User login
Woody Erythematous Induration on the Posterior Neck
The Diagnosis: Scleredema Diabeticorum
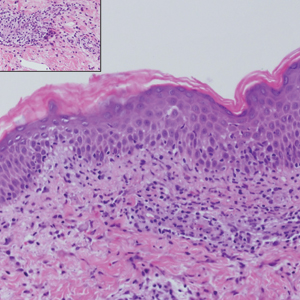
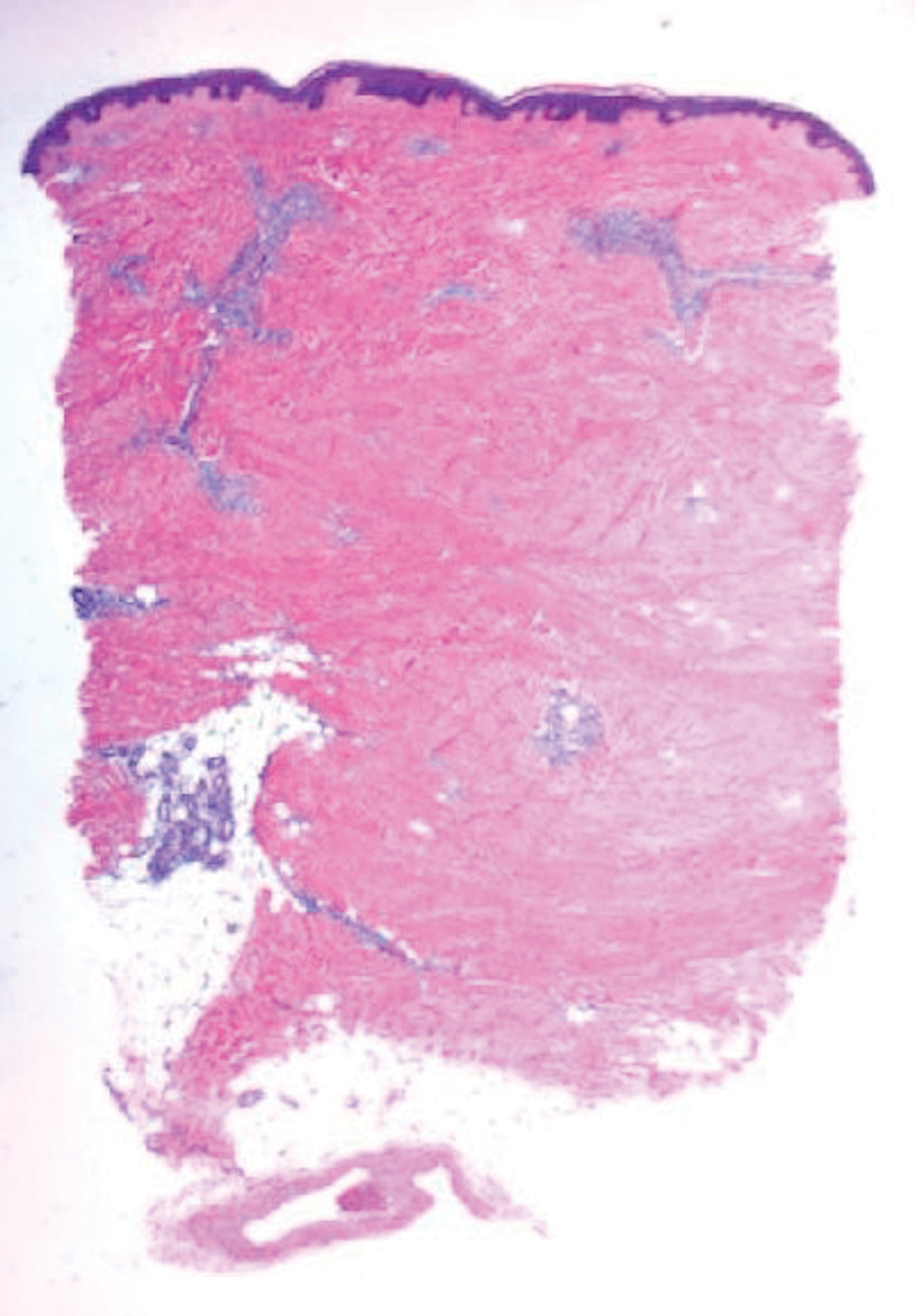
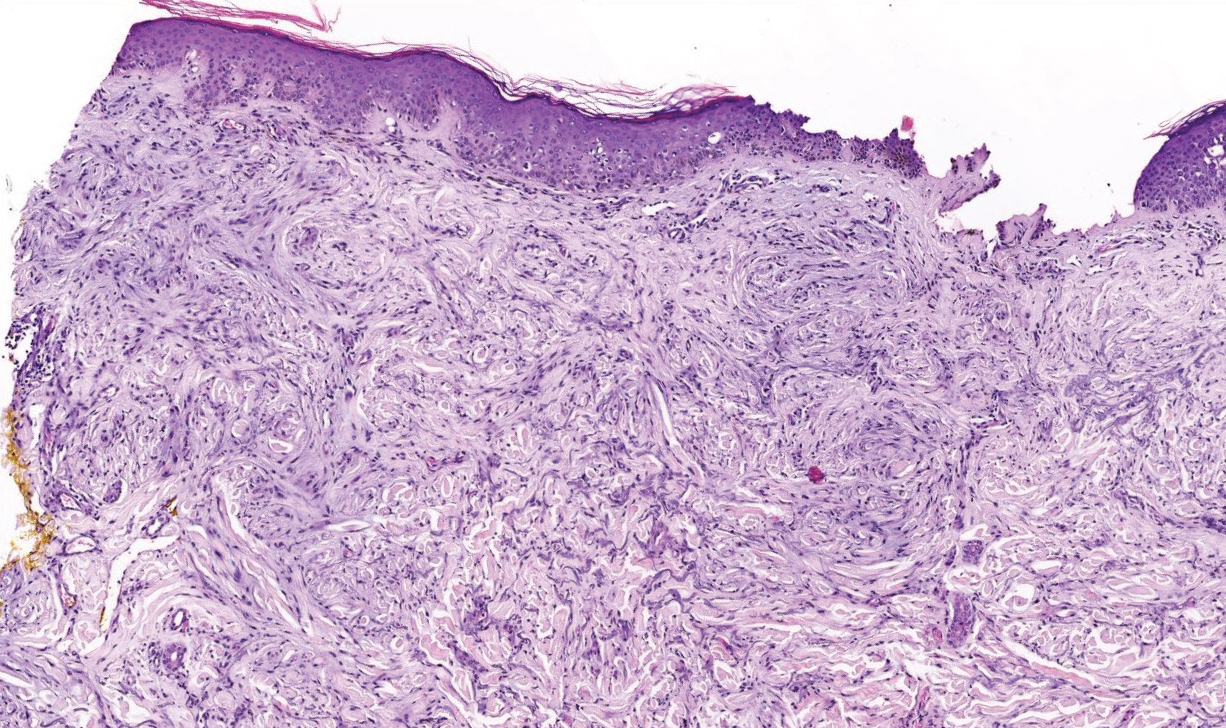
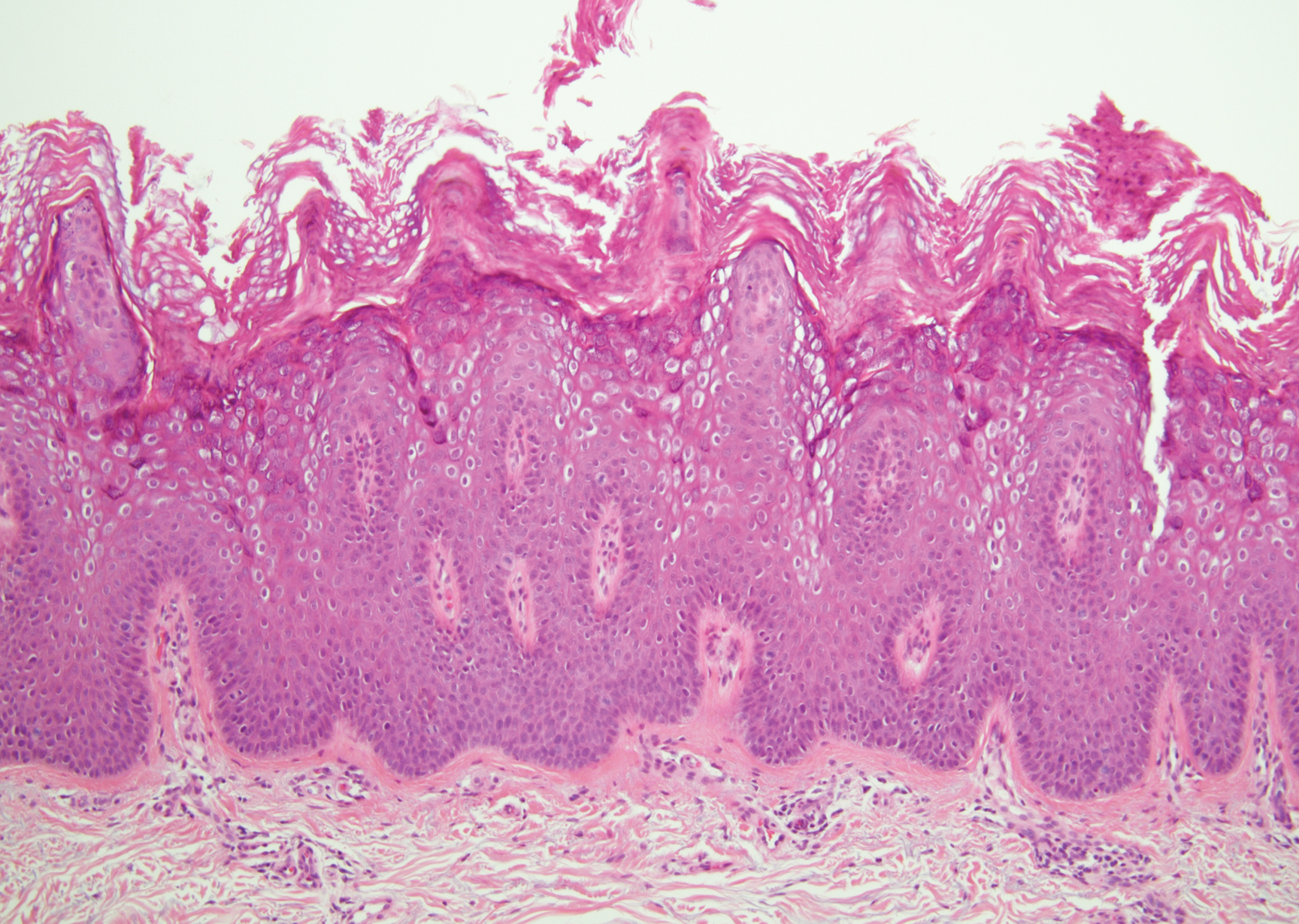
Histologically, scleredema is characterized by mucin deposition between collagen bundles in the deep dermis. Clinically, it is characterized by a progressive indurated plaque with associated stiffness of the involved area. It most commonly presents on the posterior aspect of the neck, though it can extend to involve the shoulders and upper torso.1 Scleredema is divided into 3 subtypes based on clinical associations. Type 1 often is preceded by an infection, most commonly group A Streptococcus. This type occurs acutely and often resolves completely over a few months.2 Type 2, which has progressive onset, is associated with monoclonal gammopathy.3 Type 3 is the most common type and is associated with diabetes mellitus. A study of 484 patients with type 2 diabetes mellitus demonstrated a prevalence of 2.5%.4 Although the exact pathogenesis has not been defined, it is hypothesized that irreversible glycosylation of collagen and alterations in collagenase activity may lead to accumulation of collagen and mucin in the dermis.5 Similar to type 2, type 3 scleredema appears subtly, progresses slowly, and tends to be chronic.1,6 Scleredema is characterized by marked dermal thickening and enlarged collagen bundles separated by mucin deposition (Figure 1). Fibroblast proliferation is characteristically absent.1
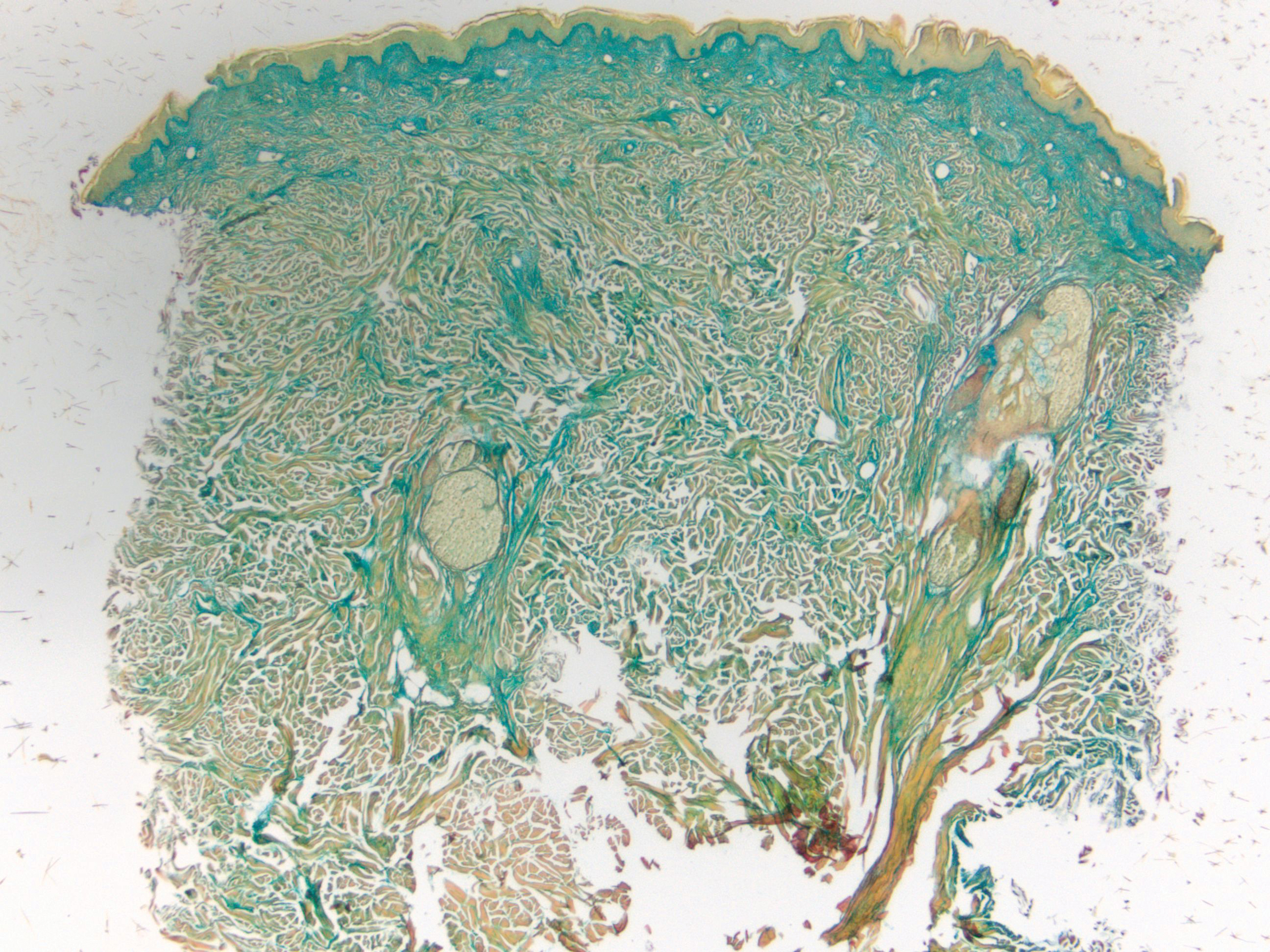
Clinically, tumid lupus erythematosus presents with erythematous edematous plaques on sun-exposed areas.7 Pretibial myxedema (PM) classically is associated with Graves disease; however, it can present in association with other types of thyroid dysfunction. Classically, PM presents on the pretibial regions as well-demarcated erythematous or hyperpigmented plaques.8 Similar to scleredema, histologic examination of tumid lupus erythematosus and PM reveals mucin deposition. Tumid lupus erythematosus also may demonstrate periadnexal and perivascular lymphocytic inflammation (Figure 2).7 The collagen bundles present in PM often are thin in comparison to scleredema (Figure 3).8

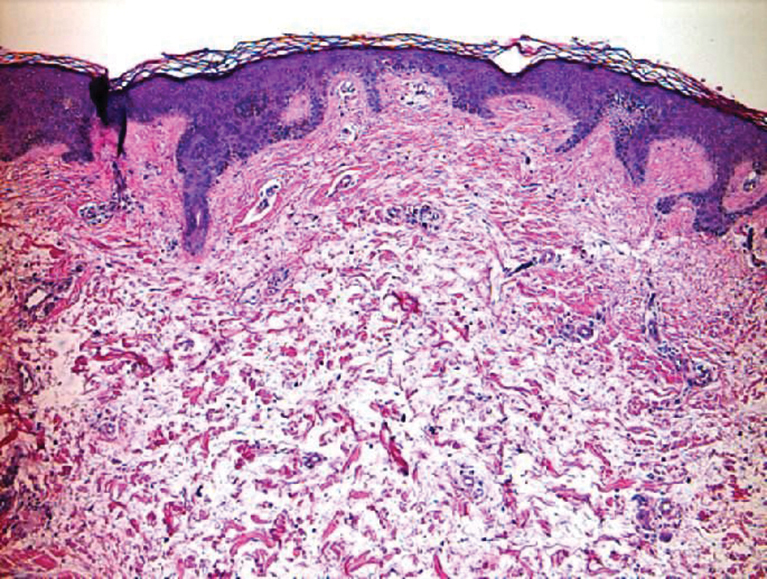
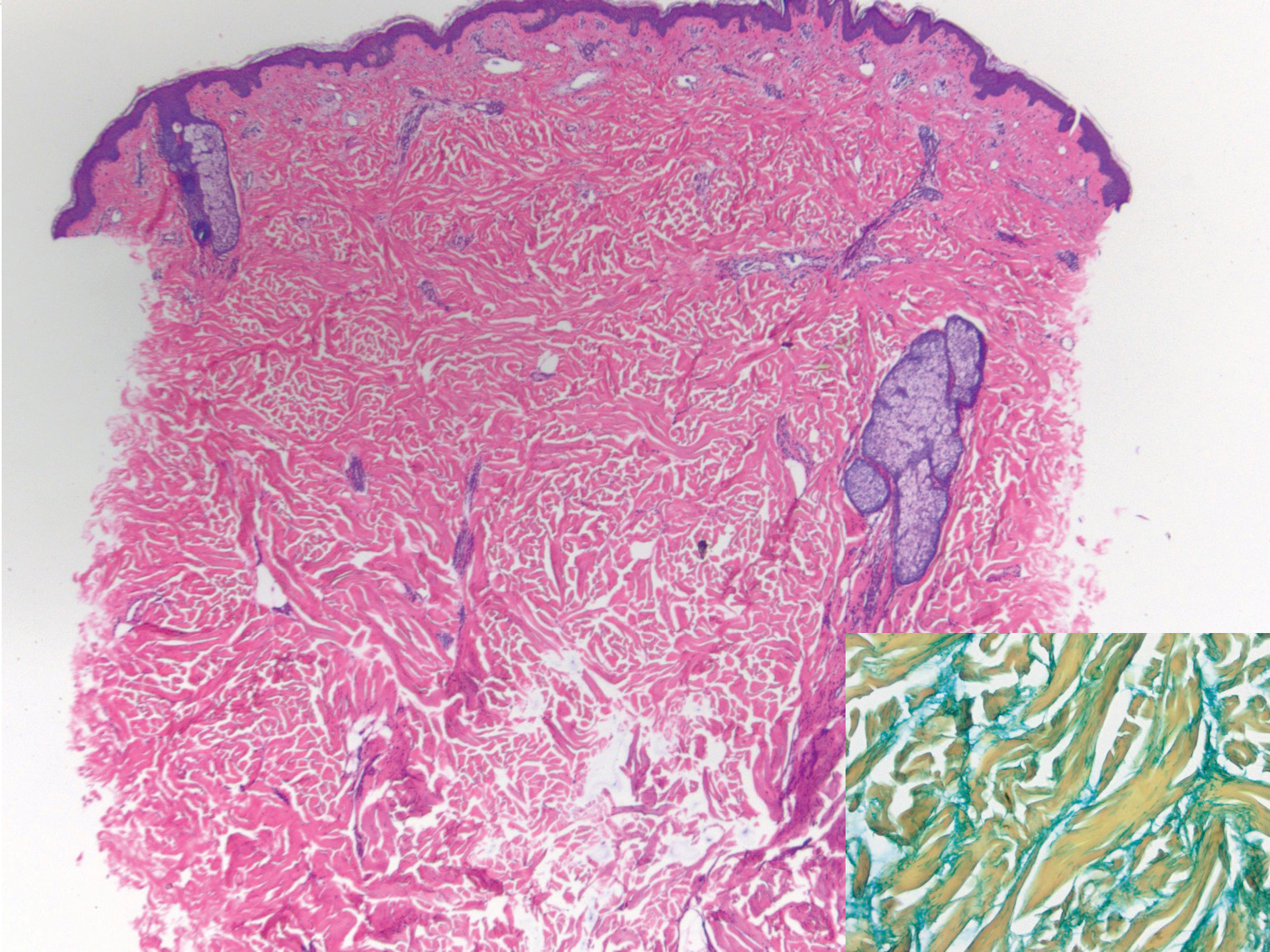
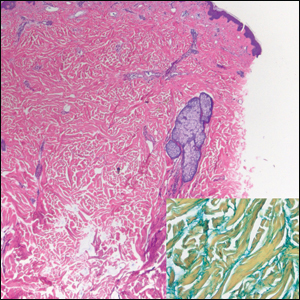
Scleroderma also presents with skin induration, erythema, and stiffening. However, unlike scleredema, scleroderma commonly involves the fingers, toes, and face. It presents with symptoms of Raynaud phenomenon, painful digital nonpitting edema, perioral skin tightening, mucocutaneous telangiectasia, and calcinosis cutis. Scleroderma also can involve organs such as the lungs, heart, kidneys, and gastrointestinal tract.9 Histologically, scleroderma is characterized by a compact dermis with closely packed collagen bundles. Other features of scleroderma can include perivascular mononuclear inflammatory cell infiltration, progressive atrophy of intradermal and perieccrine fat, and fibrosis (Figure 4).10
Scleromyxedema, also called papular mucinosis, is primary dermal mucinosis that often presents with waxy, dome-shaped papules that may coalesce into plaques. Similar to scleredema, scleromyxedema shows increased mucin deposition. However, scleromyxedema commonly is associated with fibroblast proliferation, which is characteristically absent in scleredema (Figure 5).11
- Beers WH, Ince A, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
- Cron RQ, Swetter SM. Scleredema revisited. a poststreptococcal complication. Clin Pediatr (Phila). 1994;33:606-610.
- Kövary PM, Vakilzadeh F, Macher E, et al. Monoclonal gammopathy in scleredema. observations in three cases. Arch Dermatol. 1981;117:536-539.
- Cole GW, Headley J, Skowsky R. Scleredema diabeticorum: a common and distinct cutaneous manifestation of diabetes mellitus. Diabetes Care. 1983;6:189-192.
- Namas R, Ashraf A. Scleredema of Buschke. Eur J Rheumatol. 2016;3:191-192.
- Knobler R, Moinzadeh P, Hunzelmann N, et al. European Dermatology Forum S1-guideline on the diagnosis and treatment of sclerosing diseases of the skin, part 2: scleromyxedema, scleredema and nephrogenic systemic fibrosis. J Eur Acad Dermatol Venereol. 2017;31:1581-1594.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus--a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
- van den Hoogen F, Khanna D, Fransen J, et al. 2013 Classification Criteria for Systemic Sclerosis: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. 2013;65:2737-2747.
- Ferreli C, Gasparini G, Parodi A, et al. Cutaneous manifestations of scleroderma and scleroderma-like disorders: a comprehensive review. Clin Rev Allergy Immunol. 2017;53:306-336.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
The Diagnosis: Scleredema Diabeticorum
Histologically, scleredema is characterized by mucin deposition between collagen bundles in the deep dermis. Clinically, it is characterized by a progressive indurated plaque with associated stiffness of the involved area. It most commonly presents on the posterior aspect of the neck, though it can extend to involve the shoulders and upper torso.1 Scleredema is divided into 3 subtypes based on clinical associations. Type 1 often is preceded by an infection, most commonly group A Streptococcus. This type occurs acutely and often resolves completely over a few months.2 Type 2, which has progressive onset, is associated with monoclonal gammopathy.3 Type 3 is the most common type and is associated with diabetes mellitus. A study of 484 patients with type 2 diabetes mellitus demonstrated a prevalence of 2.5%.4 Although the exact pathogenesis has not been defined, it is hypothesized that irreversible glycosylation of collagen and alterations in collagenase activity may lead to accumulation of collagen and mucin in the dermis.5 Similar to type 2, type 3 scleredema appears subtly, progresses slowly, and tends to be chronic.1,6 Scleredema is characterized by marked dermal thickening and enlarged collagen bundles separated by mucin deposition (Figure 1). Fibroblast proliferation is characteristically absent.1
Clinically, tumid lupus erythematosus presents with erythematous edematous plaques on sun-exposed areas.7 Pretibial myxedema (PM) classically is associated with Graves disease; however, it can present in association with other types of thyroid dysfunction. Classically, PM presents on the pretibial regions as well-demarcated erythematous or hyperpigmented plaques.8 Similar to scleredema, histologic examination of tumid lupus erythematosus and PM reveals mucin deposition. Tumid lupus erythematosus also may demonstrate periadnexal and perivascular lymphocytic inflammation (Figure 2).7 The collagen bundles present in PM often are thin in comparison to scleredema (Figure 3).8
Scleroderma also presents with skin induration, erythema, and stiffening. However, unlike scleredema, scleroderma commonly involves the fingers, toes, and face. It presents with symptoms of Raynaud phenomenon, painful digital nonpitting edema, perioral skin tightening, mucocutaneous telangiectasia, and calcinosis cutis. Scleroderma also can involve organs such as the lungs, heart, kidneys, and gastrointestinal tract.9 Histologically, scleroderma is characterized by a compact dermis with closely packed collagen bundles. Other features of scleroderma can include perivascular mononuclear inflammatory cell infiltration, progressive atrophy of intradermal and perieccrine fat, and fibrosis (Figure 4).10
Scleromyxedema, also called papular mucinosis, is primary dermal mucinosis that often presents with waxy, dome-shaped papules that may coalesce into plaques. Similar to scleredema, scleromyxedema shows increased mucin deposition. However, scleromyxedema commonly is associated with fibroblast proliferation, which is characteristically absent in scleredema (Figure 5).11
The Diagnosis: Scleredema Diabeticorum
Histologically, scleredema is characterized by mucin deposition between collagen bundles in the deep dermis. Clinically, it is characterized by a progressive indurated plaque with associated stiffness of the involved area. It most commonly presents on the posterior aspect of the neck, though it can extend to involve the shoulders and upper torso.1 Scleredema is divided into 3 subtypes based on clinical associations. Type 1 often is preceded by an infection, most commonly group A Streptococcus. This type occurs acutely and often resolves completely over a few months.2 Type 2, which has progressive onset, is associated with monoclonal gammopathy.3 Type 3 is the most common type and is associated with diabetes mellitus. A study of 484 patients with type 2 diabetes mellitus demonstrated a prevalence of 2.5%.4 Although the exact pathogenesis has not been defined, it is hypothesized that irreversible glycosylation of collagen and alterations in collagenase activity may lead to accumulation of collagen and mucin in the dermis.5 Similar to type 2, type 3 scleredema appears subtly, progresses slowly, and tends to be chronic.1,6 Scleredema is characterized by marked dermal thickening and enlarged collagen bundles separated by mucin deposition (Figure 1). Fibroblast proliferation is characteristically absent.1
Clinically, tumid lupus erythematosus presents with erythematous edematous plaques on sun-exposed areas.7 Pretibial myxedema (PM) classically is associated with Graves disease; however, it can present in association with other types of thyroid dysfunction. Classically, PM presents on the pretibial regions as well-demarcated erythematous or hyperpigmented plaques.8 Similar to scleredema, histologic examination of tumid lupus erythematosus and PM reveals mucin deposition. Tumid lupus erythematosus also may demonstrate periadnexal and perivascular lymphocytic inflammation (Figure 2).7 The collagen bundles present in PM often are thin in comparison to scleredema (Figure 3).8
Scleroderma also presents with skin induration, erythema, and stiffening. However, unlike scleredema, scleroderma commonly involves the fingers, toes, and face. It presents with symptoms of Raynaud phenomenon, painful digital nonpitting edema, perioral skin tightening, mucocutaneous telangiectasia, and calcinosis cutis. Scleroderma also can involve organs such as the lungs, heart, kidneys, and gastrointestinal tract.9 Histologically, scleroderma is characterized by a compact dermis with closely packed collagen bundles. Other features of scleroderma can include perivascular mononuclear inflammatory cell infiltration, progressive atrophy of intradermal and perieccrine fat, and fibrosis (Figure 4).10
Scleromyxedema, also called papular mucinosis, is primary dermal mucinosis that often presents with waxy, dome-shaped papules that may coalesce into plaques. Similar to scleredema, scleromyxedema shows increased mucin deposition. However, scleromyxedema commonly is associated with fibroblast proliferation, which is characteristically absent in scleredema (Figure 5).11
- Beers WH, Ince A, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
- Cron RQ, Swetter SM. Scleredema revisited. a poststreptococcal complication. Clin Pediatr (Phila). 1994;33:606-610.
- Kövary PM, Vakilzadeh F, Macher E, et al. Monoclonal gammopathy in scleredema. observations in three cases. Arch Dermatol. 1981;117:536-539.
- Cole GW, Headley J, Skowsky R. Scleredema diabeticorum: a common and distinct cutaneous manifestation of diabetes mellitus. Diabetes Care. 1983;6:189-192.
- Namas R, Ashraf A. Scleredema of Buschke. Eur J Rheumatol. 2016;3:191-192.
- Knobler R, Moinzadeh P, Hunzelmann N, et al. European Dermatology Forum S1-guideline on the diagnosis and treatment of sclerosing diseases of the skin, part 2: scleromyxedema, scleredema and nephrogenic systemic fibrosis. J Eur Acad Dermatol Venereol. 2017;31:1581-1594.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus--a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
- van den Hoogen F, Khanna D, Fransen J, et al. 2013 Classification Criteria for Systemic Sclerosis: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. 2013;65:2737-2747.
- Ferreli C, Gasparini G, Parodi A, et al. Cutaneous manifestations of scleroderma and scleroderma-like disorders: a comprehensive review. Clin Rev Allergy Immunol. 2017;53:306-336.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Beers WH, Ince A, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
- Cron RQ, Swetter SM. Scleredema revisited. a poststreptococcal complication. Clin Pediatr (Phila). 1994;33:606-610.
- Kövary PM, Vakilzadeh F, Macher E, et al. Monoclonal gammopathy in scleredema. observations in three cases. Arch Dermatol. 1981;117:536-539.
- Cole GW, Headley J, Skowsky R. Scleredema diabeticorum: a common and distinct cutaneous manifestation of diabetes mellitus. Diabetes Care. 1983;6:189-192.
- Namas R, Ashraf A. Scleredema of Buschke. Eur J Rheumatol. 2016;3:191-192.
- Knobler R, Moinzadeh P, Hunzelmann N, et al. European Dermatology Forum S1-guideline on the diagnosis and treatment of sclerosing diseases of the skin, part 2: scleromyxedema, scleredema and nephrogenic systemic fibrosis. J Eur Acad Dermatol Venereol. 2017;31:1581-1594.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus--a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
- van den Hoogen F, Khanna D, Fransen J, et al. 2013 Classification Criteria for Systemic Sclerosis: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. 2013;65:2737-2747.
- Ferreli C, Gasparini G, Parodi A, et al. Cutaneous manifestations of scleroderma and scleroderma-like disorders: a comprehensive review. Clin Rev Allergy Immunol. 2017;53:306-336.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
A 39-year-old white woman with a medical history of type 1 diabetes mellitus and rheumatoid arthritis presented to the dermatology clinic with pain and thickened skin on the posterior neck of 4 weeks’ duration. The patient noted stiffness in the neck and shoulders but denied any pain, pruritus, fever, chills, night sweats, fatigue, cough, dyspnea, dysphagia, weight loss, or change in appetite. Physical examination revealed a woody indurated plaque with slight erythema that was present diffusely on the posterior neck and upper back. The patient reported that a recent complete blood cell count and complete metabolic panel performed by her primary care physician were within reference range. Hemoglobin A1C was 8.6% of total hemoglobin (reference range, 4%–7%). A punch biopsy was performed.
Purpuric Bullae on the Lower Extremities
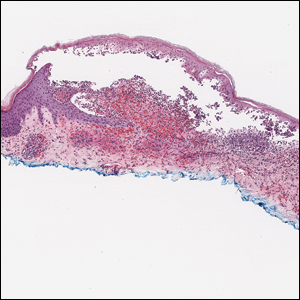
The Diagnosis: Bullous Leukocytoclastic Vasculitis
Histopathology with hematoxylin and eosin (H&E) stain showed a perivascular neutrophilic infiltrate, karyorrhexis, red blood cell extravasation, and fibrin deposition in the vessel wall (quiz images). Direct immunofluorescence (DIF) showed fibrin surrounding the vasculature, consistent with vasculitis. The clinical and histopathological evaluation supported the diagnosis of bullous leukocytoclastic vasculitis (LCV). The patient had a full LCV workup including antinuclear antibody, rheumatoid factor, hepatitis B and hepatitis C screening, erythrocyte sedimentation rate, C-reactive protein, and C3/C4/total complement level, which were all within reference range. The patient denied that she had taken any medications prior to the onset of the rash. She was started on a 12-day prednisone taper starting at 60 mg, and the rash resolved in 1 week.
Although the incidence of LCV is estimated to be 30 cases per million individuals per year,1 bullous LCV is a rarer entity with only a few cases reported in the literature.2,3 As in our patient's case, up to 50% of LCV cases are idiopathic or the etiology cannot be determined despite laboratory workup and medication review. Other cases can be secondary to medication, infection, collagen vascular disease, or malignancy.3 Despite the exact pathogenesis of bullous LCV being unknown,4 it likely is related to a type III hypersensitivity reaction with immune complex deposition in postcapillary venules leading to endothelial injury, activation of the complement cascade, and development of intraepidermal or subepidermal blister formation depending on location of inflammation and edema.2 Clinically, an intraepidermal split would be more flaccid, similar to pemphigus vulgaris, while a subepidermal split, as in our patient, would be taut bullae. The subepidermal split more commonly is seen in bullous LCV.2
Leukocytoclastic vasculitis on H&E staining characteristically has a perivascular inflammatory infiltrate, neutrophilic fragments called leukocytoclasis, and blood extravasation.3 Extravasated blood presents clinically as petechiae. In this case, the petechiae helped distinguish this entity from the differential diagnosis. Furthermore, DIF would be helpful in distinguishing bullous diseases such as bullous pemphigoid (BP) and pemphigus vulgaris from LCV.2 Direct immunofluorescence in bullous LCV would have fibrinogen surrounding the vasculature without C3 and IgG deposition (intraepidermal or subepidermal).
Mild cases of LCV often resolve with supportive measures including elevation of the legs, ice packs applied to the affected area, and removal of the inciting drug or event.4 In the few cases reported in the literature, bullous LCV presented more diffusely than classic LCV with bullous lesions on the forearms and the lower extremities. Oral steroids are efficacious for extensive bullous LCV.4
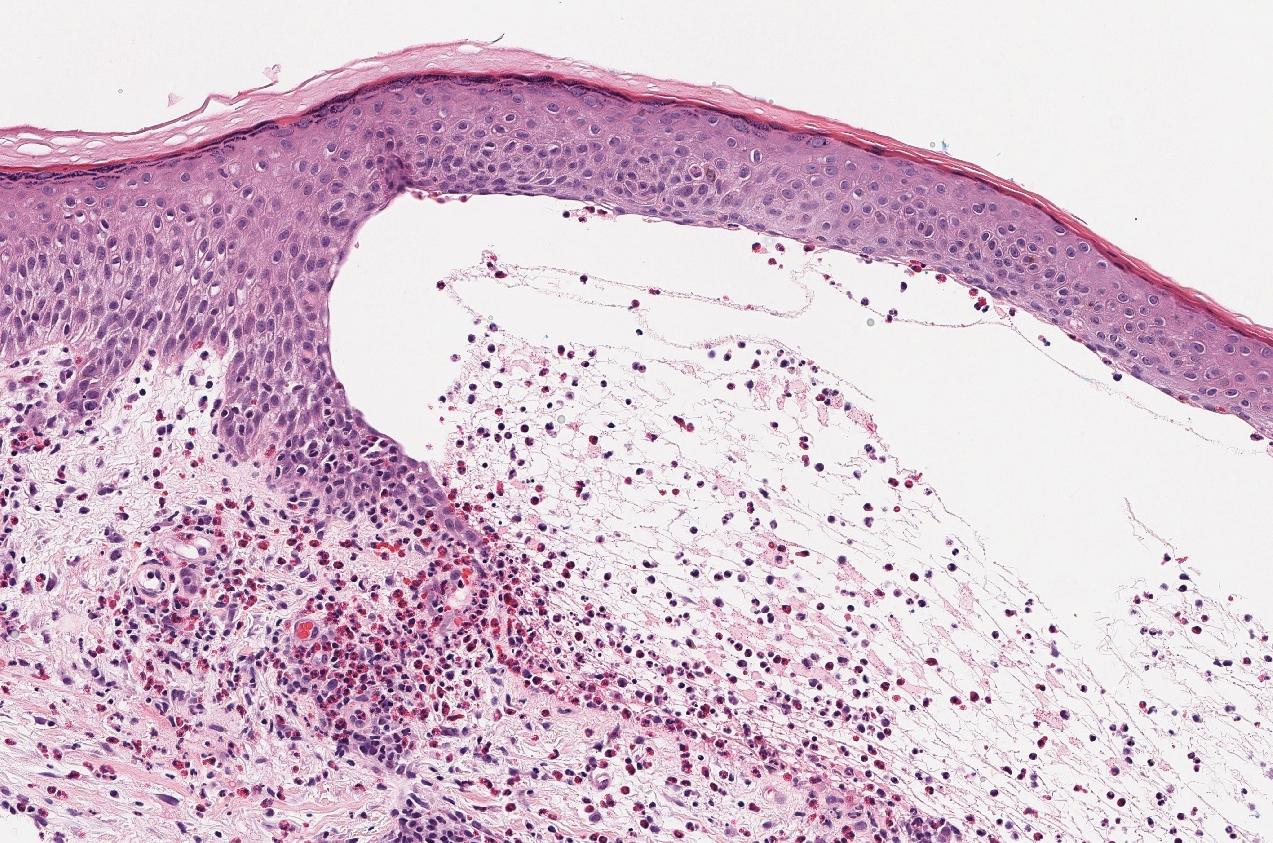
The differential diagnosis of bullous LCV includes bullous diseases with subepidermal split including BP and linear IgA bullous dermatosis (LABD). Bullous pemphigoid is an autoimmune subepidermal blistering disease typically affecting patients older than 60 years.5 The pathogenesis of BP is related to development of autoantibodies directed against hemidesmosome components, bullous pemphigoid antigen (BPAG) 1 or BPAG2.5 Bullous pemphigoid presents clinically as widespread, generally pruritic, erythematous, urticarial plaques with bullae. Histologically, BP characteristically has a subepidermal split with superficial dermal edema and eosinophils at the dermoepidermal junction (Figure 1). Direct immunofluorescence confirms the diagnosis with IgG and C3 deposition in an n-serrated pattern at the dermoepidermal junction.6 Bullous pemphigoid can be distinguished from bullous LCV by the older age of presentation, DIF findings, and the absence of purpura.
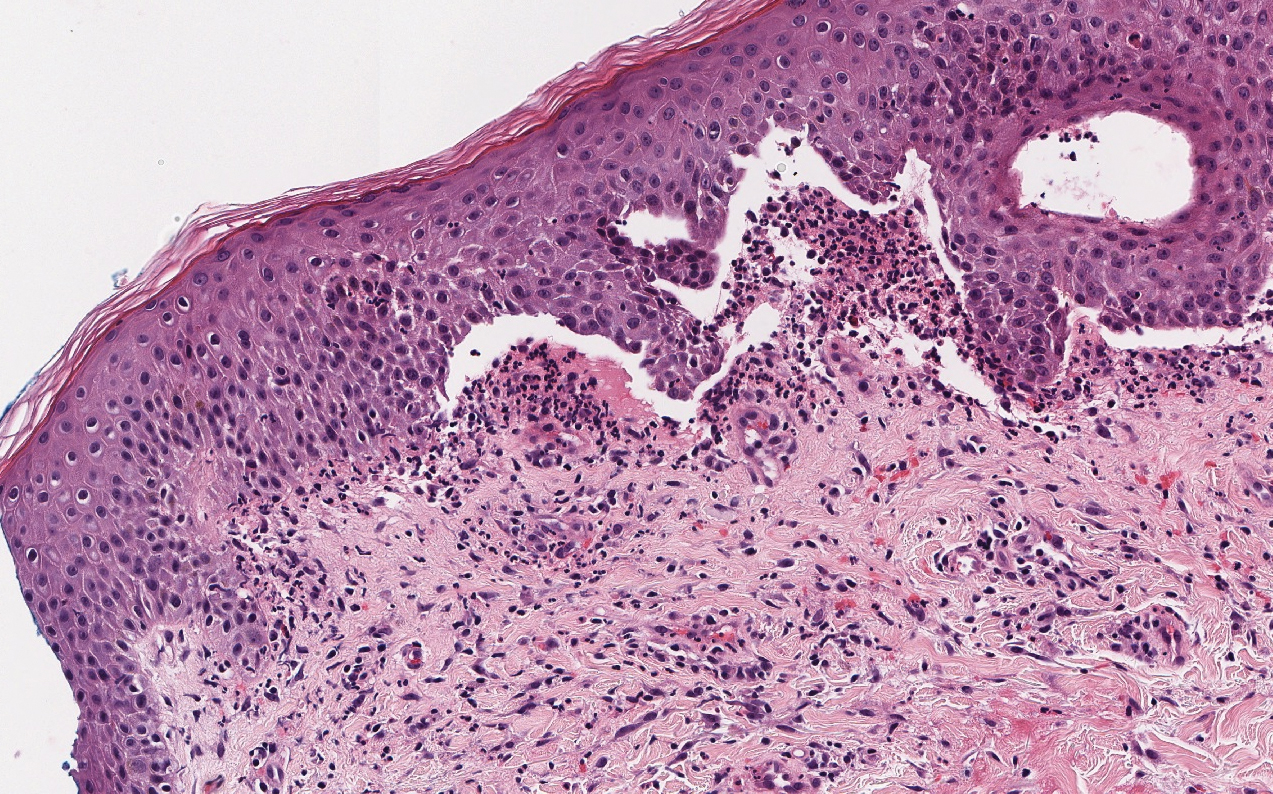
Linear IgA bullous dermatosis represents a rare subepidermal vesiculobullous disease occurring in patients in their 60s.7 Clinically, this entity presents as tense bullae often located on the periphery of an urticarial plaque, classically called the "string of pearls sign." Histologically, LABD also presents with subepidermal split; however, neutrophils are the predominant cell type vs eosinophils in BP (Figure 2).7 Direct immunofluorescence is specific with a linear deposition of IgA at the dermoepidermal junction. Linear IgA bullous dermatosis most commonly is induced by vancomycin. Unlike bullous LCV, the bullae of LABD have an annular peripheral pattern on an erythematous base and lack purpura.
