to identify people with dementia who are at an increased risk of unsafe driving, with strong evidence (level of evidence A) relating dementia stage to driving risk.4 The CDR assigns a score of 1 for mild dementia (function impaired in at least one complex activity); 2 for moderate dementia (function impaired in at least one basic activity); and 3 for severe dementia. Individuals with a CDR score of 2 or higher are considered to be at very high risk if still driving. These persons should be encouraged to surrender their driving privileges.4 Even with mild dementia (CDR score of 1), as few as 41% of drivers may drive safely.4 Most persons with mild cognitive impairment (CDR score of 0.5) are safe drivers.
Patients often have poor insight into their driving safety. However, a caregiver’s rating of driving skills as marginal or unsafe is useful in identifying unsafe drivers (level of evidence B) and can be considered a red flag.4 Predictors with less support in the literature (level of evidence C) include recent traffic citations, motor vehicle accidents, and self-reported situational avoidance, such as limiting driving to familiar roadways. Additional predictors include Mini-Mental State Examination scores of 24 or less, and/or the emergence of an aggressive or impulsive personality (Table 1). A driver evaluation is helpful when there is mild cognitive impairment or mild dementia with at least one red flag.
Clinicians who are not comfortable with staging dementia as mild, moderate, or severe may consider referring to a neurologist or geriatrician.
There is no evidence to support or refute the benefit of interventional strategies such as driver rehabilitation for drivers with dementia.
PARKINSON DISEASE
Individuals with mild motor disability from Parkinson disease may be fit drivers. As the disease progresses, drivers with Parkinson disease may make more errors than healthy elders in visual scanning, signaling, vehicle positioning, and velocity regulation (eg, traveling so slowly that it may be unsafe).5 Clinicians can consider referring a patient with Parkinson disease for a baseline driving evaluation upon diagnosis, and then every 1 to 2 years for reassessment. Alternate transportation should be arranged as the disease progresses.
EPISODIC INCAPACITATION
Approximately 1% to 3% of all motor vehicle accidents are due to sudden incapacitation of an otherwise safe driver.
Syncope. Neurally mediated (vasovagal) syncope accounts for 30% to 35% of syncopal episodes while driving.6 Cardiac arrhythmias are the next most common cause and include bradyarrhythmias (7%), supraventricular tachyarrhythmias (2%–15%), and ventricular tachyarrhythmias (5%–17%). Because neurocardiogenic syncope often recurs, consider restricting driving for those with recurrent or severe neurocardiogenic syncopal episodes until symptoms are controlled.
Arrhythmias. Driving recommendations for various arrhythmias7,8 are listed in Table 2.
Many patients who have an implantable cardioverter-defibrillator (ICD) device experience an unexpected shock. For individuals with a history of ventricular tachycardia or fibrillation, the 5-year actuarial incidence of appropriate ICD shocks ranges between 55% and 70%. However, data indicate that 90% to 100% of drivers who received ICD discharges while driving continued to drive without causing motor vehicle accidents.9,10
Seizures. States differ in their rules for reporting drivers who have epilepsy or breakthrough seizures. Physicians should refer to their state regulations when counseling these patients.
POLYPHARMACY
Polypharmacy is common in older adults. Many take psychoactive drugs that can impair tracking, alertness, coordination, and reaction time. With the “Roadwise Rx” tool (www.roadwiserx.com), health care providers and patients can enter the names of medicines to check if they affect driving ability. Nonproprietary on-line tools such as “START” (Screening Tool to Alert doctors to Right Treatment) and “STOPP” (Screening Tool of Older Persons’ Potentially Inappropriate Prescriptions) can be used to prune medication lists.
DRIVING EVALUATION
America is a nation of highways overflowing with cars. Cars provide transportation but also reflect wealth and personality, particularly for men. Practically, the ability to drive a car allows older men and women to socialize in the community, shop for essentials, and take care of themselves without being a burden. Driving cessation can cause social isolation and depressive symptoms and can strain caregiver resources.
It is therefore understandable for health care providers to feel reluctant or uncomfortable counseling older adults to give up their driving privileges. A health care provider who identifies driving safety concerns can refer a patient to a geriatrician for further risk assessment or to a certified driver rehabilitation specialist (CDRS) for a driving evaluation. A CDRS will also offer the patient and caregiver information on local resources for transportation alternatives. A list of local CDRSs can be found on the Association for Driver Rehabilitation Specialists website (www.aded.net). Many hospitals have occupational therapists who are CDRSs.
The evaluation typically involves an assessment of the driver’s knowledge of traffic signs and laws, a cognitive assessment, possibly a simulation, and finally an on-road driving evaluation if deemed appropriate. Medicare coverage depends on diagnosis and the state carrier.
- Williams AF. Teenage drivers: patterns of risk. J Safety Res 2003; 34:5–15.
- Griffith HR, Okonkwo OC, Stewart CC, et al. Lower hippocampal volume predicts decrements in lane control among drivers with amnestic mild cognitive impairment. J Geriatr Psychiatry Neurol 2013; 26:259–266.
- de Simone V, Kaplan L, Patronas N, Wassermann EM, Grafman J. Driving abilities in frontotemporal dementia patients. Dement Geriatr Cogn Disord 2007; 23:1–7.
- Iverson DJ, Gronseth GS, Reger MA, Classen S, Dubinsky RM, Rizzo M; Quality Standards Subcomittee of the American Academy of Neurology. Practice parameter update: evaluation and management of driving risk in dementia: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2010; 74:1316–1324.
- Classen S, Brumback B, Monahan M, et al. Driving errors in Parkinson’s disease: moving closer to predicting on-road outcomes. Am J Occup Ther 2014; 68:77–85.
- Blitzer ML, Saliba BC, Ghantous AE, Marieb MA, Schoenfeld MH. Causes of impaired consciousness while driving a motorized vehicle. Am J Cardiol 2003; 91:1373–1374.
- Sorajja D, Shen WK. Driving guidelines and restrictions in patients with a history of cardiac arrhythmias, syncope,or implantable devices. Curr Treat Options Cardiovasc Med 2010; 12:443–456.
- Task force members; Vijgen J, Botto G, Camm J, et al. Consensus statement of the European Heart Rhythm Association: updated recommendations for driving by patients with implantable cardioverter defibrillators. Europace 2009; 11:1097–1107.
- Conti JB, Woodard DA, Tucker KJ, Bryant B, King LC, Curtis AB. Modification of patient driving behavior after implantation of a cardioverter defibrillator. Pacing Clin Electrophysiol 1997; 20:2200–2204.
- Lerecouvreux M, Aït Saïd M, Paziaud O, et al. Automobile driving and implantable defibrillators. Arch Mal Coeur Vaiss 2005; 98:288–293. Article in French.
- Williams AF. Teenage drivers: patterns of risk. J Safety Res 2003; 34:5–15.
- Griffith HR, Okonkwo OC, Stewart CC, et al. Lower hippocampal volume predicts decrements in lane control among drivers with amnestic mild cognitive impairment. J Geriatr Psychiatry Neurol 2013; 26:259–266.
- de Simone V, Kaplan L, Patronas N, Wassermann EM, Grafman J. Driving abilities in frontotemporal dementia patients. Dement Geriatr Cogn Disord 2007; 23:1–7.
- Iverson DJ, Gronseth GS, Reger MA, Classen S, Dubinsky RM, Rizzo M; Quality Standards Subcomittee of the American Academy of Neurology. Practice parameter update: evaluation and management of driving risk in dementia: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2010; 74:1316–1324.
