User login
Squamoid Eccrine Ductal Carcinoma
Eccrine carcinomas are uncommon cutaneous neoplasms demonstrating nonuniform histologic features, behavior, and nomenclature. Given the rarity of these tumors, no known criteria by which to diagnose the tumor or guidelines for treatment have been proposed. We report a rare case of an immunocompromised patient with a primary squamoid eccrine ductal carcinoma (SEDC) who was subsequently treated with radical resection and axillary dissection. It was later determined that the patient had distant metastasis of SEDC. A review of the literature on the diagnosis, treatment, and surveillance of SEDC also is provided.
Case Report
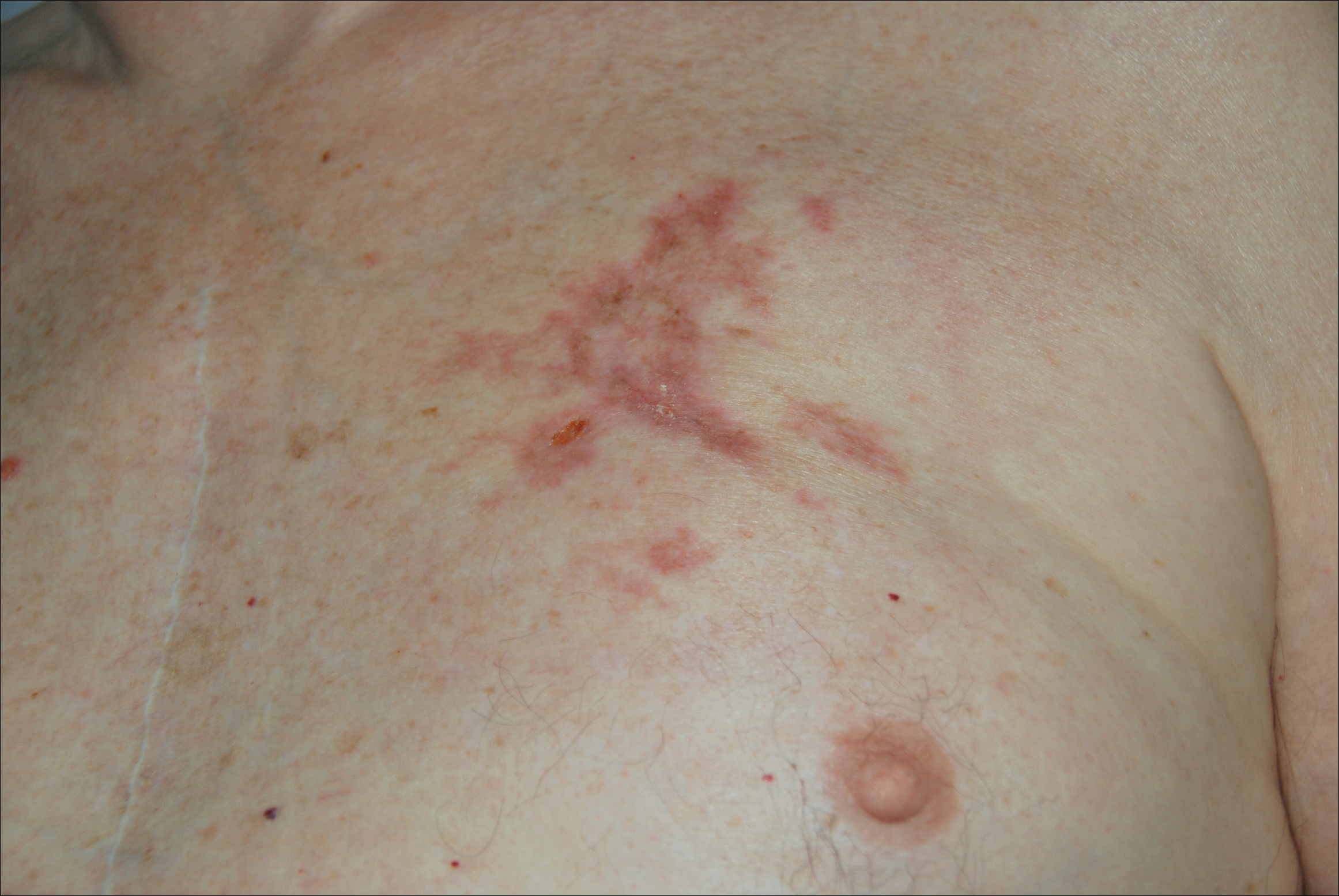
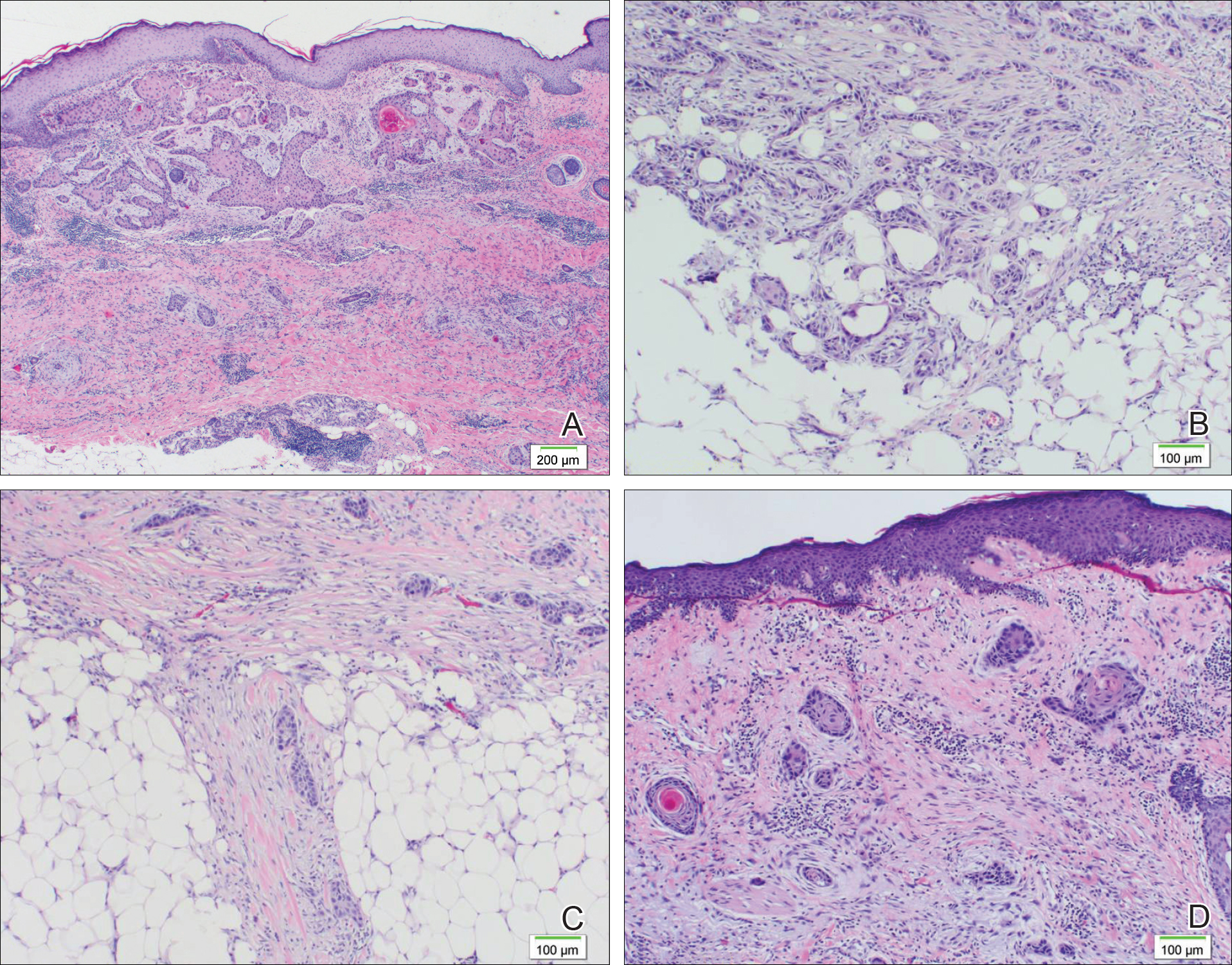
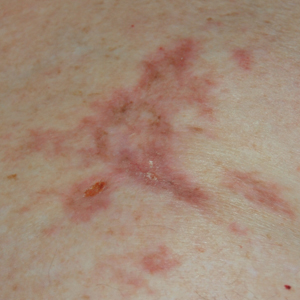
A 77-year-old man whose medical history was remarkable for chronic lymphocytic leukemia (CLL) and numerous previous basal cell carcinomas and squamous cell carcinomas (SCCs) presented with a 5-cm, stellate, sclerotic plaque on the left chest of approximately 2 years’ duration (Figure 1) and a 3-mm pink papule on the right nasal sidewall of 2 months’ duration. Initial histology of both lesions revealed carcinoma with squamous and ductal differentiation extending from the undersurface of the epidermis, favoring a diagnosis of SEDC (Figure 2). At the time of initial presentation, the patient also had a 6-mm pink papule on the right chest of several months duration that was consistent with a well-differentiated sebaceous carcinoma on histology.
Further analysis of the lesion on the left chest revealed positive staining for cytokeratin (CK) 5/14 and p63, suggestive of a cutaneous malignancy. Staining for S100 protein highlighted rare cells in the basal layer of tumor aggregates. The immunohistochemical profile showed negative staining for CK7, CK5D3, epithelial membrane antigen (EMA), estrogen receptor, progesterone receptor, and human epidermal growth factor 2.
Diagnosis of SEDC of the chest and nasal lesions was based on the morphologic architecture, which included ductal formation noted within the tumor. The chest lesion also had prominent squamoid differentiation. Another histologic feature consistent with SEDC was poorly demarcated, infiltrative neoplastic cells extending into the dermis and subcutis. Although there was some positive focal staining for carcinoembryonic antigen (CEA), variegation within the tumor and the prominent squamoid component might have contributed to this unexpected staining pattern.
The patient was admitted to the hospital for excision of the lesion on the chest wall. Initial workup revealed macrocytic anemia, which required transfusion, and an incidental finding of non–small-cell lung cancer. The chest lesion was unrelated to the non–small-cell lung cancer based on the staining profile. Material from the lung stained positive for thyroid transcription factor 1 (TTF-1) and exhibited rare staining for p63; however, the chest lesion did not stain positive for TTF-1 and had strong staining affinity for p63, indicative of a cutaneous malignancy.
The lesion on the chest wall was definitively excised. Pathologic analysis revealed a dermal-based infiltrative tumor of irregular nests and cords of squamoid cells with focal ductal formation in a fibromyxoid background stroma, suggestive of an adnexal carcinoma with a considerable degree of squamous differentiation and favoring a diagnosis of SEDC. Focal perineural invasion was noted, but no lymphovascular spread was identified; however, metastasis was identified in 1 of 26 axillary lymph nodes. The patient underwent 9 sessions of radiation therapy for the lung cancer and also was given cetuximab.
Three months later, the nasal tumor was subsequently excised in an outpatient procedure, and the final biopsy report indicated a diagnosis of basal cell carcinoma. One-and-a-half years later, in follow-up with surgery after removal of the chest lesion, a 2×3-cm mass was excised from the left neck that demonstrated lymph nodes consistent with metastatic SEDC. Careful evaluation of this patient, including family history and genetic screening, was considered. Our patient continues to follow-up with the dermatology department every 3 months. He has been doing well and has had multiple additional primary SCCs in the subsequent 5 years of follow-up.
Comment
Eccrine carcinoma is the most common subtype of adnexal carcinoma, representing 0.01% of all cutaneous tumors.1 S
Eccrine carcinoma is observed clinically as a slow-growing, nodular plaque on the scalp, arms, legs, or trunk in middle-aged and elderly individuals.1 Squamoid eccrine ductal carcinoma also has been reported in a young woman.5 Another immunocompromised patient was identified in the literature with a great toe lesion that showed follicular differentiation along with the usual SEDC features of squamoid and ductal differentiation.6 The etiology of SEDC is controversial but is thought to be an SCC arising from eccrine glands, a subtype of eccrine carcinoma with extensive squamoid differentiation, or a biphenotypic carcinoma.1,7
Histologically, SEDC is poorly circumscribed with an infiltrative growth pattern and deep extension into the dermis and subcutaneous tissue. The lesion is characterized by prominent squamous epithelial proliferation superficially with cellular atypia, keratinous cyst formation, squamous eddies, and eccrine ductal differentiation.1
The differential diagnosis of SEDC includes SCC; metastatic carcinoma with squamoid features; and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma, and porocarcinoma with squamous differentiation.1
Immunohistochemistry has a role in the diagnosis of SEDC. Findings include positive staining for S100 protein, EMA, CKs, and CEA. Glandular tissue stains positive for EMA and CEA, supporting an adnexal origin.1 Positivity for p63 and CK5/6 supports the conclusion that this is a primary cutaneous malignancy, not a metastatic disease.1
Squamoid eccrine ductal carcinoma has an indeterminate malignant potential. There is a disparity of clinical behavior between SCC and eccrine cancers; however, because squamous differentiation sometimes dominates the histological picture, eccrine carcinomas can be misdiagnosed as SCC.1,8 Eccrine adnexal tumors are characterized by multiple local recurrences (70%–80% of cases); perineural invasion; and metastasis (50% of cases) to regional lymph nodes and viscera, including the lungs, liver, bones, and brain.1 Squamous cell carcinoma, however, has a markedly lower recurrence rate (3.1%–18.7% of cases) and rate of metastasis (5.2%–37.8%).1
Squamoid eccrine ductal carcinoma is classified as one of the less aggressive eccrine tumors, although the low number of cases makes it a controversial conclusion.1 To our knowledge, no cases of SEDC metastasis have been reported with SEDC. Recurrence of SEDC has been reported locally, and perineural or perivascular invasion (or both) has been demonstrated in 3 cases.1
Since SEDC has invasive and metastatic potential, as demonstrated in our case, along with elevated local recurrence rates, physicians must be able to properly diagnose this rare entity and recommend an appropriate surgical modality. Due to the low incidence of SEDC, there are no known randomized studies comparing treatment modalities.1 O
Surgical extirpation with complete margin examination is recommended, as SEDC tends to be underestimated in size, is aggressive in its infiltration, and is predisposed to perineural and perivascular invasion. T
Along with the rarity of SEDC in our patient, the simultaneous occurrence of 3 primary malignancies also is unusual. Patients with CLL have progressive defects of cell- and humoral-mediated immunity, causing immunosuppression. In a retrospective study, Tsimberidou et al9 reviewed the records of 2028 untreated CLL patients and determined that 27% had another primary malignancy, including skin (30%) and lung cancers (6%), which were two of the malignancies seen in our patient. The investigators concluded that patients with CLL have more than twice the risk of developing a second primary malignancy and an increased frequency of certain cancer types.9 Furthermore, treatment regimens for CLL have been considered to increase cell- and humoral-mediated immune defects at specific cancer sites,10 although the exact mechanism of this action is unknown. Development of a second primary malignancy (or even a third) in patients with SEDC is increasingly being reported in CLL patients.9,10
A high index of suspicion with SEDC in the differential diagnosis should be maintained in elderly men with slow-growing, solitary, nodular lesions of the scalp, nose, arms, legs, or trunk.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. Clin Aesthet Dermatol. 2013;6:33-36.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;916:799-802.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma)[published online April 15, 2015]. J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Kavand S, Cassarino DS. Squamoid eccrine ductal carcinoma: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Tsimberidou AM, Wen S, McLaughlin P, et al. Other malignancies in chronic lymphocytic leukemia/small lymphocytic lymphoma. J Clin Oncol. 2009;27:904-910.
- Dasanu CA, Alexandrescu DT. Risk for second nonlymphoid neoplasms in chronic lymphocytic leukemia. Med Gen Med. 2007;9:35.
Eccrine carcinomas are uncommon cutaneous neoplasms demonstrating nonuniform histologic features, behavior, and nomenclature. Given the rarity of these tumors, no known criteria by which to diagnose the tumor or guidelines for treatment have been proposed. We report a rare case of an immunocompromised patient with a primary squamoid eccrine ductal carcinoma (SEDC) who was subsequently treated with radical resection and axillary dissection. It was later determined that the patient had distant metastasis of SEDC. A review of the literature on the diagnosis, treatment, and surveillance of SEDC also is provided.
Case Report
A 77-year-old man whose medical history was remarkable for chronic lymphocytic leukemia (CLL) and numerous previous basal cell carcinomas and squamous cell carcinomas (SCCs) presented with a 5-cm, stellate, sclerotic plaque on the left chest of approximately 2 years’ duration (Figure 1) and a 3-mm pink papule on the right nasal sidewall of 2 months’ duration. Initial histology of both lesions revealed carcinoma with squamous and ductal differentiation extending from the undersurface of the epidermis, favoring a diagnosis of SEDC (Figure 2). At the time of initial presentation, the patient also had a 6-mm pink papule on the right chest of several months duration that was consistent with a well-differentiated sebaceous carcinoma on histology.
Further analysis of the lesion on the left chest revealed positive staining for cytokeratin (CK) 5/14 and p63, suggestive of a cutaneous malignancy. Staining for S100 protein highlighted rare cells in the basal layer of tumor aggregates. The immunohistochemical profile showed negative staining for CK7, CK5D3, epithelial membrane antigen (EMA), estrogen receptor, progesterone receptor, and human epidermal growth factor 2.
Diagnosis of SEDC of the chest and nasal lesions was based on the morphologic architecture, which included ductal formation noted within the tumor. The chest lesion also had prominent squamoid differentiation. Another histologic feature consistent with SEDC was poorly demarcated, infiltrative neoplastic cells extending into the dermis and subcutis. Although there was some positive focal staining for carcinoembryonic antigen (CEA), variegation within the tumor and the prominent squamoid component might have contributed to this unexpected staining pattern.
The patient was admitted to the hospital for excision of the lesion on the chest wall. Initial workup revealed macrocytic anemia, which required transfusion, and an incidental finding of non–small-cell lung cancer. The chest lesion was unrelated to the non–small-cell lung cancer based on the staining profile. Material from the lung stained positive for thyroid transcription factor 1 (TTF-1) and exhibited rare staining for p63; however, the chest lesion did not stain positive for TTF-1 and had strong staining affinity for p63, indicative of a cutaneous malignancy.
The lesion on the chest wall was definitively excised. Pathologic analysis revealed a dermal-based infiltrative tumor of irregular nests and cords of squamoid cells with focal ductal formation in a fibromyxoid background stroma, suggestive of an adnexal carcinoma with a considerable degree of squamous differentiation and favoring a diagnosis of SEDC. Focal perineural invasion was noted, but no lymphovascular spread was identified; however, metastasis was identified in 1 of 26 axillary lymph nodes. The patient underwent 9 sessions of radiation therapy for the lung cancer and also was given cetuximab.
Three months later, the nasal tumor was subsequently excised in an outpatient procedure, and the final biopsy report indicated a diagnosis of basal cell carcinoma. One-and-a-half years later, in follow-up with surgery after removal of the chest lesion, a 2×3-cm mass was excised from the left neck that demonstrated lymph nodes consistent with metastatic SEDC. Careful evaluation of this patient, including family history and genetic screening, was considered. Our patient continues to follow-up with the dermatology department every 3 months. He has been doing well and has had multiple additional primary SCCs in the subsequent 5 years of follow-up.
Comment
Eccrine carcinoma is the most common subtype of adnexal carcinoma, representing 0.01% of all cutaneous tumors.1 S
Eccrine carcinoma is observed clinically as a slow-growing, nodular plaque on the scalp, arms, legs, or trunk in middle-aged and elderly individuals.1 Squamoid eccrine ductal carcinoma also has been reported in a young woman.5 Another immunocompromised patient was identified in the literature with a great toe lesion that showed follicular differentiation along with the usual SEDC features of squamoid and ductal differentiation.6 The etiology of SEDC is controversial but is thought to be an SCC arising from eccrine glands, a subtype of eccrine carcinoma with extensive squamoid differentiation, or a biphenotypic carcinoma.1,7
Histologically, SEDC is poorly circumscribed with an infiltrative growth pattern and deep extension into the dermis and subcutaneous tissue. The lesion is characterized by prominent squamous epithelial proliferation superficially with cellular atypia, keratinous cyst formation, squamous eddies, and eccrine ductal differentiation.1
The differential diagnosis of SEDC includes SCC; metastatic carcinoma with squamoid features; and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma, and porocarcinoma with squamous differentiation.1
Immunohistochemistry has a role in the diagnosis of SEDC. Findings include positive staining for S100 protein, EMA, CKs, and CEA. Glandular tissue stains positive for EMA and CEA, supporting an adnexal origin.1 Positivity for p63 and CK5/6 supports the conclusion that this is a primary cutaneous malignancy, not a metastatic disease.1
Squamoid eccrine ductal carcinoma has an indeterminate malignant potential. There is a disparity of clinical behavior between SCC and eccrine cancers; however, because squamous differentiation sometimes dominates the histological picture, eccrine carcinomas can be misdiagnosed as SCC.1,8 Eccrine adnexal tumors are characterized by multiple local recurrences (70%–80% of cases); perineural invasion; and metastasis (50% of cases) to regional lymph nodes and viscera, including the lungs, liver, bones, and brain.1 Squamous cell carcinoma, however, has a markedly lower recurrence rate (3.1%–18.7% of cases) and rate of metastasis (5.2%–37.8%).1
Squamoid eccrine ductal carcinoma is classified as one of the less aggressive eccrine tumors, although the low number of cases makes it a controversial conclusion.1 To our knowledge, no cases of SEDC metastasis have been reported with SEDC. Recurrence of SEDC has been reported locally, and perineural or perivascular invasion (or both) has been demonstrated in 3 cases.1
Since SEDC has invasive and metastatic potential, as demonstrated in our case, along with elevated local recurrence rates, physicians must be able to properly diagnose this rare entity and recommend an appropriate surgical modality. Due to the low incidence of SEDC, there are no known randomized studies comparing treatment modalities.1 O
Surgical extirpation with complete margin examination is recommended, as SEDC tends to be underestimated in size, is aggressive in its infiltration, and is predisposed to perineural and perivascular invasion. T
Along with the rarity of SEDC in our patient, the simultaneous occurrence of 3 primary malignancies also is unusual. Patients with CLL have progressive defects of cell- and humoral-mediated immunity, causing immunosuppression. In a retrospective study, Tsimberidou et al9 reviewed the records of 2028 untreated CLL patients and determined that 27% had another primary malignancy, including skin (30%) and lung cancers (6%), which were two of the malignancies seen in our patient. The investigators concluded that patients with CLL have more than twice the risk of developing a second primary malignancy and an increased frequency of certain cancer types.9 Furthermore, treatment regimens for CLL have been considered to increase cell- and humoral-mediated immune defects at specific cancer sites,10 although the exact mechanism of this action is unknown. Development of a second primary malignancy (or even a third) in patients with SEDC is increasingly being reported in CLL patients.9,10
A high index of suspicion with SEDC in the differential diagnosis should be maintained in elderly men with slow-growing, solitary, nodular lesions of the scalp, nose, arms, legs, or trunk.
Eccrine carcinomas are uncommon cutaneous neoplasms demonstrating nonuniform histologic features, behavior, and nomenclature. Given the rarity of these tumors, no known criteria by which to diagnose the tumor or guidelines for treatment have been proposed. We report a rare case of an immunocompromised patient with a primary squamoid eccrine ductal carcinoma (SEDC) who was subsequently treated with radical resection and axillary dissection. It was later determined that the patient had distant metastasis of SEDC. A review of the literature on the diagnosis, treatment, and surveillance of SEDC also is provided.
Case Report
A 77-year-old man whose medical history was remarkable for chronic lymphocytic leukemia (CLL) and numerous previous basal cell carcinomas and squamous cell carcinomas (SCCs) presented with a 5-cm, stellate, sclerotic plaque on the left chest of approximately 2 years’ duration (Figure 1) and a 3-mm pink papule on the right nasal sidewall of 2 months’ duration. Initial histology of both lesions revealed carcinoma with squamous and ductal differentiation extending from the undersurface of the epidermis, favoring a diagnosis of SEDC (Figure 2). At the time of initial presentation, the patient also had a 6-mm pink papule on the right chest of several months duration that was consistent with a well-differentiated sebaceous carcinoma on histology.
Further analysis of the lesion on the left chest revealed positive staining for cytokeratin (CK) 5/14 and p63, suggestive of a cutaneous malignancy. Staining for S100 protein highlighted rare cells in the basal layer of tumor aggregates. The immunohistochemical profile showed negative staining for CK7, CK5D3, epithelial membrane antigen (EMA), estrogen receptor, progesterone receptor, and human epidermal growth factor 2.
Diagnosis of SEDC of the chest and nasal lesions was based on the morphologic architecture, which included ductal formation noted within the tumor. The chest lesion also had prominent squamoid differentiation. Another histologic feature consistent with SEDC was poorly demarcated, infiltrative neoplastic cells extending into the dermis and subcutis. Although there was some positive focal staining for carcinoembryonic antigen (CEA), variegation within the tumor and the prominent squamoid component might have contributed to this unexpected staining pattern.
The patient was admitted to the hospital for excision of the lesion on the chest wall. Initial workup revealed macrocytic anemia, which required transfusion, and an incidental finding of non–small-cell lung cancer. The chest lesion was unrelated to the non–small-cell lung cancer based on the staining profile. Material from the lung stained positive for thyroid transcription factor 1 (TTF-1) and exhibited rare staining for p63; however, the chest lesion did not stain positive for TTF-1 and had strong staining affinity for p63, indicative of a cutaneous malignancy.
The lesion on the chest wall was definitively excised. Pathologic analysis revealed a dermal-based infiltrative tumor of irregular nests and cords of squamoid cells with focal ductal formation in a fibromyxoid background stroma, suggestive of an adnexal carcinoma with a considerable degree of squamous differentiation and favoring a diagnosis of SEDC. Focal perineural invasion was noted, but no lymphovascular spread was identified; however, metastasis was identified in 1 of 26 axillary lymph nodes. The patient underwent 9 sessions of radiation therapy for the lung cancer and also was given cetuximab.
Three months later, the nasal tumor was subsequently excised in an outpatient procedure, and the final biopsy report indicated a diagnosis of basal cell carcinoma. One-and-a-half years later, in follow-up with surgery after removal of the chest lesion, a 2×3-cm mass was excised from the left neck that demonstrated lymph nodes consistent with metastatic SEDC. Careful evaluation of this patient, including family history and genetic screening, was considered. Our patient continues to follow-up with the dermatology department every 3 months. He has been doing well and has had multiple additional primary SCCs in the subsequent 5 years of follow-up.
Comment
Eccrine carcinoma is the most common subtype of adnexal carcinoma, representing 0.01% of all cutaneous tumors.1 S
Eccrine carcinoma is observed clinically as a slow-growing, nodular plaque on the scalp, arms, legs, or trunk in middle-aged and elderly individuals.1 Squamoid eccrine ductal carcinoma also has been reported in a young woman.5 Another immunocompromised patient was identified in the literature with a great toe lesion that showed follicular differentiation along with the usual SEDC features of squamoid and ductal differentiation.6 The etiology of SEDC is controversial but is thought to be an SCC arising from eccrine glands, a subtype of eccrine carcinoma with extensive squamoid differentiation, or a biphenotypic carcinoma.1,7
Histologically, SEDC is poorly circumscribed with an infiltrative growth pattern and deep extension into the dermis and subcutaneous tissue. The lesion is characterized by prominent squamous epithelial proliferation superficially with cellular atypia, keratinous cyst formation, squamous eddies, and eccrine ductal differentiation.1
The differential diagnosis of SEDC includes SCC; metastatic carcinoma with squamoid features; and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma, and porocarcinoma with squamous differentiation.1
Immunohistochemistry has a role in the diagnosis of SEDC. Findings include positive staining for S100 protein, EMA, CKs, and CEA. Glandular tissue stains positive for EMA and CEA, supporting an adnexal origin.1 Positivity for p63 and CK5/6 supports the conclusion that this is a primary cutaneous malignancy, not a metastatic disease.1
Squamoid eccrine ductal carcinoma has an indeterminate malignant potential. There is a disparity of clinical behavior between SCC and eccrine cancers; however, because squamous differentiation sometimes dominates the histological picture, eccrine carcinomas can be misdiagnosed as SCC.1,8 Eccrine adnexal tumors are characterized by multiple local recurrences (70%–80% of cases); perineural invasion; and metastasis (50% of cases) to regional lymph nodes and viscera, including the lungs, liver, bones, and brain.1 Squamous cell carcinoma, however, has a markedly lower recurrence rate (3.1%–18.7% of cases) and rate of metastasis (5.2%–37.8%).1
Squamoid eccrine ductal carcinoma is classified as one of the less aggressive eccrine tumors, although the low number of cases makes it a controversial conclusion.1 To our knowledge, no cases of SEDC metastasis have been reported with SEDC. Recurrence of SEDC has been reported locally, and perineural or perivascular invasion (or both) has been demonstrated in 3 cases.1
Since SEDC has invasive and metastatic potential, as demonstrated in our case, along with elevated local recurrence rates, physicians must be able to properly diagnose this rare entity and recommend an appropriate surgical modality. Due to the low incidence of SEDC, there are no known randomized studies comparing treatment modalities.1 O
Surgical extirpation with complete margin examination is recommended, as SEDC tends to be underestimated in size, is aggressive in its infiltration, and is predisposed to perineural and perivascular invasion. T
Along with the rarity of SEDC in our patient, the simultaneous occurrence of 3 primary malignancies also is unusual. Patients with CLL have progressive defects of cell- and humoral-mediated immunity, causing immunosuppression. In a retrospective study, Tsimberidou et al9 reviewed the records of 2028 untreated CLL patients and determined that 27% had another primary malignancy, including skin (30%) and lung cancers (6%), which were two of the malignancies seen in our patient. The investigators concluded that patients with CLL have more than twice the risk of developing a second primary malignancy and an increased frequency of certain cancer types.9 Furthermore, treatment regimens for CLL have been considered to increase cell- and humoral-mediated immune defects at specific cancer sites,10 although the exact mechanism of this action is unknown. Development of a second primary malignancy (or even a third) in patients with SEDC is increasingly being reported in CLL patients.9,10
A high index of suspicion with SEDC in the differential diagnosis should be maintained in elderly men with slow-growing, solitary, nodular lesions of the scalp, nose, arms, legs, or trunk.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. Clin Aesthet Dermatol. 2013;6:33-36.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;916:799-802.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma)[published online April 15, 2015]. J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Kavand S, Cassarino DS. Squamoid eccrine ductal carcinoma: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Tsimberidou AM, Wen S, McLaughlin P, et al. Other malignancies in chronic lymphocytic leukemia/small lymphocytic lymphoma. J Clin Oncol. 2009;27:904-910.
- Dasanu CA, Alexandrescu DT. Risk for second nonlymphoid neoplasms in chronic lymphocytic leukemia. Med Gen Med. 2007;9:35.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. Clin Aesthet Dermatol. 2013;6:33-36.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;916:799-802.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma)[published online April 15, 2015]. J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Kavand S, Cassarino DS. Squamoid eccrine ductal carcinoma: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Tsimberidou AM, Wen S, McLaughlin P, et al. Other malignancies in chronic lymphocytic leukemia/small lymphocytic lymphoma. J Clin Oncol. 2009;27:904-910.
- Dasanu CA, Alexandrescu DT. Risk for second nonlymphoid neoplasms in chronic lymphocytic leukemia. Med Gen Med. 2007;9:35.
Practice Points
- Squamoid eccrine ductal carcinoma (SEDC) is an extremely rare cutaneous tumor of unknown etiology.
- A high index of suspicion with SEDC in the differential diagnosis should be maintained in elderly men with slow-growing, solitary, nodular lesions of the scalp, nose, arms, legs, or trunk.
- Development of a second or even a third primary malignancy in patients with SEDC is increasingly being reported in CLL patients.
Pigmented Squamous Cell Carcinoma Presenting as Longitudinal Melanonychia in a Transplant Recipient
Case Report
A 62-year-old black man presented for examination of a dark longitudinal streak located adjacent to the lateral nail fold on the third finger of the left hand. The lesion had been present for several months, during which time it had slowly expanded in size. The fingertip had recently become tender, which interfered with the patient’s ability to work. His past medical history was remarkable for end-stage renal disease secondary to glomerulonephritis with nephrotic syndrome of unclear etiology. He initially was treated by an outside physician using peritoneal dialysis for 3 years until he underwent renal transplantation in 2004 with a cadaveric organ. Other remarkable medical conditions included posttransplantation diabetes, hyperlipidemia, and gout. His multidrug regimen included 2 immunosuppressive medications: oral cyclosporine 125 mg twice daily and oral mycophenolate mofetil 250 mg twice daily.
A broad, irregular, black, pigmented, subungual band was noted on the left third finger. The lesion appeared to emanate from below the nail cuticle and traveled along the nail longitudinally toward the distal tip. The band appeared darker at the edge adjacent to the lateral nail fold and grew lighter near the middle of the nail where its free edge was noted to be irregular. A slightly thickened lateral nail fold with an irregular, small, sawtoothlike hyperkeratosis and hyperpigmentation also was noted (Figure 1).
