User login
14-Year-Old Boy With Mild Antecedent Neck Pain in Setting of Acute Trauma: A Rare Case of Benign Fibrous Histiocytoma of the Spine
Benign fibrous histiocytoma (BFH) is a rare, well-recognized, primary skeletal tumor accounting for approximately 1% of all benign bone tumors. Spinal involvement is exceedingly rare with only 11 cases reported in the literature.1,2 We present a case of BFH located in the cervical spine of a pediatric patient that was successfully treated with curretage through an anterior surgical approach, along with a review of the literature and appropriate management concerning BFH of the spine.
Case Report
A 14-year-old boy was tackled while playing football and noticed immediate neck pain and subjective paresthesia in the upper extremities. Examination revealed a nontender spine (cervical, thoracic, lumbar) and normal strength and range of motion in all extremities. Sensation was diffusely intact, long tract signs were absent, and gait was normal. On questioning, the patient endorsed mild antecedent neck pain but denied prior history of any trauma. Neck pain did not radiate and was slightly worsened by activity but was mostly intermittent and random. As the neck pain was very mild and was not interfering with daily activities, the patient had not sought care before presenting to the emergency department. He had no pertinent past medical or surgical history.
The patient presented with a computed tomography (CT) scan of his head and cervical spine and a magnetic resonance imaging (MRI) scan of the cervical spine. A magnetic resonance angiography (MRA) scan of the neck was ordered after his arrival.
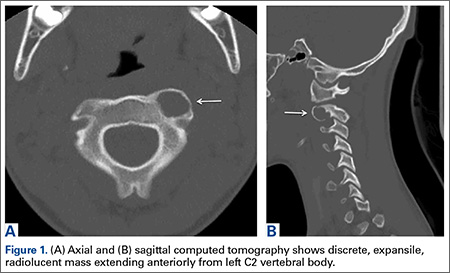
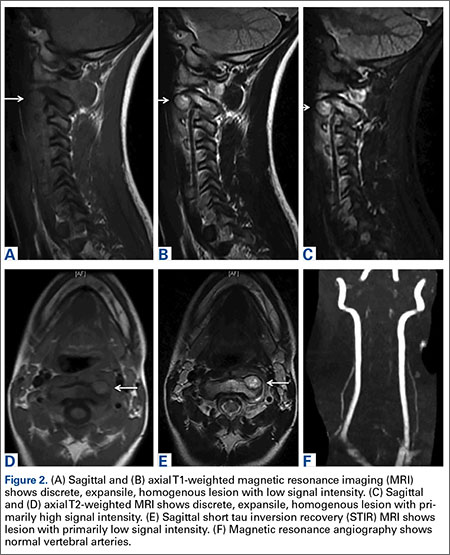
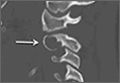
Axial and sagittal CT (Figures 1A, 1B) showed a 1×1.2-cm discrete, expansile, lytic, radiolucent mass extending anterior from the left C2 vertebral body. The mass appeared to abut the left vertebral artery foramen. The cortical bone surrounding the lesion was thin but uniform. Sagittal and axial T1-weighted MRI (Figures 2A, 2B) showed the discrete, expansile, homogenous lesion with the same intensity as normal bone marrow. Sagittal and axial T2-weighted MRI (Figures 2C, 2D) showed a discrete, expansile, homogenous lesion with primarily high signal intensity. Sagittal short tau inversion recovery (STIR) MRI (Figure 2E) again showed the lesion with primarily low intensity. Given the close proximity of the lesion to the vertebral foramen, MRA was ordered; it showed the lesion was not interfering with the vertebral artery (Figure 2F).
The tumor’s location, in the left anterior aspect of the C2 vertebral body, was not conducive to percutaneous biopsy for establishing tissue diagnosis, so the decision was made to surgically excise the lesion. A left-sided anterior incision was made 2 fingerbreadths inferior to the jaw line in a neck crease. A head and neck surgeon assisted with dissection. Dissection was carried down through the skin, subcutaneous tissue, and platysma on to the anterior part of the spine medial to the carotid sheath. Superior thyroid nerve and vessels and superior laryngeal nerve were identified and preserved. Fluoroscopy confirmed correct location at C2. The tumor was easily visualized, and the outer shell broke easily with palpation. Gentle curettage was necessary when removing the tumor off the vertebral artery. A portion of the specimen was sent during surgery for frozen section, which showed infrequent mitotic figures and no other findings concerning for malignancy. No instability was created after curettage and excision of the tumor, so no grafting or instrumentation was necessary.
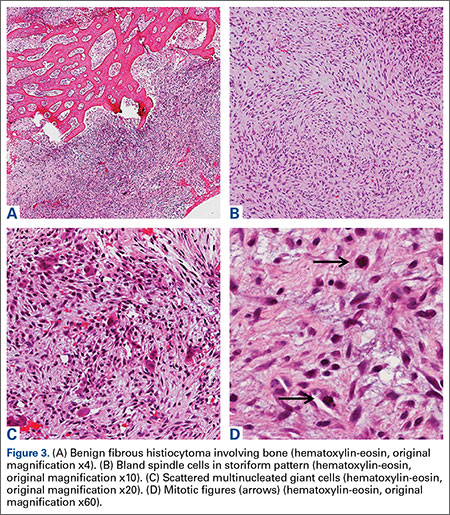
Grossly, the tumor was pale tan and firm. Histologic examination with hematoxylin-eosin staining revealed a bland spindle-cell neoplasm that focally involved bone. A storiform pattern was present. The cells had scant cytoplasm and oval to elongate nuclei with tapered ends. Significant nuclear pleomorphism was not seen. The stroma was loose, with focal myxoid change. Benign multinucleated giant cells were present. Mitotic activity was infrequent (Figures 3A–3D). Two attending pathologists reviewed the case material and the frozen and formalin-fixed specimens independently and concurred with the diagnosis of BFH. In addition, the case was reviewed at the surgical pathology consensus conference; the reviewers agreed on BFH, and additional studies were deemed unnecessary.
Given the patient’s complete clinical picture, the differential diagnosis included nonossifying fibroma (NOF), eosinophilic granuloma (EG), BFH, fibrous dysplasia, giant cell tumor (GCT), aneurysmal bone cyst (ABC), and osteoblastoma (OB).
Discussion
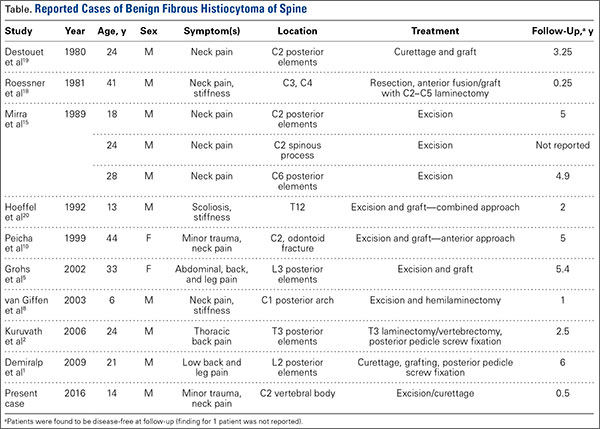
BFH is an extremely rare bone lesion, accounting for only 1% of all surgically managed bone tumors; not counting the present case, only 11 spine cases have been reported in the literature.1,2 BFH of the spine traditionally causes nonspecific, poorly localized pain. The Table lists the reported cases of spinal BFH and their presenting symptoms, location, and treatment. BFH usually occurs in young adults, but the age range is 5 to 75 years.2-4 Mean age of the 12 patients with spinal BFH in the literature (including ours) is 25 years.1 In addition, spinal BFH appears to have no predilection for sex.
Skeletal BFH presents as a discrete, well-defined, osteolytic lesion with sharp borders and potentially a sclerotic rim.4-6 Cortical expansion and even cortical disruption with invasion into adjacent tissue have occurred in flat bones.7 Histologically, BFHs contain spindle cells, multinucleated giant cells, and foam cells in storiform pattern.6
BFH shares many of its radiologic and histologic characteristics and clinical symptoms with other benign bone lesions (the tumors listed above). Therefore, accurate diagnosis of BFH requires appropriate correlation of clinical, radiographic, and histologic data.2,3,8 Below is a comparison of BFH with related bone lesions.
Spinal BFH causes a nonspecific, poorly localized pain similar to that of EG, ABC, GCT, and OB.3,9 NOF and fibrous dysplasia generally do not cause pain, unless these lesions are discovered secondary to a pathologic fracture.8,10,11 Our patient had minor antecedent neck pain, which was brought to light by his football accident. ABC and OB are more locally aggressive than BFH and can cause neurologic symptoms by mass effect and spinal cord or nerve root compression.1,8 In this case and in the 6 other cases of BFH of the cervical spine, there were no neurologic changes.4,10
Of the tumors mentioned, NOF and EG almost always occur in children. However, NOF usually occurs in the metaphyseal region of long bones, and EG is usually accompanied by systemic symptoms, such as lymphadenopathy, hepatomegaly, and increased inflammatory markers.1,8 Fibrous dysplasia usually presents in childhood but does not become symptomatic until adulthood. GCTs and OB predominantly occur in adulthood.12,13 Our patient’s age and lack of other systemic symptoms supported the diagnosis of BFH.
Appearance on MRI is reported less with BFH than with other tumors, but heterogenous signal intensity similar to that of skeletal muscle on T1-weighted images and high signal intensity on T2-weighted images is typically reported.8,14 NOF and fibrous dysplasia do not disrupt the bony cortex unless a pathologic fracture has occurred.4 GCTs are more aggressive lytic lesions with more aggressive radiologic features. GCTs generally cause cortical expansion/attenuation, and lack a sclerotic rim. GCTs also have a heterogenous appearance on MRI and give a low to intermediate signal on both T1- and T2-weighted images.12,15 The appearance of EG is similar to that of BFH as an osteolytic lesion with a sclerotic rim, though EGs typically break through the cortex and acquire a “punched-out” look.1,8 ABC typically is described as an expansile osteolytic lesion with a “soap-bubble” appearance on radiographs; periosteal elevation and cortical attenuation can also be visualized. MRI shows the typical multilobular appearance of the lesion with fluid levels.13
OB appears as a radiolucent lesion, with or without calcifications, surrounded by a thin margin of reactive bone.14,16 A distinguishing characteristic of OB was thought to be intense radioisotope uptake on bone scintigraphy, but recently a bony BFH demonstrated intense uptake.17 OBs typically demonstrate nonspecific MRI results similar to those of BFH: low to intermediate signal on T1-weighted images and intermediate to high signal on T2-weighted images.13 In our patient’s case, the radiographic appearance and lack of specific radiographic findings consistent with the other tumors supported the diagnosis of BFH.
Histologically, BFHs contain spindle cells, multinucleated giant cells, and foam cells in a storiform pattern6 which was demonstrated in our patient’s case. In addition, significant nuclear pleomorphism, mitotic activity, and necrosis were absent—a difference between BFH and malignant fibrous histiocytoma.4,15 The microscopic characteristics of BFH readily differentiate it from OB, ABC, EG, and GCT, but not from NOF on microscopic appearance alone. Clinical and radiographic findings must be consistent, as mentioned.7,18
Complete surgical excision is the reported treatment for BFH. Prognosis after resection or curettage is usually good, and recurrences have been rare.1,2 Depending on the intraspinous location of BFH, stabilization after resection or curettage may be necessary to prevent residual instability. Three of the 11 reported cases of spinal BFH required stabilization by anterior fusion or posterior pedicle screw fixation after resection.1,2 The other 8 cases underwent excision alone or excision and grafting. All 11 patients were disease-free at a mean follow-up of 3.5 years.1 In nonspinal BFH, however, both local recurrence and lung metastasis have been reported.2,5,9,19 Clarke and colleagues9 reported local recurrences in 3 of 8 cases. These recurrences involved BFH in long bones of the leg, which had been treated with curettage and grafting. There has been no reliable report of a malignant change in BFH.2,9 The only case of lung metastasis, reported by Unni and Dahlin6 in their study of 10 cases, occurred 2 years after local recurrence in the distal femur.Our patient was doing well at most recent follow-up, 6 months after surgery. He had no pain and had returned to normal activities. Although there are no reported cases of spinal BFH recurrence, we will follow this patient with imaging on an annual basis. His case is of particular interest to orthopedic surgeons because they encounter benign bone lesions every day, and many of these lesions are in difficult anatomical locations. Knowing the characteristics, differential diagnoses, and appropriate diagnostic workups for benign bone lesions is important for optimal and timely patient care.
1. Demiralp B, Kose O, Oguz E, Sanal T, Ozcan A, Sehirlioglu A. Benign fibrous histiocytoma of the lumbar vertebrae. Skeletal Radiol. 2009;38(2):187-191.
2. Kuruvath S, O’Donovan DG, Aspoas AR, David KM. Benign fibrous histiocytoma of the thoracic spine: case report and review of the literature. J Neurosurg Spine. 2006;4(3):260-264.
3. Ceroni D, Dayer R, De Coulon G, Kaelin A. Benign fibrous histiocytoma of bone in a paediatric population: a report of 6 cases. Musculoskelet Surg. 2011;95(2):107-114.
4. Dorfman HD, Czerniak B. Bone Tumors. St. Louis, MO: Mosby; 1998.
5. Grohs JG, Nicolakis M, Kainberger F, Lang S, Kotz R. Benign fibrous histiocytoma of bone: a report of ten cases and review of literature. Wien Klin Wochenschr. 2002;114(1-2):56-63.
6. Unni KK, Dahlin DC. Dahlin’s Bone Tumors. 5th ed. Philadelphia, PA: Lippincott-Raven; 1996.
7. Balasubramanian C, Rajaraman G, Singh CS, Baliga DK. Benign fibrous histiocytoma of the sacrum—diagnostic difficulties facing this rare bone tumor. Pediatr Neurosurg. 2005;41(5):253-257.
8. van Giffen NH, van Rhijn LW, van Ooij A, et al. Benign fibrous histiocytoma of the posterior arch of C1 in a 6-year old boy: a case report. Spine. 2003;28(18):E359-E363.
9. Clarke BE, Xipell JM, Thomas DP. Benign fibrous histiocytoma of bone. Am J Surg Pathol. 1985;9(11):806-815.
10. Peicha G, Siebert FJ, Bratschitsch G, Fankhauser F, Grechenig W. Pathologic odontoid fracture and benign fibrous histiocytoma of bone. Eur Spine J. 1999;8(2):161-163.
11. Unni KK, Inwards CY, Bridge JA, Kindblom LG, Wold LE. Tumors of the Bones and Joints (AFIP Atlas of Tumor Pathology Series IV). Annapolis Junction, MD: American Registry of Pathology Press; 2005.
12. Dee R. Principles of Orthopaedic Practice. 2nd ed. New York, NY: McGraw-Hill; 1997.
13. Murphey M, Andrews C, Flemming D, Temple HT, Smith WS, Smirniotopoulos JG. Primary tumors of the spine: radiologic–pathologic correlation. Radiographics. 1996;16(5):1131-1158.
14. Hamada T, Ito H, Araki Y, Fujii K, Inoue M, Ishida O. Benign fibrous histiocytoma of the femur: review of three cases. Skeletal Radiol. 1996;25(1):25-29.
15. Mirra JM, Picci P, Gold RH. Bone Tumors: Clinical, Radiologic, and Pathologic Correlations. Vol 1. Philadelphia, PA: Lea & Febiger; 1989.
16. Theodorou DJ, Theodorou SJ, Sartoris DJ. An imaging overview of primary tumors of the spine: part 1. Benign tumors. Clin Imaging. 2008;32(3):196-203.
17. Li X, Meng Z, Li D, Tan J, Song X. Benign fibrous histiocytoma of a rib. Clin Nucl Med. 2014;39(9): 837-841.
18. Roessner A, Immenkamp M, Weidner A, Hobik HP, Grundmann E. Benign fibrous histiocytoma of bone. Light- and electron-microscopic observations. J Cancer Res Clin Oncol. 1981;101(2):191-202.
19. Destouet JM, Kyriakos M, Gilula LA. Fibrous histiocytoma (fibroxanthoma) of a cervical vertebra. A report with a review of the literature. Skeletal Radiol. 1980;5(4):241-246.
20. Hoeffel JC, Bomand-Ferrand F, Tachet F, Lascombes P, Czorny A, Bernard C. So-called benign fibrous histiocytoma: report of a case. J Pediatr Surg. 1992;27(5):672-674.
Benign fibrous histiocytoma (BFH) is a rare, well-recognized, primary skeletal tumor accounting for approximately 1% of all benign bone tumors. Spinal involvement is exceedingly rare with only 11 cases reported in the literature.1,2 We present a case of BFH located in the cervical spine of a pediatric patient that was successfully treated with curretage through an anterior surgical approach, along with a review of the literature and appropriate management concerning BFH of the spine.
Case Report
A 14-year-old boy was tackled while playing football and noticed immediate neck pain and subjective paresthesia in the upper extremities. Examination revealed a nontender spine (cervical, thoracic, lumbar) and normal strength and range of motion in all extremities. Sensation was diffusely intact, long tract signs were absent, and gait was normal. On questioning, the patient endorsed mild antecedent neck pain but denied prior history of any trauma. Neck pain did not radiate and was slightly worsened by activity but was mostly intermittent and random. As the neck pain was very mild and was not interfering with daily activities, the patient had not sought care before presenting to the emergency department. He had no pertinent past medical or surgical history.
The patient presented with a computed tomography (CT) scan of his head and cervical spine and a magnetic resonance imaging (MRI) scan of the cervical spine. A magnetic resonance angiography (MRA) scan of the neck was ordered after his arrival.
Axial and sagittal CT (Figures 1A, 1B) showed a 1×1.2-cm discrete, expansile, lytic, radiolucent mass extending anterior from the left C2 vertebral body. The mass appeared to abut the left vertebral artery foramen. The cortical bone surrounding the lesion was thin but uniform. Sagittal and axial T1-weighted MRI (Figures 2A, 2B) showed the discrete, expansile, homogenous lesion with the same intensity as normal bone marrow. Sagittal and axial T2-weighted MRI (Figures 2C, 2D) showed a discrete, expansile, homogenous lesion with primarily high signal intensity. Sagittal short tau inversion recovery (STIR) MRI (Figure 2E) again showed the lesion with primarily low intensity. Given the close proximity of the lesion to the vertebral foramen, MRA was ordered; it showed the lesion was not interfering with the vertebral artery (Figure 2F).
The tumor’s location, in the left anterior aspect of the C2 vertebral body, was not conducive to percutaneous biopsy for establishing tissue diagnosis, so the decision was made to surgically excise the lesion. A left-sided anterior incision was made 2 fingerbreadths inferior to the jaw line in a neck crease. A head and neck surgeon assisted with dissection. Dissection was carried down through the skin, subcutaneous tissue, and platysma on to the anterior part of the spine medial to the carotid sheath. Superior thyroid nerve and vessels and superior laryngeal nerve were identified and preserved. Fluoroscopy confirmed correct location at C2. The tumor was easily visualized, and the outer shell broke easily with palpation. Gentle curettage was necessary when removing the tumor off the vertebral artery. A portion of the specimen was sent during surgery for frozen section, which showed infrequent mitotic figures and no other findings concerning for malignancy. No instability was created after curettage and excision of the tumor, so no grafting or instrumentation was necessary.
Grossly, the tumor was pale tan and firm. Histologic examination with hematoxylin-eosin staining revealed a bland spindle-cell neoplasm that focally involved bone. A storiform pattern was present. The cells had scant cytoplasm and oval to elongate nuclei with tapered ends. Significant nuclear pleomorphism was not seen. The stroma was loose, with focal myxoid change. Benign multinucleated giant cells were present. Mitotic activity was infrequent (Figures 3A–3D). Two attending pathologists reviewed the case material and the frozen and formalin-fixed specimens independently and concurred with the diagnosis of BFH. In addition, the case was reviewed at the surgical pathology consensus conference; the reviewers agreed on BFH, and additional studies were deemed unnecessary.
Given the patient’s complete clinical picture, the differential diagnosis included nonossifying fibroma (NOF), eosinophilic granuloma (EG), BFH, fibrous dysplasia, giant cell tumor (GCT), aneurysmal bone cyst (ABC), and osteoblastoma (OB).
Discussion
BFH is an extremely rare bone lesion, accounting for only 1% of all surgically managed bone tumors; not counting the present case, only 11 spine cases have been reported in the literature.1,2 BFH of the spine traditionally causes nonspecific, poorly localized pain. The Table lists the reported cases of spinal BFH and their presenting symptoms, location, and treatment. BFH usually occurs in young adults, but the age range is 5 to 75 years.2-4 Mean age of the 12 patients with spinal BFH in the literature (including ours) is 25 years.1 In addition, spinal BFH appears to have no predilection for sex.
Skeletal BFH presents as a discrete, well-defined, osteolytic lesion with sharp borders and potentially a sclerotic rim.4-6 Cortical expansion and even cortical disruption with invasion into adjacent tissue have occurred in flat bones.7 Histologically, BFHs contain spindle cells, multinucleated giant cells, and foam cells in storiform pattern.6
BFH shares many of its radiologic and histologic characteristics and clinical symptoms with other benign bone lesions (the tumors listed above). Therefore, accurate diagnosis of BFH requires appropriate correlation of clinical, radiographic, and histologic data.2,3,8 Below is a comparison of BFH with related bone lesions.
Spinal BFH causes a nonspecific, poorly localized pain similar to that of EG, ABC, GCT, and OB.3,9 NOF and fibrous dysplasia generally do not cause pain, unless these lesions are discovered secondary to a pathologic fracture.8,10,11 Our patient had minor antecedent neck pain, which was brought to light by his football accident. ABC and OB are more locally aggressive than BFH and can cause neurologic symptoms by mass effect and spinal cord or nerve root compression.1,8 In this case and in the 6 other cases of BFH of the cervical spine, there were no neurologic changes.4,10
Of the tumors mentioned, NOF and EG almost always occur in children. However, NOF usually occurs in the metaphyseal region of long bones, and EG is usually accompanied by systemic symptoms, such as lymphadenopathy, hepatomegaly, and increased inflammatory markers.1,8 Fibrous dysplasia usually presents in childhood but does not become symptomatic until adulthood. GCTs and OB predominantly occur in adulthood.12,13 Our patient’s age and lack of other systemic symptoms supported the diagnosis of BFH.
Appearance on MRI is reported less with BFH than with other tumors, but heterogenous signal intensity similar to that of skeletal muscle on T1-weighted images and high signal intensity on T2-weighted images is typically reported.8,14 NOF and fibrous dysplasia do not disrupt the bony cortex unless a pathologic fracture has occurred.4 GCTs are more aggressive lytic lesions with more aggressive radiologic features. GCTs generally cause cortical expansion/attenuation, and lack a sclerotic rim. GCTs also have a heterogenous appearance on MRI and give a low to intermediate signal on both T1- and T2-weighted images.12,15 The appearance of EG is similar to that of BFH as an osteolytic lesion with a sclerotic rim, though EGs typically break through the cortex and acquire a “punched-out” look.1,8 ABC typically is described as an expansile osteolytic lesion with a “soap-bubble” appearance on radiographs; periosteal elevation and cortical attenuation can also be visualized. MRI shows the typical multilobular appearance of the lesion with fluid levels.13
OB appears as a radiolucent lesion, with or without calcifications, surrounded by a thin margin of reactive bone.14,16 A distinguishing characteristic of OB was thought to be intense radioisotope uptake on bone scintigraphy, but recently a bony BFH demonstrated intense uptake.17 OBs typically demonstrate nonspecific MRI results similar to those of BFH: low to intermediate signal on T1-weighted images and intermediate to high signal on T2-weighted images.13 In our patient’s case, the radiographic appearance and lack of specific radiographic findings consistent with the other tumors supported the diagnosis of BFH.
Histologically, BFHs contain spindle cells, multinucleated giant cells, and foam cells in a storiform pattern6 which was demonstrated in our patient’s case. In addition, significant nuclear pleomorphism, mitotic activity, and necrosis were absent—a difference between BFH and malignant fibrous histiocytoma.4,15 The microscopic characteristics of BFH readily differentiate it from OB, ABC, EG, and GCT, but not from NOF on microscopic appearance alone. Clinical and radiographic findings must be consistent, as mentioned.7,18
Complete surgical excision is the reported treatment for BFH. Prognosis after resection or curettage is usually good, and recurrences have been rare.1,2 Depending on the intraspinous location of BFH, stabilization after resection or curettage may be necessary to prevent residual instability. Three of the 11 reported cases of spinal BFH required stabilization by anterior fusion or posterior pedicle screw fixation after resection.1,2 The other 8 cases underwent excision alone or excision and grafting. All 11 patients were disease-free at a mean follow-up of 3.5 years.1 In nonspinal BFH, however, both local recurrence and lung metastasis have been reported.2,5,9,19 Clarke and colleagues9 reported local recurrences in 3 of 8 cases. These recurrences involved BFH in long bones of the leg, which had been treated with curettage and grafting. There has been no reliable report of a malignant change in BFH.2,9 The only case of lung metastasis, reported by Unni and Dahlin6 in their study of 10 cases, occurred 2 years after local recurrence in the distal femur.Our patient was doing well at most recent follow-up, 6 months after surgery. He had no pain and had returned to normal activities. Although there are no reported cases of spinal BFH recurrence, we will follow this patient with imaging on an annual basis. His case is of particular interest to orthopedic surgeons because they encounter benign bone lesions every day, and many of these lesions are in difficult anatomical locations. Knowing the characteristics, differential diagnoses, and appropriate diagnostic workups for benign bone lesions is important for optimal and timely patient care.
Benign fibrous histiocytoma (BFH) is a rare, well-recognized, primary skeletal tumor accounting for approximately 1% of all benign bone tumors. Spinal involvement is exceedingly rare with only 11 cases reported in the literature.1,2 We present a case of BFH located in the cervical spine of a pediatric patient that was successfully treated with curretage through an anterior surgical approach, along with a review of the literature and appropriate management concerning BFH of the spine.
Case Report
A 14-year-old boy was tackled while playing football and noticed immediate neck pain and subjective paresthesia in the upper extremities. Examination revealed a nontender spine (cervical, thoracic, lumbar) and normal strength and range of motion in all extremities. Sensation was diffusely intact, long tract signs were absent, and gait was normal. On questioning, the patient endorsed mild antecedent neck pain but denied prior history of any trauma. Neck pain did not radiate and was slightly worsened by activity but was mostly intermittent and random. As the neck pain was very mild and was not interfering with daily activities, the patient had not sought care before presenting to the emergency department. He had no pertinent past medical or surgical history.
The patient presented with a computed tomography (CT) scan of his head and cervical spine and a magnetic resonance imaging (MRI) scan of the cervical spine. A magnetic resonance angiography (MRA) scan of the neck was ordered after his arrival.
Axial and sagittal CT (Figures 1A, 1B) showed a 1×1.2-cm discrete, expansile, lytic, radiolucent mass extending anterior from the left C2 vertebral body. The mass appeared to abut the left vertebral artery foramen. The cortical bone surrounding the lesion was thin but uniform. Sagittal and axial T1-weighted MRI (Figures 2A, 2B) showed the discrete, expansile, homogenous lesion with the same intensity as normal bone marrow. Sagittal and axial T2-weighted MRI (Figures 2C, 2D) showed a discrete, expansile, homogenous lesion with primarily high signal intensity. Sagittal short tau inversion recovery (STIR) MRI (Figure 2E) again showed the lesion with primarily low intensity. Given the close proximity of the lesion to the vertebral foramen, MRA was ordered; it showed the lesion was not interfering with the vertebral artery (Figure 2F).
The tumor’s location, in the left anterior aspect of the C2 vertebral body, was not conducive to percutaneous biopsy for establishing tissue diagnosis, so the decision was made to surgically excise the lesion. A left-sided anterior incision was made 2 fingerbreadths inferior to the jaw line in a neck crease. A head and neck surgeon assisted with dissection. Dissection was carried down through the skin, subcutaneous tissue, and platysma on to the anterior part of the spine medial to the carotid sheath. Superior thyroid nerve and vessels and superior laryngeal nerve were identified and preserved. Fluoroscopy confirmed correct location at C2. The tumor was easily visualized, and the outer shell broke easily with palpation. Gentle curettage was necessary when removing the tumor off the vertebral artery. A portion of the specimen was sent during surgery for frozen section, which showed infrequent mitotic figures and no other findings concerning for malignancy. No instability was created after curettage and excision of the tumor, so no grafting or instrumentation was necessary.
Grossly, the tumor was pale tan and firm. Histologic examination with hematoxylin-eosin staining revealed a bland spindle-cell neoplasm that focally involved bone. A storiform pattern was present. The cells had scant cytoplasm and oval to elongate nuclei with tapered ends. Significant nuclear pleomorphism was not seen. The stroma was loose, with focal myxoid change. Benign multinucleated giant cells were present. Mitotic activity was infrequent (Figures 3A–3D). Two attending pathologists reviewed the case material and the frozen and formalin-fixed specimens independently and concurred with the diagnosis of BFH. In addition, the case was reviewed at the surgical pathology consensus conference; the reviewers agreed on BFH, and additional studies were deemed unnecessary.
Given the patient’s complete clinical picture, the differential diagnosis included nonossifying fibroma (NOF), eosinophilic granuloma (EG), BFH, fibrous dysplasia, giant cell tumor (GCT), aneurysmal bone cyst (ABC), and osteoblastoma (OB).
Discussion
BFH is an extremely rare bone lesion, accounting for only 1% of all surgically managed bone tumors; not counting the present case, only 11 spine cases have been reported in the literature.1,2 BFH of the spine traditionally causes nonspecific, poorly localized pain. The Table lists the reported cases of spinal BFH and their presenting symptoms, location, and treatment. BFH usually occurs in young adults, but the age range is 5 to 75 years.2-4 Mean age of the 12 patients with spinal BFH in the literature (including ours) is 25 years.1 In addition, spinal BFH appears to have no predilection for sex.
Skeletal BFH presents as a discrete, well-defined, osteolytic lesion with sharp borders and potentially a sclerotic rim.4-6 Cortical expansion and even cortical disruption with invasion into adjacent tissue have occurred in flat bones.7 Histologically, BFHs contain spindle cells, multinucleated giant cells, and foam cells in storiform pattern.6
BFH shares many of its radiologic and histologic characteristics and clinical symptoms with other benign bone lesions (the tumors listed above). Therefore, accurate diagnosis of BFH requires appropriate correlation of clinical, radiographic, and histologic data.2,3,8 Below is a comparison of BFH with related bone lesions.
Spinal BFH causes a nonspecific, poorly localized pain similar to that of EG, ABC, GCT, and OB.3,9 NOF and fibrous dysplasia generally do not cause pain, unless these lesions are discovered secondary to a pathologic fracture.8,10,11 Our patient had minor antecedent neck pain, which was brought to light by his football accident. ABC and OB are more locally aggressive than BFH and can cause neurologic symptoms by mass effect and spinal cord or nerve root compression.1,8 In this case and in the 6 other cases of BFH of the cervical spine, there were no neurologic changes.4,10
Of the tumors mentioned, NOF and EG almost always occur in children. However, NOF usually occurs in the metaphyseal region of long bones, and EG is usually accompanied by systemic symptoms, such as lymphadenopathy, hepatomegaly, and increased inflammatory markers.1,8 Fibrous dysplasia usually presents in childhood but does not become symptomatic until adulthood. GCTs and OB predominantly occur in adulthood.12,13 Our patient’s age and lack of other systemic symptoms supported the diagnosis of BFH.
Appearance on MRI is reported less with BFH than with other tumors, but heterogenous signal intensity similar to that of skeletal muscle on T1-weighted images and high signal intensity on T2-weighted images is typically reported.8,14 NOF and fibrous dysplasia do not disrupt the bony cortex unless a pathologic fracture has occurred.4 GCTs are more aggressive lytic lesions with more aggressive radiologic features. GCTs generally cause cortical expansion/attenuation, and lack a sclerotic rim. GCTs also have a heterogenous appearance on MRI and give a low to intermediate signal on both T1- and T2-weighted images.12,15 The appearance of EG is similar to that of BFH as an osteolytic lesion with a sclerotic rim, though EGs typically break through the cortex and acquire a “punched-out” look.1,8 ABC typically is described as an expansile osteolytic lesion with a “soap-bubble” appearance on radiographs; periosteal elevation and cortical attenuation can also be visualized. MRI shows the typical multilobular appearance of the lesion with fluid levels.13
OB appears as a radiolucent lesion, with or without calcifications, surrounded by a thin margin of reactive bone.14,16 A distinguishing characteristic of OB was thought to be intense radioisotope uptake on bone scintigraphy, but recently a bony BFH demonstrated intense uptake.17 OBs typically demonstrate nonspecific MRI results similar to those of BFH: low to intermediate signal on T1-weighted images and intermediate to high signal on T2-weighted images.13 In our patient’s case, the radiographic appearance and lack of specific radiographic findings consistent with the other tumors supported the diagnosis of BFH.
Histologically, BFHs contain spindle cells, multinucleated giant cells, and foam cells in a storiform pattern6 which was demonstrated in our patient’s case. In addition, significant nuclear pleomorphism, mitotic activity, and necrosis were absent—a difference between BFH and malignant fibrous histiocytoma.4,15 The microscopic characteristics of BFH readily differentiate it from OB, ABC, EG, and GCT, but not from NOF on microscopic appearance alone. Clinical and radiographic findings must be consistent, as mentioned.7,18
Complete surgical excision is the reported treatment for BFH. Prognosis after resection or curettage is usually good, and recurrences have been rare.1,2 Depending on the intraspinous location of BFH, stabilization after resection or curettage may be necessary to prevent residual instability. Three of the 11 reported cases of spinal BFH required stabilization by anterior fusion or posterior pedicle screw fixation after resection.1,2 The other 8 cases underwent excision alone or excision and grafting. All 11 patients were disease-free at a mean follow-up of 3.5 years.1 In nonspinal BFH, however, both local recurrence and lung metastasis have been reported.2,5,9,19 Clarke and colleagues9 reported local recurrences in 3 of 8 cases. These recurrences involved BFH in long bones of the leg, which had been treated with curettage and grafting. There has been no reliable report of a malignant change in BFH.2,9 The only case of lung metastasis, reported by Unni and Dahlin6 in their study of 10 cases, occurred 2 years after local recurrence in the distal femur.Our patient was doing well at most recent follow-up, 6 months after surgery. He had no pain and had returned to normal activities. Although there are no reported cases of spinal BFH recurrence, we will follow this patient with imaging on an annual basis. His case is of particular interest to orthopedic surgeons because they encounter benign bone lesions every day, and many of these lesions are in difficult anatomical locations. Knowing the characteristics, differential diagnoses, and appropriate diagnostic workups for benign bone lesions is important for optimal and timely patient care.
1. Demiralp B, Kose O, Oguz E, Sanal T, Ozcan A, Sehirlioglu A. Benign fibrous histiocytoma of the lumbar vertebrae. Skeletal Radiol. 2009;38(2):187-191.
2. Kuruvath S, O’Donovan DG, Aspoas AR, David KM. Benign fibrous histiocytoma of the thoracic spine: case report and review of the literature. J Neurosurg Spine. 2006;4(3):260-264.
3. Ceroni D, Dayer R, De Coulon G, Kaelin A. Benign fibrous histiocytoma of bone in a paediatric population: a report of 6 cases. Musculoskelet Surg. 2011;95(2):107-114.
4. Dorfman HD, Czerniak B. Bone Tumors. St. Louis, MO: Mosby; 1998.
5. Grohs JG, Nicolakis M, Kainberger F, Lang S, Kotz R. Benign fibrous histiocytoma of bone: a report of ten cases and review of literature. Wien Klin Wochenschr. 2002;114(1-2):56-63.
6. Unni KK, Dahlin DC. Dahlin’s Bone Tumors. 5th ed. Philadelphia, PA: Lippincott-Raven; 1996.
7. Balasubramanian C, Rajaraman G, Singh CS, Baliga DK. Benign fibrous histiocytoma of the sacrum—diagnostic difficulties facing this rare bone tumor. Pediatr Neurosurg. 2005;41(5):253-257.
8. van Giffen NH, van Rhijn LW, van Ooij A, et al. Benign fibrous histiocytoma of the posterior arch of C1 in a 6-year old boy: a case report. Spine. 2003;28(18):E359-E363.
9. Clarke BE, Xipell JM, Thomas DP. Benign fibrous histiocytoma of bone. Am J Surg Pathol. 1985;9(11):806-815.
10. Peicha G, Siebert FJ, Bratschitsch G, Fankhauser F, Grechenig W. Pathologic odontoid fracture and benign fibrous histiocytoma of bone. Eur Spine J. 1999;8(2):161-163.
11. Unni KK, Inwards CY, Bridge JA, Kindblom LG, Wold LE. Tumors of the Bones and Joints (AFIP Atlas of Tumor Pathology Series IV). Annapolis Junction, MD: American Registry of Pathology Press; 2005.
12. Dee R. Principles of Orthopaedic Practice. 2nd ed. New York, NY: McGraw-Hill; 1997.
13. Murphey M, Andrews C, Flemming D, Temple HT, Smith WS, Smirniotopoulos JG. Primary tumors of the spine: radiologic–pathologic correlation. Radiographics. 1996;16(5):1131-1158.
14. Hamada T, Ito H, Araki Y, Fujii K, Inoue M, Ishida O. Benign fibrous histiocytoma of the femur: review of three cases. Skeletal Radiol. 1996;25(1):25-29.
15. Mirra JM, Picci P, Gold RH. Bone Tumors: Clinical, Radiologic, and Pathologic Correlations. Vol 1. Philadelphia, PA: Lea & Febiger; 1989.
16. Theodorou DJ, Theodorou SJ, Sartoris DJ. An imaging overview of primary tumors of the spine: part 1. Benign tumors. Clin Imaging. 2008;32(3):196-203.
17. Li X, Meng Z, Li D, Tan J, Song X. Benign fibrous histiocytoma of a rib. Clin Nucl Med. 2014;39(9): 837-841.
18. Roessner A, Immenkamp M, Weidner A, Hobik HP, Grundmann E. Benign fibrous histiocytoma of bone. Light- and electron-microscopic observations. J Cancer Res Clin Oncol. 1981;101(2):191-202.
19. Destouet JM, Kyriakos M, Gilula LA. Fibrous histiocytoma (fibroxanthoma) of a cervical vertebra. A report with a review of the literature. Skeletal Radiol. 1980;5(4):241-246.
20. Hoeffel JC, Bomand-Ferrand F, Tachet F, Lascombes P, Czorny A, Bernard C. So-called benign fibrous histiocytoma: report of a case. J Pediatr Surg. 1992;27(5):672-674.
1. Demiralp B, Kose O, Oguz E, Sanal T, Ozcan A, Sehirlioglu A. Benign fibrous histiocytoma of the lumbar vertebrae. Skeletal Radiol. 2009;38(2):187-191.