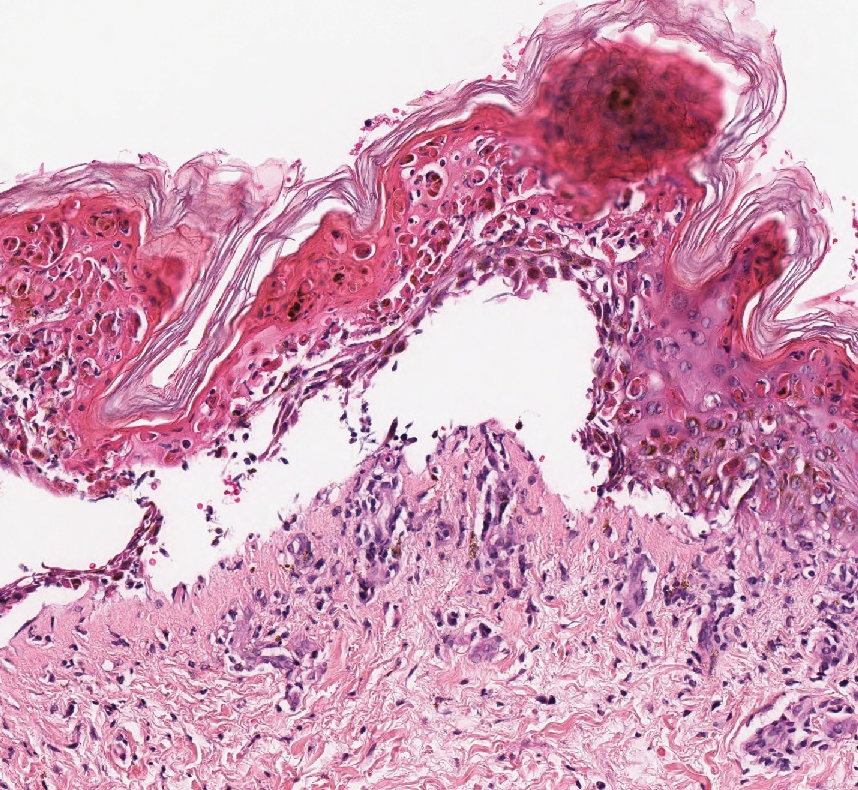
Stasis dermatitis is inflammation of the dermis due to venous insufficiency that often is present in the bilateral lower extremities. The disorder affects approximately 7% of adults older than 50 years, but it also can occur in younger patients.8 The pathophysiology of stasis dermatitis is caused by edema, which leads to extracellular fluid, plasma proteins, macrophages, and erythrocytes passing into the interstitial space. Patients with stasis dermatitis present with scaly erythematous papules and plaques or edematous blisters on the lower extremities. Diagnosis usually can be made clinically; however, a skin biopsy also can be helpful. Hematoxylin and eosin shows a pauci-inflammatory subepidermal bulla with fibrin (Figure 3).8 The overlying epidermis is intact. The dermis has cannon ball angiomatosis, red blood cell extravasation, and fibrosis typical of stasis dermatitis. Stasis dermatitis with bullae is cell poor and lacks the perivascular inflammatory infiltrate and neutrophilic fragments that often are present in LCV, making the 2 entities distinguishable.

Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) lies on a spectrum of severe cutaneous drug reactions involving the skin and mucous membranes. Cutaneous involvement typically begins on the trunk and face and later can involve the palms and soles.9 Similar drugs have been implicated in bullous LCV and SJS/TEN, including nonsteroidal anti-inflammatory drugs and antibiotics. Histologically, SJS/TEN has full-thickness epidermal necrolysis, vacuolar interface, and keratinocyte apoptosis (Figure 4).9 The clinical presentation of sloughing of skin with positive Nikolsky sign, oral involvement, and H&E and DIF findings can help differentiate this entity from bullous LCV.
- Einhorn J, Levis JT. Dermatologic diagnosis: leukocytoclastic vasculitis. Perm J. 2015;19:77-78.
- Davidson KA, Ringpfeil F, Lee JB. Ibuprofen-induced bullous leukocytoclastic vasculitis. Cutis. 2001;67:303-307.
- Lazic T, Fonder M, Robinson-Bostom L, et al. Orlistat-induced bullous leukocytoclastic vasculitis. Cutis. 2013;91:148-149.
- Mericliler M, Shnawa A, Al-Qaysi D, et al. Oxacillin-induced leukocytoclastic vasculitis. IDCases. 2019;17:E00539.
- Bernard P, Antonicelli F. Bullous pemphigoid: a review of its diagnosis, associations and treatment. Am J Clin Dermatol. 2017;18:513-528.
- High WA. Blistering disorders. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019:161-171.
- Visentainer L, Massuda JY, Cintra ML, et al. Vancomycin-induced linear IgA bullous dermatosis (LABD)--an atypical presentation. Clin Case Rep. 2019;7:1091-1093.
- Hyman DA, Cohen PR. Stasis dermatitis as a complication of recurrent levofloxacin-associated bilateral leg edema. Dermatol Online J. 2013;19:20399.
- Harr T, French LE. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;5:39.
The Diagnosis: Bullous Leukocytoclastic Vasculitis
Histopathology with hematoxylin and eosin (H&E) stain showed a perivascular neutrophilic infiltrate, karyorrhexis, red blood cell extravasation, and fibrin deposition in the vessel wall (quiz images). Direct immunofluorescence (DIF) showed fibrin surrounding the vasculature, consistent with vasculitis. The clinical and histopathological evaluation supported the diagnosis of bullous leukocytoclastic vasculitis (LCV). The patient had a full LCV workup including antinuclear antibody, rheumatoid factor, hepatitis B and hepatitis C screening, erythrocyte sedimentation rate, C-reactive protein, and C3/C4/total complement level, which were all within reference range. The patient denied that she had taken any medications prior to the onset of the rash. She was started on a 12-day prednisone taper starting at 60 mg, and the rash resolved in 1 week.
Although the incidence of LCV is estimated to be 30 cases per million individuals per year,1 bullous LCV is a rarer entity with only a few cases reported in the literature.2,3 As in our patient's case, up to 50% of LCV cases are idiopathic or the etiology cannot be determined despite laboratory workup and medication review. Other cases can be secondary to medication, infection, collagen vascular disease, or malignancy.3 Despite the exact pathogenesis of bullous LCV being unknown,4 it likely is related to a type III hypersensitivity reaction with immune complex deposition in postcapillary venules leading to endothelial injury, activation of the complement cascade, and development of intraepidermal or subepidermal blister formation depending on location of inflammation and edema.2 Clinically, an intraepidermal split would be more flaccid, similar to pemphigus vulgaris, while a subepidermal split, as in our patient, would be taut bullae. The subepidermal split more commonly is seen in bullous LCV.2
Leukocytoclastic vasculitis on H&E staining characteristically has a perivascular inflammatory infiltrate, neutrophilic fragments called leukocytoclasis, and blood extravasation.3 Extravasated blood presents clinically as petechiae. In this case, the petechiae helped distinguish this entity from the differential diagnosis. Furthermore, DIF would be helpful in distinguishing bullous diseases such as bullous pemphigoid (BP) and pemphigus vulgaris from LCV.2 Direct immunofluorescence in bullous LCV would have fibrinogen surrounding the vasculature without C3 and IgG deposition (intraepidermal or subepidermal).
Mild cases of LCV often resolve with supportive measures including elevation of the legs, ice packs applied to the affected area, and removal of the inciting drug or event.4 In the few cases reported in the literature, bullous LCV presented more diffusely than classic LCV with bullous lesions on the forearms and the lower extremities. Oral steroids are efficacious for extensive bullous LCV.4
The differential diagnosis of bullous LCV includes bullous diseases with subepidermal split including BP and linear IgA bullous dermatosis (LABD). Bullous pemphigoid is an autoimmune subepidermal blistering disease typically affecting patients older than 60 years.5 The pathogenesis of BP is related to development of autoantibodies directed against hemidesmosome components, bullous pemphigoid antigen (BPAG) 1 or BPAG2.5 Bullous pemphigoid presents clinically as widespread, generally pruritic, erythematous, urticarial plaques with bullae. Histologically, BP characteristically has a subepidermal split with superficial dermal edema and eosinophils at the dermoepidermal junction (Figure 1). Direct immunofluorescence confirms the diagnosis with IgG and C3 deposition in an n-serrated pattern at the dermoepidermal junction.6 Bullous pemphigoid can be distinguished from bullous LCV by the older age of presentation, DIF findings, and the absence of purpura.
Linear IgA bullous dermatosis represents a rare subepidermal vesiculobullous disease occurring in patients in their 60s.7 Clinically, this entity presents as tense bullae often located on the periphery of an urticarial plaque, classically called the "string of pearls sign." Histologically, LABD also presents with subepidermal split; however, neutrophils are the predominant cell type vs eosinophils in BP (Figure 2).7 Direct immunofluorescence is specific with a linear deposition of IgA at the dermoepidermal junction. Linear IgA bullous dermatosis most commonly is induced by vancomycin. Unlike bullous LCV, the bullae of LABD have an annular peripheral pattern on an erythematous base and lack purpura.
Stasis dermatitis is inflammation of the dermis due to venous insufficiency that often is present in the bilateral lower extremities. The disorder affects approximately 7% of adults older than 50 years, but it also can occur in younger patients.8 The pathophysiology of stasis dermatitis is caused by edema, which leads to extracellular fluid, plasma proteins, macrophages, and erythrocytes passing into the interstitial space. Patients with stasis dermatitis present with scaly erythematous papules and plaques or edematous blisters on the lower extremities. Diagnosis usually can be made clinically; however, a skin biopsy also can be helpful. Hematoxylin and eosin shows a pauci-inflammatory subepidermal bulla with fibrin (Figure 3).8 The overlying epidermis is intact. The dermis has cannon ball angiomatosis, red blood cell extravasation, and fibrosis typical of stasis dermatitis. Stasis dermatitis with bullae is cell poor and lacks the perivascular inflammatory infiltrate and neutrophilic fragments that often are present in LCV, making the 2 entities distinguishable.
Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) lies on a spectrum of severe cutaneous drug reactions involving the skin and mucous membranes. Cutaneous involvement typically begins on the trunk and face and later can involve the palms and soles.9 Similar drugs have been implicated in bullous LCV and SJS/TEN, including nonsteroidal anti-inflammatory drugs and antibiotics. Histologically, SJS/TEN has full-thickness epidermal necrolysis, vacuolar interface, and keratinocyte apoptosis (Figure 4).9 The clinical presentation of sloughing of skin with positive Nikolsky sign, oral involvement, and H&E and DIF findings can help differentiate this entity from bullous LCV.
The Diagnosis: Bullous Leukocytoclastic Vasculitis
Histopathology with hematoxylin and eosin (H&E) stain showed a perivascular neutrophilic infiltrate, karyorrhexis, red blood cell extravasation, and fibrin deposition in the vessel wall (quiz images). Direct immunofluorescence (DIF) showed fibrin surrounding the vasculature, consistent with vasculitis. The clinical and histopathological evaluation supported the diagnosis of bullous leukocytoclastic vasculitis (LCV). The patient had a full LCV workup including antinuclear antibody, rheumatoid factor, hepatitis B and hepatitis C screening, erythrocyte sedimentation rate, C-reactive protein, and C3/C4/total complement level, which were all within reference range. The patient denied that she had taken any medications prior to the onset of the rash. She was started on a 12-day prednisone taper starting at 60 mg, and the rash resolved in 1 week.
Although the incidence of LCV is estimated to be 30 cases per million individuals per year,1 bullous LCV is a rarer entity with only a few cases reported in the literature.2,3 As in our patient's case, up to 50% of LCV cases are idiopathic or the etiology cannot be determined despite laboratory workup and medication review. Other cases can be secondary to medication, infection, collagen vascular disease, or malignancy.3 Despite the exact pathogenesis of bullous LCV being unknown,4 it likely is related to a type III hypersensitivity reaction with immune complex deposition in postcapillary venules leading to endothelial injury, activation of the complement cascade, and development of intraepidermal or subepidermal blister formation depending on location of inflammation and edema.2 Clinically, an intraepidermal split would be more flaccid, similar to pemphigus vulgaris, while a subepidermal split, as in our patient, would be taut bullae. The subepidermal split more commonly is seen in bullous LCV.2
Leukocytoclastic vasculitis on H&E staining characteristically has a perivascular inflammatory infiltrate, neutrophilic fragments called leukocytoclasis, and blood extravasation.3 Extravasated blood presents clinically as petechiae. In this case, the petechiae helped distinguish this entity from the differential diagnosis. Furthermore, DIF would be helpful in distinguishing bullous diseases such as bullous pemphigoid (BP) and pemphigus vulgaris from LCV.2 Direct immunofluorescence in bullous LCV would have fibrinogen surrounding the vasculature without C3 and IgG deposition (intraepidermal or subepidermal).
Mild cases of LCV often resolve with supportive measures including elevation of the legs, ice packs applied to the affected area, and removal of the inciting drug or event.4 In the few cases reported in the literature, bullous LCV presented more diffusely than classic LCV with bullous lesions on the forearms and the lower extremities. Oral steroids are efficacious for extensive bullous LCV.4
The differential diagnosis of bullous LCV includes bullous diseases with subepidermal split including BP and linear IgA bullous dermatosis (LABD). Bullous pemphigoid is an autoimmune subepidermal blistering disease typically affecting patients older than 60 years.5 The pathogenesis of BP is related to development of autoantibodies directed against hemidesmosome components, bullous pemphigoid antigen (BPAG) 1 or BPAG2.5 Bullous pemphigoid presents clinically as widespread, generally pruritic, erythematous, urticarial plaques with bullae. Histologically, BP characteristically has a subepidermal split with superficial dermal edema and eosinophils at the dermoepidermal junction (Figure 1). Direct immunofluorescence confirms the diagnosis with IgG and C3 deposition in an n-serrated pattern at the dermoepidermal junction.6 Bullous pemphigoid can be distinguished from bullous LCV by the older age of presentation, DIF findings, and the absence of purpura.
Linear IgA bullous dermatosis represents a rare subepidermal vesiculobullous disease occurring in patients in their 60s.7 Clinically, this entity presents as tense bullae often located on the periphery of an urticarial plaque, classically called the "string of pearls sign." Histologically, LABD also presents with subepidermal split; however, neutrophils are the predominant cell type vs eosinophils in BP (Figure 2).7 Direct immunofluorescence is specific with a linear deposition of IgA at the dermoepidermal junction. Linear IgA bullous dermatosis most commonly is induced by vancomycin. Unlike bullous LCV, the bullae of LABD have an annular peripheral pattern on an erythematous base and lack purpura.
Stasis dermatitis is inflammation of the dermis due to venous insufficiency that often is present in the bilateral lower extremities. The disorder affects approximately 7% of adults older than 50 years, but it also can occur in younger patients.8 The pathophysiology of stasis dermatitis is caused by edema, which leads to extracellular fluid, plasma proteins, macrophages, and erythrocytes passing into the interstitial space. Patients with stasis dermatitis present with scaly erythematous papules and plaques or edematous blisters on the lower extremities. Diagnosis usually can be made clinically; however, a skin biopsy also can be helpful. Hematoxylin and eosin shows a pauci-inflammatory subepidermal bulla with fibrin (Figure 3).8 The overlying epidermis is intact. The dermis has cannon ball angiomatosis, red blood cell extravasation, and fibrosis typical of stasis dermatitis. Stasis dermatitis with bullae is cell poor and lacks the perivascular inflammatory infiltrate and neutrophilic fragments that often are present in LCV, making the 2 entities distinguishable.
Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) lies on a spectrum of severe cutaneous drug reactions involving the skin and mucous membranes. Cutaneous involvement typically begins on the trunk and face and later can involve the palms and soles.9 Similar drugs have been implicated in bullous LCV and SJS/TEN, including nonsteroidal anti-inflammatory drugs and antibiotics. Histologically, SJS/TEN has full-thickness epidermal necrolysis, vacuolar interface, and keratinocyte apoptosis (Figure 4).9 The clinical presentation of sloughing of skin with positive Nikolsky sign, oral involvement, and H&E and DIF findings can help differentiate this entity from bullous LCV.
- Einhorn J, Levis JT. Dermatologic diagnosis: leukocytoclastic vasculitis. Perm J. 2015;19:77-78.
- Davidson KA, Ringpfeil F, Lee JB. Ibuprofen-induced bullous leukocytoclastic vasculitis. Cutis. 2001;67:303-307.
- Lazic T, Fonder M, Robinson-Bostom L, et al. Orlistat-induced bullous leukocytoclastic vasculitis. Cutis. 2013;91:148-149.
- Mericliler M, Shnawa A, Al-Qaysi D, et al. Oxacillin-induced leukocytoclastic vasculitis. IDCases. 2019;17:E00539.
- Bernard P, Antonicelli F. Bullous pemphigoid: a review of its diagnosis, associations and treatment. Am J Clin Dermatol. 2017;18:513-528.
- High WA. Blistering disorders. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019:161-171.
- Visentainer L, Massuda JY, Cintra ML, et al. Vancomycin-induced linear IgA bullous dermatosis (LABD)--an atypical presentation. Clin Case Rep. 2019;7:1091-1093.
- Hyman DA, Cohen PR. Stasis dermatitis as a complication of recurrent levofloxacin-associated bilateral leg edema. Dermatol Online J. 2013;19:20399.
- Harr T, French LE. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;5:39.
- Einhorn J, Levis JT. Dermatologic diagnosis: leukocytoclastic vasculitis. Perm J. 2015;19:77-78.
- Davidson KA, Ringpfeil F, Lee JB. Ibuprofen-induced bullous leukocytoclastic vasculitis. Cutis. 2001;67:303-307.
- Lazic T, Fonder M, Robinson-Bostom L, et al. Orlistat-induced bullous leukocytoclastic vasculitis. Cutis. 2013;91:148-149.
- Mericliler M, Shnawa A, Al-Qaysi D, et al. Oxacillin-induced leukocytoclastic vasculitis. IDCases. 2019;17:E00539.
- Bernard P, Antonicelli F. Bullous pemphigoid: a review of its diagnosis, associations and treatment. Am J Clin Dermatol. 2017;18:513-528.
- High WA. Blistering disorders. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019:161-171.
- Visentainer L, Massuda JY, Cintra ML, et al. Vancomycin-induced linear IgA bullous dermatosis (LABD)--an atypical presentation. Clin Case Rep. 2019;7:1091-1093.
- Hyman DA, Cohen PR. Stasis dermatitis as a complication of recurrent levofloxacin-associated bilateral leg edema. Dermatol Online J. 2013;19:20399.
- Harr T, French LE. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;5:39.
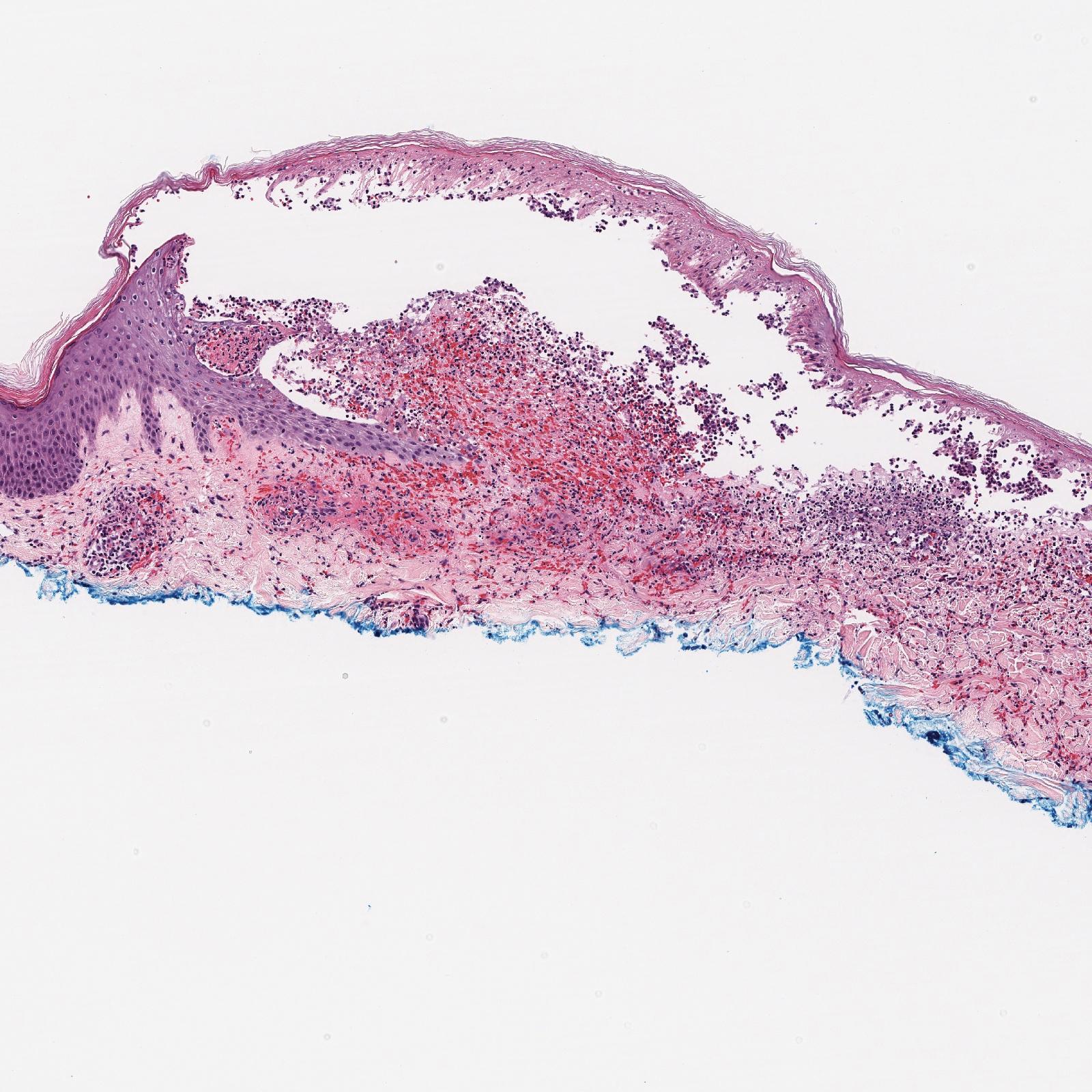
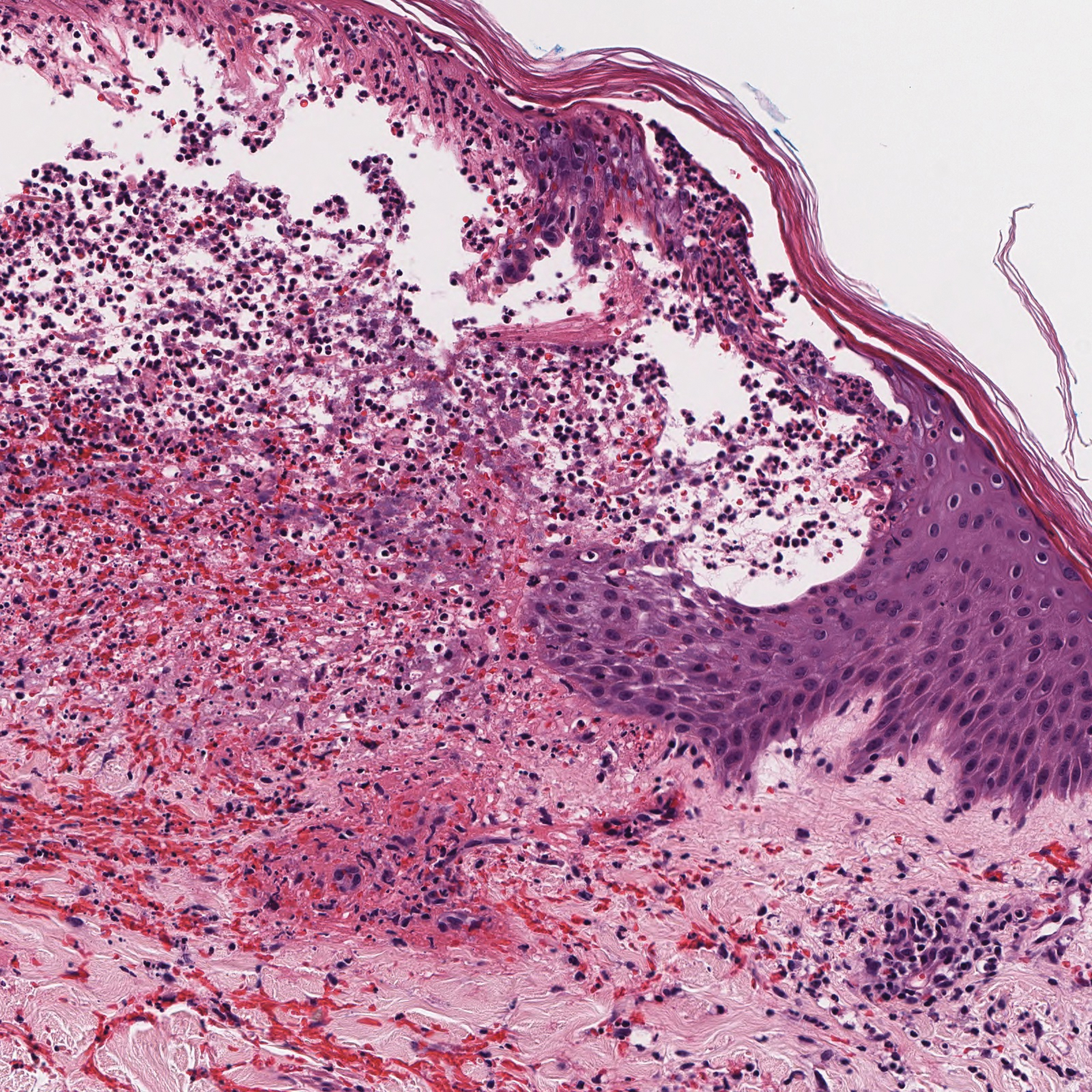
A 30-year-old woman with a medical history of uncontrolled type 2 diabetes mellitus and morbid obesity presented to the dermatology clinic with a painful blistering rash on the lower extremities with scattered red-purple papules of 1 week's duration. The rash began on the left dorsal foot. Physical examination showed nonblanching, 2- to 4-mm, violaceous papules with numerous vesiculopustular bullae on the lower extremities from the dorsal feet to the proximal knee. A shave biopsy with hematoxylin and eosin stain and a punch biopsy for direct immunofluorescence were performed.
Keratotic Papule on the Abdomen
The Diagnosis: Hypergranulotic Dyscornification
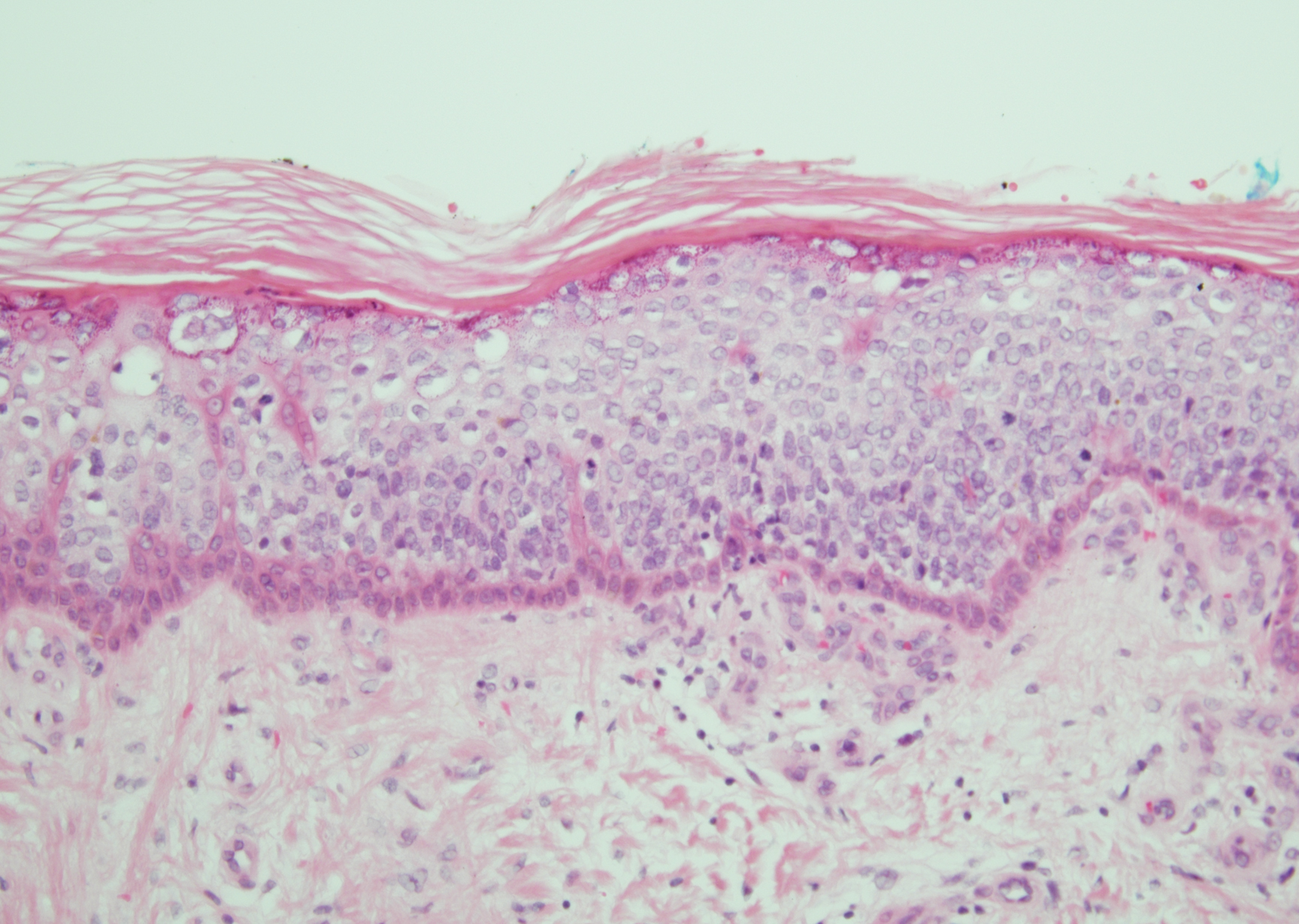
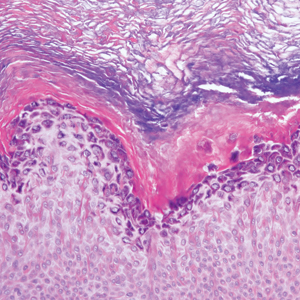
Hypergranulotic dyscornification (HD) is a rarely reported reaction pattern present in benign solitary keratoses with only few reports to date. It may be an underrecognized reaction pattern based on the paucity of reported cases as well as the histologic similarities to other entities. It has been hypothesized that this pattern reflects an underlying keratin mutation or disorder of keratinization.1
Clinically, HD most commonly presents as a waxy, tan-colored, solitary keratosis generally found on the lower limbs, trunk, or back in individuals aged 20 to 60 years.1,2 Histopathology shows marked hyperkeratosis, papillomatosis, and clumped basophilic keratohyalin granules within the corneocytes with digitated epidermal hyperplasia. There is abnormal cornification across the entire lesion with papillomatosis and marked hypergranulosis.3 There often are homogeneous orthokeratotic mounds of large, dull, eosinophilic-staining anucleate keratinocytes that are sharply demarcated from the thickened granular layer.1,2 Within the spinous, granular, and corneal layers, there is a pale, gray-staining, basophilic, cytoplasmic substance intercellularly.1
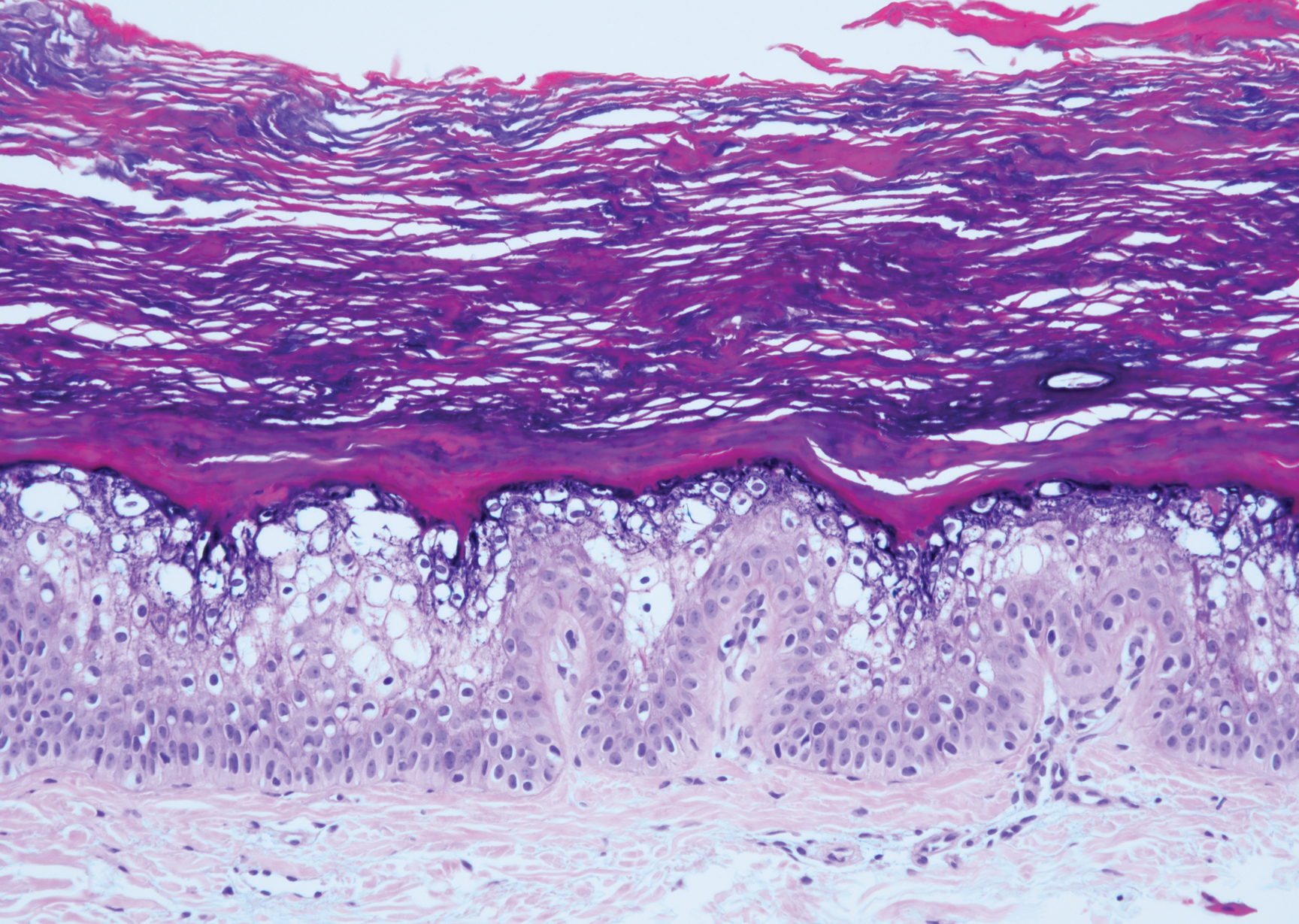
Histopathologically, HD may be mistaken for several other entities both benign and malignant.1 Epidermolytic hyperkeratosis can be a genetic disorder, an incidental finding in a variety of skin conditions, or an isolated lesion.4 The genetic syndrome, caused by mutation in keratins 1 or 10, clinically presents with hyperkeratosis, erosions, blisters, and thickening of the epidermis, often with a corrugated appearance. Epidermal nevi findings often are seen in conjunction with histologic changes of epidermolytic hyperkeratosis caused by mutation. Solitary lesions also can resemble seborrheic keratosis or verruca. In all examples of epidermolytic hyperkeratosis, the histopathologic findings are identical.4 The granular layer is thickened, and coarse keratohyalin granules aggregate in the suprabasal cells.5 There is acantholysis with perinuclear vacuolization in the spinous and granular layers with characteristic pale cytoplasmic areas devoid of keratin filaments (Figure 1). The basal layer may be hyperproliferative.5
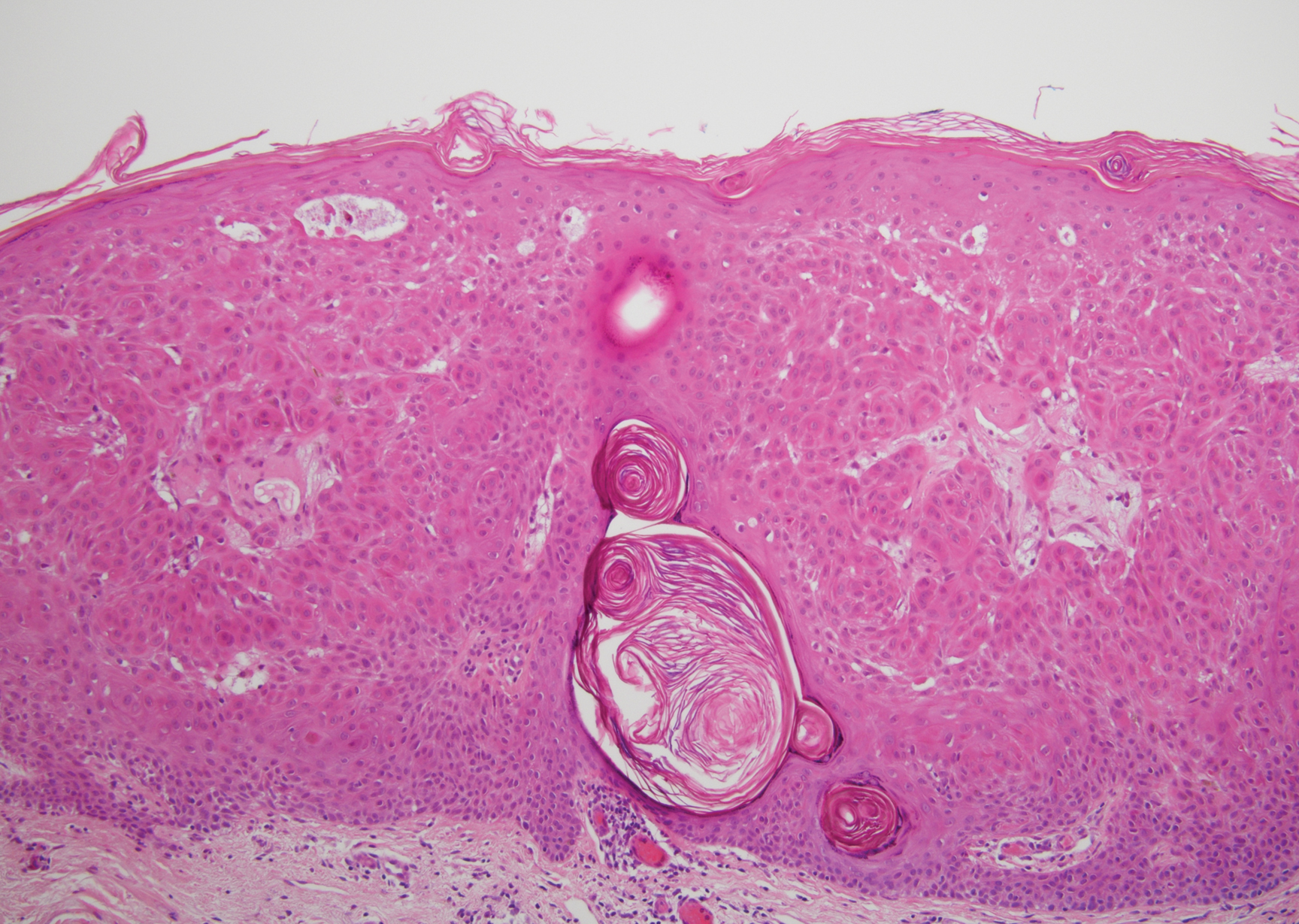
Irritated seborrheic keratosis presents as an exophytic, waxy, dark, sharply demarcated plaque with a stuck-on appearance.6 There is visible keratinization with comedolike openings, fissures and ridges, and scale; it also can contain milialike cysts. Histopathologically there is papillomatosis with prominent rete ridges, often including keratin pseudohorn cysts and squamous eddies. Enlarged capillaries can be seen in the dermal papillae. There is normal cytology with benign sheets of basaloid cells (Figure 2).7 Activating mutation in fibroblast growth factor receptor 3 leads to the growth and thickness of the epidermis that has been identified in these benign lesions.8
Verruca plana appears as a flesh-colored or reddish, warty, flat-topped papule that often forms clusters. Histopathologically it shows prominent hypergranulosis, thickened stratum spinosum, and vacuolized keratinocytes.9 The nuclei demonstrate a characteristic cytopathic effect of the virion, blurring the nuclear chromatin due to viral particle accumulation, known as koilocytes (Figure 3). The cause is the double-stranded DNA human papillomavirus types 2, 3, and 10.10
Bowen disease is a form of squamous cell carcinoma in situ characterized by an enlarging, well-demarcated, erythematous plaque with an irregular border and crusting or scaling. Histopathology reveals pleomorphic epidermal keratinization that becomes incorporated in the stratum corneum as parakeratotic nuclei. There is acanthosis, elongation of the rete ridges, and disorganized keratinocytes with atypia.11 The granular and spinous layers show an atypical honeycomb pattern with atypical cellular morphology (Figure 4).12 Bowen disease is a malignant lesion commonly found in older adults on sun-exposed skin that can evolve into invasive squamous cell carcinoma.
- Roy SF, Ko CJ, Moeckel GW, et al. Hypergranulotic dyscornification: 30 cases of a striking epithelial reaction pattern. J Cutan Pathol. 2019;46:742-747.
- Dohse L, Elston D, Lountzis N, et al. Benign hypergranulotic keratosis with dyscornification. J Am Acad Dermatol. 2010;62:AB52.
- Reichel M. Hypergranulotic dyscornification. Am J Dermatopathol. 1999;21:21-24.
- Kumar P, Kumar R, Kumar Mandal RK, et al. Systematized linear epidermolytic hyperkeratosis. Dermatol Online J. 2014;20:21248.
- Peter Rout D, Nair A, Gupta A, et al. Epidermolytic hyperkeratosis: clinical update. Clin Cosmet Investig Dermatol. 2019;12:333-344.
- Ingraffea A. Benign skin neoplasms. Facial Plast Surg Clin North Am. 2013;21:21-32.
- Braun R. Dermoscopy of pigmented seborrheic keratosis. Arch Dermatol. 2002;138:1556.
- Duperret EK, Oh SJ, McNeal A, et al. Activating FGFR3 mutations cause mild hyperplasia in human skin, but are insufficient to drive benign or malignant skin tumors. Cell Cycle. 2014;13:1551-1559.
- Liu H, Chen S, Zhang F, et al. Seborrheic keratosis or verruca plana? a pilot study with confocal laser scanning microscopy. Skin Res Technol. 2010;16:408-412.
- Prieto-Granada CN, Lobo AZC, Mihm MC. Skin infections. In: Kradin RL, ed. Diagnostic Pathology of Infectious Disease. Philadelphia, PA: Saunders Elsevier; 2010:519-616.
- DeCoste R, Moss P, Boutilier R, et al. Bowen disease with invasive mucin-secreting sweat gland differentiation: report of a case and review of the literature. J Cutan Pathol. 2019;46:425-430.
- Ulrich M, Kanitakis J, González S, et al. Evaluation of Bowen disease by in vivo reflectance confocal microscopy. Br J Dermatol. 2011;166:451-453.
The Diagnosis: Hypergranulotic Dyscornification
Hypergranulotic dyscornification (HD) is a rarely reported reaction pattern present in benign solitary keratoses with only few reports to date. It may be an underrecognized reaction pattern based on the paucity of reported cases as well as the histologic similarities to other entities. It has been hypothesized that this pattern reflects an underlying keratin mutation or disorder of keratinization.1
Clinically, HD most commonly presents as a waxy, tan-colored, solitary keratosis generally found on the lower limbs, trunk, or back in individuals aged 20 to 60 years.1,2 Histopathology shows marked hyperkeratosis, papillomatosis, and clumped basophilic keratohyalin granules within the corneocytes with digitated epidermal hyperplasia. There is abnormal cornification across the entire lesion with papillomatosis and marked hypergranulosis.3 There often are homogeneous orthokeratotic mounds of large, dull, eosinophilic-staining anucleate keratinocytes that are sharply demarcated from the thickened granular layer.1,2 Within the spinous, granular, and corneal layers, there is a pale, gray-staining, basophilic, cytoplasmic substance intercellularly.1
Histopathologically, HD may be mistaken for several other entities both benign and malignant.1 Epidermolytic hyperkeratosis can be a genetic disorder, an incidental finding in a variety of skin conditions, or an isolated lesion.4 The genetic syndrome, caused by mutation in keratins 1 or 10, clinically presents with hyperkeratosis, erosions, blisters, and thickening of the epidermis, often with a corrugated appearance. Epidermal nevi findings often are seen in conjunction with histologic changes of epidermolytic hyperkeratosis caused by mutation. Solitary lesions also can resemble seborrheic keratosis or verruca. In all examples of epidermolytic hyperkeratosis, the histopathologic findings are identical.4 The granular layer is thickened, and coarse keratohyalin granules aggregate in the suprabasal cells.5 There is acantholysis with perinuclear vacuolization in the spinous and granular layers with characteristic pale cytoplasmic areas devoid of keratin filaments (Figure 1). The basal layer may be hyperproliferative.5
Irritated seborrheic keratosis presents as an exophytic, waxy, dark, sharply demarcated plaque with a stuck-on appearance.6 There is visible keratinization with comedolike openings, fissures and ridges, and scale; it also can contain milialike cysts. Histopathologically there is papillomatosis with prominent rete ridges, often including keratin pseudohorn cysts and squamous eddies. Enlarged capillaries can be seen in the dermal papillae. There is normal cytology with benign sheets of basaloid cells (Figure 2).7 Activating mutation in fibroblast growth factor receptor 3 leads to the growth and thickness of the epidermis that has been identified in these benign lesions.8
Verruca plana appears as a flesh-colored or reddish, warty, flat-topped papule that often forms clusters. Histopathologically it shows prominent hypergranulosis, thickened stratum spinosum, and vacuolized keratinocytes.9 The nuclei demonstrate a characteristic cytopathic effect of the virion, blurring the nuclear chromatin due to viral particle accumulation, known as koilocytes (Figure 3). The cause is the double-stranded DNA human papillomavirus types 2, 3, and 10.10
Bowen disease is a form of squamous cell carcinoma in situ characterized by an enlarging, well-demarcated, erythematous plaque with an irregular border and crusting or scaling. Histopathology reveals pleomorphic epidermal keratinization that becomes incorporated in the stratum corneum as parakeratotic nuclei. There is acanthosis, elongation of the rete ridges, and disorganized keratinocytes with atypia.11 The granular and spinous layers show an atypical honeycomb pattern with atypical cellular morphology (Figure 4).12 Bowen disease is a malignant lesion commonly found in older adults on sun-exposed skin that can evolve into invasive squamous cell carcinoma.
