User login
USPSTF update: Screening for abnormal blood glucose, diabetes
In December 2015, the United States Preventive Services Task Force updated its recommendation on screening for abnormal blood glucose and diabetes to say that clinicians should screen all adults ages 40 to 70 years who are overweight or obese as part of a cardiovascular risk assessment.1 This recommendation carries a B grade signifying a moderate certainty that a moderate net benefit will be gained by detecting impaired fasting glucose (IFG), impaired glucose tolerance (IGT), or diabetes, and by implementing intensive lifestyle interventions. In this article, as in the Task Force recommendation, the term diabetes means type 2 diabetes. Obesity is defined as a body mass index (BMI) of ≥30 kg/m2, and overweight as a BMI >25.
How the Task Force recommendation evolved
The previous Task Force recommendation on this topic, made in 2008, advised screening only adults with hypertension because there was no evidence that any other group benefited from screening. In subsequent years, there were calls for the Task Force to revise its recommendation to bring it more in line with that of the American Diabetes Association (ADA).2 While this new recommendation does add more adults to the cohort of those the Task Force believes should be screened, it is still not totally in concert with the ADA, which recommends screening all adults 45 years or older and those who are younger if they have multiple risk factors.3
Both the Task Force and the ADA acknowledge there is no direct evidence for any benefit in screening for diabetes in the general, asymptomatic population. The Task Force, with its standard of making recommendations only when good evidence supports them, has opted to address screening for abnormal glucose levels in the context of cardiovascular risk reduction and persuasive evidence that lifestyle interventions can reduce cardiovascular risks and slow progression to diabetes.
The ADA is willing to rely on less rigorous evidence of benefit in screening, diagnosing, and treating undetected diabetes. It believes that morbidity and mortality from this pervasive chronic disease can be reduced with early detection and treatment.
Still the Task Force and ADA agree more than they differ
While it appears that significant differences exist between the recommendations of the Task Force and the ADA, a closer look shows they actually have much in common; and, as they pertain to daily practice, any remaining differences are primarily ones of emphasis. For instance, the Clinical Considerations section of the Task Force recommendation acknowledges that certain people are at increased risk for diabetes at younger ages and at a lower BMI, and that clinicians should “consider” screening them earlier than at age 40 years. The risks listed include a family history of diabetes or a personal history of gestational diabetes or polycystic ovarian syndrome; or being African American, Hispanic, Asian American, American Indian, Alaskan Native, or Native Hawaiian.
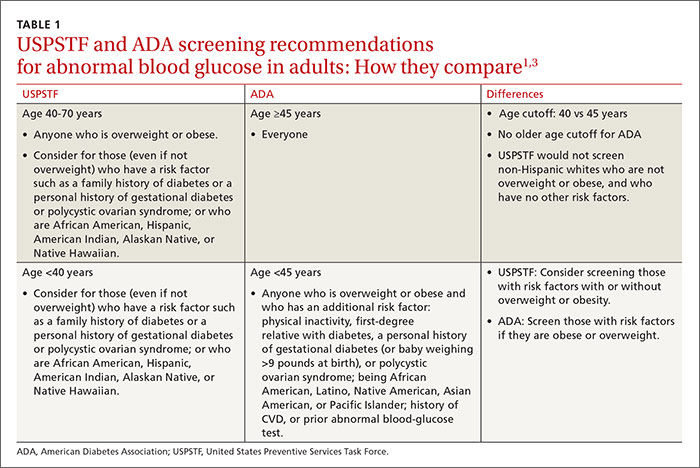
The Task Force statement seems to imply—although this is not entirely clear—that those who have these risks should also be screened if they are older than age 40 years even if they are not obese. So, although the ADA would screen everyone ages 45 and older, the Task Force would screen everyone ages 40 and older, except for non-Hispanic whites who are not overweight or obese, and who have no other risk factors. TABLE 11,3 details the Task Force and the ADA screening criteria and how they differ.
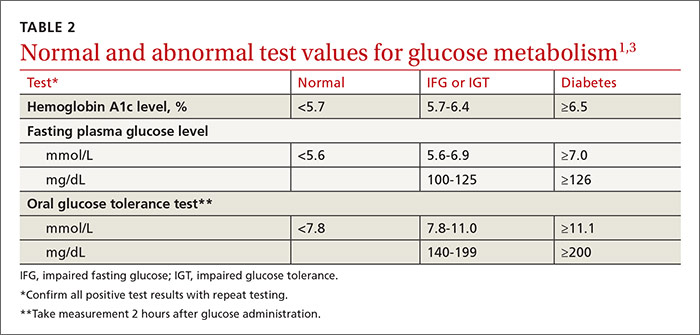
The Task Force and the ADA also agree on the 3 tests acceptable for screening and the test values that define normal glucose, IGT, IFG, and diabetes (TABLE 2).1,3 The tests are a randomly measured glycated hemoglobin level, a fasting plasma glucose level, and an oral glucose tolerance test performed in the morning after an overnight fast, with glucose measured 2 hours after a 75-g oral glucose load. If a screening result is abnormal, confirmation should be sought by repeating the same test. And both organizations suggest that, following a normal test result, the optimal interval for retesting is 3 years.
Intervening to delay progression to diabetes
For anyone with a confirmed abnormal blood glucose level, the Task Force advises referral for intensive behavioral interventions—ie, multiple counseling sessions over an extended period on a healthy diet and optimal physical activity. These types of interventions can reduce blood glucose levels and lower the risk of progression to diabetes, and can help with lowering weight, blood pressure, and lipid levels. The evidence report that preceded the recommendation pooled the results from 10 studies on lifestyle modification.4 The length of follow-up in these studies ranged from 3 to 23 years, and the number needed to treat to prevent one case of progression to diabetes ranged from about 5 to 20.4
Medications such as metformin, thiazolidinediones, and alpha-glucosidase inhibitors can also reduce blood glucose levels and slow progression to diabetes. However, the Task Force says there is insufficient evidence that pharmacologic interventions have the same multifactorial benefits—weight loss or reductions in glucose levels, blood pressure, and lipid levels—as behavioral interventions.1
As for the other modifiable risk factors for cardiovascular disease—obesity, lack of physical activity, high lipid levels, high blood pressure, and smoking—the Task Force has developed recommendations on screening for and treating each of them,5 which supplement the recommendations discussed in this article.
1. U.S. Preventive Services Task Force. Abnormal blood glucose and type 2 diabetes mellitus: screening. Available at: http://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/screening-for-abnormal-blood-glucose-and-type-2-diabetes. Accessed May 20, 2016.
2. Casagrande SS, Cowie CC, Fradkin JE. Utility of the US Preventive Services Task Force criteria for diabetes screening. Am J Prev Med. 2013;45:167-174.
3. American Diabetes Association. Standards of medical care in diabetes - 2016. Diabetes Care. 2016;39(Suppl 1):S1–S112.
4. Selph S, Dana T, Bougatsos C, et al. A systematic review to update the 2008 U.S. Preventive Services Task Force recommendation [Agency for Healthcare Research and Quality]. 2015. Available at: http://www.ncbi.nlm.nih.gov/books/NBK293871/. Accessed May 20, 2016.
5. U.S. Preventive Services Task Force. Healthful diet and physical activity for cardiovascular disease prevention in adults with cardiovascular risk factors: behavioral counseling. Available at: http://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/healthy-diet-and-physical-activity-counseling-adults-with-high-risk-of-cvd. Accessed May 20,
2016.
In December 2015, the United States Preventive Services Task Force updated its recommendation on screening for abnormal blood glucose and diabetes to say that clinicians should screen all adults ages 40 to 70 years who are overweight or obese as part of a cardiovascular risk assessment.1 This recommendation carries a B grade signifying a moderate certainty that a moderate net benefit will be gained by detecting impaired fasting glucose (IFG), impaired glucose tolerance (IGT), or diabetes, and by implementing intensive lifestyle interventions. In this article, as in the Task Force recommendation, the term diabetes means type 2 diabetes. Obesity is defined as a body mass index (BMI) of ≥30 kg/m2, and overweight as a BMI >25.
How the Task Force recommendation evolved
The previous Task Force recommendation on this topic, made in 2008, advised screening only adults with hypertension because there was no evidence that any other group benefited from screening. In subsequent years, there were calls for the Task Force to revise its recommendation to bring it more in line with that of the American Diabetes Association (ADA).2 While this new recommendation does add more adults to the cohort of those the Task Force believes should be screened, it is still not totally in concert with the ADA, which recommends screening all adults 45 years or older and those who are younger if they have multiple risk factors.3
Both the Task Force and the ADA acknowledge there is no direct evidence for any benefit in screening for diabetes in the general, asymptomatic population. The Task Force, with its standard of making recommendations only when good evidence supports them, has opted to address screening for abnormal glucose levels in the context of cardiovascular risk reduction and persuasive evidence that lifestyle interventions can reduce cardiovascular risks and slow progression to diabetes.
The ADA is willing to rely on less rigorous evidence of benefit in screening, diagnosing, and treating undetected diabetes. It believes that morbidity and mortality from this pervasive chronic disease can be reduced with early detection and treatment.
Still the Task Force and ADA agree more than they differ
While it appears that significant differences exist between the recommendations of the Task Force and the ADA, a closer look shows they actually have much in common; and, as they pertain to daily practice, any remaining differences are primarily ones of emphasis. For instance, the Clinical Considerations section of the Task Force recommendation acknowledges that certain people are at increased risk for diabetes at younger ages and at a lower BMI, and that clinicians should “consider” screening them earlier than at age 40 years. The risks listed include a family history of diabetes or a personal history of gestational diabetes or polycystic ovarian syndrome; or being African American, Hispanic, Asian American, American Indian, Alaskan Native, or Native Hawaiian.
The Task Force statement seems to imply—although this is not entirely clear—that those who have these risks should also be screened if they are older than age 40 years even if they are not obese. So, although the ADA would screen everyone ages 45 and older, the Task Force would screen everyone ages 40 and older, except for non-Hispanic whites who are not overweight or obese, and who have no other risk factors. TABLE 11,3 details the Task Force and the ADA screening criteria and how they differ.
The Task Force and the ADA also agree on the 3 tests acceptable for screening and the test values that define normal glucose, IGT, IFG, and diabetes (TABLE 2).1,3 The tests are a randomly measured glycated hemoglobin level, a fasting plasma glucose level, and an oral glucose tolerance test performed in the morning after an overnight fast, with glucose measured 2 hours after a 75-g oral glucose load. If a screening result is abnormal, confirmation should be sought by repeating the same test. And both organizations suggest that, following a normal test result, the optimal interval for retesting is 3 years.
Intervening to delay progression to diabetes
For anyone with a confirmed abnormal blood glucose level, the Task Force advises referral for intensive behavioral interventions—ie, multiple counseling sessions over an extended period on a healthy diet and optimal physical activity. These types of interventions can reduce blood glucose levels and lower the risk of progression to diabetes, and can help with lowering weight, blood pressure, and lipid levels. The evidence report that preceded the recommendation pooled the results from 10 studies on lifestyle modification.4 The length of follow-up in these studies ranged from 3 to 23 years, and the number needed to treat to prevent one case of progression to diabetes ranged from about 5 to 20.4
Medications such as metformin, thiazolidinediones, and alpha-glucosidase inhibitors can also reduce blood glucose levels and slow progression to diabetes. However, the Task Force says there is insufficient evidence that pharmacologic interventions have the same multifactorial benefits—weight loss or reductions in glucose levels, blood pressure, and lipid levels—as behavioral interventions.1
As for the other modifiable risk factors for cardiovascular disease—obesity, lack of physical activity, high lipid levels, high blood pressure, and smoking—the Task Force has developed recommendations on screening for and treating each of them,5 which supplement the recommendations discussed in this article.
In December 2015, the United States Preventive Services Task Force updated its recommendation on screening for abnormal blood glucose and diabetes to say that clinicians should screen all adults ages 40 to 70 years who are overweight or obese as part of a cardiovascular risk assessment.1 This recommendation carries a B grade signifying a moderate certainty that a moderate net benefit will be gained by detecting impaired fasting glucose (IFG), impaired glucose tolerance (IGT), or diabetes, and by implementing intensive lifestyle interventions. In this article, as in the Task Force recommendation, the term diabetes means type 2 diabetes. Obesity is defined as a body mass index (BMI) of ≥30 kg/m2, and overweight as a BMI >25.
How the Task Force recommendation evolved
The previous Task Force recommendation on this topic, made in 2008, advised screening only adults with hypertension because there was no evidence that any other group benefited from screening. In subsequent years, there were calls for the Task Force to revise its recommendation to bring it more in line with that of the American Diabetes Association (ADA).2 While this new recommendation does add more adults to the cohort of those the Task Force believes should be screened, it is still not totally in concert with the ADA, which recommends screening all adults 45 years or older and those who are younger if they have multiple risk factors.3
Both the Task Force and the ADA acknowledge there is no direct evidence for any benefit in screening for diabetes in the general, asymptomatic population. The Task Force, with its standard of making recommendations only when good evidence supports them, has opted to address screening for abnormal glucose levels in the context of cardiovascular risk reduction and persuasive evidence that lifestyle interventions can reduce cardiovascular risks and slow progression to diabetes.
The ADA is willing to rely on less rigorous evidence of benefit in screening, diagnosing, and treating undetected diabetes. It believes that morbidity and mortality from this pervasive chronic disease can be reduced with early detection and treatment.
Still the Task Force and ADA agree more than they differ
While it appears that significant differences exist between the recommendations of the Task Force and the ADA, a closer look shows they actually have much in common; and, as they pertain to daily practice, any remaining differences are primarily ones of emphasis. For instance, the Clinical Considerations section of the Task Force recommendation acknowledges that certain people are at increased risk for diabetes at younger ages and at a lower BMI, and that clinicians should “consider” screening them earlier than at age 40 years. The risks listed include a family history of diabetes or a personal history of gestational diabetes or polycystic ovarian syndrome; or being African American, Hispanic, Asian American, American Indian, Alaskan Native, or Native Hawaiian.
The Task Force statement seems to imply—although this is not entirely clear—that those who have these risks should also be screened if they are older than age 40 years even if they are not obese. So, although the ADA would screen everyone ages 45 and older, the Task Force would screen everyone ages 40 and older, except for non-Hispanic whites who are not overweight or obese, and who have no other risk factors. TABLE 11,3 details the Task Force and the ADA screening criteria and how they differ.
The Task Force and the ADA also agree on the 3 tests acceptable for screening and the test values that define normal glucose, IGT, IFG, and diabetes (TABLE 2).1,3 The tests are a randomly measured glycated hemoglobin level, a fasting plasma glucose level, and an oral glucose tolerance test performed in the morning after an overnight fast, with glucose measured 2 hours after a 75-g oral glucose load. If a screening result is abnormal, confirmation should be sought by repeating the same test. And both organizations suggest that, following a normal test result, the optimal interval for retesting is 3 years.
Intervening to delay progression to diabetes
For anyone with a confirmed abnormal blood glucose level, the Task Force advises referral for intensive behavioral interventions—ie, multiple counseling sessions over an extended period on a healthy diet and optimal physical activity. These types of interventions can reduce blood glucose levels and lower the risk of progression to diabetes, and can help with lowering weight, blood pressure, and lipid levels. The evidence report that preceded the recommendation pooled the results from 10 studies on lifestyle modification.4 The length of follow-up in these studies ranged from 3 to 23 years, and the number needed to treat to prevent one case of progression to diabetes ranged from about 5 to 20.4
Medications such as metformin, thiazolidinediones, and alpha-glucosidase inhibitors can also reduce blood glucose levels and slow progression to diabetes. However, the Task Force says there is insufficient evidence that pharmacologic interventions have the same multifactorial benefits—weight loss or reductions in glucose levels, blood pressure, and lipid levels—as behavioral interventions.1
As for the other modifiable risk factors for cardiovascular disease—obesity, lack of physical activity, high lipid levels, high blood pressure, and smoking—the Task Force has developed recommendations on screening for and treating each of them,5 which supplement the recommendations discussed in this article.
1. U.S. Preventive Services Task Force. Abnormal blood glucose and type 2 diabetes mellitus: screening. Available at: http://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/screening-for-abnormal-blood-glucose-and-type-2-diabetes. Accessed May 20, 2016.
2. Casagrande SS, Cowie CC, Fradkin JE. Utility of the US Preventive Services Task Force criteria for diabetes screening. Am J Prev Med. 2013;45:167-174.
3. American Diabetes Association. Standards of medical care in diabetes - 2016. Diabetes Care. 2016;39(Suppl 1):S1–S112.
4. Selph S, Dana T, Bougatsos C, et al. A systematic review to update the 2008 U.S. Preventive Services Task Force recommendation [Agency for Healthcare Research and Quality]. 2015. Available at: http://www.ncbi.nlm.nih.gov/books/NBK293871/. Accessed May 20, 2016.
5. U.S. Preventive Services Task Force. Healthful diet and physical activity for cardiovascular disease prevention in adults with cardiovascular risk factors: behavioral counseling. Available at: http://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/healthy-diet-and-physical-activity-counseling-adults-with-high-risk-of-cvd. Accessed May 20,
2016.
1. U.S. Preventive Services Task Force. Abnormal blood glucose and type 2 diabetes mellitus: screening. Available at: http://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/screening-for-abnormal-blood-glucose-and-type-2-diabetes. Accessed May 20, 2016.
2. Casagrande SS, Cowie CC, Fradkin JE. Utility of the US Preventive Services Task Force criteria for diabetes screening. Am J Prev Med. 2013;45:167-174.
3. American Diabetes Association. Standards of medical care in diabetes - 2016. Diabetes Care. 2016;39(Suppl 1):S1–S112.
4. Selph S, Dana T, Bougatsos C, et al. A systematic review to update the 2008 U.S. Preventive Services Task Force recommendation [Agency for Healthcare Research and Quality]. 2015. Available at: http://www.ncbi.nlm.nih.gov/books/NBK293871/. Accessed May 20, 2016.
5. U.S. Preventive Services Task Force. Healthful diet and physical activity for cardiovascular disease prevention in adults with cardiovascular risk factors: behavioral counseling. Available at: http://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/healthy-diet-and-physical-activity-counseling-adults-with-high-risk-of-cvd. Accessed May 20,
2016.

How to talk to patients and families about brain stimulation
Brain stimulation often is used for treatment-resistant depression when medications and psychotherapy are not enough to elicit a meaningful response. It is both old and new again: electroconvulsive therapy (ECT) has been used for decades, while emerging technologies, such as transcranial magnetic stimulation (TMS), are gaining acceptance.
Patients and families often arrive at the office with fears and assumptions about these types of treatments, which should be discussed openly. There are also differences between these treatment approaches that can be discussed (Table).

Electroconvulsive therapy
Although ECT has been shown to be the most efficacious treatment for treatment-resistant depression,1 the most common response from patients and families that I hear when discussing ECT use is, “Do you really still do that?” Many patients and family members associate this treatment with mass media portrayals over the past several decades, such as the motion picture One Flew Over the Cuckoo’s Nest, which paired inhumane and unnecessary use of ECT with a frontal lobotomy, thereby associating this treatment with something inherently unethical.
My approach to discussing ECT with patients and families is to convey these main points:
- Consensual. In most cases, ECT is performed with the explicit informed consent of the patient, and is not done against the patient’s will.
- Effective. ECT has a remission rate of 75% after the first 2 weeks of use in patients suffering from acute depressive illnesses.2
- Safe. ECT protocols have evolved to maximize efficacy while minimizing adverse effects. Advances in anesthesia use with paralytic agents and anti-inflammatory medications reduce convulsions and subsequent musculoskeletal discomfort.
In addition, I note that:
- Ultra-brief stimulation parameters often are used to minimize cognitive side effects.
- ECT is associated with some psychosocial limitations, including being unable to drive during acute treatment and requiring supervision for several hours after sessions.
Transcranial magnetic stimulation
The field of non-invasive brain stimulation—in particular, TMS—faces a different set of complex issues to navigate. Because TMS is relatively new (approved by the FDA in 2008 for treatment-resistant depression),3 patients and families might believe that TMS may be more effective than ECT, which has not been demonstrated.4 It is important to communicate that:
- Although TMS is a FDA-approved treatment that has helped many patients with treatment-resistant depression, ECT remains the clinical treatment of choice for severe depression.
- Among antidepressant non-responders who had stopped all other antidepressant treatment, 44% of those who received deep TMS responded to treatment after 16 weeks, compared with 26% who received sham treatment.5
- Most patients usually require TMS for 4 to 6 weeks, 5 days a week, before beginning a taper phase.
- TMS has few side effects (headache being the most common); serious adverse effects (seizures, mania) have been reported but are rare.3
- Patients usually are able to continue their daily life and other outpatient treatments without the restrictions often placed on patients receiving ECT.
- If the patient responded to ECT in the past but could not tolerate adverse cognitive effects, TMS might be a better choice than other treatments.
1. Pagnin D, de Queiroz V, Pini S, et al. Efficacy of ECT in depression: a meta-analytic review. J ECT. 2004;20(1):13-20.
2. Husain MM, Rush AJ, Fink M, et al. Speed of response and remission in major depressive disorder with acute electroconvulsive therapy (ECT): a Consortium for Research in ECT (CORE) report. J Clin Psychiatry. 2004;65(4):485-491.
3. Stern AP, Cohen D. Repetitive transcranial magnetic stimulation for treatment-resistant depression. Neuropsychiatry. 2013;3(1):107-115.
4. Micallef-Trigona B. Comparing the effects of repetitive transcranial magnetic stimulation and electroconvulsive therapy in the treatment of depression: a systematic review and meta-analysis. Depress Res Treat. 2014;2014:135049. doi: 10.1155/2014/135049.
5. Levkovitz Y, Isserles M, Padberg F. Efficacy and safety of deep transcranial magnetic stimulation for major depression: a prospective, multi-center, randomized, controlled trial. World Psychiatry. 2015;14(1):64-73.
Brain stimulation often is used for treatment-resistant depression when medications and psychotherapy are not enough to elicit a meaningful response. It is both old and new again: electroconvulsive therapy (ECT) has been used for decades, while emerging technologies, such as transcranial magnetic stimulation (TMS), are gaining acceptance.
Patients and families often arrive at the office with fears and assumptions about these types of treatments, which should be discussed openly. There are also differences between these treatment approaches that can be discussed (Table).
Electroconvulsive therapy
Although ECT has been shown to be the most efficacious treatment for treatment-resistant depression,1 the most common response from patients and families that I hear when discussing ECT use is, “Do you really still do that?” Many patients and family members associate this treatment with mass media portrayals over the past several decades, such as the motion picture One Flew Over the Cuckoo’s Nest, which paired inhumane and unnecessary use of ECT with a frontal lobotomy, thereby associating this treatment with something inherently unethical.
My approach to discussing ECT with patients and families is to convey these main points:
- Consensual. In most cases, ECT is performed with the explicit informed consent of the patient, and is not done against the patient’s will.
- Effective. ECT has a remission rate of 75% after the first 2 weeks of use in patients suffering from acute depressive illnesses.2
- Safe. ECT protocols have evolved to maximize efficacy while minimizing adverse effects. Advances in anesthesia use with paralytic agents and anti-inflammatory medications reduce convulsions and subsequent musculoskeletal discomfort.
In addition, I note that:
- Ultra-brief stimulation parameters often are used to minimize cognitive side effects.
- ECT is associated with some psychosocial limitations, including being unable to drive during acute treatment and requiring supervision for several hours after sessions.
Transcranial magnetic stimulation
The field of non-invasive brain stimulation—in particular, TMS—faces a different set of complex issues to navigate. Because TMS is relatively new (approved by the FDA in 2008 for treatment-resistant depression),3 patients and families might believe that TMS may be more effective than ECT, which has not been demonstrated.4 It is important to communicate that:
- Although TMS is a FDA-approved treatment that has helped many patients with treatment-resistant depression, ECT remains the clinical treatment of choice for severe depression.
- Among antidepressant non-responders who had stopped all other antidepressant treatment, 44% of those who received deep TMS responded to treatment after 16 weeks, compared with 26% who received sham treatment.5
- Most patients usually require TMS for 4 to 6 weeks, 5 days a week, before beginning a taper phase.
- TMS has few side effects (headache being the most common); serious adverse effects (seizures, mania) have been reported but are rare.3
- Patients usually are able to continue their daily life and other outpatient treatments without the restrictions often placed on patients receiving ECT.
- If the patient responded to ECT in the past but could not tolerate adverse cognitive effects, TMS might be a better choice than other treatments.
Brain stimulation often is used for treatment-resistant depression when medications and psychotherapy are not enough to elicit a meaningful response. It is both old and new again: electroconvulsive therapy (ECT) has been used for decades, while emerging technologies, such as transcranial magnetic stimulation (TMS), are gaining acceptance.
Patients and families often arrive at the office with fears and assumptions about these types of treatments, which should be discussed openly. There are also differences between these treatment approaches that can be discussed (Table).
Electroconvulsive therapy
Although ECT has been shown to be the most efficacious treatment for treatment-resistant depression,1 the most common response from patients and families that I hear when discussing ECT use is, “Do you really still do that?” Many patients and family members associate this treatment with mass media portrayals over the past several decades, such as the motion picture One Flew Over the Cuckoo’s Nest, which paired inhumane and unnecessary use of ECT with a frontal lobotomy, thereby associating this treatment with something inherently unethical.
My approach to discussing ECT with patients and families is to convey these main points:
- Consensual. In most cases, ECT is performed with the explicit informed consent of the patient, and is not done against the patient’s will.
- Effective. ECT has a remission rate of 75% after the first 2 weeks of use in patients suffering from acute depressive illnesses.2
- Safe. ECT protocols have evolved to maximize efficacy while minimizing adverse effects. Advances in anesthesia use with paralytic agents and anti-inflammatory medications reduce convulsions and subsequent musculoskeletal discomfort.
In addition, I note that:
- Ultra-brief stimulation parameters often are used to minimize cognitive side effects.
- ECT is associated with some psychosocial limitations, including being unable to drive during acute treatment and requiring supervision for several hours after sessions.
Transcranial magnetic stimulation
The field of non-invasive brain stimulation—in particular, TMS—faces a different set of complex issues to navigate. Because TMS is relatively new (approved by the FDA in 2008 for treatment-resistant depression),3 patients and families might believe that TMS may be more effective than ECT, which has not been demonstrated.4 It is important to communicate that:
- Although TMS is a FDA-approved treatment that has helped many patients with treatment-resistant depression, ECT remains the clinical treatment of choice for severe depression.
- Among antidepressant non-responders who had stopped all other antidepressant treatment, 44% of those who received deep TMS responded to treatment after 16 weeks, compared with 26% who received sham treatment.5
- Most patients usually require TMS for 4 to 6 weeks, 5 days a week, before beginning a taper phase.
- TMS has few side effects (headache being the most common); serious adverse effects (seizures, mania) have been reported but are rare.3
- Patients usually are able to continue their daily life and other outpatient treatments without the restrictions often placed on patients receiving ECT.
- If the patient responded to ECT in the past but could not tolerate adverse cognitive effects, TMS might be a better choice than other treatments.
1. Pagnin D, de Queiroz V, Pini S, et al. Efficacy of ECT in depression: a meta-analytic review. J ECT. 2004;20(1):13-20.
2. Husain MM, Rush AJ, Fink M, et al. Speed of response and remission in major depressive disorder with acute electroconvulsive therapy (ECT): a Consortium for Research in ECT (CORE) report. J Clin Psychiatry. 2004;65(4):485-491.
3. Stern AP, Cohen D. Repetitive transcranial magnetic stimulation for treatment-resistant depression. Neuropsychiatry. 2013;3(1):107-115.
4. Micallef-Trigona B. Comparing the effects of repetitive transcranial magnetic stimulation and electroconvulsive therapy in the treatment of depression: a systematic review and meta-analysis. Depress Res Treat. 2014;2014:135049. doi: 10.1155/2014/135049.
5. Levkovitz Y, Isserles M, Padberg F. Efficacy and safety of deep transcranial magnetic stimulation for major depression: a prospective, multi-center, randomized, controlled trial. World Psychiatry. 2015;14(1):64-73.
1. Pagnin D, de Queiroz V, Pini S, et al. Efficacy of ECT in depression: a meta-analytic review. J ECT. 2004;20(1):13-20.
2. Husain MM, Rush AJ, Fink M, et al. Speed of response and remission in major depressive disorder with acute electroconvulsive therapy (ECT): a Consortium for Research in ECT (CORE) report. J Clin Psychiatry. 2004;65(4):485-491.
3. Stern AP, Cohen D. Repetitive transcranial magnetic stimulation for treatment-resistant depression. Neuropsychiatry. 2013;3(1):107-115.
4. Micallef-Trigona B. Comparing the effects of repetitive transcranial magnetic stimulation and electroconvulsive therapy in the treatment of depression: a systematic review and meta-analysis. Depress Res Treat. 2014;2014:135049. doi: 10.1155/2014/135049.
5. Levkovitz Y, Isserles M, Padberg F. Efficacy and safety of deep transcranial magnetic stimulation for major depression: a prospective, multi-center, randomized, controlled trial. World Psychiatry. 2015;14(1):64-73.
Rediscovering clozapine: After a turbulent history, current guidance on initiating and monitoring
Although clozapine is the medication with the clearest benefits in treatment-resistant schizophrenia, many eligible patients never receive it. In the United States, 20% to 30% of patients with schizophrenia can be classified as treatment resistant, but clozapine accounts for <5% of antipsychotics prescribed.1,2 Clinicians worldwide tend to under-prescribe clozapine3—a reluctance one author coined as “clozaphobia.”4
Admittedly, clozapine has had a turbulent history—both lauded as a near-miracle drug and condemned as a deadly agent. The FDA has overhauled its prescribing and monitoring guidelines, however, offering psychiatrists a perfect opportunity to reacquaint themselves with this potentially life-changing intervention.
We begin this article with clozapine’s story, then spotlight new terrain the FDA created in 2015 when the agency introduced the Clozapine Risk Evaluation and Mitigation Strategy (REMS). Our goal in the 3 articles of this series is to deepen your appreciation for this tricyclic antipsychotic and provide practical clinical guidance for using it safely and effectively.
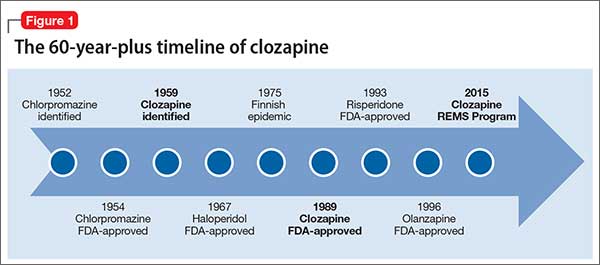
Setbacks, but the drug has an enduring presenceThe 1950s was an exciting era of exploration for new psychotropic medications. While searching for tricyclic antidepressants, Wander Laboratories discovered neuroleptic tricyclics, with clozapine identified in 1959 (Figure 1). Haloperidol’s development and release in the 1960s reinforced the prevailing dogma of the time that effective neuroleptics correlated with extrapyramidal symptoms, thus limiting interest in the newly discovered, but pharmacologically unique, clozapine. Throughout the 1960s, most research on clozapine was published in German, with less of an international presence.5
Agranulocytosis deaths. Clozapine earned its scarlet letter in 1975, when 8 patients in Finland died of agranulocytosis.6 Sandoz, its manufacturer, withdrew clozapine from the market and halted all clinical trials. The Finnish epidemic triggered detailed investigations into blood dyscrasias and early identification of agranulocytosis associated with clozapine and other antipsychotics.7
Clozapine endured only because of its unique efficacy. When psychiatrists witnessed relapses in patients who had to discontinue clozapine, some countries allowed its use with strict monitoring.5 The FDA kept clozapine minimally available in the United States by allowing so-called “compassionate need programs” to continue.7
New data, FDA approval. Two studies in 1987 and 1988 that compared clozapine with chlorpromazine for treatment-refractory schizophrenia demonstrated clozapine’s superior effect on both negative and positive symptoms.8,9 The FDA approved clozapine for refractory schizophrenia in 1989, and clozapine became clinically available in 1990.
Initially, the high annual cost of clozapine’s required “bundle” ($8,900 per patient for medication and monitoring) led to political outcry. As patients and their family struggled to afford the newly released medication, multiple states filed antitrust lawsuits. A federal court found both the manufacturer and individual states at fault and required expanded access to clozapine and its necessary monitoring. National clozapine registries were formed, and bundling was eliminated.7
The clozapine REMS programSix clozapine registries operated independently, each managed by a different manufacturer,10 until the FDA introduced REMS in September 2015. The REMS program created a centralized registry to monitor all U.S. patients treated with clozapine and made important changes to prescribing and monitoring guidelines.11,12 It also incorporated the National Non-Rechallenge Master File (NNRMF).
Initially, the REMS program was scheduled for rollout October 12, 2015, the closing date of the 6 registries. Since November 2015, pharmacies have been required to register with the program to dispense clozapine. A similar registration deadline for clozapine prescribers was extended indefinitely, however, because of technical problems. Once the deadline is finalized, all clozapine prescribers must complete 3 steps to be certified in the REMS program (Table 1).11

New requirements. Certified clozapine prescribers will have new responsibilities: enrolling patients and submitting lab results. They can designate someone else to perform these tasks on their behalf, but designees must enroll in the REMS program and the prescriber must confirm the designee. Pharmacists can no longer enroll patients for clozapine therapy unless they are confirmed as a prescriber designee. For outpatients, the absolute neutrophil count (ANC) must be reported before the pharmacy can dispense clozapine. For inpatients, the ANC must be reported within 7 days of the patient’s most recent blood draw.
Once the system is fully operational, Social Security numbers will no longer be used as patient identification for dispensing clozapine. Instead, outpatient pharmacies will obtain a predispense authorization, or PDA, from the REMS program. A person initiated on clozapine as an inpatient must be re-enrolled after discharge by their outpatient prescriber.
The REMS program includes information about clozapine patients who were maintained through the 6 registries, and these patients have been allowed to continue clozapine treatment. Data pertaining to patients last prescribed clozapine before October 1, 2012, did not transfer into the new system unless their name was on the NNRMF.
CASE
Is Mr. A a candidate for clozapine?Age 28, with schizophrenia, Mr. A is highly disorganized and psychotic when brought to the emergency room by police for inappropriate behavior. His family arrives and reports that similar events have occurred several times over the past few years. Mr. A’s outpatient psychiatrist has prescribed 3 different antipsychotic medications at adequate dosages, including 1 long-acting injectable, but Mr. A has remained consistently symptomatic.
Although disorganized and psychotic, Mr. A does not meet criteria for long-term involuntary hospitalization. His family wants to take him home, and the treatment team discusses clozapine as an antipsychotic option. Mr. A and his family agree to a trial of clozapine during voluntary hospitalization, but they would like him home within a week to attend his sister’s birthday party.
The treatment team decides to initiate clozapine and monitor his response in a controlled setting for a few days before transitioning him to outpatient care.
Initiating clozapine therapyThe case of Mr. A exemplifies a situation in which initiating clozapine is a reasonable clinical consideration. As the first step, we recommend checking baseline lab values and vital signs (Table 2), keeping in mind that the REMS program requires a baseline ANC within 7 days of initiating clozapine. When working with a highly disorganized or agitated patient, balance benefits of testing against the risk of harm to staff and patient.

REMS guidelines recommend a baseline ANC ≥1,500/µL for a new patient starting clozapine, except when benign ethnic neutropenia (BEN) has been confirmed. (Initiation guidelines for BEN are discussed later in this article.)
Dosing alternatives. We recommend following the manufacturer’s dosing guidelines when initiating clozapine (Figure 2).13,14 Three oral forms are available: tablet, disintegrating tablet, and suspension. All can be titrated using the schedule suggested with tablets. The disintegrating tablets or suspension might be beneficial for a patient with either:
- a history of “cheeking” or otherwise disposing of tablets
- a medical condition that affects swallowing or absorption.
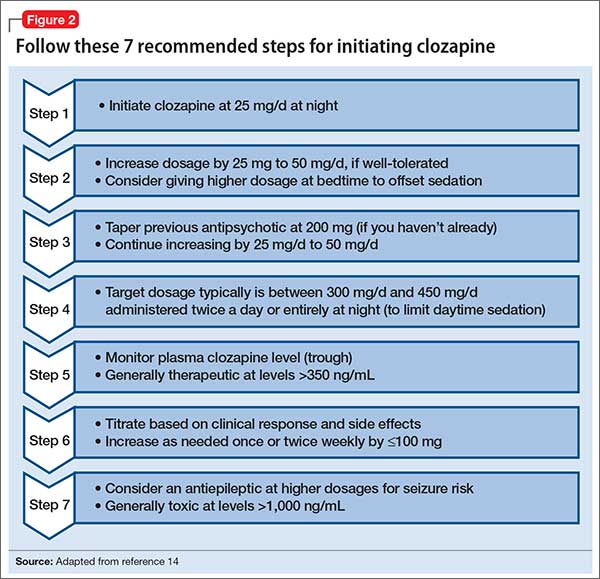
The disintegrating tablet is available in 12.5-mg, 25-mg, 100-mg, 150-mg, and 200-mg doses. It dissolves without requiring additional liquids. Each mL of the suspension contains 50 mg of clozapine.
Rapid titration? One group, working in Romania, examined the safety and efficacy of rapid titration of clozapine in 111 inpatients with schizophrenia.15 In the absence of additional studies, we do not recommend routine rapid titration of clozapine.
Monitoring: Greater flexibilityUnder the REMS program, laboratory monitoring of clozapine treatment must continue indefinitely. If not, pharmacies cannot dispense clozapine. Fortunately, the ANC is the only lab value tracked by the registry, and the frequency of required blood draws decreases over time (Figure 3).