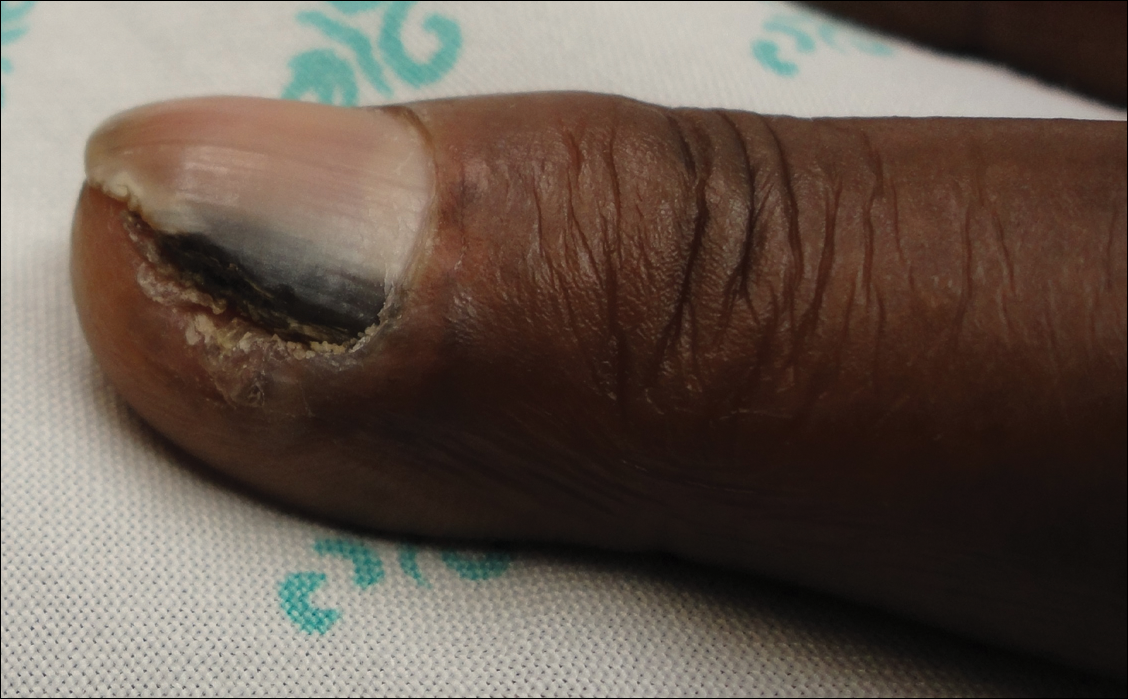
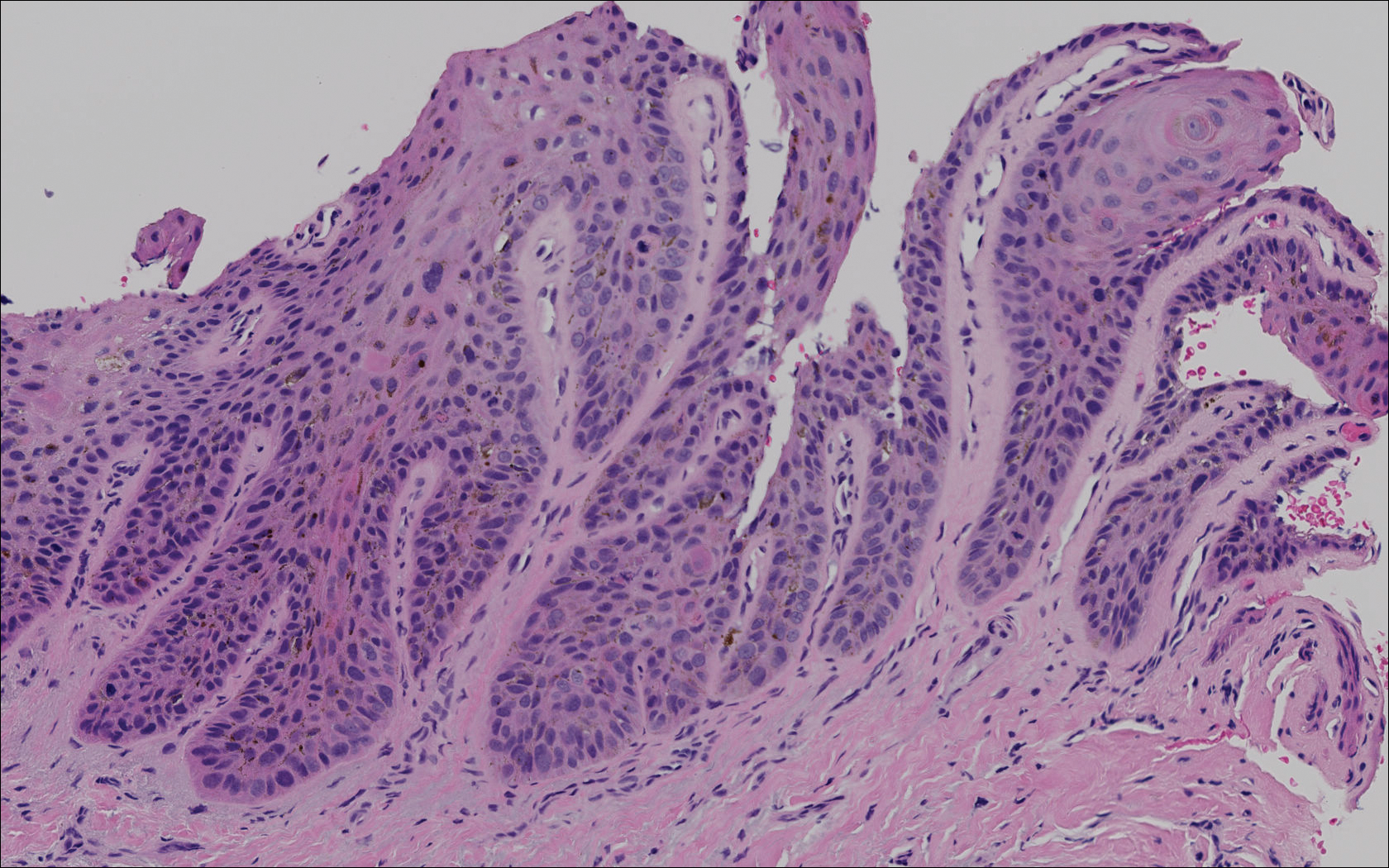
Subungual melanoma, onychomycosis, squamous cell carcinoma (SCC), and a verruca copresenting with onychomycosis were considered in the differential diagnosis. The patient underwent nail avulsion and biopsy of the nail bed as well as the nail matrix. Histopathology was notable for malignant dyskeratosis with a lack of nuclear maturation, occasional mitoses, multinucleation, and individual cell keratinization (Figure 2). Immunostaining for S100 was negative, while staining for cytokeratins AE1/AE3 was positive. Deposition of melanin pigment in the malignant dyskeratotic cells was noted. Periodic acid–Schiff staining identified pseudohyphae without invasion of the nail plate. A diagnosis of pigmented SCC (pSCC) was made. The patient’s nail also was sent for fungal cultures that later grew Candida glabrata and Candida parapsilosis.
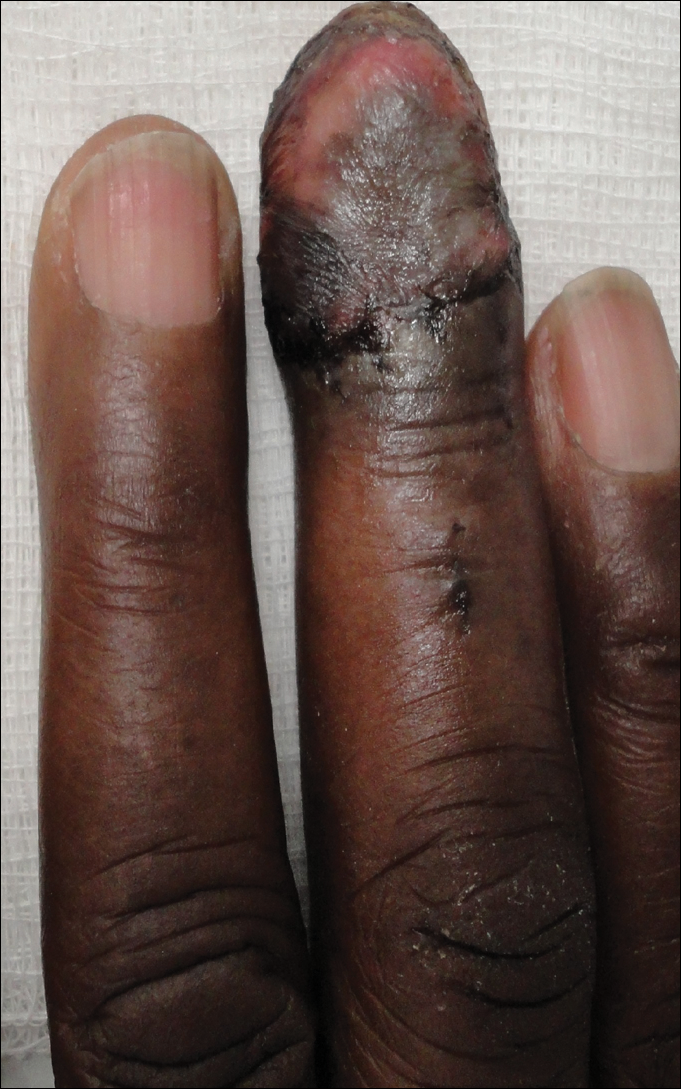
The patient underwent Mohs micrographic surgery for removal of the pSCC, which was found to be more extensive than originally suspected and required en bloc excision of the nail repaired with a full-thickness skin graft from the left forearm. The area healed well with some hyperpigmentation (Figure 3).
Comment
Among the various types of skin cancer, an estimated 700,000 patients are diagnosed with SCC annually, making it the second most common form of skin cancer in the United States.1 Basal cell carcinoma (BCC) is the most common skin cancer among whites in the United States, while in contrast SCC is the most common skin cancer in patients with skin of color.2 Only an estimated 2% to 5% of all SCCs are pigmented, and this variant is more commonly seen in patients with skin of color.3-5 One analysis of 52 cases of pSCC showed that common features included a flat or slightly raised appearance and hyperpigmentation with varying levels of scaling.6 Studies have shown an altered presentation of pSCC in black skin with increased melanin production and thickness of the stratum corneum in contrast with cases seen in white patients.7 Other potential features include scaling, erosive changes, and sharply demarcated borders. Squamous cell carcinoma typically occurs in sun-exposed areas, reflecting its association with UV light damage; however, SCC in skin of color patients has been noted to occur in sun-protected areas and in areas of chronic scarring.8 Pigmented SCC also appears to follow this distribution, as affected areas are not necessarily in direct exposure to the sun. Pigmented SCCs have been associated with pruritus and/or burning pain, which also was seen in our case when our patient complained of tenderness at the site.
We describe the case of a subungual pSCC clinically presenting as longitudinal melanonychia. Pigmented SCC presenting as longitudinal melanonychia was first described by Baran and Simon in 1988.9 Since that time, it has been reported that approximately 10% of subungual pSCCs clinically present as longitudinal melanonychia.10,11 A retrospective study reviewing 35 cases of SCC of the nail apparatus found that 5 (14.3%) cases presented as longitudinal melanonychia.10 Another retrospective study found that 6 of 51 (11.8%) cases of SCCs affecting the nail unit presented as the warty type of SCC in association with longitudinal melanonychia.12 Cases of pSCC in situ appearing as longitudinal melanonychia also have been reported.13,14
Risk factors for the development of pSCC include advanced age, male sex, presence of human papilloma virus, and use of immunosuppressants.15 Male predominance and advanced age at the time of diagnosis (mean age, 67 years) have been observed in pSCC cases.16 It is now well established that renal transplant recipients have an increased risk of SCC, with a reported incidence rate of 5% to 6%.16 When these patients develop an SCC, they typically follow a more aggressive course. Renal transplantation has a higher ratio than cardiac transplantation for SCC development (2.37:1), whereas cardiac transplantation is associated with a higher risk of BCC development.17 A study of 384 transplant recipients found that 96 (25.0%) had a postsurgical nonmelanoma skin cancer (NMSC), with a ratio of SCC to BCC of 1.2:1.16 The calculated incidence of NMSC at 10 and 20 years posttransplantation was 24.2% and 54.4%, respectively. Another study also determined that SCC rates (50.0%) in postrenal transplant recipients were approximately twice that of BCC (27.0%).18
A daily regimen of immunosuppressive medications such as cyclosporine and mycophenolate mofetil showed an increased risk for development of NMSC.15 Immunosuppressive medications play an important role in the pathogenesis of SCC due to a direct oncogenic effect as well as impairment of the immune system’s ability to fight precancerous developments.15 A 4-year study of 100 renal transplant recipients using mycophenolate mofetil as part of an immunosuppressive regimen reported 22% NMSC findings among 9 patients.19 On average, patients developed an NMSC approximately 61 months posttransplantation, with a wide range from 2 to 120 months.
Advanced age was another important risk factor, with each decade of life producing a 60% increase in instantaneous risk of SCC development for transplant recipients.15 A steady increase in risk was related to the length of time adhering to an immunosuppressive regimen, especially from 2 to 6 years, and then remaining constant in subsequent years. For older patients on immunosuppressant regimens for more than 8 years, the calculated relative risk was noted to be over 200 times greater than the normal population’s development of skin cancers.18
Conclusion
Although cases of pSCC presenting as longitudinal melanonychia have previously been reported,9-14,20 our case is unique in that it describes pSCC in a renal transplant recipient. Our patient had many of the known risk factors for the development of pSCC including male sex, advanced age, skin of color, history of renal transplantation, and immunosuppressive therapy. Although regular full-body skin examinations are an accepted part of renal transplantation follow-up due to SCC risk, our case emphasizes the need to remain vigilant due to possible atypical presentations among the immunosuppressed. The nail unit should not be overlooked during the clinical examination of renal transplant recipients as demonstrated by our patient’s rare presentation of pSCC in the nail.
- Karia PS, Han J, Schmults CD. Cutaneous squamous cell carcinoma: estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012 [published online February 1, 2013]. J Am Acad Dermatol. 2013;68:957-966.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- McCall CO, Chen SC. Squamous cell carcinoma of the legs in African Americans. J Am Acad Dermatol. 2002;47:524-529.
- Krishna R, Lewis A, Orengo IF, et al. Pigmented Bowen’s disease (squamous cell carcinoma in situ): a mimic of malignant melanoma. Dermatol Surg. 2001;27:673-674.
- Brinca A, Teixeira V, Goncalo M, et al. A large pigmented lesion mimicking malignant melanoma. Clin Exp Dermatol. 2012;37:817-818.
- Cameron A, Rosendahl C, Tschandl P, et al. Dermatoscopy of pigmented Bowen’s disease. J Am Acad Dermatol. 2010;62:597-604.
- Singh B, Bhaya M, Shaha A, et al. Presentation, course, and outcome of head and neck cancers in African Americans: a case-control study. Laryngoscope. 1998;108(8 pt 1):1159-1163.
- Cancer Facts and Figures 2006. Atlanta, GA: American Cancer Society; 2006.
- Baran R, Simon C. Longitudinal melanonychia: a symptom of Bowen’s disease. J Am Acad Dermatol. 1988;18:1359-1360.
- Dalle S, Depape L, Phan A, et al. Squamous cell carcinoma of the nail apparatus: clinicopathological study of 35 cases. Br J Dermatol. 2007;156:871-874.
- Ishida M, Iwai M, Yoshida K, et al. Subungual pigmented squamous cell carcinoma presenting as longitudinal melanonychia: a case report with review of the literature. Int J Clin Exp Pathol. 2014;7:844-847.
- Lecerf P, Richert B, Theunis A, et al. A retrospective study of squamous cell carcinoma of the nail unit diagnosed in a Belgian general hospital over a 15-year period. J Am Acad Dermatol. 2013;69:253-261.
- Saito T, Uchi H, Moroi Y, et al. Subungual Bowen disease revealed by longitudinal melanonychia. J Am Acad Dermatol. 2012;67:E240-E241.
- Saxena A, Kasper DA, Campanelli CD, et al. Pigmented Bowen’s disease clinically mimicking melanoma on the nail. Dermatol Surg. 2006;32:1522-1525.
- Mackenzie KA, Wells JE, Lynn KL, et al. First and subsequent nonmelanoma skin cancers: incidence and predictors in a population of New Zealand renal transplant recipients. Nephrol Dial Transplant. 2010;25:300-306.
- Gutiérrez-Mendoza D, Narro-Llorente R, Karam-Orantes M, et al. Dermoscopy clues in pigmented Bowen’s disease [published online ahead of print September 16, 2010]. Dermatol Res Pract. 2010;2010.
- Euvards S, Kanitakis J, Pouteil-Noble C, et al. Comparative epidemiologic study of premalignant and malignant epithelial cutaneous lesions developing after kidney and heart transplantation. J Am Acad Dermatol. 1995;33(2 pt 1):222-229.
- Moloney FJ, Comber H, O’Lorcain P, et al. A population-based study of skin cancer incidence and prevalence in renal transplant patients. Br J Dermatol. 2006;154:498-504.
- Formicone F, Fargnoli MC, Pisani F, et al. Cutaneous manifestations in Italian kidney transplant recipients. Transplant Proc. 2005;37:2527-2528.
- Fernandes Massa A, Debarbieux S, Depaepe L, et al. Pigmented squamous cell carcinoma of the nail bed presenting as a melanonychia striata: diagnosis by perioperative reflectance confocal microscopy. Br J Dermatol. 2013;169:198-199.
Case Report
A 62-year-old black man presented for examination of a dark longitudinal streak located adjacent to the lateral nail fold on the third finger of the left hand. The lesion had been present for several months, during which time it had slowly expanded in size. The fingertip had recently become tender, which interfered with the patient’s ability to work. His past medical history was remarkable for end-stage renal disease secondary to glomerulonephritis with nephrotic syndrome of unclear etiology. He initially was treated by an outside physician using peritoneal dialysis for 3 years until he underwent renal transplantation in 2004 with a cadaveric organ. Other remarkable medical conditions included posttransplantation diabetes, hyperlipidemia, and gout. His multidrug regimen included 2 immunosuppressive medications: oral cyclosporine 125 mg twice daily and oral mycophenolate mofetil 250 mg twice daily.
A broad, irregular, black, pigmented, subungual band was noted on the left third finger. The lesion appeared to emanate from below the nail cuticle and traveled along the nail longitudinally toward the distal tip. The band appeared darker at the edge adjacent to the lateral nail fold and grew lighter near the middle of the nail where its free edge was noted to be irregular. A slightly thickened lateral nail fold with an irregular, small, sawtoothlike hyperkeratosis and hyperpigmentation also was noted (Figure 1).
Subungual melanoma, onychomycosis, squamous cell carcinoma (SCC), and a verruca copresenting with onychomycosis were considered in the differential diagnosis. The patient underwent nail avulsion and biopsy of the nail bed as well as the nail matrix. Histopathology was notable for malignant dyskeratosis with a lack of nuclear maturation, occasional mitoses, multinucleation, and individual cell keratinization (Figure 2). Immunostaining for S100 was negative, while staining for cytokeratins AE1/AE3 was positive. Deposition of melanin pigment in the malignant dyskeratotic cells was noted. Periodic acid–Schiff staining identified pseudohyphae without invasion of the nail plate. A diagnosis of pigmented SCC (pSCC) was made. The patient’s nail also was sent for fungal cultures that later grew Candida glabrata and Candida parapsilosis.
The patient underwent Mohs micrographic surgery for removal of the pSCC, which was found to be more extensive than originally suspected and required en bloc excision of the nail repaired with a full-thickness skin graft from the left forearm. The area healed well with some hyperpigmentation (Figure 3).
Comment
Among the various types of skin cancer, an estimated 700,000 patients are diagnosed with SCC annually, making it the second most common form of skin cancer in the United States.1 Basal cell carcinoma (BCC) is the most common skin cancer among whites in the United States, while in contrast SCC is the most common skin cancer in patients with skin of color.2 Only an estimated 2% to 5% of all SCCs are pigmented, and this variant is more commonly seen in patients with skin of color.3-5 One analysis of 52 cases of pSCC showed that common features included a flat or slightly raised appearance and hyperpigmentation with varying levels of scaling.6 Studies have shown an altered presentation of pSCC in black skin with increased melanin production and thickness of the stratum corneum in contrast with cases seen in white patients.7 Other potential features include scaling, erosive changes, and sharply demarcated borders. Squamous cell carcinoma typically occurs in sun-exposed areas, reflecting its association with UV light damage; however, SCC in skin of color patients has been noted to occur in sun-protected areas and in areas of chronic scarring.8 Pigmented SCC also appears to follow this distribution, as affected areas are not necessarily in direct exposure to the sun. Pigmented SCCs have been associated with pruritus and/or burning pain, which also was seen in our case when our patient complained of tenderness at the site.
We describe the case of a subungual pSCC clinically presenting as longitudinal melanonychia. Pigmented SCC presenting as longitudinal melanonychia was first described by Baran and Simon in 1988.9 Since that time, it has been reported that approximately 10% of subungual pSCCs clinically present as longitudinal melanonychia.10,11 A retrospective study reviewing 35 cases of SCC of the nail apparatus found that 5 (14.3%) cases presented as longitudinal melanonychia.10 Another retrospective study found that 6 of 51 (11.8%) cases of SCCs affecting the nail unit presented as the warty type of SCC in association with longitudinal melanonychia.12 Cases of pSCC in situ appearing as longitudinal melanonychia also have been reported.13,14
Risk factors for the development of pSCC include advanced age, male sex, presence of human papilloma virus, and use of immunosuppressants.15 Male predominance and advanced age at the time of diagnosis (mean age, 67 years) have been observed in pSCC cases.16 It is now well established that renal transplant recipients have an increased risk of SCC, with a reported incidence rate of 5% to 6%.16 When these patients develop an SCC, they typically follow a more aggressive course. Renal transplantation has a higher ratio than cardiac transplantation for SCC development (2.37:1), whereas cardiac transplantation is associated with a higher risk of BCC development.17 A study of 384 transplant recipients found that 96 (25.0%) had a postsurgical nonmelanoma skin cancer (NMSC), with a ratio of SCC to BCC of 1.2:1.16 The calculated incidence of NMSC at 10 and 20 years posttransplantation was 24.2% and 54.4%, respectively. Another study also determined that SCC rates (50.0%) in postrenal transplant recipients were approximately twice that of BCC (27.0%).18
A daily regimen of immunosuppressive medications such as cyclosporine and mycophenolate mofetil showed an increased risk for development of NMSC.15 Immunosuppressive medications play an important role in the pathogenesis of SCC due to a direct oncogenic effect as well as impairment of the immune system’s ability to fight precancerous developments.15 A 4-year study of 100 renal transplant recipients using mycophenolate mofetil as part of an immunosuppressive regimen reported 22% NMSC findings among 9 patients.19 On average, patients developed an NMSC approximately 61 months posttransplantation, with a wide range from 2 to 120 months.
Advanced age was another important risk factor, with each decade of life producing a 60% increase in instantaneous risk of SCC development for transplant recipients.15 A steady increase in risk was related to the length of time adhering to an immunosuppressive regimen, especially from 2 to 6 years, and then remaining constant in subsequent years. For older patients on immunosuppressant regimens for more than 8 years, the calculated relative risk was noted to be over 200 times greater than the normal population’s development of skin cancers.18
Conclusion
Although cases of pSCC presenting as longitudinal melanonychia have previously been reported,9-14,20 our case is unique in that it describes pSCC in a renal transplant recipient. Our patient had many of the known risk factors for the development of pSCC including male sex, advanced age, skin of color, history of renal transplantation, and immunosuppressive therapy. Although regular full-body skin examinations are an accepted part of renal transplantation follow-up due to SCC risk, our case emphasizes the need to remain vigilant due to possible atypical presentations among the immunosuppressed. The nail unit should not be overlooked during the clinical examination of renal transplant recipients as demonstrated by our patient’s rare presentation of pSCC in the nail.
Case Report
A 62-year-old black man presented for examination of a dark longitudinal streak located adjacent to the lateral nail fold on the third finger of the left hand. The lesion had been present for several months, during which time it had slowly expanded in size. The fingertip had recently become tender, which interfered with the patient’s ability to work. His past medical history was remarkable for end-stage renal disease secondary to glomerulonephritis with nephrotic syndrome of unclear etiology. He initially was treated by an outside physician using peritoneal dialysis for 3 years until he underwent renal transplantation in 2004 with a cadaveric organ. Other remarkable medical conditions included posttransplantation diabetes, hyperlipidemia, and gout. His multidrug regimen included 2 immunosuppressive medications: oral cyclosporine 125 mg twice daily and oral mycophenolate mofetil 250 mg twice daily.
A broad, irregular, black, pigmented, subungual band was noted on the left third finger. The lesion appeared to emanate from below the nail cuticle and traveled along the nail longitudinally toward the distal tip. The band appeared darker at the edge adjacent to the lateral nail fold and grew lighter near the middle of the nail where its free edge was noted to be irregular. A slightly thickened lateral nail fold with an irregular, small, sawtoothlike hyperkeratosis and hyperpigmentation also was noted (Figure 1).
Subungual melanoma, onychomycosis, squamous cell carcinoma (SCC), and a verruca copresenting with onychomycosis were considered in the differential diagnosis. The patient underwent nail avulsion and biopsy of the nail bed as well as the nail matrix. Histopathology was notable for malignant dyskeratosis with a lack of nuclear maturation, occasional mitoses, multinucleation, and individual cell keratinization (Figure 2). Immunostaining for S100 was negative, while staining for cytokeratins AE1/AE3 was positive. Deposition of melanin pigment in the malignant dyskeratotic cells was noted. Periodic acid–Schiff staining identified pseudohyphae without invasion of the nail plate. A diagnosis of pigmented SCC (pSCC) was made. The patient’s nail also was sent for fungal cultures that later grew Candida glabrata and Candida parapsilosis.
The patient underwent Mohs micrographic surgery for removal of the pSCC, which was found to be more extensive than originally suspected and required en bloc excision of the nail repaired with a full-thickness skin graft from the left forearm. The area healed well with some hyperpigmentation (Figure 3).
Comment
Among the various types of skin cancer, an estimated 700,000 patients are diagnosed with SCC annually, making it the second most common form of skin cancer in the United States.1 Basal cell carcinoma (BCC) is the most common skin cancer among whites in the United States, while in contrast SCC is the most common skin cancer in patients with skin of color.2 Only an estimated 2% to 5% of all SCCs are pigmented, and this variant is more commonly seen in patients with skin of color.3-5 One analysis of 52 cases of pSCC showed that common features included a flat or slightly raised appearance and hyperpigmentation with varying levels of scaling.6 Studies have shown an altered presentation of pSCC in black skin with increased melanin production and thickness of the stratum corneum in contrast with cases seen in white patients.7 Other potential features include scaling, erosive changes, and sharply demarcated borders. Squamous cell carcinoma typically occurs in sun-exposed areas, reflecting its association with UV light damage; however, SCC in skin of color patients has been noted to occur in sun-protected areas and in areas of chronic scarring.8 Pigmented SCC also appears to follow this distribution, as affected areas are not necessarily in direct exposure to the sun. Pigmented SCCs have been associated with pruritus and/or burning pain, which also was seen in our case when our patient complained of tenderness at the site.
We describe the case of a subungual pSCC clinically presenting as longitudinal melanonychia. Pigmented SCC presenting as longitudinal melanonychia was first described by Baran and Simon in 1988.9 Since that time, it has been reported that approximately 10% of subungual pSCCs clinically present as longitudinal melanonychia.10,11 A retrospective study reviewing 35 cases of SCC of the nail apparatus found that 5 (14.3%) cases presented as longitudinal melanonychia.10 Another retrospective study found that 6 of 51 (11.8%) cases of SCCs affecting the nail unit presented as the warty type of SCC in association with longitudinal melanonychia.12 Cases of pSCC in situ appearing as longitudinal melanonychia also have been reported.13,14
Risk factors for the development of pSCC include advanced age, male sex, presence of human papilloma virus, and use of immunosuppressants.15 Male predominance and advanced age at the time of diagnosis (mean age, 67 years) have been observed in pSCC cases.16 It is now well established that renal transplant recipients have an increased risk of SCC, with a reported incidence rate of 5% to 6%.16 When these patients develop an SCC, they typically follow a more aggressive course. Renal transplantation has a higher ratio than cardiac transplantation for SCC development (2.37:1), whereas cardiac transplantation is associated with a higher risk of BCC development.17 A study of 384 transplant recipients found that 96 (25.0%) had a postsurgical nonmelanoma skin cancer (NMSC), with a ratio of SCC to BCC of 1.2:1.16 The calculated incidence of NMSC at 10 and 20 years posttransplantation was 24.2% and 54.4%, respectively. Another study also determined that SCC rates (50.0%) in postrenal transplant recipients were approximately twice that of BCC (27.0%).18
A daily regimen of immunosuppressive medications such as cyclosporine and mycophenolate mofetil showed an increased risk for development of NMSC.15 Immunosuppressive medications play an important role in the pathogenesis of SCC due to a direct oncogenic effect as well as impairment of the immune system’s ability to fight precancerous developments.15 A 4-year study of 100 renal transplant recipients using mycophenolate mofetil as part of an immunosuppressive regimen reported 22% NMSC findings among 9 patients.19 On average, patients developed an NMSC approximately 61 months posttransplantation, with a wide range from 2 to 120 months.
Advanced age was another important risk factor, with each decade of life producing a 60% increase in instantaneous risk of SCC development for transplant recipients.15 A steady increase in risk was related to the length of time adhering to an immunosuppressive regimen, especially from 2 to 6 years, and then remaining constant in subsequent years. For older patients on immunosuppressant regimens for more than 8 years, the calculated relative risk was noted to be over 200 times greater than the normal population’s development of skin cancers.18
Conclusion
Although cases of pSCC presenting as longitudinal melanonychia have previously been reported,9-14,20 our case is unique in that it describes pSCC in a renal transplant recipient. Our patient had many of the known risk factors for the development of pSCC including male sex, advanced age, skin of color, history of renal transplantation, and immunosuppressive therapy. Although regular full-body skin examinations are an accepted part of renal transplantation follow-up due to SCC risk, our case emphasizes the need to remain vigilant due to possible atypical presentations among the immunosuppressed. The nail unit should not be overlooked during the clinical examination of renal transplant recipients as demonstrated by our patient’s rare presentation of pSCC in the nail.
- Karia PS, Han J, Schmults CD. Cutaneous squamous cell carcinoma: estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012 [published online February 1, 2013]. J Am Acad Dermatol. 2013;68:957-966.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- McCall CO, Chen SC. Squamous cell carcinoma of the legs in African Americans. J Am Acad Dermatol. 2002;47:524-529.
- Krishna R, Lewis A, Orengo IF, et al. Pigmented Bowen’s disease (squamous cell carcinoma in situ): a mimic of malignant melanoma. Dermatol Surg. 2001;27:673-674.
- Brinca A, Teixeira V, Goncalo M, et al. A large pigmented lesion mimicking malignant melanoma. Clin Exp Dermatol. 2012;37:817-818.
- Cameron A, Rosendahl C, Tschandl P, et al. Dermatoscopy of pigmented Bowen’s disease. J Am Acad Dermatol. 2010;62:597-604.
- Singh B, Bhaya M, Shaha A, et al. Presentation, course, and outcome of head and neck cancers in African Americans: a case-control study. Laryngoscope. 1998;108(8 pt 1):1159-1163.
- Cancer Facts and Figures 2006. Atlanta, GA: American Cancer Society; 2006.
- Baran R, Simon C. Longitudinal melanonychia: a symptom of Bowen’s disease. J Am Acad Dermatol. 1988;18:1359-1360.
- Dalle S, Depape L, Phan A, et al. Squamous cell carcinoma of the nail apparatus: clinicopathological study of 35 cases. Br J Dermatol. 2007;156:871-874.
- Ishida M, Iwai M, Yoshida K, et al. Subungual pigmented squamous cell carcinoma presenting as longitudinal melanonychia: a case report with review of the literature. Int J Clin Exp Pathol. 2014;7:844-847.
- Lecerf P, Richert B, Theunis A, et al. A retrospective study of squamous cell carcinoma of the nail unit diagnosed in a Belgian general hospital over a 15-year period. J Am Acad Dermatol. 2013;69:253-261.
- Saito T, Uchi H, Moroi Y, et al. Subungual Bowen disease revealed by longitudinal melanonychia. J Am Acad Dermatol. 2012;67:E240-E241.
- Saxena A, Kasper DA, Campanelli CD, et al. Pigmented Bowen’s disease clinically mimicking melanoma on the nail. Dermatol Surg. 2006;32:1522-1525.
- Mackenzie KA, Wells JE, Lynn KL, et al. First and subsequent nonmelanoma skin cancers: incidence and predictors in a population of New Zealand renal transplant recipients. Nephrol Dial Transplant. 2010;25:300-306.
- Gutiérrez-Mendoza D, Narro-Llorente R, Karam-Orantes M, et al. Dermoscopy clues in pigmented Bowen’s disease [published online ahead of print September 16, 2010]. Dermatol Res Pract. 2010;2010.
- Euvards S, Kanitakis J, Pouteil-Noble C, et al. Comparative epidemiologic study of premalignant and malignant epithelial cutaneous lesions developing after kidney and heart transplantation. J Am Acad Dermatol. 1995;33(2 pt 1):222-229.
- Moloney FJ, Comber H, O’Lorcain P, et al. A population-based study of skin cancer incidence and prevalence in renal transplant patients. Br J Dermatol. 2006;154:498-504.
- Formicone F, Fargnoli MC, Pisani F, et al. Cutaneous manifestations in Italian kidney transplant recipients. Transplant Proc. 2005;37:2527-2528.
- Fernandes Massa A, Debarbieux S, Depaepe L, et al. Pigmented squamous cell carcinoma of the nail bed presenting as a melanonychia striata: diagnosis by perioperative reflectance confocal microscopy. Br J Dermatol. 2013;169:198-199.
- Karia PS, Han J, Schmults CD. Cutaneous squamous cell carcinoma: estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012 [published online February 1, 2013]. J Am Acad Dermatol. 2013;68:957-966.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- McCall CO, Chen SC. Squamous cell carcinoma of the legs in African Americans. J Am Acad Dermatol. 2002;47:524-529.
- Krishna R, Lewis A, Orengo IF, et al. Pigmented Bowen’s disease (squamous cell carcinoma in situ): a mimic of malignant melanoma. Dermatol Surg. 2001;27:673-674.
- Brinca A, Teixeira V, Goncalo M, et al. A large pigmented lesion mimicking malignant melanoma. Clin Exp Dermatol. 2012;37:817-818.
- Cameron A, Rosendahl C, Tschandl P, et al. Dermatoscopy of pigmented Bowen’s disease. J Am Acad Dermatol. 2010;62:597-604.
- Singh B, Bhaya M, Shaha A, et al. Presentation, course, and outcome of head and neck cancers in African Americans: a case-control study. Laryngoscope. 1998;108(8 pt 1):1159-1163.
- Cancer Facts and Figures 2006. Atlanta, GA: American Cancer Society; 2006.
- Baran R, Simon C. Longitudinal melanonychia: a symptom of Bowen’s disease. J Am Acad Dermatol. 1988;18:1359-1360.
- Dalle S, Depape L, Phan A, et al. Squamous cell carcinoma of the nail apparatus: clinicopathological study of 35 cases. Br J Dermatol. 2007;156:871-874.
