User login
Rapidly Progressive Necrotizing Myositis Mimicking Pyoderma Gangrenosum
To the Editor:
Necrotizing myositis (NM) is an exceedingly rare necrotizing soft-tissue infection (NSTI) that is characterized by skeletal muscle involvement. β -Hemolytic streptococci, such as Streptococcus pyogenes , are the most common causative organisms. The overall prevalence and incidence of NM is unknown. A review of the literature by Adams et al 2 identified only 21 cases between 1900 and 1985.
Timely treatment of this infection leads to improved outcomes, but diagnosis can be challenging due to the ambiguous presentation of NM and lack of specific cutaneous changes.3 Clinical manifestations including bullae, blisters, vesicles, and petechiae become more prominent as infection progresses.4 If NM is suspected due to cutaneous manifestations, it is imperative that the underlying cause be identified; for example, NM must be distinguished from the overlapping presentation of pyoderma gangrenosum (PG). Because NM has nearly 100% mortality without prompt surgical intervention, early identification is critical.5 Herein, we report a case of NM that illustrates the correlation of clinical, histological, and imaging findings required to diagnose this potentially fatal infection.
An 80-year-old man presented to the emergency department with worsening pain, edema, and spreading redness of the right wrist over the last 5 weeks. He had a history of atopic dermatitis that was refractory to topical steroids and methotrexate; he was dependent on an oral steroid (prednisone 30 mg/d) for symptom control. The patient reported minor trauma to the area after performing home renovations. He received numerous rounds of oral antibiotics as an outpatient for presumed cellulitis and reported he was “getting better” but that the signs and symptoms of the condition grew worse after outpatient arthrocentesis. Dermatology was consulted to evaluate for a necrotizing neutrophilic dermatosis such as PG.
At the current presentation, the patient was tachycardic and afebrile (temperature, 98.2 °F [36.8 °C]). Physical examination revealed large, exquisitely tender, ill-defined necrotic ulceration of the right wrist with purulent debris and diffuse edema (Figure 1). Sequential evaluation at 6-hour intervals revealed notably increasing purulence, edema, and tenderness. Interconnected sinus tracts that extended to the fascial plane were observed.

Laboratory workup was notable for a markedly elevated C-reactive protein level of 18.9 mg/dL (reference range, 0–0.8 mg/dL) and an elevated white blood cell count of 19.92×109/L (reference range, 4.5–11.0×109/L). Blood and tissue cultures were positive for methicillin-sensitive Staphylococcus aureus. Computed tomography and magnetic resonance imaging (MRI) prior to biopsy demonstrated findings consistent with extensive subcutaneous and intramuscular areas of loculation and foci of gas (Figure 2). These findings were consistent with intramuscular involvement. A punch biopsy revealed a necrotic epidermis filled with neutrophilic pustules and a dense dermal infiltrate of neutrophilic inflammation consistent with infection (Figure 3).
, contrast-enhancing erosions of the carpus (arrows), and rim-enhancing")
Emergency surgery was performed with debridement of necrotic tissue and muscle. Postoperatively, he became more clinically stable after being placed on cefazolin through a peripherally inserted central catheter. He underwent 4 additional washouts over the ensuing month, as well as tendon reconstructions, a radial forearm flap, and reverse radial forearm flap reconstruction of the forearm. At the time of publication, there has been no recurrence. The patient’s atopic dermatitis is well controlled on dupilumab and topical fluocinonide alone, with a recent IgA level of 1 g/L and a body surface area measurement of 2%. Dupilumab was started 3 months after surgery.
.")
Necrotizing myositis is a rare, rapidly progressive infection involving muscle that can manifest as superficial cutaneous involvement. The clinical manifestation of NM is harder to recognize than other NSTIs such as necrotizing fasciitis, likely due to the initial prodromal phase of NM, which consists of nonspecific constitutional symptoms.3 Systemic findings such as tachycardia, fever, hypotension, and shock occur in only 10% to 40% of NM patients.4,5
In our patient, clues of NM included fulfillment of criteria for systemic inflammatory response syndrome at admission and a presumed source of infection; taken together, these findings should lead to a diagnosis of sepsis until otherwise proven. The patient also reported pain that was not proportional to the skin findings, which suggested an NSTI. His lack of constitutional symptoms may have been due to the effects of prednisone, which was changed to dupilumab during hospitalization.
The clinical and histological findings of NM are nonspecific. Clinical findings include skin discoloration with bullae, blisters, vesicles, or petechiae.4 Our case adds to the descriptive morphology by including marked edema with ulceration, progressive purulence, and interconnected sinuses tracking to the fascial plane. Histologic findings can include confluent necrosis extending from the epidermis to the underlying muscle with dense neutrophilic inflammation. Notably, these findings can mirror necrotizing neutrophilic dermatoses in the absence of an infectious cause. Failure to recognize simple systemic inflammatory response syndrome criteria in NM patients due to slow treatment response or incorrect treatment can can lead to loss of a limb or death.
Workup reveals overlap with necrotizing neutrophilic dermatoses including PG, which is the prototypical neutrophilic dermatosis. Morphologically, PG presents as an ulcer with a purple and undermined border, often having developed from an initial papule, vesicle, or pustule. A neutrophilic infiltrate of the ulcer edge is the major criterion required to diagnose PG6; minor criteria include a positive pathergy test, history of inflammatory arthritis or inflammatory bowel disease, and exclusion of infection.6 When compared directly to an NSTI such as NM, the most important variable that sets PG apart is the absence of bacterial growth on blood and tissue cultures.7
Imaging studies can aid in the clinical diagnosis of NM and help distinguish the disease from PG. Computed tomography and MRI may demonstrate hallmarks of extensive necrotizing infection, such as gas formation and consequent fascial swelling, thickening and edema of involved muscle, and subfascial fluid collection.3,4 Distinct from NM, imaging findings in PG are more subtle, suggesting cellulitic inflammation with edema.8 A defining radiographic feature of NM can be foci of gas within muscle or fascia, though absence of this finding does not exclude NM.1,4
In conclusion, NM is a rare intramuscular infection that can be difficult to diagnose due to its nonspecific presentation and lack of constitutional symptoms. Dermatologists should maintain a high level of suspicion for NM in the setting of rapidly progressive clinical findings; accurate diagnosis requires a multimodal approach with complete correlation of clinical, histological, and imaging findings. Computed tomography and MRI can heighten the approach, even when necrotizing neutrophilic dermatoses and NM have similar clinical and histological appearances. Once a diagnosis of NM is established, prompt surgical and medical intervention improves the prognosis.
- Stevens DL, Baddour LM. Necrotizing soft tissue infections. UpToDate. Updated October 7, 2022. Accessed February 13, 2024. https://www.uptodate.com/contents/necrotizing-soft-tissue-infections?search=Necrotizing%20soft%20tissue%20infections&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1
- Adams EM, Gudmundsson S, Yocum DE, et al. Streptococcal myositis. Arch Intern Med . 1985;145:1020-1023.
- Khanna A, Gurusinghe D, Taylor D. Necrotizing myositis: highlighting the hidden depths—case series and review of the literature. ANZ J Surg . 2020;90:130-134. doi:10.1111/ans.15429
- Boinpally H, Howell RS, Ram B, et al. Necrotizing myositis: a rare necrotizing soft tissue infection involving muscle. Wounds . 2018;30:E116-E120.
- Anaya DA, Dellinger EP. Necrotizing soft-tissue infection: diagnosis and management. Clin Infect Dis . 2007;44:705-710. doi:10.1086/511638
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatol . 2018;154:461-466. doi:10.1001/jamadermatol.2017.5980
- Sanchez IM, Lowenstein S, Johnson KA, et al. Clinical features of neutrophilic dermatosis variants resembling necrotizing fasciitis. JAMA Dermatol . 2019;155:79-84. doi:10.1001/jamadermatol.2018.3890
- Demirdover C, Geyik A, Vayvada H. Necrotising fasciitis or pyoderma gangrenosum: a fatal dilemma. Int Wound J . 2019;16:1347-1353. doi:10.1111/iwj.13196
To the Editor:
Necrotizing myositis (NM) is an exceedingly rare necrotizing soft-tissue infection (NSTI) that is characterized by skeletal muscle involvement. β -Hemolytic streptococci, such as Streptococcus pyogenes , are the most common causative organisms. The overall prevalence and incidence of NM is unknown. A review of the literature by Adams et al 2 identified only 21 cases between 1900 and 1985.
Timely treatment of this infection leads to improved outcomes, but diagnosis can be challenging due to the ambiguous presentation of NM and lack of specific cutaneous changes.3 Clinical manifestations including bullae, blisters, vesicles, and petechiae become more prominent as infection progresses.4 If NM is suspected due to cutaneous manifestations, it is imperative that the underlying cause be identified; for example, NM must be distinguished from the overlapping presentation of pyoderma gangrenosum (PG). Because NM has nearly 100% mortality without prompt surgical intervention, early identification is critical.5 Herein, we report a case of NM that illustrates the correlation of clinical, histological, and imaging findings required to diagnose this potentially fatal infection.
An 80-year-old man presented to the emergency department with worsening pain, edema, and spreading redness of the right wrist over the last 5 weeks. He had a history of atopic dermatitis that was refractory to topical steroids and methotrexate; he was dependent on an oral steroid (prednisone 30 mg/d) for symptom control. The patient reported minor trauma to the area after performing home renovations. He received numerous rounds of oral antibiotics as an outpatient for presumed cellulitis and reported he was “getting better” but that the signs and symptoms of the condition grew worse after outpatient arthrocentesis. Dermatology was consulted to evaluate for a necrotizing neutrophilic dermatosis such as PG.
At the current presentation, the patient was tachycardic and afebrile (temperature, 98.2 °F [36.8 °C]). Physical examination revealed large, exquisitely tender, ill-defined necrotic ulceration of the right wrist with purulent debris and diffuse edema (Figure 1). Sequential evaluation at 6-hour intervals revealed notably increasing purulence, edema, and tenderness. Interconnected sinus tracts that extended to the fascial plane were observed.
Laboratory workup was notable for a markedly elevated C-reactive protein level of 18.9 mg/dL (reference range, 0–0.8 mg/dL) and an elevated white blood cell count of 19.92×109/L (reference range, 4.5–11.0×109/L). Blood and tissue cultures were positive for methicillin-sensitive Staphylococcus aureus. Computed tomography and magnetic resonance imaging (MRI) prior to biopsy demonstrated findings consistent with extensive subcutaneous and intramuscular areas of loculation and foci of gas (Figure 2). These findings were consistent with intramuscular involvement. A punch biopsy revealed a necrotic epidermis filled with neutrophilic pustules and a dense dermal infiltrate of neutrophilic inflammation consistent with infection (Figure 3).
Emergency surgery was performed with debridement of necrotic tissue and muscle. Postoperatively, he became more clinically stable after being placed on cefazolin through a peripherally inserted central catheter. He underwent 4 additional washouts over the ensuing month, as well as tendon reconstructions, a radial forearm flap, and reverse radial forearm flap reconstruction of the forearm. At the time of publication, there has been no recurrence. The patient’s atopic dermatitis is well controlled on dupilumab and topical fluocinonide alone, with a recent IgA level of 1 g/L and a body surface area measurement of 2%. Dupilumab was started 3 months after surgery.
Necrotizing myositis is a rare, rapidly progressive infection involving muscle that can manifest as superficial cutaneous involvement. The clinical manifestation of NM is harder to recognize than other NSTIs such as necrotizing fasciitis, likely due to the initial prodromal phase of NM, which consists of nonspecific constitutional symptoms.3 Systemic findings such as tachycardia, fever, hypotension, and shock occur in only 10% to 40% of NM patients.4,5
In our patient, clues of NM included fulfillment of criteria for systemic inflammatory response syndrome at admission and a presumed source of infection; taken together, these findings should lead to a diagnosis of sepsis until otherwise proven. The patient also reported pain that was not proportional to the skin findings, which suggested an NSTI. His lack of constitutional symptoms may have been due to the effects of prednisone, which was changed to dupilumab during hospitalization.
The clinical and histological findings of NM are nonspecific. Clinical findings include skin discoloration with bullae, blisters, vesicles, or petechiae.4 Our case adds to the descriptive morphology by including marked edema with ulceration, progressive purulence, and interconnected sinuses tracking to the fascial plane. Histologic findings can include confluent necrosis extending from the epidermis to the underlying muscle with dense neutrophilic inflammation. Notably, these findings can mirror necrotizing neutrophilic dermatoses in the absence of an infectious cause. Failure to recognize simple systemic inflammatory response syndrome criteria in NM patients due to slow treatment response or incorrect treatment can can lead to loss of a limb or death.
Workup reveals overlap with necrotizing neutrophilic dermatoses including PG, which is the prototypical neutrophilic dermatosis. Morphologically, PG presents as an ulcer with a purple and undermined border, often having developed from an initial papule, vesicle, or pustule. A neutrophilic infiltrate of the ulcer edge is the major criterion required to diagnose PG6; minor criteria include a positive pathergy test, history of inflammatory arthritis or inflammatory bowel disease, and exclusion of infection.6 When compared directly to an NSTI such as NM, the most important variable that sets PG apart is the absence of bacterial growth on blood and tissue cultures.7
Imaging studies can aid in the clinical diagnosis of NM and help distinguish the disease from PG. Computed tomography and MRI may demonstrate hallmarks of extensive necrotizing infection, such as gas formation and consequent fascial swelling, thickening and edema of involved muscle, and subfascial fluid collection.3,4 Distinct from NM, imaging findings in PG are more subtle, suggesting cellulitic inflammation with edema.8 A defining radiographic feature of NM can be foci of gas within muscle or fascia, though absence of this finding does not exclude NM.1,4
In conclusion, NM is a rare intramuscular infection that can be difficult to diagnose due to its nonspecific presentation and lack of constitutional symptoms. Dermatologists should maintain a high level of suspicion for NM in the setting of rapidly progressive clinical findings; accurate diagnosis requires a multimodal approach with complete correlation of clinical, histological, and imaging findings. Computed tomography and MRI can heighten the approach, even when necrotizing neutrophilic dermatoses and NM have similar clinical and histological appearances. Once a diagnosis of NM is established, prompt surgical and medical intervention improves the prognosis.
To the Editor:
Necrotizing myositis (NM) is an exceedingly rare necrotizing soft-tissue infection (NSTI) that is characterized by skeletal muscle involvement. β -Hemolytic streptococci, such as Streptococcus pyogenes , are the most common causative organisms. The overall prevalence and incidence of NM is unknown. A review of the literature by Adams et al 2 identified only 21 cases between 1900 and 1985.
Timely treatment of this infection leads to improved outcomes, but diagnosis can be challenging due to the ambiguous presentation of NM and lack of specific cutaneous changes.3 Clinical manifestations including bullae, blisters, vesicles, and petechiae become more prominent as infection progresses.4 If NM is suspected due to cutaneous manifestations, it is imperative that the underlying cause be identified; for example, NM must be distinguished from the overlapping presentation of pyoderma gangrenosum (PG). Because NM has nearly 100% mortality without prompt surgical intervention, early identification is critical.5 Herein, we report a case of NM that illustrates the correlation of clinical, histological, and imaging findings required to diagnose this potentially fatal infection.
An 80-year-old man presented to the emergency department with worsening pain, edema, and spreading redness of the right wrist over the last 5 weeks. He had a history of atopic dermatitis that was refractory to topical steroids and methotrexate; he was dependent on an oral steroid (prednisone 30 mg/d) for symptom control. The patient reported minor trauma to the area after performing home renovations. He received numerous rounds of oral antibiotics as an outpatient for presumed cellulitis and reported he was “getting better” but that the signs and symptoms of the condition grew worse after outpatient arthrocentesis. Dermatology was consulted to evaluate for a necrotizing neutrophilic dermatosis such as PG.
At the current presentation, the patient was tachycardic and afebrile (temperature, 98.2 °F [36.8 °C]). Physical examination revealed large, exquisitely tender, ill-defined necrotic ulceration of the right wrist with purulent debris and diffuse edema (Figure 1). Sequential evaluation at 6-hour intervals revealed notably increasing purulence, edema, and tenderness. Interconnected sinus tracts that extended to the fascial plane were observed.
Laboratory workup was notable for a markedly elevated C-reactive protein level of 18.9 mg/dL (reference range, 0–0.8 mg/dL) and an elevated white blood cell count of 19.92×109/L (reference range, 4.5–11.0×109/L). Blood and tissue cultures were positive for methicillin-sensitive Staphylococcus aureus. Computed tomography and magnetic resonance imaging (MRI) prior to biopsy demonstrated findings consistent with extensive subcutaneous and intramuscular areas of loculation and foci of gas (Figure 2). These findings were consistent with intramuscular involvement. A punch biopsy revealed a necrotic epidermis filled with neutrophilic pustules and a dense dermal infiltrate of neutrophilic inflammation consistent with infection (Figure 3).
Emergency surgery was performed with debridement of necrotic tissue and muscle. Postoperatively, he became more clinically stable after being placed on cefazolin through a peripherally inserted central catheter. He underwent 4 additional washouts over the ensuing month, as well as tendon reconstructions, a radial forearm flap, and reverse radial forearm flap reconstruction of the forearm. At the time of publication, there has been no recurrence. The patient’s atopic dermatitis is well controlled on dupilumab and topical fluocinonide alone, with a recent IgA level of 1 g/L and a body surface area measurement of 2%. Dupilumab was started 3 months after surgery.
Necrotizing myositis is a rare, rapidly progressive infection involving muscle that can manifest as superficial cutaneous involvement. The clinical manifestation of NM is harder to recognize than other NSTIs such as necrotizing fasciitis, likely due to the initial prodromal phase of NM, which consists of nonspecific constitutional symptoms.3 Systemic findings such as tachycardia, fever, hypotension, and shock occur in only 10% to 40% of NM patients.4,5
In our patient, clues of NM included fulfillment of criteria for systemic inflammatory response syndrome at admission and a presumed source of infection; taken together, these findings should lead to a diagnosis of sepsis until otherwise proven. The patient also reported pain that was not proportional to the skin findings, which suggested an NSTI. His lack of constitutional symptoms may have been due to the effects of prednisone, which was changed to dupilumab during hospitalization.
The clinical and histological findings of NM are nonspecific. Clinical findings include skin discoloration with bullae, blisters, vesicles, or petechiae.4 Our case adds to the descriptive morphology by including marked edema with ulceration, progressive purulence, and interconnected sinuses tracking to the fascial plane. Histologic findings can include confluent necrosis extending from the epidermis to the underlying muscle with dense neutrophilic inflammation. Notably, these findings can mirror necrotizing neutrophilic dermatoses in the absence of an infectious cause. Failure to recognize simple systemic inflammatory response syndrome criteria in NM patients due to slow treatment response or incorrect treatment can can lead to loss of a limb or death.
Workup reveals overlap with necrotizing neutrophilic dermatoses including PG, which is the prototypical neutrophilic dermatosis. Morphologically, PG presents as an ulcer with a purple and undermined border, often having developed from an initial papule, vesicle, or pustule. A neutrophilic infiltrate of the ulcer edge is the major criterion required to diagnose PG6; minor criteria include a positive pathergy test, history of inflammatory arthritis or inflammatory bowel disease, and exclusion of infection.6 When compared directly to an NSTI such as NM, the most important variable that sets PG apart is the absence of bacterial growth on blood and tissue cultures.7
Imaging studies can aid in the clinical diagnosis of NM and help distinguish the disease from PG. Computed tomography and MRI may demonstrate hallmarks of extensive necrotizing infection, such as gas formation and consequent fascial swelling, thickening and edema of involved muscle, and subfascial fluid collection.3,4 Distinct from NM, imaging findings in PG are more subtle, suggesting cellulitic inflammation with edema.8 A defining radiographic feature of NM can be foci of gas within muscle or fascia, though absence of this finding does not exclude NM.1,4
In conclusion, NM is a rare intramuscular infection that can be difficult to diagnose due to its nonspecific presentation and lack of constitutional symptoms. Dermatologists should maintain a high level of suspicion for NM in the setting of rapidly progressive clinical findings; accurate diagnosis requires a multimodal approach with complete correlation of clinical, histological, and imaging findings. Computed tomography and MRI can heighten the approach, even when necrotizing neutrophilic dermatoses and NM have similar clinical and histological appearances. Once a diagnosis of NM is established, prompt surgical and medical intervention improves the prognosis.
- Stevens DL, Baddour LM. Necrotizing soft tissue infections. UpToDate. Updated October 7, 2022. Accessed February 13, 2024. https://www.uptodate.com/contents/necrotizing-soft-tissue-infections?search=Necrotizing%20soft%20tissue%20infections&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1
- Adams EM, Gudmundsson S, Yocum DE, et al. Streptococcal myositis. Arch Intern Med . 1985;145:1020-1023.
- Khanna A, Gurusinghe D, Taylor D. Necrotizing myositis: highlighting the hidden depths—case series and review of the literature. ANZ J Surg . 2020;90:130-134. doi:10.1111/ans.15429
- Boinpally H, Howell RS, Ram B, et al. Necrotizing myositis: a rare necrotizing soft tissue infection involving muscle. Wounds . 2018;30:E116-E120.
- Anaya DA, Dellinger EP. Necrotizing soft-tissue infection: diagnosis and management. Clin Infect Dis . 2007;44:705-710. doi:10.1086/511638
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatol . 2018;154:461-466. doi:10.1001/jamadermatol.2017.5980
- Sanchez IM, Lowenstein S, Johnson KA, et al. Clinical features of neutrophilic dermatosis variants resembling necrotizing fasciitis. JAMA Dermatol . 2019;155:79-84. doi:10.1001/jamadermatol.2018.3890
- Demirdover C, Geyik A, Vayvada H. Necrotising fasciitis or pyoderma gangrenosum: a fatal dilemma. Int Wound J . 2019;16:1347-1353. doi:10.1111/iwj.13196
- Stevens DL, Baddour LM. Necrotizing soft tissue infections. UpToDate. Updated October 7, 2022. Accessed February 13, 2024. https://www.uptodate.com/contents/necrotizing-soft-tissue-infections?search=Necrotizing%20soft%20tissue%20infections&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1
- Adams EM, Gudmundsson S, Yocum DE, et al. Streptococcal myositis. Arch Intern Med . 1985;145:1020-1023.
- Khanna A, Gurusinghe D, Taylor D. Necrotizing myositis: highlighting the hidden depths—case series and review of the literature. ANZ J Surg . 2020;90:130-134. doi:10.1111/ans.15429
- Boinpally H, Howell RS, Ram B, et al. Necrotizing myositis: a rare necrotizing soft tissue infection involving muscle. Wounds . 2018;30:E116-E120.
- Anaya DA, Dellinger EP. Necrotizing soft-tissue infection: diagnosis and management. Clin Infect Dis . 2007;44:705-710. doi:10.1086/511638
- Maverakis E, Ma C, Shinkai K, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi consensus of international experts. JAMA Dermatol . 2018;154:461-466. doi:10.1001/jamadermatol.2017.5980
- Sanchez IM, Lowenstein S, Johnson KA, et al. Clinical features of neutrophilic dermatosis variants resembling necrotizing fasciitis. JAMA Dermatol . 2019;155:79-84. doi:10.1001/jamadermatol.2018.3890
- Demirdover C, Geyik A, Vayvada H. Necrotising fasciitis or pyoderma gangrenosum: a fatal dilemma. Int Wound J . 2019;16:1347-1353. doi:10.1111/iwj.13196
Practice Points
- The accurate diagnosis of necrotizing myositis (NM) requires a multimodal approach with complete clinical, histological, and radiographic correlation.
- Necrotizing myositis can manifest as violaceous erythematous plaques, bullae, blisters, or vesicles with petechiae, marked edema with ulceration, progressive purulence, and interconnected sinuses tracking to the fascial plane.
- The differential diagnosis of NM includes pyoderma gangrenosum.