- Classen S, Brumback B, Monahan M, et al. Driving errors in Parkinson’s disease: moving closer to predicting on-road outcomes. Am J Occup Ther 2014; 68:77–85.
- Blitzer ML, Saliba BC, Ghantous AE, Marieb MA, Schoenfeld MH. Causes of impaired consciousness while driving a motorized vehicle. Am J Cardiol 2003; 91:1373–1374.
- Sorajja D, Shen WK. Driving guidelines and restrictions in patients with a history of cardiac arrhythmias, syncope,or implantable devices. Curr Treat Options Cardiovasc Med 2010; 12:443–456.
- Task force members; Vijgen J, Botto G, Camm J, et al. Consensus statement of the European Heart Rhythm Association: updated recommendations for driving by patients with implantable cardioverter defibrillators. Europace 2009; 11:1097–1107.
- Conti JB, Woodard DA, Tucker KJ, Bryant B, King LC, Curtis AB. Modification of patient driving behavior after implantation of a cardioverter defibrillator. Pacing Clin Electrophysiol 1997; 20:2200–2204.
- Lerecouvreux M, Aït Saïd M, Paziaud O, et al. Automobile driving and implantable defibrillators. Arch Mal Coeur Vaiss 2005; 98:288–293. Article in French.

Is antibiotic treatment indicated in a patient with a positive urine culture but no symptoms?
The 2005 Infectious Diseases Society of America (IDSA) guidelines1 recommend screening pregnant women and patients who will undergo an invasive urologic procedure with a urine culture and treating them with antibiotics if bacteriuria is significant. The IDSA recommends against screening for or treating asymptomatic bacteriuria in other populations.
WHAT IS ASYMPTOMATIC BACTERIURIA?
A positive urine culture can represent three different conditions:
- Symptomatic urinary tract infection
- Contamination of the sample by organisms that are present distal to the bladder and that enter the urine at the time the specimen is collected
- Asymptomatic bacteriuria, defined as the isolation of a specified quantitative count of a single uropathogen in an appropriately collected urine specimen obtained from someone without symptoms or signs attributable to a urinary tract infection (Table 1). It represents the true presence of bacteria in the bladder and may be thought of as a state of colonization.
HOW COMMON IS ASYMPTOMATIC BACTERIURIA?

Rates vary depending on the age (higher in older persons), sex (higher in women), and presence of genitourinary abnormalities of the population studied. Prevalence rates are estimated to be 1% to 5% in healthy premenopausal women, 2% to 9% in pregnant women, 9% to 27% in diabetic women, 15% to 50% in elderly men and women in long-term care facilities, and 28% in patients undergoing hemodialysis.2–6 In patients with an indwelling urinary catheter, the rate goes up by 3% to 8% per day, and bacteriuria is nearly universal at 30 days.7,8 Asymptomatic bacteriuria can be transient, as commonly occurs in healthy young women, or it may be more prolonged, as commonly occurs in elderly patients or those with a chronic indwelling urinary catheter.
WHOM SHOULD WE SCREEN?
Screening for asymptomatic bacteriuria and treating it are strongly recommended (grade A-I recommendation) in pregnant women and in men who will undergo transurethral resection of the prostate.
Pregnant women have a risk of pyelonephritis 20 to 30 times higher if they have asymptomatic bacteriuria.9 Cohort studies and randomized clinical trials have consistently reported significant reductions in rates of pyelonephritis and low birth weight when antibiotic therapy is given for asymptomatic bacteriuria during pregnancy.
The ideal time to screen for this in pregnancy is between the 9th and 16th weeks of gestation. The appropriate screening test is a urine culture, since screening for pyuria has a low sensitivity and specificity. The choice of antibiotic is based on the results of culture. Antibiotics that have been safely used in these patients include nitrofurantoin, cephalexin, amoxicillin, and fosfomycin.10 The recommended treatment duration is between 3 and 7 days. Periodic screening for recurrent bacteriuria should be performed during the remainder of the pregnancy.
Men about to undergo transurethral resection of the prostate1 who have asymptomatic bacteriuria before the procedure have a 60% rate of bacteremia and a 6% to 10% rate of sepsis after the procedure if they do not receive antibiotic therapy. Clinical trials have documented significant reductions in these complications when antimicrobial therapy is given before the procedure.
The optimal time for obtaining the urine culture, the optimal time for starting antimicrobial therapy, and the optimal duration of antimicrobial therapy are not well defined, although some data support giving antibiotics the night before or just before the procedure.
The recommendation has been extrapolated to include not only men undergoing transurethral resection of the prostate but also any patient undergoing a urologic procedure associated with significant mucosal bleeding.
Women with catheter-acquired asymptomatic bacteriuria. If the bacteriuria persists 48 hours after catheter removal, the IDSA guidelines state that antibiotic therapy may be considered (grade B-I recommendation). However, there are no recommendations to screen women 48 hours after catheter removal.
WHAT IS THE EVIDENCE FOR NO TREATMENT?
Asymptomatic bacteriuria should not be screened for or treated in:
- Premenopausal women who are not pregnant (grade A-I recommendation)
- Diabetic women (A-I)
- Older persons residing in the community (A-II)
- Elderly residents of long-term care facilities (A-I)
- Patients with spinal cord injury (A-I)
- Patients with an indwelling urethral catheter (A-I).
Randomized controlled trials comparing antibiotic therapy with no therapy in these groups showed no benefit of antibiotic treatment in reducing the frequency of symptomatic urinary tract infection11–16 and no decrease in rates of fever or reinfection in patients with a long-term catheter.17 Moreover, in a number of trials,12,14,17 antibiotic therapy for asymptomatic bacteriuria was associated with an increase in adverse antimicrobial effects and reinfection with resistant organisms.
In transplant recipients. Because of lack of evidence, the 2005 IDSA guidelines could not make a recommendation for or against screening for or treatment of asymptomatic bacteriuria in renal transplant or other solid-organ transplant recipients (C-III). A more recent review18 noted a lack of consensus as to whether asymptomatic bacteriuria should be treated in renal transplant recipients. Based on available data, the authors recommended limiting routine screening for it to the first 1 to 3 months after renal transplantation and limiting treatment to 5 to 7 days, using the narrowest-spectrum antibiotic available.18
In prosthetic joint recipients. The 2005 IDSA guidelines recommended further research to determine if screening and treatment before surgical procedures with prosthetic implantation have clinical benefit.
Since then, two studies19,20 have suggested no benefit of screening or treatment before prosthetic joint implantation. Rates of prosthetic joint infection were not different in patients with asymptomatic bacteriuria before hip arthroplasty randomized to receive no antibiotic therapy vs those receiving antibiotic therapy specific for organisms cultured from the urine.19 Asymptomatic bacteriuria was found to be an independent risk factor for prosthetic joint infection.20 However, rates of joint infection were not different in those treated with antibiotics than in those not treated, and in no case were the microorganisms isolated in the prosthetic joint infection the same as in their preoperative urine culture.20
The authors concluded that asymptomatic bacteriuria may be a surrogate marker for increased risk of infection, but that preoperative antibiotic treatment was not beneficial.20
WHAT DOES 'ASYMPTOMATIC' MEAN?