- Ishida M, Iwai M, Yoshida K, et al. Subungual pigmented squamous cell carcinoma presenting as longitudinal melanonychia: a case report with review of the literature. Int J Clin Exp Pathol. 2014;7:844-847.
- Lecerf P, Richert B, Theunis A, et al. A retrospective study of squamous cell carcinoma of the nail unit diagnosed in a Belgian general hospital over a 15-year period. J Am Acad Dermatol. 2013;69:253-261.
- Saito T, Uchi H, Moroi Y, et al. Subungual Bowen disease revealed by longitudinal melanonychia. J Am Acad Dermatol. 2012;67:E240-E241.
- Saxena A, Kasper DA, Campanelli CD, et al. Pigmented Bowen’s disease clinically mimicking melanoma on the nail. Dermatol Surg. 2006;32:1522-1525.
- Mackenzie KA, Wells JE, Lynn KL, et al. First and subsequent nonmelanoma skin cancers: incidence and predictors in a population of New Zealand renal transplant recipients. Nephrol Dial Transplant. 2010;25:300-306.
- Gutiérrez-Mendoza D, Narro-Llorente R, Karam-Orantes M, et al. Dermoscopy clues in pigmented Bowen’s disease [published online ahead of print September 16, 2010]. Dermatol Res Pract. 2010;2010.
- Euvards S, Kanitakis J, Pouteil-Noble C, et al. Comparative epidemiologic study of premalignant and malignant epithelial cutaneous lesions developing after kidney and heart transplantation. J Am Acad Dermatol. 1995;33(2 pt 1):222-229.
- Moloney FJ, Comber H, O’Lorcain P, et al. A population-based study of skin cancer incidence and prevalence in renal transplant patients. Br J Dermatol. 2006;154:498-504.
- Formicone F, Fargnoli MC, Pisani F, et al. Cutaneous manifestations in Italian kidney transplant recipients. Transplant Proc. 2005;37:2527-2528.
- Fernandes Massa A, Debarbieux S, Depaepe L, et al. Pigmented squamous cell carcinoma of the nail bed presenting as a melanonychia striata: diagnosis by perioperative reflectance confocal microscopy. Br J Dermatol. 2013;169:198-199.
Practice Points
- Risk factors for the development of pigmented squamous cell carcinoma (pSCC) include older age, male sex, and use of immunosuppressant medications.
- Subungual pSCC can present as longitudinal melanonychia and should be considered in the differential diagnosis for melanonychia in patients with skin of color or those who are immunosuppressed.
Discoid Lupus Erythematosus Following Herpes Zoster
Cutaneous manifestations of systemic lupus erythematosus (SLE) can be classified as lupus-specific or lupus-nonspecific skin lesions. Lupus-specific lesions commonly are photodistributed, with involvement of the malar region, arms, and trunk. The development of discoid lupus erythematosus (DLE) in areas of trauma, including sun-exposed skin, is not uncommon and may be associated with an isomorphic response. We present a rare case of an isomorphic response following herpes zoster (HZ) in a young woman undergoing treatment with immunosuppressive agents for SLE and DLE. Potential prophylactic therapy also is discussed.
Case Report
A 19-year-old woman initially presented to an outside dermatologist for evaluation of new-onset scarring alopecia, crusted erythematous plaques on the face and arms, and arthralgia. A punch biopsy of a lesion on the left arm demonstrated a lichenoid and perivascular lymphocytic infiltrate with scattered necrotic keratinocytes, perifollicular inflammation, and focally thickened basement membrane at the dermoepidermal junction consistent with discoid lupus erythematosus (DLE). A laboratory workup for SLE revealed 1:1280 antinuclear antibodies (reference range, negative <1:80) with elevated titers of double-stranded DNA, Smith, ribonucleoprotein, Sjögren syndrome A, and Sjögren syndrome B autoantibodies with low complement levels. Based on these findings, a diagnosis of SLE and DLE was made.
At that time, the patient was started on hydroxychloroquine 200 mg twice daily for SLE. Four days later she developed swelling in both hands and feet, and hydroxychloroquine was stopped due to a presumed adverse reaction; however, her symptoms subsequently were determined to be polyarthritis secondary to a lupus flare. Prednisone 10 mg once daily was then initiated. The patient was encouraged to restart hydroxychloroquine, but she declined.
Over the next 13 months, the patient developed severe photosensitivity, oral ulcers, Raynaud phenomenon, anemia, and nephrotic-range proteinuria. She ultimately was diagnosed by the nephrology department at our institution with mixed diffuse proliferative and membranous glomerulonephritis. Induction therapy with oral mycophenolate mofetil 1000 mg twice daily and prednisone 60 mg once daily was started, followed by the addition of tacrolimus 1 mg twice daily. Despite immunosuppressive therapy, she continued to develop new discoid lesions on the face, chest, and arms. Th
After 4 weeks of treatment with mycophenolate mofetil, prednisone, and tacrolimus, the patient developed a painful vesicular rash on the left breast with extension over the left axilla and scapula in a T3 to T4 dermatomal distribution. A clinical diagnosis of HZ was made, and she was started on intravenous acyclovir 10 mg/kg in dextrose 5% every 8 hours for 4 days followed by oral valacyclovir 1000 mg every 8 hours for 14 days, which led to resolution of the eruption.
Over the next 4 months, the patient continued to experience pain confined to the same dermatomal area as the HZ, which was consistent with postherpetic neuralgia. Mycophenolate mofetil was discontinued after she developed acute liver toxicity attributed to the drug. Upon discontinuation, the patient developed a new pruritic rash on both arms and the back. Physical examination by the dermatology department at our institution revealed diffuse, scaly, hyperpigmented papules and annular plaques with central pink hypopigmentation on the face, ears, anterior chest, arms, hands, and back. On the left anterior chest and back, the distribution was strikingly unilateral and multidermatomal (Figure 1). Upon further questioning, the patient confirmed that the areas of the new rash coincided with areas previously affected by HZ. Histologic examination of a representative lesion from the left lateral breast revealed hyperkeratosis, follicular plugging, a patchy lichenoid and perivascular mononuclear cell infiltrate, and pigment incontinence (Figure 2A). These histologic features were subtle and were not diagnostic for lupus; however, direct immunofluorescence demonstrated a continuous granular band of IgG and C3 along the dermoepidermal junction, confirming the diagnosis of DLE (Figure 2B). The histologic findings and clinical presentation were consistent with the development of DLE in areas of previous trauma from HZ. The patient continues to follow-up with the rheumatology and nephrology departments but was lost to dermatology follow-up.
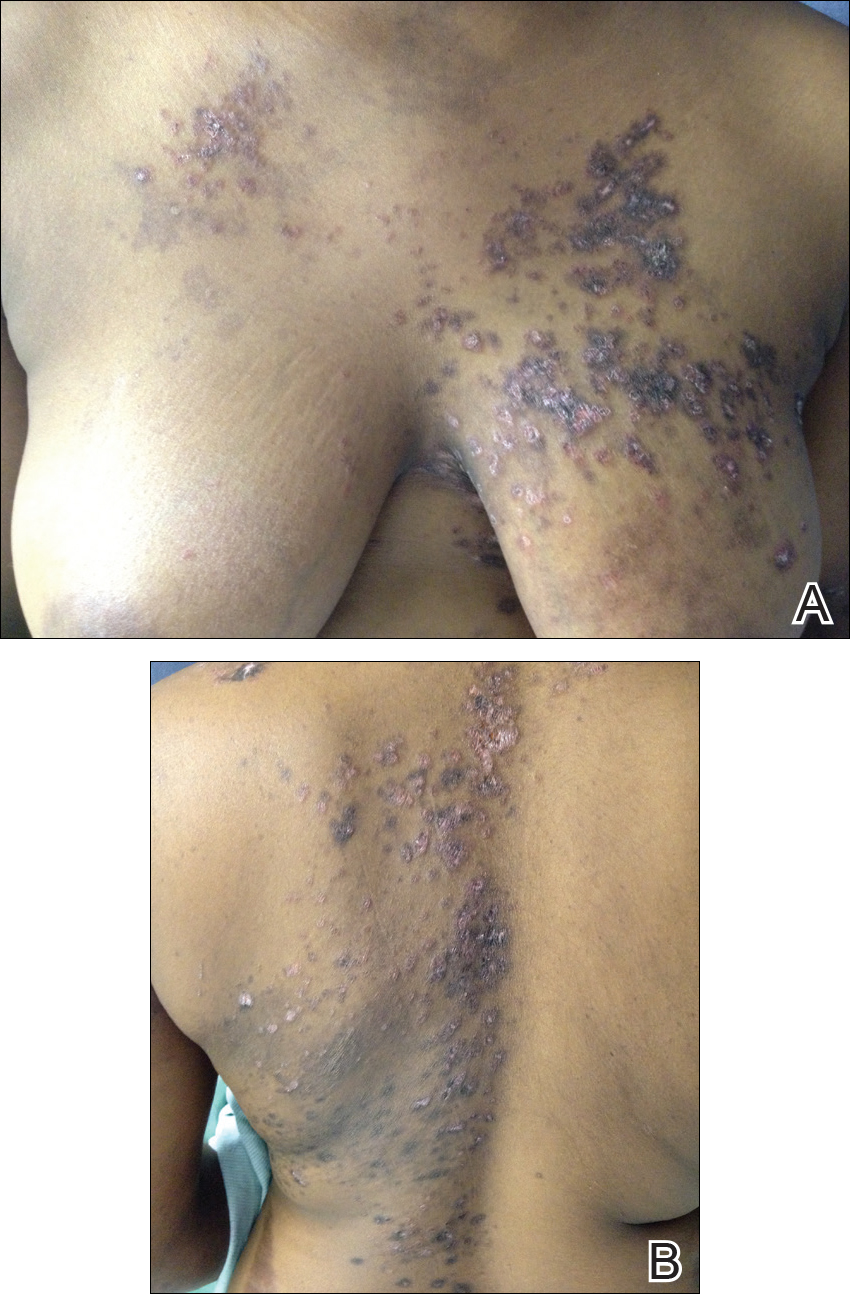
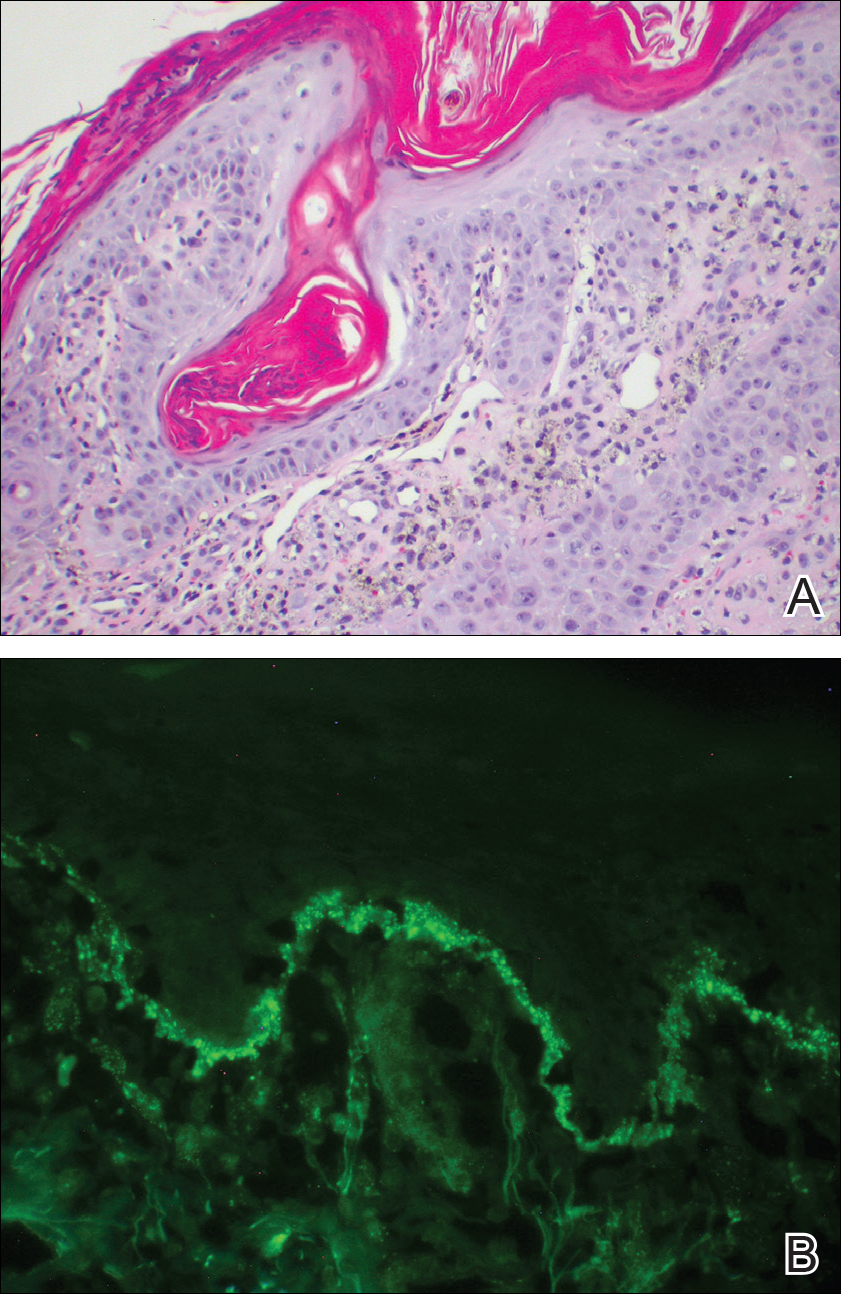
Comment
The pathogenesis of DLE is poorly understood but is thought to be multifactorial, involving genetics, sun exposure, and immune dysregulation.1 Development of DLE lesions in skin traumatized by tattoos, scratches, scars, and prolonged heat exposure has been reported.2 Clarification of the mechanism(s) underlying these traumatized areas may provide insight into the pathophysiology of DLE.
The isomorphic response, also known as the Köbner phenomenon, is the development of a preexisting skin condition at a site of trauma. This phenomenon has been observed in several dermatologic conditions including psoriasis, lichen planus, systemic sclerosis, dermatomyositis, sarcoidosis, vitiligo, and DLE.3 Koebnerization may result from trauma to the skin caused by scratches, sun exposure, radiography, prolonged heat and cold exposure, pressure, tattoos, scars, and inflammatory dermatoses.2,4 Ueki4 suggested that localized trauma to the skin stimulates an immune response that makes the traumatized site a target for a preexisting skin condition. Inflammatory mediators such as IL-1, tumor necrosis factor α, IL-6, and interferon γ have been implicated in the pathophysiology of the isomorphic response.4
Wolf isotopic response is a similar entity that refers to the development of a novel skin condition at the site of a distinct, previously resolved skin disorder. This phenomenon was described by Wolf et al5 in 1995, and since then over 170 cases have been reported.5-7 In most cases the initial skin condition is HZ, although herpes simplex virus has also been implicated. The common resulting skin conditions include granulomatous reactions, malignant tumors, lichen planus, morphea, and infections. The notion that the antecedent skin disease alters the affected site and causes it to be more susceptible to autoimmunity has been proposed as a mechanism for the isotopic response.7,8 While one might consider our presentation of DLE following HZ to be an isotopic response, we believe this case is best classified as an isomorphic response, as the patient already had an established diagnosis of DLE.
The development of DLE at the site of a previous HZ eruption has been described in 2 other cases of young women with SLE.9,10 Unique to our case is the development of a multidermatomal eruption, which may be an indication of her degree of immunosuppression, as immunosuppressed patients are more likely to present with multidermatomal reactivation of varicella zoster virus and postherpetic neuralgia.11 The similarities between our case and the 2 prior reports—including the patients’ age, sex, history of SLE, and degree of immunosuppression—are noteworthy in that they may represent a subset of SLE patients who are predisposed to developing koebnerization following HZ. Physicians should be aware of this phenomenon and consider being proactive in preventing long-term damage.
When feasible, physicians should consider administering the HZ vaccine to reduce the course and severity of HZ before prescribing immunosuppressive agents. When HZ presents in young, immunosuppressed women with a history of SLE, we suggest monitoring the affected sites closely for any evidence of DLE. Topical corticosteroids should be applied to involved areas of the face or body at the earliest appearance of such lesions, which may prevent the isomorphic response and its potentially scarring DLE lesions. This will be our therapeutic approach if we encounter a similar clinical situation in the future. Fur
Acknowledgment
We thank Carolyn E. Grotkowski, MD, from the Department of Pathology, Cooper Medical School of Rowan University, Camden, New Jersey, for her assistance in photographing the pathology slides.
- Lin JH, Dutz JP, Sontheimer RD, et al. Pathophysiology of cutaneous lupus erythematosus. Clinic Rev Allerg Immunol. 2007;33:85-106.
- Ueki H. Köbner phenomenon in lupus erythematosus [in German]. Hautarzt. 1994;45:154-160.
- Boyd AS, Neldner KH. The isomorphic response of Koebner. Int J Dermatol. 1990;29:401-410.
- Ueki H. Koebner phenomenon in lupus erythematosus with special consideration of clinical findings. Autoimmun Rev. 2005;4:219-223.
- Wolf R, Brenner S, Ruocco V, et al. Isotopic response. Int J Dermatol. 1995;34:341-348.
- Wolf R, Wolf D, Ruocco E, et al. Wolf’s isotopic response. Clin Dermatol. 2011;29:237-240.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Martires KJ, Baird K, Citrin DE, et al. Localization of sclerotic-type chronic graft-vs-host disease to sites of skin injury. Arch Dermatol. 2011;147:1081-1086.
- Lee NY, Daniel AS, Dasher DA, et al. Cutaneous lupus after herpes zoster: isomorphic, isotopic, or both [published online May 29, 2012]? Pediatr Dermatol. 2013;30:e110-e113.
- Longhi BS, Centeville M, Marini R, et al. Koebner’s phenomenon in systemic lupus erythematosus. Rheumatol Int. 2012;32:1403-1405.
- Failla V, Jacques J, Castronovo C, et al. Herpes zoster in patients treated with biologicals. Dermatology. 2012;224:251-256.
Cutaneous manifestations of systemic lupus erythematosus (SLE) can be classified as lupus-specific or lupus-nonspecific skin lesions. Lupus-specific lesions commonly are photodistributed, with involvement of the malar region, arms, and trunk. The development of discoid lupus erythematosus (DLE) in areas of trauma, including sun-exposed skin, is not uncommon and may be associated with an isomorphic response. We present a rare case of an isomorphic response following herpes zoster (HZ) in a young woman undergoing treatment with immunosuppressive agents for SLE and DLE. Potential prophylactic therapy also is discussed.
Case Report
A 19-year-old woman initially presented to an outside dermatologist for evaluation of new-onset scarring alopecia, crusted erythematous plaques on the face and arms, and arthralgia. A punch biopsy of a lesion on the left arm demonstrated a lichenoid and perivascular lymphocytic infiltrate with scattered necrotic keratinocytes, perifollicular inflammation, and focally thickened basement membrane at the dermoepidermal junction consistent with discoid lupus erythematosus (DLE). A laboratory workup for SLE revealed 1:1280 antinuclear antibodies (reference range, negative <1:80) with elevated titers of double-stranded DNA, Smith, ribonucleoprotein, Sjögren syndrome A, and Sjögren syndrome B autoantibodies with low complement levels. Based on these findings, a diagnosis of SLE and DLE was made.
At that time, the patient was started on hydroxychloroquine 200 mg twice daily for SLE. Four days later she developed swelling in both hands and feet, and hydroxychloroquine was stopped due to a presumed adverse reaction; however, her symptoms subsequently were determined to be polyarthritis secondary to a lupus flare. Prednisone 10 mg once daily was then initiated. The patient was encouraged to restart hydroxychloroquine, but she declined.
Over the next 13 months, the patient developed severe photosensitivity, oral ulcers, Raynaud phenomenon, anemia, and nephrotic-range proteinuria. She ultimately was diagnosed by the nephrology department at our institution with mixed diffuse proliferative and membranous glomerulonephritis. Induction therapy with oral mycophenolate mofetil 1000 mg twice daily and prednisone 60 mg once daily was started, followed by the addition of tacrolimus 1 mg twice daily. Despite immunosuppressive therapy, she continued to develop new discoid lesions on the face, chest, and arms. Th
After 4 weeks of treatment with mycophenolate mofetil, prednisone, and tacrolimus, the patient developed a painful vesicular rash on the left breast with extension over the left axilla and scapula in a T3 to T4 dermatomal distribution. A clinical diagnosis of HZ was made, and she was started on intravenous acyclovir 10 mg/kg in dextrose 5% every 8 hours for 4 days followed by oral valacyclovir 1000 mg every 8 hours for 14 days, which led to resolution of the eruption.
Over the next 4 months, the patient continued to experience pain confined to the same dermatomal area as the HZ, which was consistent with postherpetic neuralgia. Mycophenolate mofetil was discontinued after she developed acute liver toxicity attributed to the drug. Upon discontinuation, the patient developed a new pruritic rash on both arms and the back. Physical examination by the dermatology department at our institution revealed diffuse, scaly, hyperpigmented papules and annular plaques with central pink hypopigmentation on the face, ears, anterior chest, arms, hands, and back. On the left anterior chest and back, the distribution was strikingly unilateral and multidermatomal (Figure 1). Upon further questioning, the patient confirmed that the areas of the new rash coincided with areas previously affected by HZ. Histologic examination of a representative lesion from the left lateral breast revealed hyperkeratosis, follicular plugging, a patchy lichenoid and perivascular mononuclear cell infiltrate, and pigment incontinence (Figure 2A). These histologic features were subtle and were not diagnostic for lupus; however, direct immunofluorescence demonstrated a continuous granular band of IgG and C3 along the dermoepidermal junction, confirming the diagnosis of DLE (Figure 2B). The histologic findings and clinical presentation were consistent with the development of DLE in areas of previous trauma from HZ. The patient continues to follow-up with the rheumatology and nephrology departments but was lost to dermatology follow-up.
Comment
The pathogenesis of DLE is poorly understood but is thought to be multifactorial, involving genetics, sun exposure, and immune dysregulation.1 Development of DLE lesions in skin traumatized by tattoos, scratches, scars, and prolonged heat exposure has been reported.2 Clarification of the mechanism(s) underlying these traumatized areas may provide insight into the pathophysiology of DLE.
The isomorphic response, also known as the Köbner phenomenon, is the development of a preexisting skin condition at a site of trauma. This phenomenon has been observed in several dermatologic conditions including psoriasis, lichen planus, systemic sclerosis, dermatomyositis, sarcoidosis, vitiligo, and DLE.3 Koebnerization may result from trauma to the skin caused by scratches, sun exposure, radiography, prolonged heat and cold exposure, pressure, tattoos, scars, and inflammatory dermatoses.2,4 Ueki4 suggested that localized trauma to the skin stimulates an immune response that makes the traumatized site a target for a preexisting skin condition. Inflammatory mediators such as IL-1, tumor necrosis factor α, IL-6, and interferon γ have been implicated in the pathophysiology of the isomorphic response.4
Wolf isotopic response is a similar entity that refers to the development of a novel skin condition at the site of a distinct, previously resolved skin disorder. This phenomenon was described by Wolf et al5 in 1995, and since then over 170 cases have been reported.5-7 In most cases the initial skin condition is HZ, although herpes simplex virus has also been implicated. The common resulting skin conditions include granulomatous reactions, malignant tumors, lichen planus, morphea, and infections. The notion that the antecedent skin disease alters the affected site and causes it to be more susceptible to autoimmunity has been proposed as a mechanism for the isotopic response.7,8 While one might consider our presentation of DLE following HZ to be an isotopic response, we believe this case is best classified as an isomorphic response, as the patient already had an established diagnosis of DLE.
The development of DLE at the site of a previous HZ eruption has been described in 2 other cases of young women with SLE.9,10 Unique to our case is the development of a multidermatomal eruption, which may be an indication of her degree of immunosuppression, as immunosuppressed patients are more likely to present with multidermatomal reactivation of varicella zoster virus and postherpetic neuralgia.11 The similarities between our case and the 2 prior reports—including the patients’ age, sex, history of SLE, and degree of immunosuppression—are noteworthy in that they may represent a subset of SLE patients who are predisposed to developing koebnerization following HZ. Physicians should be aware of this phenomenon and consider being proactive in preventing long-term damage.
When feasible, physicians should consider administering the HZ vaccine to reduce the course and severity of HZ before prescribing immunosuppressive agents. When HZ presents in young, immunosuppressed women with a history of SLE, we suggest monitoring the affected sites closely for any evidence of DLE. Topical corticosteroids should be applied to involved areas of the face or body at the earliest appearance of such lesions, which may prevent the isomorphic response and its potentially scarring DLE lesions. This will be our therapeutic approach if we encounter a similar clinical situation in the future. Fur
Acknowledgment
We thank Carolyn E. Grotkowski, MD, from the Department of Pathology, Cooper Medical School of Rowan University, Camden, New Jersey, for her assistance in photographing the pathology slides.
Cutaneous manifestations of systemic lupus erythematosus (SLE) can be classified as lupus-specific or lupus-nonspecific skin lesions. Lupus-specific lesions commonly are photodistributed, with involvement of the malar region, arms, and trunk. The development of discoid lupus erythematosus (DLE) in areas of trauma, including sun-exposed skin, is not uncommon and may be associated with an isomorphic response. We present a rare case of an isomorphic response following herpes zoster (HZ) in a young woman undergoing treatment with immunosuppressive agents for SLE and DLE. Potential prophylactic therapy also is discussed.
Case Report
A 19-year-old woman initially presented to an outside dermatologist for evaluation of new-onset scarring alopecia, crusted erythematous plaques on the face and arms, and arthralgia. A punch biopsy of a lesion on the left arm demonstrated a lichenoid and perivascular lymphocytic infiltrate with scattered necrotic keratinocytes, perifollicular inflammation, and focally thickened basement membrane at the dermoepidermal junction consistent with discoid lupus erythematosus (DLE). A laboratory workup for SLE revealed 1:1280 antinuclear antibodies (reference range, negative <1:80) with elevated titers of double-stranded DNA, Smith, ribonucleoprotein, Sjögren syndrome A, and Sjögren syndrome B autoantibodies with low complement levels. Based on these findings, a diagnosis of SLE and DLE was made.
At that time, the patient was started on hydroxychloroquine 200 mg twice daily for SLE. Four days later she developed swelling in both hands and feet, and hydroxychloroquine was stopped due to a presumed adverse reaction; however, her symptoms subsequently were determined to be polyarthritis secondary to a lupus flare. Prednisone 10 mg once daily was then initiated. The patient was encouraged to restart hydroxychloroquine, but she declined.
Over the next 13 months, the patient developed severe photosensitivity, oral ulcers, Raynaud phenomenon, anemia, and nephrotic-range proteinuria. She ultimately was diagnosed by the nephrology department at our institution with mixed diffuse proliferative and membranous glomerulonephritis. Induction therapy with oral mycophenolate mofetil 1000 mg twice daily and prednisone 60 mg once daily was started, followed by the addition of tacrolimus 1 mg twice daily. Despite immunosuppressive therapy, she continued to develop new discoid lesions on the face, chest, and arms. Th
After 4 weeks of treatment with mycophenolate mofetil, prednisone, and tacrolimus, the patient developed a painful vesicular rash on the left breast with extension over the left axilla and scapula in a T3 to T4 dermatomal distribution. A clinical diagnosis of HZ was made, and she was started on intravenous acyclovir 10 mg/kg in dextrose 5% every 8 hours for 4 days followed by oral valacyclovir 1000 mg every 8 hours for 14 days, which led to resolution of the eruption.
Over the next 4 months, the patient continued to experience pain confined to the same dermatomal area as the HZ, which was consistent with postherpetic neuralgia. Mycophenolate mofetil was discontinued after she developed acute liver toxicity attributed to the drug. Upon discontinuation, the patient developed a new pruritic rash on both arms and the back. Physical examination by the dermatology department at our institution revealed diffuse, scaly, hyperpigmented papules and annular plaques with central pink hypopigmentation on the face, ears, anterior chest, arms, hands, and back. On the left anterior chest and back, the distribution was strikingly unilateral and multidermatomal (Figure 1). Upon further questioning, the patient confirmed that the areas of the new rash coincided with areas previously affected by HZ. Histologic examination of a representative lesion from the left lateral breast revealed hyperkeratosis, follicular plugging, a patchy lichenoid and perivascular mononuclear cell infiltrate, and pigment incontinence (Figure 2A). These histologic features were subtle and were not diagnostic for lupus; however, direct immunofluorescence demonstrated a continuous granular band of IgG and C3 along the dermoepidermal junction, confirming the diagnosis of DLE (Figure 2B). The histologic findings and clinical presentation were consistent with the development of DLE in areas of previous trauma from HZ. The patient continues to follow-up with the rheumatology and nephrology departments but was lost to dermatology follow-up.
Comment
The pathogenesis of DLE is poorly understood but is thought to be multifactorial, involving genetics, sun exposure, and immune dysregulation.1 Development of DLE lesions in skin traumatized by tattoos, scratches, scars, and prolonged heat exposure has been reported.2 Clarification of the mechanism(s) underlying these traumatized areas may provide insight into the pathophysiology of DLE.
The isomorphic response, also known as the Köbner phenomenon, is the development of a preexisting skin condition at a site of trauma. This phenomenon has been observed in several dermatologic conditions including psoriasis, lichen planus, systemic sclerosis, dermatomyositis, sarcoidosis, vitiligo, and DLE.3 Koebnerization may result from trauma to the skin caused by scratches, sun exposure, radiography, prolonged heat and cold exposure, pressure, tattoos, scars, and inflammatory dermatoses.2,4 Ueki4 suggested that localized trauma to the skin stimulates an immune response that makes the traumatized site a target for a preexisting skin condition. Inflammatory mediators such as IL-1, tumor necrosis factor α, IL-6, and interferon γ have been implicated in the pathophysiology of the isomorphic response.4
Wolf isotopic response is a similar entity that refers to the development of a novel skin condition at the site of a distinct, previously resolved skin disorder. This phenomenon was described by Wolf et al5 in 1995, and since then over 170 cases have been reported.5-7 In most cases the initial skin condition is HZ, although herpes simplex virus has also been implicated. The common resulting skin conditions include granulomatous reactions, malignant tumors, lichen planus, morphea, and infections. The notion that the antecedent skin disease alters the affected site and causes it to be more susceptible to autoimmunity has been proposed as a mechanism for the isotopic response.7,8 While one might consider our presentation of DLE following HZ to be an isotopic response, we believe this case is best classified as an isomorphic response, as the patient already had an established diagnosis of DLE.
The development of DLE at the site of a previous HZ eruption has been described in 2 other cases of young women with SLE.9,10 Unique to our case is the development of a multidermatomal eruption, which may be an indication of her degree of immunosuppression, as immunosuppressed patients are more likely to present with multidermatomal reactivation of varicella zoster virus and postherpetic neuralgia.11 The similarities between our case and the 2 prior reports—including the patients’ age, sex, history of SLE, and degree of immunosuppression—are noteworthy in that they may represent a subset of SLE patients who are predisposed to developing koebnerization following HZ. Physicians should be aware of this phenomenon and consider being proactive in preventing long-term damage.
When feasible, physicians should consider administering the HZ vaccine to reduce the course and severity of HZ before prescribing immunosuppressive agents. When HZ presents in young, immunosuppressed women with a history of SLE, we suggest monitoring the affected sites closely for any evidence of DLE. Topical corticosteroids should be applied to involved areas of the face or body at the earliest appearance of such lesions, which may prevent the isomorphic response and its potentially scarring DLE lesions. This will be our therapeutic approach if we encounter a similar clinical situation in the future. Fur
Acknowledgment
We thank Carolyn E. Grotkowski, MD, from the Department of Pathology, Cooper Medical School of Rowan University, Camden, New Jersey, for her assistance in photographing the pathology slides.
- Lin JH, Dutz JP, Sontheimer RD, et al. Pathophysiology of cutaneous lupus erythematosus. Clinic Rev Allerg Immunol. 2007;33:85-106.
- Ueki H. Köbner phenomenon in lupus erythematosus [in German]. Hautarzt. 1994;45:154-160.
- Boyd AS, Neldner KH. The isomorphic response of Koebner. Int J Dermatol. 1990;29:401-410.
- Ueki H. Koebner phenomenon in lupus erythematosus with special consideration of clinical findings. Autoimmun Rev. 2005;4:219-223.
- Wolf R, Brenner S, Ruocco V, et al. Isotopic response. Int J Dermatol. 1995;34:341-348.
- Wolf R, Wolf D, Ruocco E, et al. Wolf’s isotopic response. Clin Dermatol. 2011;29:237-240.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Martires KJ, Baird K, Citrin DE, et al. Localization of sclerotic-type chronic graft-vs-host disease to sites of skin injury. Arch Dermatol. 2011;147:1081-1086.
- Lee NY, Daniel AS, Dasher DA, et al. Cutaneous lupus after herpes zoster: isomorphic, isotopic, or both [published online May 29, 2012]? Pediatr Dermatol. 2013;30:e110-e113.
- Longhi BS, Centeville M, Marini R, et al. Koebner’s phenomenon in systemic lupus erythematosus. Rheumatol Int. 2012;32:1403-1405.
- Failla V, Jacques J, Castronovo C, et al. Herpes zoster in patients treated with biologicals. Dermatology. 2012;224:251-256.
- Lin JH, Dutz JP, Sontheimer RD, et al. Pathophysiology of cutaneous lupus erythematosus. Clinic Rev Allerg Immunol. 2007;33:85-106.
- Ueki H. Köbner phenomenon in lupus erythematosus [in German]. Hautarzt. 1994;45:154-160.
- Boyd AS, Neldner KH. The isomorphic response of Koebner. Int J Dermatol. 1990;29:401-410.
- Ueki H. Koebner phenomenon in lupus erythematosus with special consideration of clinical findings. Autoimmun Rev. 2005;4:219-223.
- Wolf R, Brenner S, Ruocco V, et al. Isotopic response. Int J Dermatol. 1995;34:341-348.
- Wolf R, Wolf D, Ruocco E, et al. Wolf’s isotopic response. Clin Dermatol. 2011;29:237-240.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Martires KJ, Baird K, Citrin DE, et al. Localization of sclerotic-type chronic graft-vs-host disease to sites of skin injury. Arch Dermatol. 2011;147:1081-1086.
