User login
Finger-stick lithium test
A new, FDA-approved in-office lithium test (Table) can eliminate the inconvenience and fallibility of testing venous blood samples that often discourage lithium use. The test, which measures lithium in capillary blood drawn from a finger stick, has shown reliability when compared in clinical trials with established testing methods.
Why finger-stick testing?
Periodically monitoring serum or plasma lithium minimizes side effects and toxicity, maintains therapeutic dosing, and ensures treatment adherence. Laboratories generally use flame photometry, atomic absorption (AA) spectrophotometry, or ion-selective electrode analysis to measure lithium in blood drawn via venipuncture. A colorimetric assay is also available.1
Table
Lithium fingerstick test: Fast facts
| Brand name: |
| InstaRead Lithium System |
| Indication: |
| Testing plasma lithium levels in-office |
| Manufacturer: |
| ReliaLAB |
| Recommended use: |
| Testing plasma lithium levels 12 hours after dosing; repeat test after 5 minutes to confirm abnormal reading |
| Reimbursement information: |
| 1-866-467-8273 or www.relialab.com/Reimbursement.html |
For years, researchers have investigated alternatives to venipuncture lithium testing. Aside from being inconvenient, venipuncture draws can increase risk of excessive bleeding, hematoma, infection, vasovagal syncope, and multiple punctures to locate a vein. In some cases:
- psychiatrists wait 2 or more days for a laboratory to return results
- patients forget to have blood drawn before the office visit
- samples are incorrectly timed in relation to the last dose
- results are filed away unnoticed
- or the psychiatrist needs to call the patient 3 days or so after the visit to discuss an abnormal reading.
With the new in-office test, clinicians can ensure they will obtain a valid blood sample in minutes, 12 hours after dosing. Psychiatrists then can immediately discuss the result with patients, perform a repeat test 5 minutes later to check an abnormal reading, and counsel patients on raising low lithium levels. This instant feedback can powerfully reinforce a physician’s advice and promote treatment adherence.2
How it works
A 50-μl blood sample is drawn via finger stick and converted to plasma in a lectin-coated membrane separator. The clinician then adds 0.2 μl of the plasma to a micro-cuvette containing a colorimetric reagent that is photometrically analyzed for lithium. The test takes 5 minutes or less (Figure).
The assay has been shown to be sensitive to 0.1 mEq/L of lithium and linear between 0.1 and 2.5 mEq/L.3
Figure How finger-stick lithium test works
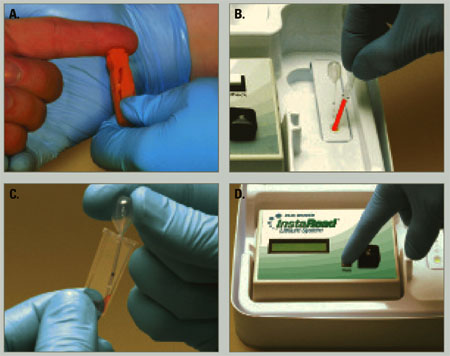
Clinician obtains blood sample (a) and empties it into a separator (b), which processes blood to plasma. Clinician then adds plasma to a reagent vial (c), which is inserted into a reader (d) to obtain a lithium level.
Reliability
In clinical trials during which patients were tested and retested, the colorimetric assay showed reliability when compared with:
- routine lithium spectrophotometry. Researchers compared venipuncture blood samples split for colorimetric and spectrophotometric testing
- atomic absorption spectrophotometry of venipuncture blood from psychiatric patients
- standard spectrophotometry of venipuncture samples to which a known amount of lithium was added.4
Colorimetric finger-stick testing also was compared with AA spectrophotometry testing of 88 matched venipuncture samples from 56 bipolar patients.5 Results were not identical, but most fingerstick results varied no more than±0.2 mEq/L from the AA results. Differences were positive and negative, indicating random variation between the two methods rather than systematic bias.
Clinical applicability
In-office finger-stick blood testing for lithium levels could improve quality of care for patients taking lithium.
The manufacturer, ReliaLAB, says the test costs $399, plus $264 for a refill kit containing 24 patient test packs. A certain volume of patients taking lithium would seem to be necessary to justify purchasing the instrument.
The test may be reimbursable under certain circumstances. ReliaLAB offers information on coding and reimbursement for in-office lithium monitoring (Table).
Also, because instant in-office creatinine and thyroid-stimulating hormone tests are not available, lithium therapy monitoring will still require laboratory visits when these tests are needed. Nonetheless, point-of-care plasma lithium level determination should improve convenience, compliance, and overall comprehensiveness of care.
Related resources
- Online information on in-office lithium test. www.relialab.com/Lith.html.
- Johnson FN. The origins of lithium therapy. Rev Contemp Pharmacother 1999;10:193-265.
Drug brand names
- Lithium • Eskalith, others
Disclosure
Dr. Jefferson reports no financial relationship with or proprietary interest in ReliaLAB.
1. Jefferson JW, Greist JH. Lithium. In: Sadock BJ, Sadock VA (eds). Comprehensive textbook of psychiatry, vol. 2 (8th ed). Philadelphia: Lippincott Williams & Wilkins; 2005;2839-51.
2. Srinivasan DP, Birch NJ. Instant lithium monitoring: A clinical revolution in the making. Br J Clin Pract 1996;50:386-88.
3. Glazer WM, Sonnenberg JG, Reinstein MJ, Akers RF. A novel, point-of-care test for lithium levels: Description and reliability. J Clin Psychiatry 2004;652-5.
4. Vrouwe EX, Luttge R, van den Berg A. Direct measurement of lithium in whole blood using microchip capillary electrophoresis with integrated conductivity detection. Electrophoresis 2004;25:1660-7.
5. Glazer WM, Sonnenberg J, Reinstein MJ. A novel, “point of care” test for lithium levels (poster presentation). Atlanta, GA: American Psychiatric Association annual meeting, 2005.
A new, FDA-approved in-office lithium test (Table) can eliminate the inconvenience and fallibility of testing venous blood samples that often discourage lithium use. The test, which measures lithium in capillary blood drawn from a finger stick, has shown reliability when compared in clinical trials with established testing methods.
Why finger-stick testing?
Periodically monitoring serum or plasma lithium minimizes side effects and toxicity, maintains therapeutic dosing, and ensures treatment adherence. Laboratories generally use flame photometry, atomic absorption (AA) spectrophotometry, or ion-selective electrode analysis to measure lithium in blood drawn via venipuncture. A colorimetric assay is also available.1
Table
Lithium fingerstick test: Fast facts
| Brand name: |
| InstaRead Lithium System |
| Indication: |
| Testing plasma lithium levels in-office |
| Manufacturer: |
| ReliaLAB |
| Recommended use: |
| Testing plasma lithium levels 12 hours after dosing; repeat test after 5 minutes to confirm abnormal reading |
| Reimbursement information: |
| 1-866-467-8273 or www.relialab.com/Reimbursement.html |
For years, researchers have investigated alternatives to venipuncture lithium testing. Aside from being inconvenient, venipuncture draws can increase risk of excessive bleeding, hematoma, infection, vasovagal syncope, and multiple punctures to locate a vein. In some cases:
- psychiatrists wait 2 or more days for a laboratory to return results
- patients forget to have blood drawn before the office visit
- samples are incorrectly timed in relation to the last dose
- results are filed away unnoticed
- or the psychiatrist needs to call the patient 3 days or so after the visit to discuss an abnormal reading.
With the new in-office test, clinicians can ensure they will obtain a valid blood sample in minutes, 12 hours after dosing. Psychiatrists then can immediately discuss the result with patients, perform a repeat test 5 minutes later to check an abnormal reading, and counsel patients on raising low lithium levels. This instant feedback can powerfully reinforce a physician’s advice and promote treatment adherence.2
How it works
A 50-μl blood sample is drawn via finger stick and converted to plasma in a lectin-coated membrane separator. The clinician then adds 0.2 μl of the plasma to a micro-cuvette containing a colorimetric reagent that is photometrically analyzed for lithium. The test takes 5 minutes or less (Figure).
The assay has been shown to be sensitive to 0.1 mEq/L of lithium and linear between 0.1 and 2.5 mEq/L.3
Figure How finger-stick lithium test works
Clinician obtains blood sample (a) and empties it into a separator (b), which processes blood to plasma. Clinician then adds plasma to a reagent vial (c), which is inserted into a reader (d) to obtain a lithium level.
Reliability
In clinical trials during which patients were tested and retested, the colorimetric assay showed reliability when compared with:
- routine lithium spectrophotometry. Researchers compared venipuncture blood samples split for colorimetric and spectrophotometric testing
- atomic absorption spectrophotometry of venipuncture blood from psychiatric patients
- standard spectrophotometry of venipuncture samples to which a known amount of lithium was added.4
Colorimetric finger-stick testing also was compared with AA spectrophotometry testing of 88 matched venipuncture samples from 56 bipolar patients.5 Results were not identical, but most fingerstick results varied no more than±0.2 mEq/L from the AA results. Differences were positive and negative, indicating random variation between the two methods rather than systematic bias.
Clinical applicability
In-office finger-stick blood testing for lithium levels could improve quality of care for patients taking lithium.
The manufacturer, ReliaLAB, says the test costs $399, plus $264 for a refill kit containing 24 patient test packs. A certain volume of patients taking lithium would seem to be necessary to justify purchasing the instrument.
The test may be reimbursable under certain circumstances. ReliaLAB offers information on coding and reimbursement for in-office lithium monitoring (Table).
Also, because instant in-office creatinine and thyroid-stimulating hormone tests are not available, lithium therapy monitoring will still require laboratory visits when these tests are needed. Nonetheless, point-of-care plasma lithium level determination should improve convenience, compliance, and overall comprehensiveness of care.
Related resources
- Online information on in-office lithium test. www.relialab.com/Lith.html.
- Johnson FN. The origins of lithium therapy. Rev Contemp Pharmacother 1999;10:193-265.
Drug brand names
- Lithium • Eskalith, others
Disclosure
Dr. Jefferson reports no financial relationship with or proprietary interest in ReliaLAB.
A new, FDA-approved in-office lithium test (Table) can eliminate the inconvenience and fallibility of testing venous blood samples that often discourage lithium use. The test, which measures lithium in capillary blood drawn from a finger stick, has shown reliability when compared in clinical trials with established testing methods.
Why finger-stick testing?
Periodically monitoring serum or plasma lithium minimizes side effects and toxicity, maintains therapeutic dosing, and ensures treatment adherence. Laboratories generally use flame photometry, atomic absorption (AA) spectrophotometry, or ion-selective electrode analysis to measure lithium in blood drawn via venipuncture. A colorimetric assay is also available.1
Table
Lithium fingerstick test: Fast facts
| Brand name: |
| InstaRead Lithium System |
| Indication: |
| Testing plasma lithium levels in-office |
| Manufacturer: |
| ReliaLAB |
| Recommended use: |
| Testing plasma lithium levels 12 hours after dosing; repeat test after 5 minutes to confirm abnormal reading |
| Reimbursement information: |
| 1-866-467-8273 or www.relialab.com/Reimbursement.html |
For years, researchers have investigated alternatives to venipuncture lithium testing. Aside from being inconvenient, venipuncture draws can increase risk of excessive bleeding, hematoma, infection, vasovagal syncope, and multiple punctures to locate a vein. In some cases:
- psychiatrists wait 2 or more days for a laboratory to return results
- patients forget to have blood drawn before the office visit
- samples are incorrectly timed in relation to the last dose
- results are filed away unnoticed
- or the psychiatrist needs to call the patient 3 days or so after the visit to discuss an abnormal reading.
With the new in-office test, clinicians can ensure they will obtain a valid blood sample in minutes, 12 hours after dosing. Psychiatrists then can immediately discuss the result with patients, perform a repeat test 5 minutes later to check an abnormal reading, and counsel patients on raising low lithium levels. This instant feedback can powerfully reinforce a physician’s advice and promote treatment adherence.2
How it works
A 50-μl blood sample is drawn via finger stick and converted to plasma in a lectin-coated membrane separator. The clinician then adds 0.2 μl of the plasma to a micro-cuvette containing a colorimetric reagent that is photometrically analyzed for lithium. The test takes 5 minutes or less (Figure).
The assay has been shown to be sensitive to 0.1 mEq/L of lithium and linear between 0.1 and 2.5 mEq/L.3
Figure How finger-stick lithium test works
Clinician obtains blood sample (a) and empties it into a separator (b), which processes blood to plasma. Clinician then adds plasma to a reagent vial (c), which is inserted into a reader (d) to obtain a lithium level.
Reliability
In clinical trials during which patients were tested and retested, the colorimetric assay showed reliability when compared with:
- routine lithium spectrophotometry. Researchers compared venipuncture blood samples split for colorimetric and spectrophotometric testing
- atomic absorption spectrophotometry of venipuncture blood from psychiatric patients
- standard spectrophotometry of venipuncture samples to which a known amount of lithium was added.4
Colorimetric finger-stick testing also was compared with AA spectrophotometry testing of 88 matched venipuncture samples from 56 bipolar patients.5 Results were not identical, but most fingerstick results varied no more than±0.2 mEq/L from the AA results. Differences were positive and negative, indicating random variation between the two methods rather than systematic bias.
Clinical applicability
In-office finger-stick blood testing for lithium levels could improve quality of care for patients taking lithium.
The manufacturer, ReliaLAB, says the test costs $399, plus $264 for a refill kit containing 24 patient test packs. A certain volume of patients taking lithium would seem to be necessary to justify purchasing the instrument.
The test may be reimbursable under certain circumstances. ReliaLAB offers information on coding and reimbursement for in-office lithium monitoring (Table).
Also, because instant in-office creatinine and thyroid-stimulating hormone tests are not available, lithium therapy monitoring will still require laboratory visits when these tests are needed. Nonetheless, point-of-care plasma lithium level determination should improve convenience, compliance, and overall comprehensiveness of care.
Related resources
- Online information on in-office lithium test. www.relialab.com/Lith.html.
- Johnson FN. The origins of lithium therapy. Rev Contemp Pharmacother 1999;10:193-265.
Drug brand names
- Lithium • Eskalith, others
Disclosure
Dr. Jefferson reports no financial relationship with or proprietary interest in ReliaLAB.
1. Jefferson JW, Greist JH. Lithium. In: Sadock BJ, Sadock VA (eds). Comprehensive textbook of psychiatry, vol. 2 (8th ed). Philadelphia: Lippincott Williams & Wilkins; 2005;2839-51.
2. Srinivasan DP, Birch NJ. Instant lithium monitoring: A clinical revolution in the making. Br J Clin Pract 1996;50:386-88.
3. Glazer WM, Sonnenberg JG, Reinstein MJ, Akers RF. A novel, point-of-care test for lithium levels: Description and reliability. J Clin Psychiatry 2004;652-5.
4. Vrouwe EX, Luttge R, van den Berg A. Direct measurement of lithium in whole blood using microchip capillary electrophoresis with integrated conductivity detection. Electrophoresis 2004;25:1660-7.
5. Glazer WM, Sonnenberg J, Reinstein MJ. A novel, “point of care” test for lithium levels (poster presentation). Atlanta, GA: American Psychiatric Association annual meeting, 2005.
1. Jefferson JW, Greist JH. Lithium. In: Sadock BJ, Sadock VA (eds). Comprehensive textbook of psychiatry, vol. 2 (8th ed). Philadelphia: Lippincott Williams & Wilkins; 2005;2839-51.
2. Srinivasan DP, Birch NJ. Instant lithium monitoring: A clinical revolution in the making. Br J Clin Pract 1996;50:386-88.
3. Glazer WM, Sonnenberg JG, Reinstein MJ, Akers RF. A novel, point-of-care test for lithium levels: Description and reliability. J Clin Psychiatry 2004;652-5.
4. Vrouwe EX, Luttge R, van den Berg A. Direct measurement of lithium in whole blood using microchip capillary electrophoresis with integrated conductivity detection. Electrophoresis 2004;25:1660-7.
5. Glazer WM, Sonnenberg J, Reinstein MJ. A novel, “point of care” test for lithium levels (poster presentation). Atlanta, GA: American Psychiatric Association annual meeting, 2005.
Vagus nerve stimulation
What is vagus nerve stimulation’s (VNS) role in treating chronic or recurrent depression? Which patients would benefit from this implant, now FDA-approved for depression as well as epilepsy?
Drawing from the evidence, this article discusses which patients with depression may be candidates for VNS, how it works, and its potential benefits and side effects.
Clinical Applicability
VNS is indicated for patients with chronic or recurrent treatment-resistant depression during an episode that has not responded to ≥4 adequate antidepressant treatment trials (defined as ≥3 on the Antidepressant Treatment History Form [ATHF]) (Table 1). Implantation theoretically promotes 100% adherence and reduces drug-drug interaction risk. Interactions between VNS and nonpsychotropics are possible but unlikely.
Paradoxically, data suggest that patients with low to moderate resistance to antidepressant treatment (≤3 antidepressant trial failures) are most likely to benefit from VNS.1 Patients who had never received electroconvulsive therapy (ECT) (indicating relatively low treatment resistance) were nearly four times more likely than ECT-treated patients to respond to VNS.2 Conversely, 13 subjects who had not responded to ≥ 7 adequate treatment trials (indicating relatively severe treatment resistance) did not respond to VNS.2
Table 1
Vagus nerve stimulation device: Fast facts
| Brand name: Cyberonics Vagus Nerve Stimulation (VNS) Therapy System |
| FDA-approved indications: Treatment-resistant depression (previously approved for treatment-refractory epilepsy) |
| Manufacturer: Cyberonics |
| Recommended use: Treating depressive episode that has not responded to ≥4 antidepressant trials or electroconvulsive therapy in a patient with chronic or recurrent depression |
| Information on VNS remote device training: 1-877-NOW-4-VNS (669-4867) or www.vnstherapy.com |
How VNS Works
The vagus (10th cranial) nerve is a main efferent outflow tract for parasympathetic innervation of the abdomen and chest, regulating heart rate, acid secretion, and bowel motility.
The largest component of the left vagus nerve—approximately 80%—conducts information about pain, hunger, and satiety. These fibers are also believed to contribute to VNS’ antidepressant effects by carrying information to the solitary nucleus of the medulla. From there, fibers project to the median raphe nucleus and locus coeruleus, key areas of serotonergic and noradrenergic innervation relevant to depression.
Positron emission tomography studies suggest that VNS also increases blood flow to the thalamus, hypothalamus, and insula—brain areas considered relevant to mood disorders.3
VNS requires subcutaneous implantation of a pacemaker-like pulse generator into the upper left chest. The generator is 6.9 mm thick and weighs 25 grams. Wires extend from the device into the left vagus nerve in the neck (Figure). A neurosurgeon usually performs the 1- to 2-hour outpatient procedure, although ENT, vascular, and general surgeons may also do the implant.
The device sends electric pulses to the left vagus nerve every few seconds (Table 2). Using an accompanying hand-held device and a computer, the clinician programs the implant and adjusts stimulation parameters to ensure the correct amount of stimulation.
FDA approved VNS in 1997 for refractory epilepsy. Clinical observations that VNS improved epilepsy patients’ mood spurred interest in its antidepressant effects.4 Preliminary data suggest VNS also could help manage anxiety disorders, obesity, pain syndromes, and Alzheimer’s disease.5
Figure How VNS device works
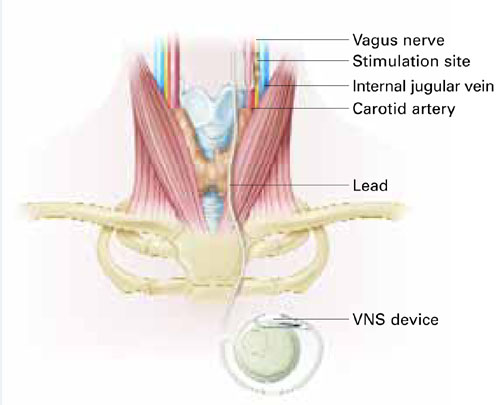
Pacemaker-like VNSdevice is implanted into the upper left chest. Wires extending from the device transport electric pulses into the left vagus nerve in the neck, which carries information to areas of serotonergic and noradrenergic innervation relevant to depression.Table 2
VNS stimulation parameters
| Frequency: 20 to 30 Hz |
| Intensity: 0.25 mA (0.25 to 3.0 mA) |
| Pulse width: 250 to 500 μs |
| Duty cycle: 30 seconds on/5 minutes off |
Cost
VNS implantation costs approximately $25,000, including the device, surgeon’s fee, and facility charge. Psychiatrists generally would initiate the referral process.
Follow-up management fees for epilepsy are $150 to $250 per visit. Several follow-up visits are required after stimulation is started to verify the device is working, evaluate treatment response and tolerability, and adjust stimulation as needed. Thereafter, periodic visits are appropriate.
Generally, insurers cover VNS as an epilepsy treatment; whether private insurers and Medicare will cover VNS for depression remains to be seen. Case mangers at Cyberonics, the device’s manufacturer, are on call to assist with VNS coverage, coding, and reimbursement issues (see Related resources).
Because the internal implant’s battery life is 6 to 11 years, VNS therapy will likely be cost-effective for many patients, although follow-up surgery would be required to replace the battery. Costs of using VNS have not been compared with other antidepressant modalities.
VNS’ Efficacy In Depression
In an open-label trial, 60 patients ages 20 to 63 received VNS with no placebo or active comparator.2 Thirty had completed an open-label pilot study that showed VNS’ potential antidepressant effects.6 Before implantation, all subjects had:
- a major depressive episode lasting >2 years or >4 lifetime major depressive episodes
- nonresponse to ECT or ≥2 adequate antidepressant trials (ATHF scores >3) during their current major depressive episode (median duration: 4.7 years)
- DSM-IV diagnosis of major depressive disorder or bipolar type I or II disorder depressed phase.
- baseline scores ≥20 on the 28-item Hamilton Rating Scale for Depression (HRSD-28) and ≤50 on the Global Assessment of Functioning (GAF) scale.
Two weeks after implantation, the stimulator was turned on and adjusted for another 2 weeks to the maximum tolerable dose. Patients then received 8 weeks of fixed-dose stimulation. Participants who had been taking an antidepressant, mood stabilizer, second-generation antipsychotic, or other psychotropic at the same dosages for ≥4 weeks before the study could continue their medications during the VNS trial (median concurrent treatments: 4).
Three months after implantation, 18 of 59 subjects (30.5%) showed clinical response (≥50% improvement in HRSD-28 scores over baseline). Nine patients (15.3%) showed depression remission (HRSD-28 score ≤10). Median time to first response was 45.5 days.
Twenty participants (34%) showed a ≥50% reduction in baseline Montgomery-Asberg Depression Rating Scale (MADRS) scores, and 22 (37%) showed Clinical Global Impression-Improvement Scale (CGI-I) scores improving to 1 or 2.
Therapeutic effects did not differ among patients with unipolar and bipolar depression. Participants with mild to moderate depression (defined as 2 to 3 failed adequate trials) showed higher response rates (50% vs. 29.1%) than did those with more-severe depression (defined as ≥4 failed adequate trials).2
Among 28 patients followed for 1 year, 13 (46%) met HRSD-28 response criteria (≥ 50% score reduction) and 8 (29%) met remission criteria (score ≤ 10), showing gradual improvement.1 After 2 years, 44% of patients met HDRS-28 response criteria, and 22% met remission criteria, showing sustained benefit.7 How many subjects were taking one or more concomitant psychotropics is unknown.
In a double-blind controlled trial, 235 subjects ages 18 to 80 received VNS or a sham comparator.8 Treatment response and remission were defined as ≥50% reduction from baseline and ≤9, respectively, on the 24-item HRSD (HRSD-24). Patient selection criteria were similar to those of the open-label study.
All patients received VNS implants, which were inactive the first 2 weeks. Patients were then randomly assigned to active treatment (stimulator turned on) or sham control (stimulator left off). After 10 weeks of treatment, HRSD-24, CGI-I, and MADRS scores were similar between the VNS and sham groups, but Inventory of Depressive Symptomatology Self Report (IDS-SR) scores improved much more in the active treatment group (P<0.03). Patients in the sham group then had their stimulators turned on.
After 1 year of active treatment for both groups, response and remission rates more than doubled among 205 evaluable subjects (response: 14.4% to 29.8%; remission: 7.3% to 17.1%). MADRS and IDS-SR scores also improved. Three percent of subjects dropped out because of adverse events.
Another analysis of these data revealed significant improvement among the VNS treatment group vs. a comparator-matched control group of treatment-resistant patients across 2 years.8
Depression treatment among patients in the comparator group followed standard clinical practice.
Side Effects
Voice alteration or hoarseness was most commonly reported after 12 weeks in the open-label trial (55% of subjects). Headache (22%), cough (17%), shortness of breath (15%), neck pain (17%), dysphagia (20%), and pain (15%) were also reported.2 These effects emerge or increase with stimulation intensity and may be ameliorated by reducing the dose.
Small risks of infection (1%) and nerve damage (1%) were reported. Leaving the stimulator off for 14 days after implantation decreases nerve damage risk. Pain at the incision site (experienced by 30%) resolved after 1 to 2 weeks.2 Other adverse events included:
- hypomania in one bipolar patient; this was resolved by adjusting medication and reducing stimulation
- leg pain in 2 subjects
- worsened depression in 5 patients (2 of these may have been related to stimulation)
- emesis and diarrhea in 1 subject.
One patient with multiple cardiac risk factors developed a myocardial infarction but completed the trial after angioplasty and stent placement.2
After 1 year in the open-label trial, no subjects dropped out because of adverse events. Common side events included voice alteration (21%), shortness of breath (7%), and neck pain (7%). More-serious adverse events reported between the acute trial and 12-month follow-up included hypomania (2 episodes), one deep venous thrombophlebitits episode, and one episode each of back pain and appendicitis.1 No cognitive effects have been reported.
In the double-blind controlled trial, 31 of 235 subjects (13%) experienced worsening of depression, and 25 of the 31 depressed subjects attempted suicide.9 Whether these effects were related to the depression or VNS stimulation is unclear. Side effects reported more frequently in the active treatment group than in the sham control group included voice alteration (68% vs. 38%), cough (29% vs. 9%), shortness of breath (23% vs. 14%), dysphagia (21% vs. 10%), and neck pain (21% vs. 10%).
If VNS Is Intolerable
Patients may deactivate the device with a magnet if they are uncomfortable. Pulse stimulation stops when a magnet is held against the left upper chest and resumes when the magnet is removed.
Training
Cyberonics plans to offer free VNS training to psychiatrists who practice at selected centers that accept treatment-resistant depression case referrals from primary care physicians, community psychiatrists, and other providers. Community psychiatrists who see treatment-resistant patients also are eligible for free training. For information, see Related resources.
- Cyberonics VNS therapy Web site. www.vnstherapy.com.
- Cyberonics reimbursement and case management support services. www.vnstherapy.com/depression/hcp/ReimbursementIns/default.aspx.
- Harden CL, Pulver MC, Ravdin LD, et al. A pilot study of mood in epilepsy patients treated with vagus nerve stimulation. Epilepsy Behav 2000;1:93-9.
Disclosure
The authors receive grant support from Neuronetics. They report no proprietary interest in the technology discussed in this article.
1. Marangell LB, Rush AJ, George MS, et al. Vagus nerve stimulation (VNS) for major depressive episodes: one year outcomes. Biol Psychiatry 2002;51:280-7.
2. Sackeim HA, Rush AJ, George MS, et al. Vagus nerve stimulation (VNS) for treatment-resistant depression: efficacy, side effects, and predictors of outcome. Neuropsychopharmacology 2001;25(5):713-28.
3. Henry TR, Bakay RA, Votaw JR, et al. Brain blood flow alterations induced in partial epilepsy I: acute effects at high and low levels of stimulation. Epilepsia 1998;39(9):983-90.
4. Elger G, Hoppe C, Falkai P, et al. Vagus nerve stimulation is associated with mood improvements in epilepsy patients. Epilepsy Res 2000;42(2):203-10.
5. George MS, Nahas Z, Bohning DE, et al. Vagus nerve stimulation therapy: a research update. Neurology 2002;59(6 suppl 4):S56-61.
6. Rush AJ, George MS, Sackeim HA, et al. Vagus nerve stimulation (VNS) for treatment-resistant depression: a multicenter study. Biol Psychiatry 2000;47:276-86.
7. Rush AJ, George MS, Sackeim HA, et al. Continuing benefit of VNS therapy over 2 years for treatment-resistant depression. San Juan, Puerto Rico: American College of Neuropsychopharmacology annual meeting, 2002.
8. Cyberonics premarket approval application supplement (D-02/D-04 clinical report, PMA-S), submitted to FDA October 2003.
9. Zwillich T. FDA panel recommends device for depression. WebMD Medical News June 17, 2004. Available at: http://my.webmd.com/content/article/89/100114.htm. Accessed August 9, 2005.
What is vagus nerve stimulation’s (VNS) role in treating chronic or recurrent depression? Which patients would benefit from this implant, now FDA-approved for depression as well as epilepsy?
Drawing from the evidence, this article discusses which patients with depression may be candidates for VNS, how it works, and its potential benefits and side effects.
Clinical Applicability
VNS is indicated for patients with chronic or recurrent treatment-resistant depression during an episode that has not responded to ≥4 adequate antidepressant treatment trials (defined as ≥3 on the Antidepressant Treatment History Form [ATHF]) (Table 1). Implantation theoretically promotes 100% adherence and reduces drug-drug interaction risk. Interactions between VNS and nonpsychotropics are possible but unlikely.
Paradoxically, data suggest that patients with low to moderate resistance to antidepressant treatment (≤3 antidepressant trial failures) are most likely to benefit from VNS.1 Patients who had never received electroconvulsive therapy (ECT) (indicating relatively low treatment resistance) were nearly four times more likely than ECT-treated patients to respond to VNS.2 Conversely, 13 subjects who had not responded to ≥ 7 adequate treatment trials (indicating relatively severe treatment resistance) did not respond to VNS.2
Table 1
Vagus nerve stimulation device: Fast facts
| Brand name: Cyberonics Vagus Nerve Stimulation (VNS) Therapy System |
| FDA-approved indications: Treatment-resistant depression (previously approved for treatment-refractory epilepsy) |
| Manufacturer: Cyberonics |
| Recommended use: Treating depressive episode that has not responded to ≥4 antidepressant trials or electroconvulsive therapy in a patient with chronic or recurrent depression |
| Information on VNS remote device training: 1-877-NOW-4-VNS (669-4867) or www.vnstherapy.com |
How VNS Works
The vagus (10th cranial) nerve is a main efferent outflow tract for parasympathetic innervation of the abdomen and chest, regulating heart rate, acid secretion, and bowel motility.
The largest component of the left vagus nerve—approximately 80%—conducts information about pain, hunger, and satiety. These fibers are also believed to contribute to VNS’ antidepressant effects by carrying information to the solitary nucleus of the medulla. From there, fibers project to the median raphe nucleus and locus coeruleus, key areas of serotonergic and noradrenergic innervation relevant to depression.
Positron emission tomography studies suggest that VNS also increases blood flow to the thalamus, hypothalamus, and insula—brain areas considered relevant to mood disorders.3
VNS requires subcutaneous implantation of a pacemaker-like pulse generator into the upper left chest. The generator is 6.9 mm thick and weighs 25 grams. Wires extend from the device into the left vagus nerve in the neck (Figure). A neurosurgeon usually performs the 1- to 2-hour outpatient procedure, although ENT, vascular, and general surgeons may also do the implant.
The device sends electric pulses to the left vagus nerve every few seconds (Table 2). Using an accompanying hand-held device and a computer, the clinician programs the implant and adjusts stimulation parameters to ensure the correct amount of stimulation.
FDA approved VNS in 1997 for refractory epilepsy. Clinical observations that VNS improved epilepsy patients’ mood spurred interest in its antidepressant effects.4 Preliminary data suggest VNS also could help manage anxiety disorders, obesity, pain syndromes, and Alzheimer’s disease.5
Figure How VNS device works
Pacemaker-like VNSdevice is implanted into the upper left chest. Wires extending from the device transport electric pulses into the left vagus nerve in the neck, which carries information to areas of serotonergic and noradrenergic innervation relevant to depression.Table 2
VNS stimulation parameters
| Frequency: 20 to 30 Hz |
| Intensity: 0.25 mA (0.25 to 3.0 mA) |
| Pulse width: 250 to 500 μs |
| Duty cycle: 30 seconds on/5 minutes off |
Cost
VNS implantation costs approximately $25,000, including the device, surgeon’s fee, and facility charge. Psychiatrists generally would initiate the referral process.
Follow-up management fees for epilepsy are $150 to $250 per visit. Several follow-up visits are required after stimulation is started to verify the device is working, evaluate treatment response and tolerability, and adjust stimulation as needed. Thereafter, periodic visits are appropriate.
Generally, insurers cover VNS as an epilepsy treatment; whether private insurers and Medicare will cover VNS for depression remains to be seen. Case mangers at Cyberonics, the device’s manufacturer, are on call to assist with VNS coverage, coding, and reimbursement issues (see Related resources).
Because the internal implant’s battery life is 6 to 11 years, VNS therapy will likely be cost-effective for many patients, although follow-up surgery would be required to replace the battery. Costs of using VNS have not been compared with other antidepressant modalities.
VNS’ Efficacy In Depression
In an open-label trial, 60 patients ages 20 to 63 received VNS with no placebo or active comparator.2 Thirty had completed an open-label pilot study that showed VNS’ potential antidepressant effects.6 Before implantation, all subjects had:
- a major depressive episode lasting >2 years or >4 lifetime major depressive episodes
- nonresponse to ECT or ≥2 adequate antidepressant trials (ATHF scores >3) during their current major depressive episode (median duration: 4.7 years)
- DSM-IV diagnosis of major depressive disorder or bipolar type I or II disorder depressed phase.
- baseline scores ≥20 on the 28-item Hamilton Rating Scale for Depression (HRSD-28) and ≤50 on the Global Assessment of Functioning (GAF) scale.
Two weeks after implantation, the stimulator was turned on and adjusted for another 2 weeks to the maximum tolerable dose. Patients then received 8 weeks of fixed-dose stimulation. Participants who had been taking an antidepressant, mood stabilizer, second-generation antipsychotic, or other psychotropic at the same dosages for ≥4 weeks before the study could continue their medications during the VNS trial (median concurrent treatments: 4).
Three months after implantation, 18 of 59 subjects (30.5%) showed clinical response (≥50% improvement in HRSD-28 scores over baseline). Nine patients (15.3%) showed depression remission (HRSD-28 score ≤10). Median time to first response was 45.5 days.
Twenty participants (34%) showed a ≥50% reduction in baseline Montgomery-Asberg Depression Rating Scale (MADRS) scores, and 22 (37%) showed Clinical Global Impression-Improvement Scale (CGI-I) scores improving to 1 or 2.
Therapeutic effects did not differ among patients with unipolar and bipolar depression. Participants with mild to moderate depression (defined as 2 to 3 failed adequate trials) showed higher response rates (50% vs. 29.1%) than did those with more-severe depression (defined as ≥4 failed adequate trials).2
Among 28 patients followed for 1 year, 13 (46%) met HRSD-28 response criteria (≥ 50% score reduction) and 8 (29%) met remission criteria (score ≤ 10), showing gradual improvement.1 After 2 years, 44% of patients met HDRS-28 response criteria, and 22% met remission criteria, showing sustained benefit.7 How many subjects were taking one or more concomitant psychotropics is unknown.
