User login
Pediatric insomnia: Assessment and diagnosis
FIRST OF 2 PARTS
A thorough evaluation can identify modifiable factors and guide treatment
Sleep problems are common among children and adolescents,1 with prevalence rates of 25% to 40%.2-4 Young children most commonly exhibit what is referred to as bedtime problems and night wakenings, whereas children in middle childhood (age 4 to 12) through adolescence (age 13 to 17) report insomnia. For many children, these problems persist.3 Insufficient sleep in children and adolescents worsens inattention, daytime fatigue, and cognitive and behavioral deficits.5 Assessment and treatment of sleep problems in children and adolescents is critical because poor sleep among youth increases the risk for depression, self-harm, and suicide,6,7 increases family stress, and decreases parental well-being.1
This 2-part article describes the assessment, diagnosis, and treatment of sleep problems among children and adolescents. In part 1, we focus on:
- sleep architecture (circadian rhythms, stages of sleep)
- sleep in healthy youth (age 6 to 17) and those with attention-deficit/hyperactivity disorder (ADHD), depressive disorders, and anxiety
- how to assess sleep, and the differential diagnosis of behavioral sleep problems in pediatric patients.
In Part 2, we will cover psychotherapeutic and psychopharmacologic interventions for youth with insomnia, and describe an effective approach to consultation with pediatric sleep medicine specialists.
How much sleep do children and adolescents need?
Throughout their development, children spend 40% to 50% of their time asleep. Sleep schedules are based on circadian rhythms, which are physical, mental, and behavioral changes that follow an approximately 24-hour cycle. Human circadian rhythm varies between 24 and 25 hours and is vital in determining our sleep patterns. Exposure to sunlight drives our circadian rhythm, sending signals to our bodies to “turn on” melatonin production at night (ie, 9
Box
Sleep architecture consists of 3 states: wake; non-rapid eye movement (NREM) sleep; and rapid eye movement (REM) sleep (“dreaming” sleep).2 These stages have distinct polysomnographic features of electroencephalographic EEG patterns, eye movements, and muscle tone.2 NREM sleep can be further divided into 3 stages: stage 1 (N1), stage 2 (N2), and stage 3 (N3). Stage 1 is the lightest stage and lasts for 30 seconds to 5 minutes; it is easy to wake up from stage 1 sleep. During stage 2 sleep, the body moves into a deeper sleep stage that is considered “true” sleep. This sleep stage is characterized by bursts of rhythmic rapid EEG activity known as spindles, as well as high-amplitude slow-wave spikes called K complexes.2 Stage 2 sleep lasts for 10 to 45 minutes. Stage 3, better known as “deep sleep,” slow-wave sleep, or delta sleep, is the most restorative sleep.2 Respiration is low and parasympathetic activity is high.2 It is difficult to be awakened during deep sleep, and if aroused, the person likely will feel confused or groggy. Deep sleep is followed by a return to lighter stage of sleep before the first REM sleep period begins.
REM sleep is the active stage of sleep. Breathing and heart rate become irregular, and the body experiences muscle atonia, or temporary paralysis, of arms and legs. When in REM sleep, individuals have the highest brain metabolic rates, and periodic bursts of eye movements.2 Most individuals move through stages of NREM and REM sleep in predicable ways, meaning they experience NREM sleep, return to a lighter stage of sleep after deep sleep, then move into REM sleep before the cycle repeats. It takes approximately 90 minutes for most adults to complete the NREM sleep cycle, and then REM sleep occurs before returning to NREM sleep.
In children, especially in infants and babies, sleep cycles are closer to 50 to 60 minutes. Newborns spend approximately 50% of their sleep in REM sleep, whereas adults spend 20% to 25% of their sleep in REM sleep. Children will spend more time in REM sleep until the third and fourth years of life, at which point REM gradually decreases to 20% to 25% by adulthood.
Sleep needs also change predictably throughout the lifespan. The National Sleep Foundation guidelines for sleep duration provide clinicians and parents with a range of recommended sleep for each stage of development. Infants require 14 to 17 hours of sleep, whereas adolescents need 8 to 10 hours by age 14 to 17.8 The key for clinicians is to determine if the child is within the recommended range, and how they are functioning on the number of hours of sleep they report. This allows for variation in how much sleep an individual child might need while acknowledging that some children within a specific age group might need more or less sleep than other children of the same age.
Sleep in healthy youth: Middle childhood
School-age children (age 6 to 12) typically need 9 to 10 hours of sleep over a 24-hour period.2 This developmental period is especially important for children to develop healthy sleep habits; however, developmentally appropriate cognitive and social/emotional factors might interfere with the quality and quantity of sleep. Middle childhood is a time when children can understand the dangers of the outside world (ie, violence, health problems) and resulting anxiety can disrupt sleep. Parents usually are less involved in bedtime as children approach adolescence, which leads to later bedtimes. At this stage, many children begin to take on more serious roles in their academics and extracurricular activities, peer relationships become more important, and use of electronics (eg, television, video games, internet, and handheld devices) increases—all of which compete with sleep.9 Frequent sleep issues during middle childhood include:
- irregular sleep-wake schedules
- later bedtimes
- decreased nighttime sleep
- increased caffeine intake
- reduced parental presence at bedtime
- daytime sleepiness.3
In school-age children, regular napping, falling asleep during short car rides, and daytime fatigue at school or home are cause for concern. When these symptoms are present, an evaluation is warranted.
Sleep in healthy youth: Adolescence
The National Sleep Foundation recommends adolescents obtain 8 to 10 hours of sleep per night; for some adolescents, as much as 11 hours of sleep per night might be appropriate.8 However, this contrasts with findings from the National Sleep Foundation’s Sleep in America Poll, which revealed that 75% of 12th graders report <8 hours of sleep nightly.10 Many adolescents experience delayed sleep phase syndrome or delayed sleep-wake phase disorder, which involves a persistent phase shift of >2 hours in the sleep-wake schedule that conflicts with the adolescent’s school, work, or lifestyle demands.11 Such circadian rhythm disorders typically result from a poor match between the sleep-wake schedule and the demands of the adolescent’s life, or a failure to synchronize their internal clock with a 24-hour circadian clock.12 Children typically become tired after sunset, but puberty is associated with reduced slow-wave sleep and changes in circadian rhythms. As a result, a 3-hour delay (delayed phase preference) is common among adolescents. At approximately age 20, people start to become tired after sunset and awaken earlier in the morning—a pattern driven by sunlight and the timing of melatonin release that will remain stable until the sixth decade of life.
Continue to: Effects of chronic sleep deprivation...
Effects of chronic sleep deprivation
Most older studies of sleep loss examined the impact of total sleep loss (sleep deprivation) rather than the effect of partial sleep loss or sleep restriction, a more commonly experienced phenomenon. More recent research shows that a cumulative sleep deficit could cause the body to override voluntary wakefulness and a sleep-deprived individual can experience brief “microsleeps” where they are unaware and lose attention/wakefulness for several seconds.2 This can be deadly if a sleep-deprived adolescent experiences microsleeps while driving.13
There is a well-studied correlation between chronic sleep deprivation and increased body mass index in children.14 This might be caused by reduction in physical activity as well as alterations in the “hunger hormones”—ghrelin and leptin—that have been observed with sleep deprivation.15-17 Other studies have noted decreased glucose tolerance, reduced insulin sensitivity, and catecholamine and cortisol secretion abnormalities, which place children at higher risk for metabolic syndrome and hypertension.13,18 Sleep deprivation also is associated with mood and anxiety disorders and is an independent risk factor for substance use and suicidal ideation among adolescents.19 Sleep deprivation increases impairments in impulse control, concentration, and attention, which could be especially problematic in school-age children.
How sleep is assessed
The sleep history is the first step in evaluating a child or adolescent for a sleep disorder. The sleep history includes exploring the chief complaint, sleep patterns and schedules, bedtime routines, and nocturnal and daytime behaviors (Table).

Chief complaint
Behavioral sleep specialists will assess the primary problem with everyone involved in the child’s bedtime.20 This might include parents (custodial and noncustodial), grandparents, or stepparents as well as the child/adolescent. This important step can reveal a sleep disorder or an inappropriately early bedtime relative to the child’s development. During this assessment, ask detailed questions about how long the sleep problem has persisted, the frequency of sleep problems, and any precipitating stressors. Parents and caregivers can review strategies they have tried, and for how long and to what extent interventions were implemented consistently to result in change.
Sleep patterns and schedules
Review the child/adolescent’s typical sleep patterns and behaviors. Ask parents and caregivers, as well as the patient, about general sleep schedules for the past few weeks or a typical 2-week time period.2 A behavioral assessment of sleep should include asking families about how the child/adolescent sleeps during the week and over the weekend, and if school-year sleep differs from summer or holiday sleep schedules. These questions can illuminate how long a sleep problem has been occurring and what sleep habits might be contributing to the problem. Bedtime
Determine if there is a set bedtime or if the child goes to bed when they wish. It is important to ascertain if the bedtime is age-appropriate, if weekday and weekend bedtimes differ, and to what extent extracurricular activities or school demands impact bedtime. Assess the consistency of the bedtime, the nature of bedtime routines (eg, is the child engaging in stimulating activities before bed), where the bedtime routine occurs (eg, sibling’s room, parents’ room, child’s room), and what role (if any) electronic devices play.2
Nocturnal behaviors
Assessment should include a series of questions and age-specific questionnaires to focus on what behaviors occur at night, including awakenings. Parents should be asked how frequent night awakenings occur, how long arousals last, and how the child signals for the parent (eg, calling out, climbing into parents’ bed).2 Additionally, ask how parents respond and what is required to help the child fall back asleep (eg, rocking, soothing, feeding). The presence of nightmares, night terrors, parasomnias, and sleep-related breathing disorders also must be assessed.20
Daytime behaviors
A sleep history should include assessment of daytime functioning, including daytime sleepiness, fatigue, morning waking, and functioning during school, extracurriculars, and homework. For children and teens, falling asleep in the car, while in school, or during passive activities (meals, conversation) suggests insufficient sleep, sleep disruption, or excessive daytime sleepiness.2
Continue to: Sleep disruption in youth with psychiatric disorders...
Sleep disruption in youth with psychiatric disorders
Disordered sleep is common across psychiatric disorders. The National Comorbidity Survey Adolescent Supplement—a nationally representative cross-sectional survey of adolescents (N = 10,123)—found that a later weeknight bedtime, shorter weeknight sleep duration, and greater weekend bedtime delay increased the risk of developing a mood, anxiety, or substance use (including nicotine) disorder, and suicidality. These risk factors also were associated with lower “perceived mental and physical health.”21 Clinicians should routinely obtain a sleep history in children and adolescents with these disorders. Consider using the sleep screening tool BEARS:
- Bedtime issues
- Excessive daytime sleepiness
- Awakenings
- Regularity and duration of sleep
- Snoring.
ADHD
Up to one-half of children and adolescents with ADHD experience sleep problems,22,23 including delayed sleep onset, bedtime resistance, daytime fatigue, and feeling groggy in the morning beyond what is typical (>20 minutes). Pharmacotherapy for ADHD contributes to sleep disturbances24,25 while sleep deprivation exacerbates inattention and hyperactivity. In youth with ADHD, restless leg syndrome, periodic limb movement disorder, and sleep-disordered breathing disorder are more common than in the general population.
Depressive disorders
Up to three-quarters of depressed children and 90% of depressed adolescents report sleep disturbances, including initial, middle, and terminal insomnia as well as hypersomnia.26 Disrupted sleep in pediatric patients with major depressive disorder could be moderated by the patient’s age, with depressive symptoms more common among adolescents (age 12 to 17) than among younger children (age 6 to 11).27 Successful treatment of depression fails to relieve dyssomnia in 10% of children. Sleep problems that persist after successfully treating a depressive episode could increase the risk of another depressive episode.28
Anxiety disorders
Sleep problems are common among children and adolescents with anxiety disorders.29 Longitudinal data from >900 children found that symptoms of sleep disturbance in early childhood were correlated with experiencing an anxiety disorder 20 years later.30 Fears related to the dark or monsters under the bed that are developmentally appropriate for younger children may interfere with sleep. However, in anxious children, fears might also be related to separation, sleeping alone, worry about the loss of a loved one, concerns about personal safety, fear of frightening dreams, or concerns about academics and social relationships. Anxious individuals ruminate about their worries, and this might be especially true for children at bedtime, when there are limited distractions from ruminative fears.31 Bedtime resistance, parental involvement in bedtime rituals, and cultural factors related to sleep also could play a role for children with anxiety symptoms and sleep problems.
Having an anxiety disorder is significantly associated with an increased risk of insomnia; however, 73% of the time anxiety symptoms precede an insomnia diagnosis.29 Sleep problems and anxiety symptoms might have a reciprocal influence on one another; tiredness that results from sleep problems could exacerbate anxiety, which further worsens sleep problems.
A bridge to treatment
A thorough assessment can help identify modifiable factors and guide treatment selections. In Part 2 of this article, we will describe healthy sleep practices, cognitive-behavioral therapy for insomnia, when pharmacotherapy might be indicated, and the evidence supporting several medications commonly used to treat pediatric insomnia. We also will discuss factors to consider when seeking consultation with a pediatric behavioral sleep specialist.
1. Meltzer LJ, Mindell JA. Systematic review and meta-analysis of behavioral interventions for pediatric insomnia. J Pediatr Psychol. 2014;39(8):932-948. doi:10.1093/jpepsy/jsu041
2. Owens JA, Mindell JA. Pediatric insomnia. Pediatr Clin North Am. 2011;58(3):555-569. doi:10.1016/j.pcl.2011.03.011
3. Meltzer LJ, Plaufcan MR, Thomas JH, et al. Sleep problems and sleep disorders in pediatric primary care: treatment recommendations, persistence, and health care utilization. J Clin Sleep Med. 2014;10(4):421-426. doi:10.5664/jcsm.3620
4. Moore M, Meltzer LJ, Mindell JA. Bedtime problems and night wakings in children. Prim Care. 2008;35(3):569-581, viii. doi:10.1016/j.pop.2008.06.002
5. Williamson AA, Mindell JA, Hiscock H, et al. Longitudinal sleep problem trajectories are associated with multiple impairments in child well-being. J Child Psychol Psychiatry. 2020;61(10):1092-1103. doi:10.1111/jcpp.13303
6. Roberts RE, Roberts CR, Chen IG. Impact of insomnia on future functioning of adolescents. J Psychosom Res. 2002; 53(1):561-569. doi:10.1016/s0022-3999(02)00446-4
7. Singareddy R, Krishnamurthy VB, Vgontzas AN, et al. Subjective and objective sleep and self-harm behaviors in young children: a general population study. Psychiatry Res. 2013;209(3):549-553. doi:10.1016/j.psychres.2013.03.036
8. Hirshkowitz M, Whiton K, Albert SM, et al. National Sleep Foundation’s updated sleep duration recommendations: final report. Sleep Health. 2015;1(4):233-243. doi:10.1016/j.sleh.2015.10.004
9. Calamaro CJ, Mason TBA, Ratcliffe SJ. Adolescents living the 24/7 lifestyle: Effects of caffeine and technology on sleep duration and daytime functioning. Pediatrics. 2009;123(6):e1005-1010. doi:10.1542/peds.2008-3641
10. Mindell JA, Owens JA, Carskadon MA. Developmental features of sleep. Child Adolesc Psychiatr Clin N Am. 1999;8(4):695-725.
11. Moore M, Meltzer LJ. The sleepy adolescent: causes and consequences of sleepiness in teens. Paediatr Respir Rev. 2008;9(2):114-120. doi:10.1016/j.prrv.2008.01.001
12. Crowley SJ, Acebo C, Carskadon MA. Sleep, circadian rhythms, and delayed phase in adolescence. Sleep Med. 2007;8(6):602-612. doi:10.1016/j.sleep.2006.12.002
13. Millman RP; Working Group on Sleepiness in Adolescents/Young Adults; AAP Committee on Adolescence. Excessive sleepiness in adolescents and young adults: causes, consequences, and treatment strategies. Pediatrics. 2005;115(6):1774-1786. doi:10.1542/peds.2005-0772
14. Kaczor M, Skalski M. Prevalence and consequences of insomnia in pediatric population. Psychiatr Pol. 2016;50(3):555-569. doi:10.12740/PP/61226
15. Gomes TN, Dos Santos FK, Santos D, et al. Correlates of sedentary time in children: a multilevel modelling approach. BMC Public Health. 2014;14:890. doi:10.1186/1471-2458-14-890
16. Stone MR, Stevens D, Faulkner GEJ. Maintaining recommended sleep throughout the week is associated with increased physical activity in children. Prev Med. 2013;56(2):112-117. doi:10.1016/j.ypmed.2012.11.015
17. Hart CN, Fava JL, Subak LL, et al. Time in bed is associated with decreased physical activity and higher BMI in women seeking weight loss treatment. ISRN Obes. 2012;2012:320157. doi:10.5402/2012/320157
18. Tasali E, Leproult R, Ehrmann DA, et al. Slow-wave sleep and the risk of type 2 diabetes in humans. Proc Natl Acad Sci U S A. 2008;105(3):1044-1049. doi:10.1073/pnas.0706446105
19. de Zambotti M, Goldstone A, Colrain IM, et al. Insomnia disorder in adolescence: diagnosis, impact, and treatment. Sleep Med Rev. 2018;39:12-24. doi:10.1016/j.smrv.2017.06.009
20. Mindell JA, Owens JA. A clinical guide to pediatric sleep: diagnosis and management of sleep problems. 3rd ed. Lippincott Williams & Wilkins; 2015.
21. Zhang J, Paksarian D, Lamers F, et al. Sleep patterns and mental health correlates in US adolescents. J Pediatr. 2017;182:137-143. doi:10.1016/j.jpeds.2016.11.007
22. Gregory AM, Agnew-Blais JC, Matthews T, et al. ADHD and sleep quality: longitudinal analyses from childhood to early adulthood in a twin cohort. J Clin Child Adolesc Psychol. 2017;46(2):284-294. doi:10.1080/15374416.2016.1183499
23. Weiss MD, Salpekar J. Sleep problems in the child with attention-deficit hyperactivity disorder: Defining aetiology and appropriate treatments. CNS Drugs. 2010;24(10):811-828. doi:10.2165/11538990-000000000-00000
24. Galland BC, Tripp EG, Taylor BJ. The sleep of children with attention deficit hyperactivity disorder on and off methylphenidate: a matched case-control study. J Sleep Res. 2010;19(2):366-373. doi:10.1111/j.1365-2869.2009.00795.x
25. Becker SP, Froehlich TE, Epstein JN. Effects of methylphenidate on sleep functioning in children with attention-deficit/hyperactivity disorder. J Dev Behav Pediatr. 2016;37(5):395-404. doi:10.1097/DBP.0000000000000285
26. Roberts RE, Duong HT. Depression and insomnia among adolescents: a prospective perspective. J Affect Disord. 2013;148(1):66-71. doi:10.1016/j.jad.2012.11.049
27. Emslie GJ, Rush AJ, Weinberg WA, et al. Sleep EEG features of adolescents with major depression. Biol Psychiatry. 1994;36(9):573-581. doi:10.1016/0006-3223(94)90067-1
28. Alfano CA, Zakem AH, Costa NM, et al. Sleep problems and their relation to cognitive factors, anxiety, and depressive symptoms in children and adolescents. Depress Anxiety. 2009;26(6):503-512. doi:10.1002/da.20443
29. Alfano CA, Ginsburg GS, Kingery JN. Sleep-related problems among children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):224-232. doi:10.1097/01.chi.0000242233.06011.8e
30. Gregory AM, Caspi A, Eley TC, et al. Prospective longitudinal associations between persistent sleep problems in childhood and anxiety and depression disorders in adulthood. J Abnorm Child Psychol. 2005;33(2):157-163. doi: 10.1007/s10802-005-1824-0
31. Chorney DB, Detweiler MF, Morris TL, et al. The interplay of sleep disturbance, anxiety, and depression in children. J Pediatr Psychol. 2008;33(4):339-348. doi:10.1093/jpepsy/jsm105
32. Sadeh A. Stress, trauma, and sleep in children. Child Adolesc Psychiatr Clin N Am. 1996;5(3):685-700. doi:10.1016/S1056-4993(18)30356-0

33. Glod CA, Teicher MH, Hartman CR, et al. Increased nocturnal activity and impaired sleep maintenance in abused children. J Am Acad Child Adolesc Psychiatry. 1997;36(9):1236-1243. doi:10.1097/00004583-199709000-00016
34. Strawn JR, Lu L, Peris TS, et al. Research review: pediatric anxiety disorders: what have we learnt in the last 10 years? J Child Psychol Psychiatry. 2021;62(2):114-139. doi:10.1111/jcpp.13262
35. Wehry AM, Beesdo-Baum K, Hennelly MM, et al. Assessment and treatment of anxiety disorders in children and adolescents. Curr Psychiatry Rep. 2015;17(7):52. doi:10.1007/s11920-015-0591-z
36. Hamill Skoch S, Mills JA, Ramsey L, et al. Letter to editor: sleep disturbances in selective serotonin reuptake inhibitor-treated youth with anxiety disorders and obsessive compulsive disorder— a bayesian hierarchical modeling meta-analysis. J Child Adolesc Psychopharmacol. 2021;31(5):387-388. doi:10.1089/cap.2020.0169
FIRST OF 2 PARTS
A thorough evaluation can identify modifiable factors and guide treatment
Sleep problems are common among children and adolescents,1 with prevalence rates of 25% to 40%.2-4 Young children most commonly exhibit what is referred to as bedtime problems and night wakenings, whereas children in middle childhood (age 4 to 12) through adolescence (age 13 to 17) report insomnia. For many children, these problems persist.3 Insufficient sleep in children and adolescents worsens inattention, daytime fatigue, and cognitive and behavioral deficits.5 Assessment and treatment of sleep problems in children and adolescents is critical because poor sleep among youth increases the risk for depression, self-harm, and suicide,6,7 increases family stress, and decreases parental well-being.1
This 2-part article describes the assessment, diagnosis, and treatment of sleep problems among children and adolescents. In part 1, we focus on:
- sleep architecture (circadian rhythms, stages of sleep)
- sleep in healthy youth (age 6 to 17) and those with attention-deficit/hyperactivity disorder (ADHD), depressive disorders, and anxiety
- how to assess sleep, and the differential diagnosis of behavioral sleep problems in pediatric patients.
In Part 2, we will cover psychotherapeutic and psychopharmacologic interventions for youth with insomnia, and describe an effective approach to consultation with pediatric sleep medicine specialists.
How much sleep do children and adolescents need?
Throughout their development, children spend 40% to 50% of their time asleep. Sleep schedules are based on circadian rhythms, which are physical, mental, and behavioral changes that follow an approximately 24-hour cycle. Human circadian rhythm varies between 24 and 25 hours and is vital in determining our sleep patterns. Exposure to sunlight drives our circadian rhythm, sending signals to our bodies to “turn on” melatonin production at night (ie, 9
Box
Sleep architecture consists of 3 states: wake; non-rapid eye movement (NREM) sleep; and rapid eye movement (REM) sleep (“dreaming” sleep).2 These stages have distinct polysomnographic features of electroencephalographic EEG patterns, eye movements, and muscle tone.2 NREM sleep can be further divided into 3 stages: stage 1 (N1), stage 2 (N2), and stage 3 (N3). Stage 1 is the lightest stage and lasts for 30 seconds to 5 minutes; it is easy to wake up from stage 1 sleep. During stage 2 sleep, the body moves into a deeper sleep stage that is considered “true” sleep. This sleep stage is characterized by bursts of rhythmic rapid EEG activity known as spindles, as well as high-amplitude slow-wave spikes called K complexes.2 Stage 2 sleep lasts for 10 to 45 minutes. Stage 3, better known as “deep sleep,” slow-wave sleep, or delta sleep, is the most restorative sleep.2 Respiration is low and parasympathetic activity is high.2 It is difficult to be awakened during deep sleep, and if aroused, the person likely will feel confused or groggy. Deep sleep is followed by a return to lighter stage of sleep before the first REM sleep period begins.
REM sleep is the active stage of sleep. Breathing and heart rate become irregular, and the body experiences muscle atonia, or temporary paralysis, of arms and legs. When in REM sleep, individuals have the highest brain metabolic rates, and periodic bursts of eye movements.2 Most individuals move through stages of NREM and REM sleep in predicable ways, meaning they experience NREM sleep, return to a lighter stage of sleep after deep sleep, then move into REM sleep before the cycle repeats. It takes approximately 90 minutes for most adults to complete the NREM sleep cycle, and then REM sleep occurs before returning to NREM sleep.
In children, especially in infants and babies, sleep cycles are closer to 50 to 60 minutes. Newborns spend approximately 50% of their sleep in REM sleep, whereas adults spend 20% to 25% of their sleep in REM sleep. Children will spend more time in REM sleep until the third and fourth years of life, at which point REM gradually decreases to 20% to 25% by adulthood.
Sleep needs also change predictably throughout the lifespan. The National Sleep Foundation guidelines for sleep duration provide clinicians and parents with a range of recommended sleep for each stage of development. Infants require 14 to 17 hours of sleep, whereas adolescents need 8 to 10 hours by age 14 to 17.8 The key for clinicians is to determine if the child is within the recommended range, and how they are functioning on the number of hours of sleep they report. This allows for variation in how much sleep an individual child might need while acknowledging that some children within a specific age group might need more or less sleep than other children of the same age.
Sleep in healthy youth: Middle childhood
School-age children (age 6 to 12) typically need 9 to 10 hours of sleep over a 24-hour period.2 This developmental period is especially important for children to develop healthy sleep habits; however, developmentally appropriate cognitive and social/emotional factors might interfere with the quality and quantity of sleep. Middle childhood is a time when children can understand the dangers of the outside world (ie, violence, health problems) and resulting anxiety can disrupt sleep. Parents usually are less involved in bedtime as children approach adolescence, which leads to later bedtimes. At this stage, many children begin to take on more serious roles in their academics and extracurricular activities, peer relationships become more important, and use of electronics (eg, television, video games, internet, and handheld devices) increases—all of which compete with sleep.9 Frequent sleep issues during middle childhood include:
- irregular sleep-wake schedules
- later bedtimes
- decreased nighttime sleep
- increased caffeine intake
- reduced parental presence at bedtime
- daytime sleepiness.3
In school-age children, regular napping, falling asleep during short car rides, and daytime fatigue at school or home are cause for concern. When these symptoms are present, an evaluation is warranted.
Sleep in healthy youth: Adolescence
The National Sleep Foundation recommends adolescents obtain 8 to 10 hours of sleep per night; for some adolescents, as much as 11 hours of sleep per night might be appropriate.8 However, this contrasts with findings from the National Sleep Foundation’s Sleep in America Poll, which revealed that 75% of 12th graders report <8 hours of sleep nightly.10 Many adolescents experience delayed sleep phase syndrome or delayed sleep-wake phase disorder, which involves a persistent phase shift of >2 hours in the sleep-wake schedule that conflicts with the adolescent’s school, work, or lifestyle demands.11 Such circadian rhythm disorders typically result from a poor match between the sleep-wake schedule and the demands of the adolescent’s life, or a failure to synchronize their internal clock with a 24-hour circadian clock.12 Children typically become tired after sunset, but puberty is associated with reduced slow-wave sleep and changes in circadian rhythms. As a result, a 3-hour delay (delayed phase preference) is common among adolescents. At approximately age 20, people start to become tired after sunset and awaken earlier in the morning—a pattern driven by sunlight and the timing of melatonin release that will remain stable until the sixth decade of life.
Continue to: Effects of chronic sleep deprivation...
Effects of chronic sleep deprivation
Most older studies of sleep loss examined the impact of total sleep loss (sleep deprivation) rather than the effect of partial sleep loss or sleep restriction, a more commonly experienced phenomenon. More recent research shows that a cumulative sleep deficit could cause the body to override voluntary wakefulness and a sleep-deprived individual can experience brief “microsleeps” where they are unaware and lose attention/wakefulness for several seconds.2 This can be deadly if a sleep-deprived adolescent experiences microsleeps while driving.13
There is a well-studied correlation between chronic sleep deprivation and increased body mass index in children.14 This might be caused by reduction in physical activity as well as alterations in the “hunger hormones”—ghrelin and leptin—that have been observed with sleep deprivation.15-17 Other studies have noted decreased glucose tolerance, reduced insulin sensitivity, and catecholamine and cortisol secretion abnormalities, which place children at higher risk for metabolic syndrome and hypertension.13,18 Sleep deprivation also is associated with mood and anxiety disorders and is an independent risk factor for substance use and suicidal ideation among adolescents.19 Sleep deprivation increases impairments in impulse control, concentration, and attention, which could be especially problematic in school-age children.
How sleep is assessed
The sleep history is the first step in evaluating a child or adolescent for a sleep disorder. The sleep history includes exploring the chief complaint, sleep patterns and schedules, bedtime routines, and nocturnal and daytime behaviors (Table).
Chief complaint
Behavioral sleep specialists will assess the primary problem with everyone involved in the child’s bedtime.20 This might include parents (custodial and noncustodial), grandparents, or stepparents as well as the child/adolescent. This important step can reveal a sleep disorder or an inappropriately early bedtime relative to the child’s development. During this assessment, ask detailed questions about how long the sleep problem has persisted, the frequency of sleep problems, and any precipitating stressors. Parents and caregivers can review strategies they have tried, and for how long and to what extent interventions were implemented consistently to result in change.
Sleep patterns and schedules
Review the child/adolescent’s typical sleep patterns and behaviors. Ask parents and caregivers, as well as the patient, about general sleep schedules for the past few weeks or a typical 2-week time period.2 A behavioral assessment of sleep should include asking families about how the child/adolescent sleeps during the week and over the weekend, and if school-year sleep differs from summer or holiday sleep schedules. These questions can illuminate how long a sleep problem has been occurring and what sleep habits might be contributing to the problem. Bedtime
Determine if there is a set bedtime or if the child goes to bed when they wish. It is important to ascertain if the bedtime is age-appropriate, if weekday and weekend bedtimes differ, and to what extent extracurricular activities or school demands impact bedtime. Assess the consistency of the bedtime, the nature of bedtime routines (eg, is the child engaging in stimulating activities before bed), where the bedtime routine occurs (eg, sibling’s room, parents’ room, child’s room), and what role (if any) electronic devices play.2
Nocturnal behaviors
Assessment should include a series of questions and age-specific questionnaires to focus on what behaviors occur at night, including awakenings. Parents should be asked how frequent night awakenings occur, how long arousals last, and how the child signals for the parent (eg, calling out, climbing into parents’ bed).2 Additionally, ask how parents respond and what is required to help the child fall back asleep (eg, rocking, soothing, feeding). The presence of nightmares, night terrors, parasomnias, and sleep-related breathing disorders also must be assessed.20
Daytime behaviors
A sleep history should include assessment of daytime functioning, including daytime sleepiness, fatigue, morning waking, and functioning during school, extracurriculars, and homework. For children and teens, falling asleep in the car, while in school, or during passive activities (meals, conversation) suggests insufficient sleep, sleep disruption, or excessive daytime sleepiness.2
Continue to: Sleep disruption in youth with psychiatric disorders...
Sleep disruption in youth with psychiatric disorders
Disordered sleep is common across psychiatric disorders. The National Comorbidity Survey Adolescent Supplement—a nationally representative cross-sectional survey of adolescents (N = 10,123)—found that a later weeknight bedtime, shorter weeknight sleep duration, and greater weekend bedtime delay increased the risk of developing a mood, anxiety, or substance use (including nicotine) disorder, and suicidality. These risk factors also were associated with lower “perceived mental and physical health.”21 Clinicians should routinely obtain a sleep history in children and adolescents with these disorders. Consider using the sleep screening tool BEARS:
- Bedtime issues
- Excessive daytime sleepiness
- Awakenings
- Regularity and duration of sleep
- Snoring.
ADHD
Up to one-half of children and adolescents with ADHD experience sleep problems,22,23 including delayed sleep onset, bedtime resistance, daytime fatigue, and feeling groggy in the morning beyond what is typical (>20 minutes). Pharmacotherapy for ADHD contributes to sleep disturbances24,25 while sleep deprivation exacerbates inattention and hyperactivity. In youth with ADHD, restless leg syndrome, periodic limb movement disorder, and sleep-disordered breathing disorder are more common than in the general population.
Depressive disorders
Up to three-quarters of depressed children and 90% of depressed adolescents report sleep disturbances, including initial, middle, and terminal insomnia as well as hypersomnia.26 Disrupted sleep in pediatric patients with major depressive disorder could be moderated by the patient’s age, with depressive symptoms more common among adolescents (age 12 to 17) than among younger children (age 6 to 11).27 Successful treatment of depression fails to relieve dyssomnia in 10% of children. Sleep problems that persist after successfully treating a depressive episode could increase the risk of another depressive episode.28
Anxiety disorders
Sleep problems are common among children and adolescents with anxiety disorders.29 Longitudinal data from >900 children found that symptoms of sleep disturbance in early childhood were correlated with experiencing an anxiety disorder 20 years later.30 Fears related to the dark or monsters under the bed that are developmentally appropriate for younger children may interfere with sleep. However, in anxious children, fears might also be related to separation, sleeping alone, worry about the loss of a loved one, concerns about personal safety, fear of frightening dreams, or concerns about academics and social relationships. Anxious individuals ruminate about their worries, and this might be especially true for children at bedtime, when there are limited distractions from ruminative fears.31 Bedtime resistance, parental involvement in bedtime rituals, and cultural factors related to sleep also could play a role for children with anxiety symptoms and sleep problems.
Having an anxiety disorder is significantly associated with an increased risk of insomnia; however, 73% of the time anxiety symptoms precede an insomnia diagnosis.29 Sleep problems and anxiety symptoms might have a reciprocal influence on one another; tiredness that results from sleep problems could exacerbate anxiety, which further worsens sleep problems.
A bridge to treatment
A thorough assessment can help identify modifiable factors and guide treatment selections. In Part 2 of this article, we will describe healthy sleep practices, cognitive-behavioral therapy for insomnia, when pharmacotherapy might be indicated, and the evidence supporting several medications commonly used to treat pediatric insomnia. We also will discuss factors to consider when seeking consultation with a pediatric behavioral sleep specialist.
FIRST OF 2 PARTS
A thorough evaluation can identify modifiable factors and guide treatment
Sleep problems are common among children and adolescents,1 with prevalence rates of 25% to 40%.2-4 Young children most commonly exhibit what is referred to as bedtime problems and night wakenings, whereas children in middle childhood (age 4 to 12) through adolescence (age 13 to 17) report insomnia. For many children, these problems persist.3 Insufficient sleep in children and adolescents worsens inattention, daytime fatigue, and cognitive and behavioral deficits.5 Assessment and treatment of sleep problems in children and adolescents is critical because poor sleep among youth increases the risk for depression, self-harm, and suicide,6,7 increases family stress, and decreases parental well-being.1
This 2-part article describes the assessment, diagnosis, and treatment of sleep problems among children and adolescents. In part 1, we focus on:
- sleep architecture (circadian rhythms, stages of sleep)
- sleep in healthy youth (age 6 to 17) and those with attention-deficit/hyperactivity disorder (ADHD), depressive disorders, and anxiety
- how to assess sleep, and the differential diagnosis of behavioral sleep problems in pediatric patients.
In Part 2, we will cover psychotherapeutic and psychopharmacologic interventions for youth with insomnia, and describe an effective approach to consultation with pediatric sleep medicine specialists.
How much sleep do children and adolescents need?
Throughout their development, children spend 40% to 50% of their time asleep. Sleep schedules are based on circadian rhythms, which are physical, mental, and behavioral changes that follow an approximately 24-hour cycle. Human circadian rhythm varies between 24 and 25 hours and is vital in determining our sleep patterns. Exposure to sunlight drives our circadian rhythm, sending signals to our bodies to “turn on” melatonin production at night (ie, 9
Box
Sleep architecture consists of 3 states: wake; non-rapid eye movement (NREM) sleep; and rapid eye movement (REM) sleep (“dreaming” sleep).2 These stages have distinct polysomnographic features of electroencephalographic EEG patterns, eye movements, and muscle tone.2 NREM sleep can be further divided into 3 stages: stage 1 (N1), stage 2 (N2), and stage 3 (N3). Stage 1 is the lightest stage and lasts for 30 seconds to 5 minutes; it is easy to wake up from stage 1 sleep. During stage 2 sleep, the body moves into a deeper sleep stage that is considered “true” sleep. This sleep stage is characterized by bursts of rhythmic rapid EEG activity known as spindles, as well as high-amplitude slow-wave spikes called K complexes.2 Stage 2 sleep lasts for 10 to 45 minutes. Stage 3, better known as “deep sleep,” slow-wave sleep, or delta sleep, is the most restorative sleep.2 Respiration is low and parasympathetic activity is high.2 It is difficult to be awakened during deep sleep, and if aroused, the person likely will feel confused or groggy. Deep sleep is followed by a return to lighter stage of sleep before the first REM sleep period begins.
REM sleep is the active stage of sleep. Breathing and heart rate become irregular, and the body experiences muscle atonia, or temporary paralysis, of arms and legs. When in REM sleep, individuals have the highest brain metabolic rates, and periodic bursts of eye movements.2 Most individuals move through stages of NREM and REM sleep in predicable ways, meaning they experience NREM sleep, return to a lighter stage of sleep after deep sleep, then move into REM sleep before the cycle repeats. It takes approximately 90 minutes for most adults to complete the NREM sleep cycle, and then REM sleep occurs before returning to NREM sleep.
In children, especially in infants and babies, sleep cycles are closer to 50 to 60 minutes. Newborns spend approximately 50% of their sleep in REM sleep, whereas adults spend 20% to 25% of their sleep in REM sleep. Children will spend more time in REM sleep until the third and fourth years of life, at which point REM gradually decreases to 20% to 25% by adulthood.
Sleep needs also change predictably throughout the lifespan. The National Sleep Foundation guidelines for sleep duration provide clinicians and parents with a range of recommended sleep for each stage of development. Infants require 14 to 17 hours of sleep, whereas adolescents need 8 to 10 hours by age 14 to 17.8 The key for clinicians is to determine if the child is within the recommended range, and how they are functioning on the number of hours of sleep they report. This allows for variation in how much sleep an individual child might need while acknowledging that some children within a specific age group might need more or less sleep than other children of the same age.
Sleep in healthy youth: Middle childhood
School-age children (age 6 to 12) typically need 9 to 10 hours of sleep over a 24-hour period.2 This developmental period is especially important for children to develop healthy sleep habits; however, developmentally appropriate cognitive and social/emotional factors might interfere with the quality and quantity of sleep. Middle childhood is a time when children can understand the dangers of the outside world (ie, violence, health problems) and resulting anxiety can disrupt sleep. Parents usually are less involved in bedtime as children approach adolescence, which leads to later bedtimes. At this stage, many children begin to take on more serious roles in their academics and extracurricular activities, peer relationships become more important, and use of electronics (eg, television, video games, internet, and handheld devices) increases—all of which compete with sleep.9 Frequent sleep issues during middle childhood include:
- irregular sleep-wake schedules
- later bedtimes
- decreased nighttime sleep
- increased caffeine intake
- reduced parental presence at bedtime
- daytime sleepiness.3
In school-age children, regular napping, falling asleep during short car rides, and daytime fatigue at school or home are cause for concern. When these symptoms are present, an evaluation is warranted.
Sleep in healthy youth: Adolescence
The National Sleep Foundation recommends adolescents obtain 8 to 10 hours of sleep per night; for some adolescents, as much as 11 hours of sleep per night might be appropriate.8 However, this contrasts with findings from the National Sleep Foundation’s Sleep in America Poll, which revealed that 75% of 12th graders report <8 hours of sleep nightly.10 Many adolescents experience delayed sleep phase syndrome or delayed sleep-wake phase disorder, which involves a persistent phase shift of >2 hours in the sleep-wake schedule that conflicts with the adolescent’s school, work, or lifestyle demands.11 Such circadian rhythm disorders typically result from a poor match between the sleep-wake schedule and the demands of the adolescent’s life, or a failure to synchronize their internal clock with a 24-hour circadian clock.12 Children typically become tired after sunset, but puberty is associated with reduced slow-wave sleep and changes in circadian rhythms. As a result, a 3-hour delay (delayed phase preference) is common among adolescents. At approximately age 20, people start to become tired after sunset and awaken earlier in the morning—a pattern driven by sunlight and the timing of melatonin release that will remain stable until the sixth decade of life.
Continue to: Effects of chronic sleep deprivation...
Effects of chronic sleep deprivation
Most older studies of sleep loss examined the impact of total sleep loss (sleep deprivation) rather than the effect of partial sleep loss or sleep restriction, a more commonly experienced phenomenon. More recent research shows that a cumulative sleep deficit could cause the body to override voluntary wakefulness and a sleep-deprived individual can experience brief “microsleeps” where they are unaware and lose attention/wakefulness for several seconds.2 This can be deadly if a sleep-deprived adolescent experiences microsleeps while driving.13
There is a well-studied correlation between chronic sleep deprivation and increased body mass index in children.14 This might be caused by reduction in physical activity as well as alterations in the “hunger hormones”—ghrelin and leptin—that have been observed with sleep deprivation.15-17 Other studies have noted decreased glucose tolerance, reduced insulin sensitivity, and catecholamine and cortisol secretion abnormalities, which place children at higher risk for metabolic syndrome and hypertension.13,18 Sleep deprivation also is associated with mood and anxiety disorders and is an independent risk factor for substance use and suicidal ideation among adolescents.19 Sleep deprivation increases impairments in impulse control, concentration, and attention, which could be especially problematic in school-age children.
How sleep is assessed
The sleep history is the first step in evaluating a child or adolescent for a sleep disorder. The sleep history includes exploring the chief complaint, sleep patterns and schedules, bedtime routines, and nocturnal and daytime behaviors (Table).
Chief complaint
Behavioral sleep specialists will assess the primary problem with everyone involved in the child’s bedtime.20 This might include parents (custodial and noncustodial), grandparents, or stepparents as well as the child/adolescent. This important step can reveal a sleep disorder or an inappropriately early bedtime relative to the child’s development. During this assessment, ask detailed questions about how long the sleep problem has persisted, the frequency of sleep problems, and any precipitating stressors. Parents and caregivers can review strategies they have tried, and for how long and to what extent interventions were implemented consistently to result in change.
Sleep patterns and schedules
Review the child/adolescent’s typical sleep patterns and behaviors. Ask parents and caregivers, as well as the patient, about general sleep schedules for the past few weeks or a typical 2-week time period.2 A behavioral assessment of sleep should include asking families about how the child/adolescent sleeps during the week and over the weekend, and if school-year sleep differs from summer or holiday sleep schedules. These questions can illuminate how long a sleep problem has been occurring and what sleep habits might be contributing to the problem. Bedtime
Determine if there is a set bedtime or if the child goes to bed when they wish. It is important to ascertain if the bedtime is age-appropriate, if weekday and weekend bedtimes differ, and to what extent extracurricular activities or school demands impact bedtime. Assess the consistency of the bedtime, the nature of bedtime routines (eg, is the child engaging in stimulating activities before bed), where the bedtime routine occurs (eg, sibling’s room, parents’ room, child’s room), and what role (if any) electronic devices play.2
Nocturnal behaviors
Assessment should include a series of questions and age-specific questionnaires to focus on what behaviors occur at night, including awakenings. Parents should be asked how frequent night awakenings occur, how long arousals last, and how the child signals for the parent (eg, calling out, climbing into parents’ bed).2 Additionally, ask how parents respond and what is required to help the child fall back asleep (eg, rocking, soothing, feeding). The presence of nightmares, night terrors, parasomnias, and sleep-related breathing disorders also must be assessed.20
Daytime behaviors
A sleep history should include assessment of daytime functioning, including daytime sleepiness, fatigue, morning waking, and functioning during school, extracurriculars, and homework. For children and teens, falling asleep in the car, while in school, or during passive activities (meals, conversation) suggests insufficient sleep, sleep disruption, or excessive daytime sleepiness.2
Continue to: Sleep disruption in youth with psychiatric disorders...
Sleep disruption in youth with psychiatric disorders
Disordered sleep is common across psychiatric disorders. The National Comorbidity Survey Adolescent Supplement—a nationally representative cross-sectional survey of adolescents (N = 10,123)—found that a later weeknight bedtime, shorter weeknight sleep duration, and greater weekend bedtime delay increased the risk of developing a mood, anxiety, or substance use (including nicotine) disorder, and suicidality. These risk factors also were associated with lower “perceived mental and physical health.”21 Clinicians should routinely obtain a sleep history in children and adolescents with these disorders. Consider using the sleep screening tool BEARS:
- Bedtime issues
- Excessive daytime sleepiness
- Awakenings
- Regularity and duration of sleep
- Snoring.
ADHD
Up to one-half of children and adolescents with ADHD experience sleep problems,22,23 including delayed sleep onset, bedtime resistance, daytime fatigue, and feeling groggy in the morning beyond what is typical (>20 minutes). Pharmacotherapy for ADHD contributes to sleep disturbances24,25 while sleep deprivation exacerbates inattention and hyperactivity. In youth with ADHD, restless leg syndrome, periodic limb movement disorder, and sleep-disordered breathing disorder are more common than in the general population.
Depressive disorders
Up to three-quarters of depressed children and 90% of depressed adolescents report sleep disturbances, including initial, middle, and terminal insomnia as well as hypersomnia.26 Disrupted sleep in pediatric patients with major depressive disorder could be moderated by the patient’s age, with depressive symptoms more common among adolescents (age 12 to 17) than among younger children (age 6 to 11).27 Successful treatment of depression fails to relieve dyssomnia in 10% of children. Sleep problems that persist after successfully treating a depressive episode could increase the risk of another depressive episode.28
Anxiety disorders
Sleep problems are common among children and adolescents with anxiety disorders.29 Longitudinal data from >900 children found that symptoms of sleep disturbance in early childhood were correlated with experiencing an anxiety disorder 20 years later.30 Fears related to the dark or monsters under the bed that are developmentally appropriate for younger children may interfere with sleep. However, in anxious children, fears might also be related to separation, sleeping alone, worry about the loss of a loved one, concerns about personal safety, fear of frightening dreams, or concerns about academics and social relationships. Anxious individuals ruminate about their worries, and this might be especially true for children at bedtime, when there are limited distractions from ruminative fears.31 Bedtime resistance, parental involvement in bedtime rituals, and cultural factors related to sleep also could play a role for children with anxiety symptoms and sleep problems.
Having an anxiety disorder is significantly associated with an increased risk of insomnia; however, 73% of the time anxiety symptoms precede an insomnia diagnosis.29 Sleep problems and anxiety symptoms might have a reciprocal influence on one another; tiredness that results from sleep problems could exacerbate anxiety, which further worsens sleep problems.
A bridge to treatment
A thorough assessment can help identify modifiable factors and guide treatment selections. In Part 2 of this article, we will describe healthy sleep practices, cognitive-behavioral therapy for insomnia, when pharmacotherapy might be indicated, and the evidence supporting several medications commonly used to treat pediatric insomnia. We also will discuss factors to consider when seeking consultation with a pediatric behavioral sleep specialist.
1. Meltzer LJ, Mindell JA. Systematic review and meta-analysis of behavioral interventions for pediatric insomnia. J Pediatr Psychol. 2014;39(8):932-948. doi:10.1093/jpepsy/jsu041
2. Owens JA, Mindell JA. Pediatric insomnia. Pediatr Clin North Am. 2011;58(3):555-569. doi:10.1016/j.pcl.2011.03.011
3. Meltzer LJ, Plaufcan MR, Thomas JH, et al. Sleep problems and sleep disorders in pediatric primary care: treatment recommendations, persistence, and health care utilization. J Clin Sleep Med. 2014;10(4):421-426. doi:10.5664/jcsm.3620
4. Moore M, Meltzer LJ, Mindell JA. Bedtime problems and night wakings in children. Prim Care. 2008;35(3):569-581, viii. doi:10.1016/j.pop.2008.06.002
5. Williamson AA, Mindell JA, Hiscock H, et al. Longitudinal sleep problem trajectories are associated with multiple impairments in child well-being. J Child Psychol Psychiatry. 2020;61(10):1092-1103. doi:10.1111/jcpp.13303
6. Roberts RE, Roberts CR, Chen IG. Impact of insomnia on future functioning of adolescents. J Psychosom Res. 2002; 53(1):561-569. doi:10.1016/s0022-3999(02)00446-4
7. Singareddy R, Krishnamurthy VB, Vgontzas AN, et al. Subjective and objective sleep and self-harm behaviors in young children: a general population study. Psychiatry Res. 2013;209(3):549-553. doi:10.1016/j.psychres.2013.03.036
8. Hirshkowitz M, Whiton K, Albert SM, et al. National Sleep Foundation’s updated sleep duration recommendations: final report. Sleep Health. 2015;1(4):233-243. doi:10.1016/j.sleh.2015.10.004
9. Calamaro CJ, Mason TBA, Ratcliffe SJ. Adolescents living the 24/7 lifestyle: Effects of caffeine and technology on sleep duration and daytime functioning. Pediatrics. 2009;123(6):e1005-1010. doi:10.1542/peds.2008-3641
10. Mindell JA, Owens JA, Carskadon MA. Developmental features of sleep. Child Adolesc Psychiatr Clin N Am. 1999;8(4):695-725.
11. Moore M, Meltzer LJ. The sleepy adolescent: causes and consequences of sleepiness in teens. Paediatr Respir Rev. 2008;9(2):114-120. doi:10.1016/j.prrv.2008.01.001
12. Crowley SJ, Acebo C, Carskadon MA. Sleep, circadian rhythms, and delayed phase in adolescence. Sleep Med. 2007;8(6):602-612. doi:10.1016/j.sleep.2006.12.002
13. Millman RP; Working Group on Sleepiness in Adolescents/Young Adults; AAP Committee on Adolescence. Excessive sleepiness in adolescents and young adults: causes, consequences, and treatment strategies. Pediatrics. 2005;115(6):1774-1786. doi:10.1542/peds.2005-0772
14. Kaczor M, Skalski M. Prevalence and consequences of insomnia in pediatric population. Psychiatr Pol. 2016;50(3):555-569. doi:10.12740/PP/61226
15. Gomes TN, Dos Santos FK, Santos D, et al. Correlates of sedentary time in children: a multilevel modelling approach. BMC Public Health. 2014;14:890. doi:10.1186/1471-2458-14-890
16. Stone MR, Stevens D, Faulkner GEJ. Maintaining recommended sleep throughout the week is associated with increased physical activity in children. Prev Med. 2013;56(2):112-117. doi:10.1016/j.ypmed.2012.11.015
17. Hart CN, Fava JL, Subak LL, et al. Time in bed is associated with decreased physical activity and higher BMI in women seeking weight loss treatment. ISRN Obes. 2012;2012:320157. doi:10.5402/2012/320157
18. Tasali E, Leproult R, Ehrmann DA, et al. Slow-wave sleep and the risk of type 2 diabetes in humans. Proc Natl Acad Sci U S A. 2008;105(3):1044-1049. doi:10.1073/pnas.0706446105
19. de Zambotti M, Goldstone A, Colrain IM, et al. Insomnia disorder in adolescence: diagnosis, impact, and treatment. Sleep Med Rev. 2018;39:12-24. doi:10.1016/j.smrv.2017.06.009
20. Mindell JA, Owens JA. A clinical guide to pediatric sleep: diagnosis and management of sleep problems. 3rd ed. Lippincott Williams & Wilkins; 2015.
21. Zhang J, Paksarian D, Lamers F, et al. Sleep patterns and mental health correlates in US adolescents. J Pediatr. 2017;182:137-143. doi:10.1016/j.jpeds.2016.11.007
22. Gregory AM, Agnew-Blais JC, Matthews T, et al. ADHD and sleep quality: longitudinal analyses from childhood to early adulthood in a twin cohort. J Clin Child Adolesc Psychol. 2017;46(2):284-294. doi:10.1080/15374416.2016.1183499
23. Weiss MD, Salpekar J. Sleep problems in the child with attention-deficit hyperactivity disorder: Defining aetiology and appropriate treatments. CNS Drugs. 2010;24(10):811-828. doi:10.2165/11538990-000000000-00000
24. Galland BC, Tripp EG, Taylor BJ. The sleep of children with attention deficit hyperactivity disorder on and off methylphenidate: a matched case-control study. J Sleep Res. 2010;19(2):366-373. doi:10.1111/j.1365-2869.2009.00795.x
25. Becker SP, Froehlich TE, Epstein JN. Effects of methylphenidate on sleep functioning in children with attention-deficit/hyperactivity disorder. J Dev Behav Pediatr. 2016;37(5):395-404. doi:10.1097/DBP.0000000000000285
26. Roberts RE, Duong HT. Depression and insomnia among adolescents: a prospective perspective. J Affect Disord. 2013;148(1):66-71. doi:10.1016/j.jad.2012.11.049
27. Emslie GJ, Rush AJ, Weinberg WA, et al. Sleep EEG features of adolescents with major depression. Biol Psychiatry. 1994;36(9):573-581. doi:10.1016/0006-3223(94)90067-1
28. Alfano CA, Zakem AH, Costa NM, et al. Sleep problems and their relation to cognitive factors, anxiety, and depressive symptoms in children and adolescents. Depress Anxiety. 2009;26(6):503-512. doi:10.1002/da.20443
29. Alfano CA, Ginsburg GS, Kingery JN. Sleep-related problems among children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):224-232. doi:10.1097/01.chi.0000242233.06011.8e
30. Gregory AM, Caspi A, Eley TC, et al. Prospective longitudinal associations between persistent sleep problems in childhood and anxiety and depression disorders in adulthood. J Abnorm Child Psychol. 2005;33(2):157-163. doi: 10.1007/s10802-005-1824-0
31. Chorney DB, Detweiler MF, Morris TL, et al. The interplay of sleep disturbance, anxiety, and depression in children. J Pediatr Psychol. 2008;33(4):339-348. doi:10.1093/jpepsy/jsm105
32. Sadeh A. Stress, trauma, and sleep in children. Child Adolesc Psychiatr Clin N Am. 1996;5(3):685-700. doi:10.1016/S1056-4993(18)30356-0
33. Glod CA, Teicher MH, Hartman CR, et al. Increased nocturnal activity and impaired sleep maintenance in abused children. J Am Acad Child Adolesc Psychiatry. 1997;36(9):1236-1243. doi:10.1097/00004583-199709000-00016
34. Strawn JR, Lu L, Peris TS, et al. Research review: pediatric anxiety disorders: what have we learnt in the last 10 years? J Child Psychol Psychiatry. 2021;62(2):114-139. doi:10.1111/jcpp.13262
35. Wehry AM, Beesdo-Baum K, Hennelly MM, et al. Assessment and treatment of anxiety disorders in children and adolescents. Curr Psychiatry Rep. 2015;17(7):52. doi:10.1007/s11920-015-0591-z
36. Hamill Skoch S, Mills JA, Ramsey L, et al. Letter to editor: sleep disturbances in selective serotonin reuptake inhibitor-treated youth with anxiety disorders and obsessive compulsive disorder— a bayesian hierarchical modeling meta-analysis. J Child Adolesc Psychopharmacol. 2021;31(5):387-388. doi:10.1089/cap.2020.0169
1. Meltzer LJ, Mindell JA. Systematic review and meta-analysis of behavioral interventions for pediatric insomnia. J Pediatr Psychol. 2014;39(8):932-948. doi:10.1093/jpepsy/jsu041
2. Owens JA, Mindell JA. Pediatric insomnia. Pediatr Clin North Am. 2011;58(3):555-569. doi:10.1016/j.pcl.2011.03.011
3. Meltzer LJ, Plaufcan MR, Thomas JH, et al. Sleep problems and sleep disorders in pediatric primary care: treatment recommendations, persistence, and health care utilization. J Clin Sleep Med. 2014;10(4):421-426. doi:10.5664/jcsm.3620
4. Moore M, Meltzer LJ, Mindell JA. Bedtime problems and night wakings in children. Prim Care. 2008;35(3):569-581, viii. doi:10.1016/j.pop.2008.06.002
5. Williamson AA, Mindell JA, Hiscock H, et al. Longitudinal sleep problem trajectories are associated with multiple impairments in child well-being. J Child Psychol Psychiatry. 2020;61(10):1092-1103. doi:10.1111/jcpp.13303
6. Roberts RE, Roberts CR, Chen IG. Impact of insomnia on future functioning of adolescents. J Psychosom Res. 2002; 53(1):561-569. doi:10.1016/s0022-3999(02)00446-4
7. Singareddy R, Krishnamurthy VB, Vgontzas AN, et al. Subjective and objective sleep and self-harm behaviors in young children: a general population study. Psychiatry Res. 2013;209(3):549-553. doi:10.1016/j.psychres.2013.03.036
8. Hirshkowitz M, Whiton K, Albert SM, et al. National Sleep Foundation’s updated sleep duration recommendations: final report. Sleep Health. 2015;1(4):233-243. doi:10.1016/j.sleh.2015.10.004
9. Calamaro CJ, Mason TBA, Ratcliffe SJ. Adolescents living the 24/7 lifestyle: Effects of caffeine and technology on sleep duration and daytime functioning. Pediatrics. 2009;123(6):e1005-1010. doi:10.1542/peds.2008-3641
10. Mindell JA, Owens JA, Carskadon MA. Developmental features of sleep. Child Adolesc Psychiatr Clin N Am. 1999;8(4):695-725.