Other guideline changes provide clinicians with greater flexibility to make patient-specific treatment decisions; for example, the allowable ANC to continue clozapine therapy has decreased. Usually, clozapine therapy should be interrupted for an ANC <1,000/µL if the prescriber suspects clozapine-induced neutropenia. Even when the ANC drops below 1,000/µL, however, prescribers can now continue clozapine treatment if they consider the benefits to outweigh risks for a given patient.
Separate guidelines now exist for patients with BEN, most commonly observed in persons of certain ethnic groups. BEN typically is diagnosed based on repeated ANC values <1,500/µL over several months. Patients with BEN do not have an increased risk of oral or systemic infections, as occur with other congenital neutropenias.16 In patients with BEN, clozapine therapy:
- can be initiated only after at least 2 baseline ANC measurements ≥1,000/µL
- should be interrupted for an ANC <500/µL if the prescriber suspects clozapine-induced neutropenia.
Substantial drops in ANC no longer require action (repeat lab draws) unless the drop causes neutropenia. Prescribers will receive an automated notification any time a patient experiences neutropenia that is considered mild (ANC 1,000 to 1,499/µL), moderate (ANC 500 to 999/µL), or severe (ANC <500/µL).
The NNRMF list is no longer definitive. All patients are now eligible for rechallenge, assuming they meet the new clozapine initiation criteria.
Next, when rediscovering clozapine: Adverse effectsDespite an intimidating list of side effects and interactions, clozapine is associated with a significant reduction in patients’ risk of overall mortality. In Part 2 of this series in the August 2016 issue, we discuss early identification of clozapine’s adverse effects and provide guidance for management.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stroup TS, Gerhard T, Crystal S, et al. Geographic and clinical variation in clozapine use in the United States. Psychiatr Serv. 2014;65(2):186-192.
2. Olfson M, Gerhard T, Crystal S, et al. Clozapine for schizophrenia: state variation in evidence-based practice. Psychiatr Serv. 2016;67(2):152.
3. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
4. Cetin M. Clozaphobia: fear of prescribers of clozapine for treatment of schizophrenia. Klinik Psikofarmakol Bulteni. 2014;24(4):295-301.
5. Hippius H. A historical perspective of clozapine. J Clin Psychiatry. 1999;60(suppl 12):22-23.
6. Amsler HA, Teerenhovi L, Barth E, et al. Agranulocytosis in patients treated with clozapine. A study of the Finnish epidemic. Acta Psychiatr Scand. 1977;56(4):241-248.
7. Crilly J. The history of clozapine and its emergence in the U.S. market: a review and analysis. Hist Psychiatry. 2007;18(1):39-60.
8. Claghorn J, Honigfeld G, Abuzzahab FS, et al. The risks and benefits of clozapine versus chlorpromazine. J Clin Psychopharmacol. 1987;7(6):377-384.
9. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
10. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA modified monitoring for neutropenia associated with schizophrenia medicine clozapine; approves new shared REMS program for all clozapine medicines. http://www.fda.gov/Drugs/DrugSafety/ucm461853.htm. Published September 15, 2015. Accessed November 23, 2015.
11. Clozapine REMS Program. What’s new with clozapine: an overview. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/WhatsNEWwithClozapine_An%20Overview.pdf. Published September 2015. Accessed November 23, 2015.
12. Clozapine REMS Program. Clozapine and the risk of neutropenia: a guide for healthcare providers. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/resources/Clozapine_REMS_HCP_Guide.pdf. Published September 2015. Accessed November 23, 2015.
13. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 16, 2016.
14. Newman WJ. Psychopharmacologic management of aggression. Psychiatr Clin North Am. 2012;35(4):957-972.
15. Ifteni P, Nielsen J, Burtea V, et al. Effectiveness and safety of rapid clozapine titration in schizophrenia. Acta Psychiatr Scand. 2014;130(1):25-29.
16. Hsieh MM, Tisdale JF, Rodgers GP, et al. Neutrophil count in African Americans: lowering the target cutoff to initiate or resume chemotherapy? J Clin Oncol. 2010;28(10):1633-1637.
Although clozapine is the medication with the clearest benefits in treatment-resistant schizophrenia, many eligible patients never receive it. In the United States, 20% to 30% of patients with schizophrenia can be classified as treatment resistant, but clozapine accounts for <5% of antipsychotics prescribed.1,2 Clinicians worldwide tend to under-prescribe clozapine3—a reluctance one author coined as “clozaphobia.”4
Admittedly, clozapine has had a turbulent history—both lauded as a near-miracle drug and condemned as a deadly agent. The FDA has overhauled its prescribing and monitoring guidelines, however, offering psychiatrists a perfect opportunity to reacquaint themselves with this potentially life-changing intervention.
We begin this article with clozapine’s story, then spotlight new terrain the FDA created in 2015 when the agency introduced the Clozapine Risk Evaluation and Mitigation Strategy (REMS). Our goal in the 3 articles of this series is to deepen your appreciation for this tricyclic antipsychotic and provide practical clinical guidance for using it safely and effectively.
Setbacks, but the drug has an enduring presenceThe 1950s was an exciting era of exploration for new psychotropic medications. While searching for tricyclic antidepressants, Wander Laboratories discovered neuroleptic tricyclics, with clozapine identified in 1959 (Figure 1). Haloperidol’s development and release in the 1960s reinforced the prevailing dogma of the time that effective neuroleptics correlated with extrapyramidal symptoms, thus limiting interest in the newly discovered, but pharmacologically unique, clozapine. Throughout the 1960s, most research on clozapine was published in German, with less of an international presence.5
Agranulocytosis deaths. Clozapine earned its scarlet letter in 1975, when 8 patients in Finland died of agranulocytosis.6 Sandoz, its manufacturer, withdrew clozapine from the market and halted all clinical trials. The Finnish epidemic triggered detailed investigations into blood dyscrasias and early identification of agranulocytosis associated with clozapine and other antipsychotics.7
Clozapine endured only because of its unique efficacy. When psychiatrists witnessed relapses in patients who had to discontinue clozapine, some countries allowed its use with strict monitoring.5 The FDA kept clozapine minimally available in the United States by allowing so-called “compassionate need programs” to continue.7
New data, FDA approval. Two studies in 1987 and 1988 that compared clozapine with chlorpromazine for treatment-refractory schizophrenia demonstrated clozapine’s superior effect on both negative and positive symptoms.8,9 The FDA approved clozapine for refractory schizophrenia in 1989, and clozapine became clinically available in 1990.
Initially, the high annual cost of clozapine’s required “bundle” ($8,900 per patient for medication and monitoring) led to political outcry. As patients and their family struggled to afford the newly released medication, multiple states filed antitrust lawsuits. A federal court found both the manufacturer and individual states at fault and required expanded access to clozapine and its necessary monitoring. National clozapine registries were formed, and bundling was eliminated.7
The clozapine REMS programSix clozapine registries operated independently, each managed by a different manufacturer,10 until the FDA introduced REMS in September 2015. The REMS program created a centralized registry to monitor all U.S. patients treated with clozapine and made important changes to prescribing and monitoring guidelines.11,12 It also incorporated the National Non-Rechallenge Master File (NNRMF).
Initially, the REMS program was scheduled for rollout October 12, 2015, the closing date of the 6 registries. Since November 2015, pharmacies have been required to register with the program to dispense clozapine. A similar registration deadline for clozapine prescribers was extended indefinitely, however, because of technical problems. Once the deadline is finalized, all clozapine prescribers must complete 3 steps to be certified in the REMS program (Table 1).11
New requirements. Certified clozapine prescribers will have new responsibilities: enrolling patients and submitting lab results. They can designate someone else to perform these tasks on their behalf, but designees must enroll in the REMS program and the prescriber must confirm the designee. Pharmacists can no longer enroll patients for clozapine therapy unless they are confirmed as a prescriber designee. For outpatients, the absolute neutrophil count (ANC) must be reported before the pharmacy can dispense clozapine. For inpatients, the ANC must be reported within 7 days of the patient’s most recent blood draw.
Once the system is fully operational, Social Security numbers will no longer be used as patient identification for dispensing clozapine. Instead, outpatient pharmacies will obtain a predispense authorization, or PDA, from the REMS program. A person initiated on clozapine as an inpatient must be re-enrolled after discharge by their outpatient prescriber.
The REMS program includes information about clozapine patients who were maintained through the 6 registries, and these patients have been allowed to continue clozapine treatment. Data pertaining to patients last prescribed clozapine before October 1, 2012, did not transfer into the new system unless their name was on the NNRMF.
CASE
Is Mr. A a candidate for clozapine?Age 28, with schizophrenia, Mr. A is highly disorganized and psychotic when brought to the emergency room by police for inappropriate behavior. His family arrives and reports that similar events have occurred several times over the past few years. Mr. A’s outpatient psychiatrist has prescribed 3 different antipsychotic medications at adequate dosages, including 1 long-acting injectable, but Mr. A has remained consistently symptomatic.
Although disorganized and psychotic, Mr. A does not meet criteria for long-term involuntary hospitalization. His family wants to take him home, and the treatment team discusses clozapine as an antipsychotic option. Mr. A and his family agree to a trial of clozapine during voluntary hospitalization, but they would like him home within a week to attend his sister’s birthday party.
The treatment team decides to initiate clozapine and monitor his response in a controlled setting for a few days before transitioning him to outpatient care.
Initiating clozapine therapyThe case of Mr. A exemplifies a situation in which initiating clozapine is a reasonable clinical consideration. As the first step, we recommend checking baseline lab values and vital signs (Table 2), keeping in mind that the REMS program requires a baseline ANC within 7 days of initiating clozapine. When working with a highly disorganized or agitated patient, balance benefits of testing against the risk of harm to staff and patient.
REMS guidelines recommend a baseline ANC ≥1,500/µL for a new patient starting clozapine, except when benign ethnic neutropenia (BEN) has been confirmed. (Initiation guidelines for BEN are discussed later in this article.)
Dosing alternatives. We recommend following the manufacturer’s dosing guidelines when initiating clozapine (Figure 2).13,14 Three oral forms are available: tablet, disintegrating tablet, and suspension. All can be titrated using the schedule suggested with tablets. The disintegrating tablets or suspension might be beneficial for a patient with either:
- a history of “cheeking” or otherwise disposing of tablets
- a medical condition that affects swallowing or absorption.
The disintegrating tablet is available in 12.5-mg, 25-mg, 100-mg, 150-mg, and 200-mg doses. It dissolves without requiring additional liquids. Each mL of the suspension contains 50 mg of clozapine.
Rapid titration? One group, working in Romania, examined the safety and efficacy of rapid titration of clozapine in 111 inpatients with schizophrenia.15 In the absence of additional studies, we do not recommend routine rapid titration of clozapine.
Monitoring: Greater flexibilityUnder the REMS program, laboratory monitoring of clozapine treatment must continue indefinitely. If not, pharmacies cannot dispense clozapine. Fortunately, the ANC is the only lab value tracked by the registry, and the frequency of required blood draws decreases over time (Figure 3).
Other guideline changes provide clinicians with greater flexibility to make patient-specific treatment decisions; for example, the allowable ANC to continue clozapine therapy has decreased. Usually, clozapine therapy should be interrupted for an ANC <1,000/µL if the prescriber suspects clozapine-induced neutropenia. Even when the ANC drops below 1,000/µL, however, prescribers can now continue clozapine treatment if they consider the benefits to outweigh risks for a given patient.
Separate guidelines now exist for patients with BEN, most commonly observed in persons of certain ethnic groups. BEN typically is diagnosed based on repeated ANC values <1,500/µL over several months. Patients with BEN do not have an increased risk of oral or systemic infections, as occur with other congenital neutropenias.16 In patients with BEN, clozapine therapy:
- can be initiated only after at least 2 baseline ANC measurements ≥1,000/µL
- should be interrupted for an ANC <500/µL if the prescriber suspects clozapine-induced neutropenia.
Substantial drops in ANC no longer require action (repeat lab draws) unless the drop causes neutropenia. Prescribers will receive an automated notification any time a patient experiences neutropenia that is considered mild (ANC 1,000 to 1,499/µL), moderate (ANC 500 to 999/µL), or severe (ANC <500/µL).
The NNRMF list is no longer definitive. All patients are now eligible for rechallenge, assuming they meet the new clozapine initiation criteria.
Next, when rediscovering clozapine: Adverse effectsDespite an intimidating list of side effects and interactions, clozapine is associated with a significant reduction in patients’ risk of overall mortality. In Part 2 of this series in the August 2016 issue, we discuss early identification of clozapine’s adverse effects and provide guidance for management.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Although clozapine is the medication with the clearest benefits in treatment-resistant schizophrenia, many eligible patients never receive it. In the United States, 20% to 30% of patients with schizophrenia can be classified as treatment resistant, but clozapine accounts for <5% of antipsychotics prescribed.1,2 Clinicians worldwide tend to under-prescribe clozapine3—a reluctance one author coined as “clozaphobia.”4
Admittedly, clozapine has had a turbulent history—both lauded as a near-miracle drug and condemned as a deadly agent. The FDA has overhauled its prescribing and monitoring guidelines, however, offering psychiatrists a perfect opportunity to reacquaint themselves with this potentially life-changing intervention.
We begin this article with clozapine’s story, then spotlight new terrain the FDA created in 2015 when the agency introduced the Clozapine Risk Evaluation and Mitigation Strategy (REMS). Our goal in the 3 articles of this series is to deepen your appreciation for this tricyclic antipsychotic and provide practical clinical guidance for using it safely and effectively.
Setbacks, but the drug has an enduring presenceThe 1950s was an exciting era of exploration for new psychotropic medications. While searching for tricyclic antidepressants, Wander Laboratories discovered neuroleptic tricyclics, with clozapine identified in 1959 (Figure 1). Haloperidol’s development and release in the 1960s reinforced the prevailing dogma of the time that effective neuroleptics correlated with extrapyramidal symptoms, thus limiting interest in the newly discovered, but pharmacologically unique, clozapine. Throughout the 1960s, most research on clozapine was published in German, with less of an international presence.5
Agranulocytosis deaths. Clozapine earned its scarlet letter in 1975, when 8 patients in Finland died of agranulocytosis.6 Sandoz, its manufacturer, withdrew clozapine from the market and halted all clinical trials. The Finnish epidemic triggered detailed investigations into blood dyscrasias and early identification of agranulocytosis associated with clozapine and other antipsychotics.7
Clozapine endured only because of its unique efficacy. When psychiatrists witnessed relapses in patients who had to discontinue clozapine, some countries allowed its use with strict monitoring.5 The FDA kept clozapine minimally available in the United States by allowing so-called “compassionate need programs” to continue.7
New data, FDA approval. Two studies in 1987 and 1988 that compared clozapine with chlorpromazine for treatment-refractory schizophrenia demonstrated clozapine’s superior effect on both negative and positive symptoms.8,9 The FDA approved clozapine for refractory schizophrenia in 1989, and clozapine became clinically available in 1990.
Initially, the high annual cost of clozapine’s required “bundle” ($8,900 per patient for medication and monitoring) led to political outcry. As patients and their family struggled to afford the newly released medication, multiple states filed antitrust lawsuits. A federal court found both the manufacturer and individual states at fault and required expanded access to clozapine and its necessary monitoring. National clozapine registries were formed, and bundling was eliminated.7
The clozapine REMS programSix clozapine registries operated independently, each managed by a different manufacturer,10 until the FDA introduced REMS in September 2015. The REMS program created a centralized registry to monitor all U.S. patients treated with clozapine and made important changes to prescribing and monitoring guidelines.11,12 It also incorporated the National Non-Rechallenge Master File (NNRMF).
Initially, the REMS program was scheduled for rollout October 12, 2015, the closing date of the 6 registries. Since November 2015, pharmacies have been required to register with the program to dispense clozapine. A similar registration deadline for clozapine prescribers was extended indefinitely, however, because of technical problems. Once the deadline is finalized, all clozapine prescribers must complete 3 steps to be certified in the REMS program (Table 1).11
New requirements. Certified clozapine prescribers will have new responsibilities: enrolling patients and submitting lab results. They can designate someone else to perform these tasks on their behalf, but designees must enroll in the REMS program and the prescriber must confirm the designee. Pharmacists can no longer enroll patients for clozapine therapy unless they are confirmed as a prescriber designee. For outpatients, the absolute neutrophil count (ANC) must be reported before the pharmacy can dispense clozapine. For inpatients, the ANC must be reported within 7 days of the patient’s most recent blood draw.
Once the system is fully operational, Social Security numbers will no longer be used as patient identification for dispensing clozapine. Instead, outpatient pharmacies will obtain a predispense authorization, or PDA, from the REMS program. A person initiated on clozapine as an inpatient must be re-enrolled after discharge by their outpatient prescriber.
The REMS program includes information about clozapine patients who were maintained through the 6 registries, and these patients have been allowed to continue clozapine treatment. Data pertaining to patients last prescribed clozapine before October 1, 2012, did not transfer into the new system unless their name was on the NNRMF.
CASE
Is Mr. A a candidate for clozapine?Age 28, with schizophrenia, Mr. A is highly disorganized and psychotic when brought to the emergency room by police for inappropriate behavior. His family arrives and reports that similar events have occurred several times over the past few years. Mr. A’s outpatient psychiatrist has prescribed 3 different antipsychotic medications at adequate dosages, including 1 long-acting injectable, but Mr. A has remained consistently symptomatic.
Although disorganized and psychotic, Mr. A does not meet criteria for long-term involuntary hospitalization. His family wants to take him home, and the treatment team discusses clozapine as an antipsychotic option. Mr. A and his family agree to a trial of clozapine during voluntary hospitalization, but they would like him home within a week to attend his sister’s birthday party.
The treatment team decides to initiate clozapine and monitor his response in a controlled setting for a few days before transitioning him to outpatient care.
Initiating clozapine therapyThe case of Mr. A exemplifies a situation in which initiating clozapine is a reasonable clinical consideration. As the first step, we recommend checking baseline lab values and vital signs (Table 2), keeping in mind that the REMS program requires a baseline ANC within 7 days of initiating clozapine. When working with a highly disorganized or agitated patient, balance benefits of testing against the risk of harm to staff and patient.
REMS guidelines recommend a baseline ANC ≥1,500/µL for a new patient starting clozapine, except when benign ethnic neutropenia (BEN) has been confirmed. (Initiation guidelines for BEN are discussed later in this article.)
Dosing alternatives. We recommend following the manufacturer’s dosing guidelines when initiating clozapine (Figure 2).13,14 Three oral forms are available: tablet, disintegrating tablet, and suspension. All can be titrated using the schedule suggested with tablets. The disintegrating tablets or suspension might be beneficial for a patient with either:
- a history of “cheeking” or otherwise disposing of tablets
- a medical condition that affects swallowing or absorption.
The disintegrating tablet is available in 12.5-mg, 25-mg, 100-mg, 150-mg, and 200-mg doses. It dissolves without requiring additional liquids. Each mL of the suspension contains 50 mg of clozapine.
Rapid titration? One group, working in Romania, examined the safety and efficacy of rapid titration of clozapine in 111 inpatients with schizophrenia.15 In the absence of additional studies, we do not recommend routine rapid titration of clozapine.
Monitoring: Greater flexibilityUnder the REMS program, laboratory monitoring of clozapine treatment must continue indefinitely. If not, pharmacies cannot dispense clozapine. Fortunately, the ANC is the only lab value tracked by the registry, and the frequency of required blood draws decreases over time (Figure 3).
Other guideline changes provide clinicians with greater flexibility to make patient-specific treatment decisions; for example, the allowable ANC to continue clozapine therapy has decreased. Usually, clozapine therapy should be interrupted for an ANC <1,000/µL if the prescriber suspects clozapine-induced neutropenia. Even when the ANC drops below 1,000/µL, however, prescribers can now continue clozapine treatment if they consider the benefits to outweigh risks for a given patient.
Separate guidelines now exist for patients with BEN, most commonly observed in persons of certain ethnic groups. BEN typically is diagnosed based on repeated ANC values <1,500/µL over several months. Patients with BEN do not have an increased risk of oral or systemic infections, as occur with other congenital neutropenias.16 In patients with BEN, clozapine therapy:
- can be initiated only after at least 2 baseline ANC measurements ≥1,000/µL
- should be interrupted for an ANC <500/µL if the prescriber suspects clozapine-induced neutropenia.
Substantial drops in ANC no longer require action (repeat lab draws) unless the drop causes neutropenia. Prescribers will receive an automated notification any time a patient experiences neutropenia that is considered mild (ANC 1,000 to 1,499/µL), moderate (ANC 500 to 999/µL), or severe (ANC <500/µL).
The NNRMF list is no longer definitive. All patients are now eligible for rechallenge, assuming they meet the new clozapine initiation criteria.
Next, when rediscovering clozapine: Adverse effectsDespite an intimidating list of side effects and interactions, clozapine is associated with a significant reduction in patients’ risk of overall mortality. In Part 2 of this series in the August 2016 issue, we discuss early identification of clozapine’s adverse effects and provide guidance for management.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stroup TS, Gerhard T, Crystal S, et al. Geographic and clinical variation in clozapine use in the United States. Psychiatr Serv. 2014;65(2):186-192.
2. Olfson M, Gerhard T, Crystal S, et al. Clozapine for schizophrenia: state variation in evidence-based practice. Psychiatr Serv. 2016;67(2):152.
3. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
4. Cetin M. Clozaphobia: fear of prescribers of clozapine for treatment of schizophrenia. Klinik Psikofarmakol Bulteni. 2014;24(4):295-301.
5. Hippius H. A historical perspective of clozapine. J Clin Psychiatry. 1999;60(suppl 12):22-23.
6. Amsler HA, Teerenhovi L, Barth E, et al. Agranulocytosis in patients treated with clozapine. A study of the Finnish epidemic. Acta Psychiatr Scand. 1977;56(4):241-248.
7. Crilly J. The history of clozapine and its emergence in the U.S. market: a review and analysis. Hist Psychiatry. 2007;18(1):39-60.
8. Claghorn J, Honigfeld G, Abuzzahab FS, et al. The risks and benefits of clozapine versus chlorpromazine. J Clin Psychopharmacol. 1987;7(6):377-384.
9. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
10. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA modified monitoring for neutropenia associated with schizophrenia medicine clozapine; approves new shared REMS program for all clozapine medicines. http://www.fda.gov/Drugs/DrugSafety/ucm461853.htm. Published September 15, 2015. Accessed November 23, 2015.
11. Clozapine REMS Program. What’s new with clozapine: an overview. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/WhatsNEWwithClozapine_An%20Overview.pdf. Published September 2015. Accessed November 23, 2015.
12. Clozapine REMS Program. Clozapine and the risk of neutropenia: a guide for healthcare providers. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/resources/Clozapine_REMS_HCP_Guide.pdf. Published September 2015. Accessed November 23, 2015.
13. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 16, 2016.
14. Newman WJ. Psychopharmacologic management of aggression. Psychiatr Clin North Am. 2012;35(4):957-972.
15. Ifteni P, Nielsen J, Burtea V, et al. Effectiveness and safety of rapid clozapine titration in schizophrenia. Acta Psychiatr Scand. 2014;130(1):25-29.
16. Hsieh MM, Tisdale JF, Rodgers GP, et al. Neutrophil count in African Americans: lowering the target cutoff to initiate or resume chemotherapy? J Clin Oncol. 2010;28(10):1633-1637.
1. Stroup TS, Gerhard T, Crystal S, et al. Geographic and clinical variation in clozapine use in the United States. Psychiatr Serv. 2014;65(2):186-192.
2. Olfson M, Gerhard T, Crystal S, et al. Clozapine for schizophrenia: state variation in evidence-based practice. Psychiatr Serv. 2016;67(2):152.
3. Warnez S, Alessi-Severini S. Clozapine: a review of clinical practice guidelines and prescribing trends. BMC Psychiatry. 2014;14:102.
4. Cetin M. Clozaphobia: fear of prescribers of clozapine for treatment of schizophrenia. Klinik Psikofarmakol Bulteni. 2014;24(4):295-301.
5. Hippius H. A historical perspective of clozapine. J Clin Psychiatry. 1999;60(suppl 12):22-23.
6. Amsler HA, Teerenhovi L, Barth E, et al. Agranulocytosis in patients treated with clozapine. A study of the Finnish epidemic. Acta Psychiatr Scand. 1977;56(4):241-248.
7. Crilly J. The history of clozapine and its emergence in the U.S. market: a review and analysis. Hist Psychiatry. 2007;18(1):39-60.
8. Claghorn J, Honigfeld G, Abuzzahab FS, et al. The risks and benefits of clozapine versus chlorpromazine. J Clin Psychopharmacol. 1987;7(6):377-384.
9. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
10. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA modified monitoring for neutropenia associated with schizophrenia medicine clozapine; approves new shared REMS program for all clozapine medicines. http://www.fda.gov/Drugs/DrugSafety/ucm461853.htm. Published September 15, 2015. Accessed November 23, 2015.
11. Clozapine REMS Program. What’s new with clozapine: an overview. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/WhatsNEWwithClozapine_An%20Overview.pdf. Published September 2015. Accessed November 23, 2015.
12. Clozapine REMS Program. Clozapine and the risk of neutropenia: a guide for healthcare providers. https://www.clozapinerems.com/CpmgClozapineUI/rems/pdf/resources/Clozapine_REMS_HCP_Guide.pdf. Published September 2015. Accessed November 23, 2015.
13. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 16, 2016.
14. Newman WJ. Psychopharmacologic management of aggression. Psychiatr Clin North Am. 2012;35(4):957-972.
15. Ifteni P, Nielsen J, Burtea V, et al. Effectiveness and safety of rapid clozapine titration in schizophrenia. Acta Psychiatr Scand. 2014;130(1):25-29.
16. Hsieh MM, Tisdale JF, Rodgers GP, et al. Neutrophil count in African Americans: lowering the target cutoff to initiate or resume chemotherapy? J Clin Oncol. 2010;28(10):1633-1637.

Long-acting injectable aripiprazole lauroxil for schizophrenia
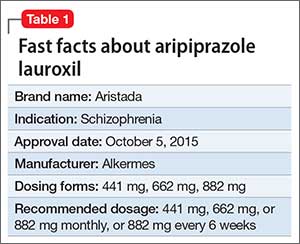
Approximately 80% of patients with schizophrenia relapse within 5 years1 despite the availability and increased use of second-generation antipsychotics. Long-acting depot formulations are a proven, effective treatment option for patients with schizophrenia. In October 2015, another long-acting injectable antipsychotic, aripiprazole lauroxil, was FDA-approved for schizophrenia.2 Aripiprazole lauroxil is administered IM every 4 to 6 weeks in the deltoid or gluteal region and is available in multiple dosages (Table 1).
Mechanism of action
Aripiprazole lauroxil is a prodrug of aripiprazole. Prodrugs are chemical compounds that exert their pharmacological effects after they undergo a biologic transformation and transform into a more active metabolite.3 The development of prodrugs is an established method used to improve physio-chemical or pharmacokinetic properties of the pharmacologically active compound.
After IM injection, aripiprazole lauroxil is most likely converted by an enzyme-mediated hydrolysis to N-hydroxymethyl aripiprazole, which is then hydrolyzed to aripiprazole. Aripiprazole’s mechanism of action is mediated through a combination of partial agonist activity at D2 and 5-HT1A receptors and antagonistic activity at 5-HT2A receptors.2,4
Dosing and administration
If your patient has never taken aripiprazole, ensure that she (he) will tolerate the drug by initiating a trial of oral aripiprazole before beginning treatment with aripiprazole lauroxil; establishing tolerability might take as long as 2 weeks because of the half-life of aripiprazole.
Aripiprazole lauroxil can be started at 441 mg, 662 mg, or 882 mg administered monthly; these dosages correspond to 300 mg, 450 mg, and 600 mg of aripiprazole, or 10 mg/d, 15 mg/d, ≥20 mg/d of oral aripiprazole, respectively (Table 2).2 Aripiprazole lauroxil can be administered either in the deltoid muscle (441 mg only) or gluteal muscle (441 mg, 662 mg, or 882 mg).2,4,5 Treatment with the 441-mg, 662-mg, or 882-mg dosages can be given every 4 weeks but the 882-mg dosage can be given every 6 weeks and only in the gluteal muscle, which provides greater dosing flexibility compared with extended-release injectable aripiprazole.2,4,5

Supplementation with oral aripiprazole is required for 21 days before the first aripiprazole lauroxil injection.2,4 The next injection should not be given earlier than 14 days after the previous dose. When a dose is missed, follow the guidelines outlined in Table 3.2

After a single injection, aripiprazole starts to appear in the systemic circulation at Day 5 or Day 6 and continues to be released for another 36 days.2 Steady-state concentration will be reached after the fourth monthly injection. The termination half-life of aripiprazole lauroxil ranged from 29 to 35 days after each monthly injection.2
Packaging. Aripiprazole lauroxil is available as single-dose, pre-filled, color-coded syringes for IM injection at 441 mg (light blue), 662 mg (green), and 882 mg (burgundy); syringes do not require refrigeration (Table 2).2 The syringe needs to be tapped at least 10 times to dislodge any material that might have settled. Shake the syringe vigorously for at least 30 seconds to ensure a uniform suspension. Shake it again for 30 seconds if the syringe is not used within 15 minutes.2
Efficacy
The efficacy of aripiprazole lauroxil for treating patients with schizophrenia has been established, in part, on the basis of efficacy data from clinical trials of oral aripiprazole. In addition, efficacy has been established in a 12-week, multicenter, randomized, placebo-controlled, double-blind, fixed-dose study of 622 individuals age 18 to 70 with schizophrenia.4,5 All eligible patients were diagnosed with schizophrenia as defined by DSM-IV-TR criteria and confirmed by the Structured Clinical Interview for DSM-IV Disorders, Clinical Trial Version and were experiencing an acute exacerbation of their illness at the time of the study. To be eligible for the study, participants had to have a Positive and Negative Syndrome Scale (PANSS) total score of 70 to 120 and score of ≥4 for ≥2 of the selected positive items (delusions, conceptual disorganization, hallucinatory behavior, and suspiciousness/persecution). Individuals also were required to have a Clinical Global Impression-Severity scale score of ≥4. Efficacy was assessed using the PANSS and Clinical Global Impression–Improvement scale (CGI-I).
Patients were randomized in a 1:1:1 ratio to receive IM aripiprazole lauroxil, 441 mg, aripiprazole lauroxil, 882 mg, or placebo once monthly in the gluteal region for 12 weeks. The gluteal muscle was selected as the injection site to maintain blinding to the study drug.4,5 After establishing tolerability to oral aripiprazole, participants received oral aripiprazole or placebo daily for the first 3 weeks. The IM injections were administered on Days 1, 29, and 57.
Efficacy was measured primarily as change in total PANSS score from the baseline to day 854,5; secondary efficacy variable was the CGI-I score at day 85. Statistically significant separation in PANSS score was observed in each aripiprazole lauroxil dosage group (441 mg and 882 mg) compared with placebo. Significant improvement in both active treatment groups was observed as early as Day 8 and continued throughout the study (P ≤ .004). The number of patients who improved much or very much on the CGI-I was significantly greater in either aripiprazole lauroxil group, compared with placebo (P < .001).
Contraindications
Allergic reactions. Patients who are hypersensitive to oral aripiprazole should not receive aripiprazole lauroxil. Hypersensitivity reactions have ranged from pruritus and urticaria to anaphylaxis.2
Drug−drug interactions. Reduce aripiprazole lauroxil dosage to the next lower dosage when used in combination with strong cytochrome P450 (CYP) 3A4 inhibitors (eg, itraconazole, clarithromycin) or strong CYP2D6 inhibitors (eg, quinidine, fluoxetine, paroxetine) for more than 2 weeks or if the patient is known to be a poor metabolizer of CYP2D6, because concentration of aripiprazole lauroxil could increase. No dose adjustment is required if the patient is already taking 441 mg/month or if CYP450 modulators are added for less than 2 weeks.2 Similarly, a dosage increase is recommended when aripiprazole lauroxil is used in combination with strong CYP3A4 inducers (eg, carbamazepine, rifampin).2
Overdose
No data are available on aripiprazole lauroxil overdose. However, there is one known case of oral aripiprazole overdose in a patient who ingested 1,260 mg of oral aripiprazole (42 times the maximum recommended daily dosage) but recovered completely.2 Common side effects reported in at least 5% of all overdose cases include vomiting, somnolence, and tremor. If an overdose occurs, call a poison control center immediately.
‘Black-box’ warning for patients with dementia
Aripiprazole lauroxil, similar to all other atypical antipsychotics, has a “black-box” warning stating that (1) it is not approved for treating dementia-related psychosis, and (2) it is associated with an increased risk of death with off-label use to treat behavioral problems in older adults with dementia-related psychosis.2 Meta-analysis of 17 placebo-controlled trials in patients taking an atypical antipsychotic (olanzapine, aripiprazole, risperidone, or quetiapine) revealed a risk of death in drug-treated patients 1.6 to 1.7 times that of placebo-treated patients.6
Adverse reactions
The overall safety profile of aripiprazole lauroxil is similar to that of oral aripiprazole. Most commonly observed adverse reaction during clinical trials of aripiprazole lauroxil was akathisia (incidence ≥5% and at least twice rate seen with placebo).2 Other common adverse reactions are shown in Table 4.2 Recently, the FDA issued a warning that compulsive or uncontrollable urges to gamble, binge eat, shop, and have sex have been reported with all formulations of aripiprazole.7 According to reports, these urges stopped when the drug was discontinued or the dosage reduced. Although rare, these impulse-control problems could result in harm if they are not recognized. See the full prescribing information for a complete set of adverse reactions.
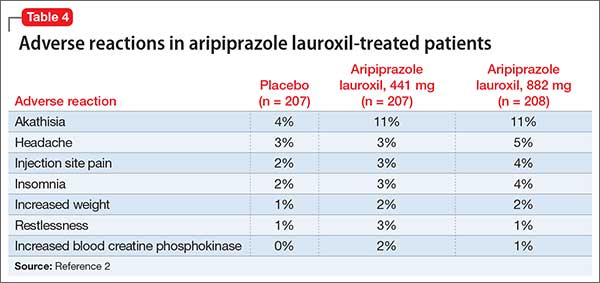
BOTTOM LINE
Aripiprazole lauroxil is a novel, long-acting second-generation antipsychotic that offers flexibility in terms of safe and effective dosing and can be administered in the deltoid (441 mg) or gluteal muscle (626 mg and 882 mg) and at dosing intervals of 4 to 6 weeks. Safety and tolerability profile of aripiprazole lauroxil are similar to that of oral aripiprazole. Aripiprazole lauroxil represents a new treatment option for patients with schizophrenia.
Related Resources
- Kennedy WK. When and how to use long-acting injectable antipsychotics. Current Psychiatry. 2012;11(8):40-43.
- Citrome L, Du Y, Risinger R, et al. Effect of aripiprazole lauroxil on agitation and hostility in patients with schizophrenia. Int Clin Psychopharmacol. 2016;31(2):69-75.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole extended-release • Abilify Maintena
Aripiprazole lauroxil • Aristada
Carbamazepine • Tegretol
Clarithromycin • Biaxin
Fluoxetine • Prozac
Itraconazole • Sporanox
Olanzapine • Zyprexa
Paroxetine • Paxil
Quetiapine • Seroquel
Quinidine • Quinidex
Rifampin • Rifadin
Risperidone • Risperdal
Acknowledgement
Maaz A. Khan, a student at the University of Oklahoma, Norman, Oklahoma, contributed to this article.
1. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry. 1999;56(3):241-247.
2. Aristada [package insert]. Waltham, MA; Alkermes; 2015.
3. Turncliff R, Hard M, Du Y, et al. Relative bioavailability and safety of aripiprazole lauroxil, a novel once-monthly, long-acting injectable atypical antipsychotic following deltoid and gluteal administration in adult subjects with schizophrenia. Schizophr Res. 2014;159(2-3):404-410.
4. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
5. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
6. U.S. Food and Drug Administration. Public health advisory: deaths with antipsychotics in elderly patients with behavioral disturbances. http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm053171. Published April 11, 2005. Accessed April 29, 2016.
7. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA warns about new impulse-control problems associated with mental health drug aripiprazole (Abilify, Abilify Maintena, Aristada). http://www.fda.gov/Drugs/DrugSafety/ucm498662.htm. Published May 3, 2016. Accessed June 20, 2016.
Approximately 80% of patients with schizophrenia relapse within 5 years1 despite the availability and increased use of second-generation antipsychotics. Long-acting depot formulations are a proven, effective treatment option for patients with schizophrenia. In October 2015, another long-acting injectable antipsychotic, aripiprazole lauroxil, was FDA-approved for schizophrenia.2 Aripiprazole lauroxil is administered IM every 4 to 6 weeks in the deltoid or gluteal region and is available in multiple dosages (Table 1).
Mechanism of action
Aripiprazole lauroxil is a prodrug of aripiprazole. Prodrugs are chemical compounds that exert their pharmacological effects after they undergo a biologic transformation and transform into a more active metabolite.3 The development of prodrugs is an established method used to improve physio-chemical or pharmacokinetic properties of the pharmacologically active compound.
After IM injection, aripiprazole lauroxil is most likely converted by an enzyme-mediated hydrolysis to N-hydroxymethyl aripiprazole, which is then hydrolyzed to aripiprazole. Aripiprazole’s mechanism of action is mediated through a combination of partial agonist activity at D2 and 5-HT1A receptors and antagonistic activity at 5-HT2A receptors.2,4
Dosing and administration
If your patient has never taken aripiprazole, ensure that she (he) will tolerate the drug by initiating a trial of oral aripiprazole before beginning treatment with aripiprazole lauroxil; establishing tolerability might take as long as 2 weeks because of the half-life of aripiprazole.
Aripiprazole lauroxil can be started at 441 mg, 662 mg, or 882 mg administered monthly; these dosages correspond to 300 mg, 450 mg, and 600 mg of aripiprazole, or 10 mg/d, 15 mg/d, ≥20 mg/d of oral aripiprazole, respectively (Table 2).2 Aripiprazole lauroxil can be administered either in the deltoid muscle (441 mg only) or gluteal muscle (441 mg, 662 mg, or 882 mg).2,4,5 Treatment with the 441-mg, 662-mg, or 882-mg dosages can be given every 4 weeks but the 882-mg dosage can be given every 6 weeks and only in the gluteal muscle, which provides greater dosing flexibility compared with extended-release injectable aripiprazole.2,4,5
Supplementation with oral aripiprazole is required for 21 days before the first aripiprazole lauroxil injection.2,4 The next injection should not be given earlier than 14 days after the previous dose. When a dose is missed, follow the guidelines outlined in Table 3.2
After a single injection, aripiprazole starts to appear in the systemic circulation at Day 5 or Day 6 and continues to be released for another 36 days.2 Steady-state concentration will be reached after the fourth monthly injection. The termination half-life of aripiprazole lauroxil ranged from 29 to 35 days after each monthly injection.2
Packaging. Aripiprazole lauroxil is available as single-dose, pre-filled, color-coded syringes for IM injection at 441 mg (light blue), 662 mg (green), and 882 mg (burgundy); syringes do not require refrigeration (Table 2).2 The syringe needs to be tapped at least 10 times to dislodge any material that might have settled. Shake the syringe vigorously for at least 30 seconds to ensure a uniform suspension. Shake it again for 30 seconds if the syringe is not used within 15 minutes.2
Efficacy
The efficacy of aripiprazole lauroxil for treating patients with schizophrenia has been established, in part, on the basis of efficacy data from clinical trials of oral aripiprazole. In addition, efficacy has been established in a 12-week, multicenter, randomized, placebo-controlled, double-blind, fixed-dose study of 622 individuals age 18 to 70 with schizophrenia.4,5 All eligible patients were diagnosed with schizophrenia as defined by DSM-IV-TR criteria and confirmed by the Structured Clinical Interview for DSM-IV Disorders, Clinical Trial Version and were experiencing an acute exacerbation of their illness at the time of the study. To be eligible for the study, participants had to have a Positive and Negative Syndrome Scale (PANSS) total score of 70 to 120 and score of ≥4 for ≥2 of the selected positive items (delusions, conceptual disorganization, hallucinatory behavior, and suspiciousness/persecution). Individuals also were required to have a Clinical Global Impression-Severity scale score of ≥4. Efficacy was assessed using the PANSS and Clinical Global Impression–Improvement scale (CGI-I).
Patients were randomized in a 1:1:1 ratio to receive IM aripiprazole lauroxil, 441 mg, aripiprazole lauroxil, 882 mg, or placebo once monthly in the gluteal region for 12 weeks. The gluteal muscle was selected as the injection site to maintain blinding to the study drug.4,5 After establishing tolerability to oral aripiprazole, participants received oral aripiprazole or placebo daily for the first 3 weeks. The IM injections were administered on Days 1, 29, and 57.
Efficacy was measured primarily as change in total PANSS score from the baseline to day 854,5; secondary efficacy variable was the CGI-I score at day 85. Statistically significant separation in PANSS score was observed in each aripiprazole lauroxil dosage group (441 mg and 882 mg) compared with placebo. Significant improvement in both active treatment groups was observed as early as Day 8 and continued throughout the study (P ≤ .004). The number of patients who improved much or very much on the CGI-I was significantly greater in either aripiprazole lauroxil group, compared with placebo (P < .001).
Contraindications
Allergic reactions. Patients who are hypersensitive to oral aripiprazole should not receive aripiprazole lauroxil. Hypersensitivity reactions have ranged from pruritus and urticaria to anaphylaxis.2
Drug−drug interactions. Reduce aripiprazole lauroxil dosage to the next lower dosage when used in combination with strong cytochrome P450 (CYP) 3A4 inhibitors (eg, itraconazole, clarithromycin) or strong CYP2D6 inhibitors (eg, quinidine, fluoxetine, paroxetine) for more than 2 weeks or if the patient is known to be a poor metabolizer of CYP2D6, because concentration of aripiprazole lauroxil could increase. No dose adjustment is required if the patient is already taking 441 mg/month or if CYP450 modulators are added for less than 2 weeks.2 Similarly, a dosage increase is recommended when aripiprazole lauroxil is used in combination with strong CYP3A4 inducers (eg, carbamazepine, rifampin).2
Overdose
No data are available on aripiprazole lauroxil overdose. However, there is one known case of oral aripiprazole overdose in a patient who ingested 1,260 mg of oral aripiprazole (42 times the maximum recommended daily dosage) but recovered completely.2 Common side effects reported in at least 5% of all overdose cases include vomiting, somnolence, and tremor. If an overdose occurs, call a poison control center immediately.
‘Black-box’ warning for patients with dementia
Aripiprazole lauroxil, similar to all other atypical antipsychotics, has a “black-box” warning stating that (1) it is not approved for treating dementia-related psychosis, and (2) it is associated with an increased risk of death with off-label use to treat behavioral problems in older adults with dementia-related psychosis.2 Meta-analysis of 17 placebo-controlled trials in patients taking an atypical antipsychotic (olanzapine, aripiprazole, risperidone, or quetiapine) revealed a risk of death in drug-treated patients 1.6 to 1.7 times that of placebo-treated patients.6
Adverse reactions
The overall safety profile of aripiprazole lauroxil is similar to that of oral aripiprazole. Most commonly observed adverse reaction during clinical trials of aripiprazole lauroxil was akathisia (incidence ≥5% and at least twice rate seen with placebo).2 Other common adverse reactions are shown in Table 4.2 Recently, the FDA issued a warning that compulsive or uncontrollable urges to gamble, binge eat, shop, and have sex have been reported with all formulations of aripiprazole.7 According to reports, these urges stopped when the drug was discontinued or the dosage reduced. Although rare, these impulse-control problems could result in harm if they are not recognized. See the full prescribing information for a complete set of adverse reactions.
BOTTOM LINE
Aripiprazole lauroxil is a novel, long-acting second-generation antipsychotic that offers flexibility in terms of safe and effective dosing and can be administered in the deltoid (441 mg) or gluteal muscle (626 mg and 882 mg) and at dosing intervals of 4 to 6 weeks. Safety and tolerability profile of aripiprazole lauroxil are similar to that of oral aripiprazole. Aripiprazole lauroxil represents a new treatment option for patients with schizophrenia.
Related Resources
- Kennedy WK. When and how to use long-acting injectable antipsychotics. Current Psychiatry. 2012;11(8):40-43.
- Citrome L, Du Y, Risinger R, et al. Effect of aripiprazole lauroxil on agitation and hostility in patients with schizophrenia. Int Clin Psychopharmacol. 2016;31(2):69-75.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole extended-release • Abilify Maintena
Aripiprazole lauroxil • Aristada
Carbamazepine • Tegretol
Clarithromycin • Biaxin
Fluoxetine • Prozac
Itraconazole • Sporanox
Olanzapine • Zyprexa
Paroxetine • Paxil
Quetiapine • Seroquel
Quinidine • Quinidex
Rifampin • Rifadin
Risperidone • Risperdal
Acknowledgement
Maaz A. Khan, a student at the University of Oklahoma, Norman, Oklahoma, contributed to this article.
Approximately 80% of patients with schizophrenia relapse within 5 years1 despite the availability and increased use of second-generation antipsychotics. Long-acting depot formulations are a proven, effective treatment option for patients with schizophrenia. In October 2015, another long-acting injectable antipsychotic, aripiprazole lauroxil, was FDA-approved for schizophrenia.2 Aripiprazole lauroxil is administered IM every 4 to 6 weeks in the deltoid or gluteal region and is available in multiple dosages (Table 1).
Mechanism of action
Aripiprazole lauroxil is a prodrug of aripiprazole. Prodrugs are chemical compounds that exert their pharmacological effects after they undergo a biologic transformation and transform into a more active metabolite.3 The development of prodrugs is an established method used to improve physio-chemical or pharmacokinetic properties of the pharmacologically active compound.
After IM injection, aripiprazole lauroxil is most likely converted by an enzyme-mediated hydrolysis to N-hydroxymethyl aripiprazole, which is then hydrolyzed to aripiprazole. Aripiprazole’s mechanism of action is mediated through a combination of partial agonist activity at D2 and 5-HT1A receptors and antagonistic activity at 5-HT2A receptors.2,4
Dosing and administration
If your patient has never taken aripiprazole, ensure that she (he) will tolerate the drug by initiating a trial of oral aripiprazole before beginning treatment with aripiprazole lauroxil; establishing tolerability might take as long as 2 weeks because of the half-life of aripiprazole.
Aripiprazole lauroxil can be started at 441 mg, 662 mg, or 882 mg administered monthly; these dosages correspond to 300 mg, 450 mg, and 600 mg of aripiprazole, or 10 mg/d, 15 mg/d, ≥20 mg/d of oral aripiprazole, respectively (Table 2).2 Aripiprazole lauroxil can be administered either in the deltoid muscle (441 mg only) or gluteal muscle (441 mg, 662 mg, or 882 mg).2,4,5 Treatment with the 441-mg, 662-mg, or 882-mg dosages can be given every 4 weeks but the 882-mg dosage can be given every 6 weeks and only in the gluteal muscle, which provides greater dosing flexibility compared with extended-release injectable aripiprazole.2,4,5
Supplementation with oral aripiprazole is required for 21 days before the first aripiprazole lauroxil injection.2,4 The next injection should not be given earlier than 14 days after the previous dose. When a dose is missed, follow the guidelines outlined in Table 3.2
After a single injection, aripiprazole starts to appear in the systemic circulation at Day 5 or Day 6 and continues to be released for another 36 days.2 Steady-state concentration will be reached after the fourth monthly injection. The termination half-life of aripiprazole lauroxil ranged from 29 to 35 days after each monthly injection.2
Packaging. Aripiprazole lauroxil is available as single-dose, pre-filled, color-coded syringes for IM injection at 441 mg (light blue), 662 mg (green), and 882 mg (burgundy); syringes do not require refrigeration (Table 2).2 The syringe needs to be tapped at least 10 times to dislodge any material that might have settled. Shake the syringe vigorously for at least 30 seconds to ensure a uniform suspension. Shake it again for 30 seconds if the syringe is not used within 15 minutes.2
Efficacy
The efficacy of aripiprazole lauroxil for treating patients with schizophrenia has been established, in part, on the basis of efficacy data from clinical trials of oral aripiprazole. In addition, efficacy has been established in a 12-week, multicenter, randomized, placebo-controlled, double-blind, fixed-dose study of 622 individuals age 18 to 70 with schizophrenia.4,5 All eligible patients were diagnosed with schizophrenia as defined by DSM-IV-TR criteria and confirmed by the Structured Clinical Interview for DSM-IV Disorders, Clinical Trial Version and were experiencing an acute exacerbation of their illness at the time of the study. To be eligible for the study, participants had to have a Positive and Negative Syndrome Scale (PANSS) total score of 70 to 120 and score of ≥4 for ≥2 of the selected positive items (delusions, conceptual disorganization, hallucinatory behavior, and suspiciousness/persecution). Individuals also were required to have a Clinical Global Impression-Severity scale score of ≥4. Efficacy was assessed using the PANSS and Clinical Global Impression–Improvement scale (CGI-I).
Patients were randomized in a 1:1:1 ratio to receive IM aripiprazole lauroxil, 441 mg, aripiprazole lauroxil, 882 mg, or placebo once monthly in the gluteal region for 12 weeks. The gluteal muscle was selected as the injection site to maintain blinding to the study drug.4,5 After establishing tolerability to oral aripiprazole, participants received oral aripiprazole or placebo daily for the first 3 weeks. The IM injections were administered on Days 1, 29, and 57.
Efficacy was measured primarily as change in total PANSS score from the baseline to day 854,5; secondary efficacy variable was the CGI-I score at day 85. Statistically significant separation in PANSS score was observed in each aripiprazole lauroxil dosage group (441 mg and 882 mg) compared with placebo. Significant improvement in both active treatment groups was observed as early as Day 8 and continued throughout the study (P ≤ .004). The number of patients who improved much or very much on the CGI-I was significantly greater in either aripiprazole lauroxil group, compared with placebo (P < .001).
Contraindications
Allergic reactions. Patients who are hypersensitive to oral aripiprazole should not receive aripiprazole lauroxil. Hypersensitivity reactions have ranged from pruritus and urticaria to anaphylaxis.2
Drug−drug interactions. Reduce aripiprazole lauroxil dosage to the next lower dosage when used in combination with strong cytochrome P450 (CYP) 3A4 inhibitors (eg, itraconazole, clarithromycin) or strong CYP2D6 inhibitors (eg, quinidine, fluoxetine, paroxetine) for more than 2 weeks or if the patient is known to be a poor metabolizer of CYP2D6, because concentration of aripiprazole lauroxil could increase. No dose adjustment is required if the patient is already taking 441 mg/month or if CYP450 modulators are added for less than 2 weeks.2 Similarly, a dosage increase is recommended when aripiprazole lauroxil is used in combination with strong CYP3A4 inducers (eg, carbamazepine, rifampin).2
Overdose
No data are available on aripiprazole lauroxil overdose. However, there is one known case of oral aripiprazole overdose in a patient who ingested 1,260 mg of oral aripiprazole (42 times the maximum recommended daily dosage) but recovered completely.2 Common side effects reported in at least 5% of all overdose cases include vomiting, somnolence, and tremor. If an overdose occurs, call a poison control center immediately.
‘Black-box’ warning for patients with dementia
Aripiprazole lauroxil, similar to all other atypical antipsychotics, has a “black-box” warning stating that (1) it is not approved for treating dementia-related psychosis, and (2) it is associated with an increased risk of death with off-label use to treat behavioral problems in older adults with dementia-related psychosis.2 Meta-analysis of 17 placebo-controlled trials in patients taking an atypical antipsychotic (olanzapine, aripiprazole, risperidone, or quetiapine) revealed a risk of death in drug-treated patients 1.6 to 1.7 times that of placebo-treated patients.6
Adverse reactions
The overall safety profile of aripiprazole lauroxil is similar to that of oral aripiprazole. Most commonly observed adverse reaction during clinical trials of aripiprazole lauroxil was akathisia (incidence ≥5% and at least twice rate seen with placebo).2 Other common adverse reactions are shown in Table 4.2 Recently, the FDA issued a warning that compulsive or uncontrollable urges to gamble, binge eat, shop, and have sex have been reported with all formulations of aripiprazole.7 According to reports, these urges stopped when the drug was discontinued or the dosage reduced. Although rare, these impulse-control problems could result in harm if they are not recognized. See the full prescribing information for a complete set of adverse reactions.
BOTTOM LINE
Aripiprazole lauroxil is a novel, long-acting second-generation antipsychotic that offers flexibility in terms of safe and effective dosing and can be administered in the deltoid (441 mg) or gluteal muscle (626 mg and 882 mg) and at dosing intervals of 4 to 6 weeks. Safety and tolerability profile of aripiprazole lauroxil are similar to that of oral aripiprazole. Aripiprazole lauroxil represents a new treatment option for patients with schizophrenia.
Related Resources
- Kennedy WK. When and how to use long-acting injectable antipsychotics. Current Psychiatry. 2012;11(8):40-43.
- Citrome L, Du Y, Risinger R, et al. Effect of aripiprazole lauroxil on agitation and hostility in patients with schizophrenia. Int Clin Psychopharmacol. 2016;31(2):69-75.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole extended-release • Abilify Maintena
Aripiprazole lauroxil • Aristada
Carbamazepine • Tegretol
Clarithromycin • Biaxin
Fluoxetine • Prozac
Itraconazole • Sporanox
Olanzapine • Zyprexa
Paroxetine • Paxil
Quetiapine • Seroquel
Quinidine • Quinidex
Rifampin • Rifadin
Risperidone • Risperdal
Acknowledgement
Maaz A. Khan, a student at the University of Oklahoma, Norman, Oklahoma, contributed to this article.
1. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry. 1999;56(3):241-247.
2. Aristada [package insert]. Waltham, MA; Alkermes; 2015.
3. Turncliff R, Hard M, Du Y, et al. Relative bioavailability and safety of aripiprazole lauroxil, a novel once-monthly, long-acting injectable atypical antipsychotic following deltoid and gluteal administration in adult subjects with schizophrenia. Schizophr Res. 2014;159(2-3):404-410.
4. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
5. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
6. U.S. Food and Drug Administration. Public health advisory: deaths with antipsychotics in elderly patients with behavioral disturbances. http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm053171. Published April 11, 2005. Accessed April 29, 2016.
7. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA warns about new impulse-control problems associated with mental health drug aripiprazole (Abilify, Abilify Maintena, Aristada). http://www.fda.gov/Drugs/DrugSafety/ucm498662.htm. Published May 3, 2016. Accessed June 20, 2016.
1. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry. 1999;56(3):241-247.
2. Aristada [package insert]. Waltham, MA; Alkermes; 2015.
3. Turncliff R, Hard M, Du Y, et al. Relative bioavailability and safety of aripiprazole lauroxil, a novel once-monthly, long-acting injectable atypical antipsychotic following deltoid and gluteal administration in adult subjects with schizophrenia. Schizophr Res. 2014;159(2-3):404-410.
4. Meltzer HY, Risinger R, Nasrallah HA, et al. A randomized, double-blind, placebo-controlled trial of aripiprazole lauroxil in acute exacerbation of schizophrenia. J Clin Psychiatry. 2015;76(8):1085-1090.
5. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
6. U.S. Food and Drug Administration. Public health advisory: deaths with antipsychotics in elderly patients with behavioral disturbances. http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm053171. Published April 11, 2005. Accessed April 29, 2016.
7. U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA warns about new impulse-control problems associated with mental health drug aripiprazole (Abilify, Abilify Maintena, Aristada). http://www.fda.gov/Drugs/DrugSafety/ucm498662.htm. Published May 3, 2016. Accessed June 20, 2016.
Counseling geriatric patients about opportunity and risk when ‘digital dating’
Baby Boomers represent a rapidly growing segment of digital device users.1 As these people age, their continued, even increasing, use of the Internet can be expected.1 At the same time, many older adults (age ≥65) are engaged in intimate relationships and regard sexuality as an important part of life.2
At this intersection, the Internet is likely to play a role in geriatric sexuality and “digital intimacy”—in that older adults can adopt patterns of using online dating sites similar to what their younger counterparts engage in. There is a need among clinicians to avoid stereotypical perceptions of “ageism” and the myth of “geriatric asexuality” as a result of older patients’ continued sexual interest and their adoption of social media technologies to facilitate the development of new intimate relationships. Acknowledgement of these realities by clinicians may assist in understanding and communication regarding these important areas of patients’ lives.
Why online dating?
Contemporary social and demographic changes (eg, higher divorce rates, increased longevity, aging of Baby Boomers) have influenced patterns of dating behaviors.3 Consistent with evolutionary theory, studies on courtship behaviors show that women remain the “choosers” of partners in relationships at all ages3; in contemporary society, however, there is an increasing ratio of women to men in later life, and the degree to which this demographic change might influence older men and women who are pursuing sexual relationships is unclear.3 Older adults might be aware of these demographic realities, and may use the Internet to increase their chances of finding a relationship.
For older homosexual men and women, demographic trends also are important because fewer available partners of similar sexual orientation might be available in their immediate communities, similarly incentivizing the use of online dating sites.
Hand in hand: Risk and vulnerability
Clinicians can discuss with geriatric patients who present with questions or concerns about sexuality and risks of online dating. Although risks associated with digital dating can involve anyone, those who are recently divorced, widowed, disabled, or elderly can be targeted by predators or fraudulent schemes, and thus become victims. Recognizing those risks and the vulnerability in the geriatric patient is crucial.
Chronic illness. Age-related physiological changes do not necessarily make one vulnerable; however, chronic diseases of aging, including major neurocognitive disorders, can impair daily function and increase disability and vulnerability. The majority of online dating sites do not discriminate among users, including those with disabilities such as incapacitating neuropsychiatric disorders. The clinician may need to assess cognitive status of patients specific to their capacity to fully understand the risks of use of social media. Inability to accomplish basic mastery of computer skills or inability to maintain appropriate boundaries and safeguards in relationships initiated and maintained using the Internet may assist in this determination. Patients with other problematic Internet use (eg, excessive devotion to online shopping or online gambling) may be prone to misusing social media and dating sites as well. Patients with clear impairment of memory or poor social judgment based on a neurocognitive disorder also might not maintain proper boundaries with social media use.
Feeling alone. Older persons might feel socially isolated, and therefore may be more willing to participate in online dating to increase their chances of establishing an intimate relationship or companionship. Research has shown that increased social ties, participation in groups, contact with friends and family, and perceived social support are associated with longer survival; on the other hand, social disengagement, low participation in leisure activities, and limited social networks are associated with higher risk of major neurocognitive disorders and increased disability.4
Little is known about social vulnerability in institutional settings, but institutional living could decrease social vulnerability in important ways (eg, access to social support, networks and activities, not living alone).4 Although the literature on older adults and “digital” or “virtual” dating is limited, there are essentially no such data from within institutional settings. It is important to separately address the issue of cognitively impaired patients’ capacity to consent to sexual activity both within institutional settings and elsewhere, as it raises numerous ethical dilemmas for clinicians.
Being sexually active. Early research into online dating focused particularly on the risks of sexually transmitted infections (STIs),5 which could be acquired through failure to use condoms with a new partner.6 Older women particularly are less likely to use condoms with new sexual partners.6 Screening at-risk adults should occur regardless of age. Effective interventions are needed to increase condom use in this age group. Research in the general population has started to investigate how the use of technology can minimize the risks associated with online dating.5 The Table5,6 lists strategies that can be used to minimize some of the risks of online dating among geriatric patients, including STIs and victimization.

