User login
Annular Atrophic Lichen Planus Responds to Hydroxychloroquine and Acitretin
Annular atrophic lichen planus (AALP) is a rare variant of lichen planus that was first described by Friedman and Hashimoto1 in 1991. Clinically, it combines the configuration and morphological features of both annular and atrophic lichen planus. It is a rare entity. We report a case of AALP in a 69-year-old black man. The clinical and histopathological presentation depicted the defining features of this entity with a characteristic loss of elastic fibers corresponding to central atrophy of active lesions.
Case Report
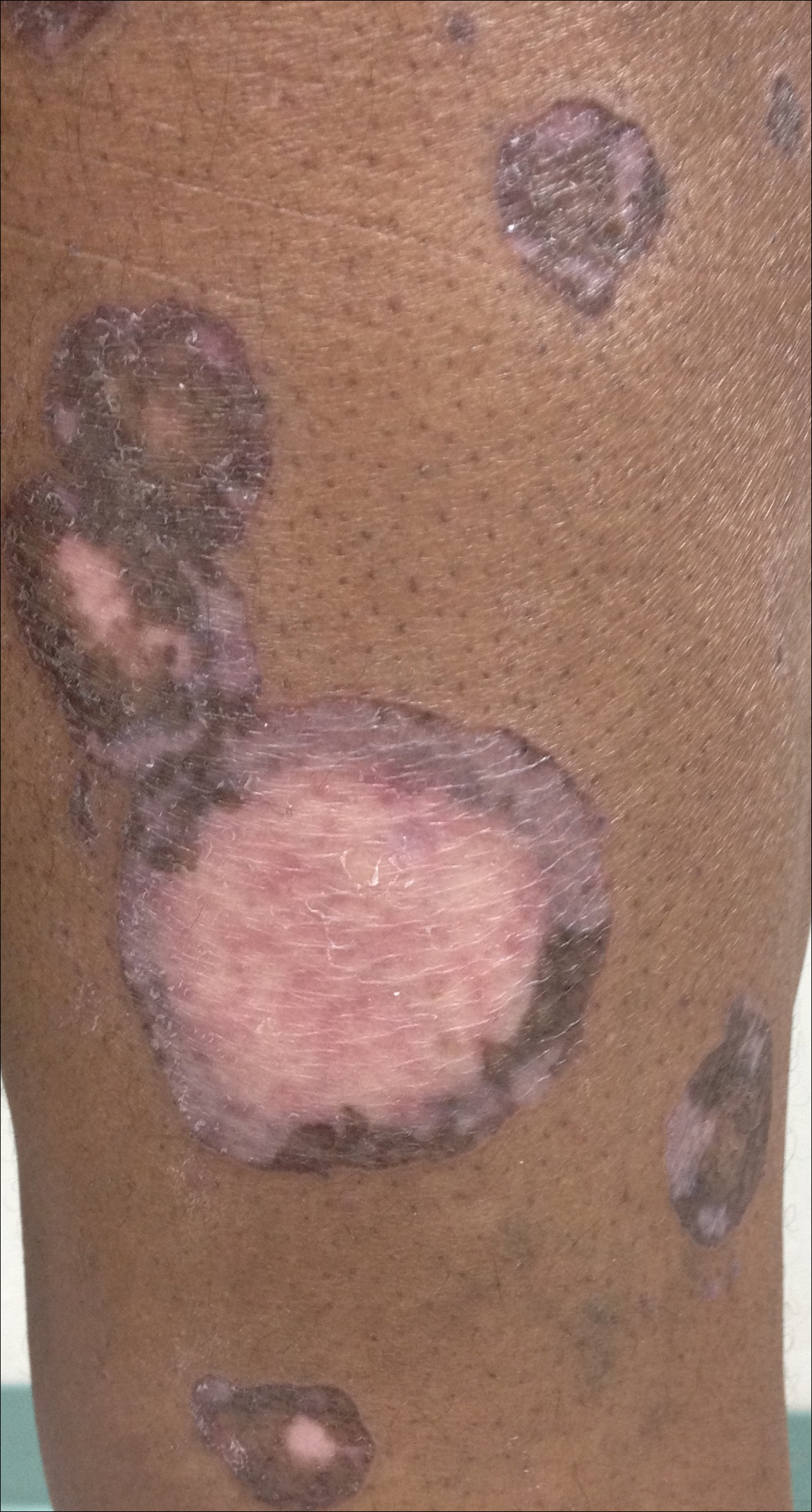
A 69-year-old black man with a history of hepatitis C virus infection and hypothyroidism presented to the dermatology clinic with a pruritic rash on the trunk, extremities, groin, and scalp of 4 months' duration. He denied any new medications, recent illnesses, or sick contacts. Physical examination demonstrated well-demarcated violaceous papules and plaques on the trunk, extensor aspect of the forearms, and thighs involving 10% of the body surface area (Figure 1A). The lesions were annular with raised borders and central depigmented atrophic scarring (Figure 1B). The examination also revealed several large hypopigmented atrophic patches and plaques in the right inguinal region and on the dorsal aspect of the penile shaft and buttocks as well as a single atrophic plaque on the scalp. No oral lesions were seen. An initial punch biopsy was consistent with a nonspecific lichenoid dermatitis (Figure 2), and the patient was prescribed triamcinolone ointment 0.1% for the trunk and extremities and tacrolimus ointment 0.1% for the groin and genital region.


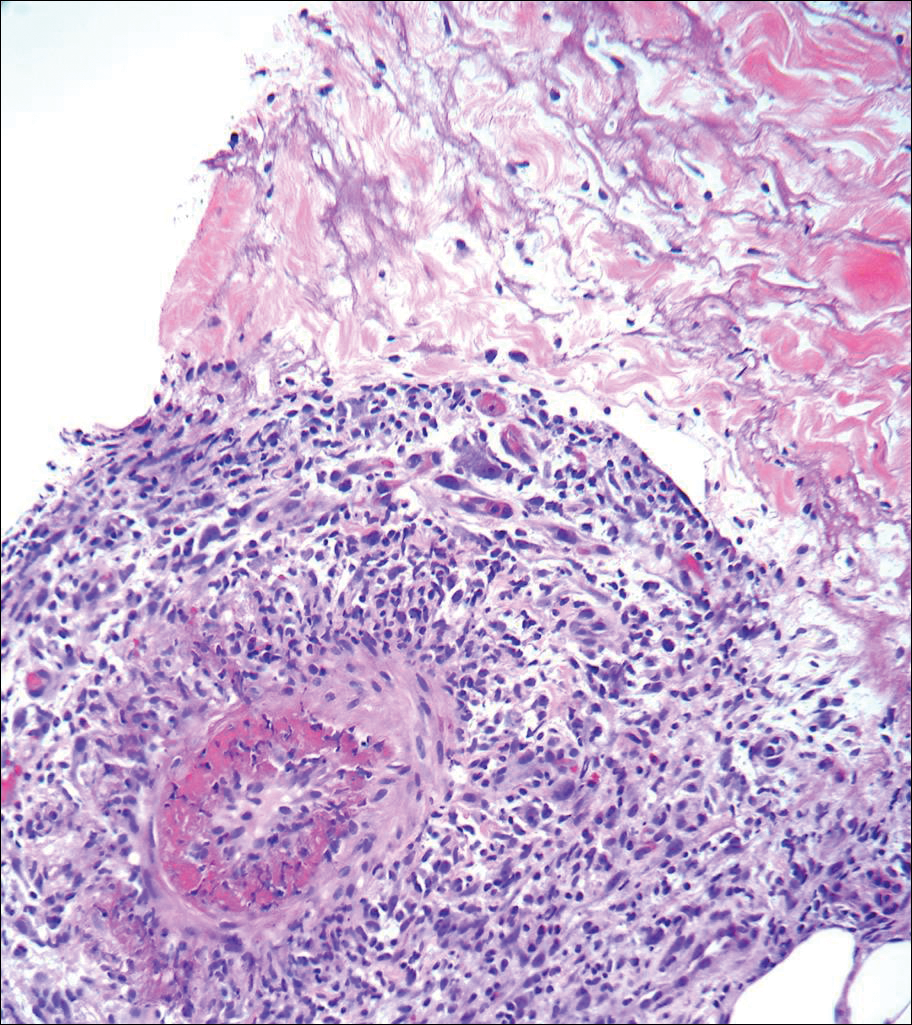
The patient continued to develop new annular atrophic skin lesions over the next several months. Repeat punch biopsies of lesional and uninvolved perilesional skin from the trunk were obtained for histopathologic confirmation and special staining. Lichenoid dermatitis again was noted on the lesional biopsy, and no notable histopathologic changes were observed on the perilesional biopsy. Verhoeff-van Gieson staining for elastic fibers was performed on both biopsies, which revealed destruction of elastic fibers in the central papillary dermis and upper reticular dermis of the lesional biopsy (Figure 3A). The elastic fibers on the perilesional biopsy were preserved (Figure 3B).

The clinical presentation and histopathological findings confirmed a diagnosis of AALP. The patient was prescribed a short taper of oral prednisone, which halted further disease progression. The patient was then started on pentoxifylline and continued on tacrolimus ointment 0.1% with minimal improvement in existing lesions. These medications were discontinued after 3 months. Hydroxychloroquine 400 mg once daily was administered, which initially resulted in some thinning of the plaques on the trunk; however, further progression of the disease was noted after 3 months. Acitretin 25 mg once daily was added to his treatment regimen. Marked thinning of active lesions, hyperpigmentation, and residual scarring was noted after 2 months of combined therapy with acitretin and hydroxychloroquine (Figure 4), with continued improvement appreciable several months later.
Comment
Lichen planus is a common pruritic inflammatory disease of the skin, mucous membranes, hair follicles, and nails with a highly variable clinical pattern and disease course that typically affects the adult population.2 There are many clinical variants of lichen planus, which all demonstrate lichenoid dermatitis on histology. Annular lichen planus is an uncommon variant most commonly seen in men with asymptomatic lesions involving the axillae and groin.2 Atrophic lichen planus is another variant demonstrating atrophic papules and plaques on the trunk and extremities.3 Annular atrophic lichen planus is the rarest variant of lichen planus, incorporating features of both annular and atrophic lichen planus.
The first case of AALP involved a 56-year-old black man with a 25-year history of annular atrophic papules and plaques on the trunk and extremities.1 The second case reported by Requena et al4 in 1994 described a 65-year-old woman with characteristic lesions on the right elbow and left knee. Lipsker et al5 reported a third case in a 41-year-old man with a history of Sneddon syndrome who had lesions typical for AALP for 20 years. In all of these cases, histopathologic examination revealed a lichenoid infiltrate with thinning of the epidermis and loss of elastic fibers in the center of the active lesions.
In more recent cases of AALP, the characteristic findings primarily occurred on the trunk and extremities.6-10 Treatment with topical corticosteroids failed in most cases and some patients noted moderate improvement with tacrolimus ointment 0.1%. Sugashima and Yamamoto11 reported a unique case in 2012 of a 32-year-old woman with AALP on the lower lip. She had notable improvement with tacrolimus ointment 0.1% after 6 months.11
All of the known cases of AALP to date have occurred in adults, both male and female, presenting with a limited number of annular plaques with slightly elevated borders and depressed atrophic centers.1,3-11 Disease duration of AALP has ranged from 2 months to 25 years.11 Histopathologic findings characteristically demonstrate a lichenoid dermatitis of the raised lesional border with a flattened epidermis, loss of rete ridges, and fibrosis of dermal papillae in the lesion center.7 The elastic fibers are destroyed in the papillary dermis of the lesion center, presumably due to elastolytic activity of inflammatory cells.1 Macrophages present in the lichenoid infiltrate of acute lesions release elastases contributing to this destruction.7 Furthermore, elastic fibers appear fragmented on electron microscopy.1
The clinical course of AALP has proven to be chronic in most cases and frequently is resistant to treatment with topical corticosteroids, retinoids, phototherapy, and immunosuppressive agents.3 Treatment administered early in the disease course may provide a more favorable outcome.11 Lesions characteristically heal with scarring and hyperpigmentation. Our case displayed more extensive involvement than has previously been reported. Our patient showed minimal improvement with topical therapy; however, he demonstrated thinning and regression of active lesions after 2 months of combined treatment with hydroxychloroquine and acitretin. Our use of oral pentoxifylline, hydroxychloroquine, and acitretin has not been previously reported in the other cases of AALP we reviewed. Acitretin is the only systemic agent for lichen planus that has achieved level A evidence, as it previously was shown to be highly effective in a placebo-controlled, double-blind study of 65 patients.12
Conclusion
Annular atrophic lichen planus is a known variant of lichen planus characterized by a loss of elastic fibers in the papillary dermis in the center of active lesions. Treatment with topical corticosteroids and phototherapy frequently is ineffective. To our knowledge, there are no studies to date regarding the efficacy of systemic therapy in treatment of AALP. Hydroxychloroquine and acitretin may prove to be beneficial treatment options for resistant AALP. Additional alternative treatments continue to be explored. We encourage reporting additional cases of AALP to further characterize its clinical presentation and response to treatments.
- Friedman DB, Hashimoto K. Annular atrophic lichen planus. J Am Acad Dermatol. 1991;25:392-394.
- James WD, Berger TG, Elston DM. Lichen planus and related conditions. In: James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:213-215.
- Kim BS, Seo SH, Jang BS, et al. A case of annular atrophic lichen planus. J Eur Acad Dermatol Venereol. 2007;21:989-990.
- Requena L, Olivares M, Pique E, et al. Annular atrophic lichen planus. Dermatology. 1994;189:95-98.
- Lipsker D, Piette JC, Laporte JL, et al. Annular atrophic lichen planus and Sneddon's syndrome. Dermatology. 1997;105:402-403.
- Mseddi M, Bouassadi S, Marrakchi S, et al. Annular atrophic lichen planus. Dermatology. 2003;207:208-209.
- Morales-Callaghan A Jr, Martinez G, Aragoneses H, et al. Annular atrophic lichen planus. J Am Acad Dermatol. 2005;52:906-908.
- Ponce-Olivera RM, Tirado-Sánchez A, Montes-de-Oca-Sánchez G, et al. Annular atrophic lichen planus. Int J Dermatol. 2007;46:490-491.
- Kim JS, Kang MS, Sagong C, et al. Annular atrophic lichen planus associated with hypertrophic lichen planus. Clin Exp Dermatol. 2008;33:195-197.
- Li B, Li JH, Xiao T, et al. Annular atrophic lichen planus. Eur J Dermatol. 2010;20:842-843.
- Sugashima Y, Yamamoto T. Annular atrophic lichen planus of the lip. Dermatol Online J. 2012;18:14.
- Manousaridis I, Manousaridis K, Peitsch WK, et al. Individualizing treatment and choice of medication in lichen planus: a step by step approach. J Dtsch Dermatol Ges. 2013;11:981-991.
Annular atrophic lichen planus (AALP) is a rare variant of lichen planus that was first described by Friedman and Hashimoto1 in 1991. Clinically, it combines the configuration and morphological features of both annular and atrophic lichen planus. It is a rare entity. We report a case of AALP in a 69-year-old black man. The clinical and histopathological presentation depicted the defining features of this entity with a characteristic loss of elastic fibers corresponding to central atrophy of active lesions.
Case Report
A 69-year-old black man with a history of hepatitis C virus infection and hypothyroidism presented to the dermatology clinic with a pruritic rash on the trunk, extremities, groin, and scalp of 4 months' duration. He denied any new medications, recent illnesses, or sick contacts. Physical examination demonstrated well-demarcated violaceous papules and plaques on the trunk, extensor aspect of the forearms, and thighs involving 10% of the body surface area (Figure 1A). The lesions were annular with raised borders and central depigmented atrophic scarring (Figure 1B). The examination also revealed several large hypopigmented atrophic patches and plaques in the right inguinal region and on the dorsal aspect of the penile shaft and buttocks as well as a single atrophic plaque on the scalp. No oral lesions were seen. An initial punch biopsy was consistent with a nonspecific lichenoid dermatitis (Figure 2), and the patient was prescribed triamcinolone ointment 0.1% for the trunk and extremities and tacrolimus ointment 0.1% for the groin and genital region.
The patient continued to develop new annular atrophic skin lesions over the next several months. Repeat punch biopsies of lesional and uninvolved perilesional skin from the trunk were obtained for histopathologic confirmation and special staining. Lichenoid dermatitis again was noted on the lesional biopsy, and no notable histopathologic changes were observed on the perilesional biopsy. Verhoeff-van Gieson staining for elastic fibers was performed on both biopsies, which revealed destruction of elastic fibers in the central papillary dermis and upper reticular dermis of the lesional biopsy (Figure 3A). The elastic fibers on the perilesional biopsy were preserved (Figure 3B).
The clinical presentation and histopathological findings confirmed a diagnosis of AALP. The patient was prescribed a short taper of oral prednisone, which halted further disease progression. The patient was then started on pentoxifylline and continued on tacrolimus ointment 0.1% with minimal improvement in existing lesions. These medications were discontinued after 3 months. Hydroxychloroquine 400 mg once daily was administered, which initially resulted in some thinning of the plaques on the trunk; however, further progression of the disease was noted after 3 months. Acitretin 25 mg once daily was added to his treatment regimen. Marked thinning of active lesions, hyperpigmentation, and residual scarring was noted after 2 months of combined therapy with acitretin and hydroxychloroquine (Figure 4), with continued improvement appreciable several months later.
Comment
Lichen planus is a common pruritic inflammatory disease of the skin, mucous membranes, hair follicles, and nails with a highly variable clinical pattern and disease course that typically affects the adult population.2 There are many clinical variants of lichen planus, which all demonstrate lichenoid dermatitis on histology. Annular lichen planus is an uncommon variant most commonly seen in men with asymptomatic lesions involving the axillae and groin.2 Atrophic lichen planus is another variant demonstrating atrophic papules and plaques on the trunk and extremities.3 Annular atrophic lichen planus is the rarest variant of lichen planus, incorporating features of both annular and atrophic lichen planus.
The first case of AALP involved a 56-year-old black man with a 25-year history of annular atrophic papules and plaques on the trunk and extremities.1 The second case reported by Requena et al4 in 1994 described a 65-year-old woman with characteristic lesions on the right elbow and left knee. Lipsker et al5 reported a third case in a 41-year-old man with a history of Sneddon syndrome who had lesions typical for AALP for 20 years. In all of these cases, histopathologic examination revealed a lichenoid infiltrate with thinning of the epidermis and loss of elastic fibers in the center of the active lesions.
In more recent cases of AALP, the characteristic findings primarily occurred on the trunk and extremities.6-10 Treatment with topical corticosteroids failed in most cases and some patients noted moderate improvement with tacrolimus ointment 0.1%. Sugashima and Yamamoto11 reported a unique case in 2012 of a 32-year-old woman with AALP on the lower lip. She had notable improvement with tacrolimus ointment 0.1% after 6 months.11
All of the known cases of AALP to date have occurred in adults, both male and female, presenting with a limited number of annular plaques with slightly elevated borders and depressed atrophic centers.1,3-11 Disease duration of AALP has ranged from 2 months to 25 years.11 Histopathologic findings characteristically demonstrate a lichenoid dermatitis of the raised lesional border with a flattened epidermis, loss of rete ridges, and fibrosis of dermal papillae in the lesion center.7 The elastic fibers are destroyed in the papillary dermis of the lesion center, presumably due to elastolytic activity of inflammatory cells.1 Macrophages present in the lichenoid infiltrate of acute lesions release elastases contributing to this destruction.7 Furthermore, elastic fibers appear fragmented on electron microscopy.1
The clinical course of AALP has proven to be chronic in most cases and frequently is resistant to treatment with topical corticosteroids, retinoids, phototherapy, and immunosuppressive agents.3 Treatment administered early in the disease course may provide a more favorable outcome.11 Lesions characteristically heal with scarring and hyperpigmentation. Our case displayed more extensive involvement than has previously been reported. Our patient showed minimal improvement with topical therapy; however, he demonstrated thinning and regression of active lesions after 2 months of combined treatment with hydroxychloroquine and acitretin. Our use of oral pentoxifylline, hydroxychloroquine, and acitretin has not been previously reported in the other cases of AALP we reviewed. Acitretin is the only systemic agent for lichen planus that has achieved level A evidence, as it previously was shown to be highly effective in a placebo-controlled, double-blind study of 65 patients.12
Conclusion
Annular atrophic lichen planus is a known variant of lichen planus characterized by a loss of elastic fibers in the papillary dermis in the center of active lesions. Treatment with topical corticosteroids and phototherapy frequently is ineffective. To our knowledge, there are no studies to date regarding the efficacy of systemic therapy in treatment of AALP. Hydroxychloroquine and acitretin may prove to be beneficial treatment options for resistant AALP. Additional alternative treatments continue to be explored. We encourage reporting additional cases of AALP to further characterize its clinical presentation and response to treatments.
Annular atrophic lichen planus (AALP) is a rare variant of lichen planus that was first described by Friedman and Hashimoto1 in 1991. Clinically, it combines the configuration and morphological features of both annular and atrophic lichen planus. It is a rare entity. We report a case of AALP in a 69-year-old black man. The clinical and histopathological presentation depicted the defining features of this entity with a characteristic loss of elastic fibers corresponding to central atrophy of active lesions.
Case Report
A 69-year-old black man with a history of hepatitis C virus infection and hypothyroidism presented to the dermatology clinic with a pruritic rash on the trunk, extremities, groin, and scalp of 4 months' duration. He denied any new medications, recent illnesses, or sick contacts. Physical examination demonstrated well-demarcated violaceous papules and plaques on the trunk, extensor aspect of the forearms, and thighs involving 10% of the body surface area (Figure 1A). The lesions were annular with raised borders and central depigmented atrophic scarring (Figure 1B). The examination also revealed several large hypopigmented atrophic patches and plaques in the right inguinal region and on the dorsal aspect of the penile shaft and buttocks as well as a single atrophic plaque on the scalp. No oral lesions were seen. An initial punch biopsy was consistent with a nonspecific lichenoid dermatitis (Figure 2), and the patient was prescribed triamcinolone ointment 0.1% for the trunk and extremities and tacrolimus ointment 0.1% for the groin and genital region.
The patient continued to develop new annular atrophic skin lesions over the next several months. Repeat punch biopsies of lesional and uninvolved perilesional skin from the trunk were obtained for histopathologic confirmation and special staining. Lichenoid dermatitis again was noted on the lesional biopsy, and no notable histopathologic changes were observed on the perilesional biopsy. Verhoeff-van Gieson staining for elastic fibers was performed on both biopsies, which revealed destruction of elastic fibers in the central papillary dermis and upper reticular dermis of the lesional biopsy (Figure 3A). The elastic fibers on the perilesional biopsy were preserved (Figure 3B).
The clinical presentation and histopathological findings confirmed a diagnosis of AALP. The patient was prescribed a short taper of oral prednisone, which halted further disease progression. The patient was then started on pentoxifylline and continued on tacrolimus ointment 0.1% with minimal improvement in existing lesions. These medications were discontinued after 3 months. Hydroxychloroquine 400 mg once daily was administered, which initially resulted in some thinning of the plaques on the trunk; however, further progression of the disease was noted after 3 months. Acitretin 25 mg once daily was added to his treatment regimen. Marked thinning of active lesions, hyperpigmentation, and residual scarring was noted after 2 months of combined therapy with acitretin and hydroxychloroquine (Figure 4), with continued improvement appreciable several months later.
Comment
Lichen planus is a common pruritic inflammatory disease of the skin, mucous membranes, hair follicles, and nails with a highly variable clinical pattern and disease course that typically affects the adult population.2 There are many clinical variants of lichen planus, which all demonstrate lichenoid dermatitis on histology. Annular lichen planus is an uncommon variant most commonly seen in men with asymptomatic lesions involving the axillae and groin.2 Atrophic lichen planus is another variant demonstrating atrophic papules and plaques on the trunk and extremities.3 Annular atrophic lichen planus is the rarest variant of lichen planus, incorporating features of both annular and atrophic lichen planus.
The first case of AALP involved a 56-year-old black man with a 25-year history of annular atrophic papules and plaques on the trunk and extremities.1 The second case reported by Requena et al4 in 1994 described a 65-year-old woman with characteristic lesions on the right elbow and left knee. Lipsker et al5 reported a third case in a 41-year-old man with a history of Sneddon syndrome who had lesions typical for AALP for 20 years. In all of these cases, histopathologic examination revealed a lichenoid infiltrate with thinning of the epidermis and loss of elastic fibers in the center of the active lesions.
In more recent cases of AALP, the characteristic findings primarily occurred on the trunk and extremities.6-10 Treatment with topical corticosteroids failed in most cases and some patients noted moderate improvement with tacrolimus ointment 0.1%. Sugashima and Yamamoto11 reported a unique case in 2012 of a 32-year-old woman with AALP on the lower lip. She had notable improvement with tacrolimus ointment 0.1% after 6 months.11
All of the known cases of AALP to date have occurred in adults, both male and female, presenting with a limited number of annular plaques with slightly elevated borders and depressed atrophic centers.1,3-11 Disease duration of AALP has ranged from 2 months to 25 years.11 Histopathologic findings characteristically demonstrate a lichenoid dermatitis of the raised lesional border with a flattened epidermis, loss of rete ridges, and fibrosis of dermal papillae in the lesion center.7 The elastic fibers are destroyed in the papillary dermis of the lesion center, presumably due to elastolytic activity of inflammatory cells.1 Macrophages present in the lichenoid infiltrate of acute lesions release elastases contributing to this destruction.7 Furthermore, elastic fibers appear fragmented on electron microscopy.1
The clinical course of AALP has proven to be chronic in most cases and frequently is resistant to treatment with topical corticosteroids, retinoids, phototherapy, and immunosuppressive agents.3 Treatment administered early in the disease course may provide a more favorable outcome.11 Lesions characteristically heal with scarring and hyperpigmentation. Our case displayed more extensive involvement than has previously been reported. Our patient showed minimal improvement with topical therapy; however, he demonstrated thinning and regression of active lesions after 2 months of combined treatment with hydroxychloroquine and acitretin. Our use of oral pentoxifylline, hydroxychloroquine, and acitretin has not been previously reported in the other cases of AALP we reviewed. Acitretin is the only systemic agent for lichen planus that has achieved level A evidence, as it previously was shown to be highly effective in a placebo-controlled, double-blind study of 65 patients.12
Conclusion
Annular atrophic lichen planus is a known variant of lichen planus characterized by a loss of elastic fibers in the papillary dermis in the center of active lesions. Treatment with topical corticosteroids and phototherapy frequently is ineffective. To our knowledge, there are no studies to date regarding the efficacy of systemic therapy in treatment of AALP. Hydroxychloroquine and acitretin may prove to be beneficial treatment options for resistant AALP. Additional alternative treatments continue to be explored. We encourage reporting additional cases of AALP to further characterize its clinical presentation and response to treatments.
- Friedman DB, Hashimoto K. Annular atrophic lichen planus. J Am Acad Dermatol. 1991;25:392-394.
- James WD, Berger TG, Elston DM. Lichen planus and related conditions. In: James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:213-215.
- Kim BS, Seo SH, Jang BS, et al. A case of annular atrophic lichen planus. J Eur Acad Dermatol Venereol. 2007;21:989-990.
- Requena L, Olivares M, Pique E, et al. Annular atrophic lichen planus. Dermatology. 1994;189:95-98.
- Lipsker D, Piette JC, Laporte JL, et al. Annular atrophic lichen planus and Sneddon's syndrome. Dermatology. 1997;105:402-403.
- Mseddi M, Bouassadi S, Marrakchi S, et al. Annular atrophic lichen planus. Dermatology. 2003;207:208-209.
- Morales-Callaghan A Jr, Martinez G, Aragoneses H, et al. Annular atrophic lichen planus. J Am Acad Dermatol. 2005;52:906-908.
- Ponce-Olivera RM, Tirado-Sánchez A, Montes-de-Oca-Sánchez G, et al. Annular atrophic lichen planus. Int J Dermatol. 2007;46:490-491.
- Kim JS, Kang MS, Sagong C, et al. Annular atrophic lichen planus associated with hypertrophic lichen planus. Clin Exp Dermatol. 2008;33:195-197.
- Li B, Li JH, Xiao T, et al. Annular atrophic lichen planus. Eur J Dermatol. 2010;20:842-843.
- Sugashima Y, Yamamoto T. Annular atrophic lichen planus of the lip. Dermatol Online J. 2012;18:14.
- Manousaridis I, Manousaridis K, Peitsch WK, et al. Individualizing treatment and choice of medication in lichen planus: a step by step approach. J Dtsch Dermatol Ges. 2013;11:981-991.
- Friedman DB, Hashimoto K. Annular atrophic lichen planus. J Am Acad Dermatol. 1991;25:392-394.
- James WD, Berger TG, Elston DM. Lichen planus and related conditions. In: James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. China: Saunders Elsevier; 2011:213-215.
- Kim BS, Seo SH, Jang BS, et al. A case of annular atrophic lichen planus. J Eur Acad Dermatol Venereol. 2007;21:989-990.
- Requena L, Olivares M, Pique E, et al. Annular atrophic lichen planus. Dermatology. 1994;189:95-98.
- Lipsker D, Piette JC, Laporte JL, et al. Annular atrophic lichen planus and Sneddon's syndrome. Dermatology. 1997;105:402-403.
- Mseddi M, Bouassadi S, Marrakchi S, et al. Annular atrophic lichen planus. Dermatology. 2003;207:208-209.
- Morales-Callaghan A Jr, Martinez G, Aragoneses H, et al. Annular atrophic lichen planus. J Am Acad Dermatol. 2005;52:906-908.
- Ponce-Olivera RM, Tirado-Sánchez A, Montes-de-Oca-Sánchez G, et al. Annular atrophic lichen planus. Int J Dermatol. 2007;46:490-491.
- Kim JS, Kang MS, Sagong C, et al. Annular atrophic lichen planus associated with hypertrophic lichen planus. Clin Exp Dermatol. 2008;33:195-197.
- Li B, Li JH, Xiao T, et al. Annular atrophic lichen planus. Eur J Dermatol. 2010;20:842-843.
- Sugashima Y, Yamamoto T. Annular atrophic lichen planus of the lip. Dermatol Online J. 2012;18:14.
- Manousaridis I, Manousaridis K, Peitsch WK, et al. Individualizing treatment and choice of medication in lichen planus: a step by step approach. J Dtsch Dermatol Ges. 2013;11:981-991.
Asymptomatic Cutaneous Polyarteritis Nodosa: Treatment Options and Therapeutic Guidelines
In 1931, Lindberg1 described a cutaneous variant of polyarteritis nodosa, which lacked visceral involvement and possessed a more favorable prognosis.2 Cutaneous polyarteritis nodosa (CPAN) is a localized small- to medium-vessel vasculitis restricted to the skin. Both benign and chronic courses have been described, and systemic involvement does not occur.3 Diagnostic criteria proposed by Nakamura et al3 in 2009 included cutaneous nodules, livedo reticularis, purpura, or ulcers; histopathologic fibrinoid necrotizing vasculitis of small- to medium-sized vessels; and exclusion of systemic symptoms (eg, fever, hypertension, weight loss, renal failure, cerebral hemorrhage, neuropathy, myocardial infarction, ischemic heart disease, pericarditis, pleuritis, arthralgia/myalgia). Nodules occur in 30% to 50% of cases and can remain for years if left untreated. Ulcerations occur in up to 30% of patients. Myositis, arthritis, and weakness also have been reported with this condition.4 Cutaneous polyarteritis nodosa has been associated with abnormal antibody testing with elevations of antiphospholipid cofactor antibody, lupus anticoagulant, anticardiolipin antibody, and anti-β2-glycoprotein I–dependent cardiolipin antibody, as well as elevated anti–phosphatidylserine-prothrombin complex antibody.5 These antibodies suggest increased risk for thrombosis and systemic diseases such as lupus or other autoimmune connective tissue disease. The distinction of this entity from systemic polyartertitis nodosa is key when determining treatment options and monitoring parameters.
Case Report
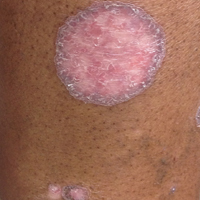
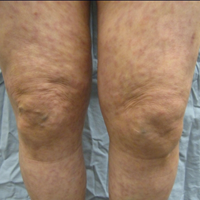
A 66-year-old woman was referred to our facility by an outside dermatologist with a mildly pruritic, blanchable, reticulated erythema on the chest and bilateral arms and legs of 3 months’ duration consistent with livedo reticularis (Figure 1). Prior systemic therapy included prednisone 10 mg 3 times daily, fexofenadine, loratadine, and hydroxyzine. When the systemic steroid was tapered, the patient developed an asymptomatic flare of her eruption. On presentation, the lesions had waxed and waned, and the patient was taking only vitamin B12 and vitamin C. Her medical history was notable for an unknown-type lymphoma of the chest wall diagnosed at 46 years of age that was treated with an unknown chemotherapeutic agent, chronic pancreatitis that resulted in a duodenectomy at 61 years of age, chronic cholecystitis, and 1 first-trimester miscarriage. Outside laboratory tests, including a comprehensive metabolic panel, complete blood cell count, urinalysis, renal function, and liver function tests were within reference range, except for the finding of mild leukocytosis (11,000/µL)(reference range, 3800–10,800/µL), which resolved after steroids were discontinued, with otherwise normal results. Punch biopsy of a specimen from the right thigh revealed medium-vessel vasculitis consistent with polyarteritis nodosa (Figure 2). Laboratory workup by our facility including hepatitis panel, perinuclear antineutrophil cytoplasmic antibody, cytoplasmic antineutrophil cytoplasmic antibody, factor V Leiden, prothrombin time/international normalized ratio, anticardiolipin antibody, and proteins C and S were all within reference range. Abnormal values included a low positive but nondiagnostic antinuclear antibody screen with negative titers, and the lupus anticoagulant titer was mildly elevated at 44 IgG binding units (reference range, <40 IgG binding units). Serum protein electrophoresis (SPEP) and urine protein electrophoresis also were performed, and SPEP was low positive for elevated κ and γ light chains. The patient was referred to oncology, and further testing revealed no underlying malignancy. The patient was monitored and no treatment was initiated; her rash completely resolved within 3 months. Laboratory monitoring at 6 months including SPEP, urine protein electrophoresis, lupus anticoagulant, and clotting studies all were within reference range.
Comment
Although the treatment of systemic polyarteritis nodosa often is necessary and typically involves high-dose corticosteroids and cyclophosphamide, the treatment of CPAN initially is less aggressive. Of the options available for treatment of CPAN, each has associated risks and side effects. Chen6 classified CPAN into 3 groups: 1 (mild), 2 (severe with no systemic involvement), and 3 (severe with progression to systemic disease)(Table). The authors performed a review of all the published treatments and their respective side effects to evaluate if treatment should be instituted for asymptomatic (group 1) disease presenting with abnormal antibody findings as demonstrated in our case.

First-line treatment of CPAN includes nonsteroidal anti-inflammatory drugs (NSAIDS) and colchicine.7 Nonsteroidal anti-inflammatory drugs are preferred; however, they also have been associated with gastrointestinal tract upset and increased risk for peptic ulcer disease with long-term use. Although colchicine often is used in conjunction with NSAIDS8 for its anti-inflammatory activity, no studies have been performed on this drug as monotherapy, and the side effect of diarrhea often limits its use.
Other therapies include dapsone, which should be monitored carefully due to the risk for dapsone hypersensitivity syndrome.8,9 Topical corticosteroids have been proven effective for mild cases of confluent erythema with remission occurring as early as 4 weeks.4 Some reports emphasize the role of streptococcal infections in CPAN, especially in children.8,10-12 Consequently it is recommended that anti–streptolysin O titers should be included in the workup for CPAN. Long-term penicillin prophylaxis and tonsillectomy have been used to prevent disease flares with limited success.8,10-12
For more severe disease, especially with neuromuscular involvement, oral methylprednisolone up to 1 mg/kg daily has been used and has proven effective in the control of acute exacerbations.7,13 However, the many adverse effects of systemic steroids limit their use long-term, and taper will often result in flare of disease.4,7 Medications used in conjunction with steroids include hydroxychloroquine, dapsone, azathioprine, cyclophosphamide, methotrexate, sulfapyridine, pentoxifylline, infliximab, etanercept, and intravenous immunoglobulin.4,9,12-17
Low-dose methotrexate has shown some improvement in skin disease with CPAN, but other case reports suggest that complete remission is not achieved with this drug.15,18 More studies are needed to assess the use of methotrexate for CPAN.
Immunomodulators have been used in multiple case reports with varying levels of success. Rogalski and Sticherling4 reported 3 cases that cleared with methylprednisolone plus azathioprine ranging from 4 weeks to 6 months; nausea limited tolerance of azathioprine in 1 case. Mycophenolate mofetil also was successfully used in 2 cases with clearance at 17 weeks and 6 months. In this series of cases, cyclosporine was ineffective for CPAN.4 Two case reports documented cutaneous clearance with cyclophosphamide in conjunction with prednisolone.9,10 No prospective trials have been performed on these medications, and immunosuppressants should only be considered in steroid-resistant cases.
The use of intravenous immunoglobulin has been reported effective in prior cases that showed resistance to more conventional trials of steroids, azathioprine, and/or cyclophosphamide.12,14 Intravenous immunoglobulin may be regarded as a treatment option for severe resistant disease. Several case reports also have documented success using tumor necrosis factor α blockers, particularly infliximab, as an adjunct to steroids and etanercept as both a steroid adjunct and monotherapy.16,17,19 More studies are necessary to evaluate these treatments.