11. Moore M, Meltzer LJ. The sleepy adolescent: causes and consequences of sleepiness in teens. Paediatr Respir Rev. 2008;9(2):114-120. doi:10.1016/j.prrv.2008.01.001
12. Crowley SJ, Acebo C, Carskadon MA. Sleep, circadian rhythms, and delayed phase in adolescence. Sleep Med. 2007;8(6):602-612. doi:10.1016/j.sleep.2006.12.002
13. Millman RP; Working Group on Sleepiness in Adolescents/Young Adults; AAP Committee on Adolescence. Excessive sleepiness in adolescents and young adults: causes, consequences, and treatment strategies. Pediatrics. 2005;115(6):1774-1786. doi:10.1542/peds.2005-0772
14. Kaczor M, Skalski M. Prevalence and consequences of insomnia in pediatric population. Psychiatr Pol. 2016;50(3):555-569. doi:10.12740/PP/61226
15. Gomes TN, Dos Santos FK, Santos D, et al. Correlates of sedentary time in children: a multilevel modelling approach. BMC Public Health. 2014;14:890. doi:10.1186/1471-2458-14-890
16. Stone MR, Stevens D, Faulkner GEJ. Maintaining recommended sleep throughout the week is associated with increased physical activity in children. Prev Med. 2013;56(2):112-117. doi:10.1016/j.ypmed.2012.11.015
17. Hart CN, Fava JL, Subak LL, et al. Time in bed is associated with decreased physical activity and higher BMI in women seeking weight loss treatment. ISRN Obes. 2012;2012:320157. doi:10.5402/2012/320157
18. Tasali E, Leproult R, Ehrmann DA, et al. Slow-wave sleep and the risk of type 2 diabetes in humans. Proc Natl Acad Sci U S A. 2008;105(3):1044-1049. doi:10.1073/pnas.0706446105
19. de Zambotti M, Goldstone A, Colrain IM, et al. Insomnia disorder in adolescence: diagnosis, impact, and treatment. Sleep Med Rev. 2018;39:12-24. doi:10.1016/j.smrv.2017.06.009
20. Mindell JA, Owens JA. A clinical guide to pediatric sleep: diagnosis and management of sleep problems. 3rd ed. Lippincott Williams & Wilkins; 2015.
21. Zhang J, Paksarian D, Lamers F, et al. Sleep patterns and mental health correlates in US adolescents. J Pediatr. 2017;182:137-143. doi:10.1016/j.jpeds.2016.11.007
22. Gregory AM, Agnew-Blais JC, Matthews T, et al. ADHD and sleep quality: longitudinal analyses from childhood to early adulthood in a twin cohort. J Clin Child Adolesc Psychol. 2017;46(2):284-294. doi:10.1080/15374416.2016.1183499
23. Weiss MD, Salpekar J. Sleep problems in the child with attention-deficit hyperactivity disorder: Defining aetiology and appropriate treatments. CNS Drugs. 2010;24(10):811-828. doi:10.2165/11538990-000000000-00000
24. Galland BC, Tripp EG, Taylor BJ. The sleep of children with attention deficit hyperactivity disorder on and off methylphenidate: a matched case-control study. J Sleep Res. 2010;19(2):366-373. doi:10.1111/j.1365-2869.2009.00795.x
25. Becker SP, Froehlich TE, Epstein JN. Effects of methylphenidate on sleep functioning in children with attention-deficit/hyperactivity disorder. J Dev Behav Pediatr. 2016;37(5):395-404. doi:10.1097/DBP.0000000000000285
26. Roberts RE, Duong HT. Depression and insomnia among adolescents: a prospective perspective. J Affect Disord. 2013;148(1):66-71. doi:10.1016/j.jad.2012.11.049
27. Emslie GJ, Rush AJ, Weinberg WA, et al. Sleep EEG features of adolescents with major depression. Biol Psychiatry. 1994;36(9):573-581. doi:10.1016/0006-3223(94)90067-1
28. Alfano CA, Zakem AH, Costa NM, et al. Sleep problems and their relation to cognitive factors, anxiety, and depressive symptoms in children and adolescents. Depress Anxiety. 2009;26(6):503-512. doi:10.1002/da.20443
29. Alfano CA, Ginsburg GS, Kingery JN. Sleep-related problems among children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):224-232. doi:10.1097/01.chi.0000242233.06011.8e
30. Gregory AM, Caspi A, Eley TC, et al. Prospective longitudinal associations between persistent sleep problems in childhood and anxiety and depression disorders in adulthood. J Abnorm Child Psychol. 2005;33(2):157-163. doi: 10.1007/s10802-005-1824-0
31. Chorney DB, Detweiler MF, Morris TL, et al. The interplay of sleep disturbance, anxiety, and depression in children. J Pediatr Psychol. 2008;33(4):339-348. doi:10.1093/jpepsy/jsm105
32. Sadeh A. Stress, trauma, and sleep in children. Child Adolesc Psychiatr Clin N Am. 1996;5(3):685-700. doi:10.1016/S1056-4993(18)30356-0
33. Glod CA, Teicher MH, Hartman CR, et al. Increased nocturnal activity and impaired sleep maintenance in abused children. J Am Acad Child Adolesc Psychiatry. 1997;36(9):1236-1243. doi:10.1097/00004583-199709000-00016
34. Strawn JR, Lu L, Peris TS, et al. Research review: pediatric anxiety disorders: what have we learnt in the last 10 years? J Child Psychol Psychiatry. 2021;62(2):114-139. doi:10.1111/jcpp.13262
35. Wehry AM, Beesdo-Baum K, Hennelly MM, et al. Assessment and treatment of anxiety disorders in children and adolescents. Curr Psychiatry Rep. 2015;17(7):52. doi:10.1007/s11920-015-0591-z
36. Hamill Skoch S, Mills JA, Ramsey L, et al. Letter to editor: sleep disturbances in selective serotonin reuptake inhibitor-treated youth with anxiety disorders and obsessive compulsive disorder— a bayesian hierarchical modeling meta-analysis. J Child Adolesc Psychopharmacol. 2021;31(5):387-388. doi:10.1089/cap.2020.0169

Lithium: An underutilized element
In clinicians and patients alike, lithium triggers reactions ranging from apprehension and fear about adverse effects and toxicity to confusion over lithium’s usefulness compared with other mood stabilizers that do not require blood monitoring. Research from the 1950s to the 1970s demonstrated that lithium is effective for prophylaxis of mood episodes in patients with bipolar disorder and could reduce the frequency of hospitalization in patients who are depressed.1 For years, lithium was commonly prescribed to treat bipolar disorder, but in recent years its use has fallen out of favor due to concerns about its risks, and the availability of newer medications. This article reviews lithium’s origins (Box1-4), pharmacology, risks, and benefits, and makes a case for why it should remain a first-line therapy for bipolar disorder.
Box
Lithium was initially used in the 1840s to treat gout. William Hammond became the first physician to prescribe lithium bromide for acute mania in 1871, and in 1894, Danish psychiatrist Frederik Lange first used lithium carbonate to treat “melancholic depression.”1 In the 20th century, lithium-containing products were used to treat rheumatologic conditions such as renal calculi and other uric acid diatheses.
Lithium experienced a revival in 1949 when John Cade expanded upon Archibald Garrod’s theory regarding uric acid and gout. As a physician during WWII, Cade observed manic and depressive behaviors among prisoners.2 Theorizing that this was caused by either an excess or lack of a metabolite, he injected urine from patients with mania, depression, and schizophrenia and from healthy individuals into guinea pigs.3 Animals who received urine from patients with mania died faster than those injected with urine from a patient with schizophrenia.2 Concluding that urea was the culprit, Cade substituted the relatively water insoluble uric acid for “the most soluble of urates,” which was lithium urate.2,3 Rather than succumbing to a quicker death, guinea pigs injected with lithium urate became placid, tranquilized, lost their natural timidity, and generally did not respond to stimulation.3
Cade administered lithium carbonate and lithium citrate to himself and, because he did not experience any unwanted effects, began testing the medication on patients. Cade’s landmark 1949 paper4 notes improvement in all 10 patients with mania but little change in 6 patients with schizophrenia and 3 with chronic depression.2
In the United States, interest in lithium did not begin until the 1960s, when Samuel Gershon introduced the medication to a psychiatric hospital in Michigan. Financed by the National Institute of Mental Health, this program bought bulk lithium from a chemical supply store, and a local pharmacy formed it into capsules. Analysis of 4 controlled studies from 1963 to 1971 showed an average response rate to lithium of 78% in 116 patients with mania.1
By the end of the 1960s, many psychiatrists were prescribing lithium. At that time, lithium was not FDA-approved, but it could be prescribed as an investigational new drug by obtaining a special permit. In 1970, the FDA approved lithium for acute mania, and for prophylaxis of mania in 1975. Lithium has not yet been approved for prophylaxis of depression, despite substantial evidence indicating efficacy.1
How lithium works
Lithium has effects on neurotransmitters implicated in mania, such as glutamate, dopamine, and gamma-aminobutyric acid.5 Quiroz et al6 provide a detailed description of lithium’s effects, which can be summarized as modulating neuronal signaling pathways, including B-cell lymphoma 2 (BCL2), cAMP-response element binding protein (CREB), and glycogen synthase kinase-3 (GSK-3). Through these signaling cascades, lithium can curtail progression of neuronal apoptosis caused by the biochemical stress commonly seen in bipolar disorder pathogenesis.6
A wide range of potential adverse effects
Lithium can cause adverse effects in several organ systems. Clinicians must be aware of these effects before prescribing lithium or continuing long-term use. The most commonly documented adverse effects and symptoms of toxicity are:
- tremor
- renal dysfunction, including renal insufficiency and polyuria or polydipsia
- hypothyroidism
- hyperparathyroidism (with subsequent hypercalcemia)
- weight gain
- gastrointestinal (GI) symptoms.
These symptoms tend to occur when lithium serum levels are outside the reference range of 0.6 to 1.2 mEq/L, typically once blood levels reach ≥1.5 mEq/L.7 However, thyroid and renal abnormalities can occur at levels below this value, and might be related to cumulative lithium exposure.7 Adverse effects usually are precipitated by inadequate water intake or inadvertently taking an extra dose. Symptoms of lithium toxicity can be mild, moderate (GI complaints, tremor, weakness, fatigue), or severe (agitation, seizures, autonomic dysregulation, confusion, coma, death).
Lithium adverse effects and toxicity are infrequent. An analysis of 17 years of data in Sweden showed the incidence of moderate to severe lithium intoxication (serum level ≥1.5 mEq/L) was .01 patients per year.8 A recently published US analysis found the prevalence rate of lithium toxicity was 2.2%.9 Results from both groups show that drug interactions were an important cause of increased lithium levels, and specifically that initiating a medication that could interact with lithium was associated with 30-fold higher risk of needing acute care for lithium toxicity.9 Possible drug interactions include nonsteroidal anti-inflammatory drugs, diuretics, and renin-angiotensin-aldosterone system inhibitors.9 Because lithium is eliminated exclusively by the kidneys, impaired or altered renal function can increase the risk of lithium retention, leading to intoxication. Other risk factors include older age, alteration of water-salt homeostasis (fever, diarrhea, vomiting), higher number of treated chronic diseases as measured by Chronic Disease Score (range: 0 to 35; higher scores denotes higher number of treated chronic diseases and increased hospitalization risk), and higher total daily lithium dosage.9
Presentation of lithium intoxication often is mild or nonspecific, and physicians should have a low threshold for checking lithium blood levels.8 Lithium intoxication can be safely managed with volume expansion, forced diuresis, and hemodialysis.
Continue to: Lithium use during pregnancy...
Lithium use during pregnancy
When considering lithium for a woman who is pregnant, it is important to weigh the potential teratogenic risks against the benefit of successful management of the mood disorder. Ebstein’s anomaly (abnormal tricuspid valve leaflets) is the most well-known teratogenic risk associated with lithium, with an estimated absolute risk of 1 in 1,000 in patients treated with lithium compared with 1 in 20,000 in controls.10,11 The risk of congenital anomalies is increased in infants exposed to lithium in utero (4% to 12% vs 2% to 4% in controls)12; exposure during the first trimester of pregnancy is associated with increased risk. Lithium levels must be adjusted during pregnancy. Pregnant patients are at higher risk of relapse to mania because renal lithium clearance increases by 30% to 50% during pregnancy, and normalizes shortly after delivery.13
Lithium exposure during pregnancy has been linked to increased risk of miscarriage and preterm delivery; however, more research is needed to define the true risk of noncardiac teratogenicity associated with lithium.11 Because there is a lack of definitive data regarding teratogenicity, and because of lithium’s well-documented effectiveness in mood disorders, lithium should be considered a first-line therapy for pregnant patients with bipolar disorder.10
Prescribing trends
Despite data showing the efficacy and benefits of lithium, there has been a paradoxical decrease in lithium prescribing. This is the result of multiple factors, including fear of adverse effects and lithium toxicity and a shift toward newer medications, such as anticonvulsants and antipsychotics, for treatment and prophylaxis of mania.
A 2011 study examined prescribing trends for bipolar disorder in the United Kingdom.14 Overall, it found increased usage of valproate, carbamazepine, and lamotrigine from 1995 to 2009. During that time, lithium prescribing mostly remained steady at approximately 30%, whereas valproate use increased from 0% to 22.7%. Overall, antipsychotic and valproate prescribing increased relative to lithium.14 A literature review15 analyzed 6 studies of lithium prescribing trends from 1950 to 2010. Four of these studies (2 in the United States, 1 in Canada, and 1 in German-Swiss-Austrian hospitals) found lithium use was declining. The increased use found in Italy and Spain was attributed to multiple factors, including a broader definition of bipolar disorders and the unavailability of valproate in Spain, lithium’s low cost, and mental health reforms in both countries that resulted in overall increased psychotropic prescribing. Decreased lithium use was attributed to increased use of valproate and second-generation antipsychotics, lack of clinician training in lithium therapy, and aggressive marketing of brand-name medications.15
Reduced suicides, possible protection against dementia
A 2013 meta-analysis of 48 randomized controlled trials (RCTs) that included a total of 6,674 patients with mood disorders indicated that compared with placebo, lithium was more effective in reducing suicides and deaths from any cause.16
Large retrospective studies have demonstrated that compared with valproate, lithium has superior anti-suicide properties.17 Researchers found that risk of suicide attempt or completion was 1.5 to 3 times higher during periods of valproate treatment compared with lithium.18 Both short- and long-term lithium use was associated with decreased non-suicide mortality compared with valproate.19 In Denmark, compared with valproate, lithium was associated with fewer psychiatric hospital admissions.19 One RCT, the BALANCE trial, showed that lithium (alone or in combination with valproate) is more likely to prevent relapse in persons with bipolar I disorder than valproate monotherapy.20
Recent research in Denmark suggests that long-term doses of naturally occurring lithium in drinking water might confer some level of protection against dementia.21 Researchers examined the Danish National Patient Register to determine where participants lived and their local water supply. Drinking water lithium levels were assessed, and the mean lithium level for each municipality was calculated. This case-control study selected patients with dementia and 10 age- and sex-matched controls.21
Researchers found that the incidence rate ratio of Alzheimer disease, vascular dementia, and dementia overall was significantly lower among individuals whose drinking water contained lithium, 15.1 to 27.0 µg/L, compared with those whose water had lithium levels 2.0 to 5.0 µg/L.21 Although this study does not prove causality, it opens the door for continued research on lithium as a neuroprotective agent involved in pathways beyond mood stabilization.
Why should you prescribe lithium?
Lithium, which is available in several formulations (Table), should continue to be first-line pharmacotherapy for treating acute mood episodes, prophylaxis, and suicide prevention in bipolar disorder. Although there are many effective medications for treating bipolar disorder—such as second-generation antipsychotics that are available as a long-acting injectable formulation or can be combined with a mood stabilizer—lithium is a thoroughly researched medication with a long history of effectiveness for managing bipolar disorder. As is the case with all psychotropic medications, lithium has adverse effects and necessary precautions, but these are outweighed by its neuroprotective benefits and efficacy. Research has demonstrated that lithium outperforms medications that have largely replaced it, specifically valproate.
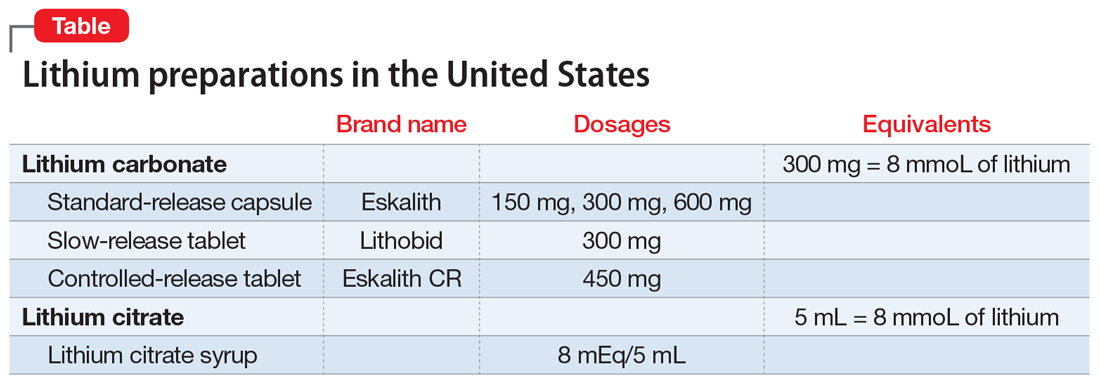
Related Resources
- Ali ZA, El-Mallakh RS. Lithium and kidney disease: Understand the risks. Current Psychiatry. 2021;20(6):34- 38,50. doi:10.12788/cp.0130
- Malhi GS, Gessler D, Outhred T. The use of lithium for the treatment of bipolar disorder: recommendations from clinical practice guidelines. J Affect Disord. 2017;217: 266-280. doi:10.1016/j.jad.2017.03.052
Drug Brand Names
Carbamazepine • Tegretol
Lamotrigine • Lamictal
Lithium • Eskalith, Lithobid
Valproate • Depacon, Depakote, Depakene
Bottom Line
Lithium is a well-researched first-line pharmacotherapy for bipolar disorder, with efficacy equivalent to—or superior to—newer pharmacotherapies such as valproate and second-generation antipsychotics. When prescribing lithium, carefully monitor patients for symptoms of adverse effects or toxicity. Despite teratogenic risks, lithium can be considered for pregnant patients with bipolar disorder.
1. Shorter E. The history of lithium therapy. Bipolar Disord. 2009;11 suppl 2(suppl 2):4-9. doi: 10.1111/j.1399-5618.2009.00706.x
2. Cole N, Parker G. Cade’s identification of lithium for manic-depressive illness—the prospector who found a gold nugget. J Nerv Ment Dis. 2012;200(12):1101-1104. doi:10.1097/NMD.0b013e318275d3cb
3. Johnson FN. Lithium research and therapy. Academic Press; 1975.
4. Cade J. Lithium salts in the treatment of psychotic excitement. Med J Aust. 1949;2(10):518-520. doi:10.1080/j.1440-1614.1999.06241.x
5. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211. doi:10.1177/0004867412437346
6. Quiroz JA, Machado-Vieira R, Zarate CA Jr, et al. Novel insights into lithium’s mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology. 2010;62(1):50-60. doi:10.1159/000314310
7. Gitlin M. Lithium side effects and toxicity: prevalence and management strategies. Int J Bipolar Disord. 2016;4(1):27. doi:10.1186/s40345-016-0068-y
8. Ott M, Stegmayr B, Salander Renberg E, et al. Lithium intoxication: incidence, clinical course and renal function - a population-based retrospective cohort study. J Psychopharmacol. 2016;30(10):1008-1019. doi:10.1177/0269881116652577
9. Heath LJ, Billups SJ, Gaughan KM, et al. Risk factors for utilization of acute care services for lithium toxicity. Psychiatr Serv. 2018;69(6):671-676. doi:10.1176/appi.ps.201700346
10. Raffi ER, Nonacs R, Cohen LS. Safety of psychotropic medications during pregnancy. Clin Perinatol. 2019;46(2):215-234. doi: 10.1016/j.clp.2019.02.004
11. McKnight RF, Adida M, Budge K, et al. Lithium toxicity profile: a systematic review and meta-analysis. Lancet. 2012;379(9817):721-728. doi:10.1016/S0140-6736(11)61516-X
12. Mohandas E, Rajmohan V. Lithium use in special populations. Indian J Psychiatry. 2007;49(3):211-8. doi: 10.4103/0019-5545.37325
13. Deligiannidis KM, Byatt N, Freeman MP. Pharmacotherapy for mood disorders in pregnancy: a review of pharmacokinetic changes and clinical recommendations for therapeutic drug monitoring. J Clin Psychopharmacol. 2014;34(2):244-55. doi: 10.1097/JCP.0000000000000087
14. Hayes J, Prah P, Nazareth I, et al. Prescribing trends in bipolar disorder: cohort study in the United Kingdom THIN primary care database 1995-2009. PLoS One. 2011;6(12):e28725. doi:10.1371/journal.pone.0028725
15. Netto I, Patil R, Kamble P, et al. Lithium prescribing trends: review. International Journal of Healthcare and Biomedical Research. 2014;2(2):95-103.
16. Cipriani A, Hawton K, Stockton S, et al. Lithium in the prevention of suicide in mood disorders: updated systematic review and meta-analysis. BMJ. 2013;346:f3646. doi: 10.1136/bmj.f3646
17. Meyer J. Lithium is regaining favor over anticonvulsants. Psychiatric News. October 2, 2015. Accessed October 12, 2021. https://psychnews.psychiatryonline.org/doi/full/10.1176/appi.pn.2015.PP10a6
18. Goodwin FK, Fireman B, Simon GE, et al. Suicide risk in bipolar disorder during treatment with lithium and divalproex. JAMA. 2003;290(11):1467-1473. doi:10.1001/jama.290.11.1467
19. Smith EG, Austin KL, Kim HM, et al. Mortality associated with lithium and valproate treatment of US Veterans Health Administration patients with mental disorders. Br J Psychiatry. 2015;207(1):55-63. doi:10.1192/bjp.bp.113.138685
20. Geddes JR, Goodwin GM, Rendell J, et al; BALANCE investigators and collaborators. Lithium plus valproate combination therapy versus monotherapy for relapse prevention in bipolar I disorder (BALANCE): a randomised open-label trial. Lancet. 2010;375(9712):385-395. doi:10.1016/S0140-6736(09)61828-6
21. Kessing LV, Gerds TA, Knudsen NN, et al. Association of lithium in drinking water with the incidence of dementia. JAMA Psychiatry. 2017;74(10):1005-1010. doi:10.1001/jamapsychiatry.2017.2362
In clinicians and patients alike, lithium triggers reactions ranging from apprehension and fear about adverse effects and toxicity to confusion over lithium’s usefulness compared with other mood stabilizers that do not require blood monitoring. Research from the 1950s to the 1970s demonstrated that lithium is effective for prophylaxis of mood episodes in patients with bipolar disorder and could reduce the frequency of hospitalization in patients who are depressed.1 For years, lithium was commonly prescribed to treat bipolar disorder, but in recent years its use has fallen out of favor due to concerns about its risks, and the availability of newer medications. This article reviews lithium’s origins (Box1-4), pharmacology, risks, and benefits, and makes a case for why it should remain a first-line therapy for bipolar disorder.
Box
Lithium was initially used in the 1840s to treat gout. William Hammond became the first physician to prescribe lithium bromide for acute mania in 1871, and in 1894, Danish psychiatrist Frederik Lange first used lithium carbonate to treat “melancholic depression.”1 In the 20th century, lithium-containing products were used to treat rheumatologic conditions such as renal calculi and other uric acid diatheses.
Lithium experienced a revival in 1949 when John Cade expanded upon Archibald Garrod’s theory regarding uric acid and gout. As a physician during WWII, Cade observed manic and depressive behaviors among prisoners.2 Theorizing that this was caused by either an excess or lack of a metabolite, he injected urine from patients with mania, depression, and schizophrenia and from healthy individuals into guinea pigs.3 Animals who received urine from patients with mania died faster than those injected with urine from a patient with schizophrenia.2 Concluding that urea was the culprit, Cade substituted the relatively water insoluble uric acid for “the most soluble of urates,” which was lithium urate.2,3 Rather than succumbing to a quicker death, guinea pigs injected with lithium urate became placid, tranquilized, lost their natural timidity, and generally did not respond to stimulation.3
Cade administered lithium carbonate and lithium citrate to himself and, because he did not experience any unwanted effects, began testing the medication on patients. Cade’s landmark 1949 paper4 notes improvement in all 10 patients with mania but little change in 6 patients with schizophrenia and 3 with chronic depression.2
In the United States, interest in lithium did not begin until the 1960s, when Samuel Gershon introduced the medication to a psychiatric hospital in Michigan. Financed by the National Institute of Mental Health, this program bought bulk lithium from a chemical supply store, and a local pharmacy formed it into capsules. Analysis of 4 controlled studies from 1963 to 1971 showed an average response rate to lithium of 78% in 116 patients with mania.1
By the end of the 1960s, many psychiatrists were prescribing lithium. At that time, lithium was not FDA-approved, but it could be prescribed as an investigational new drug by obtaining a special permit. In 1970, the FDA approved lithium for acute mania, and for prophylaxis of mania in 1975. Lithium has not yet been approved for prophylaxis of depression, despite substantial evidence indicating efficacy.1
How lithium works
Lithium has effects on neurotransmitters implicated in mania, such as glutamate, dopamine, and gamma-aminobutyric acid.5 Quiroz et al6 provide a detailed description of lithium’s effects, which can be summarized as modulating neuronal signaling pathways, including B-cell lymphoma 2 (BCL2), cAMP-response element binding protein (CREB), and glycogen synthase kinase-3 (GSK-3). Through these signaling cascades, lithium can curtail progression of neuronal apoptosis caused by the biochemical stress commonly seen in bipolar disorder pathogenesis.6
A wide range of potential adverse effects
Lithium can cause adverse effects in several organ systems. Clinicians must be aware of these effects before prescribing lithium or continuing long-term use. The most commonly documented adverse effects and symptoms of toxicity are:
- tremor
- renal dysfunction, including renal insufficiency and polyuria or polydipsia
- hypothyroidism
- hyperparathyroidism (with subsequent hypercalcemia)
- weight gain
- gastrointestinal (GI) symptoms.
These symptoms tend to occur when lithium serum levels are outside the reference range of 0.6 to 1.2 mEq/L, typically once blood levels reach ≥1.5 mEq/L.7 However, thyroid and renal abnormalities can occur at levels below this value, and might be related to cumulative lithium exposure.7 Adverse effects usually are precipitated by inadequate water intake or inadvertently taking an extra dose. Symptoms of lithium toxicity can be mild, moderate (GI complaints, tremor, weakness, fatigue), or severe (agitation, seizures, autonomic dysregulation, confusion, coma, death).
Lithium adverse effects and toxicity are infrequent. An analysis of 17 years of data in Sweden showed the incidence of moderate to severe lithium intoxication (serum level ≥1.5 mEq/L) was .01 patients per year.8 A recently published US analysis found the prevalence rate of lithium toxicity was 2.2%.9 Results from both groups show that drug interactions were an important cause of increased lithium levels, and specifically that initiating a medication that could interact with lithium was associated with 30-fold higher risk of needing acute care for lithium toxicity.9 Possible drug interactions include nonsteroidal anti-inflammatory drugs, diuretics, and renin-angiotensin-aldosterone system inhibitors.9 Because lithium is eliminated exclusively by the kidneys, impaired or altered renal function can increase the risk of lithium retention, leading to intoxication. Other risk factors include older age, alteration of water-salt homeostasis (fever, diarrhea, vomiting), higher number of treated chronic diseases as measured by Chronic Disease Score (range: 0 to 35; higher scores denotes higher number of treated chronic diseases and increased hospitalization risk), and higher total daily lithium dosage.9
Presentation of lithium intoxication often is mild or nonspecific, and physicians should have a low threshold for checking lithium blood levels.8 Lithium intoxication can be safely managed with volume expansion, forced diuresis, and hemodialysis.
Continue to: Lithium use during pregnancy...
Lithium use during pregnancy
When considering lithium for a woman who is pregnant, it is important to weigh the potential teratogenic risks against the benefit of successful management of the mood disorder. Ebstein’s anomaly (abnormal tricuspid valve leaflets) is the most well-known teratogenic risk associated with lithium, with an estimated absolute risk of 1 in 1,000 in patients treated with lithium compared with 1 in 20,000 in controls.10,11 The risk of congenital anomalies is increased in infants exposed to lithium in utero (4% to 12% vs 2% to 4% in controls)12; exposure during the first trimester of pregnancy is associated with increased risk. Lithium levels must be adjusted during pregnancy. Pregnant patients are at higher risk of relapse to mania because renal lithium clearance increases by 30% to 50% during pregnancy, and normalizes shortly after delivery.13
Lithium exposure during pregnancy has been linked to increased risk of miscarriage and preterm delivery; however, more research is needed to define the true risk of noncardiac teratogenicity associated with lithium.11 Because there is a lack of definitive data regarding teratogenicity, and because of lithium’s well-documented effectiveness in mood disorders, lithium should be considered a first-line therapy for pregnant patients with bipolar disorder.10
Prescribing trends
Despite data showing the efficacy and benefits of lithium, there has been a paradoxical decrease in lithium prescribing. This is the result of multiple factors, including fear of adverse effects and lithium toxicity and a shift toward newer medications, such as anticonvulsants and antipsychotics, for treatment and prophylaxis of mania.
A 2011 study examined prescribing trends for bipolar disorder in the United Kingdom.14 Overall, it found increased usage of valproate, carbamazepine, and lamotrigine from 1995 to 2009. During that time, lithium prescribing mostly remained steady at approximately 30%, whereas valproate use increased from 0% to 22.7%. Overall, antipsychotic and valproate prescribing increased relative to lithium.14 A literature review15 analyzed 6 studies of lithium prescribing trends from 1950 to 2010. Four of these studies (2 in the United States, 1 in Canada, and 1 in German-Swiss-Austrian hospitals) found lithium use was declining. The increased use found in Italy and Spain was attributed to multiple factors, including a broader definition of bipolar disorders and the unavailability of valproate in Spain, lithium’s low cost, and mental health reforms in both countries that resulted in overall increased psychotropic prescribing. Decreased lithium use was attributed to increased use of valproate and second-generation antipsychotics, lack of clinician training in lithium therapy, and aggressive marketing of brand-name medications.15
Reduced suicides, possible protection against dementia
A 2013 meta-analysis of 48 randomized controlled trials (RCTs) that included a total of 6,674 patients with mood disorders indicated that compared with placebo, lithium was more effective in reducing suicides and deaths from any cause.16
Large retrospective studies have demonstrated that compared with valproate, lithium has superior anti-suicide properties.17 Researchers found that risk of suicide attempt or completion was 1.5 to 3 times higher during periods of valproate treatment compared with lithium.18 Both short- and long-term lithium use was associated with decreased non-suicide mortality compared with valproate.19 In Denmark, compared with valproate, lithium was associated with fewer psychiatric hospital admissions.19 One RCT, the BALANCE trial, showed that lithium (alone or in combination with valproate) is more likely to prevent relapse in persons with bipolar I disorder than valproate monotherapy.20
Recent research in Denmark suggests that long-term doses of naturally occurring lithium in drinking water might confer some level of protection against dementia.21 Researchers examined the Danish National Patient Register to determine where participants lived and their local water supply. Drinking water lithium levels were assessed, and the mean lithium level for each municipality was calculated. This case-control study selected patients with dementia and 10 age- and sex-matched controls.21
Researchers found that the incidence rate ratio of Alzheimer disease, vascular dementia, and dementia overall was significantly lower among individuals whose drinking water contained lithium, 15.1 to 27.0 µg/L, compared with those whose water had lithium levels 2.0 to 5.0 µg/L.21 Although this study does not prove causality, it opens the door for continued research on lithium as a neuroprotective agent involved in pathways beyond mood stabilization.
Why should you prescribe lithium?
Lithium, which is available in several formulations (Table), should continue to be first-line pharmacotherapy for treating acute mood episodes, prophylaxis, and suicide prevention in bipolar disorder. Although there are many effective medications for treating bipolar disorder—such as second-generation antipsychotics that are available as a long-acting injectable formulation or can be combined with a mood stabilizer—lithium is a thoroughly researched medication with a long history of effectiveness for managing bipolar disorder. As is the case with all psychotropic medications, lithium has adverse effects and necessary precautions, but these are outweighed by its neuroprotective benefits and efficacy. Research has demonstrated that lithium outperforms medications that have largely replaced it, specifically valproate.
Related Resources
- Ali ZA, El-Mallakh RS. Lithium and kidney disease: Understand the risks. Current Psychiatry. 2021;20(6):34- 38,50. doi:10.12788/cp.0130
- Malhi GS, Gessler D, Outhred T. The use of lithium for the treatment of bipolar disorder: recommendations from clinical practice guidelines. J Affect Disord. 2017;217: 266-280. doi:10.1016/j.jad.2017.03.052
Drug Brand Names
Carbamazepine • Tegretol
Lamotrigine • Lamictal
Lithium • Eskalith, Lithobid
Valproate • Depacon, Depakote, Depakene
Bottom Line
Lithium is a well-researched first-line pharmacotherapy for bipolar disorder, with efficacy equivalent to—or superior to—newer pharmacotherapies such as valproate and second-generation antipsychotics. When prescribing lithium, carefully monitor patients for symptoms of adverse effects or toxicity. Despite teratogenic risks, lithium can be considered for pregnant patients with bipolar disorder.
In clinicians and patients alike, lithium triggers reactions ranging from apprehension and fear about adverse effects and toxicity to confusion over lithium’s usefulness compared with other mood stabilizers that do not require blood monitoring. Research from the 1950s to the 1970s demonstrated that lithium is effective for prophylaxis of mood episodes in patients with bipolar disorder and could reduce the frequency of hospitalization in patients who are depressed.1 For years, lithium was commonly prescribed to treat bipolar disorder, but in recent years its use has fallen out of favor due to concerns about its risks, and the availability of newer medications. This article reviews lithium’s origins (Box1-4), pharmacology, risks, and benefits, and makes a case for why it should remain a first-line therapy for bipolar disorder.
Box
Lithium was initially used in the 1840s to treat gout. William Hammond became the first physician to prescribe lithium bromide for acute mania in 1871, and in 1894, Danish psychiatrist Frederik Lange first used lithium carbonate to treat “melancholic depression.”1 In the 20th century, lithium-containing products were used to treat rheumatologic conditions such as renal calculi and other uric acid diatheses.
Lithium experienced a revival in 1949 when John Cade expanded upon Archibald Garrod’s theory regarding uric acid and gout. As a physician during WWII, Cade observed manic and depressive behaviors among prisoners.2 Theorizing that this was caused by either an excess or lack of a metabolite, he injected urine from patients with mania, depression, and schizophrenia and from healthy individuals into guinea pigs.3 Animals who received urine from patients with mania died faster than those injected with urine from a patient with schizophrenia.2 Concluding that urea was the culprit, Cade substituted the relatively water insoluble uric acid for “the most soluble of urates,” which was lithium urate.2,3 Rather than succumbing to a quicker death, guinea pigs injected with lithium urate became placid, tranquilized, lost their natural timidity, and generally did not respond to stimulation.3
Cade administered lithium carbonate and lithium citrate to himself and, because he did not experience any unwanted effects, began testing the medication on patients. Cade’s landmark 1949 paper4 notes improvement in all 10 patients with mania but little change in 6 patients with schizophrenia and 3 with chronic depression.2
In the United States, interest in lithium did not begin until the 1960s, when Samuel Gershon introduced the medication to a psychiatric hospital in Michigan. Financed by the National Institute of Mental Health, this program bought bulk lithium from a chemical supply store, and a local pharmacy formed it into capsules. Analysis of 4 controlled studies from 1963 to 1971 showed an average response rate to lithium of 78% in 116 patients with mania.1
By the end of the 1960s, many psychiatrists were prescribing lithium. At that time, lithium was not FDA-approved, but it could be prescribed as an investigational new drug by obtaining a special permit. In 1970, the FDA approved lithium for acute mania, and for prophylaxis of mania in 1975. Lithium has not yet been approved for prophylaxis of depression, despite substantial evidence indicating efficacy.1
How lithium works
Lithium has effects on neurotransmitters implicated in mania, such as glutamate, dopamine, and gamma-aminobutyric acid.5 Quiroz et al6 provide a detailed description of lithium’s effects, which can be summarized as modulating neuronal signaling pathways, including B-cell lymphoma 2 (BCL2), cAMP-response element binding protein (CREB), and glycogen synthase kinase-3 (GSK-3). Through these signaling cascades, lithium can curtail progression of neuronal apoptosis caused by the biochemical stress commonly seen in bipolar disorder pathogenesis.6
A wide range of potential adverse effects
Lithium can cause adverse effects in several organ systems. Clinicians must be aware of these effects before prescribing lithium or continuing long-term use. The most commonly documented adverse effects and symptoms of toxicity are:
- tremor
- renal dysfunction, including renal insufficiency and polyuria or polydipsia
- hypothyroidism
- hyperparathyroidism (with subsequent hypercalcemia)
- weight gain
- gastrointestinal (GI) symptoms.
These symptoms tend to occur when lithium serum levels are outside the reference range of 0.6 to 1.2 mEq/L, typically once blood levels reach ≥1.5 mEq/L.7 However, thyroid and renal abnormalities can occur at levels below this value, and might be related to cumulative lithium exposure.7 Adverse effects usually are precipitated by inadequate water intake or inadvertently taking an extra dose. Symptoms of lithium toxicity can be mild, moderate (GI complaints, tremor, weakness, fatigue), or severe (agitation, seizures, autonomic dysregulation, confusion, coma, death).
Lithium adverse effects and toxicity are infrequent. An analysis of 17 years of data in Sweden showed the incidence of moderate to severe lithium intoxication (serum level ≥1.5 mEq/L) was .01 patients per year.8 A recently published US analysis found the prevalence rate of lithium toxicity was 2.2%.9 Results from both groups show that drug interactions were an important cause of increased lithium levels, and specifically that initiating a medication that could interact with lithium was associated with 30-fold higher risk of needing acute care for lithium toxicity.9 Possible drug interactions include nonsteroidal anti-inflammatory drugs, diuretics, and renin-angiotensin-aldosterone system inhibitors.9 Because lithium is eliminated exclusively by the kidneys, impaired or altered renal function can increase the risk of lithium retention, leading to intoxication. Other risk factors include older age, alteration of water-salt homeostasis (fever, diarrhea, vomiting), higher number of treated chronic diseases as measured by Chronic Disease Score (range: 0 to 35; higher scores denotes higher number of treated chronic diseases and increased hospitalization risk), and higher total daily lithium dosage.9
Presentation of lithium intoxication often is mild or nonspecific, and physicians should have a low threshold for checking lithium blood levels.8 Lithium intoxication can be safely managed with volume expansion, forced diuresis, and hemodialysis.
Continue to: Lithium use during pregnancy...
Lithium use during pregnancy
When considering lithium for a woman who is pregnant, it is important to weigh the potential teratogenic risks against the benefit of successful management of the mood disorder. Ebstein’s anomaly (abnormal tricuspid valve leaflets) is the most well-known teratogenic risk associated with lithium, with an estimated absolute risk of 1 in 1,000 in patients treated with lithium compared with 1 in 20,000 in controls.10,11 The risk of congenital anomalies is increased in infants exposed to lithium in utero (4% to 12% vs 2% to 4% in controls)12; exposure during the first trimester of pregnancy is associated with increased risk. Lithium levels must be adjusted during pregnancy. Pregnant patients are at higher risk of relapse to mania because renal lithium clearance increases by 30% to 50% during pregnancy, and normalizes shortly after delivery.13
Lithium exposure during pregnancy has been linked to increased risk of miscarriage and preterm delivery; however, more research is needed to define the true risk of noncardiac teratogenicity associated with lithium.11 Because there is a lack of definitive data regarding teratogenicity, and because of lithium’s well-documented effectiveness in mood disorders, lithium should be considered a first-line therapy for pregnant patients with bipolar disorder.10
Prescribing trends
Despite data showing the efficacy and benefits of lithium, there has been a paradoxical decrease in lithium prescribing. This is the result of multiple factors, including fear of adverse effects and lithium toxicity and a shift toward newer medications, such as anticonvulsants and antipsychotics, for treatment and prophylaxis of mania.
A 2011 study examined prescribing trends for bipolar disorder in the United Kingdom.14 Overall, it found increased usage of valproate, carbamazepine, and lamotrigine from 1995 to 2009. During that time, lithium prescribing mostly remained steady at approximately 30%, whereas valproate use increased from 0% to 22.7%. Overall, antipsychotic and valproate prescribing increased relative to lithium.14 A literature review15 analyzed 6 studies of lithium prescribing trends from 1950 to 2010. Four of these studies (2 in the United States, 1 in Canada, and 1 in German-Swiss-Austrian hospitals) found lithium use was declining. The increased use found in Italy and Spain was attributed to multiple factors, including a broader definition of bipolar disorders and the unavailability of valproate in Spain, lithium’s low cost, and mental health reforms in both countries that resulted in overall increased psychotropic prescribing. Decreased lithium use was attributed to increased use of valproate and second-generation antipsychotics, lack of clinician training in lithium therapy, and aggressive marketing of brand-name medications.15
Reduced suicides, possible protection against dementia
A 2013 meta-analysis of 48 randomized controlled trials (RCTs) that included a total of 6,674 patients with mood disorders indicated that compared with placebo, lithium was more effective in reducing suicides and deaths from any cause.16
Large retrospective studies have demonstrated that compared with valproate, lithium has superior anti-suicide properties.17 Researchers found that risk of suicide attempt or completion was 1.5 to 3 times higher during periods of valproate treatment compared with lithium.18 Both short- and long-term lithium use was associated with decreased non-suicide mortality compared with valproate.19 In Denmark, compared with valproate, lithium was associated with fewer psychiatric hospital admissions.19 One RCT, the BALANCE trial, showed that lithium (alone or in combination with valproate) is more likely to prevent relapse in persons with bipolar I disorder than valproate monotherapy.20
Recent research in Denmark suggests that long-term doses of naturally occurring lithium in drinking water might confer some level of protection against dementia.21 Researchers examined the Danish National Patient Register to determine where participants lived and their local water supply. Drinking water lithium levels were assessed, and the mean lithium level for each municipality was calculated. This case-control study selected patients with dementia and 10 age- and sex-matched controls.21
Researchers found that the incidence rate ratio of Alzheimer disease, vascular dementia, and dementia overall was significantly lower among individuals whose drinking water contained lithium, 15.1 to 27.0 µg/L, compared with those whose water had lithium levels 2.0 to 5.0 µg/L.21 Although this study does not prove causality, it opens the door for continued research on lithium as a neuroprotective agent involved in pathways beyond mood stabilization.
Why should you prescribe lithium?
Lithium, which is available in several formulations (Table), should continue to be first-line pharmacotherapy for treating acute mood episodes, prophylaxis, and suicide prevention in bipolar disorder. Although there are many effective medications for treating bipolar disorder—such as second-generation antipsychotics that are available as a long-acting injectable formulation or can be combined with a mood stabilizer—lithium is a thoroughly researched medication with a long history of effectiveness for managing bipolar disorder. As is the case with all psychotropic medications, lithium has adverse effects and necessary precautions, but these are outweighed by its neuroprotective benefits and efficacy. Research has demonstrated that lithium outperforms medications that have largely replaced it, specifically valproate.
Related Resources
- Ali ZA, El-Mallakh RS. Lithium and kidney disease: Understand the risks. Current Psychiatry. 2021;20(6):34- 38,50. doi:10.12788/cp.0130
- Malhi GS, Gessler D, Outhred T. The use of lithium for the treatment of bipolar disorder: recommendations from clinical practice guidelines. J Affect Disord. 2017;217: 266-280. doi:10.1016/j.jad.2017.03.052
Drug Brand Names
Carbamazepine • Tegretol
Lamotrigine • Lamictal
Lithium • Eskalith, Lithobid
Valproate • Depacon, Depakote, Depakene
Bottom Line
Lithium is a well-researched first-line pharmacotherapy for bipolar disorder, with efficacy equivalent to—or superior to—newer pharmacotherapies such as valproate and second-generation antipsychotics. When prescribing lithium, carefully monitor patients for symptoms of adverse effects or toxicity. Despite teratogenic risks, lithium can be considered for pregnant patients with bipolar disorder.
1. Shorter E. The history of lithium therapy. Bipolar Disord. 2009;11 suppl 2(suppl 2):4-9. doi: 10.1111/j.1399-5618.2009.00706.x
2. Cole N, Parker G. Cade’s identification of lithium for manic-depressive illness—the prospector who found a gold nugget. J Nerv Ment Dis. 2012;200(12):1101-1104. doi:10.1097/NMD.0b013e318275d3cb
3. Johnson FN. Lithium research and therapy. Academic Press; 1975.
4. Cade J. Lithium salts in the treatment of psychotic excitement. Med J Aust. 1949;2(10):518-520. doi:10.1080/j.1440-1614.1999.06241.x
5. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211. doi:10.1177/0004867412437346
6. Quiroz JA, Machado-Vieira R, Zarate CA Jr, et al. Novel insights into lithium’s mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology. 2010;62(1):50-60. doi:10.1159/000314310
7. Gitlin M. Lithium side effects and toxicity: prevalence and management strategies. Int J Bipolar Disord. 2016;4(1):27. doi:10.1186/s40345-016-0068-y
8. Ott M, Stegmayr B, Salander Renberg E, et al. Lithium intoxication: incidence, clinical course and renal function - a population-based retrospective cohort study. J Psychopharmacol. 2016;30(10):1008-1019. doi:10.1177/0269881116652577
9. Heath LJ, Billups SJ, Gaughan KM, et al. Risk factors for utilization of acute care services for lithium toxicity. Psychiatr Serv. 2018;69(6):671-676. doi:10.1176/appi.ps.201700346
10. Raffi ER, Nonacs R, Cohen LS. Safety of psychotropic medications during pregnancy. Clin Perinatol. 2019;46(2):215-234. doi: 10.1016/j.clp.2019.02.004
11. McKnight RF, Adida M, Budge K, et al. Lithium toxicity profile: a systematic review and meta-analysis. Lancet. 2012;379(9817):721-728. doi:10.1016/S0140-6736(11)61516-X
12. Mohandas E, Rajmohan V. Lithium use in special populations. Indian J Psychiatry. 2007;49(3):211-8. doi: 10.4103/0019-5545.37325
13. Deligiannidis KM, Byatt N, Freeman MP. Pharmacotherapy for mood disorders in pregnancy: a review of pharmacokinetic changes and clinical recommendations for therapeutic drug monitoring. J Clin Psychopharmacol. 2014;34(2):244-55. doi: 10.1097/JCP.0000000000000087
14. Hayes J, Prah P, Nazareth I, et al. Prescribing trends in bipolar disorder: cohort study in the United Kingdom THIN primary care database 1995-2009. PLoS One. 2011;6(12):e28725. doi:10.1371/journal.pone.0028725
15. Netto I, Patil R, Kamble P, et al. Lithium prescribing trends: review. International Journal of Healthcare and Biomedical Research. 2014;2(2):95-103.
16. Cipriani A, Hawton K, Stockton S, et al. Lithium in the prevention of suicide in mood disorders: updated systematic review and meta-analysis. BMJ. 2013;346:f3646. doi: 10.1136/bmj.f3646
17. Meyer J. Lithium is regaining favor over anticonvulsants. Psychiatric News. October 2, 2015. Accessed October 12, 2021. https://psychnews.psychiatryonline.org/doi/full/10.1176/appi.pn.2015.PP10a6
18. Goodwin FK, Fireman B, Simon GE, et al. Suicide risk in bipolar disorder during treatment with lithium and divalproex. JAMA. 2003;290(11):1467-1473. doi:10.1001/jama.290.11.1467
19. Smith EG, Austin KL, Kim HM, et al. Mortality associated with lithium and valproate treatment of US Veterans Health Administration patients with mental disorders. Br J Psychiatry. 2015;207(1):55-63. doi:10.1192/bjp.bp.113.138685
20. Geddes JR, Goodwin GM, Rendell J, et al; BALANCE investigators and collaborators. Lithium plus valproate combination therapy versus monotherapy for relapse prevention in bipolar I disorder (BALANCE): a randomised open-label trial. Lancet. 2010;375(9712):385-395. doi:10.1016/S0140-6736(09)61828-6
21. Kessing LV, Gerds TA, Knudsen NN, et al. Association of lithium in drinking water with the incidence of dementia. JAMA Psychiatry. 2017;74(10):1005-1010. doi:10.1001/jamapsychiatry.2017.2362
1. Shorter E. The history of lithium therapy. Bipolar Disord. 2009;11 suppl 2(suppl 2):4-9. doi: 10.1111/j.1399-5618.2009.00706.x
2. Cole N, Parker G. Cade’s identification of lithium for manic-depressive illness—the prospector who found a gold nugget. J Nerv Ment Dis. 2012;200(12):1101-1104. doi:10.1097/NMD.0b013e318275d3cb
3. Johnson FN. Lithium research and therapy. Academic Press; 1975.
4. Cade J. Lithium salts in the treatment of psychotic excitement. Med J Aust. 1949;2(10):518-520. doi:10.1080/j.1440-1614.1999.06241.x
5. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211. doi:10.1177/0004867412437346
6. Quiroz JA, Machado-Vieira R, Zarate CA Jr, et al. Novel insights into lithium’s mechanism of action: neurotrophic and neuroprotective effects. Neuropsychobiology. 2010;62(1):50-60. doi:10.1159/000314310
7. Gitlin M. Lithium side effects and toxicity: prevalence and management strategies. Int J Bipolar Disord. 2016;4(1):27. doi:10.1186/s40345-016-0068-y
8. Ott M, Stegmayr B, Salander Renberg E, et al. Lithium intoxication: incidence, clinical course and renal function - a population-based retrospective cohort study. J Psychopharmacol. 2016;30(10):1008-1019. doi:10.1177/0269881116652577
9. Heath LJ, Billups SJ, Gaughan KM, et al. Risk factors for utilization of acute care services for lithium toxicity. Psychiatr Serv. 2018;69(6):671-676. doi:10.1176/appi.ps.201700346
10. Raffi ER, Nonacs R, Cohen LS. Safety of psychotropic medications during pregnancy. Clin Perinatol. 2019;46(2):215-234. doi: 10.1016/j.clp.2019.02.004
11. McKnight RF, Adida M, Budge K, et al. Lithium toxicity profile: a systematic review and meta-analysis. Lancet. 2012;379(9817):721-728. doi:10.1016/S0140-6736(11)61516-X
12. Mohandas E, Rajmohan V. Lithium use in special populations. Indian J Psychiatry. 2007;49(3):211-8. doi: 10.4103/0019-5545.37325
13. Deligiannidis KM, Byatt N, Freeman MP. Pharmacotherapy for mood disorders in pregnancy: a review of pharmacokinetic changes and clinical recommendations for therapeutic drug monitoring. J Clin Psychopharmacol. 2014;34(2):244-55. doi: 10.1097/JCP.0000000000000087
14. Hayes J, Prah P, Nazareth I, et al. Prescribing trends in bipolar disorder: cohort study in the United Kingdom THIN primary care database 1995-2009. PLoS One. 2011;6(12):e28725. doi:10.1371/journal.pone.0028725
15. Netto I, Patil R, Kamble P, et al. Lithium prescribing trends: review. International Journal of Healthcare and Biomedical Research. 2014;2(2):95-103.
16. Cipriani A, Hawton K, Stockton S, et al. Lithium in the prevention of suicide in mood disorders: updated systematic review and meta-analysis. BMJ. 2013;346:f3646. doi: 10.1136/bmj.f3646
17. Meyer J. Lithium is regaining favor over anticonvulsants. Psychiatric News. October 2, 2015. Accessed October 12, 2021. https://psychnews.psychiatryonline.org/doi/full/10.1176/appi.pn.2015.PP10a6
18. Goodwin FK, Fireman B, Simon GE, et al. Suicide risk in bipolar disorder during treatment with lithium and divalproex. JAMA. 2003;290(11):1467-1473. doi:10.1001/jama.290.11.1467
19. Smith EG, Austin KL, Kim HM, et al. Mortality associated with lithium and valproate treatment of US Veterans Health Administration patients with mental disorders. Br J Psychiatry. 2015;207(1):55-63. doi:10.1192/bjp.bp.113.138685
20. Geddes JR, Goodwin GM, Rendell J, et al; BALANCE investigators and collaborators. Lithium plus valproate combination therapy versus monotherapy for relapse prevention in bipolar I disorder (BALANCE): a randomised open-label trial. Lancet. 2010;375(9712):385-395. doi:10.1016/S0140-6736(09)61828-6
21. Kessing LV, Gerds TA, Knudsen NN, et al. Association of lithium in drinking water with the incidence of dementia. JAMA Psychiatry. 2017;74(10):1005-1010. doi:10.1001/jamapsychiatry.2017.2362

Serotonin-mediated anxiety: How to recognize and treat it
Sara R. Abell, MD, and Rif S. El-Mallakh, MD
Individuals with anxiety will experience frequent or chronic excessive worry, nervousness, a sense of unease, a feeling of being unfocused, and distress, which result in functional impairment.1 Frequently, anxiety is accompanied by restlessness or muscle tension. Generalized anxiety disorder is one of the most common psychiatric diagnoses in the United States and has a prevalence of 2% to 6% globally.2 Although research has been conducted regarding anxiety’s pathogenesis, to date a firm consensus on its etiology has not been reached.3 It is likely multifactorial, with environmental and biologic components.
One area of focus has been neurotransmitters and the possible role they play in the pathogenesis of anxiety. Specifically, the monoamine neurotransmitters have been implicated in the clinical manifestations of anxiety. Among the amines, normal roles include stimulating the autonomic nervous system and regulating numerous cognitive phenomena, such as volition and emotion. Many psychiatric medications modify aminergic transmission, and many current anxiety medications target amine neurotransmitters. Medications that target histamine, serotonin, norepinephrine, and dopamine all play a role in treating anxiety.
In this article, we focus on serotonin (5-hydroxytryptamine, 5-HT) as a mediator of anxiety and on excessive synaptic 5-HT as the cause of anxiety. We discuss how 5-HT–mediated anxiety can be identified and offer some solutions for its treatment.
The amine neurotransmitters
There are 6 amine neurotransmitters in the CNS. These are derived from tyrosine (dopamine [DA], norepinephrine [NE], and epinephrine), histidine (histamine), and tryptophan (serotonin [5-HT] and melatonin). In addition to their physiologic actions, amines have been implicated in both acute and chronic anxiety. Excessive DA stimulation has been linked with fear4,5; NE elevations are central to hypervigilance and hyperarousal of posttraumatic stress disorder6; and histamine may mediate emotional memories involved in fear and anxiety.7 Understanding the normal function of 5-HT will aid in understanding its potential problematic role (Box,8-18page 38).
How serotonin-mediated anxiety presents
“Anxiety” is a collection of signs and symptoms that likely represent multiple processes and have the common characteristic of being subjectively unpleasant, with a subjective wish for the feeling to end. The expression of anxiety disorders is quite diverse and ranges from brief episodes such as panic attacks (which may be mediated, in part, by epinephrine/NE19) to lifelong stereotypic obsessions and compulsions (which may be mediated, in part, by DA and modified by 5-HT20,21). Biochemical separation of the anxiety disorders is key to achieving tailored treatment.6 Towards this end, it is important to investigate the phenomenon of serotonin-mediated anxiety.
Because clinicians are familiar with reductions of anxiety as selective serotonin reuptake inhibitors (SSRIs) increase 5-HT levels in the synapse, it is difficult to conceptualize serotonin-mediated anxiety. However, many of the effects at postsynaptic 5-HT receptors may be biphasic.15-18 Serotonin-mediated anxiety appears to occur when levels of 5-HT (or stimulation of 5-HT receptors) are particularly high. This is most frequently seen in patients who genetically have high synaptic 5-HT (by virtue of the short form of the 5-HT transporter),22 whose synaptic 5-HT is further increased by treatment with an SSRI,23 and who are experiencing a stressor that yet further increases their synaptic 5-HT.24 However, it may occur in some individuals with only 2 of these 3 conditions.Clinically, individuals with serotonin-mediated anxiety will usually appear calm. The anxiety they are experiencing is not exhibited in any way in the motor system (ie, they do not appear restless, do not pace, muscle tone is not increased, etc.). However, they will generally complain of an internal agitation, a sense of a negative internal energy. Frequently, they will use descriptions such as “I feel I could jump out of my skin.” As previously mentioned, this is usually in the setting of some environmental stress, in addition to either a pharmacologic (SSRI) or genetic (short form of the 5-HT transporter) reason for increasing synaptic 5-HT, or both.
