User login
Preventing a first episode of esophageal variceal hemorrhage
Variceal hemorrhage is a medical emergency in which up to 20% of patients die.1 Even if the patient survives an initial episode of variceal bleeding, the probability of another episode is high: the rebleeding rate without treatment is 70% within 1 year. The mortality rate with rebleeding is 33%.
With such overwhelming consequences, the best strategy in any patient with cirrhosis and known varices is to try to prevent the first episode of bleeding.
WHO IS AT RISK?
Esophageal varices are present in 30% of patients with compensated cirrhosis and in up to 60% of those with decompensated cirrhosis (ie, with evidence of ascites or encephalopathy).2
The risk of variceal hemorrhage is related to three factors:
- The size of the varices. Varices 5 mm in diameter or smaller have a 7% risk of bleeding in 2 years, while those larger than 5 mm have a 30% risk of bleeding within 2 years.3
- The appearance of the varices. Morphologic features of varices, including red wale signs (red streaks of the mucosa overlying the varix), have been correlated with an increased risk of hemorrhage.
- The severity of liver dysfunction, as assessed by the Child-Pugh classification—an index of liver dysfunction based on serum albumin concentration, bilirubin level, prothrombin time, and the presence of ascites and encephalopathy. A high Child-Pugh score (ie, class B or C), representing decompensated cirrhosis, is associated with an increased risk of bleeding.
HOW VARICES DEVELOP: PORTAL HYPERTENSION
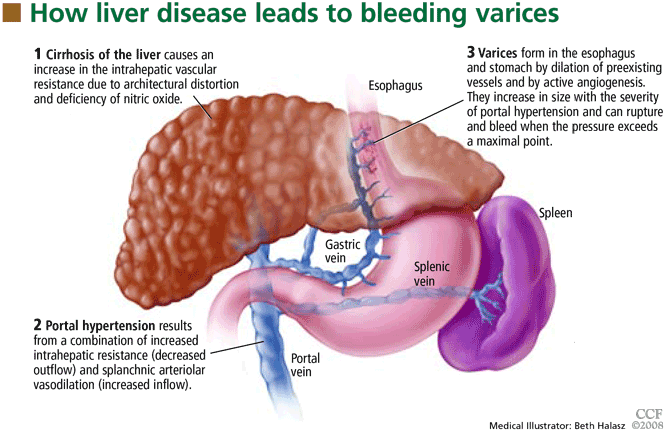
Esophageal varices form as a result of increased portal pressure, the product of increased portal venous inflow and resistance to outflow from the portal venous system. Portal hypertension is a major complication of chronic liver disease. In cirrhosis, architectural distortion of the liver causes an increase in the intrahepatic vascular resistance.
Portal venous inflow depends on mesenteric arteriolar tone, increasing when tone decreases. In cirrhotic patients, the increase in portal pressure results from a combination of increased portal blood flow secondary to splanchnic arteriolar vasodilation and elevated resistance to outflow through distorted hepatic sinusoids.
The potent vasodilator nitric oxide (NO) plays an important role in portal hypertension. In patients with cirrhosis, NO bioavailability is decreased in the intrahepatic circulation due to defects in the posttranslational regulation of endothelial NO synthase.4 This deficiency of NO, along with mechanical factors in the sinusoids, contributes to the increase in intrahepatic resistance. In the systemic and splanchnic circulation, NO bioavailability is increased due to upregulation and posttranslational regulation of endothelial NO synthase, thereby increasing splanchnic vasodilatation and leading to increased portal venous inflow.5 This results in a marked increase in cardiac output and so-called hyperdynamic circulation.
Portal hypertension results in the development of collateral circulation, including venous channels in the esophagus and stomach, by the dilation of preexisting vessels and active angiogenesis. Esophagogastric varices increase in size with the severity of portal hypertension and can rupture when the tension in their walls exceeds a maximal point.
HEPATIC VEIN PRESSURE GRADIENT: A PROXY FOR PORTAL PRESSURE
Ideally, the portal venous pressure should be directly measured. However, since direct measurement is invasive and impractical, the hepatic vein pressure gradient (HVPG) can be measured instead and correlates well with the portal pressure.6
The normal HVPG is 5 mm Hg or less; anything above this value denotes portal hypertension. However, studies have shown that varices may develop but do not bleed if the HVPG is less than 12 mm Hg.8
TWO WAYS TO PREVENT BLEEDING
Bleeding can be prevented either by reducing the portal venous pressure or by obliterating the varices. Portal pressure can be reduced by placing a portosystemic shunt either surgically or percutaneously with radiographic guidance or by giving drugs such as nonselective beta-blockers, nitrates, or a combination of these drugs. Variceal obliteration is typically done by endoscopic methods with either injection of a sclerosant or band ligation.
NONSELECTIVE BETA-BLOCKERS: THE MAINSTAY OF TREATMENT
Nonselective beta-blockers, the most commonly used drugs for preventing first esophageal variceal bleeding, decrease portal pressure by blocking both beta-1 and beta-2 adrenergic receptors.9 Beta-1 blockade decreases portal flow by decreasing the heart rate and cardiac output, while blockade of beta-2 receptors results in unopposed alpha-adrenergic-mediated vasoconstriction.
Selective beta-blockers do not appear to be as useful for primary prophylaxis. More than 2 decades ago, metoprolol (Toprol, Lopressor), a beta-1 selective antagonist, was compared with propranolol (Inderal), a nons-elective agent, in patients with cirrhosis and portal hypertension.10 Although both drugs significantly reduced the heart rate and cardiac output, only those taking propranolol showed a marked fall in portal pressure (mean decrease of 6.8 mm Hg vs 3.8 mm Hg with metoprolol) and a significant reduction in hepatic blood flow. The differences were thought to be related to beta-2 blockade of vasodilator receptors in the splanchnic circulation, which occurs only with nonselective beta-blockers such as propranolol.
The two nonselective beta-blockers most often used to prevent variceal bleeding are nadolol (Corgard) and propranolol. Both have been extensively studied in preventing a first variceal hemorrhage.
Effectiveness of beta-blockers
D’Amico et al11 performed a meta-analysis in 1995, examining nine trials (996 patients total) of the effectiveness of beta-blockers in preventing a first variceal hemorrhage. Seven trials found that bleeding risk was reduced with beta-blockers (significantly in four), one trial found that risk was unchanged, and one trial found that risk was increased—an outlier due to a small sample size. The meta-analysis showed a significant bleeding reduction with the use of a beta-blocker, either including the outlier trial (pooled odds ratio 0.54; 95% confidence interval 0.39–0.74) or excluding it (pooled odds ratio 0.48; 95% confidence interval 0.35–0.66).
Mortality rates were also reduced in seven trials, but the reduction was statistically significant in only one. However, in the pooled estimate, the mortality risk reduction approached statistical significance (pooled odds ratio 0.75; 95% confidence interval 0.57–1.06).
Ideo et al12 gave either nadolol or placebo to 79 patients with cirrhosis and large esophageal varices that had never bled. Nadolol was found to protect against a first variceal hemorrhage: at 2-year follow-up, only 1 of the 30 patients allocated to nadolol had had bleeding, vs 11 of the 49 patients in the placebo group.
Merkel et al13 found that the risk of variceal bleeding was lower in patients who started treatment with beta-blockers when their varices were small (12% at 5 years) than in those who started treatment after a diagnosis of large esophageal varices (22% at 5 years). They concluded that nadolol helps prevent small varices from growing into larger ones.
Response to beta-blockers is not uniform
Although beta-blockers decrease the portal pressure in many cirrhotic patients, the response is not uniform. In a study of 60 cirrhotic patients,14 40% showed no reduction or even a slight increase in HVPG with propranolol. Most patients showed a significant reduction in heart rate (17.5% ± 10%) after receiving 40 mg of propranolol. In the patients whose HVPG did not decrease by at least 10% with 40 mg of propranolol, increasing the dose caused a decrease in HVPG without a further decrease in heart rate. This suggests that 40 mg of propranolol successfully produced beta-1 blockade but that a higher dose was required for effective beta-2 blockade.
Failure to respond in certain patients may be due to a concurrent rise in collateral or hepatic sinusoidal resistance, or both. This was confirmed in a study in portal-hypertensive rats treated with propranolol.15 The reduction in portal blood flow expected was accompanied by a disproportionately small reduction in portal pressure, which was thought to be due to a rise in portal and collateral vascular resistance.
How to tell if beta-blocker treatment is ‘working’
An HVPG ≤ 12 mm Hg? Studies have shown that the most important predictor of efficacy of prophylaxis for variceal bleeding is a decrease in the HVPG to 12 mm Hg or less or a decrease in the initial HVPG of more than 20%.9 Although measuring the HVPG is invasive, expensive, and not routinely done in clinical practice, several studies have investigated the role of measuring hemodynamic response to medication.