The Diagnosis: Hypergranulotic Dyscornification
Hypergranulotic dyscornification (HD) is a rarely reported reaction pattern present in benign solitary keratoses with only few reports to date. It may be an underrecognized reaction pattern based on the paucity of reported cases as well as the histologic similarities to other entities. It has been hypothesized that this pattern reflects an underlying keratin mutation or disorder of keratinization.1
Clinically, HD most commonly presents as a waxy, tan-colored, solitary keratosis generally found on the lower limbs, trunk, or back in individuals aged 20 to 60 years.1,2 Histopathology shows marked hyperkeratosis, papillomatosis, and clumped basophilic keratohyalin granules within the corneocytes with digitated epidermal hyperplasia. There is abnormal cornification across the entire lesion with papillomatosis and marked hypergranulosis.3 There often are homogeneous orthokeratotic mounds of large, dull, eosinophilic-staining anucleate keratinocytes that are sharply demarcated from the thickened granular layer.1,2 Within the spinous, granular, and corneal layers, there is a pale, gray-staining, basophilic, cytoplasmic substance intercellularly.1
Histopathologically, HD may be mistaken for several other entities both benign and malignant.1 Epidermolytic hyperkeratosis can be a genetic disorder, an incidental finding in a variety of skin conditions, or an isolated lesion.4 The genetic syndrome, caused by mutation in keratins 1 or 10, clinically presents with hyperkeratosis, erosions, blisters, and thickening of the epidermis, often with a corrugated appearance. Epidermal nevi findings often are seen in conjunction with histologic changes of epidermolytic hyperkeratosis caused by mutation. Solitary lesions also can resemble seborrheic keratosis or verruca. In all examples of epidermolytic hyperkeratosis, the histopathologic findings are identical.4 The granular layer is thickened, and coarse keratohyalin granules aggregate in the suprabasal cells.5 There is acantholysis with perinuclear vacuolization in the spinous and granular layers with characteristic pale cytoplasmic areas devoid of keratin filaments (Figure 1). The basal layer may be hyperproliferative.5
Irritated seborrheic keratosis presents as an exophytic, waxy, dark, sharply demarcated plaque with a stuck-on appearance.6 There is visible keratinization with comedolike openings, fissures and ridges, and scale; it also can contain milialike cysts. Histopathologically there is papillomatosis with prominent rete ridges, often including keratin pseudohorn cysts and squamous eddies. Enlarged capillaries can be seen in the dermal papillae. There is normal cytology with benign sheets of basaloid cells (Figure 2).7 Activating mutation in fibroblast growth factor receptor 3 leads to the growth and thickness of the epidermis that has been identified in these benign lesions.8
Verruca plana appears as a flesh-colored or reddish, warty, flat-topped papule that often forms clusters. Histopathologically it shows prominent hypergranulosis, thickened stratum spinosum, and vacuolized keratinocytes.9 The nuclei demonstrate a characteristic cytopathic effect of the virion, blurring the nuclear chromatin due to viral particle accumulation, known as koilocytes (Figure 3). The cause is the double-stranded DNA human papillomavirus types 2, 3, and 10.10
Bowen disease is a form of squamous cell carcinoma in situ characterized by an enlarging, well-demarcated, erythematous plaque with an irregular border and crusting or scaling. Histopathology reveals pleomorphic epidermal keratinization that becomes incorporated in the stratum corneum as parakeratotic nuclei. There is acanthosis, elongation of the rete ridges, and disorganized keratinocytes with atypia.11 The granular and spinous layers show an atypical honeycomb pattern with atypical cellular morphology (Figure 4).12 Bowen disease is a malignant lesion commonly found in older adults on sun-exposed skin that can evolve into invasive squamous cell carcinoma.
- Roy SF, Ko CJ, Moeckel GW, et al. Hypergranulotic dyscornification: 30 cases of a striking epithelial reaction pattern. J Cutan Pathol. 2019;46:742-747.
- Dohse L, Elston D, Lountzis N, et al. Benign hypergranulotic keratosis with dyscornification. J Am Acad Dermatol. 2010;62:AB52.
- Reichel M. Hypergranulotic dyscornification. Am J Dermatopathol. 1999;21:21-24.
- Kumar P, Kumar R, Kumar Mandal RK, et al. Systematized linear epidermolytic hyperkeratosis. Dermatol Online J. 2014;20:21248.
- Peter Rout D, Nair A, Gupta A, et al. Epidermolytic hyperkeratosis: clinical update. Clin Cosmet Investig Dermatol. 2019;12:333-344.
- Ingraffea A. Benign skin neoplasms. Facial Plast Surg Clin North Am. 2013;21:21-32.
- Braun R. Dermoscopy of pigmented seborrheic keratosis. Arch Dermatol. 2002;138:1556.
- Duperret EK, Oh SJ, McNeal A, et al. Activating FGFR3 mutations cause mild hyperplasia in human skin, but are insufficient to drive benign or malignant skin tumors. Cell Cycle. 2014;13:1551-1559.
- Liu H, Chen S, Zhang F, et al. Seborrheic keratosis or verruca plana? a pilot study with confocal laser scanning microscopy. Skin Res Technol. 2010;16:408-412.
- Prieto-Granada CN, Lobo AZC, Mihm MC. Skin infections. In: Kradin RL, ed. Diagnostic Pathology of Infectious Disease. Philadelphia, PA: Saunders Elsevier; 2010:519-616.
- DeCoste R, Moss P, Boutilier R, et al. Bowen disease with invasive mucin-secreting sweat gland differentiation: report of a case and review of the literature. J Cutan Pathol. 2019;46:425-430.
- Ulrich M, Kanitakis J, González S, et al. Evaluation of Bowen disease by in vivo reflectance confocal microscopy. Br J Dermatol. 2011;166:451-453.
- Roy SF, Ko CJ, Moeckel GW, et al. Hypergranulotic dyscornification: 30 cases of a striking epithelial reaction pattern. J Cutan Pathol. 2019;46:742-747.
- Dohse L, Elston D, Lountzis N, et al. Benign hypergranulotic keratosis with dyscornification. J Am Acad Dermatol. 2010;62:AB52.
- Reichel M. Hypergranulotic dyscornification. Am J Dermatopathol. 1999;21:21-24.
- Kumar P, Kumar R, Kumar Mandal RK, et al. Systematized linear epidermolytic hyperkeratosis. Dermatol Online J. 2014;20:21248.
- Peter Rout D, Nair A, Gupta A, et al. Epidermolytic hyperkeratosis: clinical update. Clin Cosmet Investig Dermatol. 2019;12:333-344.
- Ingraffea A. Benign skin neoplasms. Facial Plast Surg Clin North Am. 2013;21:21-32.
- Braun R. Dermoscopy of pigmented seborrheic keratosis. Arch Dermatol. 2002;138:1556.
- Duperret EK, Oh SJ, McNeal A, et al. Activating FGFR3 mutations cause mild hyperplasia in human skin, but are insufficient to drive benign or malignant skin tumors. Cell Cycle. 2014;13:1551-1559.
- Liu H, Chen S, Zhang F, et al. Seborrheic keratosis or verruca plana? a pilot study with confocal laser scanning microscopy. Skin Res Technol. 2010;16:408-412.
- Prieto-Granada CN, Lobo AZC, Mihm MC. Skin infections. In: Kradin RL, ed. Diagnostic Pathology of Infectious Disease. Philadelphia, PA: Saunders Elsevier; 2010:519-616.
- DeCoste R, Moss P, Boutilier R, et al. Bowen disease with invasive mucin-secreting sweat gland differentiation: report of a case and review of the literature. J Cutan Pathol. 2019;46:425-430.
- Ulrich M, Kanitakis J, González S, et al. Evaluation of Bowen disease by in vivo reflectance confocal microscopy. Br J Dermatol. 2011;166:451-453.
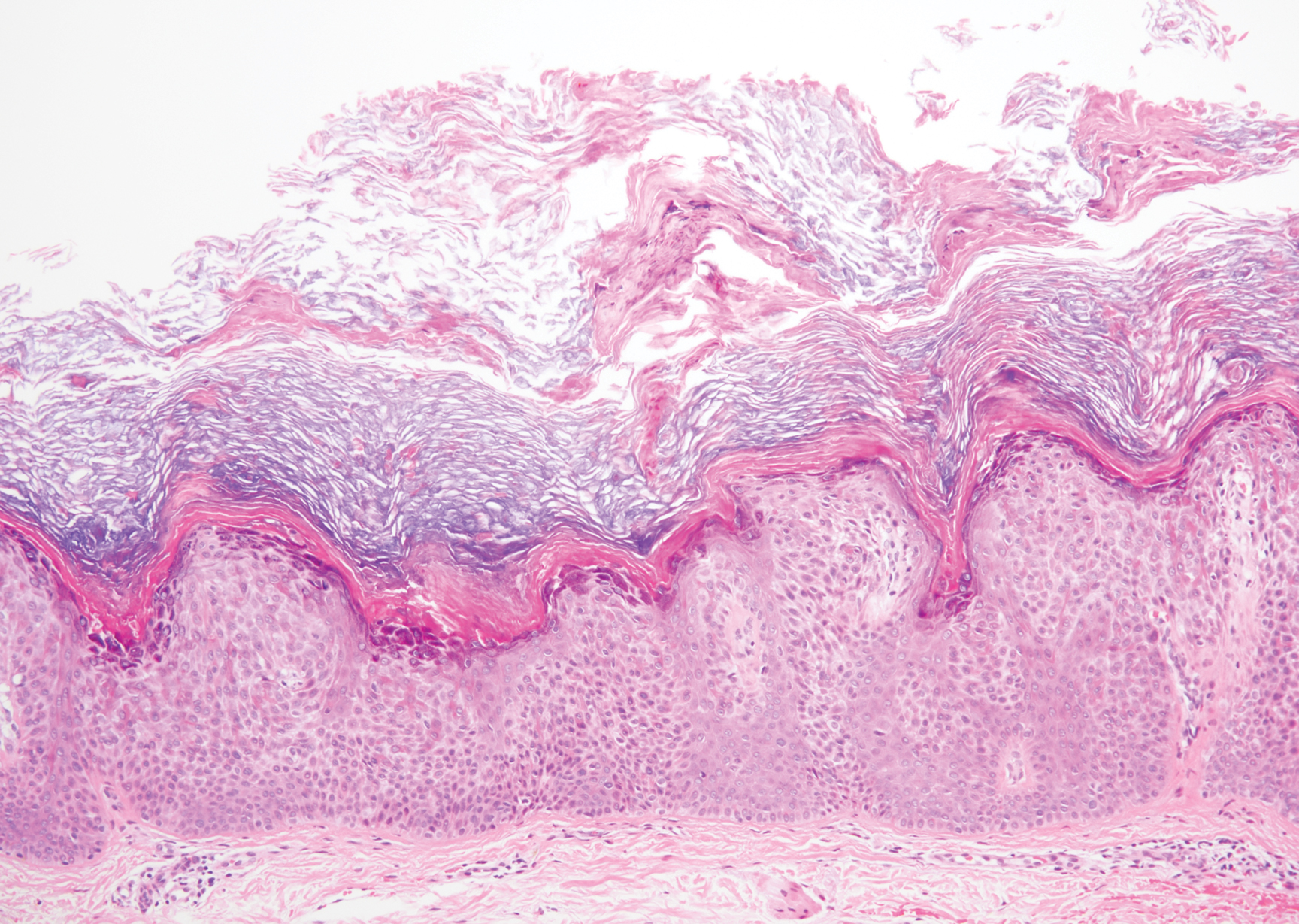
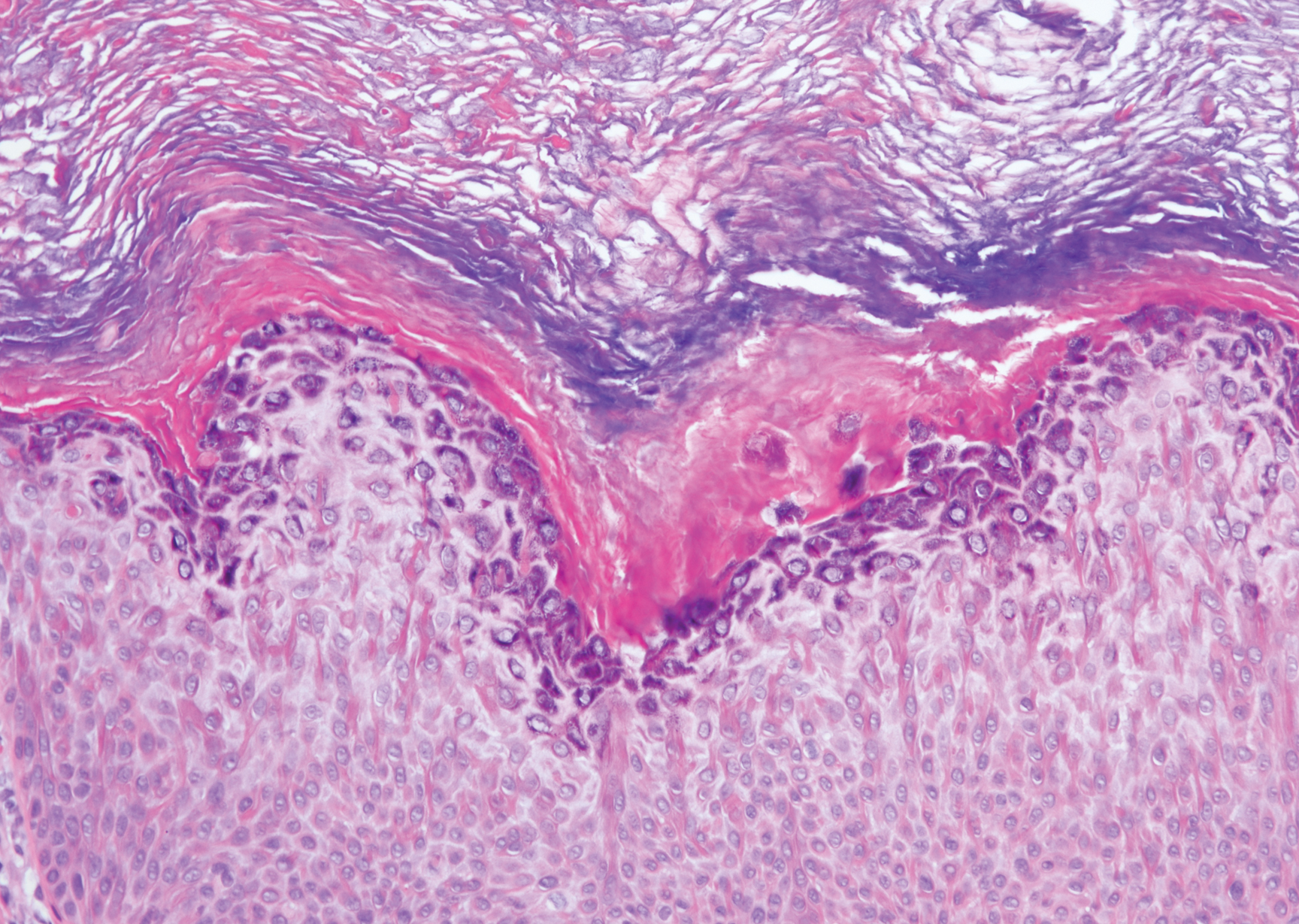
A 59-year-old woman with a history of basal cell carcinoma, uterine and ovarian cancer, and verrucae presented with an asymptomatic 3-mm lesion on the left side of the lower abdomen. Physical examination revealed a waxy, tan-colored, solitary keratosis. A shave biopsy was performed. Histopathology showed hyperkeratosis, focal parakeratosis, papillomatosis, and marked hypergranulosis with pale gray cytoplasm of the spinous-layer keratinocytes.
Edema Affecting the Penis and Scrotum
The Diagnosis: Cutaneous Crohn Disease
Crohn disease (CD) is an inflammatory bowel disease that can involve any region of the gastrointestinal (GI) tract from the mouth to the anus but most commonly presents in the terminal ileum, colon, or small bowel with transmural inflammation, fistula formation, and knife-cut fissures among the frequently described findings. Extraintestinal manifestations may be found in the liver, eyes, and joints, with cutaneous extraintestinal manifestations occurring in up to one-third of patients.1
Crohn disease can be associated with multiple cutaneous findings, including erythema nodosum, pyoderma gangrenosum, aphthous ulcers, pyodermatitis-pyostomatitis vegetans, necrotizing vasculitis, and metastatic Crohn disease (MCD).2 Typical histopathologic findings seen in MCD such as noncaseating granulomatous inflammation in the papillary and reticular dermis, possibly extending to the subcutaneous fat, are not specific to MCD. Associated genital edema is thought to be a consequence of granulomatous inflammation of lymphatics. In one study reviewing specimens from 10 cases of CD, a mean of 46% of all granulomas identified on the slides (264 granulomas in total) were located proximal to lymphatic vessels, suggesting a common pathway for development of intestinal disease and genital edema.3 The differential diagnosis for penile and scrotal swelling is broad, and the diagnosis may be missed if attention is not given to the clinical history of the patient in addition to histopathologic findings.2
Skin changes in CD also can be separated into perianal disease and true metastatic disease--the former recognized when anal lesions appear associated with segmental involvement of the GI tract and the latter as ulceration of the skin separated from the GI tract by normal tissue.1 The term sarcoidal reaction often is used to describe histopathologic findings in cutaneous CD, as it refers to the noncaseating granulomas found in approximately 60% of all cases.4 Ultimately, the location of noncaseating granulomas within the dermis of our patient's biopsy, taken in conjunction with the clinical history and the lack of defining features for other potential etiologies (eg, polarizable material, organisms on special stains), led to the diagnosis of cutaneous CD.
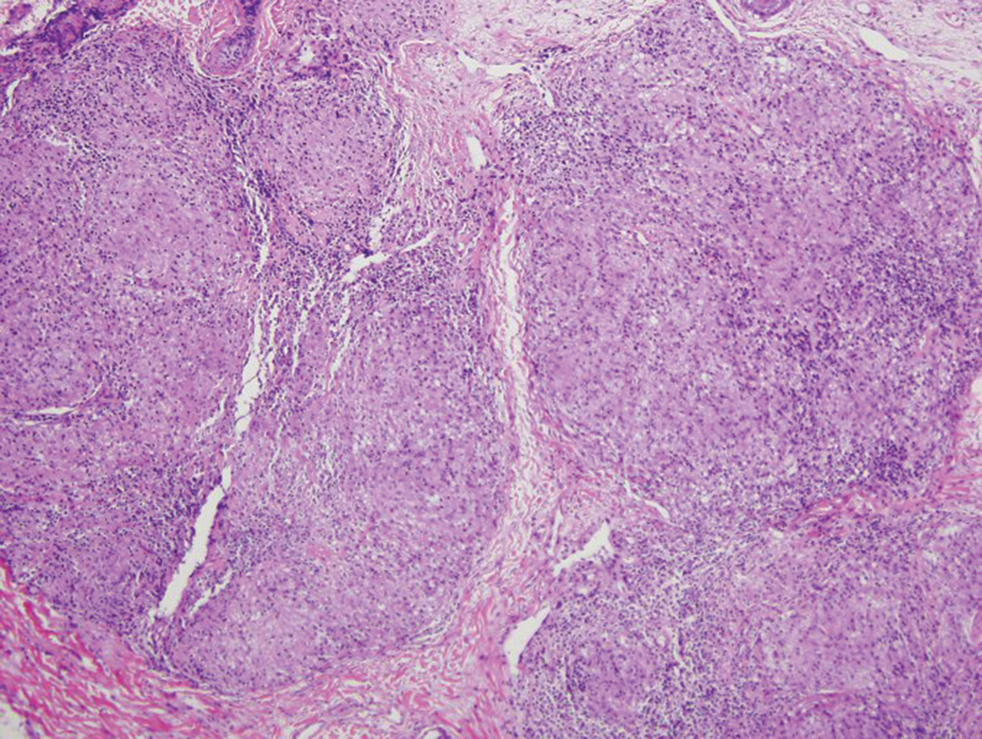
Cutaneous manifestations of sarcoidosis most commonly occur as papules, plaques, and subcutaneous nodules predominantly on the face, upper back, arms, and legs. Although the histologic features of sarcoidosis are characterized by lymphocyte-poor noncaseating granulomas (Figure 1), these findings also can be seen as a consequence of multiple granulomatous causes.5,6 In a review of 48 cutaneous specimens from patients with sarcoidosis, the granulomas were found most frequently in the deep dermis (34/48 [70.8%]), with superficial dermis (21/48) and subcutaneous fat granulomas (20/48) each present in less than 50% of biopsies.5 Although less typical, cutaneous sarcoidosis also has been noted in the literature to present in the perianal and gluteal region, demonstrating dermal noncaseating granulomas on biopsy.7 One distinction in particular to be noted between sarcoid and CD is that sarcoid lesions in the skin rarely ulcerate, while the lesions of cutaneous CD often are ulcerated.4,6
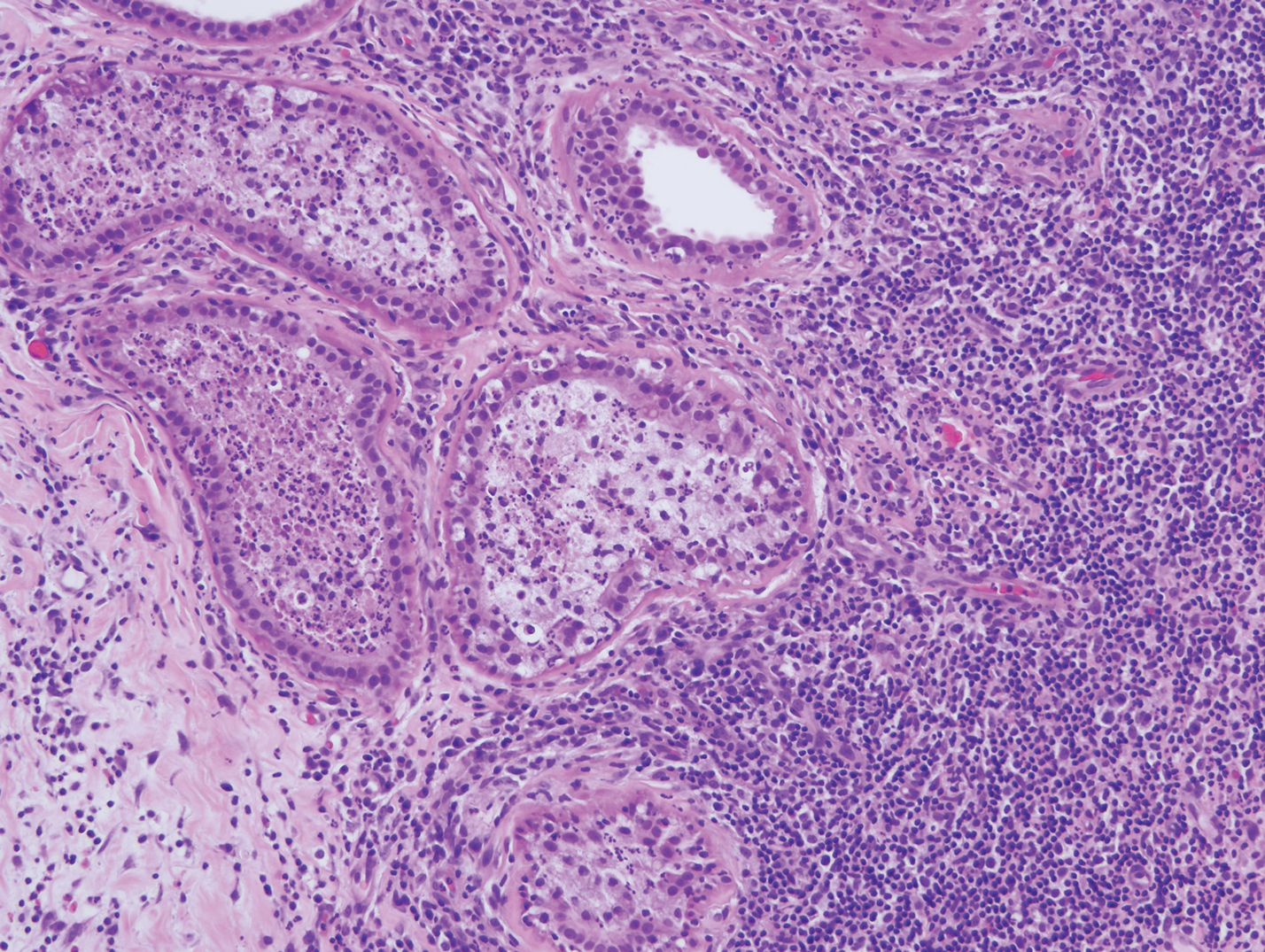
Lesions including abscesses in the groin may raise concern for hidradenitis suppurativa (HS), a disease of the apocrine gland-bearing skin. Typical lesions are tender subcutaneous erythematous nodules, cysts, and comedones that develop rapidly and may rupture to drain suppurative bloody discharge, subsequently healing with an atrophic scar.8 More persistent inflammation and rupture of nodules into the dermis may lead to formation of dermal tunnels with palpable cords and sinus tracts.8 Typical areas of disease involvement are in the axillae, inframammary folds, groin, or perigenital or perineal regions, with the diagnosis made on a combination of lesion morphology, location, and progression/recurrence frequency.9 Histologic examination of HS specimens can demonstrate a perifollicular lymphocytic infiltrate, with more advanced disease characterized by increased inflammatory cells, predominantly neutrophils, monocytes, and mast cells (Figure 2). The presence of granulomas in HS most often is of the foreign body type.9 Epithelioid granulomas noted in an area separate from inflammation in a patient with HS serve as a clue to be alert for systemic granulomatous disease.10
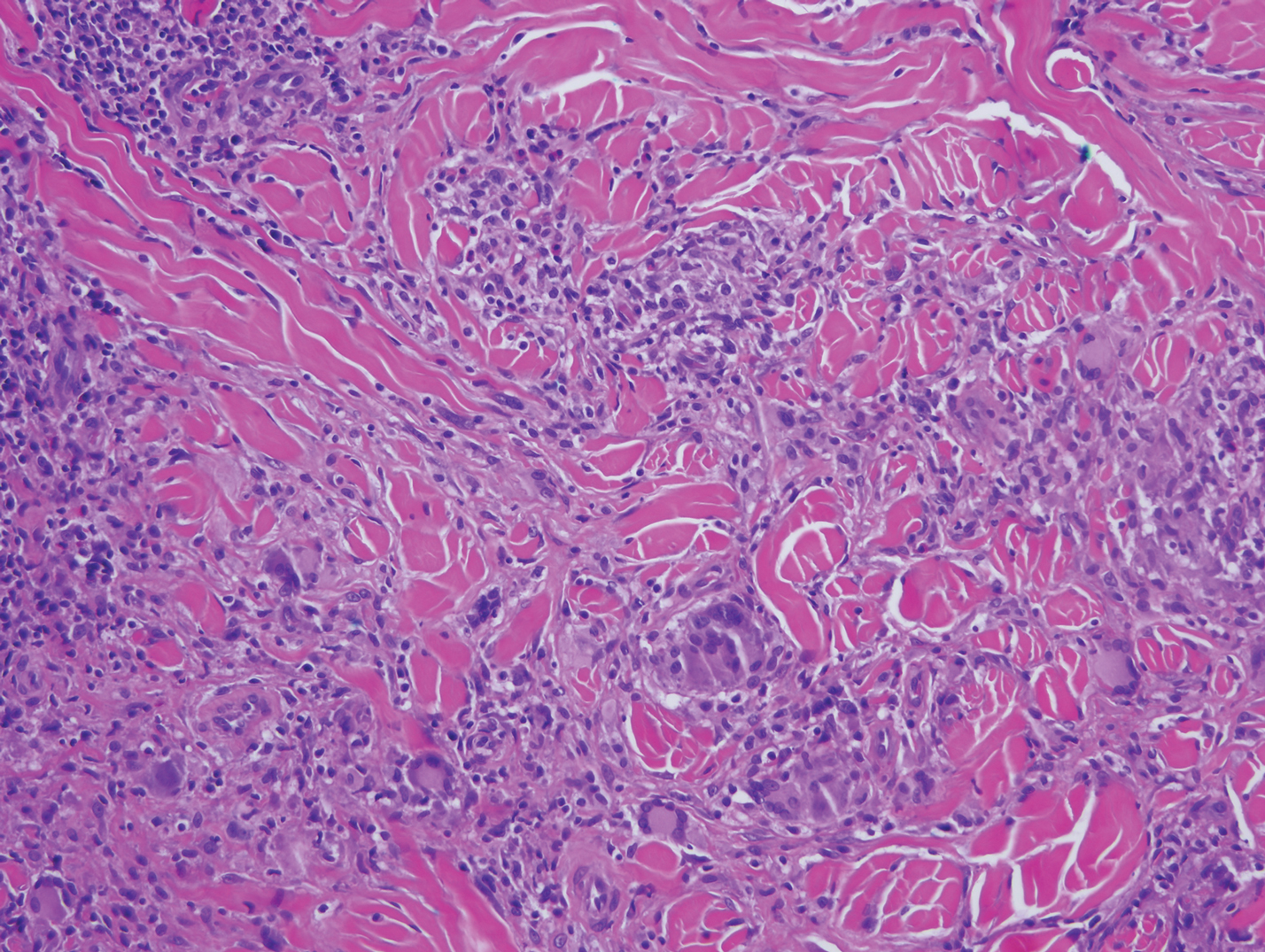
Mycosis fungoides is the most common primary cutaneous lymphoma to show a granulomatous infiltrate; the granuloma generally is sarcoidal, though other forms are described (Figure 3).11 Beyond these granulomatous foci, the key histopathologic feature of granulomatous mycosis fungoides (GMF) is diffuse dermal infiltration by atypical lymphoid cells. Epidermotropism and sparing of dermal nerves is the most critical finding in the diagnosis of GMF, especially in geographic regions where leprosy is endemic and high on the differential, as the conditions have histopathologic similarities.11,12 At the same time, lack of epidermotropism does not exclude the diagnosis of GMF.13 Clinically, GMF presentation is variable, but common findings include erythematous and hyperpigmented patches and plaques. Given the lack of clear clinical criteria, the diagnosis relies primarily on histopathologic features.11
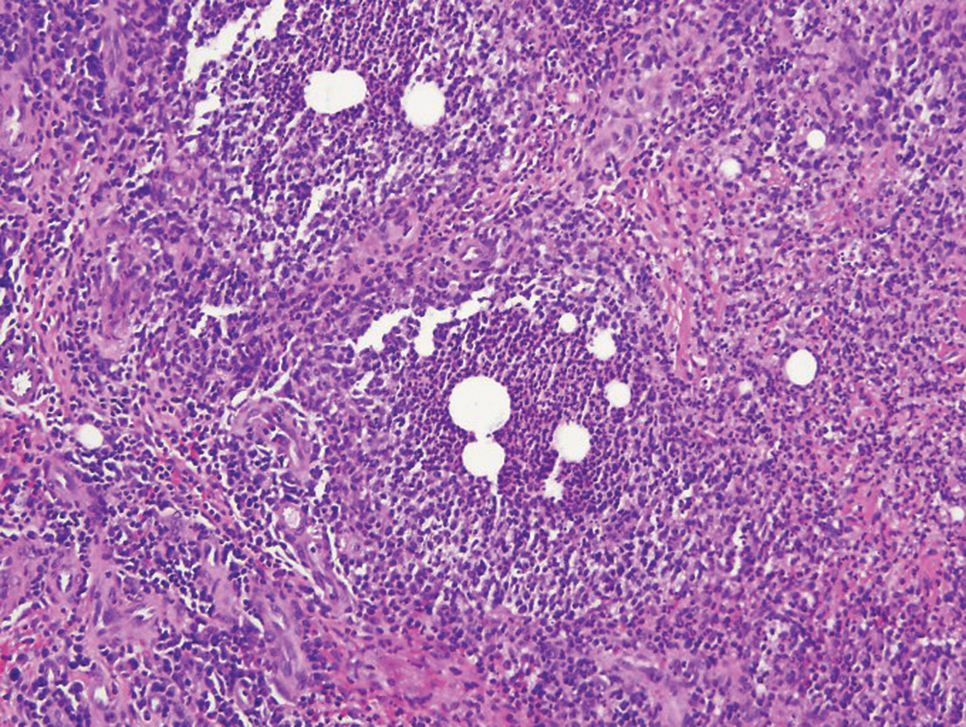
Mycobacterial skin and soft tissue infections may be attributed to both tuberculous and nontuberculous strains (atypical species).14 Clinical features range from small papules to large deformative plaques and ulcers.15 Histologic features also distinguish cutaneous tuberculosis (TB) from nontuberculous mycobacterial causes. Cutaneous TB shows caseous granulomas in the upper and mid dermis, while nontuberculous mycobacterial infections have more prominent neutrophil infiltration and interstitial granulomas (Figure 4).16
In cutaneous TB specifically, extrapulmonary manifestations may involve the skin in 1% to 1.5% of all TB cases, and although rare, ulcerative skin TB has been noted in one report as a nonhealing perianal ulcer that showed necrotizing granulomas on biopsy.17 Ultimately, diagnosis of cutaneous mycobacterial infection is confirmed with detection of acid-fast bacilli in the biopsy specimen.16
Diagnosis of cutaneous CD requires clinicopathologic correlation, as the clinical and histopathologic differential diagnoses of genital edema and noncaseating granulomas, respectively, are broad. Even though the clinical context was appropriate for cutaneous CD in this case, correct diagnosis required confirmatory histologic findings. Furthermore, taking multiple biopsies is prudent. In our patient, diagnostic findings only were present in the biopsy from the scrotum.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Barrick BJ, Tollefson MM, Schoch JJ, et al. Penile and scrotal swelling: an underrecognized presentation of Crohn's disease. Pediatr Dermatol. 2016;33:172-177.
- Mooney EE, Walker J, Hourihane DO. Relation of granulomas to lymphatic vessels in Crohn's disease. J Clin Pathol. 1995;48:335-338.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis: clinical spectrum and histological analysis of 40 cases [published online October 18, 2018]. Int J Dermatol. 2019;58:178-184.
- Yoo SS, Mimouni D, Nikolskaia OV, et al. Clinicopathologic features of ulcerative-atrophic sarcoidosis. Int J Dermatol. 2004;43:108-112.
- Cohen GF, Wolfe CM. Recalcitrant diffuse cutaneous sarcoidosis with perianal involvement responding to adalimumab. J Drugs Dermatol. 2017;16:1305-1306.
- Hoffman LK, Ghias MH, Lowes MA. Pathophysiology of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:47-54.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Attanoos RL, Appleton MA, Hughes LE, et al. Granulomatous hidradenitis suppurativa and cutaneous Crohn's disease. Histopathology. 1993;23:111-115.
- Gutte R, Kharkar V, Mahajan S, et al. Granulomatous mycosis fungoides with hypohydrosis mimicking lepromatous leprosy. Indian J Dermatol Venerol Leprol. 2010;76:686-690.
- Pousa CM, Nery NS, Mann D, et al. Granulomatous mycosis fungoides--a diagnostic challenge. An Bras Dermatol. 2015;90:554-556.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617.
- van Mechelen M, van der Hilst J, Gyssens IC, et al. Mycobacterial skin and soft tissue infections: TB or not TB? Neth J Med. 2018;76:269-274.
- van Zyl L, du Plessis J, Viljoen J. Cutaneous tuberculosis overview and current treatment regimens. Tuberculosis (Edinb). 2015;95:629-638.
- De Maio F, Trecarichi EM, Visconti E, et al. Understanding cutaneous tuberculosis: two clinical cases. JMM Case Rep. 2016;3:E005070.
- Wu S, Wang W, Chen H, et al. Perianal ulcerative skin tuberculosis: a case report. Medicine (Baltimore). 2018;97:E10836.
The Diagnosis: Cutaneous Crohn Disease
Crohn disease (CD) is an inflammatory bowel disease that can involve any region of the gastrointestinal (GI) tract from the mouth to the anus but most commonly presents in the terminal ileum, colon, or small bowel with transmural inflammation, fistula formation, and knife-cut fissures among the frequently described findings. Extraintestinal manifestations may be found in the liver, eyes, and joints, with cutaneous extraintestinal manifestations occurring in up to one-third of patients.1
Crohn disease can be associated with multiple cutaneous findings, including erythema nodosum, pyoderma gangrenosum, aphthous ulcers, pyodermatitis-pyostomatitis vegetans, necrotizing vasculitis, and metastatic Crohn disease (MCD).2 Typical histopathologic findings seen in MCD such as noncaseating granulomatous inflammation in the papillary and reticular dermis, possibly extending to the subcutaneous fat, are not specific to MCD. Associated genital edema is thought to be a consequence of granulomatous inflammation of lymphatics. In one study reviewing specimens from 10 cases of CD, a mean of 46% of all granulomas identified on the slides (264 granulomas in total) were located proximal to lymphatic vessels, suggesting a common pathway for development of intestinal disease and genital edema.3 The differential diagnosis for penile and scrotal swelling is broad, and the diagnosis may be missed if attention is not given to the clinical history of the patient in addition to histopathologic findings.2
Skin changes in CD also can be separated into perianal disease and true metastatic disease--the former recognized when anal lesions appear associated with segmental involvement of the GI tract and the latter as ulceration of the skin separated from the GI tract by normal tissue.1 The term sarcoidal reaction often is used to describe histopathologic findings in cutaneous CD, as it refers to the noncaseating granulomas found in approximately 60% of all cases.4 Ultimately, the location of noncaseating granulomas within the dermis of our patient's biopsy, taken in conjunction with the clinical history and the lack of defining features for other potential etiologies (eg, polarizable material, organisms on special stains), led to the diagnosis of cutaneous CD.
Cutaneous manifestations of sarcoidosis most commonly occur as papules, plaques, and subcutaneous nodules predominantly on the face, upper back, arms, and legs. Although the histologic features of sarcoidosis are characterized by lymphocyte-poor noncaseating granulomas (Figure 1), these findings also can be seen as a consequence of multiple granulomatous causes.5,6 In a review of 48 cutaneous specimens from patients with sarcoidosis, the granulomas were found most frequently in the deep dermis (34/48 [70.8%]), with superficial dermis (21/48) and subcutaneous fat granulomas (20/48) each present in less than 50% of biopsies.5 Although less typical, cutaneous sarcoidosis also has been noted in the literature to present in the perianal and gluteal region, demonstrating dermal noncaseating granulomas on biopsy.7 One distinction in particular to be noted between sarcoid and CD is that sarcoid lesions in the skin rarely ulcerate, while the lesions of cutaneous CD often are ulcerated.4,6
Lesions including abscesses in the groin may raise concern for hidradenitis suppurativa (HS), a disease of the apocrine gland-bearing skin. Typical lesions are tender subcutaneous erythematous nodules, cysts, and comedones that develop rapidly and may rupture to drain suppurative bloody discharge, subsequently healing with an atrophic scar.8 More persistent inflammation and rupture of nodules into the dermis may lead to formation of dermal tunnels with palpable cords and sinus tracts.8 Typical areas of disease involvement are in the axillae, inframammary folds, groin, or perigenital or perineal regions, with the diagnosis made on a combination of lesion morphology, location, and progression/recurrence frequency.9 Histologic examination of HS specimens can demonstrate a perifollicular lymphocytic infiltrate, with more advanced disease characterized by increased inflammatory cells, predominantly neutrophils, monocytes, and mast cells (Figure 2). The presence of granulomas in HS most often is of the foreign body type.9 Epithelioid granulomas noted in an area separate from inflammation in a patient with HS serve as a clue to be alert for systemic granulomatous disease.10
Mycosis fungoides is the most common primary cutaneous lymphoma to show a granulomatous infiltrate; the granuloma generally is sarcoidal, though other forms are described (Figure 3).11 Beyond these granulomatous foci, the key histopathologic feature of granulomatous mycosis fungoides (GMF) is diffuse dermal infiltration by atypical lymphoid cells. Epidermotropism and sparing of dermal nerves is the most critical finding in the diagnosis of GMF, especially in geographic regions where leprosy is endemic and high on the differential, as the conditions have histopathologic similarities.11,12 At the same time, lack of epidermotropism does not exclude the diagnosis of GMF.13 Clinically, GMF presentation is variable, but common findings include erythematous and hyperpigmented patches and plaques. Given the lack of clear clinical criteria, the diagnosis relies primarily on histopathologic features.11
Mycobacterial skin and soft tissue infections may be attributed to both tuberculous and nontuberculous strains (atypical species).14 Clinical features range from small papules to large deformative plaques and ulcers.15 Histologic features also distinguish cutaneous tuberculosis (TB) from nontuberculous mycobacterial causes. Cutaneous TB shows caseous granulomas in the upper and mid dermis, while nontuberculous mycobacterial infections have more prominent neutrophil infiltration and interstitial granulomas (Figure 4).16
In cutaneous TB specifically, extrapulmonary manifestations may involve the skin in 1% to 1.5% of all TB cases, and although rare, ulcerative skin TB has been noted in one report as a nonhealing perianal ulcer that showed necrotizing granulomas on biopsy.17 Ultimately, diagnosis of cutaneous mycobacterial infection is confirmed with detection of acid-fast bacilli in the biopsy specimen.16
Diagnosis of cutaneous CD requires clinicopathologic correlation, as the clinical and histopathologic differential diagnoses of genital edema and noncaseating granulomas, respectively, are broad. Even though the clinical context was appropriate for cutaneous CD in this case, correct diagnosis required confirmatory histologic findings. Furthermore, taking multiple biopsies is prudent. In our patient, diagnostic findings only were present in the biopsy from the scrotum.
The Diagnosis: Cutaneous Crohn Disease
Crohn disease (CD) is an inflammatory bowel disease that can involve any region of the gastrointestinal (GI) tract from the mouth to the anus but most commonly presents in the terminal ileum, colon, or small bowel with transmural inflammation, fistula formation, and knife-cut fissures among the frequently described findings. Extraintestinal manifestations may be found in the liver, eyes, and joints, with cutaneous extraintestinal manifestations occurring in up to one-third of patients.1
Crohn disease can be associated with multiple cutaneous findings, including erythema nodosum, pyoderma gangrenosum, aphthous ulcers, pyodermatitis-pyostomatitis vegetans, necrotizing vasculitis, and metastatic Crohn disease (MCD).2 Typical histopathologic findings seen in MCD such as noncaseating granulomatous inflammation in the papillary and reticular dermis, possibly extending to the subcutaneous fat, are not specific to MCD. Associated genital edema is thought to be a consequence of granulomatous inflammation of lymphatics. In one study reviewing specimens from 10 cases of CD, a mean of 46% of all granulomas identified on the slides (264 granulomas in total) were located proximal to lymphatic vessels, suggesting a common pathway for development of intestinal disease and genital edema.3 The differential diagnosis for penile and scrotal swelling is broad, and the diagnosis may be missed if attention is not given to the clinical history of the patient in addition to histopathologic findings.2
Skin changes in CD also can be separated into perianal disease and true metastatic disease--the former recognized when anal lesions appear associated with segmental involvement of the GI tract and the latter as ulceration of the skin separated from the GI tract by normal tissue.1 The term sarcoidal reaction often is used to describe histopathologic findings in cutaneous CD, as it refers to the noncaseating granulomas found in approximately 60% of all cases.4 Ultimately, the location of noncaseating granulomas within the dermis of our patient's biopsy, taken in conjunction with the clinical history and the lack of defining features for other potential etiologies (eg, polarizable material, organisms on special stains), led to the diagnosis of cutaneous CD.
