User login
Case Report: An Unusual Case of Morel-Lavallée Lesion of the Upper Extremity
Case
A 32-year-old previously healthy woman presented to the ED with right upper arm pain and swelling of 6 days duration. According to the patient, the swelling began 2 days after she sustained a work-related injury at a coin-manufacturing factory. She stated that her right arm had gotten caught inside of a rolling press while she was cleaning it. The roller had stopped over her upper arm, trapping it between the roller and the platform for several minutes before it was extricated. She was brought to the ED by emergency medical services for evaluation immediately following this incident. At this first visit to the ED, the patient complained of mild pain in her right arm. Physical examination at that time revealed mild diffuse swelling extending from her hand to her distal humerus, with mild pain on passive flexion, and extension without associated numbness or tingling. Plain films of her right upper extremity were ordered, the results of which were relatively unremarkable. She was evaluated by an orthopedist, who ruled out compartment syndrome based on her benign physical examination and soft compartments. She was ultimately discharged and told to follow up with her primary care provider.
Over the course of 48 hours from the first ED visit, the patient developed large bullae on the dorsal and volar aspect of her forearm, elbow, and upper arm with associated pain. In addition to dark discolorations of the skin over her affected arm, she noticed that the bullae had become numerous and discolored. These symptoms continued to grow progressively worse, prompting her second presentation to the ED.
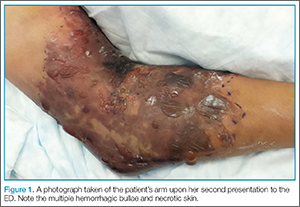
The patient was taken to the operating room and underwent debridement and resection of the circumferential necrotic skin and subcutaneous tissue in her right arm, and the placement of a skin graft with overlying wound vacuum-assisted closure. During the procedure, a large amount of serosanguinous fluid was drained and cultured, and was found to be sterile. Due to the size of her injury, she underwent two additional episodes of debridement and graft placement over the course of the next 2 weeks.
Discussion
First described in the 1850s by the French physician Maurice Morel-Lavallée, Morel-Lavallée lesion is a rare, traumatic, soft-tissue injury.1 It is an internal degloving injury wherein the skin and subcutaneous tissue have been forcibly separated from the underlying fascia as a result of shear stress. The lymphatic and blood vessels between the layers are also disrupted in this process, resulting in the accumulation of blood and lymphatic fluid as well as subcutaneous debris in the potential space that forms. Excess accumulation over time can compromise blood supply to the overlying skin and cause necrosis.2 Morel-Lavallée lesion is missed on initial evaluation in up to one-third of the cases and may have a delay in presentation ranging from hours to months after the inciting injury.3
Morel-Lavallée lesions typically involve the flank, hips, thigh, and prepatellar regions as a result of shear injuries sustained from bicycle falls and motor vehicle accidents.4 These lesions are often associated with concomitant acetabular and pelvic fractures.5 Involvement of the upper extremities is unusual. Typically, presentation consists of a fluctuant and painful mass underneath the skin which increases over time. The overlying skin may show the mechanism of the original injury, for example, as abrasions after a crush injury. The excessive skin necrosis and hemorrhagic bullae seen in this particular case is a very rare presentation.
Differential Diagnosis
The differential diagnosis includes compartment syndrome, coma blisters, a missed fracture, bullous pemphigoid, bullous drug reactions, and linear immunoglobulin A disease. Most of these conditions were easily ruled out in this case as the patient was previously healthy and not on any medications. The lesions in this case could have been confused with coma blisters, which are similar in appearance, self-limiting, and can develop on the extremities. However, coma blisters are classically associated with toxicity from various central nervous system depressants, as well as reduced consciousness from other causes—all of which were readily ruled-out based on the patient’s history. Moreover, the Morel-Lavallée lesion is a degloving injury of the subcutaneous tissue from the fascia underneath, whereas the pathology of coma blisters includes subepidermal bullae formation as well as immunoglobulin and complement deposition.6
Diagnostic Imaging
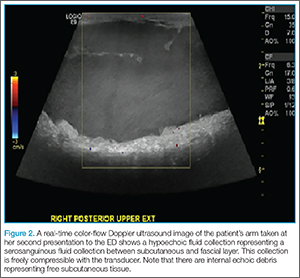
Morel-Lavallée lesion can often be confirmed via several imaging modalities, including ultrasound, CT, 3D CT, or magnetic resonance imaging (MRI).3,7 Ultrasound will usually show a well-circumscribed hypoechoic fluid collection with hyperechoic fat globules from the subcutaneous tissue, whereas CT tends to show an encapsulated mass with fluid collection underneath. In MRI, Morel-Lavallée lesion often appears as a hypointense T1-sequence and hyperintense T2-sequence similar to most other fluid collections. There may be variable T1- and T2-intensities with subcutaneous tissues in the fluid collection.2
Management
Despite recognition of this disease entity, controversies still exist regarding management. Case reports have demonstrated a relatively high rate of infected fluid collections depending on the chronicity of the injury.8 A recent algorithm to management described by Nickerson et al4 proposes that for patients with viable skin, percutaneous aspiration of more than 50 cc of fluid from these lesions should be treated with more extensive operative intervention based on the increased likelihood of recurrence. Patients without viable skin require formal debridement with possible skin grafting.
Other treatment options include conservative management, surgical drainage, sclerodesis, and extensive open surgery.8-10 Management is always case-based and dependent upon the size of the lesion and associated injuries.
Conclusion
This case represents an example of Morel-Lavallée lesions in their most severe and atypical form. It also serves as a reminder that vigilance and knowledge of this disease process is important in helping to diagnose this rare but potentially devastating condition. The key to recognizing this injury lies in keeping this disease process in the differential diagnosis of traumatic injuries with suspicious mechanism involving significant shear forces. Significant physical examination findings may not be present initially and evolve over a time period of hours to days. Once this injury is identified, management hinges on the size of the lesion and affected body part. Despite timely interventions, Morel-Lavallée lesions may result in significant morbidity and functional disability.
Dr Ye is an emergency medicine resident at the Brown Alpert Medical School in Providence, Rhode Island. Dr Rosenberg is a clinical assistant professor at Brown Alpert Medical School, and an emergency medicine attending physican at Rhode Island Hospital and The Miriam Hospital, Providence, Rhode Island.
- Morel-Lavallée M. Epanchements traumatique de serosite. Arc Générales Méd. 1853;691-731.
- Chokshi F, Jose J, Clifford P. Morel Lavallée Lesion. Am J Orthop (Belle Mead NJ). 2010;39(5): 252-253.
- Bonilla-Yoon I, Masih S, Patel DB, et al. The Morel-Lavallée lesion: pathophysiology, clinical presentation, imaging features, and treatment options. Emerg Radiol. 2014;21(1):35-43.
- Nickerson T, Zielinski M, Jenkins D, Schiller HJ. The Mayo Clinic experience with Morel-Lavallée lesions: establishment of a practice management guideline. J Trauma Acute Care Surg. 2014:76(2);493-497.
- Powers ML, Hatem SF, Sundaram M. Morel-Lavallee lesion. Orthopedics. 2007;30(4):322-323.
- Agarwal A, Bansal M, Conner K. Coma blisters with hypoxemic respiratory failure. Dermatol Online Journal. 2012:18(3);10.
- Reddix RN, Carrol E, Webb LX. Early diagnosis of a Morel-Lavallee lesion using three-dimensional computed tomography reconstructions: a case report. J Trauma. 2009;67(2):e57-e59.
- Lin HL, Lee WC, Kuo LC, Chen CW. Closed internal degloving injury with conservative treatment. Am J Emerg Med. 2008:26(2);254.e5-e6.
- Luria S, Applbaum Y,Weil Y, Liebergall M, Peyser A. Talc sclerodhesis of persistent Morel-Lavallée lesions (posttraumatic pseudocysts): case report of 4 patients. J Orthop Trauma. 2006;20(6):435-438.
- Penaud A, Quignon R, Danin A, Bahé L, Zakine G. Alcohol sclerodhesis: an innovative treatment for chronic Morel-Lavallée lesions. J Plast Reconstr Aesthet Surg. 2011;64(10): e262-264.
Case
A 32-year-old previously healthy woman presented to the ED with right upper arm pain and swelling of 6 days duration. According to the patient, the swelling began 2 days after she sustained a work-related injury at a coin-manufacturing factory. She stated that her right arm had gotten caught inside of a rolling press while she was cleaning it. The roller had stopped over her upper arm, trapping it between the roller and the platform for several minutes before it was extricated. She was brought to the ED by emergency medical services for evaluation immediately following this incident. At this first visit to the ED, the patient complained of mild pain in her right arm. Physical examination at that time revealed mild diffuse swelling extending from her hand to her distal humerus, with mild pain on passive flexion, and extension without associated numbness or tingling. Plain films of her right upper extremity were ordered, the results of which were relatively unremarkable. She was evaluated by an orthopedist, who ruled out compartment syndrome based on her benign physical examination and soft compartments. She was ultimately discharged and told to follow up with her primary care provider.
Over the course of 48 hours from the first ED visit, the patient developed large bullae on the dorsal and volar aspect of her forearm, elbow, and upper arm with associated pain. In addition to dark discolorations of the skin over her affected arm, she noticed that the bullae had become numerous and discolored. These symptoms continued to grow progressively worse, prompting her second presentation to the ED.
The patient was taken to the operating room and underwent debridement and resection of the circumferential necrotic skin and subcutaneous tissue in her right arm, and the placement of a skin graft with overlying wound vacuum-assisted closure. During the procedure, a large amount of serosanguinous fluid was drained and cultured, and was found to be sterile. Due to the size of her injury, she underwent two additional episodes of debridement and graft placement over the course of the next 2 weeks.
Discussion
First described in the 1850s by the French physician Maurice Morel-Lavallée, Morel-Lavallée lesion is a rare, traumatic, soft-tissue injury.1 It is an internal degloving injury wherein the skin and subcutaneous tissue have been forcibly separated from the underlying fascia as a result of shear stress. The lymphatic and blood vessels between the layers are also disrupted in this process, resulting in the accumulation of blood and lymphatic fluid as well as subcutaneous debris in the potential space that forms. Excess accumulation over time can compromise blood supply to the overlying skin and cause necrosis.2 Morel-Lavallée lesion is missed on initial evaluation in up to one-third of the cases and may have a delay in presentation ranging from hours to months after the inciting injury.3
Morel-Lavallée lesions typically involve the flank, hips, thigh, and prepatellar regions as a result of shear injuries sustained from bicycle falls and motor vehicle accidents.4 These lesions are often associated with concomitant acetabular and pelvic fractures.5 Involvement of the upper extremities is unusual. Typically, presentation consists of a fluctuant and painful mass underneath the skin which increases over time. The overlying skin may show the mechanism of the original injury, for example, as abrasions after a crush injury. The excessive skin necrosis and hemorrhagic bullae seen in this particular case is a very rare presentation.
Differential Diagnosis
The differential diagnosis includes compartment syndrome, coma blisters, a missed fracture, bullous pemphigoid, bullous drug reactions, and linear immunoglobulin A disease. Most of these conditions were easily ruled out in this case as the patient was previously healthy and not on any medications. The lesions in this case could have been confused with coma blisters, which are similar in appearance, self-limiting, and can develop on the extremities. However, coma blisters are classically associated with toxicity from various central nervous system depressants, as well as reduced consciousness from other causes—all of which were readily ruled-out based on the patient’s history. Moreover, the Morel-Lavallée lesion is a degloving injury of the subcutaneous tissue from the fascia underneath, whereas the pathology of coma blisters includes subepidermal bullae formation as well as immunoglobulin and complement deposition.6
Diagnostic Imaging
Morel-Lavallée lesion can often be confirmed via several imaging modalities, including ultrasound, CT, 3D CT, or magnetic resonance imaging (MRI).3,7 Ultrasound will usually show a well-circumscribed hypoechoic fluid collection with hyperechoic fat globules from the subcutaneous tissue, whereas CT tends to show an encapsulated mass with fluid collection underneath. In MRI, Morel-Lavallée lesion often appears as a hypointense T1-sequence and hyperintense T2-sequence similar to most other fluid collections. There may be variable T1- and T2-intensities with subcutaneous tissues in the fluid collection.2
Management
Despite recognition of this disease entity, controversies still exist regarding management. Case reports have demonstrated a relatively high rate of infected fluid collections depending on the chronicity of the injury.8 A recent algorithm to management described by Nickerson et al4 proposes that for patients with viable skin, percutaneous aspiration of more than 50 cc of fluid from these lesions should be treated with more extensive operative intervention based on the increased likelihood of recurrence. Patients without viable skin require formal debridement with possible skin grafting.
Other treatment options include conservative management, surgical drainage, sclerodesis, and extensive open surgery.8-10 Management is always case-based and dependent upon the size of the lesion and associated injuries.
Conclusion
This case represents an example of Morel-Lavallée lesions in their most severe and atypical form. It also serves as a reminder that vigilance and knowledge of this disease process is important in helping to diagnose this rare but potentially devastating condition. The key to recognizing this injury lies in keeping this disease process in the differential diagnosis of traumatic injuries with suspicious mechanism involving significant shear forces. Significant physical examination findings may not be present initially and evolve over a time period of hours to days. Once this injury is identified, management hinges on the size of the lesion and affected body part. Despite timely interventions, Morel-Lavallée lesions may result in significant morbidity and functional disability.
Dr Ye is an emergency medicine resident at the Brown Alpert Medical School in Providence, Rhode Island. Dr Rosenberg is a clinical assistant professor at Brown Alpert Medical School, and an emergency medicine attending physican at Rhode Island Hospital and The Miriam Hospital, Providence, Rhode Island.
Case
A 32-year-old previously healthy woman presented to the ED with right upper arm pain and swelling of 6 days duration. According to the patient, the swelling began 2 days after she sustained a work-related injury at a coin-manufacturing factory. She stated that her right arm had gotten caught inside of a rolling press while she was cleaning it. The roller had stopped over her upper arm, trapping it between the roller and the platform for several minutes before it was extricated. She was brought to the ED by emergency medical services for evaluation immediately following this incident. At this first visit to the ED, the patient complained of mild pain in her right arm. Physical examination at that time revealed mild diffuse swelling extending from her hand to her distal humerus, with mild pain on passive flexion, and extension without associated numbness or tingling. Plain films of her right upper extremity were ordered, the results of which were relatively unremarkable. She was evaluated by an orthopedist, who ruled out compartment syndrome based on her benign physical examination and soft compartments. She was ultimately discharged and told to follow up with her primary care provider.
Over the course of 48 hours from the first ED visit, the patient developed large bullae on the dorsal and volar aspect of her forearm, elbow, and upper arm with associated pain. In addition to dark discolorations of the skin over her affected arm, she noticed that the bullae had become numerous and discolored. These symptoms continued to grow progressively worse, prompting her second presentation to the ED.
The patient was taken to the operating room and underwent debridement and resection of the circumferential necrotic skin and subcutaneous tissue in her right arm, and the placement of a skin graft with overlying wound vacuum-assisted closure. During the procedure, a large amount of serosanguinous fluid was drained and cultured, and was found to be sterile. Due to the size of her injury, she underwent two additional episodes of debridement and graft placement over the course of the next 2 weeks.
Discussion
First described in the 1850s by the French physician Maurice Morel-Lavallée, Morel-Lavallée lesion is a rare, traumatic, soft-tissue injury.1 It is an internal degloving injury wherein the skin and subcutaneous tissue have been forcibly separated from the underlying fascia as a result of shear stress. The lymphatic and blood vessels between the layers are also disrupted in this process, resulting in the accumulation of blood and lymphatic fluid as well as subcutaneous debris in the potential space that forms. Excess accumulation over time can compromise blood supply to the overlying skin and cause necrosis.2 Morel-Lavallée lesion is missed on initial evaluation in up to one-third of the cases and may have a delay in presentation ranging from hours to months after the inciting injury.3
Morel-Lavallée lesions typically involve the flank, hips, thigh, and prepatellar regions as a result of shear injuries sustained from bicycle falls and motor vehicle accidents.4 These lesions are often associated with concomitant acetabular and pelvic fractures.5 Involvement of the upper extremities is unusual. Typically, presentation consists of a fluctuant and painful mass underneath the skin which increases over time. The overlying skin may show the mechanism of the original injury, for example, as abrasions after a crush injury. The excessive skin necrosis and hemorrhagic bullae seen in this particular case is a very rare presentation.
Differential Diagnosis
The differential diagnosis includes compartment syndrome, coma blisters, a missed fracture, bullous pemphigoid, bullous drug reactions, and linear immunoglobulin A disease. Most of these conditions were easily ruled out in this case as the patient was previously healthy and not on any medications. The lesions in this case could have been confused with coma blisters, which are similar in appearance, self-limiting, and can develop on the extremities. However, coma blisters are classically associated with toxicity from various central nervous system depressants, as well as reduced consciousness from other causes—all of which were readily ruled-out based on the patient’s history. Moreover, the Morel-Lavallée lesion is a degloving injury of the subcutaneous tissue from the fascia underneath, whereas the pathology of coma blisters includes subepidermal bullae formation as well as immunoglobulin and complement deposition.6
Diagnostic Imaging
Morel-Lavallée lesion can often be confirmed via several imaging modalities, including ultrasound, CT, 3D CT, or magnetic resonance imaging (MRI).3,7 Ultrasound will usually show a well-circumscribed hypoechoic fluid collection with hyperechoic fat globules from the subcutaneous tissue, whereas CT tends to show an encapsulated mass with fluid collection underneath. In MRI, Morel-Lavallée lesion often appears as a hypointense T1-sequence and hyperintense T2-sequence similar to most other fluid collections. There may be variable T1- and T2-intensities with subcutaneous tissues in the fluid collection.2
Management
Despite recognition of this disease entity, controversies still exist regarding management. Case reports have demonstrated a relatively high rate of infected fluid collections depending on the chronicity of the injury.8 A recent algorithm to management described by Nickerson et al4 proposes that for patients with viable skin, percutaneous aspiration of more than 50 cc of fluid from these lesions should be treated with more extensive operative intervention based on the increased likelihood of recurrence. Patients without viable skin require formal debridement with possible skin grafting.
Other treatment options include conservative management, surgical drainage, sclerodesis, and extensive open surgery.8-10 Management is always case-based and dependent upon the size of the lesion and associated injuries.
Conclusion
This case represents an example of Morel-Lavallée lesions in their most severe and atypical form. It also serves as a reminder that vigilance and knowledge of this disease process is important in helping to diagnose this rare but potentially devastating condition. The key to recognizing this injury lies in keeping this disease process in the differential diagnosis of traumatic injuries with suspicious mechanism involving significant shear forces. Significant physical examination findings may not be present initially and evolve over a time period of hours to days. Once this injury is identified, management hinges on the size of the lesion and affected body part. Despite timely interventions, Morel-Lavallée lesions may result in significant morbidity and functional disability.
Dr Ye is an emergency medicine resident at the Brown Alpert Medical School in Providence, Rhode Island. Dr Rosenberg is a clinical assistant professor at Brown Alpert Medical School, and an emergency medicine attending physican at Rhode Island Hospital and The Miriam Hospital, Providence, Rhode Island.
- Morel-Lavallée M. Epanchements traumatique de serosite. Arc Générales Méd. 1853;691-731.
- Chokshi F, Jose J, Clifford P. Morel Lavallée Lesion. Am J Orthop (Belle Mead NJ). 2010;39(5): 252-253.
- Bonilla-Yoon I, Masih S, Patel DB, et al. The Morel-Lavallée lesion: pathophysiology, clinical presentation, imaging features, and treatment options. Emerg Radiol. 2014;21(1):35-43.
- Nickerson T, Zielinski M, Jenkins D, Schiller HJ. The Mayo Clinic experience with Morel-Lavallée lesions: establishment of a practice management guideline. J Trauma Acute Care Surg. 2014:76(2);493-497.
- Powers ML, Hatem SF, Sundaram M. Morel-Lavallee lesion. Orthopedics. 2007;30(4):322-323.
- Agarwal A, Bansal M, Conner K. Coma blisters with hypoxemic respiratory failure. Dermatol Online Journal. 2012:18(3);10.
- Reddix RN, Carrol E, Webb LX. Early diagnosis of a Morel-Lavallee lesion using three-dimensional computed tomography reconstructions: a case report. J Trauma. 2009;67(2):e57-e59.
- Lin HL, Lee WC, Kuo LC, Chen CW. Closed internal degloving injury with conservative treatment. Am J Emerg Med. 2008:26(2);254.e5-e6.
- Luria S, Applbaum Y,Weil Y, Liebergall M, Peyser A. Talc sclerodhesis of persistent Morel-Lavallée lesions (posttraumatic pseudocysts): case report of 4 patients. J Orthop Trauma. 2006;20(6):435-438.
- Penaud A, Quignon R, Danin A, Bahé L, Zakine G. Alcohol sclerodhesis: an innovative treatment for chronic Morel-Lavallée lesions. J Plast Reconstr Aesthet Surg. 2011;64(10): e262-264.
- Morel-Lavallée M. Epanchements traumatique de serosite. Arc Générales Méd. 1853;691-731.
- Chokshi F, Jose J, Clifford P. Morel Lavallée Lesion. Am J Orthop (Belle Mead NJ). 2010;39(5): 252-253.
- Bonilla-Yoon I, Masih S, Patel DB, et al. The Morel-Lavallée lesion: pathophysiology, clinical presentation, imaging features, and treatment options. Emerg Radiol. 2014;21(1):35-43.
- Nickerson T, Zielinski M, Jenkins D, Schiller HJ. The Mayo Clinic experience with Morel-Lavallée lesions: establishment of a practice management guideline. J Trauma Acute Care Surg. 2014:76(2);493-497.
- Powers ML, Hatem SF, Sundaram M. Morel-Lavallee lesion. Orthopedics. 2007;30(4):322-323.
- Agarwal A, Bansal M, Conner K. Coma blisters with hypoxemic respiratory failure. Dermatol Online Journal. 2012:18(3);10.
- Reddix RN, Carrol E, Webb LX. Early diagnosis of a Morel-Lavallee lesion using three-dimensional computed tomography reconstructions: a case report. J Trauma. 2009;67(2):e57-e59.
- Lin HL, Lee WC, Kuo LC, Chen CW. Closed internal degloving injury with conservative treatment. Am J Emerg Med. 2008:26(2);254.e5-e6.
- Luria S, Applbaum Y,Weil Y, Liebergall M, Peyser A. Talc sclerodhesis of persistent Morel-Lavallée lesions (posttraumatic pseudocysts): case report of 4 patients. J Orthop Trauma. 2006;20(6):435-438.
- Penaud A, Quignon R, Danin A, Bahé L, Zakine G. Alcohol sclerodhesis: an innovative treatment for chronic Morel-Lavallée lesions. J Plast Reconstr Aesthet Surg. 2011;64(10): e262-264.
When it's beneficial to defer dialysis
THE CASE
A 94-year-old Hispanic man with hypertension, congestive heart failure (CHF), anemia of chronic disease, and end-stage renal disease (ESRD) presented to our facility with weakness and shortness of breath. We diagnosed a CHF exacerbation. Initially, he exhibited some respiratory distress that required observation in the coronary care unit and bi-level positive airway pressure therapy to maintain oxygen saturation. Our patient was then moved to a step-down unit where his primary caregiver, his granddaughter, told the medical team that he was limited at home in some of his instrumental activities of daily living. Specifically, he was unable to prepare meals or manage his finances on his own.
Nephrology was consulted for consideration of hemodialysis (HD) because our patient’s creatinine on admission was 7.2 mg/dL (normal for men is 0.7-1.3 mg/dL) and his estimated glomerular filtration rate (GFR) was 7 mL/min (normal is 90-120 mL/min). The patient’s family was conflicted over whether or not to start HD. Palliative Care was consulted to help establish goals of care.
A decision is made. In light of the patient’s limited functional status and his expressed desire to stay at home with his family and receive limited medical care there, the Nephrology and Palliative Care teams recommended delaying HD despite the patient’s worsening renal function. The patient was discharged home with home care services, and he and the family were instructed to follow up with Nephrology for supportive renal management.
DISCUSSION
The decision to delay HD in patients with ESRD is a difficult one that requires shared decision-making between patients and medical providers. Palliative Care consultation services are often involved in this process.
Recent literature supports an “intent-to-defer” based on an evaluation of the patient’s functionality. This represents a paradigm shift from the previous “intent-to-start-early” treatment strategy. In fact, rather than starting early, the Canadian Society of Nephrology recommends delaying initiation of HD in patients with a GFR <15 mL/min.1 Close monitoring of these patients by both a primary care physician and nephrologist is essential.
When considering initiation of HD, it’s important to look at the overall benefit of this intervention in light of the patient’s mortality risk and quality of life. Many patients who receive HD—especially the elderly—report that it takes more than 6 hours to recover following a dialysis treatment.2
Not surprisingly, depression is common in elderly HD patients. Compared to their younger cohorts, older HD patients have a 62% increased risk of developing depression.3 Also, patients who are considered frail and are receiving HD have more than 3 times the mortality risk within one year than those who are not (hazard ratio=3.42; 95% confidence interval, 2.45-4.76).4 (The researchers’ definition of frailty included poor self-reported physical function, exhaustion/fatigue, low physical activity, and undernutrition.4)
Functional status. Although a patient’s age should not be a limiting factor for HD referral, functional status should be considered. Patients with limited functionality and significant dependence have an increased risk of death during the first year of HD.5
Palliative approach gains acceptance. It is becoming more accepted within the nephrology community to consider a palliative approach to patients with ESRD. Organizations such as the Renal Physicians Association recommend effective prognostication, early advanced care planning, forgoing HD in patients with a poor prognosis, and involving Palliative Care early in the decision-making process.6 Aligning the patient’s goals of care with the appropriate treatment method—particularly in patients with comorbid conditions—is an important practice when caring for those with limited life expectancy and functionality.7
THE TAKEAWAY
Intent-to-defer HD may be a preferred strategy when caring for many patients with ESRD. Taking into consideration a patient’s comorbidities and functional status, while considering mortality risk and quality of life are essential. Involving palliative care and nephrology specialists can help patients and families understand HD and make an educated decision regarding when to start it.
1. Nesrallah GE, Mustafa RA, Clark WF, et al; Canadian Society of Nephrology. Canadian Society of Nephrology 2014 clinical practice guideline for timing the initiation of chronic dialysis. CMAJ. 2014;186:112-117.
2. Rayner HC, Zepel L, Fuller DS, et al. Recovery time, quality of life, and mortality in hemodialysis patients: the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am J Kidney Dis. 2014;64:86-94.
3. Canaud B, Tong L, Tentori F, et al. Clinical practices and outcomes in elderly hemodialysis patients: results from the Dialysis Outcomes and Practice Patterns Study (DOPPS). Clin J Am Soc Nephrol. 2011;6:1651-1662.
4. Johansen KL, Chertow GM, Jin C, et al. Significance of frailty among dialysis patients. J Am Soc Nephrol. 2007;18:2960-2967.
5. Joly D, Anglicheau D, Alberti C, et al. Octogenarians reaching end-stage renal disease: cohort study of decision-making and clinical outcomes. J Am Soc Nephrol. 2003;14:1012-1021.
6. Renal Physicians Association. Shared decision-making in the appropriate initiation of and withdrawal from dialysis: clinical practice guideline. 2nd ed. Rockville, MD: Renal Physicians Association; 2010.
7. Grubbs V, Moss AH, Cohen LM, et al; Dialysis Advisory Group of the American Society of Nephrology. A palliative approach to dialysis care: a patient-centered transition to the end of life. Clin J Am Soc Nephrol. 2014;9:2203-2209.
THE CASE
A 94-year-old Hispanic man with hypertension, congestive heart failure (CHF), anemia of chronic disease, and end-stage renal disease (ESRD) presented to our facility with weakness and shortness of breath. We diagnosed a CHF exacerbation. Initially, he exhibited some respiratory distress that required observation in the coronary care unit and bi-level positive airway pressure therapy to maintain oxygen saturation. Our patient was then moved to a step-down unit where his primary caregiver, his granddaughter, told the medical team that he was limited at home in some of his instrumental activities of daily living. Specifically, he was unable to prepare meals or manage his finances on his own.
Nephrology was consulted for consideration of hemodialysis (HD) because our patient’s creatinine on admission was 7.2 mg/dL (normal for men is 0.7-1.3 mg/dL) and his estimated glomerular filtration rate (GFR) was 7 mL/min (normal is 90-120 mL/min). The patient’s family was conflicted over whether or not to start HD. Palliative Care was consulted to help establish goals of care.
A decision is made. In light of the patient’s limited functional status and his expressed desire to stay at home with his family and receive limited medical care there, the Nephrology and Palliative Care teams recommended delaying HD despite the patient’s worsening renal function. The patient was discharged home with home care services, and he and the family were instructed to follow up with Nephrology for supportive renal management.
DISCUSSION
The decision to delay HD in patients with ESRD is a difficult one that requires shared decision-making between patients and medical providers. Palliative Care consultation services are often involved in this process.
Recent literature supports an “intent-to-defer” based on an evaluation of the patient’s functionality. This represents a paradigm shift from the previous “intent-to-start-early” treatment strategy. In fact, rather than starting early, the Canadian Society of Nephrology recommends delaying initiation of HD in patients with a GFR <15 mL/min.1 Close monitoring of these patients by both a primary care physician and nephrologist is essential.
When considering initiation of HD, it’s important to look at the overall benefit of this intervention in light of the patient’s mortality risk and quality of life. Many patients who receive HD—especially the elderly—report that it takes more than 6 hours to recover following a dialysis treatment.2
Not surprisingly, depression is common in elderly HD patients. Compared to their younger cohorts, older HD patients have a 62% increased risk of developing depression.3 Also, patients who are considered frail and are receiving HD have more than 3 times the mortality risk within one year than those who are not (hazard ratio=3.42; 95% confidence interval, 2.45-4.76).4 (The researchers’ definition of frailty included poor self-reported physical function, exhaustion/fatigue, low physical activity, and undernutrition.4)
Functional status. Although a patient’s age should not be a limiting factor for HD referral, functional status should be considered. Patients with limited functionality and significant dependence have an increased risk of death during the first year of HD.5
Palliative approach gains acceptance. It is becoming more accepted within the nephrology community to consider a palliative approach to patients with ESRD. Organizations such as the Renal Physicians Association recommend effective prognostication, early advanced care planning, forgoing HD in patients with a poor prognosis, and involving Palliative Care early in the decision-making process.6 Aligning the patient’s goals of care with the appropriate treatment method—particularly in patients with comorbid conditions—is an important practice when caring for those with limited life expectancy and functionality.7
THE TAKEAWAY
Intent-to-defer HD may be a preferred strategy when caring for many patients with ESRD. Taking into consideration a patient’s comorbidities and functional status, while considering mortality risk and quality of life are essential. Involving palliative care and nephrology specialists can help patients and families understand HD and make an educated decision regarding when to start it.
THE CASE
A 94-year-old Hispanic man with hypertension, congestive heart failure (CHF), anemia of chronic disease, and end-stage renal disease (ESRD) presented to our facility with weakness and shortness of breath. We diagnosed a CHF exacerbation. Initially, he exhibited some respiratory distress that required observation in the coronary care unit and bi-level positive airway pressure therapy to maintain oxygen saturation. Our patient was then moved to a step-down unit where his primary caregiver, his granddaughter, told the medical team that he was limited at home in some of his instrumental activities of daily living. Specifically, he was unable to prepare meals or manage his finances on his own.
Nephrology was consulted for consideration of hemodialysis (HD) because our patient’s creatinine on admission was 7.2 mg/dL (normal for men is 0.7-1.3 mg/dL) and his estimated glomerular filtration rate (GFR) was 7 mL/min (normal is 90-120 mL/min). The patient’s family was conflicted over whether or not to start HD. Palliative Care was consulted to help establish goals of care.
A decision is made. In light of the patient’s limited functional status and his expressed desire to stay at home with his family and receive limited medical care there, the Nephrology and Palliative Care teams recommended delaying HD despite the patient’s worsening renal function. The patient was discharged home with home care services, and he and the family were instructed to follow up with Nephrology for supportive renal management.
DISCUSSION
The decision to delay HD in patients with ESRD is a difficult one that requires shared decision-making between patients and medical providers. Palliative Care consultation services are often involved in this process.
Recent literature supports an “intent-to-defer” based on an evaluation of the patient’s functionality. This represents a paradigm shift from the previous “intent-to-start-early” treatment strategy. In fact, rather than starting early, the Canadian Society of Nephrology recommends delaying initiation of HD in patients with a GFR <15 mL/min.1 Close monitoring of these patients by both a primary care physician and nephrologist is essential.
When considering initiation of HD, it’s important to look at the overall benefit of this intervention in light of the patient’s mortality risk and quality of life. Many patients who receive HD—especially the elderly—report that it takes more than 6 hours to recover following a dialysis treatment.2
Not surprisingly, depression is common in elderly HD patients. Compared to their younger cohorts, older HD patients have a 62% increased risk of developing depression.3 Also, patients who are considered frail and are receiving HD have more than 3 times the mortality risk within one year than those who are not (hazard ratio=3.42; 95% confidence interval, 2.45-4.76).4 (The researchers’ definition of frailty included poor self-reported physical function, exhaustion/fatigue, low physical activity, and undernutrition.4)
Functional status. Although a patient’s age should not be a limiting factor for HD referral, functional status should be considered. Patients with limited functionality and significant dependence have an increased risk of death during the first year of HD.5
Palliative approach gains acceptance. It is becoming more accepted within the nephrology community to consider a palliative approach to patients with ESRD. Organizations such as the Renal Physicians Association recommend effective prognostication, early advanced care planning, forgoing HD in patients with a poor prognosis, and involving Palliative Care early in the decision-making process.6 Aligning the patient’s goals of care with the appropriate treatment method—particularly in patients with comorbid conditions—is an important practice when caring for those with limited life expectancy and functionality.7
THE TAKEAWAY
Intent-to-defer HD may be a preferred strategy when caring for many patients with ESRD. Taking into consideration a patient’s comorbidities and functional status, while considering mortality risk and quality of life are essential. Involving palliative care and nephrology specialists can help patients and families understand HD and make an educated decision regarding when to start it.
1. Nesrallah GE, Mustafa RA, Clark WF, et al; Canadian Society of Nephrology. Canadian Society of Nephrology 2014 clinical practice guideline for timing the initiation of chronic dialysis. CMAJ. 2014;186:112-117.
2. Rayner HC, Zepel L, Fuller DS, et al. Recovery time, quality of life, and mortality in hemodialysis patients: the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am J Kidney Dis. 2014;64:86-94.