Additionally, single case reports have outlined the use of other therapeutic agents, including tamoxifen (10 mg twice daily increased to 20 mg twice daily during episodes of breakthrough lesions),20 hyperbaric oxygen therapy (100% oxygen for 90 minutes 5 times weekly at 1.5 atm absolute followed by 2 weeks of 2 atm absolute),21 and granulocyte-macrophage colony-stimulating factor (300 µg injection in small portion to ulcer edges twice monthly for 2 months).22 All of these treatments show promise, but data are limited.
Because thrombosis is postulated to be a potential mechanism leading to CPAN, agents such as pentoxifylline, clopidogrel, and warfarin have been examined as treatment options. Pentoxifylline in combination with mycophenolate mofetil has been successful in treating a case that was resistant to other immunosuppressants.23 Clopidogrel blocks the adenosine diphosphate pathway and impairs clot retraction. Clopidogrel was reported effective in an acute flare of CPAN for clearance of skin lesions and normalization of lupus anticoagulant.24 It also was used successfully in recurrent CPAN after steroid treatments in a patient with neuromuscular symptoms. There was no recurrence in either of the patients in this case report series. Warfarin therapy at an international normalized ratio of 3.0 also has demonstrated success in halting disease progression and in facilitating the resolution of skin changes and normalization of anti–phosphatidylserine-prothrombin complex antibodies.24 Our review of the literature did not reveal evidence of a standardized length of treatment following symptom resolution or if treatment is indicated in asymptomatic disease, or as in our case, with only mild elevations of antiphospholipid antibodies.
Conclusion
Multiple treatment options exist for CPAN, but the data on their efficacies is limited and based only on anecdotal evidence, not prospective analysis. We believe that it seems reasonable to initiate treatment only for symptomatic disease or cases in which the antibody titers suggest that the patient may be at high risk for thrombosis. Mild symptoms and mild cutaneous changes would suggest the likely choice of NSAIDs, colchicine, or dapsone as treatment options versus no treatment. In patients with antibody titers, pentoxifylline, clopidogrel, or warfarin may be considered first-line therapies. With severe ulcerative lesions and neuromuscular involvement, steroids, immunosuppressants, and other investigative agents should be contemplated. In our patient, the laboratory studies were repeated and normalized on complete resolution of her livedo eruption. She remained asymptomatic and clear for 8 months without any treatment. The incidence of this presentation of CPAN is unknown and is likely underreported, as we would not expect most patients to present to their physicians for the evaluation of otherwise asymptomatic livedo reticularis. In essence, our case report suggests that it may be prudent to simply monitor patients with asymptomatic CPAN.
- Lindberg K. Ein Beitrag zur Kenntnis der Periarteritis nodosa. Acta Med Scand. 1931;76:183-225.
- Kraemer M, Linden D, Berlit P. The spectrum of differential diagnosis in neurological patients with livedo reticularis and livedo racemosa [published online August 26, 2005]. J Neurol. 2005;252:1155-1166.
- Nakamura T, Kanazawa N, Ikeda T, et al. Cutaneous polyarteritis nodosa: revisiting its definition and diagnostic criteria. Arch Dermatol Res. 2009;301:117-121.
- Rogalski C, Sticherling M. Panateritis cutanea benigna—an entity limited to the skin or cutaneous presentation of a systemic necrotizing vasculitis? report of seven cases and review of the literature. Int J Dermatol. 2007;46:817-821.
- Kawakami T, Yamazaki M, Mizoguchi M, et al. High titer of anti-phosphatidylserine-prothrombin complex antibodies in patients with cutaneous polyarteritis nodosa. Arthritis Rheum. 2007;57:1507-1513.
- Chen KR. Cutaneous polyarteritis nodosa: a clinical and histopathological study of 20 cases. J Dermatol. 1989;6:429-442.
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756.
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathologic analysis and review of the published work. J Dermatol. 2010;37:85-93.
- Flanagan N, Casey EB, Watson R, et al. Cutaneous polyartertitis nodosa with seronegative arthritis. Rheumatology (Oxford). 1999;38:1161-1162.
- Fathalla B, Miller L, Brady S, et al. Cutaneous polyarteritis nodosa in children. J Am Acad Dermatol. 2005;53:724-728.
- Misago N, Mochizuki Y, Sekiyama-Kodera H, et al. Cutaneous polyarteritis nodosa: therapy and clinical course in four cases. J Dermatol. 2001;28:719-727.
- Breda L, Franchini S, Marzetti V, et al. Intravenous immunoglobulins for cutaneous polyarteritis nodosa resistant to conventional treatment. Scand J Rheumatol. 2016;45:169-170.
- Maillard H, Szczesniak S, Martin L. Cutaneous periarteritis nodosa: diagnostic and therapeutic aspects of 9 cases. Ann Dermatol Venereol. 1999;26:125-129.
- Lobo I, Ferreira M, Silva E. Cutaneous polyarteritis nodosa treated with intravenous immunoglobulin. J Eur Acad Dermatol Venereol. 2007;22:880-882.
- Boehm I, Bauer R. Low-dose methotrexate controls a severe form of polyarteritis nodosa. Arch Dermatol. 2000;136:167-169.
- Campanilho-Marques R, Ramos F, Canhão H, et al. Remission induced by infliximab in a childhood polyarteritis nodosa refractory to conventional immunosuppression and rituximab. Joint Bone Spine. 2014;81:277-278.
- Inoue N, Shimizu M, Mizuta M, et al. Refractory cutaneous polyarteritis nodosa: successful treatment with etanercept. Pediatr Int. 2017;59:751-752.
- Schartz NE. Successful treatment in two cases of steroid dependent cutaneous polyarteritis nodosa with low-dose methotrexate. Dermatology. 2001;203:336-338.
- Valor L, Monteagudo I, de la Torre I, et al. Young male patient diagnosed with cutaneous polyarteritis nodosa successfully treated with etanercept. Mod Rheumatol. 2014;24:688-689.
- Cvancara JL, Meffert JJ, Elston DM. Estrogen sensitive cutaneous polyarteritis nodosa: response to tamoxifen. J Am Acad Dermatol. 1998;39:643-646.
- Mazokopakis E, Milkas A, Tsartsalis A, et al. Improvement of cutaneous polyarteritis nodosa with hyperbaric oxygen. Int J Dermatol. 2009;48:1017-1029.
- Tursen U, Api H, Kaya TI, et al. Rapid healing of chronic leg ulcers during perilesional injections of granulocyte-macrophage colony stimulating factor in a patient with cutaneous polyarteritis nodosa. J Eur Acad Dermatol Venereol. 2006;20:1341-1343.
- Kluger N, Guillot B, Bessis D. Ulcerative cutaneous polyarteritis nodosa treated with mycophenolate mofetil and pentoxifylline. J Dermatolog Treat. 2011;22:175-177.
- Kawakami T, Soma Y. Use of warfarin therapy at a target international normalized ratio of 3.0 for cutaneous polyarteritis nodosa. J Am Acad Dermatol. 2010;63:602-606.
In 1931, Lindberg1 described a cutaneous variant of polyarteritis nodosa, which lacked visceral involvement and possessed a more favorable prognosis.2 Cutaneous polyarteritis nodosa (CPAN) is a localized small- to medium-vessel vasculitis restricted to the skin. Both benign and chronic courses have been described, and systemic involvement does not occur.3 Diagnostic criteria proposed by Nakamura et al3 in 2009 included cutaneous nodules, livedo reticularis, purpura, or ulcers; histopathologic fibrinoid necrotizing vasculitis of small- to medium-sized vessels; and exclusion of systemic symptoms (eg, fever, hypertension, weight loss, renal failure, cerebral hemorrhage, neuropathy, myocardial infarction, ischemic heart disease, pericarditis, pleuritis, arthralgia/myalgia). Nodules occur in 30% to 50% of cases and can remain for years if left untreated. Ulcerations occur in up to 30% of patients. Myositis, arthritis, and weakness also have been reported with this condition.4 Cutaneous polyarteritis nodosa has been associated with abnormal antibody testing with elevations of antiphospholipid cofactor antibody, lupus anticoagulant, anticardiolipin antibody, and anti-β2-glycoprotein I–dependent cardiolipin antibody, as well as elevated anti–phosphatidylserine-prothrombin complex antibody.5 These antibodies suggest increased risk for thrombosis and systemic diseases such as lupus or other autoimmune connective tissue disease. The distinction of this entity from systemic polyartertitis nodosa is key when determining treatment options and monitoring parameters.
Case Report
A 66-year-old woman was referred to our facility by an outside dermatologist with a mildly pruritic, blanchable, reticulated erythema on the chest and bilateral arms and legs of 3 months’ duration consistent with livedo reticularis (Figure 1). Prior systemic therapy included prednisone 10 mg 3 times daily, fexofenadine, loratadine, and hydroxyzine. When the systemic steroid was tapered, the patient developed an asymptomatic flare of her eruption. On presentation, the lesions had waxed and waned, and the patient was taking only vitamin B12 and vitamin C. Her medical history was notable for an unknown-type lymphoma of the chest wall diagnosed at 46 years of age that was treated with an unknown chemotherapeutic agent, chronic pancreatitis that resulted in a duodenectomy at 61 years of age, chronic cholecystitis, and 1 first-trimester miscarriage. Outside laboratory tests, including a comprehensive metabolic panel, complete blood cell count, urinalysis, renal function, and liver function tests were within reference range, except for the finding of mild leukocytosis (11,000/µL)(reference range, 3800–10,800/µL), which resolved after steroids were discontinued, with otherwise normal results. Punch biopsy of a specimen from the right thigh revealed medium-vessel vasculitis consistent with polyarteritis nodosa (Figure 2). Laboratory workup by our facility including hepatitis panel, perinuclear antineutrophil cytoplasmic antibody, cytoplasmic antineutrophil cytoplasmic antibody, factor V Leiden, prothrombin time/international normalized ratio, anticardiolipin antibody, and proteins C and S were all within reference range. Abnormal values included a low positive but nondiagnostic antinuclear antibody screen with negative titers, and the lupus anticoagulant titer was mildly elevated at 44 IgG binding units (reference range, <40 IgG binding units). Serum protein electrophoresis (SPEP) and urine protein electrophoresis also were performed, and SPEP was low positive for elevated κ and γ light chains. The patient was referred to oncology, and further testing revealed no underlying malignancy. The patient was monitored and no treatment was initiated; her rash completely resolved within 3 months. Laboratory monitoring at 6 months including SPEP, urine protein electrophoresis, lupus anticoagulant, and clotting studies all were within reference range.
Comment
Although the treatment of systemic polyarteritis nodosa often is necessary and typically involves high-dose corticosteroids and cyclophosphamide, the treatment of CPAN initially is less aggressive. Of the options available for treatment of CPAN, each has associated risks and side effects. Chen6 classified CPAN into 3 groups: 1 (mild), 2 (severe with no systemic involvement), and 3 (severe with progression to systemic disease)(Table). The authors performed a review of all the published treatments and their respective side effects to evaluate if treatment should be instituted for asymptomatic (group 1) disease presenting with abnormal antibody findings as demonstrated in our case.
First-line treatment of CPAN includes nonsteroidal anti-inflammatory drugs (NSAIDS) and colchicine.7 Nonsteroidal anti-inflammatory drugs are preferred; however, they also have been associated with gastrointestinal tract upset and increased risk for peptic ulcer disease with long-term use. Although colchicine often is used in conjunction with NSAIDS8 for its anti-inflammatory activity, no studies have been performed on this drug as monotherapy, and the side effect of diarrhea often limits its use.
Other therapies include dapsone, which should be monitored carefully due to the risk for dapsone hypersensitivity syndrome.8,9 Topical corticosteroids have been proven effective for mild cases of confluent erythema with remission occurring as early as 4 weeks.4 Some reports emphasize the role of streptococcal infections in CPAN, especially in children.8,10-12 Consequently it is recommended that anti–streptolysin O titers should be included in the workup for CPAN. Long-term penicillin prophylaxis and tonsillectomy have been used to prevent disease flares with limited success.8,10-12
For more severe disease, especially with neuromuscular involvement, oral methylprednisolone up to 1 mg/kg daily has been used and has proven effective in the control of acute exacerbations.7,13 However, the many adverse effects of systemic steroids limit their use long-term, and taper will often result in flare of disease.4,7 Medications used in conjunction with steroids include hydroxychloroquine, dapsone, azathioprine, cyclophosphamide, methotrexate, sulfapyridine, pentoxifylline, infliximab, etanercept, and intravenous immunoglobulin.4,9,12-17
Low-dose methotrexate has shown some improvement in skin disease with CPAN, but other case reports suggest that complete remission is not achieved with this drug.15,18 More studies are needed to assess the use of methotrexate for CPAN.
Immunomodulators have been used in multiple case reports with varying levels of success. Rogalski and Sticherling4 reported 3 cases that cleared with methylprednisolone plus azathioprine ranging from 4 weeks to 6 months; nausea limited tolerance of azathioprine in 1 case. Mycophenolate mofetil also was successfully used in 2 cases with clearance at 17 weeks and 6 months. In this series of cases, cyclosporine was ineffective for CPAN.4 Two case reports documented cutaneous clearance with cyclophosphamide in conjunction with prednisolone.9,10 No prospective trials have been performed on these medications, and immunosuppressants should only be considered in steroid-resistant cases.
The use of intravenous immunoglobulin has been reported effective in prior cases that showed resistance to more conventional trials of steroids, azathioprine, and/or cyclophosphamide.12,14 Intravenous immunoglobulin may be regarded as a treatment option for severe resistant disease. Several case reports also have documented success using tumor necrosis factor α blockers, particularly infliximab, as an adjunct to steroids and etanercept as both a steroid adjunct and monotherapy.16,17,19 More studies are necessary to evaluate these treatments.
Additionally, single case reports have outlined the use of other therapeutic agents, including tamoxifen (10 mg twice daily increased to 20 mg twice daily during episodes of breakthrough lesions),20 hyperbaric oxygen therapy (100% oxygen for 90 minutes 5 times weekly at 1.5 atm absolute followed by 2 weeks of 2 atm absolute),21 and granulocyte-macrophage colony-stimulating factor (300 µg injection in small portion to ulcer edges twice monthly for 2 months).22 All of these treatments show promise, but data are limited.
Because thrombosis is postulated to be a potential mechanism leading to CPAN, agents such as pentoxifylline, clopidogrel, and warfarin have been examined as treatment options. Pentoxifylline in combination with mycophenolate mofetil has been successful in treating a case that was resistant to other immunosuppressants.23 Clopidogrel blocks the adenosine diphosphate pathway and impairs clot retraction. Clopidogrel was reported effective in an acute flare of CPAN for clearance of skin lesions and normalization of lupus anticoagulant.24 It also was used successfully in recurrent CPAN after steroid treatments in a patient with neuromuscular symptoms. There was no recurrence in either of the patients in this case report series. Warfarin therapy at an international normalized ratio of 3.0 also has demonstrated success in halting disease progression and in facilitating the resolution of skin changes and normalization of anti–phosphatidylserine-prothrombin complex antibodies.24 Our review of the literature did not reveal evidence of a standardized length of treatment following symptom resolution or if treatment is indicated in asymptomatic disease, or as in our case, with only mild elevations of antiphospholipid antibodies.
Conclusion
Multiple treatment options exist for CPAN, but the data on their efficacies is limited and based only on anecdotal evidence, not prospective analysis. We believe that it seems reasonable to initiate treatment only for symptomatic disease or cases in which the antibody titers suggest that the patient may be at high risk for thrombosis. Mild symptoms and mild cutaneous changes would suggest the likely choice of NSAIDs, colchicine, or dapsone as treatment options versus no treatment. In patients with antibody titers, pentoxifylline, clopidogrel, or warfarin may be considered first-line therapies. With severe ulcerative lesions and neuromuscular involvement, steroids, immunosuppressants, and other investigative agents should be contemplated. In our patient, the laboratory studies were repeated and normalized on complete resolution of her livedo eruption. She remained asymptomatic and clear for 8 months without any treatment. The incidence of this presentation of CPAN is unknown and is likely underreported, as we would not expect most patients to present to their physicians for the evaluation of otherwise asymptomatic livedo reticularis. In essence, our case report suggests that it may be prudent to simply monitor patients with asymptomatic CPAN.
In 1931, Lindberg1 described a cutaneous variant of polyarteritis nodosa, which lacked visceral involvement and possessed a more favorable prognosis.2 Cutaneous polyarteritis nodosa (CPAN) is a localized small- to medium-vessel vasculitis restricted to the skin. Both benign and chronic courses have been described, and systemic involvement does not occur.3 Diagnostic criteria proposed by Nakamura et al3 in 2009 included cutaneous nodules, livedo reticularis, purpura, or ulcers; histopathologic fibrinoid necrotizing vasculitis of small- to medium-sized vessels; and exclusion of systemic symptoms (eg, fever, hypertension, weight loss, renal failure, cerebral hemorrhage, neuropathy, myocardial infarction, ischemic heart disease, pericarditis, pleuritis, arthralgia/myalgia). Nodules occur in 30% to 50% of cases and can remain for years if left untreated. Ulcerations occur in up to 30% of patients. Myositis, arthritis, and weakness also have been reported with this condition.4 Cutaneous polyarteritis nodosa has been associated with abnormal antibody testing with elevations of antiphospholipid cofactor antibody, lupus anticoagulant, anticardiolipin antibody, and anti-β2-glycoprotein I–dependent cardiolipin antibody, as well as elevated anti–phosphatidylserine-prothrombin complex antibody.5 These antibodies suggest increased risk for thrombosis and systemic diseases such as lupus or other autoimmune connective tissue disease. The distinction of this entity from systemic polyartertitis nodosa is key when determining treatment options and monitoring parameters.
Case Report
A 66-year-old woman was referred to our facility by an outside dermatologist with a mildly pruritic, blanchable, reticulated erythema on the chest and bilateral arms and legs of 3 months’ duration consistent with livedo reticularis (Figure 1). Prior systemic therapy included prednisone 10 mg 3 times daily, fexofenadine, loratadine, and hydroxyzine. When the systemic steroid was tapered, the patient developed an asymptomatic flare of her eruption. On presentation, the lesions had waxed and waned, and the patient was taking only vitamin B12 and vitamin C. Her medical history was notable for an unknown-type lymphoma of the chest wall diagnosed at 46 years of age that was treated with an unknown chemotherapeutic agent, chronic pancreatitis that resulted in a duodenectomy at 61 years of age, chronic cholecystitis, and 1 first-trimester miscarriage. Outside laboratory tests, including a comprehensive metabolic panel, complete blood cell count, urinalysis, renal function, and liver function tests were within reference range, except for the finding of mild leukocytosis (11,000/µL)(reference range, 3800–10,800/µL), which resolved after steroids were discontinued, with otherwise normal results. Punch biopsy of a specimen from the right thigh revealed medium-vessel vasculitis consistent with polyarteritis nodosa (Figure 2). Laboratory workup by our facility including hepatitis panel, perinuclear antineutrophil cytoplasmic antibody, cytoplasmic antineutrophil cytoplasmic antibody, factor V Leiden, prothrombin time/international normalized ratio, anticardiolipin antibody, and proteins C and S were all within reference range. Abnormal values included a low positive but nondiagnostic antinuclear antibody screen with negative titers, and the lupus anticoagulant titer was mildly elevated at 44 IgG binding units (reference range, <40 IgG binding units). Serum protein electrophoresis (SPEP) and urine protein electrophoresis also were performed, and SPEP was low positive for elevated κ and γ light chains. The patient was referred to oncology, and further testing revealed no underlying malignancy. The patient was monitored and no treatment was initiated; her rash completely resolved within 3 months. Laboratory monitoring at 6 months including SPEP, urine protein electrophoresis, lupus anticoagulant, and clotting studies all were within reference range.
Comment
Although the treatment of systemic polyarteritis nodosa often is necessary and typically involves high-dose corticosteroids and cyclophosphamide, the treatment of CPAN initially is less aggressive. Of the options available for treatment of CPAN, each has associated risks and side effects. Chen6 classified CPAN into 3 groups: 1 (mild), 2 (severe with no systemic involvement), and 3 (severe with progression to systemic disease)(Table). The authors performed a review of all the published treatments and their respective side effects to evaluate if treatment should be instituted for asymptomatic (group 1) disease presenting with abnormal antibody findings as demonstrated in our case.
First-line treatment of CPAN includes nonsteroidal anti-inflammatory drugs (NSAIDS) and colchicine.7 Nonsteroidal anti-inflammatory drugs are preferred; however, they also have been associated with gastrointestinal tract upset and increased risk for peptic ulcer disease with long-term use. Although colchicine often is used in conjunction with NSAIDS8 for its anti-inflammatory activity, no studies have been performed on this drug as monotherapy, and the side effect of diarrhea often limits its use.
Other therapies include dapsone, which should be monitored carefully due to the risk for dapsone hypersensitivity syndrome.8,9 Topical corticosteroids have been proven effective for mild cases of confluent erythema with remission occurring as early as 4 weeks.4 Some reports emphasize the role of streptococcal infections in CPAN, especially in children.8,10-12 Consequently it is recommended that anti–streptolysin O titers should be included in the workup for CPAN. Long-term penicillin prophylaxis and tonsillectomy have been used to prevent disease flares with limited success.8,10-12
For more severe disease, especially with neuromuscular involvement, oral methylprednisolone up to 1 mg/kg daily has been used and has proven effective in the control of acute exacerbations.7,13 However, the many adverse effects of systemic steroids limit their use long-term, and taper will often result in flare of disease.4,7 Medications used in conjunction with steroids include hydroxychloroquine, dapsone, azathioprine, cyclophosphamide, methotrexate, sulfapyridine, pentoxifylline, infliximab, etanercept, and intravenous immunoglobulin.4,9,12-17
Low-dose methotrexate has shown some improvement in skin disease with CPAN, but other case reports suggest that complete remission is not achieved with this drug.15,18 More studies are needed to assess the use of methotrexate for CPAN.
Immunomodulators have been used in multiple case reports with varying levels of success. Rogalski and Sticherling4 reported 3 cases that cleared with methylprednisolone plus azathioprine ranging from 4 weeks to 6 months; nausea limited tolerance of azathioprine in 1 case. Mycophenolate mofetil also was successfully used in 2 cases with clearance at 17 weeks and 6 months. In this series of cases, cyclosporine was ineffective for CPAN.4 Two case reports documented cutaneous clearance with cyclophosphamide in conjunction with prednisolone.9,10 No prospective trials have been performed on these medications, and immunosuppressants should only be considered in steroid-resistant cases.
The use of intravenous immunoglobulin has been reported effective in prior cases that showed resistance to more conventional trials of steroids, azathioprine, and/or cyclophosphamide.12,14 Intravenous immunoglobulin may be regarded as a treatment option for severe resistant disease. Several case reports also have documented success using tumor necrosis factor α blockers, particularly infliximab, as an adjunct to steroids and etanercept as both a steroid adjunct and monotherapy.16,17,19 More studies are necessary to evaluate these treatments.
Additionally, single case reports have outlined the use of other therapeutic agents, including tamoxifen (10 mg twice daily increased to 20 mg twice daily during episodes of breakthrough lesions),20 hyperbaric oxygen therapy (100% oxygen for 90 minutes 5 times weekly at 1.5 atm absolute followed by 2 weeks of 2 atm absolute),21 and granulocyte-macrophage colony-stimulating factor (300 µg injection in small portion to ulcer edges twice monthly for 2 months).22 All of these treatments show promise, but data are limited.
Because thrombosis is postulated to be a potential mechanism leading to CPAN, agents such as pentoxifylline, clopidogrel, and warfarin have been examined as treatment options. Pentoxifylline in combination with mycophenolate mofetil has been successful in treating a case that was resistant to other immunosuppressants.23 Clopidogrel blocks the adenosine diphosphate pathway and impairs clot retraction. Clopidogrel was reported effective in an acute flare of CPAN for clearance of skin lesions and normalization of lupus anticoagulant.24 It also was used successfully in recurrent CPAN after steroid treatments in a patient with neuromuscular symptoms. There was no recurrence in either of the patients in this case report series. Warfarin therapy at an international normalized ratio of 3.0 also has demonstrated success in halting disease progression and in facilitating the resolution of skin changes and normalization of anti–phosphatidylserine-prothrombin complex antibodies.24 Our review of the literature did not reveal evidence of a standardized length of treatment following symptom resolution or if treatment is indicated in asymptomatic disease, or as in our case, with only mild elevations of antiphospholipid antibodies.
Conclusion
Multiple treatment options exist for CPAN, but the data on their efficacies is limited and based only on anecdotal evidence, not prospective analysis. We believe that it seems reasonable to initiate treatment only for symptomatic disease or cases in which the antibody titers suggest that the patient may be at high risk for thrombosis. Mild symptoms and mild cutaneous changes would suggest the likely choice of NSAIDs, colchicine, or dapsone as treatment options versus no treatment. In patients with antibody titers, pentoxifylline, clopidogrel, or warfarin may be considered first-line therapies. With severe ulcerative lesions and neuromuscular involvement, steroids, immunosuppressants, and other investigative agents should be contemplated. In our patient, the laboratory studies were repeated and normalized on complete resolution of her livedo eruption. She remained asymptomatic and clear for 8 months without any treatment. The incidence of this presentation of CPAN is unknown and is likely underreported, as we would not expect most patients to present to their physicians for the evaluation of otherwise asymptomatic livedo reticularis. In essence, our case report suggests that it may be prudent to simply monitor patients with asymptomatic CPAN.
- Lindberg K. Ein Beitrag zur Kenntnis der Periarteritis nodosa. Acta Med Scand. 1931;76:183-225.
- Kraemer M, Linden D, Berlit P. The spectrum of differential diagnosis in neurological patients with livedo reticularis and livedo racemosa [published online August 26, 2005]. J Neurol. 2005;252:1155-1166.
- Nakamura T, Kanazawa N, Ikeda T, et al. Cutaneous polyarteritis nodosa: revisiting its definition and diagnostic criteria. Arch Dermatol Res. 2009;301:117-121.
- Rogalski C, Sticherling M. Panateritis cutanea benigna—an entity limited to the skin or cutaneous presentation of a systemic necrotizing vasculitis? report of seven cases and review of the literature. Int J Dermatol. 2007;46:817-821.
- Kawakami T, Yamazaki M, Mizoguchi M, et al. High titer of anti-phosphatidylserine-prothrombin complex antibodies in patients with cutaneous polyarteritis nodosa. Arthritis Rheum. 2007;57:1507-1513.
- Chen KR. Cutaneous polyarteritis nodosa: a clinical and histopathological study of 20 cases. J Dermatol. 1989;6:429-442.
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756.
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathologic analysis and review of the published work. J Dermatol. 2010;37:85-93.
- Flanagan N, Casey EB, Watson R, et al. Cutaneous polyartertitis nodosa with seronegative arthritis. Rheumatology (Oxford). 1999;38:1161-1162.
- Fathalla B, Miller L, Brady S, et al. Cutaneous polyarteritis nodosa in children. J Am Acad Dermatol. 2005;53:724-728.
- Misago N, Mochizuki Y, Sekiyama-Kodera H, et al. Cutaneous polyarteritis nodosa: therapy and clinical course in four cases. J Dermatol. 2001;28:719-727.
- Breda L, Franchini S, Marzetti V, et al. Intravenous immunoglobulins for cutaneous polyarteritis nodosa resistant to conventional treatment. Scand J Rheumatol. 2016;45:169-170.
- Maillard H, Szczesniak S, Martin L. Cutaneous periarteritis nodosa: diagnostic and therapeutic aspects of 9 cases. Ann Dermatol Venereol. 1999;26:125-129.
- Lobo I, Ferreira M, Silva E. Cutaneous polyarteritis nodosa treated with intravenous immunoglobulin. J Eur Acad Dermatol Venereol. 2007;22:880-882.
- Boehm I, Bauer R. Low-dose methotrexate controls a severe form of polyarteritis nodosa. Arch Dermatol. 2000;136:167-169.
- Campanilho-Marques R, Ramos F, Canhão H, et al. Remission induced by infliximab in a childhood polyarteritis nodosa refractory to conventional immunosuppression and rituximab. Joint Bone Spine. 2014;81:277-278.
- Inoue N, Shimizu M, Mizuta M, et al. Refractory cutaneous polyarteritis nodosa: successful treatment with etanercept. Pediatr Int. 2017;59:751-752.
- Schartz NE. Successful treatment in two cases of steroid dependent cutaneous polyarteritis nodosa with low-dose methotrexate. Dermatology. 2001;203:336-338.
- Valor L, Monteagudo I, de la Torre I, et al. Young male patient diagnosed with cutaneous polyarteritis nodosa successfully treated with etanercept. Mod Rheumatol. 2014;24:688-689.
- Cvancara JL, Meffert JJ, Elston DM. Estrogen sensitive cutaneous polyarteritis nodosa: response to tamoxifen. J Am Acad Dermatol. 1998;39:643-646.
- Mazokopakis E, Milkas A, Tsartsalis A, et al. Improvement of cutaneous polyarteritis nodosa with hyperbaric oxygen. Int J Dermatol. 2009;48:1017-1029.
- Tursen U, Api H, Kaya TI, et al. Rapid healing of chronic leg ulcers during perilesional injections of granulocyte-macrophage colony stimulating factor in a patient with cutaneous polyarteritis nodosa. J Eur Acad Dermatol Venereol. 2006;20:1341-1343.
- Kluger N, Guillot B, Bessis D. Ulcerative cutaneous polyarteritis nodosa treated with mycophenolate mofetil and pentoxifylline. J Dermatolog Treat. 2011;22:175-177.
- Kawakami T, Soma Y. Use of warfarin therapy at a target international normalized ratio of 3.0 for cutaneous polyarteritis nodosa. J Am Acad Dermatol. 2010;63:602-606.
- Lindberg K. Ein Beitrag zur Kenntnis der Periarteritis nodosa. Acta Med Scand. 1931;76:183-225.
- Kraemer M, Linden D, Berlit P. The spectrum of differential diagnosis in neurological patients with livedo reticularis and livedo racemosa [published online August 26, 2005]. J Neurol. 2005;252:1155-1166.
- Nakamura T, Kanazawa N, Ikeda T, et al. Cutaneous polyarteritis nodosa: revisiting its definition and diagnostic criteria. Arch Dermatol Res. 2009;301:117-121.
- Rogalski C, Sticherling M. Panateritis cutanea benigna—an entity limited to the skin or cutaneous presentation of a systemic necrotizing vasculitis? report of seven cases and review of the literature. Int J Dermatol. 2007;46:817-821.
- Kawakami T, Yamazaki M, Mizoguchi M, et al. High titer of anti-phosphatidylserine-prothrombin complex antibodies in patients with cutaneous polyarteritis nodosa. Arthritis Rheum. 2007;57:1507-1513.
- Chen KR. Cutaneous polyarteritis nodosa: a clinical and histopathological study of 20 cases. J Dermatol. 1989;6:429-442.
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756.
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathologic analysis and review of the published work. J Dermatol. 2010;37:85-93.
- Flanagan N, Casey EB, Watson R, et al. Cutaneous polyartertitis nodosa with seronegative arthritis. Rheumatology (Oxford). 1999;38:1161-1162.
- Fathalla B, Miller L, Brady S, et al. Cutaneous polyarteritis nodosa in children. J Am Acad Dermatol. 2005;53:724-728.
- Misago N, Mochizuki Y, Sekiyama-Kodera H, et al. Cutaneous polyarteritis nodosa: therapy and clinical course in four cases. J Dermatol. 2001;28:719-727.
- Breda L, Franchini S, Marzetti V, et al. Intravenous immunoglobulins for cutaneous polyarteritis nodosa resistant to conventional treatment. Scand J Rheumatol. 2016;45:169-170.
- Maillard H, Szczesniak S, Martin L. Cutaneous periarteritis nodosa: diagnostic and therapeutic aspects of 9 cases. Ann Dermatol Venereol. 1999;26:125-129.
- Lobo I, Ferreira M, Silva E. Cutaneous polyarteritis nodosa treated with intravenous immunoglobulin. J Eur Acad Dermatol Venereol. 2007;22:880-882.
- Boehm I, Bauer R. Low-dose methotrexate controls a severe form of polyarteritis nodosa. Arch Dermatol. 2000;136:167-169.
- Campanilho-Marques R, Ramos F, Canhão H, et al. Remission induced by infliximab in a childhood polyarteritis nodosa refractory to conventional immunosuppression and rituximab. Joint Bone Spine. 2014;81:277-278.
- Inoue N, Shimizu M, Mizuta M, et al. Refractory cutaneous polyarteritis nodosa: successful treatment with etanercept. Pediatr Int. 2017;59:751-752.
- Schartz NE. Successful treatment in two cases of steroid dependent cutaneous polyarteritis nodosa with low-dose methotrexate. Dermatology. 2001;203:336-338.
- Valor L, Monteagudo I, de la Torre I, et al. Young male patient diagnosed with cutaneous polyarteritis nodosa successfully treated with etanercept. Mod Rheumatol. 2014;24:688-689.
- Cvancara JL, Meffert JJ, Elston DM. Estrogen sensitive cutaneous polyarteritis nodosa: response to tamoxifen. J Am Acad Dermatol. 1998;39:643-646.
- Mazokopakis E, Milkas A, Tsartsalis A, et al. Improvement of cutaneous polyarteritis nodosa with hyperbaric oxygen. Int J Dermatol. 2009;48:1017-1029.
- Tursen U, Api H, Kaya TI, et al. Rapid healing of chronic leg ulcers during perilesional injections of granulocyte-macrophage colony stimulating factor in a patient with cutaneous polyarteritis nodosa. J Eur Acad Dermatol Venereol. 2006;20:1341-1343.
- Kluger N, Guillot B, Bessis D. Ulcerative cutaneous polyarteritis nodosa treated with mycophenolate mofetil and pentoxifylline. J Dermatolog Treat. 2011;22:175-177.
- Kawakami T, Soma Y. Use of warfarin therapy at a target international normalized ratio of 3.0 for cutaneous polyarteritis nodosa. J Am Acad Dermatol. 2010;63:602-606.
Practice Points
- Cutaneous polyarteritis nodosa should be in the differential of new-onset livedo reticularis.
- Workup with biopsy and specific blood work is important.
- Treatment options at this time are limited.
Low-grade fever, erythematous rash in pregnant woman • Dx?
THE CASE
A 31-year-old woman presented to her obstetrician’s office at 16 weeks’ gestation with a 2-day history of low-grade fever and an erythematous rash measuring 1 x 4 cm on her right groin. She had a medical history of a penicillin allergy (urticaria) and her outdoor activities included gardening and picnicking.
We suspected that she was experiencing an allergic reaction and recommended an antihistamine (diphenhydramine). The patient returned 4 days later with new symptoms including headache, photophobia, neck pain, unilateral large joint pain, and periorbital cellulitis, as well as expansion of her rash. She was afebrile and an examination revealed that the 1 x 4 cm rash on her groin had grown; it was now a demarcated erythematous rash with faint central clearing measuring 5 x 8 cm. Right periorbital erythema and nuchal rigidity were also noted.
Because of her expanding rash and nuchal rigidity, we suspected Lyme meningitis and we referred her to the emergency department. Within 24 hours, the rash had spread to her abdomen, thigh, and wrist, and was consistent with erythema migrans.
Laboratory evaluation revealed an increased number of white bloods cells (13.5 million cells/mcL; normal range 4.5-11.0 million cells/mcL), and an increased number of neutrophils (10.8 million cells/mcL; normal range 1.8-8 million cells/mcL), indicating leukocytosis with a left shift. Lab tests also revealed a low hemoglobin level (10.6 g/dL; normal range 12-16 g/dL) and a mean corpuscular volume of 85.6 fL/red cell (normal range 80-100 fL/red cell), indicating microcytic anemia. A lumbar puncture was negative for disseminated Lyme disease by Gram stain, culture, and polymerase chain reaction.
THE DIAGNOSIS
A diagnosis of Lyme disease was confirmed with a positive Lyme titer serology via an enzyme-linked immunosorbent assay. The rash and other symptoms responded promptly to intravenous ceftriaxone 2 g, and the patient was discharged home on oral cefuroxime 500 mg bid for 14 days.
DISCUSSION
Lyme disease is the most common vector-borne illness in the United States, concentrated heavily in the Northeast and upper Midwest.1 The most recent information released by the Centers for Disease Control and Prevention (CDC) lists Vermont, Maine, Pennsylvania, Rhode Island, Connecticut, New Jersey, Massachusetts, Delaware, New Hampshire, and Minnesota as the states with the highest incidence of Lyme disease.2
The number of reported cases in the United States has increased over the past 2 decades, from approximately 11,000 in 1995 to about 28,000 in 2015.3 Over the past year, we have seen several cases of Lyme disease in the obstetric population of our own practice.