- Lee NY, Daniel AS, Dasher DA, et al. Cutaneous lupus after herpes zoster: isomorphic, isotopic, or both [published online May 29, 2012]? Pediatr Dermatol. 2013;30:e110-e113.
- Longhi BS, Centeville M, Marini R, et al. Koebner’s phenomenon in systemic lupus erythematosus. Rheumatol Int. 2012;32:1403-1405.
- Failla V, Jacques J, Castronovo C, et al. Herpes zoster in patients treated with biologicals. Dermatology. 2012;224:251-256.
Practice Points
- Discoid lupus erythematosus (DLE) most commonly presents as scaling and crusted plaques in sun-exposed areas of the face and arms. It also may present in skin traumatized by tattoos, scratches, scars, prolonged heat exposure, andherpes zoster (HZ).
- Patients with a history of DLE who subsequently develop HZ should be followed closely for the development of DLE in HZ-affected dermatomes.
- Following resolution of HZ, topical corticosteroids may have a role in prevention of DLE in HZ-affected dermatomes.
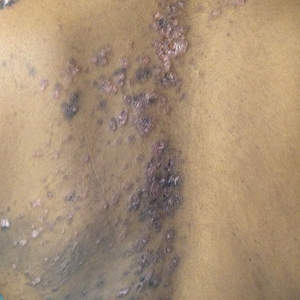
Secukinumab Emerges as a Rapidly Effective Therapy for Pityriasis Rubra Pilaris
Although there currently are no formal guidelines for the treatment of refractory pityriasis rubra pilaris (PRP), successful off-label treatment of the condition with multiple biologics approved for psoriasis has been reported.1,2 Secukinumab, an IL-17A antagonist, has shown particularly striking results in the treatment of PRP in 2 recent case reports.3,4 We report 2 additional cases of severe refractory PRP that responded rapidly to treatment with secukinumab. In both cases, the patients’ erythematous plaques resolved or had nearly resolved by week 4 of treatment. Our findings suggest that IL-17 plays an important role in PRP pathogenesis and support future clinical trials of anti–IL-17 agents for treatment of this entity.
Case Reports
Patient 1
A 60-year-old man with a history of biopsy-proven PRP presented with persistent generalized erythema, scattered patches of normal skin, and hyperkeratotic plaques on the bilateral palms of 1 year’s duration. Previous therapies included topical steroids, topical calcipotriene, adalimumab 40 mg once every other week, infliximab 5 mg/kg once every 8 weeks, ustekinumab 90 mg once every 12 weeks, acitretin 25 mg once daily, and most recently cyclosporine 200 mg twice daily. Of these treatments, infliximab was the
At 4 weeks’ follow-up, there was a marked decrease in erythema and scaling. The body surface area affected had decreased to 5%, and improvement of palmar keratoderma was noted. The patient continued with maintenance dosing of secukinumab 300 mg once every 4 weeks. By week 8, the erythema had fully resolved (Figure 1B), and he remained clear at week 24. No adverse events were noted since initiation of therapy.
Patient 2
A 74-year-old woman with a history of PRP that had previously been misdiagnosed as psoriasis by an outside physician presented for evaluation of palmoplantar keratoderma (Figure 2A), follicular hyperkeratosis, and erythematous plaques on the trunk and arms of 5 years’ duration. Previous therapies included topical steroids, topical urea, methotrexate 20 mg once weekly, adalimumab 40 mg once every other week, infliximab 10 mg/kg once every 4 weeks, ustekinumab 90 mg once every 12 weeks, and most recently acitretin 50 mg once daily.
The patient had been maintained on ustekinumab and acitretin for 2 years with only mild improvement. Ustekinumab was then discontinued, and after 3 months treatment with secukinumab was added to the once-daily acitretin. Similar to Patient 1, loading doses of secukinumab 300 mg were administered once weekly for 5 weeks. The plaques on the trunk and arms had resolved by week 4, but the palmoplantar keratoderma persisted. The patient continued with the maintenance dose of secukinumab 300 mg once every 4 weeks and reported an increase in peeling of the palms and soles at week 8.
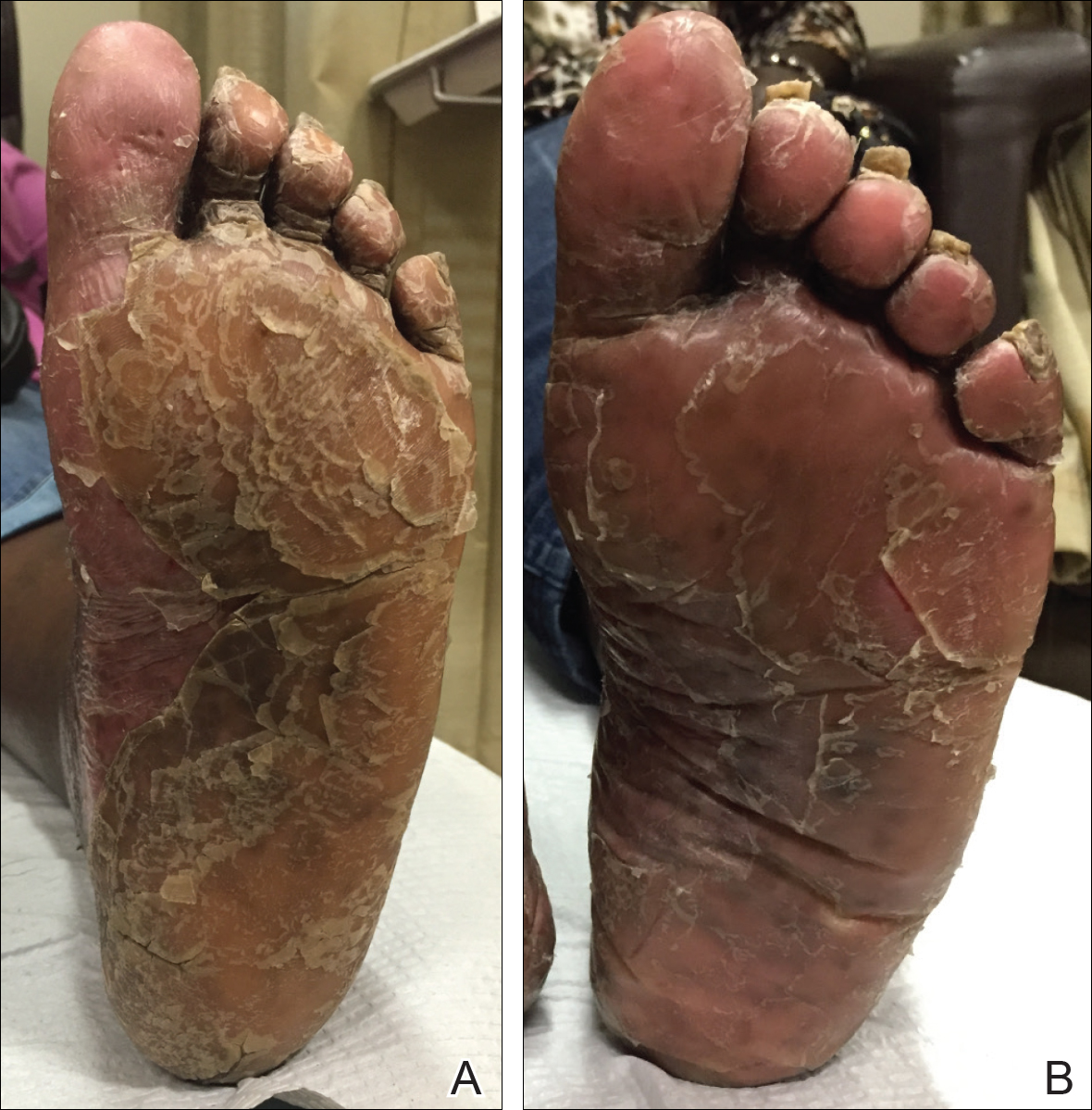
By week 12 of treatment, the palmar keratoderma had resolved, and debridement of the soles revealed patches of normal skin (Figure 2B). By week 52, no adverse events had been noted. The patient continued to experience mild keratoderma of the soles, making us reluctant to discontinue acitretin; however, she has maintained her maximal response, and her quality of life has significantly improved. The patient was continued on acitretin and secukinumab, and her condition remained stable.
Comment
Because there are no formal treatment guidelines for refractory PRP, case reports play an important role in clinical decision-making. When a patient is unresponsive to topical medications and first-line traditional systemic therapies (eg, methotrexate, cyclosporine, acitretin), biologic drugs effective in the treatment of psoriasis are widely accepted as the next therapeutic step.1 The biologic medications that are most often reported in the treatment of PRP are the TNF-α antagonists, as they have been available the longest.1-2 In a systematic review of 15 patients with PRP who were treated with TNF-α antagonists,2 80% of patients achieved complete response (mean time to maximal response, 5 months). There also are a number of reports of successful treatment of PRP with the IL-12/23 antagonist ustekinumab, which has been commercially available since 2009.5-9 Although improvement was noted in most of these patients at the time of the second injection (week 4 of therapy), maximal response with ustekinumab typically occurs between weeks 12 and 28.10
In our cases of PRP treated with secukinumab as well as 2 others that were recently reported in the literature, resolution of erythema and plaques was rapid. This superiority of the response rate parallels the performance of secukinumab relative to ustekinumab in patients with psoriasis11 In one case of a 67-year-old man with PRP treated with secukinumab, scaling and pruritus were reduced by week 3 of treatment and erythema had cleared by week 8.3 In another case of a 33-year-old woman with PRP, pruritus resolved after 1 week of treatment and erythematous plaques and palmoplantar keratoderma improved by week 2.4 In both of our cases, plaques had resolved or nearly resolved by week 4 of follow-up. Patient 1 achieved complete response at week 8 of therapy. Patient 2 never attained complete response, but by week 12 she achieved maximal response, which still resulted in markedly increased quality of life. We do not intend to make additions to her treatment plan because she is currently the clearest she has been since onset of symptoms and is happy with her present condition.
Although it is difficult to predict the long-term prognosis in our 2 patients, we will continue their current regimens indefinitely—as long as the response persists and no adverse events are experienced. This approach is consistent with guidelines for management of plaque psoriasis with secukinumab.12
This accumulation of evidence suggests the importance of the role of IL-17 in the pathogenesis of PRP. The serum level of IL-17 was not evaluated in our patients, but elevation of IL-17 has been reported in a case of PRP.13 Further studies are needed to clarify the role of IL-17 in this disease entity.
Conclusion
Given the refractory nature of PRP and the relative safety of targeted immunotherapy, trials of new biologics and potent small molecules approved for psoriasis treatment are worth exploring for PRP. In light of our reports and those in the literature and given the relative safety of anti–IL-17 agents, it may be reasonable to consider such agents as a first-line therapy for this predictably refractory disease.
- Klein A, Landthaler M, Karrer S. Pityriasis rubra pilaris. Am J Clin Dermatol. 2010;11:157-170.
- Petrof G, Almaani N, Archer CB, et al. A systematic review of the literature on the treatment of pityriasis rubra pilaris type 1 with TNF-antagonists. J Eur Acad Dermatol Venereol. 2013;27:E131-E135.
- Schuster D, Pfister-Wartha A, Bruckner-Tuderman L, et al. Successful treatment of refractory pityriasis rubra pilaris with secukinumab. JAMA Dermatol. 2016;152:1278-1280.
- Gauci ML, Jachiet M, Gottlieb J, et al. Successful treatment of type II pityriasis rubra pilaris with secukinumab. JAAD Case Rep. 2016;2:462-264.
- Chowdhary M, Davila U, Cohen DJ. Ustekinumab as an alternative treatment option for chronic pityriasis rubra pilaris. Case Rep Dermatol. 2015;7:46-50.
- Wohlrab J, Kreft B. Treatment of pityriasis rubra pilaris with ustekinumab. Br J Dermatol. 2010;163:655-656.
- Villaverde RR, Cano DS. Successful treatment of type 1 pityriasis rubra pilaris with ustekinumab therapy. Eur J Dermatol. 2010;20:630-631.
- Di Stefani A, Galluzzo M, Talamonti M, et al. Long-term ustekinumab treatment for refractory type I pityriasis rubra pilaris. J Dermatol Case Rep. 2013;7:5-9.
- Eytan O, Sarig O, Sprecher E, et al. Clinical response to ustekinumab in familial pityriasis rubra pilaris caused by a novel mutation in CARD14. Br J Dermatol. 2014;171:420-422.
- Papp KA, Langley RG, Lebwohl M, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet. 2008;371:1675-1684.
- Thaçi D, Blauvelt A, Reich K, et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: CLEAR, a randomized controlled trial. J Am Acad Dermatol. 2015;73:400-409.
- van de Kerkhof PC, Griffiths CE, Reich K, et al. Secukinumab long-term safety experience: a pooled analysis of 10 phase II and III clinical studies in patients with moderate to severe plaque psoriasis. J Am Acad Dermatol. 2016;75:83-98.
- Adnot-Desanlis L, Antonicelli F, Tabary T, et al. Effectiveness of infliximab in pityriasis rubra pilaris is associated with pro-inflammatory cytokine inhibition. Dermatology. 2013;226:41-46.
Although there currently are no formal guidelines for the treatment of refractory pityriasis rubra pilaris (PRP), successful off-label treatment of the condition with multiple biologics approved for psoriasis has been reported.1,2 Secukinumab, an IL-17A antagonist, has shown particularly striking results in the treatment of PRP in 2 recent case reports.3,4 We report 2 additional cases of severe refractory PRP that responded rapidly to treatment with secukinumab. In both cases, the patients’ erythematous plaques resolved or had nearly resolved by week 4 of treatment. Our findings suggest that IL-17 plays an important role in PRP pathogenesis and support future clinical trials of anti–IL-17 agents for treatment of this entity.
Case Reports
Patient 1
A 60-year-old man with a history of biopsy-proven PRP presented with persistent generalized erythema, scattered patches of normal skin, and hyperkeratotic plaques on the bilateral palms of 1 year’s duration. Previous therapies included topical steroids, topical calcipotriene, adalimumab 40 mg once every other week, infliximab 5 mg/kg once every 8 weeks, ustekinumab 90 mg once every 12 weeks, acitretin 25 mg once daily, and most recently cyclosporine 200 mg twice daily. Of these treatments, infliximab was the
At 4 weeks’ follow-up, there was a marked decrease in erythema and scaling. The body surface area affected had decreased to 5%, and improvement of palmar keratoderma was noted. The patient continued with maintenance dosing of secukinumab 300 mg once every 4 weeks. By week 8, the erythema had fully resolved (Figure 1B), and he remained clear at week 24. No adverse events were noted since initiation of therapy.
Patient 2
A 74-year-old woman with a history of PRP that had previously been misdiagnosed as psoriasis by an outside physician presented for evaluation of palmoplantar keratoderma (Figure 2A), follicular hyperkeratosis, and erythematous plaques on the trunk and arms of 5 years’ duration. Previous therapies included topical steroids, topical urea, methotrexate 20 mg once weekly, adalimumab 40 mg once every other week, infliximab 10 mg/kg once every 4 weeks, ustekinumab 90 mg once every 12 weeks, and most recently acitretin 50 mg once daily.
The patient had been maintained on ustekinumab and acitretin for 2 years with only mild improvement. Ustekinumab was then discontinued, and after 3 months treatment with secukinumab was added to the once-daily acitretin. Similar to Patient 1, loading doses of secukinumab 300 mg were administered once weekly for 5 weeks. The plaques on the trunk and arms had resolved by week 4, but the palmoplantar keratoderma persisted. The patient continued with the maintenance dose of secukinumab 300 mg once every 4 weeks and reported an increase in peeling of the palms and soles at week 8.
By week 12 of treatment, the palmar keratoderma had resolved, and debridement of the soles revealed patches of normal skin (Figure 2B). By week 52, no adverse events had been noted. The patient continued to experience mild keratoderma of the soles, making us reluctant to discontinue acitretin; however, she has maintained her maximal response, and her quality of life has significantly improved. The patient was continued on acitretin and secukinumab, and her condition remained stable.
Comment
Because there are no formal treatment guidelines for refractory PRP, case reports play an important role in clinical decision-making. When a patient is unresponsive to topical medications and first-line traditional systemic therapies (eg, methotrexate, cyclosporine, acitretin), biologic drugs effective in the treatment of psoriasis are widely accepted as the next therapeutic step.1 The biologic medications that are most often reported in the treatment of PRP are the TNF-α antagonists, as they have been available the longest.1-2 In a systematic review of 15 patients with PRP who were treated with TNF-α antagonists,2 80% of patients achieved complete response (mean time to maximal response, 5 months). There also are a number of reports of successful treatment of PRP with the IL-12/23 antagonist ustekinumab, which has been commercially available since 2009.5-9 Although improvement was noted in most of these patients at the time of the second injection (week 4 of therapy), maximal response with ustekinumab typically occurs between weeks 12 and 28.10
In our cases of PRP treated with secukinumab as well as 2 others that were recently reported in the literature, resolution of erythema and plaques was rapid. This superiority of the response rate parallels the performance of secukinumab relative to ustekinumab in patients with psoriasis11 In one case of a 67-year-old man with PRP treated with secukinumab, scaling and pruritus were reduced by week 3 of treatment and erythema had cleared by week 8.3 In another case of a 33-year-old woman with PRP, pruritus resolved after 1 week of treatment and erythematous plaques and palmoplantar keratoderma improved by week 2.4 In both of our cases, plaques had resolved or nearly resolved by week 4 of follow-up. Patient 1 achieved complete response at week 8 of therapy. Patient 2 never attained complete response, but by week 12 she achieved maximal response, which still resulted in markedly increased quality of life. We do not intend to make additions to her treatment plan because she is currently the clearest she has been since onset of symptoms and is happy with her present condition.
Although it is difficult to predict the long-term prognosis in our 2 patients, we will continue their current regimens indefinitely—as long as the response persists and no adverse events are experienced. This approach is consistent with guidelines for management of plaque psoriasis with secukinumab.12
This accumulation of evidence suggests the importance of the role of IL-17 in the pathogenesis of PRP. The serum level of IL-17 was not evaluated in our patients, but elevation of IL-17 has been reported in a case of PRP.13 Further studies are needed to clarify the role of IL-17 in this disease entity.
Conclusion
Given the refractory nature of PRP and the relative safety of targeted immunotherapy, trials of new biologics and potent small molecules approved for psoriasis treatment are worth exploring for PRP. In light of our reports and those in the literature and given the relative safety of anti–IL-17 agents, it may be reasonable to consider such agents as a first-line therapy for this predictably refractory disease.
Although there currently are no formal guidelines for the treatment of refractory pityriasis rubra pilaris (PRP), successful off-label treatment of the condition with multiple biologics approved for psoriasis has been reported.1,2 Secukinumab, an IL-17A antagonist, has shown particularly striking results in the treatment of PRP in 2 recent case reports.3,4 We report 2 additional cases of severe refractory PRP that responded rapidly to treatment with secukinumab. In both cases, the patients’ erythematous plaques resolved or had nearly resolved by week 4 of treatment. Our findings suggest that IL-17 plays an important role in PRP pathogenesis and support future clinical trials of anti–IL-17 agents for treatment of this entity.
Case Reports
Patient 1
A 60-year-old man with a history of biopsy-proven PRP presented with persistent generalized erythema, scattered patches of normal skin, and hyperkeratotic plaques on the bilateral palms of 1 year’s duration. Previous therapies included topical steroids, topical calcipotriene, adalimumab 40 mg once every other week, infliximab 5 mg/kg once every 8 weeks, ustekinumab 90 mg once every 12 weeks, acitretin 25 mg once daily, and most recently cyclosporine 200 mg twice daily. Of these treatments, infliximab was the
At 4 weeks’ follow-up, there was a marked decrease in erythema and scaling. The body surface area affected had decreased to 5%, and improvement of palmar keratoderma was noted. The patient continued with maintenance dosing of secukinumab 300 mg once every 4 weeks. By week 8, the erythema had fully resolved (Figure 1B), and he remained clear at week 24. No adverse events were noted since initiation of therapy.
Patient 2
A 74-year-old woman with a history of PRP that had previously been misdiagnosed as psoriasis by an outside physician presented for evaluation of palmoplantar keratoderma (Figure 2A), follicular hyperkeratosis, and erythematous plaques on the trunk and arms of 5 years’ duration. Previous therapies included topical steroids, topical urea, methotrexate 20 mg once weekly, adalimumab 40 mg once every other week, infliximab 10 mg/kg once every 4 weeks, ustekinumab 90 mg once every 12 weeks, and most recently acitretin 50 mg once daily.
The patient had been maintained on ustekinumab and acitretin for 2 years with only mild improvement. Ustekinumab was then discontinued, and after 3 months treatment with secukinumab was added to the once-daily acitretin. Similar to Patient 1, loading doses of secukinumab 300 mg were administered once weekly for 5 weeks. The plaques on the trunk and arms had resolved by week 4, but the palmoplantar keratoderma persisted. The patient continued with the maintenance dose of secukinumab 300 mg once every 4 weeks and reported an increase in peeling of the palms and soles at week 8.
By week 12 of treatment, the palmar keratoderma had resolved, and debridement of the soles revealed patches of normal skin (Figure 2B). By week 52, no adverse events had been noted. The patient continued to experience mild keratoderma of the soles, making us reluctant to discontinue acitretin; however, she has maintained her maximal response, and her quality of life has significantly improved. The patient was continued on acitretin and secukinumab, and her condition remained stable.
Comment
Because there are no formal treatment guidelines for refractory PRP, case reports play an important role in clinical decision-making. When a patient is unresponsive to topical medications and first-line traditional systemic therapies (eg, methotrexate, cyclosporine, acitretin), biologic drugs effective in the treatment of psoriasis are widely accepted as the next therapeutic step.1 The biologic medications that are most often reported in the treatment of PRP are the TNF-α antagonists, as they have been available the longest.1-2 In a systematic review of 15 patients with PRP who were treated with TNF-α antagonists,2 80% of patients achieved complete response (mean time to maximal response, 5 months). There also are a number of reports of successful treatment of PRP with the IL-12/23 antagonist ustekinumab, which has been commercially available since 2009.5-9 Although improvement was noted in most of these patients at the time of the second injection (week 4 of therapy), maximal response with ustekinumab typically occurs between weeks 12 and 28.10
In our cases of PRP treated with secukinumab as well as 2 others that were recently reported in the literature, resolution of erythema and plaques was rapid. This superiority of the response rate parallels the performance of secukinumab relative to ustekinumab in patients with psoriasis11 In one case of a 67-year-old man with PRP treated with secukinumab, scaling and pruritus were reduced by week 3 of treatment and erythema had cleared by week 8.3 In another case of a 33-year-old woman with PRP, pruritus resolved after 1 week of treatment and erythematous plaques and palmoplantar keratoderma improved by week 2.4 In both of our cases, plaques had resolved or nearly resolved by week 4 of follow-up. Patient 1 achieved complete response at week 8 of therapy. Patient 2 never attained complete response, but by week 12 she achieved maximal response, which still resulted in markedly increased quality of life. We do not intend to make additions to her treatment plan because she is currently the clearest she has been since onset of symptoms and is happy with her present condition.
Although it is difficult to predict the long-term prognosis in our 2 patients, we will continue their current regimens indefinitely—as long as the response persists and no adverse events are experienced. This approach is consistent with guidelines for management of plaque psoriasis with secukinumab.12
This accumulation of evidence suggests the importance of the role of IL-17 in the pathogenesis of PRP. The serum level of IL-17 was not evaluated in our patients, but elevation of IL-17 has been reported in a case of PRP.13 Further studies are needed to clarify the role of IL-17 in this disease entity.
Conclusion
Given the refractory nature of PRP and the relative safety of targeted immunotherapy, trials of new biologics and potent small molecules approved for psoriasis treatment are worth exploring for PRP. In light of our reports and those in the literature and given the relative safety of anti–IL-17 agents, it may be reasonable to consider such agents as a first-line therapy for this predictably refractory disease.
- Klein A, Landthaler M, Karrer S. Pityriasis rubra pilaris. Am J Clin Dermatol. 2010;11:157-170.
- Petrof G, Almaani N, Archer CB, et al. A systematic review of the literature on the treatment of pityriasis rubra pilaris type 1 with TNF-antagonists. J Eur Acad Dermatol Venereol. 2013;27:E131-E135.
- Schuster D, Pfister-Wartha A, Bruckner-Tuderman L, et al. Successful treatment of refractory pityriasis rubra pilaris with secukinumab. JAMA Dermatol. 2016;152:1278-1280.
- Gauci ML, Jachiet M, Gottlieb J, et al. Successful treatment of type II pityriasis rubra pilaris with secukinumab. JAAD Case Rep. 2016;2:462-264.
- Chowdhary M, Davila U, Cohen DJ. Ustekinumab as an alternative treatment option for chronic pityriasis rubra pilaris. Case Rep Dermatol. 2015;7:46-50.
- Wohlrab J, Kreft B. Treatment of pityriasis rubra pilaris with ustekinumab. Br J Dermatol. 2010;163:655-656.
- Villaverde RR, Cano DS. Successful treatment of type 1 pityriasis rubra pilaris with ustekinumab therapy. Eur J Dermatol. 2010;20:630-631.
- Di Stefani A, Galluzzo M, Talamonti M, et al. Long-term ustekinumab treatment for refractory type I pityriasis rubra pilaris. J Dermatol Case Rep. 2013;7:5-9.
- Eytan O, Sarig O, Sprecher E, et al. Clinical response to ustekinumab in familial pityriasis rubra pilaris caused by a novel mutation in CARD14. Br J Dermatol. 2014;171:420-422.
- Papp KA, Langley RG, Lebwohl M, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet. 2008;371:1675-1684.
- Thaçi D, Blauvelt A, Reich K, et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: CLEAR, a randomized controlled trial. J Am Acad Dermatol. 2015;73:400-409.
- van de Kerkhof PC, Griffiths CE, Reich K, et al. Secukinumab long-term safety experience: a pooled analysis of 10 phase II and III clinical studies in patients with moderate to severe plaque psoriasis. J Am Acad Dermatol. 2016;75:83-98.
- Adnot-Desanlis L, Antonicelli F, Tabary T, et al. Effectiveness of infliximab in pityriasis rubra pilaris is associated with pro-inflammatory cytokine inhibition. Dermatology. 2013;226:41-46.
- Klein A, Landthaler M, Karrer S. Pityriasis rubra pilaris. Am J Clin Dermatol. 2010;11:157-170.
- Petrof G, Almaani N, Archer CB, et al. A systematic review of the literature on the treatment of pityriasis rubra pilaris type 1 with TNF-antagonists. J Eur Acad Dermatol Venereol. 2013;27:E131-E135.
- Schuster D, Pfister-Wartha A, Bruckner-Tuderman L, et al. Successful treatment of refractory pityriasis rubra pilaris with secukinumab. JAMA Dermatol. 2016;152:1278-1280.
- Gauci ML, Jachiet M, Gottlieb J, et al. Successful treatment of type II pityriasis rubra pilaris with secukinumab. JAAD Case Rep. 2016;2:462-264.
- Chowdhary M, Davila U, Cohen DJ. Ustekinumab as an alternative treatment option for chronic pityriasis rubra pilaris. Case Rep Dermatol. 2015;7:46-50.
- Wohlrab J, Kreft B. Treatment of pityriasis rubra pilaris with ustekinumab. Br J Dermatol. 2010;163:655-656.
- Villaverde RR, Cano DS. Successful treatment of type 1 pityriasis rubra pilaris with ustekinumab therapy. Eur J Dermatol. 2010;20:630-631.
- Di Stefani A, Galluzzo M, Talamonti M, et al. Long-term ustekinumab treatment for refractory type I pityriasis rubra pilaris. J Dermatol Case Rep. 2013;7:5-9.
- Eytan O, Sarig O, Sprecher E, et al. Clinical response to ustekinumab in familial pityriasis rubra pilaris caused by a novel mutation in CARD14. Br J Dermatol. 2014;171:420-422.
- Papp KA, Langley RG, Lebwohl M, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet. 2008;371:1675-1684.
- Thaçi D, Blauvelt A, Reich K, et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: CLEAR, a randomized controlled trial. J Am Acad Dermatol. 2015;73:400-409.
- van de Kerkhof PC, Griffiths CE, Reich K, et al. Secukinumab long-term safety experience: a pooled analysis of 10 phase II and III clinical studies in patients with moderate to severe plaque psoriasis. J Am Acad Dermatol. 2016;75:83-98.
- Adnot-Desanlis L, Antonicelli F, Tabary T, et al. Effectiveness of infliximab in pityriasis rubra pilaris is associated with pro-inflammatory cytokine inhibition. Dermatology. 2013;226:41-46.
Practice Points
- In patients with pityriasis rubra pilaris (PRP) who have not responded to topical treatments, off-label treatment with systemic therapies approved for plaque psoriasis can be considered.
- Secukinumab, an IL-17A antagonist, has shown particularly striking results in the treatment of PRP.
Perianal Basal Cell Carcinoma Treated With Mohs Micrographic Surgery
Basal cell carcinoma (BCC) is the most common skin cancer in the United States1 and most commonly occurs in sun-exposed areas. Although BCCs can and do develop on other non–sun-exposed areas of the body, BCCs of the perianal or genital regions are very rare (0.27% of cases). It is estimated that perianal BCCs account for less than 0.08% of all BCCs.2
We present a case of a superficial nodular perianal BCC that was discovered following an annual total-body skin examination and was treated with Mohs micrographic surgery (MMS).
Case Report
A 76-year-old man presented to the dermatology clinic for an annual total-body skin examination as well as evaluation of a new submental skin lesion. The patient’s medical history included successfully treated malignant melanoma in situ, multiple actinic keratoses, and an eccrine carcinoma. His family history was noncontributory. Inspection of the submental lesion revealed a pearly, 1.8-cm, telangiectatic, nodular plaque that was highly suspected to be a BCC. During the examination, a 1-cm pinkish-red plaque was found on the skin in the left perianal region (Figure 1). The patient was unaware of the lesion and did not report any symptoms upon questioning.
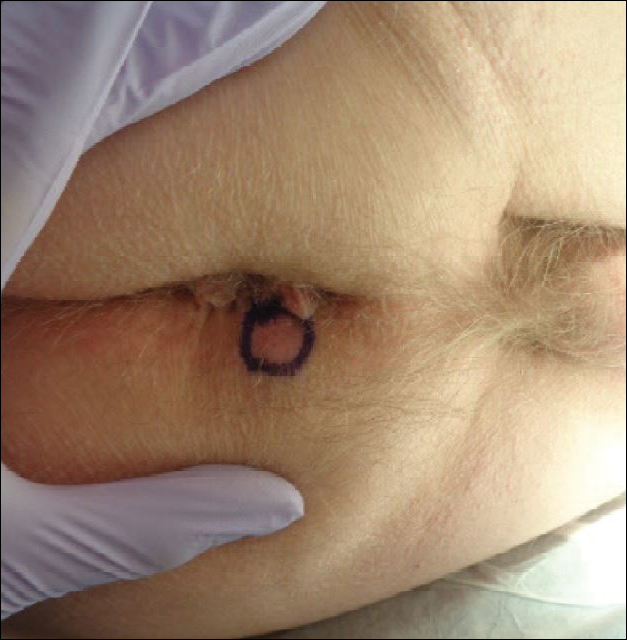
A shave biopsy of the submental lesion confirmed a diagnosis of micronodular BCC, and the patient was referred for MMS. It was decided to reevaluate the perianal lesion clinically at a follow-up appointment 2 months later and biopsy if it had not resolved. However, the patient did not attend the 2-month follow-up visit as scheduled, and it was not until the following year at his next annual total-body skin examination that the perianal lesion was rechecked. The lesion was unchanged at the time and was similar to the previous findings in both appearance and size. A punch biopsy was performed, and the pathology showed a superficial nodular perianal BCC (Figure 2). The perianal BCC was excised during a 2-stage MMS procedure with no recurrence at 6-month follow-up (Figure 3).

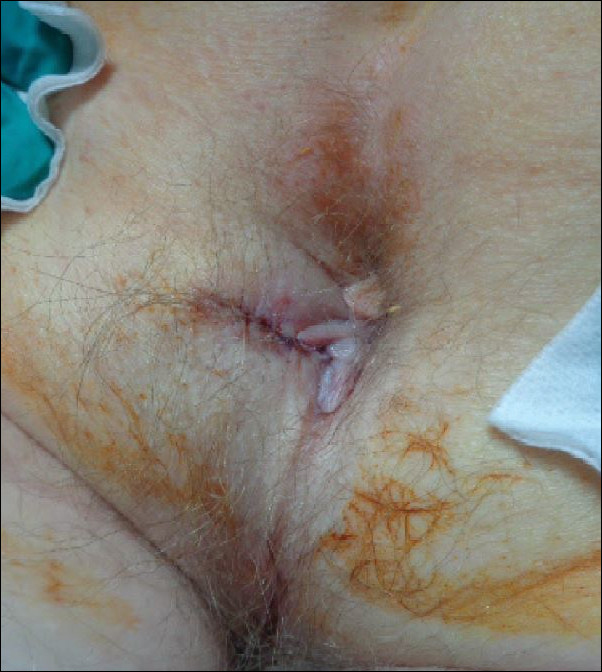
Comment
At the time of the patient’s initial visit, the differential diagnosis for this perianal lesion included an inflammatory or infectious dermatosis. Its asymptomatic nature made it difficult to determine how long it had been present. The lack of resolution on reevaluation of the lesion 1 year later raised the possibilities of amelanotic melanoma, squamous cell carcinoma, and lichen planus. Basal cell carcinoma was much lower in the differential diagnosis, as BCCs rarely are found in this area of the body; in fact, BCCs account for 0.2% of all anorectal neoplasms,3 and less than 0.08% of BCCs will occur in the perianal region.2
This challenging presentation is common for BCCs found in the perianal and perineal regions, as they are difficult to diagnose and often are overlooked as inflammatory dermatoses.4,5 The infrequency of perianal BCC reported in the literature as well as the predominance of BCC in sun-exposed areas makes it difficult for dermatologists to diagnose perianal BCC without biopsy. Another feature indicative of this diagnostic difficulty is that the average size of perianal and perineal BCCs has been found to be 1.95 cm.2 Without thorough and routine total-body skin examinations, there is no reliable way to catch asymptomatic BCCs in the perianal region until they have progressed far enough to become symptomatic. When possible, we recommend that dermatologists check the genital and anal regions during skin examinations and biopsy any suspicious lesions.