3. Canaud B, Tong L, Tentori F, et al. Clinical practices and outcomes in elderly hemodialysis patients: results from the Dialysis Outcomes and Practice Patterns Study (DOPPS). Clin J Am Soc Nephrol. 2011;6:1651-1662.
4. Johansen KL, Chertow GM, Jin C, et al. Significance of frailty among dialysis patients. J Am Soc Nephrol. 2007;18:2960-2967.
5. Joly D, Anglicheau D, Alberti C, et al. Octogenarians reaching end-stage renal disease: cohort study of decision-making and clinical outcomes. J Am Soc Nephrol. 2003;14:1012-1021.
6. Renal Physicians Association. Shared decision-making in the appropriate initiation of and withdrawal from dialysis: clinical practice guideline. 2nd ed. Rockville, MD: Renal Physicians Association; 2010.
7. Grubbs V, Moss AH, Cohen LM, et al; Dialysis Advisory Group of the American Society of Nephrology. A palliative approach to dialysis care: a patient-centered transition to the end of life. Clin J Am Soc Nephrol. 2014;9:2203-2209.
1. Nesrallah GE, Mustafa RA, Clark WF, et al; Canadian Society of Nephrology. Canadian Society of Nephrology 2014 clinical practice guideline for timing the initiation of chronic dialysis. CMAJ. 2014;186:112-117.
2. Rayner HC, Zepel L, Fuller DS, et al. Recovery time, quality of life, and mortality in hemodialysis patients: the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am J Kidney Dis. 2014;64:86-94.
3. Canaud B, Tong L, Tentori F, et al. Clinical practices and outcomes in elderly hemodialysis patients: results from the Dialysis Outcomes and Practice Patterns Study (DOPPS). Clin J Am Soc Nephrol. 2011;6:1651-1662.
4. Johansen KL, Chertow GM, Jin C, et al. Significance of frailty among dialysis patients. J Am Soc Nephrol. 2007;18:2960-2967.
5. Joly D, Anglicheau D, Alberti C, et al. Octogenarians reaching end-stage renal disease: cohort study of decision-making and clinical outcomes. J Am Soc Nephrol. 2003;14:1012-1021.
6. Renal Physicians Association. Shared decision-making in the appropriate initiation of and withdrawal from dialysis: clinical practice guideline. 2nd ed. Rockville, MD: Renal Physicians Association; 2010.
7. Grubbs V, Moss AH, Cohen LM, et al; Dialysis Advisory Group of the American Society of Nephrology. A palliative approach to dialysis care: a patient-centered transition to the end of life. Clin J Am Soc Nephrol. 2014;9:2203-2209.

Weight loss • fatigue • joint pain • Dx?
THE CASE
A 49-year-old Mexican immigrant woman was admitted to the hospital with a 5-month history of fatigue and a 30-pound unintentional weight loss. She was also experiencing arthralgia, swelling, and stiffness in her hands and feet that was worse in the morning. The patient, who was obese and suffered from type 2 diabetes and hypertension, said that at the onset of her illness 5 months earlier, she’d experienced approximately 2 weeks of night sweats and a few days of fever.
A month before being admitted to the hospital, she’d been seen in our southern New Mexico family medicine office. Her recent history of fever, joint symptoms, and weight loss raised concerns of an insidious infection, a new-onset rheumatologic condition, or an occult malignancy.
Initial lab tests revealed leukopenia (white blood cell count, 3200/mcL), microcytic anemia (hemoglobin, 9.4 g/dL), and an elevated erythrocyte sedimentation rate of 30 mm/hr (normal range, 0-20 mm/hr). A rheumatoid factor test was negative, and her thyroid, kidney, and liver function tests were all normal.
More testing… The patient frequently traveled between New Mexico and her hometown of Chihuahua, Mexico, but there had been no recent changes in her diet or environmental exposures. She denied drinking any unpasteurized milk in Chihuahua. But based on her travel history, we ordered enzyme-linked immunosorbent assay (ELISA) antibody titers for Brucella, immunoglobulin G, and immunoglobulin M, which all came back negative. Additionally, we ordered an abdominal and pelvic ultrasound and a chest x-ray that were nondiagnostic. Given the patient’s weight loss and anemia, we referred her to a gastroenterologist for upper and lower gastrointestinal endoscopic evaluations. Unfortunately, the patient was uninsured and did not go to see the gastroenterologist.
A month after seeing us, our patient’s fatigue, lack of appetite, and joint pain became debilitating and she was admitted to the hospital for further evaluation, including a consultation with an oncologist.
THE DIAGNOSIS
During our patient’s 6-day hospital stay, a bone scintigraphy showed a focus of uptake in her left parietal bone and computed tomography scans of her chest, abdomen, and pelvis revealed a left thyroid nodule, as well as multiple noncalcified pulmonary nodules. Blood cultures were also obtained.
Despite the initial negative antibody tests, the blood cultures drawn in the hospital revealed the presence of Brucella melitensis, and we diagnosed brucellosis in this patient.
DISCUSSION
Brucella melitensis is one of the 4 recognized, land-based species of the Brucella genus that can cause disease in humans. Goats, sheep, and camels are natural hosts of B melitensis and consumption of their unpasteurized, infected milk and milk products (especially soft cheeses, ice cream, milk, and butter) leads to human disease. (Once hospitalized, our patient admitted to frequently eating unpasteurized goat cheese from Chihuahua. The only other person in her household that ate the cheese was her 26-year-old daughter, who was also experiencing similar symptoms.)
Brucellosis can also result from inhaling infected, aerosolized material; therefore, individuals whose occupations involve close work with host animals or work in laboratories with the bacteria have an increased risk of infection.1 Due to the risk of acquiring the infection via inhalation, brucellosis is considered a bioterrorism threat.2 Additionally, there have been reports of human-to-human transmission via sexual intercourse, transplacental infection, blood and bone marrow transfusion, and breastfeeding.3
B melitensis is the cause of the majority of Brucella-related illnesses in the world, though symptoms of infection are similar among the different Brucella species. The pathogen can affect almost all organ systems after the initial 2- to 4-week incubation period. Symptoms of brucellosis can be highly variable, although fever is consistently present.1 Other red flags include arthritis (usually affecting the peripheral joints, the sacroiliac joints, and the lower spine), epididymo-orchitis, and hepatitis resulting in transaminase elevation. Abscess formation can be seen in the liver, spleen, and other organs.
Less common but more ominous complications include central nervous system infections and abscesses, endocarditis, and pulmonary infections. Endocarditis and the resulting aortic valve involvement is the major cause of mortality.1Brucella-related uveitis, thyroiditis, nephritis, vasculitis, and acalculous cholecystitis have also been reported.4-9
Rare in the United States. Pasteurization of dairy products and mass vaccination of livestock make Brucella infection rare in the United States. While there have only been 80 to 139 cases of brucellosis reported per year in the United States since 1993, it remains a persistent threat. International travel is common from the United States to the Middle East and other parts of the world where brucellosis is endemic.
Additionally, infection of livestock with Brucella remains widespread in Mexico and the consumption of unpasteurized Mexican dairy products from goats and sheep remains a high-risk activity for acquiring the disease.10 Consequently, Texas and California account for more than half of the brucellosis diagnoses in the United States. However, in 2010, cases were reported in 25 other states and the District of Columbia.11
Repeat serology tests are preferred for confirming the Dx
It is interesting that our patient’s initial Brucella serology by ELISA was negative, because it was ordered months after her initial symptoms. Antibodies should be seen within a month of symptom onset. The Centers for Disease Control and Prevention (CDC) recommends taking 2 serum samples to establish a serologic diagnosis of brucellosis. The first should be drawn within 7 days of symptom onset and the second should be taken 2 to 4 weeks later. A greater than 4-fold rise in the antibody titer confirms the diagnosis. While ELISA is an acceptable serologic test, the CDC recommends using a serum tube agglutination test called the Brucella microagglutination test (BMAT).12 Repeat serology was not performed on our patient because the diagnosis had been confirmed by blood culture.
A combination of antibiotics is the recommended treatment
Treatment of brucellosis should include a tetracycline for at least 6 weeks in combination with an aminoglycoside or rifampin 600 mg/d for an all-oral regimen.13 Doxycycline 100 mg twice a day is preferred due to fewer gastrointestinal adverse effects than tetracycline. Relapse is not uncommon (10%) and usually occurs within one year of completing the antibiotics.8 However, there is a case report of a patient having reactivated brucellosis manifested as acalculous cholecystitis 28 years after completing antibiotics.8
Our patient was started on oral doxycycline 100 mg twice a day and oral rifampin 600 mg/d for 6 weeks. Within days of starting the antibiotics, her joint symptoms and fatigue rapidly abated and her appetite returned. Follow-up radiological testing was not performed after her initial hospital studies due to her lack of financial resources.
The patient’s daughter had also been experiencing night sweats, chills, malaise, anorexia, joint pains, weight loss, and alopecia over the previous 2 months. Her blood cultures were positive for B melitensis as well, and she was started on the same antibiotic regimen as her mother. The daughter was also seen in our clinic by another physician and improved quickly within a week of starting treatment.
Both our patient and her daughter remained symptom-free 6 years after treatment.
THE TAKEAWAY
Brucellosis is rare in the United States, but international travel to endemic areas is commonplace and consumption of unpasteurized Mexican dairy products from goats and sheep is widespread. Brucellosis has a wide range of symptoms, but a prompt diagnosis by an ELISA or BMAT serologic test and appropriate treatment can avoid morbidity and mortality. Treatment includes a tetracycline for at least 6 weeks in combination with an aminoglycoside or rifampin.
1. Pappas G, Akritidis N, Bosilkovski M, et al. Brucellosis. N Engl J Med. 2005;352:2325-2336.
2. Centers for Disease Control and Prevention (CDC). Suspected brucellosis case prompts investigation of possible bioterrorismrelated activity—New Hampshire and Massachusetts, 1999. MMWR Morb Mortal Wkly Rep. 2000;49:509-512.
3. Chen S, Zhang H, Liu X, et al. Increasing threat of brucellosis to low-risk persons in urban settings, China. Emerg Infect Dis. 2014;20:126-130.
4. Rolando I, Vilchez G, Olarte L, et al. Brucellar uveitis: intraocular fluids and biopsy studies. Int J Infect Dis. 2009;13:e206-e211.
5. Azizi F, Katchoui A. Brucella infection of the thyroid gland. Thyroid. 1996;6:461-463.
6. Siegelmann N, Abraham AS, Rudensky B, et al. Brucellosis with nephrotic syndrome, nephritis and IgA nephropathy. Postgrad Med J. 1992;68:834-836.
7. Tanyel E, Tasdelen Fisgin N, Yildiz L, et al. Panniculitis as the initial manifestation of brucellosis: a case report. Am J Dermatopathol. 2008;30:169-171.
8. Ögredici Ö, Erb S, Langer I, et al. Brucellosis reactivation after 28 years. Emerg Infect Dis. 2010;16:2021-2022.
9. Dhand A, Ross JJ. Implantable cardioverter-defibrillator infection due to Brucella melitensis: case report and review of brucellosis of cardiac devices. Clin Infect Dis. 2007;44:e37-e39.
10. Solorio-Rivera JL, Segura-Correa JC, Sánchez-Gil LG. Seroprevalence of and risk factors for brucellosis of goats in herds of Michoacan, Mexico. Prev Vet Med. 2007;82:282-290.
11. Centers for Disease Control and Prevention. Brucellosis surveillance. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/brucellosis/resources/surveillance.html. Accessed October 30, 2015.
12. Centers for Disease Control and Prevention. Serology. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/brucellosis/clinicians/serology.html. Accessed October 30, 2015.
13. Corbel MJ. Brucellosis in humans and animals. World Health Organization (WHO);2006:36-41. Available at: http://www.who.int/csr/resources/publications/Brucellosis.pdf. Accessed November 2, 2015.
THE CASE
A 49-year-old Mexican immigrant woman was admitted to the hospital with a 5-month history of fatigue and a 30-pound unintentional weight loss. She was also experiencing arthralgia, swelling, and stiffness in her hands and feet that was worse in the morning. The patient, who was obese and suffered from type 2 diabetes and hypertension, said that at the onset of her illness 5 months earlier, she’d experienced approximately 2 weeks of night sweats and a few days of fever.
A month before being admitted to the hospital, she’d been seen in our southern New Mexico family medicine office. Her recent history of fever, joint symptoms, and weight loss raised concerns of an insidious infection, a new-onset rheumatologic condition, or an occult malignancy.
Initial lab tests revealed leukopenia (white blood cell count, 3200/mcL), microcytic anemia (hemoglobin, 9.4 g/dL), and an elevated erythrocyte sedimentation rate of 30 mm/hr (normal range, 0-20 mm/hr). A rheumatoid factor test was negative, and her thyroid, kidney, and liver function tests were all normal.
More testing… The patient frequently traveled between New Mexico and her hometown of Chihuahua, Mexico, but there had been no recent changes in her diet or environmental exposures. She denied drinking any unpasteurized milk in Chihuahua. But based on her travel history, we ordered enzyme-linked immunosorbent assay (ELISA) antibody titers for Brucella, immunoglobulin G, and immunoglobulin M, which all came back negative. Additionally, we ordered an abdominal and pelvic ultrasound and a chest x-ray that were nondiagnostic. Given the patient’s weight loss and anemia, we referred her to a gastroenterologist for upper and lower gastrointestinal endoscopic evaluations. Unfortunately, the patient was uninsured and did not go to see the gastroenterologist.
A month after seeing us, our patient’s fatigue, lack of appetite, and joint pain became debilitating and she was admitted to the hospital for further evaluation, including a consultation with an oncologist.
THE DIAGNOSIS
During our patient’s 6-day hospital stay, a bone scintigraphy showed a focus of uptake in her left parietal bone and computed tomography scans of her chest, abdomen, and pelvis revealed a left thyroid nodule, as well as multiple noncalcified pulmonary nodules. Blood cultures were also obtained.
Despite the initial negative antibody tests, the blood cultures drawn in the hospital revealed the presence of Brucella melitensis, and we diagnosed brucellosis in this patient.
DISCUSSION
Brucella melitensis is one of the 4 recognized, land-based species of the Brucella genus that can cause disease in humans. Goats, sheep, and camels are natural hosts of B melitensis and consumption of their unpasteurized, infected milk and milk products (especially soft cheeses, ice cream, milk, and butter) leads to human disease. (Once hospitalized, our patient admitted to frequently eating unpasteurized goat cheese from Chihuahua. The only other person in her household that ate the cheese was her 26-year-old daughter, who was also experiencing similar symptoms.)
Brucellosis can also result from inhaling infected, aerosolized material; therefore, individuals whose occupations involve close work with host animals or work in laboratories with the bacteria have an increased risk of infection.1 Due to the risk of acquiring the infection via inhalation, brucellosis is considered a bioterrorism threat.2 Additionally, there have been reports of human-to-human transmission via sexual intercourse, transplacental infection, blood and bone marrow transfusion, and breastfeeding.3
B melitensis is the cause of the majority of Brucella-related illnesses in the world, though symptoms of infection are similar among the different Brucella species. The pathogen can affect almost all organ systems after the initial 2- to 4-week incubation period. Symptoms of brucellosis can be highly variable, although fever is consistently present.1 Other red flags include arthritis (usually affecting the peripheral joints, the sacroiliac joints, and the lower spine), epididymo-orchitis, and hepatitis resulting in transaminase elevation. Abscess formation can be seen in the liver, spleen, and other organs.
Less common but more ominous complications include central nervous system infections and abscesses, endocarditis, and pulmonary infections. Endocarditis and the resulting aortic valve involvement is the major cause of mortality.1Brucella-related uveitis, thyroiditis, nephritis, vasculitis, and acalculous cholecystitis have also been reported.4-9
Rare in the United States. Pasteurization of dairy products and mass vaccination of livestock make Brucella infection rare in the United States. While there have only been 80 to 139 cases of brucellosis reported per year in the United States since 1993, it remains a persistent threat. International travel is common from the United States to the Middle East and other parts of the world where brucellosis is endemic.
Additionally, infection of livestock with Brucella remains widespread in Mexico and the consumption of unpasteurized Mexican dairy products from goats and sheep remains a high-risk activity for acquiring the disease.10 Consequently, Texas and California account for more than half of the brucellosis diagnoses in the United States. However, in 2010, cases were reported in 25 other states and the District of Columbia.11
Repeat serology tests are preferred for confirming the Dx
It is interesting that our patient’s initial Brucella serology by ELISA was negative, because it was ordered months after her initial symptoms. Antibodies should be seen within a month of symptom onset. The Centers for Disease Control and Prevention (CDC) recommends taking 2 serum samples to establish a serologic diagnosis of brucellosis. The first should be drawn within 7 days of symptom onset and the second should be taken 2 to 4 weeks later. A greater than 4-fold rise in the antibody titer confirms the diagnosis. While ELISA is an acceptable serologic test, the CDC recommends using a serum tube agglutination test called the Brucella microagglutination test (BMAT).12 Repeat serology was not performed on our patient because the diagnosis had been confirmed by blood culture.
A combination of antibiotics is the recommended treatment
Treatment of brucellosis should include a tetracycline for at least 6 weeks in combination with an aminoglycoside or rifampin 600 mg/d for an all-oral regimen.13 Doxycycline 100 mg twice a day is preferred due to fewer gastrointestinal adverse effects than tetracycline. Relapse is not uncommon (10%) and usually occurs within one year of completing the antibiotics.8 However, there is a case report of a patient having reactivated brucellosis manifested as acalculous cholecystitis 28 years after completing antibiotics.8
Our patient was started on oral doxycycline 100 mg twice a day and oral rifampin 600 mg/d for 6 weeks. Within days of starting the antibiotics, her joint symptoms and fatigue rapidly abated and her appetite returned. Follow-up radiological testing was not performed after her initial hospital studies due to her lack of financial resources.
The patient’s daughter had also been experiencing night sweats, chills, malaise, anorexia, joint pains, weight loss, and alopecia over the previous 2 months. Her blood cultures were positive for B melitensis as well, and she was started on the same antibiotic regimen as her mother. The daughter was also seen in our clinic by another physician and improved quickly within a week of starting treatment.
Both our patient and her daughter remained symptom-free 6 years after treatment.
THE TAKEAWAY
Brucellosis is rare in the United States, but international travel to endemic areas is commonplace and consumption of unpasteurized Mexican dairy products from goats and sheep is widespread. Brucellosis has a wide range of symptoms, but a prompt diagnosis by an ELISA or BMAT serologic test and appropriate treatment can avoid morbidity and mortality. Treatment includes a tetracycline for at least 6 weeks in combination with an aminoglycoside or rifampin.
THE CASE
A 49-year-old Mexican immigrant woman was admitted to the hospital with a 5-month history of fatigue and a 30-pound unintentional weight loss. She was also experiencing arthralgia, swelling, and stiffness in her hands and feet that was worse in the morning. The patient, who was obese and suffered from type 2 diabetes and hypertension, said that at the onset of her illness 5 months earlier, she’d experienced approximately 2 weeks of night sweats and a few days of fever.
A month before being admitted to the hospital, she’d been seen in our southern New Mexico family medicine office. Her recent history of fever, joint symptoms, and weight loss raised concerns of an insidious infection, a new-onset rheumatologic condition, or an occult malignancy.
Initial lab tests revealed leukopenia (white blood cell count, 3200/mcL), microcytic anemia (hemoglobin, 9.4 g/dL), and an elevated erythrocyte sedimentation rate of 30 mm/hr (normal range, 0-20 mm/hr). A rheumatoid factor test was negative, and her thyroid, kidney, and liver function tests were all normal.
More testing… The patient frequently traveled between New Mexico and her hometown of Chihuahua, Mexico, but there had been no recent changes in her diet or environmental exposures. She denied drinking any unpasteurized milk in Chihuahua. But based on her travel history, we ordered enzyme-linked immunosorbent assay (ELISA) antibody titers for Brucella, immunoglobulin G, and immunoglobulin M, which all came back negative. Additionally, we ordered an abdominal and pelvic ultrasound and a chest x-ray that were nondiagnostic. Given the patient’s weight loss and anemia, we referred her to a gastroenterologist for upper and lower gastrointestinal endoscopic evaluations. Unfortunately, the patient was uninsured and did not go to see the gastroenterologist.
A month after seeing us, our patient’s fatigue, lack of appetite, and joint pain became debilitating and she was admitted to the hospital for further evaluation, including a consultation with an oncologist.
THE DIAGNOSIS
During our patient’s 6-day hospital stay, a bone scintigraphy showed a focus of uptake in her left parietal bone and computed tomography scans of her chest, abdomen, and pelvis revealed a left thyroid nodule, as well as multiple noncalcified pulmonary nodules. Blood cultures were also obtained.
Despite the initial negative antibody tests, the blood cultures drawn in the hospital revealed the presence of Brucella melitensis, and we diagnosed brucellosis in this patient.
DISCUSSION
Brucella melitensis is one of the 4 recognized, land-based species of the Brucella genus that can cause disease in humans. Goats, sheep, and camels are natural hosts of B melitensis and consumption of their unpasteurized, infected milk and milk products (especially soft cheeses, ice cream, milk, and butter) leads to human disease. (Once hospitalized, our patient admitted to frequently eating unpasteurized goat cheese from Chihuahua. The only other person in her household that ate the cheese was her 26-year-old daughter, who was also experiencing similar symptoms.)
Brucellosis can also result from inhaling infected, aerosolized material; therefore, individuals whose occupations involve close work with host animals or work in laboratories with the bacteria have an increased risk of infection.1 Due to the risk of acquiring the infection via inhalation, brucellosis is considered a bioterrorism threat.2 Additionally, there have been reports of human-to-human transmission via sexual intercourse, transplacental infection, blood and bone marrow transfusion, and breastfeeding.3
B melitensis is the cause of the majority of Brucella-related illnesses in the world, though symptoms of infection are similar among the different Brucella species. The pathogen can affect almost all organ systems after the initial 2- to 4-week incubation period. Symptoms of brucellosis can be highly variable, although fever is consistently present.1 Other red flags include arthritis (usually affecting the peripheral joints, the sacroiliac joints, and the lower spine), epididymo-orchitis, and hepatitis resulting in transaminase elevation. Abscess formation can be seen in the liver, spleen, and other organs.
Less common but more ominous complications include central nervous system infections and abscesses, endocarditis, and pulmonary infections. Endocarditis and the resulting aortic valve involvement is the major cause of mortality.1Brucella-related uveitis, thyroiditis, nephritis, vasculitis, and acalculous cholecystitis have also been reported.4-9
Rare in the United States. Pasteurization of dairy products and mass vaccination of livestock make Brucella infection rare in the United States. While there have only been 80 to 139 cases of brucellosis reported per year in the United States since 1993, it remains a persistent threat. International travel is common from the United States to the Middle East and other parts of the world where brucellosis is endemic.
Additionally, infection of livestock with Brucella remains widespread in Mexico and the consumption of unpasteurized Mexican dairy products from goats and sheep remains a high-risk activity for acquiring the disease.10 Consequently, Texas and California account for more than half of the brucellosis diagnoses in the United States. However, in 2010, cases were reported in 25 other states and the District of Columbia.11
Repeat serology tests are preferred for confirming the Dx
It is interesting that our patient’s initial Brucella serology by ELISA was negative, because it was ordered months after her initial symptoms. Antibodies should be seen within a month of symptom onset. The Centers for Disease Control and Prevention (CDC) recommends taking 2 serum samples to establish a serologic diagnosis of brucellosis. The first should be drawn within 7 days of symptom onset and the second should be taken 2 to 4 weeks later. A greater than 4-fold rise in the antibody titer confirms the diagnosis. While ELISA is an acceptable serologic test, the CDC recommends using a serum tube agglutination test called the Brucella microagglutination test (BMAT).12 Repeat serology was not performed on our patient because the diagnosis had been confirmed by blood culture.
A combination of antibiotics is the recommended treatment
Treatment of brucellosis should include a tetracycline for at least 6 weeks in combination with an aminoglycoside or rifampin 600 mg/d for an all-oral regimen.13 Doxycycline 100 mg twice a day is preferred due to fewer gastrointestinal adverse effects than tetracycline. Relapse is not uncommon (10%) and usually occurs within one year of completing the antibiotics.8 However, there is a case report of a patient having reactivated brucellosis manifested as acalculous cholecystitis 28 years after completing antibiotics.8
Our patient was started on oral doxycycline 100 mg twice a day and oral rifampin 600 mg/d for 6 weeks. Within days of starting the antibiotics, her joint symptoms and fatigue rapidly abated and her appetite returned. Follow-up radiological testing was not performed after her initial hospital studies due to her lack of financial resources.
The patient’s daughter had also been experiencing night sweats, chills, malaise, anorexia, joint pains, weight loss, and alopecia over the previous 2 months. Her blood cultures were positive for B melitensis as well, and she was started on the same antibiotic regimen as her mother. The daughter was also seen in our clinic by another physician and improved quickly within a week of starting treatment.
Both our patient and her daughter remained symptom-free 6 years after treatment.
THE TAKEAWAY
Brucellosis is rare in the United States, but international travel to endemic areas is commonplace and consumption of unpasteurized Mexican dairy products from goats and sheep is widespread. Brucellosis has a wide range of symptoms, but a prompt diagnosis by an ELISA or BMAT serologic test and appropriate treatment can avoid morbidity and mortality. Treatment includes a tetracycline for at least 6 weeks in combination with an aminoglycoside or rifampin.
1. Pappas G, Akritidis N, Bosilkovski M, et al. Brucellosis. N Engl J Med. 2005;352:2325-2336.
2. Centers for Disease Control and Prevention (CDC). Suspected brucellosis case prompts investigation of possible bioterrorismrelated activity—New Hampshire and Massachusetts, 1999. MMWR Morb Mortal Wkly Rep. 2000;49:509-512.
3. Chen S, Zhang H, Liu X, et al. Increasing threat of brucellosis to low-risk persons in urban settings, China. Emerg Infect Dis. 2014;20:126-130.
4. Rolando I, Vilchez G, Olarte L, et al. Brucellar uveitis: intraocular fluids and biopsy studies. Int J Infect Dis. 2009;13:e206-e211.
5. Azizi F, Katchoui A. Brucella infection of the thyroid gland. Thyroid. 1996;6:461-463.
6. Siegelmann N, Abraham AS, Rudensky B, et al. Brucellosis with nephrotic syndrome, nephritis and IgA nephropathy. Postgrad Med J. 1992;68:834-836.
7. Tanyel E, Tasdelen Fisgin N, Yildiz L, et al. Panniculitis as the initial manifestation of brucellosis: a case report. Am J Dermatopathol. 2008;30:169-171.
8. Ögredici Ö, Erb S, Langer I, et al. Brucellosis reactivation after 28 years. Emerg Infect Dis. 2010;16:2021-2022.
9. Dhand A, Ross JJ. Implantable cardioverter-defibrillator infection due to Brucella melitensis: case report and review of brucellosis of cardiac devices. Clin Infect Dis. 2007;44:e37-e39.
10. Solorio-Rivera JL, Segura-Correa JC, Sánchez-Gil LG. Seroprevalence of and risk factors for brucellosis of goats in herds of Michoacan, Mexico. Prev Vet Med. 2007;82:282-290.
11. Centers for Disease Control and Prevention. Brucellosis surveillance. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/brucellosis/resources/surveillance.html. Accessed October 30, 2015.
12. Centers for Disease Control and Prevention. Serology. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/brucellosis/clinicians/serology.html. Accessed October 30, 2015.
13. Corbel MJ. Brucellosis in humans and animals. World Health Organization (WHO);2006:36-41. Available at: http://www.who.int/csr/resources/publications/Brucellosis.pdf. Accessed November 2, 2015.
1. Pappas G, Akritidis N, Bosilkovski M, et al. Brucellosis. N Engl J Med. 2005;352:2325-2336.
2. Centers for Disease Control and Prevention (CDC). Suspected brucellosis case prompts investigation of possible bioterrorismrelated activity—New Hampshire and Massachusetts, 1999. MMWR Morb Mortal Wkly Rep. 2000;49:509-512.
3. Chen S, Zhang H, Liu X, et al. Increasing threat of brucellosis to low-risk persons in urban settings, China. Emerg Infect Dis. 2014;20:126-130.
4. Rolando I, Vilchez G, Olarte L, et al. Brucellar uveitis: intraocular fluids and biopsy studies. Int J Infect Dis. 2009;13:e206-e211.
5. Azizi F, Katchoui A. Brucella infection of the thyroid gland. Thyroid. 1996;6:461-463.
6. Siegelmann N, Abraham AS, Rudensky B, et al. Brucellosis with nephrotic syndrome, nephritis and IgA nephropathy. Postgrad Med J. 1992;68:834-836.
7. Tanyel E, Tasdelen Fisgin N, Yildiz L, et al. Panniculitis as the initial manifestation of brucellosis: a case report. Am J Dermatopathol. 2008;30:169-171.
8. Ögredici Ö, Erb S, Langer I, et al. Brucellosis reactivation after 28 years. Emerg Infect Dis. 2010;16:2021-2022.
9. Dhand A, Ross JJ. Implantable cardioverter-defibrillator infection due to Brucella melitensis: case report and review of brucellosis of cardiac devices. Clin Infect Dis. 2007;44:e37-e39.
10. Solorio-Rivera JL, Segura-Correa JC, Sánchez-Gil LG. Seroprevalence of and risk factors for brucellosis of goats in herds of Michoacan, Mexico. Prev Vet Med. 2007;82:282-290.
11. Centers for Disease Control and Prevention. Brucellosis surveillance. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/brucellosis/resources/surveillance.html. Accessed October 30, 2015.
12. Centers for Disease Control and Prevention. Serology. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/brucellosis/clinicians/serology.html. Accessed October 30, 2015.
13. Corbel MJ. Brucellosis in humans and animals. World Health Organization (WHO);2006:36-41. Available at: http://www.who.int/csr/resources/publications/Brucellosis.pdf. Accessed November 2, 2015.

Cutaneous Leishmaniasis: An Emerging Infectious Disease in Travelers
Leishmaniasis describes any disease caused by protozoan parasites of the genus Leishmania1 and can manifest in 3 different forms: cutaneous (the most common); mucosal, a destructive metastatic sequela of the cutaneous form; and visceral, which is potentially fatal.2 According to the World Health Organization, the leishmaniases are endemic in 88 countries.3 It is estimated that 95% of cutaneous cases occur in the Americas (most notably Central and South America), the Mediterranean basin, the Middle East, and Central Asia.2 Most cutaneous cases diagnosed among nonmilitary personnel in the United States are acquired in Mexico and Central America.4 In Central and South America, the causative human pathogens include species of the Leishmania (Viannia) complex (eg, Leishmania panamensis, Leishmania braziliensis, Leishmania guyanensis, Leishmania peruviana) and the Leishmania mexicana complex (eg, Leishmania mexicana, Leishmania amazonensis, Leishmania venezuelensis). All of these species can cause localized cutaneous lesions, but only L panamensis, L braziliensis, and L guyanensis are associated with metastatic mucosal lesions. In Central and South Americas, only Leishmaniasis chagasi (also known as Leishmaniasis infantum) is known to cause visceral leishmaniasis.5
Case Report
A 26-year-old man was referred to the dermatology clinic by his primary care provider for evaluation of a nonhealing sore on the left volar forearm of 6 weeks’ duration. The patient described the initial lesion as a red bump resembling a mosquito bite. Over 6 weeks the papule evolved into an indurated plaque with painless ulceration. The patient’s primary care provider had prescribed antibiotics for a presumed Staphylococcus aureus infection of the skin 5 weeks prior to presentation; however, the lesion continued to enlarge in size, resulting in referral to our dermatology clinic.
Skin examination revealed a solitary, 4-cm, painless, ulcerated plaque on the left volar forearm (Figure 1). No lymphadenopathy was noted. The patient reported that he had returned from a mission trip to rural Costa Rica 2 weeks prior to the appearance of the lesion. His medical history was otherwise unremarkable and his vital signs were within normal limits. Our initial differential diagnosis included pyoderma gangrenosum, Sweet syndrome, cutaneous leishmaniasis, and an insect bite.

Histopathologic study of a 5-mm punch biopsy specimen from the lesion showed a dense nodular and diffuse lymphohistiocytic infiltrate containing foci of suppuration. Within these suppurative foci were histiocytes parasitized by intracellular organisms that appeared to be of uniform size and shape on Giemsa staining, all of which are considered to be pathognomonic features of cutaneous leishmaniasis6 (Figures 2 and 3). The dermatopathologist’s diagnosis of cutaneous leishmaniasis was confirmed by the Centers for Disease Control and Prevention. The species was identified by polymerase chain reaction (PCR) as L panamensis.
The patient was treated with intravenous sodium stibogluconate 20 mg/kg for 20 consecutive days as recommended by expert consensus. The decision to treat a frequently self-limited cutaneous lesion with a highly toxic systemic drug was based on the small but real risk of metastatic mucosal lesions, which is caused by the Viannia subgenus, including L panamensis. Of note, sodium stibogluconate and other antimony drugs are not sold in the United States. Sodium stibogluconate is approved by the US Food and Drug Administration to be distributed by the Centers for Disease Control and Prevention under a protocol requiring baseline and weekly electrocardiograms and monitoring of patients’ creatinine, transaminase, lipase, amylase, and complete blood count levels.7 Our patient tolerated treatment but experienced mild to moderate flulike symptoms. The patient experienced no remarkable sequelae other than scarring in the affected area. He was warned to notify his health care providers of any persistent nasal symptoms, including nasal stuffiness, mucosal bleeding, and increased secretions, heralding the possibility of mucosal metastasis.
