User login
Transdermal rivastigmine for dementia
The rivastigmine patch is the first transdermal treatment for symptoms of mild to moderate Alzheimer’s disease (AD) and mild to moderate Parkinson’s disease dementia (Table). Rivastigmine, a cholinesterase inhibitor, is the only therapy approved for both indications.
Table
Rivastigmine transdermal patch: Fast facts
| Brand name: Exelon Patch |
| Class: Cholinesterase inhibitor |
| Indication: Symptomatic treatment of mild to moderate Alzheimer’s-type dementia and mild to moderate dementia associated with Parkinson’s disease |
| Manufacturer: Novartis Pharmaceuticals, Inc. |
| Dosing forms: 4.6 and 9.5 mg/24 hours transdermal patches (5 cm2 and 10 cm2, respectively) |
| Recommended dosage: Start with 4.6 mg/24 hours patch for ≥4 weeks, followed by a one-step increase to the target dose 9.5 mg/24 hours patch* |
| *Unless the patient is taking oral rivastigmine (see ‘Transitioning to rivastigmine patch,’) |
Clinical implications
The rivastigmine patch offers continuous drug delivery through the skin into the bloodstream over 24 hours.1 This may reduce the incidence of side effects compared with oral rivastigmine,2 making optimal therapeutic doses easier to attain.3 The target dose 9.5 mg/24 hours patch provides efficacy similar to the highest recommended rivastigmine capsule dose (6 mg bid for a total of 12 mg/d).2
How it works
The rivastigmine patch uses matrix technology, which enables delivery of a large amount of drug from a small surface area.4 The patch is available in 2 dosage forms:
- a 5-cm2 size containing 9 mg of rivastigmine that delivers 4.6 mg/24 hours
- a 10-cm2 size containing 18 mg of rivastigmine that delivers 9.5 mg/24 hours.
Each patch consists of 4 layers: the backing layer, an acrylic drug matrix, a silicone adhesive matrix, and an overlapping release liner that is removed and discarded before the patch is applied.1
Cholinesterase inhibitors are believed to exert their effects by increasing available levels of the neurotransmitter acetylcholine in the brain. Two studies have demonstrated that cognitive improvements associated with rivastigmine treatment correlate significantly with cholinesterase inhibition.5,6 In 1 study, rivastigmine’s inhibitory effects on cholinesterase were sustained for 12 months.6
Pharmacokinetics
Rivastigmine is metabolized by its target cholinesterase enzymes to the decarbamylated metabolite NAP 226-90, which has minimal acetylcholinesterase inhibition and is excreted through the urine.1 As a result of its low accumulation potential and cytochrome P 450-independent metabolism, rivastigmine has low potential for pharmacokinetic drug–drug interactions. This lack of interaction has been confirmed for many drugs commonly taken by elderly patients, such as digoxin, nonsteroidal anti-inflammatory drugs, and estrogens.7
Rivastigmine has a half-life of 1 to 2 hours, so it is rapidly cleared.8 In the event of a serious reaction, significant clearance of rivastigmine from the body would occur within 3 hours of patch removal.
Centrally mediated cholinergic gastrointestinal (GI) side effects associated with oral rivastigmine are related to high maximum plasma concentrations (Cmax) and short time interval to Cmax (Tmax).9 In an open-label, parallel-group study of 51 AD patients that compared rivastigmine patches with rivastigmine capsules, transdermal administration was associated with slower increases to lower peak plasma concentrations (prolonged Tmax and reduced Cmax), and less fluctuation in plasma concentration.1 Despite these effects, the rivastigmine 9.5 mg/24 hours patch provided drug exposure comparable to the highest dose of capsules (6 mg bid for a total of 12 mg/d), with improved GI tolerability.3
Efficacy
Rivastigmine patch efficacy was evaluated in a single, 24-week, international, randomized, double-blind trial of 1,195 patients with AD.2 The study group represented typical patients with mild to moderate AD—age 50 to 85 years with Mini-Mental State Examination scores of 10 to 20 at baseline. Patients were randomly assigned to receive:
- 17.4 mg/24 hours rivastigmine patch (20-cm2 patch; n=303)
- 9.5 mg/24 hours rivastigmine patch (10-cm2 patch; n=293)
- 6 mg bid rivastigmine capsules (n=297)
- or placebo (n=302).
Data for the 17.4 mg/24 hours patch are not discussed here because this dose exceeds the FDA-approved maximum dosage (9.5 mg/24 hours) and is not available.
Patients in the 9.5 mg/24 hours patch group received a 4.6 mg/24 hours patch (5 cm2) for weeks 1 through 4, and then the 9.5 mg/24 hours patch for the remainder of the study. Patients in the capsule group started on 3 mg/d (1.5 mg bid) and were titrated every 4 weeks in steps of 3 mg/d to a maximum of 12 mg/d administered as 6 mg bid.
Primary outcomes were measured as mean change in score from baseline to endpoint on the Alzheimer’s Disease Assessment Scale–Cognitive Subscale (ADAS-Cog) and Alzheimer’s Disease Co-operative Study–Clinical Global Impression of Change (ADCS-CGIC). By study endpoint, the 9.5 mg/24 hours patch and capsules, 12 mg/d, showed comparable efficacy (Figure).2 Compared with those receiving placebo, patients in the 9.5 mg/24 hours patch and capsule groups showed significant improvements in dementia symptoms, including:
- cognition
- global performance
- attention
- activities of daily living.2
Based on my clinical experience, these improvements reflect small but clinically meaningful changes that are noted by patients and caregivers.
Figure
Efficacy of transdermal rivastigmine for Alzheimer’s symptoms
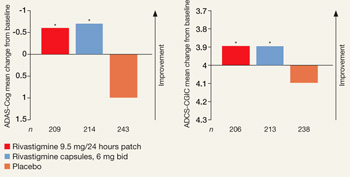
*P<0.05 vs placebo
ADAS-Cog: Alzheimer’s Disease Assessment Scale-Cognitive Subscale; ADCS-CGIC: Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change
Source: Adapted from reference 2
In a 24-week study, transdermal rivastigmine, 9.5 mg/24 hours, and the highest recommended dose of oral rivastigmine (6 mg bid) showed comparable efficacy as measured by mean change in score on scales commonly used in Alzheimer’s disease clinical trials. ADAS-Cog assesses orientation, memory, language, praxis, and visuospatial functions. ADCS-CGIC provides a single global rating of change from baseline based on interviews with the patient and caregiver.
Safety and tolerability
Adverse events associated with rivastigmine are predominantly cholinergic; GI side effects—nausea, vomiting, and diarrhea—are observed most frequently.2 These events occur less frequently with the patch than with capsules. In the efficacy trial, patients in the 9.5 mg/24 hours rivastigmine patch group had one-third as many reports of nausea (7.2% vs 23.1%) and vomiting (6.2% vs 17.0%) compared with the 6 mg bid capsule group.2
Diarrhea was reported by 6% of subjects receiving the 9.5 mg/24 hours patch, 5% of those taking 6-mg capsule bid, and 3% receiving placebo. Fewer subjects in the 9.5 mg/24 hours patch group (3%) experienced decreased weight compared with those in the capsule group (5%). The rate of decreased weight with placebo was 1%.
Dizziness affected 2% of those in the 9.5 mg/24 hours patch and placebo groups; incidence in the capsule group was significantly higher at 8%. Headache was similar with the 9.5 mg/24 hours patch (3%) and placebo (2%), with the capsule significantly higher at 6%.2
The proportion of patients who experienced no, slight, or mild skin irritation ranged from 90% to 98%.2 The most commonly reported moderate or severe skin irritations were erythema (8% rivastigmine patch vs 4% placebo) and pruritus (7% rivastigmine patch vs 3% placebo). Two percent of patients using active patch discontinued the trial because of skin irritation.
Rivastigmine appears not to produce adverse effects on cardiac function as assessed by ECG. In clinical trials of 2,791 patients, pooled 12-lead ECG data comparing oral rivastigmine and placebo groups did not differ significantly in heart rate or PR, QRS, and QTc intervals.10
Dosing
The rivastigmine patch is administered once daily, and the recommended maintenance dose is the 9.5 mg/24 hours patch. Start patients on a 4.6 mg/24 hours patch for at least 4 weeks and then increase to the 9.5 mg/24 hours target dose if the lower dose is well tolerated.
Dosage adjustment of rivastigmine is not necessary in patients with hepatic or renal disease because of minimal liver metabolism and the acetylcholinesterase-mediated hydrolysis of rivastigmine to the inactive decarbamylated metabolite NAP 226-90, which is excreted in the urine.11
Instruct patients or caregivers to apply the patch to clean, dry, hairless skin that is free of cuts, rashes, or irritation on the upper or lower back or upper arm or chest.1 The patch has shown good adhesive properties over 24 hours, remaining attached in a range of situations, including bathing and hot weather.2 In the 9.5 mg/24 hours group of the efficacy study, 96% of patches remained attached or had slight lifting of the edges (1,336 total patch evaluations).
Transitioning to rivastigmine patch
The efficacy study included an open-label extension, during which blinding was maintained. This provided information on patients beginning rivastigmine patch therapy directly from placebo2 or transitioning from rivastigmine capsules to the target dose 9.5 mg/24 hours patch.12 Based on these results, transition patients as follows:
- Patients taking oral rivastigmine, <6 mg/d: Switch to a 4.6 mg/24 hours patch for ≥4 weeks before increasing to a 9.5 mg/24 hours patch.
- Patients taking oral rivastigmine, 6 to 12 mg/d: Switch directly to a 9.5 mg/24 hours patch.
Apply the first patch the day after the last oral dose.
Related resource
- Rivastigmine transdermal system prescribing information. www.pharma.us.novartis.com/product/pi/pdf/exelonpatch.pdf.
Drug brand names
- Digoxin • Lanoxin
- Rivastigmine • Exelon
- Rivastigmine transdermal
- system • Exelon Patch
Disclosure
Dr. Sadowsky is a consultant to and speaker for Forest Pharmaceuticals and Novartis Pharmaceuticals.
Acknowledgment
The author thanks Christina Mackins, PhD, a medical writer for Alpha-Plus Medical Communications Ltd, for her editorial assistance with this article. Funding for her work was provided by Novartis Pharmaceuticals.
1. Lefèvre G, Sedek G, Jhee S, et al. Pharmacokinetics and pharmacodynamics of the novel daily rivastigmine transdermal patch compared with twice-daily capsules in Alzheimer’s disease patients. J Clin Pharmacol 2007;47:471-8.
2. Winblad B, Cummings J, Andreasen N, et al. A six-month, double-blind, randomized, placebo-controlled study of a transdermal patch in Alzheimer’s disease—rivastigmine patch versus capsule. Int J Geriatr Psychiatry 2007;22:456-67.
3. Oertel W, Ross JS, Eggert K, Adler G. Rationale for transdermal drug administration in Alzheimer disease. Neurology 2007;69(suppl 1):S4-S9.
4. Petersen TA. Transdermal drug formulations and process development. Pharmaceut Technol 2003;(suppl):18-21.
5. Giacobini E, Spiegel R, Enz A, et al. Inhibition of acetyl- and butyryl-cholinesterase in the cerebrospinal fluid of patients with Alzheimer’s disease by rivastigmine: correlation with cognitive benefit. J Neural Transm 2002;109:1053-65.
6. Darreh-Shori T, Almkvist O, Guan ZZ, et al. Sustained cholinesterase inhibition in AD patients receiving rivastigmine for 12 months. Neurology 2002;59:563-72.
7. Grossberg GT, Stahelin HB, Messina JC, et al. Lack of adverse pharmacodynamic drug interactions with rivastigmine and twenty-two classes of medications. Int J Geriatr Psychiatry 2000;15(3):242-7.
8. Polinsky RJ. Clinical pharmacology of rivastigmine: a new-generation acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease. Clin Ther 1998;20:634-47.
9. Jann MW, Shirley KL, Small GW. Clinical pharmacokinetics and pharmacodynamics of cholinesterase inhibitors. Clin Pharmacokinet 2002;41:719-39.
10. Morganroth J, Graham S, Hartman R, et al. Electrocardiographic effects of rivastigmine. J Clin Pharmacol 2002;42:558-68.
11. Exelon patch [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2007.
12. Frölich L, Barone P, Förstl H, et al. IDEAL: A 28-week open-label extension of a 24-week double-blind study of the first transdermal patch in Alzheimer’s disease. Poster presented at: 11th Congress of the European Federation of Neurological Societies; August 25-28, 2007; Brussels, Belgium.
Dr. Sadowsky is associate clinical professor of neurology, Nova Southeastern University, Fort Lauderdale, FL, and director, Premier Research Institute, Palm Beach Neurology, West Palm Beach, FL.
The rivastigmine patch is the first transdermal treatment for symptoms of mild to moderate Alzheimer’s disease (AD) and mild to moderate Parkinson’s disease dementia (Table). Rivastigmine, a cholinesterase inhibitor, is the only therapy approved for both indications.
Table
Rivastigmine transdermal patch: Fast facts
| Brand name: Exelon Patch |
| Class: Cholinesterase inhibitor |
| Indication: Symptomatic treatment of mild to moderate Alzheimer’s-type dementia and mild to moderate dementia associated with Parkinson’s disease |
| Manufacturer: Novartis Pharmaceuticals, Inc. |
| Dosing forms: 4.6 and 9.5 mg/24 hours transdermal patches (5 cm2 and 10 cm2, respectively) |
| Recommended dosage: Start with 4.6 mg/24 hours patch for ≥4 weeks, followed by a one-step increase to the target dose 9.5 mg/24 hours patch* |
| *Unless the patient is taking oral rivastigmine (see ‘Transitioning to rivastigmine patch,’) |
Clinical implications
The rivastigmine patch offers continuous drug delivery through the skin into the bloodstream over 24 hours.1 This may reduce the incidence of side effects compared with oral rivastigmine,2 making optimal therapeutic doses easier to attain.3 The target dose 9.5 mg/24 hours patch provides efficacy similar to the highest recommended rivastigmine capsule dose (6 mg bid for a total of 12 mg/d).2
How it works
The rivastigmine patch uses matrix technology, which enables delivery of a large amount of drug from a small surface area.4 The patch is available in 2 dosage forms:
- a 5-cm2 size containing 9 mg of rivastigmine that delivers 4.6 mg/24 hours
- a 10-cm2 size containing 18 mg of rivastigmine that delivers 9.5 mg/24 hours.
Each patch consists of 4 layers: the backing layer, an acrylic drug matrix, a silicone adhesive matrix, and an overlapping release liner that is removed and discarded before the patch is applied.1
Cholinesterase inhibitors are believed to exert their effects by increasing available levels of the neurotransmitter acetylcholine in the brain. Two studies have demonstrated that cognitive improvements associated with rivastigmine treatment correlate significantly with cholinesterase inhibition.5,6 In 1 study, rivastigmine’s inhibitory effects on cholinesterase were sustained for 12 months.6
Pharmacokinetics
Rivastigmine is metabolized by its target cholinesterase enzymes to the decarbamylated metabolite NAP 226-90, which has minimal acetylcholinesterase inhibition and is excreted through the urine.1 As a result of its low accumulation potential and cytochrome P 450-independent metabolism, rivastigmine has low potential for pharmacokinetic drug–drug interactions. This lack of interaction has been confirmed for many drugs commonly taken by elderly patients, such as digoxin, nonsteroidal anti-inflammatory drugs, and estrogens.7
Rivastigmine has a half-life of 1 to 2 hours, so it is rapidly cleared.8 In the event of a serious reaction, significant clearance of rivastigmine from the body would occur within 3 hours of patch removal.
Centrally mediated cholinergic gastrointestinal (GI) side effects associated with oral rivastigmine are related to high maximum plasma concentrations (Cmax) and short time interval to Cmax (Tmax).9 In an open-label, parallel-group study of 51 AD patients that compared rivastigmine patches with rivastigmine capsules, transdermal administration was associated with slower increases to lower peak plasma concentrations (prolonged Tmax and reduced Cmax), and less fluctuation in plasma concentration.1 Despite these effects, the rivastigmine 9.5 mg/24 hours patch provided drug exposure comparable to the highest dose of capsules (6 mg bid for a total of 12 mg/d), with improved GI tolerability.3
Efficacy
Rivastigmine patch efficacy was evaluated in a single, 24-week, international, randomized, double-blind trial of 1,195 patients with AD.2 The study group represented typical patients with mild to moderate AD—age 50 to 85 years with Mini-Mental State Examination scores of 10 to 20 at baseline. Patients were randomly assigned to receive:
- 17.4 mg/24 hours rivastigmine patch (20-cm2 patch; n=303)
- 9.5 mg/24 hours rivastigmine patch (10-cm2 patch; n=293)
- 6 mg bid rivastigmine capsules (n=297)
- or placebo (n=302).
Data for the 17.4 mg/24 hours patch are not discussed here because this dose exceeds the FDA-approved maximum dosage (9.5 mg/24 hours) and is not available.
Patients in the 9.5 mg/24 hours patch group received a 4.6 mg/24 hours patch (5 cm2) for weeks 1 through 4, and then the 9.5 mg/24 hours patch for the remainder of the study. Patients in the capsule group started on 3 mg/d (1.5 mg bid) and were titrated every 4 weeks in steps of 3 mg/d to a maximum of 12 mg/d administered as 6 mg bid.
Primary outcomes were measured as mean change in score from baseline to endpoint on the Alzheimer’s Disease Assessment Scale–Cognitive Subscale (ADAS-Cog) and Alzheimer’s Disease Co-operative Study–Clinical Global Impression of Change (ADCS-CGIC). By study endpoint, the 9.5 mg/24 hours patch and capsules, 12 mg/d, showed comparable efficacy (Figure).2 Compared with those receiving placebo, patients in the 9.5 mg/24 hours patch and capsule groups showed significant improvements in dementia symptoms, including:
- cognition
- global performance
- attention
- activities of daily living.2
Based on my clinical experience, these improvements reflect small but clinically meaningful changes that are noted by patients and caregivers.
Figure
Efficacy of transdermal rivastigmine for Alzheimer’s symptoms
*P<0.05 vs placebo
ADAS-Cog: Alzheimer’s Disease Assessment Scale-Cognitive Subscale; ADCS-CGIC: Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change
Source: Adapted from reference 2
In a 24-week study, transdermal rivastigmine, 9.5 mg/24 hours, and the highest recommended dose of oral rivastigmine (6 mg bid) showed comparable efficacy as measured by mean change in score on scales commonly used in Alzheimer’s disease clinical trials. ADAS-Cog assesses orientation, memory, language, praxis, and visuospatial functions. ADCS-CGIC provides a single global rating of change from baseline based on interviews with the patient and caregiver.
Safety and tolerability
Adverse events associated with rivastigmine are predominantly cholinergic; GI side effects—nausea, vomiting, and diarrhea—are observed most frequently.2 These events occur less frequently with the patch than with capsules. In the efficacy trial, patients in the 9.5 mg/24 hours rivastigmine patch group had one-third as many reports of nausea (7.2% vs 23.1%) and vomiting (6.2% vs 17.0%) compared with the 6 mg bid capsule group.2
Diarrhea was reported by 6% of subjects receiving the 9.5 mg/24 hours patch, 5% of those taking 6-mg capsule bid, and 3% receiving placebo. Fewer subjects in the 9.5 mg/24 hours patch group (3%) experienced decreased weight compared with those in the capsule group (5%). The rate of decreased weight with placebo was 1%.
Dizziness affected 2% of those in the 9.5 mg/24 hours patch and placebo groups; incidence in the capsule group was significantly higher at 8%. Headache was similar with the 9.5 mg/24 hours patch (3%) and placebo (2%), with the capsule significantly higher at 6%.2
The proportion of patients who experienced no, slight, or mild skin irritation ranged from 90% to 98%.2 The most commonly reported moderate or severe skin irritations were erythema (8% rivastigmine patch vs 4% placebo) and pruritus (7% rivastigmine patch vs 3% placebo). Two percent of patients using active patch discontinued the trial because of skin irritation.
Rivastigmine appears not to produce adverse effects on cardiac function as assessed by ECG. In clinical trials of 2,791 patients, pooled 12-lead ECG data comparing oral rivastigmine and placebo groups did not differ significantly in heart rate or PR, QRS, and QTc intervals.10
Dosing
The rivastigmine patch is administered once daily, and the recommended maintenance dose is the 9.5 mg/24 hours patch. Start patients on a 4.6 mg/24 hours patch for at least 4 weeks and then increase to the 9.5 mg/24 hours target dose if the lower dose is well tolerated.
Dosage adjustment of rivastigmine is not necessary in patients with hepatic or renal disease because of minimal liver metabolism and the acetylcholinesterase-mediated hydrolysis of rivastigmine to the inactive decarbamylated metabolite NAP 226-90, which is excreted in the urine.11
Instruct patients or caregivers to apply the patch to clean, dry, hairless skin that is free of cuts, rashes, or irritation on the upper or lower back or upper arm or chest.1 The patch has shown good adhesive properties over 24 hours, remaining attached in a range of situations, including bathing and hot weather.2 In the 9.5 mg/24 hours group of the efficacy study, 96% of patches remained attached or had slight lifting of the edges (1,336 total patch evaluations).
Transitioning to rivastigmine patch
The efficacy study included an open-label extension, during which blinding was maintained. This provided information on patients beginning rivastigmine patch therapy directly from placebo2 or transitioning from rivastigmine capsules to the target dose 9.5 mg/24 hours patch.12 Based on these results, transition patients as follows:
- Patients taking oral rivastigmine, <6 mg/d: Switch to a 4.6 mg/24 hours patch for ≥4 weeks before increasing to a 9.5 mg/24 hours patch.
- Patients taking oral rivastigmine, 6 to 12 mg/d: Switch directly to a 9.5 mg/24 hours patch.
Apply the first patch the day after the last oral dose.
Related resource
- Rivastigmine transdermal system prescribing information. www.pharma.us.novartis.com/product/pi/pdf/exelonpatch.pdf.
Drug brand names
- Digoxin • Lanoxin
- Rivastigmine • Exelon
- Rivastigmine transdermal
- system • Exelon Patch
Disclosure
Dr. Sadowsky is a consultant to and speaker for Forest Pharmaceuticals and Novartis Pharmaceuticals.
Acknowledgment
The author thanks Christina Mackins, PhD, a medical writer for Alpha-Plus Medical Communications Ltd, for her editorial assistance with this article. Funding for her work was provided by Novartis Pharmaceuticals.
The rivastigmine patch is the first transdermal treatment for symptoms of mild to moderate Alzheimer’s disease (AD) and mild to moderate Parkinson’s disease dementia (Table). Rivastigmine, a cholinesterase inhibitor, is the only therapy approved for both indications.
Table
Rivastigmine transdermal patch: Fast facts
| Brand name: Exelon Patch |
| Class: Cholinesterase inhibitor |
| Indication: Symptomatic treatment of mild to moderate Alzheimer’s-type dementia and mild to moderate dementia associated with Parkinson’s disease |
| Manufacturer: Novartis Pharmaceuticals, Inc. |
| Dosing forms: 4.6 and 9.5 mg/24 hours transdermal patches (5 cm2 and 10 cm2, respectively) |
| Recommended dosage: Start with 4.6 mg/24 hours patch for ≥4 weeks, followed by a one-step increase to the target dose 9.5 mg/24 hours patch* |
| *Unless the patient is taking oral rivastigmine (see ‘Transitioning to rivastigmine patch,’) |
Clinical implications
The rivastigmine patch offers continuous drug delivery through the skin into the bloodstream over 24 hours.1 This may reduce the incidence of side effects compared with oral rivastigmine,2 making optimal therapeutic doses easier to attain.3 The target dose 9.5 mg/24 hours patch provides efficacy similar to the highest recommended rivastigmine capsule dose (6 mg bid for a total of 12 mg/d).2
How it works
The rivastigmine patch uses matrix technology, which enables delivery of a large amount of drug from a small surface area.4 The patch is available in 2 dosage forms:
- a 5-cm2 size containing 9 mg of rivastigmine that delivers 4.6 mg/24 hours
- a 10-cm2 size containing 18 mg of rivastigmine that delivers 9.5 mg/24 hours.
Each patch consists of 4 layers: the backing layer, an acrylic drug matrix, a silicone adhesive matrix, and an overlapping release liner that is removed and discarded before the patch is applied.1
Cholinesterase inhibitors are believed to exert their effects by increasing available levels of the neurotransmitter acetylcholine in the brain. Two studies have demonstrated that cognitive improvements associated with rivastigmine treatment correlate significantly with cholinesterase inhibition.5,6 In 1 study, rivastigmine’s inhibitory effects on cholinesterase were sustained for 12 months.6
Pharmacokinetics
Rivastigmine is metabolized by its target cholinesterase enzymes to the decarbamylated metabolite NAP 226-90, which has minimal acetylcholinesterase inhibition and is excreted through the urine.1 As a result of its low accumulation potential and cytochrome P 450-independent metabolism, rivastigmine has low potential for pharmacokinetic drug–drug interactions. This lack of interaction has been confirmed for many drugs commonly taken by elderly patients, such as digoxin, nonsteroidal anti-inflammatory drugs, and estrogens.7
Rivastigmine has a half-life of 1 to 2 hours, so it is rapidly cleared.8 In the event of a serious reaction, significant clearance of rivastigmine from the body would occur within 3 hours of patch removal.
Centrally mediated cholinergic gastrointestinal (GI) side effects associated with oral rivastigmine are related to high maximum plasma concentrations (Cmax) and short time interval to Cmax (Tmax).9 In an open-label, parallel-group study of 51 AD patients that compared rivastigmine patches with rivastigmine capsules, transdermal administration was associated with slower increases to lower peak plasma concentrations (prolonged Tmax and reduced Cmax), and less fluctuation in plasma concentration.1 Despite these effects, the rivastigmine 9.5 mg/24 hours patch provided drug exposure comparable to the highest dose of capsules (6 mg bid for a total of 12 mg/d), with improved GI tolerability.3
Efficacy
Rivastigmine patch efficacy was evaluated in a single, 24-week, international, randomized, double-blind trial of 1,195 patients with AD.2 The study group represented typical patients with mild to moderate AD—age 50 to 85 years with Mini-Mental State Examination scores of 10 to 20 at baseline. Patients were randomly assigned to receive:
- 17.4 mg/24 hours rivastigmine patch (20-cm2 patch; n=303)
- 9.5 mg/24 hours rivastigmine patch (10-cm2 patch; n=293)
- 6 mg bid rivastigmine capsules (n=297)
- or placebo (n=302).
Data for the 17.4 mg/24 hours patch are not discussed here because this dose exceeds the FDA-approved maximum dosage (9.5 mg/24 hours) and is not available.
Patients in the 9.5 mg/24 hours patch group received a 4.6 mg/24 hours patch (5 cm2) for weeks 1 through 4, and then the 9.5 mg/24 hours patch for the remainder of the study. Patients in the capsule group started on 3 mg/d (1.5 mg bid) and were titrated every 4 weeks in steps of 3 mg/d to a maximum of 12 mg/d administered as 6 mg bid.
Primary outcomes were measured as mean change in score from baseline to endpoint on the Alzheimer’s Disease Assessment Scale–Cognitive Subscale (ADAS-Cog) and Alzheimer’s Disease Co-operative Study–Clinical Global Impression of Change (ADCS-CGIC). By study endpoint, the 9.5 mg/24 hours patch and capsules, 12 mg/d, showed comparable efficacy (Figure).2 Compared with those receiving placebo, patients in the 9.5 mg/24 hours patch and capsule groups showed significant improvements in dementia symptoms, including:
- cognition
- global performance
- attention
- activities of daily living.2
Based on my clinical experience, these improvements reflect small but clinically meaningful changes that are noted by patients and caregivers.
Figure
Efficacy of transdermal rivastigmine for Alzheimer’s symptoms
*P<0.05 vs placebo
ADAS-Cog: Alzheimer’s Disease Assessment Scale-Cognitive Subscale; ADCS-CGIC: Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change
Source: Adapted from reference 2
In a 24-week study, transdermal rivastigmine, 9.5 mg/24 hours, and the highest recommended dose of oral rivastigmine (6 mg bid) showed comparable efficacy as measured by mean change in score on scales commonly used in Alzheimer’s disease clinical trials. ADAS-Cog assesses orientation, memory, language, praxis, and visuospatial functions. ADCS-CGIC provides a single global rating of change from baseline based on interviews with the patient and caregiver.
Safety and tolerability
Adverse events associated with rivastigmine are predominantly cholinergic; GI side effects—nausea, vomiting, and diarrhea—are observed most frequently.2 These events occur less frequently with the patch than with capsules. In the efficacy trial, patients in the 9.5 mg/24 hours rivastigmine patch group had one-third as many reports of nausea (7.2% vs 23.1%) and vomiting (6.2% vs 17.0%) compared with the 6 mg bid capsule group.2
Diarrhea was reported by 6% of subjects receiving the 9.5 mg/24 hours patch, 5% of those taking 6-mg capsule bid, and 3% receiving placebo. Fewer subjects in the 9.5 mg/24 hours patch group (3%) experienced decreased weight compared with those in the capsule group (5%). The rate of decreased weight with placebo was 1%.
Dizziness affected 2% of those in the 9.5 mg/24 hours patch and placebo groups; incidence in the capsule group was significantly higher at 8%. Headache was similar with the 9.5 mg/24 hours patch (3%) and placebo (2%), with the capsule significantly higher at 6%.2
The proportion of patients who experienced no, slight, or mild skin irritation ranged from 90% to 98%.2 The most commonly reported moderate or severe skin irritations were erythema (8% rivastigmine patch vs 4% placebo) and pruritus (7% rivastigmine patch vs 3% placebo). Two percent of patients using active patch discontinued the trial because of skin irritation.
Rivastigmine appears not to produce adverse effects on cardiac function as assessed by ECG. In clinical trials of 2,791 patients, pooled 12-lead ECG data comparing oral rivastigmine and placebo groups did not differ significantly in heart rate or PR, QRS, and QTc intervals.10
Dosing
The rivastigmine patch is administered once daily, and the recommended maintenance dose is the 9.5 mg/24 hours patch. Start patients on a 4.6 mg/24 hours patch for at least 4 weeks and then increase to the 9.5 mg/24 hours target dose if the lower dose is well tolerated.
Dosage adjustment of rivastigmine is not necessary in patients with hepatic or renal disease because of minimal liver metabolism and the acetylcholinesterase-mediated hydrolysis of rivastigmine to the inactive decarbamylated metabolite NAP 226-90, which is excreted in the urine.11
Instruct patients or caregivers to apply the patch to clean, dry, hairless skin that is free of cuts, rashes, or irritation on the upper or lower back or upper arm or chest.1 The patch has shown good adhesive properties over 24 hours, remaining attached in a range of situations, including bathing and hot weather.2 In the 9.5 mg/24 hours group of the efficacy study, 96% of patches remained attached or had slight lifting of the edges (1,336 total patch evaluations).
Transitioning to rivastigmine patch
The efficacy study included an open-label extension, during which blinding was maintained. This provided information on patients beginning rivastigmine patch therapy directly from placebo2 or transitioning from rivastigmine capsules to the target dose 9.5 mg/24 hours patch.12 Based on these results, transition patients as follows:
- Patients taking oral rivastigmine, <6 mg/d: Switch to a 4.6 mg/24 hours patch for ≥4 weeks before increasing to a 9.5 mg/24 hours patch.
- Patients taking oral rivastigmine, 6 to 12 mg/d: Switch directly to a 9.5 mg/24 hours patch.
Apply the first patch the day after the last oral dose.
Related resource
- Rivastigmine transdermal system prescribing information. www.pharma.us.novartis.com/product/pi/pdf/exelonpatch.pdf.
Drug brand names
- Digoxin • Lanoxin
- Rivastigmine • Exelon
- Rivastigmine transdermal
- system • Exelon Patch
Disclosure
Dr. Sadowsky is a consultant to and speaker for Forest Pharmaceuticals and Novartis Pharmaceuticals.
Acknowledgment
The author thanks Christina Mackins, PhD, a medical writer for Alpha-Plus Medical Communications Ltd, for her editorial assistance with this article. Funding for her work was provided by Novartis Pharmaceuticals.
1. Lefèvre G, Sedek G, Jhee S, et al. Pharmacokinetics and pharmacodynamics of the novel daily rivastigmine transdermal patch compared with twice-daily capsules in Alzheimer’s disease patients. J Clin Pharmacol 2007;47:471-8.
2. Winblad B, Cummings J, Andreasen N, et al. A six-month, double-blind, randomized, placebo-controlled study of a transdermal patch in Alzheimer’s disease—rivastigmine patch versus capsule. Int J Geriatr Psychiatry 2007;22:456-67.
3. Oertel W, Ross JS, Eggert K, Adler G. Rationale for transdermal drug administration in Alzheimer disease. Neurology 2007;69(suppl 1):S4-S9.
4. Petersen TA. Transdermal drug formulations and process development. Pharmaceut Technol 2003;(suppl):18-21.
5. Giacobini E, Spiegel R, Enz A, et al. Inhibition of acetyl- and butyryl-cholinesterase in the cerebrospinal fluid of patients with Alzheimer’s disease by rivastigmine: correlation with cognitive benefit. J Neural Transm 2002;109:1053-65.
6. Darreh-Shori T, Almkvist O, Guan ZZ, et al. Sustained cholinesterase inhibition in AD patients receiving rivastigmine for 12 months. Neurology 2002;59:563-72.
7. Grossberg GT, Stahelin HB, Messina JC, et al. Lack of adverse pharmacodynamic drug interactions with rivastigmine and twenty-two classes of medications. Int J Geriatr Psychiatry 2000;15(3):242-7.
8. Polinsky RJ. Clinical pharmacology of rivastigmine: a new-generation acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease. Clin Ther 1998;20:634-47.
9. Jann MW, Shirley KL, Small GW. Clinical pharmacokinetics and pharmacodynamics of cholinesterase inhibitors. Clin Pharmacokinet 2002;41:719-39.
10. Morganroth J, Graham S, Hartman R, et al. Electrocardiographic effects of rivastigmine. J Clin Pharmacol 2002;42:558-68.
11. Exelon patch [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2007.
12. Frölich L, Barone P, Förstl H, et al. IDEAL: A 28-week open-label extension of a 24-week double-blind study of the first transdermal patch in Alzheimer’s disease. Poster presented at: 11th Congress of the European Federation of Neurological Societies; August 25-28, 2007; Brussels, Belgium.
Dr. Sadowsky is associate clinical professor of neurology, Nova Southeastern University, Fort Lauderdale, FL, and director, Premier Research Institute, Palm Beach Neurology, West Palm Beach, FL.
1. Lefèvre G, Sedek G, Jhee S, et al. Pharmacokinetics and pharmacodynamics of the novel daily rivastigmine transdermal patch compared with twice-daily capsules in Alzheimer’s disease patients. J Clin Pharmacol 2007;47:471-8.
2. Winblad B, Cummings J, Andreasen N, et al. A six-month, double-blind, randomized, placebo-controlled study of a transdermal patch in Alzheimer’s disease—rivastigmine patch versus capsule. Int J Geriatr Psychiatry 2007;22:456-67.
3. Oertel W, Ross JS, Eggert K, Adler G. Rationale for transdermal drug administration in Alzheimer disease. Neurology 2007;69(suppl 1):S4-S9.
4. Petersen TA. Transdermal drug formulations and process development. Pharmaceut Technol 2003;(suppl):18-21.
5. Giacobini E, Spiegel R, Enz A, et al. Inhibition of acetyl- and butyryl-cholinesterase in the cerebrospinal fluid of patients with Alzheimer’s disease by rivastigmine: correlation with cognitive benefit. J Neural Transm 2002;109:1053-65.
6. Darreh-Shori T, Almkvist O, Guan ZZ, et al. Sustained cholinesterase inhibition in AD patients receiving rivastigmine for 12 months. Neurology 2002;59:563-72.
7. Grossberg GT, Stahelin HB, Messina JC, et al. Lack of adverse pharmacodynamic drug interactions with rivastigmine and twenty-two classes of medications. Int J Geriatr Psychiatry 2000;15(3):242-7.
8. Polinsky RJ. Clinical pharmacology of rivastigmine: a new-generation acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease. Clin Ther 1998;20:634-47.
9. Jann MW, Shirley KL, Small GW. Clinical pharmacokinetics and pharmacodynamics of cholinesterase inhibitors. Clin Pharmacokinet 2002;41:719-39.
10. Morganroth J, Graham S, Hartman R, et al. Electrocardiographic effects of rivastigmine. J Clin Pharmacol 2002;42:558-68.
11. Exelon patch [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2007.
12. Frölich L, Barone P, Förstl H, et al. IDEAL: A 28-week open-label extension of a 24-week double-blind study of the first transdermal patch in Alzheimer’s disease. Poster presented at: 11th Congress of the European Federation of Neurological Societies; August 25-28, 2007; Brussels, Belgium.
Dr. Sadowsky is associate clinical professor of neurology, Nova Southeastern University, Fort Lauderdale, FL, and director, Premier Research Institute, Palm Beach Neurology, West Palm Beach, FL.
Risperidone’s 2 new pediatric indications
Risperidone is the first second-generation antipsychotic (SGA) to receive FDA approval for treating children and adolescents with bipolar mania or schizophrenia. Specifically, the SGA is indicated for treating schizophrenia in patients age 13 to 17 and as monotherapy in short-term treatment of manic or mixed episodes of bipolar I disorder in patients age 10 to 17 (Table 1).
Risperidone also is approved for:
- schizophrenia in adults
- acute mania or mixed episodes associated with bipolar I disorder in adults, alone or in combination with lithium or valproate
- irritability associated with autistic disorder in patients age 5 to 16.
Table 1
Risperidone: Fast facts
| Brand name: Risperdal |
| Class: Second-generation antipsychotic |
| New indications: Schizophrenia in adolescents age 13 to 17 and monotherapy in short-term treatment of manic or mixed episodes of bipolar I disorder in children and adolescents age 10 to 17. (Risperidone had been approved for schizophrenia and short-term treatment of acute manic or mixed episodes associated with bipolar I disorder in adults and treatment of irritability associated with autistic disorder in children and adolescents.) |
| Approval date: August 22, 2007 for pediatric schizophrenia and mania indications |
| Manufacturer: Janssen, L.P. |
| Dosing forms: 0.25-, 0.5-, 1-, 2-, 3-, and 4-mg tablets; 0.5-, 1-, 2-, 3-, and 4-mg orally disintegrating tablets; 1 mg/mL oral solution |
| Recommended target dosage: 3 mg/d (pediatric schizophrenia) or 2.5 mg/d (pediatric bipolar mania). See Table 2 for initial dosages and titration |
Clinical implications
Risperidone is widely used off-label to treat irritability in children with pervasive developmental disorders,1,2 aggressive behaviors associated with conduct disorder,3 psychotic disorders,4 and bipolar disorder.5 It also has been used off-label to treat pediatric schizophrenia and bipolar disorder for many years.
These 2 new indications give clinicians additional support for using SGAs in children and adolescents with these serious psychiatric disorders.
How it works
Risperidone’s therapeutic activity in schizophrenia seems to be mediated through a combination of dopamine type 2 (D2) and serotonin type 2 (5HT2) receptor antagonism. Antagonism at receptors other than D2 and 5HT2 may explain some of risperidone’s other therapeutic effects.
Pharmacokinetics
In children, the half-lives of risperidone and its major active metabolite 9-hydroxyrisperidone are 3±2.3 hours and 22±46 hours, respectively.6 The pharmacologic activity of 9-hydroxyrisperidone is similar to that of risperidone.
Risperidone is extensively metabolized in the liver by the cytochrome P-450 (CYP) 2D6 enzyme system. The main metabolic pathway is through hydroxylation of risperidone to 9-hydroxyrisperidone by CYP 2D6. Food does not affect the rate or extent of the drug’s absorption.6
Efficacy studies
In schizophrenia. Approval of the indication for pediatric schizophrenia was based on data from 2 short-term (6 and 8 weeks) randomized, double-blind, controlled trials involving a total of 416 patients age 13 to 17 who met DSM-IV-TR criteria for schizophrenia and were experiencing an acute episode at enrollment.7 In one study, patients received risperidone, 1 to 3 mg/d, 4 to 6 mg/d, or placebo. In the other study, dosages were 0.15 to 0.6 mg/d or 1.5 to 6 mg/d. Except for patients in the 0.15 to 0.6 mg group (who initially received 0.05 mg/d), most patients started risperidone at 0.5 mg/d. In both trials, starting dosages were titrated to the target range in approximately 7 days.
Outcomes were measured as changes in total Positive and Negative Syndrome Scale (PANSS) and Personal and Social Performance (PSP) scale scores. The multi-item PANSS inventory measures positive and negative schizophrenia symptoms, disorganized thoughts, uncontrolled hostility/excitement, and anxiety/depression. The PSP gauges personal and social functioning in socially useful activities (work and study), personal and social relationships, self-care, and disturbing/ aggressive behaviors.
Risperidone, 1 to 6 mg/d, improved schizophrenia symptoms significantly more than placebo, as measured by PANSS scores. Doses >3 mg/d did not show greater efficacy than lower doses, as evaluated by PANSS and PSP scores.
Adverse reactions experienced by >5% of patients treated with risperidone included somnolence, parkinsonism, tremor, dystonia, dizziness, akathisia, increased salivation, and anxiety.7
In bipolar I disorder. Risperidone’s efficacy for short-term treatment of mania in children and adolescents was demonstrated in a 3-week, randomized, double-blind, placebo-controlled, multi-center study of 169 patients age 10 to 17 who were experiencing a manic or mixed episode of bipolar I disorder.7 Patients were randomly assigned to risperidone, 0.5 to 2.5 mg/d or 3 to 6 mg/d, or placebo. All patients were started at 0.5 mg/d and this dose was titrated to the target dosage range in 7 days.