Prompt treatment is crucial. Pregnant women who are acutely infected with Borrelia burgdorferi (the primary cause of Lyme disease) and do not receive treatment have experienced multiple adverse pregnancy outcomes, including preterm delivery, infants born with rash, and stillbirth.4 Additional concern exists for fetal cardiac anomalies, with data showing that there are twice as many cardiac defects in children born to mothers residing in endemic regions.5
What animal studies have taught us about Lyme disease
The potential causal relationship between Lyme disease and fetal demise was first studied in 2007. This case report involved the stillbirth of a full-term baby from an acutely infected woman who did not receive treatment. She experienced erythema multiforme 6 weeks prior to delivery.6
The vast majority of research on Lyme disease in pregnancy comes from work with mice and dogs. These studies confirmed that acute infection with Lyme disease is associated with an increased risk of adverse fetal outcomes, specifically fetal death.7
Silver et al further researched the association using murine models in the 1980s. They found that fetal death occurred in 12% of acutely infected mice, compared with none of the mice that were chronically infected.7
In 2010, Lakos and Solymosi examined the effects of Lyme disease on pregnancy outcomes in acutely infected women. Seven out of 95 pregnant women acutely infected with B burgdorferi experienced fetal demise, further supporting the association seen in animal experiments.8
Treating pregnant patients
Doxycycline and tetracycline, which are routinely used to treat Lyme disease, are not appropriate in the obstetric population. The CDC recommends up to a 3-week course of antibiotics; the standard regimen is amoxicillin 500 mg by mouth tid. For women who are allergic to penicillin, as was the case with our patient, cefuroxime 500 mg by mouth bid is the treatment of choice.9
Our patient underwent a detailed ultrasound at 21 weeks, which revealed normal fetal anatomy and no evidence of cardiac malformations. The remainder of her pregnancy was uncomplicated, and she gave birth vaginally at 41 weeks to a baby boy weighing 3700 g.
THE TAKEAWAY
There is a need to increase awareness of Lyme disease in pregnancy on a national level. It is the responsibility of every practitioner caring for obstetric patients in endemic regions to address new-onset rash promptly. There have been cases of women who experienced erythema migrans and arthralgias after exposure to a tick bite, later delivering infants with cardiac anomalies such as atrial and ventricular septal defects.10 In obstetric patients acutely infected during the first trimester, a fetal echocardiogram is reasonable, given the demonstrated high potential for fetal cardiac anomalies.
Preventing adverse fetal outcomes requires early treatment with antibiotics. The CDC maintains that there have been no life-threatening adverse fetal effects from Lyme disease seen in women who are appropriately treated, as well as no transmission of Lyme disease in the breast milk of lactating mothers.9
1. Centers for Disease Control and Prevention. Lyme disease. Data and statistics. Available at: https://www.cdc.gov/lyme/stats/. Accessed July 5, 2017.
2. Centers for Disease Control and Prevention. Lyme disease data tables. Reported cases of Lyme disease by state or locality, 2005-2015. Available at: http://www.cdc.gov/lyme/stats/chartstables/reportedcases_statelocality.html. Accessed July 5, 2017.
3. Centers for Disease Control and Prevention. Lyme disease graphs. Reported cases of Lyme disease by year, United States, 1995-2015. Available at: https://www.cdc.gov/lyme/stats/graphs.html. Accessed July 5, 2017.
4. Maraspin V, Cimperman J, Lotric-Furlan S, et al. Erythema migrans in pregnancy. Wien Klin Wochenschr. 1999;111:933-940.
5. Strobino BA, Williams CL, Abid S, et al. Lyme disease and pregnancy outcome: a prospective study of two thousand prenatal patients. Am J Obstet Gynecol. 1993;169:367-374.
6. Gibbs RS, Roberts DJ. Case records of the Massachusetts General Hospital. Case 27-2007. A 30-year-old pregnant woman with intrauterine fetal death. N Engl J Med. 2007;357:918-925.
7. Silver RM, Yang L, Daynes RA, et al. Fetal outcome in murine Lyme disease. Infect Immun. 1995;63:66-72.
8. Lakos A, Solymosi N. Maternal Lyme borreliosis and pregnancy outcomes. Int J Infect Dis. 2010;14:e494-e498.
9. Centers for Disease Control and Prevention. Ticks and Lyme disease. Pregnancy and Lyme disease. Available at: https://www.cdc.gov/lyme/resources/toolkit/factsheets/10_508_Lyme%20disease_PregnantWoman_FACTSheet.pdf. Accessed July 5, 2017.
10. O’Brien JM, Martens MG. Lyme disease in pregnancy: a New Jersey medical advisory. MD Advis. 2014;7:24-27.
THE CASE
A 31-year-old woman presented to her obstetrician’s office at 16 weeks’ gestation with a 2-day history of low-grade fever and an erythematous rash measuring 1 x 4 cm on her right groin. She had a medical history of a penicillin allergy (urticaria) and her outdoor activities included gardening and picnicking.
We suspected that she was experiencing an allergic reaction and recommended an antihistamine (diphenhydramine). The patient returned 4 days later with new symptoms including headache, photophobia, neck pain, unilateral large joint pain, and periorbital cellulitis, as well as expansion of her rash. She was afebrile and an examination revealed that the 1 x 4 cm rash on her groin had grown; it was now a demarcated erythematous rash with faint central clearing measuring 5 x 8 cm. Right periorbital erythema and nuchal rigidity were also noted.
Because of her expanding rash and nuchal rigidity, we suspected Lyme meningitis and we referred her to the emergency department. Within 24 hours, the rash had spread to her abdomen, thigh, and wrist, and was consistent with erythema migrans.
Laboratory evaluation revealed an increased number of white bloods cells (13.5 million cells/mcL; normal range 4.5-11.0 million cells/mcL), and an increased number of neutrophils (10.8 million cells/mcL; normal range 1.8-8 million cells/mcL), indicating leukocytosis with a left shift. Lab tests also revealed a low hemoglobin level (10.6 g/dL; normal range 12-16 g/dL) and a mean corpuscular volume of 85.6 fL/red cell (normal range 80-100 fL/red cell), indicating microcytic anemia. A lumbar puncture was negative for disseminated Lyme disease by Gram stain, culture, and polymerase chain reaction.
THE DIAGNOSIS
A diagnosis of Lyme disease was confirmed with a positive Lyme titer serology via an enzyme-linked immunosorbent assay. The rash and other symptoms responded promptly to intravenous ceftriaxone 2 g, and the patient was discharged home on oral cefuroxime 500 mg bid for 14 days.
DISCUSSION
Lyme disease is the most common vector-borne illness in the United States, concentrated heavily in the Northeast and upper Midwest.1 The most recent information released by the Centers for Disease Control and Prevention (CDC) lists Vermont, Maine, Pennsylvania, Rhode Island, Connecticut, New Jersey, Massachusetts, Delaware, New Hampshire, and Minnesota as the states with the highest incidence of Lyme disease.2
The number of reported cases in the United States has increased over the past 2 decades, from approximately 11,000 in 1995 to about 28,000 in 2015.3 Over the past year, we have seen several cases of Lyme disease in the obstetric population of our own practice.
Prompt treatment is crucial. Pregnant women who are acutely infected with Borrelia burgdorferi (the primary cause of Lyme disease) and do not receive treatment have experienced multiple adverse pregnancy outcomes, including preterm delivery, infants born with rash, and stillbirth.4 Additional concern exists for fetal cardiac anomalies, with data showing that there are twice as many cardiac defects in children born to mothers residing in endemic regions.5
What animal studies have taught us about Lyme disease
The potential causal relationship between Lyme disease and fetal demise was first studied in 2007. This case report involved the stillbirth of a full-term baby from an acutely infected woman who did not receive treatment. She experienced erythema multiforme 6 weeks prior to delivery.6
The vast majority of research on Lyme disease in pregnancy comes from work with mice and dogs. These studies confirmed that acute infection with Lyme disease is associated with an increased risk of adverse fetal outcomes, specifically fetal death.7
Silver et al further researched the association using murine models in the 1980s. They found that fetal death occurred in 12% of acutely infected mice, compared with none of the mice that were chronically infected.7
In 2010, Lakos and Solymosi examined the effects of Lyme disease on pregnancy outcomes in acutely infected women. Seven out of 95 pregnant women acutely infected with B burgdorferi experienced fetal demise, further supporting the association seen in animal experiments.8
Treating pregnant patients
Doxycycline and tetracycline, which are routinely used to treat Lyme disease, are not appropriate in the obstetric population. The CDC recommends up to a 3-week course of antibiotics; the standard regimen is amoxicillin 500 mg by mouth tid. For women who are allergic to penicillin, as was the case with our patient, cefuroxime 500 mg by mouth bid is the treatment of choice.9
Our patient underwent a detailed ultrasound at 21 weeks, which revealed normal fetal anatomy and no evidence of cardiac malformations. The remainder of her pregnancy was uncomplicated, and she gave birth vaginally at 41 weeks to a baby boy weighing 3700 g.
THE TAKEAWAY
There is a need to increase awareness of Lyme disease in pregnancy on a national level. It is the responsibility of every practitioner caring for obstetric patients in endemic regions to address new-onset rash promptly. There have been cases of women who experienced erythema migrans and arthralgias after exposure to a tick bite, later delivering infants with cardiac anomalies such as atrial and ventricular septal defects.10 In obstetric patients acutely infected during the first trimester, a fetal echocardiogram is reasonable, given the demonstrated high potential for fetal cardiac anomalies.
Preventing adverse fetal outcomes requires early treatment with antibiotics. The CDC maintains that there have been no life-threatening adverse fetal effects from Lyme disease seen in women who are appropriately treated, as well as no transmission of Lyme disease in the breast milk of lactating mothers.9
THE CASE
A 31-year-old woman presented to her obstetrician’s office at 16 weeks’ gestation with a 2-day history of low-grade fever and an erythematous rash measuring 1 x 4 cm on her right groin. She had a medical history of a penicillin allergy (urticaria) and her outdoor activities included gardening and picnicking.
We suspected that she was experiencing an allergic reaction and recommended an antihistamine (diphenhydramine). The patient returned 4 days later with new symptoms including headache, photophobia, neck pain, unilateral large joint pain, and periorbital cellulitis, as well as expansion of her rash. She was afebrile and an examination revealed that the 1 x 4 cm rash on her groin had grown; it was now a demarcated erythematous rash with faint central clearing measuring 5 x 8 cm. Right periorbital erythema and nuchal rigidity were also noted.
Because of her expanding rash and nuchal rigidity, we suspected Lyme meningitis and we referred her to the emergency department. Within 24 hours, the rash had spread to her abdomen, thigh, and wrist, and was consistent with erythema migrans.
Laboratory evaluation revealed an increased number of white bloods cells (13.5 million cells/mcL; normal range 4.5-11.0 million cells/mcL), and an increased number of neutrophils (10.8 million cells/mcL; normal range 1.8-8 million cells/mcL), indicating leukocytosis with a left shift. Lab tests also revealed a low hemoglobin level (10.6 g/dL; normal range 12-16 g/dL) and a mean corpuscular volume of 85.6 fL/red cell (normal range 80-100 fL/red cell), indicating microcytic anemia. A lumbar puncture was negative for disseminated Lyme disease by Gram stain, culture, and polymerase chain reaction.
THE DIAGNOSIS
A diagnosis of Lyme disease was confirmed with a positive Lyme titer serology via an enzyme-linked immunosorbent assay. The rash and other symptoms responded promptly to intravenous ceftriaxone 2 g, and the patient was discharged home on oral cefuroxime 500 mg bid for 14 days.
DISCUSSION
Lyme disease is the most common vector-borne illness in the United States, concentrated heavily in the Northeast and upper Midwest.1 The most recent information released by the Centers for Disease Control and Prevention (CDC) lists Vermont, Maine, Pennsylvania, Rhode Island, Connecticut, New Jersey, Massachusetts, Delaware, New Hampshire, and Minnesota as the states with the highest incidence of Lyme disease.2
The number of reported cases in the United States has increased over the past 2 decades, from approximately 11,000 in 1995 to about 28,000 in 2015.3 Over the past year, we have seen several cases of Lyme disease in the obstetric population of our own practice.
Prompt treatment is crucial. Pregnant women who are acutely infected with Borrelia burgdorferi (the primary cause of Lyme disease) and do not receive treatment have experienced multiple adverse pregnancy outcomes, including preterm delivery, infants born with rash, and stillbirth.4 Additional concern exists for fetal cardiac anomalies, with data showing that there are twice as many cardiac defects in children born to mothers residing in endemic regions.5
What animal studies have taught us about Lyme disease
The potential causal relationship between Lyme disease and fetal demise was first studied in 2007. This case report involved the stillbirth of a full-term baby from an acutely infected woman who did not receive treatment. She experienced erythema multiforme 6 weeks prior to delivery.6
The vast majority of research on Lyme disease in pregnancy comes from work with mice and dogs. These studies confirmed that acute infection with Lyme disease is associated with an increased risk of adverse fetal outcomes, specifically fetal death.7
Silver et al further researched the association using murine models in the 1980s. They found that fetal death occurred in 12% of acutely infected mice, compared with none of the mice that were chronically infected.7
In 2010, Lakos and Solymosi examined the effects of Lyme disease on pregnancy outcomes in acutely infected women. Seven out of 95 pregnant women acutely infected with B burgdorferi experienced fetal demise, further supporting the association seen in animal experiments.8
Treating pregnant patients
Doxycycline and tetracycline, which are routinely used to treat Lyme disease, are not appropriate in the obstetric population. The CDC recommends up to a 3-week course of antibiotics; the standard regimen is amoxicillin 500 mg by mouth tid. For women who are allergic to penicillin, as was the case with our patient, cefuroxime 500 mg by mouth bid is the treatment of choice.9
Our patient underwent a detailed ultrasound at 21 weeks, which revealed normal fetal anatomy and no evidence of cardiac malformations. The remainder of her pregnancy was uncomplicated, and she gave birth vaginally at 41 weeks to a baby boy weighing 3700 g.
THE TAKEAWAY
There is a need to increase awareness of Lyme disease in pregnancy on a national level. It is the responsibility of every practitioner caring for obstetric patients in endemic regions to address new-onset rash promptly. There have been cases of women who experienced erythema migrans and arthralgias after exposure to a tick bite, later delivering infants with cardiac anomalies such as atrial and ventricular septal defects.10 In obstetric patients acutely infected during the first trimester, a fetal echocardiogram is reasonable, given the demonstrated high potential for fetal cardiac anomalies.
Preventing adverse fetal outcomes requires early treatment with antibiotics. The CDC maintains that there have been no life-threatening adverse fetal effects from Lyme disease seen in women who are appropriately treated, as well as no transmission of Lyme disease in the breast milk of lactating mothers.9
1. Centers for Disease Control and Prevention. Lyme disease. Data and statistics. Available at: https://www.cdc.gov/lyme/stats/. Accessed July 5, 2017.
2. Centers for Disease Control and Prevention. Lyme disease data tables. Reported cases of Lyme disease by state or locality, 2005-2015. Available at: http://www.cdc.gov/lyme/stats/chartstables/reportedcases_statelocality.html. Accessed July 5, 2017.
3. Centers for Disease Control and Prevention. Lyme disease graphs. Reported cases of Lyme disease by year, United States, 1995-2015. Available at: https://www.cdc.gov/lyme/stats/graphs.html. Accessed July 5, 2017.
4. Maraspin V, Cimperman J, Lotric-Furlan S, et al. Erythema migrans in pregnancy. Wien Klin Wochenschr. 1999;111:933-940.
5. Strobino BA, Williams CL, Abid S, et al. Lyme disease and pregnancy outcome: a prospective study of two thousand prenatal patients. Am J Obstet Gynecol. 1993;169:367-374.
6. Gibbs RS, Roberts DJ. Case records of the Massachusetts General Hospital. Case 27-2007. A 30-year-old pregnant woman with intrauterine fetal death. N Engl J Med. 2007;357:918-925.
7. Silver RM, Yang L, Daynes RA, et al. Fetal outcome in murine Lyme disease. Infect Immun. 1995;63:66-72.
8. Lakos A, Solymosi N. Maternal Lyme borreliosis and pregnancy outcomes. Int J Infect Dis. 2010;14:e494-e498.
9. Centers for Disease Control and Prevention. Ticks and Lyme disease. Pregnancy and Lyme disease. Available at: https://www.cdc.gov/lyme/resources/toolkit/factsheets/10_508_Lyme%20disease_PregnantWoman_FACTSheet.pdf. Accessed July 5, 2017.
10. O’Brien JM, Martens MG. Lyme disease in pregnancy: a New Jersey medical advisory. MD Advis. 2014;7:24-27.
1. Centers for Disease Control and Prevention. Lyme disease. Data and statistics. Available at: https://www.cdc.gov/lyme/stats/. Accessed July 5, 2017.
2. Centers for Disease Control and Prevention. Lyme disease data tables. Reported cases of Lyme disease by state or locality, 2005-2015. Available at: http://www.cdc.gov/lyme/stats/chartstables/reportedcases_statelocality.html. Accessed July 5, 2017.
3. Centers for Disease Control and Prevention. Lyme disease graphs. Reported cases of Lyme disease by year, United States, 1995-2015. Available at: https://www.cdc.gov/lyme/stats/graphs.html. Accessed July 5, 2017.
4. Maraspin V, Cimperman J, Lotric-Furlan S, et al. Erythema migrans in pregnancy. Wien Klin Wochenschr. 1999;111:933-940.
5. Strobino BA, Williams CL, Abid S, et al. Lyme disease and pregnancy outcome: a prospective study of two thousand prenatal patients. Am J Obstet Gynecol. 1993;169:367-374.
6. Gibbs RS, Roberts DJ. Case records of the Massachusetts General Hospital. Case 27-2007. A 30-year-old pregnant woman with intrauterine fetal death. N Engl J Med. 2007;357:918-925.
7. Silver RM, Yang L, Daynes RA, et al. Fetal outcome in murine Lyme disease. Infect Immun. 1995;63:66-72.
8. Lakos A, Solymosi N. Maternal Lyme borreliosis and pregnancy outcomes. Int J Infect Dis. 2010;14:e494-e498.
9. Centers for Disease Control and Prevention. Ticks and Lyme disease. Pregnancy and Lyme disease. Available at: https://www.cdc.gov/lyme/resources/toolkit/factsheets/10_508_Lyme%20disease_PregnantWoman_FACTSheet.pdf. Accessed July 5, 2017.
10. O’Brien JM, Martens MG. Lyme disease in pregnancy: a New Jersey medical advisory. MD Advis. 2014;7:24-27.

Active 46-year-old man with right-sided visual loss and no family history of stroke • Dx?
THE CASE
A 46-year-old man presented to the emergency department (ED) with sudden-onset right-sided visual loss. He had a history of asthma, but no family history of hypercoagulability, deep vein thrombosis (DVT), or stroke. The patient had an active lifestyle that included scuba diving, mountain biking, and hockey (coaching and playing). The physical examination revealed a right homonymous upper quadrantanopia. The neurologic examination was within normal limits, except for the visual deficit and unequal pupil size. A computerized tomography scan of the patient’s head did not reveal any lesions.
Based on the patient’s clinical picture, the ED physician prescribed alteplase, a tissue plasminogen activator (tPA), and admitted him to the intensive care unit for monitoring.
Subsequent magnetic resonance imaging (MRI) of the brain showed multiple small areas of acute infarct in the posterior circulation territory bilaterally, with involvement of small portions of the bilateral cerebellar hemispheres and parts of the left occipital lobe (FIGURE 1A and 1B).
An electrocardiogram showed no evidence of atrial fibrillation, and hypercoagulability studies were within normal limits. There was no evidence of May-Thurner anatomy, and an ultrasound of the lower extremities showed no DVT.
THE DIAGNOSIS
An echocardiogram with bubble study confirmed a diagnosis of patent foramen ovale (PFO) with bidirectional flow, a normal ejection fraction, and no evidence of left ventricular or left atrial thrombus. We started the patient on the anticoagulant enoxaparin 70 mg bid bridged with warfarin 5 mg/d.
Taking the patient’s active lifestyle into consideration, he was approved for PFO closure by the PFO committee and underwent closure. Following treatment, the patient was left with a residual 2-mm blind spot in the right visual field. At a 2-year follow-up visit, he showed no new focal deficits or recurrent symptoms.
DISCUSSION
Since 1988 when Lechat et al reported increased incidence of PFO in young stroke patients,1 many studies have supported the association between PFO and cryptogenic stroke (CS) in young adults.2 Because it remained controversial as to whether PFO is a risk factor for stroke or transient ischemic attack recurrence,3 researchers investigated PFO closure as a preventive measure to decrease stroke recurrence in patients with both CS and PFO.
A 2012 meta-analysis showed possible benefits of closure compared with medical management using antiplatelet or anticoagulation therapies.4 However, these results were not supported by results of other studies. These include the CLOSURE I trial,5 which compared device closure of PFO with medical therapy, and the RESPECT6 and PC trials,7 which did not show a significant difference in the primary end point of recurrent stroke between patients who received medical therapy and those who had PFO closure.
American Heart Association/American Stroke Association’s 2011 guidelines recommend only antiplatelet therapy for patients with CS and PFO.8 While there is consensus that surgical closure is not better than a medical approach to patients with CS and PFO, cases should be individualized, as a patient’s clinical or social factors may dictate otherwise.
Lifestyle may warrant PFO closure
No previous studies have considered occupation or hobbies as an indication for PFO closure in patients with CS. Our patient’s active lifestyle, particularly his scuba diving and participation in contact sports, made him a poor candidate for anticoagulation. Scuba diving is associated with decompression sickness and air emboli, which can be a mechanism of cerebral ischemia, especially in patients with a right-to-left shunt, such as with PFO.9
We did not observe a strong temporal relationship between diving and stroke in our patient. MRI findings suggested that he had multiple minor embolic events over time, which is consistent with a prior case report.9 This suggested air emboli as a possible source of stroke, in which case, our patient might not benefit from antiplatelet or anticoagulation therapy.
THE TAKEAWAY
This case illustrates the importance of a thorough social history and knowledge of the patient’s hobbies, occupation, and preferences in evaluating and treating individuals with CS associated with PFO. The current literature does not provide complete answers to the cause, diagnosis, and management of CS; additional research is needed.
The work-up involved in defining the etiology of stroke includes, but is not limited to, head and brain imaging, an echocardiogram, hypercoagulability tests, and vascular imaging. The work of Sanna et al showed that approximately 12% of patients with CS have atrial fibrillation when monitored over a one-year period, suggesting atrial fibrillation as a possible cause in some cases.10
As the case described here demonstrates, further research is warranted regarding how a patient’s occupation and lifestyle factor into decision-making for patients with PFO.
1. Lechat P, Mas JL, Lascault G, et al. Prevalence of patent foramen ovale in patients with stroke. N Engl J Med. 1988;318:1148-1152.
2. Ferro JM, Massaro AR, Mas JL. Aetiological diagnosis of ischaemic stroke in young adults. Lancet Neurol. 2010;9:1085-1096.
3. Cotter PE, Belham M, Martin PJ. Stroke in younger patients: the heart of the matter. J Neurol. 2010;257:1777-1787.
4. Kitsios GD, Dahabreh IJ, Abu Dabrh AM, et al. Patent foramen ovale closure and medical treatments for secondary stroke prevention: a systematic review of observational and randomized evidence. Stroke. 2012;43:422-431.
5. Furlan AJ, Reisman M, Massaro J, et al. Closure or medical therapy for cryptogenic stroke with patent foramen ovale. N Engl J Med. 2012;366:991-999.
6. Carroll JD, Saver JL, Thaler DE, et al. Closure of patent foramen ovale versus medical therapy after cryptogenic stroke. N Engl J Med. 2013;368:1092-1100.
7. Meier B, Kalesan B, Mattle HP, et al. Percutaneous closure of patent foramen ovale in cryptogenic embolism. N Engl J Med. 2013;368:1083-1091.
8. Furie KL, Kasner SE, Adams RJ, et al. Guidelines for the prevention of stroke in patients with stroke or transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:227-276.
9. Menkin M, Schwartzman RJ. Cerebral air embolism. Report of five cases and review of the literature. Arch Neurol. 1977;34:168-170.
10. Sanna T, Diener HC, Passman RS, et al. Cryptogenic stroke and underlying atrial fibrillation. N Engl J Med. 2014;370:2478-2486.
THE CASE
A 46-year-old man presented to the emergency department (ED) with sudden-onset right-sided visual loss. He had a history of asthma, but no family history of hypercoagulability, deep vein thrombosis (DVT), or stroke. The patient had an active lifestyle that included scuba diving, mountain biking, and hockey (coaching and playing). The physical examination revealed a right homonymous upper quadrantanopia. The neurologic examination was within normal limits, except for the visual deficit and unequal pupil size. A computerized tomography scan of the patient’s head did not reveal any lesions.
Based on the patient’s clinical picture, the ED physician prescribed alteplase, a tissue plasminogen activator (tPA), and admitted him to the intensive care unit for monitoring.
Subsequent magnetic resonance imaging (MRI) of the brain showed multiple small areas of acute infarct in the posterior circulation territory bilaterally, with involvement of small portions of the bilateral cerebellar hemispheres and parts of the left occipital lobe (FIGURE 1A and 1B).
An electrocardiogram showed no evidence of atrial fibrillation, and hypercoagulability studies were within normal limits. There was no evidence of May-Thurner anatomy, and an ultrasound of the lower extremities showed no DVT.
THE DIAGNOSIS
An echocardiogram with bubble study confirmed a diagnosis of patent foramen ovale (PFO) with bidirectional flow, a normal ejection fraction, and no evidence of left ventricular or left atrial thrombus. We started the patient on the anticoagulant enoxaparin 70 mg bid bridged with warfarin 5 mg/d.
Taking the patient’s active lifestyle into consideration, he was approved for PFO closure by the PFO committee and underwent closure. Following treatment, the patient was left with a residual 2-mm blind spot in the right visual field. At a 2-year follow-up visit, he showed no new focal deficits or recurrent symptoms.
DISCUSSION
Since 1988 when Lechat et al reported increased incidence of PFO in young stroke patients,1 many studies have supported the association between PFO and cryptogenic stroke (CS) in young adults.2 Because it remained controversial as to whether PFO is a risk factor for stroke or transient ischemic attack recurrence,3 researchers investigated PFO closure as a preventive measure to decrease stroke recurrence in patients with both CS and PFO.
A 2012 meta-analysis showed possible benefits of closure compared with medical management using antiplatelet or anticoagulation therapies.4 However, these results were not supported by results of other studies. These include the CLOSURE I trial,5 which compared device closure of PFO with medical therapy, and the RESPECT6 and PC trials,7 which did not show a significant difference in the primary end point of recurrent stroke between patients who received medical therapy and those who had PFO closure.
American Heart Association/American Stroke Association’s 2011 guidelines recommend only antiplatelet therapy for patients with CS and PFO.8 While there is consensus that surgical closure is not better than a medical approach to patients with CS and PFO, cases should be individualized, as a patient’s clinical or social factors may dictate otherwise.
Lifestyle may warrant PFO closure
No previous studies have considered occupation or hobbies as an indication for PFO closure in patients with CS. Our patient’s active lifestyle, particularly his scuba diving and participation in contact sports, made him a poor candidate for anticoagulation. Scuba diving is associated with decompression sickness and air emboli, which can be a mechanism of cerebral ischemia, especially in patients with a right-to-left shunt, such as with PFO.9
We did not observe a strong temporal relationship between diving and stroke in our patient. MRI findings suggested that he had multiple minor embolic events over time, which is consistent with a prior case report.9 This suggested air emboli as a possible source of stroke, in which case, our patient might not benefit from antiplatelet or anticoagulation therapy.
THE TAKEAWAY
This case illustrates the importance of a thorough social history and knowledge of the patient’s hobbies, occupation, and preferences in evaluating and treating individuals with CS associated with PFO. The current literature does not provide complete answers to the cause, diagnosis, and management of CS; additional research is needed.
The work-up involved in defining the etiology of stroke includes, but is not limited to, head and brain imaging, an echocardiogram, hypercoagulability tests, and vascular imaging. The work of Sanna et al showed that approximately 12% of patients with CS have atrial fibrillation when monitored over a one-year period, suggesting atrial fibrillation as a possible cause in some cases.10
As the case described here demonstrates, further research is warranted regarding how a patient’s occupation and lifestyle factor into decision-making for patients with PFO.
THE CASE
A 46-year-old man presented to the emergency department (ED) with sudden-onset right-sided visual loss. He had a history of asthma, but no family history of hypercoagulability, deep vein thrombosis (DVT), or stroke. The patient had an active lifestyle that included scuba diving, mountain biking, and hockey (coaching and playing). The physical examination revealed a right homonymous upper quadrantanopia. The neurologic examination was within normal limits, except for the visual deficit and unequal pupil size. A computerized tomography scan of the patient’s head did not reveal any lesions.
Based on the patient’s clinical picture, the ED physician prescribed alteplase, a tissue plasminogen activator (tPA), and admitted him to the intensive care unit for monitoring.
Subsequent magnetic resonance imaging (MRI) of the brain showed multiple small areas of acute infarct in the posterior circulation territory bilaterally, with involvement of small portions of the bilateral cerebellar hemispheres and parts of the left occipital lobe (FIGURE 1A and 1B).
An electrocardiogram showed no evidence of atrial fibrillation, and hypercoagulability studies were within normal limits. There was no evidence of May-Thurner anatomy, and an ultrasound of the lower extremities showed no DVT.
THE DIAGNOSIS
An echocardiogram with bubble study confirmed a diagnosis of patent foramen ovale (PFO) with bidirectional flow, a normal ejection fraction, and no evidence of left ventricular or left atrial thrombus. We started the patient on the anticoagulant enoxaparin 70 mg bid bridged with warfarin 5 mg/d.
Taking the patient’s active lifestyle into consideration, he was approved for PFO closure by the PFO committee and underwent closure. Following treatment, the patient was left with a residual 2-mm blind spot in the right visual field. At a 2-year follow-up visit, he showed no new focal deficits or recurrent symptoms.
DISCUSSION
Since 1988 when Lechat et al reported increased incidence of PFO in young stroke patients,1 many studies have supported the association between PFO and cryptogenic stroke (CS) in young adults.2 Because it remained controversial as to whether PFO is a risk factor for stroke or transient ischemic attack recurrence,3 researchers investigated PFO closure as a preventive measure to decrease stroke recurrence in patients with both CS and PFO.
A 2012 meta-analysis showed possible benefits of closure compared with medical management using antiplatelet or anticoagulation therapies.4 However, these results were not supported by results of other studies. These include the CLOSURE I trial,5 which compared device closure of PFO with medical therapy, and the RESPECT6 and PC trials,7 which did not show a significant difference in the primary end point of recurrent stroke between patients who received medical therapy and those who had PFO closure.
American Heart Association/American Stroke Association’s 2011 guidelines recommend only antiplatelet therapy for patients with CS and PFO.8 While there is consensus that surgical closure is not better than a medical approach to patients with CS and PFO, cases should be individualized, as a patient’s clinical or social factors may dictate otherwise.
Lifestyle may warrant PFO closure
No previous studies have considered occupation or hobbies as an indication for PFO closure in patients with CS. Our patient’s active lifestyle, particularly his scuba diving and participation in contact sports, made him a poor candidate for anticoagulation. Scuba diving is associated with decompression sickness and air emboli, which can be a mechanism of cerebral ischemia, especially in patients with a right-to-left shunt, such as with PFO.9
We did not observe a strong temporal relationship between diving and stroke in our patient. MRI findings suggested that he had multiple minor embolic events over time, which is consistent with a prior case report.9 This suggested air emboli as a possible source of stroke, in which case, our patient might not benefit from antiplatelet or anticoagulation therapy.
THE TAKEAWAY
This case illustrates the importance of a thorough social history and knowledge of the patient’s hobbies, occupation, and preferences in evaluating and treating individuals with CS associated with PFO. The current literature does not provide complete answers to the cause, diagnosis, and management of CS; additional research is needed.
The work-up involved in defining the etiology of stroke includes, but is not limited to, head and brain imaging, an echocardiogram, hypercoagulability tests, and vascular imaging. The work of Sanna et al showed that approximately 12% of patients with CS have atrial fibrillation when monitored over a one-year period, suggesting atrial fibrillation as a possible cause in some cases.10
As the case described here demonstrates, further research is warranted regarding how a patient’s occupation and lifestyle factor into decision-making for patients with PFO.
1. Lechat P, Mas JL, Lascault G, et al. Prevalence of patent foramen ovale in patients with stroke. N Engl J Med. 1988;318:1148-1152.
2. Ferro JM, Massaro AR, Mas JL. Aetiological diagnosis of ischaemic stroke in young adults. Lancet Neurol. 2010;9:1085-1096.
3. Cotter PE, Belham M, Martin PJ. Stroke in younger patients: the heart of the matter. J Neurol. 2010;257:1777-1787.
4. Kitsios GD, Dahabreh IJ, Abu Dabrh AM, et al. Patent foramen ovale closure and medical treatments for secondary stroke prevention: a systematic review of observational and randomized evidence. Stroke. 2012;43:422-431.
5. Furlan AJ, Reisman M, Massaro J, et al. Closure or medical therapy for cryptogenic stroke with patent foramen ovale. N Engl J Med. 2012;366:991-999.
6. Carroll JD, Saver JL, Thaler DE, et al. Closure of patent foramen ovale versus medical therapy after cryptogenic stroke. N Engl J Med. 2013;368:1092-1100.
7. Meier B, Kalesan B, Mattle HP, et al. Percutaneous closure of patent foramen ovale in cryptogenic embolism. N Engl J Med. 2013;368:1083-1091.
8. Furie KL, Kasner SE, Adams RJ, et al. Guidelines for the prevention of stroke in patients with stroke or transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:227-276.
9. Menkin M, Schwartzman RJ. Cerebral air embolism. Report of five cases and review of the literature. Arch Neurol. 1977;34:168-170.
10. Sanna T, Diener HC, Passman RS, et al. Cryptogenic stroke and underlying atrial fibrillation. N Engl J Med. 2014;370:2478-2486.
1. Lechat P, Mas JL, Lascault G, et al. Prevalence of patent foramen ovale in patients with stroke. N Engl J Med. 1988;318:1148-1152.
2. Ferro JM, Massaro AR, Mas JL. Aetiological diagnosis of ischaemic stroke in young adults. Lancet Neurol. 2010;9:1085-1096.
3. Cotter PE, Belham M, Martin PJ. Stroke in younger patients: the heart of the matter. J Neurol. 2010;257:1777-1787.
4. Kitsios GD, Dahabreh IJ, Abu Dabrh AM, et al. Patent foramen ovale closure and medical treatments for secondary stroke prevention: a systematic review of observational and randomized evidence. Stroke. 2012;43:422-431.
5. Furlan AJ, Reisman M, Massaro J, et al. Closure or medical therapy for cryptogenic stroke with patent foramen ovale. N Engl J Med. 2012;366:991-999.
6. Carroll JD, Saver JL, Thaler DE, et al. Closure of patent foramen ovale versus medical therapy after cryptogenic stroke. N Engl J Med. 2013;368:1092-1100.
7. Meier B, Kalesan B, Mattle HP, et al. Percutaneous closure of patent foramen ovale in cryptogenic embolism. N Engl J Med. 2013;368:1083-1091.
8. Furie KL, Kasner SE, Adams RJ, et al. Guidelines for the prevention of stroke in patients with stroke or transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:227-276.
9. Menkin M, Schwartzman RJ. Cerebral air embolism. Report of five cases and review of the literature. Arch Neurol. 1977;34:168-170.
10. Sanna T, Diener HC, Passman RS, et al. Cryptogenic stroke and underlying atrial fibrillation. N Engl J Med. 2014;370:2478-2486.
Trans-Scaphoid Transcapitate Perilunate Fracture-Dislocation
Take-Home Points
- TSTC-PLFD is a rare hyperextension wrist injury characterized by fracture of both the scaphoid and the capitate and rotation of the proximal bone fragment of the capitate.
- TSTC-PLFD is associated by a complex ligamentous injury of the wrist.
- Impaction of the wrist in extension seems to be the most important predictor of this injury.
- Optimal treatment for TSTC-PLFD is open reduction, anatomical alignment, and ligamentous and osseous stabilization.
- The most important complications of scaphoid and capitate fractures and PLFD are osteonecrosis and nonunion.
Trans-scaphoid transcapitate (TSTC) perilunate fracture-dislocation (PLFD) is a rare hyperextension wrist injury characterized by fracture of both the scaphoid and the capitate and rotation of the proximal bone fragment of the capitate.1 Isolated capitate fractures with or without rotation of its proximal fragment have been well described.2,3 Obviously, this specific type of injury represents just the osseous part of a more complex ligamentous wrist injury.2,3
TSTC-PLFD was first described by Nicholson4 in 1940. In 1956, Fenton5 coined the term scaphocapitate syndrome, which became widely known. With PLFD, accurate diagnosis may be delayed. Usually, only the scaphoid fracture is identified by radiologic examination, and thus the severity of the injury is underestimated and appropriate treatment delayed.3,6,7 The English literature includes only case reports and small series on this rare perilunate injury.6-9 In this article, we report the case of an adult with TSTC-PLFD. We describe the radiographic and intraoperative findings, review the current surgical principles for reduction and stabilization of this injury, and assess the clinical and radiologic outcomes. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 32-year-old man sustained an isolated injury of his right (dominant) hand after falling from a height of 6 feet and landing on his outstretched right arm with the wrist in extension. 

With the patient under general anesthesia and a humerus tourniquet applied, an external fixator was placed for spanning of the wrist joint. The dorsal aspect of the wrist joint was approached through a midline longitudinal 5-cm incision, centered over the Lister tubercle. For adequate exposure of the dorsal wrist, a flap of the dorsal capsule was raised with the apex at the triquetrum and a radial broad base, as previously described.9 An avulsion fracture at the insertion of the dorsal capsule to the triquetrum was observed. The dorsal surface of the hamate and lunate showed a small area of bone contusion with hemorrhagic infiltration. The scapholunate and lunotriquetral ligaments were intact. The proximal fragment of the capitate was identified deep into the space between the lunate and distal capitate fragment; the articular surface of the bone fragment was rotated 180° distally (Figure 3). 