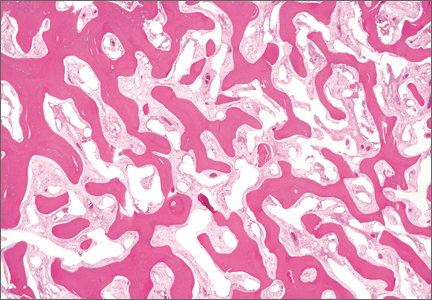
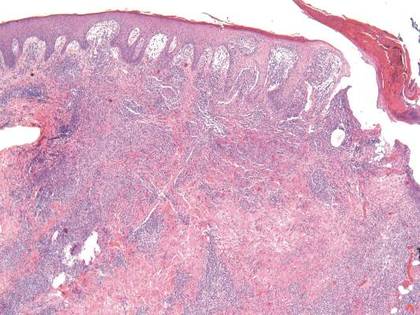 | 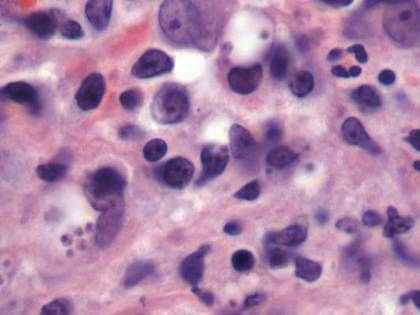 | |
Figure 2. Dense nodular and diffuse lymphohistiocytic infiltrate containing foci of suppuration (H&E, original magnification ×10). | Figure 3. Histiocytes parasitized by intracellular organisms of uniform shape and size on Giemsa staining (original magnification ×1000). |
Comment
The true incidence of cutaneous leishmaniasis in American travelers returning from Mexico and South and Central Americas is not known. The best incidence estimates are based on the number of physician requests for sodium stibogluconate and travel surveillance data collected by the Centers for Disease Control and Prevention. One study estimated the incidence of cutaneous leishmaniasis in Americans to be 1 case per every 100,000 travelers to Mexico.9 Data on the incidence of cutaneous leishmaniasis in American travelers seen in travel clinics for skin lesions gives a different perspective.10 Leishmaniasis is one of the most common dermatologic diseases seen in patients (European, North American, and other) returning from South America, accounting for 143 of every 1000 patients diagnosed with a skin disease acquired in South America.
Although males are thought to be at higher risk for cutaneous leishmaniasis infection than females, other demographic and behavioral risk factors are not well defined. In a case series of US travelers diagnosed with cutaneous leishmaniasis between January 1985 and April 1990, Herwaldt et al9 found that 46% (27/59) were conducting field studies, while 39% (23/59) were tourists, visitors, or tour guides. At least 15 of the 58 travelers interviewed (26%) were in forested areas for 1 week or less, and of these 15 respondents, at least 6 had a maximum exposure of 2 days.9
Evidence suggests that cutaneous leishmaniasis is inefficiently diagnosed in the United States. One study showed that some patients may consult up to 7 physicians before a definitive diagnosis is made, and the median time from noticing eruption of the lesions to definitive treatment was 112 days.9 Several factors may contribute to delays and inefficiencies in diagnosis. First, the lesions of cutaneous leishmaniasis are varied in morphology, and although ulcers are thought to be the most commonly presenting lesions,11 there are no specific morphologic features that are pathognomonic for cutaneous leishmaniasis. Second, the temporal association with travel to endemic countries is not necessarily apparent, with lesions developing gradually or weeks after the patient returns home. In the one study, 17% (10/58) of patients were home for more than 1 month before they noticed skin lesions.9 Finally, definitive diagnosis requires biopsy or scraping of the lesion followed by PCR, special histopathological staining (Giemsa), or culture. Polymerase chain reaction is currently the best means of identifying the causative Leishmania species.12-14 However, since skin biopsies are not routine in primary care settings and few practitioners are familiar with PCR for identification of leishmaniasis, diagnosis is typically made only after referral to a specialist.
Leishmaniasis transmission occurs in diverse geographical settings though a variety of mechanisms (Figure 4). The morphology of cutaneous leishmaniasis varies and may include papules, nodules, psoriasiform plaques, or ulcers. The differential diagnosis may include staphylococcal skin infection, insect bite, cutaneous neoplasm, pyoderma gangrenosum, sporotrichosis, blastomycosis, chromomycosis, lobomycosis, cutaneous tuberculosis, atypical mycobacterial infection, syphilis, yaws, leprosy, Hansen disease, and sarcoidosis. A definitive diagnosis can be made only after identifying the causative parasite. A scraping or punch biopsy taken from a cleaned lesion provides an adequate sample. Identification can then be accomplished by histopathology, tissue culture, or PCR.5

We present a rhyme that can be used to promote greater awareness of cutaneous leishmaniasis among US health care practitioners:
And on his leg finds an ulcerated plaque.
The possibilities are many,
Numbering far more than 20.
Leishmaniasis is a lurking issue,
So the savvy physician tests the tissue.
Although clinical resolution of cutaneous leishmaniasis usually occurs over months to years, the unsightly appearance of the lesions as well as the potential for scarring and mucosal metastasis (associated with some species) drives medical treatment.15 Pentavalent antimonial drugs, which have been the mainstay of treatment for more than 50 years, remain the most popular treatment for cutaneous leishmaniasis. Two antimony compounds, sodium stibogluconate and meglumine antimoniate, often lead to clinical cure in less than 1 month7; however, these drugs are far from ideal because of the inconvenience of obtaining them, emerging parasite resistance, long treatment course, parenteral route of administration, and serious side effects including infusion reactions, arrhythmias, pancreatitis, and liver toxicity. Moreover, the subclinical persistence of cutaneous leishmaniasis years after treatment and clinical cure is common. There have been reports of spontaneous disease reactivation in immunocompromised individuals, and Leishmania has been detected in old cutaneous leishmaniasis scars on PCR testing.16-18 Other therapies that have been used to treat cutaneous leishmaniasis include allopurinol, aminosidine sulphate, amphotericin B, the Bacillus Calmette–Guérin vaccine, cotrimoxazole, cryotherapy, dapsone, fluconazole, itraconazole, ketoconazole, laser therapy, metronidazole, miltefosine, paromomycin, pentamidine, pentoxifylline, photodynamic therapy, rifampicin, and surgical excision of the entire lesion.8 A 2009 Cochrane review of the various treatments for cutaneous leishmaniasis concluded that “no general consensus on optimal treatment has been achieved” and suggested “the creation of an international platform to improve the quality and standardization of future trials in order to develop a better evidence-based approach.”8
Conclusion
Cutaneous leishmaniasis should be included in the differential diagnosis for travelers returning from endemic areas who present with new skin lesions. Since no specific lesion types are pathognomonic for cutaneous leishmaniasis, tissue biopsy for histopathology and PCR are essential for diagnosis. Prevention of cutaneous leishmaniasis hinges on appropriate counseling of travelers to endemic regions.
1. Etymologia-Leishmaniasis. Emerg Infect Dis. 2008;14:666.
2. Burden and distribution. World Health Organization Web site. http://www.who.int/leishmaniasis/burden/en/. Accessed November 10, 2015.
3. Emergencies preparedness, response. World Health Organization Web site. http://www.who.int/csr/resources/publications/CSR_ISR_2000_1leish/en/. Accessed November 3, 2015.
4. Pavli A, Maltezou HC. Leishmaniasis, an emerging infection in travelers. Int J Infect Dis. 2010;14:e1032-e1039.
5. Magill AJ. Leishmania species: visceral (Kala-Azar), cutaneous, and mucosal leishmaniasis. In: Mandell GL, Bennett JE, Dolin R, eds. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, PA: Churchill Livingstone; 2009:3463-3480.
6. Mysore V. Invisible dermatoses. Indian J Dermatol Venereol Leprol. 2010;76:239-248.
7. Parasites – Leishmaniasis. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/parasites/leishmaniasis/health_professionals/. Updated September 14, 2015. Accessed November 13, 2015.
8. González U, Pinart M, Rengifo-Pardo M, et al. Interventions for American cutaneous and mucocutaneous leishmaniasis. Cochrane Database Syst Rev. 2009;15:CD004834.
9. Herwaldt BL, Stokes SL, Juranek DD. American cutaneous leishmaniasis in U.S. travelers. Ann Intern Med. 1993;118:779-784.
10. Freedman DO, Weld LH, Kozarsky PE, et al. Spectrum of disease and relation to place of exposure among ill returned travelers. New Engl J Med. 2006;354:119-130.
11. El Hajj L, Thellier M, Carriere J, et al. Localized cutaneous leishmaniasis imported into Paris: a review of 39 cases. Int J Dermatol. 2004;43:120-125.
12. Harris E, Kropp G, Belli A, et al. Single-step multiplex PCR assay for characterization of New World Leishmania complexes. J Clin Microbiol. 1998;36:1989-1995.
13. Marfurt J, Niederwieser I, Makia D, et al. Diagnostic genotyping of Old and New World Leishmania species by PCR-RFLP. Diagn Microbiol Infect Dis. 2003;46:115-124.
14. Schonian G, Nasereddin A, Dinse N, et al. PCR diagnosis and characterization of Leishmania in local and imported clinical samples. Diagn Microbiol Infect Dis. 2003;47:349-358.
15. Reithinger R, Aadil K, Kolaczinski J, et al. Social impact of leishmaniasis, Afghanistan. Emerg Infect Dis. 2005;11:634-636.
16. Morales MA, Cruz I, Rubio JM, et al. Relapses versus reinfections in patients coinfected with Leishmania infantum and human immunodeficiency virus type 1 [published online ahead of print April 22, 2002]. J Infect Dis. 2002;185:1533-1537.
17. Coutinho SG, Pirmez C, Da-Cruz AM. Parasitological and immunological follow-up of American tegumentary leishmaniasis patients. Trans R Soc Trop Med Hyg. 2002;96(suppl 1):S173-S178.
18. Mendonça MG, de Brito ME, Rodrigues EH, et al. Persistance of leishmania parasites in scars after clinical cure of American cutaneous leishmaniasis: is there a sterile cure [published online ahead of print March 2, 2004]? J Infect Dis. 2004;189:1018-1023.
Leishmaniasis describes any disease caused by protozoan parasites of the genus Leishmania1 and can manifest in 3 different forms: cutaneous (the most common); mucosal, a destructive metastatic sequela of the cutaneous form; and visceral, which is potentially fatal.2 According to the World Health Organization, the leishmaniases are endemic in 88 countries.3 It is estimated that 95% of cutaneous cases occur in the Americas (most notably Central and South America), the Mediterranean basin, the Middle East, and Central Asia.2 Most cutaneous cases diagnosed among nonmilitary personnel in the United States are acquired in Mexico and Central America.4 In Central and South America, the causative human pathogens include species of the Leishmania (Viannia) complex (eg, Leishmania panamensis, Leishmania braziliensis, Leishmania guyanensis, Leishmania peruviana) and the Leishmania mexicana complex (eg, Leishmania mexicana, Leishmania amazonensis, Leishmania venezuelensis). All of these species can cause localized cutaneous lesions, but only L panamensis, L braziliensis, and L guyanensis are associated with metastatic mucosal lesions. In Central and South Americas, only Leishmaniasis chagasi (also known as Leishmaniasis infantum) is known to cause visceral leishmaniasis.5
Case Report
A 26-year-old man was referred to the dermatology clinic by his primary care provider for evaluation of a nonhealing sore on the left volar forearm of 6 weeks’ duration. The patient described the initial lesion as a red bump resembling a mosquito bite. Over 6 weeks the papule evolved into an indurated plaque with painless ulceration. The patient’s primary care provider had prescribed antibiotics for a presumed Staphylococcus aureus infection of the skin 5 weeks prior to presentation; however, the lesion continued to enlarge in size, resulting in referral to our dermatology clinic.
Skin examination revealed a solitary, 4-cm, painless, ulcerated plaque on the left volar forearm (Figure 1). No lymphadenopathy was noted. The patient reported that he had returned from a mission trip to rural Costa Rica 2 weeks prior to the appearance of the lesion. His medical history was otherwise unremarkable and his vital signs were within normal limits. Our initial differential diagnosis included pyoderma gangrenosum, Sweet syndrome, cutaneous leishmaniasis, and an insect bite.
Histopathologic study of a 5-mm punch biopsy specimen from the lesion showed a dense nodular and diffuse lymphohistiocytic infiltrate containing foci of suppuration. Within these suppurative foci were histiocytes parasitized by intracellular organisms that appeared to be of uniform size and shape on Giemsa staining, all of which are considered to be pathognomonic features of cutaneous leishmaniasis6 (Figures 2 and 3). The dermatopathologist’s diagnosis of cutaneous leishmaniasis was confirmed by the Centers for Disease Control and Prevention. The species was identified by polymerase chain reaction (PCR) as L panamensis.
The patient was treated with intravenous sodium stibogluconate 20 mg/kg for 20 consecutive days as recommended by expert consensus. The decision to treat a frequently self-limited cutaneous lesion with a highly toxic systemic drug was based on the small but real risk of metastatic mucosal lesions, which is caused by the Viannia subgenus, including L panamensis. Of note, sodium stibogluconate and other antimony drugs are not sold in the United States. Sodium stibogluconate is approved by the US Food and Drug Administration to be distributed by the Centers for Disease Control and Prevention under a protocol requiring baseline and weekly electrocardiograms and monitoring of patients’ creatinine, transaminase, lipase, amylase, and complete blood count levels.7 Our patient tolerated treatment but experienced mild to moderate flulike symptoms. The patient experienced no remarkable sequelae other than scarring in the affected area. He was warned to notify his health care providers of any persistent nasal symptoms, including nasal stuffiness, mucosal bleeding, and increased secretions, heralding the possibility of mucosal metastasis.
| | |
Figure 2. Dense nodular and diffuse lymphohistiocytic infiltrate containing foci of suppuration (H&E, original magnification ×10). | Figure 3. Histiocytes parasitized by intracellular organisms of uniform shape and size on Giemsa staining (original magnification ×1000). |
Comment
The true incidence of cutaneous leishmaniasis in American travelers returning from Mexico and South and Central Americas is not known. The best incidence estimates are based on the number of physician requests for sodium stibogluconate and travel surveillance data collected by the Centers for Disease Control and Prevention. One study estimated the incidence of cutaneous leishmaniasis in Americans to be 1 case per every 100,000 travelers to Mexico.9 Data on the incidence of cutaneous leishmaniasis in American travelers seen in travel clinics for skin lesions gives a different perspective.10 Leishmaniasis is one of the most common dermatologic diseases seen in patients (European, North American, and other) returning from South America, accounting for 143 of every 1000 patients diagnosed with a skin disease acquired in South America.
Although males are thought to be at higher risk for cutaneous leishmaniasis infection than females, other demographic and behavioral risk factors are not well defined. In a case series of US travelers diagnosed with cutaneous leishmaniasis between January 1985 and April 1990, Herwaldt et al9 found that 46% (27/59) were conducting field studies, while 39% (23/59) were tourists, visitors, or tour guides. At least 15 of the 58 travelers interviewed (26%) were in forested areas for 1 week or less, and of these 15 respondents, at least 6 had a maximum exposure of 2 days.9
Evidence suggests that cutaneous leishmaniasis is inefficiently diagnosed in the United States. One study showed that some patients may consult up to 7 physicians before a definitive diagnosis is made, and the median time from noticing eruption of the lesions to definitive treatment was 112 days.9 Several factors may contribute to delays and inefficiencies in diagnosis. First, the lesions of cutaneous leishmaniasis are varied in morphology, and although ulcers are thought to be the most commonly presenting lesions,11 there are no specific morphologic features that are pathognomonic for cutaneous leishmaniasis. Second, the temporal association with travel to endemic countries is not necessarily apparent, with lesions developing gradually or weeks after the patient returns home. In the one study, 17% (10/58) of patients were home for more than 1 month before they noticed skin lesions.9 Finally, definitive diagnosis requires biopsy or scraping of the lesion followed by PCR, special histopathological staining (Giemsa), or culture. Polymerase chain reaction is currently the best means of identifying the causative Leishmania species.12-14 However, since skin biopsies are not routine in primary care settings and few practitioners are familiar with PCR for identification of leishmaniasis, diagnosis is typically made only after referral to a specialist.
Leishmaniasis transmission occurs in diverse geographical settings though a variety of mechanisms (Figure 4). The morphology of cutaneous leishmaniasis varies and may include papules, nodules, psoriasiform plaques, or ulcers. The differential diagnosis may include staphylococcal skin infection, insect bite, cutaneous neoplasm, pyoderma gangrenosum, sporotrichosis, blastomycosis, chromomycosis, lobomycosis, cutaneous tuberculosis, atypical mycobacterial infection, syphilis, yaws, leprosy, Hansen disease, and sarcoidosis. A definitive diagnosis can be made only after identifying the causative parasite. A scraping or punch biopsy taken from a cleaned lesion provides an adequate sample. Identification can then be accomplished by histopathology, tissue culture, or PCR.5
We present a rhyme that can be used to promote greater awareness of cutaneous leishmaniasis among US health care practitioners:
And on his leg finds an ulcerated plaque.
The possibilities are many,
Numbering far more than 20.
Leishmaniasis is a lurking issue,
So the savvy physician tests the tissue.
Although clinical resolution of cutaneous leishmaniasis usually occurs over months to years, the unsightly appearance of the lesions as well as the potential for scarring and mucosal metastasis (associated with some species) drives medical treatment.15 Pentavalent antimonial drugs, which have been the mainstay of treatment for more than 50 years, remain the most popular treatment for cutaneous leishmaniasis. Two antimony compounds, sodium stibogluconate and meglumine antimoniate, often lead to clinical cure in less than 1 month7; however, these drugs are far from ideal because of the inconvenience of obtaining them, emerging parasite resistance, long treatment course, parenteral route of administration, and serious side effects including infusion reactions, arrhythmias, pancreatitis, and liver toxicity. Moreover, the subclinical persistence of cutaneous leishmaniasis years after treatment and clinical cure is common. There have been reports of spontaneous disease reactivation in immunocompromised individuals, and Leishmania has been detected in old cutaneous leishmaniasis scars on PCR testing.16-18 Other therapies that have been used to treat cutaneous leishmaniasis include allopurinol, aminosidine sulphate, amphotericin B, the Bacillus Calmette–Guérin vaccine, cotrimoxazole, cryotherapy, dapsone, fluconazole, itraconazole, ketoconazole, laser therapy, metronidazole, miltefosine, paromomycin, pentamidine, pentoxifylline, photodynamic therapy, rifampicin, and surgical excision of the entire lesion.8 A 2009 Cochrane review of the various treatments for cutaneous leishmaniasis concluded that “no general consensus on optimal treatment has been achieved” and suggested “the creation of an international platform to improve the quality and standardization of future trials in order to develop a better evidence-based approach.”8
Conclusion
Cutaneous leishmaniasis should be included in the differential diagnosis for travelers returning from endemic areas who present with new skin lesions. Since no specific lesion types are pathognomonic for cutaneous leishmaniasis, tissue biopsy for histopathology and PCR are essential for diagnosis. Prevention of cutaneous leishmaniasis hinges on appropriate counseling of travelers to endemic regions.
Leishmaniasis describes any disease caused by protozoan parasites of the genus Leishmania1 and can manifest in 3 different forms: cutaneous (the most common); mucosal, a destructive metastatic sequela of the cutaneous form; and visceral, which is potentially fatal.2 According to the World Health Organization, the leishmaniases are endemic in 88 countries.3 It is estimated that 95% of cutaneous cases occur in the Americas (most notably Central and South America), the Mediterranean basin, the Middle East, and Central Asia.2 Most cutaneous cases diagnosed among nonmilitary personnel in the United States are acquired in Mexico and Central America.4 In Central and South America, the causative human pathogens include species of the Leishmania (Viannia) complex (eg, Leishmania panamensis, Leishmania braziliensis, Leishmania guyanensis, Leishmania peruviana) and the Leishmania mexicana complex (eg, Leishmania mexicana, Leishmania amazonensis, Leishmania venezuelensis). All of these species can cause localized cutaneous lesions, but only L panamensis, L braziliensis, and L guyanensis are associated with metastatic mucosal lesions. In Central and South Americas, only Leishmaniasis chagasi (also known as Leishmaniasis infantum) is known to cause visceral leishmaniasis.5
Case Report
A 26-year-old man was referred to the dermatology clinic by his primary care provider for evaluation of a nonhealing sore on the left volar forearm of 6 weeks’ duration. The patient described the initial lesion as a red bump resembling a mosquito bite. Over 6 weeks the papule evolved into an indurated plaque with painless ulceration. The patient’s primary care provider had prescribed antibiotics for a presumed Staphylococcus aureus infection of the skin 5 weeks prior to presentation; however, the lesion continued to enlarge in size, resulting in referral to our dermatology clinic.
Skin examination revealed a solitary, 4-cm, painless, ulcerated plaque on the left volar forearm (Figure 1). No lymphadenopathy was noted. The patient reported that he had returned from a mission trip to rural Costa Rica 2 weeks prior to the appearance of the lesion. His medical history was otherwise unremarkable and his vital signs were within normal limits. Our initial differential diagnosis included pyoderma gangrenosum, Sweet syndrome, cutaneous leishmaniasis, and an insect bite.
Histopathologic study of a 5-mm punch biopsy specimen from the lesion showed a dense nodular and diffuse lymphohistiocytic infiltrate containing foci of suppuration. Within these suppurative foci were histiocytes parasitized by intracellular organisms that appeared to be of uniform size and shape on Giemsa staining, all of which are considered to be pathognomonic features of cutaneous leishmaniasis6 (Figures 2 and 3). The dermatopathologist’s diagnosis of cutaneous leishmaniasis was confirmed by the Centers for Disease Control and Prevention. The species was identified by polymerase chain reaction (PCR) as L panamensis.
The patient was treated with intravenous sodium stibogluconate 20 mg/kg for 20 consecutive days as recommended by expert consensus. The decision to treat a frequently self-limited cutaneous lesion with a highly toxic systemic drug was based on the small but real risk of metastatic mucosal lesions, which is caused by the Viannia subgenus, including L panamensis. Of note, sodium stibogluconate and other antimony drugs are not sold in the United States. Sodium stibogluconate is approved by the US Food and Drug Administration to be distributed by the Centers for Disease Control and Prevention under a protocol requiring baseline and weekly electrocardiograms and monitoring of patients’ creatinine, transaminase, lipase, amylase, and complete blood count levels.7 Our patient tolerated treatment but experienced mild to moderate flulike symptoms. The patient experienced no remarkable sequelae other than scarring in the affected area. He was warned to notify his health care providers of any persistent nasal symptoms, including nasal stuffiness, mucosal bleeding, and increased secretions, heralding the possibility of mucosal metastasis.
| | |
Figure 2. Dense nodular and diffuse lymphohistiocytic infiltrate containing foci of suppuration (H&E, original magnification ×10). | Figure 3. Histiocytes parasitized by intracellular organisms of uniform shape and size on Giemsa staining (original magnification ×1000). |
Comment
The true incidence of cutaneous leishmaniasis in American travelers returning from Mexico and South and Central Americas is not known. The best incidence estimates are based on the number of physician requests for sodium stibogluconate and travel surveillance data collected by the Centers for Disease Control and Prevention. One study estimated the incidence of cutaneous leishmaniasis in Americans to be 1 case per every 100,000 travelers to Mexico.9 Data on the incidence of cutaneous leishmaniasis in American travelers seen in travel clinics for skin lesions gives a different perspective.10 Leishmaniasis is one of the most common dermatologic diseases seen in patients (European, North American, and other) returning from South America, accounting for 143 of every 1000 patients diagnosed with a skin disease acquired in South America.
Although males are thought to be at higher risk for cutaneous leishmaniasis infection than females, other demographic and behavioral risk factors are not well defined. In a case series of US travelers diagnosed with cutaneous leishmaniasis between January 1985 and April 1990, Herwaldt et al9 found that 46% (27/59) were conducting field studies, while 39% (23/59) were tourists, visitors, or tour guides. At least 15 of the 58 travelers interviewed (26%) were in forested areas for 1 week or less, and of these 15 respondents, at least 6 had a maximum exposure of 2 days.9
Evidence suggests that cutaneous leishmaniasis is inefficiently diagnosed in the United States. One study showed that some patients may consult up to 7 physicians before a definitive diagnosis is made, and the median time from noticing eruption of the lesions to definitive treatment was 112 days.9 Several factors may contribute to delays and inefficiencies in diagnosis. First, the lesions of cutaneous leishmaniasis are varied in morphology, and although ulcers are thought to be the most commonly presenting lesions,11 there are no specific morphologic features that are pathognomonic for cutaneous leishmaniasis. Second, the temporal association with travel to endemic countries is not necessarily apparent, with lesions developing gradually or weeks after the patient returns home. In the one study, 17% (10/58) of patients were home for more than 1 month before they noticed skin lesions.9 Finally, definitive diagnosis requires biopsy or scraping of the lesion followed by PCR, special histopathological staining (Giemsa), or culture. Polymerase chain reaction is currently the best means of identifying the causative Leishmania species.12-14 However, since skin biopsies are not routine in primary care settings and few practitioners are familiar with PCR for identification of leishmaniasis, diagnosis is typically made only after referral to a specialist.
Leishmaniasis transmission occurs in diverse geographical settings though a variety of mechanisms (Figure 4). The morphology of cutaneous leishmaniasis varies and may include papules, nodules, psoriasiform plaques, or ulcers. The differential diagnosis may include staphylococcal skin infection, insect bite, cutaneous neoplasm, pyoderma gangrenosum, sporotrichosis, blastomycosis, chromomycosis, lobomycosis, cutaneous tuberculosis, atypical mycobacterial infection, syphilis, yaws, leprosy, Hansen disease, and sarcoidosis. A definitive diagnosis can be made only after identifying the causative parasite. A scraping or punch biopsy taken from a cleaned lesion provides an adequate sample. Identification can then be accomplished by histopathology, tissue culture, or PCR.5
We present a rhyme that can be used to promote greater awareness of cutaneous leishmaniasis among US health care practitioners:
And on his leg finds an ulcerated plaque.
The possibilities are many,
Numbering far more than 20.
Leishmaniasis is a lurking issue,
So the savvy physician tests the tissue.
Although clinical resolution of cutaneous leishmaniasis usually occurs over months to years, the unsightly appearance of the lesions as well as the potential for scarring and mucosal metastasis (associated with some species) drives medical treatment.15 Pentavalent antimonial drugs, which have been the mainstay of treatment for more than 50 years, remain the most popular treatment for cutaneous leishmaniasis. Two antimony compounds, sodium stibogluconate and meglumine antimoniate, often lead to clinical cure in less than 1 month7; however, these drugs are far from ideal because of the inconvenience of obtaining them, emerging parasite resistance, long treatment course, parenteral route of administration, and serious side effects including infusion reactions, arrhythmias, pancreatitis, and liver toxicity. Moreover, the subclinical persistence of cutaneous leishmaniasis years after treatment and clinical cure is common. There have been reports of spontaneous disease reactivation in immunocompromised individuals, and Leishmania has been detected in old cutaneous leishmaniasis scars on PCR testing.16-18 Other therapies that have been used to treat cutaneous leishmaniasis include allopurinol, aminosidine sulphate, amphotericin B, the Bacillus Calmette–Guérin vaccine, cotrimoxazole, cryotherapy, dapsone, fluconazole, itraconazole, ketoconazole, laser therapy, metronidazole, miltefosine, paromomycin, pentamidine, pentoxifylline, photodynamic therapy, rifampicin, and surgical excision of the entire lesion.8 A 2009 Cochrane review of the various treatments for cutaneous leishmaniasis concluded that “no general consensus on optimal treatment has been achieved” and suggested “the creation of an international platform to improve the quality and standardization of future trials in order to develop a better evidence-based approach.”8
Conclusion
Cutaneous leishmaniasis should be included in the differential diagnosis for travelers returning from endemic areas who present with new skin lesions. Since no specific lesion types are pathognomonic for cutaneous leishmaniasis, tissue biopsy for histopathology and PCR are essential for diagnosis. Prevention of cutaneous leishmaniasis hinges on appropriate counseling of travelers to endemic regions.
1. Etymologia-Leishmaniasis. Emerg Infect Dis. 2008;14:666.
2. Burden and distribution. World Health Organization Web site. http://www.who.int/leishmaniasis/burden/en/. Accessed November 10, 2015.
3. Emergencies preparedness, response. World Health Organization Web site. http://www.who.int/csr/resources/publications/CSR_ISR_2000_1leish/en/. Accessed November 3, 2015.
4. Pavli A, Maltezou HC. Leishmaniasis, an emerging infection in travelers. Int J Infect Dis. 2010;14:e1032-e1039.
5. Magill AJ. Leishmania species: visceral (Kala-Azar), cutaneous, and mucosal leishmaniasis. In: Mandell GL, Bennett JE, Dolin R, eds. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, PA: Churchill Livingstone; 2009:3463-3480.
6. Mysore V. Invisible dermatoses. Indian J Dermatol Venereol Leprol. 2010;76:239-248.
7. Parasites – Leishmaniasis. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/parasites/leishmaniasis/health_professionals/. Updated September 14, 2015. Accessed November 13, 2015.
8. González U, Pinart M, Rengifo-Pardo M, et al. Interventions for American cutaneous and mucocutaneous leishmaniasis. Cochrane Database Syst Rev. 2009;15:CD004834.
9. Herwaldt BL, Stokes SL, Juranek DD. American cutaneous leishmaniasis in U.S. travelers. Ann Intern Med. 1993;118:779-784.
10. Freedman DO, Weld LH, Kozarsky PE, et al. Spectrum of disease and relation to place of exposure among ill returned travelers. New Engl J Med. 2006;354:119-130.
11. El Hajj L, Thellier M, Carriere J, et al. Localized cutaneous leishmaniasis imported into Paris: a review of 39 cases. Int J Dermatol. 2004;43:120-125.
12. Harris E, Kropp G, Belli A, et al. Single-step multiplex PCR assay for characterization of New World Leishmania complexes. J Clin Microbiol. 1998;36:1989-1995.
13. Marfurt J, Niederwieser I, Makia D, et al. Diagnostic genotyping of Old and New World Leishmania species by PCR-RFLP. Diagn Microbiol Infect Dis. 2003;46:115-124.
14. Schonian G, Nasereddin A, Dinse N, et al. PCR diagnosis and characterization of Leishmania in local and imported clinical samples. Diagn Microbiol Infect Dis. 2003;47:349-358.
15. Reithinger R, Aadil K, Kolaczinski J, et al. Social impact of leishmaniasis, Afghanistan. Emerg Infect Dis. 2005;11:634-636.
16. Morales MA, Cruz I, Rubio JM, et al. Relapses versus reinfections in patients coinfected with Leishmania infantum and human immunodeficiency virus type 1 [published online ahead of print April 22, 2002]. J Infect Dis. 2002;185:1533-1537.
17. Coutinho SG, Pirmez C, Da-Cruz AM. Parasitological and immunological follow-up of American tegumentary leishmaniasis patients. Trans R Soc Trop Med Hyg. 2002;96(suppl 1):S173-S178.
18. Mendonça MG, de Brito ME, Rodrigues EH, et al. Persistance of leishmania parasites in scars after clinical cure of American cutaneous leishmaniasis: is there a sterile cure [published online ahead of print March 2, 2004]? J Infect Dis. 2004;189:1018-1023.
1. Etymologia-Leishmaniasis. Emerg Infect Dis. 2008;14:666.
2. Burden and distribution. World Health Organization Web site. http://www.who.int/leishmaniasis/burden/en/. Accessed November 10, 2015.
3. Emergencies preparedness, response. World Health Organization Web site. http://www.who.int/csr/resources/publications/CSR_ISR_2000_1leish/en/. Accessed November 3, 2015.
4. Pavli A, Maltezou HC. Leishmaniasis, an emerging infection in travelers. Int J Infect Dis. 2010;14:e1032-e1039.
5. Magill AJ. Leishmania species: visceral (Kala-Azar), cutaneous, and mucosal leishmaniasis. In: Mandell GL, Bennett JE, Dolin R, eds. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, PA: Churchill Livingstone; 2009:3463-3480.
6. Mysore V. Invisible dermatoses. Indian J Dermatol Venereol Leprol. 2010;76:239-248.
7. Parasites – Leishmaniasis. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/parasites/leishmaniasis/health_professionals/. Updated September 14, 2015. Accessed November 13, 2015.
8. González U, Pinart M, Rengifo-Pardo M, et al. Interventions for American cutaneous and mucocutaneous leishmaniasis. Cochrane Database Syst Rev. 2009;15:CD004834.
9. Herwaldt BL, Stokes SL, Juranek DD. American cutaneous leishmaniasis in U.S. travelers. Ann Intern Med. 1993;118:779-784.
10. Freedman DO, Weld LH, Kozarsky PE, et al. Spectrum of disease and relation to place of exposure among ill returned travelers. New Engl J Med. 2006;354:119-130.
11. El Hajj L, Thellier M, Carriere J, et al. Localized cutaneous leishmaniasis imported into Paris: a review of 39 cases. Int J Dermatol. 2004;43:120-125.
12. Harris E, Kropp G, Belli A, et al. Single-step multiplex PCR assay for characterization of New World Leishmania complexes. J Clin Microbiol. 1998;36:1989-1995.
13. Marfurt J, Niederwieser I, Makia D, et al. Diagnostic genotyping of Old and New World Leishmania species by PCR-RFLP. Diagn Microbiol Infect Dis. 2003;46:115-124.
14. Schonian G, Nasereddin A, Dinse N, et al. PCR diagnosis and characterization of Leishmania in local and imported clinical samples. Diagn Microbiol Infect Dis. 2003;47:349-358.
15. Reithinger R, Aadil K, Kolaczinski J, et al. Social impact of leishmaniasis, Afghanistan. Emerg Infect Dis. 2005;11:634-636.
16. Morales MA, Cruz I, Rubio JM, et al. Relapses versus reinfections in patients coinfected with Leishmania infantum and human immunodeficiency virus type 1 [published online ahead of print April 22, 2002]. J Infect Dis. 2002;185:1533-1537.
17. Coutinho SG, Pirmez C, Da-Cruz AM. Parasitological and immunological follow-up of American tegumentary leishmaniasis patients. Trans R Soc Trop Med Hyg. 2002;96(suppl 1):S173-S178.
18. Mendonça MG, de Brito ME, Rodrigues EH, et al. Persistance of leishmania parasites in scars after clinical cure of American cutaneous leishmaniasis: is there a sterile cure [published online ahead of print March 2, 2004]? J Infect Dis. 2004;189:1018-1023.
Practice Points
- Cutaneous leishmaniasis is an emerging infectious disease that may be misdiagnosed due to its rarity and varied clinical presentation as well as the limited use of tissue biopsy in general practice.
- United States health care practitioners who evaluate patients with new isolated skin lesions and a history of recent travel to Mexico or South or Central Americas should consider cutaneous leishmaniasis in the differential diagnosis.
- Whenever possible, travelers to rural areas of Mexico and South and Central Americas should be educated about strategies to avoid arthropod bites, such as wearing protective clothing and using insect repellents.