Cutaneous manifestations of sarcoidosis most commonly occur as papules, plaques, and subcutaneous nodules predominantly on the face, upper back, arms, and legs. Although the histologic features of sarcoidosis are characterized by lymphocyte-poor noncaseating granulomas (Figure 1), these findings also can be seen as a consequence of multiple granulomatous causes.5,6 In a review of 48 cutaneous specimens from patients with sarcoidosis, the granulomas were found most frequently in the deep dermis (34/48 [70.8%]), with superficial dermis (21/48) and subcutaneous fat granulomas (20/48) each present in less than 50% of biopsies.5 Although less typical, cutaneous sarcoidosis also has been noted in the literature to present in the perianal and gluteal region, demonstrating dermal noncaseating granulomas on biopsy.7 One distinction in particular to be noted between sarcoid and CD is that sarcoid lesions in the skin rarely ulcerate, while the lesions of cutaneous CD often are ulcerated.4,6
Lesions including abscesses in the groin may raise concern for hidradenitis suppurativa (HS), a disease of the apocrine gland-bearing skin. Typical lesions are tender subcutaneous erythematous nodules, cysts, and comedones that develop rapidly and may rupture to drain suppurative bloody discharge, subsequently healing with an atrophic scar.8 More persistent inflammation and rupture of nodules into the dermis may lead to formation of dermal tunnels with palpable cords and sinus tracts.8 Typical areas of disease involvement are in the axillae, inframammary folds, groin, or perigenital or perineal regions, with the diagnosis made on a combination of lesion morphology, location, and progression/recurrence frequency.9 Histologic examination of HS specimens can demonstrate a perifollicular lymphocytic infiltrate, with more advanced disease characterized by increased inflammatory cells, predominantly neutrophils, monocytes, and mast cells (Figure 2). The presence of granulomas in HS most often is of the foreign body type.9 Epithelioid granulomas noted in an area separate from inflammation in a patient with HS serve as a clue to be alert for systemic granulomatous disease.10
Mycosis fungoides is the most common primary cutaneous lymphoma to show a granulomatous infiltrate; the granuloma generally is sarcoidal, though other forms are described (Figure 3).11 Beyond these granulomatous foci, the key histopathologic feature of granulomatous mycosis fungoides (GMF) is diffuse dermal infiltration by atypical lymphoid cells. Epidermotropism and sparing of dermal nerves is the most critical finding in the diagnosis of GMF, especially in geographic regions where leprosy is endemic and high on the differential, as the conditions have histopathologic similarities.11,12 At the same time, lack of epidermotropism does not exclude the diagnosis of GMF.13 Clinically, GMF presentation is variable, but common findings include erythematous and hyperpigmented patches and plaques. Given the lack of clear clinical criteria, the diagnosis relies primarily on histopathologic features.11
Mycobacterial skin and soft tissue infections may be attributed to both tuberculous and nontuberculous strains (atypical species).14 Clinical features range from small papules to large deformative plaques and ulcers.15 Histologic features also distinguish cutaneous tuberculosis (TB) from nontuberculous mycobacterial causes. Cutaneous TB shows caseous granulomas in the upper and mid dermis, while nontuberculous mycobacterial infections have more prominent neutrophil infiltration and interstitial granulomas (Figure 4).16
In cutaneous TB specifically, extrapulmonary manifestations may involve the skin in 1% to 1.5% of all TB cases, and although rare, ulcerative skin TB has been noted in one report as a nonhealing perianal ulcer that showed necrotizing granulomas on biopsy.17 Ultimately, diagnosis of cutaneous mycobacterial infection is confirmed with detection of acid-fast bacilli in the biopsy specimen.16
Diagnosis of cutaneous CD requires clinicopathologic correlation, as the clinical and histopathologic differential diagnoses of genital edema and noncaseating granulomas, respectively, are broad. Even though the clinical context was appropriate for cutaneous CD in this case, correct diagnosis required confirmatory histologic findings. Furthermore, taking multiple biopsies is prudent. In our patient, diagnostic findings only were present in the biopsy from the scrotum.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Barrick BJ, Tollefson MM, Schoch JJ, et al. Penile and scrotal swelling: an underrecognized presentation of Crohn's disease. Pediatr Dermatol. 2016;33:172-177.
- Mooney EE, Walker J, Hourihane DO. Relation of granulomas to lymphatic vessels in Crohn's disease. J Clin Pathol. 1995;48:335-338.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis: clinical spectrum and histological analysis of 40 cases [published online October 18, 2018]. Int J Dermatol. 2019;58:178-184.
- Yoo SS, Mimouni D, Nikolskaia OV, et al. Clinicopathologic features of ulcerative-atrophic sarcoidosis. Int J Dermatol. 2004;43:108-112.
- Cohen GF, Wolfe CM. Recalcitrant diffuse cutaneous sarcoidosis with perianal involvement responding to adalimumab. J Drugs Dermatol. 2017;16:1305-1306.
- Hoffman LK, Ghias MH, Lowes MA. Pathophysiology of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:47-54.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Attanoos RL, Appleton MA, Hughes LE, et al. Granulomatous hidradenitis suppurativa and cutaneous Crohn's disease. Histopathology. 1993;23:111-115.
- Gutte R, Kharkar V, Mahajan S, et al. Granulomatous mycosis fungoides with hypohydrosis mimicking lepromatous leprosy. Indian J Dermatol Venerol Leprol. 2010;76:686-690.
- Pousa CM, Nery NS, Mann D, et al. Granulomatous mycosis fungoides--a diagnostic challenge. An Bras Dermatol. 2015;90:554-556.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617.
- van Mechelen M, van der Hilst J, Gyssens IC, et al. Mycobacterial skin and soft tissue infections: TB or not TB? Neth J Med. 2018;76:269-274.
- van Zyl L, du Plessis J, Viljoen J. Cutaneous tuberculosis overview and current treatment regimens. Tuberculosis (Edinb). 2015;95:629-638.
- De Maio F, Trecarichi EM, Visconti E, et al. Understanding cutaneous tuberculosis: two clinical cases. JMM Case Rep. 2016;3:E005070.
- Wu S, Wang W, Chen H, et al. Perianal ulcerative skin tuberculosis: a case report. Medicine (Baltimore). 2018;97:E10836.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Barrick BJ, Tollefson MM, Schoch JJ, et al. Penile and scrotal swelling: an underrecognized presentation of Crohn's disease. Pediatr Dermatol. 2016;33:172-177.
- Mooney EE, Walker J, Hourihane DO. Relation of granulomas to lymphatic vessels in Crohn's disease. J Clin Pathol. 1995;48:335-338.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis: clinical spectrum and histological analysis of 40 cases [published online October 18, 2018]. Int J Dermatol. 2019;58:178-184.
- Yoo SS, Mimouni D, Nikolskaia OV, et al. Clinicopathologic features of ulcerative-atrophic sarcoidosis. Int J Dermatol. 2004;43:108-112.
- Cohen GF, Wolfe CM. Recalcitrant diffuse cutaneous sarcoidosis with perianal involvement responding to adalimumab. J Drugs Dermatol. 2017;16:1305-1306.
- Hoffman LK, Ghias MH, Lowes MA. Pathophysiology of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:47-54.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Attanoos RL, Appleton MA, Hughes LE, et al. Granulomatous hidradenitis suppurativa and cutaneous Crohn's disease. Histopathology. 1993;23:111-115.
- Gutte R, Kharkar V, Mahajan S, et al. Granulomatous mycosis fungoides with hypohydrosis mimicking lepromatous leprosy. Indian J Dermatol Venerol Leprol. 2010;76:686-690.
- Pousa CM, Nery NS, Mann D, et al. Granulomatous mycosis fungoides--a diagnostic challenge. An Bras Dermatol. 2015;90:554-556.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617.
- van Mechelen M, van der Hilst J, Gyssens IC, et al. Mycobacterial skin and soft tissue infections: TB or not TB? Neth J Med. 2018;76:269-274.
- van Zyl L, du Plessis J, Viljoen J. Cutaneous tuberculosis overview and current treatment regimens. Tuberculosis (Edinb). 2015;95:629-638.
- De Maio F, Trecarichi EM, Visconti E, et al. Understanding cutaneous tuberculosis: two clinical cases. JMM Case Rep. 2016;3:E005070.
- Wu S, Wang W, Chen H, et al. Perianal ulcerative skin tuberculosis: a case report. Medicine (Baltimore). 2018;97:E10836.

A 44-year-old man presented for evaluation of self-described "skin ripping" on the penis with penile and scrotal edema of 1 year's duration. He had a history of bowel symptoms and anorectal fistula of 3 years' duration. Purulent penile drainage and inguinal lymphadenopathy were noted on physical examination. Excisional biopsies of the scrotum and penis were performed. Special stains for organisms were negative.
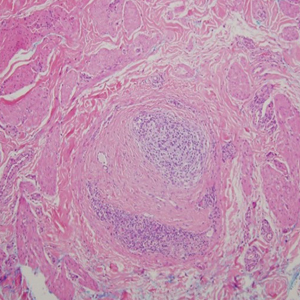
Spiky Papules on the Dorsal Feet
The Diagnosis: Hyperkeratosis Lenticularis Perstans (Flegel Disease)
Hyperkeratosis lenticularis perstans, also known as Flegel disease, is a rare dermatosis first described by Flegel1 in 1958. This benign disorder is characterized by multiple asymptomatic 1- to 5-mm keratotic papules in a symmetric distribution favoring the dorsal aspects of the feet and distal extremities in adults. An autosomal-dominant inheritance pattern has been postulated, though many cases sporadically occur.2 The characteristic spiky papules typically appear during mid to late adulthood and tend to persist. Treatment options are lacking, with reports of partial or no response to topical calcipotriol, topical 5-fluorouracil, cryotherapy, and topical and oral retinoids.3,4
The histopathology of hyperkeratosis lenticularis perstans is distinct, showing a central discrete area of orthohyperkeratosis with patchy parakeratosis flanked by a normal stratum corneum. The underlying epidermis typically shows effacement of the rete ridge pattern with subtle basal zone vacuolization and rare necrotic keratinocytes with an underlying lichenoid infiltrate within the papillary dermis comprised of lymphomononuclear cells.
In contrast, punctate porokeratosis clinically tends to involve the palms and soles, though the arms and legs also may be involved. This entity tends to occur during adolescence. A raised hyperkeratotic papule clinically is present. Histopathologically, the epidermis has a cup-shaped depression filled with hyperkeratosis and a column of parakeratosis (coronoid lamellae)(Figure 1).
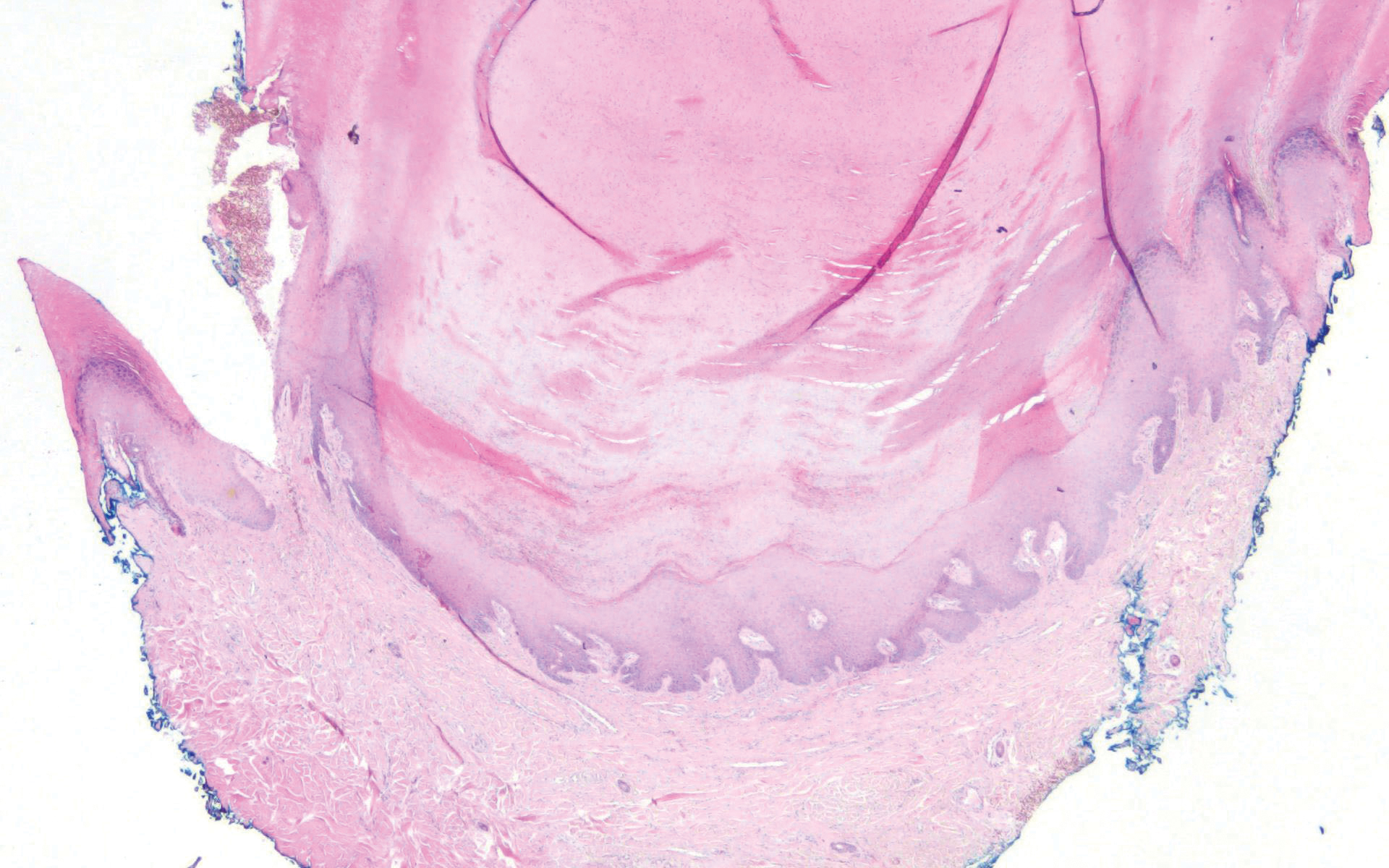
Acrokeratosis verruciformis of Hopf clinically appears on the dorsal aspects of the hands and feet as small warty papules in association with Darier disease. It typically presents during early childhood. Histopathology shows tiered hyperkeratosis, papillomatosis, and acanthosis (Figure 2).
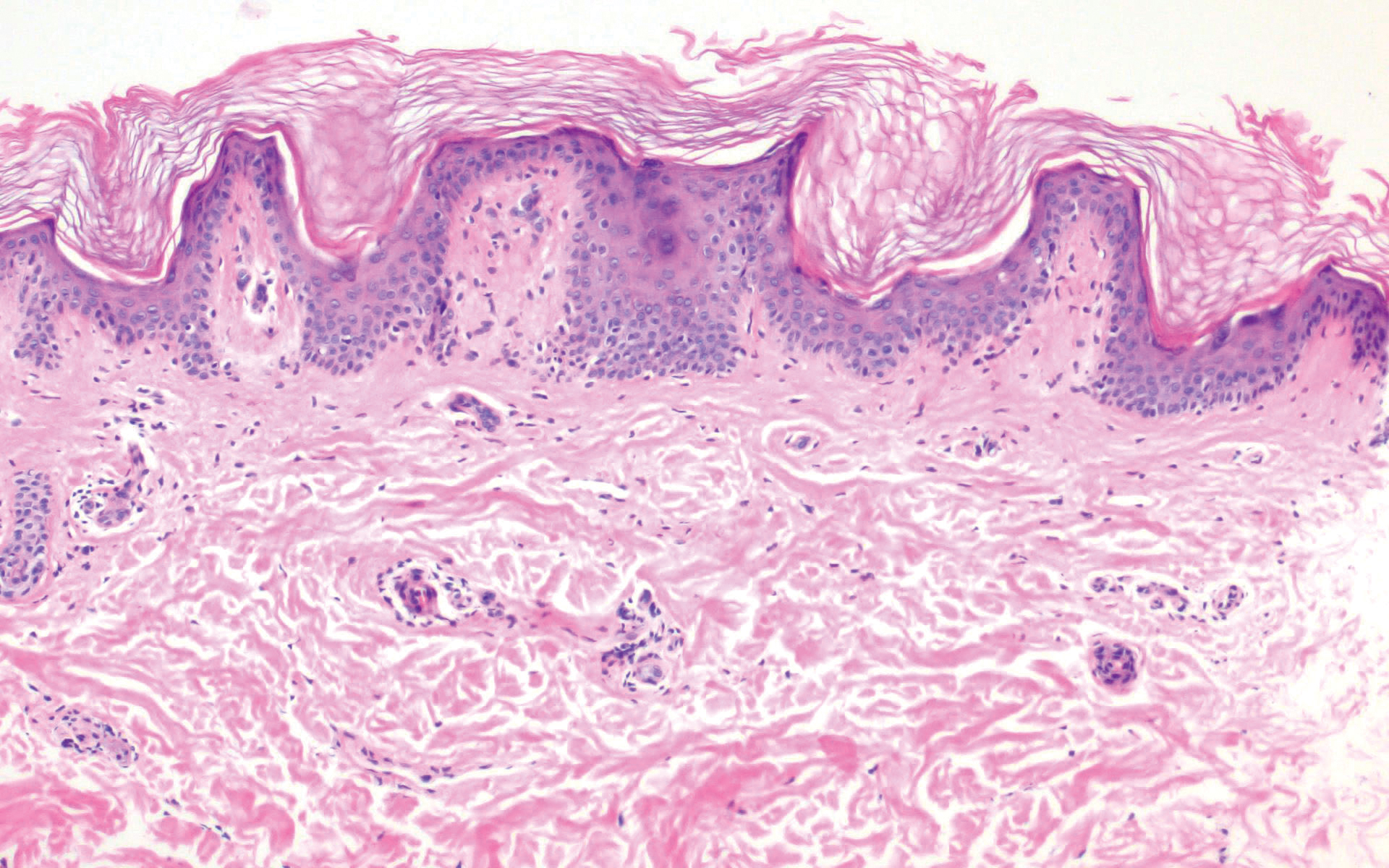
Perforating granuloma annulare presents on the dorsal aspects of the hands and fingers as scaly papules with either central umbilication or keratotic plugs. Histopathology shows transepidermal elimination of degenerated collagen (Figure 3).
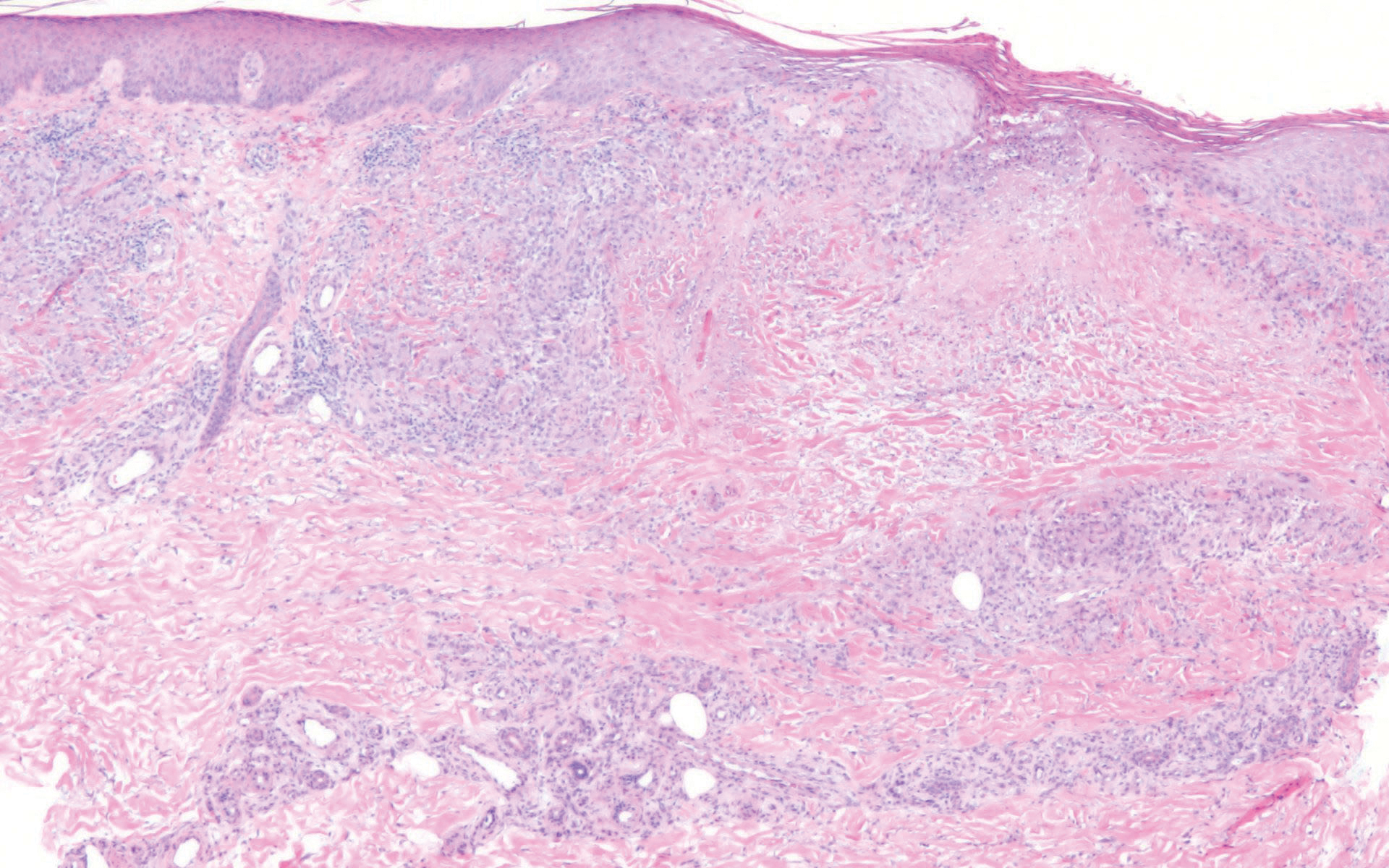
Stucco keratoses present on the dorsal aspects of the feet and ankles but are waxy smooth papules as opposed to hyperkeratotic spiky papules. Histologically, they are characterized by retention hyperkeratosis with lack of parakeratosis and regular acanthosis with a "string sign" indicating that the lesion extends to a uniform depth. (Figure 4).
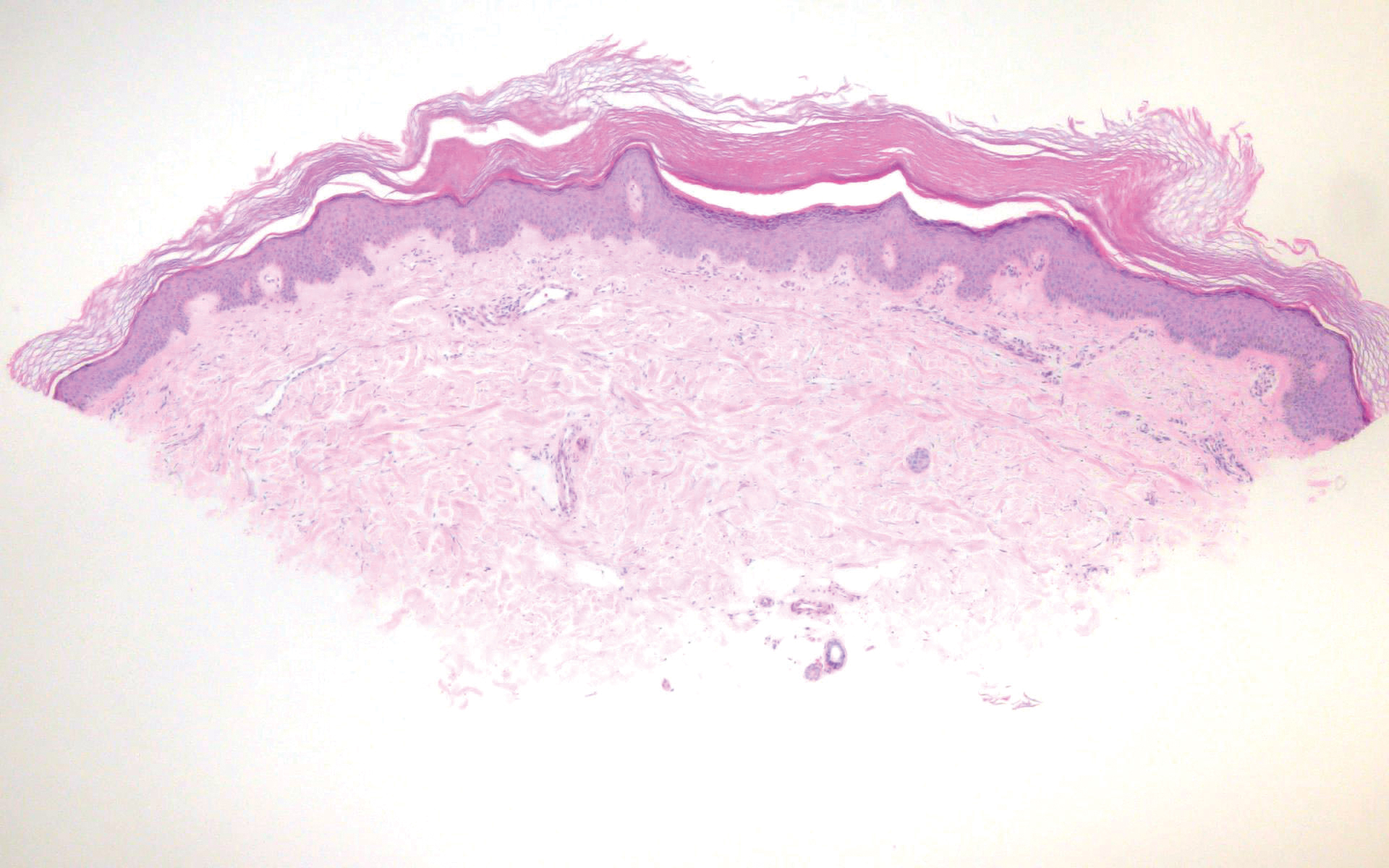
- Flegel H. Hyperkeratosis lenticularis perstans. Hautzarzt. 1958;9:363-364.
- Ando K, Hattori H, Yamauchi Y. Histopathological differences between early and old lesions of hyperkeratosis lenticularis perstans (Flegel's disease). Am J Dermatopathol. 2006;28:122-126.
- Langer K, Zonzits E, Konrad K. Hyperkeratosis lenticularis perstans (Flegel's disease). ultrastructural study of lesional and perilesional skin and therapeutic trial of topical tretinoin versus 5-fluorouracil. J Am Acad Dermatol. 1992;27:812-816.
- Blaheta HJ, Metzler G, Rassner G, et al. Hyperkeratosis lenticularis perstans (Flegel's disease)--lack of response to treatment with tacalcitol and calcipotriol. Dermatology. 2001;202:255-258.
The Diagnosis: Hyperkeratosis Lenticularis Perstans (Flegel Disease)
Hyperkeratosis lenticularis perstans, also known as Flegel disease, is a rare dermatosis first described by Flegel1 in 1958. This benign disorder is characterized by multiple asymptomatic 1- to 5-mm keratotic papules in a symmetric distribution favoring the dorsal aspects of the feet and distal extremities in adults. An autosomal-dominant inheritance pattern has been postulated, though many cases sporadically occur.2 The characteristic spiky papules typically appear during mid to late adulthood and tend to persist. Treatment options are lacking, with reports of partial or no response to topical calcipotriol, topical 5-fluorouracil, cryotherapy, and topical and oral retinoids.3,4
The histopathology of hyperkeratosis lenticularis perstans is distinct, showing a central discrete area of orthohyperkeratosis with patchy parakeratosis flanked by a normal stratum corneum. The underlying epidermis typically shows effacement of the rete ridge pattern with subtle basal zone vacuolization and rare necrotic keratinocytes with an underlying lichenoid infiltrate within the papillary dermis comprised of lymphomononuclear cells.
In contrast, punctate porokeratosis clinically tends to involve the palms and soles, though the arms and legs also may be involved. This entity tends to occur during adolescence. A raised hyperkeratotic papule clinically is present. Histopathologically, the epidermis has a cup-shaped depression filled with hyperkeratosis and a column of parakeratosis (coronoid lamellae)(Figure 1).
Acrokeratosis verruciformis of Hopf clinically appears on the dorsal aspects of the hands and feet as small warty papules in association with Darier disease. It typically presents during early childhood. Histopathology shows tiered hyperkeratosis, papillomatosis, and acanthosis (Figure 2).
Perforating granuloma annulare presents on the dorsal aspects of the hands and fingers as scaly papules with either central umbilication or keratotic plugs. Histopathology shows transepidermal elimination of degenerated collagen (Figure 3).
Stucco keratoses present on the dorsal aspects of the feet and ankles but are waxy smooth papules as opposed to hyperkeratotic spiky papules. Histologically, they are characterized by retention hyperkeratosis with lack of parakeratosis and regular acanthosis with a "string sign" indicating that the lesion extends to a uniform depth. (Figure 4).
The Diagnosis: Hyperkeratosis Lenticularis Perstans (Flegel Disease)
Hyperkeratosis lenticularis perstans, also known as Flegel disease, is a rare dermatosis first described by Flegel1 in 1958. This benign disorder is characterized by multiple asymptomatic 1- to 5-mm keratotic papules in a symmetric distribution favoring the dorsal aspects of the feet and distal extremities in adults. An autosomal-dominant inheritance pattern has been postulated, though many cases sporadically occur.2 The characteristic spiky papules typically appear during mid to late adulthood and tend to persist. Treatment options are lacking, with reports of partial or no response to topical calcipotriol, topical 5-fluorouracil, cryotherapy, and topical and oral retinoids.3,4
The histopathology of hyperkeratosis lenticularis perstans is distinct, showing a central discrete area of orthohyperkeratosis with patchy parakeratosis flanked by a normal stratum corneum. The underlying epidermis typically shows effacement of the rete ridge pattern with subtle basal zone vacuolization and rare necrotic keratinocytes with an underlying lichenoid infiltrate within the papillary dermis comprised of lymphomononuclear cells.
In contrast, punctate porokeratosis clinically tends to involve the palms and soles, though the arms and legs also may be involved. This entity tends to occur during adolescence. A raised hyperkeratotic papule clinically is present. Histopathologically, the epidermis has a cup-shaped depression filled with hyperkeratosis and a column of parakeratosis (coronoid lamellae)(Figure 1).
Acrokeratosis verruciformis of Hopf clinically appears on the dorsal aspects of the hands and feet as small warty papules in association with Darier disease. It typically presents during early childhood. Histopathology shows tiered hyperkeratosis, papillomatosis, and acanthosis (Figure 2).
Perforating granuloma annulare presents on the dorsal aspects of the hands and fingers as scaly papules with either central umbilication or keratotic plugs. Histopathology shows transepidermal elimination of degenerated collagen (Figure 3).
Stucco keratoses present on the dorsal aspects of the feet and ankles but are waxy smooth papules as opposed to hyperkeratotic spiky papules. Histologically, they are characterized by retention hyperkeratosis with lack of parakeratosis and regular acanthosis with a "string sign" indicating that the lesion extends to a uniform depth. (Figure 4).
- Flegel H. Hyperkeratosis lenticularis perstans. Hautzarzt. 1958;9:363-364.
- Ando K, Hattori H, Yamauchi Y. Histopathological differences between early and old lesions of hyperkeratosis lenticularis perstans (Flegel's disease). Am J Dermatopathol. 2006;28:122-126.
- Langer K, Zonzits E, Konrad K. Hyperkeratosis lenticularis perstans (Flegel's disease). ultrastructural study of lesional and perilesional skin and therapeutic trial of topical tretinoin versus 5-fluorouracil. J Am Acad Dermatol. 1992;27:812-816.
- Blaheta HJ, Metzler G, Rassner G, et al. Hyperkeratosis lenticularis perstans (Flegel's disease)--lack of response to treatment with tacalcitol and calcipotriol. Dermatology. 2001;202:255-258.
- Flegel H. Hyperkeratosis lenticularis perstans. Hautzarzt. 1958;9:363-364.
- Ando K, Hattori H, Yamauchi Y. Histopathological differences between early and old lesions of hyperkeratosis lenticularis perstans (Flegel's disease). Am J Dermatopathol. 2006;28:122-126.
- Langer K, Zonzits E, Konrad K. Hyperkeratosis lenticularis perstans (Flegel's disease). ultrastructural study of lesional and perilesional skin and therapeutic trial of topical tretinoin versus 5-fluorouracil. J Am Acad Dermatol. 1992;27:812-816.
- Blaheta HJ, Metzler G, Rassner G, et al. Hyperkeratosis lenticularis perstans (Flegel's disease)--lack of response to treatment with tacalcitol and calcipotriol. Dermatology. 2001;202:255-258.
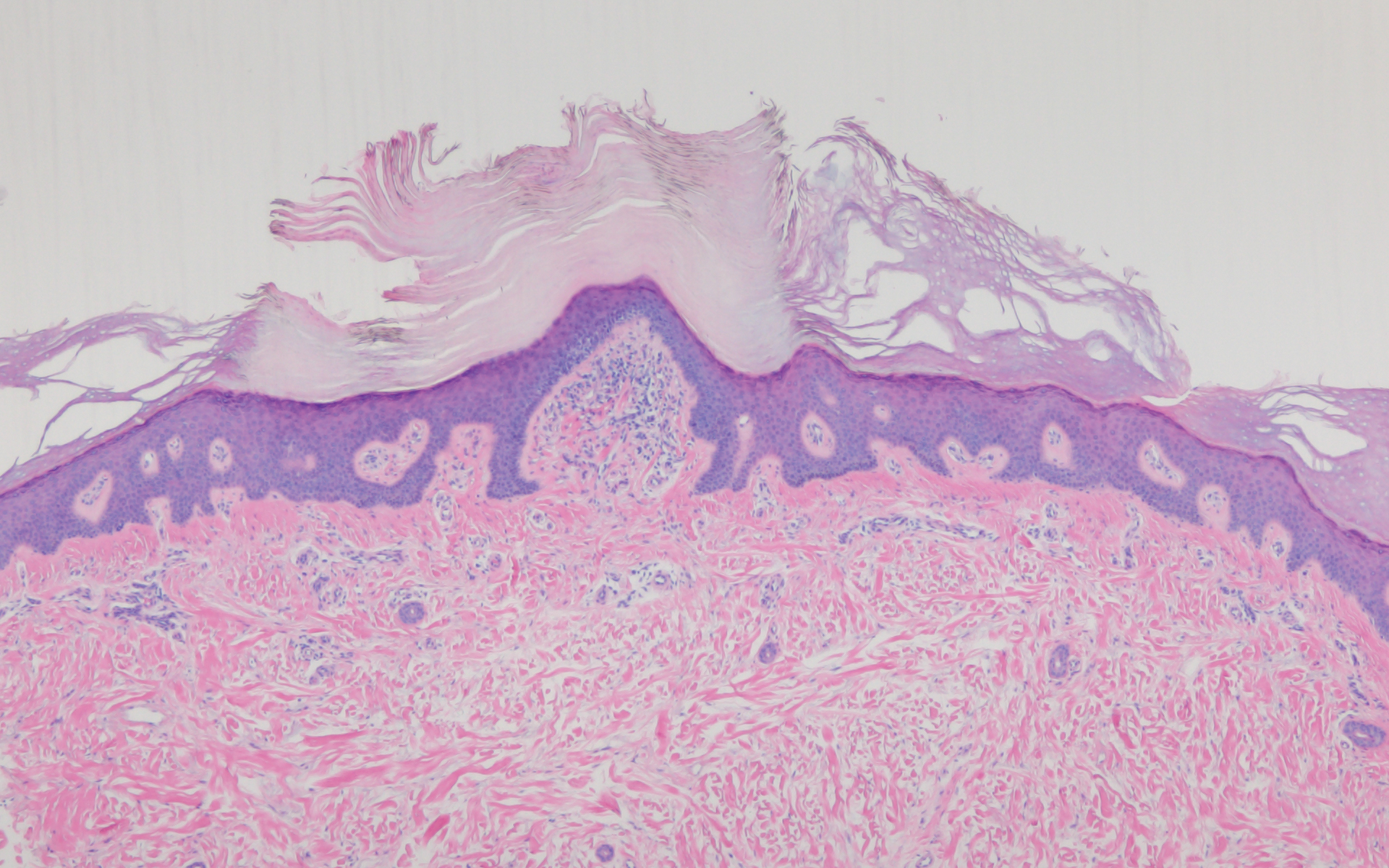
A 54-year-old man who was otherwise healthy presented with asymptomatic, discrete, rough, red-brown, hyperkeratotic papules on the dorsal aspects of the feet of several years' duration. The lesions spared the soles of the feet and hands. A diagnosis of eczema previously was made by his general practitioner, and he was using moisturizer. No prescription treatments were pursued, and no other rashes or lesions were noted on physical examination. A punch biopsy of a spiky papule was performed.
Distinct Violaceous Plaques in Conjunction With Blisters
The Diagnosis: Lichen Planus Pemphigoides
Lichen planus pemphigoides (LPP) is a rare autoimmune subepithelial blistering disorder with clinical, pathologic, and immunologic features of lichen planus (LP) and bullous pemphigoid (BP).1 It mainly arises in adults and usually is idiopathic but has been associated with certain infections,2 drugs such as angiotensin-converting enzyme inhibitors,3 phototherapy,4 and malignancy.5 Patients classically present with lichenoid lesions, tense vesiculobullae, and erosions.6 Vesiculobullae formation usually follows the development of lichenoid lesions, occurs on both lichenoid lesions and unaffected skin, and predominantly involves the lower extremities, as in our patient.1,6
The pathogenesis of LPP is not fully understood but likely represents a distinct entity rather than a subtype of BP or the simultaneous occurrence of LP and BP. Lichen planus pemphigoides generally has an earlier onset and better treatment response compared to BP.7 Further, autoantibodies in patients with LPP react to a novel epitope within the C-terminal portion of the BP-180 NC16A domain. Accordingly, it has been postulated that an inflammatory cutaneous process resulting from infection, phototherapy, or LP itself leads to damage of the epidermis and triggers a secondary blistering autoimmune dermatosis mediated by antibody formation against basement membrane (BM) antigens, such as BP-180.7
The diagnosis of LPP ultimately is confirmed with immunohistologic analysis. Biopsy of LPP shows findings consistent with both LP and BP (quiz image [top]). In the lichenoid portion, biopsy reveals orthohyperkeratosis, hypergranulosis, and acanthosis of the epidermis; a bandlike infiltrate consisting primarily of lymphocytes in the upper dermis; and apoptotic keratinocytes (colloid bodies) and vacuolar degeneration at the dermoepidermal junction (DEJ).1 Biopsy of bullae reveals eosinophilic spongiosis, a subepithelial blister plane with eosinophils, and a mixed superficial inflammatory cell infiltrate. Direct immunofluorescence from perilesional skin reveals linear deposition of IgG and/or C3 at the DEJ (quiz image [bottom]).1 Measurement of anti-BM antibodies against BP-180 and BP-230 can be useful in suspected cases, as 50% to 60% of patients have circulating antibodies against these antigens.6 Remission usually is achieved with topical and systemic corticosteroids and/or steroid-sparing agents, with rare recurrence following lesion resolution.1 More recently, successful treatment with biologics such as ustekinumab has been reported.8
The predominant differential diagnosis for LPP is bullous LP, a variant of LP in which vesiculobullous disease occurs exclusively on preexisting LP lesions, commonly on the legs due to severe vacuolar degeneration at the DEJ. On histopathology, the characteristic features of LP (eg, orthohyperkeratosis, hypergranulosis, acanthosis, bandlike lymphocytic infiltrate, colloid bodies) along with subepidermal clefting will be seen. However, in bullous LP (Figure 1) there is an absence of linear IgG and/or C3 deposition at the DEJ on direct immunofluorescence. Furthermore, patients lack circulating antibodies against BP-180 and BP-230.9
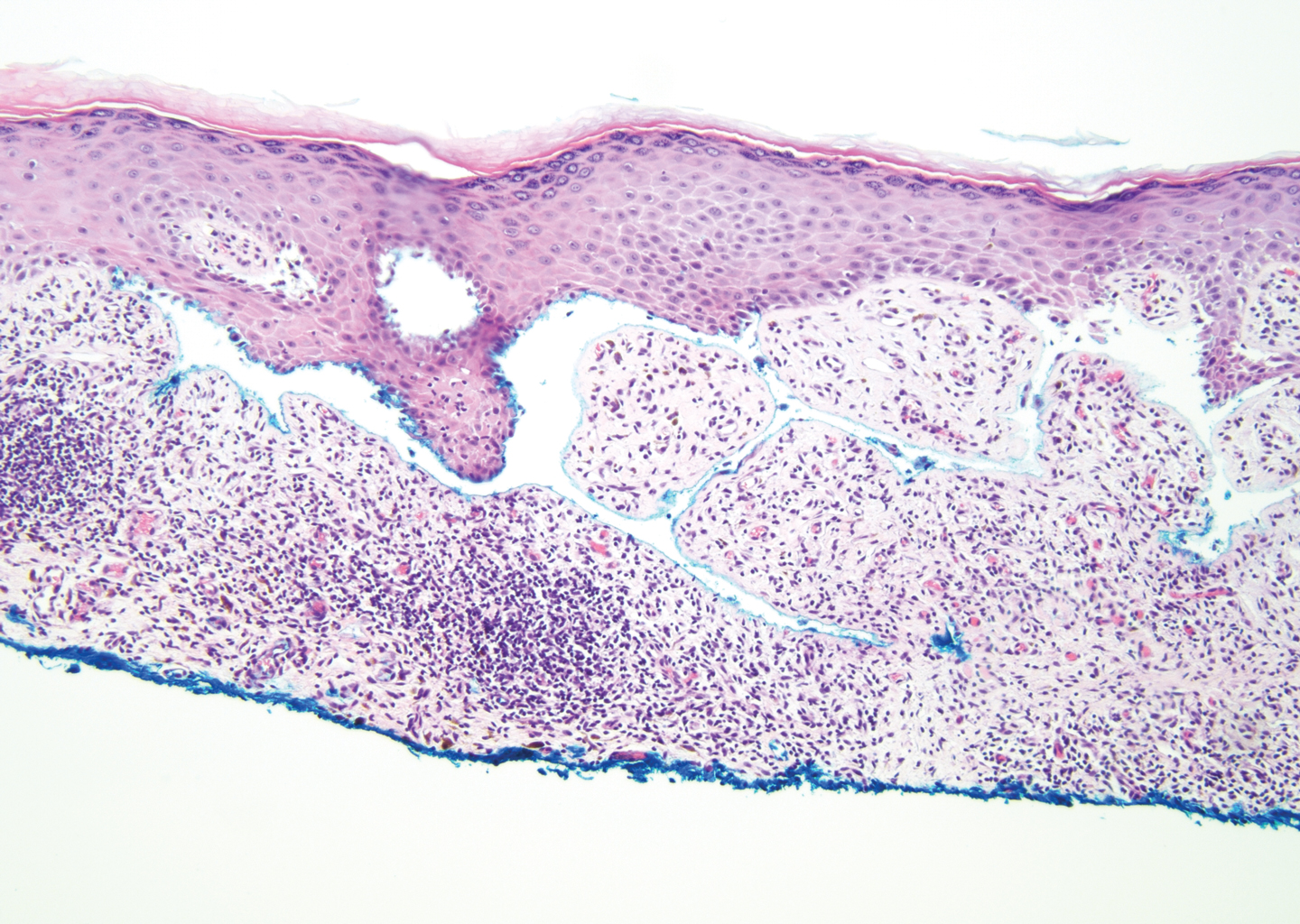
Lichen planus pemphigoides also can be confused with BP. Bullous pemphigoid is the most common autoimmune blistering disorder; typically arises in older adults; and is caused by autoantibody formation against hemidesmosomal proteins, particularly BP-180 and BP-230. Patients classically present with tense bullae and erosions on an erythematous, urticarial, or normal base. These lesions often are pruritic and concentrated on the trunk, axillary and inguinal folds, and extremity flexures. Histopathologic examination of a bulla edge reveals the classic findings seen in BP (eg, eosinophilic spongiosis, subepithelial blister plane with eosinophils)(Figure 2). Direct immunofluorescence of perilesional skin reveals linear IgG and/or C3 deposition along the DEJ. A large subset of patients also has circulating antibodies against BP-180 and BP-230. In contrast to LPP, however, patients with BP do not develop lichenoid lesions clinically or a lichenoid tissue reaction histopathologically.10
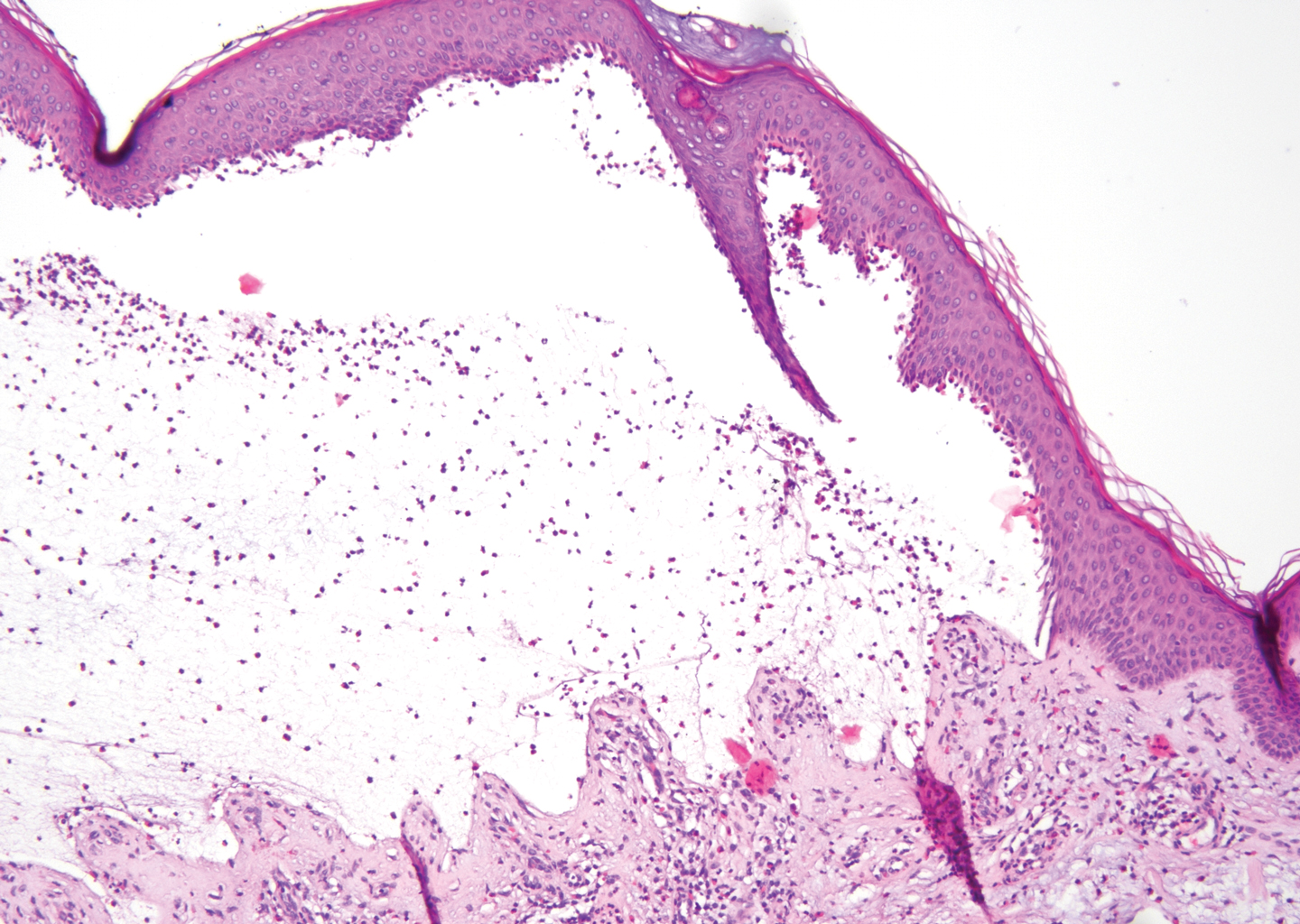
Bullous systemic lupus erythematosus (SLE), a rare cutaneous manifestation of SLE, typically arises in young women of African descent and is due to autoantibody formation against type VII collagen and other BM-zone antigens. Patients generally present with acute onset of tense vesiculobullae on a normal or erythematous base, which often are transient and heal without milia or scarring. Common sites of involvement include the trunk, arms, neck, face, and vermilion border, as well as the oral mucosa. The diagnosis of bullous SLE requires that patients fulfill the criteria for SLE and is confirmed by immunohistologic analysis. Biopsy of a bulla edge reveals a subepidermal blister containing neutrophils and increased mucin within the reticular dermis (Figure 3). Direct immunofluorescence of perilesional skin most commonly reveals linear and/or granular deposition of IgG, IgA, C3, and IgM at the DEJ.11

Bullous tinea is a manifestation of cutaneous dermatophytosis that usually occurs in the setting of tinea pedis. Common causative dermatophytes include Trichophyton mentagrophytes, Trichophyton rubrum, and Epidermophyton floccosum. Diagnosis is made by demonstration of fungal hyphae on potassium hydroxide preparation of the blister roof, biopsy with periodic acid-Schiff stain, or fungal culture. If routine histopathologic analysis is performed, epidermal spongiosis with varying degrees of papillary dermal edema is seen, along with abundant fungal elements in the stratum corneum (Figure 4). Direct immunofluorescence of perilesional skin usually is negative, but C3 deposition in a linear and/or granular pattern along the DEJ has been reported.12

Lichen planus pemphigoides is a rare disease entity and often presents a diagnostic challenge to clinicians. The differential for LPP includes bullous LP as well as other bullous disorders. Ultimately, the diagnosis is confirmed through immunohistologic analysis. Timely diagnosis of LPP is crucial, as most patients can achieve long-term remission with appropriate treatment.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Mohanarao TS, Kumar GA, Chennamsetty K, et al. Childhood lichen planus pemphigoides triggered by chickenpox. Indian Dermatol Online J. 2014;5:S98-S100.
