User login
Solitary Papule on the Leg
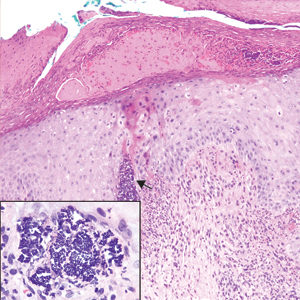
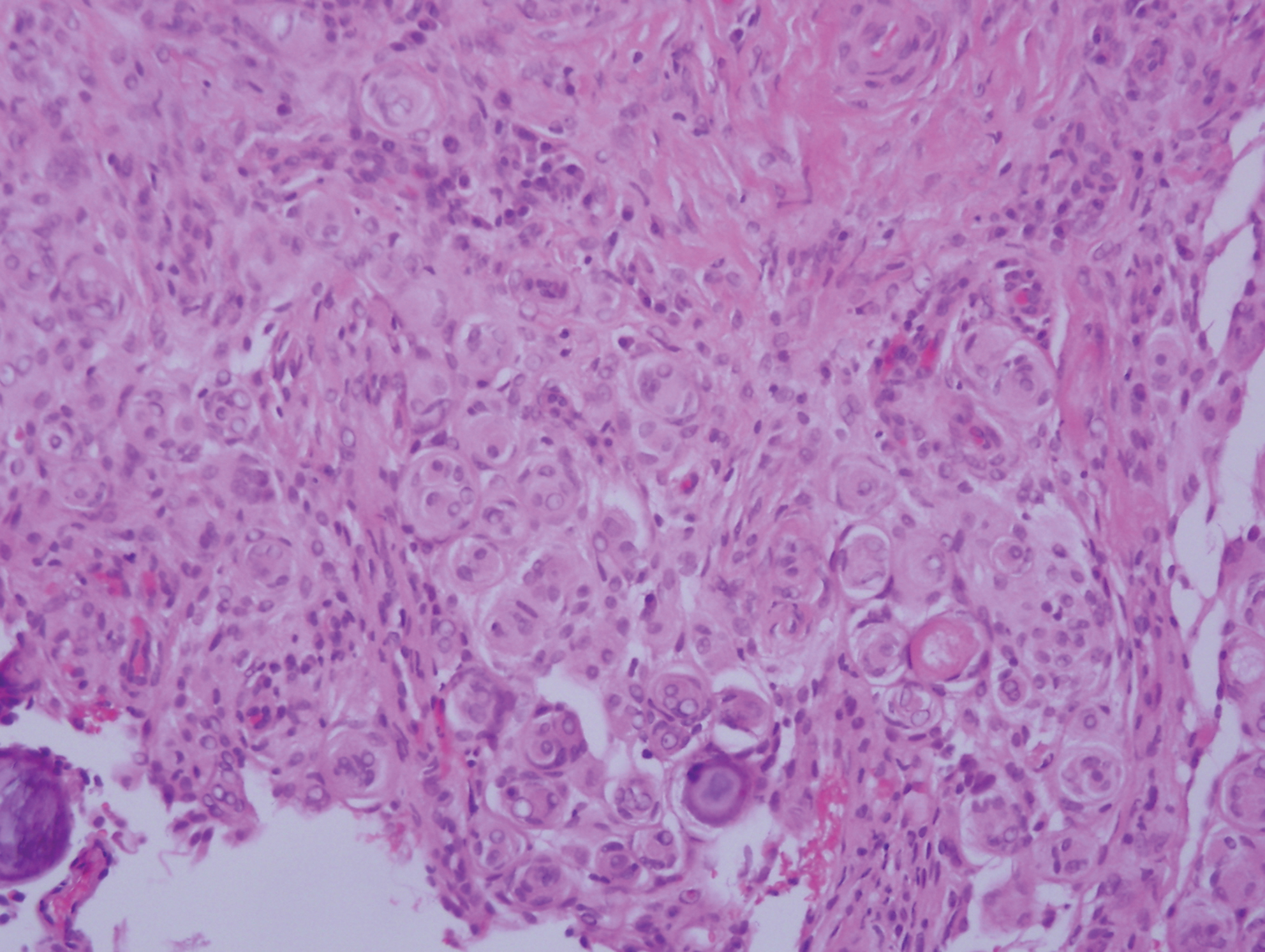
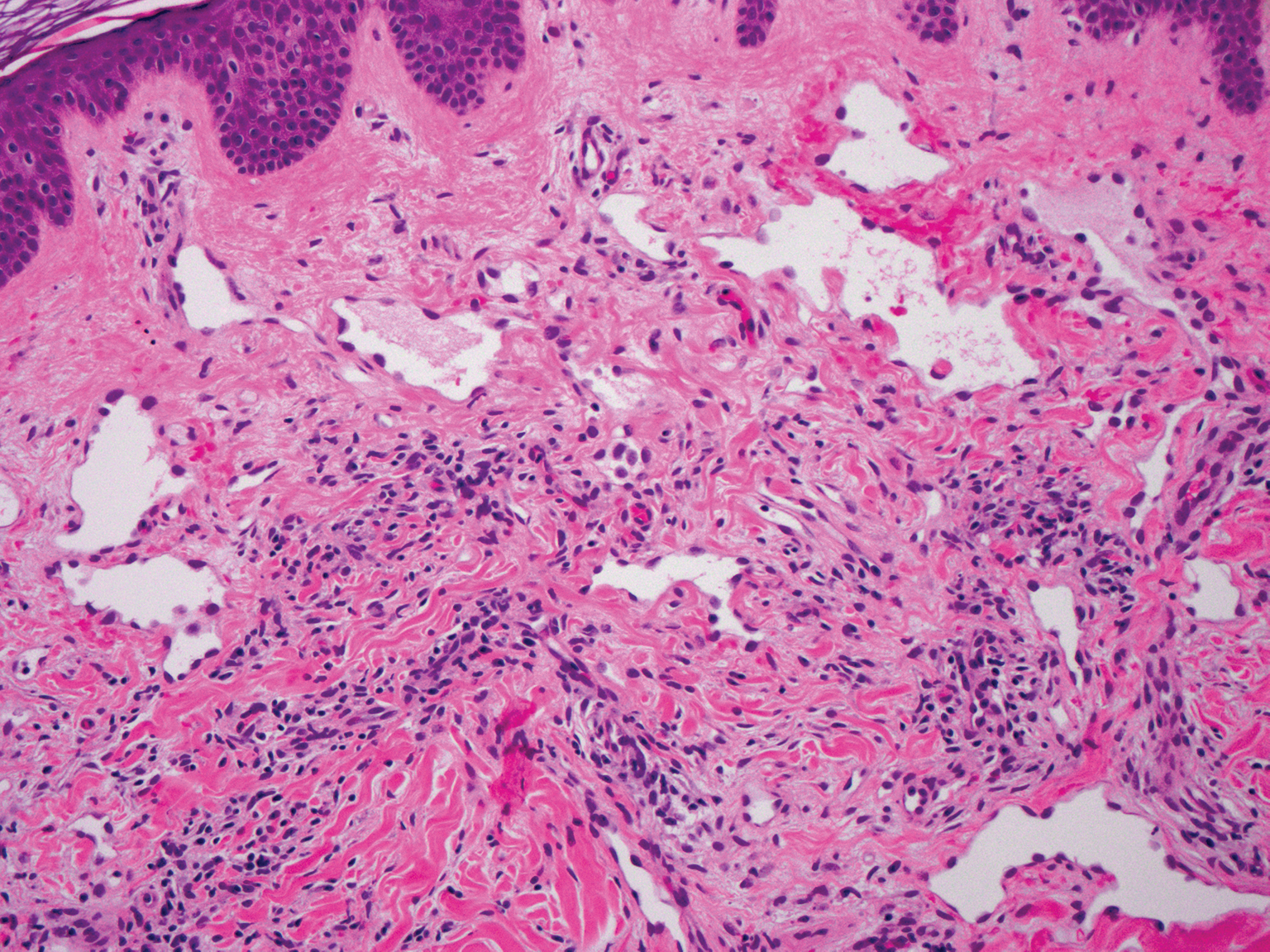
The Diagnosis: Epithelioid Histiocytoma
Epithelioid histiocytoma (EH), also known as epithelioid cell histiocytoma or epithelioid fibrous histiocytoma, is a rare benign fibrohistiocytic tumor first described in 1989.1 Epithelioid histiocytoma commonly presents in middle-aged adults with a slight predilection for males.2 The most frequently affected site is the lower extremity. The arms, trunk, head and neck, groin, and tongue also can be involved.3,4 It usually presents as a solitary asymptomatic papule or nodule, though cases with multiple lesions have been reported.5 Anaplastic lymphoma kinase rearrangement and overexpression have been confirmed and suggest that EH is distinct from conventional cutaneous fibrous histiocytoma.5
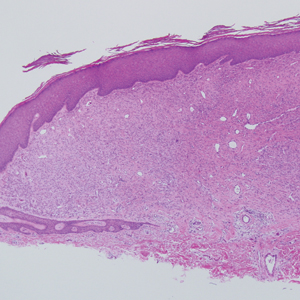
Histologically, EH appears as an exophytic, symmetric, and well-demarcated dermal nodule with a classic epidermal collarette. Prominent vascularity with perivascular accentuation of the epithelioid tumor cells is common. Older lesions may be hyalinized and sclerotic. Epithelioid cells commonly account for more than 50% of the tumor and are characterized by eosinophilic cytoplasm, vesicular nuclei, and small eosinophilic nucleoli. A small population of lymphocytes and mast cells are variably present (quiz image, bottom).1-3,7 A predominantly spindle cell variant has been reported.8 Other histopathologic variants include granular cell,9 cellular,10 and EH with perineuriomalike growth.11 Immunohistochemical staining shows anaplastic lymphoma kinase positivity in most cases, and more than half of cases stain positive for factor XIIIa and epithelial membrane antigen. Tumor cells consistently are negative for desmin and cytokeratins.6,10,12 Excision is curative.8
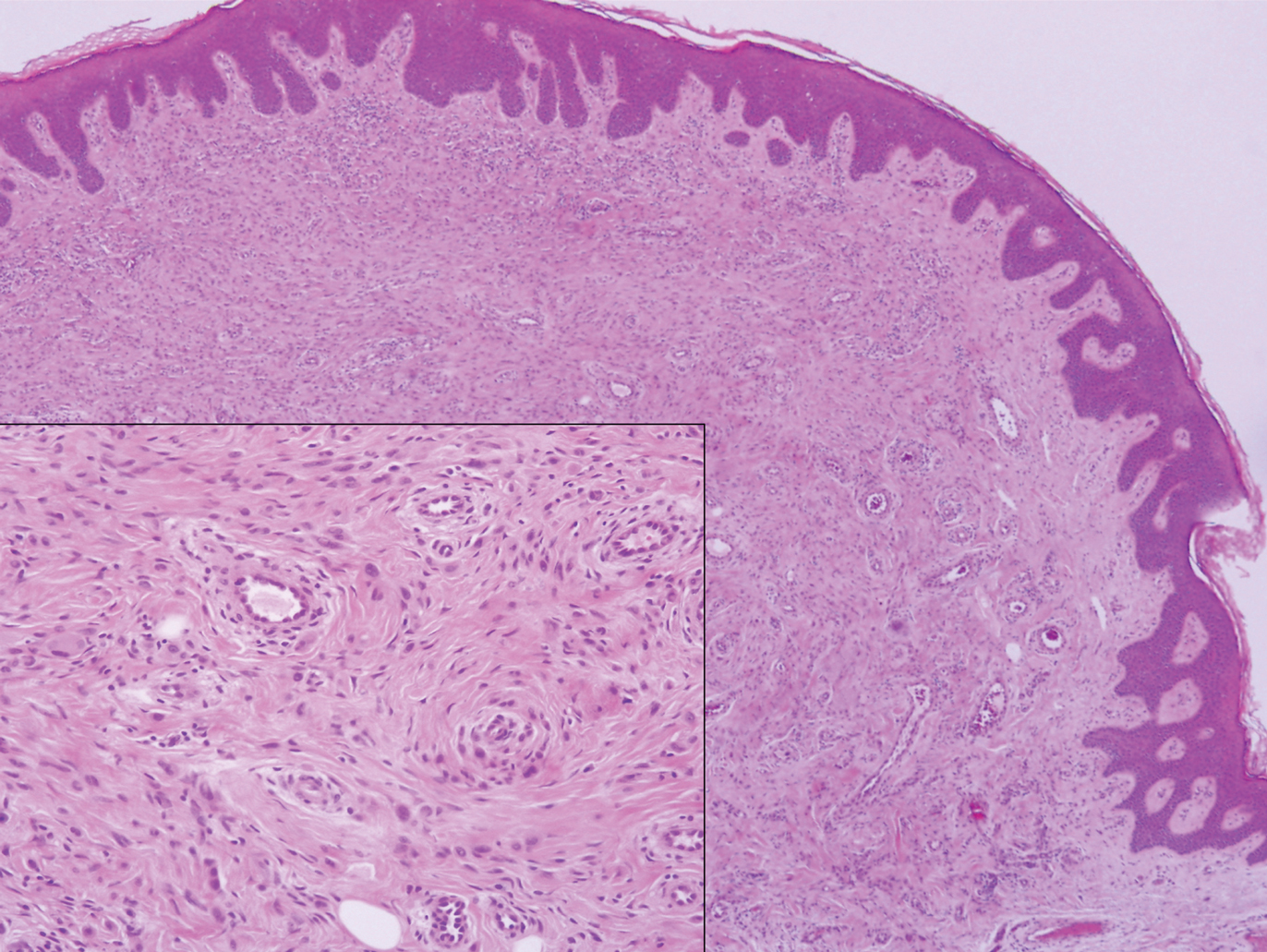
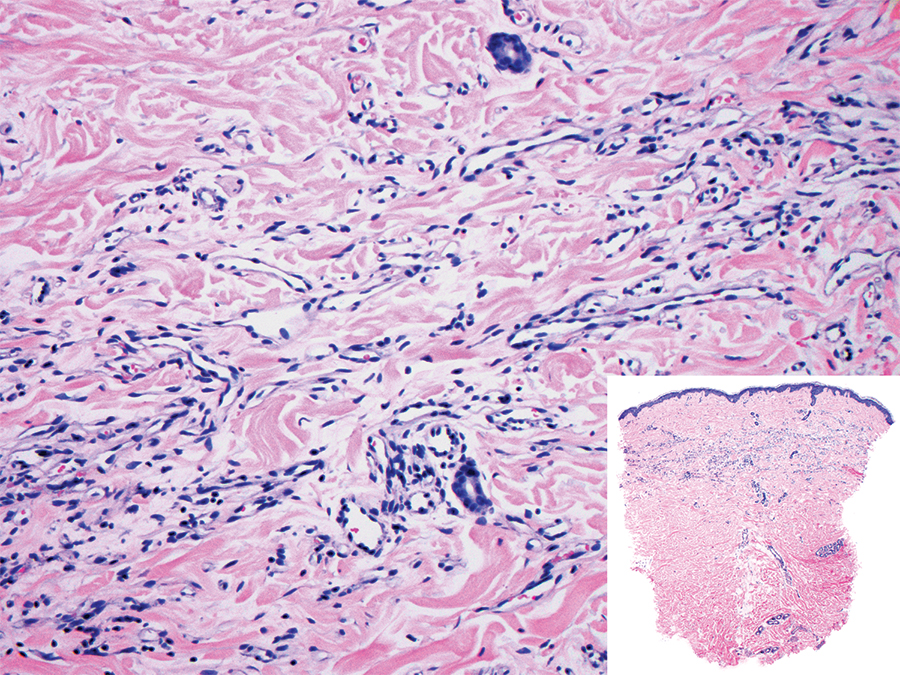
Polypoid Spitz nevus (PSN) is a benign nevus with a conspicuous polypoid or papillary exophytic architecture. The term was coined in 2000 by Fabrizi and Massi.13 Spitz nevus is a benign acquired melanocytic tumor that typically presents in children and adolescents and has a wide histologic spectrum.14 There is some debate on this entity, as some authors do not regard PSN as a distinct histologic variant; thus, it seems underreported in the literature.15 In a review of 349 cases of Spitz nevi, the authors found 7 cases of PSN.16 In another review of 74 cases of intradermal Spitz nevi, 14 cases of PSN were identified.14 This polypoid variant is easily mistaken for a polypoid melanoma because it can show cytologic atypia with large nuclei. Polypoid Spitz nevus usually lacks mitoses, notable pleomorphism, and sheetlike growth, unlike melanoma (Figure 1).13,14
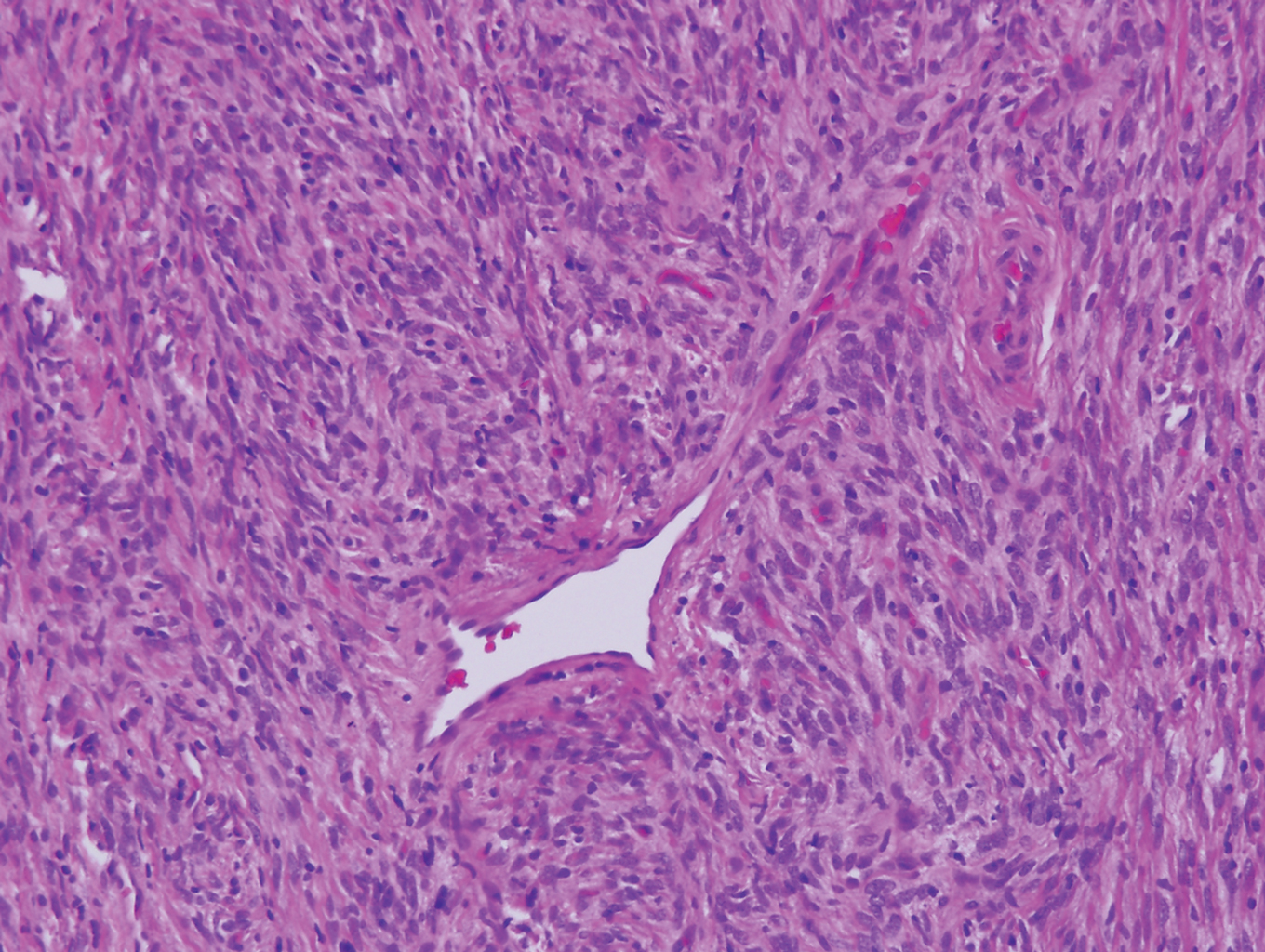
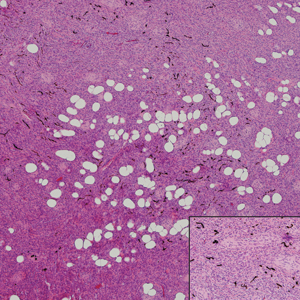
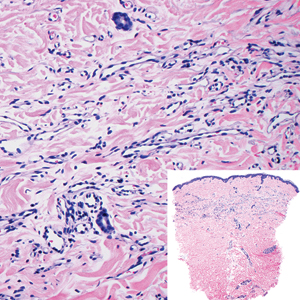
Myopericytoma is an uncommon benign mesenchymal neoplasm that typically presents as a solitary, slowly enlarging and painless nodule with a predilection for the lower extremities, usually in adult males.17-20 Histologically, it consists of a well-circumscribed nodule with numerous thin-walled vessels and a proliferation of ovoid to spindled myopericytes exhibiting a concentric perivascular growth pattern (Figure 2). Myopericytoma usually is positive for smooth muscle actin and h-caldesmon but is negative or only focally positive for desmin. The prognosis is good with rare recurrence, despite incomplete excision.17,18

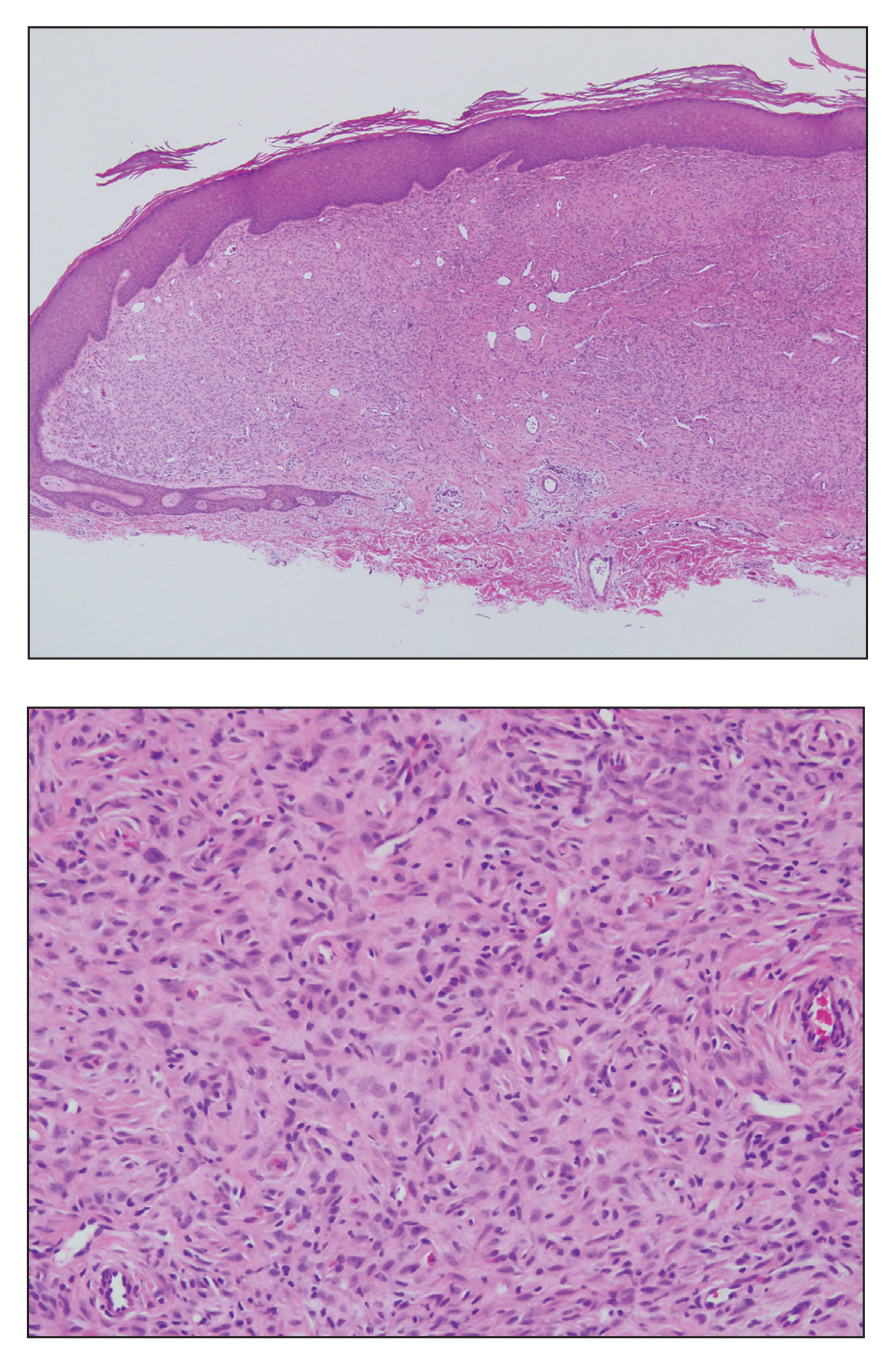
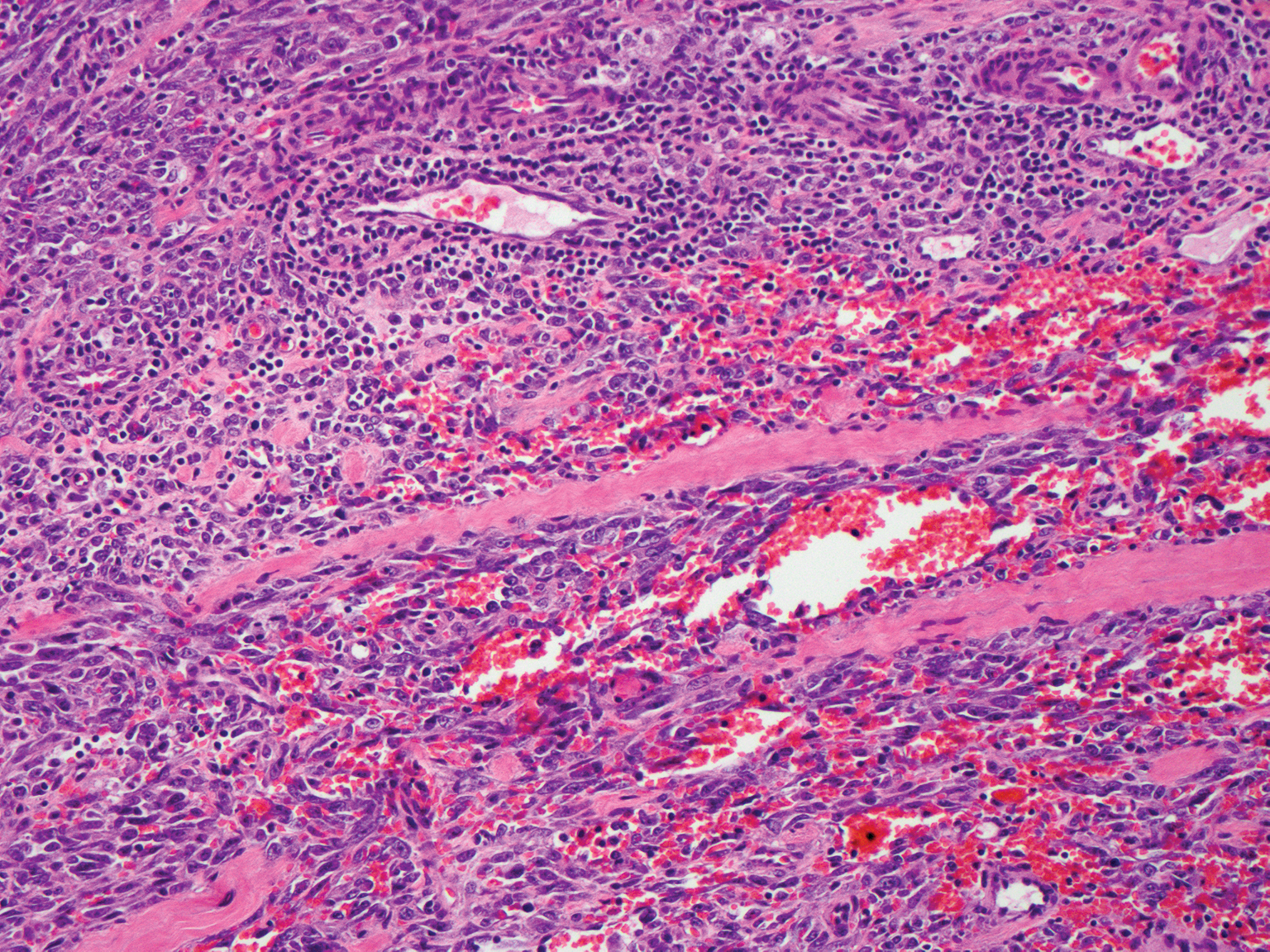
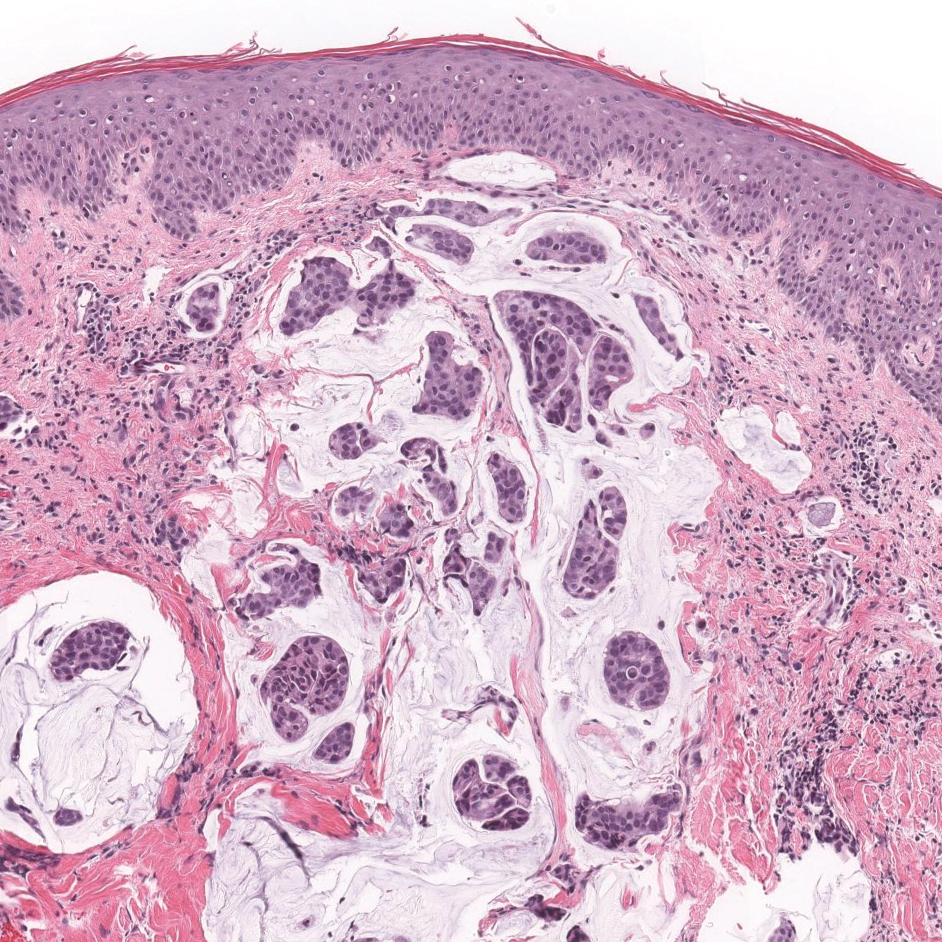
Solitary reticulohistiocytoma is a rare benign form of non-Langerhans cell histiocytosis.21,22 Unlike its multicentric counterpart, solitary reticulohistiocytoma rarely is associated with systemic disease. It presents as a small, dome-shaped, painless papule or nodule that can affect any part of the body.22,23 Solitary reticulohistiocytoma characteristically demonstrates cells with a ground glass-like appearance and 2-toned cytoplasm. A mixed inflammatory infiltrate including neutrophils, eosinophils, and lymphocytes commonly is present (Figure 3). The epithelioid histiocytes are positive for vimentin and histiocytic markers including CD68 and CD163.22

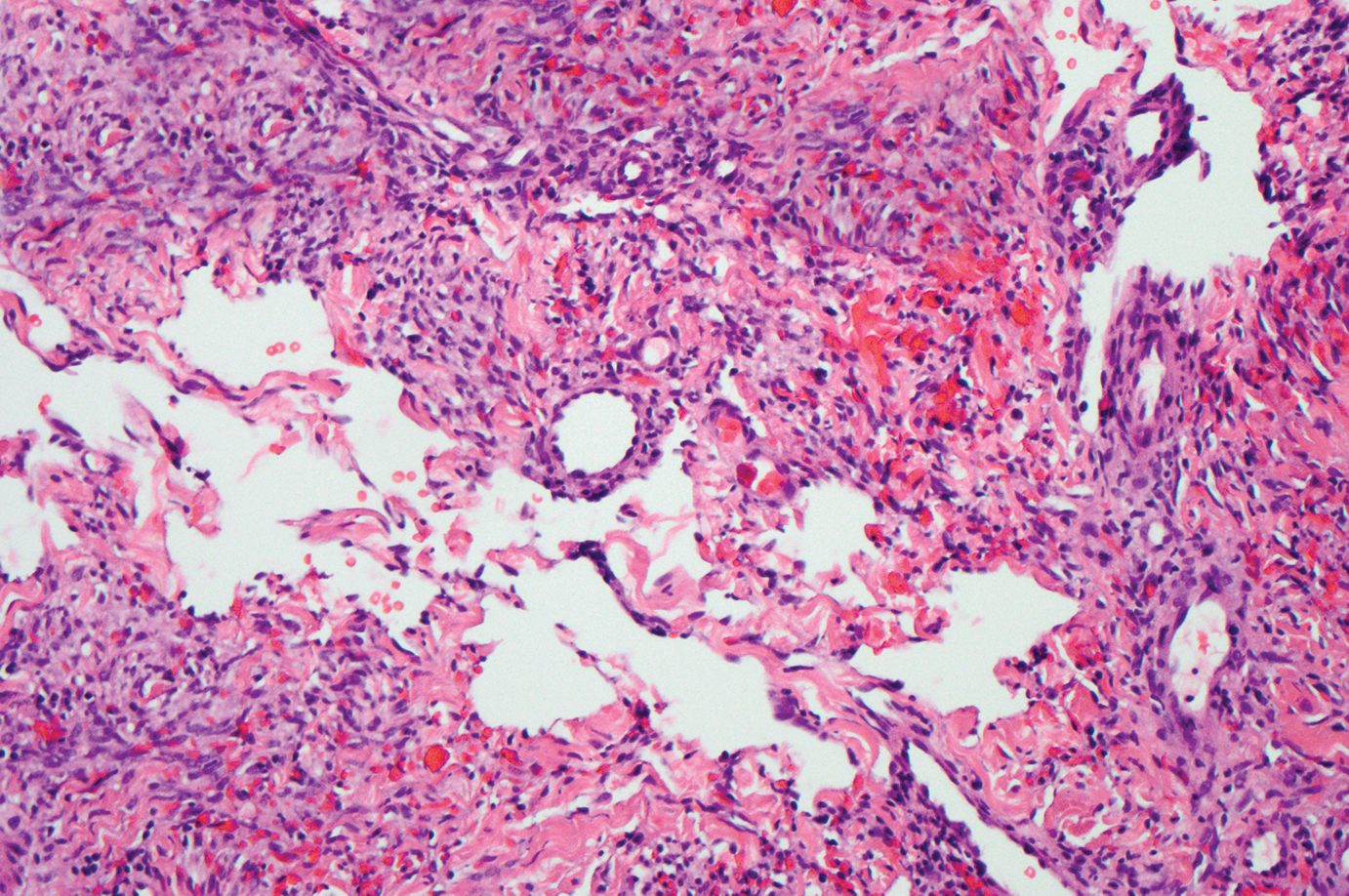
Solitary fibrous tumor (SFT) is an uncommon mesenchymal fibroblastic neoplasm that can arise at almost any anatomic site.24 Cutaneous SFTs are more common in women, most often involve the head, and appear to behave in an indolent manner.25 Solitary fibrous tumors are translocation-associated neoplasms with a NAB2-STAT6 gene fusion.26 The classic histology of SFT is a spindled fibroblastic proliferation arranged in a "patternless pattern" with interspersed stag horn-like, thin-walled blood vessels (Figure 4). Tumor cells usually are positive for CD34, CD99, and Bcl-2.27 In addition, STAT6 immunoreactivity is useful in diagnosis of SFT.25
- Jones EW, Cerio R, Smith NP. Epithelioid cell histiocytoma: a new entity. Br J Dermatol. 1989;120:185-195.
- Singh Gomez C, Calonje E, Fletcher CD. Epithelioid benign fibrous histiocytoma of skin: clinico-pathological analysis of 20 cases of a poorly known variant. Histopathology. 1994;24:123-129.
- Felty CC, Linos K. Epithelioid fibrous histiocytoma: a concise review [published online October 4, 2018]. Am J Dermatopathol. doi:10.1097/DAD.0000000000001272.
- Rawal YB, Kalmar JR, Shumway B, et al. Presentation of an epithelioid cell histiocytoma on the ventral tongue. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:75-83.
- Cangelosi JJ, Prieto VG, Baker GF, et al. Unusual presentation of multiple epithelioid cell histiocytomas. Am J Dermatopathol. 2008;30:373-376.
- Doyle LA, Marino-Enriquez A, Fletcher CD, et al. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol. 2015;28:904-912.
- Silverman JS, Glusac EJ. Epithelioid cell histiocytoma--histogenetic and kinetics analysis of dermal microvascular unit dendritic cell subpopulations. J Cutan Pathol. 2003;30:415-422.
- Murigu T, Bhatt N, Miller K, et al. Spindle cell-predominant epithelioid fibrous histiocytoma. Histopathology. 2018;72:1233-1236.
- Rabkin MS, Vukmer T. Granular cell variant of epithelioid cell histiocytoma. Am J Dermatopathol. 2012;34:766-769.
- Glusac EJ, Barr RJ, Everett MA, et al. Epithelioid cell histiocytoma. a report of 10 cases including a new cellular variant. Am J Surg Pathol. 1994;18:583-590.
- Creytens D, Ferdinande L, Van Dorpe J. ALK Rearrangement and overexpression in an unusual cutaneous epithelioid tumor with a peculiar whorled "perineurioma-like" growth pattern: epithelioid fibrous histiocytoma. Appl Immunohistochem Mol Morphol. 2017;25:E46-E48.
- Doyle LA, Fletcher CD. EMA positivity in epithelioid fibrous histiocytoma: a potential diagnostic pitfall. J Cutan Pathol. 2011;38:697-703.
- Fabrizi G, Massi G. Polypoid Spitz naevus: the benign counterpart of polypoid malignant melanoma. Br J Dermatol. 2000;142:128-132.
- Plaza JA, De Stefano D, Suster S, et al. Intradermal Spitz nevi: a rare subtype of Spitz nevi analyzed in a clinicopathologic study of 74 cases. Am J Dermatopathol. 2014;36:283-294; quiz 295-287.
- Menezes FD, Mooi WJ. Spitz tumors of the skin. Surg Pathol Clin. 2017;10:281-298.
- Requena C, Requena L, Kutzner H, et al. Spitz nevus: a clinicopathological study of 349 cases. Am J Dermatopathol. 2009;31:107-116.
- Mentzel T, Dei Tos AP, Sapi Z, et al. Myopericytoma of skin and soft tissues: clinicopathologic and immunohistochemical study of 54 cases. Am J Surg Pathol. 2006;30:104-113.
- Aung PP, Goldberg LJ, Mahalingam M, et al. Cutaneous myopericytoma: a report of 3 cases and review of the literature. Dermatopathology (Basel). 2015;2:9-14.
- Morzycki A, Joukhadar N, Murphy A, et al. Digital myopericytoma: a case report and systematic literature review. J Hand Microsurg. 2017;9:32-36.
- LeBlanc RE, Taube J. Myofibroma, myopericytoma, myoepithelioma, and myofibroblastoma of skin and soft tissue. Surg Pathol Clin. 2011;4:745-759.
- Chisolm SS, Schulman JM, Fox LP. Adult xanthogranuloma, reticulohistiocytosis, and Rosai-Dorfman disease. Dermatol Clin. 2015;33:465-472; discussion 473.
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521-528.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatol Online J. 2014. pii:doj_21725.
- Soldano AC, Meehan SA. Cutaneous solitary fibrous tumor: a report of 2 cases and review of the literature. Am J Dermatopathol. 2008;30:54-58.
- Feasel P, Al-Ibraheemi A, Fritchie K, et al. Superficial solitary fibrous tumor: a series of 26 cases. Am J Surg Pathol. 2018;42:778-785.
- Thway K, Ng W, Noujaim J, et al. The current status of solitary fibrous tumor: diagnostic features, variants, and genetics. Int J Surg Pathol. 2016;24:281-292.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
The Diagnosis: Epithelioid Histiocytoma
Epithelioid histiocytoma (EH), also known as epithelioid cell histiocytoma or epithelioid fibrous histiocytoma, is a rare benign fibrohistiocytic tumor first described in 1989.1 Epithelioid histiocytoma commonly presents in middle-aged adults with a slight predilection for males.2 The most frequently affected site is the lower extremity. The arms, trunk, head and neck, groin, and tongue also can be involved.3,4 It usually presents as a solitary asymptomatic papule or nodule, though cases with multiple lesions have been reported.5 Anaplastic lymphoma kinase rearrangement and overexpression have been confirmed and suggest that EH is distinct from conventional cutaneous fibrous histiocytoma.5
Histologically, EH appears as an exophytic, symmetric, and well-demarcated dermal nodule with a classic epidermal collarette. Prominent vascularity with perivascular accentuation of the epithelioid tumor cells is common. Older lesions may be hyalinized and sclerotic. Epithelioid cells commonly account for more than 50% of the tumor and are characterized by eosinophilic cytoplasm, vesicular nuclei, and small eosinophilic nucleoli. A small population of lymphocytes and mast cells are variably present (quiz image, bottom).1-3,7 A predominantly spindle cell variant has been reported.8 Other histopathologic variants include granular cell,9 cellular,10 and EH with perineuriomalike growth.11 Immunohistochemical staining shows anaplastic lymphoma kinase positivity in most cases, and more than half of cases stain positive for factor XIIIa and epithelial membrane antigen. Tumor cells consistently are negative for desmin and cytokeratins.6,10,12 Excision is curative.8
Polypoid Spitz nevus (PSN) is a benign nevus with a conspicuous polypoid or papillary exophytic architecture. The term was coined in 2000 by Fabrizi and Massi.13 Spitz nevus is a benign acquired melanocytic tumor that typically presents in children and adolescents and has a wide histologic spectrum.14 There is some debate on this entity, as some authors do not regard PSN as a distinct histologic variant; thus, it seems underreported in the literature.15 In a review of 349 cases of Spitz nevi, the authors found 7 cases of PSN.16 In another review of 74 cases of intradermal Spitz nevi, 14 cases of PSN were identified.14 This polypoid variant is easily mistaken for a polypoid melanoma because it can show cytologic atypia with large nuclei. Polypoid Spitz nevus usually lacks mitoses, notable pleomorphism, and sheetlike growth, unlike melanoma (Figure 1).13,14
Myopericytoma is an uncommon benign mesenchymal neoplasm that typically presents as a solitary, slowly enlarging and painless nodule with a predilection for the lower extremities, usually in adult males.17-20 Histologically, it consists of a well-circumscribed nodule with numerous thin-walled vessels and a proliferation of ovoid to spindled myopericytes exhibiting a concentric perivascular growth pattern (Figure 2). Myopericytoma usually is positive for smooth muscle actin and h-caldesmon but is negative or only focally positive for desmin. The prognosis is good with rare recurrence, despite incomplete excision.17,18
Solitary reticulohistiocytoma is a rare benign form of non-Langerhans cell histiocytosis.21,22 Unlike its multicentric counterpart, solitary reticulohistiocytoma rarely is associated with systemic disease. It presents as a small, dome-shaped, painless papule or nodule that can affect any part of the body.22,23 Solitary reticulohistiocytoma characteristically demonstrates cells with a ground glass-like appearance and 2-toned cytoplasm. A mixed inflammatory infiltrate including neutrophils, eosinophils, and lymphocytes commonly is present (Figure 3). The epithelioid histiocytes are positive for vimentin and histiocytic markers including CD68 and CD163.22
Solitary fibrous tumor (SFT) is an uncommon mesenchymal fibroblastic neoplasm that can arise at almost any anatomic site.24 Cutaneous SFTs are more common in women, most often involve the head, and appear to behave in an indolent manner.25 Solitary fibrous tumors are translocation-associated neoplasms with a NAB2-STAT6 gene fusion.26 The classic histology of SFT is a spindled fibroblastic proliferation arranged in a "patternless pattern" with interspersed stag horn-like, thin-walled blood vessels (Figure 4). Tumor cells usually are positive for CD34, CD99, and Bcl-2.27 In addition, STAT6 immunoreactivity is useful in diagnosis of SFT.25
The Diagnosis: Epithelioid Histiocytoma
Epithelioid histiocytoma (EH), also known as epithelioid cell histiocytoma or epithelioid fibrous histiocytoma, is a rare benign fibrohistiocytic tumor first described in 1989.1 Epithelioid histiocytoma commonly presents in middle-aged adults with a slight predilection for males.2 The most frequently affected site is the lower extremity. The arms, trunk, head and neck, groin, and tongue also can be involved.3,4 It usually presents as a solitary asymptomatic papule or nodule, though cases with multiple lesions have been reported.5 Anaplastic lymphoma kinase rearrangement and overexpression have been confirmed and suggest that EH is distinct from conventional cutaneous fibrous histiocytoma.5
Histologically, EH appears as an exophytic, symmetric, and well-demarcated dermal nodule with a classic epidermal collarette. Prominent vascularity with perivascular accentuation of the epithelioid tumor cells is common. Older lesions may be hyalinized and sclerotic. Epithelioid cells commonly account for more than 50% of the tumor and are characterized by eosinophilic cytoplasm, vesicular nuclei, and small eosinophilic nucleoli. A small population of lymphocytes and mast cells are variably present (quiz image, bottom).1-3,7 A predominantly spindle cell variant has been reported.8 Other histopathologic variants include granular cell,9 cellular,10 and EH with perineuriomalike growth.11 Immunohistochemical staining shows anaplastic lymphoma kinase positivity in most cases, and more than half of cases stain positive for factor XIIIa and epithelial membrane antigen. Tumor cells consistently are negative for desmin and cytokeratins.6,10,12 Excision is curative.8
Polypoid Spitz nevus (PSN) is a benign nevus with a conspicuous polypoid or papillary exophytic architecture. The term was coined in 2000 by Fabrizi and Massi.13 Spitz nevus is a benign acquired melanocytic tumor that typically presents in children and adolescents and has a wide histologic spectrum.14 There is some debate on this entity, as some authors do not regard PSN as a distinct histologic variant; thus, it seems underreported in the literature.15 In a review of 349 cases of Spitz nevi, the authors found 7 cases of PSN.16 In another review of 74 cases of intradermal Spitz nevi, 14 cases of PSN were identified.14 This polypoid variant is easily mistaken for a polypoid melanoma because it can show cytologic atypia with large nuclei. Polypoid Spitz nevus usually lacks mitoses, notable pleomorphism, and sheetlike growth, unlike melanoma (Figure 1).13,14
Myopericytoma is an uncommon benign mesenchymal neoplasm that typically presents as a solitary, slowly enlarging and painless nodule with a predilection for the lower extremities, usually in adult males.17-20 Histologically, it consists of a well-circumscribed nodule with numerous thin-walled vessels and a proliferation of ovoid to spindled myopericytes exhibiting a concentric perivascular growth pattern (Figure 2). Myopericytoma usually is positive for smooth muscle actin and h-caldesmon but is negative or only focally positive for desmin. The prognosis is good with rare recurrence, despite incomplete excision.17,18
Solitary reticulohistiocytoma is a rare benign form of non-Langerhans cell histiocytosis.21,22 Unlike its multicentric counterpart, solitary reticulohistiocytoma rarely is associated with systemic disease. It presents as a small, dome-shaped, painless papule or nodule that can affect any part of the body.22,23 Solitary reticulohistiocytoma characteristically demonstrates cells with a ground glass-like appearance and 2-toned cytoplasm. A mixed inflammatory infiltrate including neutrophils, eosinophils, and lymphocytes commonly is present (Figure 3). The epithelioid histiocytes are positive for vimentin and histiocytic markers including CD68 and CD163.22
Solitary fibrous tumor (SFT) is an uncommon mesenchymal fibroblastic neoplasm that can arise at almost any anatomic site.24 Cutaneous SFTs are more common in women, most often involve the head, and appear to behave in an indolent manner.25 Solitary fibrous tumors are translocation-associated neoplasms with a NAB2-STAT6 gene fusion.26 The classic histology of SFT is a spindled fibroblastic proliferation arranged in a "patternless pattern" with interspersed stag horn-like, thin-walled blood vessels (Figure 4). Tumor cells usually are positive for CD34, CD99, and Bcl-2.27 In addition, STAT6 immunoreactivity is useful in diagnosis of SFT.25
- Jones EW, Cerio R, Smith NP. Epithelioid cell histiocytoma: a new entity. Br J Dermatol. 1989;120:185-195.