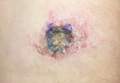
Treatment-related MDS/AML in a patient after treatment for large-cell neuroendocrine lung cancer
Secondary leukemia is a common late complication after exposure to cancer therapies such as chemotherapy and radiotherapy. With the increase in the overall survival of cancer patients over the past 3 decades, treatment-related malignant neoplasms have increased in incidence. Secondary leukemias due to breast cancer and Hodgkin lymphoma have been studied in detail, but to our knowledge only a few case studies have reported secondary leukemias with previous lung cancer.1-4 Lung cancer is the leading cause of cancer death in the United States.5
Click on the PDF icon at the top of this introduction to read the full article.
Secondary leukemia is a common late complication after exposure to cancer therapies such as chemotherapy and radiotherapy. With the increase in the overall survival of cancer patients over the past 3 decades, treatment-related malignant neoplasms have increased in incidence. Secondary leukemias due to breast cancer and Hodgkin lymphoma have been studied in detail, but to our knowledge only a few case studies have reported secondary leukemias with previous lung cancer.1-4 Lung cancer is the leading cause of cancer death in the United States.5
Click on the PDF icon at the top of this introduction to read the full article.
Secondary leukemia is a common late complication after exposure to cancer therapies such as chemotherapy and radiotherapy. With the increase in the overall survival of cancer patients over the past 3 decades, treatment-related malignant neoplasms have increased in incidence. Secondary leukemias due to breast cancer and Hodgkin lymphoma have been studied in detail, but to our knowledge only a few case studies have reported secondary leukemias with previous lung cancer.1-4 Lung cancer is the leading cause of cancer death in the United States.5
Click on the PDF icon at the top of this introduction to read the full article.
Idiopathic Intracranial Hypertension in Pregnancy
A 27-year-old white woman presented to the clinic with headaches and decreased vision through her reading glasses while performing near tasks. Her medical history was significant for herpes simplex, hyperlipidemia, and migraine headaches with aura. Her migraines began following an earlier motor vehicle accident, and her most recent magnetic resonance imaging (MRI) showed no abnormalities. Her current medications included prophylactic acyclovir for herpes and acetaminophen and caffeine tablets as needed for headache. She reported no other trauma or surgery and no known allergies. The patient’s best-corrected Snellen visual acuities in both eyes were 20/20 (distance) and 20/30 (near).
Preliminary testing, including pupils, extraocular motilities, confrontation fields, and color vision, were all within normal limits. Her slit-lamp examination was unremarkable. A dilated fundus examination revealed crowded, elevated discs without vessel obscuration, hemorrhage, hyperemia, or drusen (Figure 1). The fundus examination was otherwise unremarkable. Optical coherence tomography of the optic nerves showed increased nerve fiber layer thickness in both eyes (Figure 2). Her blood pressure (BP) at this visit was 106/77 mm/Hg.
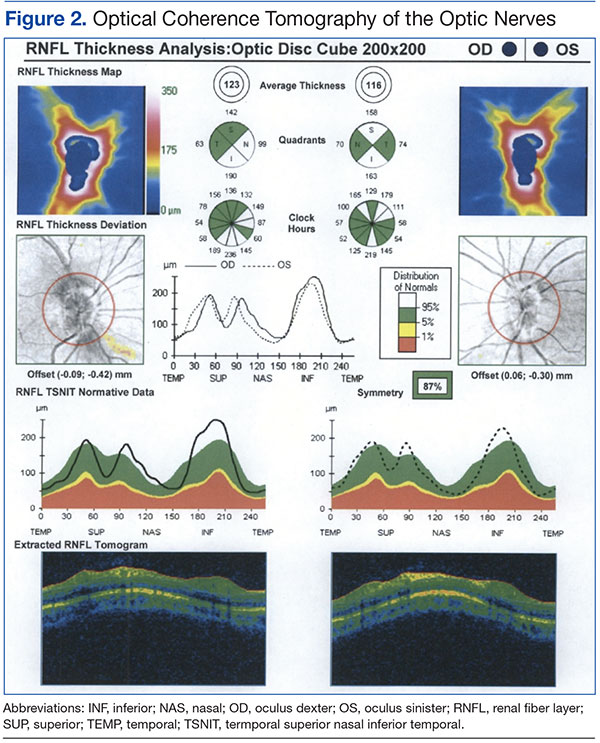
The diagnosis based on these findings was bilateral optic nerve elevation with long-standing migraine headaches. The plan was for the patient to return to the clinic for repeat visual field testing and B-scan ultrasonography to rule out buried optic nerve head drusen.
Two months later, the patient presented to the clinic 19 weeks pregnant and reported that her headaches had increased in frequency, but she had no diplopia. All preliminary testing, including visual acuities, pupil reaction, color vision, and slit-lamp examination remained normal. Fundus examination showed the patient’s nerves were unchanged in appearance from the initial presentation. Visual fields revealed an enlarged blind spot in the right eye and paracentral defects in the left eye. The B-scan testing was negative for optic nerve drusen. Due to the increased frequency of headaches, pregnancy, and suspicious optic nerves, an urgent consult was placed to neurology.
At the neurology appointment 1 month later, the patient was diagnosed with migraine headache syndrome and idiopathic intracranial hypertension (IIH). The neurologist believed her headaches might have been resulting from analgesic rebound. He suggested that the patient discontinue or decrease use of oral butalbital, acetaminophen and caffeine tablets, and other forms of caffeine. It was decided that divalproxen sodium and verapamil were not feasible due to pregnancy. The neurologist started her on oral acetazolamide 500 mg twice daily.
The patient returned to her obstetrician 1 month later for a routine follow-up; the headaches had worsened and were now accompanied by nausea and vomiting twice daily on average. Her medications still included acetaminophen and caffeine tablets, although it had been recommended she discontinue them, prochlorperazine, and acetazolamide. Due to the worsening of her symptoms and visual fields (eFigure 1), the obstetrician recommended that the patient deliver by cesarean section at 38 to 39 weeks.
(eFigure 1.Visual Fields at Follow-up 1 and 2)
Right eye
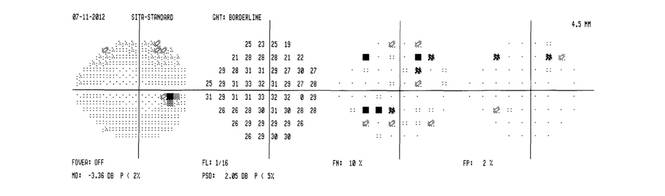
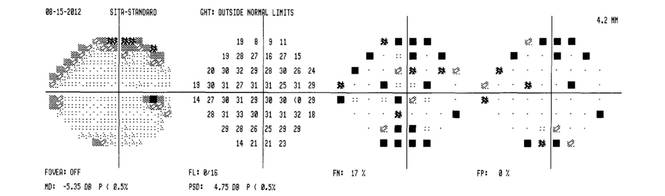
Left Eye
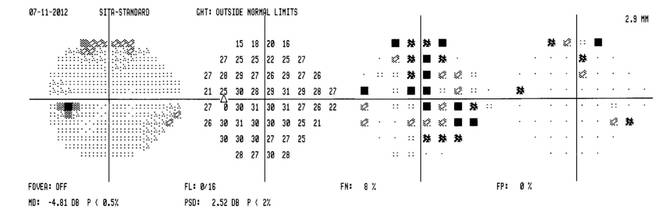
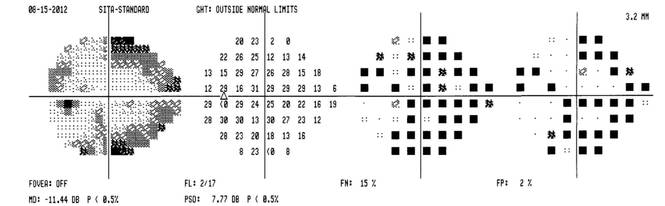
Following an uncomplicated cesarean delivery at 38 weeks, the patient returned to the clinic for visual field testing. Humphrey visual fields were full in the right eye and showed some scattered central depressions in the left. Both eyes were significantly improved from previous fields (eFigure 2) . The patient had discontinued acetazolamide and reported minor tension headaches she believed were due to lack of sleep but stated that she was no longer having migraines. There was no papilledema noted on fundus examination, and Snellen distance visual acuity measured 20/20 in both eyes. An MRI had been performed after delivery and was negative for intracranial hemorrhage, mass, or hydrocephalus).
(eFigure 2. Visual Fields Postpartum)
Right eye
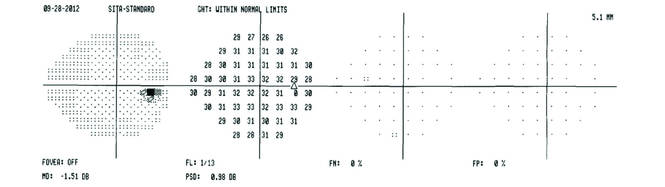
Left eye
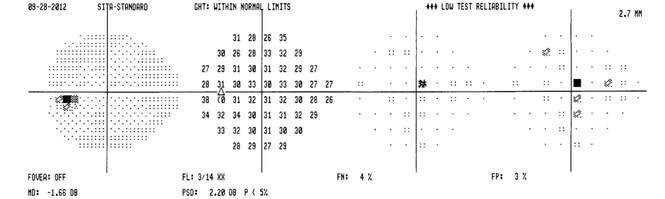
Three months later, the patient returned for her yearly comprehensive examination. At that visit, she reported a decrease in frequency of the migraine headaches. Optical coherence tomography was performed and showed a significant decrease in optic nerve head swelling.
Related: Diabetes on the Rise Among Other Pregnancy Problems
Clinical Picture
Idiopathic intracranial hypertension presents clinically with signs and symptoms of increased intracranial pressure (ICP). Headache is the most common symptom, usually presenting as daily and pulsatile.1 Nausea may be associated with the headache, although vomiting is rare, and the headache may awaken the patient. The headache may remain after resolution of elevated ICP (Table).2

Papilledema is the most common sign of IIH.1,2 Visual loss associated with papilledema is generally mild at first but progressive. Transient blur lasts usually 30 seconds and may be monocular or binocular.1 The cause is thought to be related to transient ischemia of the optic nerve.1 Vision loss is typically reversible with resolution of optic nerve swelling, but 25% of patients may develop optic atrophy, which results in permanent vision loss.2 Common patterns of visual abnormalities include enlargement of the physiologic blind spot, inferonasal and arcuate defects, and eventually severe peripheral constriction.1,2 It is imperative that all patients with IIH have visual field testing performed.
About one-third of patients with IIH experience diplopia. This binocular, horizontal diplopia is caused by a sixth nerve palsy in 10% to 20% of patients.1 Cranial nerves II, VI, and VII make a 90-degree bend and seem to be prone to damage at the site of the bend.1
Pulse-synchronous tinnitus is common in IIH as well.2,3 This generally occurs unilaterally and may be eliminated by jugular compression or the head turning to the ipsilateral side.1,3 The sound is caused by the transmission of an increase in the vascular pulse due to high pressure on the cerebrospinal fluid (CSF).1,3
Idiopathic intracranial hypertension most typically presents in obese women of childbearing age.1-3 An increasing degree of obesity is generally associated with an increased risk of vision loss.1,2 Men seem to have worse acuity and visual fields at presentation than do women.2 Men are less likely to report headaches than are women and, therefore, have double the likelihood of severe vision loss.2 Hence, closer monitoring and more aggressive intervention is recommended for men due to their lesser tendency for headaches.2 Black patients also demonstrate more aggressive disease and, therefore, require closer monitoring and early aggressive intervention.1,2
Papilledema is the most common sign of IIH and may be caused by several processes. In this case, most were ruled out given the patient’s normal visual acuities, pupillary reaction, color vision testing, BP measurement, and B-scan imaging. The patient’s systemic history was negative for thyroid-related disease, diabetes, hypertension, autoimmune disease, or infection. She had no family history of vision loss or hereditary ocular conditions. The most recent MRI was negative for any long-standing space-occupying lesion or hydrocephalus.
Pathophysiology
Several mechanisms leading to increased ICP have been proposed. These include increased brain water content, excess CSF production, reduced CSF absorption, and increased cerebral venous pressure.2,3 There is also a suspicion of the role of sex hormones in IIH due to its high predilection for females.2
The role of vitamin A metabolism has also been studied in IIH.1 Retinol levels are elevated in the CSF of patients with IIH. Patients may ingest an abnormally large amount of vitamin A, metabolize it abnormally, or be sensitive to its effects.2,4 The function of adipose tissue as an actively secreting endocrine tissue may play a role in IIH due to its release of adipose tissue-derived retinol binding protein.2 Other adipose-produced cytokines include leptin, which has been implicated in IIH due to its elevated levels found in the CSF of patients with IIH.2
Stenosis of the cerebral sinuses is another proposed mechanism of IIH.1-3 Cerebrospinal fluid exits the cranium into the venous sinuses via the arachnoid villi.2 An obstruction in these sinuses may impair CSF outflow and result in intracranial hypertension. Microthrombosis caused by hypercoaguable disorders may result in increased cerebral venous pressure and impaired CSF absorption as well.2,4
Some medications have been found in association with IIH. These include tetracycline, cyclosporine, lithium, nalidixic acid, nitrofurantoin, oral contraceptives, levonorgestrel, danaxol, and tamoxifen.1-4 Tetracycline seems to have the strongest association with IIH and should be discontinued in those patients where the association is very likely to be the causative factor.2 The link to oral contraceptives may occur simply due to their association with young women most at risk for IIH.1-3
Related:Young Man With Headache, Confusion, and Hearing Loss
Management
The goals of treatment with IIH are to preserve vision and relieve symptoms, particularly headache. The general recommendation is that pregnant women with IIH should be managed and treated the same as any other patient with IIH. However, imaging and some drug contraindications exist between these 2 groups.
The diagnostic test for IIH is a lumbar puncture, which is also the most effective treatment.1-3,5 Lumbar puncture should be performed in the relaxed lateral decubitus position without sedation.1-3 The opening pressure should be measured and is the most clinically significant diagnostic tool for diagnosis of IIH. Opening pressures of > 250 mm H2O are diagnostic of IIH.1-3,5
Weight loss is an essential part of treatment in obese patients with IIH.1-3 A low-calorie, low-salt diet with mild fluid restriction seems to reverse the symptoms of IIH. A 5% to 10% reduction in body weight may reduce symptoms and signs of IIH.2
Carbonic anhydrase inhibitors (CAIs), such as acetalzolamide, have a multifactorial role in IIH.4 They are usually prescribed in 1 to 2 grams over several doses and function by decreasing CSF production.1 Carbonic anhydrase inhibitors also are known to change the taste of foods and may, therefore, aid in weight loss.1,2 Patients prescribed CAIs commonly experience a tingling in their fingers, toes, and perioral region, an indication that the medication is working.1,2 A rare but serious adverse effect (AE) is aplastic anemia, which generally occurs in the first 6 months of treatment in elderly patients.1 The use of CAIs in pregnancy is controversial, and although rare complications are reported, it is considered a class C drug.5
In patients with rapidly progressive vision loss but with minimal headache, optic nerve sheath fenestration (ONSF) is the surgical treatment of choice.2,3,6 In this procedure, a window or series of slits are created behind the globe in the optic nerve sheath.1 About 50% of patients achieve adequate headache control with ONSF, especially for frontal headaches.1,2
For patients with vision loss, papilledema, and headache that do not respond to medical therapy, a CSF diversion procedure is the preferred treatment. Cerebrospinal fluid diversion with ventriculoperitoneal or lumboperitoneal shunts may prevent progressive loss of vision.1,4,6 However, variable response rates and shunt failure requiring subsequent revisions are common and may occur in as many as half of patients undergoing these procedures.1
Increased intracranial venous pressure due to stenosis of the venous sinuses has been thought to be a possible cause of IIH. Stenting of the transverse venous sinus stenosis has been shown to reduce cerebral venous pressure, reduce ICP, and improve symptoms in patients with IIH.1-3 It is unclear whether elevations in ICP cause transverse sinus stenosis or whether transverse sinus stenosis causes increased ICP.2 Regardless, stents have a high rate of complications, including subdural hemorrhage, venous sinus perforation, in-stent thrombosis, and recurrent stenosis proximal to the stent.2
Steroids have been used to treat IIH in the past, although their mechanism of action remains unclear.2 There may be recurrence of papilledema if they are tapered too quickly. Due to their association with long-term AEs, including weight gain, they should be avoided.2
Management in Pregnancy
Several studies agree that vision loss occurs in the same frequency in pregnant and nonpregnant patients with IIH.4,7 Idiopathic intracranial hypertension can occur in any trimester in pregnancy. It has been found that patients have the same spontaneous abortion rate and visual outcomes as the general population.6-8 It has also been concluded that treatment should be the same in both patient populations with slight variability in the use of acetazolamide.4,6,7
The use of dilating drops during pregnancy is controversial. Although there have been no teratogenic effects reported with use of topical anesthetics and dilating drops, all drugs should be avoided during the first trimester.7-10 Guidelines have been established by the American Congress of Obstetricians and Gynecologists for X-ray examination and exposure during pregnancy. It has been determined that exposure from a single diagnostic X-ray procedure does not result in harmful fetal effects.11 Magnetic resonance imaging is not associated with any known adverse fetal effects and is a better imaging option during pregnancy, because it is not associated with the use of ionizing radiation.11
The use of CAIs in the first trimester is controversial.4,7 Some believe it should be avoided because it is a Pregnancy Category C drug. However, a single case of sacrococcygeal teratoma has been reported in humans; therefore, some believe this is not a strong basis for withholding the medication in patients with the potential risk for severe vision loss.4,7 In this case, a consult to the patient’s obstetrician was made, and the use of acetazolamide had no effect on the health of the baby.
In pregnant women with IIH with progressive vision loss, failed treatment, or nonadherence, surgery may be necessary. Optic nerve sheath fenestration is preferred due to lower morbidity and mortality compared with shunting procedures.1,2,4,6 The growing fetus may be affected by the peritoneal end of the shunt.4
Related: 49-Year-Old Woman With a Broken Heart
Conclusions
Vision loss associated with IIH can be severe and permanent if left untreated. The best treatments and often the most effective involve weight loss and lumbar puncture. Acetazolamide has been a proven effective treatment in some patients, but some debate exists over the safety of its use during pregnancy. This patient did not have any AEs from its use; however, it did not prove valuable in her treatment. Studies often disagree on the use of acetazolamide in pregnancy; however, all agree that proper patient counseling on potential AEs and management by an obstetrician are important. With proper management, pregnant women with IIH have had outcomes similar to those of the general population.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Wall M. Idiopathic intracranial hypertension. Neurol Clin. 2010;28(3):593-617.
2. Bruce BB, Biousee V, Newman NJ. Update on idiopathic intracranial hypertension. Am J Ophthalmol. 2011;152(2):163-169.
3. Fields JD, Javendani PP, Falardeau J, et al. Dural venous sinus angioplasty and stenting for the treatment of idiopathic intracranial hypertension. J Neurointerv Surg. 2013;5(1):62-68.
4. Evans RW, Lee AG. Idiopathic intracranial hypertension in pregnancy. Headache. 2010;50(9):1513-1515.
5. Friedman DI, Jacobson DM. Diagnostic criteria for idiopathic intracranial hypertension. Neurology. 2002;59(10):1492-1495.
6. Martínez-Varea A, Diago-Almela VJ, Abad-Carrascosa A, Perales-Marín A. Progressive visual loss in a pregnant woman with idiopathic intracranial hypertension. Eur J Obstet Gynecol Reprod Biol. 2012;163(1):117-122.
7. Falardeau J, Lobb B, Golden S, Maxfield SD, Tanne E. The use of acetazolamide during pregnancy in intracranial hypertension patients. J Neuroophthalmol. 2013;33(1):9-12.
8. Dinn RB, Harris A, Marcus PS. Ocular changes in pregnancy. Obstet Gynecol Surg. 2003;58(2):137-144.
9. Shultz KL, Birnbaum AD, Goldstein DA. Ocular disease in pregnancy. Curr Opin Ophthalmol. 2005;16(5):308-314.
10. Chung CY, Kwok AKH, Chung KL. Use of ophthalmic medications during pregnancy. Hong Kong Med J. 2004;10(3):191-195.
11. American Congress of Obstetricians and Gynecologists. Committee Opinion. Guidelines for diagnostic imaging during pregnancy. American Congress of Obstetricians and Gynecologists Website. http://www.acog.org/-/media/Committee-Opinions/Committee-on-Obstetric-Practice/co299.pdf. Published 2004. Accessed October 9, 2015.
A 27-year-old white woman presented to the clinic with headaches and decreased vision through her reading glasses while performing near tasks. Her medical history was significant for herpes simplex, hyperlipidemia, and migraine headaches with aura. Her migraines began following an earlier motor vehicle accident, and her most recent magnetic resonance imaging (MRI) showed no abnormalities. Her current medications included prophylactic acyclovir for herpes and acetaminophen and caffeine tablets as needed for headache. She reported no other trauma or surgery and no known allergies. The patient’s best-corrected Snellen visual acuities in both eyes were 20/20 (distance) and 20/30 (near).
Preliminary testing, including pupils, extraocular motilities, confrontation fields, and color vision, were all within normal limits. Her slit-lamp examination was unremarkable. A dilated fundus examination revealed crowded, elevated discs without vessel obscuration, hemorrhage, hyperemia, or drusen (Figure 1). The fundus examination was otherwise unremarkable. Optical coherence tomography of the optic nerves showed increased nerve fiber layer thickness in both eyes (Figure 2). Her blood pressure (BP) at this visit was 106/77 mm/Hg.
The diagnosis based on these findings was bilateral optic nerve elevation with long-standing migraine headaches. The plan was for the patient to return to the clinic for repeat visual field testing and B-scan ultrasonography to rule out buried optic nerve head drusen.
Two months later, the patient presented to the clinic 19 weeks pregnant and reported that her headaches had increased in frequency, but she had no diplopia. All preliminary testing, including visual acuities, pupil reaction, color vision, and slit-lamp examination remained normal. Fundus examination showed the patient’s nerves were unchanged in appearance from the initial presentation. Visual fields revealed an enlarged blind spot in the right eye and paracentral defects in the left eye. The B-scan testing was negative for optic nerve drusen. Due to the increased frequency of headaches, pregnancy, and suspicious optic nerves, an urgent consult was placed to neurology.
At the neurology appointment 1 month later, the patient was diagnosed with migraine headache syndrome and idiopathic intracranial hypertension (IIH). The neurologist believed her headaches might have been resulting from analgesic rebound. He suggested that the patient discontinue or decrease use of oral butalbital, acetaminophen and caffeine tablets, and other forms of caffeine. It was decided that divalproxen sodium and verapamil were not feasible due to pregnancy. The neurologist started her on oral acetazolamide 500 mg twice daily.
The patient returned to her obstetrician 1 month later for a routine follow-up; the headaches had worsened and were now accompanied by nausea and vomiting twice daily on average. Her medications still included acetaminophen and caffeine tablets, although it had been recommended she discontinue them, prochlorperazine, and acetazolamide. Due to the worsening of her symptoms and visual fields (eFigure 1), the obstetrician recommended that the patient deliver by cesarean section at 38 to 39 weeks.
(eFigure 1.Visual Fields at Follow-up 1 and 2)
Right eye
Left Eye
Following an uncomplicated cesarean delivery at 38 weeks, the patient returned to the clinic for visual field testing. Humphrey visual fields were full in the right eye and showed some scattered central depressions in the left. Both eyes were significantly improved from previous fields (eFigure 2) . The patient had discontinued acetazolamide and reported minor tension headaches she believed were due to lack of sleep but stated that she was no longer having migraines. There was no papilledema noted on fundus examination, and Snellen distance visual acuity measured 20/20 in both eyes. An MRI had been performed after delivery and was negative for intracranial hemorrhage, mass, or hydrocephalus).
(eFigure 2. Visual Fields Postpartum)
Right eye
Left eye
Three months later, the patient returned for her yearly comprehensive examination. At that visit, she reported a decrease in frequency of the migraine headaches. Optical coherence tomography was performed and showed a significant decrease in optic nerve head swelling.
Related: Diabetes on the Rise Among Other Pregnancy Problems
Clinical Picture
Idiopathic intracranial hypertension presents clinically with signs and symptoms of increased intracranial pressure (ICP). Headache is the most common symptom, usually presenting as daily and pulsatile.1 Nausea may be associated with the headache, although vomiting is rare, and the headache may awaken the patient. The headache may remain after resolution of elevated ICP (Table).2
Papilledema is the most common sign of IIH.1,2 Visual loss associated with papilledema is generally mild at first but progressive. Transient blur lasts usually 30 seconds and may be monocular or binocular.1 The cause is thought to be related to transient ischemia of the optic nerve.1 Vision loss is typically reversible with resolution of optic nerve swelling, but 25% of patients may develop optic atrophy, which results in permanent vision loss.2 Common patterns of visual abnormalities include enlargement of the physiologic blind spot, inferonasal and arcuate defects, and eventually severe peripheral constriction.1,2 It is imperative that all patients with IIH have visual field testing performed.
About one-third of patients with IIH experience diplopia. This binocular, horizontal diplopia is caused by a sixth nerve palsy in 10% to 20% of patients.1 Cranial nerves II, VI, and VII make a 90-degree bend and seem to be prone to damage at the site of the bend.1
Pulse-synchronous tinnitus is common in IIH as well.2,3 This generally occurs unilaterally and may be eliminated by jugular compression or the head turning to the ipsilateral side.1,3 The sound is caused by the transmission of an increase in the vascular pulse due to high pressure on the cerebrospinal fluid (CSF).1,3
Idiopathic intracranial hypertension most typically presents in obese women of childbearing age.1-3 An increasing degree of obesity is generally associated with an increased risk of vision loss.1,2 Men seem to have worse acuity and visual fields at presentation than do women.2 Men are less likely to report headaches than are women and, therefore, have double the likelihood of severe vision loss.2 Hence, closer monitoring and more aggressive intervention is recommended for men due to their lesser tendency for headaches.2 Black patients also demonstrate more aggressive disease and, therefore, require closer monitoring and early aggressive intervention.1,2
Papilledema is the most common sign of IIH and may be caused by several processes. In this case, most were ruled out given the patient’s normal visual acuities, pupillary reaction, color vision testing, BP measurement, and B-scan imaging. The patient’s systemic history was negative for thyroid-related disease, diabetes, hypertension, autoimmune disease, or infection. She had no family history of vision loss or hereditary ocular conditions. The most recent MRI was negative for any long-standing space-occupying lesion or hydrocephalus.
Pathophysiology
Several mechanisms leading to increased ICP have been proposed. These include increased brain water content, excess CSF production, reduced CSF absorption, and increased cerebral venous pressure.2,3 There is also a suspicion of the role of sex hormones in IIH due to its high predilection for females.2
The role of vitamin A metabolism has also been studied in IIH.1 Retinol levels are elevated in the CSF of patients with IIH. Patients may ingest an abnormally large amount of vitamin A, metabolize it abnormally, or be sensitive to its effects.2,4 The function of adipose tissue as an actively secreting endocrine tissue may play a role in IIH due to its release of adipose tissue-derived retinol binding protein.2 Other adipose-produced cytokines include leptin, which has been implicated in IIH due to its elevated levels found in the CSF of patients with IIH.2
Stenosis of the cerebral sinuses is another proposed mechanism of IIH.1-3 Cerebrospinal fluid exits the cranium into the venous sinuses via the arachnoid villi.2 An obstruction in these sinuses may impair CSF outflow and result in intracranial hypertension. Microthrombosis caused by hypercoaguable disorders may result in increased cerebral venous pressure and impaired CSF absorption as well.2,4
Some medications have been found in association with IIH. These include tetracycline, cyclosporine, lithium, nalidixic acid, nitrofurantoin, oral contraceptives, levonorgestrel, danaxol, and tamoxifen.1-4 Tetracycline seems to have the strongest association with IIH and should be discontinued in those patients where the association is very likely to be the causative factor.2 The link to oral contraceptives may occur simply due to their association with young women most at risk for IIH.1-3
Related:Young Man With Headache, Confusion, and Hearing Loss
Management
The goals of treatment with IIH are to preserve vision and relieve symptoms, particularly headache. The general recommendation is that pregnant women with IIH should be managed and treated the same as any other patient with IIH. However, imaging and some drug contraindications exist between these 2 groups.
The diagnostic test for IIH is a lumbar puncture, which is also the most effective treatment.1-3,5 Lumbar puncture should be performed in the relaxed lateral decubitus position without sedation.1-3 The opening pressure should be measured and is the most clinically significant diagnostic tool for diagnosis of IIH. Opening pressures of > 250 mm H2O are diagnostic of IIH.1-3,5
Weight loss is an essential part of treatment in obese patients with IIH.1-3 A low-calorie, low-salt diet with mild fluid restriction seems to reverse the symptoms of IIH. A 5% to 10% reduction in body weight may reduce symptoms and signs of IIH.2
Carbonic anhydrase inhibitors (CAIs), such as acetalzolamide, have a multifactorial role in IIH.4 They are usually prescribed in 1 to 2 grams over several doses and function by decreasing CSF production.1 Carbonic anhydrase inhibitors also are known to change the taste of foods and may, therefore, aid in weight loss.1,2 Patients prescribed CAIs commonly experience a tingling in their fingers, toes, and perioral region, an indication that the medication is working.1,2 A rare but serious adverse effect (AE) is aplastic anemia, which generally occurs in the first 6 months of treatment in elderly patients.1 The use of CAIs in pregnancy is controversial, and although rare complications are reported, it is considered a class C drug.5
In patients with rapidly progressive vision loss but with minimal headache, optic nerve sheath fenestration (ONSF) is the surgical treatment of choice.2,3,6 In this procedure, a window or series of slits are created behind the globe in the optic nerve sheath.1 About 50% of patients achieve adequate headache control with ONSF, especially for frontal headaches.1,2
For patients with vision loss, papilledema, and headache that do not respond to medical therapy, a CSF diversion procedure is the preferred treatment. Cerebrospinal fluid diversion with ventriculoperitoneal or lumboperitoneal shunts may prevent progressive loss of vision.1,4,6 However, variable response rates and shunt failure requiring subsequent revisions are common and may occur in as many as half of patients undergoing these procedures.1
Increased intracranial venous pressure due to stenosis of the venous sinuses has been thought to be a possible cause of IIH. Stenting of the transverse venous sinus stenosis has been shown to reduce cerebral venous pressure, reduce ICP, and improve symptoms in patients with IIH.1-3 It is unclear whether elevations in ICP cause transverse sinus stenosis or whether transverse sinus stenosis causes increased ICP.2 Regardless, stents have a high rate of complications, including subdural hemorrhage, venous sinus perforation, in-stent thrombosis, and recurrent stenosis proximal to the stent.2
Steroids have been used to treat IIH in the past, although their mechanism of action remains unclear.2 There may be recurrence of papilledema if they are tapered too quickly. Due to their association with long-term AEs, including weight gain, they should be avoided.2
Management in Pregnancy
Several studies agree that vision loss occurs in the same frequency in pregnant and nonpregnant patients with IIH.4,7 Idiopathic intracranial hypertension can occur in any trimester in pregnancy. It has been found that patients have the same spontaneous abortion rate and visual outcomes as the general population.6-8 It has also been concluded that treatment should be the same in both patient populations with slight variability in the use of acetazolamide.4,6,7
The use of dilating drops during pregnancy is controversial. Although there have been no teratogenic effects reported with use of topical anesthetics and dilating drops, all drugs should be avoided during the first trimester.7-10 Guidelines have been established by the American Congress of Obstetricians and Gynecologists for X-ray examination and exposure during pregnancy. It has been determined that exposure from a single diagnostic X-ray procedure does not result in harmful fetal effects.11 Magnetic resonance imaging is not associated with any known adverse fetal effects and is a better imaging option during pregnancy, because it is not associated with the use of ionizing radiation.11
The use of CAIs in the first trimester is controversial.4,7 Some believe it should be avoided because it is a Pregnancy Category C drug. However, a single case of sacrococcygeal teratoma has been reported in humans; therefore, some believe this is not a strong basis for withholding the medication in patients with the potential risk for severe vision loss.4,7 In this case, a consult to the patient’s obstetrician was made, and the use of acetazolamide had no effect on the health of the baby.
In pregnant women with IIH with progressive vision loss, failed treatment, or nonadherence, surgery may be necessary. Optic nerve sheath fenestration is preferred due to lower morbidity and mortality compared with shunting procedures.1,2,4,6 The growing fetus may be affected by the peritoneal end of the shunt.4
Related: 49-Year-Old Woman With a Broken Heart
Conclusions
Vision loss associated with IIH can be severe and permanent if left untreated. The best treatments and often the most effective involve weight loss and lumbar puncture. Acetazolamide has been a proven effective treatment in some patients, but some debate exists over the safety of its use during pregnancy. This patient did not have any AEs from its use; however, it did not prove valuable in her treatment. Studies often disagree on the use of acetazolamide in pregnancy; however, all agree that proper patient counseling on potential AEs and management by an obstetrician are important. With proper management, pregnant women with IIH have had outcomes similar to those of the general population.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
A 27-year-old white woman presented to the clinic with headaches and decreased vision through her reading glasses while performing near tasks. Her medical history was significant for herpes simplex, hyperlipidemia, and migraine headaches with aura. Her migraines began following an earlier motor vehicle accident, and her most recent magnetic resonance imaging (MRI) showed no abnormalities. Her current medications included prophylactic acyclovir for herpes and acetaminophen and caffeine tablets as needed for headache. She reported no other trauma or surgery and no known allergies. The patient’s best-corrected Snellen visual acuities in both eyes were 20/20 (distance) and 20/30 (near).
Preliminary testing, including pupils, extraocular motilities, confrontation fields, and color vision, were all within normal limits. Her slit-lamp examination was unremarkable. A dilated fundus examination revealed crowded, elevated discs without vessel obscuration, hemorrhage, hyperemia, or drusen (Figure 1). The fundus examination was otherwise unremarkable. Optical coherence tomography of the optic nerves showed increased nerve fiber layer thickness in both eyes (Figure 2). Her blood pressure (BP) at this visit was 106/77 mm/Hg.
The diagnosis based on these findings was bilateral optic nerve elevation with long-standing migraine headaches. The plan was for the patient to return to the clinic for repeat visual field testing and B-scan ultrasonography to rule out buried optic nerve head drusen.
Two months later, the patient presented to the clinic 19 weeks pregnant and reported that her headaches had increased in frequency, but she had no diplopia. All preliminary testing, including visual acuities, pupil reaction, color vision, and slit-lamp examination remained normal. Fundus examination showed the patient’s nerves were unchanged in appearance from the initial presentation. Visual fields revealed an enlarged blind spot in the right eye and paracentral defects in the left eye. The B-scan testing was negative for optic nerve drusen. Due to the increased frequency of headaches, pregnancy, and suspicious optic nerves, an urgent consult was placed to neurology.