- Onprasert W, Chanprapaph K. Lichen planus pemphigoides induced by enalapril: a case report and a review of literature. Case Rep Dermatol. 2017;9:217-224.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Shimada H, Shono T, Sakai T, et al. Lichen planus pemphigoides concomitant with rectal adenocarcinoma: fortuitous or a true association? Eur J Dermatol. 2015;25:501-503.
- Matos-Pires E, Campos S, Lencastre A, et al. Lichen planus pemphigoides. J Dtsch Dermatol Ges. 2018;16:335-337.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Knisley RR, Petropolis AA, Mackey VT. Lichen planus pemphigoides treated with ustekinumab. Cutis. 2017;100:415-418.
- Wagner G, Rose C, Sachse MM. Clinical variants of lichen planus. J Dtsch Dermatol Ges. 2013;11:309-319.
- Bagci IS, Horvath ON, Ruzicka T, et al. Bullous pemphigoid. Autoimmun Rev. 2017;16:445-455.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Miller DD, Bhawan J. Bullous tinea pedis with direct immunofluorescence positivity: when is a positive result not autoimmune bullous disease? Am J Dermatopathol. 2013;35:587-594.
The Diagnosis: Lichen Planus Pemphigoides
Lichen planus pemphigoides (LPP) is a rare autoimmune subepithelial blistering disorder with clinical, pathologic, and immunologic features of lichen planus (LP) and bullous pemphigoid (BP).1 It mainly arises in adults and usually is idiopathic but has been associated with certain infections,2 drugs such as angiotensin-converting enzyme inhibitors,3 phototherapy,4 and malignancy.5 Patients classically present with lichenoid lesions, tense vesiculobullae, and erosions.6 Vesiculobullae formation usually follows the development of lichenoid lesions, occurs on both lichenoid lesions and unaffected skin, and predominantly involves the lower extremities, as in our patient.1,6
The pathogenesis of LPP is not fully understood but likely represents a distinct entity rather than a subtype of BP or the simultaneous occurrence of LP and BP. Lichen planus pemphigoides generally has an earlier onset and better treatment response compared to BP.7 Further, autoantibodies in patients with LPP react to a novel epitope within the C-terminal portion of the BP-180 NC16A domain. Accordingly, it has been postulated that an inflammatory cutaneous process resulting from infection, phototherapy, or LP itself leads to damage of the epidermis and triggers a secondary blistering autoimmune dermatosis mediated by antibody formation against basement membrane (BM) antigens, such as BP-180.7
The diagnosis of LPP ultimately is confirmed with immunohistologic analysis. Biopsy of LPP shows findings consistent with both LP and BP (quiz image [top]). In the lichenoid portion, biopsy reveals orthohyperkeratosis, hypergranulosis, and acanthosis of the epidermis; a bandlike infiltrate consisting primarily of lymphocytes in the upper dermis; and apoptotic keratinocytes (colloid bodies) and vacuolar degeneration at the dermoepidermal junction (DEJ).1 Biopsy of bullae reveals eosinophilic spongiosis, a subepithelial blister plane with eosinophils, and a mixed superficial inflammatory cell infiltrate. Direct immunofluorescence from perilesional skin reveals linear deposition of IgG and/or C3 at the DEJ (quiz image [bottom]).1 Measurement of anti-BM antibodies against BP-180 and BP-230 can be useful in suspected cases, as 50% to 60% of patients have circulating antibodies against these antigens.6 Remission usually is achieved with topical and systemic corticosteroids and/or steroid-sparing agents, with rare recurrence following lesion resolution.1 More recently, successful treatment with biologics such as ustekinumab has been reported.8
The predominant differential diagnosis for LPP is bullous LP, a variant of LP in which vesiculobullous disease occurs exclusively on preexisting LP lesions, commonly on the legs due to severe vacuolar degeneration at the DEJ. On histopathology, the characteristic features of LP (eg, orthohyperkeratosis, hypergranulosis, acanthosis, bandlike lymphocytic infiltrate, colloid bodies) along with subepidermal clefting will be seen. However, in bullous LP (Figure 1) there is an absence of linear IgG and/or C3 deposition at the DEJ on direct immunofluorescence. Furthermore, patients lack circulating antibodies against BP-180 and BP-230.9
Lichen planus pemphigoides also can be confused with BP. Bullous pemphigoid is the most common autoimmune blistering disorder; typically arises in older adults; and is caused by autoantibody formation against hemidesmosomal proteins, particularly BP-180 and BP-230. Patients classically present with tense bullae and erosions on an erythematous, urticarial, or normal base. These lesions often are pruritic and concentrated on the trunk, axillary and inguinal folds, and extremity flexures. Histopathologic examination of a bulla edge reveals the classic findings seen in BP (eg, eosinophilic spongiosis, subepithelial blister plane with eosinophils)(Figure 2). Direct immunofluorescence of perilesional skin reveals linear IgG and/or C3 deposition along the DEJ. A large subset of patients also has circulating antibodies against BP-180 and BP-230. In contrast to LPP, however, patients with BP do not develop lichenoid lesions clinically or a lichenoid tissue reaction histopathologically.10
Bullous systemic lupus erythematosus (SLE), a rare cutaneous manifestation of SLE, typically arises in young women of African descent and is due to autoantibody formation against type VII collagen and other BM-zone antigens. Patients generally present with acute onset of tense vesiculobullae on a normal or erythematous base, which often are transient and heal without milia or scarring. Common sites of involvement include the trunk, arms, neck, face, and vermilion border, as well as the oral mucosa. The diagnosis of bullous SLE requires that patients fulfill the criteria for SLE and is confirmed by immunohistologic analysis. Biopsy of a bulla edge reveals a subepidermal blister containing neutrophils and increased mucin within the reticular dermis (Figure 3). Direct immunofluorescence of perilesional skin most commonly reveals linear and/or granular deposition of IgG, IgA, C3, and IgM at the DEJ.11
Bullous tinea is a manifestation of cutaneous dermatophytosis that usually occurs in the setting of tinea pedis. Common causative dermatophytes include Trichophyton mentagrophytes, Trichophyton rubrum, and Epidermophyton floccosum. Diagnosis is made by demonstration of fungal hyphae on potassium hydroxide preparation of the blister roof, biopsy with periodic acid-Schiff stain, or fungal culture. If routine histopathologic analysis is performed, epidermal spongiosis with varying degrees of papillary dermal edema is seen, along with abundant fungal elements in the stratum corneum (Figure 4). Direct immunofluorescence of perilesional skin usually is negative, but C3 deposition in a linear and/or granular pattern along the DEJ has been reported.12
Lichen planus pemphigoides is a rare disease entity and often presents a diagnostic challenge to clinicians. The differential for LPP includes bullous LP as well as other bullous disorders. Ultimately, the diagnosis is confirmed through immunohistologic analysis. Timely diagnosis of LPP is crucial, as most patients can achieve long-term remission with appropriate treatment.
The Diagnosis: Lichen Planus Pemphigoides
Lichen planus pemphigoides (LPP) is a rare autoimmune subepithelial blistering disorder with clinical, pathologic, and immunologic features of lichen planus (LP) and bullous pemphigoid (BP).1 It mainly arises in adults and usually is idiopathic but has been associated with certain infections,2 drugs such as angiotensin-converting enzyme inhibitors,3 phototherapy,4 and malignancy.5 Patients classically present with lichenoid lesions, tense vesiculobullae, and erosions.6 Vesiculobullae formation usually follows the development of lichenoid lesions, occurs on both lichenoid lesions and unaffected skin, and predominantly involves the lower extremities, as in our patient.1,6
The pathogenesis of LPP is not fully understood but likely represents a distinct entity rather than a subtype of BP or the simultaneous occurrence of LP and BP. Lichen planus pemphigoides generally has an earlier onset and better treatment response compared to BP.7 Further, autoantibodies in patients with LPP react to a novel epitope within the C-terminal portion of the BP-180 NC16A domain. Accordingly, it has been postulated that an inflammatory cutaneous process resulting from infection, phototherapy, or LP itself leads to damage of the epidermis and triggers a secondary blistering autoimmune dermatosis mediated by antibody formation against basement membrane (BM) antigens, such as BP-180.7
The diagnosis of LPP ultimately is confirmed with immunohistologic analysis. Biopsy of LPP shows findings consistent with both LP and BP (quiz image [top]). In the lichenoid portion, biopsy reveals orthohyperkeratosis, hypergranulosis, and acanthosis of the epidermis; a bandlike infiltrate consisting primarily of lymphocytes in the upper dermis; and apoptotic keratinocytes (colloid bodies) and vacuolar degeneration at the dermoepidermal junction (DEJ).1 Biopsy of bullae reveals eosinophilic spongiosis, a subepithelial blister plane with eosinophils, and a mixed superficial inflammatory cell infiltrate. Direct immunofluorescence from perilesional skin reveals linear deposition of IgG and/or C3 at the DEJ (quiz image [bottom]).1 Measurement of anti-BM antibodies against BP-180 and BP-230 can be useful in suspected cases, as 50% to 60% of patients have circulating antibodies against these antigens.6 Remission usually is achieved with topical and systemic corticosteroids and/or steroid-sparing agents, with rare recurrence following lesion resolution.1 More recently, successful treatment with biologics such as ustekinumab has been reported.8
The predominant differential diagnosis for LPP is bullous LP, a variant of LP in which vesiculobullous disease occurs exclusively on preexisting LP lesions, commonly on the legs due to severe vacuolar degeneration at the DEJ. On histopathology, the characteristic features of LP (eg, orthohyperkeratosis, hypergranulosis, acanthosis, bandlike lymphocytic infiltrate, colloid bodies) along with subepidermal clefting will be seen. However, in bullous LP (Figure 1) there is an absence of linear IgG and/or C3 deposition at the DEJ on direct immunofluorescence. Furthermore, patients lack circulating antibodies against BP-180 and BP-230.9
Lichen planus pemphigoides also can be confused with BP. Bullous pemphigoid is the most common autoimmune blistering disorder; typically arises in older adults; and is caused by autoantibody formation against hemidesmosomal proteins, particularly BP-180 and BP-230. Patients classically present with tense bullae and erosions on an erythematous, urticarial, or normal base. These lesions often are pruritic and concentrated on the trunk, axillary and inguinal folds, and extremity flexures. Histopathologic examination of a bulla edge reveals the classic findings seen in BP (eg, eosinophilic spongiosis, subepithelial blister plane with eosinophils)(Figure 2). Direct immunofluorescence of perilesional skin reveals linear IgG and/or C3 deposition along the DEJ. A large subset of patients also has circulating antibodies against BP-180 and BP-230. In contrast to LPP, however, patients with BP do not develop lichenoid lesions clinically or a lichenoid tissue reaction histopathologically.10
Bullous systemic lupus erythematosus (SLE), a rare cutaneous manifestation of SLE, typically arises in young women of African descent and is due to autoantibody formation against type VII collagen and other BM-zone antigens. Patients generally present with acute onset of tense vesiculobullae on a normal or erythematous base, which often are transient and heal without milia or scarring. Common sites of involvement include the trunk, arms, neck, face, and vermilion border, as well as the oral mucosa. The diagnosis of bullous SLE requires that patients fulfill the criteria for SLE and is confirmed by immunohistologic analysis. Biopsy of a bulla edge reveals a subepidermal blister containing neutrophils and increased mucin within the reticular dermis (Figure 3). Direct immunofluorescence of perilesional skin most commonly reveals linear and/or granular deposition of IgG, IgA, C3, and IgM at the DEJ.11
Bullous tinea is a manifestation of cutaneous dermatophytosis that usually occurs in the setting of tinea pedis. Common causative dermatophytes include Trichophyton mentagrophytes, Trichophyton rubrum, and Epidermophyton floccosum. Diagnosis is made by demonstration of fungal hyphae on potassium hydroxide preparation of the blister roof, biopsy with periodic acid-Schiff stain, or fungal culture. If routine histopathologic analysis is performed, epidermal spongiosis with varying degrees of papillary dermal edema is seen, along with abundant fungal elements in the stratum corneum (Figure 4). Direct immunofluorescence of perilesional skin usually is negative, but C3 deposition in a linear and/or granular pattern along the DEJ has been reported.12
Lichen planus pemphigoides is a rare disease entity and often presents a diagnostic challenge to clinicians. The differential for LPP includes bullous LP as well as other bullous disorders. Ultimately, the diagnosis is confirmed through immunohistologic analysis. Timely diagnosis of LPP is crucial, as most patients can achieve long-term remission with appropriate treatment.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Mohanarao TS, Kumar GA, Chennamsetty K, et al. Childhood lichen planus pemphigoides triggered by chickenpox. Indian Dermatol Online J. 2014;5:S98-S100.
- Onprasert W, Chanprapaph K. Lichen planus pemphigoides induced by enalapril: a case report and a review of literature. Case Rep Dermatol. 2017;9:217-224.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Shimada H, Shono T, Sakai T, et al. Lichen planus pemphigoides concomitant with rectal adenocarcinoma: fortuitous or a true association? Eur J Dermatol. 2015;25:501-503.
- Matos-Pires E, Campos S, Lencastre A, et al. Lichen planus pemphigoides. J Dtsch Dermatol Ges. 2018;16:335-337.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Knisley RR, Petropolis AA, Mackey VT. Lichen planus pemphigoides treated with ustekinumab. Cutis. 2017;100:415-418.
- Wagner G, Rose C, Sachse MM. Clinical variants of lichen planus. J Dtsch Dermatol Ges. 2013;11:309-319.
- Bagci IS, Horvath ON, Ruzicka T, et al. Bullous pemphigoid. Autoimmun Rev. 2017;16:445-455.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Miller DD, Bhawan J. Bullous tinea pedis with direct immunofluorescence positivity: when is a positive result not autoimmune bullous disease? Am J Dermatopathol. 2013;35:587-594.
- Zaraa I, Mahfoudh A, Sellami MK, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. 2013;52:406-412.
- Mohanarao TS, Kumar GA, Chennamsetty K, et al. Childhood lichen planus pemphigoides triggered by chickenpox. Indian Dermatol Online J. 2014;5:S98-S100.
- Onprasert W, Chanprapaph K. Lichen planus pemphigoides induced by enalapril: a case report and a review of literature. Case Rep Dermatol. 2017;9:217-224.
- Kuramoto N, Kishimoto S, Shibagaki R, et al. PUVA-induced lichen planus pemphigoides. Br J Dermatol. 2000;142:509-512.
- Shimada H, Shono T, Sakai T, et al. Lichen planus pemphigoides concomitant with rectal adenocarcinoma: fortuitous or a true association? Eur J Dermatol. 2015;25:501-503.
- Matos-Pires E, Campos S, Lencastre A, et al. Lichen planus pemphigoides. J Dtsch Dermatol Ges. 2018;16:335-337.
- Zillikens D, Caux F, Mascaro JM, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. 1999;113:117-121.
- Knisley RR, Petropolis AA, Mackey VT. Lichen planus pemphigoides treated with ustekinumab. Cutis. 2017;100:415-418.
- Wagner G, Rose C, Sachse MM. Clinical variants of lichen planus. J Dtsch Dermatol Ges. 2013;11:309-319.
- Bagci IS, Horvath ON, Ruzicka T, et al. Bullous pemphigoid. Autoimmun Rev. 2017;16:445-455.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Miller DD, Bhawan J. Bullous tinea pedis with direct immunofluorescence positivity: when is a positive result not autoimmune bullous disease? Am J Dermatopathol. 2013;35:587-594.
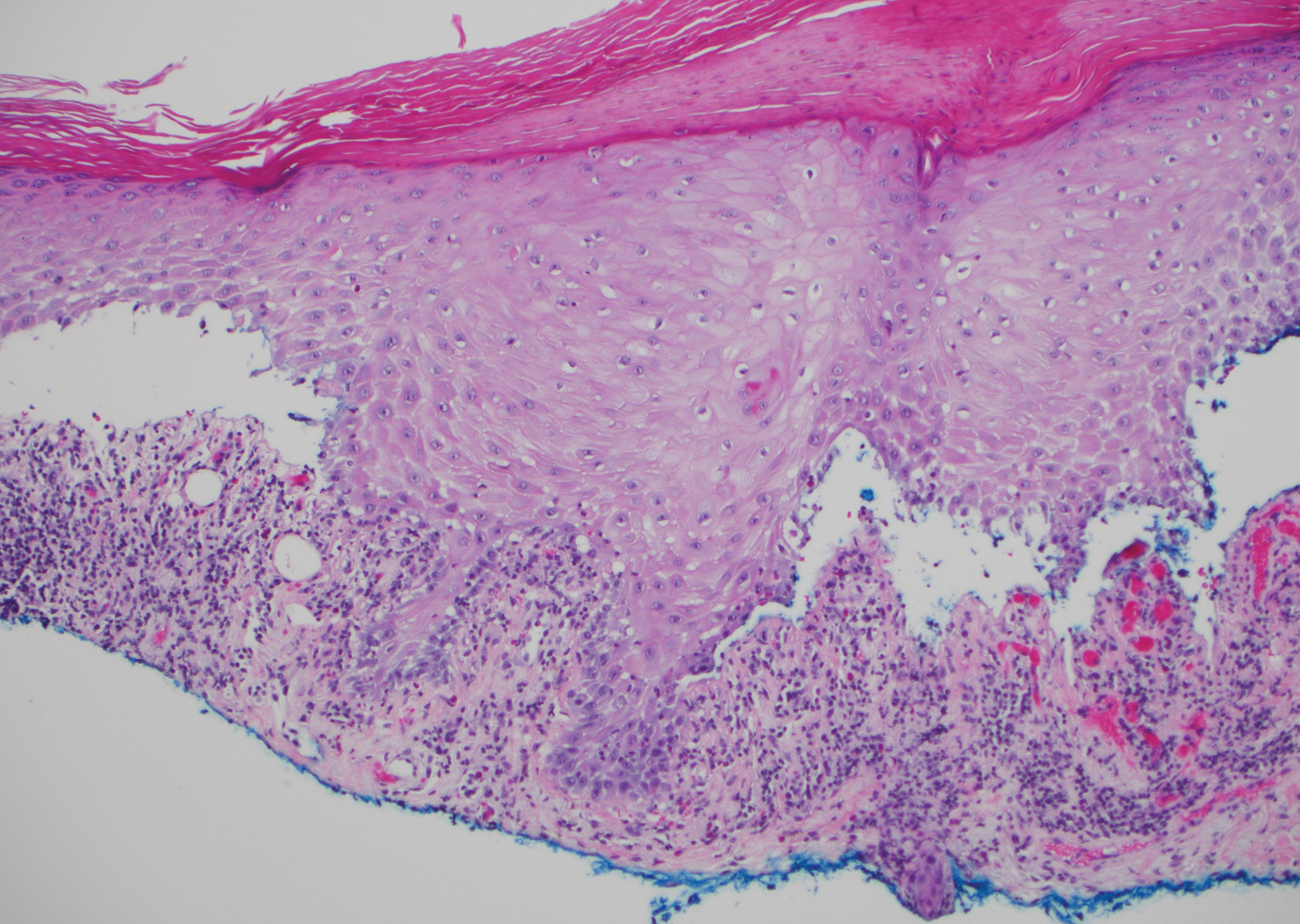
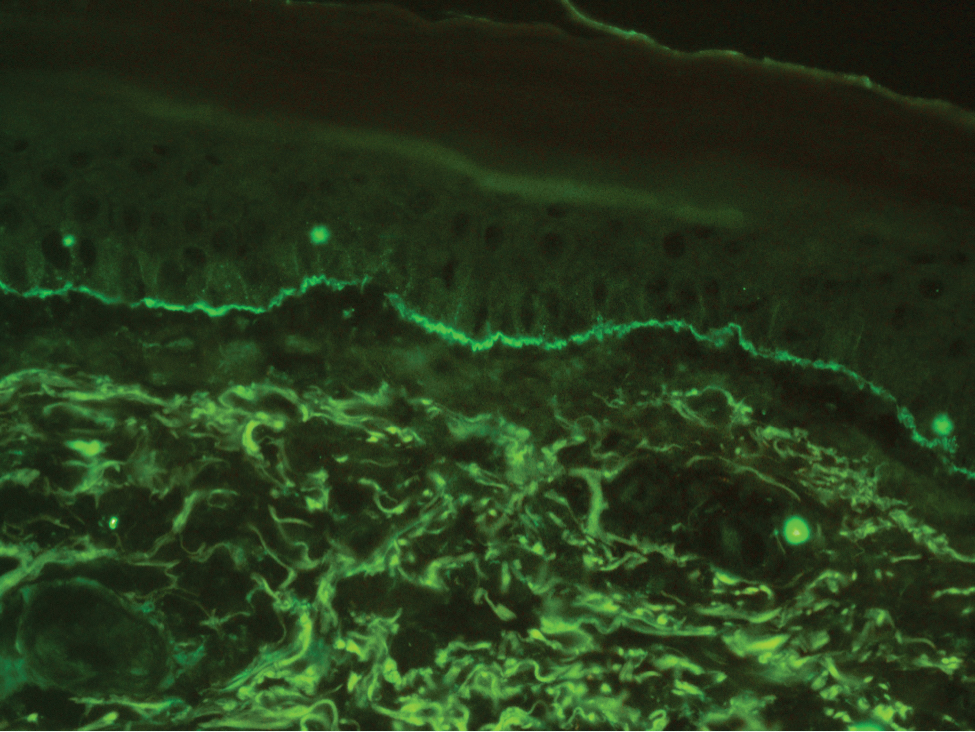
A 72-year-old woman presented to our dermatology clinic with a rash of several months' duration that began as itchy bumps on the wrists and spread to involve the legs. Approximately 2 months prior to presentation, she noted blisters on the feet and legs. She initially went to her primary care physician, who prescribed levofloxacin, cephalexin, and a 5-day course of prednisone. The prednisone initially helped; however the rash worsened on discontinuation. In our clinic, the patient had scattered tense bullae and numerous erosions with crust on the dorsum of the feet and legs, some of which were in conjunction with violaceous papules and plaques. There also was hypertrophic scale on the soles of the feet. A potassium hydroxide preparation of skin scrapings from the feet was negative for fungal elements. Two shave biopsies of a violaceous plaque and bulla as well as a perilesional punch biopsy from the leg were obtained.
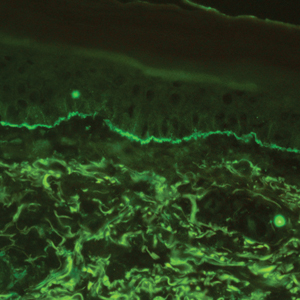
Ulcerated Nodule on the Scalp
The Diagnosis: Proliferating Pilar Tumor
Proliferating pilar tumor (PPT), or cyst, is a neoplasm of trichilemmal keratinization first described by Wilson-Jones1 in 1966. Proliferating pilar tumors lie on a spectrum with malignant PPT, which is a rare adnexal neoplasm first described by Saida et al2 in 1983. The incidence of PPT is unknown given the paucity of cases and the possible misdiagnosis as squamous cell carcinoma (SCC). Proliferating pilar tumors tend to present on the head and neck of older females as a multilobular and sometimes ulcerating nodule.3 Although PPT can occur de novo, the majority of cases are thought to develop progressively from a benign pilar cyst. Histopathologically, PPT is characterized by cords and nests of squamous cells that display trichilemmal keratinization (quiz images).
Classification of PPT as benign or malignant is challenging, though criteria have been proposed.3-7 Lesions with minimal infiltration into the surrounding dermis and scant mitosis typically behave in a benign manner, while lesions showing nuclear atypia, atypical mitosis, and irregular infiltration into the surrounding dermis can have up to a 50% locoregional recurrence rate.3 In addition, distinguishing a PPT from an SCC or trichilemmal carcinoma also can be difficult; however, SCC is favored when there is a lack of trichilemmal keratinization or when squamous atypia is present in the adjacent epidermis.8 Trichilemmal carcinoma is a rare tumor that has been questioned as a distinct entity.9-12
Pilomatricoma, also known as calcifying epithelioma of Malherbe, is a benign pilar tumor that presents as a slowly growing nodule on the head or neck area or arms.13,14 Most pilomatricomas develop by the second decade of life. Multiple lesions may be present in association with myotonic dystrophy or Gardner syndrome among other syndromes.15-17 Similar to PPT, pilomatricomas present as large dermal nodules; however, they tend to be circumscribed and have a trabecular network that consists of basophilic cells and eosinophilic keratinized shadow cells (Figure 1).18 Calcification may be seen and bone formation subsequently may occur.19
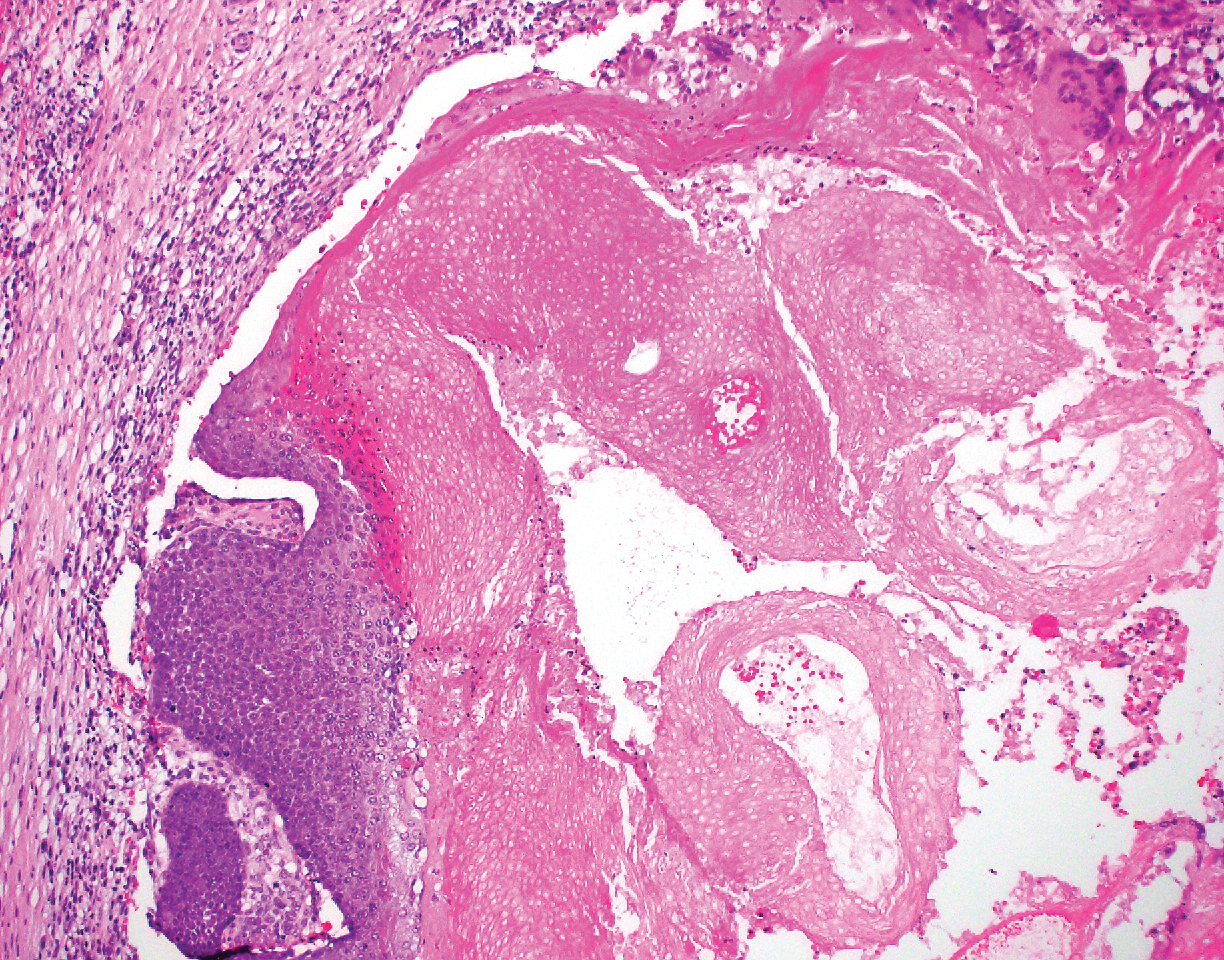
Most sources now consider keratoacanthoma (KA) as a well-differentiated SCC.20 The typical presentation consists of a rapidly growing erythematous to flesh-colored nodule with a central keratinous plug that develops over a period of weeks. If untreated, KAs may resolve over a period of months and leave a depressed scar. Local destruction can result from KAs, and they have the potential to transform into a more aggressive SCC. Accordingly, most clinicians use tissue destructive methods, excision, or Mohs micrographic surgery for treatment based on location. Histologically, a well-circumscribed proliferation of glassy cytoplasm is noted. A depressed keratin-filled center is surrounded by a lip of epithelium extending over the lesion (Figure 2).20,21 Pseudoepitheliomatous hyperplasia accompanied by hypergranulosis is seen in the center of KAs rather than at the periphery, which is typical of non-KA SCCs. Typical KAs lack acantholysis, a feature suggesting a non-KA type of SCC. Neutrophilic microabscesses and eosinophils commonly are seen in KAs.20,21
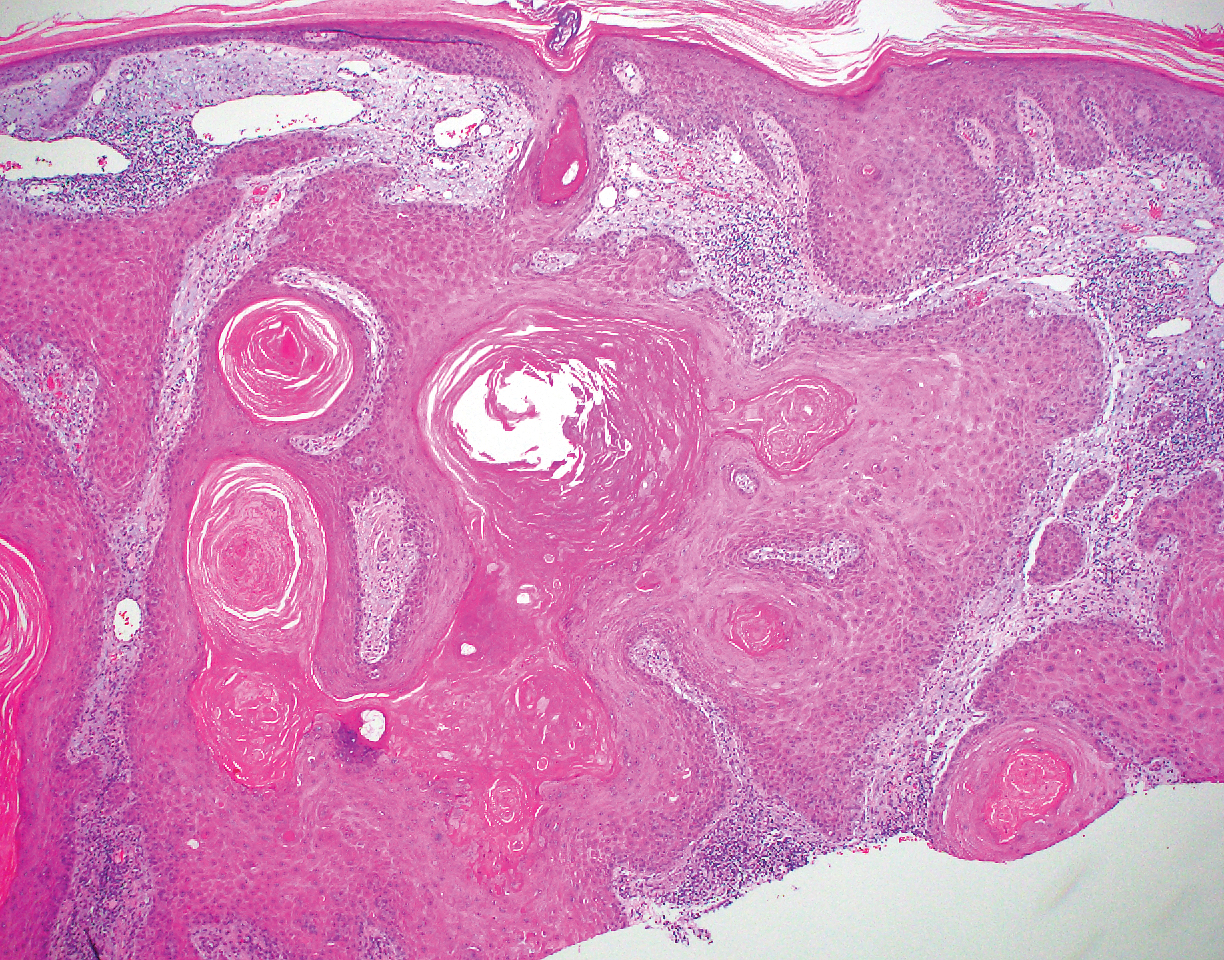
Inverted follicular keratosis is a benign tumor that gained traction as its own entity in the 1960s.22 These lesions typically develop from the follicular infundibulum, but some consider them a version of a wart or seborrheic keratosis.23 They generally are flesh-colored nodules on the upper cutaneous lip or face. Treatment usually consists of complete excision. There are many different growth patterns described, but they typically are endophytic tumors with eosinophilic squamous cells in the center and more basophilic cells at the periphery (Figure 3).24 Characteristically, there are squamous eddies throughout the tumor (Figure 3 [inset]). There also may be a scant lymphohistiocytic infiltrate within the dermis surrounding the lesion.
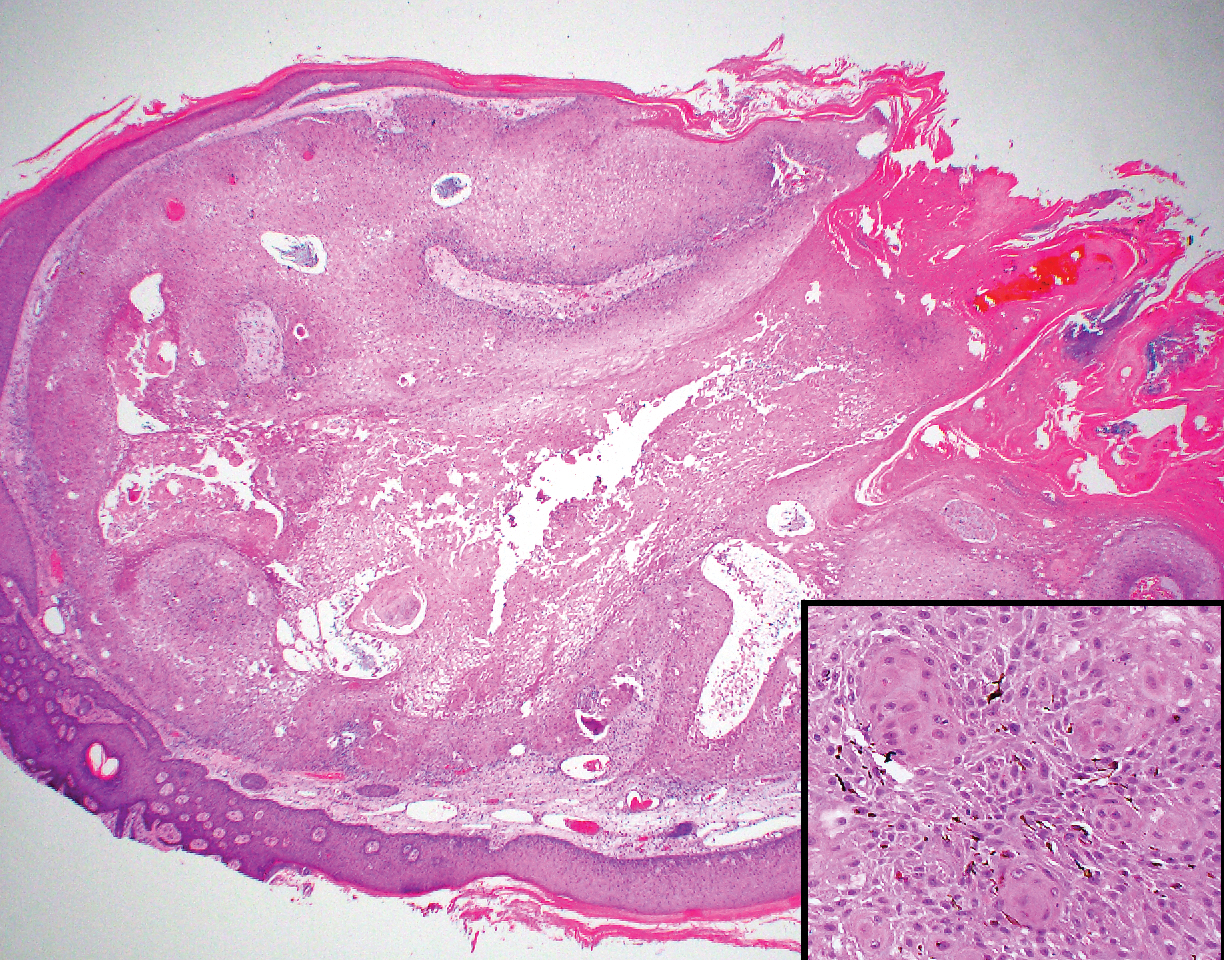
Trichilemmomas are flesh-colored adnexal neoplasms that may present as a solitary lesion or in clusters on the face. They have been reported to occur on all nonglabrous skin sites.25 Multiple lesions may occur in association with Cowden syndrome or with nevus sebaceous.26 A desmoplastic variant of trichilemmomas has been reported.27 Desmoplastic trichilemmomas appear as well-circumscribed tumors of outer root sheath differentiation with lobules extending down into the dermis.28 Vacuolated glycogen-filled keratinocytes are scattered throughout the lesion but are most prominent at the base. At the periphery of the lobules, peripheral palisading of basaloid cells is accompanied by a thickened eosinophilic basement membrane that is periodic acid-Schiff positive. Typical trichilemmomas also can display these features; however, the main differentiating feature of a desmoplastic trichilemmoma is the pink hyalinized stroma separating small islets of basophilic cells (Figure 4). Differentiation from an invasive malignant carcinoma sometimes can be challenging without a focus of typical trichilemmoma or if the biopsy specimen is too superficial.29

Pilar cysts are common tumors that typically arise on the scalp and sometimes are proliferating. Proliferating pilar tumor should be kept on the differential when secondary changes such as ulceration occur in the primary lesion of the scalp. Microscopically, and sometimes clinically, PPT can be difficult to differentiate from other mimickers.
- Wilson-Jones E. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208.
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumours: a clinicopathological study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574.
- Garg PK, Dangi A, Khurana N, et al. Malignant proliferating trichilemmal cyst: a case report with review of literature. Malaysian J Pathol. 2009;31:71-76.