Nonepidemic Kaposi Sarcoma: A Case of a Rare Epidemiologic Subtype
To the Editor:
Kaposi sarcoma (KS) is a rare angioproliferative disorder associated with human herpesvirus 8 (HHV-8) infection.1 There are 4 main recognized epidemiologic forms of KS: classic, endemic, epidemic, and iatrogenic (Table). Nonepidemic KS is a recently described rare fifth type of KS that occurs in a subset of patients who do not fit the other classifications—HIV-negative patients without detectable cellular or humoral immune deficiency. This subset has been described as clinically similar to classic KS with limited disease but occurring in younger men.2,3 We describe a case of nonepidemic KS in a Middle Eastern heterosexual immunocompetent man.

A 30-year-old man presented for evaluation of a growth on the nose of 3 months’ duration. The patient reported being otherwise healthy and was not taking long-term medications. He denied a history of malignancy, organ transplant, or immunosuppressive therapy. He was born in Syria and lived in Thailand for several years prior to moving to the United States. HIV testing 6 months prior to presentation was negative. He denied fever, chills, lymphadenopathy, shortness of breath, hemoptysis, melena, hematochezia, and intravenous drug use.

Physical examination revealed a solitary shiny, 7-mm, pink-red papule on the nasal dorsum (Figure 1). No other skin or mucosal lesions were identified. There was no cervical, axillary, or inguinal lymphadenopathy. A laboratory workup consisting of serum immunoglobulins and serum protein electrophoresis was unremarkable. Tests for HIV-1 and HIV-2 as well as human T-lymphotropic virus 1 and 2 were negative. The CD4 and CD8 counts were within reference range. Histopathology of a shave biopsy revealed a dermal spindle cell proliferation arranged in short intersecting fascicles and admixed with plasma cells and occasional mitotic figures. Immunohistochemistry showed that the spindle cells stained positive for CD34, CD31, and HHV-8 (Figure 2). The lesion resolved after treatment with cryotherapy. Repeat HIV testing 3 months later was negative. No recurrence or new lesions were identified at 3-month follow-up.

Similar to the other subtypes of KS, the nonepidemic form is dependent on HHV-8 infection, which is more commonly transmitted via saliva and sexual contact.3,4 After infecting endothelial cells, HHV-8 is believed to activate the mammalian target of rapamycin and nuclear factor κB pathways, resulting in aberrant cellular differentiation and neoangiogenesis through upregulation of vascular endothelial growth factor and basic fibroblast growth factor.2,4 Similar to what is seen with other herpesviruses, HHV-8 infection typically is lifelong due to the virus’s ability to establish latency within human B cells and endothelial cells as well as undergo sporadic bouts of lytic reactivation during its life cycle.4
Nonepidemic KS resembles other variants clinically, manifesting as erythematous or violaceous, painless, nonblanchable macules, papules, and nodules.1 Early lesions often are asymptomatic and can manifest as pigmented macules or small papules that vary from pale pink to vivid purple. Nodules also can occur and be exophytic and ulcerated with bleeding.1 Secondary lymphoproliferative disorders including Castleman disease and lymphoma have been reported.2,5
In contrast to other types of KS in which pulmonary or gastrointestinal tract lesions can develop with hemoptysis or hematochezia, mucocutaneous and visceral lesions rarely are reported in nonepidemic KS.3 Lymphedema, a feature associated with endemic KS, is notably absent in nonepidemic KS.1,3
The differential diagnosis applicable to all KS subtypes includes other vascular lesions such as angiomatosis and angiosarcoma. Histopathologic analysis is critical to differentiate KS from these conditions; visual diagnosis alone has only an 80% positive predictive value for KS.4 The histopathologic presentation of KS is a vascular proliferation in the dermis accompanied by an increased number of vessels without an endothelial cell lining.4 Spindle cell proliferation also is a common feature and is considered to be the KS tumor cell. Immunostaining for HHV-8 antigen as well as for CD31 and CD34 can be used to confirm the diagnosis.4
The management and prognosis of KS depends on the epidemiologic subtype. Classic and nonepidemic KS generally are indolent with a good prognosis. Periodic follow-up is recommended because of an increased risk for secondary malignancy such as lymphoma. The treatment of epidemic KS is highly active antiretroviral therapy. Similarly, reduction of immunosuppression is warranted for iatrogenic KS. For all types, cutaneous lesions can be treated with local excision, cryosurgery, radiation, chemotherapy, intralesional vincristine, or a topical agent such as imiquimod or alitretinoin.6
- Hinojosa T, Lewis DJ, Liu M, et al. Nonepidemic Kaposi sarcoma: a recently proposed category. J Am Acad Dermatol. 2017;3:441-443. doi: 10.1016/j.jdcr.2017.04.012
- Heymann WR. Nonepidemic Kaposi sarcoma: the fifth dimension. Dermatology World Insights and Inquiries. Published October 16, 2019. Accessed January 30, 2024. https://www.aad.org/dw/dw-insights-and-inquiries/2019-archive/october/nonepidemic-kaposi-sarcoma
- Vangipuram R, Tyring SK. Epidemiology of Kaposi sarcoma: review and description of the nonepidemic variant. Int J Dermatol. 2019;58:538-542. doi: 10.1111/ijd.14080
- Cesarman E, Damania B, Krown SE, et al. Kaposi sarcoma. Nat Rev Dis Primers. 2019;5:9. doi:10.1038/s41572-019-0060-9
- Vecerek N, Truong A, Turner R, et al. Nonepidemic Kaposi’s sarcoma: an underrecognized subtype in HIV-negative patients. J Am Acad Dermatol. 2019;81(suppl 1):AB247. doi:10.1016/j.jaad.2019.09.1096
- Schneider JW, Dittmer DP. Diagnosis and treatment of Kaposi sarcoma. Am J Clin Dermatol. 2017;18:529-539. doi:10.1007/s40257-017-0270-4
To the Editor:
Kaposi sarcoma (KS) is a rare angioproliferative disorder associated with human herpesvirus 8 (HHV-8) infection.1 There are 4 main recognized epidemiologic forms of KS: classic, endemic, epidemic, and iatrogenic (Table). Nonepidemic KS is a recently described rare fifth type of KS that occurs in a subset of patients who do not fit the other classifications—HIV-negative patients without detectable cellular or humoral immune deficiency. This subset has been described as clinically similar to classic KS with limited disease but occurring in younger men.2,3 We describe a case of nonepidemic KS in a Middle Eastern heterosexual immunocompetent man.
A 30-year-old man presented for evaluation of a growth on the nose of 3 months’ duration. The patient reported being otherwise healthy and was not taking long-term medications. He denied a history of malignancy, organ transplant, or immunosuppressive therapy. He was born in Syria and lived in Thailand for several years prior to moving to the United States. HIV testing 6 months prior to presentation was negative. He denied fever, chills, lymphadenopathy, shortness of breath, hemoptysis, melena, hematochezia, and intravenous drug use.
Physical examination revealed a solitary shiny, 7-mm, pink-red papule on the nasal dorsum (Figure 1). No other skin or mucosal lesions were identified. There was no cervical, axillary, or inguinal lymphadenopathy. A laboratory workup consisting of serum immunoglobulins and serum protein electrophoresis was unremarkable. Tests for HIV-1 and HIV-2 as well as human T-lymphotropic virus 1 and 2 were negative. The CD4 and CD8 counts were within reference range. Histopathology of a shave biopsy revealed a dermal spindle cell proliferation arranged in short intersecting fascicles and admixed with plasma cells and occasional mitotic figures. Immunohistochemistry showed that the spindle cells stained positive for CD34, CD31, and HHV-8 (Figure 2). The lesion resolved after treatment with cryotherapy. Repeat HIV testing 3 months later was negative. No recurrence or new lesions were identified at 3-month follow-up.
Similar to the other subtypes of KS, the nonepidemic form is dependent on HHV-8 infection, which is more commonly transmitted via saliva and sexual contact.3,4 After infecting endothelial cells, HHV-8 is believed to activate the mammalian target of rapamycin and nuclear factor κB pathways, resulting in aberrant cellular differentiation and neoangiogenesis through upregulation of vascular endothelial growth factor and basic fibroblast growth factor.2,4 Similar to what is seen with other herpesviruses, HHV-8 infection typically is lifelong due to the virus’s ability to establish latency within human B cells and endothelial cells as well as undergo sporadic bouts of lytic reactivation during its life cycle.4
Nonepidemic KS resembles other variants clinically, manifesting as erythematous or violaceous, painless, nonblanchable macules, papules, and nodules.1 Early lesions often are asymptomatic and can manifest as pigmented macules or small papules that vary from pale pink to vivid purple. Nodules also can occur and be exophytic and ulcerated with bleeding.1 Secondary lymphoproliferative disorders including Castleman disease and lymphoma have been reported.2,5
In contrast to other types of KS in which pulmonary or gastrointestinal tract lesions can develop with hemoptysis or hematochezia, mucocutaneous and visceral lesions rarely are reported in nonepidemic KS.3 Lymphedema, a feature associated with endemic KS, is notably absent in nonepidemic KS.1,3
The differential diagnosis applicable to all KS subtypes includes other vascular lesions such as angiomatosis and angiosarcoma. Histopathologic analysis is critical to differentiate KS from these conditions; visual diagnosis alone has only an 80% positive predictive value for KS.4 The histopathologic presentation of KS is a vascular proliferation in the dermis accompanied by an increased number of vessels without an endothelial cell lining.4 Spindle cell proliferation also is a common feature and is considered to be the KS tumor cell. Immunostaining for HHV-8 antigen as well as for CD31 and CD34 can be used to confirm the diagnosis.4
The management and prognosis of KS depends on the epidemiologic subtype. Classic and nonepidemic KS generally are indolent with a good prognosis. Periodic follow-up is recommended because of an increased risk for secondary malignancy such as lymphoma. The treatment of epidemic KS is highly active antiretroviral therapy. Similarly, reduction of immunosuppression is warranted for iatrogenic KS. For all types, cutaneous lesions can be treated with local excision, cryosurgery, radiation, chemotherapy, intralesional vincristine, or a topical agent such as imiquimod or alitretinoin.6
To the Editor:
Kaposi sarcoma (KS) is a rare angioproliferative disorder associated with human herpesvirus 8 (HHV-8) infection.1 There are 4 main recognized epidemiologic forms of KS: classic, endemic, epidemic, and iatrogenic (Table). Nonepidemic KS is a recently described rare fifth type of KS that occurs in a subset of patients who do not fit the other classifications—HIV-negative patients without detectable cellular or humoral immune deficiency. This subset has been described as clinically similar to classic KS with limited disease but occurring in younger men.2,3 We describe a case of nonepidemic KS in a Middle Eastern heterosexual immunocompetent man.
A 30-year-old man presented for evaluation of a growth on the nose of 3 months’ duration. The patient reported being otherwise healthy and was not taking long-term medications. He denied a history of malignancy, organ transplant, or immunosuppressive therapy. He was born in Syria and lived in Thailand for several years prior to moving to the United States. HIV testing 6 months prior to presentation was negative. He denied fever, chills, lymphadenopathy, shortness of breath, hemoptysis, melena, hematochezia, and intravenous drug use.
Physical examination revealed a solitary shiny, 7-mm, pink-red papule on the nasal dorsum (Figure 1). No other skin or mucosal lesions were identified. There was no cervical, axillary, or inguinal lymphadenopathy. A laboratory workup consisting of serum immunoglobulins and serum protein electrophoresis was unremarkable. Tests for HIV-1 and HIV-2 as well as human T-lymphotropic virus 1 and 2 were negative. The CD4 and CD8 counts were within reference range. Histopathology of a shave biopsy revealed a dermal spindle cell proliferation arranged in short intersecting fascicles and admixed with plasma cells and occasional mitotic figures. Immunohistochemistry showed that the spindle cells stained positive for CD34, CD31, and HHV-8 (Figure 2). The lesion resolved after treatment with cryotherapy. Repeat HIV testing 3 months later was negative. No recurrence or new lesions were identified at 3-month follow-up.
Similar to the other subtypes of KS, the nonepidemic form is dependent on HHV-8 infection, which is more commonly transmitted via saliva and sexual contact.3,4 After infecting endothelial cells, HHV-8 is believed to activate the mammalian target of rapamycin and nuclear factor κB pathways, resulting in aberrant cellular differentiation and neoangiogenesis through upregulation of vascular endothelial growth factor and basic fibroblast growth factor.2,4 Similar to what is seen with other herpesviruses, HHV-8 infection typically is lifelong due to the virus’s ability to establish latency within human B cells and endothelial cells as well as undergo sporadic bouts of lytic reactivation during its life cycle.4
Nonepidemic KS resembles other variants clinically, manifesting as erythematous or violaceous, painless, nonblanchable macules, papules, and nodules.1 Early lesions often are asymptomatic and can manifest as pigmented macules or small papules that vary from pale pink to vivid purple. Nodules also can occur and be exophytic and ulcerated with bleeding.1 Secondary lymphoproliferative disorders including Castleman disease and lymphoma have been reported.2,5
In contrast to other types of KS in which pulmonary or gastrointestinal tract lesions can develop with hemoptysis or hematochezia, mucocutaneous and visceral lesions rarely are reported in nonepidemic KS.3 Lymphedema, a feature associated with endemic KS, is notably absent in nonepidemic KS.1,3
The differential diagnosis applicable to all KS subtypes includes other vascular lesions such as angiomatosis and angiosarcoma. Histopathologic analysis is critical to differentiate KS from these conditions; visual diagnosis alone has only an 80% positive predictive value for KS.4 The histopathologic presentation of KS is a vascular proliferation in the dermis accompanied by an increased number of vessels without an endothelial cell lining.4 Spindle cell proliferation also is a common feature and is considered to be the KS tumor cell. Immunostaining for HHV-8 antigen as well as for CD31 and CD34 can be used to confirm the diagnosis.4
The management and prognosis of KS depends on the epidemiologic subtype. Classic and nonepidemic KS generally are indolent with a good prognosis. Periodic follow-up is recommended because of an increased risk for secondary malignancy such as lymphoma. The treatment of epidemic KS is highly active antiretroviral therapy. Similarly, reduction of immunosuppression is warranted for iatrogenic KS. For all types, cutaneous lesions can be treated with local excision, cryosurgery, radiation, chemotherapy, intralesional vincristine, or a topical agent such as imiquimod or alitretinoin.6
- Hinojosa T, Lewis DJ, Liu M, et al. Nonepidemic Kaposi sarcoma: a recently proposed category. J Am Acad Dermatol. 2017;3:441-443. doi: 10.1016/j.jdcr.2017.04.012
- Heymann WR. Nonepidemic Kaposi sarcoma: the fifth dimension. Dermatology World Insights and Inquiries. Published October 16, 2019. Accessed January 30, 2024. https://www.aad.org/dw/dw-insights-and-inquiries/2019-archive/october/nonepidemic-kaposi-sarcoma
- Vangipuram R, Tyring SK. Epidemiology of Kaposi sarcoma: review and description of the nonepidemic variant. Int J Dermatol. 2019;58:538-542. doi: 10.1111/ijd.14080
- Cesarman E, Damania B, Krown SE, et al. Kaposi sarcoma. Nat Rev Dis Primers. 2019;5:9. doi:10.1038/s41572-019-0060-9
- Vecerek N, Truong A, Turner R, et al. Nonepidemic Kaposi’s sarcoma: an underrecognized subtype in HIV-negative patients. J Am Acad Dermatol. 2019;81(suppl 1):AB247. doi:10.1016/j.jaad.2019.09.1096
- Schneider JW, Dittmer DP. Diagnosis and treatment of Kaposi sarcoma. Am J Clin Dermatol. 2017;18:529-539. doi:10.1007/s40257-017-0270-4
- Hinojosa T, Lewis DJ, Liu M, et al. Nonepidemic Kaposi sarcoma: a recently proposed category. J Am Acad Dermatol. 2017;3:441-443. doi: 10.1016/j.jdcr.2017.04.012
- Heymann WR. Nonepidemic Kaposi sarcoma: the fifth dimension. Dermatology World Insights and Inquiries. Published October 16, 2019. Accessed January 30, 2024. https://www.aad.org/dw/dw-insights-and-inquiries/2019-archive/october/nonepidemic-kaposi-sarcoma
- Vangipuram R, Tyring SK. Epidemiology of Kaposi sarcoma: review and description of the nonepidemic variant. Int J Dermatol. 2019;58:538-542. doi: 10.1111/ijd.14080
- Cesarman E, Damania B, Krown SE, et al. Kaposi sarcoma. Nat Rev Dis Primers. 2019;5:9. doi:10.1038/s41572-019-0060-9
- Vecerek N, Truong A, Turner R, et al. Nonepidemic Kaposi’s sarcoma: an underrecognized subtype in HIV-negative patients. J Am Acad Dermatol. 2019;81(suppl 1):AB247. doi:10.1016/j.jaad.2019.09.1096
- Schneider JW, Dittmer DP. Diagnosis and treatment of Kaposi sarcoma. Am J Clin Dermatol. 2017;18:529-539. doi:10.1007/s40257-017-0270-4
Practice Points
- Nonepidemic Kaposi sarcoma (KS) is a recently described fifth subtype of the disease that typically occurs in younger men who are HIV-negative without detectable cellular or humoral immune deficiency.
- The cutaneous manifestations of nonepidemic KS are similar to those of classic KS, except that disease extent is limited and the prognosis is favorable in nonepidemic KS.
- Dermatologists should consider KS when a patient presents with clinically representative findings, even in the absence of typical risk factors such as immunosuppression.