2. Kuruvath S, O’Donovan DG, Aspoas AR, David KM. Benign fibrous histiocytoma of the thoracic spine: case report and review of the literature. J Neurosurg Spine. 2006;4(3):260-264.
3. Ceroni D, Dayer R, De Coulon G, Kaelin A. Benign fibrous histiocytoma of bone in a paediatric population: a report of 6 cases. Musculoskelet Surg. 2011;95(2):107-114.
4. Dorfman HD, Czerniak B. Bone Tumors. St. Louis, MO: Mosby; 1998.
5. Grohs JG, Nicolakis M, Kainberger F, Lang S, Kotz R. Benign fibrous histiocytoma of bone: a report of ten cases and review of literature. Wien Klin Wochenschr. 2002;114(1-2):56-63.
6. Unni KK, Dahlin DC. Dahlin’s Bone Tumors. 5th ed. Philadelphia, PA: Lippincott-Raven; 1996.
7. Balasubramanian C, Rajaraman G, Singh CS, Baliga DK. Benign fibrous histiocytoma of the sacrum—diagnostic difficulties facing this rare bone tumor. Pediatr Neurosurg. 2005;41(5):253-257.
8. van Giffen NH, van Rhijn LW, van Ooij A, et al. Benign fibrous histiocytoma of the posterior arch of C1 in a 6-year old boy: a case report. Spine. 2003;28(18):E359-E363.
9. Clarke BE, Xipell JM, Thomas DP. Benign fibrous histiocytoma of bone. Am J Surg Pathol. 1985;9(11):806-815.
10. Peicha G, Siebert FJ, Bratschitsch G, Fankhauser F, Grechenig W. Pathologic odontoid fracture and benign fibrous histiocytoma of bone. Eur Spine J. 1999;8(2):161-163.
11. Unni KK, Inwards CY, Bridge JA, Kindblom LG, Wold LE. Tumors of the Bones and Joints (AFIP Atlas of Tumor Pathology Series IV). Annapolis Junction, MD: American Registry of Pathology Press; 2005.
12. Dee R. Principles of Orthopaedic Practice. 2nd ed. New York, NY: McGraw-Hill; 1997.
13. Murphey M, Andrews C, Flemming D, Temple HT, Smith WS, Smirniotopoulos JG. Primary tumors of the spine: radiologic–pathologic correlation. Radiographics. 1996;16(5):1131-1158.
14. Hamada T, Ito H, Araki Y, Fujii K, Inoue M, Ishida O. Benign fibrous histiocytoma of the femur: review of three cases. Skeletal Radiol. 1996;25(1):25-29.
15. Mirra JM, Picci P, Gold RH. Bone Tumors: Clinical, Radiologic, and Pathologic Correlations. Vol 1. Philadelphia, PA: Lea & Febiger; 1989.
16. Theodorou DJ, Theodorou SJ, Sartoris DJ. An imaging overview of primary tumors of the spine: part 1. Benign tumors. Clin Imaging. 2008;32(3):196-203.
17. Li X, Meng Z, Li D, Tan J, Song X. Benign fibrous histiocytoma of a rib. Clin Nucl Med. 2014;39(9): 837-841.
18. Roessner A, Immenkamp M, Weidner A, Hobik HP, Grundmann E. Benign fibrous histiocytoma of bone. Light- and electron-microscopic observations. J Cancer Res Clin Oncol. 1981;101(2):191-202.
19. Destouet JM, Kyriakos M, Gilula LA. Fibrous histiocytoma (fibroxanthoma) of a cervical vertebra. A report with a review of the literature. Skeletal Radiol. 1980;5(4):241-246.
20. Hoeffel JC, Bomand-Ferrand F, Tachet F, Lascombes P, Czorny A, Bernard C. So-called benign fibrous histiocytoma: report of a case. J Pediatr Surg. 1992;27(5):672-674.
Necrotizing Cellulitis With Multiple Abscesses on the Leg Caused by Serratia marcescens
A gram-negative bacillus of the Enterobacteriaceae family, Serratia marcescens is an organism known to cause bacteremia, pneumonia, urinary tract infection, endocarditis, meningitis, and septic arthritis.1 Unusual cases of cellulitis and necrotizing fasciitis (NF) caused by S marcescens also have been reported.2,3 This entity has been initially described in immunocompromised and nonimmunocompromised patients.4 Both community and nosocomial cases also have been reported.3
Case Report
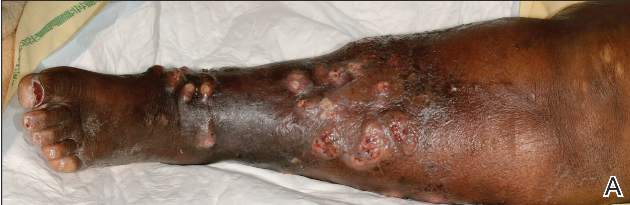
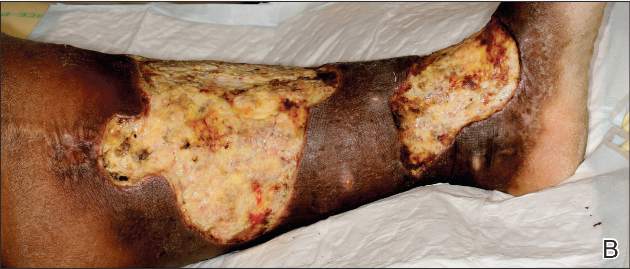
A 68-year-old morbidly obese woman with high blood pressure, diabetes mellitus, chronic renal insufficiency, chronic venous insufficiency, and left leg lymphoedema was referred to our emergency unit. She had pain and circumferential erythema with multiple abscesses of the left leg of 2 weeks’ duration. No history of trauma, ulcer, injection, or animal bite was noted. At the time of presentation she had no fever and vital parameters were normal. Empirical treatment with oral amoxicillin (6 g daily) and amoxicillin-clavulanate (375 mg daily) was started. Forty-eight hours later, inflammation, pain, and abscesses worsened (Figure 1A). Laboratory tests showed an elevated white blood cell count (15.9×109⁄L with 86% neutrophils [reference range, 4.5–11.0×109⁄L]) and an elevated C-reactive protein level (322 mg/L [reference range, <2 mg/L]). Human immunodeficiency virus serology was negative. Needle aspiration of an abscess yielded S marcescens. A second aspiration confirmed the presence of the same organism, wild-type S marcescens, which was resistant to amoxicillin and clavulanic acid, first-generation cephalosporin, and tobramycin but sensitive to piperacillin, third-generation cephalosporins, amikacin, ciprofloxacin, and co-trimoxazole. Intravenous cefepime, a third-generation cephalosporin, was started. During the next 48 hours the patient developed severe sepsis with confusion, acute renal failure (creatinine: 231 µmol/L vs 138 µmol/L at baseline [reference range, 53–106 µmol/L), and worsening of skin lesions. Blood cultures were negative and amikacin was added. Magnetic resonance imaging showed a diffuse inflammatory process involving the skin and subcutaneous tissue that extended to the soleus fascia with no other muscle involvement or deep collection (Figure 2). Surgical debridement of infected tissues was performed (Figure 1B). Histologic examination revealed spreading suppurative inflammation involving the dermis and subcutaneous tissues. Clinical healing was obtained after 21 days of antimicrobial therapy. The debrided area required skin grafting 2 months later (Figure 1C).
 |
 |
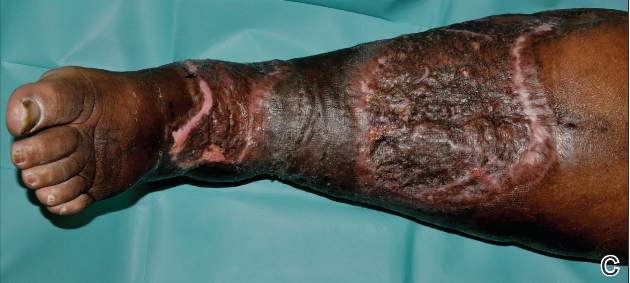 |
| Figure 1. Erythema with multiple abscesses on the left ankle and leg at presentation (A), day 1 following surgical debridement of infected tissues (B), and 2 months later with complete healing following a skin graft (C). |

Comment
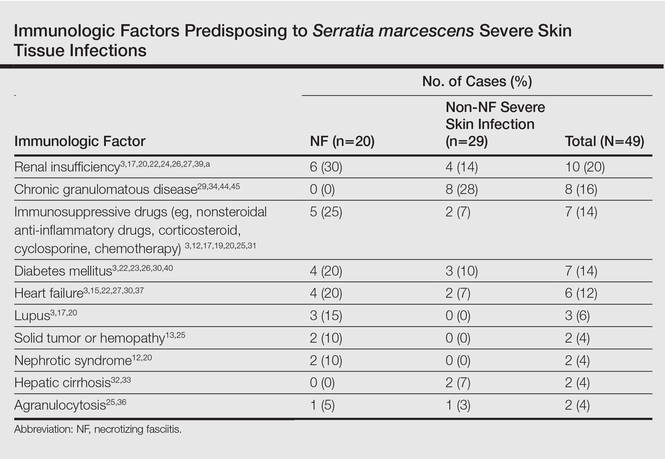
The most common causative bacteria of cellulitis are Staphylococcus aureus and group A β-hemolytic streptococci. Serratia marcescens is a rare but increasingly recognized pathogen of skin and soft tissue infections.5 The proposed pathogenic mechanism for skin necrosis during S marcescens infection is the bacterial production of large proteases (eg, deoxyribonuclease, lipase, gelatinase).6 Injection of purified proteinase from S marcescens into rat skin leads to increased vascular permeability, necrosis of epidermal tissue, dermal inflammation and edema, and infiltration of polymorphonuclear leukocytes into the subcutaneous fat and muscle.7Serratia marcescens is ubiquitous in soil and water and it also may colonize the respiratory, urinary, and digestive tracts in humans. Cellulitis due to S marcescens secondary to iguana bites8,9 and snake bites10 or leech-borne cellulitis11 suggest that the oral cavity of these animals may be colonized. To date, 49 cases of severe S marcescens skin infections have been described, according to a search of PubMed articles indexed for MEDLINE using the terms Serratia marcescens and skin, cutaneous, soft tissue, and cellulitis or necrotizing fasciitis: 20 cases with NF3,12-28 and 29 non-NF cases8-11,29-46 (typical cellulitis presentation [n=8]9,11,35-38,40; abscesses, gumma, or pyoderma gangrenosum–like lesions associated with chronic granulomatous disease in childhood [n=7]29,44,45; painful nodules with secondary abscesses [n=6]31-34,46; acute bullous cellulitis [n=4]8,10,30; secondary infections of ulcers [n=2]35,40; abscesses in immunocompetent patient [n=1]41; and necrotizing skin ulceration [n=1]36). Lower extremities were frequently involved (NF cases, n=13; non-NF cases, n=16). Underlying immunosuppression was observed in 14 NF cases and in 17 non-NF cases. Predisposing immunologic factors are summarized in the Table. Local risk factors, including chronic leg edema, trauma, surgical wound, filler injection, and ulcer, were frequently reported in NF and non-NF cases,16,20,26-28,31,32,34,35,37,38,40,46 including our case. Surgery was required in 19 NF cases and in 7 non-NF cases. Serratia marcescens–mediated NF led to higher mortality (n=12) than non-NF cases (n=1). Other nonsevere clinical manifestations of S marcescens infection reported in the literature included disseminated papular eruptions with human immunodeficiency virus infection42 and trunk folliculitis.43 Our patient had many risk factors, including chronic edema, diabetes mellitus, chronic renal insufficiency, and chronic venous insufficiency. The potential presence of abscesses and necrotic tissue hinders antibiotic penetration at the infection site, and surgery should be systematically considered as early as possible in view of the high mortality rate of S marcescens cellulitis.
Conclusion
Although uncommon, an S marcescens skin infection may be suspected in cases of cellulitis in immunocompromised patients, especially when conventional antibiotics are not effective. Serratia marcescens naturally produces a cephalosporinase that confers resistance to amoxicillin and to amoxicillin associated with clavulanic acid. Broad-spectrum antibiotics such as third-generation cephalosporins, fluoroquinolones, or imipenem-cilastatin are indicated in cases of S marcescens skin infections, and surgery should be promptly considered if appropriate antibiotic therapy does not lead to rapid clinical improvement.
1. Engel HJ, Collignon PJ, Whiting PT, et al. Serratia sp. bacteremia in Canberra, Australia: a population-based study over 10 years. Eur J Clin Microbiol Infect Dis. 2009;28:821-824.
2. Brenner DE, Lookingbill DP. Serratia marcescens cellulitis. Arch Dermatol. 1977;113:1599-1600.
3. Rehman T, Moore TA, Seoane L. Serratia marcescens necrotizing fasciitis presenting as bilateral breast necrosis. J Clin Microbiol. 2012;50:3406-3408.
4. Yu VL. Serratia marcescens: historical perspective and clinical review. N Engl J Med. 1979;300:887-893.
5. Moet GJ, Jones RN, Biedenbach DJ, et al. 2007. Contemporary causes of skin and soft tissue infections in North America, Latin America, and Europe: report from the SENTRY Antimicrobial Surveillance Program (1998–2004). Diagn Microbiol Infect Dis. 2007;57:7-13.
6. Aucken HM, Pitt TL. Antibiotic resistance and putative virulence factors of Serratia marcescens with respect to O and K serotypes. J Med Microbiol. 1998;47:1105-1113.
7. Conroy MC, Bander NH, Lepow IH. Effect in the rat of intradermal injection of purified proteinases from Streptococcus and Serratia marcescens. Proc Soc Exp Biol Med. 1975;150:801-806.
8. Grim KD, Doherty C, Rosen T. Serratia marcescens bullous cellulitis after iguana bites. J Am Acad Dermatol. 2010;62:1075-1076.
9. Hsieh S, Babl FE. Serratia marcescens cellulitis following an iguana bite. Clin Infect Dis. 1999;28:1181-1182.
10. Subramani P, Narasimhamurthy GB, Ashokan B, et al. Serratia marcescens: an unusual pathogen associated with snakebite cellulitis. J Infect Dev Ctries. 2013;7:152-154.
11. Pereira JA, Greig JR, Liddy H, et al. Leech-borne Serratia marcescens infection following complex hand injury. Br J Plast Surg. 1998;51:640-641.
12. Wen YK. Necrotizing fasciitis caused by Serratia marcescens: a fatal complication of nephrotic syndrome. Ren Fail. 2012;34:649-652.
13. Prelog T, Jereb M, Cuček I, et al. Necrotizing fasciitis caused by Serratia marcescens after venous access port implantation in a child with acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2012;34:e246-e248.
14. Meisel M, Schultz-Coulon HJ. Life-threatening necrotizing fasciitis colli caused by Serratia marcescens [in German]. HNO. 2009;57:1071-1074.
15. Campos GA, Burgos LAM, Fica CA, et al. Fatal necrotizing fasciitis due to Serratia marcescens [in Spanish]. Rev Chilena Infectol. 2007;24:319-322.
16. Bustamante Rodríguez R, Bustamante Rodríguez E, Obón Azuara B. Community-acquired necrotizing fasciitis by Serratia marcescens [in Spanish]. Med Clin (Barc). 2008;130:198-199.
17. Pascual J, Liaño F, Rivera M, et al. Necrotizing myositis secondary to Serratia marcescens in a renal allograft recipient. Nephron. 1990;55:329-331.
18. Statham MM, Vohra A, Mehta DK, et al. Serratia marcescens causing cervical necrotizing oropharyngitis. Int J Pediatr Otorhinolaryngol. 2009;73:467-473.
19. Rimailho A, Riou B, Richard C, et al. Fulminant necrotizing fasciitis and nonsteroidal anti-inflammatory drugs. J Infect Dis. 1987;155:143-146.
20. Huang JW, Fang CT, Hung KY, et al. Necrotizing fasciitis caused by Serratia marcescens in two patients receiving corticosteroid therapy. J Formos Med Assoc. 1999;98:851-854.
21. Newton CL, deLemos D, Abramo TJ, et al. Cervical necrotizing fasciitis caused by Serratia marcescens in a 2 year old. Pediatr Emerg Care. 2002;18:433-435.
22. Curtis CE, Chock S, Henderson T, et al. A fatal case of necrotizing fasciitis caused by Serratia marcescens. Am Surg. 2005;71:228-230.
23. Zipper RP, Bustamante MA, Khatib R. Serratia marcescens: a single pathogen in necrotizing fasciitis. Clin Infect Dis. 1996;23:648-649.
24. Liangpunsakul S, Pursell K. Community-acquired necrotizing fasciitis caused by Serratia marcescens: case report and review. Eur J Clin Microbiol Infect Dis. 2001;20:509-510.
25. Vano-Galvan S, Álvarez-Twose I, Moreno-Martín P, et al. Fulminant necrotizing fasciitis caused by Serratia marcescens in an immunosuppressed host. Int J Dermatol. 2014;53:e57-e58.
26. Majumdar R, Crum-Cianflone NF. Necrotizing fasciitis due to Serratia marcescens: case report and review of the literature [published online October 23, 2015]. Infection. doi:10.1007/s15010-015-0855-x.
27. Cope TE, Cope W, Beaumont DM. A case of necrotising fasciitis caused by Serratia marcescens: extreme age as functional immunosuppression? Age Ageing. 2013;42:266-268.
28. Lakhani NA, Narsinghani U, Kumar R. Necrotizing fasciitis of the abdominal wall caused by Serratia marcescens. Infect Dis Rep. 2015;157:5774.
29. Friend JC, Hilligoss DM, Marquesen M, et al. Skin ulcers and disseminated abscesses are characteristic of Serratia marcescens infection in older patients with chronic granulomatous disease [published online May 27, 2009]. J Allergy Clin Immunol. 2009;124:164-166.
30. Cooper CL, Wiseman M, Brunham R. Bullous cellulitis caused by Serratia marcescens. Int J Infect Dis. 1998;3:36-38.
31. Langrock ML, Linde HJ, Landthaler M, et al. Leg ulcers and abscesses caused by Serratia marcescens. Eur J Dermatol. 2008;18:705-707.
32. João AM, Serrano PN, Cachão MP, et al. Recurrent Serratia marcescens cutaneous infection manifesting as painful nodules and ulcers. J Am Acad Dermatol. 2008;58(2 suppl):S55-S57.
33. Friedman DN, Peterson NB, Sumner WT, et al. Spontaneous dermal abscesses and ulcers as a result of Serratia marcescens. J Am Acad Dermatol. 2003;49:S193-S194.
34. Soria X, Bielsa I, Ribera M, et al. Acute dermal abscesses caused by Serratia marcescens. J Am Acad Dermatol. 2008;58:891-893.
35. Bogaert MA, Hogan DJ, Miller AE Jr. Serratia cellulitis and secondary infection of leg ulcers by Serratia. J Am Acad Dermatol. 1991;25:565.
36. Gössl M, Eggebrecht H. Necrotizing skin ulceration in antibiotic-induced agranulocytosis. Mayo Clin Proc. 2006;81:1527.
37. Brenner DE, Lookingbill DP. Serratia marcescens cellulitis. Arch Dermatol. 1977;113:1599-1600.
38. Bonner MJ, Meharg JG Jr. Primary cellulitis due to Serratia marcescens. JAMA. 1983;250:2348-2349.
39. Bornstein PF, Ditto AM, Noskin GA. Serratia marcescens cellulitis in a patient on hemodialysis. Am J Nephrol. 1992;12:374-376.
40. Kaplan H, Sehtman L, Ricover N, et al. Serratia marcescens: cutaneous involvement. preliminary report. Med Cutan Ibero Lat Am. 1988;16:305-308.
41. Giráldez P, Mayo E, Pavón P, et al. Skin infection due to Serratia marcescens in an immunocompetent patient [in Spanish]. Actas Dermosifiliogr. 2011;102:236-237.
42. Muñoz-Pérez MA, Rodriguez-Pichardo A, Camacho F. Disseminated papular eruption caused by Serratia marcescens: a new cutaneous manifestation in HIV-positive patients. AIDS. 1996;10:1179-1180.
43. Lehrhoff S, Yost M, Robinson M, et al. Serratia marcescens folliculitis and concomitant acne vulgaris. Dermatol Online J. 2012;18:19.
44. Benajiba N, Amrani R, Rkain M, et al. Serratia marcescens cutaneous gumma and chronic septic granulomatosis. Med Mal Infect. 2014;44:39-41.
45. Barbato M, Ragusa G, Civitelli F, et al. Chronic granulomatous disease mimicking early-onset Crohn’s disease with cutaneous manifestations. BMC Pediatr. 2014;14:156.
46. Park KY, Seo SJ. Cutaneous Serratia marcescens infection in an immunocompetent patient after filler injection. Acta Derm Venereol. 2013;93:191-192.
A gram-negative bacillus of the Enterobacteriaceae family, Serratia marcescens is an organism known to cause bacteremia, pneumonia, urinary tract infection, endocarditis, meningitis, and septic arthritis.1 Unusual cases of cellulitis and necrotizing fasciitis (NF) caused by S marcescens also have been reported.2,3 This entity has been initially described in immunocompromised and nonimmunocompromised patients.4 Both community and nosocomial cases also have been reported.3
Case Report
A 68-year-old morbidly obese woman with high blood pressure, diabetes mellitus, chronic renal insufficiency, chronic venous insufficiency, and left leg lymphoedema was referred to our emergency unit. She had pain and circumferential erythema with multiple abscesses of the left leg of 2 weeks’ duration. No history of trauma, ulcer, injection, or animal bite was noted. At the time of presentation she had no fever and vital parameters were normal. Empirical treatment with oral amoxicillin (6 g daily) and amoxicillin-clavulanate (375 mg daily) was started. Forty-eight hours later, inflammation, pain, and abscesses worsened (Figure 1A). Laboratory tests showed an elevated white blood cell count (15.9×109⁄L with 86% neutrophils [reference range, 4.5–11.0×109⁄L]) and an elevated C-reactive protein level (322 mg/L [reference range, <2 mg/L]). Human immunodeficiency virus serology was negative. Needle aspiration of an abscess yielded S marcescens. A second aspiration confirmed the presence of the same organism, wild-type S marcescens, which was resistant to amoxicillin and clavulanic acid, first-generation cephalosporin, and tobramycin but sensitive to piperacillin, third-generation cephalosporins, amikacin, ciprofloxacin, and co-trimoxazole. Intravenous cefepime, a third-generation cephalosporin, was started. During the next 48 hours the patient developed severe sepsis with confusion, acute renal failure (creatinine: 231 µmol/L vs 138 µmol/L at baseline [reference range, 53–106 µmol/L), and worsening of skin lesions. Blood cultures were negative and amikacin was added. Magnetic resonance imaging showed a diffuse inflammatory process involving the skin and subcutaneous tissue that extended to the soleus fascia with no other muscle involvement or deep collection (Figure 2). Surgical debridement of infected tissues was performed (Figure 1B). Histologic examination revealed spreading suppurative inflammation involving the dermis and subcutaneous tissues. Clinical healing was obtained after 21 days of antimicrobial therapy. The debrided area required skin grafting 2 months later (Figure 1C).
|
|
|
| Figure 1. Erythema with multiple abscesses on the left ankle and leg at presentation (A), day 1 following surgical debridement of infected tissues (B), and 2 months later with complete healing following a skin graft (C). |
Comment
The most common causative bacteria of cellulitis are Staphylococcus aureus and group A β-hemolytic streptococci. Serratia marcescens is a rare but increasingly recognized pathogen of skin and soft tissue infections.5 The proposed pathogenic mechanism for skin necrosis during S marcescens infection is the bacterial production of large proteases (eg, deoxyribonuclease, lipase, gelatinase).6 Injection of purified proteinase from S marcescens into rat skin leads to increased vascular permeability, necrosis of epidermal tissue, dermal inflammation and edema, and infiltration of polymorphonuclear leukocytes into the subcutaneous fat and muscle.7Serratia marcescens is ubiquitous in soil and water and it also may colonize the respiratory, urinary, and digestive tracts in humans. Cellulitis due to S marcescens secondary to iguana bites8,9 and snake bites10 or leech-borne cellulitis11 suggest that the oral cavity of these animals may be colonized. To date, 49 cases of severe S marcescens skin infections have been described, according to a search of PubMed articles indexed for MEDLINE using the terms Serratia marcescens and skin, cutaneous, soft tissue, and cellulitis or necrotizing fasciitis: 20 cases with NF3,12-28 and 29 non-NF cases8-11,29-46 (typical cellulitis presentation [n=8]9,11,35-38,40; abscesses, gumma, or pyoderma gangrenosum–like lesions associated with chronic granulomatous disease in childhood [n=7]29,44,45; painful nodules with secondary abscesses [n=6]31-34,46; acute bullous cellulitis [n=4]8,10,30; secondary infections of ulcers [n=2]35,40; abscesses in immunocompetent patient [n=1]41; and necrotizing skin ulceration [n=1]36). Lower extremities were frequently involved (NF cases, n=13; non-NF cases, n=16). Underlying immunosuppression was observed in 14 NF cases and in 17 non-NF cases. Predisposing immunologic factors are summarized in the Table. Local risk factors, including chronic leg edema, trauma, surgical wound, filler injection, and ulcer, were frequently reported in NF and non-NF cases,16,20,26-28,31,32,34,35,37,38,40,46 including our case. Surgery was required in 19 NF cases and in 7 non-NF cases. Serratia marcescens–mediated NF led to higher mortality (n=12) than non-NF cases (n=1). Other nonsevere clinical manifestations of S marcescens infection reported in the literature included disseminated papular eruptions with human immunodeficiency virus infection42 and trunk folliculitis.43 Our patient had many risk factors, including chronic edema, diabetes mellitus, chronic renal insufficiency, and chronic venous insufficiency. The potential presence of abscesses and necrotic tissue hinders antibiotic penetration at the infection site, and surgery should be systematically considered as early as possible in view of the high mortality rate of S marcescens cellulitis.
Conclusion
Although uncommon, an S marcescens skin infection may be suspected in cases of cellulitis in immunocompromised patients, especially when conventional antibiotics are not effective. Serratia marcescens naturally produces a cephalosporinase that confers resistance to amoxicillin and to amoxicillin associated with clavulanic acid. Broad-spectrum antibiotics such as third-generation cephalosporins, fluoroquinolones, or imipenem-cilastatin are indicated in cases of S marcescens skin infections, and surgery should be promptly considered if appropriate antibiotic therapy does not lead to rapid clinical improvement.
A gram-negative bacillus of the Enterobacteriaceae family, Serratia marcescens is an organism known to cause bacteremia, pneumonia, urinary tract infection, endocarditis, meningitis, and septic arthritis.1 Unusual cases of cellulitis and necrotizing fasciitis (NF) caused by S marcescens also have been reported.2,3 This entity has been initially described in immunocompromised and nonimmunocompromised patients.4 Both community and nosocomial cases also have been reported.3
Case Report
A 68-year-old morbidly obese woman with high blood pressure, diabetes mellitus, chronic renal insufficiency, chronic venous insufficiency, and left leg lymphoedema was referred to our emergency unit. She had pain and circumferential erythema with multiple abscesses of the left leg of 2 weeks’ duration. No history of trauma, ulcer, injection, or animal bite was noted. At the time of presentation she had no fever and vital parameters were normal. Empirical treatment with oral amoxicillin (6 g daily) and amoxicillin-clavulanate (375 mg daily) was started. Forty-eight hours later, inflammation, pain, and abscesses worsened (Figure 1A). Laboratory tests showed an elevated white blood cell count (15.9×109⁄L with 86% neutrophils [reference range, 4.5–11.0×109⁄L]) and an elevated C-reactive protein level (322 mg/L [reference range, <2 mg/L]). Human immunodeficiency virus serology was negative. Needle aspiration of an abscess yielded S marcescens. A second aspiration confirmed the presence of the same organism, wild-type S marcescens, which was resistant to amoxicillin and clavulanic acid, first-generation cephalosporin, and tobramycin but sensitive to piperacillin, third-generation cephalosporins, amikacin, ciprofloxacin, and co-trimoxazole. Intravenous cefepime, a third-generation cephalosporin, was started. During the next 48 hours the patient developed severe sepsis with confusion, acute renal failure (creatinine: 231 µmol/L vs 138 µmol/L at baseline [reference range, 53–106 µmol/L), and worsening of skin lesions. Blood cultures were negative and amikacin was added. Magnetic resonance imaging showed a diffuse inflammatory process involving the skin and subcutaneous tissue that extended to the soleus fascia with no other muscle involvement or deep collection (Figure 2). Surgical debridement of infected tissues was performed (Figure 1B). Histologic examination revealed spreading suppurative inflammation involving the dermis and subcutaneous tissues. Clinical healing was obtained after 21 days of antimicrobial therapy. The debrided area required skin grafting 2 months later (Figure 1C).
|
|
|
| Figure 1. Erythema with multiple abscesses on the left ankle and leg at presentation (A), day 1 following surgical debridement of infected tissues (B), and 2 months later with complete healing following a skin graft (C). |
Comment
The most common causative bacteria of cellulitis are Staphylococcus aureus and group A β-hemolytic streptococci. Serratia marcescens is a rare but increasingly recognized pathogen of skin and soft tissue infections.5 The proposed pathogenic mechanism for skin necrosis during S marcescens infection is the bacterial production of large proteases (eg, deoxyribonuclease, lipase, gelatinase).6 Injection of purified proteinase from S marcescens into rat skin leads to increased vascular permeability, necrosis of epidermal tissue, dermal inflammation and edema, and infiltration of polymorphonuclear leukocytes into the subcutaneous fat and muscle.7Serratia marcescens is ubiquitous in soil and water and it also may colonize the respiratory, urinary, and digestive tracts in humans. Cellulitis due to S marcescens secondary to iguana bites8,9 and snake bites10 or leech-borne cellulitis11 suggest that the oral cavity of these animals may be colonized. To date, 49 cases of severe S marcescens skin infections have been described, according to a search of PubMed articles indexed for MEDLINE using the terms Serratia marcescens and skin, cutaneous, soft tissue, and cellulitis or necrotizing fasciitis: 20 cases with NF3,12-28 and 29 non-NF cases8-11,29-46 (typical cellulitis presentation [n=8]9,11,35-38,40; abscesses, gumma, or pyoderma gangrenosum–like lesions associated with chronic granulomatous disease in childhood [n=7]29,44,45; painful nodules with secondary abscesses [n=6]31-34,46; acute bullous cellulitis [n=4]8,10,30; secondary infections of ulcers [n=2]35,40; abscesses in immunocompetent patient [n=1]41; and necrotizing skin ulceration [n=1]36). Lower extremities were frequently involved (NF cases, n=13; non-NF cases, n=16). Underlying immunosuppression was observed in 14 NF cases and in 17 non-NF cases. Predisposing immunologic factors are summarized in the Table. Local risk factors, including chronic leg edema, trauma, surgical wound, filler injection, and ulcer, were frequently reported in NF and non-NF cases,16,20,26-28,31,32,34,35,37,38,40,46 including our case. Surgery was required in 19 NF cases and in 7 non-NF cases. Serratia marcescens–mediated NF led to higher mortality (n=12) than non-NF cases (n=1). Other nonsevere clinical manifestations of S marcescens infection reported in the literature included disseminated papular eruptions with human immunodeficiency virus infection42 and trunk folliculitis.43 Our patient had many risk factors, including chronic edema, diabetes mellitus, chronic renal insufficiency, and chronic venous insufficiency. The potential presence of abscesses and necrotic tissue hinders antibiotic penetration at the infection site, and surgery should be systematically considered as early as possible in view of the high mortality rate of S marcescens cellulitis.
Conclusion
Although uncommon, an S marcescens skin infection may be suspected in cases of cellulitis in immunocompromised patients, especially when conventional antibiotics are not effective. Serratia marcescens naturally produces a cephalosporinase that confers resistance to amoxicillin and to amoxicillin associated with clavulanic acid. Broad-spectrum antibiotics such as third-generation cephalosporins, fluoroquinolones, or imipenem-cilastatin are indicated in cases of S marcescens skin infections, and surgery should be promptly considered if appropriate antibiotic therapy does not lead to rapid clinical improvement.
1. Engel HJ, Collignon PJ, Whiting PT, et al. Serratia sp. bacteremia in Canberra, Australia: a population-based study over 10 years. Eur J Clin Microbiol Infect Dis. 2009;28:821-824.
2. Brenner DE, Lookingbill DP. Serratia marcescens cellulitis. Arch Dermatol. 1977;113:1599-1600.
3. Rehman T, Moore TA, Seoane L. Serratia marcescens necrotizing fasciitis presenting as bilateral breast necrosis. J Clin Microbiol. 2012;50:3406-3408.
4. Yu VL. Serratia marcescens: historical perspective and clinical review. N Engl J Med. 1979;300:887-893.
5. Moet GJ, Jones RN, Biedenbach DJ, et al. 2007. Contemporary causes of skin and soft tissue infections in North America, Latin America, and Europe: report from the SENTRY Antimicrobial Surveillance Program (1998–2004). Diagn Microbiol Infect Dis. 2007;57:7-13.
6. Aucken HM, Pitt TL. Antibiotic resistance and putative virulence factors of Serratia marcescens with respect to O and K serotypes. J Med Microbiol. 1998;47:1105-1113.
7. Conroy MC, Bander NH, Lepow IH. Effect in the rat of intradermal injection of purified proteinases from Streptococcus and Serratia marcescens. Proc Soc Exp Biol Med. 1975;150:801-806.
8. Grim KD, Doherty C, Rosen T. Serratia marcescens bullous cellulitis after iguana bites. J Am Acad Dermatol. 2010;62:1075-1076.
9. Hsieh S, Babl FE. Serratia marcescens cellulitis following an iguana bite. Clin Infect Dis. 1999;28:1181-1182.
10. Subramani P, Narasimhamurthy GB, Ashokan B, et al. Serratia marcescens: an unusual pathogen associated with snakebite cellulitis. J Infect Dev Ctries. 2013;7:152-154.
11. Pereira JA, Greig JR, Liddy H, et al. Leech-borne Serratia marcescens infection following complex hand injury. Br J Plast Surg. 1998;51:640-641.
12. Wen YK. Necrotizing fasciitis caused by Serratia marcescens: a fatal complication of nephrotic syndrome. Ren Fail. 2012;34:649-652.
13. Prelog T, Jereb M, Cuček I, et al. Necrotizing fasciitis caused by Serratia marcescens after venous access port implantation in a child with acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2012;34:e246-e248.
14. Meisel M, Schultz-Coulon HJ. Life-threatening necrotizing fasciitis colli caused by Serratia marcescens [in German]. HNO. 2009;57:1071-1074.
15. Campos GA, Burgos LAM, Fica CA, et al. Fatal necrotizing fasciitis due to Serratia marcescens [in Spanish]. Rev Chilena Infectol. 2007;24:319-322.
16. Bustamante Rodríguez R, Bustamante Rodríguez E, Obón Azuara B. Community-acquired necrotizing fasciitis by Serratia marcescens [in Spanish]. Med Clin (Barc). 2008;130:198-199.
17. Pascual J, Liaño F, Rivera M, et al. Necrotizing myositis secondary to Serratia marcescens in a renal allograft recipient. Nephron. 1990;55:329-331.
18. Statham MM, Vohra A, Mehta DK, et al. Serratia marcescens causing cervical necrotizing oropharyngitis. Int J Pediatr Otorhinolaryngol. 2009;73:467-473.
19. Rimailho A, Riou B, Richard C, et al. Fulminant necrotizing fasciitis and nonsteroidal anti-inflammatory drugs. J Infect Dis. 1987;155:143-146.
20. Huang JW, Fang CT, Hung KY, et al. Necrotizing fasciitis caused by Serratia marcescens in two patients receiving corticosteroid therapy. J Formos Med Assoc. 1999;98:851-854.
21. Newton CL, deLemos D, Abramo TJ, et al. Cervical necrotizing fasciitis caused by Serratia marcescens in a 2 year old. Pediatr Emerg Care. 2002;18:433-435.
22. Curtis CE, Chock S, Henderson T, et al. A fatal case of necrotizing fasciitis caused by Serratia marcescens. Am Surg. 2005;71:228-230.
23. Zipper RP, Bustamante MA, Khatib R. Serratia marcescens: a single pathogen in necrotizing fasciitis. Clin Infect Dis. 1996;23:648-649.
24. Liangpunsakul S, Pursell K. Community-acquired necrotizing fasciitis caused by Serratia marcescens: case report and review. Eur J Clin Microbiol Infect Dis. 2001;20:509-510.
25. Vano-Galvan S, Álvarez-Twose I, Moreno-Martín P, et al. Fulminant necrotizing fasciitis caused by Serratia marcescens in an immunosuppressed host. Int J Dermatol. 2014;53:e57-e58.
26. Majumdar R, Crum-Cianflone NF. Necrotizing fasciitis due to Serratia marcescens: case report and review of the literature [published online October 23, 2015]. Infection. doi:10.1007/s15010-015-0855-x.
27. Cope TE, Cope W, Beaumont DM. A case of necrotising fasciitis caused by Serratia marcescens: extreme age as functional immunosuppression? Age Ageing. 2013;42:266-268.
28. Lakhani NA, Narsinghani U, Kumar R. Necrotizing fasciitis of the abdominal wall caused by Serratia marcescens. Infect Dis Rep. 2015;157:5774.
29. Friend JC, Hilligoss DM, Marquesen M, et al. Skin ulcers and disseminated abscesses are characteristic of Serratia marcescens infection in older patients with chronic granulomatous disease [published online May 27, 2009]. J Allergy Clin Immunol. 2009;124:164-166.
30. Cooper CL, Wiseman M, Brunham R. Bullous cellulitis caused by Serratia marcescens. Int J Infect Dis. 1998;3:36-38.
31. Langrock ML, Linde HJ, Landthaler M, et al. Leg ulcers and abscesses caused by Serratia marcescens. Eur J Dermatol. 2008;18:705-707.
32. João AM, Serrano PN, Cachão MP, et al. Recurrent Serratia marcescens cutaneous infection manifesting as painful nodules and ulcers. J Am Acad Dermatol. 2008;58(2 suppl):S55-S57.
33. Friedman DN, Peterson NB, Sumner WT, et al. Spontaneous dermal abscesses and ulcers as a result of Serratia marcescens. J Am Acad Dermatol. 2003;49:S193-S194.
34. Soria X, Bielsa I, Ribera M, et al. Acute dermal abscesses caused by Serratia marcescens. J Am Acad Dermatol. 2008;58:891-893.
35. Bogaert MA, Hogan DJ, Miller AE Jr. Serratia cellulitis and secondary infection of leg ulcers by Serratia. J Am Acad Dermatol. 1991;25:565.
36. Gössl M, Eggebrecht H. Necrotizing skin ulceration in antibiotic-induced agranulocytosis. Mayo Clin Proc. 2006;81:1527.