In a double-blind controlled trial, 235 subjects ages 18 to 80 received VNS or a sham comparator.8 Treatment response and remission were defined as ≥50% reduction from baseline and ≤9, respectively, on the 24-item HRSD (HRSD-24). Patient selection criteria were similar to those of the open-label study.
All patients received VNS implants, which were inactive the first 2 weeks. Patients were then randomly assigned to active treatment (stimulator turned on) or sham control (stimulator left off). After 10 weeks of treatment, HRSD-24, CGI-I, and MADRS scores were similar between the VNS and sham groups, but Inventory of Depressive Symptomatology Self Report (IDS-SR) scores improved much more in the active treatment group (P<0.03). Patients in the sham group then had their stimulators turned on.
After 1 year of active treatment for both groups, response and remission rates more than doubled among 205 evaluable subjects (response: 14.4% to 29.8%; remission: 7.3% to 17.1%). MADRS and IDS-SR scores also improved. Three percent of subjects dropped out because of adverse events.
Another analysis of these data revealed significant improvement among the VNS treatment group vs. a comparator-matched control group of treatment-resistant patients across 2 years.8
Depression treatment among patients in the comparator group followed standard clinical practice.
Side Effects
Voice alteration or hoarseness was most commonly reported after 12 weeks in the open-label trial (55% of subjects). Headache (22%), cough (17%), shortness of breath (15%), neck pain (17%), dysphagia (20%), and pain (15%) were also reported.2 These effects emerge or increase with stimulation intensity and may be ameliorated by reducing the dose.
Small risks of infection (1%) and nerve damage (1%) were reported. Leaving the stimulator off for 14 days after implantation decreases nerve damage risk. Pain at the incision site (experienced by 30%) resolved after 1 to 2 weeks.2 Other adverse events included:
- hypomania in one bipolar patient; this was resolved by adjusting medication and reducing stimulation
- leg pain in 2 subjects
- worsened depression in 5 patients (2 of these may have been related to stimulation)
- emesis and diarrhea in 1 subject.
One patient with multiple cardiac risk factors developed a myocardial infarction but completed the trial after angioplasty and stent placement.2
After 1 year in the open-label trial, no subjects dropped out because of adverse events. Common side events included voice alteration (21%), shortness of breath (7%), and neck pain (7%). More-serious adverse events reported between the acute trial and 12-month follow-up included hypomania (2 episodes), one deep venous thrombophlebitits episode, and one episode each of back pain and appendicitis.1 No cognitive effects have been reported.
In the double-blind controlled trial, 31 of 235 subjects (13%) experienced worsening of depression, and 25 of the 31 depressed subjects attempted suicide.9 Whether these effects were related to the depression or VNS stimulation is unclear. Side effects reported more frequently in the active treatment group than in the sham control group included voice alteration (68% vs. 38%), cough (29% vs. 9%), shortness of breath (23% vs. 14%), dysphagia (21% vs. 10%), and neck pain (21% vs. 10%).
If VNS Is Intolerable
Patients may deactivate the device with a magnet if they are uncomfortable. Pulse stimulation stops when a magnet is held against the left upper chest and resumes when the magnet is removed.
Training
Cyberonics plans to offer free VNS training to psychiatrists who practice at selected centers that accept treatment-resistant depression case referrals from primary care physicians, community psychiatrists, and other providers. Community psychiatrists who see treatment-resistant patients also are eligible for free training. For information, see Related resources.
- Cyberonics VNS therapy Web site. www.vnstherapy.com.
- Cyberonics reimbursement and case management support services. www.vnstherapy.com/depression/hcp/ReimbursementIns/default.aspx.
- Harden CL, Pulver MC, Ravdin LD, et al. A pilot study of mood in epilepsy patients treated with vagus nerve stimulation. Epilepsy Behav 2000;1:93-9.
Disclosure
The authors receive grant support from Neuronetics. They report no proprietary interest in the technology discussed in this article.
What is vagus nerve stimulation’s (VNS) role in treating chronic or recurrent depression? Which patients would benefit from this implant, now FDA-approved for depression as well as epilepsy?
Drawing from the evidence, this article discusses which patients with depression may be candidates for VNS, how it works, and its potential benefits and side effects.
Clinical Applicability
VNS is indicated for patients with chronic or recurrent treatment-resistant depression during an episode that has not responded to ≥4 adequate antidepressant treatment trials (defined as ≥3 on the Antidepressant Treatment History Form [ATHF]) (Table 1). Implantation theoretically promotes 100% adherence and reduces drug-drug interaction risk. Interactions between VNS and nonpsychotropics are possible but unlikely.
Paradoxically, data suggest that patients with low to moderate resistance to antidepressant treatment (≤3 antidepressant trial failures) are most likely to benefit from VNS.1 Patients who had never received electroconvulsive therapy (ECT) (indicating relatively low treatment resistance) were nearly four times more likely than ECT-treated patients to respond to VNS.2 Conversely, 13 subjects who had not responded to ≥ 7 adequate treatment trials (indicating relatively severe treatment resistance) did not respond to VNS.2
Table 1
Vagus nerve stimulation device: Fast facts
| Brand name: Cyberonics Vagus Nerve Stimulation (VNS) Therapy System |
| FDA-approved indications: Treatment-resistant depression (previously approved for treatment-refractory epilepsy) |
| Manufacturer: Cyberonics |
| Recommended use: Treating depressive episode that has not responded to ≥4 antidepressant trials or electroconvulsive therapy in a patient with chronic or recurrent depression |
| Information on VNS remote device training: 1-877-NOW-4-VNS (669-4867) or www.vnstherapy.com |
How VNS Works
The vagus (10th cranial) nerve is a main efferent outflow tract for parasympathetic innervation of the abdomen and chest, regulating heart rate, acid secretion, and bowel motility.
The largest component of the left vagus nerve—approximately 80%—conducts information about pain, hunger, and satiety. These fibers are also believed to contribute to VNS’ antidepressant effects by carrying information to the solitary nucleus of the medulla. From there, fibers project to the median raphe nucleus and locus coeruleus, key areas of serotonergic and noradrenergic innervation relevant to depression.
Positron emission tomography studies suggest that VNS also increases blood flow to the thalamus, hypothalamus, and insula—brain areas considered relevant to mood disorders.3
VNS requires subcutaneous implantation of a pacemaker-like pulse generator into the upper left chest. The generator is 6.9 mm thick and weighs 25 grams. Wires extend from the device into the left vagus nerve in the neck (Figure). A neurosurgeon usually performs the 1- to 2-hour outpatient procedure, although ENT, vascular, and general surgeons may also do the implant.
The device sends electric pulses to the left vagus nerve every few seconds (Table 2). Using an accompanying hand-held device and a computer, the clinician programs the implant and adjusts stimulation parameters to ensure the correct amount of stimulation.
FDA approved VNS in 1997 for refractory epilepsy. Clinical observations that VNS improved epilepsy patients’ mood spurred interest in its antidepressant effects.4 Preliminary data suggest VNS also could help manage anxiety disorders, obesity, pain syndromes, and Alzheimer’s disease.5
Figure How VNS device works
Pacemaker-like VNSdevice is implanted into the upper left chest. Wires extending from the device transport electric pulses into the left vagus nerve in the neck, which carries information to areas of serotonergic and noradrenergic innervation relevant to depression.Table 2
VNS stimulation parameters
| Frequency: 20 to 30 Hz |
| Intensity: 0.25 mA (0.25 to 3.0 mA) |
| Pulse width: 250 to 500 μs |
| Duty cycle: 30 seconds on/5 minutes off |
Cost
VNS implantation costs approximately $25,000, including the device, surgeon’s fee, and facility charge. Psychiatrists generally would initiate the referral process.
Follow-up management fees for epilepsy are $150 to $250 per visit. Several follow-up visits are required after stimulation is started to verify the device is working, evaluate treatment response and tolerability, and adjust stimulation as needed. Thereafter, periodic visits are appropriate.
Generally, insurers cover VNS as an epilepsy treatment; whether private insurers and Medicare will cover VNS for depression remains to be seen. Case mangers at Cyberonics, the device’s manufacturer, are on call to assist with VNS coverage, coding, and reimbursement issues (see Related resources).
Because the internal implant’s battery life is 6 to 11 years, VNS therapy will likely be cost-effective for many patients, although follow-up surgery would be required to replace the battery. Costs of using VNS have not been compared with other antidepressant modalities.
VNS’ Efficacy In Depression
In an open-label trial, 60 patients ages 20 to 63 received VNS with no placebo or active comparator.2 Thirty had completed an open-label pilot study that showed VNS’ potential antidepressant effects.6 Before implantation, all subjects had:
- a major depressive episode lasting >2 years or >4 lifetime major depressive episodes
- nonresponse to ECT or ≥2 adequate antidepressant trials (ATHF scores >3) during their current major depressive episode (median duration: 4.7 years)
- DSM-IV diagnosis of major depressive disorder or bipolar type I or II disorder depressed phase.
- baseline scores ≥20 on the 28-item Hamilton Rating Scale for Depression (HRSD-28) and ≤50 on the Global Assessment of Functioning (GAF) scale.
Two weeks after implantation, the stimulator was turned on and adjusted for another 2 weeks to the maximum tolerable dose. Patients then received 8 weeks of fixed-dose stimulation. Participants who had been taking an antidepressant, mood stabilizer, second-generation antipsychotic, or other psychotropic at the same dosages for ≥4 weeks before the study could continue their medications during the VNS trial (median concurrent treatments: 4).
Three months after implantation, 18 of 59 subjects (30.5%) showed clinical response (≥50% improvement in HRSD-28 scores over baseline). Nine patients (15.3%) showed depression remission (HRSD-28 score ≤10). Median time to first response was 45.5 days.
Twenty participants (34%) showed a ≥50% reduction in baseline Montgomery-Asberg Depression Rating Scale (MADRS) scores, and 22 (37%) showed Clinical Global Impression-Improvement Scale (CGI-I) scores improving to 1 or 2.
Therapeutic effects did not differ among patients with unipolar and bipolar depression. Participants with mild to moderate depression (defined as 2 to 3 failed adequate trials) showed higher response rates (50% vs. 29.1%) than did those with more-severe depression (defined as ≥4 failed adequate trials).2
Among 28 patients followed for 1 year, 13 (46%) met HRSD-28 response criteria (≥ 50% score reduction) and 8 (29%) met remission criteria (score ≤ 10), showing gradual improvement.1 After 2 years, 44% of patients met HDRS-28 response criteria, and 22% met remission criteria, showing sustained benefit.7 How many subjects were taking one or more concomitant psychotropics is unknown.
In a double-blind controlled trial, 235 subjects ages 18 to 80 received VNS or a sham comparator.8 Treatment response and remission were defined as ≥50% reduction from baseline and ≤9, respectively, on the 24-item HRSD (HRSD-24). Patient selection criteria were similar to those of the open-label study.
All patients received VNS implants, which were inactive the first 2 weeks. Patients were then randomly assigned to active treatment (stimulator turned on) or sham control (stimulator left off). After 10 weeks of treatment, HRSD-24, CGI-I, and MADRS scores were similar between the VNS and sham groups, but Inventory of Depressive Symptomatology Self Report (IDS-SR) scores improved much more in the active treatment group (P<0.03). Patients in the sham group then had their stimulators turned on.
After 1 year of active treatment for both groups, response and remission rates more than doubled among 205 evaluable subjects (response: 14.4% to 29.8%; remission: 7.3% to 17.1%). MADRS and IDS-SR scores also improved. Three percent of subjects dropped out because of adverse events.
Another analysis of these data revealed significant improvement among the VNS treatment group vs. a comparator-matched control group of treatment-resistant patients across 2 years.8
Depression treatment among patients in the comparator group followed standard clinical practice.
Side Effects
Voice alteration or hoarseness was most commonly reported after 12 weeks in the open-label trial (55% of subjects). Headache (22%), cough (17%), shortness of breath (15%), neck pain (17%), dysphagia (20%), and pain (15%) were also reported.2 These effects emerge or increase with stimulation intensity and may be ameliorated by reducing the dose.
Small risks of infection (1%) and nerve damage (1%) were reported. Leaving the stimulator off for 14 days after implantation decreases nerve damage risk. Pain at the incision site (experienced by 30%) resolved after 1 to 2 weeks.2 Other adverse events included:
- hypomania in one bipolar patient; this was resolved by adjusting medication and reducing stimulation
- leg pain in 2 subjects
- worsened depression in 5 patients (2 of these may have been related to stimulation)
- emesis and diarrhea in 1 subject.
One patient with multiple cardiac risk factors developed a myocardial infarction but completed the trial after angioplasty and stent placement.2
After 1 year in the open-label trial, no subjects dropped out because of adverse events. Common side events included voice alteration (21%), shortness of breath (7%), and neck pain (7%). More-serious adverse events reported between the acute trial and 12-month follow-up included hypomania (2 episodes), one deep venous thrombophlebitits episode, and one episode each of back pain and appendicitis.1 No cognitive effects have been reported.
In the double-blind controlled trial, 31 of 235 subjects (13%) experienced worsening of depression, and 25 of the 31 depressed subjects attempted suicide.9 Whether these effects were related to the depression or VNS stimulation is unclear. Side effects reported more frequently in the active treatment group than in the sham control group included voice alteration (68% vs. 38%), cough (29% vs. 9%), shortness of breath (23% vs. 14%), dysphagia (21% vs. 10%), and neck pain (21% vs. 10%).
If VNS Is Intolerable
Patients may deactivate the device with a magnet if they are uncomfortable. Pulse stimulation stops when a magnet is held against the left upper chest and resumes when the magnet is removed.
Training
Cyberonics plans to offer free VNS training to psychiatrists who practice at selected centers that accept treatment-resistant depression case referrals from primary care physicians, community psychiatrists, and other providers. Community psychiatrists who see treatment-resistant patients also are eligible for free training. For information, see Related resources.
- Cyberonics VNS therapy Web site. www.vnstherapy.com.
- Cyberonics reimbursement and case management support services. www.vnstherapy.com/depression/hcp/ReimbursementIns/default.aspx.
- Harden CL, Pulver MC, Ravdin LD, et al. A pilot study of mood in epilepsy patients treated with vagus nerve stimulation. Epilepsy Behav 2000;1:93-9.
Disclosure
The authors receive grant support from Neuronetics. They report no proprietary interest in the technology discussed in this article.
1. Marangell LB, Rush AJ, George MS, et al. Vagus nerve stimulation (VNS) for major depressive episodes: one year outcomes. Biol Psychiatry 2002;51:280-7.
2. Sackeim HA, Rush AJ, George MS, et al. Vagus nerve stimulation (VNS) for treatment-resistant depression: efficacy, side effects, and predictors of outcome. Neuropsychopharmacology 2001;25(5):713-28.
3. Henry TR, Bakay RA, Votaw JR, et al. Brain blood flow alterations induced in partial epilepsy I: acute effects at high and low levels of stimulation. Epilepsia 1998;39(9):983-90.
4. Elger G, Hoppe C, Falkai P, et al. Vagus nerve stimulation is associated with mood improvements in epilepsy patients. Epilepsy Res 2000;42(2):203-10.
5. George MS, Nahas Z, Bohning DE, et al. Vagus nerve stimulation therapy: a research update. Neurology 2002;59(6 suppl 4):S56-61.
6. Rush AJ, George MS, Sackeim HA, et al. Vagus nerve stimulation (VNS) for treatment-resistant depression: a multicenter study. Biol Psychiatry 2000;47:276-86.
7. Rush AJ, George MS, Sackeim HA, et al. Continuing benefit of VNS therapy over 2 years for treatment-resistant depression. San Juan, Puerto Rico: American College of Neuropsychopharmacology annual meeting, 2002.
8. Cyberonics premarket approval application supplement (D-02/D-04 clinical report, PMA-S), submitted to FDA October 2003.
9. Zwillich T. FDA panel recommends device for depression. WebMD Medical News June 17, 2004. Available at: http://my.webmd.com/content/article/89/100114.htm. Accessed August 9, 2005.
1. Marangell LB, Rush AJ, George MS, et al. Vagus nerve stimulation (VNS) for major depressive episodes: one year outcomes. Biol Psychiatry 2002;51:280-7.
2. Sackeim HA, Rush AJ, George MS, et al. Vagus nerve stimulation (VNS) for treatment-resistant depression: efficacy, side effects, and predictors of outcome. Neuropsychopharmacology 2001;25(5):713-28.
3. Henry TR, Bakay RA, Votaw JR, et al. Brain blood flow alterations induced in partial epilepsy I: acute effects at high and low levels of stimulation. Epilepsia 1998;39(9):983-90.
4. Elger G, Hoppe C, Falkai P, et al. Vagus nerve stimulation is associated with mood improvements in epilepsy patients. Epilepsy Res 2000;42(2):203-10.
5. George MS, Nahas Z, Bohning DE, et al. Vagus nerve stimulation therapy: a research update. Neurology 2002;59(6 suppl 4):S56-61.
6. Rush AJ, George MS, Sackeim HA, et al. Vagus nerve stimulation (VNS) for treatment-resistant depression: a multicenter study. Biol Psychiatry 2000;47:276-86.
7. Rush AJ, George MS, Sackeim HA, et al. Continuing benefit of VNS therapy over 2 years for treatment-resistant depression. San Juan, Puerto Rico: American College of Neuropsychopharmacology annual meeting, 2002.
8. Cyberonics premarket approval application supplement (D-02/D-04 clinical report, PMA-S), submitted to FDA October 2003.
9. Zwillich T. FDA panel recommends device for depression. WebMD Medical News June 17, 2004. Available at: http://my.webmd.com/content/article/89/100114.htm. Accessed August 9, 2005.
Pharmacogenomic DNA chip
Genotyping for cytochrome (CYP) P-450 gene variations can identify patients who will not benefit from, or may react badly to, some psychotropics.1 Psychiatrists can then more accurately tailor initial dosages to improve response and prevent adverse reactions.
An FDA-approved pharmacogenomic diagnostic DNA chip is expected to be available to clinical laboratories this month (Table 1). The chip provides an accurate genotype for two drug-metabolizing enzymes—2D6 and 2C19.
Table 1
Pharmacogenomic DNA chip: Fast facts
| Brand name: |
| AmpliChip CYP 450 Test |
| FDA-approved indication: |
| Genotyping patients |
| Manufacturer: |
| Roche Diagnostics |
| Estimated availability: |
| July 2005 |
| Recommended use: |
| Determining cytochrome P-450 2D6 and 2C19 gene variations in patients before prescribing a psychotropic metabolized through these pathways. |
| Laboratories that process AmpliChip results: |
| Labcore, Mayo Medical Laboratories, Quest Diagnostics |
Genotyping’S Role in Psychiatry
CYP 2D6 and 2C19 enzymes help metabolize many commonly prescribed psychotropics, including:
- fluoxetine, paroxetine, and venlafaxine, which are among the psychotropics primarily metabolized by the cytochrome P-450 2D6 enzyme (Table 2).
- amitriptyline and citalopram, which are among the psychotropics metabolized in part by 2C19 (Table 3).
The chip can identify patients who are genetically predisposed to abnormal metabolism of 2D6 and 2C19 substrates. This information can help psychiatrists improve response for ultrarapid metabolizers and minimize adverse effects experienced by poor metabolizers of these substrates.
For example, if the patient is an ultrarapid metabolizer of 2D6 and/or 2C19 substrates, the psychiatrist can:
- exceed the recommended dosage to reach adequate serum levels
- or choose an antidepressant not primarily metabolized by either enzyme.
For a poor metabolizer of 2D6 and/or 2C19 substrates, the psychiatrist can:
- choose an antidepressant metabolized by a different enzyme
- or prescribe 2D6 and 2C19 substrates at very low dosages.
For example, some poor metabolizers of 2D6 substrates have been successfully treated with fluoxetine, 2 to 5 mg/d.2,3 This approach can help avoid side effects and potentially save the patient money. To prevent prescription errors, make sure the pharmacist understands your rationale for lower-than-recommended dosages.
Patients who are poor metabolizers of 2C19 and extensive metabolizers of 2D6 substrates can probably tolerate citalopram and amitriptyline dosages at the low end of the therapeutic range. Watch for high serum levels of either or both drugs if both enzyme systems are inactive.
Table 2
Evidence suggests these drugs are predominantly metabolized by the 2D6 enzyme*
| Antidepressants | Antipsychotics | Stimulants |
|---|---|---|
| Desipramine | Fluphenazine | Atomoxetine |
| Fluoxetine | Perphenazine | |
| Nortriptyline | Risperidone | |
| Paroxetine | Thioridazine | |
| Venlafaxine | ||
| *Use caution when prescribing these agents to patients who are poor 2D6 metabolizers. | ||
Table 3
Evidence suggests these drugs are predominantly metabolized by the 2C19 enzyme*
| Antidepressants | |
|---|---|
| Diazepam | Citalopram |
| Clomipramine | Escitalopram |
| Imipramine | Sertraline |
| Benzodiazepines | |
| Amitriptyline | |
| *Use caution when prescribing these agents to patients who are poor 2C19 metabolizers. | |
Pharmacogenomic Chip’s Accuracy
The 2D6 gene has more than 100 variations, many of which are very rare mutations. The pharmacogenomic DNA chip can detect 27 of these variants, allowing the chip to accurately genotype most patients. By contrast, early 2D6 genotyping techniques identified only four or five variants, resulting in too many false negatives for clinical use.4
The chip also can identify the normal form of the 2C19 gene and two of its variants. Both variants produce an inactive 2C19 enzyme form that is ineffective in metabolizing 2C19 substrates.
Clinical Use
When should a psychiatrist obtain 2D6 and 2C19 genotypes?
First, understand that the pharmacogenomic chip does not predict which medications will produce a therapeutic response. Gene chips that predict response are in development but probably will not be available before 2008.
The chip, however, can identify the relatively few ultrarapid metabolizers who will not benefit from 2D6 or 2C19 substrate medications at normal dosages, as well as “poor metabolizers” of these substrates.1 The approximately 1% of whites in the United States who have ≥3 copies of the 2D6 gene metabolize 2D6 substrates very rapidly and will not respond to recommended dosages. About 10% of whites in the United States metabolize 2D6 or 2C19 substrates poorly and face increased risk of adverse reactions from these medications.
There is some evidence that the prevalence of these genetic variations differ among ethnicities. Approximately 15% of Saudi Arabians and 20% of Ethiopians are ultrarapid metabolizers of 2D6 and 2C19 substrates.5,6
The most common 2D6 poor metabolizer allele (*4) has been found in 12% to 21% of whites, whereas 23% to 32% of Asians and 13% of whites have the most common 2C19 poor metabolizer allele (*2).6-10 Prevalence of poor 2D6 and/or 2C19 metabolism among African Americans, Hispanics, and Native Americans has not been established.
Clinical Practicality
Clinicians’ unfamiliarity with genotyping and cost concerns pose potential barriers to the test’s use.
Clinician knowledge. Pharmacogenomic 2D6 and 2C19 tests will soon be offered nationwide at reference laboratories such as Quest Diagnostics, Labcore, and Mayo Medical Laboratories. The psychiatrist can call the lab for instructions, then send a blood sample and receive results by mail within 2 to 3 days.
While I believe the test’s usefulness will soon be widely understood, courses are available to help clinicians learn about genetic testing. Mayo Clinic College of Medicine (http://www.mayo. edu/cme/genomics.html) offers an annual week-long CME course in August. The American Psychiatric Association, as part of its May 2006 annual meeting, will offer a similar half-day course led by Mayo Clinic psychiatrists.
Cost. The exact cost of using the pharmacogenomic chip varies, as each laboratory sets fees for genotyping. Even so, genotyping could offer enormous cost savings by preventing failed medication trials and reducing the need for more-intensive psychiatric care. Furthermore, many insurance companies cover genotype testing.
Related resources
- Pharmacogenomic diagnostic DNA chip product information. www.rochediagnostics.com/products_services/amplichip_cyp450.html.
- Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103(3):173-92.
Drug brand names
- Amitriptyline • Elavil
- Atomoxetine • Strattera
- Citalopram • Celexa
- Clomipramine • Anafranil
- Desipramine • Norpramin
- Diazepam • Valium
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Nortriptyline • Pamelor
- Paroxetine • Paxil
- Perphenazine • Trilafon
- Risperidone • Risperdal
Disclosure
Dr. Mrazek is a consultant to Predix Pharmaceuticals.
1. Mrazek DA. New tool: genotyping makes prescribing safer, more effective. Current Psychiatry 2004;3(9):11-23.
2. Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103:173-92.
3. Kirchheiner J, Nickchen K, Bauer M, et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol Psychiatry 2004;9:442-73.
4. Chou WH, Yan FX, Robbins-Weilert DK, et al. Comparison of two CYP2D6 genotyping methods and assessment of genotype-phenotype relationships. Clin Chem 2003;49:542-51.
5. Ingelman-Sundberg M, Oscarson M, McLellan RA. Polymorphic human cytochrome P450 enzymes: an opportunity for individualized drug treatment. Trends Pharmacol Sci 1999;20:342-9.
6. Phillips KA, Veenstra DL, Oren E, et al. Potential role of pharmacogenomics in reducing adverse drug reactions: a systematic review. JAMA 2001;286:2270-9.
7. Ingelman-Sundberg M. Pharmacogenetics: an opportunity for a safer and more efficient pharmacotherapy. J Intern Med 2001;250:186-200.
8. Mizutani T. PM frequencies of major CYPs in Asians and Caucasians. Drug Metab Rev 2003;35:99-106.
9. Griese EU, Ilett KF, Kitteringham NR, et al. Allele and genotype frequencies of polymorphic cytochromes P450 2D6, 2C19, and 2E1 in aborigines from western Australia. Pharmacogenetics 2001;11:69-76.
10. Sachse C, Brockmoller J, Bauer S, Roots I. Cytochrome P450 2D6 variants in a Caucasian population: allele frequencies and phenotypic consequences. Am J Hum Genet 1997;60:284-95.
Genotyping for cytochrome (CYP) P-450 gene variations can identify patients who will not benefit from, or may react badly to, some psychotropics.1 Psychiatrists can then more accurately tailor initial dosages to improve response and prevent adverse reactions.
An FDA-approved pharmacogenomic diagnostic DNA chip is expected to be available to clinical laboratories this month (Table 1). The chip provides an accurate genotype for two drug-metabolizing enzymes—2D6 and 2C19.
Table 1
Pharmacogenomic DNA chip: Fast facts
| Brand name: |
| AmpliChip CYP 450 Test |
| FDA-approved indication: |
| Genotyping patients |
| Manufacturer: |
| Roche Diagnostics |
| Estimated availability: |
| July 2005 |
| Recommended use: |
| Determining cytochrome P-450 2D6 and 2C19 gene variations in patients before prescribing a psychotropic metabolized through these pathways. |
| Laboratories that process AmpliChip results: |
| Labcore, Mayo Medical Laboratories, Quest Diagnostics |
Genotyping’S Role in Psychiatry
CYP 2D6 and 2C19 enzymes help metabolize many commonly prescribed psychotropics, including:
- fluoxetine, paroxetine, and venlafaxine, which are among the psychotropics primarily metabolized by the cytochrome P-450 2D6 enzyme (Table 2).
- amitriptyline and citalopram, which are among the psychotropics metabolized in part by 2C19 (Table 3).
The chip can identify patients who are genetically predisposed to abnormal metabolism of 2D6 and 2C19 substrates. This information can help psychiatrists improve response for ultrarapid metabolizers and minimize adverse effects experienced by poor metabolizers of these substrates.
For example, if the patient is an ultrarapid metabolizer of 2D6 and/or 2C19 substrates, the psychiatrist can:
- exceed the recommended dosage to reach adequate serum levels
- or choose an antidepressant not primarily metabolized by either enzyme.
For a poor metabolizer of 2D6 and/or 2C19 substrates, the psychiatrist can:
- choose an antidepressant metabolized by a different enzyme
- or prescribe 2D6 and 2C19 substrates at very low dosages.
For example, some poor metabolizers of 2D6 substrates have been successfully treated with fluoxetine, 2 to 5 mg/d.2,3 This approach can help avoid side effects and potentially save the patient money. To prevent prescription errors, make sure the pharmacist understands your rationale for lower-than-recommended dosages.
Patients who are poor metabolizers of 2C19 and extensive metabolizers of 2D6 substrates can probably tolerate citalopram and amitriptyline dosages at the low end of the therapeutic range. Watch for high serum levels of either or both drugs if both enzyme systems are inactive.
Table 2
Evidence suggests these drugs are predominantly metabolized by the 2D6 enzyme*
| Antidepressants | Antipsychotics | Stimulants |
|---|---|---|
| Desipramine | Fluphenazine | Atomoxetine |
| Fluoxetine | Perphenazine | |
| Nortriptyline | Risperidone | |
| Paroxetine | Thioridazine | |
| Venlafaxine | ||
| *Use caution when prescribing these agents to patients who are poor 2D6 metabolizers. | ||
Table 3
Evidence suggests these drugs are predominantly metabolized by the 2C19 enzyme*
| Antidepressants | |
|---|---|
| Diazepam | Citalopram |
| Clomipramine | Escitalopram |
| Imipramine | Sertraline |
| Benzodiazepines | |
| Amitriptyline | |
| *Use caution when prescribing these agents to patients who are poor 2C19 metabolizers. | |
Pharmacogenomic Chip’s Accuracy
The 2D6 gene has more than 100 variations, many of which are very rare mutations. The pharmacogenomic DNA chip can detect 27 of these variants, allowing the chip to accurately genotype most patients. By contrast, early 2D6 genotyping techniques identified only four or five variants, resulting in too many false negatives for clinical use.4
The chip also can identify the normal form of the 2C19 gene and two of its variants. Both variants produce an inactive 2C19 enzyme form that is ineffective in metabolizing 2C19 substrates.
Clinical Use
When should a psychiatrist obtain 2D6 and 2C19 genotypes?
First, understand that the pharmacogenomic chip does not predict which medications will produce a therapeutic response. Gene chips that predict response are in development but probably will not be available before 2008.
The chip, however, can identify the relatively few ultrarapid metabolizers who will not benefit from 2D6 or 2C19 substrate medications at normal dosages, as well as “poor metabolizers” of these substrates.1 The approximately 1% of whites in the United States who have ≥3 copies of the 2D6 gene metabolize 2D6 substrates very rapidly and will not respond to recommended dosages. About 10% of whites in the United States metabolize 2D6 or 2C19 substrates poorly and face increased risk of adverse reactions from these medications.
There is some evidence that the prevalence of these genetic variations differ among ethnicities. Approximately 15% of Saudi Arabians and 20% of Ethiopians are ultrarapid metabolizers of 2D6 and 2C19 substrates.5,6
The most common 2D6 poor metabolizer allele (*4) has been found in 12% to 21% of whites, whereas 23% to 32% of Asians and 13% of whites have the most common 2C19 poor metabolizer allele (*2).6-10 Prevalence of poor 2D6 and/or 2C19 metabolism among African Americans, Hispanics, and Native Americans has not been established.
Clinical Practicality
Clinicians’ unfamiliarity with genotyping and cost concerns pose potential barriers to the test’s use.
Clinician knowledge. Pharmacogenomic 2D6 and 2C19 tests will soon be offered nationwide at reference laboratories such as Quest Diagnostics, Labcore, and Mayo Medical Laboratories. The psychiatrist can call the lab for instructions, then send a blood sample and receive results by mail within 2 to 3 days.
While I believe the test’s usefulness will soon be widely understood, courses are available to help clinicians learn about genetic testing. Mayo Clinic College of Medicine (http://www.mayo. edu/cme/genomics.html) offers an annual week-long CME course in August. The American Psychiatric Association, as part of its May 2006 annual meeting, will offer a similar half-day course led by Mayo Clinic psychiatrists.
Cost. The exact cost of using the pharmacogenomic chip varies, as each laboratory sets fees for genotyping. Even so, genotyping could offer enormous cost savings by preventing failed medication trials and reducing the need for more-intensive psychiatric care. Furthermore, many insurance companies cover genotype testing.
Related resources
- Pharmacogenomic diagnostic DNA chip product information. www.rochediagnostics.com/products_services/amplichip_cyp450.html.
- Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103(3):173-92.
Drug brand names
- Amitriptyline • Elavil
- Atomoxetine • Strattera
- Citalopram • Celexa
- Clomipramine • Anafranil
- Desipramine • Norpramin
- Diazepam • Valium
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Nortriptyline • Pamelor
- Paroxetine • Paxil
- Perphenazine • Trilafon
- Risperidone • Risperdal
Disclosure
Dr. Mrazek is a consultant to Predix Pharmaceuticals.
Genotyping for cytochrome (CYP) P-450 gene variations can identify patients who will not benefit from, or may react badly to, some psychotropics.1 Psychiatrists can then more accurately tailor initial dosages to improve response and prevent adverse reactions.
An FDA-approved pharmacogenomic diagnostic DNA chip is expected to be available to clinical laboratories this month (Table 1). The chip provides an accurate genotype for two drug-metabolizing enzymes—2D6 and 2C19.
Table 1
Pharmacogenomic DNA chip: Fast facts
| Brand name: |
| AmpliChip CYP 450 Test |
| FDA-approved indication: |
| Genotyping patients |
| Manufacturer: |
| Roche Diagnostics |
| Estimated availability: |
| July 2005 |
| Recommended use: |
| Determining cytochrome P-450 2D6 and 2C19 gene variations in patients before prescribing a psychotropic metabolized through these pathways. |
| Laboratories that process AmpliChip results: |
| Labcore, Mayo Medical Laboratories, Quest Diagnostics |
Genotyping’S Role in Psychiatry
CYP 2D6 and 2C19 enzymes help metabolize many commonly prescribed psychotropics, including:
- fluoxetine, paroxetine, and venlafaxine, which are among the psychotropics primarily metabolized by the cytochrome P-450 2D6 enzyme (Table 2).
- amitriptyline and citalopram, which are among the psychotropics metabolized in part by 2C19 (Table 3).
The chip can identify patients who are genetically predisposed to abnormal metabolism of 2D6 and 2C19 substrates. This information can help psychiatrists improve response for ultrarapid metabolizers and minimize adverse effects experienced by poor metabolizers of these substrates.
For example, if the patient is an ultrarapid metabolizer of 2D6 and/or 2C19 substrates, the psychiatrist can:
- exceed the recommended dosage to reach adequate serum levels
- or choose an antidepressant not primarily metabolized by either enzyme.
For a poor metabolizer of 2D6 and/or 2C19 substrates, the psychiatrist can:
- choose an antidepressant metabolized by a different enzyme
- or prescribe 2D6 and 2C19 substrates at very low dosages.
For example, some poor metabolizers of 2D6 substrates have been successfully treated with fluoxetine, 2 to 5 mg/d.2,3 This approach can help avoid side effects and potentially save the patient money. To prevent prescription errors, make sure the pharmacist understands your rationale for lower-than-recommended dosages.
Patients who are poor metabolizers of 2C19 and extensive metabolizers of 2D6 substrates can probably tolerate citalopram and amitriptyline dosages at the low end of the therapeutic range. Watch for high serum levels of either or both drugs if both enzyme systems are inactive.