Risperidone, 0.5 to 6 mg/d, significantly decreased the total Young Mania Rating Scale score—a measure of the severity of elevated mood, increased motor activity energy, sexual interest, sleep, irritability, speech (rate/amount), language (thought disorder, content, disruptive), aggressive behavior, appearance, and insight. No evidence of increased efficacy was observed at doses >2.5 mg/d. In this trial, symptoms reported by >5% of patients included fatigue, dizziness, dystonia, parkinsonism, akathisia, abdominal pain, dyspepsia, nausea, vomiting, and diarrhea.7
Pediatric dosing. Based on these studies, the recommended starting dose for children and adolescents is 0.5 mg/d, with titration in 0.5-to 1-mg increments to targets of:
Table 2
Recommended dosing of risperidone
for pediatric schizophrenia and bipolar mania
| Indication | Initial dose | Titration | Target dose | Effective dose range |
|---|---|---|---|---|
| Schizophrenia, adolescents age 13 to 17 | 0.5 mg/d | 0.5 to 1 mg/d | 3 mg/d | 1 to 6 mg/d |
| Bipolar mania, children and adolescents age 10 to 17 | 0.5 mg/d | 0.5 to 1 mg/d | 2.5 mg/d | 0.5 to 6 mg/d |
| Source:Reference 7 | ||||
Tolerability studies
In long-term studies, the most commonly reported adverse events associated with risperidone in children and adolescents have been rhinitis, abdominal pain, increased saliva, body pain, gynecomastia, and weight increase.8 Specific adverse effects that pose long-term concerns are:
- tardive dyskinesia (TD)
- weight gain
- increased prolactin levels
Tardive dyskinesia. In clinical trials that included 1,885 children and adolescents with autistic disorder or other psychiatric disorders treated with risperidone, 2 patients (0.1%) were reported to have TD, which resolved when risperidone was discontinued.7 To monitor for TD, administer the Abnormal Involuntary Movement Scale at baseline and every 6 months while using risperidone in pediatric patients.
Weight gain. In long-term, open-label trials, patients with autistic or other psychiatric disorders gained an average 7.5
kg after 12 months of risperidone treatment. Most of the weight gain occurred in the first 6 months.9 Expected normal weight gain in children is 3 to 3.5 kg/year adjusted for age, based on Centers for Disease Control and Prevention normative data.
Follow the American Diabetes Association guidelines10 for monitoring metabolic parameters during antipsychotic
treatment, and intervene if clinically significant weight gain occurs.
In a 16-week, placebo-controlled study,11 metformin reversed weight gain associated with SGAs in children and adolescents. Metformin’s potential side effects include hypoglycemia, diarrhea, nausea/vomiting, and (rarely) lactic acidosis, but no adverse events were attributed to metformin.
Increased prolactin. As in adults, risperidone elevates serum prolactin in children and adolescents. All pediatric risperidone trials—of autism,2 disruptive behavior disorders in children with subaverage intelligence,9 schizophrenia,7 and bipolar mania—have shown increased serum prolactin. Risperidone’s long-term effects on growth and sexual maturation have not been fully evaluated, but hyperprolactinemia may inhibit reproductive function.
Findling et al12 analyzed data from 5 clinical trials (total 700 patients) in which children and adolescents age 5 to 15 years with subaverage IQs and conduct or other disruptive behavior disorders received risperidone for up to 55 weeks. Mean prolactin levels rose from 7.8 ng/mL
at baseline to 29.4 ng/mL at weeks 4 to 7, then progressively decreased to 16.1 ng/mL at weeks 40 to 48 (n=358) and 13.0 ng/mL at weeks 52 to 55 (n=42). Girls returned to a mean value within the normal range (≤30 ng/mL) by weeks 8 to 12, and boys were close to normal values (≤18 ng/mL) by weeks 16 to 24.
The researchers concluded that serum prolactin levels in children tend to rise and peak within the first 1 to 2 months of risperidone treatment and then steadily decline to values within or very close to normal range by 3 to 5 months.
The biological significance of chronic, mild prolactin elevations is unknown.13 Children entering puberty appear to be at highest risk for elevated prolactin and clinical symptoms while treated with risperidone.14 Therefore, ask all adolescents treated with risperidone about increases in breast size and galactorrhea. Switch those who develop these symptoms to an SGA that does not increase serum prolactin.
Contraindications. Risperidone is contraindicated in patients with a known hypersensitivity to the drug.
- Risperdal prescribing information. www.risperdal.com/risperdal/shared/pi/risperdal.pdf.
Drug brand names
- Lithium • Eskalith, Lithobid
- Risperidone • Risperdal
- Metformin • Glucophage, Fortamet
- Valproate • Depakote
Disclosures
Dr. Kowatch receives research support from Bristol-Meyers Squibb, Stanley Research Foundation, National Institute of Mental Health, and National Institute of Child Health and Human Development. He is a consultant for Creative Educational Concepts, Child and Adolescent Bipolar Foundation, Abbott Laboratories, and sanofi-aventis, and a speaker for Abbott Laboratories and AstraZeneca.
1. Aman MG, De Smedt G, Derivan A, et al. Double-blind, placebo-controlled study of risperidone for the treatment of disruptive behaviors in children with subaverage intelligence. Am J Psychiatry 2002;159:1337-46.
2. McCracken JT, McGough J, Shah B, et al. Risperidone in children with autism and serious behavioral problems. N Engl J Med 2002;347(5):314-21.
3. Findling RL, McNamara NK, Branicky LA, et al. A double-blind pilot study of risperidone in the treatment of conduct disorder. J Am Acad Child Adolesc Psychiatry 2000;39(4):509-16.
4. Sikich L, Hamer R, Malekpour AH, et al. Double-blind trial comparing risperidone, olanzapine, and haloperidol in the treatment of psychotic children and adolescents. Paper presented at: Society of Biological Psychiatry Annual Meeting; May 16-18, 2002; Philadelphia, PA.
5. Frazier JA, Meyer MC, Biederman J, et al. Risperidone treatment for juvenile bipolar disorder: a retrospective chart review. J Am Acad Child Adolesc Psychiatry 1999;38(8):960-5.
6. Aman MG, Vinks AA, Remmerie B, et al. Plasma pharmacokinetic characteristics of risperidone and their relationship to saliva concentrations in children with psychiatric or neurodevelopmental disorders. Clin Ther 2007;29(7):1476-86.
7. Risperdal [package insert]. Titusville, NJ: Janssen, L.P; 2007.
8. Reyes MR, Olah R, Csaba K, et al. Long-term safety and efficacy of risperidone in children with disruptive behaviour disorders. Results of a 2-year extension study. Eur Child Adolesc Psychiatry 2006;15(2):97-104.
9. Croonenberghs J, Fegert JM, Findling RL, et al. Risperidone in children with disruptive behavior disorders and subaverage intelligence: a 1-year, open-label study of 504 patients. J Am Acad Child Adolesc Psychiatry 2005;44(1):64-72.
10. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. J Clin Psychiatry 2004;65(2):267-72.
11. Klein DJ, Cottingham EM, Sorter M, et al. A randomized, double-blind, placebo-controlled trial of metformin treatment of weight gain associated with initiation of atypical antipsychotic therapy in children and adolescents. Am J Psychiatry 2006;163(12):2072-9.
12. Findling RL, Kusumakar V, Daneman D, et al. Prolactin levels during long-term risperidone treatment in children and adolescents. J Clin Psychiatry 2003;64(11):1362-9.
13. Staller J. The effect of long-term antipsychotic treatment on prolactin. J Child Adolesc Psychopharmacol 2006;16(3):317-26.
14. Holzer L, Eap CB. Risperidone-induced symptomatic hyperprolactinaemia in adolescents. J Clin Psychopharmacol 2006;26(2):167-71.
Dr. Kowatch is professor of psychiatry and pediatrics at Cincinnati Children’s Hospital Medical Center and a Section Editor for Current Psychiatry.
Risperidone is the first second-generation antipsychotic (SGA) to receive FDA approval for treating children and adolescents with bipolar mania or schizophrenia. Specifically, the SGA is indicated for treating schizophrenia in patients age 13 to 17 and as monotherapy in short-term treatment of manic or mixed episodes of bipolar I disorder in patients age 10 to 17 (Table 1).
Risperidone also is approved for:
- schizophrenia in adults
- acute mania or mixed episodes associated with bipolar I disorder in adults, alone or in combination with lithium or valproate
- irritability associated with autistic disorder in patients age 5 to 16.
Table 1
Risperidone: Fast facts
| Brand name: Risperdal |
| Class: Second-generation antipsychotic |
| New indications: Schizophrenia in adolescents age 13 to 17 and monotherapy in short-term treatment of manic or mixed episodes of bipolar I disorder in children and adolescents age 10 to 17. (Risperidone had been approved for schizophrenia and short-term treatment of acute manic or mixed episodes associated with bipolar I disorder in adults and treatment of irritability associated with autistic disorder in children and adolescents.) |
| Approval date: August 22, 2007 for pediatric schizophrenia and mania indications |
| Manufacturer: Janssen, L.P. |
| Dosing forms: 0.25-, 0.5-, 1-, 2-, 3-, and 4-mg tablets; 0.5-, 1-, 2-, 3-, and 4-mg orally disintegrating tablets; 1 mg/mL oral solution |
| Recommended target dosage: 3 mg/d (pediatric schizophrenia) or 2.5 mg/d (pediatric bipolar mania). See Table 2 for initial dosages and titration |
Clinical implications
Risperidone is widely used off-label to treat irritability in children with pervasive developmental disorders,1,2 aggressive behaviors associated with conduct disorder,3 psychotic disorders,4 and bipolar disorder.5 It also has been used off-label to treat pediatric schizophrenia and bipolar disorder for many years.
These 2 new indications give clinicians additional support for using SGAs in children and adolescents with these serious psychiatric disorders.
How it works
Risperidone’s therapeutic activity in schizophrenia seems to be mediated through a combination of dopamine type 2 (D2) and serotonin type 2 (5HT2) receptor antagonism. Antagonism at receptors other than D2 and 5HT2 may explain some of risperidone’s other therapeutic effects.
Pharmacokinetics
In children, the half-lives of risperidone and its major active metabolite 9-hydroxyrisperidone are 3±2.3 hours and 22±46 hours, respectively.6 The pharmacologic activity of 9-hydroxyrisperidone is similar to that of risperidone.
Risperidone is extensively metabolized in the liver by the cytochrome P-450 (CYP) 2D6 enzyme system. The main metabolic pathway is through hydroxylation of risperidone to 9-hydroxyrisperidone by CYP 2D6. Food does not affect the rate or extent of the drug’s absorption.6
Efficacy studies
In schizophrenia. Approval of the indication for pediatric schizophrenia was based on data from 2 short-term (6 and 8 weeks) randomized, double-blind, controlled trials involving a total of 416 patients age 13 to 17 who met DSM-IV-TR criteria for schizophrenia and were experiencing an acute episode at enrollment.7 In one study, patients received risperidone, 1 to 3 mg/d, 4 to 6 mg/d, or placebo. In the other study, dosages were 0.15 to 0.6 mg/d or 1.5 to 6 mg/d. Except for patients in the 0.15 to 0.6 mg group (who initially received 0.05 mg/d), most patients started risperidone at 0.5 mg/d. In both trials, starting dosages were titrated to the target range in approximately 7 days.
Outcomes were measured as changes in total Positive and Negative Syndrome Scale (PANSS) and Personal and Social Performance (PSP) scale scores. The multi-item PANSS inventory measures positive and negative schizophrenia symptoms, disorganized thoughts, uncontrolled hostility/excitement, and anxiety/depression. The PSP gauges personal and social functioning in socially useful activities (work and study), personal and social relationships, self-care, and disturbing/ aggressive behaviors.
Risperidone, 1 to 6 mg/d, improved schizophrenia symptoms significantly more than placebo, as measured by PANSS scores. Doses >3 mg/d did not show greater efficacy than lower doses, as evaluated by PANSS and PSP scores.
Adverse reactions experienced by >5% of patients treated with risperidone included somnolence, parkinsonism, tremor, dystonia, dizziness, akathisia, increased salivation, and anxiety.7
In bipolar I disorder. Risperidone’s efficacy for short-term treatment of mania in children and adolescents was demonstrated in a 3-week, randomized, double-blind, placebo-controlled, multi-center study of 169 patients age 10 to 17 who were experiencing a manic or mixed episode of bipolar I disorder.7 Patients were randomly assigned to risperidone, 0.5 to 2.5 mg/d or 3 to 6 mg/d, or placebo. All patients were started at 0.5 mg/d and this dose was titrated to the target dosage range in 7 days.
Risperidone, 0.5 to 6 mg/d, significantly decreased the total Young Mania Rating Scale score—a measure of the severity of elevated mood, increased motor activity energy, sexual interest, sleep, irritability, speech (rate/amount), language (thought disorder, content, disruptive), aggressive behavior, appearance, and insight. No evidence of increased efficacy was observed at doses >2.5 mg/d. In this trial, symptoms reported by >5% of patients included fatigue, dizziness, dystonia, parkinsonism, akathisia, abdominal pain, dyspepsia, nausea, vomiting, and diarrhea.7
Pediatric dosing. Based on these studies, the recommended starting dose for children and adolescents is 0.5 mg/d, with titration in 0.5-to 1-mg increments to targets of:
Table 2
Recommended dosing of risperidone
for pediatric schizophrenia and bipolar mania
| Indication | Initial dose | Titration | Target dose | Effective dose range |
|---|---|---|---|---|
| Schizophrenia, adolescents age 13 to 17 | 0.5 mg/d | 0.5 to 1 mg/d | 3 mg/d | 1 to 6 mg/d |
| Bipolar mania, children and adolescents age 10 to 17 | 0.5 mg/d | 0.5 to 1 mg/d | 2.5 mg/d | 0.5 to 6 mg/d |
| Source:Reference 7 | ||||
Tolerability studies
In long-term studies, the most commonly reported adverse events associated with risperidone in children and adolescents have been rhinitis, abdominal pain, increased saliva, body pain, gynecomastia, and weight increase.8 Specific adverse effects that pose long-term concerns are:
- tardive dyskinesia (TD)
- weight gain
- increased prolactin levels
Tardive dyskinesia. In clinical trials that included 1,885 children and adolescents with autistic disorder or other psychiatric disorders treated with risperidone, 2 patients (0.1%) were reported to have TD, which resolved when risperidone was discontinued.7 To monitor for TD, administer the Abnormal Involuntary Movement Scale at baseline and every 6 months while using risperidone in pediatric patients.
Weight gain. In long-term, open-label trials, patients with autistic or other psychiatric disorders gained an average 7.5
kg after 12 months of risperidone treatment. Most of the weight gain occurred in the first 6 months.9 Expected normal weight gain in children is 3 to 3.5 kg/year adjusted for age, based on Centers for Disease Control and Prevention normative data.
Follow the American Diabetes Association guidelines10 for monitoring metabolic parameters during antipsychotic
treatment, and intervene if clinically significant weight gain occurs.
In a 16-week, placebo-controlled study,11 metformin reversed weight gain associated with SGAs in children and adolescents. Metformin’s potential side effects include hypoglycemia, diarrhea, nausea/vomiting, and (rarely) lactic acidosis, but no adverse events were attributed to metformin.
Increased prolactin. As in adults, risperidone elevates serum prolactin in children and adolescents. All pediatric risperidone trials—of autism,2 disruptive behavior disorders in children with subaverage intelligence,9 schizophrenia,7 and bipolar mania—have shown increased serum prolactin. Risperidone’s long-term effects on growth and sexual maturation have not been fully evaluated, but hyperprolactinemia may inhibit reproductive function.
Findling et al12 analyzed data from 5 clinical trials (total 700 patients) in which children and adolescents age 5 to 15 years with subaverage IQs and conduct or other disruptive behavior disorders received risperidone for up to 55 weeks. Mean prolactin levels rose from 7.8 ng/mL
at baseline to 29.4 ng/mL at weeks 4 to 7, then progressively decreased to 16.1 ng/mL at weeks 40 to 48 (n=358) and 13.0 ng/mL at weeks 52 to 55 (n=42). Girls returned to a mean value within the normal range (≤30 ng/mL) by weeks 8 to 12, and boys were close to normal values (≤18 ng/mL) by weeks 16 to 24.
The researchers concluded that serum prolactin levels in children tend to rise and peak within the first 1 to 2 months of risperidone treatment and then steadily decline to values within or very close to normal range by 3 to 5 months.
The biological significance of chronic, mild prolactin elevations is unknown.13 Children entering puberty appear to be at highest risk for elevated prolactin and clinical symptoms while treated with risperidone.14 Therefore, ask all adolescents treated with risperidone about increases in breast size and galactorrhea. Switch those who develop these symptoms to an SGA that does not increase serum prolactin.
Contraindications. Risperidone is contraindicated in patients with a known hypersensitivity to the drug.
- Risperdal prescribing information. www.risperdal.com/risperdal/shared/pi/risperdal.pdf.
Drug brand names
- Lithium • Eskalith, Lithobid
- Risperidone • Risperdal
- Metformin • Glucophage, Fortamet
- Valproate • Depakote
Disclosures
Dr. Kowatch receives research support from Bristol-Meyers Squibb, Stanley Research Foundation, National Institute of Mental Health, and National Institute of Child Health and Human Development. He is a consultant for Creative Educational Concepts, Child and Adolescent Bipolar Foundation, Abbott Laboratories, and sanofi-aventis, and a speaker for Abbott Laboratories and AstraZeneca.
Risperidone is the first second-generation antipsychotic (SGA) to receive FDA approval for treating children and adolescents with bipolar mania or schizophrenia. Specifically, the SGA is indicated for treating schizophrenia in patients age 13 to 17 and as monotherapy in short-term treatment of manic or mixed episodes of bipolar I disorder in patients age 10 to 17 (Table 1).
Risperidone also is approved for:
- schizophrenia in adults
- acute mania or mixed episodes associated with bipolar I disorder in adults, alone or in combination with lithium or valproate
- irritability associated with autistic disorder in patients age 5 to 16.
Table 1
Risperidone: Fast facts
| Brand name: Risperdal |
| Class: Second-generation antipsychotic |
| New indications: Schizophrenia in adolescents age 13 to 17 and monotherapy in short-term treatment of manic or mixed episodes of bipolar I disorder in children and adolescents age 10 to 17. (Risperidone had been approved for schizophrenia and short-term treatment of acute manic or mixed episodes associated with bipolar I disorder in adults and treatment of irritability associated with autistic disorder in children and adolescents.) |
| Approval date: August 22, 2007 for pediatric schizophrenia and mania indications |
| Manufacturer: Janssen, L.P. |
| Dosing forms: 0.25-, 0.5-, 1-, 2-, 3-, and 4-mg tablets; 0.5-, 1-, 2-, 3-, and 4-mg orally disintegrating tablets; 1 mg/mL oral solution |
| Recommended target dosage: 3 mg/d (pediatric schizophrenia) or 2.5 mg/d (pediatric bipolar mania). See Table 2 for initial dosages and titration |
Clinical implications
Risperidone is widely used off-label to treat irritability in children with pervasive developmental disorders,1,2 aggressive behaviors associated with conduct disorder,3 psychotic disorders,4 and bipolar disorder.5 It also has been used off-label to treat pediatric schizophrenia and bipolar disorder for many years.
These 2 new indications give clinicians additional support for using SGAs in children and adolescents with these serious psychiatric disorders.
How it works
Risperidone’s therapeutic activity in schizophrenia seems to be mediated through a combination of dopamine type 2 (D2) and serotonin type 2 (5HT2) receptor antagonism. Antagonism at receptors other than D2 and 5HT2 may explain some of risperidone’s other therapeutic effects.
Pharmacokinetics
In children, the half-lives of risperidone and its major active metabolite 9-hydroxyrisperidone are 3±2.3 hours and 22±46 hours, respectively.6 The pharmacologic activity of 9-hydroxyrisperidone is similar to that of risperidone.
Risperidone is extensively metabolized in the liver by the cytochrome P-450 (CYP) 2D6 enzyme system. The main metabolic pathway is through hydroxylation of risperidone to 9-hydroxyrisperidone by CYP 2D6. Food does not affect the rate or extent of the drug’s absorption.6
Efficacy studies
In schizophrenia. Approval of the indication for pediatric schizophrenia was based on data from 2 short-term (6 and 8 weeks) randomized, double-blind, controlled trials involving a total of 416 patients age 13 to 17 who met DSM-IV-TR criteria for schizophrenia and were experiencing an acute episode at enrollment.7 In one study, patients received risperidone, 1 to 3 mg/d, 4 to 6 mg/d, or placebo. In the other study, dosages were 0.15 to 0.6 mg/d or 1.5 to 6 mg/d. Except for patients in the 0.15 to 0.6 mg group (who initially received 0.05 mg/d), most patients started risperidone at 0.5 mg/d. In both trials, starting dosages were titrated to the target range in approximately 7 days.
Outcomes were measured as changes in total Positive and Negative Syndrome Scale (PANSS) and Personal and Social Performance (PSP) scale scores. The multi-item PANSS inventory measures positive and negative schizophrenia symptoms, disorganized thoughts, uncontrolled hostility/excitement, and anxiety/depression. The PSP gauges personal and social functioning in socially useful activities (work and study), personal and social relationships, self-care, and disturbing/ aggressive behaviors.
Risperidone, 1 to 6 mg/d, improved schizophrenia symptoms significantly more than placebo, as measured by PANSS scores. Doses >3 mg/d did not show greater efficacy than lower doses, as evaluated by PANSS and PSP scores.
Adverse reactions experienced by >5% of patients treated with risperidone included somnolence, parkinsonism, tremor, dystonia, dizziness, akathisia, increased salivation, and anxiety.7
In bipolar I disorder. Risperidone’s efficacy for short-term treatment of mania in children and adolescents was demonstrated in a 3-week, randomized, double-blind, placebo-controlled, multi-center study of 169 patients age 10 to 17 who were experiencing a manic or mixed episode of bipolar I disorder.7 Patients were randomly assigned to risperidone, 0.5 to 2.5 mg/d or 3 to 6 mg/d, or placebo. All patients were started at 0.5 mg/d and this dose was titrated to the target dosage range in 7 days.
Risperidone, 0.5 to 6 mg/d, significantly decreased the total Young Mania Rating Scale score—a measure of the severity of elevated mood, increased motor activity energy, sexual interest, sleep, irritability, speech (rate/amount), language (thought disorder, content, disruptive), aggressive behavior, appearance, and insight. No evidence of increased efficacy was observed at doses >2.5 mg/d. In this trial, symptoms reported by >5% of patients included fatigue, dizziness, dystonia, parkinsonism, akathisia, abdominal pain, dyspepsia, nausea, vomiting, and diarrhea.7
Pediatric dosing. Based on these studies, the recommended starting dose for children and adolescents is 0.5 mg/d, with titration in 0.5-to 1-mg increments to targets of:
Table 2
Recommended dosing of risperidone
for pediatric schizophrenia and bipolar mania
| Indication | Initial dose | Titration | Target dose | Effective dose range |
|---|---|---|---|---|
| Schizophrenia, adolescents age 13 to 17 | 0.5 mg/d | 0.5 to 1 mg/d | 3 mg/d | 1 to 6 mg/d |
| Bipolar mania, children and adolescents age 10 to 17 | 0.5 mg/d | 0.5 to 1 mg/d | 2.5 mg/d | 0.5 to 6 mg/d |
| Source:Reference 7 | ||||
Tolerability studies
In long-term studies, the most commonly reported adverse events associated with risperidone in children and adolescents have been rhinitis, abdominal pain, increased saliva, body pain, gynecomastia, and weight increase.8 Specific adverse effects that pose long-term concerns are:
- tardive dyskinesia (TD)
- weight gain
- increased prolactin levels
Tardive dyskinesia. In clinical trials that included 1,885 children and adolescents with autistic disorder or other psychiatric disorders treated with risperidone, 2 patients (0.1%) were reported to have TD, which resolved when risperidone was discontinued.7 To monitor for TD, administer the Abnormal Involuntary Movement Scale at baseline and every 6 months while using risperidone in pediatric patients.
Weight gain. In long-term, open-label trials, patients with autistic or other psychiatric disorders gained an average 7.5
kg after 12 months of risperidone treatment. Most of the weight gain occurred in the first 6 months.9 Expected normal weight gain in children is 3 to 3.5 kg/year adjusted for age, based on Centers for Disease Control and Prevention normative data.
Follow the American Diabetes Association guidelines10 for monitoring metabolic parameters during antipsychotic
treatment, and intervene if clinically significant weight gain occurs.
In a 16-week, placebo-controlled study,11 metformin reversed weight gain associated with SGAs in children and adolescents. Metformin’s potential side effects include hypoglycemia, diarrhea, nausea/vomiting, and (rarely) lactic acidosis, but no adverse events were attributed to metformin.
Increased prolactin. As in adults, risperidone elevates serum prolactin in children and adolescents. All pediatric risperidone trials—of autism,2 disruptive behavior disorders in children with subaverage intelligence,9 schizophrenia,7 and bipolar mania—have shown increased serum prolactin. Risperidone’s long-term effects on growth and sexual maturation have not been fully evaluated, but hyperprolactinemia may inhibit reproductive function.
Findling et al12 analyzed data from 5 clinical trials (total 700 patients) in which children and adolescents age 5 to 15 years with subaverage IQs and conduct or other disruptive behavior disorders received risperidone for up to 55 weeks. Mean prolactin levels rose from 7.8 ng/mL
at baseline to 29.4 ng/mL at weeks 4 to 7, then progressively decreased to 16.1 ng/mL at weeks 40 to 48 (n=358) and 13.0 ng/mL at weeks 52 to 55 (n=42). Girls returned to a mean value within the normal range (≤30 ng/mL) by weeks 8 to 12, and boys were close to normal values (≤18 ng/mL) by weeks 16 to 24.
The researchers concluded that serum prolactin levels in children tend to rise and peak within the first 1 to 2 months of risperidone treatment and then steadily decline to values within or very close to normal range by 3 to 5 months.
The biological significance of chronic, mild prolactin elevations is unknown.13 Children entering puberty appear to be at highest risk for elevated prolactin and clinical symptoms while treated with risperidone.14 Therefore, ask all adolescents treated with risperidone about increases in breast size and galactorrhea. Switch those who develop these symptoms to an SGA that does not increase serum prolactin.
Contraindications. Risperidone is contraindicated in patients with a known hypersensitivity to the drug.
- Risperdal prescribing information. www.risperdal.com/risperdal/shared/pi/risperdal.pdf.
Drug brand names
- Lithium • Eskalith, Lithobid
- Risperidone • Risperdal
- Metformin • Glucophage, Fortamet
- Valproate • Depakote
Disclosures
Dr. Kowatch receives research support from Bristol-Meyers Squibb, Stanley Research Foundation, National Institute of Mental Health, and National Institute of Child Health and Human Development. He is a consultant for Creative Educational Concepts, Child and Adolescent Bipolar Foundation, Abbott Laboratories, and sanofi-aventis, and a speaker for Abbott Laboratories and AstraZeneca.
1. Aman MG, De Smedt G, Derivan A, et al. Double-blind, placebo-controlled study of risperidone for the treatment of disruptive behaviors in children with subaverage intelligence. Am J Psychiatry 2002;159:1337-46.
2. McCracken JT, McGough J, Shah B, et al. Risperidone in children with autism and serious behavioral problems. N Engl J Med 2002;347(5):314-21.
3. Findling RL, McNamara NK, Branicky LA, et al. A double-blind pilot study of risperidone in the treatment of conduct disorder. J Am Acad Child Adolesc Psychiatry 2000;39(4):509-16.
4. Sikich L, Hamer R, Malekpour AH, et al. Double-blind trial comparing risperidone, olanzapine, and haloperidol in the treatment of psychotic children and adolescents. Paper presented at: Society of Biological Psychiatry Annual Meeting; May 16-18, 2002; Philadelphia, PA.
5. Frazier JA, Meyer MC, Biederman J, et al. Risperidone treatment for juvenile bipolar disorder: a retrospective chart review. J Am Acad Child Adolesc Psychiatry 1999;38(8):960-5.
6. Aman MG, Vinks AA, Remmerie B, et al. Plasma pharmacokinetic characteristics of risperidone and their relationship to saliva concentrations in children with psychiatric or neurodevelopmental disorders. Clin Ther 2007;29(7):1476-86.
7. Risperdal [package insert]. Titusville, NJ: Janssen, L.P; 2007.
8. Reyes MR, Olah R, Csaba K, et al. Long-term safety and efficacy of risperidone in children with disruptive behaviour disorders. Results of a 2-year extension study. Eur Child Adolesc Psychiatry 2006;15(2):97-104.
9. Croonenberghs J, Fegert JM, Findling RL, et al. Risperidone in children with disruptive behavior disorders and subaverage intelligence: a 1-year, open-label study of 504 patients. J Am Acad Child Adolesc Psychiatry 2005;44(1):64-72.
10. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. J Clin Psychiatry 2004;65(2):267-72.
11. Klein DJ, Cottingham EM, Sorter M, et al. A randomized, double-blind, placebo-controlled trial of metformin treatment of weight gain associated with initiation of atypical antipsychotic therapy in children and adolescents. Am J Psychiatry 2006;163(12):2072-9.
12. Findling RL, Kusumakar V, Daneman D, et al. Prolactin levels during long-term risperidone treatment in children and adolescents. J Clin Psychiatry 2003;64(11):1362-9.
13. Staller J. The effect of long-term antipsychotic treatment on prolactin. J Child Adolesc Psychopharmacol 2006;16(3):317-26.
14. Holzer L, Eap CB. Risperidone-induced symptomatic hyperprolactinaemia in adolescents. J Clin Psychopharmacol 2006;26(2):167-71.
Dr. Kowatch is professor of psychiatry and pediatrics at Cincinnati Children’s Hospital Medical Center and a Section Editor for Current Psychiatry.
1. Aman MG, De Smedt G, Derivan A, et al. Double-blind, placebo-controlled study of risperidone for the treatment of disruptive behaviors in children with subaverage intelligence. Am J Psychiatry 2002;159:1337-46.
2. McCracken JT, McGough J, Shah B, et al. Risperidone in children with autism and serious behavioral problems. N Engl J Med 2002;347(5):314-21.
3. Findling RL, McNamara NK, Branicky LA, et al. A double-blind pilot study of risperidone in the treatment of conduct disorder. J Am Acad Child Adolesc Psychiatry 2000;39(4):509-16.
4. Sikich L, Hamer R, Malekpour AH, et al. Double-blind trial comparing risperidone, olanzapine, and haloperidol in the treatment of psychotic children and adolescents. Paper presented at: Society of Biological Psychiatry Annual Meeting; May 16-18, 2002; Philadelphia, PA.
5. Frazier JA, Meyer MC, Biederman J, et al. Risperidone treatment for juvenile bipolar disorder: a retrospective chart review. J Am Acad Child Adolesc Psychiatry 1999;38(8):960-5.
6. Aman MG, Vinks AA, Remmerie B, et al. Plasma pharmacokinetic characteristics of risperidone and their relationship to saliva concentrations in children with psychiatric or neurodevelopmental disorders. Clin Ther 2007;29(7):1476-86.
7. Risperdal [package insert]. Titusville, NJ: Janssen, L.P; 2007.
8. Reyes MR, Olah R, Csaba K, et al. Long-term safety and efficacy of risperidone in children with disruptive behaviour disorders. Results of a 2-year extension study. Eur Child Adolesc Psychiatry 2006;15(2):97-104.
9. Croonenberghs J, Fegert JM, Findling RL, et al. Risperidone in children with disruptive behavior disorders and subaverage intelligence: a 1-year, open-label study of 504 patients. J Am Acad Child Adolesc Psychiatry 2005;44(1):64-72.
10. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. J Clin Psychiatry 2004;65(2):267-72.
11. Klein DJ, Cottingham EM, Sorter M, et al. A randomized, double-blind, placebo-controlled trial of metformin treatment of weight gain associated with initiation of atypical antipsychotic therapy in children and adolescents. Am J Psychiatry 2006;163(12):2072-9.
12. Findling RL, Kusumakar V, Daneman D, et al. Prolactin levels during long-term risperidone treatment in children and adolescents. J Clin Psychiatry 2003;64(11):1362-9.
13. Staller J. The effect of long-term antipsychotic treatment on prolactin. J Child Adolesc Psychopharmacol 2006;16(3):317-26.
14. Holzer L, Eap CB. Risperidone-induced symptomatic hyperprolactinaemia in adolescents. J Clin Psychopharmacol 2006;26(2):167-71.
Dr. Kowatch is professor of psychiatry and pediatrics at Cincinnati Children’s Hospital Medical Center and a Section Editor for Current Psychiatry.
IM aripiprazole for acute agitation
In recent clinical trials, a new intramuscular (IM) form of the second-generation antipsychotic (SGA) aripiprazole has controlled agitation in adults with schizophrenia or bipolar mania without causing significant side effects (Table 1).1-3
Table 1
IM aripiprazole: Fast facts
| Brand name: Abilify |
| Class: Second-generation antipsychotic |
| Indication: Acute agitation associated with schizophrenia or type I bipolar disorder (mixed or manic episodes) |
| Manufacturer: Otsuka America Pharmaceutical (marketed in collaboration with Bristol-Myers Squibb) |
| Dosing forms: 1.3-mL vial of clear, aqueous solution containing 9.75 mg of active drug |
| Recommended dosage: 9.75 mg every 2 hours as needed; do not exceed 30 mg across 24 hours |
Clinical implications
Rapid intervention is critical to protecting the patient and caregivers when violent and/or destructive behavior accompanies agitation. IM aripiprazole substantially reduced agitation within 45 to 60 minutes of dosing in randomized, double-blind, placebo-controlled studies.1-3
How it works
Whereas other SGAs have relatively little effect on D2 (dopamine) receptors and relatively high 5-HT2A (serotonin) receptor affinities, aripiprazole appears to work via partial D2 receptor agonism. The medication:
- blocks D2 receptors in brain regions where dopamine is overactive in schizophrenia, such as the mesolimbic pathway. This produces an antipsychotic effect.
- maintains or moderately boosts dopamine activity as needed in regions such as the nigrostriatal pathway. This reduces the risk of motor side effects and might improve negative and cognitive schizophrenia symptoms.
Aripiprazole is a partial 5-HT1A receptor agonist and—like other SGAs—a 5-HT2A receptor antagonist. These receptor subtypes have been implicated in antipsychotic action. In particular, partial 5-HT1A receptor agonism is thought to help:
- reduce anxiety
- lessen depressive, negative, and cognitive symptoms
- decrease extrapyramidal symptom (EPS) liability.4
Aripiprazole also has moderate affinity for histaminic and alpha-adrenergic receptors and no appreciable effect on cholinergic muscarinic receptors.5-8
Pharmacokinetics
IM aripiprazole’s activity has been attributed to its parent drug and to a lesser extent its major metabolite, dehydroaripiprazole. Both moieties act on D2 receptors, and dehydroaripiprazole accounts for 40% of the parent drug’s exposure in plasma.
Mean elimination half-lives for aripiprazole and dehydroaripiprazole are approximately 75 and 94 hours, respectively, allowing for daily administration. Both active moieties reach steady-state concentration within 14 days of dosing. Because aripiprazole accumulation is predictable after a single dose and its pharmacokinetics are dose-proportional at steady state, higher doses are not always more effective and could increase side-effect risk.
Aripiprazole is metabolized mainly through the liver by cytochrome P-450 2D6 and 3A4 isozymes. This requires careful monitoring when prescribing the drug concomitantly with:
- agents that induce CYP 3A4—such as carbamazepine—which could diminish aripiprazole’s effectiveness by increasing its clearance and decreasing aripiprazole blood levels
- CYP 3A4 inhibitors such as ketoconazole or CYP 2D6 inhibitors such as quinidine, fluoxetine, or paroxetine, which can inhibit aripiprazole elimination9 and increase the risk of adverse events.
Similarly, aripiprazole could be efficacious at lower-than-therapeutic dosages when taken with medications that raise aripiprazole blood levels.
Efficacy
In 3 randomized, placebo-controlled, double-blind trials, IM aripiprazole reduced agitation in inpatients with schizophrenia, schizoaffective disorder, or type I bipolar disorder with manic or mixed episodes, with or without psychotic features.
In each trial, IM aripiprazole was as effective as comparable dosages of haloperidol or lorazepam IM preparations. Patients were moderately to severely agitated based on Positive and Negative Syndrome Scale Excited Component (PANSS-EC) assessments, which gauged impulse control, tension, hostility, uncooperativeness, and excitement.
Patients could receive up to 3 injections within 24 hours but had to wait ≥2 hours for the second injection so that investigators could record follow-up PANSS-EC scores. Clinical Global Impression of Improvement (CGI-I) scale scores were a key secondary measure.
Examination of population subsets in the studies showed no differential response based on age, race, or gender.
Tran-Johnson et al1followed 357 patients with schizophreniform disorders, schizophrenia, or schizoaffective disorders.
Two hours after initial injection, mean PANSS-EC scores decreased approximately 3 points with placebo and 4 to 6.5 points among patients receiving 7.5 mg of IM haloperidol or 5.25, 9.75, or 15 mg of IM aripiprazole. Agitation improved significantly after 45 minutes among patients receiving 9.75 mg of IM aripiprazole, compared with 105 minutes in the IM haloperidol group.
Prevalence of EPS across 24 hours with haloperidol was 19.3%, compared with an average 5.2% among all IM aripiprazole groups, suggesting that IM aripiprazole carries a substantially lower EPS risk.
Andrezina et al2followed 448 patients with schizophrenia or schizoaffective disorder. Two hours after injection, patients in both treatment groups showed much greater improvement compared with placebo based on mean PANSS-EC score decreases and mean CGI-I scores (Table 2).
Prevalence of EPS was 1.7% with IM aripiprazole, 2.3% with placebo, and 12.6% with IM haloperidol. Prevalence of EPS-related adverse events was 0% with IM aripiprazole, 1.6% with placebo, and 16.5% with IM haloperidol.
Zimbroff et al3 gave IM aripiprazole, 9.75 or 15 mg; IM lorazepam, 2 mg; or placebo to 301 patients with type I bipolar disorder with manic or mixed episodes.
Two hours later, all 3 treatment groups showed significantly greater agitation improvement as shown by PANSS-EC scores, compared with placebo (Table 3).
Across 2 hours, oversedation—defined as an Agitation-Calmness Evaluation Scale score of 8 or 9—was less prevalent among patients receiving IM aripiprazole, 9.75 mg (6.7%), compared with IM aripiprazole, 15 mg (17.3%), or IM lorazepam (19.1%).
Table 2
Agitation, symptom improvement 2 hours after aripiprazole or haloperidol injection
| Assessment scale | IM aripiprazole, 9.75 mg | IM haloperidol, 6.5 mg | Placebo |
|---|---|---|---|
| PANSS-EC mean score decrease (P | 7.27 | 7.75 | 4.78 |
| CGI-I mean score (P | 2.42 | 2.37 | 3.10 |
| PANSS-EC: Positive and Negative Syndrome Scale Excited Component; CGI-I: Clinical Global Impression of Improvement | |||
| Source: Adapted from reference 2 | |||
Table 3
Agitation improvement 2 hours after aripiprazole or lorazepam injection
| IM preparation | PANSS-EC mean score decrease |
|---|---|
| Aripiprazole, 9.75 mg | 8.7 |
| Aripiprazole, 15 mg | 8.7 |
| Lorazepam, 2 mg | 9.6 |
| Placebo | 5.6 |
| PANSS-EC: Positive and Negative Syndrome Scale Excited Component | |
| Source: Adapted from reference 3 | |
Safety and tolerability
IM aripiprazole was well tolerated in clinical trials and did not cause excessive sedation10 or injection-site pain.1-3
Most frequently reported adverse events were headache (12% with IM aripiprazole vs 7% with placebo), nausea (9% vs 3%), dizziness (8% vs 5%), and somnolence (7% vs 4%).
Prevalence of akathisia or dystonia among all IM aripiprazole groups in the 3 trials was 2% and
No clinically significant ECG abnormalities were reported among the aripiprazole groups.1-3,11
Dosing
Start at 9.75 mg every 2 hours as needed, but do not exceed 30 mg/d across 24 hours. Controlled studies have not evaluated efficacy or safety of more-frequent injections or safety of total daily doses >30 mg.
Try a lower dose (5.25 mg) for patients who are elderly or small in body size or have reacted adversely to other antipsychotics. If necessary, give another 5.25 mg in 2 hours. If the patient is still agitated 2 hours after the second dose, consider a third dose at 9.75 mg. Again, do not exceed 30 mg over 24 hours. Obtain lower doses by administering a portion of the vial.
Transitioning to oral Tx
If IM aripiprazole reduces psychotic symptoms as well as acute behaviors, switch the patient to oral aripiprazole once the risk of violence has diminished.12 If psychosis does not improve with IM aripiprazole, weigh clinical factors before choosing an oral antipsychotic.
Only one controlled trial12 has examined transitioning from IM aripiprazole to an oral antipsychotic. In the randomized study, 448 patients receiving IM aripiprazole, 9.75 mg; IM haloperidol, 6.5 mg; or placebo for agitation secondary to schizophrenia or schizoaffective disorder were transitioned to the oral preparation of the drug they were receiving: aripiprazole, 10 to 15 mg/d, or haloperidol, 7.5 to 10 mg/d. Placebo-group patients transitioned to oral aripiprazole.
Over 4 days, both oral treatments provided continued efficacy, suggesting that:
- patients receiving IM aripiprazole can be conveniently switched to the oral preparation
- IM and oral aripiprazole are equally safe.
Oral and IM aripiprazole doses are equivalent and the pharmacokinetics are comparable. For example, a patient receiving 20 mg/d of IM aripiprazole can take 20 mg of oral aripiprazole within 24 hours of the last injection.