This case also highlights the challenge of missed appointments, which dermatologists also consistently face. Nonattendance rates in US dermatology clinics have been estimated at 17%,6 18.6%,7 19.4%,8 and 23.9%9 and present a challenge for even the best-run practices. Among patients with missed appointments, the most frequently stated reason in one survey was forgetting, and 24% of those contacted reported that they had not been reminded of their appointment.8 Many of the patients surveyed also expressed that they had preferred methods of receiving reminders such as e-mail or text message, which fell outside of traditional contact methods (eg, phone calls, voicemails). Confirming appointments ahead of time can reduce the number of missed appointments due to patient forgetfulness, and incorporating multiple communication modalities may lead to more effective appointment reminders.
Conclusion
Perianal BCC is challenging to diagnose and easy to overlook. Basal cell carcinoma is rarely found in the perianal regions and accounts for a fraction of all anorectal neoplasms. We recommend thorough total-body skin examinations that include the genital region and gluteal cleft when possible and encourage physicians to biopsy suspicious lesions in these regions. Routine, thorough total-body skin examinations can reveal neoplasms when they are smaller and asymptomatic. When surgical excision is indicated, MMS is an effective way to preserve as much tissue as possible and minimize recurrence.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population, 2012. JAMA Dermatology. 2015;151:1081-1086.
- Gibson GE, Ahmed I. Perianal and genital basal cell carcinoma: a clinicopathologic review of 51 cases. J Am Acad Dermatol. 2001;45:68-71.
- Leonard D, Beddy D, Dozois EJ. Neoplasms of anal canal and perianal skin. Clin Colon Rectal Surg. 2011;24:54-63.
- Bulur I, Boyuk E, Saracoglu ZN, et al. Perianal basal cell carcinoma. Case Rep Dermatol. 2015;7:25-28.
- Collins PS, Farber GA, Hegre AM. Basal-cell carcinoma of the vulva. J Dermatol Surg Oncol. 1981;7:711-714.
- Penneys NS, Glaser DA. The incidence of cancellation and nonattendance at a dermatology clinic. J Am Acad Dermatol. 1990;40:714-718.
- Cronin P, DeCoste L, Kimball A. A multivariate analysis of dermatology missed appointment predictors. JAMA Dermatology. 2013;149:1435-1437.
- Moustafa FA, Ramsey L, Huang KE, et al. Factors associated with missed dermatology appointments. Cutis. 2015;96:E20-E23.
- Canizares MJ, Penneys NS. The incidence of nonattendance at an urgent care dermatology clinic. J Am Acad Dermatol. 2002;46:457-459.
Basal cell carcinoma (BCC) is the most common skin cancer in the United States1 and most commonly occurs in sun-exposed areas. Although BCCs can and do develop on other non–sun-exposed areas of the body, BCCs of the perianal or genital regions are very rare (0.27% of cases). It is estimated that perianal BCCs account for less than 0.08% of all BCCs.2
We present a case of a superficial nodular perianal BCC that was discovered following an annual total-body skin examination and was treated with Mohs micrographic surgery (MMS).
Case Report
A 76-year-old man presented to the dermatology clinic for an annual total-body skin examination as well as evaluation of a new submental skin lesion. The patient’s medical history included successfully treated malignant melanoma in situ, multiple actinic keratoses, and an eccrine carcinoma. His family history was noncontributory. Inspection of the submental lesion revealed a pearly, 1.8-cm, telangiectatic, nodular plaque that was highly suspected to be a BCC. During the examination, a 1-cm pinkish-red plaque was found on the skin in the left perianal region (Figure 1). The patient was unaware of the lesion and did not report any symptoms upon questioning.
A shave biopsy of the submental lesion confirmed a diagnosis of micronodular BCC, and the patient was referred for MMS. It was decided to reevaluate the perianal lesion clinically at a follow-up appointment 2 months later and biopsy if it had not resolved. However, the patient did not attend the 2-month follow-up visit as scheduled, and it was not until the following year at his next annual total-body skin examination that the perianal lesion was rechecked. The lesion was unchanged at the time and was similar to the previous findings in both appearance and size. A punch biopsy was performed, and the pathology showed a superficial nodular perianal BCC (Figure 2). The perianal BCC was excised during a 2-stage MMS procedure with no recurrence at 6-month follow-up (Figure 3).
Comment
At the time of the patient’s initial visit, the differential diagnosis for this perianal lesion included an inflammatory or infectious dermatosis. Its asymptomatic nature made it difficult to determine how long it had been present. The lack of resolution on reevaluation of the lesion 1 year later raised the possibilities of amelanotic melanoma, squamous cell carcinoma, and lichen planus. Basal cell carcinoma was much lower in the differential diagnosis, as BCCs rarely are found in this area of the body; in fact, BCCs account for 0.2% of all anorectal neoplasms,3 and less than 0.08% of BCCs will occur in the perianal region.2
This challenging presentation is common for BCCs found in the perianal and perineal regions, as they are difficult to diagnose and often are overlooked as inflammatory dermatoses.4,5 The infrequency of perianal BCC reported in the literature as well as the predominance of BCC in sun-exposed areas makes it difficult for dermatologists to diagnose perianal BCC without biopsy. Another feature indicative of this diagnostic difficulty is that the average size of perianal and perineal BCCs has been found to be 1.95 cm.2 Without thorough and routine total-body skin examinations, there is no reliable way to catch asymptomatic BCCs in the perianal region until they have progressed far enough to become symptomatic. When possible, we recommend that dermatologists check the genital and anal regions during skin examinations and biopsy any suspicious lesions.
This case also highlights the challenge of missed appointments, which dermatologists also consistently face. Nonattendance rates in US dermatology clinics have been estimated at 17%,6 18.6%,7 19.4%,8 and 23.9%9 and present a challenge for even the best-run practices. Among patients with missed appointments, the most frequently stated reason in one survey was forgetting, and 24% of those contacted reported that they had not been reminded of their appointment.8 Many of the patients surveyed also expressed that they had preferred methods of receiving reminders such as e-mail or text message, which fell outside of traditional contact methods (eg, phone calls, voicemails). Confirming appointments ahead of time can reduce the number of missed appointments due to patient forgetfulness, and incorporating multiple communication modalities may lead to more effective appointment reminders.
Conclusion
Perianal BCC is challenging to diagnose and easy to overlook. Basal cell carcinoma is rarely found in the perianal regions and accounts for a fraction of all anorectal neoplasms. We recommend thorough total-body skin examinations that include the genital region and gluteal cleft when possible and encourage physicians to biopsy suspicious lesions in these regions. Routine, thorough total-body skin examinations can reveal neoplasms when they are smaller and asymptomatic. When surgical excision is indicated, MMS is an effective way to preserve as much tissue as possible and minimize recurrence.
Basal cell carcinoma (BCC) is the most common skin cancer in the United States1 and most commonly occurs in sun-exposed areas. Although BCCs can and do develop on other non–sun-exposed areas of the body, BCCs of the perianal or genital regions are very rare (0.27% of cases). It is estimated that perianal BCCs account for less than 0.08% of all BCCs.2
We present a case of a superficial nodular perianal BCC that was discovered following an annual total-body skin examination and was treated with Mohs micrographic surgery (MMS).
Case Report
A 76-year-old man presented to the dermatology clinic for an annual total-body skin examination as well as evaluation of a new submental skin lesion. The patient’s medical history included successfully treated malignant melanoma in situ, multiple actinic keratoses, and an eccrine carcinoma. His family history was noncontributory. Inspection of the submental lesion revealed a pearly, 1.8-cm, telangiectatic, nodular plaque that was highly suspected to be a BCC. During the examination, a 1-cm pinkish-red plaque was found on the skin in the left perianal region (Figure 1). The patient was unaware of the lesion and did not report any symptoms upon questioning.
A shave biopsy of the submental lesion confirmed a diagnosis of micronodular BCC, and the patient was referred for MMS. It was decided to reevaluate the perianal lesion clinically at a follow-up appointment 2 months later and biopsy if it had not resolved. However, the patient did not attend the 2-month follow-up visit as scheduled, and it was not until the following year at his next annual total-body skin examination that the perianal lesion was rechecked. The lesion was unchanged at the time and was similar to the previous findings in both appearance and size. A punch biopsy was performed, and the pathology showed a superficial nodular perianal BCC (Figure 2). The perianal BCC was excised during a 2-stage MMS procedure with no recurrence at 6-month follow-up (Figure 3).
Comment
At the time of the patient’s initial visit, the differential diagnosis for this perianal lesion included an inflammatory or infectious dermatosis. Its asymptomatic nature made it difficult to determine how long it had been present. The lack of resolution on reevaluation of the lesion 1 year later raised the possibilities of amelanotic melanoma, squamous cell carcinoma, and lichen planus. Basal cell carcinoma was much lower in the differential diagnosis, as BCCs rarely are found in this area of the body; in fact, BCCs account for 0.2% of all anorectal neoplasms,3 and less than 0.08% of BCCs will occur in the perianal region.2
This challenging presentation is common for BCCs found in the perianal and perineal regions, as they are difficult to diagnose and often are overlooked as inflammatory dermatoses.4,5 The infrequency of perianal BCC reported in the literature as well as the predominance of BCC in sun-exposed areas makes it difficult for dermatologists to diagnose perianal BCC without biopsy. Another feature indicative of this diagnostic difficulty is that the average size of perianal and perineal BCCs has been found to be 1.95 cm.2 Without thorough and routine total-body skin examinations, there is no reliable way to catch asymptomatic BCCs in the perianal region until they have progressed far enough to become symptomatic. When possible, we recommend that dermatologists check the genital and anal regions during skin examinations and biopsy any suspicious lesions.
This case also highlights the challenge of missed appointments, which dermatologists also consistently face. Nonattendance rates in US dermatology clinics have been estimated at 17%,6 18.6%,7 19.4%,8 and 23.9%9 and present a challenge for even the best-run practices. Among patients with missed appointments, the most frequently stated reason in one survey was forgetting, and 24% of those contacted reported that they had not been reminded of their appointment.8 Many of the patients surveyed also expressed that they had preferred methods of receiving reminders such as e-mail or text message, which fell outside of traditional contact methods (eg, phone calls, voicemails). Confirming appointments ahead of time can reduce the number of missed appointments due to patient forgetfulness, and incorporating multiple communication modalities may lead to more effective appointment reminders.
Conclusion
Perianal BCC is challenging to diagnose and easy to overlook. Basal cell carcinoma is rarely found in the perianal regions and accounts for a fraction of all anorectal neoplasms. We recommend thorough total-body skin examinations that include the genital region and gluteal cleft when possible and encourage physicians to biopsy suspicious lesions in these regions. Routine, thorough total-body skin examinations can reveal neoplasms when they are smaller and asymptomatic. When surgical excision is indicated, MMS is an effective way to preserve as much tissue as possible and minimize recurrence.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population, 2012. JAMA Dermatology. 2015;151:1081-1086.
- Gibson GE, Ahmed I. Perianal and genital basal cell carcinoma: a clinicopathologic review of 51 cases. J Am Acad Dermatol. 2001;45:68-71.
- Leonard D, Beddy D, Dozois EJ. Neoplasms of anal canal and perianal skin. Clin Colon Rectal Surg. 2011;24:54-63.
- Bulur I, Boyuk E, Saracoglu ZN, et al. Perianal basal cell carcinoma. Case Rep Dermatol. 2015;7:25-28.
- Collins PS, Farber GA, Hegre AM. Basal-cell carcinoma of the vulva. J Dermatol Surg Oncol. 1981;7:711-714.
- Penneys NS, Glaser DA. The incidence of cancellation and nonattendance at a dermatology clinic. J Am Acad Dermatol. 1990;40:714-718.
- Cronin P, DeCoste L, Kimball A. A multivariate analysis of dermatology missed appointment predictors. JAMA Dermatology. 2013;149:1435-1437.
- Moustafa FA, Ramsey L, Huang KE, et al. Factors associated with missed dermatology appointments. Cutis. 2015;96:E20-E23.
- Canizares MJ, Penneys NS. The incidence of nonattendance at an urgent care dermatology clinic. J Am Acad Dermatol. 2002;46:457-459.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the US population, 2012. JAMA Dermatology. 2015;151:1081-1086.
- Gibson GE, Ahmed I. Perianal and genital basal cell carcinoma: a clinicopathologic review of 51 cases. J Am Acad Dermatol. 2001;45:68-71.
- Leonard D, Beddy D, Dozois EJ. Neoplasms of anal canal and perianal skin. Clin Colon Rectal Surg. 2011;24:54-63.
- Bulur I, Boyuk E, Saracoglu ZN, et al. Perianal basal cell carcinoma. Case Rep Dermatol. 2015;7:25-28.
- Collins PS, Farber GA, Hegre AM. Basal-cell carcinoma of the vulva. J Dermatol Surg Oncol. 1981;7:711-714.
- Penneys NS, Glaser DA. The incidence of cancellation and nonattendance at a dermatology clinic. J Am Acad Dermatol. 1990;40:714-718.
- Cronin P, DeCoste L, Kimball A. A multivariate analysis of dermatology missed appointment predictors. JAMA Dermatology. 2013;149:1435-1437.
- Moustafa FA, Ramsey L, Huang KE, et al. Factors associated with missed dermatology appointments. Cutis. 2015;96:E20-E23.
- Canizares MJ, Penneys NS. The incidence of nonattendance at an urgent care dermatology clinic. J Am Acad Dermatol. 2002;46:457-459.
Practice Points
- Basal cell carcinoma is less common in non–sun-exposed areas of the body and is exceptionally rare in the perineal and perianal regions.
- Thorough total-body skin examinations may lead to early detection of asymptomatic skin lesions, allowing for earlier and less invasive treatment.
- Appointment attendance and patient compliance are common challenges that dermatologists face. Patient reminders via their preferred method of communication may help reduce missed dermatology appointments.
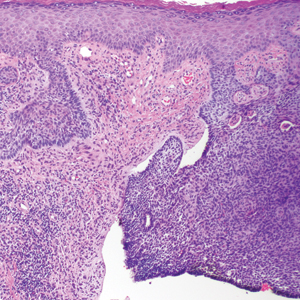
Critical anemia • light-headedness • bilateral leg swelling • Dx?
THE CASE
A 40-year-old man was referred to the emergency department (ED) with critical anemia after routine blood work at an outside clinic showed a hemoglobin level of 3.5 g/dL. On presentation, he reported symptoms of fatigue, shortness of breath, bilateral leg swelling, dizziness (characterized as light-headedness), and frequent heartburn. He said that the symptoms began 5 weeks earlier, after he was exposed to a relative with hand, foot, and mouth disease.
Additionally, the patient reported an intentional 14-lb weight loss over the 6 months prior to presentation. He denied fever, rash, chest pain, loss of consciousness, headache, abdominal pain, hematemesis, melena, and hematochezia. His medical history was significant for peptic ulcer disease (diagnosed and treated at age 8). He did not recall the specifics, and he denied any related chronic symptoms or complications. His family history (paternal) was significant for colon cancer.
The physical exam revealed conjunctival pallor, skin pallor, jaundice, +1 bilateral lower extremity edema, tachycardia, and tachypnea. Stool Hemoccult was negative. On repeat complete blood count (performed in the ED), hemoglobin was found to be 3.1 g/dL with a mean corpuscular volume of 47 fL.
THE DIAGNOSIS
The patient was admitted to the family medicine service and received 4 units of packed red blood cells, which increased his hemoglobin to the target goal of >7 g/dL. A colonoscopy and an esophagogastroduodenoscopy (EGD) were performed (FIGURE 1A-1C), with results suggestive of diverticulosis, probable Barrett’s mucosa, esophageal ulcer, huge hiatal hernia (at least one-half of the stomach was in the chest), and Cameron ulcers. Esophageal biopsies showed cardiac mucosa with chronic inflammation. Esophageal ulcer biopsies revealed Barrett’s esophagus without dysplasia. Duodenal biopsies displayed normal mucosa.
DISCUSSION
Although rare, Cameron ulcers must be considered in the differential diagnosis of patients with chronic anemia of unknown origin. The potential for these lesions to result in chronic blood loss, which could over time manifest as severe anemia or hypovolemic shock, makes proper diagnosis and prompt treatment especially important.4,5
In our patient’s case, his severe anemia was likely the result of a combination of the esophageal and Cameron ulcers evidenced on EGD, rather than any single ulcer. In our review of the literature, we found no reports of any patients with anemia and Cameron ulcers who presented with hemoglobin levels as low as our patient had.
Treat with a PPI and iron supplementation
Multiple EGDs may be needed to properly diagnose Cameron ulcers, as they can be difficult to identify. Once a patient receives the diagnosis, he or she will typically be put on a daily proton pump inhibitor (PPI) regimen, such as omeprazole 20 mg bid. However, since many patients with Cameron ulcers also have acid-related problems (as was true in this case), a multifactorial acid suppression approach may be warranted.1 This may include recommending lifestyle modifications (eg, eating small meals, avoiding foods that provoke symptoms, or losing weight) and prescribing medications in addition to a PPI, such as an H2 blocker (eg, 300 mg qid, before meals and at bedtime).
In addition, iron sulfate (325 mg/d, in this case) and blood transfusions may be required to treat the anemia. In refractory cases, endoscopic or surgical interventions, such as hemoclipping, Nissen fundoplication, or laparoscopic gastropexy, may need to be performed.2
Our patient was given a prescription for ferrous sulfate 325 mg/d and omeprazole 20 mg bid. His symptoms improved with treatment, and he was discharged on Day 5; his hemoglobin remained >7 g/dL.
THE TAKEAWAY
The association between chronic iron deficiency anemia and Cameron ulcers has been established but is commonly overlooked in patients presenting with unexplained anemia or an undiagnosed hiatal hernia. This is likely due to their rarity as a cause of anemia, in general.
Furthermore, the lesions can be missed on EGD; multiple EGDs may be needed to make the diagnosis. Once diagnosed, Cameron ulcers typically respond well to twice daily PPI treatment. Patients with refractory, recurrent, or severe lesions, or large, symptomatic hiatal hernias should be referred for surgical assessment.
CORRESPONDENCE
Megan Yee, 801 Broadward Avenue NW, Grand Rapids, MI 49504; Megan.Yee@mercyhealth.com.
1. Maganty K, Smith RL. Cameron lesions: unusual cause of gastrointestinal bleeding and anemia. Digestion. 2008;77:214-217.
2. Camus M, Jensen DM, Ohning GV, et al. Severe upper gastrointestinal hemorrhage from linear gastric ulcers in large hiatal hernias: a large prospective case series of Cameron ulcers. Endoscopy. 2013;45:397-400.
3. Kimer N, Schmidt PN, Krag, A. Cameron lesions: an often overlooked cause of iron deficiency anaemia in patients with large hiatal hernias. BMJ Case Rep. 2010;2010.
4. Kapadia S, Jagroop S, Kumar, A. Cameron ulcers: an atypical source for a massive upper gastrointestinal bleed. World J Gastroenterol. 2012;18:4959-4961.
5. Gupta P, Suryadevara M, Das A, et al. Cameron ulcer causing severe anemia in a patient with diaphragmatic hernia. Am J Case Rep. 2015;16:733-736.
THE CASE
A 40-year-old man was referred to the emergency department (ED) with critical anemia after routine blood work at an outside clinic showed a hemoglobin level of 3.5 g/dL. On presentation, he reported symptoms of fatigue, shortness of breath, bilateral leg swelling, dizziness (characterized as light-headedness), and frequent heartburn. He said that the symptoms began 5 weeks earlier, after he was exposed to a relative with hand, foot, and mouth disease.
Additionally, the patient reported an intentional 14-lb weight loss over the 6 months prior to presentation. He denied fever, rash, chest pain, loss of consciousness, headache, abdominal pain, hematemesis, melena, and hematochezia. His medical history was significant for peptic ulcer disease (diagnosed and treated at age 8). He did not recall the specifics, and he denied any related chronic symptoms or complications. His family history (paternal) was significant for colon cancer.
The physical exam revealed conjunctival pallor, skin pallor, jaundice, +1 bilateral lower extremity edema, tachycardia, and tachypnea. Stool Hemoccult was negative. On repeat complete blood count (performed in the ED), hemoglobin was found to be 3.1 g/dL with a mean corpuscular volume of 47 fL.
THE DIAGNOSIS
The patient was admitted to the family medicine service and received 4 units of packed red blood cells, which increased his hemoglobin to the target goal of >7 g/dL. A colonoscopy and an esophagogastroduodenoscopy (EGD) were performed (FIGURE 1A-1C), with results suggestive of diverticulosis, probable Barrett’s mucosa, esophageal ulcer, huge hiatal hernia (at least one-half of the stomach was in the chest), and Cameron ulcers. Esophageal biopsies showed cardiac mucosa with chronic inflammation. Esophageal ulcer biopsies revealed Barrett’s esophagus without dysplasia. Duodenal biopsies displayed normal mucosa.
DISCUSSION
Although rare, Cameron ulcers must be considered in the differential diagnosis of patients with chronic anemia of unknown origin. The potential for these lesions to result in chronic blood loss, which could over time manifest as severe anemia or hypovolemic shock, makes proper diagnosis and prompt treatment especially important.4,5
In our patient’s case, his severe anemia was likely the result of a combination of the esophageal and Cameron ulcers evidenced on EGD, rather than any single ulcer. In our review of the literature, we found no reports of any patients with anemia and Cameron ulcers who presented with hemoglobin levels as low as our patient had.
Treat with a PPI and iron supplementation
Multiple EGDs may be needed to properly diagnose Cameron ulcers, as they can be difficult to identify. Once a patient receives the diagnosis, he or she will typically be put on a daily proton pump inhibitor (PPI) regimen, such as omeprazole 20 mg bid. However, since many patients with Cameron ulcers also have acid-related problems (as was true in this case), a multifactorial acid suppression approach may be warranted.1 This may include recommending lifestyle modifications (eg, eating small meals, avoiding foods that provoke symptoms, or losing weight) and prescribing medications in addition to a PPI, such as an H2 blocker (eg, 300 mg qid, before meals and at bedtime).
In addition, iron sulfate (325 mg/d, in this case) and blood transfusions may be required to treat the anemia. In refractory cases, endoscopic or surgical interventions, such as hemoclipping, Nissen fundoplication, or laparoscopic gastropexy, may need to be performed.2
Our patient was given a prescription for ferrous sulfate 325 mg/d and omeprazole 20 mg bid. His symptoms improved with treatment, and he was discharged on Day 5; his hemoglobin remained >7 g/dL.
THE TAKEAWAY
The association between chronic iron deficiency anemia and Cameron ulcers has been established but is commonly overlooked in patients presenting with unexplained anemia or an undiagnosed hiatal hernia. This is likely due to their rarity as a cause of anemia, in general.
Furthermore, the lesions can be missed on EGD; multiple EGDs may be needed to make the diagnosis. Once diagnosed, Cameron ulcers typically respond well to twice daily PPI treatment. Patients with refractory, recurrent, or severe lesions, or large, symptomatic hiatal hernias should be referred for surgical assessment.
CORRESPONDENCE
Megan Yee, 801 Broadward Avenue NW, Grand Rapids, MI 49504; Megan.Yee@mercyhealth.com.
THE CASE
A 40-year-old man was referred to the emergency department (ED) with critical anemia after routine blood work at an outside clinic showed a hemoglobin level of 3.5 g/dL. On presentation, he reported symptoms of fatigue, shortness of breath, bilateral leg swelling, dizziness (characterized as light-headedness), and frequent heartburn. He said that the symptoms began 5 weeks earlier, after he was exposed to a relative with hand, foot, and mouth disease.
Additionally, the patient reported an intentional 14-lb weight loss over the 6 months prior to presentation. He denied fever, rash, chest pain, loss of consciousness, headache, abdominal pain, hematemesis, melena, and hematochezia. His medical history was significant for peptic ulcer disease (diagnosed and treated at age 8). He did not recall the specifics, and he denied any related chronic symptoms or complications. His family history (paternal) was significant for colon cancer.
The physical exam revealed conjunctival pallor, skin pallor, jaundice, +1 bilateral lower extremity edema, tachycardia, and tachypnea. Stool Hemoccult was negative. On repeat complete blood count (performed in the ED), hemoglobin was found to be 3.1 g/dL with a mean corpuscular volume of 47 fL.
THE DIAGNOSIS
The patient was admitted to the family medicine service and received 4 units of packed red blood cells, which increased his hemoglobin to the target goal of >7 g/dL. A colonoscopy and an esophagogastroduodenoscopy (EGD) were performed (FIGURE 1A-1C), with results suggestive of diverticulosis, probable Barrett’s mucosa, esophageal ulcer, huge hiatal hernia (at least one-half of the stomach was in the chest), and Cameron ulcers. Esophageal biopsies showed cardiac mucosa with chronic inflammation. Esophageal ulcer biopsies revealed Barrett’s esophagus without dysplasia. Duodenal biopsies displayed normal mucosa.
DISCUSSION
Although rare, Cameron ulcers must be considered in the differential diagnosis of patients with chronic anemia of unknown origin. The potential for these lesions to result in chronic blood loss, which could over time manifest as severe anemia or hypovolemic shock, makes proper diagnosis and prompt treatment especially important.4,5
In our patient’s case, his severe anemia was likely the result of a combination of the esophageal and Cameron ulcers evidenced on EGD, rather than any single ulcer. In our review of the literature, we found no reports of any patients with anemia and Cameron ulcers who presented with hemoglobin levels as low as our patient had.
Treat with a PPI and iron supplementation
Multiple EGDs may be needed to properly diagnose Cameron ulcers, as they can be difficult to identify. Once a patient receives the diagnosis, he or she will typically be put on a daily proton pump inhibitor (PPI) regimen, such as omeprazole 20 mg bid. However, since many patients with Cameron ulcers also have acid-related problems (as was true in this case), a multifactorial acid suppression approach may be warranted.1 This may include recommending lifestyle modifications (eg, eating small meals, avoiding foods that provoke symptoms, or losing weight) and prescribing medications in addition to a PPI, such as an H2 blocker (eg, 300 mg qid, before meals and at bedtime).
In addition, iron sulfate (325 mg/d, in this case) and blood transfusions may be required to treat the anemia. In refractory cases, endoscopic or surgical interventions, such as hemoclipping, Nissen fundoplication, or laparoscopic gastropexy, may need to be performed.2
Our patient was given a prescription for ferrous sulfate 325 mg/d and omeprazole 20 mg bid. His symptoms improved with treatment, and he was discharged on Day 5; his hemoglobin remained >7 g/dL.
THE TAKEAWAY
The association between chronic iron deficiency anemia and Cameron ulcers has been established but is commonly overlooked in patients presenting with unexplained anemia or an undiagnosed hiatal hernia. This is likely due to their rarity as a cause of anemia, in general.
Furthermore, the lesions can be missed on EGD; multiple EGDs may be needed to make the diagnosis. Once diagnosed, Cameron ulcers typically respond well to twice daily PPI treatment. Patients with refractory, recurrent, or severe lesions, or large, symptomatic hiatal hernias should be referred for surgical assessment.
CORRESPONDENCE
Megan Yee, 801 Broadward Avenue NW, Grand Rapids, MI 49504; Megan.Yee@mercyhealth.com.
1. Maganty K, Smith RL. Cameron lesions: unusual cause of gastrointestinal bleeding and anemia. Digestion. 2008;77:214-217.
2. Camus M, Jensen DM, Ohning GV, et al. Severe upper gastrointestinal hemorrhage from linear gastric ulcers in large hiatal hernias: a large prospective case series of Cameron ulcers. Endoscopy. 2013;45:397-400.
3. Kimer N, Schmidt PN, Krag, A. Cameron lesions: an often overlooked cause of iron deficiency anaemia in patients with large hiatal hernias. BMJ Case Rep. 2010;2010.
4. Kapadia S, Jagroop S, Kumar, A. Cameron ulcers: an atypical source for a massive upper gastrointestinal bleed. World J Gastroenterol. 2012;18:4959-4961.
5. Gupta P, Suryadevara M, Das A, et al. Cameron ulcer causing severe anemia in a patient with diaphragmatic hernia. Am J Case Rep. 2015;16:733-736.
1. Maganty K, Smith RL. Cameron lesions: unusual cause of gastrointestinal bleeding and anemia. Digestion. 2008;77:214-217.
2. Camus M, Jensen DM, Ohning GV, et al. Severe upper gastrointestinal hemorrhage from linear gastric ulcers in large hiatal hernias: a large prospective case series of Cameron ulcers. Endoscopy. 2013;45:397-400.
3. Kimer N, Schmidt PN, Krag, A. Cameron lesions: an often overlooked cause of iron deficiency anaemia in patients with large hiatal hernias. BMJ Case Rep. 2010;2010.
4. Kapadia S, Jagroop S, Kumar, A. Cameron ulcers: an atypical source for a massive upper gastrointestinal bleed. World J Gastroenterol. 2012;18:4959-4961.
5. Gupta P, Suryadevara M, Das A, et al. Cameron ulcer causing severe anemia in a patient with diaphragmatic hernia. Am J Case Rep. 2015;16:733-736.
Isolated ocular metastases from lung cancer
Non–small cell lung cancer constitutes 80%-85% of lung cancers, and 40% of NSCLC are adenocarcinoma. It is rare to find intraocular metastasis from lung cancer. In this article, we present the case of a patient who presented with complaints of diminished vision redness of the eye and was found to have intra-ocular metastases from lung cancer.
Case presentation and summary
A 60-year-old man with a 40-pack per year history of smoking presented to multiple ophthalmologists with complaints of decreased vision and redness of the left eye. He was eventually evaluated by an ophthalmologist who performed a biopsy of the anterior chamber of the eye. Histologic findings were consistent with adenocarcinoma of lung primary (Figures 1 and 2).
After the diagnosis, a chest X-ray showed that the patient had a left lower lung mass. The results of his physical exam were all within normal limits, with the exception of decreased visual acuity in the left eye. The results of his laboratory studies, including complete blood count and serum chemistries, were also within normal limits. Imaging studies – including a computed-tomography (CT) scan of the chest, abdomen, and pelvis and a full-body positron-emission tomography–CT scan – showed a hypermetabolic left lower lobe mass 4.5 cm and right lower paratracheal lymph node metastasis 2 cm with a small focus of increased uptake alone the medial aspect of the left globe (Figures 3 and 4).
An MRI orbit was performed in an attempt to better characterize the left eye mass, but no optic lesion was identified. A biopsy of the left lower lung mass was consistent with non–small-cell lung cancer (NSCLC). Aside from the isolated left eye metastases, the patient did not have evidence of other distant metastatic involvement.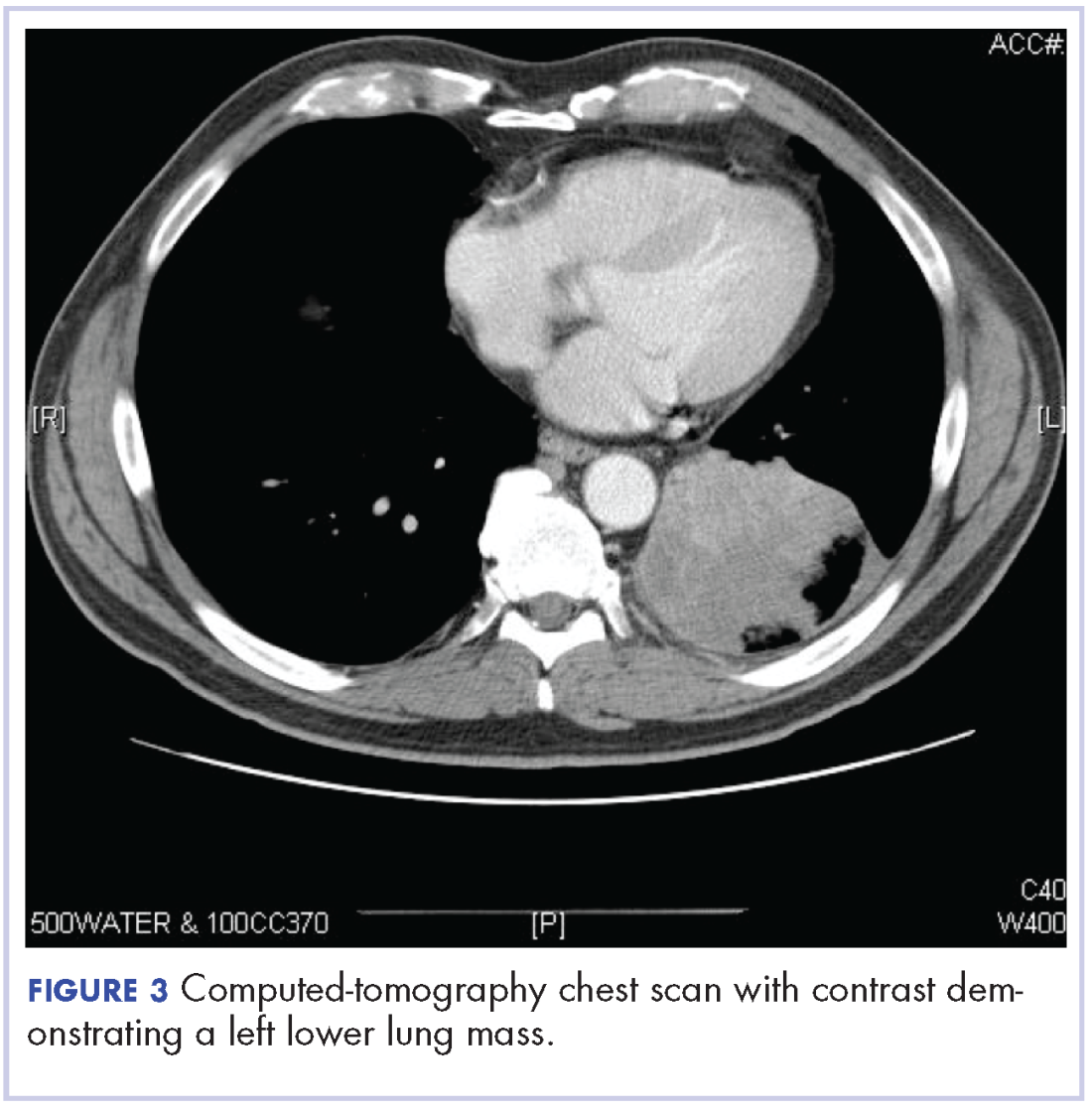

He was started on palliative chemotherapy on a clinical trial and received intravenous carboplatin AUC 6, pemetrexed 500 mg/m2, and bevacizumab 15 mg/kg every 3 weeks. He received 1 dose intraocular bevacizumab injection before initiation of systemic chemotherapy as he was symptomatic from the intraocular metastases. Within 2 weeks after intravitreal bevacizumab was administered, the patient had subjective improvement in vision. Mutational analysis to identify if the patient would benefit from targeted therapy showed no presence of EGFR mutation and ALK gene rearrangement, and that the patient was K-RAS mutant.