Table 2
Evidence suggests these drugs are predominantly metabolized by the 2D6 enzyme*
| Antidepressants | Antipsychotics | Stimulants |
|---|---|---|
| Desipramine | Fluphenazine | Atomoxetine |
| Fluoxetine | Perphenazine | |
| Nortriptyline | Risperidone | |
| Paroxetine | Thioridazine | |
| Venlafaxine | ||
| *Use caution when prescribing these agents to patients who are poor 2D6 metabolizers. | ||
Table 3
Evidence suggests these drugs are predominantly metabolized by the 2C19 enzyme*
| Antidepressants | |
|---|---|
| Diazepam | Citalopram |
| Clomipramine | Escitalopram |
| Imipramine | Sertraline |
| Benzodiazepines | |
| Amitriptyline | |
| *Use caution when prescribing these agents to patients who are poor 2C19 metabolizers. | |
Pharmacogenomic Chip’s Accuracy
The 2D6 gene has more than 100 variations, many of which are very rare mutations. The pharmacogenomic DNA chip can detect 27 of these variants, allowing the chip to accurately genotype most patients. By contrast, early 2D6 genotyping techniques identified only four or five variants, resulting in too many false negatives for clinical use.4
The chip also can identify the normal form of the 2C19 gene and two of its variants. Both variants produce an inactive 2C19 enzyme form that is ineffective in metabolizing 2C19 substrates.
Clinical Use
When should a psychiatrist obtain 2D6 and 2C19 genotypes?
First, understand that the pharmacogenomic chip does not predict which medications will produce a therapeutic response. Gene chips that predict response are in development but probably will not be available before 2008.
The chip, however, can identify the relatively few ultrarapid metabolizers who will not benefit from 2D6 or 2C19 substrate medications at normal dosages, as well as “poor metabolizers” of these substrates.1 The approximately 1% of whites in the United States who have ≥3 copies of the 2D6 gene metabolize 2D6 substrates very rapidly and will not respond to recommended dosages. About 10% of whites in the United States metabolize 2D6 or 2C19 substrates poorly and face increased risk of adverse reactions from these medications.
There is some evidence that the prevalence of these genetic variations differ among ethnicities. Approximately 15% of Saudi Arabians and 20% of Ethiopians are ultrarapid metabolizers of 2D6 and 2C19 substrates.5,6
The most common 2D6 poor metabolizer allele (*4) has been found in 12% to 21% of whites, whereas 23% to 32% of Asians and 13% of whites have the most common 2C19 poor metabolizer allele (*2).6-10 Prevalence of poor 2D6 and/or 2C19 metabolism among African Americans, Hispanics, and Native Americans has not been established.
Clinical Practicality
Clinicians’ unfamiliarity with genotyping and cost concerns pose potential barriers to the test’s use.
Clinician knowledge. Pharmacogenomic 2D6 and 2C19 tests will soon be offered nationwide at reference laboratories such as Quest Diagnostics, Labcore, and Mayo Medical Laboratories. The psychiatrist can call the lab for instructions, then send a blood sample and receive results by mail within 2 to 3 days.
While I believe the test’s usefulness will soon be widely understood, courses are available to help clinicians learn about genetic testing. Mayo Clinic College of Medicine (http://www.mayo. edu/cme/genomics.html) offers an annual week-long CME course in August. The American Psychiatric Association, as part of its May 2006 annual meeting, will offer a similar half-day course led by Mayo Clinic psychiatrists.
Cost. The exact cost of using the pharmacogenomic chip varies, as each laboratory sets fees for genotyping. Even so, genotyping could offer enormous cost savings by preventing failed medication trials and reducing the need for more-intensive psychiatric care. Furthermore, many insurance companies cover genotype testing.
Related resources
- Pharmacogenomic diagnostic DNA chip product information. www.rochediagnostics.com/products_services/amplichip_cyp450.html.
- Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103(3):173-92.
Drug brand names
- Amitriptyline • Elavil
- Atomoxetine • Strattera
- Citalopram • Celexa
- Clomipramine • Anafranil
- Desipramine • Norpramin
- Diazepam • Valium
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Nortriptyline • Pamelor
- Paroxetine • Paxil
- Perphenazine • Trilafon
- Risperidone • Risperdal
Disclosure
Dr. Mrazek is a consultant to Predix Pharmaceuticals.
1. Mrazek DA. New tool: genotyping makes prescribing safer, more effective. Current Psychiatry 2004;3(9):11-23.
2. Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103:173-92.
3. Kirchheiner J, Nickchen K, Bauer M, et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol Psychiatry 2004;9:442-73.
4. Chou WH, Yan FX, Robbins-Weilert DK, et al. Comparison of two CYP2D6 genotyping methods and assessment of genotype-phenotype relationships. Clin Chem 2003;49:542-51.
5. Ingelman-Sundberg M, Oscarson M, McLellan RA. Polymorphic human cytochrome P450 enzymes: an opportunity for individualized drug treatment. Trends Pharmacol Sci 1999;20:342-9.
6. Phillips KA, Veenstra DL, Oren E, et al. Potential role of pharmacogenomics in reducing adverse drug reactions: a systematic review. JAMA 2001;286:2270-9.
7. Ingelman-Sundberg M. Pharmacogenetics: an opportunity for a safer and more efficient pharmacotherapy. J Intern Med 2001;250:186-200.
8. Mizutani T. PM frequencies of major CYPs in Asians and Caucasians. Drug Metab Rev 2003;35:99-106.
9. Griese EU, Ilett KF, Kitteringham NR, et al. Allele and genotype frequencies of polymorphic cytochromes P450 2D6, 2C19, and 2E1 in aborigines from western Australia. Pharmacogenetics 2001;11:69-76.
10. Sachse C, Brockmoller J, Bauer S, Roots I. Cytochrome P450 2D6 variants in a Caucasian population: allele frequencies and phenotypic consequences. Am J Hum Genet 1997;60:284-95.
1. Mrazek DA. New tool: genotyping makes prescribing safer, more effective. Current Psychiatry 2004;3(9):11-23.
2. Kirchheiner J, Borsen K, Dahl ML, et al. CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a first step towards subpopulation-specific dosages. Acta Psychiatr Scand 2001;103:173-92.
3. Kirchheiner J, Nickchen K, Bauer M, et al. Pharmacogenetics of antidepressants and antipsychotics: the contribution of allelic variations to the phenotype of drug response. Mol Psychiatry 2004;9:442-73.
4. Chou WH, Yan FX, Robbins-Weilert DK, et al. Comparison of two CYP2D6 genotyping methods and assessment of genotype-phenotype relationships. Clin Chem 2003;49:542-51.
5. Ingelman-Sundberg M, Oscarson M, McLellan RA. Polymorphic human cytochrome P450 enzymes: an opportunity for individualized drug treatment. Trends Pharmacol Sci 1999;20:342-9.
6. Phillips KA, Veenstra DL, Oren E, et al. Potential role of pharmacogenomics in reducing adverse drug reactions: a systematic review. JAMA 2001;286:2270-9.
7. Ingelman-Sundberg M. Pharmacogenetics: an opportunity for a safer and more efficient pharmacotherapy. J Intern Med 2001;250:186-200.
8. Mizutani T. PM frequencies of major CYPs in Asians and Caucasians. Drug Metab Rev 2003;35:99-106.
9. Griese EU, Ilett KF, Kitteringham NR, et al. Allele and genotype frequencies of polymorphic cytochromes P450 2D6, 2C19, and 2E1 in aborigines from western Australia. Pharmacogenetics 2001;11:69-76.
10. Sachse C, Brockmoller J, Bauer S, Roots I. Cytochrome P450 2D6 variants in a Caucasian population: allele frequencies and phenotypic consequences. Am J Hum Genet 1997;60:284-95.
Carbohydrate-deficient transferrin test
Biomarkers can help psychiatrists monitor abstinence and detect relapse in patients with alcohol use disorders. A blood test that measures carbohydrate-deficient transferrin percentage (%CDT) is FDA-approved for detecting heavy drinking (Table 1)1 and has shown effectiveness in several medical and surgical uses.
Table 1
%CDT test: Fast facts
| Brand names: |
| Axis-Shield %CDT, Bio-Rad %CDT TIA, Tina-quant (a) %CDT |
| FDA-approved indication: |
| Testing for excessive alcohol use |
| Manufacturers: |
| Axis-Shield PLC, Bio-Rad Laboratories, Roche Diagnostics |
| Recommended use: |
| Detecting heavy alcohol consumption, monitoring abstinence, and identifying elapse in patients with alcohol use disorders |
| Laboratories that process %CDT results: |
|
HOW %CDT SIGNALS ALCOHOL ABUSE
In 1976, Swedish researchers detected transferrin fractions in the CSF of alcoholic patients but not in nonalcoholics.2 One of these fractions was also present in alcoholics’ serum. This discovery led to the CDT biomarker.
%CDT testing is based on the finding that consuming an average >60 grams of alcohol (about 5 standard drinks) daily for ≥ 2 weeks causes a higher percentage of transferrin—a glycoprotein that transports iron in the blood—to lack its usual carbohydrate content. Transferrin appears in 6 isoforms, and studies have shown that heavy drinking increases three of these—asialo, mono sialo, and disialo transferrin—a state collectively called “carbohydrate-deficient.”
A %CDT reading ≥ 2.6 indicates that a patient may have had on average at least 5 alcoholic drinks daily for ≥ 2 weeks. Because CDT has a short mean half-life (7 to 14 days), readings >2.6 may suggest much heavier drinking at some time before the blood sample was taken.
Microcolumn chromatography separation assays that measure CDT as a percentage of total circulating transferrin have replaced initial CDT assays that used isoelectric focusing separation and immunoassay systems. This advance corrected for individual transferrin level variations.
CLINICAL USE
The %CDT test has shown effectiveness in several medical and surgical uses, including
- screening patients with diseases possibly triggered by alcohol use, such as treatment-resistant hypertension, gastroesophageal reflux disease, or depression
- detecting alcohol-use disorders in hospitalized patients
- screening presurgical and trauma patients to predict alcohol withdrawal syndrome and/or postsurgical complications.3,4
Although no alcohol biomarker alone reliably confirms alcohol abuse/dependence, %CDT can corroborate initial clinical impressions, especially when the patient’s self-report is suspect or information from significant others is not available.
Among biomarkers, %CDT most accurately predicts alcohol withdrawal syndrome in men (mean corpuscular volume [MCV] measurements are more accurate in women).4 During psychiatric consults, the test may help confirm suspected alcohol use in patients admitted to inpatient medical, surgical, or trauma units.
%CDT may also help monitor abstinence when treating an alcohol use disorder. In a major prospective treatment outcome study, %CDT was more sensitive than gamma-glutamyltransferase (GGT) in detecting relapse in male but not female alcoholics.5 Patients who abstained from drinking for 12 weeks showed a 30% decrease in %CDT. Subjects who relapsed during treatment later showed a 60% increase in %CDT, indicating sustained heavy drinking.
%CDT decreased over the first 4 weeks of treatment and continued to decline over time for abstinent patients (Figure). After week 4, %CDT increased again for those who consumed ≥ 5 drinks per day or had a full-blown relapse.5
Together, routine %CDT and GGT testing during treatment and follow-up can help clinicians monitor a patient’s progress and provide accurate reports to courts, child welfare agencies, and programs for impaired professionals. A 30% reduction from baseline in either biomarker indicates abstinence or significantly reduced alcohol consumption, whereas a 30% increase suggests relapse.6 Consider all clinical information in the final analysis, however.
%CDT results also provide an objective basis for discussing relapses with patients. Those who are responding to treatment often welcome %CDT testing as proof they are abstaining from alcohol.
Figure %CDT changes with alcohol use or abstinence
CLINICAL PRACTICALITY
The %CDT test is an immunoassay with ion-exchange column separation followed by turbidimetric measurement. Because the procedure is complex, laboratories generally require 24 to 72 hours to test the sample.
Because the %CDT test is relatively new, pricing, reimbursement rates, and availability are not well established. Pricing will likely vary widely from state to state and among laboratories and insurers. Medicare reimburses approximately $25 for %CDT testing, compared with $10 for GGT testing.
Only select reference laboratories offer %CDT testing, but the test should become more widely available over time (Table 1).
SENSITIVITY AND SPECIFICITY
%CDT test sensitivity is 70% to 90% in male inpatient alcoholics (usually drinking within 4 to 7 days of blood draw), with specificities >90%. Sensitivity is 12% to 40% in general populations—among whom underreporting of drinking may lead to more false positives—and 40% to 60% in outpatient alcoholics.7,8 Specificity, however, remains high—80% to 90%—when sensitivity is reduced.
Overall, %CDT is as sensitive as GGT and more sensitive than other biomarkers, including MCV, aspartate transaminase, and alanine transaminase.9 %CDT is more specific than GGT (92% vs. 75%) and other biomarkers. %CDT appears to be more sensitive in men, whereas GGT is more sensitive in women (Table 2).10
Ample evidence, however, suggests that %CDT and GGT readings together may provide a comprehensive picture of recent alcohol abuse.10 Whereas frequent drinking alters %CDT, GGT signals drinking intensity and is more effective at detecting episodic binge drinking (Table 2). This probably explains why problem drinkers age <20—who typically binge-drink—show little or no change in %CDT.
Table 2
%CDT vs GGT testing for recent alcohol abuse
| %CDT | GGT | |
|---|---|---|
| Advantages |
|
|
| Disadvantages |
|
|
| Cutoff value |
|
|
| %CDT: Carbohydrate-deficient transferrin percentage | ||
| GGT: Gamma-glutamyltransferase | ||
FACTORS THAT ALTER RESULTS
Unlike GGT and MCV, few medical conditions distort %CDT,11 meaning the test is accurate in patients with most medical conditions. Only end-stage liver disease, biliary cirrhosis, and rare genetic transferrin variants alter %CDT.
Women tend to have higher and more-variable CDT values than do men, possibly because of variability in normal transferrin levels, anemia secondary to iron deficiency, pregnancy, use of oral contraceptives, or menopause. %CDT cutoff scores are not gender-specific, however, because percentage rather than absolute CDT is typically measured.
%CDT seems to decrease slightly as body mass index increases, suggesting a small but significant inverse relationship between weight and %CDT. Smoking seems to raise %CDT values slightly. The specifics of and reasons for these relationships are unclear.
Related resources
- Axis-Shield. www.axis-shield.com. Click on “Lab diagnostics,” then “Alcohol-related.”
Disclosure
Dr. Anton is a consultant to Axis-Shield.
Dr. Miller and Ms. Dominick report no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
1. Anton RF, Dominick C, Bigelow M, Westby C. CDTect Research Group. Comparison of Bio-Rad %CDT TIA and CDTect as laboratory markers of heavy alcohol use and their relationships with gamma-glutamyltransferase. Clin Chem 2001;47:1769-75.
2. Stibler H, Kjellin KG. Isoelectric focusing and electrophoresis of the CSF proteins on tremor of different origins. J Neurol 1976;30:269-85.
3. Miller PM. Recent developments in alcohol biomarkers: Implications for managing alcohol-sensitive diseases. Resid Staff Physician 2004;50:11-17.
4. Spies CD, Kissner M, Neumann T, et al. Elevated carbohydrate-deficient transferrin predicts prolonged intensive care unit stay in traumatized men. Alcohol Alcohol 1998;33:661-9.
5. Anton RF, Moak DH, Latham P. Carbohydrate-deficient transferrin as an indicator of drinking status during a treatment outcome study. Alcohol Clin Exp Res 1996;20:841-6.
6. Anton RF, Lieber C, Tabakoff B. Carbohydrate-deficient transferrin and gamma-glutamyltransferase for the detection and monitoring of alcohol use: results from a multisite study. Alcohol Clin Exp Res 2002;26:1215-22.
7. Allen JP, Litten RZ, Anton RF, Cross GM. Carbohydrate-deficient transferrin as a measure of immoderate drinking: remaining issues. Alcohol Clin Exp Res 1994;18:799-812.
8. Gronbaek M, Henriksen JH, Becker U. Carbohydrate-deficient transferrin—a valid marker of alcoholism in population studies? Results from the Copenhagen City Heart Study. Alcohol Clin Exp Res 1995;19:457-61.
9. Bell H, Tallaksen CM, Try K, Haug E. Carbohydrate-deficient transferrin and other markers of high alcohol consumption: A study of 502 patients admitted consecutively to a medical department. Alcohol Clin Exp Res 1994;18:1103-8.
10. Conigrave KM, Degenhardt LJ, Whitfield JB, et al. CDT, GGT, and AST as markers of alcohol use: The WHO/ISBRA Collaborative Project. Alcohol Clin Exp Res 2002;26:332-9.
11. Meerkerk GJ, Njoo KH, Bongers IM, et al. The specificity of the CDT assay in general practice: the influence of common chronic diseases and medication on the serum CDT concentration. Alcohol Clin Exp Res 1998;22:908-13.
Biomarkers can help psychiatrists monitor abstinence and detect relapse in patients with alcohol use disorders. A blood test that measures carbohydrate-deficient transferrin percentage (%CDT) is FDA-approved for detecting heavy drinking (Table 1)1 and has shown effectiveness in several medical and surgical uses.
Table 1
%CDT test: Fast facts
| Brand names: |
| Axis-Shield %CDT, Bio-Rad %CDT TIA, Tina-quant (a) %CDT |
| FDA-approved indication: |
| Testing for excessive alcohol use |
| Manufacturers: |
| Axis-Shield PLC, Bio-Rad Laboratories, Roche Diagnostics |
| Recommended use: |
| Detecting heavy alcohol consumption, monitoring abstinence, and identifying elapse in patients with alcohol use disorders |
| Laboratories that process %CDT results: |
|
HOW %CDT SIGNALS ALCOHOL ABUSE
In 1976, Swedish researchers detected transferrin fractions in the CSF of alcoholic patients but not in nonalcoholics.2 One of these fractions was also present in alcoholics’ serum. This discovery led to the CDT biomarker.
%CDT testing is based on the finding that consuming an average >60 grams of alcohol (about 5 standard drinks) daily for ≥ 2 weeks causes a higher percentage of transferrin—a glycoprotein that transports iron in the blood—to lack its usual carbohydrate content. Transferrin appears in 6 isoforms, and studies have shown that heavy drinking increases three of these—asialo, mono sialo, and disialo transferrin—a state collectively called “carbohydrate-deficient.”
A %CDT reading ≥ 2.6 indicates that a patient may have had on average at least 5 alcoholic drinks daily for ≥ 2 weeks. Because CDT has a short mean half-life (7 to 14 days), readings >2.6 may suggest much heavier drinking at some time before the blood sample was taken.
Microcolumn chromatography separation assays that measure CDT as a percentage of total circulating transferrin have replaced initial CDT assays that used isoelectric focusing separation and immunoassay systems. This advance corrected for individual transferrin level variations.
CLINICAL USE
The %CDT test has shown effectiveness in several medical and surgical uses, including
- screening patients with diseases possibly triggered by alcohol use, such as treatment-resistant hypertension, gastroesophageal reflux disease, or depression
- detecting alcohol-use disorders in hospitalized patients
- screening presurgical and trauma patients to predict alcohol withdrawal syndrome and/or postsurgical complications.3,4
Although no alcohol biomarker alone reliably confirms alcohol abuse/dependence, %CDT can corroborate initial clinical impressions, especially when the patient’s self-report is suspect or information from significant others is not available.
Among biomarkers, %CDT most accurately predicts alcohol withdrawal syndrome in men (mean corpuscular volume [MCV] measurements are more accurate in women).4 During psychiatric consults, the test may help confirm suspected alcohol use in patients admitted to inpatient medical, surgical, or trauma units.
%CDT may also help monitor abstinence when treating an alcohol use disorder. In a major prospective treatment outcome study, %CDT was more sensitive than gamma-glutamyltransferase (GGT) in detecting relapse in male but not female alcoholics.5 Patients who abstained from drinking for 12 weeks showed a 30% decrease in %CDT. Subjects who relapsed during treatment later showed a 60% increase in %CDT, indicating sustained heavy drinking.
%CDT decreased over the first 4 weeks of treatment and continued to decline over time for abstinent patients (Figure). After week 4, %CDT increased again for those who consumed ≥ 5 drinks per day or had a full-blown relapse.5
Together, routine %CDT and GGT testing during treatment and follow-up can help clinicians monitor a patient’s progress and provide accurate reports to courts, child welfare agencies, and programs for impaired professionals. A 30% reduction from baseline in either biomarker indicates abstinence or significantly reduced alcohol consumption, whereas a 30% increase suggests relapse.6 Consider all clinical information in the final analysis, however.
%CDT results also provide an objective basis for discussing relapses with patients. Those who are responding to treatment often welcome %CDT testing as proof they are abstaining from alcohol.
Figure %CDT changes with alcohol use or abstinence
CLINICAL PRACTICALITY
The %CDT test is an immunoassay with ion-exchange column separation followed by turbidimetric measurement. Because the procedure is complex, laboratories generally require 24 to 72 hours to test the sample.
Because the %CDT test is relatively new, pricing, reimbursement rates, and availability are not well established. Pricing will likely vary widely from state to state and among laboratories and insurers. Medicare reimburses approximately $25 for %CDT testing, compared with $10 for GGT testing.
Only select reference laboratories offer %CDT testing, but the test should become more widely available over time (Table 1).
SENSITIVITY AND SPECIFICITY
%CDT test sensitivity is 70% to 90% in male inpatient alcoholics (usually drinking within 4 to 7 days of blood draw), with specificities >90%. Sensitivity is 12% to 40% in general populations—among whom underreporting of drinking may lead to more false positives—and 40% to 60% in outpatient alcoholics.7,8 Specificity, however, remains high—80% to 90%—when sensitivity is reduced.
Overall, %CDT is as sensitive as GGT and more sensitive than other biomarkers, including MCV, aspartate transaminase, and alanine transaminase.9 %CDT is more specific than GGT (92% vs. 75%) and other biomarkers. %CDT appears to be more sensitive in men, whereas GGT is more sensitive in women (Table 2).10
Ample evidence, however, suggests that %CDT and GGT readings together may provide a comprehensive picture of recent alcohol abuse.10 Whereas frequent drinking alters %CDT, GGT signals drinking intensity and is more effective at detecting episodic binge drinking (Table 2). This probably explains why problem drinkers age <20—who typically binge-drink—show little or no change in %CDT.
Table 2
%CDT vs GGT testing for recent alcohol abuse
| %CDT | GGT | |
|---|---|---|
| Advantages |
|
|
| Disadvantages |
|
|
| Cutoff value |
|
|
| %CDT: Carbohydrate-deficient transferrin percentage | ||
| GGT: Gamma-glutamyltransferase | ||
FACTORS THAT ALTER RESULTS
Unlike GGT and MCV, few medical conditions distort %CDT,11 meaning the test is accurate in patients with most medical conditions. Only end-stage liver disease, biliary cirrhosis, and rare genetic transferrin variants alter %CDT.
Women tend to have higher and more-variable CDT values than do men, possibly because of variability in normal transferrin levels, anemia secondary to iron deficiency, pregnancy, use of oral contraceptives, or menopause. %CDT cutoff scores are not gender-specific, however, because percentage rather than absolute CDT is typically measured.
%CDT seems to decrease slightly as body mass index increases, suggesting a small but significant inverse relationship between weight and %CDT. Smoking seems to raise %CDT values slightly. The specifics of and reasons for these relationships are unclear.
Related resources
- Axis-Shield. www.axis-shield.com. Click on “Lab diagnostics,” then “Alcohol-related.”
Disclosure
Dr. Anton is a consultant to Axis-Shield.
Dr. Miller and Ms. Dominick report no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
Biomarkers can help psychiatrists monitor abstinence and detect relapse in patients with alcohol use disorders. A blood test that measures carbohydrate-deficient transferrin percentage (%CDT) is FDA-approved for detecting heavy drinking (Table 1)1 and has shown effectiveness in several medical and surgical uses.
Table 1
%CDT test: Fast facts
| Brand names: |
| Axis-Shield %CDT, Bio-Rad %CDT TIA, Tina-quant (a) %CDT |
| FDA-approved indication: |
| Testing for excessive alcohol use |
| Manufacturers: |
| Axis-Shield PLC, Bio-Rad Laboratories, Roche Diagnostics |
| Recommended use: |
| Detecting heavy alcohol consumption, monitoring abstinence, and identifying elapse in patients with alcohol use disorders |
| Laboratories that process %CDT results: |
|
HOW %CDT SIGNALS ALCOHOL ABUSE
In 1976, Swedish researchers detected transferrin fractions in the CSF of alcoholic patients but not in nonalcoholics.2 One of these fractions was also present in alcoholics’ serum. This discovery led to the CDT biomarker.
%CDT testing is based on the finding that consuming an average >60 grams of alcohol (about 5 standard drinks) daily for ≥ 2 weeks causes a higher percentage of transferrin—a glycoprotein that transports iron in the blood—to lack its usual carbohydrate content. Transferrin appears in 6 isoforms, and studies have shown that heavy drinking increases three of these—asialo, mono sialo, and disialo transferrin—a state collectively called “carbohydrate-deficient.”
A %CDT reading ≥ 2.6 indicates that a patient may have had on average at least 5 alcoholic drinks daily for ≥ 2 weeks. Because CDT has a short mean half-life (7 to 14 days), readings >2.6 may suggest much heavier drinking at some time before the blood sample was taken.
Microcolumn chromatography separation assays that measure CDT as a percentage of total circulating transferrin have replaced initial CDT assays that used isoelectric focusing separation and immunoassay systems. This advance corrected for individual transferrin level variations.
CLINICAL USE
The %CDT test has shown effectiveness in several medical and surgical uses, including
- screening patients with diseases possibly triggered by alcohol use, such as treatment-resistant hypertension, gastroesophageal reflux disease, or depression
- detecting alcohol-use disorders in hospitalized patients
- screening presurgical and trauma patients to predict alcohol withdrawal syndrome and/or postsurgical complications.3,4
Although no alcohol biomarker alone reliably confirms alcohol abuse/dependence, %CDT can corroborate initial clinical impressions, especially when the patient’s self-report is suspect or information from significant others is not available.
Among biomarkers, %CDT most accurately predicts alcohol withdrawal syndrome in men (mean corpuscular volume [MCV] measurements are more accurate in women).4 During psychiatric consults, the test may help confirm suspected alcohol use in patients admitted to inpatient medical, surgical, or trauma units.
%CDT may also help monitor abstinence when treating an alcohol use disorder. In a major prospective treatment outcome study, %CDT was more sensitive than gamma-glutamyltransferase (GGT) in detecting relapse in male but not female alcoholics.5 Patients who abstained from drinking for 12 weeks showed a 30% decrease in %CDT. Subjects who relapsed during treatment later showed a 60% increase in %CDT, indicating sustained heavy drinking.
%CDT decreased over the first 4 weeks of treatment and continued to decline over time for abstinent patients (Figure). After week 4, %CDT increased again for those who consumed ≥ 5 drinks per day or had a full-blown relapse.5
Together, routine %CDT and GGT testing during treatment and follow-up can help clinicians monitor a patient’s progress and provide accurate reports to courts, child welfare agencies, and programs for impaired professionals. A 30% reduction from baseline in either biomarker indicates abstinence or significantly reduced alcohol consumption, whereas a 30% increase suggests relapse.6 Consider all clinical information in the final analysis, however.
%CDT results also provide an objective basis for discussing relapses with patients. Those who are responding to treatment often welcome %CDT testing as proof they are abstaining from alcohol.
Figure %CDT changes with alcohol use or abstinence
CLINICAL PRACTICALITY
The %CDT test is an immunoassay with ion-exchange column separation followed by turbidimetric measurement. Because the procedure is complex, laboratories generally require 24 to 72 hours to test the sample.
Because the %CDT test is relatively new, pricing, reimbursement rates, and availability are not well established. Pricing will likely vary widely from state to state and among laboratories and insurers. Medicare reimburses approximately $25 for %CDT testing, compared with $10 for GGT testing.
Only select reference laboratories offer %CDT testing, but the test should become more widely available over time (Table 1).
SENSITIVITY AND SPECIFICITY
%CDT test sensitivity is 70% to 90% in male inpatient alcoholics (usually drinking within 4 to 7 days of blood draw), with specificities >90%. Sensitivity is 12% to 40% in general populations—among whom underreporting of drinking may lead to more false positives—and 40% to 60% in outpatient alcoholics.7,8 Specificity, however, remains high—80% to 90%—when sensitivity is reduced.
Overall, %CDT is as sensitive as GGT and more sensitive than other biomarkers, including MCV, aspartate transaminase, and alanine transaminase.9 %CDT is more specific than GGT (92% vs. 75%) and other biomarkers. %CDT appears to be more sensitive in men, whereas GGT is more sensitive in women (Table 2).10
Ample evidence, however, suggests that %CDT and GGT readings together may provide a comprehensive picture of recent alcohol abuse.10 Whereas frequent drinking alters %CDT, GGT signals drinking intensity and is more effective at detecting episodic binge drinking (Table 2). This probably explains why problem drinkers age <20—who typically binge-drink—show little or no change in %CDT.
Table 2
%CDT vs GGT testing for recent alcohol abuse
| %CDT | GGT | |
|---|---|---|
| Advantages |
|
|
| Disadvantages |
|
|
| Cutoff value |
|
|
| %CDT: Carbohydrate-deficient transferrin percentage | ||
| GGT: Gamma-glutamyltransferase | ||
FACTORS THAT ALTER RESULTS
Unlike GGT and MCV, few medical conditions distort %CDT,11 meaning the test is accurate in patients with most medical conditions. Only end-stage liver disease, biliary cirrhosis, and rare genetic transferrin variants alter %CDT.
Women tend to have higher and more-variable CDT values than do men, possibly because of variability in normal transferrin levels, anemia secondary to iron deficiency, pregnancy, use of oral contraceptives, or menopause. %CDT cutoff scores are not gender-specific, however, because percentage rather than absolute CDT is typically measured.
%CDT seems to decrease slightly as body mass index increases, suggesting a small but significant inverse relationship between weight and %CDT. Smoking seems to raise %CDT values slightly. The specifics of and reasons for these relationships are unclear.
Related resources
- Axis-Shield. www.axis-shield.com. Click on “Lab diagnostics,” then “Alcohol-related.”
Disclosure
Dr. Anton is a consultant to Axis-Shield.
Dr. Miller and Ms. Dominick report no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
1. Anton RF, Dominick C, Bigelow M, Westby C. CDTect Research Group. Comparison of Bio-Rad %CDT TIA and CDTect as laboratory markers of heavy alcohol use and their relationships with gamma-glutamyltransferase. Clin Chem 2001;47:1769-75.
2. Stibler H, Kjellin KG. Isoelectric focusing and electrophoresis of the CSF proteins on tremor of different origins. J Neurol 1976;30:269-85.
3. Miller PM. Recent developments in alcohol biomarkers: Implications for managing alcohol-sensitive diseases. Resid Staff Physician 2004;50:11-17.
4. Spies CD, Kissner M, Neumann T, et al. Elevated carbohydrate-deficient transferrin predicts prolonged intensive care unit stay in traumatized men. Alcohol Alcohol 1998;33:661-9.
5. Anton RF, Moak DH, Latham P. Carbohydrate-deficient transferrin as an indicator of drinking status during a treatment outcome study. Alcohol Clin Exp Res 1996;20:841-6.
6. Anton RF, Lieber C, Tabakoff B. Carbohydrate-deficient transferrin and gamma-glutamyltransferase for the detection and monitoring of alcohol use: results from a multisite study. Alcohol Clin Exp Res 2002;26:1215-22.
7. Allen JP, Litten RZ, Anton RF, Cross GM. Carbohydrate-deficient transferrin as a measure of immoderate drinking: remaining issues. Alcohol Clin Exp Res 1994;18:799-812.
8. Gronbaek M, Henriksen JH, Becker U. Carbohydrate-deficient transferrin—a valid marker of alcoholism in population studies? Results from the Copenhagen City Heart Study. Alcohol Clin Exp Res 1995;19:457-61.
9. Bell H, Tallaksen CM, Try K, Haug E. Carbohydrate-deficient transferrin and other markers of high alcohol consumption: A study of 502 patients admitted consecutively to a medical department. Alcohol Clin Exp Res 1994;18:1103-8.
10. Conigrave KM, Degenhardt LJ, Whitfield JB, et al. CDT, GGT, and AST as markers of alcohol use: The WHO/ISBRA Collaborative Project. Alcohol Clin Exp Res 2002;26:332-9.
11. Meerkerk GJ, Njoo KH, Bongers IM, et al. The specificity of the CDT assay in general practice: the influence of common chronic diseases and medication on the serum CDT concentration. Alcohol Clin Exp Res 1998;22:908-13.
1. Anton RF, Dominick C, Bigelow M, Westby C. CDTect Research Group. Comparison of Bio-Rad %CDT TIA and CDTect as laboratory markers of heavy alcohol use and their relationships with gamma-glutamyltransferase. Clin Chem 2001;47:1769-75.
2. Stibler H, Kjellin KG. Isoelectric focusing and electrophoresis of the CSF proteins on tremor of different origins. J Neurol 1976;30:269-85.
3. Miller PM. Recent developments in alcohol biomarkers: Implications for managing alcohol-sensitive diseases. Resid Staff Physician 2004;50:11-17.
4. Spies CD, Kissner M, Neumann T, et al. Elevated carbohydrate-deficient transferrin predicts prolonged intensive care unit stay in traumatized men. Alcohol Alcohol 1998;33:661-9.
5. Anton RF, Moak DH, Latham P. Carbohydrate-deficient transferrin as an indicator of drinking status during a treatment outcome study. Alcohol Clin Exp Res 1996;20:841-6.
6. Anton RF, Lieber C, Tabakoff B. Carbohydrate-deficient transferrin and gamma-glutamyltransferase for the detection and monitoring of alcohol use: results from a multisite study. Alcohol Clin Exp Res 2002;26:1215-22.
7. Allen JP, Litten RZ, Anton RF, Cross GM. Carbohydrate-deficient transferrin as a measure of immoderate drinking: remaining issues. Alcohol Clin Exp Res 1994;18:799-812.
8. Gronbaek M, Henriksen JH, Becker U. Carbohydrate-deficient transferrin—a valid marker of alcoholism in population studies? Results from the Copenhagen City Heart Study. Alcohol Clin Exp Res 1995;19:457-61.
9. Bell H, Tallaksen CM, Try K, Haug E. Carbohydrate-deficient transferrin and other markers of high alcohol consumption: A study of 502 patients admitted consecutively to a medical department. Alcohol Clin Exp Res 1994;18:1103-8.
10. Conigrave KM, Degenhardt LJ, Whitfield JB, et al. CDT, GGT, and AST as markers of alcohol use: The WHO/ISBRA Collaborative Project. Alcohol Clin Exp Res 2002;26:332-9.
11. Meerkerk GJ, Njoo KH, Bongers IM, et al. The specificity of the CDT assay in general practice: the influence of common chronic diseases and medication on the serum CDT concentration. Alcohol Clin Exp Res 1998;22:908-13.
Extended-release carbamazepine
Over 2 decades, carbamazepine has become a well-established, off-label alternative to lithium for treating acute mania. In December, the FDA approved an extended-release form of the anticonvulsant to treat type I bipolar disorder (Table 1).