Related resources
- Allen MH, Currier GW, Carpenter D, et al. Expert consensus guidelines: treatment of behavioral emergencies. J Psychiatr Pract 2005;11(suppl 1):5-108.
- Currier GW, Citrome LL, Zimbroff DL, et al. Intramuscular aripiprazole in the control of agitation. J Psychiatr Pract 2007;13:159-69.
Drug brand names
- Aripiprazole IM • Abilify
- Carbamazepine • Tegretol, others
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Ketoconazole • Nizoral
- Lorazepam • Ativan
- Paroxetine • Paxil
- Quinidine • Quinaglute
Disclosure
Dr. Josiassen was principal investigator and Dr. Shaughnessy a co-investigator on a pre-approval clinical trial of IM aripiprazole. Both have conducted sponsor- and investigator-initiated studies for AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Janssen, Novartis Pharmaceuticals Corp., Organon, Otsuka America Pharmaceuticals, Otsuka Maryland Research Institute, Pfizer, and Yamanuchi.
Acknowledgments
This article was supported in part by the Arthur P. Noyes Research Foundation.
The authors thank Margit Kacso, Cara Bendler, Dawn Filmyer, and Jon Weinstein for their technical and editorial assistance in preparing this article.
1. Tran-Johnson TK, Sack DA, Marcus RN, et al. Efficacy and safety of intramuscular aripiprazole inpatients with acute agitation: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 2007;68:111-9.
2. Andrezina R, Josiassen RC, Marcus R, et al. Intramuscular aripiprazole for the treatment of acute agitation in patients with schizophrenia or schizoaffective disorder: a double-blind, placebo-controlled comparison with intramuscular haloperidol. Psychopharmacology (Berl) 2006;188:281-92.
3. Zimbroff DL, Marcus RN, Manos G, et al. Management of acute agitation in patients with bipolar disorder: efficacy and safety of intramuscular aripiprazole. J Clin Psychopharmacol 2007;27:171-6.
4. Stark AD, Jordan S, Allers KA, et al. Interaction of the novel antipsychotic aripiprazole with HT1A and 5-HT2A receptors: functional receptor-binding and in vivo electrophysiological studies. Psychopharmacol 2007;190:373-82.
5. Burris KD, Molski TF, Xu C, et al. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J Pharmacol Exp Ther 2002;302:381-9.
6. Shapiro DA, Renock S, Arrington E, et al. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 2003;28:1400-11.
7. Jordan S, Koprivica V, Dunn R, et al. In vivo effects of aripiprazole on cortical and striatal dopaminergic and serotonergic function. Eur J Pharmacol 2004;483:45-53.
8. Hirose T, Uwahodo Y, Yamada S, et al. Mechanism of action of aripiprazole predicts clinical efficacy and a favourable side-effect profile. J Psychopharmacol 2004;18:375-83.
9. Physicians’ desk reference 61st ed. Montvale, NJ: Thomson PDR; 2007.
10. Currier GW, Crandall D, Archibald D, et al. Intramuscular (IM) aripiprazole controls agitation in patients with schizophrenia or bipolar I disorder without excessive sedation. Paper presented at: Annual Meeting of the American Psychiatric Association; May 20-25, 2006; Toronto, Canada.
11. Currier GW, Citrome LL, Zimbroff DL, et al. Intramuscular aripiprazole in the control of agitation. J Psychiatr Pract 2007;13:159-69.
12. Daniel DG, Currier GW, Zimbroff DL, et al. Efficacy and safety of oral aripiprazole compared with haloperidol in patients transitioning from acute treatment with intramuscular formulations. J Psychiatr Pract 2007;13:170-7.
Dr. Josiassen is chief scientific officer, Arthur P. Noyes Research Foundation, Norristown, PA, and adjunct professor of psychiatry, University of Pennsylvania, Philadelphia.
Dr. Shaughnessy is associate professor of psychiatry, Drexel University College of Medicine, Philadelphia, and medical consultant, Arthur P. Noyes Research Foundation.
In recent clinical trials, a new intramuscular (IM) form of the second-generation antipsychotic (SGA) aripiprazole has controlled agitation in adults with schizophrenia or bipolar mania without causing significant side effects (Table 1).1-3
Table 1
IM aripiprazole: Fast facts
| Brand name: Abilify |
| Class: Second-generation antipsychotic |
| Indication: Acute agitation associated with schizophrenia or type I bipolar disorder (mixed or manic episodes) |
| Manufacturer: Otsuka America Pharmaceutical (marketed in collaboration with Bristol-Myers Squibb) |
| Dosing forms: 1.3-mL vial of clear, aqueous solution containing 9.75 mg of active drug |
| Recommended dosage: 9.75 mg every 2 hours as needed; do not exceed 30 mg across 24 hours |
Clinical implications
Rapid intervention is critical to protecting the patient and caregivers when violent and/or destructive behavior accompanies agitation. IM aripiprazole substantially reduced agitation within 45 to 60 minutes of dosing in randomized, double-blind, placebo-controlled studies.1-3
How it works
Whereas other SGAs have relatively little effect on D2 (dopamine) receptors and relatively high 5-HT2A (serotonin) receptor affinities, aripiprazole appears to work via partial D2 receptor agonism. The medication:
- blocks D2 receptors in brain regions where dopamine is overactive in schizophrenia, such as the mesolimbic pathway. This produces an antipsychotic effect.
- maintains or moderately boosts dopamine activity as needed in regions such as the nigrostriatal pathway. This reduces the risk of motor side effects and might improve negative and cognitive schizophrenia symptoms.
Aripiprazole is a partial 5-HT1A receptor agonist and—like other SGAs—a 5-HT2A receptor antagonist. These receptor subtypes have been implicated in antipsychotic action. In particular, partial 5-HT1A receptor agonism is thought to help:
- reduce anxiety
- lessen depressive, negative, and cognitive symptoms
- decrease extrapyramidal symptom (EPS) liability.4
Aripiprazole also has moderate affinity for histaminic and alpha-adrenergic receptors and no appreciable effect on cholinergic muscarinic receptors.5-8
Pharmacokinetics
IM aripiprazole’s activity has been attributed to its parent drug and to a lesser extent its major metabolite, dehydroaripiprazole. Both moieties act on D2 receptors, and dehydroaripiprazole accounts for 40% of the parent drug’s exposure in plasma.
Mean elimination half-lives for aripiprazole and dehydroaripiprazole are approximately 75 and 94 hours, respectively, allowing for daily administration. Both active moieties reach steady-state concentration within 14 days of dosing. Because aripiprazole accumulation is predictable after a single dose and its pharmacokinetics are dose-proportional at steady state, higher doses are not always more effective and could increase side-effect risk.
Aripiprazole is metabolized mainly through the liver by cytochrome P-450 2D6 and 3A4 isozymes. This requires careful monitoring when prescribing the drug concomitantly with:
- agents that induce CYP 3A4—such as carbamazepine—which could diminish aripiprazole’s effectiveness by increasing its clearance and decreasing aripiprazole blood levels
- CYP 3A4 inhibitors such as ketoconazole or CYP 2D6 inhibitors such as quinidine, fluoxetine, or paroxetine, which can inhibit aripiprazole elimination9 and increase the risk of adverse events.
Similarly, aripiprazole could be efficacious at lower-than-therapeutic dosages when taken with medications that raise aripiprazole blood levels.
Efficacy
In 3 randomized, placebo-controlled, double-blind trials, IM aripiprazole reduced agitation in inpatients with schizophrenia, schizoaffective disorder, or type I bipolar disorder with manic or mixed episodes, with or without psychotic features.
In each trial, IM aripiprazole was as effective as comparable dosages of haloperidol or lorazepam IM preparations. Patients were moderately to severely agitated based on Positive and Negative Syndrome Scale Excited Component (PANSS-EC) assessments, which gauged impulse control, tension, hostility, uncooperativeness, and excitement.
Patients could receive up to 3 injections within 24 hours but had to wait ≥2 hours for the second injection so that investigators could record follow-up PANSS-EC scores. Clinical Global Impression of Improvement (CGI-I) scale scores were a key secondary measure.
Examination of population subsets in the studies showed no differential response based on age, race, or gender.
Tran-Johnson et al1followed 357 patients with schizophreniform disorders, schizophrenia, or schizoaffective disorders.
Two hours after initial injection, mean PANSS-EC scores decreased approximately 3 points with placebo and 4 to 6.5 points among patients receiving 7.5 mg of IM haloperidol or 5.25, 9.75, or 15 mg of IM aripiprazole. Agitation improved significantly after 45 minutes among patients receiving 9.75 mg of IM aripiprazole, compared with 105 minutes in the IM haloperidol group.
Prevalence of EPS across 24 hours with haloperidol was 19.3%, compared with an average 5.2% among all IM aripiprazole groups, suggesting that IM aripiprazole carries a substantially lower EPS risk.
Andrezina et al2followed 448 patients with schizophrenia or schizoaffective disorder. Two hours after injection, patients in both treatment groups showed much greater improvement compared with placebo based on mean PANSS-EC score decreases and mean CGI-I scores (Table 2).
Prevalence of EPS was 1.7% with IM aripiprazole, 2.3% with placebo, and 12.6% with IM haloperidol. Prevalence of EPS-related adverse events was 0% with IM aripiprazole, 1.6% with placebo, and 16.5% with IM haloperidol.
Zimbroff et al3 gave IM aripiprazole, 9.75 or 15 mg; IM lorazepam, 2 mg; or placebo to 301 patients with type I bipolar disorder with manic or mixed episodes.
Two hours later, all 3 treatment groups showed significantly greater agitation improvement as shown by PANSS-EC scores, compared with placebo (Table 3).
Across 2 hours, oversedation—defined as an Agitation-Calmness Evaluation Scale score of 8 or 9—was less prevalent among patients receiving IM aripiprazole, 9.75 mg (6.7%), compared with IM aripiprazole, 15 mg (17.3%), or IM lorazepam (19.1%).
Table 2
Agitation, symptom improvement 2 hours after aripiprazole or haloperidol injection
| Assessment scale | IM aripiprazole, 9.75 mg | IM haloperidol, 6.5 mg | Placebo |
|---|---|---|---|
| PANSS-EC mean score decrease (P | 7.27 | 7.75 | 4.78 |
| CGI-I mean score (P | 2.42 | 2.37 | 3.10 |
| PANSS-EC: Positive and Negative Syndrome Scale Excited Component; CGI-I: Clinical Global Impression of Improvement | |||
| Source: Adapted from reference 2 | |||
Table 3
Agitation improvement 2 hours after aripiprazole or lorazepam injection
| IM preparation | PANSS-EC mean score decrease |
|---|---|
| Aripiprazole, 9.75 mg | 8.7 |
| Aripiprazole, 15 mg | 8.7 |
| Lorazepam, 2 mg | 9.6 |
| Placebo | 5.6 |
| PANSS-EC: Positive and Negative Syndrome Scale Excited Component | |
| Source: Adapted from reference 3 | |
Safety and tolerability
IM aripiprazole was well tolerated in clinical trials and did not cause excessive sedation10 or injection-site pain.1-3
Most frequently reported adverse events were headache (12% with IM aripiprazole vs 7% with placebo), nausea (9% vs 3%), dizziness (8% vs 5%), and somnolence (7% vs 4%).
Prevalence of akathisia or dystonia among all IM aripiprazole groups in the 3 trials was 2% and
No clinically significant ECG abnormalities were reported among the aripiprazole groups.1-3,11
Dosing
Start at 9.75 mg every 2 hours as needed, but do not exceed 30 mg/d across 24 hours. Controlled studies have not evaluated efficacy or safety of more-frequent injections or safety of total daily doses >30 mg.
Try a lower dose (5.25 mg) for patients who are elderly or small in body size or have reacted adversely to other antipsychotics. If necessary, give another 5.25 mg in 2 hours. If the patient is still agitated 2 hours after the second dose, consider a third dose at 9.75 mg. Again, do not exceed 30 mg over 24 hours. Obtain lower doses by administering a portion of the vial.
Transitioning to oral Tx
If IM aripiprazole reduces psychotic symptoms as well as acute behaviors, switch the patient to oral aripiprazole once the risk of violence has diminished.12 If psychosis does not improve with IM aripiprazole, weigh clinical factors before choosing an oral antipsychotic.
Only one controlled trial12 has examined transitioning from IM aripiprazole to an oral antipsychotic. In the randomized study, 448 patients receiving IM aripiprazole, 9.75 mg; IM haloperidol, 6.5 mg; or placebo for agitation secondary to schizophrenia or schizoaffective disorder were transitioned to the oral preparation of the drug they were receiving: aripiprazole, 10 to 15 mg/d, or haloperidol, 7.5 to 10 mg/d. Placebo-group patients transitioned to oral aripiprazole.
Over 4 days, both oral treatments provided continued efficacy, suggesting that:
- patients receiving IM aripiprazole can be conveniently switched to the oral preparation
- IM and oral aripiprazole are equally safe.
Oral and IM aripiprazole doses are equivalent and the pharmacokinetics are comparable. For example, a patient receiving 20 mg/d of IM aripiprazole can take 20 mg of oral aripiprazole within 24 hours of the last injection.
Related resources
- Allen MH, Currier GW, Carpenter D, et al. Expert consensus guidelines: treatment of behavioral emergencies. J Psychiatr Pract 2005;11(suppl 1):5-108.
- Currier GW, Citrome LL, Zimbroff DL, et al. Intramuscular aripiprazole in the control of agitation. J Psychiatr Pract 2007;13:159-69.
Drug brand names
- Aripiprazole IM • Abilify
- Carbamazepine • Tegretol, others
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Ketoconazole • Nizoral
- Lorazepam • Ativan
- Paroxetine • Paxil
- Quinidine • Quinaglute
Disclosure
Dr. Josiassen was principal investigator and Dr. Shaughnessy a co-investigator on a pre-approval clinical trial of IM aripiprazole. Both have conducted sponsor- and investigator-initiated studies for AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Janssen, Novartis Pharmaceuticals Corp., Organon, Otsuka America Pharmaceuticals, Otsuka Maryland Research Institute, Pfizer, and Yamanuchi.
Acknowledgments
This article was supported in part by the Arthur P. Noyes Research Foundation.
The authors thank Margit Kacso, Cara Bendler, Dawn Filmyer, and Jon Weinstein for their technical and editorial assistance in preparing this article.
In recent clinical trials, a new intramuscular (IM) form of the second-generation antipsychotic (SGA) aripiprazole has controlled agitation in adults with schizophrenia or bipolar mania without causing significant side effects (Table 1).1-3
Table 1
IM aripiprazole: Fast facts
| Brand name: Abilify |
| Class: Second-generation antipsychotic |
| Indication: Acute agitation associated with schizophrenia or type I bipolar disorder (mixed or manic episodes) |
| Manufacturer: Otsuka America Pharmaceutical (marketed in collaboration with Bristol-Myers Squibb) |
| Dosing forms: 1.3-mL vial of clear, aqueous solution containing 9.75 mg of active drug |
| Recommended dosage: 9.75 mg every 2 hours as needed; do not exceed 30 mg across 24 hours |
Clinical implications
Rapid intervention is critical to protecting the patient and caregivers when violent and/or destructive behavior accompanies agitation. IM aripiprazole substantially reduced agitation within 45 to 60 minutes of dosing in randomized, double-blind, placebo-controlled studies.1-3
How it works
Whereas other SGAs have relatively little effect on D2 (dopamine) receptors and relatively high 5-HT2A (serotonin) receptor affinities, aripiprazole appears to work via partial D2 receptor agonism. The medication:
- blocks D2 receptors in brain regions where dopamine is overactive in schizophrenia, such as the mesolimbic pathway. This produces an antipsychotic effect.
- maintains or moderately boosts dopamine activity as needed in regions such as the nigrostriatal pathway. This reduces the risk of motor side effects and might improve negative and cognitive schizophrenia symptoms.
Aripiprazole is a partial 5-HT1A receptor agonist and—like other SGAs—a 5-HT2A receptor antagonist. These receptor subtypes have been implicated in antipsychotic action. In particular, partial 5-HT1A receptor agonism is thought to help:
- reduce anxiety
- lessen depressive, negative, and cognitive symptoms
- decrease extrapyramidal symptom (EPS) liability.4
Aripiprazole also has moderate affinity for histaminic and alpha-adrenergic receptors and no appreciable effect on cholinergic muscarinic receptors.5-8
Pharmacokinetics
IM aripiprazole’s activity has been attributed to its parent drug and to a lesser extent its major metabolite, dehydroaripiprazole. Both moieties act on D2 receptors, and dehydroaripiprazole accounts for 40% of the parent drug’s exposure in plasma.
Mean elimination half-lives for aripiprazole and dehydroaripiprazole are approximately 75 and 94 hours, respectively, allowing for daily administration. Both active moieties reach steady-state concentration within 14 days of dosing. Because aripiprazole accumulation is predictable after a single dose and its pharmacokinetics are dose-proportional at steady state, higher doses are not always more effective and could increase side-effect risk.
Aripiprazole is metabolized mainly through the liver by cytochrome P-450 2D6 and 3A4 isozymes. This requires careful monitoring when prescribing the drug concomitantly with:
- agents that induce CYP 3A4—such as carbamazepine—which could diminish aripiprazole’s effectiveness by increasing its clearance and decreasing aripiprazole blood levels
- CYP 3A4 inhibitors such as ketoconazole or CYP 2D6 inhibitors such as quinidine, fluoxetine, or paroxetine, which can inhibit aripiprazole elimination9 and increase the risk of adverse events.
Similarly, aripiprazole could be efficacious at lower-than-therapeutic dosages when taken with medications that raise aripiprazole blood levels.
Efficacy
In 3 randomized, placebo-controlled, double-blind trials, IM aripiprazole reduced agitation in inpatients with schizophrenia, schizoaffective disorder, or type I bipolar disorder with manic or mixed episodes, with or without psychotic features.
In each trial, IM aripiprazole was as effective as comparable dosages of haloperidol or lorazepam IM preparations. Patients were moderately to severely agitated based on Positive and Negative Syndrome Scale Excited Component (PANSS-EC) assessments, which gauged impulse control, tension, hostility, uncooperativeness, and excitement.
Patients could receive up to 3 injections within 24 hours but had to wait ≥2 hours for the second injection so that investigators could record follow-up PANSS-EC scores. Clinical Global Impression of Improvement (CGI-I) scale scores were a key secondary measure.
Examination of population subsets in the studies showed no differential response based on age, race, or gender.
Tran-Johnson et al1followed 357 patients with schizophreniform disorders, schizophrenia, or schizoaffective disorders.
Two hours after initial injection, mean PANSS-EC scores decreased approximately 3 points with placebo and 4 to 6.5 points among patients receiving 7.5 mg of IM haloperidol or 5.25, 9.75, or 15 mg of IM aripiprazole. Agitation improved significantly after 45 minutes among patients receiving 9.75 mg of IM aripiprazole, compared with 105 minutes in the IM haloperidol group.
Prevalence of EPS across 24 hours with haloperidol was 19.3%, compared with an average 5.2% among all IM aripiprazole groups, suggesting that IM aripiprazole carries a substantially lower EPS risk.
Andrezina et al2followed 448 patients with schizophrenia or schizoaffective disorder. Two hours after injection, patients in both treatment groups showed much greater improvement compared with placebo based on mean PANSS-EC score decreases and mean CGI-I scores (Table 2).
Prevalence of EPS was 1.7% with IM aripiprazole, 2.3% with placebo, and 12.6% with IM haloperidol. Prevalence of EPS-related adverse events was 0% with IM aripiprazole, 1.6% with placebo, and 16.5% with IM haloperidol.
Zimbroff et al3 gave IM aripiprazole, 9.75 or 15 mg; IM lorazepam, 2 mg; or placebo to 301 patients with type I bipolar disorder with manic or mixed episodes.
Two hours later, all 3 treatment groups showed significantly greater agitation improvement as shown by PANSS-EC scores, compared with placebo (Table 3).
Across 2 hours, oversedation—defined as an Agitation-Calmness Evaluation Scale score of 8 or 9—was less prevalent among patients receiving IM aripiprazole, 9.75 mg (6.7%), compared with IM aripiprazole, 15 mg (17.3%), or IM lorazepam (19.1%).
Table 2
Agitation, symptom improvement 2 hours after aripiprazole or haloperidol injection
| Assessment scale | IM aripiprazole, 9.75 mg | IM haloperidol, 6.5 mg | Placebo |
|---|---|---|---|
| PANSS-EC mean score decrease (P | 7.27 | 7.75 | 4.78 |
| CGI-I mean score (P | 2.42 | 2.37 | 3.10 |
| PANSS-EC: Positive and Negative Syndrome Scale Excited Component; CGI-I: Clinical Global Impression of Improvement | |||
| Source: Adapted from reference 2 | |||
Table 3
Agitation improvement 2 hours after aripiprazole or lorazepam injection
| IM preparation | PANSS-EC mean score decrease |
|---|---|
| Aripiprazole, 9.75 mg | 8.7 |
| Aripiprazole, 15 mg | 8.7 |
| Lorazepam, 2 mg | 9.6 |
| Placebo | 5.6 |
| PANSS-EC: Positive and Negative Syndrome Scale Excited Component | |
| Source: Adapted from reference 3 | |
Safety and tolerability
IM aripiprazole was well tolerated in clinical trials and did not cause excessive sedation10 or injection-site pain.1-3
Most frequently reported adverse events were headache (12% with IM aripiprazole vs 7% with placebo), nausea (9% vs 3%), dizziness (8% vs 5%), and somnolence (7% vs 4%).
Prevalence of akathisia or dystonia among all IM aripiprazole groups in the 3 trials was 2% and
No clinically significant ECG abnormalities were reported among the aripiprazole groups.1-3,11
Dosing
Start at 9.75 mg every 2 hours as needed, but do not exceed 30 mg/d across 24 hours. Controlled studies have not evaluated efficacy or safety of more-frequent injections or safety of total daily doses >30 mg.
Try a lower dose (5.25 mg) for patients who are elderly or small in body size or have reacted adversely to other antipsychotics. If necessary, give another 5.25 mg in 2 hours. If the patient is still agitated 2 hours after the second dose, consider a third dose at 9.75 mg. Again, do not exceed 30 mg over 24 hours. Obtain lower doses by administering a portion of the vial.
Transitioning to oral Tx
If IM aripiprazole reduces psychotic symptoms as well as acute behaviors, switch the patient to oral aripiprazole once the risk of violence has diminished.12 If psychosis does not improve with IM aripiprazole, weigh clinical factors before choosing an oral antipsychotic.
Only one controlled trial12 has examined transitioning from IM aripiprazole to an oral antipsychotic. In the randomized study, 448 patients receiving IM aripiprazole, 9.75 mg; IM haloperidol, 6.5 mg; or placebo for agitation secondary to schizophrenia or schizoaffective disorder were transitioned to the oral preparation of the drug they were receiving: aripiprazole, 10 to 15 mg/d, or haloperidol, 7.5 to 10 mg/d. Placebo-group patients transitioned to oral aripiprazole.
Over 4 days, both oral treatments provided continued efficacy, suggesting that:
- patients receiving IM aripiprazole can be conveniently switched to the oral preparation
- IM and oral aripiprazole are equally safe.
Oral and IM aripiprazole doses are equivalent and the pharmacokinetics are comparable. For example, a patient receiving 20 mg/d of IM aripiprazole can take 20 mg of oral aripiprazole within 24 hours of the last injection.
Related resources
- Allen MH, Currier GW, Carpenter D, et al. Expert consensus guidelines: treatment of behavioral emergencies. J Psychiatr Pract 2005;11(suppl 1):5-108.
- Currier GW, Citrome LL, Zimbroff DL, et al. Intramuscular aripiprazole in the control of agitation. J Psychiatr Pract 2007;13:159-69.
Drug brand names
- Aripiprazole IM • Abilify
- Carbamazepine • Tegretol, others
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Ketoconazole • Nizoral
- Lorazepam • Ativan
- Paroxetine • Paxil
- Quinidine • Quinaglute
Disclosure
Dr. Josiassen was principal investigator and Dr. Shaughnessy a co-investigator on a pre-approval clinical trial of IM aripiprazole. Both have conducted sponsor- and investigator-initiated studies for AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Janssen, Novartis Pharmaceuticals Corp., Organon, Otsuka America Pharmaceuticals, Otsuka Maryland Research Institute, Pfizer, and Yamanuchi.
Acknowledgments
This article was supported in part by the Arthur P. Noyes Research Foundation.
The authors thank Margit Kacso, Cara Bendler, Dawn Filmyer, and Jon Weinstein for their technical and editorial assistance in preparing this article.
1. Tran-Johnson TK, Sack DA, Marcus RN, et al. Efficacy and safety of intramuscular aripiprazole inpatients with acute agitation: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 2007;68:111-9.
2. Andrezina R, Josiassen RC, Marcus R, et al. Intramuscular aripiprazole for the treatment of acute agitation in patients with schizophrenia or schizoaffective disorder: a double-blind, placebo-controlled comparison with intramuscular haloperidol. Psychopharmacology (Berl) 2006;188:281-92.
3. Zimbroff DL, Marcus RN, Manos G, et al. Management of acute agitation in patients with bipolar disorder: efficacy and safety of intramuscular aripiprazole. J Clin Psychopharmacol 2007;27:171-6.
4. Stark AD, Jordan S, Allers KA, et al. Interaction of the novel antipsychotic aripiprazole with HT1A and 5-HT2A receptors: functional receptor-binding and in vivo electrophysiological studies. Psychopharmacol 2007;190:373-82.
5. Burris KD, Molski TF, Xu C, et al. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J Pharmacol Exp Ther 2002;302:381-9.
6. Shapiro DA, Renock S, Arrington E, et al. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 2003;28:1400-11.
7. Jordan S, Koprivica V, Dunn R, et al. In vivo effects of aripiprazole on cortical and striatal dopaminergic and serotonergic function. Eur J Pharmacol 2004;483:45-53.
8. Hirose T, Uwahodo Y, Yamada S, et al. Mechanism of action of aripiprazole predicts clinical efficacy and a favourable side-effect profile. J Psychopharmacol 2004;18:375-83.
9. Physicians’ desk reference 61st ed. Montvale, NJ: Thomson PDR; 2007.
10. Currier GW, Crandall D, Archibald D, et al. Intramuscular (IM) aripiprazole controls agitation in patients with schizophrenia or bipolar I disorder without excessive sedation. Paper presented at: Annual Meeting of the American Psychiatric Association; May 20-25, 2006; Toronto, Canada.
11. Currier GW, Citrome LL, Zimbroff DL, et al. Intramuscular aripiprazole in the control of agitation. J Psychiatr Pract 2007;13:159-69.
12. Daniel DG, Currier GW, Zimbroff DL, et al. Efficacy and safety of oral aripiprazole compared with haloperidol in patients transitioning from acute treatment with intramuscular formulations. J Psychiatr Pract 2007;13:170-7.
Dr. Josiassen is chief scientific officer, Arthur P. Noyes Research Foundation, Norristown, PA, and adjunct professor of psychiatry, University of Pennsylvania, Philadelphia.
Dr. Shaughnessy is associate professor of psychiatry, Drexel University College of Medicine, Philadelphia, and medical consultant, Arthur P. Noyes Research Foundation.
1. Tran-Johnson TK, Sack DA, Marcus RN, et al. Efficacy and safety of intramuscular aripiprazole inpatients with acute agitation: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 2007;68:111-9.
2. Andrezina R, Josiassen RC, Marcus R, et al. Intramuscular aripiprazole for the treatment of acute agitation in patients with schizophrenia or schizoaffective disorder: a double-blind, placebo-controlled comparison with intramuscular haloperidol. Psychopharmacology (Berl) 2006;188:281-92.
3. Zimbroff DL, Marcus RN, Manos G, et al. Management of acute agitation in patients with bipolar disorder: efficacy and safety of intramuscular aripiprazole. J Clin Psychopharmacol 2007;27:171-6.
4. Stark AD, Jordan S, Allers KA, et al. Interaction of the novel antipsychotic aripiprazole with HT1A and 5-HT2A receptors: functional receptor-binding and in vivo electrophysiological studies. Psychopharmacol 2007;190:373-82.
5. Burris KD, Molski TF, Xu C, et al. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J Pharmacol Exp Ther 2002;302:381-9.
6. Shapiro DA, Renock S, Arrington E, et al. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 2003;28:1400-11.
7. Jordan S, Koprivica V, Dunn R, et al. In vivo effects of aripiprazole on cortical and striatal dopaminergic and serotonergic function. Eur J Pharmacol 2004;483:45-53.
8. Hirose T, Uwahodo Y, Yamada S, et al. Mechanism of action of aripiprazole predicts clinical efficacy and a favourable side-effect profile. J Psychopharmacol 2004;18:375-83.
9. Physicians’ desk reference 61st ed. Montvale, NJ: Thomson PDR; 2007.
10. Currier GW, Crandall D, Archibald D, et al. Intramuscular (IM) aripiprazole controls agitation in patients with schizophrenia or bipolar I disorder without excessive sedation. Paper presented at: Annual Meeting of the American Psychiatric Association; May 20-25, 2006; Toronto, Canada.
11. Currier GW, Citrome LL, Zimbroff DL, et al. Intramuscular aripiprazole in the control of agitation. J Psychiatr Pract 2007;13:159-69.
12. Daniel DG, Currier GW, Zimbroff DL, et al. Efficacy and safety of oral aripiprazole compared with haloperidol in patients transitioning from acute treatment with intramuscular formulations. J Psychiatr Pract 2007;13:170-7.
Dr. Josiassen is chief scientific officer, Arthur P. Noyes Research Foundation, Norristown, PA, and adjunct professor of psychiatry, University of Pennsylvania, Philadelphia.
Dr. Shaughnessy is associate professor of psychiatry, Drexel University College of Medicine, Philadelphia, and medical consultant, Arthur P. Noyes Research Foundation.
Lisdexamfetamine for ADHD
Lisdexamfetamine—FDA-approved to treat attention-deficit/hyperactivity disorder (ADHD) in children ages 6 to 12 (Table 1)—reduces ADHD symptoms during and after school and may be less likely to be abused than other psychostimulants, particularly immediate-release preparations, clinical data suggest.
Table 1
Lisdexamfetamine: Fast facts
| Brand name: Vyvanse |
| Indication: ADHD in children ages 6 to 12 |
| Approval date: February 23, 2007 |
| Manufacturers: New River Pharmaceuticals and Shire |
| Dosing forms: 30-, 50-, and 70-mg capsules |
| Recommended dosage: Start at 30 mg/d. If necessary, titrate by 20 mg every 3 to 7 days to a maximum 70 mg/d. |
Clinical implications
Because it is effective for about 12 hours, lisdexamfetamine might improve the child’s ability to complete homework and participate in extracurricular activities, which in turn might enhance academic performance and/or socialization skills.
Lisdexamfetamine could help the child with ADHD who shows no contraindications to the drug —particularly if he or she needs daylong coverage.
How it works
Lisdexamfetamine—a dextroamphetamine derivative—is rapidly absorbed and converted to dextroamphetamine, which is believed to exert therapeutic effect by:
- blocking norepinephrine and dopamine reuptake into presynaptic neurons
- increasing the neurotransmitters’ release into the extraneuronal space.
The medication’s amphetamine release is highly predictable, which contributes to its therapeutic benefit in ADHD. Amphetamine is released through GI metabolism of lisdexamfetamine, which produces the active d-amphetamine moiety that reaches the bloodstream. The medication is derived from d-amphetamine, with negligible amounts of lysine cleaved.
Lisdexamfetamine requires in vivo metabolism (in the GI tract) to its active constituent d-amphetamine. As a result, the medication will not produce high d-amphetamine blood levels—and should not cause euphoria or other reinforcing effects—if injected or snorted. Its abuse potential is lower overall compared with immediate-release psychostimulant formulations.
Pharmacokinetics
Dextroamphetamine’s plasma elimination half-life is approximately 9½ hours—which accounts for lisdexamfetamine’s extended action. The drug reaches steady-state concentrations in 2 to 3 days.
Food does not affect absorption and delays maximum concentration by 1 hour or less, so taking lisdexamfetamine during breakfast should not slow its therapeutic effect. Because dextroamphetamine reaches maximum concentration in approximately 3½ hours, the medication should take effect by the time the child gets to school. In one randomized, phase-2 trial, children with ADHD who received lisdexamfetamine, 30 to 70 mg/d, showed overall improvement within 2 hours after dosing.1
Efficacy
Lisdexamfetamine reduced ADHD symptoms in 2 double-blind studies: a phase-2 crossover study and a phase-3 random-dose trial.
Phase-2 crossover study.2 Fifty-two children ages 6 to 12 with combined or hyperactive-impulsive type ADHD received extended-release mixed amphetamine salts (MAS) for 3 weeks. Subjects received 10 mg/d or dosages titrated to 20 or 30 mg/d based on response to medication.
The youths then were divided into 3 groups based on optimal MAS dosage and received 3 treatments for 1 week each:
- group 1: placebo; MAS, 10 mg/d; lisdexamfetamine, 30 mg/d
- group 2: placebo; MAS, 20 mg/d; lisdexamfetamine, 50 mg/d
- group 3: placebo; MAS, 30 mg/d; lisdexamfetamine, 70 mg/d.
While taking lisdexamfetamine or MAS, subjects showed similar improvement in behavior, based on Swanson, Kotkin, Agler, M-Flynn, and Pelham (SKAMP) scores, and inattention, based on SKAMP and Permanent Product Measure of Performance scores.
Both psychostimulants outperformed placebo in both measures, and both improved behavior more decisively than inattention. Based on post-hoc analysis, improvement 12 hours after dosing was more substantial with lisdexamfetamine than with MAS.
Phase-3 random-dose trial.3 A total of 290 children ages 6 to 12 with combined or hyperactive-impulsive type ADHD were “washed out” from prior medications over 1 week, then received lisdexamfetamine or placebo for 4 weeks. Treatment-group children were started at 30 mg/d; some received dosages titrated at random to 50 or 70 mg/d in weekly 20-mg increments.
Over 4 weeks, ADHD Rating Scale Version IV (ADHD-RS-IV) scores fell 50% to 59% among the 3 lisdexamfetamine dosage groups, compared with a 15% reduction in the placebo group. Substantial ADHD-RS-IV score improvements after 1 week of lisdexamfetamine were maintained throughout the trial, suggesting the medication sustains ADHD symptom improvement. Controlled trials have not addressed lisdexamfetamine use >4 weeks, however.
Based on parents’ and guardians’ reports, treatment-group patients’ ADHD symptoms were notably less severe at 10 AM, 2 PM, and 6 PM compared with placebo-group children.3 This suggests that lisdexamfetamine offers a daylong therapeutic effect.
Tolerability
In the phase-3 study,3 162 of 218 (74%) children receiving any dosage of lisdexamfetamine reported an adverse event, compared with 34 of 72 (47%) children in the placebo group. Overall, 39% of lisdexamfetamine-group patients reported decreased appetite. Also common were insomnia, headaches, irritability, upper abdominal pain, vomiting, and weight loss (Table 2).
Although most adverse events were mild to moderate, 9.2% of treatment-group children dropped out because of intolerability, compared with 1.4% of the placebo group. The investigators increased dosages quickly, regardless of efficacy or tolerability,3 which might have increased side-effect incidence among the treatment groups.
In the phase-2 crossover trial,2 adverse event rates were similar among the lisdexamfetamine, extended-release MAS, and placebo groups (15% to 18%). Among youths receiving lisdexamfetamine, 8% reported insomnia and 6% reported appetite loss, compared with 2% and 4% of the MAS group, respectively.
Table 2
Rates of commonly reported adverse effects
during phase-3 lisdexamfetamine (LDX) study
| Adverse effect | LDX 30 mg/d | LDX 50 mg/d* | LDX 70 mg/d* | LDX all dosages | Placebo |
|---|---|---|---|---|---|
| All adverse effects | 72% | 68% | 84% | 74% | 47% |
| Decreased appetite | 37% | 31% | 49% | 39% | 4% |
| Insomnia | 16% | 16% | 25% | 19% | 3% |
| Upper abdominal pain | 14% | 7% | 15% | 12% | 6% |
| Headache | 10% | 10% | 16% | 12% | 10% |
| Irritability | 11% | 8% | 10% | 10% | 0% |
| Vomiting | 7% | 5% | 14% | 9% | 4% |
| Weight loss | 6% | 3% | 19% | 9% | 1% |
| *Dosages were randomly titrated regardless of efficacy or tolerability. | |||||
| Source: Reference 3 | |||||
Safety
Findling et al4 found a larger change in corrected QT interval with lisdexamfetamine (7 to 14 msec) than with extended-release MAS (5 to 10 msec) 5 and 10½ hours after dosing. The authors reasoned that these findings are atypical, and no children suffered serious adverse events during the trial. Nonetheless, more research on whether lisdexamfetamine increases cardiac risk is needed.
In a lethal-dose study in rats,5 oral lisdexamfetamine doses up to 1,000 mg/kg did not result in death, suggesting the medication might undergo saturation kinetics in the GI tract that may protect against overdose or abuse at higher dosages. By comparison, the median lethal oral dosage of d-amphetamine in rats was 96.8 mg/kg.5
Abuse potential
As with other psychostimulants indicated for ADHD, the Drug Enforcement Administration has classified lisdexamfetamine as a schedule II drug, which applies to addictive prescription-only medications with an accepted medical use.
Clinical data suggest, however, that lisdexamfetamine might be less “enjoyable”—and less likely to be abused intravenously, orally, or intranasally—than equipotent d-amphetamine. In an abuse liability study,6 12 adults with histories of stimulant abuse received intravenous immediate-release (IR) d-amphetamine, 10 or 20 mg. Two days later, they received a comparable dose of IV lisdexamfetamine, 25 or 50 mg. The researchers found that:
- Plasma d-amphetamine peaked within 5 minutes after injection, compared with 2 to 3 hours after lisdexamfetamine dosing.
- Subjects who received IR d-amphetamine said they felt euphoria within 15 minutes of injection. By contrast, no one reported euphoria or amphetamine-like subjective effects after receiving lisdexamfetamine.
When asked which medication they would try again, 9 of 12 subjects chose IR d-amphetamine and 1 chose lisdexamfetamine.
In a double-blind, randomized, placebo-controlled study,7 oral lisdexamfetamine, 50 or 100 mg, was not more “likeable” than placebo. Subjects reported “liking” effects with 150 mg of lisdexamfetamine, however, suggesting the medication could be misused or abused at higher-than-therapeutic dosages.
As with other psychostimulants, do not give lisdexamfetamine to youths with preexisting serious structural cardiac abnormalities or other heart problems. Assess patient and family history of heart disease before prescribing this medication.
Do not prescribe lisdexamfetamine to patients taking a monoamine oxidase inhibitor (MAOI). By slowing amphetamine metabolism, these antidepressants intensify amphetamines’ effect on monoamine release, which can cause headaches and lead to hypertensive crisis. Before starting lisdexamfetamine, ask if the patient is taking an MAOI or has taken one within 2 weeks of presentation.
Use caution when prescribing lisdexamfetamine to patients with:
- a comorbid eating disorder or sleep disturbance. Determine whether to address the comorbidity before treating ADHD symptoms, and make sure lisdexamfetamine is not worsening the comorbid symptoms.
- untreated hypertension or other cardiovascular conditions, as stimulant medications can increase blood pressure and heart rate. Watch for significant heart rate and blood pressure changes in patients taking lisdexamfetamine, which probably would not cause sustained blood pressure increase in patients taking antihypertensives.8
Related resources
- Lisdexamfetamine Web site. www.vyvanse.com.
Drug brand names
- Extended-release mixed amphetamine salts • Adderall XR
- Lisdexamfetamine • Vyvanse
Disclosure
Dr. Wilens receives research/grant support from Abbott Laboratories, Eli Lilly and Company, National Institute on Drug Abuse, NeuroSearch, Ortho-McNeil, and Shire; is a speaker for Novartis Pharmaceuticals Corp., Ortho-McNeil, and Shire; and is a consultant to Abbott Laboratories, Eli Lilly and Company, GlaxoSmithKline, National Institute on Drug Abuse, Novartis Pharmaceuticals Corp., Ortho-McNeil, Pfizer, and Shire.
1. Lopez FA, Boellner SW, Childress A, et al. ADHD symptom improvement in children treated with lisdexamfetamine dimesylate (LDX). Poster presented at: Annual Meeting of the American Academy of Child and Adolescent Psychiatry; October 24-29, 2006; San Diego, CA.
2. Biederman J, Boellner SW, Childress A, et al. Improvements in symptoms of attention-deficit/hyperactivity disorder in school-aged children with lisdexamfetamine (NRP 104) and mixed amphetamine salts, extended-release versus placebo. Poster presented at: Annual Meeting of the American Psychiatric Association; May 20-25, 2006; Toronto, Canada.
3. Biederman J, Krishnan S, Zhang Y, et al. Efficacy and tolerability of lisdexamfetamine dimesylate (NRP 104) in children with attention-deficit/hyperactivity disorder: a phase III, multicenter, randomized, double-blind, forced-dose, parallel-group study. Clin Ther 2007;29:450-63.
4. Findling FL, Biederman J, Wilens TE, et al. Short- and long-term cardiovascular effects of mixed amphetamine salts extended release in children. J Pediatr 2005;147:348-54.
5. Krishnan S. Toxicity profile of lisdexamfetamine dimesylate (LDX NRP104) in three independent rat toxicology studies. Basic Clin Phamacol Toxicol. In press.
6. Jasinski DR. Abuse liability of intravenous L-lysine-d-amphetamine (NRP 104). Poster presented at: Annual Meeting of the College on Problems of Drug Dependence; June 17-22, 2006; Scottsdale, AZ. Available at: http://xml.10kwizard.com/filing_raw.php?repo=tenk&ipage=4234033. Accessed April 5, 2007.