Clinicians working with sexually active geriatric patients need to perform sexual risk assessments, complete capacity assessments, and provide preventive measures.
Legal issues
Criminal and civil liability issues have arisen with online dating involving cases of murder, rape, fraud, identity theft, loans, theft, domestic violence, stalking, and burglary. Online dating also raises concerns around the right to fair use of the Internet in different contexts. Flirting in cyberspace can occur with e-mail, text, Twitter, Skype, and Instant Messenger. Practices likely will vary depending on whether older adults are institutionalized or living in the community, as well as their mental status (eg, having a major neurocognitive disorder).
Some questions with legal implications worth considering include:
- To what extent is there a duty to accommodate healthy sexual relationships in institutionalized settings?
- At what point does monitoring and supervision become overly intrusive?
- Are older adults fully aware of the potential ramifications of sharing sensitive information in cyberspace?
- What is the threshold for capacity to consent among older adults to understand the sexual nature of the act and consent to the act?
Nursing homes and health care providers may become concerned about potential liability if their organization provides digital devices or electronic platforms that are not closely monitored. Clinicians have a duty to protect patients under their care from risks associated with predators who target vulnerable and lonely people, whether financially, emotionally, or physically. Some patients in nursing home settings may benefit from discussing with their family members or attorney the possibility of completing a “sexual power of attorney”7 that could be completed in conjunction with an advance health care directive that addresses or authorizes an agent to make decisions about their sexual activities if cognitively impaired in the future.
One might also consider to what extent local regulatory oversight will protect your patient. Not all jurisdictions regulate online dating services similarly; many existing regulations focus on unfair contracts and pay less heed to safety concerns.
As a result, some dissatisfied clients have been known to sue an online dating service for breach of contract or misrepresentation. One of the most significant issues, however, is making sure there are appropriate background checks. Online dating services may need to change their policies to screen and verify for criminal background checks.8 Older adults interested in online dating should be made aware of these emerging issues.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Veenhof B, Timusk P. Online activities of Canadian boomers and seniors. http://www.statcan.gc.ca/pub/11-008-x/2009002/article/10910-eng.htm#tphp. Updated April 23, 2014. Accessed April 26, 2015.
2. Lindau ST, Schumm LP, Laumann EO, et al. A study of sexuality and health among older adults in the United States. N Engl J Med. 2007;357(8):762-774.
3. Alterovitz SS, Mendelsohn GA. Partner p across the life span: online dating by older adults. Psychol Aging. 2009;24(2):513-517.
4. Andrew MK, Mitnitski AB, Rockwood K. Social vulnerability, frailty and mortality in elderly people. PLoS ONE. 2008;3(5):e2232. doi: 10.1371/journal.pone.0002232.
5. Couch D, Liamputtong P, Pitts M. Online daters and the use of technology for surveillance and risk management. International Journal of Emerging Technologies and Society. 2011;9(2):116-134.
6. Bateson DJ, Weisberg E, McCaffery KJ, et al. When online becomes offline: attitudes to safer sex practices in older and younger women using an Australian internet dating service. Sex Health. 2012;9(2):152-159.
7. Hill E. We’ll always have Shady Pines: surrogate decision-making tools for preserving sexual autonomy in elderly nursing home residents. William Mary J Women Law. 2014;20(2):468-490.
8. Doe v Match.com, 789 F Supp 2d 1197, 1199 (CD Cal 2011).
Baby Boomers represent a rapidly growing segment of digital device users.1 As these people age, their continued, even increasing, use of the Internet can be expected.1 At the same time, many older adults (age ≥65) are engaged in intimate relationships and regard sexuality as an important part of life.2
At this intersection, the Internet is likely to play a role in geriatric sexuality and “digital intimacy”—in that older adults can adopt patterns of using online dating sites similar to what their younger counterparts engage in. There is a need among clinicians to avoid stereotypical perceptions of “ageism” and the myth of “geriatric asexuality” as a result of older patients’ continued sexual interest and their adoption of social media technologies to facilitate the development of new intimate relationships. Acknowledgement of these realities by clinicians may assist in understanding and communication regarding these important areas of patients’ lives.
Why online dating?
Contemporary social and demographic changes (eg, higher divorce rates, increased longevity, aging of Baby Boomers) have influenced patterns of dating behaviors.3 Consistent with evolutionary theory, studies on courtship behaviors show that women remain the “choosers” of partners in relationships at all ages3; in contemporary society, however, there is an increasing ratio of women to men in later life, and the degree to which this demographic change might influence older men and women who are pursuing sexual relationships is unclear.3 Older adults might be aware of these demographic realities, and may use the Internet to increase their chances of finding a relationship.
For older homosexual men and women, demographic trends also are important because fewer available partners of similar sexual orientation might be available in their immediate communities, similarly incentivizing the use of online dating sites.
Hand in hand: Risk and vulnerability
Clinicians can discuss with geriatric patients who present with questions or concerns about sexuality and risks of online dating. Although risks associated with digital dating can involve anyone, those who are recently divorced, widowed, disabled, or elderly can be targeted by predators or fraudulent schemes, and thus become victims. Recognizing those risks and the vulnerability in the geriatric patient is crucial.
Chronic illness. Age-related physiological changes do not necessarily make one vulnerable; however, chronic diseases of aging, including major neurocognitive disorders, can impair daily function and increase disability and vulnerability. The majority of online dating sites do not discriminate among users, including those with disabilities such as incapacitating neuropsychiatric disorders. The clinician may need to assess cognitive status of patients specific to their capacity to fully understand the risks of use of social media. Inability to accomplish basic mastery of computer skills or inability to maintain appropriate boundaries and safeguards in relationships initiated and maintained using the Internet may assist in this determination. Patients with other problematic Internet use (eg, excessive devotion to online shopping or online gambling) may be prone to misusing social media and dating sites as well. Patients with clear impairment of memory or poor social judgment based on a neurocognitive disorder also might not maintain proper boundaries with social media use.
Feeling alone. Older persons might feel socially isolated, and therefore may be more willing to participate in online dating to increase their chances of establishing an intimate relationship or companionship. Research has shown that increased social ties, participation in groups, contact with friends and family, and perceived social support are associated with longer survival; on the other hand, social disengagement, low participation in leisure activities, and limited social networks are associated with higher risk of major neurocognitive disorders and increased disability.4
Little is known about social vulnerability in institutional settings, but institutional living could decrease social vulnerability in important ways (eg, access to social support, networks and activities, not living alone).4 Although the literature on older adults and “digital” or “virtual” dating is limited, there are essentially no such data from within institutional settings. It is important to separately address the issue of cognitively impaired patients’ capacity to consent to sexual activity both within institutional settings and elsewhere, as it raises numerous ethical dilemmas for clinicians.
Being sexually active. Early research into online dating focused particularly on the risks of sexually transmitted infections (STIs),5 which could be acquired through failure to use condoms with a new partner.6 Older women particularly are less likely to use condoms with new sexual partners.6 Screening at-risk adults should occur regardless of age. Effective interventions are needed to increase condom use in this age group. Research in the general population has started to investigate how the use of technology can minimize the risks associated with online dating.5 The Table5,6 lists strategies that can be used to minimize some of the risks of online dating among geriatric patients, including STIs and victimization.
Clinicians working with sexually active geriatric patients need to perform sexual risk assessments, complete capacity assessments, and provide preventive measures.
Legal issues
Criminal and civil liability issues have arisen with online dating involving cases of murder, rape, fraud, identity theft, loans, theft, domestic violence, stalking, and burglary. Online dating also raises concerns around the right to fair use of the Internet in different contexts. Flirting in cyberspace can occur with e-mail, text, Twitter, Skype, and Instant Messenger. Practices likely will vary depending on whether older adults are institutionalized or living in the community, as well as their mental status (eg, having a major neurocognitive disorder).
Some questions with legal implications worth considering include:
- To what extent is there a duty to accommodate healthy sexual relationships in institutionalized settings?
- At what point does monitoring and supervision become overly intrusive?
- Are older adults fully aware of the potential ramifications of sharing sensitive information in cyberspace?
- What is the threshold for capacity to consent among older adults to understand the sexual nature of the act and consent to the act?
Nursing homes and health care providers may become concerned about potential liability if their organization provides digital devices or electronic platforms that are not closely monitored. Clinicians have a duty to protect patients under their care from risks associated with predators who target vulnerable and lonely people, whether financially, emotionally, or physically. Some patients in nursing home settings may benefit from discussing with their family members or attorney the possibility of completing a “sexual power of attorney”7 that could be completed in conjunction with an advance health care directive that addresses or authorizes an agent to make decisions about their sexual activities if cognitively impaired in the future.
One might also consider to what extent local regulatory oversight will protect your patient. Not all jurisdictions regulate online dating services similarly; many existing regulations focus on unfair contracts and pay less heed to safety concerns.
As a result, some dissatisfied clients have been known to sue an online dating service for breach of contract or misrepresentation. One of the most significant issues, however, is making sure there are appropriate background checks. Online dating services may need to change their policies to screen and verify for criminal background checks.8 Older adults interested in online dating should be made aware of these emerging issues.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Baby Boomers represent a rapidly growing segment of digital device users.1 As these people age, their continued, even increasing, use of the Internet can be expected.1 At the same time, many older adults (age ≥65) are engaged in intimate relationships and regard sexuality as an important part of life.2
At this intersection, the Internet is likely to play a role in geriatric sexuality and “digital intimacy”—in that older adults can adopt patterns of using online dating sites similar to what their younger counterparts engage in. There is a need among clinicians to avoid stereotypical perceptions of “ageism” and the myth of “geriatric asexuality” as a result of older patients’ continued sexual interest and their adoption of social media technologies to facilitate the development of new intimate relationships. Acknowledgement of these realities by clinicians may assist in understanding and communication regarding these important areas of patients’ lives.
Why online dating?
Contemporary social and demographic changes (eg, higher divorce rates, increased longevity, aging of Baby Boomers) have influenced patterns of dating behaviors.3 Consistent with evolutionary theory, studies on courtship behaviors show that women remain the “choosers” of partners in relationships at all ages3; in contemporary society, however, there is an increasing ratio of women to men in later life, and the degree to which this demographic change might influence older men and women who are pursuing sexual relationships is unclear.3 Older adults might be aware of these demographic realities, and may use the Internet to increase their chances of finding a relationship.
For older homosexual men and women, demographic trends also are important because fewer available partners of similar sexual orientation might be available in their immediate communities, similarly incentivizing the use of online dating sites.
Hand in hand: Risk and vulnerability
Clinicians can discuss with geriatric patients who present with questions or concerns about sexuality and risks of online dating. Although risks associated with digital dating can involve anyone, those who are recently divorced, widowed, disabled, or elderly can be targeted by predators or fraudulent schemes, and thus become victims. Recognizing those risks and the vulnerability in the geriatric patient is crucial.
Chronic illness. Age-related physiological changes do not necessarily make one vulnerable; however, chronic diseases of aging, including major neurocognitive disorders, can impair daily function and increase disability and vulnerability. The majority of online dating sites do not discriminate among users, including those with disabilities such as incapacitating neuropsychiatric disorders. The clinician may need to assess cognitive status of patients specific to their capacity to fully understand the risks of use of social media. Inability to accomplish basic mastery of computer skills or inability to maintain appropriate boundaries and safeguards in relationships initiated and maintained using the Internet may assist in this determination. Patients with other problematic Internet use (eg, excessive devotion to online shopping or online gambling) may be prone to misusing social media and dating sites as well. Patients with clear impairment of memory or poor social judgment based on a neurocognitive disorder also might not maintain proper boundaries with social media use.
Feeling alone. Older persons might feel socially isolated, and therefore may be more willing to participate in online dating to increase their chances of establishing an intimate relationship or companionship. Research has shown that increased social ties, participation in groups, contact with friends and family, and perceived social support are associated with longer survival; on the other hand, social disengagement, low participation in leisure activities, and limited social networks are associated with higher risk of major neurocognitive disorders and increased disability.4
Little is known about social vulnerability in institutional settings, but institutional living could decrease social vulnerability in important ways (eg, access to social support, networks and activities, not living alone).4 Although the literature on older adults and “digital” or “virtual” dating is limited, there are essentially no such data from within institutional settings. It is important to separately address the issue of cognitively impaired patients’ capacity to consent to sexual activity both within institutional settings and elsewhere, as it raises numerous ethical dilemmas for clinicians.
Being sexually active. Early research into online dating focused particularly on the risks of sexually transmitted infections (STIs),5 which could be acquired through failure to use condoms with a new partner.6 Older women particularly are less likely to use condoms with new sexual partners.6 Screening at-risk adults should occur regardless of age. Effective interventions are needed to increase condom use in this age group. Research in the general population has started to investigate how the use of technology can minimize the risks associated with online dating.5 The Table5,6 lists strategies that can be used to minimize some of the risks of online dating among geriatric patients, including STIs and victimization.
Clinicians working with sexually active geriatric patients need to perform sexual risk assessments, complete capacity assessments, and provide preventive measures.
Legal issues
Criminal and civil liability issues have arisen with online dating involving cases of murder, rape, fraud, identity theft, loans, theft, domestic violence, stalking, and burglary. Online dating also raises concerns around the right to fair use of the Internet in different contexts. Flirting in cyberspace can occur with e-mail, text, Twitter, Skype, and Instant Messenger. Practices likely will vary depending on whether older adults are institutionalized or living in the community, as well as their mental status (eg, having a major neurocognitive disorder).
Some questions with legal implications worth considering include:
- To what extent is there a duty to accommodate healthy sexual relationships in institutionalized settings?
- At what point does monitoring and supervision become overly intrusive?
- Are older adults fully aware of the potential ramifications of sharing sensitive information in cyberspace?
- What is the threshold for capacity to consent among older adults to understand the sexual nature of the act and consent to the act?
Nursing homes and health care providers may become concerned about potential liability if their organization provides digital devices or electronic platforms that are not closely monitored. Clinicians have a duty to protect patients under their care from risks associated with predators who target vulnerable and lonely people, whether financially, emotionally, or physically. Some patients in nursing home settings may benefit from discussing with their family members or attorney the possibility of completing a “sexual power of attorney”7 that could be completed in conjunction with an advance health care directive that addresses or authorizes an agent to make decisions about their sexual activities if cognitively impaired in the future.
One might also consider to what extent local regulatory oversight will protect your patient. Not all jurisdictions regulate online dating services similarly; many existing regulations focus on unfair contracts and pay less heed to safety concerns.
As a result, some dissatisfied clients have been known to sue an online dating service for breach of contract or misrepresentation. One of the most significant issues, however, is making sure there are appropriate background checks. Online dating services may need to change their policies to screen and verify for criminal background checks.8 Older adults interested in online dating should be made aware of these emerging issues.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Veenhof B, Timusk P. Online activities of Canadian boomers and seniors. http://www.statcan.gc.ca/pub/11-008-x/2009002/article/10910-eng.htm#tphp. Updated April 23, 2014. Accessed April 26, 2015.
2. Lindau ST, Schumm LP, Laumann EO, et al. A study of sexuality and health among older adults in the United States. N Engl J Med. 2007;357(8):762-774.
3. Alterovitz SS, Mendelsohn GA. Partner p across the life span: online dating by older adults. Psychol Aging. 2009;24(2):513-517.
4. Andrew MK, Mitnitski AB, Rockwood K. Social vulnerability, frailty and mortality in elderly people. PLoS ONE. 2008;3(5):e2232. doi: 10.1371/journal.pone.0002232.
5. Couch D, Liamputtong P, Pitts M. Online daters and the use of technology for surveillance and risk management. International Journal of Emerging Technologies and Society. 2011;9(2):116-134.
6. Bateson DJ, Weisberg E, McCaffery KJ, et al. When online becomes offline: attitudes to safer sex practices in older and younger women using an Australian internet dating service. Sex Health. 2012;9(2):152-159.
7. Hill E. We’ll always have Shady Pines: surrogate decision-making tools for preserving sexual autonomy in elderly nursing home residents. William Mary J Women Law. 2014;20(2):468-490.
8. Doe v Match.com, 789 F Supp 2d 1197, 1199 (CD Cal 2011).
1. Veenhof B, Timusk P. Online activities of Canadian boomers and seniors. http://www.statcan.gc.ca/pub/11-008-x/2009002/article/10910-eng.htm#tphp. Updated April 23, 2014. Accessed April 26, 2015.
2. Lindau ST, Schumm LP, Laumann EO, et al. A study of sexuality and health among older adults in the United States. N Engl J Med. 2007;357(8):762-774.
3. Alterovitz SS, Mendelsohn GA. Partner p across the life span: online dating by older adults. Psychol Aging. 2009;24(2):513-517.
4. Andrew MK, Mitnitski AB, Rockwood K. Social vulnerability, frailty and mortality in elderly people. PLoS ONE. 2008;3(5):e2232. doi: 10.1371/journal.pone.0002232.
5. Couch D, Liamputtong P, Pitts M. Online daters and the use of technology for surveillance and risk management. International Journal of Emerging Technologies and Society. 2011;9(2):116-134.
6. Bateson DJ, Weisberg E, McCaffery KJ, et al. When online becomes offline: attitudes to safer sex practices in older and younger women using an Australian internet dating service. Sex Health. 2012;9(2):152-159.
7. Hill E. We’ll always have Shady Pines: surrogate decision-making tools for preserving sexual autonomy in elderly nursing home residents. William Mary J Women Law. 2014;20(2):468-490.
8. Doe v Match.com, 789 F Supp 2d 1197, 1199 (CD Cal 2011).
5 Myths of tobacco cessation
Here are 5 commonly held beliefs about stopping tobacco use, and about your role in helping these patients, that go up in smoke on close inspection.
Treating nicotine use disorder isn’t really a psychiatrist’s job. False! Smoking is the leading preventable cause of death, causing 1 in every 5 deaths in the United States and as many as 1 of every 2 deaths among patients with depression, bipolar disorder, or schizophrenia.1,2 As psychiatrists, our experience with treating addiction positions us to address nicotine use disorder more effectively than deferring exclusively to primary care.
I can’t treat my patients’ nicotine dependence until they are ready to quit. Not so! Treatment with varenicline, bupropion, or nicotine replacement therapy is likely to decrease smoking even if the patient has not made a commitment to quit. A smoker treated with pharmacotherapy is more likely to try to quit than one who is not receiving medication.3,4
Motivational interviewing is an excellent intervention to facilitate readiness to quit smoking. Many smokers want to quit—but if they don’t believe that effective treatments exist or that psychiatrists provide such care, they won’t initiate that conversation with you.
Smokeless tobacco isn’t so bad. Poppycock! Chewing and dipping tobacco contains many undesirable chemicals, including abrasives, salts, sweeteners, and carcinogens. Smokeless tobacco is a risk factor for cancer of the mouth and pancreas, as well as tooth decay, periodontal disease, hypertension, hyperlipidemia, myocardial infarction, and fatal stroke.5
Nicotine replacement products are as bad as smoking. Claptrap! You can reassure patients that nicotine is not a carcinogen. If your patients use the same amount of nicotine but replace tobacco in whole or in part with a patch, gum, or an inhaler, they will have better health even if they use nicotine replacement for the rest of their life. Nicotine replacement products are less addictive than cigarettes because they release nicotine more slowly. (Cigarettes bring peak levels of nicotine to the brain even faster than IV administration does.) Nicotine replacement is recommended for at least 3 months after quitting tobacco or for as long the patient needs it.3
Nicotine replacement products are dangerous for current smokers. Balderdash! Many patients are afraid of using nicotine from >1 source. A common myth is that using a nicotine patch while smoking increases the risk of heart attack, which discourages patients from trying a nicotine replacement product before they are sure they will stop smoking. Nicotine replacement is likely to reduce the frequency of their smoking and reduce harm, not add to it.3
1. Centers for Disease Control and Prevention. Smoking & tobacco use: tobacco-related mortality. http://www.cdc.gov/tobacco/ data_statistics/fact_sheets/ health_effects/tobacco_related_ mortality. Updated August 18, 2015. Accessed December 20, 2015.
2. Callaghan RC, Veldhuizen S, Jeysingh T, et al. Patterns of tobacco-related mortality among individuals diagnosed with schizophrenia, bipolar disorder, or depression. J Psychiatr Res. 2014;48(1):102-110.
3. Stead LF, Perera R, Bullen C, et al. Nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev. 2012;11:CD000146. doi: 10.1002/14651858.CD000146.pub4.
4. Ebbert JO, Hughes JR, West RJ, et al. Effect of varenicline on smoking cessation through smoking reduction: a randomized clinical trial. JAMA. 2015;313(7):678-694.
5. Piano MR, Benowitz NL, Fitzgerald GA, et al; American Heart Association Council on Cardiovascular Nursing. Impact of smokeless tobacco products on cardiovascular disease: implications for policy, prevention, and treatment: a policy statement from the American Heart Association. Circulation. 2010;122(15):1520-1544.
Here are 5 commonly held beliefs about stopping tobacco use, and about your role in helping these patients, that go up in smoke on close inspection.
Treating nicotine use disorder isn’t really a psychiatrist’s job. False! Smoking is the leading preventable cause of death, causing 1 in every 5 deaths in the United States and as many as 1 of every 2 deaths among patients with depression, bipolar disorder, or schizophrenia.1,2 As psychiatrists, our experience with treating addiction positions us to address nicotine use disorder more effectively than deferring exclusively to primary care.
I can’t treat my patients’ nicotine dependence until they are ready to quit. Not so! Treatment with varenicline, bupropion, or nicotine replacement therapy is likely to decrease smoking even if the patient has not made a commitment to quit. A smoker treated with pharmacotherapy is more likely to try to quit than one who is not receiving medication.3,4
Motivational interviewing is an excellent intervention to facilitate readiness to quit smoking. Many smokers want to quit—but if they don’t believe that effective treatments exist or that psychiatrists provide such care, they won’t initiate that conversation with you.
Smokeless tobacco isn’t so bad. Poppycock! Chewing and dipping tobacco contains many undesirable chemicals, including abrasives, salts, sweeteners, and carcinogens. Smokeless tobacco is a risk factor for cancer of the mouth and pancreas, as well as tooth decay, periodontal disease, hypertension, hyperlipidemia, myocardial infarction, and fatal stroke.5
Nicotine replacement products are as bad as smoking. Claptrap! You can reassure patients that nicotine is not a carcinogen. If your patients use the same amount of nicotine but replace tobacco in whole or in part with a patch, gum, or an inhaler, they will have better health even if they use nicotine replacement for the rest of their life. Nicotine replacement products are less addictive than cigarettes because they release nicotine more slowly. (Cigarettes bring peak levels of nicotine to the brain even faster than IV administration does.) Nicotine replacement is recommended for at least 3 months after quitting tobacco or for as long the patient needs it.3
Nicotine replacement products are dangerous for current smokers. Balderdash! Many patients are afraid of using nicotine from >1 source. A common myth is that using a nicotine patch while smoking increases the risk of heart attack, which discourages patients from trying a nicotine replacement product before they are sure they will stop smoking. Nicotine replacement is likely to reduce the frequency of their smoking and reduce harm, not add to it.3
Here are 5 commonly held beliefs about stopping tobacco use, and about your role in helping these patients, that go up in smoke on close inspection.
Treating nicotine use disorder isn’t really a psychiatrist’s job. False! Smoking is the leading preventable cause of death, causing 1 in every 5 deaths in the United States and as many as 1 of every 2 deaths among patients with depression, bipolar disorder, or schizophrenia.1,2 As psychiatrists, our experience with treating addiction positions us to address nicotine use disorder more effectively than deferring exclusively to primary care.
I can’t treat my patients’ nicotine dependence until they are ready to quit. Not so! Treatment with varenicline, bupropion, or nicotine replacement therapy is likely to decrease smoking even if the patient has not made a commitment to quit. A smoker treated with pharmacotherapy is more likely to try to quit than one who is not receiving medication.3,4
Motivational interviewing is an excellent intervention to facilitate readiness to quit smoking. Many smokers want to quit—but if they don’t believe that effective treatments exist or that psychiatrists provide such care, they won’t initiate that conversation with you.
Smokeless tobacco isn’t so bad. Poppycock! Chewing and dipping tobacco contains many undesirable chemicals, including abrasives, salts, sweeteners, and carcinogens. Smokeless tobacco is a risk factor for cancer of the mouth and pancreas, as well as tooth decay, periodontal disease, hypertension, hyperlipidemia, myocardial infarction, and fatal stroke.5
Nicotine replacement products are as bad as smoking. Claptrap! You can reassure patients that nicotine is not a carcinogen. If your patients use the same amount of nicotine but replace tobacco in whole or in part with a patch, gum, or an inhaler, they will have better health even if they use nicotine replacement for the rest of their life. Nicotine replacement products are less addictive than cigarettes because they release nicotine more slowly. (Cigarettes bring peak levels of nicotine to the brain even faster than IV administration does.) Nicotine replacement is recommended for at least 3 months after quitting tobacco or for as long the patient needs it.3
Nicotine replacement products are dangerous for current smokers. Balderdash! Many patients are afraid of using nicotine from >1 source. A common myth is that using a nicotine patch while smoking increases the risk of heart attack, which discourages patients from trying a nicotine replacement product before they are sure they will stop smoking. Nicotine replacement is likely to reduce the frequency of their smoking and reduce harm, not add to it.3
1. Centers for Disease Control and Prevention. Smoking & tobacco use: tobacco-related mortality. http://www.cdc.gov/tobacco/ data_statistics/fact_sheets/ health_effects/tobacco_related_ mortality. Updated August 18, 2015. Accessed December 20, 2015.
2. Callaghan RC, Veldhuizen S, Jeysingh T, et al. Patterns of tobacco-related mortality among individuals diagnosed with schizophrenia, bipolar disorder, or depression. J Psychiatr Res. 2014;48(1):102-110.
3. Stead LF, Perera R, Bullen C, et al. Nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev. 2012;11:CD000146. doi: 10.1002/14651858.CD000146.pub4.
4. Ebbert JO, Hughes JR, West RJ, et al. Effect of varenicline on smoking cessation through smoking reduction: a randomized clinical trial. JAMA. 2015;313(7):678-694.
5. Piano MR, Benowitz NL, Fitzgerald GA, et al; American Heart Association Council on Cardiovascular Nursing. Impact of smokeless tobacco products on cardiovascular disease: implications for policy, prevention, and treatment: a policy statement from the American Heart Association. Circulation. 2010;122(15):1520-1544.
1. Centers for Disease Control and Prevention. Smoking & tobacco use: tobacco-related mortality. http://www.cdc.gov/tobacco/ data_statistics/fact_sheets/ health_effects/tobacco_related_ mortality. Updated August 18, 2015. Accessed December 20, 2015.
2. Callaghan RC, Veldhuizen S, Jeysingh T, et al. Patterns of tobacco-related mortality among individuals diagnosed with schizophrenia, bipolar disorder, or depression. J Psychiatr Res. 2014;48(1):102-110.
3. Stead LF, Perera R, Bullen C, et al. Nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev. 2012;11:CD000146. doi: 10.1002/14651858.CD000146.pub4.
4. Ebbert JO, Hughes JR, West RJ, et al. Effect of varenicline on smoking cessation through smoking reduction: a randomized clinical trial. JAMA. 2015;313(7):678-694.
5. Piano MR, Benowitz NL, Fitzgerald GA, et al; American Heart Association Council on Cardiovascular Nursing. Impact of smokeless tobacco products on cardiovascular disease: implications for policy, prevention, and treatment: a policy statement from the American Heart Association. Circulation. 2010;122(15):1520-1544.
Pregnant and nursing patients benefit from ‘ambitious’ changes to drug labeling for safety
In December 2014, the FDA issued draft guidance for sweeping changes to labeling of pharmaceutical treatments in regard to pregnancy and lactation information. These changes are now in effect for use in practice.1 The undertaking has been years in the making, and is truly ambitious.
The outdated system of letter categories (A, B, C, D, X) falls short of clinical needs in several ways:
- the quality and volume of data can be lacking
- comparative risk is not described
- using letters can led to oversimplification or, in some cases, exaggeration of risk and safety (Box).