After treatment initiation, interval imaging studies (a computed-tomography scan of the chest, abdomen, pelvis; and magnetic-resonance imaging of the brain) after 3 cycles showed no evidence of disease progression, and after 4 cycles of chemotherapy with these drugs, the patient was started on maintenance chemotherapy with bevacizumab 15 mg/kg and pemetrexed 500 mg/m2.
Discussion
Choroidal metastasis is the most common site of intraocular tumor. In an autopsy study of 230 patients with carcinoma, 12% of cases demonstrated histologic evidence of ocular metastasis.1 A retrospective series of patients with malignant involvement of the eye, 66% of patients had a known history of primary cancer and in 34% of patients the ocular tumor was the first sign of cancer.2 The most common cancers that were found to have ocular metastasis were lung and breast cancer.2 Adenocarcinoma was the most common histologic type of lung cancer to result in ocular metastases and was seen in 41% of patients.3
Decreased or blurred vision with redness as the primary complaint of NSCLC is rare. Only a few case reports are available. Abundo and colleagues reported that 0.7%-12% of patients with lung cancer develop ocular metastases.4 Therefore, routine ophthalmologic screening for ocular metastases in patients with cancer has not been pursued in asymptomatic patients.5 Ophthalmological evaluation is recommended in symptomatic patients.
Metastatic involvement of two or more other organs was found to be a risk factor for development of choroidal metastasis in patients with lung cancer though in our patient no evidence of other organ involvement was found.5 The most common site of metastases in patients with NSCLC with ocular metastases was found to be the liver. Choroidal metastases was reported to be the sixth common site of metastases in patients with lung cancer.5
Treatment of ocular manifestations has been generally confined to surgical resection or radiation therapy, but advances in chemotherapy and development of novel targeted agents have shown promising results.7 Median life expectancy after a diagnosis of uveal metastases was reported to be 12 months in a retrospective study, which is similar to the reported median survival in metastatic NSCLC.8
Our patient was enrolled in a clinical trial and was treated with a regimen of carboplatin, paclitaxel, and bevacizumab. On presentation, he had significant impairment of vision with pain. He was treated with intravitreal bevacizumab yielding improvement in his visual symptoms. Bevacizumab is a vascular endothelial growth factor receptor monoclonal antibody approved for use in patients with metastatic lung cancer. Other pathways that have been reported in development of lung cancer involve the ALK gene translocation, and EGFR and K-RAS mutations, and targeted therapy has shown good results in cancer patients with these molecular defects. Randomized clinical trials in patients with advanced NSCLC and an EGFR mutation have shown significant improvement in overall survival with the use of erlotinib, a tyrosine kinase inhibitor targeting the epidermal growth factor receptor.9 Similarly, crizotinib has shown promising results in patients with metastatic NSCLC who have ELM-ALK rearrangement.10 As our patient’s tumor did not have either of these mutations, he was initiated on chemotherapy with bevacizumab. The presence of a K-RAS mutation in this patient further supported the use of front-line chemotherapy given that it may confer resistance against agents that target the EGFR pathway.
In our review of the literature, we found cases of patients with ocular metastases who responded well to therapy with targeted agents (Table).
Singh and colleagues did a systematic review of 55 cases of patients with lung cancer and choroidal metastases and found that the type of therapy depended on when the diagnosis had been made in relation to the advent of targeted therapy: cases diagnosed before targeted therapy had received radiation therapy or enucleation.6 As far as we could ascertain, there have been no randomized studies evaluating the impact of various targeted therapies or systemic chemotherapy on ocular metastases, although case reports have documented improvement in vision and regression of metastases with such therapy.
Conclusion
The goal of therapy in metastatic lung cancer is palliation of symptoms and improvement in patient quality of life with prolongation in overall survival. The newer targeted chemotherapeutic agents assist in achieving these goals and may decrease the morbidity associated from radiation or surgery with improvement in vision and regression of ocular metastatic lesions. Targeted therapies should be considered in the treatment of patients with ocular metastases from NSCLC.
1. Bloch RS, Gartner S. The incidence of ocular metastatic carcinoma. Arch Ophthalmol-Chic. 1971;85(6):673-675.
2. Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology. 1997;104(8):1265-1276.
3. Kreusel KM, Bechrakis NE, Wiegel T, Krause L, Foerster MH. Incidence and clinical characteristics of symptomatic choroidal metastasis from lung cancer. Acta Ophthalmol. 2008;86(5):515-519.
4. Abundo RE, Orenic CJ, Anderson SF, Townsend JC. Choroidal metastases resulting from carcinoma of the lung. J Am Optom Assoc. 1997;68(2):95-108.
5. Kreusel KM, Wiegel T, Stange M, Bornfeld N, Hinkelbein W, Foerster MH. Choroidal metastasis in disseminated lung cancer: frequency and risk factors. Am J Ophthalmol. 2002;134(3):445-447.
6. Singh N, Kulkarni P, Aggarwal AN, et al. Choroidal metastasis as a presenting manifestation of lung cancer: a report of 3 cases and systematic review of the literature. Medicine (Baltimore). 2012;91(4):179-194.
7. Chen CJ, McCoy AN, Brahmer J, Handa JT. Emerging treatments for choroidal metastases. Surv Ophthalmol. 2011;56(6):511-521.
8. Shah SU, Mashayekhi A, Shields CL, et al. Uveal metastasis from lung cancer: clinical features, treatment, and outcome in 194 patients. Ophthalmology. 2014;121(1):352-357.
9. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123-132.
10. Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385-2394.
11. Kim SW, Kim MJ, Huh K, Oh J. Complete regression of choroidal metastasis secondary to non-small-cell lung cancer with intravitreal bevacizumab and oral erlotinib combination therapy. Ophthalmologica. 2009;223(6):411-413.
12. George B, Wirostko WJ, Connor TB, Choong NW. Complete and durable response of choroid metastasis from non-small cell lung cancer with systemic bevacizumab and chemotherapy. J Thorac Oncol. 2009;4(5):661-662.
13. Inoue M, Watanabe Y, Yamane S, et al. Choroidal metastasis with adenocarcinoma of the lung treated with gefitinib. Eur J Ophthalmol. 2010;20(5):963-965.
14. Shimomura I, Tada Y, Miura G, et al. Choroidal metastasis of non-small cell lung cancer that responded to gefitinib. https://www.hindawi.com/journals/criopm/2013/213124/. Published 2013. Accessed May 4, 2017.
15. Feng Y, Singh AD, Lanigan C, Tubbs RR, Ma PC. Choroidal metastases responsive to crizotinib therapy in a lung adenocarcinoma patient with ALK 2p23 fusion identified by ALK immunohistochemistry. J Thorac Oncol. 2013;8(12):e109-111.
Non–small cell lung cancer constitutes 80%-85% of lung cancers, and 40% of NSCLC are adenocarcinoma. It is rare to find intraocular metastasis from lung cancer. In this article, we present the case of a patient who presented with complaints of diminished vision redness of the eye and was found to have intra-ocular metastases from lung cancer.
Case presentation and summary
A 60-year-old man with a 40-pack per year history of smoking presented to multiple ophthalmologists with complaints of decreased vision and redness of the left eye. He was eventually evaluated by an ophthalmologist who performed a biopsy of the anterior chamber of the eye. Histologic findings were consistent with adenocarcinoma of lung primary (Figures 1 and 2).
After the diagnosis, a chest X-ray showed that the patient had a left lower lung mass. The results of his physical exam were all within normal limits, with the exception of decreased visual acuity in the left eye. The results of his laboratory studies, including complete blood count and serum chemistries, were also within normal limits. Imaging studies – including a computed-tomography (CT) scan of the chest, abdomen, and pelvis and a full-body positron-emission tomography–CT scan – showed a hypermetabolic left lower lobe mass 4.5 cm and right lower paratracheal lymph node metastasis 2 cm with a small focus of increased uptake alone the medial aspect of the left globe (Figures 3 and 4).
An MRI orbit was performed in an attempt to better characterize the left eye mass, but no optic lesion was identified. A biopsy of the left lower lung mass was consistent with non–small-cell lung cancer (NSCLC). Aside from the isolated left eye metastases, the patient did not have evidence of other distant metastatic involvement.
He was started on palliative chemotherapy on a clinical trial and received intravenous carboplatin AUC 6, pemetrexed 500 mg/m2, and bevacizumab 15 mg/kg every 3 weeks. He received 1 dose intraocular bevacizumab injection before initiation of systemic chemotherapy as he was symptomatic from the intraocular metastases. Within 2 weeks after intravitreal bevacizumab was administered, the patient had subjective improvement in vision. Mutational analysis to identify if the patient would benefit from targeted therapy showed no presence of EGFR mutation and ALK gene rearrangement, and that the patient was K-RAS mutant.
After treatment initiation, interval imaging studies (a computed-tomography scan of the chest, abdomen, pelvis; and magnetic-resonance imaging of the brain) after 3 cycles showed no evidence of disease progression, and after 4 cycles of chemotherapy with these drugs, the patient was started on maintenance chemotherapy with bevacizumab 15 mg/kg and pemetrexed 500 mg/m2.
Discussion
Choroidal metastasis is the most common site of intraocular tumor. In an autopsy study of 230 patients with carcinoma, 12% of cases demonstrated histologic evidence of ocular metastasis.1 A retrospective series of patients with malignant involvement of the eye, 66% of patients had a known history of primary cancer and in 34% of patients the ocular tumor was the first sign of cancer.2 The most common cancers that were found to have ocular metastasis were lung and breast cancer.2 Adenocarcinoma was the most common histologic type of lung cancer to result in ocular metastases and was seen in 41% of patients.3
Decreased or blurred vision with redness as the primary complaint of NSCLC is rare. Only a few case reports are available. Abundo and colleagues reported that 0.7%-12% of patients with lung cancer develop ocular metastases.4 Therefore, routine ophthalmologic screening for ocular metastases in patients with cancer has not been pursued in asymptomatic patients.5 Ophthalmological evaluation is recommended in symptomatic patients.
Metastatic involvement of two or more other organs was found to be a risk factor for development of choroidal metastasis in patients with lung cancer though in our patient no evidence of other organ involvement was found.5 The most common site of metastases in patients with NSCLC with ocular metastases was found to be the liver. Choroidal metastases was reported to be the sixth common site of metastases in patients with lung cancer.5
Treatment of ocular manifestations has been generally confined to surgical resection or radiation therapy, but advances in chemotherapy and development of novel targeted agents have shown promising results.7 Median life expectancy after a diagnosis of uveal metastases was reported to be 12 months in a retrospective study, which is similar to the reported median survival in metastatic NSCLC.8
Our patient was enrolled in a clinical trial and was treated with a regimen of carboplatin, paclitaxel, and bevacizumab. On presentation, he had significant impairment of vision with pain. He was treated with intravitreal bevacizumab yielding improvement in his visual symptoms. Bevacizumab is a vascular endothelial growth factor receptor monoclonal antibody approved for use in patients with metastatic lung cancer. Other pathways that have been reported in development of lung cancer involve the ALK gene translocation, and EGFR and K-RAS mutations, and targeted therapy has shown good results in cancer patients with these molecular defects. Randomized clinical trials in patients with advanced NSCLC and an EGFR mutation have shown significant improvement in overall survival with the use of erlotinib, a tyrosine kinase inhibitor targeting the epidermal growth factor receptor.9 Similarly, crizotinib has shown promising results in patients with metastatic NSCLC who have ELM-ALK rearrangement.10 As our patient’s tumor did not have either of these mutations, he was initiated on chemotherapy with bevacizumab. The presence of a K-RAS mutation in this patient further supported the use of front-line chemotherapy given that it may confer resistance against agents that target the EGFR pathway.
In our review of the literature, we found cases of patients with ocular metastases who responded well to therapy with targeted agents (Table).
Singh and colleagues did a systematic review of 55 cases of patients with lung cancer and choroidal metastases and found that the type of therapy depended on when the diagnosis had been made in relation to the advent of targeted therapy: cases diagnosed before targeted therapy had received radiation therapy or enucleation.6 As far as we could ascertain, there have been no randomized studies evaluating the impact of various targeted therapies or systemic chemotherapy on ocular metastases, although case reports have documented improvement in vision and regression of metastases with such therapy.
Conclusion
The goal of therapy in metastatic lung cancer is palliation of symptoms and improvement in patient quality of life with prolongation in overall survival. The newer targeted chemotherapeutic agents assist in achieving these goals and may decrease the morbidity associated from radiation or surgery with improvement in vision and regression of ocular metastatic lesions. Targeted therapies should be considered in the treatment of patients with ocular metastases from NSCLC.
Non–small cell lung cancer constitutes 80%-85% of lung cancers, and 40% of NSCLC are adenocarcinoma. It is rare to find intraocular metastasis from lung cancer. In this article, we present the case of a patient who presented with complaints of diminished vision redness of the eye and was found to have intra-ocular metastases from lung cancer.
Case presentation and summary
A 60-year-old man with a 40-pack per year history of smoking presented to multiple ophthalmologists with complaints of decreased vision and redness of the left eye. He was eventually evaluated by an ophthalmologist who performed a biopsy of the anterior chamber of the eye. Histologic findings were consistent with adenocarcinoma of lung primary (Figures 1 and 2).
After the diagnosis, a chest X-ray showed that the patient had a left lower lung mass. The results of his physical exam were all within normal limits, with the exception of decreased visual acuity in the left eye. The results of his laboratory studies, including complete blood count and serum chemistries, were also within normal limits. Imaging studies – including a computed-tomography (CT) scan of the chest, abdomen, and pelvis and a full-body positron-emission tomography–CT scan – showed a hypermetabolic left lower lobe mass 4.5 cm and right lower paratracheal lymph node metastasis 2 cm with a small focus of increased uptake alone the medial aspect of the left globe (Figures 3 and 4).
An MRI orbit was performed in an attempt to better characterize the left eye mass, but no optic lesion was identified. A biopsy of the left lower lung mass was consistent with non–small-cell lung cancer (NSCLC). Aside from the isolated left eye metastases, the patient did not have evidence of other distant metastatic involvement.
He was started on palliative chemotherapy on a clinical trial and received intravenous carboplatin AUC 6, pemetrexed 500 mg/m2, and bevacizumab 15 mg/kg every 3 weeks. He received 1 dose intraocular bevacizumab injection before initiation of systemic chemotherapy as he was symptomatic from the intraocular metastases. Within 2 weeks after intravitreal bevacizumab was administered, the patient had subjective improvement in vision. Mutational analysis to identify if the patient would benefit from targeted therapy showed no presence of EGFR mutation and ALK gene rearrangement, and that the patient was K-RAS mutant.
After treatment initiation, interval imaging studies (a computed-tomography scan of the chest, abdomen, pelvis; and magnetic-resonance imaging of the brain) after 3 cycles showed no evidence of disease progression, and after 4 cycles of chemotherapy with these drugs, the patient was started on maintenance chemotherapy with bevacizumab 15 mg/kg and pemetrexed 500 mg/m2.
Discussion
Choroidal metastasis is the most common site of intraocular tumor. In an autopsy study of 230 patients with carcinoma, 12% of cases demonstrated histologic evidence of ocular metastasis.1 A retrospective series of patients with malignant involvement of the eye, 66% of patients had a known history of primary cancer and in 34% of patients the ocular tumor was the first sign of cancer.2 The most common cancers that were found to have ocular metastasis were lung and breast cancer.2 Adenocarcinoma was the most common histologic type of lung cancer to result in ocular metastases and was seen in 41% of patients.3
Decreased or blurred vision with redness as the primary complaint of NSCLC is rare. Only a few case reports are available. Abundo and colleagues reported that 0.7%-12% of patients with lung cancer develop ocular metastases.4 Therefore, routine ophthalmologic screening for ocular metastases in patients with cancer has not been pursued in asymptomatic patients.5 Ophthalmological evaluation is recommended in symptomatic patients.
Metastatic involvement of two or more other organs was found to be a risk factor for development of choroidal metastasis in patients with lung cancer though in our patient no evidence of other organ involvement was found.5 The most common site of metastases in patients with NSCLC with ocular metastases was found to be the liver. Choroidal metastases was reported to be the sixth common site of metastases in patients with lung cancer.5
Treatment of ocular manifestations has been generally confined to surgical resection or radiation therapy, but advances in chemotherapy and development of novel targeted agents have shown promising results.7 Median life expectancy after a diagnosis of uveal metastases was reported to be 12 months in a retrospective study, which is similar to the reported median survival in metastatic NSCLC.8
Our patient was enrolled in a clinical trial and was treated with a regimen of carboplatin, paclitaxel, and bevacizumab. On presentation, he had significant impairment of vision with pain. He was treated with intravitreal bevacizumab yielding improvement in his visual symptoms. Bevacizumab is a vascular endothelial growth factor receptor monoclonal antibody approved for use in patients with metastatic lung cancer. Other pathways that have been reported in development of lung cancer involve the ALK gene translocation, and EGFR and K-RAS mutations, and targeted therapy has shown good results in cancer patients with these molecular defects. Randomized clinical trials in patients with advanced NSCLC and an EGFR mutation have shown significant improvement in overall survival with the use of erlotinib, a tyrosine kinase inhibitor targeting the epidermal growth factor receptor.9 Similarly, crizotinib has shown promising results in patients with metastatic NSCLC who have ELM-ALK rearrangement.10 As our patient’s tumor did not have either of these mutations, he was initiated on chemotherapy with bevacizumab. The presence of a K-RAS mutation in this patient further supported the use of front-line chemotherapy given that it may confer resistance against agents that target the EGFR pathway.
In our review of the literature, we found cases of patients with ocular metastases who responded well to therapy with targeted agents (Table).
Singh and colleagues did a systematic review of 55 cases of patients with lung cancer and choroidal metastases and found that the type of therapy depended on when the diagnosis had been made in relation to the advent of targeted therapy: cases diagnosed before targeted therapy had received radiation therapy or enucleation.6 As far as we could ascertain, there have been no randomized studies evaluating the impact of various targeted therapies or systemic chemotherapy on ocular metastases, although case reports have documented improvement in vision and regression of metastases with such therapy.
Conclusion
The goal of therapy in metastatic lung cancer is palliation of symptoms and improvement in patient quality of life with prolongation in overall survival. The newer targeted chemotherapeutic agents assist in achieving these goals and may decrease the morbidity associated from radiation or surgery with improvement in vision and regression of ocular metastatic lesions. Targeted therapies should be considered in the treatment of patients with ocular metastases from NSCLC.
1. Bloch RS, Gartner S. The incidence of ocular metastatic carcinoma. Arch Ophthalmol-Chic. 1971;85(6):673-675.
2. Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology. 1997;104(8):1265-1276.
3. Kreusel KM, Bechrakis NE, Wiegel T, Krause L, Foerster MH. Incidence and clinical characteristics of symptomatic choroidal metastasis from lung cancer. Acta Ophthalmol. 2008;86(5):515-519.
4. Abundo RE, Orenic CJ, Anderson SF, Townsend JC. Choroidal metastases resulting from carcinoma of the lung. J Am Optom Assoc. 1997;68(2):95-108.
5. Kreusel KM, Wiegel T, Stange M, Bornfeld N, Hinkelbein W, Foerster MH. Choroidal metastasis in disseminated lung cancer: frequency and risk factors. Am J Ophthalmol. 2002;134(3):445-447.
6. Singh N, Kulkarni P, Aggarwal AN, et al. Choroidal metastasis as a presenting manifestation of lung cancer: a report of 3 cases and systematic review of the literature. Medicine (Baltimore). 2012;91(4):179-194.
7. Chen CJ, McCoy AN, Brahmer J, Handa JT. Emerging treatments for choroidal metastases. Surv Ophthalmol. 2011;56(6):511-521.
8. Shah SU, Mashayekhi A, Shields CL, et al. Uveal metastasis from lung cancer: clinical features, treatment, and outcome in 194 patients. Ophthalmology. 2014;121(1):352-357.
9. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123-132.
10. Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385-2394.
11. Kim SW, Kim MJ, Huh K, Oh J. Complete regression of choroidal metastasis secondary to non-small-cell lung cancer with intravitreal bevacizumab and oral erlotinib combination therapy. Ophthalmologica. 2009;223(6):411-413.
12. George B, Wirostko WJ, Connor TB, Choong NW. Complete and durable response of choroid metastasis from non-small cell lung cancer with systemic bevacizumab and chemotherapy. J Thorac Oncol. 2009;4(5):661-662.
13. Inoue M, Watanabe Y, Yamane S, et al. Choroidal metastasis with adenocarcinoma of the lung treated with gefitinib. Eur J Ophthalmol. 2010;20(5):963-965.
14. Shimomura I, Tada Y, Miura G, et al. Choroidal metastasis of non-small cell lung cancer that responded to gefitinib. https://www.hindawi.com/journals/criopm/2013/213124/. Published 2013. Accessed May 4, 2017.
15. Feng Y, Singh AD, Lanigan C, Tubbs RR, Ma PC. Choroidal metastases responsive to crizotinib therapy in a lung adenocarcinoma patient with ALK 2p23 fusion identified by ALK immunohistochemistry. J Thorac Oncol. 2013;8(12):e109-111.
1. Bloch RS, Gartner S. The incidence of ocular metastatic carcinoma. Arch Ophthalmol-Chic. 1971;85(6):673-675.
2. Shields CL, Shields JA, Gross NE, Schwartz GP, Lally SE. Survey of 520 eyes with uveal metastases. Ophthalmology. 1997;104(8):1265-1276.
3. Kreusel KM, Bechrakis NE, Wiegel T, Krause L, Foerster MH. Incidence and clinical characteristics of symptomatic choroidal metastasis from lung cancer. Acta Ophthalmol. 2008;86(5):515-519.
4. Abundo RE, Orenic CJ, Anderson SF, Townsend JC. Choroidal metastases resulting from carcinoma of the lung. J Am Optom Assoc. 1997;68(2):95-108.
5. Kreusel KM, Wiegel T, Stange M, Bornfeld N, Hinkelbein W, Foerster MH. Choroidal metastasis in disseminated lung cancer: frequency and risk factors. Am J Ophthalmol. 2002;134(3):445-447.
6. Singh N, Kulkarni P, Aggarwal AN, et al. Choroidal metastasis as a presenting manifestation of lung cancer: a report of 3 cases and systematic review of the literature. Medicine (Baltimore). 2012;91(4):179-194.
7. Chen CJ, McCoy AN, Brahmer J, Handa JT. Emerging treatments for choroidal metastases. Surv Ophthalmol. 2011;56(6):511-521.
8. Shah SU, Mashayekhi A, Shields CL, et al. Uveal metastasis from lung cancer: clinical features, treatment, and outcome in 194 patients. Ophthalmology. 2014;121(1):352-357.
9. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123-132.
10. Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385-2394.
11. Kim SW, Kim MJ, Huh K, Oh J. Complete regression of choroidal metastasis secondary to non-small-cell lung cancer with intravitreal bevacizumab and oral erlotinib combination therapy. Ophthalmologica. 2009;223(6):411-413.
12. George B, Wirostko WJ, Connor TB, Choong NW. Complete and durable response of choroid metastasis from non-small cell lung cancer with systemic bevacizumab and chemotherapy. J Thorac Oncol. 2009;4(5):661-662.
13. Inoue M, Watanabe Y, Yamane S, et al. Choroidal metastasis with adenocarcinoma of the lung treated with gefitinib. Eur J Ophthalmol. 2010;20(5):963-965.
14. Shimomura I, Tada Y, Miura G, et al. Choroidal metastasis of non-small cell lung cancer that responded to gefitinib. https://www.hindawi.com/journals/criopm/2013/213124/. Published 2013. Accessed May 4, 2017.
15. Feng Y, Singh AD, Lanigan C, Tubbs RR, Ma PC. Choroidal metastases responsive to crizotinib therapy in a lung adenocarcinoma patient with ALK 2p23 fusion identified by ALK immunohistochemistry. J Thorac Oncol. 2013;8(12):e109-111.
Resolution of refractory pruritus with aprepitant in a patient with microcystic adnexal carcinoma
Substance P is an important neurotransmitter implicated in itch pathways.1 After binding to its receptor, neurokinin-1 (NK-1), substance P induces release of factors including histamine, which may cause pruritus.2 Recent literature has reported successful use of aprepitant, an NK-1 antagonist that has been approved by the US Food and Drug Administration for the treatment of chemotherapy-induced nausea and vomiting, for treatment of pruritus. We report here the case of a patient with microcystic adnexal carcinoma (MAC) who presented with refractory pruritus and who had rapid and complete resolution of itch after administration of aprepitant.
Case presentation and summary
A 73-year-old man presented with a 12-year history of a small nodule on his philtrum, which had been increasing in size. He subsequently developed upper-lip numbness and nasal induration. He complained of 2.5 months of severe, debilitating, full-body pruritus. His symptoms were refractory to treatment with prednisone, gabapentin, doxycycline, doxepin, antihistamines, and topical steroids. At the time of consultation, he was being treated with hydroxyzine and topical pramocaine lotion with minimal relief.
At initial dermatologic evaluation, his tumor involved the lower two-thirds of the nose and entire upper cutaneous lip. There was a 4-mm rolled ulcer on the nasal tip and a 1-cm exophytic, smooth nodule on the left upper lip with palpable 4-cm submandibular adenopathy (Figure). Skin examination otherwise revealed linear excoriations on the upper back with no additional primary lesions. The nodule was biopsied, and the patient was diagnosed with MAC with gross nodal involvement. Laboratory findings including serum chemistries, blood urea nitrogen, complete blood cell count, thyroid, and liver function were normal. Positron emission tomography-computed tomography (PET-CT) imaging was negative for distant metastases.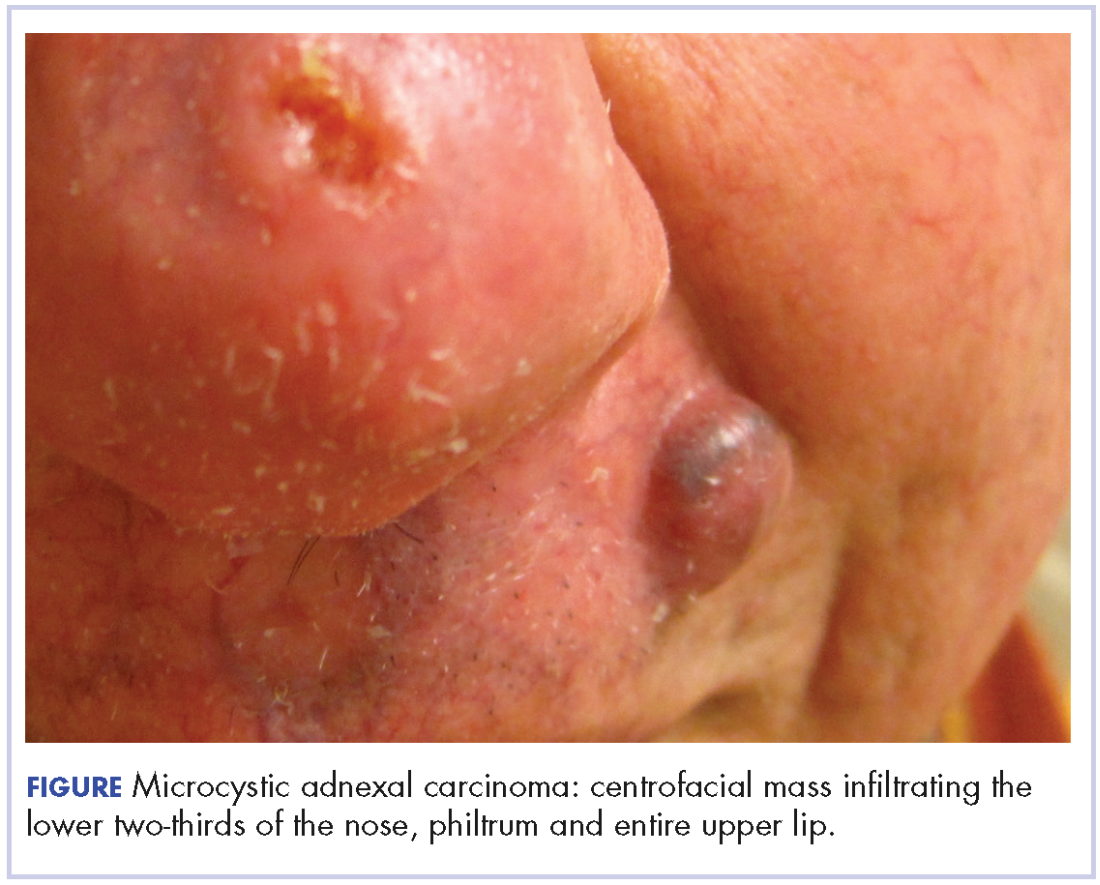
Treatment was initiated with oral aprepitant – 125 mg on day 1, 80 mg on day 2, and 80 mg on day 3 –with concomitant weekly carboplatin (AUC 1.5) and paclitaxel (30 mg/m2) as well as radiation. Within hours after the first dose of aprepitant, the patient reported a notable cessation in his pruritus. He reported that after 5 hours, his skin “finally turned off” and over the hour that followed, he had complete resolution of symptoms. He completed chemoradiation with a significant disease response. Despite persistent MAC confined to the philtrum, he has been followed for over 2 years without recurrence of itch.
Discussion
MAC is an uncommon cutaneous malignancy of sweat and eccrine gland differentiation. In all, 700 cases of MAC have been described in the literature; a 2008 review estimated the incidence of metastasis at around 2.1%.3 Though metastasis is exceedingly rare, the tumor is locally aggressive and there are reports of invasion into the muscle, perichondrium, periosteum, bone marrow, as well as perineural spaces and vascular adventitia.4
The clinical presentation of MAC includes smooth, flesh-colored or yellow papules, nodules, or plaques.3 Patients often present with numbness, paresthesia, and burning in the area of involvement because of neural infiltration with tumor. Despite the rarity of MAC, pruritus has been reported as a presenting symptom in 1 other case in the literature.4 Our case represents the first report of MAC presenting with a grossly enlarging centrofacial mass, lymph node involvement, and severe full-body pruritus. Our patient responded completely, and within hours, to treatment with aprepitant after experiencing months of failure with conventional antipruritus treatments and without recurrence in symptoms in more than 2 years of follow-up.
Aprepitant blocks the binding of substance P to its receptor NK-1 and has been approved as an anti-emetic for chemotherapy patients. Substance P has been shown to be important in both nausea and itch pathways. The largest prospective study to date on aprepitant for the indication of pruritus in 45 patients with metastatic solid tumors demonstrated a 91% response rate, defined by >50% reduction in pruritus intensity, and 13% recurrence rate that occurred at a median of 7 weeks after initial treatment.5 Aprepitant treatment has been used with success for pruritus associated with both malignant and nonmalignant conditions in at least 74 patients,6 among whom the malignant conditions included cutaneous T-cell lymphoma, Hodgkin lymphoma, and metastatic solid tumors.5-7 Aprepitant has also been used for erlotinib- and nivolumab-induced pruritus in non–small cell lung cancer, which suggests a possible future role for aprepitant in the treatment of pruritus secondary to novel cancer therapies, perhaps including immune checkpoint inhibitors.8-10
However, despite those reports, and likely owing to the multifactorial nature of pruritus, aprepitant is not unviversally effective. Mechanisms of malignancy-associated itch are yet to be elucidated, and optimal patient selection for aprepitant use needs to be determined. However, our patient’s notable response supports the increasing evidence that substance P is a key mediator of pruritus and that disruption of binding to its receptor may result in significant improvement in symptoms in certain patients. It remains to be seen whether the cell type or the tendency toward neural invasion plays a role. Large, randomized studies are needed to guide patient selection and confirm the findings reported here and in the literature, with careful documentation of and close attention paid to timing of pruritus relief and improvement in patient quality of life. Aprepitant might be an important therapeutic tool for refractory, malignancy-associated pruritus, in which patient quality of life is especially critical.
Acknowledgments
This work was presented at the Multinational Association of Supportive Care and Cancer Meeting, in Miami Florida, June 26-28, 2014. The authors are indebted to Saajar Jadeja for his assistance preparing the manuscript.
1. Wallengren J. Neuroanatomy and neurophysiology of itch. Dermatol Ther. 2005;18(4):292-303.
2. Kulka M, Sheen CH, Tancowny BP, Grammer LC, Schleimer RP. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology. 2008;123(3):398-410.
3. Wetter R, Goldstein GD. Microcystic adnexal carcinoma: a diagnostic and therapeutic challenge. Dermatol Ther. 2008;21(6):452-458.
4. Adamson T. Microcystic adnexal carcinoma. Dermatol Nurs. 2004;16(4):365.
5. Santini D, Vincenzi B, Guida FM, et al. Aprepitant for management of severe pruritus related to biological cancer treatments: a pilot study. Lancet Oncol. 2012;13(10):1020-1024.
6. Song JS, Tawa M, Chau NG, Kupper TS, LeBoeuf NR. Aprepitant for refractory cutaneous T-cell lymphoma-associated pruritus: 4 cases and a review of the literature. BMC Cancer. 2017;17.
7. Villafranca JJA, Siles MG, Casanova M, Goitia BT, Domínguez AR. Paraneoplastic pruritus presenting with Hodgkin’s lymphoma: a case report. J Med Case Reports. 2014;8:300.
8. Ito J, Fujimoto D, Nakamura A, et al. Aprepitant for refractory nivolumab-induced pruritus. Lung Cancer Amst Neth. 2017;109:58-61.