Merkel et al16 measured the HVPG in 49 cirrhotic patients with previously nonbleeding varices before starting therapy with beta-blockers with or without nitrates and after 1 to 3 months of treatment. They followed the patients for up to 5 years. The mean HVPG value at baseline was 18.8 mm Hg. At 3 years of follow-up, 7% of those who had responded well to therapy (defined as achieving an HVPG less than 13 mm Hg or a decrease of more than 20%) had experienced a bleeding episode, which was significantly less than the rate (41%) in those who did not meet those hemodynamic end points. No patient reaching an HVPG of 12 mm Hg or less during treatment had variceal bleeding during follow-up.
Groszmann et al17 also prospectively measured the HVPG in patients with cirrhosis and varices, but their patients received either propranolol or placebo. Variceal hemorrhage occurred in 13 patients (11 of 51 in the placebo group and 2 of 51 in the propranolol group), all of whom had an HVPG greater than 12 mm Hg. Again, none of the patients whose HVPG was decreased to 12 mm Hg or less bled from esophageal varices.
Unfortunately, routine HVPG measurement to guide primary prophylaxis is an expensive strategy. Data suggest that measuring the HVPG is cost-effective only when the cost of measuring the HVPG is very low, the risk of variceal bleeding is very high, or the patient is expected to survive at least 3 to 5 years.18
A heart rate of 55 to 60? An alternative to HVPG measurement to monitor the effectiveness of beta-blocker therapy is to follow the heart rate. A 25% reduction from baseline or a heart rate of 55 to 60 beats per minute is the standard goal19,20; yet, at least 40% of patients treated with enough propranolol to decrease the heart rate by 25% do not respond with significant HVPG reductions.14,21
So, although beta-blockade is effective peripherally, it may not reduce HVPG to less than 12 mm Hg or 20% from baseline, and direct HVPG measurement is still the gold standard.
Treatment should be lifelong
Once a patient is started on a beta-blocker to prevent variceal hemorrhage, the treatment should be lifelong.
In 2001, a group of patients (most of them in Child-Pugh class A or B) completing a prospective randomized controlled trial of propranolol for primary prevention of variceal hemorrhage were tapered off propranolol or placebo.22 Of the 49 patients, 9 experienced variceal hemorrhage (6 of 25 former propranolol recipients and 3 of 24 former placebo recipients), and 17 patients died (12 former propranolol and 5 former placebo recipients), suggesting that treatment should be maintained for life.
Therefore, when beta-blocker therapy is discontinued, the risk of variceal hemorrhage returns to what would be expected in an untreated population.
Beta-blockers may not prevent varices
Although many trials have shown that beta-blockers are effective as prophylaxis against a first variceal hemorrhage, there is no evidence that these drugs prevent varices from forming in cirrhotic patients.
Groszmann et al23 treated more than 200 patients who had biopsy-proven cirrhosis and portal hypertension (HVPG > 6 mm Hg) but no varices with timolol (Blocadren), a nonselective beta-blocker, or placebo. At a median follow-up of about 55 months, the groups did not differ significantly in the incidence of primary events (development of varices or variceal hemorrhage) or treatment failures (transplantation or death). Varices developed less frequently among patients with a baseline HVPG of less than 10 mm Hg and among those whose HVPG had decreased by more than 10% at 1 year. In patients whose HVPG increased by more than 10%, varices developed more frequently.
Contraindications, side effects
The major drawbacks to therapy with beta-blockers are their contraindications and side effects.
Contraindications include chronic obstructive lung disease, psychosis, atrioventricular heart blocks, and aortic-valve disease.
Side effects are reported in 15% of patients but severe events are rare.24 Still, an estimated 10% to 20% of patients discontinue treatment because they cannot tolerate it.25 The more common complaints include fatigue, shortness of breath, sexual dysfunction, and sleep disorders.
Dosage
No specific starting dose of beta-blockers is agreed upon, but nadolol 20 to 40 mg once daily or long-acting propranolol 60 mg once daily can be used as initial therapy.25 Once-daily dosing increases the likelihood of compliance.
Since portal pressure progressively declines from 12 noon to 7 PM and then increases throughout the night and back to baseline by 9 AM,26 we recommend that the medication be taken in the evening to counteract increases in portal pressure that occur in the middle of the night.
ENDOSCOPIC VARICEAL LIGATION
Endoscopic variceal ligation has been investigated extensively for use as prophylaxis against first variceal hemorrhage. The procedure involves placing a rubber band around a varix aspirated into a cylinder on the tip of an endoscope.
Effectiveness of ligation
Lay et al,27 in a prospective, randomized trial in 126 cirrhotic patients endoscopically judged to be at high risk of hemorrhage, found that ligation significantly reduced the 2-year cumulative bleeding rate (19% with ligation vs 60% in an untreated control group) and the overall mortality rate (28% vs 58%). The lower risk of bleeding in the ligation group was attributed to a rapid reduction of variceal size; 60% of those in the ligation group had complete eradication of varices and 38% had varices reduced in size.
Imperiale and Chalasani28 performed a meta-analysis in 2001 that included 601 patients in five trials comparing prophylactic ligation with untreated controls and 283 patients in four trials comparing ligation with beta-blocker therapy. Compared with no treatment, ligation reduced the risk of first variceal hemorrhage, bleeding-related death, and death from any cause. Compared with propranolol, ligation reduced the risk of first-time bleeding but had no effect on the death rate.
Schepke et al,29 in a randomized controlled multicenter trial in 152 cirrhotic patients with two or more esophageal varices, found that neither bleeding incidence nor death rate differed significantly between ligation and propranolol.
Lui et al30 followed 172 cirrhotic patients with grade II or III esophageal varices for 6 years and found that ligation was equivalent to propranolol. However, many patients reported side effects with propranolol, and 30% of patients withdrew from propranolol treatment, making ligation a more attractive option.
Khuroo et al31 performed a meta-analysis of eight randomized controlled trials including 596 patients and found that ligation significantly reduced the rates of first gastrointestinal hemorrhage by 31% and of first variceal hemorrhage by 43%. In subgroup analysis, ligation had a significant advantage compared with beta-blockers in trials with patients with a high bleeding risk, ie, trials in which more than 30% of patients were in Child-Pugh class C and more than 50% of the patients had large varices.
Jutabha et al32 performed a multicenter, prospective trial (published in 2005) in 62 patients with high-risk esophageal varices randomized to propranolol or banding. The trial was ended early after an interim analysis showed that the failure rate of propranolol was significantly higher than that of banding (6/31 vs 0/31, P = .0098). Esophageal variceal hemorrhage occurred in 4 (12.9%) of the patients in the propranolol group compared with 0 in the ligation group. Similarly, 4 patients in the propranolol group died, compared with 0 in the ligation group. All the patients in this trial were liver transplant candidates and therefore all had severe liver disease.
In another trial favoring variceal banding over beta-blockers, Psilopoulos et al33 in 2005 followed 60 patients with cirrhosis and esophageal varices with no history of bleeding. Thirty percent of the patients in the propranolol group developed variceal bleeding compared with 6.7% in the ligation group (P = .043).
Lay et al34 followed 100 cirrhotic patients for 2 years and found comparable cumulative bleeding rates with ligation vs propranolol (18% vs 16%, respectively) and also comparable rates of death (28% vs 24%, respectively).
Sarin et al35 investigated the role of propranolol in addition to ligation in the prevention of first hemorrhage in 144 patients. Adding propranolol did not further decrease the incidence of initial bleeding (7% in the combination group vs 11% in the ligation-only group). Survival rates were similar at 20 months: 92% in the combination group vs 85% in the ligation-only group. However, the rate of variceal recurrence was lower with combination therapy: 6% in the combination group vs 15% with ligation alone.
Does esophageal variceal ligation increase gastric varices?
A less researched topic is whether variceal ligation results in gastric hemodynamic changes that increase the size of fundal varices and worsen portal hypertensive gastropathy.
Yuksel et al36 found that 37 of 85 patients had fundal varices before they underwent ligation of esophageal varices, increasing to 46 after the procedure, a statistically significant increment. The severity of portal hypertensive gastropathy also increased.
Further research is required regarding the long-term consequences of these findings.
ANGIOTENSIN II RECEPTOR ANTAGONISTS: ROLE UNKNOWN
Angiotensin II increases portal pressure, and angiotensin II levels are elevated in patients with cirrhosis, suggesting that this hormone plays a role in the pathogenesis of portal hypertension.
Losartan (Cozaar), an angiotensin II receptor antagonist, was found to decrease the HVPG significantly in patients with severe and moderate portal hypertension in a pilot study37 in 1999. However, in two subsequent studies,38,39 losartan only moderately reduced the HVPG and caused hypotension and a reduction in the glomerular filtration rate. The role of angiotensin II receptor blockers in primary prevention of variceal bleeding is still unknown.