Painful Retiform Purpura in a Peritoneal Dialysis Patient
The Diagnosis: Calcific Uremic Arteriolopathy
Computed tomography of the abdomen and pelvis with contrast revealed a right complex renal cyst with peripheral calcification; computed tomography of the head without contrast revealed atherosclerotic changes with calcification of the intracranial arteries, vertebral basilar arteries, and bilateral branches of the ophthalmic artery. Histopathology revealed occlusive vasculopathy with epidermal ischemic changes as well as dermal and subcutaneous vascular congestion and small thrombi. Within the subcutis, there were tiny stippled calcium deposits within very small vascular lumina (Figure). The combination of clinical and histological findings was highly suggestive of calcific uremic arteriolopathy, and the patient was transitioned to hemodialysis against a low-calcium bath to avoid hypercalcemia. Unfortunately, she developed complications related to sepsis and experienced worsening mentation. After a discussion with palliative care, the patient was transitioned to comfort measures and discharged home on hospice 1 week after the biopsy at her family’s request.
.")
Calcific uremic arteriolopathy (also known as calciphylaxis) is a rare, life-threatening syndrome of widespread vascular calcification leading to microvascular occlusion within the dermis and subcutaneous tissues.1 Clinically, it typically manifests as severely painful, purpuric skin lesions that evolve through phases of blistering, ulceration, and ultimately visible skin necrosis.2 The pain likely is a consequence of ischemia and nociceptive activation and often may precede any visibly apparent skin lesions.3 Risk factors associated with the development of this condition include female sex; history of diabetes mellitus, obesity, rapid weight loss, or end-stage renal disease; abnormalities in calcium and phosphorus homeostasis; and vitamin K deficiency.1,3 It is more prevalent in patients on peritoneal dialysis compared to hemodialysis.4
Calciphylaxis is diagnosed with combined clinical and histopathological evidence. Laboratory test abnormalities are not specific for disease; therefore, skin biopsy is the standard confirmatory test, though its practice is contentious due to the risk for nonhealing ulceration and increasing risk for infection.1 Findings suggestive of disease include focal to diffuse calcification (intravascular, extravascular, or perieccrine), superficial fat calcium deposition, mid panniculus calcium deposition, mid panniculus vascular thrombi, and focal to diffuse angioplasia.5 The hallmark feature is diffuse calcification of small capillaries in adipose tissue.6
The mortality rate associated with this disease is high—a 6-month mortality rate of 27% to 43% has been reported from the time of diagnosis7-9—which often is related to subsequent superimposed infections patients acquire from necrotic skin tissue.2 The disease also carries high morbidity, with patients experiencing frequent hospitalizations related to pain, infections, and nonhealing wounds.6 There is no standard treatment, and trials have been limited to small sample sizes. A multidisciplinary treatment approach is essential to maximize outcomes, which includes wound care, risk factor modification, analgesia, and symptomatic management strategies.1,2,6
Some pharmacologic agents have received noteworthy attention in treating calciphylaxis, including sodium thiosulfate (STS), bisphosphonates, and vitamin K supplementation.1 The strongest evidence supporting the use of STS comes from 2 trials involving 53 and 27 dialysis patients, with complete remission in 14 (26%) and 14 (52%) patients, respectively.10,11 However, these trials did not include control groups to compare outcomes, and mortality rates were similarly high among partial responders and nonresponders compared with patients not treated with STS. A 2018 systematic review failed to assess the efficacy of STS alone for the treatment of calciphylaxis but suggested there may be a future role for it, with 251 of 358 patients (70.1%) responding to therapy.12
Erythema ab igne is a cutaneous reaction related to long-term heat exposure, often from electronic devices such as laptops, heating pads, space heaters, or hot-water bottles.13,14 Clinically, this rash appears as an erythematous, purpuric, or hyperpigmented reticular dermatosis that is below the clinical threshold to define a thermal burn.13 Lesions often are seen on the anterior thighs or across the abdomen.15 There usually are no long-term clinical sequelae; however, rare malignant transformation has been documented in cases of atrophy or nonhealing ulceration.16 Treatment is supportive with removal of the offending agent, but hyperpigmentation may persist for months to years.14
Livedo reticularis is a cutaneous pattern of mottled violaceous or hyperpigmented changes that often signifies underlying vascular dermal changes.17 It can be seen in various pathologic states, including vasculitis, autoimmune disease, connective tissue disease, neurologic disease, infection, or malignancy, or it can be drug induced.18 There are no pathognomonic microscopic changes, as the histology will drastically differ based on the etiology. Workup can be extensive; cues to the underlying pathology should be sought based on the patient’s history and concurrent presenting symptoms. Livedo reticularis is the most common dermatologic finding in patients with antiphospholipid syndrome, and workup should include antiphospholipid antibodies (eg, lupus anticoagulant, anticardiolipin, anti–beta-2-glycoproteins) as well as lupus testing (eg, antinuclear antibodies, anti– double-stranded DNA).19 Treatment is targeted at the underlying disease process.
Cryoglobulinemia is a disease characterized by abnormal serum immunoglobulins that precipitate at cold temperatures and is further subcategorized by the type of complexes that are deposited.20 Type I represents purely monoclonal cryoglobulins, type III purely polyclonal, and type II a mixed picture. Clinical manifestations arise from excessive deposition of these proteins in the skin, joints, peripheral vasculature, and kidneys leading to purpuric skin lesions, chronic ulceration, arthralgia, and glomerulonephritis. Cutaneous findings may include erythematous to purpuric macular or papular changes with or without the presence of ulceration, infarction, or hemorrhagic crusting.21 Systemic disease often underlies a diagnosis, and further investigation for hepatitis C virus, connective tissue disease, and hematologic malignancies should be considered.20 Treatment is targeted at underlying systemic disease, such as antiviral treatment for hepatitis or chemotherapeutic regimens for hematologic disease.22
Polyarteritis nodosa is a systemic necrotizing vasculitis that typically involves small- to medium-sized arteries. Cutaneous manifestations often include subcutaneous nodules, livedo reticularis, and ulcerations most found on the lower extremities.23 Systemic symptoms including fever, myalgia, arthralgia, and neuropathy often are present. Characteristic histopathology findings include inflammation and destruction of medium-sized arteries at the junctional zone of the dermis and subcutis along with microaneurysms along the vessels.24 Treatment is based on the severity of disease, with localized cutaneous disease often being controlled with topical steroids and anti-inflammatory agents, while more widespread disease requires immunosuppression with systemic steroids, hydroxychloroquine, azathioprine, methotrexate, mycophenolate mofetil, or intravenous immunoglobulins.23
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714. doi:10.1056/NEJMra1505292
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Chang JJ. Calciphylaxis: diagnosis, pathogenesis, and treatment. Adv Skin Wound Care. 2019;32:205-215. doi:10.1097/01 .ASW.0000554443.14002.13
- Zhang Y, Corapi KM, Luongo M, et al. Calciphylaxis in peritoneal dialysis patients: a single center cohort study. Int J Nephrol Renovasc Dis. 2016;9:235-241. doi:10.2147/ijnrd.S115701
- Chen TY, Lehman JS, Gibson LE, et al. Histopathology of calciphylaxis: cohort study with clinical correlations. Am J Dermatopathol. 2017;39:795-802. doi:10.1097/DAD.0000000000000824
- Kodumudi V, Jeha GM, Mydlo N, et al. Management of cutaneous calciphylaxis. Adv Ther. 2020;37:4797-4807. doi:10.1007 /s12325-020-01504-w
- Nigwekar SU, Zhao S, Wenger J, et al. A nationally representative study of calcific uremic arteriolopathy risk factors. J Am Soc Nephrol. 2016;27:3421-3429. doi:10.1681/asn.2015091065
- McCarthy JT, El-Azhary RA, Patzelt MT, et al. Survival, risk factors, and effect of treatment in 101 patients with calciphylaxis. Mayo Clin Proc. 2016;91:1384-1394. doi:10.1016/j.mayocp.2016.06.025
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217. doi:10.1046/j.1523-1755.2002.00375.x
- Nigwekar SU, Brunelli SM, Meade D, et al. Sodium thiosulfate therapy for calcific uremic arteriolopathy. Clin J Am Soc Nephrol. 2013;8:1162-1170. doi:10.2215/cjn.09880912
- Zitt E, König M, Vychytil A, et al. Use of sodium thiosulphate in a multi-interventional setting for the treatment of calciphylaxis in dialysis patients. Nephrol Dial Transplant. 2013;28:1232-1240. doi:10.1093/ndt/gfs548
- Peng T, Zhuo L, Wang Y, et al. Systematic review of sodium thiosulfate in treating calciphylaxis in chronic kidney disease patients. Nephrology (Carlton). 2018;23:669-675. doi:10.1111/nep.13081
- Miller K, Hunt R, Chu J, et al. Erythema ab igne. Dermatol Online J. 2011;17:28.
- Kettelhut EA, Traylor J, Sathe NC, et al. Erythema ab igne. StatPearls. StatPearls Publishing; 2022.
- Knöpfel N, Weibel L. Erythema Ab Igne. JAMA Dermatol. 2021;157: 106. doi:10.1001/jamadermatol.2020.3995
- Sigmon JR, Cantrell J, Teague D, et al. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35:676-678. doi:10.1097/DAD.0b013e3182871648
- Rose AE, Sagger V, Boyd KP, et al. Livedo reticularis. Dermatol Online J. 2013;19:20705.
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103/2229-5178.164493
- Uthman IW, Khamashta MA. Livedo racemosa: a striking dermatological sign for the antiphospholipid syndrome. J Rheumatol. 2006;33:2379-2382.
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j .jbspin.2019.01.016
- Cohen SJ, Pittelkow MR, Su WP. Cutaneous manifestations of cryoglobulinemia: clinical and histopathologic study of seventy-two patients. J Am Acad Dermatol. 1991;25(1, pt 1):21-27. doi:10.1016 /0190-9622(91)70168-2
- Takada S, Shimizu T, Hadano Y, et al. Cryoglobulinemia (review). Mol Med Rep. 2012;6:3-8. doi:10.3892/mmr.2012.861
- Turska M, Parada-Turska J. Cutaneous polyarteritis nodosa. Wiad Lek. 2018;71(1, pt 1):73-77.
- De Virgilio A, Greco A, Magliulo G, et al. Polyarteritis nodosa: a contemporary overview. Autoimmun Rev. 2016;15:564-570. doi:10.1016/j.autrev.2016.02.015
The Diagnosis: Calcific Uremic Arteriolopathy
Computed tomography of the abdomen and pelvis with contrast revealed a right complex renal cyst with peripheral calcification; computed tomography of the head without contrast revealed atherosclerotic changes with calcification of the intracranial arteries, vertebral basilar arteries, and bilateral branches of the ophthalmic artery. Histopathology revealed occlusive vasculopathy with epidermal ischemic changes as well as dermal and subcutaneous vascular congestion and small thrombi. Within the subcutis, there were tiny stippled calcium deposits within very small vascular lumina (Figure). The combination of clinical and histological findings was highly suggestive of calcific uremic arteriolopathy, and the patient was transitioned to hemodialysis against a low-calcium bath to avoid hypercalcemia. Unfortunately, she developed complications related to sepsis and experienced worsening mentation. After a discussion with palliative care, the patient was transitioned to comfort measures and discharged home on hospice 1 week after the biopsy at her family’s request.
Calcific uremic arteriolopathy (also known as calciphylaxis) is a rare, life-threatening syndrome of widespread vascular calcification leading to microvascular occlusion within the dermis and subcutaneous tissues.1 Clinically, it typically manifests as severely painful, purpuric skin lesions that evolve through phases of blistering, ulceration, and ultimately visible skin necrosis.2 The pain likely is a consequence of ischemia and nociceptive activation and often may precede any visibly apparent skin lesions.3 Risk factors associated with the development of this condition include female sex; history of diabetes mellitus, obesity, rapid weight loss, or end-stage renal disease; abnormalities in calcium and phosphorus homeostasis; and vitamin K deficiency.1,3 It is more prevalent in patients on peritoneal dialysis compared to hemodialysis.4
Calciphylaxis is diagnosed with combined clinical and histopathological evidence. Laboratory test abnormalities are not specific for disease; therefore, skin biopsy is the standard confirmatory test, though its practice is contentious due to the risk for nonhealing ulceration and increasing risk for infection.1 Findings suggestive of disease include focal to diffuse calcification (intravascular, extravascular, or perieccrine), superficial fat calcium deposition, mid panniculus calcium deposition, mid panniculus vascular thrombi, and focal to diffuse angioplasia.5 The hallmark feature is diffuse calcification of small capillaries in adipose tissue.6
The mortality rate associated with this disease is high—a 6-month mortality rate of 27% to 43% has been reported from the time of diagnosis7-9—which often is related to subsequent superimposed infections patients acquire from necrotic skin tissue.2 The disease also carries high morbidity, with patients experiencing frequent hospitalizations related to pain, infections, and nonhealing wounds.6 There is no standard treatment, and trials have been limited to small sample sizes. A multidisciplinary treatment approach is essential to maximize outcomes, which includes wound care, risk factor modification, analgesia, and symptomatic management strategies.1,2,6
Some pharmacologic agents have received noteworthy attention in treating calciphylaxis, including sodium thiosulfate (STS), bisphosphonates, and vitamin K supplementation.1 The strongest evidence supporting the use of STS comes from 2 trials involving 53 and 27 dialysis patients, with complete remission in 14 (26%) and 14 (52%) patients, respectively.10,11 However, these trials did not include control groups to compare outcomes, and mortality rates were similarly high among partial responders and nonresponders compared with patients not treated with STS. A 2018 systematic review failed to assess the efficacy of STS alone for the treatment of calciphylaxis but suggested there may be a future role for it, with 251 of 358 patients (70.1%) responding to therapy.12
Erythema ab igne is a cutaneous reaction related to long-term heat exposure, often from electronic devices such as laptops, heating pads, space heaters, or hot-water bottles.13,14 Clinically, this rash appears as an erythematous, purpuric, or hyperpigmented reticular dermatosis that is below the clinical threshold to define a thermal burn.13 Lesions often are seen on the anterior thighs or across the abdomen.15 There usually are no long-term clinical sequelae; however, rare malignant transformation has been documented in cases of atrophy or nonhealing ulceration.16 Treatment is supportive with removal of the offending agent, but hyperpigmentation may persist for months to years.14
Livedo reticularis is a cutaneous pattern of mottled violaceous or hyperpigmented changes that often signifies underlying vascular dermal changes.17 It can be seen in various pathologic states, including vasculitis, autoimmune disease, connective tissue disease, neurologic disease, infection, or malignancy, or it can be drug induced.18 There are no pathognomonic microscopic changes, as the histology will drastically differ based on the etiology. Workup can be extensive; cues to the underlying pathology should be sought based on the patient’s history and concurrent presenting symptoms. Livedo reticularis is the most common dermatologic finding in patients with antiphospholipid syndrome, and workup should include antiphospholipid antibodies (eg, lupus anticoagulant, anticardiolipin, anti–beta-2-glycoproteins) as well as lupus testing (eg, antinuclear antibodies, anti– double-stranded DNA).19 Treatment is targeted at the underlying disease process.
Cryoglobulinemia is a disease characterized by abnormal serum immunoglobulins that precipitate at cold temperatures and is further subcategorized by the type of complexes that are deposited.20 Type I represents purely monoclonal cryoglobulins, type III purely polyclonal, and type II a mixed picture. Clinical manifestations arise from excessive deposition of these proteins in the skin, joints, peripheral vasculature, and kidneys leading to purpuric skin lesions, chronic ulceration, arthralgia, and glomerulonephritis. Cutaneous findings may include erythematous to purpuric macular or papular changes with or without the presence of ulceration, infarction, or hemorrhagic crusting.21 Systemic disease often underlies a diagnosis, and further investigation for hepatitis C virus, connective tissue disease, and hematologic malignancies should be considered.20 Treatment is targeted at underlying systemic disease, such as antiviral treatment for hepatitis or chemotherapeutic regimens for hematologic disease.22
Polyarteritis nodosa is a systemic necrotizing vasculitis that typically involves small- to medium-sized arteries. Cutaneous manifestations often include subcutaneous nodules, livedo reticularis, and ulcerations most found on the lower extremities.23 Systemic symptoms including fever, myalgia, arthralgia, and neuropathy often are present. Characteristic histopathology findings include inflammation and destruction of medium-sized arteries at the junctional zone of the dermis and subcutis along with microaneurysms along the vessels.24 Treatment is based on the severity of disease, with localized cutaneous disease often being controlled with topical steroids and anti-inflammatory agents, while more widespread disease requires immunosuppression with systemic steroids, hydroxychloroquine, azathioprine, methotrexate, mycophenolate mofetil, or intravenous immunoglobulins.23
The Diagnosis: Calcific Uremic Arteriolopathy
Computed tomography of the abdomen and pelvis with contrast revealed a right complex renal cyst with peripheral calcification; computed tomography of the head without contrast revealed atherosclerotic changes with calcification of the intracranial arteries, vertebral basilar arteries, and bilateral branches of the ophthalmic artery. Histopathology revealed occlusive vasculopathy with epidermal ischemic changes as well as dermal and subcutaneous vascular congestion and small thrombi. Within the subcutis, there were tiny stippled calcium deposits within very small vascular lumina (Figure). The combination of clinical and histological findings was highly suggestive of calcific uremic arteriolopathy, and the patient was transitioned to hemodialysis against a low-calcium bath to avoid hypercalcemia. Unfortunately, she developed complications related to sepsis and experienced worsening mentation. After a discussion with palliative care, the patient was transitioned to comfort measures and discharged home on hospice 1 week after the biopsy at her family’s request.
Calcific uremic arteriolopathy (also known as calciphylaxis) is a rare, life-threatening syndrome of widespread vascular calcification leading to microvascular occlusion within the dermis and subcutaneous tissues.1 Clinically, it typically manifests as severely painful, purpuric skin lesions that evolve through phases of blistering, ulceration, and ultimately visible skin necrosis.2 The pain likely is a consequence of ischemia and nociceptive activation and often may precede any visibly apparent skin lesions.3 Risk factors associated with the development of this condition include female sex; history of diabetes mellitus, obesity, rapid weight loss, or end-stage renal disease; abnormalities in calcium and phosphorus homeostasis; and vitamin K deficiency.1,3 It is more prevalent in patients on peritoneal dialysis compared to hemodialysis.4
Calciphylaxis is diagnosed with combined clinical and histopathological evidence. Laboratory test abnormalities are not specific for disease; therefore, skin biopsy is the standard confirmatory test, though its practice is contentious due to the risk for nonhealing ulceration and increasing risk for infection.1 Findings suggestive of disease include focal to diffuse calcification (intravascular, extravascular, or perieccrine), superficial fat calcium deposition, mid panniculus calcium deposition, mid panniculus vascular thrombi, and focal to diffuse angioplasia.5 The hallmark feature is diffuse calcification of small capillaries in adipose tissue.6
The mortality rate associated with this disease is high—a 6-month mortality rate of 27% to 43% has been reported from the time of diagnosis7-9—which often is related to subsequent superimposed infections patients acquire from necrotic skin tissue.2 The disease also carries high morbidity, with patients experiencing frequent hospitalizations related to pain, infections, and nonhealing wounds.6 There is no standard treatment, and trials have been limited to small sample sizes. A multidisciplinary treatment approach is essential to maximize outcomes, which includes wound care, risk factor modification, analgesia, and symptomatic management strategies.1,2,6
Some pharmacologic agents have received noteworthy attention in treating calciphylaxis, including sodium thiosulfate (STS), bisphosphonates, and vitamin K supplementation.1 The strongest evidence supporting the use of STS comes from 2 trials involving 53 and 27 dialysis patients, with complete remission in 14 (26%) and 14 (52%) patients, respectively.10,11 However, these trials did not include control groups to compare outcomes, and mortality rates were similarly high among partial responders and nonresponders compared with patients not treated with STS. A 2018 systematic review failed to assess the efficacy of STS alone for the treatment of calciphylaxis but suggested there may be a future role for it, with 251 of 358 patients (70.1%) responding to therapy.12
Erythema ab igne is a cutaneous reaction related to long-term heat exposure, often from electronic devices such as laptops, heating pads, space heaters, or hot-water bottles.13,14 Clinically, this rash appears as an erythematous, purpuric, or hyperpigmented reticular dermatosis that is below the clinical threshold to define a thermal burn.13 Lesions often are seen on the anterior thighs or across the abdomen.15 There usually are no long-term clinical sequelae; however, rare malignant transformation has been documented in cases of atrophy or nonhealing ulceration.16 Treatment is supportive with removal of the offending agent, but hyperpigmentation may persist for months to years.14
Livedo reticularis is a cutaneous pattern of mottled violaceous or hyperpigmented changes that often signifies underlying vascular dermal changes.17 It can be seen in various pathologic states, including vasculitis, autoimmune disease, connective tissue disease, neurologic disease, infection, or malignancy, or it can be drug induced.18 There are no pathognomonic microscopic changes, as the histology will drastically differ based on the etiology. Workup can be extensive; cues to the underlying pathology should be sought based on the patient’s history and concurrent presenting symptoms. Livedo reticularis is the most common dermatologic finding in patients with antiphospholipid syndrome, and workup should include antiphospholipid antibodies (eg, lupus anticoagulant, anticardiolipin, anti–beta-2-glycoproteins) as well as lupus testing (eg, antinuclear antibodies, anti– double-stranded DNA).19 Treatment is targeted at the underlying disease process.
Cryoglobulinemia is a disease characterized by abnormal serum immunoglobulins that precipitate at cold temperatures and is further subcategorized by the type of complexes that are deposited.20 Type I represents purely monoclonal cryoglobulins, type III purely polyclonal, and type II a mixed picture. Clinical manifestations arise from excessive deposition of these proteins in the skin, joints, peripheral vasculature, and kidneys leading to purpuric skin lesions, chronic ulceration, arthralgia, and glomerulonephritis. Cutaneous findings may include erythematous to purpuric macular or papular changes with or without the presence of ulceration, infarction, or hemorrhagic crusting.21 Systemic disease often underlies a diagnosis, and further investigation for hepatitis C virus, connective tissue disease, and hematologic malignancies should be considered.20 Treatment is targeted at underlying systemic disease, such as antiviral treatment for hepatitis or chemotherapeutic regimens for hematologic disease.22
Polyarteritis nodosa is a systemic necrotizing vasculitis that typically involves small- to medium-sized arteries. Cutaneous manifestations often include subcutaneous nodules, livedo reticularis, and ulcerations most found on the lower extremities.23 Systemic symptoms including fever, myalgia, arthralgia, and neuropathy often are present. Characteristic histopathology findings include inflammation and destruction of medium-sized arteries at the junctional zone of the dermis and subcutis along with microaneurysms along the vessels.24 Treatment is based on the severity of disease, with localized cutaneous disease often being controlled with topical steroids and anti-inflammatory agents, while more widespread disease requires immunosuppression with systemic steroids, hydroxychloroquine, azathioprine, methotrexate, mycophenolate mofetil, or intravenous immunoglobulins.23
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714. doi:10.1056/NEJMra1505292
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Chang JJ. Calciphylaxis: diagnosis, pathogenesis, and treatment. Adv Skin Wound Care. 2019;32:205-215. doi:10.1097/01 .ASW.0000554443.14002.13
- Zhang Y, Corapi KM, Luongo M, et al. Calciphylaxis in peritoneal dialysis patients: a single center cohort study. Int J Nephrol Renovasc Dis. 2016;9:235-241. doi:10.2147/ijnrd.S115701
- Chen TY, Lehman JS, Gibson LE, et al. Histopathology of calciphylaxis: cohort study with clinical correlations. Am J Dermatopathol. 2017;39:795-802. doi:10.1097/DAD.0000000000000824
- Kodumudi V, Jeha GM, Mydlo N, et al. Management of cutaneous calciphylaxis. Adv Ther. 2020;37:4797-4807. doi:10.1007 /s12325-020-01504-w
- Nigwekar SU, Zhao S, Wenger J, et al. A nationally representative study of calcific uremic arteriolopathy risk factors. J Am Soc Nephrol. 2016;27:3421-3429. doi:10.1681/asn.2015091065
- McCarthy JT, El-Azhary RA, Patzelt MT, et al. Survival, risk factors, and effect of treatment in 101 patients with calciphylaxis. Mayo Clin Proc. 2016;91:1384-1394. doi:10.1016/j.mayocp.2016.06.025
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217. doi:10.1046/j.1523-1755.2002.00375.x
- Nigwekar SU, Brunelli SM, Meade D, et al. Sodium thiosulfate therapy for calcific uremic arteriolopathy. Clin J Am Soc Nephrol. 2013;8:1162-1170. doi:10.2215/cjn.09880912
- Zitt E, König M, Vychytil A, et al. Use of sodium thiosulphate in a multi-interventional setting for the treatment of calciphylaxis in dialysis patients. Nephrol Dial Transplant. 2013;28:1232-1240. doi:10.1093/ndt/gfs548
- Peng T, Zhuo L, Wang Y, et al. Systematic review of sodium thiosulfate in treating calciphylaxis in chronic kidney disease patients. Nephrology (Carlton). 2018;23:669-675. doi:10.1111/nep.13081
- Miller K, Hunt R, Chu J, et al. Erythema ab igne. Dermatol Online J. 2011;17:28.
- Kettelhut EA, Traylor J, Sathe NC, et al. Erythema ab igne. StatPearls. StatPearls Publishing; 2022.
- Knöpfel N, Weibel L. Erythema Ab Igne. JAMA Dermatol. 2021;157: 106. doi:10.1001/jamadermatol.2020.3995
- Sigmon JR, Cantrell J, Teague D, et al. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35:676-678. doi:10.1097/DAD.0b013e3182871648
- Rose AE, Sagger V, Boyd KP, et al. Livedo reticularis. Dermatol Online J. 2013;19:20705.
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103/2229-5178.164493
- Uthman IW, Khamashta MA. Livedo racemosa: a striking dermatological sign for the antiphospholipid syndrome. J Rheumatol. 2006;33:2379-2382.
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j .jbspin.2019.01.016
- Cohen SJ, Pittelkow MR, Su WP. Cutaneous manifestations of cryoglobulinemia: clinical and histopathologic study of seventy-two patients. J Am Acad Dermatol. 1991;25(1, pt 1):21-27. doi:10.1016 /0190-9622(91)70168-2
- Takada S, Shimizu T, Hadano Y, et al. Cryoglobulinemia (review). Mol Med Rep. 2012;6:3-8. doi:10.3892/mmr.2012.861
- Turska M, Parada-Turska J. Cutaneous polyarteritis nodosa. Wiad Lek. 2018;71(1, pt 1):73-77.
- De Virgilio A, Greco A, Magliulo G, et al. Polyarteritis nodosa: a contemporary overview. Autoimmun Rev. 2016;15:564-570. doi:10.1016/j.autrev.2016.02.015
- Nigwekar SU, Thadhani R, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714. doi:10.1056/NEJMra1505292
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146. doi:10.1053/j.ajkd.2015.01.034
- Chang JJ. Calciphylaxis: diagnosis, pathogenesis, and treatment. Adv Skin Wound Care. 2019;32:205-215. doi:10.1097/01 .ASW.0000554443.14002.13
- Zhang Y, Corapi KM, Luongo M, et al. Calciphylaxis in peritoneal dialysis patients: a single center cohort study. Int J Nephrol Renovasc Dis. 2016;9:235-241. doi:10.2147/ijnrd.S115701
- Chen TY, Lehman JS, Gibson LE, et al. Histopathology of calciphylaxis: cohort study with clinical correlations. Am J Dermatopathol. 2017;39:795-802. doi:10.1097/DAD.0000000000000824
- Kodumudi V, Jeha GM, Mydlo N, et al. Management of cutaneous calciphylaxis. Adv Ther. 2020;37:4797-4807. doi:10.1007 /s12325-020-01504-w
- Nigwekar SU, Zhao S, Wenger J, et al. A nationally representative study of calcific uremic arteriolopathy risk factors. J Am Soc Nephrol. 2016;27:3421-3429. doi:10.1681/asn.2015091065
- McCarthy JT, El-Azhary RA, Patzelt MT, et al. Survival, risk factors, and effect of treatment in 101 patients with calciphylaxis. Mayo Clin Proc. 2016;91:1384-1394. doi:10.1016/j.mayocp.2016.06.025
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217. doi:10.1046/j.1523-1755.2002.00375.x
- Nigwekar SU, Brunelli SM, Meade D, et al. Sodium thiosulfate therapy for calcific uremic arteriolopathy. Clin J Am Soc Nephrol. 2013;8:1162-1170. doi:10.2215/cjn.09880912
- Zitt E, König M, Vychytil A, et al. Use of sodium thiosulphate in a multi-interventional setting for the treatment of calciphylaxis in dialysis patients. Nephrol Dial Transplant. 2013;28:1232-1240. doi:10.1093/ndt/gfs548
- Peng T, Zhuo L, Wang Y, et al. Systematic review of sodium thiosulfate in treating calciphylaxis in chronic kidney disease patients. Nephrology (Carlton). 2018;23:669-675. doi:10.1111/nep.13081
- Miller K, Hunt R, Chu J, et al. Erythema ab igne. Dermatol Online J. 2011;17:28.
- Kettelhut EA, Traylor J, Sathe NC, et al. Erythema ab igne. StatPearls. StatPearls Publishing; 2022.
- Knöpfel N, Weibel L. Erythema Ab Igne. JAMA Dermatol. 2021;157: 106. doi:10.1001/jamadermatol.2020.3995
- Sigmon JR, Cantrell J, Teague D, et al. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35:676-678. doi:10.1097/DAD.0b013e3182871648
- Rose AE, Sagger V, Boyd KP, et al. Livedo reticularis. Dermatol Online J. 2013;19:20705.
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103/2229-5178.164493
- Uthman IW, Khamashta MA. Livedo racemosa: a striking dermatological sign for the antiphospholipid syndrome. J Rheumatol. 2006;33:2379-2382.
- Desbois AC, Cacoub P, Saadoun D. Cryoglobulinemia: an update in 2019. Joint Bone Spine. 2019;86:707-713. doi:10.1016/j .jbspin.2019.01.016
- Cohen SJ, Pittelkow MR, Su WP. Cutaneous manifestations of cryoglobulinemia: clinical and histopathologic study of seventy-two patients. J Am Acad Dermatol. 1991;25(1, pt 1):21-27. doi:10.1016 /0190-9622(91)70168-2
- Takada S, Shimizu T, Hadano Y, et al. Cryoglobulinemia (review). Mol Med Rep. 2012;6:3-8. doi:10.3892/mmr.2012.861
- Turska M, Parada-Turska J. Cutaneous polyarteritis nodosa. Wiad Lek. 2018;71(1, pt 1):73-77.
- De Virgilio A, Greco A, Magliulo G, et al. Polyarteritis nodosa: a contemporary overview. Autoimmun Rev. 2016;15:564-570. doi:10.1016/j.autrev.2016.02.015
A 72-year-old woman presented to the emergency department with concerns of confusion and lethargy during a session of peritoneal dialysis, which she had been receiving for the last 2 years for end-stage renal disease. She had a history of type 2 diabetes mellitus, diabetic retinopathy, hypertension, coronary artery disease, and peripheral vascular disease preceding a recent right below-knee amputation. A review of systems was positive for a rash on the thighs of several weeks’ duration that was preceded by several days of burning pain in the same distribution. Physical examination revealed retiform purpura with irregular contours and interspersed white stellate patterns scattered across the superomedial thighs, right lower back, and left lower abdomen. An initial laboratory workup revealed an elevated creatinine level of 5.03 mg/dL (reference range, 0.6–1.1 mg/dL; baseline level, 3.0 mg/dL) and mild leukocytosis (12.5 cells/mm3 [reference range, 4.5–11.0 cells/mm3]). Dermatology was consulted, and a 4-mm punch biopsy was obtained from the left medial thigh. Nephrology, infectious disease, and wound care consultations also were placed.