- Herrero J, Monteagudo C, Ruiz A, et al. Malignant proliferating trichilemmal tumors: a histopathological and immunohistochemical study of three cases with DNA ploidy and morphometric evaluation. Histopathology. 1998;33:542-546.
- Haas N, Audring H, Sterry W. Carcinoma arising in a proliferating trichilemmal cyst expresses fetal and trichilemmal hair phenotype. Am J Dermatopathol. 2002;24:340-344.
- Rutty GN, Richman PI, Laing JH. Malignant change in trichilemmal cysts: a study of cell proliferation and DNA content. Histopathology. 1992;21:465-468.
- Brownstein MH, Arluk DJ. Proliferating trichilemmal cyst: a simulant of squamous cell carcinoma. Cancer. 1981;48:1207-1214.
- Misago N, Ackerman AB. Tricholemmal carcinoma? Dermatopathol Pract Concept. 1999;5:205-206.
- Misago N, Narisawa Y. Tricholemmal carcinoma in continuity with trichoblastoma within nevus sebaceous. Am J Dermatopathol. 2002;24:149-155.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Swanson PE, Marrogi AJ, Williams DJ, et al. Trichilemmal carcinoma: clinicopathologic study of 10 cases. J Cutan Pathol. 1992;19:100-109.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Julian CG, Bowers PW. A clinical review of 209 pilomatricomas. J Am Acad Dermatol. 1998;39:191-195.
- Marrogi AJ, Wick MR, Dehner LP. Pilomatrical neoplasms in children and young adults. Am J Dermatopathol. 1992;14:87-94.
- Berberian BJ, Colonna TM, Battaglia M, et al. Multiple pilomatricomas in association with myotonic dystrophy and a family history of melanoma. J Am Acad Dermatol. 1997;37:268-269.
- Cooper PH, Fechner RE. Pilomatricoma-like changes in the epidermal cysts of Gardner's syndrome. J Am Acad Dermatol. 1983;8:639-644.
- Kaddu S, Soyer HP, Cerroni L, et al. Clinical and histopathologic spectrum of pilomatricomas in adults. Int J Dermatol. 1994;33:705-708.
- Sano Y, Mihara M, Miyamoto T, et al. Simultaneous occurrence of calcification and amyloid deposit in pilomatricoma. Acta Derm Venereol. 1990;70:256-259.
- Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233.
- Mehregan AH. Inverted follicular keratosis. Arch Dermatol. 1964;89:117-123.
- Spielvogel RL, Austin C, Ackerman AB. Inverted follicular keratosis is not a specific keratosis but a verruca vulgaris (or seborrheic keratosis) with squamous eddies. Am J Dermatopathol. 1983;5:427-445.
- Mehregan AH. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
- Brownstein MH. Trichilemmoma. benign follicular tumor or viral wart? Am J Dermatopathol. 1980;2:229-231.
- Brownstein MH. Multiple trichilemmomas in Cowden's syndrome. Arch Dermatol. 1979;115:111.
- Roson E, Gomez Centeno P, Sanchez Aguilar D, et al. Desmoplastic trichilemmoma arising within a nevus sebaceous. Am J Dermatopathol. 1998;20:495-497.
- Tellechea O, Reis JP, Baptista AP. Desmoplastic trichilemmoma. Am J Dermatopathol. 1992;14:107-114.
- Sharma R, Sirohi D, Sengupta P, et al. Desmoplastic trichilemmoma of the facial region mimicking invasive carcinoma. J Maxillofac Oral Surg. 2010;10:71-73.
The Diagnosis: Proliferating Pilar Tumor
Proliferating pilar tumor (PPT), or cyst, is a neoplasm of trichilemmal keratinization first described by Wilson-Jones1 in 1966. Proliferating pilar tumors lie on a spectrum with malignant PPT, which is a rare adnexal neoplasm first described by Saida et al2 in 1983. The incidence of PPT is unknown given the paucity of cases and the possible misdiagnosis as squamous cell carcinoma (SCC). Proliferating pilar tumors tend to present on the head and neck of older females as a multilobular and sometimes ulcerating nodule.3 Although PPT can occur de novo, the majority of cases are thought to develop progressively from a benign pilar cyst. Histopathologically, PPT is characterized by cords and nests of squamous cells that display trichilemmal keratinization (quiz images).
Classification of PPT as benign or malignant is challenging, though criteria have been proposed.3-7 Lesions with minimal infiltration into the surrounding dermis and scant mitosis typically behave in a benign manner, while lesions showing nuclear atypia, atypical mitosis, and irregular infiltration into the surrounding dermis can have up to a 50% locoregional recurrence rate.3 In addition, distinguishing a PPT from an SCC or trichilemmal carcinoma also can be difficult; however, SCC is favored when there is a lack of trichilemmal keratinization or when squamous atypia is present in the adjacent epidermis.8 Trichilemmal carcinoma is a rare tumor that has been questioned as a distinct entity.9-12
Pilomatricoma, also known as calcifying epithelioma of Malherbe, is a benign pilar tumor that presents as a slowly growing nodule on the head or neck area or arms.13,14 Most pilomatricomas develop by the second decade of life. Multiple lesions may be present in association with myotonic dystrophy or Gardner syndrome among other syndromes.15-17 Similar to PPT, pilomatricomas present as large dermal nodules; however, they tend to be circumscribed and have a trabecular network that consists of basophilic cells and eosinophilic keratinized shadow cells (Figure 1).18 Calcification may be seen and bone formation subsequently may occur.19
Most sources now consider keratoacanthoma (KA) as a well-differentiated SCC.20 The typical presentation consists of a rapidly growing erythematous to flesh-colored nodule with a central keratinous plug that develops over a period of weeks. If untreated, KAs may resolve over a period of months and leave a depressed scar. Local destruction can result from KAs, and they have the potential to transform into a more aggressive SCC. Accordingly, most clinicians use tissue destructive methods, excision, or Mohs micrographic surgery for treatment based on location. Histologically, a well-circumscribed proliferation of glassy cytoplasm is noted. A depressed keratin-filled center is surrounded by a lip of epithelium extending over the lesion (Figure 2).20,21 Pseudoepitheliomatous hyperplasia accompanied by hypergranulosis is seen in the center of KAs rather than at the periphery, which is typical of non-KA SCCs. Typical KAs lack acantholysis, a feature suggesting a non-KA type of SCC. Neutrophilic microabscesses and eosinophils commonly are seen in KAs.20,21
Inverted follicular keratosis is a benign tumor that gained traction as its own entity in the 1960s.22 These lesions typically develop from the follicular infundibulum, but some consider them a version of a wart or seborrheic keratosis.23 They generally are flesh-colored nodules on the upper cutaneous lip or face. Treatment usually consists of complete excision. There are many different growth patterns described, but they typically are endophytic tumors with eosinophilic squamous cells in the center and more basophilic cells at the periphery (Figure 3).24 Characteristically, there are squamous eddies throughout the tumor (Figure 3 [inset]). There also may be a scant lymphohistiocytic infiltrate within the dermis surrounding the lesion.
Trichilemmomas are flesh-colored adnexal neoplasms that may present as a solitary lesion or in clusters on the face. They have been reported to occur on all nonglabrous skin sites.25 Multiple lesions may occur in association with Cowden syndrome or with nevus sebaceous.26 A desmoplastic variant of trichilemmomas has been reported.27 Desmoplastic trichilemmomas appear as well-circumscribed tumors of outer root sheath differentiation with lobules extending down into the dermis.28 Vacuolated glycogen-filled keratinocytes are scattered throughout the lesion but are most prominent at the base. At the periphery of the lobules, peripheral palisading of basaloid cells is accompanied by a thickened eosinophilic basement membrane that is periodic acid-Schiff positive. Typical trichilemmomas also can display these features; however, the main differentiating feature of a desmoplastic trichilemmoma is the pink hyalinized stroma separating small islets of basophilic cells (Figure 4). Differentiation from an invasive malignant carcinoma sometimes can be challenging without a focus of typical trichilemmoma or if the biopsy specimen is too superficial.29
Pilar cysts are common tumors that typically arise on the scalp and sometimes are proliferating. Proliferating pilar tumor should be kept on the differential when secondary changes such as ulceration occur in the primary lesion of the scalp. Microscopically, and sometimes clinically, PPT can be difficult to differentiate from other mimickers.
The Diagnosis: Proliferating Pilar Tumor
Proliferating pilar tumor (PPT), or cyst, is a neoplasm of trichilemmal keratinization first described by Wilson-Jones1 in 1966. Proliferating pilar tumors lie on a spectrum with malignant PPT, which is a rare adnexal neoplasm first described by Saida et al2 in 1983. The incidence of PPT is unknown given the paucity of cases and the possible misdiagnosis as squamous cell carcinoma (SCC). Proliferating pilar tumors tend to present on the head and neck of older females as a multilobular and sometimes ulcerating nodule.3 Although PPT can occur de novo, the majority of cases are thought to develop progressively from a benign pilar cyst. Histopathologically, PPT is characterized by cords and nests of squamous cells that display trichilemmal keratinization (quiz images).
Classification of PPT as benign or malignant is challenging, though criteria have been proposed.3-7 Lesions with minimal infiltration into the surrounding dermis and scant mitosis typically behave in a benign manner, while lesions showing nuclear atypia, atypical mitosis, and irregular infiltration into the surrounding dermis can have up to a 50% locoregional recurrence rate.3 In addition, distinguishing a PPT from an SCC or trichilemmal carcinoma also can be difficult; however, SCC is favored when there is a lack of trichilemmal keratinization or when squamous atypia is present in the adjacent epidermis.8 Trichilemmal carcinoma is a rare tumor that has been questioned as a distinct entity.9-12
Pilomatricoma, also known as calcifying epithelioma of Malherbe, is a benign pilar tumor that presents as a slowly growing nodule on the head or neck area or arms.13,14 Most pilomatricomas develop by the second decade of life. Multiple lesions may be present in association with myotonic dystrophy or Gardner syndrome among other syndromes.15-17 Similar to PPT, pilomatricomas present as large dermal nodules; however, they tend to be circumscribed and have a trabecular network that consists of basophilic cells and eosinophilic keratinized shadow cells (Figure 1).18 Calcification may be seen and bone formation subsequently may occur.19
Most sources now consider keratoacanthoma (KA) as a well-differentiated SCC.20 The typical presentation consists of a rapidly growing erythematous to flesh-colored nodule with a central keratinous plug that develops over a period of weeks. If untreated, KAs may resolve over a period of months and leave a depressed scar. Local destruction can result from KAs, and they have the potential to transform into a more aggressive SCC. Accordingly, most clinicians use tissue destructive methods, excision, or Mohs micrographic surgery for treatment based on location. Histologically, a well-circumscribed proliferation of glassy cytoplasm is noted. A depressed keratin-filled center is surrounded by a lip of epithelium extending over the lesion (Figure 2).20,21 Pseudoepitheliomatous hyperplasia accompanied by hypergranulosis is seen in the center of KAs rather than at the periphery, which is typical of non-KA SCCs. Typical KAs lack acantholysis, a feature suggesting a non-KA type of SCC. Neutrophilic microabscesses and eosinophils commonly are seen in KAs.20,21
Inverted follicular keratosis is a benign tumor that gained traction as its own entity in the 1960s.22 These lesions typically develop from the follicular infundibulum, but some consider them a version of a wart or seborrheic keratosis.23 They generally are flesh-colored nodules on the upper cutaneous lip or face. Treatment usually consists of complete excision. There are many different growth patterns described, but they typically are endophytic tumors with eosinophilic squamous cells in the center and more basophilic cells at the periphery (Figure 3).24 Characteristically, there are squamous eddies throughout the tumor (Figure 3 [inset]). There also may be a scant lymphohistiocytic infiltrate within the dermis surrounding the lesion.
Trichilemmomas are flesh-colored adnexal neoplasms that may present as a solitary lesion or in clusters on the face. They have been reported to occur on all nonglabrous skin sites.25 Multiple lesions may occur in association with Cowden syndrome or with nevus sebaceous.26 A desmoplastic variant of trichilemmomas has been reported.27 Desmoplastic trichilemmomas appear as well-circumscribed tumors of outer root sheath differentiation with lobules extending down into the dermis.28 Vacuolated glycogen-filled keratinocytes are scattered throughout the lesion but are most prominent at the base. At the periphery of the lobules, peripheral palisading of basaloid cells is accompanied by a thickened eosinophilic basement membrane that is periodic acid-Schiff positive. Typical trichilemmomas also can display these features; however, the main differentiating feature of a desmoplastic trichilemmoma is the pink hyalinized stroma separating small islets of basophilic cells (Figure 4). Differentiation from an invasive malignant carcinoma sometimes can be challenging without a focus of typical trichilemmoma or if the biopsy specimen is too superficial.29
Pilar cysts are common tumors that typically arise on the scalp and sometimes are proliferating. Proliferating pilar tumor should be kept on the differential when secondary changes such as ulceration occur in the primary lesion of the scalp. Microscopically, and sometimes clinically, PPT can be difficult to differentiate from other mimickers.
- Wilson-Jones E. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208.
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumours: a clinicopathological study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574.
- Garg PK, Dangi A, Khurana N, et al. Malignant proliferating trichilemmal cyst: a case report with review of literature. Malaysian J Pathol. 2009;31:71-76.
- Herrero J, Monteagudo C, Ruiz A, et al. Malignant proliferating trichilemmal tumors: a histopathological and immunohistochemical study of three cases with DNA ploidy and morphometric evaluation. Histopathology. 1998;33:542-546.
- Haas N, Audring H, Sterry W. Carcinoma arising in a proliferating trichilemmal cyst expresses fetal and trichilemmal hair phenotype. Am J Dermatopathol. 2002;24:340-344.
- Rutty GN, Richman PI, Laing JH. Malignant change in trichilemmal cysts: a study of cell proliferation and DNA content. Histopathology. 1992;21:465-468.
- Brownstein MH, Arluk DJ. Proliferating trichilemmal cyst: a simulant of squamous cell carcinoma. Cancer. 1981;48:1207-1214.
- Misago N, Ackerman AB. Tricholemmal carcinoma? Dermatopathol Pract Concept. 1999;5:205-206.
- Misago N, Narisawa Y. Tricholemmal carcinoma in continuity with trichoblastoma within nevus sebaceous. Am J Dermatopathol. 2002;24:149-155.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Swanson PE, Marrogi AJ, Williams DJ, et al. Trichilemmal carcinoma: clinicopathologic study of 10 cases. J Cutan Pathol. 1992;19:100-109.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Julian CG, Bowers PW. A clinical review of 209 pilomatricomas. J Am Acad Dermatol. 1998;39:191-195.
- Marrogi AJ, Wick MR, Dehner LP. Pilomatrical neoplasms in children and young adults. Am J Dermatopathol. 1992;14:87-94.
- Berberian BJ, Colonna TM, Battaglia M, et al. Multiple pilomatricomas in association with myotonic dystrophy and a family history of melanoma. J Am Acad Dermatol. 1997;37:268-269.
- Cooper PH, Fechner RE. Pilomatricoma-like changes in the epidermal cysts of Gardner's syndrome. J Am Acad Dermatol. 1983;8:639-644.
- Kaddu S, Soyer HP, Cerroni L, et al. Clinical and histopathologic spectrum of pilomatricomas in adults. Int J Dermatol. 1994;33:705-708.
- Sano Y, Mihara M, Miyamoto T, et al. Simultaneous occurrence of calcification and amyloid deposit in pilomatricoma. Acta Derm Venereol. 1990;70:256-259.
- Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233.
- Mehregan AH. Inverted follicular keratosis. Arch Dermatol. 1964;89:117-123.
- Spielvogel RL, Austin C, Ackerman AB. Inverted follicular keratosis is not a specific keratosis but a verruca vulgaris (or seborrheic keratosis) with squamous eddies. Am J Dermatopathol. 1983;5:427-445.
- Mehregan AH. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
- Brownstein MH. Trichilemmoma. benign follicular tumor or viral wart? Am J Dermatopathol. 1980;2:229-231.
- Brownstein MH. Multiple trichilemmomas in Cowden's syndrome. Arch Dermatol. 1979;115:111.
- Roson E, Gomez Centeno P, Sanchez Aguilar D, et al. Desmoplastic trichilemmoma arising within a nevus sebaceous. Am J Dermatopathol. 1998;20:495-497.
- Tellechea O, Reis JP, Baptista AP. Desmoplastic trichilemmoma. Am J Dermatopathol. 1992;14:107-114.
- Sharma R, Sirohi D, Sengupta P, et al. Desmoplastic trichilemmoma of the facial region mimicking invasive carcinoma. J Maxillofac Oral Surg. 2010;10:71-73.
- Wilson-Jones E. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208.
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumours: a clinicopathological study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574.
- Garg PK, Dangi A, Khurana N, et al. Malignant proliferating trichilemmal cyst: a case report with review of literature. Malaysian J Pathol. 2009;31:71-76.
- Herrero J, Monteagudo C, Ruiz A, et al. Malignant proliferating trichilemmal tumors: a histopathological and immunohistochemical study of three cases with DNA ploidy and morphometric evaluation. Histopathology. 1998;33:542-546.
- Haas N, Audring H, Sterry W. Carcinoma arising in a proliferating trichilemmal cyst expresses fetal and trichilemmal hair phenotype. Am J Dermatopathol. 2002;24:340-344.
- Rutty GN, Richman PI, Laing JH. Malignant change in trichilemmal cysts: a study of cell proliferation and DNA content. Histopathology. 1992;21:465-468.
- Brownstein MH, Arluk DJ. Proliferating trichilemmal cyst: a simulant of squamous cell carcinoma. Cancer. 1981;48:1207-1214.
- Misago N, Ackerman AB. Tricholemmal carcinoma? Dermatopathol Pract Concept. 1999;5:205-206.
- Misago N, Narisawa Y. Tricholemmal carcinoma in continuity with trichoblastoma within nevus sebaceous. Am J Dermatopathol. 2002;24:149-155.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Swanson PE, Marrogi AJ, Williams DJ, et al. Trichilemmal carcinoma: clinicopathologic study of 10 cases. J Cutan Pathol. 1992;19:100-109.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Julian CG, Bowers PW. A clinical review of 209 pilomatricomas. J Am Acad Dermatol. 1998;39:191-195.
- Marrogi AJ, Wick MR, Dehner LP. Pilomatrical neoplasms in children and young adults. Am J Dermatopathol. 1992;14:87-94.
- Berberian BJ, Colonna TM, Battaglia M, et al. Multiple pilomatricomas in association with myotonic dystrophy and a family history of melanoma. J Am Acad Dermatol. 1997;37:268-269.
- Cooper PH, Fechner RE. Pilomatricoma-like changes in the epidermal cysts of Gardner's syndrome. J Am Acad Dermatol. 1983;8:639-644.
- Kaddu S, Soyer HP, Cerroni L, et al. Clinical and histopathologic spectrum of pilomatricomas in adults. Int J Dermatol. 1994;33:705-708.
- Sano Y, Mihara M, Miyamoto T, et al. Simultaneous occurrence of calcification and amyloid deposit in pilomatricoma. Acta Derm Venereol. 1990;70:256-259.
- Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233.
- Mehregan AH. Inverted follicular keratosis. Arch Dermatol. 1964;89:117-123.
- Spielvogel RL, Austin C, Ackerman AB. Inverted follicular keratosis is not a specific keratosis but a verruca vulgaris (or seborrheic keratosis) with squamous eddies. Am J Dermatopathol. 1983;5:427-445.
- Mehregan AH. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
- Brownstein MH. Trichilemmoma. benign follicular tumor or viral wart? Am J Dermatopathol. 1980;2:229-231.
- Brownstein MH. Multiple trichilemmomas in Cowden's syndrome. Arch Dermatol. 1979;115:111.
- Roson E, Gomez Centeno P, Sanchez Aguilar D, et al. Desmoplastic trichilemmoma arising within a nevus sebaceous. Am J Dermatopathol. 1998;20:495-497.
- Tellechea O, Reis JP, Baptista AP. Desmoplastic trichilemmoma. Am J Dermatopathol. 1992;14:107-114.
- Sharma R, Sirohi D, Sengupta P, et al. Desmoplastic trichilemmoma of the facial region mimicking invasive carcinoma. J Maxillofac Oral Surg. 2010;10:71-73.
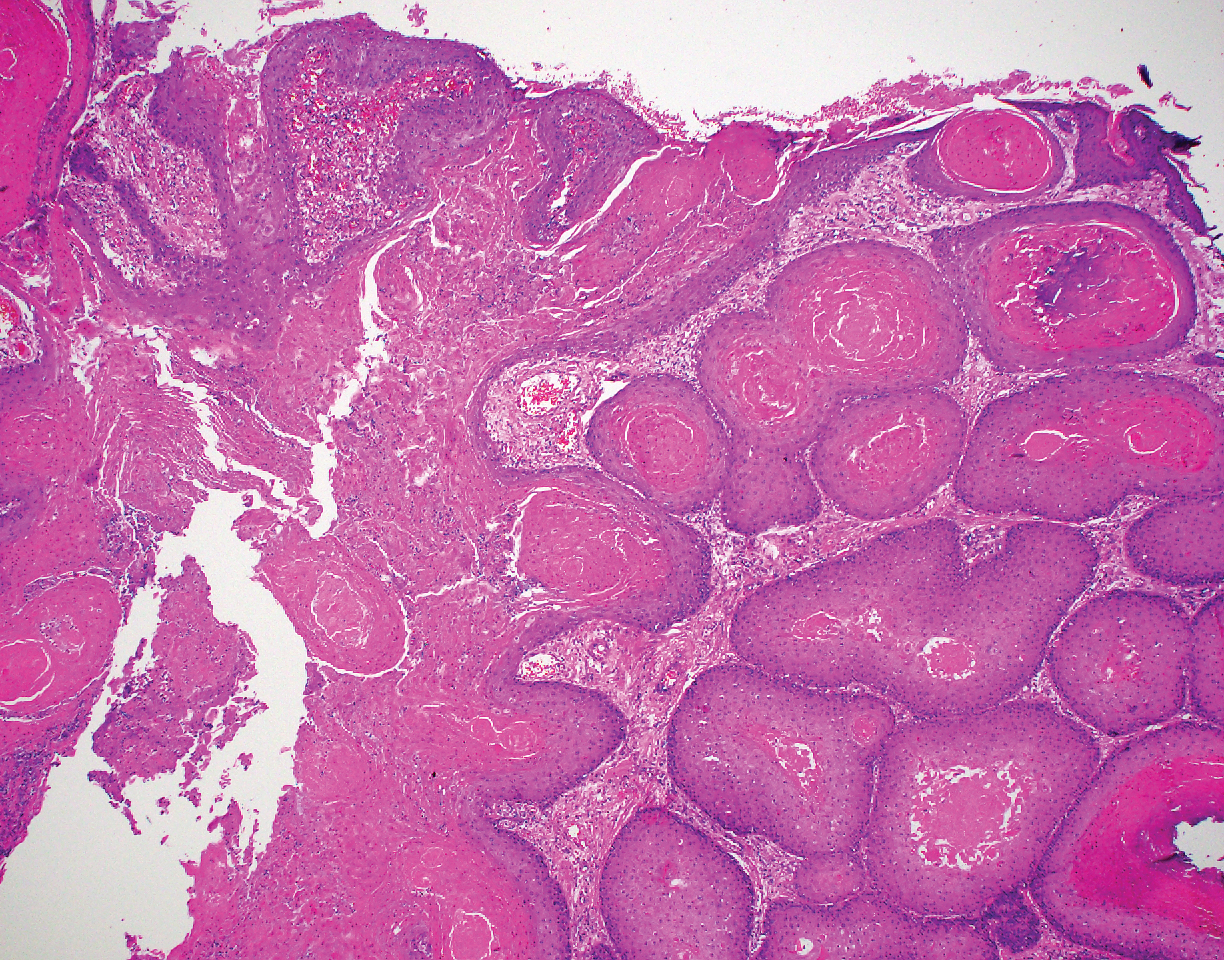
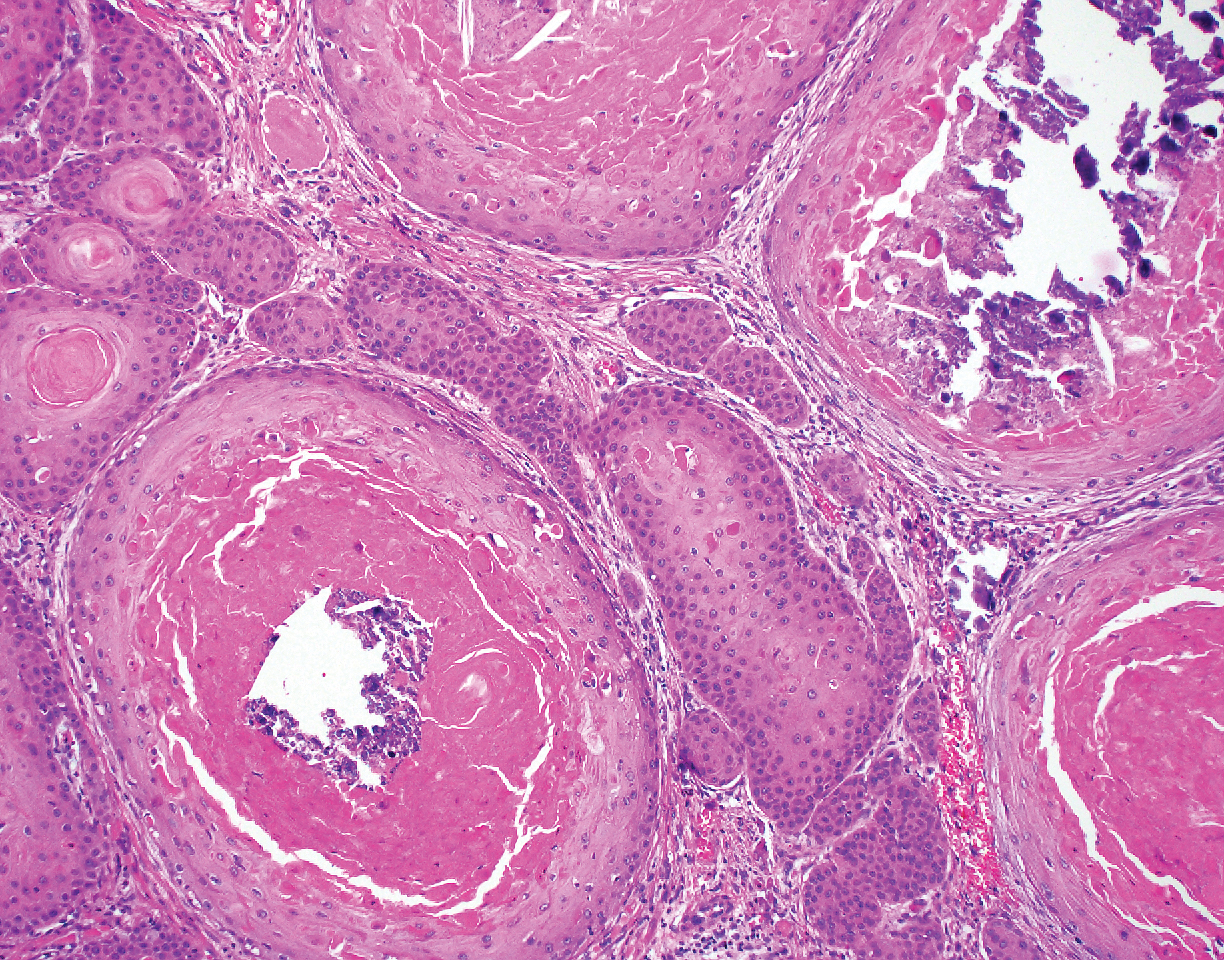
A 66-year-old woman presented to the dermatology clinic with a rapidly enlarging, draining lesion on the scalp. The lesion seemed to enlarge over the last 3 months from a lesion that had been there for years. Physical examination revealed a 2.2-cm ulcerated nodule on the right parietal scalp. A shave biopsy was obtained.
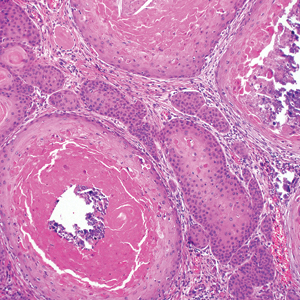
Tender Papules on the Bilateral Dorsal Hands
The Diagnosis: Interstitial Granulomatous Dermatitis
Interstitial granulomatous dermatitis (IGD) is rare, and the exact incidence is unknown, with only a few cases reported in the literature annually.1 Although IGD may arise in both children and adults, it occurs more commonly in adults, with an age of onset of 52 to 58.5 years. Interstitial granulomatous dermatitis also shows a female predominance.1
Interstitial granulomatous dermatitis may present as annular flesh-colored or erythematous to violaceous papules and plaques, or less commonly erythematous linear cordlike subcutaneous nodules (called the rope sign).1 Lesions often are asymptomatic but may be pruritic or tender. Interstitial granulomatous dermatitis has been associated with autoimmune conditions such as rheumatoid arthritis, systemic lupus erythematosus, and primary biliary cholangitis, and rarely malignancy.2 Interstitial granulomatous drug reactions can occur months to years after initiation of therapy with offending agents, and common causes include calcium channel blockers, statins, and tumor necrosis factor α inhibitors.3
Interstitial granulomatous dermatitis and palisaded neutrophilic and granulomatous dermatitis (PNGD) demonstrate overlapping clinical features and are thought to be part of the same spectrum of granulomatous dermatitis.4 Both IGD and PNGD may present with symmetric flesh-colored to erythematous papules or erythematous annular or linear plaques.5 Interstitial granulomatous dermatitis and PNGD may be differentiated through histopathologic examination.
Histopathology of IGD shows an interstitial infiltrate of epithelioid histiocytes in the dermis, often surrounding foci of degenerated collagen resembling palisading granulomas (quiz images).1 Perivascular and interstitial lymphocytic infiltrates also are present in most cases. Epidermal changes are minimal in IGD but can be associated with interstitial granulomatous drug reactions.1 There usually is no vasculitis, and mucin typically is absent, unlike granuloma annulare (GA).3,6 In comparison, histopathologic examination of PNGD shows basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris with focal areas of leukocytoclastic vasculitis and rare mucin.5
No specific treatment is recommended, and lesions may resolve without any therapy. Reported treatments include topical, intralesional, or systemic steroids; nonsteroidal anti-inflammatory drugs; methotrexate; hydroxychloroquine; and cyclosporine.6 Due to the strong association with systemic diseases, it is important to evaluate patients with IGD for autoimmune diseases and conduct age-appropriate cancer screening. Furthermore, a review of medications is warranted to assess the possibility of interstitial granulomatous drug reactions.6 In our patient, rheumatologic workup and age-appropriate cancer screenings were negative, and the rash spontaneously resolved without treatment.
Granuloma annulare presents with asymptomatic flesh-colored to erythematous papules and plaques in an annular configuration. In the localized variant of GA, plaques frequently localize to the distal extremities, especially the dorsal hands, as in our patient. Other variants include generalized GA, subcutaneous GA, and perforating GA. Mucin and a palisading or interstitial pattern of granulomatous inflammation are key features on histopathology in all subtypes of GA (Figure 1).7 Patch GA is a rare variant that presents with asymptomatic erythematous to brown patches, is associated with interstitial-type inflammation on histopathology, and can be difficult to distinguish from IGD.8 Granuloma annulare with interstitial inflammation on histology can be differentiated from IGD by the comparative lack of mucin in IGD.7
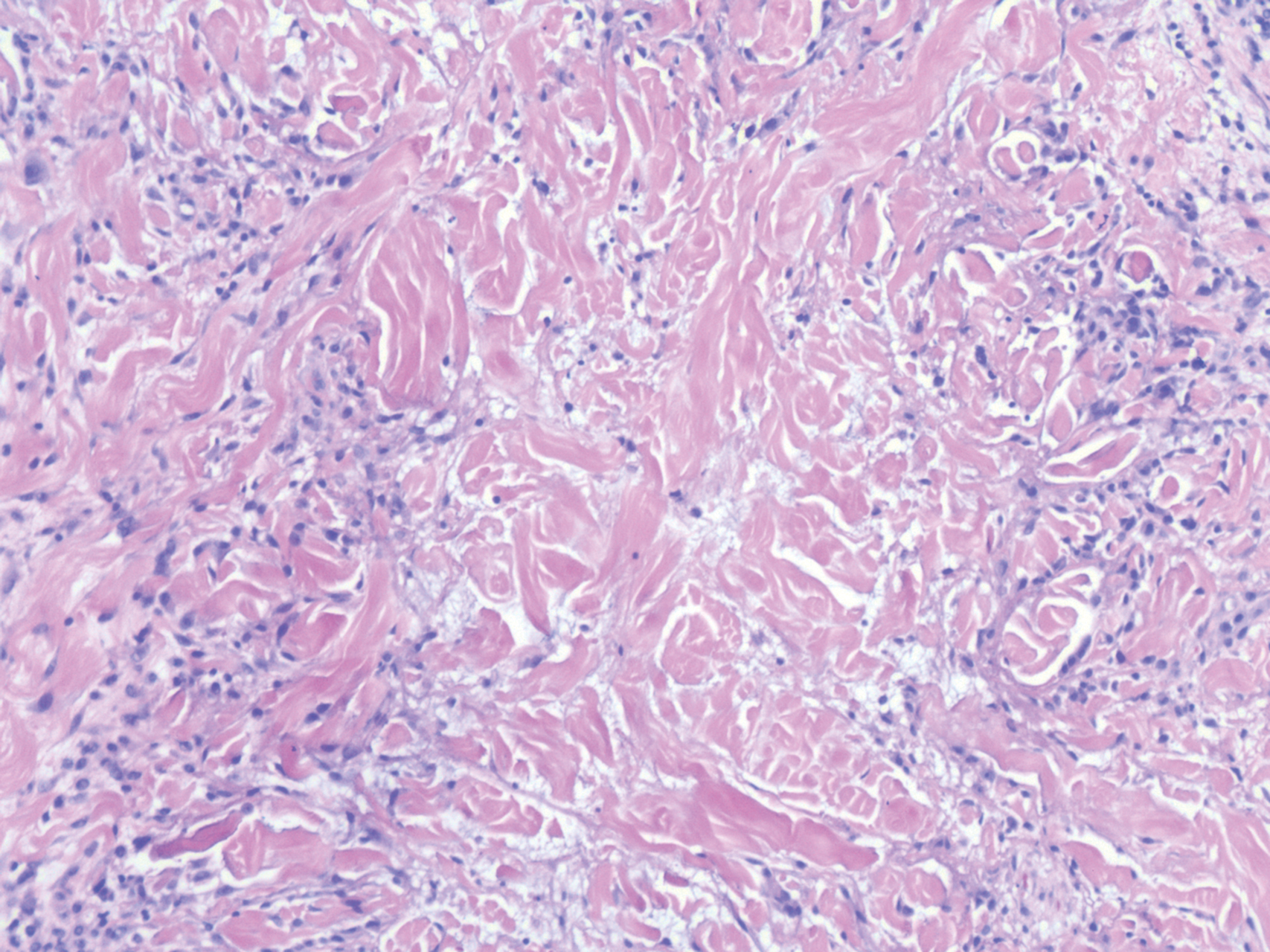
Sweet syndrome (SS) is characterized by sudden-onset, painful, erythematous plaques and/or nodules, commonly associated with fever and leukocytosis. Clinical variants of SS include pustular and bullous SS; giant cellulitis-like SS; necrotizing SS; and neutrophilic dermatosis of the dorsal hands presenting with hemorrhagic bullae, plaques, and pustules.7-9 Histopathologic examination shows dense nodular or perivascular neutrophilic infiltrate in the dermis without evidence of vasculitis (Figure 2).10 Histopathologic variants include histiocytoid, lymphocytic, subcutaneous, and cryptococcoid.9 The classic variant of SS has a bandlike, predominantly neutrophilic infiltrate with marked leukocytoclasia, which can be differentiated from the histiocytoid infiltrate of IGD.11 It has been shown that the infiltrate of the histiocytoid variant of SS is composed of myeloperoxidase-positive, immature myeloid cells rather than true histiocytes, and therefore can be differentiated from IGD.12 Lastly, all variants of SS have dermal edema, which typically is absent in IGD, and SS has no evidence of necrobiosis.
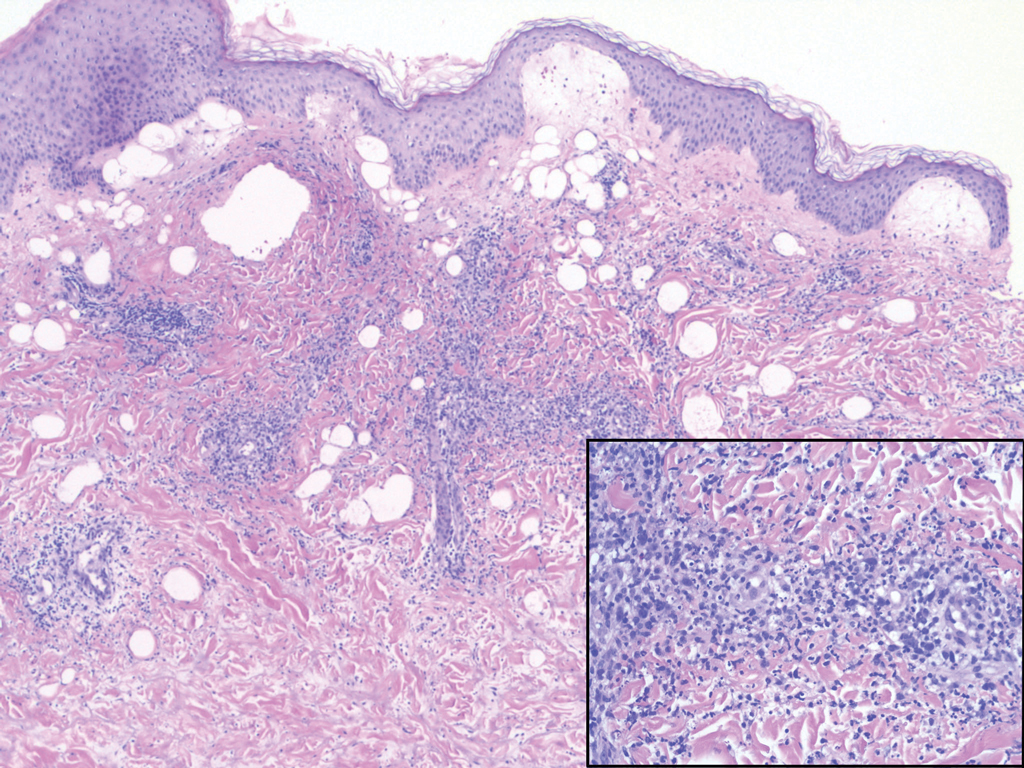
Erythema elevatum diutinum (EED) is a rare disease that presents with bilateral violaceous or erythematous to brown papules, plaques, or nodules. Lesions frequently localize to extensor surfaces, including the hands and fingers, and may be asymptomatic or associated with pruritus, burning, or tingling.13 Early EED lesions are characterized by leukocytoclastic vasculitis of the papillary and mid-dermal vessels with a perivascular neutrophilic infiltrate and perivascular fibrinoid necrosis. With older EED lesions, dermal and perivascular onion skin-like fibrosis become more prominent (Figure 3).14 The neutrophilic infiltrate, dermal fibrosis, and chronic vasculitic changes distinguish EED from IGD.
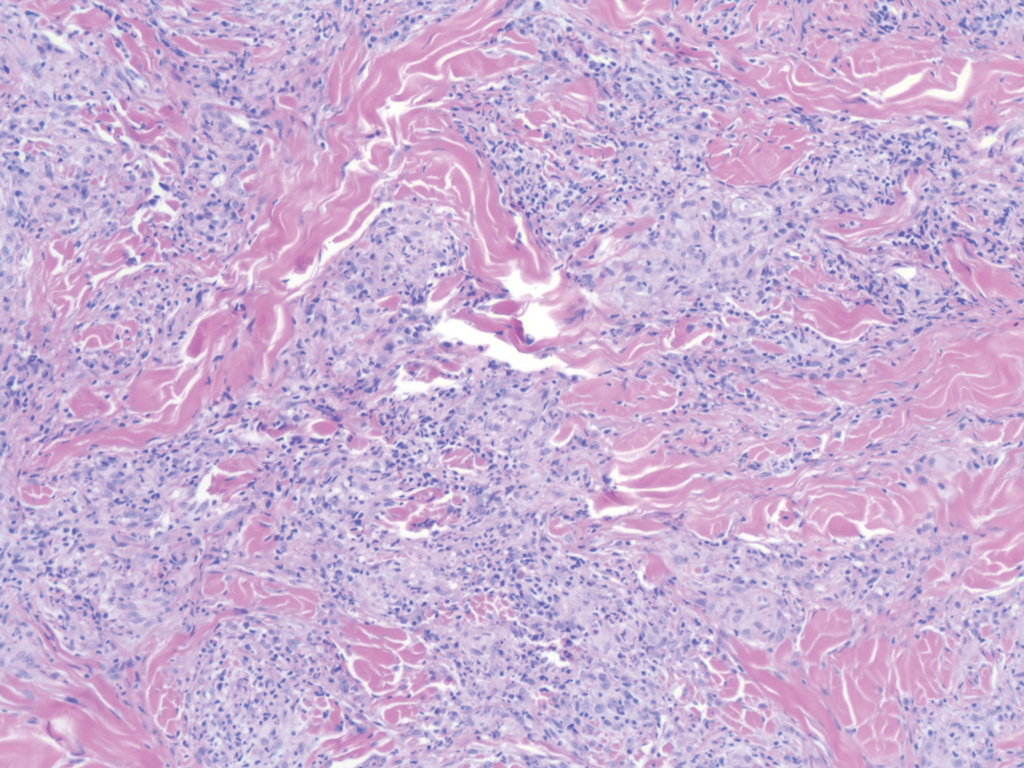
Necrobiosis lipoidica (NL) is a rare disease that presents with well-demarcated, yellow to red-brown papules and nodules most commonly localized to the bilateral lower extremities on the pretibial area. Papules and nodules evolve into plaques over time, and ulceration is common.15 On histopathology, NL primarily exhibits granulomatous inflammation with parallel palisading (Figure 4). The hallmark feature is necrobiosis--or degeneration--of collagen; the alternation of necrobiotic collagen and inflammatory infiltrate creates a layered cake-like appearance on low power.16 The clinical presentation as well as the dermal necrobiotic granuloma consisting of a large confluent area of necrobiosis centered in the superficial dermis and subcutaneous tissue of NL distinguishes it from the histiocytic infiltrate of IGD.
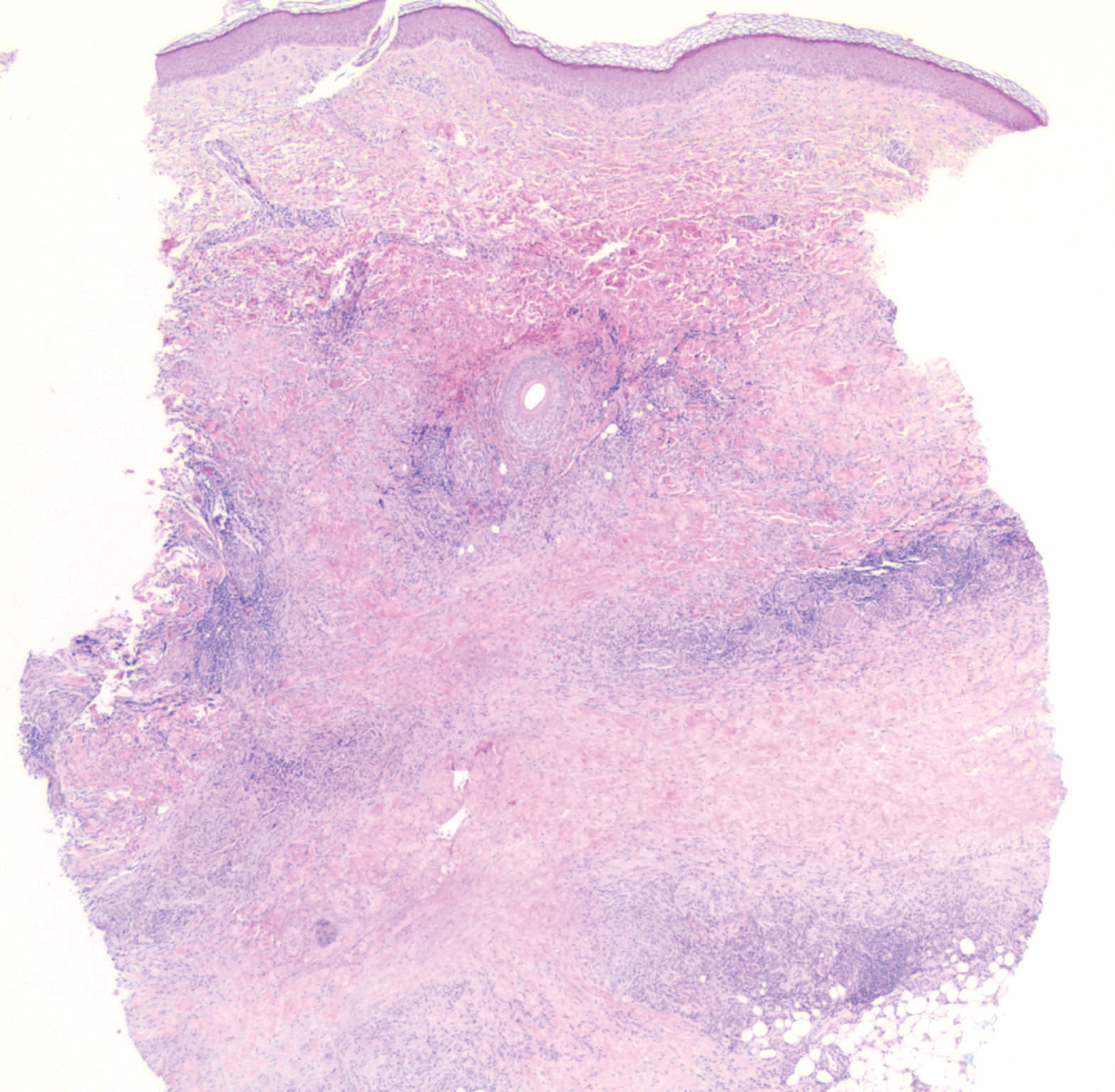
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Terziroli Beretta-Piccoli B, Mainetti C, Peeters MA, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1644-1663.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Huizenga T, Kado JA, Pellicane B, et al. Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis. Cutis. 2018;101:E19-E21.