This article addresses clinical use of extended-release capsules of carbamazepine (ERC-CBZ) and their safety, tolerability, and potential to interact with other medications.
Table 1
Extended-release capsules
of carbamazepine:
Fast facts
| Brand name: Equetro |
| Class: Anticonvulsant |
| FDA-approved indication: Bipolar mania |
| Manufacturer: Shire Pharmaceuticals Group |
| Dosing form: 100-, 200-, and 300-mg capsules |
| Recommended dosage: Manufacturer recommends starting at 400 mg/d in two divided doses.* Adjust dosage in 200-mg increments to achieve optimal response. Dosages >1,600 mg/d have not been studied. |
| *The author recommends starting at 200 mg/d using once-daily, nighttime dosing and titrating slowly based on tolerability and efficacy, continuing with nighttime dosing only. |
PHARMACOKINETICS
Because ERC-CBZ and carbamazepine yield similar molecules, the extended- and immediate-release forms have similar pharmacodynamic properties. There are notable pharmacokinetic differences, however.
With chronic use, carbamazepine autoinduces cytochrome P-450 (CYP) 3A4 hepatic enzymes. These enzymes rapidly break down carbamazepine to its active 10, 11-epoxide metabolite and the epoxide to the inactive diol. Because this sequence shortens carbamazepine’s half-life considerably—from 35 to 40 hours to 12 to 17 hours after 2 to 3 weeks of use—multiple daily dosing and minor dosage increases often are needed to maintain carbamazepine blood levels. Also, with immediate-release carbamazepine’s peak/trough variations, transient side effects such as ataxia, dizziness, or diplopia may emerge 2 to 3 hours after dosing once steady state is reached.
By contrast, ERC-CBZ should be more tolerable and easier to use because it smooths out these variations. Although studies of ERC-CBZ in mania have examined twice-daily dosing, using once-nightly dosing instead (starting at 200 mg) will harness carbamazepine’s sedative and other side effects to promote sleep onset, and lower levels throughout the day will further increase its tolerability.1 Patients who experience breakthrough afternoon or evening manic symptoms with once-nightly dosing can be returned to twice-daily dosing.
EFFICACY IN BIPOLAR DISORDER
ERC-CBZ has shown efficacy for treating bipolar disorder in three studies,2,3,4 including two large double-blind, placebo-controlled, multi-center trials that followed patients with type I bipolar disorder with current manic or mixed episodes.
In the first trial,2 204 patients received ERC-CBZ, 400 to 1,600 mg/d (mean±SD daily dosage 756.4±413.4 mg/d, mean plasma level 8.9 μg/mL), or placebo for 3 weeks. Young Mania Rating Scale (YMRS) scores decreased 50% in 41.5% of the treatment group and in 22.4% of the placebo group. Hamilton Rating Scale for Depression (HAM-D) scores decreased more in the ERC-CBZ group, but the difference was not clinically significant.
In a second trial of 239 patients,3 YMRS scores fell 50% across 3 weeks in 60.8% of those taking ERC-CBZ, 400 to 1,600 mg/d (mean±SD dosage 642±369.2 mg/d), compared with 28.7% of placebotreated patients. HAM-D total scores also improved significantly in ERC-CBZ-treated patients compared with the placebo group in a subanalysis of 188 intent-to-treat patients with a manic episode.
In a 6-month extension following 92 patients from two double-blind trials,4 mean total YMRS scores decreased among former placebo group patients switched to open-label ERC-CBZ, 200 to 1,600 mg/d (mean dosage 938 mg, mean serum level 6.6 μg/mL). Patients who had taken ERC-CBZ during the acute trial saw little change in YMRS scores during continued ERC-CBZ treatment, except in the second month. At end point, however, YMRS scores fell further for both treatment groups. HAM-D total scores differed little across 6 months, but 54 patients with mixed states maintained significant reductions.
Maintenance therapy. In many studies, carbamazepine has compared favorably with lithium for long-term bipolar maintenance in some patients. Consider patients who respond well to acute carbamazepine therapy for continuation treatment with ERC-CBZ. Patients who may be most likely to respond to carbamazepine therapy include those with:
- type II bipolar disorder
- substance abuse comorbidity
- mood-incongruent delusions
- no family history of bipolar illness among first-degree relatives.5
Contraindications (such as use during pregnancy or breast-feeding) are the same for extended- and immediate-release carbamazepine.
TOLERABILITY
Carbamazepine in any form can cause a range of common to rare side effects (Table 2), which have been reviewed elsewhere.4-6
Side effects of ERC-CBZ most commonly reported during the double-blind, placebo-controlled studies include dizziness, nausea, somnolence, headache, vomiting, dyspepsia, dry mouth, pruritus, and benign rash. Slower upward titration of single nighttime doses—instead of the twice-daily dosing used in these studies—could prevent most of these effects.
Only one patient in either study developed a serious side effect possibly related to ERC-CBZ (fever with rash); the rash resolved 6 days after the drug was stopped.
Total cholesterol in patients taking ERC-CBZ also rose 12% to 13% in the double-blind studies.2,3 Consider dietary and/or cholesterol-lowering medications in patients taking ERC-CBZ who are at high risk for cardiovascular events.
In the 6-month open-label study, headache, dizziness, and benign rash were most frequently reported. No serious adverse events related to the study drug were reported.
Table 2
Carbamazepine’s common to rare side effects
| Common | Infrequent |
| Ataxia | Hyponatremia (asymptomatic to symptomatic, reversed by demeclocycline or lithium) |
| Benign rash | Liver enzyme elevations |
| Benign white blood cell count suppression (reversed by lithium) | Tremor |
| Decreased thyroid hormones | Weight gain |
| Diplopia | Rare/serious |
| Dizziness | Agranulocytosis |
| Fatigue, sedation | Aplastic anemia |
| Increased cholesterol | Hyponatremia (symptomatic) |
| Nausea | Severe rash
|
| Spina bifida (following in utero exposure) |
INTERACTIONS WITH OTHER MEDICATIONS
Because of its potent induction of CYP 3A4 enzymes,6 carbamazepine in any form may substantially lower blood levels of several compounds metabolized principally by CYP 3A4 isoenzymes (Table 3), including typical antipsychotics such as haloperidol and the atypical antipsychotic aripiprazole. Even so, patients often improve with combination carbamazepine/haloperidol therapy despite lower haloperidol blood levels.
If a patient is taking oral contraceptives, inform her primary care physician or OB/GYN when prescribing carbamazepine. Because the anticonvulsant lowers circulating estrogen, a higher contraceptive dosage or alternate birth-control method should be considered to prevent unwanted pregnancy.
Most other drug-drug interactions have been well-delineated and can be avoided. Inform the patient and his or her primary care physician when giving carbamazepine concomitantly with any drug.
Numerous medications can also increase serum carbamazepine levels, causing problems in a patient already near his or her side-effect threshold (Table 4). Reduce the carbamazepine dosage to avoid these adverse effects.
Table 3
Carbamazepine decreases serum concentrations of these drugs
| Antipsychotics | Analgesics |
| Buprenorphine | Aripiprazole* |
| Methadone | Clozapine |
| Antimicrobials | Haloperidol* |
| Caspofungin | Olanzapine* |
| Doxycycline | Risperidone* |
| Anticoagulants | Thiothixene |
| Warfarin*† | Ziprasidone* |
| Anticonvulsants | Antivirals |
| Carbamazepine*† | Delavirdine |
| Lamotrigine*† | Protease inhibitors† |
| Oxcarbazepine | Anxiolytics/sedatives |
| Phenobarbital | Alprazolam* |
| Phenytoin | Steroids |
| Topiramate | Estrogen in hormonal contraceptives*† |
| Valproate*‡ | Mifepristone |
| Zonisamide | Prednisolone* |
| Antidepressants | Stimulants |
| Bupropion* | Methylphenidate* |
| Citalopram* | Modafinil* |
| Mirtazapine | Others |
| Tricyclics | Cisplatin |
| Doxorubicin | |
| Theophylline | |
| * Carbamazepine is often given with this medication. | |
| † Potentially serious interaction. | |
| ‡ Less-serious interaction likely with carbamazepine. | |
Table 4
These drugs increase serum carbamazepine and may cause toxicity
| Anticonvulsants | Macrolide antibiotics |
| Valproate (increases carbamazepine 10, 11-epoxide levels)*† | Clarithromycin*† |
| Antidepressants | Erythromycin*† |
| Fluoxetine*‡ | Flurithromycin*† |
| Fluvoxamine*‡ | Josamycin*† |
| Nefazodone*‡ | Ponsinomycin*† |
| Antimicrobials | Triacetyloleandromycin*† |
| Isoniazid† | Others |
| Quinupristin/dalfopristin | Acetazolamide |
| Calcium channel blockers | Cimetidine§ |
| Diltiazem*† | Danazol |
| Verapamil*† | d-Propoxyphene‡ |
| Hypolipidemics | Ketoconazole† |
| Gemfibrozil | Niacinamide |
| Nicotinamide | Omeprazole |
| Ritonavir† | |
| Ticlopidine | |
| * Carbamazepine is often given with this medication. | |
| † Potentially serious interaction. | |
| ‡ Less-serious interaction likely with carbamazepine. | |
| § Data on interactions with carbamazepine unclear. | |
CLINICAL IMPLICATIONS
Long-acting carbamazepine suitable for single nighttime dosing should facilitate adherence and reduce daytime side effects. Consider ERC-CBZ for patients not responding adequately to lithium or valproate, as individual response to any of these three drugs can vary greatly. Sideeffect tolerability (such as less weight gain with carbamazepine than with valproate) also could help guide drug choice. In patients with rapid cycling, carbamazepine plus lithium may be more effective than either drug alone.
New data suggest that carbamazepine offers acute antidepressant effects in some individuals and in long-term depression treatment.5 More research is needed to identify depressed patients most likely to respond to this agent.
For now, when using ERC-CBZ, we can draw from the larger experience with immediate-release carbamazepine to treat epilepsy, bipolar disorder, and related mood disorders. Once you master carbamazepine’s pharmacokinetic interactions with other commonly used agents, ERC-CBZ in slowly titrated, single nighttime dosages should simplify the compound’s administration and tolerability.
Related resources
- Carbamazepine (extended-release) Web site. www.equetro.com.
- Post RM, Speer AM, Obrocea GV, Leverich GS. Acute and prophylactic effects of anticonvulsants in bipolar depression. Clin Neurosci Res 2002;2:228-51.
- Denicoff KD, Smith-Jackson EE, Disney ER, et al. Comparative prophylactic efficacy of lithium, carbamazepine, and the combination in bipolar disorder. J Clin Psychiatry 1997;58:470-8.
Drug brand names
- Aripiprazole • Abilify
- Carbamazepine (extended-release) • Equetro
- Carbamazepine (immediate-release) • Tegretol, others
- Haloperidol • Haldol
- Lithium • Eskalith, others
- Valproate • Depakote
Disclosure
Dr. Post reports no current financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
1. Miller AD, Krauss GL, Hamzeh FM. Improved CNS tolerability following conversion from immediate- to extended-release carbamazepine. Acta Neurol Scand 2004;109:374-7.
2. Weisler RH, Kalali AH, Ketter TA. A multicenter, randomized, double-blind, placebo-controlled trial of extended-release carbamazepine capsules as monotherapy for bipolar disorder patients with manic or mixed episodes. J Clin Psychiatry 2004;65:478-84.
3. Weisler RH, Keck PE Jr, Swann AC, et al, for the SPD417 Study Group. Extended-release carbamazepine capsules as monotherapy for acute mania in bipolar disorder: A multicenter, randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 2005;66:323-30.
4. Ketter TA, Kalali AH, Weisler RH. A 6-month, multicenter, openlabel evaluation of beaded, extended-release carbamazepine capsule monotherapy in bipolar disorder patients with manic or mixed episodes. J Clin Psychiatry 2004;65:668-73.
5. Post RM, Frye MA. Carbamazepine. In: Sadock BJ, Sadock VA (eds). Kaplan & Sadock’s comprehensive textbook of psychiatry (9th ed). New York: Lippincott Williams & Wilkins, 2005, in press.
6. Ketter TA, Wang PW, Post RM. Carbamazepine and oxcarbazepine. In: Schatzberg AF, Nemeroff CB (eds). Textbook of psychopharmacology. Washington, DC: American Psychiatric Press, 2004;581-606.
Over 2 decades, carbamazepine has become a well-established, off-label alternative to lithium for treating acute mania. In December, the FDA approved an extended-release form of the anticonvulsant to treat type I bipolar disorder (Table 1).
This article addresses clinical use of extended-release capsules of carbamazepine (ERC-CBZ) and their safety, tolerability, and potential to interact with other medications.
Table 1
Extended-release capsules
of carbamazepine:
Fast facts
| Brand name: Equetro |
| Class: Anticonvulsant |
| FDA-approved indication: Bipolar mania |
| Manufacturer: Shire Pharmaceuticals Group |
| Dosing form: 100-, 200-, and 300-mg capsules |
| Recommended dosage: Manufacturer recommends starting at 400 mg/d in two divided doses.* Adjust dosage in 200-mg increments to achieve optimal response. Dosages >1,600 mg/d have not been studied. |
| *The author recommends starting at 200 mg/d using once-daily, nighttime dosing and titrating slowly based on tolerability and efficacy, continuing with nighttime dosing only. |
PHARMACOKINETICS
Because ERC-CBZ and carbamazepine yield similar molecules, the extended- and immediate-release forms have similar pharmacodynamic properties. There are notable pharmacokinetic differences, however.
With chronic use, carbamazepine autoinduces cytochrome P-450 (CYP) 3A4 hepatic enzymes. These enzymes rapidly break down carbamazepine to its active 10, 11-epoxide metabolite and the epoxide to the inactive diol. Because this sequence shortens carbamazepine’s half-life considerably—from 35 to 40 hours to 12 to 17 hours after 2 to 3 weeks of use—multiple daily dosing and minor dosage increases often are needed to maintain carbamazepine blood levels. Also, with immediate-release carbamazepine’s peak/trough variations, transient side effects such as ataxia, dizziness, or diplopia may emerge 2 to 3 hours after dosing once steady state is reached.
By contrast, ERC-CBZ should be more tolerable and easier to use because it smooths out these variations. Although studies of ERC-CBZ in mania have examined twice-daily dosing, using once-nightly dosing instead (starting at 200 mg) will harness carbamazepine’s sedative and other side effects to promote sleep onset, and lower levels throughout the day will further increase its tolerability.1 Patients who experience breakthrough afternoon or evening manic symptoms with once-nightly dosing can be returned to twice-daily dosing.
EFFICACY IN BIPOLAR DISORDER
ERC-CBZ has shown efficacy for treating bipolar disorder in three studies,2,3,4 including two large double-blind, placebo-controlled, multi-center trials that followed patients with type I bipolar disorder with current manic or mixed episodes.
In the first trial,2 204 patients received ERC-CBZ, 400 to 1,600 mg/d (mean±SD daily dosage 756.4±413.4 mg/d, mean plasma level 8.9 μg/mL), or placebo for 3 weeks. Young Mania Rating Scale (YMRS) scores decreased 50% in 41.5% of the treatment group and in 22.4% of the placebo group. Hamilton Rating Scale for Depression (HAM-D) scores decreased more in the ERC-CBZ group, but the difference was not clinically significant.
In a second trial of 239 patients,3 YMRS scores fell 50% across 3 weeks in 60.8% of those taking ERC-CBZ, 400 to 1,600 mg/d (mean±SD dosage 642±369.2 mg/d), compared with 28.7% of placebotreated patients. HAM-D total scores also improved significantly in ERC-CBZ-treated patients compared with the placebo group in a subanalysis of 188 intent-to-treat patients with a manic episode.
In a 6-month extension following 92 patients from two double-blind trials,4 mean total YMRS scores decreased among former placebo group patients switched to open-label ERC-CBZ, 200 to 1,600 mg/d (mean dosage 938 mg, mean serum level 6.6 μg/mL). Patients who had taken ERC-CBZ during the acute trial saw little change in YMRS scores during continued ERC-CBZ treatment, except in the second month. At end point, however, YMRS scores fell further for both treatment groups. HAM-D total scores differed little across 6 months, but 54 patients with mixed states maintained significant reductions.
Maintenance therapy. In many studies, carbamazepine has compared favorably with lithium for long-term bipolar maintenance in some patients. Consider patients who respond well to acute carbamazepine therapy for continuation treatment with ERC-CBZ. Patients who may be most likely to respond to carbamazepine therapy include those with:
- type II bipolar disorder
- substance abuse comorbidity
- mood-incongruent delusions
- no family history of bipolar illness among first-degree relatives.5
Contraindications (such as use during pregnancy or breast-feeding) are the same for extended- and immediate-release carbamazepine.
TOLERABILITY
Carbamazepine in any form can cause a range of common to rare side effects (Table 2), which have been reviewed elsewhere.4-6
Side effects of ERC-CBZ most commonly reported during the double-blind, placebo-controlled studies include dizziness, nausea, somnolence, headache, vomiting, dyspepsia, dry mouth, pruritus, and benign rash. Slower upward titration of single nighttime doses—instead of the twice-daily dosing used in these studies—could prevent most of these effects.
Only one patient in either study developed a serious side effect possibly related to ERC-CBZ (fever with rash); the rash resolved 6 days after the drug was stopped.
Total cholesterol in patients taking ERC-CBZ also rose 12% to 13% in the double-blind studies.2,3 Consider dietary and/or cholesterol-lowering medications in patients taking ERC-CBZ who are at high risk for cardiovascular events.
In the 6-month open-label study, headache, dizziness, and benign rash were most frequently reported. No serious adverse events related to the study drug were reported.
Table 2
Carbamazepine’s common to rare side effects
| Common | Infrequent |
| Ataxia | Hyponatremia (asymptomatic to symptomatic, reversed by demeclocycline or lithium) |
| Benign rash | Liver enzyme elevations |
| Benign white blood cell count suppression (reversed by lithium) | Tremor |
| Decreased thyroid hormones | Weight gain |
| Diplopia | Rare/serious |
| Dizziness | Agranulocytosis |
| Fatigue, sedation | Aplastic anemia |
| Increased cholesterol | Hyponatremia (symptomatic) |
| Nausea | Severe rash
|
| Spina bifida (following in utero exposure) |
INTERACTIONS WITH OTHER MEDICATIONS
Because of its potent induction of CYP 3A4 enzymes,6 carbamazepine in any form may substantially lower blood levels of several compounds metabolized principally by CYP 3A4 isoenzymes (Table 3), including typical antipsychotics such as haloperidol and the atypical antipsychotic aripiprazole. Even so, patients often improve with combination carbamazepine/haloperidol therapy despite lower haloperidol blood levels.
If a patient is taking oral contraceptives, inform her primary care physician or OB/GYN when prescribing carbamazepine. Because the anticonvulsant lowers circulating estrogen, a higher contraceptive dosage or alternate birth-control method should be considered to prevent unwanted pregnancy.
Most other drug-drug interactions have been well-delineated and can be avoided. Inform the patient and his or her primary care physician when giving carbamazepine concomitantly with any drug.
Numerous medications can also increase serum carbamazepine levels, causing problems in a patient already near his or her side-effect threshold (Table 4). Reduce the carbamazepine dosage to avoid these adverse effects.
Table 3
Carbamazepine decreases serum concentrations of these drugs
| Antipsychotics | Analgesics |
| Buprenorphine | Aripiprazole* |
| Methadone | Clozapine |
| Antimicrobials | Haloperidol* |
| Caspofungin | Olanzapine* |
| Doxycycline | Risperidone* |
| Anticoagulants | Thiothixene |
| Warfarin*† | Ziprasidone* |
| Anticonvulsants | Antivirals |
| Carbamazepine*† | Delavirdine |
| Lamotrigine*† | Protease inhibitors† |
| Oxcarbazepine | Anxiolytics/sedatives |
| Phenobarbital | Alprazolam* |
| Phenytoin | Steroids |
| Topiramate | Estrogen in hormonal contraceptives*† |
| Valproate*‡ | Mifepristone |
| Zonisamide | Prednisolone* |
| Antidepressants | Stimulants |
| Bupropion* | Methylphenidate* |
| Citalopram* | Modafinil* |
| Mirtazapine | Others |
| Tricyclics | Cisplatin |
| Doxorubicin | |
| Theophylline | |
| * Carbamazepine is often given with this medication. | |
| † Potentially serious interaction. | |
| ‡ Less-serious interaction likely with carbamazepine. | |
Table 4
These drugs increase serum carbamazepine and may cause toxicity
| Anticonvulsants | Macrolide antibiotics |
| Valproate (increases carbamazepine 10, 11-epoxide levels)*† | Clarithromycin*† |
| Antidepressants | Erythromycin*† |
| Fluoxetine*‡ | Flurithromycin*† |
| Fluvoxamine*‡ | Josamycin*† |
| Nefazodone*‡ | Ponsinomycin*† |
| Antimicrobials | Triacetyloleandromycin*† |
| Isoniazid† | Others |
| Quinupristin/dalfopristin | Acetazolamide |
| Calcium channel blockers | Cimetidine§ |
| Diltiazem*† | Danazol |
| Verapamil*† | d-Propoxyphene‡ |
| Hypolipidemics | Ketoconazole† |
| Gemfibrozil | Niacinamide |
| Nicotinamide | Omeprazole |
| Ritonavir† | |
| Ticlopidine | |
| * Carbamazepine is often given with this medication. | |
| † Potentially serious interaction. | |
| ‡ Less-serious interaction likely with carbamazepine. | |
| § Data on interactions with carbamazepine unclear. | |
CLINICAL IMPLICATIONS
Long-acting carbamazepine suitable for single nighttime dosing should facilitate adherence and reduce daytime side effects. Consider ERC-CBZ for patients not responding adequately to lithium or valproate, as individual response to any of these three drugs can vary greatly. Sideeffect tolerability (such as less weight gain with carbamazepine than with valproate) also could help guide drug choice. In patients with rapid cycling, carbamazepine plus lithium may be more effective than either drug alone.
New data suggest that carbamazepine offers acute antidepressant effects in some individuals and in long-term depression treatment.5 More research is needed to identify depressed patients most likely to respond to this agent.
For now, when using ERC-CBZ, we can draw from the larger experience with immediate-release carbamazepine to treat epilepsy, bipolar disorder, and related mood disorders. Once you master carbamazepine’s pharmacokinetic interactions with other commonly used agents, ERC-CBZ in slowly titrated, single nighttime dosages should simplify the compound’s administration and tolerability.
Related resources
- Carbamazepine (extended-release) Web site. www.equetro.com.
- Post RM, Speer AM, Obrocea GV, Leverich GS. Acute and prophylactic effects of anticonvulsants in bipolar depression. Clin Neurosci Res 2002;2:228-51.
- Denicoff KD, Smith-Jackson EE, Disney ER, et al. Comparative prophylactic efficacy of lithium, carbamazepine, and the combination in bipolar disorder. J Clin Psychiatry 1997;58:470-8.
Drug brand names
- Aripiprazole • Abilify
- Carbamazepine (extended-release) • Equetro
- Carbamazepine (immediate-release) • Tegretol, others
- Haloperidol • Haldol
- Lithium • Eskalith, others
- Valproate • Depakote
Disclosure
Dr. Post reports no current financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
Over 2 decades, carbamazepine has become a well-established, off-label alternative to lithium for treating acute mania. In December, the FDA approved an extended-release form of the anticonvulsant to treat type I bipolar disorder (Table 1).
This article addresses clinical use of extended-release capsules of carbamazepine (ERC-CBZ) and their safety, tolerability, and potential to interact with other medications.
Table 1
Extended-release capsules
of carbamazepine:
Fast facts
| Brand name: Equetro |
| Class: Anticonvulsant |
| FDA-approved indication: Bipolar mania |
| Manufacturer: Shire Pharmaceuticals Group |
| Dosing form: 100-, 200-, and 300-mg capsules |
| Recommended dosage: Manufacturer recommends starting at 400 mg/d in two divided doses.* Adjust dosage in 200-mg increments to achieve optimal response. Dosages >1,600 mg/d have not been studied. |
| *The author recommends starting at 200 mg/d using once-daily, nighttime dosing and titrating slowly based on tolerability and efficacy, continuing with nighttime dosing only. |
PHARMACOKINETICS
Because ERC-CBZ and carbamazepine yield similar molecules, the extended- and immediate-release forms have similar pharmacodynamic properties. There are notable pharmacokinetic differences, however.
With chronic use, carbamazepine autoinduces cytochrome P-450 (CYP) 3A4 hepatic enzymes. These enzymes rapidly break down carbamazepine to its active 10, 11-epoxide metabolite and the epoxide to the inactive diol. Because this sequence shortens carbamazepine’s half-life considerably—from 35 to 40 hours to 12 to 17 hours after 2 to 3 weeks of use—multiple daily dosing and minor dosage increases often are needed to maintain carbamazepine blood levels. Also, with immediate-release carbamazepine’s peak/trough variations, transient side effects such as ataxia, dizziness, or diplopia may emerge 2 to 3 hours after dosing once steady state is reached.
By contrast, ERC-CBZ should be more tolerable and easier to use because it smooths out these variations. Although studies of ERC-CBZ in mania have examined twice-daily dosing, using once-nightly dosing instead (starting at 200 mg) will harness carbamazepine’s sedative and other side effects to promote sleep onset, and lower levels throughout the day will further increase its tolerability.1 Patients who experience breakthrough afternoon or evening manic symptoms with once-nightly dosing can be returned to twice-daily dosing.
EFFICACY IN BIPOLAR DISORDER
ERC-CBZ has shown efficacy for treating bipolar disorder in three studies,2,3,4 including two large double-blind, placebo-controlled, multi-center trials that followed patients with type I bipolar disorder with current manic or mixed episodes.
In the first trial,2 204 patients received ERC-CBZ, 400 to 1,600 mg/d (mean±SD daily dosage 756.4±413.4 mg/d, mean plasma level 8.9 μg/mL), or placebo for 3 weeks. Young Mania Rating Scale (YMRS) scores decreased 50% in 41.5% of the treatment group and in 22.4% of the placebo group. Hamilton Rating Scale for Depression (HAM-D) scores decreased more in the ERC-CBZ group, but the difference was not clinically significant.
In a second trial of 239 patients,3 YMRS scores fell 50% across 3 weeks in 60.8% of those taking ERC-CBZ, 400 to 1,600 mg/d (mean±SD dosage 642±369.2 mg/d), compared with 28.7% of placebotreated patients. HAM-D total scores also improved significantly in ERC-CBZ-treated patients compared with the placebo group in a subanalysis of 188 intent-to-treat patients with a manic episode.
In a 6-month extension following 92 patients from two double-blind trials,4 mean total YMRS scores decreased among former placebo group patients switched to open-label ERC-CBZ, 200 to 1,600 mg/d (mean dosage 938 mg, mean serum level 6.6 μg/mL). Patients who had taken ERC-CBZ during the acute trial saw little change in YMRS scores during continued ERC-CBZ treatment, except in the second month. At end point, however, YMRS scores fell further for both treatment groups. HAM-D total scores differed little across 6 months, but 54 patients with mixed states maintained significant reductions.
Maintenance therapy. In many studies, carbamazepine has compared favorably with lithium for long-term bipolar maintenance in some patients. Consider patients who respond well to acute carbamazepine therapy for continuation treatment with ERC-CBZ. Patients who may be most likely to respond to carbamazepine therapy include those with:
- type II bipolar disorder
- substance abuse comorbidity
- mood-incongruent delusions
- no family history of bipolar illness among first-degree relatives.5
Contraindications (such as use during pregnancy or breast-feeding) are the same for extended- and immediate-release carbamazepine.
TOLERABILITY
Carbamazepine in any form can cause a range of common to rare side effects (Table 2), which have been reviewed elsewhere.4-6
Side effects of ERC-CBZ most commonly reported during the double-blind, placebo-controlled studies include dizziness, nausea, somnolence, headache, vomiting, dyspepsia, dry mouth, pruritus, and benign rash. Slower upward titration of single nighttime doses—instead of the twice-daily dosing used in these studies—could prevent most of these effects.
Only one patient in either study developed a serious side effect possibly related to ERC-CBZ (fever with rash); the rash resolved 6 days after the drug was stopped.
Total cholesterol in patients taking ERC-CBZ also rose 12% to 13% in the double-blind studies.2,3 Consider dietary and/or cholesterol-lowering medications in patients taking ERC-CBZ who are at high risk for cardiovascular events.
In the 6-month open-label study, headache, dizziness, and benign rash were most frequently reported. No serious adverse events related to the study drug were reported.
Table 2
Carbamazepine’s common to rare side effects
| Common | Infrequent |
| Ataxia | Hyponatremia (asymptomatic to symptomatic, reversed by demeclocycline or lithium) |
| Benign rash | Liver enzyme elevations |
| Benign white blood cell count suppression (reversed by lithium) | Tremor |
| Decreased thyroid hormones | Weight gain |
| Diplopia | Rare/serious |
| Dizziness | Agranulocytosis |
| Fatigue, sedation | Aplastic anemia |
| Increased cholesterol | Hyponatremia (symptomatic) |
| Nausea | Severe rash
|
| Spina bifida (following in utero exposure) |
INTERACTIONS WITH OTHER MEDICATIONS
Because of its potent induction of CYP 3A4 enzymes,6 carbamazepine in any form may substantially lower blood levels of several compounds metabolized principally by CYP 3A4 isoenzymes (Table 3), including typical antipsychotics such as haloperidol and the atypical antipsychotic aripiprazole. Even so, patients often improve with combination carbamazepine/haloperidol therapy despite lower haloperidol blood levels.
If a patient is taking oral contraceptives, inform her primary care physician or OB/GYN when prescribing carbamazepine. Because the anticonvulsant lowers circulating estrogen, a higher contraceptive dosage or alternate birth-control method should be considered to prevent unwanted pregnancy.
Most other drug-drug interactions have been well-delineated and can be avoided. Inform the patient and his or her primary care physician when giving carbamazepine concomitantly with any drug.
Numerous medications can also increase serum carbamazepine levels, causing problems in a patient already near his or her side-effect threshold (Table 4). Reduce the carbamazepine dosage to avoid these adverse effects.
Table 3
Carbamazepine decreases serum concentrations of these drugs
| Antipsychotics | Analgesics |
| Buprenorphine | Aripiprazole* |
| Methadone | Clozapine |
| Antimicrobials | Haloperidol* |
| Caspofungin | Olanzapine* |
| Doxycycline | Risperidone* |
| Anticoagulants | Thiothixene |
| Warfarin*† | Ziprasidone* |
| Anticonvulsants | Antivirals |
| Carbamazepine*† | Delavirdine |
| Lamotrigine*† | Protease inhibitors† |
| Oxcarbazepine | Anxiolytics/sedatives |
| Phenobarbital | Alprazolam* |
| Phenytoin | Steroids |
| Topiramate | Estrogen in hormonal contraceptives*† |
| Valproate*‡ | Mifepristone |
| Zonisamide | Prednisolone* |
| Antidepressants | Stimulants |
| Bupropion* | Methylphenidate* |
| Citalopram* | Modafinil* |
| Mirtazapine | Others |
| Tricyclics | Cisplatin |
| Doxorubicin | |
| Theophylline | |
| * Carbamazepine is often given with this medication. | |
| † Potentially serious interaction. | |
| ‡ Less-serious interaction likely with carbamazepine. | |
Table 4
These drugs increase serum carbamazepine and may cause toxicity
| Anticonvulsants | Macrolide antibiotics |
| Valproate (increases carbamazepine 10, 11-epoxide levels)*† | Clarithromycin*† |
| Antidepressants | Erythromycin*† |
| Fluoxetine*‡ | Flurithromycin*† |
| Fluvoxamine*‡ | Josamycin*† |
| Nefazodone*‡ | Ponsinomycin*† |
| Antimicrobials | Triacetyloleandromycin*† |
| Isoniazid† | Others |
| Quinupristin/dalfopristin | Acetazolamide |
| Calcium channel blockers | Cimetidine§ |
| Diltiazem*† | Danazol |
| Verapamil*† | d-Propoxyphene‡ |
| Hypolipidemics | Ketoconazole† |
| Gemfibrozil | Niacinamide |
| Nicotinamide | Omeprazole |
| Ritonavir† | |
| Ticlopidine | |
| * Carbamazepine is often given with this medication. | |
| † Potentially serious interaction. | |
| ‡ Less-serious interaction likely with carbamazepine. | |
| § Data on interactions with carbamazepine unclear. | |
CLINICAL IMPLICATIONS
Long-acting carbamazepine suitable for single nighttime dosing should facilitate adherence and reduce daytime side effects. Consider ERC-CBZ for patients not responding adequately to lithium or valproate, as individual response to any of these three drugs can vary greatly. Sideeffect tolerability (such as less weight gain with carbamazepine than with valproate) also could help guide drug choice. In patients with rapid cycling, carbamazepine plus lithium may be more effective than either drug alone.
New data suggest that carbamazepine offers acute antidepressant effects in some individuals and in long-term depression treatment.5 More research is needed to identify depressed patients most likely to respond to this agent.
For now, when using ERC-CBZ, we can draw from the larger experience with immediate-release carbamazepine to treat epilepsy, bipolar disorder, and related mood disorders. Once you master carbamazepine’s pharmacokinetic interactions with other commonly used agents, ERC-CBZ in slowly titrated, single nighttime dosages should simplify the compound’s administration and tolerability.
Related resources
- Carbamazepine (extended-release) Web site. www.equetro.com.
- Post RM, Speer AM, Obrocea GV, Leverich GS. Acute and prophylactic effects of anticonvulsants in bipolar depression. Clin Neurosci Res 2002;2:228-51.
- Denicoff KD, Smith-Jackson EE, Disney ER, et al. Comparative prophylactic efficacy of lithium, carbamazepine, and the combination in bipolar disorder. J Clin Psychiatry 1997;58:470-8.
Drug brand names
- Aripiprazole • Abilify
- Carbamazepine (extended-release) • Equetro
- Carbamazepine (immediate-release) • Tegretol, others
- Haloperidol • Haldol
- Lithium • Eskalith, others
- Valproate • Depakote
Disclosure
Dr. Post reports no current financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
1. Miller AD, Krauss GL, Hamzeh FM. Improved CNS tolerability following conversion from immediate- to extended-release carbamazepine. Acta Neurol Scand 2004;109:374-7.
2. Weisler RH, Kalali AH, Ketter TA. A multicenter, randomized, double-blind, placebo-controlled trial of extended-release carbamazepine capsules as monotherapy for bipolar disorder patients with manic or mixed episodes. J Clin Psychiatry 2004;65:478-84.
3. Weisler RH, Keck PE Jr, Swann AC, et al, for the SPD417 Study Group. Extended-release carbamazepine capsules as monotherapy for acute mania in bipolar disorder: A multicenter, randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 2005;66:323-30.
4. Ketter TA, Kalali AH, Weisler RH. A 6-month, multicenter, openlabel evaluation of beaded, extended-release carbamazepine capsule monotherapy in bipolar disorder patients with manic or mixed episodes. J Clin Psychiatry 2004;65:668-73.
5. Post RM, Frye MA. Carbamazepine. In: Sadock BJ, Sadock VA (eds). Kaplan & Sadock’s comprehensive textbook of psychiatry (9th ed). New York: Lippincott Williams & Wilkins, 2005, in press.