37. Brenner DE, Lookingbill DP. Serratia marcescens cellulitis. Arch Dermatol. 1977;113:1599-1600.
38. Bonner MJ, Meharg JG Jr. Primary cellulitis due to Serratia marcescens. JAMA. 1983;250:2348-2349.
39. Bornstein PF, Ditto AM, Noskin GA. Serratia marcescens cellulitis in a patient on hemodialysis. Am J Nephrol. 1992;12:374-376.
40. Kaplan H, Sehtman L, Ricover N, et al. Serratia marcescens: cutaneous involvement. preliminary report. Med Cutan Ibero Lat Am. 1988;16:305-308.
41. Giráldez P, Mayo E, Pavón P, et al. Skin infection due to Serratia marcescens in an immunocompetent patient [in Spanish]. Actas Dermosifiliogr. 2011;102:236-237.
42. Muñoz-Pérez MA, Rodriguez-Pichardo A, Camacho F. Disseminated papular eruption caused by Serratia marcescens: a new cutaneous manifestation in HIV-positive patients. AIDS. 1996;10:1179-1180.
43. Lehrhoff S, Yost M, Robinson M, et al. Serratia marcescens folliculitis and concomitant acne vulgaris. Dermatol Online J. 2012;18:19.
44. Benajiba N, Amrani R, Rkain M, et al. Serratia marcescens cutaneous gumma and chronic septic granulomatosis. Med Mal Infect. 2014;44:39-41.
45. Barbato M, Ragusa G, Civitelli F, et al. Chronic granulomatous disease mimicking early-onset Crohn’s disease with cutaneous manifestations. BMC Pediatr. 2014;14:156.
46. Park KY, Seo SJ. Cutaneous Serratia marcescens infection in an immunocompetent patient after filler injection. Acta Derm Venereol. 2013;93:191-192.
1. Engel HJ, Collignon PJ, Whiting PT, et al. Serratia sp. bacteremia in Canberra, Australia: a population-based study over 10 years. Eur J Clin Microbiol Infect Dis. 2009;28:821-824.
2. Brenner DE, Lookingbill DP. Serratia marcescens cellulitis. Arch Dermatol. 1977;113:1599-1600.
3. Rehman T, Moore TA, Seoane L. Serratia marcescens necrotizing fasciitis presenting as bilateral breast necrosis. J Clin Microbiol. 2012;50:3406-3408.
4. Yu VL. Serratia marcescens: historical perspective and clinical review. N Engl J Med. 1979;300:887-893.
5. Moet GJ, Jones RN, Biedenbach DJ, et al. 2007. Contemporary causes of skin and soft tissue infections in North America, Latin America, and Europe: report from the SENTRY Antimicrobial Surveillance Program (1998–2004). Diagn Microbiol Infect Dis. 2007;57:7-13.
6. Aucken HM, Pitt TL. Antibiotic resistance and putative virulence factors of Serratia marcescens with respect to O and K serotypes. J Med Microbiol. 1998;47:1105-1113.
7. Conroy MC, Bander NH, Lepow IH. Effect in the rat of intradermal injection of purified proteinases from Streptococcus and Serratia marcescens. Proc Soc Exp Biol Med. 1975;150:801-806.
8. Grim KD, Doherty C, Rosen T. Serratia marcescens bullous cellulitis after iguana bites. J Am Acad Dermatol. 2010;62:1075-1076.
9. Hsieh S, Babl FE. Serratia marcescens cellulitis following an iguana bite. Clin Infect Dis. 1999;28:1181-1182.
10. Subramani P, Narasimhamurthy GB, Ashokan B, et al. Serratia marcescens: an unusual pathogen associated with snakebite cellulitis. J Infect Dev Ctries. 2013;7:152-154.
11. Pereira JA, Greig JR, Liddy H, et al. Leech-borne Serratia marcescens infection following complex hand injury. Br J Plast Surg. 1998;51:640-641.
12. Wen YK. Necrotizing fasciitis caused by Serratia marcescens: a fatal complication of nephrotic syndrome. Ren Fail. 2012;34:649-652.
13. Prelog T, Jereb M, Cuček I, et al. Necrotizing fasciitis caused by Serratia marcescens after venous access port implantation in a child with acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2012;34:e246-e248.
14. Meisel M, Schultz-Coulon HJ. Life-threatening necrotizing fasciitis colli caused by Serratia marcescens [in German]. HNO. 2009;57:1071-1074.
15. Campos GA, Burgos LAM, Fica CA, et al. Fatal necrotizing fasciitis due to Serratia marcescens [in Spanish]. Rev Chilena Infectol. 2007;24:319-322.
16. Bustamante Rodríguez R, Bustamante Rodríguez E, Obón Azuara B. Community-acquired necrotizing fasciitis by Serratia marcescens [in Spanish]. Med Clin (Barc). 2008;130:198-199.
17. Pascual J, Liaño F, Rivera M, et al. Necrotizing myositis secondary to Serratia marcescens in a renal allograft recipient. Nephron. 1990;55:329-331.
18. Statham MM, Vohra A, Mehta DK, et al. Serratia marcescens causing cervical necrotizing oropharyngitis. Int J Pediatr Otorhinolaryngol. 2009;73:467-473.
19. Rimailho A, Riou B, Richard C, et al. Fulminant necrotizing fasciitis and nonsteroidal anti-inflammatory drugs. J Infect Dis. 1987;155:143-146.
20. Huang JW, Fang CT, Hung KY, et al. Necrotizing fasciitis caused by Serratia marcescens in two patients receiving corticosteroid therapy. J Formos Med Assoc. 1999;98:851-854.
21. Newton CL, deLemos D, Abramo TJ, et al. Cervical necrotizing fasciitis caused by Serratia marcescens in a 2 year old. Pediatr Emerg Care. 2002;18:433-435.
22. Curtis CE, Chock S, Henderson T, et al. A fatal case of necrotizing fasciitis caused by Serratia marcescens. Am Surg. 2005;71:228-230.
23. Zipper RP, Bustamante MA, Khatib R. Serratia marcescens: a single pathogen in necrotizing fasciitis. Clin Infect Dis. 1996;23:648-649.
24. Liangpunsakul S, Pursell K. Community-acquired necrotizing fasciitis caused by Serratia marcescens: case report and review. Eur J Clin Microbiol Infect Dis. 2001;20:509-510.
25. Vano-Galvan S, Álvarez-Twose I, Moreno-Martín P, et al. Fulminant necrotizing fasciitis caused by Serratia marcescens in an immunosuppressed host. Int J Dermatol. 2014;53:e57-e58.
26. Majumdar R, Crum-Cianflone NF. Necrotizing fasciitis due to Serratia marcescens: case report and review of the literature [published online October 23, 2015]. Infection. doi:10.1007/s15010-015-0855-x.
27. Cope TE, Cope W, Beaumont DM. A case of necrotising fasciitis caused by Serratia marcescens: extreme age as functional immunosuppression? Age Ageing. 2013;42:266-268.
28. Lakhani NA, Narsinghani U, Kumar R. Necrotizing fasciitis of the abdominal wall caused by Serratia marcescens. Infect Dis Rep. 2015;157:5774.
29. Friend JC, Hilligoss DM, Marquesen M, et al. Skin ulcers and disseminated abscesses are characteristic of Serratia marcescens infection in older patients with chronic granulomatous disease [published online May 27, 2009]. J Allergy Clin Immunol. 2009;124:164-166.
30. Cooper CL, Wiseman M, Brunham R. Bullous cellulitis caused by Serratia marcescens. Int J Infect Dis. 1998;3:36-38.
31. Langrock ML, Linde HJ, Landthaler M, et al. Leg ulcers and abscesses caused by Serratia marcescens. Eur J Dermatol. 2008;18:705-707.
32. João AM, Serrano PN, Cachão MP, et al. Recurrent Serratia marcescens cutaneous infection manifesting as painful nodules and ulcers. J Am Acad Dermatol. 2008;58(2 suppl):S55-S57.
33. Friedman DN, Peterson NB, Sumner WT, et al. Spontaneous dermal abscesses and ulcers as a result of Serratia marcescens. J Am Acad Dermatol. 2003;49:S193-S194.
34. Soria X, Bielsa I, Ribera M, et al. Acute dermal abscesses caused by Serratia marcescens. J Am Acad Dermatol. 2008;58:891-893.
35. Bogaert MA, Hogan DJ, Miller AE Jr. Serratia cellulitis and secondary infection of leg ulcers by Serratia. J Am Acad Dermatol. 1991;25:565.
36. Gössl M, Eggebrecht H. Necrotizing skin ulceration in antibiotic-induced agranulocytosis. Mayo Clin Proc. 2006;81:1527.
37. Brenner DE, Lookingbill DP. Serratia marcescens cellulitis. Arch Dermatol. 1977;113:1599-1600.
38. Bonner MJ, Meharg JG Jr. Primary cellulitis due to Serratia marcescens. JAMA. 1983;250:2348-2349.
39. Bornstein PF, Ditto AM, Noskin GA. Serratia marcescens cellulitis in a patient on hemodialysis. Am J Nephrol. 1992;12:374-376.
40. Kaplan H, Sehtman L, Ricover N, et al. Serratia marcescens: cutaneous involvement. preliminary report. Med Cutan Ibero Lat Am. 1988;16:305-308.
41. Giráldez P, Mayo E, Pavón P, et al. Skin infection due to Serratia marcescens in an immunocompetent patient [in Spanish]. Actas Dermosifiliogr. 2011;102:236-237.
42. Muñoz-Pérez MA, Rodriguez-Pichardo A, Camacho F. Disseminated papular eruption caused by Serratia marcescens: a new cutaneous manifestation in HIV-positive patients. AIDS. 1996;10:1179-1180.
43. Lehrhoff S, Yost M, Robinson M, et al. Serratia marcescens folliculitis and concomitant acne vulgaris. Dermatol Online J. 2012;18:19.
44. Benajiba N, Amrani R, Rkain M, et al. Serratia marcescens cutaneous gumma and chronic septic granulomatosis. Med Mal Infect. 2014;44:39-41.
45. Barbato M, Ragusa G, Civitelli F, et al. Chronic granulomatous disease mimicking early-onset Crohn’s disease with cutaneous manifestations. BMC Pediatr. 2014;14:156.
46. Park KY, Seo SJ. Cutaneous Serratia marcescens infection in an immunocompetent patient after filler injection. Acta Derm Venereol. 2013;93:191-192.
Practice Points
- Serratia marcescens skin infection should be considered in cases of cellulitis in immunocompromised patients when conventional antibiotics are not effective.
- Broad-spectrum antibiotics such as third-generation cephalosporins, fluoroquinolones, or imipenem-cilastatin are indicated in cases of S marcescens skin infections, and surgery should be promptly considered.
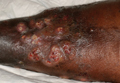
Radiation-Induced Pemphigus or Pemphigoid Disease in 3 Patients With Distinct Underlying Malignancies
A number of adverse cutaneous effects may result from radiation therapy, including radiodermatitis, alopecia, and radiation-induced neoplasms. Radiation therapy rarely induces pemphigus or pemphigoid disease, but awareness of this disorder is of clinical importance because these cutaneous lesions may resemble other skin diseases, including recurrent underlying cancer. We report 3 cases of pemphigus or pemphigoid disease that occurred after radiation therapy for in situ ductal carcinoma of the breast, cervical squamous cell carcinoma, and metastatic squamous cell carcinoma of unknown origin, respectively.
Case Reports
To identify all the patients with radiation-induced pemphigus, pemphigoid diseases, or both diagnosed and treated at Mayo Clinic (Rochester, Minnesota) from 1988 to 2009, we performed a computerized search of dermatology, laboratory medicine, and pathology medical records using the following keywords: radiation, pemphigoid, pemphigus vulgaris, pemphigus foliaceus, pemphigus erythematosus, and blistering disease. Inclusion criteria were a history of radiation therapy and subsequent development of pemphigus or pemphigoid disease within the irradiated fields. Patients with a history of immunobullous disease preceding radiation therapy and patients with a diagnosis of paraneoplastic pemphigus or paraneoplastic autoimmune multiorgan syndrome were excluded. The diagnoses were confirmed by routine pathology as well as direct and indirect immunofluorescence examinations.
We identified 3 patients with severe extensive radiation-associated pemphigus/pemphigoid disease that had developed within 14 months after they received radiation therapy for their underlying cancer. The identified patients’ medical records were reviewed for underlying malignancy, symptoms at the time of diagnosis, treatment course, and follow-up. The protocol was reviewed and approved by the Mayo Clinic institutional review board.
Patient 1—A 58-year-old woman was diagnosed with in situ ductal carcinoma of the right breast and underwent a lumpectomy with subsequent radiation therapy at an outside institution. Fourteen months after the final radiation treatment, she developed localized flaccid blisters and a superficial erosion on the right areola (Figure 1). Routine pathologic and direct immunofluorescence studies performed on shave biopsies in conjunction with serum analysis by indirect immunofluorescence confirmed the diagnosis of pemphigus vulgaris (Figure 2). Additionally, a deeper 4-mm punch biopsy ruled out metastatic breast carcinoma. The patient initially was treated with prednisone 60 mg and azathioprine 50 mg daily. The prednisone was tapered over 4 to 5 months to a dose of 5 mg every other day for another 4 to 5 months. Azathioprine was discontinued after a few months because of increased liver enzyme levels and a rapid clinical response of the pemphigus to this regimen.
Subsequently, she developed oral and ocular erosions that were compatible with pemphigus and were believed to be precipitated by trauma secondary to dental work and to the use of contact lenses. These flares were treated and stabilized with short courses of prednisone at higher doses that were successfully tapered to a maintenance dose of 5 mg every other day to control the pemphigus. With that prednisone dosage, her disease has remained clinically stable.
Patient 2—A 40-year-old woman was diagnosed with stage IIIB cervical squamous carcinoma with para-aortic adenopathy. She was initially treated with primary radiation therapy directed at the pelvis and para-aortic regions using a 4-field approach at our institution, and she received weekly cisplatin chemotherapy at another institution. Nine months later, the patient was admitted to our institution with persistent metastatic cervical carcinoma of the retroperitoneum. She was scheduled for intraoperative radiation therapy as well as aggressive surgical cytoreduction. The day before her surgery she presented to our dermatology clinic with a generalized pruritic rash of 1 month’s duration and occasional blistering without mucosal involvement. Biopsy specimens from the lower back and abdomen were sent for routine histologic studies and direct immunofluorescence. Serum was sent for analysis by indirect immunofluorescence. Pathology results were consistent with a diagnosis of bullous pemphigoid with an infiltrate of eosinophils in the papillary dermis; direct immunofluorescence revealed continuous strong linear deposition of C3, which also was consistent with pemphigoid.
At that time, we recommended application of topical clobetasol 0.05% twice daily to affected areas before initiating prednisone. Postoperatively, her rash improved dramatically with clobetasol monotherapy. However, 4 months after discharge from our hospital, her local dermatologist called us for a telephone consultation regarding clinical and laboratory evidence of pemphigoid relapse. A direct immunofluorescence study showed both linear IgG and C3 deposition. The patient had healed well from the surgery, and the metastatic cervical carcinoma was quiescent. Prednisone in combination with a second immunosuppressive agent was recommended, pending approval by her local oncologist. No further follow-up information is available at this time.
Patient 3—A 72-year-old woman presented with a blistering eruption that had developed on the neck, the upper part of the chest, and other body sites, including the oral mucosa, 6 months after radiation therapy for metastatic squamous cell carcinoma of unknown origin on the neck. On admission to the local hospital, she received a diagnosis of pemphigoid, although the outside biopsy specimens and reports were not available.
The patient was initially treated with prednisone, which was rapidly tapered because she was diabetic and her blood glucose levels were labile. Consequently, she was switched to azathioprine 50 mg 3 times daily and mycophenolate mofetil 500 mg 3 times daily. The patient was transferred to our institution with mild fatigue, dysphagia, weight loss, and generalized blistering involving the skin and lips. Otolaryngologic consultation and radiographic evaluation revealed no evidence of recurrent carcinoma. A shave biopsy was obtained for routine histologic evaluation and immunofluorescence and confirmed the diagnosis of bullous pemphigoid. The patient, however, also was found to have pancytopenia, most likely induced by the combination of azathioprine and mycophenolate mofetil. Her therapeutic regimen was switched to triamcinolone ointment 0.1% to be applied to the eroded areas twice daily and mupirocin ointment to be applied to the hemorrhagic scabs. Subsequently, her complete blood cell count returned to normal.
She continued to use topical corticosteroid therapy to control pemphigoid symptoms, but 6 months later the patient was found to have a lung mass and died secondary to respiratory failure.
 |  |
| Figure 2. Pathologic and immunofluorescence studies confirmed the diagnosis of pemphigus vulgaris. Intraepidermal acantholysis forming a suprabasal blister with a tombstone appearance was seen along the basal cell layer (A)(H&E, original magnification ×400). Intercellular IgG deposition involving the epidermis was noted with direct immunofluorescence (B)(original magnification ×600). | |
|
|
Comment
A wide range of cutaneous reactions are known to occur in conjunction with radiation therapy. Early or acute adverse effects on the skin, such as erythema, edema, and desquamation, can be observed during radiation therapy and for several weeks thereafter. They are usually followed by hair loss and postinflammatory hyperpigmentation. Pemphigus or pemphigoid disease is a rare complication of radiation therapy and has been reported in case reports and small case series.1-17 These disorders include bullous pemphigoid, pemphigus vulgaris, pemphigus foliaceus, bullous lupus erythematosus, and acquired epidermolysis bullosa.10
The mechanism by which radiation therapy induces pemphigus remains open to speculation. Ionizing radiation may alter the antigenicity of the keratinocyte surface by disrupting the sulfhydryl groups,13 thus changing the immunoreactivity of the desmogleins or unmasking certain epidermal antigens. Another possible explanation is immune surveillance interference by damaged T-suppressor cells, which are preferentially sensitive to radiation.8 Robbins et al12 presented a patient with radiation-induced mucocutaneous pemphigus. They performed immunomapping of perilesional skin for the irradiated field, which illustrated altered expression of desmoglein (Dsg) 1, a commonly targeted antigen in pemphigus. Their study also suggested that radiation changed either the distribution or the expression of Dsg1 in the epidermis.12
Approximately half the reported cases we identified were associated with breast carcinoma,1-4,8,14 as in the case of patient 1. The majority of patients initially experienced blistering confined to the irradiated area followed by a variable degree of dissemination to other sites, probably due to the epitope-spreading phenomenon.12 During the months after radiation therapy, Aguado et al1 documented that their patient, who was initially positive for only anti-Dsg3 antibody, developed anti-Dsg1 antibodies. Therefore, the unusual development of mucosal ulcers, other skin lesions, or both after radiation therapy should raise suspicion for this diagnosis.
Bullous pemphigoid primarily affects elderly patients with blister formation along the dermoepidermal junction. Various causes, such as drugs, trauma, UV light, and ionizing radiation, have been associated with this autoimmune blistering disorder. In a systemic literature review, Mul et al10 discovered 27 case reports of bullous pemphigoid that were associated with radiation. It has been suggested that the alteration of the antigenicity and damaged dermoepidermal junction by radiation is a disease-producing mechanism.15,16 Another explanation is that the patients had subclinical pemphigoid and underwent radiation therapy, which damaged the basal layer sufficiently to produce subepidermal blister formation (triggered pemphigoid).17
The patients in this analysis had clinical presentations similar to those previously reported, with a blistering rash that usually began in the irradiated field, raising the possibility of acute radiation dermatitis. However, unlike acute radiation dermatitis, the lesions extended beyond the radiation fields in all 3 cases with mucosal involvement in patients 1 and 3. Although an onset of pemphigoid was previously observed after a minimum dose of 20 Gy,10 there was no definitive correlation observed between the extent and the severity of the cutaneous eruption and the radiation dose in prior studies. Unfortunately, we could not obtain exact radiation doses in our cases because all 3 patients were treated by radiation oncologists at other institutions. We did not, however, observe in our patients that the eruptions were more severe within the irradiated areas. Our analysis demonstrated that radiation-induced pemphigus or pemphigoid disease does not differ greatly from the endogenous form of the disease in its response to therapy or clinical course.
In summary, radiation-induced pemphigus or pemphigoid disease, a rare but serious adverse effect of radiation therapy, should be considered in patients with new-onset blistering or erosive skin disease who have recently undergone irradiation. The accurate diagnosis of pemphigus or pemphigoid disease is important because such diseases often require long-term immunosuppressive therapy. A thorough history and skin examination must be obtained from all patients who receive radiation therapy and subsequently have blisters or eruptions on the skin, mucous membranes, or both. Appropriate diagnostic studies, including routine biopsy for histologic evaluation and direct immunofluorescence, serum for indirect immunofluorescence, and enzyme-linked immunosorbent assay, should be performed to exclude pemphigus or pemphigoid disease.
1. Aguado L, Marguina M, Pretel M, et al. Lesions of pemphigus vulgaris on irradiated skin [published online January 13, 2009]. Clin Exper Dermatol. 2009;34:e148-e150.
2. Ambay A, Sratman E. Ionizing radiation-induced pemphigus foliaceus. J Am Acad Dermatol. 2005;54(suppl 5):S251-S252.
3. Cianchini G, Lembo L, Colonna L, et al. Pemphigus foliaceus induced by radiotherapy and response to dapsone. J Dermatol Treat. 2006;17:244-246.
4. Correia MP, Santos D, Jorge M, et al. Radiotherapy-induced pemphigus. Acta Med Port. 1998;11:581-583.
5. Delaporte E, Piette F, Bergoend H. Pemphigus vulgaris induced by radiotherapy. Ann Dermatol Venereol. 1991;118:447-451.
6. Girolomoni G, Mazzone E, Zambrunno G. Pemphigus vulgaris following cobalt therapy for bronchial carcinoma. Dermatologica. 1989;178:37-38.
7. Krauze E, Wygledowska-Kania M, Kaminska-Budzinska G, et al. Radiotherapy induced pemphigus vulgaris [in French]. Ann Dermatol Venereol. 2003;130:549-550.
8. Low GJ, Keeling JH. Ionizing radiation-induced pemphigus. case presentations and literature review. Arch Dermatol. 1990;126:1319-1323.
9. Mseddi M, Bouassida S, Khemakhem M, et al. Radiotherapy-induced pemphigus: a case report [published online January 18, 2005]. Cancer Radiother. 2005;9:96-98.
10. Mul VE, van Geest AJ, Pijls-Johannesma MC, et al. Radiation-induced bullous pemphigoid: a systemic review of an unusual radiation side effect [published online December 11, 2006]. Radiother Oncol. 2007;82:5-9.
11. Orion E, Matz H, Wolf R. Pemphigus vulgaris induced by radiotherapy. J Eur Acad Dermatol Venereol. 2004;18:508-509.
12. Robbins AC, Lazarova Z, Janson MM, et al. Pemphigus vulgaris presenting in a radiation portal. J Am Acad Dermatol. 2007;56(suppl 5):S82-S85.
13. Rucco V, Pisani M. Induced pemphigus. Arch Dermatol Res. 1982;274:123-140.
14. Vigna-Taglianti R, Russi EG, Denaro N, et al. Radiation-induced pemphigus vulgaris of the breast [published online April 20, 2011]. Cancer Radiother. 2011;15:334-337.
15. Cliff S, Harland CC, Fallowfield ME, et al. Localised bullous pemphigoid following radiotherapy Acta Derm Venereol. 1997;76:330-331.
16. Ohata C, Shirabe H, Takagi K, et al. Localized bullous pemphigoid after radiation therapy: two cases. Acta Derm Venereol. 1997;77:157.
17. Bernhardt M. Bullous pemphigoid after irradiation therapy. J Am Acad Dermatol. 1989;20:141-142.
A number of adverse cutaneous effects may result from radiation therapy, including radiodermatitis, alopecia, and radiation-induced neoplasms. Radiation therapy rarely induces pemphigus or pemphigoid disease, but awareness of this disorder is of clinical importance because these cutaneous lesions may resemble other skin diseases, including recurrent underlying cancer. We report 3 cases of pemphigus or pemphigoid disease that occurred after radiation therapy for in situ ductal carcinoma of the breast, cervical squamous cell carcinoma, and metastatic squamous cell carcinoma of unknown origin, respectively.
Case Reports
To identify all the patients with radiation-induced pemphigus, pemphigoid diseases, or both diagnosed and treated at Mayo Clinic (Rochester, Minnesota) from 1988 to 2009, we performed a computerized search of dermatology, laboratory medicine, and pathology medical records using the following keywords: radiation, pemphigoid, pemphigus vulgaris, pemphigus foliaceus, pemphigus erythematosus, and blistering disease. Inclusion criteria were a history of radiation therapy and subsequent development of pemphigus or pemphigoid disease within the irradiated fields. Patients with a history of immunobullous disease preceding radiation therapy and patients with a diagnosis of paraneoplastic pemphigus or paraneoplastic autoimmune multiorgan syndrome were excluded. The diagnoses were confirmed by routine pathology as well as direct and indirect immunofluorescence examinations.
We identified 3 patients with severe extensive radiation-associated pemphigus/pemphigoid disease that had developed within 14 months after they received radiation therapy for their underlying cancer. The identified patients’ medical records were reviewed for underlying malignancy, symptoms at the time of diagnosis, treatment course, and follow-up. The protocol was reviewed and approved by the Mayo Clinic institutional review board.
Patient 1—A 58-year-old woman was diagnosed with in situ ductal carcinoma of the right breast and underwent a lumpectomy with subsequent radiation therapy at an outside institution. Fourteen months after the final radiation treatment, she developed localized flaccid blisters and a superficial erosion on the right areola (Figure 1). Routine pathologic and direct immunofluorescence studies performed on shave biopsies in conjunction with serum analysis by indirect immunofluorescence confirmed the diagnosis of pemphigus vulgaris (Figure 2). Additionally, a deeper 4-mm punch biopsy ruled out metastatic breast carcinoma. The patient initially was treated with prednisone 60 mg and azathioprine 50 mg daily. The prednisone was tapered over 4 to 5 months to a dose of 5 mg every other day for another 4 to 5 months. Azathioprine was discontinued after a few months because of increased liver enzyme levels and a rapid clinical response of the pemphigus to this regimen.
Subsequently, she developed oral and ocular erosions that were compatible with pemphigus and were believed to be precipitated by trauma secondary to dental work and to the use of contact lenses. These flares were treated and stabilized with short courses of prednisone at higher doses that were successfully tapered to a maintenance dose of 5 mg every other day to control the pemphigus. With that prednisone dosage, her disease has remained clinically stable.
Patient 2—A 40-year-old woman was diagnosed with stage IIIB cervical squamous carcinoma with para-aortic adenopathy. She was initially treated with primary radiation therapy directed at the pelvis and para-aortic regions using a 4-field approach at our institution, and she received weekly cisplatin chemotherapy at another institution. Nine months later, the patient was admitted to our institution with persistent metastatic cervical carcinoma of the retroperitoneum. She was scheduled for intraoperative radiation therapy as well as aggressive surgical cytoreduction. The day before her surgery she presented to our dermatology clinic with a generalized pruritic rash of 1 month’s duration and occasional blistering without mucosal involvement. Biopsy specimens from the lower back and abdomen were sent for routine histologic studies and direct immunofluorescence. Serum was sent for analysis by indirect immunofluorescence. Pathology results were consistent with a diagnosis of bullous pemphigoid with an infiltrate of eosinophils in the papillary dermis; direct immunofluorescence revealed continuous strong linear deposition of C3, which also was consistent with pemphigoid.
At that time, we recommended application of topical clobetasol 0.05% twice daily to affected areas before initiating prednisone. Postoperatively, her rash improved dramatically with clobetasol monotherapy. However, 4 months after discharge from our hospital, her local dermatologist called us for a telephone consultation regarding clinical and laboratory evidence of pemphigoid relapse. A direct immunofluorescence study showed both linear IgG and C3 deposition. The patient had healed well from the surgery, and the metastatic cervical carcinoma was quiescent. Prednisone in combination with a second immunosuppressive agent was recommended, pending approval by her local oncologist. No further follow-up information is available at this time.
Patient 3—A 72-year-old woman presented with a blistering eruption that had developed on the neck, the upper part of the chest, and other body sites, including the oral mucosa, 6 months after radiation therapy for metastatic squamous cell carcinoma of unknown origin on the neck. On admission to the local hospital, she received a diagnosis of pemphigoid, although the outside biopsy specimens and reports were not available.
The patient was initially treated with prednisone, which was rapidly tapered because she was diabetic and her blood glucose levels were labile. Consequently, she was switched to azathioprine 50 mg 3 times daily and mycophenolate mofetil 500 mg 3 times daily. The patient was transferred to our institution with mild fatigue, dysphagia, weight loss, and generalized blistering involving the skin and lips. Otolaryngologic consultation and radiographic evaluation revealed no evidence of recurrent carcinoma. A shave biopsy was obtained for routine histologic evaluation and immunofluorescence and confirmed the diagnosis of bullous pemphigoid. The patient, however, also was found to have pancytopenia, most likely induced by the combination of azathioprine and mycophenolate mofetil. Her therapeutic regimen was switched to triamcinolone ointment 0.1% to be applied to the eroded areas twice daily and mupirocin ointment to be applied to the hemorrhagic scabs. Subsequently, her complete blood cell count returned to normal.
She continued to use topical corticosteroid therapy to control pemphigoid symptoms, but 6 months later the patient was found to have a lung mass and died secondary to respiratory failure.
| |
| Figure 2. Pathologic and immunofluorescence studies confirmed the diagnosis of pemphigus vulgaris. Intraepidermal acantholysis forming a suprabasal blister with a tombstone appearance was seen along the basal cell layer (A)(H&E, original magnification ×400). Intercellular IgG deposition involving the epidermis was noted with direct immunofluorescence (B)(original magnification ×600). | |
|
|
Comment
A wide range of cutaneous reactions are known to occur in conjunction with radiation therapy. Early or acute adverse effects on the skin, such as erythema, edema, and desquamation, can be observed during radiation therapy and for several weeks thereafter. They are usually followed by hair loss and postinflammatory hyperpigmentation. Pemphigus or pemphigoid disease is a rare complication of radiation therapy and has been reported in case reports and small case series.1-17 These disorders include bullous pemphigoid, pemphigus vulgaris, pemphigus foliaceus, bullous lupus erythematosus, and acquired epidermolysis bullosa.10
The mechanism by which radiation therapy induces pemphigus remains open to speculation. Ionizing radiation may alter the antigenicity of the keratinocyte surface by disrupting the sulfhydryl groups,13 thus changing the immunoreactivity of the desmogleins or unmasking certain epidermal antigens. Another possible explanation is immune surveillance interference by damaged T-suppressor cells, which are preferentially sensitive to radiation.8 Robbins et al12 presented a patient with radiation-induced mucocutaneous pemphigus. They performed immunomapping of perilesional skin for the irradiated field, which illustrated altered expression of desmoglein (Dsg) 1, a commonly targeted antigen in pemphigus. Their study also suggested that radiation changed either the distribution or the expression of Dsg1 in the epidermis.12
Approximately half the reported cases we identified were associated with breast carcinoma,1-4,8,14 as in the case of patient 1. The majority of patients initially experienced blistering confined to the irradiated area followed by a variable degree of dissemination to other sites, probably due to the epitope-spreading phenomenon.12 During the months after radiation therapy, Aguado et al1 documented that their patient, who was initially positive for only anti-Dsg3 antibody, developed anti-Dsg1 antibodies. Therefore, the unusual development of mucosal ulcers, other skin lesions, or both after radiation therapy should raise suspicion for this diagnosis.
Bullous pemphigoid primarily affects elderly patients with blister formation along the dermoepidermal junction. Various causes, such as drugs, trauma, UV light, and ionizing radiation, have been associated with this autoimmune blistering disorder. In a systemic literature review, Mul et al10 discovered 27 case reports of bullous pemphigoid that were associated with radiation. It has been suggested that the alteration of the antigenicity and damaged dermoepidermal junction by radiation is a disease-producing mechanism.15,16 Another explanation is that the patients had subclinical pemphigoid and underwent radiation therapy, which damaged the basal layer sufficiently to produce subepidermal blister formation (triggered pemphigoid).17
The patients in this analysis had clinical presentations similar to those previously reported, with a blistering rash that usually began in the irradiated field, raising the possibility of acute radiation dermatitis. However, unlike acute radiation dermatitis, the lesions extended beyond the radiation fields in all 3 cases with mucosal involvement in patients 1 and 3. Although an onset of pemphigoid was previously observed after a minimum dose of 20 Gy,10 there was no definitive correlation observed between the extent and the severity of the cutaneous eruption and the radiation dose in prior studies. Unfortunately, we could not obtain exact radiation doses in our cases because all 3 patients were treated by radiation oncologists at other institutions. We did not, however, observe in our patients that the eruptions were more severe within the irradiated areas. Our analysis demonstrated that radiation-induced pemphigus or pemphigoid disease does not differ greatly from the endogenous form of the disease in its response to therapy or clinical course.
In summary, radiation-induced pemphigus or pemphigoid disease, a rare but serious adverse effect of radiation therapy, should be considered in patients with new-onset blistering or erosive skin disease who have recently undergone irradiation. The accurate diagnosis of pemphigus or pemphigoid disease is important because such diseases often require long-term immunosuppressive therapy. A thorough history and skin examination must be obtained from all patients who receive radiation therapy and subsequently have blisters or eruptions on the skin, mucous membranes, or both. Appropriate diagnostic studies, including routine biopsy for histologic evaluation and direct immunofluorescence, serum for indirect immunofluorescence, and enzyme-linked immunosorbent assay, should be performed to exclude pemphigus or pemphigoid disease.
A number of adverse cutaneous effects may result from radiation therapy, including radiodermatitis, alopecia, and radiation-induced neoplasms. Radiation therapy rarely induces pemphigus or pemphigoid disease, but awareness of this disorder is of clinical importance because these cutaneous lesions may resemble other skin diseases, including recurrent underlying cancer. We report 3 cases of pemphigus or pemphigoid disease that occurred after radiation therapy for in situ ductal carcinoma of the breast, cervical squamous cell carcinoma, and metastatic squamous cell carcinoma of unknown origin, respectively.
Case Reports
To identify all the patients with radiation-induced pemphigus, pemphigoid diseases, or both diagnosed and treated at Mayo Clinic (Rochester, Minnesota) from 1988 to 2009, we performed a computerized search of dermatology, laboratory medicine, and pathology medical records using the following keywords: radiation, pemphigoid, pemphigus vulgaris, pemphigus foliaceus, pemphigus erythematosus, and blistering disease. Inclusion criteria were a history of radiation therapy and subsequent development of pemphigus or pemphigoid disease within the irradiated fields. Patients with a history of immunobullous disease preceding radiation therapy and patients with a diagnosis of paraneoplastic pemphigus or paraneoplastic autoimmune multiorgan syndrome were excluded. The diagnoses were confirmed by routine pathology as well as direct and indirect immunofluorescence examinations.
We identified 3 patients with severe extensive radiation-associated pemphigus/pemphigoid disease that had developed within 14 months after they received radiation therapy for their underlying cancer. The identified patients’ medical records were reviewed for underlying malignancy, symptoms at the time of diagnosis, treatment course, and follow-up. The protocol was reviewed and approved by the Mayo Clinic institutional review board.
Patient 1—A 58-year-old woman was diagnosed with in situ ductal carcinoma of the right breast and underwent a lumpectomy with subsequent radiation therapy at an outside institution. Fourteen months after the final radiation treatment, she developed localized flaccid blisters and a superficial erosion on the right areola (Figure 1). Routine pathologic and direct immunofluorescence studies performed on shave biopsies in conjunction with serum analysis by indirect immunofluorescence confirmed the diagnosis of pemphigus vulgaris (Figure 2). Additionally, a deeper 4-mm punch biopsy ruled out metastatic breast carcinoma. The patient initially was treated with prednisone 60 mg and azathioprine 50 mg daily. The prednisone was tapered over 4 to 5 months to a dose of 5 mg every other day for another 4 to 5 months. Azathioprine was discontinued after a few months because of increased liver enzyme levels and a rapid clinical response of the pemphigus to this regimen.
Subsequently, she developed oral and ocular erosions that were compatible with pemphigus and were believed to be precipitated by trauma secondary to dental work and to the use of contact lenses. These flares were treated and stabilized with short courses of prednisone at higher doses that were successfully tapered to a maintenance dose of 5 mg every other day to control the pemphigus. With that prednisone dosage, her disease has remained clinically stable.
Patient 2—A 40-year-old woman was diagnosed with stage IIIB cervical squamous carcinoma with para-aortic adenopathy. She was initially treated with primary radiation therapy directed at the pelvis and para-aortic regions using a 4-field approach at our institution, and she received weekly cisplatin chemotherapy at another institution. Nine months later, the patient was admitted to our institution with persistent metastatic cervical carcinoma of the retroperitoneum. She was scheduled for intraoperative radiation therapy as well as aggressive surgical cytoreduction. The day before her surgery she presented to our dermatology clinic with a generalized pruritic rash of 1 month’s duration and occasional blistering without mucosal involvement. Biopsy specimens from the lower back and abdomen were sent for routine histologic studies and direct immunofluorescence. Serum was sent for analysis by indirect immunofluorescence. Pathology results were consistent with a diagnosis of bullous pemphigoid with an infiltrate of eosinophils in the papillary dermis; direct immunofluorescence revealed continuous strong linear deposition of C3, which also was consistent with pemphigoid.
At that time, we recommended application of topical clobetasol 0.05% twice daily to affected areas before initiating prednisone. Postoperatively, her rash improved dramatically with clobetasol monotherapy. However, 4 months after discharge from our hospital, her local dermatologist called us for a telephone consultation regarding clinical and laboratory evidence of pemphigoid relapse. A direct immunofluorescence study showed both linear IgG and C3 deposition. The patient had healed well from the surgery, and the metastatic cervical carcinoma was quiescent. Prednisone in combination with a second immunosuppressive agent was recommended, pending approval by her local oncologist. No further follow-up information is available at this time.
Patient 3—A 72-year-old woman presented with a blistering eruption that had developed on the neck, the upper part of the chest, and other body sites, including the oral mucosa, 6 months after radiation therapy for metastatic squamous cell carcinoma of unknown origin on the neck. On admission to the local hospital, she received a diagnosis of pemphigoid, although the outside biopsy specimens and reports were not available.
The patient was initially treated with prednisone, which was rapidly tapered because she was diabetic and her blood glucose levels were labile. Consequently, she was switched to azathioprine 50 mg 3 times daily and mycophenolate mofetil 500 mg 3 times daily. The patient was transferred to our institution with mild fatigue, dysphagia, weight loss, and generalized blistering involving the skin and lips. Otolaryngologic consultation and radiographic evaluation revealed no evidence of recurrent carcinoma. A shave biopsy was obtained for routine histologic evaluation and immunofluorescence and confirmed the diagnosis of bullous pemphigoid. The patient, however, also was found to have pancytopenia, most likely induced by the combination of azathioprine and mycophenolate mofetil. Her therapeutic regimen was switched to triamcinolone ointment 0.1% to be applied to the eroded areas twice daily and mupirocin ointment to be applied to the hemorrhagic scabs. Subsequently, her complete blood cell count returned to normal.