- Rosenbach M, English JC 3rd. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Mutasim DF, Bridges AG. Patch granuloma annulare: clinicopathologic study of 6 patients. J Am Acad Dermatol. 2000;42:417-421.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Dabade TS, Davis MD. Diagnosis and treatment of the neutrophilic dermatoses (pyoderma gangrenosum, Sweet's syndrome). Dermatol Ther. 2011;24:273-284.
- Davis M, Moschella L. Neutrophilic dermatoses. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:2102-2112.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Sardiña LA, Jour G, Piliang MP, et al. Erythema elevatum diutinum a rare and poorly understood cutaneous vasculitis: a single institution experience. J Cutan Pathol. 2019;46:97-101.
- Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
- Sibbald C, Reid S, Alavi A. Necrobiosis lipoidica. Dermatol Clin. 2015;33:343-360.
The Diagnosis: Interstitial Granulomatous Dermatitis
Interstitial granulomatous dermatitis (IGD) is rare, and the exact incidence is unknown, with only a few cases reported in the literature annually.1 Although IGD may arise in both children and adults, it occurs more commonly in adults, with an age of onset of 52 to 58.5 years. Interstitial granulomatous dermatitis also shows a female predominance.1
Interstitial granulomatous dermatitis may present as annular flesh-colored or erythematous to violaceous papules and plaques, or less commonly erythematous linear cordlike subcutaneous nodules (called the rope sign).1 Lesions often are asymptomatic but may be pruritic or tender. Interstitial granulomatous dermatitis has been associated with autoimmune conditions such as rheumatoid arthritis, systemic lupus erythematosus, and primary biliary cholangitis, and rarely malignancy.2 Interstitial granulomatous drug reactions can occur months to years after initiation of therapy with offending agents, and common causes include calcium channel blockers, statins, and tumor necrosis factor α inhibitors.3
Interstitial granulomatous dermatitis and palisaded neutrophilic and granulomatous dermatitis (PNGD) demonstrate overlapping clinical features and are thought to be part of the same spectrum of granulomatous dermatitis.4 Both IGD and PNGD may present with symmetric flesh-colored to erythematous papules or erythematous annular or linear plaques.5 Interstitial granulomatous dermatitis and PNGD may be differentiated through histopathologic examination.
Histopathology of IGD shows an interstitial infiltrate of epithelioid histiocytes in the dermis, often surrounding foci of degenerated collagen resembling palisading granulomas (quiz images).1 Perivascular and interstitial lymphocytic infiltrates also are present in most cases. Epidermal changes are minimal in IGD but can be associated with interstitial granulomatous drug reactions.1 There usually is no vasculitis, and mucin typically is absent, unlike granuloma annulare (GA).3,6 In comparison, histopathologic examination of PNGD shows basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris with focal areas of leukocytoclastic vasculitis and rare mucin.5
No specific treatment is recommended, and lesions may resolve without any therapy. Reported treatments include topical, intralesional, or systemic steroids; nonsteroidal anti-inflammatory drugs; methotrexate; hydroxychloroquine; and cyclosporine.6 Due to the strong association with systemic diseases, it is important to evaluate patients with IGD for autoimmune diseases and conduct age-appropriate cancer screening. Furthermore, a review of medications is warranted to assess the possibility of interstitial granulomatous drug reactions.6 In our patient, rheumatologic workup and age-appropriate cancer screenings were negative, and the rash spontaneously resolved without treatment.
Granuloma annulare presents with asymptomatic flesh-colored to erythematous papules and plaques in an annular configuration. In the localized variant of GA, plaques frequently localize to the distal extremities, especially the dorsal hands, as in our patient. Other variants include generalized GA, subcutaneous GA, and perforating GA. Mucin and a palisading or interstitial pattern of granulomatous inflammation are key features on histopathology in all subtypes of GA (Figure 1).7 Patch GA is a rare variant that presents with asymptomatic erythematous to brown patches, is associated with interstitial-type inflammation on histopathology, and can be difficult to distinguish from IGD.8 Granuloma annulare with interstitial inflammation on histology can be differentiated from IGD by the comparative lack of mucin in IGD.7
Sweet syndrome (SS) is characterized by sudden-onset, painful, erythematous plaques and/or nodules, commonly associated with fever and leukocytosis. Clinical variants of SS include pustular and bullous SS; giant cellulitis-like SS; necrotizing SS; and neutrophilic dermatosis of the dorsal hands presenting with hemorrhagic bullae, plaques, and pustules.7-9 Histopathologic examination shows dense nodular or perivascular neutrophilic infiltrate in the dermis without evidence of vasculitis (Figure 2).10 Histopathologic variants include histiocytoid, lymphocytic, subcutaneous, and cryptococcoid.9 The classic variant of SS has a bandlike, predominantly neutrophilic infiltrate with marked leukocytoclasia, which can be differentiated from the histiocytoid infiltrate of IGD.11 It has been shown that the infiltrate of the histiocytoid variant of SS is composed of myeloperoxidase-positive, immature myeloid cells rather than true histiocytes, and therefore can be differentiated from IGD.12 Lastly, all variants of SS have dermal edema, which typically is absent in IGD, and SS has no evidence of necrobiosis.
Erythema elevatum diutinum (EED) is a rare disease that presents with bilateral violaceous or erythematous to brown papules, plaques, or nodules. Lesions frequently localize to extensor surfaces, including the hands and fingers, and may be asymptomatic or associated with pruritus, burning, or tingling.13 Early EED lesions are characterized by leukocytoclastic vasculitis of the papillary and mid-dermal vessels with a perivascular neutrophilic infiltrate and perivascular fibrinoid necrosis. With older EED lesions, dermal and perivascular onion skin-like fibrosis become more prominent (Figure 3).14 The neutrophilic infiltrate, dermal fibrosis, and chronic vasculitic changes distinguish EED from IGD.
Necrobiosis lipoidica (NL) is a rare disease that presents with well-demarcated, yellow to red-brown papules and nodules most commonly localized to the bilateral lower extremities on the pretibial area. Papules and nodules evolve into plaques over time, and ulceration is common.15 On histopathology, NL primarily exhibits granulomatous inflammation with parallel palisading (Figure 4). The hallmark feature is necrobiosis--or degeneration--of collagen; the alternation of necrobiotic collagen and inflammatory infiltrate creates a layered cake-like appearance on low power.16 The clinical presentation as well as the dermal necrobiotic granuloma consisting of a large confluent area of necrobiosis centered in the superficial dermis and subcutaneous tissue of NL distinguishes it from the histiocytic infiltrate of IGD.
The Diagnosis: Interstitial Granulomatous Dermatitis
Interstitial granulomatous dermatitis (IGD) is rare, and the exact incidence is unknown, with only a few cases reported in the literature annually.1 Although IGD may arise in both children and adults, it occurs more commonly in adults, with an age of onset of 52 to 58.5 years. Interstitial granulomatous dermatitis also shows a female predominance.1
Interstitial granulomatous dermatitis may present as annular flesh-colored or erythematous to violaceous papules and plaques, or less commonly erythematous linear cordlike subcutaneous nodules (called the rope sign).1 Lesions often are asymptomatic but may be pruritic or tender. Interstitial granulomatous dermatitis has been associated with autoimmune conditions such as rheumatoid arthritis, systemic lupus erythematosus, and primary biliary cholangitis, and rarely malignancy.2 Interstitial granulomatous drug reactions can occur months to years after initiation of therapy with offending agents, and common causes include calcium channel blockers, statins, and tumor necrosis factor α inhibitors.3
Interstitial granulomatous dermatitis and palisaded neutrophilic and granulomatous dermatitis (PNGD) demonstrate overlapping clinical features and are thought to be part of the same spectrum of granulomatous dermatitis.4 Both IGD and PNGD may present with symmetric flesh-colored to erythematous papules or erythematous annular or linear plaques.5 Interstitial granulomatous dermatitis and PNGD may be differentiated through histopathologic examination.
Histopathology of IGD shows an interstitial infiltrate of epithelioid histiocytes in the dermis, often surrounding foci of degenerated collagen resembling palisading granulomas (quiz images).1 Perivascular and interstitial lymphocytic infiltrates also are present in most cases. Epidermal changes are minimal in IGD but can be associated with interstitial granulomatous drug reactions.1 There usually is no vasculitis, and mucin typically is absent, unlike granuloma annulare (GA).3,6 In comparison, histopathologic examination of PNGD shows basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris with focal areas of leukocytoclastic vasculitis and rare mucin.5
No specific treatment is recommended, and lesions may resolve without any therapy. Reported treatments include topical, intralesional, or systemic steroids; nonsteroidal anti-inflammatory drugs; methotrexate; hydroxychloroquine; and cyclosporine.6 Due to the strong association with systemic diseases, it is important to evaluate patients with IGD for autoimmune diseases and conduct age-appropriate cancer screening. Furthermore, a review of medications is warranted to assess the possibility of interstitial granulomatous drug reactions.6 In our patient, rheumatologic workup and age-appropriate cancer screenings were negative, and the rash spontaneously resolved without treatment.
Granuloma annulare presents with asymptomatic flesh-colored to erythematous papules and plaques in an annular configuration. In the localized variant of GA, plaques frequently localize to the distal extremities, especially the dorsal hands, as in our patient. Other variants include generalized GA, subcutaneous GA, and perforating GA. Mucin and a palisading or interstitial pattern of granulomatous inflammation are key features on histopathology in all subtypes of GA (Figure 1).7 Patch GA is a rare variant that presents with asymptomatic erythematous to brown patches, is associated with interstitial-type inflammation on histopathology, and can be difficult to distinguish from IGD.8 Granuloma annulare with interstitial inflammation on histology can be differentiated from IGD by the comparative lack of mucin in IGD.7
Sweet syndrome (SS) is characterized by sudden-onset, painful, erythematous plaques and/or nodules, commonly associated with fever and leukocytosis. Clinical variants of SS include pustular and bullous SS; giant cellulitis-like SS; necrotizing SS; and neutrophilic dermatosis of the dorsal hands presenting with hemorrhagic bullae, plaques, and pustules.7-9 Histopathologic examination shows dense nodular or perivascular neutrophilic infiltrate in the dermis without evidence of vasculitis (Figure 2).10 Histopathologic variants include histiocytoid, lymphocytic, subcutaneous, and cryptococcoid.9 The classic variant of SS has a bandlike, predominantly neutrophilic infiltrate with marked leukocytoclasia, which can be differentiated from the histiocytoid infiltrate of IGD.11 It has been shown that the infiltrate of the histiocytoid variant of SS is composed of myeloperoxidase-positive, immature myeloid cells rather than true histiocytes, and therefore can be differentiated from IGD.12 Lastly, all variants of SS have dermal edema, which typically is absent in IGD, and SS has no evidence of necrobiosis.
Erythema elevatum diutinum (EED) is a rare disease that presents with bilateral violaceous or erythematous to brown papules, plaques, or nodules. Lesions frequently localize to extensor surfaces, including the hands and fingers, and may be asymptomatic or associated with pruritus, burning, or tingling.13 Early EED lesions are characterized by leukocytoclastic vasculitis of the papillary and mid-dermal vessels with a perivascular neutrophilic infiltrate and perivascular fibrinoid necrosis. With older EED lesions, dermal and perivascular onion skin-like fibrosis become more prominent (Figure 3).14 The neutrophilic infiltrate, dermal fibrosis, and chronic vasculitic changes distinguish EED from IGD.
Necrobiosis lipoidica (NL) is a rare disease that presents with well-demarcated, yellow to red-brown papules and nodules most commonly localized to the bilateral lower extremities on the pretibial area. Papules and nodules evolve into plaques over time, and ulceration is common.15 On histopathology, NL primarily exhibits granulomatous inflammation with parallel palisading (Figure 4). The hallmark feature is necrobiosis--or degeneration--of collagen; the alternation of necrobiotic collagen and inflammatory infiltrate creates a layered cake-like appearance on low power.16 The clinical presentation as well as the dermal necrobiotic granuloma consisting of a large confluent area of necrobiosis centered in the superficial dermis and subcutaneous tissue of NL distinguishes it from the histiocytic infiltrate of IGD.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Terziroli Beretta-Piccoli B, Mainetti C, Peeters MA, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1644-1663.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Huizenga T, Kado JA, Pellicane B, et al. Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis. Cutis. 2018;101:E19-E21.
- Rosenbach M, English JC 3rd. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Mutasim DF, Bridges AG. Patch granuloma annulare: clinicopathologic study of 6 patients. J Am Acad Dermatol. 2000;42:417-421.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Dabade TS, Davis MD. Diagnosis and treatment of the neutrophilic dermatoses (pyoderma gangrenosum, Sweet's syndrome). Dermatol Ther. 2011;24:273-284.
- Davis M, Moschella L. Neutrophilic dermatoses. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:2102-2112.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Sardiña LA, Jour G, Piliang MP, et al. Erythema elevatum diutinum a rare and poorly understood cutaneous vasculitis: a single institution experience. J Cutan Pathol. 2019;46:97-101.
- Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
- Sibbald C, Reid S, Alavi A. Necrobiosis lipoidica. Dermatol Clin. 2015;33:343-360.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Terziroli Beretta-Piccoli B, Mainetti C, Peeters MA, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1644-1663.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Huizenga T, Kado JA, Pellicane B, et al. Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis. Cutis. 2018;101:E19-E21.
- Rosenbach M, English JC 3rd. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Mutasim DF, Bridges AG. Patch granuloma annulare: clinicopathologic study of 6 patients. J Am Acad Dermatol. 2000;42:417-421.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Dabade TS, Davis MD. Diagnosis and treatment of the neutrophilic dermatoses (pyoderma gangrenosum, Sweet's syndrome). Dermatol Ther. 2011;24:273-284.
- Davis M, Moschella L. Neutrophilic dermatoses. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:2102-2112.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Sardiña LA, Jour G, Piliang MP, et al. Erythema elevatum diutinum a rare and poorly understood cutaneous vasculitis: a single institution experience. J Cutan Pathol. 2019;46:97-101.
- Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
- Sibbald C, Reid S, Alavi A. Necrobiosis lipoidica. Dermatol Clin. 2015;33:343-360.
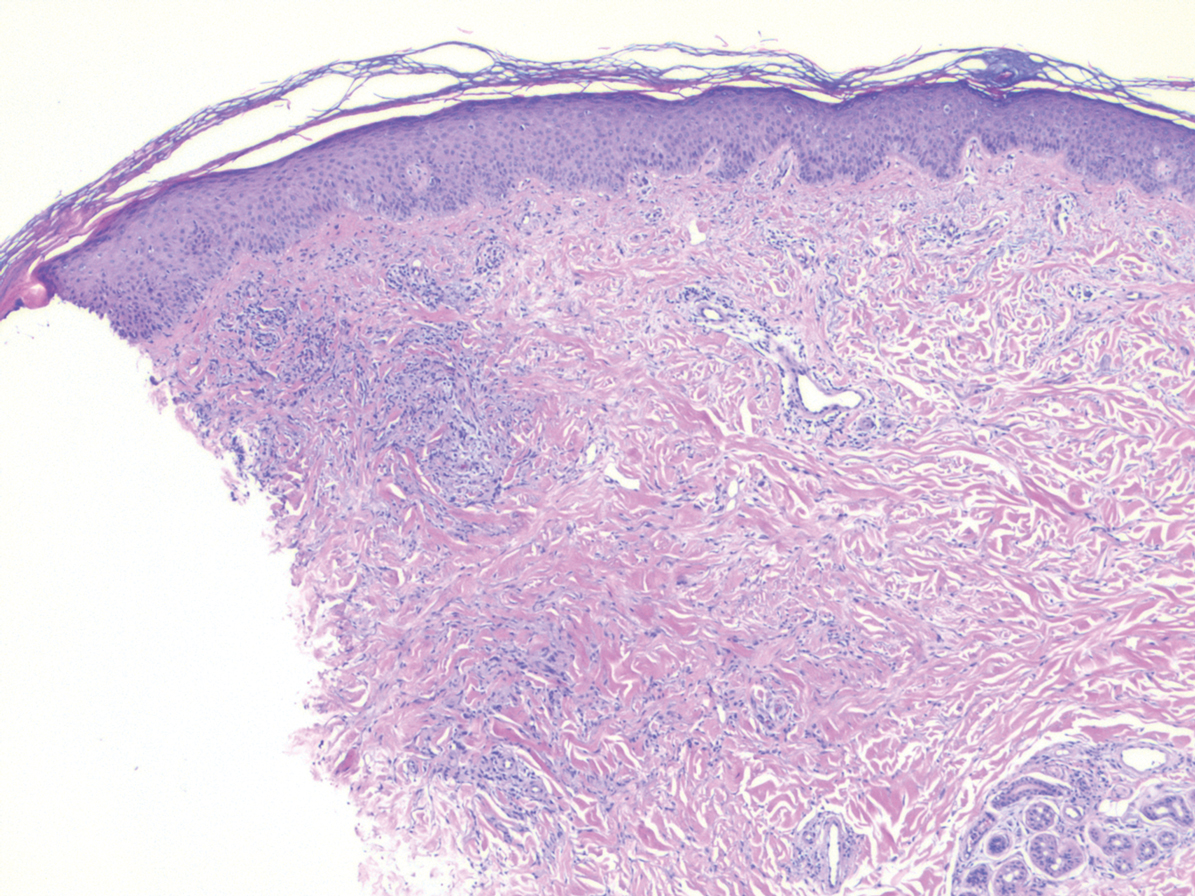
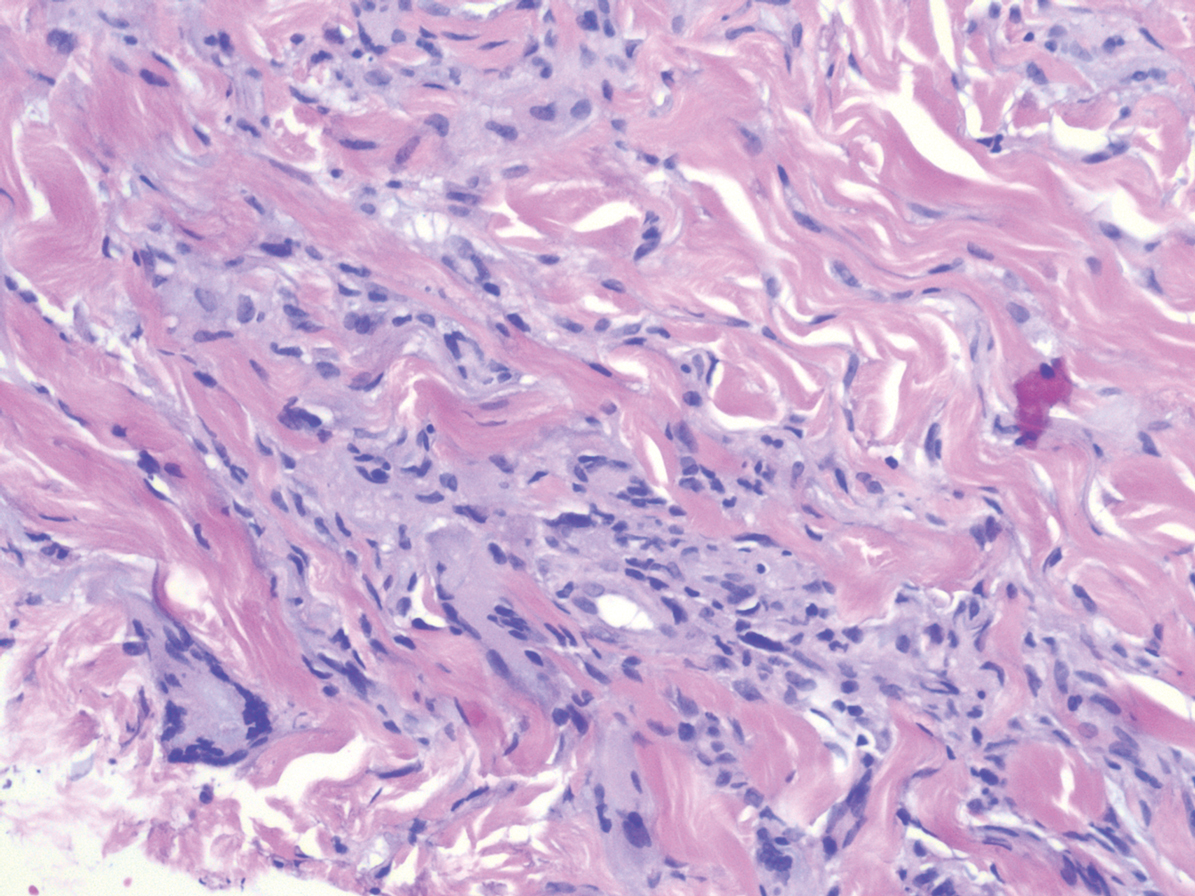
A 58-year-old woman with a medical history of asthma, hypertension, hypothyroidism, and hyperlipidemia presented with a painful rash of 10 days' duration. The rash was associated with fever at home (temperature, 38.5.2 °C), and a review of systems was positive for joint pain. Physical examination revealed numerous 8- to 10-mm, erythematous, discus-shaped papules on the bilateral dorsal hands, bilateral palms, right knee, and right dorsal foot with slight tenderness to palpation. A papule on the right dorsal hand was biopsied.
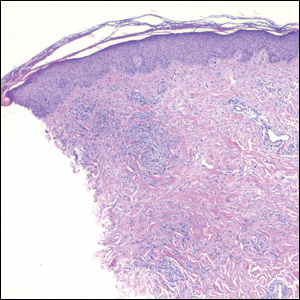
Solitary Papule on the Nose
The Diagnosis: Sclerosing Perineurioma
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
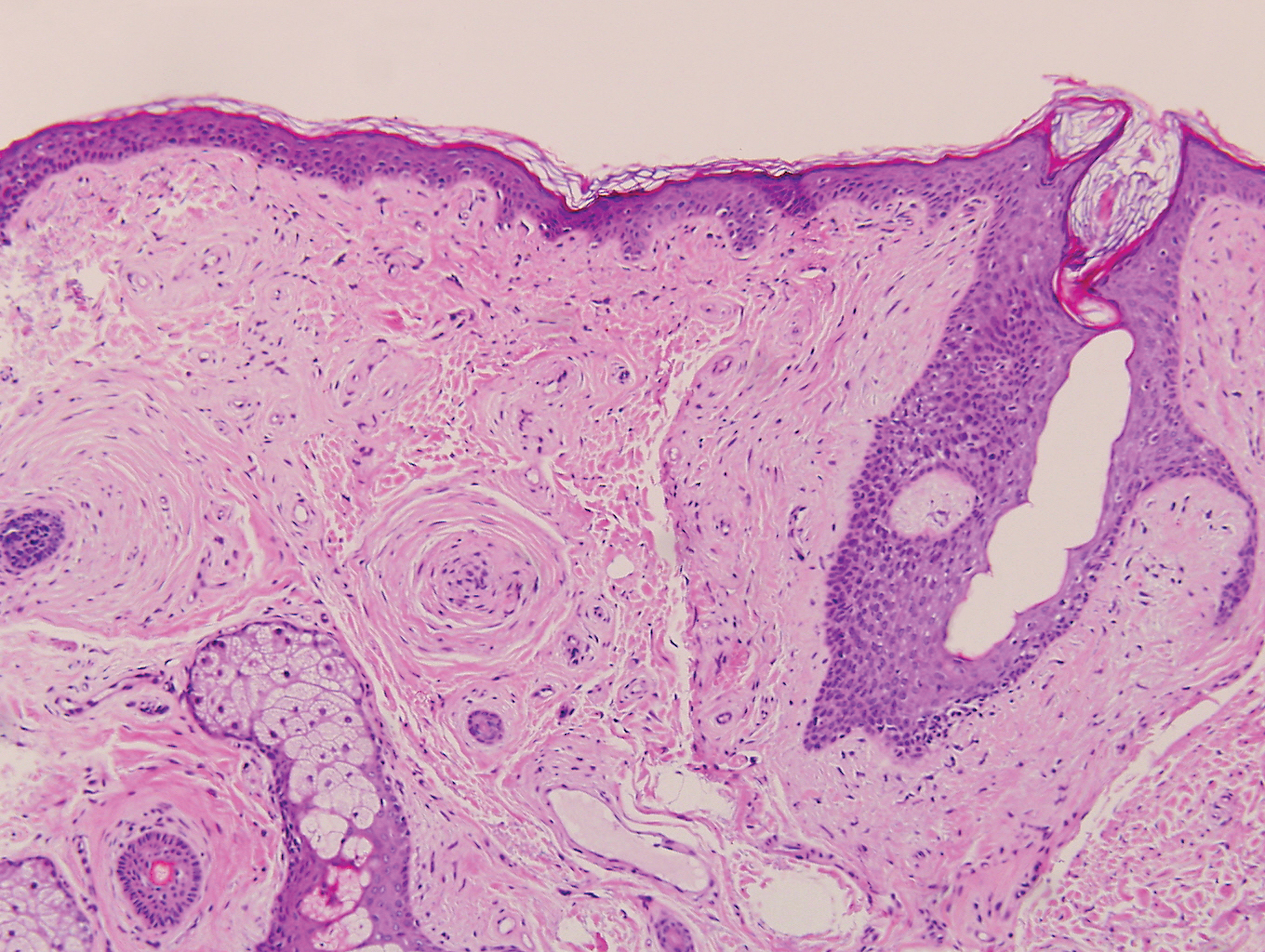
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
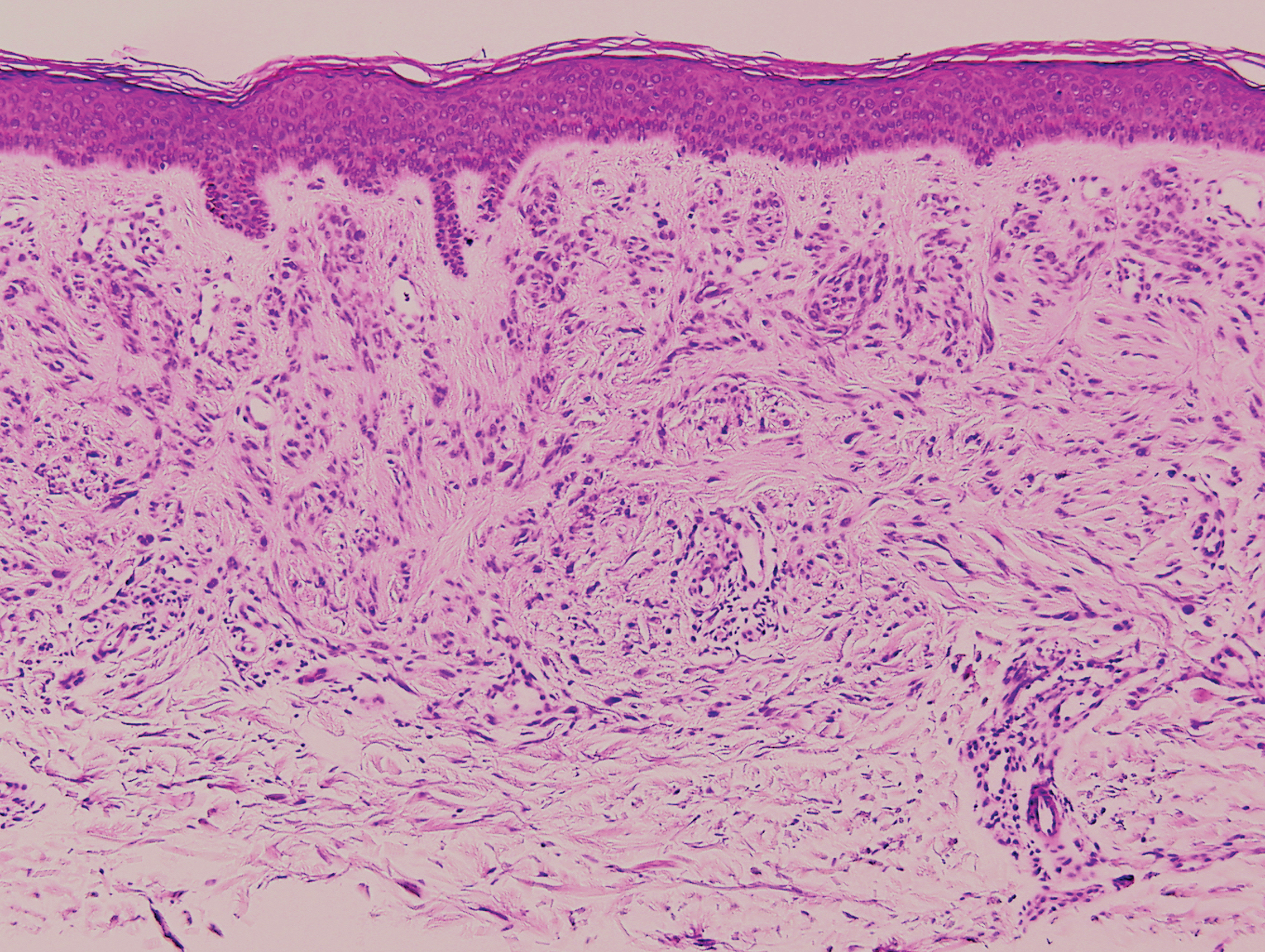
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
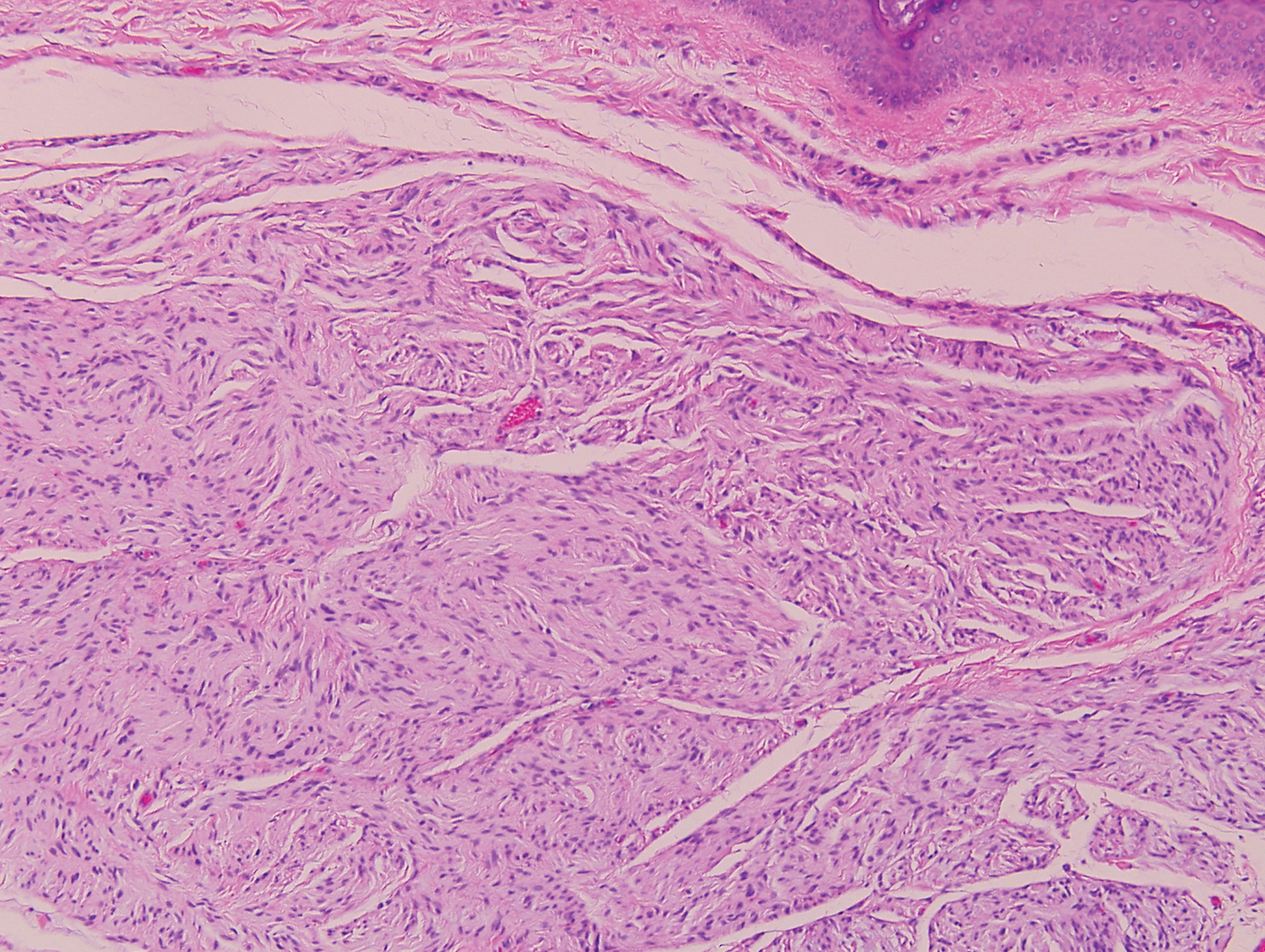
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
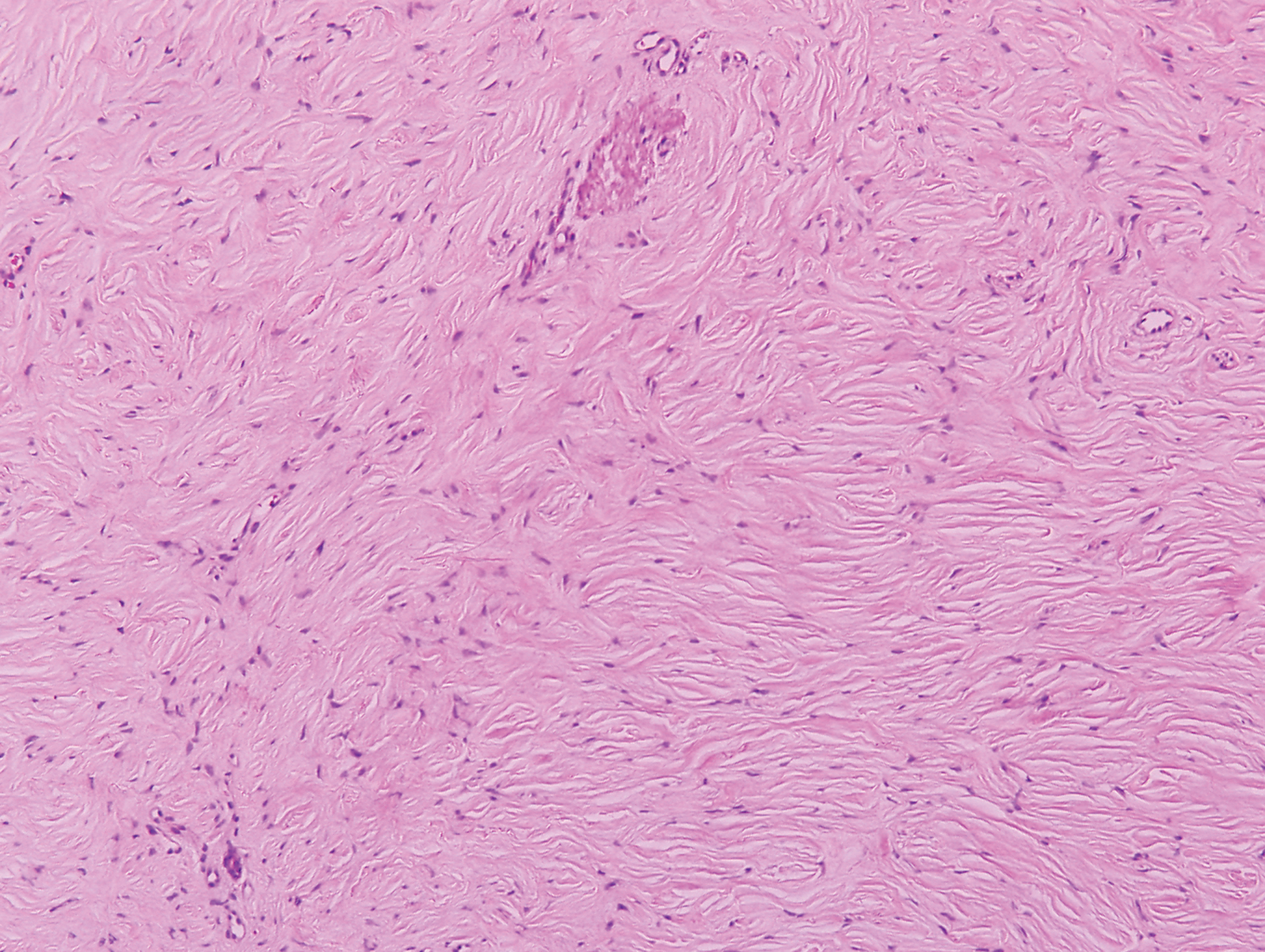
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
The Diagnosis: Sclerosing Perineurioma
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
The Diagnosis: Sclerosing Perineurioma
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
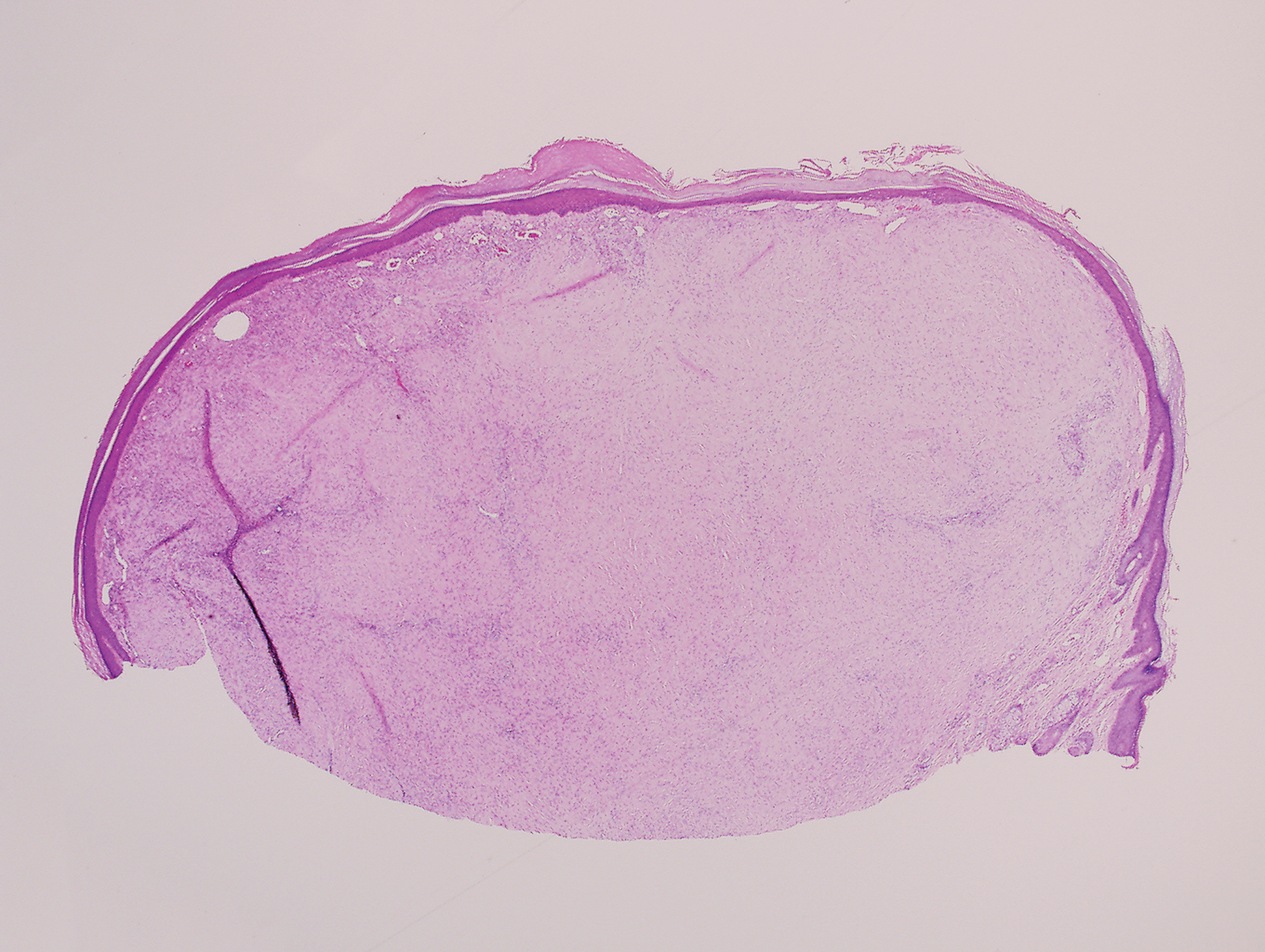
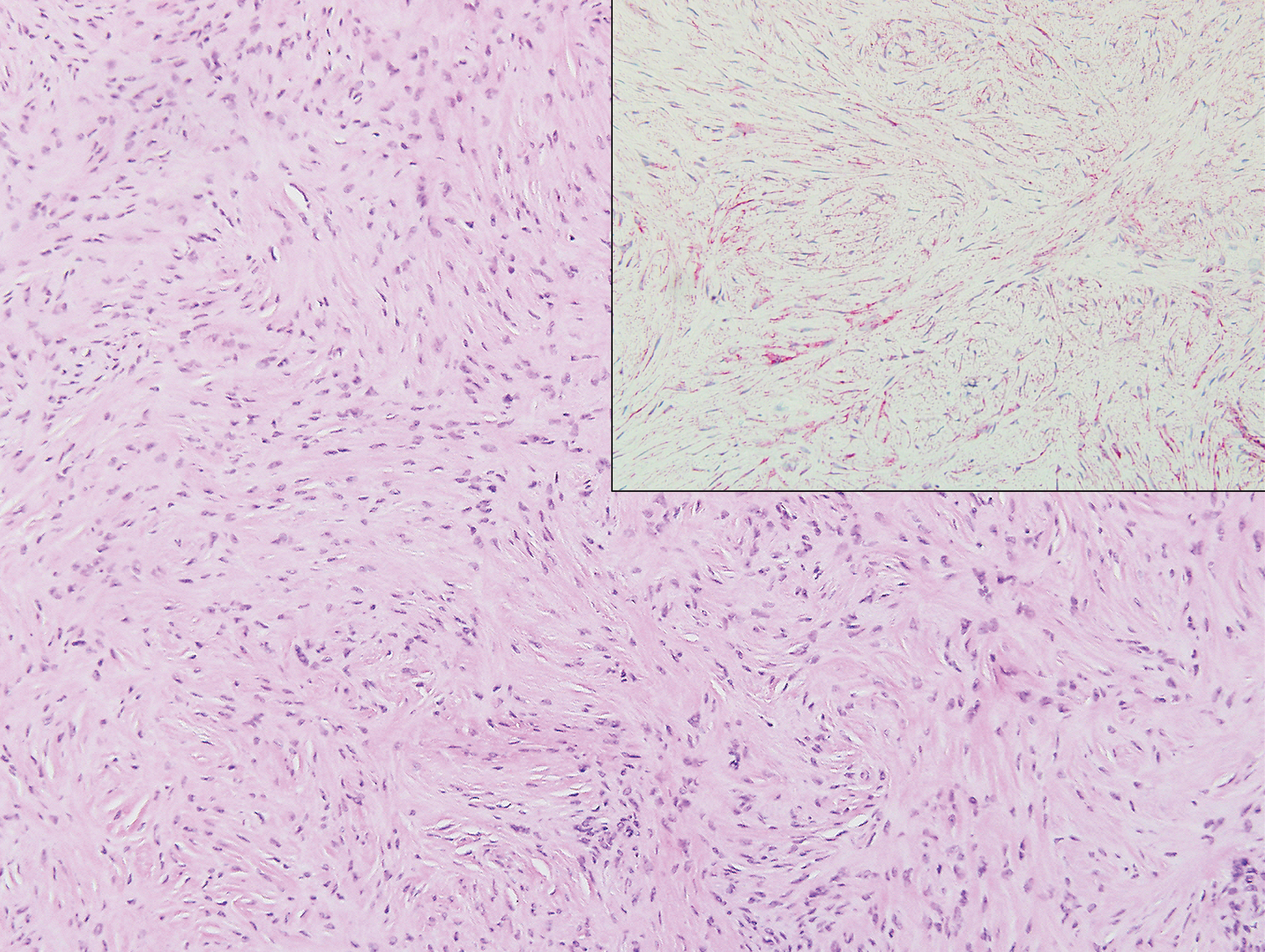
A 25-year-old man presented with a flesh-colored papule on the left side of the nose of 2 years' duration.