Noduloplaque on the Forehead
The Diagnosis: Giant Apocrine Hidrocystoma
Histopathology of the noduloplaque revealed an unremarkable epidermis with multilocular cystic spaces centered in the dermis. The cysts had a double-lined epithelium with inner columnar to cuboidal cells and outer myoepithelial cells (bottom quiz image). Columnar cells showing decapitation secretion could be appreciated at places indicating apocrine secretion (Figure). A final diagnosis of apocrine hidrocystoma was made.
.")
Hidrocystomas are rare, benign, cystic lesions derived either from apocrine or eccrine glands.1 Apocrine hidrocystoma usually manifests as asymptomatic, solitary, dome-shaped papules or nodules with a predilection for the head and neck region. Hidrocystomas can vary from flesh colored to blue, brown, or black. Pigmentation in hidrocystoma is seen in 5% to 80% of cases and is attributed to the Tyndall effect.1 The tumor usually is less than 20 mm in diameter; larger lesions are termed giant apocrine hidrocystoma.2 Apocrine hidrocystoma manifesting with multiple lesions and a size greater than 10 mm, as seen in our case, is uncommon.
Zaballos et al3 described dermoscopy of apocrine hidrocystoma in 22 patients. Hallmark dermoscopic findings were the presence of a homogeneous flesh-colored, yellowish, blue to pinkish-blue area involving the entire lesion with arborizing vessels and whitish structures.3 Similar dermoscopic findings were present in our patient. The homogeneous area histologically correlates to the multiloculated cysts located in the dermis. The exact reason for white structures is unknown; however, their visualization in apocrine hidrocystoma could be attributed to the alternation in collagen orientation secondary to the presence of large or multiple cysts in the dermis.
The presence of shiny white dots arranged in a square resembling a four-leaf clover (also known as white rosettes) was a unique dermoscopic finding in our patient. These rosettes can be appreciated only with polarized dermoscopy, and they have been described in actinic keratosis, seborrheic keratosis, squamous cell carcinoma, and basal cell carcinoma.4 The exact morphologic correlate of white rosettes is unknown but is postulated to be secondary to material inside adnexal openings in small rosettes and concentric perifollicular fibrosis in larger rosettes.4 In our patient, we believe the white rosettes can be attributed to the accumulated secretions in the dermal glands, which also were seen via histopathology. Dermoscopy also revealed increased peripheral, brown, networklike pigmentation, which was unique and could be secondary to the patient’s darker skin phenotype.
Differential diagnoses of apocrine hidrocystoma include both melanocytic and nonmelanocytic conditions such as epidermal cyst, nodular melanoma, nodular hidradenoma, syringoma, blue nevus, pilomatricoma, eccrine poroma, nodular Kaposi sarcoma, and venous lake.1 Histopathology showing large unilocular or multilocular dermal cysts with double lining comprising outer myoepithelial cells and inner columnar or cuboidal cell with decapitation secretion is paramount in confirming the diagnosis of apocrine hidrocystoma.
Dermoscopy can act as a valuable noninvasive modality in differentiating apocrine hidrocystoma from its melanocytic and nonmelanocytic differential diagnoses (Table).5-8 In our patient, the presence of a homogeneous pink to bluish area involving the entire lesion, linear branched vessels, and whitish structures on dermoscopy pointed to the diagnosis of apocrine hidrocystoma, which was further confirmed by characteristic histopathologic findings.

The treatment of apocrine hidrocystoma includes surgical excision for solitary lesions, with electrodesiccation and curettage, chemical cautery, and CO2 laser ablation employed for multiple lesions.1 Our patient was scheduled for CO2 laser ablation, considering the multiple lesions and size of the apocrine hidrocystoma but was subsequently lost to follow-up.
- Nguyen HP, Barker HS, Bloomquist L, et al. Giant pigmented apocrine hidrocystoma of the scalp [published online August 15, 2020]. Dermatol Online J. 2020;26:13030/qt7rt3s4pp.
- Anzai S, Goto M, Fujiwara S, et al. Apocrine hidrocystoma: a case report and analysis of 167 Japanese cases. Int J Dermatol. 2005;44:702-703. doi:10.1111/j.1365-4632.2005.02512.x
- Zaballos P, Bañuls J, Medina C, et al. Dermoscopy of apocrine hidrocystomas: a morphological study. J Eur Acad Dermatol Venereol. 2014;28:378-381. doi:10.1111/jdv.12044
- Haspeslagh M, Noë M, De Wispelaere I, et al. Rosettes and other white shiny structures in polarized dermoscopy: histological correlate and optical explanation. J Eur Acad Dermatol Venereol. 2016;30:311-313. doi:10.1111/jdv.13080
- Suh KS, Kang DY, Park JB, et al. Usefulness of dermoscopy in the differential diagnosis of ruptured and unruptured epidermal cysts. Ann Dermatol. 2017;29:33-38. doi:10.5021/ad.2017.29.1.33
- Serrano P, Lallas A, Del Pozo LJ, et al. Dermoscopy of nodular hidradenoma, a great masquerader: a morphological study of 28 cases. Dermatology. 2016;232:78-82. doi:10.1159/000441218
- Russo T, Piccolo V, Lallas A, et al. Dermoscopy of malignant skin tumours: what’s new? Dermatology. 2017;233:64-73. doi:10.1159/000472253
- Zaballos P, Llambrich A, Puig S, et al. Dermoscopic findings of pilomatricomas. Dermatology. 2008;217:225-230. doi:10.1159 /000148248
The Diagnosis: Giant Apocrine Hidrocystoma
Histopathology of the noduloplaque revealed an unremarkable epidermis with multilocular cystic spaces centered in the dermis. The cysts had a double-lined epithelium with inner columnar to cuboidal cells and outer myoepithelial cells (bottom quiz image). Columnar cells showing decapitation secretion could be appreciated at places indicating apocrine secretion (Figure). A final diagnosis of apocrine hidrocystoma was made.
Hidrocystomas are rare, benign, cystic lesions derived either from apocrine or eccrine glands.1 Apocrine hidrocystoma usually manifests as asymptomatic, solitary, dome-shaped papules or nodules with a predilection for the head and neck region. Hidrocystomas can vary from flesh colored to blue, brown, or black. Pigmentation in hidrocystoma is seen in 5% to 80% of cases and is attributed to the Tyndall effect.1 The tumor usually is less than 20 mm in diameter; larger lesions are termed giant apocrine hidrocystoma.2 Apocrine hidrocystoma manifesting with multiple lesions and a size greater than 10 mm, as seen in our case, is uncommon.
Zaballos et al3 described dermoscopy of apocrine hidrocystoma in 22 patients. Hallmark dermoscopic findings were the presence of a homogeneous flesh-colored, yellowish, blue to pinkish-blue area involving the entire lesion with arborizing vessels and whitish structures.3 Similar dermoscopic findings were present in our patient. The homogeneous area histologically correlates to the multiloculated cysts located in the dermis. The exact reason for white structures is unknown; however, their visualization in apocrine hidrocystoma could be attributed to the alternation in collagen orientation secondary to the presence of large or multiple cysts in the dermis.
The presence of shiny white dots arranged in a square resembling a four-leaf clover (also known as white rosettes) was a unique dermoscopic finding in our patient. These rosettes can be appreciated only with polarized dermoscopy, and they have been described in actinic keratosis, seborrheic keratosis, squamous cell carcinoma, and basal cell carcinoma.4 The exact morphologic correlate of white rosettes is unknown but is postulated to be secondary to material inside adnexal openings in small rosettes and concentric perifollicular fibrosis in larger rosettes.4 In our patient, we believe the white rosettes can be attributed to the accumulated secretions in the dermal glands, which also were seen via histopathology. Dermoscopy also revealed increased peripheral, brown, networklike pigmentation, which was unique and could be secondary to the patient’s darker skin phenotype.
Differential diagnoses of apocrine hidrocystoma include both melanocytic and nonmelanocytic conditions such as epidermal cyst, nodular melanoma, nodular hidradenoma, syringoma, blue nevus, pilomatricoma, eccrine poroma, nodular Kaposi sarcoma, and venous lake.1 Histopathology showing large unilocular or multilocular dermal cysts with double lining comprising outer myoepithelial cells and inner columnar or cuboidal cell with decapitation secretion is paramount in confirming the diagnosis of apocrine hidrocystoma.
Dermoscopy can act as a valuable noninvasive modality in differentiating apocrine hidrocystoma from its melanocytic and nonmelanocytic differential diagnoses (Table).5-8 In our patient, the presence of a homogeneous pink to bluish area involving the entire lesion, linear branched vessels, and whitish structures on dermoscopy pointed to the diagnosis of apocrine hidrocystoma, which was further confirmed by characteristic histopathologic findings.
The treatment of apocrine hidrocystoma includes surgical excision for solitary lesions, with electrodesiccation and curettage, chemical cautery, and CO2 laser ablation employed for multiple lesions.1 Our patient was scheduled for CO2 laser ablation, considering the multiple lesions and size of the apocrine hidrocystoma but was subsequently lost to follow-up.
The Diagnosis: Giant Apocrine Hidrocystoma
Histopathology of the noduloplaque revealed an unremarkable epidermis with multilocular cystic spaces centered in the dermis. The cysts had a double-lined epithelium with inner columnar to cuboidal cells and outer myoepithelial cells (bottom quiz image). Columnar cells showing decapitation secretion could be appreciated at places indicating apocrine secretion (Figure). A final diagnosis of apocrine hidrocystoma was made.
Hidrocystomas are rare, benign, cystic lesions derived either from apocrine or eccrine glands.1 Apocrine hidrocystoma usually manifests as asymptomatic, solitary, dome-shaped papules or nodules with a predilection for the head and neck region. Hidrocystomas can vary from flesh colored to blue, brown, or black. Pigmentation in hidrocystoma is seen in 5% to 80% of cases and is attributed to the Tyndall effect.1 The tumor usually is less than 20 mm in diameter; larger lesions are termed giant apocrine hidrocystoma.2 Apocrine hidrocystoma manifesting with multiple lesions and a size greater than 10 mm, as seen in our case, is uncommon.
Zaballos et al3 described dermoscopy of apocrine hidrocystoma in 22 patients. Hallmark dermoscopic findings were the presence of a homogeneous flesh-colored, yellowish, blue to pinkish-blue area involving the entire lesion with arborizing vessels and whitish structures.3 Similar dermoscopic findings were present in our patient. The homogeneous area histologically correlates to the multiloculated cysts located in the dermis. The exact reason for white structures is unknown; however, their visualization in apocrine hidrocystoma could be attributed to the alternation in collagen orientation secondary to the presence of large or multiple cysts in the dermis.
The presence of shiny white dots arranged in a square resembling a four-leaf clover (also known as white rosettes) was a unique dermoscopic finding in our patient. These rosettes can be appreciated only with polarized dermoscopy, and they have been described in actinic keratosis, seborrheic keratosis, squamous cell carcinoma, and basal cell carcinoma.4 The exact morphologic correlate of white rosettes is unknown but is postulated to be secondary to material inside adnexal openings in small rosettes and concentric perifollicular fibrosis in larger rosettes.4 In our patient, we believe the white rosettes can be attributed to the accumulated secretions in the dermal glands, which also were seen via histopathology. Dermoscopy also revealed increased peripheral, brown, networklike pigmentation, which was unique and could be secondary to the patient’s darker skin phenotype.
Differential diagnoses of apocrine hidrocystoma include both melanocytic and nonmelanocytic conditions such as epidermal cyst, nodular melanoma, nodular hidradenoma, syringoma, blue nevus, pilomatricoma, eccrine poroma, nodular Kaposi sarcoma, and venous lake.1 Histopathology showing large unilocular or multilocular dermal cysts with double lining comprising outer myoepithelial cells and inner columnar or cuboidal cell with decapitation secretion is paramount in confirming the diagnosis of apocrine hidrocystoma.
Dermoscopy can act as a valuable noninvasive modality in differentiating apocrine hidrocystoma from its melanocytic and nonmelanocytic differential diagnoses (Table).5-8 In our patient, the presence of a homogeneous pink to bluish area involving the entire lesion, linear branched vessels, and whitish structures on dermoscopy pointed to the diagnosis of apocrine hidrocystoma, which was further confirmed by characteristic histopathologic findings.
The treatment of apocrine hidrocystoma includes surgical excision for solitary lesions, with electrodesiccation and curettage, chemical cautery, and CO2 laser ablation employed for multiple lesions.1 Our patient was scheduled for CO2 laser ablation, considering the multiple lesions and size of the apocrine hidrocystoma but was subsequently lost to follow-up.
- Nguyen HP, Barker HS, Bloomquist L, et al. Giant pigmented apocrine hidrocystoma of the scalp [published online August 15, 2020]. Dermatol Online J. 2020;26:13030/qt7rt3s4pp.
- Anzai S, Goto M, Fujiwara S, et al. Apocrine hidrocystoma: a case report and analysis of 167 Japanese cases. Int J Dermatol. 2005;44:702-703. doi:10.1111/j.1365-4632.2005.02512.x
- Zaballos P, Bañuls J, Medina C, et al. Dermoscopy of apocrine hidrocystomas: a morphological study. J Eur Acad Dermatol Venereol. 2014;28:378-381. doi:10.1111/jdv.12044
- Haspeslagh M, Noë M, De Wispelaere I, et al. Rosettes and other white shiny structures in polarized dermoscopy: histological correlate and optical explanation. J Eur Acad Dermatol Venereol. 2016;30:311-313. doi:10.1111/jdv.13080
- Suh KS, Kang DY, Park JB, et al. Usefulness of dermoscopy in the differential diagnosis of ruptured and unruptured epidermal cysts. Ann Dermatol. 2017;29:33-38. doi:10.5021/ad.2017.29.1.33
- Serrano P, Lallas A, Del Pozo LJ, et al. Dermoscopy of nodular hidradenoma, a great masquerader: a morphological study of 28 cases. Dermatology. 2016;232:78-82. doi:10.1159/000441218
- Russo T, Piccolo V, Lallas A, et al. Dermoscopy of malignant skin tumours: what’s new? Dermatology. 2017;233:64-73. doi:10.1159/000472253
- Zaballos P, Llambrich A, Puig S, et al. Dermoscopic findings of pilomatricomas. Dermatology. 2008;217:225-230. doi:10.1159 /000148248
- Nguyen HP, Barker HS, Bloomquist L, et al. Giant pigmented apocrine hidrocystoma of the scalp [published online August 15, 2020]. Dermatol Online J. 2020;26:13030/qt7rt3s4pp.
- Anzai S, Goto M, Fujiwara S, et al. Apocrine hidrocystoma: a case report and analysis of 167 Japanese cases. Int J Dermatol. 2005;44:702-703. doi:10.1111/j.1365-4632.2005.02512.x
- Zaballos P, Bañuls J, Medina C, et al. Dermoscopy of apocrine hidrocystomas: a morphological study. J Eur Acad Dermatol Venereol. 2014;28:378-381. doi:10.1111/jdv.12044
- Haspeslagh M, Noë M, De Wispelaere I, et al. Rosettes and other white shiny structures in polarized dermoscopy: histological correlate and optical explanation. J Eur Acad Dermatol Venereol. 2016;30:311-313. doi:10.1111/jdv.13080
- Suh KS, Kang DY, Park JB, et al. Usefulness of dermoscopy in the differential diagnosis of ruptured and unruptured epidermal cysts. Ann Dermatol. 2017;29:33-38. doi:10.5021/ad.2017.29.1.33
- Serrano P, Lallas A, Del Pozo LJ, et al. Dermoscopy of nodular hidradenoma, a great masquerader: a morphological study of 28 cases. Dermatology. 2016;232:78-82. doi:10.1159/000441218
- Russo T, Piccolo V, Lallas A, et al. Dermoscopy of malignant skin tumours: what’s new? Dermatology. 2017;233:64-73. doi:10.1159/000472253
- Zaballos P, Llambrich A, Puig S, et al. Dermoscopic findings of pilomatricomas. Dermatology. 2008;217:225-230. doi:10.1159 /000148248
A 21-year-old man presented with a raised lesion on the forehead that had started as a single papule 16 years prior and gradually increased in number and size. There were no associated symptoms and no history of seasonal variation in the size of the lesions. Physical examination revealed multiple erythematous to slightly bluish translucent papules that coalesced to form a 3×3-cm noduloplaque with cystic consistency on the right side of the forehead (top). Dermoscopic examination (middle) (polarized noncontact mode) revealed a homogeneous pink to bluish background, scattered linear vessels with branches (black arrows), multiple chrysalislike shiny white lines (blue arrows), and dots arranged in a 4-dot pattern (black circle) resembling a four-leaf clover. Increased peripheral, brown, networklike pigmentation (black stars) also was noted on dermoscopy. Histopathologic examination of the noduloplaque was performed (bottom).