She continued to use topical corticosteroid therapy to control pemphigoid symptoms, but 6 months later the patient was found to have a lung mass and died secondary to respiratory failure.
| |
| Figure 2. Pathologic and immunofluorescence studies confirmed the diagnosis of pemphigus vulgaris. Intraepidermal acantholysis forming a suprabasal blister with a tombstone appearance was seen along the basal cell layer (A)(H&E, original magnification ×400). Intercellular IgG deposition involving the epidermis was noted with direct immunofluorescence (B)(original magnification ×600). | |
|
|
Comment
A wide range of cutaneous reactions are known to occur in conjunction with radiation therapy. Early or acute adverse effects on the skin, such as erythema, edema, and desquamation, can be observed during radiation therapy and for several weeks thereafter. They are usually followed by hair loss and postinflammatory hyperpigmentation. Pemphigus or pemphigoid disease is a rare complication of radiation therapy and has been reported in case reports and small case series.1-17 These disorders include bullous pemphigoid, pemphigus vulgaris, pemphigus foliaceus, bullous lupus erythematosus, and acquired epidermolysis bullosa.10
The mechanism by which radiation therapy induces pemphigus remains open to speculation. Ionizing radiation may alter the antigenicity of the keratinocyte surface by disrupting the sulfhydryl groups,13 thus changing the immunoreactivity of the desmogleins or unmasking certain epidermal antigens. Another possible explanation is immune surveillance interference by damaged T-suppressor cells, which are preferentially sensitive to radiation.8 Robbins et al12 presented a patient with radiation-induced mucocutaneous pemphigus. They performed immunomapping of perilesional skin for the irradiated field, which illustrated altered expression of desmoglein (Dsg) 1, a commonly targeted antigen in pemphigus. Their study also suggested that radiation changed either the distribution or the expression of Dsg1 in the epidermis.12
Approximately half the reported cases we identified were associated with breast carcinoma,1-4,8,14 as in the case of patient 1. The majority of patients initially experienced blistering confined to the irradiated area followed by a variable degree of dissemination to other sites, probably due to the epitope-spreading phenomenon.12 During the months after radiation therapy, Aguado et al1 documented that their patient, who was initially positive for only anti-Dsg3 antibody, developed anti-Dsg1 antibodies. Therefore, the unusual development of mucosal ulcers, other skin lesions, or both after radiation therapy should raise suspicion for this diagnosis.
Bullous pemphigoid primarily affects elderly patients with blister formation along the dermoepidermal junction. Various causes, such as drugs, trauma, UV light, and ionizing radiation, have been associated with this autoimmune blistering disorder. In a systemic literature review, Mul et al10 discovered 27 case reports of bullous pemphigoid that were associated with radiation. It has been suggested that the alteration of the antigenicity and damaged dermoepidermal junction by radiation is a disease-producing mechanism.15,16 Another explanation is that the patients had subclinical pemphigoid and underwent radiation therapy, which damaged the basal layer sufficiently to produce subepidermal blister formation (triggered pemphigoid).17
The patients in this analysis had clinical presentations similar to those previously reported, with a blistering rash that usually began in the irradiated field, raising the possibility of acute radiation dermatitis. However, unlike acute radiation dermatitis, the lesions extended beyond the radiation fields in all 3 cases with mucosal involvement in patients 1 and 3. Although an onset of pemphigoid was previously observed after a minimum dose of 20 Gy,10 there was no definitive correlation observed between the extent and the severity of the cutaneous eruption and the radiation dose in prior studies. Unfortunately, we could not obtain exact radiation doses in our cases because all 3 patients were treated by radiation oncologists at other institutions. We did not, however, observe in our patients that the eruptions were more severe within the irradiated areas. Our analysis demonstrated that radiation-induced pemphigus or pemphigoid disease does not differ greatly from the endogenous form of the disease in its response to therapy or clinical course.
In summary, radiation-induced pemphigus or pemphigoid disease, a rare but serious adverse effect of radiation therapy, should be considered in patients with new-onset blistering or erosive skin disease who have recently undergone irradiation. The accurate diagnosis of pemphigus or pemphigoid disease is important because such diseases often require long-term immunosuppressive therapy. A thorough history and skin examination must be obtained from all patients who receive radiation therapy and subsequently have blisters or eruptions on the skin, mucous membranes, or both. Appropriate diagnostic studies, including routine biopsy for histologic evaluation and direct immunofluorescence, serum for indirect immunofluorescence, and enzyme-linked immunosorbent assay, should be performed to exclude pemphigus or pemphigoid disease.
1. Aguado L, Marguina M, Pretel M, et al. Lesions of pemphigus vulgaris on irradiated skin [published online January 13, 2009]. Clin Exper Dermatol. 2009;34:e148-e150.
2. Ambay A, Sratman E. Ionizing radiation-induced pemphigus foliaceus. J Am Acad Dermatol. 2005;54(suppl 5):S251-S252.
3. Cianchini G, Lembo L, Colonna L, et al. Pemphigus foliaceus induced by radiotherapy and response to dapsone. J Dermatol Treat. 2006;17:244-246.
4. Correia MP, Santos D, Jorge M, et al. Radiotherapy-induced pemphigus. Acta Med Port. 1998;11:581-583.
5. Delaporte E, Piette F, Bergoend H. Pemphigus vulgaris induced by radiotherapy. Ann Dermatol Venereol. 1991;118:447-451.
6. Girolomoni G, Mazzone E, Zambrunno G. Pemphigus vulgaris following cobalt therapy for bronchial carcinoma. Dermatologica. 1989;178:37-38.
7. Krauze E, Wygledowska-Kania M, Kaminska-Budzinska G, et al. Radiotherapy induced pemphigus vulgaris [in French]. Ann Dermatol Venereol. 2003;130:549-550.
8. Low GJ, Keeling JH. Ionizing radiation-induced pemphigus. case presentations and literature review. Arch Dermatol. 1990;126:1319-1323.
9. Mseddi M, Bouassida S, Khemakhem M, et al. Radiotherapy-induced pemphigus: a case report [published online January 18, 2005]. Cancer Radiother. 2005;9:96-98.
10. Mul VE, van Geest AJ, Pijls-Johannesma MC, et al. Radiation-induced bullous pemphigoid: a systemic review of an unusual radiation side effect [published online December 11, 2006]. Radiother Oncol. 2007;82:5-9.
11. Orion E, Matz H, Wolf R. Pemphigus vulgaris induced by radiotherapy. J Eur Acad Dermatol Venereol. 2004;18:508-509.
12. Robbins AC, Lazarova Z, Janson MM, et al. Pemphigus vulgaris presenting in a radiation portal. J Am Acad Dermatol. 2007;56(suppl 5):S82-S85.
13. Rucco V, Pisani M. Induced pemphigus. Arch Dermatol Res. 1982;274:123-140.
14. Vigna-Taglianti R, Russi EG, Denaro N, et al. Radiation-induced pemphigus vulgaris of the breast [published online April 20, 2011]. Cancer Radiother. 2011;15:334-337.
15. Cliff S, Harland CC, Fallowfield ME, et al. Localised bullous pemphigoid following radiotherapy Acta Derm Venereol. 1997;76:330-331.
16. Ohata C, Shirabe H, Takagi K, et al. Localized bullous pemphigoid after radiation therapy: two cases. Acta Derm Venereol. 1997;77:157.
17. Bernhardt M. Bullous pemphigoid after irradiation therapy. J Am Acad Dermatol. 1989;20:141-142.
1. Aguado L, Marguina M, Pretel M, et al. Lesions of pemphigus vulgaris on irradiated skin [published online January 13, 2009]. Clin Exper Dermatol. 2009;34:e148-e150.
2. Ambay A, Sratman E. Ionizing radiation-induced pemphigus foliaceus. J Am Acad Dermatol. 2005;54(suppl 5):S251-S252.
3. Cianchini G, Lembo L, Colonna L, et al. Pemphigus foliaceus induced by radiotherapy and response to dapsone. J Dermatol Treat. 2006;17:244-246.
4. Correia MP, Santos D, Jorge M, et al. Radiotherapy-induced pemphigus. Acta Med Port. 1998;11:581-583.
5. Delaporte E, Piette F, Bergoend H. Pemphigus vulgaris induced by radiotherapy. Ann Dermatol Venereol. 1991;118:447-451.
6. Girolomoni G, Mazzone E, Zambrunno G. Pemphigus vulgaris following cobalt therapy for bronchial carcinoma. Dermatologica. 1989;178:37-38.
7. Krauze E, Wygledowska-Kania M, Kaminska-Budzinska G, et al. Radiotherapy induced pemphigus vulgaris [in French]. Ann Dermatol Venereol. 2003;130:549-550.
8. Low GJ, Keeling JH. Ionizing radiation-induced pemphigus. case presentations and literature review. Arch Dermatol. 1990;126:1319-1323.
9. Mseddi M, Bouassida S, Khemakhem M, et al. Radiotherapy-induced pemphigus: a case report [published online January 18, 2005]. Cancer Radiother. 2005;9:96-98.
10. Mul VE, van Geest AJ, Pijls-Johannesma MC, et al. Radiation-induced bullous pemphigoid: a systemic review of an unusual radiation side effect [published online December 11, 2006]. Radiother Oncol. 2007;82:5-9.
11. Orion E, Matz H, Wolf R. Pemphigus vulgaris induced by radiotherapy. J Eur Acad Dermatol Venereol. 2004;18:508-509.
12. Robbins AC, Lazarova Z, Janson MM, et al. Pemphigus vulgaris presenting in a radiation portal. J Am Acad Dermatol. 2007;56(suppl 5):S82-S85.
13. Rucco V, Pisani M. Induced pemphigus. Arch Dermatol Res. 1982;274:123-140.
14. Vigna-Taglianti R, Russi EG, Denaro N, et al. Radiation-induced pemphigus vulgaris of the breast [published online April 20, 2011]. Cancer Radiother. 2011;15:334-337.
15. Cliff S, Harland CC, Fallowfield ME, et al. Localised bullous pemphigoid following radiotherapy Acta Derm Venereol. 1997;76:330-331.
16. Ohata C, Shirabe H, Takagi K, et al. Localized bullous pemphigoid after radiation therapy: two cases. Acta Derm Venereol. 1997;77:157.
17. Bernhardt M. Bullous pemphigoid after irradiation therapy. J Am Acad Dermatol. 1989;20:141-142.
Practice Points
- The use of radiation therapy is increasing because of its therapeutic benefit, especially in advanced-stage cancer patients.
- Although there is a wide range of adverse effects associated with radiation therapy, pemphigus or pemphigoid disease is rare and needs to be distinguished from other skin diseases or even recurrent underlying cancer.
- The precise mechanism of radiation-induced pemphigus or pemphigoid disease is unknown, but clinicians should be alert to this potentially serious complication, and all cutaneous eruptions developing during and after radiation therapy should be evaluated with routine histologic examination in conjunction with direct immunofluorescence, serum for indirect immunofluorescence, and enzyme-linked immunosorbent assay.
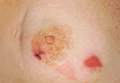
Prolonged Pustular Eruption From Hydroxychloroquine: An Unusual Case of Acute Generalized Exanthematous Pustulosis
Acute generalized exanthematous pustulosis (AGEP) is an uncommon cutaneous eruption characterized by acute, extensive, nonfollicular, sterile pustules accompanied by widespread erythema, fever, and leukocytosis. The clinical hallmark is superficial, sterile, subcorneal pustular dermatosis, which typically starts on the face, axilla, and groin and then progresses to most of the body. Approximately 90% of AGEP cases are due to drug hypersensitivity to a newly initiated medication, while the other 10% are thought to be viral in origin.1 Discontinuation of the offending agent may allow for complete resolution within 15 days. Agents commonly implicated in causing AGEP are antibiotics such as aminopenicillins, macrolides, and cephalosporins.2 Hydroxychloroquine (HCQ) also has been reported to cause AGEP,3-7 with resolution shortly after discontinuation of the drug,4,6 close to the characteristic 15 days of AGEP due to alternate medications.We report an unusual case of HCQ-induced AGEP that lasted far beyond the typical 15 days. We also review other cases of HCQ-induced AGEP and possible mechanisms to explain our patient’s symptoms.
|
|
| Figure 1. Acute generalized exanthematous pustulosis extending to the chest and upper extremities (A) as well as the shoulders and back (B). |
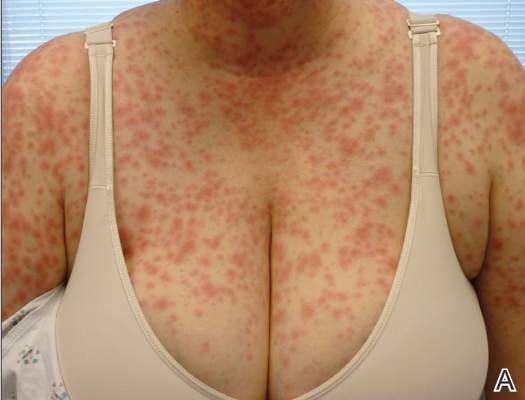

Case Report
A 50-year-old woman who was previously diagnosed with rheumatoid factor seronegative, nonerosive rheumatoid arthritis, which was only moderately controlled with low-dose prednisone (5 mg once daily) after 2 months of treatment, was started on oral HCQ 200 mg twice daily by her rheumatologist. Two weeks after starting HCQ treatment, she developed a pustular exanthem that gradually spread on the back over the next 24 to 48 hours. She described the eruption initially as pruritic, but she then developed painful stinging sensations as the eruption spread. She visited her primary care physician the next day and stopped the HCQ after 14 days following a discussion with the physician. Her prednisone dosage was increased to 50 mg daily for 5 days, but by the fifth day the lesions had spread to the face, full back, shoulders, and upper chest (Figure 1). Morphologically, she presented to the dermatology clinic with innumerable 1- to 2-mm pustules with confluent erythema on the back, extending to the forearms (Figure 2). She also had scattered erythematous macules and papules on the buttocks, legs, and plantar surfaces of the feet. A biopsy taken from the right forearm demonstrated subcorneal pustular dermatosis consistent with AGEP. Prednisone 50 mg once daily was continued. She was scheduled for a follow-up in 3 days but instead went to the emergency department 1 day later due to worsening of the eruption, fever, and malaise. On examination there were multiple discrete and confluent erythematous plaques on the face that extended to the lower extremities. Pustules and scales were noted on the back. New pustules had developed on the hands and feet with intense pruritus.
On admission, her vitals were stable with mild tachycardia. Aggressive intravenous hydration was administered. Her white blood cell count was elevated at 28.3×109/L (reference range, 4.5–10×109/L). She was started on intravenous methylprednisolone 100 mg once daily; topical steroid wet wraps with triamcinolone 0.1% were applied to the trunk, arms, legs, and abdomen twice daily; and hydrocortisone cream 2.5% was applied to the face and intertriginous areas 3 times daily. Over the next 2 days, eruptions continued to persist and the patient reported worsening of pain despite treatment. On day 3, intravenous methylprednisolone 100 mg was switched to oral prednisone 80 mg once daily.
Over the ensuing 5 days, recurrent episodes of erythema on the back had spread to the extremities. After 1 week in the hospital, the diffuse erythema had improved and she had widespread desquamation. She was discharged and prescribed oral prednisone 80 mg once daily and topical therapy twice daily. The patient followed up in the dermatology clinic 4 days after discharge with a mildly pruritic eruption on the trunk and proximal lower extremities but otherwise was doing well. She was instructed to taper the prednisone by 10 mg every 4 days.
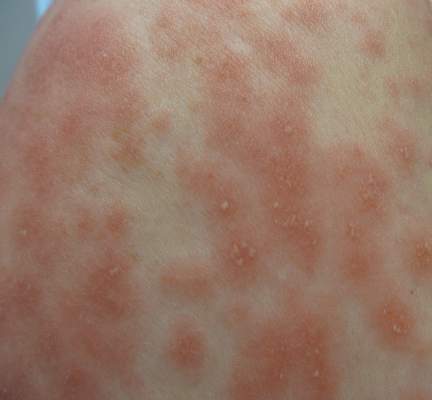
At a follow-up 3 weeks later, she had persistent stinging and tingling sensations, widespread xerosis, and diffuse patchy erythema primarily on the back and proximal extremities, which flared over the last week. The patient reported waxing and waning of the erythema and pruritus since being discharged from the hospital. Despite the recent flare, which was her fourth flare of cutaneous eruption, she showed marked improvement since her initial examination and 40 days after discontinuation of HCQ. She was taking prednisone 40 mg once daily and was advised to continue tapering the dose by 2 mg every 6 to 8 days as tolerated. At 81 days after AGEP onset, the eruption had resolved and the patient was back to her baseline prednisone dosage of 5 mg once daily.
Comment
Acute generalized exanthematous pustulosis is characterized by the sudden appearance of erythema and hundreds of sterile nonfollicular pustules, fever, and leukocytosis. Histologically, AGEP is composed of subcorneal and intraepidermal pustules, edema of the papillary dermis, and perivascular infiltrates of neutrophils and possible eosinophils. The pathogenesis of AGEP is thought to be due to the release of increased amounts of IL-8 by T cells, which attract and activate polymorphonuclear neutrophils.1 Psoriasiform changes are uncommon. Clinically, AGEP is similar to pustular psoriasis but has shown to be its own distinct entity. Unlike patients with pustular psoriasis, patients with AGEP lack a personal or family history of psoriasis or arthritis, have a shorter duration of pustules and fever, and have a history of new medication administration. Other conditions to consider in the differential diagnosis include pustular psoriasis, subcorneal pustulosis, IgA pemphigus, drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, Stevens-Johnson syndrome, and acute febrile neutrophilic dermatosis.
In AGEP, the average duration of medication exposure prior to onset varies depending on the causative agent. Antibiotics consistently have been shown to trigger symptoms after 1 day, whereas other medications, including HCQ, averaged closer to 11 days. Hydroxychloroquine is widely used to treat rheumatic and dermatologic diseases and has previously been reported to be a less common cause of AGEP3; however, a EuroSCAR study found that patients treated with HCQ were at a greater risk for AGEP.2 Acute generalized exanthematous pustulosis usually follows a benign self-limiting course. Within days the eruption gradually evolves into superficial desquamation. Characteristically, removal of the offending agent typically leads to spontaneous resolution in less than 15 days. Resolution is generally without complications and, therefore, treatment is not always necessary. Death has been reported in up to 2% of cases.8 There are no known therapies that prevent the spread of lesions or further decline of the patient’s condition. Systemic corticosteroids often are used to treat AGEP with variable results.1,5
Unique to our patient were recurring exacerbations of the cutaneous lesions beyond the typical 15 days for complete resolution. Even up to 40 days after discontinuation of medication, our patient continued to experience cutaneous symptoms. Other reported cases have not described patients with symptoms flaring or continuing for this extended period of time. A review of 7 external AGEP cases caused by HCQ (identified through a PubMed search of articles indexed for MEDLINE using the search terms acute generalized exanthematous pustulosis or eruption with hydroxychloroquine or plaquenil) showed resolution within 8 days to 3 weeks (Table).3-6,8 One case report documented disease exacerbation on day 18 after tapering the methylprednisolone dose. This patient was then treated with cyclosporine and had a prompt recovery.5 One case of AGEP due to terbinafine reported continual symptoms for approximately 4 weeks after terbinafine discontinuation.9 Our patient’s continual symptoms beyond the typical 15 days may be due to the long half-life of HCQ, which is approximately 40 to 50 days. Systemic corticosteroids often are used to control severe eruptions in AGEP and were administered to our patient; however, their utility in shortening the duration or reducing the severity of the eruption has not been proven.
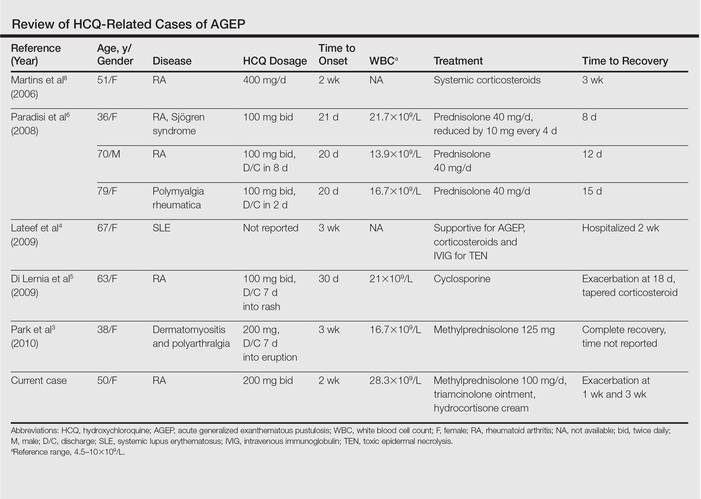
Conclusion
Hydroxychloroquine is a commonly used agent for dermatologic and rheumatologic conditions. The rare but severe acute adverse event of AGEP warrants caution in HCQ use. Correct diagnosis of AGEP with HCQ cessation generally is effective as therapy. Our patient demonstrated that not all cases of AGEP show rapid resolution of cutaneous symptoms after cessation of the drug. Hydroxychloroquine’s extended half-life of 40 to 50 days surpasses that of other medications known to cause AGEP and may explain our patient’s symptoms beyond the usual course.
1. Speeckaert MM, Speeckaert R, Lambert J, et al. Acute generalized exanthematous pustulosis: an overview of the clinical, immunological and diagnostic concepts [published online June 14, 2010]. Eur J Dermatol. 2010;20:425-433.
2. Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR) [published online September 13, 2007]. Br J Dermatol. 2007;157:989-996.
3. Park JJ, Yun SJ, Lee JB, et al. A case of hydroxy-chloroquine induced acute generalized exanthematous pustulosis confirmed by accidental oral provocation [published online February 28, 2010]. Ann Dermatol. 2010;22:102-105.
4. Lateef A, Tan KB, Lau TC. Acute generalized exanthematous pustulosis and toxic epidermal necrolysis induced by hydroxychloroquine [published online August 30, 2009]. Clin Rheumatol. 2009;28:1449-1452.
5. Di Lernia V, Grenzi L, Guareschi E, et al. Rapid clearing of acute generalized exanthematous pustulosis after administration of ciclosporin [published online July 29, 2009]. Clin Exp Dermatol. 2009;34:e757-e759.
6. Paradisi A, Bugatti L, Sisto T, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine: three cases and a review of the literature. Clin Ther. 2008;30:930-940.
7. Choi MJ, Kim HS, Park HJ, et al. Clinicopathologic manifestations of 36 Korean patients with acute generalized exanthematous pustulosis: a case series and review of the literature [published online May 17, 2010]. Ann Dermatol. 2010;22:163-169.
8. Martins A, Lopes LC, Paiva Lopes MJ, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine. Eur J Dermatol. 2006;16:317-318.
9. Lombardo M, Cerati M, Pazzaglia A, et al. Acute generalized exanthematous pustulosis induced by terbinafine. J Am Acad Dermatol. 2003;49:158-159.
Acute generalized exanthematous pustulosis (AGEP) is an uncommon cutaneous eruption characterized by acute, extensive, nonfollicular, sterile pustules accompanied by widespread erythema, fever, and leukocytosis. The clinical hallmark is superficial, sterile, subcorneal pustular dermatosis, which typically starts on the face, axilla, and groin and then progresses to most of the body. Approximately 90% of AGEP cases are due to drug hypersensitivity to a newly initiated medication, while the other 10% are thought to be viral in origin.1 Discontinuation of the offending agent may allow for complete resolution within 15 days. Agents commonly implicated in causing AGEP are antibiotics such as aminopenicillins, macrolides, and cephalosporins.2 Hydroxychloroquine (HCQ) also has been reported to cause AGEP,3-7 with resolution shortly after discontinuation of the drug,4,6 close to the characteristic 15 days of AGEP due to alternate medications.We report an unusual case of HCQ-induced AGEP that lasted far beyond the typical 15 days. We also review other cases of HCQ-induced AGEP and possible mechanisms to explain our patient’s symptoms.
|
|
| Figure 1. Acute generalized exanthematous pustulosis extending to the chest and upper extremities (A) as well as the shoulders and back (B). |
Case Report
A 50-year-old woman who was previously diagnosed with rheumatoid factor seronegative, nonerosive rheumatoid arthritis, which was only moderately controlled with low-dose prednisone (5 mg once daily) after 2 months of treatment, was started on oral HCQ 200 mg twice daily by her rheumatologist. Two weeks after starting HCQ treatment, she developed a pustular exanthem that gradually spread on the back over the next 24 to 48 hours. She described the eruption initially as pruritic, but she then developed painful stinging sensations as the eruption spread. She visited her primary care physician the next day and stopped the HCQ after 14 days following a discussion with the physician. Her prednisone dosage was increased to 50 mg daily for 5 days, but by the fifth day the lesions had spread to the face, full back, shoulders, and upper chest (Figure 1). Morphologically, she presented to the dermatology clinic with innumerable 1- to 2-mm pustules with confluent erythema on the back, extending to the forearms (Figure 2). She also had scattered erythematous macules and papules on the buttocks, legs, and plantar surfaces of the feet. A biopsy taken from the right forearm demonstrated subcorneal pustular dermatosis consistent with AGEP. Prednisone 50 mg once daily was continued. She was scheduled for a follow-up in 3 days but instead went to the emergency department 1 day later due to worsening of the eruption, fever, and malaise. On examination there were multiple discrete and confluent erythematous plaques on the face that extended to the lower extremities. Pustules and scales were noted on the back. New pustules had developed on the hands and feet with intense pruritus.
On admission, her vitals were stable with mild tachycardia. Aggressive intravenous hydration was administered. Her white blood cell count was elevated at 28.3×109/L (reference range, 4.5–10×109/L). She was started on intravenous methylprednisolone 100 mg once daily; topical steroid wet wraps with triamcinolone 0.1% were applied to the trunk, arms, legs, and abdomen twice daily; and hydrocortisone cream 2.5% was applied to the face and intertriginous areas 3 times daily. Over the next 2 days, eruptions continued to persist and the patient reported worsening of pain despite treatment. On day 3, intravenous methylprednisolone 100 mg was switched to oral prednisone 80 mg once daily.
Over the ensuing 5 days, recurrent episodes of erythema on the back had spread to the extremities. After 1 week in the hospital, the diffuse erythema had improved and she had widespread desquamation. She was discharged and prescribed oral prednisone 80 mg once daily and topical therapy twice daily. The patient followed up in the dermatology clinic 4 days after discharge with a mildly pruritic eruption on the trunk and proximal lower extremities but otherwise was doing well. She was instructed to taper the prednisone by 10 mg every 4 days.
At a follow-up 3 weeks later, she had persistent stinging and tingling sensations, widespread xerosis, and diffuse patchy erythema primarily on the back and proximal extremities, which flared over the last week. The patient reported waxing and waning of the erythema and pruritus since being discharged from the hospital. Despite the recent flare, which was her fourth flare of cutaneous eruption, she showed marked improvement since her initial examination and 40 days after discontinuation of HCQ. She was taking prednisone 40 mg once daily and was advised to continue tapering the dose by 2 mg every 6 to 8 days as tolerated. At 81 days after AGEP onset, the eruption had resolved and the patient was back to her baseline prednisone dosage of 5 mg once daily.
Comment
Acute generalized exanthematous pustulosis is characterized by the sudden appearance of erythema and hundreds of sterile nonfollicular pustules, fever, and leukocytosis. Histologically, AGEP is composed of subcorneal and intraepidermal pustules, edema of the papillary dermis, and perivascular infiltrates of neutrophils and possible eosinophils. The pathogenesis of AGEP is thought to be due to the release of increased amounts of IL-8 by T cells, which attract and activate polymorphonuclear neutrophils.1 Psoriasiform changes are uncommon. Clinically, AGEP is similar to pustular psoriasis but has shown to be its own distinct entity. Unlike patients with pustular psoriasis, patients with AGEP lack a personal or family history of psoriasis or arthritis, have a shorter duration of pustules and fever, and have a history of new medication administration. Other conditions to consider in the differential diagnosis include pustular psoriasis, subcorneal pustulosis, IgA pemphigus, drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, Stevens-Johnson syndrome, and acute febrile neutrophilic dermatosis.
In AGEP, the average duration of medication exposure prior to onset varies depending on the causative agent. Antibiotics consistently have been shown to trigger symptoms after 1 day, whereas other medications, including HCQ, averaged closer to 11 days. Hydroxychloroquine is widely used to treat rheumatic and dermatologic diseases and has previously been reported to be a less common cause of AGEP3; however, a EuroSCAR study found that patients treated with HCQ were at a greater risk for AGEP.2 Acute generalized exanthematous pustulosis usually follows a benign self-limiting course. Within days the eruption gradually evolves into superficial desquamation. Characteristically, removal of the offending agent typically leads to spontaneous resolution in less than 15 days. Resolution is generally without complications and, therefore, treatment is not always necessary. Death has been reported in up to 2% of cases.8 There are no known therapies that prevent the spread of lesions or further decline of the patient’s condition. Systemic corticosteroids often are used to treat AGEP with variable results.1,5
Unique to our patient were recurring exacerbations of the cutaneous lesions beyond the typical 15 days for complete resolution. Even up to 40 days after discontinuation of medication, our patient continued to experience cutaneous symptoms. Other reported cases have not described patients with symptoms flaring or continuing for this extended period of time. A review of 7 external AGEP cases caused by HCQ (identified through a PubMed search of articles indexed for MEDLINE using the search terms acute generalized exanthematous pustulosis or eruption with hydroxychloroquine or plaquenil) showed resolution within 8 days to 3 weeks (Table).3-6,8 One case report documented disease exacerbation on day 18 after tapering the methylprednisolone dose. This patient was then treated with cyclosporine and had a prompt recovery.5 One case of AGEP due to terbinafine reported continual symptoms for approximately 4 weeks after terbinafine discontinuation.9 Our patient’s continual symptoms beyond the typical 15 days may be due to the long half-life of HCQ, which is approximately 40 to 50 days. Systemic corticosteroids often are used to control severe eruptions in AGEP and were administered to our patient; however, their utility in shortening the duration or reducing the severity of the eruption has not been proven.
Conclusion
Hydroxychloroquine is a commonly used agent for dermatologic and rheumatologic conditions. The rare but severe acute adverse event of AGEP warrants caution in HCQ use. Correct diagnosis of AGEP with HCQ cessation generally is effective as therapy. Our patient demonstrated that not all cases of AGEP show rapid resolution of cutaneous symptoms after cessation of the drug. Hydroxychloroquine’s extended half-life of 40 to 50 days surpasses that of other medications known to cause AGEP and may explain our patient’s symptoms beyond the usual course.
Acute generalized exanthematous pustulosis (AGEP) is an uncommon cutaneous eruption characterized by acute, extensive, nonfollicular, sterile pustules accompanied by widespread erythema, fever, and leukocytosis. The clinical hallmark is superficial, sterile, subcorneal pustular dermatosis, which typically starts on the face, axilla, and groin and then progresses to most of the body. Approximately 90% of AGEP cases are due to drug hypersensitivity to a newly initiated medication, while the other 10% are thought to be viral in origin.1 Discontinuation of the offending agent may allow for complete resolution within 15 days. Agents commonly implicated in causing AGEP are antibiotics such as aminopenicillins, macrolides, and cephalosporins.2 Hydroxychloroquine (HCQ) also has been reported to cause AGEP,3-7 with resolution shortly after discontinuation of the drug,4,6 close to the characteristic 15 days of AGEP due to alternate medications.We report an unusual case of HCQ-induced AGEP that lasted far beyond the typical 15 days. We also review other cases of HCQ-induced AGEP and possible mechanisms to explain our patient’s symptoms.
|
|
| Figure 1. Acute generalized exanthematous pustulosis extending to the chest and upper extremities (A) as well as the shoulders and back (B). |
Case Report
A 50-year-old woman who was previously diagnosed with rheumatoid factor seronegative, nonerosive rheumatoid arthritis, which was only moderately controlled with low-dose prednisone (5 mg once daily) after 2 months of treatment, was started on oral HCQ 200 mg twice daily by her rheumatologist. Two weeks after starting HCQ treatment, she developed a pustular exanthem that gradually spread on the back over the next 24 to 48 hours. She described the eruption initially as pruritic, but she then developed painful stinging sensations as the eruption spread. She visited her primary care physician the next day and stopped the HCQ after 14 days following a discussion with the physician. Her prednisone dosage was increased to 50 mg daily for 5 days, but by the fifth day the lesions had spread to the face, full back, shoulders, and upper chest (Figure 1). Morphologically, she presented to the dermatology clinic with innumerable 1- to 2-mm pustules with confluent erythema on the back, extending to the forearms (Figure 2). She also had scattered erythematous macules and papules on the buttocks, legs, and plantar surfaces of the feet. A biopsy taken from the right forearm demonstrated subcorneal pustular dermatosis consistent with AGEP. Prednisone 50 mg once daily was continued. She was scheduled for a follow-up in 3 days but instead went to the emergency department 1 day later due to worsening of the eruption, fever, and malaise. On examination there were multiple discrete and confluent erythematous plaques on the face that extended to the lower extremities. Pustules and scales were noted on the back. New pustules had developed on the hands and feet with intense pruritus.
On admission, her vitals were stable with mild tachycardia. Aggressive intravenous hydration was administered. Her white blood cell count was elevated at 28.3×109/L (reference range, 4.5–10×109/L). She was started on intravenous methylprednisolone 100 mg once daily; topical steroid wet wraps with triamcinolone 0.1% were applied to the trunk, arms, legs, and abdomen twice daily; and hydrocortisone cream 2.5% was applied to the face and intertriginous areas 3 times daily. Over the next 2 days, eruptions continued to persist and the patient reported worsening of pain despite treatment. On day 3, intravenous methylprednisolone 100 mg was switched to oral prednisone 80 mg once daily.
Over the ensuing 5 days, recurrent episodes of erythema on the back had spread to the extremities. After 1 week in the hospital, the diffuse erythema had improved and she had widespread desquamation. She was discharged and prescribed oral prednisone 80 mg once daily and topical therapy twice daily. The patient followed up in the dermatology clinic 4 days after discharge with a mildly pruritic eruption on the trunk and proximal lower extremities but otherwise was doing well. She was instructed to taper the prednisone by 10 mg every 4 days.
At a follow-up 3 weeks later, she had persistent stinging and tingling sensations, widespread xerosis, and diffuse patchy erythema primarily on the back and proximal extremities, which flared over the last week. The patient reported waxing and waning of the erythema and pruritus since being discharged from the hospital. Despite the recent flare, which was her fourth flare of cutaneous eruption, she showed marked improvement since her initial examination and 40 days after discontinuation of HCQ. She was taking prednisone 40 mg once daily and was advised to continue tapering the dose by 2 mg every 6 to 8 days as tolerated. At 81 days after AGEP onset, the eruption had resolved and the patient was back to her baseline prednisone dosage of 5 mg once daily.
Comment
Acute generalized exanthematous pustulosis is characterized by the sudden appearance of erythema and hundreds of sterile nonfollicular pustules, fever, and leukocytosis. Histologically, AGEP is composed of subcorneal and intraepidermal pustules, edema of the papillary dermis, and perivascular infiltrates of neutrophils and possible eosinophils. The pathogenesis of AGEP is thought to be due to the release of increased amounts of IL-8 by T cells, which attract and activate polymorphonuclear neutrophils.1 Psoriasiform changes are uncommon. Clinically, AGEP is similar to pustular psoriasis but has shown to be its own distinct entity. Unlike patients with pustular psoriasis, patients with AGEP lack a personal or family history of psoriasis or arthritis, have a shorter duration of pustules and fever, and have a history of new medication administration. Other conditions to consider in the differential diagnosis include pustular psoriasis, subcorneal pustulosis, IgA pemphigus, drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, Stevens-Johnson syndrome, and acute febrile neutrophilic dermatosis.
In AGEP, the average duration of medication exposure prior to onset varies depending on the causative agent. Antibiotics consistently have been shown to trigger symptoms after 1 day, whereas other medications, including HCQ, averaged closer to 11 days. Hydroxychloroquine is widely used to treat rheumatic and dermatologic diseases and has previously been reported to be a less common cause of AGEP3; however, a EuroSCAR study found that patients treated with HCQ were at a greater risk for AGEP.2 Acute generalized exanthematous pustulosis usually follows a benign self-limiting course. Within days the eruption gradually evolves into superficial desquamation. Characteristically, removal of the offending agent typically leads to spontaneous resolution in less than 15 days. Resolution is generally without complications and, therefore, treatment is not always necessary. Death has been reported in up to 2% of cases.8 There are no known therapies that prevent the spread of lesions or further decline of the patient’s condition. Systemic corticosteroids often are used to treat AGEP with variable results.1,5
Unique to our patient were recurring exacerbations of the cutaneous lesions beyond the typical 15 days for complete resolution. Even up to 40 days after discontinuation of medication, our patient continued to experience cutaneous symptoms. Other reported cases have not described patients with symptoms flaring or continuing for this extended period of time. A review of 7 external AGEP cases caused by HCQ (identified through a PubMed search of articles indexed for MEDLINE using the search terms acute generalized exanthematous pustulosis or eruption with hydroxychloroquine or plaquenil) showed resolution within 8 days to 3 weeks (Table).3-6,8 One case report documented disease exacerbation on day 18 after tapering the methylprednisolone dose. This patient was then treated with cyclosporine and had a prompt recovery.5 One case of AGEP due to terbinafine reported continual symptoms for approximately 4 weeks after terbinafine discontinuation.9 Our patient’s continual symptoms beyond the typical 15 days may be due to the long half-life of HCQ, which is approximately 40 to 50 days. Systemic corticosteroids often are used to control severe eruptions in AGEP and were administered to our patient; however, their utility in shortening the duration or reducing the severity of the eruption has not been proven.
Conclusion
Hydroxychloroquine is a commonly used agent for dermatologic and rheumatologic conditions. The rare but severe acute adverse event of AGEP warrants caution in HCQ use. Correct diagnosis of AGEP with HCQ cessation generally is effective as therapy. Our patient demonstrated that not all cases of AGEP show rapid resolution of cutaneous symptoms after cessation of the drug. Hydroxychloroquine’s extended half-life of 40 to 50 days surpasses that of other medications known to cause AGEP and may explain our patient’s symptoms beyond the usual course.
1. Speeckaert MM, Speeckaert R, Lambert J, et al. Acute generalized exanthematous pustulosis: an overview of the clinical, immunological and diagnostic concepts [published online June 14, 2010]. Eur J Dermatol. 2010;20:425-433.
2. Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR) [published online September 13, 2007]. Br J Dermatol. 2007;157:989-996.
3. Park JJ, Yun SJ, Lee JB, et al. A case of hydroxy-chloroquine induced acute generalized exanthematous pustulosis confirmed by accidental oral provocation [published online February 28, 2010]. Ann Dermatol. 2010;22:102-105.
4. Lateef A, Tan KB, Lau TC. Acute generalized exanthematous pustulosis and toxic epidermal necrolysis induced by hydroxychloroquine [published online August 30, 2009]. Clin Rheumatol. 2009;28:1449-1452.
5. Di Lernia V, Grenzi L, Guareschi E, et al. Rapid clearing of acute generalized exanthematous pustulosis after administration of ciclosporin [published online July 29, 2009]. Clin Exp Dermatol. 2009;34:e757-e759.
6. Paradisi A, Bugatti L, Sisto T, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine: three cases and a review of the literature. Clin Ther. 2008;30:930-940.
7. Choi MJ, Kim HS, Park HJ, et al. Clinicopathologic manifestations of 36 Korean patients with acute generalized exanthematous pustulosis: a case series and review of the literature [published online May 17, 2010]. Ann Dermatol. 2010;22:163-169.
8. Martins A, Lopes LC, Paiva Lopes MJ, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine. Eur J Dermatol. 2006;16:317-318.
9. Lombardo M, Cerati M, Pazzaglia A, et al. Acute generalized exanthematous pustulosis induced by terbinafine. J Am Acad Dermatol. 2003;49:158-159.
1. Speeckaert MM, Speeckaert R, Lambert J, et al. Acute generalized exanthematous pustulosis: an overview of the clinical, immunological and diagnostic concepts [published online June 14, 2010]. Eur J Dermatol. 2010;20:425-433.
2. Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR) [published online September 13, 2007]. Br J Dermatol. 2007;157:989-996.
3. Park JJ, Yun SJ, Lee JB, et al. A case of hydroxy-chloroquine induced acute generalized exanthematous pustulosis confirmed by accidental oral provocation [published online February 28, 2010]. Ann Dermatol. 2010;22:102-105.
4. Lateef A, Tan KB, Lau TC. Acute generalized exanthematous pustulosis and toxic epidermal necrolysis induced by hydroxychloroquine [published online August 30, 2009]. Clin Rheumatol. 2009;28:1449-1452.
5. Di Lernia V, Grenzi L, Guareschi E, et al. Rapid clearing of acute generalized exanthematous pustulosis after administration of ciclosporin [published online July 29, 2009]. Clin Exp Dermatol. 2009;34:e757-e759.
6. Paradisi A, Bugatti L, Sisto T, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine: three cases and a review of the literature. Clin Ther. 2008;30:930-940.
7. Choi MJ, Kim HS, Park HJ, et al. Clinicopathologic manifestations of 36 Korean patients with acute generalized exanthematous pustulosis: a case series and review of the literature [published online May 17, 2010]. Ann Dermatol. 2010;22:163-169.
8. Martins A, Lopes LC, Paiva Lopes MJ, et al. Acute generalized exanthematous pustulosis induced by hydroxychloroquine. Eur J Dermatol. 2006;16:317-318.
9. Lombardo M, Cerati M, Pazzaglia A, et al. Acute generalized exanthematous pustulosis induced by terbinafine. J Am Acad Dermatol. 2003;49:158-159.
Practice Points
- Acute generalized exanthematous pustulosis (AGEP) is most commonly caused by antibiotics (eg, aminopenicillins, macrolides, cephalosporins) followed by calcium channel blockers.
- The main treatment of AGEP is discontinuation of the culprit medication, which typically results in resolution within 2 weeks. Treatment also can symptomatically include topical or systemic corticosteroids and antipyretics.
- Hydroxychloroquine (HCQ) can be a culprit of AGEP with a prolonged recovery course. It is important to inform patients with HCQ-associated AGEP that the clearance of their lesions may take longer than the typical 2 weeks.
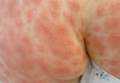
Don’t Get Hung Up on Fishhooks: A Guide to Fishhook Removal
Fishing is one of the world’s most beloved activities, enjoyed as a sport or a leisure activity. However, a common injury from fishing is embedment of the fishhook in the cutaneous tissue. Barbed fishhooks are used for their effectiveness in maintaining the fish on the hook once it is caught, but when implanted in the hand of a fisherman or fisherwoman, barbs can pose problems for removal without exacerbating internal tissue injury. Nevertheless, dermatologists should not shy away from removal of barbed fishhooks, as there are several simple methods that can be easily utilized in the outpatient setting.

Case Report
A 68-year-old man presented to an outpatient dermatology clinic after sustaining a barbed fishhook injury while fishing. The fishhook was firmly inserted into the ventral side of the third digit of the right hand (Figure 1).
Prior to presenting to dermatology, the patient went to 2 urgent care clinics the same day seeking treatment. He reported that practitioners at the first clinic were not able to remove the fishhook because they did not have pliers in stock. At the second clinic he was told the fishhook might be embedded in deeper tissues and was advised to go to the emergency department at the local hospital. When he arrived at the emergency department, a 6-hour wait time prompted him to see a local dermatologist instead.
To remove the fishhook, the area was cleaned and prepared first; lidocaine 2% was administered for local anesthesia. An 18-gauge needle was then advanced through the puncture site parallel to the fishhook’s inner shaft on the same side as the barb, which could be successfully palpated using the tip of the 18-gauge needle. The tip of the needle was then used to cap the barb beneath the skin. This technique allowed for the hook to be easily extracted in a retrograde manner without causing further destruction to the surrounding tissue. The patient then was started on prophylaxis cephalexin 500 mg 3 times daily for 3 days.
Comment
The hand is the most common site of fishhook injury, followed closely by the head and eyes.1 Barbless fishhooks usually can be removed by pushing the hook in a retrograde manner along the path of insertion. This method is simple and rarely results in complications. However, there are no guidelines for removal of barbed fishhooks. Furthermore, removing a barbed fishhook in the same retrograde manner would result in extensive internal tissue destruction and increased complications. Due to the popularity of the sport of fishing, fishhook injuries, depending on geographical location, are not uncommon.2 For this reason, trauma and emergency practitioners have become well versed in safe methods for barbed fishhook removal. However, patients are not always able or willing to seek medical care in emergency departments and may opt to seek treatment in outpatient settings, such as in our case. As a result, dermatologists should familiarize themselves with safe and effective fishhook removal methods, as they are not time consuming and do not require complex equipment. Failure to treat the patient may lead to further patient discomfort and increased risk for complications. Additionally, many of the techniques for removal may be useful with other foreign bodies embedded in cutaneous tissue (eg, splinters).
There are a number of safe and effective techniques for removing barbed fishhooks from cutaneous tissue, including the advance-and-cut method, the cut-it-out technique, the string-pull method, and the needle cover technique.1-3 The method chosen to remove the fishhook is dependent on a variety of factors, such as anatomic location, tissue depth, and provider comfort.
With the advance-and-cut method (Figure 2), the affected area is anesthetized and a small incision in the skin is created to expose the barb. The fishhook is then advanced through the incision, providing visibility of the barb and thus allowing the practitioner to cut the barbed tip without creating further damage to the surrounding tissue. The shaft of the fishhook can subsequently be removed in a retrograde fashion. The advantages of this technique include that it may be successfully used in all types of barbed fishhooks and it provides the practitioner with direct visibility of the barb, thus minimizing risk for neurovascular injury during removal.1 However, the primary disadvantage is that a second cutaneous wound is created in exposing the barb.
 |  | |
| Figure 2. The advance-and-cut method for fishhook removal. | Figure 3. The cut-it-out method for fishhook removal. |
|
The cut-it-out technique (Figure 3) is similar to the advance-and-cut method in that they both require anesthesia along with creating an incision. With this method, a scalpel is used to create a small linear incision originating at the fishhook entrance site and ending at the approximated location of the fishhook’s tip. The fishhook then is simply lifted superiorly in a retrograde fashion.
|
The string-pull method (Figure 4) has been credited to fishermen in South Australia and was first described by Cooke2 in 1961. This method is relatively painless, does not require anesthesia, and has a high success rate when properly administered. However, it does require rapid and confident motions (ie, without hesitation) by the practitioner and should not be performed on free-moving areas of the body (eg, earlobe).3 With this technique, a sturdy piece of suture (eg, 2/0 or 3/0 strength silk) is looped around the hook and is extended away from the practitioner at a 30° angle. The free end of the suture is then securely fastened around the index finger of the practitioner’s dominant hand. The index finger of the nondominant hand should apply a downward pressure to the hook shaft to disengage the barb from the tissue. Simultaneously and rather quickly and forcefully the practitioner must pull the dominant index finger with the string attached in a superior and lateral direction, as depicted by the long arrow in Figure 4. If successful, the barbed hook will pull out of the entrance site. The use of string in pulling the fishhook parallel to the site of injury is helpful for smaller fishhooks that may be difficult to grab with fingers alone. However, with larger fishhooks, the string may not be required so long as the practitioner is able to obtain a secure grasp on the fishhook shaft. The string-pull method becomes particularly useful when anesthesia is unavailable or when the barb of the hook is embedded too deeply for safe advancement through tissue to visualize and cut the barb.
|
Lastly, the needle cover technique (Figure 5) is another simple method that does not require the creation of a secondary wound. An 18-gauge needle is simply inserted parallel to the fishhook curvature into the site of entry. By using the needle to slide along the fishhook’s curve, the practitioner is able to follow its pathway while in the tissue. The tip of the 18-gauge needle is then used to cap or cover the barb, thus allowing the fishhook to be removed in a retrograde fashion from the wound. In an outpatient setting, this technique does not require the creation of additional tissue damage and practitioners who are inexperienced with fishhook removal may proceed through the motions more slowly and methodically than the string-pull method permits.
Wound care following fishhook removal should involve adequate flushing of the wound with normal saline along with the application of topical antibiotics and a simple dressing and adhesive bandage. Oral prophylactic antibiotics typically are not required for shallow cutaneous injuries unless the fishhook is dirty, the patient is immunocompromised, or the patient has a condition lending to poor wound healing (eg, diabetes mellitus, peripheral vascular disease).3 When deciding on antibiotics, it is important to note that fishhook injuries while saltwater fishing are associated with Vibrio infection, while injuries sustained during freshwater fishing are associated with gram-negative bacteria (eg, Pseudomonas and Aeromonas species).3 Lastly, it is essential to find out the immunization status of the patient, and tetanus immune globulin should be provided if necessary.
 |  | |
| Figure 4. The string-pull method for fishhook removal. | Figure 5. The needle cover technique for fishhook removal. |
Conclusion
Although guidelines for barbed fishhook removal are not available, outpatient physicians, including dermatologists, should not fear removal procedures. There are many safe and effective fishhook removal methods that are not time consuming and do not require complex equipment. Furthermore, familiarization with these same techniques may be useful for removal of other foreign bodies embedded in cutaneous tissue.
1. Khan HA, Kamal Y, Lone AU. Fish hook injury: removal by “push through and cut off” technique: a case report and brief literature review [published online March 24, 2014]. Trauma Mon. 2014;19:e17728.
2. Cooke T. How to remove fish-hooks with a bit of string. Med J Aust. 1961;48:815-816.
3. Thommasen HV, Thommasen A. The occasional removal of an embedded fish hook. Can J Rural Med. 2005;10:255-259.
Fishing is one of the world’s most beloved activities, enjoyed as a sport or a leisure activity. However, a common injury from fishing is embedment of the fishhook in the cutaneous tissue. Barbed fishhooks are used for their effectiveness in maintaining the fish on the hook once it is caught, but when implanted in the hand of a fisherman or fisherwoman, barbs can pose problems for removal without exacerbating internal tissue injury. Nevertheless, dermatologists should not shy away from removal of barbed fishhooks, as there are several simple methods that can be easily utilized in the outpatient setting.
Case Report
A 68-year-old man presented to an outpatient dermatology clinic after sustaining a barbed fishhook injury while fishing. The fishhook was firmly inserted into the ventral side of the third digit of the right hand (Figure 1).
Prior to presenting to dermatology, the patient went to 2 urgent care clinics the same day seeking treatment. He reported that practitioners at the first clinic were not able to remove the fishhook because they did not have pliers in stock. At the second clinic he was told the fishhook might be embedded in deeper tissues and was advised to go to the emergency department at the local hospital. When he arrived at the emergency department, a 6-hour wait time prompted him to see a local dermatologist instead.
To remove the fishhook, the area was cleaned and prepared first; lidocaine 2% was administered for local anesthesia. An 18-gauge needle was then advanced through the puncture site parallel to the fishhook’s inner shaft on the same side as the barb, which could be successfully palpated using the tip of the 18-gauge needle. The tip of the needle was then used to cap the barb beneath the skin. This technique allowed for the hook to be easily extracted in a retrograde manner without causing further destruction to the surrounding tissue. The patient then was started on prophylaxis cephalexin 500 mg 3 times daily for 3 days.
Comment
The hand is the most common site of fishhook injury, followed closely by the head and eyes.1 Barbless fishhooks usually can be removed by pushing the hook in a retrograde manner along the path of insertion. This method is simple and rarely results in complications. However, there are no guidelines for removal of barbed fishhooks. Furthermore, removing a barbed fishhook in the same retrograde manner would result in extensive internal tissue destruction and increased complications. Due to the popularity of the sport of fishing, fishhook injuries, depending on geographical location, are not uncommon.2 For this reason, trauma and emergency practitioners have become well versed in safe methods for barbed fishhook removal. However, patients are not always able or willing to seek medical care in emergency departments and may opt to seek treatment in outpatient settings, such as in our case. As a result, dermatologists should familiarize themselves with safe and effective fishhook removal methods, as they are not time consuming and do not require complex equipment. Failure to treat the patient may lead to further patient discomfort and increased risk for complications. Additionally, many of the techniques for removal may be useful with other foreign bodies embedded in cutaneous tissue (eg, splinters).
There are a number of safe and effective techniques for removing barbed fishhooks from cutaneous tissue, including the advance-and-cut method, the cut-it-out technique, the string-pull method, and the needle cover technique.1-3 The method chosen to remove the fishhook is dependent on a variety of factors, such as anatomic location, tissue depth, and provider comfort.
With the advance-and-cut method (Figure 2), the affected area is anesthetized and a small incision in the skin is created to expose the barb. The fishhook is then advanced through the incision, providing visibility of the barb and thus allowing the practitioner to cut the barbed tip without creating further damage to the surrounding tissue. The shaft of the fishhook can subsequently be removed in a retrograde fashion. The advantages of this technique include that it may be successfully used in all types of barbed fishhooks and it provides the practitioner with direct visibility of the barb, thus minimizing risk for neurovascular injury during removal.1 However, the primary disadvantage is that a second cutaneous wound is created in exposing the barb.
| | |
| Figure 2. The advance-and-cut method for fishhook removal. | Figure 3. The cut-it-out method for fishhook removal. |
|
The cut-it-out technique (Figure 3) is similar to the advance-and-cut method in that they both require anesthesia along with creating an incision. With this method, a scalpel is used to create a small linear incision originating at the fishhook entrance site and ending at the approximated location of the fishhook’s tip. The fishhook then is simply lifted superiorly in a retrograde fashion.
|
The string-pull method (Figure 4) has been credited to fishermen in South Australia and was first described by Cooke2 in 1961. This method is relatively painless, does not require anesthesia, and has a high success rate when properly administered. However, it does require rapid and confident motions (ie, without hesitation) by the practitioner and should not be performed on free-moving areas of the body (eg, earlobe).3 With this technique, a sturdy piece of suture (eg, 2/0 or 3/0 strength silk) is looped around the hook and is extended away from the practitioner at a 30° angle. The free end of the suture is then securely fastened around the index finger of the practitioner’s dominant hand. The index finger of the nondominant hand should apply a downward pressure to the hook shaft to disengage the barb from the tissue. Simultaneously and rather quickly and forcefully the practitioner must pull the dominant index finger with the string attached in a superior and lateral direction, as depicted by the long arrow in Figure 4. If successful, the barbed hook will pull out of the entrance site. The use of string in pulling the fishhook parallel to the site of injury is helpful for smaller fishhooks that may be difficult to grab with fingers alone. However, with larger fishhooks, the string may not be required so long as the practitioner is able to obtain a secure grasp on the fishhook shaft. The string-pull method becomes particularly useful when anesthesia is unavailable or when the barb of the hook is embedded too deeply for safe advancement through tissue to visualize and cut the barb.
|
Lastly, the needle cover technique (Figure 5) is another simple method that does not require the creation of a secondary wound. An 18-gauge needle is simply inserted parallel to the fishhook curvature into the site of entry. By using the needle to slide along the fishhook’s curve, the practitioner is able to follow its pathway while in the tissue. The tip of the 18-gauge needle is then used to cap or cover the barb, thus allowing the fishhook to be removed in a retrograde fashion from the wound. In an outpatient setting, this technique does not require the creation of additional tissue damage and practitioners who are inexperienced with fishhook removal may proceed through the motions more slowly and methodically than the string-pull method permits.
Wound care following fishhook removal should involve adequate flushing of the wound with normal saline along with the application of topical antibiotics and a simple dressing and adhesive bandage. Oral prophylactic antibiotics typically are not required for shallow cutaneous injuries unless the fishhook is dirty, the patient is immunocompromised, or the patient has a condition lending to poor wound healing (eg, diabetes mellitus, peripheral vascular disease).3 When deciding on antibiotics, it is important to note that fishhook injuries while saltwater fishing are associated with Vibrio infection, while injuries sustained during freshwater fishing are associated with gram-negative bacteria (eg, Pseudomonas and Aeromonas species).3 Lastly, it is essential to find out the immunization status of the patient, and tetanus immune globulin should be provided if necessary.
| | |
| Figure 4. The string-pull method for fishhook removal. | Figure 5. The needle cover technique for fishhook removal. |
Conclusion
Although guidelines for barbed fishhook removal are not available, outpatient physicians, including dermatologists, should not fear removal procedures. There are many safe and effective fishhook removal methods that are not time consuming and do not require complex equipment. Furthermore, familiarization with these same techniques may be useful for removal of other foreign bodies embedded in cutaneous tissue.
Fishing is one of the world’s most beloved activities, enjoyed as a sport or a leisure activity. However, a common injury from fishing is embedment of the fishhook in the cutaneous tissue. Barbed fishhooks are used for their effectiveness in maintaining the fish on the hook once it is caught, but when implanted in the hand of a fisherman or fisherwoman, barbs can pose problems for removal without exacerbating internal tissue injury. Nevertheless, dermatologists should not shy away from removal of barbed fishhooks, as there are several simple methods that can be easily utilized in the outpatient setting.
Case Report
A 68-year-old man presented to an outpatient dermatology clinic after sustaining a barbed fishhook injury while fishing. The fishhook was firmly inserted into the ventral side of the third digit of the right hand (Figure 1).
Prior to presenting to dermatology, the patient went to 2 urgent care clinics the same day seeking treatment. He reported that practitioners at the first clinic were not able to remove the fishhook because they did not have pliers in stock. At the second clinic he was told the fishhook might be embedded in deeper tissues and was advised to go to the emergency department at the local hospital. When he arrived at the emergency department, a 6-hour wait time prompted him to see a local dermatologist instead.
To remove the fishhook, the area was cleaned and prepared first; lidocaine 2% was administered for local anesthesia. An 18-gauge needle was then advanced through the puncture site parallel to the fishhook’s inner shaft on the same side as the barb, which could be successfully palpated using the tip of the 18-gauge needle. The tip of the needle was then used to cap the barb beneath the skin. This technique allowed for the hook to be easily extracted in a retrograde manner without causing further destruction to the surrounding tissue. The patient then was started on prophylaxis cephalexin 500 mg 3 times daily for 3 days.
Comment
The hand is the most common site of fishhook injury, followed closely by the head and eyes.1 Barbless fishhooks usually can be removed by pushing the hook in a retrograde manner along the path of insertion. This method is simple and rarely results in complications. However, there are no guidelines for removal of barbed fishhooks. Furthermore, removing a barbed fishhook in the same retrograde manner would result in extensive internal tissue destruction and increased complications. Due to the popularity of the sport of fishing, fishhook injuries, depending on geographical location, are not uncommon.2 For this reason, trauma and emergency practitioners have become well versed in safe methods for barbed fishhook removal. However, patients are not always able or willing to seek medical care in emergency departments and may opt to seek treatment in outpatient settings, such as in our case. As a result, dermatologists should familiarize themselves with safe and effective fishhook removal methods, as they are not time consuming and do not require complex equipment. Failure to treat the patient may lead to further patient discomfort and increased risk for complications. Additionally, many of the techniques for removal may be useful with other foreign bodies embedded in cutaneous tissue (eg, splinters).
There are a number of safe and effective techniques for removing barbed fishhooks from cutaneous tissue, including the advance-and-cut method, the cut-it-out technique, the string-pull method, and the needle cover technique.1-3 The method chosen to remove the fishhook is dependent on a variety of factors, such as anatomic location, tissue depth, and provider comfort.
With the advance-and-cut method (Figure 2), the affected area is anesthetized and a small incision in the skin is created to expose the barb. The fishhook is then advanced through the incision, providing visibility of the barb and thus allowing the practitioner to cut the barbed tip without creating further damage to the surrounding tissue. The shaft of the fishhook can subsequently be removed in a retrograde fashion. The advantages of this technique include that it may be successfully used in all types of barbed fishhooks and it provides the practitioner with direct visibility of the barb, thus minimizing risk for neurovascular injury during removal.1 However, the primary disadvantage is that a second cutaneous wound is created in exposing the barb.
| | |
| Figure 2. The advance-and-cut method for fishhook removal. | Figure 3. The cut-it-out method for fishhook removal. |
|
The cut-it-out technique (Figure 3) is similar to the advance-and-cut method in that they both require anesthesia along with creating an incision. With this method, a scalpel is used to create a small linear incision originating at the fishhook entrance site and ending at the approximated location of the fishhook’s tip. The fishhook then is simply lifted superiorly in a retrograde fashion.
|
The string-pull method (Figure 4) has been credited to fishermen in South Australia and was first described by Cooke2 in 1961. This method is relatively painless, does not require anesthesia, and has a high success rate when properly administered. However, it does require rapid and confident motions (ie, without hesitation) by the practitioner and should not be performed on free-moving areas of the body (eg, earlobe).3 With this technique, a sturdy piece of suture (eg, 2/0 or 3/0 strength silk) is looped around the hook and is extended away from the practitioner at a 30° angle. The free end of the suture is then securely fastened around the index finger of the practitioner’s dominant hand. The index finger of the nondominant hand should apply a downward pressure to the hook shaft to disengage the barb from the tissue. Simultaneously and rather quickly and forcefully the practitioner must pull the dominant index finger with the string attached in a superior and lateral direction, as depicted by the long arrow in Figure 4. If successful, the barbed hook will pull out of the entrance site. The use of string in pulling the fishhook parallel to the site of injury is helpful for smaller fishhooks that may be difficult to grab with fingers alone. However, with larger fishhooks, the string may not be required so long as the practitioner is able to obtain a secure grasp on the fishhook shaft. The string-pull method becomes particularly useful when anesthesia is unavailable or when the barb of the hook is embedded too deeply for safe advancement through tissue to visualize and cut the barb.
|
Lastly, the needle cover technique (Figure 5) is another simple method that does not require the creation of a secondary wound. An 18-gauge needle is simply inserted parallel to the fishhook curvature into the site of entry. By using the needle to slide along the fishhook’s curve, the practitioner is able to follow its pathway while in the tissue. The tip of the 18-gauge needle is then used to cap or cover the barb, thus allowing the fishhook to be removed in a retrograde fashion from the wound. In an outpatient setting, this technique does not require the creation of additional tissue damage and practitioners who are inexperienced with fishhook removal may proceed through the motions more slowly and methodically than the string-pull method permits.
Wound care following fishhook removal should involve adequate flushing of the wound with normal saline along with the application of topical antibiotics and a simple dressing and adhesive bandage. Oral prophylactic antibiotics typically are not required for shallow cutaneous injuries unless the fishhook is dirty, the patient is immunocompromised, or the patient has a condition lending to poor wound healing (eg, diabetes mellitus, peripheral vascular disease).3 When deciding on antibiotics, it is important to note that fishhook injuries while saltwater fishing are associated with Vibrio infection, while injuries sustained during freshwater fishing are associated with gram-negative bacteria (eg, Pseudomonas and Aeromonas species).3 Lastly, it is essential to find out the immunization status of the patient, and tetanus immune globulin should be provided if necessary.
| | |
| Figure 4. The string-pull method for fishhook removal. | Figure 5. The needle cover technique for fishhook removal. |
Conclusion
Although guidelines for barbed fishhook removal are not available, outpatient physicians, including dermatologists, should not fear removal procedures. There are many safe and effective fishhook removal methods that are not time consuming and do not require complex equipment. Furthermore, familiarization with these same techniques may be useful for removal of other foreign bodies embedded in cutaneous tissue.
1. Khan HA, Kamal Y, Lone AU. Fish hook injury: removal by “push through and cut off” technique: a case report and brief literature review [published online March 24, 2014]. Trauma Mon. 2014;19:e17728.
2. Cooke T. How to remove fish-hooks with a bit of string. Med J Aust. 1961;48:815-816.
3. Thommasen HV, Thommasen A. The occasional removal of an embedded fish hook. Can J Rural Med. 2005;10:255-259.
1. Khan HA, Kamal Y, Lone AU. Fish hook injury: removal by “push through and cut off” technique: a case report and brief literature review [published online March 24, 2014]. Trauma Mon. 2014;19:e17728.
2. Cooke T. How to remove fish-hooks with a bit of string. Med J Aust. 1961;48:815-816.
3. Thommasen HV, Thommasen A. The occasional removal of an embedded fish hook. Can J Rural Med. 2005;10:255-259.
Practice Points
- Barbed fishhooks should never be removed by pushing the hook in a retrograde manner along the path of insertion, as this method may result in extensive internal tissue destruction and increased complications.
- There are a number of safe and effective techniques for removing barbed fishhooks from cutaneous tissue that also may be applicable in removing other foreign bodies embedded in cutaneous tissue (eg, splinters).

Diagnosing Porokeratosis of Mibelli Every Time: A Novel Biopsy Technique to Maximize Histopathologic Confirmation
Porokeratosis of Mibelli (PM) is a lesion characterized by a surrounding cornoid lamella with variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) in the center of the lesion that typically presents in infancy to early childhood.1 We report a case of PM in which a prior biopsy from the center of the lesion demonstrated papulosquamous dermatitis. We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM.
Case Report
A 3-year-old girl presented with an erythematous, hypopigmented, scaling plaque on the posterior aspect of the left ankle surrounded by a hard rim. The plaque was first noted at 12 months of age and had slowly enlarged as the patient grew. Six months prior, a biopsy from the center of the lesion performed at another facility demonstrated a papulosquamous dermatitis.
Physical examination revealed a lesion that was 4.2-cm long, 2.2-cm wide at the superior pole, and 3.5-cm wide at the inferior pole (Figure 1). A line was drawn with a skin marker perpendicular to the rim of the lesion (Figure 2A) and a 6-mm punch biopsy was performed, centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). The tissue was then bisected at the bedside along the skin marker line with a #15 blade (Figure 2C) and submitted in formalin for histologic processing. Histologic examination revealed an invagination of the epidermis producing a tier of parakeratotic cells with its apex pointed away from the center of the lesion. Dyskeratotic cells were noted at the base of the parakeratosis (Figure 3). Verrucous hyperplasia was present in the central portion of the specimen adjacent to the cornoid lamella. Based on these histopathologic findings, the correct diagnosis of PM was made.
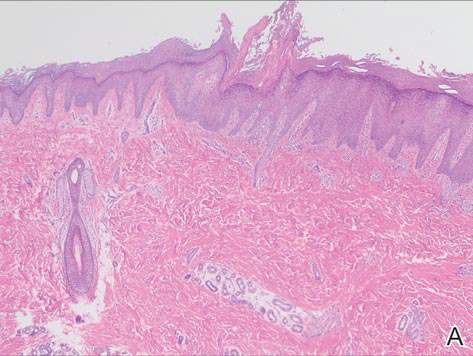
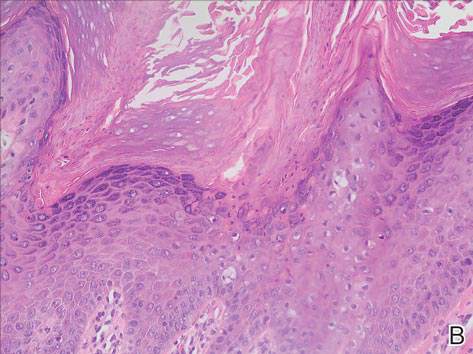
Comment
Porokeratosis of Mibelli is a rare condition that typically presents in infancy to early childhood.1 It may appear as small keratotic papules or larger plaques that reach several centimeters in diameter.2 There is a 7.5% risk for malignant transformation (eg, basal cell carcinoma, squamous cell carcinoma, Bowen disease).3 Variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) typically are present in the center of the lesion. In our case, a biopsy from the center of the plaque demonstrated verrucous hyperplasia. The incorrect diagnosis of PM as psoriasis also has been reported.4
We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM. First, draw a line perpendicular to the rim of the lesion to mark the biopsy site (Figure 2A). Second, perform a punch biopsy centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). Third, section the biopsied tissue with a #15 blade along the perpendicular line at the bedside (Figure 2C). The surgical pathology requisition should mention that the specimen has been transected and the cut edges should be placed down in the cassette, ensuring that the cornoid lamella will be present in cross-section on the slides.
If the punch biopsy specimen is not bisected, it can be difficult to orient it in the pathology laboratory, especially if the cornoid lamellae are not prominent. Furthermore, the technician processing the tissue may not be aware of the importance of sectioning the specimen perpendicular to the cornoid lamella. Following this procedure, diagnosis can be confirmed in virtually every case of PM.
- Richard G, Irvine A, Traupe H, et al. Ichthyosis and disorders of other conification. In: Schachner L, Hansen R, Krafchik B, et al, eds. Pediatric Dermatology. Philadelphia, PA: Elsevier Health Sciences; 2011:640-643.
- Pierson D, Bandel C, Ehrig, et al. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo J, Rapini R, et al, eds. Dermatology. 1st ed. Vol 2. Edinburgh, Scotland: Elsevier; 2003:1707-1709.
- Cort DF, Abdel-Aziz AH. Epithelioma arising in porokeratosis of Mibelli. Br J Plast Surg. 1972;25:318-328.
- De Simone C, Paradisi A, Massi G, et al. Giant verrucous porokeratosis of Mibelli mimicking psoriasis in a patient with psoriasis. J Am Acad Dermatol. 2007;57:665-668.
Porokeratosis of Mibelli (PM) is a lesion characterized by a surrounding cornoid lamella with variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) in the center of the lesion that typically presents in infancy to early childhood.1 We report a case of PM in which a prior biopsy from the center of the lesion demonstrated papulosquamous dermatitis. We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM.
Case Report
A 3-year-old girl presented with an erythematous, hypopigmented, scaling plaque on the posterior aspect of the left ankle surrounded by a hard rim. The plaque was first noted at 12 months of age and had slowly enlarged as the patient grew. Six months prior, a biopsy from the center of the lesion performed at another facility demonstrated a papulosquamous dermatitis.
Physical examination revealed a lesion that was 4.2-cm long, 2.2-cm wide at the superior pole, and 3.5-cm wide at the inferior pole (Figure 1). A line was drawn with a skin marker perpendicular to the rim of the lesion (Figure 2A) and a 6-mm punch biopsy was performed, centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). The tissue was then bisected at the bedside along the skin marker line with a #15 blade (Figure 2C) and submitted in formalin for histologic processing. Histologic examination revealed an invagination of the epidermis producing a tier of parakeratotic cells with its apex pointed away from the center of the lesion. Dyskeratotic cells were noted at the base of the parakeratosis (Figure 3). Verrucous hyperplasia was present in the central portion of the specimen adjacent to the cornoid lamella. Based on these histopathologic findings, the correct diagnosis of PM was made.
Comment
Porokeratosis of Mibelli is a rare condition that typically presents in infancy to early childhood.1 It may appear as small keratotic papules or larger plaques that reach several centimeters in diameter.2 There is a 7.5% risk for malignant transformation (eg, basal cell carcinoma, squamous cell carcinoma, Bowen disease).3 Variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) typically are present in the center of the lesion. In our case, a biopsy from the center of the plaque demonstrated verrucous hyperplasia. The incorrect diagnosis of PM as psoriasis also has been reported.4
We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM. First, draw a line perpendicular to the rim of the lesion to mark the biopsy site (Figure 2A). Second, perform a punch biopsy centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). Third, section the biopsied tissue with a #15 blade along the perpendicular line at the bedside (Figure 2C). The surgical pathology requisition should mention that the specimen has been transected and the cut edges should be placed down in the cassette, ensuring that the cornoid lamella will be present in cross-section on the slides.
If the punch biopsy specimen is not bisected, it can be difficult to orient it in the pathology laboratory, especially if the cornoid lamellae are not prominent. Furthermore, the technician processing the tissue may not be aware of the importance of sectioning the specimen perpendicular to the cornoid lamella. Following this procedure, diagnosis can be confirmed in virtually every case of PM.
Porokeratosis of Mibelli (PM) is a lesion characterized by a surrounding cornoid lamella with variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) in the center of the lesion that typically presents in infancy to early childhood.1 We report a case of PM in which a prior biopsy from the center of the lesion demonstrated papulosquamous dermatitis. We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM.
Case Report
A 3-year-old girl presented with an erythematous, hypopigmented, scaling plaque on the posterior aspect of the left ankle surrounded by a hard rim. The plaque was first noted at 12 months of age and had slowly enlarged as the patient grew. Six months prior, a biopsy from the center of the lesion performed at another facility demonstrated a papulosquamous dermatitis.
Physical examination revealed a lesion that was 4.2-cm long, 2.2-cm wide at the superior pole, and 3.5-cm wide at the inferior pole (Figure 1). A line was drawn with a skin marker perpendicular to the rim of the lesion (Figure 2A) and a 6-mm punch biopsy was performed, centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). The tissue was then bisected at the bedside along the skin marker line with a #15 blade (Figure 2C) and submitted in formalin for histologic processing. Histologic examination revealed an invagination of the epidermis producing a tier of parakeratotic cells with its apex pointed away from the center of the lesion. Dyskeratotic cells were noted at the base of the parakeratosis (Figure 3). Verrucous hyperplasia was present in the central portion of the specimen adjacent to the cornoid lamella. Based on these histopathologic findings, the correct diagnosis of PM was made.
Comment
Porokeratosis of Mibelli is a rare condition that typically presents in infancy to early childhood.1 It may appear as small keratotic papules or larger plaques that reach several centimeters in diameter.2 There is a 7.5% risk for malignant transformation (eg, basal cell carcinoma, squamous cell carcinoma, Bowen disease).3 Variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) typically are present in the center of the lesion. In our case, a biopsy from the center of the plaque demonstrated verrucous hyperplasia. The incorrect diagnosis of PM as psoriasis also has been reported.4
We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM. First, draw a line perpendicular to the rim of the lesion to mark the biopsy site (Figure 2A). Second, perform a punch biopsy centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). Third, section the biopsied tissue with a #15 blade along the perpendicular line at the bedside (Figure 2C). The surgical pathology requisition should mention that the specimen has been transected and the cut edges should be placed down in the cassette, ensuring that the cornoid lamella will be present in cross-section on the slides.
If the punch biopsy specimen is not bisected, it can be difficult to orient it in the pathology laboratory, especially if the cornoid lamellae are not prominent. Furthermore, the technician processing the tissue may not be aware of the importance of sectioning the specimen perpendicular to the cornoid lamella. Following this procedure, diagnosis can be confirmed in virtually every case of PM.
- Richard G, Irvine A, Traupe H, et al. Ichthyosis and disorders of other conification. In: Schachner L, Hansen R, Krafchik B, et al, eds. Pediatric Dermatology. Philadelphia, PA: Elsevier Health Sciences; 2011:640-643.
- Pierson D, Bandel C, Ehrig, et al. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo J, Rapini R, et al, eds. Dermatology. 1st ed. Vol 2. Edinburgh, Scotland: Elsevier; 2003:1707-1709.
- Cort DF, Abdel-Aziz AH. Epithelioma arising in porokeratosis of Mibelli. Br J Plast Surg. 1972;25:318-328.
- De Simone C, Paradisi A, Massi G, et al. Giant verrucous porokeratosis of Mibelli mimicking psoriasis in a patient with psoriasis. J Am Acad Dermatol. 2007;57:665-668.
- Richard G, Irvine A, Traupe H, et al. Ichthyosis and disorders of other conification. In: Schachner L, Hansen R, Krafchik B, et al, eds. Pediatric Dermatology. Philadelphia, PA: Elsevier Health Sciences; 2011:640-643.
- Pierson D, Bandel C, Ehrig, et al. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo J, Rapini R, et al, eds. Dermatology. 1st ed. Vol 2. Edinburgh, Scotland: Elsevier; 2003:1707-1709.
- Cort DF, Abdel-Aziz AH. Epithelioma arising in porokeratosis of Mibelli. Br J Plast Surg. 1972;25:318-328.
- De Simone C, Paradisi A, Massi G, et al. Giant verrucous porokeratosis of Mibelli mimicking psoriasis in a patient with psoriasis. J Am Acad Dermatol. 2007;57:665-668.
Practice Points
- A biopsy from the center of a plaque of porokeratosis will produce nonspecific findings.
- Bisecting the punch specimen at the bedside along a line drawn perpendicular to the cornoid lamella guarantees proper orientation of the specimen.
PTSD in Combat Veterans With Cognitive Decline
The number of veterans aged ≥ 65 years is expected to increase steadily as the Vietnam-era cohort ages. In 2012, the number of veterans aged ≥ 85 years was expected to peak at nearly 1.4 million. Vietnam-era veterans comprise the largest cohort of veterans, and > 15% of male and > 8% of female Vietnam veterans receiving care in the VA system have been diagnosed with posttraumatic stress disorder (PTSD). These veterans are rapidly approaching age groups in which cognitive disorders increase exponentially in prevalence.
Combat exposure has been called a common but “hidden variable” in studies of aging and health.1 Combat exposure may be even more hidden for Vietnam veterans who have pursued health care outside the VA system and less likely to announce their service to health care providers.
Even veterans who did not serve in traditional combat roles can experience chronic debilitation from the psychological stress of overseas deployment to a war zone. Indeed, cases of noncombat trauma have been presented in the context of cognitive decline and late-onset PTSD.2 It is probable that survivors of sexual assault, child abuse, crime, and natural disaster are also vulnerable to a recurrence of trauma symptoms if they experience cognitive slippage. In this article the authors report a case of delayed onset PTSD symptoms, precipitated by cognitive decline.
