User login
Bilateral Superior Labrum Anterior to Posterior (SLAP) Tears With Abnormal Anatomy of Biceps Tendon
The biceps brachii derives its name from the 2 heads of the muscle. The short head originates from the coracoid apex, with the coracobrachialis muscle. The long head of the biceps tendon (LHBT) starts within the capsule of the shoulder joint, running from the supraglenoid tubercle or labrum.1 The tendon typically runs free along its intra-articular course, but it is also extrasynovial and ensheathed by a continuation of the synovial lining of the articular capsule that extends to the inferior-most extent of the bicipital groove.2 Congenital anomalies of the LHBT are uncommon, although several atypical forms have been described. A literature search for anomalous LHBT identified several variations in anatomic descriptions, including Y-shaped variant, complete absence of tendon, extra-articular attachment, and a variety of intracapsular attachments. In all, 8 case reports of aberrant intracapsular attachment of LHBT3-12 were identified. These cases presented with a variety of clinical manifestations and pathologic changes. Often, these anatomic variations are considered innocuous, yet some present with pathologic findings.
We present the clinical, magnetic resonance imaging (MRI), and arthroscopic findings of a relatively young athletic patient who was experiencing symptoms of bilateral superior labrum anterior to posterior (SLAP) tears that were unresponsive to conservative management. A unique anatomic variant of the LHBT that involved confluence of the LHBT with the undersurface of the anterosuperior capsule at the rotator interval, as well as a Buford complex anteriorly, was identified and treated. We believe that the tethering of the biceps tendon to the capsule combined with the Buford complex created increased stress on the superior labrum and biceps anchor variant, leading to the development of bilateral symptomatic type II SLAP tears. Knowledge of this variant, though perhaps rare, may be relevant for diagnostic recognition of young athletic patients who present with recalcitrant shoulder symptoms. The patient and the patient’s parents provided written informed consent for print and electronic publication of this case report.
Case Report
A 15-year-old healthy and active athletic boy presented with pain in the right shoulder without history of trauma. He was active in both swimming and baseball. He complained of pain that was present with activities, such as lifting weights, swimming, and throwing. His treatment prior to the office visit consisted of nonsteroidal anti-inflammatory medication, rest, and a therapy program initiated by his high school athletic trainer.
Physical examination demonstrated tenderness to palpation over the posterior capsule and biceps. Motion was full, cuff strength was normal, and SLAP signs (O’Brien, Speed, and Jobe relocation) were positive. A radiograph showed no sign of fracture or dislocation, and no evidence of bony abnormality.
The patient was sent for an MRI arthrogram, which showed a SLAP tear extending from 1 o’clock anteriorly to 10 o’clock posteriorly without intra-articular displacement. No rotator cuff tear was noted. The biceps tendon was noted to be unremarkable and located within the bicipital groove, although retrospective review of the MRI showed that the intra-articular biceps tendon was somewhat confluent with the adjacent tissues.
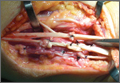
The patient underwent right shoulder arthroscopy. The shoulder was stable to ligamentous examination under anesthesia. Arthroscopic evaluation revealed that there was a type II SLAP tear extending from the 11-o’clock to the 2-o’clock positions. The superior glenohumeral ligament was identified as it arose from the upper pole of the glenoid labrum and then ran parallel and inferior to the tendon of the biceps towards the lesser tubercle. Surprisingly, there was a very unusual attachment of the intracapsular LHBT to the undersurface of the rotator interval, which restricted biceps excursion in relation to the rotator cuff. Additionally, there was a thick cord-like middle glenohumeral ligament anteriorly that lacked the normal glenoid attachments, thus representing a Buford complex. Interestingly, the labral tear could not only be displaced with a probe, but placing the shoulder through a range of motion also led to increased displacement of the labrum from the glenoid, likely because the biceps tendon was tethered to the undersurface of the capsule.
At the time of arthroscopy, the LHBT was released from its attachment to the capsule at the rotator interval with a radiofrequency wand and shaver. A labral repair was performed using three 2.9-mm bioabsorbable suture anchors, placing 2 posterior and 1 anterior to the biceps tendon. The integrity of the labral repair was observed while placing the shoulder through range of motion.
Postoperatively, the patient was kept in a sling for 5 weeks. Home exercises were initiated at 2 weeks, and outpatient physical therapy was implemented at 4 weeks. The patient resumed swimming, throwing, and other activities—with minimal discomfort—at 6 months postoperatively.
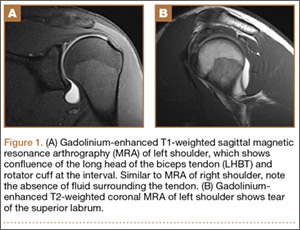
Three years after his initial visit, the patient returned to the office with a similar complaint of pain and limitation of function in his left shoulder after returning to full athletic competition. Once again, there was no history of injury, and history, physical examination, and MRI arthrogram (Figures 1A, 1B) evaluation proved to be very similar to this young athlete’s right shoulder work-up.
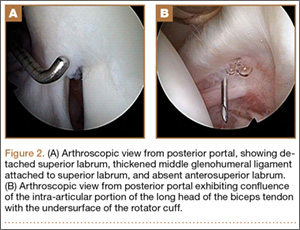
The patient once again underwent shoulder arthroscopy and treatment. Although this was now the left shoulder, the findings were essentially identical to the right shoulder. Once again, the labrum was detached from the 11-o’clock to 2-o’clock positions, and a Buford complex was present anteriorly (Figure 2A). The labral tear was easily displaceable from the glenoid with a probe, and placing the shoulder through a range of motion led to increased displacement of the labrum from the glenoid. There was also confluence of the intra-articular LHBT with the undersurface of the capsule within the rotator interval (Figure 2B). A radiofrequency wand, shaver, and elevator were used to define the biceps tendon and separate it from the undersurface of the capsule. The SLAP repair was performed using three 2.9-mm absorbable suture anchors with 2 posterior and 1 anterior to the biceps tendon insertion. The labral repair was observed while placing the shoulder through range of motion and the shoulder was seen to be free of any undue tension on the labrum.
Postoperatively, the patient’s sling and rehabilitation protocol was identical to that of the right shoulder. The patient progressed well, was released to full activity at 6 months, and has not returned with any further complaints of left or right shoulder pain. Approximately 3 years after treatment the patient was contacted via phone and asked about symptoms, pain, and activity. He denies current symptoms of clicking or instability and has no pain that he can identify as being related to previous pathology or treatment. Since the surgery, he has ceased competitive sports and weight lifting, which he attributes to deconditioning associated with postsurgical immobilization and lack of motivation.
Discussion
Of the 8 case reports in the literature that identified variable intra-articular biceps insertional anatomy, only 2 reports represented confluence of the biceps within the rotator interval.7 Interestingly, of the cases identified, the single case that presented a patient with similar pathology of a type II SLAP lesion had an almost identical anatomical variant presentation consisting of both the anomalous insertion of the LHBT into the undersurface of the rotator interval and a Buford variant of the anterosuperior glenohumeral ligament complex. To our knowledge, our bilateral case of an altered intra-articular biceps insertion and a concomitant SLAP tear supports the theory that this pattern of anomalous insertion may very well have altered the biomechanics of the tendon, resulting in acquired pathology to the superior labrum.
The literature reviewed showed the prevalence of anatomic variations of the LHBT ranged from 1.9% to 7.4%.13,14 These variations are generally considered benign; however, in some cases—as in the cases of the young athletes presented by Wahl and MacGillivray7 and in this report—anatomic variation may play an important role in pathogenesis of different injury patterns. The primary function of the LHBT is the stabilization of the glenohumeral joint during abduction and external rotation.15 When the insertion diverges from normal (eg, when the tendon is tethered to the undersurface of the rotator cuff), the biomechanical stresses on the tendon likely change. As a result of the anomalous position of the LHBT origin, there may be a change in the shoulder joint’s biomechanics, with increased strain on the glenohumeral ligament and its attachment onto the glenoid.16
This case report differs from publications on variable superior glenohumeral ligament attachments because a discrete superior glenohumeral ligament structure was isolated from the biceps tendon. Although a larger case series or patient cohort, as well as more involved biomechanical analysis, would certainly be necessary to prove our hypothesis, we believe that this case suggests certain anatomic LHBT and labral variations can contribute to the develop of SLAP tears in younger individuals.
1. Vangsness CT Jr, Jorgenson SS, Watson T, Johnson DL. The origin of the long head of the biceps from the scapula and glenoid labrum. An anatomical study of 100 shoulders. J Bone Joint Surg Br. 1994;76(6):951-954.
2. Burkhead WZ Jr. The biceps tendon. In: Rockwood CA Jr, Matsen FA III, eds. The Shoulder. Vol. 2. Philadelphia: WB Saunders; 1990:791-836.
3. Parikh SN, Bonnaig N, Zbojniewicz A. Intracapsular origin of the long head biceps tendon with glenoid avulsion of the glenohumeral ligaments. Orthopedics. 2011;34(11):781-784.
4. Gaskin CM, Golish SR, Blount KJ, Diduch DR. Anomalies of the long head of the biceps brachii tendon: clinical significance, MR arthrographic findings, and arthroscopic correlation in two patients. Skeletal Radiol. 2007;36(8):785-789.
5. Yeh L, Pedowitz R, Kwak S, et al. Intracapsular origin of the long head of the biceps tendon. Skeletal Radiol. 1999;28(3):178-181.
6. Richards DP, Schwartz M. Anomalous intraarticular origin of the long head of the biceps brachii. Clin J Sport Med. 2003;13(2):122-124.
7. Wahl CJ, MacGillivray JD. Three congenital variations in the long head of the biceps tendon: a review of the pathoanatomic considerations and case reports. J Shoulder Elbow Surg. 2007;16(6):e25-e30.I
8. Egea JM, Melguizo C, Prados J, Aránega A. Capsular origin of the long head of the biceps tendon: a clinical case. Rom J Morphol Embryol. 2010;51(2):375-377.
9. Hyman JL, Warren RF. Extra-articular origin of biceps brachii. Arthroscopy. 2001;17(7): E29.
10. Enad JG. Bifurcate origin of the long head of the biceps tendon. Arthroscopy. 2004;20(10):1081-1083.
11. Mariani PP, Bellelli A, Botticella C. Arthroscopic absence of the long head of the biceps tendon. Arthroscopy. 1997;13(4):499-501.
12. Koplas MC, Winalski CS, Ulmer WH Jr, Recht M. Bilateral congenital absence of the long head of the biceps tendon. Skeletal Radiol. 2009;38(7):715-719.
13. Kanatli U, Ozturk BY, Eisen E, Bolukbasi S. Intra-articular variations of the long head of the biceps tendon. Knee Surg Sports Traumatol Arthrosc. 2011;19(9):1576-1581.
14. Dierickx C, Ceccarelli E, Conti M, Vanlommel J, Castagna A. Variations of the intra-articular portion of the long head of the biceps tendon: a classification of embryologically explained variations. J Shoulder Elbow Surg. 2009;18(4):556-565.
15. Rodosky MW, Harner CD, Fu FH. The role of the long head of the biceps muscle and superior glenoid labrum in anterior stability of the shoulder. Am J Sports Med. 1994;22(1):121-130.
16. Bigliani LU, Kelkar R, Flatow EL, Pollock RG, Mow VC. Glenohumeral stability. Biomechanical properties of passive and active stabilizers. Clin Orthop Relat Res. 1996;(330):13-30.
The biceps brachii derives its name from the 2 heads of the muscle. The short head originates from the coracoid apex, with the coracobrachialis muscle. The long head of the biceps tendon (LHBT) starts within the capsule of the shoulder joint, running from the supraglenoid tubercle or labrum.1 The tendon typically runs free along its intra-articular course, but it is also extrasynovial and ensheathed by a continuation of the synovial lining of the articular capsule that extends to the inferior-most extent of the bicipital groove.2 Congenital anomalies of the LHBT are uncommon, although several atypical forms have been described. A literature search for anomalous LHBT identified several variations in anatomic descriptions, including Y-shaped variant, complete absence of tendon, extra-articular attachment, and a variety of intracapsular attachments. In all, 8 case reports of aberrant intracapsular attachment of LHBT3-12 were identified. These cases presented with a variety of clinical manifestations and pathologic changes. Often, these anatomic variations are considered innocuous, yet some present with pathologic findings.
We present the clinical, magnetic resonance imaging (MRI), and arthroscopic findings of a relatively young athletic patient who was experiencing symptoms of bilateral superior labrum anterior to posterior (SLAP) tears that were unresponsive to conservative management. A unique anatomic variant of the LHBT that involved confluence of the LHBT with the undersurface of the anterosuperior capsule at the rotator interval, as well as a Buford complex anteriorly, was identified and treated. We believe that the tethering of the biceps tendon to the capsule combined with the Buford complex created increased stress on the superior labrum and biceps anchor variant, leading to the development of bilateral symptomatic type II SLAP tears. Knowledge of this variant, though perhaps rare, may be relevant for diagnostic recognition of young athletic patients who present with recalcitrant shoulder symptoms. The patient and the patient’s parents provided written informed consent for print and electronic publication of this case report.
Case Report
A 15-year-old healthy and active athletic boy presented with pain in the right shoulder without history of trauma. He was active in both swimming and baseball. He complained of pain that was present with activities, such as lifting weights, swimming, and throwing. His treatment prior to the office visit consisted of nonsteroidal anti-inflammatory medication, rest, and a therapy program initiated by his high school athletic trainer.
Physical examination demonstrated tenderness to palpation over the posterior capsule and biceps. Motion was full, cuff strength was normal, and SLAP signs (O’Brien, Speed, and Jobe relocation) were positive. A radiograph showed no sign of fracture or dislocation, and no evidence of bony abnormality.
The patient was sent for an MRI arthrogram, which showed a SLAP tear extending from 1 o’clock anteriorly to 10 o’clock posteriorly without intra-articular displacement. No rotator cuff tear was noted. The biceps tendon was noted to be unremarkable and located within the bicipital groove, although retrospective review of the MRI showed that the intra-articular biceps tendon was somewhat confluent with the adjacent tissues.
The patient underwent right shoulder arthroscopy. The shoulder was stable to ligamentous examination under anesthesia. Arthroscopic evaluation revealed that there was a type II SLAP tear extending from the 11-o’clock to the 2-o’clock positions. The superior glenohumeral ligament was identified as it arose from the upper pole of the glenoid labrum and then ran parallel and inferior to the tendon of the biceps towards the lesser tubercle. Surprisingly, there was a very unusual attachment of the intracapsular LHBT to the undersurface of the rotator interval, which restricted biceps excursion in relation to the rotator cuff. Additionally, there was a thick cord-like middle glenohumeral ligament anteriorly that lacked the normal glenoid attachments, thus representing a Buford complex. Interestingly, the labral tear could not only be displaced with a probe, but placing the shoulder through a range of motion also led to increased displacement of the labrum from the glenoid, likely because the biceps tendon was tethered to the undersurface of the capsule.
At the time of arthroscopy, the LHBT was released from its attachment to the capsule at the rotator interval with a radiofrequency wand and shaver. A labral repair was performed using three 2.9-mm bioabsorbable suture anchors, placing 2 posterior and 1 anterior to the biceps tendon. The integrity of the labral repair was observed while placing the shoulder through range of motion.
Postoperatively, the patient was kept in a sling for 5 weeks. Home exercises were initiated at 2 weeks, and outpatient physical therapy was implemented at 4 weeks. The patient resumed swimming, throwing, and other activities—with minimal discomfort—at 6 months postoperatively.
Three years after his initial visit, the patient returned to the office with a similar complaint of pain and limitation of function in his left shoulder after returning to full athletic competition. Once again, there was no history of injury, and history, physical examination, and MRI arthrogram (Figures 1A, 1B) evaluation proved to be very similar to this young athlete’s right shoulder work-up.
The patient once again underwent shoulder arthroscopy and treatment. Although this was now the left shoulder, the findings were essentially identical to the right shoulder. Once again, the labrum was detached from the 11-o’clock to 2-o’clock positions, and a Buford complex was present anteriorly (Figure 2A). The labral tear was easily displaceable from the glenoid with a probe, and placing the shoulder through a range of motion led to increased displacement of the labrum from the glenoid. There was also confluence of the intra-articular LHBT with the undersurface of the capsule within the rotator interval (Figure 2B). A radiofrequency wand, shaver, and elevator were used to define the biceps tendon and separate it from the undersurface of the capsule. The SLAP repair was performed using three 2.9-mm absorbable suture anchors with 2 posterior and 1 anterior to the biceps tendon insertion. The labral repair was observed while placing the shoulder through range of motion and the shoulder was seen to be free of any undue tension on the labrum.
Postoperatively, the patient’s sling and rehabilitation protocol was identical to that of the right shoulder. The patient progressed well, was released to full activity at 6 months, and has not returned with any further complaints of left or right shoulder pain. Approximately 3 years after treatment the patient was contacted via phone and asked about symptoms, pain, and activity. He denies current symptoms of clicking or instability and has no pain that he can identify as being related to previous pathology or treatment. Since the surgery, he has ceased competitive sports and weight lifting, which he attributes to deconditioning associated with postsurgical immobilization and lack of motivation.
Discussion
Of the 8 case reports in the literature that identified variable intra-articular biceps insertional anatomy, only 2 reports represented confluence of the biceps within the rotator interval.7 Interestingly, of the cases identified, the single case that presented a patient with similar pathology of a type II SLAP lesion had an almost identical anatomical variant presentation consisting of both the anomalous insertion of the LHBT into the undersurface of the rotator interval and a Buford variant of the anterosuperior glenohumeral ligament complex. To our knowledge, our bilateral case of an altered intra-articular biceps insertion and a concomitant SLAP tear supports the theory that this pattern of anomalous insertion may very well have altered the biomechanics of the tendon, resulting in acquired pathology to the superior labrum.
The literature reviewed showed the prevalence of anatomic variations of the LHBT ranged from 1.9% to 7.4%.13,14 These variations are generally considered benign; however, in some cases—as in the cases of the young athletes presented by Wahl and MacGillivray7 and in this report—anatomic variation may play an important role in pathogenesis of different injury patterns. The primary function of the LHBT is the stabilization of the glenohumeral joint during abduction and external rotation.15 When the insertion diverges from normal (eg, when the tendon is tethered to the undersurface of the rotator cuff), the biomechanical stresses on the tendon likely change. As a result of the anomalous position of the LHBT origin, there may be a change in the shoulder joint’s biomechanics, with increased strain on the glenohumeral ligament and its attachment onto the glenoid.16
This case report differs from publications on variable superior glenohumeral ligament attachments because a discrete superior glenohumeral ligament structure was isolated from the biceps tendon. Although a larger case series or patient cohort, as well as more involved biomechanical analysis, would certainly be necessary to prove our hypothesis, we believe that this case suggests certain anatomic LHBT and labral variations can contribute to the develop of SLAP tears in younger individuals.
The biceps brachii derives its name from the 2 heads of the muscle. The short head originates from the coracoid apex, with the coracobrachialis muscle. The long head of the biceps tendon (LHBT) starts within the capsule of the shoulder joint, running from the supraglenoid tubercle or labrum.1 The tendon typically runs free along its intra-articular course, but it is also extrasynovial and ensheathed by a continuation of the synovial lining of the articular capsule that extends to the inferior-most extent of the bicipital groove.2 Congenital anomalies of the LHBT are uncommon, although several atypical forms have been described. A literature search for anomalous LHBT identified several variations in anatomic descriptions, including Y-shaped variant, complete absence of tendon, extra-articular attachment, and a variety of intracapsular attachments. In all, 8 case reports of aberrant intracapsular attachment of LHBT3-12 were identified. These cases presented with a variety of clinical manifestations and pathologic changes. Often, these anatomic variations are considered innocuous, yet some present with pathologic findings.
We present the clinical, magnetic resonance imaging (MRI), and arthroscopic findings of a relatively young athletic patient who was experiencing symptoms of bilateral superior labrum anterior to posterior (SLAP) tears that were unresponsive to conservative management. A unique anatomic variant of the LHBT that involved confluence of the LHBT with the undersurface of the anterosuperior capsule at the rotator interval, as well as a Buford complex anteriorly, was identified and treated. We believe that the tethering of the biceps tendon to the capsule combined with the Buford complex created increased stress on the superior labrum and biceps anchor variant, leading to the development of bilateral symptomatic type II SLAP tears. Knowledge of this variant, though perhaps rare, may be relevant for diagnostic recognition of young athletic patients who present with recalcitrant shoulder symptoms. The patient and the patient’s parents provided written informed consent for print and electronic publication of this case report.
Case Report
A 15-year-old healthy and active athletic boy presented with pain in the right shoulder without history of trauma. He was active in both swimming and baseball. He complained of pain that was present with activities, such as lifting weights, swimming, and throwing. His treatment prior to the office visit consisted of nonsteroidal anti-inflammatory medication, rest, and a therapy program initiated by his high school athletic trainer.
Physical examination demonstrated tenderness to palpation over the posterior capsule and biceps. Motion was full, cuff strength was normal, and SLAP signs (O’Brien, Speed, and Jobe relocation) were positive. A radiograph showed no sign of fracture or dislocation, and no evidence of bony abnormality.
The patient was sent for an MRI arthrogram, which showed a SLAP tear extending from 1 o’clock anteriorly to 10 o’clock posteriorly without intra-articular displacement. No rotator cuff tear was noted. The biceps tendon was noted to be unremarkable and located within the bicipital groove, although retrospective review of the MRI showed that the intra-articular biceps tendon was somewhat confluent with the adjacent tissues.
The patient underwent right shoulder arthroscopy. The shoulder was stable to ligamentous examination under anesthesia. Arthroscopic evaluation revealed that there was a type II SLAP tear extending from the 11-o’clock to the 2-o’clock positions. The superior glenohumeral ligament was identified as it arose from the upper pole of the glenoid labrum and then ran parallel and inferior to the tendon of the biceps towards the lesser tubercle. Surprisingly, there was a very unusual attachment of the intracapsular LHBT to the undersurface of the rotator interval, which restricted biceps excursion in relation to the rotator cuff. Additionally, there was a thick cord-like middle glenohumeral ligament anteriorly that lacked the normal glenoid attachments, thus representing a Buford complex. Interestingly, the labral tear could not only be displaced with a probe, but placing the shoulder through a range of motion also led to increased displacement of the labrum from the glenoid, likely because the biceps tendon was tethered to the undersurface of the capsule.
At the time of arthroscopy, the LHBT was released from its attachment to the capsule at the rotator interval with a radiofrequency wand and shaver. A labral repair was performed using three 2.9-mm bioabsorbable suture anchors, placing 2 posterior and 1 anterior to the biceps tendon. The integrity of the labral repair was observed while placing the shoulder through range of motion.
Postoperatively, the patient was kept in a sling for 5 weeks. Home exercises were initiated at 2 weeks, and outpatient physical therapy was implemented at 4 weeks. The patient resumed swimming, throwing, and other activities—with minimal discomfort—at 6 months postoperatively.
Three years after his initial visit, the patient returned to the office with a similar complaint of pain and limitation of function in his left shoulder after returning to full athletic competition. Once again, there was no history of injury, and history, physical examination, and MRI arthrogram (Figures 1A, 1B) evaluation proved to be very similar to this young athlete’s right shoulder work-up.
The patient once again underwent shoulder arthroscopy and treatment. Although this was now the left shoulder, the findings were essentially identical to the right shoulder. Once again, the labrum was detached from the 11-o’clock to 2-o’clock positions, and a Buford complex was present anteriorly (Figure 2A). The labral tear was easily displaceable from the glenoid with a probe, and placing the shoulder through a range of motion led to increased displacement of the labrum from the glenoid. There was also confluence of the intra-articular LHBT with the undersurface of the capsule within the rotator interval (Figure 2B). A radiofrequency wand, shaver, and elevator were used to define the biceps tendon and separate it from the undersurface of the capsule. The SLAP repair was performed using three 2.9-mm absorbable suture anchors with 2 posterior and 1 anterior to the biceps tendon insertion. The labral repair was observed while placing the shoulder through range of motion and the shoulder was seen to be free of any undue tension on the labrum.
Postoperatively, the patient’s sling and rehabilitation protocol was identical to that of the right shoulder. The patient progressed well, was released to full activity at 6 months, and has not returned with any further complaints of left or right shoulder pain. Approximately 3 years after treatment the patient was contacted via phone and asked about symptoms, pain, and activity. He denies current symptoms of clicking or instability and has no pain that he can identify as being related to previous pathology or treatment. Since the surgery, he has ceased competitive sports and weight lifting, which he attributes to deconditioning associated with postsurgical immobilization and lack of motivation.
Discussion
Of the 8 case reports in the literature that identified variable intra-articular biceps insertional anatomy, only 2 reports represented confluence of the biceps within the rotator interval.7 Interestingly, of the cases identified, the single case that presented a patient with similar pathology of a type II SLAP lesion had an almost identical anatomical variant presentation consisting of both the anomalous insertion of the LHBT into the undersurface of the rotator interval and a Buford variant of the anterosuperior glenohumeral ligament complex. To our knowledge, our bilateral case of an altered intra-articular biceps insertion and a concomitant SLAP tear supports the theory that this pattern of anomalous insertion may very well have altered the biomechanics of the tendon, resulting in acquired pathology to the superior labrum.
The literature reviewed showed the prevalence of anatomic variations of the LHBT ranged from 1.9% to 7.4%.13,14 These variations are generally considered benign; however, in some cases—as in the cases of the young athletes presented by Wahl and MacGillivray7 and in this report—anatomic variation may play an important role in pathogenesis of different injury patterns. The primary function of the LHBT is the stabilization of the glenohumeral joint during abduction and external rotation.15 When the insertion diverges from normal (eg, when the tendon is tethered to the undersurface of the rotator cuff), the biomechanical stresses on the tendon likely change. As a result of the anomalous position of the LHBT origin, there may be a change in the shoulder joint’s biomechanics, with increased strain on the glenohumeral ligament and its attachment onto the glenoid.16
This case report differs from publications on variable superior glenohumeral ligament attachments because a discrete superior glenohumeral ligament structure was isolated from the biceps tendon. Although a larger case series or patient cohort, as well as more involved biomechanical analysis, would certainly be necessary to prove our hypothesis, we believe that this case suggests certain anatomic LHBT and labral variations can contribute to the develop of SLAP tears in younger individuals.
1. Vangsness CT Jr, Jorgenson SS, Watson T, Johnson DL. The origin of the long head of the biceps from the scapula and glenoid labrum. An anatomical study of 100 shoulders. J Bone Joint Surg Br. 1994;76(6):951-954.
2. Burkhead WZ Jr. The biceps tendon. In: Rockwood CA Jr, Matsen FA III, eds. The Shoulder. Vol. 2. Philadelphia: WB Saunders; 1990:791-836.
3. Parikh SN, Bonnaig N, Zbojniewicz A. Intracapsular origin of the long head biceps tendon with glenoid avulsion of the glenohumeral ligaments. Orthopedics. 2011;34(11):781-784.
4. Gaskin CM, Golish SR, Blount KJ, Diduch DR. Anomalies of the long head of the biceps brachii tendon: clinical significance, MR arthrographic findings, and arthroscopic correlation in two patients. Skeletal Radiol. 2007;36(8):785-789.
5. Yeh L, Pedowitz R, Kwak S, et al. Intracapsular origin of the long head of the biceps tendon. Skeletal Radiol. 1999;28(3):178-181.
6. Richards DP, Schwartz M. Anomalous intraarticular origin of the long head of the biceps brachii. Clin J Sport Med. 2003;13(2):122-124.
7. Wahl CJ, MacGillivray JD. Three congenital variations in the long head of the biceps tendon: a review of the pathoanatomic considerations and case reports. J Shoulder Elbow Surg. 2007;16(6):e25-e30.I
8. Egea JM, Melguizo C, Prados J, Aránega A. Capsular origin of the long head of the biceps tendon: a clinical case. Rom J Morphol Embryol. 2010;51(2):375-377.
9. Hyman JL, Warren RF. Extra-articular origin of biceps brachii. Arthroscopy. 2001;17(7): E29.
10. Enad JG. Bifurcate origin of the long head of the biceps tendon. Arthroscopy. 2004;20(10):1081-1083.
11. Mariani PP, Bellelli A, Botticella C. Arthroscopic absence of the long head of the biceps tendon. Arthroscopy. 1997;13(4):499-501.
12. Koplas MC, Winalski CS, Ulmer WH Jr, Recht M. Bilateral congenital absence of the long head of the biceps tendon. Skeletal Radiol. 2009;38(7):715-719.
13. Kanatli U, Ozturk BY, Eisen E, Bolukbasi S. Intra-articular variations of the long head of the biceps tendon. Knee Surg Sports Traumatol Arthrosc. 2011;19(9):1576-1581.
14. Dierickx C, Ceccarelli E, Conti M, Vanlommel J, Castagna A. Variations of the intra-articular portion of the long head of the biceps tendon: a classification of embryologically explained variations. J Shoulder Elbow Surg. 2009;18(4):556-565.
15. Rodosky MW, Harner CD, Fu FH. The role of the long head of the biceps muscle and superior glenoid labrum in anterior stability of the shoulder. Am J Sports Med. 1994;22(1):121-130.
16. Bigliani LU, Kelkar R, Flatow EL, Pollock RG, Mow VC. Glenohumeral stability. Biomechanical properties of passive and active stabilizers. Clin Orthop Relat Res. 1996;(330):13-30.
1. Vangsness CT Jr, Jorgenson SS, Watson T, Johnson DL. The origin of the long head of the biceps from the scapula and glenoid labrum. An anatomical study of 100 shoulders. J Bone Joint Surg Br. 1994;76(6):951-954.
2. Burkhead WZ Jr. The biceps tendon. In: Rockwood CA Jr, Matsen FA III, eds. The Shoulder. Vol. 2. Philadelphia: WB Saunders; 1990:791-836.
3. Parikh SN, Bonnaig N, Zbojniewicz A. Intracapsular origin of the long head biceps tendon with glenoid avulsion of the glenohumeral ligaments. Orthopedics. 2011;34(11):781-784.
4. Gaskin CM, Golish SR, Blount KJ, Diduch DR. Anomalies of the long head of the biceps brachii tendon: clinical significance, MR arthrographic findings, and arthroscopic correlation in two patients. Skeletal Radiol. 2007;36(8):785-789.
5. Yeh L, Pedowitz R, Kwak S, et al. Intracapsular origin of the long head of the biceps tendon. Skeletal Radiol. 1999;28(3):178-181.
6. Richards DP, Schwartz M. Anomalous intraarticular origin of the long head of the biceps brachii. Clin J Sport Med. 2003;13(2):122-124.
7. Wahl CJ, MacGillivray JD. Three congenital variations in the long head of the biceps tendon: a review of the pathoanatomic considerations and case reports. J Shoulder Elbow Surg. 2007;16(6):e25-e30.I
8. Egea JM, Melguizo C, Prados J, Aránega A. Capsular origin of the long head of the biceps tendon: a clinical case. Rom J Morphol Embryol. 2010;51(2):375-377.
9. Hyman JL, Warren RF. Extra-articular origin of biceps brachii. Arthroscopy. 2001;17(7): E29.
10. Enad JG. Bifurcate origin of the long head of the biceps tendon. Arthroscopy. 2004;20(10):1081-1083.
11. Mariani PP, Bellelli A, Botticella C. Arthroscopic absence of the long head of the biceps tendon. Arthroscopy. 1997;13(4):499-501.