6. Ketter TA, Wang PW, Post RM. Carbamazepine and oxcarbazepine. In: Schatzberg AF, Nemeroff CB (eds). Textbook of psychopharmacology. Washington, DC: American Psychiatric Press, 2004;581-606.
1. Miller AD, Krauss GL, Hamzeh FM. Improved CNS tolerability following conversion from immediate- to extended-release carbamazepine. Acta Neurol Scand 2004;109:374-7.
2. Weisler RH, Kalali AH, Ketter TA. A multicenter, randomized, double-blind, placebo-controlled trial of extended-release carbamazepine capsules as monotherapy for bipolar disorder patients with manic or mixed episodes. J Clin Psychiatry 2004;65:478-84.
3. Weisler RH, Keck PE Jr, Swann AC, et al, for the SPD417 Study Group. Extended-release carbamazepine capsules as monotherapy for acute mania in bipolar disorder: A multicenter, randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 2005;66:323-30.
4. Ketter TA, Kalali AH, Weisler RH. A 6-month, multicenter, openlabel evaluation of beaded, extended-release carbamazepine capsule monotherapy in bipolar disorder patients with manic or mixed episodes. J Clin Psychiatry 2004;65:668-73.
5. Post RM, Frye MA. Carbamazepine. In: Sadock BJ, Sadock VA (eds). Kaplan & Sadock’s comprehensive textbook of psychiatry (9th ed). New York: Lippincott Williams & Wilkins, 2005, in press.
6. Ketter TA, Wang PW, Post RM. Carbamazepine and oxcarbazepine. In: Schatzberg AF, Nemeroff CB (eds). Textbook of psychopharmacology. Washington, DC: American Psychiatric Press, 2004;581-606.
Acamprosate: For discomfort of early alcohol abstinence
Acamprosate, a gamma-aminobutyric acid (GABA) analogue used worldwide to treat alcohol dependence, is available in this country (Table 1). The agent appears to reduce discomfort—including restlessness, anxiety, dysphoria, and insomnia—common within the first 6 months of alcohol abstinence. In clinical trials, it prolonged abstinence in alcohol-dependent patients who completed an initial detoxification and were receiving relapse prevention treatment.
HOW IT WORKS
Acamprosate’s chemical structure resembles both GABA and taurine, an endogenous amino acid derivative that enhances GABA-ergic activity.1 The drug’s synthetic structure facilitates its passage across the blood-brain barrier, and the brain mediates its major effect.
Table 1
Acamprosate: Fast facts
| Brand name: |
| Campral |
| Class: |
| GABA analogue |
| FDA-approved indication: |
| Maintaining abstinence in alcohol-dependent patients |
| Approval date: |
| July 29, 2004 |
| Manufacturer: |
| Forest Pharmaceuticals |
| Dosing form: |
| 333-mg tablets |
| Recommended dosage: |
| Adults age |
The mechanisms by which acamprosate promotes abstinence in alcohol dependence are unknown. The drug may bind to N-methyl-D-aspartate (NMDA) glutamate receptors and work as a partial antagonist, but direct ligand activity does not appear to cause most of its central actions. Rather, acamprosate interacts with glutamate and GABA to normalize the hyperexcitability that accompanies early abstinence (Table 2).
Table 2
Acamprosate’s proposed mechanisms of action*
| Neurotransmitter interactions | Pharmacologic effect | Clinical effect |
| Glutamatergic system (NMDA receptor) | Blocks increased glutamate release in nucleus accumbens during alcohol withdrawal; may bind to receptor site as partial antagonist | Decreased arousal, craving, and dysphoria associated with early abstinence |
| GABAergic system (GABAAreceptor) | Normalizes alcohol-induced decrease in basal GABA concentrations in nucleus accumbens | Same as above |
| Neuromodulator interactions | ||
| Taurine | Increases extracellular taurine concentrations in nucleus accumbens; taurine shifts the glutamate/GABA balance in favor of GABAergic activity | Mimics increase in taurine seen with acute alcohol intake, likely facilitating GABA normalization |
| * Based on animal models of alcohol dependence | ||
| Source: reference 3 | ||
PHARMACOKINETICS
Acamprosate’s bioavailability is relatively poor (11%), so it is prescribed to be taken three times daily. Although patients in clinical practice often have trouble following frequent daily dosing schedules, subjects in one study reportedly had little difficulty adhering to this regimen.2
Acamprosate’s half-life is approximately 13 hours, and it reaches peak plasma concentrations in 3.5 to 9.5 hours. Pharmaceutical studies indicate that food does not significantly affect absorption.
Although 666 mg tid has shown efficacy in clinical trials, the blood level at which acamprosate becomes therapeutic has not been determined.
The drug reaches steady-state blood levels within 1 week, meaning it will not be fully effective for 5 to 7 days but may still reach therapeutic blood levels during that time. Advise patients that adverse effects may not clear for 5 to 7 days after discontinuation.
Acamprosate does not bind with plasma proteins, so it will not interact with drugs that do. The drug, which is renally excreted in an unmetabolized state, has not been found to interact adversely with commonly prescribed antidepressants, anxiolytics, antipsychotics, alcohol, or disulfiram.3 How acamprosate interacts with renally excreted drugs such as lithium is unknown.
In two studies following 24 healthy volunteers4 and 23 alcohol-dependent patients,5 concomitant naltrexone, 50 to 100 mg/d, and acamprosate, 2 to 3 g/d, increased acamprosate plasma concentrations as much as 25%, but did not change plasma levels of naltrexone or its major metabolite. Naltrexone might delay gastric emptying, thereby increasing acamprosate absorption.
EFFICACY
Acamprosate with psychosocial treatment increased total abstinent days in:
- 15 randomized, controlled trials (RCT) conducted in Europe6
- a meta-analysis of 12 methodologically comparable RCTs conducted in Europe7
- an open-label trial in France that studied acamprosate as an adjunct to treatment-as-usual in primary care settings.2
Acamprosate may improve patient retention in substance abuse treatment, which predicts favorable outcomes.7 Patients receiving acamprosate and treatment-as-usual reported fewer alcohol-related problems and improved quality of life compared with treatment-as-usual alone.2 Reduced subjective craving for alcohol is difficult to study and has not been sufficiently shown.
Combined pharmacotherapy. It is unclear whether acamprosate and naltrexone or disulfiram are more effective than acamprosate alone.3,6,7
In one multi-center, placebo-controlled trial, a subgroup of severely alcohol-dependent patients sought acamprosate/disulfiram therapy. The combination was shown to be safe and increased total abstinent days compared with acamprosate or disulfiram alone, but effectiveness could not be determined because of the self-selection bias of those who requested combined pharmacotherapy.
In one 12-week RCT,8 naltrexone/acamprosate therapy was more effective than acamprosate alone—but not more effective than naltrexone alone—in reducing time to first drink and relapse to heavy drinking.
The multi-center COMBINE (Combining Medications and Behavioral Interventions) study,9 funded by the National Institute on Alcohol Abuse and Alcoholism, is comparing the efficacy of naltrexone, acamprosate, and both agents when given with low-intensity psychosocial treatment or moderate-intensity, alcohol-specific psychosocial treatment. Preliminary safety, tolerability, and adherence results with the acamprosate/naltrexone combination have been promising. Efficacy findings are expected later this year.
SAFETY
Acamprosate is contraindicated in patients with severely compromised renal function (creatinine clearance
The drug is safe for patients with mild to moderate alcohol-related liver disease as defined by the Child-Pugh classification of hepatic impairment.10 For a patient with severe liver disease, consult his or her gastroenterologist to gauge risks and benefits, as acamprosate can cause adverse GI effects.
Acamprosate has not been tested in children or the elderly, although one study suggests efficacy in alcohol-dependent adolescents ages 16 to 19.7. The agent’s safety during pregnancy or lactation is unknown.
TOLERABILITY
Acamprosate has been well-tolerated in clinical trials. Discontinuation rates because of adverse effects have been similar in treatment and placebo groups.7
GI side effects are most common, with overall rates of 17% and 11% among acamprosate and placebo groups, respectively.7 Diarrhea may be transient and may also resolve with a reduced dosage.6
Slightly higher rates of suicidal ideation were reported among patients taking acamprosate vs those taking placebo (1.4 % vs. 0.5% in short-term [10 Screen all patients taking acamprosate for suicidal ideation or behavior.
Other reported side effects include headache, abdominal pain, nausea and vomiting, dyspepsia, flatulence, pruritus, rash, drowsiness, and dizziness. Acamprosate has no abuse potential and low potential for toxicity in overdose. Higher acamprosate plasma levels during combined acamprosate/naltrexone treatment may increase risk of diarrhea.9
CLINICAL IMPLICATIONS
Drinking alcohol while taking acamprosate will not make a patient sick, which makes it an alternative for patients who fear the harsh effects of “slipping up” while taking disulfiram.
Also, acamprosate does not interact with prescription opioids. By contrast, naltrexone is contraindicated in patients taking opioids for pain.
Related resources
- Campral Web site. www.campral.com
- National Institute on Alcohol Abuse and Alcoholism. www.niaaa.nih.gov
- National Institute on Drug Abuse. www.nida.nih.gov
Drug brand names
- Acamprosate • Campral
- Disulfiram • Antabuse
- Naltrexone • ReVia
Disclosure
Drs. Connery and Weiss receive research/grant support from Ortho-McNeil Pharmaceutical. Dr. Weiss is also a speaker for Forest Laboratories.
1. Dahchour A, De Witte P. Ethanol and amino acids in the central nervous system: assessment of the pharmacological actions of acamprosate. Prog Neurobiol 2000;60:343-62.
2. Kiritze-Topor P, Huas D, Rosenzweig C, et al. A pragmatic trial of acamprosate in the treatment of alcohol dependence in primary care. Alcohol Alcohol 2004;39:520-7.
3. Kiefer F, Wiedemann K. Combined therapy: what does acamprosate and naltrexone combination tell us? Alcohol Alcohol 2004;39:542-7.
4. Mason BJ, Goodman AM, Dixon RM, et al. A pharmacokinetic and pharmacodynamic drug interaction study of acamprosate and naltrexone. Neuropsychopharmacology 2002;27:596-606.
5. Johnson BA, O’Malley SS, Ciraulo DA, et al. Dose-ranging kinetics and behavioral pharmacology of naltrexone and acamprosate, both alone and combined, in alcohol-dependent subjects. J Clin Psychopharmacol 2003;23:281-93.
6. Overman GP, Teter CJ, Guthrie SK. Acamprosate for the adjunctive treatment of alcohol dependence. Ann Pharmacother 2003;37:1090-9.
7. Carmen B, Angeles M, Ana M, Maria AJ. Efficacy and safety of naltrexone and acamprosate in the treatment of alcohol dependence: a systematic review. Addiction 2004;99:811-28.
8. Kiefer F, Jahn H, Tarnaske T, et al. Comparing and combining naltrexone and acamprosate in relapse prevention of alcoholism: a double-blind, placebo-controlled study. Arch Gen Psychiatry 2003;60:92-9.
9. The COMBINE study research group. Testing combined pharmacotherapies and behavioral interventions in alcohol dependence: rationale and methods. Alcohol Clin Exp Res 2003;27:1107-22.
10. Campral prescribing information. Available at: http://www.campral.com. Accessed Jan. 7, 2005.
Acamprosate, a gamma-aminobutyric acid (GABA) analogue used worldwide to treat alcohol dependence, is available in this country (Table 1). The agent appears to reduce discomfort—including restlessness, anxiety, dysphoria, and insomnia—common within the first 6 months of alcohol abstinence. In clinical trials, it prolonged abstinence in alcohol-dependent patients who completed an initial detoxification and were receiving relapse prevention treatment.
HOW IT WORKS
Acamprosate’s chemical structure resembles both GABA and taurine, an endogenous amino acid derivative that enhances GABA-ergic activity.1 The drug’s synthetic structure facilitates its passage across the blood-brain barrier, and the brain mediates its major effect.
Table 1
Acamprosate: Fast facts
| Brand name: |
| Campral |
| Class: |
| GABA analogue |
| FDA-approved indication: |
| Maintaining abstinence in alcohol-dependent patients |
| Approval date: |
| July 29, 2004 |
| Manufacturer: |
| Forest Pharmaceuticals |
| Dosing form: |
| 333-mg tablets |
| Recommended dosage: |
| Adults age |
The mechanisms by which acamprosate promotes abstinence in alcohol dependence are unknown. The drug may bind to N-methyl-D-aspartate (NMDA) glutamate receptors and work as a partial antagonist, but direct ligand activity does not appear to cause most of its central actions. Rather, acamprosate interacts with glutamate and GABA to normalize the hyperexcitability that accompanies early abstinence (Table 2).
Table 2
Acamprosate’s proposed mechanisms of action*
| Neurotransmitter interactions | Pharmacologic effect | Clinical effect |
| Glutamatergic system (NMDA receptor) | Blocks increased glutamate release in nucleus accumbens during alcohol withdrawal; may bind to receptor site as partial antagonist | Decreased arousal, craving, and dysphoria associated with early abstinence |
| GABAergic system (GABAAreceptor) | Normalizes alcohol-induced decrease in basal GABA concentrations in nucleus accumbens | Same as above |
| Neuromodulator interactions | ||
| Taurine | Increases extracellular taurine concentrations in nucleus accumbens; taurine shifts the glutamate/GABA balance in favor of GABAergic activity | Mimics increase in taurine seen with acute alcohol intake, likely facilitating GABA normalization |
| * Based on animal models of alcohol dependence | ||
| Source: reference 3 | ||
PHARMACOKINETICS
Acamprosate’s bioavailability is relatively poor (11%), so it is prescribed to be taken three times daily. Although patients in clinical practice often have trouble following frequent daily dosing schedules, subjects in one study reportedly had little difficulty adhering to this regimen.2
Acamprosate’s half-life is approximately 13 hours, and it reaches peak plasma concentrations in 3.5 to 9.5 hours. Pharmaceutical studies indicate that food does not significantly affect absorption.
Although 666 mg tid has shown efficacy in clinical trials, the blood level at which acamprosate becomes therapeutic has not been determined.
The drug reaches steady-state blood levels within 1 week, meaning it will not be fully effective for 5 to 7 days but may still reach therapeutic blood levels during that time. Advise patients that adverse effects may not clear for 5 to 7 days after discontinuation.
Acamprosate does not bind with plasma proteins, so it will not interact with drugs that do. The drug, which is renally excreted in an unmetabolized state, has not been found to interact adversely with commonly prescribed antidepressants, anxiolytics, antipsychotics, alcohol, or disulfiram.3 How acamprosate interacts with renally excreted drugs such as lithium is unknown.
In two studies following 24 healthy volunteers4 and 23 alcohol-dependent patients,5 concomitant naltrexone, 50 to 100 mg/d, and acamprosate, 2 to 3 g/d, increased acamprosate plasma concentrations as much as 25%, but did not change plasma levels of naltrexone or its major metabolite. Naltrexone might delay gastric emptying, thereby increasing acamprosate absorption.
EFFICACY
Acamprosate with psychosocial treatment increased total abstinent days in:
- 15 randomized, controlled trials (RCT) conducted in Europe6
- a meta-analysis of 12 methodologically comparable RCTs conducted in Europe7
- an open-label trial in France that studied acamprosate as an adjunct to treatment-as-usual in primary care settings.2
Acamprosate may improve patient retention in substance abuse treatment, which predicts favorable outcomes.7 Patients receiving acamprosate and treatment-as-usual reported fewer alcohol-related problems and improved quality of life compared with treatment-as-usual alone.2 Reduced subjective craving for alcohol is difficult to study and has not been sufficiently shown.
Combined pharmacotherapy. It is unclear whether acamprosate and naltrexone or disulfiram are more effective than acamprosate alone.3,6,7
In one multi-center, placebo-controlled trial, a subgroup of severely alcohol-dependent patients sought acamprosate/disulfiram therapy. The combination was shown to be safe and increased total abstinent days compared with acamprosate or disulfiram alone, but effectiveness could not be determined because of the self-selection bias of those who requested combined pharmacotherapy.
In one 12-week RCT,8 naltrexone/acamprosate therapy was more effective than acamprosate alone—but not more effective than naltrexone alone—in reducing time to first drink and relapse to heavy drinking.
The multi-center COMBINE (Combining Medications and Behavioral Interventions) study,9 funded by the National Institute on Alcohol Abuse and Alcoholism, is comparing the efficacy of naltrexone, acamprosate, and both agents when given with low-intensity psychosocial treatment or moderate-intensity, alcohol-specific psychosocial treatment. Preliminary safety, tolerability, and adherence results with the acamprosate/naltrexone combination have been promising. Efficacy findings are expected later this year.
SAFETY
Acamprosate is contraindicated in patients with severely compromised renal function (creatinine clearance
The drug is safe for patients with mild to moderate alcohol-related liver disease as defined by the Child-Pugh classification of hepatic impairment.10 For a patient with severe liver disease, consult his or her gastroenterologist to gauge risks and benefits, as acamprosate can cause adverse GI effects.
Acamprosate has not been tested in children or the elderly, although one study suggests efficacy in alcohol-dependent adolescents ages 16 to 19.7. The agent’s safety during pregnancy or lactation is unknown.
TOLERABILITY
Acamprosate has been well-tolerated in clinical trials. Discontinuation rates because of adverse effects have been similar in treatment and placebo groups.7
GI side effects are most common, with overall rates of 17% and 11% among acamprosate and placebo groups, respectively.7 Diarrhea may be transient and may also resolve with a reduced dosage.6
Slightly higher rates of suicidal ideation were reported among patients taking acamprosate vs those taking placebo (1.4 % vs. 0.5% in short-term [10 Screen all patients taking acamprosate for suicidal ideation or behavior.
Other reported side effects include headache, abdominal pain, nausea and vomiting, dyspepsia, flatulence, pruritus, rash, drowsiness, and dizziness. Acamprosate has no abuse potential and low potential for toxicity in overdose. Higher acamprosate plasma levels during combined acamprosate/naltrexone treatment may increase risk of diarrhea.9
CLINICAL IMPLICATIONS
Drinking alcohol while taking acamprosate will not make a patient sick, which makes it an alternative for patients who fear the harsh effects of “slipping up” while taking disulfiram.
Also, acamprosate does not interact with prescription opioids. By contrast, naltrexone is contraindicated in patients taking opioids for pain.
Related resources
- Campral Web site. www.campral.com
- National Institute on Alcohol Abuse and Alcoholism. www.niaaa.nih.gov
- National Institute on Drug Abuse. www.nida.nih.gov
Drug brand names
- Acamprosate • Campral
- Disulfiram • Antabuse
- Naltrexone • ReVia
Disclosure
Drs. Connery and Weiss receive research/grant support from Ortho-McNeil Pharmaceutical. Dr. Weiss is also a speaker for Forest Laboratories.
Acamprosate, a gamma-aminobutyric acid (GABA) analogue used worldwide to treat alcohol dependence, is available in this country (Table 1). The agent appears to reduce discomfort—including restlessness, anxiety, dysphoria, and insomnia—common within the first 6 months of alcohol abstinence. In clinical trials, it prolonged abstinence in alcohol-dependent patients who completed an initial detoxification and were receiving relapse prevention treatment.
HOW IT WORKS
Acamprosate’s chemical structure resembles both GABA and taurine, an endogenous amino acid derivative that enhances GABA-ergic activity.1 The drug’s synthetic structure facilitates its passage across the blood-brain barrier, and the brain mediates its major effect.
Table 1
Acamprosate: Fast facts
| Brand name: |
| Campral |
| Class: |
| GABA analogue |
| FDA-approved indication: |
| Maintaining abstinence in alcohol-dependent patients |
| Approval date: |
| July 29, 2004 |
| Manufacturer: |
| Forest Pharmaceuticals |
| Dosing form: |
| 333-mg tablets |
| Recommended dosage: |
| Adults age |
The mechanisms by which acamprosate promotes abstinence in alcohol dependence are unknown. The drug may bind to N-methyl-D-aspartate (NMDA) glutamate receptors and work as a partial antagonist, but direct ligand activity does not appear to cause most of its central actions. Rather, acamprosate interacts with glutamate and GABA to normalize the hyperexcitability that accompanies early abstinence (Table 2).
Table 2
Acamprosate’s proposed mechanisms of action*
| Neurotransmitter interactions | Pharmacologic effect | Clinical effect |
| Glutamatergic system (NMDA receptor) | Blocks increased glutamate release in nucleus accumbens during alcohol withdrawal; may bind to receptor site as partial antagonist | Decreased arousal, craving, and dysphoria associated with early abstinence |
| GABAergic system (GABAAreceptor) | Normalizes alcohol-induced decrease in basal GABA concentrations in nucleus accumbens | Same as above |
| Neuromodulator interactions | ||
| Taurine | Increases extracellular taurine concentrations in nucleus accumbens; taurine shifts the glutamate/GABA balance in favor of GABAergic activity | Mimics increase in taurine seen with acute alcohol intake, likely facilitating GABA normalization |
| * Based on animal models of alcohol dependence | ||
| Source: reference 3 | ||
PHARMACOKINETICS
Acamprosate’s bioavailability is relatively poor (11%), so it is prescribed to be taken three times daily. Although patients in clinical practice often have trouble following frequent daily dosing schedules, subjects in one study reportedly had little difficulty adhering to this regimen.2
Acamprosate’s half-life is approximately 13 hours, and it reaches peak plasma concentrations in 3.5 to 9.5 hours. Pharmaceutical studies indicate that food does not significantly affect absorption.
Although 666 mg tid has shown efficacy in clinical trials, the blood level at which acamprosate becomes therapeutic has not been determined.
The drug reaches steady-state blood levels within 1 week, meaning it will not be fully effective for 5 to 7 days but may still reach therapeutic blood levels during that time. Advise patients that adverse effects may not clear for 5 to 7 days after discontinuation.
Acamprosate does not bind with plasma proteins, so it will not interact with drugs that do. The drug, which is renally excreted in an unmetabolized state, has not been found to interact adversely with commonly prescribed antidepressants, anxiolytics, antipsychotics, alcohol, or disulfiram.3 How acamprosate interacts with renally excreted drugs such as lithium is unknown.
In two studies following 24 healthy volunteers4 and 23 alcohol-dependent patients,5 concomitant naltrexone, 50 to 100 mg/d, and acamprosate, 2 to 3 g/d, increased acamprosate plasma concentrations as much as 25%, but did not change plasma levels of naltrexone or its major metabolite. Naltrexone might delay gastric emptying, thereby increasing acamprosate absorption.
EFFICACY
Acamprosate with psychosocial treatment increased total abstinent days in:
- 15 randomized, controlled trials (RCT) conducted in Europe6
- a meta-analysis of 12 methodologically comparable RCTs conducted in Europe7
- an open-label trial in France that studied acamprosate as an adjunct to treatment-as-usual in primary care settings.2
Acamprosate may improve patient retention in substance abuse treatment, which predicts favorable outcomes.7 Patients receiving acamprosate and treatment-as-usual reported fewer alcohol-related problems and improved quality of life compared with treatment-as-usual alone.2 Reduced subjective craving for alcohol is difficult to study and has not been sufficiently shown.
Combined pharmacotherapy. It is unclear whether acamprosate and naltrexone or disulfiram are more effective than acamprosate alone.3,6,7
In one multi-center, placebo-controlled trial, a subgroup of severely alcohol-dependent patients sought acamprosate/disulfiram therapy. The combination was shown to be safe and increased total abstinent days compared with acamprosate or disulfiram alone, but effectiveness could not be determined because of the self-selection bias of those who requested combined pharmacotherapy.
In one 12-week RCT,8 naltrexone/acamprosate therapy was more effective than acamprosate alone—but not more effective than naltrexone alone—in reducing time to first drink and relapse to heavy drinking.
The multi-center COMBINE (Combining Medications and Behavioral Interventions) study,9 funded by the National Institute on Alcohol Abuse and Alcoholism, is comparing the efficacy of naltrexone, acamprosate, and both agents when given with low-intensity psychosocial treatment or moderate-intensity, alcohol-specific psychosocial treatment. Preliminary safety, tolerability, and adherence results with the acamprosate/naltrexone combination have been promising. Efficacy findings are expected later this year.
SAFETY
Acamprosate is contraindicated in patients with severely compromised renal function (creatinine clearance
The drug is safe for patients with mild to moderate alcohol-related liver disease as defined by the Child-Pugh classification of hepatic impairment.10 For a patient with severe liver disease, consult his or her gastroenterologist to gauge risks and benefits, as acamprosate can cause adverse GI effects.
Acamprosate has not been tested in children or the elderly, although one study suggests efficacy in alcohol-dependent adolescents ages 16 to 19.7. The agent’s safety during pregnancy or lactation is unknown.
TOLERABILITY
Acamprosate has been well-tolerated in clinical trials. Discontinuation rates because of adverse effects have been similar in treatment and placebo groups.7
GI side effects are most common, with overall rates of 17% and 11% among acamprosate and placebo groups, respectively.7 Diarrhea may be transient and may also resolve with a reduced dosage.6
Slightly higher rates of suicidal ideation were reported among patients taking acamprosate vs those taking placebo (1.4 % vs. 0.5% in short-term [10 Screen all patients taking acamprosate for suicidal ideation or behavior.
Other reported side effects include headache, abdominal pain, nausea and vomiting, dyspepsia, flatulence, pruritus, rash, drowsiness, and dizziness. Acamprosate has no abuse potential and low potential for toxicity in overdose. Higher acamprosate plasma levels during combined acamprosate/naltrexone treatment may increase risk of diarrhea.9
CLINICAL IMPLICATIONS
Drinking alcohol while taking acamprosate will not make a patient sick, which makes it an alternative for patients who fear the harsh effects of “slipping up” while taking disulfiram.
Also, acamprosate does not interact with prescription opioids. By contrast, naltrexone is contraindicated in patients taking opioids for pain.
Related resources
- Campral Web site. www.campral.com
- National Institute on Alcohol Abuse and Alcoholism. www.niaaa.nih.gov
- National Institute on Drug Abuse. www.nida.nih.gov
Drug brand names
- Acamprosate • Campral
- Disulfiram • Antabuse
- Naltrexone • ReVia
Disclosure
Drs. Connery and Weiss receive research/grant support from Ortho-McNeil Pharmaceutical. Dr. Weiss is also a speaker for Forest Laboratories.
1. Dahchour A, De Witte P. Ethanol and amino acids in the central nervous system: assessment of the pharmacological actions of acamprosate. Prog Neurobiol 2000;60:343-62.
2. Kiritze-Topor P, Huas D, Rosenzweig C, et al. A pragmatic trial of acamprosate in the treatment of alcohol dependence in primary care. Alcohol Alcohol 2004;39:520-7.
3. Kiefer F, Wiedemann K. Combined therapy: what does acamprosate and naltrexone combination tell us? Alcohol Alcohol 2004;39:542-7.
4. Mason BJ, Goodman AM, Dixon RM, et al. A pharmacokinetic and pharmacodynamic drug interaction study of acamprosate and naltrexone. Neuropsychopharmacology 2002;27:596-606.
5. Johnson BA, O’Malley SS, Ciraulo DA, et al. Dose-ranging kinetics and behavioral pharmacology of naltrexone and acamprosate, both alone and combined, in alcohol-dependent subjects. J Clin Psychopharmacol 2003;23:281-93.
6. Overman GP, Teter CJ, Guthrie SK. Acamprosate for the adjunctive treatment of alcohol dependence. Ann Pharmacother 2003;37:1090-9.
7. Carmen B, Angeles M, Ana M, Maria AJ. Efficacy and safety of naltrexone and acamprosate in the treatment of alcohol dependence: a systematic review. Addiction 2004;99:811-28.
8. Kiefer F, Jahn H, Tarnaske T, et al. Comparing and combining naltrexone and acamprosate in relapse prevention of alcoholism: a double-blind, placebo-controlled study. Arch Gen Psychiatry 2003;60:92-9.
9. The COMBINE study research group. Testing combined pharmacotherapies and behavioral interventions in alcohol dependence: rationale and methods. Alcohol Clin Exp Res 2003;27:1107-22.
10. Campral prescribing information. Available at: http://www.campral.com. Accessed Jan. 7, 2005.
1. Dahchour A, De Witte P. Ethanol and amino acids in the central nervous system: assessment of the pharmacological actions of acamprosate. Prog Neurobiol 2000;60:343-62.
2. Kiritze-Topor P, Huas D, Rosenzweig C, et al. A pragmatic trial of acamprosate in the treatment of alcohol dependence in primary care. Alcohol Alcohol 2004;39:520-7.
3. Kiefer F, Wiedemann K. Combined therapy: what does acamprosate and naltrexone combination tell us? Alcohol Alcohol 2004;39:542-7.
4. Mason BJ, Goodman AM, Dixon RM, et al. A pharmacokinetic and pharmacodynamic drug interaction study of acamprosate and naltrexone. Neuropsychopharmacology 2002;27:596-606.
5. Johnson BA, O’Malley SS, Ciraulo DA, et al. Dose-ranging kinetics and behavioral pharmacology of naltrexone and acamprosate, both alone and combined, in alcohol-dependent subjects. J Clin Psychopharmacol 2003;23:281-93.
6. Overman GP, Teter CJ, Guthrie SK. Acamprosate for the adjunctive treatment of alcohol dependence. Ann Pharmacother 2003;37:1090-9.
7. Carmen B, Angeles M, Ana M, Maria AJ. Efficacy and safety of naltrexone and acamprosate in the treatment of alcohol dependence: a systematic review. Addiction 2004;99:811-28.
8. Kiefer F, Jahn H, Tarnaske T, et al. Comparing and combining naltrexone and acamprosate in relapse prevention of alcoholism: a double-blind, placebo-controlled study. Arch Gen Psychiatry 2003;60:92-9.
9. The COMBINE study research group. Testing combined pharmacotherapies and behavioral interventions in alcohol dependence: rationale and methods. Alcohol Clin Exp Res 2003;27:1107-22.
10. Campral prescribing information. Available at: http://www.campral.com. Accessed Jan. 7, 2005.
Eszopiclone: Targeting chronic insomnia
Nonbenzodiazepine hypnotics have become mainstays in insomnia treatment. These agents do not interfere with cognitive function upon awakening, compared with benzodiazepines and other agents used off-label as hypnotics.1
Eszopiclone has shown efficacy in clinical trials for treating short-term and long-term (lasting ≥3 weeks) insomnia. By contrast, zaleplon and zolpidem are indicated for short-term insomnia treatment.
HOW IT WORKS
Eszopiclone, a cyclopyrrolone, is the racemic form of zopiclone, an agent used worldwide to treat insomnia but not available in the United States. The racemic zopiclone has a high affinity for benzodiazepine binding sites in the cerebral cortex, hippocampus, and cerebellum.
As with the selective benzodiazepine receptor agonists zaleplon and zolpidem, information on eszopiclone’s receptor binding profile is limited. It is unclear if the agent binds directly to the benzodiazepine receptor or to a related site on the GABA receptor complex.
Table
Eszopiclone: Fast facts
| Brand name: |
| Lunesta |
| Class |
| Novel cyclopyrrolone, nonbenzodiazepine hypnotic |
| FDA-approved indication: |
| Insomnia |
| Approval date: |
| Dec. 15, 2004 |
| Manufacturer: |
| Sepracor |
| Dosing form: |
| 1-, 2-, and 3-mg tablets |
| Recommended dosage: |
| 2 to 3 mg HS (at bedtime) for adults age ≤65 |
| 1 to 2 mg HS for adults age >65 |
PHARMACOKINETICS
Preliminary studies suggest eszopiclone is rapidly absorbed from the GI tract, mostly within 1 hour of taking it.2,3 The agent reaches peak concentration within 30 minutes to 4 hours in healthy persons. A high-fat or heavy meal may delay hypnotic onset by approximately 1 hour.
Eszopiclone is metabolized mostly through the 3A4 isoenzyme of the cytochrome P(CYP)-450 system, although the CYP 2E1 isoenzyme also plays a minor role. About 75% of the dose is excreted in urine.4 Because its elimination half-life is approximately 6 hours, eszopiclone leaves no residual effects when patients awaken after about 6 hours of sleep.1
Because they take weeks to eliminate, some older sleep-promoting medications can cause increasing daytime sedation when used daily. By contrast, eszopiclone can be taken once daily with no risk of drug accumulation.
EFFICACY
Although relatively few clinical studies of eszopiclone have been published, the new-drug application submitted to the FDA summarized 24 clinical trials totaling more than 2,700 subjects.
Zammit et al5 gave 308 patients eszopiclone, 2 or 3 mg HS (at bedtime), or placebo for 6 weeks. Eszopiclone decreased time to falling asleep, increased total sleep time, improved continuity of sleep, and increased overall sleep quality throughout the night. After 6 weeks, patients in the treatment group showed:
- no residual morning sedation based on repeated polysomnography and morning questionnaire measures
- no residual daytime sedation based on results of the Digit Symbol Substitution Test, which gauges psychomotor impairment.
Patients taking 3 mg showed reduced wakefulness at night on objective and subjective measures compared with the placebo group.
A randomized, double-blind, multicenter, placebo-controlled study (N=788)6,7 assessed eszopiclone’s safety and efficacy across 6 months in patients with chronic insomnia. Before enrollment, patients slept
SAFETY AND TOLERABILITY
Eszopiclone was well tolerated in preclinical and clinical trials. The most common adverse event was a bitter taste reported by 34% of participants; this prompted 1.7% of patients in one study4 to discontinue eszopiclone, compared with 0.5% of patients taking placebo. Other common adverse effects included:
- daytime somnolence, (8% prevalence, 2.2% dropout rate
- depression (1% dropout rate).4
Krystal et al found no clinically significant changes in vital signs, ECG results, laboratory values, and physical examination findings between the eszopiclone and placebo groups.6,7
Few significant interactions between eszopiclone and other drugs have been reported. However:
- Increased sedation and decreased psychomotor functioning were observed with eszopiclone, 3 mg, and olanzapine, 10 mg.
- Drugs that inhibit (eg, ketoconazole) or induce (eg, rifampicin) the CYP 3A4 isoenzyme may alter eszopiclone levels.8
- A possible drug-drug interaction between eszopiclone and alcohol, 0.7 g/kg, decreased psychomotor performance for up to 4 hours after alcohol use.8
No significant drug-drug interactions were reported between eszopiclone and paroxetine or lorazepam.4
In another case, the parent compound zopiclone given concomitantly with trimipramine decreased both drugs’ bioavailability but did not noticeably change either drug’s clinical effect.9 As eszopiclone and zopiclone are chemically similar, be careful when giving eszopiclone to patients taking trimipramine or similar medications, such as tricyclic antidepressants.
DOSING
Start eszopiclone at 2 mg HS for adults and titrate to 3 mg as needed. For many patients, 3 mg may suffice as maintenance therapy. The risks and benefits of dosing eszopiclone at >3 mg are not known.
Lower doses are recommended for patients age >65 because of the risk of decreased motor and/or cognitive performance. Give 2 mg for maintenance and 1 mg for difficulty falling asleep. There are no other known contraindications to eszopiclone use.
As with other hypnotics, supplement eszopiclone with sleep hygiene education and relaxation techniques.