Skin sutures were removed 2 weeks after surgery, K-wires 6 weeks after surgery, and the external fixator 8 weeks after surgery. At 8 weeks, radiographs showed healing of both fractures, scaphoid and capitate. The patient was allowed gradual passive and active-assisted range-of-motion exercises of the wrist at 8 weeks, and he returned to work 3 months after surgery. At 12-month follow-up, all fractures were completely healed, and the wrist was stable and pain-free. 
Discussion
The exact biomechanism of TSTC-PLFD is unclear. Impaction of the wrist in extension seems to be the most important predictor of this injury.5,7,9-11 According to Stein and Siegel,10 scaphoid fractures first allow hyperextension of the wrist; the lunate and the capitate rotate dorsally, and the dorsal surface of the capitate impacts the dorsal edge of the distal radius, causing a fracture of the neck of the capitate. If the wrist continues to rotate into further hyperextension, the unsupported, proximal part of the capitate rotates 90° around itself.9,10 When the carpus returns to neutral position, the bone fragment of the capitate rotates further, reaching a position of 180°, with its proximal articular surface facing distally. In this type of injury, the axis of rotation is transverse (radioulnar), in contrast to the perpendicular (anteroposterior) axis of rotation suggested by the initial report by Fenton.5 The scaphoid is fractured by impaction of the radial styloid process. Monahan and Galasko11 reported a case of capitate fracture with palmar displacement and 90° rotation of the proximal bone fragment; the fragmented surface was facing dorsally. A transverse axis of rotation, as in our patient’s case, could explain this type of displacement supporting the mechanism of injury proposed by Stein and Siegel.10 Vance and colleagues7 described various patterns of scaphocapitate fractures and concluded that no single mechanism of injury accounts for these types of injuries. Other authors have considered scaphocapitate syndrome as a specific type of TSTC-PLFD, one that reduces either spontaneously or with manipulation.1,3,12 Detailed evaluation of standard anteroposterior and lateral wrist radiographs can provide enough evidence for the diagnosis of this injury. Computed tomography may define further the type and extent of injury.7 In our patient’s case, wrist impaction caused the scaphoid and capitate fractures and the avulsion of the capsule attachment to the triquetrum. The distal fragment of the capitate subluxated dorsally in relation to the lunate. The lateral radiograph of the wrist showed its position in the lunate fossa. According to the classification of Herzberg and colleagues12 and Mayfield and colleagues,13 this represents a dorsal PLFD of the greater carpal bones arc.
Conservative treatment is not recommended for PLFD because closed reduction usually is not possible, and poor functional outcomes are common. Instead, optimal treatment is open reduction, anatomical alignment, and ligamentous and osseous stabilization.7,12,14,15 Dorsal, palmar, and combined approaches have been used in surgery for perilunate injuries. A dorsal approach through a radius-based capsular flap allows excellent exposure of the dorsal wrist and facilitates reduction of fractures.9 Capitate reduction should precede scaphoid reduction because scaphoid reduction cannot be easily maintained, especially when the fracture interface is comminuted.7 In addition, scaphoid reduction may be guided from the radial surface of the capitate. Moreover, when the scaphoid is fixated first, reduction of the rotated head of the capitate usually is difficult. In our patient’s case, traction applied through the external fixator facilitated reduction and K-wire fixation of the capitate fracture. After scaphoid fixation, the K-wires were advanced through the capitate to the lunate to stabilize the capitolunate joint. The wrist must be immobilized for 6 to 8 weeks after surgical repair of PLFD. A cast can be used, but, as with our patient, an external fixator facilitates fracture reduction and wrist stability during osteosynthesis. During immobilization, the wrist should be maintained in neutral position to avoid stretching the dorsal and palmar wrist capsule and ligaments.16The most important complications of scaphoid and capitate fractures and PLFD are osteonecrosis and nonunion.17-20 Similar to scaphoid fractures, capitate fractures proximal to the waist of the capitate are associated with increased risk of osteonecrosis. Therefore, anatomical reduction and stabilization favor revascularization of the proximal bone fragment. Moreover, any osteonecrosis that occurs in the proximal part of the capitate is not an indication for further surgery as long as wrist height is maintained. Nonunion is not common after open reduction and internal fixation of PLFD (eg, our patient’s fractures healed completely).17 Radiographically, nonunion is characterized by bone absorption and sclerosis of the ends of the bone. Treatment of capitate nonunion depends on symptom severity, bone fragment size, and radiographic evidence of arthritic changes.3,7,21-23 Treatment options include resection of sclerotic edges, bone grafting, and stabilization21 and removal of the proximal capitate fragment and limited arthrodesis,22 as arthritic changes likely are inevitable.22,23TSTC-PLFD is a rare wrist injury. Careful radiographic evaluation of the carpal bones and their relationships on both anteroposterior and lateral views is mandatory in making the correct diagnosis. Open reduction (preferably with use of an external fixator) and internal fixation are recommended for optimal healing and functional outcomes.
Am J Orthop. 2017;46(4):E230-E234. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Johnson RP. The acutely injured wrist and its residuals. Clin Orthop Relat Res. 1980;(149):33-44.
2. Volk AG, Schnall SB, Merkle P, Stevanovic M. Unusual capitate fracture: a case report. J Hand Surg Am. 1995;20(4):581-582.
3. Apergis E, Darmanis S, Kastanis G, Papanikolaou A. Does the term scaphocapitate syndrome need to be revised? A report of 6 cases. J Hand Surg Br. 2001;26(5):441-445.
4. Nicholson CB. Fracture dislocation of the os magnum. J Roy Navy Med Serv. 1940;26:289-291.
5. Fenton RL. The naviculo-capitate fracture syndrome. J Bone Joint Surg Am. 1956;38(3):681-684.
6. Strohm PC, Laier P, Müller CA, Gutorski S, Pfister U. Scaphocapitate fracture syndrome of both hands—first description of a bilateral occurrence of a rare carpal injury [in German]. Unfallchirurg. 2003;106(4):339-342.
7. Vance RM, Gelberman R, Evans EF. Scaphocapitate fractures. Patterns of dislocation, mechanisms of injury, and preliminary results of treatment. J Bone Joint Surg Am. 1980;62(2):271-276.
8. Apostolides JG, Lifchez SD, Christy MR. Complex and rare fracture patterns in perilunate dislocations. Hand. 2011;6(3):287-294.
9. Berger RA, Bishop AT, Bettinger PC. New dorsal capsulotomy for the surgical exposure of the wrist. Ann Plast Surg. 1995;35(1):54-59.
10. Stein F, Siegel MW. Naviculocapitate fracture syndrome. A case report: new thoughts on the mechanism of injury. J Bone Joint Surg Am. 1969;51(2):391-395.
11. Monahan PR, Galasko CS. The scapho-capitate fracture syndrome. A mechanism of injury. J Bone Joint Surg Br. 1972;54(1):122-124.
12. Herzberg G, Comtet JJ, Linscheid RL, Amadio PC, Cooney WP, Stalder J. Perilunate dislocations and fracture-dislocations: a multicenter study. J Hand Surg Am. 1993;18(5):768-779.
13. Mayfield JK, Johnson RP, Kilcoyne RK. Carpal dislocations: pathomechanics and progressive perilunar instability. J Hand Surg Am. 1980;5(3):226-241.
14. Moneim MS, Hofammann KE 3rd, Omer GE. Transscaphoid perilunate fracture-dislocation. Result of open reduction and pin fixation. Clin Orthop Relat Res. 1984;(190):227-235.
15. Andreasi A, Coppo M, Danda F. Trans-scapho-capitate perilunar dislocation of the carpus. Ital J Orthop Traumatol. 1986;12(4):461-466.
16. Song D, Goodman S, Gilula LA, Wollstein R. Ulnocarpal translation in perilunate dislocations. J Hand Surg Eur. 2009;34(3):388-390.
17. Rand JA, Linscheid RL, Dobyns JH. Capitate fractures: a long-term follow-up. Clin Orthop Relat Res. 1982;(165):209-216.
18. Panagis JS, Gelberman RH, Taleisnik J, Baumgaertner M. The arterial anatomy of the human carpus. Part II: the intraosseous vascularity. J Hand Surg Am. 1983;8(4):375-382.
19. Freedman DM, Botte MJ, Gelberman RH. Vascularity of the carpus. Clin Orthop Relat Res. 2001;(383):47-59.
20. Vander Grend R, Dell PC, Glowczewskie F, Leslie B, Ruby LK. Intraosseous blood supply of the capitate and its correlation with aseptic necrosis. J Hand Surg Am. 1984;9(5):677-683.
21. Rico AA, Holguin PH, Martin JG. Pseudarthrosis of the capitate. J Hand Surg Br. 1999;24(3):382-384.
22. Kumar A, Olney DB. Multiple carpometacarpal dislocations. J Accid Emerg Med. 1994;11(4):257-258.
23. Kohut GN. Extra-articular fractures of the distal radius in young adults. A technique of closed reposition and stabilisation by mono-segmental, radio-radial external fixator. Ann Chir Main Memb Super. 1995;14(1):14-19.
Take-Home Points
- TSTC-PLFD is a rare hyperextension wrist injury characterized by fracture of both the scaphoid and the capitate and rotation of the proximal bone fragment of the capitate.
- TSTC-PLFD is associated by a complex ligamentous injury of the wrist.
- Impaction of the wrist in extension seems to be the most important predictor of this injury.
- Optimal treatment for TSTC-PLFD is open reduction, anatomical alignment, and ligamentous and osseous stabilization.
- The most important complications of scaphoid and capitate fractures and PLFD are osteonecrosis and nonunion.
Trans-scaphoid transcapitate (TSTC) perilunate fracture-dislocation (PLFD) is a rare hyperextension wrist injury characterized by fracture of both the scaphoid and the capitate and rotation of the proximal bone fragment of the capitate.1 Isolated capitate fractures with or without rotation of its proximal fragment have been well described.2,3 Obviously, this specific type of injury represents just the osseous part of a more complex ligamentous wrist injury.2,3
TSTC-PLFD was first described by Nicholson4 in 1940. In 1956, Fenton5 coined the term scaphocapitate syndrome, which became widely known. With PLFD, accurate diagnosis may be delayed. Usually, only the scaphoid fracture is identified by radiologic examination, and thus the severity of the injury is underestimated and appropriate treatment delayed.3,6,7 The English literature includes only case reports and small series on this rare perilunate injury.6-9 In this article, we report the case of an adult with TSTC-PLFD. We describe the radiographic and intraoperative findings, review the current surgical principles for reduction and stabilization of this injury, and assess the clinical and radiologic outcomes. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 32-year-old man sustained an isolated injury of his right (dominant) hand after falling from a height of 6 feet and landing on his outstretched right arm with the wrist in extension.
With the patient under general anesthesia and a humerus tourniquet applied, an external fixator was placed for spanning of the wrist joint. The dorsal aspect of the wrist joint was approached through a midline longitudinal 5-cm incision, centered over the Lister tubercle. For adequate exposure of the dorsal wrist, a flap of the dorsal capsule was raised with the apex at the triquetrum and a radial broad base, as previously described.9 An avulsion fracture at the insertion of the dorsal capsule to the triquetrum was observed. The dorsal surface of the hamate and lunate showed a small area of bone contusion with hemorrhagic infiltration. The scapholunate and lunotriquetral ligaments were intact. The proximal fragment of the capitate was identified deep into the space between the lunate and distal capitate fragment; the articular surface of the bone fragment was rotated 180° distally (Figure 3).
Skin sutures were removed 2 weeks after surgery, K-wires 6 weeks after surgery, and the external fixator 8 weeks after surgery. At 8 weeks, radiographs showed healing of both fractures, scaphoid and capitate. The patient was allowed gradual passive and active-assisted range-of-motion exercises of the wrist at 8 weeks, and he returned to work 3 months after surgery. At 12-month follow-up, all fractures were completely healed, and the wrist was stable and pain-free.
Discussion
The exact biomechanism of TSTC-PLFD is unclear. Impaction of the wrist in extension seems to be the most important predictor of this injury.5,7,9-11 According to Stein and Siegel,10 scaphoid fractures first allow hyperextension of the wrist; the lunate and the capitate rotate dorsally, and the dorsal surface of the capitate impacts the dorsal edge of the distal radius, causing a fracture of the neck of the capitate. If the wrist continues to rotate into further hyperextension, the unsupported, proximal part of the capitate rotates 90° around itself.9,10 When the carpus returns to neutral position, the bone fragment of the capitate rotates further, reaching a position of 180°, with its proximal articular surface facing distally. In this type of injury, the axis of rotation is transverse (radioulnar), in contrast to the perpendicular (anteroposterior) axis of rotation suggested by the initial report by Fenton.5 The scaphoid is fractured by impaction of the radial styloid process. Monahan and Galasko11 reported a case of capitate fracture with palmar displacement and 90° rotation of the proximal bone fragment; the fragmented surface was facing dorsally. A transverse axis of rotation, as in our patient’s case, could explain this type of displacement supporting the mechanism of injury proposed by Stein and Siegel.10 Vance and colleagues7 described various patterns of scaphocapitate fractures and concluded that no single mechanism of injury accounts for these types of injuries. Other authors have considered scaphocapitate syndrome as a specific type of TSTC-PLFD, one that reduces either spontaneously or with manipulation.1,3,12 Detailed evaluation of standard anteroposterior and lateral wrist radiographs can provide enough evidence for the diagnosis of this injury. Computed tomography may define further the type and extent of injury.7 In our patient’s case, wrist impaction caused the scaphoid and capitate fractures and the avulsion of the capsule attachment to the triquetrum. The distal fragment of the capitate subluxated dorsally in relation to the lunate. The lateral radiograph of the wrist showed its position in the lunate fossa. According to the classification of Herzberg and colleagues12 and Mayfield and colleagues,13 this represents a dorsal PLFD of the greater carpal bones arc.
Conservative treatment is not recommended for PLFD because closed reduction usually is not possible, and poor functional outcomes are common. Instead, optimal treatment is open reduction, anatomical alignment, and ligamentous and osseous stabilization.7,12,14,15 Dorsal, palmar, and combined approaches have been used in surgery for perilunate injuries. A dorsal approach through a radius-based capsular flap allows excellent exposure of the dorsal wrist and facilitates reduction of fractures.9 Capitate reduction should precede scaphoid reduction because scaphoid reduction cannot be easily maintained, especially when the fracture interface is comminuted.7 In addition, scaphoid reduction may be guided from the radial surface of the capitate. Moreover, when the scaphoid is fixated first, reduction of the rotated head of the capitate usually is difficult. In our patient’s case, traction applied through the external fixator facilitated reduction and K-wire fixation of the capitate fracture. After scaphoid fixation, the K-wires were advanced through the capitate to the lunate to stabilize the capitolunate joint. The wrist must be immobilized for 6 to 8 weeks after surgical repair of PLFD. A cast can be used, but, as with our patient, an external fixator facilitates fracture reduction and wrist stability during osteosynthesis. During immobilization, the wrist should be maintained in neutral position to avoid stretching the dorsal and palmar wrist capsule and ligaments.16The most important complications of scaphoid and capitate fractures and PLFD are osteonecrosis and nonunion.17-20 Similar to scaphoid fractures, capitate fractures proximal to the waist of the capitate are associated with increased risk of osteonecrosis. Therefore, anatomical reduction and stabilization favor revascularization of the proximal bone fragment. Moreover, any osteonecrosis that occurs in the proximal part of the capitate is not an indication for further surgery as long as wrist height is maintained. Nonunion is not common after open reduction and internal fixation of PLFD (eg, our patient’s fractures healed completely).17 Radiographically, nonunion is characterized by bone absorption and sclerosis of the ends of the bone. Treatment of capitate nonunion depends on symptom severity, bone fragment size, and radiographic evidence of arthritic changes.3,7,21-23 Treatment options include resection of sclerotic edges, bone grafting, and stabilization21 and removal of the proximal capitate fragment and limited arthrodesis,22 as arthritic changes likely are inevitable.22,23TSTC-PLFD is a rare wrist injury. Careful radiographic evaluation of the carpal bones and their relationships on both anteroposterior and lateral views is mandatory in making the correct diagnosis. Open reduction (preferably with use of an external fixator) and internal fixation are recommended for optimal healing and functional outcomes.
Am J Orthop. 2017;46(4):E230-E234. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- TSTC-PLFD is a rare hyperextension wrist injury characterized by fracture of both the scaphoid and the capitate and rotation of the proximal bone fragment of the capitate.
- TSTC-PLFD is associated by a complex ligamentous injury of the wrist.
- Impaction of the wrist in extension seems to be the most important predictor of this injury.
- Optimal treatment for TSTC-PLFD is open reduction, anatomical alignment, and ligamentous and osseous stabilization.
- The most important complications of scaphoid and capitate fractures and PLFD are osteonecrosis and nonunion.
Trans-scaphoid transcapitate (TSTC) perilunate fracture-dislocation (PLFD) is a rare hyperextension wrist injury characterized by fracture of both the scaphoid and the capitate and rotation of the proximal bone fragment of the capitate.1 Isolated capitate fractures with or without rotation of its proximal fragment have been well described.2,3 Obviously, this specific type of injury represents just the osseous part of a more complex ligamentous wrist injury.2,3
TSTC-PLFD was first described by Nicholson4 in 1940. In 1956, Fenton5 coined the term scaphocapitate syndrome, which became widely known. With PLFD, accurate diagnosis may be delayed. Usually, only the scaphoid fracture is identified by radiologic examination, and thus the severity of the injury is underestimated and appropriate treatment delayed.3,6,7 The English literature includes only case reports and small series on this rare perilunate injury.6-9 In this article, we report the case of an adult with TSTC-PLFD. We describe the radiographic and intraoperative findings, review the current surgical principles for reduction and stabilization of this injury, and assess the clinical and radiologic outcomes. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 32-year-old man sustained an isolated injury of his right (dominant) hand after falling from a height of 6 feet and landing on his outstretched right arm with the wrist in extension.
With the patient under general anesthesia and a humerus tourniquet applied, an external fixator was placed for spanning of the wrist joint. The dorsal aspect of the wrist joint was approached through a midline longitudinal 5-cm incision, centered over the Lister tubercle. For adequate exposure of the dorsal wrist, a flap of the dorsal capsule was raised with the apex at the triquetrum and a radial broad base, as previously described.9 An avulsion fracture at the insertion of the dorsal capsule to the triquetrum was observed. The dorsal surface of the hamate and lunate showed a small area of bone contusion with hemorrhagic infiltration. The scapholunate and lunotriquetral ligaments were intact. The proximal fragment of the capitate was identified deep into the space between the lunate and distal capitate fragment; the articular surface of the bone fragment was rotated 180° distally (Figure 3).
Skin sutures were removed 2 weeks after surgery, K-wires 6 weeks after surgery, and the external fixator 8 weeks after surgery. At 8 weeks, radiographs showed healing of both fractures, scaphoid and capitate. The patient was allowed gradual passive and active-assisted range-of-motion exercises of the wrist at 8 weeks, and he returned to work 3 months after surgery. At 12-month follow-up, all fractures were completely healed, and the wrist was stable and pain-free.
Discussion
The exact biomechanism of TSTC-PLFD is unclear. Impaction of the wrist in extension seems to be the most important predictor of this injury.5,7,9-11 According to Stein and Siegel,10 scaphoid fractures first allow hyperextension of the wrist; the lunate and the capitate rotate dorsally, and the dorsal surface of the capitate impacts the dorsal edge of the distal radius, causing a fracture of the neck of the capitate. If the wrist continues to rotate into further hyperextension, the unsupported, proximal part of the capitate rotates 90° around itself.9,10 When the carpus returns to neutral position, the bone fragment of the capitate rotates further, reaching a position of 180°, with its proximal articular surface facing distally. In this type of injury, the axis of rotation is transverse (radioulnar), in contrast to the perpendicular (anteroposterior) axis of rotation suggested by the initial report by Fenton.5 The scaphoid is fractured by impaction of the radial styloid process. Monahan and Galasko11 reported a case of capitate fracture with palmar displacement and 90° rotation of the proximal bone fragment; the fragmented surface was facing dorsally. A transverse axis of rotation, as in our patient’s case, could explain this type of displacement supporting the mechanism of injury proposed by Stein and Siegel.10 Vance and colleagues7 described various patterns of scaphocapitate fractures and concluded that no single mechanism of injury accounts for these types of injuries. Other authors have considered scaphocapitate syndrome as a specific type of TSTC-PLFD, one that reduces either spontaneously or with manipulation.1,3,12 Detailed evaluation of standard anteroposterior and lateral wrist radiographs can provide enough evidence for the diagnosis of this injury. Computed tomography may define further the type and extent of injury.7 In our patient’s case, wrist impaction caused the scaphoid and capitate fractures and the avulsion of the capsule attachment to the triquetrum. The distal fragment of the capitate subluxated dorsally in relation to the lunate. The lateral radiograph of the wrist showed its position in the lunate fossa. According to the classification of Herzberg and colleagues12 and Mayfield and colleagues,13 this represents a dorsal PLFD of the greater carpal bones arc.
Conservative treatment is not recommended for PLFD because closed reduction usually is not possible, and poor functional outcomes are common. Instead, optimal treatment is open reduction, anatomical alignment, and ligamentous and osseous stabilization.7,12,14,15 Dorsal, palmar, and combined approaches have been used in surgery for perilunate injuries. A dorsal approach through a radius-based capsular flap allows excellent exposure of the dorsal wrist and facilitates reduction of fractures.9 Capitate reduction should precede scaphoid reduction because scaphoid reduction cannot be easily maintained, especially when the fracture interface is comminuted.7 In addition, scaphoid reduction may be guided from the radial surface of the capitate. Moreover, when the scaphoid is fixated first, reduction of the rotated head of the capitate usually is difficult. In our patient’s case, traction applied through the external fixator facilitated reduction and K-wire fixation of the capitate fracture. After scaphoid fixation, the K-wires were advanced through the capitate to the lunate to stabilize the capitolunate joint. The wrist must be immobilized for 6 to 8 weeks after surgical repair of PLFD. A cast can be used, but, as with our patient, an external fixator facilitates fracture reduction and wrist stability during osteosynthesis. During immobilization, the wrist should be maintained in neutral position to avoid stretching the dorsal and palmar wrist capsule and ligaments.16The most important complications of scaphoid and capitate fractures and PLFD are osteonecrosis and nonunion.17-20 Similar to scaphoid fractures, capitate fractures proximal to the waist of the capitate are associated with increased risk of osteonecrosis. Therefore, anatomical reduction and stabilization favor revascularization of the proximal bone fragment. Moreover, any osteonecrosis that occurs in the proximal part of the capitate is not an indication for further surgery as long as wrist height is maintained. Nonunion is not common after open reduction and internal fixation of PLFD (eg, our patient’s fractures healed completely).17 Radiographically, nonunion is characterized by bone absorption and sclerosis of the ends of the bone. Treatment of capitate nonunion depends on symptom severity, bone fragment size, and radiographic evidence of arthritic changes.3,7,21-23 Treatment options include resection of sclerotic edges, bone grafting, and stabilization21 and removal of the proximal capitate fragment and limited arthrodesis,22 as arthritic changes likely are inevitable.22,23TSTC-PLFD is a rare wrist injury. Careful radiographic evaluation of the carpal bones and their relationships on both anteroposterior and lateral views is mandatory in making the correct diagnosis. Open reduction (preferably with use of an external fixator) and internal fixation are recommended for optimal healing and functional outcomes.
Am J Orthop. 2017;46(4):E230-E234. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Johnson RP. The acutely injured wrist and its residuals. Clin Orthop Relat Res. 1980;(149):33-44.
2. Volk AG, Schnall SB, Merkle P, Stevanovic M. Unusual capitate fracture: a case report. J Hand Surg Am. 1995;20(4):581-582.
3. Apergis E, Darmanis S, Kastanis G, Papanikolaou A. Does the term scaphocapitate syndrome need to be revised? A report of 6 cases. J Hand Surg Br. 2001;26(5):441-445.
4. Nicholson CB. Fracture dislocation of the os magnum. J Roy Navy Med Serv. 1940;26:289-291.
5. Fenton RL. The naviculo-capitate fracture syndrome. J Bone Joint Surg Am. 1956;38(3):681-684.
6. Strohm PC, Laier P, Müller CA, Gutorski S, Pfister U. Scaphocapitate fracture syndrome of both hands—first description of a bilateral occurrence of a rare carpal injury [in German]. Unfallchirurg. 2003;106(4):339-342.
7. Vance RM, Gelberman R, Evans EF. Scaphocapitate fractures. Patterns of dislocation, mechanisms of injury, and preliminary results of treatment. J Bone Joint Surg Am. 1980;62(2):271-276.
8. Apostolides JG, Lifchez SD, Christy MR. Complex and rare fracture patterns in perilunate dislocations. Hand. 2011;6(3):287-294.
9. Berger RA, Bishop AT, Bettinger PC. New dorsal capsulotomy for the surgical exposure of the wrist. Ann Plast Surg. 1995;35(1):54-59.
10. Stein F, Siegel MW. Naviculocapitate fracture syndrome. A case report: new thoughts on the mechanism of injury. J Bone Joint Surg Am. 1969;51(2):391-395.
11. Monahan PR, Galasko CS. The scapho-capitate fracture syndrome. A mechanism of injury. J Bone Joint Surg Br. 1972;54(1):122-124.
12. Herzberg G, Comtet JJ, Linscheid RL, Amadio PC, Cooney WP, Stalder J. Perilunate dislocations and fracture-dislocations: a multicenter study. J Hand Surg Am. 1993;18(5):768-779.
13. Mayfield JK, Johnson RP, Kilcoyne RK. Carpal dislocations: pathomechanics and progressive perilunar instability. J Hand Surg Am. 1980;5(3):226-241.
14. Moneim MS, Hofammann KE 3rd, Omer GE. Transscaphoid perilunate fracture-dislocation. Result of open reduction and pin fixation. Clin Orthop Relat Res. 1984;(190):227-235.
15. Andreasi A, Coppo M, Danda F. Trans-scapho-capitate perilunar dislocation of the carpus. Ital J Orthop Traumatol. 1986;12(4):461-466.
16. Song D, Goodman S, Gilula LA, Wollstein R. Ulnocarpal translation in perilunate dislocations. J Hand Surg Eur. 2009;34(3):388-390.
17. Rand JA, Linscheid RL, Dobyns JH. Capitate fractures: a long-term follow-up. Clin Orthop Relat Res. 1982;(165):209-216.
18. Panagis JS, Gelberman RH, Taleisnik J, Baumgaertner M. The arterial anatomy of the human carpus. Part II: the intraosseous vascularity. J Hand Surg Am. 1983;8(4):375-382.
19. Freedman DM, Botte MJ, Gelberman RH. Vascularity of the carpus. Clin Orthop Relat Res. 2001;(383):47-59.
20. Vander Grend R, Dell PC, Glowczewskie F, Leslie B, Ruby LK. Intraosseous blood supply of the capitate and its correlation with aseptic necrosis. J Hand Surg Am. 1984;9(5):677-683.
21. Rico AA, Holguin PH, Martin JG. Pseudarthrosis of the capitate. J Hand Surg Br. 1999;24(3):382-384.
22. Kumar A, Olney DB. Multiple carpometacarpal dislocations. J Accid Emerg Med. 1994;11(4):257-258.
23. Kohut GN. Extra-articular fractures of the distal radius in young adults. A technique of closed reposition and stabilisation by mono-segmental, radio-radial external fixator. Ann Chir Main Memb Super. 1995;14(1):14-19.
1. Johnson RP. The acutely injured wrist and its residuals. Clin Orthop Relat Res. 1980;(149):33-44.
2. Volk AG, Schnall SB, Merkle P, Stevanovic M. Unusual capitate fracture: a case report. J Hand Surg Am. 1995;20(4):581-582.
3. Apergis E, Darmanis S, Kastanis G, Papanikolaou A. Does the term scaphocapitate syndrome need to be revised? A report of 6 cases. J Hand Surg Br. 2001;26(5):441-445.
4. Nicholson CB. Fracture dislocation of the os magnum. J Roy Navy Med Serv. 1940;26:289-291.
5. Fenton RL. The naviculo-capitate fracture syndrome. J Bone Joint Surg Am. 1956;38(3):681-684.
6. Strohm PC, Laier P, Müller CA, Gutorski S, Pfister U. Scaphocapitate fracture syndrome of both hands—first description of a bilateral occurrence of a rare carpal injury [in German]. Unfallchirurg. 2003;106(4):339-342.
7. Vance RM, Gelberman R, Evans EF. Scaphocapitate fractures. Patterns of dislocation, mechanisms of injury, and preliminary results of treatment. J Bone Joint Surg Am. 1980;62(2):271-276.
8. Apostolides JG, Lifchez SD, Christy MR. Complex and rare fracture patterns in perilunate dislocations. Hand. 2011;6(3):287-294.
9. Berger RA, Bishop AT, Bettinger PC. New dorsal capsulotomy for the surgical exposure of the wrist. Ann Plast Surg. 1995;35(1):54-59.
10. Stein F, Siegel MW. Naviculocapitate fracture syndrome. A case report: new thoughts on the mechanism of injury. J Bone Joint Surg Am. 1969;51(2):391-395.
11. Monahan PR, Galasko CS. The scapho-capitate fracture syndrome. A mechanism of injury. J Bone Joint Surg Br. 1972;54(1):122-124.
12. Herzberg G, Comtet JJ, Linscheid RL, Amadio PC, Cooney WP, Stalder J. Perilunate dislocations and fracture-dislocations: a multicenter study. J Hand Surg Am. 1993;18(5):768-779.
13. Mayfield JK, Johnson RP, Kilcoyne RK. Carpal dislocations: pathomechanics and progressive perilunar instability. J Hand Surg Am. 1980;5(3):226-241.
14. Moneim MS, Hofammann KE 3rd, Omer GE. Transscaphoid perilunate fracture-dislocation. Result of open reduction and pin fixation. Clin Orthop Relat Res. 1984;(190):227-235.
15. Andreasi A, Coppo M, Danda F. Trans-scapho-capitate perilunar dislocation of the carpus. Ital J Orthop Traumatol. 1986;12(4):461-466.
16. Song D, Goodman S, Gilula LA, Wollstein R. Ulnocarpal translation in perilunate dislocations. J Hand Surg Eur. 2009;34(3):388-390.
17. Rand JA, Linscheid RL, Dobyns JH. Capitate fractures: a long-term follow-up. Clin Orthop Relat Res. 1982;(165):209-216.
18. Panagis JS, Gelberman RH, Taleisnik J, Baumgaertner M. The arterial anatomy of the human carpus. Part II: the intraosseous vascularity. J Hand Surg Am. 1983;8(4):375-382.
19. Freedman DM, Botte MJ, Gelberman RH. Vascularity of the carpus. Clin Orthop Relat Res. 2001;(383):47-59.
20. Vander Grend R, Dell PC, Glowczewskie F, Leslie B, Ruby LK. Intraosseous blood supply of the capitate and its correlation with aseptic necrosis. J Hand Surg Am. 1984;9(5):677-683.
21. Rico AA, Holguin PH, Martin JG. Pseudarthrosis of the capitate. J Hand Surg Br. 1999;24(3):382-384.
22. Kumar A, Olney DB. Multiple carpometacarpal dislocations. J Accid Emerg Med. 1994;11(4):257-258.
23. Kohut GN. Extra-articular fractures of the distal radius in young adults. A technique of closed reposition and stabilisation by mono-segmental, radio-radial external fixator. Ann Chir Main Memb Super. 1995;14(1):14-19.
Co-occurrence of Steatocystoma Multiplex, Eruptive Vellus Hair Cysts, and Trichofolliculomas
An association between steatocystoma multiplex (SCM) and eruptive vellus hair cysts (EVHCs) has been recognized. They are related conditions representing nevoid malformations of the pilosebaceous junctions1-10 that have similar clinical features but distinctive histologic features. Both conditions most commonly involve the anterior aspect of the chest. Six cases of a rare facial variant of SCM have been reported,11-16 3 involving lesions limited to the forehead.13-15 Two patients with a rare facial variant of EVHC also have been reported.17 The development of separate lesions of SCM and EVHC on the trunk can uncommonly occur.5,6,10 One case of SCM and EVHC on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. The simultaneous occurrence of multiple trichoblastomas, trichoepitheliomas, and SCM on the face and trunk has been reported in 1 case.18 Milia, SCM, and EVHC on the face and trunk have been reported in 1 family.4 A report of facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus also has occurred in 1 patient.19 Here, we report the simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead.
Case Report
A 37-year-old man had an increasing number of flesh-colored to yellow papules on the forehead that had been present since puberty. Although the lesions were asymptomatic, some had recently become tender, which led him to seek medical care. There was no history of trauma, burns, irradiation, or application of topical agents to the area or use of eyeglasses or goggles. The patient’s father had similar lesions limited to the forehead, which developed during adolescence.
On evaluation at our clinic, skin examination revealed 16 discrete, 0.3- to 1-cm, flesh-colored, yellow to blue, mobile, smooth papules, as well as flesh-colored papules with a central black punctum, on the forehead (Figure 1). Similar lesions were not present on the rest of the face; around the ears; or on the scalp, neck, chest, back, abdomen, genitalia, buttocks, palms, soles, axillae, arms, or legs. There were no nail abnormalities.
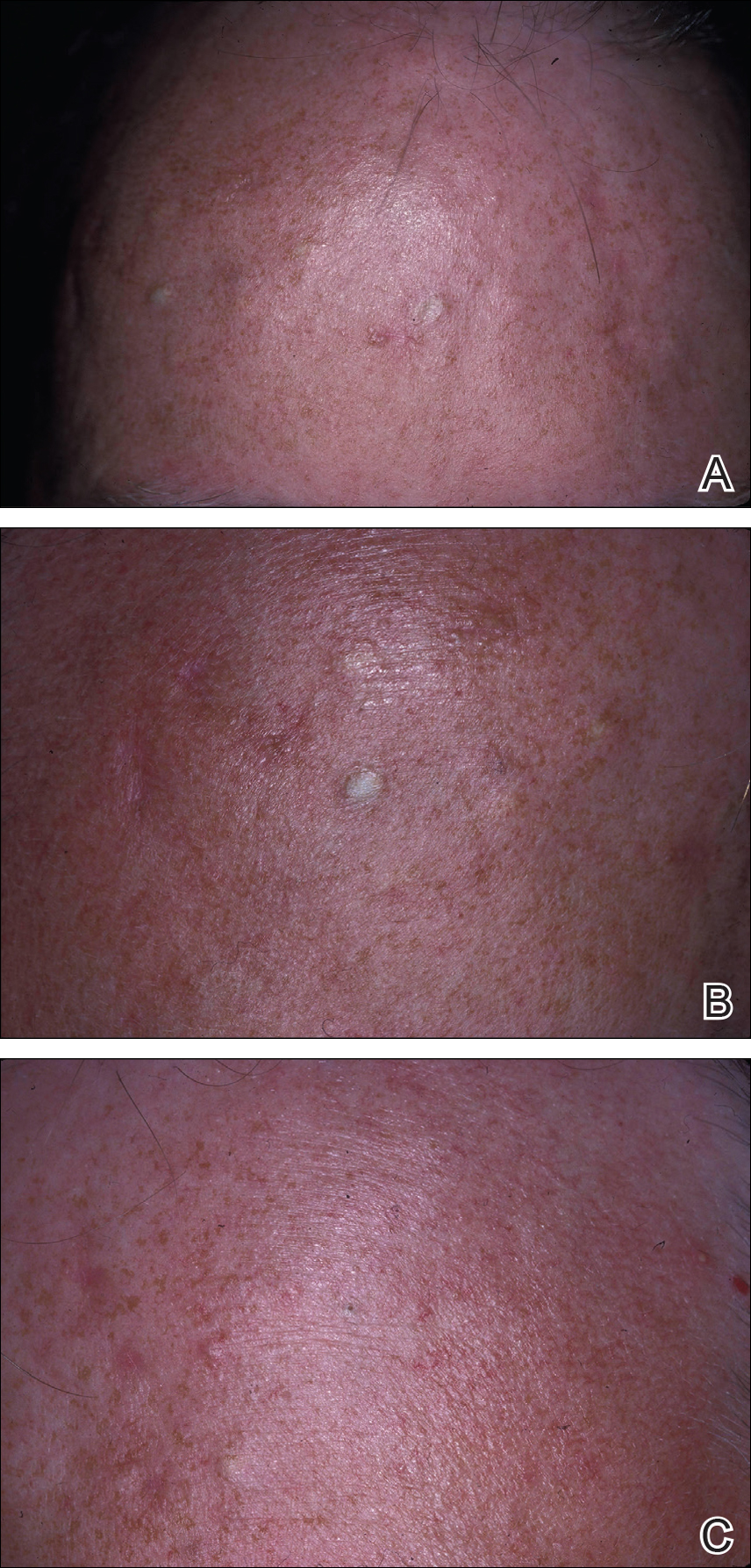
Multiple 3-, 4-, and 6-mm punch and excisional biopsies were performed to remove all 16 lesions on the forehead. Histologic examination revealed a collapsed cystic structure in the mid dermis in 10 lesions. The cysts were lined with a squamous epithelium without a granular layer but with an eosinophilic corrugated lining, and the cyst cavity contained scant homogeneous eosinophilic secretion. Mature sebaceous glands were adjacent to the outer portion of the cyst wall. These histologic findings were consistent with SCM (Figure 2).