Erythematous Papules on the Scrotum, Trunk, and Extremities
The Diagnosis: Lichenoid and Granulomatous Dermatitis in the Setting of Secondary Syphilis
Syphilis, an infectious disease that has risen in incidence and is most commonly reported in men who have sex with men, involves a vast array of clinical and histologic presentations.1 Clinically, secondary syphilis involves an erythematous maculopapular eruption on the face, trunk, palms, soles, or genital area.2 The characteristic histologic features for secondary syphilis include endothelial swelling, interstitial inflammatory array, irregular acanthosis, elongated rete ridges, and vacuolar interface dermatitis with lymphocytes and plasma cells.1 Syphilitic infection has been associated with lichenoid and granulomatous dermatitis, which is an inflammatory skin disease described by Magro and Crowson.3 Lichenoid and granulomatous dermatitis has been linked to various systemic disorders, including chronic hepatitis C, Crohn disease, rheumatoid arthritis, endocrinopathy, subacute cutaneous lupus erythematosus, secondary syphilis, prior herpes infection, tuberculoid leprosy, mycobacterial infection, and human immunodeficiency virus infection.3-7 For this patient, given histopathology findings, clinical presentation, and positive rapid plasma reagin serologies, a diagnosis of lichenoid and granulomatous dermatitis in the setting of a secondary syphilis infection was established. A comprehensive investigation should be conducted to consider secondary syphilis or other systemic diseases in patients with a histologic finding of lichenoid and granulomatous dermatitis.
Histologically, lichenoid and granulomatous dermatitis cases show a bandlike infiltrate of lymphocytes with neighboring histiocytes along the dermoepidermal junction, accompanied by epithelial changes of dyskeratosis, vasculopathy, and colloid body formation, in addition to a dermal histiocytic component.3 Our patient's biopsy showed a lichenoid reaction pattern with vacuolar interface changes, dyskeratosis, plump endothelial cells, and small collections of plasma cells. Additionally, there was a granulomatous component in the dermis with histiocytes admixed with lymphocytes and plasma cells. The presence of spirochetes was confirmed with antitreponemal immunohistochemical stain (Figure 1). Quantitative rapid plasma reagin was 1:64 (reference range, <1:1) and Treponema pallidum antibody was reactive.
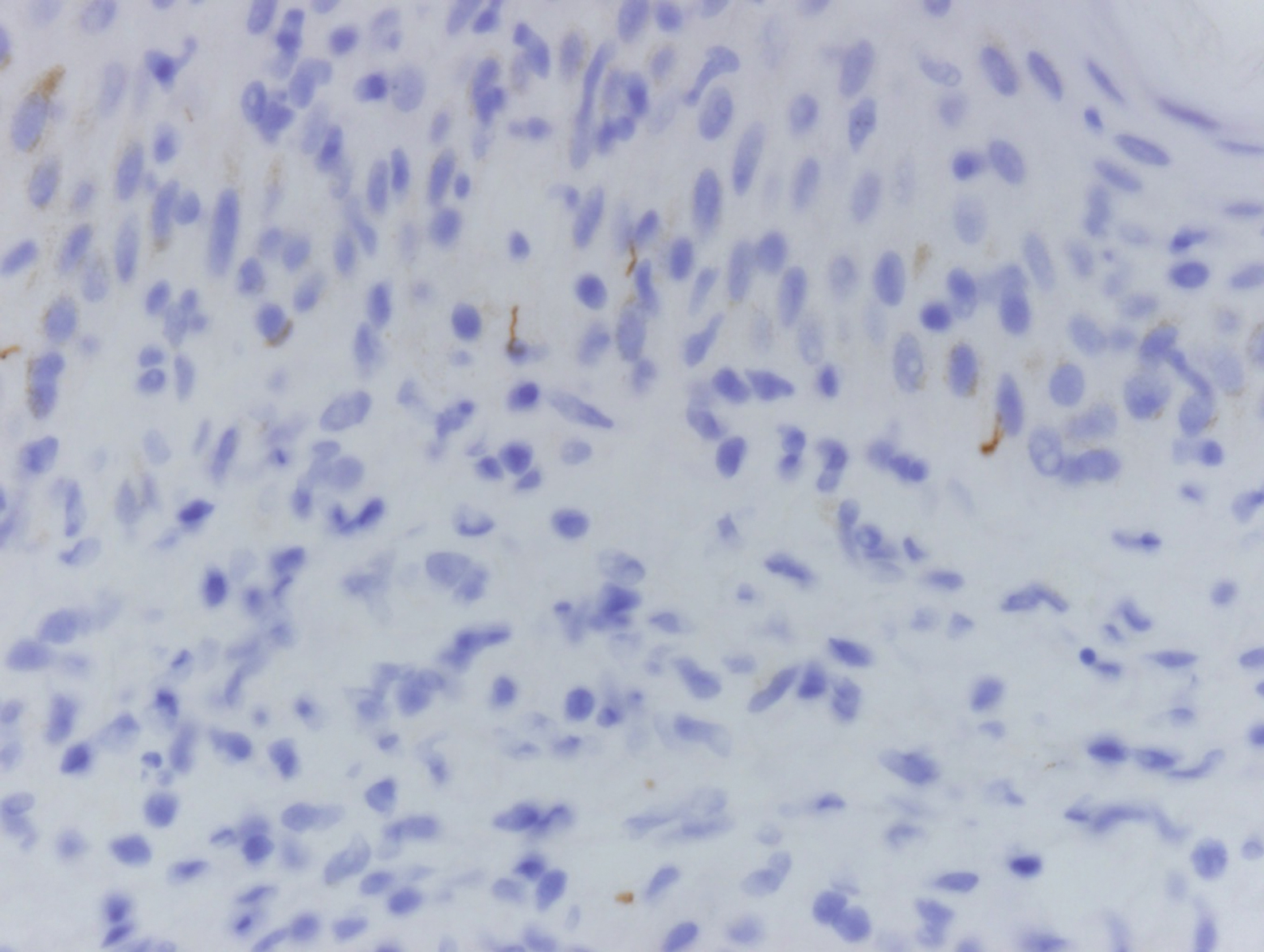
Interstitial granulomatous dermatitis has a variable clinical presentation, often with red-purple annular plaques, hyperpigmented papules, and nodules frequently in a linear arrangement and predominantly on the trunk, thighs, groin, or buttocks.8,9 On histopathology, there are histiocytes in the reticular dermis and/or a macrophage infiltrate in the mid to deep dermis with collections of degenerated collagen (Figure 2).8,10 An interstitial infiltrate of eosinophils and neutrophils also may be appreciated, but mucin generally is absent.8,11 This condition often coexists with rheumatic and systemic autoimmune diseases.8-10
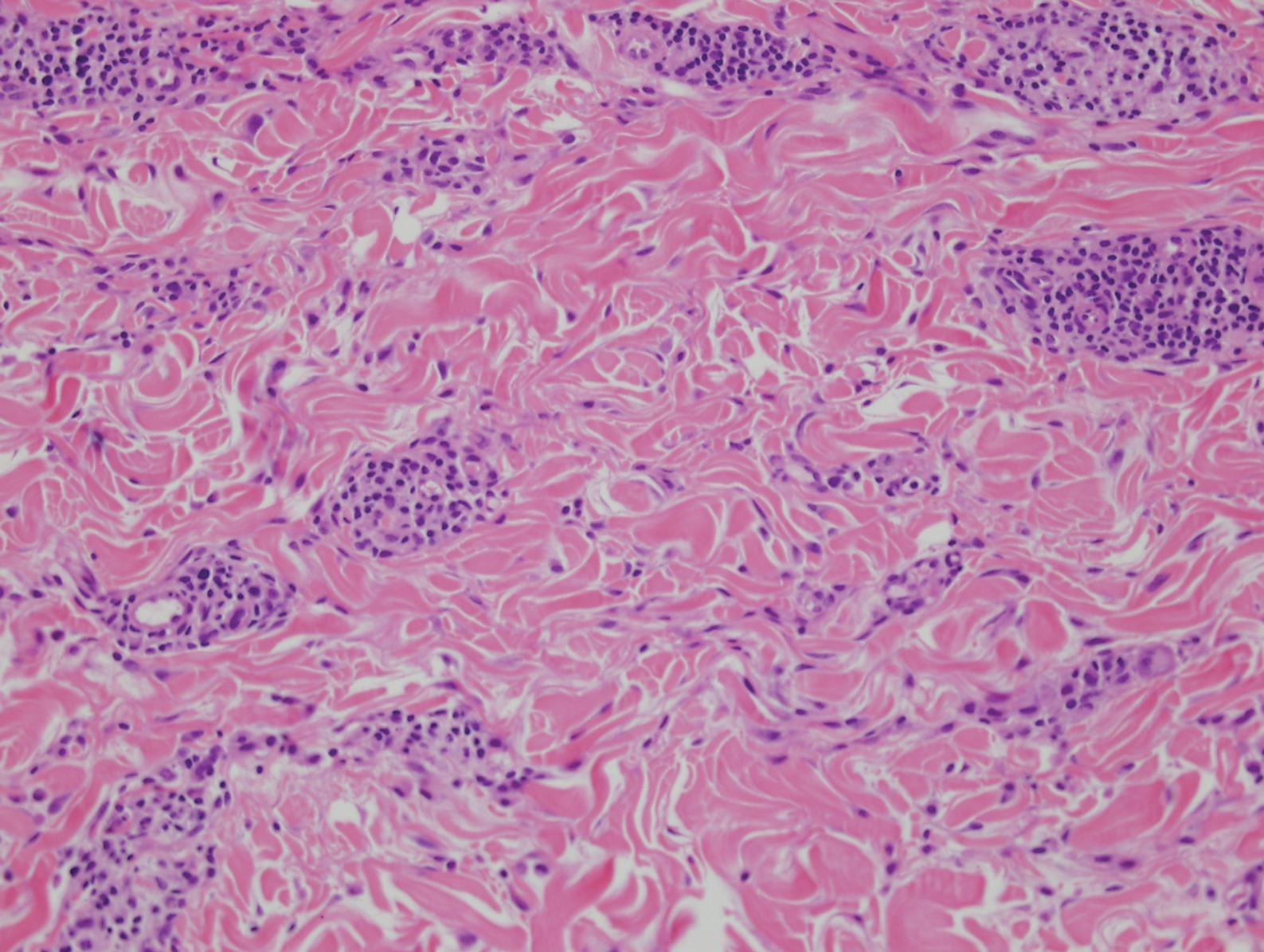
Interstitial granuloma annulare is a noninfectious granulomatous skin condition that often presents clinically as asymptomatic annular red-brown patches, usually on the extremities.11-13 On histopathology, an interstitial or palisaded inflammatory infiltrate with histiocytes and multinucleated giant cells may be seen along with collagen degeneration or collagen bundles without necrosis (Figure 3).9 Mucin often is associated with the histiocytes.11 Of note, our patient's skin biopsy shows interface dermatitis, differentiating it from both interstitial granuloma annulare and interstitial granulomatous dermatitis.
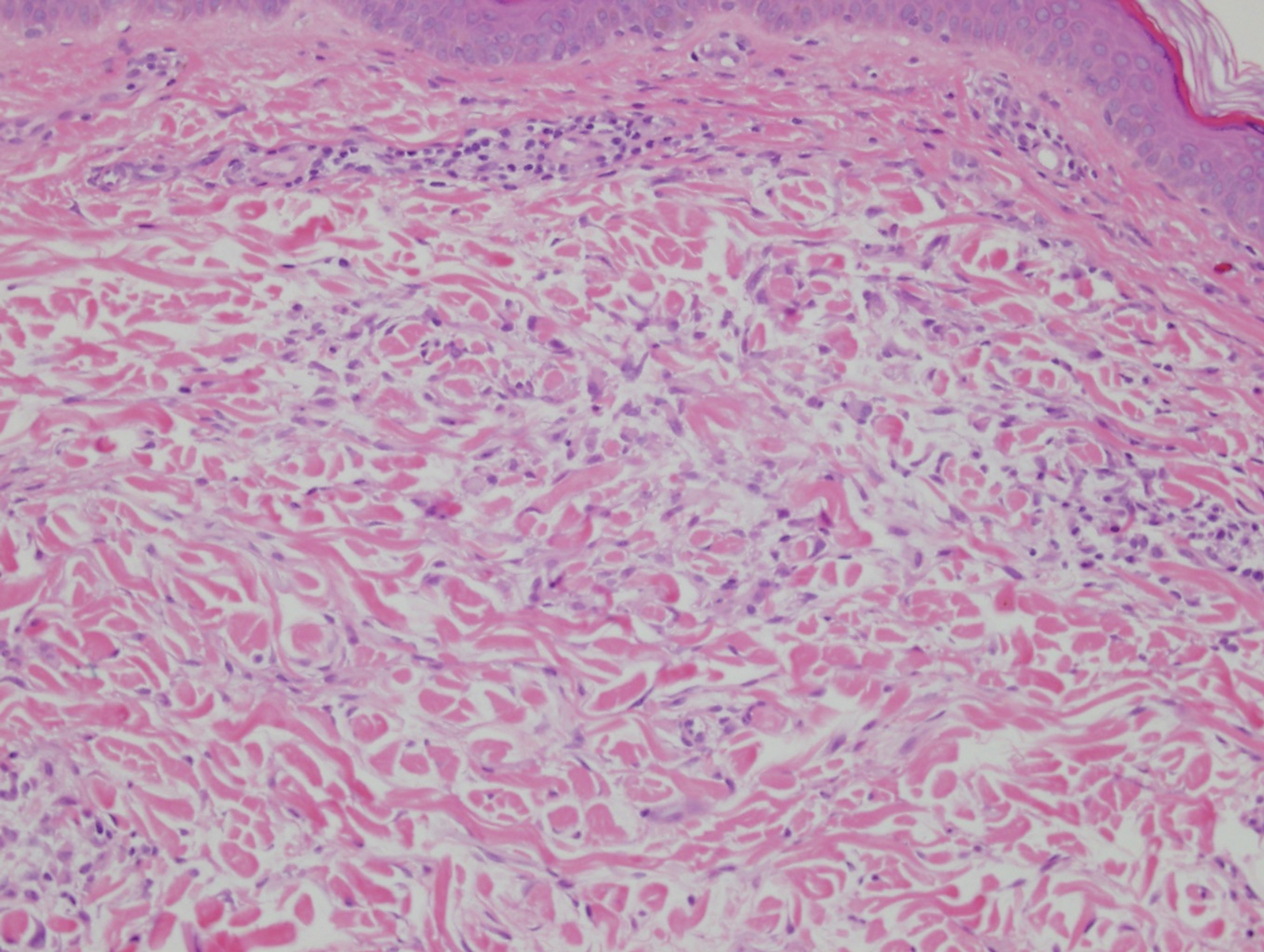
Postviral granulomatous reactions are the most frequently reported types of reactions to occur at the location of herpes zoster infection up to years after the initial disease. Wolf isotopic reaction encompasses skin reactions in the body region of formerly resolved skin disease, commonly herpesvirus infection.14,15 This manifestation may occur due to a hypersensitivity reaction from enduring viral proteins, resident memory T cells, or local neuroimmune imbalance from herpesvirus-induced injury to dermal sensory nerve fibers.14-17 Clinically, patients present with red-purple pruritic papules and plaques in a bandlike unilateral pattern, usually in the same region as the prior herpes infection and often accompanied by postherpetic neuralgia.16-19 Of note, our patient's clinical findings were more diffuse than the frequently localized and often linear distribution seen in postherpetic granulomatous reaction. On histopathology, granulomatous or lichenoid tissue reaction most commonly is appreciated.15 Specifically, interstitial granulomatous dermatitis with histiocytes, lymphocytes, and multinucleated giant cells showing elastophagocytosis and an inflammatory infiltrate with lymphocytes and plasma cells around vasculature, eccrine glands, and nerves can be noted (Figure 4).19

Lupus erythematosus is an autoimmune condition with a wide array of clinical features, including skin manifestations and systemic symptoms. Specifically, discoid lupus erythematosus presents with clearly outlined, red-pink macules or papules with scaling. Histologic features include keratotic follicular plugging, acanthosis, dermal mucin, thickening of the basement membrane zone, and dense lymphocytic infiltrate (Figure 5).20
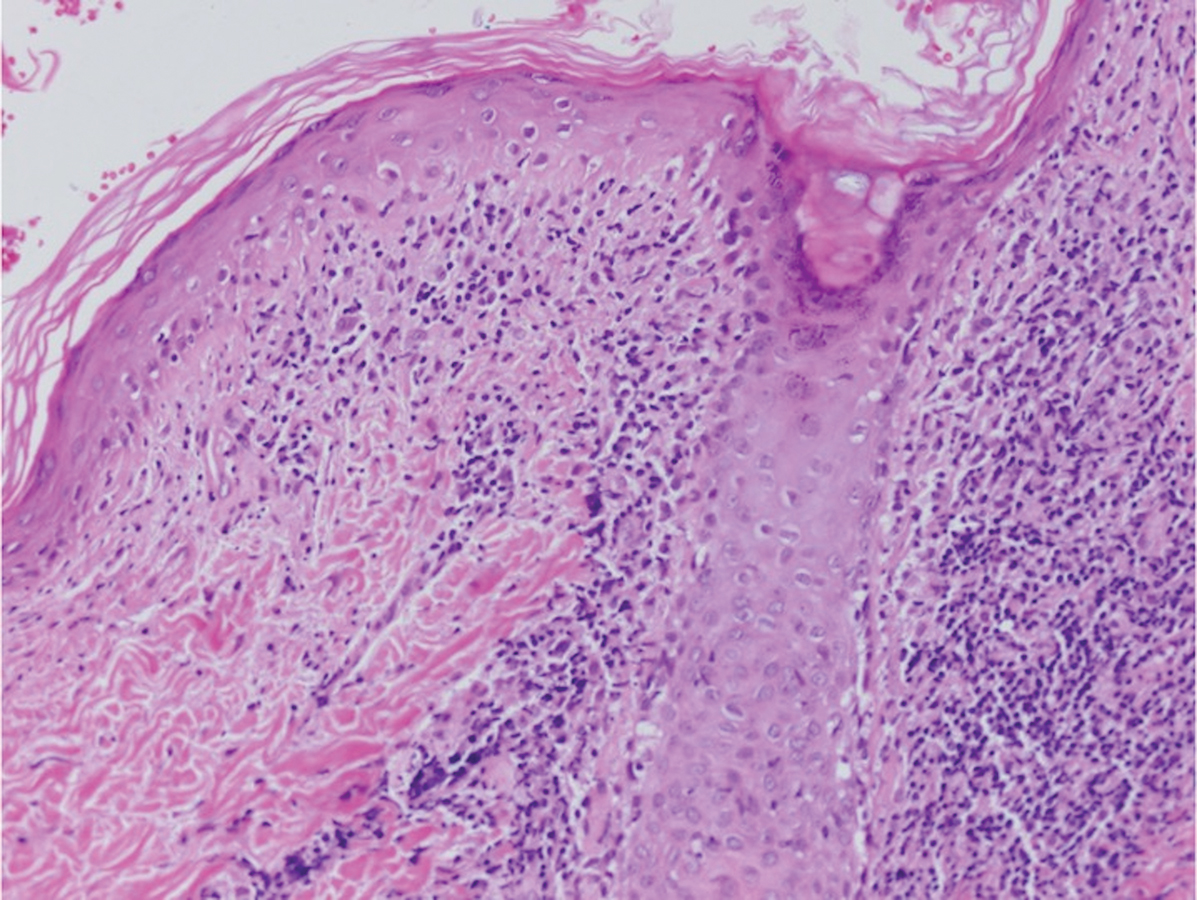
- Flamm A, Parikh K, Xie Q, et al. Histologic features of secondary syphilis: a multicenter retrospective review. J Am Acad Dermatol. 2015;73:325-330.
- Zeltser R, Kurban AK. Syphilis. Clin Dermatol. 2004;22:461-468.
- Magro CM, Crowson AN. Lichenoid and granulomatous dermatitis. Int J Dermatol. 2000;39:12-33.
- S Breza T Jr, Magro CM. Lichenoid and granulomatous dermatitis associated with atypical mycobacterium infections. J Cutan Pathol. 2006;33:512-515.
- Granel B, Serratrice J, Rey J, et al. Chronic hepatitis C virus infection associated with a generalized granuloma annulare. J Am Acad Dermatol. 2000;43(5, pt 2):918-919.
- Jorizzo JL, Gonzalez EB, Apisarnthanarax P, et al. Pigmented purpuric eruption in a patient with rheumatoid arthritis. Arch Intern Med. 1982;142:2184-2185.
- Magro CM, Crowson AN, Regauer S. Granuloma annulare and necrobiosis lipoidica tissue reactions as a manifestation of systemic disease. Hum Pathol. 1996;27:50-56.
- Błażewicz I, Szczerkowska-Dobosz A, Pęksa R, et al. Interstitial granulomatous dermatitis: a characteristic histological pattern with variable clinical manifestations. Postepy Dermatol Alergol. 2015;32:475-477.
- Sezer E, Luzar B, Calonje E. Secondary syphilis with an interstitial granuloma annulare-like histopathologic pattern. J Cutan Pathol. 2011;38:439-442.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Sakiyama T, Hirai I, Konohana A, et al. Interstitial-type granuloma annulare associated with Sjögren syndrome. J Dtsch Dermatol Ges. 2014;12:415-416.
- Spring P, Vernez M, Maniu CM, et al. Localized interstitial granuloma annulare induced by subcutaneous injections for desensitization. Dermatol Online J. 2013;19:18572.
- Kluger N, Moguelet P, Chaslin-Ferbus D, et al. Generalized interstitial granuloma annulare induced by pegylated interferon-alpha. Dermatology. 2006;213:248-249.
- Ruocco E, Baroni A, Cutrì FT, et al. Granuloma annulare in a site of healed herpes zoster: Wolf's isotopic response. J Eur Acad Dermatol Venereol. 2003;17:686-688.
- Ise M, Tanese K, Adachi T, et al. Postherpetic Wolf's isotopic response: possible contribution of resident memory T cells to the pathogenesis of lichenoid reaction. Br J Dermatol. 2015;173:1331-1334.
- Lora V, Cota C, Kanitakis J. Zosteriform lichen planus after herpes zoster: report of a new case of Wolf's isotopic phenomenon and literature review. Dermatol Online J. 2014;20. pii:13030/qt5vf99178.
- Lin CH, Chen HC, Gao HW, et al. Wolf's post-herpetic isotopic response to tocilizumab for rheumatoid arthritis. Australas J Dermatol. 2018;59:E135-E137.
- Melgar E, Henry J, Valois A, et al. Extra-facial Lever granuloma on a herpes zoster scar: Wolf's isotopic response. Ann Dermatol Venereol. 2018;145:354-358.
- Ferenczi K, Rosenberg AS, McCalmont TH, et al. Herpes zoster granulomatous dermatitis: histopathologic findings in a case series. J Cutan Pathol. 2015;42:739-745.
- Li Q, Wu H, Liao W, et al. A comprehensive review of immune-mediated dermatopathology in systemic lupus erythematosus. J Autoimmun. 2018;93:1-15.
The Diagnosis: Lichenoid and Granulomatous Dermatitis in the Setting of Secondary Syphilis
Syphilis, an infectious disease that has risen in incidence and is most commonly reported in men who have sex with men, involves a vast array of clinical and histologic presentations.1 Clinically, secondary syphilis involves an erythematous maculopapular eruption on the face, trunk, palms, soles, or genital area.2 The characteristic histologic features for secondary syphilis include endothelial swelling, interstitial inflammatory array, irregular acanthosis, elongated rete ridges, and vacuolar interface dermatitis with lymphocytes and plasma cells.1 Syphilitic infection has been associated with lichenoid and granulomatous dermatitis, which is an inflammatory skin disease described by Magro and Crowson.3 Lichenoid and granulomatous dermatitis has been linked to various systemic disorders, including chronic hepatitis C, Crohn disease, rheumatoid arthritis, endocrinopathy, subacute cutaneous lupus erythematosus, secondary syphilis, prior herpes infection, tuberculoid leprosy, mycobacterial infection, and human immunodeficiency virus infection.3-7 For this patient, given histopathology findings, clinical presentation, and positive rapid plasma reagin serologies, a diagnosis of lichenoid and granulomatous dermatitis in the setting of a secondary syphilis infection was established. A comprehensive investigation should be conducted to consider secondary syphilis or other systemic diseases in patients with a histologic finding of lichenoid and granulomatous dermatitis.
Histologically, lichenoid and granulomatous dermatitis cases show a bandlike infiltrate of lymphocytes with neighboring histiocytes along the dermoepidermal junction, accompanied by epithelial changes of dyskeratosis, vasculopathy, and colloid body formation, in addition to a dermal histiocytic component.3 Our patient's biopsy showed a lichenoid reaction pattern with vacuolar interface changes, dyskeratosis, plump endothelial cells, and small collections of plasma cells. Additionally, there was a granulomatous component in the dermis with histiocytes admixed with lymphocytes and plasma cells. The presence of spirochetes was confirmed with antitreponemal immunohistochemical stain (Figure 1). Quantitative rapid plasma reagin was 1:64 (reference range, <1:1) and Treponema pallidum antibody was reactive.
Interstitial granulomatous dermatitis has a variable clinical presentation, often with red-purple annular plaques, hyperpigmented papules, and nodules frequently in a linear arrangement and predominantly on the trunk, thighs, groin, or buttocks.8,9 On histopathology, there are histiocytes in the reticular dermis and/or a macrophage infiltrate in the mid to deep dermis with collections of degenerated collagen (Figure 2).8,10 An interstitial infiltrate of eosinophils and neutrophils also may be appreciated, but mucin generally is absent.8,11 This condition often coexists with rheumatic and systemic autoimmune diseases.8-10
Interstitial granuloma annulare is a noninfectious granulomatous skin condition that often presents clinically as asymptomatic annular red-brown patches, usually on the extremities.11-13 On histopathology, an interstitial or palisaded inflammatory infiltrate with histiocytes and multinucleated giant cells may be seen along with collagen degeneration or collagen bundles without necrosis (Figure 3).9 Mucin often is associated with the histiocytes.11 Of note, our patient's skin biopsy shows interface dermatitis, differentiating it from both interstitial granuloma annulare and interstitial granulomatous dermatitis.
Postviral granulomatous reactions are the most frequently reported types of reactions to occur at the location of herpes zoster infection up to years after the initial disease. Wolf isotopic reaction encompasses skin reactions in the body region of formerly resolved skin disease, commonly herpesvirus infection.14,15 This manifestation may occur due to a hypersensitivity reaction from enduring viral proteins, resident memory T cells, or local neuroimmune imbalance from herpesvirus-induced injury to dermal sensory nerve fibers.14-17 Clinically, patients present with red-purple pruritic papules and plaques in a bandlike unilateral pattern, usually in the same region as the prior herpes infection and often accompanied by postherpetic neuralgia.16-19 Of note, our patient's clinical findings were more diffuse than the frequently localized and often linear distribution seen in postherpetic granulomatous reaction. On histopathology, granulomatous or lichenoid tissue reaction most commonly is appreciated.15 Specifically, interstitial granulomatous dermatitis with histiocytes, lymphocytes, and multinucleated giant cells showing elastophagocytosis and an inflammatory infiltrate with lymphocytes and plasma cells around vasculature, eccrine glands, and nerves can be noted (Figure 4).19
Lupus erythematosus is an autoimmune condition with a wide array of clinical features, including skin manifestations and systemic symptoms. Specifically, discoid lupus erythematosus presents with clearly outlined, red-pink macules or papules with scaling. Histologic features include keratotic follicular plugging, acanthosis, dermal mucin, thickening of the basement membrane zone, and dense lymphocytic infiltrate (Figure 5).20
The Diagnosis: Lichenoid and Granulomatous Dermatitis in the Setting of Secondary Syphilis
Syphilis, an infectious disease that has risen in incidence and is most commonly reported in men who have sex with men, involves a vast array of clinical and histologic presentations.1 Clinically, secondary syphilis involves an erythematous maculopapular eruption on the face, trunk, palms, soles, or genital area.2 The characteristic histologic features for secondary syphilis include endothelial swelling, interstitial inflammatory array, irregular acanthosis, elongated rete ridges, and vacuolar interface dermatitis with lymphocytes and plasma cells.1 Syphilitic infection has been associated with lichenoid and granulomatous dermatitis, which is an inflammatory skin disease described by Magro and Crowson.3 Lichenoid and granulomatous dermatitis has been linked to various systemic disorders, including chronic hepatitis C, Crohn disease, rheumatoid arthritis, endocrinopathy, subacute cutaneous lupus erythematosus, secondary syphilis, prior herpes infection, tuberculoid leprosy, mycobacterial infection, and human immunodeficiency virus infection.3-7 For this patient, given histopathology findings, clinical presentation, and positive rapid plasma reagin serologies, a diagnosis of lichenoid and granulomatous dermatitis in the setting of a secondary syphilis infection was established. A comprehensive investigation should be conducted to consider secondary syphilis or other systemic diseases in patients with a histologic finding of lichenoid and granulomatous dermatitis.
Histologically, lichenoid and granulomatous dermatitis cases show a bandlike infiltrate of lymphocytes with neighboring histiocytes along the dermoepidermal junction, accompanied by epithelial changes of dyskeratosis, vasculopathy, and colloid body formation, in addition to a dermal histiocytic component.3 Our patient's biopsy showed a lichenoid reaction pattern with vacuolar interface changes, dyskeratosis, plump endothelial cells, and small collections of plasma cells. Additionally, there was a granulomatous component in the dermis with histiocytes admixed with lymphocytes and plasma cells. The presence of spirochetes was confirmed with antitreponemal immunohistochemical stain (Figure 1). Quantitative rapid plasma reagin was 1:64 (reference range, <1:1) and Treponema pallidum antibody was reactive.
Interstitial granulomatous dermatitis has a variable clinical presentation, often with red-purple annular plaques, hyperpigmented papules, and nodules frequently in a linear arrangement and predominantly on the trunk, thighs, groin, or buttocks.8,9 On histopathology, there are histiocytes in the reticular dermis and/or a macrophage infiltrate in the mid to deep dermis with collections of degenerated collagen (Figure 2).8,10 An interstitial infiltrate of eosinophils and neutrophils also may be appreciated, but mucin generally is absent.8,11 This condition often coexists with rheumatic and systemic autoimmune diseases.8-10
Interstitial granuloma annulare is a noninfectious granulomatous skin condition that often presents clinically as asymptomatic annular red-brown patches, usually on the extremities.11-13 On histopathology, an interstitial or palisaded inflammatory infiltrate with histiocytes and multinucleated giant cells may be seen along with collagen degeneration or collagen bundles without necrosis (Figure 3).9 Mucin often is associated with the histiocytes.11 Of note, our patient's skin biopsy shows interface dermatitis, differentiating it from both interstitial granuloma annulare and interstitial granulomatous dermatitis.
Postviral granulomatous reactions are the most frequently reported types of reactions to occur at the location of herpes zoster infection up to years after the initial disease. Wolf isotopic reaction encompasses skin reactions in the body region of formerly resolved skin disease, commonly herpesvirus infection.14,15 This manifestation may occur due to a hypersensitivity reaction from enduring viral proteins, resident memory T cells, or local neuroimmune imbalance from herpesvirus-induced injury to dermal sensory nerve fibers.14-17 Clinically, patients present with red-purple pruritic papules and plaques in a bandlike unilateral pattern, usually in the same region as the prior herpes infection and often accompanied by postherpetic neuralgia.16-19 Of note, our patient's clinical findings were more diffuse than the frequently localized and often linear distribution seen in postherpetic granulomatous reaction. On histopathology, granulomatous or lichenoid tissue reaction most commonly is appreciated.15 Specifically, interstitial granulomatous dermatitis with histiocytes, lymphocytes, and multinucleated giant cells showing elastophagocytosis and an inflammatory infiltrate with lymphocytes and plasma cells around vasculature, eccrine glands, and nerves can be noted (Figure 4).19
Lupus erythematosus is an autoimmune condition with a wide array of clinical features, including skin manifestations and systemic symptoms. Specifically, discoid lupus erythematosus presents with clearly outlined, red-pink macules or papules with scaling. Histologic features include keratotic follicular plugging, acanthosis, dermal mucin, thickening of the basement membrane zone, and dense lymphocytic infiltrate (Figure 5).20
- Flamm A, Parikh K, Xie Q, et al. Histologic features of secondary syphilis: a multicenter retrospective review. J Am Acad Dermatol. 2015;73:325-330.
- Zeltser R, Kurban AK. Syphilis. Clin Dermatol. 2004;22:461-468.
- Magro CM, Crowson AN. Lichenoid and granulomatous dermatitis. Int J Dermatol. 2000;39:12-33.
- S Breza T Jr, Magro CM. Lichenoid and granulomatous dermatitis associated with atypical mycobacterium infections. J Cutan Pathol. 2006;33:512-515.
- Granel B, Serratrice J, Rey J, et al. Chronic hepatitis C virus infection associated with a generalized granuloma annulare. J Am Acad Dermatol. 2000;43(5, pt 2):918-919.
- Jorizzo JL, Gonzalez EB, Apisarnthanarax P, et al. Pigmented purpuric eruption in a patient with rheumatoid arthritis. Arch Intern Med. 1982;142:2184-2185.
- Magro CM, Crowson AN, Regauer S. Granuloma annulare and necrobiosis lipoidica tissue reactions as a manifestation of systemic disease. Hum Pathol. 1996;27:50-56.
- Błażewicz I, Szczerkowska-Dobosz A, Pęksa R, et al. Interstitial granulomatous dermatitis: a characteristic histological pattern with variable clinical manifestations. Postepy Dermatol Alergol. 2015;32:475-477.
- Sezer E, Luzar B, Calonje E. Secondary syphilis with an interstitial granuloma annulare-like histopathologic pattern. J Cutan Pathol. 2011;38:439-442.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Sakiyama T, Hirai I, Konohana A, et al. Interstitial-type granuloma annulare associated with Sjögren syndrome. J Dtsch Dermatol Ges. 2014;12:415-416.
- Spring P, Vernez M, Maniu CM, et al. Localized interstitial granuloma annulare induced by subcutaneous injections for desensitization. Dermatol Online J. 2013;19:18572.
- Kluger N, Moguelet P, Chaslin-Ferbus D, et al. Generalized interstitial granuloma annulare induced by pegylated interferon-alpha. Dermatology. 2006;213:248-249.
- Ruocco E, Baroni A, Cutrì FT, et al. Granuloma annulare in a site of healed herpes zoster: Wolf's isotopic response. J Eur Acad Dermatol Venereol. 2003;17:686-688.
- Ise M, Tanese K, Adachi T, et al. Postherpetic Wolf's isotopic response: possible contribution of resident memory T cells to the pathogenesis of lichenoid reaction. Br J Dermatol. 2015;173:1331-1334.
- Lora V, Cota C, Kanitakis J. Zosteriform lichen planus after herpes zoster: report of a new case of Wolf's isotopic phenomenon and literature review. Dermatol Online J. 2014;20. pii:13030/qt5vf99178.
- Lin CH, Chen HC, Gao HW, et al. Wolf's post-herpetic isotopic response to tocilizumab for rheumatoid arthritis. Australas J Dermatol. 2018;59:E135-E137.
- Melgar E, Henry J, Valois A, et al. Extra-facial Lever granuloma on a herpes zoster scar: Wolf's isotopic response. Ann Dermatol Venereol. 2018;145:354-358.
- Ferenczi K, Rosenberg AS, McCalmont TH, et al. Herpes zoster granulomatous dermatitis: histopathologic findings in a case series. J Cutan Pathol. 2015;42:739-745.
- Li Q, Wu H, Liao W, et al. A comprehensive review of immune-mediated dermatopathology in systemic lupus erythematosus. J Autoimmun. 2018;93:1-15.
- Flamm A, Parikh K, Xie Q, et al. Histologic features of secondary syphilis: a multicenter retrospective review. J Am Acad Dermatol. 2015;73:325-330.
- Zeltser R, Kurban AK. Syphilis. Clin Dermatol. 2004;22:461-468.
- Magro CM, Crowson AN. Lichenoid and granulomatous dermatitis. Int J Dermatol. 2000;39:12-33.
- S Breza T Jr, Magro CM. Lichenoid and granulomatous dermatitis associated with atypical mycobacterium infections. J Cutan Pathol. 2006;33:512-515.
- Granel B, Serratrice J, Rey J, et al. Chronic hepatitis C virus infection associated with a generalized granuloma annulare. J Am Acad Dermatol. 2000;43(5, pt 2):918-919.
- Jorizzo JL, Gonzalez EB, Apisarnthanarax P, et al. Pigmented purpuric eruption in a patient with rheumatoid arthritis. Arch Intern Med. 1982;142:2184-2185.
- Magro CM, Crowson AN, Regauer S. Granuloma annulare and necrobiosis lipoidica tissue reactions as a manifestation of systemic disease. Hum Pathol. 1996;27:50-56.
- Błażewicz I, Szczerkowska-Dobosz A, Pęksa R, et al. Interstitial granulomatous dermatitis: a characteristic histological pattern with variable clinical manifestations. Postepy Dermatol Alergol. 2015;32:475-477.
- Sezer E, Luzar B, Calonje E. Secondary syphilis with an interstitial granuloma annulare-like histopathologic pattern. J Cutan Pathol. 2011;38:439-442.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Sakiyama T, Hirai I, Konohana A, et al. Interstitial-type granuloma annulare associated with Sjögren syndrome. J Dtsch Dermatol Ges. 2014;12:415-416.
- Spring P, Vernez M, Maniu CM, et al. Localized interstitial granuloma annulare induced by subcutaneous injections for desensitization. Dermatol Online J. 2013;19:18572.
- Kluger N, Moguelet P, Chaslin-Ferbus D, et al. Generalized interstitial granuloma annulare induced by pegylated interferon-alpha. Dermatology. 2006;213:248-249.
- Ruocco E, Baroni A, Cutrì FT, et al. Granuloma annulare in a site of healed herpes zoster: Wolf's isotopic response. J Eur Acad Dermatol Venereol. 2003;17:686-688.
- Ise M, Tanese K, Adachi T, et al. Postherpetic Wolf's isotopic response: possible contribution of resident memory T cells to the pathogenesis of lichenoid reaction. Br J Dermatol. 2015;173:1331-1334.
- Lora V, Cota C, Kanitakis J. Zosteriform lichen planus after herpes zoster: report of a new case of Wolf's isotopic phenomenon and literature review. Dermatol Online J. 2014;20. pii:13030/qt5vf99178.
- Lin CH, Chen HC, Gao HW, et al. Wolf's post-herpetic isotopic response to tocilizumab for rheumatoid arthritis. Australas J Dermatol. 2018;59:E135-E137.
- Melgar E, Henry J, Valois A, et al. Extra-facial Lever granuloma on a herpes zoster scar: Wolf's isotopic response. Ann Dermatol Venereol. 2018;145:354-358.
- Ferenczi K, Rosenberg AS, McCalmont TH, et al. Herpes zoster granulomatous dermatitis: histopathologic findings in a case series. J Cutan Pathol. 2015;42:739-745.
- Li Q, Wu H, Liao W, et al. A comprehensive review of immune-mediated dermatopathology in systemic lupus erythematosus. J Autoimmun. 2018;93:1-15.
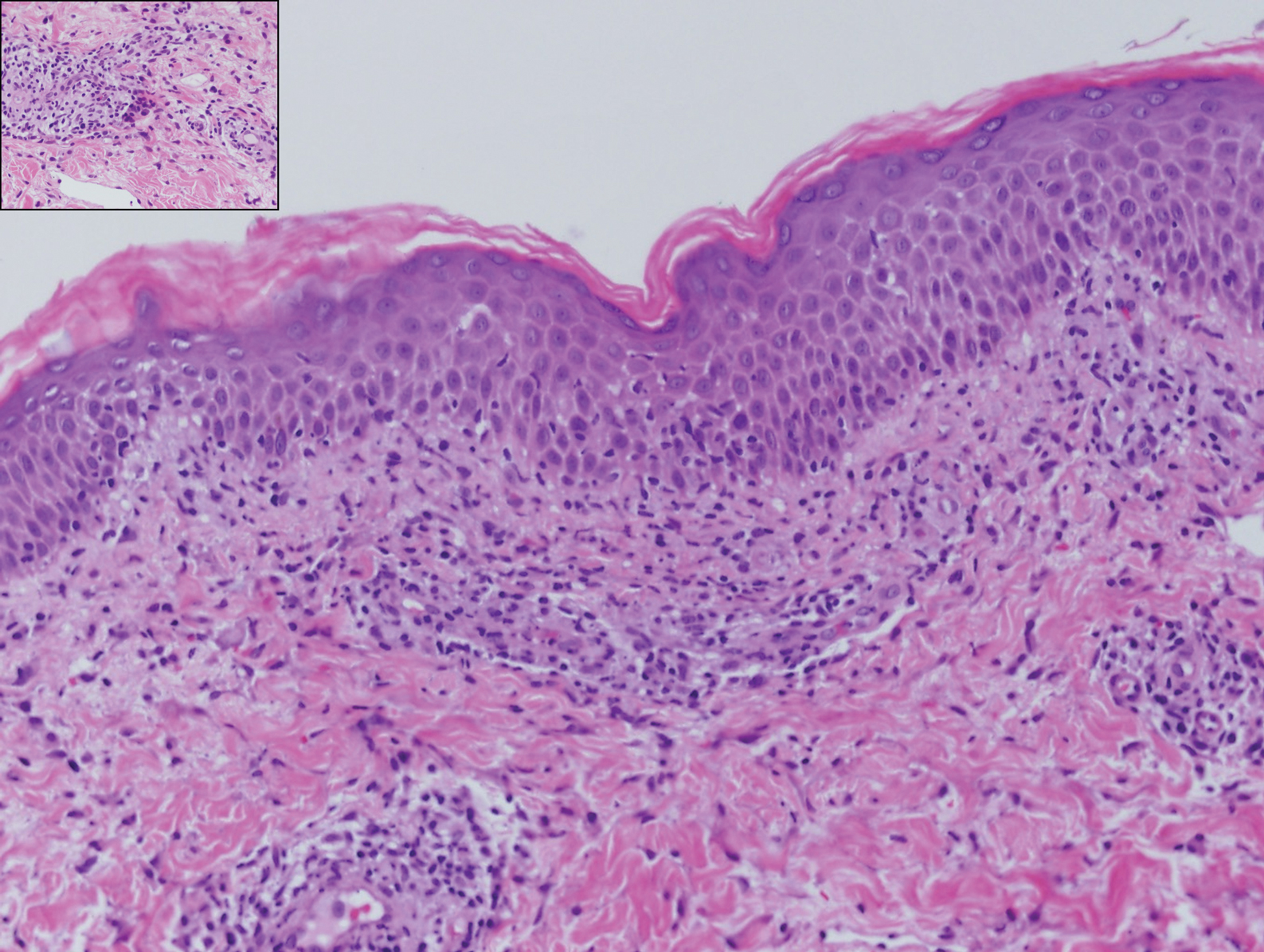
A 54-year-old man presented with painful, nonpruritic, erythematous papules that began on the scrotum. The eruption progressed to involve the trunk, arms, and legs.