Other drawbacks include infrequent updating of information and omission of information about baseline rates of reproductive-related adverse events, to provide a more meaningful context for risk assessment.
A note before we continue discussion of labeling: Recognize that pregnancy itself is inherently risky; poor outcomes are, regrettably, not uncommon. The rate of birth defects in the United States is approximately 3%, and obstetric complications, such as prematurity, are common.2,3
New system described
The new labeling content has been described in the FDA’s Pregnancy and Lactation Labeling Rule (also called the “final rule”), issued in December 2014. For each medication, there will be subsections in the labeling:
- Pregnancy
- Lactation
- Females and Males of Reproductive Potential.
In addition, FDA instructions now state that labeling:
- must be updated when new information becomes available
- needs to include evaluation of human data that becomes available mainly after the drug is approved
- needs to include information about the background rates of adverse events related to reproduction.
Labeling in pregnancy. As an example, the “Pregnancy” section of every label contains 3 subsections, all of great clinical importance. First is information about pregnancy exposure registries, with a listing of scientifically acceptable registries (if a registry is available for that drug) and contact information; this section focuses on the high value of data that are systematically and prospectively collected. The second summarizes risk associated with the drug during pregnancy, based on available human, animal, and pharmacologic data. Third is a discussion of clinical considerations.
Need for appropriate controls. Psychiatric disorders increase the risk of pregnancy complications, and often are associated with variables that might increase the risk of a poor pregnancy outcome. For example, a patient who has a psychiatric disorder might be less likely to seek prenatal care, take a prenatal vitamin, and sleep and eat well; she also might use alcohol, tobacco, or other substances of abuse.
The medical literature on the reproductive safety of psychotropic medications is fraught with confounding variables other than the medications themselves. These include variables that, taken alone, might confer a poorer outcome on the fetus or newborn of a pregnant or lactating woman who has a psychiatric illness (to the extent that she uses psychotropics during a pregnancy), compared with what would be seen in (1) a healthy woman who is not taking such medication or (2) the general population.
On the new labels, detailed statements on human data include information from clinical trials, pregnancy exposure registries, and epidemiologic studies. Labels are also to include:
- incidence of adverse events
- effect of dosage
- effect of duration of exposure
- effect of gestational timing of exposure.
The labels emphasize quantifying risk relative to the risk of the same outcome in infants born to women who have not been exposed to the particular drug, but who have the disease or condition for which the drug is indicated (ie, appropriate controls).
Clinical considerations are to include information on the following related to the specific medication (when that information is known):
- more information for prescribers, to further risk-benefit counseling
- disease-associated maternal-fetal risks
- dosage adjustments during pregnancy and postpartum
- maternal adverse reactions
- fetal and neonatal adverse reactions
- labor and delivery.
Clearly, this overdue shift in providing information regarding reproductive safety has the potential to inform clinicians and patients in a meaningful way about the risks and benefits of specific treatments during pregnancy and lactation. Translating that information into practice is daunting, however.
Important aspects of implementation
Pregnancy exposure registries will play a crucial role. For most medications, no systematic registry has been established; to do so, rigorous methodology is required to acquire prospective data and account for confounding variables.4 Appropriate control groups also are required to yield data that are useful and interpretable. Primary outcomes require verification, such as review of medical records. Last, registries must be well-conducted and therefore adequately funded, yet labeling changes have not been accompanied by funding requirements set forth by regulators to pharmaceutical manufacturers.
Labeling must be updated continually. Furthermore, it is unclear who will review data for precision and comprehensiveness.
Data need to be understandable to health care providers across disciplines and to patients with varying levels of education for the label to have a meaningful impact on clinical care.
As noted, there is no mandate for funding the meticulous pharmacovigilance required to provide definitive data for labeling. It is unclear if the potential benefits of the new labeling can be reaped without adequate financing of the pharmacovigilance mechanisms required to inform patients adequately.
Role of pregnancy registries
Over the past 2 decades, pregnancy registries have emerged as a rapid, systematic means of collecting important reproductive safety data on the risk for major malformations after prenatal exposure to a medication or a class of medications.5,6 Such registries enhance the rigor of available cohort studies and other analyses of reproductive safety data that have been derived from large administrative databases.
NPRAA and NPRAD. Recently, the National Pregnancy Registry for Atypical Antipsychotics (NPRAA) and the National Pregnancy Registry for Antidepressants (NPRAD) were established in an effort to obtain reproductive safety data about fetal exposure to second-generation antipsychotics (SGAs) and to newer antidepressants.7 Based at Massachusetts General Hospital in Boston, NPRAA and NPRAD systematically and prospectively evaluate the risk of malformations among infants who have been exposed in utero to an SGA or an antidepressant.
The structure of both registries are the same, modeled after the North American Antiepileptic Drug Registry.5,8 Data are collected prospectively from pregnant women, age 18 to 45, by means of 3 telephone interviews conducted proximate to enrollment, at 7 months’ gestation, and at 2 or 3 months’ postpartum.
Participants include (1) pregnant women who have a history of fetal exposure to an SGA or an antidepressant, or both, and (2) a comparison group of non-exposed pregnant women who have a history of a psychiatric illness. Authorization for release of medical records is obtained for obstetric care, labor and delivery, and neonatal care (≤6 months of age).
Information on the presence of major malformations is abstracted from the medical record, along with other data on neonatal and maternal health outcomes. Identified cases of a congenital malformation are sent to a dysmorphologist, who has been blinded to drug exposure, for final adjudication. Release of findings is dictated by a governing Scientific Advisory Board.
Results so far. Results are available from the NPRAA.9 As of December 2014, 487 women were enrolled: 353 who used an SGA and 134 comparison women. Medical records were obtained for 82.2% of participants. A total of 303 women completed the study and were eligible for inclusion in the analysis. Findings include:
- Of 214 live births with first-trimester exposure to an SGA, 3 major malformations were confirmed. In the control group (n = 89), 1 major malformation was confirmed
- The absolute risk of a major malformation was 1.4% for an exposed infant and 1.1% for an unexposed infant
- The odds ratio for a major malformation, comparing exposed infants with unexposed infants, was 1.25 (95% CI, 0.13–12.19).
It is reasonable, therefore, to conclude that, as a class, SGAs are not major teratogens. Although the confidence intervals around the odds ratio estimate remain wide, with the probability for change over the course of the study, it is unlikely that risk will rise to the level of known major teratogens, such as valproate and thalidomide.10,11
Help with decision-making
Given recent FDA guidance about the importance of pregnancy registries (www.fda.gov/pregnancyregistries), such carefully collected data might help clinicians and patients make informed choices about treatment. Future efforts of NPRAA and NPRAD will focus on sustaining growth in enrollment of participants so that the reproductive safety of SGAs and newer antidepressants can be delineated more clearly.
Last, you can refer potential participants to NPRAA and NPRAD by calling 1-866-961-2388. More information is available at www.womensmentalhealth.org.
1. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Pregnancy, lactation, and reproductive potential: labeling for human prescription drug and biological products—content and format: guidance for industry. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM425398.pdf. Published December 2014. Accessed June 7, 2016.
2. Centers for Disease Control and Prevention. Birth defects. http://www.cdc.gov/ncbddd/birthdefects/facts.html. Updated September 21, 2005. Accessed June 7, 2016.
3. Centers for Disease Control and Prevention. Preterm birth. http://www.cdc.gov/reproductivehealth/maternalinfanthealth/pretermbirth.htm. Updated December 4, 2015. Accessed June 7, 2016.
4. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Guidance for industry: establishing pregnancy exposure registries. http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/WomensHealthResearch/UCM133332.pdf. Published August 2002. Accessed June 7, 2016.
5. Holmes LB, Wyszynski DF. North American antiepileptic drug pregnancy registry. Epilepsia. 2004;45(11):1465.
6. Tomson T, Battino D, Craig J, et al; ILAE Commission on Therapeutic Strategies. Pregnancy registries: differences, similarities, and possible harmonization. Epilepsia. 2010;51(5):909-915.
7. Cohen LS, Viguera AC, McInerney KA, et al. Establishment of the National Pregnancy Registry for Atypical Antipsychotics. J Clin Psychiatry. 2015;76(7):986-989.
8. Holmes LB, Wyszynski DF, Lieberman E. The AED (antiepileptic drug) pregnancy registry: a 6-year experience. Arch Neurol. 2004;61(5):673-678.
9. Cohen LS, Viguera AC, McInerney KA, et al. Reproductive safety of second-generation antipsychotics: current data from the Massachusetts General Hospital National Pregnancy Registry for Atypical Antipsychotics. Am J Psychiatry. 2016;173(3):263-270.
10. McBride WG. Thalidomide and congenital abnormalities. Lancet. 1961;2(7216):1358.
11. Wyszynski DF, Nambisan M, Surve T, et al; Antiepileptic Drug Pregnancy Registry. Increased rate of major malformations in offspring exposed to valproate during pregnancy. Neurology. 2005;64(6):961-965.
In December 2014, the FDA issued draft guidance for sweeping changes to labeling of pharmaceutical treatments in regard to pregnancy and lactation information. These changes are now in effect for use in practice.1 The undertaking has been years in the making, and is truly ambitious.
The outdated system of letter categories (A, B, C, D, X) falls short of clinical needs in several ways:
- the quality and volume of data can be lacking
- comparative risk is not described
- using letters can led to oversimplification or, in some cases, exaggeration of risk and safety (Box).
Other drawbacks include infrequent updating of information and omission of information about baseline rates of reproductive-related adverse events, to provide a more meaningful context for risk assessment.
A note before we continue discussion of labeling: Recognize that pregnancy itself is inherently risky; poor outcomes are, regrettably, not uncommon. The rate of birth defects in the United States is approximately 3%, and obstetric complications, such as prematurity, are common.2,3
New system described
The new labeling content has been described in the FDA’s Pregnancy and Lactation Labeling Rule (also called the “final rule”), issued in December 2014. For each medication, there will be subsections in the labeling:
- Pregnancy
- Lactation
- Females and Males of Reproductive Potential.
In addition, FDA instructions now state that labeling:
- must be updated when new information becomes available
- needs to include evaluation of human data that becomes available mainly after the drug is approved
- needs to include information about the background rates of adverse events related to reproduction.
Labeling in pregnancy. As an example, the “Pregnancy” section of every label contains 3 subsections, all of great clinical importance. First is information about pregnancy exposure registries, with a listing of scientifically acceptable registries (if a registry is available for that drug) and contact information; this section focuses on the high value of data that are systematically and prospectively collected. The second summarizes risk associated with the drug during pregnancy, based on available human, animal, and pharmacologic data. Third is a discussion of clinical considerations.
Need for appropriate controls. Psychiatric disorders increase the risk of pregnancy complications, and often are associated with variables that might increase the risk of a poor pregnancy outcome. For example, a patient who has a psychiatric disorder might be less likely to seek prenatal care, take a prenatal vitamin, and sleep and eat well; she also might use alcohol, tobacco, or other substances of abuse.
The medical literature on the reproductive safety of psychotropic medications is fraught with confounding variables other than the medications themselves. These include variables that, taken alone, might confer a poorer outcome on the fetus or newborn of a pregnant or lactating woman who has a psychiatric illness (to the extent that she uses psychotropics during a pregnancy), compared with what would be seen in (1) a healthy woman who is not taking such medication or (2) the general population.
On the new labels, detailed statements on human data include information from clinical trials, pregnancy exposure registries, and epidemiologic studies. Labels are also to include:
- incidence of adverse events
- effect of dosage
- effect of duration of exposure
- effect of gestational timing of exposure.
The labels emphasize quantifying risk relative to the risk of the same outcome in infants born to women who have not been exposed to the particular drug, but who have the disease or condition for which the drug is indicated (ie, appropriate controls).
Clinical considerations are to include information on the following related to the specific medication (when that information is known):
- more information for prescribers, to further risk-benefit counseling
- disease-associated maternal-fetal risks
- dosage adjustments during pregnancy and postpartum
- maternal adverse reactions
- fetal and neonatal adverse reactions
- labor and delivery.
Clearly, this overdue shift in providing information regarding reproductive safety has the potential to inform clinicians and patients in a meaningful way about the risks and benefits of specific treatments during pregnancy and lactation. Translating that information into practice is daunting, however.
Important aspects of implementation
Pregnancy exposure registries will play a crucial role. For most medications, no systematic registry has been established; to do so, rigorous methodology is required to acquire prospective data and account for confounding variables.4 Appropriate control groups also are required to yield data that are useful and interpretable. Primary outcomes require verification, such as review of medical records. Last, registries must be well-conducted and therefore adequately funded, yet labeling changes have not been accompanied by funding requirements set forth by regulators to pharmaceutical manufacturers.
Labeling must be updated continually. Furthermore, it is unclear who will review data for precision and comprehensiveness.
Data need to be understandable to health care providers across disciplines and to patients with varying levels of education for the label to have a meaningful impact on clinical care.
As noted, there is no mandate for funding the meticulous pharmacovigilance required to provide definitive data for labeling. It is unclear if the potential benefits of the new labeling can be reaped without adequate financing of the pharmacovigilance mechanisms required to inform patients adequately.
Role of pregnancy registries
Over the past 2 decades, pregnancy registries have emerged as a rapid, systematic means of collecting important reproductive safety data on the risk for major malformations after prenatal exposure to a medication or a class of medications.5,6 Such registries enhance the rigor of available cohort studies and other analyses of reproductive safety data that have been derived from large administrative databases.
NPRAA and NPRAD. Recently, the National Pregnancy Registry for Atypical Antipsychotics (NPRAA) and the National Pregnancy Registry for Antidepressants (NPRAD) were established in an effort to obtain reproductive safety data about fetal exposure to second-generation antipsychotics (SGAs) and to newer antidepressants.7 Based at Massachusetts General Hospital in Boston, NPRAA and NPRAD systematically and prospectively evaluate the risk of malformations among infants who have been exposed in utero to an SGA or an antidepressant.
The structure of both registries are the same, modeled after the North American Antiepileptic Drug Registry.5,8 Data are collected prospectively from pregnant women, age 18 to 45, by means of 3 telephone interviews conducted proximate to enrollment, at 7 months’ gestation, and at 2 or 3 months’ postpartum.
Participants include (1) pregnant women who have a history of fetal exposure to an SGA or an antidepressant, or both, and (2) a comparison group of non-exposed pregnant women who have a history of a psychiatric illness. Authorization for release of medical records is obtained for obstetric care, labor and delivery, and neonatal care (≤6 months of age).
Information on the presence of major malformations is abstracted from the medical record, along with other data on neonatal and maternal health outcomes. Identified cases of a congenital malformation are sent to a dysmorphologist, who has been blinded to drug exposure, for final adjudication. Release of findings is dictated by a governing Scientific Advisory Board.
Results so far. Results are available from the NPRAA.9 As of December 2014, 487 women were enrolled: 353 who used an SGA and 134 comparison women. Medical records were obtained for 82.2% of participants. A total of 303 women completed the study and were eligible for inclusion in the analysis. Findings include:
- Of 214 live births with first-trimester exposure to an SGA, 3 major malformations were confirmed. In the control group (n = 89), 1 major malformation was confirmed
- The absolute risk of a major malformation was 1.4% for an exposed infant and 1.1% for an unexposed infant
- The odds ratio for a major malformation, comparing exposed infants with unexposed infants, was 1.25 (95% CI, 0.13–12.19).
It is reasonable, therefore, to conclude that, as a class, SGAs are not major teratogens. Although the confidence intervals around the odds ratio estimate remain wide, with the probability for change over the course of the study, it is unlikely that risk will rise to the level of known major teratogens, such as valproate and thalidomide.10,11
Help with decision-making
Given recent FDA guidance about the importance of pregnancy registries (www.fda.gov/pregnancyregistries), such carefully collected data might help clinicians and patients make informed choices about treatment. Future efforts of NPRAA and NPRAD will focus on sustaining growth in enrollment of participants so that the reproductive safety of SGAs and newer antidepressants can be delineated more clearly.
Last, you can refer potential participants to NPRAA and NPRAD by calling 1-866-961-2388. More information is available at www.womensmentalhealth.org.
In December 2014, the FDA issued draft guidance for sweeping changes to labeling of pharmaceutical treatments in regard to pregnancy and lactation information. These changes are now in effect for use in practice.1 The undertaking has been years in the making, and is truly ambitious.
The outdated system of letter categories (A, B, C, D, X) falls short of clinical needs in several ways:
- the quality and volume of data can be lacking
- comparative risk is not described
- using letters can led to oversimplification or, in some cases, exaggeration of risk and safety (Box).
Other drawbacks include infrequent updating of information and omission of information about baseline rates of reproductive-related adverse events, to provide a more meaningful context for risk assessment.
A note before we continue discussion of labeling: Recognize that pregnancy itself is inherently risky; poor outcomes are, regrettably, not uncommon. The rate of birth defects in the United States is approximately 3%, and obstetric complications, such as prematurity, are common.2,3
New system described
The new labeling content has been described in the FDA’s Pregnancy and Lactation Labeling Rule (also called the “final rule”), issued in December 2014. For each medication, there will be subsections in the labeling:
- Pregnancy
- Lactation
- Females and Males of Reproductive Potential.
In addition, FDA instructions now state that labeling:
- must be updated when new information becomes available
- needs to include evaluation of human data that becomes available mainly after the drug is approved
- needs to include information about the background rates of adverse events related to reproduction.
Labeling in pregnancy. As an example, the “Pregnancy” section of every label contains 3 subsections, all of great clinical importance. First is information about pregnancy exposure registries, with a listing of scientifically acceptable registries (if a registry is available for that drug) and contact information; this section focuses on the high value of data that are systematically and prospectively collected. The second summarizes risk associated with the drug during pregnancy, based on available human, animal, and pharmacologic data. Third is a discussion of clinical considerations.
Need for appropriate controls. Psychiatric disorders increase the risk of pregnancy complications, and often are associated with variables that might increase the risk of a poor pregnancy outcome. For example, a patient who has a psychiatric disorder might be less likely to seek prenatal care, take a prenatal vitamin, and sleep and eat well; she also might use alcohol, tobacco, or other substances of abuse.
The medical literature on the reproductive safety of psychotropic medications is fraught with confounding variables other than the medications themselves. These include variables that, taken alone, might confer a poorer outcome on the fetus or newborn of a pregnant or lactating woman who has a psychiatric illness (to the extent that she uses psychotropics during a pregnancy), compared with what would be seen in (1) a healthy woman who is not taking such medication or (2) the general population.
On the new labels, detailed statements on human data include information from clinical trials, pregnancy exposure registries, and epidemiologic studies. Labels are also to include:
- incidence of adverse events
- effect of dosage
- effect of duration of exposure
- effect of gestational timing of exposure.
The labels emphasize quantifying risk relative to the risk of the same outcome in infants born to women who have not been exposed to the particular drug, but who have the disease or condition for which the drug is indicated (ie, appropriate controls).
Clinical considerations are to include information on the following related to the specific medication (when that information is known):
- more information for prescribers, to further risk-benefit counseling
- disease-associated maternal-fetal risks
- dosage adjustments during pregnancy and postpartum
- maternal adverse reactions
- fetal and neonatal adverse reactions
- labor and delivery.
Clearly, this overdue shift in providing information regarding reproductive safety has the potential to inform clinicians and patients in a meaningful way about the risks and benefits of specific treatments during pregnancy and lactation. Translating that information into practice is daunting, however.
Important aspects of implementation
Pregnancy exposure registries will play a crucial role. For most medications, no systematic registry has been established; to do so, rigorous methodology is required to acquire prospective data and account for confounding variables.4 Appropriate control groups also are required to yield data that are useful and interpretable. Primary outcomes require verification, such as review of medical records. Last, registries must be well-conducted and therefore adequately funded, yet labeling changes have not been accompanied by funding requirements set forth by regulators to pharmaceutical manufacturers.
Labeling must be updated continually. Furthermore, it is unclear who will review data for precision and comprehensiveness.
Data need to be understandable to health care providers across disciplines and to patients with varying levels of education for the label to have a meaningful impact on clinical care.
As noted, there is no mandate for funding the meticulous pharmacovigilance required to provide definitive data for labeling. It is unclear if the potential benefits of the new labeling can be reaped without adequate financing of the pharmacovigilance mechanisms required to inform patients adequately.
Role of pregnancy registries
Over the past 2 decades, pregnancy registries have emerged as a rapid, systematic means of collecting important reproductive safety data on the risk for major malformations after prenatal exposure to a medication or a class of medications.5,6 Such registries enhance the rigor of available cohort studies and other analyses of reproductive safety data that have been derived from large administrative databases.
NPRAA and NPRAD. Recently, the National Pregnancy Registry for Atypical Antipsychotics (NPRAA) and the National Pregnancy Registry for Antidepressants (NPRAD) were established in an effort to obtain reproductive safety data about fetal exposure to second-generation antipsychotics (SGAs) and to newer antidepressants.7 Based at Massachusetts General Hospital in Boston, NPRAA and NPRAD systematically and prospectively evaluate the risk of malformations among infants who have been exposed in utero to an SGA or an antidepressant.
The structure of both registries are the same, modeled after the North American Antiepileptic Drug Registry.5,8 Data are collected prospectively from pregnant women, age 18 to 45, by means of 3 telephone interviews conducted proximate to enrollment, at 7 months’ gestation, and at 2 or 3 months’ postpartum.
Participants include (1) pregnant women who have a history of fetal exposure to an SGA or an antidepressant, or both, and (2) a comparison group of non-exposed pregnant women who have a history of a psychiatric illness. Authorization for release of medical records is obtained for obstetric care, labor and delivery, and neonatal care (≤6 months of age).
Information on the presence of major malformations is abstracted from the medical record, along with other data on neonatal and maternal health outcomes. Identified cases of a congenital malformation are sent to a dysmorphologist, who has been blinded to drug exposure, for final adjudication. Release of findings is dictated by a governing Scientific Advisory Board.
Results so far. Results are available from the NPRAA.9 As of December 2014, 487 women were enrolled: 353 who used an SGA and 134 comparison women. Medical records were obtained for 82.2% of participants. A total of 303 women completed the study and were eligible for inclusion in the analysis. Findings include:
- Of 214 live births with first-trimester exposure to an SGA, 3 major malformations were confirmed. In the control group (n = 89), 1 major malformation was confirmed
- The absolute risk of a major malformation was 1.4% for an exposed infant and 1.1% for an unexposed infant
- The odds ratio for a major malformation, comparing exposed infants with unexposed infants, was 1.25 (95% CI, 0.13–12.19).
It is reasonable, therefore, to conclude that, as a class, SGAs are not major teratogens. Although the confidence intervals around the odds ratio estimate remain wide, with the probability for change over the course of the study, it is unlikely that risk will rise to the level of known major teratogens, such as valproate and thalidomide.10,11
Help with decision-making
Given recent FDA guidance about the importance of pregnancy registries (www.fda.gov/pregnancyregistries), such carefully collected data might help clinicians and patients make informed choices about treatment. Future efforts of NPRAA and NPRAD will focus on sustaining growth in enrollment of participants so that the reproductive safety of SGAs and newer antidepressants can be delineated more clearly.
Last, you can refer potential participants to NPRAA and NPRAD by calling 1-866-961-2388. More information is available at www.womensmentalhealth.org.
1. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Pregnancy, lactation, and reproductive potential: labeling for human prescription drug and biological products—content and format: guidance for industry. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM425398.pdf. Published December 2014. Accessed June 7, 2016.
2. Centers for Disease Control and Prevention. Birth defects. http://www.cdc.gov/ncbddd/birthdefects/facts.html. Updated September 21, 2005. Accessed June 7, 2016.
3. Centers for Disease Control and Prevention. Preterm birth. http://www.cdc.gov/reproductivehealth/maternalinfanthealth/pretermbirth.htm. Updated December 4, 2015. Accessed June 7, 2016.
4. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Guidance for industry: establishing pregnancy exposure registries. http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/WomensHealthResearch/UCM133332.pdf. Published August 2002. Accessed June 7, 2016.
5. Holmes LB, Wyszynski DF. North American antiepileptic drug pregnancy registry. Epilepsia. 2004;45(11):1465.
6. Tomson T, Battino D, Craig J, et al; ILAE Commission on Therapeutic Strategies. Pregnancy registries: differences, similarities, and possible harmonization. Epilepsia. 2010;51(5):909-915.
7. Cohen LS, Viguera AC, McInerney KA, et al. Establishment of the National Pregnancy Registry for Atypical Antipsychotics. J Clin Psychiatry. 2015;76(7):986-989.
8. Holmes LB, Wyszynski DF, Lieberman E. The AED (antiepileptic drug) pregnancy registry: a 6-year experience. Arch Neurol. 2004;61(5):673-678.
9. Cohen LS, Viguera AC, McInerney KA, et al. Reproductive safety of second-generation antipsychotics: current data from the Massachusetts General Hospital National Pregnancy Registry for Atypical Antipsychotics. Am J Psychiatry. 2016;173(3):263-270.
10. McBride WG. Thalidomide and congenital abnormalities. Lancet. 1961;2(7216):1358.
11. Wyszynski DF, Nambisan M, Surve T, et al; Antiepileptic Drug Pregnancy Registry. Increased rate of major malformations in offspring exposed to valproate during pregnancy. Neurology. 2005;64(6):961-965.
1. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Pregnancy, lactation, and reproductive potential: labeling for human prescription drug and biological products—content and format: guidance for industry. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM425398.pdf. Published December 2014. Accessed June 7, 2016.
2. Centers for Disease Control and Prevention. Birth defects. http://www.cdc.gov/ncbddd/birthdefects/facts.html. Updated September 21, 2005. Accessed June 7, 2016.
3. Centers for Disease Control and Prevention. Preterm birth. http://www.cdc.gov/reproductivehealth/maternalinfanthealth/pretermbirth.htm. Updated December 4, 2015. Accessed June 7, 2016.
4. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologic Evaluation and Research (CBER). Guidance for industry: establishing pregnancy exposure registries. http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/WomensHealthResearch/UCM133332.pdf. Published August 2002. Accessed June 7, 2016.
5. Holmes LB, Wyszynski DF. North American antiepileptic drug pregnancy registry. Epilepsia. 2004;45(11):1465.
6. Tomson T, Battino D, Craig J, et al; ILAE Commission on Therapeutic Strategies. Pregnancy registries: differences, similarities, and possible harmonization. Epilepsia. 2010;51(5):909-915.
7. Cohen LS, Viguera AC, McInerney KA, et al. Establishment of the National Pregnancy Registry for Atypical Antipsychotics. J Clin Psychiatry. 2015;76(7):986-989.
8. Holmes LB, Wyszynski DF, Lieberman E. The AED (antiepileptic drug) pregnancy registry: a 6-year experience. Arch Neurol. 2004;61(5):673-678.
9. Cohen LS, Viguera AC, McInerney KA, et al. Reproductive safety of second-generation antipsychotics: current data from the Massachusetts General Hospital National Pregnancy Registry for Atypical Antipsychotics. Am J Psychiatry. 2016;173(3):263-270.
10. McBride WG. Thalidomide and congenital abnormalities. Lancet. 1961;2(7216):1358.
11. Wyszynski DF, Nambisan M, Surve T, et al; Antiepileptic Drug Pregnancy Registry. Increased rate of major malformations in offspring exposed to valproate during pregnancy. Neurology. 2005;64(6):961-965.

Dietary and medical management of recurrent nephrolithiasis
Nephrolithiasis is common and often recurs. This review focuses on measures to prevent recurrent stone formation. Some measures apply to all patients, and some apply to specific types of stones.
COMMON AND INCREASING
According to data from the 2007–2010 National Health and Nutrition Examination Survey, the prevalence of nephrolithiasis in the United States was 10.6% in men and 7.1% in women. On average, 1 in 11 Americans will develop kidney stones at least once in their lifetime.1
By race and sex, white men have the highest incidence of nephrolithiasis and Asian women have the lowest. It is less common before age 20 and peaks in incidence in the third and fourth decades of life.
The prevalence has steadily increased in the past few decades (Table 1),1,2 but the reasons are not clear. The trend may be due to changes in diet and lifestyle, increasing prevalence of obesity and diabetes, migration from rural to urban areas, and global warming, with higher temperature resulting in dehydration and high urinary concentration of calcium and other stone-forming salts.3 Nephrolithiasis is now recognized as a systemic disorder associated with chronic kidney disease, bone loss and fractures, increased risk of coronary artery disease, hypertension, type 2 diabetes mellitus, and metabolic syndrome (Table 2).4–7