9. Levêque D. Aprepitant for erlotinib-induced pruritus. N Engl J Med. 2010;363(17):1680-1681; author reply 1681.
10. Gerber PA, Buhren BA, Homey B. More on aprepitant for erlotinib-induced pruritus. N Engl J Med. 2011;364(5):486-487.
Substance P is an important neurotransmitter implicated in itch pathways.1 After binding to its receptor, neurokinin-1 (NK-1), substance P induces release of factors including histamine, which may cause pruritus.2 Recent literature has reported successful use of aprepitant, an NK-1 antagonist that has been approved by the US Food and Drug Administration for the treatment of chemotherapy-induced nausea and vomiting, for treatment of pruritus. We report here the case of a patient with microcystic adnexal carcinoma (MAC) who presented with refractory pruritus and who had rapid and complete resolution of itch after administration of aprepitant.
Case presentation and summary
A 73-year-old man presented with a 12-year history of a small nodule on his philtrum, which had been increasing in size. He subsequently developed upper-lip numbness and nasal induration. He complained of 2.5 months of severe, debilitating, full-body pruritus. His symptoms were refractory to treatment with prednisone, gabapentin, doxycycline, doxepin, antihistamines, and topical steroids. At the time of consultation, he was being treated with hydroxyzine and topical pramocaine lotion with minimal relief.
At initial dermatologic evaluation, his tumor involved the lower two-thirds of the nose and entire upper cutaneous lip. There was a 4-mm rolled ulcer on the nasal tip and a 1-cm exophytic, smooth nodule on the left upper lip with palpable 4-cm submandibular adenopathy (Figure). Skin examination otherwise revealed linear excoriations on the upper back with no additional primary lesions. The nodule was biopsied, and the patient was diagnosed with MAC with gross nodal involvement. Laboratory findings including serum chemistries, blood urea nitrogen, complete blood cell count, thyroid, and liver function were normal. Positron emission tomography-computed tomography (PET-CT) imaging was negative for distant metastases.
Treatment was initiated with oral aprepitant – 125 mg on day 1, 80 mg on day 2, and 80 mg on day 3 –with concomitant weekly carboplatin (AUC 1.5) and paclitaxel (30 mg/m2) as well as radiation. Within hours after the first dose of aprepitant, the patient reported a notable cessation in his pruritus. He reported that after 5 hours, his skin “finally turned off” and over the hour that followed, he had complete resolution of symptoms. He completed chemoradiation with a significant disease response. Despite persistent MAC confined to the philtrum, he has been followed for over 2 years without recurrence of itch.
Discussion
MAC is an uncommon cutaneous malignancy of sweat and eccrine gland differentiation. In all, 700 cases of MAC have been described in the literature; a 2008 review estimated the incidence of metastasis at around 2.1%.3 Though metastasis is exceedingly rare, the tumor is locally aggressive and there are reports of invasion into the muscle, perichondrium, periosteum, bone marrow, as well as perineural spaces and vascular adventitia.4
The clinical presentation of MAC includes smooth, flesh-colored or yellow papules, nodules, or plaques.3 Patients often present with numbness, paresthesia, and burning in the area of involvement because of neural infiltration with tumor. Despite the rarity of MAC, pruritus has been reported as a presenting symptom in 1 other case in the literature.4 Our case represents the first report of MAC presenting with a grossly enlarging centrofacial mass, lymph node involvement, and severe full-body pruritus. Our patient responded completely, and within hours, to treatment with aprepitant after experiencing months of failure with conventional antipruritus treatments and without recurrence in symptoms in more than 2 years of follow-up.
Aprepitant blocks the binding of substance P to its receptor NK-1 and has been approved as an anti-emetic for chemotherapy patients. Substance P has been shown to be important in both nausea and itch pathways. The largest prospective study to date on aprepitant for the indication of pruritus in 45 patients with metastatic solid tumors demonstrated a 91% response rate, defined by >50% reduction in pruritus intensity, and 13% recurrence rate that occurred at a median of 7 weeks after initial treatment.5 Aprepitant treatment has been used with success for pruritus associated with both malignant and nonmalignant conditions in at least 74 patients,6 among whom the malignant conditions included cutaneous T-cell lymphoma, Hodgkin lymphoma, and metastatic solid tumors.5-7 Aprepitant has also been used for erlotinib- and nivolumab-induced pruritus in non–small cell lung cancer, which suggests a possible future role for aprepitant in the treatment of pruritus secondary to novel cancer therapies, perhaps including immune checkpoint inhibitors.8-10
However, despite those reports, and likely owing to the multifactorial nature of pruritus, aprepitant is not unviversally effective. Mechanisms of malignancy-associated itch are yet to be elucidated, and optimal patient selection for aprepitant use needs to be determined. However, our patient’s notable response supports the increasing evidence that substance P is a key mediator of pruritus and that disruption of binding to its receptor may result in significant improvement in symptoms in certain patients. It remains to be seen whether the cell type or the tendency toward neural invasion plays a role. Large, randomized studies are needed to guide patient selection and confirm the findings reported here and in the literature, with careful documentation of and close attention paid to timing of pruritus relief and improvement in patient quality of life. Aprepitant might be an important therapeutic tool for refractory, malignancy-associated pruritus, in which patient quality of life is especially critical.
Acknowledgments
This work was presented at the Multinational Association of Supportive Care and Cancer Meeting, in Miami Florida, June 26-28, 2014. The authors are indebted to Saajar Jadeja for his assistance preparing the manuscript.
Substance P is an important neurotransmitter implicated in itch pathways.1 After binding to its receptor, neurokinin-1 (NK-1), substance P induces release of factors including histamine, which may cause pruritus.2 Recent literature has reported successful use of aprepitant, an NK-1 antagonist that has been approved by the US Food and Drug Administration for the treatment of chemotherapy-induced nausea and vomiting, for treatment of pruritus. We report here the case of a patient with microcystic adnexal carcinoma (MAC) who presented with refractory pruritus and who had rapid and complete resolution of itch after administration of aprepitant.
Case presentation and summary
A 73-year-old man presented with a 12-year history of a small nodule on his philtrum, which had been increasing in size. He subsequently developed upper-lip numbness and nasal induration. He complained of 2.5 months of severe, debilitating, full-body pruritus. His symptoms were refractory to treatment with prednisone, gabapentin, doxycycline, doxepin, antihistamines, and topical steroids. At the time of consultation, he was being treated with hydroxyzine and topical pramocaine lotion with minimal relief.
At initial dermatologic evaluation, his tumor involved the lower two-thirds of the nose and entire upper cutaneous lip. There was a 4-mm rolled ulcer on the nasal tip and a 1-cm exophytic, smooth nodule on the left upper lip with palpable 4-cm submandibular adenopathy (Figure). Skin examination otherwise revealed linear excoriations on the upper back with no additional primary lesions. The nodule was biopsied, and the patient was diagnosed with MAC with gross nodal involvement. Laboratory findings including serum chemistries, blood urea nitrogen, complete blood cell count, thyroid, and liver function were normal. Positron emission tomography-computed tomography (PET-CT) imaging was negative for distant metastases.
Treatment was initiated with oral aprepitant – 125 mg on day 1, 80 mg on day 2, and 80 mg on day 3 –with concomitant weekly carboplatin (AUC 1.5) and paclitaxel (30 mg/m2) as well as radiation. Within hours after the first dose of aprepitant, the patient reported a notable cessation in his pruritus. He reported that after 5 hours, his skin “finally turned off” and over the hour that followed, he had complete resolution of symptoms. He completed chemoradiation with a significant disease response. Despite persistent MAC confined to the philtrum, he has been followed for over 2 years without recurrence of itch.
Discussion
MAC is an uncommon cutaneous malignancy of sweat and eccrine gland differentiation. In all, 700 cases of MAC have been described in the literature; a 2008 review estimated the incidence of metastasis at around 2.1%.3 Though metastasis is exceedingly rare, the tumor is locally aggressive and there are reports of invasion into the muscle, perichondrium, periosteum, bone marrow, as well as perineural spaces and vascular adventitia.4
The clinical presentation of MAC includes smooth, flesh-colored or yellow papules, nodules, or plaques.3 Patients often present with numbness, paresthesia, and burning in the area of involvement because of neural infiltration with tumor. Despite the rarity of MAC, pruritus has been reported as a presenting symptom in 1 other case in the literature.4 Our case represents the first report of MAC presenting with a grossly enlarging centrofacial mass, lymph node involvement, and severe full-body pruritus. Our patient responded completely, and within hours, to treatment with aprepitant after experiencing months of failure with conventional antipruritus treatments and without recurrence in symptoms in more than 2 years of follow-up.
Aprepitant blocks the binding of substance P to its receptor NK-1 and has been approved as an anti-emetic for chemotherapy patients. Substance P has been shown to be important in both nausea and itch pathways. The largest prospective study to date on aprepitant for the indication of pruritus in 45 patients with metastatic solid tumors demonstrated a 91% response rate, defined by >50% reduction in pruritus intensity, and 13% recurrence rate that occurred at a median of 7 weeks after initial treatment.5 Aprepitant treatment has been used with success for pruritus associated with both malignant and nonmalignant conditions in at least 74 patients,6 among whom the malignant conditions included cutaneous T-cell lymphoma, Hodgkin lymphoma, and metastatic solid tumors.5-7 Aprepitant has also been used for erlotinib- and nivolumab-induced pruritus in non–small cell lung cancer, which suggests a possible future role for aprepitant in the treatment of pruritus secondary to novel cancer therapies, perhaps including immune checkpoint inhibitors.8-10
However, despite those reports, and likely owing to the multifactorial nature of pruritus, aprepitant is not unviversally effective. Mechanisms of malignancy-associated itch are yet to be elucidated, and optimal patient selection for aprepitant use needs to be determined. However, our patient’s notable response supports the increasing evidence that substance P is a key mediator of pruritus and that disruption of binding to its receptor may result in significant improvement in symptoms in certain patients. It remains to be seen whether the cell type or the tendency toward neural invasion plays a role. Large, randomized studies are needed to guide patient selection and confirm the findings reported here and in the literature, with careful documentation of and close attention paid to timing of pruritus relief and improvement in patient quality of life. Aprepitant might be an important therapeutic tool for refractory, malignancy-associated pruritus, in which patient quality of life is especially critical.
Acknowledgments
This work was presented at the Multinational Association of Supportive Care and Cancer Meeting, in Miami Florida, June 26-28, 2014. The authors are indebted to Saajar Jadeja for his assistance preparing the manuscript.
1. Wallengren J. Neuroanatomy and neurophysiology of itch. Dermatol Ther. 2005;18(4):292-303.
2. Kulka M, Sheen CH, Tancowny BP, Grammer LC, Schleimer RP. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology. 2008;123(3):398-410.
3. Wetter R, Goldstein GD. Microcystic adnexal carcinoma: a diagnostic and therapeutic challenge. Dermatol Ther. 2008;21(6):452-458.
4. Adamson T. Microcystic adnexal carcinoma. Dermatol Nurs. 2004;16(4):365.
5. Santini D, Vincenzi B, Guida FM, et al. Aprepitant for management of severe pruritus related to biological cancer treatments: a pilot study. Lancet Oncol. 2012;13(10):1020-1024.
6. Song JS, Tawa M, Chau NG, Kupper TS, LeBoeuf NR. Aprepitant for refractory cutaneous T-cell lymphoma-associated pruritus: 4 cases and a review of the literature. BMC Cancer. 2017;17.
7. Villafranca JJA, Siles MG, Casanova M, Goitia BT, Domínguez AR. Paraneoplastic pruritus presenting with Hodgkin’s lymphoma: a case report. J Med Case Reports. 2014;8:300.
8. Ito J, Fujimoto D, Nakamura A, et al. Aprepitant for refractory nivolumab-induced pruritus. Lung Cancer Amst Neth. 2017;109:58-61.
9. Levêque D. Aprepitant for erlotinib-induced pruritus. N Engl J Med. 2010;363(17):1680-1681; author reply 1681.
10. Gerber PA, Buhren BA, Homey B. More on aprepitant for erlotinib-induced pruritus. N Engl J Med. 2011;364(5):486-487.
1. Wallengren J. Neuroanatomy and neurophysiology of itch. Dermatol Ther. 2005;18(4):292-303.
2. Kulka M, Sheen CH, Tancowny BP, Grammer LC, Schleimer RP. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology. 2008;123(3):398-410.
3. Wetter R, Goldstein GD. Microcystic adnexal carcinoma: a diagnostic and therapeutic challenge. Dermatol Ther. 2008;21(6):452-458.
4. Adamson T. Microcystic adnexal carcinoma. Dermatol Nurs. 2004;16(4):365.
5. Santini D, Vincenzi B, Guida FM, et al. Aprepitant for management of severe pruritus related to biological cancer treatments: a pilot study. Lancet Oncol. 2012;13(10):1020-1024.
6. Song JS, Tawa M, Chau NG, Kupper TS, LeBoeuf NR. Aprepitant for refractory cutaneous T-cell lymphoma-associated pruritus: 4 cases and a review of the literature. BMC Cancer. 2017;17.
7. Villafranca JJA, Siles MG, Casanova M, Goitia BT, Domínguez AR. Paraneoplastic pruritus presenting with Hodgkin’s lymphoma: a case report. J Med Case Reports. 2014;8:300.
8. Ito J, Fujimoto D, Nakamura A, et al. Aprepitant for refractory nivolumab-induced pruritus. Lung Cancer Amst Neth. 2017;109:58-61.
9. Levêque D. Aprepitant for erlotinib-induced pruritus. N Engl J Med. 2010;363(17):1680-1681; author reply 1681.
10. Gerber PA, Buhren BA, Homey B. More on aprepitant for erlotinib-induced pruritus. N Engl J Med. 2011;364(5):486-487.
Rare paraneoplastic dermatomyositis secondary to high-grade bladder cancer
The clinical presentation of bladder cancer typically presents with hematuria; changes in voiding habits such as urgency, frequency, and pain; or less commonly, obstructive symptoms. Rarely does bladder cancer first present as part of a paraneoplastic syndrome with an inflammatory myopathy. Inflammatory myopathies such as dermatomyositis have been known to be associated with malignancy, however, in a meta-analysis by Yang and colleagues of 449 patients with dermatomyositis and malignancy there were only 8 cases reported of bladder cancer.1 Herein, we report a paraneoplastic dermatomyositis in the setting of a bladder cancer.
Case presentation and summary
A 65-year-old man with a medical history of hypertension and alcohol use presented to the emergency department with worsening pain, stiffness in the neck, shoulders, and inability to lift his arms above his shoulders. During the physical exam, an erythematous purple rash was noted over his chest, neck, and arms. Upon further evaluation, his creatine phosphokinase was 3,500 U/L (reference range 52-336 U/L) suggesting muscle breakdown and possible inflammatory myopathy. A biopsy of the left deltoid and quadriceps muscles was performed and yielded a diagnosis of dermatomyositis. He was treated with prednisone 60 mg daily for his inflammatory myopathy. The patient also reported an unintentional weight loss of 20 lbs. and increasing weakness and inability to swallow, which caused aspiration events without developing pneumonia.
The patient’s symptoms worsened while he was on steroids, and we became concerned about the possibility of a primary malignancy, which led to further work-up. The results of a computed-tomography (CT) scan of the abdomen and pelvis showed right-sided hydronephrosis and hydrourteter with an irregular, soft-tissue density mass of 4.7 x 3.2 x 4.2 cm along the posterior wall of the bladder (Figure 1).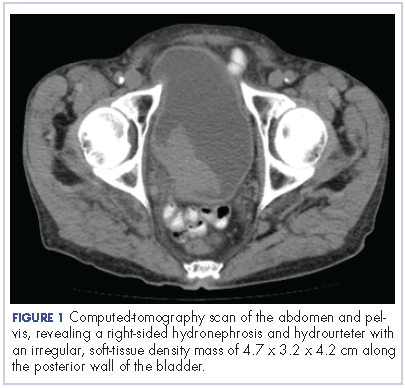
A cystoscopy was performed with transurethral resection of a bladder tumor that was more than 8 cm in diameter. Because the mass was not fully resectable, only 25% of the tumor burden was removed. The pathology report revealed an invasive, high-grade urothelial cell carcinoma (Figure 2, see PDF). Further imaging ruled out metastatic spread. The patient was continued on steroids. He was not a candidate for neoadjuvant chemotherapy because of his comorbidities and cisplatin ineligibility owing to his significant bilateral hearing deficiencies. Members of a multidisciplinary tumor board decided to move forward with definitive surgery. The patient underwent a robotic-assisted laparoscoptic cystoprostatectomy with bilateral pelvic lymph node dissection and open ileal conduit urinary diversion. Staging of tumor was determined as pT3b N1 (1/30) M0, LVI+. After the surgery, the patient had resolution of his rash and significant improvement in his muscle weakness with the ability to raise his arms over his head and climb stairs. Adjuvant chemotherapy was not given since he was cisplatin ineligible as a result of his hearing loss. Active surveillance was preferred.
Four months after his cystoprostatectomy, he experienced new-onset hip pain and further imaging, including a bone scan, was performed. It showed metastatic disease in the ischium and iliac crest (Figure 3).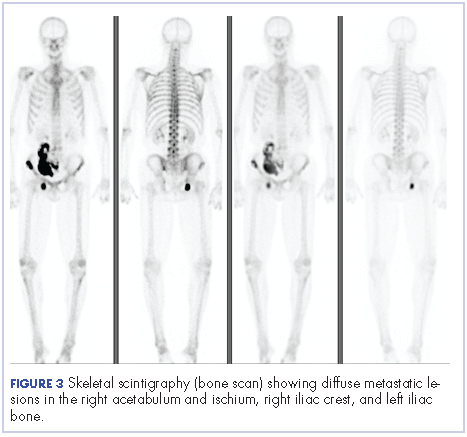
The patient decided to forgo any palliative chemotherapy and to have palliative radiation for pain and enroll in hospice. He died nine months after the initial diagnosis of urothelial cell carcinoma.
Discussion
Dermatomyositis is one of the inflammatory myopathies with a clinical presentation of proximal muscle weakness and characteristic skin findings of Gottron papules and heliotrope eruption. The most common subgroups of inflammatory myopathies are dermatomyositis, polymyositis, necrotizing autoimmune myopathy, and inclusion body myopathy. The pathogenesis of inflammatory myopathies is not well understood; however, some theories have been described, including: type 1 interferon signaling causing myofiber injury and antibody-complement mediated processes causing ischemia resulting in myofiber injury. 2,3 The diagnoses of inflammatory myopathies may be suggested based on history, physical examination findings, laboratory values showing muscle injury (creatine kinase, aldolase, ALT, AST, LDH), myositis-specific antibodies (antisynthetase autoantibodies), electromyogram, and magnetic-resonance imaging. However, muscle biopsy remains the gold standard.4
The initial treatment of inflammatory myopathies begins with glucocorticoid therapy at 0.5-1.0 mg/kg. This regimen may be titrated down over 6 weeks to a level adequate to control symptoms. Even while on glucocorticoid therapy, this patient’s symptoms continued, along with the development of dysphagia. Dysphagia is another notable symptom of dermatomyositis that may result in aspiration pneumonia with fatal outcomes.5,6,7 Not only did this patient initially respond poorly to corticosteroids, but the unintentional weight loss was another alarming feature prompting further evaluation. That led to the diagnosis of urothelial cell carcinoma, which was causing the paraneoplastic syndrome.
A paraneoplastic syndrome is a collection of symptoms that are observed in organ systems separate from the primary disease. This process is mostly caused by an autoimmune response to the tumor and nervous system.8 Inflammatory myopathies, such as dermatomyositis, have been shown to be associated with a variety of malignancies as part of a paraneoplastic syndrome. The most common cancers associated with dermatomyositis are ovarian, lung, pancreatic, stomach, colorectal, and non-Hodgkin lymphoma.9 Although an association between dermatomyositis and bladder cancer has been established, very few cases have been reported in the literature. In the Yang meta-analysis, the relative risk of malignancy for patients with dermatomyositis was 5.5%, and of the 449 patients with dermatomyositis who had malignancy, only 8 cases of bladder cancer were reported.1
After a patient has been diagnosed with an inflammatory myopathy, there should be further evaluation for an underling malignancy causing a paraneoplastic process. The risk of these patients having a malignancy overall is 4.5 times higher than patients without dermatomyositis.1 Definite screening recommendations have not been established, but screening should be based on patient’s age, gender, and clinical scenario. The European Federation of Neurological Societies formed a task force to focus on malignancy screening of paraneoplastic neurological syndromes and included dermatomyositis as one of the signs.10 Patients should have a CT scan of the chest, abdomen, and pelvis. Women should have a mammogram and a pelvis ultrasound. Men younger than 50 years should consider testes ultrasound, and patients older than 50 years should undergo usual colonoscopy screening.
The risk of malignancy is highest in the first year after diagnosis, but may extend to 5 years after the diagnosis, so repeat screening should be performed 3-6 months after diagnosis, followed with biannual testing for 4 years. If a malignancy is present, then treatment should be tailored to the neoplasm to improve symptoms of myositis; however, response is generally worse than it would be with dermatomyositis in the absence of malignancy. In the present case with bladder cancer, therapies may include platinum-based-chemotherapy, resection, and radiation. Dermatomyositis as a result of a bladder cancer paraneoplastic syndrome is associated with a poor prognosis as demonstrated in the case of this patient and others reported in the literature.11
Even though dermatomyositis is usually a chronic disease process, 87% of patients respond initially to corticosteroid treatment.12 Therefore, treatment should be escalated with an agent such as azathioprine or methotrexate, or, like in this case, an underlying malignancy should be suspected. This case emphasizes the importance of screening patients appropriately for malignancy in patients with an inflammatory myopathy and reveals the poor prognosis associated with this disease.
1. Yang Z, Lin F, Qin B, Liang Y, Zhong R. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42(2):282-291.
2. Greenberg, SA. Dermatomyositis and type 1 interferons. Curr Rheumatol Rep. 2010;12(3):198-203.
3. Dalakas, MC, Hohlfeld, R. Polymyositis and dermatomyositis. Lancet. 2003;362(9388):971-982.
4. Malik A, Hayat G, Kalia JS, Guzman MA. Idiopathic inflammatory myopathies: clinical approach and management. Front Neurol. 2016;7:64.
5. Sabio JM, Vargas-Hitos JA, Jiménez-Alonso J. Paraneoplastic dermatomyositis associated with bladder cancer. Lupus. 2006;15(9):619-620.
6. Mallon E, Osborne G, Dinneen M, Lane RJ, Glaser M, Bunker CB. Dermatomyositis in association with transitional cell carcinoma of the bladder. Clin Exp Dermatol. 1999;24(2):94-96.
7. Hafejee A, Coulson IH. Dysphagia in dermatomyositis secondary to bladder cancer: rapid response to combined immunoglobulin and methylprednisolone. Clin Exp Dermatol. 2005;30(1):93-94.
8. Dalmau J, Gultekin HS, Posner JB. Paraneoplastic neurologic syndromes: pathogenesis and physiopathology. Brain Pathol. 1999;9(2):275-284.
9. Hill CL, Zhang Y, Sigureirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357(9250):96-100.
10. Titulaer, MJ, Soffietti R, Dalmau J, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS Task Force. Eur J Neurol. 2011;18(1):19-e3.
11. Xu R, Zhong Z, Jiang H, Zhang L, Zhao X. A rare paraneoplastic dermatomyositis in bladder cancer with fatal outcome. Urol J. 2013;10(1):815-817.
12. Troyanov Y, Targoff IN, Tremblay JL, Goulet JR, Raymond Y, Senecal JL. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore), 2005;84(4):231-249.
The clinical presentation of bladder cancer typically presents with hematuria; changes in voiding habits such as urgency, frequency, and pain; or less commonly, obstructive symptoms. Rarely does bladder cancer first present as part of a paraneoplastic syndrome with an inflammatory myopathy. Inflammatory myopathies such as dermatomyositis have been known to be associated with malignancy, however, in a meta-analysis by Yang and colleagues of 449 patients with dermatomyositis and malignancy there were only 8 cases reported of bladder cancer.1 Herein, we report a paraneoplastic dermatomyositis in the setting of a bladder cancer.
Case presentation and summary
A 65-year-old man with a medical history of hypertension and alcohol use presented to the emergency department with worsening pain, stiffness in the neck, shoulders, and inability to lift his arms above his shoulders. During the physical exam, an erythematous purple rash was noted over his chest, neck, and arms. Upon further evaluation, his creatine phosphokinase was 3,500 U/L (reference range 52-336 U/L) suggesting muscle breakdown and possible inflammatory myopathy. A biopsy of the left deltoid and quadriceps muscles was performed and yielded a diagnosis of dermatomyositis. He was treated with prednisone 60 mg daily for his inflammatory myopathy. The patient also reported an unintentional weight loss of 20 lbs. and increasing weakness and inability to swallow, which caused aspiration events without developing pneumonia.
The patient’s symptoms worsened while he was on steroids, and we became concerned about the possibility of a primary malignancy, which led to further work-up. The results of a computed-tomography (CT) scan of the abdomen and pelvis showed right-sided hydronephrosis and hydrourteter with an irregular, soft-tissue density mass of 4.7 x 3.2 x 4.2 cm along the posterior wall of the bladder (Figure 1).
A cystoscopy was performed with transurethral resection of a bladder tumor that was more than 8 cm in diameter. Because the mass was not fully resectable, only 25% of the tumor burden was removed. The pathology report revealed an invasive, high-grade urothelial cell carcinoma (Figure 2, see PDF). Further imaging ruled out metastatic spread. The patient was continued on steroids. He was not a candidate for neoadjuvant chemotherapy because of his comorbidities and cisplatin ineligibility owing to his significant bilateral hearing deficiencies. Members of a multidisciplinary tumor board decided to move forward with definitive surgery. The patient underwent a robotic-assisted laparoscoptic cystoprostatectomy with bilateral pelvic lymph node dissection and open ileal conduit urinary diversion. Staging of tumor was determined as pT3b N1 (1/30) M0, LVI+. After the surgery, the patient had resolution of his rash and significant improvement in his muscle weakness with the ability to raise his arms over his head and climb stairs. Adjuvant chemotherapy was not given since he was cisplatin ineligible as a result of his hearing loss. Active surveillance was preferred.
Four months after his cystoprostatectomy, he experienced new-onset hip pain and further imaging, including a bone scan, was performed. It showed metastatic disease in the ischium and iliac crest (Figure 3).
The patient decided to forgo any palliative chemotherapy and to have palliative radiation for pain and enroll in hospice. He died nine months after the initial diagnosis of urothelial cell carcinoma.
Discussion
Dermatomyositis is one of the inflammatory myopathies with a clinical presentation of proximal muscle weakness and characteristic skin findings of Gottron papules and heliotrope eruption. The most common subgroups of inflammatory myopathies are dermatomyositis, polymyositis, necrotizing autoimmune myopathy, and inclusion body myopathy. The pathogenesis of inflammatory myopathies is not well understood; however, some theories have been described, including: type 1 interferon signaling causing myofiber injury and antibody-complement mediated processes causing ischemia resulting in myofiber injury. 2,3 The diagnoses of inflammatory myopathies may be suggested based on history, physical examination findings, laboratory values showing muscle injury (creatine kinase, aldolase, ALT, AST, LDH), myositis-specific antibodies (antisynthetase autoantibodies), electromyogram, and magnetic-resonance imaging. However, muscle biopsy remains the gold standard.4
The initial treatment of inflammatory myopathies begins with glucocorticoid therapy at 0.5-1.0 mg/kg. This regimen may be titrated down over 6 weeks to a level adequate to control symptoms. Even while on glucocorticoid therapy, this patient’s symptoms continued, along with the development of dysphagia. Dysphagia is another notable symptom of dermatomyositis that may result in aspiration pneumonia with fatal outcomes.5,6,7 Not only did this patient initially respond poorly to corticosteroids, but the unintentional weight loss was another alarming feature prompting further evaluation. That led to the diagnosis of urothelial cell carcinoma, which was causing the paraneoplastic syndrome.
A paraneoplastic syndrome is a collection of symptoms that are observed in organ systems separate from the primary disease. This process is mostly caused by an autoimmune response to the tumor and nervous system.8 Inflammatory myopathies, such as dermatomyositis, have been shown to be associated with a variety of malignancies as part of a paraneoplastic syndrome. The most common cancers associated with dermatomyositis are ovarian, lung, pancreatic, stomach, colorectal, and non-Hodgkin lymphoma.9 Although an association between dermatomyositis and bladder cancer has been established, very few cases have been reported in the literature. In the Yang meta-analysis, the relative risk of malignancy for patients with dermatomyositis was 5.5%, and of the 449 patients with dermatomyositis who had malignancy, only 8 cases of bladder cancer were reported.1
After a patient has been diagnosed with an inflammatory myopathy, there should be further evaluation for an underling malignancy causing a paraneoplastic process. The risk of these patients having a malignancy overall is 4.5 times higher than patients without dermatomyositis.1 Definite screening recommendations have not been established, but screening should be based on patient’s age, gender, and clinical scenario. The European Federation of Neurological Societies formed a task force to focus on malignancy screening of paraneoplastic neurological syndromes and included dermatomyositis as one of the signs.10 Patients should have a CT scan of the chest, abdomen, and pelvis. Women should have a mammogram and a pelvis ultrasound. Men younger than 50 years should consider testes ultrasound, and patients older than 50 years should undergo usual colonoscopy screening.
The risk of malignancy is highest in the first year after diagnosis, but may extend to 5 years after the diagnosis, so repeat screening should be performed 3-6 months after diagnosis, followed with biannual testing for 4 years. If a malignancy is present, then treatment should be tailored to the neoplasm to improve symptoms of myositis; however, response is generally worse than it would be with dermatomyositis in the absence of malignancy. In the present case with bladder cancer, therapies may include platinum-based-chemotherapy, resection, and radiation. Dermatomyositis as a result of a bladder cancer paraneoplastic syndrome is associated with a poor prognosis as demonstrated in the case of this patient and others reported in the literature.11
Even though dermatomyositis is usually a chronic disease process, 87% of patients respond initially to corticosteroid treatment.12 Therefore, treatment should be escalated with an agent such as azathioprine or methotrexate, or, like in this case, an underlying malignancy should be suspected. This case emphasizes the importance of screening patients appropriately for malignancy in patients with an inflammatory myopathy and reveals the poor prognosis associated with this disease.
The clinical presentation of bladder cancer typically presents with hematuria; changes in voiding habits such as urgency, frequency, and pain; or less commonly, obstructive symptoms. Rarely does bladder cancer first present as part of a paraneoplastic syndrome with an inflammatory myopathy. Inflammatory myopathies such as dermatomyositis have been known to be associated with malignancy, however, in a meta-analysis by Yang and colleagues of 449 patients with dermatomyositis and malignancy there were only 8 cases reported of bladder cancer.1 Herein, we report a paraneoplastic dermatomyositis in the setting of a bladder cancer.
Case presentation and summary
A 65-year-old man with a medical history of hypertension and alcohol use presented to the emergency department with worsening pain, stiffness in the neck, shoulders, and inability to lift his arms above his shoulders. During the physical exam, an erythematous purple rash was noted over his chest, neck, and arms. Upon further evaluation, his creatine phosphokinase was 3,500 U/L (reference range 52-336 U/L) suggesting muscle breakdown and possible inflammatory myopathy. A biopsy of the left deltoid and quadriceps muscles was performed and yielded a diagnosis of dermatomyositis. He was treated with prednisone 60 mg daily for his inflammatory myopathy. The patient also reported an unintentional weight loss of 20 lbs. and increasing weakness and inability to swallow, which caused aspiration events without developing pneumonia.
The patient’s symptoms worsened while he was on steroids, and we became concerned about the possibility of a primary malignancy, which led to further work-up. The results of a computed-tomography (CT) scan of the abdomen and pelvis showed right-sided hydronephrosis and hydrourteter with an irregular, soft-tissue density mass of 4.7 x 3.2 x 4.2 cm along the posterior wall of the bladder (Figure 1).
A cystoscopy was performed with transurethral resection of a bladder tumor that was more than 8 cm in diameter. Because the mass was not fully resectable, only 25% of the tumor burden was removed. The pathology report revealed an invasive, high-grade urothelial cell carcinoma (Figure 2, see PDF). Further imaging ruled out metastatic spread. The patient was continued on steroids. He was not a candidate for neoadjuvant chemotherapy because of his comorbidities and cisplatin ineligibility owing to his significant bilateral hearing deficiencies. Members of a multidisciplinary tumor board decided to move forward with definitive surgery. The patient underwent a robotic-assisted laparoscoptic cystoprostatectomy with bilateral pelvic lymph node dissection and open ileal conduit urinary diversion. Staging of tumor was determined as pT3b N1 (1/30) M0, LVI+. After the surgery, the patient had resolution of his rash and significant improvement in his muscle weakness with the ability to raise his arms over his head and climb stairs. Adjuvant chemotherapy was not given since he was cisplatin ineligible as a result of his hearing loss. Active surveillance was preferred.
Four months after his cystoprostatectomy, he experienced new-onset hip pain and further imaging, including a bone scan, was performed. It showed metastatic disease in the ischium and iliac crest (Figure 3).
The patient decided to forgo any palliative chemotherapy and to have palliative radiation for pain and enroll in hospice. He died nine months after the initial diagnosis of urothelial cell carcinoma.
Discussion
Dermatomyositis is one of the inflammatory myopathies with a clinical presentation of proximal muscle weakness and characteristic skin findings of Gottron papules and heliotrope eruption. The most common subgroups of inflammatory myopathies are dermatomyositis, polymyositis, necrotizing autoimmune myopathy, and inclusion body myopathy. The pathogenesis of inflammatory myopathies is not well understood; however, some theories have been described, including: type 1 interferon signaling causing myofiber injury and antibody-complement mediated processes causing ischemia resulting in myofiber injury. 2,3 The diagnoses of inflammatory myopathies may be suggested based on history, physical examination findings, laboratory values showing muscle injury (creatine kinase, aldolase, ALT, AST, LDH), myositis-specific antibodies (antisynthetase autoantibodies), electromyogram, and magnetic-resonance imaging. However, muscle biopsy remains the gold standard.4
The initial treatment of inflammatory myopathies begins with glucocorticoid therapy at 0.5-1.0 mg/kg. This regimen may be titrated down over 6 weeks to a level adequate to control symptoms. Even while on glucocorticoid therapy, this patient’s symptoms continued, along with the development of dysphagia. Dysphagia is another notable symptom of dermatomyositis that may result in aspiration pneumonia with fatal outcomes.5,6,7 Not only did this patient initially respond poorly to corticosteroids, but the unintentional weight loss was another alarming feature prompting further evaluation. That led to the diagnosis of urothelial cell carcinoma, which was causing the paraneoplastic syndrome.