7. Jasinski D, Krishnan S. A double-blind, randomized, placebo and active-controlled, six-period crossover study to evaluate the likeability, safety, and abuse liability of NRP 104 in healthy adult volunteers with histories of stimulant abuse (NRP104. A03). Poster presented at: Annual Meeting of the College on Problems of Drug Dependence; June 17-22, 2006; Scottsdale, AZ. Available at: http://www.secinfo.com/d12Pk6.v9Ac.d.htm. Accessed May 14, 2007.
8. Wilens TE, Zusman RM, Hammerness PG, et al. An open-label study of the tolerability of mixed amphetamine salts in adults with attention-deficit/hyperactivity disorder and treated primary essential hypertension. J Clin Psychiatry 2006;67:696-702.
Lisdexamfetamine—FDA-approved to treat attention-deficit/hyperactivity disorder (ADHD) in children ages 6 to 12 (Table 1)—reduces ADHD symptoms during and after school and may be less likely to be abused than other psychostimulants, particularly immediate-release preparations, clinical data suggest.
Table 1
Lisdexamfetamine: Fast facts
| Brand name: Vyvanse |
| Indication: ADHD in children ages 6 to 12 |
| Approval date: February 23, 2007 |
| Manufacturers: New River Pharmaceuticals and Shire |
| Dosing forms: 30-, 50-, and 70-mg capsules |
| Recommended dosage: Start at 30 mg/d. If necessary, titrate by 20 mg every 3 to 7 days to a maximum 70 mg/d. |
Clinical implications
Because it is effective for about 12 hours, lisdexamfetamine might improve the child’s ability to complete homework and participate in extracurricular activities, which in turn might enhance academic performance and/or socialization skills.
Lisdexamfetamine could help the child with ADHD who shows no contraindications to the drug —particularly if he or she needs daylong coverage.
How it works
Lisdexamfetamine—a dextroamphetamine derivative—is rapidly absorbed and converted to dextroamphetamine, which is believed to exert therapeutic effect by:
- blocking norepinephrine and dopamine reuptake into presynaptic neurons
- increasing the neurotransmitters’ release into the extraneuronal space.
The medication’s amphetamine release is highly predictable, which contributes to its therapeutic benefit in ADHD. Amphetamine is released through GI metabolism of lisdexamfetamine, which produces the active d-amphetamine moiety that reaches the bloodstream. The medication is derived from d-amphetamine, with negligible amounts of lysine cleaved.
Lisdexamfetamine requires in vivo metabolism (in the GI tract) to its active constituent d-amphetamine. As a result, the medication will not produce high d-amphetamine blood levels—and should not cause euphoria or other reinforcing effects—if injected or snorted. Its abuse potential is lower overall compared with immediate-release psychostimulant formulations.
Pharmacokinetics
Dextroamphetamine’s plasma elimination half-life is approximately 9½ hours—which accounts for lisdexamfetamine’s extended action. The drug reaches steady-state concentrations in 2 to 3 days.
Food does not affect absorption and delays maximum concentration by 1 hour or less, so taking lisdexamfetamine during breakfast should not slow its therapeutic effect. Because dextroamphetamine reaches maximum concentration in approximately 3½ hours, the medication should take effect by the time the child gets to school. In one randomized, phase-2 trial, children with ADHD who received lisdexamfetamine, 30 to 70 mg/d, showed overall improvement within 2 hours after dosing.1
Efficacy
Lisdexamfetamine reduced ADHD symptoms in 2 double-blind studies: a phase-2 crossover study and a phase-3 random-dose trial.
Phase-2 crossover study.2 Fifty-two children ages 6 to 12 with combined or hyperactive-impulsive type ADHD received extended-release mixed amphetamine salts (MAS) for 3 weeks. Subjects received 10 mg/d or dosages titrated to 20 or 30 mg/d based on response to medication.
The youths then were divided into 3 groups based on optimal MAS dosage and received 3 treatments for 1 week each:
- group 1: placebo; MAS, 10 mg/d; lisdexamfetamine, 30 mg/d
- group 2: placebo; MAS, 20 mg/d; lisdexamfetamine, 50 mg/d
- group 3: placebo; MAS, 30 mg/d; lisdexamfetamine, 70 mg/d.
While taking lisdexamfetamine or MAS, subjects showed similar improvement in behavior, based on Swanson, Kotkin, Agler, M-Flynn, and Pelham (SKAMP) scores, and inattention, based on SKAMP and Permanent Product Measure of Performance scores.
Both psychostimulants outperformed placebo in both measures, and both improved behavior more decisively than inattention. Based on post-hoc analysis, improvement 12 hours after dosing was more substantial with lisdexamfetamine than with MAS.
Phase-3 random-dose trial.3 A total of 290 children ages 6 to 12 with combined or hyperactive-impulsive type ADHD were “washed out” from prior medications over 1 week, then received lisdexamfetamine or placebo for 4 weeks. Treatment-group children were started at 30 mg/d; some received dosages titrated at random to 50 or 70 mg/d in weekly 20-mg increments.
Over 4 weeks, ADHD Rating Scale Version IV (ADHD-RS-IV) scores fell 50% to 59% among the 3 lisdexamfetamine dosage groups, compared with a 15% reduction in the placebo group. Substantial ADHD-RS-IV score improvements after 1 week of lisdexamfetamine were maintained throughout the trial, suggesting the medication sustains ADHD symptom improvement. Controlled trials have not addressed lisdexamfetamine use >4 weeks, however.
Based on parents’ and guardians’ reports, treatment-group patients’ ADHD symptoms were notably less severe at 10 AM, 2 PM, and 6 PM compared with placebo-group children.3 This suggests that lisdexamfetamine offers a daylong therapeutic effect.
Tolerability
In the phase-3 study,3 162 of 218 (74%) children receiving any dosage of lisdexamfetamine reported an adverse event, compared with 34 of 72 (47%) children in the placebo group. Overall, 39% of lisdexamfetamine-group patients reported decreased appetite. Also common were insomnia, headaches, irritability, upper abdominal pain, vomiting, and weight loss (Table 2).
Although most adverse events were mild to moderate, 9.2% of treatment-group children dropped out because of intolerability, compared with 1.4% of the placebo group. The investigators increased dosages quickly, regardless of efficacy or tolerability,3 which might have increased side-effect incidence among the treatment groups.
In the phase-2 crossover trial,2 adverse event rates were similar among the lisdexamfetamine, extended-release MAS, and placebo groups (15% to 18%). Among youths receiving lisdexamfetamine, 8% reported insomnia and 6% reported appetite loss, compared with 2% and 4% of the MAS group, respectively.
Table 2
Rates of commonly reported adverse effects
during phase-3 lisdexamfetamine (LDX) study
| Adverse effect | LDX 30 mg/d | LDX 50 mg/d* | LDX 70 mg/d* | LDX all dosages | Placebo |
|---|---|---|---|---|---|
| All adverse effects | 72% | 68% | 84% | 74% | 47% |
| Decreased appetite | 37% | 31% | 49% | 39% | 4% |
| Insomnia | 16% | 16% | 25% | 19% | 3% |
| Upper abdominal pain | 14% | 7% | 15% | 12% | 6% |
| Headache | 10% | 10% | 16% | 12% | 10% |
| Irritability | 11% | 8% | 10% | 10% | 0% |
| Vomiting | 7% | 5% | 14% | 9% | 4% |
| Weight loss | 6% | 3% | 19% | 9% | 1% |
| *Dosages were randomly titrated regardless of efficacy or tolerability. | |||||
| Source: Reference 3 | |||||
Safety
Findling et al4 found a larger change in corrected QT interval with lisdexamfetamine (7 to 14 msec) than with extended-release MAS (5 to 10 msec) 5 and 10½ hours after dosing. The authors reasoned that these findings are atypical, and no children suffered serious adverse events during the trial. Nonetheless, more research on whether lisdexamfetamine increases cardiac risk is needed.
In a lethal-dose study in rats,5 oral lisdexamfetamine doses up to 1,000 mg/kg did not result in death, suggesting the medication might undergo saturation kinetics in the GI tract that may protect against overdose or abuse at higher dosages. By comparison, the median lethal oral dosage of d-amphetamine in rats was 96.8 mg/kg.5
Abuse potential
As with other psychostimulants indicated for ADHD, the Drug Enforcement Administration has classified lisdexamfetamine as a schedule II drug, which applies to addictive prescription-only medications with an accepted medical use.
Clinical data suggest, however, that lisdexamfetamine might be less “enjoyable”—and less likely to be abused intravenously, orally, or intranasally—than equipotent d-amphetamine. In an abuse liability study,6 12 adults with histories of stimulant abuse received intravenous immediate-release (IR) d-amphetamine, 10 or 20 mg. Two days later, they received a comparable dose of IV lisdexamfetamine, 25 or 50 mg. The researchers found that:
- Plasma d-amphetamine peaked within 5 minutes after injection, compared with 2 to 3 hours after lisdexamfetamine dosing.
- Subjects who received IR d-amphetamine said they felt euphoria within 15 minutes of injection. By contrast, no one reported euphoria or amphetamine-like subjective effects after receiving lisdexamfetamine.
When asked which medication they would try again, 9 of 12 subjects chose IR d-amphetamine and 1 chose lisdexamfetamine.
In a double-blind, randomized, placebo-controlled study,7 oral lisdexamfetamine, 50 or 100 mg, was not more “likeable” than placebo. Subjects reported “liking” effects with 150 mg of lisdexamfetamine, however, suggesting the medication could be misused or abused at higher-than-therapeutic dosages.
As with other psychostimulants, do not give lisdexamfetamine to youths with preexisting serious structural cardiac abnormalities or other heart problems. Assess patient and family history of heart disease before prescribing this medication.
Do not prescribe lisdexamfetamine to patients taking a monoamine oxidase inhibitor (MAOI). By slowing amphetamine metabolism, these antidepressants intensify amphetamines’ effect on monoamine release, which can cause headaches and lead to hypertensive crisis. Before starting lisdexamfetamine, ask if the patient is taking an MAOI or has taken one within 2 weeks of presentation.
Use caution when prescribing lisdexamfetamine to patients with:
- a comorbid eating disorder or sleep disturbance. Determine whether to address the comorbidity before treating ADHD symptoms, and make sure lisdexamfetamine is not worsening the comorbid symptoms.
- untreated hypertension or other cardiovascular conditions, as stimulant medications can increase blood pressure and heart rate. Watch for significant heart rate and blood pressure changes in patients taking lisdexamfetamine, which probably would not cause sustained blood pressure increase in patients taking antihypertensives.8
Related resources
- Lisdexamfetamine Web site. www.vyvanse.com.
Drug brand names
- Extended-release mixed amphetamine salts • Adderall XR
- Lisdexamfetamine • Vyvanse
Disclosure
Dr. Wilens receives research/grant support from Abbott Laboratories, Eli Lilly and Company, National Institute on Drug Abuse, NeuroSearch, Ortho-McNeil, and Shire; is a speaker for Novartis Pharmaceuticals Corp., Ortho-McNeil, and Shire; and is a consultant to Abbott Laboratories, Eli Lilly and Company, GlaxoSmithKline, National Institute on Drug Abuse, Novartis Pharmaceuticals Corp., Ortho-McNeil, Pfizer, and Shire.
Lisdexamfetamine—FDA-approved to treat attention-deficit/hyperactivity disorder (ADHD) in children ages 6 to 12 (Table 1)—reduces ADHD symptoms during and after school and may be less likely to be abused than other psychostimulants, particularly immediate-release preparations, clinical data suggest.
Table 1
Lisdexamfetamine: Fast facts
| Brand name: Vyvanse |
| Indication: ADHD in children ages 6 to 12 |
| Approval date: February 23, 2007 |
| Manufacturers: New River Pharmaceuticals and Shire |
| Dosing forms: 30-, 50-, and 70-mg capsules |
| Recommended dosage: Start at 30 mg/d. If necessary, titrate by 20 mg every 3 to 7 days to a maximum 70 mg/d. |
Clinical implications
Because it is effective for about 12 hours, lisdexamfetamine might improve the child’s ability to complete homework and participate in extracurricular activities, which in turn might enhance academic performance and/or socialization skills.
Lisdexamfetamine could help the child with ADHD who shows no contraindications to the drug —particularly if he or she needs daylong coverage.
How it works
Lisdexamfetamine—a dextroamphetamine derivative—is rapidly absorbed and converted to dextroamphetamine, which is believed to exert therapeutic effect by:
- blocking norepinephrine and dopamine reuptake into presynaptic neurons
- increasing the neurotransmitters’ release into the extraneuronal space.
The medication’s amphetamine release is highly predictable, which contributes to its therapeutic benefit in ADHD. Amphetamine is released through GI metabolism of lisdexamfetamine, which produces the active d-amphetamine moiety that reaches the bloodstream. The medication is derived from d-amphetamine, with negligible amounts of lysine cleaved.
Lisdexamfetamine requires in vivo metabolism (in the GI tract) to its active constituent d-amphetamine. As a result, the medication will not produce high d-amphetamine blood levels—and should not cause euphoria or other reinforcing effects—if injected or snorted. Its abuse potential is lower overall compared with immediate-release psychostimulant formulations.
Pharmacokinetics
Dextroamphetamine’s plasma elimination half-life is approximately 9½ hours—which accounts for lisdexamfetamine’s extended action. The drug reaches steady-state concentrations in 2 to 3 days.
Food does not affect absorption and delays maximum concentration by 1 hour or less, so taking lisdexamfetamine during breakfast should not slow its therapeutic effect. Because dextroamphetamine reaches maximum concentration in approximately 3½ hours, the medication should take effect by the time the child gets to school. In one randomized, phase-2 trial, children with ADHD who received lisdexamfetamine, 30 to 70 mg/d, showed overall improvement within 2 hours after dosing.1
Efficacy
Lisdexamfetamine reduced ADHD symptoms in 2 double-blind studies: a phase-2 crossover study and a phase-3 random-dose trial.
Phase-2 crossover study.2 Fifty-two children ages 6 to 12 with combined or hyperactive-impulsive type ADHD received extended-release mixed amphetamine salts (MAS) for 3 weeks. Subjects received 10 mg/d or dosages titrated to 20 or 30 mg/d based on response to medication.
The youths then were divided into 3 groups based on optimal MAS dosage and received 3 treatments for 1 week each:
- group 1: placebo; MAS, 10 mg/d; lisdexamfetamine, 30 mg/d
- group 2: placebo; MAS, 20 mg/d; lisdexamfetamine, 50 mg/d
- group 3: placebo; MAS, 30 mg/d; lisdexamfetamine, 70 mg/d.
While taking lisdexamfetamine or MAS, subjects showed similar improvement in behavior, based on Swanson, Kotkin, Agler, M-Flynn, and Pelham (SKAMP) scores, and inattention, based on SKAMP and Permanent Product Measure of Performance scores.
Both psychostimulants outperformed placebo in both measures, and both improved behavior more decisively than inattention. Based on post-hoc analysis, improvement 12 hours after dosing was more substantial with lisdexamfetamine than with MAS.
Phase-3 random-dose trial.3 A total of 290 children ages 6 to 12 with combined or hyperactive-impulsive type ADHD were “washed out” from prior medications over 1 week, then received lisdexamfetamine or placebo for 4 weeks. Treatment-group children were started at 30 mg/d; some received dosages titrated at random to 50 or 70 mg/d in weekly 20-mg increments.
Over 4 weeks, ADHD Rating Scale Version IV (ADHD-RS-IV) scores fell 50% to 59% among the 3 lisdexamfetamine dosage groups, compared with a 15% reduction in the placebo group. Substantial ADHD-RS-IV score improvements after 1 week of lisdexamfetamine were maintained throughout the trial, suggesting the medication sustains ADHD symptom improvement. Controlled trials have not addressed lisdexamfetamine use >4 weeks, however.
Based on parents’ and guardians’ reports, treatment-group patients’ ADHD symptoms were notably less severe at 10 AM, 2 PM, and 6 PM compared with placebo-group children.3 This suggests that lisdexamfetamine offers a daylong therapeutic effect.
Tolerability
In the phase-3 study,3 162 of 218 (74%) children receiving any dosage of lisdexamfetamine reported an adverse event, compared with 34 of 72 (47%) children in the placebo group. Overall, 39% of lisdexamfetamine-group patients reported decreased appetite. Also common were insomnia, headaches, irritability, upper abdominal pain, vomiting, and weight loss (Table 2).
Although most adverse events were mild to moderate, 9.2% of treatment-group children dropped out because of intolerability, compared with 1.4% of the placebo group. The investigators increased dosages quickly, regardless of efficacy or tolerability,3 which might have increased side-effect incidence among the treatment groups.
In the phase-2 crossover trial,2 adverse event rates were similar among the lisdexamfetamine, extended-release MAS, and placebo groups (15% to 18%). Among youths receiving lisdexamfetamine, 8% reported insomnia and 6% reported appetite loss, compared with 2% and 4% of the MAS group, respectively.
Table 2
Rates of commonly reported adverse effects
during phase-3 lisdexamfetamine (LDX) study
| Adverse effect | LDX 30 mg/d | LDX 50 mg/d* | LDX 70 mg/d* | LDX all dosages | Placebo |
|---|---|---|---|---|---|
| All adverse effects | 72% | 68% | 84% | 74% | 47% |
| Decreased appetite | 37% | 31% | 49% | 39% | 4% |
| Insomnia | 16% | 16% | 25% | 19% | 3% |
| Upper abdominal pain | 14% | 7% | 15% | 12% | 6% |
| Headache | 10% | 10% | 16% | 12% | 10% |
| Irritability | 11% | 8% | 10% | 10% | 0% |
| Vomiting | 7% | 5% | 14% | 9% | 4% |
| Weight loss | 6% | 3% | 19% | 9% | 1% |
| *Dosages were randomly titrated regardless of efficacy or tolerability. | |||||
| Source: Reference 3 | |||||
Safety
Findling et al4 found a larger change in corrected QT interval with lisdexamfetamine (7 to 14 msec) than with extended-release MAS (5 to 10 msec) 5 and 10½ hours after dosing. The authors reasoned that these findings are atypical, and no children suffered serious adverse events during the trial. Nonetheless, more research on whether lisdexamfetamine increases cardiac risk is needed.
In a lethal-dose study in rats,5 oral lisdexamfetamine doses up to 1,000 mg/kg did not result in death, suggesting the medication might undergo saturation kinetics in the GI tract that may protect against overdose or abuse at higher dosages. By comparison, the median lethal oral dosage of d-amphetamine in rats was 96.8 mg/kg.5
Abuse potential
As with other psychostimulants indicated for ADHD, the Drug Enforcement Administration has classified lisdexamfetamine as a schedule II drug, which applies to addictive prescription-only medications with an accepted medical use.
Clinical data suggest, however, that lisdexamfetamine might be less “enjoyable”—and less likely to be abused intravenously, orally, or intranasally—than equipotent d-amphetamine. In an abuse liability study,6 12 adults with histories of stimulant abuse received intravenous immediate-release (IR) d-amphetamine, 10 or 20 mg. Two days later, they received a comparable dose of IV lisdexamfetamine, 25 or 50 mg. The researchers found that:
- Plasma d-amphetamine peaked within 5 minutes after injection, compared with 2 to 3 hours after lisdexamfetamine dosing.
- Subjects who received IR d-amphetamine said they felt euphoria within 15 minutes of injection. By contrast, no one reported euphoria or amphetamine-like subjective effects after receiving lisdexamfetamine.
When asked which medication they would try again, 9 of 12 subjects chose IR d-amphetamine and 1 chose lisdexamfetamine.
In a double-blind, randomized, placebo-controlled study,7 oral lisdexamfetamine, 50 or 100 mg, was not more “likeable” than placebo. Subjects reported “liking” effects with 150 mg of lisdexamfetamine, however, suggesting the medication could be misused or abused at higher-than-therapeutic dosages.
As with other psychostimulants, do not give lisdexamfetamine to youths with preexisting serious structural cardiac abnormalities or other heart problems. Assess patient and family history of heart disease before prescribing this medication.
Do not prescribe lisdexamfetamine to patients taking a monoamine oxidase inhibitor (MAOI). By slowing amphetamine metabolism, these antidepressants intensify amphetamines’ effect on monoamine release, which can cause headaches and lead to hypertensive crisis. Before starting lisdexamfetamine, ask if the patient is taking an MAOI or has taken one within 2 weeks of presentation.
Use caution when prescribing lisdexamfetamine to patients with:
- a comorbid eating disorder or sleep disturbance. Determine whether to address the comorbidity before treating ADHD symptoms, and make sure lisdexamfetamine is not worsening the comorbid symptoms.
- untreated hypertension or other cardiovascular conditions, as stimulant medications can increase blood pressure and heart rate. Watch for significant heart rate and blood pressure changes in patients taking lisdexamfetamine, which probably would not cause sustained blood pressure increase in patients taking antihypertensives.8
Related resources
- Lisdexamfetamine Web site. www.vyvanse.com.
Drug brand names
- Extended-release mixed amphetamine salts • Adderall XR
- Lisdexamfetamine • Vyvanse
Disclosure
Dr. Wilens receives research/grant support from Abbott Laboratories, Eli Lilly and Company, National Institute on Drug Abuse, NeuroSearch, Ortho-McNeil, and Shire; is a speaker for Novartis Pharmaceuticals Corp., Ortho-McNeil, and Shire; and is a consultant to Abbott Laboratories, Eli Lilly and Company, GlaxoSmithKline, National Institute on Drug Abuse, Novartis Pharmaceuticals Corp., Ortho-McNeil, Pfizer, and Shire.
1. Lopez FA, Boellner SW, Childress A, et al. ADHD symptom improvement in children treated with lisdexamfetamine dimesylate (LDX). Poster presented at: Annual Meeting of the American Academy of Child and Adolescent Psychiatry; October 24-29, 2006; San Diego, CA.
2. Biederman J, Boellner SW, Childress A, et al. Improvements in symptoms of attention-deficit/hyperactivity disorder in school-aged children with lisdexamfetamine (NRP 104) and mixed amphetamine salts, extended-release versus placebo. Poster presented at: Annual Meeting of the American Psychiatric Association; May 20-25, 2006; Toronto, Canada.
3. Biederman J, Krishnan S, Zhang Y, et al. Efficacy and tolerability of lisdexamfetamine dimesylate (NRP 104) in children with attention-deficit/hyperactivity disorder: a phase III, multicenter, randomized, double-blind, forced-dose, parallel-group study. Clin Ther 2007;29:450-63.
4. Findling FL, Biederman J, Wilens TE, et al. Short- and long-term cardiovascular effects of mixed amphetamine salts extended release in children. J Pediatr 2005;147:348-54.
5. Krishnan S. Toxicity profile of lisdexamfetamine dimesylate (LDX NRP104) in three independent rat toxicology studies. Basic Clin Phamacol Toxicol. In press.
6. Jasinski DR. Abuse liability of intravenous L-lysine-d-amphetamine (NRP 104). Poster presented at: Annual Meeting of the College on Problems of Drug Dependence; June 17-22, 2006; Scottsdale, AZ. Available at: http://xml.10kwizard.com/filing_raw.php?repo=tenk&ipage=4234033. Accessed April 5, 2007.
7. Jasinski D, Krishnan S. A double-blind, randomized, placebo and active-controlled, six-period crossover study to evaluate the likeability, safety, and abuse liability of NRP 104 in healthy adult volunteers with histories of stimulant abuse (NRP104. A03). Poster presented at: Annual Meeting of the College on Problems of Drug Dependence; June 17-22, 2006; Scottsdale, AZ. Available at: http://www.secinfo.com/d12Pk6.v9Ac.d.htm. Accessed May 14, 2007.
8. Wilens TE, Zusman RM, Hammerness PG, et al. An open-label study of the tolerability of mixed amphetamine salts in adults with attention-deficit/hyperactivity disorder and treated primary essential hypertension. J Clin Psychiatry 2006;67:696-702.
1. Lopez FA, Boellner SW, Childress A, et al. ADHD symptom improvement in children treated with lisdexamfetamine dimesylate (LDX). Poster presented at: Annual Meeting of the American Academy of Child and Adolescent Psychiatry; October 24-29, 2006; San Diego, CA.
2. Biederman J, Boellner SW, Childress A, et al. Improvements in symptoms of attention-deficit/hyperactivity disorder in school-aged children with lisdexamfetamine (NRP 104) and mixed amphetamine salts, extended-release versus placebo. Poster presented at: Annual Meeting of the American Psychiatric Association; May 20-25, 2006; Toronto, Canada.
3. Biederman J, Krishnan S, Zhang Y, et al. Efficacy and tolerability of lisdexamfetamine dimesylate (NRP 104) in children with attention-deficit/hyperactivity disorder: a phase III, multicenter, randomized, double-blind, forced-dose, parallel-group study. Clin Ther 2007;29:450-63.
4. Findling FL, Biederman J, Wilens TE, et al. Short- and long-term cardiovascular effects of mixed amphetamine salts extended release in children. J Pediatr 2005;147:348-54.
5. Krishnan S. Toxicity profile of lisdexamfetamine dimesylate (LDX NRP104) in three independent rat toxicology studies. Basic Clin Phamacol Toxicol. In press.
6. Jasinski DR. Abuse liability of intravenous L-lysine-d-amphetamine (NRP 104). Poster presented at: Annual Meeting of the College on Problems of Drug Dependence; June 17-22, 2006; Scottsdale, AZ. Available at: http://xml.10kwizard.com/filing_raw.php?repo=tenk&ipage=4234033. Accessed April 5, 2007.
7. Jasinski D, Krishnan S. A double-blind, randomized, placebo and active-controlled, six-period crossover study to evaluate the likeability, safety, and abuse liability of NRP 104 in healthy adult volunteers with histories of stimulant abuse (NRP104. A03). Poster presented at: Annual Meeting of the College on Problems of Drug Dependence; June 17-22, 2006; Scottsdale, AZ. Available at: http://www.secinfo.com/d12Pk6.v9Ac.d.htm. Accessed May 14, 2007.
8. Wilens TE, Zusman RM, Hammerness PG, et al. An open-label study of the tolerability of mixed amphetamine salts in adults with attention-deficit/hyperactivity disorder and treated primary essential hypertension. J Clin Psychiatry 2006;67:696-702.
Varenicline
Varenicline tartrate—the first nicotine-free medication FDA-approved for smoking cessation in nearly a decade (Table 1)—has helped patients stop smoking and remain smoke-free for up to 1 year in clinical trials. Its selective action on the receptor subtype that makes tobacco enjoyable offers a novel approach to antismoking therapy.
Table 1
Varenicline: Fast facts
| Brand name: Chantix |
| Class: Partial nicotinic acetylcholine receptor agonist |
| Indication: Tobacco dependence |
| Approval date: May 10, 2006 |
| Manufacturer: Pfizer |
| Dosing forms: 0.5- and 1-mg tablets |
| Recommended dosage: 0.5 mg/d for 3 days, 0.5 mg bid for next 4 days, then 1 mg bid for 11 weeks. Patients who quit successfully should receive an additional 12-week course to reduce relapse risk. |
How it works
Unlike other FDA-approved smoking cessation treatments such as nicotine replacement therapy and sustained-release bupropion, varenicline selectively targets the α4β2 nicotinic acetylcholine receptor (nAChR),1 which helps mediate nicotine’s reinforcing effects.2-4 By targeting this receptor subtype, varenicline ultimately diminishes these effects in the mesocorticolimbic dopamine system—the brain’s “reward center.”
As a partial α4β2 nAChR agonist, varenicline offers a two-pronged approach to smoking cessation:1
- During abstinence, varenicline stimulates low-level dopamine release by binding to α4β2 receptors located on dopamine neurons. This action, which compensates for loss of exogenous nicotine after quitting, can help counteract craving and other signs and symptoms of nicotine withdrawal caused by dopamine depletion.
- If the patient resumes smoking, varenicline makes tobacco less pleasurable by competitively binding at the α4β2 receptor.1,5
Pharmacokinetics
Varenicline is rapidly absorbed across the gut mucosa and reaches maximum concentration in approximately 4 hours. After repeated dosing, the drug reaches steady-state concentrations within 4 days, and its elimination half-life is 17 to 24 hours.
Because varenicline’s simple benzazepine structure lacks bulky moieties that would promote hepatic biotransformation,5 90% of the drug is excreted through the kidneys. To date, no clinically relevant drug-drug interactions have been reported.6
Efficacy
Varenicline showed a dose-dependent effect in phase-2 clinical trials,7,8 with 1 mg bid providing optimal efficacy and tolerability. Compared with placebo, varenicline was significantly more effective in initiating:
- continuous abstinence for ≥4 weeks during active treatment, confirmed by measuring carbon monoxide (CO) in exhaled breath
- long-term abstinence, evidenced by self-report and exhaled CO ≤10 ppm at 24 and 52 weeks.7,8
Odds ratios calculated for patients who stayed smoke-free for ≥4 weeks during 7 to 12 weeks of active treatment suggest that smokers who use varenicline, 1 mg bid, are approximately 4 to 8 times more likely to achieve short-term abstinence during this active treatment period than those who received placebo.7,8
In phase-3 trials,9-11 patients were also followed for up to 1 year and received brief, standardized counseling along with medication or placebo—as recommended in the U.S. Department of Health and Human Services Clinical Practice Guideline, Treating Tobacco Use and Dependence.12
12-week treatment trials.9,10 A total of 2,052 adults in two randomized, double-blind trials received varenicline, sustained-release (SR) bupropion, or placebo for 12 weeks. Based on phase-2 trial results—which showed that varenicline was better tolerated after a 1-week dosage titration period—varenicline was given at:
- 0.5 mg/d for days 1 through 3
- 0.5 mg bid for days 4 through 7
- 1 mg bid through week 12.
Bupropion SR was given at 150 mg/d for days 1 through 3, then 150 mg bid through week 12.
Patients were then followed for up to 40 weeks after drug discontinuation. Patients had been smoking ≥10 cigarettes/day at baseline and were motivated to stop smoking.
Overall, varenicline was associated with higher short- and long-term abstinence rates compared with bupropion SR or placebo (Table 2), although the comparison with bupropion SR was not statistically significant (P=0.057) for weeks 9 through 52 in one study.9 As in the phase-2 studies, abstinence was confirmed by measuring CO in exhaled breath.
Compared with placebo, varenicline also reduced cravings and other signs and symptoms of tobacco withdrawal as measured with the Brief Questionnaire of Smoking Urges and Minnesota Nicotine Withdrawal Scale.9,10
Relapse prevention study. Tonstad et al11 investigated whether extended varenicline treatment prolongs smoking abstinence. A total of 1,210 patients who quit smoking after 12 weeks of open-label varenicline treatment continued taking varenicline at 1 mg bid or were switched to placebo during a 3-month, double-blind phase.
Compared with placebo, rates of continuous smoking abstinence were significantly higher among the varenicline group during the double-blind active treatment phase (70.5% vs. 49.6%) and for 6 months after drug discontinuation (43.6% vs. 36.9%).11 These data suggest that an extended varenicline regimen might promote long-term abstinence.6,13
Table 2
Smoking abstinence* rates among patients
during varenicline phase-3 clinical trials
| 4 weeks of continued abstinence, weeks 9 through 12 | Continued abstinence, weeks 9 through 24 | Continued abstinence, weeks 9 through 52 | |
|---|---|---|---|
| Gonzales et al 20069 | |||
| Varenicline, 1 mg bid | 44% | 29.5% | 21.9% |
| Bupropion SR, 150 mg bid | 29.5% | 20.7% | 16.1% |
| Placebo | 17.7% | 10.5% | 8.4% |
| Jorenby et al 200610 | |||
| Varenicline, 1 mg bid | 43.9% | 29.7% | 23% |
| Bupropion SR, 150 mg bid | 29.8% | 20.2% | 14.6% |
| Placebo | 17.6% | 13.2% | 10.3% |
| * Confirmed by self-report and exhaled carbon monoxide ≤10 ppm. | |||
Tolerability
Overall, varenicline was safe and well tolerated in clinical trials.
Nausea was the most commonly reported adverse event in fixed-dose, placebo-controlled studies.6 Although approximately 3% of patients stopped varenicline prematurely because of upset stomach,6 most rated their nausea as mild to moderate and reported reduced nausea with continued varenicline use. For patients with intolerable nausea, consider reducing the dosage.
Sleep disturbance, constipation, flatulence, and vomiting were twice as prevalent among the varenicline groups compared with placebo.6 Overall treatment discontinuation rates were similar with varenicline, 1 mg bid, and placebo (12% vs. 10%) in 12-week phase-2 and phase-3 clinical trials.6
To improve tolerability, the FDA recommends splitting varenicline into twice-daily doses.6,13
Dosing
Start varenicline at 0.5 mg/d for 3 days, 0.5 mg bid for the next 4 days, then 1 mg bid through week 12. To improve tolerability, advise patients to take varenicline after eating and with a full glass of water.
Setting a target quit date (TQD) is a critical element of smoking cessation treatment. Schedule the TQD for the same day the patient begins 1 mg bid varenicline dosing so that the medication is approaching maximal steady-state concentrations during the quit attempt to help counter withdrawal. Allow patients to continue smoking during the 1-week titration period, but stress the importance of trying to quit on the TQD.
Because varenicline is primarily eliminated through the kidneys, limit dosages to 0.5 mg bid in patients with severe renal impairment (estimated creatinine clearance 6,13
Varenicline has not been studied in patients with substance use and other psychiatric disorders—patients who account for most of a psychiatrist’s caseload and whose nicotine dependence is difficult to treat. Even so, the medication’s lack of discernible drug-drug interactions and selectivity of α4β2 nAChR action make varenicline worth considering for these patients.
Varenicline also has not been tested or approved for use in adolescent or pregnant smokers; research is needed on how the medication works in these patients.
Role of behavioral treatment
The Clinical Practice Guideline, Treating Tobacco Use and Dependence12 suggests combining antismoking pharmacotherapy with counseling to maximize outcome. To that end, varenicline’s manufacturer has developed a personalized behavioral support program for patients taking the medication.14 Adjunctive therapy via the Internet, telephone, or direct mail can complement other extra-treatment supports—such as toll-free quit lines and classes offered through health organizations—and more-formal, intensive behavioral interventions.
Related resources
- Varenicline Web site. www.chantix.com.
- World Health Organization. Causes of death. In: Epidemiology and burden of disease. Geneva: World Health Organization, 2003.
- Fiore MC, Bailey WC, Cohen SJ, et al. Clinical practice guideline. Treating tobacco use and dependence. Rockville, MD: U.S. Department of Health and Human Services, 2000. www.surgeongeneral.gov/tobacco/treating_tobacco_use.pdf.
Drug brand names
- Bupropion SR • Wellbutrin, Zyban
- Varenicline • Chantix
Disclosures
Dr. Anthenelli is a consultant for Alkermes and Cephalon and a speaker and consultant for Pfizer and sanofi-aventis.
The Tri-State Tobacco and Alcohol Research Center receives research grants from Addex, Ortho-McNeil Neurologics, and sanofi-aventis.
Dr. Anthenelli and the Tri-State Tobacco and Alcohol Research Center were members of the Varenicline Study Group.
Acknowledgments
The writing of this article was supported in part by National Institutes of Health Grant Awards Nos. AA013957 and AA013307.
The author thanks Reene Cantwell for her technical assistance in preparing this article.
1. Coe JW, Brooks PR, Vetelino MG, et al. Varenicline: an alpha4beta2 nicotinic receptor partial agonist for smoking cessation. J Med Chem 2005;48:3474-7.
2. Marubio LM, Gardier AM, Durier S, et al. Effects of nicotine in the dopaminergic system of mice lacking the alpha4 subunit of neuronal nicotinic acetylcholine receptors. Eur J Neurosci 2003;17:1329-37.
3. Picciotto MR, Zoli M, Rimondini R, et al. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature 1998;391:173-7.
4. Tapper AR, McKinney SL, Nashmi R, et al. Nicotine activation of α4* receptors: sufficient for reward, tolerance, and sensitization. Science 2004;306:1029-32.
5. Obach RS, Reed-Hagen AE, Krueger SS, et al. Metabolism and disposition of varenicline, a selective alpha4beta2 acetylcholine receptor partial agonist, in vivo and in vitro. Drug Metab Dispos 2006;34:121-30.
6. Chantix (varenicline) package insert. Available at: http://www.pfizer.com/pfizer/download/uspi_chantix.pdf. Accessed November 28, 2006.
7. Oncken C, Gonzales D, Nides M, et al. Efficacy and safety of the novel selective nicotinic acetylcholine receptor partial agonist, varenicline, for smoking cessation. Arch Intern Med 2006;166:1571-7.
8. Nides M, Oncken C, Gonzales D, et al. Smoking cessation with varenicline, a selective alpha4beta2 nicotinic receptor partial agonist: results from a 7-week, randomized, placebo- and bupropion-controlled trial with 1-year follow-up. Arch Intern Med 2006;166:1561-8.
9. Gonzales D, Rennard SI, Nides M, et al. Varenicline Phase 3 Study Group. Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs sustained-release bupropion and placebo for smoking cessation: a randomized controlled trial. JAMA 2006;296:47-55.
10. Jorenby DE, Hays JT, Rigotti NA, et al. Varenicline Phase 3 Study Group. Efficacy of varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs placebo or sustained-release bupropion for smoking cessation: a randomized controlled trial. JAMA 2006;296:56-63.
11. Tonstad S, Tonnesen P, Hajek P, et al. Effect of maintenance therapy with varenicline on smoking cessation: a randomized controlled trial. JAMA 2006;296:64-71.
12. Fiore MC, Bailey WC, Cohen SJ, et al. Clinical practice guideline. Treating tobacco use and dependence. Rockville, MD: U.S. Department of Health and Human Services, 2000. Available at: http://www.surgeongeneral.gov/tobacco/treating_tobacco_use.pdf. Accessed November 30, 2006.
13. Varenicline (Chantix) for tobacco dependence. Med Lett Drugs Ther 2006;48:66-8.
14. Varenicline. Web site. Available at: http://www.chantix.com. Accessed November 30, 2006.
Varenicline tartrate—the first nicotine-free medication FDA-approved for smoking cessation in nearly a decade (Table 1)—has helped patients stop smoking and remain smoke-free for up to 1 year in clinical trials. Its selective action on the receptor subtype that makes tobacco enjoyable offers a novel approach to antismoking therapy.
Table 1
Varenicline: Fast facts
| Brand name: Chantix |
| Class: Partial nicotinic acetylcholine receptor agonist |
| Indication: Tobacco dependence |
| Approval date: May 10, 2006 |
| Manufacturer: Pfizer |
| Dosing forms: 0.5- and 1-mg tablets |
| Recommended dosage: 0.5 mg/d for 3 days, 0.5 mg bid for next 4 days, then 1 mg bid for 11 weeks. Patients who quit successfully should receive an additional 12-week course to reduce relapse risk. |
How it works
Unlike other FDA-approved smoking cessation treatments such as nicotine replacement therapy and sustained-release bupropion, varenicline selectively targets the α4β2 nicotinic acetylcholine receptor (nAChR),1 which helps mediate nicotine’s reinforcing effects.2-4 By targeting this receptor subtype, varenicline ultimately diminishes these effects in the mesocorticolimbic dopamine system—the brain’s “reward center.”
As a partial α4β2 nAChR agonist, varenicline offers a two-pronged approach to smoking cessation:1
- During abstinence, varenicline stimulates low-level dopamine release by binding to α4β2 receptors located on dopamine neurons. This action, which compensates for loss of exogenous nicotine after quitting, can help counteract craving and other signs and symptoms of nicotine withdrawal caused by dopamine depletion.
- If the patient resumes smoking, varenicline makes tobacco less pleasurable by competitively binding at the α4β2 receptor.1,5
Pharmacokinetics
Varenicline is rapidly absorbed across the gut mucosa and reaches maximum concentration in approximately 4 hours. After repeated dosing, the drug reaches steady-state concentrations within 4 days, and its elimination half-life is 17 to 24 hours.
Because varenicline’s simple benzazepine structure lacks bulky moieties that would promote hepatic biotransformation,5 90% of the drug is excreted through the kidneys. To date, no clinically relevant drug-drug interactions have been reported.6
Efficacy
Varenicline showed a dose-dependent effect in phase-2 clinical trials,7,8 with 1 mg bid providing optimal efficacy and tolerability. Compared with placebo, varenicline was significantly more effective in initiating:
- continuous abstinence for ≥4 weeks during active treatment, confirmed by measuring carbon monoxide (CO) in exhaled breath
- long-term abstinence, evidenced by self-report and exhaled CO ≤10 ppm at 24 and 52 weeks.7,8
Odds ratios calculated for patients who stayed smoke-free for ≥4 weeks during 7 to 12 weeks of active treatment suggest that smokers who use varenicline, 1 mg bid, are approximately 4 to 8 times more likely to achieve short-term abstinence during this active treatment period than those who received placebo.7,8
In phase-3 trials,9-11 patients were also followed for up to 1 year and received brief, standardized counseling along with medication or placebo—as recommended in the U.S. Department of Health and Human Services Clinical Practice Guideline, Treating Tobacco Use and Dependence.12
12-week treatment trials.9,10 A total of 2,052 adults in two randomized, double-blind trials received varenicline, sustained-release (SR) bupropion, or placebo for 12 weeks. Based on phase-2 trial results—which showed that varenicline was better tolerated after a 1-week dosage titration period—varenicline was given at:
- 0.5 mg/d for days 1 through 3
- 0.5 mg bid for days 4 through 7
- 1 mg bid through week 12.
Bupropion SR was given at 150 mg/d for days 1 through 3, then 150 mg bid through week 12.
Patients were then followed for up to 40 weeks after drug discontinuation. Patients had been smoking ≥10 cigarettes/day at baseline and were motivated to stop smoking.
Overall, varenicline was associated with higher short- and long-term abstinence rates compared with bupropion SR or placebo (Table 2), although the comparison with bupropion SR was not statistically significant (P=0.057) for weeks 9 through 52 in one study.9 As in the phase-2 studies, abstinence was confirmed by measuring CO in exhaled breath.