Case Report
Mr. B was a 72-year-old Korean War veteran referred for neuropsychological evaluation to establish baseline cognitive status before elective cardiac surgery. Mr. B relied on his wife to fill in many details of his personal history. His wife reported that the patient’s memory problems had increased significantly over the previous 12 months. Mr. B had been treated with donepezil 10 mg daily for about 1 year, with no observed benefit. His wife described life at home as “tense” due to his increased irritability and poor insight into his condition. Mr. B reported that he was often afraid of noises at night and needed to go outside and look around. His wife reported that he was very afraid of “strangers coming into the house.”
Mr. B was born in Arizona and experienced significant physical abuse while under the care of an alcoholic foster parent. He dropped out of high school and enlisted in the U.S. Marine Corps. Upon his discharge from military service, he worked as a truck driver for 23 years. He retired after experiencing hip problems. He drank heavily for many years after the war and, according to his wife, was “very violent,” but stopped 27 years previously, after injuring his wife while intoxicated. The patient’s medical history included hospitalization about 1 year prior to the evaluation following a fall associated with altered level of consciousness and confusion, which lasted several hours. He was discharged the same day and was thought to have had a stroke. The patient also had hypertension, hyperlipidemia, and sciatica. A carotid ultrasound showed bilateral carotid stenosis > 50%.
Mr. B was married for 45 years and had 5 children and 12 grandchildren. He enlisted in the U.S. Marine Corps at age 19 and served as a tank gunner during the Korean War. He experienced extremely heavy combat, was wounded several times (including loss of consciousness due to an explosion), and was hospitalizedfor 4 m onths in Japan. When he returned to the frontline, he found that many of the men in his unit had been killed. He was promoted to staff sergeant and tank commander. Mr. B received an honorable discharge after the war and a 50% service-connected disability pension for PTSD. He reported having received group psychotherapy at a VA hospital soon after the war but no other psychiatric treatment. He avoided watching the news because the Gulf War news reminded him of Korea.
Mr. B was smiling, pleasant, and cooperative throughout the 2 hours of testing and interviewing. He wore a Korean War veteran baseball cap festooned with military pins and ribbons, including a Purple Heart ribbon that he proudly showed to the test administrators. Unbidden, he also presented for inspection an assortment of life membership cards in various veterans service organizations. Mr. B reported frequent nightmares, night sweats, and intrusive thoughts about his combat experiences. During testing, he was repeatedly triggered by innocuous items and launched into a discourse on his combat experiences. When asked to memorize a short list of words that included the word fire, he said, “You know what that reminds me of...we had to fire big guns, 90 millimeter, that’s what it was…killing and how to kill.” When shown an abstract design that resembled the number 44, he said, “You know what that is? It was the radio call sign of our tank—‘This is 44, come in, we need some help.’”
Mr. B’s memory problems were marked by rapid forgetting, impaired ability to learn new information, and impaired ability to recall previously learned information. Language problems were also present, including difficulty recognizing and naming common objects, impaired auditory comprehension, and problems with verbal associative fluency during timed tasks. He also showed difficulties with executive functioning, attention, and working memory. His mini-mental state examination score was 21/30. He stated the year was 2020, did not know the day of the week, registered 2/3 words and recalled 0/3, he counted 3/5 in serial 7s, and was unable to repeat the phrase, “no ifs, ands, or buts.”
Discussion
Posttraumatic stress symptoms were present during the immediate aftermath of the initial trauma exposure for this patient. He managed to lead a relatively successful and productive life, sustained a marriage, and raised a family. The onset of cognitive decline precipitated a recrudescence of PTSD symptomology. In fact, the effects of combat trauma seem more malignant and extreme at the time of the memory disorders evaluation than at any prior time in his life.
A number of case reports have been published in recent years that describe comorbid presentations of cognitive disorder and PTSD symptomatology.3-6 A clinicalconsensus that cognitive decline can exacerbate previously well-managed symptoms of earlier psychological trauma seems to be emerging. Several published casestudies have noted that comorbid presentation of dementia and PTSD is often marked by violence, psychotic symptoms, and increased risk of hospitalization.7-9
PTSD Research
Unfortunately, systematic investigation into the relationship between PTSD and cognitive decline is in its infancy. Previous authors have posited various mechanisms to explain the exacerbation of dormant PTSD symptoms after cognitive decline.10,11 Some have attributed the phenomenon to an age-related failure of either repression or avoidance or to a compromised ability to actively focus their attention elsewhere.2,12 A finding of preservative errors on neuropsychological tests has been associated with an inability to organize and inhibit intrusive thought.13 In one case, the effects of combat trauma were purported to be denied, repressed, and largely forgotten for 30 years until rekindled by the patient’s deteriorating health and loss of employment.14 Several case examples have been presented in which physical illness, interpersonal loss, retirement, or losses of social support were other factors.15-18 Two major studies of veterans with PTSD, found that subjects were twice as likely to develop dementia.3,4 There is a strong association between chronic psychological stress and later development
of dementia. In a study by Wilson and colleagues, subjects with higher baseline stress had twice the chance of developing Alzheimer disease.19 Similar findings of
accelerated or higher cognitive decline were found by other studies, too.17,20
Hippocampal damage associated with prolonged, intense psychological stress has been cited as a possible contributor to PTSD symptom recrudescence in older adults.21 It is well known that emotional arousal leads to better-encoded memories. In the context of a cognitive disorder marked by gradual memory loss, traumatic memories might be the last to go.22 Another proposed biologic mechanism is a reduction in hippocampal volume and decreased inhibition of the amgydala, which results in preferential recall of the nondeclarative, amygdaloidal traumatic memories.8
Research on selective area damage in the hippocampus opens a new era of understanding of consequences of stress. The dentate gyrus (DG) is the main area of hippocampus that helps in neurogenesis and cornus ammonis 3 (CA3) for dendritic branching.23-25 In recent studies by Wang and colleagues, PTSD has been found to be associated with selective volume loss of the CA3/DG subfields, consistent with animal studies.24-28 Abundance of glucocorticoid receptors in the hippocampus, especially at CA3,29,30 may make it more vulnerable to the neurotoxic effect of glucocorticoids, causing suppression of neurogenesis,29 diminished dendritic branching,30 loss of synapses,26,31 and eventually diminished neuroplasticity,32 because CA3/DG is the main target of neurotoxicity by glucocorticoid and inflammatory damage.
The results of neuroimaging studies suggest that decreased integration of the prefrontal cortex and the hippocampus results in impaired short-term memory and perhaps increasing the prominence of long-term distressing memories.33 Clinical observation confirms that patients with PTSD experience vivid, intense, detailed, and realistic recollections of remote memories at a time when their ability to recall nontraumatic autobiographical detail is severely compromised.
Symptom Reemergence
Both prospective and retrospective studies have shown that PTSD symptoms can evolve, even after a 20-year long symptom-free period, and reemergence of PTSD
symptoms is not uncommon.34,35 A longer delay usually presents with less severe symptoms.36 The unavailability of complete information regarding a patient’s past adjustment to psychological trauma has encouraged some experts to label exacerbation of PTSD symptoms precipitated by cognitive disorder as delayed onset PTSD. In most cases, it seems that this is more accurately described as a recrudescence of symptoms that were better managed previously. The picture is clouded by the often bizarre and extreme manifestation of PTSD symptoms in patients with memory disorders. The course of PTSD often does involve a delay between the time of exposure to trauma and symptom manifestation. In addition, symptom intensity can fluctuate significantly over the course of this often chronic illness.
The suffering associated with PTSD is often personal and concealed. Family and other collateral sources may be able to report only on social and occupational functioning. The authors recommend increased attention to proper assessment of (1) remote trauma history in patients being evaluated for memory disorders; and (2) cognitive decline in patients with history of PTSD. The problem of underreported cognitive decline is well known, although its extent is not. Early detection may help to mitigate the combined effects of these conditions. Aggressive early treatment of symptoms during the onset of cognitive dysfunction may prolong patients’ ability to remain at home.
Patient Care
Mr. B’s case was marked by significant tension in the home. Education and support of caregivers is essential to maintaining care in the least restrictive setting, such as the patient’s home. Families might be utterly bewildered by a patient’s apparently sudden preoccupation with traumatic memories. For many, this might be the first time they have ever heard the patient speak at length about the traumatic events. Simple strategies to limit exposure to distressing stimuli, improve grounding, and understand the effects of trauma can be taught. Psychopharmacologic intervention to improve sleep, slow cognitive decline, and decrease behavioral disturbances may be indicated.
Behavioral disturbance is frequently encountered when treating patients with cognitive impairment. In the limited literature on the subject, patients with both PTSD and cognitive impairment do not seem to be more prone to behavioral disturbance than patients with cognitive impairment alone.9 However, the case reports cited here demonstrate a high incidence of violence or potential violence in these comorbid patients. Routine assessment of potential harm from firearms or other weapons should be conducted assiduously.
It is possible that Vietnam War veterans may be more likely than previous veterans to exhibit behavioral disturbances in the context of cognitive decline and PTSD. A higher incidence of aggression, violence, and resistance to authority has been documented in this group.37 Substance abuse and dependence also occurs with higher frequency in this cohort and may complicate treatment of cognitive impairment and PTSD.38,39 A large number of these veterans may initially present to non-VA health care providers and these clinicians may be unaware of a patient’s prior combat exposure and thus fail to accurately assess PTSD.
Although the relation of PTSD and vulnerability to dementia has been well established, it is unknown how the presence of PTSD symptomatology impacts dementia symptoms or how the presence of dementia impacts PTSD symptoms. Posttraumatic stress disorder and dementia share similar risks like traumatic brain injury, low IQ, poor education, substance abuse, precipitated by stressful life events and impairment of coping, physical health and related risk factors. Unmasking PTSD symptoms resulting from dementia is a well-known phenomenon described in recent studies on late-onset stress symptomatology (LOSS).5,10,40
Since PTSD is a major risk factor that doubles the chance of developing dementia, mandatory screening for dementia in older patients along with assessment of other risk factors as a standard of care may help physicians in the early detection and initiation of care. Recognition of LOSS may be an important milestone in the treatment of delayed onset PTSD, which is considered a normal aging process and a premorbid stage of PTSD.10,40
Although there is no established treatment, early psychotherapeutic approaches like reminiscent therapy along with psychoeducation may be beneficial in patients with LOSS.40-42 Effective treatments for PTSD with patients with dementia may be challenging, though dementia was not found to be a barrier to implement prolonged exposure therapy in patients with mild cognitive impairment.43 Patient aligned care teams can be an ideal approach for the care of these veterans.
Conclusion
Posttraumatic stress disorder and dementia are well studied and documented disorders, although PTSD has been studied far more extensively in younger populations. Accounts of comorbidity of the 2 disorders are limited in the literature. Individuals may exhibit PTSD symptoms prior to the onset of dementia. They also may develop or uncover long quiescent symptoms of the disease. The populations of patients with PTSD and dementia are recognized, but their characteristics are largely unstudied and thus unknown.
Although the authors believe this to be a phenomenon of unrecognized coexistence of the 2 disorders, a disproportionate number of patients may be found in certain populations, especially among veterans. There is good evidence to expect increased numbers of these patients in the VA system, especially given the relative frequency of PTSD symptoms in aging cohorts of VA patients.
Click here to continue reading.
1. Spiro A 3rd, Schurr PP, Aldwin CM. Combat-related posttraumatic stress disorder symptoms in older men. Psychol Aging. 1994;9(1):17-26.
2. Van Achterberg ME, Rohrbaugh RM, Southwich SM. Emergence of PTSD in trauma survivors with dementia. J Clin Psychiatry. 2001;62(3):206-207.
3. Yaffe K, Vittinghoff E, Lindquist K, et al. Posttraumatic stress disorder and risk of dementia among US veterans. Arch Gen Psychiatry. 2010;67(6):608-613.
4. Qureshi SU, Kimbrell T, Pyne JM, et al. Greater prevalence and incidence of dementia in older veterans with posttraumatic stress disorder. J Am Geriatr Soc. 2010;58(9):1627-1633.
5. Barman R, Detweiler MB. Late onset stress symptomatology, subclinical PTSD or mixed etiologies in previously symptom free aging combat veterans. J Trauma Stress Disor Treat. 2014;3:4.
6. Barman R, Detweiler MB. The case for early psychotherapy in aging combat veterans experiencing late onset stress symptomatology. J Psychol Psychother. 2015;5:200.
7. Johnston D. A series of cases of dementia presenting with PTSD symptoms in World War II combat veterans. J Am Geriatr Soc. 2000;48(1):70-72.
8. Mittal D, Torres R, Abashidze A, Jimerson N. Worsening of post-traumatic stress disorder symptoms with cognitive decline: case series. J Geriatr Psychiatry Neurol. 2001;14(1):17-20.
9. Verma S, Orengo CA, Maxwell R, et al. Contribution of PTSD/POW history to behavioral disturbances in dementia. Int J Geriatr Psychiatry. 2001;16(4):356-360.
10. Potter CM, Kaiser AP, King LA, et al. Distinguishing late-onset stress symptomatology from posttraumatic stress disorder in older combat veterans. Aging Ment Health. 2013;17(2):173-179.
11. Cook JM. Post-traumatic stress disorder in older adults. PTSD Res Q. 2001;12(3):1-8.
12. Horowitz MJ. Stress Response Syndromes. 2nd ed. New York, NY: Jason Aronson; 1978.
13. Grossman AB, Levin BE, Katzen HL, Lechner S. PTSD symptoms and onset of neurologic disease in elderly trauma survivors. J Clin Exp Neuropsych. 2004;26(5):698-705.
14. Van Dyke C, Zilberg NJ, McKinnon JA. Posttraumatic stress disorder: a thirty-year delay in a World War II veteran. Am J Psychiatry. 1985;142(9):1070-1073.
15. Yehuda R. Status of glucocorticoid alterations in post-traumatic stress disorder. Ann N Y Acad Sci. 2009;1179:56-69.
16. Elder GH Jr, Clipp EC. Combat experience and emotional health: impairment and resilience in later life. J Pers. 1989;57(2):311-341.
17. Peavy GM, Salmon DP, Jacobson MW, et al. Effects of chronic stress on memory decline in cognitively normal and mildly impaired older adults. Am J Psychiatry. 2009;166(12):1384-1391.
18. Deng J, Lian Y, Shen C, et al. Adverse life event and risk of cognitive impairment: a 5-year prospective longitudinal study in Chongqing, China. Eur J Neurol. 2012;19(4):631-637.
19. Wilson RS, Arnold SE, Schneider JA, Kelly JF, Tang Y, Bennett DA. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology. 2006;27(3):143-153.
20. Johansson L, Guo X, Waern M, et al. Midlife psychological stress and risk of
dementia: a 35-year longitudinal population study. Brain. 2010;133(pt 8):2217-2224.
Note: Page numbers differ between the print issue and digital edition.
The number of veterans aged ≥ 65 years is expected to increase steadily as the Vietnam-era cohort ages. In 2012, the number of veterans aged ≥ 85 years was expected to peak at nearly 1.4 million. Vietnam-era veterans comprise the largest cohort of veterans, and > 15% of male and > 8% of female Vietnam veterans receiving care in the VA system have been diagnosed with posttraumatic stress disorder (PTSD). These veterans are rapidly approaching age groups in which cognitive disorders increase exponentially in prevalence.
Combat exposure has been called a common but “hidden variable” in studies of aging and health.1 Combat exposure may be even more hidden for Vietnam veterans who have pursued health care outside the VA system and less likely to announce their service to health care providers.
Even veterans who did not serve in traditional combat roles can experience chronic debilitation from the psychological stress of overseas deployment to a war zone. Indeed, cases of noncombat trauma have been presented in the context of cognitive decline and late-onset PTSD.2 It is probable that survivors of sexual assault, child abuse, crime, and natural disaster are also vulnerable to a recurrence of trauma symptoms if they experience cognitive slippage. In this article the authors report a case of delayed onset PTSD symptoms, precipitated by cognitive decline.
Case Report
Mr. B was a 72-year-old Korean War veteran referred for neuropsychological evaluation to establish baseline cognitive status before elective cardiac surgery. Mr. B relied on his wife to fill in many details of his personal history. His wife reported that the patient’s memory problems had increased significantly over the previous 12 months. Mr. B had been treated with donepezil 10 mg daily for about 1 year, with no observed benefit. His wife described life at home as “tense” due to his increased irritability and poor insight into his condition. Mr. B reported that he was often afraid of noises at night and needed to go outside and look around. His wife reported that he was very afraid of “strangers coming into the house.”
Mr. B was born in Arizona and experienced significant physical abuse while under the care of an alcoholic foster parent. He dropped out of high school and enlisted in the U.S. Marine Corps. Upon his discharge from military service, he worked as a truck driver for 23 years. He retired after experiencing hip problems. He drank heavily for many years after the war and, according to his wife, was “very violent,” but stopped 27 years previously, after injuring his wife while intoxicated. The patient’s medical history included hospitalization about 1 year prior to the evaluation following a fall associated with altered level of consciousness and confusion, which lasted several hours. He was discharged the same day and was thought to have had a stroke. The patient also had hypertension, hyperlipidemia, and sciatica. A carotid ultrasound showed bilateral carotid stenosis > 50%.
Mr. B was married for 45 years and had 5 children and 12 grandchildren. He enlisted in the U.S. Marine Corps at age 19 and served as a tank gunner during the Korean War. He experienced extremely heavy combat, was wounded several times (including loss of consciousness due to an explosion), and was hospitalizedfor 4 m onths in Japan. When he returned to the frontline, he found that many of the men in his unit had been killed. He was promoted to staff sergeant and tank commander. Mr. B received an honorable discharge after the war and a 50% service-connected disability pension for PTSD. He reported having received group psychotherapy at a VA hospital soon after the war but no other psychiatric treatment. He avoided watching the news because the Gulf War news reminded him of Korea.
Mr. B was smiling, pleasant, and cooperative throughout the 2 hours of testing and interviewing. He wore a Korean War veteran baseball cap festooned with military pins and ribbons, including a Purple Heart ribbon that he proudly showed to the test administrators. Unbidden, he also presented for inspection an assortment of life membership cards in various veterans service organizations. Mr. B reported frequent nightmares, night sweats, and intrusive thoughts about his combat experiences. During testing, he was repeatedly triggered by innocuous items and launched into a discourse on his combat experiences. When asked to memorize a short list of words that included the word fire, he said, “You know what that reminds me of...we had to fire big guns, 90 millimeter, that’s what it was…killing and how to kill.” When shown an abstract design that resembled the number 44, he said, “You know what that is? It was the radio call sign of our tank—‘This is 44, come in, we need some help.’”
Mr. B’s memory problems were marked by rapid forgetting, impaired ability to learn new information, and impaired ability to recall previously learned information. Language problems were also present, including difficulty recognizing and naming common objects, impaired auditory comprehension, and problems with verbal associative fluency during timed tasks. He also showed difficulties with executive functioning, attention, and working memory. His mini-mental state examination score was 21/30. He stated the year was 2020, did not know the day of the week, registered 2/3 words and recalled 0/3, he counted 3/5 in serial 7s, and was unable to repeat the phrase, “no ifs, ands, or buts.”
Discussion
Posttraumatic stress symptoms were present during the immediate aftermath of the initial trauma exposure for this patient. He managed to lead a relatively successful and productive life, sustained a marriage, and raised a family. The onset of cognitive decline precipitated a recrudescence of PTSD symptomology. In fact, the effects of combat trauma seem more malignant and extreme at the time of the memory disorders evaluation than at any prior time in his life.
A number of case reports have been published in recent years that describe comorbid presentations of cognitive disorder and PTSD symptomatology.3-6 A clinicalconsensus that cognitive decline can exacerbate previously well-managed symptoms of earlier psychological trauma seems to be emerging. Several published casestudies have noted that comorbid presentation of dementia and PTSD is often marked by violence, psychotic symptoms, and increased risk of hospitalization.7-9
PTSD Research
Unfortunately, systematic investigation into the relationship between PTSD and cognitive decline is in its infancy. Previous authors have posited various mechanisms to explain the exacerbation of dormant PTSD symptoms after cognitive decline.10,11 Some have attributed the phenomenon to an age-related failure of either repression or avoidance or to a compromised ability to actively focus their attention elsewhere.2,12 A finding of preservative errors on neuropsychological tests has been associated with an inability to organize and inhibit intrusive thought.13 In one case, the effects of combat trauma were purported to be denied, repressed, and largely forgotten for 30 years until rekindled by the patient’s deteriorating health and loss of employment.14 Several case examples have been presented in which physical illness, interpersonal loss, retirement, or losses of social support were other factors.15-18 Two major studies of veterans with PTSD, found that subjects were twice as likely to develop dementia.3,4 There is a strong association between chronic psychological stress and later development
of dementia. In a study by Wilson and colleagues, subjects with higher baseline stress had twice the chance of developing Alzheimer disease.19 Similar findings of
accelerated or higher cognitive decline were found by other studies, too.17,20
Hippocampal damage associated with prolonged, intense psychological stress has been cited as a possible contributor to PTSD symptom recrudescence in older adults.21 It is well known that emotional arousal leads to better-encoded memories. In the context of a cognitive disorder marked by gradual memory loss, traumatic memories might be the last to go.22 Another proposed biologic mechanism is a reduction in hippocampal volume and decreased inhibition of the amgydala, which results in preferential recall of the nondeclarative, amygdaloidal traumatic memories.8
Research on selective area damage in the hippocampus opens a new era of understanding of consequences of stress. The dentate gyrus (DG) is the main area of hippocampus that helps in neurogenesis and cornus ammonis 3 (CA3) for dendritic branching.23-25 In recent studies by Wang and colleagues, PTSD has been found to be associated with selective volume loss of the CA3/DG subfields, consistent with animal studies.24-28 Abundance of glucocorticoid receptors in the hippocampus, especially at CA3,29,30 may make it more vulnerable to the neurotoxic effect of glucocorticoids, causing suppression of neurogenesis,29 diminished dendritic branching,30 loss of synapses,26,31 and eventually diminished neuroplasticity,32 because CA3/DG is the main target of neurotoxicity by glucocorticoid and inflammatory damage.
The results of neuroimaging studies suggest that decreased integration of the prefrontal cortex and the hippocampus results in impaired short-term memory and perhaps increasing the prominence of long-term distressing memories.33 Clinical observation confirms that patients with PTSD experience vivid, intense, detailed, and realistic recollections of remote memories at a time when their ability to recall nontraumatic autobiographical detail is severely compromised.
Symptom Reemergence
Both prospective and retrospective studies have shown that PTSD symptoms can evolve, even after a 20-year long symptom-free period, and reemergence of PTSD
symptoms is not uncommon.34,35 A longer delay usually presents with less severe symptoms.36 The unavailability of complete information regarding a patient’s past adjustment to psychological trauma has encouraged some experts to label exacerbation of PTSD symptoms precipitated by cognitive disorder as delayed onset PTSD. In most cases, it seems that this is more accurately described as a recrudescence of symptoms that were better managed previously. The picture is clouded by the often bizarre and extreme manifestation of PTSD symptoms in patients with memory disorders. The course of PTSD often does involve a delay between the time of exposure to trauma and symptom manifestation. In addition, symptom intensity can fluctuate significantly over the course of this often chronic illness.
The suffering associated with PTSD is often personal and concealed. Family and other collateral sources may be able to report only on social and occupational functioning. The authors recommend increased attention to proper assessment of (1) remote trauma history in patients being evaluated for memory disorders; and (2) cognitive decline in patients with history of PTSD. The problem of underreported cognitive decline is well known, although its extent is not. Early detection may help to mitigate the combined effects of these conditions. Aggressive early treatment of symptoms during the onset of cognitive dysfunction may prolong patients’ ability to remain at home.
Patient Care
Mr. B’s case was marked by significant tension in the home. Education and support of caregivers is essential to maintaining care in the least restrictive setting, such as the patient’s home. Families might be utterly bewildered by a patient’s apparently sudden preoccupation with traumatic memories. For many, this might be the first time they have ever heard the patient speak at length about the traumatic events. Simple strategies to limit exposure to distressing stimuli, improve grounding, and understand the effects of trauma can be taught. Psychopharmacologic intervention to improve sleep, slow cognitive decline, and decrease behavioral disturbances may be indicated.
Behavioral disturbance is frequently encountered when treating patients with cognitive impairment. In the limited literature on the subject, patients with both PTSD and cognitive impairment do not seem to be more prone to behavioral disturbance than patients with cognitive impairment alone.9 However, the case reports cited here demonstrate a high incidence of violence or potential violence in these comorbid patients. Routine assessment of potential harm from firearms or other weapons should be conducted assiduously.
It is possible that Vietnam War veterans may be more likely than previous veterans to exhibit behavioral disturbances in the context of cognitive decline and PTSD. A higher incidence of aggression, violence, and resistance to authority has been documented in this group.37 Substance abuse and dependence also occurs with higher frequency in this cohort and may complicate treatment of cognitive impairment and PTSD.38,39 A large number of these veterans may initially present to non-VA health care providers and these clinicians may be unaware of a patient’s prior combat exposure and thus fail to accurately assess PTSD.
Although the relation of PTSD and vulnerability to dementia has been well established, it is unknown how the presence of PTSD symptomatology impacts dementia symptoms or how the presence of dementia impacts PTSD symptoms. Posttraumatic stress disorder and dementia share similar risks like traumatic brain injury, low IQ, poor education, substance abuse, precipitated by stressful life events and impairment of coping, physical health and related risk factors. Unmasking PTSD symptoms resulting from dementia is a well-known phenomenon described in recent studies on late-onset stress symptomatology (LOSS).5,10,40
Since PTSD is a major risk factor that doubles the chance of developing dementia, mandatory screening for dementia in older patients along with assessment of other risk factors as a standard of care may help physicians in the early detection and initiation of care. Recognition of LOSS may be an important milestone in the treatment of delayed onset PTSD, which is considered a normal aging process and a premorbid stage of PTSD.10,40
Although there is no established treatment, early psychotherapeutic approaches like reminiscent therapy along with psychoeducation may be beneficial in patients with LOSS.40-42 Effective treatments for PTSD with patients with dementia may be challenging, though dementia was not found to be a barrier to implement prolonged exposure therapy in patients with mild cognitive impairment.43 Patient aligned care teams can be an ideal approach for the care of these veterans.
Conclusion
Posttraumatic stress disorder and dementia are well studied and documented disorders, although PTSD has been studied far more extensively in younger populations. Accounts of comorbidity of the 2 disorders are limited in the literature. Individuals may exhibit PTSD symptoms prior to the onset of dementia. They also may develop or uncover long quiescent symptoms of the disease. The populations of patients with PTSD and dementia are recognized, but their characteristics are largely unstudied and thus unknown.
Although the authors believe this to be a phenomenon of unrecognized coexistence of the 2 disorders, a disproportionate number of patients may be found in certain populations, especially among veterans. There is good evidence to expect increased numbers of these patients in the VA system, especially given the relative frequency of PTSD symptoms in aging cohorts of VA patients.
Click here to continue reading.
The number of veterans aged ≥ 65 years is expected to increase steadily as the Vietnam-era cohort ages. In 2012, the number of veterans aged ≥ 85 years was expected to peak at nearly 1.4 million. Vietnam-era veterans comprise the largest cohort of veterans, and > 15% of male and > 8% of female Vietnam veterans receiving care in the VA system have been diagnosed with posttraumatic stress disorder (PTSD). These veterans are rapidly approaching age groups in which cognitive disorders increase exponentially in prevalence.
Combat exposure has been called a common but “hidden variable” in studies of aging and health.1 Combat exposure may be even more hidden for Vietnam veterans who have pursued health care outside the VA system and less likely to announce their service to health care providers.
Even veterans who did not serve in traditional combat roles can experience chronic debilitation from the psychological stress of overseas deployment to a war zone. Indeed, cases of noncombat trauma have been presented in the context of cognitive decline and late-onset PTSD.2 It is probable that survivors of sexual assault, child abuse, crime, and natural disaster are also vulnerable to a recurrence of trauma symptoms if they experience cognitive slippage. In this article the authors report a case of delayed onset PTSD symptoms, precipitated by cognitive decline.
Case Report
Mr. B was a 72-year-old Korean War veteran referred for neuropsychological evaluation to establish baseline cognitive status before elective cardiac surgery. Mr. B relied on his wife to fill in many details of his personal history. His wife reported that the patient’s memory problems had increased significantly over the previous 12 months. Mr. B had been treated with donepezil 10 mg daily for about 1 year, with no observed benefit. His wife described life at home as “tense” due to his increased irritability and poor insight into his condition. Mr. B reported that he was often afraid of noises at night and needed to go outside and look around. His wife reported that he was very afraid of “strangers coming into the house.”
Mr. B was born in Arizona and experienced significant physical abuse while under the care of an alcoholic foster parent. He dropped out of high school and enlisted in the U.S. Marine Corps. Upon his discharge from military service, he worked as a truck driver for 23 years. He retired after experiencing hip problems. He drank heavily for many years after the war and, according to his wife, was “very violent,” but stopped 27 years previously, after injuring his wife while intoxicated. The patient’s medical history included hospitalization about 1 year prior to the evaluation following a fall associated with altered level of consciousness and confusion, which lasted several hours. He was discharged the same day and was thought to have had a stroke. The patient also had hypertension, hyperlipidemia, and sciatica. A carotid ultrasound showed bilateral carotid stenosis > 50%.
Mr. B was married for 45 years and had 5 children and 12 grandchildren. He enlisted in the U.S. Marine Corps at age 19 and served as a tank gunner during the Korean War. He experienced extremely heavy combat, was wounded several times (including loss of consciousness due to an explosion), and was hospitalizedfor 4 m onths in Japan. When he returned to the frontline, he found that many of the men in his unit had been killed. He was promoted to staff sergeant and tank commander. Mr. B received an honorable discharge after the war and a 50% service-connected disability pension for PTSD. He reported having received group psychotherapy at a VA hospital soon after the war but no other psychiatric treatment. He avoided watching the news because the Gulf War news reminded him of Korea.
Mr. B was smiling, pleasant, and cooperative throughout the 2 hours of testing and interviewing. He wore a Korean War veteran baseball cap festooned with military pins and ribbons, including a Purple Heart ribbon that he proudly showed to the test administrators. Unbidden, he also presented for inspection an assortment of life membership cards in various veterans service organizations. Mr. B reported frequent nightmares, night sweats, and intrusive thoughts about his combat experiences. During testing, he was repeatedly triggered by innocuous items and launched into a discourse on his combat experiences. When asked to memorize a short list of words that included the word fire, he said, “You know what that reminds me of...we had to fire big guns, 90 millimeter, that’s what it was…killing and how to kill.” When shown an abstract design that resembled the number 44, he said, “You know what that is? It was the radio call sign of our tank—‘This is 44, come in, we need some help.’”
Mr. B’s memory problems were marked by rapid forgetting, impaired ability to learn new information, and impaired ability to recall previously learned information. Language problems were also present, including difficulty recognizing and naming common objects, impaired auditory comprehension, and problems with verbal associative fluency during timed tasks. He also showed difficulties with executive functioning, attention, and working memory. His mini-mental state examination score was 21/30. He stated the year was 2020, did not know the day of the week, registered 2/3 words and recalled 0/3, he counted 3/5 in serial 7s, and was unable to repeat the phrase, “no ifs, ands, or buts.”
Discussion
Posttraumatic stress symptoms were present during the immediate aftermath of the initial trauma exposure for this patient. He managed to lead a relatively successful and productive life, sustained a marriage, and raised a family. The onset of cognitive decline precipitated a recrudescence of PTSD symptomology. In fact, the effects of combat trauma seem more malignant and extreme at the time of the memory disorders evaluation than at any prior time in his life.
A number of case reports have been published in recent years that describe comorbid presentations of cognitive disorder and PTSD symptomatology.3-6 A clinicalconsensus that cognitive decline can exacerbate previously well-managed symptoms of earlier psychological trauma seems to be emerging. Several published casestudies have noted that comorbid presentation of dementia and PTSD is often marked by violence, psychotic symptoms, and increased risk of hospitalization.7-9
PTSD Research
Unfortunately, systematic investigation into the relationship between PTSD and cognitive decline is in its infancy. Previous authors have posited various mechanisms to explain the exacerbation of dormant PTSD symptoms after cognitive decline.10,11 Some have attributed the phenomenon to an age-related failure of either repression or avoidance or to a compromised ability to actively focus their attention elsewhere.2,12 A finding of preservative errors on neuropsychological tests has been associated with an inability to organize and inhibit intrusive thought.13 In one case, the effects of combat trauma were purported to be denied, repressed, and largely forgotten for 30 years until rekindled by the patient’s deteriorating health and loss of employment.14 Several case examples have been presented in which physical illness, interpersonal loss, retirement, or losses of social support were other factors.15-18 Two major studies of veterans with PTSD, found that subjects were twice as likely to develop dementia.3,4 There is a strong association between chronic psychological stress and later development
of dementia. In a study by Wilson and colleagues, subjects with higher baseline stress had twice the chance of developing Alzheimer disease.19 Similar findings of
accelerated or higher cognitive decline were found by other studies, too.17,20
Hippocampal damage associated with prolonged, intense psychological stress has been cited as a possible contributor to PTSD symptom recrudescence in older adults.21 It is well known that emotional arousal leads to better-encoded memories. In the context of a cognitive disorder marked by gradual memory loss, traumatic memories might be the last to go.22 Another proposed biologic mechanism is a reduction in hippocampal volume and decreased inhibition of the amgydala, which results in preferential recall of the nondeclarative, amygdaloidal traumatic memories.8
Research on selective area damage in the hippocampus opens a new era of understanding of consequences of stress. The dentate gyrus (DG) is the main area of hippocampus that helps in neurogenesis and cornus ammonis 3 (CA3) for dendritic branching.23-25 In recent studies by Wang and colleagues, PTSD has been found to be associated with selective volume loss of the CA3/DG subfields, consistent with animal studies.24-28 Abundance of glucocorticoid receptors in the hippocampus, especially at CA3,29,30 may make it more vulnerable to the neurotoxic effect of glucocorticoids, causing suppression of neurogenesis,29 diminished dendritic branching,30 loss of synapses,26,31 and eventually diminished neuroplasticity,32 because CA3/DG is the main target of neurotoxicity by glucocorticoid and inflammatory damage.
The results of neuroimaging studies suggest that decreased integration of the prefrontal cortex and the hippocampus results in impaired short-term memory and perhaps increasing the prominence of long-term distressing memories.33 Clinical observation confirms that patients with PTSD experience vivid, intense, detailed, and realistic recollections of remote memories at a time when their ability to recall nontraumatic autobiographical detail is severely compromised.
Symptom Reemergence
Both prospective and retrospective studies have shown that PTSD symptoms can evolve, even after a 20-year long symptom-free period, and reemergence of PTSD
symptoms is not uncommon.34,35 A longer delay usually presents with less severe symptoms.36 The unavailability of complete information regarding a patient’s past adjustment to psychological trauma has encouraged some experts to label exacerbation of PTSD symptoms precipitated by cognitive disorder as delayed onset PTSD. In most cases, it seems that this is more accurately described as a recrudescence of symptoms that were better managed previously. The picture is clouded by the often bizarre and extreme manifestation of PTSD symptoms in patients with memory disorders. The course of PTSD often does involve a delay between the time of exposure to trauma and symptom manifestation. In addition, symptom intensity can fluctuate significantly over the course of this often chronic illness.
The suffering associated with PTSD is often personal and concealed. Family and other collateral sources may be able to report only on social and occupational functioning. The authors recommend increased attention to proper assessment of (1) remote trauma history in patients being evaluated for memory disorders; and (2) cognitive decline in patients with history of PTSD. The problem of underreported cognitive decline is well known, although its extent is not. Early detection may help to mitigate the combined effects of these conditions. Aggressive early treatment of symptoms during the onset of cognitive dysfunction may prolong patients’ ability to remain at home.
Patient Care
Mr. B’s case was marked by significant tension in the home. Education and support of caregivers is essential to maintaining care in the least restrictive setting, such as the patient’s home. Families might be utterly bewildered by a patient’s apparently sudden preoccupation with traumatic memories. For many, this might be the first time they have ever heard the patient speak at length about the traumatic events. Simple strategies to limit exposure to distressing stimuli, improve grounding, and understand the effects of trauma can be taught. Psychopharmacologic intervention to improve sleep, slow cognitive decline, and decrease behavioral disturbances may be indicated.
Behavioral disturbance is frequently encountered when treating patients with cognitive impairment. In the limited literature on the subject, patients with both PTSD and cognitive impairment do not seem to be more prone to behavioral disturbance than patients with cognitive impairment alone.9 However, the case reports cited here demonstrate a high incidence of violence or potential violence in these comorbid patients. Routine assessment of potential harm from firearms or other weapons should be conducted assiduously.
It is possible that Vietnam War veterans may be more likely than previous veterans to exhibit behavioral disturbances in the context of cognitive decline and PTSD. A higher incidence of aggression, violence, and resistance to authority has been documented in this group.37 Substance abuse and dependence also occurs with higher frequency in this cohort and may complicate treatment of cognitive impairment and PTSD.38,39 A large number of these veterans may initially present to non-VA health care providers and these clinicians may be unaware of a patient’s prior combat exposure and thus fail to accurately assess PTSD.
Although the relation of PTSD and vulnerability to dementia has been well established, it is unknown how the presence of PTSD symptomatology impacts dementia symptoms or how the presence of dementia impacts PTSD symptoms. Posttraumatic stress disorder and dementia share similar risks like traumatic brain injury, low IQ, poor education, substance abuse, precipitated by stressful life events and impairment of coping, physical health and related risk factors. Unmasking PTSD symptoms resulting from dementia is a well-known phenomenon described in recent studies on late-onset stress symptomatology (LOSS).5,10,40
Since PTSD is a major risk factor that doubles the chance of developing dementia, mandatory screening for dementia in older patients along with assessment of other risk factors as a standard of care may help physicians in the early detection and initiation of care. Recognition of LOSS may be an important milestone in the treatment of delayed onset PTSD, which is considered a normal aging process and a premorbid stage of PTSD.10,40
Although there is no established treatment, early psychotherapeutic approaches like reminiscent therapy along with psychoeducation may be beneficial in patients with LOSS.40-42 Effective treatments for PTSD with patients with dementia may be challenging, though dementia was not found to be a barrier to implement prolonged exposure therapy in patients with mild cognitive impairment.43 Patient aligned care teams can be an ideal approach for the care of these veterans.
Conclusion
Posttraumatic stress disorder and dementia are well studied and documented disorders, although PTSD has been studied far more extensively in younger populations. Accounts of comorbidity of the 2 disorders are limited in the literature. Individuals may exhibit PTSD symptoms prior to the onset of dementia. They also may develop or uncover long quiescent symptoms of the disease. The populations of patients with PTSD and dementia are recognized, but their characteristics are largely unstudied and thus unknown.