Almost always, interventions that block multiple postsynaptic 5-HT receptors or discontinuation of the SSRI (if applicable) will alleviate the anxiety, quickly or more slowly, respectively. Sublingual asenapine, which at low doses can block 5-HT2C (Ki = 0.03 nM), 5-HT2A (Ki = 0.07 nM), 5-HT7 (Ki = 0.11 nM), 5-HT2B (Ki = 0.18 nM), and 5-HT6 (Ki = 0.25 nM),25,26 and which will produce peak plasma levels within 10 minutes,27 usually is quite effective.
Box
Serotonin (5-HT) arises from neurons in the raphe nuclei of the rostral pons and projects superiorly to the cerebral cortex and inferiorly to the spinal cord.8 It works in an inhibitory or excitatory manner depending on which receptors are activated. In the periphery, 5-HT influences intestinal peristalsis, sensory modulation, gland function, thermoregulation, blood pressure, platelet aggregation, and sexual behavior,9 all actions that produce potential adverse effects of serotonin reuptake– inhibiting antidepressants. In the CNS, 5-HT plays a role in attention bias; decision-making; sleep and wakefulness; and mood regulation. In short, serotonin can be viewed as mediating emotional motivation.10
Serotonin alters neuroplasticity. During development, 5-HT stimulates creation of new synapses and increases the density of synaptic webs. It has a direct stimulatory effect on the length of dendrites, their branching, and their myelination.11 In the CNS, it plays a role in dendritic arborization. Animal studies with rats have shown that lesioning highly concentrated 5-HT areas at early ages resulted in an adult brain with a lower number of neurons and a less complex web of dendrites.12,13 In situations of emotional stress, it is theorized that low levels of 5-HT lead to a reduced ability to deal with emotional stressors due to lower levels of complexity in synaptic connections.
Serotonin has also been implicated in mediating some aspects of dopamine-related actions, such as locomotion, reward, and threat avoidance. This is believed to contribute to the beneficial effect of 5-HT2A blockade by secondgeneration antipsychotics (SGAs).14 Blockade of other 5-HT receptors, such as 5-HT1A, 5-HT2C, 5-HT6, and 5-HT7, may also contribute to the antipsychotic action of SGAs.14
Serotonin receptors are found throughout the body, and 14 subtypes have been identified.9 Excitatory and inhibitory action of 5-HT depends on the receptor, and the actions of 5-HT can differ with the same receptor at different concentrations. This is because serotonin’s effects are biphasic and concentration-dependent, meaning that levels of 5-HT in the synapse will dictate the downstream effect of receptor agonism or antagonism. Animal models have shown that low-dose agonism of 5-HT receptors causes vasoconstriction of the coronary arteries, and high doses cause relaxation. This response has also been demonstrated in the vasculature of the kidneys and the smooth muscle of the trachea. Additionally, 5-HT works in conjunction with histamine to produce a biphasic response in the colonic arteries and veins in situations of endothelial damage.15
Most relevant to this discussion are 5-HT’s actions in mood regulation and behavior. Low 5-HT states result in less behavioral inhibition, leading to higher impulse control failures and aggression. Experiments in mice with deficient serotonergic brain regions show hypoactivity, extended daytime sleep, anxiety, and depressive behaviors.13 Serotonin’s behavioral effects are also biphasic. For example, lowdose antagonism with trazodone of 5-HT receptors demonstrated a pro-aggressive behavioral effect, while high-dose antagonism is anti-aggressive.15 Similar biphasic effects may result in either induction or reduction of anxiety with agents that block or excite certain 5-HT receptors.16-18
Continue to: A key difference: No motor system involvement...
A key difference: No motor system involvement
What distinguishes 5-HT from the other amine transmitters as a mediator of anxiety is the lack of involvement of the motor system. Multiple studies in rats illustrate that exogenously augmenting 5-HT has no effect on levels of locomotor activity. Dopamine depletion is well-characterized in the motor dysfunction of Parkinson’s disease, and DA excess can cause repetitive, stereotyped movements, such as seen in tardive dyskinesia or Huntington’s disease.8 In humans, serotonin-mediated anxiety is usually without a motoric component; patients appear calm but complain of extreme anxiety or agitation. Agitation has been reported after initiation of an SSRI,29 and is more likely to occur in patients with the short form of the 5-HT transporter.30 Motoric activation has been reported in some of these studies, but does not seem to cluster with the complaint of agitation.29 The reduced number of available transporters means a chronic steady-state elevation of serotonin, because less serotonin is being removed from the synapse after it is released. This is one of the reasons patients with the short form of the 5-HT transporter may be more susceptible to serotonin-mediated anxiety.
What you need to keep in mind
Pharmacologic treatment of anxiety begins with an SSRI, a serotonin-norepinephrine reuptake inhibitor (SNRI), or buspirone. Second-line treatments include hydroxyzine, gabapentin, pregabalin, and quetiapine.3,31 However, clinicians need to be aware that a fraction of their patients will report anxiety that will not have any external manifestations, but will be experienced as an unpleasant internal energy. These patients may report an increase in their anxiety levels when started on an SSRI or SNRI.29,30 This anxiety is most likely mediated by increases of synaptic 5-HT. This occurs because many serotonergic receptors may have a biphasic response, so that too much stimulation is experienced as excessive internal energy.16-18 In such patients, blockade of key 5-HT receptors may reduce that internal agitation. The advantage of recognizing serotonin-mediated anxiety is that one can specifically tailor treatment to address the patient’s specific physiology.
It is important to note that the anxiolytic effect of asenapine is specific to patients with serotonin-mediated anxiety. Unlike quetiapine, which is effective as augmentation therapy in generalized anxiety disorder,31 asenapine does not appear to reduce anxiety in patients with schizophrenia32 or borderline personality disorder33 when administered for other reasons. However, it may reduce anxiety in patients with the short form of the 5-HT transporter.30,34
Bottom Line
Serotonin-mediated anxiety occurs when levels of synaptic serotonin (5-HT) are high. Patients with serotonin-mediated anxiety appear calm but will report experiencing an unpleasant internal energy. Interventions that block multiple postsynaptic 5-HT receptors or discontinuation of a selective serotonin reuptake inhibitor (if applicable) will alleviate the anxiety.
Related Resource
• Bhatt NV. Anxiety disorders. https://emedicine.medscape. com/article/286227-overview
Drug Brand Names
Asenapine • Saphris, Secuado
Gabapentin • Neurontin
Hydroxyzine • Vistaril
Pregabalin • Lyrica
Quetiapine • Seroquel
Trazodone • Oleptro
1. Shelton CI. Diagnosis and management of anxiety disorders. J Am Osteopath Assoc. 2004;104(3 Suppl 3):S2-S5.
2. Ruscio AM, Hallion LS, Lim CCW, et al. Cross-sectional comparison of the epidemiology of DSM-5 generalized anxiety disorder across the globe. JAMA Psychiatry. 2017;74(5):465-475.
3. Locke AB, Kirst N, Shultz CG. Diagnosis and management of generalized anxiety disorder and panic disorder in adults. Am Fam Physician. 2015;91(9):617-624.
4. Hariri AR, Mattay VS, Tessitore A, et al. Dextroamphetamine modulates the response of the human amygdala. Neuropsychopharmacology. 2002;27(6):1036-1040.
5. Colombo AC, de Oliveira AR, Reimer AE, et al. Dopaminergic mechanisms underlying catalepsy, fear and anxiety: do they interact? Behav Brain Res. 2013;257:201-207.
6. Togay B, El-Mallakh RS. Posttraumatic stress disorder: from pathophysiology to pharmacology. Curr Psychiatry. 2020;19(5):33-39.
7. Provensi G, Passani MB, Costa A, et al. Neuronal histamine and the memory of emotionally salient events. Br J Pharmacol. 2020;177(3):557-569.
8. Purves D, Augustine GJ, Fitzpatrick D, et al (eds). Neuroscience. 2nd ed. Sinauer Associates; 2001.
9. Pytliak M, Vargová V, Mechírová V, et al. Serotonin receptors – from molecular biology to clinical applications. Physiol Res. 2011;60(1):15-25.
10. Meneses A, Liy-Salmeron G. Serotonin and emotion, learning and memory. Rev Neurosci. 2012;23(5-6):543-553.
11. Whitaker-Azmitia PM. Serotonin and brain development: role in human developmental diseases. Brain Res Bull. 2001;56(5):479-485.
12. Towle AC, Breese GR, Mueller RA, et al. Early postnatal administration of 5,7-DHT: effects on serotonergic neurons and terminals. Brain Res. 1984;310(1):67-75.
13. Rok-Bujko P, Krzs´cik P, Szyndler J, et al. The influence of neonatal serotonin depletion on emotional and exploratory behaviours in rats. Behav Brain Res. 2012;226(1):87-95.
14. Meltzer HY. The role of serotonin in antipsychotic drug action. Neuropsychopharmacology. 1999;21(2 Suppl):106S-115S.
15. Calabrese EJ. 5-Hydroxytryptamine (serotonin): biphasic dose responses. Crit Rev Toxicol. 2001;31(4-5):553-561.
16. Zuardi AW. 5-HT-related drugs and human experimental anxiety. Neurosci Biobehav Rev. 1990;14(4):507-510.
17. Sánchez C, Meier E. Behavioral profiles of SSRIs in animal models of depression, anxiety and aggression. Are they all alike? Psychopharmacology (Berl). 1997;129(3):197-205.
18. Koek W, Mitchell NC, Daws LC. Biphasic effects of selective serotonin reuptake inhibitors on anxiety: rapid reversal of escitalopram’s anxiogenic effects in the novelty-induced hypophagia test in mice? Behav Pharmacol. 2018;29(4):365-369.
19. van Zijderveld GA, Veltman DJ, van Dyck R, et al. Epinephrine-induced panic attacks and hyperventilation. J Psychiatr Res. 1999;33(1):73-78.
20. Ho EV, Thompson SL, Katzka WR, et al. Clinically effective OCD treatment prevents 5-HT1B receptor-induced repetitive behavior and striatal activation. Psychopharmacology (Berl). 2016;233(1):57-70.
21. Stein DJ, Costa DLC, Lochner C, et al. Obsessive-compulsive disorder. Nat Rev Dis Primers. 2019;5(1):52.
22. Luddington NS, Mandadapu A, Husk M, et al. Clinical implications of genetic variation in the serotonin transporter promoter region: a review. Prim Care Companion J Clin Psychiatry. 2009;11(3):93-102.
23. Stahl SM. Mechanism of action of serotonin selective reuptake inhibitors. Serotonin receptors and pathways mediate therapeutic effects and side effects. J Affect Disord. 1998;51(3):215-235.
24. Chaouloff F, Berton O, Mormède P. Serotonin and stress. Neuropsychopharmacology. 1999;21(2 Suppl):28S-32S.
25. Siafis S, Tzachanis D, Samara M, et al. Antipsychotic drugs: From receptor-binding profiles to metabolic side effects. Curr Neuropharmacol. 2018;16(8):1210-1223.
26. Carrithers B, El-Mallakh RS. Transdermal asenapine in schizophrenia: a systematic review. Patient Prefer Adherence. 2020;14:1541-1551.
27. Citrome L. Asenapine review, part I: chemistry, receptor affinity profile, pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. 2014;10(6):893-903.
28. Pratts M, Citrome L, Grant W, et al. A single-dose, randomized, double-blind, placebo-controlled trial of sublingual asenapine for acute agitation. Acta Psychiatr Scand. 2014;130(1):61-68.
29. Biswas AB, Bhaumik S, Branford D. Treatment-emergent behavioural side effects with selective serotonin re-uptake inhibitors in adults with learning disabilities. Hum Psychopharmacol. 2001;16(2):133-137.
30. Perlis RH, Mischoulon D, Smoller JW, et al. Serotonin transporter polymorphisms and adverse effects with fluoxetine treatment. Biol Psychiatry. 2003;54(9):879-883.
31. Ipser JC, Carey P, Dhansay Y, et al. Pharmacotherapy augmentation strategies in treatment-resistant anxiety disorders. Cochrane Database Syst Rev. 2006;(4):CD005473.
32. Kane JM, Mackle M, Snow-Adami L, et al. A randomized placebo-controlled trial of asenapine for the prevention of relapse of schizophrenia after long-term treatment. J Clin Psychiatry. 2011;72(3):349-355.
33. Bozzatello P, Rocca P, Uscinska M, et al. Efficacy and tolerability of asenapine compared with olanzapine in borderline personality disorder: an open-label randomized controlled trial. CNS Drugs. 2017;31(9):809-819.
34. El-Mallakh RS, Nuss S, Gao D, et al. Asenapine in the treatment of bipolar depression. Psychopharmacol Bull. 2020;50(1):8-18.
Sara R. Abell, MD, and Rif S. El-Mallakh, MD
Individuals with anxiety will experience frequent or chronic excessive worry, nervousness, a sense of unease, a feeling of being unfocused, and distress, which result in functional impairment.1 Frequently, anxiety is accompanied by restlessness or muscle tension. Generalized anxiety disorder is one of the most common psychiatric diagnoses in the United States and has a prevalence of 2% to 6% globally.2 Although research has been conducted regarding anxiety’s pathogenesis, to date a firm consensus on its etiology has not been reached.3 It is likely multifactorial, with environmental and biologic components.
One area of focus has been neurotransmitters and the possible role they play in the pathogenesis of anxiety. Specifically, the monoamine neurotransmitters have been implicated in the clinical manifestations of anxiety. Among the amines, normal roles include stimulating the autonomic nervous system and regulating numerous cognitive phenomena, such as volition and emotion. Many psychiatric medications modify aminergic transmission, and many current anxiety medications target amine neurotransmitters. Medications that target histamine, serotonin, norepinephrine, and dopamine all play a role in treating anxiety.
In this article, we focus on serotonin (5-hydroxytryptamine, 5-HT) as a mediator of anxiety and on excessive synaptic 5-HT as the cause of anxiety. We discuss how 5-HT–mediated anxiety can be identified and offer some solutions for its treatment.
The amine neurotransmitters
There are 6 amine neurotransmitters in the CNS. These are derived from tyrosine (dopamine [DA], norepinephrine [NE], and epinephrine), histidine (histamine), and tryptophan (serotonin [5-HT] and melatonin). In addition to their physiologic actions, amines have been implicated in both acute and chronic anxiety. Excessive DA stimulation has been linked with fear4,5; NE elevations are central to hypervigilance and hyperarousal of posttraumatic stress disorder6; and histamine may mediate emotional memories involved in fear and anxiety.7 Understanding the normal function of 5-HT will aid in understanding its potential problematic role (Box,8-18page 38).
How serotonin-mediated anxiety presents
“Anxiety” is a collection of signs and symptoms that likely represent multiple processes and have the common characteristic of being subjectively unpleasant, with a subjective wish for the feeling to end. The expression of anxiety disorders is quite diverse and ranges from brief episodes such as panic attacks (which may be mediated, in part, by epinephrine/NE19) to lifelong stereotypic obsessions and compulsions (which may be mediated, in part, by DA and modified by 5-HT20,21). Biochemical separation of the anxiety disorders is key to achieving tailored treatment.6 Towards this end, it is important to investigate the phenomenon of serotonin-mediated anxiety.
Because clinicians are familiar with reductions of anxiety as selective serotonin reuptake inhibitors (SSRIs) increase 5-HT levels in the synapse, it is difficult to conceptualize serotonin-mediated anxiety. However, many of the effects at postsynaptic 5-HT receptors may be biphasic.15-18 Serotonin-mediated anxiety appears to occur when levels of 5-HT (or stimulation of 5-HT receptors) are particularly high. This is most frequently seen in patients who genetically have high synaptic 5-HT (by virtue of the short form of the 5-HT transporter),22 whose synaptic 5-HT is further increased by treatment with an SSRI,23 and who are experiencing a stressor that yet further increases their synaptic 5-HT.24 However, it may occur in some individuals with only 2 of these 3 conditions.Clinically, individuals with serotonin-mediated anxiety will usually appear calm. The anxiety they are experiencing is not exhibited in any way in the motor system (ie, they do not appear restless, do not pace, muscle tone is not increased, etc.). However, they will generally complain of an internal agitation, a sense of a negative internal energy. Frequently, they will use descriptions such as “I feel I could jump out of my skin.” As previously mentioned, this is usually in the setting of some environmental stress, in addition to either a pharmacologic (SSRI) or genetic (short form of the 5-HT transporter) reason for increasing synaptic 5-HT, or both.
Almost always, interventions that block multiple postsynaptic 5-HT receptors or discontinuation of the SSRI (if applicable) will alleviate the anxiety, quickly or more slowly, respectively. Sublingual asenapine, which at low doses can block 5-HT2C (Ki = 0.03 nM), 5-HT2A (Ki = 0.07 nM), 5-HT7 (Ki = 0.11 nM), 5-HT2B (Ki = 0.18 nM), and 5-HT6 (Ki = 0.25 nM),25,26 and which will produce peak plasma levels within 10 minutes,27 usually is quite effective.
Box
Serotonin (5-HT) arises from neurons in the raphe nuclei of the rostral pons and projects superiorly to the cerebral cortex and inferiorly to the spinal cord.8 It works in an inhibitory or excitatory manner depending on which receptors are activated. In the periphery, 5-HT influences intestinal peristalsis, sensory modulation, gland function, thermoregulation, blood pressure, platelet aggregation, and sexual behavior,9 all actions that produce potential adverse effects of serotonin reuptake– inhibiting antidepressants. In the CNS, 5-HT plays a role in attention bias; decision-making; sleep and wakefulness; and mood regulation. In short, serotonin can be viewed as mediating emotional motivation.10
Serotonin alters neuroplasticity. During development, 5-HT stimulates creation of new synapses and increases the density of synaptic webs. It has a direct stimulatory effect on the length of dendrites, their branching, and their myelination.11 In the CNS, it plays a role in dendritic arborization. Animal studies with rats have shown that lesioning highly concentrated 5-HT areas at early ages resulted in an adult brain with a lower number of neurons and a less complex web of dendrites.12,13 In situations of emotional stress, it is theorized that low levels of 5-HT lead to a reduced ability to deal with emotional stressors due to lower levels of complexity in synaptic connections.
Serotonin has also been implicated in mediating some aspects of dopamine-related actions, such as locomotion, reward, and threat avoidance. This is believed to contribute to the beneficial effect of 5-HT2A blockade by secondgeneration antipsychotics (SGAs).14 Blockade of other 5-HT receptors, such as 5-HT1A, 5-HT2C, 5-HT6, and 5-HT7, may also contribute to the antipsychotic action of SGAs.14
Serotonin receptors are found throughout the body, and 14 subtypes have been identified.9 Excitatory and inhibitory action of 5-HT depends on the receptor, and the actions of 5-HT can differ with the same receptor at different concentrations. This is because serotonin’s effects are biphasic and concentration-dependent, meaning that levels of 5-HT in the synapse will dictate the downstream effect of receptor agonism or antagonism. Animal models have shown that low-dose agonism of 5-HT receptors causes vasoconstriction of the coronary arteries, and high doses cause relaxation. This response has also been demonstrated in the vasculature of the kidneys and the smooth muscle of the trachea. Additionally, 5-HT works in conjunction with histamine to produce a biphasic response in the colonic arteries and veins in situations of endothelial damage.15
Most relevant to this discussion are 5-HT’s actions in mood regulation and behavior. Low 5-HT states result in less behavioral inhibition, leading to higher impulse control failures and aggression. Experiments in mice with deficient serotonergic brain regions show hypoactivity, extended daytime sleep, anxiety, and depressive behaviors.13 Serotonin’s behavioral effects are also biphasic. For example, lowdose antagonism with trazodone of 5-HT receptors demonstrated a pro-aggressive behavioral effect, while high-dose antagonism is anti-aggressive.15 Similar biphasic effects may result in either induction or reduction of anxiety with agents that block or excite certain 5-HT receptors.16-18
Continue to: A key difference: No motor system involvement...
A key difference: No motor system involvement
What distinguishes 5-HT from the other amine transmitters as a mediator of anxiety is the lack of involvement of the motor system. Multiple studies in rats illustrate that exogenously augmenting 5-HT has no effect on levels of locomotor activity. Dopamine depletion is well-characterized in the motor dysfunction of Parkinson’s disease, and DA excess can cause repetitive, stereotyped movements, such as seen in tardive dyskinesia or Huntington’s disease.8 In humans, serotonin-mediated anxiety is usually without a motoric component; patients appear calm but complain of extreme anxiety or agitation. Agitation has been reported after initiation of an SSRI,29 and is more likely to occur in patients with the short form of the 5-HT transporter.30 Motoric activation has been reported in some of these studies, but does not seem to cluster with the complaint of agitation.29 The reduced number of available transporters means a chronic steady-state elevation of serotonin, because less serotonin is being removed from the synapse after it is released. This is one of the reasons patients with the short form of the 5-HT transporter may be more susceptible to serotonin-mediated anxiety.
What you need to keep in mind
Pharmacologic treatment of anxiety begins with an SSRI, a serotonin-norepinephrine reuptake inhibitor (SNRI), or buspirone. Second-line treatments include hydroxyzine, gabapentin, pregabalin, and quetiapine.3,31 However, clinicians need to be aware that a fraction of their patients will report anxiety that will not have any external manifestations, but will be experienced as an unpleasant internal energy. These patients may report an increase in their anxiety levels when started on an SSRI or SNRI.29,30 This anxiety is most likely mediated by increases of synaptic 5-HT. This occurs because many serotonergic receptors may have a biphasic response, so that too much stimulation is experienced as excessive internal energy.16-18 In such patients, blockade of key 5-HT receptors may reduce that internal agitation. The advantage of recognizing serotonin-mediated anxiety is that one can specifically tailor treatment to address the patient’s specific physiology.
It is important to note that the anxiolytic effect of asenapine is specific to patients with serotonin-mediated anxiety. Unlike quetiapine, which is effective as augmentation therapy in generalized anxiety disorder,31 asenapine does not appear to reduce anxiety in patients with schizophrenia32 or borderline personality disorder33 when administered for other reasons. However, it may reduce anxiety in patients with the short form of the 5-HT transporter.30,34
Bottom Line
Serotonin-mediated anxiety occurs when levels of synaptic serotonin (5-HT) are high. Patients with serotonin-mediated anxiety appear calm but will report experiencing an unpleasant internal energy. Interventions that block multiple postsynaptic 5-HT receptors or discontinuation of a selective serotonin reuptake inhibitor (if applicable) will alleviate the anxiety.
Related Resource
• Bhatt NV. Anxiety disorders. https://emedicine.medscape. com/article/286227-overview
Drug Brand Names
Asenapine • Saphris, Secuado
Gabapentin • Neurontin
Hydroxyzine • Vistaril
Pregabalin • Lyrica
Quetiapine • Seroquel
Trazodone • Oleptro
Sara R. Abell, MD, and Rif S. El-Mallakh, MD
Individuals with anxiety will experience frequent or chronic excessive worry, nervousness, a sense of unease, a feeling of being unfocused, and distress, which result in functional impairment.1 Frequently, anxiety is accompanied by restlessness or muscle tension. Generalized anxiety disorder is one of the most common psychiatric diagnoses in the United States and has a prevalence of 2% to 6% globally.2 Although research has been conducted regarding anxiety’s pathogenesis, to date a firm consensus on its etiology has not been reached.3 It is likely multifactorial, with environmental and biologic components.
One area of focus has been neurotransmitters and the possible role they play in the pathogenesis of anxiety. Specifically, the monoamine neurotransmitters have been implicated in the clinical manifestations of anxiety. Among the amines, normal roles include stimulating the autonomic nervous system and regulating numerous cognitive phenomena, such as volition and emotion. Many psychiatric medications modify aminergic transmission, and many current anxiety medications target amine neurotransmitters. Medications that target histamine, serotonin, norepinephrine, and dopamine all play a role in treating anxiety.
In this article, we focus on serotonin (5-hydroxytryptamine, 5-HT) as a mediator of anxiety and on excessive synaptic 5-HT as the cause of anxiety. We discuss how 5-HT–mediated anxiety can be identified and offer some solutions for its treatment.
The amine neurotransmitters
There are 6 amine neurotransmitters in the CNS. These are derived from tyrosine (dopamine [DA], norepinephrine [NE], and epinephrine), histidine (histamine), and tryptophan (serotonin [5-HT] and melatonin). In addition to their physiologic actions, amines have been implicated in both acute and chronic anxiety. Excessive DA stimulation has been linked with fear4,5; NE elevations are central to hypervigilance and hyperarousal of posttraumatic stress disorder6; and histamine may mediate emotional memories involved in fear and anxiety.7 Understanding the normal function of 5-HT will aid in understanding its potential problematic role (Box,8-18page 38).
How serotonin-mediated anxiety presents
“Anxiety” is a collection of signs and symptoms that likely represent multiple processes and have the common characteristic of being subjectively unpleasant, with a subjective wish for the feeling to end. The expression of anxiety disorders is quite diverse and ranges from brief episodes such as panic attacks (which may be mediated, in part, by epinephrine/NE19) to lifelong stereotypic obsessions and compulsions (which may be mediated, in part, by DA and modified by 5-HT20,21). Biochemical separation of the anxiety disorders is key to achieving tailored treatment.6 Towards this end, it is important to investigate the phenomenon of serotonin-mediated anxiety.
Because clinicians are familiar with reductions of anxiety as selective serotonin reuptake inhibitors (SSRIs) increase 5-HT levels in the synapse, it is difficult to conceptualize serotonin-mediated anxiety. However, many of the effects at postsynaptic 5-HT receptors may be biphasic.15-18 Serotonin-mediated anxiety appears to occur when levels of 5-HT (or stimulation of 5-HT receptors) are particularly high. This is most frequently seen in patients who genetically have high synaptic 5-HT (by virtue of the short form of the 5-HT transporter),22 whose synaptic 5-HT is further increased by treatment with an SSRI,23 and who are experiencing a stressor that yet further increases their synaptic 5-HT.24 However, it may occur in some individuals with only 2 of these 3 conditions.Clinically, individuals with serotonin-mediated anxiety will usually appear calm. The anxiety they are experiencing is not exhibited in any way in the motor system (ie, they do not appear restless, do not pace, muscle tone is not increased, etc.). However, they will generally complain of an internal agitation, a sense of a negative internal energy. Frequently, they will use descriptions such as “I feel I could jump out of my skin.” As previously mentioned, this is usually in the setting of some environmental stress, in addition to either a pharmacologic (SSRI) or genetic (short form of the 5-HT transporter) reason for increasing synaptic 5-HT, or both.
Almost always, interventions that block multiple postsynaptic 5-HT receptors or discontinuation of the SSRI (if applicable) will alleviate the anxiety, quickly or more slowly, respectively. Sublingual asenapine, which at low doses can block 5-HT2C (Ki = 0.03 nM), 5-HT2A (Ki = 0.07 nM), 5-HT7 (Ki = 0.11 nM), 5-HT2B (Ki = 0.18 nM), and 5-HT6 (Ki = 0.25 nM),25,26 and which will produce peak plasma levels within 10 minutes,27 usually is quite effective.
Box
Serotonin (5-HT) arises from neurons in the raphe nuclei of the rostral pons and projects superiorly to the cerebral cortex and inferiorly to the spinal cord.8 It works in an inhibitory or excitatory manner depending on which receptors are activated. In the periphery, 5-HT influences intestinal peristalsis, sensory modulation, gland function, thermoregulation, blood pressure, platelet aggregation, and sexual behavior,9 all actions that produce potential adverse effects of serotonin reuptake– inhibiting antidepressants. In the CNS, 5-HT plays a role in attention bias; decision-making; sleep and wakefulness; and mood regulation. In short, serotonin can be viewed as mediating emotional motivation.10
Serotonin alters neuroplasticity. During development, 5-HT stimulates creation of new synapses and increases the density of synaptic webs. It has a direct stimulatory effect on the length of dendrites, their branching, and their myelination.11 In the CNS, it plays a role in dendritic arborization. Animal studies with rats have shown that lesioning highly concentrated 5-HT areas at early ages resulted in an adult brain with a lower number of neurons and a less complex web of dendrites.12,13 In situations of emotional stress, it is theorized that low levels of 5-HT lead to a reduced ability to deal with emotional stressors due to lower levels of complexity in synaptic connections.
Serotonin has also been implicated in mediating some aspects of dopamine-related actions, such as locomotion, reward, and threat avoidance. This is believed to contribute to the beneficial effect of 5-HT2A blockade by secondgeneration antipsychotics (SGAs).14 Blockade of other 5-HT receptors, such as 5-HT1A, 5-HT2C, 5-HT6, and 5-HT7, may also contribute to the antipsychotic action of SGAs.14
Serotonin receptors are found throughout the body, and 14 subtypes have been identified.9 Excitatory and inhibitory action of 5-HT depends on the receptor, and the actions of 5-HT can differ with the same receptor at different concentrations. This is because serotonin’s effects are biphasic and concentration-dependent, meaning that levels of 5-HT in the synapse will dictate the downstream effect of receptor agonism or antagonism. Animal models have shown that low-dose agonism of 5-HT receptors causes vasoconstriction of the coronary arteries, and high doses cause relaxation. This response has also been demonstrated in the vasculature of the kidneys and the smooth muscle of the trachea. Additionally, 5-HT works in conjunction with histamine to produce a biphasic response in the colonic arteries and veins in situations of endothelial damage.15
Most relevant to this discussion are 5-HT’s actions in mood regulation and behavior. Low 5-HT states result in less behavioral inhibition, leading to higher impulse control failures and aggression. Experiments in mice with deficient serotonergic brain regions show hypoactivity, extended daytime sleep, anxiety, and depressive behaviors.13 Serotonin’s behavioral effects are also biphasic. For example, lowdose antagonism with trazodone of 5-HT receptors demonstrated a pro-aggressive behavioral effect, while high-dose antagonism is anti-aggressive.15 Similar biphasic effects may result in either induction or reduction of anxiety with agents that block or excite certain 5-HT receptors.16-18
Continue to: A key difference: No motor system involvement...
A key difference: No motor system involvement
What distinguishes 5-HT from the other amine transmitters as a mediator of anxiety is the lack of involvement of the motor system. Multiple studies in rats illustrate that exogenously augmenting 5-HT has no effect on levels of locomotor activity. Dopamine depletion is well-characterized in the motor dysfunction of Parkinson’s disease, and DA excess can cause repetitive, stereotyped movements, such as seen in tardive dyskinesia or Huntington’s disease.8 In humans, serotonin-mediated anxiety is usually without a motoric component; patients appear calm but complain of extreme anxiety or agitation. Agitation has been reported after initiation of an SSRI,29 and is more likely to occur in patients with the short form of the 5-HT transporter.30 Motoric activation has been reported in some of these studies, but does not seem to cluster with the complaint of agitation.29 The reduced number of available transporters means a chronic steady-state elevation of serotonin, because less serotonin is being removed from the synapse after it is released. This is one of the reasons patients with the short form of the 5-HT transporter may be more susceptible to serotonin-mediated anxiety.
What you need to keep in mind
Pharmacologic treatment of anxiety begins with an SSRI, a serotonin-norepinephrine reuptake inhibitor (SNRI), or buspirone. Second-line treatments include hydroxyzine, gabapentin, pregabalin, and quetiapine.3,31 However, clinicians need to be aware that a fraction of their patients will report anxiety that will not have any external manifestations, but will be experienced as an unpleasant internal energy. These patients may report an increase in their anxiety levels when started on an SSRI or SNRI.29,30 This anxiety is most likely mediated by increases of synaptic 5-HT. This occurs because many serotonergic receptors may have a biphasic response, so that too much stimulation is experienced as excessive internal energy.16-18 In such patients, blockade of key 5-HT receptors may reduce that internal agitation. The advantage of recognizing serotonin-mediated anxiety is that one can specifically tailor treatment to address the patient’s specific physiology.
It is important to note that the anxiolytic effect of asenapine is specific to patients with serotonin-mediated anxiety. Unlike quetiapine, which is effective as augmentation therapy in generalized anxiety disorder,31 asenapine does not appear to reduce anxiety in patients with schizophrenia32 or borderline personality disorder33 when administered for other reasons. However, it may reduce anxiety in patients with the short form of the 5-HT transporter.30,34
Bottom Line
Serotonin-mediated anxiety occurs when levels of synaptic serotonin (5-HT) are high. Patients with serotonin-mediated anxiety appear calm but will report experiencing an unpleasant internal energy. Interventions that block multiple postsynaptic 5-HT receptors or discontinuation of a selective serotonin reuptake inhibitor (if applicable) will alleviate the anxiety.
Related Resource
• Bhatt NV. Anxiety disorders. https://emedicine.medscape. com/article/286227-overview
Drug Brand Names
Asenapine • Saphris, Secuado
Gabapentin • Neurontin
Hydroxyzine • Vistaril
Pregabalin • Lyrica
Quetiapine • Seroquel
Trazodone • Oleptro
1. Shelton CI. Diagnosis and management of anxiety disorders. J Am Osteopath Assoc. 2004;104(3 Suppl 3):S2-S5.
2. Ruscio AM, Hallion LS, Lim CCW, et al. Cross-sectional comparison of the epidemiology of DSM-5 generalized anxiety disorder across the globe. JAMA Psychiatry. 2017;74(5):465-475.
3. Locke AB, Kirst N, Shultz CG. Diagnosis and management of generalized anxiety disorder and panic disorder in adults. Am Fam Physician. 2015;91(9):617-624.
4. Hariri AR, Mattay VS, Tessitore A, et al. Dextroamphetamine modulates the response of the human amygdala. Neuropsychopharmacology. 2002;27(6):1036-1040.
5. Colombo AC, de Oliveira AR, Reimer AE, et al. Dopaminergic mechanisms underlying catalepsy, fear and anxiety: do they interact? Behav Brain Res. 2013;257:201-207.
6. Togay B, El-Mallakh RS. Posttraumatic stress disorder: from pathophysiology to pharmacology. Curr Psychiatry. 2020;19(5):33-39.
7. Provensi G, Passani MB, Costa A, et al. Neuronal histamine and the memory of emotionally salient events. Br J Pharmacol. 2020;177(3):557-569.
8. Purves D, Augustine GJ, Fitzpatrick D, et al (eds). Neuroscience. 2nd ed. Sinauer Associates; 2001.
9. Pytliak M, Vargová V, Mechírová V, et al. Serotonin receptors – from molecular biology to clinical applications. Physiol Res. 2011;60(1):15-25.
10. Meneses A, Liy-Salmeron G. Serotonin and emotion, learning and memory. Rev Neurosci. 2012;23(5-6):543-553.
11. Whitaker-Azmitia PM. Serotonin and brain development: role in human developmental diseases. Brain Res Bull. 2001;56(5):479-485.
12. Towle AC, Breese GR, Mueller RA, et al. Early postnatal administration of 5,7-DHT: effects on serotonergic neurons and terminals. Brain Res. 1984;310(1):67-75.
13. Rok-Bujko P, Krzs´cik P, Szyndler J, et al. The influence of neonatal serotonin depletion on emotional and exploratory behaviours in rats. Behav Brain Res. 2012;226(1):87-95.
14. Meltzer HY. The role of serotonin in antipsychotic drug action. Neuropsychopharmacology. 1999;21(2 Suppl):106S-115S.
15. Calabrese EJ. 5-Hydroxytryptamine (serotonin): biphasic dose responses. Crit Rev Toxicol. 2001;31(4-5):553-561.
16. Zuardi AW. 5-HT-related drugs and human experimental anxiety. Neurosci Biobehav Rev. 1990;14(4):507-510.
17. Sánchez C, Meier E. Behavioral profiles of SSRIs in animal models of depression, anxiety and aggression. Are they all alike? Psychopharmacology (Berl). 1997;129(3):197-205.
18. Koek W, Mitchell NC, Daws LC. Biphasic effects of selective serotonin reuptake inhibitors on anxiety: rapid reversal of escitalopram’s anxiogenic effects in the novelty-induced hypophagia test in mice? Behav Pharmacol. 2018;29(4):365-369.
19. van Zijderveld GA, Veltman DJ, van Dyck R, et al. Epinephrine-induced panic attacks and hyperventilation. J Psychiatr Res. 1999;33(1):73-78.
20. Ho EV, Thompson SL, Katzka WR, et al. Clinically effective OCD treatment prevents 5-HT1B receptor-induced repetitive behavior and striatal activation. Psychopharmacology (Berl). 2016;233(1):57-70.
21. Stein DJ, Costa DLC, Lochner C, et al. Obsessive-compulsive disorder. Nat Rev Dis Primers. 2019;5(1):52.
22. Luddington NS, Mandadapu A, Husk M, et al. Clinical implications of genetic variation in the serotonin transporter promoter region: a review. Prim Care Companion J Clin Psychiatry. 2009;11(3):93-102.
23. Stahl SM. Mechanism of action of serotonin selective reuptake inhibitors. Serotonin receptors and pathways mediate therapeutic effects and side effects. J Affect Disord. 1998;51(3):215-235.
24. Chaouloff F, Berton O, Mormède P. Serotonin and stress. Neuropsychopharmacology. 1999;21(2 Suppl):28S-32S.
25. Siafis S, Tzachanis D, Samara M, et al. Antipsychotic drugs: From receptor-binding profiles to metabolic side effects. Curr Neuropharmacol. 2018;16(8):1210-1223.
26. Carrithers B, El-Mallakh RS. Transdermal asenapine in schizophrenia: a systematic review. Patient Prefer Adherence. 2020;14:1541-1551.
27. Citrome L. Asenapine review, part I: chemistry, receptor affinity profile, pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. 2014;10(6):893-903.
28. Pratts M, Citrome L, Grant W, et al. A single-dose, randomized, double-blind, placebo-controlled trial of sublingual asenapine for acute agitation. Acta Psychiatr Scand. 2014;130(1):61-68.
29. Biswas AB, Bhaumik S, Branford D. Treatment-emergent behavioural side effects with selective serotonin re-uptake inhibitors in adults with learning disabilities. Hum Psychopharmacol. 2001;16(2):133-137.
30. Perlis RH, Mischoulon D, Smoller JW, et al. Serotonin transporter polymorphisms and adverse effects with fluoxetine treatment. Biol Psychiatry. 2003;54(9):879-883.
31. Ipser JC, Carey P, Dhansay Y, et al. Pharmacotherapy augmentation strategies in treatment-resistant anxiety disorders. Cochrane Database Syst Rev. 2006;(4):CD005473.
32. Kane JM, Mackle M, Snow-Adami L, et al. A randomized placebo-controlled trial of asenapine for the prevention of relapse of schizophrenia after long-term treatment. J Clin Psychiatry. 2011;72(3):349-355.
33. Bozzatello P, Rocca P, Uscinska M, et al. Efficacy and tolerability of asenapine compared with olanzapine in borderline personality disorder: an open-label randomized controlled trial. CNS Drugs. 2017;31(9):809-819.
34. El-Mallakh RS, Nuss S, Gao D, et al. Asenapine in the treatment of bipolar depression. Psychopharmacol Bull. 2020;50(1):8-18.
1. Shelton CI. Diagnosis and management of anxiety disorders. J Am Osteopath Assoc. 2004;104(3 Suppl 3):S2-S5.
2. Ruscio AM, Hallion LS, Lim CCW, et al. Cross-sectional comparison of the epidemiology of DSM-5 generalized anxiety disorder across the globe. JAMA Psychiatry. 2017;74(5):465-475.
3. Locke AB, Kirst N, Shultz CG. Diagnosis and management of generalized anxiety disorder and panic disorder in adults. Am Fam Physician. 2015;91(9):617-624.
4. Hariri AR, Mattay VS, Tessitore A, et al. Dextroamphetamine modulates the response of the human amygdala. Neuropsychopharmacology. 2002;27(6):1036-1040.
5. Colombo AC, de Oliveira AR, Reimer AE, et al. Dopaminergic mechanisms underlying catalepsy, fear and anxiety: do they interact? Behav Brain Res. 2013;257:201-207.
6. Togay B, El-Mallakh RS. Posttraumatic stress disorder: from pathophysiology to pharmacology. Curr Psychiatry. 2020;19(5):33-39.
7. Provensi G, Passani MB, Costa A, et al. Neuronal histamine and the memory of emotionally salient events. Br J Pharmacol. 2020;177(3):557-569.
8. Purves D, Augustine GJ, Fitzpatrick D, et al (eds). Neuroscience. 2nd ed. Sinauer Associates; 2001.
9. Pytliak M, Vargová V, Mechírová V, et al. Serotonin receptors – from molecular biology to clinical applications. Physiol Res. 2011;60(1):15-25.
10. Meneses A, Liy-Salmeron G. Serotonin and emotion, learning and memory. Rev Neurosci. 2012;23(5-6):543-553.
11. Whitaker-Azmitia PM. Serotonin and brain development: role in human developmental diseases. Brain Res Bull. 2001;56(5):479-485.
12. Towle AC, Breese GR, Mueller RA, et al. Early postnatal administration of 5,7-DHT: effects on serotonergic neurons and terminals. Brain Res. 1984;310(1):67-75.
13. Rok-Bujko P, Krzs´cik P, Szyndler J, et al. The influence of neonatal serotonin depletion on emotional and exploratory behaviours in rats. Behav Brain Res. 2012;226(1):87-95.
14. Meltzer HY. The role of serotonin in antipsychotic drug action. Neuropsychopharmacology. 1999;21(2 Suppl):106S-115S.
15. Calabrese EJ. 5-Hydroxytryptamine (serotonin): biphasic dose responses. Crit Rev Toxicol. 2001;31(4-5):553-561.
16. Zuardi AW. 5-HT-related drugs and human experimental anxiety. Neurosci Biobehav Rev. 1990;14(4):507-510.
17. Sánchez C, Meier E. Behavioral profiles of SSRIs in animal models of depression, anxiety and aggression. Are they all alike? Psychopharmacology (Berl). 1997;129(3):197-205.
18. Koek W, Mitchell NC, Daws LC. Biphasic effects of selective serotonin reuptake inhibitors on anxiety: rapid reversal of escitalopram’s anxiogenic effects in the novelty-induced hypophagia test in mice? Behav Pharmacol. 2018;29(4):365-369.
19. van Zijderveld GA, Veltman DJ, van Dyck R, et al. Epinephrine-induced panic attacks and hyperventilation. J Psychiatr Res. 1999;33(1):73-78.
20. Ho EV, Thompson SL, Katzka WR, et al. Clinically effective OCD treatment prevents 5-HT1B receptor-induced repetitive behavior and striatal activation. Psychopharmacology (Berl). 2016;233(1):57-70.
21. Stein DJ, Costa DLC, Lochner C, et al. Obsessive-compulsive disorder. Nat Rev Dis Primers. 2019;5(1):52.
22. Luddington NS, Mandadapu A, Husk M, et al. Clinical implications of genetic variation in the serotonin transporter promoter region: a review. Prim Care Companion J Clin Psychiatry. 2009;11(3):93-102.
23. Stahl SM. Mechanism of action of serotonin selective reuptake inhibitors. Serotonin receptors and pathways mediate therapeutic effects and side effects. J Affect Disord. 1998;51(3):215-235.
24. Chaouloff F, Berton O, Mormède P. Serotonin and stress. Neuropsychopharmacology. 1999;21(2 Suppl):28S-32S.
25. Siafis S, Tzachanis D, Samara M, et al. Antipsychotic drugs: From receptor-binding profiles to metabolic side effects. Curr Neuropharmacol. 2018;16(8):1210-1223.
26. Carrithers B, El-Mallakh RS. Transdermal asenapine in schizophrenia: a systematic review. Patient Prefer Adherence. 2020;14:1541-1551.
27. Citrome L. Asenapine review, part I: chemistry, receptor affinity profile, pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. 2014;10(6):893-903.
28. Pratts M, Citrome L, Grant W, et al. A single-dose, randomized, double-blind, placebo-controlled trial of sublingual asenapine for acute agitation. Acta Psychiatr Scand. 2014;130(1):61-68.
29. Biswas AB, Bhaumik S, Branford D. Treatment-emergent behavioural side effects with selective serotonin re-uptake inhibitors in adults with learning disabilities. Hum Psychopharmacol. 2001;16(2):133-137.
30. Perlis RH, Mischoulon D, Smoller JW, et al. Serotonin transporter polymorphisms and adverse effects with fluoxetine treatment. Biol Psychiatry. 2003;54(9):879-883.
31. Ipser JC, Carey P, Dhansay Y, et al. Pharmacotherapy augmentation strategies in treatment-resistant anxiety disorders. Cochrane Database Syst Rev. 2006;(4):CD005473.
32. Kane JM, Mackle M, Snow-Adami L, et al. A randomized placebo-controlled trial of asenapine for the prevention of relapse of schizophrenia after long-term treatment. J Clin Psychiatry. 2011;72(3):349-355.
33. Bozzatello P, Rocca P, Uscinska M, et al. Efficacy and tolerability of asenapine compared with olanzapine in borderline personality disorder: an open-label randomized controlled trial. CNS Drugs. 2017;31(9):809-819.
34. El-Mallakh RS, Nuss S, Gao D, et al. Asenapine in the treatment of bipolar depression. Psychopharmacol Bull. 2020;50(1):8-18.

Borderline personality disorder: 6 studies of biological interventions
FIRST OF 2 PARTS
Borderline personality disorder (BPD) is marked by an ongoing pattern of mood instability, cognitive distortions, problems with self-image, and impulsive behavior, often resulting in problems in relationships. BPD is associated with serious impairment in psychosocial functioning.1 Patients with BPD tend to use more mental health services than patients with other personality disorders or those with major depressive disorder (MDD).2 However, there has been little consensus on the best treatment(s) for this serious and debilitating disorder, and some clinicians view BPD as difficult to treat.
Current treatments for BPD include psychological and pharmacological interventions. Neuromodulation techniques, such as repetitive transcranial magnetic stimulation, may also positively affect BPD symptomatology. In recent years, there have been some promising findings in the treatment of BPD. In this 2-part article, we focus on current (within the last 5 years) findings from randomized controlled trials (RCTs) of BPD treatments. Here in Part 1, we focus on 6 studies that evaluated biological interventions (Table,3-8). In Part 2, we will focus on RCTs that investigated psychological interventions.
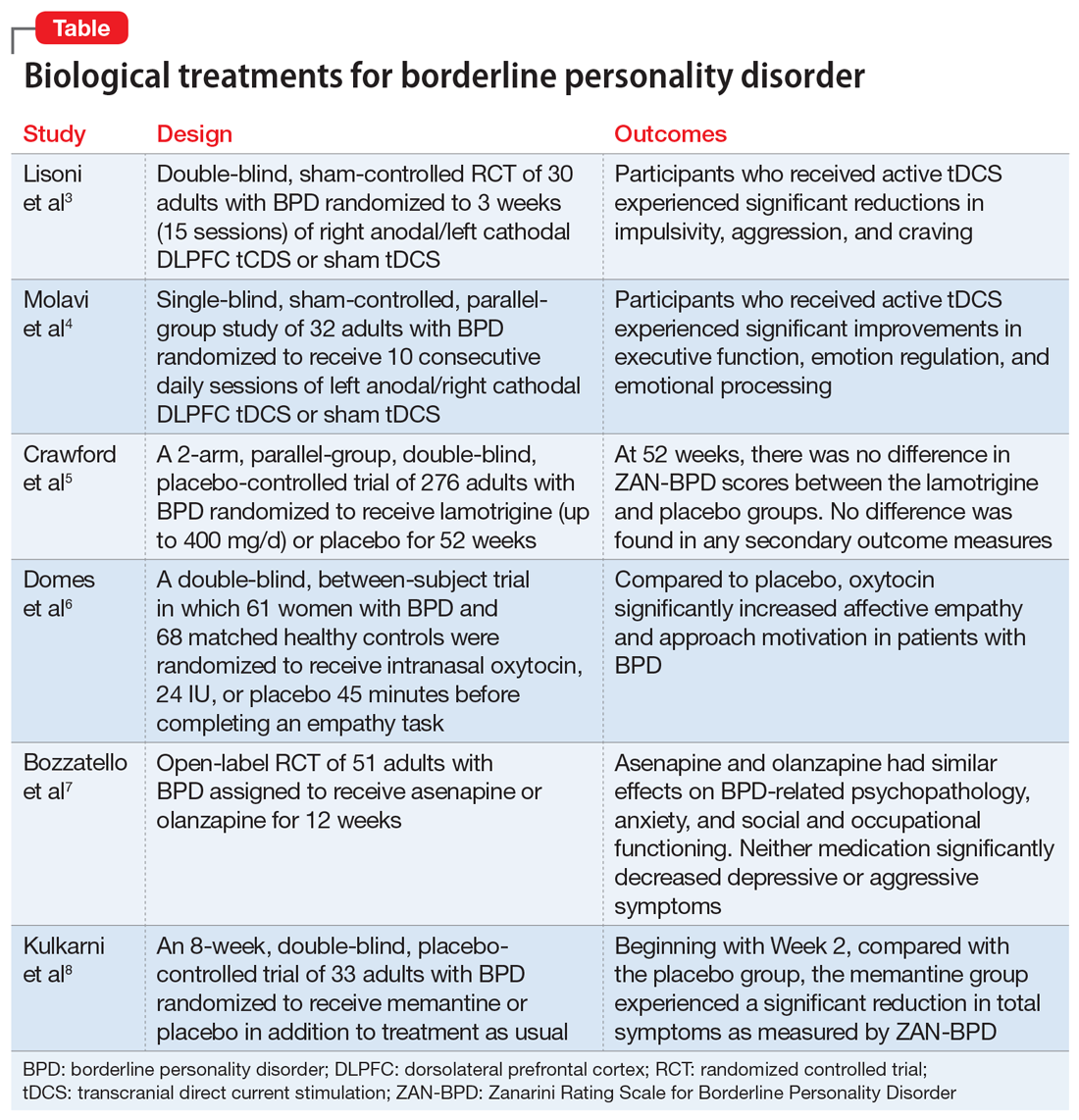
1. Lisoni J, Miotto P, Barlati S, et al. Change in core symptoms of borderline personality disorder by tDCS: a pilot study. Psychiatry Res. 2020;291:113261. doi: 10.1016/j.psychres.2020.113261
Impulsivity has been described as the core feature of BPD that best explains its behavioral, cognitive, and clinical manifestations. Studies have repeatedly demonstrated the role of the prefrontal cortex in modulating impulsivity. Dysfunction of the
Continue to: Study design...
Study design
- In a double-blind, sham-controlled trial, adults who met DSM-IV-TR criteria for BPD were randomized to 3 weeks (15 sessions) of right anodal/left cathodal DLPFC tCDS (n = 15) or sham tDCS (n = 15). This study included patients with comorbid psychiatric disorders, including substance use disorders. Discontinuation or alteration of existing medications was not allowed.
- The presence, severity, and change over time of BPD core symptoms was assessed at baseline and after 3 weeks using several clinical scales, self-questionnaires, and neuropsychological tests, including the Barratt Impulsiveness Scale-11 (BIS-11), Buss-Perry Aggression Questionnaire (BP-AQ), Difficulties in Emotion Regulation Scale (DERS), Hamilton Depression Rating Scale (HAM-D), Beck Depression Inventory (BDI), Hamilton Anxiety Rating Scale (HAM-A), Irritability-Depression Anxiety Scale (IDA), Visual Analog Scales (VAS), and Iowa Gambling Task.
Outcomes
- Participants in the active tDCS group experienced significant reductions in impulsivity, aggression, and craving as measured by the BIS-11, BP-AQ, and VAS.
- Compared to the sham group, the active tDCS group had greater reductions in HAM-D and BDI scores.
- HAM-A and IDA scores were improved in both groups, although the active tDCS group showed greater reductions in IDA scores compared with the sham group.
- As measured by DERS, active tDCS did not improve affective dysregulation more than sham tDCS.
Conclusions/limitations
- Bilateral tDCS targeting the right DLPFC with anodal stimulation is a safe, well-tolerated technique that may modulate core dimensions of BPD, including impulsivity, aggression, and craving.
- Excitatory anodal stimulation of the right DLFPC coupled with inhibitory cathodal stimulation on the left DLPFC may be an effective montage for targeting impulsivity in patients with BPD.
- Study limitations include a small sample size, use of targeted questionnaires only, inclusion of patients with BPD who also had certain comorbid psychiatric disorders, lack of analysis of the contributions of medications, lack of functional neuroimaging, and lack of a follow-up phase.
2. Molavi P, Aziziaram S, Basharpoor S, et al. Repeated transcranial direct current stimulation of dorsolateral-prefrontal cortex improves executive functions, cognitive reappraisal emotion regulation, and control over emotional processing in borderline personality disorder: a randomized, sham-controlled, parallel-group study. J Affect Disord. 2020;274:93-102. doi: 10.1016/j.jad.2020.05.007
Emotional dysregulation is considered a core feature of BPD psychopathology and is closely associated with executive dysfunction and cognitive control. Manifestations of executive dysfunction include aggressiveness, impulsive decision-making, disinhibition, and self-destructive behaviors. Neuroimaging of patients with BPD has shown enhanced activity in the insula, posterior cingulate cortex, and amygdala, with reduced activity in the medial PFC, subgenual anterior cingulate cortex, and DLPFC. Molavi et al4 postulated that increasing DLPFC activation with left anodal tDCS would result in improved executive functioning and emotion dysregulation in patients with BPD.
Study design
- In this single-blind, sham-controlled, parallel-group study, adults who met DSM-5 criteria for BPD were randomized to receive 10 consecutive daily sessions of left anodal/right cathodal DLPFC tDCS (n = 16) or sham tDCS (n = 16).
- The effect of tDCS on executive dysfunction, emotion dysregulation, and emotional processing was measured using the Executive Skills Questionnaire for Adults (ESQ), Emotion Regulation Questionnaire (ERQ), and Emotional Processing Scale (EPS). Measurements occurred at baseline and after 10 sessions of active or sham tDCS.
Outcomes
- Participants who received active tDCS experienced significant improvements in ESQ overall score and most of the executive function domains measured by the ESQ.
- Those in the active tDCS group also experienced significant improvement in emotion regulation as measured by the cognitive reappraisal subscale (but not the expressive suppression subscale) of the ERQ after the intervention.
- Overall emotional processing as measured by the EPS was significantly improved in the active tDCS group following the intervention.
Conclusions/limitations
- Repeated bilateral left anodal/right cathodal tDCS stimulation of the DLPFC significantly improved executive functioning and aspects of emotion regulation and emotional processing in patients with BPD. This improvement was presumed to be the result of increased activity of left DLPFC.
- Study limitations include a single-blind design, lack of follow-up to assess durability and stability of response over time, reliance on self-report measures, lack of functional neuroimaging, and limited focality of tDCS.
3. Crawford MJ, Sanatinia R, Barrett B, et al; LABILE study team. The clinical effectiveness and cost-effectiveness of lamotrigine in borderline personality disorder: a randomized placebo-controlled trial. Am J Psychiatry. 2018;175(8):756-764. doi: 10.1176/appi.ajp.2018.17091006
One of the hallmark symptoms of BPD is mood dysregulation. Current treatment guidelines recommend the use of mood stabilizers for BPD despite limited quality evidence of effectiveness and a lack of FDA-approved medications with this indication. In this RCT, Crawford et al5 examined whether lamotrigine is a clinically effective and cost-effective treatment for people with BPD.
Continue to: Study design...
Study design
- In this 2-arm, parallel-group, double-blind, placebo-controlled trial, 276 adults who met DSM-IV criteria for BPD were randomized to receive lamotrigine (up to 400 mg/d) or placebo for 52 weeks.
- The primary outcome was the score on the Zanarini Rating Scale for Borderline Personality Disorder (ZAN-BPD) at 52 weeks. Secondary outcomes included depressive symptoms, deliberate self-harm, social functioning, health-related quality of life, resource use and costs, treatment adverse effects, and adverse events. These were assessed using the BDI; Acts of Deliberate Self-Harm Inventory; Social Functioning Questionnaire; Alcohol, Smoking, and Substance Involvement Screening Test; and the EQ-5D-3L.
Outcomes
- Mean ZAN-BPD score decreased at 12 weeks in both groups, after which time the score remained stable.
- There was no difference in ZAN-BPD scores at 52 weeks between treatment arms. No difference was found in any secondary outcome measures.
- Difference in costs between groups was not significant.
Conclusions/limitations
- There was no evidence that lamotrigine led to clinical improvements in BPD symptomatology, social functioning, health-related quality of life, or substance use.
- Lamotrigine is neither clinically effective nor a cost-effective use of resources in the treatment of BPD.
- Limitations include a low level of adherence.