SURGICAL PORTAL DECOMPRESSION HAS BEEN ABANDONED
The first method investigated to prevent variceal bleeding was surgical portal decompression.
A meta-analysis of four randomized controlled trials in 302 patients with varices of all sizes compared portocaval shunt surgery and medical therapy.11 Although shunt surgery was very effective in preventing variceal bleeding, the risk of chronic or recurrent encephalopathy was significantly increased (odds ratio 2.0), as was the risk of death (odds ratio 1.6).
These poor results, combined with advances in endoscopic procedures, led to the abandonment of surgical shunting for primary prophylaxis.
TIPS PROCEDURE: NO ROLE AT PRESENT
The transjugular intrahepatic portosystemic shunt (TIPS) procedure is used to treat the main consequences of portal hypertension, including ascites and variceal hemorrhage. The procedure entails accessing the hepatic vein via the right jugular vein and placing a stent to the portal vein, forming a low-resistance channel and allowing blood to return to the systemic circulation.
TIPS placement increases the risk of encephalopathy; liver failure is a rare complication, and procedural complications (ie, shunt dysfunction) also occur. Trials comparing the TIPS procedure with other forms of therapy to prevent first variceal hemorrhages have not been performed.40 Research to improve the outcome of the TIPS procedure is ongoing, but currently this procedure has no role in primary prevention of variceal bleeding.
ENDOSCOPIC SCLEROTHERAPY MAY INCREASE THE RISK OF DEATH
Numerous clinical trials evaluated sclerotherapy as prophylaxis against a first esophageal variceal hemorrhage. The procedure involves injecting a sclerosant in and around varices.
In a large Veterans Administration study,41 sclerotherapy was compared with sham treatment in 281 men with alcoholic liver disease who had documented varices but no history of bleeding. The trial was terminated after 22.5 months because the rate of all-cause mortality was significantly higher in the sclerotherapy group (32.5%) than in the sham therapy group (17.4%). The higher death rate did not persist after the treatment was discontinued, and it was speculated that, although sclerotherapy had reduced new episodes of variceal hemorrhage, the procedure might have caused bleeding from esophageal ulcers, leading to an increased mortality rate in that group.
The PROVA Study Group from Norway and Denmark found similar results when 286 cirrhotic patients were randomized to receive sclerotherapy, propranolol, combination sclerotherapy and propranolol, or no treatment to prevent a first variceal hemorrhage.42 The incidence of variceal bleeding was almost identical in the four groups, but the mortality rate with variceal bleeding was 2.75 times higher in the sclerotherapy groups than in the other groups (P = .002). It was speculated that repeated sclerotherapy sessions might be poorly tolerated by patients in Child-Pugh classes B and C and might have contributed to the precipitation of liver failure and other common complications of cirrhosis.
A meta-analysis by D’Amico et al11 evaluated 19 trials (1,630 patients) comparing sclerotherapy with nonactive treatment. Sclerotherapy tended to be favorable in trials with a high bleeding rate in the control patients and unfavorable in trials with a low bleeding rate. The benefit seen in patients at high risk is consistent with the efficacy of sclerotherapy for preventing rebleeding, whereas the harmful effect in the low-risk patients points towards side effects and complications exceeding the potential benefits.
In general, currently available evidence suggests that the benefits of prophylactic sclerotherapy are marginal, and therefore sclerotherapy is not recommended as primary prophylaxis for variceal hemorrhage.
NITRATES: NO LONGER USED AS MONOTHERAPY
Unlike vasoconstrictors, which decrease portal pressure by decreasing blood flow, vasodilators reduce hepatic pressure by decreasing intrahepatic and portocollateral vascular resistance.43 In addition, larger doses directly affect the arterial circulation, lowering systemic and therefore splanchnic perfusion pressure.44 Unfortunately, the systemic vasodilatory effects of nitrates exacerbate the hyperdynamic state that is characteristic of cirrhosis, thereby limiting their use and tolerability in many patients.
A trial comparing propranolol vs isosorbide mononitrate initially found that the groups did not differ significantly with regard to bleeding rates and 2-year survival rates,45 but a 6-year follow-up found the likelihood of death greater in patients older than 50 years in the nitrate group.46 In an additional study comparing isosorbide mononitrate vs placebo in patients with contraindications to or intolerance of beta-blockers, no difference in the relative risk of first variceal hemorrhage was found between the two groups.47 Therefore, nitrates are no longer used as monotherapy to prevent variceal bleeding.
Combination therapy with beta-blockers plus nitrates is controversial. In a trial in 1996, Merkel et al48 found the cumulative risk of variceal bleeding was 18% at 40 months with nadolol alone vs 7.5% with nadolol plus isosorbide mononitrate. However, in a later trial, Garcia-Pagán et al49 found no significant advantage to combination therapy. The incidence of variceal bleeding at 1 year was 8.3% in the group receiving propranolol plus placebo and 5% in the group receiving propranolol plus isosorbide mononitrate; at 2 years, the rates were 10.6% vs 12.5%.
RECOMMENDATIONS FOR SCREENING AND PROPHYLAXIS
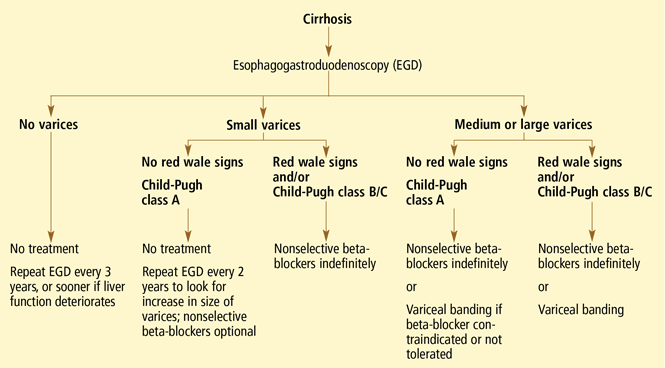
- All patients with cirrhosis should be screened for varices at the time of diagnosis.
- The size of the varices, including small (≤ 5 mm) and large (> 5 mm), and the presence of red wale marks on the varices should be recorded.
- Patients who have no varices on screening endoscopy should be rescreened every 3 years if their liver function is stable or every year if their liver function deteriorates. (Varices grow at a rate proportional to the severity of the liver disease.)
- Patients with portal hypertension but without varices do not need treatment with nonselective beta-blockers. Endoscopy should be performed at the intervals suggested above.
- Those who are found to have small varices on screening endoscopy but who have well-compensated liver disease (Child-Pugh class A) and no red wale marks should be rescreened every other year because the development of large varices is greater in patients with small varices on initial endoscopy than in patients with no varices. Emerging data support the use of beta-blockers to prevent varices from increasing in size.
- Patients who have small varices with red wale signs or who are in Child-Pugh class B or C have an increased risk of bleeding and should be treated with beta-blockers. If beta-blockers are not used, endoscopy should be done every year to look for an increase in variceal size.
- Patients who have large varices without red wale signs or who are in Child-Pugh class B or C should be treated with nonselective beta-blockers. The dose should be adjusted to achieve maximal tolerable decrease in heart rate to a minimum of 55 beats per minute, and treatment should be continued indefinitely.
- Endoscopic variceal ligation is an acceptable alternative to beta-blocker treatment as first-line therapy in those who cannot tolerate beta-blockers or who have contraindications to their use, or in those who have red wale marking or who are in Child-Pugh class B or C.
- D’Amico G, De Franchis R Cooperative Study Group. Upper digestive bleeding in cirrhosis. Post-therapeutic outcome and prognostic indicators. Hepatology 2003; 38:599–612.
- D’Amico G, Luca A. Natural history. Clinicalhaemodynamic correlations. Prediction of the risk of bleeding. Baillieres Clin Gastroenterol 1997; 11:243–256.
- Kamath PS. Esophageal variceal bleeding: primary prophylaxis. Clin Gastroenterol Hepatol 2005; 3:90–93.
- Shah V, Toruner M, Haddad F, et al. Impaired endothelial nitric oxide synthase activity associated with enhanced caveolin binding in experimental cirrhosis in the rat. Gastroenterology 1999; 117:1222–1228.
- Morales-Ruiz M, Jimenez W, Perez-Sala D, et al. Increased nitric oxide synthase expression in arterial vessels of cirrhotic rats with ascites. Hepatology 1996; 24:1481–1486.
- De Franchis R, Dell’Era A, Iannuzzi F. Diagnosis and treatment of portal hypertension. Dig Liver Dis 2004; 36:787–798.
- Groszmann RJ, Wongcharatrawee S. The hepatic venous pressure gradient: anything worth doing should be done right. Hepatology 2004; 39:280–282.