Annular Erythematous Plaques on the Back
The Diagnosis: Granuloma Annulare
The biopsies revealed palisading granulomatous dermatitis consistent with granuloma annulare (GA). This diagnosis was supported by the clinical presentation and histopathologic findings. Although the pathogenesis of GA is unclear, it is a benign, self-limiting condition. Primarily affected sites include the trunk and forearms. Generalized GA (or GA with ≥10 lesions) may warrant workup for malignancy, as it may represent a paraneoplastic process.1 Histopathology reveals granulomas comprising a dermal lymphohistiocytic infiltrate as well as central mucin and nuclear debris. There are a few histologic subtypes of GA, including palisading and interstitial, which refer to the distribution of the histiocytic infiltrate.2,3 This case—with palisading histiocytes lining the collection of necrobiosis and mucin (bottom quiz image)—features palisading GA. Notably, GA exhibits central rather than diffuse mucin.4
Erythema gyratum repens is a paraneoplastic arcuate erythema that manifests as erythematous figurate, gyrate, or annular plaques exhibiting a trailing scale. Clinically, erythema gyratum repens spreads rapidly—as quickly as 1 cm/d—and can be extensive (as in this case). Histopathology ruled out this diagnosis in our patient. Nonspecific findings of acanthosis, parakeratosis, and superficial spongiosis can be found in erythema gyratum repens. A superficial and deep perivascular lymphohistiocytic infiltrate may be seen in figurate erythemas (Figure 1).5 Unlike GA, this infiltrate does not form granulomas, is more superficial, and does not contain mucin.
.")
Histopathology also can help establish the diagnosis of leprosy and its specific subtype, as leprosy exists on a spectrum from tuberculoid to lepromatous, with a great deal of overlap in between.6 Lepromatous leprosy has many cutaneous clinical presentations but typically manifests as erythematous papules or nodules. It is multibacillary, and these mycobacteria form clumps known as globi that can be seen on Fite stain.7 In lepromatous leprosy, there is a characteristic dense lymphohistiocytic infiltrate (Figure 2) above which a Grenz zone can be seen.4,8 There are no well-formed granulomas in lepromatous leprosy, unlike in tuberculoid leprosy, which is paucibacillary and creates a granulomatous response surrounding nerves and adnexal structures.6
.")
Mycosis fungoides (MF) is the most common cutaneous lymphoma. There are patch, plaque, and tumor stages of MF, each of which exhibits various histopathologic findings.9 In early patch-stage MF, lymphocytes have perinuclear clearing, and the degree of lymphocytic infiltrate is out of proportion to the spongiosis present. Epidermotropism and Pautrier microabscesses often are present in the epidermis (Figure 3). In the plaque stage, there is a denser lymphoid infiltrate in a lichenoid pattern with epidermotropism and Pautrier microabscesses. The tumor stage shows a dense dermal lymphoid infiltrate with more atypia and typically a lack of epidermotropism. Rarely, MF can exhibit a granulomatous variant in which epithelioid histiocytes collect to form granulomas along with atypical lymphocytes.10
.")
The diagnosis of cutaneous sarcoidosis requires clinicopathologic corroboration. Histopathology demonstrates epithelioid histiocytes forming noncaseating granulomas with little to no lymphocytic infiltrate (Figure 4). There typically is no necrosis or necrobiosis as there is in GA. The diagnosis of sarcoidosis can be challenging histopathologically, and stains should be used to rule out infectious processes.4 Asteroid bodies— star-shaped eosinophilic inclusions within giant cells—may be present but are nonspecific for sarcoidosis.11 Schaumann bodies—inclusions of calcifications within giant cells—also may be present and can aid in diagnosis.12
.")
- Kovich O, Burgin S. Generalized granuloma annulare [published online December 30, 2005]. Dermatol Online J. 2005;11:23.
- Al Ameer MA, Al-Natour SH, Alsahaf HAA, et al. Eruptive granuloma annulare in an elderly man with diabetes [published online January 14, 2022]. Cureus. 2022;14:E21242. doi:10.7759/cureus.21242
- Howard A, White CR Jr. Non-infectious granulomas. In: Bolognia JL, et al, eds. Dermatology. Mosby; 2003:1455.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 3rd ed. Elsevier; 2018.
- Gore M, Winters ME. Erythema gyratum repens: a rare paraneoplastic rash. West J Emerg Med. 2011;12:556-558. doi:10.5811/westjem.2010.11.2090
- Maymone MBC, Laughter M, Venkatesh S, et al. Leprosy: clinical aspects and diagnostic techniques. J Am Acad Dermatol. 2020;83:1-14. doi:10.1016/j.jaad.2019.12.080
- Pedley JC, Harman DJ, Waudby H, et al. Leprosy in peripheral nerves: histopathological findings in 119 untreated patients in Nepal. J Neurol Neurosurg Psychiatry. 1980;43:198-204. doi:10.1136/jnnp.43.3.198
- Booth AV, Kovich OI. Lepromatous leprosy [published online January 27, 2007]. Dermatol Online J. 2007;13:9.
- Robson A. The pathology of cutaneous T-cell lymphoma. Oncology (Williston Park). 2007;21(2 suppl 1):9-12.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617. doi:10.1001/archdermatol.2008.46
- Azar HA, Lunardelli C. Collagen nature of asteroid bodies of giant cells in sarcoidosis. Am J Pathol. 1969;57:81-92.
- Sreeja C, Priyadarshini A, Premika, et al. Sarcoidosis—a review article. J Oral Maxillofac Pathol. 2022;26:242-253. doi:10.4103 /jomfp.jomfp_373_21
The Diagnosis: Granuloma Annulare
The biopsies revealed palisading granulomatous dermatitis consistent with granuloma annulare (GA). This diagnosis was supported by the clinical presentation and histopathologic findings. Although the pathogenesis of GA is unclear, it is a benign, self-limiting condition. Primarily affected sites include the trunk and forearms. Generalized GA (or GA with ≥10 lesions) may warrant workup for malignancy, as it may represent a paraneoplastic process.1 Histopathology reveals granulomas comprising a dermal lymphohistiocytic infiltrate as well as central mucin and nuclear debris. There are a few histologic subtypes of GA, including palisading and interstitial, which refer to the distribution of the histiocytic infiltrate.2,3 This case—with palisading histiocytes lining the collection of necrobiosis and mucin (bottom quiz image)—features palisading GA. Notably, GA exhibits central rather than diffuse mucin.4
Erythema gyratum repens is a paraneoplastic arcuate erythema that manifests as erythematous figurate, gyrate, or annular plaques exhibiting a trailing scale. Clinically, erythema gyratum repens spreads rapidly—as quickly as 1 cm/d—and can be extensive (as in this case). Histopathology ruled out this diagnosis in our patient. Nonspecific findings of acanthosis, parakeratosis, and superficial spongiosis can be found in erythema gyratum repens. A superficial and deep perivascular lymphohistiocytic infiltrate may be seen in figurate erythemas (Figure 1).5 Unlike GA, this infiltrate does not form granulomas, is more superficial, and does not contain mucin.
Histopathology also can help establish the diagnosis of leprosy and its specific subtype, as leprosy exists on a spectrum from tuberculoid to lepromatous, with a great deal of overlap in between.6 Lepromatous leprosy has many cutaneous clinical presentations but typically manifests as erythematous papules or nodules. It is multibacillary, and these mycobacteria form clumps known as globi that can be seen on Fite stain.7 In lepromatous leprosy, there is a characteristic dense lymphohistiocytic infiltrate (Figure 2) above which a Grenz zone can be seen.4,8 There are no well-formed granulomas in lepromatous leprosy, unlike in tuberculoid leprosy, which is paucibacillary and creates a granulomatous response surrounding nerves and adnexal structures.6
Mycosis fungoides (MF) is the most common cutaneous lymphoma. There are patch, plaque, and tumor stages of MF, each of which exhibits various histopathologic findings.9 In early patch-stage MF, lymphocytes have perinuclear clearing, and the degree of lymphocytic infiltrate is out of proportion to the spongiosis present. Epidermotropism and Pautrier microabscesses often are present in the epidermis (Figure 3). In the plaque stage, there is a denser lymphoid infiltrate in a lichenoid pattern with epidermotropism and Pautrier microabscesses. The tumor stage shows a dense dermal lymphoid infiltrate with more atypia and typically a lack of epidermotropism. Rarely, MF can exhibit a granulomatous variant in which epithelioid histiocytes collect to form granulomas along with atypical lymphocytes.10
The diagnosis of cutaneous sarcoidosis requires clinicopathologic corroboration. Histopathology demonstrates epithelioid histiocytes forming noncaseating granulomas with little to no lymphocytic infiltrate (Figure 4). There typically is no necrosis or necrobiosis as there is in GA. The diagnosis of sarcoidosis can be challenging histopathologically, and stains should be used to rule out infectious processes.4 Asteroid bodies— star-shaped eosinophilic inclusions within giant cells—may be present but are nonspecific for sarcoidosis.11 Schaumann bodies—inclusions of calcifications within giant cells—also may be present and can aid in diagnosis.12
The Diagnosis: Granuloma Annulare
The biopsies revealed palisading granulomatous dermatitis consistent with granuloma annulare (GA). This diagnosis was supported by the clinical presentation and histopathologic findings. Although the pathogenesis of GA is unclear, it is a benign, self-limiting condition. Primarily affected sites include the trunk and forearms. Generalized GA (or GA with ≥10 lesions) may warrant workup for malignancy, as it may represent a paraneoplastic process.1 Histopathology reveals granulomas comprising a dermal lymphohistiocytic infiltrate as well as central mucin and nuclear debris. There are a few histologic subtypes of GA, including palisading and interstitial, which refer to the distribution of the histiocytic infiltrate.2,3 This case—with palisading histiocytes lining the collection of necrobiosis and mucin (bottom quiz image)—features palisading GA. Notably, GA exhibits central rather than diffuse mucin.4
Erythema gyratum repens is a paraneoplastic arcuate erythema that manifests as erythematous figurate, gyrate, or annular plaques exhibiting a trailing scale. Clinically, erythema gyratum repens spreads rapidly—as quickly as 1 cm/d—and can be extensive (as in this case). Histopathology ruled out this diagnosis in our patient. Nonspecific findings of acanthosis, parakeratosis, and superficial spongiosis can be found in erythema gyratum repens. A superficial and deep perivascular lymphohistiocytic infiltrate may be seen in figurate erythemas (Figure 1).5 Unlike GA, this infiltrate does not form granulomas, is more superficial, and does not contain mucin.
Histopathology also can help establish the diagnosis of leprosy and its specific subtype, as leprosy exists on a spectrum from tuberculoid to lepromatous, with a great deal of overlap in between.6 Lepromatous leprosy has many cutaneous clinical presentations but typically manifests as erythematous papules or nodules. It is multibacillary, and these mycobacteria form clumps known as globi that can be seen on Fite stain.7 In lepromatous leprosy, there is a characteristic dense lymphohistiocytic infiltrate (Figure 2) above which a Grenz zone can be seen.4,8 There are no well-formed granulomas in lepromatous leprosy, unlike in tuberculoid leprosy, which is paucibacillary and creates a granulomatous response surrounding nerves and adnexal structures.6
Mycosis fungoides (MF) is the most common cutaneous lymphoma. There are patch, plaque, and tumor stages of MF, each of which exhibits various histopathologic findings.9 In early patch-stage MF, lymphocytes have perinuclear clearing, and the degree of lymphocytic infiltrate is out of proportion to the spongiosis present. Epidermotropism and Pautrier microabscesses often are present in the epidermis (Figure 3). In the plaque stage, there is a denser lymphoid infiltrate in a lichenoid pattern with epidermotropism and Pautrier microabscesses. The tumor stage shows a dense dermal lymphoid infiltrate with more atypia and typically a lack of epidermotropism. Rarely, MF can exhibit a granulomatous variant in which epithelioid histiocytes collect to form granulomas along with atypical lymphocytes.10
The diagnosis of cutaneous sarcoidosis requires clinicopathologic corroboration. Histopathology demonstrates epithelioid histiocytes forming noncaseating granulomas with little to no lymphocytic infiltrate (Figure 4). There typically is no necrosis or necrobiosis as there is in GA. The diagnosis of sarcoidosis can be challenging histopathologically, and stains should be used to rule out infectious processes.4 Asteroid bodies— star-shaped eosinophilic inclusions within giant cells—may be present but are nonspecific for sarcoidosis.11 Schaumann bodies—inclusions of calcifications within giant cells—also may be present and can aid in diagnosis.12
- Kovich O, Burgin S. Generalized granuloma annulare [published online December 30, 2005]. Dermatol Online J. 2005;11:23.
- Al Ameer MA, Al-Natour SH, Alsahaf HAA, et al. Eruptive granuloma annulare in an elderly man with diabetes [published online January 14, 2022]. Cureus. 2022;14:E21242. doi:10.7759/cureus.21242
- Howard A, White CR Jr. Non-infectious granulomas. In: Bolognia JL, et al, eds. Dermatology. Mosby; 2003:1455.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 3rd ed. Elsevier; 2018.
- Gore M, Winters ME. Erythema gyratum repens: a rare paraneoplastic rash. West J Emerg Med. 2011;12:556-558. doi:10.5811/westjem.2010.11.2090
- Maymone MBC, Laughter M, Venkatesh S, et al. Leprosy: clinical aspects and diagnostic techniques. J Am Acad Dermatol. 2020;83:1-14. doi:10.1016/j.jaad.2019.12.080
- Pedley JC, Harman DJ, Waudby H, et al. Leprosy in peripheral nerves: histopathological findings in 119 untreated patients in Nepal. J Neurol Neurosurg Psychiatry. 1980;43:198-204. doi:10.1136/jnnp.43.3.198
- Booth AV, Kovich OI. Lepromatous leprosy [published online January 27, 2007]. Dermatol Online J. 2007;13:9.
- Robson A. The pathology of cutaneous T-cell lymphoma. Oncology (Williston Park). 2007;21(2 suppl 1):9-12.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617. doi:10.1001/archdermatol.2008.46
- Azar HA, Lunardelli C. Collagen nature of asteroid bodies of giant cells in sarcoidosis. Am J Pathol. 1969;57:81-92.
- Sreeja C, Priyadarshini A, Premika, et al. Sarcoidosis—a review article. J Oral Maxillofac Pathol. 2022;26:242-253. doi:10.4103 /jomfp.jomfp_373_21
- Kovich O, Burgin S. Generalized granuloma annulare [published online December 30, 2005]. Dermatol Online J. 2005;11:23.
- Al Ameer MA, Al-Natour SH, Alsahaf HAA, et al. Eruptive granuloma annulare in an elderly man with diabetes [published online January 14, 2022]. Cureus. 2022;14:E21242. doi:10.7759/cureus.21242
- Howard A, White CR Jr. Non-infectious granulomas. In: Bolognia JL, et al, eds. Dermatology. Mosby; 2003:1455.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 3rd ed. Elsevier; 2018.
- Gore M, Winters ME. Erythema gyratum repens: a rare paraneoplastic rash. West J Emerg Med. 2011;12:556-558. doi:10.5811/westjem.2010.11.2090
- Maymone MBC, Laughter M, Venkatesh S, et al. Leprosy: clinical aspects and diagnostic techniques. J Am Acad Dermatol. 2020;83:1-14. doi:10.1016/j.jaad.2019.12.080
- Pedley JC, Harman DJ, Waudby H, et al. Leprosy in peripheral nerves: histopathological findings in 119 untreated patients in Nepal. J Neurol Neurosurg Psychiatry. 1980;43:198-204. doi:10.1136/jnnp.43.3.198
- Booth AV, Kovich OI. Lepromatous leprosy [published online January 27, 2007]. Dermatol Online J. 2007;13:9.
- Robson A. The pathology of cutaneous T-cell lymphoma. Oncology (Williston Park). 2007;21(2 suppl 1):9-12.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617. doi:10.1001/archdermatol.2008.46
- Azar HA, Lunardelli C. Collagen nature of asteroid bodies of giant cells in sarcoidosis. Am J Pathol. 1969;57:81-92.
- Sreeja C, Priyadarshini A, Premika, et al. Sarcoidosis—a review article. J Oral Maxillofac Pathol. 2022;26:242-253. doi:10.4103 /jomfp.jomfp_373_21
An 84-year-old man presented to the clinic for evaluation of a pruritic rash on the back of 6 months’ duration that spread to the neck and chest over the past 2 months and then to the abdomen and thighs more recently. His primary care provider prescribed a 1-week course of oral steroids and steroid cream. The oral medication did not help, but the cream alleviated the pruritus. He had a medical history of coronary artery disease, hypertension, and diabetes mellitus. He also had a rash on the forearms that had waxed and waned for many years but was not associated with pruritus. He had not sought medical care for the rash and had never treated it. Physical examination revealed pink to violaceous annular plaques with central clearing and raised borders that coalesced into larger plaques on the trunk (top). Dusky, scaly, pink plaques were present on the dorsal forearms. Three punch biopsies—2 from the upper back (bottom) and 1 from the left forearm—all demonstrated consistent findings.


Punch Biopsy Extraction With Fingers
Practice Gap
Punch biopsies are utilized frequently by dermatologists to aid in the diagnosis of various skin diseases.1 When performing a punch biopsy, dermatologists are taught to use either forceps or skin hooks in addition to scissors to extract the tissue from the skin.2 However, the use of these sterile instruments for a simple biopsy adds extra costs to the procedure. Herein, a cheaper and often faster method of obtaining the specimen from the patient is described.
The Technique
A 3- or 4-mm disposable punch biopsy tool is employed for this method. After locally anesthetizing the skin, the skin is punched to a subcutaneous depth utilizing the full length of the blade and a little extra pressure is applied downward while stretching the skin around (Figure, A). This may be helpful to dislodge the punch specimen from the surrounding skin. The specimen now can be easily removed by gently grasping it with the thumb and index finger (Figure, B and C). It then can be transferred immediately to the formalin container.
Practice Implications
This technique saves time as well as financial and environmental costs associated with the use of sterile instruments. An additional advantage to this simple method is avoiding specimen crush injuries, which are common when using forceps. This solution works in most cases but may not be suitable for certain special anatomic locations such as the scalp, nose, and ears.
- Gronbeck C, Feng H. Volume and distribution of skin biopsies performed by dermatologists and other health care providers in the Medicare population in 2019. J Am Acad Dermatol. 2022;87:675-678.
- Bolognia JL, Schaffer JV, Cerroni L. Dermatology. Elsevier; 2018.
Practice Gap
Punch biopsies are utilized frequently by dermatologists to aid in the diagnosis of various skin diseases.1 When performing a punch biopsy, dermatologists are taught to use either forceps or skin hooks in addition to scissors to extract the tissue from the skin.2 However, the use of these sterile instruments for a simple biopsy adds extra costs to the procedure. Herein, a cheaper and often faster method of obtaining the specimen from the patient is described.
The Technique
A 3- or 4-mm disposable punch biopsy tool is employed for this method. After locally anesthetizing the skin, the skin is punched to a subcutaneous depth utilizing the full length of the blade and a little extra pressure is applied downward while stretching the skin around (Figure, A). This may be helpful to dislodge the punch specimen from the surrounding skin. The specimen now can be easily removed by gently grasping it with the thumb and index finger (Figure, B and C). It then can be transferred immediately to the formalin container.
Practice Implications
This technique saves time as well as financial and environmental costs associated with the use of sterile instruments. An additional advantage to this simple method is avoiding specimen crush injuries, which are common when using forceps. This solution works in most cases but may not be suitable for certain special anatomic locations such as the scalp, nose, and ears.
Practice Gap
Punch biopsies are utilized frequently by dermatologists to aid in the diagnosis of various skin diseases.1 When performing a punch biopsy, dermatologists are taught to use either forceps or skin hooks in addition to scissors to extract the tissue from the skin.2 However, the use of these sterile instruments for a simple biopsy adds extra costs to the procedure. Herein, a cheaper and often faster method of obtaining the specimen from the patient is described.
The Technique
A 3- or 4-mm disposable punch biopsy tool is employed for this method. After locally anesthetizing the skin, the skin is punched to a subcutaneous depth utilizing the full length of the blade and a little extra pressure is applied downward while stretching the skin around (Figure, A). This may be helpful to dislodge the punch specimen from the surrounding skin. The specimen now can be easily removed by gently grasping it with the thumb and index finger (Figure, B and C). It then can be transferred immediately to the formalin container.
Practice Implications
This technique saves time as well as financial and environmental costs associated with the use of sterile instruments. An additional advantage to this simple method is avoiding specimen crush injuries, which are common when using forceps. This solution works in most cases but may not be suitable for certain special anatomic locations such as the scalp, nose, and ears.
- Gronbeck C, Feng H. Volume and distribution of skin biopsies performed by dermatologists and other health care providers in the Medicare population in 2019. J Am Acad Dermatol. 2022;87:675-678.
- Bolognia JL, Schaffer JV, Cerroni L. Dermatology. Elsevier; 2018.
- Gronbeck C, Feng H. Volume and distribution of skin biopsies performed by dermatologists and other health care providers in the Medicare population in 2019. J Am Acad Dermatol. 2022;87:675-678.
- Bolognia JL, Schaffer JV, Cerroni L. Dermatology. Elsevier; 2018.

Ectatic Vessels on the Chest
The Diagnosis: Superior Vena Cava Syndrome
Computed tomography angiography of the chest confirmed a diagnosis of superior vena cava (SVC) syndrome due to external pressure of the indwelling catheter. Upon diagnosis, the left indwelling catheter was removed. Further testing to assess for a potential pulmonary embolism was negative. Resolution of the ectatic spider veins and patientreported intermittent facial swelling was achieved after catheter removal.
Superior vena cava syndrome occurs when the SVC is occluded due to extrinsic pressure or thrombosis. Although classically thought to be due to underlying bronchogenic carcinomas, all pathologies that cause compression of the SVC also can lead to vessel occlusion.1 Superior vena cava syndrome initially can be detected on physical examination. The most prominent skin finding includes diffusely dilated blood vessels on the central chest wall, which indicate the presence of collateral blood vessels.1 Imaging studies such as abdominal computed tomography can provide information on the etiology of the condition but are not required for diagnosis. Given the high correlation of SVC syndrome with underlying lung and mediastinal carcinomas, imaging was warranted in our patient. Imaging also can distinguish if the condition is due to external pressure or thrombosis.2 For SVC syndrome due to thrombosis, endovascular therapy is first-line management; however, mechanical thrombectomy may be preferred in patients with absolute contraindication to thrombolytic agents.3 In the setting of increased external pressure on the SVC, treatment includes the removal of the source of pressure.4
In a case series including 78 patients, ports and indwelling catheters accounted for 71% of benign SVC cases.5 Our patient’s SVC syndrome most likely was due to the indwelling catheter pressing on the SVC. The goal of treatment is to address the underlying cause—whether it be pressure or thrombosis. In the setting of increased external pressure, treatment includes removal of the source of pressure from the SVC.4
Other differential diagnoses to consider for newonset ectatic vessels on the chest wall include generalized essential telangiectasia, scleroderma, poikiloderma vasculare atrophicans, and caput medusae. Generalized essential telangiectasia is characterized by red or pink dilated capillary blood vessels in a branch or lacelike pattern predominantly on the lower limbs. The eruption primarily is asymptomatic, though tingling or numbness may be reported.6 The diagnosis can be made with a punch biopsy, with histopathology showing dilated vessels in the dermis.7
Scleroderma is a connective tissue fibrosis disorder with variable clinical presentations. The systemic sclerosis subset can be divided into localized systemic sclerosis and diffuse systemic sclerosis. Physical examination reveals cutaneous sclerosis in various areas of the body. Localized systemic sclerosis includes sclerosis of the fingers and face, while diffuse systemic sclerosis is notable for progression to the arms, legs, and trunk.8 In addition to sclerosis, diffuse telangiectases also can be observed. Systemic sclerosis is a clinical diagnosis based on physical examination and laboratory studies to identify antibodies such as antinuclear antibodies.
Poikiloderma vasculare atrophicans is a variant of cutaneous T-cell lymphoma. The initial presentation is characterized by plaques of hypopigmentation and hyperpigmentation with atrophy and telangiectases. The lesions may be asymptomatic or mildly pruritic and classically involve the trunk and flexural areas.9 The diagnosis is made with skin biopsy and immunohistochemical studies, with findings reflective of mycosis fungoides.
Caput medusae (palm tree sign) is a cardinal feature of portal hypertension characterized by grossly dilated and engorged periumbilical veins. To shunt blood from the portal venous system, cutaneous collateral veins between the umbilical veins and abdominal wall veins are used, resulting in the appearance of engorged veins in the anterior abdominal wall.10 The diagnosis can be made with abdominal ultrasonography showing the direction of blood flow through abdominal vessels.
- Drouin L, Pistorius MA, Lafforgue A, et al. Upper-extremity venous thrombosis: a retrospective study about 160 cases [in French]. Rev Med Interne. 2019;40:9-15.
- Richie E. Clinical pearl: diagnosing superior vena cava syndrome. Emergency Medicine News. 2017;39:22. doi:10.1097/01 .EEM.0000522220.37441.d2
- Azizi A, Shafi I, Shah N, et al. Superior vena cava syndrome. JACC Cardiovasc Interv. 2020;13:2896-2910. doi:10.1016/j.jcin.2020.08.038
- Dumantepe M, Tarhan A, Ozler A. Successful treatment of central venous catheter induced superior vena cava syndrome with ultrasound accelerated catheter-directed thrombolysis. Catheter Cardiovasc Interv. 2013;81:E269-E273.
- Rice TW, Rodriguez RM, Light RW. The superior vena cava syndrome: clinical characteristics and evolving etiology. Medicine (Baltimore) 2006;85:37-42. doi:10.1097/01.md.0000198474.99876.f0
- Long D, Marshman G. Generalized essential telangiectasia. Australas J Dermatol. 2004;45:67-69. doi:10.1111/j.1440-0960.2004.00033.x
- Braverman IM. Ultrastructure and organization of the cutaneous microvasculature in normal and pathologic states. J Invest Dermatol. 1989;93(2 suppl):2S-9S.
- Ferreli C, Gasparini G, Parodi A, et al. Cutaneous manifestations of scleroderma and scleroderma-like disorders: a comprehensive review. Clin Rev Allergy Immunol. 2017;53:306-336. doi:10.1007 /s12016-017-8625-4
- Bloom B, Marchbein S, Fischer M, et al. Poikilodermatous mycosis fungoides. Dermatol Online J. 2012;18:4.
- Sharma B, Raina S. Caput medusae. Indian J Med Res. 2015;141:494. doi:10.4103/0971-5916.159322
The Diagnosis: Superior Vena Cava Syndrome
Computed tomography angiography of the chest confirmed a diagnosis of superior vena cava (SVC) syndrome due to external pressure of the indwelling catheter. Upon diagnosis, the left indwelling catheter was removed. Further testing to assess for a potential pulmonary embolism was negative. Resolution of the ectatic spider veins and patientreported intermittent facial swelling was achieved after catheter removal.
Superior vena cava syndrome occurs when the SVC is occluded due to extrinsic pressure or thrombosis. Although classically thought to be due to underlying bronchogenic carcinomas, all pathologies that cause compression of the SVC also can lead to vessel occlusion.1 Superior vena cava syndrome initially can be detected on physical examination. The most prominent skin finding includes diffusely dilated blood vessels on the central chest wall, which indicate the presence of collateral blood vessels.1 Imaging studies such as abdominal computed tomography can provide information on the etiology of the condition but are not required for diagnosis. Given the high correlation of SVC syndrome with underlying lung and mediastinal carcinomas, imaging was warranted in our patient. Imaging also can distinguish if the condition is due to external pressure or thrombosis.2 For SVC syndrome due to thrombosis, endovascular therapy is first-line management; however, mechanical thrombectomy may be preferred in patients with absolute contraindication to thrombolytic agents.3 In the setting of increased external pressure on the SVC, treatment includes the removal of the source of pressure.4
In a case series including 78 patients, ports and indwelling catheters accounted for 71% of benign SVC cases.5 Our patient’s SVC syndrome most likely was due to the indwelling catheter pressing on the SVC. The goal of treatment is to address the underlying cause—whether it be pressure or thrombosis. In the setting of increased external pressure, treatment includes removal of the source of pressure from the SVC.4
Other differential diagnoses to consider for newonset ectatic vessels on the chest wall include generalized essential telangiectasia, scleroderma, poikiloderma vasculare atrophicans, and caput medusae. Generalized essential telangiectasia is characterized by red or pink dilated capillary blood vessels in a branch or lacelike pattern predominantly on the lower limbs. The eruption primarily is asymptomatic, though tingling or numbness may be reported.6 The diagnosis can be made with a punch biopsy, with histopathology showing dilated vessels in the dermis.7
Scleroderma is a connective tissue fibrosis disorder with variable clinical presentations. The systemic sclerosis subset can be divided into localized systemic sclerosis and diffuse systemic sclerosis. Physical examination reveals cutaneous sclerosis in various areas of the body. Localized systemic sclerosis includes sclerosis of the fingers and face, while diffuse systemic sclerosis is notable for progression to the arms, legs, and trunk.8 In addition to sclerosis, diffuse telangiectases also can be observed. Systemic sclerosis is a clinical diagnosis based on physical examination and laboratory studies to identify antibodies such as antinuclear antibodies.
Poikiloderma vasculare atrophicans is a variant of cutaneous T-cell lymphoma. The initial presentation is characterized by plaques of hypopigmentation and hyperpigmentation with atrophy and telangiectases. The lesions may be asymptomatic or mildly pruritic and classically involve the trunk and flexural areas.9 The diagnosis is made with skin biopsy and immunohistochemical studies, with findings reflective of mycosis fungoides.
Caput medusae (palm tree sign) is a cardinal feature of portal hypertension characterized by grossly dilated and engorged periumbilical veins. To shunt blood from the portal venous system, cutaneous collateral veins between the umbilical veins and abdominal wall veins are used, resulting in the appearance of engorged veins in the anterior abdominal wall.10 The diagnosis can be made with abdominal ultrasonography showing the direction of blood flow through abdominal vessels.
The Diagnosis: Superior Vena Cava Syndrome
Computed tomography angiography of the chest confirmed a diagnosis of superior vena cava (SVC) syndrome due to external pressure of the indwelling catheter. Upon diagnosis, the left indwelling catheter was removed. Further testing to assess for a potential pulmonary embolism was negative. Resolution of the ectatic spider veins and patientreported intermittent facial swelling was achieved after catheter removal.
Superior vena cava syndrome occurs when the SVC is occluded due to extrinsic pressure or thrombosis. Although classically thought to be due to underlying bronchogenic carcinomas, all pathologies that cause compression of the SVC also can lead to vessel occlusion.1 Superior vena cava syndrome initially can be detected on physical examination. The most prominent skin finding includes diffusely dilated blood vessels on the central chest wall, which indicate the presence of collateral blood vessels.1 Imaging studies such as abdominal computed tomography can provide information on the etiology of the condition but are not required for diagnosis. Given the high correlation of SVC syndrome with underlying lung and mediastinal carcinomas, imaging was warranted in our patient. Imaging also can distinguish if the condition is due to external pressure or thrombosis.2 For SVC syndrome due to thrombosis, endovascular therapy is first-line management; however, mechanical thrombectomy may be preferred in patients with absolute contraindication to thrombolytic agents.3 In the setting of increased external pressure on the SVC, treatment includes the removal of the source of pressure.4
In a case series including 78 patients, ports and indwelling catheters accounted for 71% of benign SVC cases.5 Our patient’s SVC syndrome most likely was due to the indwelling catheter pressing on the SVC. The goal of treatment is to address the underlying cause—whether it be pressure or thrombosis. In the setting of increased external pressure, treatment includes removal of the source of pressure from the SVC.4
Other differential diagnoses to consider for newonset ectatic vessels on the chest wall include generalized essential telangiectasia, scleroderma, poikiloderma vasculare atrophicans, and caput medusae. Generalized essential telangiectasia is characterized by red or pink dilated capillary blood vessels in a branch or lacelike pattern predominantly on the lower limbs. The eruption primarily is asymptomatic, though tingling or numbness may be reported.6 The diagnosis can be made with a punch biopsy, with histopathology showing dilated vessels in the dermis.7
Scleroderma is a connective tissue fibrosis disorder with variable clinical presentations. The systemic sclerosis subset can be divided into localized systemic sclerosis and diffuse systemic sclerosis. Physical examination reveals cutaneous sclerosis in various areas of the body. Localized systemic sclerosis includes sclerosis of the fingers and face, while diffuse systemic sclerosis is notable for progression to the arms, legs, and trunk.8 In addition to sclerosis, diffuse telangiectases also can be observed. Systemic sclerosis is a clinical diagnosis based on physical examination and laboratory studies to identify antibodies such as antinuclear antibodies.
Poikiloderma vasculare atrophicans is a variant of cutaneous T-cell lymphoma. The initial presentation is characterized by plaques of hypopigmentation and hyperpigmentation with atrophy and telangiectases. The lesions may be asymptomatic or mildly pruritic and classically involve the trunk and flexural areas.9 The diagnosis is made with skin biopsy and immunohistochemical studies, with findings reflective of mycosis fungoides.
Caput medusae (palm tree sign) is a cardinal feature of portal hypertension characterized by grossly dilated and engorged periumbilical veins. To shunt blood from the portal venous system, cutaneous collateral veins between the umbilical veins and abdominal wall veins are used, resulting in the appearance of engorged veins in the anterior abdominal wall.10 The diagnosis can be made with abdominal ultrasonography showing the direction of blood flow through abdominal vessels.
- Drouin L, Pistorius MA, Lafforgue A, et al. Upper-extremity venous thrombosis: a retrospective study about 160 cases [in French]. Rev Med Interne. 2019;40:9-15.
- Richie E. Clinical pearl: diagnosing superior vena cava syndrome. Emergency Medicine News. 2017;39:22. doi:10.1097/01 .EEM.0000522220.37441.d2
- Azizi A, Shafi I, Shah N, et al. Superior vena cava syndrome. JACC Cardiovasc Interv. 2020;13:2896-2910. doi:10.1016/j.jcin.2020.08.038
- Dumantepe M, Tarhan A, Ozler A. Successful treatment of central venous catheter induced superior vena cava syndrome with ultrasound accelerated catheter-directed thrombolysis. Catheter Cardiovasc Interv. 2013;81:E269-E273.
- Rice TW, Rodriguez RM, Light RW. The superior vena cava syndrome: clinical characteristics and evolving etiology. Medicine (Baltimore) 2006;85:37-42. doi:10.1097/01.md.0000198474.99876.f0
- Long D, Marshman G. Generalized essential telangiectasia. Australas J Dermatol. 2004;45:67-69. doi:10.1111/j.1440-0960.2004.00033.x
- Braverman IM. Ultrastructure and organization of the cutaneous microvasculature in normal and pathologic states. J Invest Dermatol. 1989;93(2 suppl):2S-9S.
- Ferreli C, Gasparini G, Parodi A, et al. Cutaneous manifestations of scleroderma and scleroderma-like disorders: a comprehensive review. Clin Rev Allergy Immunol. 2017;53:306-336. doi:10.1007 /s12016-017-8625-4
- Bloom B, Marchbein S, Fischer M, et al. Poikilodermatous mycosis fungoides. Dermatol Online J. 2012;18:4.
- Sharma B, Raina S. Caput medusae. Indian J Med Res. 2015;141:494. doi:10.4103/0971-5916.159322
- Drouin L, Pistorius MA, Lafforgue A, et al. Upper-extremity venous thrombosis: a retrospective study about 160 cases [in French]. Rev Med Interne. 2019;40:9-15.
- Richie E. Clinical pearl: diagnosing superior vena cava syndrome. Emergency Medicine News. 2017;39:22. doi:10.1097/01 .EEM.0000522220.37441.d2
- Azizi A, Shafi I, Shah N, et al. Superior vena cava syndrome. JACC Cardiovasc Interv. 2020;13:2896-2910. doi:10.1016/j.jcin.2020.08.038
- Dumantepe M, Tarhan A, Ozler A. Successful treatment of central venous catheter induced superior vena cava syndrome with ultrasound accelerated catheter-directed thrombolysis. Catheter Cardiovasc Interv. 2013;81:E269-E273.
- Rice TW, Rodriguez RM, Light RW. The superior vena cava syndrome: clinical characteristics and evolving etiology. Medicine (Baltimore) 2006;85:37-42. doi:10.1097/01.md.0000198474.99876.f0
- Long D, Marshman G. Generalized essential telangiectasia. Australas J Dermatol. 2004;45:67-69. doi:10.1111/j.1440-0960.2004.00033.x
- Braverman IM. Ultrastructure and organization of the cutaneous microvasculature in normal and pathologic states. J Invest Dermatol. 1989;93(2 suppl):2S-9S.
- Ferreli C, Gasparini G, Parodi A, et al. Cutaneous manifestations of scleroderma and scleroderma-like disorders: a comprehensive review. Clin Rev Allergy Immunol. 2017;53:306-336. doi:10.1007 /s12016-017-8625-4
- Bloom B, Marchbein S, Fischer M, et al. Poikilodermatous mycosis fungoides. Dermatol Online J. 2012;18:4.
- Sharma B, Raina S. Caput medusae. Indian J Med Res. 2015;141:494. doi:10.4103/0971-5916.159322
A 32-year-old woman presented to vascular surgery for evaluation of spider veins of 2 years’ duration that originated on the breasts but later spread to include the central chest, inframammary folds, and back. She reported associated pain and discomfort as well as intermittent facial swelling and tachycardia but denied pruritus and bleeding. The patient had a history of a kidney transplant 6 months prior, Langerhans cell histiocytosis, and Sjögren syndrome with a left indwelling catheter. Her current medications included systemic immunosuppressive agents. Physical examination revealed blue-purple ectatic vessels on the inframammary folds and central chest extending to the back. Erythema on the face, neck, and arms was not appreciated. No palpable cervical, supraclavicular, or axillary lymph nodes were noted.