CLINICAL IMPLICATIONS
Eszopiclone has shown efficacy for >2 weeks in primary insomnia, suggesting the agent may help treat chronic insomnia.
As with other nonbenzodiazepine hypnotics, off-label use of eszopiclone with antidepressants may help treat insomnia secondary to depressive or anxiety disorders. Research is needed to gauge the drug’s effectiveness for this use.
Related resources
- American Academy of Sleep Medicine. www.aasmnet.org
- Lunesta Web site. www.lunesta.com
Drug brand names
- Eszopiclone • Lunesta
- Ketoconazole • Nizoral
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Trimipramine • Surmontil
- Zaleplon • Sonata
- Zolpidem • Ambien
Disclosure
Dr. Krahn reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Terzano MG, Rossi M, Palomba V, et al. New drugs for insomnia: comparative tolerability of zopiclone, zolpidem, and zaleplon. Drug Safety 2003;26:261-82.
2. Fernandez C, Martin C, Giminez F, Farinotti R. Clinical pharmacokinetics of zopiclone. Clin Pharmacokinet 1995;29:431-41.
3. Leese P, Maier G. Eszopiclone: Pharmacokinetic (PK) and pharmacodynamic effects of a novel sedative anti-insomnia agent after daytime administration in healthy subjects. Sleep 2002;25 (suppl):A45.-
4. Lunesta (eszopiclone) prescribing information. Available at: http://www.lunesta.com. Accessed Jan. 6, 2005.
5. Zammit GK, Gillin JC, McNabb L, et al. Eszopiclone, a novel non-benzodiazepine anti-insomnia agent: a six-week efficacy and safety study in adult patients with chronic insomnia. Sleep 2003;26(suppl):A297.-
6. Krystal A, Walsh J, Roth T, et al. The sustained efficacy and safety of eszopiclone over six months of nightly treatment: a placebo controlled study in patients with chronic insomnia. Sleep 2003;26(suppl):0779.-
7. Krystal A, Walsh J, Laska E, et al. Sustained efficacy of eszopiclone over six months of nightly treatment: results of a randomized, double-blind, placebo-controlled study in adults with chronic insomnia. Sleep 2003;26:793-9.
8. Hesse LM, von Moltke LL, Greenblatt DJ. Clinically important drug interactions with zopiclone, zolpidem, and zaleplon. CNS Drugs 2003;17:513-32.
9. Caille G, du Souich P, Spenard J, et al. Pharmacokinetic and clinical parameters of zopiclone and trimipramine when administered simultaneously to volunteers. Biopharm Drug Dispos 1984;5:117-25.
Nonbenzodiazepine hypnotics have become mainstays in insomnia treatment. These agents do not interfere with cognitive function upon awakening, compared with benzodiazepines and other agents used off-label as hypnotics.1
Eszopiclone has shown efficacy in clinical trials for treating short-term and long-term (lasting ≥3 weeks) insomnia. By contrast, zaleplon and zolpidem are indicated for short-term insomnia treatment.
HOW IT WORKS
Eszopiclone, a cyclopyrrolone, is the racemic form of zopiclone, an agent used worldwide to treat insomnia but not available in the United States. The racemic zopiclone has a high affinity for benzodiazepine binding sites in the cerebral cortex, hippocampus, and cerebellum.
As with the selective benzodiazepine receptor agonists zaleplon and zolpidem, information on eszopiclone’s receptor binding profile is limited. It is unclear if the agent binds directly to the benzodiazepine receptor or to a related site on the GABA receptor complex.
Table
Eszopiclone: Fast facts
| Brand name: |
| Lunesta |
| Class |
| Novel cyclopyrrolone, nonbenzodiazepine hypnotic |
| FDA-approved indication: |
| Insomnia |
| Approval date: |
| Dec. 15, 2004 |
| Manufacturer: |
| Sepracor |
| Dosing form: |
| 1-, 2-, and 3-mg tablets |
| Recommended dosage: |
| 2 to 3 mg HS (at bedtime) for adults age ≤65 |
| 1 to 2 mg HS for adults age >65 |
PHARMACOKINETICS
Preliminary studies suggest eszopiclone is rapidly absorbed from the GI tract, mostly within 1 hour of taking it.2,3 The agent reaches peak concentration within 30 minutes to 4 hours in healthy persons. A high-fat or heavy meal may delay hypnotic onset by approximately 1 hour.
Eszopiclone is metabolized mostly through the 3A4 isoenzyme of the cytochrome P(CYP)-450 system, although the CYP 2E1 isoenzyme also plays a minor role. About 75% of the dose is excreted in urine.4 Because its elimination half-life is approximately 6 hours, eszopiclone leaves no residual effects when patients awaken after about 6 hours of sleep.1
Because they take weeks to eliminate, some older sleep-promoting medications can cause increasing daytime sedation when used daily. By contrast, eszopiclone can be taken once daily with no risk of drug accumulation.
EFFICACY
Although relatively few clinical studies of eszopiclone have been published, the new-drug application submitted to the FDA summarized 24 clinical trials totaling more than 2,700 subjects.
Zammit et al5 gave 308 patients eszopiclone, 2 or 3 mg HS (at bedtime), or placebo for 6 weeks. Eszopiclone decreased time to falling asleep, increased total sleep time, improved continuity of sleep, and increased overall sleep quality throughout the night. After 6 weeks, patients in the treatment group showed:
- no residual morning sedation based on repeated polysomnography and morning questionnaire measures
- no residual daytime sedation based on results of the Digit Symbol Substitution Test, which gauges psychomotor impairment.
Patients taking 3 mg showed reduced wakefulness at night on objective and subjective measures compared with the placebo group.
A randomized, double-blind, multicenter, placebo-controlled study (N=788)6,7 assessed eszopiclone’s safety and efficacy across 6 months in patients with chronic insomnia. Before enrollment, patients slept
SAFETY AND TOLERABILITY
Eszopiclone was well tolerated in preclinical and clinical trials. The most common adverse event was a bitter taste reported by 34% of participants; this prompted 1.7% of patients in one study4 to discontinue eszopiclone, compared with 0.5% of patients taking placebo. Other common adverse effects included:
- daytime somnolence, (8% prevalence, 2.2% dropout rate
- depression (1% dropout rate).4
Krystal et al found no clinically significant changes in vital signs, ECG results, laboratory values, and physical examination findings between the eszopiclone and placebo groups.6,7
Few significant interactions between eszopiclone and other drugs have been reported. However:
- Increased sedation and decreased psychomotor functioning were observed with eszopiclone, 3 mg, and olanzapine, 10 mg.
- Drugs that inhibit (eg, ketoconazole) or induce (eg, rifampicin) the CYP 3A4 isoenzyme may alter eszopiclone levels.8
- A possible drug-drug interaction between eszopiclone and alcohol, 0.7 g/kg, decreased psychomotor performance for up to 4 hours after alcohol use.8
No significant drug-drug interactions were reported between eszopiclone and paroxetine or lorazepam.4
In another case, the parent compound zopiclone given concomitantly with trimipramine decreased both drugs’ bioavailability but did not noticeably change either drug’s clinical effect.9 As eszopiclone and zopiclone are chemically similar, be careful when giving eszopiclone to patients taking trimipramine or similar medications, such as tricyclic antidepressants.
DOSING
Start eszopiclone at 2 mg HS for adults and titrate to 3 mg as needed. For many patients, 3 mg may suffice as maintenance therapy. The risks and benefits of dosing eszopiclone at >3 mg are not known.
Lower doses are recommended for patients age >65 because of the risk of decreased motor and/or cognitive performance. Give 2 mg for maintenance and 1 mg for difficulty falling asleep. There are no other known contraindications to eszopiclone use.
As with other hypnotics, supplement eszopiclone with sleep hygiene education and relaxation techniques.
CLINICAL IMPLICATIONS
Eszopiclone has shown efficacy for >2 weeks in primary insomnia, suggesting the agent may help treat chronic insomnia.
As with other nonbenzodiazepine hypnotics, off-label use of eszopiclone with antidepressants may help treat insomnia secondary to depressive or anxiety disorders. Research is needed to gauge the drug’s effectiveness for this use.
Related resources
- American Academy of Sleep Medicine. www.aasmnet.org
- Lunesta Web site. www.lunesta.com
Drug brand names
- Eszopiclone • Lunesta
- Ketoconazole • Nizoral
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Trimipramine • Surmontil
- Zaleplon • Sonata
- Zolpidem • Ambien
Disclosure
Dr. Krahn reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Nonbenzodiazepine hypnotics have become mainstays in insomnia treatment. These agents do not interfere with cognitive function upon awakening, compared with benzodiazepines and other agents used off-label as hypnotics.1
Eszopiclone has shown efficacy in clinical trials for treating short-term and long-term (lasting ≥3 weeks) insomnia. By contrast, zaleplon and zolpidem are indicated for short-term insomnia treatment.
HOW IT WORKS
Eszopiclone, a cyclopyrrolone, is the racemic form of zopiclone, an agent used worldwide to treat insomnia but not available in the United States. The racemic zopiclone has a high affinity for benzodiazepine binding sites in the cerebral cortex, hippocampus, and cerebellum.
As with the selective benzodiazepine receptor agonists zaleplon and zolpidem, information on eszopiclone’s receptor binding profile is limited. It is unclear if the agent binds directly to the benzodiazepine receptor or to a related site on the GABA receptor complex.
Table
Eszopiclone: Fast facts
| Brand name: |
| Lunesta |
| Class |
| Novel cyclopyrrolone, nonbenzodiazepine hypnotic |
| FDA-approved indication: |
| Insomnia |
| Approval date: |
| Dec. 15, 2004 |
| Manufacturer: |
| Sepracor |
| Dosing form: |
| 1-, 2-, and 3-mg tablets |
| Recommended dosage: |
| 2 to 3 mg HS (at bedtime) for adults age ≤65 |
| 1 to 2 mg HS for adults age >65 |
PHARMACOKINETICS
Preliminary studies suggest eszopiclone is rapidly absorbed from the GI tract, mostly within 1 hour of taking it.2,3 The agent reaches peak concentration within 30 minutes to 4 hours in healthy persons. A high-fat or heavy meal may delay hypnotic onset by approximately 1 hour.
Eszopiclone is metabolized mostly through the 3A4 isoenzyme of the cytochrome P(CYP)-450 system, although the CYP 2E1 isoenzyme also plays a minor role. About 75% of the dose is excreted in urine.4 Because its elimination half-life is approximately 6 hours, eszopiclone leaves no residual effects when patients awaken after about 6 hours of sleep.1
Because they take weeks to eliminate, some older sleep-promoting medications can cause increasing daytime sedation when used daily. By contrast, eszopiclone can be taken once daily with no risk of drug accumulation.
EFFICACY
Although relatively few clinical studies of eszopiclone have been published, the new-drug application submitted to the FDA summarized 24 clinical trials totaling more than 2,700 subjects.
Zammit et al5 gave 308 patients eszopiclone, 2 or 3 mg HS (at bedtime), or placebo for 6 weeks. Eszopiclone decreased time to falling asleep, increased total sleep time, improved continuity of sleep, and increased overall sleep quality throughout the night. After 6 weeks, patients in the treatment group showed:
- no residual morning sedation based on repeated polysomnography and morning questionnaire measures
- no residual daytime sedation based on results of the Digit Symbol Substitution Test, which gauges psychomotor impairment.
Patients taking 3 mg showed reduced wakefulness at night on objective and subjective measures compared with the placebo group.
A randomized, double-blind, multicenter, placebo-controlled study (N=788)6,7 assessed eszopiclone’s safety and efficacy across 6 months in patients with chronic insomnia. Before enrollment, patients slept
SAFETY AND TOLERABILITY
Eszopiclone was well tolerated in preclinical and clinical trials. The most common adverse event was a bitter taste reported by 34% of participants; this prompted 1.7% of patients in one study4 to discontinue eszopiclone, compared with 0.5% of patients taking placebo. Other common adverse effects included:
- daytime somnolence, (8% prevalence, 2.2% dropout rate
- depression (1% dropout rate).4
Krystal et al found no clinically significant changes in vital signs, ECG results, laboratory values, and physical examination findings between the eszopiclone and placebo groups.6,7
Few significant interactions between eszopiclone and other drugs have been reported. However:
- Increased sedation and decreased psychomotor functioning were observed with eszopiclone, 3 mg, and olanzapine, 10 mg.
- Drugs that inhibit (eg, ketoconazole) or induce (eg, rifampicin) the CYP 3A4 isoenzyme may alter eszopiclone levels.8
- A possible drug-drug interaction between eszopiclone and alcohol, 0.7 g/kg, decreased psychomotor performance for up to 4 hours after alcohol use.8
No significant drug-drug interactions were reported between eszopiclone and paroxetine or lorazepam.4
In another case, the parent compound zopiclone given concomitantly with trimipramine decreased both drugs’ bioavailability but did not noticeably change either drug’s clinical effect.9 As eszopiclone and zopiclone are chemically similar, be careful when giving eszopiclone to patients taking trimipramine or similar medications, such as tricyclic antidepressants.
DOSING
Start eszopiclone at 2 mg HS for adults and titrate to 3 mg as needed. For many patients, 3 mg may suffice as maintenance therapy. The risks and benefits of dosing eszopiclone at >3 mg are not known.
Lower doses are recommended for patients age >65 because of the risk of decreased motor and/or cognitive performance. Give 2 mg for maintenance and 1 mg for difficulty falling asleep. There are no other known contraindications to eszopiclone use.
As with other hypnotics, supplement eszopiclone with sleep hygiene education and relaxation techniques.
CLINICAL IMPLICATIONS
Eszopiclone has shown efficacy for >2 weeks in primary insomnia, suggesting the agent may help treat chronic insomnia.
As with other nonbenzodiazepine hypnotics, off-label use of eszopiclone with antidepressants may help treat insomnia secondary to depressive or anxiety disorders. Research is needed to gauge the drug’s effectiveness for this use.
Related resources
- American Academy of Sleep Medicine. www.aasmnet.org
- Lunesta Web site. www.lunesta.com
Drug brand names
- Eszopiclone • Lunesta
- Ketoconazole • Nizoral
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Trimipramine • Surmontil
- Zaleplon • Sonata
- Zolpidem • Ambien
Disclosure
Dr. Krahn reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Terzano MG, Rossi M, Palomba V, et al. New drugs for insomnia: comparative tolerability of zopiclone, zolpidem, and zaleplon. Drug Safety 2003;26:261-82.
2. Fernandez C, Martin C, Giminez F, Farinotti R. Clinical pharmacokinetics of zopiclone. Clin Pharmacokinet 1995;29:431-41.
3. Leese P, Maier G. Eszopiclone: Pharmacokinetic (PK) and pharmacodynamic effects of a novel sedative anti-insomnia agent after daytime administration in healthy subjects. Sleep 2002;25 (suppl):A45.-
4. Lunesta (eszopiclone) prescribing information. Available at: http://www.lunesta.com. Accessed Jan. 6, 2005.
5. Zammit GK, Gillin JC, McNabb L, et al. Eszopiclone, a novel non-benzodiazepine anti-insomnia agent: a six-week efficacy and safety study in adult patients with chronic insomnia. Sleep 2003;26(suppl):A297.-
6. Krystal A, Walsh J, Roth T, et al. The sustained efficacy and safety of eszopiclone over six months of nightly treatment: a placebo controlled study in patients with chronic insomnia. Sleep 2003;26(suppl):0779.-
7. Krystal A, Walsh J, Laska E, et al. Sustained efficacy of eszopiclone over six months of nightly treatment: results of a randomized, double-blind, placebo-controlled study in adults with chronic insomnia. Sleep 2003;26:793-9.
8. Hesse LM, von Moltke LL, Greenblatt DJ. Clinically important drug interactions with zopiclone, zolpidem, and zaleplon. CNS Drugs 2003;17:513-32.
9. Caille G, du Souich P, Spenard J, et al. Pharmacokinetic and clinical parameters of zopiclone and trimipramine when administered simultaneously to volunteers. Biopharm Drug Dispos 1984;5:117-25.
1. Terzano MG, Rossi M, Palomba V, et al. New drugs for insomnia: comparative tolerability of zopiclone, zolpidem, and zaleplon. Drug Safety 2003;26:261-82.
2. Fernandez C, Martin C, Giminez F, Farinotti R. Clinical pharmacokinetics of zopiclone. Clin Pharmacokinet 1995;29:431-41.
3. Leese P, Maier G. Eszopiclone: Pharmacokinetic (PK) and pharmacodynamic effects of a novel sedative anti-insomnia agent after daytime administration in healthy subjects. Sleep 2002;25 (suppl):A45.-
4. Lunesta (eszopiclone) prescribing information. Available at: http://www.lunesta.com. Accessed Jan. 6, 2005.
5. Zammit GK, Gillin JC, McNabb L, et al. Eszopiclone, a novel non-benzodiazepine anti-insomnia agent: a six-week efficacy and safety study in adult patients with chronic insomnia. Sleep 2003;26(suppl):A297.-
6. Krystal A, Walsh J, Roth T, et al. The sustained efficacy and safety of eszopiclone over six months of nightly treatment: a placebo controlled study in patients with chronic insomnia. Sleep 2003;26(suppl):0779.-
7. Krystal A, Walsh J, Laska E, et al. Sustained efficacy of eszopiclone over six months of nightly treatment: results of a randomized, double-blind, placebo-controlled study in adults with chronic insomnia. Sleep 2003;26:793-9.
8. Hesse LM, von Moltke LL, Greenblatt DJ. Clinically important drug interactions with zopiclone, zolpidem, and zaleplon. CNS Drugs 2003;17:513-32.
9. Caille G, du Souich P, Spenard J, et al. Pharmacokinetic and clinical parameters of zopiclone and trimipramine when administered simultaneously to volunteers. Biopharm Drug Dispos 1984;5:117-25.
Duloxetine: Dual-action antidepressant
Depression’s remission rates remain low,1 and its common somatic symptoms (aches and pains, headaches, backaches) often complicate diagnosis and treatment.2
Duloxetine, recently FDA-approved for treating major depression Table 1, has shown efficacy against depression’s emotional and somatic symptoms in clinical trials.
HOW IT WORKS
Duloxetine inhibits both serotonin and norepinephrine reuptake. Researchers suggest that antidepressants exhibiting this dual action may be more effective and act faster than single-action selective serotonin reuptake inhibitors.3,4 Newer dual-action antidepressants also are more tolerable than dual-action tricyclic antidepressants.
Table 1
Duloxetine: Fast facts
| Drug brand name: Cymbalta |
| Class Serotonin and norepinephrine reuptake inhibitor |
| FDA-approved indication: Treatment of major depressive episodes |
| Approval date: August 3 2004 |
| Manufacturer: Eli Lilly and Co. |
| Dosing form: 20 mg, 30 mg, 60 mg capsules |
| Recommended dosage: 40 to 60 mg/d |
| Maximum dosage(studied in major depression): 120 mg/d |
Researchers have seen synergism between serotonergic and noradrenergic pain modulation at the spinal cord,4,5 suggesting that dual-action antidepressants may ameliorate major depression’s somatic symptoms.
Table 2
Plasma levels of these agents may affect—or be affected by— duloxetine coadministration
| CYP 2D6 substrates |
| Amitriptyline |
| Beta blockers Propranolol, metoprolol, timolol |
| Desipramine |
| Fluoxetine |
| Fluvoxamine |
| Haloperidol |
| Nortriptyline |
| Risperidone |
| Thioridazine |
| Type 1C antiarrhythmics Propafenone, flecainide |
| Venlafaxine |
| CYP 2D6 inhibitors |
| Cimetidine |
| Fluoxetine |
| Haloperidol |
| Paroxetine |
| Quinidine |
| CYP 1A2 inhibitors |
| Cimetidine |
| Ciprofloxacin |
| Enoxacin |
| Source: Reference 7 |
PHARMACOKINETICS
Despite its 12-hour plasma half-life, duloxetine has shown efficacy in clinical trials when given once daily. Mean plasma clearance is approximately 101 L/hr, with a mean volume of distribution of about 1640 L, meaning that duloxetine is distributed throughout the body.
The agent is more than 90% protein bound; thus, giving duloxetine concomitantly with another highly protein-bound agent could increase the side-effect risk of either drug.
Food does not alter duloxetine’s absorption but delays maximum concentration by about 4 hours Duloxetine may be taken before or after meals, though taking it after meals could reduce the risk of nausea—a common early side effect.
Duloxetine is metabolized by the 2D6 and 1A2 isoenzymes of the cytochrome P-450 system. It inhibits the CYP 2D6 isoenzyme but to a lesser extent than fluoxetine does.6 Co-administering duloxetine with a CYP 2D6 substrate or inhibitor or a CYP 1A2 inhibitor Table 27 could elevate plasma levels of duloxetine or the other agent, possibly increasing adverse effects.
EFFICACY
In an 8-week, placebo-controlled trial, Goldstein et al8 compared fluoxetine, 20 mg/d, and duloxetine, 40 mg/d titrated to 120 mg/d over 3 weeks, in 173 patients with major depressive disorder. Participants’ scores at baseline were 15 on the 17-item Hamilton Rating Scale for Depression (HAM-D-17) and 4 on the Clinical Global Improvement-Severity scale. Estimated probability of remission was 56% with duloxetine, 30% with fluoxetine, and 32% with placebo, with remission defined as achieving a HAM-D-17 score 7.
In two prospective, double-blind, placebocontrolled trials of 512 patients with major depression,9,10 duloxetine, 60 mg/d, reduced body, back, and shoulder pain based on Visual Analog Scale (VAS) scores; pre- and posttreatment VAS scores were not listed in the published studies. Estimated probability of remission in these two studies was 44% and 43% among patients taking duloxetine vs 16% and 28% in the placebo groups. Remission again was defined as HAM-D-17 score 7.
TOLERABILITY
In the two double-blind studies just mentioned,9,10 Detke et al reported adverse event-related drop out rates of 12.5% and 13.8% for duloxetine, 60 mg/d, vs 4.3% and 2.3% for placebo. Nausea, insomnia, headaches, somnolence, dry mouth, and sweating were most frequently reported.
Dizziness. Mild dizziness was reported in 11.3% of patients who abruptly stopped duloxetine after 9 weeks.9
Headaches. In one comparator trial,8 fewer headaches (20%) were reported among patients taking duloxetine, 40 to 120 mg/d, vs those taking fluoxetine, 20 mg/d (33.3%), or placebo (31.4%).
Hypertension. Detke et al10 found no statistical separation in systolic and diastolic blood pressures between the duloxetine (60 mg/d) and placebo groups. Likewise, Goldstein et al8 found a similar incidence of hypertension among patients taking duloxetine, 40 to 120 mg/d, or placebo. In clinical trials,11 duloxetine increased blood pressure by a mean of 2.0 mm Hg (systolic) and 0.5 mm Hg (diastolic).
As with several other noradrenergic medications, FDA recommends that clinicians check blood pressures before starting duloxetine therapy and periodically thereafter.
Duloxetine has not been studied in persons with poorly controlled hypertension.
Nausea. Mild to moderate nausea was the most common adverse event in one study;9 the effect dissipated after a median of 7 days. One patient reported severe nausea, and 1 patient out of 123 stopped the medication because of nausea.
Sexual dysfunction. Using the Arizona Sexual Experiences Scale (ASEX), Goldstein et al8 prospectively assessed sexual function in 70 men and women taking duloxetine or placebo. No statistical difference was seen between the two groups from baseline to endpoint.
In another study,12 duloxetine showed worsening only in ASEX item 4 (“How easily can you reach an orgasm?”), indicating some adverse sexual effects in men. No such differences were found in women. Duloxetine’s effect on sexual function needs to be studied further.
Duloxetine is an FDA Use-in-Pregnancy category C medication, meaning that risk to the fetus has not been ruled out. The agent is contraindicated in patients taking monoamine oxidase inhibitors and in those with narrow-angle glaucoma. FDA recommends not using duloxetine in patients with hepatic insufficiency, endstage renal disease, and substantial alcohol use.
DOSING STRATEGIES
Duloxetine, 40 to 120 mg/d, appears to be safe and effective for most adults.8-10 FDA recommends starting at 40 mg/d (20 mg bid) to 60 mg/d (once-daily or 30 mg bid) with no regard to meals. Dosages >60 mg/d have not shown additional benefit. Age and tolerability should drive initial dosing and titration. Side-effect incidence has not been directly compared at 60, 90, and 120 mg/d.
CLINICAL IMPLICATIONS
In clinical trials, duloxetine has shown a high estimated probability of remission in major depression and has shown efficacy against depression’s physical and emotional symptoms. Based on efficacy and safety data, duloxetine appears to be a first-line treatment option for major depression.
Related resources
- Gray GE. Concise guide to evidence-based psychiatry. Washington, DC: American Psychiatric Publishing, 2004.
- Schatzberg AF, Nemeroff CB (eds). Textbook of psychopharmacology (3rd ed). Washington, DC: American Psychiatric Publishing, 2004.
Drug brand names
- Amitriptyline • Elavil
- Cimetidine • Tagamet
- Ciprofloxacin • Cipro
- Desipramine • Norpramin
- Duloxetine • Cymbalta
- Enoxacin • Penetrex
- Fluoxetine • Prozac
- Flecainide • Tambocor
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Metoprolol • Toprol
- Nortriptyline • Pamelor
- Paroxetine • Paxil
- Propafenone • Rythmol
- Propranolol • Inderal
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Timolol • Blocadren, others
- Venlafaxine • Effexor
Disclosure
Dr. Rakesh Jain receives research grants from Eli Lilly and Co., Forest Pharmaceuticals, GlaxoSmithKline, Merck and Co., Organon, Pfizer Inc., and Sepracor. He is a consultant to and speaker for Eli Lilly and Co., and is a speaker for GlaxoSmithKline, Pfizer Inc., and Wyeth Pharmaceuticals.
Dr. Shailesh Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of compefting products.
1. Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or serotonin reuptake inhibitors. Br J Psychiatry 2001;178:234-41.
2. Fava M. The role of the serotonergic and noradrenergic neurotransmitter systems in the treatment of psychological and physical symptoms of depression. J Clin Psychiatry 2003;64(suppl 13):26-9.
3. Tran PV, Bymaster FP, McNamara RK, Potter WZ. Dual monoamine modulation for improved treatment of major depressive disorder. J Clin Psychopharmacol 2003;23:78-86.
4. Ansari A. The efficacy of newer antidepressants in the treatment of chronic pain: a review of current literature. Harv Rev Psychiatry 2000;7:257-77.
5. Willis WD, Westlund KN. Neuroanatomy of the pain system and the pathways that modulate pain. J Clin Neurophysiol 1997;14:2-31.
6. Skinner MH, Kuan HY, Pan A, et al. Duloxetine is both an inhibitor and a substrate of cytochrome P4502D6 in healthy volunteers. Clin Pharmacol Ther 2003;73:170-7.
7. Sadock BJ, Sadock VA. Kaplan and Sadock's pocket handbook of psychotropic drug treatment (3rd ed). Baltimore, MD: Lippincott Williams and Wilkins, 2001.
8. Goldstein DJ, Mallinckrodt C, Lu Y, Demitrack M. Duloxetine in the treatment of major depression disorder: a double-blind clinical trial. J Clin Psychiatry 2002;63:225-31.
9. Detke MJ, Lu Y, Goldstein DJ, et al. Duloxetine 60 mg once-daily for major depressive disorder: a randomized double-blind placebocontrolled trial. J Clin Psychiatry 2002;63:308-15.
10. Detke MJ, Lu Y, Goldstein DJ, et al. Duloxetine 60 mg once daily dosing versus placebo in the treatment of major depression. J Psychiatr Res 2002;36:383-90.
11. Cymbalta prescribing information Eli Lilly and Co 2004.
12. Goldstein DJ, Lu Y, Detke MJ, et al. Duloxetine in the treatment of depression: a double-blind placebo-controlled comparison with paroxetine. J Clin Psychopharmacol 2004;24:389-99.
Depression’s remission rates remain low,1 and its common somatic symptoms (aches and pains, headaches, backaches) often complicate diagnosis and treatment.2
Duloxetine, recently FDA-approved for treating major depression Table 1, has shown efficacy against depression’s emotional and somatic symptoms in clinical trials.
HOW IT WORKS
Duloxetine inhibits both serotonin and norepinephrine reuptake. Researchers suggest that antidepressants exhibiting this dual action may be more effective and act faster than single-action selective serotonin reuptake inhibitors.3,4 Newer dual-action antidepressants also are more tolerable than dual-action tricyclic antidepressants.
Table 1
Duloxetine: Fast facts
| Drug brand name: Cymbalta |
| Class Serotonin and norepinephrine reuptake inhibitor |
| FDA-approved indication: Treatment of major depressive episodes |
| Approval date: August 3 2004 |
| Manufacturer: Eli Lilly and Co. |
| Dosing form: 20 mg, 30 mg, 60 mg capsules |
| Recommended dosage: 40 to 60 mg/d |
| Maximum dosage(studied in major depression): 120 mg/d |
Researchers have seen synergism between serotonergic and noradrenergic pain modulation at the spinal cord,4,5 suggesting that dual-action antidepressants may ameliorate major depression’s somatic symptoms.
Table 2
Plasma levels of these agents may affect—or be affected by— duloxetine coadministration
| CYP 2D6 substrates |
| Amitriptyline |
| Beta blockers Propranolol, metoprolol, timolol |
| Desipramine |
| Fluoxetine |
| Fluvoxamine |
| Haloperidol |
| Nortriptyline |
| Risperidone |
| Thioridazine |
| Type 1C antiarrhythmics Propafenone, flecainide |
| Venlafaxine |
| CYP 2D6 inhibitors |
| Cimetidine |
| Fluoxetine |
| Haloperidol |
| Paroxetine |
| Quinidine |
| CYP 1A2 inhibitors |
| Cimetidine |
| Ciprofloxacin |
| Enoxacin |
| Source: Reference 7 |
PHARMACOKINETICS
Despite its 12-hour plasma half-life, duloxetine has shown efficacy in clinical trials when given once daily. Mean plasma clearance is approximately 101 L/hr, with a mean volume of distribution of about 1640 L, meaning that duloxetine is distributed throughout the body.
The agent is more than 90% protein bound; thus, giving duloxetine concomitantly with another highly protein-bound agent could increase the side-effect risk of either drug.
Food does not alter duloxetine’s absorption but delays maximum concentration by about 4 hours Duloxetine may be taken before or after meals, though taking it after meals could reduce the risk of nausea—a common early side effect.
Duloxetine is metabolized by the 2D6 and 1A2 isoenzymes of the cytochrome P-450 system. It inhibits the CYP 2D6 isoenzyme but to a lesser extent than fluoxetine does.6 Co-administering duloxetine with a CYP 2D6 substrate or inhibitor or a CYP 1A2 inhibitor Table 27 could elevate plasma levels of duloxetine or the other agent, possibly increasing adverse effects.
EFFICACY
In an 8-week, placebo-controlled trial, Goldstein et al8 compared fluoxetine, 20 mg/d, and duloxetine, 40 mg/d titrated to 120 mg/d over 3 weeks, in 173 patients with major depressive disorder. Participants’ scores at baseline were 15 on the 17-item Hamilton Rating Scale for Depression (HAM-D-17) and 4 on the Clinical Global Improvement-Severity scale. Estimated probability of remission was 56% with duloxetine, 30% with fluoxetine, and 32% with placebo, with remission defined as achieving a HAM-D-17 score 7.
In two prospective, double-blind, placebocontrolled trials of 512 patients with major depression,9,10 duloxetine, 60 mg/d, reduced body, back, and shoulder pain based on Visual Analog Scale (VAS) scores; pre- and posttreatment VAS scores were not listed in the published studies. Estimated probability of remission in these two studies was 44% and 43% among patients taking duloxetine vs 16% and 28% in the placebo groups. Remission again was defined as HAM-D-17 score 7.
TOLERABILITY
In the two double-blind studies just mentioned,9,10 Detke et al reported adverse event-related drop out rates of 12.5% and 13.8% for duloxetine, 60 mg/d, vs 4.3% and 2.3% for placebo. Nausea, insomnia, headaches, somnolence, dry mouth, and sweating were most frequently reported.
Dizziness. Mild dizziness was reported in 11.3% of patients who abruptly stopped duloxetine after 9 weeks.9
Headaches. In one comparator trial,8 fewer headaches (20%) were reported among patients taking duloxetine, 40 to 120 mg/d, vs those taking fluoxetine, 20 mg/d (33.3%), or placebo (31.4%).
Hypertension. Detke et al10 found no statistical separation in systolic and diastolic blood pressures between the duloxetine (60 mg/d) and placebo groups. Likewise, Goldstein et al8 found a similar incidence of hypertension among patients taking duloxetine, 40 to 120 mg/d, or placebo. In clinical trials,11 duloxetine increased blood pressure by a mean of 2.0 mm Hg (systolic) and 0.5 mm Hg (diastolic).
As with several other noradrenergic medications, FDA recommends that clinicians check blood pressures before starting duloxetine therapy and periodically thereafter.
Duloxetine has not been studied in persons with poorly controlled hypertension.
Nausea. Mild to moderate nausea was the most common adverse event in one study;9 the effect dissipated after a median of 7 days. One patient reported severe nausea, and 1 patient out of 123 stopped the medication because of nausea.
Sexual dysfunction. Using the Arizona Sexual Experiences Scale (ASEX), Goldstein et al8 prospectively assessed sexual function in 70 men and women taking duloxetine or placebo. No statistical difference was seen between the two groups from baseline to endpoint.
In another study,12 duloxetine showed worsening only in ASEX item 4 (“How easily can you reach an orgasm?”), indicating some adverse sexual effects in men. No such differences were found in women. Duloxetine’s effect on sexual function needs to be studied further.
Duloxetine is an FDA Use-in-Pregnancy category C medication, meaning that risk to the fetus has not been ruled out. The agent is contraindicated in patients taking monoamine oxidase inhibitors and in those with narrow-angle glaucoma. FDA recommends not using duloxetine in patients with hepatic insufficiency, endstage renal disease, and substantial alcohol use.
DOSING STRATEGIES
Duloxetine, 40 to 120 mg/d, appears to be safe and effective for most adults.8-10 FDA recommends starting at 40 mg/d (20 mg bid) to 60 mg/d (once-daily or 30 mg bid) with no regard to meals. Dosages >60 mg/d have not shown additional benefit. Age and tolerability should drive initial dosing and titration. Side-effect incidence has not been directly compared at 60, 90, and 120 mg/d.
CLINICAL IMPLICATIONS
In clinical trials, duloxetine has shown a high estimated probability of remission in major depression and has shown efficacy against depression’s physical and emotional symptoms. Based on efficacy and safety data, duloxetine appears to be a first-line treatment option for major depression.
Related resources
- Gray GE. Concise guide to evidence-based psychiatry. Washington, DC: American Psychiatric Publishing, 2004.
- Schatzberg AF, Nemeroff CB (eds). Textbook of psychopharmacology (3rd ed). Washington, DC: American Psychiatric Publishing, 2004.