Without medical treatment, the 5-year recurrence rate is high, ranging from 35% to 50% after an initial stone event.8 Annual medical costs of care for kidney stones in the United States exceed $4.5 billion, with additional costs from missed work. Therefore, this condition has a considerable economic and social burden, which underscores the importance of prevention.9
MOST STONES CONTAIN CALCIUM
About 80% of kidney stones in adults contain calcium, and calcium oxalate stones are more common than calcium phosphate stones. Uric acid and struvite stones account for 5% to 15%, and cystine, protease inhibitor, triamterene, 2,8-dihydroxyadenine (2,8-DHA) and xanthine stones each account for less than 1%.10
Stones form when the urinary concentration of stone-forming salts, which is inversely proportional to urine volume, is higher than their saturation point, which is affected by urine pH. Acidic urine (low pH) predisposes to the formation of uric acid and cystine stones, whereas alkaline urine (high pH) favors calcium phosphate stones.
INCREASED FLUID INTAKE FOR ALL
High fluid intake, enough to produce at least 2.5 L of urine per day, should be the initial therapy to prevent stone recurrence.11
Borghi et al12 randomly assigned 199 patients who had a first calcium stone to high oral fluid intake or no intervention and followed them prospectively for 5 years. The recurrence rate was 12% in the treated group and 27% in the control group. Another study, in patients who had undergone shock wave lithotripsy, found a recurrence rate of 8% in those randomized to increase fluid intake to achieve urine output greater than 2.5 L/day, compared with 56% in those assigned to no treatment.13
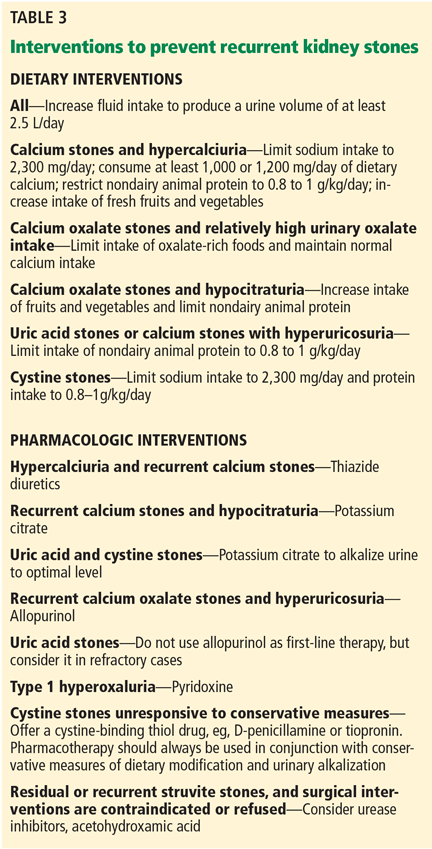
Certain beverages increase the risk of stones and should be avoided. Sugar-sweetened noncola soda and punch are associated with a 33% higher risk of kidney stones, and cola sodas are associated with a 23% higher risk.14 Prospective studies have shown that the consumption of coffee, beer, wine, and orange juice is associated with a lower likelihood of stone formation.13,15
Table 3 is a brief summary of the dietary and pharmacologic interventions in the management of recurrent nephrolithiasis.
PREVENTING CALCIUM OXALATE STONES
Major urinary risk factors associated with calcium oxalate stones are hypercalciuria, hyperoxaluria, hyperuricosuria, hypocitraturia, and low urine volume.16 Preventing calcium stones therefore depends on reducing the urinary concentration of calcium and oxalate, increasing urinary levels of inhibitors such as citrate, and increasing urine volume.
Reducing calcium excretion
Hypercalciuria has been traditionally defined as 24-hour urinary calcium excretion greater than 300 mg/day in men, greater than 250 mg in women, or greater than 4 mg/kg in men or women.17 It is a graded risk factor, and the cut points used in published research and clinical laboratories vary substantially. Some institutions use the same value for hypercalciuria in both sexes, eg, greater than 200 mg/day.18
Excessive sodium intake is the most common cause of hypercalciuria. Systemic conditions such as primary hyperparathyroidism, sarcoidosis, and renal tubular acidosis also cause hypercalciuria but are uncommon.19 Management depends on the underlying cause and includes dietary modifications and pharmacologic therapy.
Dietary modifications have a pivotal role in the management of recurrent stones that are due to hypercalciuria.
Dietary calcium should not be restricted, since calcium reduces the excretion of urinary oxalate by decreasing intestinal absorption of oxalate. Guidelines from the American Urological Association recommend a daily calcium intake of 1,000 to 1,200 mg.11–20 Moreover, restriction of dietary calcium to less than 800 mg/day (the current recommended daily allowance for adults) can lead to negative calcium balance and bone loss.
Sodium intake also influences hypercalciuria. Calcium is reabsorbed passively in the proximal tubule due to the concentration gradient created by active reabsorption of sodium. A high sodium intake causes volume expansion, leading to a decrease in proximal sodium and calcium reabsorption and enhancing calcium excretion. A low-sodium diet (80–100 mmol/day, or 1,800–2,300 mg/day) is recommended. This enhances proximal sodium and passive calcium absorption and leads to a decrease in calcium excretion.21
Dietary protein increases the acid load by production of sulfuric acid and leads to hypercalciuria by its action on bone and kidney. Animal protein has a higher content of sulfur and generates a higher acid load compared with vegetable protein and has been associated with an increased incidence of stone formation, at least in men.20,22 Borghi et al23 reported that the combination of restricted intake of animal protein (52 g/day), restricted salt intake (50 mmol, or 2,900 mg/day of sodium chloride), and normal calcium intake (30 mmol/day, or 1,200 mg/day) was associated with a lower incidence of stone recurrence in men with hypercalciuria compared with traditional low-calcium intake (10 mmol, or 400 mg/day). Patients should therefore be advised to avoid excessive intake of animal protein.
Increasing the dietary intake of fruits and vegetables as in the Dietary Approach to Stop Hypertension (DASH) diet is beneficial and reduces the risk of stone recurrence, mainly by increasing citrate excretion.24
Pharmacologic therapy in hypercalciuria. Thiazide diuretics are the mainstay of pharmacotherapy for preventing recurrent stones in patients with idiopathic hypercalciuria. They reduce the risk of stone recurrence by about 50%, as reported in a recent meta-analysis that looked at five trials comparing thiazide diuretics with placebo.25 They lower calcium excretion by causing volume depletion, thereby increasing proximal sodium and passive calcium reabsorption.
Chlorthalidone and hydrochlorothiazide are the thiazides commonly used to treat hypercalciuria. The dosage is titrated to the urinary calcium excretion, and a common mistake is to use doses that are too low. They are usually started at 25 mg/day, but often require an increase to 50 to 100 mg/day for adequate lowering of urinary calcium.
Care should be taken to avoid hypokalemia. If it occurs, it can be corrected by adding the potassium-sparing diuretic amiloride (5–10 mg/day), which increases calcium reabsorption in collecting ducts or, in patients with hypocitraturia, potassium citrate-potassium bicarbonate. (Sodium salts should be avoided, since they increase renal calcium excretion.)26
Management of hypercalciuria with metabolic causes, which include primary hyperparathyroidism and chronic acidemia. Patients who have hypercalciuria from primary hyperparathyroidism are treated with parathyroidectomy.27 Chronic metabolic acidosis causes hypercalciuria by loss of bone calcium and hypocitraturia by increasing active proximal absorption of citrate. Potassium citrate or potassium bicarbonate is used to prevent stones in such patients; sodium salts should be avoided.28
Reducing oxalate excretion
Hyperoxaluria has traditionally been defined as urinary oxalate excretion of more than 45 mg/day. However, the optimal cutoff point for urinary oxalate excretion is unclear, as is the optimal cutoff for hypercalciuria. The risk of stone formation has been shown to increase with oxalate excretion even above 25 mg/day, which is within the normal limit.18
Idiopathic hyperoxaluria. High dietary oxalate intake, especially when associated with low calcium intake, leads to idiopathic hyperoxaluria. However, the contribution of abnormal endogenous oxalate metabolism is uncertain. Ingested calcium binds to oxalate in the intestinal tract and reduces both the absorption of intestinal oxalate absorption and the excretion of urinary oxalate.29 High dietary oxalate intake has usually been regarded as a major risk factor for kidney stones.
Taylor and Curhan,30 in a prospective study, reported a mild increase in the risk of stones in the highest quintile of dietary oxalate intake compared with the lowest quintile for men (relative risk [RR] 1.22, 95% confidence interval [CI] 1.03–1.45) and older women (RR 1.21, 95% CI 1.01–1.44). They also demonstrated that eating eight or more servings of spinach per month compared with fewer than one serving per month was associated with a similar increase of stone risk in men (RR 1.30, 95% CI 1.08–1.58) and older women (RR 1.34 95% CI 1.1–1.64). In contrast, spinach and dietary oxalate intake did not increase the risk of nephrolithiasis in young women. The authors concluded that the risk associated with oxalate intake was modest, and their data did not support the contention that dietary oxalate is a major risk factor for kidney stones.
Higher oxalate intake increases urinary oxalate excretion and presumably the risk of nephrolithiasis. Limiting dietary oxalate to prevent stones is recommended if habitually high dietary intake of oxalate is identified or follow-up urine measurements show a decrease in oxalate excretion.31 Foods rich in oxalate include spinach, rhubarb, nuts, legumes, cocoa, okra, and chocolate.
The DASH diet, which is high in fruits and vegetables, moderate in low-fat dairy products, and low in animal protein, is an effective dietary alternative and has been associated with a lower risk of calcium oxalate stones.24 Consuming fruits and vegetables increases the excretion of urinary citrate, which is an inhibitor of stone formation. Also, it has been proposed that the DASH diet contains unknown factors that reduce stone risk.
Taylor et al32 prospectively examined the relationship between the DASH diet and the incidence of kidney stones and found that the diet significantly reduced the risk of kidney stones. The relative risks of occurrence of kidney stones in participants in the highest quintile of the DASH score (a measure of adherence to the DASH diet) compared with the lowest quintile were 0.55 (95% CI 0.46–0.65) for men, 0.58 (95% CI 0.49–0.68) for older women, and 0.60 (95% CI 0.52–0.70) for younger women, which the authors characterized as “a marked decrease in kidney stone risk.”
Vitamin C intake should be restricted to 90 mg/day in patients who have a history of calcium oxalate stones. Urivetzky et al33 found that urinary oxalate excretion increased by 6 to 13 mg/day at doses of ascorbic acid greater than 500 mg.
Pyridoxine (vitamin B6), a coenzyme of alanine-glyoxylate aminotransferase (AGT), increases the conversion of glyoxylate to glycine instead of oxalate and is used in the treatment of type 1 primary hyperoxaluria (see below).34 However, its effect in preventing stones in idiopathic hyperoxaluria is not well known, and it has not been studied in a randomized controlled trial. In a prospective study, Curhan et al35 reported that high intake of pyridoxine (> 40 mg/day) was associated with a lower risk of stone formation in women, but no such benefit was found in men.
Enteric hyperoxaluria. About 90% of dietary oxalate binds to calcium in the small intestine and is excreted in the stool. The remaining 10% is absorbed in the colon and is secreted in urine. Hyperoxaluria is frequently seen with fat malabsorption from inflammatory bowel disease, short gut syndrome, and gastric bypass surgery. In these conditions, excess fat binds to dietary calcium, leading to increased absorption of free oxalate in the colon.36
Treatment is directed at decreasing intestinal oxalate absorption and should include high fluid intake and oral calcium supplements. Calcium carbonate or citrate causes precipitation of oxalate in the intestinal lumen and is prescribed as 1 to 4 g in three to four divided doses, always with meals. Calcium citrate is preferred over calcium carbonate in stone-formers because of the benefit of citrate and calcium citrate’s higher solubility and greater effectiveness in the presence of achlorhydria.37 Patients should be advised to avoid foods high in oxalate and fat.
Primary hyperoxaluria is caused by inherited inborn errors of glyoxylate metabolism that cause overproduction of oxalate and urinary oxalate excretion above 135 to 270 mg/day.
Type 1 primary hyperoxaluria is the most common (accounting for 90% of cases) and is caused by reduced activity of hepatic peroxisomal AGT.
Type 2 is from a deficiency of glyoxylate reductase-hydroxypyruvate reductase (GRHPR).
Type 3 is from mutations in the HOGA1 gene, which codes for the liver-specific mitochondrial 4-hydroxy-2-oxoglutarate aldolase enzyme involved in degradation of hydroxyproline to pyruvate and glyoxalate.38
High fluid intake to produce a urinary volume of 3 L/day reduces intratubular oxalate deposition and should be encouraged. Potassium citrate (0.15 mg/kg), oral phosphate supplements (30–40 mg/kg of orthophosphate), and magnesium oxide (500 mg/day/m2) inhibit precipitation of calcium oxalate in the urine.39,40 Pyridoxine, a coenzyme of AGT, increases the conversion of glyoxylate to glycine instead of oxalate and is prescribed at a starting dose of 5 mg/kg (which can be titrated up to 20 mg/kg if there is no response) in patients with type 1 primary hyperoxaluria. About 50% of patients with type 1 respond successfully to pyridoxine, and a 3- to 6-month trial should be given in all patients in this category.34 AGT is present only in hepatocytes, and GRHPR is found in multiple tissues; therefore, combined liver-kidney transplant is the treatment of choice in patients with type 1 primary hyperoxaluria, whereas isolated kidney transplant is recommended in patients with type 2.41
Reducing uric acid excretion
Hyperuricosuria is defined as uric acid excretion of greater than 800 mg/day in men and greater than 750 mg/day in women.
The association of hyperuricosuria with increased risk of calcium oxalate stone formation is controversial. Curhan and Taylor,18 in a cross-sectional study of 3,350 men and women, reported that there was no difference in mean 24-hour uric acid excretion in individuals with and without a history of stones.
The mechanism by which uric acid leads to calcium oxalate stones is not completely known and could be the “salting out” of calcium oxalate from the urine.42
Dietary purine restriction, ie, limiting intake of nondairy animal protein to 0.8 to 1 g/kg/day, is the initial dietary intervention.11 Allopurinol is the alternative approach if the patient is not compliant or if dietary restriction fails.43
In a study by Ettinger et al,44 60 patients with hyperuricosuria and normocalciuria were randomized to receive allopurinol (100 mg three times daily) or a placebo. The allopurinol group had a rate of calculus events of 0.12 per patient per year, compared with 0.26 in the placebo group.
Increasing citrate excretion
Hypocitraturia is a well-known risk factor for the formation of kidney stones. It is usually defined as a citrate excretion of less than 320 mg/day for adults.
Citrate prevents formation of calcium crystals by binding to calcium, thereby lowering the concentration of calcium oxalate below the saturation point.45
Diet therapy. Patients with calcium oxalate stones and hypocitraturia should be encouraged to increase their intake of fruits and vegetables, which enhances urinary citrate excretion, and to limit their intake of nondairy animal protein.11
The use of citrus products in preventing stones in patients with hypocitraturia is controversial, however, and needs to be studied more.
One study46 demonstrated that lemon juice was beneficial in hypocitraturic nephrolithiasis: 4 oz/day of lemon juice concentrate in the form of lemonade was associated with an increase in urinary citrate excretion to 346 mg/day from 142 mg/day in 11 of 12 patients who participated.
Odvina47 compared the effects of orange juice with those of lemonade on the acid-base profile and urinary stone risk under controlled metabolic conditions in 13 volunteers. Orange juice was reported to have greater alkalinizing and citraturic effects and was associated with lower calculated calcium oxalate supersaturation compared with lemonade.
Lemonade therapy may be used as adjunctive treatment in patients who do not comply with or cannot tolerate alkali therapy. However, we advise caution about recommending citrus products, as they can increase oxalate excretion.
Pharmacotherapy includes alkali therapy. Barcelo et al48 compared the effects of potassium citrate and placebo in 57 patients with calcium oxalate stones and hypocitraturia. Patients treated with potassium citrate had a rate of stone formation of 0.1 event per patient per year, compared with 1.1 in the placebo group.
Many forms of alkaline citrate are available. Potassium citrate is preferred over sodium citrate since the latter may increase urine calcium excretion.49 Treatment is usually started at 30 mEq/day and is titrated to a maximal dose of 60 mEq/day for a urinary citrate excretion greater than 500 mg/day.
Common side effects are abdominal bloating and hyperkalemia (especially with renal insufficiency), and in such cases sodium-based alkali, sodium citrate, or sodium bicarbonate can be prescribed.
PREVENTING CALCIUM PHOSPHATE STONES
Risk factors for calcium phosphate stones are similar to those for calcium oxalate stones (other than hyperoxaluria), but calcium phosphate stones are formed in alkaline urine (usually urine pH > 6.0), often the result of distal renal tubular acidosis. Preventive measures are similar to those for calcium oxalate stones.
Alkali therapy should be used with caution because of its effect on urinary pH and the risk of precipitation of calcium phosphate crystals.50 Use of potassium citrate was found to be associated with increases in both urinary citrate excretion and calcium phosphate supersaturation in hypercalciuric stone-forming rats.51 It is therefore challenging to manage patients with calcium phosphate stones and hypocitraturia. Alkali administration in this setting may diminish the formation of new stones by correcting hypocitraturia, but at the same time it may increase the likelihood of calcium phosphate stone formation by increasing the urinary pH. When the urine pH increases to above 6.5 with no significant change in urine citrate or urine calcium excretion, we recommend stopping alkali therapy.
PREVENTING URIC ACID STONES
Clinical conditions associated with uric acid stones include metabolic syndrome, diabetes mellitus, gout, chronic diarrheal illness, and conditions that increase tissue turnover and uric acid production, such as malignancies. Other risk factors for uric acid stone formation are low urine volume, low uric pH, and hyperuricosuria.
Abnormally acidic urine is the most common risk factor. Metabolic syndrome and diabetes mellitus reduce ammonia production, resulting in a lower urinary pH, which predisposes to uric acid stone formation. Chronic diarrhea also acidifies the urine by loss of bicarbonate. Similarly, in gout, the predisposing factor in uric acid stone formation is the persistently acidic urine due to impaired ammonium excretion.52 Uric acid precipitates to form uric acid stones in a low urinary pH even with normal excretion rates of 600 to 800 mg/day and a urinary volume of 1 to 1.5 L.53
Therefore, apart from increasing fluid intake, urinary alkalization is the cornerstone of management of uric acid stones. Potassium citrate is the preferred alkali salt and is started at a dose of 30 mEq/day for a goal urinary pH of 6 to 6.5.47
Patients with hyperuricosuria are also advised to restrict their protein intake to no more than 0.8 to 1 mg/kg/day.
If the above measures fail, patients are treated with a xanthine oxidase inhibitor, ie, allopurinol or febuxostat, even if their uric acid excretion is normal.54
PREVENTING STRUVITE STONES
Struvite stones contain magnesium ammonium phosphate and are due to chronic upper urinary tract infection with urea-splitting bacteria such as Proteus, Klebsiella, Pseudomonas, and enterococci. Urea hydrolysis releases hydroxyl ions, resulting in alkaline urine that promotes struvite stone formation. Early detection and treatment are important, since struvite stones are associated with morbidity and rapid progression.
Medical treatment of struvite stones is usually unsuccessful, and the patient is referred to a urologist for surgical removal of the stones, the gold standard treatment.55 Long-term use of culture-specific antibiotics to prevent new stone growth is not well studied. Medical therapy by itself is preferred in patients who refuse stone removal or cannot tolerate it. Urease inhibitors such as acetohydroxamic acid have been successful in preventing or slowing stone growth, but their use is limited by frequent side effects such as nausea, headache, rash, and thrombophlebitis.56
CYSTINE STONES
Cystine stones occur in people with inherited defects of renal tubular and intestinal transport of cysteine and dibasic amino acids that cause excessive excretion of urinary cystine, ie, 480 to 3,600 mg/day.
Cystine is formed from two cysteine molecules linked by a disulfide bond. The solubility of cystine is pH-dependent, with increased solubility at higher urinary pH. The goal is to maintain a urinary cystine concentration below its solubility level by keeping the cystine concentration below 243 mg/L and the urine cystine supersaturation (the ratio of the urine cysteine concentration to the cysteine solubility in the same sample) less than 0.6.57 Therapy is aimed at increasing daily urinary volume to 3 L and urine alkalization to pH above 7, in order to increase cystine solubility by 300%.58
Overnight dehydration should be prevented, and patients should be encouraged to wake up at least once a night to void and drink additional water. Sodium restriction to 100 mmol/day (2,300 mg/day) and moderate protein restriction to 0.8 to 1 g/kg/day are associated with decreased cystine excretion, but long-term studies demonstrating their benefit in preventing cystine stones are lacking.59
A thiol-containing drug, eg, D-penicillamine (0.5–2 g/day) or tiopronin (400–1,200 mg/day), should be added to the conservative measures if they have not been effective for 3 months or if there is history of noncompliance.60 Thiol-containing drugs have a sulfhydryl group that reduces the disulfide bond, and they form soluble disulfide cysteine-drug complexes with greater ability to solubilize cystine in alkaline urine. They must always be used in conjunction with fluid and alkali therapy.61
Both drugs have severe and common adverse effects including leukopenia, aplastic anemia, fever, rash, arthritis, hepatotoxicity, pyridoxine deficiency, and proteinuria (membranous nephropathy). However, tiopronin seems to have a lesser incidence of side effects.62 Regular monitoring of complete blood cell counts, liver enzymes, and urine protein should be done.
Captopril contains a sulfhydryl group, and the captopril-cysteine disulfide is more soluble than cysteine alone. The amount of captopril that appears in the urine is low, and doses of 150 mg/day are usually required to reduce cysteine excretion, which can lead to hypotension. The efficacy of captopril in treating cystine stones is unproven, and this drug is used only if patients cannot tolerate other thiol-containing drugs.63
- Scales CD Jr, Smith AC, Hanley JM, Saigal CS; Urologic Diseases in America Project. Prevalence of kidney stones in the United States. Eur Urol 2012; 62:160–165.
- Stamatelou KK, Francis ME, Jones CA, Nyberg LM Jr, Curhan GC. Time trends in reported prevalence of kidney stones in the United States: 1976–1994. Kidney Int 2003; 63:1817–1823.
- Romero V, Akpinar H, Assimos DG. Kidney stones: a global picture of prevalence, incidence, and associated risk factors. Rev Urol 2010; 12:e86–e96.
- Sakhaee K, Maalouf NM, Kumar R, Pasch A, Moe OW. Nephrolithiasis-associated bone disease: pathogenesis and treatment options. Kidney Int 2011; 79:393–403.
- Sakhaee K. Nephrolithiasis as a systemic disorder. Curr Opin Nephrol Hypertens 2008; 17:304–309.
- Hamano S, Nakatsu H, Suzuki N, Tomioka S, Tanaka M, Murakami S. Kidney stone disease and risk factors for coronary heart disease. Int J Urol 2005; 12:859–863.
- Ritz E. Metabolic syndrome: an emerging threat to renal function. Clin J Am Soc Nephrol 2007; 2:869–871.
- Uribarri J, Oh MS, Carroll HJ. The first kidney stone. Ann Intern Med 1989; 111:1006–1009.
- Saigal CS, Joyce G, Timilsina AR; Urologic Diseases in America Project. Direct and indirect costs of nephrolithiasis in an employed population: opportunity for disease management? Kidney Int 2005; 68:1808–1814.
- Moe OW. Kidney stones: pathophysiology and medical management. Lancet 2006; 367:333–344.
- Pearle MS, Goldfarb DS, Assimos DG, et al; American Urological Assocation. Medical management of kidney stones: AUA guideline. J Urol 2014; 192:316–324.
- Borghi L, Meschi T, Amato F, Briganti A, Novarini A, Giannini A. Urinary volume, water and recurrences in idiopathic calcium nephrolithiasis: a 5-year randomized prospective study. J Urol 1996; 155:839–843.
- Sarica K, Inal Y, Erturhan S, Yagci F. The effect of calcium channel blockers on stone regrowth and recurrence after shock wave lithotripsy. Urol Res 2006; 34:184–189.
- Ferraro PM, Taylor EN, Gambaro G, Curhan GC. Soda and other beverages and the risk of kidney stones. Clin J Am Soc Nephrol 2013; 8:1389–1395.
- Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Beverage use and risk for kidney stones in women. Ann Intern Med 1998; 128:534–540.
- Pak CY, Britton F, Peterson R, et al. Ambulatory evaluation of nephrolithiasis. Classification, clinical presentation and diagnostic criteria. Am J Med 1980; 69:19–30.
- Hall PM. Nephrolithiasis: treatment, causes, and prevention. Cleve Clin J Med 2009; 76:583–591.
- Curhan GC, Taylor EN. 24-h uric acid excretion and the risk of kidney stones. Kidney Int 2008; 73:489–496.
- Coe FL, Evan A, Worcester E. Kidney stone disease. J Clin Invest 2005; 115:2598–2608.
- Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med 1993; 328:833–838.
- Muldowney FP, Freaney R, Moloney MF. Importance of dietary sodium in the hypercalciuria syndrome. Kidney Int 1982; 22:292–296.
- Breslau NA, Brinkley L, Hill KD, Pak CY. Relationship of animal protein-rich diet to kidney stone formation and calcium metabolism. J Clin Endocrinol Metab 1988; 66:140–146.
- Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med 2002; 346:77–84.
- Noori N, Honarkar E, Goldfarb DS, et al. Urinary lithogenic risk profile in recurrent stone formers with hyperoxaluria: a randomized controlled trial comparing DASH (Dietary Approaches to Stop Hypertension)-style and low-oxalate diets. Am J Kidney Dis 2014; 63:456–463.
- Fink HA, Wilt TJ, Eidman KE, et al. Medical management to prevent recurrent nephrolithiasis in adults: a systematic review for an American College of Physicians Clinical Guideline. Ann Intern Med 2013; 158:535–543.
- Alon U, Costanzo LS, Chan JC. Additive hypocalciuric effects of amiloride and hydrochlorothiazide in patients treated with calcitriol. Miner Electrolyte Metab 1984; 10:379–386.
- Corbetta S, Baccarelli A, Aroldi A, et al. Risk factors associated to kidney stones in primary hyperparathyroidism. J Endocrinol Invest 2005; 28:122–128.
- Haymann JP. Metabolic disorders: stones as first clinical manifestation of significant diseases. World J Urol 2015; 33:187–192.
- Jaeger P, Portmann L, Jacquet AF, Burckhardt P. Influence of the calcium content of the diet on the incidence of mild hyperoxaluria in idiopathic renal stone formers. Am J Nephrol 1985; 5:40–44.
- Taylor EN, Curhan GC. Oxalate intake and the risk for nephrolithiasis. J Am Soc Nephrol 2007; 18:2198–2204.
- Lieske JC, Tremaine WJ, De Simone C, et al. Diet, but not oral probiotics, effectively reduces urinary oxalate excretion and calcium oxalate supersaturation. Kidney Int 2010; 78:1178–1185.
- Taylor EN, Fung TT, Curhan GC. DASH-style diet associates with reduced risk for kidney stones. J Am Soc Nephrol 2009; 20:2253–2259.
- Urivetzky M, Kessaris D, Smith AD. Ascorbic acid overdosing: a risk factor for calcium oxalate nephrolithiasis. J Urol 1992; 147:1215–1218.
- Hoyer-Kuhn H, Kohbrok S, Volland R, et al. Vitamin B6 in primary hyperoxaluria I: first prospective trial after 40 years of practice. Clin J Am Soc Nephrol 2014; 9:468–477.
- Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Intake of vitamins B6 and C and the risk of kidney stones in women. J Am Soc Nephrol 1999; 10:840–845.
- Parks JH, Worcester EM, O'Connor RC, Coe FL. Urine stone risk factors in nephrolithiasis patients with and without bowel disease. Kidney Int 2003; 63:255–265.
- Hess B, Jost C, Zipperle L, Takkinen R, Jaeger P. High-calcium intake abolishes hyperoxaluria and reduces urinary crystallization during a 20-fold normal oxalate load in humans. Nephrol Dial Transplant 1998; 13:2241–2247.
- Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int 2009; 75:1264–1271.
- Cochat P, Hulton SA, Acquaviva C, et al; OxalEurope. Primary hyperoxaluria type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant 2012; 27:1729–1736.
- Leumann E, Hoppe B, Neuhaus T. Management of primary hyperoxaluria: efficacy of oral citrate administration. Pediatr Nephrol 1993; 7:207–211.
- Bergstralh EJ, Monico CG, Lieske JC, et al; IPHR Investigators. Transplantation outcomes in primary hyperoxaluria. Am J Transplant 2010; 10:2493–2501.
- Grover PK, Marshall VR, Ryall RL. Dissolved urate salts out calcium oxalate in undiluted human urine in vitro: implications for calcium oxalate stone genesis. Chem Biol 2003; 10:271–278.
- Coe FL, Parks JH. Hyperuricosuria and calcium nephrolithiasis. Urol Clin North Am 1981; 8:227–244.
- Ettinger B, Tang A, Citron JT, Livermore B, Williams T. Randomized trial of allopurinol in the prevention of calcium oxalate calculi. N Engl J Med 1986; 315:1386–1389.
- Zuckerman JM, Assimos DG. Hypocitraturia: pathophysiology and medical management. Rev Urol 2009; 11:134–144.
- Seltzer MA, Low RK, McDonald M, Shami GS, Stoller ML. Dietary manipulation with lemonade to treat hypocitraturic calcium nephrolithiasis. J Urol 1996; 156:907–909.
- Odvina CV. Comparative value of orange juice versus lemonade in reducing stone-forming risk. Clin J Am Soc Nephrol 2006; 1:1269–1274.
- Barcelo P, Wuhl O, Servitge E, Rousaud A, Pak CY. Randomized double-blind study of potassium citrate in idiopathic hypocitraturic calcium nephrolithiasis. J Urol 1993; 150:1761–1764.
- Lemann J Jr, Gray RW, Pleuss JA. Potassium bicarbonate, but not sodium bicarbonate, reduces urinary calcium excretion and improves calcium balance in healthy men. Kidney Int 1989; 35:688–695.
- Gault MH, Chafe LL, Morgan JM, et al. Comparison of patients with idiopathic calcium phosphate and calcium oxalate stones. Medicine (Baltimore) 1991; 70:345–359.
- Krieger NS, Asplin JR, Frick KK, et al. Effect of potassium citrate on calcium phosphate stones in a model of hypercalciuria. J Am Soc Nephrol 2015; 26:3001–3008.
- Falls WF Jr. Comparison of urinary acidification and ammonium excretion in normal and gouty subjects. Metabolism 1972; 21:433–445.
- Coe FL, Parks JH, Asplin JR. The pathogenesis and treatment of kidney stones. N Engl J Med 1992; 327:1141–1152.
- Kenny JE, Goldfarb DS. Update on the pathophysiology and management of uric acid renal stones. Curr Rheumatol Rep 2010; 12:125–129.
- Preminger GM, Assimos DG, Lingeman JE, Nakada SY, Pearle MS, Wolf JS Jr (AUA Nephrolithiasis Guideline Panel). Chapter 1: AUA guideline on management of staghorn calculi: diagnosis and treatment recommendations. J Urol 2005; 173:1991–2000.
- Williams JJ, Rodman JS, Peterson CM. A randomized double-blind study of acetohydroxamic acid in struvite nephrolithiasis. N Engl J Med 1984; 311:760–764.
- Nakagawa Y, Asplin JR, Goldfarb DS, Parks JH, Coe FL. Clinical use of cystine supersaturation measurements. J Urol 2000; 164:1481–1485.
- Palacın MGP, Nunes V, Gasparini P. Cystinuria. In: Shriver CR, editor. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill; 2001:4909–4932.
- Goldfarb DS, Coe FL, Asplin JR. Urinary cystine excretion and capacity in patients with cystinuria. Kidney Int 2006; 69:1041–1047.
- Barbey F, Joly D, Rieu P, Mejean A, Daudon M, Jungers P. Medical treatment of cystinuria: critical reappraisal of long-term results. J Urol 2000; 163:1419–1423.
- Asplin DM, Asplin JR. The Interaction of thiol drugs and urine pH in the treatment of cystinuria. J Urol 2013; 189:2147–2151.
- Habib GS, Saliba W, Nashashibi M, Armali Z. Penicillamine and nephrotic syndrome. Eur J Intern Med 2006; 17:343–348.
- Sloand JA, Izzo JL Jr. Captopril reduces urinary cystine excretion in cystinuria. Arch Intern Med 1987; 147:1409–1412.
Nephrolithiasis is common and often recurs. This review focuses on measures to prevent recurrent stone formation. Some measures apply to all patients, and some apply to specific types of stones.
COMMON AND INCREASING
According to data from the 2007–2010 National Health and Nutrition Examination Survey, the prevalence of nephrolithiasis in the United States was 10.6% in men and 7.1% in women. On average, 1 in 11 Americans will develop kidney stones at least once in their lifetime.1
By race and sex, white men have the highest incidence of nephrolithiasis and Asian women have the lowest. It is less common before age 20 and peaks in incidence in the third and fourth decades of life.
The prevalence has steadily increased in the past few decades (Table 1),1,2 but the reasons are not clear. The trend may be due to changes in diet and lifestyle, increasing prevalence of obesity and diabetes, migration from rural to urban areas, and global warming, with higher temperature resulting in dehydration and high urinary concentration of calcium and other stone-forming salts.3 Nephrolithiasis is now recognized as a systemic disorder associated with chronic kidney disease, bone loss and fractures, increased risk of coronary artery disease, hypertension, type 2 diabetes mellitus, and metabolic syndrome (Table 2).4–7
Without medical treatment, the 5-year recurrence rate is high, ranging from 35% to 50% after an initial stone event.8 Annual medical costs of care for kidney stones in the United States exceed $4.5 billion, with additional costs from missed work. Therefore, this condition has a considerable economic and social burden, which underscores the importance of prevention.9
MOST STONES CONTAIN CALCIUM
About 80% of kidney stones in adults contain calcium, and calcium oxalate stones are more common than calcium phosphate stones. Uric acid and struvite stones account for 5% to 15%, and cystine, protease inhibitor, triamterene, 2,8-dihydroxyadenine (2,8-DHA) and xanthine stones each account for less than 1%.10
Stones form when the urinary concentration of stone-forming salts, which is inversely proportional to urine volume, is higher than their saturation point, which is affected by urine pH. Acidic urine (low pH) predisposes to the formation of uric acid and cystine stones, whereas alkaline urine (high pH) favors calcium phosphate stones.
INCREASED FLUID INTAKE FOR ALL
High fluid intake, enough to produce at least 2.5 L of urine per day, should be the initial therapy to prevent stone recurrence.11
Borghi et al12 randomly assigned 199 patients who had a first calcium stone to high oral fluid intake or no intervention and followed them prospectively for 5 years. The recurrence rate was 12% in the treated group and 27% in the control group. Another study, in patients who had undergone shock wave lithotripsy, found a recurrence rate of 8% in those randomized to increase fluid intake to achieve urine output greater than 2.5 L/day, compared with 56% in those assigned to no treatment.13
Certain beverages increase the risk of stones and should be avoided. Sugar-sweetened noncola soda and punch are associated with a 33% higher risk of kidney stones, and cola sodas are associated with a 23% higher risk.14 Prospective studies have shown that the consumption of coffee, beer, wine, and orange juice is associated with a lower likelihood of stone formation.13,15
Table 3 is a brief summary of the dietary and pharmacologic interventions in the management of recurrent nephrolithiasis.
PREVENTING CALCIUM OXALATE STONES
Major urinary risk factors associated with calcium oxalate stones are hypercalciuria, hyperoxaluria, hyperuricosuria, hypocitraturia, and low urine volume.16 Preventing calcium stones therefore depends on reducing the urinary concentration of calcium and oxalate, increasing urinary levels of inhibitors such as citrate, and increasing urine volume.
Reducing calcium excretion
Hypercalciuria has been traditionally defined as 24-hour urinary calcium excretion greater than 300 mg/day in men, greater than 250 mg in women, or greater than 4 mg/kg in men or women.17 It is a graded risk factor, and the cut points used in published research and clinical laboratories vary substantially. Some institutions use the same value for hypercalciuria in both sexes, eg, greater than 200 mg/day.18
Excessive sodium intake is the most common cause of hypercalciuria. Systemic conditions such as primary hyperparathyroidism, sarcoidosis, and renal tubular acidosis also cause hypercalciuria but are uncommon.19 Management depends on the underlying cause and includes dietary modifications and pharmacologic therapy.
Dietary modifications have a pivotal role in the management of recurrent stones that are due to hypercalciuria.
Dietary calcium should not be restricted, since calcium reduces the excretion of urinary oxalate by decreasing intestinal absorption of oxalate. Guidelines from the American Urological Association recommend a daily calcium intake of 1,000 to 1,200 mg.11–20 Moreover, restriction of dietary calcium to less than 800 mg/day (the current recommended daily allowance for adults) can lead to negative calcium balance and bone loss.
Sodium intake also influences hypercalciuria. Calcium is reabsorbed passively in the proximal tubule due to the concentration gradient created by active reabsorption of sodium. A high sodium intake causes volume expansion, leading to a decrease in proximal sodium and calcium reabsorption and enhancing calcium excretion. A low-sodium diet (80–100 mmol/day, or 1,800–2,300 mg/day) is recommended. This enhances proximal sodium and passive calcium absorption and leads to a decrease in calcium excretion.21
Dietary protein increases the acid load by production of sulfuric acid and leads to hypercalciuria by its action on bone and kidney. Animal protein has a higher content of sulfur and generates a higher acid load compared with vegetable protein and has been associated with an increased incidence of stone formation, at least in men.20,22 Borghi et al23 reported that the combination of restricted intake of animal protein (52 g/day), restricted salt intake (50 mmol, or 2,900 mg/day of sodium chloride), and normal calcium intake (30 mmol/day, or 1,200 mg/day) was associated with a lower incidence of stone recurrence in men with hypercalciuria compared with traditional low-calcium intake (10 mmol, or 400 mg/day). Patients should therefore be advised to avoid excessive intake of animal protein.
Increasing the dietary intake of fruits and vegetables as in the Dietary Approach to Stop Hypertension (DASH) diet is beneficial and reduces the risk of stone recurrence, mainly by increasing citrate excretion.24
Pharmacologic therapy in hypercalciuria. Thiazide diuretics are the mainstay of pharmacotherapy for preventing recurrent stones in patients with idiopathic hypercalciuria. They reduce the risk of stone recurrence by about 50%, as reported in a recent meta-analysis that looked at five trials comparing thiazide diuretics with placebo.25 They lower calcium excretion by causing volume depletion, thereby increasing proximal sodium and passive calcium reabsorption.
Chlorthalidone and hydrochlorothiazide are the thiazides commonly used to treat hypercalciuria. The dosage is titrated to the urinary calcium excretion, and a common mistake is to use doses that are too low. They are usually started at 25 mg/day, but often require an increase to 50 to 100 mg/day for adequate lowering of urinary calcium.
Care should be taken to avoid hypokalemia. If it occurs, it can be corrected by adding the potassium-sparing diuretic amiloride (5–10 mg/day), which increases calcium reabsorption in collecting ducts or, in patients with hypocitraturia, potassium citrate-potassium bicarbonate. (Sodium salts should be avoided, since they increase renal calcium excretion.)26
Management of hypercalciuria with metabolic causes, which include primary hyperparathyroidism and chronic acidemia. Patients who have hypercalciuria from primary hyperparathyroidism are treated with parathyroidectomy.27 Chronic metabolic acidosis causes hypercalciuria by loss of bone calcium and hypocitraturia by increasing active proximal absorption of citrate. Potassium citrate or potassium bicarbonate is used to prevent stones in such patients; sodium salts should be avoided.28
Reducing oxalate excretion
Hyperoxaluria has traditionally been defined as urinary oxalate excretion of more than 45 mg/day. However, the optimal cutoff point for urinary oxalate excretion is unclear, as is the optimal cutoff for hypercalciuria. The risk of stone formation has been shown to increase with oxalate excretion even above 25 mg/day, which is within the normal limit.18
Idiopathic hyperoxaluria. High dietary oxalate intake, especially when associated with low calcium intake, leads to idiopathic hyperoxaluria. However, the contribution of abnormal endogenous oxalate metabolism is uncertain. Ingested calcium binds to oxalate in the intestinal tract and reduces both the absorption of intestinal oxalate absorption and the excretion of urinary oxalate.29 High dietary oxalate intake has usually been regarded as a major risk factor for kidney stones.
Taylor and Curhan,30 in a prospective study, reported a mild increase in the risk of stones in the highest quintile of dietary oxalate intake compared with the lowest quintile for men (relative risk [RR] 1.22, 95% confidence interval [CI] 1.03–1.45) and older women (RR 1.21, 95% CI 1.01–1.44). They also demonstrated that eating eight or more servings of spinach per month compared with fewer than one serving per month was associated with a similar increase of stone risk in men (RR 1.30, 95% CI 1.08–1.58) and older women (RR 1.34 95% CI 1.1–1.64). In contrast, spinach and dietary oxalate intake did not increase the risk of nephrolithiasis in young women. The authors concluded that the risk associated with oxalate intake was modest, and their data did not support the contention that dietary oxalate is a major risk factor for kidney stones.
Higher oxalate intake increases urinary oxalate excretion and presumably the risk of nephrolithiasis. Limiting dietary oxalate to prevent stones is recommended if habitually high dietary intake of oxalate is identified or follow-up urine measurements show a decrease in oxalate excretion.31 Foods rich in oxalate include spinach, rhubarb, nuts, legumes, cocoa, okra, and chocolate.
The DASH diet, which is high in fruits and vegetables, moderate in low-fat dairy products, and low in animal protein, is an effective dietary alternative and has been associated with a lower risk of calcium oxalate stones.24 Consuming fruits and vegetables increases the excretion of urinary citrate, which is an inhibitor of stone formation. Also, it has been proposed that the DASH diet contains unknown factors that reduce stone risk.
Taylor et al32 prospectively examined the relationship between the DASH diet and the incidence of kidney stones and found that the diet significantly reduced the risk of kidney stones. The relative risks of occurrence of kidney stones in participants in the highest quintile of the DASH score (a measure of adherence to the DASH diet) compared with the lowest quintile were 0.55 (95% CI 0.46–0.65) for men, 0.58 (95% CI 0.49–0.68) for older women, and 0.60 (95% CI 0.52–0.70) for younger women, which the authors characterized as “a marked decrease in kidney stone risk.”
Vitamin C intake should be restricted to 90 mg/day in patients who have a history of calcium oxalate stones. Urivetzky et al33 found that urinary oxalate excretion increased by 6 to 13 mg/day at doses of ascorbic acid greater than 500 mg.
Pyridoxine (vitamin B6), a coenzyme of alanine-glyoxylate aminotransferase (AGT), increases the conversion of glyoxylate to glycine instead of oxalate and is used in the treatment of type 1 primary hyperoxaluria (see below).34 However, its effect in preventing stones in idiopathic hyperoxaluria is not well known, and it has not been studied in a randomized controlled trial. In a prospective study, Curhan et al35 reported that high intake of pyridoxine (> 40 mg/day) was associated with a lower risk of stone formation in women, but no such benefit was found in men.
Enteric hyperoxaluria. About 90% of dietary oxalate binds to calcium in the small intestine and is excreted in the stool. The remaining 10% is absorbed in the colon and is secreted in urine. Hyperoxaluria is frequently seen with fat malabsorption from inflammatory bowel disease, short gut syndrome, and gastric bypass surgery. In these conditions, excess fat binds to dietary calcium, leading to increased absorption of free oxalate in the colon.36
Treatment is directed at decreasing intestinal oxalate absorption and should include high fluid intake and oral calcium supplements. Calcium carbonate or citrate causes precipitation of oxalate in the intestinal lumen and is prescribed as 1 to 4 g in three to four divided doses, always with meals. Calcium citrate is preferred over calcium carbonate in stone-formers because of the benefit of citrate and calcium citrate’s higher solubility and greater effectiveness in the presence of achlorhydria.37 Patients should be advised to avoid foods high in oxalate and fat.
Primary hyperoxaluria is caused by inherited inborn errors of glyoxylate metabolism that cause overproduction of oxalate and urinary oxalate excretion above 135 to 270 mg/day.
Type 1 primary hyperoxaluria is the most common (accounting for 90% of cases) and is caused by reduced activity of hepatic peroxisomal AGT.
Type 2 is from a deficiency of glyoxylate reductase-hydroxypyruvate reductase (GRHPR).
Type 3 is from mutations in the HOGA1 gene, which codes for the liver-specific mitochondrial 4-hydroxy-2-oxoglutarate aldolase enzyme involved in degradation of hydroxyproline to pyruvate and glyoxalate.38
High fluid intake to produce a urinary volume of 3 L/day reduces intratubular oxalate deposition and should be encouraged. Potassium citrate (0.15 mg/kg), oral phosphate supplements (30–40 mg/kg of orthophosphate), and magnesium oxide (500 mg/day/m2) inhibit precipitation of calcium oxalate in the urine.39,40 Pyridoxine, a coenzyme of AGT, increases the conversion of glyoxylate to glycine instead of oxalate and is prescribed at a starting dose of 5 mg/kg (which can be titrated up to 20 mg/kg if there is no response) in patients with type 1 primary hyperoxaluria. About 50% of patients with type 1 respond successfully to pyridoxine, and a 3- to 6-month trial should be given in all patients in this category.34 AGT is present only in hepatocytes, and GRHPR is found in multiple tissues; therefore, combined liver-kidney transplant is the treatment of choice in patients with type 1 primary hyperoxaluria, whereas isolated kidney transplant is recommended in patients with type 2.41
Reducing uric acid excretion
Hyperuricosuria is defined as uric acid excretion of greater than 800 mg/day in men and greater than 750 mg/day in women.
The association of hyperuricosuria with increased risk of calcium oxalate stone formation is controversial. Curhan and Taylor,18 in a cross-sectional study of 3,350 men and women, reported that there was no difference in mean 24-hour uric acid excretion in individuals with and without a history of stones.
The mechanism by which uric acid leads to calcium oxalate stones is not completely known and could be the “salting out” of calcium oxalate from the urine.42
Dietary purine restriction, ie, limiting intake of nondairy animal protein to 0.8 to 1 g/kg/day, is the initial dietary intervention.11 Allopurinol is the alternative approach if the patient is not compliant or if dietary restriction fails.43
In a study by Ettinger et al,44 60 patients with hyperuricosuria and normocalciuria were randomized to receive allopurinol (100 mg three times daily) or a placebo. The allopurinol group had a rate of calculus events of 0.12 per patient per year, compared with 0.26 in the placebo group.
Increasing citrate excretion
Hypocitraturia is a well-known risk factor for the formation of kidney stones. It is usually defined as a citrate excretion of less than 320 mg/day for adults.
Citrate prevents formation of calcium crystals by binding to calcium, thereby lowering the concentration of calcium oxalate below the saturation point.45
Diet therapy. Patients with calcium oxalate stones and hypocitraturia should be encouraged to increase their intake of fruits and vegetables, which enhances urinary citrate excretion, and to limit their intake of nondairy animal protein.11
The use of citrus products in preventing stones in patients with hypocitraturia is controversial, however, and needs to be studied more.
One study46 demonstrated that lemon juice was beneficial in hypocitraturic nephrolithiasis: 4 oz/day of lemon juice concentrate in the form of lemonade was associated with an increase in urinary citrate excretion to 346 mg/day from 142 mg/day in 11 of 12 patients who participated.
Odvina47 compared the effects of orange juice with those of lemonade on the acid-base profile and urinary stone risk under controlled metabolic conditions in 13 volunteers. Orange juice was reported to have greater alkalinizing and citraturic effects and was associated with lower calculated calcium oxalate supersaturation compared with lemonade.
Lemonade therapy may be used as adjunctive treatment in patients who do not comply with or cannot tolerate alkali therapy. However, we advise caution about recommending citrus products, as they can increase oxalate excretion.
Pharmacotherapy includes alkali therapy. Barcelo et al48 compared the effects of potassium citrate and placebo in 57 patients with calcium oxalate stones and hypocitraturia. Patients treated with potassium citrate had a rate of stone formation of 0.1 event per patient per year, compared with 1.1 in the placebo group.