Pink Papules on the Cheek
The Diagnosis: Cutaneous Rosai-Dorfman Disease
Rosai-Dorfman disease is a rare benign non- Langerhans cell histiocytopathy that can manifest initially with lymph node involvement—classically, massive painless cervical lymphadenopathy.1 Cutaneous Rosai-Dorfman disease (CRDD) is a variant that can be associated with lymph node and internal involvement, but more than 80% of cases lack extracutaneous involvement.2,3 In cases with extracutaneous involvement, lymph node disease is most frequent.3 Cutaneous Rosai-Dorfman disease unassociated with extracutaneous disease is a benign self-limiting histiocytopathy that manifests as painless red-brown, yellow, or fleshcolored nodules, plaques, or papules that may become tender or ulcerated.4
Cutaneous Rosai-Dorfman disease represents a benign histiocytopathy of resident dendritic cell derivation.3 A characteristic immunohistochemical finding is S-100 positivity, which might suggest a Langerhans cell transdifferentiation phenotype, but other markers corroborative of a Langerhans cell phenotype—namely CD1a and langerin—will be negative. Biopsies typically show a mid to deep dermal histiocytic infiltration in a variably dense polymorphous inflammatory cell background comprised of a mixture of lymphocytes, plasma cells, and neutrophils.3 At times the extent of lymphocytic infiltration can be to a magnitude that resembles a lymphoma on histopathology. In our patient, lymphoma was excluded based on clinical presentation, as this patient lacked the typical symptoms of lymphadenopathy or B symptoms that come with B-cell lymphoma.5
The histiocytes in CRDD are characteristically large mononuclear cells exhibiting a low nuclear to cytoplasmic ratio reflective of the voluminous, nonvacuolated, watery cytoplasm. They have ill-defined cytoplasmic membranes resulting in a seemingly syncytial growth pattern. A hallmark of the histiocytes is emperipolesis characterized by intracytoplasmic localization of intact inflammatory cells including neutrophils, lymphocytes, and plasma cells.3
The differential diagnosis of CRDD includes Langerhans cell histiocytosis (LCH), indeterminate cell histiocytosis, xanthogranuloma, and reticulohistiocytoma. All of these conditions can be differentiated by their unique histopathologic and phenotypic characteristics.
Langerhans cell histiocytosis is a distinct clonal histiocytopathy that has a varied presentation ranging from cutaneous confined cases manifesting as a solitary lesion to one of disseminated cutaneous disease with the potential for multiorgan involvement. Regardless of the variant of LCH, the hallmark cell is one showing an eccentrically disposed, reniform nucleus with an open chromatin and abundant eosinophilic cytoplasm (Figure 1).6 Both LCH and CRDD stain positive for S-100. However, unlike the histiocytes in CRDD, those seen in LCH stain positive for CD1a and langerin and would not express factor XIIIA. Additionally, the neoplastic cells would not exhibit the same extent of CD68 positivity seen in CRDD.6 Treatment of LCH depends on the extent of disease, especially for the presence or absence of extracutaneous disease.7

A variant of LCH is indeterminate cell histiocytosis, which can be seen in neonates or adults. It represents a neoplastic proliferation of Langerhans cells that are devoid of Birbeck granules, reflective of an immature early phase of differentiation in the skin prior to the cells acquiring the Birbeck granule (as would be seen in neonates) or a later phase of differentiation after the mature Langerhans cell has encountered antigen and is en route to the lymph node (typically seen in adults).8 The phenotypic profile is identical to conventional LCH except the cells do not express langerin. Microscopically, the infiltrates are composed of Langerhans cells that are morphologically indistinguishable from classic LCH but without epidermotropism and exhibit a dominant localization in the dermis typically unassociated with other inflammatory cell elements (Figure 2).9

Xanthogranuloma is seen in young children (juvenile xanthogranuloma) as a solitary lesion, though a multifocal cutaneous variant and extracutaneous presentations have been described. Similar lesions can be seen in adults.10 These lesions are evolutionary in their morphology. In the inception of a juvenile xanthogranuloma, the lesions are highly cellular, and the histiocytes typically are poorly lipidized; there may be a dearth of other inflammatory cell elements. As the lesions mature, the histiocytes become lipidized, and characteristic Touton giant cells that exhibit a wreath of nuclei with peripheral lipidization may develop (Figure 3). In the later stages, there is considerable hyalinizing fibrosis, and the cells can acquire a spindled appearance. The absence of emperipolesis and the presence of notable lipidization are useful light microscopy features differentiating xanthogranuloma from CRDD.11 Treatment of xanthogranuloma can range from a conservative monitoring approach to an aggressive approach combining various antineoplastic therapies with immunosuppressive agents.12

Solitary and multicentric reticulohistiocytoma is another form of resident dendritic cell histiocytopathy that can resemble Rosai-Dorfman disease. It is a dermal histiocytic infiltrate accompanied by a polymorphous inflammatory cell infiltrate (Figure 4) and can show variable fibrosis.13 One of the hallmarks is the distinct amphophilic cytoplasms, possibly attributable to nuclear DNA released into the cytoplasm from effete nuclei.13 The solitary form is unassociated with systemic disease, whereas the multicentric variant can be a paraneoplastic syndrome in the setting of solid and hematologic malignancies.14 In addition, in the multicentric variant, similar lesions can affect any organ but there can be a proclivity to involve the hand and knee joints, leading to a crippling arthritis.15 We presented a case of CRDD, a benign resident dendritic cell histiocytopathy that can manifest as a cutaneous confined process in the skin where the clinical course is characteristically benign. It potentially can be confused with LCH, indeterminate cell histiocytosis, xanthogranuloma, and reticulohistiocytoma. For a solitary lesion, intralesional triamcinolone injection and excision are options. Multifocal cutaneous disease or CRDD with notable extracutaneous disease may require systemic treatment.16 In our patient, one intralesional triamcinolone injection was performed with notable resolution.

- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002;24:385.
- Ahmed A, Crowson N, Magro CM. A comprehensive assessment of cutaneous Rosai-Dorfman disease. Ann Diagn Pathol. 2019;40:166-173.
- Frater JL, Maddox JS, Obadiah JM, et al. Cutaneous Rosai-Dorfman disease: comprehensive review of cases reported in the medical literature since 1990 and presentation of an illustrative case. J Cutan Med Surg. 2006;10:281-290.
- Friedberg JW, Fisher RI. Diffuse large B-cell lymphoma. Hematol Oncol Clin North Am. 2008;22:941-952. Doi:10.1016/j.hoc.2008.07.002
- Allen CE, Merad M, McClain KL. Langerhans-cell histiocytosis. N Engl J Med. 2018;379:856-868.
- Board PPTE. Langerhans cell histiocytosis treatment (PDQ®). In: PDQ Cancer Information Summaries [Internet]. National Cancer Institute (US); 2009.
- Chu A, Eisinger M, Lee JS, et al. Immunoelectron microscopic identification of Langerhans cells using a new antigenic marker. J Invest Dermatol. 1982;78:177-180. doi:10.1111/1523-1747.ep12506352
- Berti E, Gianotti R, Alessi E. Unusual cutaneous histiocytosis expressing an intermediate immunophenotype between Langerhans’ cells and dermal macrophages. Arch Dermatol. 1988;124:1250-1253. doi:10.1001/archderm.1988.01670080062020
- Cypel TKS, Zuker RM. Juvenile xanthogranuloma: case report and review of the literature. Can J Plast Surg. 2008;16:175-177.
- Rodriguez J, Ackerman AB. Xanthogranuloma in adults. Arch Dermatol. 1976;112:43-44.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. In: StatPearls [Internet]. StatPearls Publishing; 2022.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492. doi:10.1016/j. clindermatol.2006.07.010
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521.
- Gold RH, Metzger AL, Mirra JM, et al. Multicentric reticulohistiocytosis (lipoid dermato-arthritis). An erosive polyarthritis with distinctive clinical, roentgenographic and pathologic features. Am J Roentgenol Radium Ther Nucl Med. 1975;124:610-624. doi:10.2214/ajr.124.4.610
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327. doi:10.1177/107327481402100408
The Diagnosis: Cutaneous Rosai-Dorfman Disease
Rosai-Dorfman disease is a rare benign non- Langerhans cell histiocytopathy that can manifest initially with lymph node involvement—classically, massive painless cervical lymphadenopathy.1 Cutaneous Rosai-Dorfman disease (CRDD) is a variant that can be associated with lymph node and internal involvement, but more than 80% of cases lack extracutaneous involvement.2,3 In cases with extracutaneous involvement, lymph node disease is most frequent.3 Cutaneous Rosai-Dorfman disease unassociated with extracutaneous disease is a benign self-limiting histiocytopathy that manifests as painless red-brown, yellow, or fleshcolored nodules, plaques, or papules that may become tender or ulcerated.4
Cutaneous Rosai-Dorfman disease represents a benign histiocytopathy of resident dendritic cell derivation.3 A characteristic immunohistochemical finding is S-100 positivity, which might suggest a Langerhans cell transdifferentiation phenotype, but other markers corroborative of a Langerhans cell phenotype—namely CD1a and langerin—will be negative. Biopsies typically show a mid to deep dermal histiocytic infiltration in a variably dense polymorphous inflammatory cell background comprised of a mixture of lymphocytes, plasma cells, and neutrophils.3 At times the extent of lymphocytic infiltration can be to a magnitude that resembles a lymphoma on histopathology. In our patient, lymphoma was excluded based on clinical presentation, as this patient lacked the typical symptoms of lymphadenopathy or B symptoms that come with B-cell lymphoma.5
The histiocytes in CRDD are characteristically large mononuclear cells exhibiting a low nuclear to cytoplasmic ratio reflective of the voluminous, nonvacuolated, watery cytoplasm. They have ill-defined cytoplasmic membranes resulting in a seemingly syncytial growth pattern. A hallmark of the histiocytes is emperipolesis characterized by intracytoplasmic localization of intact inflammatory cells including neutrophils, lymphocytes, and plasma cells.3
The differential diagnosis of CRDD includes Langerhans cell histiocytosis (LCH), indeterminate cell histiocytosis, xanthogranuloma, and reticulohistiocytoma. All of these conditions can be differentiated by their unique histopathologic and phenotypic characteristics.
Langerhans cell histiocytosis is a distinct clonal histiocytopathy that has a varied presentation ranging from cutaneous confined cases manifesting as a solitary lesion to one of disseminated cutaneous disease with the potential for multiorgan involvement. Regardless of the variant of LCH, the hallmark cell is one showing an eccentrically disposed, reniform nucleus with an open chromatin and abundant eosinophilic cytoplasm (Figure 1).6 Both LCH and CRDD stain positive for S-100. However, unlike the histiocytes in CRDD, those seen in LCH stain positive for CD1a and langerin and would not express factor XIIIA. Additionally, the neoplastic cells would not exhibit the same extent of CD68 positivity seen in CRDD.6 Treatment of LCH depends on the extent of disease, especially for the presence or absence of extracutaneous disease.7
A variant of LCH is indeterminate cell histiocytosis, which can be seen in neonates or adults. It represents a neoplastic proliferation of Langerhans cells that are devoid of Birbeck granules, reflective of an immature early phase of differentiation in the skin prior to the cells acquiring the Birbeck granule (as would be seen in neonates) or a later phase of differentiation after the mature Langerhans cell has encountered antigen and is en route to the lymph node (typically seen in adults).8 The phenotypic profile is identical to conventional LCH except the cells do not express langerin. Microscopically, the infiltrates are composed of Langerhans cells that are morphologically indistinguishable from classic LCH but without epidermotropism and exhibit a dominant localization in the dermis typically unassociated with other inflammatory cell elements (Figure 2).9
Xanthogranuloma is seen in young children (juvenile xanthogranuloma) as a solitary lesion, though a multifocal cutaneous variant and extracutaneous presentations have been described. Similar lesions can be seen in adults.10 These lesions are evolutionary in their morphology. In the inception of a juvenile xanthogranuloma, the lesions are highly cellular, and the histiocytes typically are poorly lipidized; there may be a dearth of other inflammatory cell elements. As the lesions mature, the histiocytes become lipidized, and characteristic Touton giant cells that exhibit a wreath of nuclei with peripheral lipidization may develop (Figure 3). In the later stages, there is considerable hyalinizing fibrosis, and the cells can acquire a spindled appearance. The absence of emperipolesis and the presence of notable lipidization are useful light microscopy features differentiating xanthogranuloma from CRDD.11 Treatment of xanthogranuloma can range from a conservative monitoring approach to an aggressive approach combining various antineoplastic therapies with immunosuppressive agents.12
Solitary and multicentric reticulohistiocytoma is another form of resident dendritic cell histiocytopathy that can resemble Rosai-Dorfman disease. It is a dermal histiocytic infiltrate accompanied by a polymorphous inflammatory cell infiltrate (Figure 4) and can show variable fibrosis.13 One of the hallmarks is the distinct amphophilic cytoplasms, possibly attributable to nuclear DNA released into the cytoplasm from effete nuclei.13 The solitary form is unassociated with systemic disease, whereas the multicentric variant can be a paraneoplastic syndrome in the setting of solid and hematologic malignancies.14 In addition, in the multicentric variant, similar lesions can affect any organ but there can be a proclivity to involve the hand and knee joints, leading to a crippling arthritis.15 We presented a case of CRDD, a benign resident dendritic cell histiocytopathy that can manifest as a cutaneous confined process in the skin where the clinical course is characteristically benign. It potentially can be confused with LCH, indeterminate cell histiocytosis, xanthogranuloma, and reticulohistiocytoma. For a solitary lesion, intralesional triamcinolone injection and excision are options. Multifocal cutaneous disease or CRDD with notable extracutaneous disease may require systemic treatment.16 In our patient, one intralesional triamcinolone injection was performed with notable resolution.
The Diagnosis: Cutaneous Rosai-Dorfman Disease
Rosai-Dorfman disease is a rare benign non- Langerhans cell histiocytopathy that can manifest initially with lymph node involvement—classically, massive painless cervical lymphadenopathy.1 Cutaneous Rosai-Dorfman disease (CRDD) is a variant that can be associated with lymph node and internal involvement, but more than 80% of cases lack extracutaneous involvement.2,3 In cases with extracutaneous involvement, lymph node disease is most frequent.3 Cutaneous Rosai-Dorfman disease unassociated with extracutaneous disease is a benign self-limiting histiocytopathy that manifests as painless red-brown, yellow, or fleshcolored nodules, plaques, or papules that may become tender or ulcerated.4
Cutaneous Rosai-Dorfman disease represents a benign histiocytopathy of resident dendritic cell derivation.3 A characteristic immunohistochemical finding is S-100 positivity, which might suggest a Langerhans cell transdifferentiation phenotype, but other markers corroborative of a Langerhans cell phenotype—namely CD1a and langerin—will be negative. Biopsies typically show a mid to deep dermal histiocytic infiltration in a variably dense polymorphous inflammatory cell background comprised of a mixture of lymphocytes, plasma cells, and neutrophils.3 At times the extent of lymphocytic infiltration can be to a magnitude that resembles a lymphoma on histopathology. In our patient, lymphoma was excluded based on clinical presentation, as this patient lacked the typical symptoms of lymphadenopathy or B symptoms that come with B-cell lymphoma.5
The histiocytes in CRDD are characteristically large mononuclear cells exhibiting a low nuclear to cytoplasmic ratio reflective of the voluminous, nonvacuolated, watery cytoplasm. They have ill-defined cytoplasmic membranes resulting in a seemingly syncytial growth pattern. A hallmark of the histiocytes is emperipolesis characterized by intracytoplasmic localization of intact inflammatory cells including neutrophils, lymphocytes, and plasma cells.3
The differential diagnosis of CRDD includes Langerhans cell histiocytosis (LCH), indeterminate cell histiocytosis, xanthogranuloma, and reticulohistiocytoma. All of these conditions can be differentiated by their unique histopathologic and phenotypic characteristics.
Langerhans cell histiocytosis is a distinct clonal histiocytopathy that has a varied presentation ranging from cutaneous confined cases manifesting as a solitary lesion to one of disseminated cutaneous disease with the potential for multiorgan involvement. Regardless of the variant of LCH, the hallmark cell is one showing an eccentrically disposed, reniform nucleus with an open chromatin and abundant eosinophilic cytoplasm (Figure 1).6 Both LCH and CRDD stain positive for S-100. However, unlike the histiocytes in CRDD, those seen in LCH stain positive for CD1a and langerin and would not express factor XIIIA. Additionally, the neoplastic cells would not exhibit the same extent of CD68 positivity seen in CRDD.6 Treatment of LCH depends on the extent of disease, especially for the presence or absence of extracutaneous disease.7
A variant of LCH is indeterminate cell histiocytosis, which can be seen in neonates or adults. It represents a neoplastic proliferation of Langerhans cells that are devoid of Birbeck granules, reflective of an immature early phase of differentiation in the skin prior to the cells acquiring the Birbeck granule (as would be seen in neonates) or a later phase of differentiation after the mature Langerhans cell has encountered antigen and is en route to the lymph node (typically seen in adults).8 The phenotypic profile is identical to conventional LCH except the cells do not express langerin. Microscopically, the infiltrates are composed of Langerhans cells that are morphologically indistinguishable from classic LCH but without epidermotropism and exhibit a dominant localization in the dermis typically unassociated with other inflammatory cell elements (Figure 2).9
Xanthogranuloma is seen in young children (juvenile xanthogranuloma) as a solitary lesion, though a multifocal cutaneous variant and extracutaneous presentations have been described. Similar lesions can be seen in adults.10 These lesions are evolutionary in their morphology. In the inception of a juvenile xanthogranuloma, the lesions are highly cellular, and the histiocytes typically are poorly lipidized; there may be a dearth of other inflammatory cell elements. As the lesions mature, the histiocytes become lipidized, and characteristic Touton giant cells that exhibit a wreath of nuclei with peripheral lipidization may develop (Figure 3). In the later stages, there is considerable hyalinizing fibrosis, and the cells can acquire a spindled appearance. The absence of emperipolesis and the presence of notable lipidization are useful light microscopy features differentiating xanthogranuloma from CRDD.11 Treatment of xanthogranuloma can range from a conservative monitoring approach to an aggressive approach combining various antineoplastic therapies with immunosuppressive agents.12
Solitary and multicentric reticulohistiocytoma is another form of resident dendritic cell histiocytopathy that can resemble Rosai-Dorfman disease. It is a dermal histiocytic infiltrate accompanied by a polymorphous inflammatory cell infiltrate (Figure 4) and can show variable fibrosis.13 One of the hallmarks is the distinct amphophilic cytoplasms, possibly attributable to nuclear DNA released into the cytoplasm from effete nuclei.13 The solitary form is unassociated with systemic disease, whereas the multicentric variant can be a paraneoplastic syndrome in the setting of solid and hematologic malignancies.14 In addition, in the multicentric variant, similar lesions can affect any organ but there can be a proclivity to involve the hand and knee joints, leading to a crippling arthritis.15 We presented a case of CRDD, a benign resident dendritic cell histiocytopathy that can manifest as a cutaneous confined process in the skin where the clinical course is characteristically benign. It potentially can be confused with LCH, indeterminate cell histiocytosis, xanthogranuloma, and reticulohistiocytoma. For a solitary lesion, intralesional triamcinolone injection and excision are options. Multifocal cutaneous disease or CRDD with notable extracutaneous disease may require systemic treatment.16 In our patient, one intralesional triamcinolone injection was performed with notable resolution.
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002;24:385.
- Ahmed A, Crowson N, Magro CM. A comprehensive assessment of cutaneous Rosai-Dorfman disease. Ann Diagn Pathol. 2019;40:166-173.
- Frater JL, Maddox JS, Obadiah JM, et al. Cutaneous Rosai-Dorfman disease: comprehensive review of cases reported in the medical literature since 1990 and presentation of an illustrative case. J Cutan Med Surg. 2006;10:281-290.
- Friedberg JW, Fisher RI. Diffuse large B-cell lymphoma. Hematol Oncol Clin North Am. 2008;22:941-952. Doi:10.1016/j.hoc.2008.07.002
- Allen CE, Merad M, McClain KL. Langerhans-cell histiocytosis. N Engl J Med. 2018;379:856-868.
- Board PPTE. Langerhans cell histiocytosis treatment (PDQ®). In: PDQ Cancer Information Summaries [Internet]. National Cancer Institute (US); 2009.
- Chu A, Eisinger M, Lee JS, et al. Immunoelectron microscopic identification of Langerhans cells using a new antigenic marker. J Invest Dermatol. 1982;78:177-180. doi:10.1111/1523-1747.ep12506352
- Berti E, Gianotti R, Alessi E. Unusual cutaneous histiocytosis expressing an intermediate immunophenotype between Langerhans’ cells and dermal macrophages. Arch Dermatol. 1988;124:1250-1253. doi:10.1001/archderm.1988.01670080062020
- Cypel TKS, Zuker RM. Juvenile xanthogranuloma: case report and review of the literature. Can J Plast Surg. 2008;16:175-177.
- Rodriguez J, Ackerman AB. Xanthogranuloma in adults. Arch Dermatol. 1976;112:43-44.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. In: StatPearls [Internet]. StatPearls Publishing; 2022.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492. doi:10.1016/j. clindermatol.2006.07.010
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521.
- Gold RH, Metzger AL, Mirra JM, et al. Multicentric reticulohistiocytosis (lipoid dermato-arthritis). An erosive polyarthritis with distinctive clinical, roentgenographic and pathologic features. Am J Roentgenol Radium Ther Nucl Med. 1975;124:610-624. doi:10.2214/ajr.124.4.610
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327. doi:10.1177/107327481402100408
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002;24:385.
- Ahmed A, Crowson N, Magro CM. A comprehensive assessment of cutaneous Rosai-Dorfman disease. Ann Diagn Pathol. 2019;40:166-173.
- Frater JL, Maddox JS, Obadiah JM, et al. Cutaneous Rosai-Dorfman disease: comprehensive review of cases reported in the medical literature since 1990 and presentation of an illustrative case. J Cutan Med Surg. 2006;10:281-290.
- Friedberg JW, Fisher RI. Diffuse large B-cell lymphoma. Hematol Oncol Clin North Am. 2008;22:941-952. Doi:10.1016/j.hoc.2008.07.002
- Allen CE, Merad M, McClain KL. Langerhans-cell histiocytosis. N Engl J Med. 2018;379:856-868.
- Board PPTE. Langerhans cell histiocytosis treatment (PDQ®). In: PDQ Cancer Information Summaries [Internet]. National Cancer Institute (US); 2009.
- Chu A, Eisinger M, Lee JS, et al. Immunoelectron microscopic identification of Langerhans cells using a new antigenic marker. J Invest Dermatol. 1982;78:177-180. doi:10.1111/1523-1747.ep12506352
- Berti E, Gianotti R, Alessi E. Unusual cutaneous histiocytosis expressing an intermediate immunophenotype between Langerhans’ cells and dermal macrophages. Arch Dermatol. 1988;124:1250-1253. doi:10.1001/archderm.1988.01670080062020
- Cypel TKS, Zuker RM. Juvenile xanthogranuloma: case report and review of the literature. Can J Plast Surg. 2008;16:175-177.
- Rodriguez J, Ackerman AB. Xanthogranuloma in adults. Arch Dermatol. 1976;112:43-44.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. In: StatPearls [Internet]. StatPearls Publishing; 2022.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492. doi:10.1016/j. clindermatol.2006.07.010
- Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol. 2006;30:521.
- Gold RH, Metzger AL, Mirra JM, et al. Multicentric reticulohistiocytosis (lipoid dermato-arthritis). An erosive polyarthritis with distinctive clinical, roentgenographic and pathologic features. Am J Roentgenol Radium Ther Nucl Med. 1975;124:610-624. doi:10.2214/ajr.124.4.610
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327. doi:10.1177/107327481402100408
A 31-year-old woman presented with a slow-growing, tender, pruritic lesion on the right cheek of 4 to 5 months’ duration. She had been applying petroleum jelly and hydrocortisone cream 2.5% without any improvement. Physical examination revealed a 1×5-mm, pearly pink, erythematous, crusted papule with arborizing vessels surrounded by scattered pink papules with white dots within. No cervical lymphadenopathy was appreciated on physical examination, and the patient denied any other systemic symptoms. Shave and punch biopsies of the lesion were performed; stains for microorganisms were negative. The biopsy showed a dense reticular mixed inflammatory cell infiltrate comprised of a mixture of histiocytes (top), lymphocytes, neutrophils, and plasma cells that assumed a diffuse growth pattern within the dermis. The histiocytes exhibited abundant watery cytoplasms with ill-defined cytoplasmic membranes; intact leukocytes were found within the cytoplasms. The histiocytes demonstrated a unique phenotype characterized by S-100 (bottom) and CD68 positivity.