- Singh Gomez C, Calonje E, Fletcher CD. Epithelioid benign fibrous histiocytoma of skin: clinico-pathological analysis of 20 cases of a poorly known variant. Histopathology. 1994;24:123-129.
- Felty CC, Linos K. Epithelioid fibrous histiocytoma: a concise review [published online October 4, 2018]. Am J Dermatopathol. doi:10.1097/DAD.0000000000001272.
- Rawal YB, Kalmar JR, Shumway B, et al. Presentation of an epithelioid cell histiocytoma on the ventral tongue. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:75-83.
- Cangelosi JJ, Prieto VG, Baker GF, et al. Unusual presentation of multiple epithelioid cell histiocytomas. Am J Dermatopathol. 2008;30:373-376.
- Doyle LA, Marino-Enriquez A, Fletcher CD, et al. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol. 2015;28:904-912.
- Silverman JS, Glusac EJ. Epithelioid cell histiocytoma--histogenetic and kinetics analysis of dermal microvascular unit dendritic cell subpopulations. J Cutan Pathol. 2003;30:415-422.
- Murigu T, Bhatt N, Miller K, et al. Spindle cell-predominant epithelioid fibrous histiocytoma. Histopathology. 2018;72:1233-1236.
- Rabkin MS, Vukmer T. Granular cell variant of epithelioid cell histiocytoma. Am J Dermatopathol. 2012;34:766-769.
- Glusac EJ, Barr RJ, Everett MA, et al. Epithelioid cell histiocytoma. a report of 10 cases including a new cellular variant. Am J Surg Pathol. 1994;18:583-590.
- Creytens D, Ferdinande L, Van Dorpe J. ALK Rearrangement and overexpression in an unusual cutaneous epithelioid tumor with a peculiar whorled "perineurioma-like" growth pattern: epithelioid fibrous histiocytoma. Appl Immunohistochem Mol Morphol. 2017;25:E46-E48.
- Doyle LA, Fletcher CD. EMA positivity in epithelioid fibrous histiocytoma: a potential diagnostic pitfall. J Cutan Pathol. 2011;38:697-703.
- Fabrizi G, Massi G. Polypoid Spitz naevus: the benign counterpart of polypoid malignant melanoma. Br J Dermatol. 2000;142:128-132.
- Plaza JA, De Stefano D, Suster S, et al. Intradermal Spitz nevi: a rare subtype of Spitz nevi analyzed in a clinicopathologic study of 74 cases. Am J Dermatopathol. 2014;36:283-294; quiz 295-287.
- Menezes FD, Mooi WJ. Spitz tumors of the skin. Surg Pathol Clin. 2017;10:281-298.
- Requena C, Requena L, Kutzner H, et al. Spitz nevus: a clinicopathological study of 349 cases. Am J Dermatopathol. 2009;31:107-116.
- Mentzel T, Dei Tos AP, Sapi Z, et al. Myopericytoma of skin and soft tissues: clinicopathologic and immunohistochemical study of 54 cases. Am J Surg Pathol. 2006;30:104-113.
- Aung PP, Goldberg LJ, Mahalingam M, et al. Cutaneous myopericytoma: a report of 3 cases and review of the literature. Dermatopathology (Basel). 2015;2:9-14.
- Morzycki A, Joukhadar N, Murphy A, et al. Digital myopericytoma: a case report and systematic literature review. J Hand Microsurg. 2017;9:32-36.
- LeBlanc RE, Taube J. Myofibroma, myopericytoma, myoepithelioma, and myofibroblastoma of skin and soft tissue. Surg Pathol Clin. 2011;4:745-759.
- Chisolm SS, Schulman JM, Fox LP. Adult xanthogranuloma, reticulohistiocytosis, and Rosai-Dorfman disease. Dermatol Clin. 2015;33:465-472; discussion 473.
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521-528.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatol Online J. 2014. pii:doj_21725.
- Soldano AC, Meehan SA. Cutaneous solitary fibrous tumor: a report of 2 cases and review of the literature. Am J Dermatopathol. 2008;30:54-58.
- Feasel P, Al-Ibraheemi A, Fritchie K, et al. Superficial solitary fibrous tumor: a series of 26 cases. Am J Surg Pathol. 2018;42:778-785.
- Thway K, Ng W, Noujaim J, et al. The current status of solitary fibrous tumor: diagnostic features, variants, and genetics. Int J Surg Pathol. 2016;24:281-292.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
- Jones EW, Cerio R, Smith NP. Epithelioid cell histiocytoma: a new entity. Br J Dermatol. 1989;120:185-195.
- Singh Gomez C, Calonje E, Fletcher CD. Epithelioid benign fibrous histiocytoma of skin: clinico-pathological analysis of 20 cases of a poorly known variant. Histopathology. 1994;24:123-129.
- Felty CC, Linos K. Epithelioid fibrous histiocytoma: a concise review [published online October 4, 2018]. Am J Dermatopathol. doi:10.1097/DAD.0000000000001272.
- Rawal YB, Kalmar JR, Shumway B, et al. Presentation of an epithelioid cell histiocytoma on the ventral tongue. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:75-83.
- Cangelosi JJ, Prieto VG, Baker GF, et al. Unusual presentation of multiple epithelioid cell histiocytomas. Am J Dermatopathol. 2008;30:373-376.
- Doyle LA, Marino-Enriquez A, Fletcher CD, et al. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod Pathol. 2015;28:904-912.
- Silverman JS, Glusac EJ. Epithelioid cell histiocytoma--histogenetic and kinetics analysis of dermal microvascular unit dendritic cell subpopulations. J Cutan Pathol. 2003;30:415-422.
- Murigu T, Bhatt N, Miller K, et al. Spindle cell-predominant epithelioid fibrous histiocytoma. Histopathology. 2018;72:1233-1236.
- Rabkin MS, Vukmer T. Granular cell variant of epithelioid cell histiocytoma. Am J Dermatopathol. 2012;34:766-769.
- Glusac EJ, Barr RJ, Everett MA, et al. Epithelioid cell histiocytoma. a report of 10 cases including a new cellular variant. Am J Surg Pathol. 1994;18:583-590.
- Creytens D, Ferdinande L, Van Dorpe J. ALK Rearrangement and overexpression in an unusual cutaneous epithelioid tumor with a peculiar whorled "perineurioma-like" growth pattern: epithelioid fibrous histiocytoma. Appl Immunohistochem Mol Morphol. 2017;25:E46-E48.
- Doyle LA, Fletcher CD. EMA positivity in epithelioid fibrous histiocytoma: a potential diagnostic pitfall. J Cutan Pathol. 2011;38:697-703.
- Fabrizi G, Massi G. Polypoid Spitz naevus: the benign counterpart of polypoid malignant melanoma. Br J Dermatol. 2000;142:128-132.
- Plaza JA, De Stefano D, Suster S, et al. Intradermal Spitz nevi: a rare subtype of Spitz nevi analyzed in a clinicopathologic study of 74 cases. Am J Dermatopathol. 2014;36:283-294; quiz 295-287.
- Menezes FD, Mooi WJ. Spitz tumors of the skin. Surg Pathol Clin. 2017;10:281-298.
- Requena C, Requena L, Kutzner H, et al. Spitz nevus: a clinicopathological study of 349 cases. Am J Dermatopathol. 2009;31:107-116.
- Mentzel T, Dei Tos AP, Sapi Z, et al. Myopericytoma of skin and soft tissues: clinicopathologic and immunohistochemical study of 54 cases. Am J Surg Pathol. 2006;30:104-113.
- Aung PP, Goldberg LJ, Mahalingam M, et al. Cutaneous myopericytoma: a report of 3 cases and review of the literature. Dermatopathology (Basel). 2015;2:9-14.
- Morzycki A, Joukhadar N, Murphy A, et al. Digital myopericytoma: a case report and systematic literature review. J Hand Microsurg. 2017;9:32-36.
- LeBlanc RE, Taube J. Myofibroma, myopericytoma, myoepithelioma, and myofibroblastoma of skin and soft tissue. Surg Pathol Clin. 2011;4:745-759.
- Chisolm SS, Schulman JM, Fox LP. Adult xanthogranuloma, reticulohistiocytosis, and Rosai-Dorfman disease. Dermatol Clin. 2015;33:465-472; discussion 473.
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521-528.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatol Online J. 2014. pii:doj_21725.
- Soldano AC, Meehan SA. Cutaneous solitary fibrous tumor: a report of 2 cases and review of the literature. Am J Dermatopathol. 2008;30:54-58.
- Feasel P, Al-Ibraheemi A, Fritchie K, et al. Superficial solitary fibrous tumor: a series of 26 cases. Am J Surg Pathol. 2018;42:778-785.
- Thway K, Ng W, Noujaim J, et al. The current status of solitary fibrous tumor: diagnostic features, variants, and genetics. Int J Surg Pathol. 2016;24:281-292.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
A 28-year-old man presented with a growing asymptomatic papule on the right leg.
Pigmented Mass on the Shoulder
The Diagnosis: Pigmented Dermatofibrosarcoma Protuberans
Pigmented dermatofibrosarcoma protuberans (PDFSP), also known as Bednar tumor, is an uncommon variant of dermatofibrosarcoma protuberans (DFSP). Pigmented dermatofibrosarcoma protuberans constitutes 1% to 5% of all DFSP cases and most commonly is seen in nonwhite adults in the fourth decade of life, with occasional cases seen in pediatric patients, including some congenital cases. Typical sites of involvement include the shoulders, trunk, arms, legs, head, and neck.1,2 It also has been reported at sites of prior immunization, trauma, and insect bites.3
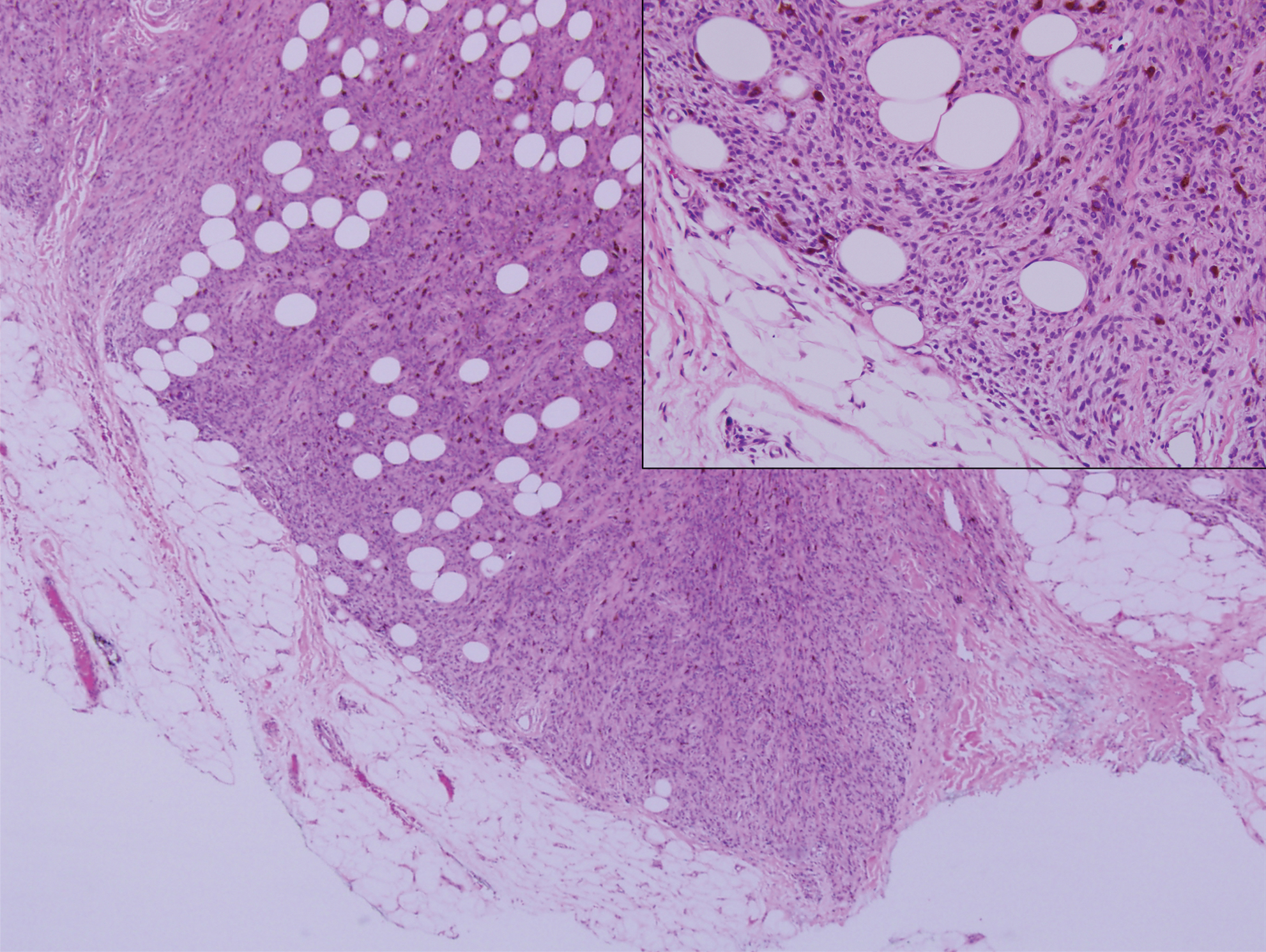
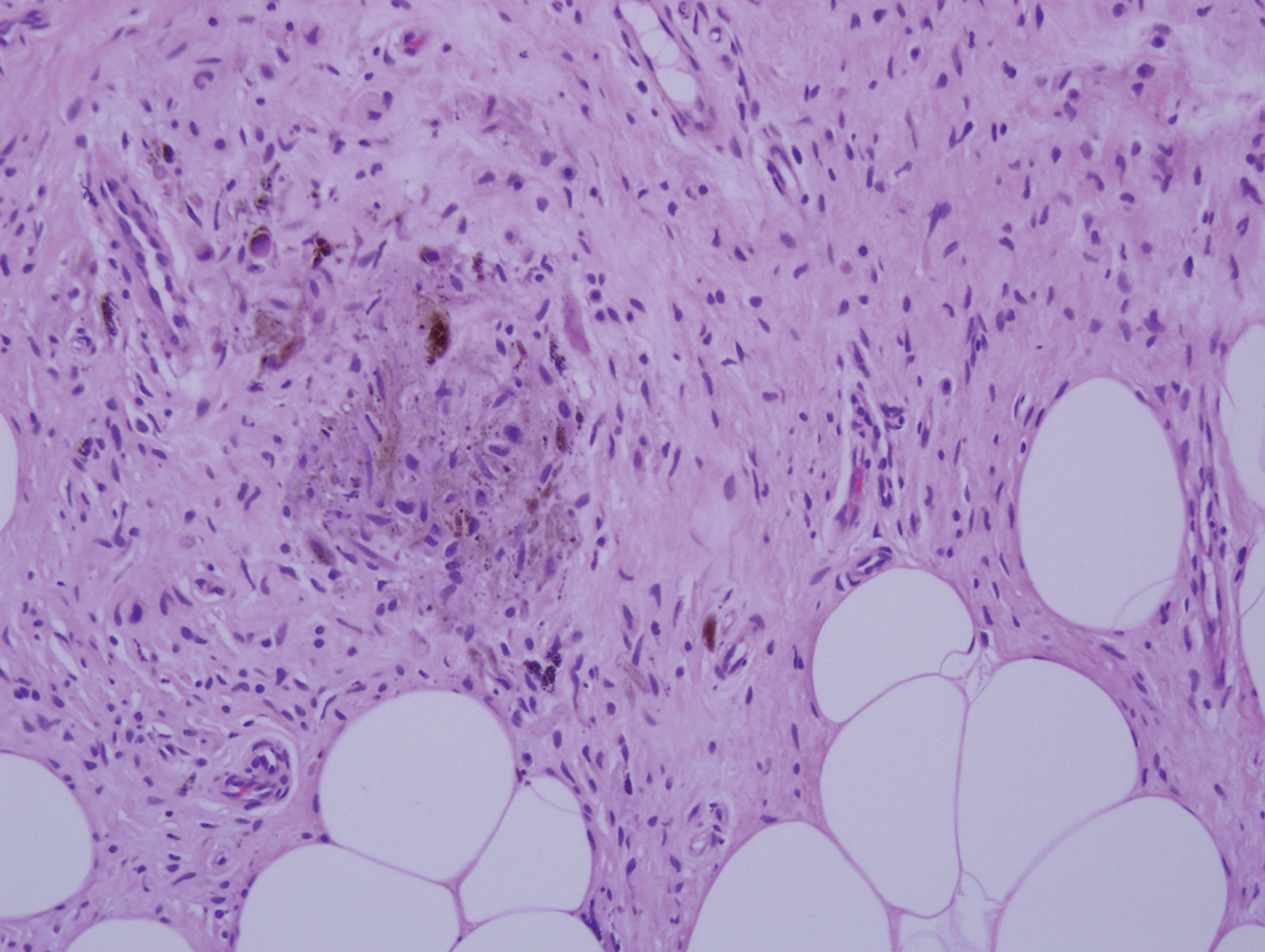
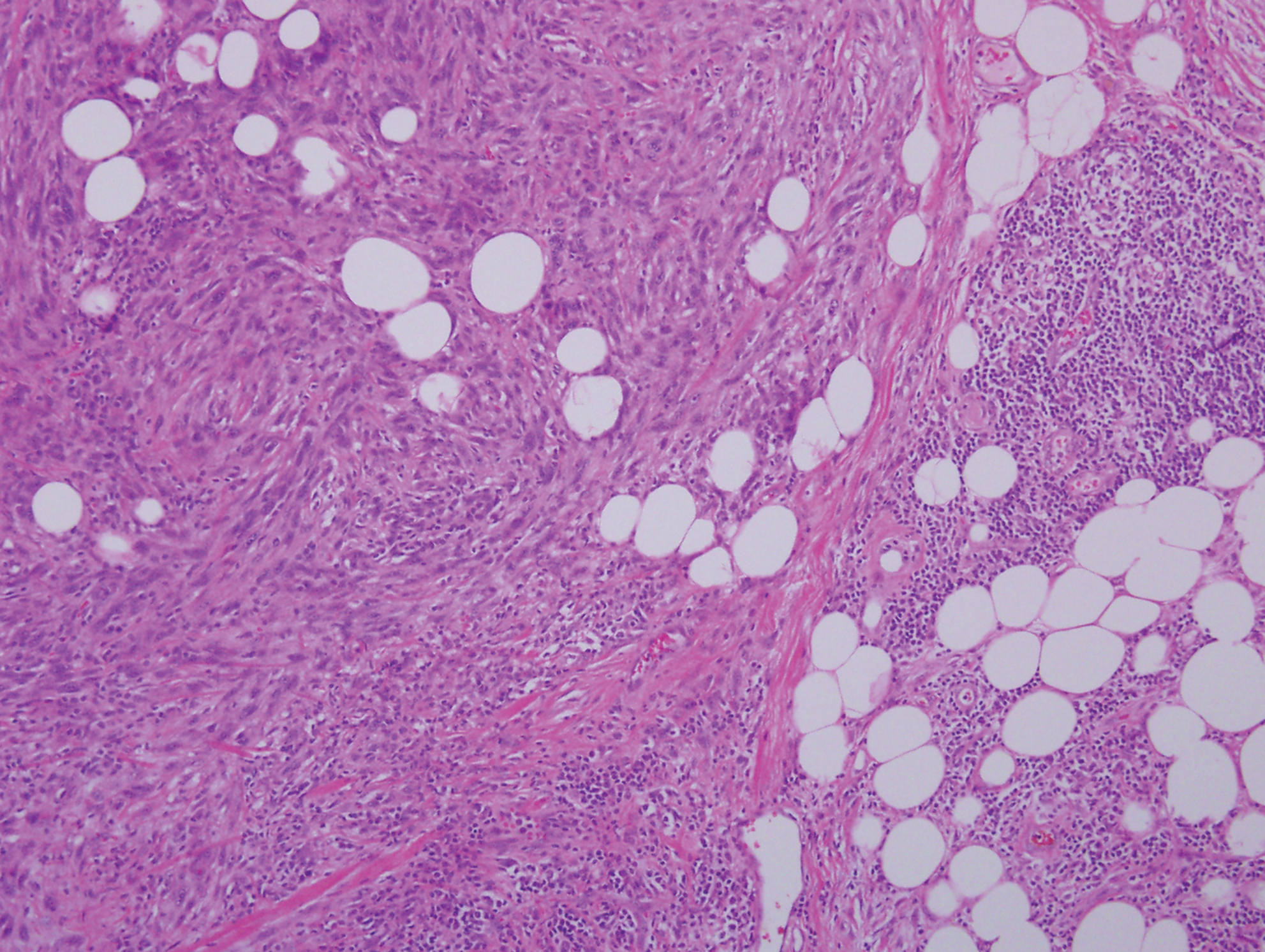
Histopathologic examination of our patient's shoulder nodule revealed an infiltrative neoplasm in the dermis and subcutaneous tissue composed of spindled cells with a storiform pattern and foci of scattered elongated dendritic pigmented cells. A narrow grenz zone separated the tumor from the epidermis, and characteristic honeycomb infiltration by tumor cells was noted in the subcutaneous fat. The nuclei were bland and monomorphous with areas of neuroid differentiation containing whorls and nerve cord-like structures (quiz image). The tumor cells were diffusely CD34 and vimentin positive, while S-100, SOX-10, neurofilament, smooth muscle actin, desmin, epithelial membrane antigen, and cytokeratins were negative. The immunophenotype excluded the possibility of neurogenic, pericytic, myofibroblastic, and myoid differentiation.
Wang and Yang4 previously reported a case of PDFSP with prominent meningothelial-like whorls focally resembling extracranial meningioma; however, the tumor cells were CD34 positive and epithelial membrane antigen negative, weighing against a diagnosis of meningioma. Most cases of PDFSP demonstrate the COL1A1-PDGFB (collagen type I α; 1/platelet-derived growth factor B-chain) fusion protein caused by the translocation t(17;22)(q22;q13), as in classic DFSP.5
Cellular blue nevus (CBN) is a benign melanocytic neoplasm that can present at any age and often occurs on the buttocks and in the sacrococcygeal region. Clinically, CBN presents as a firm, bluish black to bluish gray, dome-shaped nodule. The size varies from a few millimeters to several centimeters.6,7 Histologically, CBN is located completely in the dermis, extending along the adnexae into the subcutaneous tissue with a dumbbell-shaped outline (Figure 1).6-8 The tumor demonstrates oval epithelioid melanocytes with vesicular nuclei and prominent nucleoli. Immunohistochemically, tumor cells stain positively for melanocytic markers such as S-100, SOX-10, MART-1, and human melanoma black 45. CD34 expression rarely is reported in a subset of CBN.9
Pigmented neurofibroma is a rare variant of neurofibroma that produces melanin pigment and has a strong association with neurofibromatosis.10 It occurs most frequently in dark-skinned populations (Fitzpatrick skin types IV-VI). The most common location is the head and neck region.11,12 Histologically, pigmented neurofibroma resembles a diffuse neurofibroma admixed with melanin-producing cells (Figure 2).12 Immunostaining shows positivity for S-100 in both pigmented and Schwann cells; however, the pigmented cells stain positively for human melanoma black 45, Melan-A, and tyrosinase.10 CD34 can be fingerprint positive in neurofibroma, but a distinction from DFSP can be made by S-100 and SOX-10 immunostaining.13
Desmoplastic melanoma (DM) is an uncommon variant of malignant melanoma and has a higher tendency for persistent local growth and less frequent metastases than other variants of melanoma. It has a predilection for chronically sun-exposed areas such as the head and neck and occurs later in life. Clinically, DM appears as nonspecific, often amelanotic nodules or plaques or as scarlike lesions.14 Histologically, DM can be classified as mixed or pure based on the degree of desmoplasia and cellularity. A paucicellular proliferation of malignant spindled melanocytes within a densely fibrotic stroma with lymphoid nodules in the dermis is characteristic (Figure 3); perineural involvement is common.14,15 The most reliable confirmative stains are S-100 and SOX-10.16
Cutaneous meningioma is a rare tumor and could be subtyped into 3 groups. Type I is primary cutaneous meningioma and usually is present at birth on the scalp and paravertebral regions with a relatively good prognosis. Type II is ectopic soft-tissue meningioma that extends into the skin from around the sensory organs on the face. Type III is local invasion or true metastasis from a central nervous system meningioma. Types II and III develop later in life and the prognosis is poor.17,18 Clinically, lesions present as firm subcutaneous nodules or swellings. Cutaneous meningioma has several histopathologic variants. The classic presentation reveals concentric wrapping of tumor cells with round-oval nuclei containing delicate chromatin. Psammoma bodies are a common finding (Figure 4). Immunohistochemically, tumor cells are diffusely positive for epithelial membrane antigen and vimentin.18,19
- Amonkar GP, Rupani A, Shah A, et al. Bednar tumor: an uncommon entity. Dermatopathology (Basel). 2016;3:36-38.
- El Hachem M, Diociaiuti A, Latella E, et al. Congenital myxoid and pigmented dermatofibrosarcoma protuberans: a case report. Pediatr Dermatol. 2013;30:E74-E77.
- Anon-Requena MJ, Pico-Valimana M, Munoz-Arias G. Bednar tumor (pigmented dermatofibrosarcoma protuberans). Actas Dermosifiliogr. 2016;107:618-620.
- Wang J, Yang W. Pigmented dermatofibrosarcoma protuberans with prominent meningothelial-like whorls. J Cutan Pathol. 2008;35(suppl 1):65-69.
- Zardawi IM, Kattampallil J, Rode J. An unusual pigmented skin tumour. Bednar tumour, dorsum of left foot (pigmented dermatofibrosarcoma protuberans). Pathology. 2004;36:358-361.
- Sugianto JZ, Ralston JS, Metcalf JS, et al. Blue nevus and "malignant blue nevus": a concise review. Semin Diagn Pathol. 2016;33:219-224.
- Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2017;37:401-415.
- Zembowicz A, Granter SR, McKee PH, et al. Amelanotic cellular blue nevus: a hypopigmented variant of the cellular blue nevus: clinicopathologic analysis of 20 cases. Am J Surg Pathol. 2002;26:1493-1500.
- Smith K, Germain M, Williams J, et al. CD34-positive cellular blue nevi. J Cutan Pathol. 2001;28:145-150.
- Inaba M, Yamamoto T, Minami R, et al. Pigmented neurofibroma: report of two cases and literature review. Pathol Int. 2001;51:565-569.
- Fetsch JF, Michal M, Miettinen M. Pigmented (melanotic) neurofibroma: a clinicopathologic and immunohistochemical analysis of 19 lesions from 17 patients. Am J Surg Pathol. 2000;24:331-343.
- Motoi T, Ishida T, Kawato A, et al. Pigmented neurofibroma: review of Japanese patients with an analysis of melanogenesis demonstrating coexpression of c-met protooncogene and microphthalmia-associated transcription factor. Hum Pathol. 2005;36:871-877.
- Yeh I, McCalmont TH. Distinguishing neurofibroma from desmoplastic melanoma: the value of the CD34 fingerprint. J Cutan Pathol. 2011;38:625-630.
- Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
- Busam KJ. Desmoplastic melanoma. Clin Lab Med. 2011;31:321-330.
- Schleich C, Ferringer T. Desmoplastic melanoma. Cutis. 2015;96:306, 313-314, 335.
- Lopez DA, Silvers DN, Helwig EB. Cutaneous meningiomas--a clinicopathologic study. Cancer. 1974;34:728-744.
- Miedema JR, Zedek D. Cutaneous meningioma. Arch Pathol Lab Med. 2012;136:208-211.
- Bhanusali DG, Heath C, Gur D, et al. Metastatic meningioma of the scalp. Cutis. 2018;101:386-389.
The Diagnosis: Pigmented Dermatofibrosarcoma Protuberans
Pigmented dermatofibrosarcoma protuberans (PDFSP), also known as Bednar tumor, is an uncommon variant of dermatofibrosarcoma protuberans (DFSP). Pigmented dermatofibrosarcoma protuberans constitutes 1% to 5% of all DFSP cases and most commonly is seen in nonwhite adults in the fourth decade of life, with occasional cases seen in pediatric patients, including some congenital cases. Typical sites of involvement include the shoulders, trunk, arms, legs, head, and neck.1,2 It also has been reported at sites of prior immunization, trauma, and insect bites.3
Histopathologic examination of our patient's shoulder nodule revealed an infiltrative neoplasm in the dermis and subcutaneous tissue composed of spindled cells with a storiform pattern and foci of scattered elongated dendritic pigmented cells. A narrow grenz zone separated the tumor from the epidermis, and characteristic honeycomb infiltration by tumor cells was noted in the subcutaneous fat. The nuclei were bland and monomorphous with areas of neuroid differentiation containing whorls and nerve cord-like structures (quiz image). The tumor cells were diffusely CD34 and vimentin positive, while S-100, SOX-10, neurofilament, smooth muscle actin, desmin, epithelial membrane antigen, and cytokeratins were negative. The immunophenotype excluded the possibility of neurogenic, pericytic, myofibroblastic, and myoid differentiation.
Wang and Yang4 previously reported a case of PDFSP with prominent meningothelial-like whorls focally resembling extracranial meningioma; however, the tumor cells were CD34 positive and epithelial membrane antigen negative, weighing against a diagnosis of meningioma. Most cases of PDFSP demonstrate the COL1A1-PDGFB (collagen type I α; 1/platelet-derived growth factor B-chain) fusion protein caused by the translocation t(17;22)(q22;q13), as in classic DFSP.5
Cellular blue nevus (CBN) is a benign melanocytic neoplasm that can present at any age and often occurs on the buttocks and in the sacrococcygeal region. Clinically, CBN presents as a firm, bluish black to bluish gray, dome-shaped nodule. The size varies from a few millimeters to several centimeters.6,7 Histologically, CBN is located completely in the dermis, extending along the adnexae into the subcutaneous tissue with a dumbbell-shaped outline (Figure 1).6-8 The tumor demonstrates oval epithelioid melanocytes with vesicular nuclei and prominent nucleoli. Immunohistochemically, tumor cells stain positively for melanocytic markers such as S-100, SOX-10, MART-1, and human melanoma black 45. CD34 expression rarely is reported in a subset of CBN.9
Pigmented neurofibroma is a rare variant of neurofibroma that produces melanin pigment and has a strong association with neurofibromatosis.10 It occurs most frequently in dark-skinned populations (Fitzpatrick skin types IV-VI). The most common location is the head and neck region.11,12 Histologically, pigmented neurofibroma resembles a diffuse neurofibroma admixed with melanin-producing cells (Figure 2).12 Immunostaining shows positivity for S-100 in both pigmented and Schwann cells; however, the pigmented cells stain positively for human melanoma black 45, Melan-A, and tyrosinase.10 CD34 can be fingerprint positive in neurofibroma, but a distinction from DFSP can be made by S-100 and SOX-10 immunostaining.13
Desmoplastic melanoma (DM) is an uncommon variant of malignant melanoma and has a higher tendency for persistent local growth and less frequent metastases than other variants of melanoma. It has a predilection for chronically sun-exposed areas such as the head and neck and occurs later in life. Clinically, DM appears as nonspecific, often amelanotic nodules or plaques or as scarlike lesions.14 Histologically, DM can be classified as mixed or pure based on the degree of desmoplasia and cellularity. A paucicellular proliferation of malignant spindled melanocytes within a densely fibrotic stroma with lymphoid nodules in the dermis is characteristic (Figure 3); perineural involvement is common.14,15 The most reliable confirmative stains are S-100 and SOX-10.16
Cutaneous meningioma is a rare tumor and could be subtyped into 3 groups. Type I is primary cutaneous meningioma and usually is present at birth on the scalp and paravertebral regions with a relatively good prognosis. Type II is ectopic soft-tissue meningioma that extends into the skin from around the sensory organs on the face. Type III is local invasion or true metastasis from a central nervous system meningioma. Types II and III develop later in life and the prognosis is poor.17,18 Clinically, lesions present as firm subcutaneous nodules or swellings. Cutaneous meningioma has several histopathologic variants. The classic presentation reveals concentric wrapping of tumor cells with round-oval nuclei containing delicate chromatin. Psammoma bodies are a common finding (Figure 4). Immunohistochemically, tumor cells are diffusely positive for epithelial membrane antigen and vimentin.18,19
The Diagnosis: Pigmented Dermatofibrosarcoma Protuberans
Pigmented dermatofibrosarcoma protuberans (PDFSP), also known as Bednar tumor, is an uncommon variant of dermatofibrosarcoma protuberans (DFSP). Pigmented dermatofibrosarcoma protuberans constitutes 1% to 5% of all DFSP cases and most commonly is seen in nonwhite adults in the fourth decade of life, with occasional cases seen in pediatric patients, including some congenital cases. Typical sites of involvement include the shoulders, trunk, arms, legs, head, and neck.1,2 It also has been reported at sites of prior immunization, trauma, and insect bites.3
Histopathologic examination of our patient's shoulder nodule revealed an infiltrative neoplasm in the dermis and subcutaneous tissue composed of spindled cells with a storiform pattern and foci of scattered elongated dendritic pigmented cells. A narrow grenz zone separated the tumor from the epidermis, and characteristic honeycomb infiltration by tumor cells was noted in the subcutaneous fat. The nuclei were bland and monomorphous with areas of neuroid differentiation containing whorls and nerve cord-like structures (quiz image). The tumor cells were diffusely CD34 and vimentin positive, while S-100, SOX-10, neurofilament, smooth muscle actin, desmin, epithelial membrane antigen, and cytokeratins were negative. The immunophenotype excluded the possibility of neurogenic, pericytic, myofibroblastic, and myoid differentiation.
Wang and Yang4 previously reported a case of PDFSP with prominent meningothelial-like whorls focally resembling extracranial meningioma; however, the tumor cells were CD34 positive and epithelial membrane antigen negative, weighing against a diagnosis of meningioma. Most cases of PDFSP demonstrate the COL1A1-PDGFB (collagen type I α; 1/platelet-derived growth factor B-chain) fusion protein caused by the translocation t(17;22)(q22;q13), as in classic DFSP.5
Cellular blue nevus (CBN) is a benign melanocytic neoplasm that can present at any age and often occurs on the buttocks and in the sacrococcygeal region. Clinically, CBN presents as a firm, bluish black to bluish gray, dome-shaped nodule. The size varies from a few millimeters to several centimeters.6,7 Histologically, CBN is located completely in the dermis, extending along the adnexae into the subcutaneous tissue with a dumbbell-shaped outline (Figure 1).6-8 The tumor demonstrates oval epithelioid melanocytes with vesicular nuclei and prominent nucleoli. Immunohistochemically, tumor cells stain positively for melanocytic markers such as S-100, SOX-10, MART-1, and human melanoma black 45. CD34 expression rarely is reported in a subset of CBN.9
Pigmented neurofibroma is a rare variant of neurofibroma that produces melanin pigment and has a strong association with neurofibromatosis.10 It occurs most frequently in dark-skinned populations (Fitzpatrick skin types IV-VI). The most common location is the head and neck region.11,12 Histologically, pigmented neurofibroma resembles a diffuse neurofibroma admixed with melanin-producing cells (Figure 2).12 Immunostaining shows positivity for S-100 in both pigmented and Schwann cells; however, the pigmented cells stain positively for human melanoma black 45, Melan-A, and tyrosinase.10 CD34 can be fingerprint positive in neurofibroma, but a distinction from DFSP can be made by S-100 and SOX-10 immunostaining.13
Desmoplastic melanoma (DM) is an uncommon variant of malignant melanoma and has a higher tendency for persistent local growth and less frequent metastases than other variants of melanoma. It has a predilection for chronically sun-exposed areas such as the head and neck and occurs later in life. Clinically, DM appears as nonspecific, often amelanotic nodules or plaques or as scarlike lesions.14 Histologically, DM can be classified as mixed or pure based on the degree of desmoplasia and cellularity. A paucicellular proliferation of malignant spindled melanocytes within a densely fibrotic stroma with lymphoid nodules in the dermis is characteristic (Figure 3); perineural involvement is common.14,15 The most reliable confirmative stains are S-100 and SOX-10.16
Cutaneous meningioma is a rare tumor and could be subtyped into 3 groups. Type I is primary cutaneous meningioma and usually is present at birth on the scalp and paravertebral regions with a relatively good prognosis. Type II is ectopic soft-tissue meningioma that extends into the skin from around the sensory organs on the face. Type III is local invasion or true metastasis from a central nervous system meningioma. Types II and III develop later in life and the prognosis is poor.17,18 Clinically, lesions present as firm subcutaneous nodules or swellings. Cutaneous meningioma has several histopathologic variants. The classic presentation reveals concentric wrapping of tumor cells with round-oval nuclei containing delicate chromatin. Psammoma bodies are a common finding (Figure 4). Immunohistochemically, tumor cells are diffusely positive for epithelial membrane antigen and vimentin.18,19
- Amonkar GP, Rupani A, Shah A, et al. Bednar tumor: an uncommon entity. Dermatopathology (Basel). 2016;3:36-38.
- El Hachem M, Diociaiuti A, Latella E, et al. Congenital myxoid and pigmented dermatofibrosarcoma protuberans: a case report. Pediatr Dermatol. 2013;30:E74-E77.
- Anon-Requena MJ, Pico-Valimana M, Munoz-Arias G. Bednar tumor (pigmented dermatofibrosarcoma protuberans). Actas Dermosifiliogr. 2016;107:618-620.
- Wang J, Yang W. Pigmented dermatofibrosarcoma protuberans with prominent meningothelial-like whorls. J Cutan Pathol. 2008;35(suppl 1):65-69.
- Zardawi IM, Kattampallil J, Rode J. An unusual pigmented skin tumour. Bednar tumour, dorsum of left foot (pigmented dermatofibrosarcoma protuberans). Pathology. 2004;36:358-361.
- Sugianto JZ, Ralston JS, Metcalf JS, et al. Blue nevus and "malignant blue nevus": a concise review. Semin Diagn Pathol. 2016;33:219-224.
- Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2017;37:401-415.
- Zembowicz A, Granter SR, McKee PH, et al. Amelanotic cellular blue nevus: a hypopigmented variant of the cellular blue nevus: clinicopathologic analysis of 20 cases. Am J Surg Pathol. 2002;26:1493-1500.
- Smith K, Germain M, Williams J, et al. CD34-positive cellular blue nevi. J Cutan Pathol. 2001;28:145-150.
- Inaba M, Yamamoto T, Minami R, et al. Pigmented neurofibroma: report of two cases and literature review. Pathol Int. 2001;51:565-569.
- Fetsch JF, Michal M, Miettinen M. Pigmented (melanotic) neurofibroma: a clinicopathologic and immunohistochemical analysis of 19 lesions from 17 patients. Am J Surg Pathol. 2000;24:331-343.
- Motoi T, Ishida T, Kawato A, et al. Pigmented neurofibroma: review of Japanese patients with an analysis of melanogenesis demonstrating coexpression of c-met protooncogene and microphthalmia-associated transcription factor. Hum Pathol. 2005;36:871-877.
- Yeh I, McCalmont TH. Distinguishing neurofibroma from desmoplastic melanoma: the value of the CD34 fingerprint. J Cutan Pathol. 2011;38:625-630.
- Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
- Busam KJ. Desmoplastic melanoma. Clin Lab Med. 2011;31:321-330.
- Schleich C, Ferringer T. Desmoplastic melanoma. Cutis. 2015;96:306, 313-314, 335.
- Lopez DA, Silvers DN, Helwig EB. Cutaneous meningiomas--a clinicopathologic study. Cancer. 1974;34:728-744.
- Miedema JR, Zedek D. Cutaneous meningioma. Arch Pathol Lab Med. 2012;136:208-211.
- Bhanusali DG, Heath C, Gur D, et al. Metastatic meningioma of the scalp. Cutis. 2018;101:386-389.
- Amonkar GP, Rupani A, Shah A, et al. Bednar tumor: an uncommon entity. Dermatopathology (Basel). 2016;3:36-38.
- El Hachem M, Diociaiuti A, Latella E, et al. Congenital myxoid and pigmented dermatofibrosarcoma protuberans: a case report. Pediatr Dermatol. 2013;30:E74-E77.
- Anon-Requena MJ, Pico-Valimana M, Munoz-Arias G. Bednar tumor (pigmented dermatofibrosarcoma protuberans). Actas Dermosifiliogr. 2016;107:618-620.
- Wang J, Yang W. Pigmented dermatofibrosarcoma protuberans with prominent meningothelial-like whorls. J Cutan Pathol. 2008;35(suppl 1):65-69.
- Zardawi IM, Kattampallil J, Rode J. An unusual pigmented skin tumour. Bednar tumour, dorsum of left foot (pigmented dermatofibrosarcoma protuberans). Pathology. 2004;36:358-361.
- Sugianto JZ, Ralston JS, Metcalf JS, et al. Blue nevus and "malignant blue nevus": a concise review. Semin Diagn Pathol. 2016;33:219-224.
- Zembowicz A. Blue nevi and related tumors. Clin Lab Med. 2017;37:401-415.
- Zembowicz A, Granter SR, McKee PH, et al. Amelanotic cellular blue nevus: a hypopigmented variant of the cellular blue nevus: clinicopathologic analysis of 20 cases. Am J Surg Pathol. 2002;26:1493-1500.
- Smith K, Germain M, Williams J, et al. CD34-positive cellular blue nevi. J Cutan Pathol. 2001;28:145-150.