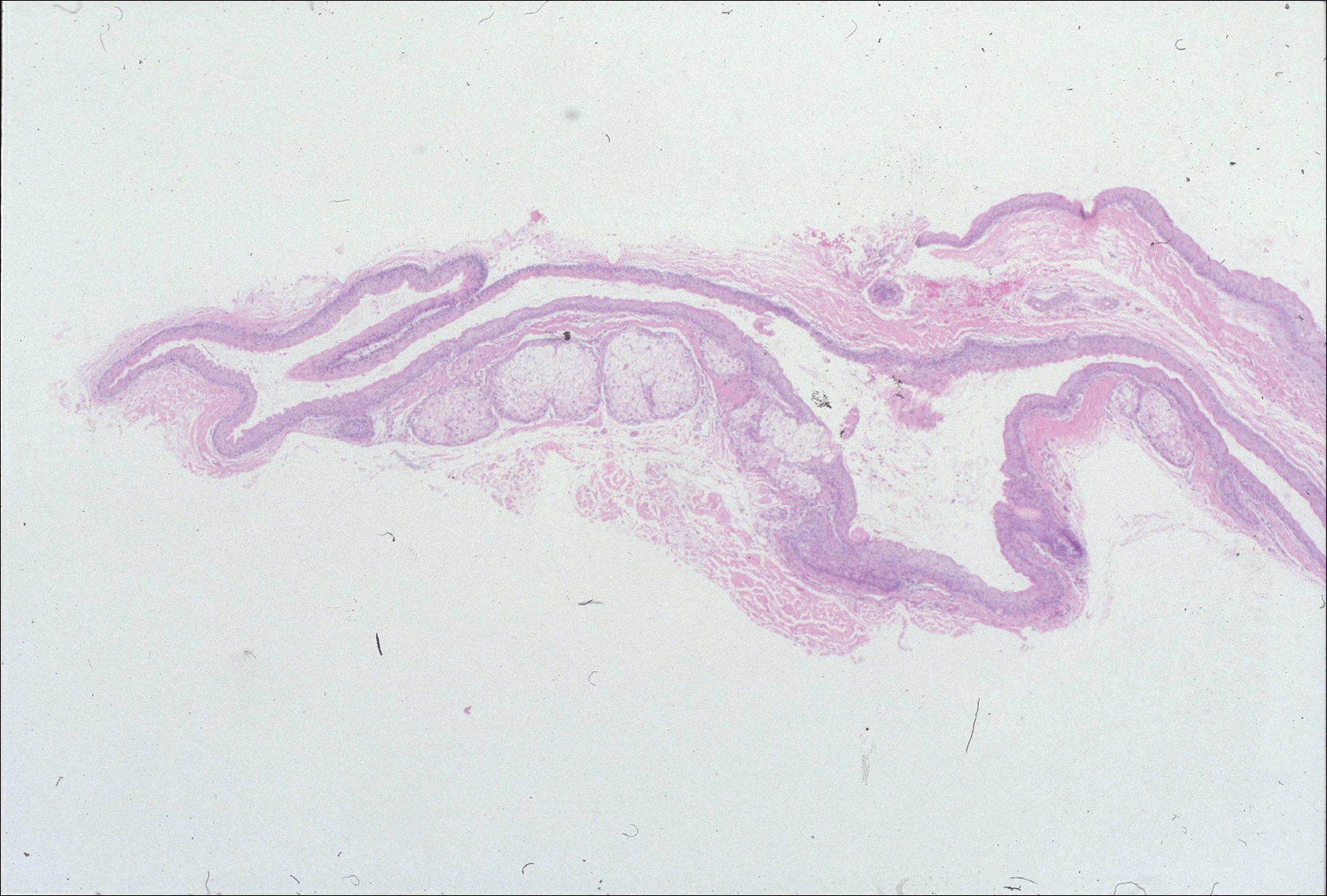
In 3 lesions, histologic examination revealed a cystic structure lined by a few layers of stratified squamous epithelium in the mid dermis. The cyst cavity contained numerous small vellus hairs and laminated keratin. These histologic findings were consistent with EVHC (Figure 3).
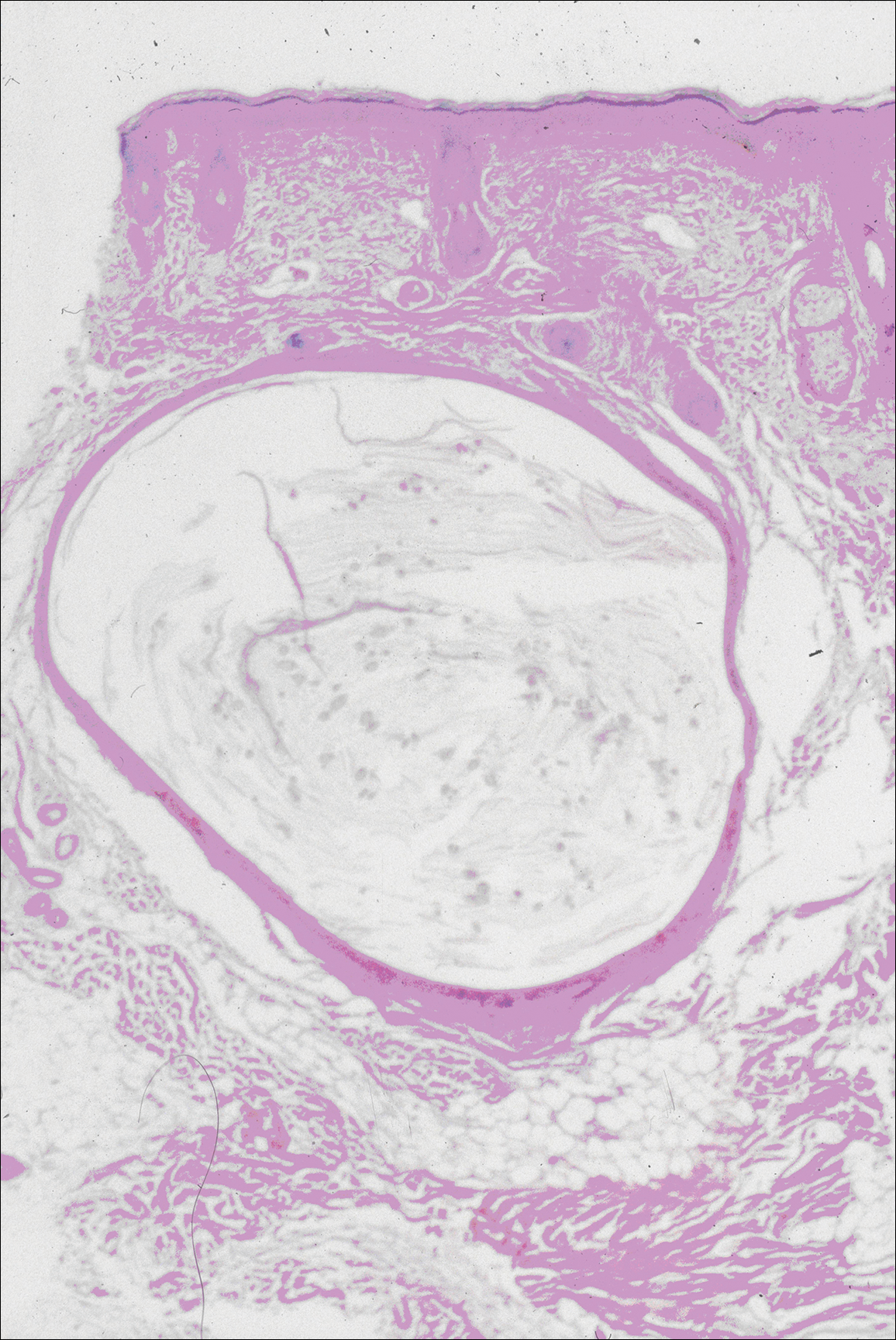
In the other 3 lesions, histologic examination revealed a dilated central cystic cavity filled with laminated keratin in the mid dermis. Multiple small follicles arose from the cysts and showed differentiation toward germinative epithelium. The surrounding stroma was fibrotic and contained a patchy lymphocytic infiltrate. These histologic findings were consistent with trichofolliculomas (Figure 4).
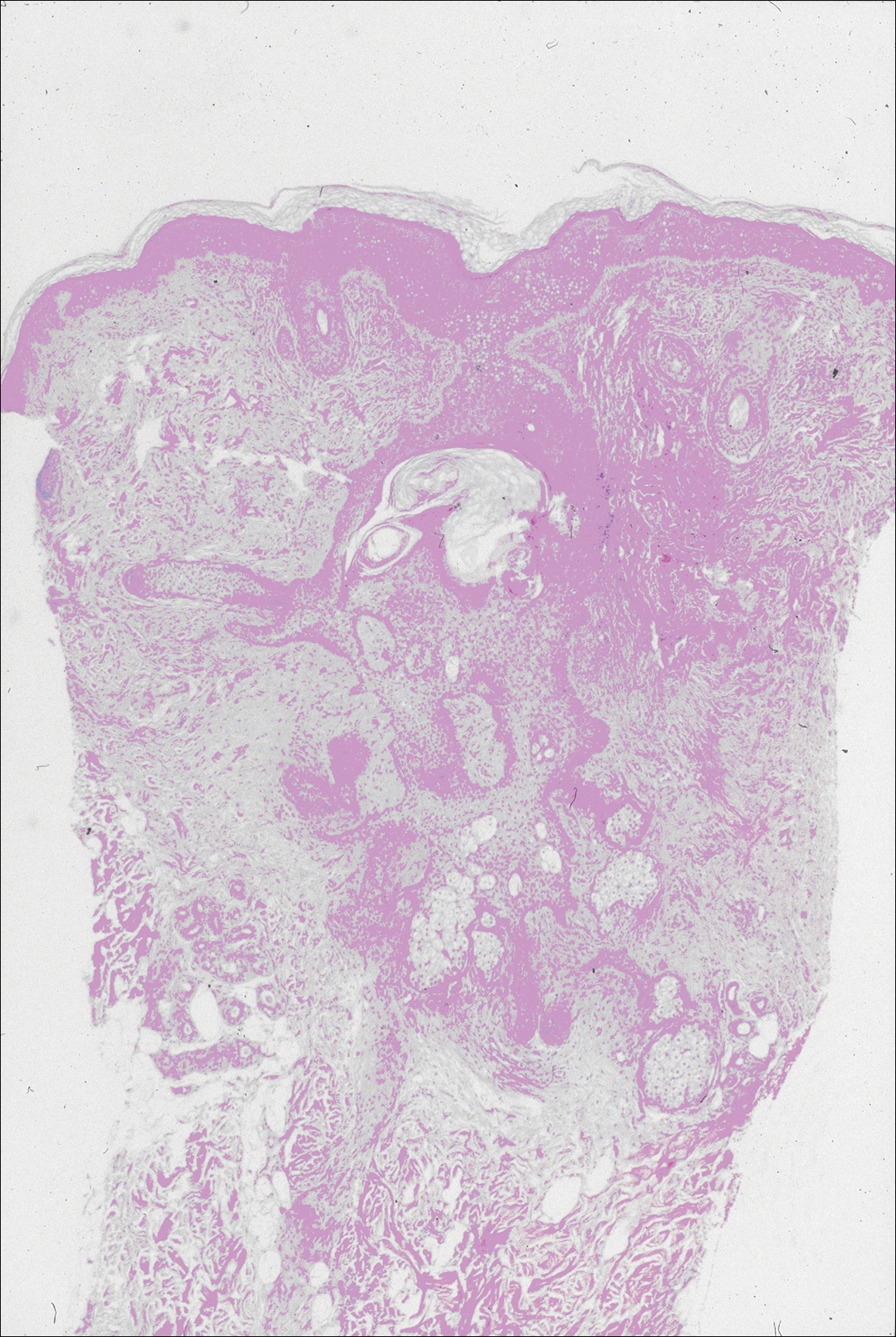
Comment
Characteristics of SCM
Steatocystoma multiplex is an uncommon condition characterized by the formation of asymptomatic, 0.2- to 2-cm, yellow to flesh-colored, soft, mobile papules or nodules on the trunk, extremities, axillae, genitalia, and/or chest. The lesions contain a clear or opaque, oily, milky or yellow, odorless fluid and most commonly are located on the anterior aspect of the chest. The face is not a commonly involved site in this condition. Six cases of a rare facial variant of SCM have been reported,11-16 with lesions limited to the forehead in 3 cases.13-15
In 1937, Mount20 credited Bozellini for describing the first case, though 3 cases reported in the late 1800s probably were SCM.21 In 1899, Pringle22 coined the term steatocystoma multiplex for this condition. It can be sporadic or have an autosomal-dominant inheritance pattern. Steatocystoma multiplex can occur at any age, though lesions develop most frequently in adolescence or young adulthood. There is no sex predilection.
Steatocystoma multiplex with pachyonychia congenita has been reported in a familial case.23 Other findings reported in patients with SCM include ichthyosis, koilonychia, acrokeratosis verruciformis of Hopf and hypertrophic lichen planus, hidradenitis suppurativa, hypotrichosis, multiple keratoacanthomas, and rheumatoid arthritis.12,24-26
Steatocystoma multiplex is a cyst lined by stratified squamous epithelium without a granular layer but with a thick eosinophilic cuticle. Mature sebaceous lobules are closely associated with the cyst wall. Steatocystoma multiplex arises from the sebaceous duct because the lining of the lumen is composed of undulating eosinophilic cuticle.
Characteristics of EVHCs
Eruptive vellus hair cysts, which were first described by Esterly et al,27 can occur at any age but develop most frequently in adolescents or young adults. Sometimes the lesions are congenital or appear in childhood. There is no sex predilection. They can be sporadic or have an autosomal-dominant inheritance pattern.
Eruptive vellus hair cysts are asymptomatic, 1- to 2-mm, smooth, crusted, or umbilicated papules on the chest or arms and legs. Eruptive vellus hair cysts most commonly involve the anterior aspect of the chest. The lesions are flesh-colored to yellow, though they have a slate gray color in darker-skinned individuals. A rare facial variant has been reported in 2 patients of Asian descent.17
Eruptive vellus hair cysts are small cystic structures lined by a stratified squamous epithelium with a granular layer. The cyst cavity contains numerous small vellus hair shafts and laminated keratin. Eruptive vellus hair cysts originate from the infundibulum or less frequently the isthmus or infundibular-isthmic junction of the hair follicle.
Characteristics of Trichofolliculomas
Trichofolliculomas are solitary, 3- to 5-mm, flesh-colored papules that occur on the face. They are highly differentiated, benign, neoplastic proliferations of an actively trichogenic epithelium, with structural components reflecting all portions of the pilosebaceous unit. Trichofolliculomas consist of a central dilated primary follicle contiguous with the surface epidermis embedded in a fibrous stroma. Multiple small secondary follicles with varying degrees of follicular differentiation arise from the primary follicle.
Co-occurrence of Lesions
An association between SCM and EVHC has been recognized.5-10 Steatocystoma multiplex and EVHC have similar clinical features but distinctive histologic features. They also have a similar age of onset, location/appearance of lesions, and mode of inheritance. Steatocystoma multiplex and EVHC can be distinguished by immunohistochemical techniques: SCM shows expression of keratin 10 and keratin 17, whereas EVHCs express only keratin 17.28
Steatocystoma multiplex and EVHC have only rarely been reported to occur together on the trunk. One case of SCM and EVHC occurring on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. Milia, SCM, and EVHC on the face and trunk have been reported in 1 family,4 and facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus was reported in 1 patient.19 Although trichofolliculomas have not been reported to occur with SCM or EVHC, 2 related follicular neoplasms—trichoepitheliomas and trichoblastomas—have been reported to occur in association with SCM on the face and chest and around the ears in 1 case.18
Differential Diagnosis
The clinical differential diagnosis includes multiple epidermoid cysts, dermoid cysts, Gardner syndrome, sebaceous adenomas, Muir-Torre syndrome, syringomas, milia, leiomyomas, lipomas, acneiform folliculitis, multiple familial and nonfamilial trichoepitheliomas, cylindromas, and angiofibromas.3,29
Conclusion
Our patient represents a rare case of simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead. The patient had multiple neoplasms involving differentiation toward various regions of the pilosebaceous unit. This case gives further support to the hypothesis that these benign follicular neoplasms are closely related but are distinct conditions within the spectrum of the same disease process. They represent nevoid malformations of the pilosebaceous unit that can be sporadic or inherited in an autosomal-dominant pattern. Pure types of these lesions may represent one end of the spectrum, but in some patients, there are overlapping features or hybrids of each condition. Several biopsies from patients with multiple lesions should be performed to establish an accurate diagnosis.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Ogawa Y, Nogita T, Kawashima M. The coexistence of eruptive vellus hair cysts and steatocystoma multiplex. J Dermatol. 1992;19:570-571.
- Sanchez Yus E, Requena L. Eruptive vellus hair cyst and steatocystoma multiplex. Am J Dermatopathol. 1990;12:536-537.
- Patrizi A, Neri I, Guerrini V, et al. Persistent milia, steatocystoma multiplex and eruptive vellus hair cysts: variable expression of multiple pilosebaceous cysts within an affected family. Dermatology. 1998;196:392-396.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5, pt 2):876-878.
- Kiene P, Hauschild A, Christophers E. Eruptive vellus hair cysts and steatocystoma multiplex: variants of one entity? Br J Dermatol. 1996;134:365-367.
- Hurlimann AF, Panizzon RG, Burg G. Eruptive vellus hair cyst and steatocystoma multiplex: hybrid cysts. Dermatology. 1996;192:64-66.
- Sexton M, Murdock DK. Eruptive vellus hair cysts: a follicular cyst of the sebaceous duct (sometimes). Am J Dermatopathol. 1989;11:364-368.
- Sanchez-Yus E, Aguilar-Martinez A, Cristobal-Gil MC, et al. Eruptive vellus hair cyst and steatocystoma multiplex: two related conditions? J Cutan Pathol. 1988;15:40-42.
- Ahn SK, Chung J, Lee WS, et al. Hybrid cysts showing alternate combination of eruptive vellus hair cyst, steatocystoma multiplex, and epidermoid cyst, and an association among the three conditions. Am J Dermatopathol. 1996;18:645-649.
- Ahn SK, Hwang SM, Lee SH, et al. Steatocystoma multiplex localized only in the face. Int J Dermatol. 1997;36:372-373.
- Cole LA. Steatocystoma multiplex. Arch Dermatol. 1976;112:1437-1439.
- Hansen KK, Troy JL, Fairley JA. Multiple papules of the scalp and forehead. steatocystoma multiplex (facial papular variant). Arch Dermatol. 1995;131:835-838.
- Nishimura M, Kohda H, Urabe A. Steatocystoma multiplex: a facial popular variant. Arch Dermatol. 1986;122:205-207.
- Requena L, Martin L, Renedo G, et al. A facial variant of steatocystoma multiplex. Cutis. 1993;51:449-452.
- Holmes R, Black MM. Steatocystoma multiplex with unusually prominent cysts on the face. Br J Dermatol. 1980;102:711-713.
- Kumakiri M, Takashima I, Iju M, et al. Eruptive vellus hair cysts: a facial variant. J Am Acad Dermatol. 1982;7:461-467.
- Gianotti R, Cavicchini S, Alessi E. Simultaneous occurrence of multiple trichoblastomas and steatocystoma multiplex. Am J Dermatopathol. 1997;19:294-298.
- Sardana K, Sharma RC, Jain A, et al. Facial steatocystoma multiplex associated with pilar cyst and bilateral preauricular sinus. J Dermatol. 2002;29:157-159.
- Mount LB. Steatocystoma multiplex. Arch Dermatol Syphilol. 1937;36:31-39.
- Dubreuilh W, Auche B. Kystes grassieux sudoripares. Arch Clin de Bordeaux. 1896;5:387-391.
- Pringle JJ. A case of peculiar multiple sebaceous cysts (steatocystoma multiplex). Br J Dermatol. 1899;11:381-88.
- Vineyard WR, Scott RA. Steatocystoma multiplex with pachyonychia congenital: eight cases in four generations. Arch Dermatol. 1961;84:824-827.
- Contreras MA, Costello MJ. Steatocystoma multiplex with embryonal hair formation: case presentation and consideration of pathogenesis. AMA Arch Derm. 1957;76:720-725.
- Sohn D, Chin TC, Fellner MJ. Multiple keratoacanthomas associated with steatocystoma multiplex and rheumatoid arthritis: a case report. Arch Dermatol. 1980;116:913-915.
- Verbov J. Acrokeratosis verruciformis of Hopf with steatocystoma multiplex and hypertrophic lichen planus. Br J Dermatol. 1972;86:91-94.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Tomkova H, Fujimoto W, Arata J. Expression of keratins (K10 and K17) in steatocystoma multiplex, eruptive vellus hair cysts, and epidermoid and trichilemmal cysts. Am J Dermatopathol. 1997;19:250-253.
- Feinstein A, Trau H, Movshovitz M, et al. Steatocystoma multiplex. Cutis. 1983;31:425-427.
An association between steatocystoma multiplex (SCM) and eruptive vellus hair cysts (EVHCs) has been recognized. They are related conditions representing nevoid malformations of the pilosebaceous junctions1-10 that have similar clinical features but distinctive histologic features. Both conditions most commonly involve the anterior aspect of the chest. Six cases of a rare facial variant of SCM have been reported,11-16 3 involving lesions limited to the forehead.13-15 Two patients with a rare facial variant of EVHC also have been reported.17 The development of separate lesions of SCM and EVHC on the trunk can uncommonly occur.5,6,10 One case of SCM and EVHC on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. The simultaneous occurrence of multiple trichoblastomas, trichoepitheliomas, and SCM on the face and trunk has been reported in 1 case.18 Milia, SCM, and EVHC on the face and trunk have been reported in 1 family.4 A report of facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus also has occurred in 1 patient.19 Here, we report the simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead.
Case Report
A 37-year-old man had an increasing number of flesh-colored to yellow papules on the forehead that had been present since puberty. Although the lesions were asymptomatic, some had recently become tender, which led him to seek medical care. There was no history of trauma, burns, irradiation, or application of topical agents to the area or use of eyeglasses or goggles. The patient’s father had similar lesions limited to the forehead, which developed during adolescence.
On evaluation at our clinic, skin examination revealed 16 discrete, 0.3- to 1-cm, flesh-colored, yellow to blue, mobile, smooth papules, as well as flesh-colored papules with a central black punctum, on the forehead (Figure 1). Similar lesions were not present on the rest of the face; around the ears; or on the scalp, neck, chest, back, abdomen, genitalia, buttocks, palms, soles, axillae, arms, or legs. There were no nail abnormalities.
Multiple 3-, 4-, and 6-mm punch and excisional biopsies were performed to remove all 16 lesions on the forehead. Histologic examination revealed a collapsed cystic structure in the mid dermis in 10 lesions. The cysts were lined with a squamous epithelium without a granular layer but with an eosinophilic corrugated lining, and the cyst cavity contained scant homogeneous eosinophilic secretion. Mature sebaceous glands were adjacent to the outer portion of the cyst wall. These histologic findings were consistent with SCM (Figure 2).
In 3 lesions, histologic examination revealed a cystic structure lined by a few layers of stratified squamous epithelium in the mid dermis. The cyst cavity contained numerous small vellus hairs and laminated keratin. These histologic findings were consistent with EVHC (Figure 3).
In the other 3 lesions, histologic examination revealed a dilated central cystic cavity filled with laminated keratin in the mid dermis. Multiple small follicles arose from the cysts and showed differentiation toward germinative epithelium. The surrounding stroma was fibrotic and contained a patchy lymphocytic infiltrate. These histologic findings were consistent with trichofolliculomas (Figure 4).
Comment
Characteristics of SCM
Steatocystoma multiplex is an uncommon condition characterized by the formation of asymptomatic, 0.2- to 2-cm, yellow to flesh-colored, soft, mobile papules or nodules on the trunk, extremities, axillae, genitalia, and/or chest. The lesions contain a clear or opaque, oily, milky or yellow, odorless fluid and most commonly are located on the anterior aspect of the chest. The face is not a commonly involved site in this condition. Six cases of a rare facial variant of SCM have been reported,11-16 with lesions limited to the forehead in 3 cases.13-15
In 1937, Mount20 credited Bozellini for describing the first case, though 3 cases reported in the late 1800s probably were SCM.21 In 1899, Pringle22 coined the term steatocystoma multiplex for this condition. It can be sporadic or have an autosomal-dominant inheritance pattern. Steatocystoma multiplex can occur at any age, though lesions develop most frequently in adolescence or young adulthood. There is no sex predilection.
Steatocystoma multiplex with pachyonychia congenita has been reported in a familial case.23 Other findings reported in patients with SCM include ichthyosis, koilonychia, acrokeratosis verruciformis of Hopf and hypertrophic lichen planus, hidradenitis suppurativa, hypotrichosis, multiple keratoacanthomas, and rheumatoid arthritis.12,24-26
Steatocystoma multiplex is a cyst lined by stratified squamous epithelium without a granular layer but with a thick eosinophilic cuticle. Mature sebaceous lobules are closely associated with the cyst wall. Steatocystoma multiplex arises from the sebaceous duct because the lining of the lumen is composed of undulating eosinophilic cuticle.
Characteristics of EVHCs
Eruptive vellus hair cysts, which were first described by Esterly et al,27 can occur at any age but develop most frequently in adolescents or young adults. Sometimes the lesions are congenital or appear in childhood. There is no sex predilection. They can be sporadic or have an autosomal-dominant inheritance pattern.
Eruptive vellus hair cysts are asymptomatic, 1- to 2-mm, smooth, crusted, or umbilicated papules on the chest or arms and legs. Eruptive vellus hair cysts most commonly involve the anterior aspect of the chest. The lesions are flesh-colored to yellow, though they have a slate gray color in darker-skinned individuals. A rare facial variant has been reported in 2 patients of Asian descent.17
Eruptive vellus hair cysts are small cystic structures lined by a stratified squamous epithelium with a granular layer. The cyst cavity contains numerous small vellus hair shafts and laminated keratin. Eruptive vellus hair cysts originate from the infundibulum or less frequently the isthmus or infundibular-isthmic junction of the hair follicle.
Characteristics of Trichofolliculomas
Trichofolliculomas are solitary, 3- to 5-mm, flesh-colored papules that occur on the face. They are highly differentiated, benign, neoplastic proliferations of an actively trichogenic epithelium, with structural components reflecting all portions of the pilosebaceous unit. Trichofolliculomas consist of a central dilated primary follicle contiguous with the surface epidermis embedded in a fibrous stroma. Multiple small secondary follicles with varying degrees of follicular differentiation arise from the primary follicle.
Co-occurrence of Lesions
An association between SCM and EVHC has been recognized.5-10 Steatocystoma multiplex and EVHC have similar clinical features but distinctive histologic features. They also have a similar age of onset, location/appearance of lesions, and mode of inheritance. Steatocystoma multiplex and EVHC can be distinguished by immunohistochemical techniques: SCM shows expression of keratin 10 and keratin 17, whereas EVHCs express only keratin 17.28
Steatocystoma multiplex and EVHC have only rarely been reported to occur together on the trunk. One case of SCM and EVHC occurring on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. Milia, SCM, and EVHC on the face and trunk have been reported in 1 family,4 and facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus was reported in 1 patient.19 Although trichofolliculomas have not been reported to occur with SCM or EVHC, 2 related follicular neoplasms—trichoepitheliomas and trichoblastomas—have been reported to occur in association with SCM on the face and chest and around the ears in 1 case.18
Differential Diagnosis
The clinical differential diagnosis includes multiple epidermoid cysts, dermoid cysts, Gardner syndrome, sebaceous adenomas, Muir-Torre syndrome, syringomas, milia, leiomyomas, lipomas, acneiform folliculitis, multiple familial and nonfamilial trichoepitheliomas, cylindromas, and angiofibromas.3,29
Conclusion
Our patient represents a rare case of simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead. The patient had multiple neoplasms involving differentiation toward various regions of the pilosebaceous unit. This case gives further support to the hypothesis that these benign follicular neoplasms are closely related but are distinct conditions within the spectrum of the same disease process. They represent nevoid malformations of the pilosebaceous unit that can be sporadic or inherited in an autosomal-dominant pattern. Pure types of these lesions may represent one end of the spectrum, but in some patients, there are overlapping features or hybrids of each condition. Several biopsies from patients with multiple lesions should be performed to establish an accurate diagnosis.
An association between steatocystoma multiplex (SCM) and eruptive vellus hair cysts (EVHCs) has been recognized. They are related conditions representing nevoid malformations of the pilosebaceous junctions1-10 that have similar clinical features but distinctive histologic features. Both conditions most commonly involve the anterior aspect of the chest. Six cases of a rare facial variant of SCM have been reported,11-16 3 involving lesions limited to the forehead.13-15 Two patients with a rare facial variant of EVHC also have been reported.17 The development of separate lesions of SCM and EVHC on the trunk can uncommonly occur.5,6,10 One case of SCM and EVHC on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. The simultaneous occurrence of multiple trichoblastomas, trichoepitheliomas, and SCM on the face and trunk has been reported in 1 case.18 Milia, SCM, and EVHC on the face and trunk have been reported in 1 family.4 A report of facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus also has occurred in 1 patient.19 Here, we report the simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead.
Case Report
A 37-year-old man had an increasing number of flesh-colored to yellow papules on the forehead that had been present since puberty. Although the lesions were asymptomatic, some had recently become tender, which led him to seek medical care. There was no history of trauma, burns, irradiation, or application of topical agents to the area or use of eyeglasses or goggles. The patient’s father had similar lesions limited to the forehead, which developed during adolescence.
On evaluation at our clinic, skin examination revealed 16 discrete, 0.3- to 1-cm, flesh-colored, yellow to blue, mobile, smooth papules, as well as flesh-colored papules with a central black punctum, on the forehead (Figure 1). Similar lesions were not present on the rest of the face; around the ears; or on the scalp, neck, chest, back, abdomen, genitalia, buttocks, palms, soles, axillae, arms, or legs. There were no nail abnormalities.
Multiple 3-, 4-, and 6-mm punch and excisional biopsies were performed to remove all 16 lesions on the forehead. Histologic examination revealed a collapsed cystic structure in the mid dermis in 10 lesions. The cysts were lined with a squamous epithelium without a granular layer but with an eosinophilic corrugated lining, and the cyst cavity contained scant homogeneous eosinophilic secretion. Mature sebaceous glands were adjacent to the outer portion of the cyst wall. These histologic findings were consistent with SCM (Figure 2).
In 3 lesions, histologic examination revealed a cystic structure lined by a few layers of stratified squamous epithelium in the mid dermis. The cyst cavity contained numerous small vellus hairs and laminated keratin. These histologic findings were consistent with EVHC (Figure 3).
In the other 3 lesions, histologic examination revealed a dilated central cystic cavity filled with laminated keratin in the mid dermis. Multiple small follicles arose from the cysts and showed differentiation toward germinative epithelium. The surrounding stroma was fibrotic and contained a patchy lymphocytic infiltrate. These histologic findings were consistent with trichofolliculomas (Figure 4).
Comment
Characteristics of SCM
Steatocystoma multiplex is an uncommon condition characterized by the formation of asymptomatic, 0.2- to 2-cm, yellow to flesh-colored, soft, mobile papules or nodules on the trunk, extremities, axillae, genitalia, and/or chest. The lesions contain a clear or opaque, oily, milky or yellow, odorless fluid and most commonly are located on the anterior aspect of the chest. The face is not a commonly involved site in this condition. Six cases of a rare facial variant of SCM have been reported,11-16 with lesions limited to the forehead in 3 cases.13-15
In 1937, Mount20 credited Bozellini for describing the first case, though 3 cases reported in the late 1800s probably were SCM.21 In 1899, Pringle22 coined the term steatocystoma multiplex for this condition. It can be sporadic or have an autosomal-dominant inheritance pattern. Steatocystoma multiplex can occur at any age, though lesions develop most frequently in adolescence or young adulthood. There is no sex predilection.
Steatocystoma multiplex with pachyonychia congenita has been reported in a familial case.23 Other findings reported in patients with SCM include ichthyosis, koilonychia, acrokeratosis verruciformis of Hopf and hypertrophic lichen planus, hidradenitis suppurativa, hypotrichosis, multiple keratoacanthomas, and rheumatoid arthritis.12,24-26
Steatocystoma multiplex is a cyst lined by stratified squamous epithelium without a granular layer but with a thick eosinophilic cuticle. Mature sebaceous lobules are closely associated with the cyst wall. Steatocystoma multiplex arises from the sebaceous duct because the lining of the lumen is composed of undulating eosinophilic cuticle.
Characteristics of EVHCs
Eruptive vellus hair cysts, which were first described by Esterly et al,27 can occur at any age but develop most frequently in adolescents or young adults. Sometimes the lesions are congenital or appear in childhood. There is no sex predilection. They can be sporadic or have an autosomal-dominant inheritance pattern.
Eruptive vellus hair cysts are asymptomatic, 1- to 2-mm, smooth, crusted, or umbilicated papules on the chest or arms and legs. Eruptive vellus hair cysts most commonly involve the anterior aspect of the chest. The lesions are flesh-colored to yellow, though they have a slate gray color in darker-skinned individuals. A rare facial variant has been reported in 2 patients of Asian descent.17
Eruptive vellus hair cysts are small cystic structures lined by a stratified squamous epithelium with a granular layer. The cyst cavity contains numerous small vellus hair shafts and laminated keratin. Eruptive vellus hair cysts originate from the infundibulum or less frequently the isthmus or infundibular-isthmic junction of the hair follicle.
Characteristics of Trichofolliculomas
Trichofolliculomas are solitary, 3- to 5-mm, flesh-colored papules that occur on the face. They are highly differentiated, benign, neoplastic proliferations of an actively trichogenic epithelium, with structural components reflecting all portions of the pilosebaceous unit. Trichofolliculomas consist of a central dilated primary follicle contiguous with the surface epidermis embedded in a fibrous stroma. Multiple small secondary follicles with varying degrees of follicular differentiation arise from the primary follicle.
Co-occurrence of Lesions
An association between SCM and EVHC has been recognized.5-10 Steatocystoma multiplex and EVHC have similar clinical features but distinctive histologic features. They also have a similar age of onset, location/appearance of lesions, and mode of inheritance. Steatocystoma multiplex and EVHC can be distinguished by immunohistochemical techniques: SCM shows expression of keratin 10 and keratin 17, whereas EVHCs express only keratin 17.28
Steatocystoma multiplex and EVHC have only rarely been reported to occur together on the trunk. One case of SCM and EVHC occurring on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. Milia, SCM, and EVHC on the face and trunk have been reported in 1 family,4 and facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus was reported in 1 patient.19 Although trichofolliculomas have not been reported to occur with SCM or EVHC, 2 related follicular neoplasms—trichoepitheliomas and trichoblastomas—have been reported to occur in association with SCM on the face and chest and around the ears in 1 case.18
Differential Diagnosis
The clinical differential diagnosis includes multiple epidermoid cysts, dermoid cysts, Gardner syndrome, sebaceous adenomas, Muir-Torre syndrome, syringomas, milia, leiomyomas, lipomas, acneiform folliculitis, multiple familial and nonfamilial trichoepitheliomas, cylindromas, and angiofibromas.3,29
Conclusion
Our patient represents a rare case of simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead. The patient had multiple neoplasms involving differentiation toward various regions of the pilosebaceous unit. This case gives further support to the hypothesis that these benign follicular neoplasms are closely related but are distinct conditions within the spectrum of the same disease process. They represent nevoid malformations of the pilosebaceous unit that can be sporadic or inherited in an autosomal-dominant pattern. Pure types of these lesions may represent one end of the spectrum, but in some patients, there are overlapping features or hybrids of each condition. Several biopsies from patients with multiple lesions should be performed to establish an accurate diagnosis.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Ogawa Y, Nogita T, Kawashima M. The coexistence of eruptive vellus hair cysts and steatocystoma multiplex. J Dermatol. 1992;19:570-571.
- Sanchez Yus E, Requena L. Eruptive vellus hair cyst and steatocystoma multiplex. Am J Dermatopathol. 1990;12:536-537.
- Patrizi A, Neri I, Guerrini V, et al. Persistent milia, steatocystoma multiplex and eruptive vellus hair cysts: variable expression of multiple pilosebaceous cysts within an affected family. Dermatology. 1998;196:392-396.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5, pt 2):876-878.
- Kiene P, Hauschild A, Christophers E. Eruptive vellus hair cysts and steatocystoma multiplex: variants of one entity? Br J Dermatol. 1996;134:365-367.
- Hurlimann AF, Panizzon RG, Burg G. Eruptive vellus hair cyst and steatocystoma multiplex: hybrid cysts. Dermatology. 1996;192:64-66.
- Sexton M, Murdock DK. Eruptive vellus hair cysts: a follicular cyst of the sebaceous duct (sometimes). Am J Dermatopathol. 1989;11:364-368.
- Sanchez-Yus E, Aguilar-Martinez A, Cristobal-Gil MC, et al. Eruptive vellus hair cyst and steatocystoma multiplex: two related conditions? J Cutan Pathol. 1988;15:40-42.
- Ahn SK, Chung J, Lee WS, et al. Hybrid cysts showing alternate combination of eruptive vellus hair cyst, steatocystoma multiplex, and epidermoid cyst, and an association among the three conditions. Am J Dermatopathol. 1996;18:645-649.