Although the authors believe this to be a phenomenon of unrecognized coexistence of the 2 disorders, a disproportionate number of patients may be found in certain populations, especially among veterans. There is good evidence to expect increased numbers of these patients in the VA system, especially given the relative frequency of PTSD symptoms in aging cohorts of VA patients.
Click here to continue reading.
1. Spiro A 3rd, Schurr PP, Aldwin CM. Combat-related posttraumatic stress disorder symptoms in older men. Psychol Aging. 1994;9(1):17-26.
2. Van Achterberg ME, Rohrbaugh RM, Southwich SM. Emergence of PTSD in trauma survivors with dementia. J Clin Psychiatry. 2001;62(3):206-207.
3. Yaffe K, Vittinghoff E, Lindquist K, et al. Posttraumatic stress disorder and risk of dementia among US veterans. Arch Gen Psychiatry. 2010;67(6):608-613.
4. Qureshi SU, Kimbrell T, Pyne JM, et al. Greater prevalence and incidence of dementia in older veterans with posttraumatic stress disorder. J Am Geriatr Soc. 2010;58(9):1627-1633.
5. Barman R, Detweiler MB. Late onset stress symptomatology, subclinical PTSD or mixed etiologies in previously symptom free aging combat veterans. J Trauma Stress Disor Treat. 2014;3:4.
6. Barman R, Detweiler MB. The case for early psychotherapy in aging combat veterans experiencing late onset stress symptomatology. J Psychol Psychother. 2015;5:200.
7. Johnston D. A series of cases of dementia presenting with PTSD symptoms in World War II combat veterans. J Am Geriatr Soc. 2000;48(1):70-72.
8. Mittal D, Torres R, Abashidze A, Jimerson N. Worsening of post-traumatic stress disorder symptoms with cognitive decline: case series. J Geriatr Psychiatry Neurol. 2001;14(1):17-20.
9. Verma S, Orengo CA, Maxwell R, et al. Contribution of PTSD/POW history to behavioral disturbances in dementia. Int J Geriatr Psychiatry. 2001;16(4):356-360.
10. Potter CM, Kaiser AP, King LA, et al. Distinguishing late-onset stress symptomatology from posttraumatic stress disorder in older combat veterans. Aging Ment Health. 2013;17(2):173-179.
11. Cook JM. Post-traumatic stress disorder in older adults. PTSD Res Q. 2001;12(3):1-8.
12. Horowitz MJ. Stress Response Syndromes. 2nd ed. New York, NY: Jason Aronson; 1978.
13. Grossman AB, Levin BE, Katzen HL, Lechner S. PTSD symptoms and onset of neurologic disease in elderly trauma survivors. J Clin Exp Neuropsych. 2004;26(5):698-705.
14. Van Dyke C, Zilberg NJ, McKinnon JA. Posttraumatic stress disorder: a thirty-year delay in a World War II veteran. Am J Psychiatry. 1985;142(9):1070-1073.
15. Yehuda R. Status of glucocorticoid alterations in post-traumatic stress disorder. Ann N Y Acad Sci. 2009;1179:56-69.
16. Elder GH Jr, Clipp EC. Combat experience and emotional health: impairment and resilience in later life. J Pers. 1989;57(2):311-341.
17. Peavy GM, Salmon DP, Jacobson MW, et al. Effects of chronic stress on memory decline in cognitively normal and mildly impaired older adults. Am J Psychiatry. 2009;166(12):1384-1391.
18. Deng J, Lian Y, Shen C, et al. Adverse life event and risk of cognitive impairment: a 5-year prospective longitudinal study in Chongqing, China. Eur J Neurol. 2012;19(4):631-637.
19. Wilson RS, Arnold SE, Schneider JA, Kelly JF, Tang Y, Bennett DA. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology. 2006;27(3):143-153.
20. Johansson L, Guo X, Waern M, et al. Midlife psychological stress and risk of
dementia: a 35-year longitudinal population study. Brain. 2010;133(pt 8):2217-2224.
Note: Page numbers differ between the print issue and digital edition.
1. Spiro A 3rd, Schurr PP, Aldwin CM. Combat-related posttraumatic stress disorder symptoms in older men. Psychol Aging. 1994;9(1):17-26.
2. Van Achterberg ME, Rohrbaugh RM, Southwich SM. Emergence of PTSD in trauma survivors with dementia. J Clin Psychiatry. 2001;62(3):206-207.
3. Yaffe K, Vittinghoff E, Lindquist K, et al. Posttraumatic stress disorder and risk of dementia among US veterans. Arch Gen Psychiatry. 2010;67(6):608-613.
4. Qureshi SU, Kimbrell T, Pyne JM, et al. Greater prevalence and incidence of dementia in older veterans with posttraumatic stress disorder. J Am Geriatr Soc. 2010;58(9):1627-1633.
5. Barman R, Detweiler MB. Late onset stress symptomatology, subclinical PTSD or mixed etiologies in previously symptom free aging combat veterans. J Trauma Stress Disor Treat. 2014;3:4.
6. Barman R, Detweiler MB. The case for early psychotherapy in aging combat veterans experiencing late onset stress symptomatology. J Psychol Psychother. 2015;5:200.
7. Johnston D. A series of cases of dementia presenting with PTSD symptoms in World War II combat veterans. J Am Geriatr Soc. 2000;48(1):70-72.
8. Mittal D, Torres R, Abashidze A, Jimerson N. Worsening of post-traumatic stress disorder symptoms with cognitive decline: case series. J Geriatr Psychiatry Neurol. 2001;14(1):17-20.
9. Verma S, Orengo CA, Maxwell R, et al. Contribution of PTSD/POW history to behavioral disturbances in dementia. Int J Geriatr Psychiatry. 2001;16(4):356-360.
10. Potter CM, Kaiser AP, King LA, et al. Distinguishing late-onset stress symptomatology from posttraumatic stress disorder in older combat veterans. Aging Ment Health. 2013;17(2):173-179.
11. Cook JM. Post-traumatic stress disorder in older adults. PTSD Res Q. 2001;12(3):1-8.
12. Horowitz MJ. Stress Response Syndromes. 2nd ed. New York, NY: Jason Aronson; 1978.
13. Grossman AB, Levin BE, Katzen HL, Lechner S. PTSD symptoms and onset of neurologic disease in elderly trauma survivors. J Clin Exp Neuropsych. 2004;26(5):698-705.
14. Van Dyke C, Zilberg NJ, McKinnon JA. Posttraumatic stress disorder: a thirty-year delay in a World War II veteran. Am J Psychiatry. 1985;142(9):1070-1073.
15. Yehuda R. Status of glucocorticoid alterations in post-traumatic stress disorder. Ann N Y Acad Sci. 2009;1179:56-69.
16. Elder GH Jr, Clipp EC. Combat experience and emotional health: impairment and resilience in later life. J Pers. 1989;57(2):311-341.
17. Peavy GM, Salmon DP, Jacobson MW, et al. Effects of chronic stress on memory decline in cognitively normal and mildly impaired older adults. Am J Psychiatry. 2009;166(12):1384-1391.
18. Deng J, Lian Y, Shen C, et al. Adverse life event and risk of cognitive impairment: a 5-year prospective longitudinal study in Chongqing, China. Eur J Neurol. 2012;19(4):631-637.
19. Wilson RS, Arnold SE, Schneider JA, Kelly JF, Tang Y, Bennett DA. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology. 2006;27(3):143-153.
20. Johansson L, Guo X, Waern M, et al. Midlife psychological stress and risk of
dementia: a 35-year longitudinal population study. Brain. 2010;133(pt 8):2217-2224.
Note: Page numbers differ between the print issue and digital edition.
Case Report: Acute Intermittent Porphyria
Case
A 34-year-old woman presented to the ED with severe, persistent abdominal pain that had begun 18 days earlier. She was 7 weeks pregnant and had been seen in the same ED the day before. During that visit, ultrasound had shown a single pregnancy of doubtful viability. Abdominal magnetic resonance imaging was normal. She was given multiple doses of hydromorphone. The discharge diagnosis was “missed abortion.”
Since the onset of her pain, she had been hospitalized twice elsewhere, with no clear diagnosis to explain her pain. Treatment consisted of repeat doses of hydromorphone. During the second hospitalization, a sodium level of 109 mEq/L had been corrected with hypertonic saline, and a urinary tract infection (UTI) had been treated with cephalexin.
Our patient had never experienced similar abdominal pain. Her medical history included depression and asthma. Her family history was notable for an aunt who had died of lung cancer.
On this ED visit, the patient’s vital signs were normal. On examination, she was moaning in pain and clutching her abdomen. The abdomen was tender in both lower quadrants, with guarding but no rebound. Her sodium level was 125 mEq/L; the day before it had been 132 mEq/L.
Urine dipstick testing showed 2+ glucose and 2+ bilirubin; both had been within normal range (negative) the day before. An abdominal/pelvic computed tomography scan with intravenous (IV) and oral contrast did not reveal any potential cause of the patient’s pain. Multiple doses of IV hydromorphone were given, but her pain persisted.
We revisited our patient’s family history. With prompting, she remembered that as a teenager, her mother had had an illness that caused “problems with her nerves and blood vessels and turned her urine red.” When reached by phone, the patient’s mother, who lives outside the United States, said she was familiar with the term “porphyria,” but curiously, she did not state she carried the diagnosis, and had not advised her children they could be at risk.
The patient was admitted to the intensive care unit (ICU) for treatment of hyponatremia. Her mother’s history led us to suspect porphyria, so we sent a urine sample from the ED for porphobilinogen (PBG) testing. Her urine was not red at that time (on further questioning, she remembered she had had an episode of “red urine” recently). Two days later, after the PBG result came back positive, treatment was initiated for porphyria. With further stool and serum testing, the diagnosis of acute intermittent porphyria (AIP) was made.
The patient was treated with glucose loading and hemin therapy. In the ICU, 2 ampules of 50% dextrose in water solution (D50W) was administered, and she was transferred to the hematology-oncology service for hemin therapy. Soon after, she underwent a dilation and curettage. Three weeks later, she was discharged in good condition.
Discussion
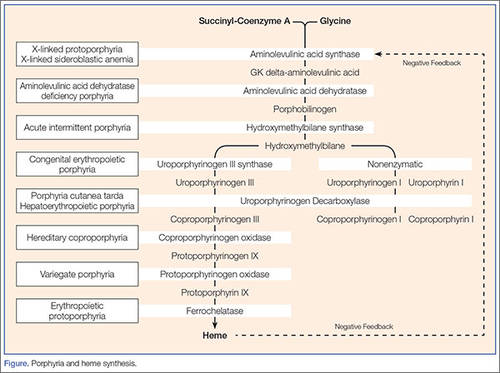
The unifying diagnostic concept is that toxins damage all components of the nervous system: intestinal, central, peripheral, and autonomic. Hence, any combination of abdominal pain, vomiting, psychiatric symptoms, vital sign instability, weakness, or sensory loss may occur.1 To further confuse the diagnostician, the constellation of symptoms may vary with each episode.
Two critical laboratory clues are red urine—which often is mistaken for a UTI or hematuria—and hyponatremia.1 Another porphyria hallmark is triggers. These include drugs, carbohydrate deprivation, smoking, and stress. Common chemical inciters include alcohol, ketamine, etomidate, macrodantin, nifedipine, progesterone, and phenytoin.1 The Atkins diet (zero carbohydrates) reportedly caused an uptick in new porphyria cases.1
Attacks usually start after puberty. Women tend to experience flares during the luteal (progesterone) phase of the menstrual cycle.1 Acute intermittent porphyria can mimic Guillain-Barré syndrome and psychosis.2 Delayed diagnosis may lead to irreversible neurological damage or death.2
Despite AIP’s complexity, initial diagnostic testing is simple: a urinary PBG level obtained during an attack is virtually 100% sensitive and specific for AIP and two other acute porphyrias: hereditary coproporphyria (HCP), and variegate porphyria (VP). A positive urine PBG mandates immediate treatment—even while awaiting porphyrin and GK delta-aminolevulinic acid (ALA) levels in stool and serum to identify which porphyria (AIP, HCP, or VP) is present. The fourth (and least common) acute porphyria, ALA dehydratase porphyria (ADP), may produce no PBG elevation and requires separate porphyrin and ALA testing to make the diagnosis.
Treatment
Treatment targets runaway heme precursor synthesis at its start and finish (Figure). Glucose-loading suppresses the initial enzyme, ALA synthase. Since the absence of normal end-product (heme) drives the enzymatic cascade, addition of IV hemin provides the substrate—and negative feedback—to stop it.
Conclusion
This case represents an example of AIP in which a patient presented with the characteristic abdominal pain and hyponatremia, complicated by the fact that she was pregnant and her urine was not red.
Intractable abdominal pain with negative imaging must prompt a search for red urine, neurological symptoms, porphyria medication triggers, and a family history of porphyria. Any constellation of findings should prompt immediate urine PBG testing.
In the appropriate clinical setting, it may be prudent to glucose-load a patient while waiting for confirmatory testing (which can take 1-2 days). Hemin therapy is best instituted by the hematology service after high urine PBG levels are confirmed.
- Sood GK, Anderson KE. Pathogenesis, clinical manifestations, and diagnosis of acute intermittent porphyria.” Available at: http://www.uptodate.com/contents/pathogenesis-clinical-manifestations-and-diagnosis-of-acute-intermittent-porphyria. Accessed February 22, 2016
- Bonkovsky HL, Siao P, Roig Z, Hedley-Whyte ET, Flotte TJ. Case records of the Massachusetts General Hospital. Case 20-2008. A 57-year-old woman with abdominal pain and weakness after gastric bypass surgery. N Engl J Med. 2008;358(26):2813-2825.
Case
A 34-year-old woman presented to the ED with severe, persistent abdominal pain that had begun 18 days earlier. She was 7 weeks pregnant and had been seen in the same ED the day before. During that visit, ultrasound had shown a single pregnancy of doubtful viability. Abdominal magnetic resonance imaging was normal. She was given multiple doses of hydromorphone. The discharge diagnosis was “missed abortion.”
Since the onset of her pain, she had been hospitalized twice elsewhere, with no clear diagnosis to explain her pain. Treatment consisted of repeat doses of hydromorphone. During the second hospitalization, a sodium level of 109 mEq/L had been corrected with hypertonic saline, and a urinary tract infection (UTI) had been treated with cephalexin.
Our patient had never experienced similar abdominal pain. Her medical history included depression and asthma. Her family history was notable for an aunt who had died of lung cancer.
On this ED visit, the patient’s vital signs were normal. On examination, she was moaning in pain and clutching her abdomen. The abdomen was tender in both lower quadrants, with guarding but no rebound. Her sodium level was 125 mEq/L; the day before it had been 132 mEq/L.
Urine dipstick testing showed 2+ glucose and 2+ bilirubin; both had been within normal range (negative) the day before. An abdominal/pelvic computed tomography scan with intravenous (IV) and oral contrast did not reveal any potential cause of the patient’s pain. Multiple doses of IV hydromorphone were given, but her pain persisted.
We revisited our patient’s family history. With prompting, she remembered that as a teenager, her mother had had an illness that caused “problems with her nerves and blood vessels and turned her urine red.” When reached by phone, the patient’s mother, who lives outside the United States, said she was familiar with the term “porphyria,” but curiously, she did not state she carried the diagnosis, and had not advised her children they could be at risk.
The patient was admitted to the intensive care unit (ICU) for treatment of hyponatremia. Her mother’s history led us to suspect porphyria, so we sent a urine sample from the ED for porphobilinogen (PBG) testing. Her urine was not red at that time (on further questioning, she remembered she had had an episode of “red urine” recently). Two days later, after the PBG result came back positive, treatment was initiated for porphyria. With further stool and serum testing, the diagnosis of acute intermittent porphyria (AIP) was made.
The patient was treated with glucose loading and hemin therapy. In the ICU, 2 ampules of 50% dextrose in water solution (D50W) was administered, and she was transferred to the hematology-oncology service for hemin therapy. Soon after, she underwent a dilation and curettage. Three weeks later, she was discharged in good condition.
Discussion
The unifying diagnostic concept is that toxins damage all components of the nervous system: intestinal, central, peripheral, and autonomic. Hence, any combination of abdominal pain, vomiting, psychiatric symptoms, vital sign instability, weakness, or sensory loss may occur.1 To further confuse the diagnostician, the constellation of symptoms may vary with each episode.
Two critical laboratory clues are red urine—which often is mistaken for a UTI or hematuria—and hyponatremia.1 Another porphyria hallmark is triggers. These include drugs, carbohydrate deprivation, smoking, and stress. Common chemical inciters include alcohol, ketamine, etomidate, macrodantin, nifedipine, progesterone, and phenytoin.1 The Atkins diet (zero carbohydrates) reportedly caused an uptick in new porphyria cases.1
Attacks usually start after puberty. Women tend to experience flares during the luteal (progesterone) phase of the menstrual cycle.1 Acute intermittent porphyria can mimic Guillain-Barré syndrome and psychosis.2 Delayed diagnosis may lead to irreversible neurological damage or death.2
Despite AIP’s complexity, initial diagnostic testing is simple: a urinary PBG level obtained during an attack is virtually 100% sensitive and specific for AIP and two other acute porphyrias: hereditary coproporphyria (HCP), and variegate porphyria (VP). A positive urine PBG mandates immediate treatment—even while awaiting porphyrin and GK delta-aminolevulinic acid (ALA) levels in stool and serum to identify which porphyria (AIP, HCP, or VP) is present. The fourth (and least common) acute porphyria, ALA dehydratase porphyria (ADP), may produce no PBG elevation and requires separate porphyrin and ALA testing to make the diagnosis.
Treatment
Treatment targets runaway heme precursor synthesis at its start and finish (Figure). Glucose-loading suppresses the initial enzyme, ALA synthase. Since the absence of normal end-product (heme) drives the enzymatic cascade, addition of IV hemin provides the substrate—and negative feedback—to stop it.
Conclusion
This case represents an example of AIP in which a patient presented with the characteristic abdominal pain and hyponatremia, complicated by the fact that she was pregnant and her urine was not red.
Intractable abdominal pain with negative imaging must prompt a search for red urine, neurological symptoms, porphyria medication triggers, and a family history of porphyria. Any constellation of findings should prompt immediate urine PBG testing.
In the appropriate clinical setting, it may be prudent to glucose-load a patient while waiting for confirmatory testing (which can take 1-2 days). Hemin therapy is best instituted by the hematology service after high urine PBG levels are confirmed.
Case
A 34-year-old woman presented to the ED with severe, persistent abdominal pain that had begun 18 days earlier. She was 7 weeks pregnant and had been seen in the same ED the day before. During that visit, ultrasound had shown a single pregnancy of doubtful viability. Abdominal magnetic resonance imaging was normal. She was given multiple doses of hydromorphone. The discharge diagnosis was “missed abortion.”
Since the onset of her pain, she had been hospitalized twice elsewhere, with no clear diagnosis to explain her pain. Treatment consisted of repeat doses of hydromorphone. During the second hospitalization, a sodium level of 109 mEq/L had been corrected with hypertonic saline, and a urinary tract infection (UTI) had been treated with cephalexin.
Our patient had never experienced similar abdominal pain. Her medical history included depression and asthma. Her family history was notable for an aunt who had died of lung cancer.
On this ED visit, the patient’s vital signs were normal. On examination, she was moaning in pain and clutching her abdomen. The abdomen was tender in both lower quadrants, with guarding but no rebound. Her sodium level was 125 mEq/L; the day before it had been 132 mEq/L.
Urine dipstick testing showed 2+ glucose and 2+ bilirubin; both had been within normal range (negative) the day before. An abdominal/pelvic computed tomography scan with intravenous (IV) and oral contrast did not reveal any potential cause of the patient’s pain. Multiple doses of IV hydromorphone were given, but her pain persisted.
We revisited our patient’s family history. With prompting, she remembered that as a teenager, her mother had had an illness that caused “problems with her nerves and blood vessels and turned her urine red.” When reached by phone, the patient’s mother, who lives outside the United States, said she was familiar with the term “porphyria,” but curiously, she did not state she carried the diagnosis, and had not advised her children they could be at risk.
The patient was admitted to the intensive care unit (ICU) for treatment of hyponatremia. Her mother’s history led us to suspect porphyria, so we sent a urine sample from the ED for porphobilinogen (PBG) testing. Her urine was not red at that time (on further questioning, she remembered she had had an episode of “red urine” recently). Two days later, after the PBG result came back positive, treatment was initiated for porphyria. With further stool and serum testing, the diagnosis of acute intermittent porphyria (AIP) was made.
The patient was treated with glucose loading and hemin therapy. In the ICU, 2 ampules of 50% dextrose in water solution (D50W) was administered, and she was transferred to the hematology-oncology service for hemin therapy. Soon after, she underwent a dilation and curettage. Three weeks later, she was discharged in good condition.
Discussion
The unifying diagnostic concept is that toxins damage all components of the nervous system: intestinal, central, peripheral, and autonomic. Hence, any combination of abdominal pain, vomiting, psychiatric symptoms, vital sign instability, weakness, or sensory loss may occur.1 To further confuse the diagnostician, the constellation of symptoms may vary with each episode.
Two critical laboratory clues are red urine—which often is mistaken for a UTI or hematuria—and hyponatremia.1 Another porphyria hallmark is triggers. These include drugs, carbohydrate deprivation, smoking, and stress. Common chemical inciters include alcohol, ketamine, etomidate, macrodantin, nifedipine, progesterone, and phenytoin.1 The Atkins diet (zero carbohydrates) reportedly caused an uptick in new porphyria cases.1
Attacks usually start after puberty. Women tend to experience flares during the luteal (progesterone) phase of the menstrual cycle.1 Acute intermittent porphyria can mimic Guillain-Barré syndrome and psychosis.2 Delayed diagnosis may lead to irreversible neurological damage or death.2
Despite AIP’s complexity, initial diagnostic testing is simple: a urinary PBG level obtained during an attack is virtually 100% sensitive and specific for AIP and two other acute porphyrias: hereditary coproporphyria (HCP), and variegate porphyria (VP). A positive urine PBG mandates immediate treatment—even while awaiting porphyrin and GK delta-aminolevulinic acid (ALA) levels in stool and serum to identify which porphyria (AIP, HCP, or VP) is present. The fourth (and least common) acute porphyria, ALA dehydratase porphyria (ADP), may produce no PBG elevation and requires separate porphyrin and ALA testing to make the diagnosis.
Treatment
Treatment targets runaway heme precursor synthesis at its start and finish (Figure). Glucose-loading suppresses the initial enzyme, ALA synthase. Since the absence of normal end-product (heme) drives the enzymatic cascade, addition of IV hemin provides the substrate—and negative feedback—to stop it.
Conclusion
This case represents an example of AIP in which a patient presented with the characteristic abdominal pain and hyponatremia, complicated by the fact that she was pregnant and her urine was not red.
Intractable abdominal pain with negative imaging must prompt a search for red urine, neurological symptoms, porphyria medication triggers, and a family history of porphyria. Any constellation of findings should prompt immediate urine PBG testing.
In the appropriate clinical setting, it may be prudent to glucose-load a patient while waiting for confirmatory testing (which can take 1-2 days). Hemin therapy is best instituted by the hematology service after high urine PBG levels are confirmed.
- Sood GK, Anderson KE. Pathogenesis, clinical manifestations, and diagnosis of acute intermittent porphyria.” Available at: http://www.uptodate.com/contents/pathogenesis-clinical-manifestations-and-diagnosis-of-acute-intermittent-porphyria. Accessed February 22, 2016
- Bonkovsky HL, Siao P, Roig Z, Hedley-Whyte ET, Flotte TJ. Case records of the Massachusetts General Hospital. Case 20-2008. A 57-year-old woman with abdominal pain and weakness after gastric bypass surgery. N Engl J Med. 2008;358(26):2813-2825.
- Sood GK, Anderson KE. Pathogenesis, clinical manifestations, and diagnosis of acute intermittent porphyria.” Available at: http://www.uptodate.com/contents/pathogenesis-clinical-manifestations-and-diagnosis-of-acute-intermittent-porphyria. Accessed February 22, 2016
- Bonkovsky HL, Siao P, Roig Z, Hedley-Whyte ET, Flotte TJ. Case records of the Massachusetts General Hospital. Case 20-2008. A 57-year-old woman with abdominal pain and weakness after gastric bypass surgery. N Engl J Med. 2008;358(26):2813-2825.

Swollen lymph nodes • patient is otherwise "healthy" • Dx?
THE CASE
A 52-year-old woman presented to our family clinic for a well woman exam. The only complaints she had were fatigue, which she attributed to a work day that began at 4 am, and hot flashes. She denied fever, weight loss, abdominal pain, medication use, or recent foreign travel. She had a history of hyperlipidemia and surgical removal of a cutaneous melanoma at age 12.
Her vital signs and physical exam were normal with the exception of 3 enlarged left inguinal lymph nodes and approximately 5 enlarged right inguinal lymph nodes. The nodes were freely moveable and non-tender. No additional lymphadenopathy or splenomegaly was found.
THE DIAGNOSIS
The patient’s work-up included a Pap smear, complete blood count (CBC), comprehensive metabolic panel (CMP), and pelvic and inguinal ultrasound. All tests were normal, except the ultrasound, which revealed 3 solid left inguinal lymph nodes measuring 1.2 to 1.6 cm and 6 solid right inguinal lymph nodes measuring 1.1 to 1.8 cm. An abdominal and pelvic computed tomography (CT) scan with contrast identified nonspecific mesenteric, inguinal, retrocrural, and retroperitoneal adenopathy. An open biopsy of the largest inguinal lymph node revealed follicular lymphoma, a form of non-Hodgkin’s lymphoma. (Hodgkin’s and non-Hodgkin’s lymphoma (NHL) are uncommon causes of inguinal lymphadenopathy.1)
We consulted Oncology and they recommended a positron emission tomography (PET)/CT scan, which showed widespread lymphadenopathy. A bone marrow biopsy confirmed follicular lymphoma grade II, Ann Arbor stage III.
DISCUSSION
Generalized lymphadenopathy involves lymph node enlargement in more than one region of the body. Lymph nodes >1 cm in adults are considered abnormal and the differential diagnosis is broad (TABLE2-5). A patient’s age is a significant factor in the evaluation of peripheral lymphadenopathy.2-5 Results from one study of 628 patients who underwent nodal biopsy for peripheral lymphadenopathy revealed approximately 80% of nodes in patients under age 30 were noncancerous and likely had an infectious cause.3 However, among patients over age 50, only 40% were noncancerous.3
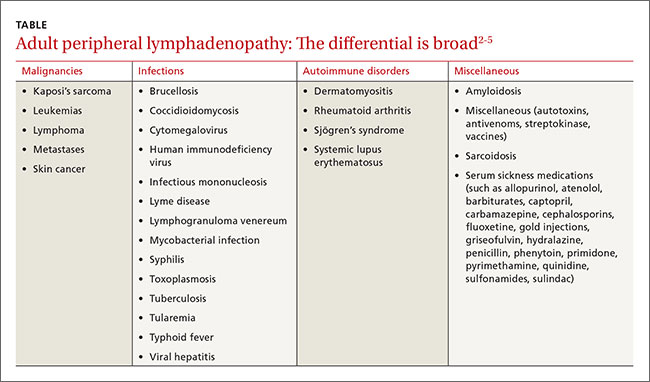
Node enlargement can be palpated in the head, neck, axilla, inguinal, and popliteal areas. Inguinal lymph nodes up to 2 cm in size may be palpable in healthy patients who spend time barefoot outdoors, have chronic leg trauma or infections, or have sexually transmitted infections.6 However, any lymph node >1 cm in adults should be considered abnormal.2-5
Method of diagnosis depends on malignancy risk
A definitive diagnosis in patients with lymph nodes >1 cm can be made by open lymph node biopsy (the gold standard) or fine needle aspiration (FNA); however, these procedures are rarely needed if malignancy risk is low.
Data on the prevalence of malignant peripheral lymphadenopathy is limited.4 Fijten et al reported that among 2556 patients who presented to a family medicine clinic with unexplained lymphadenopathy, the prevalence of malignancy was as low as 1.1%.7 However, the prevalence of malignant lymph nodes among patients referred to a surgical center for biopsy by primary care physicians was approximately 40% to 60%.3 This highlights the importance of a thorough history, physical exam, and referral when appropriate to increase the yield of diagnostic biopsies.
Low risk for malignancy is suggested when lymphadenopathy is present for less than 2 weeks or persists for more than one year with no increase in size.2 Benign causes such as sexually transmitted infections, Epstein-Barr virus, or medications should be treated appropriately. With no cause identified, 4 weeks of observation is recommended before biopsy.2,4,5,8 CT, PET, and biopsy should be considered early for large, concerning masses. No evidence supports empiric antibiotic use for unknown causes.2,5
High risk for malignancy is suggested in patients who are ≥50 years, present with constitutional symptoms, have lymphadenopathy >1 cm in >2 regions of the body, history of cancer, or have nodes that are rapidly enlarging, firm, fixed, or painless.2,3,5,7,9 Supraclavicular lymphadenopathy has the highest risk for malignancy, especially in patients ≥40 years.7 Enlarged iliac, popliteal, epitrochlear, and umbilical lymph nodes are never normal.2,4,5,7,10 Biopsy should be considered early in these patients.2-4,7 FNA or core needle biopsy is acceptable for an initial diagnosis, but negative results may require open biopsy.1,5,8 Prior to biopsy, imaging with ultrasound is recommended.1,2,8,11
Our patient was offered rituximab alone or rituximab in addition to cyclophosphamide, hydroxydoxorubicin, vincristine, and prednisone (R-CHOP). The patient chose rituximab alone, which resulted in a 30% reduction in the size of her intra-abdominal disease. At this point, the patient and her oncologist chose to stop treatment and monitor her clinically.
Three months later, the patient returned to our family clinic complaining of postnasal drip, throat pain, and neck fullness that she’d had for one month that weren’t responsive to over-the-counter remedies and antibiotics. A supervised osteopathic medical student’s exam revealed right tonsillar enlargement (grade 3+) with minimal erythema and no exudates. A neck CT confirmed right tonsillar enlargement. The patient was referred to Otolaryngology, and the surgeon performed a tonsillectomy that demonstrated disease progression to follicular lymphoma grade IIIa. Given the new findings, Oncology recommended R-CHOP and the patient agreed.
The patient completed R-CHOP and her cancer was in remission one year later.
THE TAKEAWAY
Peripheral lymphadenopathy presents a diagnostic challenge that requires a thorough history and physical exam. General wellness exams should incorporate a comprehensive physical that includes the palpation of lymph nodes. Exam challenges include distinguishing benign lymphadenopathy (reactive lymphadenitis) from malignant lymphadenopathy.
In patients with low risk for malignancy, a period of 4 weeks of observation is reasonable. Biopsy should be considered early for risk factors including patient’s age ≥50, constitutional symptoms, lymphadenopathy >1 cm in >2 regions of the body, history of cancer, or rapidly enlarging nodes.
1. Metzgeroth G, Schneider S, Walz C, et al. Fine needle aspiration and core needle biopsy in the diagnosis of lymphadenopathy of unknown aetiology. Ann Hematol. 2012;91:1477-1484.
2. Bazemore AW, Smucker DR. Lymphadenopathy and malignancy. Am Fam Physician. 2002;66:2103-2110.
3. Lee Y, Terry R, Lukes RJ. Lymph node biopsy for diagnosis: a statistical study. J Surg Oncol. 1980;14:53-60.
4. Ferrer R. Lymphadenopathy: differential diagnosis and evaluation. Am Fam Physician. 1998;58:1313-1320.
5. Motyckova G, Steensma DP. Why does my patient have lymphadenopathy or splenomegaly? Hematol Oncol Clin North Am. 2012;26:395-408.
6. Habermann TM, Steensma DP. Lymphadenopathy. Mayo Clin Proc. 2000;75:723-732.
7. Fijten GH, Blijham GH. Unexplained lymphadenopathy in family practice. An evaluation of the probability of malignant causes and the effectiveness of physicians’ workup. J Fam Pract. 1988;27:373-376.
8. Chau I, Kelleher MT, Cunningham D, et al. Rapid access multidisciplinary lymph node diagnostic clinic: analysis of 550 patients. Br J Cancer. 2003;88:354-361.
9. Vassilakopoulos TP, Pangalis GA. Application of a prediction rule to select which patients presenting with lymphadenopathy should undergo a lymph node biopsy. Medicine (Baltimore). 2000;79:338-347.
10. Dar IH, Kamili MA, Dar SH, et al. Sister Mary Joseph nodule-A case report with review of literature. J Res Med Sci. 2009;14:385-387.
11. Cui XW, Jenssen C, Saftoiu A, et al. New ultrasound techniques for lymph node evaluation. World J Gastroenterol. 2013;19:4850-4860.
THE CASE
A 52-year-old woman presented to our family clinic for a well woman exam. The only complaints she had were fatigue, which she attributed to a work day that began at 4 am, and hot flashes. She denied fever, weight loss, abdominal pain, medication use, or recent foreign travel. She had a history of hyperlipidemia and surgical removal of a cutaneous melanoma at age 12.
Her vital signs and physical exam were normal with the exception of 3 enlarged left inguinal lymph nodes and approximately 5 enlarged right inguinal lymph nodes. The nodes were freely moveable and non-tender. No additional lymphadenopathy or splenomegaly was found.
THE DIAGNOSIS
The patient’s work-up included a Pap smear, complete blood count (CBC), comprehensive metabolic panel (CMP), and pelvic and inguinal ultrasound. All tests were normal, except the ultrasound, which revealed 3 solid left inguinal lymph nodes measuring 1.2 to 1.6 cm and 6 solid right inguinal lymph nodes measuring 1.1 to 1.8 cm. An abdominal and pelvic computed tomography (CT) scan with contrast identified nonspecific mesenteric, inguinal, retrocrural, and retroperitoneal adenopathy. An open biopsy of the largest inguinal lymph node revealed follicular lymphoma, a form of non-Hodgkin’s lymphoma. (Hodgkin’s and non-Hodgkin’s lymphoma (NHL) are uncommon causes of inguinal lymphadenopathy.1)
We consulted Oncology and they recommended a positron emission tomography (PET)/CT scan, which showed widespread lymphadenopathy. A bone marrow biopsy confirmed follicular lymphoma grade II, Ann Arbor stage III.
DISCUSSION
Generalized lymphadenopathy involves lymph node enlargement in more than one region of the body. Lymph nodes >1 cm in adults are considered abnormal and the differential diagnosis is broad (TABLE2-5). A patient’s age is a significant factor in the evaluation of peripheral lymphadenopathy.2-5 Results from one study of 628 patients who underwent nodal biopsy for peripheral lymphadenopathy revealed approximately 80% of nodes in patients under age 30 were noncancerous and likely had an infectious cause.3 However, among patients over age 50, only 40% were noncancerous.3
Node enlargement can be palpated in the head, neck, axilla, inguinal, and popliteal areas. Inguinal lymph nodes up to 2 cm in size may be palpable in healthy patients who spend time barefoot outdoors, have chronic leg trauma or infections, or have sexually transmitted infections.6 However, any lymph node >1 cm in adults should be considered abnormal.2-5
Method of diagnosis depends on malignancy risk
A definitive diagnosis in patients with lymph nodes >1 cm can be made by open lymph node biopsy (the gold standard) or fine needle aspiration (FNA); however, these procedures are rarely needed if malignancy risk is low.
Data on the prevalence of malignant peripheral lymphadenopathy is limited.4 Fijten et al reported that among 2556 patients who presented to a family medicine clinic with unexplained lymphadenopathy, the prevalence of malignancy was as low as 1.1%.7 However, the prevalence of malignant lymph nodes among patients referred to a surgical center for biopsy by primary care physicians was approximately 40% to 60%.3 This highlights the importance of a thorough history, physical exam, and referral when appropriate to increase the yield of diagnostic biopsies.
Low risk for malignancy is suggested when lymphadenopathy is present for less than 2 weeks or persists for more than one year with no increase in size.2 Benign causes such as sexually transmitted infections, Epstein-Barr virus, or medications should be treated appropriately. With no cause identified, 4 weeks of observation is recommended before biopsy.2,4,5,8 CT, PET, and biopsy should be considered early for large, concerning masses. No evidence supports empiric antibiotic use for unknown causes.2,5
High risk for malignancy is suggested in patients who are ≥50 years, present with constitutional symptoms, have lymphadenopathy >1 cm in >2 regions of the body, history of cancer, or have nodes that are rapidly enlarging, firm, fixed, or painless.2,3,5,7,9 Supraclavicular lymphadenopathy has the highest risk for malignancy, especially in patients ≥40 years.7 Enlarged iliac, popliteal, epitrochlear, and umbilical lymph nodes are never normal.2,4,5,7,10 Biopsy should be considered early in these patients.2-4,7 FNA or core needle biopsy is acceptable for an initial diagnosis, but negative results may require open biopsy.1,5,8 Prior to biopsy, imaging with ultrasound is recommended.1,2,8,11
Our patient was offered rituximab alone or rituximab in addition to cyclophosphamide, hydroxydoxorubicin, vincristine, and prednisone (R-CHOP). The patient chose rituximab alone, which resulted in a 30% reduction in the size of her intra-abdominal disease. At this point, the patient and her oncologist chose to stop treatment and monitor her clinically.
Three months later, the patient returned to our family clinic complaining of postnasal drip, throat pain, and neck fullness that she’d had for one month that weren’t responsive to over-the-counter remedies and antibiotics. A supervised osteopathic medical student’s exam revealed right tonsillar enlargement (grade 3+) with minimal erythema and no exudates. A neck CT confirmed right tonsillar enlargement. The patient was referred to Otolaryngology, and the surgeon performed a tonsillectomy that demonstrated disease progression to follicular lymphoma grade IIIa. Given the new findings, Oncology recommended R-CHOP and the patient agreed.
The patient completed R-CHOP and her cancer was in remission one year later.
THE TAKEAWAY
Peripheral lymphadenopathy presents a diagnostic challenge that requires a thorough history and physical exam. General wellness exams should incorporate a comprehensive physical that includes the palpation of lymph nodes. Exam challenges include distinguishing benign lymphadenopathy (reactive lymphadenitis) from malignant lymphadenopathy.
In patients with low risk for malignancy, a period of 4 weeks of observation is reasonable. Biopsy should be considered early for risk factors including patient’s age ≥50, constitutional symptoms, lymphadenopathy >1 cm in >2 regions of the body, history of cancer, or rapidly enlarging nodes.
THE CASE
A 52-year-old woman presented to our family clinic for a well woman exam. The only complaints she had were fatigue, which she attributed to a work day that began at 4 am, and hot flashes. She denied fever, weight loss, abdominal pain, medication use, or recent foreign travel. She had a history of hyperlipidemia and surgical removal of a cutaneous melanoma at age 12.