Compared with placebo, varenicline also reduced cravings and other signs and symptoms of tobacco withdrawal as measured with the Brief Questionnaire of Smoking Urges and Minnesota Nicotine Withdrawal Scale.9,10
Relapse prevention study. Tonstad et al11 investigated whether extended varenicline treatment prolongs smoking abstinence. A total of 1,210 patients who quit smoking after 12 weeks of open-label varenicline treatment continued taking varenicline at 1 mg bid or were switched to placebo during a 3-month, double-blind phase.
Compared with placebo, rates of continuous smoking abstinence were significantly higher among the varenicline group during the double-blind active treatment phase (70.5% vs. 49.6%) and for 6 months after drug discontinuation (43.6% vs. 36.9%).11 These data suggest that an extended varenicline regimen might promote long-term abstinence.6,13
Table 2
Smoking abstinence* rates among patients
during varenicline phase-3 clinical trials
| 4 weeks of continued abstinence, weeks 9 through 12 | Continued abstinence, weeks 9 through 24 | Continued abstinence, weeks 9 through 52 | |
|---|---|---|---|
| Gonzales et al 20069 | |||
| Varenicline, 1 mg bid | 44% | 29.5% | 21.9% |
| Bupropion SR, 150 mg bid | 29.5% | 20.7% | 16.1% |
| Placebo | 17.7% | 10.5% | 8.4% |
| Jorenby et al 200610 | |||
| Varenicline, 1 mg bid | 43.9% | 29.7% | 23% |
| Bupropion SR, 150 mg bid | 29.8% | 20.2% | 14.6% |
| Placebo | 17.6% | 13.2% | 10.3% |
| * Confirmed by self-report and exhaled carbon monoxide ≤10 ppm. | |||
Tolerability
Overall, varenicline was safe and well tolerated in clinical trials.
Nausea was the most commonly reported adverse event in fixed-dose, placebo-controlled studies.6 Although approximately 3% of patients stopped varenicline prematurely because of upset stomach,6 most rated their nausea as mild to moderate and reported reduced nausea with continued varenicline use. For patients with intolerable nausea, consider reducing the dosage.
Sleep disturbance, constipation, flatulence, and vomiting were twice as prevalent among the varenicline groups compared with placebo.6 Overall treatment discontinuation rates were similar with varenicline, 1 mg bid, and placebo (12% vs. 10%) in 12-week phase-2 and phase-3 clinical trials.6
To improve tolerability, the FDA recommends splitting varenicline into twice-daily doses.6,13
Dosing
Start varenicline at 0.5 mg/d for 3 days, 0.5 mg bid for the next 4 days, then 1 mg bid through week 12. To improve tolerability, advise patients to take varenicline after eating and with a full glass of water.
Setting a target quit date (TQD) is a critical element of smoking cessation treatment. Schedule the TQD for the same day the patient begins 1 mg bid varenicline dosing so that the medication is approaching maximal steady-state concentrations during the quit attempt to help counter withdrawal. Allow patients to continue smoking during the 1-week titration period, but stress the importance of trying to quit on the TQD.
Because varenicline is primarily eliminated through the kidneys, limit dosages to 0.5 mg bid in patients with severe renal impairment (estimated creatinine clearance 6,13
Varenicline has not been studied in patients with substance use and other psychiatric disorders—patients who account for most of a psychiatrist’s caseload and whose nicotine dependence is difficult to treat. Even so, the medication’s lack of discernible drug-drug interactions and selectivity of α4β2 nAChR action make varenicline worth considering for these patients.
Varenicline also has not been tested or approved for use in adolescent or pregnant smokers; research is needed on how the medication works in these patients.
Role of behavioral treatment
The Clinical Practice Guideline, Treating Tobacco Use and Dependence12 suggests combining antismoking pharmacotherapy with counseling to maximize outcome. To that end, varenicline’s manufacturer has developed a personalized behavioral support program for patients taking the medication.14 Adjunctive therapy via the Internet, telephone, or direct mail can complement other extra-treatment supports—such as toll-free quit lines and classes offered through health organizations—and more-formal, intensive behavioral interventions.
Related resources
- Varenicline Web site. www.chantix.com.
- World Health Organization. Causes of death. In: Epidemiology and burden of disease. Geneva: World Health Organization, 2003.
- Fiore MC, Bailey WC, Cohen SJ, et al. Clinical practice guideline. Treating tobacco use and dependence. Rockville, MD: U.S. Department of Health and Human Services, 2000. www.surgeongeneral.gov/tobacco/treating_tobacco_use.pdf.
Drug brand names
- Bupropion SR • Wellbutrin, Zyban
- Varenicline • Chantix
Disclosures
Dr. Anthenelli is a consultant for Alkermes and Cephalon and a speaker and consultant for Pfizer and sanofi-aventis.
The Tri-State Tobacco and Alcohol Research Center receives research grants from Addex, Ortho-McNeil Neurologics, and sanofi-aventis.
Dr. Anthenelli and the Tri-State Tobacco and Alcohol Research Center were members of the Varenicline Study Group.
Acknowledgments
The writing of this article was supported in part by National Institutes of Health Grant Awards Nos. AA013957 and AA013307.
The author thanks Reene Cantwell for her technical assistance in preparing this article.
Varenicline tartrate—the first nicotine-free medication FDA-approved for smoking cessation in nearly a decade (Table 1)—has helped patients stop smoking and remain smoke-free for up to 1 year in clinical trials. Its selective action on the receptor subtype that makes tobacco enjoyable offers a novel approach to antismoking therapy.
Table 1
Varenicline: Fast facts
| Brand name: Chantix |
| Class: Partial nicotinic acetylcholine receptor agonist |
| Indication: Tobacco dependence |
| Approval date: May 10, 2006 |
| Manufacturer: Pfizer |
| Dosing forms: 0.5- and 1-mg tablets |
| Recommended dosage: 0.5 mg/d for 3 days, 0.5 mg bid for next 4 days, then 1 mg bid for 11 weeks. Patients who quit successfully should receive an additional 12-week course to reduce relapse risk. |
How it works
Unlike other FDA-approved smoking cessation treatments such as nicotine replacement therapy and sustained-release bupropion, varenicline selectively targets the α4β2 nicotinic acetylcholine receptor (nAChR),1 which helps mediate nicotine’s reinforcing effects.2-4 By targeting this receptor subtype, varenicline ultimately diminishes these effects in the mesocorticolimbic dopamine system—the brain’s “reward center.”
As a partial α4β2 nAChR agonist, varenicline offers a two-pronged approach to smoking cessation:1
- During abstinence, varenicline stimulates low-level dopamine release by binding to α4β2 receptors located on dopamine neurons. This action, which compensates for loss of exogenous nicotine after quitting, can help counteract craving and other signs and symptoms of nicotine withdrawal caused by dopamine depletion.
- If the patient resumes smoking, varenicline makes tobacco less pleasurable by competitively binding at the α4β2 receptor.1,5
Pharmacokinetics
Varenicline is rapidly absorbed across the gut mucosa and reaches maximum concentration in approximately 4 hours. After repeated dosing, the drug reaches steady-state concentrations within 4 days, and its elimination half-life is 17 to 24 hours.
Because varenicline’s simple benzazepine structure lacks bulky moieties that would promote hepatic biotransformation,5 90% of the drug is excreted through the kidneys. To date, no clinically relevant drug-drug interactions have been reported.6
Efficacy
Varenicline showed a dose-dependent effect in phase-2 clinical trials,7,8 with 1 mg bid providing optimal efficacy and tolerability. Compared with placebo, varenicline was significantly more effective in initiating:
- continuous abstinence for ≥4 weeks during active treatment, confirmed by measuring carbon monoxide (CO) in exhaled breath
- long-term abstinence, evidenced by self-report and exhaled CO ≤10 ppm at 24 and 52 weeks.7,8
Odds ratios calculated for patients who stayed smoke-free for ≥4 weeks during 7 to 12 weeks of active treatment suggest that smokers who use varenicline, 1 mg bid, are approximately 4 to 8 times more likely to achieve short-term abstinence during this active treatment period than those who received placebo.7,8
In phase-3 trials,9-11 patients were also followed for up to 1 year and received brief, standardized counseling along with medication or placebo—as recommended in the U.S. Department of Health and Human Services Clinical Practice Guideline, Treating Tobacco Use and Dependence.12
12-week treatment trials.9,10 A total of 2,052 adults in two randomized, double-blind trials received varenicline, sustained-release (SR) bupropion, or placebo for 12 weeks. Based on phase-2 trial results—which showed that varenicline was better tolerated after a 1-week dosage titration period—varenicline was given at:
- 0.5 mg/d for days 1 through 3
- 0.5 mg bid for days 4 through 7
- 1 mg bid through week 12.
Bupropion SR was given at 150 mg/d for days 1 through 3, then 150 mg bid through week 12.
Patients were then followed for up to 40 weeks after drug discontinuation. Patients had been smoking ≥10 cigarettes/day at baseline and were motivated to stop smoking.
Overall, varenicline was associated with higher short- and long-term abstinence rates compared with bupropion SR or placebo (Table 2), although the comparison with bupropion SR was not statistically significant (P=0.057) for weeks 9 through 52 in one study.9 As in the phase-2 studies, abstinence was confirmed by measuring CO in exhaled breath.
Compared with placebo, varenicline also reduced cravings and other signs and symptoms of tobacco withdrawal as measured with the Brief Questionnaire of Smoking Urges and Minnesota Nicotine Withdrawal Scale.9,10
Relapse prevention study. Tonstad et al11 investigated whether extended varenicline treatment prolongs smoking abstinence. A total of 1,210 patients who quit smoking after 12 weeks of open-label varenicline treatment continued taking varenicline at 1 mg bid or were switched to placebo during a 3-month, double-blind phase.
Compared with placebo, rates of continuous smoking abstinence were significantly higher among the varenicline group during the double-blind active treatment phase (70.5% vs. 49.6%) and for 6 months after drug discontinuation (43.6% vs. 36.9%).11 These data suggest that an extended varenicline regimen might promote long-term abstinence.6,13
Table 2
Smoking abstinence* rates among patients
during varenicline phase-3 clinical trials
| 4 weeks of continued abstinence, weeks 9 through 12 | Continued abstinence, weeks 9 through 24 | Continued abstinence, weeks 9 through 52 | |
|---|---|---|---|
| Gonzales et al 20069 | |||
| Varenicline, 1 mg bid | 44% | 29.5% | 21.9% |
| Bupropion SR, 150 mg bid | 29.5% | 20.7% | 16.1% |
| Placebo | 17.7% | 10.5% | 8.4% |
| Jorenby et al 200610 | |||
| Varenicline, 1 mg bid | 43.9% | 29.7% | 23% |
| Bupropion SR, 150 mg bid | 29.8% | 20.2% | 14.6% |
| Placebo | 17.6% | 13.2% | 10.3% |
| * Confirmed by self-report and exhaled carbon monoxide ≤10 ppm. | |||
Tolerability
Overall, varenicline was safe and well tolerated in clinical trials.
Nausea was the most commonly reported adverse event in fixed-dose, placebo-controlled studies.6 Although approximately 3% of patients stopped varenicline prematurely because of upset stomach,6 most rated their nausea as mild to moderate and reported reduced nausea with continued varenicline use. For patients with intolerable nausea, consider reducing the dosage.
Sleep disturbance, constipation, flatulence, and vomiting were twice as prevalent among the varenicline groups compared with placebo.6 Overall treatment discontinuation rates were similar with varenicline, 1 mg bid, and placebo (12% vs. 10%) in 12-week phase-2 and phase-3 clinical trials.6
To improve tolerability, the FDA recommends splitting varenicline into twice-daily doses.6,13
Dosing
Start varenicline at 0.5 mg/d for 3 days, 0.5 mg bid for the next 4 days, then 1 mg bid through week 12. To improve tolerability, advise patients to take varenicline after eating and with a full glass of water.
Setting a target quit date (TQD) is a critical element of smoking cessation treatment. Schedule the TQD for the same day the patient begins 1 mg bid varenicline dosing so that the medication is approaching maximal steady-state concentrations during the quit attempt to help counter withdrawal. Allow patients to continue smoking during the 1-week titration period, but stress the importance of trying to quit on the TQD.
Because varenicline is primarily eliminated through the kidneys, limit dosages to 0.5 mg bid in patients with severe renal impairment (estimated creatinine clearance 6,13
Varenicline has not been studied in patients with substance use and other psychiatric disorders—patients who account for most of a psychiatrist’s caseload and whose nicotine dependence is difficult to treat. Even so, the medication’s lack of discernible drug-drug interactions and selectivity of α4β2 nAChR action make varenicline worth considering for these patients.
Varenicline also has not been tested or approved for use in adolescent or pregnant smokers; research is needed on how the medication works in these patients.
Role of behavioral treatment
The Clinical Practice Guideline, Treating Tobacco Use and Dependence12 suggests combining antismoking pharmacotherapy with counseling to maximize outcome. To that end, varenicline’s manufacturer has developed a personalized behavioral support program for patients taking the medication.14 Adjunctive therapy via the Internet, telephone, or direct mail can complement other extra-treatment supports—such as toll-free quit lines and classes offered through health organizations—and more-formal, intensive behavioral interventions.
Related resources
- Varenicline Web site. www.chantix.com.
- World Health Organization. Causes of death. In: Epidemiology and burden of disease. Geneva: World Health Organization, 2003.
- Fiore MC, Bailey WC, Cohen SJ, et al. Clinical practice guideline. Treating tobacco use and dependence. Rockville, MD: U.S. Department of Health and Human Services, 2000. www.surgeongeneral.gov/tobacco/treating_tobacco_use.pdf.
Drug brand names
- Bupropion SR • Wellbutrin, Zyban
- Varenicline • Chantix
Disclosures
Dr. Anthenelli is a consultant for Alkermes and Cephalon and a speaker and consultant for Pfizer and sanofi-aventis.
The Tri-State Tobacco and Alcohol Research Center receives research grants from Addex, Ortho-McNeil Neurologics, and sanofi-aventis.
Dr. Anthenelli and the Tri-State Tobacco and Alcohol Research Center were members of the Varenicline Study Group.
Acknowledgments
The writing of this article was supported in part by National Institutes of Health Grant Awards Nos. AA013957 and AA013307.
The author thanks Reene Cantwell for her technical assistance in preparing this article.
1. Coe JW, Brooks PR, Vetelino MG, et al. Varenicline: an alpha4beta2 nicotinic receptor partial agonist for smoking cessation. J Med Chem 2005;48:3474-7.
2. Marubio LM, Gardier AM, Durier S, et al. Effects of nicotine in the dopaminergic system of mice lacking the alpha4 subunit of neuronal nicotinic acetylcholine receptors. Eur J Neurosci 2003;17:1329-37.
3. Picciotto MR, Zoli M, Rimondini R, et al. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature 1998;391:173-7.
4. Tapper AR, McKinney SL, Nashmi R, et al. Nicotine activation of α4* receptors: sufficient for reward, tolerance, and sensitization. Science 2004;306:1029-32.
5. Obach RS, Reed-Hagen AE, Krueger SS, et al. Metabolism and disposition of varenicline, a selective alpha4beta2 acetylcholine receptor partial agonist, in vivo and in vitro. Drug Metab Dispos 2006;34:121-30.
6. Chantix (varenicline) package insert. Available at: http://www.pfizer.com/pfizer/download/uspi_chantix.pdf. Accessed November 28, 2006.
7. Oncken C, Gonzales D, Nides M, et al. Efficacy and safety of the novel selective nicotinic acetylcholine receptor partial agonist, varenicline, for smoking cessation. Arch Intern Med 2006;166:1571-7.
8. Nides M, Oncken C, Gonzales D, et al. Smoking cessation with varenicline, a selective alpha4beta2 nicotinic receptor partial agonist: results from a 7-week, randomized, placebo- and bupropion-controlled trial with 1-year follow-up. Arch Intern Med 2006;166:1561-8.
9. Gonzales D, Rennard SI, Nides M, et al. Varenicline Phase 3 Study Group. Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs sustained-release bupropion and placebo for smoking cessation: a randomized controlled trial. JAMA 2006;296:47-55.
10. Jorenby DE, Hays JT, Rigotti NA, et al. Varenicline Phase 3 Study Group. Efficacy of varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs placebo or sustained-release bupropion for smoking cessation: a randomized controlled trial. JAMA 2006;296:56-63.
11. Tonstad S, Tonnesen P, Hajek P, et al. Effect of maintenance therapy with varenicline on smoking cessation: a randomized controlled trial. JAMA 2006;296:64-71.
12. Fiore MC, Bailey WC, Cohen SJ, et al. Clinical practice guideline. Treating tobacco use and dependence. Rockville, MD: U.S. Department of Health and Human Services, 2000. Available at: http://www.surgeongeneral.gov/tobacco/treating_tobacco_use.pdf. Accessed November 30, 2006.
13. Varenicline (Chantix) for tobacco dependence. Med Lett Drugs Ther 2006;48:66-8.
14. Varenicline. Web site. Available at: http://www.chantix.com. Accessed November 30, 2006.
1. Coe JW, Brooks PR, Vetelino MG, et al. Varenicline: an alpha4beta2 nicotinic receptor partial agonist for smoking cessation. J Med Chem 2005;48:3474-7.
2. Marubio LM, Gardier AM, Durier S, et al. Effects of nicotine in the dopaminergic system of mice lacking the alpha4 subunit of neuronal nicotinic acetylcholine receptors. Eur J Neurosci 2003;17:1329-37.
3. Picciotto MR, Zoli M, Rimondini R, et al. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature 1998;391:173-7.
4. Tapper AR, McKinney SL, Nashmi R, et al. Nicotine activation of α4* receptors: sufficient for reward, tolerance, and sensitization. Science 2004;306:1029-32.
5. Obach RS, Reed-Hagen AE, Krueger SS, et al. Metabolism and disposition of varenicline, a selective alpha4beta2 acetylcholine receptor partial agonist, in vivo and in vitro. Drug Metab Dispos 2006;34:121-30.
6. Chantix (varenicline) package insert. Available at: http://www.pfizer.com/pfizer/download/uspi_chantix.pdf. Accessed November 28, 2006.
7. Oncken C, Gonzales D, Nides M, et al. Efficacy and safety of the novel selective nicotinic acetylcholine receptor partial agonist, varenicline, for smoking cessation. Arch Intern Med 2006;166:1571-7.
8. Nides M, Oncken C, Gonzales D, et al. Smoking cessation with varenicline, a selective alpha4beta2 nicotinic receptor partial agonist: results from a 7-week, randomized, placebo- and bupropion-controlled trial with 1-year follow-up. Arch Intern Med 2006;166:1561-8.
9. Gonzales D, Rennard SI, Nides M, et al. Varenicline Phase 3 Study Group. Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs sustained-release bupropion and placebo for smoking cessation: a randomized controlled trial. JAMA 2006;296:47-55.
10. Jorenby DE, Hays JT, Rigotti NA, et al. Varenicline Phase 3 Study Group. Efficacy of varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs placebo or sustained-release bupropion for smoking cessation: a randomized controlled trial. JAMA 2006;296:56-63.
11. Tonstad S, Tonnesen P, Hajek P, et al. Effect of maintenance therapy with varenicline on smoking cessation: a randomized controlled trial. JAMA 2006;296:64-71.
12. Fiore MC, Bailey WC, Cohen SJ, et al. Clinical practice guideline. Treating tobacco use and dependence. Rockville, MD: U.S. Department of Health and Human Services, 2000. Available at: http://www.surgeongeneral.gov/tobacco/treating_tobacco_use.pdf. Accessed November 30, 2006.
13. Varenicline (Chantix) for tobacco dependence. Med Lett Drugs Ther 2006;48:66-8.
14. Varenicline. Web site. Available at: http://www.chantix.com. Accessed November 30, 2006.
Paliperidone
Paliperidone, a second-generation antipsychotic (SGA), was awaiting FDA approval for treating schizophrenia (Table 1) at press time. FDA issued an approvable letter September 29.
The long-acting oral medication can be given once daily, without the plasma level peaks and troughs associated with other SGAs.
Table 1
Paliperidone: Fast facts
| Class: Second-generation antipsychotic Prospective indication: Schizophrenia FDA action: Issued approvable letter September 29, 2006 Manufacturer: Janssen Pharmaceutica Dosing forms: Not determined Recommended dosage: Data suggest that 6 mg/d is a suitable starting dosage for most patients.1-3 No formal recommendation issued |
Clinical implications
Paliperidone—the active metabolite of risperidone (9-hydroxy risperidone)—produces the same effects as its parent compound. Because it is metabolized less by the liver, however, paliperidone will likely have a lower risk of drug-drug interactions than risperidone.
Paliperidone can help patients with hallucinations, delusions, and other florid psychotic symptoms. Once-daily dosing could also make it easier for patients with schizophrenia to adhere to treatment.
Paliperidone caused relatively few side effects in clinical trials, indicating that the drug could be started at therapeutic dosages. A 6-mg dose reaches clinically effective plasma levels in approximately 22 hours; a higher dosage might take less time.
How it works
As with all antipsychotics, paliperidone blocks dopamine uptake (D2 receptors) and—as with other newer SGAs—it has a high affinity for 5-HT (serotonin) receptors. This serotonergic action may help modulate side effects.
Paliperidone has shown antipsychotic effectiveness at 3 to 15 mg/d, and a 6-mg dosage is no more likely to cause extrapyramidal symptoms (EPS) or other adverse events than olanzapine, 10 mg/d, or placebo.1-3 This finding suggests that 6 mg/d might be a suitable starting dosage for most patients.
Clinical findings
Efficacy. The drug showed efficacy for treating acute schizophrenia in three randomized, double-blind, controlled trials.
In a 6-week study,1 444 patients who were experiencing acute schizophrenia episodes and had Positive and Negative Syndrome Scale (PANSS) scores between 70 and 120 received paliperidone, 6 or 12 mg/d, olanzapine, 10 mg/d, or placebo. The study was powered to compare paliperidone and placebo; the olanzapine group was included to confirm study sensitivity.
Mean baseline total and negative symptom PANSS scores improved twice as much among the paliperidone and olanzapine groups compared with placebo (Table 2). Personal and Social Performance (PSP) scale scores also improved significantly among patients receiving paliperidone, 6 mg/d. The PSP scale gauges function, ability to perform socially useful activities such as self-care and work, and disturbing and aggressive behavior.
In two similarly designed, 6-week studies (N=1,248),2,3 paliperidone at 3, 6, 9, or 12 mg/d produced statistically significant improvement in total and negative symptom PANSS scores and PSP scale scores compared with placebo.
Table 2
Mean PANSS score reductions among patients taking paliperidone, olanzapine, or placebo
| Paliperidone, 6 mg/d | Paliperidone, 12 mg/d | Olanzapine, 10 mg/d | Placebo | |
|---|---|---|---|---|
| Total PANSS score | 17.5 | 17.5 | 18.4 | 8.0 |
| Negative symptom PANSS score | 4.4 | 3.9 | 4.4 | 2.2 |
| Source: Reference 1 | ||||
Relapse prevention. Kramer et al4 measured paliperidone’s ability to prevent or delay schizophrenia recurrence among 205 patients experiencing an episode. Participants had been diagnosed with schizophrenia at least 1 year earlier and had PANSS total scores between 70 and 120. Patients with substance dependence or other axis I disorders were excluded.
Over an 8-week run-in period, patients were hospitalized for 2 weeks and started on open-label paliperidone, 9 mg/d; dosages then were titrated to 3 to 15 mg/d depending on efficacy and tolerability. Once stable for 2 weeks, subjects were discharged and maintained on paliperidone for 6 weeks, then were randomized to a double-blind phase during which they received a similar dosage of paliperidone or placebo. Regimen duration varied during the double-blind phase.
The study was stopped after 2 months when an interim analysis showed significant efficacy for paliperidone. Among patients who relapsed, mean time to relapse was 68 days among patients taking paliperidone (n=14, 25%), compared with 25 days in the placebo group (n=29, 53%).
Recurrence was defined as:
- psychiatric hospitalization
- total PANSS score ≥40 at randomization decreased by 25% for 2 days, or total PANSS score
- Clinical Global Impression of Severity (CGI-S) score ≥3 at randomization increased to ≥4 for 2 days, or CGI-S score ≥4 at randomization rose to ≥5
- individual-item PANSS baseline score ≥3 increased to ≥5 for 2 days, or baseline score ≥4 rose to ≥6
- self injury, suicidal or homicidal thoughts, or clinically significant aggressive behavior.
A final analysis showed a 22% relapse rate among patients taking paliperidone vs. 52% of the placebo group. Paliperidone was associated with improvements in PANSS, CGI-S, PSP, and quality of life measures at all dosages.
Safety
Compared with placebo, mean prolactin levels in the long-term study were four times as high among men (40 vs. 10 ng/mL) and five times as high among women (100 vs. 20 ng/mL) who received paliperidone at any dosage.4 Also, EPS such as dystonia and hyperkinesis were more prevalent at 12 mg/d (10%) than at 6 mg/d (5%).1,2
During paliperidone therapy, patients should be asked if they are experiencing abnormal movements, sexual dysfunction, breast enlargement, or irregular menstruation (women). If so, decrease the dosage by 3 mg and monitor for side effects and clinical efficacy.
Among other reported adverse effects in the efficacy studies:
- somnolence was less prevalent among patients receiving either paliperidone, 3 to 12 mg/d, or placebo (13%) than among those receiving olanzapine, 10 mg/d (25%)3
- tachycardia was more prevalent among the paliperidone and olanzapine groups (14% to 20%) compared with placebo (0%)1-3
- headache prevalence (10% to 20%) was similar in all groups1-3
- mean body weight changes after 6 weeks were more pronounced in the olanzapine group (1.3±2.8 kg) than among patients receiving placebo (–0.7±2.4 kg) or paliperidone at 6 mg/d (0.2±2.4 kg), 9 mg/d (0.6±2.7 kg), or 12 mg/d (0.6±2.6 kg).2
In the relapse prevention study,4 potential neuroleptic malignant syndrome was reported in 1 patient taking paliperidone 3 days after the patient withdrew from the study. The patient had received paliperidone for 19 days and was treated for EPS with an unknown medication during the run-in period. Medical status returned to normal at endpoint except for elevated creatine kinase.
Discontinuation rates in the relapse prevention study’s double-blind phase were greater among the paliperidone group (n=20, 19%) compared with placebo (n=8, 8%).4 The reasons paliperidone group patients withdrew consent were not available.
Related resources
- Karlsson P, Dencker E, Nyberg S, et al. Pharmacokinetics, dopamine D2 and serotonin 5-HT2A receptor occupancy and safety profile of paliperidone extended-release in healthy subjects. Poster presented at: Winter Workshop on Schizophrenia (WWS); February 5-11, 2006; Davos, Switzerland.
Drug brand names
- Olanzapine • Zyprexa
- Risperidone • Risperdal
Disclosures
Dr. Simpson receives grant support from AstraZeneca Pharmaceuticals, GlaxoSmithKline, Janssen Pharmaceutica, and Johnson & Johnson. He is a consultant to Janssen Pharmaceutica, Johnson & Johnson, Merck and Co., and Pfizer, and is a speaker for Janssen Pharmaceutica and Pfizer.
1. Marder S, Kramer M, Ford L, et al. Efficacy and safety of oral paliperidone extended-release tablets in treatment of schizophrenia: a 6-week placebo control study. Poster presented at: Winter Workshop on Schizophrenia (WWS); February 5-11, 2006; Davos, Switzerland.
2. Kane J, Kramer J, Ford L, et al. Treatment of schizophrenia using oral paliperidone extended-release tablets: a 6-week placebo-controlled study. Poster presented at: WWS; February 5-11, 2006; Davos, Switzerland.
3. Davidson M, Emsley R, Kramer M, et al. Efficacy, safety and effect on functioning of paliperidone extended-release in healthy subjects. Poster presented at: WWS; February 5-11, 2006; Davos, Switzerland.
4. Kramer M, Simpson G, Maciulis V, et al. Paliperidone extended release tablets for prevention of symptom recurrence in patients with schizophrenia: A randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol (in press).
Paliperidone, a second-generation antipsychotic (SGA), was awaiting FDA approval for treating schizophrenia (Table 1) at press time. FDA issued an approvable letter September 29.
The long-acting oral medication can be given once daily, without the plasma level peaks and troughs associated with other SGAs.
Table 1
Paliperidone: Fast facts
| Class: Second-generation antipsychotic Prospective indication: Schizophrenia FDA action: Issued approvable letter September 29, 2006 Manufacturer: Janssen Pharmaceutica Dosing forms: Not determined Recommended dosage: Data suggest that 6 mg/d is a suitable starting dosage for most patients.1-3 No formal recommendation issued |
Clinical implications
Paliperidone—the active metabolite of risperidone (9-hydroxy risperidone)—produces the same effects as its parent compound. Because it is metabolized less by the liver, however, paliperidone will likely have a lower risk of drug-drug interactions than risperidone.
Paliperidone can help patients with hallucinations, delusions, and other florid psychotic symptoms. Once-daily dosing could also make it easier for patients with schizophrenia to adhere to treatment.
Paliperidone caused relatively few side effects in clinical trials, indicating that the drug could be started at therapeutic dosages. A 6-mg dose reaches clinically effective plasma levels in approximately 22 hours; a higher dosage might take less time.
How it works
As with all antipsychotics, paliperidone blocks dopamine uptake (D2 receptors) and—as with other newer SGAs—it has a high affinity for 5-HT (serotonin) receptors. This serotonergic action may help modulate side effects.
Paliperidone has shown antipsychotic effectiveness at 3 to 15 mg/d, and a 6-mg dosage is no more likely to cause extrapyramidal symptoms (EPS) or other adverse events than olanzapine, 10 mg/d, or placebo.1-3 This finding suggests that 6 mg/d might be a suitable starting dosage for most patients.
Clinical findings
Efficacy. The drug showed efficacy for treating acute schizophrenia in three randomized, double-blind, controlled trials.
In a 6-week study,1 444 patients who were experiencing acute schizophrenia episodes and had Positive and Negative Syndrome Scale (PANSS) scores between 70 and 120 received paliperidone, 6 or 12 mg/d, olanzapine, 10 mg/d, or placebo. The study was powered to compare paliperidone and placebo; the olanzapine group was included to confirm study sensitivity.
Mean baseline total and negative symptom PANSS scores improved twice as much among the paliperidone and olanzapine groups compared with placebo (Table 2). Personal and Social Performance (PSP) scale scores also improved significantly among patients receiving paliperidone, 6 mg/d. The PSP scale gauges function, ability to perform socially useful activities such as self-care and work, and disturbing and aggressive behavior.
In two similarly designed, 6-week studies (N=1,248),2,3 paliperidone at 3, 6, 9, or 12 mg/d produced statistically significant improvement in total and negative symptom PANSS scores and PSP scale scores compared with placebo.
Table 2
Mean PANSS score reductions among patients taking paliperidone, olanzapine, or placebo
| Paliperidone, 6 mg/d | Paliperidone, 12 mg/d | Olanzapine, 10 mg/d | Placebo | |
|---|---|---|---|---|
| Total PANSS score | 17.5 | 17.5 | 18.4 | 8.0 |
| Negative symptom PANSS score | 4.4 | 3.9 | 4.4 | 2.2 |
| Source: Reference 1 | ||||
Relapse prevention. Kramer et al4 measured paliperidone’s ability to prevent or delay schizophrenia recurrence among 205 patients experiencing an episode. Participants had been diagnosed with schizophrenia at least 1 year earlier and had PANSS total scores between 70 and 120. Patients with substance dependence or other axis I disorders were excluded.
Over an 8-week run-in period, patients were hospitalized for 2 weeks and started on open-label paliperidone, 9 mg/d; dosages then were titrated to 3 to 15 mg/d depending on efficacy and tolerability. Once stable for 2 weeks, subjects were discharged and maintained on paliperidone for 6 weeks, then were randomized to a double-blind phase during which they received a similar dosage of paliperidone or placebo. Regimen duration varied during the double-blind phase.
The study was stopped after 2 months when an interim analysis showed significant efficacy for paliperidone. Among patients who relapsed, mean time to relapse was 68 days among patients taking paliperidone (n=14, 25%), compared with 25 days in the placebo group (n=29, 53%).
Recurrence was defined as:
- psychiatric hospitalization
- total PANSS score ≥40 at randomization decreased by 25% for 2 days, or total PANSS score
- Clinical Global Impression of Severity (CGI-S) score ≥3 at randomization increased to ≥4 for 2 days, or CGI-S score ≥4 at randomization rose to ≥5
- individual-item PANSS baseline score ≥3 increased to ≥5 for 2 days, or baseline score ≥4 rose to ≥6
- self injury, suicidal or homicidal thoughts, or clinically significant aggressive behavior.
A final analysis showed a 22% relapse rate among patients taking paliperidone vs. 52% of the placebo group. Paliperidone was associated with improvements in PANSS, CGI-S, PSP, and quality of life measures at all dosages.
Safety
Compared with placebo, mean prolactin levels in the long-term study were four times as high among men (40 vs. 10 ng/mL) and five times as high among women (100 vs. 20 ng/mL) who received paliperidone at any dosage.4 Also, EPS such as dystonia and hyperkinesis were more prevalent at 12 mg/d (10%) than at 6 mg/d (5%).1,2
During paliperidone therapy, patients should be asked if they are experiencing abnormal movements, sexual dysfunction, breast enlargement, or irregular menstruation (women). If so, decrease the dosage by 3 mg and monitor for side effects and clinical efficacy.
Among other reported adverse effects in the efficacy studies:
- somnolence was less prevalent among patients receiving either paliperidone, 3 to 12 mg/d, or placebo (13%) than among those receiving olanzapine, 10 mg/d (25%)3
- tachycardia was more prevalent among the paliperidone and olanzapine groups (14% to 20%) compared with placebo (0%)1-3
- headache prevalence (10% to 20%) was similar in all groups1-3
- mean body weight changes after 6 weeks were more pronounced in the olanzapine group (1.3±2.8 kg) than among patients receiving placebo (–0.7±2.4 kg) or paliperidone at 6 mg/d (0.2±2.4 kg), 9 mg/d (0.6±2.7 kg), or 12 mg/d (0.6±2.6 kg).2
In the relapse prevention study,4 potential neuroleptic malignant syndrome was reported in 1 patient taking paliperidone 3 days after the patient withdrew from the study. The patient had received paliperidone for 19 days and was treated for EPS with an unknown medication during the run-in period. Medical status returned to normal at endpoint except for elevated creatine kinase.
Discontinuation rates in the relapse prevention study’s double-blind phase were greater among the paliperidone group (n=20, 19%) compared with placebo (n=8, 8%).4 The reasons paliperidone group patients withdrew consent were not available.
Related resources
- Karlsson P, Dencker E, Nyberg S, et al. Pharmacokinetics, dopamine D2 and serotonin 5-HT2A receptor occupancy and safety profile of paliperidone extended-release in healthy subjects. Poster presented at: Winter Workshop on Schizophrenia (WWS); February 5-11, 2006; Davos, Switzerland.
Drug brand names
- Olanzapine • Zyprexa
- Risperidone • Risperdal
Disclosures
Dr. Simpson receives grant support from AstraZeneca Pharmaceuticals, GlaxoSmithKline, Janssen Pharmaceutica, and Johnson & Johnson. He is a consultant to Janssen Pharmaceutica, Johnson & Johnson, Merck and Co., and Pfizer, and is a speaker for Janssen Pharmaceutica and Pfizer.
Paliperidone, a second-generation antipsychotic (SGA), was awaiting FDA approval for treating schizophrenia (Table 1) at press time. FDA issued an approvable letter September 29.
The long-acting oral medication can be given once daily, without the plasma level peaks and troughs associated with other SGAs.
Table 1
Paliperidone: Fast facts
| Class: Second-generation antipsychotic Prospective indication: Schizophrenia FDA action: Issued approvable letter September 29, 2006 Manufacturer: Janssen Pharmaceutica Dosing forms: Not determined Recommended dosage: Data suggest that 6 mg/d is a suitable starting dosage for most patients.1-3 No formal recommendation issued |
Clinical implications
Paliperidone—the active metabolite of risperidone (9-hydroxy risperidone)—produces the same effects as its parent compound. Because it is metabolized less by the liver, however, paliperidone will likely have a lower risk of drug-drug interactions than risperidone.
Paliperidone can help patients with hallucinations, delusions, and other florid psychotic symptoms. Once-daily dosing could also make it easier for patients with schizophrenia to adhere to treatment.
Paliperidone caused relatively few side effects in clinical trials, indicating that the drug could be started at therapeutic dosages. A 6-mg dose reaches clinically effective plasma levels in approximately 22 hours; a higher dosage might take less time.
How it works
As with all antipsychotics, paliperidone blocks dopamine uptake (D2 receptors) and—as with other newer SGAs—it has a high affinity for 5-HT (serotonin) receptors. This serotonergic action may help modulate side effects.
Paliperidone has shown antipsychotic effectiveness at 3 to 15 mg/d, and a 6-mg dosage is no more likely to cause extrapyramidal symptoms (EPS) or other adverse events than olanzapine, 10 mg/d, or placebo.1-3 This finding suggests that 6 mg/d might be a suitable starting dosage for most patients.
Clinical findings
Efficacy. The drug showed efficacy for treating acute schizophrenia in three randomized, double-blind, controlled trials.
In a 6-week study,1 444 patients who were experiencing acute schizophrenia episodes and had Positive and Negative Syndrome Scale (PANSS) scores between 70 and 120 received paliperidone, 6 or 12 mg/d, olanzapine, 10 mg/d, or placebo. The study was powered to compare paliperidone and placebo; the olanzapine group was included to confirm study sensitivity.
Mean baseline total and negative symptom PANSS scores improved twice as much among the paliperidone and olanzapine groups compared with placebo (Table 2). Personal and Social Performance (PSP) scale scores also improved significantly among patients receiving paliperidone, 6 mg/d. The PSP scale gauges function, ability to perform socially useful activities such as self-care and work, and disturbing and aggressive behavior.
In two similarly designed, 6-week studies (N=1,248),2,3 paliperidone at 3, 6, 9, or 12 mg/d produced statistically significant improvement in total and negative symptom PANSS scores and PSP scale scores compared with placebo.
Table 2
Mean PANSS score reductions among patients taking paliperidone, olanzapine, or placebo
| Paliperidone, 6 mg/d | Paliperidone, 12 mg/d | Olanzapine, 10 mg/d | Placebo | |
|---|---|---|---|---|
| Total PANSS score | 17.5 | 17.5 | 18.4 | 8.0 |
| Negative symptom PANSS score | 4.4 | 3.9 | 4.4 | 2.2 |
| Source: Reference 1 | ||||
Relapse prevention. Kramer et al4 measured paliperidone’s ability to prevent or delay schizophrenia recurrence among 205 patients experiencing an episode. Participants had been diagnosed with schizophrenia at least 1 year earlier and had PANSS total scores between 70 and 120. Patients with substance dependence or other axis I disorders were excluded.
Over an 8-week run-in period, patients were hospitalized for 2 weeks and started on open-label paliperidone, 9 mg/d; dosages then were titrated to 3 to 15 mg/d depending on efficacy and tolerability. Once stable for 2 weeks, subjects were discharged and maintained on paliperidone for 6 weeks, then were randomized to a double-blind phase during which they received a similar dosage of paliperidone or placebo. Regimen duration varied during the double-blind phase.
The study was stopped after 2 months when an interim analysis showed significant efficacy for paliperidone. Among patients who relapsed, mean time to relapse was 68 days among patients taking paliperidone (n=14, 25%), compared with 25 days in the placebo group (n=29, 53%).
Recurrence was defined as:
- psychiatric hospitalization
- total PANSS score ≥40 at randomization decreased by 25% for 2 days, or total PANSS score
- Clinical Global Impression of Severity (CGI-S) score ≥3 at randomization increased to ≥4 for 2 days, or CGI-S score ≥4 at randomization rose to ≥5
- individual-item PANSS baseline score ≥3 increased to ≥5 for 2 days, or baseline score ≥4 rose to ≥6
- self injury, suicidal or homicidal thoughts, or clinically significant aggressive behavior.
A final analysis showed a 22% relapse rate among patients taking paliperidone vs. 52% of the placebo group. Paliperidone was associated with improvements in PANSS, CGI-S, PSP, and quality of life measures at all dosages.
Safety
Compared with placebo, mean prolactin levels in the long-term study were four times as high among men (40 vs. 10 ng/mL) and five times as high among women (100 vs. 20 ng/mL) who received paliperidone at any dosage.4 Also, EPS such as dystonia and hyperkinesis were more prevalent at 12 mg/d (10%) than at 6 mg/d (5%).1,2
During paliperidone therapy, patients should be asked if they are experiencing abnormal movements, sexual dysfunction, breast enlargement, or irregular menstruation (women). If so, decrease the dosage by 3 mg and monitor for side effects and clinical efficacy.
Among other reported adverse effects in the efficacy studies:
- somnolence was less prevalent among patients receiving either paliperidone, 3 to 12 mg/d, or placebo (13%) than among those receiving olanzapine, 10 mg/d (25%)3
- tachycardia was more prevalent among the paliperidone and olanzapine groups (14% to 20%) compared with placebo (0%)1-3
- headache prevalence (10% to 20%) was similar in all groups1-3
- mean body weight changes after 6 weeks were more pronounced in the olanzapine group (1.3±2.8 kg) than among patients receiving placebo (–0.7±2.4 kg) or paliperidone at 6 mg/d (0.2±2.4 kg), 9 mg/d (0.6±2.7 kg), or 12 mg/d (0.6±2.6 kg).2
In the relapse prevention study,4 potential neuroleptic malignant syndrome was reported in 1 patient taking paliperidone 3 days after the patient withdrew from the study. The patient had received paliperidone for 19 days and was treated for EPS with an unknown medication during the run-in period. Medical status returned to normal at endpoint except for elevated creatine kinase.