According to the definition, asymptomatic refers to patients who do not have symptoms or signs attributable to a urinary tract infection. Thus, in patients who have symptoms or signs clearly attributable to another condition, screening with urine culture testing and treatment are not indicated. In nursing home residents, nonspecific symptoms such as a change in mental status, fever, and leukocytosis should not automatically be attributed to a positive urine culture without a careful evaluation for another cause, given the high prevalence of asymptomatic bacteriuria in this population.21 Screening with urine culture testing in this population is also not recommended for isolated foul-smelling or cloudy urine, after every urethral catheter change, upon admission, or after treatment to document cure.22
Finally, pyuria (defined as the presence of at least 5 to 10 white blood cells per high-power field) is not by itself a reason to perform a urine culture or to treat a positive urine culture, since pyuria is common in asymptomatic bacteriuria, as well as in other conditions associated with inflammation in the genitourinary system.1
TAKE-HOME POINTS
- Screening for and treating asymptomatic bacteriuria is recommended for pregnant women and for patients about to undergo an invasive urologic procedure associated with significant mucosal injury
- Screening and treatment are not recommended for premenopausal nonpregnant women, diabetic women, older persons residing in the community, elderly residents of long-term care facilities, patients with spinal cord injury, or patients with an indwelling urethral catheter.
- A urine culture should not be ordered, but if it is ordered, a positive culture should not be treated in a patient whose symptoms are attributable to another cause.
- Pyuria is not helpful in distinguishing symptomatic from asymptomatic bacteriuria.
- Nicolle LE, Bradley S, Colgan R, Rice JC, Schaeffer A, Hooton TM. Infectious Diseases Society of America guidelines for the diagnosis and treatment of asymptomatic bacteriuria in adults. Clin Infect Dis 2005; 40:643–654.
- Hooton TM, Scholes D, Stapleton AE, et al. A prospective study of asymptomatic bacteriuria in sexually active young women. N Engl J Med 2000; 343:992–997.
- Whalley P. Bacteriuria of pregnancy. Am J Obstet Gynecol 1967; 97:723–738.
- Zhanel GG, Nicolle LE, Harding GK. Prevalence of asymptomatic bacteriuria and associated host factors in women with diabetes mellitus. The Manitoba Diabetic Urinary Infection Study Group. Clin Infect Dis 1995; 21:316–322.
- Nicolle LE. Asymptomatic bacteriuria in the elderly. Infect Dis Clin North Am 1997; 11:647–662.
- Chaudhry A, Stone WJ, Breyer JA. Occurrence of pyuria and bacteriuria in asymptomatic hemodialysis patients. Am J Kidney Dis 1993; 21:180–183.
- Garibaldi RA, Burke JP, Dickman ML, Smith CB. Factors predisposing to bacteriuria during indwelling urethral catheterization. N Engl J Med 1974; 291:215–219.
- Warren JW, Tenney JH, Hoopes JM, Muncie HL, Anthony WC. A prospective microbiologic study of bacteriuria in patients with chronic indwelling urethral catheters. J Infect Dis 1982; 146:719–723.
- Smaill F, Vazquez JC. Antibiotics for asymptomatic bacteriuria in pregnancy. Cochrane Database Syst Rev 2007; 2:CD000490.
- Guinto VT, De Guia B, Festin MR, Dowswell T. Different antibiotic regimens for treating asymptomatic bacteriuria in pregnancy. Cochrane Database Syst Rev 2010; 9:CD007855.
- Asscher AW, Sussman M, Waters WE, et al. Asymptomatic significant bacteriuria in the non-pregnant woman. II. Response to treatment and follow-up. Br Med J 1969; 1:804–806.
- Harding GK, Zhanel GG, Nicolle LE, Cheang M; Manitoba Diabetes Urinary Tract Infection Study Group. Antimicrobial treatment in diabetic women with asymptomatic bacteriuria. N Engl J Med 2002; 347:1576–1583.
- Boscia JA, Kobasa WD, Knight RA, Abrutyn E, Levison ME, Kaye D. Therapy vs no therapy for bacteriuria in elderly ambulatory nonhospitalized women. JAMA 1987; 257:1067–1071.
- Nicolle LE, Mayhew WJ, Bryan L. Prospective randomized comparison of therapy and no therapy for asymptomatic bacteriuria in institutionalized elderly women. Am J Med 1987; 83:27–33.
- Nicolle LE, Bjornson J, Harding GK, MacDonell JA. Bacteriuria in elderly institutionalized men. N Engl J Med 1983; 309:1420–1425
- Mohler JL, Cowen DL, Flanigan RC. Suppression and treatment of urinary tract infection in patients with an intermittently catheterized neurogenic bladder. J Urol 1987; 138:336–340.
- Warren JW, Anthony WC, Hoopes JM, Muncie HL Jr. Cephalexin for susceptible bacteriuria in afebrile, long-term catheterized patients. JAMA 1982; 248:454–458.
- Parasuraman R, Julian K; AST Infectious Diseases Community of Practice. Urinary tract infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):327–336.
- Cordero-Ampuero J, González-Fernández E, Martínez-Vélez D, Esteban J. Are antibiotics necessary in hip arthroplasty with asymptomatic bacteriuria? Seeding risk with/without treatment. Clin Orthop Relat Res 2013; 471:3822–3829.
- Sousa R, Muñoz-Mahamud E, Quayle J, et al. Is asymptomatic bacteriuria a risk factor for prosthetic joint infection? Clin Infect Dis 2014: ciu235. Epub ahead of print.
- Orr PH, Nicolle LE, Duckworth H, et al. Febrile urinary infection in the institutionalized elderly. Am J Med 1996; 100:71–77.
- Zabarsky TF, Sethi AK, Donskey CJ. Sustained reduction in inappropriate treatment of asymptomatic bacteriuria in a long-term care facility through an educational intervention. Am J Infect Control 2008; 36:476–480.
The 2005 Infectious Diseases Society of America (IDSA) guidelines1 recommend screening pregnant women and patients who will undergo an invasive urologic procedure with a urine culture and treating them with antibiotics if bacteriuria is significant. The IDSA recommends against screening for or treating asymptomatic bacteriuria in other populations.
WHAT IS ASYMPTOMATIC BACTERIURIA?
A positive urine culture can represent three different conditions:
- Symptomatic urinary tract infection
- Contamination of the sample by organisms that are present distal to the bladder and that enter the urine at the time the specimen is collected
- Asymptomatic bacteriuria, defined as the isolation of a specified quantitative count of a single uropathogen in an appropriately collected urine specimen obtained from someone without symptoms or signs attributable to a urinary tract infection (Table 1). It represents the true presence of bacteria in the bladder and may be thought of as a state of colonization.
HOW COMMON IS ASYMPTOMATIC BACTERIURIA?
Rates vary depending on the age (higher in older persons), sex (higher in women), and presence of genitourinary abnormalities of the population studied. Prevalence rates are estimated to be 1% to 5% in healthy premenopausal women, 2% to 9% in pregnant women, 9% to 27% in diabetic women, 15% to 50% in elderly men and women in long-term care facilities, and 28% in patients undergoing hemodialysis.2–6 In patients with an indwelling urinary catheter, the rate goes up by 3% to 8% per day, and bacteriuria is nearly universal at 30 days.7,8 Asymptomatic bacteriuria can be transient, as commonly occurs in healthy young women, or it may be more prolonged, as commonly occurs in elderly patients or those with a chronic indwelling urinary catheter.
WHOM SHOULD WE SCREEN?
Screening for asymptomatic bacteriuria and treating it are strongly recommended (grade A-I recommendation) in pregnant women and in men who will undergo transurethral resection of the prostate.
Pregnant women have a risk of pyelonephritis 20 to 30 times higher if they have asymptomatic bacteriuria.9 Cohort studies and randomized clinical trials have consistently reported significant reductions in rates of pyelonephritis and low birth weight when antibiotic therapy is given for asymptomatic bacteriuria during pregnancy.