Her vital signs and physical exam were normal with the exception of 3 enlarged left inguinal lymph nodes and approximately 5 enlarged right inguinal lymph nodes. The nodes were freely moveable and non-tender. No additional lymphadenopathy or splenomegaly was found.
THE DIAGNOSIS
The patient’s work-up included a Pap smear, complete blood count (CBC), comprehensive metabolic panel (CMP), and pelvic and inguinal ultrasound. All tests were normal, except the ultrasound, which revealed 3 solid left inguinal lymph nodes measuring 1.2 to 1.6 cm and 6 solid right inguinal lymph nodes measuring 1.1 to 1.8 cm. An abdominal and pelvic computed tomography (CT) scan with contrast identified nonspecific mesenteric, inguinal, retrocrural, and retroperitoneal adenopathy. An open biopsy of the largest inguinal lymph node revealed follicular lymphoma, a form of non-Hodgkin’s lymphoma. (Hodgkin’s and non-Hodgkin’s lymphoma (NHL) are uncommon causes of inguinal lymphadenopathy.1)
We consulted Oncology and they recommended a positron emission tomography (PET)/CT scan, which showed widespread lymphadenopathy. A bone marrow biopsy confirmed follicular lymphoma grade II, Ann Arbor stage III.
DISCUSSION
Generalized lymphadenopathy involves lymph node enlargement in more than one region of the body. Lymph nodes >1 cm in adults are considered abnormal and the differential diagnosis is broad (TABLE2-5). A patient’s age is a significant factor in the evaluation of peripheral lymphadenopathy.2-5 Results from one study of 628 patients who underwent nodal biopsy for peripheral lymphadenopathy revealed approximately 80% of nodes in patients under age 30 were noncancerous and likely had an infectious cause.3 However, among patients over age 50, only 40% were noncancerous.3
Node enlargement can be palpated in the head, neck, axilla, inguinal, and popliteal areas. Inguinal lymph nodes up to 2 cm in size may be palpable in healthy patients who spend time barefoot outdoors, have chronic leg trauma or infections, or have sexually transmitted infections.6 However, any lymph node >1 cm in adults should be considered abnormal.2-5
Method of diagnosis depends on malignancy risk
A definitive diagnosis in patients with lymph nodes >1 cm can be made by open lymph node biopsy (the gold standard) or fine needle aspiration (FNA); however, these procedures are rarely needed if malignancy risk is low.
Data on the prevalence of malignant peripheral lymphadenopathy is limited.4 Fijten et al reported that among 2556 patients who presented to a family medicine clinic with unexplained lymphadenopathy, the prevalence of malignancy was as low as 1.1%.7 However, the prevalence of malignant lymph nodes among patients referred to a surgical center for biopsy by primary care physicians was approximately 40% to 60%.3 This highlights the importance of a thorough history, physical exam, and referral when appropriate to increase the yield of diagnostic biopsies.
Low risk for malignancy is suggested when lymphadenopathy is present for less than 2 weeks or persists for more than one year with no increase in size.2 Benign causes such as sexually transmitted infections, Epstein-Barr virus, or medications should be treated appropriately. With no cause identified, 4 weeks of observation is recommended before biopsy.2,4,5,8 CT, PET, and biopsy should be considered early for large, concerning masses. No evidence supports empiric antibiotic use for unknown causes.2,5
High risk for malignancy is suggested in patients who are ≥50 years, present with constitutional symptoms, have lymphadenopathy >1 cm in >2 regions of the body, history of cancer, or have nodes that are rapidly enlarging, firm, fixed, or painless.2,3,5,7,9 Supraclavicular lymphadenopathy has the highest risk for malignancy, especially in patients ≥40 years.7 Enlarged iliac, popliteal, epitrochlear, and umbilical lymph nodes are never normal.2,4,5,7,10 Biopsy should be considered early in these patients.2-4,7 FNA or core needle biopsy is acceptable for an initial diagnosis, but negative results may require open biopsy.1,5,8 Prior to biopsy, imaging with ultrasound is recommended.1,2,8,11
Our patient was offered rituximab alone or rituximab in addition to cyclophosphamide, hydroxydoxorubicin, vincristine, and prednisone (R-CHOP). The patient chose rituximab alone, which resulted in a 30% reduction in the size of her intra-abdominal disease. At this point, the patient and her oncologist chose to stop treatment and monitor her clinically.
Three months later, the patient returned to our family clinic complaining of postnasal drip, throat pain, and neck fullness that she’d had for one month that weren’t responsive to over-the-counter remedies and antibiotics. A supervised osteopathic medical student’s exam revealed right tonsillar enlargement (grade 3+) with minimal erythema and no exudates. A neck CT confirmed right tonsillar enlargement. The patient was referred to Otolaryngology, and the surgeon performed a tonsillectomy that demonstrated disease progression to follicular lymphoma grade IIIa. Given the new findings, Oncology recommended R-CHOP and the patient agreed.
The patient completed R-CHOP and her cancer was in remission one year later.
THE TAKEAWAY
Peripheral lymphadenopathy presents a diagnostic challenge that requires a thorough history and physical exam. General wellness exams should incorporate a comprehensive physical that includes the palpation of lymph nodes. Exam challenges include distinguishing benign lymphadenopathy (reactive lymphadenitis) from malignant lymphadenopathy.
In patients with low risk for malignancy, a period of 4 weeks of observation is reasonable. Biopsy should be considered early for risk factors including patient’s age ≥50, constitutional symptoms, lymphadenopathy >1 cm in >2 regions of the body, history of cancer, or rapidly enlarging nodes.
1. Metzgeroth G, Schneider S, Walz C, et al. Fine needle aspiration and core needle biopsy in the diagnosis of lymphadenopathy of unknown aetiology. Ann Hematol. 2012;91:1477-1484.
2. Bazemore AW, Smucker DR. Lymphadenopathy and malignancy. Am Fam Physician. 2002;66:2103-2110.
3. Lee Y, Terry R, Lukes RJ. Lymph node biopsy for diagnosis: a statistical study. J Surg Oncol. 1980;14:53-60.
4. Ferrer R. Lymphadenopathy: differential diagnosis and evaluation. Am Fam Physician. 1998;58:1313-1320.
5. Motyckova G, Steensma DP. Why does my patient have lymphadenopathy or splenomegaly? Hematol Oncol Clin North Am. 2012;26:395-408.
6. Habermann TM, Steensma DP. Lymphadenopathy. Mayo Clin Proc. 2000;75:723-732.
7. Fijten GH, Blijham GH. Unexplained lymphadenopathy in family practice. An evaluation of the probability of malignant causes and the effectiveness of physicians’ workup. J Fam Pract. 1988;27:373-376.
8. Chau I, Kelleher MT, Cunningham D, et al. Rapid access multidisciplinary lymph node diagnostic clinic: analysis of 550 patients. Br J Cancer. 2003;88:354-361.
9. Vassilakopoulos TP, Pangalis GA. Application of a prediction rule to select which patients presenting with lymphadenopathy should undergo a lymph node biopsy. Medicine (Baltimore). 2000;79:338-347.
10. Dar IH, Kamili MA, Dar SH, et al. Sister Mary Joseph nodule-A case report with review of literature. J Res Med Sci. 2009;14:385-387.
11. Cui XW, Jenssen C, Saftoiu A, et al. New ultrasound techniques for lymph node evaluation. World J Gastroenterol. 2013;19:4850-4860.
1. Metzgeroth G, Schneider S, Walz C, et al. Fine needle aspiration and core needle biopsy in the diagnosis of lymphadenopathy of unknown aetiology. Ann Hematol. 2012;91:1477-1484.
2. Bazemore AW, Smucker DR. Lymphadenopathy and malignancy. Am Fam Physician. 2002;66:2103-2110.
3. Lee Y, Terry R, Lukes RJ. Lymph node biopsy for diagnosis: a statistical study. J Surg Oncol. 1980;14:53-60.
4. Ferrer R. Lymphadenopathy: differential diagnosis and evaluation. Am Fam Physician. 1998;58:1313-1320.
5. Motyckova G, Steensma DP. Why does my patient have lymphadenopathy or splenomegaly? Hematol Oncol Clin North Am. 2012;26:395-408.
6. Habermann TM, Steensma DP. Lymphadenopathy. Mayo Clin Proc. 2000;75:723-732.
7. Fijten GH, Blijham GH. Unexplained lymphadenopathy in family practice. An evaluation of the probability of malignant causes and the effectiveness of physicians’ workup. J Fam Pract. 1988;27:373-376.
8. Chau I, Kelleher MT, Cunningham D, et al. Rapid access multidisciplinary lymph node diagnostic clinic: analysis of 550 patients. Br J Cancer. 2003;88:354-361.
9. Vassilakopoulos TP, Pangalis GA. Application of a prediction rule to select which patients presenting with lymphadenopathy should undergo a lymph node biopsy. Medicine (Baltimore). 2000;79:338-347.
10. Dar IH, Kamili MA, Dar SH, et al. Sister Mary Joseph nodule-A case report with review of literature. J Res Med Sci. 2009;14:385-387.
11. Cui XW, Jenssen C, Saftoiu A, et al. New ultrasound techniques for lymph node evaluation. World J Gastroenterol. 2013;19:4850-4860.

Extreme Postinjection Flare in Response to Intra-Articular Triamcinolone Acetonide (Kenalog)
Intra-articular corticosteroid injections (CSIs) have been a common treatment for osteoarthritis since the 1950s and continue to be an option for patients who prefer nonoperative management.1 Although CSIs may improve pain secondary to osteoarthritis temporarily, they do not slow articular cartilage degradation, and many patients request multiple CSIs before total joint arthroplasty ultimately is required.1,2 Therefore, acute and chronic side effects of CSI must be considered when repeatedly administering corticosteroids.
A postinjection flare, the most common acute side effect of intra-articular CSI, is characterized by a localized inflammatory response that can last 2 to 3 days. The flare occurs in 2% to 25% of CSI cases.3-5 Symptoms can range from mild joint effusion to disabling pain.6 In the present case, a severe postinjection flare occurred after intra-articular administration of triamcinolone acetonide (Kenalog). This case is novel in that its acuity of onset, severity of symptoms, and synovial fluid analysis mimicked septic arthritis, which was ultimately ruled out with negative cultures and confirmation of triamcinolone acetonide crystals in the synovial aspirate, viewed by polarized light microscopy. To date, only one other case of an immediate (<2 hours) and severe postinjection flare in response to triamcinolone has been reported.7 As CSIs are often used in the nonoperative treatment of osteoarthritis, it is imperative for the treating physician to be aware of this potential side effect in order to appropriately inform the patient of this risk and guide treatment should the scenario arise. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 56-year-old woman with a history of hypertension, hypothyroidism, and moderate bilateral knee osteoarthritis presented with left knee pain. She had been receiving annual hylan injections for 5 years and had no adverse reactions, but the pain gradually worsened over the past 3 months. She was given an intra-articular injection of 2 mL of 1% lidocaine and 2 mL (40 mg) of triamcinolone acetonide in the left knee.
Two hours later, she experienced swelling and intense pain in the knee and was unable to ambulate. Physical examination revealed she was afebrile but was having severe pain in the knee through all range of motion. The knee had no appreciable erythema or warmth. Laboratory data were significant: White blood cell (WBC) count was 14,600, and erythrocyte sedimentation rate was 1 mm/h. The knee was aspirated with a return of 25 mL of “butterscotch”-colored fluid (Figure 1). The patient was admitted to rule out iatrogenic septic arthritis, or chronic, indolent septic arthritis acutely worsened by CSI, until synovial fluid analysis and cultures could be performed (Table 1).

She was treated overnight with a compressive wrap, elevation, ice, and nonsteroidal anti-inflammatory drugs, which provided significant pain relief. Polarized light microscopy revealed polymorphic intracellular and extracellular crystals with crystal morphology consistent with the injection of triamcinolone ester (Figure 2). Gram stain showed many WBCs but no organisms. These findings were thought to represent an exogenous crystal-induced acute inflammatory response. Given the patient’s improving clinical course, she was discharged the next morning.
Twelve days later, at clinic follow-up, she was still experiencing pain above her baseline level. Given the continued effusion, 8 mL of synovial fluid was aspirated, which appeared clear and only slightly blood-tinged. Synovial analysis showed resolution of leukocytosis, confirming a severe postinjection flare in response to triamcinolone acetonide.
Discussion
Although rare, side effects from repeated intra-articular CSIs include hypothalamic-pituitary-adrenal axis dysfunction and steroid-induced myopathy.8,9 Acute side effects are more common and include postinjection flare, iatrogenic septic arthritis, local tissue atrophy, cartilage damage, tendon rupture, nerve atrophy, increased blood glucose, and osteonecrosis.10,11 The present case report describes an extreme example of a postinjection flare in response to triamcinolone acetonide and summarizes the characteristics of injections that cause flares.
The physical properties of corticosteroids have a significant impact on their efficacy and on their potential for adverse events. Corticosteroid preparations can be water-soluble or water-insoluble. Most commonly, water-insoluble preparations that contain insoluble corticosteroid esters (eg, triamcinolone, methylprednisolone) are used in intra-articular injections. These form microcrystalline aggregates in solution, which require the patient’s own hydrolytic enzymes (esterases) to release the active moiety and thus have a longer duration of action. However, they are more commonly associated with postinjection flares compared with their more soluble and faster- acting counterparts (eg. dexamethasone, betamethasone).10 Microcrystalline aggregates, which are larger in size, induce a stronger inflammatory response, and in a dose-dependent manner.6A sterile inflammatory reaction to hydrocortisone, cortisone, dexamethasone, triamcinolone, and prednisolone crystals in normal joints has been previously described,6,12,13 and crystals of the various preparations have been demonstrated within leukocytes by both polarized light and electron microscopy.12,13 Table 2 summarizes previous synovial fluid analyses after intra-articular injections of various corticosteroid preparations in normal healthy joints and in patients experiencing a postinjection flare. To date, there have been no reports of an immediate (<2 hours) and severe postinjection flare in response to triamcinolone acetonide, though there was a report of a postinjection flare in response to triamcinolone hexacetonide (Aristospan),7 and here the synovial fluid WBC count (30,000) was much lower.
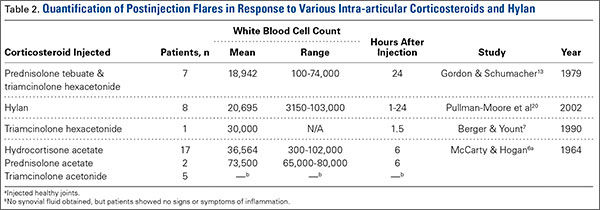
Although many cases of corticosteroid hypersensitivity have been reported, in rare cases intra-articular administration of triamcinolone has caused anaphylactic reactions and shock.14,15 Multiple case studies have determined that the specific excipient carboxymethylcellulose (found in many triamcinolone preparations), and not the corticosteroid itself, can cause an immunoglobulin E–mediated anaphylactic reaction.16-18 Therefore, performing skin-prick tests for potential corticosteroids and their excipients in patients with known postinjection flares might help prevent serious adverse reactions.18,19
The present case involved an extreme postinjection flare in response to intra-articular administration of triamcinolone acetonide. Postinjection flares are rare but significant events, and physicians using CSIs in the treatment of arthritis need to be aware of this potential reaction in order to appropriately inform patients of this risk and guide treatment should the scenario arise.
1. Hollander JL, Brown EM Jr, Jessar RA, Brown CY. Hydrocortisone and cortisone injected into arthritic joints; comparative effects of and use of hydrocortisone as a local antiarthritic agent. J Am Med Assoc. 1951;147(17):1629-1635.
2. Bellamy N, Campbell J, Robinson V, Gee T, Bourne R, Wells G. Intraarticular corticosteroid for treatment of osteoarthritis of the knee. Cochrane Database Syst Rev. 2006;19(2):CD005328.
3. Friedman DM, Moore ME. The efficacy of intraarticular steroids in osteoarthritis: a double-blind study. J Rheumatol. 1980;7(6):850-856.
4. Brown EM Jr, Frain JB, Udell L, Hollander JL. Locally administered hydrocortisone in the rheumatic diseases; a summary of its use in 547 patients. Am J Med. 1953;15(5):656-665.
5. Hollander JL, Jessar RA, Brown EM Jr. Intra-synovial corticosteroid therapy: a decade of use. Bull Rheum Dis. 1961;11:239-240.
6. McCarty DJ Jr, Hogan JM. Inflammatory reaction after intrasynovial injection of microcrystalline adrenocorticosteroid esters. Arthritis Rheum. 1964;7(4):359-367.
7. Berger RG, Yount WJ. Immediate “steroid flare” from intraarticular triamcinolone hexacetonide injection: case report and review of the literature. Arthritis Rheum. 1990;33(8):1284-1286.
8. Mader R, Lavi I, Luboshitzky R. Evaluation of the pituitary-adrenal axis function following single intraarticular injection of methylprednisolone. Arthritis Rheum. 2005;52(3):924-928.
9. Raynauld JP, Buckland-Wright C, Ward R, et al. Safety and efficacy of long-term intraarticular steroid injections in osteoarthritis of the knee: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2003;48(2):370-377.
10. MacMahon PJ, Eustace SJ, Kavanagh EC. Injectable corticosteroid and local anesthetic preparations: a review for radiologists. Radiology. 2009;252(3):647-661.
11. Sparling M, Malleson P, Wood B, Petty R. Radiographic followup of joints injected with triamcinolone hexacetonide for the management of childhood arthritis. Arthritis Rheum. 1990;33(6):821-826.
12. Eymontt MJ, Gordon GV, Schumacher HR, Hansell JR. The effects on synovial permeability and synovial fluid leukocyte counts in symptomatic osteoarthritis after intraarticular corticosteroid administration. J Rheumatol. 1982;9(2):198-203.
13. Gordon GV, Schumacher HR. Electron microscopic study of depot corticosteroid crystals with clinical studies after intra-articular injection. J Rheumatol. 1979;6(1):7-14.
14. Karsh J, Yang WH. An anaphylactic reaction to intra-articular triamcinolone: a case report and review of the literature. Ann Allergy Asthma Immunol. 2003;90(2):254-258.
15. Larsson LG. Anaphylactic shock after i.a. administration of triamcinolone acetonide in a 35-year-old female. Scand J Rheumatol. 1989;18(6):441-442.
16. García-Ortega P, Corominas M, Badia M. Carboxymethylcellulose allergy as a cause of suspected corticosteroid anaphylaxis. Ann Allergy Asthma Immunol. 2003;91(4):421.
17. Patterson DL, Yunginger JW, Dunn WF, Jones RT, Hunt LW. Anaphylaxis induced by the carboxymethylcellulose component of injectable triamcinolone acetonide suspension (Kenalog). Ann Allergy Asthma Immunol. 1995;74(2):163-166.
18. Steiner UC, Gentinetta T, Hausmann O, Pichler WJ. IgE-mediated anaphylaxis to intraarticular glucocorticoid preparations. AJR Am J Roentgenol. 2009;193(2):W156-W157.
19. Ijsselmuiden OE, Knegt-Junk KJ, van Wijk RG, van Joost T. Cutaneous adverse reactions after intra-articular injection of triamcinolone acetonide. Acta Derm Venereol. 1995;75(1):57-58.
20. Pullman-Mooar S, Mooar P, Sieck M, Clayburne G, Schumacher HR. Are there distinctive inflammatory flares after hylan g-f 20 intraarticular injections? J Rheumatol. 2002;29(12):2611-2614.
Intra-articular corticosteroid injections (CSIs) have been a common treatment for osteoarthritis since the 1950s and continue to be an option for patients who prefer nonoperative management.1 Although CSIs may improve pain secondary to osteoarthritis temporarily, they do not slow articular cartilage degradation, and many patients request multiple CSIs before total joint arthroplasty ultimately is required.1,2 Therefore, acute and chronic side effects of CSI must be considered when repeatedly administering corticosteroids.
A postinjection flare, the most common acute side effect of intra-articular CSI, is characterized by a localized inflammatory response that can last 2 to 3 days. The flare occurs in 2% to 25% of CSI cases.3-5 Symptoms can range from mild joint effusion to disabling pain.6 In the present case, a severe postinjection flare occurred after intra-articular administration of triamcinolone acetonide (Kenalog). This case is novel in that its acuity of onset, severity of symptoms, and synovial fluid analysis mimicked septic arthritis, which was ultimately ruled out with negative cultures and confirmation of triamcinolone acetonide crystals in the synovial aspirate, viewed by polarized light microscopy. To date, only one other case of an immediate (<2 hours) and severe postinjection flare in response to triamcinolone has been reported.7 As CSIs are often used in the nonoperative treatment of osteoarthritis, it is imperative for the treating physician to be aware of this potential side effect in order to appropriately inform the patient of this risk and guide treatment should the scenario arise. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 56-year-old woman with a history of hypertension, hypothyroidism, and moderate bilateral knee osteoarthritis presented with left knee pain. She had been receiving annual hylan injections for 5 years and had no adverse reactions, but the pain gradually worsened over the past 3 months. She was given an intra-articular injection of 2 mL of 1% lidocaine and 2 mL (40 mg) of triamcinolone acetonide in the left knee.
Two hours later, she experienced swelling and intense pain in the knee and was unable to ambulate. Physical examination revealed she was afebrile but was having severe pain in the knee through all range of motion. The knee had no appreciable erythema or warmth. Laboratory data were significant: White blood cell (WBC) count was 14,600, and erythrocyte sedimentation rate was 1 mm/h. The knee was aspirated with a return of 25 mL of “butterscotch”-colored fluid (Figure 1). The patient was admitted to rule out iatrogenic septic arthritis, or chronic, indolent septic arthritis acutely worsened by CSI, until synovial fluid analysis and cultures could be performed (Table 1).
She was treated overnight with a compressive wrap, elevation, ice, and nonsteroidal anti-inflammatory drugs, which provided significant pain relief. Polarized light microscopy revealed polymorphic intracellular and extracellular crystals with crystal morphology consistent with the injection of triamcinolone ester (Figure 2). Gram stain showed many WBCs but no organisms. These findings were thought to represent an exogenous crystal-induced acute inflammatory response. Given the patient’s improving clinical course, she was discharged the next morning.
Twelve days later, at clinic follow-up, she was still experiencing pain above her baseline level. Given the continued effusion, 8 mL of synovial fluid was aspirated, which appeared clear and only slightly blood-tinged. Synovial analysis showed resolution of leukocytosis, confirming a severe postinjection flare in response to triamcinolone acetonide.
Discussion
Although rare, side effects from repeated intra-articular CSIs include hypothalamic-pituitary-adrenal axis dysfunction and steroid-induced myopathy.8,9 Acute side effects are more common and include postinjection flare, iatrogenic septic arthritis, local tissue atrophy, cartilage damage, tendon rupture, nerve atrophy, increased blood glucose, and osteonecrosis.10,11 The present case report describes an extreme example of a postinjection flare in response to triamcinolone acetonide and summarizes the characteristics of injections that cause flares.
The physical properties of corticosteroids have a significant impact on their efficacy and on their potential for adverse events. Corticosteroid preparations can be water-soluble or water-insoluble. Most commonly, water-insoluble preparations that contain insoluble corticosteroid esters (eg, triamcinolone, methylprednisolone) are used in intra-articular injections. These form microcrystalline aggregates in solution, which require the patient’s own hydrolytic enzymes (esterases) to release the active moiety and thus have a longer duration of action. However, they are more commonly associated with postinjection flares compared with their more soluble and faster- acting counterparts (eg. dexamethasone, betamethasone).10 Microcrystalline aggregates, which are larger in size, induce a stronger inflammatory response, and in a dose-dependent manner.6A sterile inflammatory reaction to hydrocortisone, cortisone, dexamethasone, triamcinolone, and prednisolone crystals in normal joints has been previously described,6,12,13 and crystals of the various preparations have been demonstrated within leukocytes by both polarized light and electron microscopy.12,13 Table 2 summarizes previous synovial fluid analyses after intra-articular injections of various corticosteroid preparations in normal healthy joints and in patients experiencing a postinjection flare. To date, there have been no reports of an immediate (<2 hours) and severe postinjection flare in response to triamcinolone acetonide, though there was a report of a postinjection flare in response to triamcinolone hexacetonide (Aristospan),7 and here the synovial fluid WBC count (30,000) was much lower.
Although many cases of corticosteroid hypersensitivity have been reported, in rare cases intra-articular administration of triamcinolone has caused anaphylactic reactions and shock.14,15 Multiple case studies have determined that the specific excipient carboxymethylcellulose (found in many triamcinolone preparations), and not the corticosteroid itself, can cause an immunoglobulin E–mediated anaphylactic reaction.16-18 Therefore, performing skin-prick tests for potential corticosteroids and their excipients in patients with known postinjection flares might help prevent serious adverse reactions.18,19
The present case involved an extreme postinjection flare in response to intra-articular administration of triamcinolone acetonide. Postinjection flares are rare but significant events, and physicians using CSIs in the treatment of arthritis need to be aware of this potential reaction in order to appropriately inform patients of this risk and guide treatment should the scenario arise.
Intra-articular corticosteroid injections (CSIs) have been a common treatment for osteoarthritis since the 1950s and continue to be an option for patients who prefer nonoperative management.1 Although CSIs may improve pain secondary to osteoarthritis temporarily, they do not slow articular cartilage degradation, and many patients request multiple CSIs before total joint arthroplasty ultimately is required.1,2 Therefore, acute and chronic side effects of CSI must be considered when repeatedly administering corticosteroids.
A postinjection flare, the most common acute side effect of intra-articular CSI, is characterized by a localized inflammatory response that can last 2 to 3 days. The flare occurs in 2% to 25% of CSI cases.3-5 Symptoms can range from mild joint effusion to disabling pain.6 In the present case, a severe postinjection flare occurred after intra-articular administration of triamcinolone acetonide (Kenalog). This case is novel in that its acuity of onset, severity of symptoms, and synovial fluid analysis mimicked septic arthritis, which was ultimately ruled out with negative cultures and confirmation of triamcinolone acetonide crystals in the synovial aspirate, viewed by polarized light microscopy. To date, only one other case of an immediate (<2 hours) and severe postinjection flare in response to triamcinolone has been reported.7 As CSIs are often used in the nonoperative treatment of osteoarthritis, it is imperative for the treating physician to be aware of this potential side effect in order to appropriately inform the patient of this risk and guide treatment should the scenario arise. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 56-year-old woman with a history of hypertension, hypothyroidism, and moderate bilateral knee osteoarthritis presented with left knee pain. She had been receiving annual hylan injections for 5 years and had no adverse reactions, but the pain gradually worsened over the past 3 months. She was given an intra-articular injection of 2 mL of 1% lidocaine and 2 mL (40 mg) of triamcinolone acetonide in the left knee.
Two hours later, she experienced swelling and intense pain in the knee and was unable to ambulate. Physical examination revealed she was afebrile but was having severe pain in the knee through all range of motion. The knee had no appreciable erythema or warmth. Laboratory data were significant: White blood cell (WBC) count was 14,600, and erythrocyte sedimentation rate was 1 mm/h. The knee was aspirated with a return of 25 mL of “butterscotch”-colored fluid (Figure 1). The patient was admitted to rule out iatrogenic septic arthritis, or chronic, indolent septic arthritis acutely worsened by CSI, until synovial fluid analysis and cultures could be performed (Table 1).
She was treated overnight with a compressive wrap, elevation, ice, and nonsteroidal anti-inflammatory drugs, which provided significant pain relief. Polarized light microscopy revealed polymorphic intracellular and extracellular crystals with crystal morphology consistent with the injection of triamcinolone ester (Figure 2). Gram stain showed many WBCs but no organisms. These findings were thought to represent an exogenous crystal-induced acute inflammatory response. Given the patient’s improving clinical course, she was discharged the next morning.
Twelve days later, at clinic follow-up, she was still experiencing pain above her baseline level. Given the continued effusion, 8 mL of synovial fluid was aspirated, which appeared clear and only slightly blood-tinged. Synovial analysis showed resolution of leukocytosis, confirming a severe postinjection flare in response to triamcinolone acetonide.
Discussion
Although rare, side effects from repeated intra-articular CSIs include hypothalamic-pituitary-adrenal axis dysfunction and steroid-induced myopathy.8,9 Acute side effects are more common and include postinjection flare, iatrogenic septic arthritis, local tissue atrophy, cartilage damage, tendon rupture, nerve atrophy, increased blood glucose, and osteonecrosis.10,11 The present case report describes an extreme example of a postinjection flare in response to triamcinolone acetonide and summarizes the characteristics of injections that cause flares.
The physical properties of corticosteroids have a significant impact on their efficacy and on their potential for adverse events. Corticosteroid preparations can be water-soluble or water-insoluble. Most commonly, water-insoluble preparations that contain insoluble corticosteroid esters (eg, triamcinolone, methylprednisolone) are used in intra-articular injections. These form microcrystalline aggregates in solution, which require the patient’s own hydrolytic enzymes (esterases) to release the active moiety and thus have a longer duration of action. However, they are more commonly associated with postinjection flares compared with their more soluble and faster- acting counterparts (eg. dexamethasone, betamethasone).10 Microcrystalline aggregates, which are larger in size, induce a stronger inflammatory response, and in a dose-dependent manner.6A sterile inflammatory reaction to hydrocortisone, cortisone, dexamethasone, triamcinolone, and prednisolone crystals in normal joints has been previously described,6,12,13 and crystals of the various preparations have been demonstrated within leukocytes by both polarized light and electron microscopy.12,13 Table 2 summarizes previous synovial fluid analyses after intra-articular injections of various corticosteroid preparations in normal healthy joints and in patients experiencing a postinjection flare. To date, there have been no reports of an immediate (<2 hours) and severe postinjection flare in response to triamcinolone acetonide, though there was a report of a postinjection flare in response to triamcinolone hexacetonide (Aristospan),7 and here the synovial fluid WBC count (30,000) was much lower.
Although many cases of corticosteroid hypersensitivity have been reported, in rare cases intra-articular administration of triamcinolone has caused anaphylactic reactions and shock.14,15 Multiple case studies have determined that the specific excipient carboxymethylcellulose (found in many triamcinolone preparations), and not the corticosteroid itself, can cause an immunoglobulin E–mediated anaphylactic reaction.16-18 Therefore, performing skin-prick tests for potential corticosteroids and their excipients in patients with known postinjection flares might help prevent serious adverse reactions.18,19
The present case involved an extreme postinjection flare in response to intra-articular administration of triamcinolone acetonide. Postinjection flares are rare but significant events, and physicians using CSIs in the treatment of arthritis need to be aware of this potential reaction in order to appropriately inform patients of this risk and guide treatment should the scenario arise.
1. Hollander JL, Brown EM Jr, Jessar RA, Brown CY. Hydrocortisone and cortisone injected into arthritic joints; comparative effects of and use of hydrocortisone as a local antiarthritic agent. J Am Med Assoc. 1951;147(17):1629-1635.
2. Bellamy N, Campbell J, Robinson V, Gee T, Bourne R, Wells G. Intraarticular corticosteroid for treatment of osteoarthritis of the knee. Cochrane Database Syst Rev. 2006;19(2):CD005328.
3. Friedman DM, Moore ME. The efficacy of intraarticular steroids in osteoarthritis: a double-blind study. J Rheumatol. 1980;7(6):850-856.
4. Brown EM Jr, Frain JB, Udell L, Hollander JL. Locally administered hydrocortisone in the rheumatic diseases; a summary of its use in 547 patients. Am J Med. 1953;15(5):656-665.
5. Hollander JL, Jessar RA, Brown EM Jr. Intra-synovial corticosteroid therapy: a decade of use. Bull Rheum Dis. 1961;11:239-240.
6. McCarty DJ Jr, Hogan JM. Inflammatory reaction after intrasynovial injection of microcrystalline adrenocorticosteroid esters. Arthritis Rheum. 1964;7(4):359-367.
7. Berger RG, Yount WJ. Immediate “steroid flare” from intraarticular triamcinolone hexacetonide injection: case report and review of the literature. Arthritis Rheum. 1990;33(8):1284-1286.
8. Mader R, Lavi I, Luboshitzky R. Evaluation of the pituitary-adrenal axis function following single intraarticular injection of methylprednisolone. Arthritis Rheum. 2005;52(3):924-928.
9. Raynauld JP, Buckland-Wright C, Ward R, et al. Safety and efficacy of long-term intraarticular steroid injections in osteoarthritis of the knee: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2003;48(2):370-377.
10. MacMahon PJ, Eustace SJ, Kavanagh EC. Injectable corticosteroid and local anesthetic preparations: a review for radiologists. Radiology. 2009;252(3):647-661.
11. Sparling M, Malleson P, Wood B, Petty R. Radiographic followup of joints injected with triamcinolone hexacetonide for the management of childhood arthritis. Arthritis Rheum. 1990;33(6):821-826.
12. Eymontt MJ, Gordon GV, Schumacher HR, Hansell JR. The effects on synovial permeability and synovial fluid leukocyte counts in symptomatic osteoarthritis after intraarticular corticosteroid administration. J Rheumatol. 1982;9(2):198-203.
13. Gordon GV, Schumacher HR. Electron microscopic study of depot corticosteroid crystals with clinical studies after intra-articular injection. J Rheumatol. 1979;6(1):7-14.
14. Karsh J, Yang WH. An anaphylactic reaction to intra-articular triamcinolone: a case report and review of the literature. Ann Allergy Asthma Immunol. 2003;90(2):254-258.
15. Larsson LG. Anaphylactic shock after i.a. administration of triamcinolone acetonide in a 35-year-old female. Scand J Rheumatol. 1989;18(6):441-442.
16. García-Ortega P, Corominas M, Badia M. Carboxymethylcellulose allergy as a cause of suspected corticosteroid anaphylaxis. Ann Allergy Asthma Immunol. 2003;91(4):421.
17. Patterson DL, Yunginger JW, Dunn WF, Jones RT, Hunt LW. Anaphylaxis induced by the carboxymethylcellulose component of injectable triamcinolone acetonide suspension (Kenalog). Ann Allergy Asthma Immunol. 1995;74(2):163-166.
18. Steiner UC, Gentinetta T, Hausmann O, Pichler WJ. IgE-mediated anaphylaxis to intraarticular glucocorticoid preparations. AJR Am J Roentgenol. 2009;193(2):W156-W157.
19. Ijsselmuiden OE, Knegt-Junk KJ, van Wijk RG, van Joost T. Cutaneous adverse reactions after intra-articular injection of triamcinolone acetonide. Acta Derm Venereol. 1995;75(1):57-58.
20. Pullman-Mooar S, Mooar P, Sieck M, Clayburne G, Schumacher HR. Are there distinctive inflammatory flares after hylan g-f 20 intraarticular injections? J Rheumatol. 2002;29(12):2611-2614.
1. Hollander JL, Brown EM Jr, Jessar RA, Brown CY. Hydrocortisone and cortisone injected into arthritic joints; comparative effects of and use of hydrocortisone as a local antiarthritic agent. J Am Med Assoc. 1951;147(17):1629-1635.
2. Bellamy N, Campbell J, Robinson V, Gee T, Bourne R, Wells G. Intraarticular corticosteroid for treatment of osteoarthritis of the knee. Cochrane Database Syst Rev. 2006;19(2):CD005328.
3. Friedman DM, Moore ME. The efficacy of intraarticular steroids in osteoarthritis: a double-blind study. J Rheumatol. 1980;7(6):850-856.
4. Brown EM Jr, Frain JB, Udell L, Hollander JL. Locally administered hydrocortisone in the rheumatic diseases; a summary of its use in 547 patients. Am J Med. 1953;15(5):656-665.
5. Hollander JL, Jessar RA, Brown EM Jr. Intra-synovial corticosteroid therapy: a decade of use. Bull Rheum Dis. 1961;11:239-240.
6. McCarty DJ Jr, Hogan JM. Inflammatory reaction after intrasynovial injection of microcrystalline adrenocorticosteroid esters. Arthritis Rheum. 1964;7(4):359-367.
7. Berger RG, Yount WJ. Immediate “steroid flare” from intraarticular triamcinolone hexacetonide injection: case report and review of the literature. Arthritis Rheum. 1990;33(8):1284-1286.
8. Mader R, Lavi I, Luboshitzky R. Evaluation of the pituitary-adrenal axis function following single intraarticular injection of methylprednisolone. Arthritis Rheum. 2005;52(3):924-928.
9. Raynauld JP, Buckland-Wright C, Ward R, et al. Safety and efficacy of long-term intraarticular steroid injections in osteoarthritis of the knee: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2003;48(2):370-377.
10. MacMahon PJ, Eustace SJ, Kavanagh EC. Injectable corticosteroid and local anesthetic preparations: a review for radiologists. Radiology. 2009;252(3):647-661.
11. Sparling M, Malleson P, Wood B, Petty R. Radiographic followup of joints injected with triamcinolone hexacetonide for the management of childhood arthritis. Arthritis Rheum. 1990;33(6):821-826.
12. Eymontt MJ, Gordon GV, Schumacher HR, Hansell JR. The effects on synovial permeability and synovial fluid leukocyte counts in symptomatic osteoarthritis after intraarticular corticosteroid administration. J Rheumatol. 1982;9(2):198-203.
13. Gordon GV, Schumacher HR. Electron microscopic study of depot corticosteroid crystals with clinical studies after intra-articular injection. J Rheumatol. 1979;6(1):7-14.
14. Karsh J, Yang WH. An anaphylactic reaction to intra-articular triamcinolone: a case report and review of the literature. Ann Allergy Asthma Immunol. 2003;90(2):254-258.
15. Larsson LG. Anaphylactic shock after i.a. administration of triamcinolone acetonide in a 35-year-old female. Scand J Rheumatol. 1989;18(6):441-442.
16. García-Ortega P, Corominas M, Badia M. Carboxymethylcellulose allergy as a cause of suspected corticosteroid anaphylaxis. Ann Allergy Asthma Immunol. 2003;91(4):421.
17. Patterson DL, Yunginger JW, Dunn WF, Jones RT, Hunt LW. Anaphylaxis induced by the carboxymethylcellulose component of injectable triamcinolone acetonide suspension (Kenalog). Ann Allergy Asthma Immunol. 1995;74(2):163-166.
18. Steiner UC, Gentinetta T, Hausmann O, Pichler WJ. IgE-mediated anaphylaxis to intraarticular glucocorticoid preparations. AJR Am J Roentgenol. 2009;193(2):W156-W157.
19. Ijsselmuiden OE, Knegt-Junk KJ, van Wijk RG, van Joost T. Cutaneous adverse reactions after intra-articular injection of triamcinolone acetonide. Acta Derm Venereol. 1995;75(1):57-58.
20. Pullman-Mooar S, Mooar P, Sieck M, Clayburne G, Schumacher HR. Are there distinctive inflammatory flares after hylan g-f 20 intraarticular injections? J Rheumatol. 2002;29(12):2611-2614.