- Inaba M, Yamamoto T, Minami R, et al. Pigmented neurofibroma: report of two cases and literature review. Pathol Int. 2001;51:565-569.
- Fetsch JF, Michal M, Miettinen M. Pigmented (melanotic) neurofibroma: a clinicopathologic and immunohistochemical analysis of 19 lesions from 17 patients. Am J Surg Pathol. 2000;24:331-343.
- Motoi T, Ishida T, Kawato A, et al. Pigmented neurofibroma: review of Japanese patients with an analysis of melanogenesis demonstrating coexpression of c-met protooncogene and microphthalmia-associated transcription factor. Hum Pathol. 2005;36:871-877.
- Yeh I, McCalmont TH. Distinguishing neurofibroma from desmoplastic melanoma: the value of the CD34 fingerprint. J Cutan Pathol. 2011;38:625-630.
- Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
- Busam KJ. Desmoplastic melanoma. Clin Lab Med. 2011;31:321-330.
- Schleich C, Ferringer T. Desmoplastic melanoma. Cutis. 2015;96:306, 313-314, 335.
- Lopez DA, Silvers DN, Helwig EB. Cutaneous meningiomas--a clinicopathologic study. Cancer. 1974;34:728-744.
- Miedema JR, Zedek D. Cutaneous meningioma. Arch Pathol Lab Med. 2012;136:208-211.
- Bhanusali DG, Heath C, Gur D, et al. Metastatic meningioma of the scalp. Cutis. 2018;101:386-389.

A 37-year-old woman presented with an asymptomatic, indurated, pigmented, subcutaneous nodule on the right shoulder of more than 3 years' duration. The lesion had gradually increased in size with no associated symptoms. The patient had a history of endometrial adenocarcinoma and papillary thyroid carcinoma, which had been treated by hysterectomy-oophorectomy and right thyroidectomy, respectively. She had no other notable systemic abnormalities, and there was no family history of genetic disease or cancer. Physical examination demonstrated a 1.2×1.8-cm nontender, pigmented, subcutaneous nodule with a rough surface and indistinct borders. An excisional biopsy was performed.
Ill-Defined Macule on the Abdomen
The Diagnosis: Microvenular Hemangioma
Microvenular hemangioma is an acquired benign vascular neoplasm that was described by Hunt et al1 in 1991, though Bantel et al2 reported a similar entity termed micropapillary angioma in 1989. Microvenular hemangioma typically presents as a solitary, slowly enlarging, red to violaceous, asymptomatic papule, plaque, or nodule measuring 5 to 20 mm in diameter. It usually is located on the trunk, arms, or legs of young adults without any gender predilection. Microvenular hemangioma is rare.3 The etiology has not been elucidated, though a relationship with hormonal factors such as pregnancy or hormonal contraceptives has been described.2
Histopathologically, microvenular hemangioma has a characteristic morphology. It is comprised of a well-circumscribed collection of thin-walled blood vessels with narrow lumens (quiz image).4 The blood vessels tend to infiltrate the superficial and deep dermis and are surrounded by a collagenous or desmoplastic stroma. The endothelial cells are normal in size without atypia, mitotic figures, or pleomorphism. A mild lymphoplasmacytic inflammatory infiltrate sometimes is present. Microvenular hemangioma expresses many vascular markers confirming its endothelial origin, including CD34, CD31, WT1, factor VIII-related antigen, and von Willebrand factor.3 Moreover, WT1 staining suggests the lesion is a vascular proliferative growth, as it usually is negative in vascular malformations due to errors of endothelial development.5 In addition, it lacks expression of podoplanin (D2-40), which also supports a vascular as opposed to a lymphatic origin.4
Cutaneous angiosarcoma is a rare and highly aggressive malignant neoplasm of the vascular endothelium with a predilection for the skin and superficial soft tissue. Clinical presentation is variable, as it can arise sporadically, commonly on the scalp and face of elderly patients, in areas of chronic radiation therapy, or in association with chronic lymphedema (Stewart-Treves syndrome).6 Sporadic neoplasms appear clinically as purpuric macules, plaques, or nodules and are more common in elderly men than women. They are aggressive tumors that tend to recur and metastasize despite aggressive therapy and therefore carry a poor prognosis.7 Histopathologically, well-differentiated tumors are characterized by irregular dissecting vessels lined with crowded inconspicuous endothelial cells (Figure 1). Cutaneous angiosarcoma is poorly circumscribed with marked cytologic atypia, and the vessels can take on a sinusoidal growth pattern.8
Kaposi sarcoma (KS) is a virally induced lymphoangioproliferative disease, with human herpesvirus 8 as the implicated agent. There are 4 principal clinical variants of KS: epidemic or AIDS-associated KS, endemic or African KS, KS due to iatrogenic immunosuppression, and Mediterranean or classic KS.9 Cutaneous lesions vary from pink patches to dark purple plaques or nodules that commonly occur on the lower legs10; however, the clinical appearance of KS varies depending on the clinical variant and stage. Histopathologically, early lesions of KS exhibit a superficial dermal proliferation of small angulated and jagged vessels that tend to separate into collagen bundles and are surrounded by a lymphoplasmacytic perivascular infiltrate. These native vascular structures often are surrounded by more ectatic neoplastic channels with plump endothelial cells, known as the promontory sign (Figure 2).11 With more advanced lesions, the proliferation of slitlike vessels becomes more cellular and extends deeper into the dermis and subcutis. Although the histopathologic features vary with the stage of the lesion, they do not notably vary between clinical subtypes.
Targetoid hemosiderotic hemangioma, also known as hobnail hemangioma, is a small, benign, vascular tumor that usually affects the trunk, arms, and legs in young to middle-aged adults without a gender predilection. Clinically, it appears as a small, solitary, red to purple papule or macule that typically is surrounded by a pale thin area and a peripheral ecchymotic ring, creating a targetoid appearance, thus the term targetoid hemosiderotic hemangioma.12 Histopathologically, there is a prominent dermal vascular proliferation. In the papillary dermis, there are dilated superficial vessels lined with a single layer of endothelial cells characterized by a plump, hobnail-like appearance that protrude into the lumen (Figure 3). In the deeper dermis, the vascular spaces are angulated and slitlike and appear to dissect through collagen bundles. Hemosiderin, thrombi, extravasated erythrocytes, and a lymphocytic infiltrate also are often seen.13
Tufted angioma is a rare benign vascular lesion that usually presents as an acquired lesion in children and young adults, though it may be congenital. It is commonly localized to the skin and subcutaneous tissues. Clinically, the lesions appear as red to purple patches and plaques that typically are located on the neck or trunk. More than 50% of cases present during the first year of life and slowly spread to involve large areas before stabilizing in size.14 Partial spontaneous regression may occur, but complete regression is rare.15 Lesions usually are asymptomatic but may be painful during periods of platelet trapping (Kasabach-Merritt phenomenon), which may develop in congenital cases. Tufted angioma is named for its characteristic histopathologic appearance, which consists of multiple discrete lobules or tufts of tightly packed capillaries in a cannonball-like appearance throughout the dermis and subcutis (Figure 4).14,15

- Hunt SJ, Santa Cruz DJ, Barr RJ. Microvenular hemangioma. J Cutan Pathol. 1991;18:235-240.
- Bantel E, Grosshans E, Ortonne JP. Understanding microcapillary angioma, observations in pregnant patients and in females treated with hormonal contraceptives [in German]. Z Hautkr. 1989;64:1071-1074.
- Mansur AT, Demirci GT, Ozbal Koc E, et al. An unusual lesion on the nose: microvenular hemangioma. Dermatol Pract Concept. 2018;8:7-11.
- Napekoski KM, Fernandez AP, Billings SD. Microvenular hemangioma: a clinicopathologic review of 13 cases. J Cutan Pathol. 2014;41:816-822.
- Trinidade F, Tellechea O, Torrelo A, et al. Wilms tumor 1 expression in vascular neoplasms and vascular malformations. Am J Dermatopathol. 2011;33:569-572.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
- Morgan M, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Shon W, Billings SD. Cutaneous malignant vascular neoplasms. Clin Lab Med. 2017;37:633-646.
- Régnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol. 2013;68:313-331.
- Tappero JW, Conant MA, Wolfe SF, et al. Kaposi's sarcoma: epidemiology, pathogenesis, histology, clinical spectrum, staging criteria and therapy. J Am Acad Dermatol. 1993;28:371-395.
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Mentzel T, Partanen TA, Kutzner H. Hobnail hemangioma ("targetoid hemosiderotic hemangioma"): clinicopathologic and immunohistochemical analysis of 62 cases. J Cutan Pathol. 1999;26:279-286.
- Morales-Callaghan AM, Martinez-Garcia G, Aragoneses-Fraile H, et al. Targetoid hemosiderotic hemangioma: clinical and dermoscopical findings. J Eur Acad Dermatol Venereol. 2007;21:267-269.
- Kamath GH, Bhat RM, Kumar S. Tufted angioma. Int J Dermatol. 2005;44:1045-1047.
- Prasuna A, Rao P. A tufted angioma. Indian Dermatol Online J. 2015;6:266-268.
The Diagnosis: Microvenular Hemangioma
Microvenular hemangioma is an acquired benign vascular neoplasm that was described by Hunt et al1 in 1991, though Bantel et al2 reported a similar entity termed micropapillary angioma in 1989. Microvenular hemangioma typically presents as a solitary, slowly enlarging, red to violaceous, asymptomatic papule, plaque, or nodule measuring 5 to 20 mm in diameter. It usually is located on the trunk, arms, or legs of young adults without any gender predilection. Microvenular hemangioma is rare.3 The etiology has not been elucidated, though a relationship with hormonal factors such as pregnancy or hormonal contraceptives has been described.2
Histopathologically, microvenular hemangioma has a characteristic morphology. It is comprised of a well-circumscribed collection of thin-walled blood vessels with narrow lumens (quiz image).4 The blood vessels tend to infiltrate the superficial and deep dermis and are surrounded by a collagenous or desmoplastic stroma. The endothelial cells are normal in size without atypia, mitotic figures, or pleomorphism. A mild lymphoplasmacytic inflammatory infiltrate sometimes is present. Microvenular hemangioma expresses many vascular markers confirming its endothelial origin, including CD34, CD31, WT1, factor VIII-related antigen, and von Willebrand factor.3 Moreover, WT1 staining suggests the lesion is a vascular proliferative growth, as it usually is negative in vascular malformations due to errors of endothelial development.5 In addition, it lacks expression of podoplanin (D2-40), which also supports a vascular as opposed to a lymphatic origin.4
Cutaneous angiosarcoma is a rare and highly aggressive malignant neoplasm of the vascular endothelium with a predilection for the skin and superficial soft tissue. Clinical presentation is variable, as it can arise sporadically, commonly on the scalp and face of elderly patients, in areas of chronic radiation therapy, or in association with chronic lymphedema (Stewart-Treves syndrome).6 Sporadic neoplasms appear clinically as purpuric macules, plaques, or nodules and are more common in elderly men than women. They are aggressive tumors that tend to recur and metastasize despite aggressive therapy and therefore carry a poor prognosis.7 Histopathologically, well-differentiated tumors are characterized by irregular dissecting vessels lined with crowded inconspicuous endothelial cells (Figure 1). Cutaneous angiosarcoma is poorly circumscribed with marked cytologic atypia, and the vessels can take on a sinusoidal growth pattern.8
Kaposi sarcoma (KS) is a virally induced lymphoangioproliferative disease, with human herpesvirus 8 as the implicated agent. There are 4 principal clinical variants of KS: epidemic or AIDS-associated KS, endemic or African KS, KS due to iatrogenic immunosuppression, and Mediterranean or classic KS.9 Cutaneous lesions vary from pink patches to dark purple plaques or nodules that commonly occur on the lower legs10; however, the clinical appearance of KS varies depending on the clinical variant and stage. Histopathologically, early lesions of KS exhibit a superficial dermal proliferation of small angulated and jagged vessels that tend to separate into collagen bundles and are surrounded by a lymphoplasmacytic perivascular infiltrate. These native vascular structures often are surrounded by more ectatic neoplastic channels with plump endothelial cells, known as the promontory sign (Figure 2).11 With more advanced lesions, the proliferation of slitlike vessels becomes more cellular and extends deeper into the dermis and subcutis. Although the histopathologic features vary with the stage of the lesion, they do not notably vary between clinical subtypes.
Targetoid hemosiderotic hemangioma, also known as hobnail hemangioma, is a small, benign, vascular tumor that usually affects the trunk, arms, and legs in young to middle-aged adults without a gender predilection. Clinically, it appears as a small, solitary, red to purple papule or macule that typically is surrounded by a pale thin area and a peripheral ecchymotic ring, creating a targetoid appearance, thus the term targetoid hemosiderotic hemangioma.12 Histopathologically, there is a prominent dermal vascular proliferation. In the papillary dermis, there are dilated superficial vessels lined with a single layer of endothelial cells characterized by a plump, hobnail-like appearance that protrude into the lumen (Figure 3). In the deeper dermis, the vascular spaces are angulated and slitlike and appear to dissect through collagen bundles. Hemosiderin, thrombi, extravasated erythrocytes, and a lymphocytic infiltrate also are often seen.13
Tufted angioma is a rare benign vascular lesion that usually presents as an acquired lesion in children and young adults, though it may be congenital. It is commonly localized to the skin and subcutaneous tissues. Clinically, the lesions appear as red to purple patches and plaques that typically are located on the neck or trunk. More than 50% of cases present during the first year of life and slowly spread to involve large areas before stabilizing in size.14 Partial spontaneous regression may occur, but complete regression is rare.15 Lesions usually are asymptomatic but may be painful during periods of platelet trapping (Kasabach-Merritt phenomenon), which may develop in congenital cases. Tufted angioma is named for its characteristic histopathologic appearance, which consists of multiple discrete lobules or tufts of tightly packed capillaries in a cannonball-like appearance throughout the dermis and subcutis (Figure 4).14,15
The Diagnosis: Microvenular Hemangioma
Microvenular hemangioma is an acquired benign vascular neoplasm that was described by Hunt et al1 in 1991, though Bantel et al2 reported a similar entity termed micropapillary angioma in 1989. Microvenular hemangioma typically presents as a solitary, slowly enlarging, red to violaceous, asymptomatic papule, plaque, or nodule measuring 5 to 20 mm in diameter. It usually is located on the trunk, arms, or legs of young adults without any gender predilection. Microvenular hemangioma is rare.3 The etiology has not been elucidated, though a relationship with hormonal factors such as pregnancy or hormonal contraceptives has been described.2
Histopathologically, microvenular hemangioma has a characteristic morphology. It is comprised of a well-circumscribed collection of thin-walled blood vessels with narrow lumens (quiz image).4 The blood vessels tend to infiltrate the superficial and deep dermis and are surrounded by a collagenous or desmoplastic stroma. The endothelial cells are normal in size without atypia, mitotic figures, or pleomorphism. A mild lymphoplasmacytic inflammatory infiltrate sometimes is present. Microvenular hemangioma expresses many vascular markers confirming its endothelial origin, including CD34, CD31, WT1, factor VIII-related antigen, and von Willebrand factor.3 Moreover, WT1 staining suggests the lesion is a vascular proliferative growth, as it usually is negative in vascular malformations due to errors of endothelial development.5 In addition, it lacks expression of podoplanin (D2-40), which also supports a vascular as opposed to a lymphatic origin.4
Cutaneous angiosarcoma is a rare and highly aggressive malignant neoplasm of the vascular endothelium with a predilection for the skin and superficial soft tissue. Clinical presentation is variable, as it can arise sporadically, commonly on the scalp and face of elderly patients, in areas of chronic radiation therapy, or in association with chronic lymphedema (Stewart-Treves syndrome).6 Sporadic neoplasms appear clinically as purpuric macules, plaques, or nodules and are more common in elderly men than women. They are aggressive tumors that tend to recur and metastasize despite aggressive therapy and therefore carry a poor prognosis.7 Histopathologically, well-differentiated tumors are characterized by irregular dissecting vessels lined with crowded inconspicuous endothelial cells (Figure 1). Cutaneous angiosarcoma is poorly circumscribed with marked cytologic atypia, and the vessels can take on a sinusoidal growth pattern.8
Kaposi sarcoma (KS) is a virally induced lymphoangioproliferative disease, with human herpesvirus 8 as the implicated agent. There are 4 principal clinical variants of KS: epidemic or AIDS-associated KS, endemic or African KS, KS due to iatrogenic immunosuppression, and Mediterranean or classic KS.9 Cutaneous lesions vary from pink patches to dark purple plaques or nodules that commonly occur on the lower legs10; however, the clinical appearance of KS varies depending on the clinical variant and stage. Histopathologically, early lesions of KS exhibit a superficial dermal proliferation of small angulated and jagged vessels that tend to separate into collagen bundles and are surrounded by a lymphoplasmacytic perivascular infiltrate. These native vascular structures often are surrounded by more ectatic neoplastic channels with plump endothelial cells, known as the promontory sign (Figure 2).11 With more advanced lesions, the proliferation of slitlike vessels becomes more cellular and extends deeper into the dermis and subcutis. Although the histopathologic features vary with the stage of the lesion, they do not notably vary between clinical subtypes.
Targetoid hemosiderotic hemangioma, also known as hobnail hemangioma, is a small, benign, vascular tumor that usually affects the trunk, arms, and legs in young to middle-aged adults without a gender predilection. Clinically, it appears as a small, solitary, red to purple papule or macule that typically is surrounded by a pale thin area and a peripheral ecchymotic ring, creating a targetoid appearance, thus the term targetoid hemosiderotic hemangioma.12 Histopathologically, there is a prominent dermal vascular proliferation. In the papillary dermis, there are dilated superficial vessels lined with a single layer of endothelial cells characterized by a plump, hobnail-like appearance that protrude into the lumen (Figure 3). In the deeper dermis, the vascular spaces are angulated and slitlike and appear to dissect through collagen bundles. Hemosiderin, thrombi, extravasated erythrocytes, and a lymphocytic infiltrate also are often seen.13
Tufted angioma is a rare benign vascular lesion that usually presents as an acquired lesion in children and young adults, though it may be congenital. It is commonly localized to the skin and subcutaneous tissues. Clinically, the lesions appear as red to purple patches and plaques that typically are located on the neck or trunk. More than 50% of cases present during the first year of life and slowly spread to involve large areas before stabilizing in size.14 Partial spontaneous regression may occur, but complete regression is rare.15 Lesions usually are asymptomatic but may be painful during periods of platelet trapping (Kasabach-Merritt phenomenon), which may develop in congenital cases. Tufted angioma is named for its characteristic histopathologic appearance, which consists of multiple discrete lobules or tufts of tightly packed capillaries in a cannonball-like appearance throughout the dermis and subcutis (Figure 4).14,15
- Hunt SJ, Santa Cruz DJ, Barr RJ. Microvenular hemangioma. J Cutan Pathol. 1991;18:235-240.
- Bantel E, Grosshans E, Ortonne JP. Understanding microcapillary angioma, observations in pregnant patients and in females treated with hormonal contraceptives [in German]. Z Hautkr. 1989;64:1071-1074.
- Mansur AT, Demirci GT, Ozbal Koc E, et al. An unusual lesion on the nose: microvenular hemangioma. Dermatol Pract Concept. 2018;8:7-11.
- Napekoski KM, Fernandez AP, Billings SD. Microvenular hemangioma: a clinicopathologic review of 13 cases. J Cutan Pathol. 2014;41:816-822.
- Trinidade F, Tellechea O, Torrelo A, et al. Wilms tumor 1 expression in vascular neoplasms and vascular malformations. Am J Dermatopathol. 2011;33:569-572.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
- Morgan M, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Shon W, Billings SD. Cutaneous malignant vascular neoplasms. Clin Lab Med. 2017;37:633-646.
- Régnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol. 2013;68:313-331.
- Tappero JW, Conant MA, Wolfe SF, et al. Kaposi's sarcoma: epidemiology, pathogenesis, histology, clinical spectrum, staging criteria and therapy. J Am Acad Dermatol. 1993;28:371-395.
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Mentzel T, Partanen TA, Kutzner H. Hobnail hemangioma ("targetoid hemosiderotic hemangioma"): clinicopathologic and immunohistochemical analysis of 62 cases. J Cutan Pathol. 1999;26:279-286.
- Morales-Callaghan AM, Martinez-Garcia G, Aragoneses-Fraile H, et al. Targetoid hemosiderotic hemangioma: clinical and dermoscopical findings. J Eur Acad Dermatol Venereol. 2007;21:267-269.
- Kamath GH, Bhat RM, Kumar S. Tufted angioma. Int J Dermatol. 2005;44:1045-1047.
- Prasuna A, Rao P. A tufted angioma. Indian Dermatol Online J. 2015;6:266-268.
- Hunt SJ, Santa Cruz DJ, Barr RJ. Microvenular hemangioma. J Cutan Pathol. 1991;18:235-240.
- Bantel E, Grosshans E, Ortonne JP. Understanding microcapillary angioma, observations in pregnant patients and in females treated with hormonal contraceptives [in German]. Z Hautkr. 1989;64:1071-1074.
- Mansur AT, Demirci GT, Ozbal Koc E, et al. An unusual lesion on the nose: microvenular hemangioma. Dermatol Pract Concept. 2018;8:7-11.
- Napekoski KM, Fernandez AP, Billings SD. Microvenular hemangioma: a clinicopathologic review of 13 cases. J Cutan Pathol. 2014;41:816-822.
- Trinidade F, Tellechea O, Torrelo A, et al. Wilms tumor 1 expression in vascular neoplasms and vascular malformations. Am J Dermatopathol. 2011;33:569-572.
- Shustef E, Kazlouskaya V, Prieto VG, et al. Cutaneous angiosarcoma: a current update. J Clin Pathol. 2017;70:917-925.
- Morgan M, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Shon W, Billings SD. Cutaneous malignant vascular neoplasms. Clin Lab Med. 2017;37:633-646.
- Régnier-Rosencher E, Guillot B, Dupin N. Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol. 2013;68:313-331.
- Tappero JW, Conant MA, Wolfe SF, et al. Kaposi's sarcoma: epidemiology, pathogenesis, histology, clinical spectrum, staging criteria and therapy. J Am Acad Dermatol. 1993;28:371-395.
- Grayson W, Pantanowitz L. Histological variants of cutaneous Kaposi sarcoma. Diagn Pathol. 2008;3:31.
- Mentzel T, Partanen TA, Kutzner H. Hobnail hemangioma ("targetoid hemosiderotic hemangioma"): clinicopathologic and immunohistochemical analysis of 62 cases. J Cutan Pathol. 1999;26:279-286.
- Morales-Callaghan AM, Martinez-Garcia G, Aragoneses-Fraile H, et al. Targetoid hemosiderotic hemangioma: clinical and dermoscopical findings. J Eur Acad Dermatol Venereol. 2007;21:267-269.
- Kamath GH, Bhat RM, Kumar S. Tufted angioma. Int J Dermatol. 2005;44:1045-1047.
- Prasuna A, Rao P. A tufted angioma. Indian Dermatol Online J. 2015;6:266-268.
A 38-year-old woman presented with an asymptomatic lesion on the abdomen. On physical examination, there was a 5×2-mm, solitary, ill-defined pink macule on the right side of the abdomen. The patient denied recent change in size or color of the lesion, prior trauma, or a personal or family history of similar lesions. Due to the uncertain diagnostic appearance, a punch biopsy was performed.
Enlarging Nodule on the Thigh
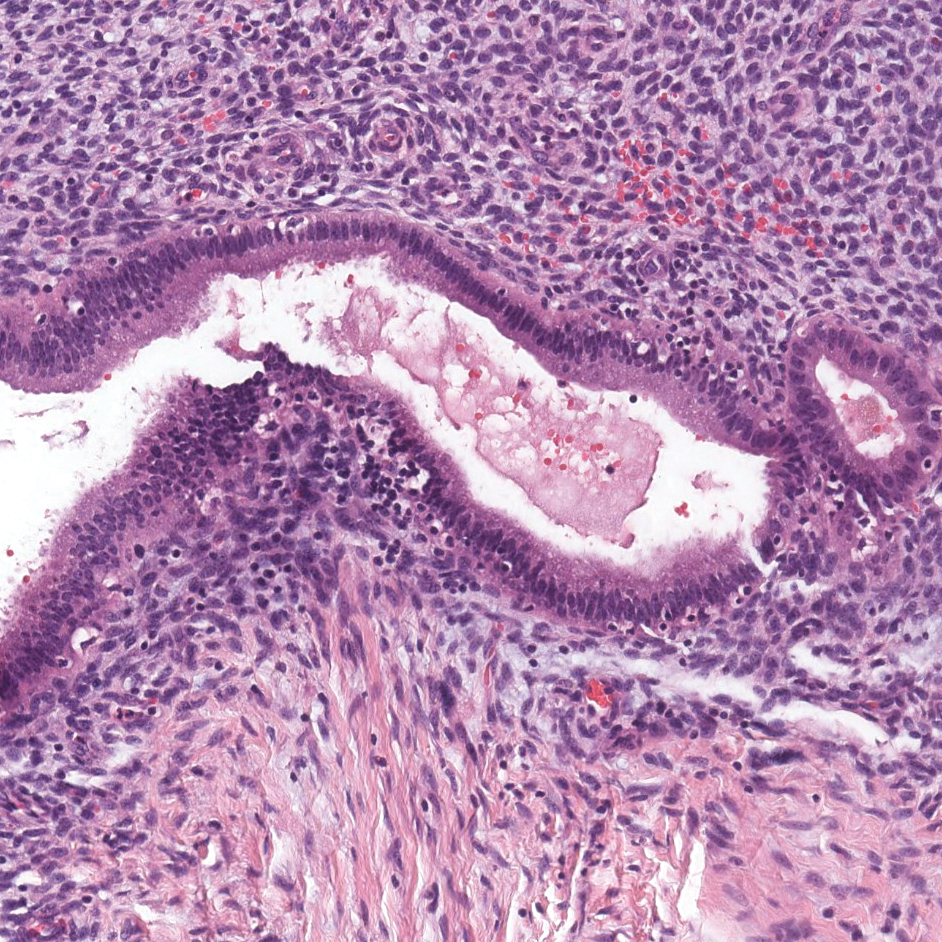
The Diagnosis: Metastatic Adenocarcinoma of the Colon
Cutaneous adenocarcinomas are uncommon, whether they present as a primary lesion or metastatic disease. In our patient, the histologic findings and immunohistochemical staining pattern were consistent with metastatic adenocarcinoma of the colon, an uncommon clinical presentation.
Colonic adenocarcinoma can cause cutaneous metastasis in 3% of cases. The most common sites of metastases include the abdomen, chest, and back.1 On histologic examination, hematoxylin and eosin (H&E)-stained sections of cutaneous metastatic adenocarcinoma illustrate a malignant gland-forming neoplasm in the dermis with luminal mucin and necrotic debris (quiz image). The glands are lined by tall columnar epithelial cells with hyperchromatic nuclei. Alternatively, poorly differentiated morphology can be seen with fewer glands and more infiltrating nests of tumor cells.2 Immunohistochemically, colonic adenocarcinoma typically is negative for cytokeratin (CK) 7 and positive for CK20 and caudal type homeobox transcription factor 2 (CDX-2).3
Primary cutaneous mucinous carcinoma is characterized by islands of neoplastic cells floating in pools of mucin (Figure 1). It may be indistinguishable from metastatic mucinous carcinomas of the colon or breast. Immunohistochemistry can be helpful in differentiating metastatic breast vs colon carcinoma. Cytokeratin 7, GATA binding protein 3, gross cystic disease fluid protein 15, and estrogen receptor will be positive in carcinomas of the breast and will be negative in colonic adenocarcinomas.4-6 Furthermore, lesional cells in metastatic adenocarcinoma of the colon are positive for CDX-2 and CK20, while those in metastatic carcinoma of the breast are negative.2 Immunohistochemistry also can differentiate primary cutaneous carcinoma from metastatic adenocarcinoma. When used in combination, p63 and podoplanin (D2-40) offer a highly sensitive and specific indicator of a primary cutaneous neoplasm, as both demonstrate either focal or diffuse positivity in this setting. In contrast, these stains typically are negative in metastatic adenocarcinomas of the skin.7
Endometriosis affects 1% to 2% of all reproductive-age females, of which extrapelvic manifestations account for only 0.5% to 1.0% of cases.8 Histologically, extrapelvic endometriosis is characterized by the triad of endometrial-type glands, endometrial stroma, and hemorrhage or hemosiderin deposition (Figure 2). The glands can enlarge and demonstrate architectural distortion with partial lack of polarity. These features initially can be concerning for adenocarcinoma, but on closer examination, nuclear morphology is regular and mitoses are absent.8,9 The diagnosis usually can be rendered with H&E alone; however, immunohistochemical stains for CD10 and estrogen receptor can highlight the endometrial stroma.10 Furthermore, endometrial glands will stain positive for paired box gene 8 (PAX8), a marker that is not expressed within the gastrointestinal tract and associated malignancies.11
cytoplasm (H&E, original magnification ×100).
Primary cutaneous angiosarcoma may mimic adenocarcinoma, as the endothelial-lined vessels can be confused as malignant glands (Figure 3). Angiosarcoma often is seen in 1 of 3 clinical presentations: the head and neck of elderly patients, postradiation treatment, and chronic lymphedema.12,13 Regardless of the location, the disease carries a poor prognosis, with a 5-year survival rate of 12% following initial diagnosis.13 Angiosarcoma is characterized by malignant endothelial cells dissecting through the dermis. Although the histology can be deceptively bland in some cases, the neoplasm most commonly demonstrates notable atypia with a multilayered endothelium and occasional intravascular atypical cells ("fish in the creek appearance").13,14 There can be frequent mitoses, and the atypical cells may show intracytoplasmic lumina containing red blood cells. The lesional cells are positive for endothelial markers such as erythroblast transformation specific related gene (ERG), CD31, CD34, and friend leukemia integration factor 1 (FLI-1).15,16
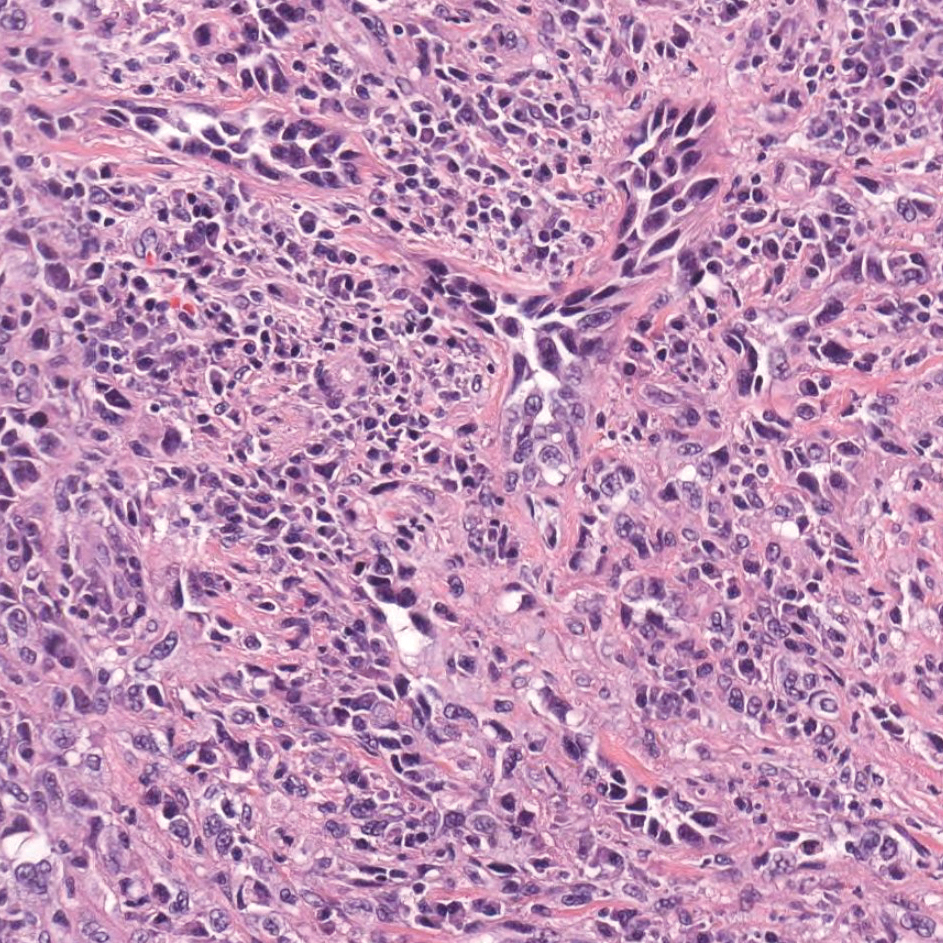
Breast cancer also can cause cutaneous metastases in approximately 20% of cases, with the most common presenting site being the anterior chest wall.17 Macroscopically, these lesions appear most commonly as painless nodules but also as telangiectatic, erysipeloid, fibrotic, and alopecic lesions.17-19 The histologic findings from H&E-stained sections of a cutaneous metastasis of breast cancer are variable and depend on the specific tumor subtype (eg, ductal, lobular, mucinous). However, the classic histologic presentation is that of nests and cords of malignant epithelial cells with variable gland formation. Often, tumor cells infiltrate in a single-file fashion (Figure 4).17 Although inflammatory breast carcinoma is a strictly clinical diagnosis, the presence of tumor cells in the lymphovascular spaces is a histologic clue to this diagnosis. Immunohistochemically, GATA binding protein 3 is helpful in identifying both hormone receptor-positive and -negative breast cancer subtypes that have metastasized.20
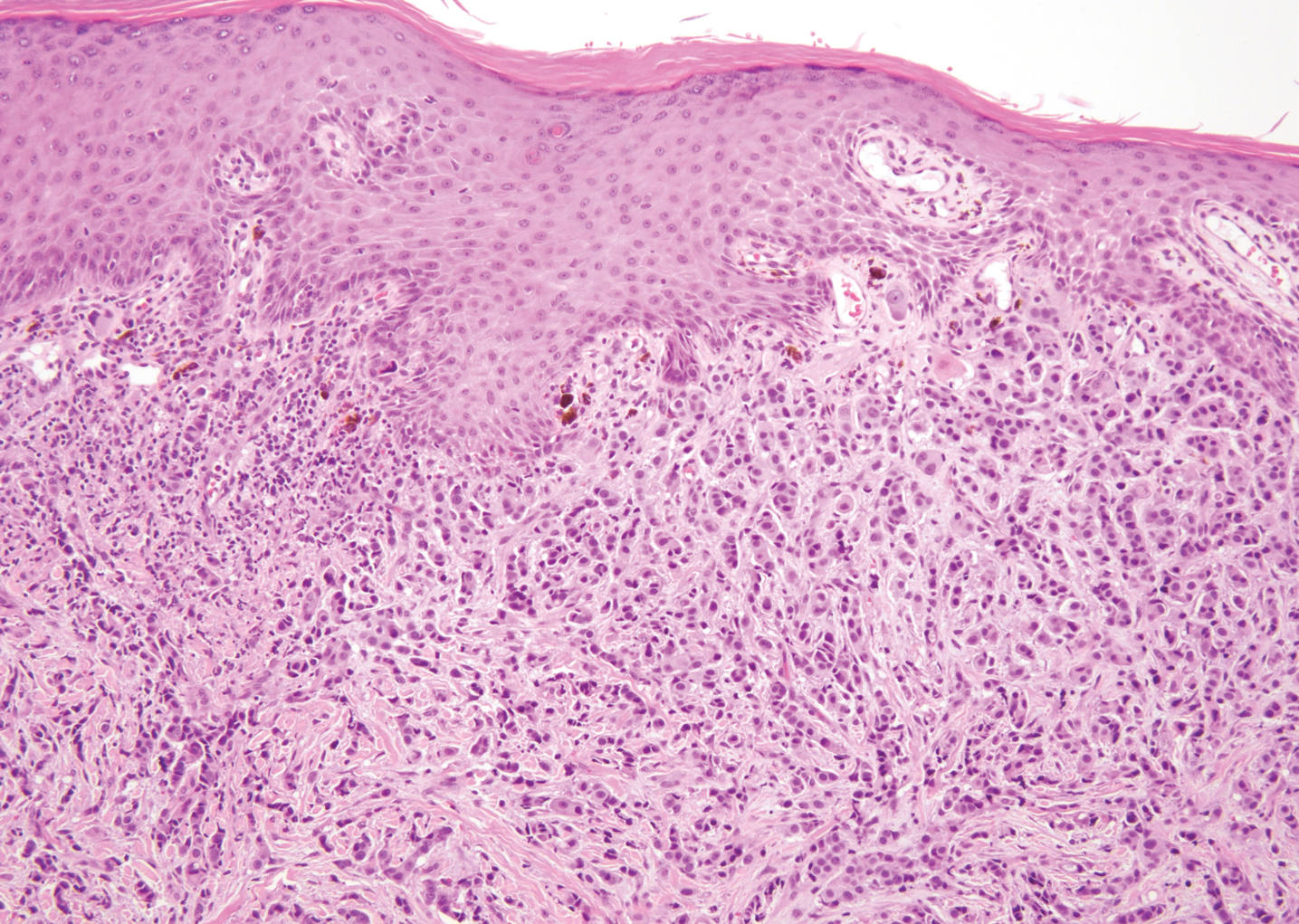
Within the histologic differential diagnoses, the most useful tool to diagnose metastatic adenocarcinoma of the colon often is a thorough clinical history. In the absence of a clinical history of adenocarcinoma, immunohistochemistry can be a useful adjunct to aid in the correct characterization and classification of a malignant gland-forming tumor.2,3,6
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Kumar V, Robbins SL. Robbins Basic Pathology. 8th ed. Philadelphia, PA: Saunders/Elsevier; 2007.
- Taliano RJ, LeGolvan M, Resnick MB. Immunohistochemistry of colorectal carcinoma: current practice and evolving applications. Hum Pathol. 2013;44:151-163.
- Kamalpour L, Brindise RT, Nodzenski M, et al. Primary cutaneous mucinous carcinoma: a systematic review and meta-analysis of outcomes after surgery. JAMA Dermatol. 2014;150:380-384.
- Roshan MH, Tambo A, Pace NP. The role of testosterone in colorectal carcinoma: pathomechanisms and open questions. EPMA J. 2016;7:22.
- Mazoujian G, Pinkus GS, Davis S, et al. Immunohistochemistry of a gross cystic disease fluid protein (GCDFP-15) of the breast. a marker of apocrine epithelium and breast carcinomas with apocrine features. Am J Pathol. 1983;110:105-112.
- Plaza JA, Ortega PF, Stockman DL, et al. Value of p63 and podoplanin (D2-40) immunoreactivity in the distinction between primary cutaneous tumors and adenocarcinomas metastatic to the skin: a clinicopathologic and immunohistochemical study of 79 cases. J Cutan Pathol. 2010;37:403-410.
- Machairiotis N, Stylianaki A, Dryllis G, et al. Extrapelvic endometriosis: a rare entity or an under diagnosed condition? Diagn Pathol. 2013;8:194.
- Chen H, Luo Q, Liu S, et al. Rectal mucosal endometriosis primarily misinterpreted as adenocarcinoma: a case report and review of literature. Int J Clin Exp Pathol. 2015;8:5902-5907.
- Terada S, Miyata Y, Nakazawa H, et al. Immunohistochemical analysis of an ectopic endometriosis in the uterine round ligament. Diagn Pathol. 2006;1:27.
- Yemelyanova A, Gown AM, Wu LS, et al. PAX8 expression in uterine adenocarcinomas and mesonephric proliferations. Int J Gynecol Pathol. 2014;33:492-499.
- Farid M, Ong WS, Lee MJ, et al. Cutaneous versus non-cutaneous angiosarcoma: clinicopathologic features and treatment outcomes in 60 patients at a single Asian cancer centre. Oncology. 2013;85:182-190.
- Requena C, Sendra E, Llombart B, et al. Cutaneous angiosarcoma: clinical and pathology study of 16 cases. Actas Dermosifiliogr. 2017;108:457-465.
- Schmidt AP, Tjarks BJ, Lynch DW. Gone fishing: a unique histologic pattern in cutaneous angiosarcoma. Cutis. 2018;101:270-272.
- Sullivan HC, Edgar MA, Cohen C, et al. The utility of ERG, CD31 and CD34 in the cytological diagnosis of angiosarcoma: an analysis of 25 cases. J Clin Pathol. 2015;68:44-50.
- Rossi S, Orvieto E, Furlanetto A, et al. Utility of the immunohistochemical detection of FLI-1 expression in round cell and vascular neoplasm using a monoclonal antibody. Mod Pathol. 2004;17:547-552.
- Tan AR. Cutaneous manifestations of breast cancer. Semin Oncol. 2016;43:331-334.
- Schwartz RA, Wiederkehr M, Lambert WC. Secondary mucinous carcinoma of the skin: metastatic breast cancer. Dermatol Surg. 2004;30(2, pt 1):234-235.
- Mallon E, Dawber RP. Alopecia neoplastica without alopecia: a unique presentation of breast carcinoma scalp metastasis. J Am Acad Dermatol. 1994;31(2, pt 2):319-321.
- Braxton DR, Cohen C, Siddiqui MT. Utility of GATA3 immunohistochemistry for diagnosis of metastatic breast carcinoma in cytology specimens. Diagn Cytopathol. 2015;43:271-277.
The Diagnosis: Metastatic Adenocarcinoma of the Colon
Cutaneous adenocarcinomas are uncommon, whether they present as a primary lesion or metastatic disease. In our patient, the histologic findings and immunohistochemical staining pattern were consistent with metastatic adenocarcinoma of the colon, an uncommon clinical presentation.
Colonic adenocarcinoma can cause cutaneous metastasis in 3% of cases. The most common sites of metastases include the abdomen, chest, and back.1 On histologic examination, hematoxylin and eosin (H&E)-stained sections of cutaneous metastatic adenocarcinoma illustrate a malignant gland-forming neoplasm in the dermis with luminal mucin and necrotic debris (quiz image). The glands are lined by tall columnar epithelial cells with hyperchromatic nuclei. Alternatively, poorly differentiated morphology can be seen with fewer glands and more infiltrating nests of tumor cells.2 Immunohistochemically, colonic adenocarcinoma typically is negative for cytokeratin (CK) 7 and positive for CK20 and caudal type homeobox transcription factor 2 (CDX-2).3
Primary cutaneous mucinous carcinoma is characterized by islands of neoplastic cells floating in pools of mucin (Figure 1). It may be indistinguishable from metastatic mucinous carcinomas of the colon or breast. Immunohistochemistry can be helpful in differentiating metastatic breast vs colon carcinoma. Cytokeratin 7, GATA binding protein 3, gross cystic disease fluid protein 15, and estrogen receptor will be positive in carcinomas of the breast and will be negative in colonic adenocarcinomas.4-6 Furthermore, lesional cells in metastatic adenocarcinoma of the colon are positive for CDX-2 and CK20, while those in metastatic carcinoma of the breast are negative.2 Immunohistochemistry also can differentiate primary cutaneous carcinoma from metastatic adenocarcinoma. When used in combination, p63 and podoplanin (D2-40) offer a highly sensitive and specific indicator of a primary cutaneous neoplasm, as both demonstrate either focal or diffuse positivity in this setting. In contrast, these stains typically are negative in metastatic adenocarcinomas of the skin.7
Endometriosis affects 1% to 2% of all reproductive-age females, of which extrapelvic manifestations account for only 0.5% to 1.0% of cases.8 Histologically, extrapelvic endometriosis is characterized by the triad of endometrial-type glands, endometrial stroma, and hemorrhage or hemosiderin deposition (Figure 2). The glands can enlarge and demonstrate architectural distortion with partial lack of polarity. These features initially can be concerning for adenocarcinoma, but on closer examination, nuclear morphology is regular and mitoses are absent.8,9 The diagnosis usually can be rendered with H&E alone; however, immunohistochemical stains for CD10 and estrogen receptor can highlight the endometrial stroma.10 Furthermore, endometrial glands will stain positive for paired box gene 8 (PAX8), a marker that is not expressed within the gastrointestinal tract and associated malignancies.11
cytoplasm (H&E, original magnification ×100).
Primary cutaneous angiosarcoma may mimic adenocarcinoma, as the endothelial-lined vessels can be confused as malignant glands (Figure 3). Angiosarcoma often is seen in 1 of 3 clinical presentations: the head and neck of elderly patients, postradiation treatment, and chronic lymphedema.12,13 Regardless of the location, the disease carries a poor prognosis, with a 5-year survival rate of 12% following initial diagnosis.13 Angiosarcoma is characterized by malignant endothelial cells dissecting through the dermis. Although the histology can be deceptively bland in some cases, the neoplasm most commonly demonstrates notable atypia with a multilayered endothelium and occasional intravascular atypical cells ("fish in the creek appearance").13,14 There can be frequent mitoses, and the atypical cells may show intracytoplasmic lumina containing red blood cells. The lesional cells are positive for endothelial markers such as erythroblast transformation specific related gene (ERG), CD31, CD34, and friend leukemia integration factor 1 (FLI-1).15,16
Breast cancer also can cause cutaneous metastases in approximately 20% of cases, with the most common presenting site being the anterior chest wall.17 Macroscopically, these lesions appear most commonly as painless nodules but also as telangiectatic, erysipeloid, fibrotic, and alopecic lesions.17-19 The histologic findings from H&E-stained sections of a cutaneous metastasis of breast cancer are variable and depend on the specific tumor subtype (eg, ductal, lobular, mucinous). However, the classic histologic presentation is that of nests and cords of malignant epithelial cells with variable gland formation. Often, tumor cells infiltrate in a single-file fashion (Figure 4).17 Although inflammatory breast carcinoma is a strictly clinical diagnosis, the presence of tumor cells in the lymphovascular spaces is a histologic clue to this diagnosis. Immunohistochemically, GATA binding protein 3 is helpful in identifying both hormone receptor-positive and -negative breast cancer subtypes that have metastasized.20
Within the histologic differential diagnoses, the most useful tool to diagnose metastatic adenocarcinoma of the colon often is a thorough clinical history. In the absence of a clinical history of adenocarcinoma, immunohistochemistry can be a useful adjunct to aid in the correct characterization and classification of a malignant gland-forming tumor.2,3,6
The Diagnosis: Metastatic Adenocarcinoma of the Colon
Cutaneous adenocarcinomas are uncommon, whether they present as a primary lesion or metastatic disease. In our patient, the histologic findings and immunohistochemical staining pattern were consistent with metastatic adenocarcinoma of the colon, an uncommon clinical presentation.