The ideal time to screen for this in pregnancy is between the 9th and 16th weeks of gestation. The appropriate screening test is a urine culture, since screening for pyuria has a low sensitivity and specificity. The choice of antibiotic is based on the results of culture. Antibiotics that have been safely used in these patients include nitrofurantoin, cephalexin, amoxicillin, and fosfomycin.10 The recommended treatment duration is between 3 and 7 days. Periodic screening for recurrent bacteriuria should be performed during the remainder of the pregnancy.
Men about to undergo transurethral resection of the prostate1 who have asymptomatic bacteriuria before the procedure have a 60% rate of bacteremia and a 6% to 10% rate of sepsis after the procedure if they do not receive antibiotic therapy. Clinical trials have documented significant reductions in these complications when antimicrobial therapy is given before the procedure.
The optimal time for obtaining the urine culture, the optimal time for starting antimicrobial therapy, and the optimal duration of antimicrobial therapy are not well defined, although some data support giving antibiotics the night before or just before the procedure.
The recommendation has been extrapolated to include not only men undergoing transurethral resection of the prostate but also any patient undergoing a urologic procedure associated with significant mucosal bleeding.
Women with catheter-acquired asymptomatic bacteriuria. If the bacteriuria persists 48 hours after catheter removal, the IDSA guidelines state that antibiotic therapy may be considered (grade B-I recommendation). However, there are no recommendations to screen women 48 hours after catheter removal.
WHAT IS THE EVIDENCE FOR NO TREATMENT?
Asymptomatic bacteriuria should not be screened for or treated in:
- Premenopausal women who are not pregnant (grade A-I recommendation)
- Diabetic women (A-I)
- Older persons residing in the community (A-II)
- Elderly residents of long-term care facilities (A-I)
- Patients with spinal cord injury (A-I)
- Patients with an indwelling urethral catheter (A-I).
Randomized controlled trials comparing antibiotic therapy with no therapy in these groups showed no benefit of antibiotic treatment in reducing the frequency of symptomatic urinary tract infection11–16 and no decrease in rates of fever or reinfection in patients with a long-term catheter.17 Moreover, in a number of trials,12,14,17 antibiotic therapy for asymptomatic bacteriuria was associated with an increase in adverse antimicrobial effects and reinfection with resistant organisms.
In transplant recipients. Because of lack of evidence, the 2005 IDSA guidelines could not make a recommendation for or against screening for or treatment of asymptomatic bacteriuria in renal transplant or other solid-organ transplant recipients (C-III). A more recent review18 noted a lack of consensus as to whether asymptomatic bacteriuria should be treated in renal transplant recipients. Based on available data, the authors recommended limiting routine screening for it to the first 1 to 3 months after renal transplantation and limiting treatment to 5 to 7 days, using the narrowest-spectrum antibiotic available.18
In prosthetic joint recipients. The 2005 IDSA guidelines recommended further research to determine if screening and treatment before surgical procedures with prosthetic implantation have clinical benefit.
Since then, two studies19,20 have suggested no benefit of screening or treatment before prosthetic joint implantation. Rates of prosthetic joint infection were not different in patients with asymptomatic bacteriuria before hip arthroplasty randomized to receive no antibiotic therapy vs those receiving antibiotic therapy specific for organisms cultured from the urine.19 Asymptomatic bacteriuria was found to be an independent risk factor for prosthetic joint infection.20 However, rates of joint infection were not different in those treated with antibiotics than in those not treated, and in no case were the microorganisms isolated in the prosthetic joint infection the same as in their preoperative urine culture.20
The authors concluded that asymptomatic bacteriuria may be a surrogate marker for increased risk of infection, but that preoperative antibiotic treatment was not beneficial.20
WHAT DOES 'ASYMPTOMATIC' MEAN?
According to the definition, asymptomatic refers to patients who do not have symptoms or signs attributable to a urinary tract infection. Thus, in patients who have symptoms or signs clearly attributable to another condition, screening with urine culture testing and treatment are not indicated. In nursing home residents, nonspecific symptoms such as a change in mental status, fever, and leukocytosis should not automatically be attributed to a positive urine culture without a careful evaluation for another cause, given the high prevalence of asymptomatic bacteriuria in this population.21 Screening with urine culture testing in this population is also not recommended for isolated foul-smelling or cloudy urine, after every urethral catheter change, upon admission, or after treatment to document cure.22
Finally, pyuria (defined as the presence of at least 5 to 10 white blood cells per high-power field) is not by itself a reason to perform a urine culture or to treat a positive urine culture, since pyuria is common in asymptomatic bacteriuria, as well as in other conditions associated with inflammation in the genitourinary system.1
TAKE-HOME POINTS
- Screening for and treating asymptomatic bacteriuria is recommended for pregnant women and for patients about to undergo an invasive urologic procedure associated with significant mucosal injury
- Screening and treatment are not recommended for premenopausal nonpregnant women, diabetic women, older persons residing in the community, elderly residents of long-term care facilities, patients with spinal cord injury, or patients with an indwelling urethral catheter.
- A urine culture should not be ordered, but if it is ordered, a positive culture should not be treated in a patient whose symptoms are attributable to another cause.
- Pyuria is not helpful in distinguishing symptomatic from asymptomatic bacteriuria.
The 2005 Infectious Diseases Society of America (IDSA) guidelines1 recommend screening pregnant women and patients who will undergo an invasive urologic procedure with a urine culture and treating them with antibiotics if bacteriuria is significant. The IDSA recommends against screening for or treating asymptomatic bacteriuria in other populations.
WHAT IS ASYMPTOMATIC BACTERIURIA?
A positive urine culture can represent three different conditions:
- Symptomatic urinary tract infection
- Contamination of the sample by organisms that are present distal to the bladder and that enter the urine at the time the specimen is collected
- Asymptomatic bacteriuria, defined as the isolation of a specified quantitative count of a single uropathogen in an appropriately collected urine specimen obtained from someone without symptoms or signs attributable to a urinary tract infection (Table 1). It represents the true presence of bacteria in the bladder and may be thought of as a state of colonization.
HOW COMMON IS ASYMPTOMATIC BACTERIURIA?
Rates vary depending on the age (higher in older persons), sex (higher in women), and presence of genitourinary abnormalities of the population studied. Prevalence rates are estimated to be 1% to 5% in healthy premenopausal women, 2% to 9% in pregnant women, 9% to 27% in diabetic women, 15% to 50% in elderly men and women in long-term care facilities, and 28% in patients undergoing hemodialysis.2–6 In patients with an indwelling urinary catheter, the rate goes up by 3% to 8% per day, and bacteriuria is nearly universal at 30 days.7,8 Asymptomatic bacteriuria can be transient, as commonly occurs in healthy young women, or it may be more prolonged, as commonly occurs in elderly patients or those with a chronic indwelling urinary catheter.
WHOM SHOULD WE SCREEN?
Screening for asymptomatic bacteriuria and treating it are strongly recommended (grade A-I recommendation) in pregnant women and in men who will undergo transurethral resection of the prostate.
Pregnant women have a risk of pyelonephritis 20 to 30 times higher if they have asymptomatic bacteriuria.9 Cohort studies and randomized clinical trials have consistently reported significant reductions in rates of pyelonephritis and low birth weight when antibiotic therapy is given for asymptomatic bacteriuria during pregnancy.
The ideal time to screen for this in pregnancy is between the 9th and 16th weeks of gestation. The appropriate screening test is a urine culture, since screening for pyuria has a low sensitivity and specificity. The choice of antibiotic is based on the results of culture. Antibiotics that have been safely used in these patients include nitrofurantoin, cephalexin, amoxicillin, and fosfomycin.10 The recommended treatment duration is between 3 and 7 days. Periodic screening for recurrent bacteriuria should be performed during the remainder of the pregnancy.