- Garcia-Tsao G, Groszmann RJ, Fisher RL, Conn HO, Atterbury CE, Glickman M. Portal pressure, presence of gastroesophageal varices and variceal bleeding. Hepatology 1985; 5:419–424.
- Bosch J, Masti R, Kravetz D, et al. Effects of propranolol on azygos venous blood flow and hepatic and systemic hemodynamics in cirrhosis. Hepatology 1984; 4:1200–1205.
- Westaby D, Bihari DJ, Gimson AE, Crossley IR, Williams R. Selective and non-selective beta receptor blockade in the reduction of portal pressure in patients with cirrhosis and portal hypertension. Gut 1984; 25:121–124.
- D’Amico G, Pagliaro L, Bosch J. The treatment of portal hypertension: a meta-analytic review. Hepatology 1995; 22:332–354.
- Ideo G, Bellati G, Fesce E, Grimoldi D. Nadolol can prevent the first gastrointestinal bleeding in cirrhotics: a prospective, randomized study. Hepatology 1988; 8:6–9.
- Merkel C, Marin R, Angeli P, et al. Gruppo Triveneto per l’Ipertensione Portale. A placebo-controlled clinical trial of nadolol in the prophylaxis of growth of small esophageal varices in cirrhosis. Gastroenterology 2004; 127:476–484.
- Garcia-Tsao G, Grace ND, Groszmann RJ, et al. Short-term effects of propranolol on portal venous pressure. Hepatology 1986; 6:101–106.
- Kroeger RJ, Groszmann RJ. Increased portal venous resistance hinders portal pressure reduction during the administration of beta-adrenergic blocking agents in a portal hypertensive model. Hepatology 1985; 5:97–101.
- Merkel C, Bolognesi M, Sacerdoti D, et al. The hemodynamic response to medical treatment of portal hypertension as a predictor of clinical effectiveness in the primary prophylaxis of variceal bleeding in cirrhosis. Hepatology 2000; 32:930–934.
- Groszmann RJ, Bosch J, Grace ND, et al. Hemodynamic events in a prospective randomized trial of propranolol versus placebo in the prevention of a first variceal hemorrhage. Gastroenterology 1990; 99:1401–1407.
- Hicken BL, Sharara AI, Abrams GA, Eloubeidi M, Fallon MB, Arguedas MR. Hepatic venous pressure gradient measurements to assess response to primary prophylaxis in patients with cirrhosis: a decision analytical study. Aliment Pharmacol Ther 2003; 17:145–153.
- Grace ND. Diagnosis and treatment of gastrointestinal bleeding secondary to portal hypertension. American College of Gastroenterology Practice Parameters Committee. Am J Gastroenterol 1997; 92:1081–1091.
- Grace ND, Groszmann RJ, Garcia-Tsao G, et al. Portal hypertension and variceal bleeding: an AASLD single topic symposium. Hepatology 1998; 28:868–880.
- Feu F, Garcia-Pagán JC, Bosch J, et al. Relation between portal pressure response to pharmacotherapy and risk of recurrent variceal haemorrhage in patients with cirrhosis. Lancet 1995; 346:1056–1059.
- Abraczinskas DR, Ookubo R, Grace ND, et al. Propranolol for the prevention of first esophageal variceal hemorrhage: a lifetime commitment? Hepatology 2001; 34:1096–1102.
- Groszmann RJ, Garcia-Tsao G, Bosch J, et al Portal Hypertension Collaborative Group. Beta-blockers to prevent gastroesophageal varices in patients with cirrhosis. N Engl J Med 2005; 353:2254–2261.
- Garcia-Pagán JC, Bosch J. Pharmacological prevention of variceal bleeding. New developments. Baillier Clin Gastroenterol 1997; 11:271–287.
- Talwalkar JA, Kamath PS. An evidence-based medicine approach to beta-blocker therapy in patients with cirrhosis. Am J Med 2004; 116:759–766.
- Garcia-Pagan JC, Feu F, Castells A, et al. Circadian variations of portal pressure and variceal hemorrhage in patients with cirrhosis. Hepatology 1994; 19:595–601.
- Lay CS, Tsai YT, Teg CY, et al. Endoscopic variceal ligation in prophylaxis of first variceal bleeding in cirrhotic patients with high-risk esophageal varices. Hepatology 1997; 25:1346–1350.
- Imperiale TF, Chalasani N. A meta-analysis of endoscopic variceal ligation for primary prophylaxis of esophageal variceal bleeding. Hepatology 2001; 33:802–807.
- Schepke M, Kleber G, Nurnberg D, et al. Ligation versus propranolol for the primary prophylaxis of variceal bleeding in cirrhosis. Hepatology 2004; 40:65–72.
- Lui HF, Stanley AJ, Forrest EH, et al. Primary prophylaxis of variceal hemorrhage: a randomized controlled trial comparing band ligation, propranolol, and isosorbide mononitrate. Gastroenterology 2002; 123:735–744.
- Khuroo MS, Khuroo NS, Farahat KL, Khuroo YS, Sofi AA, Dahab ST. Meta-analysis: endoscopic variceal ligation for primary prophylaxis of oesophageal variceal bleeding. Aliment Pharmacol Ther 2005; 21:347–361.
- Jutabha R, Jensen DM, Martin P, Savides T, Han SH, Gornbein J. Randomized study comparing banding and propranolol to prevent initial variceal hemorrhage in cirrhotics with high-risk esophageal varices. Gastroenterology 2005; 128:870–881.
- Psilopoulos D, Galanis P, Goulas S, et al. Endoscopic variceal ligation vs. propranolol for prevention of first variceal bleeding: a randomized controlled trial. Eur J Gastroenterol Hepatol 2005; 17:1111–1117.
- Lay CS, Tsai YT, Lee FY, et al. Endoscopic variceal ligation versus propranolol in prophylaxis of first variceal bleeding in patients with cirrhosis. J Gastroenterol Hepatol 2006; 21:413–419.
- Sarin SK, Wadhawan M, Agarwal SR, Tyagi P, Sharma BC. Endoscopic variceal ligation plus propranolol versus endoscopic variceal ligation alone in primary prophylaxis of variceal bleeding. Am J Gastroenterol 2005; 100:797–804.
- Yuksel O, Koklu S, Arhan M, Yolcu OF, et al. Effects of esophageal variceal eradication on portal hypertensive gastropathy and fundal varices: a retrospective and comparative study. Dig Dis Sci 2006; 51:27–30.
- Schneider AW, Kalk JF, Klein CP. Effect of losartan, an angiotensin II receptor antagonist, on portal pressure in cirrhosis. Hepatology 1999; 29:334–339.
- Schepke M, Werner E, Biecker E, et al. Hemodynamic effects of the angiotensin II receptor antagonist irbesartan in patients with cirrhosis and portal hypertension. Gastroenterology 2001; 121:389–395.
- Gonzalez-Abraldes J, Albillos A, Banares R, et al. Randomized comparison of long-term losartan versus propranolol in lowering portal pressure in cirrhosis. Gastroenterology 2001; 121:382–388.
- Boyer TD, Haskal ZJ American Association for the Study of Liver Diseases. The role of transjugular intrahepatic portosystemic shunt in the management of portal hypertension. Hepatology 2005; 41:386–400.
- Prophylactic sclerotherapy for esophageal varices in men with alcoholic liver disease. A randomized, single-blind, multicenter clinical trial. The Veterans Affairs Cooperative Variceal Sclerotherapy Group. N Engl J Med 1991; 324:1779–1784.
- The PROVA Study Group. Prophylaxis of first hemorrhage from esophageal varices by sclerotherapy, propranolol or both in cirrhotic patients: a randomized multicenter trial. Hepatology 1991; 14:1016–1024.
- Escorsell A, Feu F, Bordas JM, et al. Effects of isosorbide-5-mononitrate on variceal pressure and systemic and splanchnic haemodynamics in patients with cirrhosis. J Hepatol 1996; 24:423–429.
- Hayes PC, Westaby D, Williams R. Effect and mechanism of action of isosorbide-5-mononitrate. Gut 1988; 29:752–755.
- Angelico M, Carli L, Piat C, et al. Isosorbide-5-mononitrate versus propranolol in the prevention of first bleeding in cirrhosis. Gastroenterology 1993; 104:1460–1465.
- Angelico M, Carli L, Piat C, Gentile S, Capocaccia L. Effects of isosorbide-5-mononitrate compared with propranolol on first bleeding and long-term survival in cirrhosis. Gastroenterology 1997; 113:1632–1639.
- Garcia-Pagan JC, Villanueva C, Vila MC, et al. MOVE Group. Mononitrato Varices Esofagicas. Isosorbide mononitrate in the prevention of first variceal bleed in patients who cannot receive beta-blockers. Gastroenterology 2001; 121:908–914.