At the neurology appointment 1 month later, the patient was diagnosed with migraine headache syndrome and idiopathic intracranial hypertension (IIH). The neurologist believed her headaches might have been resulting from analgesic rebound. He suggested that the patient discontinue or decrease use of oral butalbital, acetaminophen and caffeine tablets, and other forms of caffeine. It was decided that divalproxen sodium and verapamil were not feasible due to pregnancy. The neurologist started her on oral acetazolamide 500 mg twice daily.
The patient returned to her obstetrician 1 month later for a routine follow-up; the headaches had worsened and were now accompanied by nausea and vomiting twice daily on average. Her medications still included acetaminophen and caffeine tablets, although it had been recommended she discontinue them, prochlorperazine, and acetazolamide. Due to the worsening of her symptoms and visual fields (eFigure 1), the obstetrician recommended that the patient deliver by cesarean section at 38 to 39 weeks.
(eFigure 1.Visual Fields at Follow-up 1 and 2)
Right eye
Left Eye
Following an uncomplicated cesarean delivery at 38 weeks, the patient returned to the clinic for visual field testing. Humphrey visual fields were full in the right eye and showed some scattered central depressions in the left. Both eyes were significantly improved from previous fields (eFigure 2) . The patient had discontinued acetazolamide and reported minor tension headaches she believed were due to lack of sleep but stated that she was no longer having migraines. There was no papilledema noted on fundus examination, and Snellen distance visual acuity measured 20/20 in both eyes. An MRI had been performed after delivery and was negative for intracranial hemorrhage, mass, or hydrocephalus).
(eFigure 2. Visual Fields Postpartum)
Right eye
Left eye
Three months later, the patient returned for her yearly comprehensive examination. At that visit, she reported a decrease in frequency of the migraine headaches. Optical coherence tomography was performed and showed a significant decrease in optic nerve head swelling.
Related: Diabetes on the Rise Among Other Pregnancy Problems
Clinical Picture
Idiopathic intracranial hypertension presents clinically with signs and symptoms of increased intracranial pressure (ICP). Headache is the most common symptom, usually presenting as daily and pulsatile.1 Nausea may be associated with the headache, although vomiting is rare, and the headache may awaken the patient. The headache may remain after resolution of elevated ICP (Table).2
Papilledema is the most common sign of IIH.1,2 Visual loss associated with papilledema is generally mild at first but progressive. Transient blur lasts usually 30 seconds and may be monocular or binocular.1 The cause is thought to be related to transient ischemia of the optic nerve.1 Vision loss is typically reversible with resolution of optic nerve swelling, but 25% of patients may develop optic atrophy, which results in permanent vision loss.2 Common patterns of visual abnormalities include enlargement of the physiologic blind spot, inferonasal and arcuate defects, and eventually severe peripheral constriction.1,2 It is imperative that all patients with IIH have visual field testing performed.
About one-third of patients with IIH experience diplopia. This binocular, horizontal diplopia is caused by a sixth nerve palsy in 10% to 20% of patients.1 Cranial nerves II, VI, and VII make a 90-degree bend and seem to be prone to damage at the site of the bend.1
Pulse-synchronous tinnitus is common in IIH as well.2,3 This generally occurs unilaterally and may be eliminated by jugular compression or the head turning to the ipsilateral side.1,3 The sound is caused by the transmission of an increase in the vascular pulse due to high pressure on the cerebrospinal fluid (CSF).1,3
Idiopathic intracranial hypertension most typically presents in obese women of childbearing age.1-3 An increasing degree of obesity is generally associated with an increased risk of vision loss.1,2 Men seem to have worse acuity and visual fields at presentation than do women.2 Men are less likely to report headaches than are women and, therefore, have double the likelihood of severe vision loss.2 Hence, closer monitoring and more aggressive intervention is recommended for men due to their lesser tendency for headaches.2 Black patients also demonstrate more aggressive disease and, therefore, require closer monitoring and early aggressive intervention.1,2
Papilledema is the most common sign of IIH and may be caused by several processes. In this case, most were ruled out given the patient’s normal visual acuities, pupillary reaction, color vision testing, BP measurement, and B-scan imaging. The patient’s systemic history was negative for thyroid-related disease, diabetes, hypertension, autoimmune disease, or infection. She had no family history of vision loss or hereditary ocular conditions. The most recent MRI was negative for any long-standing space-occupying lesion or hydrocephalus.
Pathophysiology
Several mechanisms leading to increased ICP have been proposed. These include increased brain water content, excess CSF production, reduced CSF absorption, and increased cerebral venous pressure.2,3 There is also a suspicion of the role of sex hormones in IIH due to its high predilection for females.2
The role of vitamin A metabolism has also been studied in IIH.1 Retinol levels are elevated in the CSF of patients with IIH. Patients may ingest an abnormally large amount of vitamin A, metabolize it abnormally, or be sensitive to its effects.2,4 The function of adipose tissue as an actively secreting endocrine tissue may play a role in IIH due to its release of adipose tissue-derived retinol binding protein.2 Other adipose-produced cytokines include leptin, which has been implicated in IIH due to its elevated levels found in the CSF of patients with IIH.2
Stenosis of the cerebral sinuses is another proposed mechanism of IIH.1-3 Cerebrospinal fluid exits the cranium into the venous sinuses via the arachnoid villi.2 An obstruction in these sinuses may impair CSF outflow and result in intracranial hypertension. Microthrombosis caused by hypercoaguable disorders may result in increased cerebral venous pressure and impaired CSF absorption as well.2,4
Some medications have been found in association with IIH. These include tetracycline, cyclosporine, lithium, nalidixic acid, nitrofurantoin, oral contraceptives, levonorgestrel, danaxol, and tamoxifen.1-4 Tetracycline seems to have the strongest association with IIH and should be discontinued in those patients where the association is very likely to be the causative factor.2 The link to oral contraceptives may occur simply due to their association with young women most at risk for IIH.1-3
Related:Young Man With Headache, Confusion, and Hearing Loss
Management
The goals of treatment with IIH are to preserve vision and relieve symptoms, particularly headache. The general recommendation is that pregnant women with IIH should be managed and treated the same as any other patient with IIH. However, imaging and some drug contraindications exist between these 2 groups.
The diagnostic test for IIH is a lumbar puncture, which is also the most effective treatment.1-3,5 Lumbar puncture should be performed in the relaxed lateral decubitus position without sedation.1-3 The opening pressure should be measured and is the most clinically significant diagnostic tool for diagnosis of IIH. Opening pressures of > 250 mm H2O are diagnostic of IIH.1-3,5
Weight loss is an essential part of treatment in obese patients with IIH.1-3 A low-calorie, low-salt diet with mild fluid restriction seems to reverse the symptoms of IIH. A 5% to 10% reduction in body weight may reduce symptoms and signs of IIH.2
Carbonic anhydrase inhibitors (CAIs), such as acetalzolamide, have a multifactorial role in IIH.4 They are usually prescribed in 1 to 2 grams over several doses and function by decreasing CSF production.1 Carbonic anhydrase inhibitors also are known to change the taste of foods and may, therefore, aid in weight loss.1,2 Patients prescribed CAIs commonly experience a tingling in their fingers, toes, and perioral region, an indication that the medication is working.1,2 A rare but serious adverse effect (AE) is aplastic anemia, which generally occurs in the first 6 months of treatment in elderly patients.1 The use of CAIs in pregnancy is controversial, and although rare complications are reported, it is considered a class C drug.5
In patients with rapidly progressive vision loss but with minimal headache, optic nerve sheath fenestration (ONSF) is the surgical treatment of choice.2,3,6 In this procedure, a window or series of slits are created behind the globe in the optic nerve sheath.1 About 50% of patients achieve adequate headache control with ONSF, especially for frontal headaches.1,2
For patients with vision loss, papilledema, and headache that do not respond to medical therapy, a CSF diversion procedure is the preferred treatment. Cerebrospinal fluid diversion with ventriculoperitoneal or lumboperitoneal shunts may prevent progressive loss of vision.1,4,6 However, variable response rates and shunt failure requiring subsequent revisions are common and may occur in as many as half of patients undergoing these procedures.1
Increased intracranial venous pressure due to stenosis of the venous sinuses has been thought to be a possible cause of IIH. Stenting of the transverse venous sinus stenosis has been shown to reduce cerebral venous pressure, reduce ICP, and improve symptoms in patients with IIH.1-3 It is unclear whether elevations in ICP cause transverse sinus stenosis or whether transverse sinus stenosis causes increased ICP.2 Regardless, stents have a high rate of complications, including subdural hemorrhage, venous sinus perforation, in-stent thrombosis, and recurrent stenosis proximal to the stent.2
Steroids have been used to treat IIH in the past, although their mechanism of action remains unclear.2 There may be recurrence of papilledema if they are tapered too quickly. Due to their association with long-term AEs, including weight gain, they should be avoided.2
Management in Pregnancy
Several studies agree that vision loss occurs in the same frequency in pregnant and nonpregnant patients with IIH.4,7 Idiopathic intracranial hypertension can occur in any trimester in pregnancy. It has been found that patients have the same spontaneous abortion rate and visual outcomes as the general population.6-8 It has also been concluded that treatment should be the same in both patient populations with slight variability in the use of acetazolamide.4,6,7
The use of dilating drops during pregnancy is controversial. Although there have been no teratogenic effects reported with use of topical anesthetics and dilating drops, all drugs should be avoided during the first trimester.7-10 Guidelines have been established by the American Congress of Obstetricians and Gynecologists for X-ray examination and exposure during pregnancy. It has been determined that exposure from a single diagnostic X-ray procedure does not result in harmful fetal effects.11 Magnetic resonance imaging is not associated with any known adverse fetal effects and is a better imaging option during pregnancy, because it is not associated with the use of ionizing radiation.11
The use of CAIs in the first trimester is controversial.4,7 Some believe it should be avoided because it is a Pregnancy Category C drug. However, a single case of sacrococcygeal teratoma has been reported in humans; therefore, some believe this is not a strong basis for withholding the medication in patients with the potential risk for severe vision loss.4,7 In this case, a consult to the patient’s obstetrician was made, and the use of acetazolamide had no effect on the health of the baby.
In pregnant women with IIH with progressive vision loss, failed treatment, or nonadherence, surgery may be necessary. Optic nerve sheath fenestration is preferred due to lower morbidity and mortality compared with shunting procedures.1,2,4,6 The growing fetus may be affected by the peritoneal end of the shunt.4
Related: 49-Year-Old Woman With a Broken Heart
Conclusions
Vision loss associated with IIH can be severe and permanent if left untreated. The best treatments and often the most effective involve weight loss and lumbar puncture. Acetazolamide has been a proven effective treatment in some patients, but some debate exists over the safety of its use during pregnancy. This patient did not have any AEs from its use; however, it did not prove valuable in her treatment. Studies often disagree on the use of acetazolamide in pregnancy; however, all agree that proper patient counseling on potential AEs and management by an obstetrician are important. With proper management, pregnant women with IIH have had outcomes similar to those of the general population.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Wall M. Idiopathic intracranial hypertension. Neurol Clin. 2010;28(3):593-617.
2. Bruce BB, Biousee V, Newman NJ. Update on idiopathic intracranial hypertension. Am J Ophthalmol. 2011;152(2):163-169.
3. Fields JD, Javendani PP, Falardeau J, et al. Dural venous sinus angioplasty and stenting for the treatment of idiopathic intracranial hypertension. J Neurointerv Surg. 2013;5(1):62-68.
4. Evans RW, Lee AG. Idiopathic intracranial hypertension in pregnancy. Headache. 2010;50(9):1513-1515.
5. Friedman DI, Jacobson DM. Diagnostic criteria for idiopathic intracranial hypertension. Neurology. 2002;59(10):1492-1495.
6. Martínez-Varea A, Diago-Almela VJ, Abad-Carrascosa A, Perales-Marín A. Progressive visual loss in a pregnant woman with idiopathic intracranial hypertension. Eur J Obstet Gynecol Reprod Biol. 2012;163(1):117-122.
7. Falardeau J, Lobb B, Golden S, Maxfield SD, Tanne E. The use of acetazolamide during pregnancy in intracranial hypertension patients. J Neuroophthalmol. 2013;33(1):9-12.
8. Dinn RB, Harris A, Marcus PS. Ocular changes in pregnancy. Obstet Gynecol Surg. 2003;58(2):137-144.
9. Shultz KL, Birnbaum AD, Goldstein DA. Ocular disease in pregnancy. Curr Opin Ophthalmol. 2005;16(5):308-314.
10. Chung CY, Kwok AKH, Chung KL. Use of ophthalmic medications during pregnancy. Hong Kong Med J. 2004;10(3):191-195.
11. American Congress of Obstetricians and Gynecologists. Committee Opinion. Guidelines for diagnostic imaging during pregnancy. American Congress of Obstetricians and Gynecologists Website. http://www.acog.org/-/media/Committee-Opinions/Committee-on-Obstetric-Practice/co299.pdf. Published 2004. Accessed October 9, 2015.
1. Wall M. Idiopathic intracranial hypertension. Neurol Clin. 2010;28(3):593-617.
2. Bruce BB, Biousee V, Newman NJ. Update on idiopathic intracranial hypertension. Am J Ophthalmol. 2011;152(2):163-169.
3. Fields JD, Javendani PP, Falardeau J, et al. Dural venous sinus angioplasty and stenting for the treatment of idiopathic intracranial hypertension. J Neurointerv Surg. 2013;5(1):62-68.
4. Evans RW, Lee AG. Idiopathic intracranial hypertension in pregnancy. Headache. 2010;50(9):1513-1515.
5. Friedman DI, Jacobson DM. Diagnostic criteria for idiopathic intracranial hypertension. Neurology. 2002;59(10):1492-1495.
6. Martínez-Varea A, Diago-Almela VJ, Abad-Carrascosa A, Perales-Marín A. Progressive visual loss in a pregnant woman with idiopathic intracranial hypertension. Eur J Obstet Gynecol Reprod Biol. 2012;163(1):117-122.
7. Falardeau J, Lobb B, Golden S, Maxfield SD, Tanne E. The use of acetazolamide during pregnancy in intracranial hypertension patients. J Neuroophthalmol. 2013;33(1):9-12.
8. Dinn RB, Harris A, Marcus PS. Ocular changes in pregnancy. Obstet Gynecol Surg. 2003;58(2):137-144.
9. Shultz KL, Birnbaum AD, Goldstein DA. Ocular disease in pregnancy. Curr Opin Ophthalmol. 2005;16(5):308-314.
10. Chung CY, Kwok AKH, Chung KL. Use of ophthalmic medications during pregnancy. Hong Kong Med J. 2004;10(3):191-195.
11. American Congress of Obstetricians and Gynecologists. Committee Opinion. Guidelines for diagnostic imaging during pregnancy. American Congress of Obstetricians and Gynecologists Website. http://www.acog.org/-/media/Committee-Opinions/Committee-on-Obstetric-Practice/co299.pdf. Published 2004. Accessed October 9, 2015.
Acute Generalized Exanthematous Pustulosis Associated With Ranolazine
Acute generalized exanthematous pustulosis (AGEP) is a potentially widespread, pustular, cutaneous eruption. In 90% of cases, AGEP results from drug administration.1,2 It manifests as numerous subcorneal, nonfollicular, sterile pustules of rapid onset on an erythematous base,2 often in conjunction with fever, peripheral leukocytosis, and neutrophilia.3 Numerous drug therapies have been implicated in the etiology of AGEP, most commonly the β-lactam antibiotics, such as the penicillin derivatives and cephalosporins.2 Typically, AGEP occurs soon after drug ingestion and resolves spontaneously, shortly after the causative drug is discontinued.
Ranolazine is an antianginal, anti-ischemic medication with an undetermined mechanism of action. Its antianginal and anti-ischemic effects do not depend on reduced heart rate or blood pressure. At therapeutic levels, it inhibits the cardiac late sodium current (INa), reducing the sodium-induced calcium overload in ischemic cardiac myocytes. Severe adverse reactions include angioedema; paresthesia; pancytopenia; and, in animal studies, tumorigenicity.4 Herein we report a case of AGEP associated with the use of ranolazine.
Case Report
An 83-year-old man presented with a generalized rash of approximately 12 days’ duration. The patient reported that the small “pimple-like” bumps initially erupted on the back of the neck but gradually spread to the chest, back, and extremities. The lesions were asymptomatic at the outset and became pruritic over time. For the last several years, the patient had been taking tamsulosin for benign prostatic hypertrophy and rosuvastatin for hyperlipidemia. Twelve days prior to the exanthem, he had started taking ranolazine for symptomatic ischemia until coronary angiography could be performed. He reported having no associated fevers, chills, or malaise and had no personal history of psoriasis, though he had a maternal history of the disorder.
Examination revealed numerous nonfollicular-based pustules on diffuse erythematous patches (Figure 1). There was no mucosal involvement and the skin was negative for the Nikolsky sign. Spongiform intracorneal collections of neutrophils were visible on punch biopsy (Figures 2 and 3). Periodic acid–Schiff stains for fungi were negative.

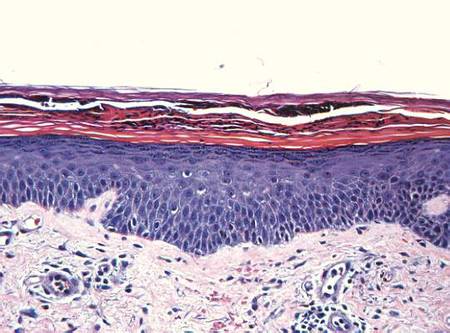 | 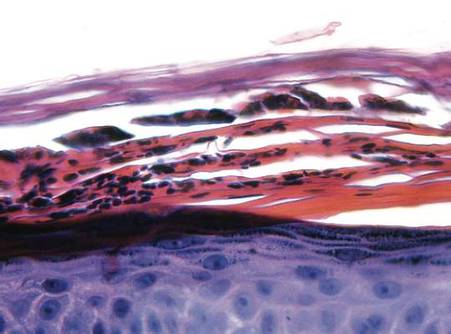 | |
Figure 2. A punch biopsy showed spongiform intracorneal collections of neutrophils (H&E, original magnification ×200). | Figure 3. A cornified layer of epidermis with neutrophils, as visible on punch biopsy (H&E, original magnification ×630). |
The patient’s primary care physician had initiated a course of oral prednisone 5 mg daily, 3 days before he presented to our outpatient dermatology clinic, but it had little effect on the rash. Upon dermatologic evaluation, we discontinued ranolazine therapy and prescribed the following tapered course of oral prednisone: 60 mg daily for 4 days; 40 mg daily for 3 days; 30 mg daily for 3 days; 20 mg daily for 3 days; 10 mg daily for 3 days; and 5 mg daily for 3 days). Within a week after this regimen was initiated, the rash showed improvement with eventual resolution and desquamation (Figure 4). Subsequently, the patient underwent successful angioplasty and multiple stent placement, which ultimately alleviated his angina.
Comment
Since its original description in 1968,5 AGEP has been misdiagnosed and underreported. Due to its rarity and clinical resemblance to more common pustular eruptions, such as exanthematous pustular psoriasis, the typical characteristics of AGEP were not clearly delineated until Beylot et al3 coined the term AGEP in 1980. Since that time, formalized criteria for the diagnosis and characterization of AGEP have been published.1,2,6-8
Numerous drug therapies have been implicated in the etiology of AGEP, most commonly antimicrobial agents, such as β-lactam antibiotics. Many other drugs, however, also have been identified as potential causative agents,8 including but not limited to antifungal, anticonvulsant, and antihypertensive agents. Other less common etiologies include viral infections,6,9-11 UV radiation, contrast media, heavy metal exposure (eg, to mercury), ingestion of urushiol (eg, in lacquered chicken), and spider bites.2,8,12-16 Nevertheless, more than 90% of AGEP cases are attributed to drug exposure, with 80% of drug-induced cases believed to be caused by antibiotics.1,8
The incidence of AGEP is estimated to be between 1 and 5 cases per million per year, using inclusion criteria from the EuroSCAR study, a multinational, case-controlled, pharmacoepidemiologic study of severe cutaneous adverse reactions.8,16 The condition seems to affect males and females equally.1,4 There are no reports of age or racial predilection.1,6,17 It has been suggested that those with AGEP may have some form of psoriatic background.1 Our patient had no personal history of inflammatory skin disease, although his mother had psoriasis.
The dermatitis presents as the sudden onset of a diffuse exanthematous eruption, which typically produces dozens to hundreds of sterile, nonfollicular, superficial pustules on an erythematous and possibly edematous base. Atypical presentations include target lesions, purpura, and vesicles. The reaction usually begins on the face or intertriginous areas of flexural surfaces and quickly disseminates. Patients may experience burning or pruritus. Acute generalized exanthematous pustulosis may involve mucous membranes but is usually limited to 1 location, most often the oral mucosa.1,8,16,18 Systemic signs and symptoms include fever, lymphadenopathy, pharyngitis, and hepatosplenomegaly. Unlike most drug allergies that demonstrate eosinophilia, AGEP is associated with leukocytosis and neutrophilic predominance. Only 25% of affected patients exhibit eosinophilia.1 Approximately 30% of patients in a retrospective analysis demonstrated abnormal renal function,2 and there have been reports of mildly elevated transaminases.8,19
In the EuroSCAR study, for reasons that were not apparent, symptoms developed within 24 hours of exposure to triggering antibiotics, whereas the median time to rash onset in response to non–anti-infective agents was 11 days.8 This finding is consistent with the delayed onset of symptoms experienced by our patient after initiating ranolazine therapy.
The differential diagnosis of AGEP primarily includes pustular psoriasis, subcorneal pustulosis, pustular folliculitis, DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome, bullous impetigo, and occasionally erythema multiforme and toxic epidermal necrolysis, with the latter typically characterized by more mucous membrane involvement.20 Biopsy does not always support a definitive diagnosis; clinical correlation is often necessary. Because of the EuroSCAR study, Sidoroff et al8 devised a clinical validation score based on morphology (presence of pustules and erythema, distribution, and eventual desquamation), histopathology (presence of intraepidermal pustules, spongiosis, and papillary edema), and disease course (duration of symptoms, neutrophilia, fever, acute onset, and time to resolution). A definitive score is 8 to 12 (out of 12), and our patient’s score was 10; the score may have been higher had blood work been performed, but by the time the diagnosis was made the patient’s condition had improved enough to make laboratory workup unnecessary.
Several theories have been proposed to explain the pathophysiology of AGEP. Some hold that the causative agent induces the formation of antigen-antibody complexes, thereby activating the complement system, which in turn produces neutrophil chemotaxis.3,21 A more recent theory suggests that drug exposure causes drug-specific CD4 and CD8 cells to migrate into dermal and epidermal layers of the skin.17 Both T cells and keratinocytes express IL-8, which attracts polymorphonuclear leukocytes, causing them to accumulate in the dermis and then the epidermis. The different clinical presentations of AGEP may be attributed to other cytokines and interleukins that T cells express during this process. In the epidermis, CD8 cells kill keratinocytes, causing focal necrosis and prompting the formation of subcorneal vesicles filled primarily with CD4 cells. CD4 and CD8 cells are then localized to the dermis where neutrophils enter the vesicles, transforming them into sterile pustules.6,16,17
Acute generalized exanthematous pustulosis has been characterized as a type IV delayed hypersensitivity reaction, with affected patients often demonstrating positive patch testing or a history of prior sensitization to the perpetrating agent.18,19,21 Although there have been reports of positive patch testing for certain drugs, the unknown sensitivity and specificity of such testing as well as preparation-dependent variables may limit the diagnostic utility of this approach.21 The additional risk for inducing AGEP by patch testing the suspected drug also is a consideration. Due to our patient’s definitive clinical validation score, we did not perform this test.21
The AGEP eruption is typically self-limited and tends to resolve within 4 to 10 days after cessation of the triggering agent. Postpustular desquamation often occurs upon resolution of the primary lesions. Treatment usually involves discontinuation of the suspected causative agent and the use of antihistamines, antipyretics, topical corticosteroids, and emollients. Although there are reports of AGEP responsiveness to oral and intravenous steroids, such treatment rarely is required.8,16,22 We prescribed a tapered course of oral prednisone due to our patient’s imminent need for angioplasty.
Conclusion
This case of AGEP induced by ranolazine is notable. Given the potential widespread use of this antianginal medication and the severity of this potential adverse reaction, it is important for clinicians to recognize AGEP, discontinue ranolazine if determined to be a causative agent, and then initiate an appropriate alternative antianginal therapy.
1. Roujeau JC, Bioulac-Sage P, Bourseau C, et al. Acute generalized exanthematous pustulosis. analysis of 63 cases. Arch Dermatol. 1991;127:1333-1338.
2. Sidoroff A, Halevy S, Bavnick JN, et al. Acute generalized exanthematous pustulosis (AGEP)–a clinical reaction pattern. J Cutan Pathol. 2001;28:113-119.
3. Beylot C, Bioulac P, Doutre MS. Acute generalized exanthematic pustuloses (four cases) [in French]. Ann Dermatol Venereol. 1980;107:37-48.
4. Ranexa [package insert]. Foster City, CA: Gilead Sciences, Inc; December 2013.
5. Baker H, Ryan TJ. Generalized pustular psoriasis. a clinical and epidemiological study of 104 cases. Br J Dermatol. 1968;80:771-793.
6. Guevara-Gutierrez E, Uribe-Jimenez E, Diaz-Canchola M, et al. Acute generalized exanthematous pustulosis: report of 12 cases and literature review. Int J Dermatol. 2009;48:253-258.
7. Chang SL, Huang YH, Yang CH, et al. Clinical manifestations and characteristics of patients with acute generalized exanthematous pustulosis in Asia. Acta Derm Venereol. 2008;88:363-365.
8. Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR) [published online ahead of print September 13, 2007]. Br J Dermatol. 2007;157:989-996.
9. Rouchouse B, Bonnefoy M, Pallot B, et al. Acute generalized exanthematous pustular dermatitis and viral infection. Dermatologica. 1986;173:180-184.
10. Naides SJ, Piette W, Veach LA, et al. Human parvovirus B19-induced vesiculopustular skin eruption. Am J Med. 1988;84:968-972.
11. Feio AB, Apetato M, Costa MM, et al. Acute generalized exanthematous pustulosis due to Coxsackie B4 virus [in Portuguese]. Acta Med Port. 1997;10:487-491.
12. Goh TK, Pang SM, Thirumoorthy T, et al. Acute generalised exanthematous pustulosis and toxic epidermal necrolysis induced by carbamazepine. Singapore Med J. 2008;49:507-510.
13. Ofuji S, Yamamoto O. Acute generalized exanthematous pustulosis associated with a human parvovirus B19 infection. J Dermatol. 2007;34:121-123.
14. Davidovici BB, Pavel D, Cagnano E, et al. Acute generalized exanthematous pustulosis following a spider bite: report of 3 cases. J Am Acad Dermatol. 2006;55:525-529.
15. Park YM, Park JG, Kang H, et al. Acute generalized exanthematous pustulosis induced by ingestion of lacquer chicken. Br J Dermatol. 2000;143:230-232.
16. Hammerbeck AA, Daniels NH, Callen JP. Ioversol-induced acute generalized exanthematous pustulosis: a case report. Arch Dermatol. 2009;145:683-687.17. Halevy S. Acute generalized exanthematous pustulosis. Curr Opin Allergy Clin Immunol. 2009;9:322-328.
18. Kim HJ, Jung KD, Lee KT, et al. Acute generalized exanthematous pustulosis caused by diltiazem [published online ahead of print February 28, 2011]. Ann Dermatol. 2011;23:108-110.
19. Speck LM, Wilkerson MG, Perri AJ, et al. Acute generalized exanthematous pustulosis caused by terazosin hydrochloride. J Drugs Dermatol. 2008;7:395-397.
20. Sidoroff A. Acute generalized exanthematous pustulosis (AGEP). UpToDate Web site. http://www.uptodate.com /contents/acute-generalized-exanthematous-pustulosis -agep?source=search_result&search=agep&selected Title=1~85. Updated March 18, 2015. Accessed October 6, 2015.
21. Mashiah J, Brenner S. A systemic reaction to patch testing for the evaluation of acute generalized exanthematous pustulosis. Arch Dermatol. 2003;139:1181-1183.
22. Ibrahimi O, Gunawardane N, Sepehr A, et al. Terbinafine-induced acute generalized exanthematous pustulosis (AGEP) responsive to high dose intravenous corticosteroid. Dermatol Online J. 2009;15:8.
Acute generalized exanthematous pustulosis (AGEP) is a potentially widespread, pustular, cutaneous eruption. In 90% of cases, AGEP results from drug administration.1,2 It manifests as numerous subcorneal, nonfollicular, sterile pustules of rapid onset on an erythematous base,2 often in conjunction with fever, peripheral leukocytosis, and neutrophilia.3 Numerous drug therapies have been implicated in the etiology of AGEP, most commonly the β-lactam antibiotics, such as the penicillin derivatives and cephalosporins.2 Typically, AGEP occurs soon after drug ingestion and resolves spontaneously, shortly after the causative drug is discontinued.
Ranolazine is an antianginal, anti-ischemic medication with an undetermined mechanism of action. Its antianginal and anti-ischemic effects do not depend on reduced heart rate or blood pressure. At therapeutic levels, it inhibits the cardiac late sodium current (INa), reducing the sodium-induced calcium overload in ischemic cardiac myocytes. Severe adverse reactions include angioedema; paresthesia; pancytopenia; and, in animal studies, tumorigenicity.4 Herein we report a case of AGEP associated with the use of ranolazine.
Case Report
An 83-year-old man presented with a generalized rash of approximately 12 days’ duration. The patient reported that the small “pimple-like” bumps initially erupted on the back of the neck but gradually spread to the chest, back, and extremities. The lesions were asymptomatic at the outset and became pruritic over time. For the last several years, the patient had been taking tamsulosin for benign prostatic hypertrophy and rosuvastatin for hyperlipidemia. Twelve days prior to the exanthem, he had started taking ranolazine for symptomatic ischemia until coronary angiography could be performed. He reported having no associated fevers, chills, or malaise and had no personal history of psoriasis, though he had a maternal history of the disorder.
Examination revealed numerous nonfollicular-based pustules on diffuse erythematous patches (Figure 1). There was no mucosal involvement and the skin was negative for the Nikolsky sign. Spongiform intracorneal collections of neutrophils were visible on punch biopsy (Figures 2 and 3). Periodic acid–Schiff stains for fungi were negative.
| | |
Figure 2. A punch biopsy showed spongiform intracorneal collections of neutrophils (H&E, original magnification ×200). | Figure 3. A cornified layer of epidermis with neutrophils, as visible on punch biopsy (H&E, original magnification ×630). |
The patient’s primary care physician had initiated a course of oral prednisone 5 mg daily, 3 days before he presented to our outpatient dermatology clinic, but it had little effect on the rash. Upon dermatologic evaluation, we discontinued ranolazine therapy and prescribed the following tapered course of oral prednisone: 60 mg daily for 4 days; 40 mg daily for 3 days; 30 mg daily for 3 days; 20 mg daily for 3 days; 10 mg daily for 3 days; and 5 mg daily for 3 days). Within a week after this regimen was initiated, the rash showed improvement with eventual resolution and desquamation (Figure 4). Subsequently, the patient underwent successful angioplasty and multiple stent placement, which ultimately alleviated his angina.
Comment
Since its original description in 1968,5 AGEP has been misdiagnosed and underreported. Due to its rarity and clinical resemblance to more common pustular eruptions, such as exanthematous pustular psoriasis, the typical characteristics of AGEP were not clearly delineated until Beylot et al3 coined the term AGEP in 1980. Since that time, formalized criteria for the diagnosis and characterization of AGEP have been published.1,2,6-8
Numerous drug therapies have been implicated in the etiology of AGEP, most commonly antimicrobial agents, such as β-lactam antibiotics. Many other drugs, however, also have been identified as potential causative agents,8 including but not limited to antifungal, anticonvulsant, and antihypertensive agents. Other less common etiologies include viral infections,6,9-11 UV radiation, contrast media, heavy metal exposure (eg, to mercury), ingestion of urushiol (eg, in lacquered chicken), and spider bites.2,8,12-16 Nevertheless, more than 90% of AGEP cases are attributed to drug exposure, with 80% of drug-induced cases believed to be caused by antibiotics.1,8
The incidence of AGEP is estimated to be between 1 and 5 cases per million per year, using inclusion criteria from the EuroSCAR study, a multinational, case-controlled, pharmacoepidemiologic study of severe cutaneous adverse reactions.8,16 The condition seems to affect males and females equally.1,4 There are no reports of age or racial predilection.1,6,17 It has been suggested that those with AGEP may have some form of psoriatic background.1 Our patient had no personal history of inflammatory skin disease, although his mother had psoriasis.
The dermatitis presents as the sudden onset of a diffuse exanthematous eruption, which typically produces dozens to hundreds of sterile, nonfollicular, superficial pustules on an erythematous and possibly edematous base. Atypical presentations include target lesions, purpura, and vesicles. The reaction usually begins on the face or intertriginous areas of flexural surfaces and quickly disseminates. Patients may experience burning or pruritus. Acute generalized exanthematous pustulosis may involve mucous membranes but is usually limited to 1 location, most often the oral mucosa.1,8,16,18 Systemic signs and symptoms include fever, lymphadenopathy, pharyngitis, and hepatosplenomegaly. Unlike most drug allergies that demonstrate eosinophilia, AGEP is associated with leukocytosis and neutrophilic predominance. Only 25% of affected patients exhibit eosinophilia.1 Approximately 30% of patients in a retrospective analysis demonstrated abnormal renal function,2 and there have been reports of mildly elevated transaminases.8,19
In the EuroSCAR study, for reasons that were not apparent, symptoms developed within 24 hours of exposure to triggering antibiotics, whereas the median time to rash onset in response to non–anti-infective agents was 11 days.8 This finding is consistent with the delayed onset of symptoms experienced by our patient after initiating ranolazine therapy.
The differential diagnosis of AGEP primarily includes pustular psoriasis, subcorneal pustulosis, pustular folliculitis, DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome, bullous impetigo, and occasionally erythema multiforme and toxic epidermal necrolysis, with the latter typically characterized by more mucous membrane involvement.20 Biopsy does not always support a definitive diagnosis; clinical correlation is often necessary. Because of the EuroSCAR study, Sidoroff et al8 devised a clinical validation score based on morphology (presence of pustules and erythema, distribution, and eventual desquamation), histopathology (presence of intraepidermal pustules, spongiosis, and papillary edema), and disease course (duration of symptoms, neutrophilia, fever, acute onset, and time to resolution). A definitive score is 8 to 12 (out of 12), and our patient’s score was 10; the score may have been higher had blood work been performed, but by the time the diagnosis was made the patient’s condition had improved enough to make laboratory workup unnecessary.