12. Koplas MC, Winalski CS, Ulmer WH Jr, Recht M. Bilateral congenital absence of the long head of the biceps tendon. Skeletal Radiol. 2009;38(7):715-719.
13. Kanatli U, Ozturk BY, Eisen E, Bolukbasi S. Intra-articular variations of the long head of the biceps tendon. Knee Surg Sports Traumatol Arthrosc. 2011;19(9):1576-1581.
14. Dierickx C, Ceccarelli E, Conti M, Vanlommel J, Castagna A. Variations of the intra-articular portion of the long head of the biceps tendon: a classification of embryologically explained variations. J Shoulder Elbow Surg. 2009;18(4):556-565.
15. Rodosky MW, Harner CD, Fu FH. The role of the long head of the biceps muscle and superior glenoid labrum in anterior stability of the shoulder. Am J Sports Med. 1994;22(1):121-130.
16. Bigliani LU, Kelkar R, Flatow EL, Pollock RG, Mow VC. Glenohumeral stability. Biomechanical properties of passive and active stabilizers. Clin Orthop Relat Res. 1996;(330):13-30.
Xanthogranulomatous Osteomyelitis of Proximal Femur Masquerading as Benign Bone Tumor
Xanthogranulomatous osteomyelitis (XO) is a type of chronic inflammatory process that is characterized by the collection of foamy macrophages along with mononuclear cells in the tissue.1 Xanthogranulomatous osteomyelitis is characterized by the presence of granular, eosinophilic, periodic acid–Schiff–positive histiocytes in the initial stages, followed by the mixture of foamy macrophages and activated plasma cells and, last, by the presence of suppurative foci and hemorrhage. This is an uncommon process best known to occur in the gallbladder, kidney, urinary bladder, fallopian tube, ovary, vagina, prostate, testis, epididymis, colon, and appendix.2-4 Very rarely, it can affect lungs, brain, or bone. Only 5 cases of XO have been reported in the literature.5-8
We report XO of the proximal femur in a 65-year-old woman who initially had a clinical and radiologic diagnosis of aneurysmal bone cyst; however, histopathologic examination confirmed the diagnosis of XO. Xanthogranulomatous osteomyelitis mimics a neoplastic pathology in gallbladder, kidney, and prostrate on gross clinical and radiologic examination.9 The pathogenesis of XO is best characterized by a delayed type of hypersensitivity reaction.10 The differential diagnosis includes chronic recurrent multifocal osteomyelitis, xanthoma, infiltrative storage disorder, malakoplakia, Langerhans cell histiocytosis, fibrohistiocytic tumor, Erdheim-Chester disease, and metastatic renal cell carcinoma.11-14 The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 65-year-old hypertensive woman presented with complaints of pain in the right hip for a duration of 6 months. Pain was radiating from the right hip region to the anteromedial aspect of the knee and progressively increasing, with a history of pain at rest suggestive of a nonmechanical pathology in the hip. There was no history of fever, weight loss, loss of appetite, pain in any other joint, or morning stiffness. The patient was mobile without support and was able to squat and sit cross-legged; however, the stance phase on the right side was less than on the left side, suggestive of an antalgic component in the gait.
On examining the patient, there was anterior hip joint tenderness with no local sign of any infective or inflammatory pathology. Trochanteric tenderness was present, but there was no irregularity, broadening, or thickening of the trochanter. There was no restriction in the range of motion, and no coronal or sagittal plane deformity in the right hip. There was no limb-length discrepancy. However, the patient was not able to raise her leg actively, probably because of pain in the right hip.
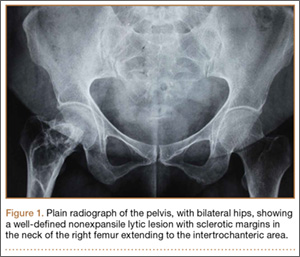
On plain radiographs of the pelvis with bilateral hips, a well-defined nonexpansile uniloculated lytic lesion with sclerotic margins was present in the neck of the right femur, extending to the intertrochanteric area (Figure 1). Ground-glass appearance was also noted. Considering the benign nature of the lesion radiologically and clinically, a differential diagnosis of hyperparathyroidism, renal osteodystrophy, multiple myeloma, and fibrous dysplasia was considered. Hematologic investigations, skeletal survey, and magnetic resonance imaging (MRI) of the bilateral hips were performed to rule out the differential diagnosis.
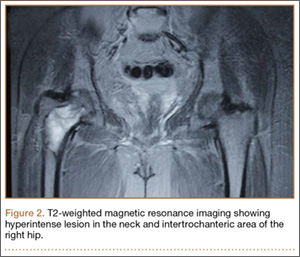
The patient’s hemoglobin level was 11.8 g/dL with total white blood cell count of 10,300/µL. Renal and hepatic functions were within normal limit. Serum erythrocyte sedimentation rate (ESR) was 12 mm/h and C-reactive protein level was normal. Serum parathyroid level was 32 pg/mL, which was within normal limits, with an alkaline phosphatase level of 101 U/L. The skeletal survey showed no other bony lesion in the body. T1-weighted MRI of both hips showed a well-defined hypointense lesion in the neck and intertrochanteric area of the right hip, which was hyperintense on T2-weighted MRI, suggestive of aneurysmal bone cyst (Figure 2).
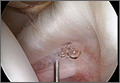
Normal ESR, hemoglobin, alkaline phosphatase, and serum parathyroid levels and normal skeletal survey almost ruled out multiple myeloma and hyperparathyroidism. Normal renal profile ruled out renal osteodystrophy and the osteitis fibrosa cystica lesion associated with it. We planned for prophylactic internal fixation of the lesion to prevent a pathologic fracture. According to Mirels,15 if there is a lytic lesion covering more than two-thirds of the circumference of the bone in the peritrochanteric area, the chances of a pathologic fracture are high and such fractures should be fixed.
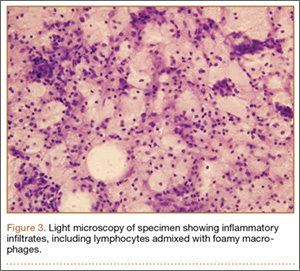
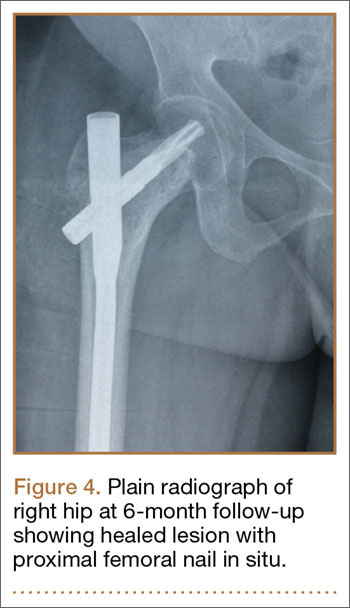
We planned for curettage of the lesion with bone grafting and in situ intramedullary fixation of the lesion. Curettage was done according to the plan and the sample was sent for histopathologic examination. In situ internal fixation and bone grafting were performed by using a proximal femoral intramedullary nail. To our surprise, the biopsy sample was reported as xanthogranuloma, with multiple foamy macrophages mixed with inflammatory cells and aggregates of lymphocytes (Figure 3). Mycobacterial and routine bacterial cultures were reported as negative. The patient was kept on oral antibiotics (cefixime and moxifloxacin) for 6 weeks, and she made an uneventful recovery. At 6-month follow-up, a radiograph of the right hip showed a healed lesion with proximal femoral nail in situ (Figure 4).
Discussion
To the best of our knowledge, a total of 5 cases of XO have been reported in the literature. The earliest of these reports were by Cozzutto and Carbone,1 who reported 2 cases of XO of the first rib and of the epiphysis of the tibia, respectively. The importance of these lesions to diagnosis is their confusion with a neoplastic disease, as XO is itself a benign disorder. These lesions can mimic a neoplastic lesion in clinical and radiologic presentation and the only way to differentiate the lesion from a neoplastic disease is by histopathologic examination of the tissue. Hypothetically, xanthogranulomatous disorders can be related to trauma or infection.
In 2007, Vankalakunti and colleagues6 reported XO of the ulna in a 50-year-old postmenopausal woman. In that case, progressive swelling was present on the extensor aspect of her right forearm for a period of 2 years, for which curettage and bone grafting were performed, using autograft from the ipsilateral iliac crest. The tissue culture was sterile, and XO was diagnosed as a result of the histopathologic examination. In 2009, Cennimo and colleagues7 reported XO of the index finger and wrist of a man complaining of pain and swelling for 1 year, which was unresponsive to antibiotics. The diagnosis of XO was confirmed histopathologically, when the culture of the same tissue grew Mycobacterium marinum. Radical synovectomy of the lesion was performed, after which minocycline, clarithromycin, and ethambutol were administered. In 2012, Borjian and colleagues8 reported a case of XO of the proximal humerus and proximal fibula in a 14-year-old child. The child, who presented with fever, pain, and restriction of shoulder movements, was started on oral antibiotics as the tissue culture grew Staphylococcus aureus; the patient did not complete the course of treatment in the hospital. No surgical intervention was done in this case. The diagnosis of XO was confirmed by microscopic examination of the tissue.
An association between bacterial infection and xanthogranulomatous inflammation has existed in several organs, such as the kidneys, and in the gastrointestinal system, but such an association of the 2 is yet to be determined for bone.5,10,16-19 Because of the paucity of literature on the disease, a management protocol for XO of bone has not been defined, and decisions have to be made considering the natural history of the disease in other organs. We present this case primarily because of its rarity, curability, and its close resemblance to bone tumors. While XO is benign, it can mimic a neoplastic bone lesion in its imaging and clinical manifestations, and appropriate differentiation is crucial. Currently, histopathologic examination of lesions is the most specific and is the gold standard for diagnosis.
Conclusion
Xanthogranulomatous osteomyelitis is a very rare entity, and only a few cases have been reported in the English-language literature. Though rare, XO warrants greater emphasis than it receives in the literature. It is a chronic inflammatory disease having a close resemblance to bone tumors. A high index of suspicion must be practiced to differentiate XO from tumors. Histopathologic examination is mandatory to establish definitive diagnosis and correct treatment.
1. Cozzutto C, Carbone A. The xanthogranulomatous process. Xanthogranulomatous inflammation. Pathol Res Pract. 1988;183(4):395-402.
2. Ladefoged C, Lorentzen M. Xanthogranulomatous cholecystitis. A clinicopathological study of 20 cases and review of the literature. APMIS. 1993;101(11):869-875.
3. Nistal M, Gonzalez-Peramato P, Serrano A, Regadera J. Xanthogranulomatous funiculitis and orchiepididymitis: report of 2 cases with immunohistochemical study and literature review. Arch Pathol Lab Med. 2004;128(8):911-914.
4. Oh YH, Seong SS, Jang KS, et al. Xanthogranulomatous inflammation presenting as a submucosal mass of the sigmoid colon. Pathol Int. 2005;55(7):440-444.
5. Cozzutto C. Xanthogranulomatous osteomyelitis. Arch Pathol Lab Med. 1984;108(12):973-6.
6. Vankalakunti M, Saikia UN, Mathew M, Kang M. Xanthogranulomatous osteomyelitis of ulna mimicking neoplasm. World J Surg Oncol. 2007;30(5):46.
7. Cennimo DJ, Agag R, Fleegler E, et al. Mycobacterium marinum hand infection in a “sushi chef.” Eplasty. 2009;14(9):e43.
8. Borjian A, Rezaei F, Eshaghi MA, Shemshaki H. Xanthogranulomatous osteomyelitis. J Orthop Traumatol. 2012;13(4):217-220.
9. Rafique M, Yaqoob N. Xanthogranulomatous prostatitis: a mimic of carcinoma of prostate. World J Surg Oncol. 2006;4:30.
10. Nakashiro H, Haraoka S, Fujiwara K, Harada S, Hisatsugu T, Watanabe T. Xanthogranulomatous cholecystis. Cell composition and a possible pathogenetic role of cell-mediated immunity. Pathol Res Pract. 1995;191(11):1078-1086.
11. Hamada T, Ito H, Araki Y, Fujii K, Inoue M, Ishida O. Benign fibrous histiocytoma of the femur: review of three cases. Skeletal Radiol. 1996;25(1):25-29.
12. Kossard S, Chow E, Wilkinson B, Killingsworth M. Lipid and giant cell poor necrobiotic xanthogranuloma. J Cutan Pathol. 2000;27(7):374-378.
13. Girschick HJ, Huppertz HI, Harmsen D, Krauspe R, Müller-Hermelink HK, Papadopoulos T. Chronic recurrent multifocal osteomyelitis in children: diagnostic value of histopathology and microbial testing. Hum Pathol. 1999;30(1):59-65.
14. Kayser R, Mahlfeld K, Grasshoff H. Vertebral Langerhans-cell histiocytosis in childhood – a differential diagnosis of spinal osteomyelitis. Klin Padiatr. 1999;211(5):399-402.
15. Mirels H. Metastatic disease in long bones. A proposed scoring system for diagnosing impending pathologic fractures. Clin Orthop Relat Res. 1989;249:256-264.
16. Machiz S, Gordon J, Block N, Politano VA. Salmonella typhosa urinary tract infection and xanthogranulomatous pyelonephritis. Case report and review of literature. J Fla Med Assoc. 1974;61(9):703-705.
17. Gauperaa T, Stalsberg H. Renal endometriosis. A case report. Scand J Urol Nephrol. 1977;11(2):189-191.
18. Goodman M, Curry T, Russell T. Xanthogranulomatous pyelonephritis (XGP): a local disease with systemic manifestations. Report of 23 patients and review of the literature. Medicine. 1979;58(2):171-181.
19. Guarino M, Reale D, Micoli G, Tricomi P, Cristofori E. Xanthogranulomatous gastritis: association with xanthogranulomatous cholecystitis. J Clin Pathol. 1993;46(1):88-90.
Xanthogranulomatous osteomyelitis (XO) is a type of chronic inflammatory process that is characterized by the collection of foamy macrophages along with mononuclear cells in the tissue.1 Xanthogranulomatous osteomyelitis is characterized by the presence of granular, eosinophilic, periodic acid–Schiff–positive histiocytes in the initial stages, followed by the mixture of foamy macrophages and activated plasma cells and, last, by the presence of suppurative foci and hemorrhage. This is an uncommon process best known to occur in the gallbladder, kidney, urinary bladder, fallopian tube, ovary, vagina, prostate, testis, epididymis, colon, and appendix.2-4 Very rarely, it can affect lungs, brain, or bone. Only 5 cases of XO have been reported in the literature.5-8
We report XO of the proximal femur in a 65-year-old woman who initially had a clinical and radiologic diagnosis of aneurysmal bone cyst; however, histopathologic examination confirmed the diagnosis of XO. Xanthogranulomatous osteomyelitis mimics a neoplastic pathology in gallbladder, kidney, and prostrate on gross clinical and radiologic examination.9 The pathogenesis of XO is best characterized by a delayed type of hypersensitivity reaction.10 The differential diagnosis includes chronic recurrent multifocal osteomyelitis, xanthoma, infiltrative storage disorder, malakoplakia, Langerhans cell histiocytosis, fibrohistiocytic tumor, Erdheim-Chester disease, and metastatic renal cell carcinoma.11-14 The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 65-year-old hypertensive woman presented with complaints of pain in the right hip for a duration of 6 months. Pain was radiating from the right hip region to the anteromedial aspect of the knee and progressively increasing, with a history of pain at rest suggestive of a nonmechanical pathology in the hip. There was no history of fever, weight loss, loss of appetite, pain in any other joint, or morning stiffness. The patient was mobile without support and was able to squat and sit cross-legged; however, the stance phase on the right side was less than on the left side, suggestive of an antalgic component in the gait.
On examining the patient, there was anterior hip joint tenderness with no local sign of any infective or inflammatory pathology. Trochanteric tenderness was present, but there was no irregularity, broadening, or thickening of the trochanter. There was no restriction in the range of motion, and no coronal or sagittal plane deformity in the right hip. There was no limb-length discrepancy. However, the patient was not able to raise her leg actively, probably because of pain in the right hip.
On plain radiographs of the pelvis with bilateral hips, a well-defined nonexpansile uniloculated lytic lesion with sclerotic margins was present in the neck of the right femur, extending to the intertrochanteric area (Figure 1). Ground-glass appearance was also noted. Considering the benign nature of the lesion radiologically and clinically, a differential diagnosis of hyperparathyroidism, renal osteodystrophy, multiple myeloma, and fibrous dysplasia was considered. Hematologic investigations, skeletal survey, and magnetic resonance imaging (MRI) of the bilateral hips were performed to rule out the differential diagnosis.
The patient’s hemoglobin level was 11.8 g/dL with total white blood cell count of 10,300/µL. Renal and hepatic functions were within normal limit. Serum erythrocyte sedimentation rate (ESR) was 12 mm/h and C-reactive protein level was normal. Serum parathyroid level was 32 pg/mL, which was within normal limits, with an alkaline phosphatase level of 101 U/L. The skeletal survey showed no other bony lesion in the body. T1-weighted MRI of both hips showed a well-defined hypointense lesion in the neck and intertrochanteric area of the right hip, which was hyperintense on T2-weighted MRI, suggestive of aneurysmal bone cyst (Figure 2).
Normal ESR, hemoglobin, alkaline phosphatase, and serum parathyroid levels and normal skeletal survey almost ruled out multiple myeloma and hyperparathyroidism. Normal renal profile ruled out renal osteodystrophy and the osteitis fibrosa cystica lesion associated with it. We planned for prophylactic internal fixation of the lesion to prevent a pathologic fracture. According to Mirels,15 if there is a lytic lesion covering more than two-thirds of the circumference of the bone in the peritrochanteric area, the chances of a pathologic fracture are high and such fractures should be fixed.
We planned for curettage of the lesion with bone grafting and in situ intramedullary fixation of the lesion. Curettage was done according to the plan and the sample was sent for histopathologic examination. In situ internal fixation and bone grafting were performed by using a proximal femoral intramedullary nail. To our surprise, the biopsy sample was reported as xanthogranuloma, with multiple foamy macrophages mixed with inflammatory cells and aggregates of lymphocytes (Figure 3). Mycobacterial and routine bacterial cultures were reported as negative. The patient was kept on oral antibiotics (cefixime and moxifloxacin) for 6 weeks, and she made an uneventful recovery. At 6-month follow-up, a radiograph of the right hip showed a healed lesion with proximal femoral nail in situ (Figure 4).
Discussion
To the best of our knowledge, a total of 5 cases of XO have been reported in the literature. The earliest of these reports were by Cozzutto and Carbone,1 who reported 2 cases of XO of the first rib and of the epiphysis of the tibia, respectively. The importance of these lesions to diagnosis is their confusion with a neoplastic disease, as XO is itself a benign disorder. These lesions can mimic a neoplastic lesion in clinical and radiologic presentation and the only way to differentiate the lesion from a neoplastic disease is by histopathologic examination of the tissue. Hypothetically, xanthogranulomatous disorders can be related to trauma or infection.
In 2007, Vankalakunti and colleagues6 reported XO of the ulna in a 50-year-old postmenopausal woman. In that case, progressive swelling was present on the extensor aspect of her right forearm for a period of 2 years, for which curettage and bone grafting were performed, using autograft from the ipsilateral iliac crest. The tissue culture was sterile, and XO was diagnosed as a result of the histopathologic examination. In 2009, Cennimo and colleagues7 reported XO of the index finger and wrist of a man complaining of pain and swelling for 1 year, which was unresponsive to antibiotics. The diagnosis of XO was confirmed histopathologically, when the culture of the same tissue grew Mycobacterium marinum. Radical synovectomy of the lesion was performed, after which minocycline, clarithromycin, and ethambutol were administered. In 2012, Borjian and colleagues8 reported a case of XO of the proximal humerus and proximal fibula in a 14-year-old child. The child, who presented with fever, pain, and restriction of shoulder movements, was started on oral antibiotics as the tissue culture grew Staphylococcus aureus; the patient did not complete the course of treatment in the hospital. No surgical intervention was done in this case. The diagnosis of XO was confirmed by microscopic examination of the tissue.
An association between bacterial infection and xanthogranulomatous inflammation has existed in several organs, such as the kidneys, and in the gastrointestinal system, but such an association of the 2 is yet to be determined for bone.5,10,16-19 Because of the paucity of literature on the disease, a management protocol for XO of bone has not been defined, and decisions have to be made considering the natural history of the disease in other organs. We present this case primarily because of its rarity, curability, and its close resemblance to bone tumors. While XO is benign, it can mimic a neoplastic bone lesion in its imaging and clinical manifestations, and appropriate differentiation is crucial. Currently, histopathologic examination of lesions is the most specific and is the gold standard for diagnosis.
Conclusion
Xanthogranulomatous osteomyelitis is a very rare entity, and only a few cases have been reported in the English-language literature. Though rare, XO warrants greater emphasis than it receives in the literature. It is a chronic inflammatory disease having a close resemblance to bone tumors. A high index of suspicion must be practiced to differentiate XO from tumors. Histopathologic examination is mandatory to establish definitive diagnosis and correct treatment.
Xanthogranulomatous osteomyelitis (XO) is a type of chronic inflammatory process that is characterized by the collection of foamy macrophages along with mononuclear cells in the tissue.1 Xanthogranulomatous osteomyelitis is characterized by the presence of granular, eosinophilic, periodic acid–Schiff–positive histiocytes in the initial stages, followed by the mixture of foamy macrophages and activated plasma cells and, last, by the presence of suppurative foci and hemorrhage. This is an uncommon process best known to occur in the gallbladder, kidney, urinary bladder, fallopian tube, ovary, vagina, prostate, testis, epididymis, colon, and appendix.2-4 Very rarely, it can affect lungs, brain, or bone. Only 5 cases of XO have been reported in the literature.5-8
We report XO of the proximal femur in a 65-year-old woman who initially had a clinical and radiologic diagnosis of aneurysmal bone cyst; however, histopathologic examination confirmed the diagnosis of XO. Xanthogranulomatous osteomyelitis mimics a neoplastic pathology in gallbladder, kidney, and prostrate on gross clinical and radiologic examination.9 The pathogenesis of XO is best characterized by a delayed type of hypersensitivity reaction.10 The differential diagnosis includes chronic recurrent multifocal osteomyelitis, xanthoma, infiltrative storage disorder, malakoplakia, Langerhans cell histiocytosis, fibrohistiocytic tumor, Erdheim-Chester disease, and metastatic renal cell carcinoma.11-14 The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 65-year-old hypertensive woman presented with complaints of pain in the right hip for a duration of 6 months. Pain was radiating from the right hip region to the anteromedial aspect of the knee and progressively increasing, with a history of pain at rest suggestive of a nonmechanical pathology in the hip. There was no history of fever, weight loss, loss of appetite, pain in any other joint, or morning stiffness. The patient was mobile without support and was able to squat and sit cross-legged; however, the stance phase on the right side was less than on the left side, suggestive of an antalgic component in the gait.
On examining the patient, there was anterior hip joint tenderness with no local sign of any infective or inflammatory pathology. Trochanteric tenderness was present, but there was no irregularity, broadening, or thickening of the trochanter. There was no restriction in the range of motion, and no coronal or sagittal plane deformity in the right hip. There was no limb-length discrepancy. However, the patient was not able to raise her leg actively, probably because of pain in the right hip.
On plain radiographs of the pelvis with bilateral hips, a well-defined nonexpansile uniloculated lytic lesion with sclerotic margins was present in the neck of the right femur, extending to the intertrochanteric area (Figure 1). Ground-glass appearance was also noted. Considering the benign nature of the lesion radiologically and clinically, a differential diagnosis of hyperparathyroidism, renal osteodystrophy, multiple myeloma, and fibrous dysplasia was considered. Hematologic investigations, skeletal survey, and magnetic resonance imaging (MRI) of the bilateral hips were performed to rule out the differential diagnosis.
The patient’s hemoglobin level was 11.8 g/dL with total white blood cell count of 10,300/µL. Renal and hepatic functions were within normal limit. Serum erythrocyte sedimentation rate (ESR) was 12 mm/h and C-reactive protein level was normal. Serum parathyroid level was 32 pg/mL, which was within normal limits, with an alkaline phosphatase level of 101 U/L. The skeletal survey showed no other bony lesion in the body. T1-weighted MRI of both hips showed a well-defined hypointense lesion in the neck and intertrochanteric area of the right hip, which was hyperintense on T2-weighted MRI, suggestive of aneurysmal bone cyst (Figure 2).
Normal ESR, hemoglobin, alkaline phosphatase, and serum parathyroid levels and normal skeletal survey almost ruled out multiple myeloma and hyperparathyroidism. Normal renal profile ruled out renal osteodystrophy and the osteitis fibrosa cystica lesion associated with it. We planned for prophylactic internal fixation of the lesion to prevent a pathologic fracture. According to Mirels,15 if there is a lytic lesion covering more than two-thirds of the circumference of the bone in the peritrochanteric area, the chances of a pathologic fracture are high and such fractures should be fixed.
We planned for curettage of the lesion with bone grafting and in situ intramedullary fixation of the lesion. Curettage was done according to the plan and the sample was sent for histopathologic examination. In situ internal fixation and bone grafting were performed by using a proximal femoral intramedullary nail. To our surprise, the biopsy sample was reported as xanthogranuloma, with multiple foamy macrophages mixed with inflammatory cells and aggregates of lymphocytes (Figure 3). Mycobacterial and routine bacterial cultures were reported as negative. The patient was kept on oral antibiotics (cefixime and moxifloxacin) for 6 weeks, and she made an uneventful recovery. At 6-month follow-up, a radiograph of the right hip showed a healed lesion with proximal femoral nail in situ (Figure 4).
Discussion
To the best of our knowledge, a total of 5 cases of XO have been reported in the literature. The earliest of these reports were by Cozzutto and Carbone,1 who reported 2 cases of XO of the first rib and of the epiphysis of the tibia, respectively. The importance of these lesions to diagnosis is their confusion with a neoplastic disease, as XO is itself a benign disorder. These lesions can mimic a neoplastic lesion in clinical and radiologic presentation and the only way to differentiate the lesion from a neoplastic disease is by histopathologic examination of the tissue. Hypothetically, xanthogranulomatous disorders can be related to trauma or infection.
In 2007, Vankalakunti and colleagues6 reported XO of the ulna in a 50-year-old postmenopausal woman. In that case, progressive swelling was present on the extensor aspect of her right forearm for a period of 2 years, for which curettage and bone grafting were performed, using autograft from the ipsilateral iliac crest. The tissue culture was sterile, and XO was diagnosed as a result of the histopathologic examination. In 2009, Cennimo and colleagues7 reported XO of the index finger and wrist of a man complaining of pain and swelling for 1 year, which was unresponsive to antibiotics. The diagnosis of XO was confirmed histopathologically, when the culture of the same tissue grew Mycobacterium marinum. Radical synovectomy of the lesion was performed, after which minocycline, clarithromycin, and ethambutol were administered. In 2012, Borjian and colleagues8 reported a case of XO of the proximal humerus and proximal fibula in a 14-year-old child. The child, who presented with fever, pain, and restriction of shoulder movements, was started on oral antibiotics as the tissue culture grew Staphylococcus aureus; the patient did not complete the course of treatment in the hospital. No surgical intervention was done in this case. The diagnosis of XO was confirmed by microscopic examination of the tissue.
An association between bacterial infection and xanthogranulomatous inflammation has existed in several organs, such as the kidneys, and in the gastrointestinal system, but such an association of the 2 is yet to be determined for bone.5,10,16-19 Because of the paucity of literature on the disease, a management protocol for XO of bone has not been defined, and decisions have to be made considering the natural history of the disease in other organs. We present this case primarily because of its rarity, curability, and its close resemblance to bone tumors. While XO is benign, it can mimic a neoplastic bone lesion in its imaging and clinical manifestations, and appropriate differentiation is crucial. Currently, histopathologic examination of lesions is the most specific and is the gold standard for diagnosis.
Conclusion
Xanthogranulomatous osteomyelitis is a very rare entity, and only a few cases have been reported in the English-language literature. Though rare, XO warrants greater emphasis than it receives in the literature. It is a chronic inflammatory disease having a close resemblance to bone tumors. A high index of suspicion must be practiced to differentiate XO from tumors. Histopathologic examination is mandatory to establish definitive diagnosis and correct treatment.
1. Cozzutto C, Carbone A. The xanthogranulomatous process. Xanthogranulomatous inflammation. Pathol Res Pract. 1988;183(4):395-402.
2. Ladefoged C, Lorentzen M. Xanthogranulomatous cholecystitis. A clinicopathological study of 20 cases and review of the literature. APMIS. 1993;101(11):869-875.
3. Nistal M, Gonzalez-Peramato P, Serrano A, Regadera J. Xanthogranulomatous funiculitis and orchiepididymitis: report of 2 cases with immunohistochemical study and literature review. Arch Pathol Lab Med. 2004;128(8):911-914.
4. Oh YH, Seong SS, Jang KS, et al. Xanthogranulomatous inflammation presenting as a submucosal mass of the sigmoid colon. Pathol Int. 2005;55(7):440-444.
5. Cozzutto C. Xanthogranulomatous osteomyelitis. Arch Pathol Lab Med. 1984;108(12):973-6.
6. Vankalakunti M, Saikia UN, Mathew M, Kang M. Xanthogranulomatous osteomyelitis of ulna mimicking neoplasm. World J Surg Oncol. 2007;30(5):46.
7. Cennimo DJ, Agag R, Fleegler E, et al. Mycobacterium marinum hand infection in a “sushi chef.” Eplasty. 2009;14(9):e43.
8. Borjian A, Rezaei F, Eshaghi MA, Shemshaki H. Xanthogranulomatous osteomyelitis. J Orthop Traumatol. 2012;13(4):217-220.
9. Rafique M, Yaqoob N. Xanthogranulomatous prostatitis: a mimic of carcinoma of prostate. World J Surg Oncol. 2006;4:30.
10. Nakashiro H, Haraoka S, Fujiwara K, Harada S, Hisatsugu T, Watanabe T. Xanthogranulomatous cholecystis. Cell composition and a possible pathogenetic role of cell-mediated immunity. Pathol Res Pract. 1995;191(11):1078-1086.
11. Hamada T, Ito H, Araki Y, Fujii K, Inoue M, Ishida O. Benign fibrous histiocytoma of the femur: review of three cases. Skeletal Radiol. 1996;25(1):25-29.
12. Kossard S, Chow E, Wilkinson B, Killingsworth M. Lipid and giant cell poor necrobiotic xanthogranuloma. J Cutan Pathol. 2000;27(7):374-378.
13. Girschick HJ, Huppertz HI, Harmsen D, Krauspe R, Müller-Hermelink HK, Papadopoulos T. Chronic recurrent multifocal osteomyelitis in children: diagnostic value of histopathology and microbial testing. Hum Pathol. 1999;30(1):59-65.
14. Kayser R, Mahlfeld K, Grasshoff H. Vertebral Langerhans-cell histiocytosis in childhood – a differential diagnosis of spinal osteomyelitis. Klin Padiatr. 1999;211(5):399-402.
15. Mirels H. Metastatic disease in long bones. A proposed scoring system for diagnosing impending pathologic fractures. Clin Orthop Relat Res. 1989;249:256-264.
16. Machiz S, Gordon J, Block N, Politano VA. Salmonella typhosa urinary tract infection and xanthogranulomatous pyelonephritis. Case report and review of literature. J Fla Med Assoc. 1974;61(9):703-705.