4. Domes G, Ower N, von Dawans B, et al. Effects of intranasal oxytocin administration on empathy and approach motivation in women with borderline personality disorder: a randomized controlled trial. Transl Psychiatry. 2019;9(1):328. doi: 10.1038/s41398-019-0658-4
A core feature of BPD is impairment in empathy; adequate empathy is required for intact social functioning. Oxytocin is a neuropeptide that helps regulate complex social cognition and behavior. Prior research has found that oxytocin administration enhances emotion regulation and empathy. Women with BPD have been observed to have lower levels of oxytocin. Domes et al6 conducted an RCT to see if oxytocin could have a beneficial effect on social approach and social cognition in women with BPD.
Study design
- In a double-blind, placebo-controlled, between-subject trial, 61 women who met DSM-IV criteria for BPD and 68 matched healthy controls were randomized to receive intranasal oxytocin, 24 IU, or placebo 45 minutes before completing an empathy task.
- An extended version of the Multifaceted Empathy Test was used to assess empathy and approach motivation.
Outcomes
- For cognitive empathy, patients with BPD exhibited significantly lower overall performance compared to controls. There was no effect of oxytocin on this performance in either group.
- Patients with BPD had significantly lower affective empathy compared with controls. After oxytocin administration, patients with BPD had significantly higher affective empathy than those with BPD who received placebo, reaching the level of healthy controls who received placebo.
- For positive stimuli, patients with BPD showed lower affective empathy than controls. Oxytocin treatment increased affective empathy in both groups.
- For negative stimuli, oxytocin increased affective empathy more in patients with BPD than in controls.
- Patients with BPD demonstrated less approach motivation than controls. Oxytocin increased approach motivation more in patients with BPD than in controls. For approach motivation toward positive stimuli, oxytocin had a significant effect on patients with BPD.
Continue to: Conclusions/limitations...
Conclusions/limitations
- Patients with BPD showed reduced cognitive and affective empathy and less approach behavior motivation than healthy controls.
- Patients with BPD who received oxytocin attained a level of affective empathy and approach motivation similar to that of healthy controls who received placebo. For positive stimuli, both groups exhibited comparable improvements from oxytocin. For negative stimuli, patients with BPD patients showed significant improvement with oxytocin, whereas healthy controls received no such benefit.
- Limitations include the use of self-report scales, lack of a control group, and inclusion of patients using psychotherapeutic medications. The study lacks generalizability because only women were included; the effect of exogenous oxytocin on men may differ.
5. Bozzatello P, Rocca P, Uscinska M, et al. Efficacy and tolerability of asenapine compared with olanzapine in borderline personality disorder: an open-label randomized controlled trial. CNS Drugs. 2017;31(9):809-819. doi: 10.1007/s40263-017-0458-4
The last decade has seen a noticeable shift in clinical practice from the use of antidepressants to mood stabilizers and second-generation antipsychotics (SGAs) in the treatment of BPD. Studies have demonstrated therapeutic effects of antipsychotic drugs across a wide range of BPD symptoms. Among SGAs, olanzapine is the most extensively studied across case reports, open-label studies, and RCTs of patients with BPD. In an RCT, Bozzatello et al7 compared the efficacy and tolerability of asenapine to olanzapine.
Study design
- In this open-label RCT, adults who met DSM-5 criteria for BPD were assigned to receive asenapine (n = 25) or olanzapine (n = 26) for 12 weeks.
- Study measurements included the Clinical Global Impression Scale, Severity item, HAM-D, HAM-A, Social and Occupational Functioning Assessment Scale, Borderline Personality Disorder Severity Index (BPDSI), BIS-11, Modified Overt Aggression Scale, and Dosage Record Treatment Emergent Symptom Scale.
Outcomes
- Asenapine and olanzapine had similar effects on BPD-related psychopathology, anxiety, and social and occupational functioning.
- Neither medication significantly decreased depressive or aggressive symptoms.
- Asenapine was superior to olanzapine in reducing the affective instability score of the BPDSI.
- Akathisia and restlessness/anxiety were more common with asenapine, and somnolence and fatigue were more common with olanzapine.
Conclusions/limitations
- The overall efficacy of asenapine was not different from olanzapine, and both medications were well-tolerated.
- Neither medication led to an improvement in depression or aggression, but asenapine was superior to olanzapine in reducing the severity of affective instability.
- Limitations include an open-label design, lack of placebo group, small sample size, high drop-out rate, exclusion of participants with co-occurring MDD and substance abuse/dependence, lack of data on prior pharmacotherapies and psychotherapies, and lack of power to detect a difference on the dissociation/paranoid ideation item of BPDSI.
6. Kulkarni J, Thomas N, Hudaib AR, et al. Effect of the glutamate NMDA receptor antagonist memantine as adjunctive treatment in borderline personality disorder: an exploratory, randomised, double-blind, placebo-controlled trial. CNS Drugs. 2018;32(2):179-187. doi: 10.1007/s40263-018-0506-8
It has been hypothesized that glutamate dysregulation and excitotoxicity are crucial to the development of the cognitive disturbances that underlie BPD. As such, glutamate modulators such as memantine hold promise for the treatment of BPD. In this RCT, Kulkarni et al8 examined the efficacy and tolerability of memantine compared with treatment as usual in patients with BPD.
Continue to: Study design...
Study design
- In an 8-week, double-blind, placebo-controlled trial, adults diagnosed with BPD according to the Diagnostic Interview for Borderline Patients were randomized to receive memantine (n = 17) or placebo (n = 16) in addition to treatment as usual. Treatment as usual included the use of antidepressants, mood stabilizers, and antipsychotics as well as psychotherapy and other psychosocial interventions.
- Patients were initiated on placebo or memantine, 10 mg/d. Memantine was increased to 20 mg/d after 7 days.
- ZAN-BPD score was the primary outcome and was measured at baseline and 2, 4, 6, and 8 weeks. An adverse effects questionnaire was administered every 2 weeks to assess tolerability.
Outcomes
- During the first 2 weeks of treatment, there were no significant improvements in ZAN-BPD score in the memantine group compared with the placebo group.
- Beginning with Week 2, compared with the placebo group, the memantine group experienced a significant reduction in total symptoms as measured by ZAN-BPD.
- There were no statistically significant differences in adverse events between groups.
Conclusions/limitations
- Memantine appears to be a well-tolerated treatment option for patients with BPD and merits further study.
- Limitations include a small sample size, and an inability to reach plateau of ZAN-BPD total score in either group. Also, there is considerable individual variability in memantine steady-state plasma concentrations, but plasma levels were not measured in this study.
Bottom Line
Findings from small randomized controlled trials suggest that transcranial direct current stimulation, oxytocin, asenapine, olanzapine, and memantine may have beneficial effects on some core symptoms of borderline personality disorder. These findings need to be replicated in larger studies.
1. Skodol AE, Gunderson JG, McGlashan TM, et al. Functional impairment in patients with schizotypal, borderline, avoidant, or obsessive-compulsive personality disorder. Am J Psychiatry. 2002; 159:276-283.
2. Bender DS, Dolan RT, Skodol AE, et al. Treatment utilization by patients with personality disorders. Am J Psychiatry. 2001;158:295-302.
3. Lisoni J, Miotto P, Barlati S, et al. Change in core symptoms of borderline personality disorder by tDCS: a pilot study. Psychiatry Res. 2020;291:113261. doi: 10.1016/j.psychres.2020.113261
4. Molavi P, Aziziaram S, Basharpoor S, et al. Repeated transcranial direct current stimulation of dorsolateral-prefrontal cortex improves executive functions, cognitive reappraisal emotion regulation, and control over emotional processing in borderline personality disorder: a randomized, sham-controlled, parallel-group study. J Affect Disord. 2020;274:93-102. doi: 10.1016/j.jad.2020.05.007
5. Crawford MJ, Sanatinia R, Barrett B, et al; LABILE study team. The clinical effectiveness and cost-effectiveness of lamotrigine in borderline personality disorder: a randomized placebo-controlled trial. Am J Psychiatry. 2018;175(8):756-764. doi: 10.1176/appi.ajp.2018.17091006
6. Domes G, Ower N, von Dawans B, et al. Effects of intranasal oxytocin administration on empathy and approach motivation in women with borderline personality disorder: a randomized controlled trial. Transl Psychiatry. 2019;9(1):328. doi: 10.1038/s41398-019-0658-4
7. Bozzatello P, Rocca P, Uscinska M, et al. Efficacy and tolerability of asenapine compared with olanzapine in borderline personality disorder: an open-label randomized controlled trial. CNS Drugs. 2017;31(9):809-819. doi: 10.1007/s40263-017-0458-4
8. Kulkarni J, Thomas N, Hudaib AR, et al. Effect of the glutamate NMDA receptor antagonist memantine as adjunctive treatment in borderline personality disorder: an exploratory, randomised, double-blind, placebo-controlled trial. CNS Drugs. 2018;32(2):179-187. doi: 10.1007/s40263-018-0506-8
FIRST OF 2 PARTS
Borderline personality disorder (BPD) is marked by an ongoing pattern of mood instability, cognitive distortions, problems with self-image, and impulsive behavior, often resulting in problems in relationships. BPD is associated with serious impairment in psychosocial functioning.1 Patients with BPD tend to use more mental health services than patients with other personality disorders or those with major depressive disorder (MDD).2 However, there has been little consensus on the best treatment(s) for this serious and debilitating disorder, and some clinicians view BPD as difficult to treat.
Current treatments for BPD include psychological and pharmacological interventions. Neuromodulation techniques, such as repetitive transcranial magnetic stimulation, may also positively affect BPD symptomatology. In recent years, there have been some promising findings in the treatment of BPD. In this 2-part article, we focus on current (within the last 5 years) findings from randomized controlled trials (RCTs) of BPD treatments. Here in Part 1, we focus on 6 studies that evaluated biological interventions (Table,3-8). In Part 2, we will focus on RCTs that investigated psychological interventions.
1. Lisoni J, Miotto P, Barlati S, et al. Change in core symptoms of borderline personality disorder by tDCS: a pilot study. Psychiatry Res. 2020;291:113261. doi: 10.1016/j.psychres.2020.113261
Impulsivity has been described as the core feature of BPD that best explains its behavioral, cognitive, and clinical manifestations. Studies have repeatedly demonstrated the role of the prefrontal cortex in modulating impulsivity. Dysfunction of the
Continue to: Study design...
Study design
- In a double-blind, sham-controlled trial, adults who met DSM-IV-TR criteria for BPD were randomized to 3 weeks (15 sessions) of right anodal/left cathodal DLPFC tCDS (n = 15) or sham tDCS (n = 15). This study included patients with comorbid psychiatric disorders, including substance use disorders. Discontinuation or alteration of existing medications was not allowed.
- The presence, severity, and change over time of BPD core symptoms was assessed at baseline and after 3 weeks using several clinical scales, self-questionnaires, and neuropsychological tests, including the Barratt Impulsiveness Scale-11 (BIS-11), Buss-Perry Aggression Questionnaire (BP-AQ), Difficulties in Emotion Regulation Scale (DERS), Hamilton Depression Rating Scale (HAM-D), Beck Depression Inventory (BDI), Hamilton Anxiety Rating Scale (HAM-A), Irritability-Depression Anxiety Scale (IDA), Visual Analog Scales (VAS), and Iowa Gambling Task.
Outcomes
- Participants in the active tDCS group experienced significant reductions in impulsivity, aggression, and craving as measured by the BIS-11, BP-AQ, and VAS.
- Compared to the sham group, the active tDCS group had greater reductions in HAM-D and BDI scores.
- HAM-A and IDA scores were improved in both groups, although the active tDCS group showed greater reductions in IDA scores compared with the sham group.
- As measured by DERS, active tDCS did not improve affective dysregulation more than sham tDCS.
Conclusions/limitations
- Bilateral tDCS targeting the right DLPFC with anodal stimulation is a safe, well-tolerated technique that may modulate core dimensions of BPD, including impulsivity, aggression, and craving.
- Excitatory anodal stimulation of the right DLFPC coupled with inhibitory cathodal stimulation on the left DLPFC may be an effective montage for targeting impulsivity in patients with BPD.
- Study limitations include a small sample size, use of targeted questionnaires only, inclusion of patients with BPD who also had certain comorbid psychiatric disorders, lack of analysis of the contributions of medications, lack of functional neuroimaging, and lack of a follow-up phase.
2. Molavi P, Aziziaram S, Basharpoor S, et al. Repeated transcranial direct current stimulation of dorsolateral-prefrontal cortex improves executive functions, cognitive reappraisal emotion regulation, and control over emotional processing in borderline personality disorder: a randomized, sham-controlled, parallel-group study. J Affect Disord. 2020;274:93-102. doi: 10.1016/j.jad.2020.05.007
Emotional dysregulation is considered a core feature of BPD psychopathology and is closely associated with executive dysfunction and cognitive control. Manifestations of executive dysfunction include aggressiveness, impulsive decision-making, disinhibition, and self-destructive behaviors. Neuroimaging of patients with BPD has shown enhanced activity in the insula, posterior cingulate cortex, and amygdala, with reduced activity in the medial PFC, subgenual anterior cingulate cortex, and DLPFC. Molavi et al4 postulated that increasing DLPFC activation with left anodal tDCS would result in improved executive functioning and emotion dysregulation in patients with BPD.
Study design
- In this single-blind, sham-controlled, parallel-group study, adults who met DSM-5 criteria for BPD were randomized to receive 10 consecutive daily sessions of left anodal/right cathodal DLPFC tDCS (n = 16) or sham tDCS (n = 16).
- The effect of tDCS on executive dysfunction, emotion dysregulation, and emotional processing was measured using the Executive Skills Questionnaire for Adults (ESQ), Emotion Regulation Questionnaire (ERQ), and Emotional Processing Scale (EPS). Measurements occurred at baseline and after 10 sessions of active or sham tDCS.
Outcomes
- Participants who received active tDCS experienced significant improvements in ESQ overall score and most of the executive function domains measured by the ESQ.
- Those in the active tDCS group also experienced significant improvement in emotion regulation as measured by the cognitive reappraisal subscale (but not the expressive suppression subscale) of the ERQ after the intervention.
- Overall emotional processing as measured by the EPS was significantly improved in the active tDCS group following the intervention.
Conclusions/limitations
- Repeated bilateral left anodal/right cathodal tDCS stimulation of the DLPFC significantly improved executive functioning and aspects of emotion regulation and emotional processing in patients with BPD. This improvement was presumed to be the result of increased activity of left DLPFC.
- Study limitations include a single-blind design, lack of follow-up to assess durability and stability of response over time, reliance on self-report measures, lack of functional neuroimaging, and limited focality of tDCS.
3. Crawford MJ, Sanatinia R, Barrett B, et al; LABILE study team. The clinical effectiveness and cost-effectiveness of lamotrigine in borderline personality disorder: a randomized placebo-controlled trial. Am J Psychiatry. 2018;175(8):756-764. doi: 10.1176/appi.ajp.2018.17091006
One of the hallmark symptoms of BPD is mood dysregulation. Current treatment guidelines recommend the use of mood stabilizers for BPD despite limited quality evidence of effectiveness and a lack of FDA-approved medications with this indication. In this RCT, Crawford et al5 examined whether lamotrigine is a clinically effective and cost-effective treatment for people with BPD.
Continue to: Study design...
Study design
- In this 2-arm, parallel-group, double-blind, placebo-controlled trial, 276 adults who met DSM-IV criteria for BPD were randomized to receive lamotrigine (up to 400 mg/d) or placebo for 52 weeks.
- The primary outcome was the score on the Zanarini Rating Scale for Borderline Personality Disorder (ZAN-BPD) at 52 weeks. Secondary outcomes included depressive symptoms, deliberate self-harm, social functioning, health-related quality of life, resource use and costs, treatment adverse effects, and adverse events. These were assessed using the BDI; Acts of Deliberate Self-Harm Inventory; Social Functioning Questionnaire; Alcohol, Smoking, and Substance Involvement Screening Test; and the EQ-5D-3L.
Outcomes
- Mean ZAN-BPD score decreased at 12 weeks in both groups, after which time the score remained stable.
- There was no difference in ZAN-BPD scores at 52 weeks between treatment arms. No difference was found in any secondary outcome measures.
- Difference in costs between groups was not significant.
Conclusions/limitations
- There was no evidence that lamotrigine led to clinical improvements in BPD symptomatology, social functioning, health-related quality of life, or substance use.
- Lamotrigine is neither clinically effective nor a cost-effective use of resources in the treatment of BPD.
- Limitations include a low level of adherence.
4. Domes G, Ower N, von Dawans B, et al. Effects of intranasal oxytocin administration on empathy and approach motivation in women with borderline personality disorder: a randomized controlled trial. Transl Psychiatry. 2019;9(1):328. doi: 10.1038/s41398-019-0658-4
A core feature of BPD is impairment in empathy; adequate empathy is required for intact social functioning. Oxytocin is a neuropeptide that helps regulate complex social cognition and behavior. Prior research has found that oxytocin administration enhances emotion regulation and empathy. Women with BPD have been observed to have lower levels of oxytocin. Domes et al6 conducted an RCT to see if oxytocin could have a beneficial effect on social approach and social cognition in women with BPD.
Study design
- In a double-blind, placebo-controlled, between-subject trial, 61 women who met DSM-IV criteria for BPD and 68 matched healthy controls were randomized to receive intranasal oxytocin, 24 IU, or placebo 45 minutes before completing an empathy task.
- An extended version of the Multifaceted Empathy Test was used to assess empathy and approach motivation.
Outcomes
- For cognitive empathy, patients with BPD exhibited significantly lower overall performance compared to controls. There was no effect of oxytocin on this performance in either group.
- Patients with BPD had significantly lower affective empathy compared with controls. After oxytocin administration, patients with BPD had significantly higher affective empathy than those with BPD who received placebo, reaching the level of healthy controls who received placebo.
- For positive stimuli, patients with BPD showed lower affective empathy than controls. Oxytocin treatment increased affective empathy in both groups.
- For negative stimuli, oxytocin increased affective empathy more in patients with BPD than in controls.
- Patients with BPD demonstrated less approach motivation than controls. Oxytocin increased approach motivation more in patients with BPD than in controls. For approach motivation toward positive stimuli, oxytocin had a significant effect on patients with BPD.
Continue to: Conclusions/limitations...
Conclusions/limitations
- Patients with BPD showed reduced cognitive and affective empathy and less approach behavior motivation than healthy controls.
- Patients with BPD who received oxytocin attained a level of affective empathy and approach motivation similar to that of healthy controls who received placebo. For positive stimuli, both groups exhibited comparable improvements from oxytocin. For negative stimuli, patients with BPD patients showed significant improvement with oxytocin, whereas healthy controls received no such benefit.
- Limitations include the use of self-report scales, lack of a control group, and inclusion of patients using psychotherapeutic medications. The study lacks generalizability because only women were included; the effect of exogenous oxytocin on men may differ.
5. Bozzatello P, Rocca P, Uscinska M, et al. Efficacy and tolerability of asenapine compared with olanzapine in borderline personality disorder: an open-label randomized controlled trial. CNS Drugs. 2017;31(9):809-819. doi: 10.1007/s40263-017-0458-4
The last decade has seen a noticeable shift in clinical practice from the use of antidepressants to mood stabilizers and second-generation antipsychotics (SGAs) in the treatment of BPD. Studies have demonstrated therapeutic effects of antipsychotic drugs across a wide range of BPD symptoms. Among SGAs, olanzapine is the most extensively studied across case reports, open-label studies, and RCTs of patients with BPD. In an RCT, Bozzatello et al7 compared the efficacy and tolerability of asenapine to olanzapine.
Study design
- In this open-label RCT, adults who met DSM-5 criteria for BPD were assigned to receive asenapine (n = 25) or olanzapine (n = 26) for 12 weeks.
- Study measurements included the Clinical Global Impression Scale, Severity item, HAM-D, HAM-A, Social and Occupational Functioning Assessment Scale, Borderline Personality Disorder Severity Index (BPDSI), BIS-11, Modified Overt Aggression Scale, and Dosage Record Treatment Emergent Symptom Scale.
Outcomes
- Asenapine and olanzapine had similar effects on BPD-related psychopathology, anxiety, and social and occupational functioning.
- Neither medication significantly decreased depressive or aggressive symptoms.
- Asenapine was superior to olanzapine in reducing the affective instability score of the BPDSI.
- Akathisia and restlessness/anxiety were more common with asenapine, and somnolence and fatigue were more common with olanzapine.
Conclusions/limitations
- The overall efficacy of asenapine was not different from olanzapine, and both medications were well-tolerated.
- Neither medication led to an improvement in depression or aggression, but asenapine was superior to olanzapine in reducing the severity of affective instability.
- Limitations include an open-label design, lack of placebo group, small sample size, high drop-out rate, exclusion of participants with co-occurring MDD and substance abuse/dependence, lack of data on prior pharmacotherapies and psychotherapies, and lack of power to detect a difference on the dissociation/paranoid ideation item of BPDSI.
6. Kulkarni J, Thomas N, Hudaib AR, et al. Effect of the glutamate NMDA receptor antagonist memantine as adjunctive treatment in borderline personality disorder: an exploratory, randomised, double-blind, placebo-controlled trial. CNS Drugs. 2018;32(2):179-187. doi: 10.1007/s40263-018-0506-8
It has been hypothesized that glutamate dysregulation and excitotoxicity are crucial to the development of the cognitive disturbances that underlie BPD. As such, glutamate modulators such as memantine hold promise for the treatment of BPD. In this RCT, Kulkarni et al8 examined the efficacy and tolerability of memantine compared with treatment as usual in patients with BPD.
Continue to: Study design...
Study design
- In an 8-week, double-blind, placebo-controlled trial, adults diagnosed with BPD according to the Diagnostic Interview for Borderline Patients were randomized to receive memantine (n = 17) or placebo (n = 16) in addition to treatment as usual. Treatment as usual included the use of antidepressants, mood stabilizers, and antipsychotics as well as psychotherapy and other psychosocial interventions.
- Patients were initiated on placebo or memantine, 10 mg/d. Memantine was increased to 20 mg/d after 7 days.
- ZAN-BPD score was the primary outcome and was measured at baseline and 2, 4, 6, and 8 weeks. An adverse effects questionnaire was administered every 2 weeks to assess tolerability.
Outcomes
- During the first 2 weeks of treatment, there were no significant improvements in ZAN-BPD score in the memantine group compared with the placebo group.
- Beginning with Week 2, compared with the placebo group, the memantine group experienced a significant reduction in total symptoms as measured by ZAN-BPD.
- There were no statistically significant differences in adverse events between groups.
Conclusions/limitations
- Memantine appears to be a well-tolerated treatment option for patients with BPD and merits further study.
- Limitations include a small sample size, and an inability to reach plateau of ZAN-BPD total score in either group. Also, there is considerable individual variability in memantine steady-state plasma concentrations, but plasma levels were not measured in this study.
Bottom Line
Findings from small randomized controlled trials suggest that transcranial direct current stimulation, oxytocin, asenapine, olanzapine, and memantine may have beneficial effects on some core symptoms of borderline personality disorder. These findings need to be replicated in larger studies.
FIRST OF 2 PARTS
Borderline personality disorder (BPD) is marked by an ongoing pattern of mood instability, cognitive distortions, problems with self-image, and impulsive behavior, often resulting in problems in relationships. BPD is associated with serious impairment in psychosocial functioning.1 Patients with BPD tend to use more mental health services than patients with other personality disorders or those with major depressive disorder (MDD).2 However, there has been little consensus on the best treatment(s) for this serious and debilitating disorder, and some clinicians view BPD as difficult to treat.
Current treatments for BPD include psychological and pharmacological interventions. Neuromodulation techniques, such as repetitive transcranial magnetic stimulation, may also positively affect BPD symptomatology. In recent years, there have been some promising findings in the treatment of BPD. In this 2-part article, we focus on current (within the last 5 years) findings from randomized controlled trials (RCTs) of BPD treatments. Here in Part 1, we focus on 6 studies that evaluated biological interventions (Table,3-8). In Part 2, we will focus on RCTs that investigated psychological interventions.
1. Lisoni J, Miotto P, Barlati S, et al. Change in core symptoms of borderline personality disorder by tDCS: a pilot study. Psychiatry Res. 2020;291:113261. doi: 10.1016/j.psychres.2020.113261
Impulsivity has been described as the core feature of BPD that best explains its behavioral, cognitive, and clinical manifestations. Studies have repeatedly demonstrated the role of the prefrontal cortex in modulating impulsivity. Dysfunction of the
Continue to: Study design...
Study design
- In a double-blind, sham-controlled trial, adults who met DSM-IV-TR criteria for BPD were randomized to 3 weeks (15 sessions) of right anodal/left cathodal DLPFC tCDS (n = 15) or sham tDCS (n = 15). This study included patients with comorbid psychiatric disorders, including substance use disorders. Discontinuation or alteration of existing medications was not allowed.
- The presence, severity, and change over time of BPD core symptoms was assessed at baseline and after 3 weeks using several clinical scales, self-questionnaires, and neuropsychological tests, including the Barratt Impulsiveness Scale-11 (BIS-11), Buss-Perry Aggression Questionnaire (BP-AQ), Difficulties in Emotion Regulation Scale (DERS), Hamilton Depression Rating Scale (HAM-D), Beck Depression Inventory (BDI), Hamilton Anxiety Rating Scale (HAM-A), Irritability-Depression Anxiety Scale (IDA), Visual Analog Scales (VAS), and Iowa Gambling Task.
Outcomes
- Participants in the active tDCS group experienced significant reductions in impulsivity, aggression, and craving as measured by the BIS-11, BP-AQ, and VAS.
- Compared to the sham group, the active tDCS group had greater reductions in HAM-D and BDI scores.
- HAM-A and IDA scores were improved in both groups, although the active tDCS group showed greater reductions in IDA scores compared with the sham group.
- As measured by DERS, active tDCS did not improve affective dysregulation more than sham tDCS.
Conclusions/limitations
- Bilateral tDCS targeting the right DLPFC with anodal stimulation is a safe, well-tolerated technique that may modulate core dimensions of BPD, including impulsivity, aggression, and craving.
- Excitatory anodal stimulation of the right DLFPC coupled with inhibitory cathodal stimulation on the left DLPFC may be an effective montage for targeting impulsivity in patients with BPD.
- Study limitations include a small sample size, use of targeted questionnaires only, inclusion of patients with BPD who also had certain comorbid psychiatric disorders, lack of analysis of the contributions of medications, lack of functional neuroimaging, and lack of a follow-up phase.
2. Molavi P, Aziziaram S, Basharpoor S, et al. Repeated transcranial direct current stimulation of dorsolateral-prefrontal cortex improves executive functions, cognitive reappraisal emotion regulation, and control over emotional processing in borderline personality disorder: a randomized, sham-controlled, parallel-group study. J Affect Disord. 2020;274:93-102. doi: 10.1016/j.jad.2020.05.007
Emotional dysregulation is considered a core feature of BPD psychopathology and is closely associated with executive dysfunction and cognitive control. Manifestations of executive dysfunction include aggressiveness, impulsive decision-making, disinhibition, and self-destructive behaviors. Neuroimaging of patients with BPD has shown enhanced activity in the insula, posterior cingulate cortex, and amygdala, with reduced activity in the medial PFC, subgenual anterior cingulate cortex, and DLPFC. Molavi et al4 postulated that increasing DLPFC activation with left anodal tDCS would result in improved executive functioning and emotion dysregulation in patients with BPD.
Study design
- In this single-blind, sham-controlled, parallel-group study, adults who met DSM-5 criteria for BPD were randomized to receive 10 consecutive daily sessions of left anodal/right cathodal DLPFC tDCS (n = 16) or sham tDCS (n = 16).
- The effect of tDCS on executive dysfunction, emotion dysregulation, and emotional processing was measured using the Executive Skills Questionnaire for Adults (ESQ), Emotion Regulation Questionnaire (ERQ), and Emotional Processing Scale (EPS). Measurements occurred at baseline and after 10 sessions of active or sham tDCS.
Outcomes
- Participants who received active tDCS experienced significant improvements in ESQ overall score and most of the executive function domains measured by the ESQ.
- Those in the active tDCS group also experienced significant improvement in emotion regulation as measured by the cognitive reappraisal subscale (but not the expressive suppression subscale) of the ERQ after the intervention.
- Overall emotional processing as measured by the EPS was significantly improved in the active tDCS group following the intervention.
Conclusions/limitations
- Repeated bilateral left anodal/right cathodal tDCS stimulation of the DLPFC significantly improved executive functioning and aspects of emotion regulation and emotional processing in patients with BPD. This improvement was presumed to be the result of increased activity of left DLPFC.
- Study limitations include a single-blind design, lack of follow-up to assess durability and stability of response over time, reliance on self-report measures, lack of functional neuroimaging, and limited focality of tDCS.
3. Crawford MJ, Sanatinia R, Barrett B, et al; LABILE study team. The clinical effectiveness and cost-effectiveness of lamotrigine in borderline personality disorder: a randomized placebo-controlled trial. Am J Psychiatry. 2018;175(8):756-764. doi: 10.1176/appi.ajp.2018.17091006
One of the hallmark symptoms of BPD is mood dysregulation. Current treatment guidelines recommend the use of mood stabilizers for BPD despite limited quality evidence of effectiveness and a lack of FDA-approved medications with this indication. In this RCT, Crawford et al5 examined whether lamotrigine is a clinically effective and cost-effective treatment for people with BPD.
Continue to: Study design...
Study design
- In this 2-arm, parallel-group, double-blind, placebo-controlled trial, 276 adults who met DSM-IV criteria for BPD were randomized to receive lamotrigine (up to 400 mg/d) or placebo for 52 weeks.
- The primary outcome was the score on the Zanarini Rating Scale for Borderline Personality Disorder (ZAN-BPD) at 52 weeks. Secondary outcomes included depressive symptoms, deliberate self-harm, social functioning, health-related quality of life, resource use and costs, treatment adverse effects, and adverse events. These were assessed using the BDI; Acts of Deliberate Self-Harm Inventory; Social Functioning Questionnaire; Alcohol, Smoking, and Substance Involvement Screening Test; and the EQ-5D-3L.
Outcomes
- Mean ZAN-BPD score decreased at 12 weeks in both groups, after which time the score remained stable.
- There was no difference in ZAN-BPD scores at 52 weeks between treatment arms. No difference was found in any secondary outcome measures.
- Difference in costs between groups was not significant.
Conclusions/limitations
- There was no evidence that lamotrigine led to clinical improvements in BPD symptomatology, social functioning, health-related quality of life, or substance use.
- Lamotrigine is neither clinically effective nor a cost-effective use of resources in the treatment of BPD.
- Limitations include a low level of adherence.
4. Domes G, Ower N, von Dawans B, et al. Effects of intranasal oxytocin administration on empathy and approach motivation in women with borderline personality disorder: a randomized controlled trial. Transl Psychiatry. 2019;9(1):328. doi: 10.1038/s41398-019-0658-4
A core feature of BPD is impairment in empathy; adequate empathy is required for intact social functioning. Oxytocin is a neuropeptide that helps regulate complex social cognition and behavior. Prior research has found that oxytocin administration enhances emotion regulation and empathy. Women with BPD have been observed to have lower levels of oxytocin. Domes et al6 conducted an RCT to see if oxytocin could have a beneficial effect on social approach and social cognition in women with BPD.
Study design
- In a double-blind, placebo-controlled, between-subject trial, 61 women who met DSM-IV criteria for BPD and 68 matched healthy controls were randomized to receive intranasal oxytocin, 24 IU, or placebo 45 minutes before completing an empathy task.
- An extended version of the Multifaceted Empathy Test was used to assess empathy and approach motivation.
Outcomes
- For cognitive empathy, patients with BPD exhibited significantly lower overall performance compared to controls. There was no effect of oxytocin on this performance in either group.
- Patients with BPD had significantly lower affective empathy compared with controls. After oxytocin administration, patients with BPD had significantly higher affective empathy than those with BPD who received placebo, reaching the level of healthy controls who received placebo.
- For positive stimuli, patients with BPD showed lower affective empathy than controls. Oxytocin treatment increased affective empathy in both groups.
- For negative stimuli, oxytocin increased affective empathy more in patients with BPD than in controls.
- Patients with BPD demonstrated less approach motivation than controls. Oxytocin increased approach motivation more in patients with BPD than in controls. For approach motivation toward positive stimuli, oxytocin had a significant effect on patients with BPD.
Continue to: Conclusions/limitations...
Conclusions/limitations
- Patients with BPD showed reduced cognitive and affective empathy and less approach behavior motivation than healthy controls.
- Patients with BPD who received oxytocin attained a level of affective empathy and approach motivation similar to that of healthy controls who received placebo. For positive stimuli, both groups exhibited comparable improvements from oxytocin. For negative stimuli, patients with BPD patients showed significant improvement with oxytocin, whereas healthy controls received no such benefit.
- Limitations include the use of self-report scales, lack of a control group, and inclusion of patients using psychotherapeutic medications. The study lacks generalizability because only women were included; the effect of exogenous oxytocin on men may differ.
5. Bozzatello P, Rocca P, Uscinska M, et al. Efficacy and tolerability of asenapine compared with olanzapine in borderline personality disorder: an open-label randomized controlled trial. CNS Drugs. 2017;31(9):809-819. doi: 10.1007/s40263-017-0458-4
The last decade has seen a noticeable shift in clinical practice from the use of antidepressants to mood stabilizers and second-generation antipsychotics (SGAs) in the treatment of BPD. Studies have demonstrated therapeutic effects of antipsychotic drugs across a wide range of BPD symptoms. Among SGAs, olanzapine is the most extensively studied across case reports, open-label studies, and RCTs of patients with BPD. In an RCT, Bozzatello et al7 compared the efficacy and tolerability of asenapine to olanzapine.
Study design
- In this open-label RCT, adults who met DSM-5 criteria for BPD were assigned to receive asenapine (n = 25) or olanzapine (n = 26) for 12 weeks.
- Study measurements included the Clinical Global Impression Scale, Severity item, HAM-D, HAM-A, Social and Occupational Functioning Assessment Scale, Borderline Personality Disorder Severity Index (BPDSI), BIS-11, Modified Overt Aggression Scale, and Dosage Record Treatment Emergent Symptom Scale.
Outcomes
- Asenapine and olanzapine had similar effects on BPD-related psychopathology, anxiety, and social and occupational functioning.
- Neither medication significantly decreased depressive or aggressive symptoms.
- Asenapine was superior to olanzapine in reducing the affective instability score of the BPDSI.
- Akathisia and restlessness/anxiety were more common with asenapine, and somnolence and fatigue were more common with olanzapine.
Conclusions/limitations
- The overall efficacy of asenapine was not different from olanzapine, and both medications were well-tolerated.
- Neither medication led to an improvement in depression or aggression, but asenapine was superior to olanzapine in reducing the severity of affective instability.
- Limitations include an open-label design, lack of placebo group, small sample size, high drop-out rate, exclusion of participants with co-occurring MDD and substance abuse/dependence, lack of data on prior pharmacotherapies and psychotherapies, and lack of power to detect a difference on the dissociation/paranoid ideation item of BPDSI.
6. Kulkarni J, Thomas N, Hudaib AR, et al. Effect of the glutamate NMDA receptor antagonist memantine as adjunctive treatment in borderline personality disorder: an exploratory, randomised, double-blind, placebo-controlled trial. CNS Drugs. 2018;32(2):179-187. doi: 10.1007/s40263-018-0506-8
It has been hypothesized that glutamate dysregulation and excitotoxicity are crucial to the development of the cognitive disturbances that underlie BPD. As such, glutamate modulators such as memantine hold promise for the treatment of BPD. In this RCT, Kulkarni et al8 examined the efficacy and tolerability of memantine compared with treatment as usual in patients with BPD.
Continue to: Study design...
Study design
- In an 8-week, double-blind, placebo-controlled trial, adults diagnosed with BPD according to the Diagnostic Interview for Borderline Patients were randomized to receive memantine (n = 17) or placebo (n = 16) in addition to treatment as usual. Treatment as usual included the use of antidepressants, mood stabilizers, and antipsychotics as well as psychotherapy and other psychosocial interventions.
- Patients were initiated on placebo or memantine, 10 mg/d. Memantine was increased to 20 mg/d after 7 days.
- ZAN-BPD score was the primary outcome and was measured at baseline and 2, 4, 6, and 8 weeks. An adverse effects questionnaire was administered every 2 weeks to assess tolerability.
Outcomes
- During the first 2 weeks of treatment, there were no significant improvements in ZAN-BPD score in the memantine group compared with the placebo group.
- Beginning with Week 2, compared with the placebo group, the memantine group experienced a significant reduction in total symptoms as measured by ZAN-BPD.
- There were no statistically significant differences in adverse events between groups.
Conclusions/limitations
- Memantine appears to be a well-tolerated treatment option for patients with BPD and merits further study.
- Limitations include a small sample size, and an inability to reach plateau of ZAN-BPD total score in either group. Also, there is considerable individual variability in memantine steady-state plasma concentrations, but plasma levels were not measured in this study.
Bottom Line
Findings from small randomized controlled trials suggest that transcranial direct current stimulation, oxytocin, asenapine, olanzapine, and memantine may have beneficial effects on some core symptoms of borderline personality disorder. These findings need to be replicated in larger studies.
1. Skodol AE, Gunderson JG, McGlashan TM, et al. Functional impairment in patients with schizotypal, borderline, avoidant, or obsessive-compulsive personality disorder. Am J Psychiatry. 2002; 159:276-283.
2. Bender DS, Dolan RT, Skodol AE, et al. Treatment utilization by patients with personality disorders. Am J Psychiatry. 2001;158:295-302.
3. Lisoni J, Miotto P, Barlati S, et al. Change in core symptoms of borderline personality disorder by tDCS: a pilot study. Psychiatry Res. 2020;291:113261. doi: 10.1016/j.psychres.2020.113261
4. Molavi P, Aziziaram S, Basharpoor S, et al. Repeated transcranial direct current stimulation of dorsolateral-prefrontal cortex improves executive functions, cognitive reappraisal emotion regulation, and control over emotional processing in borderline personality disorder: a randomized, sham-controlled, parallel-group study. J Affect Disord. 2020;274:93-102. doi: 10.1016/j.jad.2020.05.007
5. Crawford MJ, Sanatinia R, Barrett B, et al; LABILE study team. The clinical effectiveness and cost-effectiveness of lamotrigine in borderline personality disorder: a randomized placebo-controlled trial. Am J Psychiatry. 2018;175(8):756-764. doi: 10.1176/appi.ajp.2018.17091006
6. Domes G, Ower N, von Dawans B, et al. Effects of intranasal oxytocin administration on empathy and approach motivation in women with borderline personality disorder: a randomized controlled trial. Transl Psychiatry. 2019;9(1):328. doi: 10.1038/s41398-019-0658-4
7. Bozzatello P, Rocca P, Uscinska M, et al. Efficacy and tolerability of asenapine compared with olanzapine in borderline personality disorder: an open-label randomized controlled trial. CNS Drugs. 2017;31(9):809-819. doi: 10.1007/s40263-017-0458-4
8. Kulkarni J, Thomas N, Hudaib AR, et al. Effect of the glutamate NMDA receptor antagonist memantine as adjunctive treatment in borderline personality disorder: an exploratory, randomised, double-blind, placebo-controlled trial. CNS Drugs. 2018;32(2):179-187. doi: 10.1007/s40263-018-0506-8
1. Skodol AE, Gunderson JG, McGlashan TM, et al. Functional impairment in patients with schizotypal, borderline, avoidant, or obsessive-compulsive personality disorder. Am J Psychiatry. 2002; 159:276-283.
2. Bender DS, Dolan RT, Skodol AE, et al. Treatment utilization by patients with personality disorders. Am J Psychiatry. 2001;158:295-302.
3. Lisoni J, Miotto P, Barlati S, et al. Change in core symptoms of borderline personality disorder by tDCS: a pilot study. Psychiatry Res. 2020;291:113261. doi: 10.1016/j.psychres.2020.113261
4. Molavi P, Aziziaram S, Basharpoor S, et al. Repeated transcranial direct current stimulation of dorsolateral-prefrontal cortex improves executive functions, cognitive reappraisal emotion regulation, and control over emotional processing in borderline personality disorder: a randomized, sham-controlled, parallel-group study. J Affect Disord. 2020;274:93-102. doi: 10.1016/j.jad.2020.05.007
5. Crawford MJ, Sanatinia R, Barrett B, et al; LABILE study team. The clinical effectiveness and cost-effectiveness of lamotrigine in borderline personality disorder: a randomized placebo-controlled trial. Am J Psychiatry. 2018;175(8):756-764. doi: 10.1176/appi.ajp.2018.17091006
6. Domes G, Ower N, von Dawans B, et al. Effects of intranasal oxytocin administration on empathy and approach motivation in women with borderline personality disorder: a randomized controlled trial. Transl Psychiatry. 2019;9(1):328. doi: 10.1038/s41398-019-0658-4
7. Bozzatello P, Rocca P, Uscinska M, et al. Efficacy and tolerability of asenapine compared with olanzapine in borderline personality disorder: an open-label randomized controlled trial. CNS Drugs. 2017;31(9):809-819. doi: 10.1007/s40263-017-0458-4
8. Kulkarni J, Thomas N, Hudaib AR, et al. Effect of the glutamate NMDA receptor antagonist memantine as adjunctive treatment in borderline personality disorder: an exploratory, randomised, double-blind, placebo-controlled trial. CNS Drugs. 2018;32(2):179-187. doi: 10.1007/s40263-018-0506-8

Service animals and emotional support animals: Should you write that letter?
For centuries, animals, especially dogs, have assisted humans in a variety of ways in their daily lives. Animals that assist people with disabilities fall into 2 broad categories: disability service animals, and emotional support animals (ESAs). Often there is confusion in how these categories differ because of the animal’s role and the laws related to them.
This article describes the differences between disability service animals and ESAs, and outlines the forensic and ethical concerns you should consider before agreeing to write a letter for a patient outlining their need for a disability service animal or ESA. A letter may protect a patient and their service animal or ESA in situations where laws and regulations typically prohibit animals, such as on a flight or when renting an apartment or house. Note that a description of how to conduct the formal patient evaluation before writing a verification letter is beyond the scope of this article.
The differences between disability service animals and ESAs
Purpose and training. Disability service animals, or service animals, are dogs of any breed (and in some cases miniature horses) that are specially trained to perform tasks for an individual with a disability (physical, sensory, psychiatric, intellectual, or other mental disability).1-3 These tasks must be directly related to the individual’s disability.1,2 On the other hand, ESAs, which can be any species of animal, provide support and minimize the impact of an individual’s emotional or psychological disability based on their presence alone. Unlike disability service animals, ESAs are not trained to perform a specific task or duty.2,3
There is no legal requirement for service animals to know specific commands, and professional training is not required—individuals can train the animals themselves.1 Service animals, mainly dogs, can be trained to perform numerous tasks, including4:
- attending to an individual’s mobility and activities of daily living
- guiding an individual who is deaf or hearing impaired
- helping to remind an individual to take their medications
- assisting an individual during and/or after a seizure
- alerting individuals with diabetes in advance of low or high blood sugar episodes
- supporting an individual with autism
- assisting an individual with a psychiatric or mental disability
- applying sensory commands such as lying on the person or resting their head on the individual’s lap to help the individual regain behavioral control.
Service dog verification works via an honor system, which can be problematic, especially in the case of psychiatric service dogs, whose handlers may not have a visible disability (Box 11,5).
Box 1
In the United States, there is no national service dog certification program—meaning there is no official test that a dog has to pass in order to obtain formal recognition as a service animal—nor is there a central and mandatory service dog registry.5 Instead, service dog verification works through an honor system, which can be problematic.5 In many states, misrepresenting one’s dog as a service dog is considered a misdemeanor.5 Unfortunately, other than the guidance set forth by the Americans with Disabilities Act, there are no criteria by which one can recognize a genuine service dog vs one being passed off as a service dog.5
In situations in public settings where it is not obvious or there’s doubt that the dog is a service animal (such as when a person visits a restaurant or store), employees are not allowed to request documentation for the dog, require the dog demonstrate its task, or inquire about the nature of the person’s disability.1
However, they can ask 2 questions1:
1. Is the animal required because of a disability?
2. What work or task has the animal been trained to perform?
Legal protections. Under the Americans with Disabilities Act (ADA), individuals with disabilities can bring their service animals into buildings or facilities where members of the public, program participants, clients, customers, patrons, or invitees are allowed.2 This does not include private clubs, religious organizations, or places of worship that are not open to the public.6,7 ESAs do not qualify as service animals under the ADA and are not given the same legal accommodations as service animals.1,3 Although ESAs were initially covered by the Air Carrier Access Act, they are no longer allowed in aircraft cabins after the US Department of Transportation revised this Act’s regulations in December 2020. ESAs are covered under the Fair Housing Act. Box 21-3,6-15 further discusses these laws and protections.
Evidence.
Due to the difficulty in reconciling inconsistent definitions for ESAs, there is limited high-quality data pertaining to the potential benefits and risks of ESAs.9 Currently, ESAs are not an evidence-based treatment for psychiatric disorders. To date, a handful of small studies have focused on ESAs. However, data from actual tests of the clinical risks and benefits of ESAs do not exist.9 In practice, ESAs are equivalent to pets. It stands to reason that similar to pets, ESAs could reduce loneliness, improve life satisfaction, and provide a sense of well-being.9 A systematic review suggested that pets provide benefits to patients with mental health conditions “through the intensity of connectivity with their owners and the contribution they make to emotional support in times of crises together with their ability to help manage symptoms when they arise.”18 In response to a congressional mandate, the US Department of Veterans Affairs launched a multi-site study from December 2014 to June 2019 to examine how limitations on activity and quality of life in veterans with posttraumatic stress disorder are impacted by the provision of a service dog vs an emotional support dog.19 As of October 14, 2021, results had not been published.19
Continue to: What’s in a disability service animal/ESA letter?
What’s in a disability service animal/ESA letter?
If you decide to write a letter advocating for your patient to have a service animal or ESA, the letter should appear on letterhead, be written by a licensed mental health professional, and include the following2,20:
- statement that the letter is being written at the patient’s request and is being given directly to the patient for use as the patient sees fit
- confirmation of the patient’s DSM-5 mental health diagnosis
- explanation of how the animal helps alleviate symptoms of the patient’s condition, briefly describing any interaction(s) between the animal and patient that you may have observed, and if applicable, a mention of any training the animal may have received from a qualified trainer if applicable
- explanation of the possible negative effects of the patient not having the animal with him or her
- statement that you are not vouching for the animal’s behavior
- verification of your involvement in your patient’s treatment and your assessment of the patient as their licensed mental health professional (including details such as date and type of license you have and the state/other jurisdiction where it was issued).
In a letter for a service animal, also indicate that your patient is psychiatrically disabled to the extent that your patient is not able to perform at least one major life task without the daily assistance of a service animal.2Should you write your patient a letter?
Writing a letter advocating for a patient to have a service animal or ESA may appear innocuous, but doing so may have serious ramifications. Writing a letter certifying a dog as a service animal does not make that animal a service animal; the dog must be specifically trained for a task or tasks directly related to that individual’s disability. There are no current standards for conducting evaluations to determine the need a patient has for a service animal or ESA. How to conduct such evaluations is beyond the scope of this article. There are meager opportunities for formal education and training on how to conduct these evaluations.9 Online resources may be incomplete or inaccurate, and this information is often produced by lay animal enthusiasts and organizations, which can lead to a biased depiction of these animals.9
If you decide to write a letter for your patient, consider the following forensic and ethical concerns.
Remain objective. As an advocate for your patient, you may find it difficult to remain neutral and objective when asked to determine if your patient has a disability, the severity of the disability, the impact of the disability on your patient’s life, and the need for a service animal or ESA. Ensure that your advocacy for your patient does not impair your objectivity; if that is difficult, consider referring your patient to a third party who can conduct an objective evaluation.
Understand the risks. If you make written recommendations for special accommodations in a letter and those recommendations are disputed by an agency, that agency could initiate legal action and you may be called to justify your recommendations in a deposition or open court.9,21 Before writing the letter, ask yourself, “Can I defend my determination that my patient is disabled by a DSM-5 disorder and that this disability requires the presence of an animal in exception to existing policy?”21 Be prepared to state in a legal proceeding that the presence of a service animal or ESA is necessary. If you are unwilling to risk exposure to a legal action, then you should likely refrain from writing the letter. It is a crime to fraudulently certify an animal as a service animal in some jurisdictions, and such conduct could result in disciplinary action by your licensing board.21
Conduct a systematic examination. When you write a letter for your patient, you are explicitly declaring your patient has a disability or condition. Comprehensive disability determinations are complex and are best conducted by assessing for objective evidence of psychiatric disorders and impairment through the use of standard, systematic examination methods.22 Unstandardized measures (eg, asking patients open-ended questions and then relying on your clinical judgement and interpretation in arriving at conclusions) are not as effective.22 In addition, consider the possibility that your patient may malinger their symptoms in an effort to obtain a letter supporting a service animal or ESA. Assessing for malingering is essential to making a disability determination, especially if a disability claim is based primarily on self-report.22
Anticipate pushback. Problems can arise when a patient wants a letter that you cannot or will not provide due to your scope of practice. Consider how you would resolve the situation when you do not believe your patient has a disability that requires the presence of a service animal or ESA—or you believe that your patient no longer needs a service animal or ESA—and the patient disagrees.21 Disagreeing with your patient’s assessment could result in a conflict of interest that could damage the therapeutic relationship.21
Box 2
The Americans with Disabilities Act (ADA) of 1990, as amended by the ADA Amendments Act of 2008, prohibits discrimination on the basis of disability in several areas, including state and local governments (under Title II of the ADA) and places of public accommodations, commercial facilities, and private entities (under Title III of the ADA).6,7 Thus, individuals with disabilities can bring their service animals into the building or facility where members of the public, program participants, clients, customers, patrons, or invitees are allowed.2 This does not include private clubs not open to the public, religious organizations, or places of worship.6,7
Service animals. Although the ADA recognizes miniature horses as service animals, only dogs are recognized as service animals in regards to Title II and Title III protections under the ADA as of March 15, 2011.2 Federal agencies do not have to comply with the ADA1; however, Section 504 of the Rehabilitation Act of 1973 is the federal law that protects the rights of people with disabilities to participate in federal programs and services.1,8 It states that no qualified individual with a disability shall be excluded from, denied the benefits of, or be subjected to discrimination under any program or activity that receives federal funding or is conducted by federal agencies.8 Courts have strived to interpret the Rehabilitation Act and the ADA in a consistent manner, specifically applying the ADA regulations regarding service animals (including its narrow definition regarding specifically trained tasks and emotional support) to the Rehabilitation Act.9-11
Similarly, commercial airlines do not have to comply with the ADA1 ; however, the Air Carrier Access Act (ACAA) of 1986 is the federal law that protects the rights of people with disabilities in air travel.1,12 On December 2, 2020, the US Department of Transportation announced that it was revising its ACAA regulation regarding service animals on aircraft (this final rule will be effective 30 days after date of publication in the Federal Register).13 Among the many revisions, the US Department of Transportation narrowed the definition of service animals to only dogs that were individually trained to work or perform tasks for the benefits of a person with a disability.13 It requires airlines to treat psychiatric service animals the same as other service animals.13 Although the US Department of Transportation has chosen to closely align its ACAA service animal definition with US Department of Justice service animal definition under the ADA, the substantive requirements in this final rule differ from US Department of Justice’s requirements for service animals under the ADA in various areas (for example, by allowing airlines to require service animal documentation and prohibiting the use of voice control over a service animal).13
Emotional support animals. Regulations regarding ESAs are primarily set by individual states1,3; however, ESAs may qualify for a waiver of a no-pet rule or a pet deposit under the Fair Housing Amendments Act (FHAA) of 1988.2,14 Under the FHAA, if an individual has a disability, as defined by the ADA, that requires the presence of an ESA, or if they have symptoms that are ameliorated by the presence of an ESA, the landlord must comply with this request and allow the animal into the facility without charging pet fees.15
Bottom Line
Disability service animals and emotional support animals (ESAs) differ in their roles and legal protections. Before writing a letter in support of a patient’s request for a service animal or ESA, take into account the forensic and ethical implications of doing so.
Related Resources
- US Department of Justice. Civil Rights Division. Disability Rights Section. ADA requirements. Service animals. Updated February 24, 2020. https://www.ada.gov/service_ animals_2010.htm
American Veterinary Medical Association. Service, emotional support and therapy animals. https://www. avma.org/resources-tools/animal-health-welfare/ service-emotional-support-and-therapy-animals
US Department of Transportation. US Department of Transportation announces final rule on traveling by air with service animals. https://www.transportation.gov/briefingroom/us-department-transportation-announces-finalrule-traveling-air-service-animals
1. US Department of Justice. Frequently asked questions about service animals and the ADA. Published July 20, 2015. Accessed on July 28, 2021. https://www.ada.gov/regs2010/service_animal_qa.pdf
2. ADA National Network. Service animals and emotional support animals: where are they allowed and under what conditions? Published 2014. Accessed July 28, 2021. https://adata.org/sites/adata.org/files/files/Service_Animal_Booklet_2014(2).pdf
3. Huben-Kearney A. What to do if patients want service or emotional support animals. Psychiatric News. Published September 28, 2020. Accessed July 28, 2021. https://psychnews.psychiatryonline.org/doi/10.1176/appi.pn.2020.10a24
4. Fine AH. The role of therapy and service animals in the lives of persons with disabilities. Rev Sci Tech. 2018;37(1):141-149.
5. Wlodarczyk J. When pigs fly: emotional support animals, service dogs and the politics of legitimacy across species boundaries. Med Humanit. 2019;45(1):82-91.
6. Americans with Disabilities Act of 1990. Pub L. 101-336, 104 Stat. 327.
7. ADA Amendments Act of 2008. Pub L. 110-325.
8. Rehabilitation Act of 1973. Pub L. 93-112, 87 Stat 355.
9. Carroll JD, Mohlenhoff BS, Kersten CM, et al. Laws and ethics related to emotional support animals. J Am Acad Psychiatry Law. 2020;48(4):509-518.
10. Sanchez v US Dept of Energy. 870 F3d 1185 (10th Circuit 2017).
11. Berardelli v Allied Services Inst. of Rehab. Med., 900 F3d 104 (3rd Circuit 2018).
12. Air Carrier Access Act of 1986. 49 USC §41705.
13. US Department of Transportation. US Department of Transportation announces final rule on traveling by air with service animals. Published December 2, 2020. Accessed July 28, 2021. https://www.transportation.gov/briefing-room/us-department-transportation-announces-final-rule-traveling-air-service-animals
14. Fair Housing Amendments Act of 1988. Pub. L. 100-430. https://www.govinfo.gov/content/pkg/STATUTE-102/pdf/STATUTE-102-Pg1619.pdf
15. Boness CL, Younggren JN, Frumkin IB. The certification of emotional support animals: difference between clinical and forensic mental health practitioners. Professional Psychology: Research and Practice. 2017;48(3):216-223.
16. Lane DR, McNicholas J, Collis GM. Dogs for the disabled: benefits to recipients and welfare of the dog. Applied Animal Behaviour Science. 1998;59(1-3):49-60.
17. Hall SS, MacMichael J, Turner A, et al. A survey of the impact of owning a service dog on quality of life for individuals with physical and hearing disability: a pilot study. Health Qual Life Outcomes. 2017;15(1):59. doi:10.1186/s12955-017-0640-x
18. Brooks HL, Rushton K, Lovell K, et al. The power of support from companion animals for people living with mental health problems: a systematic review and narrative synthesis of the evidence. BMC Psychiatry. 2018;18(1):31. doi: 10.1186/s12888-018-1613-2
19. US National Library of Medicine: ClinicalTrials.gov. Can service dogs improve activity and quality of life in veterans with PTSD? (SDPTSD). Updated August 15, 2019. Accessed October 14, 2021. https://clinicaltrials.gov/ct2/show/study/NCT02039843
20. Clay RA. Is that a pet or therapeutic aid? American Psychological Association. 2016;47(8):38. https://www.apa.org/monitor/2016/09/pet-aid
21. Younggren JN, Boisvert JA, Boness CL. Examining emotional support animals and role conflicts in professional psychology. Prof Psychol Res Pr. 2016;47(4):255-260.
22. Gold LH, Anfang SA, Drukteinis AM, et al. AAPL practice guideline for the forensic evaluation of psychiatric disability. J Am Acad Psychiatry Law. 2008;36(4 Suppl):S3-S50. https://www.aapl.org/docs/pdf/Evaluation%20of%20Psychiatric%20Disability.pdf
For centuries, animals, especially dogs, have assisted humans in a variety of ways in their daily lives. Animals that assist people with disabilities fall into 2 broad categories: disability service animals, and emotional support animals (ESAs). Often there is confusion in how these categories differ because of the animal’s role and the laws related to them.