Colonic adenocarcinoma can cause cutaneous metastasis in 3% of cases. The most common sites of metastases include the abdomen, chest, and back.1 On histologic examination, hematoxylin and eosin (H&E)-stained sections of cutaneous metastatic adenocarcinoma illustrate a malignant gland-forming neoplasm in the dermis with luminal mucin and necrotic debris (quiz image). The glands are lined by tall columnar epithelial cells with hyperchromatic nuclei. Alternatively, poorly differentiated morphology can be seen with fewer glands and more infiltrating nests of tumor cells.2 Immunohistochemically, colonic adenocarcinoma typically is negative for cytokeratin (CK) 7 and positive for CK20 and caudal type homeobox transcription factor 2 (CDX-2).3
Primary cutaneous mucinous carcinoma is characterized by islands of neoplastic cells floating in pools of mucin (Figure 1). It may be indistinguishable from metastatic mucinous carcinomas of the colon or breast. Immunohistochemistry can be helpful in differentiating metastatic breast vs colon carcinoma. Cytokeratin 7, GATA binding protein 3, gross cystic disease fluid protein 15, and estrogen receptor will be positive in carcinomas of the breast and will be negative in colonic adenocarcinomas.4-6 Furthermore, lesional cells in metastatic adenocarcinoma of the colon are positive for CDX-2 and CK20, while those in metastatic carcinoma of the breast are negative.2 Immunohistochemistry also can differentiate primary cutaneous carcinoma from metastatic adenocarcinoma. When used in combination, p63 and podoplanin (D2-40) offer a highly sensitive and specific indicator of a primary cutaneous neoplasm, as both demonstrate either focal or diffuse positivity in this setting. In contrast, these stains typically are negative in metastatic adenocarcinomas of the skin.7
Endometriosis affects 1% to 2% of all reproductive-age females, of which extrapelvic manifestations account for only 0.5% to 1.0% of cases.8 Histologically, extrapelvic endometriosis is characterized by the triad of endometrial-type glands, endometrial stroma, and hemorrhage or hemosiderin deposition (Figure 2). The glands can enlarge and demonstrate architectural distortion with partial lack of polarity. These features initially can be concerning for adenocarcinoma, but on closer examination, nuclear morphology is regular and mitoses are absent.8,9 The diagnosis usually can be rendered with H&E alone; however, immunohistochemical stains for CD10 and estrogen receptor can highlight the endometrial stroma.10 Furthermore, endometrial glands will stain positive for paired box gene 8 (PAX8), a marker that is not expressed within the gastrointestinal tract and associated malignancies.11
cytoplasm (H&E, original magnification ×100).
Primary cutaneous angiosarcoma may mimic adenocarcinoma, as the endothelial-lined vessels can be confused as malignant glands (Figure 3). Angiosarcoma often is seen in 1 of 3 clinical presentations: the head and neck of elderly patients, postradiation treatment, and chronic lymphedema.12,13 Regardless of the location, the disease carries a poor prognosis, with a 5-year survival rate of 12% following initial diagnosis.13 Angiosarcoma is characterized by malignant endothelial cells dissecting through the dermis. Although the histology can be deceptively bland in some cases, the neoplasm most commonly demonstrates notable atypia with a multilayered endothelium and occasional intravascular atypical cells ("fish in the creek appearance").13,14 There can be frequent mitoses, and the atypical cells may show intracytoplasmic lumina containing red blood cells. The lesional cells are positive for endothelial markers such as erythroblast transformation specific related gene (ERG), CD31, CD34, and friend leukemia integration factor 1 (FLI-1).15,16
Breast cancer also can cause cutaneous metastases in approximately 20% of cases, with the most common presenting site being the anterior chest wall.17 Macroscopically, these lesions appear most commonly as painless nodules but also as telangiectatic, erysipeloid, fibrotic, and alopecic lesions.17-19 The histologic findings from H&E-stained sections of a cutaneous metastasis of breast cancer are variable and depend on the specific tumor subtype (eg, ductal, lobular, mucinous). However, the classic histologic presentation is that of nests and cords of malignant epithelial cells with variable gland formation. Often, tumor cells infiltrate in a single-file fashion (Figure 4).17 Although inflammatory breast carcinoma is a strictly clinical diagnosis, the presence of tumor cells in the lymphovascular spaces is a histologic clue to this diagnosis. Immunohistochemically, GATA binding protein 3 is helpful in identifying both hormone receptor-positive and -negative breast cancer subtypes that have metastasized.20
Within the histologic differential diagnoses, the most useful tool to diagnose metastatic adenocarcinoma of the colon often is a thorough clinical history. In the absence of a clinical history of adenocarcinoma, immunohistochemistry can be a useful adjunct to aid in the correct characterization and classification of a malignant gland-forming tumor.2,3,6
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Kumar V, Robbins SL. Robbins Basic Pathology. 8th ed. Philadelphia, PA: Saunders/Elsevier; 2007.
- Taliano RJ, LeGolvan M, Resnick MB. Immunohistochemistry of colorectal carcinoma: current practice and evolving applications. Hum Pathol. 2013;44:151-163.
- Kamalpour L, Brindise RT, Nodzenski M, et al. Primary cutaneous mucinous carcinoma: a systematic review and meta-analysis of outcomes after surgery. JAMA Dermatol. 2014;150:380-384.
- Roshan MH, Tambo A, Pace NP. The role of testosterone in colorectal carcinoma: pathomechanisms and open questions. EPMA J. 2016;7:22.
- Mazoujian G, Pinkus GS, Davis S, et al. Immunohistochemistry of a gross cystic disease fluid protein (GCDFP-15) of the breast. a marker of apocrine epithelium and breast carcinomas with apocrine features. Am J Pathol. 1983;110:105-112.
- Plaza JA, Ortega PF, Stockman DL, et al. Value of p63 and podoplanin (D2-40) immunoreactivity in the distinction between primary cutaneous tumors and adenocarcinomas metastatic to the skin: a clinicopathologic and immunohistochemical study of 79 cases. J Cutan Pathol. 2010;37:403-410.
- Machairiotis N, Stylianaki A, Dryllis G, et al. Extrapelvic endometriosis: a rare entity or an under diagnosed condition? Diagn Pathol. 2013;8:194.
- Chen H, Luo Q, Liu S, et al. Rectal mucosal endometriosis primarily misinterpreted as adenocarcinoma: a case report and review of literature. Int J Clin Exp Pathol. 2015;8:5902-5907.
- Terada S, Miyata Y, Nakazawa H, et al. Immunohistochemical analysis of an ectopic endometriosis in the uterine round ligament. Diagn Pathol. 2006;1:27.
- Yemelyanova A, Gown AM, Wu LS, et al. PAX8 expression in uterine adenocarcinomas and mesonephric proliferations. Int J Gynecol Pathol. 2014;33:492-499.
- Farid M, Ong WS, Lee MJ, et al. Cutaneous versus non-cutaneous angiosarcoma: clinicopathologic features and treatment outcomes in 60 patients at a single Asian cancer centre. Oncology. 2013;85:182-190.
- Requena C, Sendra E, Llombart B, et al. Cutaneous angiosarcoma: clinical and pathology study of 16 cases. Actas Dermosifiliogr. 2017;108:457-465.
- Schmidt AP, Tjarks BJ, Lynch DW. Gone fishing: a unique histologic pattern in cutaneous angiosarcoma. Cutis. 2018;101:270-272.
- Sullivan HC, Edgar MA, Cohen C, et al. The utility of ERG, CD31 and CD34 in the cytological diagnosis of angiosarcoma: an analysis of 25 cases. J Clin Pathol. 2015;68:44-50.
- Rossi S, Orvieto E, Furlanetto A, et al. Utility of the immunohistochemical detection of FLI-1 expression in round cell and vascular neoplasm using a monoclonal antibody. Mod Pathol. 2004;17:547-552.
- Tan AR. Cutaneous manifestations of breast cancer. Semin Oncol. 2016;43:331-334.
- Schwartz RA, Wiederkehr M, Lambert WC. Secondary mucinous carcinoma of the skin: metastatic breast cancer. Dermatol Surg. 2004;30(2, pt 1):234-235.
- Mallon E, Dawber RP. Alopecia neoplastica without alopecia: a unique presentation of breast carcinoma scalp metastasis. J Am Acad Dermatol. 1994;31(2, pt 2):319-321.
- Braxton DR, Cohen C, Siddiqui MT. Utility of GATA3 immunohistochemistry for diagnosis of metastatic breast carcinoma in cytology specimens. Diagn Cytopathol. 2015;43:271-277.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Kumar V, Robbins SL. Robbins Basic Pathology. 8th ed. Philadelphia, PA: Saunders/Elsevier; 2007.
- Taliano RJ, LeGolvan M, Resnick MB. Immunohistochemistry of colorectal carcinoma: current practice and evolving applications. Hum Pathol. 2013;44:151-163.
- Kamalpour L, Brindise RT, Nodzenski M, et al. Primary cutaneous mucinous carcinoma: a systematic review and meta-analysis of outcomes after surgery. JAMA Dermatol. 2014;150:380-384.
- Roshan MH, Tambo A, Pace NP. The role of testosterone in colorectal carcinoma: pathomechanisms and open questions. EPMA J. 2016;7:22.
- Mazoujian G, Pinkus GS, Davis S, et al. Immunohistochemistry of a gross cystic disease fluid protein (GCDFP-15) of the breast. a marker of apocrine epithelium and breast carcinomas with apocrine features. Am J Pathol. 1983;110:105-112.
- Plaza JA, Ortega PF, Stockman DL, et al. Value of p63 and podoplanin (D2-40) immunoreactivity in the distinction between primary cutaneous tumors and adenocarcinomas metastatic to the skin: a clinicopathologic and immunohistochemical study of 79 cases. J Cutan Pathol. 2010;37:403-410.
- Machairiotis N, Stylianaki A, Dryllis G, et al. Extrapelvic endometriosis: a rare entity or an under diagnosed condition? Diagn Pathol. 2013;8:194.
- Chen H, Luo Q, Liu S, et al. Rectal mucosal endometriosis primarily misinterpreted as adenocarcinoma: a case report and review of literature. Int J Clin Exp Pathol. 2015;8:5902-5907.
- Terada S, Miyata Y, Nakazawa H, et al. Immunohistochemical analysis of an ectopic endometriosis in the uterine round ligament. Diagn Pathol. 2006;1:27.
- Yemelyanova A, Gown AM, Wu LS, et al. PAX8 expression in uterine adenocarcinomas and mesonephric proliferations. Int J Gynecol Pathol. 2014;33:492-499.
- Farid M, Ong WS, Lee MJ, et al. Cutaneous versus non-cutaneous angiosarcoma: clinicopathologic features and treatment outcomes in 60 patients at a single Asian cancer centre. Oncology. 2013;85:182-190.
- Requena C, Sendra E, Llombart B, et al. Cutaneous angiosarcoma: clinical and pathology study of 16 cases. Actas Dermosifiliogr. 2017;108:457-465.
- Schmidt AP, Tjarks BJ, Lynch DW. Gone fishing: a unique histologic pattern in cutaneous angiosarcoma. Cutis. 2018;101:270-272.
- Sullivan HC, Edgar MA, Cohen C, et al. The utility of ERG, CD31 and CD34 in the cytological diagnosis of angiosarcoma: an analysis of 25 cases. J Clin Pathol. 2015;68:44-50.
- Rossi S, Orvieto E, Furlanetto A, et al. Utility of the immunohistochemical detection of FLI-1 expression in round cell and vascular neoplasm using a monoclonal antibody. Mod Pathol. 2004;17:547-552.
- Tan AR. Cutaneous manifestations of breast cancer. Semin Oncol. 2016;43:331-334.
- Schwartz RA, Wiederkehr M, Lambert WC. Secondary mucinous carcinoma of the skin: metastatic breast cancer. Dermatol Surg. 2004;30(2, pt 1):234-235.
- Mallon E, Dawber RP. Alopecia neoplastica without alopecia: a unique presentation of breast carcinoma scalp metastasis. J Am Acad Dermatol. 1994;31(2, pt 2):319-321.
- Braxton DR, Cohen C, Siddiqui MT. Utility of GATA3 immunohistochemistry for diagnosis of metastatic breast carcinoma in cytology specimens. Diagn Cytopathol. 2015;43:271-277.
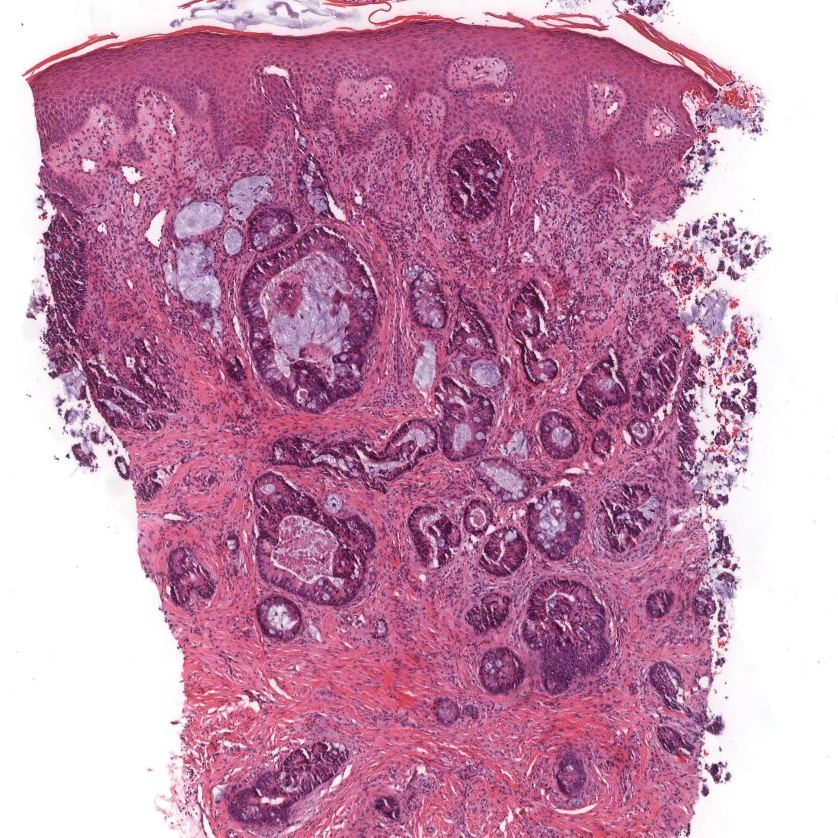
A 68-year-old patient presented with an enlarging flesh-colored nodule on the thigh that was positive for cytokeratin 20 and negative for cytokeratin 7.
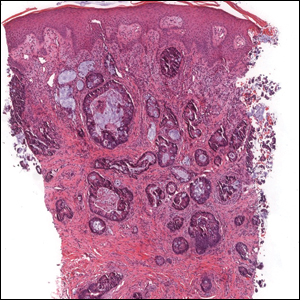
Unilateral Facial Papules and Plaques
The Diagnosis: Unilateral Dermatomal Trichoepithelioma
Adnexal lesions presenting with a linear and/or dermatomal pattern rarely have been reported. Bolognia et al1 performed a comprehensive review of Blaschko lines and skin conditions that follow such a pattern. The authors found that adnexal-related lesions included linear nevus comedonicus, linear basal cell nevus with comedones (linear basaloid follicular hamartoma), unilateral nevoid basal cell carcinoma (BCC), linear trichoepithelioma, linear trichodiscoma, linear hamartoma of the follicular infundibulum, nevus sebaceous, syringocystadenoma papilliferum, porokeratotic eccrine ostial and dermal duct nevus, linear eccrine poroma, linear spiradenoma, linear syringoma, and linear eccrine syringofibroadenoma.1
Trichoepithelioma is a hair follicle-related neoplastic lesion presenting most commonly as the autosomal-dominant multiple familial type with lesions mainly centered on the face. Initial genetic studies associated the disease with loss of heterozygosity in the 9p21 region and further studies identified mutations in the CYLD (cylindromatosis [turban tumor syndrome]) gene on chromosome 16q12-q13.2,3 Unilateral, linear, and dermatomal forms of trichoepithelioma rarely are reported. In 1986, Geffner et al4 reported a case of linear and dermatomal trichoepithelioma in a 10-year-old girl. In addition to discrete solitary lesions affecting the face, she developed lesions on the left shoulder, left side of the trunk, and left lower leg following dermatomal distribution. In 2006, 2 cases of dermatomal trichoepitheliomas affecting the face in children, as in our case, were reported.5,6 Another case involving the neck was reported in 2016.7 Although classic multiple familial trichoepithelioma can be part of conditions such as Brooke-Spiegler8 and Rombo syndromes,9 no syndromal association has been reported thus far with the unilateral, linear, or dermatomal variants.
Our case showed typical histopathologic features of trichoepithelioma, including discrete islands of basaloid cells in the dermis set in a conspicuous fibroblastic stroma. Focal connection with the epidermis was present. Most of the islands showed peripheral palisading and horn cysts lined by eosinophilic cells. The fibroblastic component was tightly adherent to the epithelial component, and only stromal clefts were detected. Papillary mesenchymal bodies also were detected as oval aggregates of fibroblastic cells invaginating into epithelial islands to form hair papillae.
Histopathologically, the 2 most important differential diagnoses of trichoepithelioma include BCC and basaloid follicular hamartoma. In differentiating BCC from trichoepithelioma, the presence of dense fibroblastic stroma and papillary mesenchymal bodies characterize trichoepithelioma, while a fibromucinous stroma with mucinous retraction artifacts and clefting between the basaloid islands and the stroma characterize BCC (Figure 1).10 Immunohistochemical studies using antibodies against Bcl-2, CD34, CD10, androgen receptor, Ki-67, cytokeratin 19, and PHLDA1 (pleckstrin homologylike domain family A member 1) have reportedly been utilized to differentiate trichoepithelioma from BCC.11,12 Basaloid follicular hamartoma is characterized by thin anastomosing strands and branching cords of undifferentiated basaloid cells that replace or associate hair follicles in a latticelike pattern (Figure 2). The strands usually are vertically oriented perpendicular to the epidermis. Peripheral palisading is possible, and the basaloid strands are surrounded with cellular connective tissue stroma.13 Tumor islands in eccrine poroma show broad connections with the epidermis and are composed of poroid cells that show evident ductal differentiation with eosinophilic cuticles (Figure 3).14 Spiradenoma is characterized by capsulated deep-seated tumorous nodules not connected with the epidermis and composed of light and dark cells with ductal differentiation and vascular stroma (Figure 4). Scattered lymphocytes within the tumor lobules and in the stroma also are seen. Eosinophilic hyaline globules rarely can be present.15

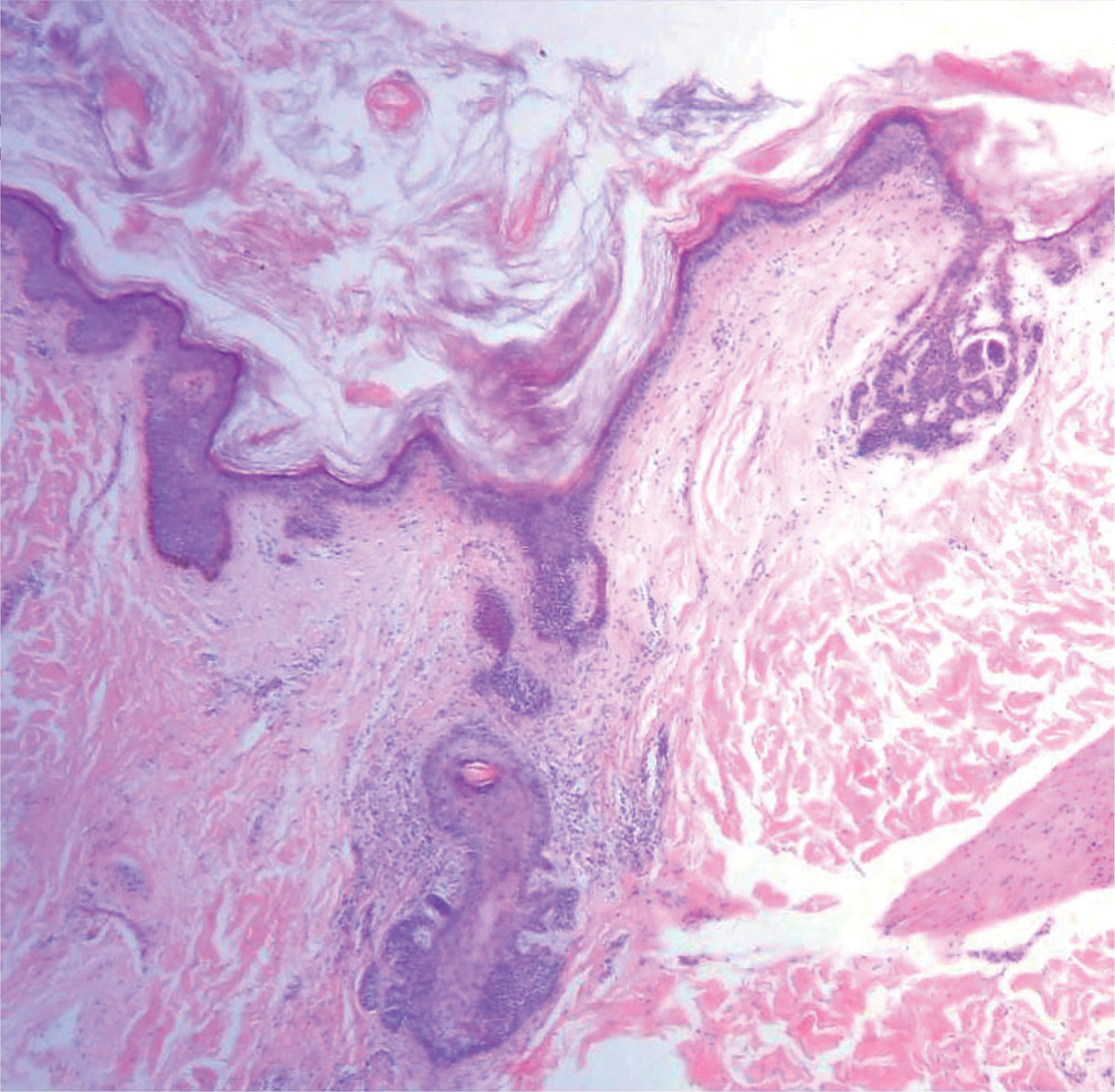

Many pathologists consider trichoepithelioma as the superficial variant of trichoblastoma. According to the recent World Health Organization classification of benign tumors with follicular differentiation, trichoepithelioma is considered synonymous with trichoblastoma.16
Trichoepitheliomas are benign tumors, and therapy is mainly directed at removal for cosmetic purposes. Several methods of removal are available including electrocautery, laser therapy, and surgery. Awareness of the possible dermatomal distribution of hair follicle and other adnexal-related conditions is important, and such lesions should be thought of in the differential diagnosis of unilateral and/or dermatomal lesions.
- Bolognia JL, Orlow SJ, Glick SA. Lines of Blaschko. J Am Acad Dermatol. 1994;31(2, pt 1):157-190.
- Harada H, Hashimoto K, Ko MS. The gene for multiple familial trichoepithelioma maps to chromosome 9p21. J Invest Dermatol. 1996;107:41-43.
- Zheng G, Hu L, Huang W, et al. CYLD mutation causes multiple familial trichoepithelioma in three Chinese families. Hum Mutat. 2004;23:400.
- Geffner RE, Goslen JB, Santa Cruz DJ. Linear and dermatomal trichoepitheliomas. J Am Acad Dermatol. 1986;14(5, pt 2):927-930.
- Chang YC, Colome-Grimmer M, Kelly E. Multiple trichoepitheliomas in the lines of Blaschko. Pediatr Dermatol. 2006;23:149-151.
- Strauss RM, Merchant WJ, Stainforth JM, et al. Unilateral naevoid trichoepitheliomas on the face of a child. Clin Exp Dermatol. 2006;6:778-780.
- Laska AJ, Belli RA, Kobayashi TT. Linear trichoepithelioma on the neck of a 15-year-old girl. Dermatol Online J. 2016;22. pii:13030/qt87b6h4q8.
- Rasmussen JE. A syndrome of trichoepitheliomas, milia and cylindroma. Arch Dermatol. 1975;111:610-614.
- Michaelson G, Olsson E, Westermark P. The Rombo syndrome. Acta Derm Venereol. 1981;61:497-503.
- Brooke JD, Fitzpatrick JE, Golitz LE. Papillary mesenchymal bodies: a histologic finding useful in differentiating trichoepitheliomas from basal cell carcinomas. J Am Acad Dermatol. 1989;21(3, pt 1):523-528.
- Mostafa NA, Assaf M, Elhakim S, et al. Diagnostic accuracy of immunohistochemical markers in differentiation between basal cell carcinoma and trichoepithelioma in small biopsy specimens. J Cutan Pathol. 2018;45:807-816.
- Poniecka AW, Alexis JB. An immunohistochemical study of basal cell carcinoma and trichoepithelioma. Am J Dermatopathol. 1999;21:332-336.
- Abdel-Halim MRE, Fawzy M, Saleh M, et al. Linear unilateral basal cell nevus with comedones (linear nevoid basaloid follicular hamartoma): a case report. J Egypt Womens Dermatol Soc. 2016;13:46-48.
- Hyman AB, Brownstein MH. Eccrine poroma: analysis of 45 new cases. Dermatologica. 1969;138:28-38.
- Mambo NC. Eccrine spiradenoma: clinical and pathologic study of 49 tumors. J Cutan Pathol. 1983;10:312-320.
- Kutzner H, Kaddu S, Kanitakis J, et al. Trichoblastoma. In: Elder D, Massi D, Scolyer RA, et al, eds. WHO Classification of Skin Tumours. 4th ed. Lyon, France: IARC; 2018.
The Diagnosis: Unilateral Dermatomal Trichoepithelioma
Adnexal lesions presenting with a linear and/or dermatomal pattern rarely have been reported. Bolognia et al1 performed a comprehensive review of Blaschko lines and skin conditions that follow such a pattern. The authors found that adnexal-related lesions included linear nevus comedonicus, linear basal cell nevus with comedones (linear basaloid follicular hamartoma), unilateral nevoid basal cell carcinoma (BCC), linear trichoepithelioma, linear trichodiscoma, linear hamartoma of the follicular infundibulum, nevus sebaceous, syringocystadenoma papilliferum, porokeratotic eccrine ostial and dermal duct nevus, linear eccrine poroma, linear spiradenoma, linear syringoma, and linear eccrine syringofibroadenoma.1
Trichoepithelioma is a hair follicle-related neoplastic lesion presenting most commonly as the autosomal-dominant multiple familial type with lesions mainly centered on the face. Initial genetic studies associated the disease with loss of heterozygosity in the 9p21 region and further studies identified mutations in the CYLD (cylindromatosis [turban tumor syndrome]) gene on chromosome 16q12-q13.2,3 Unilateral, linear, and dermatomal forms of trichoepithelioma rarely are reported. In 1986, Geffner et al4 reported a case of linear and dermatomal trichoepithelioma in a 10-year-old girl. In addition to discrete solitary lesions affecting the face, she developed lesions on the left shoulder, left side of the trunk, and left lower leg following dermatomal distribution. In 2006, 2 cases of dermatomal trichoepitheliomas affecting the face in children, as in our case, were reported.5,6 Another case involving the neck was reported in 2016.7 Although classic multiple familial trichoepithelioma can be part of conditions such as Brooke-Spiegler8 and Rombo syndromes,9 no syndromal association has been reported thus far with the unilateral, linear, or dermatomal variants.
Our case showed typical histopathologic features of trichoepithelioma, including discrete islands of basaloid cells in the dermis set in a conspicuous fibroblastic stroma. Focal connection with the epidermis was present. Most of the islands showed peripheral palisading and horn cysts lined by eosinophilic cells. The fibroblastic component was tightly adherent to the epithelial component, and only stromal clefts were detected. Papillary mesenchymal bodies also were detected as oval aggregates of fibroblastic cells invaginating into epithelial islands to form hair papillae.
Histopathologically, the 2 most important differential diagnoses of trichoepithelioma include BCC and basaloid follicular hamartoma. In differentiating BCC from trichoepithelioma, the presence of dense fibroblastic stroma and papillary mesenchymal bodies characterize trichoepithelioma, while a fibromucinous stroma with mucinous retraction artifacts and clefting between the basaloid islands and the stroma characterize BCC (Figure 1).10 Immunohistochemical studies using antibodies against Bcl-2, CD34, CD10, androgen receptor, Ki-67, cytokeratin 19, and PHLDA1 (pleckstrin homologylike domain family A member 1) have reportedly been utilized to differentiate trichoepithelioma from BCC.11,12 Basaloid follicular hamartoma is characterized by thin anastomosing strands and branching cords of undifferentiated basaloid cells that replace or associate hair follicles in a latticelike pattern (Figure 2). The strands usually are vertically oriented perpendicular to the epidermis. Peripheral palisading is possible, and the basaloid strands are surrounded with cellular connective tissue stroma.13 Tumor islands in eccrine poroma show broad connections with the epidermis and are composed of poroid cells that show evident ductal differentiation with eosinophilic cuticles (Figure 3).14 Spiradenoma is characterized by capsulated deep-seated tumorous nodules not connected with the epidermis and composed of light and dark cells with ductal differentiation and vascular stroma (Figure 4). Scattered lymphocytes within the tumor lobules and in the stroma also are seen. Eosinophilic hyaline globules rarely can be present.15
Many pathologists consider trichoepithelioma as the superficial variant of trichoblastoma. According to the recent World Health Organization classification of benign tumors with follicular differentiation, trichoepithelioma is considered synonymous with trichoblastoma.16
Trichoepitheliomas are benign tumors, and therapy is mainly directed at removal for cosmetic purposes. Several methods of removal are available including electrocautery, laser therapy, and surgery. Awareness of the possible dermatomal distribution of hair follicle and other adnexal-related conditions is important, and such lesions should be thought of in the differential diagnosis of unilateral and/or dermatomal lesions.
The Diagnosis: Unilateral Dermatomal Trichoepithelioma
Adnexal lesions presenting with a linear and/or dermatomal pattern rarely have been reported. Bolognia et al1 performed a comprehensive review of Blaschko lines and skin conditions that follow such a pattern. The authors found that adnexal-related lesions included linear nevus comedonicus, linear basal cell nevus with comedones (linear basaloid follicular hamartoma), unilateral nevoid basal cell carcinoma (BCC), linear trichoepithelioma, linear trichodiscoma, linear hamartoma of the follicular infundibulum, nevus sebaceous, syringocystadenoma papilliferum, porokeratotic eccrine ostial and dermal duct nevus, linear eccrine poroma, linear spiradenoma, linear syringoma, and linear eccrine syringofibroadenoma.1
Trichoepithelioma is a hair follicle-related neoplastic lesion presenting most commonly as the autosomal-dominant multiple familial type with lesions mainly centered on the face. Initial genetic studies associated the disease with loss of heterozygosity in the 9p21 region and further studies identified mutations in the CYLD (cylindromatosis [turban tumor syndrome]) gene on chromosome 16q12-q13.2,3 Unilateral, linear, and dermatomal forms of trichoepithelioma rarely are reported. In 1986, Geffner et al4 reported a case of linear and dermatomal trichoepithelioma in a 10-year-old girl. In addition to discrete solitary lesions affecting the face, she developed lesions on the left shoulder, left side of the trunk, and left lower leg following dermatomal distribution. In 2006, 2 cases of dermatomal trichoepitheliomas affecting the face in children, as in our case, were reported.5,6 Another case involving the neck was reported in 2016.7 Although classic multiple familial trichoepithelioma can be part of conditions such as Brooke-Spiegler8 and Rombo syndromes,9 no syndromal association has been reported thus far with the unilateral, linear, or dermatomal variants.
Our case showed typical histopathologic features of trichoepithelioma, including discrete islands of basaloid cells in the dermis set in a conspicuous fibroblastic stroma. Focal connection with the epidermis was present. Most of the islands showed peripheral palisading and horn cysts lined by eosinophilic cells. The fibroblastic component was tightly adherent to the epithelial component, and only stromal clefts were detected. Papillary mesenchymal bodies also were detected as oval aggregates of fibroblastic cells invaginating into epithelial islands to form hair papillae.
Histopathologically, the 2 most important differential diagnoses of trichoepithelioma include BCC and basaloid follicular hamartoma. In differentiating BCC from trichoepithelioma, the presence of dense fibroblastic stroma and papillary mesenchymal bodies characterize trichoepithelioma, while a fibromucinous stroma with mucinous retraction artifacts and clefting between the basaloid islands and the stroma characterize BCC (Figure 1).10 Immunohistochemical studies using antibodies against Bcl-2, CD34, CD10, androgen receptor, Ki-67, cytokeratin 19, and PHLDA1 (pleckstrin homologylike domain family A member 1) have reportedly been utilized to differentiate trichoepithelioma from BCC.11,12 Basaloid follicular hamartoma is characterized by thin anastomosing strands and branching cords of undifferentiated basaloid cells that replace or associate hair follicles in a latticelike pattern (Figure 2). The strands usually are vertically oriented perpendicular to the epidermis. Peripheral palisading is possible, and the basaloid strands are surrounded with cellular connective tissue stroma.13 Tumor islands in eccrine poroma show broad connections with the epidermis and are composed of poroid cells that show evident ductal differentiation with eosinophilic cuticles (Figure 3).14 Spiradenoma is characterized by capsulated deep-seated tumorous nodules not connected with the epidermis and composed of light and dark cells with ductal differentiation and vascular stroma (Figure 4). Scattered lymphocytes within the tumor lobules and in the stroma also are seen. Eosinophilic hyaline globules rarely can be present.15
Many pathologists consider trichoepithelioma as the superficial variant of trichoblastoma. According to the recent World Health Organization classification of benign tumors with follicular differentiation, trichoepithelioma is considered synonymous with trichoblastoma.16
Trichoepitheliomas are benign tumors, and therapy is mainly directed at removal for cosmetic purposes. Several methods of removal are available including electrocautery, laser therapy, and surgery. Awareness of the possible dermatomal distribution of hair follicle and other adnexal-related conditions is important, and such lesions should be thought of in the differential diagnosis of unilateral and/or dermatomal lesions.
- Bolognia JL, Orlow SJ, Glick SA. Lines of Blaschko. J Am Acad Dermatol. 1994;31(2, pt 1):157-190.
- Harada H, Hashimoto K, Ko MS. The gene for multiple familial trichoepithelioma maps to chromosome 9p21. J Invest Dermatol. 1996;107:41-43.
- Zheng G, Hu L, Huang W, et al. CYLD mutation causes multiple familial trichoepithelioma in three Chinese families. Hum Mutat. 2004;23:400.
- Geffner RE, Goslen JB, Santa Cruz DJ. Linear and dermatomal trichoepitheliomas. J Am Acad Dermatol. 1986;14(5, pt 2):927-930.
- Chang YC, Colome-Grimmer M, Kelly E. Multiple trichoepitheliomas in the lines of Blaschko. Pediatr Dermatol. 2006;23:149-151.
- Strauss RM, Merchant WJ, Stainforth JM, et al. Unilateral naevoid trichoepitheliomas on the face of a child. Clin Exp Dermatol. 2006;6:778-780.
- Laska AJ, Belli RA, Kobayashi TT. Linear trichoepithelioma on the neck of a 15-year-old girl. Dermatol Online J. 2016;22. pii:13030/qt87b6h4q8.
- Rasmussen JE. A syndrome of trichoepitheliomas, milia and cylindroma. Arch Dermatol. 1975;111:610-614.
- Michaelson G, Olsson E, Westermark P. The Rombo syndrome. Acta Derm Venereol. 1981;61:497-503.
- Brooke JD, Fitzpatrick JE, Golitz LE. Papillary mesenchymal bodies: a histologic finding useful in differentiating trichoepitheliomas from basal cell carcinomas. J Am Acad Dermatol. 1989;21(3, pt 1):523-528.
- Mostafa NA, Assaf M, Elhakim S, et al. Diagnostic accuracy of immunohistochemical markers in differentiation between basal cell carcinoma and trichoepithelioma in small biopsy specimens. J Cutan Pathol. 2018;45:807-816.
- Poniecka AW, Alexis JB. An immunohistochemical study of basal cell carcinoma and trichoepithelioma. Am J Dermatopathol. 1999;21:332-336.
- Abdel-Halim MRE, Fawzy M, Saleh M, et al. Linear unilateral basal cell nevus with comedones (linear nevoid basaloid follicular hamartoma): a case report. J Egypt Womens Dermatol Soc. 2016;13:46-48.
- Hyman AB, Brownstein MH. Eccrine poroma: analysis of 45 new cases. Dermatologica. 1969;138:28-38.
- Mambo NC. Eccrine spiradenoma: clinical and pathologic study of 49 tumors. J Cutan Pathol. 1983;10:312-320.
- Kutzner H, Kaddu S, Kanitakis J, et al. Trichoblastoma. In: Elder D, Massi D, Scolyer RA, et al, eds. WHO Classification of Skin Tumours. 4th ed. Lyon, France: IARC; 2018.
- Bolognia JL, Orlow SJ, Glick SA. Lines of Blaschko. J Am Acad Dermatol. 1994;31(2, pt 1):157-190.
- Harada H, Hashimoto K, Ko MS. The gene for multiple familial trichoepithelioma maps to chromosome 9p21. J Invest Dermatol. 1996;107:41-43.
- Zheng G, Hu L, Huang W, et al. CYLD mutation causes multiple familial trichoepithelioma in three Chinese families. Hum Mutat. 2004;23:400.
- Geffner RE, Goslen JB, Santa Cruz DJ. Linear and dermatomal trichoepitheliomas. J Am Acad Dermatol. 1986;14(5, pt 2):927-930.
- Chang YC, Colome-Grimmer M, Kelly E. Multiple trichoepitheliomas in the lines of Blaschko. Pediatr Dermatol. 2006;23:149-151.
- Strauss RM, Merchant WJ, Stainforth JM, et al. Unilateral naevoid trichoepitheliomas on the face of a child. Clin Exp Dermatol. 2006;6:778-780.
- Laska AJ, Belli RA, Kobayashi TT. Linear trichoepithelioma on the neck of a 15-year-old girl. Dermatol Online J. 2016;22. pii:13030/qt87b6h4q8.
- Rasmussen JE. A syndrome of trichoepitheliomas, milia and cylindroma. Arch Dermatol. 1975;111:610-614.
- Michaelson G, Olsson E, Westermark P. The Rombo syndrome. Acta Derm Venereol. 1981;61:497-503.
- Brooke JD, Fitzpatrick JE, Golitz LE. Papillary mesenchymal bodies: a histologic finding useful in differentiating trichoepitheliomas from basal cell carcinomas. J Am Acad Dermatol. 1989;21(3, pt 1):523-528.
- Mostafa NA, Assaf M, Elhakim S, et al. Diagnostic accuracy of immunohistochemical markers in differentiation between basal cell carcinoma and trichoepithelioma in small biopsy specimens. J Cutan Pathol. 2018;45:807-816.
- Poniecka AW, Alexis JB. An immunohistochemical study of basal cell carcinoma and trichoepithelioma. Am J Dermatopathol. 1999;21:332-336.
- Abdel-Halim MRE, Fawzy M, Saleh M, et al. Linear unilateral basal cell nevus with comedones (linear nevoid basaloid follicular hamartoma): a case report. J Egypt Womens Dermatol Soc. 2016;13:46-48.
- Hyman AB, Brownstein MH. Eccrine poroma: analysis of 45 new cases. Dermatologica. 1969;138:28-38.
- Mambo NC. Eccrine spiradenoma: clinical and pathologic study of 49 tumors. J Cutan Pathol. 1983;10:312-320.
- Kutzner H, Kaddu S, Kanitakis J, et al. Trichoblastoma. In: Elder D, Massi D, Scolyer RA, et al, eds. WHO Classification of Skin Tumours. 4th ed. Lyon, France: IARC; 2018.

A 9-year-old boy presented with a slowly progressive lesion of 5 years’ duration affecting only the left side of the face in a dermatomal pattern. The patient denied any symptoms and had no other anomalies or family history of similar lesions. On physical examination the lesion was found to span a 12×7-cm area of the lateral half of the left cheek and was composed of multiple variable-sized, pinkish to flesh-colored papules that coalesced in some areas to form small plaques. Few milialike cysts were present. One papule was biopsied.