17. Gauperaa T, Stalsberg H. Renal endometriosis. A case report. Scand J Urol Nephrol. 1977;11(2):189-191.
18. Goodman M, Curry T, Russell T. Xanthogranulomatous pyelonephritis (XGP): a local disease with systemic manifestations. Report of 23 patients and review of the literature. Medicine. 1979;58(2):171-181.
19. Guarino M, Reale D, Micoli G, Tricomi P, Cristofori E. Xanthogranulomatous gastritis: association with xanthogranulomatous cholecystitis. J Clin Pathol. 1993;46(1):88-90.
1. Cozzutto C, Carbone A. The xanthogranulomatous process. Xanthogranulomatous inflammation. Pathol Res Pract. 1988;183(4):395-402.
2. Ladefoged C, Lorentzen M. Xanthogranulomatous cholecystitis. A clinicopathological study of 20 cases and review of the literature. APMIS. 1993;101(11):869-875.
3. Nistal M, Gonzalez-Peramato P, Serrano A, Regadera J. Xanthogranulomatous funiculitis and orchiepididymitis: report of 2 cases with immunohistochemical study and literature review. Arch Pathol Lab Med. 2004;128(8):911-914.
4. Oh YH, Seong SS, Jang KS, et al. Xanthogranulomatous inflammation presenting as a submucosal mass of the sigmoid colon. Pathol Int. 2005;55(7):440-444.
5. Cozzutto C. Xanthogranulomatous osteomyelitis. Arch Pathol Lab Med. 1984;108(12):973-6.
6. Vankalakunti M, Saikia UN, Mathew M, Kang M. Xanthogranulomatous osteomyelitis of ulna mimicking neoplasm. World J Surg Oncol. 2007;30(5):46.
7. Cennimo DJ, Agag R, Fleegler E, et al. Mycobacterium marinum hand infection in a “sushi chef.” Eplasty. 2009;14(9):e43.
8. Borjian A, Rezaei F, Eshaghi MA, Shemshaki H. Xanthogranulomatous osteomyelitis. J Orthop Traumatol. 2012;13(4):217-220.
9. Rafique M, Yaqoob N. Xanthogranulomatous prostatitis: a mimic of carcinoma of prostate. World J Surg Oncol. 2006;4:30.
10. Nakashiro H, Haraoka S, Fujiwara K, Harada S, Hisatsugu T, Watanabe T. Xanthogranulomatous cholecystis. Cell composition and a possible pathogenetic role of cell-mediated immunity. Pathol Res Pract. 1995;191(11):1078-1086.
11. Hamada T, Ito H, Araki Y, Fujii K, Inoue M, Ishida O. Benign fibrous histiocytoma of the femur: review of three cases. Skeletal Radiol. 1996;25(1):25-29.
12. Kossard S, Chow E, Wilkinson B, Killingsworth M. Lipid and giant cell poor necrobiotic xanthogranuloma. J Cutan Pathol. 2000;27(7):374-378.
13. Girschick HJ, Huppertz HI, Harmsen D, Krauspe R, Müller-Hermelink HK, Papadopoulos T. Chronic recurrent multifocal osteomyelitis in children: diagnostic value of histopathology and microbial testing. Hum Pathol. 1999;30(1):59-65.
14. Kayser R, Mahlfeld K, Grasshoff H. Vertebral Langerhans-cell histiocytosis in childhood – a differential diagnosis of spinal osteomyelitis. Klin Padiatr. 1999;211(5):399-402.
15. Mirels H. Metastatic disease in long bones. A proposed scoring system for diagnosing impending pathologic fractures. Clin Orthop Relat Res. 1989;249:256-264.
16. Machiz S, Gordon J, Block N, Politano VA. Salmonella typhosa urinary tract infection and xanthogranulomatous pyelonephritis. Case report and review of literature. J Fla Med Assoc. 1974;61(9):703-705.
17. Gauperaa T, Stalsberg H. Renal endometriosis. A case report. Scand J Urol Nephrol. 1977;11(2):189-191.
18. Goodman M, Curry T, Russell T. Xanthogranulomatous pyelonephritis (XGP): a local disease with systemic manifestations. Report of 23 patients and review of the literature. Medicine. 1979;58(2):171-181.
19. Guarino M, Reale D, Micoli G, Tricomi P, Cristofori E. Xanthogranulomatous gastritis: association with xanthogranulomatous cholecystitis. J Clin Pathol. 1993;46(1):88-90.
Case Studies in Toxicology: Managing Missed Methadone
A 53-year-old woman presented to the ED after experiencing a fall. Her medical history was significant for chronic obstructive pulmonary disease, hepatitis, and a remote history of intravenous drug use, for which she had been maintained on methadone for the past 20 years. She reported that she had suffered several “fainting episodes” over the past month, and the morning prior to arrival, had sustained what she thought was a mechanical fall outside of the methadone program she attended. She complained of tenderness on her head but denied any other injuries.

The methadone program had referred the patient to the ED for evaluation, noting to the ED staff that her daily methadone dose of 185 mg had not been dispensed prior to transfer. During evaluation, the patient requested that the emergency physician (EP) provide the methadone dose since the clinic would close prior to her discharge from the ED.
How can requests for methadone be managed in the ED?
Methadone is a long-acting oral opioid that is used for both opioid replacement therapy and pain management. When used to reduce craving in opioid-dependent patients, methadone is administered daily through federally sanctioned methadone maintenance treatment (MMT) programs. Patients who consistently adhere to the required guidelines are given “take home” doses. When used for pain management, methadone is typically administered several times daily and may be prescribed by any provider with an appropriate DEA registration.
When given for MMT, methadone saturates the µ-opioid receptors and hinders their binding and agonism by other opioids such as heroin or oxycodone. Patients in MMT programs are started on a low initial dose and slowly titrated upward as tolerance to the adverse effects (eg, sedation) develop.
How are symptomatic patients with methadone withdrawal treated?
Most methadone programs have limited hours and require that patients who miss a dose wait until the following day to return to the program. This is typically without medical consequence because the high dose dispensed by these programs maintains a therapeutic blood concentration for far longer than the expected delay. Although the half-life of methadone exhibits wide interindividual variability, it generally ranges from 12 hours to more than 40 hours.1 Regardless, patients may feel anxious about potential opioid withdrawal, and this often leads them to access the ED for a missed dose.
The neuropsychiatric symptoms attending withdrawal may precede the objective signs of opioid withdrawal. Patients with objective signs of opioid withdrawal (eg, piloerection, vomiting, diarrhea, dilated pupils) may be sufficiently treated with supportive care alone, using antiemetics, hydration, and sometimes clonidine.
Administration of substitute opioids is problematic due to the patient’s underlying tolerance necessitating careful dose titration. Therefore, direct replacement of methadone in the ED remains controversial, and some EDs have strict policies prohibiting the administration of methadone to patients who have missed an MMT dose. Such policies, which are intended to discourage patients from using the ED as a convenience, may be appropriate given the generally benign—though uncomfortable—course of opioid withdrawal due to abstinence.
Other EDs provide replacement methadone for asymptomatic, treat-and-release patients confirmed to be enrolled in an MMT program when the time to the next dose is likely to be 24 hours or greater from the missed dose. Typically, a dose of no more than 10 mg orally or 10 mg intramuscularly (IM) is recommended, and patients should be advised that they will be receiving only a low dose to sufficient to prevent withdrawal—one that may not have the equivalent effects of the outpatient dose.
Whenever possible, a patient’s MMT program should be contacted and informed of the ED visit. For patients who display objective signs of withdrawal and who cannot be confirmed or who do not participate in an MMT program, 10 mg of methadone IM will prevent uncertainty of drug absorption in the setting of nausea or vomiting. All patients receiving oral methadone should be observed for 1 hour, and those receiving IM methadone should be observed for at least 90 minutes to assess for unexpected sedation.2
Patients encountering circumstances that prevent opioid access (eg, incarceration) and who are not in withdrawal but have gone without opioids for more than 5 days may have a loss of tolerance to their usual doses—whether the medication was obtained through an MMT program or illicitly. Harm-reduction strategies aimed at educating patients on the potential vulnerability to their familiar dosing regimens are warranted to avert inadvertent overdoses in chronic opioid users who are likely to resume illicit opoiod use.
Does this patient need syncope evaluation?
Further complicating the decision regarding ED dispensing of methadone are the effects of the drug on myocardial repolarization. Methadone affects conduction across the hERG potassium rectifier current and can prolong the QTc interval on the surface electrocardiogram (ECG), predisposing a patient to torsade de pointes (TdP). Although there is controversy regarding the role of ECG screening during the enrollment of patients in methadone maintenance clinics, doses above 60 mg, underlying myocardial disease, female sex, and electrolyte disturbances may increase the risk of QT prolongation and TdP.3
Whether there is value in obtaining a screening ECG in a patient receiving an initial dose of methadone in the ED is unclear, and this practice is controversial even among methadone clinics. However, some of the excess death in patients taking methadone may be explained by the dysrhythmogenic potential of methadone.4 An ECG therefore may elucidate a correctable cause in methadone patients presenting with syncope.
Administering methadone to patients with documented QT prolongation must weigh the risk of methadone’s conduction effects against the substantial risks of illicit opioid self-administration. For some patients at-risk for TdP, it may be preferable to use buprenorphine if possible, since it does not carry the same cardiac effects as methadone.1,5 Such therapy requires referral to a physician licensed to prescribe this medication.
How should admitted patients be managed?
While administration of methadone for withdrawal or maintenance therapy in the ED is acceptable, outpatient prescribing of methadone for these reasons is not legal, and only federally regulated clinics may engage in this practice. Hospitalized patients who are enrolled in an MMT program should have their daily methadone dose confirmed and continued—as long as the patient has not lost tolerance. Patients not participating in an MMT program can receive up to 3 days of methadone in the hospital, even if the practitioner is not registered to provide methadone.6 For these patients, it is recommended that the physician order a low dose of methadone and also consult with an addiction specialist to determine whether the patient should continue on MMT maintenance or undergo detoxification.
It is important to note that methadone may be prescribed for pain, but its use in the ED for this purpose is strongly discouraged, especially in patients who have never received methadone previously. For admitted patients requiring such potent opioid analgesia, consultation with a pain service or, when indicated, a palliative care/hospice specialist is warranted as the dosing intervals are different in each setting, and the risk of respiratory depression is high.
Case Conclusion
As requested by the MMT clinic, the patient was administered methadone 185 mg orally in the ED, though a dose of 10 mg would have been sufficient to prevent withdrawal. Unfortunately, the EP did not appreciate the relationship of the markedly prolonged QTc and the methadone, which should have prompted a dose reduction.
Evaluation of the patient’s electrolyte levels, which included magnesium and potassium, were normal. An ECG was repeated 24 hours later and revealed a persistent, but improved, QT interval at 505 ms. The remainder of the syncope workup was negative. Because the patient had no additional symptoms or events during her stay, she was discharged. At discharge, the EP followed up with the MMT clinic to discuss lowering the patient’s daily methadone dose, as well as close cardiology follow-up.
Dr Rao is the chief of the division of medical toxicology at New York Presbyterian Hospital/Weill Cornell Medical Center, New York. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Chou R, Weimer MB, Dana T. Methadone overdose and cardiac arrhythmia potential: findings from a review of the evidence for an American Pain Society and College on Problems of Drug Dependence clinical practice guideline. J Pain. 2014;15(4):338-365.
- National Highway Traffic Safety Administration Web site. Methadone. http://www.nhtsa.gov/people/injury/research/job185drugs/methadone.htm. Accessed August 3, 2015.
- Martin JA, Campbell A, Killip T, et al; Substance Abuse and Mental Health Services Administration. QT interval screening in methadone maintenance treatment: report of a SAMHSA expert panel. J Addict Dis. 2011;30(4):283-306. Erratum in: J Addict Dis. 2012;31(1):91.
- Ray WA, Chung CP, Murray KT, Cooper WO, Hall K, Stein CM. Out-of-hospital mortality among patients receiving methadone for noncancer pain. JAMA Intern Med. 2015;175(3):420-427.
- Davis MP. Twelve reasons for considering buprenorphine as a frontline analgesic in the management of pain. J Support Oncol. 2012;10(6):209-219.
- US Government Printing Office. Federal Digital System. Administering or dispensing of narcotic drugs. Code of Federal Regulations. Title 21 CFR §1306.07. http://www.gpo.gov/fdsys/pkg/CFR-1998-title21-vol9/pdf/CFR-1998-title21-vol9-sec1306-07.pdf. Accessed August 4, 2015.
A 53-year-old woman presented to the ED after experiencing a fall. Her medical history was significant for chronic obstructive pulmonary disease, hepatitis, and a remote history of intravenous drug use, for which she had been maintained on methadone for the past 20 years. She reported that she had suffered several “fainting episodes” over the past month, and the morning prior to arrival, had sustained what she thought was a mechanical fall outside of the methadone program she attended. She complained of tenderness on her head but denied any other injuries.
The methadone program had referred the patient to the ED for evaluation, noting to the ED staff that her daily methadone dose of 185 mg had not been dispensed prior to transfer. During evaluation, the patient requested that the emergency physician (EP) provide the methadone dose since the clinic would close prior to her discharge from the ED.
How can requests for methadone be managed in the ED?
Methadone is a long-acting oral opioid that is used for both opioid replacement therapy and pain management. When used to reduce craving in opioid-dependent patients, methadone is administered daily through federally sanctioned methadone maintenance treatment (MMT) programs. Patients who consistently adhere to the required guidelines are given “take home” doses. When used for pain management, methadone is typically administered several times daily and may be prescribed by any provider with an appropriate DEA registration.
When given for MMT, methadone saturates the µ-opioid receptors and hinders their binding and agonism by other opioids such as heroin or oxycodone. Patients in MMT programs are started on a low initial dose and slowly titrated upward as tolerance to the adverse effects (eg, sedation) develop.
How are symptomatic patients with methadone withdrawal treated?
Most methadone programs have limited hours and require that patients who miss a dose wait until the following day to return to the program. This is typically without medical consequence because the high dose dispensed by these programs maintains a therapeutic blood concentration for far longer than the expected delay. Although the half-life of methadone exhibits wide interindividual variability, it generally ranges from 12 hours to more than 40 hours.1 Regardless, patients may feel anxious about potential opioid withdrawal, and this often leads them to access the ED for a missed dose.
The neuropsychiatric symptoms attending withdrawal may precede the objective signs of opioid withdrawal. Patients with objective signs of opioid withdrawal (eg, piloerection, vomiting, diarrhea, dilated pupils) may be sufficiently treated with supportive care alone, using antiemetics, hydration, and sometimes clonidine.
Administration of substitute opioids is problematic due to the patient’s underlying tolerance necessitating careful dose titration. Therefore, direct replacement of methadone in the ED remains controversial, and some EDs have strict policies prohibiting the administration of methadone to patients who have missed an MMT dose. Such policies, which are intended to discourage patients from using the ED as a convenience, may be appropriate given the generally benign—though uncomfortable—course of opioid withdrawal due to abstinence.
Other EDs provide replacement methadone for asymptomatic, treat-and-release patients confirmed to be enrolled in an MMT program when the time to the next dose is likely to be 24 hours or greater from the missed dose. Typically, a dose of no more than 10 mg orally or 10 mg intramuscularly (IM) is recommended, and patients should be advised that they will be receiving only a low dose to sufficient to prevent withdrawal—one that may not have the equivalent effects of the outpatient dose.
Whenever possible, a patient’s MMT program should be contacted and informed of the ED visit. For patients who display objective signs of withdrawal and who cannot be confirmed or who do not participate in an MMT program, 10 mg of methadone IM will prevent uncertainty of drug absorption in the setting of nausea or vomiting. All patients receiving oral methadone should be observed for 1 hour, and those receiving IM methadone should be observed for at least 90 minutes to assess for unexpected sedation.2
Patients encountering circumstances that prevent opioid access (eg, incarceration) and who are not in withdrawal but have gone without opioids for more than 5 days may have a loss of tolerance to their usual doses—whether the medication was obtained through an MMT program or illicitly. Harm-reduction strategies aimed at educating patients on the potential vulnerability to their familiar dosing regimens are warranted to avert inadvertent overdoses in chronic opioid users who are likely to resume illicit opoiod use.
Does this patient need syncope evaluation?
Further complicating the decision regarding ED dispensing of methadone are the effects of the drug on myocardial repolarization. Methadone affects conduction across the hERG potassium rectifier current and can prolong the QTc interval on the surface electrocardiogram (ECG), predisposing a patient to torsade de pointes (TdP). Although there is controversy regarding the role of ECG screening during the enrollment of patients in methadone maintenance clinics, doses above 60 mg, underlying myocardial disease, female sex, and electrolyte disturbances may increase the risk of QT prolongation and TdP.3
Whether there is value in obtaining a screening ECG in a patient receiving an initial dose of methadone in the ED is unclear, and this practice is controversial even among methadone clinics. However, some of the excess death in patients taking methadone may be explained by the dysrhythmogenic potential of methadone.4 An ECG therefore may elucidate a correctable cause in methadone patients presenting with syncope.
Administering methadone to patients with documented QT prolongation must weigh the risk of methadone’s conduction effects against the substantial risks of illicit opioid self-administration. For some patients at-risk for TdP, it may be preferable to use buprenorphine if possible, since it does not carry the same cardiac effects as methadone.1,5 Such therapy requires referral to a physician licensed to prescribe this medication.
How should admitted patients be managed?
While administration of methadone for withdrawal or maintenance therapy in the ED is acceptable, outpatient prescribing of methadone for these reasons is not legal, and only federally regulated clinics may engage in this practice. Hospitalized patients who are enrolled in an MMT program should have their daily methadone dose confirmed and continued—as long as the patient has not lost tolerance. Patients not participating in an MMT program can receive up to 3 days of methadone in the hospital, even if the practitioner is not registered to provide methadone.6 For these patients, it is recommended that the physician order a low dose of methadone and also consult with an addiction specialist to determine whether the patient should continue on MMT maintenance or undergo detoxification.
It is important to note that methadone may be prescribed for pain, but its use in the ED for this purpose is strongly discouraged, especially in patients who have never received methadone previously. For admitted patients requiring such potent opioid analgesia, consultation with a pain service or, when indicated, a palliative care/hospice specialist is warranted as the dosing intervals are different in each setting, and the risk of respiratory depression is high.
Case Conclusion
As requested by the MMT clinic, the patient was administered methadone 185 mg orally in the ED, though a dose of 10 mg would have been sufficient to prevent withdrawal. Unfortunately, the EP did not appreciate the relationship of the markedly prolonged QTc and the methadone, which should have prompted a dose reduction.
Evaluation of the patient’s electrolyte levels, which included magnesium and potassium, were normal. An ECG was repeated 24 hours later and revealed a persistent, but improved, QT interval at 505 ms. The remainder of the syncope workup was negative. Because the patient had no additional symptoms or events during her stay, she was discharged. At discharge, the EP followed up with the MMT clinic to discuss lowering the patient’s daily methadone dose, as well as close cardiology follow-up.
Dr Rao is the chief of the division of medical toxicology at New York Presbyterian Hospital/Weill Cornell Medical Center, New York. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
A 53-year-old woman presented to the ED after experiencing a fall. Her medical history was significant for chronic obstructive pulmonary disease, hepatitis, and a remote history of intravenous drug use, for which she had been maintained on methadone for the past 20 years. She reported that she had suffered several “fainting episodes” over the past month, and the morning prior to arrival, had sustained what she thought was a mechanical fall outside of the methadone program she attended. She complained of tenderness on her head but denied any other injuries.
The methadone program had referred the patient to the ED for evaluation, noting to the ED staff that her daily methadone dose of 185 mg had not been dispensed prior to transfer. During evaluation, the patient requested that the emergency physician (EP) provide the methadone dose since the clinic would close prior to her discharge from the ED.
How can requests for methadone be managed in the ED?
Methadone is a long-acting oral opioid that is used for both opioid replacement therapy and pain management. When used to reduce craving in opioid-dependent patients, methadone is administered daily through federally sanctioned methadone maintenance treatment (MMT) programs. Patients who consistently adhere to the required guidelines are given “take home” doses. When used for pain management, methadone is typically administered several times daily and may be prescribed by any provider with an appropriate DEA registration.
When given for MMT, methadone saturates the µ-opioid receptors and hinders their binding and agonism by other opioids such as heroin or oxycodone. Patients in MMT programs are started on a low initial dose and slowly titrated upward as tolerance to the adverse effects (eg, sedation) develop.
How are symptomatic patients with methadone withdrawal treated?
Most methadone programs have limited hours and require that patients who miss a dose wait until the following day to return to the program. This is typically without medical consequence because the high dose dispensed by these programs maintains a therapeutic blood concentration for far longer than the expected delay. Although the half-life of methadone exhibits wide interindividual variability, it generally ranges from 12 hours to more than 40 hours.1 Regardless, patients may feel anxious about potential opioid withdrawal, and this often leads them to access the ED for a missed dose.
The neuropsychiatric symptoms attending withdrawal may precede the objective signs of opioid withdrawal. Patients with objective signs of opioid withdrawal (eg, piloerection, vomiting, diarrhea, dilated pupils) may be sufficiently treated with supportive care alone, using antiemetics, hydration, and sometimes clonidine.
Administration of substitute opioids is problematic due to the patient’s underlying tolerance necessitating careful dose titration. Therefore, direct replacement of methadone in the ED remains controversial, and some EDs have strict policies prohibiting the administration of methadone to patients who have missed an MMT dose. Such policies, which are intended to discourage patients from using the ED as a convenience, may be appropriate given the generally benign—though uncomfortable—course of opioid withdrawal due to abstinence.
Other EDs provide replacement methadone for asymptomatic, treat-and-release patients confirmed to be enrolled in an MMT program when the time to the next dose is likely to be 24 hours or greater from the missed dose. Typically, a dose of no more than 10 mg orally or 10 mg intramuscularly (IM) is recommended, and patients should be advised that they will be receiving only a low dose to sufficient to prevent withdrawal—one that may not have the equivalent effects of the outpatient dose.
Whenever possible, a patient’s MMT program should be contacted and informed of the ED visit. For patients who display objective signs of withdrawal and who cannot be confirmed or who do not participate in an MMT program, 10 mg of methadone IM will prevent uncertainty of drug absorption in the setting of nausea or vomiting. All patients receiving oral methadone should be observed for 1 hour, and those receiving IM methadone should be observed for at least 90 minutes to assess for unexpected sedation.2
Patients encountering circumstances that prevent opioid access (eg, incarceration) and who are not in withdrawal but have gone without opioids for more than 5 days may have a loss of tolerance to their usual doses—whether the medication was obtained through an MMT program or illicitly. Harm-reduction strategies aimed at educating patients on the potential vulnerability to their familiar dosing regimens are warranted to avert inadvertent overdoses in chronic opioid users who are likely to resume illicit opoiod use.
Does this patient need syncope evaluation?
Further complicating the decision regarding ED dispensing of methadone are the effects of the drug on myocardial repolarization. Methadone affects conduction across the hERG potassium rectifier current and can prolong the QTc interval on the surface electrocardiogram (ECG), predisposing a patient to torsade de pointes (TdP). Although there is controversy regarding the role of ECG screening during the enrollment of patients in methadone maintenance clinics, doses above 60 mg, underlying myocardial disease, female sex, and electrolyte disturbances may increase the risk of QT prolongation and TdP.3
Whether there is value in obtaining a screening ECG in a patient receiving an initial dose of methadone in the ED is unclear, and this practice is controversial even among methadone clinics. However, some of the excess death in patients taking methadone may be explained by the dysrhythmogenic potential of methadone.4 An ECG therefore may elucidate a correctable cause in methadone patients presenting with syncope.
Administering methadone to patients with documented QT prolongation must weigh the risk of methadone’s conduction effects against the substantial risks of illicit opioid self-administration. For some patients at-risk for TdP, it may be preferable to use buprenorphine if possible, since it does not carry the same cardiac effects as methadone.1,5 Such therapy requires referral to a physician licensed to prescribe this medication.
How should admitted patients be managed?
While administration of methadone for withdrawal or maintenance therapy in the ED is acceptable, outpatient prescribing of methadone for these reasons is not legal, and only federally regulated clinics may engage in this practice. Hospitalized patients who are enrolled in an MMT program should have their daily methadone dose confirmed and continued—as long as the patient has not lost tolerance. Patients not participating in an MMT program can receive up to 3 days of methadone in the hospital, even if the practitioner is not registered to provide methadone.6 For these patients, it is recommended that the physician order a low dose of methadone and also consult with an addiction specialist to determine whether the patient should continue on MMT maintenance or undergo detoxification.
It is important to note that methadone may be prescribed for pain, but its use in the ED for this purpose is strongly discouraged, especially in patients who have never received methadone previously. For admitted patients requiring such potent opioid analgesia, consultation with a pain service or, when indicated, a palliative care/hospice specialist is warranted as the dosing intervals are different in each setting, and the risk of respiratory depression is high.
Case Conclusion
As requested by the MMT clinic, the patient was administered methadone 185 mg orally in the ED, though a dose of 10 mg would have been sufficient to prevent withdrawal. Unfortunately, the EP did not appreciate the relationship of the markedly prolonged QTc and the methadone, which should have prompted a dose reduction.
Evaluation of the patient’s electrolyte levels, which included magnesium and potassium, were normal. An ECG was repeated 24 hours later and revealed a persistent, but improved, QT interval at 505 ms. The remainder of the syncope workup was negative. Because the patient had no additional symptoms or events during her stay, she was discharged. At discharge, the EP followed up with the MMT clinic to discuss lowering the patient’s daily methadone dose, as well as close cardiology follow-up.
Dr Rao is the chief of the division of medical toxicology at New York Presbyterian Hospital/Weill Cornell Medical Center, New York. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Chou R, Weimer MB, Dana T. Methadone overdose and cardiac arrhythmia potential: findings from a review of the evidence for an American Pain Society and College on Problems of Drug Dependence clinical practice guideline. J Pain. 2014;15(4):338-365.
- National Highway Traffic Safety Administration Web site. Methadone. http://www.nhtsa.gov/people/injury/research/job185drugs/methadone.htm. Accessed August 3, 2015.
- Martin JA, Campbell A, Killip T, et al; Substance Abuse and Mental Health Services Administration. QT interval screening in methadone maintenance treatment: report of a SAMHSA expert panel. J Addict Dis. 2011;30(4):283-306. Erratum in: J Addict Dis. 2012;31(1):91.
- Ray WA, Chung CP, Murray KT, Cooper WO, Hall K, Stein CM. Out-of-hospital mortality among patients receiving methadone for noncancer pain. JAMA Intern Med. 2015;175(3):420-427.
- Davis MP. Twelve reasons for considering buprenorphine as a frontline analgesic in the management of pain. J Support Oncol. 2012;10(6):209-219.
- US Government Printing Office. Federal Digital System. Administering or dispensing of narcotic drugs. Code of Federal Regulations. Title 21 CFR §1306.07. http://www.gpo.gov/fdsys/pkg/CFR-1998-title21-vol9/pdf/CFR-1998-title21-vol9-sec1306-07.pdf. Accessed August 4, 2015.
- Chou R, Weimer MB, Dana T. Methadone overdose and cardiac arrhythmia potential: findings from a review of the evidence for an American Pain Society and College on Problems of Drug Dependence clinical practice guideline. J Pain. 2014;15(4):338-365.
- National Highway Traffic Safety Administration Web site. Methadone. http://www.nhtsa.gov/people/injury/research/job185drugs/methadone.htm. Accessed August 3, 2015.
- Martin JA, Campbell A, Killip T, et al; Substance Abuse and Mental Health Services Administration. QT interval screening in methadone maintenance treatment: report of a SAMHSA expert panel. J Addict Dis. 2011;30(4):283-306. Erratum in: J Addict Dis. 2012;31(1):91.
- Ray WA, Chung CP, Murray KT, Cooper WO, Hall K, Stein CM. Out-of-hospital mortality among patients receiving methadone for noncancer pain. JAMA Intern Med. 2015;175(3):420-427.
- Davis MP. Twelve reasons for considering buprenorphine as a frontline analgesic in the management of pain. J Support Oncol. 2012;10(6):209-219.
- US Government Printing Office. Federal Digital System. Administering or dispensing of narcotic drugs. Code of Federal Regulations. Title 21 CFR §1306.07. http://www.gpo.gov/fdsys/pkg/CFR-1998-title21-vol9/pdf/CFR-1998-title21-vol9-sec1306-07.pdf. Accessed August 4, 2015.

Case Report: The Hungry, Hungry Haustra: The Case of a Missing Feeding Tube
Introduction
Percutaneous endoscopic gastrostomy (PEG) tubes are a common method employed for long-term feeding in patients who are unable to tolerate oral feedings.1 Though PEG-tube placement is a common, safe, and well-studied practice, there are known complications, including wound infection, dislodgement, and peritonitis.2 Dislodgement and recurrent ED visits are increasingly becoming a burden on both patients and healthcare providers, as up to 12.8% of patients will require ED replacement of a dislodged tube, totaling an estimated $1,200 per visit.3
Newer techniques include Roux-en-Y feeding jejunostomy tubes, which are anticipated to reduce long-term complications.4,5 However, dislodgement, sinus tracts, and superimposed infections still occur, also leading to ED visits.6 Foley catheters are a readily available and low-cost alternative to replace commercial feeding-tubes in the ED, and are commonly used when the original feeding-tube is not suitable for reuse.7 In the following presentation, the authors describe a previously unseen case of a fully intussuscepted Foley catheter though a Roux-en-Y jejunostomy.
Case Report
A 69-year-old man, recently diagnosed with invasive squamous cell carcinoma of the distal esophagus, presented to the ED with a chief complaint of “J-tube fell out.” One month prior to presentation, the patient had undergone a laparoscopic Janeway Roux-en-Y nipple jejunostomy. He had been previously evaluated several times in the ED for a displaced J-tube, and his commercial feeding tube had been replaced with a Foley catheter without incident.
On this visit, the patient’s wife reported that the Foley catheter had become displaced 3 days prior to presentation, and she assumed that the patient had accidentally pulled it out. According to the patient’s wife, he had attempted oral feedings, but had difficulty swallowing as well as coughing episodes.
Upon initial evaluation, the patient complained of diffuse abdominal pain and cramping. He denied any nausea or vomiting, and reported normal bowel movements. The physical examination was remarkable for the following: hypotension (blood pressure, 64/46 mm Hg); heart rate, 94 beats/minute; temperature, 96.4°F; cachexia; a diffusely tender abdomen; and viable stoma on the anterior abdominal wall. Purulent and malodorous drainage was noted at the stoma site. There was no Foley catheter or J-tube in place, and neither the patient nor his wife had brought the dislodged tube to the ED.
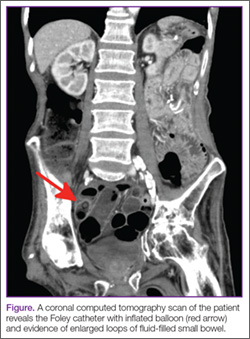
A computed tomography scan of the patient’s abdomen and pelvis were ordered with IV and oral contrast. The imaging studies revealed multiple dilated, fluid-filled loops of small bowel, and a Foley catheter proximal to the ileocecal valve, with the balloon still inflated (Figure).