This article describes the differences between disability service animals and ESAs, and outlines the forensic and ethical concerns you should consider before agreeing to write a letter for a patient outlining their need for a disability service animal or ESA. A letter may protect a patient and their service animal or ESA in situations where laws and regulations typically prohibit animals, such as on a flight or when renting an apartment or house. Note that a description of how to conduct the formal patient evaluation before writing a verification letter is beyond the scope of this article.
The differences between disability service animals and ESAs
Purpose and training. Disability service animals, or service animals, are dogs of any breed (and in some cases miniature horses) that are specially trained to perform tasks for an individual with a disability (physical, sensory, psychiatric, intellectual, or other mental disability).1-3 These tasks must be directly related to the individual’s disability.1,2 On the other hand, ESAs, which can be any species of animal, provide support and minimize the impact of an individual’s emotional or psychological disability based on their presence alone. Unlike disability service animals, ESAs are not trained to perform a specific task or duty.2,3
There is no legal requirement for service animals to know specific commands, and professional training is not required—individuals can train the animals themselves.1 Service animals, mainly dogs, can be trained to perform numerous tasks, including4:
- attending to an individual’s mobility and activities of daily living
- guiding an individual who is deaf or hearing impaired
- helping to remind an individual to take their medications
- assisting an individual during and/or after a seizure
- alerting individuals with diabetes in advance of low or high blood sugar episodes
- supporting an individual with autism
- assisting an individual with a psychiatric or mental disability
- applying sensory commands such as lying on the person or resting their head on the individual’s lap to help the individual regain behavioral control.
Service dog verification works via an honor system, which can be problematic, especially in the case of psychiatric service dogs, whose handlers may not have a visible disability (Box 11,5).
Box 1
In the United States, there is no national service dog certification program—meaning there is no official test that a dog has to pass in order to obtain formal recognition as a service animal—nor is there a central and mandatory service dog registry.5 Instead, service dog verification works through an honor system, which can be problematic.5 In many states, misrepresenting one’s dog as a service dog is considered a misdemeanor.5 Unfortunately, other than the guidance set forth by the Americans with Disabilities Act, there are no criteria by which one can recognize a genuine service dog vs one being passed off as a service dog.5
In situations in public settings where it is not obvious or there’s doubt that the dog is a service animal (such as when a person visits a restaurant or store), employees are not allowed to request documentation for the dog, require the dog demonstrate its task, or inquire about the nature of the person’s disability.1
However, they can ask 2 questions1:
1. Is the animal required because of a disability?
2. What work or task has the animal been trained to perform?
Legal protections. Under the Americans with Disabilities Act (ADA), individuals with disabilities can bring their service animals into buildings or facilities where members of the public, program participants, clients, customers, patrons, or invitees are allowed.2 This does not include private clubs, religious organizations, or places of worship that are not open to the public.6,7 ESAs do not qualify as service animals under the ADA and are not given the same legal accommodations as service animals.1,3 Although ESAs were initially covered by the Air Carrier Access Act, they are no longer allowed in aircraft cabins after the US Department of Transportation revised this Act’s regulations in December 2020. ESAs are covered under the Fair Housing Act. Box 21-3,6-15 further discusses these laws and protections.
Evidence.
Due to the difficulty in reconciling inconsistent definitions for ESAs, there is limited high-quality data pertaining to the potential benefits and risks of ESAs.9 Currently, ESAs are not an evidence-based treatment for psychiatric disorders. To date, a handful of small studies have focused on ESAs. However, data from actual tests of the clinical risks and benefits of ESAs do not exist.9 In practice, ESAs are equivalent to pets. It stands to reason that similar to pets, ESAs could reduce loneliness, improve life satisfaction, and provide a sense of well-being.9 A systematic review suggested that pets provide benefits to patients with mental health conditions “through the intensity of connectivity with their owners and the contribution they make to emotional support in times of crises together with their ability to help manage symptoms when they arise.”18 In response to a congressional mandate, the US Department of Veterans Affairs launched a multi-site study from December 2014 to June 2019 to examine how limitations on activity and quality of life in veterans with posttraumatic stress disorder are impacted by the provision of a service dog vs an emotional support dog.19 As of October 14, 2021, results had not been published.19
Continue to: What’s in a disability service animal/ESA letter?
What’s in a disability service animal/ESA letter?
If you decide to write a letter advocating for your patient to have a service animal or ESA, the letter should appear on letterhead, be written by a licensed mental health professional, and include the following2,20:
- statement that the letter is being written at the patient’s request and is being given directly to the patient for use as the patient sees fit
- confirmation of the patient’s DSM-5 mental health diagnosis
- explanation of how the animal helps alleviate symptoms of the patient’s condition, briefly describing any interaction(s) between the animal and patient that you may have observed, and if applicable, a mention of any training the animal may have received from a qualified trainer if applicable
- explanation of the possible negative effects of the patient not having the animal with him or her
- statement that you are not vouching for the animal’s behavior
- verification of your involvement in your patient’s treatment and your assessment of the patient as their licensed mental health professional (including details such as date and type of license you have and the state/other jurisdiction where it was issued).
In a letter for a service animal, also indicate that your patient is psychiatrically disabled to the extent that your patient is not able to perform at least one major life task without the daily assistance of a service animal.2Should you write your patient a letter?
Writing a letter advocating for a patient to have a service animal or ESA may appear innocuous, but doing so may have serious ramifications. Writing a letter certifying a dog as a service animal does not make that animal a service animal; the dog must be specifically trained for a task or tasks directly related to that individual’s disability. There are no current standards for conducting evaluations to determine the need a patient has for a service animal or ESA. How to conduct such evaluations is beyond the scope of this article. There are meager opportunities for formal education and training on how to conduct these evaluations.9 Online resources may be incomplete or inaccurate, and this information is often produced by lay animal enthusiasts and organizations, which can lead to a biased depiction of these animals.9
If you decide to write a letter for your patient, consider the following forensic and ethical concerns.
Remain objective. As an advocate for your patient, you may find it difficult to remain neutral and objective when asked to determine if your patient has a disability, the severity of the disability, the impact of the disability on your patient’s life, and the need for a service animal or ESA. Ensure that your advocacy for your patient does not impair your objectivity; if that is difficult, consider referring your patient to a third party who can conduct an objective evaluation.
Understand the risks. If you make written recommendations for special accommodations in a letter and those recommendations are disputed by an agency, that agency could initiate legal action and you may be called to justify your recommendations in a deposition or open court.9,21 Before writing the letter, ask yourself, “Can I defend my determination that my patient is disabled by a DSM-5 disorder and that this disability requires the presence of an animal in exception to existing policy?”21 Be prepared to state in a legal proceeding that the presence of a service animal or ESA is necessary. If you are unwilling to risk exposure to a legal action, then you should likely refrain from writing the letter. It is a crime to fraudulently certify an animal as a service animal in some jurisdictions, and such conduct could result in disciplinary action by your licensing board.21
Conduct a systematic examination. When you write a letter for your patient, you are explicitly declaring your patient has a disability or condition. Comprehensive disability determinations are complex and are best conducted by assessing for objective evidence of psychiatric disorders and impairment through the use of standard, systematic examination methods.22 Unstandardized measures (eg, asking patients open-ended questions and then relying on your clinical judgement and interpretation in arriving at conclusions) are not as effective.22 In addition, consider the possibility that your patient may malinger their symptoms in an effort to obtain a letter supporting a service animal or ESA. Assessing for malingering is essential to making a disability determination, especially if a disability claim is based primarily on self-report.22
Anticipate pushback. Problems can arise when a patient wants a letter that you cannot or will not provide due to your scope of practice. Consider how you would resolve the situation when you do not believe your patient has a disability that requires the presence of a service animal or ESA—or you believe that your patient no longer needs a service animal or ESA—and the patient disagrees.21 Disagreeing with your patient’s assessment could result in a conflict of interest that could damage the therapeutic relationship.21
Box 2
The Americans with Disabilities Act (ADA) of 1990, as amended by the ADA Amendments Act of 2008, prohibits discrimination on the basis of disability in several areas, including state and local governments (under Title II of the ADA) and places of public accommodations, commercial facilities, and private entities (under Title III of the ADA).6,7 Thus, individuals with disabilities can bring their service animals into the building or facility where members of the public, program participants, clients, customers, patrons, or invitees are allowed.2 This does not include private clubs not open to the public, religious organizations, or places of worship.6,7
Service animals. Although the ADA recognizes miniature horses as service animals, only dogs are recognized as service animals in regards to Title II and Title III protections under the ADA as of March 15, 2011.2 Federal agencies do not have to comply with the ADA1; however, Section 504 of the Rehabilitation Act of 1973 is the federal law that protects the rights of people with disabilities to participate in federal programs and services.1,8 It states that no qualified individual with a disability shall be excluded from, denied the benefits of, or be subjected to discrimination under any program or activity that receives federal funding or is conducted by federal agencies.8 Courts have strived to interpret the Rehabilitation Act and the ADA in a consistent manner, specifically applying the ADA regulations regarding service animals (including its narrow definition regarding specifically trained tasks and emotional support) to the Rehabilitation Act.9-11
Similarly, commercial airlines do not have to comply with the ADA1 ; however, the Air Carrier Access Act (ACAA) of 1986 is the federal law that protects the rights of people with disabilities in air travel.1,12 On December 2, 2020, the US Department of Transportation announced that it was revising its ACAA regulation regarding service animals on aircraft (this final rule will be effective 30 days after date of publication in the Federal Register).13 Among the many revisions, the US Department of Transportation narrowed the definition of service animals to only dogs that were individually trained to work or perform tasks for the benefits of a person with a disability.13 It requires airlines to treat psychiatric service animals the same as other service animals.13 Although the US Department of Transportation has chosen to closely align its ACAA service animal definition with US Department of Justice service animal definition under the ADA, the substantive requirements in this final rule differ from US Department of Justice’s requirements for service animals under the ADA in various areas (for example, by allowing airlines to require service animal documentation and prohibiting the use of voice control over a service animal).13
Emotional support animals. Regulations regarding ESAs are primarily set by individual states1,3; however, ESAs may qualify for a waiver of a no-pet rule or a pet deposit under the Fair Housing Amendments Act (FHAA) of 1988.2,14 Under the FHAA, if an individual has a disability, as defined by the ADA, that requires the presence of an ESA, or if they have symptoms that are ameliorated by the presence of an ESA, the landlord must comply with this request and allow the animal into the facility without charging pet fees.15
Bottom Line
Disability service animals and emotional support animals (ESAs) differ in their roles and legal protections. Before writing a letter in support of a patient’s request for a service animal or ESA, take into account the forensic and ethical implications of doing so.
Related Resources
- US Department of Justice. Civil Rights Division. Disability Rights Section. ADA requirements. Service animals. Updated February 24, 2020. https://www.ada.gov/service_ animals_2010.htm
American Veterinary Medical Association. Service, emotional support and therapy animals. https://www. avma.org/resources-tools/animal-health-welfare/ service-emotional-support-and-therapy-animals
US Department of Transportation. US Department of Transportation announces final rule on traveling by air with service animals. https://www.transportation.gov/briefingroom/us-department-transportation-announces-finalrule-traveling-air-service-animals
For centuries, animals, especially dogs, have assisted humans in a variety of ways in their daily lives. Animals that assist people with disabilities fall into 2 broad categories: disability service animals, and emotional support animals (ESAs). Often there is confusion in how these categories differ because of the animal’s role and the laws related to them.
This article describes the differences between disability service animals and ESAs, and outlines the forensic and ethical concerns you should consider before agreeing to write a letter for a patient outlining their need for a disability service animal or ESA. A letter may protect a patient and their service animal or ESA in situations where laws and regulations typically prohibit animals, such as on a flight or when renting an apartment or house. Note that a description of how to conduct the formal patient evaluation before writing a verification letter is beyond the scope of this article.
The differences between disability service animals and ESAs
Purpose and training. Disability service animals, or service animals, are dogs of any breed (and in some cases miniature horses) that are specially trained to perform tasks for an individual with a disability (physical, sensory, psychiatric, intellectual, or other mental disability).1-3 These tasks must be directly related to the individual’s disability.1,2 On the other hand, ESAs, which can be any species of animal, provide support and minimize the impact of an individual’s emotional or psychological disability based on their presence alone. Unlike disability service animals, ESAs are not trained to perform a specific task or duty.2,3
There is no legal requirement for service animals to know specific commands, and professional training is not required—individuals can train the animals themselves.1 Service animals, mainly dogs, can be trained to perform numerous tasks, including4:
- attending to an individual’s mobility and activities of daily living
- guiding an individual who is deaf or hearing impaired
- helping to remind an individual to take their medications
- assisting an individual during and/or after a seizure
- alerting individuals with diabetes in advance of low or high blood sugar episodes
- supporting an individual with autism
- assisting an individual with a psychiatric or mental disability
- applying sensory commands such as lying on the person or resting their head on the individual’s lap to help the individual regain behavioral control.
Service dog verification works via an honor system, which can be problematic, especially in the case of psychiatric service dogs, whose handlers may not have a visible disability (Box 11,5).
Box 1
In the United States, there is no national service dog certification program—meaning there is no official test that a dog has to pass in order to obtain formal recognition as a service animal—nor is there a central and mandatory service dog registry.5 Instead, service dog verification works through an honor system, which can be problematic.5 In many states, misrepresenting one’s dog as a service dog is considered a misdemeanor.5 Unfortunately, other than the guidance set forth by the Americans with Disabilities Act, there are no criteria by which one can recognize a genuine service dog vs one being passed off as a service dog.5
In situations in public settings where it is not obvious or there’s doubt that the dog is a service animal (such as when a person visits a restaurant or store), employees are not allowed to request documentation for the dog, require the dog demonstrate its task, or inquire about the nature of the person’s disability.1
However, they can ask 2 questions1:
1. Is the animal required because of a disability?
2. What work or task has the animal been trained to perform?
Legal protections. Under the Americans with Disabilities Act (ADA), individuals with disabilities can bring their service animals into buildings or facilities where members of the public, program participants, clients, customers, patrons, or invitees are allowed.2 This does not include private clubs, religious organizations, or places of worship that are not open to the public.6,7 ESAs do not qualify as service animals under the ADA and are not given the same legal accommodations as service animals.1,3 Although ESAs were initially covered by the Air Carrier Access Act, they are no longer allowed in aircraft cabins after the US Department of Transportation revised this Act’s regulations in December 2020. ESAs are covered under the Fair Housing Act. Box 21-3,6-15 further discusses these laws and protections.
Evidence.
Due to the difficulty in reconciling inconsistent definitions for ESAs, there is limited high-quality data pertaining to the potential benefits and risks of ESAs.9 Currently, ESAs are not an evidence-based treatment for psychiatric disorders. To date, a handful of small studies have focused on ESAs. However, data from actual tests of the clinical risks and benefits of ESAs do not exist.9 In practice, ESAs are equivalent to pets. It stands to reason that similar to pets, ESAs could reduce loneliness, improve life satisfaction, and provide a sense of well-being.9 A systematic review suggested that pets provide benefits to patients with mental health conditions “through the intensity of connectivity with their owners and the contribution they make to emotional support in times of crises together with their ability to help manage symptoms when they arise.”18 In response to a congressional mandate, the US Department of Veterans Affairs launched a multi-site study from December 2014 to June 2019 to examine how limitations on activity and quality of life in veterans with posttraumatic stress disorder are impacted by the provision of a service dog vs an emotional support dog.19 As of October 14, 2021, results had not been published.19
Continue to: What’s in a disability service animal/ESA letter?
What’s in a disability service animal/ESA letter?
If you decide to write a letter advocating for your patient to have a service animal or ESA, the letter should appear on letterhead, be written by a licensed mental health professional, and include the following2,20:
- statement that the letter is being written at the patient’s request and is being given directly to the patient for use as the patient sees fit
- confirmation of the patient’s DSM-5 mental health diagnosis
- explanation of how the animal helps alleviate symptoms of the patient’s condition, briefly describing any interaction(s) between the animal and patient that you may have observed, and if applicable, a mention of any training the animal may have received from a qualified trainer if applicable
- explanation of the possible negative effects of the patient not having the animal with him or her
- statement that you are not vouching for the animal’s behavior
- verification of your involvement in your patient’s treatment and your assessment of the patient as their licensed mental health professional (including details such as date and type of license you have and the state/other jurisdiction where it was issued).
In a letter for a service animal, also indicate that your patient is psychiatrically disabled to the extent that your patient is not able to perform at least one major life task without the daily assistance of a service animal.2Should you write your patient a letter?
Writing a letter advocating for a patient to have a service animal or ESA may appear innocuous, but doing so may have serious ramifications. Writing a letter certifying a dog as a service animal does not make that animal a service animal; the dog must be specifically trained for a task or tasks directly related to that individual’s disability. There are no current standards for conducting evaluations to determine the need a patient has for a service animal or ESA. How to conduct such evaluations is beyond the scope of this article. There are meager opportunities for formal education and training on how to conduct these evaluations.9 Online resources may be incomplete or inaccurate, and this information is often produced by lay animal enthusiasts and organizations, which can lead to a biased depiction of these animals.9
If you decide to write a letter for your patient, consider the following forensic and ethical concerns.
Remain objective. As an advocate for your patient, you may find it difficult to remain neutral and objective when asked to determine if your patient has a disability, the severity of the disability, the impact of the disability on your patient’s life, and the need for a service animal or ESA. Ensure that your advocacy for your patient does not impair your objectivity; if that is difficult, consider referring your patient to a third party who can conduct an objective evaluation.
Understand the risks. If you make written recommendations for special accommodations in a letter and those recommendations are disputed by an agency, that agency could initiate legal action and you may be called to justify your recommendations in a deposition or open court.9,21 Before writing the letter, ask yourself, “Can I defend my determination that my patient is disabled by a DSM-5 disorder and that this disability requires the presence of an animal in exception to existing policy?”21 Be prepared to state in a legal proceeding that the presence of a service animal or ESA is necessary. If you are unwilling to risk exposure to a legal action, then you should likely refrain from writing the letter. It is a crime to fraudulently certify an animal as a service animal in some jurisdictions, and such conduct could result in disciplinary action by your licensing board.21
Conduct a systematic examination. When you write a letter for your patient, you are explicitly declaring your patient has a disability or condition. Comprehensive disability determinations are complex and are best conducted by assessing for objective evidence of psychiatric disorders and impairment through the use of standard, systematic examination methods.22 Unstandardized measures (eg, asking patients open-ended questions and then relying on your clinical judgement and interpretation in arriving at conclusions) are not as effective.22 In addition, consider the possibility that your patient may malinger their symptoms in an effort to obtain a letter supporting a service animal or ESA. Assessing for malingering is essential to making a disability determination, especially if a disability claim is based primarily on self-report.22
Anticipate pushback. Problems can arise when a patient wants a letter that you cannot or will not provide due to your scope of practice. Consider how you would resolve the situation when you do not believe your patient has a disability that requires the presence of a service animal or ESA—or you believe that your patient no longer needs a service animal or ESA—and the patient disagrees.21 Disagreeing with your patient’s assessment could result in a conflict of interest that could damage the therapeutic relationship.21
Box 2
The Americans with Disabilities Act (ADA) of 1990, as amended by the ADA Amendments Act of 2008, prohibits discrimination on the basis of disability in several areas, including state and local governments (under Title II of the ADA) and places of public accommodations, commercial facilities, and private entities (under Title III of the ADA).6,7 Thus, individuals with disabilities can bring their service animals into the building or facility where members of the public, program participants, clients, customers, patrons, or invitees are allowed.2 This does not include private clubs not open to the public, religious organizations, or places of worship.6,7
Service animals. Although the ADA recognizes miniature horses as service animals, only dogs are recognized as service animals in regards to Title II and Title III protections under the ADA as of March 15, 2011.2 Federal agencies do not have to comply with the ADA1; however, Section 504 of the Rehabilitation Act of 1973 is the federal law that protects the rights of people with disabilities to participate in federal programs and services.1,8 It states that no qualified individual with a disability shall be excluded from, denied the benefits of, or be subjected to discrimination under any program or activity that receives federal funding or is conducted by federal agencies.8 Courts have strived to interpret the Rehabilitation Act and the ADA in a consistent manner, specifically applying the ADA regulations regarding service animals (including its narrow definition regarding specifically trained tasks and emotional support) to the Rehabilitation Act.9-11
Similarly, commercial airlines do not have to comply with the ADA1 ; however, the Air Carrier Access Act (ACAA) of 1986 is the federal law that protects the rights of people with disabilities in air travel.1,12 On December 2, 2020, the US Department of Transportation announced that it was revising its ACAA regulation regarding service animals on aircraft (this final rule will be effective 30 days after date of publication in the Federal Register).13 Among the many revisions, the US Department of Transportation narrowed the definition of service animals to only dogs that were individually trained to work or perform tasks for the benefits of a person with a disability.13 It requires airlines to treat psychiatric service animals the same as other service animals.13 Although the US Department of Transportation has chosen to closely align its ACAA service animal definition with US Department of Justice service animal definition under the ADA, the substantive requirements in this final rule differ from US Department of Justice’s requirements for service animals under the ADA in various areas (for example, by allowing airlines to require service animal documentation and prohibiting the use of voice control over a service animal).13
Emotional support animals. Regulations regarding ESAs are primarily set by individual states1,3; however, ESAs may qualify for a waiver of a no-pet rule or a pet deposit under the Fair Housing Amendments Act (FHAA) of 1988.2,14 Under the FHAA, if an individual has a disability, as defined by the ADA, that requires the presence of an ESA, or if they have symptoms that are ameliorated by the presence of an ESA, the landlord must comply with this request and allow the animal into the facility without charging pet fees.15
Bottom Line
Disability service animals and emotional support animals (ESAs) differ in their roles and legal protections. Before writing a letter in support of a patient’s request for a service animal or ESA, take into account the forensic and ethical implications of doing so.
Related Resources
- US Department of Justice. Civil Rights Division. Disability Rights Section. ADA requirements. Service animals. Updated February 24, 2020. https://www.ada.gov/service_ animals_2010.htm
American Veterinary Medical Association. Service, emotional support and therapy animals. https://www. avma.org/resources-tools/animal-health-welfare/ service-emotional-support-and-therapy-animals
US Department of Transportation. US Department of Transportation announces final rule on traveling by air with service animals. https://www.transportation.gov/briefingroom/us-department-transportation-announces-finalrule-traveling-air-service-animals
1. US Department of Justice. Frequently asked questions about service animals and the ADA. Published July 20, 2015. Accessed on July 28, 2021. https://www.ada.gov/regs2010/service_animal_qa.pdf
2. ADA National Network. Service animals and emotional support animals: where are they allowed and under what conditions? Published 2014. Accessed July 28, 2021. https://adata.org/sites/adata.org/files/files/Service_Animal_Booklet_2014(2).pdf
3. Huben-Kearney A. What to do if patients want service or emotional support animals. Psychiatric News. Published September 28, 2020. Accessed July 28, 2021. https://psychnews.psychiatryonline.org/doi/10.1176/appi.pn.2020.10a24
4. Fine AH. The role of therapy and service animals in the lives of persons with disabilities. Rev Sci Tech. 2018;37(1):141-149.
5. Wlodarczyk J. When pigs fly: emotional support animals, service dogs and the politics of legitimacy across species boundaries. Med Humanit. 2019;45(1):82-91.
6. Americans with Disabilities Act of 1990. Pub L. 101-336, 104 Stat. 327.
7. ADA Amendments Act of 2008. Pub L. 110-325.
8. Rehabilitation Act of 1973. Pub L. 93-112, 87 Stat 355.
9. Carroll JD, Mohlenhoff BS, Kersten CM, et al. Laws and ethics related to emotional support animals. J Am Acad Psychiatry Law. 2020;48(4):509-518.
10. Sanchez v US Dept of Energy. 870 F3d 1185 (10th Circuit 2017).
11. Berardelli v Allied Services Inst. of Rehab. Med., 900 F3d 104 (3rd Circuit 2018).
12. Air Carrier Access Act of 1986. 49 USC §41705.
13. US Department of Transportation. US Department of Transportation announces final rule on traveling by air with service animals. Published December 2, 2020. Accessed July 28, 2021. https://www.transportation.gov/briefing-room/us-department-transportation-announces-final-rule-traveling-air-service-animals
14. Fair Housing Amendments Act of 1988. Pub. L. 100-430. https://www.govinfo.gov/content/pkg/STATUTE-102/pdf/STATUTE-102-Pg1619.pdf
15. Boness CL, Younggren JN, Frumkin IB. The certification of emotional support animals: difference between clinical and forensic mental health practitioners. Professional Psychology: Research and Practice. 2017;48(3):216-223.
16. Lane DR, McNicholas J, Collis GM. Dogs for the disabled: benefits to recipients and welfare of the dog. Applied Animal Behaviour Science. 1998;59(1-3):49-60.
17. Hall SS, MacMichael J, Turner A, et al. A survey of the impact of owning a service dog on quality of life for individuals with physical and hearing disability: a pilot study. Health Qual Life Outcomes. 2017;15(1):59. doi:10.1186/s12955-017-0640-x
18. Brooks HL, Rushton K, Lovell K, et al. The power of support from companion animals for people living with mental health problems: a systematic review and narrative synthesis of the evidence. BMC Psychiatry. 2018;18(1):31. doi: 10.1186/s12888-018-1613-2
19. US National Library of Medicine: ClinicalTrials.gov. Can service dogs improve activity and quality of life in veterans with PTSD? (SDPTSD). Updated August 15, 2019. Accessed October 14, 2021. https://clinicaltrials.gov/ct2/show/study/NCT02039843
20. Clay RA. Is that a pet or therapeutic aid? American Psychological Association. 2016;47(8):38. https://www.apa.org/monitor/2016/09/pet-aid
21. Younggren JN, Boisvert JA, Boness CL. Examining emotional support animals and role conflicts in professional psychology. Prof Psychol Res Pr. 2016;47(4):255-260.
22. Gold LH, Anfang SA, Drukteinis AM, et al. AAPL practice guideline for the forensic evaluation of psychiatric disability. J Am Acad Psychiatry Law. 2008;36(4 Suppl):S3-S50. https://www.aapl.org/docs/pdf/Evaluation%20of%20Psychiatric%20Disability.pdf
1. US Department of Justice. Frequently asked questions about service animals and the ADA. Published July 20, 2015. Accessed on July 28, 2021. https://www.ada.gov/regs2010/service_animal_qa.pdf
2. ADA National Network. Service animals and emotional support animals: where are they allowed and under what conditions? Published 2014. Accessed July 28, 2021. https://adata.org/sites/adata.org/files/files/Service_Animal_Booklet_2014(2).pdf
3. Huben-Kearney A. What to do if patients want service or emotional support animals. Psychiatric News. Published September 28, 2020. Accessed July 28, 2021. https://psychnews.psychiatryonline.org/doi/10.1176/appi.pn.2020.10a24
4. Fine AH. The role of therapy and service animals in the lives of persons with disabilities. Rev Sci Tech. 2018;37(1):141-149.
5. Wlodarczyk J. When pigs fly: emotional support animals, service dogs and the politics of legitimacy across species boundaries. Med Humanit. 2019;45(1):82-91.
6. Americans with Disabilities Act of 1990. Pub L. 101-336, 104 Stat. 327.
7. ADA Amendments Act of 2008. Pub L. 110-325.
8. Rehabilitation Act of 1973. Pub L. 93-112, 87 Stat 355.
9. Carroll JD, Mohlenhoff BS, Kersten CM, et al. Laws and ethics related to emotional support animals. J Am Acad Psychiatry Law. 2020;48(4):509-518.
10. Sanchez v US Dept of Energy. 870 F3d 1185 (10th Circuit 2017).
11. Berardelli v Allied Services Inst. of Rehab. Med., 900 F3d 104 (3rd Circuit 2018).
12. Air Carrier Access Act of 1986. 49 USC §41705.
13. US Department of Transportation. US Department of Transportation announces final rule on traveling by air with service animals. Published December 2, 2020. Accessed July 28, 2021. https://www.transportation.gov/briefing-room/us-department-transportation-announces-final-rule-traveling-air-service-animals
14. Fair Housing Amendments Act of 1988. Pub. L. 100-430. https://www.govinfo.gov/content/pkg/STATUTE-102/pdf/STATUTE-102-Pg1619.pdf
15. Boness CL, Younggren JN, Frumkin IB. The certification of emotional support animals: difference between clinical and forensic mental health practitioners. Professional Psychology: Research and Practice. 2017;48(3):216-223.
16. Lane DR, McNicholas J, Collis GM. Dogs for the disabled: benefits to recipients and welfare of the dog. Applied Animal Behaviour Science. 1998;59(1-3):49-60.
17. Hall SS, MacMichael J, Turner A, et al. A survey of the impact of owning a service dog on quality of life for individuals with physical and hearing disability: a pilot study. Health Qual Life Outcomes. 2017;15(1):59. doi:10.1186/s12955-017-0640-x
18. Brooks HL, Rushton K, Lovell K, et al. The power of support from companion animals for people living with mental health problems: a systematic review and narrative synthesis of the evidence. BMC Psychiatry. 2018;18(1):31. doi: 10.1186/s12888-018-1613-2
19. US National Library of Medicine: ClinicalTrials.gov. Can service dogs improve activity and quality of life in veterans with PTSD? (SDPTSD). Updated August 15, 2019. Accessed October 14, 2021. https://clinicaltrials.gov/ct2/show/study/NCT02039843
20. Clay RA. Is that a pet or therapeutic aid? American Psychological Association. 2016;47(8):38. https://www.apa.org/monitor/2016/09/pet-aid
21. Younggren JN, Boisvert JA, Boness CL. Examining emotional support animals and role conflicts in professional psychology. Prof Psychol Res Pr. 2016;47(4):255-260.
22. Gold LH, Anfang SA, Drukteinis AM, et al. AAPL practice guideline for the forensic evaluation of psychiatric disability. J Am Acad Psychiatry Law. 2008;36(4 Suppl):S3-S50. https://www.aapl.org/docs/pdf/Evaluation%20of%20Psychiatric%20Disability.pdf
Early interventions for psychosis
Neuroscience research over the past half century has failed to significantly advance the treatment of severe mental illness.1,2 Hence, evidence that a longer duration of untreated psychosis (DUP) aggravates—and early intervention with medication and social supports ameliorates—the long-term adverse consequences of psychotic disorders generated a great deal of interest.3,4 This knowledge led to the development of diverse early intervention services worldwide aimed at this putative “critical window.” It raised the possibility that appropriate interventions could prevent the long-term disability that makes chronic psychosis one of the most debilitating disorders.5,6 However, even beyond the varied cultural and economic confounds, it is difficult to assess, compare, and optimize program effectiveness.7 Obstacles include paucity of sufficiently powered, well-designed randomized controlled trials (RCTs), the absence of diagnostic biomarkers or other prognostic indicators to better account for the inherent heterogeneity in the population and associated outcomes, and the absence of modifiable risk factors that can guide interventions and provide intermediate outcomes.4,8-10
To better appreciate these issues, it is important to distinguish whether a program is designed to prevent psychosis, or to mitigate the effects of psychosis. Two models include the:
- Prevention model, which focuses on young individuals who are not yet overtly psychotic but at high risk
- First-episode recovery model, which focuses on those who have experienced a first episode of psychosis (FEP) but have not yet developed a chronic disorder.
Both models share long-term goals and are hampered by many of the same issues summarized above. They both deviate markedly from the standard medical model by including psychosocial services designed to promote restoration of a self-defined trajectory to greater independence.11-14 The 2 differ, however, in the challenges they must overcome to produce their sample populations and establish effective interventions.10,15,16
In this article, we provide a succinct overview of these issues and a set of recommendations based on a “strength-based” approach. This approach focuses on finding common ground between patients, their support system, and the treatment team in the service of empowering patients to resume responsibility for transition to adulthood.
The prevention model
While most prevention initiatives in medicine rely on the growing ability to target specific pathophysiologic pathways,3 preventing psychosis relies on clinical evidence showing that DUP and early interventions predict a better course of severe mental illness.17 In contrast, initiatives such as normalizing neonatal neuronal pathways are more consistent with the strategy utilized in other fields but have yet to yield a pathophysiologic target for psychosis.3,18
Initial efforts to identify ‘at-risk’ individuals
The prevention model of psychosis is based on the ability to identify young individuals at high risk for developing a psychotic disorder (Figure). The first screening measures were focused on prodromal psychosis (eg, significant loss of function, family history, and “intermittent” and “attenuated” psychotic symptoms). When applied to referred (ie, pre-screened) samples, 30% to 40% of this group who met criteria transitioned to psychosis over the next 1 to 3 years despite antidepressant and psychosocial interventions.19 Comprising 8 academic medical centers, the North American Prodrome Longitudinal Study (NAPLS) produced similar results using the Structured Interview for Prodromal Syndromes (SIPS).17 Thus, 30% to 50% of pre-screened individuals referred by school counselors and mental health professionals met SIPS criteria, and 35% of these individuals transitioned to psychosis over 30 months. The validity of this measure was further supported by the fact that higher baseline levels of unusual thought content, suspicion/paranoia, social impairment, and substance abuse successfully distinguished approximately 80% of those who transitioned to psychosis. The results of this first generation of screening studies were exciting because they seemed to demonstrate that highly concentrated samples of young persons at high risk of developing psychosis could be identified, and that fine-tuning the screening criteria could produce even more enriched samples (ie, positive predictive power).

Initial interventions produced promising results
The development of effective screening measures led to reports of effective treatment interventions. These were largely applied in a clinical staging model that restricted antipsychotic medications to those who failed to improve after receiving potentially “less toxic” interventions (eg, omega-3 polyunsaturated fatty acids and other antioxidants; psychotherapy; cognitive-behavioral therapy [CBT]; family therapy).5 While study designs were typically quasi-experimental, the interventions appeared to dramatically diminish the transition to psychosis (ie, approximately 50%).
Continue to: The first generation...
The first generation of RCTs appeared to confirm these results, although sample sizes were small, and most study designs assessed only a single intervention. Initial meta-analyses of these data reported that both CBT and antipsychotics appeared to prevent approximately one-half of individuals from becoming psychotic at 12 months, and more than one-third at 2 to 4 years, compared with treatment as usual.20
While some researchers challenged the validity of these findings,21-23 the results generated tremendous international enthusiasm and calls for widespread implementation.6 The number of early intervention services (EIS) centers increased dramatically worldwide, and in 2014 the National Institute for Health and Care Excellence released standards for interventions to prevent transition to psychosis.24 These included close monitoring, CBT and family interventions, and avoiding antipsychotics when possible.24
Focusing on sensitivity over specificity
The first generation of studies generated by the prevention model relied on outreach programs or referrals, which produced small samples of carefully selected, pre-screened individuals (Figure, Pre-screened) who were then screened again to establish the high-risk sample.25 While approximately 33% of these individuals became psychotic, the screening process required a very efficient means of eliminating those not at high-risk (given the ultimate target population represented only approximately .5% of young people) (Figure). The pre-screening and screening processes in these first-generation studies were labor-intensive but could only identify approximately 5% of those individuals destined to become psychotic over the next 2 or 3 years. Thus, alternative methods to enhance sensitivity were needed to extend programming to the general population.
Second-generation pre-screening (Figure; Step 1). New pre-screening methods were identified that captured more individuals destined to become psychotic. For example, approximately 90% of this population were registered in health care organizations (eg, health maintenance organizations) and received a psychiatric diagnosis in the year prior to the onset of psychosis (true positives).8 These samples, however, contained a much higher percentage of persons not destined to become psychotic, and somehow the issue of specificity (decreasing false positives) was minimized.8,9 For example, pre-screened samples contained 20 to 50 individuals not destined to become psychotic for each one who did.26 Since screening measures could only eliminate approximately 20% of this group (Figure, Step 2, page 25), second-generation transition rates fell from 30% to 40% to 2% to 10%.27,28
Other pre-screening approaches were introduced, but they also focused on capturing more of those destined to become psychotic (sensitivity) than eliminating those who would not (specificity). For instance, Australia opened more than 100 “Headspace” community centers nationwide designed to promote engagement and self-esteem in youth experiencing anxiety; depression; stress; relationship, work, or school problems; or bullying.13 Most services were free and included mental health staff who screened for psychosis and provided a wide range of services in a destigmatized setting. These methods identified at least an additional 5% to 7% of individuals destined to become psychotic, but to our knowledge, no data have been published on whether they helped eliminate those who did not.
Continue to: Second-generation screening
Second-generation screening (Figure, Step 2). A second screening aims to retain those pre-screened individuals who will become psychotic (ie, minimizing false negatives) while further minimizing those who do not (ie, minimizing false positives). The addition of cognitive, neural (eg, structural MRI; neurophysiologic), and biochemical (eg, inflammatory immune and stress) markers to the risk calculators have produced a sensitivity close to 100%.8,9 Unfortunately, these studies downplayed specificity, which remained approximately 20%.8,9 Specificity is critical not just because of concerns about stigma (ie, labeling people as pre-psychotic when they are not) but also because of the adverse effects of antipsychotic medications and the effects on future program development (interventions are costly and labor-intensive). Also, diluting the pool with individuals not at risk makes it nearly impossible to identify effective interventions (ie, power).27,28
While some studies focused on increasing specificity (to approximately 75%), this leads to an unacceptable loss of sensitivity (from 90% to 60%),29 with 40% of pre-screened individuals who would become psychotic being eliminated from the study population. The addition of other biological markers (eg, salivary cortisol)30 and use of learning health systems may be able to enhance these numbers (initial reports of specificity = 87% and sensitivity = 85%).8,9 This is accomplished by integrating artificial and human intelligence measures of clinical (symptom and neurocognitive measures) and biological (eg, polygenetic risk scores; gray matter volume) variables.31 However, even if these results are replicated, more effective pre-screening measures will be required.
Identifying a suitable sample population for prevention program studies is clearly more complicated than for FEP studies, where one can usually identify many of those in the at-risk population by their first hospitalization for psychotic symptoms. The issues of false positives (eg, substance-induced psychosis) and negatives (eg, slow deterioration, prominent negative symptoms) are important concerns, but proportionately far less significant.
Prevention and FEP interventions
Once a study sample is constituted, 1 to 3 years of treatment interventions are initiated. Interventions for prevention programs typically include CBT directed at attenuated psychosis (eg, reframing or de-catastrophizing unusual thoughts and minimizing distress associated with unusual perceptions); case management to facilitate personal, educational, and vocational goals; and family therapy in single or multi-group formats to educate one’s support system about the risk state and to minimize adverse familial responses.14 Many programs also include supported education or employment services to promote reintegration in age-appropriate activities; group therapy focused on substance abuse and social skills training; cognitive remediation to ameliorate the cognitive dysfunction; and an array of pharmacologic interventions designed to delay or prevent transition to psychosis or to alleviate symptoms. While most interventions are similar, FEP programs have recently included peer support staff. This appears to instill hope in newly diagnosed patients, provide role models, and provide peer supporters an opportunity to use their experiences to help others and earn income.32
The breadth and depth of these services are critical because retention in the program is highly dependent on participant engagement, which in turn is highly dependent on whether the program can help individuals get what they want (eg, friends, employment, education, more autonomy, physical health). The setting and atmosphere of the treatment program and the willingness/ability of staff to meet participants in the community are also important elements.11,12 In this context, the Headspace community centers are having an impact far beyond Australia and may prove to be a particularly good model.13
Continue to: Assessing prevention and FEP interventions
Assessing prevention and FEP interventions
The second generation of studies of prevention programs has not confirmed, let alone extended, the earlier findings and meta-analyses. A 2020 report concluded CBT was still the most promising intervention; it was more effective than control treatments at 12 and 18 months, although not at 6, 24, or 48 months.33 This review included controlled, open-label, and naturalistic studies that assessed family therapy; omega-3 polyunsaturated fatty acids; integrated psychological therapy (a package of interventions that included family education, CBT, social skills training, and cognitive remediation); N-methyl-
While these disappointing findings are at least partly attributable to the methodological challenges described above and in the Figure, other factors may hinder establishing effective interventions. In contrast to FEP studies, those focused on prevention had a very ambitious agenda (eliminating psychosis) and tended to downplay more modest intermediate outcomes. These studies also tended to assess new ideas with small samples rather than pursue promising findings with larger multi-site studies focused on a group of interventions. The authors of a Cochrane review observed “There is the impression that in this whole area there is a triumph of hope over adversity. There is the repeated hope invested in another—often unique—study question and then a study of fewer than 100 participants are completed. This results in the set of comparisons reported here, all 9 of which are too underpowered to really highlight clear differences.”34 To use a baseball analogy, it seems that investigators are “swinging for the fence” when a few singles are what’s really needed.
From the outset, the goals of FEP studies were more modest, largely ignoring the task of developing consensus definitions of recovery that require following patients for up to 5 to 10 years. Instead, they use intermediate endpoints based on adapting treatments that already appeared effective in patients with chronic mental disorders.35 As a consequence, researchers examining FEP demonstrated clear, albeit limited, salutary effects using large multi-site trials and previously established outcome measures.3,10,36 For instance, the Recovery After an Initial Schizophrenia Episode-Early Treatment Program (RAISE-ETP) study was a 2-year, multi-site RCT (N = 404) funded by the National Institute of Mental Health (NIMH). The investigators reported improved indices of social function (eg, quality of life; education and work participation) and total ratings of psychopathology and depression compared with treatment as usual. Furthermore, they established that DUP predicted treatment response.35 The latter finding was underscored by improvement being limited to the 50% with <74 weeks DUP. Annual costs of the program per 1 standard deviation improvement in quality of life were approximately $1,000 for patients with <74 weeks DUP and $40,000 for those with >74 weeks DUP. Concurrent meta-analyses confirmed and extended these findings,16 showing higher remission rates; diminished relapses and hospital admissions; greater engagement in programming; greater involvement in work and school; improved quality of life; and other steps toward recovery. These studies were also able to establish a clear benefit of antipsychotic medications, particularly a high acceptance of long-acting injectable antipsychotic formulations, which promoted adherence and decreased some adverse events37; and early use of clozapine therapy, which improved remission rates and longer-term outcomes.38 Other findings underscored the need to anticipate and address new problems associated with effective antipsychotic therapy (eg, antipsychotic response correlates with weight gain, a particularly intolerable adverse event for this age group).39 Providing pre-emptive strategies such as exercise groups and nutritional education may be necessary to maintain adherence.
Limitations of FEP studies
The effect sizes in these FEP studies were small to medium on outcome measures tracking recovery and associated indicators (eg, global functioning, school/work participation, treatment engagement); the number needed to treat for each of these was >10. There is no clear evidence that recovery programs such as RAISE-ETP actually reduce longer-term disability. Most studies showed disability payments increased while clinical benefits tended to fade over time. In addition, by grouping interventions together, the studies made it difficult to identify effective vs ineffective treatments, let alone determine how best to personalize therapy for participants in future studies.
The next generation of FEP studies
While limited in scope, the results of the recent FEP studies justify a next generation of recovery interventions designed to address these shortcomings and optimize program outcomes.39 Most previous FEP studies were conducted in community mental health center settings, thus eliminating the need to transition services developed in academia into the “real world.” The next generation of NIMH studies will be primarily conducted in analogous settings under the Early Psychosis Intervention Network (EPINET).40 EPINET’s study design echoes that responsible for the stepwise successes in the late 20th century that produced cures for the deadliest childhood cancer, acute lymphoblastic leukemia (ALL). This disease was successfully treated by modifying diverse evidence-based practices without relying on pharmacologic or other major treatment breakthroughs. Despite this, the effort yielded successful personalized interventions that were not obtainable for other severe childhood conditions.40 EPINET hopes to automate much of these stepwise advances with a learning health system. This program relies on data routinely collected in clinical practice to drive the process of scientific discovery. Specifically, it determines the relationships between clinical features, biologic measures, treatment characteristics, and symptomatic and functional outcomes. EPINET aims to accelerate our understanding of biomarkers of psychosis risk and onset, as well as factors associated with recovery and cure. Dashboard displays of outcomes will allow for real-time comparisons within and across early intervention clinics. This in turn identifies performance gaps and drives continuous quality improvement.
Continue to: Barriers to optimizing program efficacy for both models
Barriers to optimizing program efficacy for both models
Unfortunately, there are stark differences between ALL and severe mental disorders that potentially jeopardize the achievement of these aims, despite the advances in data analytic abilities that drive the learning health system. Specifically, the heterogeneity of psychotic illnesses and the absence of reliable prognostic and modifiable risk markers (responsible for failed efforts to enhance treatment of serious mental illness over the last half century1,2,41) are unlikely to be resolved by a learning health system. These measures are vital to determine whether specific interventions are effective, particularly given the absence of a randomized control group in the EPINET/learning health system design. Fortunately, however, the National Institutes for Health has recently initiated the Accelerating Medicines Partnership–Schizophrenia (AMP-SCZ). This approach seeks “promising biological markers that can help identify those at risk of developing schizophrenia as early as possible, track the progression of symptoms and other outcomes and ultimately define targets for treatment development.”42 The Box1,4,9,10,36,41,43-45 describes some of the challenges involved in identifying biomarkers of severe mental illness.
Box
Biomarkers and modifiable risk factors4,9,10,41,43 are at the core of personalized medicine and its ultimate objective (ie, theragnostics). This is the ability to identify the correct intervention for a disorder based on a biomarker of the illness.10,36 The inability to identify biomarkers of severe mental illness is multifactorial but in part may be attributable to “looking in all the wrong places.”41 By focusing on neural processes that generate psychiatric symptomatology, investigators are assuming they can bridge the “mind gap”1 and specifically distinguish between pathological, compensatory, or collateral measures of poorly characterized limbic neural functions.41
It may be more productive to identify a pathological process within the limbic system that produces a medical condition as well as the mental disorder. If one can isolate the pathologic limbic circuit activity responsible for a medical condition, one may be able to reproduce this in animal models and determine whether analogous processes contribute to the core features of the mental illness. Characterization of the aberrant neural circuit in animal models also could yield targets for future therapies. For example, episodic water intoxication in a discrete subset of patients with schizophrenia44 appears to arise from a stress diathesis produced by anterior hippocampal pathology that disrupts regulation of antidiuretic hormone, oxytocin, and hypothalamic-pituitary-adrenal axis secretion. These patients also exhibit psychogenic polydipsia that may be a consequence of the same hippocampal pathology that disrupts ventral striatal and lateral hypothalamic circuits. These circuits, in turn, also modulate motivated behaviors and cognitive processes likely relevant to psychosis.45
A strength-based approach
The absence of sufficiently powered RCTs for prevention studies and the reliance on intermediate outcomes for FEP studies leaves unanswered whether such programs can effectively prevent chronic psychosis at a cost society is willing to pay. Still, substantial evidence indicates that outreach, long-acting injectable antipsychotics, early consideration of clozapine, family therapy, CBT for psychosis/attenuated psychosis, and services focused on competitive employment can preserve social and occupational functioning.16,34 Until these broader questions are more definitively addressed, it seems reasonable to apply what we have learned (Table11,12,35,37-39,46).

Simply avoiding the most divisive aspects of the medical model that inadvertently promote stigma and undercut self-confidence may help maintain patients’ willingness to learn how best to apply their strengths and manage their limitations.11 The progression to enduring psychotic features (eg, fixed delusions) may reflect ongoing social isolation and alienation. A strength-based approach seeks first to establish common goals (eg, school, work, friends, family support, housing, leaving home) and then works to empower the patient to successfully reach those goals.35 This typically involves giving them the opportunity to fail, avoiding criticism when they do, and focusing on these experiences as learning opportunities from which success can ultimately result.
It is difficult to offer all these services in a typical private practice setting. Instead, it may make more sense to use one of the hundreds of early intervention services programs in the United States.46 If a psychiatric clinician is dedicated to working with this population, it may also be possible to establish ongoing relationships with primary care physicians, family and CBT therapists, family support services (eg, National Alliance on Mental Illness), caseworkers and employment counselors. In essence, a psychiatrist may be able re-create a multidisciplinary effort by taking advantage of the expertise of these various professionals. The challenge is to create a consistent message for patients and families in the absence of regular meetings with the clinical team, although the recent reliance on and improved sophistication of virtual meetings may help. Psychiatrists often play a critical role even when the patient is not prescribed medication, partly because they are most comfortable handling the risks and may have the most comprehensive understanding of the issues at play. When medications are appropriate and patients with FEP are willing to take them, early consideration of long-acting injectable antipsychotics and clozapine may provide better stabilization and diminish the risk of earlier and more frequent relapses.
Bottom Line
Early interventions for psychosis include the prevention model and the first-episode recovery model. It is difficult to assess, compare, and optimize the effectiveness of such programs. Current evidence supports a ‘strength-based’ approach focused on finding common ground between patients, their support system, and the treatment team.
Related Resources
- Early Assessment and Support Alliance. National Early Psychosis Directory. https://easacommunity.org/nationaldirectory.php
- Kane JM, Robinson DG, Schooler NR, et al. Comprehensive versus usual community care for first-episode psychosis: 2-year outcomes from the NIMH RAISE Early Treatment Program. Am J Psychiatry. 2016 ;173(4):362-372
Drug Brand Name
Clozapine • Clozaril
1. Hyman SE. Revolution stalled. Sci Transl Med. 2012;4(155):155cm11. doi: 10.1126/scitranslmed.3003142
2. Harrington A. Mind fixers: psychiatry’s troubled search for the biology of mental illness. W.W. Norton & Company; 2019.
3. Millan MJ, Andrieux A, Bartzokis G, et al. Altering the course of schizophrenia: progress and perspectives. Nat Rev Drug Discov. 2016;15(7):485-515.
4. Lieberman JA, Small SA, Girgis RR. Early detection and preventive intervention in schizophrenia: from fantasy to reality. Am J Psychiatry. 2019;176(10):794-810.
5. McGorry PD, Nelson B, Nordentoft M, et al. Intervention in individuals at ultra-high risk for psychosis: a review and future directions. J Clin Psychiatry. 2009;70(9):1206-1212.
6. Csillag C, Nordentoft M, Mizuno M, et al. Early intervention in psychosis: From clinical intervention to health system implementation. Early Interv Psychiatry. 2018;12(4):757-764.
7. McGorry PD, Ratheesh A, O’Donoghue B. Early intervention—an implementation challenge for 21st century mental health care. JAMA Psychiatry. 2018;75(6):545-546.
8. Rosenheck R. Toward dissemination of secondary prevention for psychosis. Am J Psychiatry. 2018;175(5):393-394.
9. Fusar-Poli P, Salazar de Pablo G, Correll CU, et al. Prevention of psychosis: advances in detection, prognosis, and intervention. JAMA Psychiatry. 2020;77(7):755-765.
10. Oliver D, Reilly TJ, Baccaredda Boy O, et al. What causes the onset of psychosis in individuals at clinical high risk? A meta-analysis of risk and protective factors. Schizophr Bull. 2020;46(1):110-120.
11. Tindall R, Simmons M, Allott K, et al. Disengagement processes within an early intervention service for first-episode psychosis: a longitudinal, qualitative, multi-perspective study. Front Psychiatry. 2020;11:565-565.
12. Dixon LB, Holoshitz Y, Nossel I. Treatment engagement of individuals experiencing mental illness: review and update. World Psychiatry. 2016;15(1):13-20.
13. Rickwood D, Paraskakis M, Quin D, et al. Australia’s innovation in youth mental health care: The headspace centre model. Early Interv Psychiatry. 2019;13(1):159-166.
14. Woodberry KA, Shapiro DI, Bryant C, et al. Progress and future directions in research on the psychosis prodrome: a review for clinicians. Harv Rev Psychiatry. 2016;24(2):87-103.
15. Gupta T, Mittal VA. Advances in clinical staging, early intervention, and the prevention of psychosis. F1000Res. 2019;8:F1000 Faculty Rev-2027. doi: 10.12688/f1000research.20346.1
16. Correll CU, Galling B, Pawar A, et al. Comparison of early intervention services vs treatment as usual for early-phase psychosis: a systematic review, meta-analysis, and meta-regression. JAMA Psychiatry. 2018;75(6):555-565.
17. Cannon TD, Cadenhead K, Cornblatt B, et al. Prediction of psychosis in youth at high clinical risk: a multisite longitudinal study in North America. Arch Gen Psychiatry. 2008;65(1):28-37.
18. Sommer IE, Bearden CE, van Dellen E, et al. Early interventions in risk groups for schizophrenia: what are we waiting for? NPJ Schizophr. 2016;2(1):16003-16003.
19. McGorry PD, Nelson B. Clinical high risk for psychosis—not seeing the trees for the wood. JAMA Psychiatry. 2020;77(7):559-560.
20. van der Gaag M, Smit F, Bechdolf A, et al. Preventing a first episode of psychosis: meta-analysis of randomized controlled prevention trials of 12 month and longer-term follow-ups. Schizophr Res. 2013;149(1):56-62.
21. Marshall M, Rathbone J. Early intervention for psychosis. Cochrane Database Syst Rev. 2011;(6):CD004718. doi: 10.1002/14651858.CD004718.pub3
22. Heinssen RK, Insel TR. Preventing the onset of psychosis: not quite there yet. Schizophr Bull. 2015;41(1):28-29.
23. Amos AJ. Evidence that treatment prevents transition to psychosis in ultra-high-risk patients remains questionable. Schizophr Res. 2014;153(1):240.
24. National Institute for Health and Care Excellence. Psychosis and schizophrenia in adults: prevention and management. Clinical guideline [CG178]. 1.3.7 How to deliver psychological interventions. Published February 12, 2014. Updated March 1, 2014. Accessed August 30, 2021. https://www.nice.org.uk/guidance/cg178/chapter/recommendations#how-to-deliver-psychological-interventions
25. Fusar-Poli P, Werbeloff N, Rutigliano G, et al. Transdiagnostic risk calculator for the automatic detection of individuals at risk and the prediction of psychosis: second replication in an independent National Health Service Trust. Schizophr Bull. 2019;45(3):562-570.
26. Fusar-Poli P, Oliver D, Spada G, et al. The case for improved transdiagnostic detection of first-episode psychosis: electronic health record cohort study. Schizophr Res. 2021;228:547-554.
27. Fusar-Poli P. Negative psychosis prevention trials. JAMA Psychiatry. 2017;74(6):651.
28. Cuijpers P, Smit F, Furukawa TA. Most at‐risk individuals will not develop a mental disorder: the limited predictive strength of risk factors. World Psychiatry. 2021;20(2):224-225.
29. Carrión RE, Cornblatt BA, Burton CZ, et al. Personalized prediction of psychosis: external validation of the NAPLS-2 psychosis risk calculator with the EDIPPP Project. Am J Psychiatry. 2016;173(10):989-996.
30. Worthington MA, Walker EF, Addington J, et al. Incorporating cortisol into the NAPLS2 individualized risk calculator for prediction of psychosis. Schizophr Res. 2021;227:95-100.
31. Koutsouleris N, Dwyer DB, Degenhardt F, et al. Multimodal machine learning workflows for prediction of psychosis in patients with clinical high-risk syndromes and recent-onset depression. JAMA Psychiatry. 2021;78(2):195-209.
32. Simmons MB, Grace D, Fava NJ, et al. The experiences of youth mental health peer workers over time: a qualitative study with longitudinal analysis. Community Ment Health J. 2020;56(5):906-914.
33. Devoe DJ, Farris MS, Townes P, et al. Interventions and transition in youth at risk of psychosis: a systematic review and meta-analyses. J Clin Psychiatry. 2020;81(3):17r12053. doi: 10.4088/JCP.17r12053
34. Bosnjak Kuharic D, Kekin I, Hew J, et al. Interventions for prodromal stage of psychosis. Cochrane Database Syst Rev. 2019;2019(11):CD012236
35. Dixon LB, Goldman HH, Srihari VH, et al. Transforming the treatment of schizophrenia in the United States: The RAISE Initiative. Annu Rev Clin Psychol. 2018;14:237-258.
36. Friedman-Yakoobian MS, Parrish EM, Eack SM, et al. Neurocognitive and social cognitive training for youth at clinical high risk (CHR) for psychosis: a randomized controlled feasibility trial. Schizophr Res. 2020;S0920-9964(20)30461-8. doi: 10.1016/j.schres.2020.09.005
37. Kane JM, Schooler NR, Marcy P, et al. Effect of long-acting injectable antipsychotics vs usual care on time to first hospitalization in early-phase schizophrenia: a randomized clinical trial. JAMA Psychiatry. 2020;77(12):1217-1224.
38. Morrison AP, Pyle M, Maughan D, et al. Antipsychotic medication versus psychological intervention versus a combination of both in adolescents with first-episode psychosis (MAPS): a multicentre, three-arm, randomised controlled pilot and feasibility study. Lancet Psychiatry. 2020;7(9):788-800.
39. Chen YQ, Li XR, Zhang L, et al. Therapeutic response is associated with antipsychotic-induced weight gain in drug-naive first-episode patients with schizophrenia: an 8-week prospective study. J Clin Psychiatry. 2021;82(3):20m13469. doi: 10.4088/JCP.20m13469
40. Insel TR. RAISE-ing our expectations for first-episode psychosis. Am J Psychiatry. 2016;173(4):311-312.
41. Tandon R, Goldman M. Overview of neurobiology. In: Janicak PG, Marder SR, Tandon R, et al, eds. Schizophrenia: recent advances in diagnosis and treatment. Springer; 2014:27-33.