Many forms of alkaline citrate are available. Potassium citrate is preferred over sodium citrate since the latter may increase urine calcium excretion.49 Treatment is usually started at 30 mEq/day and is titrated to a maximal dose of 60 mEq/day for a urinary citrate excretion greater than 500 mg/day.
Common side effects are abdominal bloating and hyperkalemia (especially with renal insufficiency), and in such cases sodium-based alkali, sodium citrate, or sodium bicarbonate can be prescribed.
PREVENTING CALCIUM PHOSPHATE STONES
Risk factors for calcium phosphate stones are similar to those for calcium oxalate stones (other than hyperoxaluria), but calcium phosphate stones are formed in alkaline urine (usually urine pH > 6.0), often the result of distal renal tubular acidosis. Preventive measures are similar to those for calcium oxalate stones.
Alkali therapy should be used with caution because of its effect on urinary pH and the risk of precipitation of calcium phosphate crystals.50 Use of potassium citrate was found to be associated with increases in both urinary citrate excretion and calcium phosphate supersaturation in hypercalciuric stone-forming rats.51 It is therefore challenging to manage patients with calcium phosphate stones and hypocitraturia. Alkali administration in this setting may diminish the formation of new stones by correcting hypocitraturia, but at the same time it may increase the likelihood of calcium phosphate stone formation by increasing the urinary pH. When the urine pH increases to above 6.5 with no significant change in urine citrate or urine calcium excretion, we recommend stopping alkali therapy.
PREVENTING URIC ACID STONES
Clinical conditions associated with uric acid stones include metabolic syndrome, diabetes mellitus, gout, chronic diarrheal illness, and conditions that increase tissue turnover and uric acid production, such as malignancies. Other risk factors for uric acid stone formation are low urine volume, low uric pH, and hyperuricosuria.
Abnormally acidic urine is the most common risk factor. Metabolic syndrome and diabetes mellitus reduce ammonia production, resulting in a lower urinary pH, which predisposes to uric acid stone formation. Chronic diarrhea also acidifies the urine by loss of bicarbonate. Similarly, in gout, the predisposing factor in uric acid stone formation is the persistently acidic urine due to impaired ammonium excretion.52 Uric acid precipitates to form uric acid stones in a low urinary pH even with normal excretion rates of 600 to 800 mg/day and a urinary volume of 1 to 1.5 L.53
Therefore, apart from increasing fluid intake, urinary alkalization is the cornerstone of management of uric acid stones. Potassium citrate is the preferred alkali salt and is started at a dose of 30 mEq/day for a goal urinary pH of 6 to 6.5.47
Patients with hyperuricosuria are also advised to restrict their protein intake to no more than 0.8 to 1 mg/kg/day.
If the above measures fail, patients are treated with a xanthine oxidase inhibitor, ie, allopurinol or febuxostat, even if their uric acid excretion is normal.54
PREVENTING STRUVITE STONES
Struvite stones contain magnesium ammonium phosphate and are due to chronic upper urinary tract infection with urea-splitting bacteria such as Proteus, Klebsiella, Pseudomonas, and enterococci. Urea hydrolysis releases hydroxyl ions, resulting in alkaline urine that promotes struvite stone formation. Early detection and treatment are important, since struvite stones are associated with morbidity and rapid progression.
Medical treatment of struvite stones is usually unsuccessful, and the patient is referred to a urologist for surgical removal of the stones, the gold standard treatment.55 Long-term use of culture-specific antibiotics to prevent new stone growth is not well studied. Medical therapy by itself is preferred in patients who refuse stone removal or cannot tolerate it. Urease inhibitors such as acetohydroxamic acid have been successful in preventing or slowing stone growth, but their use is limited by frequent side effects such as nausea, headache, rash, and thrombophlebitis.56
CYSTINE STONES
Cystine stones occur in people with inherited defects of renal tubular and intestinal transport of cysteine and dibasic amino acids that cause excessive excretion of urinary cystine, ie, 480 to 3,600 mg/day.
Cystine is formed from two cysteine molecules linked by a disulfide bond. The solubility of cystine is pH-dependent, with increased solubility at higher urinary pH. The goal is to maintain a urinary cystine concentration below its solubility level by keeping the cystine concentration below 243 mg/L and the urine cystine supersaturation (the ratio of the urine cysteine concentration to the cysteine solubility in the same sample) less than 0.6.57 Therapy is aimed at increasing daily urinary volume to 3 L and urine alkalization to pH above 7, in order to increase cystine solubility by 300%.58
Overnight dehydration should be prevented, and patients should be encouraged to wake up at least once a night to void and drink additional water. Sodium restriction to 100 mmol/day (2,300 mg/day) and moderate protein restriction to 0.8 to 1 g/kg/day are associated with decreased cystine excretion, but long-term studies demonstrating their benefit in preventing cystine stones are lacking.59
A thiol-containing drug, eg, D-penicillamine (0.5–2 g/day) or tiopronin (400–1,200 mg/day), should be added to the conservative measures if they have not been effective for 3 months or if there is history of noncompliance.60 Thiol-containing drugs have a sulfhydryl group that reduces the disulfide bond, and they form soluble disulfide cysteine-drug complexes with greater ability to solubilize cystine in alkaline urine. They must always be used in conjunction with fluid and alkali therapy.61
Both drugs have severe and common adverse effects including leukopenia, aplastic anemia, fever, rash, arthritis, hepatotoxicity, pyridoxine deficiency, and proteinuria (membranous nephropathy). However, tiopronin seems to have a lesser incidence of side effects.62 Regular monitoring of complete blood cell counts, liver enzymes, and urine protein should be done.
Captopril contains a sulfhydryl group, and the captopril-cysteine disulfide is more soluble than cysteine alone. The amount of captopril that appears in the urine is low, and doses of 150 mg/day are usually required to reduce cysteine excretion, which can lead to hypotension. The efficacy of captopril in treating cystine stones is unproven, and this drug is used only if patients cannot tolerate other thiol-containing drugs.63
Nephrolithiasis is common and often recurs. This review focuses on measures to prevent recurrent stone formation. Some measures apply to all patients, and some apply to specific types of stones.
COMMON AND INCREASING
According to data from the 2007–2010 National Health and Nutrition Examination Survey, the prevalence of nephrolithiasis in the United States was 10.6% in men and 7.1% in women. On average, 1 in 11 Americans will develop kidney stones at least once in their lifetime.1
By race and sex, white men have the highest incidence of nephrolithiasis and Asian women have the lowest. It is less common before age 20 and peaks in incidence in the third and fourth decades of life.
The prevalence has steadily increased in the past few decades (Table 1),1,2 but the reasons are not clear. The trend may be due to changes in diet and lifestyle, increasing prevalence of obesity and diabetes, migration from rural to urban areas, and global warming, with higher temperature resulting in dehydration and high urinary concentration of calcium and other stone-forming salts.3 Nephrolithiasis is now recognized as a systemic disorder associated with chronic kidney disease, bone loss and fractures, increased risk of coronary artery disease, hypertension, type 2 diabetes mellitus, and metabolic syndrome (Table 2).4–7
Without medical treatment, the 5-year recurrence rate is high, ranging from 35% to 50% after an initial stone event.8 Annual medical costs of care for kidney stones in the United States exceed $4.5 billion, with additional costs from missed work. Therefore, this condition has a considerable economic and social burden, which underscores the importance of prevention.9
MOST STONES CONTAIN CALCIUM
About 80% of kidney stones in adults contain calcium, and calcium oxalate stones are more common than calcium phosphate stones. Uric acid and struvite stones account for 5% to 15%, and cystine, protease inhibitor, triamterene, 2,8-dihydroxyadenine (2,8-DHA) and xanthine stones each account for less than 1%.10
Stones form when the urinary concentration of stone-forming salts, which is inversely proportional to urine volume, is higher than their saturation point, which is affected by urine pH. Acidic urine (low pH) predisposes to the formation of uric acid and cystine stones, whereas alkaline urine (high pH) favors calcium phosphate stones.
INCREASED FLUID INTAKE FOR ALL
High fluid intake, enough to produce at least 2.5 L of urine per day, should be the initial therapy to prevent stone recurrence.11
Borghi et al12 randomly assigned 199 patients who had a first calcium stone to high oral fluid intake or no intervention and followed them prospectively for 5 years. The recurrence rate was 12% in the treated group and 27% in the control group. Another study, in patients who had undergone shock wave lithotripsy, found a recurrence rate of 8% in those randomized to increase fluid intake to achieve urine output greater than 2.5 L/day, compared with 56% in those assigned to no treatment.13
Certain beverages increase the risk of stones and should be avoided. Sugar-sweetened noncola soda and punch are associated with a 33% higher risk of kidney stones, and cola sodas are associated with a 23% higher risk.14 Prospective studies have shown that the consumption of coffee, beer, wine, and orange juice is associated with a lower likelihood of stone formation.13,15
Table 3 is a brief summary of the dietary and pharmacologic interventions in the management of recurrent nephrolithiasis.
PREVENTING CALCIUM OXALATE STONES
Major urinary risk factors associated with calcium oxalate stones are hypercalciuria, hyperoxaluria, hyperuricosuria, hypocitraturia, and low urine volume.16 Preventing calcium stones therefore depends on reducing the urinary concentration of calcium and oxalate, increasing urinary levels of inhibitors such as citrate, and increasing urine volume.
Reducing calcium excretion
Hypercalciuria has been traditionally defined as 24-hour urinary calcium excretion greater than 300 mg/day in men, greater than 250 mg in women, or greater than 4 mg/kg in men or women.17 It is a graded risk factor, and the cut points used in published research and clinical laboratories vary substantially. Some institutions use the same value for hypercalciuria in both sexes, eg, greater than 200 mg/day.18
Excessive sodium intake is the most common cause of hypercalciuria. Systemic conditions such as primary hyperparathyroidism, sarcoidosis, and renal tubular acidosis also cause hypercalciuria but are uncommon.19 Management depends on the underlying cause and includes dietary modifications and pharmacologic therapy.
Dietary modifications have a pivotal role in the management of recurrent stones that are due to hypercalciuria.
Dietary calcium should not be restricted, since calcium reduces the excretion of urinary oxalate by decreasing intestinal absorption of oxalate. Guidelines from the American Urological Association recommend a daily calcium intake of 1,000 to 1,200 mg.11–20 Moreover, restriction of dietary calcium to less than 800 mg/day (the current recommended daily allowance for adults) can lead to negative calcium balance and bone loss.
Sodium intake also influences hypercalciuria. Calcium is reabsorbed passively in the proximal tubule due to the concentration gradient created by active reabsorption of sodium. A high sodium intake causes volume expansion, leading to a decrease in proximal sodium and calcium reabsorption and enhancing calcium excretion. A low-sodium diet (80–100 mmol/day, or 1,800–2,300 mg/day) is recommended. This enhances proximal sodium and passive calcium absorption and leads to a decrease in calcium excretion.21
Dietary protein increases the acid load by production of sulfuric acid and leads to hypercalciuria by its action on bone and kidney. Animal protein has a higher content of sulfur and generates a higher acid load compared with vegetable protein and has been associated with an increased incidence of stone formation, at least in men.20,22 Borghi et al23 reported that the combination of restricted intake of animal protein (52 g/day), restricted salt intake (50 mmol, or 2,900 mg/day of sodium chloride), and normal calcium intake (30 mmol/day, or 1,200 mg/day) was associated with a lower incidence of stone recurrence in men with hypercalciuria compared with traditional low-calcium intake (10 mmol, or 400 mg/day). Patients should therefore be advised to avoid excessive intake of animal protein.
Increasing the dietary intake of fruits and vegetables as in the Dietary Approach to Stop Hypertension (DASH) diet is beneficial and reduces the risk of stone recurrence, mainly by increasing citrate excretion.24
Pharmacologic therapy in hypercalciuria. Thiazide diuretics are the mainstay of pharmacotherapy for preventing recurrent stones in patients with idiopathic hypercalciuria. They reduce the risk of stone recurrence by about 50%, as reported in a recent meta-analysis that looked at five trials comparing thiazide diuretics with placebo.25 They lower calcium excretion by causing volume depletion, thereby increasing proximal sodium and passive calcium reabsorption.
Chlorthalidone and hydrochlorothiazide are the thiazides commonly used to treat hypercalciuria. The dosage is titrated to the urinary calcium excretion, and a common mistake is to use doses that are too low. They are usually started at 25 mg/day, but often require an increase to 50 to 100 mg/day for adequate lowering of urinary calcium.
Care should be taken to avoid hypokalemia. If it occurs, it can be corrected by adding the potassium-sparing diuretic amiloride (5–10 mg/day), which increases calcium reabsorption in collecting ducts or, in patients with hypocitraturia, potassium citrate-potassium bicarbonate. (Sodium salts should be avoided, since they increase renal calcium excretion.)26
Management of hypercalciuria with metabolic causes, which include primary hyperparathyroidism and chronic acidemia. Patients who have hypercalciuria from primary hyperparathyroidism are treated with parathyroidectomy.27 Chronic metabolic acidosis causes hypercalciuria by loss of bone calcium and hypocitraturia by increasing active proximal absorption of citrate. Potassium citrate or potassium bicarbonate is used to prevent stones in such patients; sodium salts should be avoided.28
Reducing oxalate excretion
Hyperoxaluria has traditionally been defined as urinary oxalate excretion of more than 45 mg/day. However, the optimal cutoff point for urinary oxalate excretion is unclear, as is the optimal cutoff for hypercalciuria. The risk of stone formation has been shown to increase with oxalate excretion even above 25 mg/day, which is within the normal limit.18
Idiopathic hyperoxaluria. High dietary oxalate intake, especially when associated with low calcium intake, leads to idiopathic hyperoxaluria. However, the contribution of abnormal endogenous oxalate metabolism is uncertain. Ingested calcium binds to oxalate in the intestinal tract and reduces both the absorption of intestinal oxalate absorption and the excretion of urinary oxalate.29 High dietary oxalate intake has usually been regarded as a major risk factor for kidney stones.
Taylor and Curhan,30 in a prospective study, reported a mild increase in the risk of stones in the highest quintile of dietary oxalate intake compared with the lowest quintile for men (relative risk [RR] 1.22, 95% confidence interval [CI] 1.03–1.45) and older women (RR 1.21, 95% CI 1.01–1.44). They also demonstrated that eating eight or more servings of spinach per month compared with fewer than one serving per month was associated with a similar increase of stone risk in men (RR 1.30, 95% CI 1.08–1.58) and older women (RR 1.34 95% CI 1.1–1.64). In contrast, spinach and dietary oxalate intake did not increase the risk of nephrolithiasis in young women. The authors concluded that the risk associated with oxalate intake was modest, and their data did not support the contention that dietary oxalate is a major risk factor for kidney stones.
Higher oxalate intake increases urinary oxalate excretion and presumably the risk of nephrolithiasis. Limiting dietary oxalate to prevent stones is recommended if habitually high dietary intake of oxalate is identified or follow-up urine measurements show a decrease in oxalate excretion.31 Foods rich in oxalate include spinach, rhubarb, nuts, legumes, cocoa, okra, and chocolate.
The DASH diet, which is high in fruits and vegetables, moderate in low-fat dairy products, and low in animal protein, is an effective dietary alternative and has been associated with a lower risk of calcium oxalate stones.24 Consuming fruits and vegetables increases the excretion of urinary citrate, which is an inhibitor of stone formation. Also, it has been proposed that the DASH diet contains unknown factors that reduce stone risk.
Taylor et al32 prospectively examined the relationship between the DASH diet and the incidence of kidney stones and found that the diet significantly reduced the risk of kidney stones. The relative risks of occurrence of kidney stones in participants in the highest quintile of the DASH score (a measure of adherence to the DASH diet) compared with the lowest quintile were 0.55 (95% CI 0.46–0.65) for men, 0.58 (95% CI 0.49–0.68) for older women, and 0.60 (95% CI 0.52–0.70) for younger women, which the authors characterized as “a marked decrease in kidney stone risk.”
Vitamin C intake should be restricted to 90 mg/day in patients who have a history of calcium oxalate stones. Urivetzky et al33 found that urinary oxalate excretion increased by 6 to 13 mg/day at doses of ascorbic acid greater than 500 mg.
Pyridoxine (vitamin B6), a coenzyme of alanine-glyoxylate aminotransferase (AGT), increases the conversion of glyoxylate to glycine instead of oxalate and is used in the treatment of type 1 primary hyperoxaluria (see below).34 However, its effect in preventing stones in idiopathic hyperoxaluria is not well known, and it has not been studied in a randomized controlled trial. In a prospective study, Curhan et al35 reported that high intake of pyridoxine (> 40 mg/day) was associated with a lower risk of stone formation in women, but no such benefit was found in men.
Enteric hyperoxaluria. About 90% of dietary oxalate binds to calcium in the small intestine and is excreted in the stool. The remaining 10% is absorbed in the colon and is secreted in urine. Hyperoxaluria is frequently seen with fat malabsorption from inflammatory bowel disease, short gut syndrome, and gastric bypass surgery. In these conditions, excess fat binds to dietary calcium, leading to increased absorption of free oxalate in the colon.36
Treatment is directed at decreasing intestinal oxalate absorption and should include high fluid intake and oral calcium supplements. Calcium carbonate or citrate causes precipitation of oxalate in the intestinal lumen and is prescribed as 1 to 4 g in three to four divided doses, always with meals. Calcium citrate is preferred over calcium carbonate in stone-formers because of the benefit of citrate and calcium citrate’s higher solubility and greater effectiveness in the presence of achlorhydria.37 Patients should be advised to avoid foods high in oxalate and fat.
Primary hyperoxaluria is caused by inherited inborn errors of glyoxylate metabolism that cause overproduction of oxalate and urinary oxalate excretion above 135 to 270 mg/day.
Type 1 primary hyperoxaluria is the most common (accounting for 90% of cases) and is caused by reduced activity of hepatic peroxisomal AGT.
Type 2 is from a deficiency of glyoxylate reductase-hydroxypyruvate reductase (GRHPR).
Type 3 is from mutations in the HOGA1 gene, which codes for the liver-specific mitochondrial 4-hydroxy-2-oxoglutarate aldolase enzyme involved in degradation of hydroxyproline to pyruvate and glyoxalate.38
High fluid intake to produce a urinary volume of 3 L/day reduces intratubular oxalate deposition and should be encouraged. Potassium citrate (0.15 mg/kg), oral phosphate supplements (30–40 mg/kg of orthophosphate), and magnesium oxide (500 mg/day/m2) inhibit precipitation of calcium oxalate in the urine.39,40 Pyridoxine, a coenzyme of AGT, increases the conversion of glyoxylate to glycine instead of oxalate and is prescribed at a starting dose of 5 mg/kg (which can be titrated up to 20 mg/kg if there is no response) in patients with type 1 primary hyperoxaluria. About 50% of patients with type 1 respond successfully to pyridoxine, and a 3- to 6-month trial should be given in all patients in this category.34 AGT is present only in hepatocytes, and GRHPR is found in multiple tissues; therefore, combined liver-kidney transplant is the treatment of choice in patients with type 1 primary hyperoxaluria, whereas isolated kidney transplant is recommended in patients with type 2.41
Reducing uric acid excretion
Hyperuricosuria is defined as uric acid excretion of greater than 800 mg/day in men and greater than 750 mg/day in women.
The association of hyperuricosuria with increased risk of calcium oxalate stone formation is controversial. Curhan and Taylor,18 in a cross-sectional study of 3,350 men and women, reported that there was no difference in mean 24-hour uric acid excretion in individuals with and without a history of stones.
The mechanism by which uric acid leads to calcium oxalate stones is not completely known and could be the “salting out” of calcium oxalate from the urine.42
Dietary purine restriction, ie, limiting intake of nondairy animal protein to 0.8 to 1 g/kg/day, is the initial dietary intervention.11 Allopurinol is the alternative approach if the patient is not compliant or if dietary restriction fails.43
In a study by Ettinger et al,44 60 patients with hyperuricosuria and normocalciuria were randomized to receive allopurinol (100 mg three times daily) or a placebo. The allopurinol group had a rate of calculus events of 0.12 per patient per year, compared with 0.26 in the placebo group.
Increasing citrate excretion
Hypocitraturia is a well-known risk factor for the formation of kidney stones. It is usually defined as a citrate excretion of less than 320 mg/day for adults.
Citrate prevents formation of calcium crystals by binding to calcium, thereby lowering the concentration of calcium oxalate below the saturation point.45
Diet therapy. Patients with calcium oxalate stones and hypocitraturia should be encouraged to increase their intake of fruits and vegetables, which enhances urinary citrate excretion, and to limit their intake of nondairy animal protein.11
The use of citrus products in preventing stones in patients with hypocitraturia is controversial, however, and needs to be studied more.
One study46 demonstrated that lemon juice was beneficial in hypocitraturic nephrolithiasis: 4 oz/day of lemon juice concentrate in the form of lemonade was associated with an increase in urinary citrate excretion to 346 mg/day from 142 mg/day in 11 of 12 patients who participated.
Odvina47 compared the effects of orange juice with those of lemonade on the acid-base profile and urinary stone risk under controlled metabolic conditions in 13 volunteers. Orange juice was reported to have greater alkalinizing and citraturic effects and was associated with lower calculated calcium oxalate supersaturation compared with lemonade.
Lemonade therapy may be used as adjunctive treatment in patients who do not comply with or cannot tolerate alkali therapy. However, we advise caution about recommending citrus products, as they can increase oxalate excretion.
Pharmacotherapy includes alkali therapy. Barcelo et al48 compared the effects of potassium citrate and placebo in 57 patients with calcium oxalate stones and hypocitraturia. Patients treated with potassium citrate had a rate of stone formation of 0.1 event per patient per year, compared with 1.1 in the placebo group.
Many forms of alkaline citrate are available. Potassium citrate is preferred over sodium citrate since the latter may increase urine calcium excretion.49 Treatment is usually started at 30 mEq/day and is titrated to a maximal dose of 60 mEq/day for a urinary citrate excretion greater than 500 mg/day.
Common side effects are abdominal bloating and hyperkalemia (especially with renal insufficiency), and in such cases sodium-based alkali, sodium citrate, or sodium bicarbonate can be prescribed.
PREVENTING CALCIUM PHOSPHATE STONES
Risk factors for calcium phosphate stones are similar to those for calcium oxalate stones (other than hyperoxaluria), but calcium phosphate stones are formed in alkaline urine (usually urine pH > 6.0), often the result of distal renal tubular acidosis. Preventive measures are similar to those for calcium oxalate stones.
Alkali therapy should be used with caution because of its effect on urinary pH and the risk of precipitation of calcium phosphate crystals.50 Use of potassium citrate was found to be associated with increases in both urinary citrate excretion and calcium phosphate supersaturation in hypercalciuric stone-forming rats.51 It is therefore challenging to manage patients with calcium phosphate stones and hypocitraturia. Alkali administration in this setting may diminish the formation of new stones by correcting hypocitraturia, but at the same time it may increase the likelihood of calcium phosphate stone formation by increasing the urinary pH. When the urine pH increases to above 6.5 with no significant change in urine citrate or urine calcium excretion, we recommend stopping alkali therapy.
PREVENTING URIC ACID STONES
Clinical conditions associated with uric acid stones include metabolic syndrome, diabetes mellitus, gout, chronic diarrheal illness, and conditions that increase tissue turnover and uric acid production, such as malignancies. Other risk factors for uric acid stone formation are low urine volume, low uric pH, and hyperuricosuria.
Abnormally acidic urine is the most common risk factor. Metabolic syndrome and diabetes mellitus reduce ammonia production, resulting in a lower urinary pH, which predisposes to uric acid stone formation. Chronic diarrhea also acidifies the urine by loss of bicarbonate. Similarly, in gout, the predisposing factor in uric acid stone formation is the persistently acidic urine due to impaired ammonium excretion.52 Uric acid precipitates to form uric acid stones in a low urinary pH even with normal excretion rates of 600 to 800 mg/day and a urinary volume of 1 to 1.5 L.53
Therefore, apart from increasing fluid intake, urinary alkalization is the cornerstone of management of uric acid stones. Potassium citrate is the preferred alkali salt and is started at a dose of 30 mEq/day for a goal urinary pH of 6 to 6.5.47
Patients with hyperuricosuria are also advised to restrict their protein intake to no more than 0.8 to 1 mg/kg/day.
If the above measures fail, patients are treated with a xanthine oxidase inhibitor, ie, allopurinol or febuxostat, even if their uric acid excretion is normal.54
PREVENTING STRUVITE STONES
Struvite stones contain magnesium ammonium phosphate and are due to chronic upper urinary tract infection with urea-splitting bacteria such as Proteus, Klebsiella, Pseudomonas, and enterococci. Urea hydrolysis releases hydroxyl ions, resulting in alkaline urine that promotes struvite stone formation. Early detection and treatment are important, since struvite stones are associated with morbidity and rapid progression.
Medical treatment of struvite stones is usually unsuccessful, and the patient is referred to a urologist for surgical removal of the stones, the gold standard treatment.55 Long-term use of culture-specific antibiotics to prevent new stone growth is not well studied. Medical therapy by itself is preferred in patients who refuse stone removal or cannot tolerate it. Urease inhibitors such as acetohydroxamic acid have been successful in preventing or slowing stone growth, but their use is limited by frequent side effects such as nausea, headache, rash, and thrombophlebitis.56
CYSTINE STONES
Cystine stones occur in people with inherited defects of renal tubular and intestinal transport of cysteine and dibasic amino acids that cause excessive excretion of urinary cystine, ie, 480 to 3,600 mg/day.
Cystine is formed from two cysteine molecules linked by a disulfide bond. The solubility of cystine is pH-dependent, with increased solubility at higher urinary pH. The goal is to maintain a urinary cystine concentration below its solubility level by keeping the cystine concentration below 243 mg/L and the urine cystine supersaturation (the ratio of the urine cysteine concentration to the cysteine solubility in the same sample) less than 0.6.57 Therapy is aimed at increasing daily urinary volume to 3 L and urine alkalization to pH above 7, in order to increase cystine solubility by 300%.58
Overnight dehydration should be prevented, and patients should be encouraged to wake up at least once a night to void and drink additional water. Sodium restriction to 100 mmol/day (2,300 mg/day) and moderate protein restriction to 0.8 to 1 g/kg/day are associated with decreased cystine excretion, but long-term studies demonstrating their benefit in preventing cystine stones are lacking.59
A thiol-containing drug, eg, D-penicillamine (0.5–2 g/day) or tiopronin (400–1,200 mg/day), should be added to the conservative measures if they have not been effective for 3 months or if there is history of noncompliance.60 Thiol-containing drugs have a sulfhydryl group that reduces the disulfide bond, and they form soluble disulfide cysteine-drug complexes with greater ability to solubilize cystine in alkaline urine. They must always be used in conjunction with fluid and alkali therapy.61
Both drugs have severe and common adverse effects including leukopenia, aplastic anemia, fever, rash, arthritis, hepatotoxicity, pyridoxine deficiency, and proteinuria (membranous nephropathy). However, tiopronin seems to have a lesser incidence of side effects.62 Regular monitoring of complete blood cell counts, liver enzymes, and urine protein should be done.
Captopril contains a sulfhydryl group, and the captopril-cysteine disulfide is more soluble than cysteine alone. The amount of captopril that appears in the urine is low, and doses of 150 mg/day are usually required to reduce cysteine excretion, which can lead to hypotension. The efficacy of captopril in treating cystine stones is unproven, and this drug is used only if patients cannot tolerate other thiol-containing drugs.63
- Scales CD Jr, Smith AC, Hanley JM, Saigal CS; Urologic Diseases in America Project. Prevalence of kidney stones in the United States. Eur Urol 2012; 62:160–165.
- Stamatelou KK, Francis ME, Jones CA, Nyberg LM Jr, Curhan GC. Time trends in reported prevalence of kidney stones in the United States: 1976–1994. Kidney Int 2003; 63:1817–1823.
- Romero V, Akpinar H, Assimos DG. Kidney stones: a global picture of prevalence, incidence, and associated risk factors. Rev Urol 2010; 12:e86–e96.
- Sakhaee K, Maalouf NM, Kumar R, Pasch A, Moe OW. Nephrolithiasis-associated bone disease: pathogenesis and treatment options. Kidney Int 2011; 79:393–403.
- Sakhaee K. Nephrolithiasis as a systemic disorder. Curr Opin Nephrol Hypertens 2008; 17:304–309.
- Hamano S, Nakatsu H, Suzuki N, Tomioka S, Tanaka M, Murakami S. Kidney stone disease and risk factors for coronary heart disease. Int J Urol 2005; 12:859–863.
- Ritz E. Metabolic syndrome: an emerging threat to renal function. Clin J Am Soc Nephrol 2007; 2:869–871.
- Uribarri J, Oh MS, Carroll HJ. The first kidney stone. Ann Intern Med 1989; 111:1006–1009.
- Saigal CS, Joyce G, Timilsina AR; Urologic Diseases in America Project. Direct and indirect costs of nephrolithiasis in an employed population: opportunity for disease management? Kidney Int 2005; 68:1808–1814.
- Moe OW. Kidney stones: pathophysiology and medical management. Lancet 2006; 367:333–344.
- Pearle MS, Goldfarb DS, Assimos DG, et al; American Urological Assocation. Medical management of kidney stones: AUA guideline. J Urol 2014; 192:316–324.
- Borghi L, Meschi T, Amato F, Briganti A, Novarini A, Giannini A. Urinary volume, water and recurrences in idiopathic calcium nephrolithiasis: a 5-year randomized prospective study. J Urol 1996; 155:839–843.
- Sarica K, Inal Y, Erturhan S, Yagci F. The effect of calcium channel blockers on stone regrowth and recurrence after shock wave lithotripsy. Urol Res 2006; 34:184–189.
- Ferraro PM, Taylor EN, Gambaro G, Curhan GC. Soda and other beverages and the risk of kidney stones. Clin J Am Soc Nephrol 2013; 8:1389–1395.
- Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Beverage use and risk for kidney stones in women. Ann Intern Med 1998; 128:534–540.
- Pak CY, Britton F, Peterson R, et al. Ambulatory evaluation of nephrolithiasis. Classification, clinical presentation and diagnostic criteria. Am J Med 1980; 69:19–30.
- Hall PM. Nephrolithiasis: treatment, causes, and prevention. Cleve Clin J Med 2009; 76:583–591.
- Curhan GC, Taylor EN. 24-h uric acid excretion and the risk of kidney stones. Kidney Int 2008; 73:489–496.
- Coe FL, Evan A, Worcester E. Kidney stone disease. J Clin Invest 2005; 115:2598–2608.
- Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med 1993; 328:833–838.
- Muldowney FP, Freaney R, Moloney MF. Importance of dietary sodium in the hypercalciuria syndrome. Kidney Int 1982; 22:292–296.
- Breslau NA, Brinkley L, Hill KD, Pak CY. Relationship of animal protein-rich diet to kidney stone formation and calcium metabolism. J Clin Endocrinol Metab 1988; 66:140–146.
- Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med 2002; 346:77–84.
- Noori N, Honarkar E, Goldfarb DS, et al. Urinary lithogenic risk profile in recurrent stone formers with hyperoxaluria: a randomized controlled trial comparing DASH (Dietary Approaches to Stop Hypertension)-style and low-oxalate diets. Am J Kidney Dis 2014; 63:456–463.
- Fink HA, Wilt TJ, Eidman KE, et al. Medical management to prevent recurrent nephrolithiasis in adults: a systematic review for an American College of Physicians Clinical Guideline. Ann Intern Med 2013; 158:535–543.
- Alon U, Costanzo LS, Chan JC. Additive hypocalciuric effects of amiloride and hydrochlorothiazide in patients treated with calcitriol. Miner Electrolyte Metab 1984; 10:379–386.
- Corbetta S, Baccarelli A, Aroldi A, et al. Risk factors associated to kidney stones in primary hyperparathyroidism. J Endocrinol Invest 2005; 28:122–128.
- Haymann JP. Metabolic disorders: stones as first clinical manifestation of significant diseases. World J Urol 2015; 33:187–192.
- Jaeger P, Portmann L, Jacquet AF, Burckhardt P. Influence of the calcium content of the diet on the incidence of mild hyperoxaluria in idiopathic renal stone formers. Am J Nephrol 1985; 5:40–44.
- Taylor EN, Curhan GC. Oxalate intake and the risk for nephrolithiasis. J Am Soc Nephrol 2007; 18:2198–2204.
- Lieske JC, Tremaine WJ, De Simone C, et al. Diet, but not oral probiotics, effectively reduces urinary oxalate excretion and calcium oxalate supersaturation. Kidney Int 2010; 78:1178–1185.
- Taylor EN, Fung TT, Curhan GC. DASH-style diet associates with reduced risk for kidney stones. J Am Soc Nephrol 2009; 20:2253–2259.
- Urivetzky M, Kessaris D, Smith AD. Ascorbic acid overdosing: a risk factor for calcium oxalate nephrolithiasis. J Urol 1992; 147:1215–1218.
- Hoyer-Kuhn H, Kohbrok S, Volland R, et al. Vitamin B6 in primary hyperoxaluria I: first prospective trial after 40 years of practice. Clin J Am Soc Nephrol 2014; 9:468–477.
- Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Intake of vitamins B6 and C and the risk of kidney stones in women. J Am Soc Nephrol 1999; 10:840–845.
- Parks JH, Worcester EM, O'Connor RC, Coe FL. Urine stone risk factors in nephrolithiasis patients with and without bowel disease. Kidney Int 2003; 63:255–265.
- Hess B, Jost C, Zipperle L, Takkinen R, Jaeger P. High-calcium intake abolishes hyperoxaluria and reduces urinary crystallization during a 20-fold normal oxalate load in humans. Nephrol Dial Transplant 1998; 13:2241–2247.
- Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int 2009; 75:1264–1271.
- Cochat P, Hulton SA, Acquaviva C, et al; OxalEurope. Primary hyperoxaluria type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant 2012; 27:1729–1736.
- Leumann E, Hoppe B, Neuhaus T. Management of primary hyperoxaluria: efficacy of oral citrate administration. Pediatr Nephrol 1993; 7:207–211.
- Bergstralh EJ, Monico CG, Lieske JC, et al; IPHR Investigators. Transplantation outcomes in primary hyperoxaluria. Am J Transplant 2010; 10:2493–2501.
- Grover PK, Marshall VR, Ryall RL. Dissolved urate salts out calcium oxalate in undiluted human urine in vitro: implications for calcium oxalate stone genesis. Chem Biol 2003; 10:271–278.
- Coe FL, Parks JH. Hyperuricosuria and calcium nephrolithiasis. Urol Clin North Am 1981; 8:227–244.
- Ettinger B, Tang A, Citron JT, Livermore B, Williams T. Randomized trial of allopurinol in the prevention of calcium oxalate calculi. N Engl J Med 1986; 315:1386–1389.
- Zuckerman JM, Assimos DG. Hypocitraturia: pathophysiology and medical management. Rev Urol 2009; 11:134–144.
- Seltzer MA, Low RK, McDonald M, Shami GS, Stoller ML. Dietary manipulation with lemonade to treat hypocitraturic calcium nephrolithiasis. J Urol 1996; 156:907–909.
- Odvina CV. Comparative value of orange juice versus lemonade in reducing stone-forming risk. Clin J Am Soc Nephrol 2006; 1:1269–1274.
- Barcelo P, Wuhl O, Servitge E, Rousaud A, Pak CY. Randomized double-blind study of potassium citrate in idiopathic hypocitraturic calcium nephrolithiasis. J Urol 1993; 150:1761–1764.
- Lemann J Jr, Gray RW, Pleuss JA. Potassium bicarbonate, but not sodium bicarbonate, reduces urinary calcium excretion and improves calcium balance in healthy men. Kidney Int 1989; 35:688–695.
- Gault MH, Chafe LL, Morgan JM, et al. Comparison of patients with idiopathic calcium phosphate and calcium oxalate stones. Medicine (Baltimore) 1991; 70:345–359.
- Krieger NS, Asplin JR, Frick KK, et al. Effect of potassium citrate on calcium phosphate stones in a model of hypercalciuria. J Am Soc Nephrol 2015; 26:3001–3008.
- Falls WF Jr. Comparison of urinary acidification and ammonium excretion in normal and gouty subjects. Metabolism 1972; 21:433–445.
- Coe FL, Parks JH, Asplin JR. The pathogenesis and treatment of kidney stones. N Engl J Med 1992; 327:1141–1152.
- Kenny JE, Goldfarb DS. Update on the pathophysiology and management of uric acid renal stones. Curr Rheumatol Rep 2010; 12:125–129.
- Preminger GM, Assimos DG, Lingeman JE, Nakada SY, Pearle MS, Wolf JS Jr (AUA Nephrolithiasis Guideline Panel). Chapter 1: AUA guideline on management of staghorn calculi: diagnosis and treatment recommendations. J Urol 2005; 173:1991–2000.
- Williams JJ, Rodman JS, Peterson CM. A randomized double-blind study of acetohydroxamic acid in struvite nephrolithiasis. N Engl J Med 1984; 311:760–764.
- Nakagawa Y, Asplin JR, Goldfarb DS, Parks JH, Coe FL. Clinical use of cystine supersaturation measurements. J Urol 2000; 164:1481–1485.
- Palacın MGP, Nunes V, Gasparini P. Cystinuria. In: Shriver CR, editor. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill; 2001:4909–4932.
- Goldfarb DS, Coe FL, Asplin JR. Urinary cystine excretion and capacity in patients with cystinuria. Kidney Int 2006; 69:1041–1047.
- Barbey F, Joly D, Rieu P, Mejean A, Daudon M, Jungers P. Medical treatment of cystinuria: critical reappraisal of long-term results. J Urol 2000; 163:1419–1423.
- Asplin DM, Asplin JR. The Interaction of thiol drugs and urine pH in the treatment of cystinuria. J Urol 2013; 189:2147–2151.
- Habib GS, Saliba W, Nashashibi M, Armali Z. Penicillamine and nephrotic syndrome. Eur J Intern Med 2006; 17:343–348.
- Sloand JA, Izzo JL Jr. Captopril reduces urinary cystine excretion in cystinuria. Arch Intern Med 1987; 147:1409–1412.
- Scales CD Jr, Smith AC, Hanley JM, Saigal CS; Urologic Diseases in America Project. Prevalence of kidney stones in the United States. Eur Urol 2012; 62:160–165.
- Stamatelou KK, Francis ME, Jones CA, Nyberg LM Jr, Curhan GC. Time trends in reported prevalence of kidney stones in the United States: 1976–1994. Kidney Int 2003; 63:1817–1823.
- Romero V, Akpinar H, Assimos DG. Kidney stones: a global picture of prevalence, incidence, and associated risk factors. Rev Urol 2010; 12:e86–e96.
- Sakhaee K, Maalouf NM, Kumar R, Pasch A, Moe OW. Nephrolithiasis-associated bone disease: pathogenesis and treatment options. Kidney Int 2011; 79:393–403.
- Sakhaee K. Nephrolithiasis as a systemic disorder. Curr Opin Nephrol Hypertens 2008; 17:304–309.
- Hamano S, Nakatsu H, Suzuki N, Tomioka S, Tanaka M, Murakami S. Kidney stone disease and risk factors for coronary heart disease. Int J Urol 2005; 12:859–863.
- Ritz E. Metabolic syndrome: an emerging threat to renal function. Clin J Am Soc Nephrol 2007; 2:869–871.
- Uribarri J, Oh MS, Carroll HJ. The first kidney stone. Ann Intern Med 1989; 111:1006–1009.
- Saigal CS, Joyce G, Timilsina AR; Urologic Diseases in America Project. Direct and indirect costs of nephrolithiasis in an employed population: opportunity for disease management? Kidney Int 2005; 68:1808–1814.
- Moe OW. Kidney stones: pathophysiology and medical management. Lancet 2006; 367:333–344.
- Pearle MS, Goldfarb DS, Assimos DG, et al; American Urological Assocation. Medical management of kidney stones: AUA guideline. J Urol 2014; 192:316–324.
- Borghi L, Meschi T, Amato F, Briganti A, Novarini A, Giannini A. Urinary volume, water and recurrences in idiopathic calcium nephrolithiasis: a 5-year randomized prospective study. J Urol 1996; 155:839–843.
- Sarica K, Inal Y, Erturhan S, Yagci F. The effect of calcium channel blockers on stone regrowth and recurrence after shock wave lithotripsy. Urol Res 2006; 34:184–189.
- Ferraro PM, Taylor EN, Gambaro G, Curhan GC. Soda and other beverages and the risk of kidney stones. Clin J Am Soc Nephrol 2013; 8:1389–1395.
- Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Beverage use and risk for kidney stones in women. Ann Intern Med 1998; 128:534–540.
- Pak CY, Britton F, Peterson R, et al. Ambulatory evaluation of nephrolithiasis. Classification, clinical presentation and diagnostic criteria. Am J Med 1980; 69:19–30.
- Hall PM. Nephrolithiasis: treatment, causes, and prevention. Cleve Clin J Med 2009; 76:583–591.
- Curhan GC, Taylor EN. 24-h uric acid excretion and the risk of kidney stones. Kidney Int 2008; 73:489–496.
- Coe FL, Evan A, Worcester E. Kidney stone disease. J Clin Invest 2005; 115:2598–2608.
- Curhan GC, Willett WC, Rimm EB, Stampfer MJ. A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N Engl J Med 1993; 328:833–838.
- Muldowney FP, Freaney R, Moloney MF. Importance of dietary sodium in the hypercalciuria syndrome. Kidney Int 1982; 22:292–296.
- Breslau NA, Brinkley L, Hill KD, Pak CY. Relationship of animal protein-rich diet to kidney stone formation and calcium metabolism. J Clin Endocrinol Metab 1988; 66:140–146.
- Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med 2002; 346:77–84.
- Noori N, Honarkar E, Goldfarb DS, et al. Urinary lithogenic risk profile in recurrent stone formers with hyperoxaluria: a randomized controlled trial comparing DASH (Dietary Approaches to Stop Hypertension)-style and low-oxalate diets. Am J Kidney Dis 2014; 63:456–463.
- Fink HA, Wilt TJ, Eidman KE, et al. Medical management to prevent recurrent nephrolithiasis in adults: a systematic review for an American College of Physicians Clinical Guideline. Ann Intern Med 2013; 158:535–543.
- Alon U, Costanzo LS, Chan JC. Additive hypocalciuric effects of amiloride and hydrochlorothiazide in patients treated with calcitriol. Miner Electrolyte Metab 1984; 10:379–386.
- Corbetta S, Baccarelli A, Aroldi A, et al. Risk factors associated to kidney stones in primary hyperparathyroidism. J Endocrinol Invest 2005; 28:122–128.
- Haymann JP. Metabolic disorders: stones as first clinical manifestation of significant diseases. World J Urol 2015; 33:187–192.
- Jaeger P, Portmann L, Jacquet AF, Burckhardt P. Influence of the calcium content of the diet on the incidence of mild hyperoxaluria in idiopathic renal stone formers. Am J Nephrol 1985; 5:40–44.
- Taylor EN, Curhan GC. Oxalate intake and the risk for nephrolithiasis. J Am Soc Nephrol 2007; 18:2198–2204.
- Lieske JC, Tremaine WJ, De Simone C, et al. Diet, but not oral probiotics, effectively reduces urinary oxalate excretion and calcium oxalate supersaturation. Kidney Int 2010; 78:1178–1185.
- Taylor EN, Fung TT, Curhan GC. DASH-style diet associates with reduced risk for kidney stones. J Am Soc Nephrol 2009; 20:2253–2259.
- Urivetzky M, Kessaris D, Smith AD. Ascorbic acid overdosing: a risk factor for calcium oxalate nephrolithiasis. J Urol 1992; 147:1215–1218.
- Hoyer-Kuhn H, Kohbrok S, Volland R, et al. Vitamin B6 in primary hyperoxaluria I: first prospective trial after 40 years of practice. Clin J Am Soc Nephrol 2014; 9:468–477.
- Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Intake of vitamins B6 and C and the risk of kidney stones in women. J Am Soc Nephrol 1999; 10:840–845.
- Parks JH, Worcester EM, O'Connor RC, Coe FL. Urine stone risk factors in nephrolithiasis patients with and without bowel disease. Kidney Int 2003; 63:255–265.
- Hess B, Jost C, Zipperle L, Takkinen R, Jaeger P. High-calcium intake abolishes hyperoxaluria and reduces urinary crystallization during a 20-fold normal oxalate load in humans. Nephrol Dial Transplant 1998; 13:2241–2247.
- Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int 2009; 75:1264–1271.
- Cochat P, Hulton SA, Acquaviva C, et al; OxalEurope. Primary hyperoxaluria type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant 2012; 27:1729–1736.
- Leumann E, Hoppe B, Neuhaus T. Management of primary hyperoxaluria: efficacy of oral citrate administration. Pediatr Nephrol 1993; 7:207–211.
- Bergstralh EJ, Monico CG, Lieske JC, et al; IPHR Investigators. Transplantation outcomes in primary hyperoxaluria. Am J Transplant 2010; 10:2493–2501.
- Grover PK, Marshall VR, Ryall RL. Dissolved urate salts out calcium oxalate in undiluted human urine in vitro: implications for calcium oxalate stone genesis. Chem Biol 2003; 10:271–278.
- Coe FL, Parks JH. Hyperuricosuria and calcium nephrolithiasis. Urol Clin North Am 1981; 8:227–244.
- Ettinger B, Tang A, Citron JT, Livermore B, Williams T. Randomized trial of allopurinol in the prevention of calcium oxalate calculi. N Engl J Med 1986; 315:1386–1389.
- Zuckerman JM, Assimos DG. Hypocitraturia: pathophysiology and medical management. Rev Urol 2009; 11:134–144.
- Seltzer MA, Low RK, McDonald M, Shami GS, Stoller ML. Dietary manipulation with lemonade to treat hypocitraturic calcium nephrolithiasis. J Urol 1996; 156:907–909.
- Odvina CV. Comparative value of orange juice versus lemonade in reducing stone-forming risk. Clin J Am Soc Nephrol 2006; 1:1269–1274.
- Barcelo P, Wuhl O, Servitge E, Rousaud A, Pak CY. Randomized double-blind study of potassium citrate in idiopathic hypocitraturic calcium nephrolithiasis. J Urol 1993; 150:1761–1764.
- Lemann J Jr, Gray RW, Pleuss JA. Potassium bicarbonate, but not sodium bicarbonate, reduces urinary calcium excretion and improves calcium balance in healthy men. Kidney Int 1989; 35:688–695.
- Gault MH, Chafe LL, Morgan JM, et al. Comparison of patients with idiopathic calcium phosphate and calcium oxalate stones. Medicine (Baltimore) 1991; 70:345–359.
- Krieger NS, Asplin JR, Frick KK, et al. Effect of potassium citrate on calcium phosphate stones in a model of hypercalciuria. J Am Soc Nephrol 2015; 26:3001–3008.
- Falls WF Jr. Comparison of urinary acidification and ammonium excretion in normal and gouty subjects. Metabolism 1972; 21:433–445.
- Coe FL, Parks JH, Asplin JR. The pathogenesis and treatment of kidney stones. N Engl J Med 1992; 327:1141–1152.
- Kenny JE, Goldfarb DS. Update on the pathophysiology and management of uric acid renal stones. Curr Rheumatol Rep 2010; 12:125–129.
- Preminger GM, Assimos DG, Lingeman JE, Nakada SY, Pearle MS, Wolf JS Jr (AUA Nephrolithiasis Guideline Panel). Chapter 1: AUA guideline on management of staghorn calculi: diagnosis and treatment recommendations. J Urol 2005; 173:1991–2000.
- Williams JJ, Rodman JS, Peterson CM. A randomized double-blind study of acetohydroxamic acid in struvite nephrolithiasis. N Engl J Med 1984; 311:760–764.
- Nakagawa Y, Asplin JR, Goldfarb DS, Parks JH, Coe FL. Clinical use of cystine supersaturation measurements. J Urol 2000; 164:1481–1485.
- Palacın MGP, Nunes V, Gasparini P. Cystinuria. In: Shriver CR, editor. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill; 2001:4909–4932.
- Goldfarb DS, Coe FL, Asplin JR. Urinary cystine excretion and capacity in patients with cystinuria. Kidney Int 2006; 69:1041–1047.
- Barbey F, Joly D, Rieu P, Mejean A, Daudon M, Jungers P. Medical treatment of cystinuria: critical reappraisal of long-term results. J Urol 2000; 163:1419–1423.
- Asplin DM, Asplin JR. The Interaction of thiol drugs and urine pH in the treatment of cystinuria. J Urol 2013; 189:2147–2151.
- Habib GS, Saliba W, Nashashibi M, Armali Z. Penicillamine and nephrotic syndrome. Eur J Intern Med 2006; 17:343–348.
- Sloand JA, Izzo JL Jr. Captopril reduces urinary cystine excretion in cystinuria. Arch Intern Med 1987; 147:1409–1412.
KEY POINTS
- Nephrolithiasis is common and widespread, and its incidence and prevalence are increasing.
- Calcium stones are the most common type, and of these, calcium oxalate stones predominate.
- The most common risk factors for recurrent calcium stones are low urinary output, hypercalciuria, hyperoxaluria, hypocitraturia, and hyperuricosuria.
- Less common types of stones are usually associated with genetic abnormalities, infections, or medications.
A guide to managing acute liver failure
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.

Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13

Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements

A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT

Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
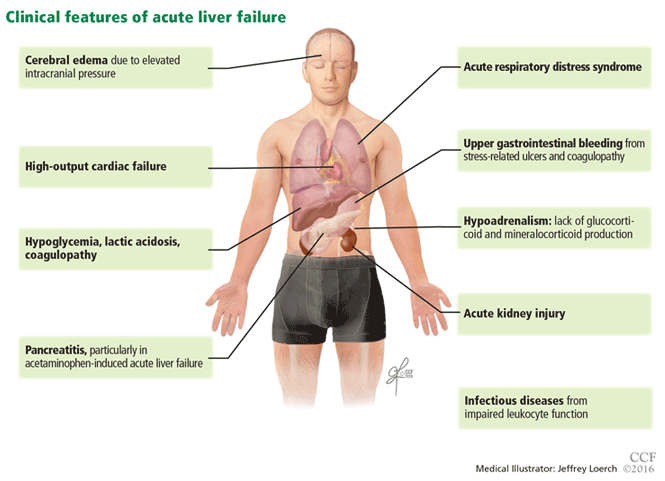
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
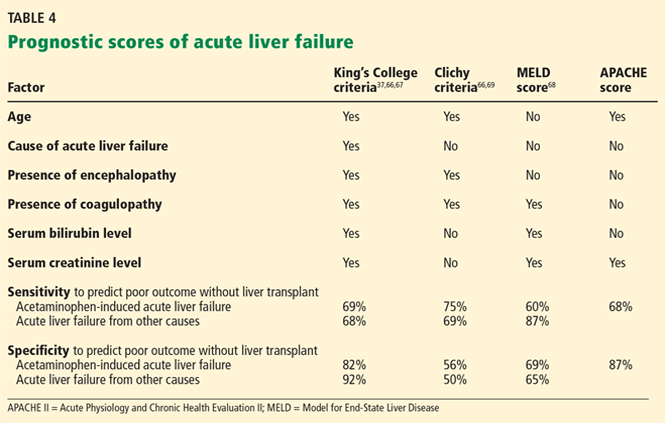
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.
Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13
Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements
A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT
Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.
Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13
Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements
A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT
Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
KEY POINTS
- In the United States, the most common cause of acute liver failure is acetaminophen toxicity, followed by viral hepatitis.
- Testing for the cause of acute liver failure needs to start as soon as possible so that specific treatment can be initiated and the patient can be placed on the transplant list if needed.
- Acetylcysteine and either a proton pump inhibitor or a histamine H2 receptor blocker should be given to all patients with acute liver failure. Liver transplant is the cornerstone of therapy in patients not responding to other treatments.
- There are a number of prognostic scores for acute liver failure, but each has limitations.
Opioids for persistent pain in older adults
The use of opioid analgesics is widely accepted for treating severe acute pain, cancer pain, and pain at the end of life.1 However, their long-term use for other types of persistent pain (Table 1) remains controversial. Clinicians and regulators need to work together to achieve a balanced approach to the use of opioids, recognizing the legitimate medical need for these medications for persistent pain while acknowledging their increasing misuse and the morbidity and mortality related to them. Finding this balance is particularly challenging in older patients.2
PAIN IN OLDER PEOPLE: COMPLICATED, OFTEN UNDERTREATED
Persistent pain is a multifaceted manifestation of an unpleasant sensation that continues for a prolonged time and may or may not be related to a distinct disease process.3 (The term “persistent pain” is preferred as it does not have the negative connotations of “chronic pain.”4) “Older” has been defined as age 65 and older. As our population ages, especially to age 85 and older, more people will be living with persistent pain due to a variety of conditions.5
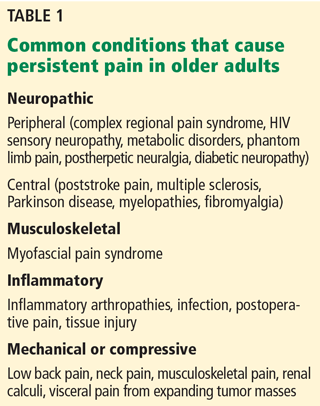
Persistent pain is more complicated in older than in younger patients. Many older people have more than one illness, making them more susceptible to adverse drug interactions such as altered pharmacokinetics and pharmacodynamics.6 Up to 40% of older outpatients report pain,7 and pain affects 70% to 80% of patients with advanced malignant disease.8 Pain is also prevalent in nonmalignant, progressive, life-limiting illnesses that are common in the geriatric population, affecting 41% to 77% of patients with advanced heart disease, 34% to 77% with advanced chronic obstructive pulmonary disease, and 47% to 50% with advanced renal disease.9
Pain is underrecognized in nursing home residents, who may have multiple somatic complaints and multiple causes of pain.10,11 From 27% to 83% of older adults in an institutionalized setting are affected by pain.12 Caregiver stress and attitudes towards pain may influence patients’ experiences with pain. This aspect should also be assessed and evaluated, if present.3
Pain in older adults is often undertreated, as evidenced by the findings of a study in which only one-third of older patients with persistent pain were receiving treatment that was consistent with current guidelines.13 Approximately 40% to 80% of older adults in the community with pain do not receive any treatment for it.14,15 Of those residing in institutions, 16% to 27% of older adults in pain do not receive any treatment for it.16,17 Inadequate treatment of persistent pain is associated with many adverse outcomes, including functional decline, falls, mood changes, decreased socialization, sleep and appetite difficulties, and increased healthcare utilization.18
GOALS: BETTER QUALITY OF LIFE AND FUNCTION
Persistent pain is multifactorial and so requires an approach that addresses a variety of causes and includes both nonpharmacologic and pharmacologic strategies. Opioids are part of a multipronged approach to pain management.
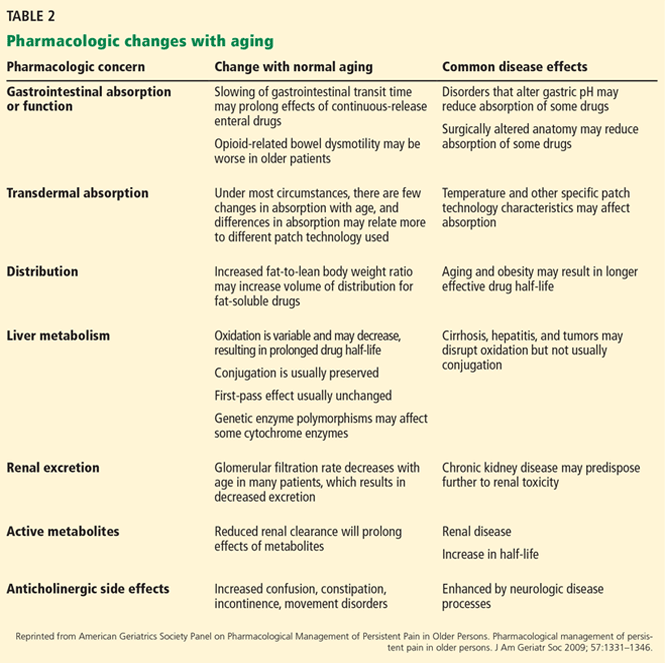
To avoid adverse effects, opioids for persistent pain in an older adult should be prescribed at the lowest possible dose that provides adequate analgesia. Due to age-related changes, finding the best treatments may be a challenge, and understanding the pharmacokinetic implications in this population is key (Table 2).
Complete pain relief is uncommon and is not the goal when using opioids in older patients. Rather, treatment goals should focus on quality of life and function. Patients need to be continually educated about these goals and regularly reassessed during treatment.
APPROACH TO PAIN MANAGEMENT
Initial steps in managing pain should always include a detailed pain assessment, ideally by an interdisciplinary team.19,20 Physical therapy, cognitive behavioral therapy, and patient and caregiver education are some effective nonpharmacologic strategies.3 If nonpharmacologic treatments are ineffective, pharmacologic strategies should be used. Often, both nonpharmacologic and pharmacologic treatments work well for persistent pain.
The World Health Organization’s three-step ladder approach, originally developed for cancer pain, has subsequently been adopted for all types of pain.
- Step 1 of the ladder is nonopioid analgesics, with or without adjuvant agents.
- Step 2 if the pain persists or increases, is a weak opioid (eg, codeine, tramadol), with or without a nonopioid analgesic and with or without an adjuvant agent.
- Step 3 is a strong opioid (eg, morphine, oxycodone, hydromorphone, fentanyl, or methadone), with or without nonopioid and adjuvant agents.
The European Association for Palliative Care recommendations state that there is no significant difference between morphine, oxycodone, and hydromorphone when given orally.21 Although this ladder has been modernized somewhat,22 it still provides a conceptual and practical guide.
FIRST STEP: NONOPIOID ANALGESICS
Acetaminophen is first-line
Acetaminophen is the first-line drug for persistent pain, as it is effective and safe. It does not have the same gastrointestinal and renal side effects that nonsteroidal anti-inflammatory drugs (NSAIDs) do. It also has fewer drug interactions, and its clearance does not decline with age.23
However, older adults should not take more than 3 g of acetaminophen in 24 hours.24 It should be used with extreme caution, if at all, in patients who have hepatic insufficiency or chronic alcohol abuse or dependence.
Topical therapies
Topical NSAIDs allow local analgesia with less risk of systemic side effects than with oral NSAIDs, which have a limited role in the older population.
Capsaicin, which depletes substance P, has primarily been studied for neuropathic pain.
Lidocaine 5% topical patch has been found effective for postherpetic neuralgia; however, there is limited evidence for using it in other painful conditions, such as osteoarthritis and back pain.25
Adjuvants
Duloxetine is a serotonin and norepinephrine reuptake inhibitor. Studies have found it effective in treating diabetic peripheral neuropathy, fibromyalgia, chronic low back pain, and osteoarthritis knee pain. However, except for the knee study, most of the patients enrolled were younger.
Antiepileptic medications. Gabapentin and pregabalin have been found to be effective in painful neuropathic conditions that commonly occur in older adults.25
Avoid oral NSAIDs
NSAIDs, both nonselective and cyclooxygenase 2-selective, should only rarely be considered for long-term use in older adults in view of increased risk of conditions such as congestive heart failure, acute kidney injury, and gastrointestinal bleeding.25 These adverse effects seem to be related to inhibition of prostaglandin, which plays a physiologic role in the gastrointestinal, renal, and cardiovascular systems.26 Oral NSAIDs should be used with extreme caution.
OPIOIDS