The emergency physician notified the original surgical team of the patient’s status. The surgical team placed a new, 14 French (Fr)-Foley catheter through the stoma, sutured it in place, and admitted the patient to their service. The patient was maintained on IV antibiotics and fluids. As he continued to pass flatus and stool, a diet was advanced through the replacement Foley catheter. The intussuscepted Foley was subsequently passed naturally on day 4 of his hospital admission. The patient unfortunately died several days later of hypoxic respiratory failure, which was not thought to be related to the ingested catheter.
Discussion
Percutaneous Foley catheters, either pre- or postpyloric, have been used for decades as permanent feeding tubes for patients unable to tolerate oral feedings. These catheters are well-known to be inexpensive and safe replacements for commercial gastrostomy tubes.7 However, a number of complications unique to Foley feeding tubes have been described in case reports, including mechanical obstruction leading to pancreatitis, duodenal obstruction, bowel ischemia secondary to balloon overfilling, pyloric obstruction, bowel infection, as well as broken and digested catheters.8-10
Interestingly, despite multiple case reports demonstrating tube migration, prospective studies have shown this to be a relatively uncommon complication.11 In 2012, a patient in Israel ingested a Foley catheter via the gastrostomy stoma, resulting in small bowel obstruction relieved only by enterotomy and removal of the catheter. There have been no previous documented reports of ingested tubes via jejunostomy stoma.12 Significant forces exerted on Foley catheters have been described, resulting in skin necrosis at the hub and stretching of the catheter from the proximal small bowel to the terminal ileum. In this case presentation, bowel peristalsis was able to advance the entire tube through the skin.13
Management of feeding-Foley-catheter complications typically involves deflation of the balloon and removal and replacement of the offending catheter—usually with a smaller sized Foley catheter (eg, 12Fr, 14Fr, 16Fr). Complicated cases with catheter malfunction have been successfully managed endoscopically.14 The patient in this case was likely at higher risk of complication given the abnormally large wound surrounding the stoma and skin breakdown secondary to superimposed infection.
Conclusion
This case highlights the potent peristaltic forces that are exerted upon a feeding Foley catheter and reinforces the importance of proper tube anchorage. Although this patient did well with direct skin suturing of the replacement catheter, previous studies recommend using a plastic retention ring. Placing a mark on the outside of the catheter as a means to continuously visualize its proper anchorage and placement has also been suggested in the literature. Additionally, whenever a patient presents with a displaced feeding tube (Foley catheter or commercial tube), providers should not assume that the tube has been displaced externally and should maintain a low-threshold for advanced imaging and/or endoscopy if the tube cannot otherwise be located.
Dr Lefkove is an attending physician in the department of emergency medicine, DeKalb Medical Center, Atlanta, Georgia. Dr Meloy is an assistant professor of emergency medicine at Emory University School of Medicine, Atlanta, Georgia.
- Vanis N, Saray A, Gornjakovic S, et al. Percutaneous endoscopic gastrostomy (PEG): retrospective analysis of a 7-year clinical experience. Acta Inform Med. 2012;20(4):235-237.
- Schapiro GD, Edmundowicz SA. Complications of percutaneous endoscopic gastrostomy. Gastrointest Endosc Clin N Am. 1996;6(2):409-422.
- Rosenberger LH, Newhook T, Schirmer B, Sawyer RG. Late accidental dislodgement of a percutaneous endoscopic gastrostomy tube: an underestimated burden on patients and the health care system. Surg Endosc. 2011;25(10):3307-3311.
- Neuman HB, Phillips JD. Laparoscopic Roux-en-Y feeding jejunostomy: a new minimally invasive surgical procedure for permanent feeding access in children with gastric dysfunction. J Laparoendosc Adv Surg Tech A. 2005;15(1):71-74.
- Arnal E, Voiglio EJ, Robert M, Schreiber V, Ceruze P, Caillot JL. Laparoscopic Janeway gastrostomy: an advantageous solution for self-sufficient enteral feeding. Ann Chir. 2005;130(10):613-617.
- Maple JT, Petersen BT, Baron TH, Gostout CJ, Wong Kee Song LM, Buttar NS. Direct percutaneous endoscopic jejunostomy: outcomes in 307 consecutive attempts. Am J Gastroenterol. 2005;100(12):2681-2688.
- Kadakia SC, Cassaday M, Shaffer RT. Comparison of Foley catheter as a replacement gastrostomy tube with commercial replacement gastrostomy tube: a prospective randomized trial. Gastrointest Endosc. 1994;40(2 Pt 1):188-193.
- Brauner E, Kluger Y. Gastrostomy tube dislodgment acute pancreatitis. World J Emerg Surg. 2014;9(1):23.
- Hopens T, Schwesinger WH. Complications of tube gastrostomy: radiologic manifestations. South Med J. 1983;76(1):9-11.
- Martel G, Lingas RI, Gutauskas A, Clark HD. Complication of a percutaneous endoscopic gastrostomy tube causing duodenal ischemia. Surg Laparosc Endosc Percutan Tech. 2006;16(6):445-446.
- Kadakia SC, Cassaday M, Shaffer RT. Prospective evaluation of Foley catheter as a replacement gastrostomy tube. Am J Gastroenterol. 1992;87(11):1594-1597.
- Netz U, Perry ZH, Mizrahi S. The lost foley catheter. Am Surg. 2012;78(9):E407-E408.
- Date RS, Das N, Bateson PG. Unusual complications of ballooned feeding tubes. Ir Med J. 2002;95(6):181-182.
- O’Keefe KP, Dula DJ, Varano V. Duodenal obstruction by a nondeflating Foley catheter gastrostomy tube. Ann Emerg Med. 1990;19(12):1454-1457.
Introduction
Percutaneous endoscopic gastrostomy (PEG) tubes are a common method employed for long-term feeding in patients who are unable to tolerate oral feedings.1 Though PEG-tube placement is a common, safe, and well-studied practice, there are known complications, including wound infection, dislodgement, and peritonitis.2 Dislodgement and recurrent ED visits are increasingly becoming a burden on both patients and healthcare providers, as up to 12.8% of patients will require ED replacement of a dislodged tube, totaling an estimated $1,200 per visit.3
Newer techniques include Roux-en-Y feeding jejunostomy tubes, which are anticipated to reduce long-term complications.4,5 However, dislodgement, sinus tracts, and superimposed infections still occur, also leading to ED visits.6 Foley catheters are a readily available and low-cost alternative to replace commercial feeding-tubes in the ED, and are commonly used when the original feeding-tube is not suitable for reuse.7 In the following presentation, the authors describe a previously unseen case of a fully intussuscepted Foley catheter though a Roux-en-Y jejunostomy.
Case Report
A 69-year-old man, recently diagnosed with invasive squamous cell carcinoma of the distal esophagus, presented to the ED with a chief complaint of “J-tube fell out.” One month prior to presentation, the patient had undergone a laparoscopic Janeway Roux-en-Y nipple jejunostomy. He had been previously evaluated several times in the ED for a displaced J-tube, and his commercial feeding tube had been replaced with a Foley catheter without incident.
On this visit, the patient’s wife reported that the Foley catheter had become displaced 3 days prior to presentation, and she assumed that the patient had accidentally pulled it out. According to the patient’s wife, he had attempted oral feedings, but had difficulty swallowing as well as coughing episodes.
Upon initial evaluation, the patient complained of diffuse abdominal pain and cramping. He denied any nausea or vomiting, and reported normal bowel movements. The physical examination was remarkable for the following: hypotension (blood pressure, 64/46 mm Hg); heart rate, 94 beats/minute; temperature, 96.4°F; cachexia; a diffusely tender abdomen; and viable stoma on the anterior abdominal wall. Purulent and malodorous drainage was noted at the stoma site. There was no Foley catheter or J-tube in place, and neither the patient nor his wife had brought the dislodged tube to the ED.
A computed tomography scan of the patient’s abdomen and pelvis were ordered with IV and oral contrast. The imaging studies revealed multiple dilated, fluid-filled loops of small bowel, and a Foley catheter proximal to the ileocecal valve, with the balloon still inflated (Figure).
The emergency physician notified the original surgical team of the patient’s status. The surgical team placed a new, 14 French (Fr)-Foley catheter through the stoma, sutured it in place, and admitted the patient to their service. The patient was maintained on IV antibiotics and fluids. As he continued to pass flatus and stool, a diet was advanced through the replacement Foley catheter. The intussuscepted Foley was subsequently passed naturally on day 4 of his hospital admission. The patient unfortunately died several days later of hypoxic respiratory failure, which was not thought to be related to the ingested catheter.
Discussion
Percutaneous Foley catheters, either pre- or postpyloric, have been used for decades as permanent feeding tubes for patients unable to tolerate oral feedings. These catheters are well-known to be inexpensive and safe replacements for commercial gastrostomy tubes.7 However, a number of complications unique to Foley feeding tubes have been described in case reports, including mechanical obstruction leading to pancreatitis, duodenal obstruction, bowel ischemia secondary to balloon overfilling, pyloric obstruction, bowel infection, as well as broken and digested catheters.8-10
Interestingly, despite multiple case reports demonstrating tube migration, prospective studies have shown this to be a relatively uncommon complication.11 In 2012, a patient in Israel ingested a Foley catheter via the gastrostomy stoma, resulting in small bowel obstruction relieved only by enterotomy and removal of the catheter. There have been no previous documented reports of ingested tubes via jejunostomy stoma.12 Significant forces exerted on Foley catheters have been described, resulting in skin necrosis at the hub and stretching of the catheter from the proximal small bowel to the terminal ileum. In this case presentation, bowel peristalsis was able to advance the entire tube through the skin.13
Management of feeding-Foley-catheter complications typically involves deflation of the balloon and removal and replacement of the offending catheter—usually with a smaller sized Foley catheter (eg, 12Fr, 14Fr, 16Fr). Complicated cases with catheter malfunction have been successfully managed endoscopically.14 The patient in this case was likely at higher risk of complication given the abnormally large wound surrounding the stoma and skin breakdown secondary to superimposed infection.
Conclusion
This case highlights the potent peristaltic forces that are exerted upon a feeding Foley catheter and reinforces the importance of proper tube anchorage. Although this patient did well with direct skin suturing of the replacement catheter, previous studies recommend using a plastic retention ring. Placing a mark on the outside of the catheter as a means to continuously visualize its proper anchorage and placement has also been suggested in the literature. Additionally, whenever a patient presents with a displaced feeding tube (Foley catheter or commercial tube), providers should not assume that the tube has been displaced externally and should maintain a low-threshold for advanced imaging and/or endoscopy if the tube cannot otherwise be located.
Dr Lefkove is an attending physician in the department of emergency medicine, DeKalb Medical Center, Atlanta, Georgia. Dr Meloy is an assistant professor of emergency medicine at Emory University School of Medicine, Atlanta, Georgia.
Introduction
Percutaneous endoscopic gastrostomy (PEG) tubes are a common method employed for long-term feeding in patients who are unable to tolerate oral feedings.1 Though PEG-tube placement is a common, safe, and well-studied practice, there are known complications, including wound infection, dislodgement, and peritonitis.2 Dislodgement and recurrent ED visits are increasingly becoming a burden on both patients and healthcare providers, as up to 12.8% of patients will require ED replacement of a dislodged tube, totaling an estimated $1,200 per visit.3
Newer techniques include Roux-en-Y feeding jejunostomy tubes, which are anticipated to reduce long-term complications.4,5 However, dislodgement, sinus tracts, and superimposed infections still occur, also leading to ED visits.6 Foley catheters are a readily available and low-cost alternative to replace commercial feeding-tubes in the ED, and are commonly used when the original feeding-tube is not suitable for reuse.7 In the following presentation, the authors describe a previously unseen case of a fully intussuscepted Foley catheter though a Roux-en-Y jejunostomy.
Case Report
A 69-year-old man, recently diagnosed with invasive squamous cell carcinoma of the distal esophagus, presented to the ED with a chief complaint of “J-tube fell out.” One month prior to presentation, the patient had undergone a laparoscopic Janeway Roux-en-Y nipple jejunostomy. He had been previously evaluated several times in the ED for a displaced J-tube, and his commercial feeding tube had been replaced with a Foley catheter without incident.
On this visit, the patient’s wife reported that the Foley catheter had become displaced 3 days prior to presentation, and she assumed that the patient had accidentally pulled it out. According to the patient’s wife, he had attempted oral feedings, but had difficulty swallowing as well as coughing episodes.
Upon initial evaluation, the patient complained of diffuse abdominal pain and cramping. He denied any nausea or vomiting, and reported normal bowel movements. The physical examination was remarkable for the following: hypotension (blood pressure, 64/46 mm Hg); heart rate, 94 beats/minute; temperature, 96.4°F; cachexia; a diffusely tender abdomen; and viable stoma on the anterior abdominal wall. Purulent and malodorous drainage was noted at the stoma site. There was no Foley catheter or J-tube in place, and neither the patient nor his wife had brought the dislodged tube to the ED.
A computed tomography scan of the patient’s abdomen and pelvis were ordered with IV and oral contrast. The imaging studies revealed multiple dilated, fluid-filled loops of small bowel, and a Foley catheter proximal to the ileocecal valve, with the balloon still inflated (Figure).
The emergency physician notified the original surgical team of the patient’s status. The surgical team placed a new, 14 French (Fr)-Foley catheter through the stoma, sutured it in place, and admitted the patient to their service. The patient was maintained on IV antibiotics and fluids. As he continued to pass flatus and stool, a diet was advanced through the replacement Foley catheter. The intussuscepted Foley was subsequently passed naturally on day 4 of his hospital admission. The patient unfortunately died several days later of hypoxic respiratory failure, which was not thought to be related to the ingested catheter.
Discussion
Percutaneous Foley catheters, either pre- or postpyloric, have been used for decades as permanent feeding tubes for patients unable to tolerate oral feedings. These catheters are well-known to be inexpensive and safe replacements for commercial gastrostomy tubes.7 However, a number of complications unique to Foley feeding tubes have been described in case reports, including mechanical obstruction leading to pancreatitis, duodenal obstruction, bowel ischemia secondary to balloon overfilling, pyloric obstruction, bowel infection, as well as broken and digested catheters.8-10
Interestingly, despite multiple case reports demonstrating tube migration, prospective studies have shown this to be a relatively uncommon complication.11 In 2012, a patient in Israel ingested a Foley catheter via the gastrostomy stoma, resulting in small bowel obstruction relieved only by enterotomy and removal of the catheter. There have been no previous documented reports of ingested tubes via jejunostomy stoma.12 Significant forces exerted on Foley catheters have been described, resulting in skin necrosis at the hub and stretching of the catheter from the proximal small bowel to the terminal ileum. In this case presentation, bowel peristalsis was able to advance the entire tube through the skin.13
Management of feeding-Foley-catheter complications typically involves deflation of the balloon and removal and replacement of the offending catheter—usually with a smaller sized Foley catheter (eg, 12Fr, 14Fr, 16Fr). Complicated cases with catheter malfunction have been successfully managed endoscopically.14 The patient in this case was likely at higher risk of complication given the abnormally large wound surrounding the stoma and skin breakdown secondary to superimposed infection.
Conclusion
This case highlights the potent peristaltic forces that are exerted upon a feeding Foley catheter and reinforces the importance of proper tube anchorage. Although this patient did well with direct skin suturing of the replacement catheter, previous studies recommend using a plastic retention ring. Placing a mark on the outside of the catheter as a means to continuously visualize its proper anchorage and placement has also been suggested in the literature. Additionally, whenever a patient presents with a displaced feeding tube (Foley catheter or commercial tube), providers should not assume that the tube has been displaced externally and should maintain a low-threshold for advanced imaging and/or endoscopy if the tube cannot otherwise be located.
Dr Lefkove is an attending physician in the department of emergency medicine, DeKalb Medical Center, Atlanta, Georgia. Dr Meloy is an assistant professor of emergency medicine at Emory University School of Medicine, Atlanta, Georgia.
- Vanis N, Saray A, Gornjakovic S, et al. Percutaneous endoscopic gastrostomy (PEG): retrospective analysis of a 7-year clinical experience. Acta Inform Med. 2012;20(4):235-237.
- Schapiro GD, Edmundowicz SA. Complications of percutaneous endoscopic gastrostomy. Gastrointest Endosc Clin N Am. 1996;6(2):409-422.
- Rosenberger LH, Newhook T, Schirmer B, Sawyer RG. Late accidental dislodgement of a percutaneous endoscopic gastrostomy tube: an underestimated burden on patients and the health care system. Surg Endosc. 2011;25(10):3307-3311.
- Neuman HB, Phillips JD. Laparoscopic Roux-en-Y feeding jejunostomy: a new minimally invasive surgical procedure for permanent feeding access in children with gastric dysfunction. J Laparoendosc Adv Surg Tech A. 2005;15(1):71-74.
- Arnal E, Voiglio EJ, Robert M, Schreiber V, Ceruze P, Caillot JL. Laparoscopic Janeway gastrostomy: an advantageous solution for self-sufficient enteral feeding. Ann Chir. 2005;130(10):613-617.
- Maple JT, Petersen BT, Baron TH, Gostout CJ, Wong Kee Song LM, Buttar NS. Direct percutaneous endoscopic jejunostomy: outcomes in 307 consecutive attempts. Am J Gastroenterol. 2005;100(12):2681-2688.
- Kadakia SC, Cassaday M, Shaffer RT. Comparison of Foley catheter as a replacement gastrostomy tube with commercial replacement gastrostomy tube: a prospective randomized trial. Gastrointest Endosc. 1994;40(2 Pt 1):188-193.
- Brauner E, Kluger Y. Gastrostomy tube dislodgment acute pancreatitis. World J Emerg Surg. 2014;9(1):23.
- Hopens T, Schwesinger WH. Complications of tube gastrostomy: radiologic manifestations. South Med J. 1983;76(1):9-11.
- Martel G, Lingas RI, Gutauskas A, Clark HD. Complication of a percutaneous endoscopic gastrostomy tube causing duodenal ischemia. Surg Laparosc Endosc Percutan Tech. 2006;16(6):445-446.
- Kadakia SC, Cassaday M, Shaffer RT. Prospective evaluation of Foley catheter as a replacement gastrostomy tube. Am J Gastroenterol. 1992;87(11):1594-1597.
- Netz U, Perry ZH, Mizrahi S. The lost foley catheter. Am Surg. 2012;78(9):E407-E408.
- Date RS, Das N, Bateson PG. Unusual complications of ballooned feeding tubes. Ir Med J. 2002;95(6):181-182.
- O’Keefe KP, Dula DJ, Varano V. Duodenal obstruction by a nondeflating Foley catheter gastrostomy tube. Ann Emerg Med. 1990;19(12):1454-1457.
- Vanis N, Saray A, Gornjakovic S, et al. Percutaneous endoscopic gastrostomy (PEG): retrospective analysis of a 7-year clinical experience. Acta Inform Med. 2012;20(4):235-237.
- Schapiro GD, Edmundowicz SA. Complications of percutaneous endoscopic gastrostomy. Gastrointest Endosc Clin N Am. 1996;6(2):409-422.
- Rosenberger LH, Newhook T, Schirmer B, Sawyer RG. Late accidental dislodgement of a percutaneous endoscopic gastrostomy tube: an underestimated burden on patients and the health care system. Surg Endosc. 2011;25(10):3307-3311.
- Neuman HB, Phillips JD. Laparoscopic Roux-en-Y feeding jejunostomy: a new minimally invasive surgical procedure for permanent feeding access in children with gastric dysfunction. J Laparoendosc Adv Surg Tech A. 2005;15(1):71-74.
- Arnal E, Voiglio EJ, Robert M, Schreiber V, Ceruze P, Caillot JL. Laparoscopic Janeway gastrostomy: an advantageous solution for self-sufficient enteral feeding. Ann Chir. 2005;130(10):613-617.
- Maple JT, Petersen BT, Baron TH, Gostout CJ, Wong Kee Song LM, Buttar NS. Direct percutaneous endoscopic jejunostomy: outcomes in 307 consecutive attempts. Am J Gastroenterol. 2005;100(12):2681-2688.
- Kadakia SC, Cassaday M, Shaffer RT. Comparison of Foley catheter as a replacement gastrostomy tube with commercial replacement gastrostomy tube: a prospective randomized trial. Gastrointest Endosc. 1994;40(2 Pt 1):188-193.
- Brauner E, Kluger Y. Gastrostomy tube dislodgment acute pancreatitis. World J Emerg Surg. 2014;9(1):23.
- Hopens T, Schwesinger WH. Complications of tube gastrostomy: radiologic manifestations. South Med J. 1983;76(1):9-11.
- Martel G, Lingas RI, Gutauskas A, Clark HD. Complication of a percutaneous endoscopic gastrostomy tube causing duodenal ischemia. Surg Laparosc Endosc Percutan Tech. 2006;16(6):445-446.
- Kadakia SC, Cassaday M, Shaffer RT. Prospective evaluation of Foley catheter as a replacement gastrostomy tube. Am J Gastroenterol. 1992;87(11):1594-1597.
- Netz U, Perry ZH, Mizrahi S. The lost foley catheter. Am Surg. 2012;78(9):E407-E408.
- Date RS, Das N, Bateson PG. Unusual complications of ballooned feeding tubes. Ir Med J. 2002;95(6):181-182.
- O’Keefe KP, Dula DJ, Varano V. Duodenal obstruction by a nondeflating Foley catheter gastrostomy tube. Ann Emerg Med. 1990;19(12):1454-1457.
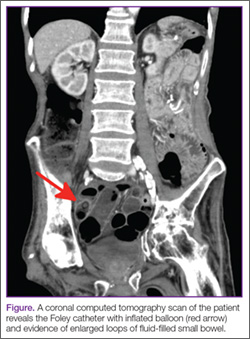
Right foot pain while walking • no erythema or edema • no evidence of structural abnormalities • Dx?
THE CASE
A 24-year-old woman came to our clinic because she had pain in her right foot. Over the previous 4 weeks, she’d noticed increasing pain in the ball of her foot while walking and climbing stairs, particularly in the push-off portion of her gait. She described it as a nagging, localized pain that she rated as a 2 or 3 out of 10. It was an annoyance, but not unbearable. She felt no pain when standing in place or in a non-weight-bearing position.
She denied any trauma to the foot or change in activity, and had been exercising her usual amount (running 2-5 miles per week). Her medical and social histories were unremarkable, and her family history was negative for relevant conditions.
An examination of the right foot revealed no evidence of pes planus, pes cavus, hallux valgus, hammertoes, or other structural abnormalities of the foot or toes. She had no calluses, nor any erythema or edema of the foot or toes. Direct palpation of the medial sesamoid reproduced the patient’s symptoms. Passive dorsiflexion and plantar flexion of the first hallux elicited pain only at the extreme ends of range of motion. Active dorsiflexion and plantar flexion of the right first hallux showed 5 out of 5 strength. A mid-foot squeeze test was negative, and the remainder of the exam was normal.
THE DIAGNOSIS
Pain on palpation of the sesamoids prompted us to gather a more detailed history. The patient had never been a dancer or a long-distance or competitive runner. However, upon delving into possible causes of the pain, she admitted that she was a frequent “knuckle cracker,” and cracked many joints regularly, including the right first metatarsophalangeal joint (MTPJ). She explained that she cracked this joint by hyper-plantarflexing her big toe against the ground, and had been doing this multiple times a day for many years. In the past 4 weeks, she had noticed significant pain in the right first MTPJ while cracking the joint, but she was having difficulty breaking the longstanding habit.
The patient’s description of right foot pain associated with the push-off portion of her gait, and the fact that the pain was exacerbated by the extremes of dorsiflexion and plantar flexion of the great toe, was consistent with MTPJ pain. This, paired with our ability to reproduce the pain by direct palpation of the medial sesamoid, prompted us to make a clinical diagnosis of sesamoiditis. To our knowledge, this is the first case report of sesamoiditis caused by knuckle cracking.
DISCUSSION
Sesamoiditis—chronic pain and inflammation of the hallucal sesamoids—is an overuse or misuse injury that’s typically seen in runners and dancers.1 The hallucal sesamoids are 2 small bones located underneath the head of the first metatarsal and encased within the flexor hallucus brevis tendon that disperses weight from the head of the first metatarsal during the push-off portion of gait.2 Runners and dancers place significant, repetitive axial loading on the sesamoids, which often leads to injury.1 Although our patient initially seemed to have no typical risk factors for developing sesamoiditis, she later revealed that she regularly cracked the MTPJ, which we believe led to her injury.
Interestingly, despite the common assumption that long-term “cracking” of joints can lead to adverse effects such as osteoarthritis, research has not supported this assumption.3,4 A retrospective case-control study of patients with and without hand osteoarthritis found no association between knuckle cracking and osteoarthritis, and the prevalence of osteoarthritis was not higher in patients who cracked their knuckles more frequently and for more years.4
Nonetheless, there have been reports of acute injuries associated with knuckle cracking, consistent with forcing a joint past its normal range of motion, as is typically done in knuckle cracking.5 In forcefully plantarflexing her great toe against a surface until a “crack” was elicited, our patient may have injured the sesamoid by forcing it along the head of the first metatarsal. Conversely, her injury may have been caused by the repetitive displacement of the sesamoid past its usual location, resulting in chronic irritation.
Differential diagnosis includes fracture and stress injury
The differential diagnosis for subacute to chronic pain localized to the sesamoids includes repetitive stress injury (sesamoiditis or capsular strain), fracture or stress fracture, osteoarthritis, osteonecrosis, and gout.1,2 Given our patient’s age and lack of erythema and edema, osteoarthritis and gout were unlikely.
To treat the injury, eliminate the behavior that caused it
Imaging studies may not be necessary in cases of suspected sesamoiditis because such studies are often negative for sesamoiditis and stress fractures of the sesamoids, and because they typically would not affect how the injury is initially treated.1,2,6 In cases in which radiographic confirmation of sesamoiditis is necessary to rule out more serious pathology, 99mTc-methylene diphosphonate (99mTc-MDP) bone scan and magnetic resonance imaging (MRI) are far more sensitive than plain films.1 While a 99mTc-MDP bone scan will show increased uptake at the sesamoids, it has been replaced by MRI, which will show bone marrow edema of the sesamoids and can rule out fracture or osteoarthritis.1
Sesamoiditis is typically managed with a combination of ice, analgesics, activity modification, and/or orthoses.2 Of course, the key to successfully treating sesamoiditis (and all musculoskeletal injuries) is to not only make the diagnosis, but to find the underlying cause in order to prevent continued—or worsening—pain.
Our patient agreed to close follow-up rather than imaging. We established that the only inciting event was the cracking of her MTPJ, and that she should try to eliminate this action before trying other interventions. Our patient stopped cracking her MTPJ and her pain completely resolved in 2 weeks. She remains symptom-free.
THE TAKEAWAY
Ask about knuckle cracking when taking the history of a patient who presents with sesamoiditis, which is characterized by chronic pain and inflammation of the hallucal sesamoids.
1. Nwawka OK, Hayashi D, Diaz LE, et al. Sesamoids and accessory ossicles of the foot: anatomical variability and related pathology. Insights Imaging. 2013;4:581-593.
2. Boike A, Schnirring-Judge M, McMillin S. Sesamoid disorders of the first metatarsophalangeal joint. Clin Podiatr Med Surg. 2011;28:269-285.
3. Castellanos J, Axelrod D. Effect of habitual knuckle cracking on hand function. Ann Rheum Dis. 1990:49:308-309.
4. Deweber K, Olszewski M, Ortolano R. Knuckle cracking and hand osteoarthritis. J Am Board Fam Med. 2011;24:169-174.
5. Chan PS, Steinberg DR, Bozentka DJ. Consequences of knuckle cracking: a report of two acute injuries. Am J Orthop. 1999;28:113-114.
6. Yang RH, Chu YK. Hallucal sesamoiditis manifested on bone scan. Clin Nucl Med. 2013;38:1019-1021.
THE CASE
A 24-year-old woman came to our clinic because she had pain in her right foot. Over the previous 4 weeks, she’d noticed increasing pain in the ball of her foot while walking and climbing stairs, particularly in the push-off portion of her gait. She described it as a nagging, localized pain that she rated as a 2 or 3 out of 10. It was an annoyance, but not unbearable. She felt no pain when standing in place or in a non-weight-bearing position.
She denied any trauma to the foot or change in activity, and had been exercising her usual amount (running 2-5 miles per week). Her medical and social histories were unremarkable, and her family history was negative for relevant conditions.
An examination of the right foot revealed no evidence of pes planus, pes cavus, hallux valgus, hammertoes, or other structural abnormalities of the foot or toes. She had no calluses, nor any erythema or edema of the foot or toes. Direct palpation of the medial sesamoid reproduced the patient’s symptoms. Passive dorsiflexion and plantar flexion of the first hallux elicited pain only at the extreme ends of range of motion. Active dorsiflexion and plantar flexion of the right first hallux showed 5 out of 5 strength. A mid-foot squeeze test was negative, and the remainder of the exam was normal.
THE DIAGNOSIS
Pain on palpation of the sesamoids prompted us to gather a more detailed history. The patient had never been a dancer or a long-distance or competitive runner. However, upon delving into possible causes of the pain, she admitted that she was a frequent “knuckle cracker,” and cracked many joints regularly, including the right first metatarsophalangeal joint (MTPJ). She explained that she cracked this joint by hyper-plantarflexing her big toe against the ground, and had been doing this multiple times a day for many years. In the past 4 weeks, she had noticed significant pain in the right first MTPJ while cracking the joint, but she was having difficulty breaking the longstanding habit.
The patient’s description of right foot pain associated with the push-off portion of her gait, and the fact that the pain was exacerbated by the extremes of dorsiflexion and plantar flexion of the great toe, was consistent with MTPJ pain. This, paired with our ability to reproduce the pain by direct palpation of the medial sesamoid, prompted us to make a clinical diagnosis of sesamoiditis. To our knowledge, this is the first case report of sesamoiditis caused by knuckle cracking.
DISCUSSION
Sesamoiditis—chronic pain and inflammation of the hallucal sesamoids—is an overuse or misuse injury that’s typically seen in runners and dancers.1 The hallucal sesamoids are 2 small bones located underneath the head of the first metatarsal and encased within the flexor hallucus brevis tendon that disperses weight from the head of the first metatarsal during the push-off portion of gait.2 Runners and dancers place significant, repetitive axial loading on the sesamoids, which often leads to injury.1 Although our patient initially seemed to have no typical risk factors for developing sesamoiditis, she later revealed that she regularly cracked the MTPJ, which we believe led to her injury.
Interestingly, despite the common assumption that long-term “cracking” of joints can lead to adverse effects such as osteoarthritis, research has not supported this assumption.3,4 A retrospective case-control study of patients with and without hand osteoarthritis found no association between knuckle cracking and osteoarthritis, and the prevalence of osteoarthritis was not higher in patients who cracked their knuckles more frequently and for more years.4
Nonetheless, there have been reports of acute injuries associated with knuckle cracking, consistent with forcing a joint past its normal range of motion, as is typically done in knuckle cracking.5 In forcefully plantarflexing her great toe against a surface until a “crack” was elicited, our patient may have injured the sesamoid by forcing it along the head of the first metatarsal. Conversely, her injury may have been caused by the repetitive displacement of the sesamoid past its usual location, resulting in chronic irritation.
Differential diagnosis includes fracture and stress injury
The differential diagnosis for subacute to chronic pain localized to the sesamoids includes repetitive stress injury (sesamoiditis or capsular strain), fracture or stress fracture, osteoarthritis, osteonecrosis, and gout.1,2 Given our patient’s age and lack of erythema and edema, osteoarthritis and gout were unlikely.