Men about to undergo transurethral resection of the prostate1 who have asymptomatic bacteriuria before the procedure have a 60% rate of bacteremia and a 6% to 10% rate of sepsis after the procedure if they do not receive antibiotic therapy. Clinical trials have documented significant reductions in these complications when antimicrobial therapy is given before the procedure.
The optimal time for obtaining the urine culture, the optimal time for starting antimicrobial therapy, and the optimal duration of antimicrobial therapy are not well defined, although some data support giving antibiotics the night before or just before the procedure.
The recommendation has been extrapolated to include not only men undergoing transurethral resection of the prostate but also any patient undergoing a urologic procedure associated with significant mucosal bleeding.
Women with catheter-acquired asymptomatic bacteriuria. If the bacteriuria persists 48 hours after catheter removal, the IDSA guidelines state that antibiotic therapy may be considered (grade B-I recommendation). However, there are no recommendations to screen women 48 hours after catheter removal.
WHAT IS THE EVIDENCE FOR NO TREATMENT?
Asymptomatic bacteriuria should not be screened for or treated in:
- Premenopausal women who are not pregnant (grade A-I recommendation)
- Diabetic women (A-I)
- Older persons residing in the community (A-II)
- Elderly residents of long-term care facilities (A-I)
- Patients with spinal cord injury (A-I)
- Patients with an indwelling urethral catheter (A-I).
Randomized controlled trials comparing antibiotic therapy with no therapy in these groups showed no benefit of antibiotic treatment in reducing the frequency of symptomatic urinary tract infection11–16 and no decrease in rates of fever or reinfection in patients with a long-term catheter.17 Moreover, in a number of trials,12,14,17 antibiotic therapy for asymptomatic bacteriuria was associated with an increase in adverse antimicrobial effects and reinfection with resistant organisms.
In transplant recipients. Because of lack of evidence, the 2005 IDSA guidelines could not make a recommendation for or against screening for or treatment of asymptomatic bacteriuria in renal transplant or other solid-organ transplant recipients (C-III). A more recent review18 noted a lack of consensus as to whether asymptomatic bacteriuria should be treated in renal transplant recipients. Based on available data, the authors recommended limiting routine screening for it to the first 1 to 3 months after renal transplantation and limiting treatment to 5 to 7 days, using the narrowest-spectrum antibiotic available.18
In prosthetic joint recipients. The 2005 IDSA guidelines recommended further research to determine if screening and treatment before surgical procedures with prosthetic implantation have clinical benefit.
Since then, two studies19,20 have suggested no benefit of screening or treatment before prosthetic joint implantation. Rates of prosthetic joint infection were not different in patients with asymptomatic bacteriuria before hip arthroplasty randomized to receive no antibiotic therapy vs those receiving antibiotic therapy specific for organisms cultured from the urine.19 Asymptomatic bacteriuria was found to be an independent risk factor for prosthetic joint infection.20 However, rates of joint infection were not different in those treated with antibiotics than in those not treated, and in no case were the microorganisms isolated in the prosthetic joint infection the same as in their preoperative urine culture.20
The authors concluded that asymptomatic bacteriuria may be a surrogate marker for increased risk of infection, but that preoperative antibiotic treatment was not beneficial.20
WHAT DOES 'ASYMPTOMATIC' MEAN?
According to the definition, asymptomatic refers to patients who do not have symptoms or signs attributable to a urinary tract infection. Thus, in patients who have symptoms or signs clearly attributable to another condition, screening with urine culture testing and treatment are not indicated. In nursing home residents, nonspecific symptoms such as a change in mental status, fever, and leukocytosis should not automatically be attributed to a positive urine culture without a careful evaluation for another cause, given the high prevalence of asymptomatic bacteriuria in this population.21 Screening with urine culture testing in this population is also not recommended for isolated foul-smelling or cloudy urine, after every urethral catheter change, upon admission, or after treatment to document cure.22
Finally, pyuria (defined as the presence of at least 5 to 10 white blood cells per high-power field) is not by itself a reason to perform a urine culture or to treat a positive urine culture, since pyuria is common in asymptomatic bacteriuria, as well as in other conditions associated with inflammation in the genitourinary system.1
TAKE-HOME POINTS
- Screening for and treating asymptomatic bacteriuria is recommended for pregnant women and for patients about to undergo an invasive urologic procedure associated with significant mucosal injury
- Screening and treatment are not recommended for premenopausal nonpregnant women, diabetic women, older persons residing in the community, elderly residents of long-term care facilities, patients with spinal cord injury, or patients with an indwelling urethral catheter.
- A urine culture should not be ordered, but if it is ordered, a positive culture should not be treated in a patient whose symptoms are attributable to another cause.
- Pyuria is not helpful in distinguishing symptomatic from asymptomatic bacteriuria.
- Nicolle LE, Bradley S, Colgan R, Rice JC, Schaeffer A, Hooton TM. Infectious Diseases Society of America guidelines for the diagnosis and treatment of asymptomatic bacteriuria in adults. Clin Infect Dis 2005; 40:643–654.
- Hooton TM, Scholes D, Stapleton AE, et al. A prospective study of asymptomatic bacteriuria in sexually active young women. N Engl J Med 2000; 343:992–997.
- Whalley P. Bacteriuria of pregnancy. Am J Obstet Gynecol 1967; 97:723–738.
- Zhanel GG, Nicolle LE, Harding GK. Prevalence of asymptomatic bacteriuria and associated host factors in women with diabetes mellitus. The Manitoba Diabetic Urinary Infection Study Group. Clin Infect Dis 1995; 21:316–322.
- Nicolle LE. Asymptomatic bacteriuria in the elderly. Infect Dis Clin North Am 1997; 11:647–662.
- Chaudhry A, Stone WJ, Breyer JA. Occurrence of pyuria and bacteriuria in asymptomatic hemodialysis patients. Am J Kidney Dis 1993; 21:180–183.
- Garibaldi RA, Burke JP, Dickman ML, Smith CB. Factors predisposing to bacteriuria during indwelling urethral catheterization. N Engl J Med 1974; 291:215–219.
- Warren JW, Tenney JH, Hoopes JM, Muncie HL, Anthony WC. A prospective microbiologic study of bacteriuria in patients with chronic indwelling urethral catheters. J Infect Dis 1982; 146:719–723.
- Smaill F, Vazquez JC. Antibiotics for asymptomatic bacteriuria in pregnancy. Cochrane Database Syst Rev 2007; 2:CD000490.
- Guinto VT, De Guia B, Festin MR, Dowswell T. Different antibiotic regimens for treating asymptomatic bacteriuria in pregnancy. Cochrane Database Syst Rev 2010; 9:CD007855.
- Asscher AW, Sussman M, Waters WE, et al. Asymptomatic significant bacteriuria in the non-pregnant woman. II. Response to treatment and follow-up. Br Med J 1969; 1:804–806.
- Harding GK, Zhanel GG, Nicolle LE, Cheang M; Manitoba Diabetes Urinary Tract Infection Study Group. Antimicrobial treatment in diabetic women with asymptomatic bacteriuria. N Engl J Med 2002; 347:1576–1583.
- Boscia JA, Kobasa WD, Knight RA, Abrutyn E, Levison ME, Kaye D. Therapy vs no therapy for bacteriuria in elderly ambulatory nonhospitalized women. JAMA 1987; 257:1067–1071.
- Nicolle LE, Mayhew WJ, Bryan L. Prospective randomized comparison of therapy and no therapy for asymptomatic bacteriuria in institutionalized elderly women. Am J Med 1987; 83:27–33.