- Merkel C, Marin R, Enzo E, et al. Randomised trial of nadolol alone or with isosorbide mononitrate for primary prophylaxis of variceal bleeding in cirrhosis. Gruppo-Triveneto per L’ipertensione portale (GTIP). Lancet 1996; 348:1677–1681.
- Garcia-Pagán JC, Morillas R, Banares R, et al Spanish Variceal Bleeding Study Group. Propranolol plus placebo versus propranolol plus isosorbide-5-mononitrate in the prevention of a first variceal bleed: a double-blind RCT. Hepatology 2003; 37:1260–1266.
- Garcia-Tsao G, Sanyal A, Grace N, et al. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology 2007; 46:922–938.
Variceal hemorrhage is a medical emergency in which up to 20% of patients die.1 Even if the patient survives an initial episode of variceal bleeding, the probability of another episode is high: the rebleeding rate without treatment is 70% within 1 year. The mortality rate with rebleeding is 33%.
With such overwhelming consequences, the best strategy in any patient with cirrhosis and known varices is to try to prevent the first episode of bleeding.
WHO IS AT RISK?
Esophageal varices are present in 30% of patients with compensated cirrhosis and in up to 60% of those with decompensated cirrhosis (ie, with evidence of ascites or encephalopathy).2
The risk of variceal hemorrhage is related to three factors:
- The size of the varices. Varices 5 mm in diameter or smaller have a 7% risk of bleeding in 2 years, while those larger than 5 mm have a 30% risk of bleeding within 2 years.3
- The appearance of the varices. Morphologic features of varices, including red wale signs (red streaks of the mucosa overlying the varix), have been correlated with an increased risk of hemorrhage.
- The severity of liver dysfunction, as assessed by the Child-Pugh classification—an index of liver dysfunction based on serum albumin concentration, bilirubin level, prothrombin time, and the presence of ascites and encephalopathy. A high Child-Pugh score (ie, class B or C), representing decompensated cirrhosis, is associated with an increased risk of bleeding.
HOW VARICES DEVELOP: PORTAL HYPERTENSION
Esophageal varices form as a result of increased portal pressure, the product of increased portal venous inflow and resistance to outflow from the portal venous system. Portal hypertension is a major complication of chronic liver disease. In cirrhosis, architectural distortion of the liver causes an increase in the intrahepatic vascular resistance.
Portal venous inflow depends on mesenteric arteriolar tone, increasing when tone decreases. In cirrhotic patients, the increase in portal pressure results from a combination of increased portal blood flow secondary to splanchnic arteriolar vasodilation and elevated resistance to outflow through distorted hepatic sinusoids.
The potent vasodilator nitric oxide (NO) plays an important role in portal hypertension. In patients with cirrhosis, NO bioavailability is decreased in the intrahepatic circulation due to defects in the posttranslational regulation of endothelial NO synthase.4 This deficiency of NO, along with mechanical factors in the sinusoids, contributes to the increase in intrahepatic resistance. In the systemic and splanchnic circulation, NO bioavailability is increased due to upregulation and posttranslational regulation of endothelial NO synthase, thereby increasing splanchnic vasodilatation and leading to increased portal venous inflow.5 This results in a marked increase in cardiac output and so-called hyperdynamic circulation.
Portal hypertension results in the development of collateral circulation, including venous channels in the esophagus and stomach, by the dilation of preexisting vessels and active angiogenesis. Esophagogastric varices increase in size with the severity of portal hypertension and can rupture when the tension in their walls exceeds a maximal point.
HEPATIC VEIN PRESSURE GRADIENT: A PROXY FOR PORTAL PRESSURE
Ideally, the portal venous pressure should be directly measured. However, since direct measurement is invasive and impractical, the hepatic vein pressure gradient (HVPG) can be measured instead and correlates well with the portal pressure.6
The normal HVPG is 5 mm Hg or less; anything above this value denotes portal hypertension. However, studies have shown that varices may develop but do not bleed if the HVPG is less than 12 mm Hg.8
TWO WAYS TO PREVENT BLEEDING
Bleeding can be prevented either by reducing the portal venous pressure or by obliterating the varices. Portal pressure can be reduced by placing a portosystemic shunt either surgically or percutaneously with radiographic guidance or by giving drugs such as nonselective beta-blockers, nitrates, or a combination of these drugs. Variceal obliteration is typically done by endoscopic methods with either injection of a sclerosant or band ligation.
NONSELECTIVE BETA-BLOCKERS: THE MAINSTAY OF TREATMENT
Nonselective beta-blockers, the most commonly used drugs for preventing first esophageal variceal bleeding, decrease portal pressure by blocking both beta-1 and beta-2 adrenergic receptors.9 Beta-1 blockade decreases portal flow by decreasing the heart rate and cardiac output, while blockade of beta-2 receptors results in unopposed alpha-adrenergic-mediated vasoconstriction.
Selective beta-blockers do not appear to be as useful for primary prophylaxis. More than 2 decades ago, metoprolol (Toprol, Lopressor), a beta-1 selective antagonist, was compared with propranolol (Inderal), a nons-elective agent, in patients with cirrhosis and portal hypertension.10 Although both drugs significantly reduced the heart rate and cardiac output, only those taking propranolol showed a marked fall in portal pressure (mean decrease of 6.8 mm Hg vs 3.8 mm Hg with metoprolol) and a significant reduction in hepatic blood flow. The differences were thought to be related to beta-2 blockade of vasodilator receptors in the splanchnic circulation, which occurs only with nonselective beta-blockers such as propranolol.
The two nonselective beta-blockers most often used to prevent variceal bleeding are nadolol (Corgard) and propranolol. Both have been extensively studied in preventing a first variceal hemorrhage.
Effectiveness of beta-blockers
D’Amico et al11 performed a meta-analysis in 1995, examining nine trials (996 patients total) of the effectiveness of beta-blockers in preventing a first variceal hemorrhage. Seven trials found that bleeding risk was reduced with beta-blockers (significantly in four), one trial found that risk was unchanged, and one trial found that risk was increased—an outlier due to a small sample size. The meta-analysis showed a significant bleeding reduction with the use of a beta-blocker, either including the outlier trial (pooled odds ratio 0.54; 95% confidence interval 0.39–0.74) or excluding it (pooled odds ratio 0.48; 95% confidence interval 0.35–0.66).
Mortality rates were also reduced in seven trials, but the reduction was statistically significant in only one. However, in the pooled estimate, the mortality risk reduction approached statistical significance (pooled odds ratio 0.75; 95% confidence interval 0.57–1.06).
Ideo et al12 gave either nadolol or placebo to 79 patients with cirrhosis and large esophageal varices that had never bled. Nadolol was found to protect against a first variceal hemorrhage: at 2-year follow-up, only 1 of the 30 patients allocated to nadolol had had bleeding, vs 11 of the 49 patients in the placebo group.
Merkel et al13 found that the risk of variceal bleeding was lower in patients who started treatment with beta-blockers when their varices were small (12% at 5 years) than in those who started treatment after a diagnosis of large esophageal varices (22% at 5 years). They concluded that nadolol helps prevent small varices from growing into larger ones.
Response to beta-blockers is not uniform
Although beta-blockers decrease the portal pressure in many cirrhotic patients, the response is not uniform. In a study of 60 cirrhotic patients,14 40% showed no reduction or even a slight increase in HVPG with propranolol. Most patients showed a significant reduction in heart rate (17.5% ± 10%) after receiving 40 mg of propranolol. In the patients whose HVPG did not decrease by at least 10% with 40 mg of propranolol, increasing the dose caused a decrease in HVPG without a further decrease in heart rate. This suggests that 40 mg of propranolol successfully produced beta-1 blockade but that a higher dose was required for effective beta-2 blockade.
Failure to respond in certain patients may be due to a concurrent rise in collateral or hepatic sinusoidal resistance, or both. This was confirmed in a study in portal-hypertensive rats treated with propranolol.15 The reduction in portal blood flow expected was accompanied by a disproportionately small reduction in portal pressure, which was thought to be due to a rise in portal and collateral vascular resistance.
How to tell if beta-blocker treatment is ‘working’
An HVPG ≤ 12 mm Hg? Studies have shown that the most important predictor of efficacy of prophylaxis for variceal bleeding is a decrease in the HVPG to 12 mm Hg or less or a decrease in the initial HVPG of more than 20%.9 Although measuring the HVPG is invasive, expensive, and not routinely done in clinical practice, several studies have investigated the role of measuring hemodynamic response to medication.
Merkel et al16 measured the HVPG in 49 cirrhotic patients with previously nonbleeding varices before starting therapy with beta-blockers with or without nitrates and after 1 to 3 months of treatment. They followed the patients for up to 5 years. The mean HVPG value at baseline was 18.8 mm Hg. At 3 years of follow-up, 7% of those who had responded well to therapy (defined as achieving an HVPG less than 13 mm Hg or a decrease of more than 20%) had experienced a bleeding episode, which was significantly less than the rate (41%) in those who did not meet those hemodynamic end points. No patient reaching an HVPG of 12 mm Hg or less during treatment had variceal bleeding during follow-up.