Discontinuation rates in the relapse prevention study’s double-blind phase were greater among the paliperidone group (n=20, 19%) compared with placebo (n=8, 8%).4 The reasons paliperidone group patients withdrew consent were not available.
Related resources
- Karlsson P, Dencker E, Nyberg S, et al. Pharmacokinetics, dopamine D2 and serotonin 5-HT2A receptor occupancy and safety profile of paliperidone extended-release in healthy subjects. Poster presented at: Winter Workshop on Schizophrenia (WWS); February 5-11, 2006; Davos, Switzerland.
Drug brand names
- Olanzapine • Zyprexa
- Risperidone • Risperdal
Disclosures
Dr. Simpson receives grant support from AstraZeneca Pharmaceuticals, GlaxoSmithKline, Janssen Pharmaceutica, and Johnson & Johnson. He is a consultant to Janssen Pharmaceutica, Johnson & Johnson, Merck and Co., and Pfizer, and is a speaker for Janssen Pharmaceutica and Pfizer.
1. Marder S, Kramer M, Ford L, et al. Efficacy and safety of oral paliperidone extended-release tablets in treatment of schizophrenia: a 6-week placebo control study. Poster presented at: Winter Workshop on Schizophrenia (WWS); February 5-11, 2006; Davos, Switzerland.
2. Kane J, Kramer J, Ford L, et al. Treatment of schizophrenia using oral paliperidone extended-release tablets: a 6-week placebo-controlled study. Poster presented at: WWS; February 5-11, 2006; Davos, Switzerland.
3. Davidson M, Emsley R, Kramer M, et al. Efficacy, safety and effect on functioning of paliperidone extended-release in healthy subjects. Poster presented at: WWS; February 5-11, 2006; Davos, Switzerland.
4. Kramer M, Simpson G, Maciulis V, et al. Paliperidone extended release tablets for prevention of symptom recurrence in patients with schizophrenia: A randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol (in press).
1. Marder S, Kramer M, Ford L, et al. Efficacy and safety of oral paliperidone extended-release tablets in treatment of schizophrenia: a 6-week placebo control study. Poster presented at: Winter Workshop on Schizophrenia (WWS); February 5-11, 2006; Davos, Switzerland.
2. Kane J, Kramer J, Ford L, et al. Treatment of schizophrenia using oral paliperidone extended-release tablets: a 6-week placebo-controlled study. Poster presented at: WWS; February 5-11, 2006; Davos, Switzerland.
3. Davidson M, Emsley R, Kramer M, et al. Efficacy, safety and effect on functioning of paliperidone extended-release in healthy subjects. Poster presented at: WWS; February 5-11, 2006; Davos, Switzerland.
4. Kramer M, Simpson G, Maciulis V, et al. Paliperidone extended release tablets for prevention of symptom recurrence in patients with schizophrenia: A randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol (in press).
Transdermal methylphenidate
When prescribing methylphenidate to children with attention-deficit/hyperactivity disorder (ADHD), psychiatrists have had two options:
- immediate-release oral methylphenidate, which works for 3 to 5 hours, necessitating multiple daily doses
- extended-release oral methylphenidate, which can prevent irritability and other rebound symptoms caused by multiple daily dosing.1 Because its effects last 12 hours, however, once-daily dosing with this formulation is inflexible.
A new option—a transdermal methylphenidate patch FDA-approved for treating ADHD in children ages 6 to 12 (Table 1)—offers flexible methylphenidate coverage based on response to or need for the medication.
Table 1
Transdermal methylphenidate: Fast facts
| Brand name: Daytrana |
| Class: CNS stimulant |
| FDA-approved indication: ADHD in children ages 6 to 12 |
| Manufacturer: Noven Pharmaceuticals (marketed by Shire PLC) |
| Dosing forms: 10-, 15-, 20-, and 30-mg patches |
| Recommended dosage: One 10- to 30-mg patch daily, worn on the hip for 9 hours. Patient can remove patch sooner if side effects become problematic. |
Clinical implications
The transdermal patch allows dosing to be tailored—or changed day to day as needed—to maximize effectiveness and reduce side-effect risk. The manufacturer recommends that the patch be worn for 9 hours daily, but it can be removed sooner if children experience appetite loss, insomnia, or other adverse effects with 9 hours of exposure to methylphenidate.
Minimizing daily exposure to methylphenidate can also reduce the risk of long-term effects. Findings from one large, randomized clinical trial2 suggest that chronic exposure to high-dose stimulant medications might suppress growth in height and weight, although other data indicate that initial height reductions found in children receiving methylphenidate for ADHD were no longer significant in adulthood.3
The patch also could benefit youths who have trouble following dosing schedules and young children who are unable to swallow pills.
How it works
The patch contains methylphenidate dispersed in an acrylic multipolymeric adhesive that is further dispersed in a silicone adhesive.4 The methylphenidate within the acrylic adhesive flows into the skin, then into the bloodstream. The patch is worn on the hip—where it is covered by clothing and unlikely to be dislodged—and changed daily.
The patch comes in four sizes—12.5, 18.75, 25, and 37.5 cm2—which, respectively, deliver 10, 15, 20, and 30 mg of methylphenidate over 9 hours.4 Methylphenidate concentration is the same for all four sizes, so patch size and duration of use determine dose delivery.
In clinical trials, therapeutic effect was seen 2 hours after patch placement and continued through 12 hours.5 Methylphenidate is delivered continuously while the patch is in place and for as long as 2 hours after it is removed.
Pharmacokinetics
Methylphenidate, a known CNS stimulant, blocks norepinephrine and dopamine reuptake in the presynaptic neuron, thereby releasing more of these neuro-transmitters into the extraneuronal space.4 Methylphenidate’s precise therapeutic action in ADHD is not known.
Methylphenidate is a racemic mixture of d- and l-enantiomers, the first of which is believed to be more active. Whereas the liver removes the l-enantiomer from oral methylphenidate, the transdermal formulation bypasses the liver and preserves the l-enantiomer, thus increasing exposure to racemic methylphenidate. This means that optimal dosages of transdermal methylphenidate (10 to 30 mg/d) may be lower compared with the oral formulation5-7 (Table 2).
Methylphenidate’s d-enantiomer has a mean 3- to 4-hour elimination half-life, approximately twice that of the l-enantiomer. This is why transdermal methylphenidate continues to exert therapeutic effect several hours after the patch is removed.5
Table 2
Transdermal methylphenidate dosage delivery in children ages 6 to 12
| Dose delivered over 9 hours (mg) | Patch size (cm2) | Dosage rate (mg/hr) | Methylphenidate content per patch (mg) |
|---|---|---|---|
| 10 | 12.5 | 1.1 | 27.5 |
| 15 | 18.75 | 1.6 | 41.3 |
| 20 | 25 | 2.2 | 55.0 |
| 30 | 37.5 | 3.3 | 82.5 |
| Source: Reference 4 | |||
Efficacy
Results from randomized, double-blind, placebo-controlled trials support short-term use of transdermal methylphenidate in ADHD. The following studies recruited children ages 6 to 12 with the disorder.
Dose-ranging study. Thirty-three children participating in a summer treatment program received transdermal methylphenidate, 6.25, 12.5, or 25 cm2 12 hours daily for 8 days.6 All three patch sizes were associated with improved academic, social, and behavioral functioning based on a range of measures.
Dose response rate diminished with higher dosages, and significant further improvements were difficult to detect as dosages increased. The children also received intensive behavioral treatment during the study, which might have accounted for some therapeutic gains and diminished the researchers’ ability to detect subtle improvements with increased dosages.
Children also had fewer negative behaviors during the first hour when the patch was applied at 6 AM instead of 7 AM. This suggests that the patch might produce optimal effect when placed first thing in the morning.
Randomized crossover trial.7 Across 6 weeks, 27 children were given placebo or transdermal methylphenidate, 12.5, 25, or 37.5 cm2/d. The children also received behavior modification treatment on alternating weeks. Medication was randomly assigned and varied daily for 4 days per week over 6 weeks, and behavioral treatment was varied weekly for 4 weeks. Each subject took each dosage for 2 days without behavioral treatment and for 4 days with behavioral treatment.
Academic productivity, interactions with peers and adults, and compliance during class improved with all dosages compared with placebo. Although most children removed the patch by 3:30 PM after 9 hours of use, parents reported that positive behavioral effects lasted into the evening.
As in the dose-ranging study, dose response diminished with higher dosages. Optimal effects were achieved on some measures with 12.5 cm2 of transdermal methylphenidate, which produces the same plasma drug level as 10 mg of oral methylphenidate.7
Multisite crossover study.5 Eighty children received transdermal methylphenidate—12.5, 18.75, 25, or 37.5 cm2 based on response to medication—over 5 weeks for 9 hours daily. Dosages were titrated by changing patch sizes until each child reached his or her optimal dosage. Children then received their optimal dosage or placebo for 1 week, then received the opposite treatment for another week. Results were measured in a simulated classroom.
Overall, children showed statistically significant improvement in Swanson, Kotkin, Agler, M-Flynn, and Pelham (SKAMP) Teacher Rating Scale scores while receiving their optimal dosage. Improvement was seen 2 hours after patches were applied and continued for 12 hours. Children also were more able to solve math problems during optimal dosage periods than while using placebo.
Nearly 80% of children were rated as significantly improved while receiving transdermal methylphenidate based on Clinical Global Impressions of Improvement scores. Roughly 12% of children showed significant improvement with placebo. Parents reported that their children were markedly less hyperactive and impulsive and more attentive during optimal dosage periods.
Tolerability
No serious side effects were reported during clinical trials of transdermal methylphenidate in children ages 6 to 12.5-7 Side effects commonly associated with oral methylphenidate—anorexia, decreased appetite, headache, insomnia, and abdominal pain—were most frequently reported with the patch.5
In one study,6 61% of children who wore the patch for 12 hours/day reported appetite loss and 47% reported insomnia. Insomnia prevalence diminished substantially when daily wear was limited to 9 hours.2,5 Loss of appetite was reported less often with lower-dose patches (12.5, 18.75 cm2) than with higher-dose patches (25, 37.5 cm2).
Although many children complained of erythema at the patch site,5-7 most reported minimal irritation or discomfort.5 Redness usually dissipated about 8 hours after the patch was removed.6
Despite concerns that youths with impulsive behaviors might remove the patches prematurely, very few children did so during clinical trials.5-7 Those who did had comorbid symptomatic conduct disturbances. Compliance with patch placement and maintenance was very high during dose optimization (98%) and analog classroom analysis (97%).5-7
Dosing
Start transdermal methylphenidate at 12.5 cm2 (10 mg) for children who have never taken methylphenidate or were previously stabilized on the drug. If the child does not respond after 1 week, switch to the 18.75 cm2 (15-mg) patch; keep switching to the next largest patch each week until optimal response is achieved. In clinical trials,5-7 18.75 or 25 cm2 (10 mg) of transdermal methylphenidate produced optimal response for most children.
Advise the child and parents to place the patch on the right and left hip on alternate days to minimize irritation. Counsel children to inform parents if the patch causes itching, burning, or irritation; tell parents to call you if they notice or the child complains of irritation. Children who experience intolerable skin sensitivity with the patch can resume taking oral methylphenidate the day after the patch is removed.
As with oral methylphenidate, the transdermal formulation may be discontinued without a taper and another formulation or medication may be started the next day. Ask the child and parents about side effects at each visit. See the child every 2 to 4 weeks during the titration period and monthly after symptoms are stabilized.
Consider trying a “drug holiday” for at least 2 weeks during the summer to see how the child behaves without methylphenidate, then evaluate the need for medication and determine the optimal dosage close to when the school year begins.
Related resources
- Transdermal methylphenidate Web site. www.daytrana.com.
- Greenhill LL, Pliszka S, Dulcan MK, et al. Practice parameter for the use of stimulant medications in the treatment of children, adolescents, and adults. J Am Acad Child Adolesc Psychiatry 2002;41:26S-49S.
Drug brand names
- Methylphenidate (oral) • Concerta, Ritalin, Metadate
- Methylphenidate (transdermal) • Daytrana
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Spencer TJ. ADHD treatment across the life cycle. J Clin Psychiatry 2004;65S3:22-6.
2. MTA Cooperative Group. National Institute of Mental Health multimodal treatment study of ADHD follow-up: changes in effectiveness and growth at the end of treatment. Pediatrics 2004;113:762-9.
3. Klein RG, Mannuzza S. Hyperactive boys almost grown up. III. Methylphenidate effects on ultimate height. Arch Gen Psychiatry 1988;45:1131-4.
4. Transdermal methylphenidate Web site. Available at: http://www.daytrana.com. Accessed April 11, 2006.
5. McGough JJ, Wigal SB, Abikoff H, et al. A randomized, double-blind, placebo controlled laboratory classroom assessment of methylphenidate transdermal system in children with ADHD. J Atten Disord 2006;9:476-85.
6. Pelham WE, Jr, Manos MJ, Ezzell CE, et al. A dose-ranging study of a methylphenidate transdermal system in children with ADHD. J Am Acad Child Adolesc Psychiatry 2005;44:522-9.
7. Pelham WE, Burrows-Maclean L, Gnagy EM, et al. Transdermal methylphenidate, behavioral, and combined treatment for children with ADHD. Exp Clin Pharmacol 2005;13:111-26.
When prescribing methylphenidate to children with attention-deficit/hyperactivity disorder (ADHD), psychiatrists have had two options:
- immediate-release oral methylphenidate, which works for 3 to 5 hours, necessitating multiple daily doses
- extended-release oral methylphenidate, which can prevent irritability and other rebound symptoms caused by multiple daily dosing.1 Because its effects last 12 hours, however, once-daily dosing with this formulation is inflexible.
A new option—a transdermal methylphenidate patch FDA-approved for treating ADHD in children ages 6 to 12 (Table 1)—offers flexible methylphenidate coverage based on response to or need for the medication.
Table 1
Transdermal methylphenidate: Fast facts
| Brand name: Daytrana |
| Class: CNS stimulant |
| FDA-approved indication: ADHD in children ages 6 to 12 |
| Manufacturer: Noven Pharmaceuticals (marketed by Shire PLC) |
| Dosing forms: 10-, 15-, 20-, and 30-mg patches |
| Recommended dosage: One 10- to 30-mg patch daily, worn on the hip for 9 hours. Patient can remove patch sooner if side effects become problematic. |
Clinical implications
The transdermal patch allows dosing to be tailored—or changed day to day as needed—to maximize effectiveness and reduce side-effect risk. The manufacturer recommends that the patch be worn for 9 hours daily, but it can be removed sooner if children experience appetite loss, insomnia, or other adverse effects with 9 hours of exposure to methylphenidate.
Minimizing daily exposure to methylphenidate can also reduce the risk of long-term effects. Findings from one large, randomized clinical trial2 suggest that chronic exposure to high-dose stimulant medications might suppress growth in height and weight, although other data indicate that initial height reductions found in children receiving methylphenidate for ADHD were no longer significant in adulthood.3
The patch also could benefit youths who have trouble following dosing schedules and young children who are unable to swallow pills.
How it works
The patch contains methylphenidate dispersed in an acrylic multipolymeric adhesive that is further dispersed in a silicone adhesive.4 The methylphenidate within the acrylic adhesive flows into the skin, then into the bloodstream. The patch is worn on the hip—where it is covered by clothing and unlikely to be dislodged—and changed daily.
The patch comes in four sizes—12.5, 18.75, 25, and 37.5 cm2—which, respectively, deliver 10, 15, 20, and 30 mg of methylphenidate over 9 hours.4 Methylphenidate concentration is the same for all four sizes, so patch size and duration of use determine dose delivery.
In clinical trials, therapeutic effect was seen 2 hours after patch placement and continued through 12 hours.5 Methylphenidate is delivered continuously while the patch is in place and for as long as 2 hours after it is removed.
Pharmacokinetics
Methylphenidate, a known CNS stimulant, blocks norepinephrine and dopamine reuptake in the presynaptic neuron, thereby releasing more of these neuro-transmitters into the extraneuronal space.4 Methylphenidate’s precise therapeutic action in ADHD is not known.
Methylphenidate is a racemic mixture of d- and l-enantiomers, the first of which is believed to be more active. Whereas the liver removes the l-enantiomer from oral methylphenidate, the transdermal formulation bypasses the liver and preserves the l-enantiomer, thus increasing exposure to racemic methylphenidate. This means that optimal dosages of transdermal methylphenidate (10 to 30 mg/d) may be lower compared with the oral formulation5-7 (Table 2).
Methylphenidate’s d-enantiomer has a mean 3- to 4-hour elimination half-life, approximately twice that of the l-enantiomer. This is why transdermal methylphenidate continues to exert therapeutic effect several hours after the patch is removed.5
Table 2
Transdermal methylphenidate dosage delivery in children ages 6 to 12
| Dose delivered over 9 hours (mg) | Patch size (cm2) | Dosage rate (mg/hr) | Methylphenidate content per patch (mg) |
|---|---|---|---|
| 10 | 12.5 | 1.1 | 27.5 |
| 15 | 18.75 | 1.6 | 41.3 |
| 20 | 25 | 2.2 | 55.0 |
| 30 | 37.5 | 3.3 | 82.5 |
| Source: Reference 4 | |||
Efficacy
Results from randomized, double-blind, placebo-controlled trials support short-term use of transdermal methylphenidate in ADHD. The following studies recruited children ages 6 to 12 with the disorder.
Dose-ranging study. Thirty-three children participating in a summer treatment program received transdermal methylphenidate, 6.25, 12.5, or 25 cm2 12 hours daily for 8 days.6 All three patch sizes were associated with improved academic, social, and behavioral functioning based on a range of measures.
Dose response rate diminished with higher dosages, and significant further improvements were difficult to detect as dosages increased. The children also received intensive behavioral treatment during the study, which might have accounted for some therapeutic gains and diminished the researchers’ ability to detect subtle improvements with increased dosages.
Children also had fewer negative behaviors during the first hour when the patch was applied at 6 AM instead of 7 AM. This suggests that the patch might produce optimal effect when placed first thing in the morning.
Randomized crossover trial.7 Across 6 weeks, 27 children were given placebo or transdermal methylphenidate, 12.5, 25, or 37.5 cm2/d. The children also received behavior modification treatment on alternating weeks. Medication was randomly assigned and varied daily for 4 days per week over 6 weeks, and behavioral treatment was varied weekly for 4 weeks. Each subject took each dosage for 2 days without behavioral treatment and for 4 days with behavioral treatment.
Academic productivity, interactions with peers and adults, and compliance during class improved with all dosages compared with placebo. Although most children removed the patch by 3:30 PM after 9 hours of use, parents reported that positive behavioral effects lasted into the evening.
As in the dose-ranging study, dose response diminished with higher dosages. Optimal effects were achieved on some measures with 12.5 cm2 of transdermal methylphenidate, which produces the same plasma drug level as 10 mg of oral methylphenidate.7
Multisite crossover study.5 Eighty children received transdermal methylphenidate—12.5, 18.75, 25, or 37.5 cm2 based on response to medication—over 5 weeks for 9 hours daily. Dosages were titrated by changing patch sizes until each child reached his or her optimal dosage. Children then received their optimal dosage or placebo for 1 week, then received the opposite treatment for another week. Results were measured in a simulated classroom.
Overall, children showed statistically significant improvement in Swanson, Kotkin, Agler, M-Flynn, and Pelham (SKAMP) Teacher Rating Scale scores while receiving their optimal dosage. Improvement was seen 2 hours after patches were applied and continued for 12 hours. Children also were more able to solve math problems during optimal dosage periods than while using placebo.
Nearly 80% of children were rated as significantly improved while receiving transdermal methylphenidate based on Clinical Global Impressions of Improvement scores. Roughly 12% of children showed significant improvement with placebo. Parents reported that their children were markedly less hyperactive and impulsive and more attentive during optimal dosage periods.
Tolerability
No serious side effects were reported during clinical trials of transdermal methylphenidate in children ages 6 to 12.5-7 Side effects commonly associated with oral methylphenidate—anorexia, decreased appetite, headache, insomnia, and abdominal pain—were most frequently reported with the patch.5
In one study,6 61% of children who wore the patch for 12 hours/day reported appetite loss and 47% reported insomnia. Insomnia prevalence diminished substantially when daily wear was limited to 9 hours.2,5 Loss of appetite was reported less often with lower-dose patches (12.5, 18.75 cm2) than with higher-dose patches (25, 37.5 cm2).
Although many children complained of erythema at the patch site,5-7 most reported minimal irritation or discomfort.5 Redness usually dissipated about 8 hours after the patch was removed.6
Despite concerns that youths with impulsive behaviors might remove the patches prematurely, very few children did so during clinical trials.5-7 Those who did had comorbid symptomatic conduct disturbances. Compliance with patch placement and maintenance was very high during dose optimization (98%) and analog classroom analysis (97%).5-7
Dosing
Start transdermal methylphenidate at 12.5 cm2 (10 mg) for children who have never taken methylphenidate or were previously stabilized on the drug. If the child does not respond after 1 week, switch to the 18.75 cm2 (15-mg) patch; keep switching to the next largest patch each week until optimal response is achieved. In clinical trials,5-7 18.75 or 25 cm2 (10 mg) of transdermal methylphenidate produced optimal response for most children.
Advise the child and parents to place the patch on the right and left hip on alternate days to minimize irritation. Counsel children to inform parents if the patch causes itching, burning, or irritation; tell parents to call you if they notice or the child complains of irritation. Children who experience intolerable skin sensitivity with the patch can resume taking oral methylphenidate the day after the patch is removed.
As with oral methylphenidate, the transdermal formulation may be discontinued without a taper and another formulation or medication may be started the next day. Ask the child and parents about side effects at each visit. See the child every 2 to 4 weeks during the titration period and monthly after symptoms are stabilized.
Consider trying a “drug holiday” for at least 2 weeks during the summer to see how the child behaves without methylphenidate, then evaluate the need for medication and determine the optimal dosage close to when the school year begins.
Related resources
- Transdermal methylphenidate Web site. www.daytrana.com.
- Greenhill LL, Pliszka S, Dulcan MK, et al. Practice parameter for the use of stimulant medications in the treatment of children, adolescents, and adults. J Am Acad Child Adolesc Psychiatry 2002;41:26S-49S.
Drug brand names
- Methylphenidate (oral) • Concerta, Ritalin, Metadate
- Methylphenidate (transdermal) • Daytrana
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
When prescribing methylphenidate to children with attention-deficit/hyperactivity disorder (ADHD), psychiatrists have had two options:
- immediate-release oral methylphenidate, which works for 3 to 5 hours, necessitating multiple daily doses
- extended-release oral methylphenidate, which can prevent irritability and other rebound symptoms caused by multiple daily dosing.1 Because its effects last 12 hours, however, once-daily dosing with this formulation is inflexible.
A new option—a transdermal methylphenidate patch FDA-approved for treating ADHD in children ages 6 to 12 (Table 1)—offers flexible methylphenidate coverage based on response to or need for the medication.
Table 1
Transdermal methylphenidate: Fast facts
| Brand name: Daytrana |
| Class: CNS stimulant |
| FDA-approved indication: ADHD in children ages 6 to 12 |
| Manufacturer: Noven Pharmaceuticals (marketed by Shire PLC) |
| Dosing forms: 10-, 15-, 20-, and 30-mg patches |
| Recommended dosage: One 10- to 30-mg patch daily, worn on the hip for 9 hours. Patient can remove patch sooner if side effects become problematic. |
Clinical implications
The transdermal patch allows dosing to be tailored—or changed day to day as needed—to maximize effectiveness and reduce side-effect risk. The manufacturer recommends that the patch be worn for 9 hours daily, but it can be removed sooner if children experience appetite loss, insomnia, or other adverse effects with 9 hours of exposure to methylphenidate.
Minimizing daily exposure to methylphenidate can also reduce the risk of long-term effects. Findings from one large, randomized clinical trial2 suggest that chronic exposure to high-dose stimulant medications might suppress growth in height and weight, although other data indicate that initial height reductions found in children receiving methylphenidate for ADHD were no longer significant in adulthood.3
The patch also could benefit youths who have trouble following dosing schedules and young children who are unable to swallow pills.
How it works
The patch contains methylphenidate dispersed in an acrylic multipolymeric adhesive that is further dispersed in a silicone adhesive.4 The methylphenidate within the acrylic adhesive flows into the skin, then into the bloodstream. The patch is worn on the hip—where it is covered by clothing and unlikely to be dislodged—and changed daily.
The patch comes in four sizes—12.5, 18.75, 25, and 37.5 cm2—which, respectively, deliver 10, 15, 20, and 30 mg of methylphenidate over 9 hours.4 Methylphenidate concentration is the same for all four sizes, so patch size and duration of use determine dose delivery.
In clinical trials, therapeutic effect was seen 2 hours after patch placement and continued through 12 hours.5 Methylphenidate is delivered continuously while the patch is in place and for as long as 2 hours after it is removed.
Pharmacokinetics
Methylphenidate, a known CNS stimulant, blocks norepinephrine and dopamine reuptake in the presynaptic neuron, thereby releasing more of these neuro-transmitters into the extraneuronal space.4 Methylphenidate’s precise therapeutic action in ADHD is not known.
Methylphenidate is a racemic mixture of d- and l-enantiomers, the first of which is believed to be more active. Whereas the liver removes the l-enantiomer from oral methylphenidate, the transdermal formulation bypasses the liver and preserves the l-enantiomer, thus increasing exposure to racemic methylphenidate. This means that optimal dosages of transdermal methylphenidate (10 to 30 mg/d) may be lower compared with the oral formulation5-7 (Table 2).
Methylphenidate’s d-enantiomer has a mean 3- to 4-hour elimination half-life, approximately twice that of the l-enantiomer. This is why transdermal methylphenidate continues to exert therapeutic effect several hours after the patch is removed.5
Table 2
Transdermal methylphenidate dosage delivery in children ages 6 to 12
| Dose delivered over 9 hours (mg) | Patch size (cm2) | Dosage rate (mg/hr) | Methylphenidate content per patch (mg) |
|---|---|---|---|
| 10 | 12.5 | 1.1 | 27.5 |
| 15 | 18.75 | 1.6 | 41.3 |
| 20 | 25 | 2.2 | 55.0 |
| 30 | 37.5 | 3.3 | 82.5 |
| Source: Reference 4 | |||
Efficacy
Results from randomized, double-blind, placebo-controlled trials support short-term use of transdermal methylphenidate in ADHD. The following studies recruited children ages 6 to 12 with the disorder.
Dose-ranging study. Thirty-three children participating in a summer treatment program received transdermal methylphenidate, 6.25, 12.5, or 25 cm2 12 hours daily for 8 days.6 All three patch sizes were associated with improved academic, social, and behavioral functioning based on a range of measures.
Dose response rate diminished with higher dosages, and significant further improvements were difficult to detect as dosages increased. The children also received intensive behavioral treatment during the study, which might have accounted for some therapeutic gains and diminished the researchers’ ability to detect subtle improvements with increased dosages.
Children also had fewer negative behaviors during the first hour when the patch was applied at 6 AM instead of 7 AM. This suggests that the patch might produce optimal effect when placed first thing in the morning.
Randomized crossover trial.7 Across 6 weeks, 27 children were given placebo or transdermal methylphenidate, 12.5, 25, or 37.5 cm2/d. The children also received behavior modification treatment on alternating weeks. Medication was randomly assigned and varied daily for 4 days per week over 6 weeks, and behavioral treatment was varied weekly for 4 weeks. Each subject took each dosage for 2 days without behavioral treatment and for 4 days with behavioral treatment.
Academic productivity, interactions with peers and adults, and compliance during class improved with all dosages compared with placebo. Although most children removed the patch by 3:30 PM after 9 hours of use, parents reported that positive behavioral effects lasted into the evening.
As in the dose-ranging study, dose response diminished with higher dosages. Optimal effects were achieved on some measures with 12.5 cm2 of transdermal methylphenidate, which produces the same plasma drug level as 10 mg of oral methylphenidate.7
Multisite crossover study.5 Eighty children received transdermal methylphenidate—12.5, 18.75, 25, or 37.5 cm2 based on response to medication—over 5 weeks for 9 hours daily. Dosages were titrated by changing patch sizes until each child reached his or her optimal dosage. Children then received their optimal dosage or placebo for 1 week, then received the opposite treatment for another week. Results were measured in a simulated classroom.
Overall, children showed statistically significant improvement in Swanson, Kotkin, Agler, M-Flynn, and Pelham (SKAMP) Teacher Rating Scale scores while receiving their optimal dosage. Improvement was seen 2 hours after patches were applied and continued for 12 hours. Children also were more able to solve math problems during optimal dosage periods than while using placebo.
Nearly 80% of children were rated as significantly improved while receiving transdermal methylphenidate based on Clinical Global Impressions of Improvement scores. Roughly 12% of children showed significant improvement with placebo. Parents reported that their children were markedly less hyperactive and impulsive and more attentive during optimal dosage periods.
Tolerability
No serious side effects were reported during clinical trials of transdermal methylphenidate in children ages 6 to 12.5-7 Side effects commonly associated with oral methylphenidate—anorexia, decreased appetite, headache, insomnia, and abdominal pain—were most frequently reported with the patch.5
In one study,6 61% of children who wore the patch for 12 hours/day reported appetite loss and 47% reported insomnia. Insomnia prevalence diminished substantially when daily wear was limited to 9 hours.2,5 Loss of appetite was reported less often with lower-dose patches (12.5, 18.75 cm2) than with higher-dose patches (25, 37.5 cm2).
Although many children complained of erythema at the patch site,5-7 most reported minimal irritation or discomfort.5 Redness usually dissipated about 8 hours after the patch was removed.6
Despite concerns that youths with impulsive behaviors might remove the patches prematurely, very few children did so during clinical trials.5-7 Those who did had comorbid symptomatic conduct disturbances. Compliance with patch placement and maintenance was very high during dose optimization (98%) and analog classroom analysis (97%).5-7
Dosing
Start transdermal methylphenidate at 12.5 cm2 (10 mg) for children who have never taken methylphenidate or were previously stabilized on the drug. If the child does not respond after 1 week, switch to the 18.75 cm2 (15-mg) patch; keep switching to the next largest patch each week until optimal response is achieved. In clinical trials,5-7 18.75 or 25 cm2 (10 mg) of transdermal methylphenidate produced optimal response for most children.
Advise the child and parents to place the patch on the right and left hip on alternate days to minimize irritation. Counsel children to inform parents if the patch causes itching, burning, or irritation; tell parents to call you if they notice or the child complains of irritation. Children who experience intolerable skin sensitivity with the patch can resume taking oral methylphenidate the day after the patch is removed.
As with oral methylphenidate, the transdermal formulation may be discontinued without a taper and another formulation or medication may be started the next day. Ask the child and parents about side effects at each visit. See the child every 2 to 4 weeks during the titration period and monthly after symptoms are stabilized.
Consider trying a “drug holiday” for at least 2 weeks during the summer to see how the child behaves without methylphenidate, then evaluate the need for medication and determine the optimal dosage close to when the school year begins.
Related resources
- Transdermal methylphenidate Web site. www.daytrana.com.
- Greenhill LL, Pliszka S, Dulcan MK, et al. Practice parameter for the use of stimulant medications in the treatment of children, adolescents, and adults. J Am Acad Child Adolesc Psychiatry 2002;41:26S-49S.
Drug brand names
- Methylphenidate (oral) • Concerta, Ritalin, Metadate
- Methylphenidate (transdermal) • Daytrana
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Spencer TJ. ADHD treatment across the life cycle. J Clin Psychiatry 2004;65S3:22-6.
2. MTA Cooperative Group. National Institute of Mental Health multimodal treatment study of ADHD follow-up: changes in effectiveness and growth at the end of treatment. Pediatrics 2004;113:762-9.
3. Klein RG, Mannuzza S. Hyperactive boys almost grown up. III. Methylphenidate effects on ultimate height. Arch Gen Psychiatry 1988;45:1131-4.
4. Transdermal methylphenidate Web site. Available at: http://www.daytrana.com. Accessed April 11, 2006.
5. McGough JJ, Wigal SB, Abikoff H, et al. A randomized, double-blind, placebo controlled laboratory classroom assessment of methylphenidate transdermal system in children with ADHD. J Atten Disord 2006;9:476-85.
6. Pelham WE, Jr, Manos MJ, Ezzell CE, et al. A dose-ranging study of a methylphenidate transdermal system in children with ADHD. J Am Acad Child Adolesc Psychiatry 2005;44:522-9.
7. Pelham WE, Burrows-Maclean L, Gnagy EM, et al. Transdermal methylphenidate, behavioral, and combined treatment for children with ADHD. Exp Clin Pharmacol 2005;13:111-26.
1. Spencer TJ. ADHD treatment across the life cycle. J Clin Psychiatry 2004;65S3:22-6.
2. MTA Cooperative Group. National Institute of Mental Health multimodal treatment study of ADHD follow-up: changes in effectiveness and growth at the end of treatment. Pediatrics 2004;113:762-9.
3. Klein RG, Mannuzza S. Hyperactive boys almost grown up. III. Methylphenidate effects on ultimate height. Arch Gen Psychiatry 1988;45:1131-4.
4. Transdermal methylphenidate Web site. Available at: http://www.daytrana.com. Accessed April 11, 2006.
5. McGough JJ, Wigal SB, Abikoff H, et al. A randomized, double-blind, placebo controlled laboratory classroom assessment of methylphenidate transdermal system in children with ADHD. J Atten Disord 2006;9:476-85.
6. Pelham WE, Jr, Manos MJ, Ezzell CE, et al. A dose-ranging study of a methylphenidate transdermal system in children with ADHD. J Am Acad Child Adolesc Psychiatry 2005;44:522-9.
7. Pelham WE, Burrows-Maclean L, Gnagy EM, et al. Transdermal methylphenidate, behavioral, and combined treatment for children with ADHD. Exp Clin Pharmacol 2005;13:111-26.
Transdermal selegiline
Many psychiatrists do not prescribe monoamine oxidase inhibitors (MAOIs) for fear of causing a potentially fatal hypertensive reaction, even though restricting foods high in the amino acid tyramine usually prevents this effect.1 Consequently, most depressed patients who might respond well to MAOIs do not receive them.2,3
Transdermal selegiline, FDA-approved for treating major depressive disorder (MDD) (Table 1), offers the clinical efficacy of an MAOI but without adverse interactions with food at the 6-mg strength. Transdermal selegiline may inhibit too much gastrointestinal MAO-A at 9 mg/d and 12 mg/d to clear tyramine from foods, so tyramine-rich foods must be restricted at these dosages (Table 2).
Table 1
Transdermal selegiline: Fast facts
| Brand name: EMSAM |
| Class: Monoamine oxidase inhibitor |
| FDA-approved indication: Major depressive disorder |
| Manufacturer: Somerset Pharmaceuticals (marketed by Bristol-Myers Squibb Co.) |
| Dosing forms: 6-, 9-, and 12-mg patches |
| Recommended dosage: One 6-mg patch every 24 hours, worn on the chest, back, or stomach. Increase dosage after 2 to 3 months if clinical response is inadequate |
Table 2
Restrict these foods when prescribing transdermal selegiline at 9 or 12 mg/d
| Food/beverage class | Foods to avoid |
|---|---|
| Beverages | Tap beer |
| Beer that has not been pasteurized* | |
| Red wines | |
| Dairy | Aged cheeses |
| Meat, poultry, fish | Air-dried, aged, and fermented meats, sausages, and salamis (including cacciatore and mortadella) |
| Pickled herring | |
| Spoiled or improperly stored fish, meat, poultry, or animal livers (check for mold, discoloration, or odor) | |
| Vegetables | Broad bean pods (fava beans) |
| Miscellaneous | Concentrated yeast extract (such as Marmite) |
| Fermented soybean products (including soy sauce) | |
| Over-the-counter supplements containing tyramine | |
| Sauerkraut | |
| *Bottled and canned beer and white wine contain little or no tyramine, but more than moderate alcohol use while taking selegiline is not recommended. | |
| Source: Shulman KI, Walker SE. A reevaluation of dietary restrictions for irreversible monoamine oxidase inhibitors. Psychiatr Ann 2001;31:378-84. | |
How it works
MAO enzyme subtypes A and B metabolize CNS monoamines, but primarily MAO-A metabolizes tyramine in the gut before the amino acid enters systemic circulation. At low concentrations, selegiline selectively inhibits MAO-B.4
Oral selegiline, approved as a adjunct to levodopa/carbidopa for patients with Parkinson’s disease,5 has been shown to be effective for treating depression at ≥30 mg/d.6 Because the drug does not selectively inhibit MAOB at ≥20 mg/d, dietary tyramine must be restricted when oral selegiline is used off-label at therapeutic dosages for depression. Otherwise, selegiline has been well-tolerated up to 60 mg/d.7
The 6-mg “patch” delivers more selegiline to the bloodstream than does low-dose oral selegiline but without inhibiting gut MAO-A. This provides the brain MAO-A and MAO-B inhibition necessary for an antidepressant effect while eliminating the need for dietary restrictions at this lowest dosage.
Clinical implications
Transdermal selegiline offers an MAOI antidepressant option that might help:
- patients whose depression has not responded satisfactorily to selective serotonin reuptake inhibitors (SSRIs) or serotonin and norepinephrine reuptake inhibitors (SNRIs)
- adults and children with chronic depression marked by atypical features, including reactive mood, rejection sensitivity, anergia, and reversed vegetative symptoms—such as oversleeping, overeating, and psychomotor retardation. Although transdermal selegiline’s efficacy against these features has not been studied, patients with this depressive subtype tend to respond preferentially to MAOIs.
Pharmacokinetics
Transdermal selegiline achieves therapeutic blood levels and reaches sustained concentration within 4 to 8 hours of administration. Compared with oral selegiline, transdermal delivery results in higher plasma selegiline concentrations (1,500 pg/mL with the 6-mg patch) with much lower exposure to metabolites.8 The concentration is maintained with successive doses.
Transdermal selegiline clears rapidly upon discontinuation but MAO inhibition persists for 2 weeks, so wait 2 weeks after the last dose before starting a new antidepressant or stopping food restrictions with the 9-mg and 12-mg patches.
Efficacy
In two randomized, double-blind clinical trials,9,10 a total of 466 adults ages 18 to 65 who met DSM-IV-TR criteria for MDD received transdermal selegiline, 6 mg/d, or placebo for 6 to 8 weeks. Participants had 17-item Hamilton Rating Scale for Depression (HAM-D-17) scores ≥20 at baseline.
In the 6-week study,9 transdermal selegiline produced a 46% greater reduction in HAM-D-17 scores, a 52% greater decrease in HAM-D-28 scores, and a 79% greater drop in Montgomery-Asburg Depression Rating Scale (MADRS) scores compared with placebo. In the 8-week trial,10 HAM-D-28 and MADRS scores among the treatment group were significantly improved at endpoint compared with placebo, but HAM-D-17 scores were not.
In a 1-year, double-blind study,11 322 subjects with MDD—who had been rated as responders in a 10-week, open-label transdermal selegiline trial—received the 6-mg patch or placebo. At 6 months and 1 year, relapse was much less frequent among the treatment group compared with placebo. Relapse was defined as:
- HAM-D-17 ≥14
- Clinical Global Impressions of Severity score ≥3 with a ≥2-point increase from baseline
- and meeting DSM-IV criteria for MDD on two consecutive visits ≥11 days apart.
Side effects
Transdermal selegiline, 6 mg/d, has been well-tolerated in clinical trials. Inflammation at the application site was the most commonly reported side effect, occurring in 32% to 36% of treatment group subjects compared with 15% to 17% of the placebo groups.9,10,12 Inflammation was usually mild, but approximately 3% of patients dropped out of one study,12 citing this effect as the reason. Fair-skinned women are at highest risk for this reaction.
In the 1-year relapse prevention study,11 12% of treatment group patients reported insomnia compared with 7% of the placebo group. Insomnia incidence was the same in the selegiline and placebo groups during the 6- to 8-week clinical trials.9,10
Unlike conventional oral MAOIs,13 the 6-mg selegiline patch has not been found to impair sexual function, alter appetite, or change body weight or blood pressure compared with placebo.10-12 The toxicity of the 9- and 12-mg patches has not been studied in humans, but 8 mg/d and 12 mg/d of transdermal selegiline across 3 months were shown not to cause drug toxicity in dogs.14
Pediatric use
Although transdermal selegiline has not been studied in children and adolescents, the 6-mg patch could benefit some youths with depression. Before starting the drug, discuss with the child’s parents/guardians the FDA’s black box warning describing a possible association between selegiline and increased suicidal behavior in youths. This applies to all antidepressants.
Geriatric use
The patch might also help some older patients with depression. In a double-blind trial of high-dose oral selegiline (60 mg/d) involving 16 older patients (mean age 65.6), both the treatment and placebo groups remained almost free of side effects across 3 weeks.7 Although the sample was small, the findings suggest that older patients can tolerate selegiline at high dosages. Side effects also were minimal among treatment-group patients age ≥65 in the yearlong relapse prevention study.11
Treatment adherence rates with transdermal selegiline have been high in published studies, suggesting that the patch’s visibility might reduce the risk of forgetting to take the medication. Observing whether the patch has been changed might help older patients and family members/caregivers keep track of dosing.
Contraindications
As with the oral form, do not prescribe transdermal selegiline to patients taking SSRIs, SNRIs, tricyclic antidepressants, mirtazapine, or bupropion.
When switching antidepressants, allow enough time for the previous agent to “wash out” before starting transdermal selegiline. How much time to allow for wash-out depends on the previous agent’s half-life.