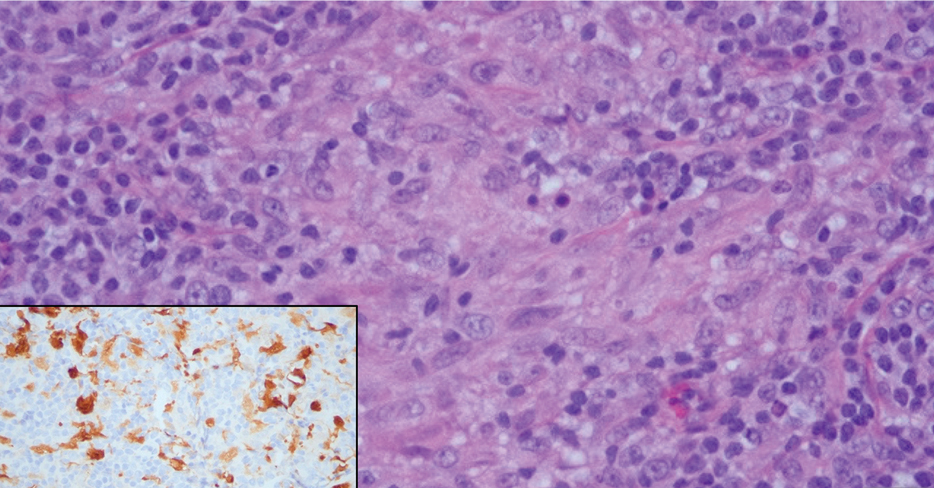

The Many Uses of the Humble Alcohol Swab
Practice Gap
In light of inflation, rising costs of procedures, and decreased reimbursements,1 there is an increased need to identify and utilize inexpensive multitasking tools that can serve the dermatologic surgeon from preoperative to postoperative care. The 70% isopropyl alcohol swab may be the dermatologist’s most cost-effective and versatile surgical tool.
The Technique
When assessing a lesion, alcohol swabs can remove scale, crust, or residue from personal care products to help reveal primary morphology. They aid in the diagnosis of porokeratosis by highlighting the cornoid lamella when used following application of gentian violet.2 The alcohol swab also can lay down a liquid interface to facilitate contact dermoscopy and improve visualization while also reducing the transmission of pathogens by the dermatoscope.3 Rubbing an area with an alcohol swab can induce vasodilation of scar tissue, which also may help localize a prior biopsy or surgical site (Figure).

Before a surgical site is marked, an initial cleanse with an alcohol swab serves to both remove debris and provide antisepsis ahead of the procedure. Additionally, the swab may improve adherence of skin markers by clearing excess lipid from the skin surface. Assessing the amount of debris and oil removed in the process can help determine a patient’s baseline level of hygiene, which can aid postoperative wound care planning. In extreme cases, use of an alcohol swab may help diagnose dermatitis neglecta or terra firma-forme dermatosis by completely removing any pigmentation.4
After surgery, the alcohol swab can remove skin marker(s) and blood and prepare the site for the surgical dressing. There also is some evidence to suggest that cleansing the surgical site with an alcohol swab as part of routine postoperative wound care may decrease incidence of surgical-site infection.5 At follow-up, the swab can remove crust and clean the skin before suture removal. If infection is suspected, the swab can cleanse skin before a wound culture is obtained to remove skin commensals and flora on the outer surface of the wound.
Practice Implications
The 70% isopropyl alcohol swab can assist the dermatologist in numerous tasks related to everyday procedures. It is readily available in every clinic and costs only a few cents.
- Pollock JR, Chen JY, Dorius DA, et al. Decreasing physician Medicare reimbursement for dermatology services. J Am Acad Dermatol. 2022;86:1154-1156.
- Thomas CJ, Elston DM. Medical pearl: Gentian violet to highlight the cornoid lamella in disseminated superficial actinic porokeratosis.J Am Acad Dermatol. 2005;52(3 pt 1):513-514.
- Kelly SC, Purcell SM. Prevention of nosocomial infection during dermoscopy? Dermatol Surg. 2006;32:552-555.
- Blattner CM, Perry B, Snider K, et al. Clinical pearl: increasing utility of isopropyl alcohol for cutaneous dyschromia. Cutis. 2016;97:287;301.
- Vogt KN, Chadi S, Parry N, et al. Daily incision cleansing with alcohol reduces the rate of surgical site infections: a pilot study. Am Surg. 2015;81:1182-1186.
Practice Gap
In light of inflation, rising costs of procedures, and decreased reimbursements,1 there is an increased need to identify and utilize inexpensive multitasking tools that can serve the dermatologic surgeon from preoperative to postoperative care. The 70% isopropyl alcohol swab may be the dermatologist’s most cost-effective and versatile surgical tool.
The Technique
When assessing a lesion, alcohol swabs can remove scale, crust, or residue from personal care products to help reveal primary morphology. They aid in the diagnosis of porokeratosis by highlighting the cornoid lamella when used following application of gentian violet.2 The alcohol swab also can lay down a liquid interface to facilitate contact dermoscopy and improve visualization while also reducing the transmission of pathogens by the dermatoscope.3 Rubbing an area with an alcohol swab can induce vasodilation of scar tissue, which also may help localize a prior biopsy or surgical site (Figure).
Before a surgical site is marked, an initial cleanse with an alcohol swab serves to both remove debris and provide antisepsis ahead of the procedure. Additionally, the swab may improve adherence of skin markers by clearing excess lipid from the skin surface. Assessing the amount of debris and oil removed in the process can help determine a patient’s baseline level of hygiene, which can aid postoperative wound care planning. In extreme cases, use of an alcohol swab may help diagnose dermatitis neglecta or terra firma-forme dermatosis by completely removing any pigmentation.4
After surgery, the alcohol swab can remove skin marker(s) and blood and prepare the site for the surgical dressing. There also is some evidence to suggest that cleansing the surgical site with an alcohol swab as part of routine postoperative wound care may decrease incidence of surgical-site infection.5 At follow-up, the swab can remove crust and clean the skin before suture removal. If infection is suspected, the swab can cleanse skin before a wound culture is obtained to remove skin commensals and flora on the outer surface of the wound.
Practice Implications
The 70% isopropyl alcohol swab can assist the dermatologist in numerous tasks related to everyday procedures. It is readily available in every clinic and costs only a few cents.
Practice Gap
In light of inflation, rising costs of procedures, and decreased reimbursements,1 there is an increased need to identify and utilize inexpensive multitasking tools that can serve the dermatologic surgeon from preoperative to postoperative care. The 70% isopropyl alcohol swab may be the dermatologist’s most cost-effective and versatile surgical tool.
The Technique
When assessing a lesion, alcohol swabs can remove scale, crust, or residue from personal care products to help reveal primary morphology. They aid in the diagnosis of porokeratosis by highlighting the cornoid lamella when used following application of gentian violet.2 The alcohol swab also can lay down a liquid interface to facilitate contact dermoscopy and improve visualization while also reducing the transmission of pathogens by the dermatoscope.3 Rubbing an area with an alcohol swab can induce vasodilation of scar tissue, which also may help localize a prior biopsy or surgical site (Figure).
Before a surgical site is marked, an initial cleanse with an alcohol swab serves to both remove debris and provide antisepsis ahead of the procedure. Additionally, the swab may improve adherence of skin markers by clearing excess lipid from the skin surface. Assessing the amount of debris and oil removed in the process can help determine a patient’s baseline level of hygiene, which can aid postoperative wound care planning. In extreme cases, use of an alcohol swab may help diagnose dermatitis neglecta or terra firma-forme dermatosis by completely removing any pigmentation.4
After surgery, the alcohol swab can remove skin marker(s) and blood and prepare the site for the surgical dressing. There also is some evidence to suggest that cleansing the surgical site with an alcohol swab as part of routine postoperative wound care may decrease incidence of surgical-site infection.5 At follow-up, the swab can remove crust and clean the skin before suture removal. If infection is suspected, the swab can cleanse skin before a wound culture is obtained to remove skin commensals and flora on the outer surface of the wound.
Practice Implications
The 70% isopropyl alcohol swab can assist the dermatologist in numerous tasks related to everyday procedures. It is readily available in every clinic and costs only a few cents.
- Pollock JR, Chen JY, Dorius DA, et al. Decreasing physician Medicare reimbursement for dermatology services. J Am Acad Dermatol. 2022;86:1154-1156.
- Thomas CJ, Elston DM. Medical pearl: Gentian violet to highlight the cornoid lamella in disseminated superficial actinic porokeratosis.J Am Acad Dermatol. 2005;52(3 pt 1):513-514.
- Kelly SC, Purcell SM. Prevention of nosocomial infection during dermoscopy? Dermatol Surg. 2006;32:552-555.
- Blattner CM, Perry B, Snider K, et al. Clinical pearl: increasing utility of isopropyl alcohol for cutaneous dyschromia. Cutis. 2016;97:287;301.
- Vogt KN, Chadi S, Parry N, et al. Daily incision cleansing with alcohol reduces the rate of surgical site infections: a pilot study. Am Surg. 2015;81:1182-1186.
- Pollock JR, Chen JY, Dorius DA, et al. Decreasing physician Medicare reimbursement for dermatology services. J Am Acad Dermatol. 2022;86:1154-1156.
- Thomas CJ, Elston DM. Medical pearl: Gentian violet to highlight the cornoid lamella in disseminated superficial actinic porokeratosis.J Am Acad Dermatol. 2005;52(3 pt 1):513-514.
- Kelly SC, Purcell SM. Prevention of nosocomial infection during dermoscopy? Dermatol Surg. 2006;32:552-555.
- Blattner CM, Perry B, Snider K, et al. Clinical pearl: increasing utility of isopropyl alcohol for cutaneous dyschromia. Cutis. 2016;97:287;301.
- Vogt KN, Chadi S, Parry N, et al. Daily incision cleansing with alcohol reduces the rate of surgical site infections: a pilot study. Am Surg. 2015;81:1182-1186.

PRAME Expression in Melanocytic Proliferations in Special Sites
The assessment and diagnosis of melanocytic lesions can present a formidable challenge to even a seasoned pathologist, which is especially true when dealing with the subset of nevi occurring at special sites—where baseline variations inherent to particular locations on the body can preclude the use of features routinely used to diagnose malignancy elsewhere. These so-called special-site nevi previously have been described in the literature along with suggested criteria for differentiating malignant lesions from their benign counterparts.1 Locations generally considered to be special sites include the acral skin, anogenital region, breast, ear, and flexural regions.1,2
When evaluating non–special-site melanocytic lesions, general characteristics associated with a malignant diagnosis include confluence or pagetoid spread of melanocytes, nuclear pleomorphism, cytologic atypia, and irregular architecture3; however, these features can be compatible with a benign diagnosis in special-site nevi depending on their extent and the site in question. Although they can be atypical, special-site nevi tend to have the bulk of their architectural distortion and cytologic atypia in the center of the lesion as opposed to the edges.1 If a given lesion is from a special site but lacks this reassuring feature, special care should be taken to rule out malignancy.
Preferentially expressed antigen in melanoma (PRAME) is an antigen first identified in tumor-reactive T-cell populations in patients with malignant melanoma. It is the product of an oncogene that frequently is overexpressed in melanomas, lung squamous cell carcinomas, sarcomas, and acute leukemias.4 It functions as an antagonist of the retinoic acid signaling pathway, which normally serves to induce further cell differentiation, senescence, or apoptosis.5 PRAME inhibits retinoid signaling by forming a complex with both the ligand-bound retinoic acid holoreceptor and the polycomb protein EZH2, which blocks retinoid-dependent gene expression by encouraging chromatin condensation at the RARβ promoter site5; therefore, expressing PRAME allows lesional cells a substantial growth advantage.
PRAME expression has been extensively characterized in non–special-site nevi and has filled the need for a rather specific marker of melanoma.6-10 Although PRAME has been studied in acral nevi,11 the expression pattern in nevi of special sites has yet to be elucidated. Herein, we present a dataset characterizing PRAME expression in these challenging lesions.
Methods
We performed a retrospective case review at the University of Virginia (Charlottesville, Virginia) and collected a panel of 36 special-site nevi that previously were diagnosed as benign by a trained dermatopathologist from January 2020 through December 2022. Special-site nevi were identified using a natural language filter for the following terms: acral, palm, sole, ear, auricular, lip, axilla, armpit, breast, groin, labia, vulva, umbilicus, and penis. This study was approved by the University of Virginia institutional review board.
The original hematoxylin and eosin slides used for primary diagnosis were re-examined to verify the prior diagnosis of benign nevus at a special site. We performed a detailed microscopic examination of all benign nevi in our cohort to determine the frequency of various characteristics at each special site. Sections were prepared from the formalin-fixed and paraffin-embedded tissue blocks and stained with a commercial PRAME antibody (#219650 [Abcam] at a 1:50 dilution) and counterstain. A trained dermatopathologist (S.S.R.) examined the stained sections and recorded the percentage of tumor cells with nuclear PRAME staining. We reported our results using previously established criteria for scoring PRAME immunohistochemistry7: 0 for no expression, 1+ for 1% to 25% expression, 2+ for 26% to 50% expression, 3+ for 51% to 75% expression, and 4+ for diffuse or 76% to 100% expression. Only strong clonal expression within a population of cells was graded.
Data handling and statistical testing were performed using the R Project for Statistical Computing (https://www.r-project.org/). Significance testing was performed using the Fisher exact test. Plot construction was performed using ggplot2 (https://ggplot2.tidyverse.org/).
Results
Our study cohort included 36 special-site nevi, and the control cohort comprised 25 melanoma in situ (MIS) or invasive melanoma (IM) lesions occurring at special sites. Table 1 provides a breakdown of the study and control cohorts by lesion site. Table 2 details the results of our microscopic examination, describing frequency of various characteristics of special-site nevi stratified by site.

Of the 36 special-site nevi in our cohort, 20 (56%) had no staining (0) for PRAME, 11 (31%) demonstrated 1+ PRAME expression, 3 (8%) demonstrated 2+ PRAME expression, and 2 (6%) demonstrated 3+ PRAME expression. No nevi showed 4+ expression. In the control cohort, 24 of 25 (96%) MIS and IM showed 3+ or 4+ expression, with 21 (84%) demonstrating diffuse/4+ expression. One control case (4%) demonstrated 0 PRAME expression. These data are summarized in Table 3 and Figure 1. There is a significant difference in diffuse (4+) PRAME expression between special-site nevi and MIS/IM occurring at special sites (P=1.039×10-12).

 expression score by special-site lesion type (0=no expression; 1+=1%–25% expression; 2+=26%– 50% expression; 3+=51%–75% expression; 4+=diffuse or 76%–100% expression)")
Based on our cohort, a positivity threshold of 3+ for PRAME expression for the diagnosis of melanoma in a special-site lesion would have a sensitivity of 96% and a specificity of 94%, while a positivity threshold of 4+ for PRAME expression would have a sensitivity of 84% and a specificity of 100%. Figures 2 through 4 show photomicrographs of a special-site nevus of the breast, which appropriately does not stain for PRAME; Figures 5 and 6 show an MIS at a special site that appropriately stains for PRAME.

Comment
The distinction between benign and malignant pigmented lesions at special sites presents a fair challenge for pathologists due to the larger degree of leniency for architectural distortion and cytologic atypia in benign lesions at these sites. The presence of architectural distortion or cytologic atypia at the lesion’s edge makes rendering a benign diagnosis especially difficult, and the need for a validated immunohistochemical stain is apparent. In our cohort, strong clonal PRAME expression provided a reliable immunohistochemical marker, allowing for the distinction of malignant lesions from benign nevi at special sites. Diffuse faint PRAME expression was present in several benign nevi within our cohort, and these lesions were considered negative (0) in our analysis.