- Ahn SK, Hwang SM, Lee SH, et al. Steatocystoma multiplex localized only in the face. Int J Dermatol. 1997;36:372-373.
- Cole LA. Steatocystoma multiplex. Arch Dermatol. 1976;112:1437-1439.
- Hansen KK, Troy JL, Fairley JA. Multiple papules of the scalp and forehead. steatocystoma multiplex (facial papular variant). Arch Dermatol. 1995;131:835-838.
- Nishimura M, Kohda H, Urabe A. Steatocystoma multiplex: a facial popular variant. Arch Dermatol. 1986;122:205-207.
- Requena L, Martin L, Renedo G, et al. A facial variant of steatocystoma multiplex. Cutis. 1993;51:449-452.
- Holmes R, Black MM. Steatocystoma multiplex with unusually prominent cysts on the face. Br J Dermatol. 1980;102:711-713.
- Kumakiri M, Takashima I, Iju M, et al. Eruptive vellus hair cysts: a facial variant. J Am Acad Dermatol. 1982;7:461-467.
- Gianotti R, Cavicchini S, Alessi E. Simultaneous occurrence of multiple trichoblastomas and steatocystoma multiplex. Am J Dermatopathol. 1997;19:294-298.
- Sardana K, Sharma RC, Jain A, et al. Facial steatocystoma multiplex associated with pilar cyst and bilateral preauricular sinus. J Dermatol. 2002;29:157-159.
- Mount LB. Steatocystoma multiplex. Arch Dermatol Syphilol. 1937;36:31-39.
- Dubreuilh W, Auche B. Kystes grassieux sudoripares. Arch Clin de Bordeaux. 1896;5:387-391.
- Pringle JJ. A case of peculiar multiple sebaceous cysts (steatocystoma multiplex). Br J Dermatol. 1899;11:381-88.
- Vineyard WR, Scott RA. Steatocystoma multiplex with pachyonychia congenital: eight cases in four generations. Arch Dermatol. 1961;84:824-827.
- Contreras MA, Costello MJ. Steatocystoma multiplex with embryonal hair formation: case presentation and consideration of pathogenesis. AMA Arch Derm. 1957;76:720-725.
- Sohn D, Chin TC, Fellner MJ. Multiple keratoacanthomas associated with steatocystoma multiplex and rheumatoid arthritis: a case report. Arch Dermatol. 1980;116:913-915.
- Verbov J. Acrokeratosis verruciformis of Hopf with steatocystoma multiplex and hypertrophic lichen planus. Br J Dermatol. 1972;86:91-94.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Tomkova H, Fujimoto W, Arata J. Expression of keratins (K10 and K17) in steatocystoma multiplex, eruptive vellus hair cysts, and epidermoid and trichilemmal cysts. Am J Dermatopathol. 1997;19:250-253.
- Feinstein A, Trau H, Movshovitz M, et al. Steatocystoma multiplex. Cutis. 1983;31:425-427.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Ogawa Y, Nogita T, Kawashima M. The coexistence of eruptive vellus hair cysts and steatocystoma multiplex. J Dermatol. 1992;19:570-571.
- Sanchez Yus E, Requena L. Eruptive vellus hair cyst and steatocystoma multiplex. Am J Dermatopathol. 1990;12:536-537.
- Patrizi A, Neri I, Guerrini V, et al. Persistent milia, steatocystoma multiplex and eruptive vellus hair cysts: variable expression of multiple pilosebaceous cysts within an affected family. Dermatology. 1998;196:392-396.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5, pt 2):876-878.
- Kiene P, Hauschild A, Christophers E. Eruptive vellus hair cysts and steatocystoma multiplex: variants of one entity? Br J Dermatol. 1996;134:365-367.
- Hurlimann AF, Panizzon RG, Burg G. Eruptive vellus hair cyst and steatocystoma multiplex: hybrid cysts. Dermatology. 1996;192:64-66.
- Sexton M, Murdock DK. Eruptive vellus hair cysts: a follicular cyst of the sebaceous duct (sometimes). Am J Dermatopathol. 1989;11:364-368.
- Sanchez-Yus E, Aguilar-Martinez A, Cristobal-Gil MC, et al. Eruptive vellus hair cyst and steatocystoma multiplex: two related conditions? J Cutan Pathol. 1988;15:40-42.
- Ahn SK, Chung J, Lee WS, et al. Hybrid cysts showing alternate combination of eruptive vellus hair cyst, steatocystoma multiplex, and epidermoid cyst, and an association among the three conditions. Am J Dermatopathol. 1996;18:645-649.
- Ahn SK, Hwang SM, Lee SH, et al. Steatocystoma multiplex localized only in the face. Int J Dermatol. 1997;36:372-373.
- Cole LA. Steatocystoma multiplex. Arch Dermatol. 1976;112:1437-1439.
- Hansen KK, Troy JL, Fairley JA. Multiple papules of the scalp and forehead. steatocystoma multiplex (facial papular variant). Arch Dermatol. 1995;131:835-838.
- Nishimura M, Kohda H, Urabe A. Steatocystoma multiplex: a facial popular variant. Arch Dermatol. 1986;122:205-207.
- Requena L, Martin L, Renedo G, et al. A facial variant of steatocystoma multiplex. Cutis. 1993;51:449-452.
- Holmes R, Black MM. Steatocystoma multiplex with unusually prominent cysts on the face. Br J Dermatol. 1980;102:711-713.
- Kumakiri M, Takashima I, Iju M, et al. Eruptive vellus hair cysts: a facial variant. J Am Acad Dermatol. 1982;7:461-467.
- Gianotti R, Cavicchini S, Alessi E. Simultaneous occurrence of multiple trichoblastomas and steatocystoma multiplex. Am J Dermatopathol. 1997;19:294-298.
- Sardana K, Sharma RC, Jain A, et al. Facial steatocystoma multiplex associated with pilar cyst and bilateral preauricular sinus. J Dermatol. 2002;29:157-159.
- Mount LB. Steatocystoma multiplex. Arch Dermatol Syphilol. 1937;36:31-39.
- Dubreuilh W, Auche B. Kystes grassieux sudoripares. Arch Clin de Bordeaux. 1896;5:387-391.
- Pringle JJ. A case of peculiar multiple sebaceous cysts (steatocystoma multiplex). Br J Dermatol. 1899;11:381-88.
- Vineyard WR, Scott RA. Steatocystoma multiplex with pachyonychia congenital: eight cases in four generations. Arch Dermatol. 1961;84:824-827.
- Contreras MA, Costello MJ. Steatocystoma multiplex with embryonal hair formation: case presentation and consideration of pathogenesis. AMA Arch Derm. 1957;76:720-725.
- Sohn D, Chin TC, Fellner MJ. Multiple keratoacanthomas associated with steatocystoma multiplex and rheumatoid arthritis: a case report. Arch Dermatol. 1980;116:913-915.
- Verbov J. Acrokeratosis verruciformis of Hopf with steatocystoma multiplex and hypertrophic lichen planus. Br J Dermatol. 1972;86:91-94.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Tomkova H, Fujimoto W, Arata J. Expression of keratins (K10 and K17) in steatocystoma multiplex, eruptive vellus hair cysts, and epidermoid and trichilemmal cysts. Am J Dermatopathol. 1997;19:250-253.
- Feinstein A, Trau H, Movshovitz M, et al. Steatocystoma multiplex. Cutis. 1983;31:425-427.
Practice Points
- Steatocystoma multiplex (SCM) and eruptive vellus hair cysts (EVHCs) have similar clinical features but distinctive histologic features.
- Milia, pilar cyst, trichoepitheliomas, and trichoblastomas simultaneously developing in association with SCM or EVHC on the face are rare.
- This case supports the hypothesis that these benign follicular neoplasms are related but distinct nevoid malformations of the pilosebaceous unit within the same disease spectrum.
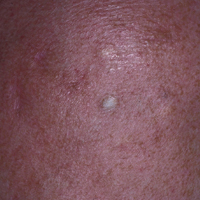
Evaluating the Clinical and Demographic Features of Extrafacial Granuloma Faciale
Granuloma faciale (GF) is a chronic benign leukocytoclastic vasculitis that can be difficult to treat. It is characterized by single or multiple, soft, well-circumscribed papules, plaques, or nodules ranging in color from red, violet, or yellow to brown that may darken with sun exposure.1 Lesions usually are smooth with follicular orifices that are accentuated, thus producing a peau d’orange appearance. Lesions generally are slow to develop and asymptomatic, though some patients report pruritus or burning.2,3 Diagnosis of GF is based on the presence of distinct histologic features. The epidermis usually is spared, with a prominent grenz zone of normal collagen separating the epidermis from a dense infiltrate of neutrophils, lymphocytes, and eosinophils. This mixed inflammatory infiltrate is seen mainly in the superficial dermis but occasionally spreads to the lower dermis and subcutaneous tissues.4
As the name implies, GF usually is confined to the face but occasionally involves extrafacial sites.5-15 The clinical characteristics of these rare extrafacial lesions are not well understood. The purpose of this study was to identify the clinical and demographic features of extrafacial GF in patients treated at Mayo Clinic (Rochester, Minnesota) during a 54-year period.
Methods
This study was approved by the Mayo institutional review board. We searched the Mayo Clinic Rochester dermatology database for all patients with a diagnosis of GF from 1959 through 2013. All histopathology slides were reviewed by a board-certified dermatologist (A.G.B.) and dermatopathologist (A.G.B.) before inclusion in this study. Histologic criteria for diagnosis of GF included the presence of a mixed inflammatory infiltrate of neutrophils, eosinophils, lymphocytes, and histiocytes in the superficial or deep dermis; a prominent grenz zone separating the uninvolved epidermis; and the presence of vascular damage, as seen by fibrin deposition in dermal blood vessels.
Medical records were reviewed for patient demographics and for history pertinent to the diagnosis of GF, including sites involved, appearance, histopathology reports, symptoms, treatments, and outcomes.
Literature Search Strategy
A computerized Ovid MEDLINE database search was undertaken to identify English-language articles concerning GF in humans using the search terms granuloma faciale with extrafacial or disseminated. To ensure that no articles were overlooked, we conducted another search for English-language articles in the Embase database (1946-2013) using the terms granuloma faciale and extrafacial or disseminated.
Statistical Analysis
Descriptive clinical and histopathologic data were summarized using means, medians, and ranges or proportions as appropriate; statistical analysis was performed using SAS software (JMP package).
Results
Ninety-six patients with a diagnosis of GF were identified, and 12 (13%) had a diagnosis of extrafacial GF. Of them, 2 patients had a diagnosis of extrafacial GF supported only by histopathology slides without accompanying clinical records and therefore were excluded from the study. Thus, 10 cases of extrafacial GF were identified from our search and were included in the study group. Clinical data for these patients are summarized in Table 1. The mean age was 58.7 years (range, 26–87 years). Six (60%) patients were male, and all patients were white. Seven patients (70%) had facial GF in addition to extrafacial GF. Six patients reported no symptoms (60%), and 4 (40%) reported pruritus, discomfort, or both associated with their GF lesions.

Extrafacial GF was diagnosed in the following anatomic locations: scalp (n=3 [30%]), posterior auricular area (n=3 [30%]), mid upper back (n=1 [10%]), right shoulder (n=1 [10%]), both ears (n=1 [10%]), right elbow (n=1 [10%]), and left infra-auricular area (n=1 [10%]). Only 1 (10%) patient had multiple extrafacial sites identified.
The lesions were characterized clinically as violet, red, and yellow to brown smooth papules, plaques, and nodules (Figure 1). Biopsies from these lesions showed a subepidermal and adnexal grenz zone; a polymorphous perivascular and periadnexal dermal infiltrate composed of neutrophils, eosinophils, lymphocytes, histiocytes, and plasma cells; and a mild subtle leukocytoclastic vasculitis with subtle mild vascular necrosis (Figure 2).
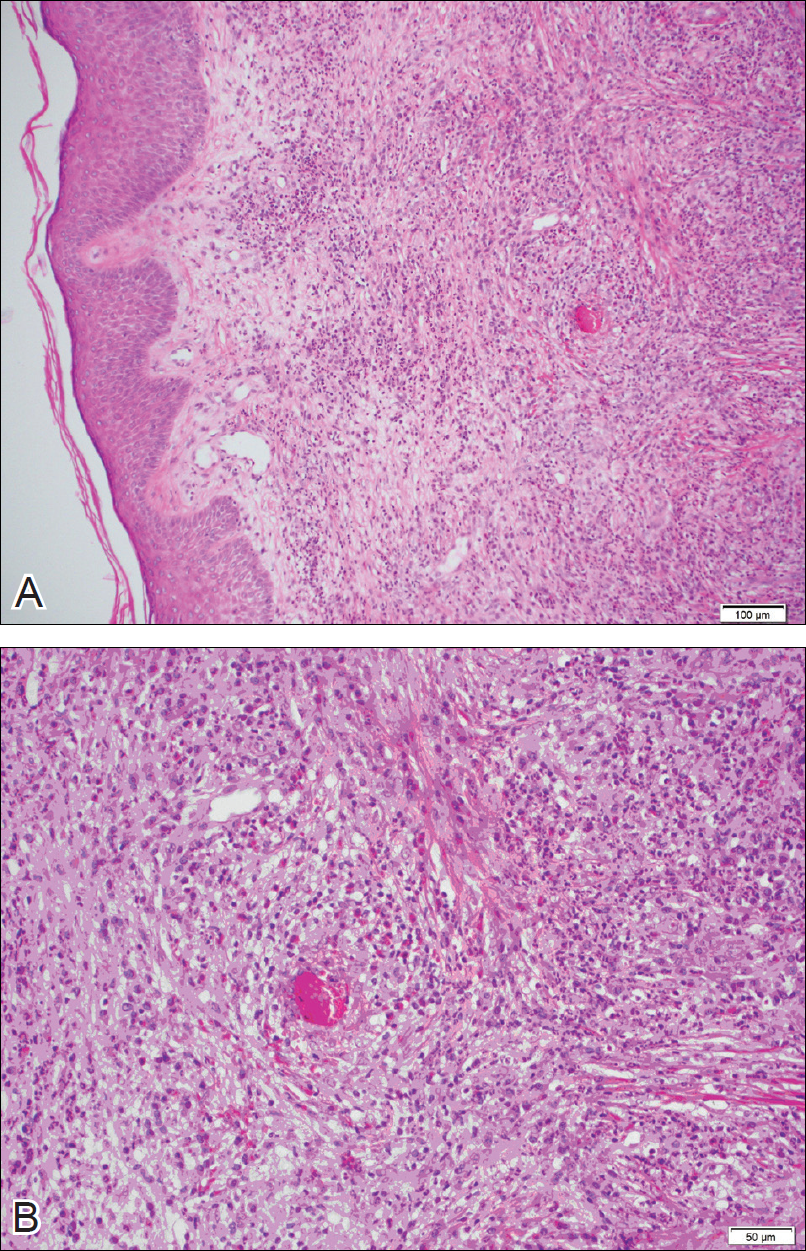
For the 9 patients who elected to undergo GF treatment, the average number of treatments attempted was 2.8 (range, 1–5). The most common method of treatment was a combination of intralesional and topical corticosteroids (n=5 [50%]). Other methods included surgery (n=3 [30%]), dapsone (n=2 [20%]), radiation therapy (n=2 [20%]), cryosurgery (n=1 [10%]), nitrogen mustard (n=1 [10%]), liquid nitrogen (n=1 [10%]), and tar shampoo and fluocinolone acetonide solution 0.01% (n=1 [10%]).
Treatment outcomes were available for 8 of 9 treated patients. Three patients (patients 7, 8, and 10) had long-term successful resolution of their lesions. Patient 7 had an extrafacial lesion that was successfully treated with intralesional and topical corticosteroids, but the facial lesions recurred. The extrafacial GF lesion in patient 8 was found adjacent to a squamous cell carcinoma and was removed with a wide surgical excision that included both lesions. Patient 10 was successfully treated with a combination of liquid nitrogen and topical corticosteroid. Patients 2 and 4 were well controlled while on dapsone; however, once the treatment was discontinued, primarily due to adverse effects, the lesions returned.
Literature Search
Our search of the English-language literature identified 20 patients with extrafacial GF (Table 2). Fifteen (75%) patients were male, which was similar to our study (6/10 [60%]). Our patient population was slightly older with a mean age of 58.7 years compared to a median age of 54 years among those identified in the literature. Additionally, 3 (30%) patients in our study had no facial lesions, as seen in classic GF, which is comparable to 8 (40%) patients identified in the literature.
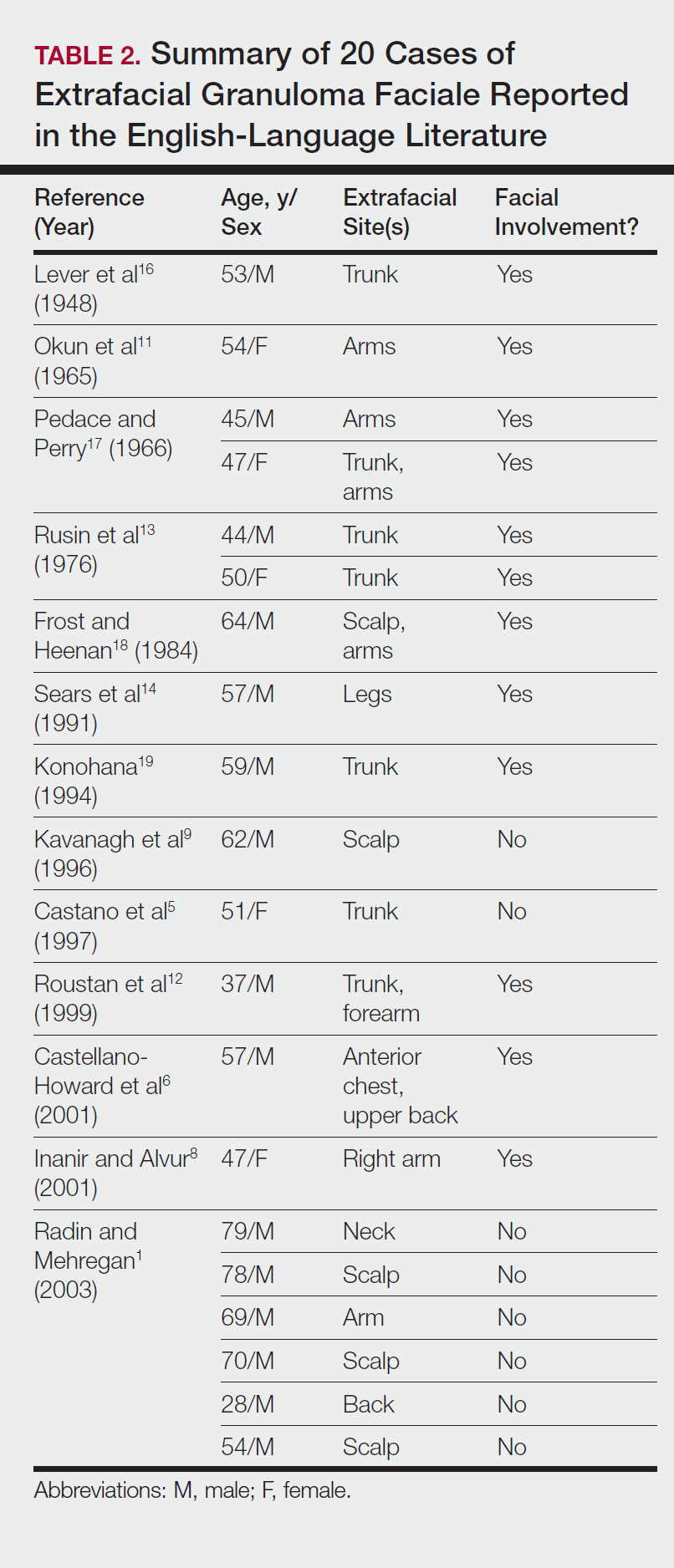
Comment
Extrafacial GF primarily affects white individuals and is more prevalent in men, as demonstrated in our study. Extrafacial GF was most often found in association with facial lesions, with only 3 patients having exclusively extrafacial sites.
Data from the current study indicate that diverse modalities were used to treat extrafacial GF with variable outcomes (chronic recurrence to complete resolution). The most common first-line treatment, intralesional corticosteroid injection, was used in 5 (50%) patients but resulted in only 1 (10%) successful resolution. Other methods frequently used in our study and prior studies were surgical excision, cryotherapy, electrosurgery, and dermabrasion.1,20 These treatments do not appear to be uniformly definitive, and the ablative methods may result in scarring.1 Different laser treatments are emerging for the management of GF lesions. Prior reports of treating facial GF with argon and CO2 lasers have indicated minimized residual scarring and pigmentation.21-23 The use of pulsed dye lasers has resulted in complete clearance of facial GF lesions, without recurrence on long-term follow-up.20,24-26
The latest investigations of immunomodulatory drugs indicate these agents are promising for the management of facial GF. Eetam et al27 reported the successful use of topical tacrolimus to treat facial GF. The relatively low cost and ease of use make these topical medications a competitive alternative to currently available surgical and laser methods. The appearance of all of these novel therapeutic modalities creates the necessity for a randomized trial to establish their efficacy on extrafacial GF lesions.
The wide array of treatments reflects the recalcitrant nature of extrafacial GF lesions. Further insight into the etiology of these lesions is needed to understand their tendency to recur. The important contribution of our study is the observed
Conclusion
The findings from this study and the cases reviewed in the literature provide a unique contribution to the understanding of the clinical and demographic characteristics of extrafacial GF. The rarity of this condition is the single most important constraint of our study, reflected in the emblematic limitations of a retrospective analysis in a select group of patients. The results of analysis of data from our patients were similar to the findings reported in the English-language medical literature. Serious consideration should be given to the development of a national registry for patients with GF. A database containing the clinicopathologic features, treatments, and outcomes for patients with both facial and extrafacial manifestations of GF may be invaluable in evaluating various treatment options and increasing understanding of the etiology and epidemiology of the disease.
- Radin DA, Mehregan DR. Granuloma faciale: distribution of the lesions and review of the literature. Cutis. 2003;72:213-219.
- Dowlati B, Firooz A, Dowlati Y. Granuloma faciale: successful treatment of nine cases with a combination of cryotherapy and intralesional corticosteroid injection. Int J Dermatol. 1997;36:548-551.
- Guill MA, Aton JK. Facial granuloma responsive to dapsone therapy. Arch Dermatol. 1982;118:332-335.
- Ryan TJ. Cutaneous vasculitis. In: Champion RH, Burton JL, Burns DA, et al, eds. Rook/Wilkins/Ebling Textbook of Dermatology. 7th ed. Malden, MA: Blackwell Science; 2004.
- Castano E, Segurado A, Iglesias L, et al. Granuloma faciale entirely in an extrafacial location. Br J Dermatol. 1997;136:978-979.
- Castellano-Howard L, Fairbee SI, Hogan DJ, et al. Extrafacial granuloma faciale: report of a case and response to treatment. Cutis. 2001;67:413-415.
- Cecchi R, Paoli S, Giomi A. Granuloma faciale with extrafacial lesions. Eur J Dermatol. 2002;12:438.
- Inanir I, Alvur Y. Granuloma faciale with extrafacial lesions. Br J Dermatol. 2001;14:360-362.
- Kavanagh GM, McLaren KM, Hunter JA. Extensive extrafacial granuloma faciale of the scalp. Br J Dermatol. 1996;134:595-596.
- Marcoval J, Moreno A, Peyr J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Okun MR, Bauman L, Minor D. Granuloma faciale with lesions on the face and hand. Arch Dermatol. 1965;92:78-80.
- Roustan G, Sanchez Yus E, Salas C, et al. Granuloma faciale with extrafacial lesions. Dermatology. 1999;198:79-82.
- Rusin LJ, Dubin HV, Taylor WB. Disseminated granuloma faciale. Arch Dermatol. 1976;112:1575-1577.
- Sears JK, Gitter DG, Stone MS. Extrafacial granuloma faciale. Arch Dermatol. 1991;127:742-743.
- Zargari O. Disseminated granuloma faciale. Int J Dermatol. 2004;43:210-212.
- Lever WF, Lane CG, Downing JG, et al. Eosinophilic granuloma of the skin: report of three cases. Arch Derm Syphilol. 1948;58:430-438.
- Pedace FJ, Perry HO. Granuloma faciale: a clinical and histopathologic review. Arch Dermatol. 1966;94:387-395.
- Frost FA, Heenan PJ. Facial granuloma. Australas J Dermatol. 1984;25:121-124.
Konohana A. Extrafacial granuloma faciale. J Dermatol. 1994;21:680-682.- Ludwig E, Allam JP, Bieber T, et al. New treatment modalities for granuloma faciale. Br J Dermatol. 2003;149:634-637.
- Apfelberg DB, Druker D, Maser MR, et al. Granuloma faciale: treatment with the argon laser. Arch Dermatol. 1983;119:573-576.
- Apfelberg DB, Maser MR, Lash H, et al. Expanded role of the argon laser in plastic surgery. J Dermatol Surg Oncol. 1983;9:145-151.
- Wheeland RG, Ashley JR, Smith DA, et al. Carbon dioxide laser treatment of granuloma faciale. J Dermatol Surg Oncol. 1984;10:730-733.
- Cheung ST, Lanigan SW. Granuloma faciale treated with the pulsed-dye laser: a case series. Clin Exp Dermatol. 2005;30:373-375.
- Chatrath V, Rohrer TE. Granuloma faciale successfully treated with long-pulsed tunable dye laser. Dermatol Surg. 2002;28:527-529.
- Elston DM. Treatment of granuloma faciale with the pulsed dye laser. Cutis. 2000;65:97-98.
- Eetam I, Ertekin B, Unal I, et al. Granuloma faciale: is it a new indication for pimecrolimus? a case report. J Dermatolog Treat. 2006;17:238-240.
- Johnson WC, Higdon RS, Helwig EB. Granuloma faciale. AMA Arch Derm. 1959;79:42-52.
Granuloma faciale (GF) is a chronic benign leukocytoclastic vasculitis that can be difficult to treat. It is characterized by single or multiple, soft, well-circumscribed papules, plaques, or nodules ranging in color from red, violet, or yellow to brown that may darken with sun exposure.1 Lesions usually are smooth with follicular orifices that are accentuated, thus producing a peau d’orange appearance. Lesions generally are slow to develop and asymptomatic, though some patients report pruritus or burning.2,3 Diagnosis of GF is based on the presence of distinct histologic features. The epidermis usually is spared, with a prominent grenz zone of normal collagen separating the epidermis from a dense infiltrate of neutrophils, lymphocytes, and eosinophils. This mixed inflammatory infiltrate is seen mainly in the superficial dermis but occasionally spreads to the lower dermis and subcutaneous tissues.4
As the name implies, GF usually is confined to the face but occasionally involves extrafacial sites.5-15 The clinical characteristics of these rare extrafacial lesions are not well understood. The purpose of this study was to identify the clinical and demographic features of extrafacial GF in patients treated at Mayo Clinic (Rochester, Minnesota) during a 54-year period.
Methods
This study was approved by the Mayo institutional review board. We searched the Mayo Clinic Rochester dermatology database for all patients with a diagnosis of GF from 1959 through 2013. All histopathology slides were reviewed by a board-certified dermatologist (A.G.B.) and dermatopathologist (A.G.B.) before inclusion in this study. Histologic criteria for diagnosis of GF included the presence of a mixed inflammatory infiltrate of neutrophils, eosinophils, lymphocytes, and histiocytes in the superficial or deep dermis; a prominent grenz zone separating the uninvolved epidermis; and the presence of vascular damage, as seen by fibrin deposition in dermal blood vessels.
Medical records were reviewed for patient demographics and for history pertinent to the diagnosis of GF, including sites involved, appearance, histopathology reports, symptoms, treatments, and outcomes.
Literature Search Strategy
A computerized Ovid MEDLINE database search was undertaken to identify English-language articles concerning GF in humans using the search terms granuloma faciale with extrafacial or disseminated. To ensure that no articles were overlooked, we conducted another search for English-language articles in the Embase database (1946-2013) using the terms granuloma faciale and extrafacial or disseminated.
Statistical Analysis
Descriptive clinical and histopathologic data were summarized using means, medians, and ranges or proportions as appropriate; statistical analysis was performed using SAS software (JMP package).
Results
Ninety-six patients with a diagnosis of GF were identified, and 12 (13%) had a diagnosis of extrafacial GF. Of them, 2 patients had a diagnosis of extrafacial GF supported only by histopathology slides without accompanying clinical records and therefore were excluded from the study. Thus, 10 cases of extrafacial GF were identified from our search and were included in the study group. Clinical data for these patients are summarized in Table 1. The mean age was 58.7 years (range, 26–87 years). Six (60%) patients were male, and all patients were white. Seven patients (70%) had facial GF in addition to extrafacial GF. Six patients reported no symptoms (60%), and 4 (40%) reported pruritus, discomfort, or both associated with their GF lesions.
Extrafacial GF was diagnosed in the following anatomic locations: scalp (n=3 [30%]), posterior auricular area (n=3 [30%]), mid upper back (n=1 [10%]), right shoulder (n=1 [10%]), both ears (n=1 [10%]), right elbow (n=1 [10%]), and left infra-auricular area (n=1 [10%]). Only 1 (10%) patient had multiple extrafacial sites identified.
The lesions were characterized clinically as violet, red, and yellow to brown smooth papules, plaques, and nodules (Figure 1). Biopsies from these lesions showed a subepidermal and adnexal grenz zone; a polymorphous perivascular and periadnexal dermal infiltrate composed of neutrophils, eosinophils, lymphocytes, histiocytes, and plasma cells; and a mild subtle leukocytoclastic vasculitis with subtle mild vascular necrosis (Figure 2).
For the 9 patients who elected to undergo GF treatment, the average number of treatments attempted was 2.8 (range, 1–5). The most common method of treatment was a combination of intralesional and topical corticosteroids (n=5 [50%]). Other methods included surgery (n=3 [30%]), dapsone (n=2 [20%]), radiation therapy (n=2 [20%]), cryosurgery (n=1 [10%]), nitrogen mustard (n=1 [10%]), liquid nitrogen (n=1 [10%]), and tar shampoo and fluocinolone acetonide solution 0.01% (n=1 [10%]).
Treatment outcomes were available for 8 of 9 treated patients. Three patients (patients 7, 8, and 10) had long-term successful resolution of their lesions. Patient 7 had an extrafacial lesion that was successfully treated with intralesional and topical corticosteroids, but the facial lesions recurred. The extrafacial GF lesion in patient 8 was found adjacent to a squamous cell carcinoma and was removed with a wide surgical excision that included both lesions. Patient 10 was successfully treated with a combination of liquid nitrogen and topical corticosteroid. Patients 2 and 4 were well controlled while on dapsone; however, once the treatment was discontinued, primarily due to adverse effects, the lesions returned.
Literature Search
Our search of the English-language literature identified 20 patients with extrafacial GF (Table 2). Fifteen (75%) patients were male, which was similar to our study (6/10 [60%]). Our patient population was slightly older with a mean age of 58.7 years compared to a median age of 54 years among those identified in the literature. Additionally, 3 (30%) patients in our study had no facial lesions, as seen in classic GF, which is comparable to 8 (40%) patients identified in the literature.
Comment
Extrafacial GF primarily affects white individuals and is more prevalent in men, as demonstrated in our study. Extrafacial GF was most often found in association with facial lesions, with only 3 patients having exclusively extrafacial sites.
Data from the current study indicate that diverse modalities were used to treat extrafacial GF with variable outcomes (chronic recurrence to complete resolution). The most common first-line treatment, intralesional corticosteroid injection, was used in 5 (50%) patients but resulted in only 1 (10%) successful resolution. Other methods frequently used in our study and prior studies were surgical excision, cryotherapy, electrosurgery, and dermabrasion.1,20 These treatments do not appear to be uniformly definitive, and the ablative methods may result in scarring.1 Different laser treatments are emerging for the management of GF lesions. Prior reports of treating facial GF with argon and CO2 lasers have indicated minimized residual scarring and pigmentation.21-23 The use of pulsed dye lasers has resulted in complete clearance of facial GF lesions, without recurrence on long-term follow-up.20,24-26
The latest investigations of immunomodulatory drugs indicate these agents are promising for the management of facial GF. Eetam et al27 reported the successful use of topical tacrolimus to treat facial GF. The relatively low cost and ease of use make these topical medications a competitive alternative to currently available surgical and laser methods. The appearance of all of these novel therapeutic modalities creates the necessity for a randomized trial to establish their efficacy on extrafacial GF lesions.
The wide array of treatments reflects the recalcitrant nature of extrafacial GF lesions. Further insight into the etiology of these lesions is needed to understand their tendency to recur. The important contribution of our study is the observed
Conclusion
The findings from this study and the cases reviewed in the literature provide a unique contribution to the understanding of the clinical and demographic characteristics of extrafacial GF. The rarity of this condition is the single most important constraint of our study, reflected in the emblematic limitations of a retrospective analysis in a select group of patients. The results of analysis of data from our patients were similar to the findings reported in the English-language medical literature. Serious consideration should be given to the development of a national registry for patients with GF. A database containing the clinicopathologic features, treatments, and outcomes for patients with both facial and extrafacial manifestations of GF may be invaluable in evaluating various treatment options and increasing understanding of the etiology and epidemiology of the disease.
Granuloma faciale (GF) is a chronic benign leukocytoclastic vasculitis that can be difficult to treat. It is characterized by single or multiple, soft, well-circumscribed papules, plaques, or nodules ranging in color from red, violet, or yellow to brown that may darken with sun exposure.1 Lesions usually are smooth with follicular orifices that are accentuated, thus producing a peau d’orange appearance. Lesions generally are slow to develop and asymptomatic, though some patients report pruritus or burning.2,3 Diagnosis of GF is based on the presence of distinct histologic features. The epidermis usually is spared, with a prominent grenz zone of normal collagen separating the epidermis from a dense infiltrate of neutrophils, lymphocytes, and eosinophils. This mixed inflammatory infiltrate is seen mainly in the superficial dermis but occasionally spreads to the lower dermis and subcutaneous tissues.4
As the name implies, GF usually is confined to the face but occasionally involves extrafacial sites.5-15 The clinical characteristics of these rare extrafacial lesions are not well understood. The purpose of this study was to identify the clinical and demographic features of extrafacial GF in patients treated at Mayo Clinic (Rochester, Minnesota) during a 54-year period.
Methods
This study was approved by the Mayo institutional review board. We searched the Mayo Clinic Rochester dermatology database for all patients with a diagnosis of GF from 1959 through 2013. All histopathology slides were reviewed by a board-certified dermatologist (A.G.B.) and dermatopathologist (A.G.B.) before inclusion in this study. Histologic criteria for diagnosis of GF included the presence of a mixed inflammatory infiltrate of neutrophils, eosinophils, lymphocytes, and histiocytes in the superficial or deep dermis; a prominent grenz zone separating the uninvolved epidermis; and the presence of vascular damage, as seen by fibrin deposition in dermal blood vessels.
Medical records were reviewed for patient demographics and for history pertinent to the diagnosis of GF, including sites involved, appearance, histopathology reports, symptoms, treatments, and outcomes.
Literature Search Strategy
A computerized Ovid MEDLINE database search was undertaken to identify English-language articles concerning GF in humans using the search terms granuloma faciale with extrafacial or disseminated. To ensure that no articles were overlooked, we conducted another search for English-language articles in the Embase database (1946-2013) using the terms granuloma faciale and extrafacial or disseminated.
Statistical Analysis
Descriptive clinical and histopathologic data were summarized using means, medians, and ranges or proportions as appropriate; statistical analysis was performed using SAS software (JMP package).
Results
Ninety-six patients with a diagnosis of GF were identified, and 12 (13%) had a diagnosis of extrafacial GF. Of them, 2 patients had a diagnosis of extrafacial GF supported only by histopathology slides without accompanying clinical records and therefore were excluded from the study. Thus, 10 cases of extrafacial GF were identified from our search and were included in the study group. Clinical data for these patients are summarized in Table 1. The mean age was 58.7 years (range, 26–87 years). Six (60%) patients were male, and all patients were white. Seven patients (70%) had facial GF in addition to extrafacial GF. Six patients reported no symptoms (60%), and 4 (40%) reported pruritus, discomfort, or both associated with their GF lesions.
Extrafacial GF was diagnosed in the following anatomic locations: scalp (n=3 [30%]), posterior auricular area (n=3 [30%]), mid upper back (n=1 [10%]), right shoulder (n=1 [10%]), both ears (n=1 [10%]), right elbow (n=1 [10%]), and left infra-auricular area (n=1 [10%]). Only 1 (10%) patient had multiple extrafacial sites identified.
The lesions were characterized clinically as violet, red, and yellow to brown smooth papules, plaques, and nodules (Figure 1). Biopsies from these lesions showed a subepidermal and adnexal grenz zone; a polymorphous perivascular and periadnexal dermal infiltrate composed of neutrophils, eosinophils, lymphocytes, histiocytes, and plasma cells; and a mild subtle leukocytoclastic vasculitis with subtle mild vascular necrosis (Figure 2).
For the 9 patients who elected to undergo GF treatment, the average number of treatments attempted was 2.8 (range, 1–5). The most common method of treatment was a combination of intralesional and topical corticosteroids (n=5 [50%]). Other methods included surgery (n=3 [30%]), dapsone (n=2 [20%]), radiation therapy (n=2 [20%]), cryosurgery (n=1 [10%]), nitrogen mustard (n=1 [10%]), liquid nitrogen (n=1 [10%]), and tar shampoo and fluocinolone acetonide solution 0.01% (n=1 [10%]).