Several theories have been proposed to explain the pathophysiology of AGEP. Some hold that the causative agent induces the formation of antigen-antibody complexes, thereby activating the complement system, which in turn produces neutrophil chemotaxis.3,21 A more recent theory suggests that drug exposure causes drug-specific CD4 and CD8 cells to migrate into dermal and epidermal layers of the skin.17 Both T cells and keratinocytes express IL-8, which attracts polymorphonuclear leukocytes, causing them to accumulate in the dermis and then the epidermis. The different clinical presentations of AGEP may be attributed to other cytokines and interleukins that T cells express during this process. In the epidermis, CD8 cells kill keratinocytes, causing focal necrosis and prompting the formation of subcorneal vesicles filled primarily with CD4 cells. CD4 and CD8 cells are then localized to the dermis where neutrophils enter the vesicles, transforming them into sterile pustules.6,16,17
Acute generalized exanthematous pustulosis has been characterized as a type IV delayed hypersensitivity reaction, with affected patients often demonstrating positive patch testing or a history of prior sensitization to the perpetrating agent.18,19,21 Although there have been reports of positive patch testing for certain drugs, the unknown sensitivity and specificity of such testing as well as preparation-dependent variables may limit the diagnostic utility of this approach.21 The additional risk for inducing AGEP by patch testing the suspected drug also is a consideration. Due to our patient’s definitive clinical validation score, we did not perform this test.21
The AGEP eruption is typically self-limited and tends to resolve within 4 to 10 days after cessation of the triggering agent. Postpustular desquamation often occurs upon resolution of the primary lesions. Treatment usually involves discontinuation of the suspected causative agent and the use of antihistamines, antipyretics, topical corticosteroids, and emollients. Although there are reports of AGEP responsiveness to oral and intravenous steroids, such treatment rarely is required.8,16,22 We prescribed a tapered course of oral prednisone due to our patient’s imminent need for angioplasty.
Conclusion
This case of AGEP induced by ranolazine is notable. Given the potential widespread use of this antianginal medication and the severity of this potential adverse reaction, it is important for clinicians to recognize AGEP, discontinue ranolazine if determined to be a causative agent, and then initiate an appropriate alternative antianginal therapy.
Acute generalized exanthematous pustulosis (AGEP) is a potentially widespread, pustular, cutaneous eruption. In 90% of cases, AGEP results from drug administration.1,2 It manifests as numerous subcorneal, nonfollicular, sterile pustules of rapid onset on an erythematous base,2 often in conjunction with fever, peripheral leukocytosis, and neutrophilia.3 Numerous drug therapies have been implicated in the etiology of AGEP, most commonly the β-lactam antibiotics, such as the penicillin derivatives and cephalosporins.2 Typically, AGEP occurs soon after drug ingestion and resolves spontaneously, shortly after the causative drug is discontinued.
Ranolazine is an antianginal, anti-ischemic medication with an undetermined mechanism of action. Its antianginal and anti-ischemic effects do not depend on reduced heart rate or blood pressure. At therapeutic levels, it inhibits the cardiac late sodium current (INa), reducing the sodium-induced calcium overload in ischemic cardiac myocytes. Severe adverse reactions include angioedema; paresthesia; pancytopenia; and, in animal studies, tumorigenicity.4 Herein we report a case of AGEP associated with the use of ranolazine.
Case Report
An 83-year-old man presented with a generalized rash of approximately 12 days’ duration. The patient reported that the small “pimple-like” bumps initially erupted on the back of the neck but gradually spread to the chest, back, and extremities. The lesions were asymptomatic at the outset and became pruritic over time. For the last several years, the patient had been taking tamsulosin for benign prostatic hypertrophy and rosuvastatin for hyperlipidemia. Twelve days prior to the exanthem, he had started taking ranolazine for symptomatic ischemia until coronary angiography could be performed. He reported having no associated fevers, chills, or malaise and had no personal history of psoriasis, though he had a maternal history of the disorder.
Examination revealed numerous nonfollicular-based pustules on diffuse erythematous patches (Figure 1). There was no mucosal involvement and the skin was negative for the Nikolsky sign. Spongiform intracorneal collections of neutrophils were visible on punch biopsy (Figures 2 and 3). Periodic acid–Schiff stains for fungi were negative.
| | |
Figure 2. A punch biopsy showed spongiform intracorneal collections of neutrophils (H&E, original magnification ×200). | Figure 3. A cornified layer of epidermis with neutrophils, as visible on punch biopsy (H&E, original magnification ×630). |
The patient’s primary care physician had initiated a course of oral prednisone 5 mg daily, 3 days before he presented to our outpatient dermatology clinic, but it had little effect on the rash. Upon dermatologic evaluation, we discontinued ranolazine therapy and prescribed the following tapered course of oral prednisone: 60 mg daily for 4 days; 40 mg daily for 3 days; 30 mg daily for 3 days; 20 mg daily for 3 days; 10 mg daily for 3 days; and 5 mg daily for 3 days). Within a week after this regimen was initiated, the rash showed improvement with eventual resolution and desquamation (Figure 4). Subsequently, the patient underwent successful angioplasty and multiple stent placement, which ultimately alleviated his angina.
Comment
Since its original description in 1968,5 AGEP has been misdiagnosed and underreported. Due to its rarity and clinical resemblance to more common pustular eruptions, such as exanthematous pustular psoriasis, the typical characteristics of AGEP were not clearly delineated until Beylot et al3 coined the term AGEP in 1980. Since that time, formalized criteria for the diagnosis and characterization of AGEP have been published.1,2,6-8
Numerous drug therapies have been implicated in the etiology of AGEP, most commonly antimicrobial agents, such as β-lactam antibiotics. Many other drugs, however, also have been identified as potential causative agents,8 including but not limited to antifungal, anticonvulsant, and antihypertensive agents. Other less common etiologies include viral infections,6,9-11 UV radiation, contrast media, heavy metal exposure (eg, to mercury), ingestion of urushiol (eg, in lacquered chicken), and spider bites.2,8,12-16 Nevertheless, more than 90% of AGEP cases are attributed to drug exposure, with 80% of drug-induced cases believed to be caused by antibiotics.1,8
The incidence of AGEP is estimated to be between 1 and 5 cases per million per year, using inclusion criteria from the EuroSCAR study, a multinational, case-controlled, pharmacoepidemiologic study of severe cutaneous adverse reactions.8,16 The condition seems to affect males and females equally.1,4 There are no reports of age or racial predilection.1,6,17 It has been suggested that those with AGEP may have some form of psoriatic background.1 Our patient had no personal history of inflammatory skin disease, although his mother had psoriasis.
The dermatitis presents as the sudden onset of a diffuse exanthematous eruption, which typically produces dozens to hundreds of sterile, nonfollicular, superficial pustules on an erythematous and possibly edematous base. Atypical presentations include target lesions, purpura, and vesicles. The reaction usually begins on the face or intertriginous areas of flexural surfaces and quickly disseminates. Patients may experience burning or pruritus. Acute generalized exanthematous pustulosis may involve mucous membranes but is usually limited to 1 location, most often the oral mucosa.1,8,16,18 Systemic signs and symptoms include fever, lymphadenopathy, pharyngitis, and hepatosplenomegaly. Unlike most drug allergies that demonstrate eosinophilia, AGEP is associated with leukocytosis and neutrophilic predominance. Only 25% of affected patients exhibit eosinophilia.1 Approximately 30% of patients in a retrospective analysis demonstrated abnormal renal function,2 and there have been reports of mildly elevated transaminases.8,19
In the EuroSCAR study, for reasons that were not apparent, symptoms developed within 24 hours of exposure to triggering antibiotics, whereas the median time to rash onset in response to non–anti-infective agents was 11 days.8 This finding is consistent with the delayed onset of symptoms experienced by our patient after initiating ranolazine therapy.
The differential diagnosis of AGEP primarily includes pustular psoriasis, subcorneal pustulosis, pustular folliculitis, DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome, bullous impetigo, and occasionally erythema multiforme and toxic epidermal necrolysis, with the latter typically characterized by more mucous membrane involvement.20 Biopsy does not always support a definitive diagnosis; clinical correlation is often necessary. Because of the EuroSCAR study, Sidoroff et al8 devised a clinical validation score based on morphology (presence of pustules and erythema, distribution, and eventual desquamation), histopathology (presence of intraepidermal pustules, spongiosis, and papillary edema), and disease course (duration of symptoms, neutrophilia, fever, acute onset, and time to resolution). A definitive score is 8 to 12 (out of 12), and our patient’s score was 10; the score may have been higher had blood work been performed, but by the time the diagnosis was made the patient’s condition had improved enough to make laboratory workup unnecessary.
Several theories have been proposed to explain the pathophysiology of AGEP. Some hold that the causative agent induces the formation of antigen-antibody complexes, thereby activating the complement system, which in turn produces neutrophil chemotaxis.3,21 A more recent theory suggests that drug exposure causes drug-specific CD4 and CD8 cells to migrate into dermal and epidermal layers of the skin.17 Both T cells and keratinocytes express IL-8, which attracts polymorphonuclear leukocytes, causing them to accumulate in the dermis and then the epidermis. The different clinical presentations of AGEP may be attributed to other cytokines and interleukins that T cells express during this process. In the epidermis, CD8 cells kill keratinocytes, causing focal necrosis and prompting the formation of subcorneal vesicles filled primarily with CD4 cells. CD4 and CD8 cells are then localized to the dermis where neutrophils enter the vesicles, transforming them into sterile pustules.6,16,17
Acute generalized exanthematous pustulosis has been characterized as a type IV delayed hypersensitivity reaction, with affected patients often demonstrating positive patch testing or a history of prior sensitization to the perpetrating agent.18,19,21 Although there have been reports of positive patch testing for certain drugs, the unknown sensitivity and specificity of such testing as well as preparation-dependent variables may limit the diagnostic utility of this approach.21 The additional risk for inducing AGEP by patch testing the suspected drug also is a consideration. Due to our patient’s definitive clinical validation score, we did not perform this test.21
The AGEP eruption is typically self-limited and tends to resolve within 4 to 10 days after cessation of the triggering agent. Postpustular desquamation often occurs upon resolution of the primary lesions. Treatment usually involves discontinuation of the suspected causative agent and the use of antihistamines, antipyretics, topical corticosteroids, and emollients. Although there are reports of AGEP responsiveness to oral and intravenous steroids, such treatment rarely is required.8,16,22 We prescribed a tapered course of oral prednisone due to our patient’s imminent need for angioplasty.
Conclusion
This case of AGEP induced by ranolazine is notable. Given the potential widespread use of this antianginal medication and the severity of this potential adverse reaction, it is important for clinicians to recognize AGEP, discontinue ranolazine if determined to be a causative agent, and then initiate an appropriate alternative antianginal therapy.
1. Roujeau JC, Bioulac-Sage P, Bourseau C, et al. Acute generalized exanthematous pustulosis. analysis of 63 cases. Arch Dermatol. 1991;127:1333-1338.
2. Sidoroff A, Halevy S, Bavnick JN, et al. Acute generalized exanthematous pustulosis (AGEP)–a clinical reaction pattern. J Cutan Pathol. 2001;28:113-119.
3. Beylot C, Bioulac P, Doutre MS. Acute generalized exanthematic pustuloses (four cases) [in French]. Ann Dermatol Venereol. 1980;107:37-48.
4. Ranexa [package insert]. Foster City, CA: Gilead Sciences, Inc; December 2013.
5. Baker H, Ryan TJ. Generalized pustular psoriasis. a clinical and epidemiological study of 104 cases. Br J Dermatol. 1968;80:771-793.
6. Guevara-Gutierrez E, Uribe-Jimenez E, Diaz-Canchola M, et al. Acute generalized exanthematous pustulosis: report of 12 cases and literature review. Int J Dermatol. 2009;48:253-258.
7. Chang SL, Huang YH, Yang CH, et al. Clinical manifestations and characteristics of patients with acute generalized exanthematous pustulosis in Asia. Acta Derm Venereol. 2008;88:363-365.
8. Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR) [published online ahead of print September 13, 2007]. Br J Dermatol. 2007;157:989-996.
9. Rouchouse B, Bonnefoy M, Pallot B, et al. Acute generalized exanthematous pustular dermatitis and viral infection. Dermatologica. 1986;173:180-184.
10. Naides SJ, Piette W, Veach LA, et al. Human parvovirus B19-induced vesiculopustular skin eruption. Am J Med. 1988;84:968-972.
11. Feio AB, Apetato M, Costa MM, et al. Acute generalized exanthematous pustulosis due to Coxsackie B4 virus [in Portuguese]. Acta Med Port. 1997;10:487-491.
12. Goh TK, Pang SM, Thirumoorthy T, et al. Acute generalised exanthematous pustulosis and toxic epidermal necrolysis induced by carbamazepine. Singapore Med J. 2008;49:507-510.
13. Ofuji S, Yamamoto O. Acute generalized exanthematous pustulosis associated with a human parvovirus B19 infection. J Dermatol. 2007;34:121-123.
14. Davidovici BB, Pavel D, Cagnano E, et al. Acute generalized exanthematous pustulosis following a spider bite: report of 3 cases. J Am Acad Dermatol. 2006;55:525-529.
15. Park YM, Park JG, Kang H, et al. Acute generalized exanthematous pustulosis induced by ingestion of lacquer chicken. Br J Dermatol. 2000;143:230-232.
16. Hammerbeck AA, Daniels NH, Callen JP. Ioversol-induced acute generalized exanthematous pustulosis: a case report. Arch Dermatol. 2009;145:683-687.17. Halevy S. Acute generalized exanthematous pustulosis. Curr Opin Allergy Clin Immunol. 2009;9:322-328.
18. Kim HJ, Jung KD, Lee KT, et al. Acute generalized exanthematous pustulosis caused by diltiazem [published online ahead of print February 28, 2011]. Ann Dermatol. 2011;23:108-110.
19. Speck LM, Wilkerson MG, Perri AJ, et al. Acute generalized exanthematous pustulosis caused by terazosin hydrochloride. J Drugs Dermatol. 2008;7:395-397.
20. Sidoroff A. Acute generalized exanthematous pustulosis (AGEP). UpToDate Web site. http://www.uptodate.com /contents/acute-generalized-exanthematous-pustulosis -agep?source=search_result&search=agep&selected Title=1~85. Updated March 18, 2015. Accessed October 6, 2015.
21. Mashiah J, Brenner S. A systemic reaction to patch testing for the evaluation of acute generalized exanthematous pustulosis. Arch Dermatol. 2003;139:1181-1183.
22. Ibrahimi O, Gunawardane N, Sepehr A, et al. Terbinafine-induced acute generalized exanthematous pustulosis (AGEP) responsive to high dose intravenous corticosteroid. Dermatol Online J. 2009;15:8.
1. Roujeau JC, Bioulac-Sage P, Bourseau C, et al. Acute generalized exanthematous pustulosis. analysis of 63 cases. Arch Dermatol. 1991;127:1333-1338.
2. Sidoroff A, Halevy S, Bavnick JN, et al. Acute generalized exanthematous pustulosis (AGEP)–a clinical reaction pattern. J Cutan Pathol. 2001;28:113-119.
3. Beylot C, Bioulac P, Doutre MS. Acute generalized exanthematic pustuloses (four cases) [in French]. Ann Dermatol Venereol. 1980;107:37-48.
4. Ranexa [package insert]. Foster City, CA: Gilead Sciences, Inc; December 2013.
5. Baker H, Ryan TJ. Generalized pustular psoriasis. a clinical and epidemiological study of 104 cases. Br J Dermatol. 1968;80:771-793.
6. Guevara-Gutierrez E, Uribe-Jimenez E, Diaz-Canchola M, et al. Acute generalized exanthematous pustulosis: report of 12 cases and literature review. Int J Dermatol. 2009;48:253-258.
7. Chang SL, Huang YH, Yang CH, et al. Clinical manifestations and characteristics of patients with acute generalized exanthematous pustulosis in Asia. Acta Derm Venereol. 2008;88:363-365.
8. Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR) [published online ahead of print September 13, 2007]. Br J Dermatol. 2007;157:989-996.
9. Rouchouse B, Bonnefoy M, Pallot B, et al. Acute generalized exanthematous pustular dermatitis and viral infection. Dermatologica. 1986;173:180-184.
10. Naides SJ, Piette W, Veach LA, et al. Human parvovirus B19-induced vesiculopustular skin eruption. Am J Med. 1988;84:968-972.
11. Feio AB, Apetato M, Costa MM, et al. Acute generalized exanthematous pustulosis due to Coxsackie B4 virus [in Portuguese]. Acta Med Port. 1997;10:487-491.
12. Goh TK, Pang SM, Thirumoorthy T, et al. Acute generalised exanthematous pustulosis and toxic epidermal necrolysis induced by carbamazepine. Singapore Med J. 2008;49:507-510.
13. Ofuji S, Yamamoto O. Acute generalized exanthematous pustulosis associated with a human parvovirus B19 infection. J Dermatol. 2007;34:121-123.
14. Davidovici BB, Pavel D, Cagnano E, et al. Acute generalized exanthematous pustulosis following a spider bite: report of 3 cases. J Am Acad Dermatol. 2006;55:525-529.
15. Park YM, Park JG, Kang H, et al. Acute generalized exanthematous pustulosis induced by ingestion of lacquer chicken. Br J Dermatol. 2000;143:230-232.
16. Hammerbeck AA, Daniels NH, Callen JP. Ioversol-induced acute generalized exanthematous pustulosis: a case report. Arch Dermatol. 2009;145:683-687.17. Halevy S. Acute generalized exanthematous pustulosis. Curr Opin Allergy Clin Immunol. 2009;9:322-328.
18. Kim HJ, Jung KD, Lee KT, et al. Acute generalized exanthematous pustulosis caused by diltiazem [published online ahead of print February 28, 2011]. Ann Dermatol. 2011;23:108-110.
19. Speck LM, Wilkerson MG, Perri AJ, et al. Acute generalized exanthematous pustulosis caused by terazosin hydrochloride. J Drugs Dermatol. 2008;7:395-397.
20. Sidoroff A. Acute generalized exanthematous pustulosis (AGEP). UpToDate Web site. http://www.uptodate.com /contents/acute-generalized-exanthematous-pustulosis -agep?source=search_result&search=agep&selected Title=1~85. Updated March 18, 2015. Accessed October 6, 2015.
21. Mashiah J, Brenner S. A systemic reaction to patch testing for the evaluation of acute generalized exanthematous pustulosis. Arch Dermatol. 2003;139:1181-1183.
22. Ibrahimi O, Gunawardane N, Sepehr A, et al. Terbinafine-induced acute generalized exanthematous pustulosis (AGEP) responsive to high dose intravenous corticosteroid. Dermatol Online J. 2009;15:8.
Practice Points
- Encountering an acute pustular reaction pattern should trigger the clinician to rule out acute generalized exanthematous pustulosis (AGEP).
- Ranolazine, a new antianginal therapy, has been associated with AGEP.
- Upon confirmation of AGEP, the patient’s recent medication history should be reviewed so the potential causative agent can be identified and withdrawn.
Fever • eschars on right leg and groin • inguinal lymphadenopathy • Dx?
THE CASE
A 76-year-old man with a history of coronary artery disease presented with a fever, headache, and malaise one week after returning from a big game hunting trip in South Africa. Five days after his return, he noticed lesions on his right leg that eventually scabbed over. He sought care at his local emergency department and with his primary care physician, and completed an empiric trial of azithromycin. His symptoms, however, persisted and he was referred to our institution for evaluation and treatment.
On exam, he had a temperature of 100.5° F, inguinal lymphadenopathy, and 2 eschars: a 1.5 cm one on his right groin and an identical one on the medial aspect of the right popliteal fossa (FIGURE 1).
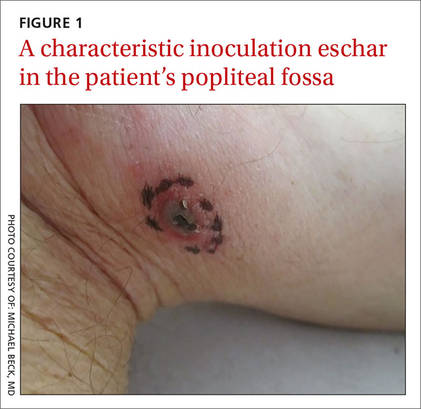
THE DIAGNOSIS
Laboratory studies showed a white blood cell count of 3000/mcL, hemoglobin of 14.1 g/dL, and platelet count of 142,000/mcL; peripheral blood smear was normal. Blood and urine cultures showed no growth. A malaria smear and antibodies for Lyme disease, dengue fever, Chikungunya virus, and Q fever were also negative. A biopsy of the eschar demonstrated epidermal and dermal necrosis consistent with infectious vasculitis caused by rickettsial disease (FIGURE 2). A polymerase chain reaction (PCR) for the spotted fever group (R rickettsii, R akari, and R conorii) and typhus fever group of rickettsial agents (R typhi and R prowazekii) were negative. However, a PCR was positive for R africae, confirming the diagnosis of African tick-bite fever (ATBF).
DISCUSSION
Two common reasons patients returning from international travel seek medical attention are fever and rash.1 Initial assessment should include a detailed travel history of urban and rural exposures and any possible exposure to ticks or fleas. The time course of symptoms is important because some tropical infections can have long incubation periods.2
Rickettsial diseases are the most common febrile illness in patients returning from international travel.1 ATBF caused by R africae is the most common rickettsiosis among returning travelers1 and may be the most widespread of all spotted fever group rickettsiae that are known to be pathogenic to humans.3R africae is endemic to South Africa. The risk of contracting R africae is 4 to 5 times higher than the risk of contracting malaria in South Africa.1
R africae is transmitted through cattle and game ticks (Amblyomma species),1-7 and tends to cause mild illness with rare progression to complicated disease.3 The risk of infection is particularly high from November to April,7 and our patient had traveled during April.
Most patients with ATBF present with fever, headache, and malaise, and 50% develop a variable rash.1,2 Local lymphadenopathy often develops, and marked neck stiffness can occur.2 An eschar is present in 95% of cases.2 The finding of an eschar is often indicative of rickettsial infection; however, not all rickettsioses show eschars, and the absence of an eschar does not exclude rickettsial infection.1
ATBF is usually benign and self-limiting, and no fatalities have been reported.2,4,8 Complications such as peripheral nerve involvement, encephalitis, and myocarditis are rare.5,8 Since rickettsial diseases may be more severe in elderly patients with underlying diseases, empiric treatment with inpatient monitoring is justifiable.5
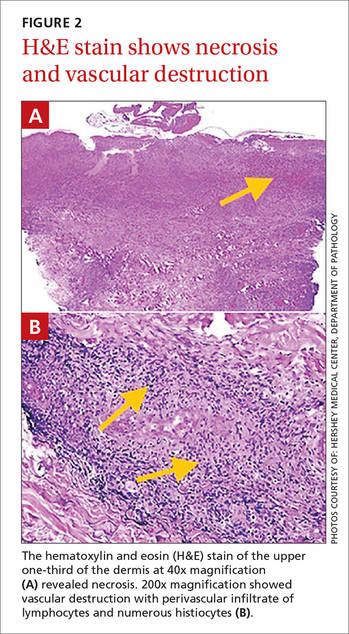
Don’t wait for lab confirmation to begin antibiotics
Laboratory findings in a patient with ATBF include pancytopenia, elevated serum C-reactive protein, and abnormal liver function tests.2,4 A blood PCR detects R africae,1,3 and if a rash or eschar is present, a biopsy can confirm the diagnosis.1 However, confirming the diagnosis is difficult if seroconversion has not occurred and PCR is not readily available.1 Also, antibodies may not be detected in patients who have a mild case of ATBF or those who are immunocompromised.5
If you suspect your patient has ATBF, don’t wait for laboratory confirmation; instead, initiate empiric treatment with doxycycline 100 mg bid twice daily for 5 to 7 days.1,2,4
Preventive measures include repellant lotions, clothing, and gear
Since there are no vaccines or prophylactic treatments for ATBF, counsel travelers on preventive measures.9 Instruct patients who plan to visit an endemic area such as sub-Saharan Africa or the West Indies to wear long-sleeved shirts, long pants, and hats. Individuals should also tuck their shirts into their pants and their pants into socks, as well as wear closed-toe shoes. When possible, it’s advisable to avoid woody and brushy areas.
Over-the-counter repellant lotions that contain ≥20% DEET are effective at preventing tick bites for several hours after an application, but should be reapplied as directed.9 These lotions should be applied after sunscreen. Advise patients that they can purchase clothing and gear that have been treated with the pesticide permethrin, or they can treat clothing and gear themselves. Explain, however, that permethrin should not be applied directly to skin.
Finally, instruct travelers to perform daily tick inspections and shower or bathe as soon as possible after returning from the outdoors.9 Educate patients on the proper technique for tick removal, which is described on the Centers for Disease Control and Prevention’s Web site at http://www.cdc.gov/ticks/removing_a_tick.html.
Our patient completed a 2-week course of doxycycline 100 mg bid. His symptoms and laboratory abnormalities completely resolved.
THE TAKEAWAY
ATBF is the most common rickettsiosis among patients returning from international travel. Because patients may present with several nonspecific symptoms, maintain a high index of suspicion for rickettsial infection among patients returning from sub-Saharan Africa or the West Indies. Though the disease usually can be successfully managed in the outpatient setting with doxycycline, elderly patients or those with comorbid conditions may require inpatient care. Educate patients who plan to travel to an endemic area about measures they can take to prevent exposure to ticks and subsequent infection.
1. Neumayr A, Hatz C, Blum J. Not to be missed! Differential diagnoses of common dermatological problems in returning travellers. Travel Med Infect Dis. 2013;11:337-349.
2. Yates J, Smith P. Fever and rash. Medicine. 2014;42:96-99.
3. Jensenius M, Fournier PE, Kelly P, et al. African tick bite fever. Lancet Infect Dis. 2003;3:557-564.
4. Frean J, Blumberg L, Ogunbanjo GA. Tick bite fever in South Africa. S Afr Fam Pract. 2008;50:33-35.
5. Roch N, Epaulard O, Pelloux I, et al. African tick bite fever in elderly patients: 8 cases in French tourists returning from South Africa. Clin Infect Dis. 2008;47:e28-e35.
6. Caruso G, Zasio C, Guzzo F, et al. Outbreak of African tick-bite fever in six Italian tourists returning from South Africa. Eur J Clin Microbiol Infect Dis. 2002;21:133-136.
7. Tsai YS, Wu YH, Kao PT, et al. African tick bite fever. J Formos Med Assoc. 2008;107:73-76.
8. Jensenius M, Fournier PE, Fladby T, et al. Sub-acute neuropathy in patients with African tick bite fever. Scand J Infect Dis. 2006;38:114-118.
9. Centers for Disease Control and Prevention. African tick-bite fever. Centers for Disease Control and Prevention Web site. Available at: http://wwwnc.cdc.gov/travel/diseases/african-tick-bite-fever. Accessed September 24, 2015.
THE CASE
A 76-year-old man with a history of coronary artery disease presented with a fever, headache, and malaise one week after returning from a big game hunting trip in South Africa. Five days after his return, he noticed lesions on his right leg that eventually scabbed over. He sought care at his local emergency department and with his primary care physician, and completed an empiric trial of azithromycin. His symptoms, however, persisted and he was referred to our institution for evaluation and treatment.
On exam, he had a temperature of 100.5° F, inguinal lymphadenopathy, and 2 eschars: a 1.5 cm one on his right groin and an identical one on the medial aspect of the right popliteal fossa (FIGURE 1).
THE DIAGNOSIS
Laboratory studies showed a white blood cell count of 3000/mcL, hemoglobin of 14.1 g/dL, and platelet count of 142,000/mcL; peripheral blood smear was normal. Blood and urine cultures showed no growth. A malaria smear and antibodies for Lyme disease, dengue fever, Chikungunya virus, and Q fever were also negative. A biopsy of the eschar demonstrated epidermal and dermal necrosis consistent with infectious vasculitis caused by rickettsial disease (FIGURE 2). A polymerase chain reaction (PCR) for the spotted fever group (R rickettsii, R akari, and R conorii) and typhus fever group of rickettsial agents (R typhi and R prowazekii) were negative. However, a PCR was positive for R africae, confirming the diagnosis of African tick-bite fever (ATBF).
DISCUSSION
Two common reasons patients returning from international travel seek medical attention are fever and rash.1 Initial assessment should include a detailed travel history of urban and rural exposures and any possible exposure to ticks or fleas. The time course of symptoms is important because some tropical infections can have long incubation periods.2
Rickettsial diseases are the most common febrile illness in patients returning from international travel.1 ATBF caused by R africae is the most common rickettsiosis among returning travelers1 and may be the most widespread of all spotted fever group rickettsiae that are known to be pathogenic to humans.3R africae is endemic to South Africa. The risk of contracting R africae is 4 to 5 times higher than the risk of contracting malaria in South Africa.1
R africae is transmitted through cattle and game ticks (Amblyomma species),1-7 and tends to cause mild illness with rare progression to complicated disease.3 The risk of infection is particularly high from November to April,7 and our patient had traveled during April.
Most patients with ATBF present with fever, headache, and malaise, and 50% develop a variable rash.1,2 Local lymphadenopathy often develops, and marked neck stiffness can occur.2 An eschar is present in 95% of cases.2 The finding of an eschar is often indicative of rickettsial infection; however, not all rickettsioses show eschars, and the absence of an eschar does not exclude rickettsial infection.1
ATBF is usually benign and self-limiting, and no fatalities have been reported.2,4,8 Complications such as peripheral nerve involvement, encephalitis, and myocarditis are rare.5,8 Since rickettsial diseases may be more severe in elderly patients with underlying diseases, empiric treatment with inpatient monitoring is justifiable.5
Don’t wait for lab confirmation to begin antibiotics
Laboratory findings in a patient with ATBF include pancytopenia, elevated serum C-reactive protein, and abnormal liver function tests.2,4 A blood PCR detects R africae,1,3 and if a rash or eschar is present, a biopsy can confirm the diagnosis.1 However, confirming the diagnosis is difficult if seroconversion has not occurred and PCR is not readily available.1 Also, antibodies may not be detected in patients who have a mild case of ATBF or those who are immunocompromised.5
If you suspect your patient has ATBF, don’t wait for laboratory confirmation; instead, initiate empiric treatment with doxycycline 100 mg bid twice daily for 5 to 7 days.1,2,4
Preventive measures include repellant lotions, clothing, and gear
Since there are no vaccines or prophylactic treatments for ATBF, counsel travelers on preventive measures.9 Instruct patients who plan to visit an endemic area such as sub-Saharan Africa or the West Indies to wear long-sleeved shirts, long pants, and hats. Individuals should also tuck their shirts into their pants and their pants into socks, as well as wear closed-toe shoes. When possible, it’s advisable to avoid woody and brushy areas.
Over-the-counter repellant lotions that contain ≥20% DEET are effective at preventing tick bites for several hours after an application, but should be reapplied as directed.9 These lotions should be applied after sunscreen. Advise patients that they can purchase clothing and gear that have been treated with the pesticide permethrin, or they can treat clothing and gear themselves. Explain, however, that permethrin should not be applied directly to skin.
Finally, instruct travelers to perform daily tick inspections and shower or bathe as soon as possible after returning from the outdoors.9 Educate patients on the proper technique for tick removal, which is described on the Centers for Disease Control and Prevention’s Web site at http://www.cdc.gov/ticks/removing_a_tick.html.
Our patient completed a 2-week course of doxycycline 100 mg bid. His symptoms and laboratory abnormalities completely resolved.
THE TAKEAWAY
ATBF is the most common rickettsiosis among patients returning from international travel. Because patients may present with several nonspecific symptoms, maintain a high index of suspicion for rickettsial infection among patients returning from sub-Saharan Africa or the West Indies. Though the disease usually can be successfully managed in the outpatient setting with doxycycline, elderly patients or those with comorbid conditions may require inpatient care. Educate patients who plan to travel to an endemic area about measures they can take to prevent exposure to ticks and subsequent infection.
THE CASE
A 76-year-old man with a history of coronary artery disease presented with a fever, headache, and malaise one week after returning from a big game hunting trip in South Africa. Five days after his return, he noticed lesions on his right leg that eventually scabbed over. He sought care at his local emergency department and with his primary care physician, and completed an empiric trial of azithromycin. His symptoms, however, persisted and he was referred to our institution for evaluation and treatment.
On exam, he had a temperature of 100.5° F, inguinal lymphadenopathy, and 2 eschars: a 1.5 cm one on his right groin and an identical one on the medial aspect of the right popliteal fossa (FIGURE 1).
THE DIAGNOSIS
Laboratory studies showed a white blood cell count of 3000/mcL, hemoglobin of 14.1 g/dL, and platelet count of 142,000/mcL; peripheral blood smear was normal. Blood and urine cultures showed no growth. A malaria smear and antibodies for Lyme disease, dengue fever, Chikungunya virus, and Q fever were also negative. A biopsy of the eschar demonstrated epidermal and dermal necrosis consistent with infectious vasculitis caused by rickettsial disease (FIGURE 2). A polymerase chain reaction (PCR) for the spotted fever group (R rickettsii, R akari, and R conorii) and typhus fever group of rickettsial agents (R typhi and R prowazekii) were negative. However, a PCR was positive for R africae, confirming the diagnosis of African tick-bite fever (ATBF).
DISCUSSION
Two common reasons patients returning from international travel seek medical attention are fever and rash.1 Initial assessment should include a detailed travel history of urban and rural exposures and any possible exposure to ticks or fleas. The time course of symptoms is important because some tropical infections can have long incubation periods.2
Rickettsial diseases are the most common febrile illness in patients returning from international travel.1 ATBF caused by R africae is the most common rickettsiosis among returning travelers1 and may be the most widespread of all spotted fever group rickettsiae that are known to be pathogenic to humans.3R africae is endemic to South Africa. The risk of contracting R africae is 4 to 5 times higher than the risk of contracting malaria in South Africa.1
R africae is transmitted through cattle and game ticks (Amblyomma species),1-7 and tends to cause mild illness with rare progression to complicated disease.3 The risk of infection is particularly high from November to April,7 and our patient had traveled during April.