Groszmann et al17 also prospectively measured the HVPG in patients with cirrhosis and varices, but their patients received either propranolol or placebo. Variceal hemorrhage occurred in 13 patients (11 of 51 in the placebo group and 2 of 51 in the propranolol group), all of whom had an HVPG greater than 12 mm Hg. Again, none of the patients whose HVPG was decreased to 12 mm Hg or less bled from esophageal varices.
Unfortunately, routine HVPG measurement to guide primary prophylaxis is an expensive strategy. Data suggest that measuring the HVPG is cost-effective only when the cost of measuring the HVPG is very low, the risk of variceal bleeding is very high, or the patient is expected to survive at least 3 to 5 years.18
A heart rate of 55 to 60? An alternative to HVPG measurement to monitor the effectiveness of beta-blocker therapy is to follow the heart rate. A 25% reduction from baseline or a heart rate of 55 to 60 beats per minute is the standard goal19,20; yet, at least 40% of patients treated with enough propranolol to decrease the heart rate by 25% do not respond with significant HVPG reductions.14,21
So, although beta-blockade is effective peripherally, it may not reduce HVPG to less than 12 mm Hg or 20% from baseline, and direct HVPG measurement is still the gold standard.
Treatment should be lifelong
Once a patient is started on a beta-blocker to prevent variceal hemorrhage, the treatment should be lifelong.
In 2001, a group of patients (most of them in Child-Pugh class A or B) completing a prospective randomized controlled trial of propranolol for primary prevention of variceal hemorrhage were tapered off propranolol or placebo.22 Of the 49 patients, 9 experienced variceal hemorrhage (6 of 25 former propranolol recipients and 3 of 24 former placebo recipients), and 17 patients died (12 former propranolol and 5 former placebo recipients), suggesting that treatment should be maintained for life.
Therefore, when beta-blocker therapy is discontinued, the risk of variceal hemorrhage returns to what would be expected in an untreated population.
Beta-blockers may not prevent varices
Although many trials have shown that beta-blockers are effective as prophylaxis against a first variceal hemorrhage, there is no evidence that these drugs prevent varices from forming in cirrhotic patients.
Groszmann et al23 treated more than 200 patients who had biopsy-proven cirrhosis and portal hypertension (HVPG > 6 mm Hg) but no varices with timolol (Blocadren), a nonselective beta-blocker, or placebo. At a median follow-up of about 55 months, the groups did not differ significantly in the incidence of primary events (development of varices or variceal hemorrhage) or treatment failures (transplantation or death). Varices developed less frequently among patients with a baseline HVPG of less than 10 mm Hg and among those whose HVPG had decreased by more than 10% at 1 year. In patients whose HVPG increased by more than 10%, varices developed more frequently.
Contraindications, side effects
The major drawbacks to therapy with beta-blockers are their contraindications and side effects.
Contraindications include chronic obstructive lung disease, psychosis, atrioventricular heart blocks, and aortic-valve disease.
Side effects are reported in 15% of patients but severe events are rare.24 Still, an estimated 10% to 20% of patients discontinue treatment because they cannot tolerate it.25 The more common complaints include fatigue, shortness of breath, sexual dysfunction, and sleep disorders.
Dosage
No specific starting dose of beta-blockers is agreed upon, but nadolol 20 to 40 mg once daily or long-acting propranolol 60 mg once daily can be used as initial therapy.25 Once-daily dosing increases the likelihood of compliance.
Since portal pressure progressively declines from 12 noon to 7 PM and then increases throughout the night and back to baseline by 9 AM,26 we recommend that the medication be taken in the evening to counteract increases in portal pressure that occur in the middle of the night.
ENDOSCOPIC VARICEAL LIGATION
Endoscopic variceal ligation has been investigated extensively for use as prophylaxis against first variceal hemorrhage. The procedure involves placing a rubber band around a varix aspirated into a cylinder on the tip of an endoscope.
Effectiveness of ligation
Lay et al,27 in a prospective, randomized trial in 126 cirrhotic patients endoscopically judged to be at high risk of hemorrhage, found that ligation significantly reduced the 2-year cumulative bleeding rate (19% with ligation vs 60% in an untreated control group) and the overall mortality rate (28% vs 58%). The lower risk of bleeding in the ligation group was attributed to a rapid reduction of variceal size; 60% of those in the ligation group had complete eradication of varices and 38% had varices reduced in size.
Imperiale and Chalasani28 performed a meta-analysis in 2001 that included 601 patients in five trials comparing prophylactic ligation with untreated controls and 283 patients in four trials comparing ligation with beta-blocker therapy. Compared with no treatment, ligation reduced the risk of first variceal hemorrhage, bleeding-related death, and death from any cause. Compared with propranolol, ligation reduced the risk of first-time bleeding but had no effect on the death rate.
Schepke et al,29 in a randomized controlled multicenter trial in 152 cirrhotic patients with two or more esophageal varices, found that neither bleeding incidence nor death rate differed significantly between ligation and propranolol.
Lui et al30 followed 172 cirrhotic patients with grade II or III esophageal varices for 6 years and found that ligation was equivalent to propranolol. However, many patients reported side effects with propranolol, and 30% of patients withdrew from propranolol treatment, making ligation a more attractive option.
Khuroo et al31 performed a meta-analysis of eight randomized controlled trials including 596 patients and found that ligation significantly reduced the rates of first gastrointestinal hemorrhage by 31% and of first variceal hemorrhage by 43%. In subgroup analysis, ligation had a significant advantage compared with beta-blockers in trials with patients with a high bleeding risk, ie, trials in which more than 30% of patients were in Child-Pugh class C and more than 50% of the patients had large varices.
Jutabha et al32 performed a multicenter, prospective trial (published in 2005) in 62 patients with high-risk esophageal varices randomized to propranolol or banding. The trial was ended early after an interim analysis showed that the failure rate of propranolol was significantly higher than that of banding (6/31 vs 0/31, P = .0098). Esophageal variceal hemorrhage occurred in 4 (12.9%) of the patients in the propranolol group compared with 0 in the ligation group. Similarly, 4 patients in the propranolol group died, compared with 0 in the ligation group. All the patients in this trial were liver transplant candidates and therefore all had severe liver disease.
In another trial favoring variceal banding over beta-blockers, Psilopoulos et al33 in 2005 followed 60 patients with cirrhosis and esophageal varices with no history of bleeding. Thirty percent of the patients in the propranolol group developed variceal bleeding compared with 6.7% in the ligation group (P = .043).
Lay et al34 followed 100 cirrhotic patients for 2 years and found comparable cumulative bleeding rates with ligation vs propranolol (18% vs 16%, respectively) and also comparable rates of death (28% vs 24%, respectively).
Sarin et al35 investigated the role of propranolol in addition to ligation in the prevention of first hemorrhage in 144 patients. Adding propranolol did not further decrease the incidence of initial bleeding (7% in the combination group vs 11% in the ligation-only group). Survival rates were similar at 20 months: 92% in the combination group vs 85% in the ligation-only group. However, the rate of variceal recurrence was lower with combination therapy: 6% in the combination group vs 15% with ligation alone.
Does esophageal variceal ligation increase gastric varices?
A less researched topic is whether variceal ligation results in gastric hemodynamic changes that increase the size of fundal varices and worsen portal hypertensive gastropathy.
Yuksel et al36 found that 37 of 85 patients had fundal varices before they underwent ligation of esophageal varices, increasing to 46 after the procedure, a statistically significant increment. The severity of portal hypertensive gastropathy also increased.
Further research is required regarding the long-term consequences of these findings.
ANGIOTENSIN II RECEPTOR ANTAGONISTS: ROLE UNKNOWN
Angiotensin II increases portal pressure, and angiotensin II levels are elevated in patients with cirrhosis, suggesting that this hormone plays a role in the pathogenesis of portal hypertension.
Losartan (Cozaar), an angiotensin II receptor antagonist, was found to decrease the HVPG significantly in patients with severe and moderate portal hypertension in a pilot study37 in 1999. However, in two subsequent studies,38,39 losartan only moderately reduced the HVPG and caused hypotension and a reduction in the glomerular filtration rate. The role of angiotensin II receptor blockers in primary prevention of variceal bleeding is still unknown.
SURGICAL PORTAL DECOMPRESSION HAS BEEN ABANDONED
The first method investigated to prevent variceal bleeding was surgical portal decompression.