Drug brand names
- Amitriptyline • Elavil
- Cimetidine • Tagamet
- Ciprofloxacin • Cipro
- Desipramine • Norpramin
- Duloxetine • Cymbalta
- Enoxacin • Penetrex
- Fluoxetine • Prozac
- Flecainide • Tambocor
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Metoprolol • Toprol
- Nortriptyline • Pamelor
- Paroxetine • Paxil
- Propafenone • Rythmol
- Propranolol • Inderal
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Timolol • Blocadren, others
- Venlafaxine • Effexor
Disclosure
Dr. Rakesh Jain receives research grants from Eli Lilly and Co., Forest Pharmaceuticals, GlaxoSmithKline, Merck and Co., Organon, Pfizer Inc., and Sepracor. He is a consultant to and speaker for Eli Lilly and Co., and is a speaker for GlaxoSmithKline, Pfizer Inc., and Wyeth Pharmaceuticals.
Dr. Shailesh Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of compefting products.
Depression’s remission rates remain low,1 and its common somatic symptoms (aches and pains, headaches, backaches) often complicate diagnosis and treatment.2
Duloxetine, recently FDA-approved for treating major depression Table 1, has shown efficacy against depression’s emotional and somatic symptoms in clinical trials.
HOW IT WORKS
Duloxetine inhibits both serotonin and norepinephrine reuptake. Researchers suggest that antidepressants exhibiting this dual action may be more effective and act faster than single-action selective serotonin reuptake inhibitors.3,4 Newer dual-action antidepressants also are more tolerable than dual-action tricyclic antidepressants.
Table 1
Duloxetine: Fast facts
| Drug brand name: Cymbalta |
| Class Serotonin and norepinephrine reuptake inhibitor |
| FDA-approved indication: Treatment of major depressive episodes |
| Approval date: August 3 2004 |
| Manufacturer: Eli Lilly and Co. |
| Dosing form: 20 mg, 30 mg, 60 mg capsules |
| Recommended dosage: 40 to 60 mg/d |
| Maximum dosage(studied in major depression): 120 mg/d |
Researchers have seen synergism between serotonergic and noradrenergic pain modulation at the spinal cord,4,5 suggesting that dual-action antidepressants may ameliorate major depression’s somatic symptoms.
Table 2
Plasma levels of these agents may affect—or be affected by— duloxetine coadministration
| CYP 2D6 substrates |
| Amitriptyline |
| Beta blockers Propranolol, metoprolol, timolol |
| Desipramine |
| Fluoxetine |
| Fluvoxamine |
| Haloperidol |
| Nortriptyline |
| Risperidone |
| Thioridazine |
| Type 1C antiarrhythmics Propafenone, flecainide |
| Venlafaxine |
| CYP 2D6 inhibitors |
| Cimetidine |
| Fluoxetine |
| Haloperidol |
| Paroxetine |
| Quinidine |
| CYP 1A2 inhibitors |
| Cimetidine |
| Ciprofloxacin |
| Enoxacin |
| Source: Reference 7 |
PHARMACOKINETICS
Despite its 12-hour plasma half-life, duloxetine has shown efficacy in clinical trials when given once daily. Mean plasma clearance is approximately 101 L/hr, with a mean volume of distribution of about 1640 L, meaning that duloxetine is distributed throughout the body.
The agent is more than 90% protein bound; thus, giving duloxetine concomitantly with another highly protein-bound agent could increase the side-effect risk of either drug.
Food does not alter duloxetine’s absorption but delays maximum concentration by about 4 hours Duloxetine may be taken before or after meals, though taking it after meals could reduce the risk of nausea—a common early side effect.
Duloxetine is metabolized by the 2D6 and 1A2 isoenzymes of the cytochrome P-450 system. It inhibits the CYP 2D6 isoenzyme but to a lesser extent than fluoxetine does.6 Co-administering duloxetine with a CYP 2D6 substrate or inhibitor or a CYP 1A2 inhibitor Table 27 could elevate plasma levels of duloxetine or the other agent, possibly increasing adverse effects.
EFFICACY
In an 8-week, placebo-controlled trial, Goldstein et al8 compared fluoxetine, 20 mg/d, and duloxetine, 40 mg/d titrated to 120 mg/d over 3 weeks, in 173 patients with major depressive disorder. Participants’ scores at baseline were 15 on the 17-item Hamilton Rating Scale for Depression (HAM-D-17) and 4 on the Clinical Global Improvement-Severity scale. Estimated probability of remission was 56% with duloxetine, 30% with fluoxetine, and 32% with placebo, with remission defined as achieving a HAM-D-17 score 7.
In two prospective, double-blind, placebocontrolled trials of 512 patients with major depression,9,10 duloxetine, 60 mg/d, reduced body, back, and shoulder pain based on Visual Analog Scale (VAS) scores; pre- and posttreatment VAS scores were not listed in the published studies. Estimated probability of remission in these two studies was 44% and 43% among patients taking duloxetine vs 16% and 28% in the placebo groups. Remission again was defined as HAM-D-17 score 7.
TOLERABILITY
In the two double-blind studies just mentioned,9,10 Detke et al reported adverse event-related drop out rates of 12.5% and 13.8% for duloxetine, 60 mg/d, vs 4.3% and 2.3% for placebo. Nausea, insomnia, headaches, somnolence, dry mouth, and sweating were most frequently reported.
Dizziness. Mild dizziness was reported in 11.3% of patients who abruptly stopped duloxetine after 9 weeks.9
Headaches. In one comparator trial,8 fewer headaches (20%) were reported among patients taking duloxetine, 40 to 120 mg/d, vs those taking fluoxetine, 20 mg/d (33.3%), or placebo (31.4%).
Hypertension. Detke et al10 found no statistical separation in systolic and diastolic blood pressures between the duloxetine (60 mg/d) and placebo groups. Likewise, Goldstein et al8 found a similar incidence of hypertension among patients taking duloxetine, 40 to 120 mg/d, or placebo. In clinical trials,11 duloxetine increased blood pressure by a mean of 2.0 mm Hg (systolic) and 0.5 mm Hg (diastolic).
As with several other noradrenergic medications, FDA recommends that clinicians check blood pressures before starting duloxetine therapy and periodically thereafter.
Duloxetine has not been studied in persons with poorly controlled hypertension.
Nausea. Mild to moderate nausea was the most common adverse event in one study;9 the effect dissipated after a median of 7 days. One patient reported severe nausea, and 1 patient out of 123 stopped the medication because of nausea.
Sexual dysfunction. Using the Arizona Sexual Experiences Scale (ASEX), Goldstein et al8 prospectively assessed sexual function in 70 men and women taking duloxetine or placebo. No statistical difference was seen between the two groups from baseline to endpoint.
In another study,12 duloxetine showed worsening only in ASEX item 4 (“How easily can you reach an orgasm?”), indicating some adverse sexual effects in men. No such differences were found in women. Duloxetine’s effect on sexual function needs to be studied further.
Duloxetine is an FDA Use-in-Pregnancy category C medication, meaning that risk to the fetus has not been ruled out. The agent is contraindicated in patients taking monoamine oxidase inhibitors and in those with narrow-angle glaucoma. FDA recommends not using duloxetine in patients with hepatic insufficiency, endstage renal disease, and substantial alcohol use.
DOSING STRATEGIES
Duloxetine, 40 to 120 mg/d, appears to be safe and effective for most adults.8-10 FDA recommends starting at 40 mg/d (20 mg bid) to 60 mg/d (once-daily or 30 mg bid) with no regard to meals. Dosages >60 mg/d have not shown additional benefit. Age and tolerability should drive initial dosing and titration. Side-effect incidence has not been directly compared at 60, 90, and 120 mg/d.
CLINICAL IMPLICATIONS
In clinical trials, duloxetine has shown a high estimated probability of remission in major depression and has shown efficacy against depression’s physical and emotional symptoms. Based on efficacy and safety data, duloxetine appears to be a first-line treatment option for major depression.
Related resources
- Gray GE. Concise guide to evidence-based psychiatry. Washington, DC: American Psychiatric Publishing, 2004.
- Schatzberg AF, Nemeroff CB (eds). Textbook of psychopharmacology (3rd ed). Washington, DC: American Psychiatric Publishing, 2004.
Drug brand names
- Amitriptyline • Elavil
- Cimetidine • Tagamet
- Ciprofloxacin • Cipro
- Desipramine • Norpramin
- Duloxetine • Cymbalta
- Enoxacin • Penetrex
- Fluoxetine • Prozac
- Flecainide • Tambocor
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Metoprolol • Toprol
- Nortriptyline • Pamelor
- Paroxetine • Paxil
- Propafenone • Rythmol
- Propranolol • Inderal
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Timolol • Blocadren, others
- Venlafaxine • Effexor
Disclosure
Dr. Rakesh Jain receives research grants from Eli Lilly and Co., Forest Pharmaceuticals, GlaxoSmithKline, Merck and Co., Organon, Pfizer Inc., and Sepracor. He is a consultant to and speaker for Eli Lilly and Co., and is a speaker for GlaxoSmithKline, Pfizer Inc., and Wyeth Pharmaceuticals.
Dr. Shailesh Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of compefting products.
1. Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or serotonin reuptake inhibitors. Br J Psychiatry 2001;178:234-41.
2. Fava M. The role of the serotonergic and noradrenergic neurotransmitter systems in the treatment of psychological and physical symptoms of depression. J Clin Psychiatry 2003;64(suppl 13):26-9.
3. Tran PV, Bymaster FP, McNamara RK, Potter WZ. Dual monoamine modulation for improved treatment of major depressive disorder. J Clin Psychopharmacol 2003;23:78-86.
4. Ansari A. The efficacy of newer antidepressants in the treatment of chronic pain: a review of current literature. Harv Rev Psychiatry 2000;7:257-77.
5. Willis WD, Westlund KN. Neuroanatomy of the pain system and the pathways that modulate pain. J Clin Neurophysiol 1997;14:2-31.
6. Skinner MH, Kuan HY, Pan A, et al. Duloxetine is both an inhibitor and a substrate of cytochrome P4502D6 in healthy volunteers. Clin Pharmacol Ther 2003;73:170-7.
7. Sadock BJ, Sadock VA. Kaplan and Sadock's pocket handbook of psychotropic drug treatment (3rd ed). Baltimore, MD: Lippincott Williams and Wilkins, 2001.
8. Goldstein DJ, Mallinckrodt C, Lu Y, Demitrack M. Duloxetine in the treatment of major depression disorder: a double-blind clinical trial. J Clin Psychiatry 2002;63:225-31.
9. Detke MJ, Lu Y, Goldstein DJ, et al. Duloxetine 60 mg once-daily for major depressive disorder: a randomized double-blind placebocontrolled trial. J Clin Psychiatry 2002;63:308-15.
10. Detke MJ, Lu Y, Goldstein DJ, et al. Duloxetine 60 mg once daily dosing versus placebo in the treatment of major depression. J Psychiatr Res 2002;36:383-90.
11. Cymbalta prescribing information Eli Lilly and Co 2004.
12. Goldstein DJ, Lu Y, Detke MJ, et al. Duloxetine in the treatment of depression: a double-blind placebo-controlled comparison with paroxetine. J Clin Psychopharmacol 2004;24:389-99.
1. Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or serotonin reuptake inhibitors. Br J Psychiatry 2001;178:234-41.
2. Fava M. The role of the serotonergic and noradrenergic neurotransmitter systems in the treatment of psychological and physical symptoms of depression. J Clin Psychiatry 2003;64(suppl 13):26-9.
3. Tran PV, Bymaster FP, McNamara RK, Potter WZ. Dual monoamine modulation for improved treatment of major depressive disorder. J Clin Psychopharmacol 2003;23:78-86.
4. Ansari A. The efficacy of newer antidepressants in the treatment of chronic pain: a review of current literature. Harv Rev Psychiatry 2000;7:257-77.
5. Willis WD, Westlund KN. Neuroanatomy of the pain system and the pathways that modulate pain. J Clin Neurophysiol 1997;14:2-31.
6. Skinner MH, Kuan HY, Pan A, et al. Duloxetine is both an inhibitor and a substrate of cytochrome P4502D6 in healthy volunteers. Clin Pharmacol Ther 2003;73:170-7.
7. Sadock BJ, Sadock VA. Kaplan and Sadock's pocket handbook of psychotropic drug treatment (3rd ed). Baltimore, MD: Lippincott Williams and Wilkins, 2001.
8. Goldstein DJ, Mallinckrodt C, Lu Y, Demitrack M. Duloxetine in the treatment of major depression disorder: a double-blind clinical trial. J Clin Psychiatry 2002;63:225-31.
9. Detke MJ, Lu Y, Goldstein DJ, et al. Duloxetine 60 mg once-daily for major depressive disorder: a randomized double-blind placebocontrolled trial. J Clin Psychiatry 2002;63:308-15.
10. Detke MJ, Lu Y, Goldstein DJ, et al. Duloxetine 60 mg once daily dosing versus placebo in the treatment of major depression. J Psychiatr Res 2002;36:383-90.
11. Cymbalta prescribing information Eli Lilly and Co 2004.
12. Goldstein DJ, Lu Y, Detke MJ, et al. Duloxetine in the treatment of depression: a double-blind placebo-controlled comparison with paroxetine. J Clin Psychopharmacol 2004;24:389-99.
Intramuscular olanzapine: Treating acute agitation in psychosis and bipolar mania
Oral atypical antipsychotics are given to treat a variety of psychiatric illnesses. Intramuscular (IM) preparations of atypicals are increasingly becoming available for emergency use, such as treating acute agitation.
The FDA has approved IM olanzapine for treating acute agitation associated with schizophrenia and bipolar type I mania.
How it works
As with the agent’s oral formulations (tablets, capsules, wafers), IM olanzapine is primarily an antagonist at serotonergic (5-HT2A) and dopaminergic (D2) receptors. Olanzapine is about twice as active at 5-HT2A compared with D2 receptors, which may underlie the agent’s efficacy as an antipsychotic and mood stabilizer without significant extrapyramidal effects.
Olanzapine also shows primarily antagonistic binding affinity at the 5-HT2B/2C, D1/D3/D4/D5, muscarinic, histamine H1 and alpha1-adrenergic receptors.1 This binding profile is comparable to that of clozapine and predicts a similar clinical response.
Pharmacokinetics
On most pharmacokinetic measures, IM olanzapine is nearly identical to its oral formulations, allowing easy comparison when switching to oral dosing as the patient improves.2
Plasma clearance (linear pharmacokinetics), half-life (approximately 30 hours), and volume of distribution are similar for IM and oral olanzapine. Maximum plasma concentrations after one, two, or three 10-mg injections given over 24 hours were similar to steady-state concentrations after daily administration of oral olanzapine, 20 mg.
The one key difference between IM and oral olanzapine is rate of absorption, which influences onset of action. IM olanzapine generally reaches maximum concentration in 15 to 45 minutes, compared with 4 hours after an oral dose. This rapid peak absorption could prove valuable in the first hour of a psychiatric emergency.
Efficacy
Three double-blind, randomized, placebo and active comparator-controlled studies demonstrated IM olanzapine’s safety and efficacy for treating acute agitation in patients with schizophrenia and bipolar type I mania. A fourth study gauged its efficacy in treating acute agitation in dementia.
Schizophrenia. In a study of 285 patients,3 IM olanzapine, 10 mg, was significantly more effective in reducing agitation than IM haloperidol, 7.5 mg, and IM placebo 15, 30, and 45 minutes after injection. Agitation was measured with the Positive and Negative Symptom Scale-Excited Component (PANSS-EC), Agitated Behavior Scale, and Agitation-Calmness Evaluation scale. Olanzapine and haloperidol were similar in efficacy 1 and 2 hours after injection, and both were more effective than placebo.
In another study,4 270 acutely agitated inpatients with schizophrenia received 1 to 3 IM injections of olanzapine (2.5, 5, 7.5, or 10 mg), haloperidol (7.5 mg), or placebo. The higher the olanzapine dose, the greater the PANSS-EC score reduction 2 hours after the first injection. Olanzapine was more effective than haloperidol on some agitation measures at 7.5 and 10 mg, and olanzapine was significantly more effective than haloperidol 24 hours post-injection, based on Agitated Behavior Scale scores.4 Both agents were similarly effective 2 hours after injection.
Bipolar type I mania. Agitated patients (N = 201) received 1 to 3 IM injections of olanzapine (10 mg for the first two injections, 5 mg for the third), lorazepam (2 mg first two, 1 mg third), or placebo.
Two hours after the first injection, agitation was more greatly reduced within the olanzapine group than in the lorazepam or placebo groups based on PANSS-EC, Agitated Behavior Scale, and Agitation-Calmness Evaluation Scale scores. At 24 hours, olanzapine was more effective than placebo but similar in efficacy to lorazepam.5
Table
IM olanzapine: Fast facts
| Drug brand name: Zyprexa IntraMuscular |
| Class Atypical antipsychotic |
| FDA-approved indication: Acute agitation associated with bipolar type I mania and schizophrenia |
| Approval date: March 29, 2004 |
| Manufacturer: Eli Lilly and Co. |
| Dosing form: 10 mg |
| Dosing recommendations: 10 mg for adults with schizophrenia and bipolar type I mania (5 mg ages 65 and older); 2.5 mg for patients who are debilitated, predisposed to hypotensive reactions, or sensitive to olanzapine. Consider 5- or 7.5-mg doses if clinical factors warrant, such as reduced clearance/slower metabolism in older, nonsmoking women. |
Dementia. A total of 272 patients with Alzheimer’s dementia, mixed dementia, or both received IM olanzapine (2.5 mg or 5 mg), IM lorazepam (1 mg), or IM placebo. The 5-mg olanzapine dose significantly reduced agitation 30 minutes post-injection, whereas lorazepam separated from placebo 60 minutes post-injection based on PANSS-EC scores. At 24 hours, both olanzapine doses were more effective than lorazepam or placebo.6
Tolerability
No clinically significant side effects have been reported with IM olanzapine. Incidence of extrapyramidal symptoms and QTc interval changes has been similar to that reported with placebo. Most studies have reported little change in vital signs, although a 7-bpm increase in heart rate and 5- to 7-mm Hg decrease in systolic blood pressure have been noted (Eli Lilly and Co., data on file).
Differences in treatment-emergent somnolence rates among patients receiving IM olanzapine (4% to 13%) and placebo (3% to 6%) were not statistically significant. Analyses of patients without treatment-emergent somnolence suggest that IM olanzapine retains a specific calming effect (as opposed to nonspecific sedation).7
Clinical implications
IM olanzapine offers psychiatrists a fast-acting option for treating agitation in patients with schizophrenia and bipolar type I mania. Its onset of action, measurable at 15 minutes post-injection, should prove valuable in the critical first hour of emergency psychiatric treatment. IM olanzapine’s efficacy and safety profile compare favorably with those of IM haloperidol and IM lorazepam.
IM olanzapine has shown safety and efficacy in treating agitation associated with dementia. Though the FDA has not approved this indication, the agent will likely be used for this purpose.
The only other fast-acting, injectable atypical antipsychotic—IM ziprasidone—is indicated for treatment of acute agitation in schizophrenia. Head-to-head comparisons between IM olanzapine and IM ziprasidone have not been conducted.
Clinical use and research will determine IM olanzapine’s role in treating patients with severe agitation (such as nonconsenting patients), those who are medically compromised, or patients in drug-induced psychotic states.
Related resources
- Zyprexa Web site. www.zyprexa.com.
- American Association for Emergency Psychiatry. http://www.emergencypsychiatry.org
Drug brand names
- Clozapine • Clozaril
- Haloperidol • Haldol
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Ziprasidone • Geodon
Disclosure
Dr. Battaglia is a consultant to and speaker for Eli Lilly and Co.
1. Bymaster FP, Calligaro DO, Falcone JF, et al. Radioreceptor binding profile of the atypical antipsychotic olanzapine. Neuropsychopharmacology 1996;14:87-96.
2. FDA Psychopharmacological Drugs Advisory Committee. Briefing document for Zyprexa (intramuscular olanzapine), February 13, 2001.
3. Wright P, Birkett M, David SR, et al. Double-blind, placebo-controlled comparison of intramuscular olanzapine and intramuscular haloperidol in the treatment of acute agitation in schizophrenia. Am J Psychiatry 2001;158:1149-51.
4. Breier A, Meehan K, Birkett M, et al. A double-blind, placebo-controlled dose-response comparison of intramuscular olanzapine and haloperidol in the treatment of acute agitation in schizophrenia. Arch Gen Psychiatry 2002;59:441-8.
5. Meehan K, Zhang F, David S, et al. A double-blind, randomized comparison of the efficacy and safety of intramuscular injections of olanzapine, lorazepam, or placebo in treating patients diagnosed with bipolar mania. J Clin Psychopharmacol 2001;21:389-97.
6. Meehan KM, Wang J, David S, et al. Comparison of rapidly acting intramuscular olanzapine, lorazepam, and placebo: A double blind, randomized study in acutely agitated patients with dementia. Neuropsychopharmacology 2002;26:494-504.
7. Battaglia J, Lindborg S, Alaka K, et al. To sleep or not to sleep? Calming versus sedative effects of intramuscular olanzapine in agitated patients. Am J Emerg Med 2003;21:192-8.
Oral atypical antipsychotics are given to treat a variety of psychiatric illnesses. Intramuscular (IM) preparations of atypicals are increasingly becoming available for emergency use, such as treating acute agitation.
The FDA has approved IM olanzapine for treating acute agitation associated with schizophrenia and bipolar type I mania.
How it works
As with the agent’s oral formulations (tablets, capsules, wafers), IM olanzapine is primarily an antagonist at serotonergic (5-HT2A) and dopaminergic (D2) receptors. Olanzapine is about twice as active at 5-HT2A compared with D2 receptors, which may underlie the agent’s efficacy as an antipsychotic and mood stabilizer without significant extrapyramidal effects.
Olanzapine also shows primarily antagonistic binding affinity at the 5-HT2B/2C, D1/D3/D4/D5, muscarinic, histamine H1 and alpha1-adrenergic receptors.1 This binding profile is comparable to that of clozapine and predicts a similar clinical response.
Pharmacokinetics
On most pharmacokinetic measures, IM olanzapine is nearly identical to its oral formulations, allowing easy comparison when switching to oral dosing as the patient improves.2
Plasma clearance (linear pharmacokinetics), half-life (approximately 30 hours), and volume of distribution are similar for IM and oral olanzapine. Maximum plasma concentrations after one, two, or three 10-mg injections given over 24 hours were similar to steady-state concentrations after daily administration of oral olanzapine, 20 mg.
The one key difference between IM and oral olanzapine is rate of absorption, which influences onset of action. IM olanzapine generally reaches maximum concentration in 15 to 45 minutes, compared with 4 hours after an oral dose. This rapid peak absorption could prove valuable in the first hour of a psychiatric emergency.
Efficacy
Three double-blind, randomized, placebo and active comparator-controlled studies demonstrated IM olanzapine’s safety and efficacy for treating acute agitation in patients with schizophrenia and bipolar type I mania. A fourth study gauged its efficacy in treating acute agitation in dementia.
Schizophrenia. In a study of 285 patients,3 IM olanzapine, 10 mg, was significantly more effective in reducing agitation than IM haloperidol, 7.5 mg, and IM placebo 15, 30, and 45 minutes after injection. Agitation was measured with the Positive and Negative Symptom Scale-Excited Component (PANSS-EC), Agitated Behavior Scale, and Agitation-Calmness Evaluation scale. Olanzapine and haloperidol were similar in efficacy 1 and 2 hours after injection, and both were more effective than placebo.
In another study,4 270 acutely agitated inpatients with schizophrenia received 1 to 3 IM injections of olanzapine (2.5, 5, 7.5, or 10 mg), haloperidol (7.5 mg), or placebo. The higher the olanzapine dose, the greater the PANSS-EC score reduction 2 hours after the first injection. Olanzapine was more effective than haloperidol on some agitation measures at 7.5 and 10 mg, and olanzapine was significantly more effective than haloperidol 24 hours post-injection, based on Agitated Behavior Scale scores.4 Both agents were similarly effective 2 hours after injection.
Bipolar type I mania. Agitated patients (N = 201) received 1 to 3 IM injections of olanzapine (10 mg for the first two injections, 5 mg for the third), lorazepam (2 mg first two, 1 mg third), or placebo.
Two hours after the first injection, agitation was more greatly reduced within the olanzapine group than in the lorazepam or placebo groups based on PANSS-EC, Agitated Behavior Scale, and Agitation-Calmness Evaluation Scale scores. At 24 hours, olanzapine was more effective than placebo but similar in efficacy to lorazepam.5
Table
IM olanzapine: Fast facts
| Drug brand name: Zyprexa IntraMuscular |
| Class Atypical antipsychotic |
| FDA-approved indication: Acute agitation associated with bipolar type I mania and schizophrenia |
| Approval date: March 29, 2004 |
| Manufacturer: Eli Lilly and Co. |
| Dosing form: 10 mg |
| Dosing recommendations: 10 mg for adults with schizophrenia and bipolar type I mania (5 mg ages 65 and older); 2.5 mg for patients who are debilitated, predisposed to hypotensive reactions, or sensitive to olanzapine. Consider 5- or 7.5-mg doses if clinical factors warrant, such as reduced clearance/slower metabolism in older, nonsmoking women. |
Dementia. A total of 272 patients with Alzheimer’s dementia, mixed dementia, or both received IM olanzapine (2.5 mg or 5 mg), IM lorazepam (1 mg), or IM placebo. The 5-mg olanzapine dose significantly reduced agitation 30 minutes post-injection, whereas lorazepam separated from placebo 60 minutes post-injection based on PANSS-EC scores. At 24 hours, both olanzapine doses were more effective than lorazepam or placebo.6
Tolerability
No clinically significant side effects have been reported with IM olanzapine. Incidence of extrapyramidal symptoms and QTc interval changes has been similar to that reported with placebo. Most studies have reported little change in vital signs, although a 7-bpm increase in heart rate and 5- to 7-mm Hg decrease in systolic blood pressure have been noted (Eli Lilly and Co., data on file).
Differences in treatment-emergent somnolence rates among patients receiving IM olanzapine (4% to 13%) and placebo (3% to 6%) were not statistically significant. Analyses of patients without treatment-emergent somnolence suggest that IM olanzapine retains a specific calming effect (as opposed to nonspecific sedation).7
Clinical implications
IM olanzapine offers psychiatrists a fast-acting option for treating agitation in patients with schizophrenia and bipolar type I mania. Its onset of action, measurable at 15 minutes post-injection, should prove valuable in the critical first hour of emergency psychiatric treatment. IM olanzapine’s efficacy and safety profile compare favorably with those of IM haloperidol and IM lorazepam.
IM olanzapine has shown safety and efficacy in treating agitation associated with dementia. Though the FDA has not approved this indication, the agent will likely be used for this purpose.
The only other fast-acting, injectable atypical antipsychotic—IM ziprasidone—is indicated for treatment of acute agitation in schizophrenia. Head-to-head comparisons between IM olanzapine and IM ziprasidone have not been conducted.
Clinical use and research will determine IM olanzapine’s role in treating patients with severe agitation (such as nonconsenting patients), those who are medically compromised, or patients in drug-induced psychotic states.
Related resources
- Zyprexa Web site. www.zyprexa.com.
- American Association for Emergency Psychiatry. http://www.emergencypsychiatry.org
Drug brand names
- Clozapine • Clozaril
- Haloperidol • Haldol
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Ziprasidone • Geodon
Disclosure
Dr. Battaglia is a consultant to and speaker for Eli Lilly and Co.
Oral atypical antipsychotics are given to treat a variety of psychiatric illnesses. Intramuscular (IM) preparations of atypicals are increasingly becoming available for emergency use, such as treating acute agitation.
The FDA has approved IM olanzapine for treating acute agitation associated with schizophrenia and bipolar type I mania.
How it works
As with the agent’s oral formulations (tablets, capsules, wafers), IM olanzapine is primarily an antagonist at serotonergic (5-HT2A) and dopaminergic (D2) receptors. Olanzapine is about twice as active at 5-HT2A compared with D2 receptors, which may underlie the agent’s efficacy as an antipsychotic and mood stabilizer without significant extrapyramidal effects.
Olanzapine also shows primarily antagonistic binding affinity at the 5-HT2B/2C, D1/D3/D4/D5, muscarinic, histamine H1 and alpha1-adrenergic receptors.1 This binding profile is comparable to that of clozapine and predicts a similar clinical response.
Pharmacokinetics
On most pharmacokinetic measures, IM olanzapine is nearly identical to its oral formulations, allowing easy comparison when switching to oral dosing as the patient improves.2
Plasma clearance (linear pharmacokinetics), half-life (approximately 30 hours), and volume of distribution are similar for IM and oral olanzapine. Maximum plasma concentrations after one, two, or three 10-mg injections given over 24 hours were similar to steady-state concentrations after daily administration of oral olanzapine, 20 mg.
The one key difference between IM and oral olanzapine is rate of absorption, which influences onset of action. IM olanzapine generally reaches maximum concentration in 15 to 45 minutes, compared with 4 hours after an oral dose. This rapid peak absorption could prove valuable in the first hour of a psychiatric emergency.
Efficacy
Three double-blind, randomized, placebo and active comparator-controlled studies demonstrated IM olanzapine’s safety and efficacy for treating acute agitation in patients with schizophrenia and bipolar type I mania. A fourth study gauged its efficacy in treating acute agitation in dementia.
Schizophrenia. In a study of 285 patients,3 IM olanzapine, 10 mg, was significantly more effective in reducing agitation than IM haloperidol, 7.5 mg, and IM placebo 15, 30, and 45 minutes after injection. Agitation was measured with the Positive and Negative Symptom Scale-Excited Component (PANSS-EC), Agitated Behavior Scale, and Agitation-Calmness Evaluation scale. Olanzapine and haloperidol were similar in efficacy 1 and 2 hours after injection, and both were more effective than placebo.
In another study,4 270 acutely agitated inpatients with schizophrenia received 1 to 3 IM injections of olanzapine (2.5, 5, 7.5, or 10 mg), haloperidol (7.5 mg), or placebo. The higher the olanzapine dose, the greater the PANSS-EC score reduction 2 hours after the first injection. Olanzapine was more effective than haloperidol on some agitation measures at 7.5 and 10 mg, and olanzapine was significantly more effective than haloperidol 24 hours post-injection, based on Agitated Behavior Scale scores.4 Both agents were similarly effective 2 hours after injection.
Bipolar type I mania. Agitated patients (N = 201) received 1 to 3 IM injections of olanzapine (10 mg for the first two injections, 5 mg for the third), lorazepam (2 mg first two, 1 mg third), or placebo.
Two hours after the first injection, agitation was more greatly reduced within the olanzapine group than in the lorazepam or placebo groups based on PANSS-EC, Agitated Behavior Scale, and Agitation-Calmness Evaluation Scale scores. At 24 hours, olanzapine was more effective than placebo but similar in efficacy to lorazepam.5
Table
IM olanzapine: Fast facts
| Drug brand name: Zyprexa IntraMuscular |
| Class Atypical antipsychotic |
| FDA-approved indication: Acute agitation associated with bipolar type I mania and schizophrenia |
| Approval date: March 29, 2004 |
| Manufacturer: Eli Lilly and Co. |
| Dosing form: 10 mg |
| Dosing recommendations: 10 mg for adults with schizophrenia and bipolar type I mania (5 mg ages 65 and older); 2.5 mg for patients who are debilitated, predisposed to hypotensive reactions, or sensitive to olanzapine. Consider 5- or 7.5-mg doses if clinical factors warrant, such as reduced clearance/slower metabolism in older, nonsmoking women. |
Dementia. A total of 272 patients with Alzheimer’s dementia, mixed dementia, or both received IM olanzapine (2.5 mg or 5 mg), IM lorazepam (1 mg), or IM placebo. The 5-mg olanzapine dose significantly reduced agitation 30 minutes post-injection, whereas lorazepam separated from placebo 60 minutes post-injection based on PANSS-EC scores. At 24 hours, both olanzapine doses were more effective than lorazepam or placebo.6
Tolerability
No clinically significant side effects have been reported with IM olanzapine. Incidence of extrapyramidal symptoms and QTc interval changes has been similar to that reported with placebo. Most studies have reported little change in vital signs, although a 7-bpm increase in heart rate and 5- to 7-mm Hg decrease in systolic blood pressure have been noted (Eli Lilly and Co., data on file).
Differences in treatment-emergent somnolence rates among patients receiving IM olanzapine (4% to 13%) and placebo (3% to 6%) were not statistically significant. Analyses of patients without treatment-emergent somnolence suggest that IM olanzapine retains a specific calming effect (as opposed to nonspecific sedation).7
Clinical implications
IM olanzapine offers psychiatrists a fast-acting option for treating agitation in patients with schizophrenia and bipolar type I mania. Its onset of action, measurable at 15 minutes post-injection, should prove valuable in the critical first hour of emergency psychiatric treatment. IM olanzapine’s efficacy and safety profile compare favorably with those of IM haloperidol and IM lorazepam.
IM olanzapine has shown safety and efficacy in treating agitation associated with dementia. Though the FDA has not approved this indication, the agent will likely be used for this purpose.
The only other fast-acting, injectable atypical antipsychotic—IM ziprasidone—is indicated for treatment of acute agitation in schizophrenia. Head-to-head comparisons between IM olanzapine and IM ziprasidone have not been conducted.
Clinical use and research will determine IM olanzapine’s role in treating patients with severe agitation (such as nonconsenting patients), those who are medically compromised, or patients in drug-induced psychotic states.
Related resources
- Zyprexa Web site. www.zyprexa.com.
- American Association for Emergency Psychiatry. http://www.emergencypsychiatry.org
Drug brand names
- Clozapine • Clozaril
- Haloperidol • Haldol
- Lorazepam • Ativan
- Olanzapine • Zyprexa
- Ziprasidone • Geodon
Disclosure
Dr. Battaglia is a consultant to and speaker for Eli Lilly and Co.
1. Bymaster FP, Calligaro DO, Falcone JF, et al. Radioreceptor binding profile of the atypical antipsychotic olanzapine. Neuropsychopharmacology 1996;14:87-96.
2. FDA Psychopharmacological Drugs Advisory Committee. Briefing document for Zyprexa (intramuscular olanzapine), February 13, 2001.
3. Wright P, Birkett M, David SR, et al. Double-blind, placebo-controlled comparison of intramuscular olanzapine and intramuscular haloperidol in the treatment of acute agitation in schizophrenia. Am J Psychiatry 2001;158:1149-51.
4. Breier A, Meehan K, Birkett M, et al. A double-blind, placebo-controlled dose-response comparison of intramuscular olanzapine and haloperidol in the treatment of acute agitation in schizophrenia. Arch Gen Psychiatry 2002;59:441-8.
5. Meehan K, Zhang F, David S, et al. A double-blind, randomized comparison of the efficacy and safety of intramuscular injections of olanzapine, lorazepam, or placebo in treating patients diagnosed with bipolar mania. J Clin Psychopharmacol 2001;21:389-97.
6. Meehan KM, Wang J, David S, et al. Comparison of rapidly acting intramuscular olanzapine, lorazepam, and placebo: A double blind, randomized study in acutely agitated patients with dementia. Neuropsychopharmacology 2002;26:494-504.
7. Battaglia J, Lindborg S, Alaka K, et al. To sleep or not to sleep? Calming versus sedative effects of intramuscular olanzapine in agitated patients. Am J Emerg Med 2003;21:192-8.
1. Bymaster FP, Calligaro DO, Falcone JF, et al. Radioreceptor binding profile of the atypical antipsychotic olanzapine. Neuropsychopharmacology 1996;14:87-96.