To treat the injury, eliminate the behavior that caused it
Imaging studies may not be necessary in cases of suspected sesamoiditis because such studies are often negative for sesamoiditis and stress fractures of the sesamoids, and because they typically would not affect how the injury is initially treated.1,2,6 In cases in which radiographic confirmation of sesamoiditis is necessary to rule out more serious pathology, 99mTc-methylene diphosphonate (99mTc-MDP) bone scan and magnetic resonance imaging (MRI) are far more sensitive than plain films.1 While a 99mTc-MDP bone scan will show increased uptake at the sesamoids, it has been replaced by MRI, which will show bone marrow edema of the sesamoids and can rule out fracture or osteoarthritis.1
Sesamoiditis is typically managed with a combination of ice, analgesics, activity modification, and/or orthoses.2 Of course, the key to successfully treating sesamoiditis (and all musculoskeletal injuries) is to not only make the diagnosis, but to find the underlying cause in order to prevent continued—or worsening—pain.
Our patient agreed to close follow-up rather than imaging. We established that the only inciting event was the cracking of her MTPJ, and that she should try to eliminate this action before trying other interventions. Our patient stopped cracking her MTPJ and her pain completely resolved in 2 weeks. She remains symptom-free.
THE TAKEAWAY
Ask about knuckle cracking when taking the history of a patient who presents with sesamoiditis, which is characterized by chronic pain and inflammation of the hallucal sesamoids.
THE CASE
A 24-year-old woman came to our clinic because she had pain in her right foot. Over the previous 4 weeks, she’d noticed increasing pain in the ball of her foot while walking and climbing stairs, particularly in the push-off portion of her gait. She described it as a nagging, localized pain that she rated as a 2 or 3 out of 10. It was an annoyance, but not unbearable. She felt no pain when standing in place or in a non-weight-bearing position.
She denied any trauma to the foot or change in activity, and had been exercising her usual amount (running 2-5 miles per week). Her medical and social histories were unremarkable, and her family history was negative for relevant conditions.
An examination of the right foot revealed no evidence of pes planus, pes cavus, hallux valgus, hammertoes, or other structural abnormalities of the foot or toes. She had no calluses, nor any erythema or edema of the foot or toes. Direct palpation of the medial sesamoid reproduced the patient’s symptoms. Passive dorsiflexion and plantar flexion of the first hallux elicited pain only at the extreme ends of range of motion. Active dorsiflexion and plantar flexion of the right first hallux showed 5 out of 5 strength. A mid-foot squeeze test was negative, and the remainder of the exam was normal.
THE DIAGNOSIS
Pain on palpation of the sesamoids prompted us to gather a more detailed history. The patient had never been a dancer or a long-distance or competitive runner. However, upon delving into possible causes of the pain, she admitted that she was a frequent “knuckle cracker,” and cracked many joints regularly, including the right first metatarsophalangeal joint (MTPJ). She explained that she cracked this joint by hyper-plantarflexing her big toe against the ground, and had been doing this multiple times a day for many years. In the past 4 weeks, she had noticed significant pain in the right first MTPJ while cracking the joint, but she was having difficulty breaking the longstanding habit.
The patient’s description of right foot pain associated with the push-off portion of her gait, and the fact that the pain was exacerbated by the extremes of dorsiflexion and plantar flexion of the great toe, was consistent with MTPJ pain. This, paired with our ability to reproduce the pain by direct palpation of the medial sesamoid, prompted us to make a clinical diagnosis of sesamoiditis. To our knowledge, this is the first case report of sesamoiditis caused by knuckle cracking.
DISCUSSION
Sesamoiditis—chronic pain and inflammation of the hallucal sesamoids—is an overuse or misuse injury that’s typically seen in runners and dancers.1 The hallucal sesamoids are 2 small bones located underneath the head of the first metatarsal and encased within the flexor hallucus brevis tendon that disperses weight from the head of the first metatarsal during the push-off portion of gait.2 Runners and dancers place significant, repetitive axial loading on the sesamoids, which often leads to injury.1 Although our patient initially seemed to have no typical risk factors for developing sesamoiditis, she later revealed that she regularly cracked the MTPJ, which we believe led to her injury.
Interestingly, despite the common assumption that long-term “cracking” of joints can lead to adverse effects such as osteoarthritis, research has not supported this assumption.3,4 A retrospective case-control study of patients with and without hand osteoarthritis found no association between knuckle cracking and osteoarthritis, and the prevalence of osteoarthritis was not higher in patients who cracked their knuckles more frequently and for more years.4
Nonetheless, there have been reports of acute injuries associated with knuckle cracking, consistent with forcing a joint past its normal range of motion, as is typically done in knuckle cracking.5 In forcefully plantarflexing her great toe against a surface until a “crack” was elicited, our patient may have injured the sesamoid by forcing it along the head of the first metatarsal. Conversely, her injury may have been caused by the repetitive displacement of the sesamoid past its usual location, resulting in chronic irritation.
Differential diagnosis includes fracture and stress injury
The differential diagnosis for subacute to chronic pain localized to the sesamoids includes repetitive stress injury (sesamoiditis or capsular strain), fracture or stress fracture, osteoarthritis, osteonecrosis, and gout.1,2 Given our patient’s age and lack of erythema and edema, osteoarthritis and gout were unlikely.
To treat the injury, eliminate the behavior that caused it
Imaging studies may not be necessary in cases of suspected sesamoiditis because such studies are often negative for sesamoiditis and stress fractures of the sesamoids, and because they typically would not affect how the injury is initially treated.1,2,6 In cases in which radiographic confirmation of sesamoiditis is necessary to rule out more serious pathology, 99mTc-methylene diphosphonate (99mTc-MDP) bone scan and magnetic resonance imaging (MRI) are far more sensitive than plain films.1 While a 99mTc-MDP bone scan will show increased uptake at the sesamoids, it has been replaced by MRI, which will show bone marrow edema of the sesamoids and can rule out fracture or osteoarthritis.1
Sesamoiditis is typically managed with a combination of ice, analgesics, activity modification, and/or orthoses.2 Of course, the key to successfully treating sesamoiditis (and all musculoskeletal injuries) is to not only make the diagnosis, but to find the underlying cause in order to prevent continued—or worsening—pain.
Our patient agreed to close follow-up rather than imaging. We established that the only inciting event was the cracking of her MTPJ, and that she should try to eliminate this action before trying other interventions. Our patient stopped cracking her MTPJ and her pain completely resolved in 2 weeks. She remains symptom-free.
THE TAKEAWAY
Ask about knuckle cracking when taking the history of a patient who presents with sesamoiditis, which is characterized by chronic pain and inflammation of the hallucal sesamoids.
1. Nwawka OK, Hayashi D, Diaz LE, et al. Sesamoids and accessory ossicles of the foot: anatomical variability and related pathology. Insights Imaging. 2013;4:581-593.
2. Boike A, Schnirring-Judge M, McMillin S. Sesamoid disorders of the first metatarsophalangeal joint. Clin Podiatr Med Surg. 2011;28:269-285.
3. Castellanos J, Axelrod D. Effect of habitual knuckle cracking on hand function. Ann Rheum Dis. 1990:49:308-309.
4. Deweber K, Olszewski M, Ortolano R. Knuckle cracking and hand osteoarthritis. J Am Board Fam Med. 2011;24:169-174.
5. Chan PS, Steinberg DR, Bozentka DJ. Consequences of knuckle cracking: a report of two acute injuries. Am J Orthop. 1999;28:113-114.
6. Yang RH, Chu YK. Hallucal sesamoiditis manifested on bone scan. Clin Nucl Med. 2013;38:1019-1021.
1. Nwawka OK, Hayashi D, Diaz LE, et al. Sesamoids and accessory ossicles of the foot: anatomical variability and related pathology. Insights Imaging. 2013;4:581-593.
2. Boike A, Schnirring-Judge M, McMillin S. Sesamoid disorders of the first metatarsophalangeal joint. Clin Podiatr Med Surg. 2011;28:269-285.
3. Castellanos J, Axelrod D. Effect of habitual knuckle cracking on hand function. Ann Rheum Dis. 1990:49:308-309.
4. Deweber K, Olszewski M, Ortolano R. Knuckle cracking and hand osteoarthritis. J Am Board Fam Med. 2011;24:169-174.
5. Chan PS, Steinberg DR, Bozentka DJ. Consequences of knuckle cracking: a report of two acute injuries. Am J Orthop. 1999;28:113-114.
6. Yang RH, Chu YK. Hallucal sesamoiditis manifested on bone scan. Clin Nucl Med. 2013;38:1019-1021.

Closed Rupture of the Flexor Profundus Tendon of Ring Finger: Case Report and Treatment Recommendations
Flexor tendons are considered the strongest component of the musculotendinous unit; they generally do not rupture unless weakened by an underlying pathologic condition.1 According to traditional teaching, when the musculotendinous unit is subjected to excessive forces, failure invariably occurs at the tendon insertion, at the musculotendinous junction, within the muscle substance, or at its origin from the bone before the tendon itself ruptures.1
Midsubstance tears in nonrheumatoid patients are less frequent and are typically attributable to an underlying cause.2 Possible pathologic conditions include, but are not limited to, osteoarthritis of the pisotriquetral joint,3 nonunion fracture of the hook of the hamate,4 lunate dislocation,5 accessory carpal bone,6 gouty infiltration of the flexor tendon,7 and tumor.8 In 1960, Boyes and colleagues9 presented a series of 80 flexor tendon ruptures in 78 patients over a 13-year period. Only 3 cases had no identifiable cause. The authors recommended using the term spontaneous for those ruptures that occur within the tendon substance without underlying or associated pathologic changes.
We describe a patient with spontaneous rupture of the flexor digitorum profundus (FDP) tendon at zone III, satisfying Boyes’ definition of the term spontaneous. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 65-year-old, right-handed manual worker was assessed in our hand clinic 3 days after he felt a cramp in his left palm while lifting a heavy object. Shortly thereafter, he noted he could not flex his ring finger distal interphalangeal (DIP) joint. He could not recall any previous injury to his finger. No predisposing pathologic conditions or bone abnormalities were identified. Clinically, there was no tenderness, swelling, or ecchymosis evident. He had full passive range of motion (ROM) of his ring finger, and proximal interphalangeal (PIP) joint active ROM was 0/110º; however, he had no activity of the FDP of the ring finger. Preoperative radiographs were normal. The hook of the hamate was clinically and radiographically normal.
A preoperative diagnosis of FDP avulsion from the distal phalanx was made, and the operation was carried out 16 days after injury. Surgical exploration started in zone II and extended proximally into the distal palmar crease, but no stump was found in either location. Therefore, exploration was carried out to the midpalmar region, revealing the tendon rupture in zone III, in the region of the origin of the ring finger lumbrical muscle (Figure 1). The flexor digitorum superficialis tendon was intact. Macroscopically, both tendon and carpal tunnel appeared normal, with no evidence of tendon attrition; thus, the tendon was not sent for histologic examination. The ends of the ruptured FDP tendon to the ring finger were at the level of the superficial palmar arch, with the distal end appearing as though it had been cut sharply with a knife. Because of the short period of time from injury to exploration, delayed primary tendon repair was possible, along with side-to-side tenodesis to the intact ring finger flexor superficialis tendon in the palm (Figure 2). Two days after surgery, the patient started a controlled mobilization program using the Duran method.10
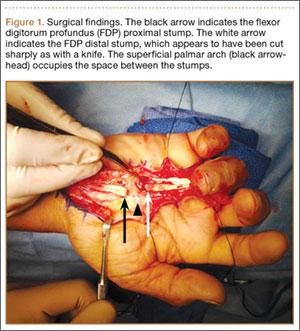
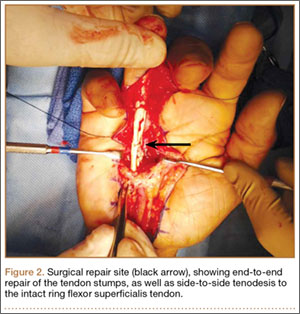
At final follow-up of 18 months, total active motion was 126°, which corresponds to a good outcome, according to the Strickland and Glogovac criteria.11 Grip strength was 50 kg, which was 84% of grip strength on the uninjured side. The patient was back to recreational activity but had not returned to work.
Discussion
Most flexor tendon ruptures result from avulsion of the FDP tendon at its distal phalanx insertion, commonly known as Jersey finger. However, true midsubstance spontaneous ruptures are infrequent. Reports of spontaneous tendon ruptures of all types, including those of the hand, have increased in incidence in most countries.12 Bois and colleagues,13 who have reviewed the literature over a 50-year period, found a total of 50 spontaneous ruptures of “normal” flexor tendon in 43 cases. The authors point to unique historical and physical examinaton findings that help differentiate spontaneous tendon ruptures from the more common FDP avulsions. Such findings include the sensation of a pop or snap, or a sudden sharp pain or cramp within the palmar region. In contrast, most avulsion ruptures cause discomfort within the region of the digit. In type I avulsion injuries of the FDP tendon, the proximal tendon stump usually retracts proximal to the digital tendon sheath, causing a tender mass in the palm.14 Flexor digitorum profundus tendon avulsions, however, are not typically associated with a snap or pop in the palm. When spontaneous ruptures of the hand occur, they typically involve the profundus tendon of the small finger, in the area of the lumbrical origin.13
In equivocal cases when the site of rupture is uncertain, ultrasound and magnetic resonance imaging may assist in making the diagnosis and provide important preoperative information for surgical decision-making and planning; this information may decrease postoperative morbidity by minimizing surgical dissection.
The etiology of spontaneous ruptures is incompletely understood. For any rupture of the ulnar flexor tendons, the hook of the hamate should be examined to rule out a previous fracture as a cause of tendon attrition.15 Tendon vascularization may be a cause for tendon rupture in the hand. When the blood supply of the lumbrical muscles was examined in 100 upper extremities from human cadavers using vascular injection studies,16 it was discovered that each lumbrical muscle received its arterial supply from 4 sources: the superficial palmar arch, the common palmar digital artery, the deep palmar arch, and the dorsal digital artery. There were no anastomoses between the networks supplying the lumbrical muscles and the FDP tendons within the palm, suggesting a possible watershed zone between the FDP tendon and lumbrical muscle origin. The patient described in this case had the tendon rupture in the area of potential hypovascularity at the lumbrical origin.
Important factors in the decision-making process for surgical treatment include the length of time between rupture and treatment, the site of rupture, and the condition of the ruptured tendon ends. Patients who present in the first 3 weeks of injury can be treated by primary tendon repair, provided that the ruptured tendon ends are not significantly frayed or attenuated. For patients presenting more than 3 weeks after injury, interposition tendon grafts or tendon transfers are suitable options for ruptures in zone III. Distal interphalangeal joint arthrodesis is another alternative in specific cases where reconstruction is not possible. In this case, direct end-to-end repair was possible, as well as tenodesis to the intact ring finger superficialis in order to prevent stretching of the repair.
Localizing the level of the tendon rupture clinically is difficult. When the site of the profundus tendon rupture is uncertain, and there is no tenderness in zone I or the PIP joint, the first incision should be made at the metacarpophalangeal joint level. This first incision will indicate if the rupture occurred in zone III. If the tendon is intact at that location, then the next incision should be at the level of the PIP joint.
Conclusion
We report a patient treated for spontaneous rupture of the flexor tendon in zone III. He was treated in the acute setting with direct tendon repair. It is important to consider spontaneous rupture of the tendon in patients presenting with a snap/pop and the sudden inability to flex a finger. A tendon rupture can be diagnosed as spontaneous in the absence of an underlying pathologic condition such as rheumatoid arthritis, gout, or occult carpal fractures. In the acute setting, these may be repaired primarily; however, if presenting after a few weeks, alternative surgical options, including interposition tendon grafts, tendon transfer, and DIP joint arthrodesis, should be considered.
1. McMaster PE. Tendon and muscle ruptures, clinical and experimental studies on the causes and location of subcutaneous ruptures. J Bone Joint Surg Am. 1933;15(3):705-722.
2. Folmar RC, Nelson CL, Phalen GS. Ruptures of the flexor tendons in hands of non-rheumatoid patients. J Bone Joint Surg Am. 1972;54(3):579-584.
3. Grant I, Berger AC, Ireland DC. Rupture of the flexor digitorum profundus tendon to the small finger within the carpal tunnel. Hand Surg. 2005;10(1):109-114.
4. Hartford JM, Murphy JM. Flexor digitorum profundus rupture of the small finger secondary to nonunion of the hook of the hamate: a case report. J Hand Surg Am. 1996;21(14):621-623.
5. Johnston GH, Bowen CV. Attritional flexor tendon ruptures by an old lunate dislocation. J Hand Surg Am. 1988;13(5):701-703.
6. Koizumi M, Kanda T, Satoh S, Yoshizu T, Maki Y, Tsubokawa N. Attritional rupture of the flexor digitorum profundus tendon to the index finger caused by accessory carpal bone in the carpal tunnel: a case report. J Hand Surg Am. 2005;30(1):142-146.
7. Wurapa RK, Zelouf DS. Flexor tendon rupture caused by gout: a case report. J Hand Surg Am. 2002;27(4):591-593.
8. Masada K, Kanazawa M, Fuji T. Flexor tendon ruptures caused by an intraosseous ganglion of the hook of the hamate. J Hand Surg Br. 1997;22(3)383-385.
9. Boyes JH, Wilson JN, Smith JW. Flexor-tendon ruptures in the forearm and hand. J Bone Joint Surg Am. 1960;42(4):637-646.
10. Duran R, Houser R, Coleman C, et al. A preliminary report in the use of controlled passive motion following flexor tendon repair in zones II and III [abstract]. J Hand Surg. 1976;1(1):79.
11. Strickland JW, Glogovac SV. Digital function following flexor tendon repair in Zone II: A comparison of immobilization and controlled passive motion techniques. J Hand Surg Am. 1980;5(6):537-543.
12. Kannus P, Jozsa L. Histopathological changes preceding spontaneous rupture of a tendon. A controlled study of 891 patients. J Bone Joint Surg Am. 1991;73(10):1507-1525.
13. Bois AJ, Johnston G, Classen D. Spontaneous flexor tendon ruptures of the hand: case series and review of the literature. J Hand Surg Am. 2007;32(7):1061-1071.
14. Leddy JP, Packer JW. Avulsion of the profundus tendon insertion in athletes. J Hand Surg Am. 1977;2(1):66-69.
15. Jebson PJ, Ferlic RJ, Engber WF. Spontaneous rupture of ulnar-sided digital flexor tendons: don’t forget the hamate. Iowa Orthop J. 1995;15:225-227.
16. Zbrodowski A, Mariéthoz E, Bednarkiewicz M, Gajisin S. The blood supply of the lumbrical muscles. J Hand Surg Br. 1998;23(3):384-388.
Flexor tendons are considered the strongest component of the musculotendinous unit; they generally do not rupture unless weakened by an underlying pathologic condition.1 According to traditional teaching, when the musculotendinous unit is subjected to excessive forces, failure invariably occurs at the tendon insertion, at the musculotendinous junction, within the muscle substance, or at its origin from the bone before the tendon itself ruptures.1
Midsubstance tears in nonrheumatoid patients are less frequent and are typically attributable to an underlying cause.2 Possible pathologic conditions include, but are not limited to, osteoarthritis of the pisotriquetral joint,3 nonunion fracture of the hook of the hamate,4 lunate dislocation,5 accessory carpal bone,6 gouty infiltration of the flexor tendon,7 and tumor.8 In 1960, Boyes and colleagues9 presented a series of 80 flexor tendon ruptures in 78 patients over a 13-year period. Only 3 cases had no identifiable cause. The authors recommended using the term spontaneous for those ruptures that occur within the tendon substance without underlying or associated pathologic changes.
We describe a patient with spontaneous rupture of the flexor digitorum profundus (FDP) tendon at zone III, satisfying Boyes’ definition of the term spontaneous. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 65-year-old, right-handed manual worker was assessed in our hand clinic 3 days after he felt a cramp in his left palm while lifting a heavy object. Shortly thereafter, he noted he could not flex his ring finger distal interphalangeal (DIP) joint. He could not recall any previous injury to his finger. No predisposing pathologic conditions or bone abnormalities were identified. Clinically, there was no tenderness, swelling, or ecchymosis evident. He had full passive range of motion (ROM) of his ring finger, and proximal interphalangeal (PIP) joint active ROM was 0/110º; however, he had no activity of the FDP of the ring finger. Preoperative radiographs were normal. The hook of the hamate was clinically and radiographically normal.
A preoperative diagnosis of FDP avulsion from the distal phalanx was made, and the operation was carried out 16 days after injury. Surgical exploration started in zone II and extended proximally into the distal palmar crease, but no stump was found in either location. Therefore, exploration was carried out to the midpalmar region, revealing the tendon rupture in zone III, in the region of the origin of the ring finger lumbrical muscle (Figure 1). The flexor digitorum superficialis tendon was intact. Macroscopically, both tendon and carpal tunnel appeared normal, with no evidence of tendon attrition; thus, the tendon was not sent for histologic examination. The ends of the ruptured FDP tendon to the ring finger were at the level of the superficial palmar arch, with the distal end appearing as though it had been cut sharply with a knife. Because of the short period of time from injury to exploration, delayed primary tendon repair was possible, along with side-to-side tenodesis to the intact ring finger flexor superficialis tendon in the palm (Figure 2). Two days after surgery, the patient started a controlled mobilization program using the Duran method.10
At final follow-up of 18 months, total active motion was 126°, which corresponds to a good outcome, according to the Strickland and Glogovac criteria.11 Grip strength was 50 kg, which was 84% of grip strength on the uninjured side. The patient was back to recreational activity but had not returned to work.
Discussion
Most flexor tendon ruptures result from avulsion of the FDP tendon at its distal phalanx insertion, commonly known as Jersey finger. However, true midsubstance spontaneous ruptures are infrequent. Reports of spontaneous tendon ruptures of all types, including those of the hand, have increased in incidence in most countries.12 Bois and colleagues,13 who have reviewed the literature over a 50-year period, found a total of 50 spontaneous ruptures of “normal” flexor tendon in 43 cases. The authors point to unique historical and physical examinaton findings that help differentiate spontaneous tendon ruptures from the more common FDP avulsions. Such findings include the sensation of a pop or snap, or a sudden sharp pain or cramp within the palmar region. In contrast, most avulsion ruptures cause discomfort within the region of the digit. In type I avulsion injuries of the FDP tendon, the proximal tendon stump usually retracts proximal to the digital tendon sheath, causing a tender mass in the palm.14 Flexor digitorum profundus tendon avulsions, however, are not typically associated with a snap or pop in the palm. When spontaneous ruptures of the hand occur, they typically involve the profundus tendon of the small finger, in the area of the lumbrical origin.13
In equivocal cases when the site of rupture is uncertain, ultrasound and magnetic resonance imaging may assist in making the diagnosis and provide important preoperative information for surgical decision-making and planning; this information may decrease postoperative morbidity by minimizing surgical dissection.
The etiology of spontaneous ruptures is incompletely understood. For any rupture of the ulnar flexor tendons, the hook of the hamate should be examined to rule out a previous fracture as a cause of tendon attrition.15 Tendon vascularization may be a cause for tendon rupture in the hand. When the blood supply of the lumbrical muscles was examined in 100 upper extremities from human cadavers using vascular injection studies,16 it was discovered that each lumbrical muscle received its arterial supply from 4 sources: the superficial palmar arch, the common palmar digital artery, the deep palmar arch, and the dorsal digital artery. There were no anastomoses between the networks supplying the lumbrical muscles and the FDP tendons within the palm, suggesting a possible watershed zone between the FDP tendon and lumbrical muscle origin. The patient described in this case had the tendon rupture in the area of potential hypovascularity at the lumbrical origin.
Important factors in the decision-making process for surgical treatment include the length of time between rupture and treatment, the site of rupture, and the condition of the ruptured tendon ends. Patients who present in the first 3 weeks of injury can be treated by primary tendon repair, provided that the ruptured tendon ends are not significantly frayed or attenuated. For patients presenting more than 3 weeks after injury, interposition tendon grafts or tendon transfers are suitable options for ruptures in zone III. Distal interphalangeal joint arthrodesis is another alternative in specific cases where reconstruction is not possible. In this case, direct end-to-end repair was possible, as well as tenodesis to the intact ring finger superficialis in order to prevent stretching of the repair.
Localizing the level of the tendon rupture clinically is difficult. When the site of the profundus tendon rupture is uncertain, and there is no tenderness in zone I or the PIP joint, the first incision should be made at the metacarpophalangeal joint level. This first incision will indicate if the rupture occurred in zone III. If the tendon is intact at that location, then the next incision should be at the level of the PIP joint.
Conclusion
We report a patient treated for spontaneous rupture of the flexor tendon in zone III. He was treated in the acute setting with direct tendon repair. It is important to consider spontaneous rupture of the tendon in patients presenting with a snap/pop and the sudden inability to flex a finger. A tendon rupture can be diagnosed as spontaneous in the absence of an underlying pathologic condition such as rheumatoid arthritis, gout, or occult carpal fractures. In the acute setting, these may be repaired primarily; however, if presenting after a few weeks, alternative surgical options, including interposition tendon grafts, tendon transfer, and DIP joint arthrodesis, should be considered.
Flexor tendons are considered the strongest component of the musculotendinous unit; they generally do not rupture unless weakened by an underlying pathologic condition.1 According to traditional teaching, when the musculotendinous unit is subjected to excessive forces, failure invariably occurs at the tendon insertion, at the musculotendinous junction, within the muscle substance, or at its origin from the bone before the tendon itself ruptures.1
Midsubstance tears in nonrheumatoid patients are less frequent and are typically attributable to an underlying cause.2 Possible pathologic conditions include, but are not limited to, osteoarthritis of the pisotriquetral joint,3 nonunion fracture of the hook of the hamate,4 lunate dislocation,5 accessory carpal bone,6 gouty infiltration of the flexor tendon,7 and tumor.8 In 1960, Boyes and colleagues9 presented a series of 80 flexor tendon ruptures in 78 patients over a 13-year period. Only 3 cases had no identifiable cause. The authors recommended using the term spontaneous for those ruptures that occur within the tendon substance without underlying or associated pathologic changes.
We describe a patient with spontaneous rupture of the flexor digitorum profundus (FDP) tendon at zone III, satisfying Boyes’ definition of the term spontaneous. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 65-year-old, right-handed manual worker was assessed in our hand clinic 3 days after he felt a cramp in his left palm while lifting a heavy object. Shortly thereafter, he noted he could not flex his ring finger distal interphalangeal (DIP) joint. He could not recall any previous injury to his finger. No predisposing pathologic conditions or bone abnormalities were identified. Clinically, there was no tenderness, swelling, or ecchymosis evident. He had full passive range of motion (ROM) of his ring finger, and proximal interphalangeal (PIP) joint active ROM was 0/110º; however, he had no activity of the FDP of the ring finger. Preoperative radiographs were normal. The hook of the hamate was clinically and radiographically normal.
A preoperative diagnosis of FDP avulsion from the distal phalanx was made, and the operation was carried out 16 days after injury. Surgical exploration started in zone II and extended proximally into the distal palmar crease, but no stump was found in either location. Therefore, exploration was carried out to the midpalmar region, revealing the tendon rupture in zone III, in the region of the origin of the ring finger lumbrical muscle (Figure 1). The flexor digitorum superficialis tendon was intact. Macroscopically, both tendon and carpal tunnel appeared normal, with no evidence of tendon attrition; thus, the tendon was not sent for histologic examination. The ends of the ruptured FDP tendon to the ring finger were at the level of the superficial palmar arch, with the distal end appearing as though it had been cut sharply with a knife. Because of the short period of time from injury to exploration, delayed primary tendon repair was possible, along with side-to-side tenodesis to the intact ring finger flexor superficialis tendon in the palm (Figure 2). Two days after surgery, the patient started a controlled mobilization program using the Duran method.10
At final follow-up of 18 months, total active motion was 126°, which corresponds to a good outcome, according to the Strickland and Glogovac criteria.11 Grip strength was 50 kg, which was 84% of grip strength on the uninjured side. The patient was back to recreational activity but had not returned to work.
Discussion
Most flexor tendon ruptures result from avulsion of the FDP tendon at its distal phalanx insertion, commonly known as Jersey finger. However, true midsubstance spontaneous ruptures are infrequent. Reports of spontaneous tendon ruptures of all types, including those of the hand, have increased in incidence in most countries.12 Bois and colleagues,13 who have reviewed the literature over a 50-year period, found a total of 50 spontaneous ruptures of “normal” flexor tendon in 43 cases. The authors point to unique historical and physical examinaton findings that help differentiate spontaneous tendon ruptures from the more common FDP avulsions. Such findings include the sensation of a pop or snap, or a sudden sharp pain or cramp within the palmar region. In contrast, most avulsion ruptures cause discomfort within the region of the digit. In type I avulsion injuries of the FDP tendon, the proximal tendon stump usually retracts proximal to the digital tendon sheath, causing a tender mass in the palm.14 Flexor digitorum profundus tendon avulsions, however, are not typically associated with a snap or pop in the palm. When spontaneous ruptures of the hand occur, they typically involve the profundus tendon of the small finger, in the area of the lumbrical origin.13
In equivocal cases when the site of rupture is uncertain, ultrasound and magnetic resonance imaging may assist in making the diagnosis and provide important preoperative information for surgical decision-making and planning; this information may decrease postoperative morbidity by minimizing surgical dissection.
The etiology of spontaneous ruptures is incompletely understood. For any rupture of the ulnar flexor tendons, the hook of the hamate should be examined to rule out a previous fracture as a cause of tendon attrition.15 Tendon vascularization may be a cause for tendon rupture in the hand. When the blood supply of the lumbrical muscles was examined in 100 upper extremities from human cadavers using vascular injection studies,16 it was discovered that each lumbrical muscle received its arterial supply from 4 sources: the superficial palmar arch, the common palmar digital artery, the deep palmar arch, and the dorsal digital artery. There were no anastomoses between the networks supplying the lumbrical muscles and the FDP tendons within the palm, suggesting a possible watershed zone between the FDP tendon and lumbrical muscle origin. The patient described in this case had the tendon rupture in the area of potential hypovascularity at the lumbrical origin.
Important factors in the decision-making process for surgical treatment include the length of time between rupture and treatment, the site of rupture, and the condition of the ruptured tendon ends. Patients who present in the first 3 weeks of injury can be treated by primary tendon repair, provided that the ruptured tendon ends are not significantly frayed or attenuated. For patients presenting more than 3 weeks after injury, interposition tendon grafts or tendon transfers are suitable options for ruptures in zone III. Distal interphalangeal joint arthrodesis is another alternative in specific cases where reconstruction is not possible. In this case, direct end-to-end repair was possible, as well as tenodesis to the intact ring finger superficialis in order to prevent stretching of the repair.
Localizing the level of the tendon rupture clinically is difficult. When the site of the profundus tendon rupture is uncertain, and there is no tenderness in zone I or the PIP joint, the first incision should be made at the metacarpophalangeal joint level. This first incision will indicate if the rupture occurred in zone III. If the tendon is intact at that location, then the next incision should be at the level of the PIP joint.