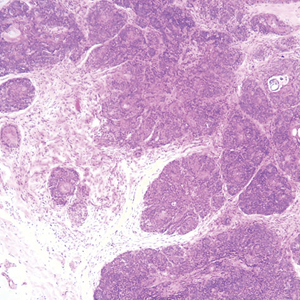
Indurated Plaque on the Shoulder
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
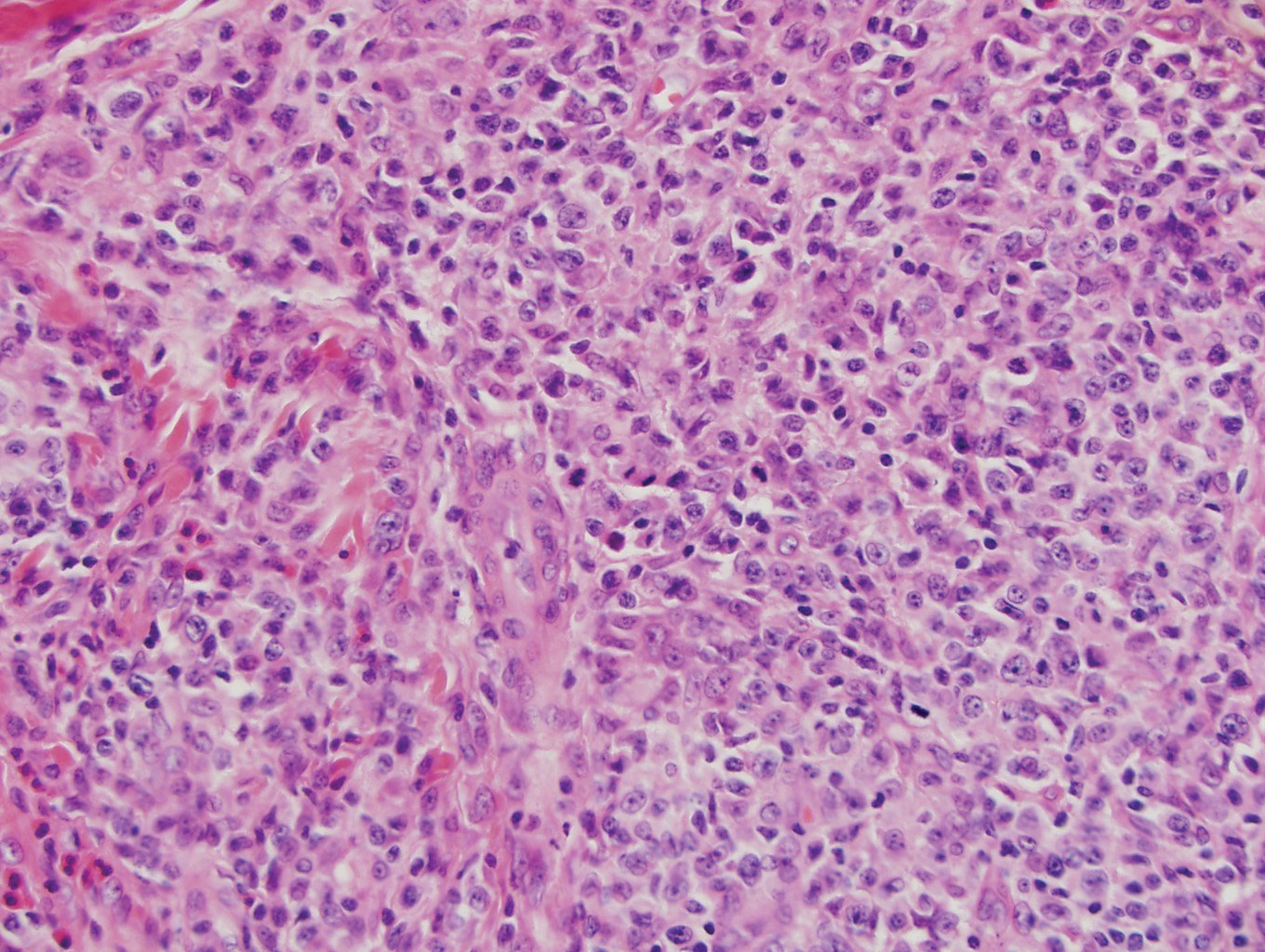
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
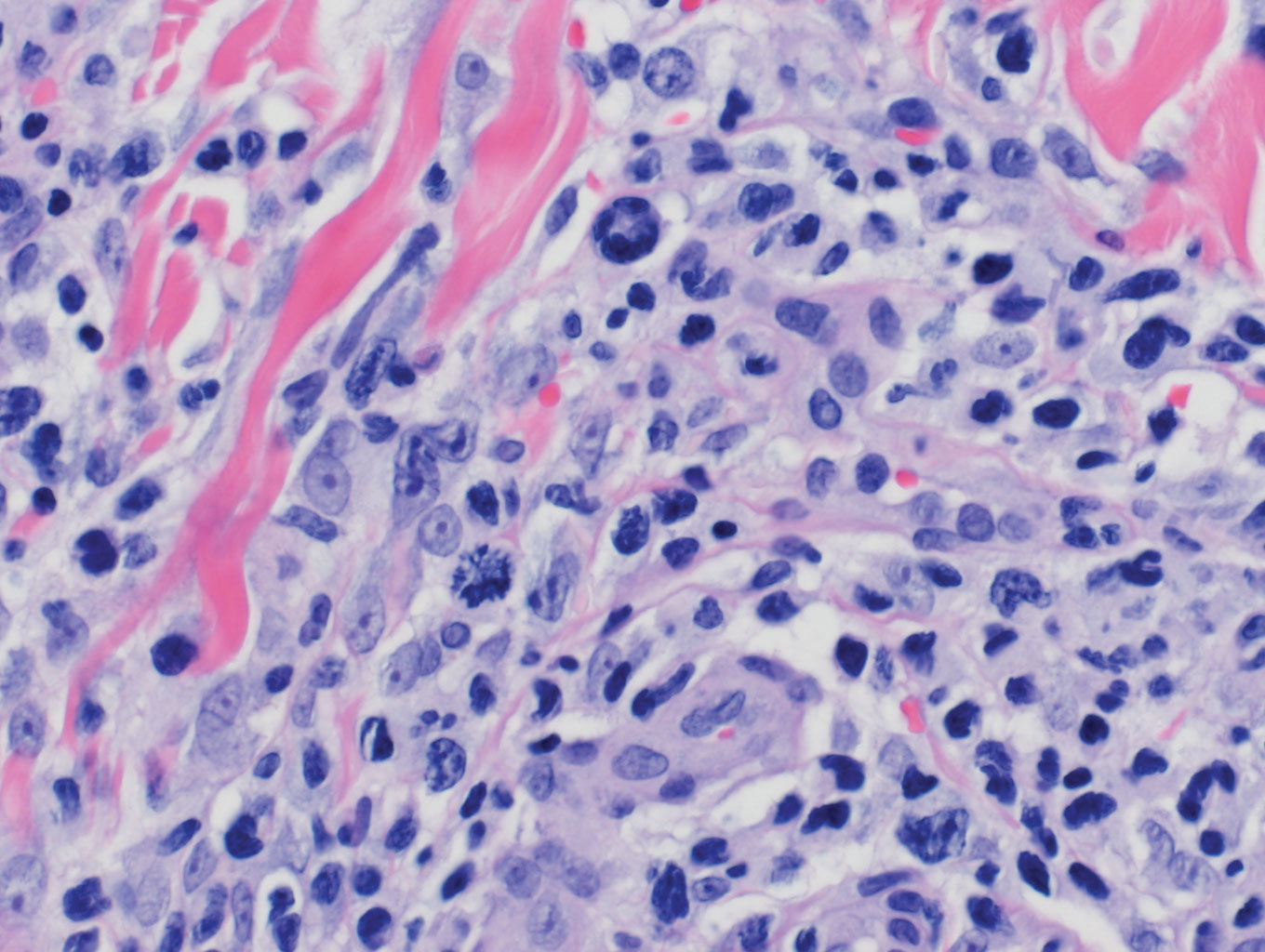
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
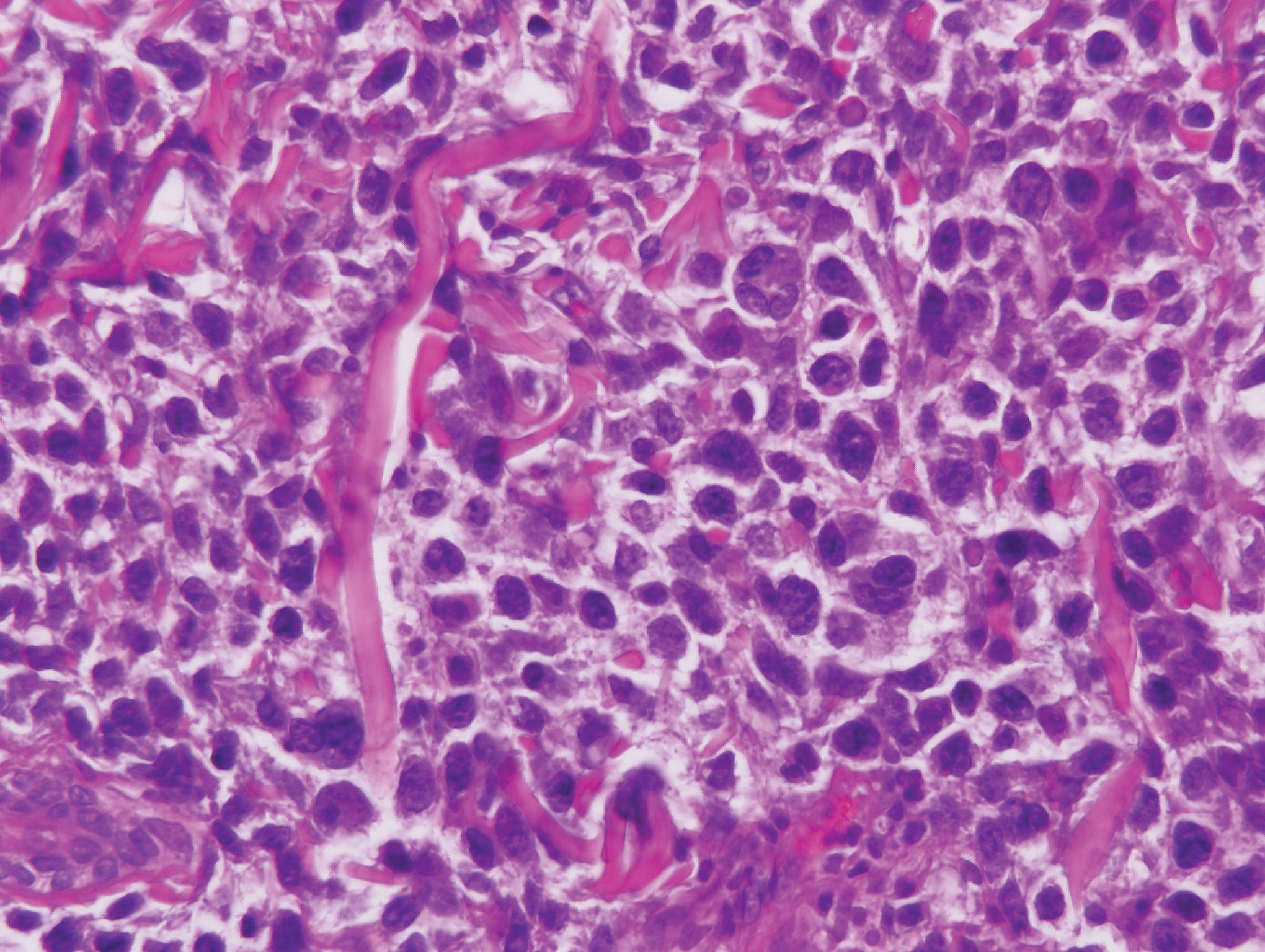
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
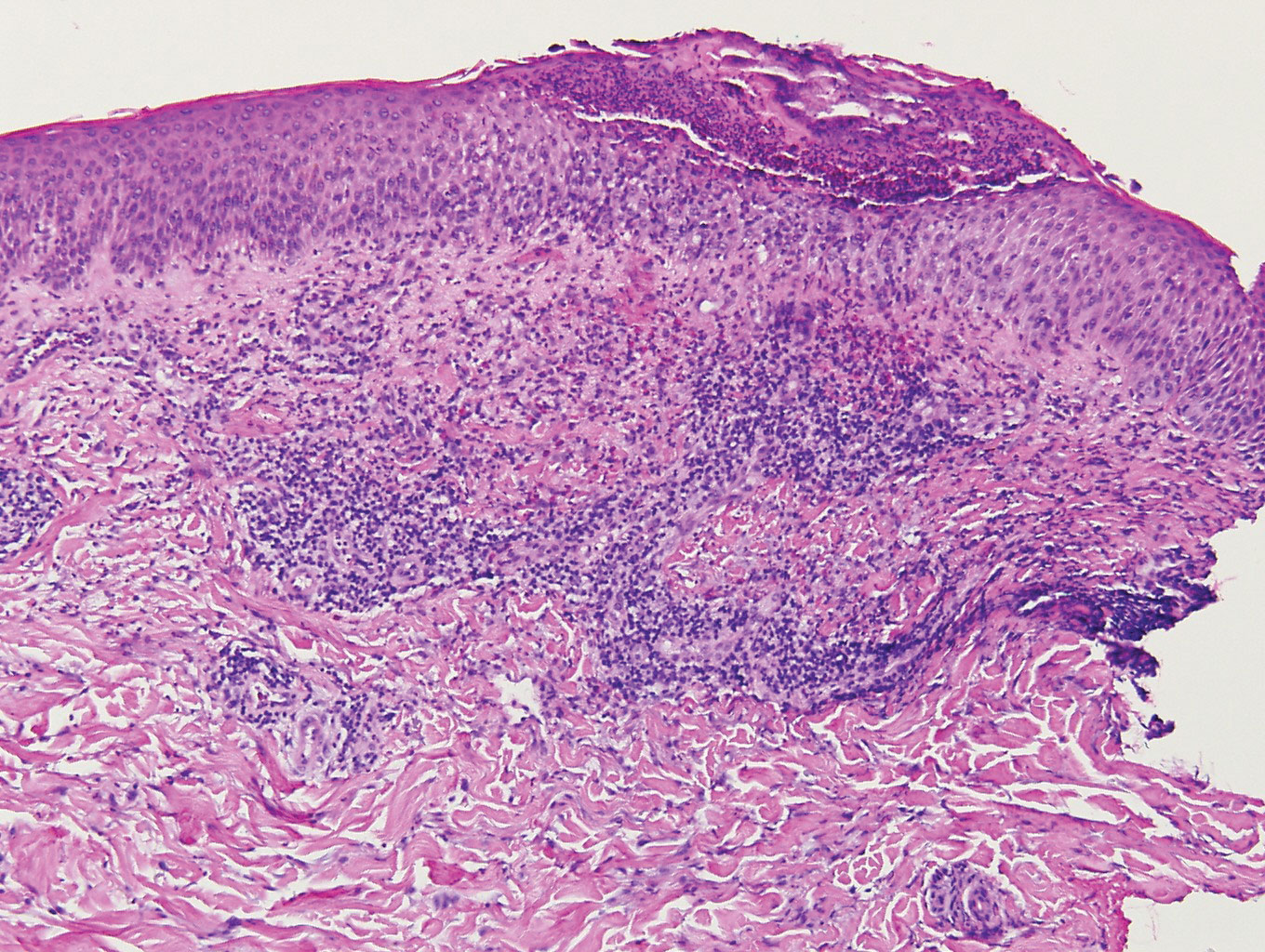
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
Herpes zoster (HZ) is a painful skin condition caused by reactivation of latent varicella-zoster virus (VZV) in dorsal root ganglion cells.1 Upon reactivation, VZV replicates in the dorsal root ganglion, which ultimately results in inflammation and necrosis of the neuron and intense neuralgia. Reactivation of latent VZV may occur spontaneously or may be induced by various factors including immunosuppression, stress, illness, and trauma. Prior to the development of skin lesions, many patients experience a prodrome of tingling, pain, or pruritus. Herpes zoster classically presents with grouped vesicles on an erythematous base in a unilateral dermatomal distribution; however, more than one adjacent dermatome may be involved, and the lesions can cross the midline. Furthermore, the development of vesicles may be preceded by the development of edematous papules or plaques.1
On histology, VZV closely resembles herpes simplex virus type 1 and herpes simplex virus type 2 infections.2 Classic histologic findings include ballooning degeneration of keratinocytes, acantholysis, nuclear molding, ground-glass nuclear inclusions, marginated chromatin, and multinucleated keratinocytes, as well as necrosis of follicles and sebaceous glands.2 Varicella-zoster virus polymerase chain reaction or immunostaining can be used to confirm the diagnosis.2
Classic mycosis fungoides (MF) presents with well-circumscribed erythematous patches in non–sun-exposed areas and eventually may progress to plaques and tumors.3 Patients with cutaneous T-cell lymphomas, such as MF, are at a higher risk for skin infections including HZ4,5; however, immunocompromised patients, such as those with cutaneous lymphomas, can have atypical clinical presentations of HZ that may be concerning for cutaneous lymphoma.6 Furthermore, cutaneous malignancies can occur in dermatomal distributions that may mimic HZ.7 Therefore, the threshold for biopsy should be lowered in those patients with dermatomal lesions and history concerning for possible malignancy.
Classically, histologic examination of MF demonstrates an infiltrate of haloed cells at the dermoepidermal junction, which are atypical T cells with hyperchromatic cerebriform nuclei that are larger, darker, and more angulated than the benign recruited lymphocytes in the perivascular infiltrate seen in VZV infection (Figure 1).3 Papillary dermal fibrosis typically is present, and the perivascular infiltrate is denser above the postcapillary venule rather than being symmetrical around the vessel (bare underbelly sign). Clusters of these cells may form within the epidermis, which are called Pautrier microabscesses.3 Mycosis fungoides also can exhibit large cell transformation in which small lymphocytes transform into larger cells, thereby associated with a poorer prognosis.8
Lymphomatoid papulosis is a CD30+-predominant form of cutaneous T-cell lymphoma characterized by papules and nodules that spontaneously involute.9 This condition is most commonly associated with MF but can be associated with other lymphomas. This condition may be mistaken for HZ clinically, but histology classically demonstrates large atypical lymphocytes resembling Reed-Sternberg cells in small clusters rather than follicular necrosis (Figure 2).9
Patients with lymphoma may sequentially develop a secondary lymphoma. There have been reports of secondary B-cell lymphomas associated with MF, but this phenomenon is rare.10 The histology depends on the type of B-cell lymphoma present, but follicular necrosis would not be expected (Figure 3).
Unusual hypersensitivity reactions to arthropod attacks have been described in patients with lymphoproliferative disorders and could be mistaken for HZ. Histology may demonstrate a wedge-shaped perivascular and/or interstitial infiltrate containing eosinophils with endothelial swelling (Figure 4), but these findings may vary depending on the type of arthropod involved.11
Our case provided a unique example of HZ in a patient with a known history of MF. Clinically, there was concern for progression of the patient’s underlying disease; however, histology demonstrated ballooning keratinocytes and follicular necrosis, which are classically seen in HZ infection.
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
- Downing C, Medoza N, Sra K, et al. Human herpesviruses. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. China: Elsevier; 2018:1400-1424.
- Chisholm C, Lopez L. Cutaneous infections caused by Herpesviridae: a review. Arch Pathol Lab Med. 2011;135:1357-1362.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70: 205.e1-205.e16.
- Vonderheid EC, van Voorst Vader PC. Herpes zoster-varicella in cutaneous T-cell lymphomas. Arch Dermatol. 1980;116:408-412.
- Lebas E, Arrese JE, Nikkels AF. Risk factors for skin infections in mycosis fungoides. Dermatology. 2016;232:731-737.
- Leinweber B, Kerl H, Cerroni L. Histopathologic features of cutaneous herpes virus infections (herpes simplex, herpes varicella/zoster): a broad spectrum of presentations with common pseudolymphomatous aspects. Am J Surg Pathol. 2006;30:50-58.
- Niiyama S, Satoh K, Kaneko S, et al. Zosteriform skin involvement of nodal T-cell lymphoma: a review of the published work of cutaneous malignancies mimicking herpes zoster. J Dermatol. 2007;34:68-73.
- Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation:clinicopathological features and prognostic factors. Pathology. 2014;46:610-616.
- Zackheim HS, Jones C, Leboit PE, et al. Lymphomatoid papulosis associated with mycosis fungoides: a study of 21 patients including analyses for clonality. J Am Acad Dermatol. 2003;49:620-623.
- Barzilai A, Trau H, David M, et al. Mycosis fungoides associated with B-cell malignancies. Br J Dermatol. 2006;155:379-386.
- Vassallo C, Passamonti F, Cananzi R, et al. Exaggerated insect bite-like reaction in patients affected by oncohaematological diseases. Acta Derm Venereol. 2005;85:76-77.
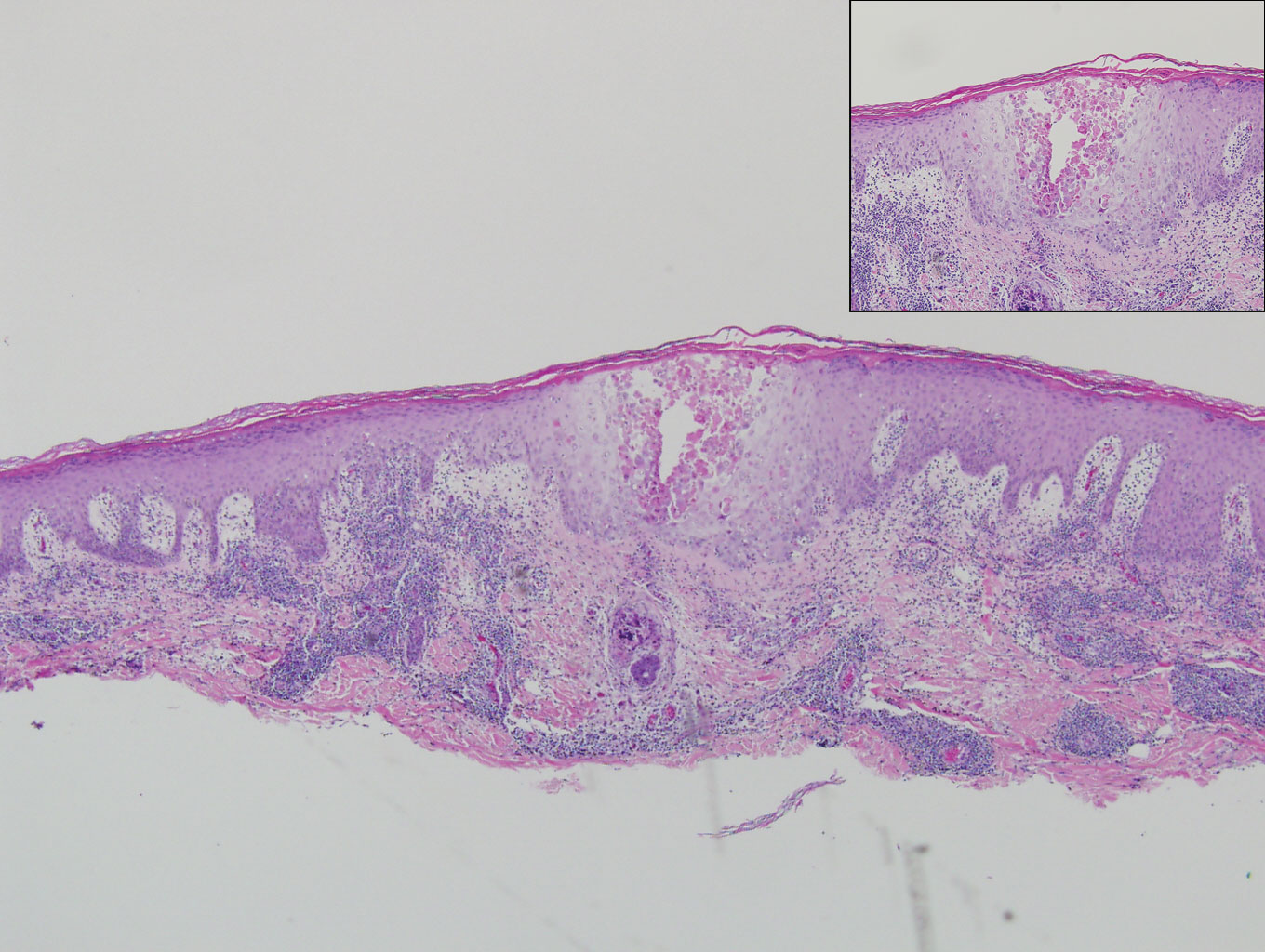
A 66-year-old man with mycosis fungoides presented with a new indurated plaque on the left shoulder. Biopsies of the left shoulder and back lesions were obtained.
Solitary Nodule on the Thigh
The Diagnosis: Ruptured Molluscum
Molluscum contagiosum (MC) is caused by a DNA virus (MC virus) belonging to the poxvirus family. Molluscum contagiosum is common and predominantly seen in children and young adults. In sexually active adults, the lesions commonly occur in the genital region, abdomen, and inner thighs. In immunocompromised individuals, including those with AIDS, the lesions are more extensive and may cause disfigurement.1 Molluscum contagiosum involving epidermoid cysts has been reported.2
Histopathologically, MC can be classified as noninflammatory or inflammatory. In noninflamed lesions, multiple large, intracytoplasmic, eosinophilic inclusions (Henderson-Paterson bodies) appear within the lobulated endophytic and hyperplastic epidermis. Ultrastructurally, these bodies show membrane-bound collections of MC virus.1 Replicating Henderson-Paterson bodies can result in rupture and inflammation. This case demonstrates a palisading granuloma containing keratin with few Henderson-Paterson bodies (quiz image) due to prior rupture of a molluscum or molluscoid cyst.
Rheumatoid nodules, the most characteristic histopathologic lesions of rheumatoid arthritis, are most commonly found in the subcutis at points of pressure and may occur in connective tissue of numerous organs. Rheumatoid nodules are firm, nontender, and mobile within the subcutaneous tissue but may be fixed to underlying structures including the periosteum, tendons, or bursae.3,4 Occasionally, superficial nodules may perforate the epidermis.5 The inner central necrobiotic zone appears as intensely eosinophilic, amorphous fibrin and other cellular debris. This central area is surrounded by histiocytes in a palisaded configuration (Figure 1). Multinucleated foreign body giant cells also may be present. Occasionally, mast cells, eosinophils, and neutrophils are present.6,7
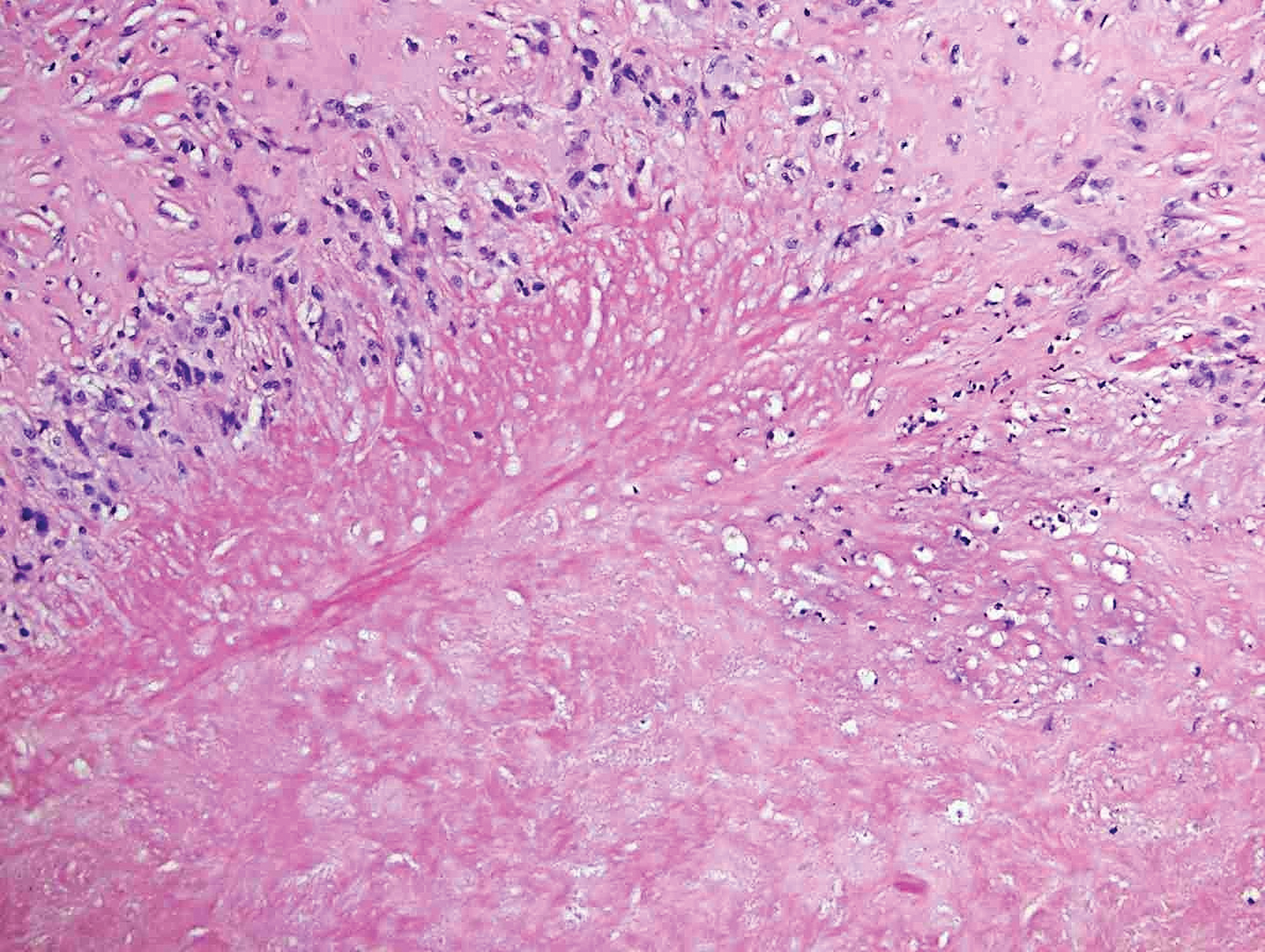
Lupus miliaris disseminatus faciei presents with multiple discrete, smooth, yellow-brown to red, dome-shaped papules. The lesions typically are located on the central and lateral sides of the face and infrequently involve the neck. Other sites including the axillae, arms, hands, legs, and groin occasionally can be involved. Diascopy may reveal an apple jelly color.8,9 The histopathologic hallmark of lupus miliaris disseminatus faciei is an epithelioid cell granuloma with central necrosis (Figure 2).

Epithelioid sarcoma (ES) is a soft tissue tumor with a known propensity for local recurrence, regional lymph node involvement, sporotrichoid spread, and distant metastases.10 The name was coined by Enzinger11 in 1970 during a review of 62 cases of a “peculiar form of sarcoma that has repeatedly been confused with a chronic inflammatory process, a necrotizing granuloma, and a squamous cell carcinoma.” Epithelioid sarcoma tends to grow slowly in a nodular or multinodular manner along fascial structures and tendons, often with central necrosis and ulceration of the overlying skin. Histopathologically, classic ES shows nodular masses of uniform plump epithelioid cells with abundant eosinophilic cytoplasm and prominent central necrosis. A biphasic pattern is typical with spindle cells merging with epithelioid cells. Cellular atypia is relatively mild and mitoses are rare (Figure 3). Recurrent or metastatic lesions can show a greater degree of pleomorphism.12 Given the low-grade atypia in early lesions, this sarcoma is easily misdiagnosed as granulomatous dermatitis. Immunohistochemically, the majority of ES cases are positive for cytokeratins and epithelial membrane antigen; SMARCB1/INI-1 expression is characteristically lost.13
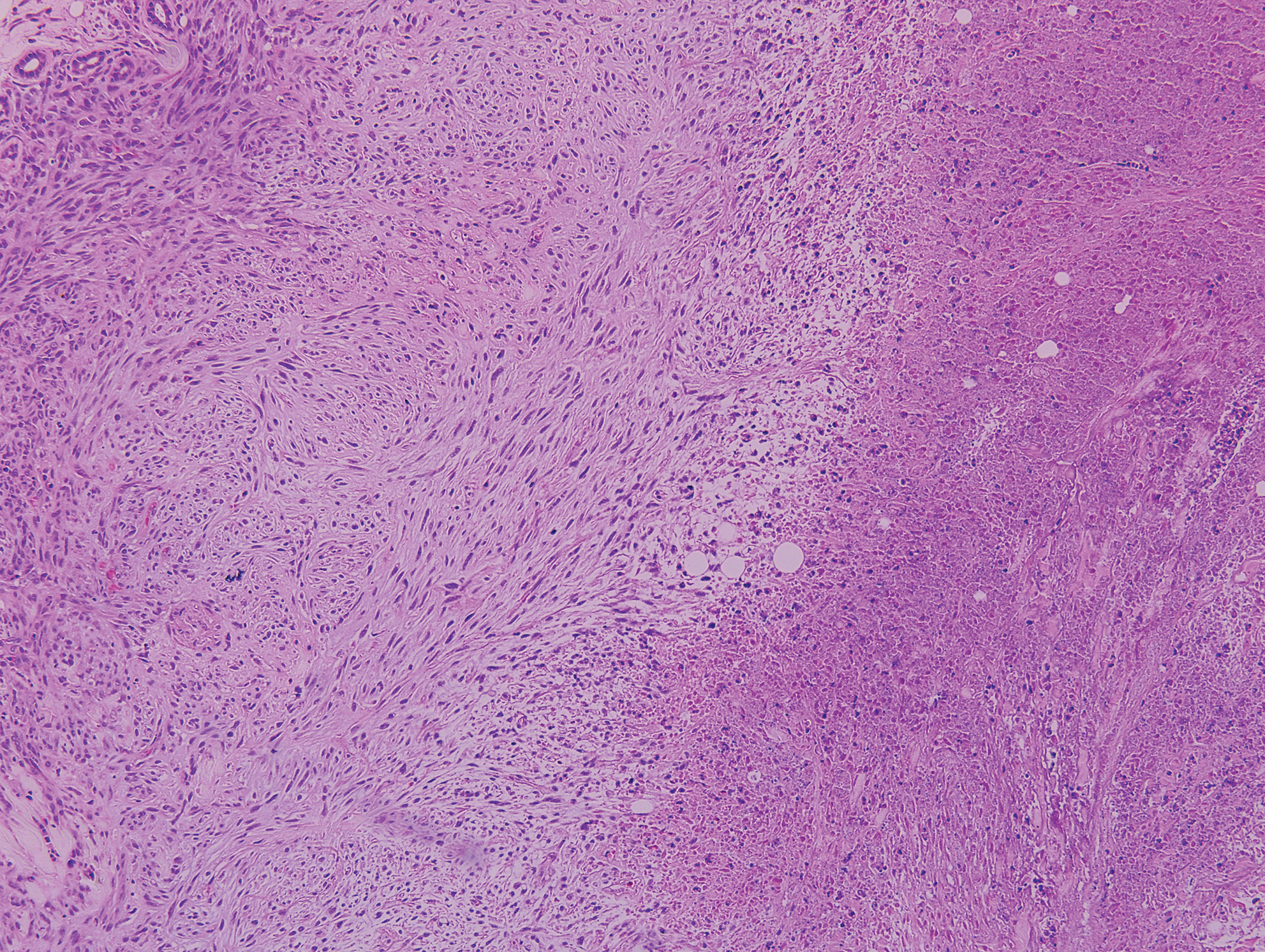
Granulomatosis with polyangiitis (formerly Wegener granulomatosis) is an autoimmune vasculitis highly associated with antineutrophil cytoplasmic antibodies. Clinical manifestations include systemic necrotizing vasculitis; necrotizing glomerulonephritis; and granulomatous inflammation, which predominantly involves the upper respiratory tract, skin, and mucosa.14,15 Skin involvement may be the initial manifestation of the disease and consists of palpable purpura, papules, ulcerations, vesicles, subcutaneous nodules, necrotizing ulcerations, papulonecrotic lesions, and petechiae. None of the findings are pathognomonic. The cutaneous histopathologic spectrum includes leukocytoclastic vasculitis, extravascular palisading granulomas, and granulomatous vasculitis.16 In the acute lesions of granulomatosis with polyangiitis, the predominant pattern of inflammation is not granulomatous but purulent with the appearance of an abscess. As it evolves, it develops a central zone of necrosis with extensive karyorrhectic debris and palisades of macrophages with scattered multinucleated giant cells (Figure 4).17
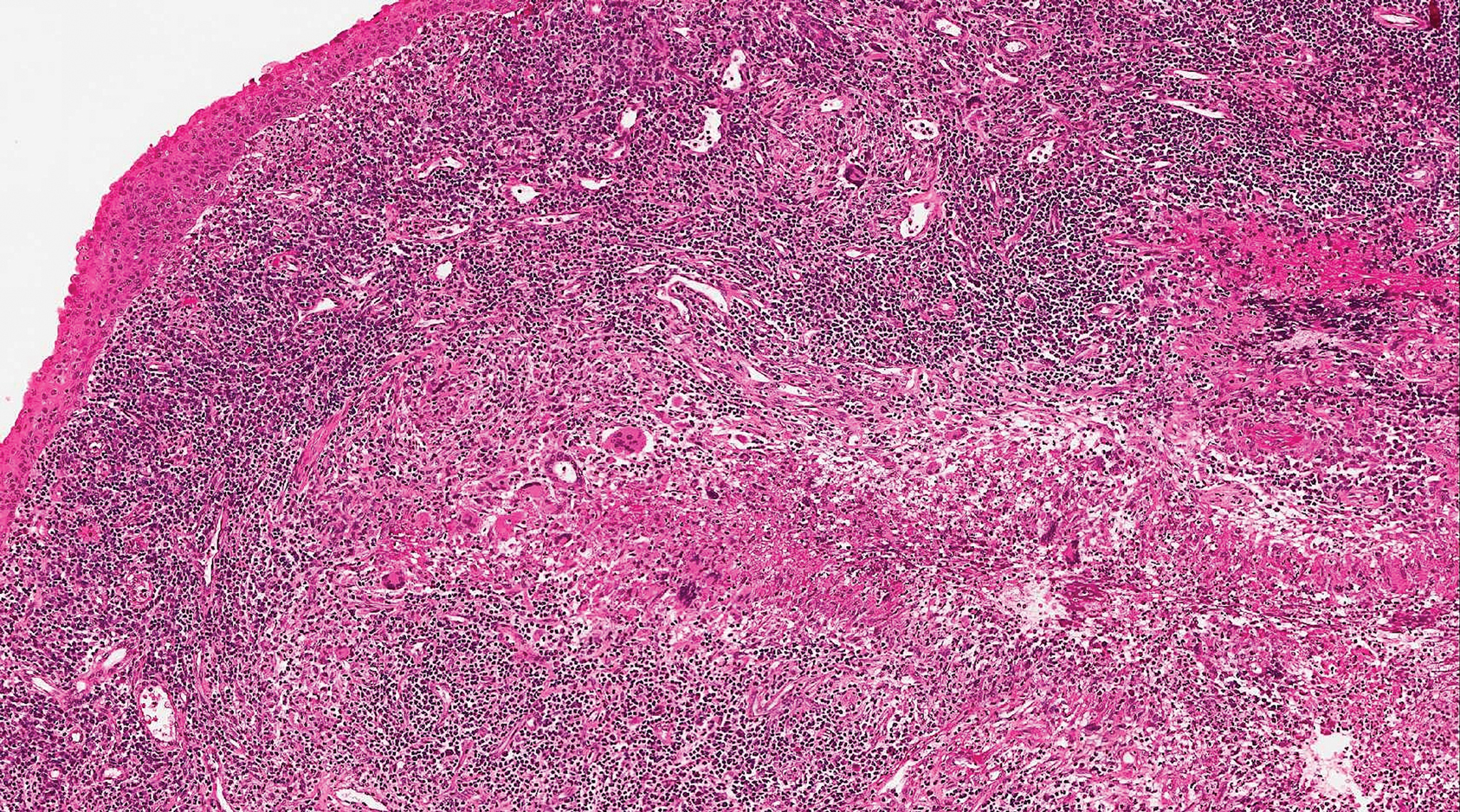
1. Nandhini G, Rajkumar K, Kanth KS, et al. Molluscum contagiosum in a 12-year-old child—report of a case and review of literature. J Int Oral Health. 2015;7:63-66.
2. Phelps A, Murphy M, Elaba Z, et al. Molluscum contagiosum virus infection in benign cutaneous epithelial cystic lesions-report of 2 cases with different pathogenesis? Am J Dermatopathol. 2010;32:740-742.
3. Sayah A, English JC 3rd. Rheumatoid arthritis: a review of the cutaneous manifestations. J Am Acad Dermatol. 2005;53:191-209; quiz 210-192.
4. Sibbitt WL Jr, Williams RC Jr. Cutaneous manifestations of rheumatoid arthritis. Int J Dermatol. 1982;21:563-572.
5. Barzilai A, Huszar M, Shpiro D, et al. Pseudorheumatoid nodules in adults: a juxta-articular form of nodular granuloma annulare. Am J Dermatopathol. 2005;27:1-5.
6. Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg. 2007;26:100-107.
7. Patterson JW. Rheumatoid nodule and subcutaneous granuloma annulare. a comparative histologic study. Am J Dermatopathol. 1988;10:1-8.
8. Sehgal VN, Srivastava G, Aggarwal AK, et al. Lupus miliaris disseminatus faciei part II: an overview. Skinmed. 2005;4:234-238.
9. Cymerman R, Rosenstein R, Shvartsbeyn M, et al. Lupus miliaris disseminatus faciei. Dermatol Online J. 2015;21. pii:13030/qt6b83q5gp.
10. Sobanko JF, Meijer L, Nigra TP. Epithelioid sarcoma: a review and update. J Clin Aesthet Dermatol. 2009;2:49-54.
11. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
12. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
13. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
14. Lutalo PM, D’Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis)[published online January 29, 2014]. J Autoimmun. 2014;48-49:94-98.
15. Frances C, Du LT, Piette JC, et al. Wegener’s granulomatosis. dermatological manifestations in 75 cases with clinicopathologic correlation. Arch Dermatol. 1994;130:861-867.
16. Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener’s granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
17. Jennette JC. Nomenclature and classification of vasculitis: lessons learned from granulomatosis with polyangiitis (Wegener’s granulomatosis). Clin Exp Immunol. 2011;164 (suppl 1):7-10.
The Diagnosis: Ruptured Molluscum
Molluscum contagiosum (MC) is caused by a DNA virus (MC virus) belonging to the poxvirus family. Molluscum contagiosum is common and predominantly seen in children and young adults. In sexually active adults, the lesions commonly occur in the genital region, abdomen, and inner thighs. In immunocompromised individuals, including those with AIDS, the lesions are more extensive and may cause disfigurement.1 Molluscum contagiosum involving epidermoid cysts has been reported.2
Histopathologically, MC can be classified as noninflammatory or inflammatory. In noninflamed lesions, multiple large, intracytoplasmic, eosinophilic inclusions (Henderson-Paterson bodies) appear within the lobulated endophytic and hyperplastic epidermis. Ultrastructurally, these bodies show membrane-bound collections of MC virus.1 Replicating Henderson-Paterson bodies can result in rupture and inflammation. This case demonstrates a palisading granuloma containing keratin with few Henderson-Paterson bodies (quiz image) due to prior rupture of a molluscum or molluscoid cyst.
Rheumatoid nodules, the most characteristic histopathologic lesions of rheumatoid arthritis, are most commonly found in the subcutis at points of pressure and may occur in connective tissue of numerous organs. Rheumatoid nodules are firm, nontender, and mobile within the subcutaneous tissue but may be fixed to underlying structures including the periosteum, tendons, or bursae.3,4 Occasionally, superficial nodules may perforate the epidermis.5 The inner central necrobiotic zone appears as intensely eosinophilic, amorphous fibrin and other cellular debris. This central area is surrounded by histiocytes in a palisaded configuration (Figure 1). Multinucleated foreign body giant cells also may be present. Occasionally, mast cells, eosinophils, and neutrophils are present.6,7
Lupus miliaris disseminatus faciei presents with multiple discrete, smooth, yellow-brown to red, dome-shaped papules. The lesions typically are located on the central and lateral sides of the face and infrequently involve the neck. Other sites including the axillae, arms, hands, legs, and groin occasionally can be involved. Diascopy may reveal an apple jelly color.8,9 The histopathologic hallmark of lupus miliaris disseminatus faciei is an epithelioid cell granuloma with central necrosis (Figure 2).
Epithelioid sarcoma (ES) is a soft tissue tumor with a known propensity for local recurrence, regional lymph node involvement, sporotrichoid spread, and distant metastases.10 The name was coined by Enzinger11 in 1970 during a review of 62 cases of a “peculiar form of sarcoma that has repeatedly been confused with a chronic inflammatory process, a necrotizing granuloma, and a squamous cell carcinoma.” Epithelioid sarcoma tends to grow slowly in a nodular or multinodular manner along fascial structures and tendons, often with central necrosis and ulceration of the overlying skin. Histopathologically, classic ES shows nodular masses of uniform plump epithelioid cells with abundant eosinophilic cytoplasm and prominent central necrosis. A biphasic pattern is typical with spindle cells merging with epithelioid cells. Cellular atypia is relatively mild and mitoses are rare (Figure 3). Recurrent or metastatic lesions can show a greater degree of pleomorphism.12 Given the low-grade atypia in early lesions, this sarcoma is easily misdiagnosed as granulomatous dermatitis. Immunohistochemically, the majority of ES cases are positive for cytokeratins and epithelial membrane antigen; SMARCB1/INI-1 expression is characteristically lost.13
Granulomatosis with polyangiitis (formerly Wegener granulomatosis) is an autoimmune vasculitis highly associated with antineutrophil cytoplasmic antibodies. Clinical manifestations include systemic necrotizing vasculitis; necrotizing glomerulonephritis; and granulomatous inflammation, which predominantly involves the upper respiratory tract, skin, and mucosa.14,15 Skin involvement may be the initial manifestation of the disease and consists of palpable purpura, papules, ulcerations, vesicles, subcutaneous nodules, necrotizing ulcerations, papulonecrotic lesions, and petechiae. None of the findings are pathognomonic. The cutaneous histopathologic spectrum includes leukocytoclastic vasculitis, extravascular palisading granulomas, and granulomatous vasculitis.16 In the acute lesions of granulomatosis with polyangiitis, the predominant pattern of inflammation is not granulomatous but purulent with the appearance of an abscess. As it evolves, it develops a central zone of necrosis with extensive karyorrhectic debris and palisades of macrophages with scattered multinucleated giant cells (Figure 4).17
The Diagnosis: Ruptured Molluscum
Molluscum contagiosum (MC) is caused by a DNA virus (MC virus) belonging to the poxvirus family. Molluscum contagiosum is common and predominantly seen in children and young adults. In sexually active adults, the lesions commonly occur in the genital region, abdomen, and inner thighs. In immunocompromised individuals, including those with AIDS, the lesions are more extensive and may cause disfigurement.1 Molluscum contagiosum involving epidermoid cysts has been reported.2
Histopathologically, MC can be classified as noninflammatory or inflammatory. In noninflamed lesions, multiple large, intracytoplasmic, eosinophilic inclusions (Henderson-Paterson bodies) appear within the lobulated endophytic and hyperplastic epidermis. Ultrastructurally, these bodies show membrane-bound collections of MC virus.1 Replicating Henderson-Paterson bodies can result in rupture and inflammation. This case demonstrates a palisading granuloma containing keratin with few Henderson-Paterson bodies (quiz image) due to prior rupture of a molluscum or molluscoid cyst.
Rheumatoid nodules, the most characteristic histopathologic lesions of rheumatoid arthritis, are most commonly found in the subcutis at points of pressure and may occur in connective tissue of numerous organs. Rheumatoid nodules are firm, nontender, and mobile within the subcutaneous tissue but may be fixed to underlying structures including the periosteum, tendons, or bursae.3,4 Occasionally, superficial nodules may perforate the epidermis.5 The inner central necrobiotic zone appears as intensely eosinophilic, amorphous fibrin and other cellular debris. This central area is surrounded by histiocytes in a palisaded configuration (Figure 1). Multinucleated foreign body giant cells also may be present. Occasionally, mast cells, eosinophils, and neutrophils are present.6,7
Lupus miliaris disseminatus faciei presents with multiple discrete, smooth, yellow-brown to red, dome-shaped papules. The lesions typically are located on the central and lateral sides of the face and infrequently involve the neck. Other sites including the axillae, arms, hands, legs, and groin occasionally can be involved. Diascopy may reveal an apple jelly color.8,9 The histopathologic hallmark of lupus miliaris disseminatus faciei is an epithelioid cell granuloma with central necrosis (Figure 2).