- Nicolle LE, Bjornson J, Harding GK, MacDonell JA. Bacteriuria in elderly institutionalized men. N Engl J Med 1983; 309:1420–1425
- Mohler JL, Cowen DL, Flanigan RC. Suppression and treatment of urinary tract infection in patients with an intermittently catheterized neurogenic bladder. J Urol 1987; 138:336–340.
- Warren JW, Anthony WC, Hoopes JM, Muncie HL Jr. Cephalexin for susceptible bacteriuria in afebrile, long-term catheterized patients. JAMA 1982; 248:454–458.
- Parasuraman R, Julian K; AST Infectious Diseases Community of Practice. Urinary tract infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):327–336.
- Cordero-Ampuero J, González-Fernández E, Martínez-Vélez D, Esteban J. Are antibiotics necessary in hip arthroplasty with asymptomatic bacteriuria? Seeding risk with/without treatment. Clin Orthop Relat Res 2013; 471:3822–3829.
- Sousa R, Muñoz-Mahamud E, Quayle J, et al. Is asymptomatic bacteriuria a risk factor for prosthetic joint infection? Clin Infect Dis 2014: ciu235. Epub ahead of print.
- Orr PH, Nicolle LE, Duckworth H, et al. Febrile urinary infection in the institutionalized elderly. Am J Med 1996; 100:71–77.
- Zabarsky TF, Sethi AK, Donskey CJ. Sustained reduction in inappropriate treatment of asymptomatic bacteriuria in a long-term care facility through an educational intervention. Am J Infect Control 2008; 36:476–480.
- Nicolle LE, Bradley S, Colgan R, Rice JC, Schaeffer A, Hooton TM. Infectious Diseases Society of America guidelines for the diagnosis and treatment of asymptomatic bacteriuria in adults. Clin Infect Dis 2005; 40:643–654.
- Hooton TM, Scholes D, Stapleton AE, et al. A prospective study of asymptomatic bacteriuria in sexually active young women. N Engl J Med 2000; 343:992–997.
- Whalley P. Bacteriuria of pregnancy. Am J Obstet Gynecol 1967; 97:723–738.
- Zhanel GG, Nicolle LE, Harding GK. Prevalence of asymptomatic bacteriuria and associated host factors in women with diabetes mellitus. The Manitoba Diabetic Urinary Infection Study Group. Clin Infect Dis 1995; 21:316–322.
- Nicolle LE. Asymptomatic bacteriuria in the elderly. Infect Dis Clin North Am 1997; 11:647–662.
- Chaudhry A, Stone WJ, Breyer JA. Occurrence of pyuria and bacteriuria in asymptomatic hemodialysis patients. Am J Kidney Dis 1993; 21:180–183.
- Garibaldi RA, Burke JP, Dickman ML, Smith CB. Factors predisposing to bacteriuria during indwelling urethral catheterization. N Engl J Med 1974; 291:215–219.
- Warren JW, Tenney JH, Hoopes JM, Muncie HL, Anthony WC. A prospective microbiologic study of bacteriuria in patients with chronic indwelling urethral catheters. J Infect Dis 1982; 146:719–723.
- Smaill F, Vazquez JC. Antibiotics for asymptomatic bacteriuria in pregnancy. Cochrane Database Syst Rev 2007; 2:CD000490.
- Guinto VT, De Guia B, Festin MR, Dowswell T. Different antibiotic regimens for treating asymptomatic bacteriuria in pregnancy. Cochrane Database Syst Rev 2010; 9:CD007855.
- Asscher AW, Sussman M, Waters WE, et al. Asymptomatic significant bacteriuria in the non-pregnant woman. II. Response to treatment and follow-up. Br Med J 1969; 1:804–806.
- Harding GK, Zhanel GG, Nicolle LE, Cheang M; Manitoba Diabetes Urinary Tract Infection Study Group. Antimicrobial treatment in diabetic women with asymptomatic bacteriuria. N Engl J Med 2002; 347:1576–1583.
- Boscia JA, Kobasa WD, Knight RA, Abrutyn E, Levison ME, Kaye D. Therapy vs no therapy for bacteriuria in elderly ambulatory nonhospitalized women. JAMA 1987; 257:1067–1071.
- Nicolle LE, Mayhew WJ, Bryan L. Prospective randomized comparison of therapy and no therapy for asymptomatic bacteriuria in institutionalized elderly women. Am J Med 1987; 83:27–33.
- Nicolle LE, Bjornson J, Harding GK, MacDonell JA. Bacteriuria in elderly institutionalized men. N Engl J Med 1983; 309:1420–1425
- Mohler JL, Cowen DL, Flanigan RC. Suppression and treatment of urinary tract infection in patients with an intermittently catheterized neurogenic bladder. J Urol 1987; 138:336–340.
- Warren JW, Anthony WC, Hoopes JM, Muncie HL Jr. Cephalexin for susceptible bacteriuria in afebrile, long-term catheterized patients. JAMA 1982; 248:454–458.
- Parasuraman R, Julian K; AST Infectious Diseases Community of Practice. Urinary tract infections in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):327–336.
- Cordero-Ampuero J, González-Fernández E, Martínez-Vélez D, Esteban J. Are antibiotics necessary in hip arthroplasty with asymptomatic bacteriuria? Seeding risk with/without treatment. Clin Orthop Relat Res 2013; 471:3822–3829.
- Sousa R, Muñoz-Mahamud E, Quayle J, et al. Is asymptomatic bacteriuria a risk factor for prosthetic joint infection? Clin Infect Dis 2014: ciu235. Epub ahead of print.
- Orr PH, Nicolle LE, Duckworth H, et al. Febrile urinary infection in the institutionalized elderly. Am J Med 1996; 100:71–77.
- Zabarsky TF, Sethi AK, Donskey CJ. Sustained reduction in inappropriate treatment of asymptomatic bacteriuria in a long-term care facility through an educational intervention. Am J Infect Control 2008; 36:476–480.
Does massive hemoptysis always merit diagnostic bronchoscopy?
Yes, all patients with massive hemoptysis should undergo diagnostic bronchoscopy. The procedure plays an important role in protecting the airway, maintaining ventilation, finding the site and underlying cause of the bleeding, and in some cases stopping the bleeding, either temporarily or definitively.

Frightening to patients, massive hemoptysis is a medical emergency and demands immediate attention by an experienced pulmonary team.1 Hemoptysis can be the initial presentation of an underlying infectious, autoimmune, or malignant disorder (Table 1).2 Fortunately, most cases of hemoptysis are not massive or life-threatening.1
WHAT IS ‘MASSIVE’ HEMOPTYSIS?
Numerous studies have defined massive hemoptysis on the basis of the volume of blood lost over time, eg, more than 1 L in 24 hours or more than 400 mL in 6 hours.
Ibrahim3 has proposed that we move away from using the word “massive,” which is not useful, and instead think in terms of “life-threatening” hemoptysis, defined as any of the following:
- More than 100 mL of blood lost in 24 hours (a low number, but blood loss is hard to estimate accurately)
- Causing abnormal gas exchange due to airway obstruction
- Causing hemodynamic instability.
In this article, we use the traditional “massive” terminology.
BRONCHOSCOPY IS SUPERIOR TO IMAGING FOR DIAGNOSIS
Radiography can help identify the side or the site of bleeding in 33% to 82% of patients, and computed tomography can in 70% to 88.5%.4 Magnetic resonance imaging may also have a role; one study found it useful in cases of thoracic endometriosis during the quiescent stage.5 However, transferring a patient who is actively bleeding out of the intensive care unit for imaging can be challenging.