Conclusion
We report a patient treated for spontaneous rupture of the flexor tendon in zone III. He was treated in the acute setting with direct tendon repair. It is important to consider spontaneous rupture of the tendon in patients presenting with a snap/pop and the sudden inability to flex a finger. A tendon rupture can be diagnosed as spontaneous in the absence of an underlying pathologic condition such as rheumatoid arthritis, gout, or occult carpal fractures. In the acute setting, these may be repaired primarily; however, if presenting after a few weeks, alternative surgical options, including interposition tendon grafts, tendon transfer, and DIP joint arthrodesis, should be considered.
1. McMaster PE. Tendon and muscle ruptures, clinical and experimental studies on the causes and location of subcutaneous ruptures. J Bone Joint Surg Am. 1933;15(3):705-722.
2. Folmar RC, Nelson CL, Phalen GS. Ruptures of the flexor tendons in hands of non-rheumatoid patients. J Bone Joint Surg Am. 1972;54(3):579-584.
3. Grant I, Berger AC, Ireland DC. Rupture of the flexor digitorum profundus tendon to the small finger within the carpal tunnel. Hand Surg. 2005;10(1):109-114.
4. Hartford JM, Murphy JM. Flexor digitorum profundus rupture of the small finger secondary to nonunion of the hook of the hamate: a case report. J Hand Surg Am. 1996;21(14):621-623.
5. Johnston GH, Bowen CV. Attritional flexor tendon ruptures by an old lunate dislocation. J Hand Surg Am. 1988;13(5):701-703.
6. Koizumi M, Kanda T, Satoh S, Yoshizu T, Maki Y, Tsubokawa N. Attritional rupture of the flexor digitorum profundus tendon to the index finger caused by accessory carpal bone in the carpal tunnel: a case report. J Hand Surg Am. 2005;30(1):142-146.
7. Wurapa RK, Zelouf DS. Flexor tendon rupture caused by gout: a case report. J Hand Surg Am. 2002;27(4):591-593.
8. Masada K, Kanazawa M, Fuji T. Flexor tendon ruptures caused by an intraosseous ganglion of the hook of the hamate. J Hand Surg Br. 1997;22(3)383-385.
9. Boyes JH, Wilson JN, Smith JW. Flexor-tendon ruptures in the forearm and hand. J Bone Joint Surg Am. 1960;42(4):637-646.
10. Duran R, Houser R, Coleman C, et al. A preliminary report in the use of controlled passive motion following flexor tendon repair in zones II and III [abstract]. J Hand Surg. 1976;1(1):79.
11. Strickland JW, Glogovac SV. Digital function following flexor tendon repair in Zone II: A comparison of immobilization and controlled passive motion techniques. J Hand Surg Am. 1980;5(6):537-543.
12. Kannus P, Jozsa L. Histopathological changes preceding spontaneous rupture of a tendon. A controlled study of 891 patients. J Bone Joint Surg Am. 1991;73(10):1507-1525.
13. Bois AJ, Johnston G, Classen D. Spontaneous flexor tendon ruptures of the hand: case series and review of the literature. J Hand Surg Am. 2007;32(7):1061-1071.
14. Leddy JP, Packer JW. Avulsion of the profundus tendon insertion in athletes. J Hand Surg Am. 1977;2(1):66-69.
15. Jebson PJ, Ferlic RJ, Engber WF. Spontaneous rupture of ulnar-sided digital flexor tendons: don’t forget the hamate. Iowa Orthop J. 1995;15:225-227.
16. Zbrodowski A, Mariéthoz E, Bednarkiewicz M, Gajisin S. The blood supply of the lumbrical muscles. J Hand Surg Br. 1998;23(3):384-388.
1. McMaster PE. Tendon and muscle ruptures, clinical and experimental studies on the causes and location of subcutaneous ruptures. J Bone Joint Surg Am. 1933;15(3):705-722.
2. Folmar RC, Nelson CL, Phalen GS. Ruptures of the flexor tendons in hands of non-rheumatoid patients. J Bone Joint Surg Am. 1972;54(3):579-584.
3. Grant I, Berger AC, Ireland DC. Rupture of the flexor digitorum profundus tendon to the small finger within the carpal tunnel. Hand Surg. 2005;10(1):109-114.
4. Hartford JM, Murphy JM. Flexor digitorum profundus rupture of the small finger secondary to nonunion of the hook of the hamate: a case report. J Hand Surg Am. 1996;21(14):621-623.
5. Johnston GH, Bowen CV. Attritional flexor tendon ruptures by an old lunate dislocation. J Hand Surg Am. 1988;13(5):701-703.
6. Koizumi M, Kanda T, Satoh S, Yoshizu T, Maki Y, Tsubokawa N. Attritional rupture of the flexor digitorum profundus tendon to the index finger caused by accessory carpal bone in the carpal tunnel: a case report. J Hand Surg Am. 2005;30(1):142-146.
7. Wurapa RK, Zelouf DS. Flexor tendon rupture caused by gout: a case report. J Hand Surg Am. 2002;27(4):591-593.
8. Masada K, Kanazawa M, Fuji T. Flexor tendon ruptures caused by an intraosseous ganglion of the hook of the hamate. J Hand Surg Br. 1997;22(3)383-385.
9. Boyes JH, Wilson JN, Smith JW. Flexor-tendon ruptures in the forearm and hand. J Bone Joint Surg Am. 1960;42(4):637-646.
10. Duran R, Houser R, Coleman C, et al. A preliminary report in the use of controlled passive motion following flexor tendon repair in zones II and III [abstract]. J Hand Surg. 1976;1(1):79.
11. Strickland JW, Glogovac SV. Digital function following flexor tendon repair in Zone II: A comparison of immobilization and controlled passive motion techniques. J Hand Surg Am. 1980;5(6):537-543.
12. Kannus P, Jozsa L. Histopathological changes preceding spontaneous rupture of a tendon. A controlled study of 891 patients. J Bone Joint Surg Am. 1991;73(10):1507-1525.
13. Bois AJ, Johnston G, Classen D. Spontaneous flexor tendon ruptures of the hand: case series and review of the literature. J Hand Surg Am. 2007;32(7):1061-1071.
14. Leddy JP, Packer JW. Avulsion of the profundus tendon insertion in athletes. J Hand Surg Am. 1977;2(1):66-69.
15. Jebson PJ, Ferlic RJ, Engber WF. Spontaneous rupture of ulnar-sided digital flexor tendons: don’t forget the hamate. Iowa Orthop J. 1995;15:225-227.
16. Zbrodowski A, Mariéthoz E, Bednarkiewicz M, Gajisin S. The blood supply of the lumbrical muscles. J Hand Surg Br. 1998;23(3):384-388.
Inflammatory metastatic breast cancer with gallbladder metastasis: an incidental finding
Breast cancer is the most frequently diagnosed cancer in women, with an estimated 231,840 new cases representing 14.0% of all new cancer cases in the United States in 2015.1 Early screening and modern techniques of imaging and diagnosis have led to a significant improvement in detecting early-stage breast cancers and to a decrease in the incidence of metastatic breast cancer (MBC).2 About 20%-30% of patients who are initially diagnosed with an early-stage, nonmetastatic breast cancer will subsequently develop a distant metastatic disease. Between 6%-10% of the new breast cancer cases present initially as stage IV, referred to as de novo MBC.
Click on the PDF icon at the top of this introduction to read the full article.
Breast cancer is the most frequently diagnosed cancer in women, with an estimated 231,840 new cases representing 14.0% of all new cancer cases in the United States in 2015.1 Early screening and modern techniques of imaging and diagnosis have led to a significant improvement in detecting early-stage breast cancers and to a decrease in the incidence of metastatic breast cancer (MBC).2 About 20%-30% of patients who are initially diagnosed with an early-stage, nonmetastatic breast cancer will subsequently develop a distant metastatic disease. Between 6%-10% of the new breast cancer cases present initially as stage IV, referred to as de novo MBC.
Click on the PDF icon at the top of this introduction to read the full article.
Breast cancer is the most frequently diagnosed cancer in women, with an estimated 231,840 new cases representing 14.0% of all new cancer cases in the United States in 2015.1 Early screening and modern techniques of imaging and diagnosis have led to a significant improvement in detecting early-stage breast cancers and to a decrease in the incidence of metastatic breast cancer (MBC).2 About 20%-30% of patients who are initially diagnosed with an early-stage, nonmetastatic breast cancer will subsequently develop a distant metastatic disease. Between 6%-10% of the new breast cancer cases present initially as stage IV, referred to as de novo MBC.
Click on the PDF icon at the top of this introduction to read the full article.
Primary Capsule-Deficient Cutaneous Cryptococcosis in a Sporotrichoid Pattern in an Immunocompetent Host
Cryptococcosis is an opportunistic yeast infection caused by Cryptococcus neoformans that remains the most common systemic fungal infection in immunosuppressed patients and often presents with signs of meningitis. Cutaneous cryptococcosis occurs in 10% to 20% of systemic Cryptococcus infections and usually is secondary to hematogenous dissemination in patients with an underlying disease, particularly human immunodeficiency virus. Primary cutaneous cryptococcosis (PCC) is a more rare clinical identity that is characterized by skin lesions confined to 1 body region, often presenting as a whitlow or phlegmon with positive culture for C neoformans and no evidence of simultaneous dissemination. We report a rare case of PCC in a 73-year-old man with intact cell-mediated immunity.
Case Report
A 73-year-old man who was a beef farmer presented on primary care referral with multiple red nodules and ulcers on the right third and fourth digits and distal forearm following abrasion to the region. The patient reported that the lesions had started as painful nodules that would open and drain. He had been taking oral ciprofloxacin and oral ketoconazole for 3 days as prescribed by his primary care physician but had not begun to see results. He denied any travel or exposure to roses, fish tanks, or any sick contacts. A review of systems was negative for fever, night sweats, malaise, headache, or any other systemic symptoms. Physical examination revealed multiple 2- to 6-mm nodules and ulcers distributed in a sporotrichoid pattern on the right hand (Figure 1) and arm (Figure 2). Lymphadenopathy was absent and the rest of the examination revealed no abnormalities.
|
Initially, 4 punch biopsies of the right hand and arm were obtained and sent for Gram staining, tissue culture (bacterial and fungal), and histopathologic review. A presumptive diagnosis of sporotrichosis was made, with change of treatment pending culture. On routine hematoxylin and eosin staining, marked acute and chronic granulomatous inflammation with microabscesses was noted. Acid-fast bacilli staining was negative. Follow-up Gomori methenamine-silver (GMS) staining showed numerous fungal spores with narrow base budding (Figure 3). Subsequent mucicarmine staining did not reveal dark red capsules characteristic of Cryptococcus (Figure 4). The pathology report indicated that the findings may represent sporotrichosis in the appropriate clinical setting, but GMS staining could not definitively classify Sporothrix schenckii or rule out other fungal infections without tissue culture. Before culture results could be obtained, the patient returned 2 weeks later for suture removal at which point the prior medications were stopped and itraconazole 200 mg once daily was initiated.
Upon receiving the culture results, a diagnosis of primary capsule-deficient cutaneous cryptococcosis was made. The lesions showed clinical improvement at 1-month follow-up, and treatment with itraconazole was continued with monthly liver function tests. After 5 months of continued improvement, the itraconazole dose was decreased to 100 mg once daily for 1 month. The patient was free of lesions and any sequelae at 6-month and 1-year follow-up.
Comment
Cryptococcosis is caused by C neoformans, an opportunistic, basidiomycetous, yeastlike fungus1 that presents as a yeast in both the environment and tissue and normally is associated with immunocompromised host infection, especially in individuals with human immunodeficiency virus. The most common route of infection is through the lungs as respiratory droplets followed by hematogenous dissemination to the central nervous system and skin, with meningitis being the most common clinical manifestation and Cryptococcus being the most common cause of fungal meningitis worldwide.2 Cutaneous involvement after hematogenous spread (secondary cutaneous cryptococcosis) is reported in 10% to 20% of systemic Cryptococcus cases, while PCC is limited to rare cases in which trauma or abrasions to the affected site are notable risk factors.2,3
|
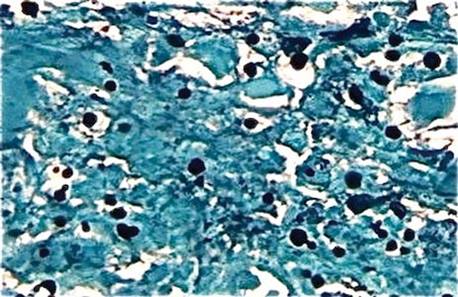
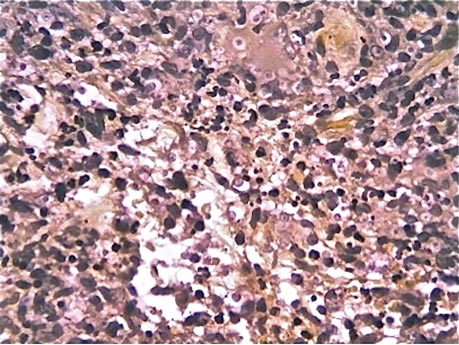
Cryptococcus can produce a myriad of skin manifestations including but not limited to nodules, ulcers, plaques, pustules, vesicobullous lesions, and draining sinuses. Neuville et al1 found that cellulitis, cutaneous ulcers, and whitlows were the most common presenting clinical features in PCC. Whitlows also have been reported as a rare presentation in secondary cutaneous cryptococcosis yielding to the much more prevalent presentation of umbilicated papules resembling molluscum contagiosum.1 This polymorphic identity can therefore mimic not only other dermatoses and neoplasms but other infections such as bacterial cellulitis, herpes simplex virus, and molluscum contagiosum, especially in disseminated cryptococcosis, making microscopic assessment crucial for the diagnostic confirmation of cutaneous cryptococcosis. The differential diagnosis includes sporotrichosis and Mycobacterium marinum due to the lymphatic distribution of the lesions as well as squamous cell carcinoma. Our initial diagnosis of sporotrichosis was assumptive until mycological data could be obtained.
The histopathology patterns characteristic of C neoformans infection fall into either a paucireactive pattern with myriads of densely packed organisms with mucoid gelatinous capsules that cause minimal tissue reaction or a mixed suppurative and granulomatous reaction with varying degrees of necrosis.4 The granulomatous form can affect histiocytes, giant cells, lymphocytes, and fibroblasts. These findings along with the characteristic carminophilic capsule of C neoformans allows for a prompt diagnosis. However, the C neoformans spore somewhat characteristically measures 3 to 20 mm in diameter and stains well with periodic acid–Schiff stain and GMS.4,5 Therefore, the lack of capsule broadened our early differential to include Histoplasma capsulatum, S schenckii, Paracoccidioides brasiliensis, and even Blastomyces dermatitidis.
Neuville et al1 proposed the following criteria for the diagnosis of PCC: the absence of dissemination and predominantly a solitary skin lesion on unclothed areas presenting as a whitlow or phlegmon, a history of skin injury or damage leading to direct inoculation, participation in outdoor activities, exposure to bird droppings, and isolation of C neoformans serotype D. Other factors that strongly support PCC diagnosis over squamous cell carcinoma (based on a review of the literature) are rural residential environment, older age, equal prevalence among men and women, and lack of underlying disease. Presence of these factors seem to favor PCC over squamous cell carcinoma, as some still consider the existence of PCC in general to be controversial because skin manifestations represent a sentinel finding indicative of disseminated disease.1,3
The fungus can be found worldwide as an ubiquitous saprophyte of soil, especially if the soil is enriched with pigeon droppings. A link between C neoformans and pigeons has been suggested, with dried avian excreta allowing the yeast to abundantly grow because of its high nitrogen content.5,6 Other possible sites include decaying wood, fruits, vegetables, and dust.1 There are 4 main serotypes and 3 varieties of C neoformans: C neoformans var grubii (serotype A; worldwide distribution), C neoformans var gattii (serotypes B and C; more circumscribed diffusion and distribution including subtropical regions of Australia, Central Africa, South Asia, and California), and C neoformans var neoformans (serotype D; worldwide distribution).3,7 A literature review indicated that known cases of serotype D (global incidence, 9%) tended to produce cutaneous lesions without systemic involvement.7 Microscopically, the most important characteristic feature found in all serotypes is the polysaccharide capsule, which normally acts as an important virulence factor.6 This capsule as well as detection of the budding yeast can be visualized with india ink (cerebrospinal fluid), methylene blue, or mucicarmine staining. The latex agglutination test for cryptococcal antigen has been used as a serologic test for cerebrospinal fluid, blood, and urine with a sensitivity of 86% to 95%.8
Treatment of cryptococcal disease depends on location and severity of lesions. Many cases of PCC spontaneously resolve, but it is a recommended practice to treat the lesions via incision, local irrigation and debridement, and anti-inflammatory and antifungal agents.9 Antifungal therapy with amphotericin B with or without flucytosine was the standard of therapy. The newer oral azole compounds (eg, ketoconazole, fluconazole, itraconazole) are effective against Cryptococcus, making them the probable treatment in immunocompetent patients because of fewer side effects. Nonetheless, these drugs should be maintained for several weeks or even months to achieve complete resolution of PCC.10
Our patient’s clinical presentation, physical findings, and treatment response seemed to fit well with a diagnosis of PCC, particularly the solitary skin lesions on unclothed areas of the skin; history of skin injury, participation in farming, or exposure to bird droppings (eg, contaminated soil, manure); isolation of C neoformans; and lack of evidence of disseminated disease. Once a diagnosis of PCC is made, however, evaluation of a patient’s immune system and other systemic involvement must be performed, as solitary skin lesions can be the only symptom and an early marker of disseminated disease. Inclusion of a lumbar puncture in the absence of localizing signs is not required in the workup of PCC, with the emergence of more cases of PCC being required before conclusive recommendations can be made. A strong history and physical examination, including pertinent details such as local trauma and exposure to bird droppings, along with the criteria provided by Neuville et al1 and laboratory information may be sufficient to diagnose PCC; close monitoring should be continued.1 Luckily, of the reported cases of PCC in immunocompetent individuals, oral antifungal therapy usually has been curative.2,3 The fact that our patient did not develop generalized disease could be explained by the presence of the possible serotype D, low virulence of the capsule-deficient strain, or perhaps some other immunologic mechanism of defense.
1. Neuville S, Dromer F, Morin O, et al. Primary cutaneous cryptococcosis: a distinct clinical entity [published online ahead of print January 17, 2003]. Clin Infect Dis. 2003;36:337-347.
2. Werchniak AE, Baughman RD. Primary cutaneous cryptococcosis in an elderly man. Clin Exp Dermatol. 2004;29:159-160.
3. Pau M, Lallai C, Aste N, et al. Primary cutaneous cryptococcosis in an immunocompetent host [published online ahead of print March 14, 2009]. Mycoses. 2010;53:256-258.
4. Ramdial PK, Calonje E, Sing Y, et al. Molluscum-like cutaneous cryptococcosis: a histopathological and pathogenetic appraisal [published online ahead of print June 4, 2008]. J Cutan Pathol. 2008;35:1007-1013.
5. Vogelaers D, Petrovic M, Deroo M, et al. A case of primary cutaneous cryptococcosis. Eur J Clin Microbiol Infect Dis. 1997;16:150-152.
6. Naka W, Masuda M, Konohana A, et al. Primary cutaneous cryptococcosis and Cryptococcus neoformans serotype D. Clin Exp Dermatol. 1995;20:221-225.
7. Xiujiao X, Aie X. Two cases of cutaneous cryptococcosis. Mycoses. 2005;48:238-241.
8. Murray PR, Rosenthal KS, Pfaller MA, eds. Medical Microbiology. 6th ed. Philadelphia, PA: Mosby Elsevier; 2009.
9. Moreno Castillo JL, Del Negro G, Heins-Vaccari E, et al. Primary cutaneous cryptococcosis. Mycopathologia. 1986;96:25-28.
10. Joshi S, Wattal C, Duggal L, et al. Cutaneous cryptococcosis. J Assoc Physicians India. 2004;52:242-243.
Cryptococcosis is an opportunistic yeast infection caused by Cryptococcus neoformans that remains the most common systemic fungal infection in immunosuppressed patients and often presents with signs of meningitis. Cutaneous cryptococcosis occurs in 10% to 20% of systemic Cryptococcus infections and usually is secondary to hematogenous dissemination in patients with an underlying disease, particularly human immunodeficiency virus. Primary cutaneous cryptococcosis (PCC) is a more rare clinical identity that is characterized by skin lesions confined to 1 body region, often presenting as a whitlow or phlegmon with positive culture for C neoformans and no evidence of simultaneous dissemination. We report a rare case of PCC in a 73-year-old man with intact cell-mediated immunity.
Case Report
A 73-year-old man who was a beef farmer presented on primary care referral with multiple red nodules and ulcers on the right third and fourth digits and distal forearm following abrasion to the region. The patient reported that the lesions had started as painful nodules that would open and drain. He had been taking oral ciprofloxacin and oral ketoconazole for 3 days as prescribed by his primary care physician but had not begun to see results. He denied any travel or exposure to roses, fish tanks, or any sick contacts. A review of systems was negative for fever, night sweats, malaise, headache, or any other systemic symptoms. Physical examination revealed multiple 2- to 6-mm nodules and ulcers distributed in a sporotrichoid pattern on the right hand (Figure 1) and arm (Figure 2). Lymphadenopathy was absent and the rest of the examination revealed no abnormalities.
|
Initially, 4 punch biopsies of the right hand and arm were obtained and sent for Gram staining, tissue culture (bacterial and fungal), and histopathologic review. A presumptive diagnosis of sporotrichosis was made, with change of treatment pending culture. On routine hematoxylin and eosin staining, marked acute and chronic granulomatous inflammation with microabscesses was noted. Acid-fast bacilli staining was negative. Follow-up Gomori methenamine-silver (GMS) staining showed numerous fungal spores with narrow base budding (Figure 3). Subsequent mucicarmine staining did not reveal dark red capsules characteristic of Cryptococcus (Figure 4). The pathology report indicated that the findings may represent sporotrichosis in the appropriate clinical setting, but GMS staining could not definitively classify Sporothrix schenckii or rule out other fungal infections without tissue culture. Before culture results could be obtained, the patient returned 2 weeks later for suture removal at which point the prior medications were stopped and itraconazole 200 mg once daily was initiated.
Upon receiving the culture results, a diagnosis of primary capsule-deficient cutaneous cryptococcosis was made. The lesions showed clinical improvement at 1-month follow-up, and treatment with itraconazole was continued with monthly liver function tests. After 5 months of continued improvement, the itraconazole dose was decreased to 100 mg once daily for 1 month. The patient was free of lesions and any sequelae at 6-month and 1-year follow-up.
Comment
Cryptococcosis is caused by C neoformans, an opportunistic, basidiomycetous, yeastlike fungus1 that presents as a yeast in both the environment and tissue and normally is associated with immunocompromised host infection, especially in individuals with human immunodeficiency virus. The most common route of infection is through the lungs as respiratory droplets followed by hematogenous dissemination to the central nervous system and skin, with meningitis being the most common clinical manifestation and Cryptococcus being the most common cause of fungal meningitis worldwide.2 Cutaneous involvement after hematogenous spread (secondary cutaneous cryptococcosis) is reported in 10% to 20% of systemic Cryptococcus cases, while PCC is limited to rare cases in which trauma or abrasions to the affected site are notable risk factors.2,3
|
Cryptococcus can produce a myriad of skin manifestations including but not limited to nodules, ulcers, plaques, pustules, vesicobullous lesions, and draining sinuses. Neuville et al1 found that cellulitis, cutaneous ulcers, and whitlows were the most common presenting clinical features in PCC. Whitlows also have been reported as a rare presentation in secondary cutaneous cryptococcosis yielding to the much more prevalent presentation of umbilicated papules resembling molluscum contagiosum.1 This polymorphic identity can therefore mimic not only other dermatoses and neoplasms but other infections such as bacterial cellulitis, herpes simplex virus, and molluscum contagiosum, especially in disseminated cryptococcosis, making microscopic assessment crucial for the diagnostic confirmation of cutaneous cryptococcosis. The differential diagnosis includes sporotrichosis and Mycobacterium marinum due to the lymphatic distribution of the lesions as well as squamous cell carcinoma. Our initial diagnosis of sporotrichosis was assumptive until mycological data could be obtained.
The histopathology patterns characteristic of C neoformans infection fall into either a paucireactive pattern with myriads of densely packed organisms with mucoid gelatinous capsules that cause minimal tissue reaction or a mixed suppurative and granulomatous reaction with varying degrees of necrosis.4 The granulomatous form can affect histiocytes, giant cells, lymphocytes, and fibroblasts. These findings along with the characteristic carminophilic capsule of C neoformans allows for a prompt diagnosis. However, the C neoformans spore somewhat characteristically measures 3 to 20 mm in diameter and stains well with periodic acid–Schiff stain and GMS.4,5 Therefore, the lack of capsule broadened our early differential to include Histoplasma capsulatum, S schenckii, Paracoccidioides brasiliensis, and even Blastomyces dermatitidis.
Neuville et al1 proposed the following criteria for the diagnosis of PCC: the absence of dissemination and predominantly a solitary skin lesion on unclothed areas presenting as a whitlow or phlegmon, a history of skin injury or damage leading to direct inoculation, participation in outdoor activities, exposure to bird droppings, and isolation of C neoformans serotype D. Other factors that strongly support PCC diagnosis over squamous cell carcinoma (based on a review of the literature) are rural residential environment, older age, equal prevalence among men and women, and lack of underlying disease. Presence of these factors seem to favor PCC over squamous cell carcinoma, as some still consider the existence of PCC in general to be controversial because skin manifestations represent a sentinel finding indicative of disseminated disease.1,3
The fungus can be found worldwide as an ubiquitous saprophyte of soil, especially if the soil is enriched with pigeon droppings. A link between C neoformans and pigeons has been suggested, with dried avian excreta allowing the yeast to abundantly grow because of its high nitrogen content.5,6 Other possible sites include decaying wood, fruits, vegetables, and dust.1 There are 4 main serotypes and 3 varieties of C neoformans: C neoformans var grubii (serotype A; worldwide distribution), C neoformans var gattii (serotypes B and C; more circumscribed diffusion and distribution including subtropical regions of Australia, Central Africa, South Asia, and California), and C neoformans var neoformans (serotype D; worldwide distribution).3,7 A literature review indicated that known cases of serotype D (global incidence, 9%) tended to produce cutaneous lesions without systemic involvement.7 Microscopically, the most important characteristic feature found in all serotypes is the polysaccharide capsule, which normally acts as an important virulence factor.6 This capsule as well as detection of the budding yeast can be visualized with india ink (cerebrospinal fluid), methylene blue, or mucicarmine staining. The latex agglutination test for cryptococcal antigen has been used as a serologic test for cerebrospinal fluid, blood, and urine with a sensitivity of 86% to 95%.8
Treatment of cryptococcal disease depends on location and severity of lesions. Many cases of PCC spontaneously resolve, but it is a recommended practice to treat the lesions via incision, local irrigation and debridement, and anti-inflammatory and antifungal agents.9 Antifungal therapy with amphotericin B with or without flucytosine was the standard of therapy. The newer oral azole compounds (eg, ketoconazole, fluconazole, itraconazole) are effective against Cryptococcus, making them the probable treatment in immunocompetent patients because of fewer side effects. Nonetheless, these drugs should be maintained for several weeks or even months to achieve complete resolution of PCC.10
Our patient’s clinical presentation, physical findings, and treatment response seemed to fit well with a diagnosis of PCC, particularly the solitary skin lesions on unclothed areas of the skin; history of skin injury, participation in farming, or exposure to bird droppings (eg, contaminated soil, manure); isolation of C neoformans; and lack of evidence of disseminated disease. Once a diagnosis of PCC is made, however, evaluation of a patient’s immune system and other systemic involvement must be performed, as solitary skin lesions can be the only symptom and an early marker of disseminated disease. Inclusion of a lumbar puncture in the absence of localizing signs is not required in the workup of PCC, with the emergence of more cases of PCC being required before conclusive recommendations can be made. A strong history and physical examination, including pertinent details such as local trauma and exposure to bird droppings, along with the criteria provided by Neuville et al1 and laboratory information may be sufficient to diagnose PCC; close monitoring should be continued.1 Luckily, of the reported cases of PCC in immunocompetent individuals, oral antifungal therapy usually has been curative.2,3 The fact that our patient did not develop generalized disease could be explained by the presence of the possible serotype D, low virulence of the capsule-deficient strain, or perhaps some other immunologic mechanism of defense.
Cryptococcosis is an opportunistic yeast infection caused by Cryptococcus neoformans that remains the most common systemic fungal infection in immunosuppressed patients and often presents with signs of meningitis. Cutaneous cryptococcosis occurs in 10% to 20% of systemic Cryptococcus infections and usually is secondary to hematogenous dissemination in patients with an underlying disease, particularly human immunodeficiency virus. Primary cutaneous cryptococcosis (PCC) is a more rare clinical identity that is characterized by skin lesions confined to 1 body region, often presenting as a whitlow or phlegmon with positive culture for C neoformans and no evidence of simultaneous dissemination. We report a rare case of PCC in a 73-year-old man with intact cell-mediated immunity.
Case Report
A 73-year-old man who was a beef farmer presented on primary care referral with multiple red nodules and ulcers on the right third and fourth digits and distal forearm following abrasion to the region. The patient reported that the lesions had started as painful nodules that would open and drain. He had been taking oral ciprofloxacin and oral ketoconazole for 3 days as prescribed by his primary care physician but had not begun to see results. He denied any travel or exposure to roses, fish tanks, or any sick contacts. A review of systems was negative for fever, night sweats, malaise, headache, or any other systemic symptoms. Physical examination revealed multiple 2- to 6-mm nodules and ulcers distributed in a sporotrichoid pattern on the right hand (Figure 1) and arm (Figure 2). Lymphadenopathy was absent and the rest of the examination revealed no abnormalities.
|
Initially, 4 punch biopsies of the right hand and arm were obtained and sent for Gram staining, tissue culture (bacterial and fungal), and histopathologic review. A presumptive diagnosis of sporotrichosis was made, with change of treatment pending culture. On routine hematoxylin and eosin staining, marked acute and chronic granulomatous inflammation with microabscesses was noted. Acid-fast bacilli staining was negative. Follow-up Gomori methenamine-silver (GMS) staining showed numerous fungal spores with narrow base budding (Figure 3). Subsequent mucicarmine staining did not reveal dark red capsules characteristic of Cryptococcus (Figure 4). The pathology report indicated that the findings may represent sporotrichosis in the appropriate clinical setting, but GMS staining could not definitively classify Sporothrix schenckii or rule out other fungal infections without tissue culture. Before culture results could be obtained, the patient returned 2 weeks later for suture removal at which point the prior medications were stopped and itraconazole 200 mg once daily was initiated.
Upon receiving the culture results, a diagnosis of primary capsule-deficient cutaneous cryptococcosis was made. The lesions showed clinical improvement at 1-month follow-up, and treatment with itraconazole was continued with monthly liver function tests. After 5 months of continued improvement, the itraconazole dose was decreased to 100 mg once daily for 1 month. The patient was free of lesions and any sequelae at 6-month and 1-year follow-up.