The American Geriatrics Society, American Pain Society, and American Academy of Pain Medicine made recommendations in 2009 supporting the use of opioids to treat persistent pain in patients who are carefully selected and monitored.4,6 An international expert panel in 2008 issued a consensus statement27 of evidence that also supported the use of opioids for those over age 65. The Federation of State Medical Boards of the United States also supports the use of opioids, particularly for adults who have refractory pain, and it recognizes undertreatment of pain as a public health issue.28
Clinicians are most comfortable with using opioids to manage cancer pain, but these drugs also provide an acceptable and effective means of analgesia in nonmalignant, persistent pain syndromes.24 The American Geriatrics Society Panel on Pharmacological Management of Persistent Pain in Older Persons recommends treatment with opioids in all patients with moderate-to-severe pain, pain-related functional impairment, or decreased quality of life due to pain, even though the evidence base is not robust.3
Unlike NSAIDs and acetaminophen, opioids do not have a presumed ceiling effect. However, in patients ages 15 to 64, the greatest benefits have been observed at lower doses of opioids, and the risk of death increases with dose.29 The dose can be raised gradually until pain is relieved.
Start low and go slow
When starting opioid therapy:
- Choose a short-acting agent
- Give it on a trial basis
- Start at a low dose and titrate up slowly.
No data are available to tell us how much to give an older adult, but a reasonable starting dose is 30% to 50% of the recommended dose for a younger adult.24 Short-acting opioids should be titrated by increasing the total daily dose by 25% to 50% every 24 hours until adequate analgesia is reached.24
Older adults who have frequent or continuous pain should receive scheduled (around-the-clock) dosing in an effort to achieve a steady state.3 The half-lives of opioids may be longer in older adults who have renal or hepatic insufficiency; therefore, their doses should be lower and the intervals between doses longer.27
When long-acting opioid preparations are used, it is important to also prescribe breakthrough (short-acting) pain management.2 Breakthrough pain includes end-of-dose failure, incident pain (ie, due to an identifiable cause, such as movement), and spontaneous pain; these can be prevented or treated with short-acting, immediate-release opioid formulations.3
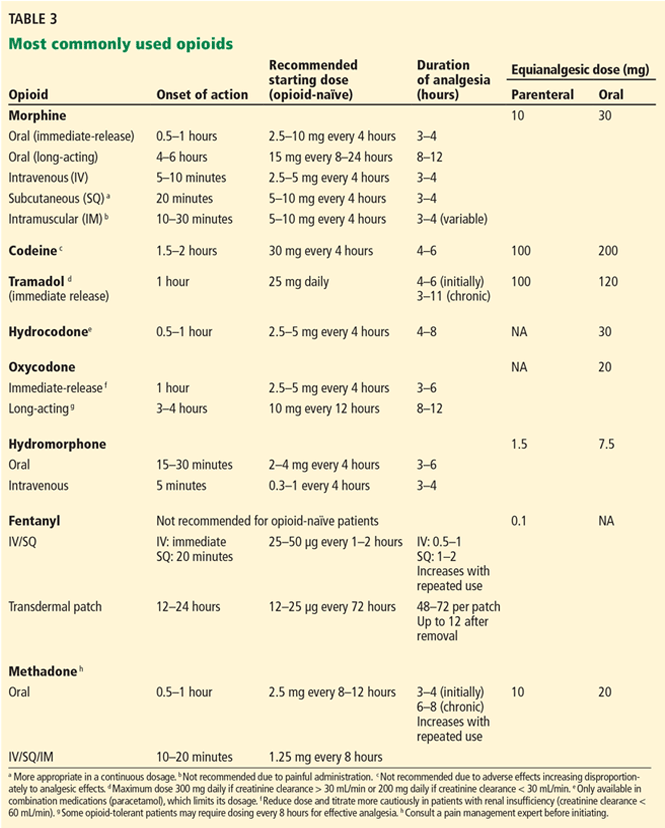
Once therapy is initiated, its safety and efficacy should be continually monitored.2 With long-term use, patients should be reassessed for ongoing attainment of therapeutic goals, adverse effects, and safe and responsible medication use.3
Table 3 lists common opioids and their initial dosing.
SIDE EFFECTS
Constipation
This is one of the most common side effects of opioids,30 and although many opioid side effects wane within days of starting as tolerance develops, this one does not.
A bowel regimen should be initiated when starting any opioid regimen. Although most of the evidence for bowel regimens is anecdotal, increasing fluid and fiber intake and taking stool softeners and laxatives are effective.31
For very difficult cases of opioid constipation, randomized trials suggest that specific agents with opioid antagonist activity that specifically target the gastrointestinal system can help.32,33 Opioid antagonists are not used as routine prophylaxis, but rather for constipation that is refractory to laxatives.34,35 A meta-analysis demonstrated that methylnaltrexone, naloxone, and alvimopan were generally well tolerated, with no significant difference in adverse effects compared with placebo.36
Sedation
Sedation due to opioids in opioid-naïve patients is well documented,37 but it decreases over time. When starting or changing the dose of opioids, it is important to counsel patients about driving and safety at work and home.
For persistent opioid-related sedation, three options are available: dose reduction, opioid rotation, and use of psychostimulants.38 Although it does not carry a US Food and Drug Administration indication for this use, methylphenidate has been studied in cancer patients, in whom it has been associated with less drowsiness, decreased pain, and less need for rescue doses of pain medications.39–41
Nausea and vomiting
Nausea and vomiting are common in opioid recipients. These adverse effects usually decrease over days to weeks with continued exposure.
A number of antiemetic therapies are available in oral, rectal, and intravenous formulations, but there is no evidence-based recommendation for antiemetic choice for opioid-induced nausea in patients with cancer.42 It is important to always rule out constipation as the cause of nausea. There is also some evidence that reducing the opioid dose or changing the route of administration may help with symptoms.42–45
Respiratory depression
Although respiratory depression is the most feared adverse effect of opioids, it is rare with low starting doses and appropriate dose titration. Sedation precedes respiratory depression, which occurs when initial opioid dosages are too high, titration is too rapid, or opioids are combined with other drugs associated with respiratory depression or that may potentiate opioid-induced respiratory depression, such as benzodiazepines.46–51
Patients with sleep apnea may be at higher risk. In addition, in a study that specifically reviewed patients who had persistent pain, specific factors that contributed to opioid-induced respiratory depression were use of methadone and transdermal fentanyl, renal impairment, and sensory deafferentation.52 Buprenorphine was found to have a ceiling effect for respiratory depression, but not for analgesia.49
Central sleep apnea
Chronic opioid use has been associated with sleep-disordered breathing, notably central sleep apnea. This is often unrecognized. The prevalence of central sleep apnea in this population is 24%.53
Although continuous positive airway pressure is the standard of care for obstructive sleep apnea, it is ineffective for central sleep apnea and possibly may make it worse. Adaptive servoventilation is a therapy that may be effective.54
Urinary retention
Opioids can cause urinary retention, which is most noted in a postoperative setting. Changes in bladder function have been found to be partially due to a peripheral opioid effect.55
Initial management: catheterize the bladder for prompt relief and try to reduce the dose of opioids.
Impaired balance and falls
Use of opioids, especially when combined with other medications active in the central nervous system, may lead to impaired balance and falls, especially in the elderly.56 In this group, all opioids are associated with falls except for buprenorphine.27,57 Older adults need to be assessed and educated about the risk of falls before they are given opioids. Physical therapy and mobility aids may help in these cases.
Dependence
The prevalence of dependence is low in patients who have no prior history of substance abuse.6 Older age is also associated with a significantly lower risk of opioid misuse and abuse.6
Opioid-induced hyperalgesia
Opioid-induced hyperalgesia should be considered if pain continues to worsen in spite of increasing doses, tolerance to opioids appears to develop rapidly, or pain becomes more diffuse and extends past the distribution of preexisting pain.58 Although the exact mechanism is unclear, exposure to opioids causes nociceptive sensitization, as measured by several techniques.59,60
Opioid-induced hyperalgesia is distinct from opioid analgesia tolerance. A key difference is that opioid tolerance can be overcome by increasing the dose, while opioid-induced hyperalgesia can be exacerbated by it.
Management of opioid-induced hyperalgesia includes decreasing the dose, switching to a different opioid, and maximizing nonopioid analgesia.58 The plan should be clearly communicated to patients and families to avoid misunderstanding.
Other adverse effects
Long-term use of opioids may suppress production of several hypothalamic, pituitary, gonadal, and adrenal hormones.3 Long-term use of opioids is also associated with bone loss.61 Opioids have also demonstrated immunodepressant effects.38,62
OPIOID ROTATION
Trying a different opioid (opioid rotation) may be required if pain remains poorly controlled despite increasing doses or if intolerable side effects occur.
According to consensus guidelines on opioid rotation,63 if the originally prescribed opioid is not providing the appropriate therapeutic effect or the patient cannot tolerate the regimen, an equianalgesic dose (Table 3) of the new opioid is calculated based on the original opioid and then decreased in two safety steps. The first safety step is a 25% to 50% reduction in the calculated equianalgesic dose to account for incomplete cross-tolerance. There are two exceptions: methadone requires a 75% to 90% reduction, and transdermal fentanyl does not require an adjustment. The next step is an adjustment of 15% to 30% based on pain severity and the patient’s medical or psychosocial aspects.63
SPECIAL POPULATION: PATIENTS WITH DEMENTIA
There is little scientific data on pain management in older adults with dementia. Many patients with mild to moderate dementia can verbally communicate pain reliably,64 but more challenging are those who are nonverbal, for whom providers depend on caregiver reports and observational scales.65
Prescribing in patients with dementia who are verbal and nonverbal mirrors the strategies used in those older adults who are cognitively intact,66 eg:
- Use scheduled (around-the-clock) dosing
- Start with nonopioid medications initially, but advance to opioids as needed, guided by the WHO ladder
- Carefully monitor the risks and benefits of pain treatment vs persistent pain.
When uncertain about whether a demented patient is in pain, a trial of analgesics is warranted. Signs of pain include not socializing, disturbed sleep, and a vegetative state.
SAFE PRESCRIBING PRACTICES
With the use of opioids to treat persistent pain comes the risk of abuse. A universal precautions approach helps establish reasonable limits before initiating therapy.
A thorough evaluation is required, including description and documentation of pain, disease processes, comorbidities, and effects on function; physical examination; and diagnostic testing. It is also important to inquire about a history of substance abuse. Tools such as the Opioid Risk Tool and the Screener and Opioid Assessment for Patients with Pain-Revised can help gauge risk of misuse or abuse.67,68
Ongoing screening and monitoring are necessary to minimize misuse and diversion. This also involves adhering to federal and state government regulatory policies and participating state prescription drug monitoring programs.69
- Chou R, Fanciullo GJ, Fine PG, et al; American Pain Society-American Academy of Pain Medicine Opioids Guidelines Panel. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J Pain 2009; 10:113–130.
- West NA, Severtson SG, Green JL, Dart RC. Trends in abuse and misuse of prescription opioids among older adults. Drug Alcohol Depend 2015; 149:117–121.
- American Geriatrics Society Panel on Pharmacological Management of Persistent Pain in Older Persons. Pharmacological management of persistent pain in older persons. J Am Geriatr Soc 2009; 57:1331–1346.
- Weiner DK, Herr K. Comprehensive interdisciplinary assessment and treatment planning: an integrative overview. In: Weiner DK, Herr K, Rudy TE, editors. Persistent pain in older adults: an interdisciplinary guide for treatment. New York, NY: Springer Publishing Company; 2002.
- He W, Sengupta M, Velkoff V; US Census Bureau. 65+ in the United States: 2005. Washington, DC: US Government Printing Office; 2005. www.census.gov/prod/2006pubs/p23-209.pdf. Accessed March 30, 2016.
- American Geriatrics Society Panel on the Pharmacological Management of Persistent Pain in Older Persons. Pharmacological management of persistent pain in older persons. Pain Med 2009; 10:1062–1083.
- Thomas E, Peat G, Harris L, Wilkie R, Croft PR. The prevalence of pain and pain interference in a general population of older adults: cross-sectional findings from the North Staffordshire Osteoarthritis Project (NorStOP). Pain 2004; 110:361–368.
- Caraceni A, Hanks G, Kaasa S, et al; European Palliative Care Research Collaborative (EPCRC); European Association for Palliative Care (EAPC). Use of opioid analgesics in the treatment of cancer pain: evidence-based recommendations from the EAPC. Lancet Oncol 2012; 13:e58–e68.
- Solano JP, Gomes B, Higginson IJ. A comparison of symptom prevalence in far advanced cancer, AIDS, heart disease, chronic obstructive pulmonary disease and renal disease. J Pain Symptom Manage 2006; 31:58–69.
- Ferrell BA, Ferrell BR, Osterweil D. Pain in the nursing home. J Am Geriatr Soc 1990; 38:409–414.
- Ferrell BA, Ferrell BR, Rivera L. Pain in cognitively impaired nursing home patients. J Pain Symptom Manage 1995; 10:591–598.
- Fox PL, Raina P, Jadad AR. Prevalence and treatment of pain in older adults in nursing homes and other long-term care institutions: a systematic review. CMAJ 1999; 160:329–333.
- Stewart C, Leveille SG, Shmerling RH, Samelson EJ, Bean JF, Schofield P. Management of persistent pain in older adults: the MOBILIZE Boston Study. J Am Geriatr Soc 2012; 60:2081–2086.
- Woo J, Ho SC, Lau J, Leung PC. Musculoskeletal complaints and associated consequences in elderly Chinese aged 70 years and over. J Rheumatol 1994; 21:1927–1931.
- Pahor M, Guralnik JM, Wan JY, et al. Lower body osteoarticular pain and dose of analgesic medications in older disabled women: the Women’s Health and Aging Study. Am J Public Health 1999; 89:930–934.
- Marzinski LR. The tragedy of dementia: clinically assessing pain in the confused nonverbal elderly. J Gerontol Nurs 1991; 17:25–28.
- Roy R, Thomas M. A survey of chronic pain in an elderly population. Can Fam Physician 1986; 32:513–516.
- AGS Panel on Persistent Pain in Older Persons. The management of persistent pain in older persons. J Am Geriatr Soc 2002; 50(suppl 6): S205–S224.
- Stanos S, Houle TT. Multidisciplinary and interdisciplinary management of chronic pain. Phys Med Rehabil Clin N Am 2006; 17:435–450.
- Helme RD, Katz B, Gibson SJ, et al. Multidisciplinary pain clinics for older people. Do they have a role? Clin Geriatr Med 1996; 12:563–582.
- Harris DG. Management of pain in advanced disease. Br Med Bull 2014; 110:117–128.
- Raffa RB, Pergolizzi JV. A modern analgesics pain ‘pyramid’. J Clin Pharm Ther 2014; 39:4–6.
- Fine PG, Herr KA. Pharmacologic management of persistent pain in older persons. Clin Geriatr 2009; 17:25–32.
- Tracy B, Sean Morrison R. Pain management in older adults. Clin Ther 2013; 35:1659–1668.
- Malec M, Shega JW. Pain management in the elderly. Med Clin North Am 2015; 99:337–350.
- Abdulla A, Adams N, Bone M, et al; British Geriatric Society. Guidance on the management of pain in older people. Age Ageing 2013; 42(suppl 1):i1–i57.
- Pergolizzi J, Böger RH, Budd K, et al. Opioids and the management of chronic severe pain in the elderly: consensus statement of an International Expert Panel with focus on the six clinically most often used World Health Organization Step III opioids (buprenorphine, fentanyl, hydromorphone, methadone, morphine, oxycodone). Pain Pract 2008; 8:287–313.
- Gloth FM 3rd. Pharmacological management of persistent pain in older persons: focus on opioids and nonopioids. J Pain 2011; 12(suppl 1):S14–S20.
- Gomes T, Mamdani MM, Dhalla IA, Paterson JM, Juurlink DN. Opioid dose and drug-related mortality in patients with nonmalignant pain. Arch Intern Med 2011; 171:686–691.
- Moore RA, McQuay HJ. Prevalence of opioid adverse events in chronic non-malignant pain: systematic review of randomised trials of oral opioids. Arthritis Res Ther 2005; 7:R1046–R1051.
- Candy B, Jones L, Larkin PJ, Vickerstaff V, Tookman A, Stone P. Laxatives for the management of constipation in people receiving palliative care. Cochrane Database Syst Rev 2015; 5:CD003448.
- Webster LR, Butera PG, Moran LV, Wu N, Burns LH, Friedmann N. Oxytrex minimizes physical dependence while providing effective analgesia: a randomized controlled trial in low back pain. J Pain 2006; 7:937–946.
- Paulson DM, Kennedy DT, Donovick RA, et al. Alvimopan: an oral, peripherally acting, mu-opioid receptor antagonist for the treatment of opioid-induced bowel dysfunction—a 21-day treatment-randomized clinical trial. J Pain 2005; 6:184–192.
- Nalamachu SR, Pergolizzi J, Taylor R, et al. Efficacy and tolerability of subcutaneous methylnaltrexone in patients with advanced illness and opioid-induced constipation: a responder analysis of 2 randomized, placebo-controlled trials. Pain Pract 2015; 15:564–571.
- Brick N. Laxatives or methylnaltrexone for the management of constipation in palliative care patients. Clin J Oncol Nurs 2013; 17:91–92.
- Ford AC, Brenner DM, Schoenfeld PS. Efficacy of pharmacological therapies for the treatment of opioid-induced constipation: systematic review and meta-analysis. Am J Gastroenterolt 2013; 108:1566–1575.
- Byas-Smith MG, Chapman SL, Reed B, Cotsonis G. The effect of opioids on driving and psychomotor performance in patients with chronic pain. Clin J Pain 2005; 21:345–352.
- Benyamin R, Trescot AM, Datta S, et al. Opioid complications and side effects. Pain Physician 2008; 11(suppl 2):S105–S120.
- Wilwerding MB, Loprinzi CL, Mailliard JA, et al. A randomized, crossover evaluation of methylphenidate in cancer patients receiving strong narcotics. Support Care Cancer 1995; 3:135–138.
- Bruera E, Miller MJ, Macmillan K, Kuehn N. Neuropsychological effects of methylphenidate in patients receiving a continuous infusion of narcotics for cancer pain. Pain 1992; 48:163–166.
- Ahmedzai S. New approaches to pain control in patients with cancer. Eur J Cancer 1997; 33:S8–S14.
- Laugsand EA, Kaasa S, Klepstad P. Management of opioid-induced nausea and vomiting in cancer patients: systematic review and evidence-based recommendations. Palliat Med 2011; 25:442–453.
- Hardy J, Daly S, McQuade B, et al. A double-blind, randomised, parallel group, multinational, multicentre study comparing a single dose of ondansetron 24 mg p.o. with placebo and metoclopramide 10 mg t.d.s. p.o. in the treatment of opioid-induced nausea and emesis in cancer patients. Support Care Cancer 2002; 10:231–236.
- Apfel CC, Jalota L. Can central antiemetic effects of opioids counter-balance opioid-induced nausea and vomiting? Acta Anaesthesiol Scand 2010; 54:129–131.
- Okamoto Y, Tsuneto S, Matsuda Y, et al. A retrospective chart review of the antiemetic effectiveness of risperidone in refractory opioid-induced nausea and vomiting in advanced cancer patients. J Pain Symptom Manage 2007; 34:217–222.
- Overdyk F, Dahan A, Roozekrans M, van der Schrier R, Aarts L, Niesters M. Opioid-induced respiratory depression in the acute care setting: a compendium of case reports. Pain Manag 2014; 4:317–325.
- Niesters M, Overdyk F, Smith T, Aarts L, Dahan A. Buprenorphine-induced respiratory depression and involvement of ABCB1 SNPs in opioid-induced respiratory depression in paediatrics. Br J Anaesth 2013; 110:842–843.
- Niesters M, Mahajan RP, Aarts L, Dahan A. High-inspired oxygen concentration further impairs opioid-induced respiratory depression. Br J Anaesth 2013; 110:837–841.
- Dahan A, Yassen A, Romberg R, et al. Buprenorphine induces ceiling in respiratory depression but not in analgesia. Br J Anaesth 2006; 96:627–632.
- van Dorp E, Yassen A, Sarton E, et al. Naloxone reversal of buprenorphine-induced respiratory depression. Anesthesiology 2006; 105:51–57.
- Macintyre PE, Loadsman JA, Scott DA. Opioids, ventilation and acute pain management. Anaesth Intensive Care 2011; 39:545–558.
- Dahan A, Overdyk F, Smith T, Aarts L, Niesters M. Pharmacovigilance: a review of opioid-induced respiratory depression in chronic pain patients. Pain Physician 2013; 16:E85–E94.
- Correa D, Farney RJ, Chung F, Prasad A, Lam D, Wong J. Chronic opioid use and central sleep apnea: a review of the prevalence, mechanisms, and perioperative considerations. Anesth Analg 2015; 120:1273–1285.
- Randerath WJ, George S. Opioid-induced sleep apnea: is it a real problem? J Clin Sleep Med 2012; 8:577–578.
- Rosow CE, Gomery P, Chen TY, Stefanovich P, Stambler N, Israel R. Reversal of opioid-induced bladder dysfunction by intravenous naloxone and methylnaltrexone. Clin Pharmacol Ther 2007; 82:48–53.
- Weiner DK, Hanlon JT, Studenski SA. Effects of central nervous system polypharmacy on falls liability in community-dwelling elderly. Gerontology 1998; 44:217–221.
- Wolff ML, Kewley R, Hassett M, Collins J, Brodeur MR, Nokes S. Falls in skilled nursing facilities associated with opioid use. J Am Geriatr Soc 2012; 60:987.
- Zylicz Z, Twycross R. Opioid-induced hyperalgesia may be more frequent than previously thought. J Clin Oncol 2008; 26:1564; author reply 1565.
- Lee M, Silverman SM, Hansen H, Patel VB, Manchikanti L. A comprehensive review of opioid-induced hyperalgesia. Pain Physician 2011 2011; 14:145–161.
- Chen L, Sein M, Vo T, et al. Clinical interpretation of opioid tolerance versus opioid-induced hyperalgesia. J Opioid Manag 2014; 10:383–393.
- Vestergaard P, Hermann P, Jensen JE, Eiken P, Mosekilde L. Effects of paracetamol, non-steroidal anti-inflammatory drugs, acetylsalicylic acid, and opioids on bone mineral density and risk of fracture: results of the Danish Osteoporosis Prevention Study (DOPS). Osteoporos Int 2012; 23:1255–1265.
- Sacerdote P, Franchi S, Panerai AE. Non-analgesic effects of opioids: mechanisms and potential clinical relevance of opioid-induced immunodepression. Curr Pharm Des 2012; 18:6034–6042.
- Fine PG, Portenoy RK; Ad Hoc Expert Panel on Evidence Review and Guidelines for Opioid Rotation. Establishing “best practices” for opioid rotation: conclusions of an expert panel. J Pain Symptom Manage 2009; 38:418–425.
- Chibnall JT, Tait RC. Pain assessment in cognitively impaired and unimpaired older adults: a comparison of four scales. Pain 2001; 92:173–186.
- Andrade DC, Faria JW, Caramelli P, et al. The assessment and management of pain in the demented and non-demented elderly patient. Arq Neuropsiquiatr 2011; 69:387–394.
- Scherder E, Herr K, Pickering G, Gibson S, Benedetti F, Lautenbacher S. Pain in dementia. Pain 2009; 145:276–278.
- Chou R, Fanciullo GJ, Fine PG, Miaskowski C, Passik SD, Portenoy RK. Opioids for chronic noncancer pain: prediction and identification of aberrant drug-related behaviors: a review of the evidence for an American Pain Society and American Academy of Pain Medicine clinical practice guideline. J Pain 2009; 10:131–146.
- Butler SF, Budman SH, Fernandez KC, Fanciullo GJ, Jamison RN. Cross-validation of a screener to predict opioid misuse in chronic pain patients (SOAPP-R). J Addict Med 2009; 3:66–73.
- de Leon-Casasola OA. Opioids for chronic pain: new evidence, new strategies, safe prescribing. Am J Med 2013; 126(suppl 1):S3–S11.
- CDC guideline for prescribing opioids for chronic pain—United States, 2016. MMWR Recomm Rep 2016 Mar 18; 65(1):1–49.
The use of opioid analgesics is widely accepted for treating severe acute pain, cancer pain, and pain at the end of life.1 However, their long-term use for other types of persistent pain (Table 1) remains controversial. Clinicians and regulators need to work together to achieve a balanced approach to the use of opioids, recognizing the legitimate medical need for these medications for persistent pain while acknowledging their increasing misuse and the morbidity and mortality related to them. Finding this balance is particularly challenging in older patients.2
PAIN IN OLDER PEOPLE: COMPLICATED, OFTEN UNDERTREATED
Persistent pain is a multifaceted manifestation of an unpleasant sensation that continues for a prolonged time and may or may not be related to a distinct disease process.3 (The term “persistent pain” is preferred as it does not have the negative connotations of “chronic pain.”4) “Older” has been defined as age 65 and older. As our population ages, especially to age 85 and older, more people will be living with persistent pain due to a variety of conditions.5
Persistent pain is more complicated in older than in younger patients. Many older people have more than one illness, making them more susceptible to adverse drug interactions such as altered pharmacokinetics and pharmacodynamics.6 Up to 40% of older outpatients report pain,7 and pain affects 70% to 80% of patients with advanced malignant disease.8 Pain is also prevalent in nonmalignant, progressive, life-limiting illnesses that are common in the geriatric population, affecting 41% to 77% of patients with advanced heart disease, 34% to 77% with advanced chronic obstructive pulmonary disease, and 47% to 50% with advanced renal disease.9
Pain is underrecognized in nursing home residents, who may have multiple somatic complaints and multiple causes of pain.10,11 From 27% to 83% of older adults in an institutionalized setting are affected by pain.12 Caregiver stress and attitudes towards pain may influence patients’ experiences with pain. This aspect should also be assessed and evaluated, if present.3
Pain in older adults is often undertreated, as evidenced by the findings of a study in which only one-third of older patients with persistent pain were receiving treatment that was consistent with current guidelines.13 Approximately 40% to 80% of older adults in the community with pain do not receive any treatment for it.14,15 Of those residing in institutions, 16% to 27% of older adults in pain do not receive any treatment for it.16,17 Inadequate treatment of persistent pain is associated with many adverse outcomes, including functional decline, falls, mood changes, decreased socialization, sleep and appetite difficulties, and increased healthcare utilization.18
GOALS: BETTER QUALITY OF LIFE AND FUNCTION
Persistent pain is multifactorial and so requires an approach that addresses a variety of causes and includes both nonpharmacologic and pharmacologic strategies. Opioids are part of a multipronged approach to pain management.
To avoid adverse effects, opioids for persistent pain in an older adult should be prescribed at the lowest possible dose that provides adequate analgesia. Due to age-related changes, finding the best treatments may be a challenge, and understanding the pharmacokinetic implications in this population is key (Table 2).
Complete pain relief is uncommon and is not the goal when using opioids in older patients. Rather, treatment goals should focus on quality of life and function. Patients need to be continually educated about these goals and regularly reassessed during treatment.
APPROACH TO PAIN MANAGEMENT
Initial steps in managing pain should always include a detailed pain assessment, ideally by an interdisciplinary team.19,20 Physical therapy, cognitive behavioral therapy, and patient and caregiver education are some effective nonpharmacologic strategies.3 If nonpharmacologic treatments are ineffective, pharmacologic strategies should be used. Often, both nonpharmacologic and pharmacologic treatments work well for persistent pain.
The World Health Organization’s three-step ladder approach, originally developed for cancer pain, has subsequently been adopted for all types of pain.
- Step 1 of the ladder is nonopioid analgesics, with or without adjuvant agents.
- Step 2 if the pain persists or increases, is a weak opioid (eg, codeine, tramadol), with or without a nonopioid analgesic and with or without an adjuvant agent.
- Step 3 is a strong opioid (eg, morphine, oxycodone, hydromorphone, fentanyl, or methadone), with or without nonopioid and adjuvant agents.
The European Association for Palliative Care recommendations state that there is no significant difference between morphine, oxycodone, and hydromorphone when given orally.21 Although this ladder has been modernized somewhat,22 it still provides a conceptual and practical guide.
FIRST STEP: NONOPIOID ANALGESICS
Acetaminophen is first-line
Acetaminophen is the first-line drug for persistent pain, as it is effective and safe. It does not have the same gastrointestinal and renal side effects that nonsteroidal anti-inflammatory drugs (NSAIDs) do. It also has fewer drug interactions, and its clearance does not decline with age.23
However, older adults should not take more than 3 g of acetaminophen in 24 hours.24 It should be used with extreme caution, if at all, in patients who have hepatic insufficiency or chronic alcohol abuse or dependence.
Topical therapies
Topical NSAIDs allow local analgesia with less risk of systemic side effects than with oral NSAIDs, which have a limited role in the older population.
Capsaicin, which depletes substance P, has primarily been studied for neuropathic pain.
Lidocaine 5% topical patch has been found effective for postherpetic neuralgia; however, there is limited evidence for using it in other painful conditions, such as osteoarthritis and back pain.25
Adjuvants
Duloxetine is a serotonin and norepinephrine reuptake inhibitor. Studies have found it effective in treating diabetic peripheral neuropathy, fibromyalgia, chronic low back pain, and osteoarthritis knee pain. However, except for the knee study, most of the patients enrolled were younger.
Antiepileptic medications. Gabapentin and pregabalin have been found to be effective in painful neuropathic conditions that commonly occur in older adults.25
Avoid oral NSAIDs
NSAIDs, both nonselective and cyclooxygenase 2-selective, should only rarely be considered for long-term use in older adults in view of increased risk of conditions such as congestive heart failure, acute kidney injury, and gastrointestinal bleeding.25 These adverse effects seem to be related to inhibition of prostaglandin, which plays a physiologic role in the gastrointestinal, renal, and cardiovascular systems.26 Oral NSAIDs should be used with extreme caution.
OPIOIDS
The American Geriatrics Society, American Pain Society, and American Academy of Pain Medicine made recommendations in 2009 supporting the use of opioids to treat persistent pain in patients who are carefully selected and monitored.4,6 An international expert panel in 2008 issued a consensus statement27 of evidence that also supported the use of opioids for those over age 65. The Federation of State Medical Boards of the United States also supports the use of opioids, particularly for adults who have refractory pain, and it recognizes undertreatment of pain as a public health issue.28
Clinicians are most comfortable with using opioids to manage cancer pain, but these drugs also provide an acceptable and effective means of analgesia in nonmalignant, persistent pain syndromes.24 The American Geriatrics Society Panel on Pharmacological Management of Persistent Pain in Older Persons recommends treatment with opioids in all patients with moderate-to-severe pain, pain-related functional impairment, or decreased quality of life due to pain, even though the evidence base is not robust.3
Unlike NSAIDs and acetaminophen, opioids do not have a presumed ceiling effect. However, in patients ages 15 to 64, the greatest benefits have been observed at lower doses of opioids, and the risk of death increases with dose.29 The dose can be raised gradually until pain is relieved.
Start low and go slow
When starting opioid therapy:
- Choose a short-acting agent
- Give it on a trial basis
- Start at a low dose and titrate up slowly.
No data are available to tell us how much to give an older adult, but a reasonable starting dose is 30% to 50% of the recommended dose for a younger adult.24 Short-acting opioids should be titrated by increasing the total daily dose by 25% to 50% every 24 hours until adequate analgesia is reached.24
Older adults who have frequent or continuous pain should receive scheduled (around-the-clock) dosing in an effort to achieve a steady state.3 The half-lives of opioids may be longer in older adults who have renal or hepatic insufficiency; therefore, their doses should be lower and the intervals between doses longer.27
When long-acting opioid preparations are used, it is important to also prescribe breakthrough (short-acting) pain management.2 Breakthrough pain includes end-of-dose failure, incident pain (ie, due to an identifiable cause, such as movement), and spontaneous pain; these can be prevented or treated with short-acting, immediate-release opioid formulations.3
Once therapy is initiated, its safety and efficacy should be continually monitored.2 With long-term use, patients should be reassessed for ongoing attainment of therapeutic goals, adverse effects, and safe and responsible medication use.3
Table 3 lists common opioids and their initial dosing.
SIDE EFFECTS
Constipation
This is one of the most common side effects of opioids,30 and although many opioid side effects wane within days of starting as tolerance develops, this one does not.
A bowel regimen should be initiated when starting any opioid regimen. Although most of the evidence for bowel regimens is anecdotal, increasing fluid and fiber intake and taking stool softeners and laxatives are effective.31
For very difficult cases of opioid constipation, randomized trials suggest that specific agents with opioid antagonist activity that specifically target the gastrointestinal system can help.32,33 Opioid antagonists are not used as routine prophylaxis, but rather for constipation that is refractory to laxatives.34,35 A meta-analysis demonstrated that methylnaltrexone, naloxone, and alvimopan were generally well tolerated, with no significant difference in adverse effects compared with placebo.36
Sedation
Sedation due to opioids in opioid-naïve patients is well documented,37 but it decreases over time. When starting or changing the dose of opioids, it is important to counsel patients about driving and safety at work and home.
For persistent opioid-related sedation, three options are available: dose reduction, opioid rotation, and use of psychostimulants.38 Although it does not carry a US Food and Drug Administration indication for this use, methylphenidate has been studied in cancer patients, in whom it has been associated with less drowsiness, decreased pain, and less need for rescue doses of pain medications.39–41
Nausea and vomiting
Nausea and vomiting are common in opioid recipients. These adverse effects usually decrease over days to weeks with continued exposure.
A number of antiemetic therapies are available in oral, rectal, and intravenous formulations, but there is no evidence-based recommendation for antiemetic choice for opioid-induced nausea in patients with cancer.42 It is important to always rule out constipation as the cause of nausea. There is also some evidence that reducing the opioid dose or changing the route of administration may help with symptoms.42–45
Respiratory depression
Although respiratory depression is the most feared adverse effect of opioids, it is rare with low starting doses and appropriate dose titration. Sedation precedes respiratory depression, which occurs when initial opioid dosages are too high, titration is too rapid, or opioids are combined with other drugs associated with respiratory depression or that may potentiate opioid-induced respiratory depression, such as benzodiazepines.46–51
Patients with sleep apnea may be at higher risk. In addition, in a study that specifically reviewed patients who had persistent pain, specific factors that contributed to opioid-induced respiratory depression were use of methadone and transdermal fentanyl, renal impairment, and sensory deafferentation.52 Buprenorphine was found to have a ceiling effect for respiratory depression, but not for analgesia.49
Central sleep apnea
Chronic opioid use has been associated with sleep-disordered breathing, notably central sleep apnea. This is often unrecognized. The prevalence of central sleep apnea in this population is 24%.53
Although continuous positive airway pressure is the standard of care for obstructive sleep apnea, it is ineffective for central sleep apnea and possibly may make it worse. Adaptive servoventilation is a therapy that may be effective.54
Urinary retention
Opioids can cause urinary retention, which is most noted in a postoperative setting. Changes in bladder function have been found to be partially due to a peripheral opioid effect.55
Initial management: catheterize the bladder for prompt relief and try to reduce the dose of opioids.
Impaired balance and falls
Use of opioids, especially when combined with other medications active in the central nervous system, may lead to impaired balance and falls, especially in the elderly.56 In this group, all opioids are associated with falls except for buprenorphine.27,57 Older adults need to be assessed and educated about the risk of falls before they are given opioids. Physical therapy and mobility aids may help in these cases.
Dependence
The prevalence of dependence is low in patients who have no prior history of substance abuse.6 Older age is also associated with a significantly lower risk of opioid misuse and abuse.6
Opioid-induced hyperalgesia
Opioid-induced hyperalgesia should be considered if pain continues to worsen in spite of increasing doses, tolerance to opioids appears to develop rapidly, or pain becomes more diffuse and extends past the distribution of preexisting pain.58 Although the exact mechanism is unclear, exposure to opioids causes nociceptive sensitization, as measured by several techniques.59,60
Opioid-induced hyperalgesia is distinct from opioid analgesia tolerance. A key difference is that opioid tolerance can be overcome by increasing the dose, while opioid-induced hyperalgesia can be exacerbated by it.
Management of opioid-induced hyperalgesia includes decreasing the dose, switching to a different opioid, and maximizing nonopioid analgesia.58 The plan should be clearly communicated to patients and families to avoid misunderstanding.
Other adverse effects
Long-term use of opioids may suppress production of several hypothalamic, pituitary, gonadal, and adrenal hormones.3 Long-term use of opioids is also associated with bone loss.61 Opioids have also demonstrated immunodepressant effects.38,62
OPIOID ROTATION
Trying a different opioid (opioid rotation) may be required if pain remains poorly controlled despite increasing doses or if intolerable side effects occur.
According to consensus guidelines on opioid rotation,63 if the originally prescribed opioid is not providing the appropriate therapeutic effect or the patient cannot tolerate the regimen, an equianalgesic dose (Table 3) of the new opioid is calculated based on the original opioid and then decreased in two safety steps. The first safety step is a 25% to 50% reduction in the calculated equianalgesic dose to account for incomplete cross-tolerance. There are two exceptions: methadone requires a 75% to 90% reduction, and transdermal fentanyl does not require an adjustment. The next step is an adjustment of 15% to 30% based on pain severity and the patient’s medical or psychosocial aspects.63
SPECIAL POPULATION: PATIENTS WITH DEMENTIA
There is little scientific data on pain management in older adults with dementia. Many patients with mild to moderate dementia can verbally communicate pain reliably,64 but more challenging are those who are nonverbal, for whom providers depend on caregiver reports and observational scales.65
Prescribing in patients with dementia who are verbal and nonverbal mirrors the strategies used in those older adults who are cognitively intact,66 eg:
- Use scheduled (around-the-clock) dosing
- Start with nonopioid medications initially, but advance to opioids as needed, guided by the WHO ladder
- Carefully monitor the risks and benefits of pain treatment vs persistent pain.
When uncertain about whether a demented patient is in pain, a trial of analgesics is warranted. Signs of pain include not socializing, disturbed sleep, and a vegetative state.
SAFE PRESCRIBING PRACTICES
With the use of opioids to treat persistent pain comes the risk of abuse. A universal precautions approach helps establish reasonable limits before initiating therapy.
A thorough evaluation is required, including description and documentation of pain, disease processes, comorbidities, and effects on function; physical examination; and diagnostic testing. It is also important to inquire about a history of substance abuse. Tools such as the Opioid Risk Tool and the Screener and Opioid Assessment for Patients with Pain-Revised can help gauge risk of misuse or abuse.67,68
Ongoing screening and monitoring are necessary to minimize misuse and diversion. This also involves adhering to federal and state government regulatory policies and participating state prescription drug monitoring programs.69
The use of opioid analgesics is widely accepted for treating severe acute pain, cancer pain, and pain at the end of life.1 However, their long-term use for other types of persistent pain (Table 1) remains controversial. Clinicians and regulators need to work together to achieve a balanced approach to the use of opioids, recognizing the legitimate medical need for these medications for persistent pain while acknowledging their increasing misuse and the morbidity and mortality related to them. Finding this balance is particularly challenging in older patients.2
PAIN IN OLDER PEOPLE: COMPLICATED, OFTEN UNDERTREATED
Persistent pain is a multifaceted manifestation of an unpleasant sensation that continues for a prolonged time and may or may not be related to a distinct disease process.3 (The term “persistent pain” is preferred as it does not have the negative connotations of “chronic pain.”4) “Older” has been defined as age 65 and older. As our population ages, especially to age 85 and older, more people will be living with persistent pain due to a variety of conditions.5
Persistent pain is more complicated in older than in younger patients. Many older people have more than one illness, making them more susceptible to adverse drug interactions such as altered pharmacokinetics and pharmacodynamics.6 Up to 40% of older outpatients report pain,7 and pain affects 70% to 80% of patients with advanced malignant disease.8 Pain is also prevalent in nonmalignant, progressive, life-limiting illnesses that are common in the geriatric population, affecting 41% to 77% of patients with advanced heart disease, 34% to 77% with advanced chronic obstructive pulmonary disease, and 47% to 50% with advanced renal disease.9
Pain is underrecognized in nursing home residents, who may have multiple somatic complaints and multiple causes of pain.10,11 From 27% to 83% of older adults in an institutionalized setting are affected by pain.12 Caregiver stress and attitudes towards pain may influence patients’ experiences with pain. This aspect should also be assessed and evaluated, if present.3
Pain in older adults is often undertreated, as evidenced by the findings of a study in which only one-third of older patients with persistent pain were receiving treatment that was consistent with current guidelines.13 Approximately 40% to 80% of older adults in the community with pain do not receive any treatment for it.14,15 Of those residing in institutions, 16% to 27% of older adults in pain do not receive any treatment for it.16,17 Inadequate treatment of persistent pain is associated with many adverse outcomes, including functional decline, falls, mood changes, decreased socialization, sleep and appetite difficulties, and increased healthcare utilization.18
GOALS: BETTER QUALITY OF LIFE AND FUNCTION
Persistent pain is multifactorial and so requires an approach that addresses a variety of causes and includes both nonpharmacologic and pharmacologic strategies. Opioids are part of a multipronged approach to pain management.
To avoid adverse effects, opioids for persistent pain in an older adult should be prescribed at the lowest possible dose that provides adequate analgesia. Due to age-related changes, finding the best treatments may be a challenge, and understanding the pharmacokinetic implications in this population is key (Table 2).
Complete pain relief is uncommon and is not the goal when using opioids in older patients. Rather, treatment goals should focus on quality of life and function. Patients need to be continually educated about these goals and regularly reassessed during treatment.
APPROACH TO PAIN MANAGEMENT
Initial steps in managing pain should always include a detailed pain assessment, ideally by an interdisciplinary team.19,20 Physical therapy, cognitive behavioral therapy, and patient and caregiver education are some effective nonpharmacologic strategies.3 If nonpharmacologic treatments are ineffective, pharmacologic strategies should be used. Often, both nonpharmacologic and pharmacologic treatments work well for persistent pain.
The World Health Organization’s three-step ladder approach, originally developed for cancer pain, has subsequently been adopted for all types of pain.
- Step 1 of the ladder is nonopioid analgesics, with or without adjuvant agents.
- Step 2 if the pain persists or increases, is a weak opioid (eg, codeine, tramadol), with or without a nonopioid analgesic and with or without an adjuvant agent.
- Step 3 is a strong opioid (eg, morphine, oxycodone, hydromorphone, fentanyl, or methadone), with or without nonopioid and adjuvant agents.
The European Association for Palliative Care recommendations state that there is no significant difference between morphine, oxycodone, and hydromorphone when given orally.21 Although this ladder has been modernized somewhat,22 it still provides a conceptual and practical guide.
FIRST STEP: NONOPIOID ANALGESICS
Acetaminophen is first-line
Acetaminophen is the first-line drug for persistent pain, as it is effective and safe. It does not have the same gastrointestinal and renal side effects that nonsteroidal anti-inflammatory drugs (NSAIDs) do. It also has fewer drug interactions, and its clearance does not decline with age.23
However, older adults should not take more than 3 g of acetaminophen in 24 hours.24 It should be used with extreme caution, if at all, in patients who have hepatic insufficiency or chronic alcohol abuse or dependence.
Topical therapies
Topical NSAIDs allow local analgesia with less risk of systemic side effects than with oral NSAIDs, which have a limited role in the older population.
Capsaicin, which depletes substance P, has primarily been studied for neuropathic pain.
Lidocaine 5% topical patch has been found effective for postherpetic neuralgia; however, there is limited evidence for using it in other painful conditions, such as osteoarthritis and back pain.25
Adjuvants
Duloxetine is a serotonin and norepinephrine reuptake inhibitor. Studies have found it effective in treating diabetic peripheral neuropathy, fibromyalgia, chronic low back pain, and osteoarthritis knee pain. However, except for the knee study, most of the patients enrolled were younger.
Antiepileptic medications. Gabapentin and pregabalin have been found to be effective in painful neuropathic conditions that commonly occur in older adults.25
Avoid oral NSAIDs
NSAIDs, both nonselective and cyclooxygenase 2-selective, should only rarely be considered for long-term use in older adults in view of increased risk of conditions such as congestive heart failure, acute kidney injury, and gastrointestinal bleeding.25 These adverse effects seem to be related to inhibition of prostaglandin, which plays a physiologic role in the gastrointestinal, renal, and cardiovascular systems.26 Oral NSAIDs should be used with extreme caution.
OPIOIDS
The American Geriatrics Society, American Pain Society, and American Academy of Pain Medicine made recommendations in 2009 supporting the use of opioids to treat persistent pain in patients who are carefully selected and monitored.4,6 An international expert panel in 2008 issued a consensus statement27 of evidence that also supported the use of opioids for those over age 65. The Federation of State Medical Boards of the United States also supports the use of opioids, particularly for adults who have refractory pain, and it recognizes undertreatment of pain as a public health issue.28
Clinicians are most comfortable with using opioids to manage cancer pain, but these drugs also provide an acceptable and effective means of analgesia in nonmalignant, persistent pain syndromes.24 The American Geriatrics Society Panel on Pharmacological Management of Persistent Pain in Older Persons recommends treatment with opioids in all patients with moderate-to-severe pain, pain-related functional impairment, or decreased quality of life due to pain, even though the evidence base is not robust.3
Unlike NSAIDs and acetaminophen, opioids do not have a presumed ceiling effect. However, in patients ages 15 to 64, the greatest benefits have been observed at lower doses of opioids, and the risk of death increases with dose.29 The dose can be raised gradually until pain is relieved.
Start low and go slow
When starting opioid therapy:
- Choose a short-acting agent
- Give it on a trial basis
- Start at a low dose and titrate up slowly.
No data are available to tell us how much to give an older adult, but a reasonable starting dose is 30% to 50% of the recommended dose for a younger adult.24 Short-acting opioids should be titrated by increasing the total daily dose by 25% to 50% every 24 hours until adequate analgesia is reached.24
Older adults who have frequent or continuous pain should receive scheduled (around-the-clock) dosing in an effort to achieve a steady state.3 The half-lives of opioids may be longer in older adults who have renal or hepatic insufficiency; therefore, their doses should be lower and the intervals between doses longer.27
When long-acting opioid preparations are used, it is important to also prescribe breakthrough (short-acting) pain management.2 Breakthrough pain includes end-of-dose failure, incident pain (ie, due to an identifiable cause, such as movement), and spontaneous pain; these can be prevented or treated with short-acting, immediate-release opioid formulations.3
Once therapy is initiated, its safety and efficacy should be continually monitored.2 With long-term use, patients should be reassessed for ongoing attainment of therapeutic goals, adverse effects, and safe and responsible medication use.3
Table 3 lists common opioids and their initial dosing.
SIDE EFFECTS
Constipation
This is one of the most common side effects of opioids,30 and although many opioid side effects wane within days of starting as tolerance develops, this one does not.
A bowel regimen should be initiated when starting any opioid regimen. Although most of the evidence for bowel regimens is anecdotal, increasing fluid and fiber intake and taking stool softeners and laxatives are effective.31
For very difficult cases of opioid constipation, randomized trials suggest that specific agents with opioid antagonist activity that specifically target the gastrointestinal system can help.32,33 Opioid antagonists are not used as routine prophylaxis, but rather for constipation that is refractory to laxatives.34,35 A meta-analysis demonstrated that methylnaltrexone, naloxone, and alvimopan were generally well tolerated, with no significant difference in adverse effects compared with placebo.36
Sedation
Sedation due to opioids in opioid-naïve patients is well documented,37 but it decreases over time. When starting or changing the dose of opioids, it is important to counsel patients about driving and safety at work and home.
For persistent opioid-related sedation, three options are available: dose reduction, opioid rotation, and use of psychostimulants.38 Although it does not carry a US Food and Drug Administration indication for this use, methylphenidate has been studied in cancer patients, in whom it has been associated with less drowsiness, decreased pain, and less need for rescue doses of pain medications.39–41
Nausea and vomiting
Nausea and vomiting are common in opioid recipients. These adverse effects usually decrease over days to weeks with continued exposure.
A number of antiemetic therapies are available in oral, rectal, and intravenous formulations, but there is no evidence-based recommendation for antiemetic choice for opioid-induced nausea in patients with cancer.42 It is important to always rule out constipation as the cause of nausea. There is also some evidence that reducing the opioid dose or changing the route of administration may help with symptoms.42–45
Respiratory depression
Although respiratory depression is the most feared adverse effect of opioids, it is rare with low starting doses and appropriate dose titration. Sedation precedes respiratory depression, which occurs when initial opioid dosages are too high, titration is too rapid, or opioids are combined with other drugs associated with respiratory depression or that may potentiate opioid-induced respiratory depression, such as benzodiazepines.46–51
Patients with sleep apnea may be at higher risk. In addition, in a study that specifically reviewed patients who had persistent pain, specific factors that contributed to opioid-induced respiratory depression were use of methadone and transdermal fentanyl, renal impairment, and sensory deafferentation.52 Buprenorphine was found to have a ceiling effect for respiratory depression, but not for analgesia.49
Central sleep apnea
Chronic opioid use has been associated with sleep-disordered breathing, notably central sleep apnea. This is often unrecognized. The prevalence of central sleep apnea in this population is 24%.53
Although continuous positive airway pressure is the standard of care for obstructive sleep apnea, it is ineffective for central sleep apnea and possibly may make it worse. Adaptive servoventilation is a therapy that may be effective.54
Urinary retention
Opioids can cause urinary retention, which is most noted in a postoperative setting. Changes in bladder function have been found to be partially due to a peripheral opioid effect.55
Initial management: catheterize the bladder for prompt relief and try to reduce the dose of opioids.
Impaired balance and falls
Use of opioids, especially when combined with other medications active in the central nervous system, may lead to impaired balance and falls, especially in the elderly.56 In this group, all opioids are associated with falls except for buprenorphine.27,57 Older adults need to be assessed and educated about the risk of falls before they are given opioids. Physical therapy and mobility aids may help in these cases.
Dependence
The prevalence of dependence is low in patients who have no prior history of substance abuse.6 Older age is also associated with a significantly lower risk of opioid misuse and abuse.6
Opioid-induced hyperalgesia
Opioid-induced hyperalgesia should be considered if pain continues to worsen in spite of increasing doses, tolerance to opioids appears to develop rapidly, or pain becomes more diffuse and extends past the distribution of preexisting pain.58 Although the exact mechanism is unclear, exposure to opioids causes nociceptive sensitization, as measured by several techniques.59,60
Opioid-induced hyperalgesia is distinct from opioid analgesia tolerance. A key difference is that opioid tolerance can be overcome by increasing the dose, while opioid-induced hyperalgesia can be exacerbated by it.
Management of opioid-induced hyperalgesia includes decreasing the dose, switching to a different opioid, and maximizing nonopioid analgesia.58 The plan should be clearly communicated to patients and families to avoid misunderstanding.
Other adverse effects
Long-term use of opioids may suppress production of several hypothalamic, pituitary, gonadal, and adrenal hormones.3 Long-term use of opioids is also associated with bone loss.61 Opioids have also demonstrated immunodepressant effects.38,62
OPIOID ROTATION
Trying a different opioid (opioid rotation) may be required if pain remains poorly controlled despite increasing doses or if intolerable side effects occur.
According to consensus guidelines on opioid rotation,63 if the originally prescribed opioid is not providing the appropriate therapeutic effect or the patient cannot tolerate the regimen, an equianalgesic dose (Table 3) of the new opioid is calculated based on the original opioid and then decreased in two safety steps. The first safety step is a 25% to 50% reduction in the calculated equianalgesic dose to account for incomplete cross-tolerance. There are two exceptions: methadone requires a 75% to 90% reduction, and transdermal fentanyl does not require an adjustment. The next step is an adjustment of 15% to 30% based on pain severity and the patient’s medical or psychosocial aspects.63
SPECIAL POPULATION: PATIENTS WITH DEMENTIA
There is little scientific data on pain management in older adults with dementia. Many patients with mild to moderate dementia can verbally communicate pain reliably,64 but more challenging are those who are nonverbal, for whom providers depend on caregiver reports and observational scales.65
Prescribing in patients with dementia who are verbal and nonverbal mirrors the strategies used in those older adults who are cognitively intact,66 eg:
- Use scheduled (around-the-clock) dosing
- Start with nonopioid medications initially, but advance to opioids as needed, guided by the WHO ladder
- Carefully monitor the risks and benefits of pain treatment vs persistent pain.
When uncertain about whether a demented patient is in pain, a trial of analgesics is warranted. Signs of pain include not socializing, disturbed sleep, and a vegetative state.
SAFE PRESCRIBING PRACTICES
With the use of opioids to treat persistent pain comes the risk of abuse. A universal precautions approach helps establish reasonable limits before initiating therapy.
A thorough evaluation is required, including description and documentation of pain, disease processes, comorbidities, and effects on function; physical examination; and diagnostic testing. It is also important to inquire about a history of substance abuse. Tools such as the Opioid Risk Tool and the Screener and Opioid Assessment for Patients with Pain-Revised can help gauge risk of misuse or abuse.67,68
Ongoing screening and monitoring are necessary to minimize misuse and diversion. This also involves adhering to federal and state government regulatory policies and participating state prescription drug monitoring programs.69
- Chou R, Fanciullo GJ, Fine PG, et al; American Pain Society-American Academy of Pain Medicine Opioids Guidelines Panel. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J Pain 2009; 10:113–130.
- West NA, Severtson SG, Green JL, Dart RC. Trends in abuse and misuse of prescription opioids among older adults. Drug Alcohol Depend 2015; 149:117–121.
- American Geriatrics Society Panel on Pharmacological Management of Persistent Pain in Older Persons. Pharmacological management of persistent pain in older persons. J Am Geriatr Soc 2009; 57:1331–1346.
- Weiner DK, Herr K. Comprehensive interdisciplinary assessment and treatment planning: an integrative overview. In: Weiner DK, Herr K, Rudy TE, editors. Persistent pain in older adults: an interdisciplinary guide for treatment. New York, NY: Springer Publishing Company; 2002.
- He W, Sengupta M, Velkoff V; US Census Bureau. 65+ in the United States: 2005. Washington, DC: US Government Printing Office; 2005. www.census.gov/prod/2006pubs/p23-209.pdf. Accessed March 30, 2016.
- American Geriatrics Society Panel on the Pharmacological Management of Persistent Pain in Older Persons. Pharmacological management of persistent pain in older persons. Pain Med 2009; 10:1062–1083.
- Thomas E, Peat G, Harris L, Wilkie R, Croft PR. The prevalence of pain and pain interference in a general population of older adults: cross-sectional findings from the North Staffordshire Osteoarthritis Project (NorStOP). Pain 2004; 110:361–368.
- Caraceni A, Hanks G, Kaasa S, et al; European Palliative Care Research Collaborative (EPCRC); European Association for Palliative Care (EAPC). Use of opioid analgesics in the treatment of cancer pain: evidence-based recommendations from the EAPC. Lancet Oncol 2012; 13:e58–e68.
- Solano JP, Gomes B, Higginson IJ. A comparison of symptom prevalence in far advanced cancer, AIDS, heart disease, chronic obstructive pulmonary disease and renal disease. J Pain Symptom Manage 2006; 31:58–69.
- Ferrell BA, Ferrell BR, Osterweil D. Pain in the nursing home. J Am Geriatr Soc 1990; 38:409–414.
- Ferrell BA, Ferrell BR, Rivera L. Pain in cognitively impaired nursing home patients. J Pain Symptom Manage 1995; 10:591–598.
- Fox PL, Raina P, Jadad AR. Prevalence and treatment of pain in older adults in nursing homes and other long-term care institutions: a systematic review. CMAJ 1999; 160:329–333.
- Stewart C, Leveille SG, Shmerling RH, Samelson EJ, Bean JF, Schofield P. Management of persistent pain in older adults: the MOBILIZE Boston Study. J Am Geriatr Soc 2012; 60:2081–2086.
- Woo J, Ho SC, Lau J, Leung PC. Musculoskeletal complaints and associated consequences in elderly Chinese aged 70 years and over. J Rheumatol 1994; 21:1927–1931.
- Pahor M, Guralnik JM, Wan JY, et al. Lower body osteoarticular pain and dose of analgesic medications in older disabled women: the Women’s Health and Aging Study. Am J Public Health 1999; 89:930–934.
- Marzinski LR. The tragedy of dementia: clinically assessing pain in the confused nonverbal elderly. J Gerontol Nurs 1991; 17:25–28.
- Roy R, Thomas M. A survey of chronic pain in an elderly population. Can Fam Physician 1986; 32:513–516.
- AGS Panel on Persistent Pain in Older Persons. The management of persistent pain in older persons. J Am Geriatr Soc 2002; 50(suppl 6): S205–S224.
- Stanos S, Houle TT. Multidisciplinary and interdisciplinary management of chronic pain. Phys Med Rehabil Clin N Am 2006; 17:435–450.
- Helme RD, Katz B, Gibson SJ, et al. Multidisciplinary pain clinics for older people. Do they have a role? Clin Geriatr Med 1996; 12:563–582.
- Harris DG. Management of pain in advanced disease. Br Med Bull 2014; 110:117–128.
- Raffa RB, Pergolizzi JV. A modern analgesics pain ‘pyramid’. J Clin Pharm Ther 2014; 39:4–6.
- Fine PG, Herr KA. Pharmacologic management of persistent pain in older persons. Clin Geriatr 2009; 17:25–32.
- Tracy B, Sean Morrison R. Pain management in older adults. Clin Ther 2013; 35:1659–1668.
- Malec M, Shega JW. Pain management in the elderly. Med Clin North Am 2015; 99:337–350.
- Abdulla A, Adams N, Bone M, et al; British Geriatric Society. Guidance on the management of pain in older people. Age Ageing 2013; 42(suppl 1):i1–i57.
- Pergolizzi J, Böger RH, Budd K, et al. Opioids and the management of chronic severe pain in the elderly: consensus statement of an International Expert Panel with focus on the six clinically most often used World Health Organization Step III opioids (buprenorphine, fentanyl, hydromorphone, methadone, morphine, oxycodone). Pain Pract 2008; 8:287–313.
- Gloth FM 3rd. Pharmacological management of persistent pain in older persons: focus on opioids and nonopioids. J Pain 2011; 12(suppl 1):S14–S20.
- Gomes T, Mamdani MM, Dhalla IA, Paterson JM, Juurlink DN. Opioid dose and drug-related mortality in patients with nonmalignant pain. Arch Intern Med 2011; 171:686–691.
- Moore RA, McQuay HJ. Prevalence of opioid adverse events in chronic non-malignant pain: systematic review of randomised trials of oral opioids. Arthritis Res Ther 2005; 7:R1046–R1051.
- Candy B, Jones L, Larkin PJ, Vickerstaff V, Tookman A, Stone P. Laxatives for the management of constipation in people receiving palliative care. Cochrane Database Syst Rev 2015; 5:CD003448.
- Webster LR, Butera PG, Moran LV, Wu N, Burns LH, Friedmann N. Oxytrex minimizes physical dependence while providing effective analgesia: a randomized controlled trial in low back pain. J Pain 2006; 7:937–946.
- Paulson DM, Kennedy DT, Donovick RA, et al. Alvimopan: an oral, peripherally acting, mu-opioid receptor antagonist for the treatment of opioid-induced bowel dysfunction—a 21-day treatment-randomized clinical trial. J Pain 2005; 6:184–192.
- Nalamachu SR, Pergolizzi J, Taylor R, et al. Efficacy and tolerability of subcutaneous methylnaltrexone in patients with advanced illness and opioid-induced constipation: a responder analysis of 2 randomized, placebo-controlled trials. Pain Pract 2015; 15:564–571.
- Brick N. Laxatives or methylnaltrexone for the management of constipation in palliative care patients. Clin J Oncol Nurs 2013; 17:91–92.
- Ford AC, Brenner DM, Schoenfeld PS. Efficacy of pharmacological therapies for the treatment of opioid-induced constipation: systematic review and meta-analysis. Am J Gastroenterolt 2013; 108:1566–1575.
- Byas-Smith MG, Chapman SL, Reed B, Cotsonis G. The effect of opioids on driving and psychomotor performance in patients with chronic pain. Clin J Pain 2005; 21:345–352.
- Benyamin R, Trescot AM, Datta S, et al. Opioid complications and side effects. Pain Physician 2008; 11(suppl 2):S105–S120.
- Wilwerding MB, Loprinzi CL, Mailliard JA, et al. A randomized, crossover evaluation of methylphenidate in cancer patients receiving strong narcotics. Support Care Cancer 1995; 3:135–138.
- Bruera E, Miller MJ, Macmillan K, Kuehn N. Neuropsychological effects of methylphenidate in patients receiving a continuous infusion of narcotics for cancer pain. Pain 1992; 48:163–166.
- Ahmedzai S. New approaches to pain control in patients with cancer. Eur J Cancer 1997; 33:S8–S14.
- Laugsand EA, Kaasa S, Klepstad P. Management of opioid-induced nausea and vomiting in cancer patients: systematic review and evidence-based recommendations. Palliat Med 2011; 25:442–453.
- Hardy J, Daly S, McQuade B, et al. A double-blind, randomised, parallel group, multinational, multicentre study comparing a single dose of ondansetron 24 mg p.o. with placebo and metoclopramide 10 mg t.d.s. p.o. in the treatment of opioid-induced nausea and emesis in cancer patients. Support Care Cancer 2002; 10:231–236.
- Apfel CC, Jalota L. Can central antiemetic effects of opioids counter-balance opioid-induced nausea and vomiting? Acta Anaesthesiol Scand 2010; 54:129–131.
- Okamoto Y, Tsuneto S, Matsuda Y, et al. A retrospective chart review of the antiemetic effectiveness of risperidone in refractory opioid-induced nausea and vomiting in advanced cancer patients. J Pain Symptom Manage 2007; 34:217–222.
- Overdyk F, Dahan A, Roozekrans M, van der Schrier R, Aarts L, Niesters M. Opioid-induced respiratory depression in the acute care setting: a compendium of case reports. Pain Manag 2014; 4:317–325.
- Niesters M, Overdyk F, Smith T, Aarts L, Dahan A. Buprenorphine-induced respiratory depression and involvement of ABCB1 SNPs in opioid-induced respiratory depression in paediatrics. Br J Anaesth 2013; 110:842–843.
- Niesters M, Mahajan RP, Aarts L, Dahan A. High-inspired oxygen concentration further impairs opioid-induced respiratory depression. Br J Anaesth 2013; 110:837–841.
- Dahan A, Yassen A, Romberg R, et al. Buprenorphine induces ceiling in respiratory depression but not in analgesia. Br J Anaesth 2006; 96:627–632.
- van Dorp E, Yassen A, Sarton E, et al. Naloxone reversal of buprenorphine-induced respiratory depression. Anesthesiology 2006; 105:51–57.
- Macintyre PE, Loadsman JA, Scott DA. Opioids, ventilation and acute pain management. Anaesth Intensive Care 2011; 39:545–558.
- Dahan A, Overdyk F, Smith T, Aarts L, Niesters M. Pharmacovigilance: a review of opioid-induced respiratory depression in chronic pain patients. Pain Physician 2013; 16:E85–E94.
- Correa D, Farney RJ, Chung F, Prasad A, Lam D, Wong J. Chronic opioid use and central sleep apnea: a review of the prevalence, mechanisms, and perioperative considerations. Anesth Analg 2015; 120:1273–1285.
- Randerath WJ, George S. Opioid-induced sleep apnea: is it a real problem? J Clin Sleep Med 2012; 8:577–578.
- Rosow CE, Gomery P, Chen TY, Stefanovich P, Stambler N, Israel R. Reversal of opioid-induced bladder dysfunction by intravenous naloxone and methylnaltrexone. Clin Pharmacol Ther 2007; 82:48–53.
- Weiner DK, Hanlon JT, Studenski SA. Effects of central nervous system polypharmacy on falls liability in community-dwelling elderly. Gerontology 1998; 44:217–221.
- Wolff ML, Kewley R, Hassett M, Collins J, Brodeur MR, Nokes S. Falls in skilled nursing facilities associated with opioid use. J Am Geriatr Soc 2012; 60:987.
- Zylicz Z, Twycross R. Opioid-induced hyperalgesia may be more frequent than previously thought. J Clin Oncol 2008; 26:1564; author reply 1565.
- Lee M, Silverman SM, Hansen H, Patel VB, Manchikanti L. A comprehensive review of opioid-induced hyperalgesia. Pain Physician 2011 2011; 14:145–161.
- Chen L, Sein M, Vo T, et al. Clinical interpretation of opioid tolerance versus opioid-induced hyperalgesia. J Opioid Manag 2014; 10:383–393.
- Vestergaard P, Hermann P, Jensen JE, Eiken P, Mosekilde L. Effects of paracetamol, non-steroidal anti-inflammatory drugs, acetylsalicylic acid, and opioids on bone mineral density and risk of fracture: results of the Danish Osteoporosis Prevention Study (DOPS). Osteoporos Int 2012; 23:1255–1265.
- Sacerdote P, Franchi S, Panerai AE. Non-analgesic effects of opioids: mechanisms and potential clinical relevance of opioid-induced immunodepression. Curr Pharm Des 2012; 18:6034–6042.
- Fine PG, Portenoy RK; Ad Hoc Expert Panel on Evidence Review and Guidelines for Opioid Rotation. Establishing “best practices” for opioid rotation: conclusions of an expert panel. J Pain Symptom Manage 2009; 38:418–425.
- Chibnall JT, Tait RC. Pain assessment in cognitively impaired and unimpaired older adults: a comparison of four scales. Pain 2001; 92:173–186.
- Andrade DC, Faria JW, Caramelli P, et al. The assessment and management of pain in the demented and non-demented elderly patient. Arq Neuropsiquiatr 2011; 69:387–394.
- Scherder E, Herr K, Pickering G, Gibson S, Benedetti F, Lautenbacher S. Pain in dementia. Pain 2009; 145:276–278.
- Chou R, Fanciullo GJ, Fine PG, Miaskowski C, Passik SD, Portenoy RK. Opioids for chronic noncancer pain: prediction and identification of aberrant drug-related behaviors: a review of the evidence for an American Pain Society and American Academy of Pain Medicine clinical practice guideline. J Pain 2009; 10:131–146.
- Butler SF, Budman SH, Fernandez KC, Fanciullo GJ, Jamison RN. Cross-validation of a screener to predict opioid misuse in chronic pain patients (SOAPP-R). J Addict Med 2009; 3:66–73.
- de Leon-Casasola OA. Opioids for chronic pain: new evidence, new strategies, safe prescribing. Am J Med 2013; 126(suppl 1):S3–S11.
- CDC guideline for prescribing opioids for chronic pain—United States, 2016. MMWR Recomm Rep 2016 Mar 18; 65(1):1–49.
- Chou R, Fanciullo GJ, Fine PG, et al; American Pain Society-American Academy of Pain Medicine Opioids Guidelines Panel. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J Pain 2009; 10:113–130.
- West NA, Severtson SG, Green JL, Dart RC. Trends in abuse and misuse of prescription opioids among older adults. Drug Alcohol Depend 2015; 149:117–121.
- American Geriatrics Society Panel on Pharmacological Management of Persistent Pain in Older Persons. Pharmacological management of persistent pain in older persons. J Am Geriatr Soc 2009; 57:1331–1346.
- Weiner DK, Herr K. Comprehensive interdisciplinary assessment and treatment planning: an integrative overview. In: Weiner DK, Herr K, Rudy TE, editors. Persistent pain in older adults: an interdisciplinary guide for treatment. New York, NY: Springer Publishing Company; 2002.
- He W, Sengupta M, Velkoff V; US Census Bureau. 65+ in the United States: 2005. Washington, DC: US Government Printing Office; 2005. www.census.gov/prod/2006pubs/p23-209.pdf. Accessed March 30, 2016.
- American Geriatrics Society Panel on the Pharmacological Management of Persistent Pain in Older Persons. Pharmacological management of persistent pain in older persons. Pain Med 2009; 10:1062–1083.
- Thomas E, Peat G, Harris L, Wilkie R, Croft PR. The prevalence of pain and pain interference in a general population of older adults: cross-sectional findings from the North Staffordshire Osteoarthritis Project (NorStOP). Pain 2004; 110:361–368.
- Caraceni A, Hanks G, Kaasa S, et al; European Palliative Care Research Collaborative (EPCRC); European Association for Palliative Care (EAPC). Use of opioid analgesics in the treatment of cancer pain: evidence-based recommendations from the EAPC. Lancet Oncol 2012; 13:e58–e68.
- Solano JP, Gomes B, Higginson IJ. A comparison of symptom prevalence in far advanced cancer, AIDS, heart disease, chronic obstructive pulmonary disease and renal disease. J Pain Symptom Manage 2006; 31:58–69.
- Ferrell BA, Ferrell BR, Osterweil D. Pain in the nursing home. J Am Geriatr Soc 1990; 38:409–414.
- Ferrell BA, Ferrell BR, Rivera L. Pain in cognitively impaired nursing home patients. J Pain Symptom Manage 1995; 10:591–598.
- Fox PL, Raina P, Jadad AR. Prevalence and treatment of pain in older adults in nursing homes and other long-term care institutions: a systematic review. CMAJ 1999; 160:329–333.
- Stewart C, Leveille SG, Shmerling RH, Samelson EJ, Bean JF, Schofield P. Management of persistent pain in older adults: the MOBILIZE Boston Study. J Am Geriatr Soc 2012; 60:2081–2086.
- Woo J, Ho SC, Lau J, Leung PC. Musculoskeletal complaints and associated consequences in elderly Chinese aged 70 years and over. J Rheumatol 1994; 21:1927–1931.
- Pahor M, Guralnik JM, Wan JY, et al. Lower body osteoarticular pain and dose of analgesic medications in older disabled women: the Women’s Health and Aging Study. Am J Public Health 1999; 89:930–934.
- Marzinski LR. The tragedy of dementia: clinically assessing pain in the confused nonverbal elderly. J Gerontol Nurs 1991; 17:25–28.
- Roy R, Thomas M. A survey of chronic pain in an elderly population. Can Fam Physician 1986; 32:513–516.
- AGS Panel on Persistent Pain in Older Persons. The management of persistent pain in older persons. J Am Geriatr Soc 2002; 50(suppl 6): S205–S224.
- Stanos S, Houle TT. Multidisciplinary and interdisciplinary management of chronic pain. Phys Med Rehabil Clin N Am 2006; 17:435–450.
- Helme RD, Katz B, Gibson SJ, et al. Multidisciplinary pain clinics for older people. Do they have a role? Clin Geriatr Med 1996; 12:563–582.
- Harris DG. Management of pain in advanced disease. Br Med Bull 2014; 110:117–128.
- Raffa RB, Pergolizzi JV. A modern analgesics pain ‘pyramid’. J Clin Pharm Ther 2014; 39:4–6.
- Fine PG, Herr KA. Pharmacologic management of persistent pain in older persons. Clin Geriatr 2009; 17:25–32.
- Tracy B, Sean Morrison R. Pain management in older adults. Clin Ther 2013; 35:1659–1668.
- Malec M, Shega JW. Pain management in the elderly. Med Clin North Am 2015; 99:337–350.
- Abdulla A, Adams N, Bone M, et al; British Geriatric Society. Guidance on the management of pain in older people. Age Ageing 2013; 42(suppl 1):i1–i57.
- Pergolizzi J, Böger RH, Budd K, et al. Opioids and the management of chronic severe pain in the elderly: consensus statement of an International Expert Panel with focus on the six clinically most often used World Health Organization Step III opioids (buprenorphine, fentanyl, hydromorphone, methadone, morphine, oxycodone). Pain Pract 2008; 8:287–313.
- Gloth FM 3rd. Pharmacological management of persistent pain in older persons: focus on opioids and nonopioids. J Pain 2011; 12(suppl 1):S14–S20.
- Gomes T, Mamdani MM, Dhalla IA, Paterson JM, Juurlink DN. Opioid dose and drug-related mortality in patients with nonmalignant pain. Arch Intern Med 2011; 171:686–691.
- Moore RA, McQuay HJ. Prevalence of opioid adverse events in chronic non-malignant pain: systematic review of randomised trials of oral opioids. Arthritis Res Ther 2005; 7:R1046–R1051.
- Candy B, Jones L, Larkin PJ, Vickerstaff V, Tookman A, Stone P. Laxatives for the management of constipation in people receiving palliative care. Cochrane Database Syst Rev 2015; 5:CD003448.
- Webster LR, Butera PG, Moran LV, Wu N, Burns LH, Friedmann N. Oxytrex minimizes physical dependence while providing effective analgesia: a randomized controlled trial in low back pain. J Pain 2006; 7:937–946.
- Paulson DM, Kennedy DT, Donovick RA, et al. Alvimopan: an oral, peripherally acting, mu-opioid receptor antagonist for the treatment of opioid-induced bowel dysfunction—a 21-day treatment-randomized clinical trial. J Pain 2005; 6:184–192.
- Nalamachu SR, Pergolizzi J, Taylor R, et al. Efficacy and tolerability of subcutaneous methylnaltrexone in patients with advanced illness and opioid-induced constipation: a responder analysis of 2 randomized, placebo-controlled trials. Pain Pract 2015; 15:564–571.
- Brick N. Laxatives or methylnaltrexone for the management of constipation in palliative care patients. Clin J Oncol Nurs 2013; 17:91–92.
- Ford AC, Brenner DM, Schoenfeld PS. Efficacy of pharmacological therapies for the treatment of opioid-induced constipation: systematic review and meta-analysis. Am J Gastroenterolt 2013; 108:1566–1575.
- Byas-Smith MG, Chapman SL, Reed B, Cotsonis G. The effect of opioids on driving and psychomotor performance in patients with chronic pain. Clin J Pain 2005; 21:345–352.
- Benyamin R, Trescot AM, Datta S, et al. Opioid complications and side effects. Pain Physician 2008; 11(suppl 2):S105–S120.
- Wilwerding MB, Loprinzi CL, Mailliard JA, et al. A randomized, crossover evaluation of methylphenidate in cancer patients receiving strong narcotics. Support Care Cancer 1995; 3:135–138.
- Bruera E, Miller MJ, Macmillan K, Kuehn N. Neuropsychological effects of methylphenidate in patients receiving a continuous infusion of narcotics for cancer pain. Pain 1992; 48:163–166.
- Ahmedzai S. New approaches to pain control in patients with cancer. Eur J Cancer 1997; 33:S8–S14.
- Laugsand EA, Kaasa S, Klepstad P. Management of opioid-induced nausea and vomiting in cancer patients: systematic review and evidence-based recommendations. Palliat Med 2011; 25:442–453.
- Hardy J, Daly S, McQuade B, et al. A double-blind, randomised, parallel group, multinational, multicentre study comparing a single dose of ondansetron 24 mg p.o. with placebo and metoclopramide 10 mg t.d.s. p.o. in the treatment of opioid-induced nausea and emesis in cancer patients. Support Care Cancer 2002; 10:231–236.
- Apfel CC, Jalota L. Can central antiemetic effects of opioids counter-balance opioid-induced nausea and vomiting? Acta Anaesthesiol Scand 2010; 54:129–131.
- Okamoto Y, Tsuneto S, Matsuda Y, et al. A retrospective chart review of the antiemetic effectiveness of risperidone in refractory opioid-induced nausea and vomiting in advanced cancer patients. J Pain Symptom Manage 2007; 34:217–222.
- Overdyk F, Dahan A, Roozekrans M, van der Schrier R, Aarts L, Niesters M. Opioid-induced respiratory depression in the acute care setting: a compendium of case reports. Pain Manag 2014; 4:317–325.
- Niesters M, Overdyk F, Smith T, Aarts L, Dahan A. Buprenorphine-induced respiratory depression and involvement of ABCB1 SNPs in opioid-induced respiratory depression in paediatrics. Br J Anaesth 2013; 110:842–843.
- Niesters M, Mahajan RP, Aarts L, Dahan A. High-inspired oxygen concentration further impairs opioid-induced respiratory depression. Br J Anaesth 2013; 110:837–841.
- Dahan A, Yassen A, Romberg R, et al. Buprenorphine induces ceiling in respiratory depression but not in analgesia. Br J Anaesth 2006; 96:627–632.
- van Dorp E, Yassen A, Sarton E, et al. Naloxone reversal of buprenorphine-induced respiratory depression. Anesthesiology 2006; 105:51–57.
- Macintyre PE, Loadsman JA, Scott DA. Opioids, ventilation and acute pain management. Anaesth Intensive Care 2011; 39:545–558.
- Dahan A, Overdyk F, Smith T, Aarts L, Niesters M. Pharmacovigilance: a review of opioid-induced respiratory depression in chronic pain patients. Pain Physician 2013; 16:E85–E94.
- Correa D, Farney RJ, Chung F, Prasad A, Lam D, Wong J. Chronic opioid use and central sleep apnea: a review of the prevalence, mechanisms, and perioperative considerations. Anesth Analg 2015; 120:1273–1285.
- Randerath WJ, George S. Opioid-induced sleep apnea: is it a real problem? J Clin Sleep Med 2012; 8:577–578.
- Rosow CE, Gomery P, Chen TY, Stefanovich P, Stambler N, Israel R. Reversal of opioid-induced bladder dysfunction by intravenous naloxone and methylnaltrexone. Clin Pharmacol Ther 2007; 82:48–53.
- Weiner DK, Hanlon JT, Studenski SA. Effects of central nervous system polypharmacy on falls liability in community-dwelling elderly. Gerontology 1998; 44:217–221.
- Wolff ML, Kewley R, Hassett M, Collins J, Brodeur MR, Nokes S. Falls in skilled nursing facilities associated with opioid use. J Am Geriatr Soc 2012; 60:987.
- Zylicz Z, Twycross R. Opioid-induced hyperalgesia may be more frequent than previously thought. J Clin Oncol 2008; 26:1564; author reply 1565.
- Lee M, Silverman SM, Hansen H, Patel VB, Manchikanti L. A comprehensive review of opioid-induced hyperalgesia. Pain Physician 2011 2011; 14:145–161.
- Chen L, Sein M, Vo T, et al. Clinical interpretation of opioid tolerance versus opioid-induced hyperalgesia. J Opioid Manag 2014; 10:383–393.
- Vestergaard P, Hermann P, Jensen JE, Eiken P, Mosekilde L. Effects of paracetamol, non-steroidal anti-inflammatory drugs, acetylsalicylic acid, and opioids on bone mineral density and risk of fracture: results of the Danish Osteoporosis Prevention Study (DOPS). Osteoporos Int 2012; 23:1255–1265.
- Sacerdote P, Franchi S, Panerai AE. Non-analgesic effects of opioids: mechanisms and potential clinical relevance of opioid-induced immunodepression. Curr Pharm Des 2012; 18:6034–6042.
- Fine PG, Portenoy RK; Ad Hoc Expert Panel on Evidence Review and Guidelines for Opioid Rotation. Establishing “best practices” for opioid rotation: conclusions of an expert panel. J Pain Symptom Manage 2009; 38:418–425.
- Chibnall JT, Tait RC. Pain assessment in cognitively impaired and unimpaired older adults: a comparison of four scales. Pain 2001; 92:173–186.
- Andrade DC, Faria JW, Caramelli P, et al. The assessment and management of pain in the demented and non-demented elderly patient. Arq Neuropsiquiatr 2011; 69:387–394.
- Scherder E, Herr K, Pickering G, Gibson S, Benedetti F, Lautenbacher S. Pain in dementia. Pain 2009; 145:276–278.
- Chou R, Fanciullo GJ, Fine PG, Miaskowski C, Passik SD, Portenoy RK. Opioids for chronic noncancer pain: prediction and identification of aberrant drug-related behaviors: a review of the evidence for an American Pain Society and American Academy of Pain Medicine clinical practice guideline. J Pain 2009; 10:131–146.
- Butler SF, Budman SH, Fernandez KC, Fanciullo GJ, Jamison RN. Cross-validation of a screener to predict opioid misuse in chronic pain patients (SOAPP-R). J Addict Med 2009; 3:66–73.
- de Leon-Casasola OA. Opioids for chronic pain: new evidence, new strategies, safe prescribing. Am J Med 2013; 126(suppl 1):S3–S11.
- CDC guideline for prescribing opioids for chronic pain—United States, 2016. MMWR Recomm Rep 2016 Mar 18; 65(1):1–49.
KEY POINTS
- Treatment of persistent pain in older adults presents several challenges.
- Often, persistent pain is underrecognized and undertreated, impairing function and reducing quality of life.
- A combination of pharmacologic and nonpharmacologic strategies is needed to address the multiple factors contributing to pain and manage it effectively.
- The World Health Organization’s three-step ladder is valuable for treating persistent pain in older adults.
- Although nonopioids are the first-line treatments for persistent pain, opioids are also important to provide safe and effective pain management in older adults.