Most patients with ATBF present with fever, headache, and malaise, and 50% develop a variable rash.1,2 Local lymphadenopathy often develops, and marked neck stiffness can occur.2 An eschar is present in 95% of cases.2 The finding of an eschar is often indicative of rickettsial infection; however, not all rickettsioses show eschars, and the absence of an eschar does not exclude rickettsial infection.1
ATBF is usually benign and self-limiting, and no fatalities have been reported.2,4,8 Complications such as peripheral nerve involvement, encephalitis, and myocarditis are rare.5,8 Since rickettsial diseases may be more severe in elderly patients with underlying diseases, empiric treatment with inpatient monitoring is justifiable.5
Don’t wait for lab confirmation to begin antibiotics
Laboratory findings in a patient with ATBF include pancytopenia, elevated serum C-reactive protein, and abnormal liver function tests.2,4 A blood PCR detects R africae,1,3 and if a rash or eschar is present, a biopsy can confirm the diagnosis.1 However, confirming the diagnosis is difficult if seroconversion has not occurred and PCR is not readily available.1 Also, antibodies may not be detected in patients who have a mild case of ATBF or those who are immunocompromised.5
If you suspect your patient has ATBF, don’t wait for laboratory confirmation; instead, initiate empiric treatment with doxycycline 100 mg bid twice daily for 5 to 7 days.1,2,4
Preventive measures include repellant lotions, clothing, and gear
Since there are no vaccines or prophylactic treatments for ATBF, counsel travelers on preventive measures.9 Instruct patients who plan to visit an endemic area such as sub-Saharan Africa or the West Indies to wear long-sleeved shirts, long pants, and hats. Individuals should also tuck their shirts into their pants and their pants into socks, as well as wear closed-toe shoes. When possible, it’s advisable to avoid woody and brushy areas.
Over-the-counter repellant lotions that contain ≥20% DEET are effective at preventing tick bites for several hours after an application, but should be reapplied as directed.9 These lotions should be applied after sunscreen. Advise patients that they can purchase clothing and gear that have been treated with the pesticide permethrin, or they can treat clothing and gear themselves. Explain, however, that permethrin should not be applied directly to skin.
Finally, instruct travelers to perform daily tick inspections and shower or bathe as soon as possible after returning from the outdoors.9 Educate patients on the proper technique for tick removal, which is described on the Centers for Disease Control and Prevention’s Web site at http://www.cdc.gov/ticks/removing_a_tick.html.
Our patient completed a 2-week course of doxycycline 100 mg bid. His symptoms and laboratory abnormalities completely resolved.
THE TAKEAWAY
ATBF is the most common rickettsiosis among patients returning from international travel. Because patients may present with several nonspecific symptoms, maintain a high index of suspicion for rickettsial infection among patients returning from sub-Saharan Africa or the West Indies. Though the disease usually can be successfully managed in the outpatient setting with doxycycline, elderly patients or those with comorbid conditions may require inpatient care. Educate patients who plan to travel to an endemic area about measures they can take to prevent exposure to ticks and subsequent infection.
1. Neumayr A, Hatz C, Blum J. Not to be missed! Differential diagnoses of common dermatological problems in returning travellers. Travel Med Infect Dis. 2013;11:337-349.
2. Yates J, Smith P. Fever and rash. Medicine. 2014;42:96-99.
3. Jensenius M, Fournier PE, Kelly P, et al. African tick bite fever. Lancet Infect Dis. 2003;3:557-564.
4. Frean J, Blumberg L, Ogunbanjo GA. Tick bite fever in South Africa. S Afr Fam Pract. 2008;50:33-35.
5. Roch N, Epaulard O, Pelloux I, et al. African tick bite fever in elderly patients: 8 cases in French tourists returning from South Africa. Clin Infect Dis. 2008;47:e28-e35.
6. Caruso G, Zasio C, Guzzo F, et al. Outbreak of African tick-bite fever in six Italian tourists returning from South Africa. Eur J Clin Microbiol Infect Dis. 2002;21:133-136.
7. Tsai YS, Wu YH, Kao PT, et al. African tick bite fever. J Formos Med Assoc. 2008;107:73-76.
8. Jensenius M, Fournier PE, Fladby T, et al. Sub-acute neuropathy in patients with African tick bite fever. Scand J Infect Dis. 2006;38:114-118.
9. Centers for Disease Control and Prevention. African tick-bite fever. Centers for Disease Control and Prevention Web site. Available at: http://wwwnc.cdc.gov/travel/diseases/african-tick-bite-fever. Accessed September 24, 2015.
1. Neumayr A, Hatz C, Blum J. Not to be missed! Differential diagnoses of common dermatological problems in returning travellers. Travel Med Infect Dis. 2013;11:337-349.
2. Yates J, Smith P. Fever and rash. Medicine. 2014;42:96-99.
3. Jensenius M, Fournier PE, Kelly P, et al. African tick bite fever. Lancet Infect Dis. 2003;3:557-564.
4. Frean J, Blumberg L, Ogunbanjo GA. Tick bite fever in South Africa. S Afr Fam Pract. 2008;50:33-35.
5. Roch N, Epaulard O, Pelloux I, et al. African tick bite fever in elderly patients: 8 cases in French tourists returning from South Africa. Clin Infect Dis. 2008;47:e28-e35.
6. Caruso G, Zasio C, Guzzo F, et al. Outbreak of African tick-bite fever in six Italian tourists returning from South Africa. Eur J Clin Microbiol Infect Dis. 2002;21:133-136.
7. Tsai YS, Wu YH, Kao PT, et al. African tick bite fever. J Formos Med Assoc. 2008;107:73-76.
8. Jensenius M, Fournier PE, Fladby T, et al. Sub-acute neuropathy in patients with African tick bite fever. Scand J Infect Dis. 2006;38:114-118.
9. Centers for Disease Control and Prevention. African tick-bite fever. Centers for Disease Control and Prevention Web site. Available at: http://wwwnc.cdc.gov/travel/diseases/african-tick-bite-fever. Accessed September 24, 2015.

Case Report: Not Just Another Kidney Stone
Case
A 36-year-old woman with a 2-week history of left flank pain presented to the ED via emergency medical services. The patient, who had a history of nephrolithiasis, assumed her pain was due to another kidney stone. She stated that while waiting for the presumed stone to pass, the pain in her left flank worsened and she felt lightheaded and weak.
The patient’s vital signs at presentation were: heart rate, 96 beats/minute; blood pressure, 133/76 mm Hg; respiratory rate, 20 breaths/minute; and temperature, 98.9˚F. Oxygen saturation was 98% on room air. On physical examination, the patient had left lower quadrant pain and left costovertebral angle tenderness. Laboratory studies were remarkable for a negative urine pregnancy test, a hemoglobin level of 6.8 g/dL, and a hematocrit of 21.1%. Based on the patient’s history and symptoms, axial and coronal computed tomography (CT) scans were ordered, revealing a ruptured left renal calyx with hemorrhage from ureterolithiasis (Figures 1a and 1b).
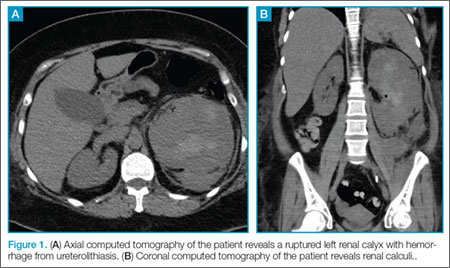
Rupture of renal calyx and extravasation of blood or urine is a potential complication of nephrolithiasis. Stone size, degree of obstruction, and length of symptomatic presentation presumably contribute to complications from nephrolithiasis. Stones that are symptomatic for more than 4 weeks are estimated to have an increased complication rate of up to 20%.1
Calyx or fornix rupture results from increased intraluminal pressure. Rupture of these structures is thought to be a type of “safety-valve” function to relieve obstructive uropathy.2
Obstructions from small leaks to large urinomas can cause extravasation of urine. In most cases, urinary extravasation is confined to the subcapsular space or perirenal space within the Gerota’s fascia;3 however, as seen in this patient, mixed hematoma/urinomas can form.
Causes
In cases of nontraumatic calyx rupture, the cause of the obstruction is most often a distal obstructing ureteral stone.4 Other causes of rupture include extrinsic compression from malignant and benign masses, ureteric junction obstructions, or iatrogenic causes.4 Interestingly, in one small study, the median size of the obstructing stone was only 4 mm. The same study also noted that proximal ureteral obstruction occurred when larger stones where present.4
Conservative Versus Nonconservative Management
Potential complications of urinomas include abscess formation, sepsis, hydronephrosis, and paralytic ileus.3 Despite possible adverse sequelae, uncomplicated urinomas may be managed conservatively with supportive care. According to a study by Chapman et al,5 about 40% of patients managed conservatively recover without complications. In addition, in a retrospective study by Doehn et al6 involving 160 cases of fornix rupture treated with endoscopic therapy or nephrostomy tube supplemented with antibiotics, no instances of perinephric abscess or other complications requiring a second procedure were noted.
Management of suspected ureterolithiasis in the ED is focused on analgesia and supportive care. Acute analgesia is often provided parenterally with opioids alone or with an opioid/nonsteroidal anti-inflammatory drug (NSAID) combination.7 Frequent reassessment of the patient is required to ensure adequate pain control and to prevent sedation. Other symptoms, such as nausea, vomiting, and dehydration, may be treated with intravenous (IV) fluids and antiemetic medications. Further radiographic evaluation is needed once analgesia is achieved.7,8
Imaging Studies
Radiological evaluation of patients with suspected ureterolithiasis may involve several imaging modalities. Noncontrast helical CT scan is the standard for rapid and efficient identification of ureteral stones while allowing visualization of other potential pathology (eg, urinoma).7-9 Other modalities, such as ultrasonography; radiography of the kidneys, ureters, and bladder; and an IV pyelogram with contrasted CT, may be ordered if noncontrast helical CT scan is not available on-site or if there are comorbidities. In addition to imaging studies, basic laboratory studies (eg, serum creatinine and blood urea nitrogen testing) are indicated to assess overall renal function and direct the choice of radiological study.7
Disposition
Clinical decision-making is key when recommending inpatient versus outpatient treatment in patients with ureterolithiasis. Patients with uncontrolled pain or vomiting may require inpatient admission for supportive care, while those demonstrating acute renal failure, pyuria with bacteriuria, complete bilateral ureteral obstruction, urinoma, or signs of sepsis demand emergent urology consultation. Specifically, patients with urinoma require ureteroscopy versus nephrostomy6,10 to allow drainage while carefully monitoring for development of subsequent bleeding and infection.
When discharging patients from the ED, expulsive therapy using tamsulosin9 and analgesia with combination of oral opioids and NSAIDs are most commonly effective.11 Outpatient urology referrals are recommended for ureteral stones greater than 5 mm in size or if the stones have been present in the ureter for greater than 4 weeks.1 Proper evaluation and management of ureterolithiasis in the ED is crucial for positive outcomes and to reduce long-term complications.
Case Conclusion
Computed tomography revealed a ruptured renal calyx on the left side with free fluid in the abdomen. Urology services were consulted and the patient was taken to the operating room for cystoscopy, ureteral stent placement, and laser lithotripsy. Following surgery, she subsequently developed urosepsis for which she was successfully treated with IV antibiotics and discharged on hospital day 15.
Mr Eisenstat is a fourth-year medical student at the University of South Carolina School of Medicine, Greenville. Dr Fabiano is an emergency physician, department of emergency medicine, Greenville Health Systems, Greenville, South Carolina. Dr Collins is family medicine physician, department of emergency medicine, Greenville Health Systems, Greenville, South Carolina.
- Hübner WA, Irby P, Stoller M. Natural history and current concepts for the treatment of small ureteral calculi. Eur Urol. 1993;24(2):172-176.
- Lin DY, Fang YC, Huang DY, Lin SP. Spontaneous rupture of the ureter secondary to urolithiasis and extravasation of calyceal fornix due to acute urinary bladder distension: four case reports. Chin J Radiology. 2004;29:269-275.
- Behzad-Noori M, Blandon JA, Negrin Exposito JE, Sarmiento JL, Dias AL, Hernandez GT. Urinoma: a rare complication from being between a rock and soft organ. El Paso Physician. 2010;33(6):5-6.
- Gershman B, Kulkarni N, Sahani DV, Eisner BH. Causes of renal forniceal rupture. BJU Int. 2011;108(11):1909-1911.
- Chapman JP, Gonzalez J, Diokno AC. Significance of urinary extravasation during renal colic. Urology. 1987;30(6):541-545.
- Doehn C, Fiola L, Peter M, Jocham D. Outcome analysis of fornix ruptures in 162 consecutive patients. J Endourol. 2010;24(11):1869-1873
- Portis AJ, Sundaram CP. Diagnosis and initial management of kidney stones. Am Fam Physician. 2001;63(7):1329-1338
- Smith RC, Verga M, Dalrymple N, McCarthy S, Rosenfield AT. Acute ureteral obstruction: value of secondary signs of helical unenhanced CT. AJR Am J Roentgenol. 1996;167(5):1109-1113.Burke TA, Wisniewski T, Ernst FR. Resource utilization and costs associated with chemotherapy-induced nausea and vomiting (CINV) following highly or moderately emetogenic chemotherapy administered in the US outpatient hospital setting. Support Care Cancer. 2011;19(1):131-140.
- Ha M, MacDonald RD. Impact of CT scan in patients with first episode of suspected nephrolithiasis. J Emerg Med. 2004;27(3):225-231.
- Tawfiek ER, Bagley DH. Management of upper urinary tract calculi with ureteroscopic techniques. Urology. 1999;53(1):25-31.
- Larkin GL, Peacock WF 4th, Pearl SM, Blair GA, D'Amico F. Efficacy of ketorolac tromethamine versus meperidine in the ED treatment of acute renal colic. Am J Emerg Med. 1999;17(1):6-10.
Case
A 36-year-old woman with a 2-week history of left flank pain presented to the ED via emergency medical services. The patient, who had a history of nephrolithiasis, assumed her pain was due to another kidney stone. She stated that while waiting for the presumed stone to pass, the pain in her left flank worsened and she felt lightheaded and weak.
The patient’s vital signs at presentation were: heart rate, 96 beats/minute; blood pressure, 133/76 mm Hg; respiratory rate, 20 breaths/minute; and temperature, 98.9˚F. Oxygen saturation was 98% on room air. On physical examination, the patient had left lower quadrant pain and left costovertebral angle tenderness. Laboratory studies were remarkable for a negative urine pregnancy test, a hemoglobin level of 6.8 g/dL, and a hematocrit of 21.1%. Based on the patient’s history and symptoms, axial and coronal computed tomography (CT) scans were ordered, revealing a ruptured left renal calyx with hemorrhage from ureterolithiasis (Figures 1a and 1b).
Rupture of renal calyx and extravasation of blood or urine is a potential complication of nephrolithiasis. Stone size, degree of obstruction, and length of symptomatic presentation presumably contribute to complications from nephrolithiasis. Stones that are symptomatic for more than 4 weeks are estimated to have an increased complication rate of up to 20%.1
Calyx or fornix rupture results from increased intraluminal pressure. Rupture of these structures is thought to be a type of “safety-valve” function to relieve obstructive uropathy.2
Obstructions from small leaks to large urinomas can cause extravasation of urine. In most cases, urinary extravasation is confined to the subcapsular space or perirenal space within the Gerota’s fascia;3 however, as seen in this patient, mixed hematoma/urinomas can form.
Causes
In cases of nontraumatic calyx rupture, the cause of the obstruction is most often a distal obstructing ureteral stone.4 Other causes of rupture include extrinsic compression from malignant and benign masses, ureteric junction obstructions, or iatrogenic causes.4 Interestingly, in one small study, the median size of the obstructing stone was only 4 mm. The same study also noted that proximal ureteral obstruction occurred when larger stones where present.4
Conservative Versus Nonconservative Management
Potential complications of urinomas include abscess formation, sepsis, hydronephrosis, and paralytic ileus.3 Despite possible adverse sequelae, uncomplicated urinomas may be managed conservatively with supportive care. According to a study by Chapman et al,5 about 40% of patients managed conservatively recover without complications. In addition, in a retrospective study by Doehn et al6 involving 160 cases of fornix rupture treated with endoscopic therapy or nephrostomy tube supplemented with antibiotics, no instances of perinephric abscess or other complications requiring a second procedure were noted.
Management of suspected ureterolithiasis in the ED is focused on analgesia and supportive care. Acute analgesia is often provided parenterally with opioids alone or with an opioid/nonsteroidal anti-inflammatory drug (NSAID) combination.7 Frequent reassessment of the patient is required to ensure adequate pain control and to prevent sedation. Other symptoms, such as nausea, vomiting, and dehydration, may be treated with intravenous (IV) fluids and antiemetic medications. Further radiographic evaluation is needed once analgesia is achieved.7,8
Imaging Studies
Radiological evaluation of patients with suspected ureterolithiasis may involve several imaging modalities. Noncontrast helical CT scan is the standard for rapid and efficient identification of ureteral stones while allowing visualization of other potential pathology (eg, urinoma).7-9 Other modalities, such as ultrasonography; radiography of the kidneys, ureters, and bladder; and an IV pyelogram with contrasted CT, may be ordered if noncontrast helical CT scan is not available on-site or if there are comorbidities. In addition to imaging studies, basic laboratory studies (eg, serum creatinine and blood urea nitrogen testing) are indicated to assess overall renal function and direct the choice of radiological study.7
Disposition
Clinical decision-making is key when recommending inpatient versus outpatient treatment in patients with ureterolithiasis. Patients with uncontrolled pain or vomiting may require inpatient admission for supportive care, while those demonstrating acute renal failure, pyuria with bacteriuria, complete bilateral ureteral obstruction, urinoma, or signs of sepsis demand emergent urology consultation. Specifically, patients with urinoma require ureteroscopy versus nephrostomy6,10 to allow drainage while carefully monitoring for development of subsequent bleeding and infection.
When discharging patients from the ED, expulsive therapy using tamsulosin9 and analgesia with combination of oral opioids and NSAIDs are most commonly effective.11 Outpatient urology referrals are recommended for ureteral stones greater than 5 mm in size or if the stones have been present in the ureter for greater than 4 weeks.1 Proper evaluation and management of ureterolithiasis in the ED is crucial for positive outcomes and to reduce long-term complications.
Case Conclusion
Computed tomography revealed a ruptured renal calyx on the left side with free fluid in the abdomen. Urology services were consulted and the patient was taken to the operating room for cystoscopy, ureteral stent placement, and laser lithotripsy. Following surgery, she subsequently developed urosepsis for which she was successfully treated with IV antibiotics and discharged on hospital day 15.
Mr Eisenstat is a fourth-year medical student at the University of South Carolina School of Medicine, Greenville. Dr Fabiano is an emergency physician, department of emergency medicine, Greenville Health Systems, Greenville, South Carolina. Dr Collins is family medicine physician, department of emergency medicine, Greenville Health Systems, Greenville, South Carolina.
Case
A 36-year-old woman with a 2-week history of left flank pain presented to the ED via emergency medical services. The patient, who had a history of nephrolithiasis, assumed her pain was due to another kidney stone. She stated that while waiting for the presumed stone to pass, the pain in her left flank worsened and she felt lightheaded and weak.
The patient’s vital signs at presentation were: heart rate, 96 beats/minute; blood pressure, 133/76 mm Hg; respiratory rate, 20 breaths/minute; and temperature, 98.9˚F. Oxygen saturation was 98% on room air. On physical examination, the patient had left lower quadrant pain and left costovertebral angle tenderness. Laboratory studies were remarkable for a negative urine pregnancy test, a hemoglobin level of 6.8 g/dL, and a hematocrit of 21.1%. Based on the patient’s history and symptoms, axial and coronal computed tomography (CT) scans were ordered, revealing a ruptured left renal calyx with hemorrhage from ureterolithiasis (Figures 1a and 1b).
Rupture of renal calyx and extravasation of blood or urine is a potential complication of nephrolithiasis. Stone size, degree of obstruction, and length of symptomatic presentation presumably contribute to complications from nephrolithiasis. Stones that are symptomatic for more than 4 weeks are estimated to have an increased complication rate of up to 20%.1
Calyx or fornix rupture results from increased intraluminal pressure. Rupture of these structures is thought to be a type of “safety-valve” function to relieve obstructive uropathy.2
Obstructions from small leaks to large urinomas can cause extravasation of urine. In most cases, urinary extravasation is confined to the subcapsular space or perirenal space within the Gerota’s fascia;3 however, as seen in this patient, mixed hematoma/urinomas can form.
Causes
In cases of nontraumatic calyx rupture, the cause of the obstruction is most often a distal obstructing ureteral stone.4 Other causes of rupture include extrinsic compression from malignant and benign masses, ureteric junction obstructions, or iatrogenic causes.4 Interestingly, in one small study, the median size of the obstructing stone was only 4 mm. The same study also noted that proximal ureteral obstruction occurred when larger stones where present.4
Conservative Versus Nonconservative Management
Potential complications of urinomas include abscess formation, sepsis, hydronephrosis, and paralytic ileus.3 Despite possible adverse sequelae, uncomplicated urinomas may be managed conservatively with supportive care. According to a study by Chapman et al,5 about 40% of patients managed conservatively recover without complications. In addition, in a retrospective study by Doehn et al6 involving 160 cases of fornix rupture treated with endoscopic therapy or nephrostomy tube supplemented with antibiotics, no instances of perinephric abscess or other complications requiring a second procedure were noted.
Management of suspected ureterolithiasis in the ED is focused on analgesia and supportive care. Acute analgesia is often provided parenterally with opioids alone or with an opioid/nonsteroidal anti-inflammatory drug (NSAID) combination.7 Frequent reassessment of the patient is required to ensure adequate pain control and to prevent sedation. Other symptoms, such as nausea, vomiting, and dehydration, may be treated with intravenous (IV) fluids and antiemetic medications. Further radiographic evaluation is needed once analgesia is achieved.7,8
Imaging Studies
Radiological evaluation of patients with suspected ureterolithiasis may involve several imaging modalities. Noncontrast helical CT scan is the standard for rapid and efficient identification of ureteral stones while allowing visualization of other potential pathology (eg, urinoma).7-9 Other modalities, such as ultrasonography; radiography of the kidneys, ureters, and bladder; and an IV pyelogram with contrasted CT, may be ordered if noncontrast helical CT scan is not available on-site or if there are comorbidities. In addition to imaging studies, basic laboratory studies (eg, serum creatinine and blood urea nitrogen testing) are indicated to assess overall renal function and direct the choice of radiological study.7
Disposition
Clinical decision-making is key when recommending inpatient versus outpatient treatment in patients with ureterolithiasis. Patients with uncontrolled pain or vomiting may require inpatient admission for supportive care, while those demonstrating acute renal failure, pyuria with bacteriuria, complete bilateral ureteral obstruction, urinoma, or signs of sepsis demand emergent urology consultation. Specifically, patients with urinoma require ureteroscopy versus nephrostomy6,10 to allow drainage while carefully monitoring for development of subsequent bleeding and infection.
When discharging patients from the ED, expulsive therapy using tamsulosin9 and analgesia with combination of oral opioids and NSAIDs are most commonly effective.11 Outpatient urology referrals are recommended for ureteral stones greater than 5 mm in size or if the stones have been present in the ureter for greater than 4 weeks.1 Proper evaluation and management of ureterolithiasis in the ED is crucial for positive outcomes and to reduce long-term complications.
Case Conclusion
Computed tomography revealed a ruptured renal calyx on the left side with free fluid in the abdomen. Urology services were consulted and the patient was taken to the operating room for cystoscopy, ureteral stent placement, and laser lithotripsy. Following surgery, she subsequently developed urosepsis for which she was successfully treated with IV antibiotics and discharged on hospital day 15.
Mr Eisenstat is a fourth-year medical student at the University of South Carolina School of Medicine, Greenville. Dr Fabiano is an emergency physician, department of emergency medicine, Greenville Health Systems, Greenville, South Carolina. Dr Collins is family medicine physician, department of emergency medicine, Greenville Health Systems, Greenville, South Carolina.
- Hübner WA, Irby P, Stoller M. Natural history and current concepts for the treatment of small ureteral calculi. Eur Urol. 1993;24(2):172-176.
- Lin DY, Fang YC, Huang DY, Lin SP. Spontaneous rupture of the ureter secondary to urolithiasis and extravasation of calyceal fornix due to acute urinary bladder distension: four case reports. Chin J Radiology. 2004;29:269-275.
- Behzad-Noori M, Blandon JA, Negrin Exposito JE, Sarmiento JL, Dias AL, Hernandez GT. Urinoma: a rare complication from being between a rock and soft organ. El Paso Physician. 2010;33(6):5-6.
- Gershman B, Kulkarni N, Sahani DV, Eisner BH. Causes of renal forniceal rupture. BJU Int. 2011;108(11):1909-1911.
- Chapman JP, Gonzalez J, Diokno AC. Significance of urinary extravasation during renal colic. Urology. 1987;30(6):541-545.
- Doehn C, Fiola L, Peter M, Jocham D. Outcome analysis of fornix ruptures in 162 consecutive patients. J Endourol. 2010;24(11):1869-1873
- Portis AJ, Sundaram CP. Diagnosis and initial management of kidney stones. Am Fam Physician. 2001;63(7):1329-1338
- Smith RC, Verga M, Dalrymple N, McCarthy S, Rosenfield AT. Acute ureteral obstruction: value of secondary signs of helical unenhanced CT. AJR Am J Roentgenol. 1996;167(5):1109-1113.Burke TA, Wisniewski T, Ernst FR. Resource utilization and costs associated with chemotherapy-induced nausea and vomiting (CINV) following highly or moderately emetogenic chemotherapy administered in the US outpatient hospital setting. Support Care Cancer. 2011;19(1):131-140.
- Ha M, MacDonald RD. Impact of CT scan in patients with first episode of suspected nephrolithiasis. J Emerg Med. 2004;27(3):225-231.
- Tawfiek ER, Bagley DH. Management of upper urinary tract calculi with ureteroscopic techniques. Urology. 1999;53(1):25-31.
- Larkin GL, Peacock WF 4th, Pearl SM, Blair GA, D'Amico F. Efficacy of ketorolac tromethamine versus meperidine in the ED treatment of acute renal colic. Am J Emerg Med. 1999;17(1):6-10.
- Hübner WA, Irby P, Stoller M. Natural history and current concepts for the treatment of small ureteral calculi. Eur Urol. 1993;24(2):172-176.
- Lin DY, Fang YC, Huang DY, Lin SP. Spontaneous rupture of the ureter secondary to urolithiasis and extravasation of calyceal fornix due to acute urinary bladder distension: four case reports. Chin J Radiology. 2004;29:269-275.
- Behzad-Noori M, Blandon JA, Negrin Exposito JE, Sarmiento JL, Dias AL, Hernandez GT. Urinoma: a rare complication from being between a rock and soft organ. El Paso Physician. 2010;33(6):5-6.
- Gershman B, Kulkarni N, Sahani DV, Eisner BH. Causes of renal forniceal rupture. BJU Int. 2011;108(11):1909-1911.
- Chapman JP, Gonzalez J, Diokno AC. Significance of urinary extravasation during renal colic. Urology. 1987;30(6):541-545.
- Doehn C, Fiola L, Peter M, Jocham D. Outcome analysis of fornix ruptures in 162 consecutive patients. J Endourol. 2010;24(11):1869-1873
- Portis AJ, Sundaram CP. Diagnosis and initial management of kidney stones. Am Fam Physician. 2001;63(7):1329-1338
- Smith RC, Verga M, Dalrymple N, McCarthy S, Rosenfield AT. Acute ureteral obstruction: value of secondary signs of helical unenhanced CT. AJR Am J Roentgenol. 1996;167(5):1109-1113.Burke TA, Wisniewski T, Ernst FR. Resource utilization and costs associated with chemotherapy-induced nausea and vomiting (CINV) following highly or moderately emetogenic chemotherapy administered in the US outpatient hospital setting. Support Care Cancer. 2011;19(1):131-140.
- Ha M, MacDonald RD. Impact of CT scan in patients with first episode of suspected nephrolithiasis. J Emerg Med. 2004;27(3):225-231.
- Tawfiek ER, Bagley DH. Management of upper urinary tract calculi with ureteroscopic techniques. Urology. 1999;53(1):25-31.
- Larkin GL, Peacock WF 4th, Pearl SM, Blair GA, D'Amico F. Efficacy of ketorolac tromethamine versus meperidine in the ED treatment of acute renal colic. Am J Emerg Med. 1999;17(1):6-10.
Surgical Management of Gorham-Stout Disease of the Pelvis Refractory to Medical and Radiation Therapy
Gorham-Stout disease (GSD) is a rare condition characterized by spontaneous idiopathic resorption of bone with lymphovascular proliferation and an absence of malignant features. It was originally described by Jackson1 in an 1838 report of a 36-year-old man whose “arm bone, between the shoulder and elbow” had completely vanished after 2 fractures. The disease was defined and its pathology characterized by Gorham and Stout2 in 1955 in a series of 24 patients. Despite about 200 reported cases in the literature,3 its etiology remains unclear. Any bone in the skeleton may be affected by GSD, although there is a predilection for the skull, humerus, clavicle, ribs, pelvis, and femur.4-6 It commonly manifests within the first 3 decades of life, but case reports range from as early as 2 months of age to the eighth decade.5,7
Gorham-Stout disease is a diagnosis of exclusion that requires careful consideration of the clinical context, radiographic findings, and histopathology. Typical histopathologic findings include benign lymphatic or vascular proliferation, involution of adipose tissue within the bone marrow, and thinning of bony trabeculae.6 Fibrous tissue may replace vascular tissue after the initial vasoproliferative, osteolytic phase.6 Some authors describe the disease as having 2 phases, the first with massive osteolysis followed by relative dormancy and the second without progression or re-ossification.8,9 Treatment remains controversial and is guided by management of the disease’s complications. Options range from careful observation and supportive management to aggressive surgical resection and reconstruction, with positive outcomes reported using many different modalities.10 Most treatment successes, however, hinge on halting bony resorption using medical and radiation therapy. Surgery is usually reserved as a salvage option for patients who have failed medical modalities and have residual symptoms or functional limitations.6
This case report describes the successful surgical management of a patient with pelvic GSD who had progressive pain and functional limitation despite exhaustive medical and radiation therapy. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A healthy 27-year-old man sought medical attention after a fall while mowing his lawn that resulted in difficulty ambulating. Radiographic studies showed discontinuous lytic lesions in the right periacetabular region and the right sacroiliac (SI) joint. Biopsy at an outside institution revealed an infiltration of thin-walled branching vascular channels involving intertrabecular marrow spaces and periosteal connective tissue. The vessels were devoid of a muscular coat and lined by flattened epithelium; these features were seen as consistent with GSD.
The patient was managed medically at the outside institution for approximately 2 years, with regimens consisting of zoledronate, denosumab, sorafenib, vincristine, sirolimus, and bevacizumab. Because there is no standard chemotherapy protocol for GSD, this broad regimen was likely an attempt by treating physicians to control disease progression before considering radiation or surgery. Zoledronate, a bisphosphonate, and denosumab, a monoclonal antibody against the receptor activator of nuclear factor κβ ligand (RANKL), both inhibit bone resorption, making them logical choices in treating an osteolytic disease. Sorafenib, vincristine, sirolimus, and bevacizumab may be of clinical benefit in GSD via inhibition of vascular proliferation, which is a key histologic feature in GSD. Sorafenib inhibits the vascular endothelial growth factor (VEGF) receptor, vincristine and sirolimus inhibit VEGF production, and bevacizumab is a monoclonal antibody targeting VEGF.
The patient’s disease continued to involve more of his right hemipelvis despite this extensive regimen of chemotherapy, and he experienced significant functional decline about 2 years after initial presentation, when he was no longer able to ambulate unassisted. Radiation therapy to the pelvis was attempted at the outside institution (6/15 MV photons, 5040 cGy, 28 fractions) without improvement. Three years after his initial injury, he presented to our clinic.
Now age 30 years, the patient ambulated only with crutches and endorsed minimal improvement in his pain over 3 years of treatment. Physical examination of the patient revealed that he was a tall, thin man in visible discomfort. Sensation was intact to light touch in the bilateral L1 to S1 nerve distributions. There was marked weakness of the right lower extremity, and his examination was limited by pain. He could not perform a straight leg raise on the right side. Right quadriceps strength was 4/5, and right hamstrings strength was 3/5. There was no weakness in the left leg. Reflexes were normal and symmetric bilaterally at the patellar and gastrocnemius soleus tendons. Distal circulatory status in both extremities was normal, and there were no deformities of the skin.
Figure 1 shows the patient’s computed tomography (CT) scan. Figures 1A and 1B reveal fragmentation of the posterior ilia and sacrum along both SI joints. Dislocation of the pubic symphysis is shown in Figures 1C and 1D, and discontinuous involvement of the ischium and posterior wall of the acetabulum is visible in Figure 1E.
Serum studies, including C-reactive protein, erythrocyte sedimentation rate, and a complete blood count, were within normal limits. A CT-guided core needle biopsy and aspiration of the right SI joint revealed no infection; pathology was nondiagnostic. Anesthetic injection of the hip joint resulted in no relief. As this man was severely functionally limited and had exhausted all medical and radiation treatment options, a collaborative decision was made to proceed with surgical management. Surgical options included spinopelvic fusion unilaterally or bilaterally, hip arthroplasty, or sacropelvic resection with or without reconstruction. The patient opted for intralesional surgery and spinopelvic fusion in place of more radical options.
Thirty-seven months after his initial presentation, he underwent posterior spinal fusion L5 to S1, SI fusion, and anterior locking plate fixation of the pubic symphysis, as seen in Figure 2. Pathology from surgical specimens, seen at original magnification ×20 and ×100 in Figures 3A and 3B, respectively, showed prominent vascular proliferation in the right ilium, with reactive bone changes in the left ilium and right sacrum. A lytic lesion showed fibrous tissue with an embedded fragment of necrotic bone.