42. National Institutes of Health. Accelerating Medicines Partnership. Schizophrenia. Accessed August 30, 2021. https://www.nih.gov/research-training/accelerating-medicines-partnership-amp/schizophrenia
43. Guloksuz S, van Os J. The slow death of the concept of schizophrenia and the painful birth of the psychosis spectrum. Psychol Med. 2018;48(2):229-244.
44. Christ-Crain M, Bichet DG, Fenske WK, et al. Diabetes insipidus. Nat Rev Dis Primers. 2019;5(1):54.
45. Ahmadi L, Goldman MB. Primary polydipsia: update. Best Pract Res Clin Endocrinol Metab. 2020;34(5):101469. doi: 10.1016/j.beem.2020.101469
46. Early Assessment and Support Alliance. National Early Psychosis Directory. Accessed August 30, 2021. https://easacommunity.org/national-directory.php
Neuroscience research over the past half century has failed to significantly advance the treatment of severe mental illness.1,2 Hence, evidence that a longer duration of untreated psychosis (DUP) aggravates—and early intervention with medication and social supports ameliorates—the long-term adverse consequences of psychotic disorders generated a great deal of interest.3,4 This knowledge led to the development of diverse early intervention services worldwide aimed at this putative “critical window.” It raised the possibility that appropriate interventions could prevent the long-term disability that makes chronic psychosis one of the most debilitating disorders.5,6 However, even beyond the varied cultural and economic confounds, it is difficult to assess, compare, and optimize program effectiveness.7 Obstacles include paucity of sufficiently powered, well-designed randomized controlled trials (RCTs), the absence of diagnostic biomarkers or other prognostic indicators to better account for the inherent heterogeneity in the population and associated outcomes, and the absence of modifiable risk factors that can guide interventions and provide intermediate outcomes.4,8-10
To better appreciate these issues, it is important to distinguish whether a program is designed to prevent psychosis, or to mitigate the effects of psychosis. Two models include the:
- Prevention model, which focuses on young individuals who are not yet overtly psychotic but at high risk
- First-episode recovery model, which focuses on those who have experienced a first episode of psychosis (FEP) but have not yet developed a chronic disorder.
Both models share long-term goals and are hampered by many of the same issues summarized above. They both deviate markedly from the standard medical model by including psychosocial services designed to promote restoration of a self-defined trajectory to greater independence.11-14 The 2 differ, however, in the challenges they must overcome to produce their sample populations and establish effective interventions.10,15,16
In this article, we provide a succinct overview of these issues and a set of recommendations based on a “strength-based” approach. This approach focuses on finding common ground between patients, their support system, and the treatment team in the service of empowering patients to resume responsibility for transition to adulthood.
The prevention model
While most prevention initiatives in medicine rely on the growing ability to target specific pathophysiologic pathways,3 preventing psychosis relies on clinical evidence showing that DUP and early interventions predict a better course of severe mental illness.17 In contrast, initiatives such as normalizing neonatal neuronal pathways are more consistent with the strategy utilized in other fields but have yet to yield a pathophysiologic target for psychosis.3,18
Initial efforts to identify ‘at-risk’ individuals
The prevention model of psychosis is based on the ability to identify young individuals at high risk for developing a psychotic disorder (Figure). The first screening measures were focused on prodromal psychosis (eg, significant loss of function, family history, and “intermittent” and “attenuated” psychotic symptoms). When applied to referred (ie, pre-screened) samples, 30% to 40% of this group who met criteria transitioned to psychosis over the next 1 to 3 years despite antidepressant and psychosocial interventions.19 Comprising 8 academic medical centers, the North American Prodrome Longitudinal Study (NAPLS) produced similar results using the Structured Interview for Prodromal Syndromes (SIPS).17 Thus, 30% to 50% of pre-screened individuals referred by school counselors and mental health professionals met SIPS criteria, and 35% of these individuals transitioned to psychosis over 30 months. The validity of this measure was further supported by the fact that higher baseline levels of unusual thought content, suspicion/paranoia, social impairment, and substance abuse successfully distinguished approximately 80% of those who transitioned to psychosis. The results of this first generation of screening studies were exciting because they seemed to demonstrate that highly concentrated samples of young persons at high risk of developing psychosis could be identified, and that fine-tuning the screening criteria could produce even more enriched samples (ie, positive predictive power).
Initial interventions produced promising results
The development of effective screening measures led to reports of effective treatment interventions. These were largely applied in a clinical staging model that restricted antipsychotic medications to those who failed to improve after receiving potentially “less toxic” interventions (eg, omega-3 polyunsaturated fatty acids and other antioxidants; psychotherapy; cognitive-behavioral therapy [CBT]; family therapy).5 While study designs were typically quasi-experimental, the interventions appeared to dramatically diminish the transition to psychosis (ie, approximately 50%).
Continue to: The first generation...
The first generation of RCTs appeared to confirm these results, although sample sizes were small, and most study designs assessed only a single intervention. Initial meta-analyses of these data reported that both CBT and antipsychotics appeared to prevent approximately one-half of individuals from becoming psychotic at 12 months, and more than one-third at 2 to 4 years, compared with treatment as usual.20
While some researchers challenged the validity of these findings,21-23 the results generated tremendous international enthusiasm and calls for widespread implementation.6 The number of early intervention services (EIS) centers increased dramatically worldwide, and in 2014 the National Institute for Health and Care Excellence released standards for interventions to prevent transition to psychosis.24 These included close monitoring, CBT and family interventions, and avoiding antipsychotics when possible.24
Focusing on sensitivity over specificity
The first generation of studies generated by the prevention model relied on outreach programs or referrals, which produced small samples of carefully selected, pre-screened individuals (Figure, Pre-screened) who were then screened again to establish the high-risk sample.25 While approximately 33% of these individuals became psychotic, the screening process required a very efficient means of eliminating those not at high-risk (given the ultimate target population represented only approximately .5% of young people) (Figure). The pre-screening and screening processes in these first-generation studies were labor-intensive but could only identify approximately 5% of those individuals destined to become psychotic over the next 2 or 3 years. Thus, alternative methods to enhance sensitivity were needed to extend programming to the general population.
Second-generation pre-screening (Figure; Step 1). New pre-screening methods were identified that captured more individuals destined to become psychotic. For example, approximately 90% of this population were registered in health care organizations (eg, health maintenance organizations) and received a psychiatric diagnosis in the year prior to the onset of psychosis (true positives).8 These samples, however, contained a much higher percentage of persons not destined to become psychotic, and somehow the issue of specificity (decreasing false positives) was minimized.8,9 For example, pre-screened samples contained 20 to 50 individuals not destined to become psychotic for each one who did.26 Since screening measures could only eliminate approximately 20% of this group (Figure, Step 2, page 25), second-generation transition rates fell from 30% to 40% to 2% to 10%.27,28
Other pre-screening approaches were introduced, but they also focused on capturing more of those destined to become psychotic (sensitivity) than eliminating those who would not (specificity). For instance, Australia opened more than 100 “Headspace” community centers nationwide designed to promote engagement and self-esteem in youth experiencing anxiety; depression; stress; relationship, work, or school problems; or bullying.13 Most services were free and included mental health staff who screened for psychosis and provided a wide range of services in a destigmatized setting. These methods identified at least an additional 5% to 7% of individuals destined to become psychotic, but to our knowledge, no data have been published on whether they helped eliminate those who did not.
Continue to: Second-generation screening
Second-generation screening (Figure, Step 2). A second screening aims to retain those pre-screened individuals who will become psychotic (ie, minimizing false negatives) while further minimizing those who do not (ie, minimizing false positives). The addition of cognitive, neural (eg, structural MRI; neurophysiologic), and biochemical (eg, inflammatory immune and stress) markers to the risk calculators have produced a sensitivity close to 100%.8,9 Unfortunately, these studies downplayed specificity, which remained approximately 20%.8,9 Specificity is critical not just because of concerns about stigma (ie, labeling people as pre-psychotic when they are not) but also because of the adverse effects of antipsychotic medications and the effects on future program development (interventions are costly and labor-intensive). Also, diluting the pool with individuals not at risk makes it nearly impossible to identify effective interventions (ie, power).27,28
While some studies focused on increasing specificity (to approximately 75%), this leads to an unacceptable loss of sensitivity (from 90% to 60%),29 with 40% of pre-screened individuals who would become psychotic being eliminated from the study population. The addition of other biological markers (eg, salivary cortisol)30 and use of learning health systems may be able to enhance these numbers (initial reports of specificity = 87% and sensitivity = 85%).8,9 This is accomplished by integrating artificial and human intelligence measures of clinical (symptom and neurocognitive measures) and biological (eg, polygenetic risk scores; gray matter volume) variables.31 However, even if these results are replicated, more effective pre-screening measures will be required.
Identifying a suitable sample population for prevention program studies is clearly more complicated than for FEP studies, where one can usually identify many of those in the at-risk population by their first hospitalization for psychotic symptoms. The issues of false positives (eg, substance-induced psychosis) and negatives (eg, slow deterioration, prominent negative symptoms) are important concerns, but proportionately far less significant.
Prevention and FEP interventions
Once a study sample is constituted, 1 to 3 years of treatment interventions are initiated. Interventions for prevention programs typically include CBT directed at attenuated psychosis (eg, reframing or de-catastrophizing unusual thoughts and minimizing distress associated with unusual perceptions); case management to facilitate personal, educational, and vocational goals; and family therapy in single or multi-group formats to educate one’s support system about the risk state and to minimize adverse familial responses.14 Many programs also include supported education or employment services to promote reintegration in age-appropriate activities; group therapy focused on substance abuse and social skills training; cognitive remediation to ameliorate the cognitive dysfunction; and an array of pharmacologic interventions designed to delay or prevent transition to psychosis or to alleviate symptoms. While most interventions are similar, FEP programs have recently included peer support staff. This appears to instill hope in newly diagnosed patients, provide role models, and provide peer supporters an opportunity to use their experiences to help others and earn income.32
The breadth and depth of these services are critical because retention in the program is highly dependent on participant engagement, which in turn is highly dependent on whether the program can help individuals get what they want (eg, friends, employment, education, more autonomy, physical health). The setting and atmosphere of the treatment program and the willingness/ability of staff to meet participants in the community are also important elements.11,12 In this context, the Headspace community centers are having an impact far beyond Australia and may prove to be a particularly good model.13
Continue to: Assessing prevention and FEP interventions
Assessing prevention and FEP interventions
The second generation of studies of prevention programs has not confirmed, let alone extended, the earlier findings and meta-analyses. A 2020 report concluded CBT was still the most promising intervention; it was more effective than control treatments at 12 and 18 months, although not at 6, 24, or 48 months.33 This review included controlled, open-label, and naturalistic studies that assessed family therapy; omega-3 polyunsaturated fatty acids; integrated psychological therapy (a package of interventions that included family education, CBT, social skills training, and cognitive remediation); N-methyl-
While these disappointing findings are at least partly attributable to the methodological challenges described above and in the Figure, other factors may hinder establishing effective interventions. In contrast to FEP studies, those focused on prevention had a very ambitious agenda (eliminating psychosis) and tended to downplay more modest intermediate outcomes. These studies also tended to assess new ideas with small samples rather than pursue promising findings with larger multi-site studies focused on a group of interventions. The authors of a Cochrane review observed “There is the impression that in this whole area there is a triumph of hope over adversity. There is the repeated hope invested in another—often unique—study question and then a study of fewer than 100 participants are completed. This results in the set of comparisons reported here, all 9 of which are too underpowered to really highlight clear differences.”34 To use a baseball analogy, it seems that investigators are “swinging for the fence” when a few singles are what’s really needed.
From the outset, the goals of FEP studies were more modest, largely ignoring the task of developing consensus definitions of recovery that require following patients for up to 5 to 10 years. Instead, they use intermediate endpoints based on adapting treatments that already appeared effective in patients with chronic mental disorders.35 As a consequence, researchers examining FEP demonstrated clear, albeit limited, salutary effects using large multi-site trials and previously established outcome measures.3,10,36 For instance, the Recovery After an Initial Schizophrenia Episode-Early Treatment Program (RAISE-ETP) study was a 2-year, multi-site RCT (N = 404) funded by the National Institute of Mental Health (NIMH). The investigators reported improved indices of social function (eg, quality of life; education and work participation) and total ratings of psychopathology and depression compared with treatment as usual. Furthermore, they established that DUP predicted treatment response.35 The latter finding was underscored by improvement being limited to the 50% with <74 weeks DUP. Annual costs of the program per 1 standard deviation improvement in quality of life were approximately $1,000 for patients with <74 weeks DUP and $40,000 for those with >74 weeks DUP. Concurrent meta-analyses confirmed and extended these findings,16 showing higher remission rates; diminished relapses and hospital admissions; greater engagement in programming; greater involvement in work and school; improved quality of life; and other steps toward recovery. These studies were also able to establish a clear benefit of antipsychotic medications, particularly a high acceptance of long-acting injectable antipsychotic formulations, which promoted adherence and decreased some adverse events37; and early use of clozapine therapy, which improved remission rates and longer-term outcomes.38 Other findings underscored the need to anticipate and address new problems associated with effective antipsychotic therapy (eg, antipsychotic response correlates with weight gain, a particularly intolerable adverse event for this age group).39 Providing pre-emptive strategies such as exercise groups and nutritional education may be necessary to maintain adherence.
Limitations of FEP studies
The effect sizes in these FEP studies were small to medium on outcome measures tracking recovery and associated indicators (eg, global functioning, school/work participation, treatment engagement); the number needed to treat for each of these was >10. There is no clear evidence that recovery programs such as RAISE-ETP actually reduce longer-term disability. Most studies showed disability payments increased while clinical benefits tended to fade over time. In addition, by grouping interventions together, the studies made it difficult to identify effective vs ineffective treatments, let alone determine how best to personalize therapy for participants in future studies.
The next generation of FEP studies
While limited in scope, the results of the recent FEP studies justify a next generation of recovery interventions designed to address these shortcomings and optimize program outcomes.39 Most previous FEP studies were conducted in community mental health center settings, thus eliminating the need to transition services developed in academia into the “real world.” The next generation of NIMH studies will be primarily conducted in analogous settings under the Early Psychosis Intervention Network (EPINET).40 EPINET’s study design echoes that responsible for the stepwise successes in the late 20th century that produced cures for the deadliest childhood cancer, acute lymphoblastic leukemia (ALL). This disease was successfully treated by modifying diverse evidence-based practices without relying on pharmacologic or other major treatment breakthroughs. Despite this, the effort yielded successful personalized interventions that were not obtainable for other severe childhood conditions.40 EPINET hopes to automate much of these stepwise advances with a learning health system. This program relies on data routinely collected in clinical practice to drive the process of scientific discovery. Specifically, it determines the relationships between clinical features, biologic measures, treatment characteristics, and symptomatic and functional outcomes. EPINET aims to accelerate our understanding of biomarkers of psychosis risk and onset, as well as factors associated with recovery and cure. Dashboard displays of outcomes will allow for real-time comparisons within and across early intervention clinics. This in turn identifies performance gaps and drives continuous quality improvement.
Continue to: Barriers to optimizing program efficacy for both models
Barriers to optimizing program efficacy for both models
Unfortunately, there are stark differences between ALL and severe mental disorders that potentially jeopardize the achievement of these aims, despite the advances in data analytic abilities that drive the learning health system. Specifically, the heterogeneity of psychotic illnesses and the absence of reliable prognostic and modifiable risk markers (responsible for failed efforts to enhance treatment of serious mental illness over the last half century1,2,41) are unlikely to be resolved by a learning health system. These measures are vital to determine whether specific interventions are effective, particularly given the absence of a randomized control group in the EPINET/learning health system design. Fortunately, however, the National Institutes for Health has recently initiated the Accelerating Medicines Partnership–Schizophrenia (AMP-SCZ). This approach seeks “promising biological markers that can help identify those at risk of developing schizophrenia as early as possible, track the progression of symptoms and other outcomes and ultimately define targets for treatment development.”42 The Box1,4,9,10,36,41,43-45 describes some of the challenges involved in identifying biomarkers of severe mental illness.
Box
Biomarkers and modifiable risk factors4,9,10,41,43 are at the core of personalized medicine and its ultimate objective (ie, theragnostics). This is the ability to identify the correct intervention for a disorder based on a biomarker of the illness.10,36 The inability to identify biomarkers of severe mental illness is multifactorial but in part may be attributable to “looking in all the wrong places.”41 By focusing on neural processes that generate psychiatric symptomatology, investigators are assuming they can bridge the “mind gap”1 and specifically distinguish between pathological, compensatory, or collateral measures of poorly characterized limbic neural functions.41
It may be more productive to identify a pathological process within the limbic system that produces a medical condition as well as the mental disorder. If one can isolate the pathologic limbic circuit activity responsible for a medical condition, one may be able to reproduce this in animal models and determine whether analogous processes contribute to the core features of the mental illness. Characterization of the aberrant neural circuit in animal models also could yield targets for future therapies. For example, episodic water intoxication in a discrete subset of patients with schizophrenia44 appears to arise from a stress diathesis produced by anterior hippocampal pathology that disrupts regulation of antidiuretic hormone, oxytocin, and hypothalamic-pituitary-adrenal axis secretion. These patients also exhibit psychogenic polydipsia that may be a consequence of the same hippocampal pathology that disrupts ventral striatal and lateral hypothalamic circuits. These circuits, in turn, also modulate motivated behaviors and cognitive processes likely relevant to psychosis.45
A strength-based approach
The absence of sufficiently powered RCTs for prevention studies and the reliance on intermediate outcomes for FEP studies leaves unanswered whether such programs can effectively prevent chronic psychosis at a cost society is willing to pay. Still, substantial evidence indicates that outreach, long-acting injectable antipsychotics, early consideration of clozapine, family therapy, CBT for psychosis/attenuated psychosis, and services focused on competitive employment can preserve social and occupational functioning.16,34 Until these broader questions are more definitively addressed, it seems reasonable to apply what we have learned (Table11,12,35,37-39,46).
Simply avoiding the most divisive aspects of the medical model that inadvertently promote stigma and undercut self-confidence may help maintain patients’ willingness to learn how best to apply their strengths and manage their limitations.11 The progression to enduring psychotic features (eg, fixed delusions) may reflect ongoing social isolation and alienation. A strength-based approach seeks first to establish common goals (eg, school, work, friends, family support, housing, leaving home) and then works to empower the patient to successfully reach those goals.35 This typically involves giving them the opportunity to fail, avoiding criticism when they do, and focusing on these experiences as learning opportunities from which success can ultimately result.
It is difficult to offer all these services in a typical private practice setting. Instead, it may make more sense to use one of the hundreds of early intervention services programs in the United States.46 If a psychiatric clinician is dedicated to working with this population, it may also be possible to establish ongoing relationships with primary care physicians, family and CBT therapists, family support services (eg, National Alliance on Mental Illness), caseworkers and employment counselors. In essence, a psychiatrist may be able re-create a multidisciplinary effort by taking advantage of the expertise of these various professionals. The challenge is to create a consistent message for patients and families in the absence of regular meetings with the clinical team, although the recent reliance on and improved sophistication of virtual meetings may help. Psychiatrists often play a critical role even when the patient is not prescribed medication, partly because they are most comfortable handling the risks and may have the most comprehensive understanding of the issues at play. When medications are appropriate and patients with FEP are willing to take them, early consideration of long-acting injectable antipsychotics and clozapine may provide better stabilization and diminish the risk of earlier and more frequent relapses.
Bottom Line
Early interventions for psychosis include the prevention model and the first-episode recovery model. It is difficult to assess, compare, and optimize the effectiveness of such programs. Current evidence supports a ‘strength-based’ approach focused on finding common ground between patients, their support system, and the treatment team.
Related Resources
- Early Assessment and Support Alliance. National Early Psychosis Directory. https://easacommunity.org/nationaldirectory.php
- Kane JM, Robinson DG, Schooler NR, et al. Comprehensive versus usual community care for first-episode psychosis: 2-year outcomes from the NIMH RAISE Early Treatment Program. Am J Psychiatry. 2016 ;173(4):362-372
Drug Brand Name
Clozapine • Clozaril
Neuroscience research over the past half century has failed to significantly advance the treatment of severe mental illness.1,2 Hence, evidence that a longer duration of untreated psychosis (DUP) aggravates—and early intervention with medication and social supports ameliorates—the long-term adverse consequences of psychotic disorders generated a great deal of interest.3,4 This knowledge led to the development of diverse early intervention services worldwide aimed at this putative “critical window.” It raised the possibility that appropriate interventions could prevent the long-term disability that makes chronic psychosis one of the most debilitating disorders.5,6 However, even beyond the varied cultural and economic confounds, it is difficult to assess, compare, and optimize program effectiveness.7 Obstacles include paucity of sufficiently powered, well-designed randomized controlled trials (RCTs), the absence of diagnostic biomarkers or other prognostic indicators to better account for the inherent heterogeneity in the population and associated outcomes, and the absence of modifiable risk factors that can guide interventions and provide intermediate outcomes.4,8-10
To better appreciate these issues, it is important to distinguish whether a program is designed to prevent psychosis, or to mitigate the effects of psychosis. Two models include the:
- Prevention model, which focuses on young individuals who are not yet overtly psychotic but at high risk
- First-episode recovery model, which focuses on those who have experienced a first episode of psychosis (FEP) but have not yet developed a chronic disorder.
Both models share long-term goals and are hampered by many of the same issues summarized above. They both deviate markedly from the standard medical model by including psychosocial services designed to promote restoration of a self-defined trajectory to greater independence.11-14 The 2 differ, however, in the challenges they must overcome to produce their sample populations and establish effective interventions.10,15,16
In this article, we provide a succinct overview of these issues and a set of recommendations based on a “strength-based” approach. This approach focuses on finding common ground between patients, their support system, and the treatment team in the service of empowering patients to resume responsibility for transition to adulthood.
The prevention model
While most prevention initiatives in medicine rely on the growing ability to target specific pathophysiologic pathways,3 preventing psychosis relies on clinical evidence showing that DUP and early interventions predict a better course of severe mental illness.17 In contrast, initiatives such as normalizing neonatal neuronal pathways are more consistent with the strategy utilized in other fields but have yet to yield a pathophysiologic target for psychosis.3,18
Initial efforts to identify ‘at-risk’ individuals
The prevention model of psychosis is based on the ability to identify young individuals at high risk for developing a psychotic disorder (Figure). The first screening measures were focused on prodromal psychosis (eg, significant loss of function, family history, and “intermittent” and “attenuated” psychotic symptoms). When applied to referred (ie, pre-screened) samples, 30% to 40% of this group who met criteria transitioned to psychosis over the next 1 to 3 years despite antidepressant and psychosocial interventions.19 Comprising 8 academic medical centers, the North American Prodrome Longitudinal Study (NAPLS) produced similar results using the Structured Interview for Prodromal Syndromes (SIPS).17 Thus, 30% to 50% of pre-screened individuals referred by school counselors and mental health professionals met SIPS criteria, and 35% of these individuals transitioned to psychosis over 30 months. The validity of this measure was further supported by the fact that higher baseline levels of unusual thought content, suspicion/paranoia, social impairment, and substance abuse successfully distinguished approximately 80% of those who transitioned to psychosis. The results of this first generation of screening studies were exciting because they seemed to demonstrate that highly concentrated samples of young persons at high risk of developing psychosis could be identified, and that fine-tuning the screening criteria could produce even more enriched samples (ie, positive predictive power).
Initial interventions produced promising results
The development of effective screening measures led to reports of effective treatment interventions. These were largely applied in a clinical staging model that restricted antipsychotic medications to those who failed to improve after receiving potentially “less toxic” interventions (eg, omega-3 polyunsaturated fatty acids and other antioxidants; psychotherapy; cognitive-behavioral therapy [CBT]; family therapy).5 While study designs were typically quasi-experimental, the interventions appeared to dramatically diminish the transition to psychosis (ie, approximately 50%).
Continue to: The first generation...
The first generation of RCTs appeared to confirm these results, although sample sizes were small, and most study designs assessed only a single intervention. Initial meta-analyses of these data reported that both CBT and antipsychotics appeared to prevent approximately one-half of individuals from becoming psychotic at 12 months, and more than one-third at 2 to 4 years, compared with treatment as usual.20
While some researchers challenged the validity of these findings,21-23 the results generated tremendous international enthusiasm and calls for widespread implementation.6 The number of early intervention services (EIS) centers increased dramatically worldwide, and in 2014 the National Institute for Health and Care Excellence released standards for interventions to prevent transition to psychosis.24 These included close monitoring, CBT and family interventions, and avoiding antipsychotics when possible.24
Focusing on sensitivity over specificity
The first generation of studies generated by the prevention model relied on outreach programs or referrals, which produced small samples of carefully selected, pre-screened individuals (Figure, Pre-screened) who were then screened again to establish the high-risk sample.25 While approximately 33% of these individuals became psychotic, the screening process required a very efficient means of eliminating those not at high-risk (given the ultimate target population represented only approximately .5% of young people) (Figure). The pre-screening and screening processes in these first-generation studies were labor-intensive but could only identify approximately 5% of those individuals destined to become psychotic over the next 2 or 3 years. Thus, alternative methods to enhance sensitivity were needed to extend programming to the general population.
Second-generation pre-screening (Figure; Step 1). New pre-screening methods were identified that captured more individuals destined to become psychotic. For example, approximately 90% of this population were registered in health care organizations (eg, health maintenance organizations) and received a psychiatric diagnosis in the year prior to the onset of psychosis (true positives).8 These samples, however, contained a much higher percentage of persons not destined to become psychotic, and somehow the issue of specificity (decreasing false positives) was minimized.8,9 For example, pre-screened samples contained 20 to 50 individuals not destined to become psychotic for each one who did.26 Since screening measures could only eliminate approximately 20% of this group (Figure, Step 2, page 25), second-generation transition rates fell from 30% to 40% to 2% to 10%.27,28
Other pre-screening approaches were introduced, but they also focused on capturing more of those destined to become psychotic (sensitivity) than eliminating those who would not (specificity). For instance, Australia opened more than 100 “Headspace” community centers nationwide designed to promote engagement and self-esteem in youth experiencing anxiety; depression; stress; relationship, work, or school problems; or bullying.13 Most services were free and included mental health staff who screened for psychosis and provided a wide range of services in a destigmatized setting. These methods identified at least an additional 5% to 7% of individuals destined to become psychotic, but to our knowledge, no data have been published on whether they helped eliminate those who did not.
Continue to: Second-generation screening
Second-generation screening (Figure, Step 2). A second screening aims to retain those pre-screened individuals who will become psychotic (ie, minimizing false negatives) while further minimizing those who do not (ie, minimizing false positives). The addition of cognitive, neural (eg, structural MRI; neurophysiologic), and biochemical (eg, inflammatory immune and stress) markers to the risk calculators have produced a sensitivity close to 100%.8,9 Unfortunately, these studies downplayed specificity, which remained approximately 20%.8,9 Specificity is critical not just because of concerns about stigma (ie, labeling people as pre-psychotic when they are not) but also because of the adverse effects of antipsychotic medications and the effects on future program development (interventions are costly and labor-intensive). Also, diluting the pool with individuals not at risk makes it nearly impossible to identify effective interventions (ie, power).27,28
While some studies focused on increasing specificity (to approximately 75%), this leads to an unacceptable loss of sensitivity (from 90% to 60%),29 with 40% of pre-screened individuals who would become psychotic being eliminated from the study population. The addition of other biological markers (eg, salivary cortisol)30 and use of learning health systems may be able to enhance these numbers (initial reports of specificity = 87% and sensitivity = 85%).8,9 This is accomplished by integrating artificial and human intelligence measures of clinical (symptom and neurocognitive measures) and biological (eg, polygenetic risk scores; gray matter volume) variables.31 However, even if these results are replicated, more effective pre-screening measures will be required.
Identifying a suitable sample population for prevention program studies is clearly more complicated than for FEP studies, where one can usually identify many of those in the at-risk population by their first hospitalization for psychotic symptoms. The issues of false positives (eg, substance-induced psychosis) and negatives (eg, slow deterioration, prominent negative symptoms) are important concerns, but proportionately far less significant.
Prevention and FEP interventions
Once a study sample is constituted, 1 to 3 years of treatment interventions are initiated. Interventions for prevention programs typically include CBT directed at attenuated psychosis (eg, reframing or de-catastrophizing unusual thoughts and minimizing distress associated with unusual perceptions); case management to facilitate personal, educational, and vocational goals; and family therapy in single or multi-group formats to educate one’s support system about the risk state and to minimize adverse familial responses.14 Many programs also include supported education or employment services to promote reintegration in age-appropriate activities; group therapy focused on substance abuse and social skills training; cognitive remediation to ameliorate the cognitive dysfunction; and an array of pharmacologic interventions designed to delay or prevent transition to psychosis or to alleviate symptoms. While most interventions are similar, FEP programs have recently included peer support staff. This appears to instill hope in newly diagnosed patients, provide role models, and provide peer supporters an opportunity to use their experiences to help others and earn income.32
The breadth and depth of these services are critical because retention in the program is highly dependent on participant engagement, which in turn is highly dependent on whether the program can help individuals get what they want (eg, friends, employment, education, more autonomy, physical health). The setting and atmosphere of the treatment program and the willingness/ability of staff to meet participants in the community are also important elements.11,12 In this context, the Headspace community centers are having an impact far beyond Australia and may prove to be a particularly good model.13
Continue to: Assessing prevention and FEP interventions
Assessing prevention and FEP interventions
The second generation of studies of prevention programs has not confirmed, let alone extended, the earlier findings and meta-analyses. A 2020 report concluded CBT was still the most promising intervention; it was more effective than control treatments at 12 and 18 months, although not at 6, 24, or 48 months.33 This review included controlled, open-label, and naturalistic studies that assessed family therapy; omega-3 polyunsaturated fatty acids; integrated psychological therapy (a package of interventions that included family education, CBT, social skills training, and cognitive remediation); N-methyl-
While these disappointing findings are at least partly attributable to the methodological challenges described above and in the Figure, other factors may hinder establishing effective interventions. In contrast to FEP studies, those focused on prevention had a very ambitious agenda (eliminating psychosis) and tended to downplay more modest intermediate outcomes. These studies also tended to assess new ideas with small samples rather than pursue promising findings with larger multi-site studies focused on a group of interventions. The authors of a Cochrane review observed “There is the impression that in this whole area there is a triumph of hope over adversity. There is the repeated hope invested in another—often unique—study question and then a study of fewer than 100 participants are completed. This results in the set of comparisons reported here, all 9 of which are too underpowered to really highlight clear differences.”34 To use a baseball analogy, it seems that investigators are “swinging for the fence” when a few singles are what’s really needed.
From the outset, the goals of FEP studies were more modest, largely ignoring the task of developing consensus definitions of recovery that require following patients for up to 5 to 10 years. Instead, they use intermediate endpoints based on adapting treatments that already appeared effective in patients with chronic mental disorders.35 As a consequence, researchers examining FEP demonstrated clear, albeit limited, salutary effects using large multi-site trials and previously established outcome measures.3,10,36 For instance, the Recovery After an Initial Schizophrenia Episode-Early Treatment Program (RAISE-ETP) study was a 2-year, multi-site RCT (N = 404) funded by the National Institute of Mental Health (NIMH). The investigators reported improved indices of social function (eg, quality of life; education and work participation) and total ratings of psychopathology and depression compared with treatment as usual. Furthermore, they established that DUP predicted treatment response.35 The latter finding was underscored by improvement being limited to the 50% with <74 weeks DUP. Annual costs of the program per 1 standard deviation improvement in quality of life were approximately $1,000 for patients with <74 weeks DUP and $40,000 for those with >74 weeks DUP. Concurrent meta-analyses confirmed and extended these findings,16 showing higher remission rates; diminished relapses and hospital admissions; greater engagement in programming; greater involvement in work and school; improved quality of life; and other steps toward recovery. These studies were also able to establish a clear benefit of antipsychotic medications, particularly a high acceptance of long-acting injectable antipsychotic formulations, which promoted adherence and decreased some adverse events37; and early use of clozapine therapy, which improved remission rates and longer-term outcomes.38 Other findings underscored the need to anticipate and address new problems associated with effective antipsychotic therapy (eg, antipsychotic response correlates with weight gain, a particularly intolerable adverse event for this age group).39 Providing pre-emptive strategies such as exercise groups and nutritional education may be necessary to maintain adherence.
Limitations of FEP studies
The effect sizes in these FEP studies were small to medium on outcome measures tracking recovery and associated indicators (eg, global functioning, school/work participation, treatment engagement); the number needed to treat for each of these was >10. There is no clear evidence that recovery programs such as RAISE-ETP actually reduce longer-term disability. Most studies showed disability payments increased while clinical benefits tended to fade over time. In addition, by grouping interventions together, the studies made it difficult to identify effective vs ineffective treatments, let alone determine how best to personalize therapy for participants in future studies.
The next generation of FEP studies
While limited in scope, the results of the recent FEP studies justify a next generation of recovery interventions designed to address these shortcomings and optimize program outcomes.39 Most previous FEP studies were conducted in community mental health center settings, thus eliminating the need to transition services developed in academia into the “real world.” The next generation of NIMH studies will be primarily conducted in analogous settings under the Early Psychosis Intervention Network (EPINET).40 EPINET’s study design echoes that responsible for the stepwise successes in the late 20th century that produced cures for the deadliest childhood cancer, acute lymphoblastic leukemia (ALL). This disease was successfully treated by modifying diverse evidence-based practices without relying on pharmacologic or other major treatment breakthroughs. Despite this, the effort yielded successful personalized interventions that were not obtainable for other severe childhood conditions.40 EPINET hopes to automate much of these stepwise advances with a learning health system. This program relies on data routinely collected in clinical practice to drive the process of scientific discovery. Specifically, it determines the relationships between clinical features, biologic measures, treatment characteristics, and symptomatic and functional outcomes. EPINET aims to accelerate our understanding of biomarkers of psychosis risk and onset, as well as factors associated with recovery and cure. Dashboard displays of outcomes will allow for real-time comparisons within and across early intervention clinics. This in turn identifies performance gaps and drives continuous quality improvement.
Continue to: Barriers to optimizing program efficacy for both models
Barriers to optimizing program efficacy for both models
Unfortunately, there are stark differences between ALL and severe mental disorders that potentially jeopardize the achievement of these aims, despite the advances in data analytic abilities that drive the learning health system. Specifically, the heterogeneity of psychotic illnesses and the absence of reliable prognostic and modifiable risk markers (responsible for failed efforts to enhance treatment of serious mental illness over the last half century1,2,41) are unlikely to be resolved by a learning health system. These measures are vital to determine whether specific interventions are effective, particularly given the absence of a randomized control group in the EPINET/learning health system design. Fortunately, however, the National Institutes for Health has recently initiated the Accelerating Medicines Partnership–Schizophrenia (AMP-SCZ). This approach seeks “promising biological markers that can help identify those at risk of developing schizophrenia as early as possible, track the progression of symptoms and other outcomes and ultimately define targets for treatment development.”42 The Box1,4,9,10,36,41,43-45 describes some of the challenges involved in identifying biomarkers of severe mental illness.
Box
Biomarkers and modifiable risk factors4,9,10,41,43 are at the core of personalized medicine and its ultimate objective (ie, theragnostics). This is the ability to identify the correct intervention for a disorder based on a biomarker of the illness.10,36 The inability to identify biomarkers of severe mental illness is multifactorial but in part may be attributable to “looking in all the wrong places.”41 By focusing on neural processes that generate psychiatric symptomatology, investigators are assuming they can bridge the “mind gap”1 and specifically distinguish between pathological, compensatory, or collateral measures of poorly characterized limbic neural functions.41
It may be more productive to identify a pathological process within the limbic system that produces a medical condition as well as the mental disorder. If one can isolate the pathologic limbic circuit activity responsible for a medical condition, one may be able to reproduce this in animal models and determine whether analogous processes contribute to the core features of the mental illness. Characterization of the aberrant neural circuit in animal models also could yield targets for future therapies. For example, episodic water intoxication in a discrete subset of patients with schizophrenia44 appears to arise from a stress diathesis produced by anterior hippocampal pathology that disrupts regulation of antidiuretic hormone, oxytocin, and hypothalamic-pituitary-adrenal axis secretion. These patients also exhibit psychogenic polydipsia that may be a consequence of the same hippocampal pathology that disrupts ventral striatal and lateral hypothalamic circuits. These circuits, in turn, also modulate motivated behaviors and cognitive processes likely relevant to psychosis.45
A strength-based approach
The absence of sufficiently powered RCTs for prevention studies and the reliance on intermediate outcomes for FEP studies leaves unanswered whether such programs can effectively prevent chronic psychosis at a cost society is willing to pay. Still, substantial evidence indicates that outreach, long-acting injectable antipsychotics, early consideration of clozapine, family therapy, CBT for psychosis/attenuated psychosis, and services focused on competitive employment can preserve social and occupational functioning.16,34 Until these broader questions are more definitively addressed, it seems reasonable to apply what we have learned (Table11,12,35,37-39,46).
Simply avoiding the most divisive aspects of the medical model that inadvertently promote stigma and undercut self-confidence may help maintain patients’ willingness to learn how best to apply their strengths and manage their limitations.11 The progression to enduring psychotic features (eg, fixed delusions) may reflect ongoing social isolation and alienation. A strength-based approach seeks first to establish common goals (eg, school, work, friends, family support, housing, leaving home) and then works to empower the patient to successfully reach those goals.35 This typically involves giving them the opportunity to fail, avoiding criticism when they do, and focusing on these experiences as learning opportunities from which success can ultimately result.
It is difficult to offer all these services in a typical private practice setting. Instead, it may make more sense to use one of the hundreds of early intervention services programs in the United States.46 If a psychiatric clinician is dedicated to working with this population, it may also be possible to establish ongoing relationships with primary care physicians, family and CBT therapists, family support services (eg, National Alliance on Mental Illness), caseworkers and employment counselors. In essence, a psychiatrist may be able re-create a multidisciplinary effort by taking advantage of the expertise of these various professionals. The challenge is to create a consistent message for patients and families in the absence of regular meetings with the clinical team, although the recent reliance on and improved sophistication of virtual meetings may help. Psychiatrists often play a critical role even when the patient is not prescribed medication, partly because they are most comfortable handling the risks and may have the most comprehensive understanding of the issues at play. When medications are appropriate and patients with FEP are willing to take them, early consideration of long-acting injectable antipsychotics and clozapine may provide better stabilization and diminish the risk of earlier and more frequent relapses.
Bottom Line
Early interventions for psychosis include the prevention model and the first-episode recovery model. It is difficult to assess, compare, and optimize the effectiveness of such programs. Current evidence supports a ‘strength-based’ approach focused on finding common ground between patients, their support system, and the treatment team.
Related Resources
- Early Assessment and Support Alliance. National Early Psychosis Directory. https://easacommunity.org/nationaldirectory.php
- Kane JM, Robinson DG, Schooler NR, et al. Comprehensive versus usual community care for first-episode psychosis: 2-year outcomes from the NIMH RAISE Early Treatment Program. Am J Psychiatry. 2016 ;173(4):362-372
Drug Brand Name
Clozapine • Clozaril
1. Hyman SE. Revolution stalled. Sci Transl Med. 2012;4(155):155cm11. doi: 10.1126/scitranslmed.3003142
2. Harrington A. Mind fixers: psychiatry’s troubled search for the biology of mental illness. W.W. Norton & Company; 2019.
3. Millan MJ, Andrieux A, Bartzokis G, et al. Altering the course of schizophrenia: progress and perspectives. Nat Rev Drug Discov. 2016;15(7):485-515.
4. Lieberman JA, Small SA, Girgis RR. Early detection and preventive intervention in schizophrenia: from fantasy to reality. Am J Psychiatry. 2019;176(10):794-810.
5. McGorry PD, Nelson B, Nordentoft M, et al. Intervention in individuals at ultra-high risk for psychosis: a review and future directions. J Clin Psychiatry. 2009;70(9):1206-1212.
6. Csillag C, Nordentoft M, Mizuno M, et al. Early intervention in psychosis: From clinical intervention to health system implementation. Early Interv Psychiatry. 2018;12(4):757-764.
7. McGorry PD, Ratheesh A, O’Donoghue B. Early intervention—an implementation challenge for 21st century mental health care. JAMA Psychiatry. 2018;75(6):545-546.
8. Rosenheck R. Toward dissemination of secondary prevention for psychosis. Am J Psychiatry. 2018;175(5):393-394.
9. Fusar-Poli P, Salazar de Pablo G, Correll CU, et al. Prevention of psychosis: advances in detection, prognosis, and intervention. JAMA Psychiatry. 2020;77(7):755-765.
10. Oliver D, Reilly TJ, Baccaredda Boy O, et al. What causes the onset of psychosis in individuals at clinical high risk? A meta-analysis of risk and protective factors. Schizophr Bull. 2020;46(1):110-120.
11. Tindall R, Simmons M, Allott K, et al. Disengagement processes within an early intervention service for first-episode psychosis: a longitudinal, qualitative, multi-perspective study. Front Psychiatry. 2020;11:565-565.
12. Dixon LB, Holoshitz Y, Nossel I. Treatment engagement of individuals experiencing mental illness: review and update. World Psychiatry. 2016;15(1):13-20.
13. Rickwood D, Paraskakis M, Quin D, et al. Australia’s innovation in youth mental health care: The headspace centre model. Early Interv Psychiatry. 2019;13(1):159-166.
14. Woodberry KA, Shapiro DI, Bryant C, et al. Progress and future directions in research on the psychosis prodrome: a review for clinicians. Harv Rev Psychiatry. 2016;24(2):87-103.
15. Gupta T, Mittal VA. Advances in clinical staging, early intervention, and the prevention of psychosis. F1000Res. 2019;8:F1000 Faculty Rev-2027. doi: 10.12688/f1000research.20346.1
16. Correll CU, Galling B, Pawar A, et al. Comparison of early intervention services vs treatment as usual for early-phase psychosis: a systematic review, meta-analysis, and meta-regression. JAMA Psychiatry. 2018;75(6):555-565.
17. Cannon TD, Cadenhead K, Cornblatt B, et al. Prediction of psychosis in youth at high clinical risk: a multisite longitudinal study in North America. Arch Gen Psychiatry. 2008;65(1):28-37.
18. Sommer IE, Bearden CE, van Dellen E, et al. Early interventions in risk groups for schizophrenia: what are we waiting for? NPJ Schizophr. 2016;2(1):16003-16003.
19. McGorry PD, Nelson B. Clinical high risk for psychosis—not seeing the trees for the wood. JAMA Psychiatry. 2020;77(7):559-560.
20. van der Gaag M, Smit F, Bechdolf A, et al. Preventing a first episode of psychosis: meta-analysis of randomized controlled prevention trials of 12 month and longer-term follow-ups. Schizophr Res. 2013;149(1):56-62.
21. Marshall M, Rathbone J. Early intervention for psychosis. Cochrane Database Syst Rev. 2011;(6):CD004718. doi: 10.1002/14651858.CD004718.pub3
22. Heinssen RK, Insel TR. Preventing the onset of psychosis: not quite there yet. Schizophr Bull. 2015;41(1):28-29.
23. Amos AJ. Evidence that treatment prevents transition to psychosis in ultra-high-risk patients remains questionable. Schizophr Res. 2014;153(1):240.
24. National Institute for Health and Care Excellence. Psychosis and schizophrenia in adults: prevention and management. Clinical guideline [CG178]. 1.3.7 How to deliver psychological interventions. Published February 12, 2014. Updated March 1, 2014. Accessed August 30, 2021. https://www.nice.org.uk/guidance/cg178/chapter/recommendations#how-to-deliver-psychological-interventions
25. Fusar-Poli P, Werbeloff N, Rutigliano G, et al. Transdiagnostic risk calculator for the automatic detection of individuals at risk and the prediction of psychosis: second replication in an independent National Health Service Trust. Schizophr Bull. 2019;45(3):562-570.
26. Fusar-Poli P, Oliver D, Spada G, et al. The case for improved transdiagnostic detection of first-episode psychosis: electronic health record cohort study. Schizophr Res. 2021;228:547-554.
27. Fusar-Poli P. Negative psychosis prevention trials. JAMA Psychiatry. 2017;74(6):651.
28. Cuijpers P, Smit F, Furukawa TA. Most at‐risk individuals will not develop a mental disorder: the limited predictive strength of risk factors. World Psychiatry. 2021;20(2):224-225.
29. Carrión RE, Cornblatt BA, Burton CZ, et al. Personalized prediction of psychosis: external validation of the NAPLS-2 psychosis risk calculator with the EDIPPP Project. Am J Psychiatry. 2016;173(10):989-996.
30. Worthington MA, Walker EF, Addington J, et al. Incorporating cortisol into the NAPLS2 individualized risk calculator for prediction of psychosis. Schizophr Res. 2021;227:95-100.
31. Koutsouleris N, Dwyer DB, Degenhardt F, et al. Multimodal machine learning workflows for prediction of psychosis in patients with clinical high-risk syndromes and recent-onset depression. JAMA Psychiatry. 2021;78(2):195-209.
32. Simmons MB, Grace D, Fava NJ, et al. The experiences of youth mental health peer workers over time: a qualitative study with longitudinal analysis. Community Ment Health J. 2020;56(5):906-914.
33. Devoe DJ, Farris MS, Townes P, et al. Interventions and transition in youth at risk of psychosis: a systematic review and meta-analyses. J Clin Psychiatry. 2020;81(3):17r12053. doi: 10.4088/JCP.17r12053
34. Bosnjak Kuharic D, Kekin I, Hew J, et al. Interventions for prodromal stage of psychosis. Cochrane Database Syst Rev. 2019;2019(11):CD012236
35. Dixon LB, Goldman HH, Srihari VH, et al. Transforming the treatment of schizophrenia in the United States: The RAISE Initiative. Annu Rev Clin Psychol. 2018;14:237-258.
36. Friedman-Yakoobian MS, Parrish EM, Eack SM, et al. Neurocognitive and social cognitive training for youth at clinical high risk (CHR) for psychosis: a randomized controlled feasibility trial. Schizophr Res. 2020;S0920-9964(20)30461-8. doi: 10.1016/j.schres.2020.09.005
37. Kane JM, Schooler NR, Marcy P, et al. Effect of long-acting injectable antipsychotics vs usual care on time to first hospitalization in early-phase schizophrenia: a randomized clinical trial. JAMA Psychiatry. 2020;77(12):1217-1224.
38. Morrison AP, Pyle M, Maughan D, et al. Antipsychotic medication versus psychological intervention versus a combination of both in adolescents with first-episode psychosis (MAPS): a multicentre, three-arm, randomised controlled pilot and feasibility study. Lancet Psychiatry. 2020;7(9):788-800.
39. Chen YQ, Li XR, Zhang L, et al. Therapeutic response is associated with antipsychotic-induced weight gain in drug-naive first-episode patients with schizophrenia: an 8-week prospective study. J Clin Psychiatry. 2021;82(3):20m13469. doi: 10.4088/JCP.20m13469
40. Insel TR. RAISE-ing our expectations for first-episode psychosis. Am J Psychiatry. 2016;173(4):311-312.
41. Tandon R, Goldman M. Overview of neurobiology. In: Janicak PG, Marder SR, Tandon R, et al, eds. Schizophrenia: recent advances in diagnosis and treatment. Springer; 2014:27-33.
42. National Institutes of Health. Accelerating Medicines Partnership. Schizophrenia. Accessed August 30, 2021. https://www.nih.gov/research-training/accelerating-medicines-partnership-amp/schizophrenia
43. Guloksuz S, van Os J. The slow death of the concept of schizophrenia and the painful birth of the psychosis spectrum. Psychol Med. 2018;48(2):229-244.
44. Christ-Crain M, Bichet DG, Fenske WK, et al. Diabetes insipidus. Nat Rev Dis Primers. 2019;5(1):54.
45. Ahmadi L, Goldman MB. Primary polydipsia: update. Best Pract Res Clin Endocrinol Metab. 2020;34(5):101469. doi: 10.1016/j.beem.2020.101469
46. Early Assessment and Support Alliance. National Early Psychosis Directory. Accessed August 30, 2021. https://easacommunity.org/national-directory.php
1. Hyman SE. Revolution stalled. Sci Transl Med. 2012;4(155):155cm11. doi: 10.1126/scitranslmed.3003142
2. Harrington A. Mind fixers: psychiatry’s troubled search for the biology of mental illness. W.W. Norton & Company; 2019.
3. Millan MJ, Andrieux A, Bartzokis G, et al. Altering the course of schizophrenia: progress and perspectives. Nat Rev Drug Discov. 2016;15(7):485-515.
4. Lieberman JA, Small SA, Girgis RR. Early detection and preventive intervention in schizophrenia: from fantasy to reality. Am J Psychiatry. 2019;176(10):794-810.
5. McGorry PD, Nelson B, Nordentoft M, et al. Intervention in individuals at ultra-high risk for psychosis: a review and future directions. J Clin Psychiatry. 2009;70(9):1206-1212.
6. Csillag C, Nordentoft M, Mizuno M, et al. Early intervention in psychosis: From clinical intervention to health system implementation. Early Interv Psychiatry. 2018;12(4):757-764.
7. McGorry PD, Ratheesh A, O’Donoghue B. Early intervention—an implementation challenge for 21st century mental health care. JAMA Psychiatry. 2018;75(6):545-546.
8. Rosenheck R. Toward dissemination of secondary prevention for psychosis. Am J Psychiatry. 2018;175(5):393-394.
9. Fusar-Poli P, Salazar de Pablo G, Correll CU, et al. Prevention of psychosis: advances in detection, prognosis, and intervention. JAMA Psychiatry. 2020;77(7):755-765.
10. Oliver D, Reilly TJ, Baccaredda Boy O, et al. What causes the onset of psychosis in individuals at clinical high risk? A meta-analysis of risk and protective factors. Schizophr Bull. 2020;46(1):110-120.
11. Tindall R, Simmons M, Allott K, et al. Disengagement processes within an early intervention service for first-episode psychosis: a longitudinal, qualitative, multi-perspective study. Front Psychiatry. 2020;11:565-565.
12. Dixon LB, Holoshitz Y, Nossel I. Treatment engagement of individuals experiencing mental illness: review and update. World Psychiatry. 2016;15(1):13-20.
13. Rickwood D, Paraskakis M, Quin D, et al. Australia’s innovation in youth mental health care: The headspace centre model. Early Interv Psychiatry. 2019;13(1):159-166.
14. Woodberry KA, Shapiro DI, Bryant C, et al. Progress and future directions in research on the psychosis prodrome: a review for clinicians. Harv Rev Psychiatry. 2016;24(2):87-103.
15. Gupta T, Mittal VA. Advances in clinical staging, early intervention, and the prevention of psychosis. F1000Res. 2019;8:F1000 Faculty Rev-2027. doi: 10.12688/f1000research.20346.1
16. Correll CU, Galling B, Pawar A, et al. Comparison of early intervention services vs treatment as usual for early-phase psychosis: a systematic review, meta-analysis, and meta-regression. JAMA Psychiatry. 2018;75(6):555-565.
17. Cannon TD, Cadenhead K, Cornblatt B, et al. Prediction of psychosis in youth at high clinical risk: a multisite longitudinal study in North America. Arch Gen Psychiatry. 2008;65(1):28-37.
18. Sommer IE, Bearden CE, van Dellen E, et al. Early interventions in risk groups for schizophrenia: what are we waiting for? NPJ Schizophr. 2016;2(1):16003-16003.
19. McGorry PD, Nelson B. Clinical high risk for psychosis—not seeing the trees for the wood. JAMA Psychiatry. 2020;77(7):559-560.
20. van der Gaag M, Smit F, Bechdolf A, et al. Preventing a first episode of psychosis: meta-analysis of randomized controlled prevention trials of 12 month and longer-term follow-ups. Schizophr Res. 2013;149(1):56-62.
21. Marshall M, Rathbone J. Early intervention for psychosis. Cochrane Database Syst Rev. 2011;(6):CD004718. doi: 10.1002/14651858.CD004718.pub3
22. Heinssen RK, Insel TR. Preventing the onset of psychosis: not quite there yet. Schizophr Bull. 2015;41(1):28-29.
23. Amos AJ. Evidence that treatment prevents transition to psychosis in ultra-high-risk patients remains questionable. Schizophr Res. 2014;153(1):240.
24. National Institute for Health and Care Excellence. Psychosis and schizophrenia in adults: prevention and management. Clinical guideline [CG178]. 1.3.7 How to deliver psychological interventions. Published February 12, 2014. Updated March 1, 2014. Accessed August 30, 2021. https://www.nice.org.uk/guidance/cg178/chapter/recommendations#how-to-deliver-psychological-interventions
25. Fusar-Poli P, Werbeloff N, Rutigliano G, et al. Transdiagnostic risk calculator for the automatic detection of individuals at risk and the prediction of psychosis: second replication in an independent National Health Service Trust. Schizophr Bull. 2019;45(3):562-570.
26. Fusar-Poli P, Oliver D, Spada G, et al. The case for improved transdiagnostic detection of first-episode psychosis: electronic health record cohort study. Schizophr Res. 2021;228:547-554.
27. Fusar-Poli P. Negative psychosis prevention trials. JAMA Psychiatry. 2017;74(6):651.
28. Cuijpers P, Smit F, Furukawa TA. Most at‐risk individuals will not develop a mental disorder: the limited predictive strength of risk factors. World Psychiatry. 2021;20(2):224-225.
29. Carrión RE, Cornblatt BA, Burton CZ, et al. Personalized prediction of psychosis: external validation of the NAPLS-2 psychosis risk calculator with the EDIPPP Project. Am J Psychiatry. 2016;173(10):989-996.
30. Worthington MA, Walker EF, Addington J, et al. Incorporating cortisol into the NAPLS2 individualized risk calculator for prediction of psychosis. Schizophr Res. 2021;227:95-100.
31. Koutsouleris N, Dwyer DB, Degenhardt F, et al. Multimodal machine learning workflows for prediction of psychosis in patients with clinical high-risk syndromes and recent-onset depression. JAMA Psychiatry. 2021;78(2):195-209.
32. Simmons MB, Grace D, Fava NJ, et al. The experiences of youth mental health peer workers over time: a qualitative study with longitudinal analysis. Community Ment Health J. 2020;56(5):906-914.
33. Devoe DJ, Farris MS, Townes P, et al. Interventions and transition in youth at risk of psychosis: a systematic review and meta-analyses. J Clin Psychiatry. 2020;81(3):17r12053. doi: 10.4088/JCP.17r12053
34. Bosnjak Kuharic D, Kekin I, Hew J, et al. Interventions for prodromal stage of psychosis. Cochrane Database Syst Rev. 2019;2019(11):CD012236
35. Dixon LB, Goldman HH, Srihari VH, et al. Transforming the treatment of schizophrenia in the United States: The RAISE Initiative. Annu Rev Clin Psychol. 2018;14:237-258.
36. Friedman-Yakoobian MS, Parrish EM, Eack SM, et al. Neurocognitive and social cognitive training for youth at clinical high risk (CHR) for psychosis: a randomized controlled feasibility trial. Schizophr Res. 2020;S0920-9964(20)30461-8. doi: 10.1016/j.schres.2020.09.005
37. Kane JM, Schooler NR, Marcy P, et al. Effect of long-acting injectable antipsychotics vs usual care on time to first hospitalization in early-phase schizophrenia: a randomized clinical trial. JAMA Psychiatry. 2020;77(12):1217-1224.
38. Morrison AP, Pyle M, Maughan D, et al. Antipsychotic medication versus psychological intervention versus a combination of both in adolescents with first-episode psychosis (MAPS): a multicentre, three-arm, randomised controlled pilot and feasibility study. Lancet Psychiatry. 2020;7(9):788-800.
39. Chen YQ, Li XR, Zhang L, et al. Therapeutic response is associated with antipsychotic-induced weight gain in drug-naive first-episode patients with schizophrenia: an 8-week prospective study. J Clin Psychiatry. 2021;82(3):20m13469. doi: 10.4088/JCP.20m13469
40. Insel TR. RAISE-ing our expectations for first-episode psychosis. Am J Psychiatry. 2016;173(4):311-312.
41. Tandon R, Goldman M. Overview of neurobiology. In: Janicak PG, Marder SR, Tandon R, et al, eds. Schizophrenia: recent advances in diagnosis and treatment. Springer; 2014:27-33.
42. National Institutes of Health. Accelerating Medicines Partnership. Schizophrenia. Accessed August 30, 2021. https://www.nih.gov/research-training/accelerating-medicines-partnership-amp/schizophrenia
43. Guloksuz S, van Os J. The slow death of the concept of schizophrenia and the painful birth of the psychosis spectrum. Psychol Med. 2018;48(2):229-244.