2. FDA Psychopharmacological Drugs Advisory Committee. Briefing document for Zyprexa (intramuscular olanzapine), February 13, 2001.
3. Wright P, Birkett M, David SR, et al. Double-blind, placebo-controlled comparison of intramuscular olanzapine and intramuscular haloperidol in the treatment of acute agitation in schizophrenia. Am J Psychiatry 2001;158:1149-51.
4. Breier A, Meehan K, Birkett M, et al. A double-blind, placebo-controlled dose-response comparison of intramuscular olanzapine and haloperidol in the treatment of acute agitation in schizophrenia. Arch Gen Psychiatry 2002;59:441-8.
5. Meehan K, Zhang F, David S, et al. A double-blind, randomized comparison of the efficacy and safety of intramuscular injections of olanzapine, lorazepam, or placebo in treating patients diagnosed with bipolar mania. J Clin Psychopharmacol 2001;21:389-97.
6. Meehan KM, Wang J, David S, et al. Comparison of rapidly acting intramuscular olanzapine, lorazepam, and placebo: A double blind, randomized study in acutely agitated patients with dementia. Neuropsychopharmacology 2002;26:494-504.
7. Battaglia J, Lindborg S, Alaka K, et al. To sleep or not to sleep? Calming versus sedative effects of intramuscular olanzapine in agitated patients. Am J Emerg Med 2003;21:192-8.
Olanzapine/fluoxetine combination: Evidence for using the first treatment indicated for bipolar depression
Patients with bipolar disorder spend half their lives significantly symptomatic, mainly in the depressive phase.1 Treating bipolar depression poses a clinical challenge, although new research is starting to uncover some answers. Antidepressant drugs are commonly used, but recent data question the effectiveness of this practice.2
An olanzapine-fluoxetine combination (OFC), FDA-approved for treating bipolar type I depression, has demonstrated efficacy in clinical trials.
How it works
Most atypical antipsychotics—including olanzapine—are potent 5-HT2A (serotonin) receptor antagonists. This effect is similar to that of some antidepressants and may mediate some antidepressant and antianxiety effects of these drugs.3
Like most atypicals, olanzapine is also a potent 5-HT2C blocker. While binding to this receptor, serotonin inhibits dopamine release in the nucleus accumbens and frontal cortex.4 Thus, serotonin blockade would increase dopamine release in these areas. One study showed that olanzapine and fluoxetine together increased dopamine and norepinephrine in the frontal cortex of rats, compared with either drug given individually.5 Dopamine is critical to regulating motivation, defined as the ability to exert energy to obtain rewards.6 Olanzapine also interacts with dopaminergic (D1-5), muscarinic (M1-5), alpha1 adrenergic, histamine1, serotonin (5-HT2B,2C,3,6), and glutamate and other receptors.
Pharmacokinetics
Combining olanzapine and fluoxetine in one capsule raises potential kinetic problems. Olanzapine’s mean half-life is 30 hours,7 but fluoxetine’s is 24 to 72 hours and its principal active metabolite, norfluoxetine, has a half-life of 4 to 16 hours.7 Because fluoxetine and norfluoxetine inhibit the cytochrome P (CYP)-450 2D6 enzyme—which is involved in their metabolism—autoinhibition of degradation occurs with chronic dosing, thereby increasing the relative half-life of fluoxetine and norfluoxetine. Therefore, maximum steady-state plasma levels will be achieved with olanzapine and fluoxetine at very different rates, although this has not posed a problem in clinical trials. Still, consider this disparity when evaluating potential side effects or drug-drug interactions.
Table 1
Drugs that may interact with OFC
| Drugs metabolized by CYP 2D6 isoenzymes | Drugs metabolized by CYP 2C isoenzymes |
|---|---|
| Citalopram | Citalopram |
| Codeine | Clomipramine |
| Dextromethorphan | Diazepam |
| Haloperidol | Imipramine |
| Metoprolol | Nonsteroidal |
| Other SSRIs | anti-inflammatory drugs |
| Perphenazine | Omeprazole |
| Propafenone | Phenytoin |
| Propranolol | Proguanil |
| Risperidone | Tolbutamide |
| Thioridazine | Tricyclic antidepressants |
| Trazodone | Warfarin |
| Tricyclic antidepressants (most) | |
| Venlafaxine | |
| Source: reference 8 | |
Both compounds reach maximum concentration in 4 to 6 hours.7 Although food’s effect on OFC’s absorption has not been tested, a clinically important effect is unlikely. Food does not significantly alter absorption kinetics of olanzapine or fluoxetine.7
Avoid giving OFC concomitantly with drugs metabolized by CYP 2D6 and 2C (Table 1), because fluoxetine is a potent inhibitor of these isoenzymes. The resulting altered plasma concentrations could lead to drug-drug interactions.8
Efficacy
In an 8-week, double-blind, multinational trial,9 833 patients with bipolar I disorder in the depressive phase randomly received placebo, olanzapine alone (5 to 20 mg/d), or OFC in several fixed combinations (all shown as olanzapine/fluoxetine): 6/25 mg/d, 6/50 mg/d, or 12/50 mg/d. Dosage titration was allowed.
The researchers found that:
- OFC was significantly more effective than placebo. A mean 18.5-point improvement in Montgomery-Asberg Depression Rating Scale (MADRS) scores was reported in the OFC group, compared with a mean 11.9-point improvement in the placebo group.
- Olanzapine alone produced a mean 15-point MADRS score reduction. Remission criteria were achieved in 24.5%, 32.8%, and 48.8% of patients treated with placebo, olanzapine, and OFC, respectively.
- Both OFC and olanzapine alone produced greater MADRS score reductions than did placebo at every follow-up week. Mania induction rates were low in the olanzapine and OFC treatment groups (5.7% and 6.4%, respectively) as measured with the Young Mania Rating Scale.
Shelton et al3 also compared OFC to olanzapine and fluoxetine alone in treatment-resistant unipolar depression. Thirty-two patients with major depression who responded inadequately to two types of antidepressants were treated with fluoxetine, up to 60 mg/d. After 7 weeks, 28 patients who did not respond to fluoxetine then received fluoxetine alone (mean modal dose: 52 mg/d), olanzapine alone (12.5 mg/d), or OFC (13.5 mg/52 mg/d) for another 8 weeks.
Olanzapine alone produced a transient effect at week 3 with relapse thereafter, possibly because of interactions between olanzapine and falling fluoxetine plasma concentrations over the first 3 weeks. Fluoxetine monotherapy produced minimal results across the 8-week random phase.
The OFC group, however, achieved significant improvement in MADRS scores compared with the placebo group after week one. The effect continued throughout the trial and during a subsequent 8-week open-label phase.3
Recent data suggest continued benefit in treatment- and nontreatment-resistant depressed patients for up to 1 year.10 Two follow-up trials—one using a lead-in with venlafaxine, the second with nortriptyline—produced negative results. In both studies, however, patients achieved a robust effect while continuing the same drug during the double-blind phase, suggesting that initial trials were inadequate.11,12 OFC showed early onset of effect in both studies. Other large-scale attempts at replication are anticipated.
Tolerability
Common side effects of OFC include increased appetite, weight gain, somnolence, fatigue, nausea, diarrhea, and dry mouth—the same effects associated with olanzapine or fluoxetine.
Combining the agents does not lessen the side effects, particularly olanzapine-induced weight gain. Simple, assertive dietary and exercise counseling can counteract olanzapine-induced weight gain.13 Sexual dysfunction was reported infrequently in clinical trials but is possible with exposure to fluoxetine.
Extrapyramidal side effects, including akathisia, appear to be relatively infrequent. Tardive dyskinesia (TD) is unlikely, although cases putatively associated with olanzapine have been reported.5 Many patients with TD have taken conventional antipsychotics, however, so the causal link with olanzapine is obscure. Still, alert patients and families to the possibility of TD and its emerging features.
Table 2
Olanzapine-fluoxetine: Fast facts
| Drug brand name: Symbyax |
| Class: Combined atypical antipsychotic/selective serotonin reuptake inhibitor |
| FDA-approved indication: Bipolar type I depression |
| Approval date: Dec. 24, 2003 |
| Manufacturer: Eli Lilly and Co. |
| Dosing forms: 6/25 mg/d, 12/50 mg/d, 12/25 mg/d, 12/50 mg/d |
| Dosing recommendations: Start at 6/25 mg at bedtime. Titrate according to tolerability and therapeutic benefit. Once the antidepressant effect is achieved, continue dosage indefinitely if no adverse effects occur. Dosages up to 18/75 mg/d have been used in clinical trials. |
Although considered rare, isolated cases of neuroleptic malignant syndrome have been attributed to olanzapine.14 Cycle induction has not been reported in clinical trials, but be mindful of this possibility with long-term treatment.
Clinical implications
Taking olanzapine and fluoxetine as a single capsule could save the patient substantial cost. OFC comes in four dosing forms (Table 2), allowing for some flexibility.
It is unclear whether clinicians will prefer the single combination capsule or prescribe each drug separately to increase flexibility. Starting treatment with olanzapine and fluoxetine individually allows the psychiatrist to change the dosages independently and in smaller increments. Taken as separate agents, however, the two products are more expensive than the combined formula. OFC costs about the same as olanzapine alone. On the other hand, if the clinician begins the compounds individually, converting to the dosages in the combined product probably will not be exactly 1:1.
Tolerability is another major advantage of OFC; the combined agent exhibited a 10% dropout rate because of adverse effects compared with 4.6% for placebo.7 Moreover, some patients will prefer the convenience of using a single capsule instead of two medications.
Related resources
- Tollefson GD, Sanger TM. Anxious-depressive symptoms in schizophrenia: a new treatment target for pharmacotherapy? Schizophr Res 1999;35(suppl):S13-S21.
- Symbyax Web site. www.symbyax.com
Drug brand names
- Citalopram • Celexa
- Clomipramine • Anafranil
- Diazepam • Valium
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Imipramine • Tofranil
- Metoprolol succinate • Toprol
- Nortriptyline • Aventyl
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Phenytoin • Dilantin
- Proguanil • Malarone
- Propafenone • Rythmol
- Propranolol • Inderal
- Risperidone • Risperdal
- Tolbutamide • Orinase
- Venlafaxine • Effexor
- Warfarin • Coumadin
Disclosure
Dr. Shelton receives research grants from Abbott Laboratories, Eli Lilly and Co., GlaxoSmithKline, Janssen Pharmaceutica, Pfizer Inc., and Wyeth Pharmaceuticals; is a consultant to Janssen Pharmaceutica, Pfizer Inc., and Wyeth Pharmaceuticals; and is a speaker for Abbott Laboratories, Eli Lilly and Co., Forest Pharmaceuticals, GlaxoSmithKline, Janssen Pharmaceutica, Pfizer Inc., and Wyeth Pharmaceuticals
1. Judd LL, Akiskal HS, Schettler PJ, et al. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry 2002;59:530-7.
2. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry 2001;158:906-12.
3. Shelton RC, Tollefson GD, Tohen M, et al. A novel augmentation strategy for treating resistant major depression. Am J Psychiatry 2001;158:131-4.
4. Shelton RC. The combination of olanzapine and fluoxetine in mood disorders. Expert Opin Pharmacother 2003;4:1175-83.
5. Zhang W, Perry KW, Wong DT, et al. Synergistic effects of olanzapine and other antipsychotic agents in combination with fluoxetine on norepinephrine and dopamine release in rat prefrontal cortex. Neuropsychopharmacology 2000;23:250-62.
6. Salamone JD, Cousins MS, Snyder BJ. Behavioral functions of nucleus accumbens dopamine: empirical and conceptual problems with the anhedonia hypothesis. Neurosci Biobehav Rev 1997;21:341-59.
7. Symbyax package insert. Eli Lilly and Co., 2003.
8. Nemeroff CB, DeVane CL, Pollock BG. Newer antidepressants and the cytochrome P450 system. Am J Psychiatry 1996;153:311-20.
9. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 2003;60:1079-88.
10. Corya SA, Andersen SW, Detke HC, et al. Long-term antidepressant efficacy and safety of olanzapine/fluoxetine combination: a 76-week open-label study. J Clin Psychiatry 2003;64:1349-56.
11. Dube S. Olanzapine-fluoxetine combination in treatment-resistant depression. Eur Psychiatry 2002;17(suppl 1):98.-
12. Dube S, Corya SA, Andersen SW, et al. Efficacy of olanzapine/fluoxetine combination in treatment resistant depression (presentation). San Juan, PR: American College of Neuropsychopharmacology annual meeting, 2002.
13. Ball MP, Coons VB, Buchanan RW. A program for treating olanzapine-related weight gain. Psychiatr Serv 2001;52:967-9.
14. Kogoj A, Velikonja I. Olanzapine-induced neuroleptic malignant syndrome—a case review. Hum Psychopharmacol 2003;18:301-9.
Patients with bipolar disorder spend half their lives significantly symptomatic, mainly in the depressive phase.1 Treating bipolar depression poses a clinical challenge, although new research is starting to uncover some answers. Antidepressant drugs are commonly used, but recent data question the effectiveness of this practice.2
An olanzapine-fluoxetine combination (OFC), FDA-approved for treating bipolar type I depression, has demonstrated efficacy in clinical trials.
How it works
Most atypical antipsychotics—including olanzapine—are potent 5-HT2A (serotonin) receptor antagonists. This effect is similar to that of some antidepressants and may mediate some antidepressant and antianxiety effects of these drugs.3
Like most atypicals, olanzapine is also a potent 5-HT2C blocker. While binding to this receptor, serotonin inhibits dopamine release in the nucleus accumbens and frontal cortex.4 Thus, serotonin blockade would increase dopamine release in these areas. One study showed that olanzapine and fluoxetine together increased dopamine and norepinephrine in the frontal cortex of rats, compared with either drug given individually.5 Dopamine is critical to regulating motivation, defined as the ability to exert energy to obtain rewards.6 Olanzapine also interacts with dopaminergic (D1-5), muscarinic (M1-5), alpha1 adrenergic, histamine1, serotonin (5-HT2B,2C,3,6), and glutamate and other receptors.
Pharmacokinetics
Combining olanzapine and fluoxetine in one capsule raises potential kinetic problems. Olanzapine’s mean half-life is 30 hours,7 but fluoxetine’s is 24 to 72 hours and its principal active metabolite, norfluoxetine, has a half-life of 4 to 16 hours.7 Because fluoxetine and norfluoxetine inhibit the cytochrome P (CYP)-450 2D6 enzyme—which is involved in their metabolism—autoinhibition of degradation occurs with chronic dosing, thereby increasing the relative half-life of fluoxetine and norfluoxetine. Therefore, maximum steady-state plasma levels will be achieved with olanzapine and fluoxetine at very different rates, although this has not posed a problem in clinical trials. Still, consider this disparity when evaluating potential side effects or drug-drug interactions.
Table 1
Drugs that may interact with OFC
| Drugs metabolized by CYP 2D6 isoenzymes | Drugs metabolized by CYP 2C isoenzymes |
|---|---|
| Citalopram | Citalopram |
| Codeine | Clomipramine |
| Dextromethorphan | Diazepam |
| Haloperidol | Imipramine |
| Metoprolol | Nonsteroidal |
| Other SSRIs | anti-inflammatory drugs |
| Perphenazine | Omeprazole |
| Propafenone | Phenytoin |
| Propranolol | Proguanil |
| Risperidone | Tolbutamide |
| Thioridazine | Tricyclic antidepressants |
| Trazodone | Warfarin |
| Tricyclic antidepressants (most) | |
| Venlafaxine | |
| Source: reference 8 | |
Both compounds reach maximum concentration in 4 to 6 hours.7 Although food’s effect on OFC’s absorption has not been tested, a clinically important effect is unlikely. Food does not significantly alter absorption kinetics of olanzapine or fluoxetine.7
Avoid giving OFC concomitantly with drugs metabolized by CYP 2D6 and 2C (Table 1), because fluoxetine is a potent inhibitor of these isoenzymes. The resulting altered plasma concentrations could lead to drug-drug interactions.8
Efficacy
In an 8-week, double-blind, multinational trial,9 833 patients with bipolar I disorder in the depressive phase randomly received placebo, olanzapine alone (5 to 20 mg/d), or OFC in several fixed combinations (all shown as olanzapine/fluoxetine): 6/25 mg/d, 6/50 mg/d, or 12/50 mg/d. Dosage titration was allowed.
The researchers found that:
- OFC was significantly more effective than placebo. A mean 18.5-point improvement in Montgomery-Asberg Depression Rating Scale (MADRS) scores was reported in the OFC group, compared with a mean 11.9-point improvement in the placebo group.
- Olanzapine alone produced a mean 15-point MADRS score reduction. Remission criteria were achieved in 24.5%, 32.8%, and 48.8% of patients treated with placebo, olanzapine, and OFC, respectively.
- Both OFC and olanzapine alone produced greater MADRS score reductions than did placebo at every follow-up week. Mania induction rates were low in the olanzapine and OFC treatment groups (5.7% and 6.4%, respectively) as measured with the Young Mania Rating Scale.
Shelton et al3 also compared OFC to olanzapine and fluoxetine alone in treatment-resistant unipolar depression. Thirty-two patients with major depression who responded inadequately to two types of antidepressants were treated with fluoxetine, up to 60 mg/d. After 7 weeks, 28 patients who did not respond to fluoxetine then received fluoxetine alone (mean modal dose: 52 mg/d), olanzapine alone (12.5 mg/d), or OFC (13.5 mg/52 mg/d) for another 8 weeks.
Olanzapine alone produced a transient effect at week 3 with relapse thereafter, possibly because of interactions between olanzapine and falling fluoxetine plasma concentrations over the first 3 weeks. Fluoxetine monotherapy produced minimal results across the 8-week random phase.
The OFC group, however, achieved significant improvement in MADRS scores compared with the placebo group after week one. The effect continued throughout the trial and during a subsequent 8-week open-label phase.3
Recent data suggest continued benefit in treatment- and nontreatment-resistant depressed patients for up to 1 year.10 Two follow-up trials—one using a lead-in with venlafaxine, the second with nortriptyline—produced negative results. In both studies, however, patients achieved a robust effect while continuing the same drug during the double-blind phase, suggesting that initial trials were inadequate.11,12 OFC showed early onset of effect in both studies. Other large-scale attempts at replication are anticipated.
Tolerability
Common side effects of OFC include increased appetite, weight gain, somnolence, fatigue, nausea, diarrhea, and dry mouth—the same effects associated with olanzapine or fluoxetine.
Combining the agents does not lessen the side effects, particularly olanzapine-induced weight gain. Simple, assertive dietary and exercise counseling can counteract olanzapine-induced weight gain.13 Sexual dysfunction was reported infrequently in clinical trials but is possible with exposure to fluoxetine.
Extrapyramidal side effects, including akathisia, appear to be relatively infrequent. Tardive dyskinesia (TD) is unlikely, although cases putatively associated with olanzapine have been reported.5 Many patients with TD have taken conventional antipsychotics, however, so the causal link with olanzapine is obscure. Still, alert patients and families to the possibility of TD and its emerging features.
Table 2
Olanzapine-fluoxetine: Fast facts
| Drug brand name: Symbyax |
| Class: Combined atypical antipsychotic/selective serotonin reuptake inhibitor |
| FDA-approved indication: Bipolar type I depression |
| Approval date: Dec. 24, 2003 |
| Manufacturer: Eli Lilly and Co. |
| Dosing forms: 6/25 mg/d, 12/50 mg/d, 12/25 mg/d, 12/50 mg/d |
| Dosing recommendations: Start at 6/25 mg at bedtime. Titrate according to tolerability and therapeutic benefit. Once the antidepressant effect is achieved, continue dosage indefinitely if no adverse effects occur. Dosages up to 18/75 mg/d have been used in clinical trials. |
Although considered rare, isolated cases of neuroleptic malignant syndrome have been attributed to olanzapine.14 Cycle induction has not been reported in clinical trials, but be mindful of this possibility with long-term treatment.
Clinical implications
Taking olanzapine and fluoxetine as a single capsule could save the patient substantial cost. OFC comes in four dosing forms (Table 2), allowing for some flexibility.
It is unclear whether clinicians will prefer the single combination capsule or prescribe each drug separately to increase flexibility. Starting treatment with olanzapine and fluoxetine individually allows the psychiatrist to change the dosages independently and in smaller increments. Taken as separate agents, however, the two products are more expensive than the combined formula. OFC costs about the same as olanzapine alone. On the other hand, if the clinician begins the compounds individually, converting to the dosages in the combined product probably will not be exactly 1:1.
Tolerability is another major advantage of OFC; the combined agent exhibited a 10% dropout rate because of adverse effects compared with 4.6% for placebo.7 Moreover, some patients will prefer the convenience of using a single capsule instead of two medications.
Related resources
- Tollefson GD, Sanger TM. Anxious-depressive symptoms in schizophrenia: a new treatment target for pharmacotherapy? Schizophr Res 1999;35(suppl):S13-S21.
- Symbyax Web site. www.symbyax.com
Drug brand names
- Citalopram • Celexa
- Clomipramine • Anafranil
- Diazepam • Valium
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Imipramine • Tofranil
- Metoprolol succinate • Toprol
- Nortriptyline • Aventyl
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Phenytoin • Dilantin
- Proguanil • Malarone
- Propafenone • Rythmol
- Propranolol • Inderal
- Risperidone • Risperdal
- Tolbutamide • Orinase
- Venlafaxine • Effexor
- Warfarin • Coumadin
Disclosure
Dr. Shelton receives research grants from Abbott Laboratories, Eli Lilly and Co., GlaxoSmithKline, Janssen Pharmaceutica, Pfizer Inc., and Wyeth Pharmaceuticals; is a consultant to Janssen Pharmaceutica, Pfizer Inc., and Wyeth Pharmaceuticals; and is a speaker for Abbott Laboratories, Eli Lilly and Co., Forest Pharmaceuticals, GlaxoSmithKline, Janssen Pharmaceutica, Pfizer Inc., and Wyeth Pharmaceuticals
Patients with bipolar disorder spend half their lives significantly symptomatic, mainly in the depressive phase.1 Treating bipolar depression poses a clinical challenge, although new research is starting to uncover some answers. Antidepressant drugs are commonly used, but recent data question the effectiveness of this practice.2
An olanzapine-fluoxetine combination (OFC), FDA-approved for treating bipolar type I depression, has demonstrated efficacy in clinical trials.
How it works
Most atypical antipsychotics—including olanzapine—are potent 5-HT2A (serotonin) receptor antagonists. This effect is similar to that of some antidepressants and may mediate some antidepressant and antianxiety effects of these drugs.3
Like most atypicals, olanzapine is also a potent 5-HT2C blocker. While binding to this receptor, serotonin inhibits dopamine release in the nucleus accumbens and frontal cortex.4 Thus, serotonin blockade would increase dopamine release in these areas. One study showed that olanzapine and fluoxetine together increased dopamine and norepinephrine in the frontal cortex of rats, compared with either drug given individually.5 Dopamine is critical to regulating motivation, defined as the ability to exert energy to obtain rewards.6 Olanzapine also interacts with dopaminergic (D1-5), muscarinic (M1-5), alpha1 adrenergic, histamine1, serotonin (5-HT2B,2C,3,6), and glutamate and other receptors.
Pharmacokinetics
Combining olanzapine and fluoxetine in one capsule raises potential kinetic problems. Olanzapine’s mean half-life is 30 hours,7 but fluoxetine’s is 24 to 72 hours and its principal active metabolite, norfluoxetine, has a half-life of 4 to 16 hours.7 Because fluoxetine and norfluoxetine inhibit the cytochrome P (CYP)-450 2D6 enzyme—which is involved in their metabolism—autoinhibition of degradation occurs with chronic dosing, thereby increasing the relative half-life of fluoxetine and norfluoxetine. Therefore, maximum steady-state plasma levels will be achieved with olanzapine and fluoxetine at very different rates, although this has not posed a problem in clinical trials. Still, consider this disparity when evaluating potential side effects or drug-drug interactions.
Table 1
Drugs that may interact with OFC
| Drugs metabolized by CYP 2D6 isoenzymes | Drugs metabolized by CYP 2C isoenzymes |
|---|---|
| Citalopram | Citalopram |
| Codeine | Clomipramine |
| Dextromethorphan | Diazepam |
| Haloperidol | Imipramine |
| Metoprolol | Nonsteroidal |
| Other SSRIs | anti-inflammatory drugs |
| Perphenazine | Omeprazole |
| Propafenone | Phenytoin |
| Propranolol | Proguanil |
| Risperidone | Tolbutamide |
| Thioridazine | Tricyclic antidepressants |
| Trazodone | Warfarin |
| Tricyclic antidepressants (most) | |
| Venlafaxine | |
| Source: reference 8 | |
Both compounds reach maximum concentration in 4 to 6 hours.7 Although food’s effect on OFC’s absorption has not been tested, a clinically important effect is unlikely. Food does not significantly alter absorption kinetics of olanzapine or fluoxetine.7
Avoid giving OFC concomitantly with drugs metabolized by CYP 2D6 and 2C (Table 1), because fluoxetine is a potent inhibitor of these isoenzymes. The resulting altered plasma concentrations could lead to drug-drug interactions.8
Efficacy
In an 8-week, double-blind, multinational trial,9 833 patients with bipolar I disorder in the depressive phase randomly received placebo, olanzapine alone (5 to 20 mg/d), or OFC in several fixed combinations (all shown as olanzapine/fluoxetine): 6/25 mg/d, 6/50 mg/d, or 12/50 mg/d. Dosage titration was allowed.
The researchers found that:
- OFC was significantly more effective than placebo. A mean 18.5-point improvement in Montgomery-Asberg Depression Rating Scale (MADRS) scores was reported in the OFC group, compared with a mean 11.9-point improvement in the placebo group.
- Olanzapine alone produced a mean 15-point MADRS score reduction. Remission criteria were achieved in 24.5%, 32.8%, and 48.8% of patients treated with placebo, olanzapine, and OFC, respectively.
- Both OFC and olanzapine alone produced greater MADRS score reductions than did placebo at every follow-up week. Mania induction rates were low in the olanzapine and OFC treatment groups (5.7% and 6.4%, respectively) as measured with the Young Mania Rating Scale.
Shelton et al3 also compared OFC to olanzapine and fluoxetine alone in treatment-resistant unipolar depression. Thirty-two patients with major depression who responded inadequately to two types of antidepressants were treated with fluoxetine, up to 60 mg/d. After 7 weeks, 28 patients who did not respond to fluoxetine then received fluoxetine alone (mean modal dose: 52 mg/d), olanzapine alone (12.5 mg/d), or OFC (13.5 mg/52 mg/d) for another 8 weeks.
Olanzapine alone produced a transient effect at week 3 with relapse thereafter, possibly because of interactions between olanzapine and falling fluoxetine plasma concentrations over the first 3 weeks. Fluoxetine monotherapy produced minimal results across the 8-week random phase.
The OFC group, however, achieved significant improvement in MADRS scores compared with the placebo group after week one. The effect continued throughout the trial and during a subsequent 8-week open-label phase.3
Recent data suggest continued benefit in treatment- and nontreatment-resistant depressed patients for up to 1 year.10 Two follow-up trials—one using a lead-in with venlafaxine, the second with nortriptyline—produced negative results. In both studies, however, patients achieved a robust effect while continuing the same drug during the double-blind phase, suggesting that initial trials were inadequate.11,12 OFC showed early onset of effect in both studies. Other large-scale attempts at replication are anticipated.
Tolerability
Common side effects of OFC include increased appetite, weight gain, somnolence, fatigue, nausea, diarrhea, and dry mouth—the same effects associated with olanzapine or fluoxetine.
Combining the agents does not lessen the side effects, particularly olanzapine-induced weight gain. Simple, assertive dietary and exercise counseling can counteract olanzapine-induced weight gain.13 Sexual dysfunction was reported infrequently in clinical trials but is possible with exposure to fluoxetine.
Extrapyramidal side effects, including akathisia, appear to be relatively infrequent. Tardive dyskinesia (TD) is unlikely, although cases putatively associated with olanzapine have been reported.5 Many patients with TD have taken conventional antipsychotics, however, so the causal link with olanzapine is obscure. Still, alert patients and families to the possibility of TD and its emerging features.
Table 2
Olanzapine-fluoxetine: Fast facts
| Drug brand name: Symbyax |
| Class: Combined atypical antipsychotic/selective serotonin reuptake inhibitor |
| FDA-approved indication: Bipolar type I depression |
| Approval date: Dec. 24, 2003 |
| Manufacturer: Eli Lilly and Co. |
| Dosing forms: 6/25 mg/d, 12/50 mg/d, 12/25 mg/d, 12/50 mg/d |
| Dosing recommendations: Start at 6/25 mg at bedtime. Titrate according to tolerability and therapeutic benefit. Once the antidepressant effect is achieved, continue dosage indefinitely if no adverse effects occur. Dosages up to 18/75 mg/d have been used in clinical trials. |
Although considered rare, isolated cases of neuroleptic malignant syndrome have been attributed to olanzapine.14 Cycle induction has not been reported in clinical trials, but be mindful of this possibility with long-term treatment.
Clinical implications
Taking olanzapine and fluoxetine as a single capsule could save the patient substantial cost. OFC comes in four dosing forms (Table 2), allowing for some flexibility.
It is unclear whether clinicians will prefer the single combination capsule or prescribe each drug separately to increase flexibility. Starting treatment with olanzapine and fluoxetine individually allows the psychiatrist to change the dosages independently and in smaller increments. Taken as separate agents, however, the two products are more expensive than the combined formula. OFC costs about the same as olanzapine alone. On the other hand, if the clinician begins the compounds individually, converting to the dosages in the combined product probably will not be exactly 1:1.
Tolerability is another major advantage of OFC; the combined agent exhibited a 10% dropout rate because of adverse effects compared with 4.6% for placebo.7 Moreover, some patients will prefer the convenience of using a single capsule instead of two medications.
Related resources
- Tollefson GD, Sanger TM. Anxious-depressive symptoms in schizophrenia: a new treatment target for pharmacotherapy? Schizophr Res 1999;35(suppl):S13-S21.
- Symbyax Web site. www.symbyax.com
Drug brand names
- Citalopram • Celexa
- Clomipramine • Anafranil
- Diazepam • Valium
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Imipramine • Tofranil
- Metoprolol succinate • Toprol
- Nortriptyline • Aventyl
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Phenytoin • Dilantin
- Proguanil • Malarone
- Propafenone • Rythmol
- Propranolol • Inderal
- Risperidone • Risperdal
- Tolbutamide • Orinase
- Venlafaxine • Effexor
- Warfarin • Coumadin
Disclosure
Dr. Shelton receives research grants from Abbott Laboratories, Eli Lilly and Co., GlaxoSmithKline, Janssen Pharmaceutica, Pfizer Inc., and Wyeth Pharmaceuticals; is a consultant to Janssen Pharmaceutica, Pfizer Inc., and Wyeth Pharmaceuticals; and is a speaker for Abbott Laboratories, Eli Lilly and Co., Forest Pharmaceuticals, GlaxoSmithKline, Janssen Pharmaceutica, Pfizer Inc., and Wyeth Pharmaceuticals
1. Judd LL, Akiskal HS, Schettler PJ, et al. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry 2002;59:530-7.
2. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry 2001;158:906-12.
3. Shelton RC, Tollefson GD, Tohen M, et al. A novel augmentation strategy for treating resistant major depression. Am J Psychiatry 2001;158:131-4.
4. Shelton RC. The combination of olanzapine and fluoxetine in mood disorders. Expert Opin Pharmacother 2003;4:1175-83.
5. Zhang W, Perry KW, Wong DT, et al. Synergistic effects of olanzapine and other antipsychotic agents in combination with fluoxetine on norepinephrine and dopamine release in rat prefrontal cortex. Neuropsychopharmacology 2000;23:250-62.
6. Salamone JD, Cousins MS, Snyder BJ. Behavioral functions of nucleus accumbens dopamine: empirical and conceptual problems with the anhedonia hypothesis. Neurosci Biobehav Rev 1997;21:341-59.
7. Symbyax package insert. Eli Lilly and Co., 2003.
8. Nemeroff CB, DeVane CL, Pollock BG. Newer antidepressants and the cytochrome P450 system. Am J Psychiatry 1996;153:311-20.
9. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 2003;60:1079-88.
10. Corya SA, Andersen SW, Detke HC, et al. Long-term antidepressant efficacy and safety of olanzapine/fluoxetine combination: a 76-week open-label study. J Clin Psychiatry 2003;64:1349-56.
11. Dube S. Olanzapine-fluoxetine combination in treatment-resistant depression. Eur Psychiatry 2002;17(suppl 1):98.-
12. Dube S, Corya SA, Andersen SW, et al. Efficacy of olanzapine/fluoxetine combination in treatment resistant depression (presentation). San Juan, PR: American College of Neuropsychopharmacology annual meeting, 2002.
13. Ball MP, Coons VB, Buchanan RW. A program for treating olanzapine-related weight gain. Psychiatr Serv 2001;52:967-9.
14. Kogoj A, Velikonja I. Olanzapine-induced neuroleptic malignant syndrome—a case review. Hum Psychopharmacol 2003;18:301-9.
1. Judd LL, Akiskal HS, Schettler PJ, et al. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry 2002;59:530-7.
2. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry 2001;158:906-12.
3. Shelton RC, Tollefson GD, Tohen M, et al. A novel augmentation strategy for treating resistant major depression. Am J Psychiatry 2001;158:131-4.
4. Shelton RC. The combination of olanzapine and fluoxetine in mood disorders. Expert Opin Pharmacother 2003;4:1175-83.
5. Zhang W, Perry KW, Wong DT, et al. Synergistic effects of olanzapine and other antipsychotic agents in combination with fluoxetine on norepinephrine and dopamine release in rat prefrontal cortex. Neuropsychopharmacology 2000;23:250-62.
6. Salamone JD, Cousins MS, Snyder BJ. Behavioral functions of nucleus accumbens dopamine: empirical and conceptual problems with the anhedonia hypothesis. Neurosci Biobehav Rev 1997;21:341-59.
7. Symbyax package insert. Eli Lilly and Co., 2003.
8. Nemeroff CB, DeVane CL, Pollock BG. Newer antidepressants and the cytochrome P450 system. Am J Psychiatry 1996;153:311-20.
9. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 2003;60:1079-88.
10. Corya SA, Andersen SW, Detke HC, et al. Long-term antidepressant efficacy and safety of olanzapine/fluoxetine combination: a 76-week open-label study. J Clin Psychiatry 2003;64:1349-56.
11. Dube S. Olanzapine-fluoxetine combination in treatment-resistant depression. Eur Psychiatry 2002;17(suppl 1):98.-
12. Dube S, Corya SA, Andersen SW, et al. Efficacy of olanzapine/fluoxetine combination in treatment resistant depression (presentation). San Juan, PR: American College of Neuropsychopharmacology annual meeting, 2002.
13. Ball MP, Coons VB, Buchanan RW. A program for treating olanzapine-related weight gain. Psychiatr Serv 2001;52:967-9.
14. Kogoj A, Velikonja I. Olanzapine-induced neuroleptic malignant syndrome—a case review. Hum Psychopharmacol 2003;18:301-9.