A meta-analysis of four randomized controlled trials in 302 patients with varices of all sizes compared portocaval shunt surgery and medical therapy.11 Although shunt surgery was very effective in preventing variceal bleeding, the risk of chronic or recurrent encephalopathy was significantly increased (odds ratio 2.0), as was the risk of death (odds ratio 1.6).
These poor results, combined with advances in endoscopic procedures, led to the abandonment of surgical shunting for primary prophylaxis.
TIPS PROCEDURE: NO ROLE AT PRESENT
The transjugular intrahepatic portosystemic shunt (TIPS) procedure is used to treat the main consequences of portal hypertension, including ascites and variceal hemorrhage. The procedure entails accessing the hepatic vein via the right jugular vein and placing a stent to the portal vein, forming a low-resistance channel and allowing blood to return to the systemic circulation.
TIPS placement increases the risk of encephalopathy; liver failure is a rare complication, and procedural complications (ie, shunt dysfunction) also occur. Trials comparing the TIPS procedure with other forms of therapy to prevent first variceal hemorrhages have not been performed.40 Research to improve the outcome of the TIPS procedure is ongoing, but currently this procedure has no role in primary prevention of variceal bleeding.
ENDOSCOPIC SCLEROTHERAPY MAY INCREASE THE RISK OF DEATH
Numerous clinical trials evaluated sclerotherapy as prophylaxis against a first esophageal variceal hemorrhage. The procedure involves injecting a sclerosant in and around varices.
In a large Veterans Administration study,41 sclerotherapy was compared with sham treatment in 281 men with alcoholic liver disease who had documented varices but no history of bleeding. The trial was terminated after 22.5 months because the rate of all-cause mortality was significantly higher in the sclerotherapy group (32.5%) than in the sham therapy group (17.4%). The higher death rate did not persist after the treatment was discontinued, and it was speculated that, although sclerotherapy had reduced new episodes of variceal hemorrhage, the procedure might have caused bleeding from esophageal ulcers, leading to an increased mortality rate in that group.
The PROVA Study Group from Norway and Denmark found similar results when 286 cirrhotic patients were randomized to receive sclerotherapy, propranolol, combination sclerotherapy and propranolol, or no treatment to prevent a first variceal hemorrhage.42 The incidence of variceal bleeding was almost identical in the four groups, but the mortality rate with variceal bleeding was 2.75 times higher in the sclerotherapy groups than in the other groups (P = .002). It was speculated that repeated sclerotherapy sessions might be poorly tolerated by patients in Child-Pugh classes B and C and might have contributed to the precipitation of liver failure and other common complications of cirrhosis.
A meta-analysis by D’Amico et al11 evaluated 19 trials (1,630 patients) comparing sclerotherapy with nonactive treatment. Sclerotherapy tended to be favorable in trials with a high bleeding rate in the control patients and unfavorable in trials with a low bleeding rate. The benefit seen in patients at high risk is consistent with the efficacy of sclerotherapy for preventing rebleeding, whereas the harmful effect in the low-risk patients points towards side effects and complications exceeding the potential benefits.
In general, currently available evidence suggests that the benefits of prophylactic sclerotherapy are marginal, and therefore sclerotherapy is not recommended as primary prophylaxis for variceal hemorrhage.
NITRATES: NO LONGER USED AS MONOTHERAPY
Unlike vasoconstrictors, which decrease portal pressure by decreasing blood flow, vasodilators reduce hepatic pressure by decreasing intrahepatic and portocollateral vascular resistance.43 In addition, larger doses directly affect the arterial circulation, lowering systemic and therefore splanchnic perfusion pressure.44 Unfortunately, the systemic vasodilatory effects of nitrates exacerbate the hyperdynamic state that is characteristic of cirrhosis, thereby limiting their use and tolerability in many patients.
A trial comparing propranolol vs isosorbide mononitrate initially found that the groups did not differ significantly with regard to bleeding rates and 2-year survival rates,45 but a 6-year follow-up found the likelihood of death greater in patients older than 50 years in the nitrate group.46 In an additional study comparing isosorbide mononitrate vs placebo in patients with contraindications to or intolerance of beta-blockers, no difference in the relative risk of first variceal hemorrhage was found between the two groups.47 Therefore, nitrates are no longer used as monotherapy to prevent variceal bleeding.
Combination therapy with beta-blockers plus nitrates is controversial. In a trial in 1996, Merkel et al48 found the cumulative risk of variceal bleeding was 18% at 40 months with nadolol alone vs 7.5% with nadolol plus isosorbide mononitrate. However, in a later trial, Garcia-Pagán et al49 found no significant advantage to combination therapy. The incidence of variceal bleeding at 1 year was 8.3% in the group receiving propranolol plus placebo and 5% in the group receiving propranolol plus isosorbide mononitrate; at 2 years, the rates were 10.6% vs 12.5%.
RECOMMENDATIONS FOR SCREENING AND PROPHYLAXIS
- All patients with cirrhosis should be screened for varices at the time of diagnosis.
- The size of the varices, including small (≤ 5 mm) and large (> 5 mm), and the presence of red wale marks on the varices should be recorded.
- Patients who have no varices on screening endoscopy should be rescreened every 3 years if their liver function is stable or every year if their liver function deteriorates. (Varices grow at a rate proportional to the severity of the liver disease.)
- Patients with portal hypertension but without varices do not need treatment with nonselective beta-blockers. Endoscopy should be performed at the intervals suggested above.
- Those who are found to have small varices on screening endoscopy but who have well-compensated liver disease (Child-Pugh class A) and no red wale marks should be rescreened every other year because the development of large varices is greater in patients with small varices on initial endoscopy than in patients with no varices. Emerging data support the use of beta-blockers to prevent varices from increasing in size.
- Patients who have small varices with red wale signs or who are in Child-Pugh class B or C have an increased risk of bleeding and should be treated with beta-blockers. If beta-blockers are not used, endoscopy should be done every year to look for an increase in variceal size.
- Patients who have large varices without red wale signs or who are in Child-Pugh class B or C should be treated with nonselective beta-blockers. The dose should be adjusted to achieve maximal tolerable decrease in heart rate to a minimum of 55 beats per minute, and treatment should be continued indefinitely.
- Endoscopic variceal ligation is an acceptable alternative to beta-blocker treatment as first-line therapy in those who cannot tolerate beta-blockers or who have contraindications to their use, or in those who have red wale marking or who are in Child-Pugh class B or C.
Variceal hemorrhage is a medical emergency in which up to 20% of patients die.1 Even if the patient survives an initial episode of variceal bleeding, the probability of another episode is high: the rebleeding rate without treatment is 70% within 1 year. The mortality rate with rebleeding is 33%.
With such overwhelming consequences, the best strategy in any patient with cirrhosis and known varices is to try to prevent the first episode of bleeding.
WHO IS AT RISK?
Esophageal varices are present in 30% of patients with compensated cirrhosis and in up to 60% of those with decompensated cirrhosis (ie, with evidence of ascites or encephalopathy).2
The risk of variceal hemorrhage is related to three factors:
- The size of the varices. Varices 5 mm in diameter or smaller have a 7% risk of bleeding in 2 years, while those larger than 5 mm have a 30% risk of bleeding within 2 years.3
- The appearance of the varices. Morphologic features of varices, including red wale signs (red streaks of the mucosa overlying the varix), have been correlated with an increased risk of hemorrhage.
- The severity of liver dysfunction, as assessed by the Child-Pugh classification—an index of liver dysfunction based on serum albumin concentration, bilirubin level, prothrombin time, and the presence of ascites and encephalopathy. A high Child-Pugh score (ie, class B or C), representing decompensated cirrhosis, is associated with an increased risk of bleeding.
HOW VARICES DEVELOP: PORTAL HYPERTENSION
Esophageal varices form as a result of increased portal pressure, the product of increased portal venous inflow and resistance to outflow from the portal venous system. Portal hypertension is a major complication of chronic liver disease. In cirrhosis, architectural distortion of the liver causes an increase in the intrahepatic vascular resistance.
Portal venous inflow depends on mesenteric arteriolar tone, increasing when tone decreases. In cirrhotic patients, the increase in portal pressure results from a combination of increased portal blood flow secondary to splanchnic arteriolar vasodilation and elevated resistance to outflow through distorted hepatic sinusoids.
The potent vasodilator nitric oxide (NO) plays an important role in portal hypertension. In patients with cirrhosis, NO bioavailability is decreased in the intrahepatic circulation due to defects in the posttranslational regulation of endothelial NO synthase.4 This deficiency of NO, along with mechanical factors in the sinusoids, contributes to the increase in intrahepatic resistance. In the systemic and splanchnic circulation, NO bioavailability is increased due to upregulation and posttranslational regulation of endothelial NO synthase, thereby increasing splanchnic vasodilatation and leading to increased portal venous inflow.5 This results in a marked increase in cardiac output and so-called hyperdynamic circulation.