Epithelioid sarcoma (ES) is a soft tissue tumor with a known propensity for local recurrence, regional lymph node involvement, sporotrichoid spread, and distant metastases.10 The name was coined by Enzinger11 in 1970 during a review of 62 cases of a “peculiar form of sarcoma that has repeatedly been confused with a chronic inflammatory process, a necrotizing granuloma, and a squamous cell carcinoma.” Epithelioid sarcoma tends to grow slowly in a nodular or multinodular manner along fascial structures and tendons, often with central necrosis and ulceration of the overlying skin. Histopathologically, classic ES shows nodular masses of uniform plump epithelioid cells with abundant eosinophilic cytoplasm and prominent central necrosis. A biphasic pattern is typical with spindle cells merging with epithelioid cells. Cellular atypia is relatively mild and mitoses are rare (Figure 3). Recurrent or metastatic lesions can show a greater degree of pleomorphism.12 Given the low-grade atypia in early lesions, this sarcoma is easily misdiagnosed as granulomatous dermatitis. Immunohistochemically, the majority of ES cases are positive for cytokeratins and epithelial membrane antigen; SMARCB1/INI-1 expression is characteristically lost.13
Granulomatosis with polyangiitis (formerly Wegener granulomatosis) is an autoimmune vasculitis highly associated with antineutrophil cytoplasmic antibodies. Clinical manifestations include systemic necrotizing vasculitis; necrotizing glomerulonephritis; and granulomatous inflammation, which predominantly involves the upper respiratory tract, skin, and mucosa.14,15 Skin involvement may be the initial manifestation of the disease and consists of palpable purpura, papules, ulcerations, vesicles, subcutaneous nodules, necrotizing ulcerations, papulonecrotic lesions, and petechiae. None of the findings are pathognomonic. The cutaneous histopathologic spectrum includes leukocytoclastic vasculitis, extravascular palisading granulomas, and granulomatous vasculitis.16 In the acute lesions of granulomatosis with polyangiitis, the predominant pattern of inflammation is not granulomatous but purulent with the appearance of an abscess. As it evolves, it develops a central zone of necrosis with extensive karyorrhectic debris and palisades of macrophages with scattered multinucleated giant cells (Figure 4).17
1. Nandhini G, Rajkumar K, Kanth KS, et al. Molluscum contagiosum in a 12-year-old child—report of a case and review of literature. J Int Oral Health. 2015;7:63-66.
2. Phelps A, Murphy M, Elaba Z, et al. Molluscum contagiosum virus infection in benign cutaneous epithelial cystic lesions-report of 2 cases with different pathogenesis? Am J Dermatopathol. 2010;32:740-742.
3. Sayah A, English JC 3rd. Rheumatoid arthritis: a review of the cutaneous manifestations. J Am Acad Dermatol. 2005;53:191-209; quiz 210-192.
4. Sibbitt WL Jr, Williams RC Jr. Cutaneous manifestations of rheumatoid arthritis. Int J Dermatol. 1982;21:563-572.
5. Barzilai A, Huszar M, Shpiro D, et al. Pseudorheumatoid nodules in adults: a juxta-articular form of nodular granuloma annulare. Am J Dermatopathol. 2005;27:1-5.
6. Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg. 2007;26:100-107.
7. Patterson JW. Rheumatoid nodule and subcutaneous granuloma annulare. a comparative histologic study. Am J Dermatopathol. 1988;10:1-8.
8. Sehgal VN, Srivastava G, Aggarwal AK, et al. Lupus miliaris disseminatus faciei part II: an overview. Skinmed. 2005;4:234-238.
9. Cymerman R, Rosenstein R, Shvartsbeyn M, et al. Lupus miliaris disseminatus faciei. Dermatol Online J. 2015;21. pii:13030/qt6b83q5gp.
10. Sobanko JF, Meijer L, Nigra TP. Epithelioid sarcoma: a review and update. J Clin Aesthet Dermatol. 2009;2:49-54.
11. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
12. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
13. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
14. Lutalo PM, D’Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis)[published online January 29, 2014]. J Autoimmun. 2014;48-49:94-98.
15. Frances C, Du LT, Piette JC, et al. Wegener’s granulomatosis. dermatological manifestations in 75 cases with clinicopathologic correlation. Arch Dermatol. 1994;130:861-867.
16. Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener’s granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
17. Jennette JC. Nomenclature and classification of vasculitis: lessons learned from granulomatosis with polyangiitis (Wegener’s granulomatosis). Clin Exp Immunol. 2011;164 (suppl 1):7-10.
1. Nandhini G, Rajkumar K, Kanth KS, et al. Molluscum contagiosum in a 12-year-old child—report of a case and review of literature. J Int Oral Health. 2015;7:63-66.
2. Phelps A, Murphy M, Elaba Z, et al. Molluscum contagiosum virus infection in benign cutaneous epithelial cystic lesions-report of 2 cases with different pathogenesis? Am J Dermatopathol. 2010;32:740-742.
3. Sayah A, English JC 3rd. Rheumatoid arthritis: a review of the cutaneous manifestations. J Am Acad Dermatol. 2005;53:191-209; quiz 210-192.
4. Sibbitt WL Jr, Williams RC Jr. Cutaneous manifestations of rheumatoid arthritis. Int J Dermatol. 1982;21:563-572.
5. Barzilai A, Huszar M, Shpiro D, et al. Pseudorheumatoid nodules in adults: a juxta-articular form of nodular granuloma annulare. Am J Dermatopathol. 2005;27:1-5.
6. Garcia-Patos V. Rheumatoid nodule. Semin Cutan Med Surg. 2007;26:100-107.
7. Patterson JW. Rheumatoid nodule and subcutaneous granuloma annulare. a comparative histologic study. Am J Dermatopathol. 1988;10:1-8.
8. Sehgal VN, Srivastava G, Aggarwal AK, et al. Lupus miliaris disseminatus faciei part II: an overview. Skinmed. 2005;4:234-238.
9. Cymerman R, Rosenstein R, Shvartsbeyn M, et al. Lupus miliaris disseminatus faciei. Dermatol Online J. 2015;21. pii:13030/qt6b83q5gp.
10. Sobanko JF, Meijer L, Nigra TP. Epithelioid sarcoma: a review and update. J Clin Aesthet Dermatol. 2009;2:49-54.
11. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
12. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
13. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
14. Lutalo PM, D’Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis)[published online January 29, 2014]. J Autoimmun. 2014;48-49:94-98.
15. Frances C, Du LT, Piette JC, et al. Wegener’s granulomatosis. dermatological manifestations in 75 cases with clinicopathologic correlation. Arch Dermatol. 1994;130:861-867.
16. Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener’s granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31:605-612.
17. Jennette JC. Nomenclature and classification of vasculitis: lessons learned from granulomatosis with polyangiitis (Wegener’s granulomatosis). Clin Exp Immunol. 2011;164 (suppl 1):7-10.
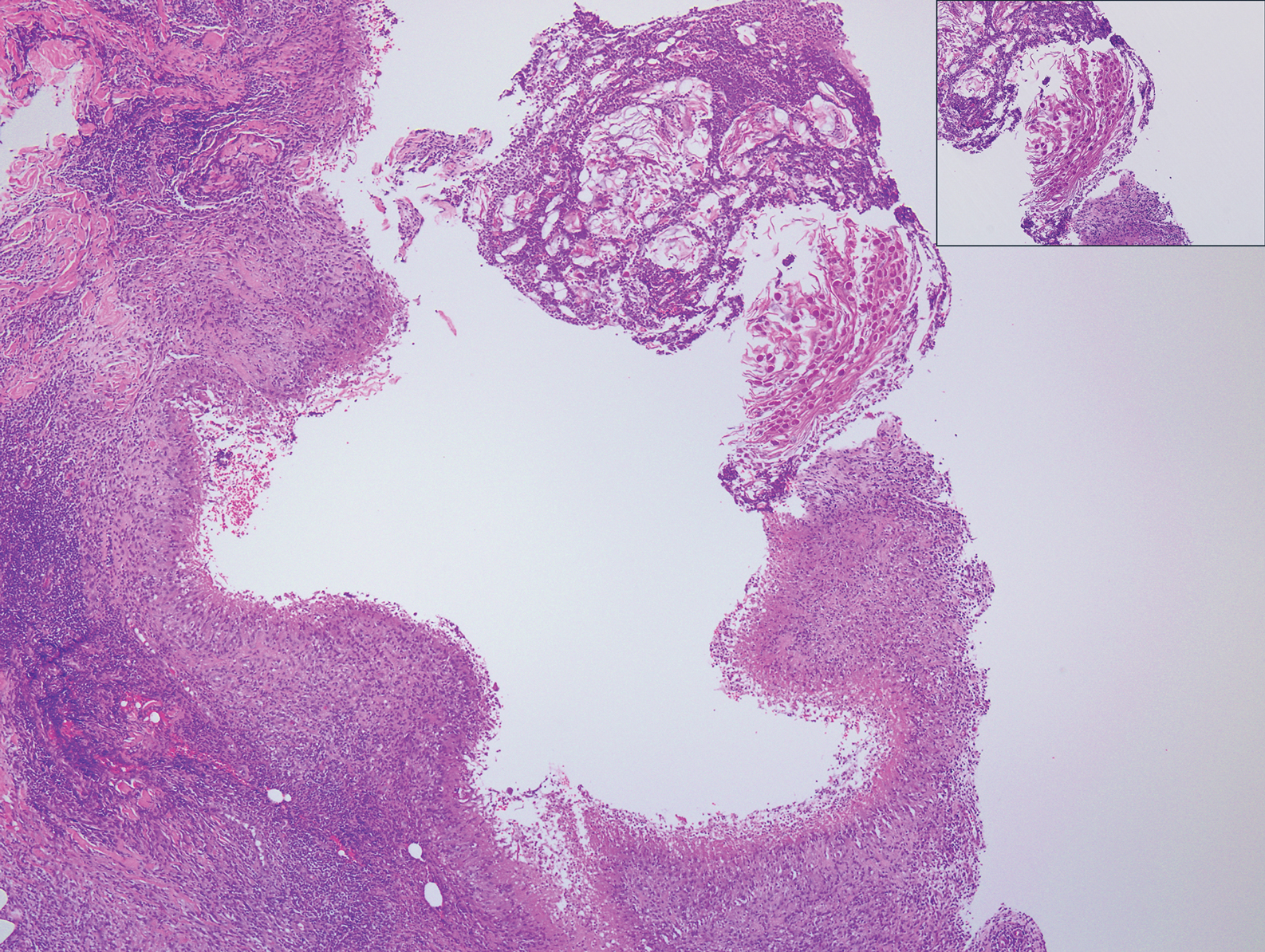
A 17-year-old adolescent girl presented with a discrete nodule on the thigh.
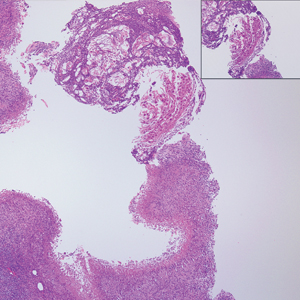
Progressive and Translucent Plaques on the Soles
The Diagnosis: Cutaneous Macroglobulinosis
Waldenström macroglobulinemia is a lymphoplasmacytic lymphoma that produces a circulating monoclonal IgM. Incidence in the United States is 1500 patients annually, most commonly men in their 70s.1 The disease process is largely indolent, with early symptoms consisting of generalized weakness, weight loss, and fatigue. Signs of lymphadenopathy, hepatosplenomegaly, and cytopenia may emerge as the disease progresses. Diagnostic criteria include bone marrow biopsy with plasmacytoid/plasmacellular infiltrate; IgM monoclonal gammopathy; and end-organ damage, which may include cutaneous manifestations.2
Cutaneous findings in Waldenström macroglobulinemia are nonspecific and secondary to the disease's hematologic manifestations, presenting as livedo reticularis, purpura, and mucosal bleeding.3 True cutaneous involvement of the disease is rare and was first described in 1978 by Tichenor.4 Specific cutaneous lesions have 2 separate clinical presentations: (1) a primary cutaneous infiltrate of lymphoplasmacytic cells, and (2) deposition of IgM in the dermis.5 Although the primary infiltrate of neoplastic cells appears as erythematous firm papules or plaques on the face and trunk, similar to other manifestations of leukemia cutis, deposition of IgM presents as translucent papules and plaques and is located more distally, particularly on the extensor extremities.6 These depositional plaques are not pruritic but may be tender if located over sites of pressure, as seen with the plantar presentation in our patient.
Histologically, cutaneous macroglobulinosis demonstrates IgM deposition in perieccrine, perivascular, or intravascular tissue that is periodic acid-Schiff (PAS) positive.7 Staining with Congo red and Alcian blue is negative. In our case, biopsy showed a nodular deposition of hypocellular globular material that stained brightly with PAS and PAS diastase. With Masson trichome stain, intensity of staining diminished, suggesting that the deposition was not composed of collagen; rather, this deposition appeared to consist of IgM storage papules on immunohistochemistry (Figure 1). Further workup revealed borderline pancytopenia and elevated globulins with a monoclonal peak on serum protein electrophoresis, confirming the diagnosis of cutaneous macroglobulinosis secondary to Waldenström macroglobulinemia.
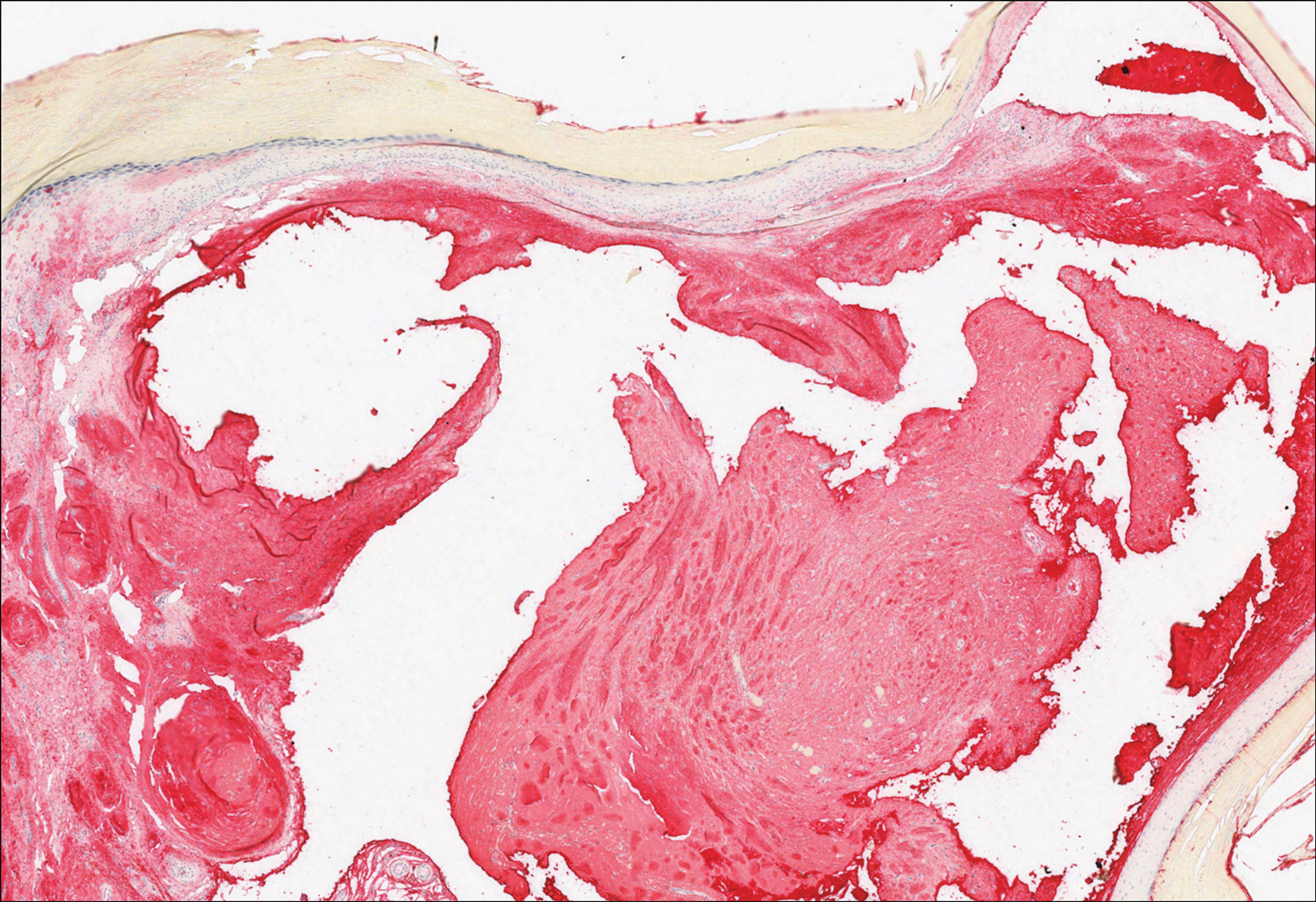
A PubMed search of articles indexed for MEDLINE using the terms cutaneous, macroglobulinosis, macroglobulinemia, Waldenström's macroglobulinemia, Waldenström's macroglobulinaemia, and macroglobulinemia cutis revealed a total of 19 cases of cutaneous macroglobulinosis (including this case). The average age of presentation in these cases is 60 years (range, 29-83 years) with a predisposition for men (68% [13/19]). The development of cutaneous macroglobulinosis primarily has been noted following diagnosis of Waldenström macroglobulinemia (53% [10/19]), with some cases prior to diagnosis (37% [7/19]) or at the time of diagnosis (11% [2/19]). The presence of cutaneous lesions does not correlate with prognosis of the underlying malignancy.5,8,9
Systemic treatment of the underlying macroglobulinemia has been suggested for symptomatic cases of cutaneous macroglobulinosis.3 Prior therapy has consisted primarily of chlorambucil; however, treatment with rituximab, occasionally in conjunction with the proteasome inhibitor bortezomib, recently has been reported.10 Because of the symptomatic nature of our patient's lesions, she was referred to the oncology department and started on rituximab therapy. The lesions improved with therapy and have remained stable following treatment.
The differential diagnosis for tender pink papules and plaques on the arms and legs includes tophaceous gout, plantar fibromatosis, erythropoietic protoporphyria, and acral fibrokeratoma.
Gouty tophi commonly accumulate as painful, edematous, yellow to whitish nodules and tumors with erythema, often overlying joints or extensor surfaces. Histopathologic examination after formalin fixation shows needle-shaped clefts within feathery amorphous pink areas surrounded by granuloma (Figure 2).11 Yellow, needle-shaped, negatively birefringent crystals can be viewed under polarized microscopy in alcohol-fixed samples.
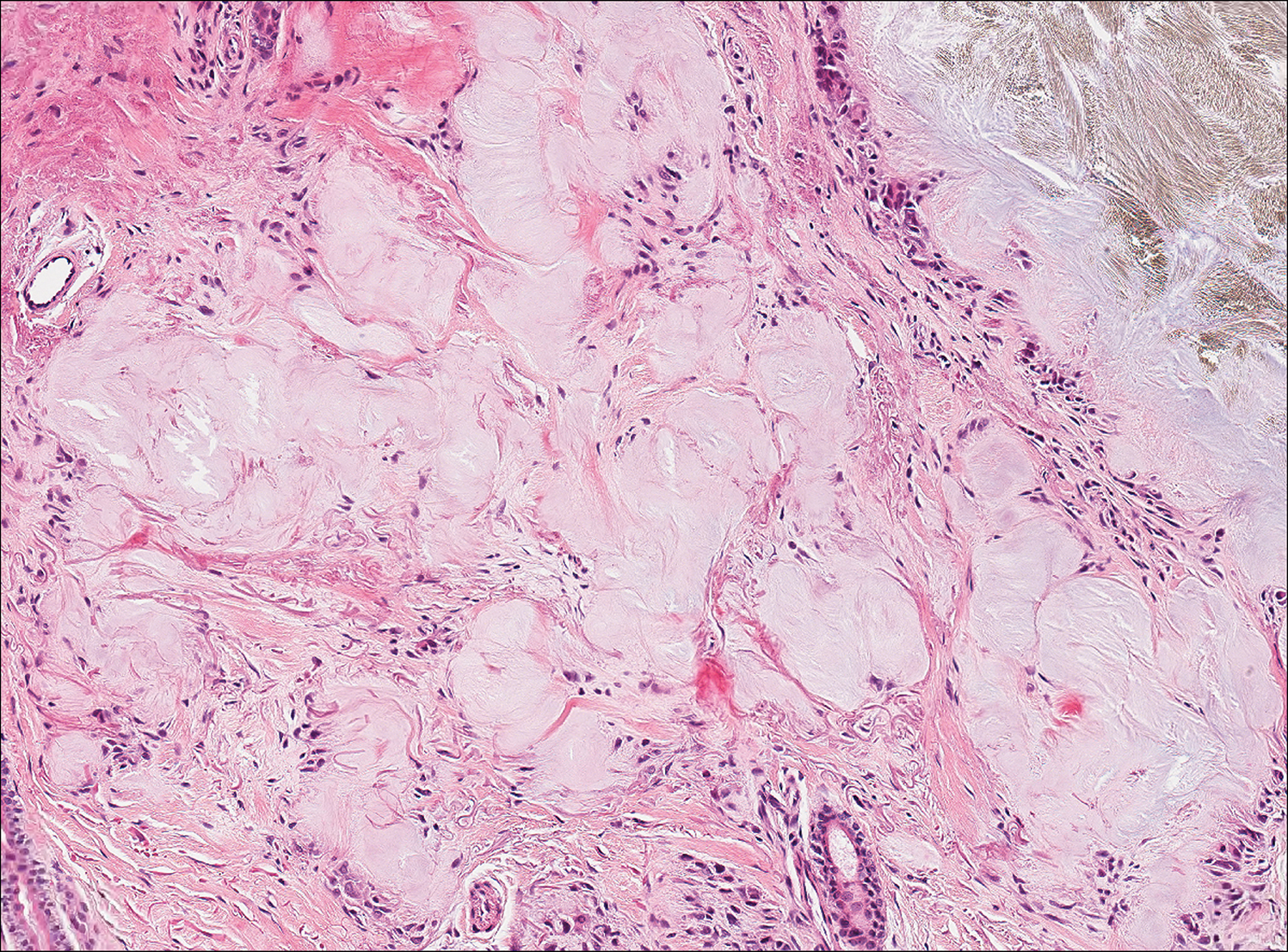
Plantar fibromatosis (Ledderhose disease) is a benign proliferation of the plantar aponeurosis linked to alcohol use; liver disease; and notably epilepsy,12 a component of our patient's medical history. Large nodules appear grossly on the plantar feet and may progress to contractures in more advanced lesions. Biopsy reveals bland hyperproliferation of fibroblasts in a background of fascial fibrous tissue (Figure 3).12 Clinically, this diagnosis is part of the differential diagnosis of plantar nodules but appears histologically different than cutaneous macroglobulinosis because there are no hyaline deposits in plantar fibromatosis.
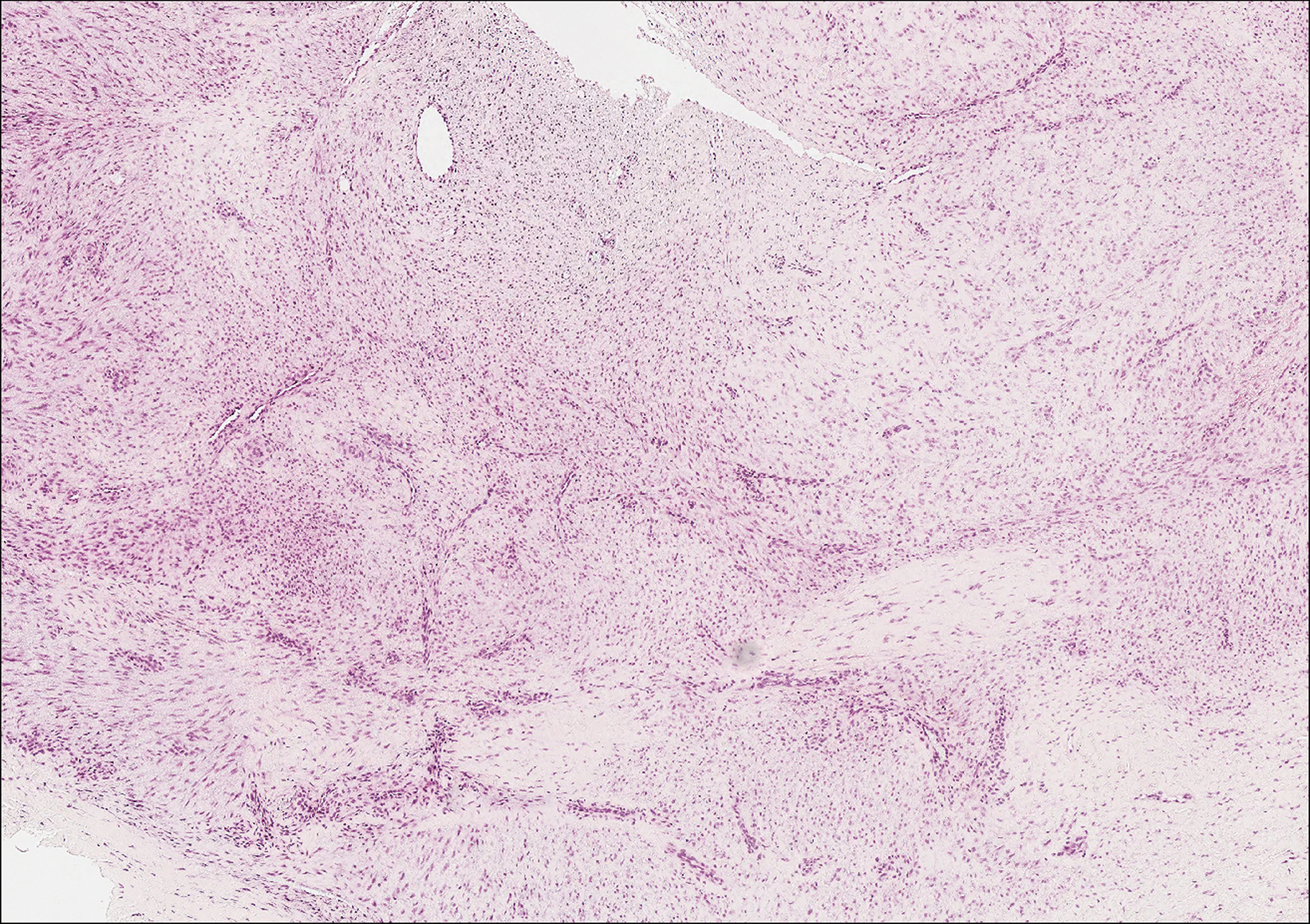
Erythropoietic protoporphyria is a rare disorder that primarily arises due to a congenital deficiency in the ferrochelatase enzyme involved in heme biosynthesis. Erythropoietic protoporphyria is the most common porphyria among children and typically presents in infancy or early childhood as a painful photosensitivity with ensuing cutaneous manifestations and possible hepatobiliary disease. Edema and severe burning pain can be noted within minutes of sun exposure in a dose-response relationship.13 Histologic findings of erythropoietic protoporphyria differ based on acute or chronic skin changes. Acute lesions exhibit a predominantly neutrophilic interstitial dermal infiltrate with vacuoles and intercellular edema. Chronic changes include the accumulation of a PAS-positive, amorphous, hyalinelike substance, similar to the microscopic findings of cutaneous macroglobulinosis (Figure 4).13
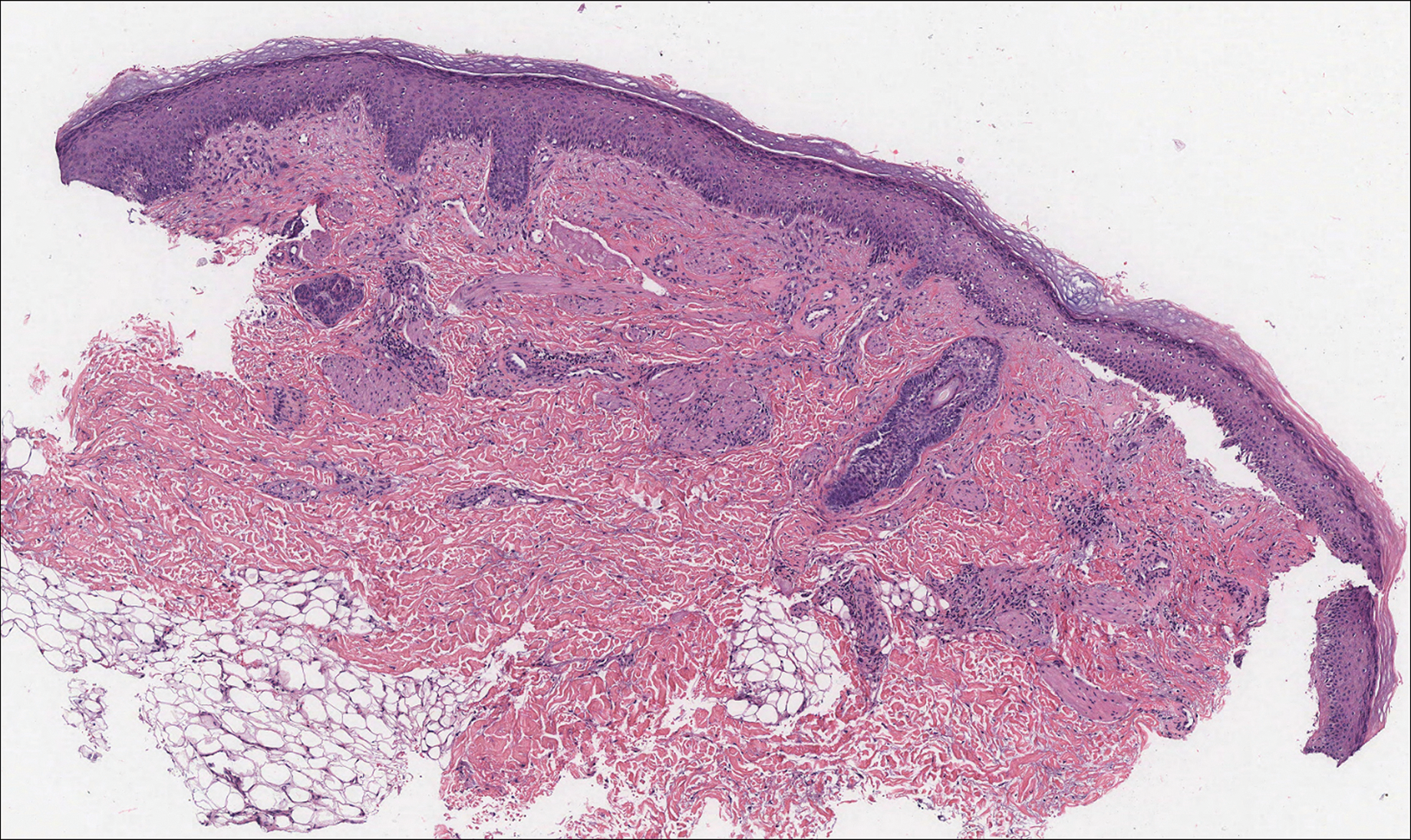
An acral fibrokeratoma is a benign fibroepithelial tumor that clinically appears as a flesh-colored or slightly erythematous exophytic nodule that most commonly is found on the fingers or toes. Thought to arise from trauma to the affected area, it is histologically characterized by interwoven collagenous bundles with overlying epidermal hyperkeratosis, acanthosis, and deep thickened rete ridges14 (Figure 5). Although multiple acral fibrokeratomas have been reported (similar to presentations of prurigo nodularis),15 they more commonly appear as solitary lesions as opposed to the numerous translucent papules seen in our patient.
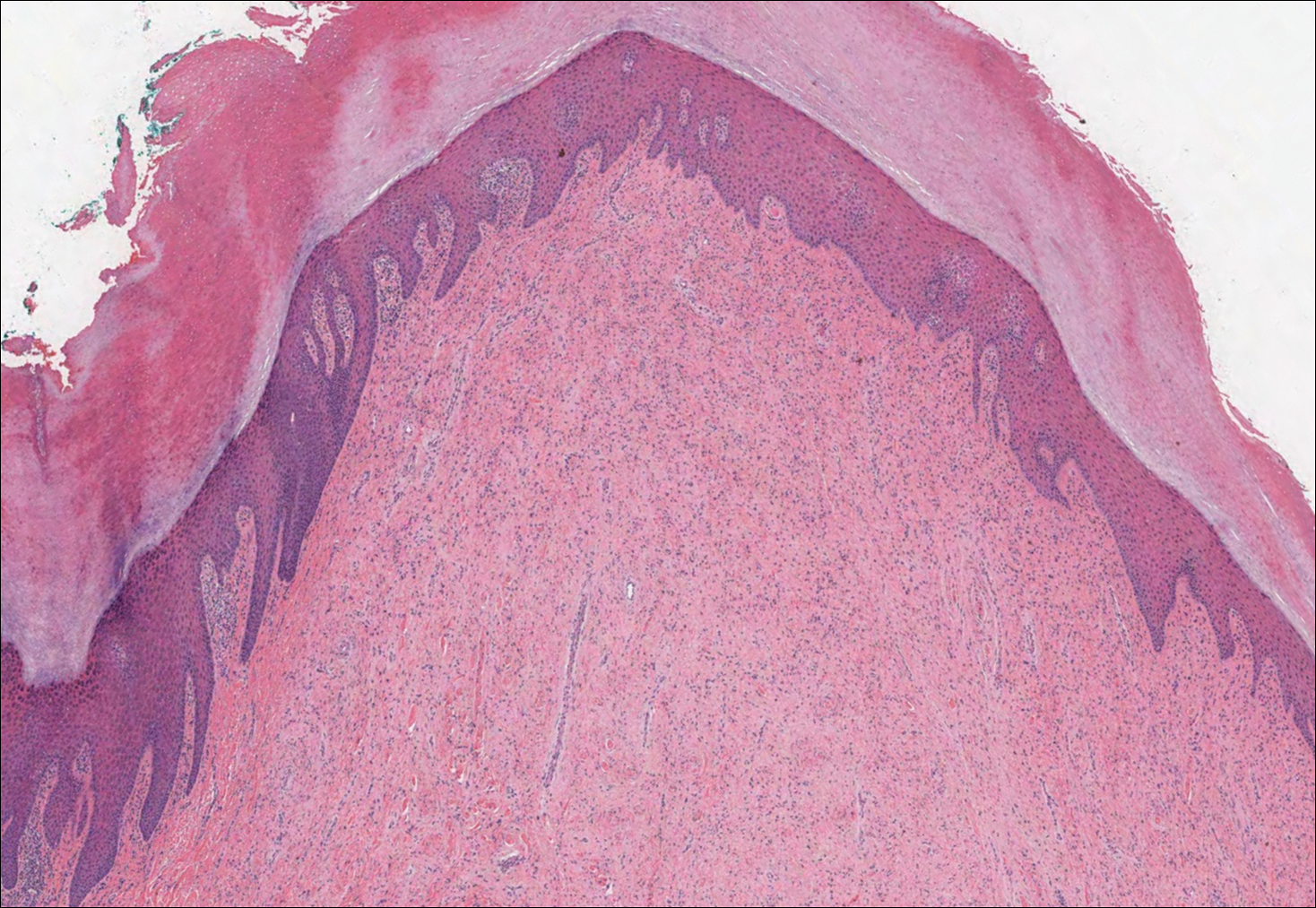
- Camp BJ, Magro CM. Cutaneous macroglobulinosis: a case series. J Cutan Pathol. 2012;39:962-970.
- Dimopoulos MA, Alexanian R. Waldenstrom's macroglobulinemia. Blood. 1994;83:1452-1459.
- D'Acunto C, Nigrisoli E, Liardo EV, et al. Painful plantar nodules: a specific manifestation of cutaneous macroglobulinosis. J Am Acad Dermatol. 2014;71:E251-E252.
- Tichenor RE. Macroglobulinemia cutis. Arch Dermatol. 1978;114:280-281.
- Gressier L, Hotz C, Lelièvre JD, et al. Cutaneous macroglobulinosis: a report of 2 cases. Arch Dermatol. 2010;146:165-169.
- Spicknall KE, Dubas LE, Mutasim DF. Cutaneous macroglobulinosis with monotypic plasma cells: a specific manifestation of Waldenström macroglobulinemia. J Cutan Pathol. 2013;40:442-444.
- Lüftl M, Sauter-Jenne B, Gramatzki M, et al. Cutaneous macroglobulinosis deposits in a patient with IgM paraproteinemia/incipient Waldenström macroglobulinemia. J Dtsch Dermatol Ges. 2010;8:1000-1003.
- Mascaro JM, Montserrat E, Estrach T, et al. Specific cutaneous manifestations of Waldenstrom macroglobulinaemia: a report of two cases. Br J Dermatol. 1982;106:217-222.
- Hanke CW, Steck WD, Bergfeld WF, et al. Cutaneous macroglobulinosis. Arch Dermatol. 1980;116:575-577.
- Oshio-Yoshii A, Fujimoto N, Shiba Y, et al. Cutaneous macroglobulinosis: successful treatment with rituximab. J Eur Acad Dermatol Venereol. 2017;31:E30-E31.
- Gupta A, Rai S, Sinha R, et al. Tophi as an initial manifestation of gout. J Cytol. 2009;26:165-166.
- Carroll P, Henshaw RM, Garwood C, et al. Plantar fibromatosis: pathophysiology, surgical and nonsurgical therapies: an evidence-based review. Foot Ankle Spec. 2018;11:168-176.
- Michaels BD, Del Rosso JQ, Mobini N, et al. Erythropoietic protoporphyria: a case report and literature review. J Clin Aesthet Dermatol. 2010;3:44-48.
- Boffeli TJ, Abben KW. Acral fibrokeratoma of the foot treated with excision and trap door flap closure: a case report. J Foot Ankle Surg. 2014;53:449-452.
- Reed RJ. Multiple acral fibrokeratomas (a variant of prurigo nodularis). discussion of classification of acral fibrous nodules and of histogenesis of acral fibrokeratomas. Arch Dermatol. 1971;103:287-297.
The Diagnosis: Cutaneous Macroglobulinosis
Waldenström macroglobulinemia is a lymphoplasmacytic lymphoma that produces a circulating monoclonal IgM. Incidence in the United States is 1500 patients annually, most commonly men in their 70s.1 The disease process is largely indolent, with early symptoms consisting of generalized weakness, weight loss, and fatigue. Signs of lymphadenopathy, hepatosplenomegaly, and cytopenia may emerge as the disease progresses. Diagnostic criteria include bone marrow biopsy with plasmacytoid/plasmacellular infiltrate; IgM monoclonal gammopathy; and end-organ damage, which may include cutaneous manifestations.2
Cutaneous findings in Waldenström macroglobulinemia are nonspecific and secondary to the disease's hematologic manifestations, presenting as livedo reticularis, purpura, and mucosal bleeding.3 True cutaneous involvement of the disease is rare and was first described in 1978 by Tichenor.4 Specific cutaneous lesions have 2 separate clinical presentations: (1) a primary cutaneous infiltrate of lymphoplasmacytic cells, and (2) deposition of IgM in the dermis.5 Although the primary infiltrate of neoplastic cells appears as erythematous firm papules or plaques on the face and trunk, similar to other manifestations of leukemia cutis, deposition of IgM presents as translucent papules and plaques and is located more distally, particularly on the extensor extremities.6 These depositional plaques are not pruritic but may be tender if located over sites of pressure, as seen with the plantar presentation in our patient.
Histologically, cutaneous macroglobulinosis demonstrates IgM deposition in perieccrine, perivascular, or intravascular tissue that is periodic acid-Schiff (PAS) positive.7 Staining with Congo red and Alcian blue is negative. In our case, biopsy showed a nodular deposition of hypocellular globular material that stained brightly with PAS and PAS diastase. With Masson trichome stain, intensity of staining diminished, suggesting that the deposition was not composed of collagen; rather, this deposition appeared to consist of IgM storage papules on immunohistochemistry (Figure 1). Further workup revealed borderline pancytopenia and elevated globulins with a monoclonal peak on serum protein electrophoresis, confirming the diagnosis of cutaneous macroglobulinosis secondary to Waldenström macroglobulinemia.
A PubMed search of articles indexed for MEDLINE using the terms cutaneous, macroglobulinosis, macroglobulinemia, Waldenström's macroglobulinemia, Waldenström's macroglobulinaemia, and macroglobulinemia cutis revealed a total of 19 cases of cutaneous macroglobulinosis (including this case). The average age of presentation in these cases is 60 years (range, 29-83 years) with a predisposition for men (68% [13/19]). The development of cutaneous macroglobulinosis primarily has been noted following diagnosis of Waldenström macroglobulinemia (53% [10/19]), with some cases prior to diagnosis (37% [7/19]) or at the time of diagnosis (11% [2/19]). The presence of cutaneous lesions does not correlate with prognosis of the underlying malignancy.5,8,9
Systemic treatment of the underlying macroglobulinemia has been suggested for symptomatic cases of cutaneous macroglobulinosis.3 Prior therapy has consisted primarily of chlorambucil; however, treatment with rituximab, occasionally in conjunction with the proteasome inhibitor bortezomib, recently has been reported.10 Because of the symptomatic nature of our patient's lesions, she was referred to the oncology department and started on rituximab therapy. The lesions improved with therapy and have remained stable following treatment.
The differential diagnosis for tender pink papules and plaques on the arms and legs includes tophaceous gout, plantar fibromatosis, erythropoietic protoporphyria, and acral fibrokeratoma.
Gouty tophi commonly accumulate as painful, edematous, yellow to whitish nodules and tumors with erythema, often overlying joints or extensor surfaces. Histopathologic examination after formalin fixation shows needle-shaped clefts within feathery amorphous pink areas surrounded by granuloma (Figure 2).11 Yellow, needle-shaped, negatively birefringent crystals can be viewed under polarized microscopy in alcohol-fixed samples.
Plantar fibromatosis (Ledderhose disease) is a benign proliferation of the plantar aponeurosis linked to alcohol use; liver disease; and notably epilepsy,12 a component of our patient's medical history. Large nodules appear grossly on the plantar feet and may progress to contractures in more advanced lesions. Biopsy reveals bland hyperproliferation of fibroblasts in a background of fascial fibrous tissue (Figure 3).12 Clinically, this diagnosis is part of the differential diagnosis of plantar nodules but appears histologically different than cutaneous macroglobulinosis because there are no hyaline deposits in plantar fibromatosis.
Erythropoietic protoporphyria is a rare disorder that primarily arises due to a congenital deficiency in the ferrochelatase enzyme involved in heme biosynthesis. Erythropoietic protoporphyria is the most common porphyria among children and typically presents in infancy or early childhood as a painful photosensitivity with ensuing cutaneous manifestations and possible hepatobiliary disease. Edema and severe burning pain can be noted within minutes of sun exposure in a dose-response relationship.13 Histologic findings of erythropoietic protoporphyria differ based on acute or chronic skin changes. Acute lesions exhibit a predominantly neutrophilic interstitial dermal infiltrate with vacuoles and intercellular edema. Chronic changes include the accumulation of a PAS-positive, amorphous, hyalinelike substance, similar to the microscopic findings of cutaneous macroglobulinosis (Figure 4).13
An acral fibrokeratoma is a benign fibroepithelial tumor that clinically appears as a flesh-colored or slightly erythematous exophytic nodule that most commonly is found on the fingers or toes. Thought to arise from trauma to the affected area, it is histologically characterized by interwoven collagenous bundles with overlying epidermal hyperkeratosis, acanthosis, and deep thickened rete ridges14 (Figure 5). Although multiple acral fibrokeratomas have been reported (similar to presentations of prurigo nodularis),15 they more commonly appear as solitary lesions as opposed to the numerous translucent papules seen in our patient.
The Diagnosis: Cutaneous Macroglobulinosis
Waldenström macroglobulinemia is a lymphoplasmacytic lymphoma that produces a circulating monoclonal IgM. Incidence in the United States is 1500 patients annually, most commonly men in their 70s.1 The disease process is largely indolent, with early symptoms consisting of generalized weakness, weight loss, and fatigue. Signs of lymphadenopathy, hepatosplenomegaly, and cytopenia may emerge as the disease progresses. Diagnostic criteria include bone marrow biopsy with plasmacytoid/plasmacellular infiltrate; IgM monoclonal gammopathy; and end-organ damage, which may include cutaneous manifestations.2
Cutaneous findings in Waldenström macroglobulinemia are nonspecific and secondary to the disease's hematologic manifestations, presenting as livedo reticularis, purpura, and mucosal bleeding.3 True cutaneous involvement of the disease is rare and was first described in 1978 by Tichenor.4 Specific cutaneous lesions have 2 separate clinical presentations: (1) a primary cutaneous infiltrate of lymphoplasmacytic cells, and (2) deposition of IgM in the dermis.5 Although the primary infiltrate of neoplastic cells appears as erythematous firm papules or plaques on the face and trunk, similar to other manifestations of leukemia cutis, deposition of IgM presents as translucent papules and plaques and is located more distally, particularly on the extensor extremities.6 These depositional plaques are not pruritic but may be tender if located over sites of pressure, as seen with the plantar presentation in our patient.
Histologically, cutaneous macroglobulinosis demonstrates IgM deposition in perieccrine, perivascular, or intravascular tissue that is periodic acid-Schiff (PAS) positive.7 Staining with Congo red and Alcian blue is negative. In our case, biopsy showed a nodular deposition of hypocellular globular material that stained brightly with PAS and PAS diastase. With Masson trichome stain, intensity of staining diminished, suggesting that the deposition was not composed of collagen; rather, this deposition appeared to consist of IgM storage papules on immunohistochemistry (Figure 1). Further workup revealed borderline pancytopenia and elevated globulins with a monoclonal peak on serum protein electrophoresis, confirming the diagnosis of cutaneous macroglobulinosis secondary to Waldenström macroglobulinemia.