44. Christ-Crain M, Bichet DG, Fenske WK, et al. Diabetes insipidus. Nat Rev Dis Primers. 2019;5(1):54.
45. Ahmadi L, Goldman MB. Primary polydipsia: update. Best Pract Res Clin Endocrinol Metab. 2020;34(5):101469. doi: 10.1016/j.beem.2020.101469
46. Early Assessment and Support Alliance. National Early Psychosis Directory. Accessed August 30, 2021. https://easacommunity.org/national-directory.php

Nontraditional therapies for treatment-resistant depression: Part 2
When patients with major depressive disorder (MDD) do not achieve optimal outcomes after FDA-approved first-line treatments and standard adjunctive strategies, clinicians look for additional approaches to alleviate their patients’ symptoms. Recent research suggests that several “nontraditional” treatments used primarily as adjuncts to standard antidepressants have promise for treatment-resistant depression.
In Part 1 of this article (

Herbal/nutraceutical agents
This category encompasses a variety of commonly available “natural” options patients often ask about and at times self-prescribe. Examples evaluated in clinical trials include:
- vitamin D
- essential fatty acids (omega-3, omega-6)
- S-adenosyl-L-methionine (SAMe)
- hypericum perforatum (St. John’s Wort)
- probiotics.
Vitamin D deficiency has been linked to depression, possibly by lowering serotonin, norepinephrine, and dopamine concentrations.1-3
A meta-analysis of 3 prospective, observational studies (N = 8,815) found an elevated risk of affective disorders in patients with low vitamin D levels.4 In addition, a systematic review and meta-analysis supported a potential role for vitamin D supplementation for patients with treatment-resistant depresssion.5
Toxicity can occur at levels >100 ng/mL, and resulting adverse effects may include weakness, fatigue, sleepiness, headache, loss of appetite, dry mouth, metallic taste, nausea, and vomiting. This vitamin can be considered as an adjunct to standard antidepressants, particularly in patients with treatment-resistant depression who have low vitamin D levels, but regular monitoring is necessary to avoid toxicity.
Essential fatty acids. Protein receptors embedded in lipid membranes and their binding affinities are influenced by omega-3 and omega-6 polyunsaturated fatty acids. Thus, essential fatty acids may benefit depression by maintaining membrane integrity and fluidity, as well as via their anti-inflammatory activity.
Continue to: Although results from...
Although results from controlled trials are mixed, a systematic review and meta-analysis of adjunctive nutraceuticals supported a potential role for essential fatty acids, primarily eicosapentaenoic acid (EPA), by itself or in combination with docosahexaenoic acid (DHA), with total EPA >60%.5 A second meta-analysis of 26 studies (N = 2,160) that considered only essential fatty acids concluded that EPA ≥60% at ≤1 g/d could benefit depression.6 Furthermore, omega-3 fatty acids may be helpful as an add-on agent for postpartum depression.7
Be aware that a diet rich in omega-6 greatly increases oxidized low-density lipoprotein levels in adipose tissue, potentially posing a cardiac risk factor. Clinicians need to be aware that self-prescribed use of essential fatty acids is common, and to ask about and monitor their patients’ use of these agents.
S-adenosyl-L-methionine (SAMe) is an intracellular amino acid and methyl donor. Among other actions, it is involved in the biosynthesis of hormones and neurotransmitters. There is promising but limited preliminary evidence of its efficacy and safety as a monotherapy or for antidepressant augmentation.
- Five out of 6 earlier controlled studies reported SAMe IV (200 to 400 mg/d) or IM (45 to 50 mg/d) was more effective than placebo
- When the above studies were added to 14 subsequent studies for a meta-analysis, 12 of 19 RCTs reported that parenteral or oral SAMe was significantly more effective than placebo for depression (P < .05).
Overall, the safety and tolerability of SAMe are good. Common adverse effects include nausea, mild insomnia, dizziness, irritability, and anxiety. This is another compound widely available without a prescription and at times self-prescribed. It carries an acceptable risk/benefit balance, with decades of experience.
Hypericum perforatum (St. John’s Wort) is widely prescribed for depression in China and Europe, typically in doses ranging from 500 to 900 mg/d. Its mechanism of action in depression may relate to inhibition of serotonin, dopamine, and norepinephrine uptake from the synaptic cleft of these interconnecting neurotransmitter systems.
Continue to: A meta-analysis of 7 clinical trials...
A meta-analysis of 7 clinical trials (N = 3,808) comparing St. John’s Wort with various selective serotonin reuptake inhibitors (SSRIs) reported comparable rates of response (pooled relative risk .983, 95% CI .924 to 1.042; P < .001) and remission (pooled relative risk 1.013, 95% CI .892 to 1.134; P < .001).9 Further, there were significantly lower discontinuation/dropout rates (pooled odds ratio .587, 95% CI .478 to 0.697; P < .001) for St. John’s Wort compared with the SSRIs.
Existing evidence on the long-term efficacy and safety is limited (studies ranged from 4 to 12 weeks), as is evidence for patients with more severe depression or high suicidality.
Serious drug interactions include the potential for serotonin syndrome when St. John’s Wort is combined with certain antidepressants, compromised efficacy of benzodiazepines and standard antidepressants, and severe skin reactions to sun exposure. In addition, St. John’s Wort may not be safe to use during pregnancy or while breastfeeding. Because potential drug interactions can be serious and individuals often self-prescribe this agent, it is important to ask patients about their use of St. John’s Wort, and to be vigilant for such potential adverse interactions.
Probiotics. These agents produce neuroactive substances that act on the brain/gut axis. Preliminary evidence suggests that these “psychobiotics” confer mental health benefits.10-12 Relative to other approaches, their low-risk profile make them an attractive option for some patients.
Anti-inflammatory/immune system therapies
Inflammation is linked to various medical and brain disorders. For example, patients with depression often demonstrate increased levels of peripheral blood inflammatory biomarkers (such as C-reactive protein and interleukin-6 and -17) that are known to alter norepinephrine, neuroendocrine (eg, the hypothalamic-pituitary-adrenal axis), and microglia function in addition to neuroplasticity. Thus, targeting inflammation may facilitate the development of novel antidepressants. In addition, these agents may benefit depression associated with comorbid autoimmune disorders, such as psoriasis or rheumatoid arthritis. A systematic review and meta-analysis of 36 RCTs (N = 10,000) found 5 out of 6 anti-inflammatory agents improved depression.13,14 In general, reported disadvantages of anti-inflammatories/immunosuppressants include the potential to block the antidepressant effect of some agents, the risk of opportunistic infections, and an increased risk of suicide.
Continue to: Statins
Statins
In a meta-analysis of 3 randomized, double-blind trials, 3 statins (lovastatin, atorvastatin, and simvastatin) significantly improved depression scores when used as an adjunctive therapy to fluoxetine and citalopram, compared with adjunctive placebo (N = 165, P < .001).15
Specific adverse effects of statins include headaches, muscle pain (rarely rhabdomyolysis), dizziness, rash, and liver damage. Statins also have the potential for adverse interactions with other medications. Given the limited efficacy literature on statins for depression and the potential for serious adverse effects, these agents probably should be limited to patients with treatment-resistant depression for whom a statin is indicated for a comorbid medical disorder, such as hypercholesteremia.
Neurosteroids
Brexanolone is FDA-approved for the treatment of postpartum depression. It is an IV formulation of the neuroactive steroid hormone allopregnanolone (a metabolite of progesterone), which acts as a positive allosteric modulator of the GABA-A receptor. Unfortunately, the infusion needs to occur over a 60-hour period.
Ganaxolone is an oral analog formulation of allopregnanolone. In an uncontrolled, open-label pilot study, this medication was administered for 8 weeks as an adjunct to an adequately dosed antidepressant to 10 postmenopausal women with persistent MDD.16 Of the 9 women who completed the study, 4 (44%) improved significantly (P < .019) and the benefit was sustained for 2 additional weeks.16 Adverse effects of ganaxolone included dizziness in 60% of participants, and sleepiness and fatigue in all of them with twice-daily dosing. If the FDA approves ganaxolone, it would become an easier-to-administer option to brexanolone.
Zuranolone is an investigational agent being studied as a treatment for postpartum depression. In a double-blind RCT that evaluated 151 women with postpartum depression, those who took oral zuranolone, 30 mg daily at bedtime for 2 weeks, experienced significant reductions in Hamilton Depression Rating Scale-17 (HDRS-17) scores compared with placebo (P < .003).17 Improvement in core depression symptom ratings was seen as early as Day 3 and persisted through Day 45.
Continue to: The most common...
The most common (≥5%) treatment-emergent adverse effects were somnolence (15%), headache (9%), dizziness (8%), upper respiratory tract infection (8%), diarrhea (6%), and sedation (5%). Two patients experienced a serious adverse event: one who received zuranolone (confusional state) and one who received placebo (pancreatitis). One patient discontinued zuranolone due to adverse effects vs no discontinuations among those who received placebo. The risk of taking zuranolone while breastfeeding is not known.
Device-based strategies
In addition to FDA-cleared approaches (eg, electroconvulsive therapy [ECT], vagus nerve stimulation [VNS], transcranial magnetic stimulation [TMS]), other devices have also demonstrated promising results.
Transcranial direct current stimulation (tDCS) involves delivering weak electrical current to the cerebral cortex through small scalp electrodes to produce the following effects:
- anodal tDCS enhances cortical excitability
- cathodal tDCS reduces cortical excitability.
A typical protocol consists of delivering 1 to 2 mA over 20 minutes with scalp electrodes placed in different configurations based on the targeted symptom(s).
While tDCS has been evaluated as a treatment for various neuropsychiatric disorders, including bipolar depression, Parkinson’s disease, and schizophrenia, most trials have looked at its use for treating depression. Results have been promising but mixed. For example, 1 meta-analysis of 6 RCTs (comprising 96 active and 80 sham tDCS courses) reported that active tDCS was superior to a sham procedure (Hedges’ g = 0.743) for symptoms of depression.18 By contrast, another meta-analysis of 6 RCTs (N = 200) did not find a significant difference between active and sham tDCS for response and remission rates.19 More recently, a group of experts created an evidence-based guideline using a systematic review of the controlled trial literature. These authors concluded there is “probable efficacy for anodal tDCS of the left dorsolateral prefrontal cortex (DLPFC) (with right orbitofrontal cathode) in major depressive episodes without drug resistance but probable inefficacy for drug-resistant major depressive episodes.”20
Continue to: Adverse effects of tDCS...
Adverse effects of tDCS are typically mild but may include persistent skin lesions similar to burns; mania or hypomania; and one reported seizure in a pediatric patient.
Because various over-the-counter direct current stimulation devices are available for purchase at modest cost, clinicians should ask patients if they have been self-administering this treatment.
Chronotherapy strategies
Agomelatine combines serotonergic (5-HT2B and 5-HT2C antagonist) and melatonergic (MT1-MT2 agonist in the suprachiasmatic nucleus) actions that contribute to stabilization of circadian rhythms and subsequent improvement in sleep patterns. Agomelatine (n = 1,274) significantly lowered depression symptoms compared with placebo (n = 689) (standardized mean difference −0.26; P < 3.48×10-11), but the clinical relevance was questionable.21 A recent review of the literature and expert opinion suggest this agent may also have efficacy for anhedonia; however, in placebo-controlled, relapse prevention studies, its long-term efficacy was not consistent.22
Common adverse effects include anxiety; nausea, vomiting, and stomach pain; abnormal dreams and insomnia; dizziness; drowsiness and fatigue; and weight gain. Some reviewers have expressed concerns about agomelatine’s potential for hepatotoxicity and the need for repeated clinical laboratory tests. Although agomelatine is approved outside of the United States, limited efficacy data and the potential for serious adverse effects have precluded FDA approval of this agent.
Sleep deprivation as a treatment technique for depression has been developed over the past 50 years. With total sleep deprivation (TSD) over 1 cycle, patients stay awake for approximately 36 hours, from daytime until the next day’s evening. While 1 to 6 cycles can produce acute antidepressant effects, prompt relapse after sleep recovery is common.
Continue to: In a systematic review...
In a systematic review and meta-analysis of 7 studies that included a total of 311 patients with bipolar depression23:
- TSD plus medications resulted in a significant decrease in depressive symptoms at 1 week compared with medications alone
- higher response rates were maintained after 3 months with lithium.
Adverse effects commonly include general fatigue and headaches; possible switch into mania with bipolar depression; and rarely, seizures or other unexpected medical conditions (eg, acute coronary syndrome). Presently, this approach is limited to research laboratories with the appropriate sophistication to safely conduct such trials.
Other nontraditional strategies
Cardiovascular exercise, resistance training, mindfulness, and yoga have been shown to decrease severe depressive symptoms when used as adjuncts for patients with treatment-resistant depression, or as monotherapy to treat patients with milder depression.
Exercise. The significant benefits of exercise in various forms as treatment for mild to moderate depression are well described in the literature, but it is less clear if it is effective for treatment-resistant depression. A 2013 Cochrane report24 (39 studies with 2,326 participants total) and 2 meta-analyses undertaken in 2015 (Kvam et al25 included 23 studies with 977 participants, and Schuh et al26 included 25 trials with 1,487 participants) reported that various types of exercise ameliorate depression of differing subtypes and severity, with effect sizes ranging from small to large. Schuh et al26 found that publication bias underestimated effect size. Also, not surprisingly, separate analysis of only higher-quality trials decreased effect size.24-26 A meta-analysis that included tai chi and yoga in addition to aerobic exercise and strength training (25 trials with 2,083 participants) found low to moderate benefit for exercise and yoga.27 Finally, a meta-analysis by Cramer et al28 that included 12 RCTs (N = 619) supported the use of yoga plus controlled breathing techniques as an ancillary treatment for depression.
Two small exercise trials specifically evaluated patients with treatment-resistant depression.29,30 Mota-Pereira et al29 compared 22 participants who walked for 30 to 45 minutes, 5 days a week for 12 weeks in addition to pharmacotherapy with 11 patients who received pharmacotherapy only. Exercise improved all outcomes, including HDRS score (both compared to baseline and to the control group). Moreover, 26% of the exercise group went into remission. Pilu et al30 evaluated strength training as an adjunctive treatment. Participants received 1 hour of strength training twice weekly for 8 months (n = 10), or pharmacotherapy only (n = 20). The adjunct strength training group had a statistically significant (P < .0001) improvement in HDRS scores at the end of the 8 months, whereas the control group did not (P < .28).
Continue to: Adverse effects...
Adverse effects of exercise are typically limited to sprains or strains; rarely, participants experience serious injuries.
Mindfulness-based interventions involve purposely paying attention in the present moment to enhance self-understanding and decrease anxiety about the future and regrets about the past, both of which complicate depression. A meta-analysis of 12 RCTs (N = 578) found this approach significantly reduced depression severity when used as an adjunctive therapy.31 There may be risks if mindfulness-based interventions are practiced incorrectly. For example, some reports have linked mindfulness-based interventions to psychotic episodes, meditation addiction, and antisocial or asocial behavior.32
Bottom Line
Nonpharmacologic options for patients with treatment-resistant depression include herbal/nutraceuticals, anti-inflammatory/immune system therapies, and devices. While research suggests some of these approaches are promising, clinicians need to carefully consider potential adverse effects, some of which may be serious.
Related Resources
- Kaur M, Sanches M. Experimental therapeutics in treatmentresistant major depressive disorder. J Exp Pharmacol. 2021;13:181-196.
- Janicak PG. What’s new in transcranial magnetic stimulation. Current Psychiatry. 2019;18(3):10-16.
Drug Brand Names
Atorvastatin • Lipitor
Brexanolone • Zulresso
Citalopram • Celexa
Fluoxetine • Prozac
Lithium • Eskalith, Lithobid
Lovastatin • Altoprev, Mevacor
Minocycline • Dynacin, Minocin
Simvastatin • Flolipid, Zocor
1. Pittampalli S, Mekala HM, Upadhyayula, S, et al. Does vitamin D deficiency cause depression? Prim Care Companion CNS Disord. 2018;20(5):17l02263.
2. Parker GB, Brotchie H, Graham RK. Vitamin D and depression. J Affect Disord. 2017;208:56-61.
3. Berridge MJ. Vitamin D and depression: cellular and regulatory mechanisms. Pharmacol Rev. 2017;69(2):80-92.
4. Anglin RE, Samaan Z, Walter SD, et al. Vitamin D deficiency and depression in adults: systematic review and meta-analysis. Br J Psychiatry. 2013;202:100-107.
5. Sarris J, Murphy J, Mischoulon D, et al. Adjunctive nutraceuticals for depression: a systematic review and meta-analyses. Am J Psychiatry 2016;173(6);575-587.
6. Liao Y, Xie B, Zhang H, et al. Efficacy of omega-3 PUFAs in depression: a meta-analysis. Transl Psychiatry. 2019;9(1):190.
7. Mocking RJT, Steijn K, Roos C, et al. Omega-3 fatty acid supplementation for perinatal depression: a meta-analysis. J Clin Psychiatry. 2020;81(5):19r13106.
8. Sharma A, Gerbarg P, Bottiglieri T, et al; Work Group of the American Psychiatric Association Council on Research. S-Adenosylmethionine (SAMe) for neuropsychiatric disorders: a clinician-oriented review of research. J Clin Psychiatry. 2017;78(6):e656-e667.
9. Ng QX, Venkatanarayanan N, Ho CY. Clinical use of hypericum perforatum (St John’s wort) in depression: a meta-analysis. J Affect Disord 2017;210:211-221.
10. Huang R, Wang K, Hu J. Effect of probiotics on depression: a systematic review and meta-analysis of randomized controlled trials. Nutrients. 2016;8(8):483.
11. Liu RT, Walsh RFL, Sheehan AE. Prebiotics and probiotics for depression and anxiety: a systematic review and meta-analysis of controlled clinical trials. Neurosci Biobehav Rev. 2019;102:13-23.
12. Wallace CJK, Milev RV. The efficacy, safety, and tolerability of probiotics on depression: clinical results from an open-label pilot study. Front Psychiatry. 2021;12(132):618279.
13. Köhler-Forsberg O, N Lyndholm C, Hjorthøj C, et al. Efficacy of anti-inflammatory treatment on major depressive disorder or depressive symptoms: meta-analysis of clinical trials. Acta Psychiatr Scand. 2019;139(5):404-419.
14. Jha MK. Anti-inflammatory treatments for major depressive disorder: what’s on the horizon? J Clin Psychiatry. 2019;80(6)18ac12630.
15. Salagre E, Fernandes BS, Dodd S, et al. Statins for the treatment of depression: a meta-analysis of randomized, double-blind, placebo-controlled trials. J Affect Disord. 2016;200:235-242.
16. Dichtel LE, Nyer M, Dording C, et al. Effects of open-label, adjunctive ganaxolone on persistent depression despite adequate antidepressant treatment in postmenopausal women: a pilot study. J Clin Psychiatry. 2020;81(4):19m12887.
17. Deligiannidis KM, Meltzer-Brody S, Gunduz-Bruce H, et al. Effect of zuranolone vs placebo in postpartum depression: a randomized clinical trial. JAMA Psychiatry. 2021;78(9):951-959.
18. Kalu UG, Sexton CE, Loo CK, et al. Transcranial direct current stimulation in the treatment of major depression: a meta-analysis. Psychol Med. 2012;42(9):1791-800.
19. Berlim MT, Van den Eynde F, Daskalakis ZJ. Clinical utility of transcranial direct current stimulation (tDCS) for treating major depression: a systematic review and meta-analysis of randomized, double-blind and sham-controlled trials. J Psychiatr Res. 2013;47(1):1-7.
20. Lefaucheur JP, Antal A, Ayache SS, et al. Evidence-based guidelines on the therapeutic use of transcranial direct current stimulation (tDCS). Clin Neurophysiol. 2017;128(1):56-92.
21. Singh SP, Singh V, Kar N. Efficacy of agomelatine in major depressive disorder: meta-analysis and appraisal. Int J Neuropsychopharmacol. 2012;15(3):417-428.
22. Norman TR, Olver JS. Agomelatine for depression: expanding the horizons? Expert Opin Pharmacother. 2019;20(6):647-656.
23. Ramirez-Mahaluf JP, Rozas-Serri E, Ivanovic-Zuvic F, et al. Effectiveness of sleep deprivation in treating acute bipolar depression as augmentation strategy: a systematic review and meta-analysis. Front Psychiatry. 2020;11:70.
24. Cooney GM, Dwan K, Greig CA, et al. Exercise for depression. Cochrane Database Syst Rev. 2013;(9):CD004366.
25. Kvam S, Kleppe CL, Nordhus IH, et al. Exercise as a treatment for depression: a meta-analysis. J Affect Disord. 2016;202:67-86.
26. Schuch FB, Vancampfort D, Richards J, et al. Exercise as a treatment for depression: a meta-analysis adjusting for publication bias. J Psychiatr Res. 2016;77:42-51.
27. Seshadri A, Adaji A, Orth SS, et al. Exercise, yoga, and tai chi for treatment of major depressive disorder in outpatient settings: a systematic review and meta-analysis. Prim Care Companion CNS Disord. 2020;23(1):20r02722.
28. Cramer H, Lauche R, Langhorst J, et al. Yoga for depression: a systematic review and meta-analysis. Depress Anxiety. 2013;30(11):1068-1083.
29. Mota-Pereira J, Silverio J, Carvalho S, et al. Moderate exercise improves depression parameters in treatment-resistant patients with major depressive disorder. J Psychiatr Res. 2011;45(8):1005-1011.
30. Pilu A, Sorba M, Hardoy MC, et al. Efficacy of physical activity in the adjunctive treatment of major depressive disorders: preliminary results. Clin Pract Epidemiol Ment Health. 2007;3:8.
31. Strauss C, Cavanagh K, Oliver A, et al. Mindfulness-based interventions for people diagnosed with a current episode of an anxiety or depressive disorder: a meta-analysis of randomised controlled trials. PLoS One. 2014;9(4):e96110.
32. Shonin E, Van Gordon W, Griffiths MD. Are there risks associated with using mindfulness for the treatment of psychopathology? Clinical Practice. 2014;11(4):389-392.
When patients with major depressive disorder (MDD) do not achieve optimal outcomes after FDA-approved first-line treatments and standard adjunctive strategies, clinicians look for additional approaches to alleviate their patients’ symptoms. Recent research suggests that several “nontraditional” treatments used primarily as adjuncts to standard antidepressants have promise for treatment-resistant depression.
In Part 1 of this article (
Herbal/nutraceutical agents
This category encompasses a variety of commonly available “natural” options patients often ask about and at times self-prescribe. Examples evaluated in clinical trials include:
- vitamin D
- essential fatty acids (omega-3, omega-6)
- S-adenosyl-L-methionine (SAMe)
- hypericum perforatum (St. John’s Wort)
- probiotics.
Vitamin D deficiency has been linked to depression, possibly by lowering serotonin, norepinephrine, and dopamine concentrations.1-3
A meta-analysis of 3 prospective, observational studies (N = 8,815) found an elevated risk of affective disorders in patients with low vitamin D levels.4 In addition, a systematic review and meta-analysis supported a potential role for vitamin D supplementation for patients with treatment-resistant depresssion.5
Toxicity can occur at levels >100 ng/mL, and resulting adverse effects may include weakness, fatigue, sleepiness, headache, loss of appetite, dry mouth, metallic taste, nausea, and vomiting. This vitamin can be considered as an adjunct to standard antidepressants, particularly in patients with treatment-resistant depression who have low vitamin D levels, but regular monitoring is necessary to avoid toxicity.
Essential fatty acids. Protein receptors embedded in lipid membranes and their binding affinities are influenced by omega-3 and omega-6 polyunsaturated fatty acids. Thus, essential fatty acids may benefit depression by maintaining membrane integrity and fluidity, as well as via their anti-inflammatory activity.
Continue to: Although results from...
Although results from controlled trials are mixed, a systematic review and meta-analysis of adjunctive nutraceuticals supported a potential role for essential fatty acids, primarily eicosapentaenoic acid (EPA), by itself or in combination with docosahexaenoic acid (DHA), with total EPA >60%.5 A second meta-analysis of 26 studies (N = 2,160) that considered only essential fatty acids concluded that EPA ≥60% at ≤1 g/d could benefit depression.6 Furthermore, omega-3 fatty acids may be helpful as an add-on agent for postpartum depression.7
Be aware that a diet rich in omega-6 greatly increases oxidized low-density lipoprotein levels in adipose tissue, potentially posing a cardiac risk factor. Clinicians need to be aware that self-prescribed use of essential fatty acids is common, and to ask about and monitor their patients’ use of these agents.
S-adenosyl-L-methionine (SAMe) is an intracellular amino acid and methyl donor. Among other actions, it is involved in the biosynthesis of hormones and neurotransmitters. There is promising but limited preliminary evidence of its efficacy and safety as a monotherapy or for antidepressant augmentation.
- Five out of 6 earlier controlled studies reported SAMe IV (200 to 400 mg/d) or IM (45 to 50 mg/d) was more effective than placebo
- When the above studies were added to 14 subsequent studies for a meta-analysis, 12 of 19 RCTs reported that parenteral or oral SAMe was significantly more effective than placebo for depression (P < .05).
Overall, the safety and tolerability of SAMe are good. Common adverse effects include nausea, mild insomnia, dizziness, irritability, and anxiety. This is another compound widely available without a prescription and at times self-prescribed. It carries an acceptable risk/benefit balance, with decades of experience.
Hypericum perforatum (St. John’s Wort) is widely prescribed for depression in China and Europe, typically in doses ranging from 500 to 900 mg/d. Its mechanism of action in depression may relate to inhibition of serotonin, dopamine, and norepinephrine uptake from the synaptic cleft of these interconnecting neurotransmitter systems.
Continue to: A meta-analysis of 7 clinical trials...
A meta-analysis of 7 clinical trials (N = 3,808) comparing St. John’s Wort with various selective serotonin reuptake inhibitors (SSRIs) reported comparable rates of response (pooled relative risk .983, 95% CI .924 to 1.042; P < .001) and remission (pooled relative risk 1.013, 95% CI .892 to 1.134; P < .001).9 Further, there were significantly lower discontinuation/dropout rates (pooled odds ratio .587, 95% CI .478 to 0.697; P < .001) for St. John’s Wort compared with the SSRIs.
Existing evidence on the long-term efficacy and safety is limited (studies ranged from 4 to 12 weeks), as is evidence for patients with more severe depression or high suicidality.
Serious drug interactions include the potential for serotonin syndrome when St. John’s Wort is combined with certain antidepressants, compromised efficacy of benzodiazepines and standard antidepressants, and severe skin reactions to sun exposure. In addition, St. John’s Wort may not be safe to use during pregnancy or while breastfeeding. Because potential drug interactions can be serious and individuals often self-prescribe this agent, it is important to ask patients about their use of St. John’s Wort, and to be vigilant for such potential adverse interactions.
Probiotics. These agents produce neuroactive substances that act on the brain/gut axis. Preliminary evidence suggests that these “psychobiotics” confer mental health benefits.10-12 Relative to other approaches, their low-risk profile make them an attractive option for some patients.
Anti-inflammatory/immune system therapies
Inflammation is linked to various medical and brain disorders. For example, patients with depression often demonstrate increased levels of peripheral blood inflammatory biomarkers (such as C-reactive protein and interleukin-6 and -17) that are known to alter norepinephrine, neuroendocrine (eg, the hypothalamic-pituitary-adrenal axis), and microglia function in addition to neuroplasticity. Thus, targeting inflammation may facilitate the development of novel antidepressants. In addition, these agents may benefit depression associated with comorbid autoimmune disorders, such as psoriasis or rheumatoid arthritis. A systematic review and meta-analysis of 36 RCTs (N = 10,000) found 5 out of 6 anti-inflammatory agents improved depression.13,14 In general, reported disadvantages of anti-inflammatories/immunosuppressants include the potential to block the antidepressant effect of some agents, the risk of opportunistic infections, and an increased risk of suicide.
Continue to: Statins
Statins
In a meta-analysis of 3 randomized, double-blind trials, 3 statins (lovastatin, atorvastatin, and simvastatin) significantly improved depression scores when used as an adjunctive therapy to fluoxetine and citalopram, compared with adjunctive placebo (N = 165, P < .001).15
Specific adverse effects of statins include headaches, muscle pain (rarely rhabdomyolysis), dizziness, rash, and liver damage. Statins also have the potential for adverse interactions with other medications. Given the limited efficacy literature on statins for depression and the potential for serious adverse effects, these agents probably should be limited to patients with treatment-resistant depression for whom a statin is indicated for a comorbid medical disorder, such as hypercholesteremia.
Neurosteroids
Brexanolone is FDA-approved for the treatment of postpartum depression. It is an IV formulation of the neuroactive steroid hormone allopregnanolone (a metabolite of progesterone), which acts as a positive allosteric modulator of the GABA-A receptor. Unfortunately, the infusion needs to occur over a 60-hour period.
Ganaxolone is an oral analog formulation of allopregnanolone. In an uncontrolled, open-label pilot study, this medication was administered for 8 weeks as an adjunct to an adequately dosed antidepressant to 10 postmenopausal women with persistent MDD.16 Of the 9 women who completed the study, 4 (44%) improved significantly (P < .019) and the benefit was sustained for 2 additional weeks.16 Adverse effects of ganaxolone included dizziness in 60% of participants, and sleepiness and fatigue in all of them with twice-daily dosing. If the FDA approves ganaxolone, it would become an easier-to-administer option to brexanolone.
Zuranolone is an investigational agent being studied as a treatment for postpartum depression. In a double-blind RCT that evaluated 151 women with postpartum depression, those who took oral zuranolone, 30 mg daily at bedtime for 2 weeks, experienced significant reductions in Hamilton Depression Rating Scale-17 (HDRS-17) scores compared with placebo (P < .003).17 Improvement in core depression symptom ratings was seen as early as Day 3 and persisted through Day 45.
Continue to: The most common...
The most common (≥5%) treatment-emergent adverse effects were somnolence (15%), headache (9%), dizziness (8%), upper respiratory tract infection (8%), diarrhea (6%), and sedation (5%). Two patients experienced a serious adverse event: one who received zuranolone (confusional state) and one who received placebo (pancreatitis). One patient discontinued zuranolone due to adverse effects vs no discontinuations among those who received placebo. The risk of taking zuranolone while breastfeeding is not known.
Device-based strategies
In addition to FDA-cleared approaches (eg, electroconvulsive therapy [ECT], vagus nerve stimulation [VNS], transcranial magnetic stimulation [TMS]), other devices have also demonstrated promising results.
Transcranial direct current stimulation (tDCS) involves delivering weak electrical current to the cerebral cortex through small scalp electrodes to produce the following effects:
- anodal tDCS enhances cortical excitability
- cathodal tDCS reduces cortical excitability.
A typical protocol consists of delivering 1 to 2 mA over 20 minutes with scalp electrodes placed in different configurations based on the targeted symptom(s).
While tDCS has been evaluated as a treatment for various neuropsychiatric disorders, including bipolar depression, Parkinson’s disease, and schizophrenia, most trials have looked at its use for treating depression. Results have been promising but mixed. For example, 1 meta-analysis of 6 RCTs (comprising 96 active and 80 sham tDCS courses) reported that active tDCS was superior to a sham procedure (Hedges’ g = 0.743) for symptoms of depression.18 By contrast, another meta-analysis of 6 RCTs (N = 200) did not find a significant difference between active and sham tDCS for response and remission rates.19 More recently, a group of experts created an evidence-based guideline using a systematic review of the controlled trial literature. These authors concluded there is “probable efficacy for anodal tDCS of the left dorsolateral prefrontal cortex (DLPFC) (with right orbitofrontal cathode) in major depressive episodes without drug resistance but probable inefficacy for drug-resistant major depressive episodes.”20
Continue to: Adverse effects of tDCS...
Adverse effects of tDCS are typically mild but may include persistent skin lesions similar to burns; mania or hypomania; and one reported seizure in a pediatric patient.
Because various over-the-counter direct current stimulation devices are available for purchase at modest cost, clinicians should ask patients if they have been self-administering this treatment.
Chronotherapy strategies
Agomelatine combines serotonergic (5-HT2B and 5-HT2C antagonist) and melatonergic (MT1-MT2 agonist in the suprachiasmatic nucleus) actions that contribute to stabilization of circadian rhythms and subsequent improvement in sleep patterns. Agomelatine (n = 1,274) significantly lowered depression symptoms compared with placebo (n = 689) (standardized mean difference −0.26; P < 3.48×10-11), but the clinical relevance was questionable.21 A recent review of the literature and expert opinion suggest this agent may also have efficacy for anhedonia; however, in placebo-controlled, relapse prevention studies, its long-term efficacy was not consistent.22
Common adverse effects include anxiety; nausea, vomiting, and stomach pain; abnormal dreams and insomnia; dizziness; drowsiness and fatigue; and weight gain. Some reviewers have expressed concerns about agomelatine’s potential for hepatotoxicity and the need for repeated clinical laboratory tests. Although agomelatine is approved outside of the United States, limited efficacy data and the potential for serious adverse effects have precluded FDA approval of this agent.
Sleep deprivation as a treatment technique for depression has been developed over the past 50 years. With total sleep deprivation (TSD) over 1 cycle, patients stay awake for approximately 36 hours, from daytime until the next day’s evening. While 1 to 6 cycles can produce acute antidepressant effects, prompt relapse after sleep recovery is common.
Continue to: In a systematic review...
In a systematic review and meta-analysis of 7 studies that included a total of 311 patients with bipolar depression23:
- TSD plus medications resulted in a significant decrease in depressive symptoms at 1 week compared with medications alone
- higher response rates were maintained after 3 months with lithium.
Adverse effects commonly include general fatigue and headaches; possible switch into mania with bipolar depression; and rarely, seizures or other unexpected medical conditions (eg, acute coronary syndrome). Presently, this approach is limited to research laboratories with the appropriate sophistication to safely conduct such trials.
Other nontraditional strategies
Cardiovascular exercise, resistance training, mindfulness, and yoga have been shown to decrease severe depressive symptoms when used as adjuncts for patients with treatment-resistant depression, or as monotherapy to treat patients with milder depression.
Exercise. The significant benefits of exercise in various forms as treatment for mild to moderate depression are well described in the literature, but it is less clear if it is effective for treatment-resistant depression. A 2013 Cochrane report24 (39 studies with 2,326 participants total) and 2 meta-analyses undertaken in 2015 (Kvam et al25 included 23 studies with 977 participants, and Schuh et al26 included 25 trials with 1,487 participants) reported that various types of exercise ameliorate depression of differing subtypes and severity, with effect sizes ranging from small to large. Schuh et al26 found that publication bias underestimated effect size. Also, not surprisingly, separate analysis of only higher-quality trials decreased effect size.24-26 A meta-analysis that included tai chi and yoga in addition to aerobic exercise and strength training (25 trials with 2,083 participants) found low to moderate benefit for exercise and yoga.27 Finally, a meta-analysis by Cramer et al28 that included 12 RCTs (N = 619) supported the use of yoga plus controlled breathing techniques as an ancillary treatment for depression.
Two small exercise trials specifically evaluated patients with treatment-resistant depression.29,30 Mota-Pereira et al29 compared 22 participants who walked for 30 to 45 minutes, 5 days a week for 12 weeks in addition to pharmacotherapy with 11 patients who received pharmacotherapy only. Exercise improved all outcomes, including HDRS score (both compared to baseline and to the control group). Moreover, 26% of the exercise group went into remission. Pilu et al30 evaluated strength training as an adjunctive treatment. Participants received 1 hour of strength training twice weekly for 8 months (n = 10), or pharmacotherapy only (n = 20). The adjunct strength training group had a statistically significant (P < .0001) improvement in HDRS scores at the end of the 8 months, whereas the control group did not (P < .28).
Continue to: Adverse effects...
Adverse effects of exercise are typically limited to sprains or strains; rarely, participants experience serious injuries.
Mindfulness-based interventions involve purposely paying attention in the present moment to enhance self-understanding and decrease anxiety about the future and regrets about the past, both of which complicate depression. A meta-analysis of 12 RCTs (N = 578) found this approach significantly reduced depression severity when used as an adjunctive therapy.31 There may be risks if mindfulness-based interventions are practiced incorrectly. For example, some reports have linked mindfulness-based interventions to psychotic episodes, meditation addiction, and antisocial or asocial behavior.32
Bottom Line
Nonpharmacologic options for patients with treatment-resistant depression include herbal/nutraceuticals, anti-inflammatory/immune system therapies, and devices. While research suggests some of these approaches are promising, clinicians need to carefully consider potential adverse effects, some of which may be serious.
Related Resources
- Kaur M, Sanches M. Experimental therapeutics in treatmentresistant major depressive disorder. J Exp Pharmacol. 2021;13:181-196.
- Janicak PG. What’s new in transcranial magnetic stimulation. Current Psychiatry. 2019;18(3):10-16.
Drug Brand Names
Atorvastatin • Lipitor
Brexanolone • Zulresso
Citalopram • Celexa
Fluoxetine • Prozac
Lithium • Eskalith, Lithobid
Lovastatin • Altoprev, Mevacor
Minocycline • Dynacin, Minocin
Simvastatin • Flolipid, Zocor
When patients with major depressive disorder (MDD) do not achieve optimal outcomes after FDA-approved first-line treatments and standard adjunctive strategies, clinicians look for additional approaches to alleviate their patients’ symptoms. Recent research suggests that several “nontraditional” treatments used primarily as adjuncts to standard antidepressants have promise for treatment-resistant depression.
In Part 1 of this article (
Herbal/nutraceutical agents
This category encompasses a variety of commonly available “natural” options patients often ask about and at times self-prescribe. Examples evaluated in clinical trials include:
- vitamin D
- essential fatty acids (omega-3, omega-6)
- S-adenosyl-L-methionine (SAMe)
- hypericum perforatum (St. John’s Wort)
- probiotics.
Vitamin D deficiency has been linked to depression, possibly by lowering serotonin, norepinephrine, and dopamine concentrations.1-3
A meta-analysis of 3 prospective, observational studies (N = 8,815) found an elevated risk of affective disorders in patients with low vitamin D levels.4 In addition, a systematic review and meta-analysis supported a potential role for vitamin D supplementation for patients with treatment-resistant depresssion.5
Toxicity can occur at levels >100 ng/mL, and resulting adverse effects may include weakness, fatigue, sleepiness, headache, loss of appetite, dry mouth, metallic taste, nausea, and vomiting. This vitamin can be considered as an adjunct to standard antidepressants, particularly in patients with treatment-resistant depression who have low vitamin D levels, but regular monitoring is necessary to avoid toxicity.
Essential fatty acids. Protein receptors embedded in lipid membranes and their binding affinities are influenced by omega-3 and omega-6 polyunsaturated fatty acids. Thus, essential fatty acids may benefit depression by maintaining membrane integrity and fluidity, as well as via their anti-inflammatory activity.
Continue to: Although results from...
Although results from controlled trials are mixed, a systematic review and meta-analysis of adjunctive nutraceuticals supported a potential role for essential fatty acids, primarily eicosapentaenoic acid (EPA), by itself or in combination with docosahexaenoic acid (DHA), with total EPA >60%.5 A second meta-analysis of 26 studies (N = 2,160) that considered only essential fatty acids concluded that EPA ≥60% at ≤1 g/d could benefit depression.6 Furthermore, omega-3 fatty acids may be helpful as an add-on agent for postpartum depression.7
Be aware that a diet rich in omega-6 greatly increases oxidized low-density lipoprotein levels in adipose tissue, potentially posing a cardiac risk factor. Clinicians need to be aware that self-prescribed use of essential fatty acids is common, and to ask about and monitor their patients’ use of these agents.
S-adenosyl-L-methionine (SAMe) is an intracellular amino acid and methyl donor. Among other actions, it is involved in the biosynthesis of hormones and neurotransmitters. There is promising but limited preliminary evidence of its efficacy and safety as a monotherapy or for antidepressant augmentation.
- Five out of 6 earlier controlled studies reported SAMe IV (200 to 400 mg/d) or IM (45 to 50 mg/d) was more effective than placebo
- When the above studies were added to 14 subsequent studies for a meta-analysis, 12 of 19 RCTs reported that parenteral or oral SAMe was significantly more effective than placebo for depression (P < .05).
Overall, the safety and tolerability of SAMe are good. Common adverse effects include nausea, mild insomnia, dizziness, irritability, and anxiety. This is another compound widely available without a prescription and at times self-prescribed. It carries an acceptable risk/benefit balance, with decades of experience.
Hypericum perforatum (St. John’s Wort) is widely prescribed for depression in China and Europe, typically in doses ranging from 500 to 900 mg/d. Its mechanism of action in depression may relate to inhibition of serotonin, dopamine, and norepinephrine uptake from the synaptic cleft of these interconnecting neurotransmitter systems.
Continue to: A meta-analysis of 7 clinical trials...
A meta-analysis of 7 clinical trials (N = 3,808) comparing St. John’s Wort with various selective serotonin reuptake inhibitors (SSRIs) reported comparable rates of response (pooled relative risk .983, 95% CI .924 to 1.042; P < .001) and remission (pooled relative risk 1.013, 95% CI .892 to 1.134; P < .001).9 Further, there were significantly lower discontinuation/dropout rates (pooled odds ratio .587, 95% CI .478 to 0.697; P < .001) for St. John’s Wort compared with the SSRIs.
Existing evidence on the long-term efficacy and safety is limited (studies ranged from 4 to 12 weeks), as is evidence for patients with more severe depression or high suicidality.
Serious drug interactions include the potential for serotonin syndrome when St. John’s Wort is combined with certain antidepressants, compromised efficacy of benzodiazepines and standard antidepressants, and severe skin reactions to sun exposure. In addition, St. John’s Wort may not be safe to use during pregnancy or while breastfeeding. Because potential drug interactions can be serious and individuals often self-prescribe this agent, it is important to ask patients about their use of St. John’s Wort, and to be vigilant for such potential adverse interactions.
Probiotics. These agents produce neuroactive substances that act on the brain/gut axis. Preliminary evidence suggests that these “psychobiotics” confer mental health benefits.10-12 Relative to other approaches, their low-risk profile make them an attractive option for some patients.
Anti-inflammatory/immune system therapies
Inflammation is linked to various medical and brain disorders. For example, patients with depression often demonstrate increased levels of peripheral blood inflammatory biomarkers (such as C-reactive protein and interleukin-6 and -17) that are known to alter norepinephrine, neuroendocrine (eg, the hypothalamic-pituitary-adrenal axis), and microglia function in addition to neuroplasticity. Thus, targeting inflammation may facilitate the development of novel antidepressants. In addition, these agents may benefit depression associated with comorbid autoimmune disorders, such as psoriasis or rheumatoid arthritis. A systematic review and meta-analysis of 36 RCTs (N = 10,000) found 5 out of 6 anti-inflammatory agents improved depression.13,14 In general, reported disadvantages of anti-inflammatories/immunosuppressants include the potential to block the antidepressant effect of some agents, the risk of opportunistic infections, and an increased risk of suicide.
Continue to: Statins
Statins
In a meta-analysis of 3 randomized, double-blind trials, 3 statins (lovastatin, atorvastatin, and simvastatin) significantly improved depression scores when used as an adjunctive therapy to fluoxetine and citalopram, compared with adjunctive placebo (N = 165, P < .001).15
Specific adverse effects of statins include headaches, muscle pain (rarely rhabdomyolysis), dizziness, rash, and liver damage. Statins also have the potential for adverse interactions with other medications. Given the limited efficacy literature on statins for depression and the potential for serious adverse effects, these agents probably should be limited to patients with treatment-resistant depression for whom a statin is indicated for a comorbid medical disorder, such as hypercholesteremia.
Neurosteroids
Brexanolone is FDA-approved for the treatment of postpartum depression. It is an IV formulation of the neuroactive steroid hormone allopregnanolone (a metabolite of progesterone), which acts as a positive allosteric modulator of the GABA-A receptor. Unfortunately, the infusion needs to occur over a 60-hour period.
Ganaxolone is an oral analog formulation of allopregnanolone. In an uncontrolled, open-label pilot study, this medication was administered for 8 weeks as an adjunct to an adequately dosed antidepressant to 10 postmenopausal women with persistent MDD.16 Of the 9 women who completed the study, 4 (44%) improved significantly (P < .019) and the benefit was sustained for 2 additional weeks.16 Adverse effects of ganaxolone included dizziness in 60% of participants, and sleepiness and fatigue in all of them with twice-daily dosing. If the FDA approves ganaxolone, it would become an easier-to-administer option to brexanolone.
Zuranolone is an investigational agent being studied as a treatment for postpartum depression. In a double-blind RCT that evaluated 151 women with postpartum depression, those who took oral zuranolone, 30 mg daily at bedtime for 2 weeks, experienced significant reductions in Hamilton Depression Rating Scale-17 (HDRS-17) scores compared with placebo (P < .003).17 Improvement in core depression symptom ratings was seen as early as Day 3 and persisted through Day 45.
Continue to: The most common...
The most common (≥5%) treatment-emergent adverse effects were somnolence (15%), headache (9%), dizziness (8%), upper respiratory tract infection (8%), diarrhea (6%), and sedation (5%). Two patients experienced a serious adverse event: one who received zuranolone (confusional state) and one who received placebo (pancreatitis). One patient discontinued zuranolone due to adverse effects vs no discontinuations among those who received placebo. The risk of taking zuranolone while breastfeeding is not known.
Device-based strategies
In addition to FDA-cleared approaches (eg, electroconvulsive therapy [ECT], vagus nerve stimulation [VNS], transcranial magnetic stimulation [TMS]), other devices have also demonstrated promising results.
Transcranial direct current stimulation (tDCS) involves delivering weak electrical current to the cerebral cortex through small scalp electrodes to produce the following effects:
- anodal tDCS enhances cortical excitability
- cathodal tDCS reduces cortical excitability.
A typical protocol consists of delivering 1 to 2 mA over 20 minutes with scalp electrodes placed in different configurations based on the targeted symptom(s).
While tDCS has been evaluated as a treatment for various neuropsychiatric disorders, including bipolar depression, Parkinson’s disease, and schizophrenia, most trials have looked at its use for treating depression. Results have been promising but mixed. For example, 1 meta-analysis of 6 RCTs (comprising 96 active and 80 sham tDCS courses) reported that active tDCS was superior to a sham procedure (Hedges’ g = 0.743) for symptoms of depression.18 By contrast, another meta-analysis of 6 RCTs (N = 200) did not find a significant difference between active and sham tDCS for response and remission rates.19 More recently, a group of experts created an evidence-based guideline using a systematic review of the controlled trial literature. These authors concluded there is “probable efficacy for anodal tDCS of the left dorsolateral prefrontal cortex (DLPFC) (with right orbitofrontal cathode) in major depressive episodes without drug resistance but probable inefficacy for drug-resistant major depressive episodes.”20
Continue to: Adverse effects of tDCS...
Adverse effects of tDCS are typically mild but may include persistent skin lesions similar to burns; mania or hypomania; and one reported seizure in a pediatric patient.
Because various over-the-counter direct current stimulation devices are available for purchase at modest cost, clinicians should ask patients if they have been self-administering this treatment.
Chronotherapy strategies
Agomelatine combines serotonergic (5-HT2B and 5-HT2C antagonist) and melatonergic (MT1-MT2 agonist in the suprachiasmatic nucleus) actions that contribute to stabilization of circadian rhythms and subsequent improvement in sleep patterns. Agomelatine (n = 1,274) significantly lowered depression symptoms compared with placebo (n = 689) (standardized mean difference −0.26; P < 3.48×10-11), but the clinical relevance was questionable.21 A recent review of the literature and expert opinion suggest this agent may also have efficacy for anhedonia; however, in placebo-controlled, relapse prevention studies, its long-term efficacy was not consistent.22
Common adverse effects include anxiety; nausea, vomiting, and stomach pain; abnormal dreams and insomnia; dizziness; drowsiness and fatigue; and weight gain. Some reviewers have expressed concerns about agomelatine’s potential for hepatotoxicity and the need for repeated clinical laboratory tests. Although agomelatine is approved outside of the United States, limited efficacy data and the potential for serious adverse effects have precluded FDA approval of this agent.
Sleep deprivation as a treatment technique for depression has been developed over the past 50 years. With total sleep deprivation (TSD) over 1 cycle, patients stay awake for approximately 36 hours, from daytime until the next day’s evening. While 1 to 6 cycles can produce acute antidepressant effects, prompt relapse after sleep recovery is common.
Continue to: In a systematic review...
In a systematic review and meta-analysis of 7 studies that included a total of 311 patients with bipolar depression23:
- TSD plus medications resulted in a significant decrease in depressive symptoms at 1 week compared with medications alone
- higher response rates were maintained after 3 months with lithium.
Adverse effects commonly include general fatigue and headaches; possible switch into mania with bipolar depression; and rarely, seizures or other unexpected medical conditions (eg, acute coronary syndrome). Presently, this approach is limited to research laboratories with the appropriate sophistication to safely conduct such trials.
Other nontraditional strategies
Cardiovascular exercise, resistance training, mindfulness, and yoga have been shown to decrease severe depressive symptoms when used as adjuncts for patients with treatment-resistant depression, or as monotherapy to treat patients with milder depression.
Exercise. The significant benefits of exercise in various forms as treatment for mild to moderate depression are well described in the literature, but it is less clear if it is effective for treatment-resistant depression. A 2013 Cochrane report24 (39 studies with 2,326 participants total) and 2 meta-analyses undertaken in 2015 (Kvam et al25 included 23 studies with 977 participants, and Schuh et al26 included 25 trials with 1,487 participants) reported that various types of exercise ameliorate depression of differing subtypes and severity, with effect sizes ranging from small to large. Schuh et al26 found that publication bias underestimated effect size. Also, not surprisingly, separate analysis of only higher-quality trials decreased effect size.24-26 A meta-analysis that included tai chi and yoga in addition to aerobic exercise and strength training (25 trials with 2,083 participants) found low to moderate benefit for exercise and yoga.27 Finally, a meta-analysis by Cramer et al28 that included 12 RCTs (N = 619) supported the use of yoga plus controlled breathing techniques as an ancillary treatment for depression.
Two small exercise trials specifically evaluated patients with treatment-resistant depression.29,30 Mota-Pereira et al29 compared 22 participants who walked for 30 to 45 minutes, 5 days a week for 12 weeks in addition to pharmacotherapy with 11 patients who received pharmacotherapy only. Exercise improved all outcomes, including HDRS score (both compared to baseline and to the control group). Moreover, 26% of the exercise group went into remission. Pilu et al30 evaluated strength training as an adjunctive treatment. Participants received 1 hour of strength training twice weekly for 8 months (n = 10), or pharmacotherapy only (n = 20). The adjunct strength training group had a statistically significant (P < .0001) improvement in HDRS scores at the end of the 8 months, whereas the control group did not (P < .28).
Continue to: Adverse effects...
Adverse effects of exercise are typically limited to sprains or strains; rarely, participants experience serious injuries.
Mindfulness-based interventions involve purposely paying attention in the present moment to enhance self-understanding and decrease anxiety about the future and regrets about the past, both of which complicate depression. A meta-analysis of 12 RCTs (N = 578) found this approach significantly reduced depression severity when used as an adjunctive therapy.31 There may be risks if mindfulness-based interventions are practiced incorrectly. For example, some reports have linked mindfulness-based interventions to psychotic episodes, meditation addiction, and antisocial or asocial behavior.32
Bottom Line
Nonpharmacologic options for patients with treatment-resistant depression include herbal/nutraceuticals, anti-inflammatory/immune system therapies, and devices. While research suggests some of these approaches are promising, clinicians need to carefully consider potential adverse effects, some of which may be serious.
Related Resources
- Kaur M, Sanches M. Experimental therapeutics in treatmentresistant major depressive disorder. J Exp Pharmacol. 2021;13:181-196.
- Janicak PG. What’s new in transcranial magnetic stimulation. Current Psychiatry. 2019;18(3):10-16.
Drug Brand Names
Atorvastatin • Lipitor
Brexanolone • Zulresso
Citalopram • Celexa
Fluoxetine • Prozac
Lithium • Eskalith, Lithobid
Lovastatin • Altoprev, Mevacor
Minocycline • Dynacin, Minocin
Simvastatin • Flolipid, Zocor
1. Pittampalli S, Mekala HM, Upadhyayula, S, et al. Does vitamin D deficiency cause depression? Prim Care Companion CNS Disord. 2018;20(5):17l02263.
2. Parker GB, Brotchie H, Graham RK. Vitamin D and depression. J Affect Disord. 2017;208:56-61.
3. Berridge MJ. Vitamin D and depression: cellular and regulatory mechanisms. Pharmacol Rev. 2017;69(2):80-92.
4. Anglin RE, Samaan Z, Walter SD, et al. Vitamin D deficiency and depression in adults: systematic review and meta-analysis. Br J Psychiatry. 2013;202:100-107.
5. Sarris J, Murphy J, Mischoulon D, et al. Adjunctive nutraceuticals for depression: a systematic review and meta-analyses. Am J Psychiatry 2016;173(6);575-587.
6. Liao Y, Xie B, Zhang H, et al. Efficacy of omega-3 PUFAs in depression: a meta-analysis. Transl Psychiatry. 2019;9(1):190.
7. Mocking RJT, Steijn K, Roos C, et al. Omega-3 fatty acid supplementation for perinatal depression: a meta-analysis. J Clin Psychiatry. 2020;81(5):19r13106.
8. Sharma A, Gerbarg P, Bottiglieri T, et al; Work Group of the American Psychiatric Association Council on Research. S-Adenosylmethionine (SAMe) for neuropsychiatric disorders: a clinician-oriented review of research. J Clin Psychiatry. 2017;78(6):e656-e667.
9. Ng QX, Venkatanarayanan N, Ho CY. Clinical use of hypericum perforatum (St John’s wort) in depression: a meta-analysis. J Affect Disord 2017;210:211-221.
10. Huang R, Wang K, Hu J. Effect of probiotics on depression: a systematic review and meta-analysis of randomized controlled trials. Nutrients. 2016;8(8):483.
11. Liu RT, Walsh RFL, Sheehan AE. Prebiotics and probiotics for depression and anxiety: a systematic review and meta-analysis of controlled clinical trials. Neurosci Biobehav Rev. 2019;102:13-23.
12. Wallace CJK, Milev RV. The efficacy, safety, and tolerability of probiotics on depression: clinical results from an open-label pilot study. Front Psychiatry. 2021;12(132):618279.
13. Köhler-Forsberg O, N Lyndholm C, Hjorthøj C, et al. Efficacy of anti-inflammatory treatment on major depressive disorder or depressive symptoms: meta-analysis of clinical trials. Acta Psychiatr Scand. 2019;139(5):404-419.
14. Jha MK. Anti-inflammatory treatments for major depressive disorder: what’s on the horizon? J Clin Psychiatry. 2019;80(6)18ac12630.
15. Salagre E, Fernandes BS, Dodd S, et al. Statins for the treatment of depression: a meta-analysis of randomized, double-blind, placebo-controlled trials. J Affect Disord. 2016;200:235-242.
16. Dichtel LE, Nyer M, Dording C, et al. Effects of open-label, adjunctive ganaxolone on persistent depression despite adequate antidepressant treatment in postmenopausal women: a pilot study. J Clin Psychiatry. 2020;81(4):19m12887.
17. Deligiannidis KM, Meltzer-Brody S, Gunduz-Bruce H, et al. Effect of zuranolone vs placebo in postpartum depression: a randomized clinical trial. JAMA Psychiatry. 2021;78(9):951-959.
18. Kalu UG, Sexton CE, Loo CK, et al. Transcranial direct current stimulation in the treatment of major depression: a meta-analysis. Psychol Med. 2012;42(9):1791-800.
19. Berlim MT, Van den Eynde F, Daskalakis ZJ. Clinical utility of transcranial direct current stimulation (tDCS) for treating major depression: a systematic review and meta-analysis of randomized, double-blind and sham-controlled trials. J Psychiatr Res. 2013;47(1):1-7.
20. Lefaucheur JP, Antal A, Ayache SS, et al. Evidence-based guidelines on the therapeutic use of transcranial direct current stimulation (tDCS). Clin Neurophysiol. 2017;128(1):56-92.
21. Singh SP, Singh V, Kar N. Efficacy of agomelatine in major depressive disorder: meta-analysis and appraisal. Int J Neuropsychopharmacol. 2012;15(3):417-428.
22. Norman TR, Olver JS. Agomelatine for depression: expanding the horizons? Expert Opin Pharmacother. 2019;20(6):647-656.
23. Ramirez-Mahaluf JP, Rozas-Serri E, Ivanovic-Zuvic F, et al. Effectiveness of sleep deprivation in treating acute bipolar depression as augmentation strategy: a systematic review and meta-analysis. Front Psychiatry. 2020;11:70.