A paraneoplastic syndrome is a collection of symptoms that are observed in organ systems separate from the primary disease. This process is mostly caused by an autoimmune response to the tumor and nervous system.8 Inflammatory myopathies, such as dermatomyositis, have been shown to be associated with a variety of malignancies as part of a paraneoplastic syndrome. The most common cancers associated with dermatomyositis are ovarian, lung, pancreatic, stomach, colorectal, and non-Hodgkin lymphoma.9 Although an association between dermatomyositis and bladder cancer has been established, very few cases have been reported in the literature. In the Yang meta-analysis, the relative risk of malignancy for patients with dermatomyositis was 5.5%, and of the 449 patients with dermatomyositis who had malignancy, only 8 cases of bladder cancer were reported.1
After a patient has been diagnosed with an inflammatory myopathy, there should be further evaluation for an underling malignancy causing a paraneoplastic process. The risk of these patients having a malignancy overall is 4.5 times higher than patients without dermatomyositis.1 Definite screening recommendations have not been established, but screening should be based on patient’s age, gender, and clinical scenario. The European Federation of Neurological Societies formed a task force to focus on malignancy screening of paraneoplastic neurological syndromes and included dermatomyositis as one of the signs.10 Patients should have a CT scan of the chest, abdomen, and pelvis. Women should have a mammogram and a pelvis ultrasound. Men younger than 50 years should consider testes ultrasound, and patients older than 50 years should undergo usual colonoscopy screening.
The risk of malignancy is highest in the first year after diagnosis, but may extend to 5 years after the diagnosis, so repeat screening should be performed 3-6 months after diagnosis, followed with biannual testing for 4 years. If a malignancy is present, then treatment should be tailored to the neoplasm to improve symptoms of myositis; however, response is generally worse than it would be with dermatomyositis in the absence of malignancy. In the present case with bladder cancer, therapies may include platinum-based-chemotherapy, resection, and radiation. Dermatomyositis as a result of a bladder cancer paraneoplastic syndrome is associated with a poor prognosis as demonstrated in the case of this patient and others reported in the literature.11
Even though dermatomyositis is usually a chronic disease process, 87% of patients respond initially to corticosteroid treatment.12 Therefore, treatment should be escalated with an agent such as azathioprine or methotrexate, or, like in this case, an underlying malignancy should be suspected. This case emphasizes the importance of screening patients appropriately for malignancy in patients with an inflammatory myopathy and reveals the poor prognosis associated with this disease.
1. Yang Z, Lin F, Qin B, Liang Y, Zhong R. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42(2):282-291.
2. Greenberg, SA. Dermatomyositis and type 1 interferons. Curr Rheumatol Rep. 2010;12(3):198-203.
3. Dalakas, MC, Hohlfeld, R. Polymyositis and dermatomyositis. Lancet. 2003;362(9388):971-982.
4. Malik A, Hayat G, Kalia JS, Guzman MA. Idiopathic inflammatory myopathies: clinical approach and management. Front Neurol. 2016;7:64.
5. Sabio JM, Vargas-Hitos JA, Jiménez-Alonso J. Paraneoplastic dermatomyositis associated with bladder cancer. Lupus. 2006;15(9):619-620.
6. Mallon E, Osborne G, Dinneen M, Lane RJ, Glaser M, Bunker CB. Dermatomyositis in association with transitional cell carcinoma of the bladder. Clin Exp Dermatol. 1999;24(2):94-96.
7. Hafejee A, Coulson IH. Dysphagia in dermatomyositis secondary to bladder cancer: rapid response to combined immunoglobulin and methylprednisolone. Clin Exp Dermatol. 2005;30(1):93-94.
8. Dalmau J, Gultekin HS, Posner JB. Paraneoplastic neurologic syndromes: pathogenesis and physiopathology. Brain Pathol. 1999;9(2):275-284.
9. Hill CL, Zhang Y, Sigureirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357(9250):96-100.
10. Titulaer, MJ, Soffietti R, Dalmau J, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS Task Force. Eur J Neurol. 2011;18(1):19-e3.
11. Xu R, Zhong Z, Jiang H, Zhang L, Zhao X. A rare paraneoplastic dermatomyositis in bladder cancer with fatal outcome. Urol J. 2013;10(1):815-817.
12. Troyanov Y, Targoff IN, Tremblay JL, Goulet JR, Raymond Y, Senecal JL. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore), 2005;84(4):231-249.
1. Yang Z, Lin F, Qin B, Liang Y, Zhong R. Polymyositis/dermatomyositis and malignancy risk: a metaanalysis study. J Rheumatol. 2015;42(2):282-291.
2. Greenberg, SA. Dermatomyositis and type 1 interferons. Curr Rheumatol Rep. 2010;12(3):198-203.
3. Dalakas, MC, Hohlfeld, R. Polymyositis and dermatomyositis. Lancet. 2003;362(9388):971-982.
4. Malik A, Hayat G, Kalia JS, Guzman MA. Idiopathic inflammatory myopathies: clinical approach and management. Front Neurol. 2016;7:64.
5. Sabio JM, Vargas-Hitos JA, Jiménez-Alonso J. Paraneoplastic dermatomyositis associated with bladder cancer. Lupus. 2006;15(9):619-620.
6. Mallon E, Osborne G, Dinneen M, Lane RJ, Glaser M, Bunker CB. Dermatomyositis in association with transitional cell carcinoma of the bladder. Clin Exp Dermatol. 1999;24(2):94-96.
7. Hafejee A, Coulson IH. Dysphagia in dermatomyositis secondary to bladder cancer: rapid response to combined immunoglobulin and methylprednisolone. Clin Exp Dermatol. 2005;30(1):93-94.
8. Dalmau J, Gultekin HS, Posner JB. Paraneoplastic neurologic syndromes: pathogenesis and physiopathology. Brain Pathol. 1999;9(2):275-284.
9. Hill CL, Zhang Y, Sigureirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357(9250):96-100.
10. Titulaer, MJ, Soffietti R, Dalmau J, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS Task Force. Eur J Neurol. 2011;18(1):19-e3.
11. Xu R, Zhong Z, Jiang H, Zhang L, Zhao X. A rare paraneoplastic dermatomyositis in bladder cancer with fatal outcome. Urol J. 2013;10(1):815-817.
12. Troyanov Y, Targoff IN, Tremblay JL, Goulet JR, Raymond Y, Senecal JL. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Baltimore), 2005;84(4):231-249.
Rapid Development of Life-Threatening Emamectin Benzoate Poisoning
Emamectin benzoate (EB) is a semisynthetic derivative of avermectin that has acaricidal, nematicidal, and insecticidal action. Avermectin analogs are natural products from soil fungi (Streptomyces avermitilis).1 Emamectin benzoate was initially developed to eradicate lepidopteran larvae, particularly armyworms, and is registered in the United States and Japan for use on vegetable crops.2-4 In addition to its agricultural use, EB also has antiparasitic effects on sea lice (Lepeophtheirus salmonis) that affect Atlantic salmon, and has been registered for use in several countries since 1999.5-7 Although a few studies have evaluated the toxic effects of avermectin on humans, there is a paucity of information regarding human toxicity associated with EB.7 This case report describes rapid deterioration of a patient following ingestion of EB.
Case
A 75-year-old man presented to the ED 20 minutes after intentionally ingesting an agricultural insecticide. Upon presentation, the patient stated that he drank a whole bottle (100 mL) of insecticide after consuming alcohol, but denied coingestion of other toxic substances or any medications. The patient provided the empty bottle upon presentation, and the ingested product was identified as Affirm, an insecticide containing 2.15% EB as the active ingredient.
The patient’s medical history was significant for major depressive disorder, for which he was on alprazolam, donepezil, paroxetine, and quetiapine. The patient stated that he also suffered from chronic back pain, noting that he only took analgesics intermittently as needed.
On examination, the patient was alert and oriented to time and place. Initially, he did not experience any physical discomfort. His vital signs were: blood pressure (BP), 126/74 mm Hg; pulse rate, 67 beats/minute; respiratory rate, mildly tachypneic at 23 breaths/minute; and temperature, 97.9°F. Oxygen saturation was 96% on room air.
Ocular examination revealed both pupils to be equally round, 3 mm in diameter, and reactive to light. Examination of the oropharynx was normal and without signs of mucosal injury. The lung sounds were clear bilaterally, and the heart was a regular rate and rhythm and without murmur. The patient’s abdomen was soft and nontender. No deficits, such as ataxia, dysarthria, or tremor were found on the neurological examination.
Prompt gastric lavage via a nasogastric tube was performed, and activated charcoal was administered. Laboratory evaluation was significant for the following: white blood cell count, 22.77 x 109/L with 78% neutrophils and 16% lymphocytes; sodium, 138 mEq/L; potassium, 3.1 mEq/L; chloride, 109 mEq/L; blood urea nitrogen, 19 mg/dL; and creatinine, 0.7 mg/dL. Arterial blood gas (ABG) results revealed a pH, 7.37; partial pressure of carbon dioxide, 25 mm Hg; partial pressure of oxygen, 93 mm Hg; bicarbonate, 14.5 mEq/L; base excess, –8.9 mEq/L; and an oxygen saturation, 97%. Serum creatine kinase (CK), CK-MB and troponin levels were both within normal range. Lactic acid, serum osmolality, and serum ethanol levels were not obtained. The patient’s electrocardiogram (ECG) and chest radiograph findings were normal.
Approximately 1 hour after presentation, the patient complained of an epigastric burning sensation and continued to exhibit mild tachypnea. A subsequent ABG test revealed progressive metabolic acidosis (Table). Although the patient was given a total of 800 mL of normal saline intravenously (IV) upon arrival at the ED, his total urinary output was less than 100 mL 7 hours afterward. Attempts to increase urinary output with IV furosemide were ineffective.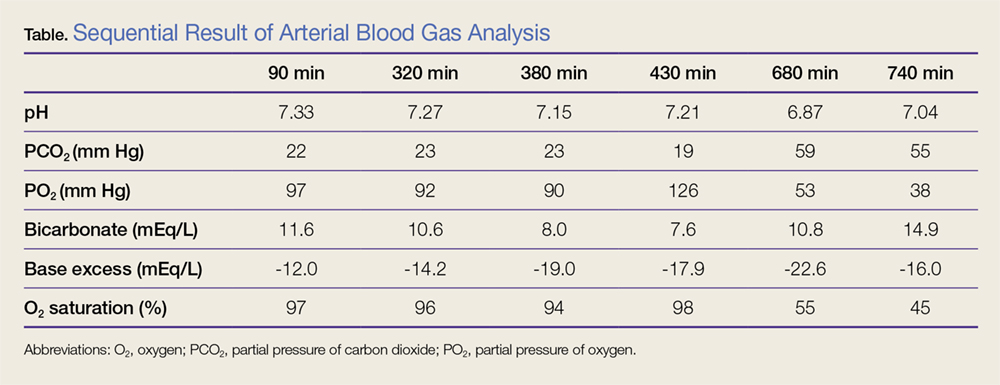
Along with the progressive metabolic acidosis, the patient became hypotensive, and did not respond to IV fluid resuscitation. A norepinephrine infusion was started to improve BP, but this was likewise ineffective. Serial ECGs did not show any specific abnormalities such as dysrhythmia or ischemia.
The patient was admitted to the intensive care unit approximately 10.5 hours after presentation where he received continuous renal replacement therapy (CRRT) to correct the severe metabolic acidosis and poor circulation. Metabolic acidosis persisted despite CRRT, and the patient remained hypotensive even after receiving high-dose IV norepinephrine (Figure).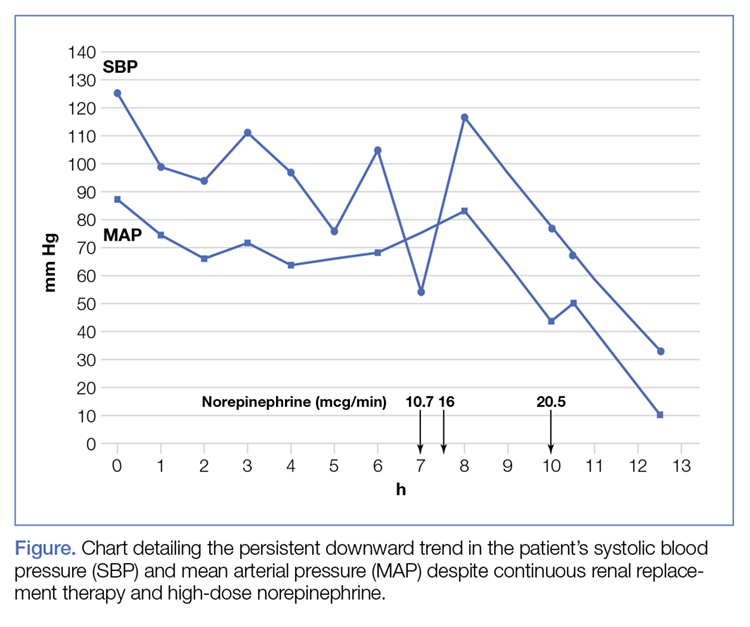
About 12.5 hours after his presentation to the ED, the patient began to vomit profusely and went into cardiac arrest. The cardiac monitor demonstrated pulseless ventricular tachycardia. Aggressive resuscitative efforts were initiated, but failed to restore spontaneous circulation.
Discussion
As an avermectin analog, EB interacts with γ-aminobutyric acid (GABA) receptors and enhances membrane chloride permeability.8 In mammals, GABA-containing neurons and receptors are found in the central nervous system (CNS), but not in the peripheral nervous system. In cases of high-dose avermectin ingestion in humans, CNS toxicity, including agitation and depressed mental status, have been reported, as well as death resulting from respiratory failure.9
With respect to human EB toxicity, there is only one other documented case in the literature by Yen and Lin.7 In their case, the authors report on a patient who ingested 100 mL of Proclaim, which contained 2.15% EB diluted with 400 mL of tap water.7 They note that the patient in their case presented with mild confusion and gastrointestinal (GI) symptoms of nausea, vomiting, and cramping discomfort. Following laboratory and radiological investigation, the patient was found to have aspiration pneumonitis and admitted to the inpatient hospital. On hospital day 2, the patient’s GI symptoms abated and he became alert and oriented. He was discharged 1 week from initial presentation and experienced no sequelae.
In our case, the patient ingested 100 mL of 2.15% EB without dilution. He also experienced GI symptoms, but did not have any CNS depression. The metabolic acidosis rapidly worsened, and could not be corrected, even with intensive therapy. This rapid life-threatening course has not previously been reported with avermectin or EB poisoning. In the avermectin poisoning cases in the literature, seven out of 19 patients (37%) exhibited severe effects, such as hypotension, coma, and aspiration with respiratory failure.9 Six of the seven patients experienced a full recovery; the remaining patient died 18 days after ingestion from multiple organ failure.
The reason for our patient’s rapid progression to metabolic acidosis and progressive deterioration (hypotension and hypoxemia) is not clear. One possible theory is that the solvents or other additives aside from EB in the ingested insecticide might make EB more toxic. In our case, the patient’s rapid deterioration alone or asphyxia by vomitus might have been the cause of the cardiac arrest. Future reports and studies about EB toxicity in humans are warranted to investigate the pathogenesis of toxicity and appropriate treatment.
Conclusion
This is the first report of a human death caused by EB poisoning; the patient experienced severe metabolic acidosis without CNS depression, ultimately leading to death. Emergency physicians should be aware of the possibility of rapid deterioration in patients who present after ingestion of EB and related substances.
1. Lasota JA, Dybas RA. Avermectins, a novel class of compounds: implications for use in arthropod pest control. Annu Rev Entomol. 1991;36:91-117. doi:10.1146/annurev.en.36.010191.000515.
2. Kuo JN, Buday C, van Aggelen G, Ikonomou MG, Pasternak J. Acute toxicity of emamectin benzoate and its desmethyl metabolite to Eohaustorius estuarius. Environ Toxicol Chem. 2010;29(8):1816-1820. doi:10.1002/etc.209.
3. Takai K, Soejima T, Suzuki T, et al. Development of a water-soluble preparation of emamectin benzoate and its preventative effect against the wilting of pot-grown pine trees inoculated with the pine wood nematode, Bursaphelenchus xylophilus. Pest Manag Sci. 2001;57(5):463-466. doi:10.1002/ps.30.
4. Chukwudebe AC, Beavers JB, Jaber M, Wislocki PG. Toxicity of emamectin benzoate to mallard duck and northern bobwhite quail. Environ Toxicol and Chem. 1998;17(6):1118-1123. doi:10.1002/etc.5620170619.
5. Armstrong R, MacPhee D, Katz T, Endris R. A field efficacy evaluation of emamectin benzoate for the control of sea lice on Atlantic salmon. Can Vet J. 2000;41(8):607-612.
6. Ramstad A, Colquhoun DJ, Nordmo R, Sutherland IH, Simmons R. Field trials in Norway with SLICE (0.2% emamectin benzoate) for the oral treatment of sea lice infestation in farmed Atlantic salmon Salmo salar. Dis Aquat Organ. 2002;21;50(1):29-33. doi:10.3354/dao050029.
7. Yen TH, Lin JL. Acute poisoning with emamectin benzoate. J Toxicol Clin Toxicol. 2004;42(5):657-661.
8. Campbell WC, Fisher MH, Stapley EO, Albers-Schönberg G, Jacob TA. Ivermectin: a potent new antiparasitic agent. Science. 1983; 221(4613):823-838.
9. Chung K, Yang CC, Wu ML, Deng JF, Tsai WJ. Agricultural avermectins: an uncommon but potentially fatal cause of pesticide poisoning. Ann Emerg Med. 1999;34(1):51-57.
Emamectin benzoate (EB) is a semisynthetic derivative of avermectin that has acaricidal, nematicidal, and insecticidal action. Avermectin analogs are natural products from soil fungi (Streptomyces avermitilis).1 Emamectin benzoate was initially developed to eradicate lepidopteran larvae, particularly armyworms, and is registered in the United States and Japan for use on vegetable crops.2-4 In addition to its agricultural use, EB also has antiparasitic effects on sea lice (Lepeophtheirus salmonis) that affect Atlantic salmon, and has been registered for use in several countries since 1999.5-7 Although a few studies have evaluated the toxic effects of avermectin on humans, there is a paucity of information regarding human toxicity associated with EB.7 This case report describes rapid deterioration of a patient following ingestion of EB.
Case
A 75-year-old man presented to the ED 20 minutes after intentionally ingesting an agricultural insecticide. Upon presentation, the patient stated that he drank a whole bottle (100 mL) of insecticide after consuming alcohol, but denied coingestion of other toxic substances or any medications. The patient provided the empty bottle upon presentation, and the ingested product was identified as Affirm, an insecticide containing 2.15% EB as the active ingredient.
The patient’s medical history was significant for major depressive disorder, for which he was on alprazolam, donepezil, paroxetine, and quetiapine. The patient stated that he also suffered from chronic back pain, noting that he only took analgesics intermittently as needed.
On examination, the patient was alert and oriented to time and place. Initially, he did not experience any physical discomfort. His vital signs were: blood pressure (BP), 126/74 mm Hg; pulse rate, 67 beats/minute; respiratory rate, mildly tachypneic at 23 breaths/minute; and temperature, 97.9°F. Oxygen saturation was 96% on room air.
Ocular examination revealed both pupils to be equally round, 3 mm in diameter, and reactive to light. Examination of the oropharynx was normal and without signs of mucosal injury. The lung sounds were clear bilaterally, and the heart was a regular rate and rhythm and without murmur. The patient’s abdomen was soft and nontender. No deficits, such as ataxia, dysarthria, or tremor were found on the neurological examination.
Prompt gastric lavage via a nasogastric tube was performed, and activated charcoal was administered. Laboratory evaluation was significant for the following: white blood cell count, 22.77 x 109/L with 78% neutrophils and 16% lymphocytes; sodium, 138 mEq/L; potassium, 3.1 mEq/L; chloride, 109 mEq/L; blood urea nitrogen, 19 mg/dL; and creatinine, 0.7 mg/dL. Arterial blood gas (ABG) results revealed a pH, 7.37; partial pressure of carbon dioxide, 25 mm Hg; partial pressure of oxygen, 93 mm Hg; bicarbonate, 14.5 mEq/L; base excess, –8.9 mEq/L; and an oxygen saturation, 97%. Serum creatine kinase (CK), CK-MB and troponin levels were both within normal range. Lactic acid, serum osmolality, and serum ethanol levels were not obtained. The patient’s electrocardiogram (ECG) and chest radiograph findings were normal.
Approximately 1 hour after presentation, the patient complained of an epigastric burning sensation and continued to exhibit mild tachypnea. A subsequent ABG test revealed progressive metabolic acidosis (Table). Although the patient was given a total of 800 mL of normal saline intravenously (IV) upon arrival at the ED, his total urinary output was less than 100 mL 7 hours afterward. Attempts to increase urinary output with IV furosemide were ineffective.
Along with the progressive metabolic acidosis, the patient became hypotensive, and did not respond to IV fluid resuscitation. A norepinephrine infusion was started to improve BP, but this was likewise ineffective. Serial ECGs did not show any specific abnormalities such as dysrhythmia or ischemia.
The patient was admitted to the intensive care unit approximately 10.5 hours after presentation where he received continuous renal replacement therapy (CRRT) to correct the severe metabolic acidosis and poor circulation. Metabolic acidosis persisted despite CRRT, and the patient remained hypotensive even after receiving high-dose IV norepinephrine (Figure).
About 12.5 hours after his presentation to the ED, the patient began to vomit profusely and went into cardiac arrest. The cardiac monitor demonstrated pulseless ventricular tachycardia. Aggressive resuscitative efforts were initiated, but failed to restore spontaneous circulation.
Discussion
As an avermectin analog, EB interacts with γ-aminobutyric acid (GABA) receptors and enhances membrane chloride permeability.8 In mammals, GABA-containing neurons and receptors are found in the central nervous system (CNS), but not in the peripheral nervous system. In cases of high-dose avermectin ingestion in humans, CNS toxicity, including agitation and depressed mental status, have been reported, as well as death resulting from respiratory failure.9
With respect to human EB toxicity, there is only one other documented case in the literature by Yen and Lin.7 In their case, the authors report on a patient who ingested 100 mL of Proclaim, which contained 2.15% EB diluted with 400 mL of tap water.7 They note that the patient in their case presented with mild confusion and gastrointestinal (GI) symptoms of nausea, vomiting, and cramping discomfort. Following laboratory and radiological investigation, the patient was found to have aspiration pneumonitis and admitted to the inpatient hospital. On hospital day 2, the patient’s GI symptoms abated and he became alert and oriented. He was discharged 1 week from initial presentation and experienced no sequelae.
In our case, the patient ingested 100 mL of 2.15% EB without dilution. He also experienced GI symptoms, but did not have any CNS depression. The metabolic acidosis rapidly worsened, and could not be corrected, even with intensive therapy. This rapid life-threatening course has not previously been reported with avermectin or EB poisoning. In the avermectin poisoning cases in the literature, seven out of 19 patients (37%) exhibited severe effects, such as hypotension, coma, and aspiration with respiratory failure.9 Six of the seven patients experienced a full recovery; the remaining patient died 18 days after ingestion from multiple organ failure.
The reason for our patient’s rapid progression to metabolic acidosis and progressive deterioration (hypotension and hypoxemia) is not clear. One possible theory is that the solvents or other additives aside from EB in the ingested insecticide might make EB more toxic. In our case, the patient’s rapid deterioration alone or asphyxia by vomitus might have been the cause of the cardiac arrest. Future reports and studies about EB toxicity in humans are warranted to investigate the pathogenesis of toxicity and appropriate treatment.
Conclusion
This is the first report of a human death caused by EB poisoning; the patient experienced severe metabolic acidosis without CNS depression, ultimately leading to death. Emergency physicians should be aware of the possibility of rapid deterioration in patients who present after ingestion of EB and related substances.
Emamectin benzoate (EB) is a semisynthetic derivative of avermectin that has acaricidal, nematicidal, and insecticidal action. Avermectin analogs are natural products from soil fungi (Streptomyces avermitilis).1 Emamectin benzoate was initially developed to eradicate lepidopteran larvae, particularly armyworms, and is registered in the United States and Japan for use on vegetable crops.2-4 In addition to its agricultural use, EB also has antiparasitic effects on sea lice (Lepeophtheirus salmonis) that affect Atlantic salmon, and has been registered for use in several countries since 1999.5-7 Although a few studies have evaluated the toxic effects of avermectin on humans, there is a paucity of information regarding human toxicity associated with EB.7 This case report describes rapid deterioration of a patient following ingestion of EB.
Case
A 75-year-old man presented to the ED 20 minutes after intentionally ingesting an agricultural insecticide. Upon presentation, the patient stated that he drank a whole bottle (100 mL) of insecticide after consuming alcohol, but denied coingestion of other toxic substances or any medications. The patient provided the empty bottle upon presentation, and the ingested product was identified as Affirm, an insecticide containing 2.15% EB as the active ingredient.
The patient’s medical history was significant for major depressive disorder, for which he was on alprazolam, donepezil, paroxetine, and quetiapine. The patient stated that he also suffered from chronic back pain, noting that he only took analgesics intermittently as needed.
On examination, the patient was alert and oriented to time and place. Initially, he did not experience any physical discomfort. His vital signs were: blood pressure (BP), 126/74 mm Hg; pulse rate, 67 beats/minute; respiratory rate, mildly tachypneic at 23 breaths/minute; and temperature, 97.9°F. Oxygen saturation was 96% on room air.
Ocular examination revealed both pupils to be equally round, 3 mm in diameter, and reactive to light. Examination of the oropharynx was normal and without signs of mucosal injury. The lung sounds were clear bilaterally, and the heart was a regular rate and rhythm and without murmur. The patient’s abdomen was soft and nontender. No deficits, such as ataxia, dysarthria, or tremor were found on the neurological examination.
Prompt gastric lavage via a nasogastric tube was performed, and activated charcoal was administered. Laboratory evaluation was significant for the following: white blood cell count, 22.77 x 109/L with 78% neutrophils and 16% lymphocytes; sodium, 138 mEq/L; potassium, 3.1 mEq/L; chloride, 109 mEq/L; blood urea nitrogen, 19 mg/dL; and creatinine, 0.7 mg/dL. Arterial blood gas (ABG) results revealed a pH, 7.37; partial pressure of carbon dioxide, 25 mm Hg; partial pressure of oxygen, 93 mm Hg; bicarbonate, 14.5 mEq/L; base excess, –8.9 mEq/L; and an oxygen saturation, 97%. Serum creatine kinase (CK), CK-MB and troponin levels were both within normal range. Lactic acid, serum osmolality, and serum ethanol levels were not obtained. The patient’s electrocardiogram (ECG) and chest radiograph findings were normal.
Approximately 1 hour after presentation, the patient complained of an epigastric burning sensation and continued to exhibit mild tachypnea. A subsequent ABG test revealed progressive metabolic acidosis (Table). Although the patient was given a total of 800 mL of normal saline intravenously (IV) upon arrival at the ED, his total urinary output was less than 100 mL 7 hours afterward. Attempts to increase urinary output with IV furosemide were ineffective.
Along with the progressive metabolic acidosis, the patient became hypotensive, and did not respond to IV fluid resuscitation. A norepinephrine infusion was started to improve BP, but this was likewise ineffective. Serial ECGs did not show any specific abnormalities such as dysrhythmia or ischemia.
The patient was admitted to the intensive care unit approximately 10.5 hours after presentation where he received continuous renal replacement therapy (CRRT) to correct the severe metabolic acidosis and poor circulation. Metabolic acidosis persisted despite CRRT, and the patient remained hypotensive even after receiving high-dose IV norepinephrine (Figure).
About 12.5 hours after his presentation to the ED, the patient began to vomit profusely and went into cardiac arrest. The cardiac monitor demonstrated pulseless ventricular tachycardia. Aggressive resuscitative efforts were initiated, but failed to restore spontaneous circulation.
Discussion
As an avermectin analog, EB interacts with γ-aminobutyric acid (GABA) receptors and enhances membrane chloride permeability.8 In mammals, GABA-containing neurons and receptors are found in the central nervous system (CNS), but not in the peripheral nervous system. In cases of high-dose avermectin ingestion in humans, CNS toxicity, including agitation and depressed mental status, have been reported, as well as death resulting from respiratory failure.9
With respect to human EB toxicity, there is only one other documented case in the literature by Yen and Lin.7 In their case, the authors report on a patient who ingested 100 mL of Proclaim, which contained 2.15% EB diluted with 400 mL of tap water.7 They note that the patient in their case presented with mild confusion and gastrointestinal (GI) symptoms of nausea, vomiting, and cramping discomfort. Following laboratory and radiological investigation, the patient was found to have aspiration pneumonitis and admitted to the inpatient hospital. On hospital day 2, the patient’s GI symptoms abated and he became alert and oriented. He was discharged 1 week from initial presentation and experienced no sequelae.
In our case, the patient ingested 100 mL of 2.15% EB without dilution. He also experienced GI symptoms, but did not have any CNS depression. The metabolic acidosis rapidly worsened, and could not be corrected, even with intensive therapy. This rapid life-threatening course has not previously been reported with avermectin or EB poisoning. In the avermectin poisoning cases in the literature, seven out of 19 patients (37%) exhibited severe effects, such as hypotension, coma, and aspiration with respiratory failure.9 Six of the seven patients experienced a full recovery; the remaining patient died 18 days after ingestion from multiple organ failure.
The reason for our patient’s rapid progression to metabolic acidosis and progressive deterioration (hypotension and hypoxemia) is not clear. One possible theory is that the solvents or other additives aside from EB in the ingested insecticide might make EB more toxic. In our case, the patient’s rapid deterioration alone or asphyxia by vomitus might have been the cause of the cardiac arrest. Future reports and studies about EB toxicity in humans are warranted to investigate the pathogenesis of toxicity and appropriate treatment.
Conclusion
This is the first report of a human death caused by EB poisoning; the patient experienced severe metabolic acidosis without CNS depression, ultimately leading to death. Emergency physicians should be aware of the possibility of rapid deterioration in patients who present after ingestion of EB and related substances.
1. Lasota JA, Dybas RA. Avermectins, a novel class of compounds: implications for use in arthropod pest control. Annu Rev Entomol. 1991;36:91-117. doi:10.1146/annurev.en.36.010191.000515.
2. Kuo JN, Buday C, van Aggelen G, Ikonomou MG, Pasternak J. Acute toxicity of emamectin benzoate and its desmethyl metabolite to Eohaustorius estuarius. Environ Toxicol Chem. 2010;29(8):1816-1820. doi:10.1002/etc.209.
3. Takai K, Soejima T, Suzuki T, et al. Development of a water-soluble preparation of emamectin benzoate and its preventative effect against the wilting of pot-grown pine trees inoculated with the pine wood nematode, Bursaphelenchus xylophilus. Pest Manag Sci. 2001;57(5):463-466. doi:10.1002/ps.30.
4. Chukwudebe AC, Beavers JB, Jaber M, Wislocki PG. Toxicity of emamectin benzoate to mallard duck and northern bobwhite quail. Environ Toxicol and Chem. 1998;17(6):1118-1123. doi:10.1002/etc.5620170619.
5. Armstrong R, MacPhee D, Katz T, Endris R. A field efficacy evaluation of emamectin benzoate for the control of sea lice on Atlantic salmon. Can Vet J. 2000;41(8):607-612.
6. Ramstad A, Colquhoun DJ, Nordmo R, Sutherland IH, Simmons R. Field trials in Norway with SLICE (0.2% emamectin benzoate) for the oral treatment of sea lice infestation in farmed Atlantic salmon Salmo salar. Dis Aquat Organ. 2002;21;50(1):29-33. doi:10.3354/dao050029.
7. Yen TH, Lin JL. Acute poisoning with emamectin benzoate. J Toxicol Clin Toxicol. 2004;42(5):657-661.
8. Campbell WC, Fisher MH, Stapley EO, Albers-Schönberg G, Jacob TA. Ivermectin: a potent new antiparasitic agent. Science. 1983; 221(4613):823-838.
9. Chung K, Yang CC, Wu ML, Deng JF, Tsai WJ. Agricultural avermectins: an uncommon but potentially fatal cause of pesticide poisoning. Ann Emerg Med. 1999;34(1):51-57.
1. Lasota JA, Dybas RA. Avermectins, a novel class of compounds: implications for use in arthropod pest control. Annu Rev Entomol. 1991;36:91-117. doi:10.1146/annurev.en.36.010191.000515.
2. Kuo JN, Buday C, van Aggelen G, Ikonomou MG, Pasternak J. Acute toxicity of emamectin benzoate and its desmethyl metabolite to Eohaustorius estuarius. Environ Toxicol Chem. 2010;29(8):1816-1820. doi:10.1002/etc.209.
3. Takai K, Soejima T, Suzuki T, et al. Development of a water-soluble preparation of emamectin benzoate and its preventative effect against the wilting of pot-grown pine trees inoculated with the pine wood nematode, Bursaphelenchus xylophilus. Pest Manag Sci. 2001;57(5):463-466. doi:10.1002/ps.30.
4. Chukwudebe AC, Beavers JB, Jaber M, Wislocki PG. Toxicity of emamectin benzoate to mallard duck and northern bobwhite quail. Environ Toxicol and Chem. 1998;17(6):1118-1123. doi:10.1002/etc.5620170619.
5. Armstrong R, MacPhee D, Katz T, Endris R. A field efficacy evaluation of emamectin benzoate for the control of sea lice on Atlantic salmon. Can Vet J. 2000;41(8):607-612.
6. Ramstad A, Colquhoun DJ, Nordmo R, Sutherland IH, Simmons R. Field trials in Norway with SLICE (0.2% emamectin benzoate) for the oral treatment of sea lice infestation in farmed Atlantic salmon Salmo salar. Dis Aquat Organ. 2002;21;50(1):29-33. doi:10.3354/dao050029.
7. Yen TH, Lin JL. Acute poisoning with emamectin benzoate. J Toxicol Clin Toxicol. 2004;42(5):657-661.
8. Campbell WC, Fisher MH, Stapley EO, Albers-Schönberg G, Jacob TA. Ivermectin: a potent new antiparasitic agent. Science. 1983; 221(4613):823-838.
9. Chung K, Yang CC, Wu ML, Deng JF, Tsai WJ. Agricultural avermectins: an uncommon but potentially fatal cause of pesticide poisoning. Ann Emerg Med. 1999;34(1):51-57.