A PubMed search of articles indexed for MEDLINE using the terms cutaneous, macroglobulinosis, macroglobulinemia, Waldenström's macroglobulinemia, Waldenström's macroglobulinaemia, and macroglobulinemia cutis revealed a total of 19 cases of cutaneous macroglobulinosis (including this case). The average age of presentation in these cases is 60 years (range, 29-83 years) with a predisposition for men (68% [13/19]). The development of cutaneous macroglobulinosis primarily has been noted following diagnosis of Waldenström macroglobulinemia (53% [10/19]), with some cases prior to diagnosis (37% [7/19]) or at the time of diagnosis (11% [2/19]). The presence of cutaneous lesions does not correlate with prognosis of the underlying malignancy.5,8,9
Systemic treatment of the underlying macroglobulinemia has been suggested for symptomatic cases of cutaneous macroglobulinosis.3 Prior therapy has consisted primarily of chlorambucil; however, treatment with rituximab, occasionally in conjunction with the proteasome inhibitor bortezomib, recently has been reported.10 Because of the symptomatic nature of our patient's lesions, she was referred to the oncology department and started on rituximab therapy. The lesions improved with therapy and have remained stable following treatment.
The differential diagnosis for tender pink papules and plaques on the arms and legs includes tophaceous gout, plantar fibromatosis, erythropoietic protoporphyria, and acral fibrokeratoma.
Gouty tophi commonly accumulate as painful, edematous, yellow to whitish nodules and tumors with erythema, often overlying joints or extensor surfaces. Histopathologic examination after formalin fixation shows needle-shaped clefts within feathery amorphous pink areas surrounded by granuloma (Figure 2).11 Yellow, needle-shaped, negatively birefringent crystals can be viewed under polarized microscopy in alcohol-fixed samples.
Plantar fibromatosis (Ledderhose disease) is a benign proliferation of the plantar aponeurosis linked to alcohol use; liver disease; and notably epilepsy,12 a component of our patient's medical history. Large nodules appear grossly on the plantar feet and may progress to contractures in more advanced lesions. Biopsy reveals bland hyperproliferation of fibroblasts in a background of fascial fibrous tissue (Figure 3).12 Clinically, this diagnosis is part of the differential diagnosis of plantar nodules but appears histologically different than cutaneous macroglobulinosis because there are no hyaline deposits in plantar fibromatosis.
Erythropoietic protoporphyria is a rare disorder that primarily arises due to a congenital deficiency in the ferrochelatase enzyme involved in heme biosynthesis. Erythropoietic protoporphyria is the most common porphyria among children and typically presents in infancy or early childhood as a painful photosensitivity with ensuing cutaneous manifestations and possible hepatobiliary disease. Edema and severe burning pain can be noted within minutes of sun exposure in a dose-response relationship.13 Histologic findings of erythropoietic protoporphyria differ based on acute or chronic skin changes. Acute lesions exhibit a predominantly neutrophilic interstitial dermal infiltrate with vacuoles and intercellular edema. Chronic changes include the accumulation of a PAS-positive, amorphous, hyalinelike substance, similar to the microscopic findings of cutaneous macroglobulinosis (Figure 4).13
An acral fibrokeratoma is a benign fibroepithelial tumor that clinically appears as a flesh-colored or slightly erythematous exophytic nodule that most commonly is found on the fingers or toes. Thought to arise from trauma to the affected area, it is histologically characterized by interwoven collagenous bundles with overlying epidermal hyperkeratosis, acanthosis, and deep thickened rete ridges14 (Figure 5). Although multiple acral fibrokeratomas have been reported (similar to presentations of prurigo nodularis),15 they more commonly appear as solitary lesions as opposed to the numerous translucent papules seen in our patient.
- Camp BJ, Magro CM. Cutaneous macroglobulinosis: a case series. J Cutan Pathol. 2012;39:962-970.
- Dimopoulos MA, Alexanian R. Waldenstrom's macroglobulinemia. Blood. 1994;83:1452-1459.
- D'Acunto C, Nigrisoli E, Liardo EV, et al. Painful plantar nodules: a specific manifestation of cutaneous macroglobulinosis. J Am Acad Dermatol. 2014;71:E251-E252.
- Tichenor RE. Macroglobulinemia cutis. Arch Dermatol. 1978;114:280-281.
- Gressier L, Hotz C, Lelièvre JD, et al. Cutaneous macroglobulinosis: a report of 2 cases. Arch Dermatol. 2010;146:165-169.
- Spicknall KE, Dubas LE, Mutasim DF. Cutaneous macroglobulinosis with monotypic plasma cells: a specific manifestation of Waldenström macroglobulinemia. J Cutan Pathol. 2013;40:442-444.
- Lüftl M, Sauter-Jenne B, Gramatzki M, et al. Cutaneous macroglobulinosis deposits in a patient with IgM paraproteinemia/incipient Waldenström macroglobulinemia. J Dtsch Dermatol Ges. 2010;8:1000-1003.
- Mascaro JM, Montserrat E, Estrach T, et al. Specific cutaneous manifestations of Waldenstrom macroglobulinaemia: a report of two cases. Br J Dermatol. 1982;106:217-222.
- Hanke CW, Steck WD, Bergfeld WF, et al. Cutaneous macroglobulinosis. Arch Dermatol. 1980;116:575-577.
- Oshio-Yoshii A, Fujimoto N, Shiba Y, et al. Cutaneous macroglobulinosis: successful treatment with rituximab. J Eur Acad Dermatol Venereol. 2017;31:E30-E31.
- Gupta A, Rai S, Sinha R, et al. Tophi as an initial manifestation of gout. J Cytol. 2009;26:165-166.
- Carroll P, Henshaw RM, Garwood C, et al. Plantar fibromatosis: pathophysiology, surgical and nonsurgical therapies: an evidence-based review. Foot Ankle Spec. 2018;11:168-176.
- Michaels BD, Del Rosso JQ, Mobini N, et al. Erythropoietic protoporphyria: a case report and literature review. J Clin Aesthet Dermatol. 2010;3:44-48.
- Boffeli TJ, Abben KW. Acral fibrokeratoma of the foot treated with excision and trap door flap closure: a case report. J Foot Ankle Surg. 2014;53:449-452.
- Reed RJ. Multiple acral fibrokeratomas (a variant of prurigo nodularis). discussion of classification of acral fibrous nodules and of histogenesis of acral fibrokeratomas. Arch Dermatol. 1971;103:287-297.
- Camp BJ, Magro CM. Cutaneous macroglobulinosis: a case series. J Cutan Pathol. 2012;39:962-970.
- Dimopoulos MA, Alexanian R. Waldenstrom's macroglobulinemia. Blood. 1994;83:1452-1459.
- D'Acunto C, Nigrisoli E, Liardo EV, et al. Painful plantar nodules: a specific manifestation of cutaneous macroglobulinosis. J Am Acad Dermatol. 2014;71:E251-E252.
- Tichenor RE. Macroglobulinemia cutis. Arch Dermatol. 1978;114:280-281.
- Gressier L, Hotz C, Lelièvre JD, et al. Cutaneous macroglobulinosis: a report of 2 cases. Arch Dermatol. 2010;146:165-169.
- Spicknall KE, Dubas LE, Mutasim DF. Cutaneous macroglobulinosis with monotypic plasma cells: a specific manifestation of Waldenström macroglobulinemia. J Cutan Pathol. 2013;40:442-444.
- Lüftl M, Sauter-Jenne B, Gramatzki M, et al. Cutaneous macroglobulinosis deposits in a patient with IgM paraproteinemia/incipient Waldenström macroglobulinemia. J Dtsch Dermatol Ges. 2010;8:1000-1003.
- Mascaro JM, Montserrat E, Estrach T, et al. Specific cutaneous manifestations of Waldenstrom macroglobulinaemia: a report of two cases. Br J Dermatol. 1982;106:217-222.
- Hanke CW, Steck WD, Bergfeld WF, et al. Cutaneous macroglobulinosis. Arch Dermatol. 1980;116:575-577.
- Oshio-Yoshii A, Fujimoto N, Shiba Y, et al. Cutaneous macroglobulinosis: successful treatment with rituximab. J Eur Acad Dermatol Venereol. 2017;31:E30-E31.
- Gupta A, Rai S, Sinha R, et al. Tophi as an initial manifestation of gout. J Cytol. 2009;26:165-166.
- Carroll P, Henshaw RM, Garwood C, et al. Plantar fibromatosis: pathophysiology, surgical and nonsurgical therapies: an evidence-based review. Foot Ankle Spec. 2018;11:168-176.
- Michaels BD, Del Rosso JQ, Mobini N, et al. Erythropoietic protoporphyria: a case report and literature review. J Clin Aesthet Dermatol. 2010;3:44-48.
- Boffeli TJ, Abben KW. Acral fibrokeratoma of the foot treated with excision and trap door flap closure: a case report. J Foot Ankle Surg. 2014;53:449-452.
- Reed RJ. Multiple acral fibrokeratomas (a variant of prurigo nodularis). discussion of classification of acral fibrous nodules and of histogenesis of acral fibrokeratomas. Arch Dermatol. 1971;103:287-297.
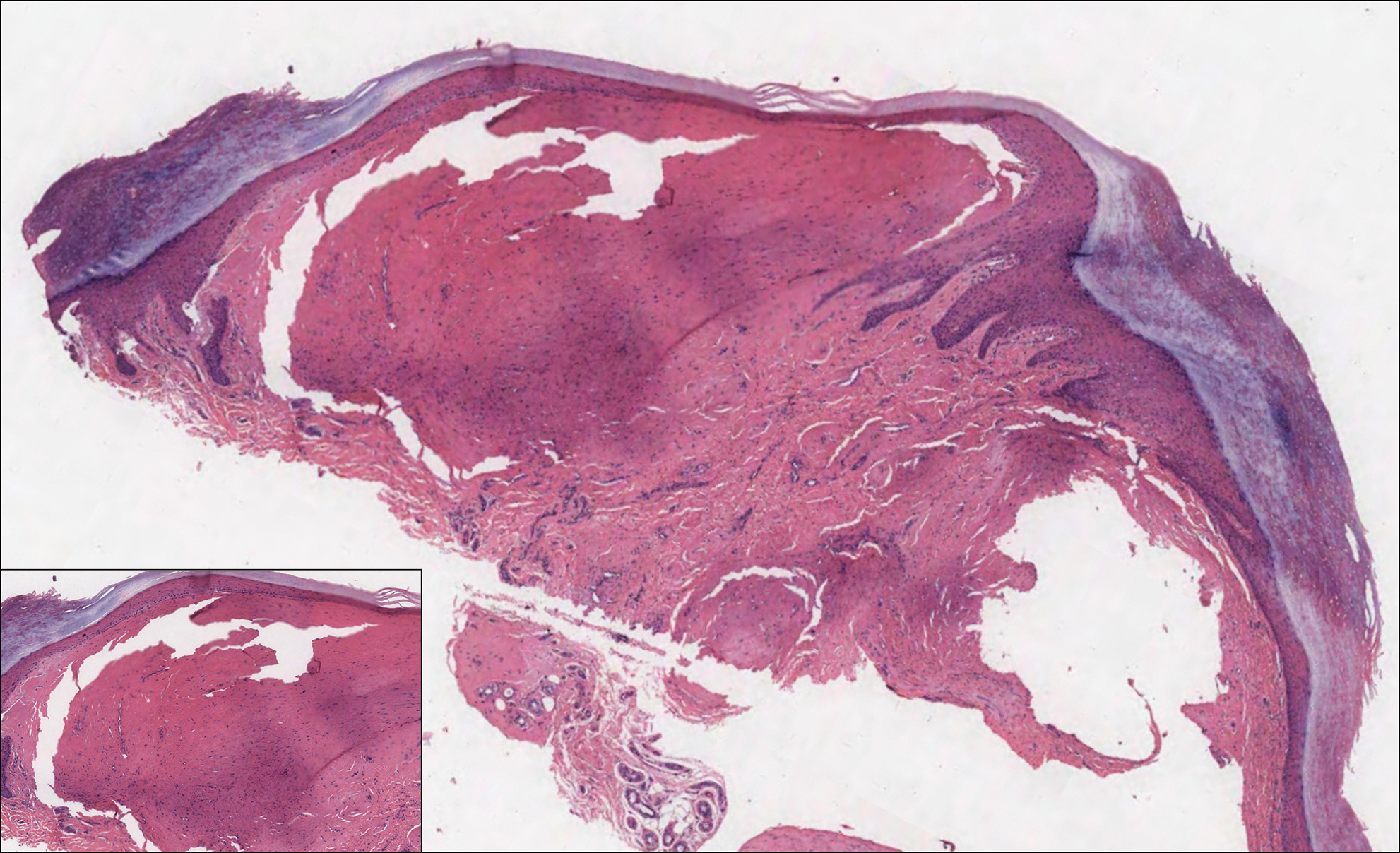
A 64-year-old woman with a medical history of Waldenström macroglobulinemia, multiple sclerosis, and epilepsy presented with slowly growing papules on the plantar feet of 21 months' duration. She was diagnosed with Waldenström macroglobulinemia incidentally on routine blood work 3 years prior and declined treatment because she was asymptomatic. Physical examination revealed a total of 20 firm, variably sized, light pink to purple, partially translucent and telangiectatic papules and plaques bilaterally on the plantar feet. A plaque from the right sole was biopsied.
Multiple Pink Papules on the Chest and Upper Abdomen
The Diagnosis: Cutaneous Metastases
Cutaneous metastases (CMs) can present in an otherwise asymptomatic patient as the only sign of an underlying disease process. In women, the most common cause of CM is breast carcinoma.1-3 Cutaneous metastases are found in approximately 25% of all patients with breast carcinoma,1 and breast carcinomas represent approximately 69% of all CMs found in women (Table 1).2 Cutaneous metastatic breast carcinoma (CMBC) is associated with a poor prognosis with a mean survival of approximately 6 months at the time of diagnosis.1,3 It commonly presents as a collection of flesh-colored, firm, asymptomatic, and rapidly appearing papules and nodules that can resemble cysts or fibrous tumors.1,3,4 They typically are located on the chest wall or abdomen near the site of the underlying malignancy.1-3 The histologic features of CMBC can include hyperchromatic tumor cells infiltrating between the collagen fibers in a characteristic single file manner,3,5 giving the appearance of a busy dermis, a nonspecific term to describe a focally hypercellular dermis at low-power magnification (Table 2).5,6 Cords and clusters of atypical cells with intracytoplasmic vacuoles or well-developed ducts also can be seen (quiz image [inset]). The carcinoma en cuirasse subtype of CMBC is characterized by a fibrotic scarlike plaque on the chest wall.1,3 If a punch biopsy is obtained, the specimen typically appears rectangular rather than tapered because of the sclerotic dermal collagen.6 In contrast, inflammatory carcinoma (carcinoma erysipelatoides) presents as an erythematous plaque resembling cellulitis due to the lymphatics being congested by tumor cells.3 Immunohistochemistry is a valuable tool in diagnosis. Positive staining is seen with cytokeratin 7, gross cystic disease fluid protein-15, mammaglobin, and GATA-3.1,3,6


Kaposi sarcoma (KS) is a low-grade endothelial malignancy associated with human herpesvirus 8.3,4 Kaposi sarcoma can be divided into 4 main subtypes: classic KS, African KS, AIDS-related KS, and immunosuppression-associated KS that occurs in patients with diseases such as human immunodeficiency virus. The cutaneous lesions are similar between subtypes and present as dark reddish purple macules that may enlarge or become nodular lesions.3,4 Histologically, 3 distinct stages of progression are described: patch, plaque, and tumor. The plaque stage has the appearance of a busy dermis due to the rapid proliferation of vascular structures within the dermis.3,6 A useful histologic feature known as the promontory sign can be seen as the proliferating tumor causes preexisting structures to project into vascular spaces (Figure 1).6 Immunohistochemistry for the endothelial and lymphatic markers CD31 and D2-40, respectively, are positive and may aid in the diagnosis.3 Staining for the latent nuclear antigen-1 of human herpesvirus 8 is a highly specific marker used to diagnose KS and can further distinguish it from the other busy dermis lesions.3
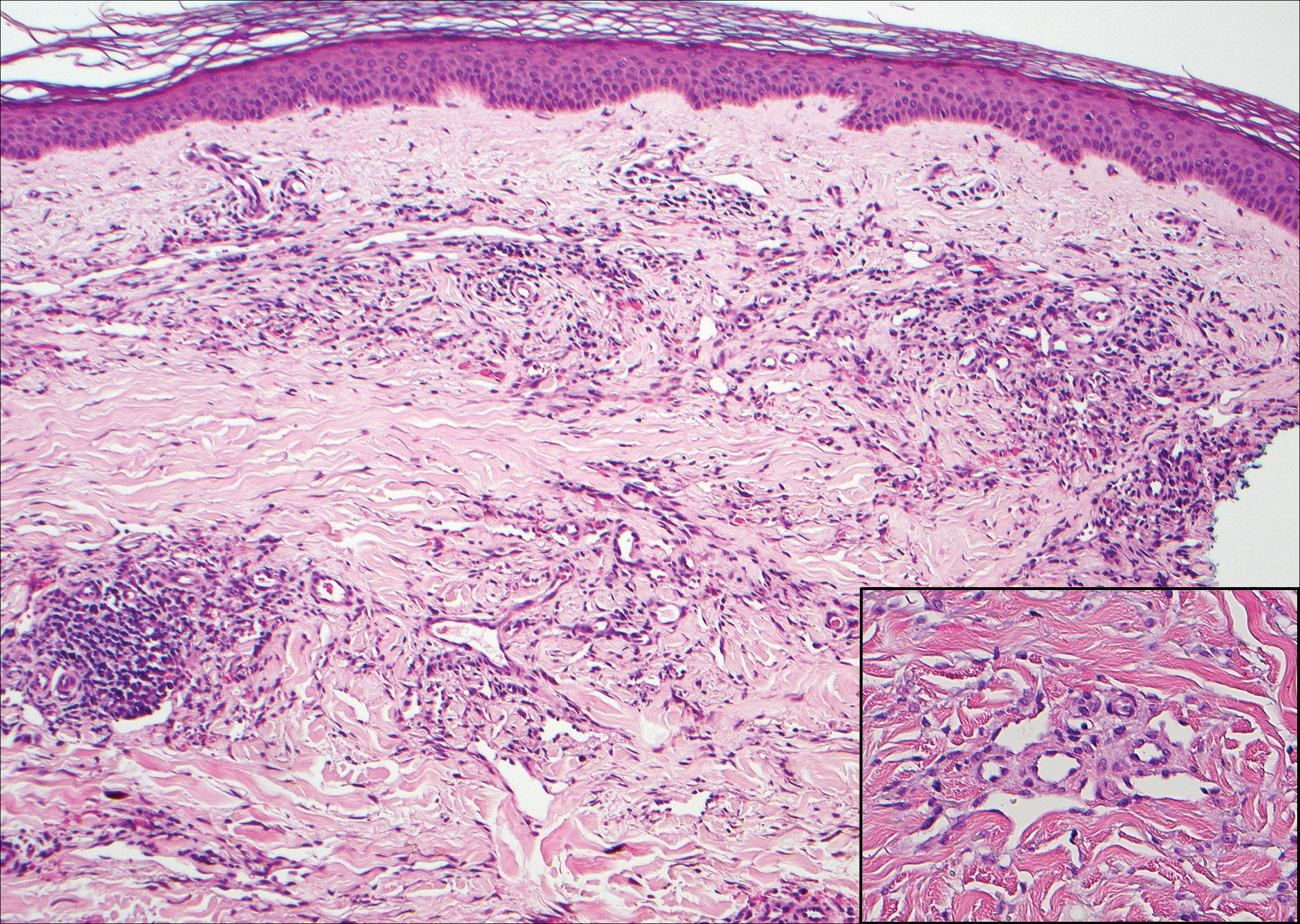
Granuloma annulare (GA) is characterized by rings of small, firm, pink to flesh-colored papules with a variable disease duration.4 Histologically, the interstitial variant of GA is characterized by a scattered inflammatory infiltrate consisting of histiocytes and lymphocytes located between altered collagen fibers in the superficial to mid dermis (Figure 2).3,6 Occasional eosinophils and increased dermal mucin are useful features to distinguish interstitial GA from other entities in the busy dermis differential.7
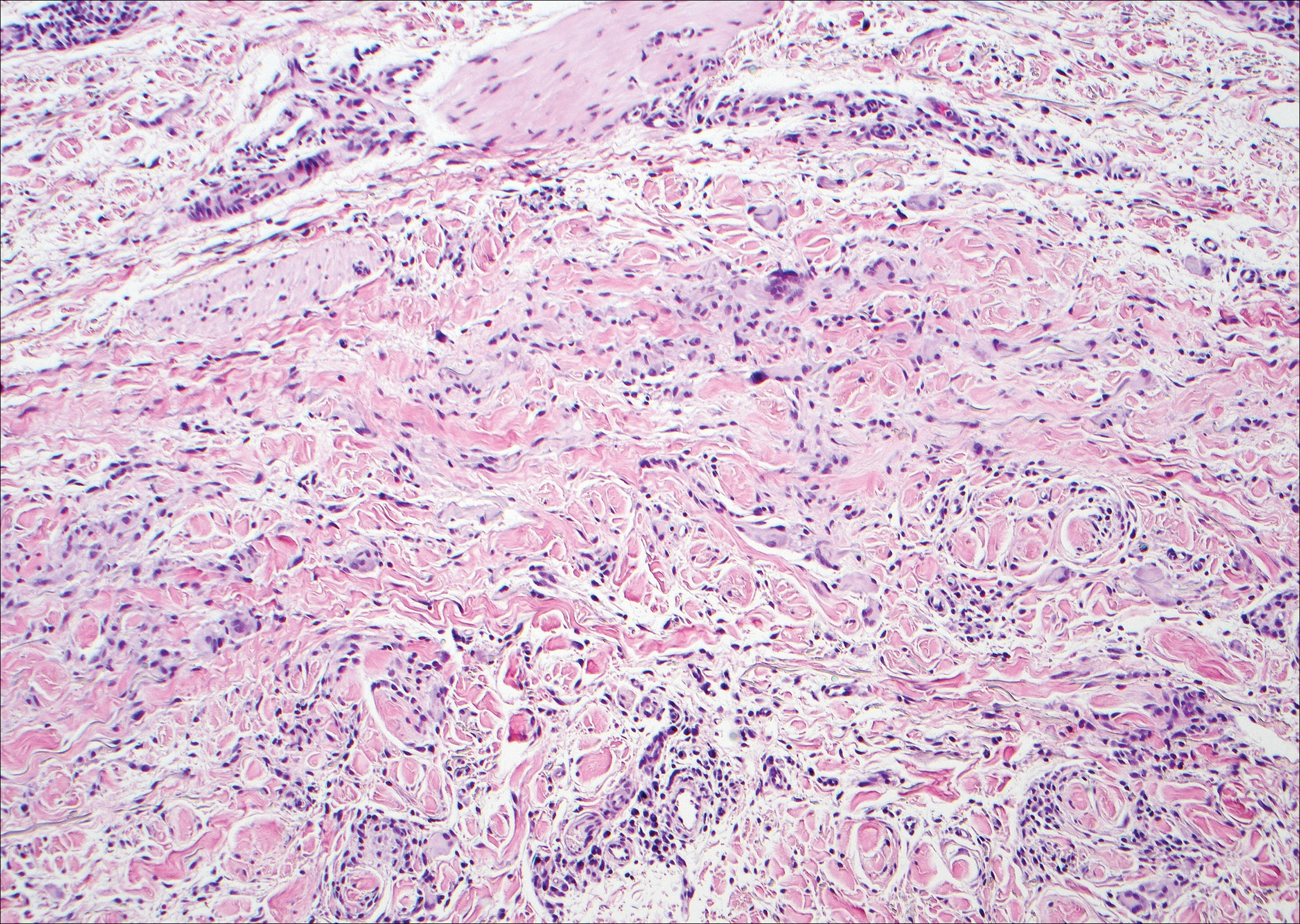
Scleromyxedema, also known as generalized lichen myxedematosus, is a rare mucinosis.3,8 Although its pathogenesis is unknown, it has been suggested that paraproteins related to the underlying gammopathy act to stimulate fibroblast proliferation and mucin overproduction.8 Clinically, characteristic widespread firm, waxy, dome-shaped papules are present over the head, upper trunk, and extremities.3,8 Histologically, scleromyxedema is characterized by increased dermal fibroblasts, mucin, and fibrosis, leading to the appearance of a busy dermis (Figure 3).3,6
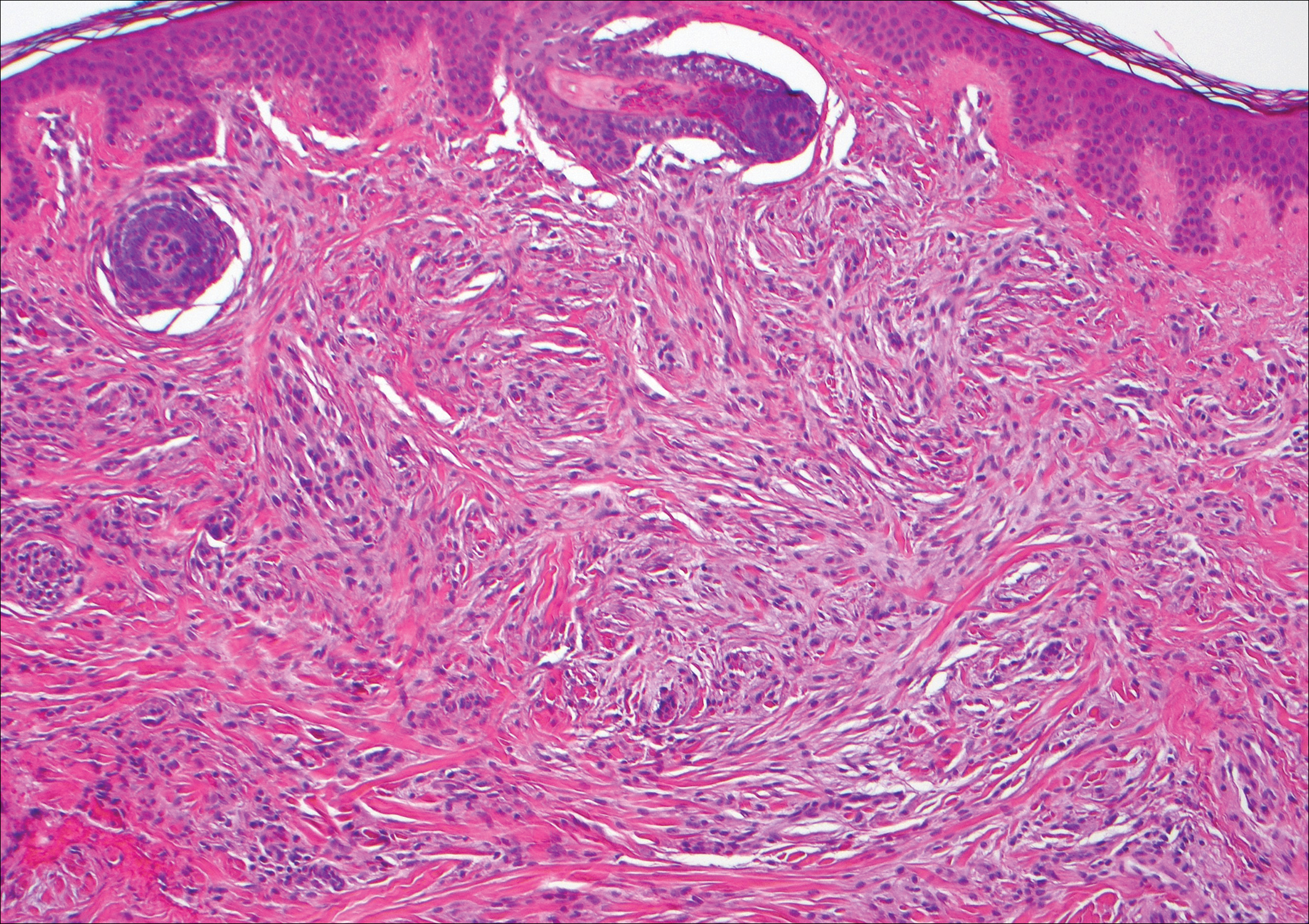
Neurofibromas are common benign peripheral nerve sheath tumors that can occur sporadically or in the setting of neurofibromatosis.3-5 They present as soft, flesh-colored papules or nodules most commonly located on the trunk and limbs.4 Histologically, neurofibromas are nonencapsulated tumors composed of abundant spindle cells with comma-shaped nuclei diffusely arranged in a pale myxoid stroma (Figure 4). Scattered mast cells can be visualized at higher magnification.3,6
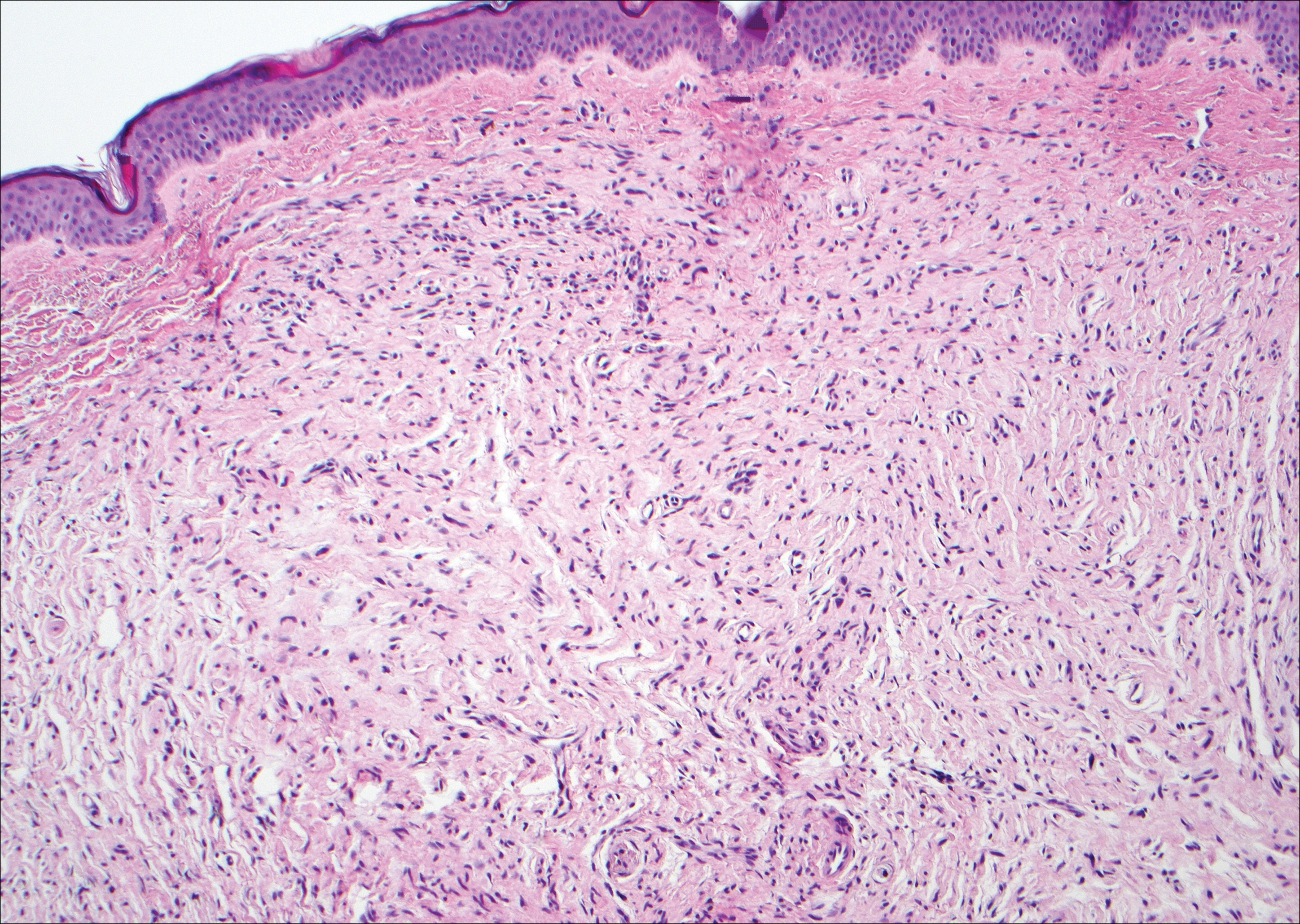
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Habif TP, Dinulos JGH, Chapman MS, et al. Skin Disease: Diagnosis and Treatment. 4th ed. Edinburgh, Scotland: Elsevier; 2017.
- Calonje JE, Brenn T, Lazar AJ, et al, eds. McKee's Pathology of the Skin. 4th ed. St. Louis, MO: Elsevier Saunders; 2012.
- Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 6th ed. Philadelphia, PA: Elsevier; 2015.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Silverman RA, Rabinowitz AD. Eosinophils in the cellular infiltrate of granuloma annulare. J Cutan Pathol. 1985;12:13-17.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
The Diagnosis: Cutaneous Metastases
Cutaneous metastases (CMs) can present in an otherwise asymptomatic patient as the only sign of an underlying disease process. In women, the most common cause of CM is breast carcinoma.1-3 Cutaneous metastases are found in approximately 25% of all patients with breast carcinoma,1 and breast carcinomas represent approximately 69% of all CMs found in women (Table 1).2 Cutaneous metastatic breast carcinoma (CMBC) is associated with a poor prognosis with a mean survival of approximately 6 months at the time of diagnosis.1,3 It commonly presents as a collection of flesh-colored, firm, asymptomatic, and rapidly appearing papules and nodules that can resemble cysts or fibrous tumors.1,3,4 They typically are located on the chest wall or abdomen near the site of the underlying malignancy.1-3 The histologic features of CMBC can include hyperchromatic tumor cells infiltrating between the collagen fibers in a characteristic single file manner,3,5 giving the appearance of a busy dermis, a nonspecific term to describe a focally hypercellular dermis at low-power magnification (Table 2).5,6 Cords and clusters of atypical cells with intracytoplasmic vacuoles or well-developed ducts also can be seen (quiz image [inset]). The carcinoma en cuirasse subtype of CMBC is characterized by a fibrotic scarlike plaque on the chest wall.1,3 If a punch biopsy is obtained, the specimen typically appears rectangular rather than tapered because of the sclerotic dermal collagen.6 In contrast, inflammatory carcinoma (carcinoma erysipelatoides) presents as an erythematous plaque resembling cellulitis due to the lymphatics being congested by tumor cells.3 Immunohistochemistry is a valuable tool in diagnosis. Positive staining is seen with cytokeratin 7, gross cystic disease fluid protein-15, mammaglobin, and GATA-3.1,3,6
Kaposi sarcoma (KS) is a low-grade endothelial malignancy associated with human herpesvirus 8.3,4 Kaposi sarcoma can be divided into 4 main subtypes: classic KS, African KS, AIDS-related KS, and immunosuppression-associated KS that occurs in patients with diseases such as human immunodeficiency virus. The cutaneous lesions are similar between subtypes and present as dark reddish purple macules that may enlarge or become nodular lesions.3,4 Histologically, 3 distinct stages of progression are described: patch, plaque, and tumor. The plaque stage has the appearance of a busy dermis due to the rapid proliferation of vascular structures within the dermis.3,6 A useful histologic feature known as the promontory sign can be seen as the proliferating tumor causes preexisting structures to project into vascular spaces (Figure 1).6 Immunohistochemistry for the endothelial and lymphatic markers CD31 and D2-40, respectively, are positive and may aid in the diagnosis.3 Staining for the latent nuclear antigen-1 of human herpesvirus 8 is a highly specific marker used to diagnose KS and can further distinguish it from the other busy dermis lesions.3
Granuloma annulare (GA) is characterized by rings of small, firm, pink to flesh-colored papules with a variable disease duration.4 Histologically, the interstitial variant of GA is characterized by a scattered inflammatory infiltrate consisting of histiocytes and lymphocytes located between altered collagen fibers in the superficial to mid dermis (Figure 2).3,6 Occasional eosinophils and increased dermal mucin are useful features to distinguish interstitial GA from other entities in the busy dermis differential.7
Scleromyxedema, also known as generalized lichen myxedematosus, is a rare mucinosis.3,8 Although its pathogenesis is unknown, it has been suggested that paraproteins related to the underlying gammopathy act to stimulate fibroblast proliferation and mucin overproduction.8 Clinically, characteristic widespread firm, waxy, dome-shaped papules are present over the head, upper trunk, and extremities.3,8 Histologically, scleromyxedema is characterized by increased dermal fibroblasts, mucin, and fibrosis, leading to the appearance of a busy dermis (Figure 3).3,6
Neurofibromas are common benign peripheral nerve sheath tumors that can occur sporadically or in the setting of neurofibromatosis.3-5 They present as soft, flesh-colored papules or nodules most commonly located on the trunk and limbs.4 Histologically, neurofibromas are nonencapsulated tumors composed of abundant spindle cells with comma-shaped nuclei diffusely arranged in a pale myxoid stroma (Figure 4). Scattered mast cells can be visualized at higher magnification.3,6
The Diagnosis: Cutaneous Metastases
Cutaneous metastases (CMs) can present in an otherwise asymptomatic patient as the only sign of an underlying disease process. In women, the most common cause of CM is breast carcinoma.1-3 Cutaneous metastases are found in approximately 25% of all patients with breast carcinoma,1 and breast carcinomas represent approximately 69% of all CMs found in women (Table 1).2 Cutaneous metastatic breast carcinoma (CMBC) is associated with a poor prognosis with a mean survival of approximately 6 months at the time of diagnosis.1,3 It commonly presents as a collection of flesh-colored, firm, asymptomatic, and rapidly appearing papules and nodules that can resemble cysts or fibrous tumors.1,3,4 They typically are located on the chest wall or abdomen near the site of the underlying malignancy.1-3 The histologic features of CMBC can include hyperchromatic tumor cells infiltrating between the collagen fibers in a characteristic single file manner,3,5 giving the appearance of a busy dermis, a nonspecific term to describe a focally hypercellular dermis at low-power magnification (Table 2).5,6 Cords and clusters of atypical cells with intracytoplasmic vacuoles or well-developed ducts also can be seen (quiz image [inset]). The carcinoma en cuirasse subtype of CMBC is characterized by a fibrotic scarlike plaque on the chest wall.1,3 If a punch biopsy is obtained, the specimen typically appears rectangular rather than tapered because of the sclerotic dermal collagen.6 In contrast, inflammatory carcinoma (carcinoma erysipelatoides) presents as an erythematous plaque resembling cellulitis due to the lymphatics being congested by tumor cells.3 Immunohistochemistry is a valuable tool in diagnosis. Positive staining is seen with cytokeratin 7, gross cystic disease fluid protein-15, mammaglobin, and GATA-3.1,3,6
Kaposi sarcoma (KS) is a low-grade endothelial malignancy associated with human herpesvirus 8.3,4 Kaposi sarcoma can be divided into 4 main subtypes: classic KS, African KS, AIDS-related KS, and immunosuppression-associated KS that occurs in patients with diseases such as human immunodeficiency virus. The cutaneous lesions are similar between subtypes and present as dark reddish purple macules that may enlarge or become nodular lesions.3,4 Histologically, 3 distinct stages of progression are described: patch, plaque, and tumor. The plaque stage has the appearance of a busy dermis due to the rapid proliferation of vascular structures within the dermis.3,6 A useful histologic feature known as the promontory sign can be seen as the proliferating tumor causes preexisting structures to project into vascular spaces (Figure 1).6 Immunohistochemistry for the endothelial and lymphatic markers CD31 and D2-40, respectively, are positive and may aid in the diagnosis.3 Staining for the latent nuclear antigen-1 of human herpesvirus 8 is a highly specific marker used to diagnose KS and can further distinguish it from the other busy dermis lesions.3
Granuloma annulare (GA) is characterized by rings of small, firm, pink to flesh-colored papules with a variable disease duration.4 Histologically, the interstitial variant of GA is characterized by a scattered inflammatory infiltrate consisting of histiocytes and lymphocytes located between altered collagen fibers in the superficial to mid dermis (Figure 2).3,6 Occasional eosinophils and increased dermal mucin are useful features to distinguish interstitial GA from other entities in the busy dermis differential.7
Scleromyxedema, also known as generalized lichen myxedematosus, is a rare mucinosis.3,8 Although its pathogenesis is unknown, it has been suggested that paraproteins related to the underlying gammopathy act to stimulate fibroblast proliferation and mucin overproduction.8 Clinically, characteristic widespread firm, waxy, dome-shaped papules are present over the head, upper trunk, and extremities.3,8 Histologically, scleromyxedema is characterized by increased dermal fibroblasts, mucin, and fibrosis, leading to the appearance of a busy dermis (Figure 3).3,6
Neurofibromas are common benign peripheral nerve sheath tumors that can occur sporadically or in the setting of neurofibromatosis.3-5 They present as soft, flesh-colored papules or nodules most commonly located on the trunk and limbs.4 Histologically, neurofibromas are nonencapsulated tumors composed of abundant spindle cells with comma-shaped nuclei diffusely arranged in a pale myxoid stroma (Figure 4). Scattered mast cells can be visualized at higher magnification.3,6
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Habif TP, Dinulos JGH, Chapman MS, et al. Skin Disease: Diagnosis and Treatment. 4th ed. Edinburgh, Scotland: Elsevier; 2017.
- Calonje JE, Brenn T, Lazar AJ, et al, eds. McKee's Pathology of the Skin. 4th ed. St. Louis, MO: Elsevier Saunders; 2012.
- Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 6th ed. Philadelphia, PA: Elsevier; 2015.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Silverman RA, Rabinowitz AD. Eosinophils in the cellular infiltrate of granuloma annulare. J Cutan Pathol. 1985;12:13-17.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Habif TP, Dinulos JGH, Chapman MS, et al. Skin Disease: Diagnosis and Treatment. 4th ed. Edinburgh, Scotland: Elsevier; 2017.
- Calonje JE, Brenn T, Lazar AJ, et al, eds. McKee's Pathology of the Skin. 4th ed. St. Louis, MO: Elsevier Saunders; 2012.
- Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 6th ed. Philadelphia, PA: Elsevier; 2015.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Silverman RA, Rabinowitz AD. Eosinophils in the cellular infiltrate of granuloma annulare. J Cutan Pathol. 1985;12:13-17.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
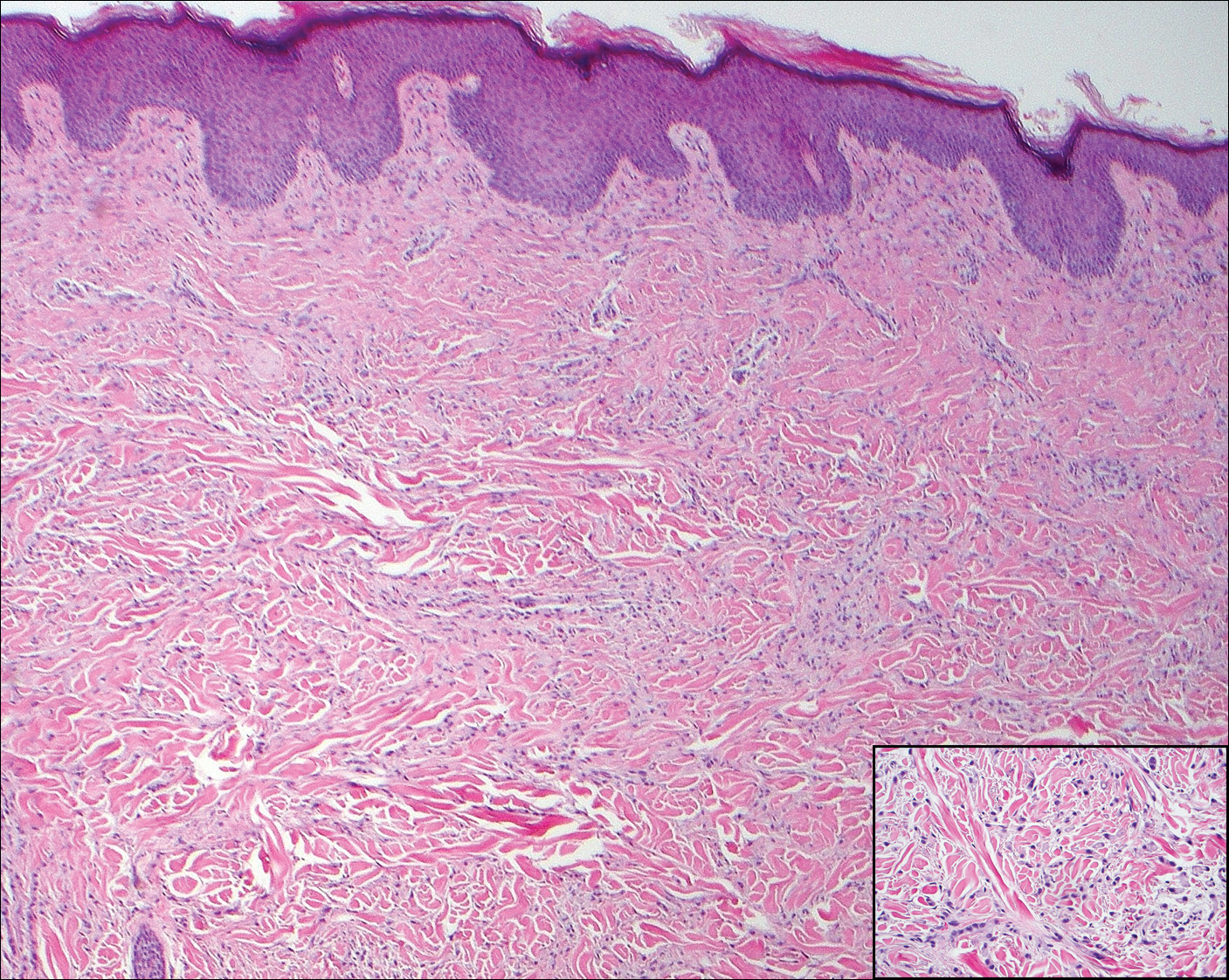
A 56-year-old woman presented with multiple asymptomatic lesions of 2 months' duration. On physical examination firm pink papules were noted dispersed across the upper abdomen, chest, and back. A 5-mm punch biopsy was obtained.