The patch is also contraindicated for patients taking:
- carbamazepine or oxcarbazepine
- meperidine
- analgesics such as tramadol, methadone, and propoxyphene
- St. John’s wort
- cough syrups containing dextromethorphan
- amphetamines, such as mixed amphetamine salts
- cyclobenzaprine
- or cold remedies or weight-loss products that contain vasoconstrictors, such as pseudoephedrine, phenylephrine, phenylpropanolamine, or ephedrine.
Do not give transdermal selegiline during pregnancy, as its effect on fetal development has not been studied.
Dosing
Start transdermal selegiline at 6 mg/d. Instruct the patient to wear the patch on the upper torso, where vascularity is richer compared with the buttocks and legs. Tell the patient to change the patch daily and to apply it to a different spot each day to prevent inflammation. Consider increasing the dosage after 2 or 3 months if response is unsatisfactory.
For treating first, second, and some third depressive episodes, continue transdermal selegiline for 6 months to 1 year of sustained recovery; consider longer-term maintenance treatment for highly recurrent depression. Transdermal selegiline has not been tapered in clinical trials, and subjects have not reported withdrawal symptoms after 1 year of continuous treatment.
Related resources
- Deniker P. The search for new antidepressants and related drugs. In: Tipton KF, Doster P, Benedetti M (eds). Monoamine oxidase and disease. London: Academic Press; 1984:2-8.
Drug brand names
- Amphetamine salts, mixed • Adderall
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol, Equetro, others
- Cyclobenzaprine • Flexeril
- Meperidine • Demerol
- Mirtazapine • Remeron
- Oxcarbazepine • Trileptal
- Propoxyphene hydrochloride • Darvon
- Propoxyphene napsylate • Darvocet
- Selegiline (oral) • Eldepryl
- Selegiline (transdermal) • EMSAM
- Tramadol • Ultracet
Disclosure
Dr. Bodkin receives grant support from the National Institute of Mental Health, Eli Lilly & Co, Jazz Pharmaceuticals, Merck & Co., Organon, Sanofi-Aventis, and Somerset Pharmaceuticals; is a consultant to Bristol-Myers Squibb Co. and Somerset Pharmaceuticals; and is a speaker for Bristol-Myers Squibb Co. He has been principal investigator in several multicenter clinical trials of selegiline.
1. Blackwell B, Mabbitt LA. Tyramine in cheese related to hypertensive crises after monoamine-oxidase inhibition. Lancet 1965;62:938-40.
2. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (2nd ed). Available at: http://www.psych.org/psych_pract/treatg/pg/Practice%20Guidelines8904/MajorDepressiveDisorder_2e.pdf. Accessed March 15, 2006.
3. IMS Health National Prescription Audit; 12/04-11/05. Available at: http://www.imshealth.com. Accessed March 15, 2006.
4. Johnston JP. Some observations on a new form of MAO in brain tissue. Biochem Pharmacol 1968;17:1285-97.
5. Youdim MB. Monoamine oxidase inhibitors as antidepressant drugs and as adjunct to L-dopa therapy of Parkinson’s disease. J Neural Transm Suppl 1980;(16):157-61.
6. Bodkin JA, Kwon AE. Selegiline and other atypical MAO inhibitors in depression. Ann Psychiatry 2001;31:385-91.
7. Sunderland T, Cohen RM, Molchan S, et al. High-dose selegiline in treatment-resistant older depressive patients. Arch Gen Psychiatry 1994;51:607-15.
8. Ziemniak JA, Kemper EM, Goodhear M, Azzaro AJ. Pharmacokinetics of selegiline administered via the patch, single oral dose, or intravenous infusion. Poster presented at: Annual Meeting, National Institute of Mental Health, New Clinical Drug Evaluation Unit, May 29, 2001, Phoenix, AZ.
9. Bodkin JA, Amsterdam JD. Transdermal selegiline in major depression: a double-blind, placebo-controlled, parallel-group study in outpatients. Am J Psychiatry 2002;159:1869-75.
10. Amsterdam JD. A double-blind, placebo-controlled trial of the safety and efficacy of selegiline transdermal system without dietary restrictions in patients with major depressive disorder. J Clin Psychiatry 2003;64:208-14.
11. Robinson DS, Moonsammy G, Azzaro AJ. Relapse prevention study shows the long-term safety and efficacy of transdermal selegiline, a new generation MAOI. Poster presented at: Annual Meeting, American College of Neuropsychopharmacology, Dec 11, 2002; San Juan, PR.
12. Robinson DS, Amsterdam JD. Safety and tolerability of selegiline transdermal system 20 mg for treatment of major depression. Poster presented at: Annual Meeting, American College of Neuropsychopharmacology, Dec. 13, 2005, Waikalo, HI.
13. Cole JO, Bodkin JA. Antidepressant drug side effects. J Clin Psychiatry 1990;51(Suppl):21-6.
14. Barrett JS, DiSanto AR, Thomford PJ, et al. Toxicokinetic evaluation of a selegiline transdermal system in the dog. Biopharm Drug Dispos 1997;18:165-84.
Many psychiatrists do not prescribe monoamine oxidase inhibitors (MAOIs) for fear of causing a potentially fatal hypertensive reaction, even though restricting foods high in the amino acid tyramine usually prevents this effect.1 Consequently, most depressed patients who might respond well to MAOIs do not receive them.2,3
Transdermal selegiline, FDA-approved for treating major depressive disorder (MDD) (Table 1), offers the clinical efficacy of an MAOI but without adverse interactions with food at the 6-mg strength. Transdermal selegiline may inhibit too much gastrointestinal MAO-A at 9 mg/d and 12 mg/d to clear tyramine from foods, so tyramine-rich foods must be restricted at these dosages (Table 2).
Table 1
Transdermal selegiline: Fast facts
| Brand name: EMSAM |
| Class: Monoamine oxidase inhibitor |
| FDA-approved indication: Major depressive disorder |
| Manufacturer: Somerset Pharmaceuticals (marketed by Bristol-Myers Squibb Co.) |
| Dosing forms: 6-, 9-, and 12-mg patches |
| Recommended dosage: One 6-mg patch every 24 hours, worn on the chest, back, or stomach. Increase dosage after 2 to 3 months if clinical response is inadequate |
Table 2
Restrict these foods when prescribing transdermal selegiline at 9 or 12 mg/d
| Food/beverage class | Foods to avoid |
|---|---|
| Beverages | Tap beer |
| Beer that has not been pasteurized* | |
| Red wines | |
| Dairy | Aged cheeses |
| Meat, poultry, fish | Air-dried, aged, and fermented meats, sausages, and salamis (including cacciatore and mortadella) |
| Pickled herring | |
| Spoiled or improperly stored fish, meat, poultry, or animal livers (check for mold, discoloration, or odor) | |
| Vegetables | Broad bean pods (fava beans) |
| Miscellaneous | Concentrated yeast extract (such as Marmite) |
| Fermented soybean products (including soy sauce) | |
| Over-the-counter supplements containing tyramine | |
| Sauerkraut | |
| *Bottled and canned beer and white wine contain little or no tyramine, but more than moderate alcohol use while taking selegiline is not recommended. | |
| Source: Shulman KI, Walker SE. A reevaluation of dietary restrictions for irreversible monoamine oxidase inhibitors. Psychiatr Ann 2001;31:378-84. | |
How it works
MAO enzyme subtypes A and B metabolize CNS monoamines, but primarily MAO-A metabolizes tyramine in the gut before the amino acid enters systemic circulation. At low concentrations, selegiline selectively inhibits MAO-B.4
Oral selegiline, approved as a adjunct to levodopa/carbidopa for patients with Parkinson’s disease,5 has been shown to be effective for treating depression at ≥30 mg/d.6 Because the drug does not selectively inhibit MAOB at ≥20 mg/d, dietary tyramine must be restricted when oral selegiline is used off-label at therapeutic dosages for depression. Otherwise, selegiline has been well-tolerated up to 60 mg/d.7
The 6-mg “patch” delivers more selegiline to the bloodstream than does low-dose oral selegiline but without inhibiting gut MAO-A. This provides the brain MAO-A and MAO-B inhibition necessary for an antidepressant effect while eliminating the need for dietary restrictions at this lowest dosage.
Clinical implications
Transdermal selegiline offers an MAOI antidepressant option that might help:
- patients whose depression has not responded satisfactorily to selective serotonin reuptake inhibitors (SSRIs) or serotonin and norepinephrine reuptake inhibitors (SNRIs)
- adults and children with chronic depression marked by atypical features, including reactive mood, rejection sensitivity, anergia, and reversed vegetative symptoms—such as oversleeping, overeating, and psychomotor retardation. Although transdermal selegiline’s efficacy against these features has not been studied, patients with this depressive subtype tend to respond preferentially to MAOIs.
Pharmacokinetics
Transdermal selegiline achieves therapeutic blood levels and reaches sustained concentration within 4 to 8 hours of administration. Compared with oral selegiline, transdermal delivery results in higher plasma selegiline concentrations (1,500 pg/mL with the 6-mg patch) with much lower exposure to metabolites.8 The concentration is maintained with successive doses.
Transdermal selegiline clears rapidly upon discontinuation but MAO inhibition persists for 2 weeks, so wait 2 weeks after the last dose before starting a new antidepressant or stopping food restrictions with the 9-mg and 12-mg patches.
Efficacy
In two randomized, double-blind clinical trials,9,10 a total of 466 adults ages 18 to 65 who met DSM-IV-TR criteria for MDD received transdermal selegiline, 6 mg/d, or placebo for 6 to 8 weeks. Participants had 17-item Hamilton Rating Scale for Depression (HAM-D-17) scores ≥20 at baseline.
In the 6-week study,9 transdermal selegiline produced a 46% greater reduction in HAM-D-17 scores, a 52% greater decrease in HAM-D-28 scores, and a 79% greater drop in Montgomery-Asburg Depression Rating Scale (MADRS) scores compared with placebo. In the 8-week trial,10 HAM-D-28 and MADRS scores among the treatment group were significantly improved at endpoint compared with placebo, but HAM-D-17 scores were not.
In a 1-year, double-blind study,11 322 subjects with MDD—who had been rated as responders in a 10-week, open-label transdermal selegiline trial—received the 6-mg patch or placebo. At 6 months and 1 year, relapse was much less frequent among the treatment group compared with placebo. Relapse was defined as:
- HAM-D-17 ≥14
- Clinical Global Impressions of Severity score ≥3 with a ≥2-point increase from baseline
- and meeting DSM-IV criteria for MDD on two consecutive visits ≥11 days apart.
Side effects
Transdermal selegiline, 6 mg/d, has been well-tolerated in clinical trials. Inflammation at the application site was the most commonly reported side effect, occurring in 32% to 36% of treatment group subjects compared with 15% to 17% of the placebo groups.9,10,12 Inflammation was usually mild, but approximately 3% of patients dropped out of one study,12 citing this effect as the reason. Fair-skinned women are at highest risk for this reaction.
In the 1-year relapse prevention study,11 12% of treatment group patients reported insomnia compared with 7% of the placebo group. Insomnia incidence was the same in the selegiline and placebo groups during the 6- to 8-week clinical trials.9,10
Unlike conventional oral MAOIs,13 the 6-mg selegiline patch has not been found to impair sexual function, alter appetite, or change body weight or blood pressure compared with placebo.10-12 The toxicity of the 9- and 12-mg patches has not been studied in humans, but 8 mg/d and 12 mg/d of transdermal selegiline across 3 months were shown not to cause drug toxicity in dogs.14
Pediatric use
Although transdermal selegiline has not been studied in children and adolescents, the 6-mg patch could benefit some youths with depression. Before starting the drug, discuss with the child’s parents/guardians the FDA’s black box warning describing a possible association between selegiline and increased suicidal behavior in youths. This applies to all antidepressants.
Geriatric use
The patch might also help some older patients with depression. In a double-blind trial of high-dose oral selegiline (60 mg/d) involving 16 older patients (mean age 65.6), both the treatment and placebo groups remained almost free of side effects across 3 weeks.7 Although the sample was small, the findings suggest that older patients can tolerate selegiline at high dosages. Side effects also were minimal among treatment-group patients age ≥65 in the yearlong relapse prevention study.11
Treatment adherence rates with transdermal selegiline have been high in published studies, suggesting that the patch’s visibility might reduce the risk of forgetting to take the medication. Observing whether the patch has been changed might help older patients and family members/caregivers keep track of dosing.
Contraindications
As with the oral form, do not prescribe transdermal selegiline to patients taking SSRIs, SNRIs, tricyclic antidepressants, mirtazapine, or bupropion.
When switching antidepressants, allow enough time for the previous agent to “wash out” before starting transdermal selegiline. How much time to allow for wash-out depends on the previous agent’s half-life.
The patch is also contraindicated for patients taking:
- carbamazepine or oxcarbazepine
- meperidine
- analgesics such as tramadol, methadone, and propoxyphene
- St. John’s wort
- cough syrups containing dextromethorphan
- amphetamines, such as mixed amphetamine salts
- cyclobenzaprine
- or cold remedies or weight-loss products that contain vasoconstrictors, such as pseudoephedrine, phenylephrine, phenylpropanolamine, or ephedrine.
Do not give transdermal selegiline during pregnancy, as its effect on fetal development has not been studied.
Dosing
Start transdermal selegiline at 6 mg/d. Instruct the patient to wear the patch on the upper torso, where vascularity is richer compared with the buttocks and legs. Tell the patient to change the patch daily and to apply it to a different spot each day to prevent inflammation. Consider increasing the dosage after 2 or 3 months if response is unsatisfactory.
For treating first, second, and some third depressive episodes, continue transdermal selegiline for 6 months to 1 year of sustained recovery; consider longer-term maintenance treatment for highly recurrent depression. Transdermal selegiline has not been tapered in clinical trials, and subjects have not reported withdrawal symptoms after 1 year of continuous treatment.
Related resources
- Deniker P. The search for new antidepressants and related drugs. In: Tipton KF, Doster P, Benedetti M (eds). Monoamine oxidase and disease. London: Academic Press; 1984:2-8.
Drug brand names
- Amphetamine salts, mixed • Adderall
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol, Equetro, others
- Cyclobenzaprine • Flexeril
- Meperidine • Demerol
- Mirtazapine • Remeron
- Oxcarbazepine • Trileptal
- Propoxyphene hydrochloride • Darvon
- Propoxyphene napsylate • Darvocet
- Selegiline (oral) • Eldepryl
- Selegiline (transdermal) • EMSAM
- Tramadol • Ultracet
Disclosure
Dr. Bodkin receives grant support from the National Institute of Mental Health, Eli Lilly & Co, Jazz Pharmaceuticals, Merck & Co., Organon, Sanofi-Aventis, and Somerset Pharmaceuticals; is a consultant to Bristol-Myers Squibb Co. and Somerset Pharmaceuticals; and is a speaker for Bristol-Myers Squibb Co. He has been principal investigator in several multicenter clinical trials of selegiline.
Many psychiatrists do not prescribe monoamine oxidase inhibitors (MAOIs) for fear of causing a potentially fatal hypertensive reaction, even though restricting foods high in the amino acid tyramine usually prevents this effect.1 Consequently, most depressed patients who might respond well to MAOIs do not receive them.2,3
Transdermal selegiline, FDA-approved for treating major depressive disorder (MDD) (Table 1), offers the clinical efficacy of an MAOI but without adverse interactions with food at the 6-mg strength. Transdermal selegiline may inhibit too much gastrointestinal MAO-A at 9 mg/d and 12 mg/d to clear tyramine from foods, so tyramine-rich foods must be restricted at these dosages (Table 2).
Table 1
Transdermal selegiline: Fast facts
| Brand name: EMSAM |
| Class: Monoamine oxidase inhibitor |
| FDA-approved indication: Major depressive disorder |
| Manufacturer: Somerset Pharmaceuticals (marketed by Bristol-Myers Squibb Co.) |
| Dosing forms: 6-, 9-, and 12-mg patches |
| Recommended dosage: One 6-mg patch every 24 hours, worn on the chest, back, or stomach. Increase dosage after 2 to 3 months if clinical response is inadequate |
Table 2
Restrict these foods when prescribing transdermal selegiline at 9 or 12 mg/d
| Food/beverage class | Foods to avoid |
|---|---|
| Beverages | Tap beer |
| Beer that has not been pasteurized* | |
| Red wines | |
| Dairy | Aged cheeses |
| Meat, poultry, fish | Air-dried, aged, and fermented meats, sausages, and salamis (including cacciatore and mortadella) |
| Pickled herring | |
| Spoiled or improperly stored fish, meat, poultry, or animal livers (check for mold, discoloration, or odor) | |
| Vegetables | Broad bean pods (fava beans) |
| Miscellaneous | Concentrated yeast extract (such as Marmite) |
| Fermented soybean products (including soy sauce) | |
| Over-the-counter supplements containing tyramine | |
| Sauerkraut | |
| *Bottled and canned beer and white wine contain little or no tyramine, but more than moderate alcohol use while taking selegiline is not recommended. | |
| Source: Shulman KI, Walker SE. A reevaluation of dietary restrictions for irreversible monoamine oxidase inhibitors. Psychiatr Ann 2001;31:378-84. | |
How it works
MAO enzyme subtypes A and B metabolize CNS monoamines, but primarily MAO-A metabolizes tyramine in the gut before the amino acid enters systemic circulation. At low concentrations, selegiline selectively inhibits MAO-B.4
Oral selegiline, approved as a adjunct to levodopa/carbidopa for patients with Parkinson’s disease,5 has been shown to be effective for treating depression at ≥30 mg/d.6 Because the drug does not selectively inhibit MAOB at ≥20 mg/d, dietary tyramine must be restricted when oral selegiline is used off-label at therapeutic dosages for depression. Otherwise, selegiline has been well-tolerated up to 60 mg/d.7
The 6-mg “patch” delivers more selegiline to the bloodstream than does low-dose oral selegiline but without inhibiting gut MAO-A. This provides the brain MAO-A and MAO-B inhibition necessary for an antidepressant effect while eliminating the need for dietary restrictions at this lowest dosage.
Clinical implications
Transdermal selegiline offers an MAOI antidepressant option that might help:
- patients whose depression has not responded satisfactorily to selective serotonin reuptake inhibitors (SSRIs) or serotonin and norepinephrine reuptake inhibitors (SNRIs)
- adults and children with chronic depression marked by atypical features, including reactive mood, rejection sensitivity, anergia, and reversed vegetative symptoms—such as oversleeping, overeating, and psychomotor retardation. Although transdermal selegiline’s efficacy against these features has not been studied, patients with this depressive subtype tend to respond preferentially to MAOIs.
Pharmacokinetics
Transdermal selegiline achieves therapeutic blood levels and reaches sustained concentration within 4 to 8 hours of administration. Compared with oral selegiline, transdermal delivery results in higher plasma selegiline concentrations (1,500 pg/mL with the 6-mg patch) with much lower exposure to metabolites.8 The concentration is maintained with successive doses.
Transdermal selegiline clears rapidly upon discontinuation but MAO inhibition persists for 2 weeks, so wait 2 weeks after the last dose before starting a new antidepressant or stopping food restrictions with the 9-mg and 12-mg patches.
Efficacy
In two randomized, double-blind clinical trials,9,10 a total of 466 adults ages 18 to 65 who met DSM-IV-TR criteria for MDD received transdermal selegiline, 6 mg/d, or placebo for 6 to 8 weeks. Participants had 17-item Hamilton Rating Scale for Depression (HAM-D-17) scores ≥20 at baseline.
In the 6-week study,9 transdermal selegiline produced a 46% greater reduction in HAM-D-17 scores, a 52% greater decrease in HAM-D-28 scores, and a 79% greater drop in Montgomery-Asburg Depression Rating Scale (MADRS) scores compared with placebo. In the 8-week trial,10 HAM-D-28 and MADRS scores among the treatment group were significantly improved at endpoint compared with placebo, but HAM-D-17 scores were not.
In a 1-year, double-blind study,11 322 subjects with MDD—who had been rated as responders in a 10-week, open-label transdermal selegiline trial—received the 6-mg patch or placebo. At 6 months and 1 year, relapse was much less frequent among the treatment group compared with placebo. Relapse was defined as:
- HAM-D-17 ≥14
- Clinical Global Impressions of Severity score ≥3 with a ≥2-point increase from baseline
- and meeting DSM-IV criteria for MDD on two consecutive visits ≥11 days apart.
Side effects
Transdermal selegiline, 6 mg/d, has been well-tolerated in clinical trials. Inflammation at the application site was the most commonly reported side effect, occurring in 32% to 36% of treatment group subjects compared with 15% to 17% of the placebo groups.9,10,12 Inflammation was usually mild, but approximately 3% of patients dropped out of one study,12 citing this effect as the reason. Fair-skinned women are at highest risk for this reaction.
In the 1-year relapse prevention study,11 12% of treatment group patients reported insomnia compared with 7% of the placebo group. Insomnia incidence was the same in the selegiline and placebo groups during the 6- to 8-week clinical trials.9,10
Unlike conventional oral MAOIs,13 the 6-mg selegiline patch has not been found to impair sexual function, alter appetite, or change body weight or blood pressure compared with placebo.10-12 The toxicity of the 9- and 12-mg patches has not been studied in humans, but 8 mg/d and 12 mg/d of transdermal selegiline across 3 months were shown not to cause drug toxicity in dogs.14
Pediatric use
Although transdermal selegiline has not been studied in children and adolescents, the 6-mg patch could benefit some youths with depression. Before starting the drug, discuss with the child’s parents/guardians the FDA’s black box warning describing a possible association between selegiline and increased suicidal behavior in youths. This applies to all antidepressants.
Geriatric use
The patch might also help some older patients with depression. In a double-blind trial of high-dose oral selegiline (60 mg/d) involving 16 older patients (mean age 65.6), both the treatment and placebo groups remained almost free of side effects across 3 weeks.7 Although the sample was small, the findings suggest that older patients can tolerate selegiline at high dosages. Side effects also were minimal among treatment-group patients age ≥65 in the yearlong relapse prevention study.11
Treatment adherence rates with transdermal selegiline have been high in published studies, suggesting that the patch’s visibility might reduce the risk of forgetting to take the medication. Observing whether the patch has been changed might help older patients and family members/caregivers keep track of dosing.
Contraindications
As with the oral form, do not prescribe transdermal selegiline to patients taking SSRIs, SNRIs, tricyclic antidepressants, mirtazapine, or bupropion.
When switching antidepressants, allow enough time for the previous agent to “wash out” before starting transdermal selegiline. How much time to allow for wash-out depends on the previous agent’s half-life.
The patch is also contraindicated for patients taking:
- carbamazepine or oxcarbazepine
- meperidine
- analgesics such as tramadol, methadone, and propoxyphene
- St. John’s wort
- cough syrups containing dextromethorphan
- amphetamines, such as mixed amphetamine salts
- cyclobenzaprine
- or cold remedies or weight-loss products that contain vasoconstrictors, such as pseudoephedrine, phenylephrine, phenylpropanolamine, or ephedrine.
Do not give transdermal selegiline during pregnancy, as its effect on fetal development has not been studied.
Dosing
Start transdermal selegiline at 6 mg/d. Instruct the patient to wear the patch on the upper torso, where vascularity is richer compared with the buttocks and legs. Tell the patient to change the patch daily and to apply it to a different spot each day to prevent inflammation. Consider increasing the dosage after 2 or 3 months if response is unsatisfactory.
For treating first, second, and some third depressive episodes, continue transdermal selegiline for 6 months to 1 year of sustained recovery; consider longer-term maintenance treatment for highly recurrent depression. Transdermal selegiline has not been tapered in clinical trials, and subjects have not reported withdrawal symptoms after 1 year of continuous treatment.
Related resources
- Deniker P. The search for new antidepressants and related drugs. In: Tipton KF, Doster P, Benedetti M (eds). Monoamine oxidase and disease. London: Academic Press; 1984:2-8.
Drug brand names
- Amphetamine salts, mixed • Adderall
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol, Equetro, others
- Cyclobenzaprine • Flexeril
- Meperidine • Demerol
- Mirtazapine • Remeron
- Oxcarbazepine • Trileptal
- Propoxyphene hydrochloride • Darvon
- Propoxyphene napsylate • Darvocet
- Selegiline (oral) • Eldepryl
- Selegiline (transdermal) • EMSAM
- Tramadol • Ultracet
Disclosure
Dr. Bodkin receives grant support from the National Institute of Mental Health, Eli Lilly & Co, Jazz Pharmaceuticals, Merck & Co., Organon, Sanofi-Aventis, and Somerset Pharmaceuticals; is a consultant to Bristol-Myers Squibb Co. and Somerset Pharmaceuticals; and is a speaker for Bristol-Myers Squibb Co. He has been principal investigator in several multicenter clinical trials of selegiline.
1. Blackwell B, Mabbitt LA. Tyramine in cheese related to hypertensive crises after monoamine-oxidase inhibition. Lancet 1965;62:938-40.
2. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (2nd ed). Available at: http://www.psych.org/psych_pract/treatg/pg/Practice%20Guidelines8904/MajorDepressiveDisorder_2e.pdf. Accessed March 15, 2006.
3. IMS Health National Prescription Audit; 12/04-11/05. Available at: http://www.imshealth.com. Accessed March 15, 2006.
4. Johnston JP. Some observations on a new form of MAO in brain tissue. Biochem Pharmacol 1968;17:1285-97.
5. Youdim MB. Monoamine oxidase inhibitors as antidepressant drugs and as adjunct to L-dopa therapy of Parkinson’s disease. J Neural Transm Suppl 1980;(16):157-61.
6. Bodkin JA, Kwon AE. Selegiline and other atypical MAO inhibitors in depression. Ann Psychiatry 2001;31:385-91.
7. Sunderland T, Cohen RM, Molchan S, et al. High-dose selegiline in treatment-resistant older depressive patients. Arch Gen Psychiatry 1994;51:607-15.
8. Ziemniak JA, Kemper EM, Goodhear M, Azzaro AJ. Pharmacokinetics of selegiline administered via the patch, single oral dose, or intravenous infusion. Poster presented at: Annual Meeting, National Institute of Mental Health, New Clinical Drug Evaluation Unit, May 29, 2001, Phoenix, AZ.
9. Bodkin JA, Amsterdam JD. Transdermal selegiline in major depression: a double-blind, placebo-controlled, parallel-group study in outpatients. Am J Psychiatry 2002;159:1869-75.
10. Amsterdam JD. A double-blind, placebo-controlled trial of the safety and efficacy of selegiline transdermal system without dietary restrictions in patients with major depressive disorder. J Clin Psychiatry 2003;64:208-14.
11. Robinson DS, Moonsammy G, Azzaro AJ. Relapse prevention study shows the long-term safety and efficacy of transdermal selegiline, a new generation MAOI. Poster presented at: Annual Meeting, American College of Neuropsychopharmacology, Dec 11, 2002; San Juan, PR.
12. Robinson DS, Amsterdam JD. Safety and tolerability of selegiline transdermal system 20 mg for treatment of major depression. Poster presented at: Annual Meeting, American College of Neuropsychopharmacology, Dec. 13, 2005, Waikalo, HI.
13. Cole JO, Bodkin JA. Antidepressant drug side effects. J Clin Psychiatry 1990;51(Suppl):21-6.
14. Barrett JS, DiSanto AR, Thomford PJ, et al. Toxicokinetic evaluation of a selegiline transdermal system in the dog. Biopharm Drug Dispos 1997;18:165-84.
1. Blackwell B, Mabbitt LA. Tyramine in cheese related to hypertensive crises after monoamine-oxidase inhibition. Lancet 1965;62:938-40.
2. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (2nd ed). Available at: http://www.psych.org/psych_pract/treatg/pg/Practice%20Guidelines8904/MajorDepressiveDisorder_2e.pdf. Accessed March 15, 2006.
3. IMS Health National Prescription Audit; 12/04-11/05. Available at: http://www.imshealth.com. Accessed March 15, 2006.
4. Johnston JP. Some observations on a new form of MAO in brain tissue. Biochem Pharmacol 1968;17:1285-97.
5. Youdim MB. Monoamine oxidase inhibitors as antidepressant drugs and as adjunct to L-dopa therapy of Parkinson’s disease. J Neural Transm Suppl 1980;(16):157-61.
6. Bodkin JA, Kwon AE. Selegiline and other atypical MAO inhibitors in depression. Ann Psychiatry 2001;31:385-91.
7. Sunderland T, Cohen RM, Molchan S, et al. High-dose selegiline in treatment-resistant older depressive patients. Arch Gen Psychiatry 1994;51:607-15.
8. Ziemniak JA, Kemper EM, Goodhear M, Azzaro AJ. Pharmacokinetics of selegiline administered via the patch, single oral dose, or intravenous infusion. Poster presented at: Annual Meeting, National Institute of Mental Health, New Clinical Drug Evaluation Unit, May 29, 2001, Phoenix, AZ.
9. Bodkin JA, Amsterdam JD. Transdermal selegiline in major depression: a double-blind, placebo-controlled, parallel-group study in outpatients. Am J Psychiatry 2002;159:1869-75.
10. Amsterdam JD. A double-blind, placebo-controlled trial of the safety and efficacy of selegiline transdermal system without dietary restrictions in patients with major depressive disorder. J Clin Psychiatry 2003;64:208-14.
11. Robinson DS, Moonsammy G, Azzaro AJ. Relapse prevention study shows the long-term safety and efficacy of transdermal selegiline, a new generation MAOI. Poster presented at: Annual Meeting, American College of Neuropsychopharmacology, Dec 11, 2002; San Juan, PR.
12. Robinson DS, Amsterdam JD. Safety and tolerability of selegiline transdermal system 20 mg for treatment of major depression. Poster presented at: Annual Meeting, American College of Neuropsychopharmacology, Dec. 13, 2005, Waikalo, HI.
13. Cole JO, Bodkin JA. Antidepressant drug side effects. J Clin Psychiatry 1990;51(Suppl):21-6.
14. Barrett JS, DiSanto AR, Thomford PJ, et al. Toxicokinetic evaluation of a selegiline transdermal system in the dog. Biopharm Drug Dispos 1997;18:165-84.
Intramuscular naltrexone
A long-acting, intramuscular (IM) naltrexone formulation—which at press time awaited FDA approval (Table)—could improve adherence to alcohol dependency pharmacotherapy.
Oral naltrexone can reduce alcohol consumption1 and relapse rates,1,2 but patients often stop taking it3 and increase their risk of relapse.2 Once-daily dosing, inconsistent motivation toward treatment, and cognitive impairment secondary to chronic alcohol dependence often thwart oral naltrexone therapy.
By contrast, IM naltrexone surmounts most compliance issues because you or a clinical assistant administer the drug. Short-term side effects—such as nausea for 2 days—are less likely to affect adherence because the medication keeps working weeks after side effects abate. This gives you time before the next dose to reassure the patient and gives the patient the benefits of continued treatment.
Table
IM naltrexone: Fast facts
| Drug brand name: Vivitrol |
| Class: Opioid antagonist |
| Prospective indication: Alcohol dependence |
| FDA action: Issued approvable letter Dec. 28, 2005 |
| Manufacturer: Alkermes |
| Dosing forms: 380 mg suspension via IM injection |
| Recommended dosage: 380 mg once monthly |
| Estimated date of availability: Spring 2006 |
How naltrexone works
Alcohol stimulates release of endogenous opioids, which in turn stimulate release of dopamine, which mediates reinforcement.4 Opioid receptor stimulation not associated with dopamine also reinforces alcohol use.5 Persons vulnerable to alcohol dependence generally have lower basal levels of opioid secretion and are stimulated at higher levels.6 Opioids also increase dopamine by inhibiting GABA neurons, which suppress dopamine release when uninhibited.
As an opioid antagonist, naltrexone prevents opioids from binding with μ-opioid receptors and modulates dopamine production. This may make drinking less “rewarding” and may reduce craving triggered by conditioned cues associated with alcohol use.
IM naltrexone is packaged in biodegradable microspheres that slowly release naltrexone for 1 month after injection. The microspheres are made of a polyactide-co-glycolide polymer used in other extended-release drugs and in absorbable sutures.
Pharmacokinetics
IM naltrexone plasma levels peak 2 to 3 days after injection, then decline gradually over 30 days. Oral naltrexone dosed at 50 mg/d for 30 days—a cumulative dose of 1,500 mg/month—produces daily peak plasma levels of approximately 10 ng/mL and troughs approaching zero. A once-monthly IM naltrexone injection results in a lower net dose but more-sustained naltrexone levels.
Efficacy
IM naltrexone significantly reduced heavy drinking among alcohol-dependent patients in a phase 3 randomized, placebo-controlled, multicenter trial.7 Actively drinking adults who met DSM-IV criteria for alcohol dependence (N=624) received IM naltrexone, 190 or 380 mg, or placebo every 4 weeks for 6 months. Oral naltrexone lead-in doses were not given. All patients also received 12 sessions of standardized supportive psychosocial therapy during the study.
The primary efficacy measure was event rate of heavy drinking, defined as number of heavy drinking days (≥5 drinks/day for men, ≥4 drinks/day for women) divided by number of days in the study. An event rate ratio (treatment-group to placebo-group event rate) was then estimated over time, taking into account patients who discontinued the study.
After 6 months, event rate of heavy drinking fell 25% among patients receiving 380 mg of IM naltrexone and supportive therapy, compared with patients receiving placebo and supportive therapy (P=0.02). That rate decreased 17% among patients who received 190 mg of IM naltrexone compared with placebo, but the difference between the two treatment groups was not statistically significant (P=0.07).
The median number of heavy drinking days per month decreased substantially across 6 months among all study groups. The decrease was more substantial among patients taking IM naltrexone, 380 mg, than among the placebo group (Figure).
Roughly 8% of patients abstained from drinking for 7 days before entering the study. Among patients who received 380 mg of IM naltrexone:
- those who were abstinent before the study had an 80% greater reduction in event rate of heavy drinking compared with placebo
- nonabstinent patients showed a 21% greater reduction in event rate of heavy drinking compared with placebo.
These findings suggest that IM naltrexone is more effective in persons abstaining from drinking but can also help actively drinking patients.
IM naltrexone also reduced heavy drinking among patients who entered a 1-year open-label extension study after completing the 6-month study.8 Drinking reductions were greater among patients who received 380 mg of naltrexone during both the 6-month and 1-year trials than among those who received placebo for 6 months and were switched to naltrexone, 380 mg, in the 1-year extension.
Figure Median heavy drinking days after 6 months of IM naltrexone or placebo
Source: Reference 7
Tolerability
IM naltrexone was well-tolerated in the phase 3 trial.7 Most-common adverse effects included
- nausea (reported by 33% of patients receiving 380 mg [n=205] and 25% of those receiving 190 mg [n=210])
- headache (22%, 16%)
- fatigue (20%, 16%).
At 380 mg, decreased appetite (13%), dizziness (13%), and injection site pain (12%) also differed significantly from placebo. Nausea was rated as mild or moderate in 95% of cases, usually occurred only after the first injection, and lasted 1 to 2 days on average.
Nine percent of patients taking naltrexone, 190 mg, or placebo also reported injection site pain. Approximately 1% of all patients dropped out because of injection site reactions.
Patients generally adhered to treatment, with 64% receiving 6 injections and 74% receiving at least 4. By comparison, a meta-analysis3 of oral naltrexone clinical trials showed an average 50% retention rate across studies, most of which lasted only 3 months. Study withdrawals because of adverse events were more prevalent among patients receiving IM naltrexone, 380 mg (14.1%), than among the placebo group (6.7%), but the number of serious adverse events differed little.7
Liver enzymes (AST and ALT) did not change significantly during the study. Gamma-glutamyltransferase decreased in all patients, consistent with reduced drinking.
Interactions between IM naltrexone and other medications are probably similar to those observed with oral naltrexone.
Contraindications
Although product labeling was not available when this article was written, IM naltrexone, like its oral form, will likely be contraindicated for patients who:
- are taking opioid analgesics
- are in acute opioid withdrawal
- test positive on urine screen for opioids
- have acute hepatitis or liver failure
- are taking maintenance methadone or buprenorphine or are opioid-dependent.
Patients should be opioid-free for 7 to 10 days before starting IM naltrexone to avoid acute withdrawal symptoms.
Before starting IM naltrexone in patients with a history of opioid abuse, give naloxone, 0.8 mg, to test for withdrawal. Do not start naltrexone if the patient shows signs of opioid withdrawal within 20 minutes of receiving naloxone.
Clinical implications
Long-acting IM naltrexone will make it easier to ensure treatment adherence, compared with oral naltrexone. Giving the drug during the office visit will change your practice patterns, but this increase in hands-on care could strengthen the therapeutic alliance. Compared with interpreting patient self-reports, you can also more accurately document adherence to IM naltrexone therapy.
All alcohol-dependence medications work best when combined with psychosocial treatment, and monthly medication visits alone will not provide patients the cognitive and skill-building work they need to recover. Patients early in recovery need to be seen much more often by you and/or another provider of recovery-oriented psychosocial treatment.
Which patients will be more receptive to in-office treatment is unclear. Patients who have relapsed because of nonadherence to oral medications may be more willing to try IM therapy after you explain its benefits. Similarly, IM naltrexone may be more beneficial to patients who:
- cannot adhere to oral medication because of cognitive problems or impulsivity
- face severe consequences—such as legal problems, loss of parental custody, or loss of employment—if treatment fails.
The optimal duration of IM naltrexone therapy is not known, but the injectable has shown efficacy after 6 months7 and 1 year.8 Some patients have taken it for more than 3 years.8 Before stopping IM naltrexone, consider whether the patient:
- has achieved sobriety
- has developed skills and external support to maintain sobriety
- has reduced craving intensity or time spent preoccupied with alcohol
- shows improved global psychosocial function as reflected in improved relationships, work performance, and general health.
Patients with family histories of alcohol dependence and who reduce days of heavy drinking but do not achieve sobriety on IM naltrexone are probably at higher risk of relapse to heavy drinking after stopping the medication.
Related resources
- Injectable naltrexone Web site. http://alkermes2005.ifactory.com/products/naltrexone.html.
Drug brand names
- IM naltrexone • Vivitrol
- Oral naltrexone • Depade
- Naloxone • Narcan
Disclosure
Dr. Rosenthal is a consultant for Forest Laboratories and Alkermes.
1. Anton RF, Moak DH, Waid LR, et al. Naltrexone and cognitive behavioral therapy for the treatment of outpatient alcoholics: results of a placebo-controlled trial. Am J Psychiatry 1999;156:1758-64.
2. Pettinati HM, Volpicelli JR, Pierce JD, Jr, O’Brien CP. Improving naltrexone response: An intervention for medical practitioners toenhance medication compliance in alcohol dependent patients. J Addict Dis 2000;19:71-83.
3. Bouza C, Magro A, Munoz A, Amate J. Efficacy and safety of naltrexone and acamprosate in the treatment of alcohol dependence: a systematic review. Addiction 2004;99:811-28.
4. Weiss F, Lorang MT, Bloom FE, Koob GF. Oral alcohol self-administration stimulates dopamine release in the rat nucleus accumbens: genetic and motivational determinants. J Pharmacol Exp Ther 1993;267:250-8.
5. Pettit HO, Ettenberg A, Bloom FE, Koob GF. Destruction of dopamine in the nucleus accumbens selectively attenuates cocaine but not heroin self-administration in rats. Psychopharmacology (Berl) 1984;84:167-73.
6. Gianoulakis C. Characterization of the effects of acute ethanol administration on the release of beta-endorphin peptides by the rat hypothalamus. Eur J Pharmacol 1990;180:21-9.
7. Garbutt JC, Kranzler HR, O’Malley SS, et al. Vivitrex Study Group. Efficacy and tolerability of long-acting injectable naltrexone for alcohol dependence: a randomized controlled trial. JAMA 2005;293:1617-25.
8. Gastfriend DR, Dong Q, Loewy J, et al. Durability of effect of long-acting injectable naltrexone. Presented at: Annual Meeting of the American Psychiatric Association; 2005; Atlanta, GA.
A long-acting, intramuscular (IM) naltrexone formulation—which at press time awaited FDA approval (Table)—could improve adherence to alcohol dependency pharmacotherapy.
Oral naltrexone can reduce alcohol consumption1 and relapse rates,1,2 but patients often stop taking it3 and increase their risk of relapse.2 Once-daily dosing, inconsistent motivation toward treatment, and cognitive impairment secondary to chronic alcohol dependence often thwart oral naltrexone therapy.
By contrast, IM naltrexone surmounts most compliance issues because you or a clinical assistant administer the drug. Short-term side effects—such as nausea for 2 days—are less likely to affect adherence because the medication keeps working weeks after side effects abate. This gives you time before the next dose to reassure the patient and gives the patient the benefits of continued treatment.
Table
IM naltrexone: Fast facts
| Drug brand name: Vivitrol |
| Class: Opioid antagonist |
| Prospective indication: Alcohol dependence |
| FDA action: Issued approvable letter Dec. 28, 2005 |
| Manufacturer: Alkermes |
| Dosing forms: 380 mg suspension via IM injection |
| Recommended dosage: 380 mg once monthly |
| Estimated date of availability: Spring 2006 |
How naltrexone works
Alcohol stimulates release of endogenous opioids, which in turn stimulate release of dopamine, which mediates reinforcement.4 Opioid receptor stimulation not associated with dopamine also reinforces alcohol use.5 Persons vulnerable to alcohol dependence generally have lower basal levels of opioid secretion and are stimulated at higher levels.6 Opioids also increase dopamine by inhibiting GABA neurons, which suppress dopamine release when uninhibited.
As an opioid antagonist, naltrexone prevents opioids from binding with μ-opioid receptors and modulates dopamine production. This may make drinking less “rewarding” and may reduce craving triggered by conditioned cues associated with alcohol use.
IM naltrexone is packaged in biodegradable microspheres that slowly release naltrexone for 1 month after injection. The microspheres are made of a polyactide-co-glycolide polymer used in other extended-release drugs and in absorbable sutures.