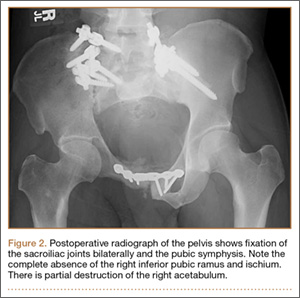
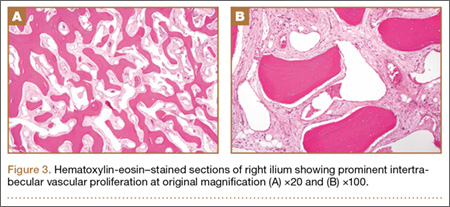
Six weeks after surgery, the patient had substantial improvement in his pain and was partially weight-bearing. He was able to ambulate with crutches and returned to work. The patient’s overall clinical status continued to improve throughout the postoperative course. He developed low back pain 7 months after surgery and was found to have a sacrococcygeal abscess and coccygeal fracture anterior to the sacrum. He underwent irrigation and débridement of the abscess and distal coccygectomy and was treated with 6 weeks of intravenous cefazolin and long-term suppression with levofloxacin and rifampin for methicillin-sensitive Staphylococcus aureus hardware infection and osteomyelitis. The patient’s clinical course subsequently improved. At latest follow-up 16 months after the index operation, pain was reported as manageable and mostly an annoyance. He was prescribed up to 40 mg of oxycodone daily for pain. The patient returned to work, ambulates with a cane (no other assistive devices), and reports being able to get around without any difficulty.
Discussion
Gorham-Stout disease is an exceedingly rare condition resulting in spontaneous osteolysis. Approximately 200 cases have been reported with no apparent gender, race, or familial predilection or systemic symptoms differentiating it from other etiologies of idiopathic osteolysis.6 These patients often seek medical attention after sustaining a pathologic fracture,6 when a broad differential diagnosis narrows to GSD only after biopsy excludes other possibilities and demonstrates characteristic angiomatosis without malignant features.2,4,6,8,10 Gorham-Stout disease appears more frequently at particular sites within the skeleton, and pelvic involvement is common—more than 20% of cases in 1 review.5,10 Limitations in the patient’s ability to ambulate invariably result from osteolysis of the pelvis, which is concerning considering the young age at which GSD typically presents. A variety of treatment modalities have been described for pelvic GSD, but surgery has been undertaken in relatively few cases.5
The diagnosis is one of exclusion after considering the clinical context and radiologic and pathologic findings. In this case, a pathologic fracture was discovered with osteolytic lesions throughout the hemipelvis. Biopsy excluded malignancy and demonstrated the key hemangiomatous vascular proliferation with thin-walled vessels that is classic for GSD. While our patient initially appeared to have 2 sites of disease, the surgical specimen revealed a primary site of vascular proliferation in the right ilium from which 2 apparent foci had spread, consistent with the typical monocentric presentation of GSD.11 A broad differential diagnosis must be considered at initial presentation, including osteomyelitis, metastatic disease, multiple myeloma, and primary bone sarcoma. Upon identifying a primary osteolytic process, several considerations besides GSD remain, such as Hajdu-Cheney syndrome, Winchester syndrome, multicentric osteolysis with nephropathy, familial osteolysis, Farber disease, and neurogenic osteolysis; most of these etiologies involve familial predispositions and/or systemic symptoms.
Treatment options for GSD include supportive care, medical therapy, radiation, and surgery. For pelvic GSD, numerous reports have demonstrated good outcomes with supportive management, since osteolysis often spontaneously arrests.8,9,12 Others have had success with medical treatments in attempts to halt bone resorption.6,13-15 Bisphosphonates are the cornerstone of medical therapy in GSD, as they appear to halt further osteoclastic bone breakdown. The levels of VEGF have been shown to be elevated in GSD,13 likely consistent with the vascular proliferation evident on pathology, and therapies such as bevacizumab and interferon α-2b have been used to target osteolysis via this pathway with good outcome.13,14,16 External beam-radiation therapy has been shown to prevent local progression of osteolysis in up to 80% of cases.4 However, even with arrest of bone resorption, damage to affected bone may have progressed to the point of significant functional limitation. This may be especially true in the pelvis.
We present a case of a patient who continued to deteriorate after maximal medical and radiation therapy. Many reported cases of pelvic GSD have had good outcomes with some combination of conservative management, medical therapy, and radiation. However, in our patient, the pelvis and lumbosacral spine were unstable as a result of significant bone loss and fracture, and his clinical deterioration was dramatic. We considered reasonable surgical approaches, including local intralesional débridement and massive en bloc resection with structural allograft. We chose the less radical procedure given the patient’s age, minimal surgical history, and personal preference. Although structural pelvic allograft has been successful in a few cases, there remains a high risk of complications, such as fracture, resorption, or infection.17 We considered the addition of hip arthroplasty with either scenario, but we elected not to perform this component given his young age and lack of symptomatic improvement with diagnostic anesthetic hip injection. The key to this patient’s surgical reconstruction, aside from eliminating gross disease, was the stabilization of the spinopelvic junction and pelvic ring. His functional improvement as early as 6 weeks after surgery demonstrates that surgery can have an important role for patients with pelvic GSD who fail medical and radiation therapy.
1. Jackson JBS. A boneless arm. Boston Med Surg J. 1838;18:368-369.
2. Gorham LW, Stout AP. Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone): its relation to hemangiomatosis. J Bone Joint Surg Am. 1955;37(5):985-1004.
3. Lehmann G, Pfeil A, Böttcher J, et al. Benefit of a 17-year long-term bisphosphonate therapy in a patient with Gorham-Stout syndrome. Arch Orthop Trauma Surg. 2009;129(7):967-972.
4. Heyd R, Micke O, Surholt C, et al; German Cooperative Group on Radiotherapy for Benign Diseases (GCG-BD). Radiation therapy for Gorham-Stout syndrome: results of a national patterns-of-care study and literature review. Int J Radiat Oncol Biol Phys. 2011;81(3):e179-e185.
5. Kulenkampff HA, Richter GM, Hasse WE, Adler CP. Massive pelvic osteolysis in the Gorham-Stout syndrome. Int Orthop. 1990;14(4):361-366.
6. Ruggieri P, Montalti M, Angelini A, Alberghini M, Mercuri M. Gorham-Stout disease: the experience of the Rizzoli Institute and review of the literature. Skeletal Radiol. 2011;40(11):1391-1397.
7. Vinée P, Tanyü MO, Hauenstein KH, Sigmund G, Stöver B, Adler CP. CT and MRI of Gorham syndrome. J Comput Assist Tomogr. 1994;18(6):985-989.
8. Boyer P, Bourgeois P, Boyer O, Catonné Y, Saillant G. Massive Gorham-Stout syndrome of the pelvis. Clin Rheumatol. 2005;24(5):551-555.
9. Malde R, Agrawal HM, Ghosh SL, Dinshaw KA. Vanishing bone disease involving the pelvis. J Cancer Res Ther. 2005;1(4):227-228.
10. Kuriyama DK, McElligott SC, Glaser DW, Thompson KS. Treatment of Gorham-Stout disease with zoledronic acid and interferon-α: a case report and literature review. J Pediatr Hematol Oncol. 2010;32(8):579-584.
11. Tie ML, Poland GA, Rosenow EC III. Chylothorax in Gorham’s syndrome. A common complication of a rare disease. Chest. 1994;105(1):208-213.
12. Möller G, Priemel M, Amling M, Werner M, Kuhlmey AS, Delling G. The Gorham-Stout syndrome (Gorham’s massive osteolysis). A report of six cases with histopathological findings. J Bone Joint Surg Br. 1999;81(3):501-506.
13. Dupond JL, Bermont L, Runge M, de Billy M. Plasma VEGF determination in disseminated lymphangiomatosis—Gorham-Stout syndrome: a marker of activity? A case report with a 5-year follow-up. Bone. 2010;46(3):873-876.
14. Wang JD, Chang TK, Cheng YY, et al. A child with dyspnea and unstable gait. Pediatr Hemat Oncol. 2007;24(4):321-324.
15. Zheng MW, Yang M, Qiu JX, et al. Gorham-Stout syndrome presenting in a 5-year-old girl with a successful bisphosphonate therapeutic effect. Exp Ther Med. 2012;4(3):449-451.
16. Timke C, Krause MF, Oppermann HC, Leuschner I, Claviez A. Interferon alpha 2b treatment in an eleven-year-old boy with disseminated lymphangiomatosis. Pediatr Blood Cancer. 2007;48(1):108-111.
17. Stöve J, Reichelt A. Massive osteolysis of the pelvis, femur and sacral bone with a Gorham-Stout syndrome. Arch Orthop Trauma Surg. 1995;114(4):207-210.
Gorham-Stout disease (GSD) is a rare condition characterized by spontaneous idiopathic resorption of bone with lymphovascular proliferation and an absence of malignant features. It was originally described by Jackson1 in an 1838 report of a 36-year-old man whose “arm bone, between the shoulder and elbow” had completely vanished after 2 fractures. The disease was defined and its pathology characterized by Gorham and Stout2 in 1955 in a series of 24 patients. Despite about 200 reported cases in the literature,3 its etiology remains unclear. Any bone in the skeleton may be affected by GSD, although there is a predilection for the skull, humerus, clavicle, ribs, pelvis, and femur.4-6 It commonly manifests within the first 3 decades of life, but case reports range from as early as 2 months of age to the eighth decade.5,7
Gorham-Stout disease is a diagnosis of exclusion that requires careful consideration of the clinical context, radiographic findings, and histopathology. Typical histopathologic findings include benign lymphatic or vascular proliferation, involution of adipose tissue within the bone marrow, and thinning of bony trabeculae.6 Fibrous tissue may replace vascular tissue after the initial vasoproliferative, osteolytic phase.6 Some authors describe the disease as having 2 phases, the first with massive osteolysis followed by relative dormancy and the second without progression or re-ossification.8,9 Treatment remains controversial and is guided by management of the disease’s complications. Options range from careful observation and supportive management to aggressive surgical resection and reconstruction, with positive outcomes reported using many different modalities.10 Most treatment successes, however, hinge on halting bony resorption using medical and radiation therapy. Surgery is usually reserved as a salvage option for patients who have failed medical modalities and have residual symptoms or functional limitations.6
This case report describes the successful surgical management of a patient with pelvic GSD who had progressive pain and functional limitation despite exhaustive medical and radiation therapy. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A healthy 27-year-old man sought medical attention after a fall while mowing his lawn that resulted in difficulty ambulating. Radiographic studies showed discontinuous lytic lesions in the right periacetabular region and the right sacroiliac (SI) joint. Biopsy at an outside institution revealed an infiltration of thin-walled branching vascular channels involving intertrabecular marrow spaces and periosteal connective tissue. The vessels were devoid of a muscular coat and lined by flattened epithelium; these features were seen as consistent with GSD.
The patient was managed medically at the outside institution for approximately 2 years, with regimens consisting of zoledronate, denosumab, sorafenib, vincristine, sirolimus, and bevacizumab. Because there is no standard chemotherapy protocol for GSD, this broad regimen was likely an attempt by treating physicians to control disease progression before considering radiation or surgery. Zoledronate, a bisphosphonate, and denosumab, a monoclonal antibody against the receptor activator of nuclear factor κβ ligand (RANKL), both inhibit bone resorption, making them logical choices in treating an osteolytic disease. Sorafenib, vincristine, sirolimus, and bevacizumab may be of clinical benefit in GSD via inhibition of vascular proliferation, which is a key histologic feature in GSD. Sorafenib inhibits the vascular endothelial growth factor (VEGF) receptor, vincristine and sirolimus inhibit VEGF production, and bevacizumab is a monoclonal antibody targeting VEGF.
The patient’s disease continued to involve more of his right hemipelvis despite this extensive regimen of chemotherapy, and he experienced significant functional decline about 2 years after initial presentation, when he was no longer able to ambulate unassisted. Radiation therapy to the pelvis was attempted at the outside institution (6/15 MV photons, 5040 cGy, 28 fractions) without improvement. Three years after his initial injury, he presented to our clinic.
Now age 30 years, the patient ambulated only with crutches and endorsed minimal improvement in his pain over 3 years of treatment. Physical examination of the patient revealed that he was a tall, thin man in visible discomfort. Sensation was intact to light touch in the bilateral L1 to S1 nerve distributions. There was marked weakness of the right lower extremity, and his examination was limited by pain. He could not perform a straight leg raise on the right side. Right quadriceps strength was 4/5, and right hamstrings strength was 3/5. There was no weakness in the left leg. Reflexes were normal and symmetric bilaterally at the patellar and gastrocnemius soleus tendons. Distal circulatory status in both extremities was normal, and there were no deformities of the skin.
Figure 1 shows the patient’s computed tomography (CT) scan. Figures 1A and 1B reveal fragmentation of the posterior ilia and sacrum along both SI joints. Dislocation of the pubic symphysis is shown in Figures 1C and 1D, and discontinuous involvement of the ischium and posterior wall of the acetabulum is visible in Figure 1E.
Serum studies, including C-reactive protein, erythrocyte sedimentation rate, and a complete blood count, were within normal limits. A CT-guided core needle biopsy and aspiration of the right SI joint revealed no infection; pathology was nondiagnostic. Anesthetic injection of the hip joint resulted in no relief. As this man was severely functionally limited and had exhausted all medical and radiation treatment options, a collaborative decision was made to proceed with surgical management. Surgical options included spinopelvic fusion unilaterally or bilaterally, hip arthroplasty, or sacropelvic resection with or without reconstruction. The patient opted for intralesional surgery and spinopelvic fusion in place of more radical options.
Thirty-seven months after his initial presentation, he underwent posterior spinal fusion L5 to S1, SI fusion, and anterior locking plate fixation of the pubic symphysis, as seen in Figure 2. Pathology from surgical specimens, seen at original magnification ×20 and ×100 in Figures 3A and 3B, respectively, showed prominent vascular proliferation in the right ilium, with reactive bone changes in the left ilium and right sacrum. A lytic lesion showed fibrous tissue with an embedded fragment of necrotic bone.
Six weeks after surgery, the patient had substantial improvement in his pain and was partially weight-bearing. He was able to ambulate with crutches and returned to work. The patient’s overall clinical status continued to improve throughout the postoperative course. He developed low back pain 7 months after surgery and was found to have a sacrococcygeal abscess and coccygeal fracture anterior to the sacrum. He underwent irrigation and débridement of the abscess and distal coccygectomy and was treated with 6 weeks of intravenous cefazolin and long-term suppression with levofloxacin and rifampin for methicillin-sensitive Staphylococcus aureus hardware infection and osteomyelitis. The patient’s clinical course subsequently improved. At latest follow-up 16 months after the index operation, pain was reported as manageable and mostly an annoyance. He was prescribed up to 40 mg of oxycodone daily for pain. The patient returned to work, ambulates with a cane (no other assistive devices), and reports being able to get around without any difficulty.
Discussion
Gorham-Stout disease is an exceedingly rare condition resulting in spontaneous osteolysis. Approximately 200 cases have been reported with no apparent gender, race, or familial predilection or systemic symptoms differentiating it from other etiologies of idiopathic osteolysis.6 These patients often seek medical attention after sustaining a pathologic fracture,6 when a broad differential diagnosis narrows to GSD only after biopsy excludes other possibilities and demonstrates characteristic angiomatosis without malignant features.2,4,6,8,10 Gorham-Stout disease appears more frequently at particular sites within the skeleton, and pelvic involvement is common—more than 20% of cases in 1 review.5,10 Limitations in the patient’s ability to ambulate invariably result from osteolysis of the pelvis, which is concerning considering the young age at which GSD typically presents. A variety of treatment modalities have been described for pelvic GSD, but surgery has been undertaken in relatively few cases.5
The diagnosis is one of exclusion after considering the clinical context and radiologic and pathologic findings. In this case, a pathologic fracture was discovered with osteolytic lesions throughout the hemipelvis. Biopsy excluded malignancy and demonstrated the key hemangiomatous vascular proliferation with thin-walled vessels that is classic for GSD. While our patient initially appeared to have 2 sites of disease, the surgical specimen revealed a primary site of vascular proliferation in the right ilium from which 2 apparent foci had spread, consistent with the typical monocentric presentation of GSD.11 A broad differential diagnosis must be considered at initial presentation, including osteomyelitis, metastatic disease, multiple myeloma, and primary bone sarcoma. Upon identifying a primary osteolytic process, several considerations besides GSD remain, such as Hajdu-Cheney syndrome, Winchester syndrome, multicentric osteolysis with nephropathy, familial osteolysis, Farber disease, and neurogenic osteolysis; most of these etiologies involve familial predispositions and/or systemic symptoms.
Treatment options for GSD include supportive care, medical therapy, radiation, and surgery. For pelvic GSD, numerous reports have demonstrated good outcomes with supportive management, since osteolysis often spontaneously arrests.8,9,12 Others have had success with medical treatments in attempts to halt bone resorption.6,13-15 Bisphosphonates are the cornerstone of medical therapy in GSD, as they appear to halt further osteoclastic bone breakdown. The levels of VEGF have been shown to be elevated in GSD,13 likely consistent with the vascular proliferation evident on pathology, and therapies such as bevacizumab and interferon α-2b have been used to target osteolysis via this pathway with good outcome.13,14,16 External beam-radiation therapy has been shown to prevent local progression of osteolysis in up to 80% of cases.4 However, even with arrest of bone resorption, damage to affected bone may have progressed to the point of significant functional limitation. This may be especially true in the pelvis.
We present a case of a patient who continued to deteriorate after maximal medical and radiation therapy. Many reported cases of pelvic GSD have had good outcomes with some combination of conservative management, medical therapy, and radiation. However, in our patient, the pelvis and lumbosacral spine were unstable as a result of significant bone loss and fracture, and his clinical deterioration was dramatic. We considered reasonable surgical approaches, including local intralesional débridement and massive en bloc resection with structural allograft. We chose the less radical procedure given the patient’s age, minimal surgical history, and personal preference. Although structural pelvic allograft has been successful in a few cases, there remains a high risk of complications, such as fracture, resorption, or infection.17 We considered the addition of hip arthroplasty with either scenario, but we elected not to perform this component given his young age and lack of symptomatic improvement with diagnostic anesthetic hip injection. The key to this patient’s surgical reconstruction, aside from eliminating gross disease, was the stabilization of the spinopelvic junction and pelvic ring. His functional improvement as early as 6 weeks after surgery demonstrates that surgery can have an important role for patients with pelvic GSD who fail medical and radiation therapy.
Gorham-Stout disease (GSD) is a rare condition characterized by spontaneous idiopathic resorption of bone with lymphovascular proliferation and an absence of malignant features. It was originally described by Jackson1 in an 1838 report of a 36-year-old man whose “arm bone, between the shoulder and elbow” had completely vanished after 2 fractures. The disease was defined and its pathology characterized by Gorham and Stout2 in 1955 in a series of 24 patients. Despite about 200 reported cases in the literature,3 its etiology remains unclear. Any bone in the skeleton may be affected by GSD, although there is a predilection for the skull, humerus, clavicle, ribs, pelvis, and femur.4-6 It commonly manifests within the first 3 decades of life, but case reports range from as early as 2 months of age to the eighth decade.5,7
Gorham-Stout disease is a diagnosis of exclusion that requires careful consideration of the clinical context, radiographic findings, and histopathology. Typical histopathologic findings include benign lymphatic or vascular proliferation, involution of adipose tissue within the bone marrow, and thinning of bony trabeculae.6 Fibrous tissue may replace vascular tissue after the initial vasoproliferative, osteolytic phase.6 Some authors describe the disease as having 2 phases, the first with massive osteolysis followed by relative dormancy and the second without progression or re-ossification.8,9 Treatment remains controversial and is guided by management of the disease’s complications. Options range from careful observation and supportive management to aggressive surgical resection and reconstruction, with positive outcomes reported using many different modalities.10 Most treatment successes, however, hinge on halting bony resorption using medical and radiation therapy. Surgery is usually reserved as a salvage option for patients who have failed medical modalities and have residual symptoms or functional limitations.6
This case report describes the successful surgical management of a patient with pelvic GSD who had progressive pain and functional limitation despite exhaustive medical and radiation therapy. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A healthy 27-year-old man sought medical attention after a fall while mowing his lawn that resulted in difficulty ambulating. Radiographic studies showed discontinuous lytic lesions in the right periacetabular region and the right sacroiliac (SI) joint. Biopsy at an outside institution revealed an infiltration of thin-walled branching vascular channels involving intertrabecular marrow spaces and periosteal connective tissue. The vessels were devoid of a muscular coat and lined by flattened epithelium; these features were seen as consistent with GSD.
The patient was managed medically at the outside institution for approximately 2 years, with regimens consisting of zoledronate, denosumab, sorafenib, vincristine, sirolimus, and bevacizumab. Because there is no standard chemotherapy protocol for GSD, this broad regimen was likely an attempt by treating physicians to control disease progression before considering radiation or surgery. Zoledronate, a bisphosphonate, and denosumab, a monoclonal antibody against the receptor activator of nuclear factor κβ ligand (RANKL), both inhibit bone resorption, making them logical choices in treating an osteolytic disease. Sorafenib, vincristine, sirolimus, and bevacizumab may be of clinical benefit in GSD via inhibition of vascular proliferation, which is a key histologic feature in GSD. Sorafenib inhibits the vascular endothelial growth factor (VEGF) receptor, vincristine and sirolimus inhibit VEGF production, and bevacizumab is a monoclonal antibody targeting VEGF.
The patient’s disease continued to involve more of his right hemipelvis despite this extensive regimen of chemotherapy, and he experienced significant functional decline about 2 years after initial presentation, when he was no longer able to ambulate unassisted. Radiation therapy to the pelvis was attempted at the outside institution (6/15 MV photons, 5040 cGy, 28 fractions) without improvement. Three years after his initial injury, he presented to our clinic.
Now age 30 years, the patient ambulated only with crutches and endorsed minimal improvement in his pain over 3 years of treatment. Physical examination of the patient revealed that he was a tall, thin man in visible discomfort. Sensation was intact to light touch in the bilateral L1 to S1 nerve distributions. There was marked weakness of the right lower extremity, and his examination was limited by pain. He could not perform a straight leg raise on the right side. Right quadriceps strength was 4/5, and right hamstrings strength was 3/5. There was no weakness in the left leg. Reflexes were normal and symmetric bilaterally at the patellar and gastrocnemius soleus tendons. Distal circulatory status in both extremities was normal, and there were no deformities of the skin.
Figure 1 shows the patient’s computed tomography (CT) scan. Figures 1A and 1B reveal fragmentation of the posterior ilia and sacrum along both SI joints. Dislocation of the pubic symphysis is shown in Figures 1C and 1D, and discontinuous involvement of the ischium and posterior wall of the acetabulum is visible in Figure 1E.
Serum studies, including C-reactive protein, erythrocyte sedimentation rate, and a complete blood count, were within normal limits. A CT-guided core needle biopsy and aspiration of the right SI joint revealed no infection; pathology was nondiagnostic. Anesthetic injection of the hip joint resulted in no relief. As this man was severely functionally limited and had exhausted all medical and radiation treatment options, a collaborative decision was made to proceed with surgical management. Surgical options included spinopelvic fusion unilaterally or bilaterally, hip arthroplasty, or sacropelvic resection with or without reconstruction. The patient opted for intralesional surgery and spinopelvic fusion in place of more radical options.
Thirty-seven months after his initial presentation, he underwent posterior spinal fusion L5 to S1, SI fusion, and anterior locking plate fixation of the pubic symphysis, as seen in Figure 2. Pathology from surgical specimens, seen at original magnification ×20 and ×100 in Figures 3A and 3B, respectively, showed prominent vascular proliferation in the right ilium, with reactive bone changes in the left ilium and right sacrum. A lytic lesion showed fibrous tissue with an embedded fragment of necrotic bone.
Six weeks after surgery, the patient had substantial improvement in his pain and was partially weight-bearing. He was able to ambulate with crutches and returned to work. The patient’s overall clinical status continued to improve throughout the postoperative course. He developed low back pain 7 months after surgery and was found to have a sacrococcygeal abscess and coccygeal fracture anterior to the sacrum. He underwent irrigation and débridement of the abscess and distal coccygectomy and was treated with 6 weeks of intravenous cefazolin and long-term suppression with levofloxacin and rifampin for methicillin-sensitive Staphylococcus aureus hardware infection and osteomyelitis. The patient’s clinical course subsequently improved. At latest follow-up 16 months after the index operation, pain was reported as manageable and mostly an annoyance. He was prescribed up to 40 mg of oxycodone daily for pain. The patient returned to work, ambulates with a cane (no other assistive devices), and reports being able to get around without any difficulty.
Discussion
Gorham-Stout disease is an exceedingly rare condition resulting in spontaneous osteolysis. Approximately 200 cases have been reported with no apparent gender, race, or familial predilection or systemic symptoms differentiating it from other etiologies of idiopathic osteolysis.6 These patients often seek medical attention after sustaining a pathologic fracture,6 when a broad differential diagnosis narrows to GSD only after biopsy excludes other possibilities and demonstrates characteristic angiomatosis without malignant features.2,4,6,8,10 Gorham-Stout disease appears more frequently at particular sites within the skeleton, and pelvic involvement is common—more than 20% of cases in 1 review.5,10 Limitations in the patient’s ability to ambulate invariably result from osteolysis of the pelvis, which is concerning considering the young age at which GSD typically presents. A variety of treatment modalities have been described for pelvic GSD, but surgery has been undertaken in relatively few cases.5
The diagnosis is one of exclusion after considering the clinical context and radiologic and pathologic findings. In this case, a pathologic fracture was discovered with osteolytic lesions throughout the hemipelvis. Biopsy excluded malignancy and demonstrated the key hemangiomatous vascular proliferation with thin-walled vessels that is classic for GSD. While our patient initially appeared to have 2 sites of disease, the surgical specimen revealed a primary site of vascular proliferation in the right ilium from which 2 apparent foci had spread, consistent with the typical monocentric presentation of GSD.11 A broad differential diagnosis must be considered at initial presentation, including osteomyelitis, metastatic disease, multiple myeloma, and primary bone sarcoma. Upon identifying a primary osteolytic process, several considerations besides GSD remain, such as Hajdu-Cheney syndrome, Winchester syndrome, multicentric osteolysis with nephropathy, familial osteolysis, Farber disease, and neurogenic osteolysis; most of these etiologies involve familial predispositions and/or systemic symptoms.
Treatment options for GSD include supportive care, medical therapy, radiation, and surgery. For pelvic GSD, numerous reports have demonstrated good outcomes with supportive management, since osteolysis often spontaneously arrests.8,9,12 Others have had success with medical treatments in attempts to halt bone resorption.6,13-15 Bisphosphonates are the cornerstone of medical therapy in GSD, as they appear to halt further osteoclastic bone breakdown. The levels of VEGF have been shown to be elevated in GSD,13 likely consistent with the vascular proliferation evident on pathology, and therapies such as bevacizumab and interferon α-2b have been used to target osteolysis via this pathway with good outcome.13,14,16 External beam-radiation therapy has been shown to prevent local progression of osteolysis in up to 80% of cases.4 However, even with arrest of bone resorption, damage to affected bone may have progressed to the point of significant functional limitation. This may be especially true in the pelvis.
We present a case of a patient who continued to deteriorate after maximal medical and radiation therapy. Many reported cases of pelvic GSD have had good outcomes with some combination of conservative management, medical therapy, and radiation. However, in our patient, the pelvis and lumbosacral spine were unstable as a result of significant bone loss and fracture, and his clinical deterioration was dramatic. We considered reasonable surgical approaches, including local intralesional débridement and massive en bloc resection with structural allograft. We chose the less radical procedure given the patient’s age, minimal surgical history, and personal preference. Although structural pelvic allograft has been successful in a few cases, there remains a high risk of complications, such as fracture, resorption, or infection.17 We considered the addition of hip arthroplasty with either scenario, but we elected not to perform this component given his young age and lack of symptomatic improvement with diagnostic anesthetic hip injection. The key to this patient’s surgical reconstruction, aside from eliminating gross disease, was the stabilization of the spinopelvic junction and pelvic ring. His functional improvement as early as 6 weeks after surgery demonstrates that surgery can have an important role for patients with pelvic GSD who fail medical and radiation therapy.
1. Jackson JBS. A boneless arm. Boston Med Surg J. 1838;18:368-369.
2. Gorham LW, Stout AP. Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone): its relation to hemangiomatosis. J Bone Joint Surg Am. 1955;37(5):985-1004.
3. Lehmann G, Pfeil A, Böttcher J, et al. Benefit of a 17-year long-term bisphosphonate therapy in a patient with Gorham-Stout syndrome. Arch Orthop Trauma Surg. 2009;129(7):967-972.
4. Heyd R, Micke O, Surholt C, et al; German Cooperative Group on Radiotherapy for Benign Diseases (GCG-BD). Radiation therapy for Gorham-Stout syndrome: results of a national patterns-of-care study and literature review. Int J Radiat Oncol Biol Phys. 2011;81(3):e179-e185.
5. Kulenkampff HA, Richter GM, Hasse WE, Adler CP. Massive pelvic osteolysis in the Gorham-Stout syndrome. Int Orthop. 1990;14(4):361-366.
6. Ruggieri P, Montalti M, Angelini A, Alberghini M, Mercuri M. Gorham-Stout disease: the experience of the Rizzoli Institute and review of the literature. Skeletal Radiol. 2011;40(11):1391-1397.
7. Vinée P, Tanyü MO, Hauenstein KH, Sigmund G, Stöver B, Adler CP. CT and MRI of Gorham syndrome. J Comput Assist Tomogr. 1994;18(6):985-989.
8. Boyer P, Bourgeois P, Boyer O, Catonné Y, Saillant G. Massive Gorham-Stout syndrome of the pelvis. Clin Rheumatol. 2005;24(5):551-555.
9. Malde R, Agrawal HM, Ghosh SL, Dinshaw KA. Vanishing bone disease involving the pelvis. J Cancer Res Ther. 2005;1(4):227-228.
10. Kuriyama DK, McElligott SC, Glaser DW, Thompson KS. Treatment of Gorham-Stout disease with zoledronic acid and interferon-α: a case report and literature review. J Pediatr Hematol Oncol. 2010;32(8):579-584.
11. Tie ML, Poland GA, Rosenow EC III. Chylothorax in Gorham’s syndrome. A common complication of a rare disease. Chest. 1994;105(1):208-213.
12. Möller G, Priemel M, Amling M, Werner M, Kuhlmey AS, Delling G. The Gorham-Stout syndrome (Gorham’s massive osteolysis). A report of six cases with histopathological findings. J Bone Joint Surg Br. 1999;81(3):501-506.
13. Dupond JL, Bermont L, Runge M, de Billy M. Plasma VEGF determination in disseminated lymphangiomatosis—Gorham-Stout syndrome: a marker of activity? A case report with a 5-year follow-up. Bone. 2010;46(3):873-876.
14. Wang JD, Chang TK, Cheng YY, et al. A child with dyspnea and unstable gait. Pediatr Hemat Oncol. 2007;24(4):321-324.
15. Zheng MW, Yang M, Qiu JX, et al. Gorham-Stout syndrome presenting in a 5-year-old girl with a successful bisphosphonate therapeutic effect. Exp Ther Med. 2012;4(3):449-451.
16. Timke C, Krause MF, Oppermann HC, Leuschner I, Claviez A. Interferon alpha 2b treatment in an eleven-year-old boy with disseminated lymphangiomatosis. Pediatr Blood Cancer. 2007;48(1):108-111.
17. Stöve J, Reichelt A. Massive osteolysis of the pelvis, femur and sacral bone with a Gorham-Stout syndrome. Arch Orthop Trauma Surg. 1995;114(4):207-210.
1. Jackson JBS. A boneless arm. Boston Med Surg J. 1838;18:368-369.
2. Gorham LW, Stout AP. Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone): its relation to hemangiomatosis. J Bone Joint Surg Am. 1955;37(5):985-1004.
3. Lehmann G, Pfeil A, Böttcher J, et al. Benefit of a 17-year long-term bisphosphonate therapy in a patient with Gorham-Stout syndrome. Arch Orthop Trauma Surg. 2009;129(7):967-972.
4. Heyd R, Micke O, Surholt C, et al; German Cooperative Group on Radiotherapy for Benign Diseases (GCG-BD). Radiation therapy for Gorham-Stout syndrome: results of a national patterns-of-care study and literature review. Int J Radiat Oncol Biol Phys. 2011;81(3):e179-e185.
5. Kulenkampff HA, Richter GM, Hasse WE, Adler CP. Massive pelvic osteolysis in the Gorham-Stout syndrome. Int Orthop. 1990;14(4):361-366.
6. Ruggieri P, Montalti M, Angelini A, Alberghini M, Mercuri M. Gorham-Stout disease: the experience of the Rizzoli Institute and review of the literature. Skeletal Radiol. 2011;40(11):1391-1397.
7. Vinée P, Tanyü MO, Hauenstein KH, Sigmund G, Stöver B, Adler CP. CT and MRI of Gorham syndrome. J Comput Assist Tomogr. 1994;18(6):985-989.
8. Boyer P, Bourgeois P, Boyer O, Catonné Y, Saillant G. Massive Gorham-Stout syndrome of the pelvis. Clin Rheumatol. 2005;24(5):551-555.
9. Malde R, Agrawal HM, Ghosh SL, Dinshaw KA. Vanishing bone disease involving the pelvis. J Cancer Res Ther. 2005;1(4):227-228.
10. Kuriyama DK, McElligott SC, Glaser DW, Thompson KS. Treatment of Gorham-Stout disease with zoledronic acid and interferon-α: a case report and literature review. J Pediatr Hematol Oncol. 2010;32(8):579-584.
11. Tie ML, Poland GA, Rosenow EC III. Chylothorax in Gorham’s syndrome. A common complication of a rare disease. Chest. 1994;105(1):208-213.
12. Möller G, Priemel M, Amling M, Werner M, Kuhlmey AS, Delling G. The Gorham-Stout syndrome (Gorham’s massive osteolysis). A report of six cases with histopathological findings. J Bone Joint Surg Br. 1999;81(3):501-506.
13. Dupond JL, Bermont L, Runge M, de Billy M. Plasma VEGF determination in disseminated lymphangiomatosis—Gorham-Stout syndrome: a marker of activity? A case report with a 5-year follow-up. Bone. 2010;46(3):873-876.
14. Wang JD, Chang TK, Cheng YY, et al. A child with dyspnea and unstable gait. Pediatr Hemat Oncol. 2007;24(4):321-324.
15. Zheng MW, Yang M, Qiu JX, et al. Gorham-Stout syndrome presenting in a 5-year-old girl with a successful bisphosphonate therapeutic effect. Exp Ther Med. 2012;4(3):449-451.
16. Timke C, Krause MF, Oppermann HC, Leuschner I, Claviez A. Interferon alpha 2b treatment in an eleven-year-old boy with disseminated lymphangiomatosis. Pediatr Blood Cancer. 2007;48(1):108-111.
17. Stöve J, Reichelt A. Massive osteolysis of the pelvis, femur and sacral bone with a Gorham-Stout syndrome. Arch Orthop Trauma Surg. 1995;114(4):207-210.