Comment
Cryptococcosis is caused by C neoformans, an opportunistic, basidiomycetous, yeastlike fungus1 that presents as a yeast in both the environment and tissue and normally is associated with immunocompromised host infection, especially in individuals with human immunodeficiency virus. The most common route of infection is through the lungs as respiratory droplets followed by hematogenous dissemination to the central nervous system and skin, with meningitis being the most common clinical manifestation and Cryptococcus being the most common cause of fungal meningitis worldwide.2 Cutaneous involvement after hematogenous spread (secondary cutaneous cryptococcosis) is reported in 10% to 20% of systemic Cryptococcus cases, while PCC is limited to rare cases in which trauma or abrasions to the affected site are notable risk factors.2,3
|
Cryptococcus can produce a myriad of skin manifestations including but not limited to nodules, ulcers, plaques, pustules, vesicobullous lesions, and draining sinuses. Neuville et al1 found that cellulitis, cutaneous ulcers, and whitlows were the most common presenting clinical features in PCC. Whitlows also have been reported as a rare presentation in secondary cutaneous cryptococcosis yielding to the much more prevalent presentation of umbilicated papules resembling molluscum contagiosum.1 This polymorphic identity can therefore mimic not only other dermatoses and neoplasms but other infections such as bacterial cellulitis, herpes simplex virus, and molluscum contagiosum, especially in disseminated cryptococcosis, making microscopic assessment crucial for the diagnostic confirmation of cutaneous cryptococcosis. The differential diagnosis includes sporotrichosis and Mycobacterium marinum due to the lymphatic distribution of the lesions as well as squamous cell carcinoma. Our initial diagnosis of sporotrichosis was assumptive until mycological data could be obtained.
The histopathology patterns characteristic of C neoformans infection fall into either a paucireactive pattern with myriads of densely packed organisms with mucoid gelatinous capsules that cause minimal tissue reaction or a mixed suppurative and granulomatous reaction with varying degrees of necrosis.4 The granulomatous form can affect histiocytes, giant cells, lymphocytes, and fibroblasts. These findings along with the characteristic carminophilic capsule of C neoformans allows for a prompt diagnosis. However, the C neoformans spore somewhat characteristically measures 3 to 20 mm in diameter and stains well with periodic acid–Schiff stain and GMS.4,5 Therefore, the lack of capsule broadened our early differential to include Histoplasma capsulatum, S schenckii, Paracoccidioides brasiliensis, and even Blastomyces dermatitidis.
Neuville et al1 proposed the following criteria for the diagnosis of PCC: the absence of dissemination and predominantly a solitary skin lesion on unclothed areas presenting as a whitlow or phlegmon, a history of skin injury or damage leading to direct inoculation, participation in outdoor activities, exposure to bird droppings, and isolation of C neoformans serotype D. Other factors that strongly support PCC diagnosis over squamous cell carcinoma (based on a review of the literature) are rural residential environment, older age, equal prevalence among men and women, and lack of underlying disease. Presence of these factors seem to favor PCC over squamous cell carcinoma, as some still consider the existence of PCC in general to be controversial because skin manifestations represent a sentinel finding indicative of disseminated disease.1,3
The fungus can be found worldwide as an ubiquitous saprophyte of soil, especially if the soil is enriched with pigeon droppings. A link between C neoformans and pigeons has been suggested, with dried avian excreta allowing the yeast to abundantly grow because of its high nitrogen content.5,6 Other possible sites include decaying wood, fruits, vegetables, and dust.1 There are 4 main serotypes and 3 varieties of C neoformans: C neoformans var grubii (serotype A; worldwide distribution), C neoformans var gattii (serotypes B and C; more circumscribed diffusion and distribution including subtropical regions of Australia, Central Africa, South Asia, and California), and C neoformans var neoformans (serotype D; worldwide distribution).3,7 A literature review indicated that known cases of serotype D (global incidence, 9%) tended to produce cutaneous lesions without systemic involvement.7 Microscopically, the most important characteristic feature found in all serotypes is the polysaccharide capsule, which normally acts as an important virulence factor.6 This capsule as well as detection of the budding yeast can be visualized with india ink (cerebrospinal fluid), methylene blue, or mucicarmine staining. The latex agglutination test for cryptococcal antigen has been used as a serologic test for cerebrospinal fluid, blood, and urine with a sensitivity of 86% to 95%.8
Treatment of cryptococcal disease depends on location and severity of lesions. Many cases of PCC spontaneously resolve, but it is a recommended practice to treat the lesions via incision, local irrigation and debridement, and anti-inflammatory and antifungal agents.9 Antifungal therapy with amphotericin B with or without flucytosine was the standard of therapy. The newer oral azole compounds (eg, ketoconazole, fluconazole, itraconazole) are effective against Cryptococcus, making them the probable treatment in immunocompetent patients because of fewer side effects. Nonetheless, these drugs should be maintained for several weeks or even months to achieve complete resolution of PCC.10
Our patient’s clinical presentation, physical findings, and treatment response seemed to fit well with a diagnosis of PCC, particularly the solitary skin lesions on unclothed areas of the skin; history of skin injury, participation in farming, or exposure to bird droppings (eg, contaminated soil, manure); isolation of C neoformans; and lack of evidence of disseminated disease. Once a diagnosis of PCC is made, however, evaluation of a patient’s immune system and other systemic involvement must be performed, as solitary skin lesions can be the only symptom and an early marker of disseminated disease. Inclusion of a lumbar puncture in the absence of localizing signs is not required in the workup of PCC, with the emergence of more cases of PCC being required before conclusive recommendations can be made. A strong history and physical examination, including pertinent details such as local trauma and exposure to bird droppings, along with the criteria provided by Neuville et al1 and laboratory information may be sufficient to diagnose PCC; close monitoring should be continued.1 Luckily, of the reported cases of PCC in immunocompetent individuals, oral antifungal therapy usually has been curative.2,3 The fact that our patient did not develop generalized disease could be explained by the presence of the possible serotype D, low virulence of the capsule-deficient strain, or perhaps some other immunologic mechanism of defense.
1. Neuville S, Dromer F, Morin O, et al. Primary cutaneous cryptococcosis: a distinct clinical entity [published online ahead of print January 17, 2003]. Clin Infect Dis. 2003;36:337-347.
2. Werchniak AE, Baughman RD. Primary cutaneous cryptococcosis in an elderly man. Clin Exp Dermatol. 2004;29:159-160.
3. Pau M, Lallai C, Aste N, et al. Primary cutaneous cryptococcosis in an immunocompetent host [published online ahead of print March 14, 2009]. Mycoses. 2010;53:256-258.
4. Ramdial PK, Calonje E, Sing Y, et al. Molluscum-like cutaneous cryptococcosis: a histopathological and pathogenetic appraisal [published online ahead of print June 4, 2008]. J Cutan Pathol. 2008;35:1007-1013.
5. Vogelaers D, Petrovic M, Deroo M, et al. A case of primary cutaneous cryptococcosis. Eur J Clin Microbiol Infect Dis. 1997;16:150-152.
6. Naka W, Masuda M, Konohana A, et al. Primary cutaneous cryptococcosis and Cryptococcus neoformans serotype D. Clin Exp Dermatol. 1995;20:221-225.
7. Xiujiao X, Aie X. Two cases of cutaneous cryptococcosis. Mycoses. 2005;48:238-241.
8. Murray PR, Rosenthal KS, Pfaller MA, eds. Medical Microbiology. 6th ed. Philadelphia, PA: Mosby Elsevier; 2009.
9. Moreno Castillo JL, Del Negro G, Heins-Vaccari E, et al. Primary cutaneous cryptococcosis. Mycopathologia. 1986;96:25-28.
10. Joshi S, Wattal C, Duggal L, et al. Cutaneous cryptococcosis. J Assoc Physicians India. 2004;52:242-243.
1. Neuville S, Dromer F, Morin O, et al. Primary cutaneous cryptococcosis: a distinct clinical entity [published online ahead of print January 17, 2003]. Clin Infect Dis. 2003;36:337-347.
2. Werchniak AE, Baughman RD. Primary cutaneous cryptococcosis in an elderly man. Clin Exp Dermatol. 2004;29:159-160.
3. Pau M, Lallai C, Aste N, et al. Primary cutaneous cryptococcosis in an immunocompetent host [published online ahead of print March 14, 2009]. Mycoses. 2010;53:256-258.
4. Ramdial PK, Calonje E, Sing Y, et al. Molluscum-like cutaneous cryptococcosis: a histopathological and pathogenetic appraisal [published online ahead of print June 4, 2008]. J Cutan Pathol. 2008;35:1007-1013.
5. Vogelaers D, Petrovic M, Deroo M, et al. A case of primary cutaneous cryptococcosis. Eur J Clin Microbiol Infect Dis. 1997;16:150-152.
6. Naka W, Masuda M, Konohana A, et al. Primary cutaneous cryptococcosis and Cryptococcus neoformans serotype D. Clin Exp Dermatol. 1995;20:221-225.
7. Xiujiao X, Aie X. Two cases of cutaneous cryptococcosis. Mycoses. 2005;48:238-241.
8. Murray PR, Rosenthal KS, Pfaller MA, eds. Medical Microbiology. 6th ed. Philadelphia, PA: Mosby Elsevier; 2009.
9. Moreno Castillo JL, Del Negro G, Heins-Vaccari E, et al. Primary cutaneous cryptococcosis. Mycopathologia. 1986;96:25-28.
10. Joshi S, Wattal C, Duggal L, et al. Cutaneous cryptococcosis. J Assoc Physicians India. 2004;52:242-243.
Practice Points
- Cryptococcus neoformans is an encapsulated yeast that is ubiquitous in the environment and is especially abundant in soil enriched with pigeon droppings.
- Immunocompetent hosts often are asymptomatic or have only mild pulmonary disease, while disseminated disease affects the lungs, central nervous system, bones, and skin in immunocompromised hosts.
- Diagnostic tests include india ink or mucicarmine staining to highlight characteristic capsules or the latex agglutination test to measure circulating capsular antigen.
Repigmentation of Gray Hair in Lesions of Annular Elastolytic Giant Cell Granuloma
Hair pigmentation is a complex phenomenon that involves many hormones, neurotransmitters, cytokines, growth factors, eicosanoids, cyclic nucleotides, nutrients, and a physicochemical milieu.1 Repigmentation of gray hair has been associated with herpes zoster infection,2 use of systemic corticosteroids,3 thyroid hormone therapy,4 or treatment with interferon and ribavirin.5 We report a case of repigmentation of gray hairs in lesions of annular elastolytic giant cell granuloma (AEGCG) on the scalp of a 67-year-old man.
Case Report
A 67-year-old man presented to the dermatology department for evaluation of pruritic lesions on the face and scalp of 1 year’s duration. The patient reported that hairs in the involved areas of the scalp had turned from gray to a dark color since the appearance of the lesions. The patient had a history of hypertension and type 2 diabetes mellitus. His current medications included irbesartan, atorvastatin, metformin, acetylsalicylic acid, omeprazole, and repaglinide.
Physical examination revealed plaques on the scalp and cheeks that were 2 to 10 mm in diameter. Some of the plaques had an atrophic center and a desquamative peripheral border. The patient had androgenetic alopecia. The remaining hair was dark in the areas affected by the inflammatory plaques while it remained white-gray in the uninvolved areas (Figure 1).
A biopsy of one of the lesions was performed. Histopathology revealed a granulomatous dermatitis involving mostly the upper and mid dermis (Figure 2). Granulomas were epithelioid with many giant cells, some of which contained many nuclei. A ringed array of nuclei was noted in some histiocytes. Elastic fibers were absent in the central zone of the granulomas, a finding that was better evidenced on orcein staining (Figure 3). On the contrary, the peripheral zone of the granulomas showed an increased amount of thick elastotic material. Elastophagocytosis was observed, but no asteroid bodies, Schaumann bodies, or mucin deposits were noted. Histochemistry for microorganisms with Ziehl-Neelsen and periodic acid–Schiff staining was negative. Other findings included a mild infiltrate of melanophages in the papillary dermis as well as a mild superficial dermal inflammatory infiltrate that was rich in plasma cells. Immunostaining for Treponema pallidum was negative. The lymphocytic infiltrate was CD4+predominant. A prominent dermal elastosis also was noted. Hair follicles within the plaques were small in size, penetrating just the dermis. Immunostaining for HMB-45, melan-A, and S-100 demonstrated preserved melanocytes in the hair bulbs (Figure 4). CD68 immunostaining made the infiltrate of macrophages stand out. Based on the results of the histopathologic evaluation, a diagnosis of AEGCG was made.
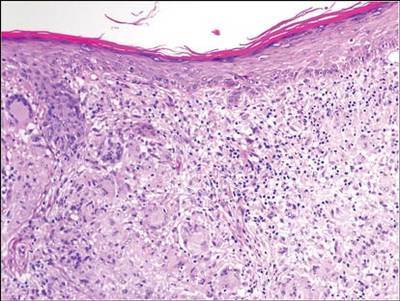 | 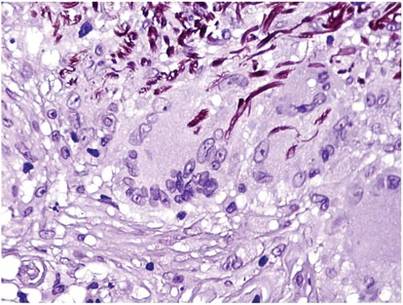 |
Comment
Annular elastolytic giant cell granuloma is a controversial entity that was first described by O’Brien6 in 1975 as actinic granuloma. Hanke et al7 proposed the term annular elastolytic giant cell granuloma to encompass lesions previously called actinic granuloma, atypical necrobiosis lipoidica, and Miescher granuloma. Some researchers have claimed that AEGCG is an independent entity, therefore separate and distinguishable from granuloma annulare. Histopathologic clues to distinguish AEGCG from granuloma annulare have been noted in the literature.7-9 Other investigators believe AEGCG is a type of granuloma annulare that appears on exposed skin.10 There are several variants of the classic clinical presentation of AEGCG, such as cases including presentation in unexposed areas of the skin,11 a papular variant,12 a rapidly regressive variant,13 a reticular variant,14 a variant of early childhood,15 a generalized variant,16 presentation in a necklace distribution,17 presentation as alopecia,18 a sarcoid variant,19 or presentation as reticulate erythema.20 However, no variant has been associated with hair repigmentation.
Melanin units from the proximal hair bulb are responsible for pigmentation in adult hair follicles and are integrated by the hair matrix, melanocytes, keratinocytes, and fibroblasts.21 Hair bulb melanocytes are larger and more dendritic than epidermal melanocytes (Figure 5). The hair only pigments during the anagen phase; therefore, its pigmentation is cyclic, as opposed to epidermal pigmentation, which is ongoing. Hair pigmentation is the result of a complex interaction between the epithelium, the mesenchyme, and the neuroectoderm. This complex pigmentation results from the interaction between follicular melanocytes, keratinocytes, and the fibroblasts from the hair papilla.22 Hair pigmentation involves many hormones, neurotransmitters, cytokines, growth factors, eicosanoids, cyclic nucleotides, nutrients, and a physicochemical milieu1,23-25 (Table), and it is regulated by autocrines, paracrines, or intracrines.21 Therefore, it is likely that many environmental factors may affect hair pigmentation, which may explain why repigmentation of the hair has been seen in the setting of herpes zoster infection,2 use of systemic corticosteroids in the treatment of bullous pemphigoid,3 thyroid hormone therapy,4 treatment with interferon and ribavirin,5 porphyria cutanea tarda,26 or lentigo maligna.27 In our patient, AEGCG might have induced some changes in the dermal environment that were responsible for the repigmentation of the patient’s gray hair. It is speculated that solar radiation and other factors can transform the antigenicity of elastic fibers and induce an immune response in AEGCG.12,15 The lymphocytic infiltrate in these lesions is predominantly CD4+, as seen in our patient, which is consistent with an autoimmune hypothesis.15 Nevertheless, it most likely is too simplistic to attribute the repigmentation to the influence of just these cells.


1. Slominski A, Tobin DJ, Shibahara S, et al. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev. 2004;84:1155-1228.
2. Adiga GU, Rehman KL, Wiernik PH. Permanent localized hair repigmentation following herpes zoster infection. Arch Dermatol. 2010;146:569-570.
3. Khaled A, Trojjets S, Zeglaoui F, et al. Repigmentation of the white hair after systemic corticosteroids for bullous pemphigoid. J Eur Acad Dermatol Venereol. 2008;22:1018-1020.
4. Redondo P, Guzmán M, Marquina M, et al. Repigmentation of gray hair after thyroid hormone treatment [in Spanish]. Actas Dermosifiliogr. 2007;98:603-610.
5. Kavak A, Akcan Y, Korkmaz U. Hair repigmentation in a hepatitis C patient treated with interferon and ribavirin. Dermatology. 2005;211:171-172.
6. O’Brien JP. Actinic granuloma. an annular connective tissue disorder affecting sun- and heat-damaged (elastotic) skin. Arch Dermatol. 1975;111:460-466.
7. Hanke CW, Bailin PL, Roenigk HH Jr. Annular elastolytic giant cell granuloma. a clinicopathologic study of five cases and a review of similar entities. J Am Acad Dermatol. 1979;1:413-421.
8. Al-Hoqail IA, Al-Ghamdi AM, Martinka M, et al. Actinic granuloma is a unique and distinct entity: a comparative study with granuloma annulare. Am J Dermatopathol. 2002;24:209-212.
9. Limas C. The spectrum of primary cutaneous elastolytic granulomas and their distinction from granuloma annulare: a clinicopathological analysis. Histopathology. 2004;44:277-282.
10. Ragaz A, Ackerman AB. Is actinic granuloma a specific condition? Am J Dermatopathol. 1979;1:43-50.
11. Muramatsu T, Shirai T, Yamashina Y, et al. Annular elastolytic giant cell granuloma: an unusual case with lesions arising in non-sun-exposed areas. J Dermatol. 1987;14:54-58.
12. Kelly BJ, Mrstik ME, Ramos-Caro FA, et al. Papular elastolytic giant cell granuloma responding to hydroxychloroquine and quinacrine. Int J Dermatol. 2004;43:964-966.
13. Misago N, Ohtsuka Y, Ishii K, et al. Papular and reticular elastolytic giant cell granuloma: rapid spontaneous regression. Acta Derm Venereol. 2007;87:89-90.
14. Hinrichs R, Weiss T, Peschke E, et al. A reticular variant of elastolytic giant cell granuloma. Clin Exp Dermatol. 2006;31:42-44.
15. Lee HW, Lee MW, Choi JH, et al. Annular elastolytic giant cell granuloma in an infant: improvement after treatment with oral tranilast andtopical pimecrolimus. J Am Acad Dermatol. 2005;53(5, suppl 1):S244-S246.
16. Klemke CD, Siebold D, Dippel E, et al. Generalised annular elastolytic giant cell granuloma. Dermatology. 2003;207:420-422.
17. Meadows KP, O’Reilly MA, Harris RM, et al. Erythematous annular plaques in a necklace distribution. annular elastolytic giant cell granuloma. Arch Dermatol. 2001;137:1647-1652.
18. Delgado-Jimenez Y, Perez-Gala S, Peñas PF, et al. O’Brien actinic granuloma presenting as alopecia. J Eur Acad Dermatol Venereol. 2006;20:226-227.
19. Gambichler T, Herde M, Hoffmann K, et al. Sarcoid variant of actinic granuloma: is it annular sarcoidosis? Dermatology. 2001;203:353-354.
20. Bannister MJ, Rubel DM, Kossard S. Mid-dermal elastophagocytosis presenting as a persistent reticulate erythema. Australas J Dermatol. 2001;42:50-54.
21. Slominski A, Paus R. Melanogenesis is coupled to murine anagen: toward new concepts for the role of melanocytes and the regulation of melanogenesis in hair growth. J Invest Dermatol. 1993;101(1 suppl):90S-97S.
22. Slominski A, Wortsman J, Plonka PM, et al. Hair follicle pigmentation. J Invest Dermatol. 2005;124:13-21.
23. Hearing VJ. Biochemical control of melanogenesis and melanosomal organization. J Investig Dermatol Symp Proc. 1999;4:24-28.
24. Slominski A, Wortsman J. Neuroendocrinology of the skin [published correction appears in Endocr Rev. 2002;23:364]. Endocr Rev. 2000;21:457-487.
25. Slominski A, Wortsman J, Luger T, et al. Corticotropin releasing hormone and proopiomelanocortin involvement in the cutaneous response to stress. Physiol Rev. 2000;80:979-1020.
26. Shaffrali FC, McDonagh AJ, Messenger AG. Hair darkening in porphyria cutanea tarda. Br J Dermatol. 2002;146:325-329.
27. Dummer R. Clinical picture: hair repigmentation in lentigo maligna. Lancet. 2001;357:598.
Hair pigmentation is a complex phenomenon that involves many hormones, neurotransmitters, cytokines, growth factors, eicosanoids, cyclic nucleotides, nutrients, and a physicochemical milieu.1 Repigmentation of gray hair has been associated with herpes zoster infection,2 use of systemic corticosteroids,3 thyroid hormone therapy,4 or treatment with interferon and ribavirin.5 We report a case of repigmentation of gray hairs in lesions of annular elastolytic giant cell granuloma (AEGCG) on the scalp of a 67-year-old man.
Case Report
A 67-year-old man presented to the dermatology department for evaluation of pruritic lesions on the face and scalp of 1 year’s duration. The patient reported that hairs in the involved areas of the scalp had turned from gray to a dark color since the appearance of the lesions. The patient had a history of hypertension and type 2 diabetes mellitus. His current medications included irbesartan, atorvastatin, metformin, acetylsalicylic acid, omeprazole, and repaglinide.
Physical examination revealed plaques on the scalp and cheeks that were 2 to 10 mm in diameter. Some of the plaques had an atrophic center and a desquamative peripheral border. The patient had androgenetic alopecia. The remaining hair was dark in the areas affected by the inflammatory plaques while it remained white-gray in the uninvolved areas (Figure 1).
A biopsy of one of the lesions was performed. Histopathology revealed a granulomatous dermatitis involving mostly the upper and mid dermis (Figure 2). Granulomas were epithelioid with many giant cells, some of which contained many nuclei. A ringed array of nuclei was noted in some histiocytes. Elastic fibers were absent in the central zone of the granulomas, a finding that was better evidenced on orcein staining (Figure 3). On the contrary, the peripheral zone of the granulomas showed an increased amount of thick elastotic material. Elastophagocytosis was observed, but no asteroid bodies, Schaumann bodies, or mucin deposits were noted. Histochemistry for microorganisms with Ziehl-Neelsen and periodic acid–Schiff staining was negative. Other findings included a mild infiltrate of melanophages in the papillary dermis as well as a mild superficial dermal inflammatory infiltrate that was rich in plasma cells. Immunostaining for Treponema pallidum was negative. The lymphocytic infiltrate was CD4+predominant. A prominent dermal elastosis also was noted. Hair follicles within the plaques were small in size, penetrating just the dermis. Immunostaining for HMB-45, melan-A, and S-100 demonstrated preserved melanocytes in the hair bulbs (Figure 4). CD68 immunostaining made the infiltrate of macrophages stand out. Based on the results of the histopathologic evaluation, a diagnosis of AEGCG was made.
| |
Comment
Annular elastolytic giant cell granuloma is a controversial entity that was first described by O’Brien6 in 1975 as actinic granuloma. Hanke et al7 proposed the term annular elastolytic giant cell granuloma to encompass lesions previously called actinic granuloma, atypical necrobiosis lipoidica, and Miescher granuloma. Some researchers have claimed that AEGCG is an independent entity, therefore separate and distinguishable from granuloma annulare. Histopathologic clues to distinguish AEGCG from granuloma annulare have been noted in the literature.7-9 Other investigators believe AEGCG is a type of granuloma annulare that appears on exposed skin.10 There are several variants of the classic clinical presentation of AEGCG, such as cases including presentation in unexposed areas of the skin,11 a papular variant,12 a rapidly regressive variant,13 a reticular variant,14 a variant of early childhood,15 a generalized variant,16 presentation in a necklace distribution,17 presentation as alopecia,18 a sarcoid variant,19 or presentation as reticulate erythema.20 However, no variant has been associated with hair repigmentation.
Melanin units from the proximal hair bulb are responsible for pigmentation in adult hair follicles and are integrated by the hair matrix, melanocytes, keratinocytes, and fibroblasts.21 Hair bulb melanocytes are larger and more dendritic than epidermal melanocytes (Figure 5). The hair only pigments during the anagen phase; therefore, its pigmentation is cyclic, as opposed to epidermal pigmentation, which is ongoing. Hair pigmentation is the result of a complex interaction between the epithelium, the mesenchyme, and the neuroectoderm. This complex pigmentation results from the interaction between follicular melanocytes, keratinocytes, and the fibroblasts from the hair papilla.22 Hair pigmentation involves many hormones, neurotransmitters, cytokines, growth factors, eicosanoids, cyclic nucleotides, nutrients, and a physicochemical milieu1,23-25 (Table), and it is regulated by autocrines, paracrines, or intracrines.21 Therefore, it is likely that many environmental factors may affect hair pigmentation, which may explain why repigmentation of the hair has been seen in the setting of herpes zoster infection,2 use of systemic corticosteroids in the treatment of bullous pemphigoid,3 thyroid hormone therapy,4 treatment with interferon and ribavirin,5 porphyria cutanea tarda,26 or lentigo maligna.27 In our patient, AEGCG might have induced some changes in the dermal environment that were responsible for the repigmentation of the patient’s gray hair. It is speculated that solar radiation and other factors can transform the antigenicity of elastic fibers and induce an immune response in AEGCG.12,15 The lymphocytic infiltrate in these lesions is predominantly CD4+, as seen in our patient, which is consistent with an autoimmune hypothesis.15 Nevertheless, it most likely is too simplistic to attribute the repigmentation to the influence of just these cells.
Hair pigmentation is a complex phenomenon that involves many hormones, neurotransmitters, cytokines, growth factors, eicosanoids, cyclic nucleotides, nutrients, and a physicochemical milieu.1 Repigmentation of gray hair has been associated with herpes zoster infection,2 use of systemic corticosteroids,3 thyroid hormone therapy,4 or treatment with interferon and ribavirin.5 We report a case of repigmentation of gray hairs in lesions of annular elastolytic giant cell granuloma (AEGCG) on the scalp of a 67-year-old man.
Case Report
A 67-year-old man presented to the dermatology department for evaluation of pruritic lesions on the face and scalp of 1 year’s duration. The patient reported that hairs in the involved areas of the scalp had turned from gray to a dark color since the appearance of the lesions. The patient had a history of hypertension and type 2 diabetes mellitus. His current medications included irbesartan, atorvastatin, metformin, acetylsalicylic acid, omeprazole, and repaglinide.
Physical examination revealed plaques on the scalp and cheeks that were 2 to 10 mm in diameter. Some of the plaques had an atrophic center and a desquamative peripheral border. The patient had androgenetic alopecia. The remaining hair was dark in the areas affected by the inflammatory plaques while it remained white-gray in the uninvolved areas (Figure 1).
A biopsy of one of the lesions was performed. Histopathology revealed a granulomatous dermatitis involving mostly the upper and mid dermis (Figure 2). Granulomas were epithelioid with many giant cells, some of which contained many nuclei. A ringed array of nuclei was noted in some histiocytes. Elastic fibers were absent in the central zone of the granulomas, a finding that was better evidenced on orcein staining (Figure 3). On the contrary, the peripheral zone of the granulomas showed an increased amount of thick elastotic material. Elastophagocytosis was observed, but no asteroid bodies, Schaumann bodies, or mucin deposits were noted. Histochemistry for microorganisms with Ziehl-Neelsen and periodic acid–Schiff staining was negative. Other findings included a mild infiltrate of melanophages in the papillary dermis as well as a mild superficial dermal inflammatory infiltrate that was rich in plasma cells. Immunostaining for Treponema pallidum was negative. The lymphocytic infiltrate was CD4+predominant. A prominent dermal elastosis also was noted. Hair follicles within the plaques were small in size, penetrating just the dermis. Immunostaining for HMB-45, melan-A, and S-100 demonstrated preserved melanocytes in the hair bulbs (Figure 4). CD68 immunostaining made the infiltrate of macrophages stand out. Based on the results of the histopathologic evaluation, a diagnosis of AEGCG was made.
| |
Comment
Annular elastolytic giant cell granuloma is a controversial entity that was first described by O’Brien6 in 1975 as actinic granuloma. Hanke et al7 proposed the term annular elastolytic giant cell granuloma to encompass lesions previously called actinic granuloma, atypical necrobiosis lipoidica, and Miescher granuloma. Some researchers have claimed that AEGCG is an independent entity, therefore separate and distinguishable from granuloma annulare. Histopathologic clues to distinguish AEGCG from granuloma annulare have been noted in the literature.7-9 Other investigators believe AEGCG is a type of granuloma annulare that appears on exposed skin.10 There are several variants of the classic clinical presentation of AEGCG, such as cases including presentation in unexposed areas of the skin,11 a papular variant,12 a rapidly regressive variant,13 a reticular variant,14 a variant of early childhood,15 a generalized variant,16 presentation in a necklace distribution,17 presentation as alopecia,18 a sarcoid variant,19 or presentation as reticulate erythema.20 However, no variant has been associated with hair repigmentation.
Melanin units from the proximal hair bulb are responsible for pigmentation in adult hair follicles and are integrated by the hair matrix, melanocytes, keratinocytes, and fibroblasts.21 Hair bulb melanocytes are larger and more dendritic than epidermal melanocytes (Figure 5). The hair only pigments during the anagen phase; therefore, its pigmentation is cyclic, as opposed to epidermal pigmentation, which is ongoing. Hair pigmentation is the result of a complex interaction between the epithelium, the mesenchyme, and the neuroectoderm. This complex pigmentation results from the interaction between follicular melanocytes, keratinocytes, and the fibroblasts from the hair papilla.22 Hair pigmentation involves many hormones, neurotransmitters, cytokines, growth factors, eicosanoids, cyclic nucleotides, nutrients, and a physicochemical milieu1,23-25 (Table), and it is regulated by autocrines, paracrines, or intracrines.21 Therefore, it is likely that many environmental factors may affect hair pigmentation, which may explain why repigmentation of the hair has been seen in the setting of herpes zoster infection,2 use of systemic corticosteroids in the treatment of bullous pemphigoid,3 thyroid hormone therapy,4 treatment with interferon and ribavirin,5 porphyria cutanea tarda,26 or lentigo maligna.27 In our patient, AEGCG might have induced some changes in the dermal environment that were responsible for the repigmentation of the patient’s gray hair. It is speculated that solar radiation and other factors can transform the antigenicity of elastic fibers and induce an immune response in AEGCG.12,15 The lymphocytic infiltrate in these lesions is predominantly CD4+, as seen in our patient, which is consistent with an autoimmune hypothesis.15 Nevertheless, it most likely is too simplistic to attribute the repigmentation to the influence of just these cells.
1. Slominski A, Tobin DJ, Shibahara S, et al. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev. 2004;84:1155-1228.
2. Adiga GU, Rehman KL, Wiernik PH. Permanent localized hair repigmentation following herpes zoster infection. Arch Dermatol. 2010;146:569-570.
3. Khaled A, Trojjets S, Zeglaoui F, et al. Repigmentation of the white hair after systemic corticosteroids for bullous pemphigoid. J Eur Acad Dermatol Venereol. 2008;22:1018-1020.
4. Redondo P, Guzmán M, Marquina M, et al. Repigmentation of gray hair after thyroid hormone treatment [in Spanish]. Actas Dermosifiliogr. 2007;98:603-610.
5. Kavak A, Akcan Y, Korkmaz U. Hair repigmentation in a hepatitis C patient treated with interferon and ribavirin. Dermatology. 2005;211:171-172.