Treatment outcomes were available for 8 of 9 treated patients. Three patients (patients 7, 8, and 10) had long-term successful resolution of their lesions. Patient 7 had an extrafacial lesion that was successfully treated with intralesional and topical corticosteroids, but the facial lesions recurred. The extrafacial GF lesion in patient 8 was found adjacent to a squamous cell carcinoma and was removed with a wide surgical excision that included both lesions. Patient 10 was successfully treated with a combination of liquid nitrogen and topical corticosteroid. Patients 2 and 4 were well controlled while on dapsone; however, once the treatment was discontinued, primarily due to adverse effects, the lesions returned.
Literature Search
Our search of the English-language literature identified 20 patients with extrafacial GF (Table 2). Fifteen (75%) patients were male, which was similar to our study (6/10 [60%]). Our patient population was slightly older with a mean age of 58.7 years compared to a median age of 54 years among those identified in the literature. Additionally, 3 (30%) patients in our study had no facial lesions, as seen in classic GF, which is comparable to 8 (40%) patients identified in the literature.
Comment
Extrafacial GF primarily affects white individuals and is more prevalent in men, as demonstrated in our study. Extrafacial GF was most often found in association with facial lesions, with only 3 patients having exclusively extrafacial sites.
Data from the current study indicate that diverse modalities were used to treat extrafacial GF with variable outcomes (chronic recurrence to complete resolution). The most common first-line treatment, intralesional corticosteroid injection, was used in 5 (50%) patients but resulted in only 1 (10%) successful resolution. Other methods frequently used in our study and prior studies were surgical excision, cryotherapy, electrosurgery, and dermabrasion.1,20 These treatments do not appear to be uniformly definitive, and the ablative methods may result in scarring.1 Different laser treatments are emerging for the management of GF lesions. Prior reports of treating facial GF with argon and CO2 lasers have indicated minimized residual scarring and pigmentation.21-23 The use of pulsed dye lasers has resulted in complete clearance of facial GF lesions, without recurrence on long-term follow-up.20,24-26
The latest investigations of immunomodulatory drugs indicate these agents are promising for the management of facial GF. Eetam et al27 reported the successful use of topical tacrolimus to treat facial GF. The relatively low cost and ease of use make these topical medications a competitive alternative to currently available surgical and laser methods. The appearance of all of these novel therapeutic modalities creates the necessity for a randomized trial to establish their efficacy on extrafacial GF lesions.
The wide array of treatments reflects the recalcitrant nature of extrafacial GF lesions. Further insight into the etiology of these lesions is needed to understand their tendency to recur. The important contribution of our study is the observed
Conclusion
The findings from this study and the cases reviewed in the literature provide a unique contribution to the understanding of the clinical and demographic characteristics of extrafacial GF. The rarity of this condition is the single most important constraint of our study, reflected in the emblematic limitations of a retrospective analysis in a select group of patients. The results of analysis of data from our patients were similar to the findings reported in the English-language medical literature. Serious consideration should be given to the development of a national registry for patients with GF. A database containing the clinicopathologic features, treatments, and outcomes for patients with both facial and extrafacial manifestations of GF may be invaluable in evaluating various treatment options and increasing understanding of the etiology and epidemiology of the disease.
- Radin DA, Mehregan DR. Granuloma faciale: distribution of the lesions and review of the literature. Cutis. 2003;72:213-219.
- Dowlati B, Firooz A, Dowlati Y. Granuloma faciale: successful treatment of nine cases with a combination of cryotherapy and intralesional corticosteroid injection. Int J Dermatol. 1997;36:548-551.
- Guill MA, Aton JK. Facial granuloma responsive to dapsone therapy. Arch Dermatol. 1982;118:332-335.
- Ryan TJ. Cutaneous vasculitis. In: Champion RH, Burton JL, Burns DA, et al, eds. Rook/Wilkins/Ebling Textbook of Dermatology. 7th ed. Malden, MA: Blackwell Science; 2004.
- Castano E, Segurado A, Iglesias L, et al. Granuloma faciale entirely in an extrafacial location. Br J Dermatol. 1997;136:978-979.
- Castellano-Howard L, Fairbee SI, Hogan DJ, et al. Extrafacial granuloma faciale: report of a case and response to treatment. Cutis. 2001;67:413-415.
- Cecchi R, Paoli S, Giomi A. Granuloma faciale with extrafacial lesions. Eur J Dermatol. 2002;12:438.
- Inanir I, Alvur Y. Granuloma faciale with extrafacial lesions. Br J Dermatol. 2001;14:360-362.
- Kavanagh GM, McLaren KM, Hunter JA. Extensive extrafacial granuloma faciale of the scalp. Br J Dermatol. 1996;134:595-596.
- Marcoval J, Moreno A, Peyr J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Okun MR, Bauman L, Minor D. Granuloma faciale with lesions on the face and hand. Arch Dermatol. 1965;92:78-80.
- Roustan G, Sanchez Yus E, Salas C, et al. Granuloma faciale with extrafacial lesions. Dermatology. 1999;198:79-82.
- Rusin LJ, Dubin HV, Taylor WB. Disseminated granuloma faciale. Arch Dermatol. 1976;112:1575-1577.
- Sears JK, Gitter DG, Stone MS. Extrafacial granuloma faciale. Arch Dermatol. 1991;127:742-743.
- Zargari O. Disseminated granuloma faciale. Int J Dermatol. 2004;43:210-212.
- Lever WF, Lane CG, Downing JG, et al. Eosinophilic granuloma of the skin: report of three cases. Arch Derm Syphilol. 1948;58:430-438.
- Pedace FJ, Perry HO. Granuloma faciale: a clinical and histopathologic review. Arch Dermatol. 1966;94:387-395.
- Frost FA, Heenan PJ. Facial granuloma. Australas J Dermatol. 1984;25:121-124.
Konohana A. Extrafacial granuloma faciale. J Dermatol. 1994;21:680-682.- Ludwig E, Allam JP, Bieber T, et al. New treatment modalities for granuloma faciale. Br J Dermatol. 2003;149:634-637.
- Apfelberg DB, Druker D, Maser MR, et al. Granuloma faciale: treatment with the argon laser. Arch Dermatol. 1983;119:573-576.
- Apfelberg DB, Maser MR, Lash H, et al. Expanded role of the argon laser in plastic surgery. J Dermatol Surg Oncol. 1983;9:145-151.
- Wheeland RG, Ashley JR, Smith DA, et al. Carbon dioxide laser treatment of granuloma faciale. J Dermatol Surg Oncol. 1984;10:730-733.
- Cheung ST, Lanigan SW. Granuloma faciale treated with the pulsed-dye laser: a case series. Clin Exp Dermatol. 2005;30:373-375.
- Chatrath V, Rohrer TE. Granuloma faciale successfully treated with long-pulsed tunable dye laser. Dermatol Surg. 2002;28:527-529.
- Elston DM. Treatment of granuloma faciale with the pulsed dye laser. Cutis. 2000;65:97-98.
- Eetam I, Ertekin B, Unal I, et al. Granuloma faciale: is it a new indication for pimecrolimus? a case report. J Dermatolog Treat. 2006;17:238-240.
- Johnson WC, Higdon RS, Helwig EB. Granuloma faciale. AMA Arch Derm. 1959;79:42-52.
- Radin DA, Mehregan DR. Granuloma faciale: distribution of the lesions and review of the literature. Cutis. 2003;72:213-219.
- Dowlati B, Firooz A, Dowlati Y. Granuloma faciale: successful treatment of nine cases with a combination of cryotherapy and intralesional corticosteroid injection. Int J Dermatol. 1997;36:548-551.
- Guill MA, Aton JK. Facial granuloma responsive to dapsone therapy. Arch Dermatol. 1982;118:332-335.
- Ryan TJ. Cutaneous vasculitis. In: Champion RH, Burton JL, Burns DA, et al, eds. Rook/Wilkins/Ebling Textbook of Dermatology. 7th ed. Malden, MA: Blackwell Science; 2004.
- Castano E, Segurado A, Iglesias L, et al. Granuloma faciale entirely in an extrafacial location. Br J Dermatol. 1997;136:978-979.
- Castellano-Howard L, Fairbee SI, Hogan DJ, et al. Extrafacial granuloma faciale: report of a case and response to treatment. Cutis. 2001;67:413-415.
- Cecchi R, Paoli S, Giomi A. Granuloma faciale with extrafacial lesions. Eur J Dermatol. 2002;12:438.
- Inanir I, Alvur Y. Granuloma faciale with extrafacial lesions. Br J Dermatol. 2001;14:360-362.
- Kavanagh GM, McLaren KM, Hunter JA. Extensive extrafacial granuloma faciale of the scalp. Br J Dermatol. 1996;134:595-596.
- Marcoval J, Moreno A, Peyr J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Okun MR, Bauman L, Minor D. Granuloma faciale with lesions on the face and hand. Arch Dermatol. 1965;92:78-80.
- Roustan G, Sanchez Yus E, Salas C, et al. Granuloma faciale with extrafacial lesions. Dermatology. 1999;198:79-82.
- Rusin LJ, Dubin HV, Taylor WB. Disseminated granuloma faciale. Arch Dermatol. 1976;112:1575-1577.
- Sears JK, Gitter DG, Stone MS. Extrafacial granuloma faciale. Arch Dermatol. 1991;127:742-743.
- Zargari O. Disseminated granuloma faciale. Int J Dermatol. 2004;43:210-212.
- Lever WF, Lane CG, Downing JG, et al. Eosinophilic granuloma of the skin: report of three cases. Arch Derm Syphilol. 1948;58:430-438.
- Pedace FJ, Perry HO. Granuloma faciale: a clinical and histopathologic review. Arch Dermatol. 1966;94:387-395.
- Frost FA, Heenan PJ. Facial granuloma. Australas J Dermatol. 1984;25:121-124.
Konohana A. Extrafacial granuloma faciale. J Dermatol. 1994;21:680-682.- Ludwig E, Allam JP, Bieber T, et al. New treatment modalities for granuloma faciale. Br J Dermatol. 2003;149:634-637.
- Apfelberg DB, Druker D, Maser MR, et al. Granuloma faciale: treatment with the argon laser. Arch Dermatol. 1983;119:573-576.
- Apfelberg DB, Maser MR, Lash H, et al. Expanded role of the argon laser in plastic surgery. J Dermatol Surg Oncol. 1983;9:145-151.
- Wheeland RG, Ashley JR, Smith DA, et al. Carbon dioxide laser treatment of granuloma faciale. J Dermatol Surg Oncol. 1984;10:730-733.
- Cheung ST, Lanigan SW. Granuloma faciale treated with the pulsed-dye laser: a case series. Clin Exp Dermatol. 2005;30:373-375.
- Chatrath V, Rohrer TE. Granuloma faciale successfully treated with long-pulsed tunable dye laser. Dermatol Surg. 2002;28:527-529.
- Elston DM. Treatment of granuloma faciale with the pulsed dye laser. Cutis. 2000;65:97-98.
- Eetam I, Ertekin B, Unal I, et al. Granuloma faciale: is it a new indication for pimecrolimus? a case report. J Dermatolog Treat. 2006;17:238-240.
- Johnson WC, Higdon RS, Helwig EB. Granuloma faciale. AMA Arch Derm. 1959;79:42-52.
Practice Points
- Extrafacial lesions are rare in granuloma faciale (GF).
- Extrafacial GF should be included in the differential diagnosis of well-demarcated plaques and nodules found on the trunk or extremities.
- Diagnosis of extrafacial GF is based on the presence of distinct histologic features identical to GF.
- Granuloma faciale is a chronic benign leukocytoclastic vasculitis that can be difficult to treat.
Rowell Syndrome: Targeting a True Definition
Case Report
A 37-year-old woman was admitted to the intensive care unit secondary to the acute development of an erythematous rash with tissue sloughing that involved acral sites and mucosal surfaces. Her medical history was notable for anti-Ro/Sjögren syndrome antigen A (SS-A)–positive lupus erythematosus (LE) with a morphologic semblance to subacute cutaneous LE (SCLE). Prior treatment had included oral corticosteroids. In addition, she reported a concurrent history of acral and mucosal lesions that appeared to flare with her lupus. The nature of these lesions was not clear to the patient or her physicians. Before this particular episode, her primary care physician had attempted to wean her off of the corticosteroids. As she dropped below 20 mg of prednisone daily, new lesions developed. The patient stated that her social situation was poor and that these lesions did seem to develop more frequently during times of physical and emotional stress. She recounted her first episode developing during her second pregnancy. Oral prednisone and over-the-counter calcium with vitamin D were her only reported medications. She denied the use of any other medications, including nonsteroidal anti-inflammatory drugs, acetaminophen, and recent antibiotic therapy.
Dermatology was called in for consultation, and physical examination revealed areas of epidermal sloughing on the hands and feet. Complete clinical exposure of the underlying dermis was noted with remarkable tenderness. These lesions were noted to be in various stages of healing (Figure 1). Figure 2 displays a lesion in early development. The mucosal surfaces of the lips and eyes demonstrated hemorrhagic crusting, and some tissue sloughing was noted on the ears. A widespread erythematous exanthema with fine scaling was noted on the face, neck, chest, back, abdomen, arms, and legs (Figure 3).
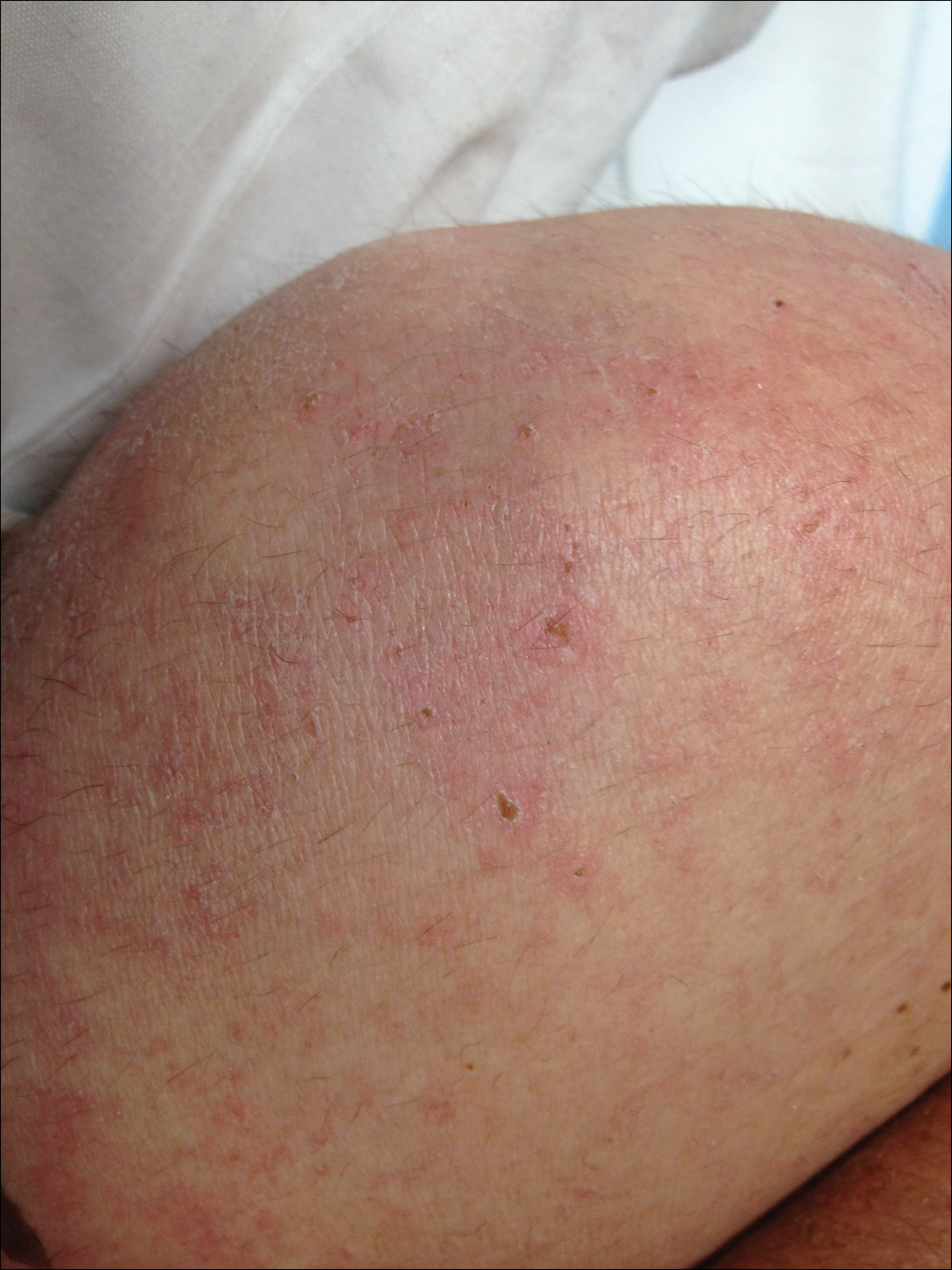
Laboratory evaluation revealed positive antinuclear antibodies (ANAs), anti-Ro/SS-A antibodies, anti-La/Sjögren syndrome antigen B (SS-B) antibodies, and anti–double-stranded DNA. The hemoglobin level was 9.4 g/dL (reference range, 12–15 g/dL) and hematocrit was 28.8% (reference range, 36%–47%). The mean corpuscular hemoglobin level was 32 pg/cell (reference range, 27–31 pg/cell), and the mean corpuscular hemoglobin concentration was 32.5 g/dL (reference range, 30–35 g/dL). Rheumatoid factor (RF) and herpes simplex virus types 1 and 2 IgM were all found to be negative.
A deep shave biopsy obtained from the patient’s right knee revealed an atrophic interface dermatitis associated with a lymphocytic eccrine hidradenitis accompanied by abundant mesenchymal mucin deposition (Figure 4). Direct immunofluorescence (DIF) from the same area demonstrated IgG and IgM along the dermoepidermal junction with some granular deposition. Frozen sections performed on acral lesions demonstrated epidermal necrosis (Figure 5). Direct immunofluorescence of acral lesions was negative. In light of these findings, a diagnosis of Rowell syndrome (RS) was suspected to be the most likely explanation for the presentation.
Intravenous corticosteroids and antibiotics were administered, and over a 2-week hospitalization, the lesions on the feet and hands slowly reepithelialized. Physical therapy was required to aid in ambulation. The patient was discharged on a tapering course of oral prednisone and hydroxychloroquine. After 6 months of therapy with hydroxychloroquine 200 mg twice daily, the patient continued to experience recurrent bouts of acral lesions, and pulse doses of oral prednisone were required. The lesions currently are controlled with azathioprine 50 mg twice daily and prednisone 10 mg by mouth daily.

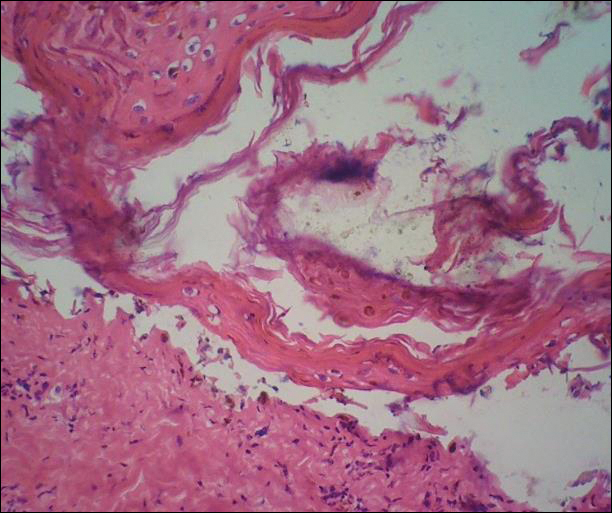
Comment
The 4 prototypical patients identified by Rowell et al1 in 1963 in the first account of the eponymous syndrome were all females with discoid lupus erythematosus (DLE) and perniosis. In addition, they all displayed positive RF and saline extract of human tissue antibodies (analogous to anti-Ro/SS-A and anti-La/SS-B).2 Since then, at least 132 patients with clinical symptoms suspicious of RS have been identified with variations on these original criteria.3 The reported permutations of the lupus component of the disease include cutaneous LE (CLE), bullous systemic LE, necrotic lesions associated with antiphospholipid syndrome, annular/polycyclic SCLE, systemic LE (SLE) without CLE, SLE with lupus nephritis, SLE with pericarditis, SLE with systemic vasculitis, Sjögren syndrome, rheumatoid arthritis, and necrotizing lymphadenitis.2 In addition, variations of the erythema multiforme (EM)–like lesions found in reported cases include changes to their gross appearance (flat vs raised), location (acral or mucosal involvement), and resemblance to other conditions (Stevens-Johnson syndrome or toxic epidermal necrolysis).2,3 From this information alone, it is clear that, as further cases have been chronicled, defining exact criteria for the disease has been challenging.
The essential question concerning the existence of RS hinges on the strength of its distinctiveness: Is it a unique disorder or merely another variant of lupus? Antiga et al2 concluded that it should be characterized as a variant of SCLE. Lee at al4 agreed, stating that “[i]n view of the lack of specific features that distinguish RS from LE, Kuhn et al5 suggested that [RS] is probably not a distinct entity and is now widely considered to be a variant of SCLE.” One of the primary contributors to this conclusion is that the laboratory findings of reported patients with SCLE have more closely mirrored the original cases from Rowell et al’s1 report than those of typical LE. Patients with SCLE have demonstrated positive ANA antibodies in 60% to 80% of cases, positive anti-Ro/SS-A antibodies in 40% to 100% of cases, positive anti-La/SS-B antibodies in 12% to 42% of cases, positive anti–double-stranded DNA in 1.2% to 10% of cases, and positive RF antibodies in 33% of cases.2 An argument could certainly be made to ascribe our patient’s condition to an SCLE variant, as 4 of 5 preceding laboratory findings were found to be positive; however, the majority of reported cases of SCLE have been linked to drugs (ie, hydrochlorothiazide, angiotensin-converting enzyme inhibitors, calcium channel blockers, terbinafine),2 which has not commonly been the attributable etiology of other cases of RS, including the 4 cases reported by Rowell et al.1
In a review of the literature on RS since 2010 in addition to their report of 132 new cases, Torchia et al3 outlined a set of diagnostic standards for the condition consisting of major and minor criteria. According to the authors, if all 4 major and 1 minor criteria are met, the patient meets the standards for true RS. The major criteria include the following: (1) presence of chronic CLE [DLE and/or chilblain]; (2) presence of EM-like lesions [typical or atypical targets]; (3) at least 1 positivity among speckled ANA, anti-Ro/SS-A, and anti-La/SS-B antibodies; and (4) negative DIF on lesional EM-like targetoid lesions. The minor criteria include the following: (1) absence of infectious or pharmacologic triggers; (2) absence of typical EM location (acral and mucosal); and (3) presence of at least 1 additional American College of Rheumatology criterion for diagnosis of SLE8 besides discoid rash and positive ANA antibodies and excluding photosensitivity, malar rash, and oral ulcers. Using these criteria, the patient in our case met the standards for diagnosis of RS.
One area of disagreement that has been encountered in the literature is the exact histologic determination of true RS, specifically related to the microscopic findings of the EM-like lesions. Two cases presented by Modi et al6 were interpreted under the stipulation that true RS must contain histologic LE and histologic EM. Because the EM-appearing lesions revealed LE histology, the cases were concluded to be variants of LE. These cases are similar to our case in that the EM-like lesions in our patient demonstrated LE pathology. Torchia et al,3 as demonstrated in the above criteria, seemed to be less concerned about the histology of the EM-like lesions, only requiring them to show negative DIF.
Conclusion
In the search for answers concerning RS, many unanswered questions remain: Where should the line be drawn in the inclusion of so many variations of both the LE and EM components of the condition? Also, should these elements even be approached as distinct components in the first place? Viewing the majority of RS cases as simply simultaneous LE and EM, Shteyngarts et al7 concluded that “the concomitant occurrence of EM with LE did not change the course, therapy, or prognosis of either disease. SLE and DLE can coexist with EM, but the coexistence does not impart any unusual characteristic to either illness. Rowell’s syndrome is not reproducible, and the immunologic disturbances in such patients are probably coincidental.”
If the condition is a genuine pathological individuality, should we not view the seemingly separate LE and EM as the product of a single underlying biochemical process? These questions and others in the search for a true definition of the disease should continue to be debated. It is clear that further investigation is warranted in the understanding of the underlying mechanism of the pathology.
- Rowell NR, Beck JS, Anderson JR. Lupus erythematosus and erythema multiforme-like lesions: a syndrome with characteristic immunological abnormalities. Arch Dermatol. 1963;88:176-180.
- Antiga E, Caproni M, Bonciani D, et al. The last word on the so-called ‘Rowell’s syndrome’? Lupus. 2012;21:577-585.
- Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67:417-421.
- Lee A, Batra P, Furer V, et al. Rowell syndrome (systemic lupus erythematosus + erythema multiforme). Dermatol Online J. 2009;15:1.
- Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1140.
- Modi GM, Shen A, Mazloom A, et al. Lupus erythematosus masquerading as erythema multiforme: does Rowell syndrome really exist? Dermatol Online J. 2009;15:5.
- Shteyngarts AR, Warner MR, Camisa C. Lupus erythematosus associated with erythema multiforme: does Rowell’s syndrome exist? J Am Acad Dermatol. 1999;40(5 pt 1):773-777.
- Lupus diagnosis. Lupus Research Alliance website. http://lupusresearchinstitute.org/lupus-facts/lupus-diagnosis. Accessed July 11, 2017.
Case Report
A 37-year-old woman was admitted to the intensive care unit secondary to the acute development of an erythematous rash with tissue sloughing that involved acral sites and mucosal surfaces. Her medical history was notable for anti-Ro/Sjögren syndrome antigen A (SS-A)–positive lupus erythematosus (LE) with a morphologic semblance to subacute cutaneous LE (SCLE). Prior treatment had included oral corticosteroids. In addition, she reported a concurrent history of acral and mucosal lesions that appeared to flare with her lupus. The nature of these lesions was not clear to the patient or her physicians. Before this particular episode, her primary care physician had attempted to wean her off of the corticosteroids. As she dropped below 20 mg of prednisone daily, new lesions developed. The patient stated that her social situation was poor and that these lesions did seem to develop more frequently during times of physical and emotional stress. She recounted her first episode developing during her second pregnancy. Oral prednisone and over-the-counter calcium with vitamin D were her only reported medications. She denied the use of any other medications, including nonsteroidal anti-inflammatory drugs, acetaminophen, and recent antibiotic therapy.
Dermatology was called in for consultation, and physical examination revealed areas of epidermal sloughing on the hands and feet. Complete clinical exposure of the underlying dermis was noted with remarkable tenderness. These lesions were noted to be in various stages of healing (Figure 1). Figure 2 displays a lesion in early development. The mucosal surfaces of the lips and eyes demonstrated hemorrhagic crusting, and some tissue sloughing was noted on the ears. A widespread erythematous exanthema with fine scaling was noted on the face, neck, chest, back, abdomen, arms, and legs (Figure 3).
Laboratory evaluation revealed positive antinuclear antibodies (ANAs), anti-Ro/SS-A antibodies, anti-La/Sjögren syndrome antigen B (SS-B) antibodies, and anti–double-stranded DNA. The hemoglobin level was 9.4 g/dL (reference range, 12–15 g/dL) and hematocrit was 28.8% (reference range, 36%–47%). The mean corpuscular hemoglobin level was 32 pg/cell (reference range, 27–31 pg/cell), and the mean corpuscular hemoglobin concentration was 32.5 g/dL (reference range, 30–35 g/dL). Rheumatoid factor (RF) and herpes simplex virus types 1 and 2 IgM were all found to be negative.
A deep shave biopsy obtained from the patient’s right knee revealed an atrophic interface dermatitis associated with a lymphocytic eccrine hidradenitis accompanied by abundant mesenchymal mucin deposition (Figure 4). Direct immunofluorescence (DIF) from the same area demonstrated IgG and IgM along the dermoepidermal junction with some granular deposition. Frozen sections performed on acral lesions demonstrated epidermal necrosis (Figure 5). Direct immunofluorescence of acral lesions was negative. In light of these findings, a diagnosis of Rowell syndrome (RS) was suspected to be the most likely explanation for the presentation.
Intravenous corticosteroids and antibiotics were administered, and over a 2-week hospitalization, the lesions on the feet and hands slowly reepithelialized. Physical therapy was required to aid in ambulation. The patient was discharged on a tapering course of oral prednisone and hydroxychloroquine. After 6 months of therapy with hydroxychloroquine 200 mg twice daily, the patient continued to experience recurrent bouts of acral lesions, and pulse doses of oral prednisone were required. The lesions currently are controlled with azathioprine 50 mg twice daily and prednisone 10 mg by mouth daily.
Comment
The 4 prototypical patients identified by Rowell et al1 in 1963 in the first account of the eponymous syndrome were all females with discoid lupus erythematosus (DLE) and perniosis. In addition, they all displayed positive RF and saline extract of human tissue antibodies (analogous to anti-Ro/SS-A and anti-La/SS-B).2 Since then, at least 132 patients with clinical symptoms suspicious of RS have been identified with variations on these original criteria.3 The reported permutations of the lupus component of the disease include cutaneous LE (CLE), bullous systemic LE, necrotic lesions associated with antiphospholipid syndrome, annular/polycyclic SCLE, systemic LE (SLE) without CLE, SLE with lupus nephritis, SLE with pericarditis, SLE with systemic vasculitis, Sjögren syndrome, rheumatoid arthritis, and necrotizing lymphadenitis.2 In addition, variations of the erythema multiforme (EM)–like lesions found in reported cases include changes to their gross appearance (flat vs raised), location (acral or mucosal involvement), and resemblance to other conditions (Stevens-Johnson syndrome or toxic epidermal necrolysis).2,3 From this information alone, it is clear that, as further cases have been chronicled, defining exact criteria for the disease has been challenging.
The essential question concerning the existence of RS hinges on the strength of its distinctiveness: Is it a unique disorder or merely another variant of lupus? Antiga et al2 concluded that it should be characterized as a variant of SCLE. Lee at al4 agreed, stating that “[i]n view of the lack of specific features that distinguish RS from LE, Kuhn et al5 suggested that [RS] is probably not a distinct entity and is now widely considered to be a variant of SCLE.” One of the primary contributors to this conclusion is that the laboratory findings of reported patients with SCLE have more closely mirrored the original cases from Rowell et al’s1 report than those of typical LE. Patients with SCLE have demonstrated positive ANA antibodies in 60% to 80% of cases, positive anti-Ro/SS-A antibodies in 40% to 100% of cases, positive anti-La/SS-B antibodies in 12% to 42% of cases, positive anti–double-stranded DNA in 1.2% to 10% of cases, and positive RF antibodies in 33% of cases.2 An argument could certainly be made to ascribe our patient’s condition to an SCLE variant, as 4 of 5 preceding laboratory findings were found to be positive; however, the majority of reported cases of SCLE have been linked to drugs (ie, hydrochlorothiazide, angiotensin-converting enzyme inhibitors, calcium channel blockers, terbinafine),2 which has not commonly been the attributable etiology of other cases of RS, including the 4 cases reported by Rowell et al.1
In a review of the literature on RS since 2010 in addition to their report of 132 new cases, Torchia et al3 outlined a set of diagnostic standards for the condition consisting of major and minor criteria. According to the authors, if all 4 major and 1 minor criteria are met, the patient meets the standards for true RS. The major criteria include the following: (1) presence of chronic CLE [DLE and/or chilblain]; (2) presence of EM-like lesions [typical or atypical targets]; (3) at least 1 positivity among speckled ANA, anti-Ro/SS-A, and anti-La/SS-B antibodies; and (4) negative DIF on lesional EM-like targetoid lesions. The minor criteria include the following: (1) absence of infectious or pharmacologic triggers; (2) absence of typical EM location (acral and mucosal); and (3) presence of at least 1 additional American College of Rheumatology criterion for diagnosis of SLE8 besides discoid rash and positive ANA antibodies and excluding photosensitivity, malar rash, and oral ulcers. Using these criteria, the patient in our case met the standards for diagnosis of RS.
One area of disagreement that has been encountered in the literature is the exact histologic determination of true RS, specifically related to the microscopic findings of the EM-like lesions. Two cases presented by Modi et al6 were interpreted under the stipulation that true RS must contain histologic LE and histologic EM. Because the EM-appearing lesions revealed LE histology, the cases were concluded to be variants of LE. These cases are similar to our case in that the EM-like lesions in our patient demonstrated LE pathology. Torchia et al,3 as demonstrated in the above criteria, seemed to be less concerned about the histology of the EM-like lesions, only requiring them to show negative DIF.
Conclusion
In the search for answers concerning RS, many unanswered questions remain: Where should the line be drawn in the inclusion of so many variations of both the LE and EM components of the condition? Also, should these elements even be approached as distinct components in the first place? Viewing the majority of RS cases as simply simultaneous LE and EM, Shteyngarts et al7 concluded that “the concomitant occurrence of EM with LE did not change the course, therapy, or prognosis of either disease. SLE and DLE can coexist with EM, but the coexistence does not impart any unusual characteristic to either illness. Rowell’s syndrome is not reproducible, and the immunologic disturbances in such patients are probably coincidental.”
If the condition is a genuine pathological individuality, should we not view the seemingly separate LE and EM as the product of a single underlying biochemical process? These questions and others in the search for a true definition of the disease should continue to be debated. It is clear that further investigation is warranted in the understanding of the underlying mechanism of the pathology.
Case Report
A 37-year-old woman was admitted to the intensive care unit secondary to the acute development of an erythematous rash with tissue sloughing that involved acral sites and mucosal surfaces. Her medical history was notable for anti-Ro/Sjögren syndrome antigen A (SS-A)–positive lupus erythematosus (LE) with a morphologic semblance to subacute cutaneous LE (SCLE). Prior treatment had included oral corticosteroids. In addition, she reported a concurrent history of acral and mucosal lesions that appeared to flare with her lupus. The nature of these lesions was not clear to the patient or her physicians. Before this particular episode, her primary care physician had attempted to wean her off of the corticosteroids. As she dropped below 20 mg of prednisone daily, new lesions developed. The patient stated that her social situation was poor and that these lesions did seem to develop more frequently during times of physical and emotional stress. She recounted her first episode developing during her second pregnancy. Oral prednisone and over-the-counter calcium with vitamin D were her only reported medications. She denied the use of any other medications, including nonsteroidal anti-inflammatory drugs, acetaminophen, and recent antibiotic therapy.
Dermatology was called in for consultation, and physical examination revealed areas of epidermal sloughing on the hands and feet. Complete clinical exposure of the underlying dermis was noted with remarkable tenderness. These lesions were noted to be in various stages of healing (Figure 1). Figure 2 displays a lesion in early development. The mucosal surfaces of the lips and eyes demonstrated hemorrhagic crusting, and some tissue sloughing was noted on the ears. A widespread erythematous exanthema with fine scaling was noted on the face, neck, chest, back, abdomen, arms, and legs (Figure 3).
Laboratory evaluation revealed positive antinuclear antibodies (ANAs), anti-Ro/SS-A antibodies, anti-La/Sjögren syndrome antigen B (SS-B) antibodies, and anti–double-stranded DNA. The hemoglobin level was 9.4 g/dL (reference range, 12–15 g/dL) and hematocrit was 28.8% (reference range, 36%–47%). The mean corpuscular hemoglobin level was 32 pg/cell (reference range, 27–31 pg/cell), and the mean corpuscular hemoglobin concentration was 32.5 g/dL (reference range, 30–35 g/dL). Rheumatoid factor (RF) and herpes simplex virus types 1 and 2 IgM were all found to be negative.
A deep shave biopsy obtained from the patient’s right knee revealed an atrophic interface dermatitis associated with a lymphocytic eccrine hidradenitis accompanied by abundant mesenchymal mucin deposition (Figure 4). Direct immunofluorescence (DIF) from the same area demonstrated IgG and IgM along the dermoepidermal junction with some granular deposition. Frozen sections performed on acral lesions demonstrated epidermal necrosis (Figure 5). Direct immunofluorescence of acral lesions was negative. In light of these findings, a diagnosis of Rowell syndrome (RS) was suspected to be the most likely explanation for the presentation.
Intravenous corticosteroids and antibiotics were administered, and over a 2-week hospitalization, the lesions on the feet and hands slowly reepithelialized. Physical therapy was required to aid in ambulation. The patient was discharged on a tapering course of oral prednisone and hydroxychloroquine. After 6 months of therapy with hydroxychloroquine 200 mg twice daily, the patient continued to experience recurrent bouts of acral lesions, and pulse doses of oral prednisone were required. The lesions currently are controlled with azathioprine 50 mg twice daily and prednisone 10 mg by mouth daily.
Comment
The 4 prototypical patients identified by Rowell et al1 in 1963 in the first account of the eponymous syndrome were all females with discoid lupus erythematosus (DLE) and perniosis. In addition, they all displayed positive RF and saline extract of human tissue antibodies (analogous to anti-Ro/SS-A and anti-La/SS-B).2 Since then, at least 132 patients with clinical symptoms suspicious of RS have been identified with variations on these original criteria.3 The reported permutations of the lupus component of the disease include cutaneous LE (CLE), bullous systemic LE, necrotic lesions associated with antiphospholipid syndrome, annular/polycyclic SCLE, systemic LE (SLE) without CLE, SLE with lupus nephritis, SLE with pericarditis, SLE with systemic vasculitis, Sjögren syndrome, rheumatoid arthritis, and necrotizing lymphadenitis.2 In addition, variations of the erythema multiforme (EM)–like lesions found in reported cases include changes to their gross appearance (flat vs raised), location (acral or mucosal involvement), and resemblance to other conditions (Stevens-Johnson syndrome or toxic epidermal necrolysis).2,3 From this information alone, it is clear that, as further cases have been chronicled, defining exact criteria for the disease has been challenging.