Given the described test characteristics, we support the implementation of PRAME immunohistochemistry with a positivity threshold of 4+ expression as an ancillary test supporting the diagnosis of IM or MIS in special sites, which would allow clinicians to leverage the high specificity of 4+ PRAME expression to distinguish an IM or MIS from a benign nevus occurring at a special site. We do not recommend the use of 4+ PRAME expression as a screening test for melanoma or MIS among special-site nevi due to its comparatively low sensitivity; however, no one marker is always reliable, and we recommend continued clinicopathologic correlation for all cases.

Although our case series included nevi and MIS/IM from all special sites, we were limited in the number of acrogenital and ear nevi included due to a relative paucity of biopsied benign nevi from these locations at the University of Virginia. Additionally, although the magnitude of the difference in PRAME expression between the study and control groups is sufficient to demonstrate statistical significance, the overall strength of our argument would be increased with a larger study group. We were limited by the number of cases available at our institution, which did not utilize PRAME during the initial diagnosis of the case; including these cases in the study group would have undermined the integrity of our argument because the differentiation of benign vs malignant initially was made using PRAME immunohistochemistry.

Conclusion
Due to their atypical features, special-site nevi can be challenging to assess. In this study, we showed that PRAME expression can be a reliable marker to distinguish benign from malignant lesions. Our results showed that 100% of benign special-site nevi demonstrated 3+ expression or less, with 56% (20/36) demonstrating no expression at all. The presence of diffuse PRAME expression (4+ PRAME staining) appears to be a specific indicator of a malignant lesion, but results should always be interpreted with respect to the patient’s clinical history and the lesion’s histomorphologic features. Further study of a larger sample size would allow refinement of the sensitivity and specificity of diffuse PRAME expression in the determination of malignancy for special-site lesions.

, which highlights the malignant melanocytes in the epidermis, supporting the diagnosis of melanoma in situ")
Acknowledgment—The authors thank the pathologistsat the University of Virginia Biorepository and Tissue Research Facility (Charlottesville, Virginia) for their skill and expertise in performing immunohistochemical staining for this study.
- VandenBoom T, Gerami P. Melanocytic nevi of special sites. In: Pathology of Melanocytic Tumors. Elsevier; 2019:90-100. doi:10.1016/B978-0-323-37457-6.00007-9
- Hosler GA, Moresi JM, Barrett TL. Nevi with site-related atypia: a review of melanocytic nevi with atypical histologic features based on anatomic site. J Cutan Pathol. 2008;35:889-898. doi:10.1111/j.1600-0560.2008.01041.x.
- Brenn T. Melanocytic lesions—staying out of trouble. Ann Diagn Pathol. 2018;37:91-102. doi:10.1016/j.anndiagpath.2018.09.010
- Ikeda H, Lethé B, Lehmann F, et al. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity. 1997;6:199-208. doi:10.1016/s1074-7613(00)80426-4
- Epping MT, Wang L, Edel MJ, et al. The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signaling. Cell. 2005;122:835-847. doi:10.1016/j.cell.2005.07.003
- Alomari AK, Tharp AW, Umphress B, et al. The utility of PRAME immunohistochemistry in the evaluation of challenging melanocytic tumors. J Cutan Pathol. 2021;48:1115-1123. doi:10.1111/cup.14000
- Lezcano C, Jungbluth AA, Nehal KS, et al. PRAME expression in melanocytic tumors. Am J Surg Pathol. 2018;42:1456-1465. doi:10.1097/PAS.0000000000001134
- Gill P, Prieto VG, Austin MT, et al. Diagnostic utility of PRAME in distinguishing proliferative nodules from melanoma in giant congenital melanocytic nevi. J Cutan Pathol. 2021;48:1410-1415. doi:10.1111/cup.14091
- Googe PB, Flanigan KL, Miedema JR. Preferentially expressed antigen in melanoma immunostaining in a series of melanocytic neoplasms. Am J Dermatopathol. 2021;43):794-800. doi:10.1097/DAD.0000000000001885
- Raghavan SS, Wang JY, Kwok S, et al. PRAME expression in melanocytic proliferations with intermediate histopathologic or spitzoid features. J Cutan Pathol. 2020;47:1123-1131. doi:10.1111/cup.13818
- McBride JD, McAfee JL, Piliang M, et al. Preferentially expressed antigen in melanoma and p16 expression in acral melanocytic neoplasms. J Cutan Pathol. 2022;49:220-230. doi:10.1111/cup.14130
The assessment and diagnosis of melanocytic lesions can present a formidable challenge to even a seasoned pathologist, which is especially true when dealing with the subset of nevi occurring at special sites—where baseline variations inherent to particular locations on the body can preclude the use of features routinely used to diagnose malignancy elsewhere. These so-called special-site nevi previously have been described in the literature along with suggested criteria for differentiating malignant lesions from their benign counterparts.1 Locations generally considered to be special sites include the acral skin, anogenital region, breast, ear, and flexural regions.1,2
When evaluating non–special-site melanocytic lesions, general characteristics associated with a malignant diagnosis include confluence or pagetoid spread of melanocytes, nuclear pleomorphism, cytologic atypia, and irregular architecture3; however, these features can be compatible with a benign diagnosis in special-site nevi depending on their extent and the site in question. Although they can be atypical, special-site nevi tend to have the bulk of their architectural distortion and cytologic atypia in the center of the lesion as opposed to the edges.1 If a given lesion is from a special site but lacks this reassuring feature, special care should be taken to rule out malignancy.
Preferentially expressed antigen in melanoma (PRAME) is an antigen first identified in tumor-reactive T-cell populations in patients with malignant melanoma. It is the product of an oncogene that frequently is overexpressed in melanomas, lung squamous cell carcinomas, sarcomas, and acute leukemias.4 It functions as an antagonist of the retinoic acid signaling pathway, which normally serves to induce further cell differentiation, senescence, or apoptosis.5 PRAME inhibits retinoid signaling by forming a complex with both the ligand-bound retinoic acid holoreceptor and the polycomb protein EZH2, which blocks retinoid-dependent gene expression by encouraging chromatin condensation at the RARβ promoter site5; therefore, expressing PRAME allows lesional cells a substantial growth advantage.
PRAME expression has been extensively characterized in non–special-site nevi and has filled the need for a rather specific marker of melanoma.6-10 Although PRAME has been studied in acral nevi,11 the expression pattern in nevi of special sites has yet to be elucidated. Herein, we present a dataset characterizing PRAME expression in these challenging lesions.
Methods
We performed a retrospective case review at the University of Virginia (Charlottesville, Virginia) and collected a panel of 36 special-site nevi that previously were diagnosed as benign by a trained dermatopathologist from January 2020 through December 2022. Special-site nevi were identified using a natural language filter for the following terms: acral, palm, sole, ear, auricular, lip, axilla, armpit, breast, groin, labia, vulva, umbilicus, and penis. This study was approved by the University of Virginia institutional review board.
The original hematoxylin and eosin slides used for primary diagnosis were re-examined to verify the prior diagnosis of benign nevus at a special site. We performed a detailed microscopic examination of all benign nevi in our cohort to determine the frequency of various characteristics at each special site. Sections were prepared from the formalin-fixed and paraffin-embedded tissue blocks and stained with a commercial PRAME antibody (#219650 [Abcam] at a 1:50 dilution) and counterstain. A trained dermatopathologist (S.S.R.) examined the stained sections and recorded the percentage of tumor cells with nuclear PRAME staining. We reported our results using previously established criteria for scoring PRAME immunohistochemistry7: 0 for no expression, 1+ for 1% to 25% expression, 2+ for 26% to 50% expression, 3+ for 51% to 75% expression, and 4+ for diffuse or 76% to 100% expression. Only strong clonal expression within a population of cells was graded.
Data handling and statistical testing were performed using the R Project for Statistical Computing (https://www.r-project.org/). Significance testing was performed using the Fisher exact test. Plot construction was performed using ggplot2 (https://ggplot2.tidyverse.org/).
Results
Our study cohort included 36 special-site nevi, and the control cohort comprised 25 melanoma in situ (MIS) or invasive melanoma (IM) lesions occurring at special sites. Table 1 provides a breakdown of the study and control cohorts by lesion site. Table 2 details the results of our microscopic examination, describing frequency of various characteristics of special-site nevi stratified by site.
Of the 36 special-site nevi in our cohort, 20 (56%) had no staining (0) for PRAME, 11 (31%) demonstrated 1+ PRAME expression, 3 (8%) demonstrated 2+ PRAME expression, and 2 (6%) demonstrated 3+ PRAME expression. No nevi showed 4+ expression. In the control cohort, 24 of 25 (96%) MIS and IM showed 3+ or 4+ expression, with 21 (84%) demonstrating diffuse/4+ expression. One control case (4%) demonstrated 0 PRAME expression. These data are summarized in Table 3 and Figure 1. There is a significant difference in diffuse (4+) PRAME expression between special-site nevi and MIS/IM occurring at special sites (P=1.039×10-12).
Based on our cohort, a positivity threshold of 3+ for PRAME expression for the diagnosis of melanoma in a special-site lesion would have a sensitivity of 96% and a specificity of 94%, while a positivity threshold of 4+ for PRAME expression would have a sensitivity of 84% and a specificity of 100%. Figures 2 through 4 show photomicrographs of a special-site nevus of the breast, which appropriately does not stain for PRAME; Figures 5 and 6 show an MIS at a special site that appropriately stains for PRAME.
Comment
The distinction between benign and malignant pigmented lesions at special sites presents a fair challenge for pathologists due to the larger degree of leniency for architectural distortion and cytologic atypia in benign lesions at these sites. The presence of architectural distortion or cytologic atypia at the lesion’s edge makes rendering a benign diagnosis especially difficult, and the need for a validated immunohistochemical stain is apparent. In our cohort, strong clonal PRAME expression provided a reliable immunohistochemical marker, allowing for the distinction of malignant lesions from benign nevi at special sites. Diffuse faint PRAME expression was present in several benign nevi within our cohort, and these lesions were considered negative (0) in our analysis.
Given the described test characteristics, we support the implementation of PRAME immunohistochemistry with a positivity threshold of 4+ expression as an ancillary test supporting the diagnosis of IM or MIS in special sites, which would allow clinicians to leverage the high specificity of 4+ PRAME expression to distinguish an IM or MIS from a benign nevus occurring at a special site. We do not recommend the use of 4+ PRAME expression as a screening test for melanoma or MIS among special-site nevi due to its comparatively low sensitivity; however, no one marker is always reliable, and we recommend continued clinicopathologic correlation for all cases.
Although our case series included nevi and MIS/IM from all special sites, we were limited in the number of acrogenital and ear nevi included due to a relative paucity of biopsied benign nevi from these locations at the University of Virginia. Additionally, although the magnitude of the difference in PRAME expression between the study and control groups is sufficient to demonstrate statistical significance, the overall strength of our argument would be increased with a larger study group. We were limited by the number of cases available at our institution, which did not utilize PRAME during the initial diagnosis of the case; including these cases in the study group would have undermined the integrity of our argument because the differentiation of benign vs malignant initially was made using PRAME immunohistochemistry.
Conclusion
Due to their atypical features, special-site nevi can be challenging to assess. In this study, we showed that PRAME expression can be a reliable marker to distinguish benign from malignant lesions. Our results showed that 100% of benign special-site nevi demonstrated 3+ expression or less, with 56% (20/36) demonstrating no expression at all. The presence of diffuse PRAME expression (4+ PRAME staining) appears to be a specific indicator of a malignant lesion, but results should always be interpreted with respect to the patient’s clinical history and the lesion’s histomorphologic features. Further study of a larger sample size would allow refinement of the sensitivity and specificity of diffuse PRAME expression in the determination of malignancy for special-site lesions.
Acknowledgment—The authors thank the pathologistsat the University of Virginia Biorepository and Tissue Research Facility (Charlottesville, Virginia) for their skill and expertise in performing immunohistochemical staining for this study.
The assessment and diagnosis of melanocytic lesions can present a formidable challenge to even a seasoned pathologist, which is especially true when dealing with the subset of nevi occurring at special sites—where baseline variations inherent to particular locations on the body can preclude the use of features routinely used to diagnose malignancy elsewhere. These so-called special-site nevi previously have been described in the literature along with suggested criteria for differentiating malignant lesions from their benign counterparts.1 Locations generally considered to be special sites include the acral skin, anogenital region, breast, ear, and flexural regions.1,2
When evaluating non–special-site melanocytic lesions, general characteristics associated with a malignant diagnosis include confluence or pagetoid spread of melanocytes, nuclear pleomorphism, cytologic atypia, and irregular architecture3; however, these features can be compatible with a benign diagnosis in special-site nevi depending on their extent and the site in question. Although they can be atypical, special-site nevi tend to have the bulk of their architectural distortion and cytologic atypia in the center of the lesion as opposed to the edges.1 If a given lesion is from a special site but lacks this reassuring feature, special care should be taken to rule out malignancy.
Preferentially expressed antigen in melanoma (PRAME) is an antigen first identified in tumor-reactive T-cell populations in patients with malignant melanoma. It is the product of an oncogene that frequently is overexpressed in melanomas, lung squamous cell carcinomas, sarcomas, and acute leukemias.4 It functions as an antagonist of the retinoic acid signaling pathway, which normally serves to induce further cell differentiation, senescence, or apoptosis.5 PRAME inhibits retinoid signaling by forming a complex with both the ligand-bound retinoic acid holoreceptor and the polycomb protein EZH2, which blocks retinoid-dependent gene expression by encouraging chromatin condensation at the RARβ promoter site5; therefore, expressing PRAME allows lesional cells a substantial growth advantage.
PRAME expression has been extensively characterized in non–special-site nevi and has filled the need for a rather specific marker of melanoma.6-10 Although PRAME has been studied in acral nevi,11 the expression pattern in nevi of special sites has yet to be elucidated. Herein, we present a dataset characterizing PRAME expression in these challenging lesions.
Methods
We performed a retrospective case review at the University of Virginia (Charlottesville, Virginia) and collected a panel of 36 special-site nevi that previously were diagnosed as benign by a trained dermatopathologist from January 2020 through December 2022. Special-site nevi were identified using a natural language filter for the following terms: acral, palm, sole, ear, auricular, lip, axilla, armpit, breast, groin, labia, vulva, umbilicus, and penis. This study was approved by the University of Virginia institutional review board.
The original hematoxylin and eosin slides used for primary diagnosis were re-examined to verify the prior diagnosis of benign nevus at a special site. We performed a detailed microscopic examination of all benign nevi in our cohort to determine the frequency of various characteristics at each special site. Sections were prepared from the formalin-fixed and paraffin-embedded tissue blocks and stained with a commercial PRAME antibody (#219650 [Abcam] at a 1:50 dilution) and counterstain. A trained dermatopathologist (S.S.R.) examined the stained sections and recorded the percentage of tumor cells with nuclear PRAME staining. We reported our results using previously established criteria for scoring PRAME immunohistochemistry7: 0 for no expression, 1+ for 1% to 25% expression, 2+ for 26% to 50% expression, 3+ for 51% to 75% expression, and 4+ for diffuse or 76% to 100% expression. Only strong clonal expression within a population of cells was graded.
Data handling and statistical testing were performed using the R Project for Statistical Computing (https://www.r-project.org/). Significance testing was performed using the Fisher exact test. Plot construction was performed using ggplot2 (https://ggplot2.tidyverse.org/).
Results
Our study cohort included 36 special-site nevi, and the control cohort comprised 25 melanoma in situ (MIS) or invasive melanoma (IM) lesions occurring at special sites. Table 1 provides a breakdown of the study and control cohorts by lesion site. Table 2 details the results of our microscopic examination, describing frequency of various characteristics of special-site nevi stratified by site.
Of the 36 special-site nevi in our cohort, 20 (56%) had no staining (0) for PRAME, 11 (31%) demonstrated 1+ PRAME expression, 3 (8%) demonstrated 2+ PRAME expression, and 2 (6%) demonstrated 3+ PRAME expression. No nevi showed 4+ expression. In the control cohort, 24 of 25 (96%) MIS and IM showed 3+ or 4+ expression, with 21 (84%) demonstrating diffuse/4+ expression. One control case (4%) demonstrated 0 PRAME expression. These data are summarized in Table 3 and Figure 1. There is a significant difference in diffuse (4+) PRAME expression between special-site nevi and MIS/IM occurring at special sites (P=1.039×10-12).
Based on our cohort, a positivity threshold of 3+ for PRAME expression for the diagnosis of melanoma in a special-site lesion would have a sensitivity of 96% and a specificity of 94%, while a positivity threshold of 4+ for PRAME expression would have a sensitivity of 84% and a specificity of 100%. Figures 2 through 4 show photomicrographs of a special-site nevus of the breast, which appropriately does not stain for PRAME; Figures 5 and 6 show an MIS at a special site that appropriately stains for PRAME.
Comment
The distinction between benign and malignant pigmented lesions at special sites presents a fair challenge for pathologists due to the larger degree of leniency for architectural distortion and cytologic atypia in benign lesions at these sites. The presence of architectural distortion or cytologic atypia at the lesion’s edge makes rendering a benign diagnosis especially difficult, and the need for a validated immunohistochemical stain is apparent. In our cohort, strong clonal PRAME expression provided a reliable immunohistochemical marker, allowing for the distinction of malignant lesions from benign nevi at special sites. Diffuse faint PRAME expression was present in several benign nevi within our cohort, and these lesions were considered negative (0) in our analysis.
Given the described test characteristics, we support the implementation of PRAME immunohistochemistry with a positivity threshold of 4+ expression as an ancillary test supporting the diagnosis of IM or MIS in special sites, which would allow clinicians to leverage the high specificity of 4+ PRAME expression to distinguish an IM or MIS from a benign nevus occurring at a special site. We do not recommend the use of 4+ PRAME expression as a screening test for melanoma or MIS among special-site nevi due to its comparatively low sensitivity; however, no one marker is always reliable, and we recommend continued clinicopathologic correlation for all cases.
Although our case series included nevi and MIS/IM from all special sites, we were limited in the number of acrogenital and ear nevi included due to a relative paucity of biopsied benign nevi from these locations at the University of Virginia. Additionally, although the magnitude of the difference in PRAME expression between the study and control groups is sufficient to demonstrate statistical significance, the overall strength of our argument would be increased with a larger study group. We were limited by the number of cases available at our institution, which did not utilize PRAME during the initial diagnosis of the case; including these cases in the study group would have undermined the integrity of our argument because the differentiation of benign vs malignant initially was made using PRAME immunohistochemistry.
Conclusion
Due to their atypical features, special-site nevi can be challenging to assess. In this study, we showed that PRAME expression can be a reliable marker to distinguish benign from malignant lesions. Our results showed that 100% of benign special-site nevi demonstrated 3+ expression or less, with 56% (20/36) demonstrating no expression at all. The presence of diffuse PRAME expression (4+ PRAME staining) appears to be a specific indicator of a malignant lesion, but results should always be interpreted with respect to the patient’s clinical history and the lesion’s histomorphologic features. Further study of a larger sample size would allow refinement of the sensitivity and specificity of diffuse PRAME expression in the determination of malignancy for special-site lesions.
Acknowledgment—The authors thank the pathologistsat the University of Virginia Biorepository and Tissue Research Facility (Charlottesville, Virginia) for their skill and expertise in performing immunohistochemical staining for this study.
- VandenBoom T, Gerami P. Melanocytic nevi of special sites. In: Pathology of Melanocytic Tumors. Elsevier; 2019:90-100. doi:10.1016/B978-0-323-37457-6.00007-9
- Hosler GA, Moresi JM, Barrett TL. Nevi with site-related atypia: a review of melanocytic nevi with atypical histologic features based on anatomic site. J Cutan Pathol. 2008;35:889-898. doi:10.1111/j.1600-0560.2008.01041.x.
- Brenn T. Melanocytic lesions—staying out of trouble. Ann Diagn Pathol. 2018;37:91-102. doi:10.1016/j.anndiagpath.2018.09.010
- Ikeda H, Lethé B, Lehmann F, et al. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity. 1997;6:199-208. doi:10.1016/s1074-7613(00)80426-4
- Epping MT, Wang L, Edel MJ, et al. The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signaling. Cell. 2005;122:835-847. doi:10.1016/j.cell.2005.07.003
- Alomari AK, Tharp AW, Umphress B, et al. The utility of PRAME immunohistochemistry in the evaluation of challenging melanocytic tumors. J Cutan Pathol. 2021;48:1115-1123. doi:10.1111/cup.14000
- Lezcano C, Jungbluth AA, Nehal KS, et al. PRAME expression in melanocytic tumors. Am J Surg Pathol. 2018;42:1456-1465. doi:10.1097/PAS.0000000000001134
- Gill P, Prieto VG, Austin MT, et al. Diagnostic utility of PRAME in distinguishing proliferative nodules from melanoma in giant congenital melanocytic nevi. J Cutan Pathol. 2021;48:1410-1415. doi:10.1111/cup.14091
- Googe PB, Flanigan KL, Miedema JR. Preferentially expressed antigen in melanoma immunostaining in a series of melanocytic neoplasms. Am J Dermatopathol. 2021;43):794-800. doi:10.1097/DAD.0000000000001885
- Raghavan SS, Wang JY, Kwok S, et al. PRAME expression in melanocytic proliferations with intermediate histopathologic or spitzoid features. J Cutan Pathol. 2020;47:1123-1131. doi:10.1111/cup.13818
- McBride JD, McAfee JL, Piliang M, et al. Preferentially expressed antigen in melanoma and p16 expression in acral melanocytic neoplasms. J Cutan Pathol. 2022;49:220-230. doi:10.1111/cup.14130
- VandenBoom T, Gerami P. Melanocytic nevi of special sites. In: Pathology of Melanocytic Tumors. Elsevier; 2019:90-100. doi:10.1016/B978-0-323-37457-6.00007-9
- Hosler GA, Moresi JM, Barrett TL. Nevi with site-related atypia: a review of melanocytic nevi with atypical histologic features based on anatomic site. J Cutan Pathol. 2008;35:889-898. doi:10.1111/j.1600-0560.2008.01041.x.
- Brenn T. Melanocytic lesions—staying out of trouble. Ann Diagn Pathol. 2018;37:91-102. doi:10.1016/j.anndiagpath.2018.09.010
- Ikeda H, Lethé B, Lehmann F, et al. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity. 1997;6:199-208. doi:10.1016/s1074-7613(00)80426-4
- Epping MT, Wang L, Edel MJ, et al. The human tumor antigen PRAME is a dominant repressor of retinoic acid receptor signaling. Cell. 2005;122:835-847. doi:10.1016/j.cell.2005.07.003
- Alomari AK, Tharp AW, Umphress B, et al. The utility of PRAME immunohistochemistry in the evaluation of challenging melanocytic tumors. J Cutan Pathol. 2021;48:1115-1123. doi:10.1111/cup.14000
- Lezcano C, Jungbluth AA, Nehal KS, et al. PRAME expression in melanocytic tumors. Am J Surg Pathol. 2018;42:1456-1465. doi:10.1097/PAS.0000000000001134
- Gill P, Prieto VG, Austin MT, et al. Diagnostic utility of PRAME in distinguishing proliferative nodules from melanoma in giant congenital melanocytic nevi. J Cutan Pathol. 2021;48:1410-1415. doi:10.1111/cup.14091
- Googe PB, Flanigan KL, Miedema JR. Preferentially expressed antigen in melanoma immunostaining in a series of melanocytic neoplasms. Am J Dermatopathol. 2021;43):794-800. doi:10.1097/DAD.0000000000001885
- Raghavan SS, Wang JY, Kwok S, et al. PRAME expression in melanocytic proliferations with intermediate histopathologic or spitzoid features. J Cutan Pathol. 2020;47:1123-1131. doi:10.1111/cup.13818
- McBride JD, McAfee JL, Piliang M, et al. Preferentially expressed antigen in melanoma and p16 expression in acral melanocytic neoplasms. J Cutan Pathol. 2022;49:220-230. doi:10.1111/cup.14130
Practice Points
- Special-site nevi are benign melanocytic proliferations at special anatomic sites. Although cytologic atypia and architectural distortion may be present, they are centrally located and should not be present at the borders of the lesion.
- Strong expression of the preferentially expressed antigen in melanoma (PRAME) via immunohistochemistry provides a reliable indicator for benignity in differentiating a special-site nevus from a malignant melanoma occurring at a special site.