Pharmacokinetics
IM naltrexone plasma levels peak 2 to 3 days after injection, then decline gradually over 30 days. Oral naltrexone dosed at 50 mg/d for 30 days—a cumulative dose of 1,500 mg/month—produces daily peak plasma levels of approximately 10 ng/mL and troughs approaching zero. A once-monthly IM naltrexone injection results in a lower net dose but more-sustained naltrexone levels.
Efficacy
IM naltrexone significantly reduced heavy drinking among alcohol-dependent patients in a phase 3 randomized, placebo-controlled, multicenter trial.7 Actively drinking adults who met DSM-IV criteria for alcohol dependence (N=624) received IM naltrexone, 190 or 380 mg, or placebo every 4 weeks for 6 months. Oral naltrexone lead-in doses were not given. All patients also received 12 sessions of standardized supportive psychosocial therapy during the study.
The primary efficacy measure was event rate of heavy drinking, defined as number of heavy drinking days (≥5 drinks/day for men, ≥4 drinks/day for women) divided by number of days in the study. An event rate ratio (treatment-group to placebo-group event rate) was then estimated over time, taking into account patients who discontinued the study.
After 6 months, event rate of heavy drinking fell 25% among patients receiving 380 mg of IM naltrexone and supportive therapy, compared with patients receiving placebo and supportive therapy (P=0.02). That rate decreased 17% among patients who received 190 mg of IM naltrexone compared with placebo, but the difference between the two treatment groups was not statistically significant (P=0.07).
The median number of heavy drinking days per month decreased substantially across 6 months among all study groups. The decrease was more substantial among patients taking IM naltrexone, 380 mg, than among the placebo group (Figure).
Roughly 8% of patients abstained from drinking for 7 days before entering the study. Among patients who received 380 mg of IM naltrexone:
- those who were abstinent before the study had an 80% greater reduction in event rate of heavy drinking compared with placebo
- nonabstinent patients showed a 21% greater reduction in event rate of heavy drinking compared with placebo.
These findings suggest that IM naltrexone is more effective in persons abstaining from drinking but can also help actively drinking patients.
IM naltrexone also reduced heavy drinking among patients who entered a 1-year open-label extension study after completing the 6-month study.8 Drinking reductions were greater among patients who received 380 mg of naltrexone during both the 6-month and 1-year trials than among those who received placebo for 6 months and were switched to naltrexone, 380 mg, in the 1-year extension.
Figure Median heavy drinking days after 6 months of IM naltrexone or placebo
Source: Reference 7
Tolerability
IM naltrexone was well-tolerated in the phase 3 trial.7 Most-common adverse effects included
- nausea (reported by 33% of patients receiving 380 mg [n=205] and 25% of those receiving 190 mg [n=210])
- headache (22%, 16%)
- fatigue (20%, 16%).
At 380 mg, decreased appetite (13%), dizziness (13%), and injection site pain (12%) also differed significantly from placebo. Nausea was rated as mild or moderate in 95% of cases, usually occurred only after the first injection, and lasted 1 to 2 days on average.
Nine percent of patients taking naltrexone, 190 mg, or placebo also reported injection site pain. Approximately 1% of all patients dropped out because of injection site reactions.
Patients generally adhered to treatment, with 64% receiving 6 injections and 74% receiving at least 4. By comparison, a meta-analysis3 of oral naltrexone clinical trials showed an average 50% retention rate across studies, most of which lasted only 3 months. Study withdrawals because of adverse events were more prevalent among patients receiving IM naltrexone, 380 mg (14.1%), than among the placebo group (6.7%), but the number of serious adverse events differed little.7
Liver enzymes (AST and ALT) did not change significantly during the study. Gamma-glutamyltransferase decreased in all patients, consistent with reduced drinking.
Interactions between IM naltrexone and other medications are probably similar to those observed with oral naltrexone.
Contraindications
Although product labeling was not available when this article was written, IM naltrexone, like its oral form, will likely be contraindicated for patients who:
- are taking opioid analgesics
- are in acute opioid withdrawal
- test positive on urine screen for opioids
- have acute hepatitis or liver failure
- are taking maintenance methadone or buprenorphine or are opioid-dependent.
Patients should be opioid-free for 7 to 10 days before starting IM naltrexone to avoid acute withdrawal symptoms.
Before starting IM naltrexone in patients with a history of opioid abuse, give naloxone, 0.8 mg, to test for withdrawal. Do not start naltrexone if the patient shows signs of opioid withdrawal within 20 minutes of receiving naloxone.
Clinical implications
Long-acting IM naltrexone will make it easier to ensure treatment adherence, compared with oral naltrexone. Giving the drug during the office visit will change your practice patterns, but this increase in hands-on care could strengthen the therapeutic alliance. Compared with interpreting patient self-reports, you can also more accurately document adherence to IM naltrexone therapy.
All alcohol-dependence medications work best when combined with psychosocial treatment, and monthly medication visits alone will not provide patients the cognitive and skill-building work they need to recover. Patients early in recovery need to be seen much more often by you and/or another provider of recovery-oriented psychosocial treatment.
Which patients will be more receptive to in-office treatment is unclear. Patients who have relapsed because of nonadherence to oral medications may be more willing to try IM therapy after you explain its benefits. Similarly, IM naltrexone may be more beneficial to patients who:
- cannot adhere to oral medication because of cognitive problems or impulsivity
- face severe consequences—such as legal problems, loss of parental custody, or loss of employment—if treatment fails.
The optimal duration of IM naltrexone therapy is not known, but the injectable has shown efficacy after 6 months7 and 1 year.8 Some patients have taken it for more than 3 years.8 Before stopping IM naltrexone, consider whether the patient:
- has achieved sobriety
- has developed skills and external support to maintain sobriety
- has reduced craving intensity or time spent preoccupied with alcohol
- shows improved global psychosocial function as reflected in improved relationships, work performance, and general health.
Patients with family histories of alcohol dependence and who reduce days of heavy drinking but do not achieve sobriety on IM naltrexone are probably at higher risk of relapse to heavy drinking after stopping the medication.
Related resources
- Injectable naltrexone Web site. http://alkermes2005.ifactory.com/products/naltrexone.html.
Drug brand names
- IM naltrexone • Vivitrol
- Oral naltrexone • Depade
- Naloxone • Narcan
Disclosure
Dr. Rosenthal is a consultant for Forest Laboratories and Alkermes.
A long-acting, intramuscular (IM) naltrexone formulation—which at press time awaited FDA approval (Table)—could improve adherence to alcohol dependency pharmacotherapy.
Oral naltrexone can reduce alcohol consumption1 and relapse rates,1,2 but patients often stop taking it3 and increase their risk of relapse.2 Once-daily dosing, inconsistent motivation toward treatment, and cognitive impairment secondary to chronic alcohol dependence often thwart oral naltrexone therapy.
By contrast, IM naltrexone surmounts most compliance issues because you or a clinical assistant administer the drug. Short-term side effects—such as nausea for 2 days—are less likely to affect adherence because the medication keeps working weeks after side effects abate. This gives you time before the next dose to reassure the patient and gives the patient the benefits of continued treatment.
Table
IM naltrexone: Fast facts
| Drug brand name: Vivitrol |
| Class: Opioid antagonist |
| Prospective indication: Alcohol dependence |
| FDA action: Issued approvable letter Dec. 28, 2005 |
| Manufacturer: Alkermes |
| Dosing forms: 380 mg suspension via IM injection |
| Recommended dosage: 380 mg once monthly |
| Estimated date of availability: Spring 2006 |
How naltrexone works
Alcohol stimulates release of endogenous opioids, which in turn stimulate release of dopamine, which mediates reinforcement.4 Opioid receptor stimulation not associated with dopamine also reinforces alcohol use.5 Persons vulnerable to alcohol dependence generally have lower basal levels of opioid secretion and are stimulated at higher levels.6 Opioids also increase dopamine by inhibiting GABA neurons, which suppress dopamine release when uninhibited.
As an opioid antagonist, naltrexone prevents opioids from binding with μ-opioid receptors and modulates dopamine production. This may make drinking less “rewarding” and may reduce craving triggered by conditioned cues associated with alcohol use.
IM naltrexone is packaged in biodegradable microspheres that slowly release naltrexone for 1 month after injection. The microspheres are made of a polyactide-co-glycolide polymer used in other extended-release drugs and in absorbable sutures.
Pharmacokinetics
IM naltrexone plasma levels peak 2 to 3 days after injection, then decline gradually over 30 days. Oral naltrexone dosed at 50 mg/d for 30 days—a cumulative dose of 1,500 mg/month—produces daily peak plasma levels of approximately 10 ng/mL and troughs approaching zero. A once-monthly IM naltrexone injection results in a lower net dose but more-sustained naltrexone levels.
Efficacy
IM naltrexone significantly reduced heavy drinking among alcohol-dependent patients in a phase 3 randomized, placebo-controlled, multicenter trial.7 Actively drinking adults who met DSM-IV criteria for alcohol dependence (N=624) received IM naltrexone, 190 or 380 mg, or placebo every 4 weeks for 6 months. Oral naltrexone lead-in doses were not given. All patients also received 12 sessions of standardized supportive psychosocial therapy during the study.
The primary efficacy measure was event rate of heavy drinking, defined as number of heavy drinking days (≥5 drinks/day for men, ≥4 drinks/day for women) divided by number of days in the study. An event rate ratio (treatment-group to placebo-group event rate) was then estimated over time, taking into account patients who discontinued the study.
After 6 months, event rate of heavy drinking fell 25% among patients receiving 380 mg of IM naltrexone and supportive therapy, compared with patients receiving placebo and supportive therapy (P=0.02). That rate decreased 17% among patients who received 190 mg of IM naltrexone compared with placebo, but the difference between the two treatment groups was not statistically significant (P=0.07).
The median number of heavy drinking days per month decreased substantially across 6 months among all study groups. The decrease was more substantial among patients taking IM naltrexone, 380 mg, than among the placebo group (Figure).
Roughly 8% of patients abstained from drinking for 7 days before entering the study. Among patients who received 380 mg of IM naltrexone:
- those who were abstinent before the study had an 80% greater reduction in event rate of heavy drinking compared with placebo
- nonabstinent patients showed a 21% greater reduction in event rate of heavy drinking compared with placebo.
These findings suggest that IM naltrexone is more effective in persons abstaining from drinking but can also help actively drinking patients.
IM naltrexone also reduced heavy drinking among patients who entered a 1-year open-label extension study after completing the 6-month study.8 Drinking reductions were greater among patients who received 380 mg of naltrexone during both the 6-month and 1-year trials than among those who received placebo for 6 months and were switched to naltrexone, 380 mg, in the 1-year extension.
Figure Median heavy drinking days after 6 months of IM naltrexone or placebo
Source: Reference 7
Tolerability
IM naltrexone was well-tolerated in the phase 3 trial.7 Most-common adverse effects included
- nausea (reported by 33% of patients receiving 380 mg [n=205] and 25% of those receiving 190 mg [n=210])
- headache (22%, 16%)
- fatigue (20%, 16%).
At 380 mg, decreased appetite (13%), dizziness (13%), and injection site pain (12%) also differed significantly from placebo. Nausea was rated as mild or moderate in 95% of cases, usually occurred only after the first injection, and lasted 1 to 2 days on average.
Nine percent of patients taking naltrexone, 190 mg, or placebo also reported injection site pain. Approximately 1% of all patients dropped out because of injection site reactions.
Patients generally adhered to treatment, with 64% receiving 6 injections and 74% receiving at least 4. By comparison, a meta-analysis3 of oral naltrexone clinical trials showed an average 50% retention rate across studies, most of which lasted only 3 months. Study withdrawals because of adverse events were more prevalent among patients receiving IM naltrexone, 380 mg (14.1%), than among the placebo group (6.7%), but the number of serious adverse events differed little.7
Liver enzymes (AST and ALT) did not change significantly during the study. Gamma-glutamyltransferase decreased in all patients, consistent with reduced drinking.
Interactions between IM naltrexone and other medications are probably similar to those observed with oral naltrexone.
Contraindications
Although product labeling was not available when this article was written, IM naltrexone, like its oral form, will likely be contraindicated for patients who:
- are taking opioid analgesics
- are in acute opioid withdrawal
- test positive on urine screen for opioids
- have acute hepatitis or liver failure
- are taking maintenance methadone or buprenorphine or are opioid-dependent.
Patients should be opioid-free for 7 to 10 days before starting IM naltrexone to avoid acute withdrawal symptoms.
Before starting IM naltrexone in patients with a history of opioid abuse, give naloxone, 0.8 mg, to test for withdrawal. Do not start naltrexone if the patient shows signs of opioid withdrawal within 20 minutes of receiving naloxone.
Clinical implications
Long-acting IM naltrexone will make it easier to ensure treatment adherence, compared with oral naltrexone. Giving the drug during the office visit will change your practice patterns, but this increase in hands-on care could strengthen the therapeutic alliance. Compared with interpreting patient self-reports, you can also more accurately document adherence to IM naltrexone therapy.
All alcohol-dependence medications work best when combined with psychosocial treatment, and monthly medication visits alone will not provide patients the cognitive and skill-building work they need to recover. Patients early in recovery need to be seen much more often by you and/or another provider of recovery-oriented psychosocial treatment.
Which patients will be more receptive to in-office treatment is unclear. Patients who have relapsed because of nonadherence to oral medications may be more willing to try IM therapy after you explain its benefits. Similarly, IM naltrexone may be more beneficial to patients who:
- cannot adhere to oral medication because of cognitive problems or impulsivity
- face severe consequences—such as legal problems, loss of parental custody, or loss of employment—if treatment fails.
The optimal duration of IM naltrexone therapy is not known, but the injectable has shown efficacy after 6 months7 and 1 year.8 Some patients have taken it for more than 3 years.8 Before stopping IM naltrexone, consider whether the patient:
- has achieved sobriety
- has developed skills and external support to maintain sobriety
- has reduced craving intensity or time spent preoccupied with alcohol
- shows improved global psychosocial function as reflected in improved relationships, work performance, and general health.
Patients with family histories of alcohol dependence and who reduce days of heavy drinking but do not achieve sobriety on IM naltrexone are probably at higher risk of relapse to heavy drinking after stopping the medication.
Related resources
- Injectable naltrexone Web site. http://alkermes2005.ifactory.com/products/naltrexone.html.
Drug brand names
- IM naltrexone • Vivitrol
- Oral naltrexone • Depade
- Naloxone • Narcan
Disclosure
Dr. Rosenthal is a consultant for Forest Laboratories and Alkermes.
1. Anton RF, Moak DH, Waid LR, et al. Naltrexone and cognitive behavioral therapy for the treatment of outpatient alcoholics: results of a placebo-controlled trial. Am J Psychiatry 1999;156:1758-64.
2. Pettinati HM, Volpicelli JR, Pierce JD, Jr, O’Brien CP. Improving naltrexone response: An intervention for medical practitioners toenhance medication compliance in alcohol dependent patients. J Addict Dis 2000;19:71-83.
3. Bouza C, Magro A, Munoz A, Amate J. Efficacy and safety of naltrexone and acamprosate in the treatment of alcohol dependence: a systematic review. Addiction 2004;99:811-28.
4. Weiss F, Lorang MT, Bloom FE, Koob GF. Oral alcohol self-administration stimulates dopamine release in the rat nucleus accumbens: genetic and motivational determinants. J Pharmacol Exp Ther 1993;267:250-8.
5. Pettit HO, Ettenberg A, Bloom FE, Koob GF. Destruction of dopamine in the nucleus accumbens selectively attenuates cocaine but not heroin self-administration in rats. Psychopharmacology (Berl) 1984;84:167-73.
6. Gianoulakis C. Characterization of the effects of acute ethanol administration on the release of beta-endorphin peptides by the rat hypothalamus. Eur J Pharmacol 1990;180:21-9.
7. Garbutt JC, Kranzler HR, O’Malley SS, et al. Vivitrex Study Group. Efficacy and tolerability of long-acting injectable naltrexone for alcohol dependence: a randomized controlled trial. JAMA 2005;293:1617-25.
8. Gastfriend DR, Dong Q, Loewy J, et al. Durability of effect of long-acting injectable naltrexone. Presented at: Annual Meeting of the American Psychiatric Association; 2005; Atlanta, GA.
1. Anton RF, Moak DH, Waid LR, et al. Naltrexone and cognitive behavioral therapy for the treatment of outpatient alcoholics: results of a placebo-controlled trial. Am J Psychiatry 1999;156:1758-64.
2. Pettinati HM, Volpicelli JR, Pierce JD, Jr, O’Brien CP. Improving naltrexone response: An intervention for medical practitioners toenhance medication compliance in alcohol dependent patients. J Addict Dis 2000;19:71-83.
3. Bouza C, Magro A, Munoz A, Amate J. Efficacy and safety of naltrexone and acamprosate in the treatment of alcohol dependence: a systematic review. Addiction 2004;99:811-28.
4. Weiss F, Lorang MT, Bloom FE, Koob GF. Oral alcohol self-administration stimulates dopamine release in the rat nucleus accumbens: genetic and motivational determinants. J Pharmacol Exp Ther 1993;267:250-8.
5. Pettit HO, Ettenberg A, Bloom FE, Koob GF. Destruction of dopamine in the nucleus accumbens selectively attenuates cocaine but not heroin self-administration in rats. Psychopharmacology (Berl) 1984;84:167-73.
6. Gianoulakis C. Characterization of the effects of acute ethanol administration on the release of beta-endorphin peptides by the rat hypothalamus. Eur J Pharmacol 1990;180:21-9.
7. Garbutt JC, Kranzler HR, O’Malley SS, et al. Vivitrex Study Group. Efficacy and tolerability of long-acting injectable naltrexone for alcohol dependence: a randomized controlled trial. JAMA 2005;293:1617-25.
8. Gastfriend DR, Dong Q, Loewy J, et al. Durability of effect of long-acting injectable naltrexone. Presented at: Annual Meeting of the American Psychiatric Association; 2005; Atlanta, GA.
Ramelteon
In clinical trials, ramelteon has helped patients fall asleep more quickly. Whereas other sleep-promoting medications sedate through effects on gamma-butyric acid (GABA) receptors, ramelteon interacts with melatonin receptors to regulate sleep patterns. It is FDA-approved for treating insomnia characterized by sleep-onset difficulty (Table 1)
Table 1
Ramelteon: Fast facts
| Brand name: |
| Rozerem |
| Class: |
| Nonbenzodiazepine hypnotic |
| FDA-approved indication: |
| Insomnia characterized by sleep-onset difficulty |
| Approval date: |
| August 18, 2005 |
| Manufacturer: |
| Takeda Pharmaceuticals North America |
| Dosing form: |
| 8-mg tablets |
| Recommended dosage: |
| 8 mg within 30 minutes of going to bed |
| Additional prescribing information: |
| www.rozerem.com |
How it works
Ramelteon, a melatonin receptor agonist, has high affinity for the MT1 and MT2 (melatonin) receptors. Although the precise mechanism by which ramelteon affects sleep remains unknown, its effect on sleep is hypothesized to be similar to that of the neurohormone melatonin.
Melatonin is important to maintaining the circadian rhythm that underlies the sleep-wake cycle. Sunlight influences neurohormones that mediate daytime-specific physiologic events. An increase in melatonin—a change that accompanies darkness—is believed to mediate changes in physiology that are characteristic of nighttime. Melatonin thus may be more of a circadian “clock” regulator than a sedative.
Ramelteon shows some features of melatonin that differentiate it from the GABA-related sedating agents. Both ramelteon and melatonin lack abuse potential and a dose-response relationship.
Pharmacokinetics
Ramelteon is absorbed rapidly from the GI tract and reaches median peak concentrations within 30 to 90 minutes of dosing. Taking ramelteon with a high-fat meal reduces its maximum concentration by 22% and slows hypnotic onset by approximately 45 minutes.
The drug is metabolized mostly through the 1A2 isoenzyme of the cytochrome P (CYP)-450 system, although CYP 2C and 3A4 isoenzymes are also involved. About 90% of the dose is excreted.
Ramelteon’s elimination half-life averages 1 to 2.6 hours, so blood levels upon awakening will likely be too low to cause residual effects. Interestingly, in one placebo-controlled study,1 subjects who received a single 64-mg dose reported significantly reduced alertness and diminished ability to concentrate upon awakening. Subjects who took a 16-mg dose did not report this effect. Whether this finding is clinically relevant or relates to a residual effect, sedation, or cognitive impairment is unclear.
Efficacy
In a randomized, double-blind, placebo-controlled trial, ramelteon shortened sleep latency (time between going to bed and falling asleep) among patients with transient insomnia.
Roth et al1 studied 375 healthy adults ages 35 to 60 who reported sleeping 6.5 to 8.5 hours nightly and usually taking ≥30 minutes to fall asleep. In sleep research centers, subjects received one dose of ramelteon, 16 or 64 mg, or placebo 30 minutes before bedtime.
Mean latency to persistent sleep, measured with polysomnography, was 10 minutes shorter among both ramelteon dosage groups than among the placebo group. Mean total sleep time was 11 to 14 minutes longer among both ramelteon groups based on polysomnography, although subjective sleep estimates the next morning were similar among all three groups.
Roth et al2 also assessed efficacy of ramelteon across 5 weeks among 829 older patients (mean age 72) with insomnia (as defined by DSM-IV-TR) for ≥3 months, total nightly sleep time ≤6.5 hours for 3 nights, and self-reported sleep latency ≥45 minutes nightly for ≥3 nights.
Mean sleep latency decreased 25 to 30 minutes among subjects taking ramelteon, 4 or 8 mg nightly, compared with a mean 15-minute decrease among the placebo group. Average total sleep time was 5 to 8 minutes longer among both ramelteon groups compared with placebo.
Subjects in both ramelteon groups then received placebo for 1 week, during which time their mean latency to persistent sleep improved further or stayed the same. This suggests that ramelteon did not cause rebound insomnia.
Safety and tolerability
Ramelteon was generally well tolerated in clinical and preclinical trials. Headaches (7% of subjects), somnolence (5%), dizziness (5%), fatigue (4%), nausea (3%), exacerbated insomnia (3%), and upper respiratory tract infection (3%) were most commonly reported.3 Less-common effects included diarrhea, myalgia, depression, dysgeusia, arthralgia, influenza, and blood cortisol decrease.
The most common side effects among subjects age ≥65 were dizziness, dysgeusia, headaches, myalgia, and somnolence (Table 2). These occurred less frequently over 5 weeks among patients taking 4 mg/d than among those who took 8 mg/d, the FDA-approved dosage.
Ramelteon also showed no abuse potential compared with triazolam and placebo in a trial of 14 patients with a history of anxiolytic or sedative/hypnotic abuse.4

Contraindications
Do not give ramelteon to patients taking fluvoxamine. The antidepressant has been shown to raise serum ramelteon approximately 70-fold, thus substantially increasing the risk of ramelteon-associated adverse events.3
Ramelteon has shown teratogenicity in animals, though at doses far exceeding human levels. Still, as with other sleep-promoting medications, avoid prescribing ramelteon to expectant mothers.
Concomitant use of a strong CYP enzyme inducer such as rifampin may increase ramelteon metabolism and reduce serum ramelteon, which might decrease its efficacy in some cases. Whether increasing the ramelteon dosage counters this interaction is unknown.
Strong CYP 2C9 inhibitors such as fluconazole or strong CYP 3A4 inhibitors such as ketoconazole can raise serum ramelteon and might increase the risk of adverse events in some persons.
Dosing
Start ramelteon at 8 mg nightly, and tell patients to take it within 30 minutes of going to bed.
Because high-fat food slows its absorption, advise patients not to take ramelteon within 1 hour of eating a high-fat meal.
Ramelteon’s efficacy and side effects do not appear to be dose-dependent when given at 8 to 64 mg/d. Whether dosages >64 mg/d increase side-effect risk or therapeutic effect is unknown.
As with other hypnotics, supplement ramelteon therapy with sleep hygiene education and relaxation techniques.
Clinical implications
Ramelteon appears to help patients who have trouble falling asleep.
Because no other prescription medication targets melatonin neurotransmitters, no precedent and little data exist to guide patient choice, dosing, and treatment duration. Effects of ramelteon use >5 weeks are unknown. Clinical use and future research should uncover more information about ramelteon’s properties.
Related resources
- Ramelteon Web site. www.rozerem.com.
- Arendt J, Skene DJ. Melatonin as a chronobiotic. Sleep Med Rev 2005;9:25-39.
- Scheer FA, Czeisler CA. Melatonin, sleep, and circadian rhythms. Sleep Med Rev 2005;9:5-9.
Drug brand names
- Fluconazole • Diflucan
- Fluvoxamine • Luvox
- Ketoconazole • Nizoral
- Ramelteon • Rozerem
- Rifampin • Rifadin
- Triazolam • Halcion
Disclosures
Dr. Krystal receives research/grant support, is a consultant to, or is a speaker for Cephalon, Cyberonics, GlaxoSmithKline, Johnson & Johnson, King Pharmaceuticals, Mecta Corp., Merck and Co., Neurocrine Biosciences, Neurogen Corp., Neuronetics, Organon, Pfizer, Respironics, Sanofi-Aventis, Sepracor, Somaxon Pharmaceuticals, Takeda Pharmaceuticals North America, and TransOral Pharmaceuticals.
1. Roth T, Stubbs C, Walsh JK. Ramelteon (TAK-375), a selective MT1/MT2-receptor agonist, reduces latency to persistent sleep in a model of transient insomnia related to a novel sleep environment. Sleep 2005;28:303-7.
2. Roth T, Seiden D, Sainati S, et al. Phase III outpatient trial of ramelteon for the treatment of chronic insomnia in elderly patients (poster presentation). Orlando, FL: American Geriatric Society annual meeting, 2005.
3. Rozerem prescribing information. Takeda Pharmaceuticals North America, 2005.
4. Griffiths R, Seuss P. Ramelteon and triazolam in humans: behavioral effects and abuse potential (poster). Atlanta, GA: American Psychiatric Association annual meeting, 2005.
In clinical trials, ramelteon has helped patients fall asleep more quickly. Whereas other sleep-promoting medications sedate through effects on gamma-butyric acid (GABA) receptors, ramelteon interacts with melatonin receptors to regulate sleep patterns. It is FDA-approved for treating insomnia characterized by sleep-onset difficulty (Table 1)
Table 1
Ramelteon: Fast facts
| Brand name: |
| Rozerem |
| Class: |
| Nonbenzodiazepine hypnotic |
| FDA-approved indication: |
| Insomnia characterized by sleep-onset difficulty |
| Approval date: |
| August 18, 2005 |
| Manufacturer: |
| Takeda Pharmaceuticals North America |
| Dosing form: |
| 8-mg tablets |
| Recommended dosage: |
| 8 mg within 30 minutes of going to bed |
| Additional prescribing information: |
| www.rozerem.com |
How it works
Ramelteon, a melatonin receptor agonist, has high affinity for the MT1 and MT2 (melatonin) receptors. Although the precise mechanism by which ramelteon affects sleep remains unknown, its effect on sleep is hypothesized to be similar to that of the neurohormone melatonin.
Melatonin is important to maintaining the circadian rhythm that underlies the sleep-wake cycle. Sunlight influences neurohormones that mediate daytime-specific physiologic events. An increase in melatonin—a change that accompanies darkness—is believed to mediate changes in physiology that are characteristic of nighttime. Melatonin thus may be more of a circadian “clock” regulator than a sedative.
Ramelteon shows some features of melatonin that differentiate it from the GABA-related sedating agents. Both ramelteon and melatonin lack abuse potential and a dose-response relationship.
Pharmacokinetics
Ramelteon is absorbed rapidly from the GI tract and reaches median peak concentrations within 30 to 90 minutes of dosing. Taking ramelteon with a high-fat meal reduces its maximum concentration by 22% and slows hypnotic onset by approximately 45 minutes.
The drug is metabolized mostly through the 1A2 isoenzyme of the cytochrome P (CYP)-450 system, although CYP 2C and 3A4 isoenzymes are also involved. About 90% of the dose is excreted.
Ramelteon’s elimination half-life averages 1 to 2.6 hours, so blood levels upon awakening will likely be too low to cause residual effects. Interestingly, in one placebo-controlled study,1 subjects who received a single 64-mg dose reported significantly reduced alertness and diminished ability to concentrate upon awakening. Subjects who took a 16-mg dose did not report this effect. Whether this finding is clinically relevant or relates to a residual effect, sedation, or cognitive impairment is unclear.
Efficacy
In a randomized, double-blind, placebo-controlled trial, ramelteon shortened sleep latency (time between going to bed and falling asleep) among patients with transient insomnia.
Roth et al1 studied 375 healthy adults ages 35 to 60 who reported sleeping 6.5 to 8.5 hours nightly and usually taking ≥30 minutes to fall asleep. In sleep research centers, subjects received one dose of ramelteon, 16 or 64 mg, or placebo 30 minutes before bedtime.
Mean latency to persistent sleep, measured with polysomnography, was 10 minutes shorter among both ramelteon dosage groups than among the placebo group. Mean total sleep time was 11 to 14 minutes longer among both ramelteon groups based on polysomnography, although subjective sleep estimates the next morning were similar among all three groups.
Roth et al2 also assessed efficacy of ramelteon across 5 weeks among 829 older patients (mean age 72) with insomnia (as defined by DSM-IV-TR) for ≥3 months, total nightly sleep time ≤6.5 hours for 3 nights, and self-reported sleep latency ≥45 minutes nightly for ≥3 nights.
Mean sleep latency decreased 25 to 30 minutes among subjects taking ramelteon, 4 or 8 mg nightly, compared with a mean 15-minute decrease among the placebo group. Average total sleep time was 5 to 8 minutes longer among both ramelteon groups compared with placebo.
Subjects in both ramelteon groups then received placebo for 1 week, during which time their mean latency to persistent sleep improved further or stayed the same. This suggests that ramelteon did not cause rebound insomnia.
Safety and tolerability
Ramelteon was generally well tolerated in clinical and preclinical trials. Headaches (7% of subjects), somnolence (5%), dizziness (5%), fatigue (4%), nausea (3%), exacerbated insomnia (3%), and upper respiratory tract infection (3%) were most commonly reported.3 Less-common effects included diarrhea, myalgia, depression, dysgeusia, arthralgia, influenza, and blood cortisol decrease.
The most common side effects among subjects age ≥65 were dizziness, dysgeusia, headaches, myalgia, and somnolence (Table 2). These occurred less frequently over 5 weeks among patients taking 4 mg/d than among those who took 8 mg/d, the FDA-approved dosage.
Ramelteon also showed no abuse potential compared with triazolam and placebo in a trial of 14 patients with a history of anxiolytic or sedative/hypnotic abuse.4
Contraindications
Do not give ramelteon to patients taking fluvoxamine. The antidepressant has been shown to raise serum ramelteon approximately 70-fold, thus substantially increasing the risk of ramelteon-associated adverse events.3
Ramelteon has shown teratogenicity in animals, though at doses far exceeding human levels. Still, as with other sleep-promoting medications, avoid prescribing ramelteon to expectant mothers.
Concomitant use of a strong CYP enzyme inducer such as rifampin may increase ramelteon metabolism and reduce serum ramelteon, which might decrease its efficacy in some cases. Whether increasing the ramelteon dosage counters this interaction is unknown.
Strong CYP 2C9 inhibitors such as fluconazole or strong CYP 3A4 inhibitors such as ketoconazole can raise serum ramelteon and might increase the risk of adverse events in some persons.
Dosing
Start ramelteon at 8 mg nightly, and tell patients to take it within 30 minutes of going to bed.
Because high-fat food slows its absorption, advise patients not to take ramelteon within 1 hour of eating a high-fat meal.
Ramelteon’s efficacy and side effects do not appear to be dose-dependent when given at 8 to 64 mg/d. Whether dosages >64 mg/d increase side-effect risk or therapeutic effect is unknown.
As with other hypnotics, supplement ramelteon therapy with sleep hygiene education and relaxation techniques.
Clinical implications
Ramelteon appears to help patients who have trouble falling asleep.
Because no other prescription medication targets melatonin neurotransmitters, no precedent and little data exist to guide patient choice, dosing, and treatment duration. Effects of ramelteon use >5 weeks are unknown. Clinical use and future research should uncover more information about ramelteon’s properties.
Related resources
- Ramelteon Web site. www.rozerem.com.
- Arendt J, Skene DJ. Melatonin as a chronobiotic. Sleep Med Rev 2005;9:25-39.
- Scheer FA, Czeisler CA. Melatonin, sleep, and circadian rhythms. Sleep Med Rev 2005;9:5-9.
Drug brand names
- Fluconazole • Diflucan
- Fluvoxamine • Luvox
- Ketoconazole • Nizoral
- Ramelteon • Rozerem
- Rifampin • Rifadin
- Triazolam • Halcion
Disclosures
Dr. Krystal receives research/grant support, is a consultant to, or is a speaker for Cephalon, Cyberonics, GlaxoSmithKline, Johnson & Johnson, King Pharmaceuticals, Mecta Corp., Merck and Co., Neurocrine Biosciences, Neurogen Corp., Neuronetics, Organon, Pfizer, Respironics, Sanofi-Aventis, Sepracor, Somaxon Pharmaceuticals, Takeda Pharmaceuticals North America, and TransOral Pharmaceuticals.
In clinical trials, ramelteon has helped patients fall asleep more quickly. Whereas other sleep-promoting medications sedate through effects on gamma-butyric acid (GABA) receptors, ramelteon interacts with melatonin receptors to regulate sleep patterns. It is FDA-approved for treating insomnia characterized by sleep-onset difficulty (Table 1)
Table 1
Ramelteon: Fast facts
| Brand name: |
| Rozerem |
| Class: |
| Nonbenzodiazepine hypnotic |
| FDA-approved indication: |
| Insomnia characterized by sleep-onset difficulty |
| Approval date: |
| August 18, 2005 |
| Manufacturer: |
| Takeda Pharmaceuticals North America |
| Dosing form: |
| 8-mg tablets |
| Recommended dosage: |
| 8 mg within 30 minutes of going to bed |
| Additional prescribing information: |
| www.rozerem.com |
How it works
Ramelteon, a melatonin receptor agonist, has high affinity for the MT1 and MT2 (melatonin) receptors. Although the precise mechanism by which ramelteon affects sleep remains unknown, its effect on sleep is hypothesized to be similar to that of the neurohormone melatonin.
Melatonin is important to maintaining the circadian rhythm that underlies the sleep-wake cycle. Sunlight influences neurohormones that mediate daytime-specific physiologic events. An increase in melatonin—a change that accompanies darkness—is believed to mediate changes in physiology that are characteristic of nighttime. Melatonin thus may be more of a circadian “clock” regulator than a sedative.
Ramelteon shows some features of melatonin that differentiate it from the GABA-related sedating agents. Both ramelteon and melatonin lack abuse potential and a dose-response relationship.
Pharmacokinetics
Ramelteon is absorbed rapidly from the GI tract and reaches median peak concentrations within 30 to 90 minutes of dosing. Taking ramelteon with a high-fat meal reduces its maximum concentration by 22% and slows hypnotic onset by approximately 45 minutes.
The drug is metabolized mostly through the 1A2 isoenzyme of the cytochrome P (CYP)-450 system, although CYP 2C and 3A4 isoenzymes are also involved. About 90% of the dose is excreted.
Ramelteon’s elimination half-life averages 1 to 2.6 hours, so blood levels upon awakening will likely be too low to cause residual effects. Interestingly, in one placebo-controlled study,1 subjects who received a single 64-mg dose reported significantly reduced alertness and diminished ability to concentrate upon awakening. Subjects who took a 16-mg dose did not report this effect. Whether this finding is clinically relevant or relates to a residual effect, sedation, or cognitive impairment is unclear.
Efficacy
In a randomized, double-blind, placebo-controlled trial, ramelteon shortened sleep latency (time between going to bed and falling asleep) among patients with transient insomnia.
Roth et al1 studied 375 healthy adults ages 35 to 60 who reported sleeping 6.5 to 8.5 hours nightly and usually taking ≥30 minutes to fall asleep. In sleep research centers, subjects received one dose of ramelteon, 16 or 64 mg, or placebo 30 minutes before bedtime.
Mean latency to persistent sleep, measured with polysomnography, was 10 minutes shorter among both ramelteon dosage groups than among the placebo group. Mean total sleep time was 11 to 14 minutes longer among both ramelteon groups based on polysomnography, although subjective sleep estimates the next morning were similar among all three groups.
Roth et al2 also assessed efficacy of ramelteon across 5 weeks among 829 older patients (mean age 72) with insomnia (as defined by DSM-IV-TR) for ≥3 months, total nightly sleep time ≤6.5 hours for 3 nights, and self-reported sleep latency ≥45 minutes nightly for ≥3 nights.
Mean sleep latency decreased 25 to 30 minutes among subjects taking ramelteon, 4 or 8 mg nightly, compared with a mean 15-minute decrease among the placebo group. Average total sleep time was 5 to 8 minutes longer among both ramelteon groups compared with placebo.
Subjects in both ramelteon groups then received placebo for 1 week, during which time their mean latency to persistent sleep improved further or stayed the same. This suggests that ramelteon did not cause rebound insomnia.
Safety and tolerability
Ramelteon was generally well tolerated in clinical and preclinical trials. Headaches (7% of subjects), somnolence (5%), dizziness (5%), fatigue (4%), nausea (3%), exacerbated insomnia (3%), and upper respiratory tract infection (3%) were most commonly reported.3 Less-common effects included diarrhea, myalgia, depression, dysgeusia, arthralgia, influenza, and blood cortisol decrease.
The most common side effects among subjects age ≥65 were dizziness, dysgeusia, headaches, myalgia, and somnolence (Table 2). These occurred less frequently over 5 weeks among patients taking 4 mg/d than among those who took 8 mg/d, the FDA-approved dosage.
Ramelteon also showed no abuse potential compared with triazolam and placebo in a trial of 14 patients with a history of anxiolytic or sedative/hypnotic abuse.4
Contraindications
Do not give ramelteon to patients taking fluvoxamine. The antidepressant has been shown to raise serum ramelteon approximately 70-fold, thus substantially increasing the risk of ramelteon-associated adverse events.3
Ramelteon has shown teratogenicity in animals, though at doses far exceeding human levels. Still, as with other sleep-promoting medications, avoid prescribing ramelteon to expectant mothers.
Concomitant use of a strong CYP enzyme inducer such as rifampin may increase ramelteon metabolism and reduce serum ramelteon, which might decrease its efficacy in some cases. Whether increasing the ramelteon dosage counters this interaction is unknown.
Strong CYP 2C9 inhibitors such as fluconazole or strong CYP 3A4 inhibitors such as ketoconazole can raise serum ramelteon and might increase the risk of adverse events in some persons.
Dosing
Start ramelteon at 8 mg nightly, and tell patients to take it within 30 minutes of going to bed.
Because high-fat food slows its absorption, advise patients not to take ramelteon within 1 hour of eating a high-fat meal.
Ramelteon’s efficacy and side effects do not appear to be dose-dependent when given at 8 to 64 mg/d. Whether dosages >64 mg/d increase side-effect risk or therapeutic effect is unknown.
As with other hypnotics, supplement ramelteon therapy with sleep hygiene education and relaxation techniques.
Clinical implications
Ramelteon appears to help patients who have trouble falling asleep.
Because no other prescription medication targets melatonin neurotransmitters, no precedent and little data exist to guide patient choice, dosing, and treatment duration. Effects of ramelteon use >5 weeks are unknown. Clinical use and future research should uncover more information about ramelteon’s properties.
Related resources
- Ramelteon Web site. www.rozerem.com.
- Arendt J, Skene DJ. Melatonin as a chronobiotic. Sleep Med Rev 2005;9:25-39.
- Scheer FA, Czeisler CA. Melatonin, sleep, and circadian rhythms. Sleep Med Rev 2005;9:5-9.
Drug brand names
- Fluconazole • Diflucan
- Fluvoxamine • Luvox
- Ketoconazole • Nizoral
- Ramelteon • Rozerem
- Rifampin • Rifadin
- Triazolam • Halcion
Disclosures
Dr. Krystal receives research/grant support, is a consultant to, or is a speaker for Cephalon, Cyberonics, GlaxoSmithKline, Johnson & Johnson, King Pharmaceuticals, Mecta Corp., Merck and Co., Neurocrine Biosciences, Neurogen Corp., Neuronetics, Organon, Pfizer, Respironics, Sanofi-Aventis, Sepracor, Somaxon Pharmaceuticals, Takeda Pharmaceuticals North America, and TransOral Pharmaceuticals.
1. Roth T, Stubbs C, Walsh JK. Ramelteon (TAK-375), a selective MT1/MT2-receptor agonist, reduces latency to persistent sleep in a model of transient insomnia related to a novel sleep environment. Sleep 2005;28:303-7.
2. Roth T, Seiden D, Sainati S, et al. Phase III outpatient trial of ramelteon for the treatment of chronic insomnia in elderly patients (poster presentation). Orlando, FL: American Geriatric Society annual meeting, 2005.
3. Rozerem prescribing information. Takeda Pharmaceuticals North America, 2005.
4. Griffiths R, Seuss P. Ramelteon and triazolam in humans: behavioral effects and abuse potential (poster). Atlanta, GA: American Psychiatric Association annual meeting, 2005.
1. Roth T, Stubbs C, Walsh JK. Ramelteon (TAK-375), a selective MT1/MT2-receptor agonist, reduces latency to persistent sleep in a model of transient insomnia related to a novel sleep environment. Sleep 2005;28:303-7.
2. Roth T, Seiden D, Sainati S, et al. Phase III outpatient trial of ramelteon for the treatment of chronic insomnia in elderly patients (poster presentation). Orlando, FL: American Geriatric Society annual meeting, 2005.
3. Rozerem prescribing information. Takeda Pharmaceuticals North America, 2005.
4. Griffiths R, Seuss P. Ramelteon and triazolam in humans: behavioral effects and abuse potential (poster). Atlanta, GA: American Psychiatric Association annual meeting, 2005.