Flexible bronchoscopy is superior to radiographic imaging in evaluating massive hemoptysis: it can be performed at the bed-side and can include therapeutic procedures to control the bleeding until the patient can undergo a definitive therapeutic procedure.6 It has been found helpful in identifying the side of bleeding in 73% to 93% of cases of massive hemoptysis.6
However, one should consider starting the procedure with a rigid bronchoscope, which protects the airway better and allows for better ventilation during the procedure than a flexible one. One can use it to isolate the nonbleeding lung and to apply pressure to the bleeding site if it is in the main bronchus.7 Measuring 12 mm in diameter, a rigid scope cannot go as far into the lung as a flexible bronchoscope (measuring 6.4 mm), but a flexible bronchoscope can be introduced through the rigid bronchoscope to go further in.
MANAGEMENT OPTIONS
The management team should include an anesthesiologist, an intensivist, a thoracic surgeon, an interventional radiologist, and an interventional pulmonologist.
In the intensive care unit, the patient should be placed in the lateral decubitus position on the bleeding side. To maintain ventilation, the nonbleeding lung should be intubated with a large-bore endotracheal tube (internal diameter 8.5–9.0 mm) or, ideally, with a rigid bronchoscope.6 Meanwhile, the patient’s circulatory status should be stabilized with adequate fluid resuscitation and transfusion of blood products, with close monitoring.
Once the bleeding site is found, a bronchoscopic treatment is selected based on the cause of the bleeding. Massive hemoptysis usually arises from high-pressure bronchial vessels (90%) or, less commonly, from non-bronchial vessels or capillaries (10%).8 A variety of agents (eg, cold saline lavage, epinephrine 1:20,000) can be instilled through the bronchoscope to slow the bleeding and offer better visualization of the airway.6
If a bleeding intrabronchial lesion is identified, such as a malignant tracheobronchial tumor, local coagulation therapy can be applied through the bronchoscope. Options include laser treatment, argon plasma coagulation, cryotherapy, and electrocautery (Figure 1).9,10
If the bleeding persists or cannot be localized to a particular subsegment, an endobronchial balloon plug can be placed proximally (Figure 2). This can be left in place to isolate the bleeding and apply tamponade until a definitive procedure can be performed, such as bronchial artery embolization, radiation therapy, or surgery.
- Jean-Baptiste E. Clinical assessment and management of massive hemoptysis. Crit Care Med 2000; 28:1642–1647.
- Abi Khalil S, Gourdier AL, Aoun N, et al. Cystic and cavitary lesions of the lung: imaging characteristics and differential diagnosis [in French]. J Radiol 2010; 91:465–473.
- Ibrahim WH. Massive haemoptysis: the definition should be revised. Eur Respir J 2008; 32:1131–1132.
- Khalil A, Soussan M, Mangiapan G, Fartoukh M, Parrot A, Carette MF. Utility of high-resolution chest CT scan in the emergency management of haemoptysis in the intensive care unit: severity, localization and aetiology. Br J Radiol 2007; 80:21–25.
- Cassina PC, Hauser M, Kacl G, Imthurn B, Schröder S, Weder W. Catamenial hemoptysis. Diagnosis with MRI. Chest 1997; 111:1447–1450.
- Sakr L, Dutau H. Massive hemoptysis: an update on the role of bronchoscopy in diagnosis and management. Respiration 2010; 80:38–58.
- Conlan AA, Hurwitz SS. Management of massive haemoptysis with the rigid bronchoscope and cold saline lavage. Thorax 1980; 35:901–904.
- Deffebach ME, Charan NB, Lakshminarayan S, Butler J. The bronchial circulation. Small, but a vital attribute of the lung. Am Rev Respir Dis 1987; 135:463–481.
- Morice RC, Ece T, Ece F, Keus L. Endobronchial argon plasma coagulation for treatment of hemoptysis and neoplastic airway obstruction. Chest 2001; 119:781–787.
- Sheski FD, Mathur PN. Cryotherapy, electrocautery, and brachytherapy. Clin Chest Med 1999; 20:123–138.
Yes, all patients with massive hemoptysis should undergo diagnostic bronchoscopy. The procedure plays an important role in protecting the airway, maintaining ventilation, finding the site and underlying cause of the bleeding, and in some cases stopping the bleeding, either temporarily or definitively.
Frightening to patients, massive hemoptysis is a medical emergency and demands immediate attention by an experienced pulmonary team.1 Hemoptysis can be the initial presentation of an underlying infectious, autoimmune, or malignant disorder (Table 1).2 Fortunately, most cases of hemoptysis are not massive or life-threatening.1
WHAT IS ‘MASSIVE’ HEMOPTYSIS?
Numerous studies have defined massive hemoptysis on the basis of the volume of blood lost over time, eg, more than 1 L in 24 hours or more than 400 mL in 6 hours.
Ibrahim3 has proposed that we move away from using the word “massive,” which is not useful, and instead think in terms of “life-threatening” hemoptysis, defined as any of the following:
- More than 100 mL of blood lost in 24 hours (a low number, but blood loss is hard to estimate accurately)
- Causing abnormal gas exchange due to airway obstruction
- Causing hemodynamic instability.
In this article, we use the traditional “massive” terminology.
BRONCHOSCOPY IS SUPERIOR TO IMAGING FOR DIAGNOSIS
Radiography can help identify the side or the site of bleeding in 33% to 82% of patients, and computed tomography can in 70% to 88.5%.4 Magnetic resonance imaging may also have a role; one study found it useful in cases of thoracic endometriosis during the quiescent stage.5 However, transferring a patient who is actively bleeding out of the intensive care unit for imaging can be challenging.
Flexible bronchoscopy is superior to radiographic imaging in evaluating massive hemoptysis: it can be performed at the bed-side and can include therapeutic procedures to control the bleeding until the patient can undergo a definitive therapeutic procedure.6 It has been found helpful in identifying the side of bleeding in 73% to 93% of cases of massive hemoptysis.6
However, one should consider starting the procedure with a rigid bronchoscope, which protects the airway better and allows for better ventilation during the procedure than a flexible one. One can use it to isolate the nonbleeding lung and to apply pressure to the bleeding site if it is in the main bronchus.7 Measuring 12 mm in diameter, a rigid scope cannot go as far into the lung as a flexible bronchoscope (measuring 6.4 mm), but a flexible bronchoscope can be introduced through the rigid bronchoscope to go further in.
MANAGEMENT OPTIONS
The management team should include an anesthesiologist, an intensivist, a thoracic surgeon, an interventional radiologist, and an interventional pulmonologist.
In the intensive care unit, the patient should be placed in the lateral decubitus position on the bleeding side. To maintain ventilation, the nonbleeding lung should be intubated with a large-bore endotracheal tube (internal diameter 8.5–9.0 mm) or, ideally, with a rigid bronchoscope.6 Meanwhile, the patient’s circulatory status should be stabilized with adequate fluid resuscitation and transfusion of blood products, with close monitoring.
Once the bleeding site is found, a bronchoscopic treatment is selected based on the cause of the bleeding. Massive hemoptysis usually arises from high-pressure bronchial vessels (90%) or, less commonly, from non-bronchial vessels or capillaries (10%).8 A variety of agents (eg, cold saline lavage, epinephrine 1:20,000) can be instilled through the bronchoscope to slow the bleeding and offer better visualization of the airway.6
If a bleeding intrabronchial lesion is identified, such as a malignant tracheobronchial tumor, local coagulation therapy can be applied through the bronchoscope. Options include laser treatment, argon plasma coagulation, cryotherapy, and electrocautery (Figure 1).9,10
If the bleeding persists or cannot be localized to a particular subsegment, an endobronchial balloon plug can be placed proximally (Figure 2). This can be left in place to isolate the bleeding and apply tamponade until a definitive procedure can be performed, such as bronchial artery embolization, radiation therapy, or surgery.