6. O’Brien JP. Actinic granuloma. an annular connective tissue disorder affecting sun- and heat-damaged (elastotic) skin. Arch Dermatol. 1975;111:460-466.
7. Hanke CW, Bailin PL, Roenigk HH Jr. Annular elastolytic giant cell granuloma. a clinicopathologic study of five cases and a review of similar entities. J Am Acad Dermatol. 1979;1:413-421.
8. Al-Hoqail IA, Al-Ghamdi AM, Martinka M, et al. Actinic granuloma is a unique and distinct entity: a comparative study with granuloma annulare. Am J Dermatopathol. 2002;24:209-212.
9. Limas C. The spectrum of primary cutaneous elastolytic granulomas and their distinction from granuloma annulare: a clinicopathological analysis. Histopathology. 2004;44:277-282.
10. Ragaz A, Ackerman AB. Is actinic granuloma a specific condition? Am J Dermatopathol. 1979;1:43-50.
11. Muramatsu T, Shirai T, Yamashina Y, et al. Annular elastolytic giant cell granuloma: an unusual case with lesions arising in non-sun-exposed areas. J Dermatol. 1987;14:54-58.
12. Kelly BJ, Mrstik ME, Ramos-Caro FA, et al. Papular elastolytic giant cell granuloma responding to hydroxychloroquine and quinacrine. Int J Dermatol. 2004;43:964-966.
13. Misago N, Ohtsuka Y, Ishii K, et al. Papular and reticular elastolytic giant cell granuloma: rapid spontaneous regression. Acta Derm Venereol. 2007;87:89-90.
14. Hinrichs R, Weiss T, Peschke E, et al. A reticular variant of elastolytic giant cell granuloma. Clin Exp Dermatol. 2006;31:42-44.
15. Lee HW, Lee MW, Choi JH, et al. Annular elastolytic giant cell granuloma in an infant: improvement after treatment with oral tranilast andtopical pimecrolimus. J Am Acad Dermatol. 2005;53(5, suppl 1):S244-S246.
16. Klemke CD, Siebold D, Dippel E, et al. Generalised annular elastolytic giant cell granuloma. Dermatology. 2003;207:420-422.
17. Meadows KP, O’Reilly MA, Harris RM, et al. Erythematous annular plaques in a necklace distribution. annular elastolytic giant cell granuloma. Arch Dermatol. 2001;137:1647-1652.
18. Delgado-Jimenez Y, Perez-Gala S, Peñas PF, et al. O’Brien actinic granuloma presenting as alopecia. J Eur Acad Dermatol Venereol. 2006;20:226-227.
19. Gambichler T, Herde M, Hoffmann K, et al. Sarcoid variant of actinic granuloma: is it annular sarcoidosis? Dermatology. 2001;203:353-354.
20. Bannister MJ, Rubel DM, Kossard S. Mid-dermal elastophagocytosis presenting as a persistent reticulate erythema. Australas J Dermatol. 2001;42:50-54.
21. Slominski A, Paus R. Melanogenesis is coupled to murine anagen: toward new concepts for the role of melanocytes and the regulation of melanogenesis in hair growth. J Invest Dermatol. 1993;101(1 suppl):90S-97S.
22. Slominski A, Wortsman J, Plonka PM, et al. Hair follicle pigmentation. J Invest Dermatol. 2005;124:13-21.
23. Hearing VJ. Biochemical control of melanogenesis and melanosomal organization. J Investig Dermatol Symp Proc. 1999;4:24-28.
24. Slominski A, Wortsman J. Neuroendocrinology of the skin [published correction appears in Endocr Rev. 2002;23:364]. Endocr Rev. 2000;21:457-487.
25. Slominski A, Wortsman J, Luger T, et al. Corticotropin releasing hormone and proopiomelanocortin involvement in the cutaneous response to stress. Physiol Rev. 2000;80:979-1020.
26. Shaffrali FC, McDonagh AJ, Messenger AG. Hair darkening in porphyria cutanea tarda. Br J Dermatol. 2002;146:325-329.
27. Dummer R. Clinical picture: hair repigmentation in lentigo maligna. Lancet. 2001;357:598.
1. Slominski A, Tobin DJ, Shibahara S, et al. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev. 2004;84:1155-1228.
2. Adiga GU, Rehman KL, Wiernik PH. Permanent localized hair repigmentation following herpes zoster infection. Arch Dermatol. 2010;146:569-570.
3. Khaled A, Trojjets S, Zeglaoui F, et al. Repigmentation of the white hair after systemic corticosteroids for bullous pemphigoid. J Eur Acad Dermatol Venereol. 2008;22:1018-1020.
4. Redondo P, Guzmán M, Marquina M, et al. Repigmentation of gray hair after thyroid hormone treatment [in Spanish]. Actas Dermosifiliogr. 2007;98:603-610.
5. Kavak A, Akcan Y, Korkmaz U. Hair repigmentation in a hepatitis C patient treated with interferon and ribavirin. Dermatology. 2005;211:171-172.
6. O’Brien JP. Actinic granuloma. an annular connective tissue disorder affecting sun- and heat-damaged (elastotic) skin. Arch Dermatol. 1975;111:460-466.
7. Hanke CW, Bailin PL, Roenigk HH Jr. Annular elastolytic giant cell granuloma. a clinicopathologic study of five cases and a review of similar entities. J Am Acad Dermatol. 1979;1:413-421.
8. Al-Hoqail IA, Al-Ghamdi AM, Martinka M, et al. Actinic granuloma is a unique and distinct entity: a comparative study with granuloma annulare. Am J Dermatopathol. 2002;24:209-212.
9. Limas C. The spectrum of primary cutaneous elastolytic granulomas and their distinction from granuloma annulare: a clinicopathological analysis. Histopathology. 2004;44:277-282.
10. Ragaz A, Ackerman AB. Is actinic granuloma a specific condition? Am J Dermatopathol. 1979;1:43-50.
11. Muramatsu T, Shirai T, Yamashina Y, et al. Annular elastolytic giant cell granuloma: an unusual case with lesions arising in non-sun-exposed areas. J Dermatol. 1987;14:54-58.
12. Kelly BJ, Mrstik ME, Ramos-Caro FA, et al. Papular elastolytic giant cell granuloma responding to hydroxychloroquine and quinacrine. Int J Dermatol. 2004;43:964-966.
13. Misago N, Ohtsuka Y, Ishii K, et al. Papular and reticular elastolytic giant cell granuloma: rapid spontaneous regression. Acta Derm Venereol. 2007;87:89-90.
14. Hinrichs R, Weiss T, Peschke E, et al. A reticular variant of elastolytic giant cell granuloma. Clin Exp Dermatol. 2006;31:42-44.
15. Lee HW, Lee MW, Choi JH, et al. Annular elastolytic giant cell granuloma in an infant: improvement after treatment with oral tranilast andtopical pimecrolimus. J Am Acad Dermatol. 2005;53(5, suppl 1):S244-S246.
16. Klemke CD, Siebold D, Dippel E, et al. Generalised annular elastolytic giant cell granuloma. Dermatology. 2003;207:420-422.
17. Meadows KP, O’Reilly MA, Harris RM, et al. Erythematous annular plaques in a necklace distribution. annular elastolytic giant cell granuloma. Arch Dermatol. 2001;137:1647-1652.
18. Delgado-Jimenez Y, Perez-Gala S, Peñas PF, et al. O’Brien actinic granuloma presenting as alopecia. J Eur Acad Dermatol Venereol. 2006;20:226-227.
19. Gambichler T, Herde M, Hoffmann K, et al. Sarcoid variant of actinic granuloma: is it annular sarcoidosis? Dermatology. 2001;203:353-354.
20. Bannister MJ, Rubel DM, Kossard S. Mid-dermal elastophagocytosis presenting as a persistent reticulate erythema. Australas J Dermatol. 2001;42:50-54.
21. Slominski A, Paus R. Melanogenesis is coupled to murine anagen: toward new concepts for the role of melanocytes and the regulation of melanogenesis in hair growth. J Invest Dermatol. 1993;101(1 suppl):90S-97S.
22. Slominski A, Wortsman J, Plonka PM, et al. Hair follicle pigmentation. J Invest Dermatol. 2005;124:13-21.
23. Hearing VJ. Biochemical control of melanogenesis and melanosomal organization. J Investig Dermatol Symp Proc. 1999;4:24-28.
24. Slominski A, Wortsman J. Neuroendocrinology of the skin [published correction appears in Endocr Rev. 2002;23:364]. Endocr Rev. 2000;21:457-487.
25. Slominski A, Wortsman J, Luger T, et al. Corticotropin releasing hormone and proopiomelanocortin involvement in the cutaneous response to stress. Physiol Rev. 2000;80:979-1020.
26. Shaffrali FC, McDonagh AJ, Messenger AG. Hair darkening in porphyria cutanea tarda. Br J Dermatol. 2002;146:325-329.
27. Dummer R. Clinical picture: hair repigmentation in lentigo maligna. Lancet. 2001;357:598.
Practice Points
- Hair repigmentation can be a clinical clue to a subjacent inflammatory disease.
- Hair depigmentation associated with aging may be a reversible condition under proper stimulation.

Madelung Deformity and Extensor Tendon Rupture
Extensor tendon rupture in chronic Madelung deformity, as a result of tendon attrition on the dislocated distal ulna, occurs infrequently. However, it is often seen in patients with rheumatoid arthritis. This issue has been reported in only a few English-language case reports. Here we report a case of multiple tendon ruptures in a previously undiagnosed Madelung deformity. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 56-year-old active woman presented with 50 days’ inability to extend the fourth and fifth fingers of her dominant right hand. The loss of finger extension progressed, over several weeks, to involve the third finger as well. The first 2 tendon ruptures had been triggered by lifting a light grocery bag, when she noticed a sharp sudden pain and “pop.” The third rupture occurred spontaneously with a snapping sound the night before surgery.
The patient had observed some prominence on the ulnar side of her right wrist since childhood but had never experienced any pain or functional disability. There was neither history of trauma, inflammatory disease, diabetes mellitus, or infection, nor positive family history of similar wrist deformity.
The physical examination showed a dorsally subluxated distal radioulnar joint, prominent ulnar styloid, and mild ulnar and volar deviation of the wrist along with limitation of wrist dorsiflexion. Complete loss of active extension of the 3 ulnar fingers was demonstrated, while neurovascular status and all other hand evaluations were normal. The wrist radiographs confirmed the typical findings of Madelung deformity (Figure 1).
Repair of the ruptured tendons and resection of the prominent distal ulna (Darrach procedure) was planned. (Given the patient’s age and evidence of degenerative changes in the radiocarpal joint, correction of the Madelung deformity did not seem necessary). At time of surgery, the recently ruptured third finger extensor tendon was easily found and approximated, and end-to-end repair was performed. The fourth and fifth fingers, however, had to be fished out more proximally from dense granulation tissue. After the distal ulna was resected for a distance of 1.5 cm, meticulous repair of the ulnar collateral ligament and the capsule and periosteum over the end of the ulna was performed. Then, for grafting of the ruptured tendons, the extensor indicis proprius tendon was isolated and transected at the second metacarpophalangeal joint level. A piece of this tendon was used as interpositional graft for the fourth extensor tendon, and the main tendon unit was transferred to the fifth finger extensor. The extensor digiti quinti tendon, which was about to rupture, was further reinforced by suturing it side to side to the muscle and tendon of the extensor indicis proprius (Figure 2).

Postoperatively, the wrist was kept in extension in a cast for 3 weeks while the fingers were free for active movement. A removable wrist splint was used for an additional month. At 3-month follow-up, the patient had regained full and strong finger extension and wrist motion.
At 3-year follow-up, the patient was pain-free, and had full extension of all fingers, full forearm rotation, and near-normal motion (better than her preoperative motion). The grip power on the operated right hand was 215 N, and pinch power was 93 N. (The values for the left side were 254 N and 83 N, respectively, using the Jamar hydraulic hand dynamometer [Patterson Medical].) The patient has had no additional tendon rupture (Figure 3).
Discussion
Madelung deformity was first described by Madelung in 1878 and several cases have reported this deformity. However, extensor tendon rupture caused by Madelung deformity is very rare, reported in few cases.1
Extensor tendon rupture caused by chronic Madelung deformity has been reported few times in the English literature. Goodwin1 apparently published the first report of such an occurrence in 1979. Ducloyer and colleagues2 from France reported 6 cases of extensor tendon rupture as a result of inferior distal radioulnar joint deformity of Madelung. Jebson and colleagues3 reported bilateral spontaneous extensor tendon ruptures in Madelung deformity in 1992.
The mechanism of tendon rupture seems to be mechanical, resulting from continuous rubbing and erosion of tendons over the deformed ulnar head, which has a rough irregular surface4 and leads to fraying of the tendons and eventual rupture and retraction of the severed tendon ends. This rupture usually progresses stepwise from more medial to the lateral tendons.2 Older patients are, therefore, subject to chronic repetitive attritional trauma leading to tendon rupture.
Tendons may rupture as a result of a variety of conditions, such as chronic synovitis in rheumatoid arthritis, systemic lupus erythematosus, mixed connective tissue disease, or crystal deposition in gout.5-8 Some other metabolic or endocrine conditions that involve tendon ruptures include diabetes mellitus, chronic renal failure, and hyperparathyroidism. Steroid injection into the tendons also has a detrimental effect on tendon integrity and may cause tendon tear.9 Mechanical factors, such as erosion on bony prominences, are well-known etiologies for tendon rupture, as commonly seen in rheumatoid arthritis, and have been reported in Kienböck disease,10 thumb carpometacarpal arthritis,11 Colles fracture, scaphoid fracture nonunion,12 and Madelung deformity.
Conclusion
Our case reflects the usual middle-aged female presentation of such a tendon rupture. The tendon ruptures were spontaneous in the reported order of ulnar to radial, beginning with the little and ring fingers, and progressed radially. The patient had isolated Madelung deformity with no other sign of dyschondrosteosis13 or dwarfism, conditions commonly mentioned in association with Madelung deformity. This case report should raise awareness about possible tendon rupture in any chronic case of Madelung deformity.
1. Goodwin DR, Michels CH, Weissman SL. Spontaneous rupture of extensor tendons in Madelung’s deformity. Hand. 1979;11(1):72-75.
2. Ducloyer P, Leclercq C, Lisfrance R, Saffar P. Spontaneous rupture of the extensor tendons of the fingers in Madelung’s deformity. J Hand Surg Br. 1991;16(3):329-333.
3. Jebson PJ, Blair WF. Bilateral spontaneous extensor tendon ruptures in Madelung’s deformity. J Hand Surg Am. 1992;17(2):277-280.
4. Schulstad I. Madelung’s deformity with extensor tendon rupture. Case report. Scand J Plast Reconstr Surg. 1971;5(2):153-155.
5. Gong HS, Lee JO, Baek GH, et al. Extensor tendon rupture in rheumatoid arthritis: a survey of patients between 2005 and 2010 at five Korean hospitals. Hand Surg. 2012;17(1):43-47.
6. Oishi H, Oda R, Morisaki S, Fujiwara H, Tokunaga D, Kubo T. Spontaneous tendon rupture of the extensor digitrum communis in systemic lupus erythematosus. Mod Rheumatol. 2013;23(3);608-610.
7. Kobayashi A, Futami T, Tadano I, Fujita M. Spontaneous rupture of extensor tendons at the wrist in a patient with mixed connective tissue disease. Mod Rheumatol. 2002;12(3):256-258.
8. Iwamoto T, Toki H, Ikari K, Yamanaka H, Momohara S. Multiple extensor tendon ruptures caused by tophaceous gout. Mod Rheumatol. 2010;20(2):210-212.
9. Nquyen ML, Jones NF. Rupture of both abductor pollicis longus and extensor pollicis brevis tendon after steroid injection for de quervain tenosynovitis. Plast Reconstr Surg. 2012;129(5):883e-886e.
10. Hernández-Cortés P, Pajares-López M, Gómez-Sánchez R, Garrido-Gómez, Lara-Garcia F. Rupture of extensor tendon secondary to previously undiagnosed Kienböck disease. J Plast Surg Hand Surg. 2012;46(3-4):291-293.
11. Apard T, Marcucci L, Jarriges J. Spontaneous rupture of extensor pollicis longus in isolated trapeziometacarpal arthritis. Chir Main. 2011;30(5):349-351.
12. Harvey FJ, Harvey PM. Three rare causes of extensor tendon rupture. J Hand Surg Am. 1989;14(6):957-962.
13. Duro EA, Prado GS. Clinical variations in Léri-Weill dyschondrosteosis. An Esp Pediatr. 1990;33(5):461-463.
Extensor tendon rupture in chronic Madelung deformity, as a result of tendon attrition on the dislocated distal ulna, occurs infrequently. However, it is often seen in patients with rheumatoid arthritis. This issue has been reported in only a few English-language case reports. Here we report a case of multiple tendon ruptures in a previously undiagnosed Madelung deformity. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 56-year-old active woman presented with 50 days’ inability to extend the fourth and fifth fingers of her dominant right hand. The loss of finger extension progressed, over several weeks, to involve the third finger as well. The first 2 tendon ruptures had been triggered by lifting a light grocery bag, when she noticed a sharp sudden pain and “pop.” The third rupture occurred spontaneously with a snapping sound the night before surgery.
The patient had observed some prominence on the ulnar side of her right wrist since childhood but had never experienced any pain or functional disability. There was neither history of trauma, inflammatory disease, diabetes mellitus, or infection, nor positive family history of similar wrist deformity.
The physical examination showed a dorsally subluxated distal radioulnar joint, prominent ulnar styloid, and mild ulnar and volar deviation of the wrist along with limitation of wrist dorsiflexion. Complete loss of active extension of the 3 ulnar fingers was demonstrated, while neurovascular status and all other hand evaluations were normal. The wrist radiographs confirmed the typical findings of Madelung deformity (Figure 1).
Repair of the ruptured tendons and resection of the prominent distal ulna (Darrach procedure) was planned. (Given the patient’s age and evidence of degenerative changes in the radiocarpal joint, correction of the Madelung deformity did not seem necessary). At time of surgery, the recently ruptured third finger extensor tendon was easily found and approximated, and end-to-end repair was performed. The fourth and fifth fingers, however, had to be fished out more proximally from dense granulation tissue. After the distal ulna was resected for a distance of 1.5 cm, meticulous repair of the ulnar collateral ligament and the capsule and periosteum over the end of the ulna was performed. Then, for grafting of the ruptured tendons, the extensor indicis proprius tendon was isolated and transected at the second metacarpophalangeal joint level. A piece of this tendon was used as interpositional graft for the fourth extensor tendon, and the main tendon unit was transferred to the fifth finger extensor. The extensor digiti quinti tendon, which was about to rupture, was further reinforced by suturing it side to side to the muscle and tendon of the extensor indicis proprius (Figure 2).
Postoperatively, the wrist was kept in extension in a cast for 3 weeks while the fingers were free for active movement. A removable wrist splint was used for an additional month. At 3-month follow-up, the patient had regained full and strong finger extension and wrist motion.
At 3-year follow-up, the patient was pain-free, and had full extension of all fingers, full forearm rotation, and near-normal motion (better than her preoperative motion). The grip power on the operated right hand was 215 N, and pinch power was 93 N. (The values for the left side were 254 N and 83 N, respectively, using the Jamar hydraulic hand dynamometer [Patterson Medical].) The patient has had no additional tendon rupture (Figure 3).
Discussion
Madelung deformity was first described by Madelung in 1878 and several cases have reported this deformity. However, extensor tendon rupture caused by Madelung deformity is very rare, reported in few cases.1
Extensor tendon rupture caused by chronic Madelung deformity has been reported few times in the English literature. Goodwin1 apparently published the first report of such an occurrence in 1979. Ducloyer and colleagues2 from France reported 6 cases of extensor tendon rupture as a result of inferior distal radioulnar joint deformity of Madelung. Jebson and colleagues3 reported bilateral spontaneous extensor tendon ruptures in Madelung deformity in 1992.
The mechanism of tendon rupture seems to be mechanical, resulting from continuous rubbing and erosion of tendons over the deformed ulnar head, which has a rough irregular surface4 and leads to fraying of the tendons and eventual rupture and retraction of the severed tendon ends. This rupture usually progresses stepwise from more medial to the lateral tendons.2 Older patients are, therefore, subject to chronic repetitive attritional trauma leading to tendon rupture.
Tendons may rupture as a result of a variety of conditions, such as chronic synovitis in rheumatoid arthritis, systemic lupus erythematosus, mixed connective tissue disease, or crystal deposition in gout.5-8 Some other metabolic or endocrine conditions that involve tendon ruptures include diabetes mellitus, chronic renal failure, and hyperparathyroidism. Steroid injection into the tendons also has a detrimental effect on tendon integrity and may cause tendon tear.9 Mechanical factors, such as erosion on bony prominences, are well-known etiologies for tendon rupture, as commonly seen in rheumatoid arthritis, and have been reported in Kienböck disease,10 thumb carpometacarpal arthritis,11 Colles fracture, scaphoid fracture nonunion,12 and Madelung deformity.
Conclusion
Our case reflects the usual middle-aged female presentation of such a tendon rupture. The tendon ruptures were spontaneous in the reported order of ulnar to radial, beginning with the little and ring fingers, and progressed radially. The patient had isolated Madelung deformity with no other sign of dyschondrosteosis13 or dwarfism, conditions commonly mentioned in association with Madelung deformity. This case report should raise awareness about possible tendon rupture in any chronic case of Madelung deformity.
Extensor tendon rupture in chronic Madelung deformity, as a result of tendon attrition on the dislocated distal ulna, occurs infrequently. However, it is often seen in patients with rheumatoid arthritis. This issue has been reported in only a few English-language case reports. Here we report a case of multiple tendon ruptures in a previously undiagnosed Madelung deformity. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 56-year-old active woman presented with 50 days’ inability to extend the fourth and fifth fingers of her dominant right hand. The loss of finger extension progressed, over several weeks, to involve the third finger as well. The first 2 tendon ruptures had been triggered by lifting a light grocery bag, when she noticed a sharp sudden pain and “pop.” The third rupture occurred spontaneously with a snapping sound the night before surgery.
The patient had observed some prominence on the ulnar side of her right wrist since childhood but had never experienced any pain or functional disability. There was neither history of trauma, inflammatory disease, diabetes mellitus, or infection, nor positive family history of similar wrist deformity.
The physical examination showed a dorsally subluxated distal radioulnar joint, prominent ulnar styloid, and mild ulnar and volar deviation of the wrist along with limitation of wrist dorsiflexion. Complete loss of active extension of the 3 ulnar fingers was demonstrated, while neurovascular status and all other hand evaluations were normal. The wrist radiographs confirmed the typical findings of Madelung deformity (Figure 1).
Repair of the ruptured tendons and resection of the prominent distal ulna (Darrach procedure) was planned. (Given the patient’s age and evidence of degenerative changes in the radiocarpal joint, correction of the Madelung deformity did not seem necessary). At time of surgery, the recently ruptured third finger extensor tendon was easily found and approximated, and end-to-end repair was performed. The fourth and fifth fingers, however, had to be fished out more proximally from dense granulation tissue. After the distal ulna was resected for a distance of 1.5 cm, meticulous repair of the ulnar collateral ligament and the capsule and periosteum over the end of the ulna was performed. Then, for grafting of the ruptured tendons, the extensor indicis proprius tendon was isolated and transected at the second metacarpophalangeal joint level. A piece of this tendon was used as interpositional graft for the fourth extensor tendon, and the main tendon unit was transferred to the fifth finger extensor. The extensor digiti quinti tendon, which was about to rupture, was further reinforced by suturing it side to side to the muscle and tendon of the extensor indicis proprius (Figure 2).
Postoperatively, the wrist was kept in extension in a cast for 3 weeks while the fingers were free for active movement. A removable wrist splint was used for an additional month. At 3-month follow-up, the patient had regained full and strong finger extension and wrist motion.
At 3-year follow-up, the patient was pain-free, and had full extension of all fingers, full forearm rotation, and near-normal motion (better than her preoperative motion). The grip power on the operated right hand was 215 N, and pinch power was 93 N. (The values for the left side were 254 N and 83 N, respectively, using the Jamar hydraulic hand dynamometer [Patterson Medical].) The patient has had no additional tendon rupture (Figure 3).
Discussion
Madelung deformity was first described by Madelung in 1878 and several cases have reported this deformity. However, extensor tendon rupture caused by Madelung deformity is very rare, reported in few cases.1
Extensor tendon rupture caused by chronic Madelung deformity has been reported few times in the English literature. Goodwin1 apparently published the first report of such an occurrence in 1979. Ducloyer and colleagues2 from France reported 6 cases of extensor tendon rupture as a result of inferior distal radioulnar joint deformity of Madelung. Jebson and colleagues3 reported bilateral spontaneous extensor tendon ruptures in Madelung deformity in 1992.
The mechanism of tendon rupture seems to be mechanical, resulting from continuous rubbing and erosion of tendons over the deformed ulnar head, which has a rough irregular surface4 and leads to fraying of the tendons and eventual rupture and retraction of the severed tendon ends. This rupture usually progresses stepwise from more medial to the lateral tendons.2 Older patients are, therefore, subject to chronic repetitive attritional trauma leading to tendon rupture.
Tendons may rupture as a result of a variety of conditions, such as chronic synovitis in rheumatoid arthritis, systemic lupus erythematosus, mixed connective tissue disease, or crystal deposition in gout.5-8 Some other metabolic or endocrine conditions that involve tendon ruptures include diabetes mellitus, chronic renal failure, and hyperparathyroidism. Steroid injection into the tendons also has a detrimental effect on tendon integrity and may cause tendon tear.9 Mechanical factors, such as erosion on bony prominences, are well-known etiologies for tendon rupture, as commonly seen in rheumatoid arthritis, and have been reported in Kienböck disease,10 thumb carpometacarpal arthritis,11 Colles fracture, scaphoid fracture nonunion,12 and Madelung deformity.
Conclusion
Our case reflects the usual middle-aged female presentation of such a tendon rupture. The tendon ruptures were spontaneous in the reported order of ulnar to radial, beginning with the little and ring fingers, and progressed radially. The patient had isolated Madelung deformity with no other sign of dyschondrosteosis13 or dwarfism, conditions commonly mentioned in association with Madelung deformity. This case report should raise awareness about possible tendon rupture in any chronic case of Madelung deformity.
1. Goodwin DR, Michels CH, Weissman SL. Spontaneous rupture of extensor tendons in Madelung’s deformity. Hand. 1979;11(1):72-75.
2. Ducloyer P, Leclercq C, Lisfrance R, Saffar P. Spontaneous rupture of the extensor tendons of the fingers in Madelung’s deformity. J Hand Surg Br. 1991;16(3):329-333.
3. Jebson PJ, Blair WF. Bilateral spontaneous extensor tendon ruptures in Madelung’s deformity. J Hand Surg Am. 1992;17(2):277-280.
4. Schulstad I. Madelung’s deformity with extensor tendon rupture. Case report. Scand J Plast Reconstr Surg. 1971;5(2):153-155.
5. Gong HS, Lee JO, Baek GH, et al. Extensor tendon rupture in rheumatoid arthritis: a survey of patients between 2005 and 2010 at five Korean hospitals. Hand Surg. 2012;17(1):43-47.
6. Oishi H, Oda R, Morisaki S, Fujiwara H, Tokunaga D, Kubo T. Spontaneous tendon rupture of the extensor digitrum communis in systemic lupus erythematosus. Mod Rheumatol. 2013;23(3);608-610.
7. Kobayashi A, Futami T, Tadano I, Fujita M. Spontaneous rupture of extensor tendons at the wrist in a patient with mixed connective tissue disease. Mod Rheumatol. 2002;12(3):256-258.
8. Iwamoto T, Toki H, Ikari K, Yamanaka H, Momohara S. Multiple extensor tendon ruptures caused by tophaceous gout. Mod Rheumatol. 2010;20(2):210-212.
9. Nquyen ML, Jones NF. Rupture of both abductor pollicis longus and extensor pollicis brevis tendon after steroid injection for de quervain tenosynovitis. Plast Reconstr Surg. 2012;129(5):883e-886e.
10. Hernández-Cortés P, Pajares-López M, Gómez-Sánchez R, Garrido-Gómez, Lara-Garcia F. Rupture of extensor tendon secondary to previously undiagnosed Kienböck disease. J Plast Surg Hand Surg. 2012;46(3-4):291-293.
11. Apard T, Marcucci L, Jarriges J. Spontaneous rupture of extensor pollicis longus in isolated trapeziometacarpal arthritis. Chir Main. 2011;30(5):349-351.
12. Harvey FJ, Harvey PM. Three rare causes of extensor tendon rupture. J Hand Surg Am. 1989;14(6):957-962.
13. Duro EA, Prado GS. Clinical variations in Léri-Weill dyschondrosteosis. An Esp Pediatr. 1990;33(5):461-463.
1. Goodwin DR, Michels CH, Weissman SL. Spontaneous rupture of extensor tendons in Madelung’s deformity. Hand. 1979;11(1):72-75.
2. Ducloyer P, Leclercq C, Lisfrance R, Saffar P. Spontaneous rupture of the extensor tendons of the fingers in Madelung’s deformity. J Hand Surg Br. 1991;16(3):329-333.
3. Jebson PJ, Blair WF. Bilateral spontaneous extensor tendon ruptures in Madelung’s deformity. J Hand Surg Am. 1992;17(2):277-280.
4. Schulstad I. Madelung’s deformity with extensor tendon rupture. Case report. Scand J Plast Reconstr Surg. 1971;5(2):153-155.
5. Gong HS, Lee JO, Baek GH, et al. Extensor tendon rupture in rheumatoid arthritis: a survey of patients between 2005 and 2010 at five Korean hospitals. Hand Surg. 2012;17(1):43-47.
6. Oishi H, Oda R, Morisaki S, Fujiwara H, Tokunaga D, Kubo T. Spontaneous tendon rupture of the extensor digitrum communis in systemic lupus erythematosus. Mod Rheumatol. 2013;23(3);608-610.
7. Kobayashi A, Futami T, Tadano I, Fujita M. Spontaneous rupture of extensor tendons at the wrist in a patient with mixed connective tissue disease. Mod Rheumatol. 2002;12(3):256-258.
8. Iwamoto T, Toki H, Ikari K, Yamanaka H, Momohara S. Multiple extensor tendon ruptures caused by tophaceous gout. Mod Rheumatol. 2010;20(2):210-212.
9. Nquyen ML, Jones NF. Rupture of both abductor pollicis longus and extensor pollicis brevis tendon after steroid injection for de quervain tenosynovitis. Plast Reconstr Surg. 2012;129(5):883e-886e.
10. Hernández-Cortés P, Pajares-López M, Gómez-Sánchez R, Garrido-Gómez, Lara-Garcia F. Rupture of extensor tendon secondary to previously undiagnosed Kienböck disease. J Plast Surg Hand Surg. 2012;46(3-4):291-293.
11. Apard T, Marcucci L, Jarriges J. Spontaneous rupture of extensor pollicis longus in isolated trapeziometacarpal arthritis. Chir Main. 2011;30(5):349-351.
12. Harvey FJ, Harvey PM. Three rare causes of extensor tendon rupture. J Hand Surg Am. 1989;14(6):957-962.
13. Duro EA, Prado GS. Clinical variations in Léri-Weill dyschondrosteosis. An Esp Pediatr. 1990;33(5):461-463.