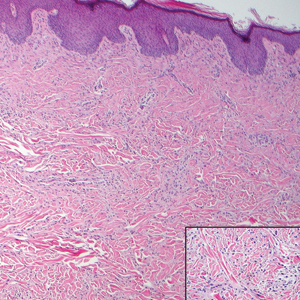
Tetrad Bodies in Skin
The Diagnosis: Bacterial Infection
The tetrad arrangement of organisms seen in this case was classic for Micrococcus and Sarcina species. Both are gram-positive cocci that occur in tetrads, but Micrococcus is aerobic and catalase positive, whereas Sarcina species are anaerobic, catalase negative, acidophilic, and form spores in alkaline pH.1 Although difficult to definitively differentiate on light microscopy, micrococci are smaller in size, ranging from 0.5 to 2.0 μm, and occur in tight clusters, as seen in this case (quiz images), in contrast to Sarcina species, which are relatively larger (1.8-3.0 μm).2 Sarcinae typically are found in soil and air, are considered pathogenic, and are associated with gastric symptoms (Sarcina ventriculi).1 Sarcina species also are reported to colonize the skin of patients with diabetes mellitus, but no pathogenic activity is known in the skin.3 Micrococcus species, with the majority being Micrococcus luteus, are part of the normal flora of the human skin as well as the oral and nasal cavities. Occasional reports of pneumonia, endocarditis, meningitis, arthritis, endophthalmitis, and sepsis have been reported in immunocompromised individuals.4 In the skin, Micrococcus is a commensal organism; however, Micrococcus sedentarius has been associated with pitted keratolysis, and reports of Micrococcus folliculitis in human immunodeficiency virus patients also are described in the literature.5,6 Micrococci are considered opportunistic bacteria and may worsen and prolong a localized cutaneous infection caused by other organisms under favorable conditions.7 Micrococcus luteus is one of the most common bacteria cultured from skin and soft tissue infections caused by fungal organisms.8 Depending on the immune status of an individual, use of broad-spectrum antibiotic and/or elimination of favorable milieu (ie, primary pathogen, breaks in skin) usually treats the infection.
Because of the rarity of infections caused and being part of the normal flora, the clinical implications of subtyping and sensitivity studies via culture or molecular studies may not be important; however, incidental presence of these organisms with unfamiliar morphology may cause confusion for the dermatopathologist. An extremely small size (0.5-2.0 μm) compared to red blood cells (7-8 μm) and white blood cells (10-12 μm) in a tight tetrad arrangement should raise the suspicion for Micrococcus.1 The refractive nature of these organisms from a thick extracellular layer can mimic fungus or plant matter; a negative Grocott-Gomori methenamine-silver stain in this case helped in not only differentiating but also ruling out secondary fungal infection. Finally, a Gram stain with violet staining of these organisms reaffirmed the diagnosis of gram-positive bacterial organisms, most consistent with Micrococcus species (Figure 1). Culture studies were not performed because of contamination of the tissue specimen and resolution of the patient's symptoms.
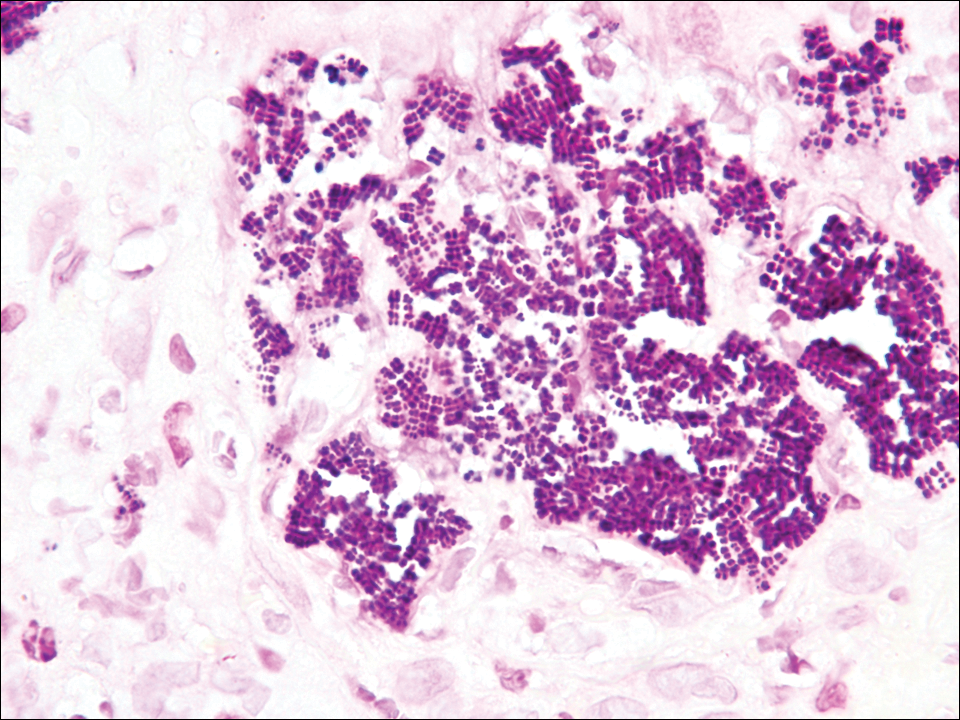
The presence of foreign material in the skin may be traumatic, occupational, cosmetic, iatrogenic, or self-inflicted, including a wide variety of substances that appear in different morphological forms on hematoxylin and eosin (H&E)-stained sections, depending on their structure and physiochemical properties.9 Although not all foreign bodies may polarize, examining the sample under polarized light is considered an important step to narrow down the differential diagnosis. The tissue reaction is primarily dependent on the nature of the substance and duration, consisting of histiocytes, macrophages, plasma cells, lymphocytes, and fibrosis.9 Activated histiocytes, multinucleated giant cells, and granulomas are classic findings that generally are seen surrounding and engulfing the foreign material (Figure 2). In addition to foreign material, substances such as calcium salts, urate crystals, extruded keratin, ruptured cysts, and hair follicles may act as foreign materials and can incite a tissue response.9 Absence of histiocytic response, granuloma formation, and fibrosis in a lesion of 1 month's duration made the tetrad bodies unlikely to be foreign material.
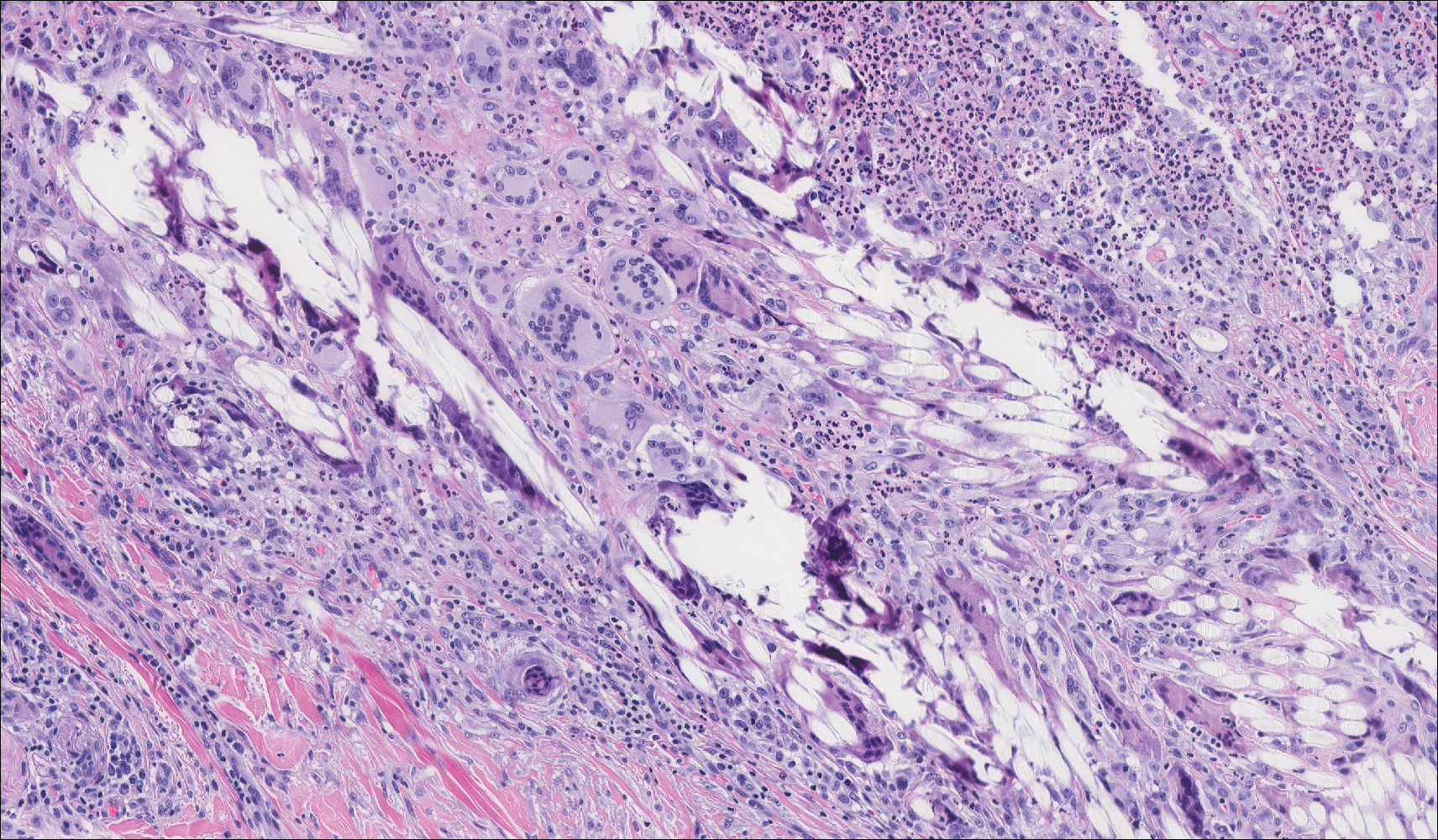
Demodex mites are superficial inhabitants of human skin that are acquired shortly after birth, live in or near pilosebaceous units, and obtain nourishment from skin cells and sebum.10,11 The mites can be recovered on 10% of skin biopsies, most commonly on the face due to high sebum production.10 Adult mites range from 0.1 to 0.4 mm in length and are round to oval in shape. Females lay eggs inside the hair follicle or sebaceous glands.11 They usually are asymptomatic, but their infestation may become pathogenic, especially in immunocompromised individuals.10 The clinical picture may resemble bacterial folliculitis, rosacea, and perioral dermatitis, while histology typically is characterized by spongiosis, lymphohistiocytic inflammation around infested follicles, and mite(s) in follicular infundibula (Figure 3). Sometimes the protrusion of mites and keratin from the follicles is seen as follicular spines on histology and referred to as pityriasis folliculorum.
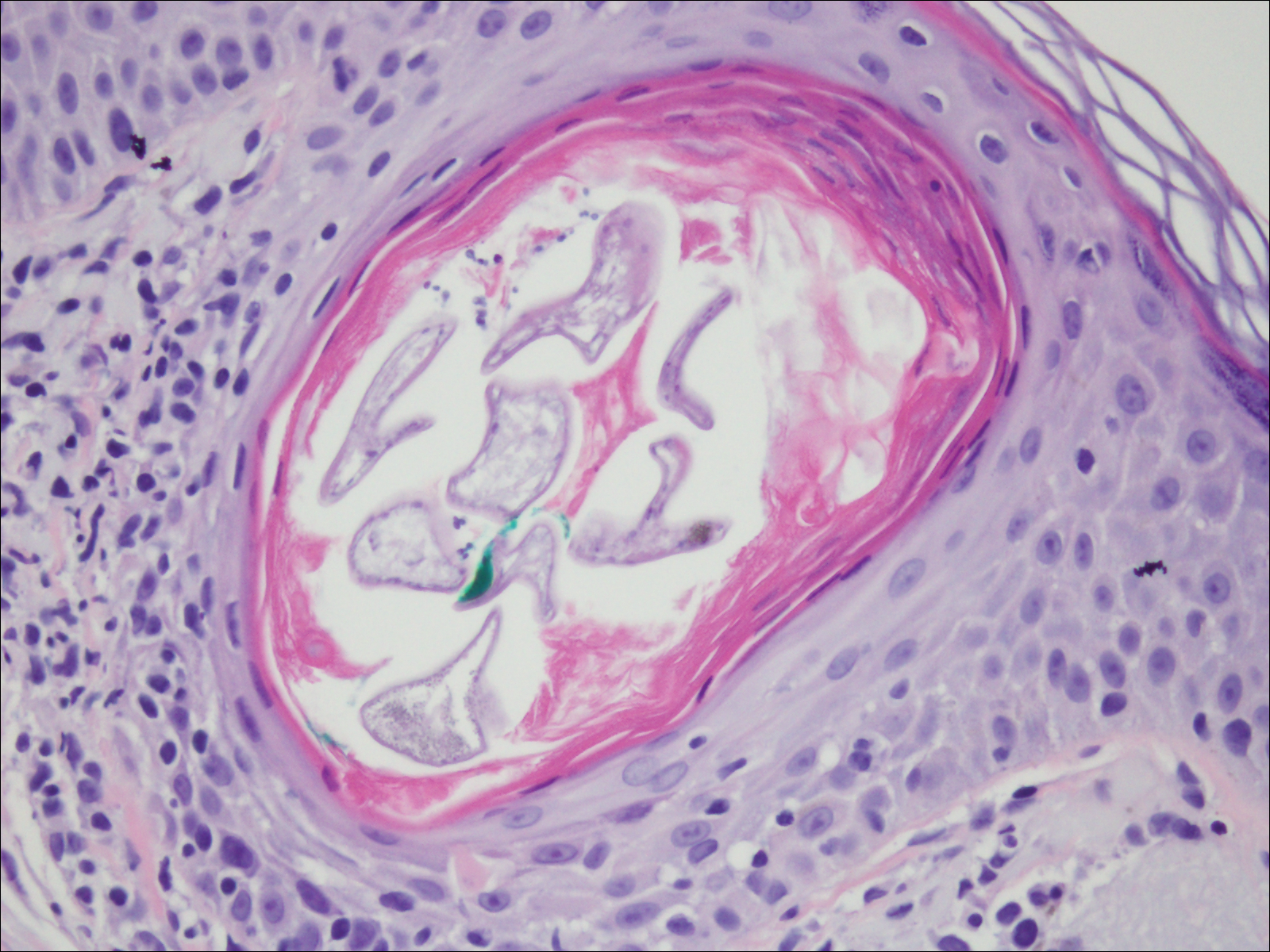
Deposits of urate crystals in skin occur from the elevated serum uric acid levels in gout. The cutaneous deposits are mainly in the dermis and subcutaneous tissue and are extremely painful.12 Urate crystals get dissolved during formalin fixation and leave needlelike clefts in a homogenous, lightly basophilic material on H&E slide (Figure 4). For the same reason, polarized microscopy also is not helpful despite the birefringent nature of urate crystals.12
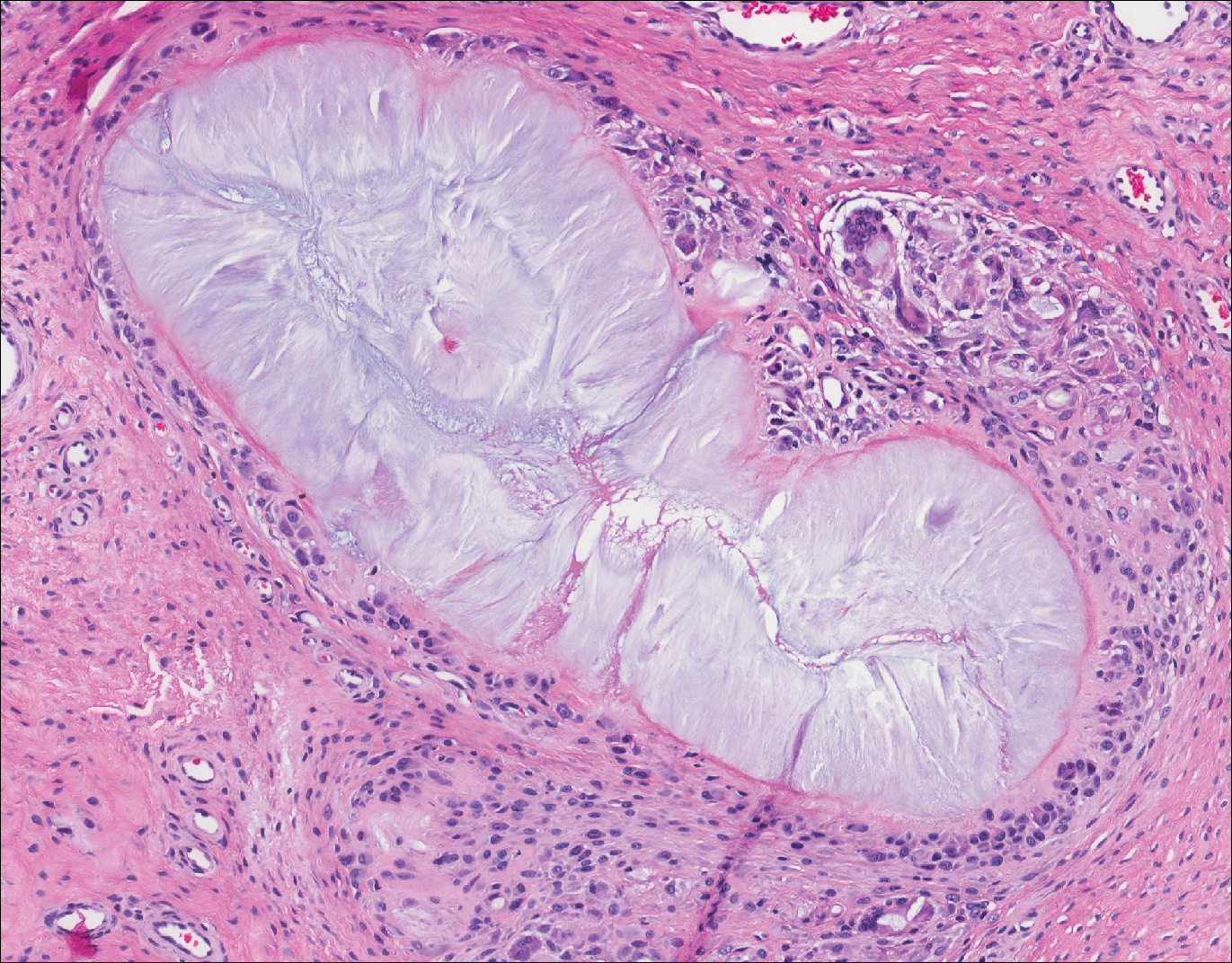
Fungal yeast forms appear round to oval under light microscopy, ranging from 2 to 100 μm in size.13 The common superficial forms involving the epidermis or hair follicles similar to the current case of bacterial infection include Malassezia and dermatophyte infections. Malassezia is part of the normal flora of sebum-rich areas of skin and is associated with superficial infections such as folliculitis, atopic dermatitis, psoriasis, seborrheic dermatitis, and dandruff.14 Malassezia appear as clusters of yeast cells that are pleomorphic and round to oval in shape, ranging from 2 to 6 μm in size. It forms hyphae in its pathogenic form and gives rise to the classic spaghetti and meatball-like appearance that can be highlighted by periodic acid-Schiff (Figure 5) and Grocott-Gomori methenamine-silver special stains. Dermatophytes include 3 genera--Trichophyton, Microsporum, and Epidermophyton--with at least 40 species that causes skin infections in humans.14 Fungal spores and hyphae forms are restricted to the stratum corneum. The hyphae forms may not be apparent on H&E stain, and periodic acid-Schiff staining is helpful in visualizing the fungal elements. The presence of neutrophils in the corneal layer, basket weave hyperkeratosis, and presence of fungal hyphae within the corneal layer fissures (sandwich sign) are clues to the dermatophyte infection.15 Other smaller fungi such as Histoplasma capsulatum (2-4 μm), Candida (3-5 μm), and Pneumocystis (2-5 μm) species can be found in skin in disseminated infections, usually affecting immunocompromised individuals.13 Histoplasma is a basophilic yeast that exhibits narrow-based budding and appears clustered within or outside of macrophages. Candida species generally are dimorphic, and yeasts are found intermingled with filamentous forms. Pneumocystis infection in skin is extremely rare, and the fungi appear as spherical or crescent-shaped bodies in a foamy amorphous material.16

- Al Rasheed MR, Senseng CG. Sarcina ventriculi: review of the literature. Arch Pathol Lab Med. 2016;140:1441-1445.
- Lam-Himlin D, Tsiatis AC, Montgomery E, et al. Sarcina organisms in the gastrointestinal tract: a clinicopathologic and molecular study. Am J Surg Pathol. 2011;35:1700-1705.
- Somerville DA, Lancaster-Smith M. The aerobic cutaneous microflora of diabetic subjects. Br J Dermatol. 1973;89:395-400.
- Hetem DJ, Rooijakkers S, Ekkelenkamp MB. Staphylococci and Micrococci. In: Cohen J, Powderly WG, Opal SM, eds. Infectious Diseases. 4th ed. Vol 2. New York, NY: Elsevier; 2017:1509-1522.
- Nordstrom KM, McGinley KJ, Cappiello L, et al. Pitted keratolysis. the role of Micrococcus sedentarius. Arch Dermatol. 1987;123:1320-1325.
- Smith KJ, Neafie R, Yeager J, et al. Micrococcus folliculitis in HIV-1 disease. Br J Dermatol. 1999;141:558-561.
- van Rensburg JJ, Lin H, Gao X, et al. The human skin microbiome associates with the outcome of and is influenced by bacterial infection. mBio. 2015;6:E01315-15. doi:10.1128/mBio.01315-15.
- Chuku A, Nwankiti OO. Association of bacteria with fungal infection of skin and soft tissue lesions in plateau state, Nigeria. Br Microbiol Res J. 2013;3:470-477.
- Molina-Ruiz AM, Requena L. Foreign body granulomas. Dermatol Clin. 2015;33:497-523.
- Elston CA, Elston DM. Demodex mites. Clin Dermatol. 2014;32:739-743.
- Rather PA, Hassan I. Human Demodex mite: the versatile mite of dermatological importance. Indian J Dermatol. 2014;59:60-66.
- Gaviria JL, Ortega VG, Gaona J, et al. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445.
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280.
- White TC, Findley K, Dawson TL Jr, et al. Fungi on the skin: dermatophytes and Malassezia. Cold Spring Harb Perspect Med. 2014;4. pii:a019802. doi:10.1101/cshperspect.a019802.
- Gottlieb GJ, Ackerman AB. The "sandwich sign" of dermatophytosis. Am J Dermatopathol. 1986;8:347.
- Hennessey NP, Parro EL, Cockerell CJ. Cutaneous Pneumocystis carinii infection in patients with acquired immunodeficiency syndrome. Arch Dermatol. 1991;127:1699-1701.
The Diagnosis: Bacterial Infection
The tetrad arrangement of organisms seen in this case was classic for Micrococcus and Sarcina species. Both are gram-positive cocci that occur in tetrads, but Micrococcus is aerobic and catalase positive, whereas Sarcina species are anaerobic, catalase negative, acidophilic, and form spores in alkaline pH.1 Although difficult to definitively differentiate on light microscopy, micrococci are smaller in size, ranging from 0.5 to 2.0 μm, and occur in tight clusters, as seen in this case (quiz images), in contrast to Sarcina species, which are relatively larger (1.8-3.0 μm).2 Sarcinae typically are found in soil and air, are considered pathogenic, and are associated with gastric symptoms (Sarcina ventriculi).1 Sarcina species also are reported to colonize the skin of patients with diabetes mellitus, but no pathogenic activity is known in the skin.3 Micrococcus species, with the majority being Micrococcus luteus, are part of the normal flora of the human skin as well as the oral and nasal cavities. Occasional reports of pneumonia, endocarditis, meningitis, arthritis, endophthalmitis, and sepsis have been reported in immunocompromised individuals.4 In the skin, Micrococcus is a commensal organism; however, Micrococcus sedentarius has been associated with pitted keratolysis, and reports of Micrococcus folliculitis in human immunodeficiency virus patients also are described in the literature.5,6 Micrococci are considered opportunistic bacteria and may worsen and prolong a localized cutaneous infection caused by other organisms under favorable conditions.7 Micrococcus luteus is one of the most common bacteria cultured from skin and soft tissue infections caused by fungal organisms.8 Depending on the immune status of an individual, use of broad-spectrum antibiotic and/or elimination of favorable milieu (ie, primary pathogen, breaks in skin) usually treats the infection.
Because of the rarity of infections caused and being part of the normal flora, the clinical implications of subtyping and sensitivity studies via culture or molecular studies may not be important; however, incidental presence of these organisms with unfamiliar morphology may cause confusion for the dermatopathologist. An extremely small size (0.5-2.0 μm) compared to red blood cells (7-8 μm) and white blood cells (10-12 μm) in a tight tetrad arrangement should raise the suspicion for Micrococcus.1 The refractive nature of these organisms from a thick extracellular layer can mimic fungus or plant matter; a negative Grocott-Gomori methenamine-silver stain in this case helped in not only differentiating but also ruling out secondary fungal infection. Finally, a Gram stain with violet staining of these organisms reaffirmed the diagnosis of gram-positive bacterial organisms, most consistent with Micrococcus species (Figure 1). Culture studies were not performed because of contamination of the tissue specimen and resolution of the patient's symptoms.
The presence of foreign material in the skin may be traumatic, occupational, cosmetic, iatrogenic, or self-inflicted, including a wide variety of substances that appear in different morphological forms on hematoxylin and eosin (H&E)-stained sections, depending on their structure and physiochemical properties.9 Although not all foreign bodies may polarize, examining the sample under polarized light is considered an important step to narrow down the differential diagnosis. The tissue reaction is primarily dependent on the nature of the substance and duration, consisting of histiocytes, macrophages, plasma cells, lymphocytes, and fibrosis.9 Activated histiocytes, multinucleated giant cells, and granulomas are classic findings that generally are seen surrounding and engulfing the foreign material (Figure 2). In addition to foreign material, substances such as calcium salts, urate crystals, extruded keratin, ruptured cysts, and hair follicles may act as foreign materials and can incite a tissue response.9 Absence of histiocytic response, granuloma formation, and fibrosis in a lesion of 1 month's duration made the tetrad bodies unlikely to be foreign material.
Demodex mites are superficial inhabitants of human skin that are acquired shortly after birth, live in or near pilosebaceous units, and obtain nourishment from skin cells and sebum.10,11 The mites can be recovered on 10% of skin biopsies, most commonly on the face due to high sebum production.10 Adult mites range from 0.1 to 0.4 mm in length and are round to oval in shape. Females lay eggs inside the hair follicle or sebaceous glands.11 They usually are asymptomatic, but their infestation may become pathogenic, especially in immunocompromised individuals.10 The clinical picture may resemble bacterial folliculitis, rosacea, and perioral dermatitis, while histology typically is characterized by spongiosis, lymphohistiocytic inflammation around infested follicles, and mite(s) in follicular infundibula (Figure 3). Sometimes the protrusion of mites and keratin from the follicles is seen as follicular spines on histology and referred to as pityriasis folliculorum.
Deposits of urate crystals in skin occur from the elevated serum uric acid levels in gout. The cutaneous deposits are mainly in the dermis and subcutaneous tissue and are extremely painful.12 Urate crystals get dissolved during formalin fixation and leave needlelike clefts in a homogenous, lightly basophilic material on H&E slide (Figure 4). For the same reason, polarized microscopy also is not helpful despite the birefringent nature of urate crystals.12
Fungal yeast forms appear round to oval under light microscopy, ranging from 2 to 100 μm in size.13 The common superficial forms involving the epidermis or hair follicles similar to the current case of bacterial infection include Malassezia and dermatophyte infections. Malassezia is part of the normal flora of sebum-rich areas of skin and is associated with superficial infections such as folliculitis, atopic dermatitis, psoriasis, seborrheic dermatitis, and dandruff.14 Malassezia appear as clusters of yeast cells that are pleomorphic and round to oval in shape, ranging from 2 to 6 μm in size. It forms hyphae in its pathogenic form and gives rise to the classic spaghetti and meatball-like appearance that can be highlighted by periodic acid-Schiff (Figure 5) and Grocott-Gomori methenamine-silver special stains. Dermatophytes include 3 genera--Trichophyton, Microsporum, and Epidermophyton--with at least 40 species that causes skin infections in humans.14 Fungal spores and hyphae forms are restricted to the stratum corneum. The hyphae forms may not be apparent on H&E stain, and periodic acid-Schiff staining is helpful in visualizing the fungal elements. The presence of neutrophils in the corneal layer, basket weave hyperkeratosis, and presence of fungal hyphae within the corneal layer fissures (sandwich sign) are clues to the dermatophyte infection.15 Other smaller fungi such as Histoplasma capsulatum (2-4 μm), Candida (3-5 μm), and Pneumocystis (2-5 μm) species can be found in skin in disseminated infections, usually affecting immunocompromised individuals.13 Histoplasma is a basophilic yeast that exhibits narrow-based budding and appears clustered within or outside of macrophages. Candida species generally are dimorphic, and yeasts are found intermingled with filamentous forms. Pneumocystis infection in skin is extremely rare, and the fungi appear as spherical or crescent-shaped bodies in a foamy amorphous material.16
The Diagnosis: Bacterial Infection
The tetrad arrangement of organisms seen in this case was classic for Micrococcus and Sarcina species. Both are gram-positive cocci that occur in tetrads, but Micrococcus is aerobic and catalase positive, whereas Sarcina species are anaerobic, catalase negative, acidophilic, and form spores in alkaline pH.1 Although difficult to definitively differentiate on light microscopy, micrococci are smaller in size, ranging from 0.5 to 2.0 μm, and occur in tight clusters, as seen in this case (quiz images), in contrast to Sarcina species, which are relatively larger (1.8-3.0 μm).2 Sarcinae typically are found in soil and air, are considered pathogenic, and are associated with gastric symptoms (Sarcina ventriculi).1 Sarcina species also are reported to colonize the skin of patients with diabetes mellitus, but no pathogenic activity is known in the skin.3 Micrococcus species, with the majority being Micrococcus luteus, are part of the normal flora of the human skin as well as the oral and nasal cavities. Occasional reports of pneumonia, endocarditis, meningitis, arthritis, endophthalmitis, and sepsis have been reported in immunocompromised individuals.4 In the skin, Micrococcus is a commensal organism; however, Micrococcus sedentarius has been associated with pitted keratolysis, and reports of Micrococcus folliculitis in human immunodeficiency virus patients also are described in the literature.5,6 Micrococci are considered opportunistic bacteria and may worsen and prolong a localized cutaneous infection caused by other organisms under favorable conditions.7 Micrococcus luteus is one of the most common bacteria cultured from skin and soft tissue infections caused by fungal organisms.8 Depending on the immune status of an individual, use of broad-spectrum antibiotic and/or elimination of favorable milieu (ie, primary pathogen, breaks in skin) usually treats the infection.
Because of the rarity of infections caused and being part of the normal flora, the clinical implications of subtyping and sensitivity studies via culture or molecular studies may not be important; however, incidental presence of these organisms with unfamiliar morphology may cause confusion for the dermatopathologist. An extremely small size (0.5-2.0 μm) compared to red blood cells (7-8 μm) and white blood cells (10-12 μm) in a tight tetrad arrangement should raise the suspicion for Micrococcus.1 The refractive nature of these organisms from a thick extracellular layer can mimic fungus or plant matter; a negative Grocott-Gomori methenamine-silver stain in this case helped in not only differentiating but also ruling out secondary fungal infection. Finally, a Gram stain with violet staining of these organisms reaffirmed the diagnosis of gram-positive bacterial organisms, most consistent with Micrococcus species (Figure 1). Culture studies were not performed because of contamination of the tissue specimen and resolution of the patient's symptoms.
The presence of foreign material in the skin may be traumatic, occupational, cosmetic, iatrogenic, or self-inflicted, including a wide variety of substances that appear in different morphological forms on hematoxylin and eosin (H&E)-stained sections, depending on their structure and physiochemical properties.9 Although not all foreign bodies may polarize, examining the sample under polarized light is considered an important step to narrow down the differential diagnosis. The tissue reaction is primarily dependent on the nature of the substance and duration, consisting of histiocytes, macrophages, plasma cells, lymphocytes, and fibrosis.9 Activated histiocytes, multinucleated giant cells, and granulomas are classic findings that generally are seen surrounding and engulfing the foreign material (Figure 2). In addition to foreign material, substances such as calcium salts, urate crystals, extruded keratin, ruptured cysts, and hair follicles may act as foreign materials and can incite a tissue response.9 Absence of histiocytic response, granuloma formation, and fibrosis in a lesion of 1 month's duration made the tetrad bodies unlikely to be foreign material.
Demodex mites are superficial inhabitants of human skin that are acquired shortly after birth, live in or near pilosebaceous units, and obtain nourishment from skin cells and sebum.10,11 The mites can be recovered on 10% of skin biopsies, most commonly on the face due to high sebum production.10 Adult mites range from 0.1 to 0.4 mm in length and are round to oval in shape. Females lay eggs inside the hair follicle or sebaceous glands.11 They usually are asymptomatic, but their infestation may become pathogenic, especially in immunocompromised individuals.10 The clinical picture may resemble bacterial folliculitis, rosacea, and perioral dermatitis, while histology typically is characterized by spongiosis, lymphohistiocytic inflammation around infested follicles, and mite(s) in follicular infundibula (Figure 3). Sometimes the protrusion of mites and keratin from the follicles is seen as follicular spines on histology and referred to as pityriasis folliculorum.
Deposits of urate crystals in skin occur from the elevated serum uric acid levels in gout. The cutaneous deposits are mainly in the dermis and subcutaneous tissue and are extremely painful.12 Urate crystals get dissolved during formalin fixation and leave needlelike clefts in a homogenous, lightly basophilic material on H&E slide (Figure 4). For the same reason, polarized microscopy also is not helpful despite the birefringent nature of urate crystals.12
Fungal yeast forms appear round to oval under light microscopy, ranging from 2 to 100 μm in size.13 The common superficial forms involving the epidermis or hair follicles similar to the current case of bacterial infection include Malassezia and dermatophyte infections. Malassezia is part of the normal flora of sebum-rich areas of skin and is associated with superficial infections such as folliculitis, atopic dermatitis, psoriasis, seborrheic dermatitis, and dandruff.14 Malassezia appear as clusters of yeast cells that are pleomorphic and round to oval in shape, ranging from 2 to 6 μm in size. It forms hyphae in its pathogenic form and gives rise to the classic spaghetti and meatball-like appearance that can be highlighted by periodic acid-Schiff (Figure 5) and Grocott-Gomori methenamine-silver special stains. Dermatophytes include 3 genera--Trichophyton, Microsporum, and Epidermophyton--with at least 40 species that causes skin infections in humans.14 Fungal spores and hyphae forms are restricted to the stratum corneum. The hyphae forms may not be apparent on H&E stain, and periodic acid-Schiff staining is helpful in visualizing the fungal elements. The presence of neutrophils in the corneal layer, basket weave hyperkeratosis, and presence of fungal hyphae within the corneal layer fissures (sandwich sign) are clues to the dermatophyte infection.15 Other smaller fungi such as Histoplasma capsulatum (2-4 μm), Candida (3-5 μm), and Pneumocystis (2-5 μm) species can be found in skin in disseminated infections, usually affecting immunocompromised individuals.13 Histoplasma is a basophilic yeast that exhibits narrow-based budding and appears clustered within or outside of macrophages. Candida species generally are dimorphic, and yeasts are found intermingled with filamentous forms. Pneumocystis infection in skin is extremely rare, and the fungi appear as spherical or crescent-shaped bodies in a foamy amorphous material.16
- Al Rasheed MR, Senseng CG. Sarcina ventriculi: review of the literature. Arch Pathol Lab Med. 2016;140:1441-1445.
- Lam-Himlin D, Tsiatis AC, Montgomery E, et al. Sarcina organisms in the gastrointestinal tract: a clinicopathologic and molecular study. Am J Surg Pathol. 2011;35:1700-1705.
- Somerville DA, Lancaster-Smith M. The aerobic cutaneous microflora of diabetic subjects. Br J Dermatol. 1973;89:395-400.
- Hetem DJ, Rooijakkers S, Ekkelenkamp MB. Staphylococci and Micrococci. In: Cohen J, Powderly WG, Opal SM, eds. Infectious Diseases. 4th ed. Vol 2. New York, NY: Elsevier; 2017:1509-1522.
- Nordstrom KM, McGinley KJ, Cappiello L, et al. Pitted keratolysis. the role of Micrococcus sedentarius. Arch Dermatol. 1987;123:1320-1325.
- Smith KJ, Neafie R, Yeager J, et al. Micrococcus folliculitis in HIV-1 disease. Br J Dermatol. 1999;141:558-561.
- van Rensburg JJ, Lin H, Gao X, et al. The human skin microbiome associates with the outcome of and is influenced by bacterial infection. mBio. 2015;6:E01315-15. doi:10.1128/mBio.01315-15.
- Chuku A, Nwankiti OO. Association of bacteria with fungal infection of skin and soft tissue lesions in plateau state, Nigeria. Br Microbiol Res J. 2013;3:470-477.
- Molina-Ruiz AM, Requena L. Foreign body granulomas. Dermatol Clin. 2015;33:497-523.
- Elston CA, Elston DM. Demodex mites. Clin Dermatol. 2014;32:739-743.
- Rather PA, Hassan I. Human Demodex mite: the versatile mite of dermatological importance. Indian J Dermatol. 2014;59:60-66.
- Gaviria JL, Ortega VG, Gaona J, et al. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445.
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280.
- White TC, Findley K, Dawson TL Jr, et al. Fungi on the skin: dermatophytes and Malassezia. Cold Spring Harb Perspect Med. 2014;4. pii:a019802. doi:10.1101/cshperspect.a019802.
- Gottlieb GJ, Ackerman AB. The "sandwich sign" of dermatophytosis. Am J Dermatopathol. 1986;8:347.
- Hennessey NP, Parro EL, Cockerell CJ. Cutaneous Pneumocystis carinii infection in patients with acquired immunodeficiency syndrome. Arch Dermatol. 1991;127:1699-1701.
- Al Rasheed MR, Senseng CG. Sarcina ventriculi: review of the literature. Arch Pathol Lab Med. 2016;140:1441-1445.
- Lam-Himlin D, Tsiatis AC, Montgomery E, et al. Sarcina organisms in the gastrointestinal tract: a clinicopathologic and molecular study. Am J Surg Pathol. 2011;35:1700-1705.
- Somerville DA, Lancaster-Smith M. The aerobic cutaneous microflora of diabetic subjects. Br J Dermatol. 1973;89:395-400.
- Hetem DJ, Rooijakkers S, Ekkelenkamp MB. Staphylococci and Micrococci. In: Cohen J, Powderly WG, Opal SM, eds. Infectious Diseases. 4th ed. Vol 2. New York, NY: Elsevier; 2017:1509-1522.
- Nordstrom KM, McGinley KJ, Cappiello L, et al. Pitted keratolysis. the role of Micrococcus sedentarius. Arch Dermatol. 1987;123:1320-1325.
- Smith KJ, Neafie R, Yeager J, et al. Micrococcus folliculitis in HIV-1 disease. Br J Dermatol. 1999;141:558-561.
- van Rensburg JJ, Lin H, Gao X, et al. The human skin microbiome associates with the outcome of and is influenced by bacterial infection. mBio. 2015;6:E01315-15. doi:10.1128/mBio.01315-15.
- Chuku A, Nwankiti OO. Association of bacteria with fungal infection of skin and soft tissue lesions in plateau state, Nigeria. Br Microbiol Res J. 2013;3:470-477.
- Molina-Ruiz AM, Requena L. Foreign body granulomas. Dermatol Clin. 2015;33:497-523.
- Elston CA, Elston DM. Demodex mites. Clin Dermatol. 2014;32:739-743.
- Rather PA, Hassan I. Human Demodex mite: the versatile mite of dermatological importance. Indian J Dermatol. 2014;59:60-66.
- Gaviria JL, Ortega VG, Gaona J, et al. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445.
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280.
- White TC, Findley K, Dawson TL Jr, et al. Fungi on the skin: dermatophytes and Malassezia. Cold Spring Harb Perspect Med. 2014;4. pii:a019802. doi:10.1101/cshperspect.a019802.
- Gottlieb GJ, Ackerman AB. The "sandwich sign" of dermatophytosis. Am J Dermatopathol. 1986;8:347.
- Hennessey NP, Parro EL, Cockerell CJ. Cutaneous Pneumocystis carinii infection in patients with acquired immunodeficiency syndrome. Arch Dermatol. 1991;127:1699-1701.
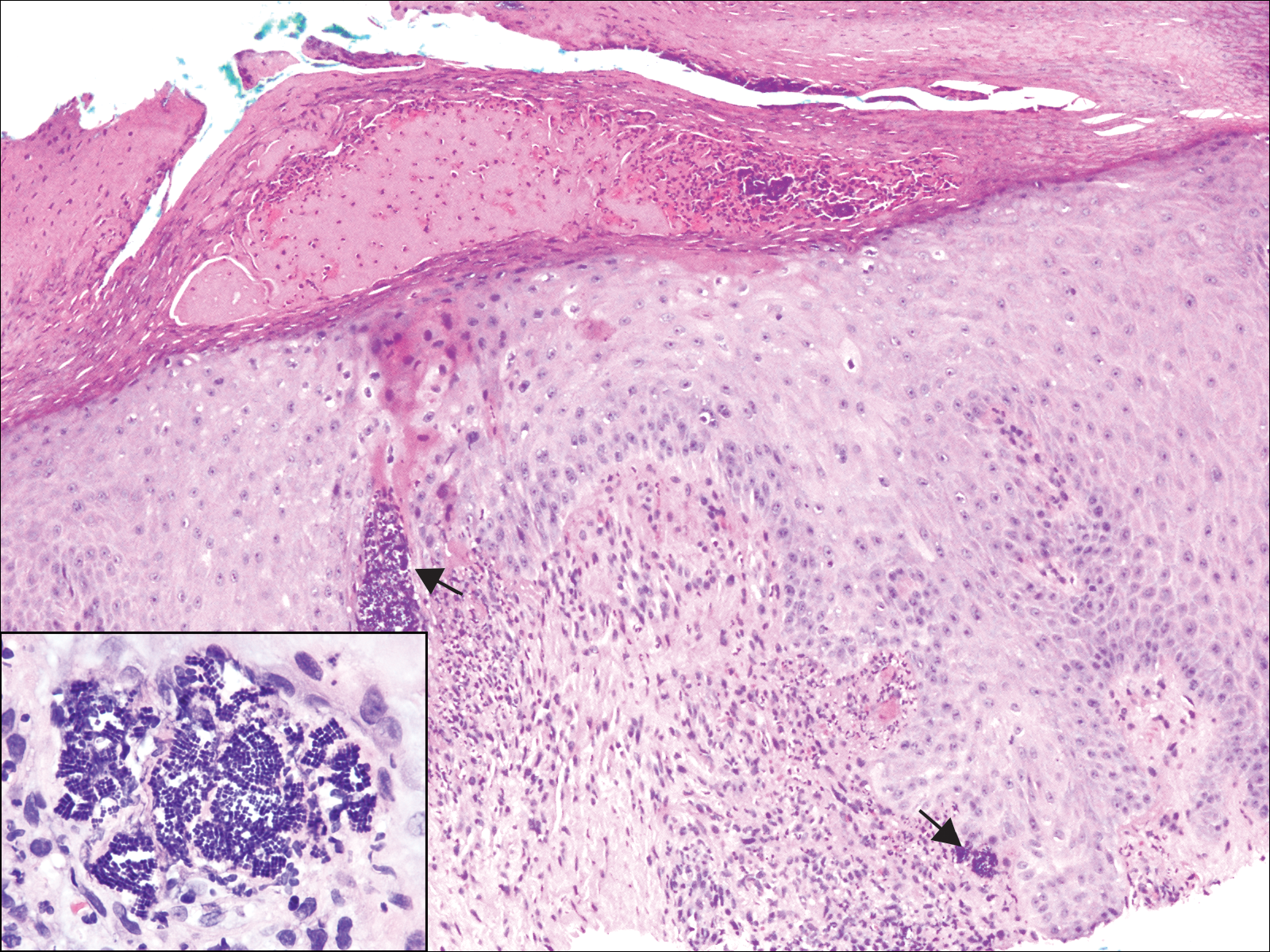
A 72-year-old woman with a medical history notable for multiple sclerosis and intravenous drug abuse presented to the dermatology clinic with a 0.6×0.5-cm, pruritic, wartlike, inflamed, keratotic papule on the palmar aspect of the right finger of more than 1 month's duration. A shave biopsy was performed that showed excoriation with serum crust, parakeratosis, and neutrophilic infiltrate in the papillary dermis. Within the serum crust and at the dermoepidermal junction, clusters of refractive basophilic bodies (arrows) in tetrad arrangement also were noted (inset). The papule resolved after the biopsy without any additional treatment.