Yes, all patients with massive hemoptysis should undergo diagnostic bronchoscopy. The procedure plays an important role in protecting the airway, maintaining ventilation, finding the site and underlying cause of the bleeding, and in some cases stopping the bleeding, either temporarily or definitively.
Frightening to patients, massive hemoptysis is a medical emergency and demands immediate attention by an experienced pulmonary team.1 Hemoptysis can be the initial presentation of an underlying infectious, autoimmune, or malignant disorder (Table 1).2 Fortunately, most cases of hemoptysis are not massive or life-threatening.1
WHAT IS ‘MASSIVE’ HEMOPTYSIS?
Numerous studies have defined massive hemoptysis on the basis of the volume of blood lost over time, eg, more than 1 L in 24 hours or more than 400 mL in 6 hours.
Ibrahim3 has proposed that we move away from using the word “massive,” which is not useful, and instead think in terms of “life-threatening” hemoptysis, defined as any of the following:
- More than 100 mL of blood lost in 24 hours (a low number, but blood loss is hard to estimate accurately)
- Causing abnormal gas exchange due to airway obstruction
- Causing hemodynamic instability.
In this article, we use the traditional “massive” terminology.
BRONCHOSCOPY IS SUPERIOR TO IMAGING FOR DIAGNOSIS
Radiography can help identify the side or the site of bleeding in 33% to 82% of patients, and computed tomography can in 70% to 88.5%.4 Magnetic resonance imaging may also have a role; one study found it useful in cases of thoracic endometriosis during the quiescent stage.5 However, transferring a patient who is actively bleeding out of the intensive care unit for imaging can be challenging.
Flexible bronchoscopy is superior to radiographic imaging in evaluating massive hemoptysis: it can be performed at the bed-side and can include therapeutic procedures to control the bleeding until the patient can undergo a definitive therapeutic procedure.6 It has been found helpful in identifying the side of bleeding in 73% to 93% of cases of massive hemoptysis.6
However, one should consider starting the procedure with a rigid bronchoscope, which protects the airway better and allows for better ventilation during the procedure than a flexible one. One can use it to isolate the nonbleeding lung and to apply pressure to the bleeding site if it is in the main bronchus.7 Measuring 12 mm in diameter, a rigid scope cannot go as far into the lung as a flexible bronchoscope (measuring 6.4 mm), but a flexible bronchoscope can be introduced through the rigid bronchoscope to go further in.
MANAGEMENT OPTIONS
The management team should include an anesthesiologist, an intensivist, a thoracic surgeon, an interventional radiologist, and an interventional pulmonologist.
In the intensive care unit, the patient should be placed in the lateral decubitus position on the bleeding side. To maintain ventilation, the nonbleeding lung should be intubated with a large-bore endotracheal tube (internal diameter 8.5–9.0 mm) or, ideally, with a rigid bronchoscope.6 Meanwhile, the patient’s circulatory status should be stabilized with adequate fluid resuscitation and transfusion of blood products, with close monitoring.
Once the bleeding site is found, a bronchoscopic treatment is selected based on the cause of the bleeding. Massive hemoptysis usually arises from high-pressure bronchial vessels (90%) or, less commonly, from non-bronchial vessels or capillaries (10%).8 A variety of agents (eg, cold saline lavage, epinephrine 1:20,000) can be instilled through the bronchoscope to slow the bleeding and offer better visualization of the airway.6
If a bleeding intrabronchial lesion is identified, such as a malignant tracheobronchial tumor, local coagulation therapy can be applied through the bronchoscope. Options include laser treatment, argon plasma coagulation, cryotherapy, and electrocautery (Figure 1).9,10
If the bleeding persists or cannot be localized to a particular subsegment, an endobronchial balloon plug can be placed proximally (Figure 2). This can be left in place to isolate the bleeding and apply tamponade until a definitive procedure can be performed, such as bronchial artery embolization, radiation therapy, or surgery.
- Jean-Baptiste E. Clinical assessment and management of massive hemoptysis. Crit Care Med 2000; 28:1642–1647.
- Abi Khalil S, Gourdier AL, Aoun N, et al. Cystic and cavitary lesions of the lung: imaging characteristics and differential diagnosis [in French]. J Radiol 2010; 91:465–473.
- Ibrahim WH. Massive haemoptysis: the definition should be revised. Eur Respir J 2008; 32:1131–1132.
- Khalil A, Soussan M, Mangiapan G, Fartoukh M, Parrot A, Carette MF. Utility of high-resolution chest CT scan in the emergency management of haemoptysis in the intensive care unit: severity, localization and aetiology. Br J Radiol 2007; 80:21–25.
- Cassina PC, Hauser M, Kacl G, Imthurn B, Schröder S, Weder W. Catamenial hemoptysis. Diagnosis with MRI. Chest 1997; 111:1447–1450.
- Sakr L, Dutau H. Massive hemoptysis: an update on the role of bronchoscopy in diagnosis and management. Respiration 2010; 80:38–58.
- Conlan AA, Hurwitz SS. Management of massive haemoptysis with the rigid bronchoscope and cold saline lavage. Thorax 1980; 35:901–904.
- Deffebach ME, Charan NB, Lakshminarayan S, Butler J. The bronchial circulation. Small, but a vital attribute of the lung. Am Rev Respir Dis 1987; 135:463–481.
- Morice RC, Ece T, Ece F, Keus L. Endobronchial argon plasma coagulation for treatment of hemoptysis and neoplastic airway obstruction. Chest 2001; 119:781–787.
- Sheski FD, Mathur PN. Cryotherapy, electrocautery, and brachytherapy. Clin Chest Med 1999; 20:123–138.
- Jean-Baptiste E. Clinical assessment and management of massive hemoptysis. Crit Care Med 2000; 28:1642–1647.
- Abi Khalil S, Gourdier AL, Aoun N, et al. Cystic and cavitary lesions of the lung: imaging characteristics and differential diagnosis [in French]. J Radiol 2010; 91:465–473.
- Ibrahim WH. Massive haemoptysis: the definition should be revised. Eur Respir J 2008; 32:1131–1132.
- Khalil A, Soussan M, Mangiapan G, Fartoukh M, Parrot A, Carette MF. Utility of high-resolution chest CT scan in the emergency management of haemoptysis in the intensive care unit: severity, localization and aetiology. Br J Radiol 2007; 80:21–25.
- Cassina PC, Hauser M, Kacl G, Imthurn B, Schröder S, Weder W. Catamenial hemoptysis. Diagnosis with MRI. Chest 1997; 111:1447–1450.
- Sakr L, Dutau H. Massive hemoptysis: an update on the role of bronchoscopy in diagnosis and management. Respiration 2010; 80:38–58.
- Conlan AA, Hurwitz SS. Management of massive haemoptysis with the rigid bronchoscope and cold saline lavage. Thorax 1980; 35:901–904.
- Deffebach ME, Charan NB, Lakshminarayan S, Butler J. The bronchial circulation. Small, but a vital attribute of the lung. Am Rev Respir Dis 1987; 135:463–481.
- Morice RC, Ece T, Ece F, Keus L. Endobronchial argon plasma coagulation for treatment of hemoptysis and neoplastic airway obstruction. Chest 2001; 119:781–787.
- Sheski FD, Mathur PN. Cryotherapy, electrocautery, and brachytherapy. Clin Chest Med 1999; 20:123–138.