The essential question concerning the existence of RS hinges on the strength of its distinctiveness: Is it a unique disorder or merely another variant of lupus? Antiga et al2 concluded that it should be characterized as a variant of SCLE. Lee at al4 agreed, stating that “[i]n view of the lack of specific features that distinguish RS from LE, Kuhn et al5 suggested that [RS] is probably not a distinct entity and is now widely considered to be a variant of SCLE.” One of the primary contributors to this conclusion is that the laboratory findings of reported patients with SCLE have more closely mirrored the original cases from Rowell et al’s1 report than those of typical LE. Patients with SCLE have demonstrated positive ANA antibodies in 60% to 80% of cases, positive anti-Ro/SS-A antibodies in 40% to 100% of cases, positive anti-La/SS-B antibodies in 12% to 42% of cases, positive anti–double-stranded DNA in 1.2% to 10% of cases, and positive RF antibodies in 33% of cases.2 An argument could certainly be made to ascribe our patient’s condition to an SCLE variant, as 4 of 5 preceding laboratory findings were found to be positive; however, the majority of reported cases of SCLE have been linked to drugs (ie, hydrochlorothiazide, angiotensin-converting enzyme inhibitors, calcium channel blockers, terbinafine),2 which has not commonly been the attributable etiology of other cases of RS, including the 4 cases reported by Rowell et al.1
In a review of the literature on RS since 2010 in addition to their report of 132 new cases, Torchia et al3 outlined a set of diagnostic standards for the condition consisting of major and minor criteria. According to the authors, if all 4 major and 1 minor criteria are met, the patient meets the standards for true RS. The major criteria include the following: (1) presence of chronic CLE [DLE and/or chilblain]; (2) presence of EM-like lesions [typical or atypical targets]; (3) at least 1 positivity among speckled ANA, anti-Ro/SS-A, and anti-La/SS-B antibodies; and (4) negative DIF on lesional EM-like targetoid lesions. The minor criteria include the following: (1) absence of infectious or pharmacologic triggers; (2) absence of typical EM location (acral and mucosal); and (3) presence of at least 1 additional American College of Rheumatology criterion for diagnosis of SLE8 besides discoid rash and positive ANA antibodies and excluding photosensitivity, malar rash, and oral ulcers. Using these criteria, the patient in our case met the standards for diagnosis of RS.
One area of disagreement that has been encountered in the literature is the exact histologic determination of true RS, specifically related to the microscopic findings of the EM-like lesions. Two cases presented by Modi et al6 were interpreted under the stipulation that true RS must contain histologic LE and histologic EM. Because the EM-appearing lesions revealed LE histology, the cases were concluded to be variants of LE. These cases are similar to our case in that the EM-like lesions in our patient demonstrated LE pathology. Torchia et al,3 as demonstrated in the above criteria, seemed to be less concerned about the histology of the EM-like lesions, only requiring them to show negative DIF.
Conclusion
In the search for answers concerning RS, many unanswered questions remain: Where should the line be drawn in the inclusion of so many variations of both the LE and EM components of the condition? Also, should these elements even be approached as distinct components in the first place? Viewing the majority of RS cases as simply simultaneous LE and EM, Shteyngarts et al7 concluded that “the concomitant occurrence of EM with LE did not change the course, therapy, or prognosis of either disease. SLE and DLE can coexist with EM, but the coexistence does not impart any unusual characteristic to either illness. Rowell’s syndrome is not reproducible, and the immunologic disturbances in such patients are probably coincidental.”
If the condition is a genuine pathological individuality, should we not view the seemingly separate LE and EM as the product of a single underlying biochemical process? These questions and others in the search for a true definition of the disease should continue to be debated. It is clear that further investigation is warranted in the understanding of the underlying mechanism of the pathology.
- Rowell NR, Beck JS, Anderson JR. Lupus erythematosus and erythema multiforme-like lesions: a syndrome with characteristic immunological abnormalities. Arch Dermatol. 1963;88:176-180.
- Antiga E, Caproni M, Bonciani D, et al. The last word on the so-called ‘Rowell’s syndrome’? Lupus. 2012;21:577-585.
- Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67:417-421.
- Lee A, Batra P, Furer V, et al. Rowell syndrome (systemic lupus erythematosus + erythema multiforme). Dermatol Online J. 2009;15:1.
- Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1140.
- Modi GM, Shen A, Mazloom A, et al. Lupus erythematosus masquerading as erythema multiforme: does Rowell syndrome really exist? Dermatol Online J. 2009;15:5.
- Shteyngarts AR, Warner MR, Camisa C. Lupus erythematosus associated with erythema multiforme: does Rowell’s syndrome exist? J Am Acad Dermatol. 1999;40(5 pt 1):773-777.
- Lupus diagnosis. Lupus Research Alliance website. http://lupusresearchinstitute.org/lupus-facts/lupus-diagnosis. Accessed July 11, 2017.
- Rowell NR, Beck JS, Anderson JR. Lupus erythematosus and erythema multiforme-like lesions: a syndrome with characteristic immunological abnormalities. Arch Dermatol. 1963;88:176-180.
- Antiga E, Caproni M, Bonciani D, et al. The last word on the so-called ‘Rowell’s syndrome’? Lupus. 2012;21:577-585.
- Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67:417-421.
- Lee A, Batra P, Furer V, et al. Rowell syndrome (systemic lupus erythematosus + erythema multiforme). Dermatol Online J. 2009;15:1.
- Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1140.
- Modi GM, Shen A, Mazloom A, et al. Lupus erythematosus masquerading as erythema multiforme: does Rowell syndrome really exist? Dermatol Online J. 2009;15:5.
- Shteyngarts AR, Warner MR, Camisa C. Lupus erythematosus associated with erythema multiforme: does Rowell’s syndrome exist? J Am Acad Dermatol. 1999;40(5 pt 1):773-777.
- Lupus diagnosis. Lupus Research Alliance website. http://lupusresearchinstitute.org/lupus-facts/lupus-diagnosis. Accessed July 11, 2017.
Practice Points
- Rowell syndrome (RS) is an often unrecognized unique presentation of lupus erythematosus.
- There have been a variety of historical criteria that have sought to characterize RS.
Lower Limb Morel-Lavallée Lesion Treated With Short-Stretch Compression Bandaging
Take-Home Points
- Have a high-index of suspicion for MLLs and initiate treatment early.
- Compression needs to occur through short-stretch bandaging over a conventional Ace wrap in order to be successful.
- Apply the short-stretch compression with care to avoid shearing underlying tissue.
- Nonoperative treatment modalities require high patient compliance.
- MLLs need close monitoring until final healing occurs.
Morel-Lavallée lesions (MLLs) are traumatic degloving injuries resulting from separation of subcutaneous fat from underlying fascia. MLLs occur in association with acetabular fractures and are also associated with low-velocity crush injuries.1,2 Shearing creates a “false” space that is filled with hemorrhaged blood, fat, and lymphatic tissue.3 Disruption of the lymphatics leads to cavity formation and, eventually, a fibrotic pseudocapsule.4The pseudocapsule prevents resorption, leading to a chronic fluid collection, which potentiates the risk of infection or tissue necrosis.3,5,6 Skin necrosis may occur through direct-pressure compromise of the dermal vascular plexus.4 Necrotic skin may require multiple débridements, negative-pressure wound therapy or soft-tissue coverage, and may ultimately result in infection. MLLs classically occur in the greater trochanteric region, lateral thigh, buttocks, and back but also appear in the prepatellar region.1,3 Patients present with soft-tissue swelling, bruising, bulging, decreased cutaneous sensation over the region, and a palpable, fluctuant subcutaneous fluid collection with mobile skin.2,4,7 The mechanism of injury may cause a concomitant fracture. Magnetic resonance imaging (MRI), the preferred imaging modality, shows a discrete fluid collection between subcutaneous fat and underlying fascia. Ultrasonography may reveal a thickened capsule surrounding either a hypoechoic area or an anechoic area but its accuracy is user-dependent.7
Large MLLs may be treated with open serial débridement and healing by secondary intention; infection rates, however, are high. Authors have described several other treatment modalities, including percutaneous débridement with a brush followed by use of a large-bore drain and antibiotics; open débridement with meticulous dead-space closure; elastic compression bandaging; aspiration; and doxycycline sclerodesis.1,5,6,8,9 Modifications of short-stretch compression bandaging were recently described in edema control for hindfoot trauma, ankle trauma, and total ankle arthroplasty, but not for MLLs.10,11 Nickerson and colleagues4 retrospectively reviewed 87 MLLs, found that fluid aspirate of >50 mL predicted recurrence and failure with conservative measures, and recommended operative intervention for any MLL with >50 mL of fluid aspirated.
We report the case of an MLL that occurred in an unusual anatomical region, and we describe a novel application of a conservative treatment, which was selected on the basis of its success in lymphedema management. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 66-year-old man was injured when a parked vehicle began moving, pulled him under, and ran over his lower right leg. In the emergency department, no fractures or major injuries were noted (Figures 1A, 1B), and the patient was discharged.
About 10 days after injury, profuse ecchymosis and swelling were noted running from the distal medial thigh to the proximal medial calf (Figures 2A-2C).
Given the size of the MLL, the fluid collection reaccumulated. The patient was evaluated by an orthopedic traumatologist 3 days after the aspiration (17 days after injury).
Another orthopedic traumatologist confirmed the low likelihood that compression would resolve the MLL, given its size (Figures 4A, 4B). 

After the second orthopedic consultation, the patient saw a physical therapist trained in complete decongestive therapy. The therapist suggested placing short-stretch bandage wraps over the conventional long-stretch Ace bandage currently being used—a treatment common in lymphedema. The patient was wrapped from toe to groin without an initial layer of padding (Figures 6A, 6B), and the response was immediate. 
Nine weeks after injury, the leg was significantly improved, and clinical signs resolved (Figure 7). 
Discussion
Short-stretch bandaging has been performed mainly in lymphedema and ulcer management. 
Compression bandaging reduces volume in lymphedematous limbs by reducing capillary filtration, shifting fluid into noncompressed parts of the body, increasing lymphatic reabsorption and lymphatic transport stimulation, improving venous pumping, and breaking down fibrosclerotic tissue.15 We think containment, improved venous flow, and enhanced muscle contraction contributed to the effectiveness of short-stretch bandaging as treatment for our patient’s MLL. Because MLLs also contain disrupted lymphatics, lymphedema management strategies (eg, short-stretch bandages) can be used. Our patient rapidly improved after conversion to short-stretch bandages.
These bandages are applied with 50% overlap to ensure even pressures throughout.16 Multiple layers are applied using a combination of spiral and figure-of-8 techniques, first clockwise and then counterclockwise, to avoid shearing underlying tissue.17 This method is very important in MLL treatment, given the degloving involved and the highly mobile skin and subcutaneous fat.
In standard lymphedema management, a foam padding layer is applied before the short-stretch bandage in order to reshape the limb and avoid proximal constrictions.13 In our patient’s case, the short-stretch wrap was applied without padding. Because his condition was acute, and the limb contour was preserved, limb reshaping and thus padding were not necessary.
Given the rapid, high-volume reduction that occurs within the first 1 to 2 weeks, bandages are reapplied daily to effectively adjust for the decreased swelling and altered limb shape.17 Most improvement is expected within the first few weeks—consistent with our patient’s case. Bandages usually are applied to the entire limb. For partial cases, the bandaging must extend past the area of swelling and incorporate the knee to prevent displacement of fluid into the joint.17 Feet and ankles are bandaged in dorsiflexion.17Several factors must be considered with short-stretch wraps. For example, pressure may need to be adjusted in patients with peripheral vascular disease. In patients with ankle-brachial indexes >0.5, it is safe to apply pressure up to 40 mm Hg.12 Reduced pressure is recommended for patients with arterial disease, sensory disturbance, lipoedema, poor mobility, frailty, or palliative needs.13The unusual location of our patient’s MLL accounts for the delay in diagnosis. To our knowledge, no other authors have reported such a large MLL in this location. A few series and case reports have listed MLLs in the calf near the gastrocnemius muscle, in the ankle, in the prepatellar area, and in the suprapatellar region, including the thigh,1,3,18-20 but there are no reports of MLLs running from medial thigh to proximal calf. MLLs of this size classically are treated surgically, but our patient selected nonoperative management.
To our knowledge, there are no earlier reports of using this nonoperative technique to treat MLLs. Conservative treatment with compression has been discussed, but no case involved short-stretch bandages. Large MLLs are thought to require surgery plus some type of drainage. The success of using short-stretch bandages in our patient’s case should prompt further investigation of use in adherent patients—which could ultimately result in reduced surgical needs, improved wound care (surgery is avoided), and a maintained low risk of infection. Although more work is needed to come to a more definitive verdict on this treatment method, it is a promising option that warrants consideration.
Am J Orthop. 2017;46(4):E213-E218. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Tejwani SG, Cohen SB, Bradley JP. Management of Morel-Lavallee lesion of the knee: twenty-seven cases in the National Football League. Am J Sports Med. 2007;35(7):1162-1167.
2. Tsur A, Galin A, Kogan L, Loberant N. Morel-Lavallee syndrome after crush injury [in Hebrew]. Harefuah. 2006;145(2):111-113.
3. Ciaschini M, Sundaram M. Radiologic case study. Prepatellar Morel-Lavallée lesion. Orthopedics. 2008;31(7):626, 719-721.
4. Nickerson TP, Zielinski MD, Jenkins DH, Schiller HJ. The Mayo Clinic experience with Morel-Lavallée lesions: establishment of a practice management guideline. J Trauma Acute Care Surg. 2014;76(2):493-497.
5. Bansal A, Bhatia N, Singh A, Singh AK. Doxycycline sclerodesis as a treatment option for persistent Morel-Lavallée lesions. Injury. 2013;44(1):66-69.
6. Carlson DA, Simmons J, Sando W, Weber T, Clements B. Morel-Lavalée lesions treated with debridement and meticulous dead space closure: surgical technique. J Orthop Trauma. 2007;21(2):140-144.
7. Miller J, Daggett J, Ambay R, Payne WG. Morel-Lavallée lesion. Eplasty. 2014;14:ic12.
8. Tseng S, Tornetta P 3rd. Percutaneous management of Morel-Lavallee lesions. J Bone Joint Surg Am. 2006;88(1):92-96.
9. Harma A, Inan M, Ertem K. The Morel-Lavallée lesion: a conservative approach to closed degloving injuries [in Turkish]. Acta Orthop Traumatol Turc. 2004;38(4):270-273.
10. Hsu A, Franceschina D, Haddad SL. A novel method of postoperative wound care following total ankle arthroplasty. Foot Ankle Int. 2014;35(7):719-724.
11. Rohner-Spengler M, Frotzler A, Honigmann P, Babst R. Effective treatment of posttraumatic and postoperative edema in patients with ankle and hindfoot fractures: a randomized controlled trial comparing multilayer compression therapy and intermittent impulse compression with the standard treatment with ice. J Bone Joint Surg Am. 2014;96(15):1263-1271.
12. Bjork R. The long and short of it: understanding compression bandaging. Wound Care Advisor. 2013;2(6):12-15.
13. Partsch H. Assessing the effectiveness of multilayer inelastic bandaging. J Lymphoedema. 2007;2(2):55-61.
14. Hafner J, Botonakis I, Burg G. A comparison of multilayer bandage systems during rest, exercise, and over 2 days of wear time. Arch Dermatol. 2000;136(7):857-863.
15. Földi E, Jünger M, Partsch H. The science of lymphoedema bandaging. In: Lymphoedema Bandaging in Practice [European Wound Management Association focus document]. London, England: Medical Education Partnership; 2005:2-4.
16. King TI, Droessler JL. Physical properties of short-stretch compression bandages used to treat lymphedema. Am J Occup Ther. 2001;55(5):573-576.
17. Williams AF, Keller M. Practical guidance on lymphoedema bandaging of the upper and lower limbs. In: Lymphoedema Bandaging in Practice [European Wound Management Association focus document]. London, England: Medical Education Partnership; 2005:10-14.
18. Moriarty JM, Borrero CG, Kavanagh EC. A rare cause of calf swelling: the Morel-Lavallee lesion. Ir J Med Sci. 2011;180(1):265-268.
19. Anakwenze OA, Trivedi V, Goodman AM, Ganley TJ. Knee Morel-Lavallee lesion after a football injury in an 11-year-old boy: case report and review of the literature. Univ Pa Orthop J. 2011;21:56-58.
20. Hudson DA, Knottenbelt JD, Krige JE. Closed degloving injuries: results following conservative surgery. Plast Reconstr Surg. 1992;89(5):853-855.
Take-Home Points
- Have a high-index of suspicion for MLLs and initiate treatment early.
- Compression needs to occur through short-stretch bandaging over a conventional Ace wrap in order to be successful.
- Apply the short-stretch compression with care to avoid shearing underlying tissue.
- Nonoperative treatment modalities require high patient compliance.
- MLLs need close monitoring until final healing occurs.
Morel-Lavallée lesions (MLLs) are traumatic degloving injuries resulting from separation of subcutaneous fat from underlying fascia. MLLs occur in association with acetabular fractures and are also associated with low-velocity crush injuries.1,2 Shearing creates a “false” space that is filled with hemorrhaged blood, fat, and lymphatic tissue.3 Disruption of the lymphatics leads to cavity formation and, eventually, a fibrotic pseudocapsule.4The pseudocapsule prevents resorption, leading to a chronic fluid collection, which potentiates the risk of infection or tissue necrosis.3,5,6 Skin necrosis may occur through direct-pressure compromise of the dermal vascular plexus.4 Necrotic skin may require multiple débridements, negative-pressure wound therapy or soft-tissue coverage, and may ultimately result in infection. MLLs classically occur in the greater trochanteric region, lateral thigh, buttocks, and back but also appear in the prepatellar region.1,3 Patients present with soft-tissue swelling, bruising, bulging, decreased cutaneous sensation over the region, and a palpable, fluctuant subcutaneous fluid collection with mobile skin.2,4,7 The mechanism of injury may cause a concomitant fracture. Magnetic resonance imaging (MRI), the preferred imaging modality, shows a discrete fluid collection between subcutaneous fat and underlying fascia. Ultrasonography may reveal a thickened capsule surrounding either a hypoechoic area or an anechoic area but its accuracy is user-dependent.7
Large MLLs may be treated with open serial débridement and healing by secondary intention; infection rates, however, are high. Authors have described several other treatment modalities, including percutaneous débridement with a brush followed by use of a large-bore drain and antibiotics; open débridement with meticulous dead-space closure; elastic compression bandaging; aspiration; and doxycycline sclerodesis.1,5,6,8,9 Modifications of short-stretch compression bandaging were recently described in edema control for hindfoot trauma, ankle trauma, and total ankle arthroplasty, but not for MLLs.10,11 Nickerson and colleagues4 retrospectively reviewed 87 MLLs, found that fluid aspirate of >50 mL predicted recurrence and failure with conservative measures, and recommended operative intervention for any MLL with >50 mL of fluid aspirated.
We report the case of an MLL that occurred in an unusual anatomical region, and we describe a novel application of a conservative treatment, which was selected on the basis of its success in lymphedema management. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 66-year-old man was injured when a parked vehicle began moving, pulled him under, and ran over his lower right leg. In the emergency department, no fractures or major injuries were noted (Figures 1A, 1B), and the patient was discharged.
About 10 days after injury, profuse ecchymosis and swelling were noted running from the distal medial thigh to the proximal medial calf (Figures 2A-2C).
Given the size of the MLL, the fluid collection reaccumulated. The patient was evaluated by an orthopedic traumatologist 3 days after the aspiration (17 days after injury).
Another orthopedic traumatologist confirmed the low likelihood that compression would resolve the MLL, given its size (Figures 4A, 4B).
After the second orthopedic consultation, the patient saw a physical therapist trained in complete decongestive therapy. The therapist suggested placing short-stretch bandage wraps over the conventional long-stretch Ace bandage currently being used—a treatment common in lymphedema. The patient was wrapped from toe to groin without an initial layer of padding (Figures 6A, 6B), and the response was immediate.
Nine weeks after injury, the leg was significantly improved, and clinical signs resolved (Figure 7).
Discussion
Short-stretch bandaging has been performed mainly in lymphedema and ulcer management.
Compression bandaging reduces volume in lymphedematous limbs by reducing capillary filtration, shifting fluid into noncompressed parts of the body, increasing lymphatic reabsorption and lymphatic transport stimulation, improving venous pumping, and breaking down fibrosclerotic tissue.15 We think containment, improved venous flow, and enhanced muscle contraction contributed to the effectiveness of short-stretch bandaging as treatment for our patient’s MLL. Because MLLs also contain disrupted lymphatics, lymphedema management strategies (eg, short-stretch bandages) can be used. Our patient rapidly improved after conversion to short-stretch bandages.
These bandages are applied with 50% overlap to ensure even pressures throughout.16 Multiple layers are applied using a combination of spiral and figure-of-8 techniques, first clockwise and then counterclockwise, to avoid shearing underlying tissue.17 This method is very important in MLL treatment, given the degloving involved and the highly mobile skin and subcutaneous fat.
In standard lymphedema management, a foam padding layer is applied before the short-stretch bandage in order to reshape the limb and avoid proximal constrictions.13 In our patient’s case, the short-stretch wrap was applied without padding. Because his condition was acute, and the limb contour was preserved, limb reshaping and thus padding were not necessary.
Given the rapid, high-volume reduction that occurs within the first 1 to 2 weeks, bandages are reapplied daily to effectively adjust for the decreased swelling and altered limb shape.17 Most improvement is expected within the first few weeks—consistent with our patient’s case. Bandages usually are applied to the entire limb. For partial cases, the bandaging must extend past the area of swelling and incorporate the knee to prevent displacement of fluid into the joint.17 Feet and ankles are bandaged in dorsiflexion.17Several factors must be considered with short-stretch wraps. For example, pressure may need to be adjusted in patients with peripheral vascular disease. In patients with ankle-brachial indexes >0.5, it is safe to apply pressure up to 40 mm Hg.12 Reduced pressure is recommended for patients with arterial disease, sensory disturbance, lipoedema, poor mobility, frailty, or palliative needs.13The unusual location of our patient’s MLL accounts for the delay in diagnosis. To our knowledge, no other authors have reported such a large MLL in this location. A few series and case reports have listed MLLs in the calf near the gastrocnemius muscle, in the ankle, in the prepatellar area, and in the suprapatellar region, including the thigh,1,3,18-20 but there are no reports of MLLs running from medial thigh to proximal calf. MLLs of this size classically are treated surgically, but our patient selected nonoperative management.
To our knowledge, there are no earlier reports of using this nonoperative technique to treat MLLs. Conservative treatment with compression has been discussed, but no case involved short-stretch bandages. Large MLLs are thought to require surgery plus some type of drainage. The success of using short-stretch bandages in our patient’s case should prompt further investigation of use in adherent patients—which could ultimately result in reduced surgical needs, improved wound care (surgery is avoided), and a maintained low risk of infection. Although more work is needed to come to a more definitive verdict on this treatment method, it is a promising option that warrants consideration.
Am J Orthop. 2017;46(4):E213-E218. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- Have a high-index of suspicion for MLLs and initiate treatment early.
- Compression needs to occur through short-stretch bandaging over a conventional Ace wrap in order to be successful.
- Apply the short-stretch compression with care to avoid shearing underlying tissue.
- Nonoperative treatment modalities require high patient compliance.
- MLLs need close monitoring until final healing occurs.
Morel-Lavallée lesions (MLLs) are traumatic degloving injuries resulting from separation of subcutaneous fat from underlying fascia. MLLs occur in association with acetabular fractures and are also associated with low-velocity crush injuries.1,2 Shearing creates a “false” space that is filled with hemorrhaged blood, fat, and lymphatic tissue.3 Disruption of the lymphatics leads to cavity formation and, eventually, a fibrotic pseudocapsule.4The pseudocapsule prevents resorption, leading to a chronic fluid collection, which potentiates the risk of infection or tissue necrosis.3,5,6 Skin necrosis may occur through direct-pressure compromise of the dermal vascular plexus.4 Necrotic skin may require multiple débridements, negative-pressure wound therapy or soft-tissue coverage, and may ultimately result in infection. MLLs classically occur in the greater trochanteric region, lateral thigh, buttocks, and back but also appear in the prepatellar region.1,3 Patients present with soft-tissue swelling, bruising, bulging, decreased cutaneous sensation over the region, and a palpable, fluctuant subcutaneous fluid collection with mobile skin.2,4,7 The mechanism of injury may cause a concomitant fracture. Magnetic resonance imaging (MRI), the preferred imaging modality, shows a discrete fluid collection between subcutaneous fat and underlying fascia. Ultrasonography may reveal a thickened capsule surrounding either a hypoechoic area or an anechoic area but its accuracy is user-dependent.7
Large MLLs may be treated with open serial débridement and healing by secondary intention; infection rates, however, are high. Authors have described several other treatment modalities, including percutaneous débridement with a brush followed by use of a large-bore drain and antibiotics; open débridement with meticulous dead-space closure; elastic compression bandaging; aspiration; and doxycycline sclerodesis.1,5,6,8,9 Modifications of short-stretch compression bandaging were recently described in edema control for hindfoot trauma, ankle trauma, and total ankle arthroplasty, but not for MLLs.10,11 Nickerson and colleagues4 retrospectively reviewed 87 MLLs, found that fluid aspirate of >50 mL predicted recurrence and failure with conservative measures, and recommended operative intervention for any MLL with >50 mL of fluid aspirated.
We report the case of an MLL that occurred in an unusual anatomical region, and we describe a novel application of a conservative treatment, which was selected on the basis of its success in lymphedema management. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 66-year-old man was injured when a parked vehicle began moving, pulled him under, and ran over his lower right leg. In the emergency department, no fractures or major injuries were noted (Figures 1A, 1B), and the patient was discharged.
About 10 days after injury, profuse ecchymosis and swelling were noted running from the distal medial thigh to the proximal medial calf (Figures 2A-2C).
Given the size of the MLL, the fluid collection reaccumulated. The patient was evaluated by an orthopedic traumatologist 3 days after the aspiration (17 days after injury).
Another orthopedic traumatologist confirmed the low likelihood that compression would resolve the MLL, given its size (Figures 4A, 4B).
After the second orthopedic consultation, the patient saw a physical therapist trained in complete decongestive therapy. The therapist suggested placing short-stretch bandage wraps over the conventional long-stretch Ace bandage currently being used—a treatment common in lymphedema. The patient was wrapped from toe to groin without an initial layer of padding (Figures 6A, 6B), and the response was immediate.
Nine weeks after injury, the leg was significantly improved, and clinical signs resolved (Figure 7).
Discussion
Short-stretch bandaging has been performed mainly in lymphedema and ulcer management.
Compression bandaging reduces volume in lymphedematous limbs by reducing capillary filtration, shifting fluid into noncompressed parts of the body, increasing lymphatic reabsorption and lymphatic transport stimulation, improving venous pumping, and breaking down fibrosclerotic tissue.15 We think containment, improved venous flow, and enhanced muscle contraction contributed to the effectiveness of short-stretch bandaging as treatment for our patient’s MLL. Because MLLs also contain disrupted lymphatics, lymphedema management strategies (eg, short-stretch bandages) can be used. Our patient rapidly improved after conversion to short-stretch bandages.
These bandages are applied with 50% overlap to ensure even pressures throughout.16 Multiple layers are applied using a combination of spiral and figure-of-8 techniques, first clockwise and then counterclockwise, to avoid shearing underlying tissue.17 This method is very important in MLL treatment, given the degloving involved and the highly mobile skin and subcutaneous fat.
In standard lymphedema management, a foam padding layer is applied before the short-stretch bandage in order to reshape the limb and avoid proximal constrictions.13 In our patient’s case, the short-stretch wrap was applied without padding. Because his condition was acute, and the limb contour was preserved, limb reshaping and thus padding were not necessary.
Given the rapid, high-volume reduction that occurs within the first 1 to 2 weeks, bandages are reapplied daily to effectively adjust for the decreased swelling and altered limb shape.17 Most improvement is expected within the first few weeks—consistent with our patient’s case. Bandages usually are applied to the entire limb. For partial cases, the bandaging must extend past the area of swelling and incorporate the knee to prevent displacement of fluid into the joint.17 Feet and ankles are bandaged in dorsiflexion.17Several factors must be considered with short-stretch wraps. For example, pressure may need to be adjusted in patients with peripheral vascular disease. In patients with ankle-brachial indexes >0.5, it is safe to apply pressure up to 40 mm Hg.12 Reduced pressure is recommended for patients with arterial disease, sensory disturbance, lipoedema, poor mobility, frailty, or palliative needs.13The unusual location of our patient’s MLL accounts for the delay in diagnosis. To our knowledge, no other authors have reported such a large MLL in this location. A few series and case reports have listed MLLs in the calf near the gastrocnemius muscle, in the ankle, in the prepatellar area, and in the suprapatellar region, including the thigh,1,3,18-20 but there are no reports of MLLs running from medial thigh to proximal calf. MLLs of this size classically are treated surgically, but our patient selected nonoperative management.
To our knowledge, there are no earlier reports of using this nonoperative technique to treat MLLs. Conservative treatment with compression has been discussed, but no case involved short-stretch bandages. Large MLLs are thought to require surgery plus some type of drainage. The success of using short-stretch bandages in our patient’s case should prompt further investigation of use in adherent patients—which could ultimately result in reduced surgical needs, improved wound care (surgery is avoided), and a maintained low risk of infection. Although more work is needed to come to a more definitive verdict on this treatment method, it is a promising option that warrants consideration.
Am J Orthop. 2017;46(4):E213-E218. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Tejwani SG, Cohen SB, Bradley JP. Management of Morel-Lavallee lesion of the knee: twenty-seven cases in the National Football League. Am J Sports Med. 2007;35(7):1162-1167.
2. Tsur A, Galin A, Kogan L, Loberant N. Morel-Lavallee syndrome after crush injury [in Hebrew]. Harefuah. 2006;145(2):111-113.
3. Ciaschini M, Sundaram M. Radiologic case study. Prepatellar Morel-Lavallée lesion. Orthopedics. 2008;31(7):626, 719-721.
4. Nickerson TP, Zielinski MD, Jenkins DH, Schiller HJ. The Mayo Clinic experience with Morel-Lavallée lesions: establishment of a practice management guideline. J Trauma Acute Care Surg. 2014;76(2):493-497.
5. Bansal A, Bhatia N, Singh A, Singh AK. Doxycycline sclerodesis as a treatment option for persistent Morel-Lavallée lesions. Injury. 2013;44(1):66-69.
6. Carlson DA, Simmons J, Sando W, Weber T, Clements B. Morel-Lavalée lesions treated with debridement and meticulous dead space closure: surgical technique. J Orthop Trauma. 2007;21(2):140-144.
7. Miller J, Daggett J, Ambay R, Payne WG. Morel-Lavallée lesion. Eplasty. 2014;14:ic12.
8. Tseng S, Tornetta P 3rd. Percutaneous management of Morel-Lavallee lesions. J Bone Joint Surg Am. 2006;88(1):92-96.
9. Harma A, Inan M, Ertem K. The Morel-Lavallée lesion: a conservative approach to closed degloving injuries [in Turkish]. Acta Orthop Traumatol Turc. 2004;38(4):270-273.
10. Hsu A, Franceschina D, Haddad SL. A novel method of postoperative wound care following total ankle arthroplasty. Foot Ankle Int. 2014;35(7):719-724.
11. Rohner-Spengler M, Frotzler A, Honigmann P, Babst R. Effective treatment of posttraumatic and postoperative edema in patients with ankle and hindfoot fractures: a randomized controlled trial comparing multilayer compression therapy and intermittent impulse compression with the standard treatment with ice. J Bone Joint Surg Am. 2014;96(15):1263-1271.
12. Bjork R. The long and short of it: understanding compression bandaging. Wound Care Advisor. 2013;2(6):12-15.
13. Partsch H. Assessing the effectiveness of multilayer inelastic bandaging. J Lymphoedema. 2007;2(2):55-61.
14. Hafner J, Botonakis I, Burg G. A comparison of multilayer bandage systems during rest, exercise, and over 2 days of wear time. Arch Dermatol. 2000;136(7):857-863.
15. Földi E, Jünger M, Partsch H. The science of lymphoedema bandaging. In: Lymphoedema Bandaging in Practice [European Wound Management Association focus document]. London, England: Medical Education Partnership; 2005:2-4.
16. King TI, Droessler JL. Physical properties of short-stretch compression bandages used to treat lymphedema. Am J Occup Ther. 2001;55(5):573-576.
17. Williams AF, Keller M. Practical guidance on lymphoedema bandaging of the upper and lower limbs. In: Lymphoedema Bandaging in Practice [European Wound Management Association focus document]. London, England: Medical Education Partnership; 2005:10-14.
18. Moriarty JM, Borrero CG, Kavanagh EC. A rare cause of calf swelling: the Morel-Lavallee lesion. Ir J Med Sci. 2011;180(1):265-268.
19. Anakwenze OA, Trivedi V, Goodman AM, Ganley TJ. Knee Morel-Lavallee lesion after a football injury in an 11-year-old boy: case report and review of the literature. Univ Pa Orthop J. 2011;21:56-58.
20. Hudson DA, Knottenbelt JD, Krige JE. Closed degloving injuries: results following conservative surgery. Plast Reconstr Surg. 1992;89(5):853-855.
1. Tejwani SG, Cohen SB, Bradley JP. Management of Morel-Lavallee lesion of the knee: twenty-seven cases in the National Football League. Am J Sports Med. 2007;35(7):1162-1167.
2. Tsur A, Galin A, Kogan L, Loberant N. Morel-Lavallee syndrome after crush injury [in Hebrew]. Harefuah. 2006;145(2):111-113.
3. Ciaschini M, Sundaram M. Radiologic case study. Prepatellar Morel-Lavallée lesion. Orthopedics. 2008;31(7):626, 719-721.
4. Nickerson TP, Zielinski MD, Jenkins DH, Schiller HJ. The Mayo Clinic experience with Morel-Lavallée lesions: establishment of a practice management guideline. J Trauma Acute Care Surg. 2014;76(2):493-497.
5. Bansal A, Bhatia N, Singh A, Singh AK. Doxycycline sclerodesis as a treatment option for persistent Morel-Lavallée lesions. Injury. 2013;44(1):66-69.
6. Carlson DA, Simmons J, Sando W, Weber T, Clements B. Morel-Lavalée lesions treated with debridement and meticulous dead space closure: surgical technique. J Orthop Trauma. 2007;21(2):140-144.
7. Miller J, Daggett J, Ambay R, Payne WG. Morel-Lavallée lesion. Eplasty. 2014;14:ic12.
8. Tseng S, Tornetta P 3rd. Percutaneous management of Morel-Lavallee lesions. J Bone Joint Surg Am. 2006;88(1):92-96.
9. Harma A, Inan M, Ertem K. The Morel-Lavallée lesion: a conservative approach to closed degloving injuries [in Turkish]. Acta Orthop Traumatol Turc. 2004;38(4):270-273.
10. Hsu A, Franceschina D, Haddad SL. A novel method of postoperative wound care following total ankle arthroplasty. Foot Ankle Int. 2014;35(7):719-724.
11. Rohner-Spengler M, Frotzler A, Honigmann P, Babst R. Effective treatment of posttraumatic and postoperative edema in patients with ankle and hindfoot fractures: a randomized controlled trial comparing multilayer compression therapy and intermittent impulse compression with the standard treatment with ice. J Bone Joint Surg Am. 2014;96(15):1263-1271.
12. Bjork R. The long and short of it: understanding compression bandaging. Wound Care Advisor. 2013;2(6):12-15.
13. Partsch H. Assessing the effectiveness of multilayer inelastic bandaging. J Lymphoedema. 2007;2(2):55-61.
14. Hafner J, Botonakis I, Burg G. A comparison of multilayer bandage systems during rest, exercise, and over 2 days of wear time. Arch Dermatol. 2000;136(7):857-863.
15. Földi E, Jünger M, Partsch H. The science of lymphoedema bandaging. In: Lymphoedema Bandaging in Practice [European Wound Management Association focus document]. London, England: Medical Education Partnership; 2005:2-4.
16. King TI, Droessler JL. Physical properties of short-stretch compression bandages used to treat lymphedema. Am J Occup Ther. 2001;55(5):573-576.
17. Williams AF, Keller M. Practical guidance on lymphoedema bandaging of the upper and lower limbs. In: Lymphoedema Bandaging in Practice [European Wound Management Association focus document]. London, England: Medical Education Partnership; 2005:10-14.
18. Moriarty JM, Borrero CG, Kavanagh EC. A rare cause of calf swelling: the Morel-Lavallee lesion. Ir J Med Sci. 2011;180(1):265-268.
19. Anakwenze OA, Trivedi V, Goodman AM, Ganley TJ. Knee Morel-Lavallee lesion after a football injury in an 11-year-old boy: case report and review of the literature. Univ Pa Orthop J. 2011;21:56-58.
20. Hudson DA, Knottenbelt JD, Krige JE. Closed degloving injuries: results following conservative surgery. Plast Reconstr Surg. 1992;89(5):853-855.