24. Cooney GM, Dwan K, Greig CA, et al. Exercise for depression. Cochrane Database Syst Rev. 2013;(9):CD004366.
25. Kvam S, Kleppe CL, Nordhus IH, et al. Exercise as a treatment for depression: a meta-analysis. J Affect Disord. 2016;202:67-86.
26. Schuch FB, Vancampfort D, Richards J, et al. Exercise as a treatment for depression: a meta-analysis adjusting for publication bias. J Psychiatr Res. 2016;77:42-51.
27. Seshadri A, Adaji A, Orth SS, et al. Exercise, yoga, and tai chi for treatment of major depressive disorder in outpatient settings: a systematic review and meta-analysis. Prim Care Companion CNS Disord. 2020;23(1):20r02722.
28. Cramer H, Lauche R, Langhorst J, et al. Yoga for depression: a systematic review and meta-analysis. Depress Anxiety. 2013;30(11):1068-1083.
29. Mota-Pereira J, Silverio J, Carvalho S, et al. Moderate exercise improves depression parameters in treatment-resistant patients with major depressive disorder. J Psychiatr Res. 2011;45(8):1005-1011.
30. Pilu A, Sorba M, Hardoy MC, et al. Efficacy of physical activity in the adjunctive treatment of major depressive disorders: preliminary results. Clin Pract Epidemiol Ment Health. 2007;3:8.
31. Strauss C, Cavanagh K, Oliver A, et al. Mindfulness-based interventions for people diagnosed with a current episode of an anxiety or depressive disorder: a meta-analysis of randomised controlled trials. PLoS One. 2014;9(4):e96110.
32. Shonin E, Van Gordon W, Griffiths MD. Are there risks associated with using mindfulness for the treatment of psychopathology? Clinical Practice. 2014;11(4):389-392.
1. Pittampalli S, Mekala HM, Upadhyayula, S, et al. Does vitamin D deficiency cause depression? Prim Care Companion CNS Disord. 2018;20(5):17l02263.
2. Parker GB, Brotchie H, Graham RK. Vitamin D and depression. J Affect Disord. 2017;208:56-61.
3. Berridge MJ. Vitamin D and depression: cellular and regulatory mechanisms. Pharmacol Rev. 2017;69(2):80-92.
4. Anglin RE, Samaan Z, Walter SD, et al. Vitamin D deficiency and depression in adults: systematic review and meta-analysis. Br J Psychiatry. 2013;202:100-107.
5. Sarris J, Murphy J, Mischoulon D, et al. Adjunctive nutraceuticals for depression: a systematic review and meta-analyses. Am J Psychiatry 2016;173(6);575-587.
6. Liao Y, Xie B, Zhang H, et al. Efficacy of omega-3 PUFAs in depression: a meta-analysis. Transl Psychiatry. 2019;9(1):190.
7. Mocking RJT, Steijn K, Roos C, et al. Omega-3 fatty acid supplementation for perinatal depression: a meta-analysis. J Clin Psychiatry. 2020;81(5):19r13106.
8. Sharma A, Gerbarg P, Bottiglieri T, et al; Work Group of the American Psychiatric Association Council on Research. S-Adenosylmethionine (SAMe) for neuropsychiatric disorders: a clinician-oriented review of research. J Clin Psychiatry. 2017;78(6):e656-e667.
9. Ng QX, Venkatanarayanan N, Ho CY. Clinical use of hypericum perforatum (St John’s wort) in depression: a meta-analysis. J Affect Disord 2017;210:211-221.
10. Huang R, Wang K, Hu J. Effect of probiotics on depression: a systematic review and meta-analysis of randomized controlled trials. Nutrients. 2016;8(8):483.
11. Liu RT, Walsh RFL, Sheehan AE. Prebiotics and probiotics for depression and anxiety: a systematic review and meta-analysis of controlled clinical trials. Neurosci Biobehav Rev. 2019;102:13-23.
12. Wallace CJK, Milev RV. The efficacy, safety, and tolerability of probiotics on depression: clinical results from an open-label pilot study. Front Psychiatry. 2021;12(132):618279.
13. Köhler-Forsberg O, N Lyndholm C, Hjorthøj C, et al. Efficacy of anti-inflammatory treatment on major depressive disorder or depressive symptoms: meta-analysis of clinical trials. Acta Psychiatr Scand. 2019;139(5):404-419.
14. Jha MK. Anti-inflammatory treatments for major depressive disorder: what’s on the horizon? J Clin Psychiatry. 2019;80(6)18ac12630.
15. Salagre E, Fernandes BS, Dodd S, et al. Statins for the treatment of depression: a meta-analysis of randomized, double-blind, placebo-controlled trials. J Affect Disord. 2016;200:235-242.
16. Dichtel LE, Nyer M, Dording C, et al. Effects of open-label, adjunctive ganaxolone on persistent depression despite adequate antidepressant treatment in postmenopausal women: a pilot study. J Clin Psychiatry. 2020;81(4):19m12887.
17. Deligiannidis KM, Meltzer-Brody S, Gunduz-Bruce H, et al. Effect of zuranolone vs placebo in postpartum depression: a randomized clinical trial. JAMA Psychiatry. 2021;78(9):951-959.
18. Kalu UG, Sexton CE, Loo CK, et al. Transcranial direct current stimulation in the treatment of major depression: a meta-analysis. Psychol Med. 2012;42(9):1791-800.
19. Berlim MT, Van den Eynde F, Daskalakis ZJ. Clinical utility of transcranial direct current stimulation (tDCS) for treating major depression: a systematic review and meta-analysis of randomized, double-blind and sham-controlled trials. J Psychiatr Res. 2013;47(1):1-7.
20. Lefaucheur JP, Antal A, Ayache SS, et al. Evidence-based guidelines on the therapeutic use of transcranial direct current stimulation (tDCS). Clin Neurophysiol. 2017;128(1):56-92.
21. Singh SP, Singh V, Kar N. Efficacy of agomelatine in major depressive disorder: meta-analysis and appraisal. Int J Neuropsychopharmacol. 2012;15(3):417-428.
22. Norman TR, Olver JS. Agomelatine for depression: expanding the horizons? Expert Opin Pharmacother. 2019;20(6):647-656.
23. Ramirez-Mahaluf JP, Rozas-Serri E, Ivanovic-Zuvic F, et al. Effectiveness of sleep deprivation in treating acute bipolar depression as augmentation strategy: a systematic review and meta-analysis. Front Psychiatry. 2020;11:70.
24. Cooney GM, Dwan K, Greig CA, et al. Exercise for depression. Cochrane Database Syst Rev. 2013;(9):CD004366.
25. Kvam S, Kleppe CL, Nordhus IH, et al. Exercise as a treatment for depression: a meta-analysis. J Affect Disord. 2016;202:67-86.
26. Schuch FB, Vancampfort D, Richards J, et al. Exercise as a treatment for depression: a meta-analysis adjusting for publication bias. J Psychiatr Res. 2016;77:42-51.
27. Seshadri A, Adaji A, Orth SS, et al. Exercise, yoga, and tai chi for treatment of major depressive disorder in outpatient settings: a systematic review and meta-analysis. Prim Care Companion CNS Disord. 2020;23(1):20r02722.
28. Cramer H, Lauche R, Langhorst J, et al. Yoga for depression: a systematic review and meta-analysis. Depress Anxiety. 2013;30(11):1068-1083.
29. Mota-Pereira J, Silverio J, Carvalho S, et al. Moderate exercise improves depression parameters in treatment-resistant patients with major depressive disorder. J Psychiatr Res. 2011;45(8):1005-1011.
30. Pilu A, Sorba M, Hardoy MC, et al. Efficacy of physical activity in the adjunctive treatment of major depressive disorders: preliminary results. Clin Pract Epidemiol Ment Health. 2007;3:8.
31. Strauss C, Cavanagh K, Oliver A, et al. Mindfulness-based interventions for people diagnosed with a current episode of an anxiety or depressive disorder: a meta-analysis of randomised controlled trials. PLoS One. 2014;9(4):e96110.
32. Shonin E, Van Gordon W, Griffiths MD. Are there risks associated with using mindfulness for the treatment of psychopathology? Clinical Practice. 2014;11(4):389-392.

Nontraditional therapies for treatment-resistant depression
Presently, FDA-approved first-line treatments and standard adjunctive strategies (eg, lithium, thyroid supplementation, stimulants, second-generation antipsychotics) for major depressive disorder (MDD) often produce less-than-desired outcomes while carrying a potentially substantial safety and tolerability burden. The lack of clinically useful and individual-based biomarkers (eg, genetic, neurophysiological, imaging) is a major obstacle to enhancing treatment efficacy and/or decreasing associated adverse effects (AEs). While the discovery of such tools is being aggressively pursued and ultimately will facilitate a more precision-based choice of therapy, empirical strategies remain our primary approach.
In controlled trials, several nontraditional treatments used primarily as adjuncts to standard antidepressants have shown promise. These include “repurposed” (off-label) medications, herbal/nutraceuticals, anti-inflammatory/immune system therapies, device-based treatments, and other alternative approaches.
Importantly, some nontraditional treatments also demonstrate AEs (Table1-16). With a careful consideration of the risk/benefit balance, this article reviews some of the better-studied treatment options for patients with treatment-resistant depression (TRD). In Part 1, we will examine off-label medications. In Part 2, we will review other nontraditional approaches to TRD, including herbal/nutraceuticals, anti-inflammatory/immune system therapies, device-based treatments, and other alternative approaches.

We believe this review will help clinicians who need to formulate a different approach after their patient with depression is not helped by traditional first-, second-, and third-line treatments. The potential options discussed in Part 1 of this article are categorized based on their putative mechanism of action (MOA) for depression.
Serotonergic and noradrenergic strategies
Pimavanserin is FDA-approved for treatment of Parkinson’s psychosis. Its potential MOA as an adjunctive strategy for MDD may involve 5-HT2A antagonist and inverse agonist receptor activity, as well as lesser effects at the 5-HT2Creceptor.
A 2-stage, 5-week randomized controlled trial (RCT) (CLARITY; N = 207) found adjunctive pimavanserin (34 mg/d) produced a robust antidepressant effect vs placebo in patients whose depression did not respond to selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs).1 Furthermore, a secondary analysis of the data suggested that pimavanserin also improved sleepiness (P < .0003) and daily functioning (P < .014) at Week 5.2
Unfortunately, two 6-week, Phase III RCTs (CLARITY-2 and -3; N = 298) did not find a statistically significant difference between active treatment and placebo. This was based on change in the primary outcome measure (Hamilton Depression Rating Scale-17 score) when adjunctive pimavanserin (34 mg/d) was added to an SSRI or SNRI in patients with TRD.3 There was, however, a significant difference favoring active treatment over placebo based on the Clinical Global Impression–Severity score.
Continue to: In these trials...
In these trials, pimavanserin was generally well-tolerated. The most common AEs were dry mouth, nausea, and headache. Pimavanserin has minimal activity at norepinephrine, dopamine, histamine, or acetylcholine receptors, thus avoiding AEs associated with these receptor interactions.
Given the mixed efficacy results of existing trials, further studies are needed to clarify this agent’s overall risk/benefit in the context of TRD.
Antihypertensive medications
Emerging data suggest that some beta-adrenergic blockers, angiotensin-inhibiting agents, and calcium antagonists are associated with a decreased incidence of depression. A large 2020 study (N = 3,747,190) used population-based Danish registries (2005 to 2015) to evaluate if any of the 41 most commonly prescribed antihypertensive medications were associated with the diagnosis of depressive disorder or use of antidepressants.4 These researchers found that enalapril, ramipril, amlodipine, propranolol, atenolol, bisoprolol, carvedilol (P < .001), and verapamil (P < .004) were strongly associated with a decreased risk of depression.4
Adverse effects across these different classes of antihypertensives are well characterized, can be substantial, and commonly are related to their impact on cardiovascular function (eg, hypotension). Clinically, these agents may be potential adjuncts for patients with TRD who need antihypertensive therapy. Their use and the choice of specific agent should only be determined in consultation with the patient’s primary care physician (PCP) or appropriate specialist.
Glutamatergic strategies
Ketamine is a dissociative anesthetic and analgesic. Its MOA for treating depression appears to occur primarily through antagonist activity at the N-methyl-
Continue to: Many published studies...
Many published studies and reviews have described ketamine’s role for treating MDD. Several studies have reported that low-dose (0.5 mg/kg) IV ketamine infusions can rapidly attenuate severe episodes of MDD as well as associated suicidality. For example, a meta-analysis of 9 RCTs (N = 368) comparing ketamine to placebo for acute treatment of unipolar and bipolar depression reported superior therapeutic effects with active treatment at 24 hours, 72 hours, and 7 days.6 The response and remission rates for ketamine were 52% and 21% at 24 hours; 48% and 24% at 72 hours; and 40% and 26% at 7 days, respectively.6
The most commonly reported AEs during the 4 hours after ketamine infusion included7:
- drowsiness, dizziness, poor coordination
- blurred vision, feeling strange or unreal
- hemodynamic changes (approximately 33%)
- small but significant (P < .05) increases in psychotomimetic and dissociative symptoms.
Because some individuals use ketamine recreationally, this agent also carries the risk of abuse.
Research is ongoing on strategies for long-term maintenance ketamine treatment, and the results of both short- and long-term trials will require careful scrutiny to better assess this agent’s safety and tolerability. Clinicians should first consider esketamine—the S-enantiomer of ketamine—because an intranasal formulation of this agent is FDA-approved for treating patients with TRD or MDD with suicidality when administered in a Risk Evaluation and Mitigation Strategy–certified setting.
Cholinergic strategies
Scopolamine is a potent muscarinic receptor antagonist used to prevent nausea and vomiting caused by motion sickness or medications used during surgery. Its use for MDD is based on the theory that muscarinic receptors may be hypersensitive in mood disorders.
Continue to: Several double-blind RCTs...
Several double-blind RCTs of patients with unipolar or bipolar depression that used 3 pulsed IV infusions (4.0 mcg/kg) over 15 minutes found a rapid, robust antidepressant effect with scopolamine vs placebo.8,9 The oral formulation might also be effective, but would not have a rapid onset.
Common adverse effects of scopolamine include agitation, dry mouth, urinary retention, and cognitive clouding. Given scopolamine’s substantial AE profile, it should be considered only for patients with TRD who could also benefit from the oral formulation for the medical indications noted above, should generally be avoided in older patients, and should be prescribed in consultation with the patient’s PCP.
Botulinum toxin. This neurotoxin inhibits acetylcholine release. It is used to treat disorders characterized by abnormal muscular contraction, such as strabismus, blepharospasm, and chronic pain syndromes. Its MOA for depression may involve its paralytic effects after injection into the glabella forehead muscle (based on the facial feedback hypothesis), as well as modulation of neurotransmitters implicated in the pathophysiology of depression.
In several small trials, injectable botulinum toxin type A (BTA) (29 units) demonstrated antidepressant effects. A recent review that considered 6 trials (N = 235; 4 of the 6 studies were RCTs, 3 of which were rated as high quality) concluded that BTA may be a promising treatment for MDD.10 Limitations of this review included lack of a priori hypotheses, small sample sizes, gender bias, and difficulty in blinding.
In clinical trials, the most common AEs included local irritation at the injection site and transient headache. This agent’s relatively mild AE profile and possible overlap when used for some of the medical indications noted above opens its potential use as an adjunct in patients with comorbid TRD.
Continue to: Endocrine strategies
Endocrine strategies
Mifepristone (RU486). This anti-glucocorticoid receptor antagonist is used as an abortifacient. Based on the theory that hyperactivity of the hypothalamic-pituitary-adrenal axis is implicated in the pathophysiology of MDD with psychotic features (psychotic depression), this agent has been studied as a treatment for this indication.
An analysis of 5 double-blind RCTs (N = 1,460) found that 7 days of mifepristone, 1,200 mg/d, was superior to placebo (P < .004) in reducing psychotic symptoms of depression.11 Plasma concentrations ≥1,600 ng/mL may be required to maximize benefit.11
Overall, this agent demonstrated a good safety profile in clinical trials, with treatment-emergent AEs reported in 556 (66.7%) patients who received mifepristone vs 386 (61.6%) patients who received placebo.11 Common AEs included gastrointestinal (GI) symptoms, headache, and dizziness. However, 3 deaths occurred: 2 patients who received mifepristone and 1 patient who received placebo. Given this potential for a fatal outcome, clinicians should first consider prescribing an adjunctive antipsychotic agent or electroconvulsive therapy.
Estrogens. These hormones are important for sexual and reproductive development and are used to treat various sexual/reproductive disorders, primarily in women. Their role in treating depression is based on the observation that perimenopause is accompanied by an increased risk of new and recurrent depression coincident with declining ovarian function.
Evidence supports the antidepressant efficacy of transdermal estradiol plus progesterone for perimenopausal depression, but not for postmenopausal depression.12-14 However, estrogens carry significant risks that must be carefully considered in relationship to their potential benefits. These risks include:
- vaginal bleeding, dysmenorrhea
- fibroid enlargement
- galactorrhea
- ovarian cancer, endometrial cancer, breast cancer
- deep vein thrombosis, pulmonary embolism
- hypertension, chest pain, myocardial infarction, stroke.
Continue to: The use of estrogens...
The use of estrogens as an adjunctive therapy for women with treatment-resistant perimenopausal depression should only be undertaken when standard strategies have failed, and in consultation with an endocrine specialist who can monitor for potentially serious AEs.
Opioid medications
Buprenorphine is used to treat opioid use disorder (OUD) as well as acute and chronic pain. The opioid system is involved in the regulation of mood and may be an appropriate target for novel antidepressants. The use of buprenorphine in combination with samidorphan (a preferential mu-opioid receptor antagonist) has shown initial promise for TRD while minimizing abuse potential.
Although earlier results were mixed, a pooled analysis of 2 recent large RCTs (N = 760) of patients with MDD who had not responded to antidepressants reported greater reduction in Montgomery-Åsberg Depression Rating Scale scores from baseline for active treatment (buprenorphine/samidorphan; 2 mg/2 mg) vs placebo at multiple timepoints, including end of treatment (-1.8; P < .010).15
The most common AEs included nausea, constipation, dizziness, vomiting, somnolence, fatigue, and sedation. There was minimal evidence of abuse, dependence, or opioid withdrawal. Due to the opioid crisis in the United States, the resulting relaxation of regulations regarding prescribing buprenorphine, and the high rates of depression among patients with OUD, buprenorphine/samidorphan, which is an investigational agent that is not FDA-approved, may be particularly helpful for patients with OUD who also experience comorbid TRD.
Antioxidant agents
N-acetylcysteine (NAC) is an amino acid that can treat acetaminophen toxicity and moderate hepatic damage by increasing glutathione levels. Glutathione is also the primary antioxidant in the CNS. NAC may protect against oxidative stress, chelate heavy metals, reduce inflammation, protect against mitochondrial dysfunction, inhibit apoptosis, and enhance neurogenesis, all potential pathophysiological processes that may contribute to depression.16
Continue to: A systematic review...
A systematic review and meta-analysis of 5 RCTs (N = 574) considered patients with various depression diagnoses who were randomized to adjunctive NAC, 1,000 mg twice a day, or placebo. Over 12 to 24 weeks, there was a significantly greater improvement in mood symptoms and functionality with NAC vs placebo.16
Overall, NAC was well-tolerated. The most common AEs were GI symptoms, musculoskeletal complaints, decreased energy, and headache. While NAC has been touted as a potential adjunct therapy for several psychiatric disorders, including TRD, the evidence for benefit remains limited. Given its favorable AE profile, however, and over-the-counter availability, it remains an option for select patients. It is important to ask patients if they are already taking NAC.
Options beyond off-label medications
There are a multitude of options available for addressing TRD. Many FDA-approved medications are repurposed and prescribed off-label for other indications when the risk/benefit balance is favorable. In Part 1 of this article, we reviewed several off-label medications that have supportive controlled data for treating TRD. In Part 2, we will review other nontraditional therapies for TRD, including herbal/nutraceuticals, anti-inflammatory/immune system therapies, device-based treatments, and other alternative approaches.
Bottom Line
Off-label medications that may offer benefit for patients with treatment-resistant depression (TRD) include pimavanserin, antihypertensive agents, ketamine, scopolamine, botulinum toxin, mifepristone, estrogens, buprenorphine, and N-acetylcysteine. Although some evidence supports use of these agents as adjuncts for TRD, an individualized risk/benefit analysis is required.
Related Resource
- Joshi KG, Frierson RL. Off-label prescribing: How to limit your liability. Current Psychiatry. 2020;19(9):12,39.
Drug Brand Names
Amlodipine • Katerzia, Norvasc
Atenolol • Tenormin
Bisoprolol • Zebeta
Buprenorphine • Sublocade, Subutex
Carvedilol • Coreg
Enalapril • Vasotec
Esketamine • Spravato
Estradiol transdermal • Estraderm
Ketamine • Ketalar
Mifepristone • Mifeprex
Pimavanserin • Nuplazid
Progesterone • Prometrium
Propranolol • Inderal
Ramipril • Altace
Verapamil • Calan, Verelan
1. Fava M, Dirks B, Freeman M, et al. A phase 2, randomized, double-blind, placebo-controlled study of adjunctive pimavanserin in patients with major depressive disorder and an inadequate response to therapy (CLARITY). J Clin Psychiatry. 2019;80(6):19m12928.
2. Jha MK, Fava M, Freeman MP, et al. Effect of adjunctive pimavanserin on sleep/wakefulness in patients with major depressive disorder: secondary analysis from CLARITY. J Clin Psychiatry. 2020;82(1):20m13425.
3. ACADIA Pharmaceuticals announces top-line results from the Phase 3 CLARITY study evaluating pimavanserin for the adjunctive treatment of major depressive disorder. News release. Acadia Pharmaceuticals Inc. Published July 20, 2020. https://ir.acadia-pharm.com/news-releases/news-release-details/acadia-pharmaceuticals-announces-top-line-results-phase-3-0
4. Kessing LV, Rytgaard HC, Ekstrom CT, et al. Antihypertensive drugs and risk of depression: a nationwide population-based study. Hypertension. 2020;76(4):1263-1279.
5. Williams NR, Heifets BD, Blasey C, et al. Attenuation of antidepressant effects of ketamine by opioid receptor antagonism. Am J Psychiatry. 2018;175(12):1205-1215.
6. Han Y, Chen J, Zou D, et al. Efficacy of ketamine in the rapid treatment of major depressive disorder: a meta-analysis of randomized, double-blind, placebo-controlled studies. Neuropsychiatr Dis Treat. 2016;12:2859-2867.
7. Wan LB, Levitch CF, Perez AM, et al. Ketamine safety and tolerability in clinical trials for treatment-resistant depression. J Clin Psychiatry. 2015;76(3):247-252.
8. Hasselmann, H. Scopolamine and depression: a role for muscarinic antagonism? CNS Neurol Disord Drug Targets. 2014;13(4):673-683.
9. Drevets WC, Zarate CA Jr, Furey ML. Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol Psychiatry. 2013;73(12):1156-1163.
10. Stearns TP, Shad MU, Guzman GC. Glabellar botulinum toxin injections in major depressive disorder: a critical review. Prim Care Companion CNS Disord. 2018;20(5): 18r02298.
11. Block TS, Kushner H, Kalin N, et al. Combined analysis of mifepristone for psychotic depression: plasma levels associated with clinical response. Biol Psychiatry. 2018;84(1):46-54.
12. Rubinow DR, Johnson SL, Schmidt PJ, et al. Efficacy of estradiol in perimenopausal depression: so much promise and so few answers. Depress Anxiety. 2015;32(8):539-549.
13. Schmidt PJ, Ben Dor R, Martinez PE, et al. Effects of estradiol withdrawal on mood in women with past perimenopausal depression: a randomized clinical trial. JAMA Psychiatry. 2015;72(7):714-726.
14. Gordon JL, Rubinow DR, Eisenlohr-Moul TA, et al. Efficacy of transdermal estradiol and micronized progesterone in the prevention of depressive symptoms in the menopause transition: a randomized clinical trial. JAMA Psychiatry. 2018;75(2):149-157.
15. Fava M, Thase ME, Trivedi MH, et al. Opioid system modulation with buprenorphine/samidorphan combination for major depressive disorder: two randomized controlled studies. Mol Psychiatry. 2020;25(7):1580-1591.
16. Fernandes BS, Dean OM, Dodd S, et al. N-Acetylcysteine in depressive symptoms and functionality: a systematic review and meta-analysis. J Clin Psychiatry. 2016;77(4):e457-466.
Presently, FDA-approved first-line treatments and standard adjunctive strategies (eg, lithium, thyroid supplementation, stimulants, second-generation antipsychotics) for major depressive disorder (MDD) often produce less-than-desired outcomes while carrying a potentially substantial safety and tolerability burden. The lack of clinically useful and individual-based biomarkers (eg, genetic, neurophysiological, imaging) is a major obstacle to enhancing treatment efficacy and/or decreasing associated adverse effects (AEs). While the discovery of such tools is being aggressively pursued and ultimately will facilitate a more precision-based choice of therapy, empirical strategies remain our primary approach.
In controlled trials, several nontraditional treatments used primarily as adjuncts to standard antidepressants have shown promise. These include “repurposed” (off-label) medications, herbal/nutraceuticals, anti-inflammatory/immune system therapies, device-based treatments, and other alternative approaches.
Importantly, some nontraditional treatments also demonstrate AEs (Table1-16). With a careful consideration of the risk/benefit balance, this article reviews some of the better-studied treatment options for patients with treatment-resistant depression (TRD). In Part 1, we will examine off-label medications. In Part 2, we will review other nontraditional approaches to TRD, including herbal/nutraceuticals, anti-inflammatory/immune system therapies, device-based treatments, and other alternative approaches.
We believe this review will help clinicians who need to formulate a different approach after their patient with depression is not helped by traditional first-, second-, and third-line treatments. The potential options discussed in Part 1 of this article are categorized based on their putative mechanism of action (MOA) for depression.
Serotonergic and noradrenergic strategies
Pimavanserin is FDA-approved for treatment of Parkinson’s psychosis. Its potential MOA as an adjunctive strategy for MDD may involve 5-HT2A antagonist and inverse agonist receptor activity, as well as lesser effects at the 5-HT2Creceptor.
A 2-stage, 5-week randomized controlled trial (RCT) (CLARITY; N = 207) found adjunctive pimavanserin (34 mg/d) produced a robust antidepressant effect vs placebo in patients whose depression did not respond to selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs).1 Furthermore, a secondary analysis of the data suggested that pimavanserin also improved sleepiness (P < .0003) and daily functioning (P < .014) at Week 5.2
Unfortunately, two 6-week, Phase III RCTs (CLARITY-2 and -3; N = 298) did not find a statistically significant difference between active treatment and placebo. This was based on change in the primary outcome measure (Hamilton Depression Rating Scale-17 score) when adjunctive pimavanserin (34 mg/d) was added to an SSRI or SNRI in patients with TRD.3 There was, however, a significant difference favoring active treatment over placebo based on the Clinical Global Impression–Severity score.
Continue to: In these trials...
In these trials, pimavanserin was generally well-tolerated. The most common AEs were dry mouth, nausea, and headache. Pimavanserin has minimal activity at norepinephrine, dopamine, histamine, or acetylcholine receptors, thus avoiding AEs associated with these receptor interactions.
Given the mixed efficacy results of existing trials, further studies are needed to clarify this agent’s overall risk/benefit in the context of TRD.
Antihypertensive medications
Emerging data suggest that some beta-adrenergic blockers, angiotensin-inhibiting agents, and calcium antagonists are associated with a decreased incidence of depression. A large 2020 study (N = 3,747,190) used population-based Danish registries (2005 to 2015) to evaluate if any of the 41 most commonly prescribed antihypertensive medications were associated with the diagnosis of depressive disorder or use of antidepressants.4 These researchers found that enalapril, ramipril, amlodipine, propranolol, atenolol, bisoprolol, carvedilol (P < .001), and verapamil (P < .004) were strongly associated with a decreased risk of depression.4
Adverse effects across these different classes of antihypertensives are well characterized, can be substantial, and commonly are related to their impact on cardiovascular function (eg, hypotension). Clinically, these agents may be potential adjuncts for patients with TRD who need antihypertensive therapy. Their use and the choice of specific agent should only be determined in consultation with the patient’s primary care physician (PCP) or appropriate specialist.
Glutamatergic strategies
Ketamine is a dissociative anesthetic and analgesic. Its MOA for treating depression appears to occur primarily through antagonist activity at the N-methyl-
Continue to: Many published studies...
Many published studies and reviews have described ketamine’s role for treating MDD. Several studies have reported that low-dose (0.5 mg/kg) IV ketamine infusions can rapidly attenuate severe episodes of MDD as well as associated suicidality. For example, a meta-analysis of 9 RCTs (N = 368) comparing ketamine to placebo for acute treatment of unipolar and bipolar depression reported superior therapeutic effects with active treatment at 24 hours, 72 hours, and 7 days.6 The response and remission rates for ketamine were 52% and 21% at 24 hours; 48% and 24% at 72 hours; and 40% and 26% at 7 days, respectively.6
The most commonly reported AEs during the 4 hours after ketamine infusion included7:
- drowsiness, dizziness, poor coordination
- blurred vision, feeling strange or unreal
- hemodynamic changes (approximately 33%)
- small but significant (P < .05) increases in psychotomimetic and dissociative symptoms.
Because some individuals use ketamine recreationally, this agent also carries the risk of abuse.
Research is ongoing on strategies for long-term maintenance ketamine treatment, and the results of both short- and long-term trials will require careful scrutiny to better assess this agent’s safety and tolerability. Clinicians should first consider esketamine—the S-enantiomer of ketamine—because an intranasal formulation of this agent is FDA-approved for treating patients with TRD or MDD with suicidality when administered in a Risk Evaluation and Mitigation Strategy–certified setting.
Cholinergic strategies
Scopolamine is a potent muscarinic receptor antagonist used to prevent nausea and vomiting caused by motion sickness or medications used during surgery. Its use for MDD is based on the theory that muscarinic receptors may be hypersensitive in mood disorders.
Continue to: Several double-blind RCTs...
Several double-blind RCTs of patients with unipolar or bipolar depression that used 3 pulsed IV infusions (4.0 mcg/kg) over 15 minutes found a rapid, robust antidepressant effect with scopolamine vs placebo.8,9 The oral formulation might also be effective, but would not have a rapid onset.
Common adverse effects of scopolamine include agitation, dry mouth, urinary retention, and cognitive clouding. Given scopolamine’s substantial AE profile, it should be considered only for patients with TRD who could also benefit from the oral formulation for the medical indications noted above, should generally be avoided in older patients, and should be prescribed in consultation with the patient’s PCP.
Botulinum toxin. This neurotoxin inhibits acetylcholine release. It is used to treat disorders characterized by abnormal muscular contraction, such as strabismus, blepharospasm, and chronic pain syndromes. Its MOA for depression may involve its paralytic effects after injection into the glabella forehead muscle (based on the facial feedback hypothesis), as well as modulation of neurotransmitters implicated in the pathophysiology of depression.
In several small trials, injectable botulinum toxin type A (BTA) (29 units) demonstrated antidepressant effects. A recent review that considered 6 trials (N = 235; 4 of the 6 studies were RCTs, 3 of which were rated as high quality) concluded that BTA may be a promising treatment for MDD.10 Limitations of this review included lack of a priori hypotheses, small sample sizes, gender bias, and difficulty in blinding.
In clinical trials, the most common AEs included local irritation at the injection site and transient headache. This agent’s relatively mild AE profile and possible overlap when used for some of the medical indications noted above opens its potential use as an adjunct in patients with comorbid TRD.
Continue to: Endocrine strategies
Endocrine strategies
Mifepristone (RU486). This anti-glucocorticoid receptor antagonist is used as an abortifacient. Based on the theory that hyperactivity of the hypothalamic-pituitary-adrenal axis is implicated in the pathophysiology of MDD with psychotic features (psychotic depression), this agent has been studied as a treatment for this indication.
An analysis of 5 double-blind RCTs (N = 1,460) found that 7 days of mifepristone, 1,200 mg/d, was superior to placebo (P < .004) in reducing psychotic symptoms of depression.11 Plasma concentrations ≥1,600 ng/mL may be required to maximize benefit.11
Overall, this agent demonstrated a good safety profile in clinical trials, with treatment-emergent AEs reported in 556 (66.7%) patients who received mifepristone vs 386 (61.6%) patients who received placebo.11 Common AEs included gastrointestinal (GI) symptoms, headache, and dizziness. However, 3 deaths occurred: 2 patients who received mifepristone and 1 patient who received placebo. Given this potential for a fatal outcome, clinicians should first consider prescribing an adjunctive antipsychotic agent or electroconvulsive therapy.
Estrogens. These hormones are important for sexual and reproductive development and are used to treat various sexual/reproductive disorders, primarily in women. Their role in treating depression is based on the observation that perimenopause is accompanied by an increased risk of new and recurrent depression coincident with declining ovarian function.
Evidence supports the antidepressant efficacy of transdermal estradiol plus progesterone for perimenopausal depression, but not for postmenopausal depression.12-14 However, estrogens carry significant risks that must be carefully considered in relationship to their potential benefits. These risks include:
- vaginal bleeding, dysmenorrhea
- fibroid enlargement
- galactorrhea
- ovarian cancer, endometrial cancer, breast cancer
- deep vein thrombosis, pulmonary embolism
- hypertension, chest pain, myocardial infarction, stroke.
Continue to: The use of estrogens...
The use of estrogens as an adjunctive therapy for women with treatment-resistant perimenopausal depression should only be undertaken when standard strategies have failed, and in consultation with an endocrine specialist who can monitor for potentially serious AEs.
Opioid medications
Buprenorphine is used to treat opioid use disorder (OUD) as well as acute and chronic pain. The opioid system is involved in the regulation of mood and may be an appropriate target for novel antidepressants. The use of buprenorphine in combination with samidorphan (a preferential mu-opioid receptor antagonist) has shown initial promise for TRD while minimizing abuse potential.
Although earlier results were mixed, a pooled analysis of 2 recent large RCTs (N = 760) of patients with MDD who had not responded to antidepressants reported greater reduction in Montgomery-Åsberg Depression Rating Scale scores from baseline for active treatment (buprenorphine/samidorphan; 2 mg/2 mg) vs placebo at multiple timepoints, including end of treatment (-1.8; P < .010).15
The most common AEs included nausea, constipation, dizziness, vomiting, somnolence, fatigue, and sedation. There was minimal evidence of abuse, dependence, or opioid withdrawal. Due to the opioid crisis in the United States, the resulting relaxation of regulations regarding prescribing buprenorphine, and the high rates of depression among patients with OUD, buprenorphine/samidorphan, which is an investigational agent that is not FDA-approved, may be particularly helpful for patients with OUD who also experience comorbid TRD.
Antioxidant agents
N-acetylcysteine (NAC) is an amino acid that can treat acetaminophen toxicity and moderate hepatic damage by increasing glutathione levels. Glutathione is also the primary antioxidant in the CNS. NAC may protect against oxidative stress, chelate heavy metals, reduce inflammation, protect against mitochondrial dysfunction, inhibit apoptosis, and enhance neurogenesis, all potential pathophysiological processes that may contribute to depression.16
Continue to: A systematic review...
A systematic review and meta-analysis of 5 RCTs (N = 574) considered patients with various depression diagnoses who were randomized to adjunctive NAC, 1,000 mg twice a day, or placebo. Over 12 to 24 weeks, there was a significantly greater improvement in mood symptoms and functionality with NAC vs placebo.16
Overall, NAC was well-tolerated. The most common AEs were GI symptoms, musculoskeletal complaints, decreased energy, and headache. While NAC has been touted as a potential adjunct therapy for several psychiatric disorders, including TRD, the evidence for benefit remains limited. Given its favorable AE profile, however, and over-the-counter availability, it remains an option for select patients. It is important to ask patients if they are already taking NAC.
Options beyond off-label medications
There are a multitude of options available for addressing TRD. Many FDA-approved medications are repurposed and prescribed off-label for other indications when the risk/benefit balance is favorable. In Part 1 of this article, we reviewed several off-label medications that have supportive controlled data for treating TRD. In Part 2, we will review other nontraditional therapies for TRD, including herbal/nutraceuticals, anti-inflammatory/immune system therapies, device-based treatments, and other alternative approaches.
Bottom Line
Off-label medications that may offer benefit for patients with treatment-resistant depression (TRD) include pimavanserin, antihypertensive agents, ketamine, scopolamine, botulinum toxin, mifepristone, estrogens, buprenorphine, and N-acetylcysteine. Although some evidence supports use of these agents as adjuncts for TRD, an individualized risk/benefit analysis is required.
Related Resource
- Joshi KG, Frierson RL. Off-label prescribing: How to limit your liability. Current Psychiatry. 2020;19(9):12,39.
Drug Brand Names
Amlodipine • Katerzia, Norvasc
Atenolol • Tenormin
Bisoprolol • Zebeta
Buprenorphine • Sublocade, Subutex
Carvedilol • Coreg
Enalapril • Vasotec
Esketamine • Spravato
Estradiol transdermal • Estraderm
Ketamine • Ketalar
Mifepristone • Mifeprex
Pimavanserin • Nuplazid
Progesterone • Prometrium
Propranolol • Inderal
Ramipril • Altace
Verapamil • Calan, Verelan
Presently, FDA-approved first-line treatments and standard adjunctive strategies (eg, lithium, thyroid supplementation, stimulants, second-generation antipsychotics) for major depressive disorder (MDD) often produce less-than-desired outcomes while carrying a potentially substantial safety and tolerability burden. The lack of clinically useful and individual-based biomarkers (eg, genetic, neurophysiological, imaging) is a major obstacle to enhancing treatment efficacy and/or decreasing associated adverse effects (AEs). While the discovery of such tools is being aggressively pursued and ultimately will facilitate a more precision-based choice of therapy, empirical strategies remain our primary approach.
In controlled trials, several nontraditional treatments used primarily as adjuncts to standard antidepressants have shown promise. These include “repurposed” (off-label) medications, herbal/nutraceuticals, anti-inflammatory/immune system therapies, device-based treatments, and other alternative approaches.
Importantly, some nontraditional treatments also demonstrate AEs (Table1-16). With a careful consideration of the risk/benefit balance, this article reviews some of the better-studied treatment options for patients with treatment-resistant depression (TRD). In Part 1, we will examine off-label medications. In Part 2, we will review other nontraditional approaches to TRD, including herbal/nutraceuticals, anti-inflammatory/immune system therapies, device-based treatments, and other alternative approaches.
We believe this review will help clinicians who need to formulate a different approach after their patient with depression is not helped by traditional first-, second-, and third-line treatments. The potential options discussed in Part 1 of this article are categorized based on their putative mechanism of action (MOA) for depression.
Serotonergic and noradrenergic strategies
Pimavanserin is FDA-approved for treatment of Parkinson’s psychosis. Its potential MOA as an adjunctive strategy for MDD may involve 5-HT2A antagonist and inverse agonist receptor activity, as well as lesser effects at the 5-HT2Creceptor.
A 2-stage, 5-week randomized controlled trial (RCT) (CLARITY; N = 207) found adjunctive pimavanserin (34 mg/d) produced a robust antidepressant effect vs placebo in patients whose depression did not respond to selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs).1 Furthermore, a secondary analysis of the data suggested that pimavanserin also improved sleepiness (P < .0003) and daily functioning (P < .014) at Week 5.2
Unfortunately, two 6-week, Phase III RCTs (CLARITY-2 and -3; N = 298) did not find a statistically significant difference between active treatment and placebo. This was based on change in the primary outcome measure (Hamilton Depression Rating Scale-17 score) when adjunctive pimavanserin (34 mg/d) was added to an SSRI or SNRI in patients with TRD.3 There was, however, a significant difference favoring active treatment over placebo based on the Clinical Global Impression–Severity score.
Continue to: In these trials...
In these trials, pimavanserin was generally well-tolerated. The most common AEs were dry mouth, nausea, and headache. Pimavanserin has minimal activity at norepinephrine, dopamine, histamine, or acetylcholine receptors, thus avoiding AEs associated with these receptor interactions.
Given the mixed efficacy results of existing trials, further studies are needed to clarify this agent’s overall risk/benefit in the context of TRD.
Antihypertensive medications
Emerging data suggest that some beta-adrenergic blockers, angiotensin-inhibiting agents, and calcium antagonists are associated with a decreased incidence of depression. A large 2020 study (N = 3,747,190) used population-based Danish registries (2005 to 2015) to evaluate if any of the 41 most commonly prescribed antihypertensive medications were associated with the diagnosis of depressive disorder or use of antidepressants.4 These researchers found that enalapril, ramipril, amlodipine, propranolol, atenolol, bisoprolol, carvedilol (P < .001), and verapamil (P < .004) were strongly associated with a decreased risk of depression.4
Adverse effects across these different classes of antihypertensives are well characterized, can be substantial, and commonly are related to their impact on cardiovascular function (eg, hypotension). Clinically, these agents may be potential adjuncts for patients with TRD who need antihypertensive therapy. Their use and the choice of specific agent should only be determined in consultation with the patient’s primary care physician (PCP) or appropriate specialist.
Glutamatergic strategies
Ketamine is a dissociative anesthetic and analgesic. Its MOA for treating depression appears to occur primarily through antagonist activity at the N-methyl-
Continue to: Many published studies...
Many published studies and reviews have described ketamine’s role for treating MDD. Several studies have reported that low-dose (0.5 mg/kg) IV ketamine infusions can rapidly attenuate severe episodes of MDD as well as associated suicidality. For example, a meta-analysis of 9 RCTs (N = 368) comparing ketamine to placebo for acute treatment of unipolar and bipolar depression reported superior therapeutic effects with active treatment at 24 hours, 72 hours, and 7 days.6 The response and remission rates for ketamine were 52% and 21% at 24 hours; 48% and 24% at 72 hours; and 40% and 26% at 7 days, respectively.6
The most commonly reported AEs during the 4 hours after ketamine infusion included7:
- drowsiness, dizziness, poor coordination
- blurred vision, feeling strange or unreal
- hemodynamic changes (approximately 33%)
- small but significant (P < .05) increases in psychotomimetic and dissociative symptoms.
Because some individuals use ketamine recreationally, this agent also carries the risk of abuse.
Research is ongoing on strategies for long-term maintenance ketamine treatment, and the results of both short- and long-term trials will require careful scrutiny to better assess this agent’s safety and tolerability. Clinicians should first consider esketamine—the S-enantiomer of ketamine—because an intranasal formulation of this agent is FDA-approved for treating patients with TRD or MDD with suicidality when administered in a Risk Evaluation and Mitigation Strategy–certified setting.
Cholinergic strategies
Scopolamine is a potent muscarinic receptor antagonist used to prevent nausea and vomiting caused by motion sickness or medications used during surgery. Its use for MDD is based on the theory that muscarinic receptors may be hypersensitive in mood disorders.
Continue to: Several double-blind RCTs...
Several double-blind RCTs of patients with unipolar or bipolar depression that used 3 pulsed IV infusions (4.0 mcg/kg) over 15 minutes found a rapid, robust antidepressant effect with scopolamine vs placebo.8,9 The oral formulation might also be effective, but would not have a rapid onset.
Common adverse effects of scopolamine include agitation, dry mouth, urinary retention, and cognitive clouding. Given scopolamine’s substantial AE profile, it should be considered only for patients with TRD who could also benefit from the oral formulation for the medical indications noted above, should generally be avoided in older patients, and should be prescribed in consultation with the patient’s PCP.
Botulinum toxin. This neurotoxin inhibits acetylcholine release. It is used to treat disorders characterized by abnormal muscular contraction, such as strabismus, blepharospasm, and chronic pain syndromes. Its MOA for depression may involve its paralytic effects after injection into the glabella forehead muscle (based on the facial feedback hypothesis), as well as modulation of neurotransmitters implicated in the pathophysiology of depression.
In several small trials, injectable botulinum toxin type A (BTA) (29 units) demonstrated antidepressant effects. A recent review that considered 6 trials (N = 235; 4 of the 6 studies were RCTs, 3 of which were rated as high quality) concluded that BTA may be a promising treatment for MDD.10 Limitations of this review included lack of a priori hypotheses, small sample sizes, gender bias, and difficulty in blinding.
In clinical trials, the most common AEs included local irritation at the injection site and transient headache. This agent’s relatively mild AE profile and possible overlap when used for some of the medical indications noted above opens its potential use as an adjunct in patients with comorbid TRD.
Continue to: Endocrine strategies
Endocrine strategies
Mifepristone (RU486). This anti-glucocorticoid receptor antagonist is used as an abortifacient. Based on the theory that hyperactivity of the hypothalamic-pituitary-adrenal axis is implicated in the pathophysiology of MDD with psychotic features (psychotic depression), this agent has been studied as a treatment for this indication.
An analysis of 5 double-blind RCTs (N = 1,460) found that 7 days of mifepristone, 1,200 mg/d, was superior to placebo (P < .004) in reducing psychotic symptoms of depression.11 Plasma concentrations ≥1,600 ng/mL may be required to maximize benefit.11
Overall, this agent demonstrated a good safety profile in clinical trials, with treatment-emergent AEs reported in 556 (66.7%) patients who received mifepristone vs 386 (61.6%) patients who received placebo.11 Common AEs included gastrointestinal (GI) symptoms, headache, and dizziness. However, 3 deaths occurred: 2 patients who received mifepristone and 1 patient who received placebo. Given this potential for a fatal outcome, clinicians should first consider prescribing an adjunctive antipsychotic agent or electroconvulsive therapy.
Estrogens. These hormones are important for sexual and reproductive development and are used to treat various sexual/reproductive disorders, primarily in women. Their role in treating depression is based on the observation that perimenopause is accompanied by an increased risk of new and recurrent depression coincident with declining ovarian function.
Evidence supports the antidepressant efficacy of transdermal estradiol plus progesterone for perimenopausal depression, but not for postmenopausal depression.12-14 However, estrogens carry significant risks that must be carefully considered in relationship to their potential benefits. These risks include:
- vaginal bleeding, dysmenorrhea
- fibroid enlargement
- galactorrhea
- ovarian cancer, endometrial cancer, breast cancer
- deep vein thrombosis, pulmonary embolism
- hypertension, chest pain, myocardial infarction, stroke.
Continue to: The use of estrogens...
The use of estrogens as an adjunctive therapy for women with treatment-resistant perimenopausal depression should only be undertaken when standard strategies have failed, and in consultation with an endocrine specialist who can monitor for potentially serious AEs.
Opioid medications
Buprenorphine is used to treat opioid use disorder (OUD) as well as acute and chronic pain. The opioid system is involved in the regulation of mood and may be an appropriate target for novel antidepressants. The use of buprenorphine in combination with samidorphan (a preferential mu-opioid receptor antagonist) has shown initial promise for TRD while minimizing abuse potential.
Although earlier results were mixed, a pooled analysis of 2 recent large RCTs (N = 760) of patients with MDD who had not responded to antidepressants reported greater reduction in Montgomery-Åsberg Depression Rating Scale scores from baseline for active treatment (buprenorphine/samidorphan; 2 mg/2 mg) vs placebo at multiple timepoints, including end of treatment (-1.8; P < .010).15
The most common AEs included nausea, constipation, dizziness, vomiting, somnolence, fatigue, and sedation. There was minimal evidence of abuse, dependence, or opioid withdrawal. Due to the opioid crisis in the United States, the resulting relaxation of regulations regarding prescribing buprenorphine, and the high rates of depression among patients with OUD, buprenorphine/samidorphan, which is an investigational agent that is not FDA-approved, may be particularly helpful for patients with OUD who also experience comorbid TRD.
Antioxidant agents
N-acetylcysteine (NAC) is an amino acid that can treat acetaminophen toxicity and moderate hepatic damage by increasing glutathione levels. Glutathione is also the primary antioxidant in the CNS. NAC may protect against oxidative stress, chelate heavy metals, reduce inflammation, protect against mitochondrial dysfunction, inhibit apoptosis, and enhance neurogenesis, all potential pathophysiological processes that may contribute to depression.16
Continue to: A systematic review...
A systematic review and meta-analysis of 5 RCTs (N = 574) considered patients with various depression diagnoses who were randomized to adjunctive NAC, 1,000 mg twice a day, or placebo. Over 12 to 24 weeks, there was a significantly greater improvement in mood symptoms and functionality with NAC vs placebo.16
Overall, NAC was well-tolerated. The most common AEs were GI symptoms, musculoskeletal complaints, decreased energy, and headache. While NAC has been touted as a potential adjunct therapy for several psychiatric disorders, including TRD, the evidence for benefit remains limited. Given its favorable AE profile, however, and over-the-counter availability, it remains an option for select patients. It is important to ask patients if they are already taking NAC.
Options beyond off-label medications
There are a multitude of options available for addressing TRD. Many FDA-approved medications are repurposed and prescribed off-label for other indications when the risk/benefit balance is favorable. In Part 1 of this article, we reviewed several off-label medications that have supportive controlled data for treating TRD. In Part 2, we will review other nontraditional therapies for TRD, including herbal/nutraceuticals, anti-inflammatory/immune system therapies, device-based treatments, and other alternative approaches.
Bottom Line
Off-label medications that may offer benefit for patients with treatment-resistant depression (TRD) include pimavanserin, antihypertensive agents, ketamine, scopolamine, botulinum toxin, mifepristone, estrogens, buprenorphine, and N-acetylcysteine. Although some evidence supports use of these agents as adjuncts for TRD, an individualized risk/benefit analysis is required.
Related Resource
- Joshi KG, Frierson RL. Off-label prescribing: How to limit your liability. Current Psychiatry. 2020;19(9):12,39.
Drug Brand Names
Amlodipine • Katerzia, Norvasc
Atenolol • Tenormin
Bisoprolol • Zebeta
Buprenorphine • Sublocade, Subutex
Carvedilol • Coreg
Enalapril • Vasotec
Esketamine • Spravato
Estradiol transdermal • Estraderm
Ketamine • Ketalar
Mifepristone • Mifeprex
Pimavanserin • Nuplazid
Progesterone • Prometrium
Propranolol • Inderal
Ramipril • Altace
Verapamil • Calan, Verelan
1. Fava M, Dirks B, Freeman M, et al. A phase 2, randomized, double-blind, placebo-controlled study of adjunctive pimavanserin in patients with major depressive disorder and an inadequate response to therapy (CLARITY). J Clin Psychiatry. 2019;80(6):19m12928.
2. Jha MK, Fava M, Freeman MP, et al. Effect of adjunctive pimavanserin on sleep/wakefulness in patients with major depressive disorder: secondary analysis from CLARITY. J Clin Psychiatry. 2020;82(1):20m13425.
3. ACADIA Pharmaceuticals announces top-line results from the Phase 3 CLARITY study evaluating pimavanserin for the adjunctive treatment of major depressive disorder. News release. Acadia Pharmaceuticals Inc. Published July 20, 2020. https://ir.acadia-pharm.com/news-releases/news-release-details/acadia-pharmaceuticals-announces-top-line-results-phase-3-0
4. Kessing LV, Rytgaard HC, Ekstrom CT, et al. Antihypertensive drugs and risk of depression: a nationwide population-based study. Hypertension. 2020;76(4):1263-1279.
5. Williams NR, Heifets BD, Blasey C, et al. Attenuation of antidepressant effects of ketamine by opioid receptor antagonism. Am J Psychiatry. 2018;175(12):1205-1215.
6. Han Y, Chen J, Zou D, et al. Efficacy of ketamine in the rapid treatment of major depressive disorder: a meta-analysis of randomized, double-blind, placebo-controlled studies. Neuropsychiatr Dis Treat. 2016;12:2859-2867.
7. Wan LB, Levitch CF, Perez AM, et al. Ketamine safety and tolerability in clinical trials for treatment-resistant depression. J Clin Psychiatry. 2015;76(3):247-252.
8. Hasselmann, H. Scopolamine and depression: a role for muscarinic antagonism? CNS Neurol Disord Drug Targets. 2014;13(4):673-683.
9. Drevets WC, Zarate CA Jr, Furey ML. Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol Psychiatry. 2013;73(12):1156-1163.
10. Stearns TP, Shad MU, Guzman GC. Glabellar botulinum toxin injections in major depressive disorder: a critical review. Prim Care Companion CNS Disord. 2018;20(5): 18r02298.
11. Block TS, Kushner H, Kalin N, et al. Combined analysis of mifepristone for psychotic depression: plasma levels associated with clinical response. Biol Psychiatry. 2018;84(1):46-54.
12. Rubinow DR, Johnson SL, Schmidt PJ, et al. Efficacy of estradiol in perimenopausal depression: so much promise and so few answers. Depress Anxiety. 2015;32(8):539-549.
13. Schmidt PJ, Ben Dor R, Martinez PE, et al. Effects of estradiol withdrawal on mood in women with past perimenopausal depression: a randomized clinical trial. JAMA Psychiatry. 2015;72(7):714-726.
14. Gordon JL, Rubinow DR, Eisenlohr-Moul TA, et al. Efficacy of transdermal estradiol and micronized progesterone in the prevention of depressive symptoms in the menopause transition: a randomized clinical trial. JAMA Psychiatry. 2018;75(2):149-157.
15. Fava M, Thase ME, Trivedi MH, et al. Opioid system modulation with buprenorphine/samidorphan combination for major depressive disorder: two randomized controlled studies. Mol Psychiatry. 2020;25(7):1580-1591.
16. Fernandes BS, Dean OM, Dodd S, et al. N-Acetylcysteine in depressive symptoms and functionality: a systematic review and meta-analysis. J Clin Psychiatry. 2016;77(4):e457-466.
1. Fava M, Dirks B, Freeman M, et al. A phase 2, randomized, double-blind, placebo-controlled study of adjunctive pimavanserin in patients with major depressive disorder and an inadequate response to therapy (CLARITY). J Clin Psychiatry. 2019;80(6):19m12928.
2. Jha MK, Fava M, Freeman MP, et al. Effect of adjunctive pimavanserin on sleep/wakefulness in patients with major depressive disorder: secondary analysis from CLARITY. J Clin Psychiatry. 2020;82(1):20m13425.
3. ACADIA Pharmaceuticals announces top-line results from the Phase 3 CLARITY study evaluating pimavanserin for the adjunctive treatment of major depressive disorder. News release. Acadia Pharmaceuticals Inc. Published July 20, 2020. https://ir.acadia-pharm.com/news-releases/news-release-details/acadia-pharmaceuticals-announces-top-line-results-phase-3-0
4. Kessing LV, Rytgaard HC, Ekstrom CT, et al. Antihypertensive drugs and risk of depression: a nationwide population-based study. Hypertension. 2020;76(4):1263-1279.
5. Williams NR, Heifets BD, Blasey C, et al. Attenuation of antidepressant effects of ketamine by opioid receptor antagonism. Am J Psychiatry. 2018;175(12):1205-1215.
6. Han Y, Chen J, Zou D, et al. Efficacy of ketamine in the rapid treatment of major depressive disorder: a meta-analysis of randomized, double-blind, placebo-controlled studies. Neuropsychiatr Dis Treat. 2016;12:2859-2867.
7. Wan LB, Levitch CF, Perez AM, et al. Ketamine safety and tolerability in clinical trials for treatment-resistant depression. J Clin Psychiatry. 2015;76(3):247-252.
8. Hasselmann, H. Scopolamine and depression: a role for muscarinic antagonism? CNS Neurol Disord Drug Targets. 2014;13(4):673-683.
9. Drevets WC, Zarate CA Jr, Furey ML. Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol Psychiatry. 2013;73(12):1156-1163.
10. Stearns TP, Shad MU, Guzman GC. Glabellar botulinum toxin injections in major depressive disorder: a critical review. Prim Care Companion CNS Disord. 2018;20(5): 18r02298.
11. Block TS, Kushner H, Kalin N, et al. Combined analysis of mifepristone for psychotic depression: plasma levels associated with clinical response. Biol Psychiatry. 2018;84(1):46-54.
12. Rubinow DR, Johnson SL, Schmidt PJ, et al. Efficacy of estradiol in perimenopausal depression: so much promise and so few answers. Depress Anxiety. 2015;32(8):539-549.
13. Schmidt PJ, Ben Dor R, Martinez PE, et al. Effects of estradiol withdrawal on mood in women with past perimenopausal depression: a randomized clinical trial. JAMA Psychiatry. 2015;72(7):714-726.
14. Gordon JL, Rubinow DR, Eisenlohr-Moul TA, et al. Efficacy of transdermal estradiol and micronized progesterone in the prevention of depressive symptoms in the menopause transition: a randomized clinical trial. JAMA Psychiatry. 2018;75(2):149-157.
15. Fava M, Thase ME, Trivedi MH, et al. Opioid system modulation with buprenorphine/samidorphan combination for major depressive disorder: two randomized controlled studies. Mol Psychiatry. 2020;25(7):1580-1591.
16. Fernandes BS, Dean OM, Dodd S, et al. N-Acetylcysteine in depressive symptoms and functionality: a systematic review and meta-analysis. J Clin Psychiatry. 2016;77(4):e457-466.