Portal hypertension results in the development of collateral circulation, including venous channels in the esophagus and stomach, by the dilation of preexisting vessels and active angiogenesis. Esophagogastric varices increase in size with the severity of portal hypertension and can rupture when the tension in their walls exceeds a maximal point.
HEPATIC VEIN PRESSURE GRADIENT: A PROXY FOR PORTAL PRESSURE
Ideally, the portal venous pressure should be directly measured. However, since direct measurement is invasive and impractical, the hepatic vein pressure gradient (HVPG) can be measured instead and correlates well with the portal pressure.6
The normal HVPG is 5 mm Hg or less; anything above this value denotes portal hypertension. However, studies have shown that varices may develop but do not bleed if the HVPG is less than 12 mm Hg.8
TWO WAYS TO PREVENT BLEEDING
Bleeding can be prevented either by reducing the portal venous pressure or by obliterating the varices. Portal pressure can be reduced by placing a portosystemic shunt either surgically or percutaneously with radiographic guidance or by giving drugs such as nonselective beta-blockers, nitrates, or a combination of these drugs. Variceal obliteration is typically done by endoscopic methods with either injection of a sclerosant or band ligation.
NONSELECTIVE BETA-BLOCKERS: THE MAINSTAY OF TREATMENT
Nonselective beta-blockers, the most commonly used drugs for preventing first esophageal variceal bleeding, decrease portal pressure by blocking both beta-1 and beta-2 adrenergic receptors.9 Beta-1 blockade decreases portal flow by decreasing the heart rate and cardiac output, while blockade of beta-2 receptors results in unopposed alpha-adrenergic-mediated vasoconstriction.
Selective beta-blockers do not appear to be as useful for primary prophylaxis. More than 2 decades ago, metoprolol (Toprol, Lopressor), a beta-1 selective antagonist, was compared with propranolol (Inderal), a nons-elective agent, in patients with cirrhosis and portal hypertension.10 Although both drugs significantly reduced the heart rate and cardiac output, only those taking propranolol showed a marked fall in portal pressure (mean decrease of 6.8 mm Hg vs 3.8 mm Hg with metoprolol) and a significant reduction in hepatic blood flow. The differences were thought to be related to beta-2 blockade of vasodilator receptors in the splanchnic circulation, which occurs only with nonselective beta-blockers such as propranolol.
The two nonselective beta-blockers most often used to prevent variceal bleeding are nadolol (Corgard) and propranolol. Both have been extensively studied in preventing a first variceal hemorrhage.
Effectiveness of beta-blockers
D’Amico et al11 performed a meta-analysis in 1995, examining nine trials (996 patients total) of the effectiveness of beta-blockers in preventing a first variceal hemorrhage. Seven trials found that bleeding risk was reduced with beta-blockers (significantly in four), one trial found that risk was unchanged, and one trial found that risk was increased—an outlier due to a small sample size. The meta-analysis showed a significant bleeding reduction with the use of a beta-blocker, either including the outlier trial (pooled odds ratio 0.54; 95% confidence interval 0.39–0.74) or excluding it (pooled odds ratio 0.48; 95% confidence interval 0.35–0.66).
Mortality rates were also reduced in seven trials, but the reduction was statistically significant in only one. However, in the pooled estimate, the mortality risk reduction approached statistical significance (pooled odds ratio 0.75; 95% confidence interval 0.57–1.06).
Ideo et al12 gave either nadolol or placebo to 79 patients with cirrhosis and large esophageal varices that had never bled. Nadolol was found to protect against a first variceal hemorrhage: at 2-year follow-up, only 1 of the 30 patients allocated to nadolol had had bleeding, vs 11 of the 49 patients in the placebo group.
Merkel et al13 found that the risk of variceal bleeding was lower in patients who started treatment with beta-blockers when their varices were small (12% at 5 years) than in those who started treatment after a diagnosis of large esophageal varices (22% at 5 years). They concluded that nadolol helps prevent small varices from growing into larger ones.
Response to beta-blockers is not uniform
Although beta-blockers decrease the portal pressure in many cirrhotic patients, the response is not uniform. In a study of 60 cirrhotic patients,14 40% showed no reduction or even a slight increase in HVPG with propranolol. Most patients showed a significant reduction in heart rate (17.5% ± 10%) after receiving 40 mg of propranolol. In the patients whose HVPG did not decrease by at least 10% with 40 mg of propranolol, increasing the dose caused a decrease in HVPG without a further decrease in heart rate. This suggests that 40 mg of propranolol successfully produced beta-1 blockade but that a higher dose was required for effective beta-2 blockade.
Failure to respond in certain patients may be due to a concurrent rise in collateral or hepatic sinusoidal resistance, or both. This was confirmed in a study in portal-hypertensive rats treated with propranolol.15 The reduction in portal blood flow expected was accompanied by a disproportionately small reduction in portal pressure, which was thought to be due to a rise in portal and collateral vascular resistance.
How to tell if beta-blocker treatment is ‘working’
An HVPG ≤ 12 mm Hg? Studies have shown that the most important predictor of efficacy of prophylaxis for variceal bleeding is a decrease in the HVPG to 12 mm Hg or less or a decrease in the initial HVPG of more than 20%.9 Although measuring the HVPG is invasive, expensive, and not routinely done in clinical practice, several studies have investigated the role of measuring hemodynamic response to medication.
Merkel et al16 measured the HVPG in 49 cirrhotic patients with previously nonbleeding varices before starting therapy with beta-blockers with or without nitrates and after 1 to 3 months of treatment. They followed the patients for up to 5 years. The mean HVPG value at baseline was 18.8 mm Hg. At 3 years of follow-up, 7% of those who had responded well to therapy (defined as achieving an HVPG less than 13 mm Hg or a decrease of more than 20%) had experienced a bleeding episode, which was significantly less than the rate (41%) in those who did not meet those hemodynamic end points. No patient reaching an HVPG of 12 mm Hg or less during treatment had variceal bleeding during follow-up.
Groszmann et al17 also prospectively measured the HVPG in patients with cirrhosis and varices, but their patients received either propranolol or placebo. Variceal hemorrhage occurred in 13 patients (11 of 51 in the placebo group and 2 of 51 in the propranolol group), all of whom had an HVPG greater than 12 mm Hg. Again, none of the patients whose HVPG was decreased to 12 mm Hg or less bled from esophageal varices.
Unfortunately, routine HVPG measurement to guide primary prophylaxis is an expensive strategy. Data suggest that measuring the HVPG is cost-effective only when the cost of measuring the HVPG is very low, the risk of variceal bleeding is very high, or the patient is expected to survive at least 3 to 5 years.18
A heart rate of 55 to 60? An alternative to HVPG measurement to monitor the effectiveness of beta-blocker therapy is to follow the heart rate. A 25% reduction from baseline or a heart rate of 55 to 60 beats per minute is the standard goal19,20; yet, at least 40% of patients treated with enough propranolol to decrease the heart rate by 25% do not respond with significant HVPG reductions.14,21
So, although beta-blockade is effective peripherally, it may not reduce HVPG to less than 12 mm Hg or 20% from baseline, and direct HVPG measurement is still the gold standard.
Treatment should be lifelong
Once a patient is started on a beta-blocker to prevent variceal hemorrhage, the treatment should be lifelong.
In 2001, a group of patients (most of them in Child-Pugh class A or B) completing a prospective randomized controlled trial of propranolol for primary prevention of variceal hemorrhage were tapered off propranolol or placebo.22 Of the 49 patients, 9 experienced variceal hemorrhage (6 of 25 former propranolol recipients and 3 of 24 former placebo recipients), and 17 patients died (12 former propranolol and 5 former placebo recipients), suggesting that treatment should be maintained for life.
Therefore, when beta-blocker therapy is discontinued, the risk of variceal hemorrhage returns to what would be expected in an untreated population.
Beta-blockers may not prevent varices
Although many trials have shown that beta-blockers are effective as prophylaxis against a first variceal hemorrhage, there is no evidence that these drugs prevent varices from forming in cirrhotic patients.
Groszmann et al23 treated more than 200 patients who had biopsy-proven cirrhosis and portal hypertension (HVPG > 6 mm Hg) but no varices with timolol (Blocadren), a nonselective beta-blocker, or placebo. At a median follow-up of about 55 months, the groups did not differ significantly in the incidence of primary events (development of varices or variceal hemorrhage) or treatment failures (transplantation or death). Varices developed less frequently among patients with a baseline HVPG of less than 10 mm Hg and among those whose HVPG had decreased by more than 10% at 1 year. In patients whose HVPG increased by more than 10%, varices developed more frequently.
Contraindications, side effects
The major drawbacks to therapy with beta-blockers are their contraindications and side effects.
Contraindications include chronic obstructive lung disease, psychosis, atrioventricular heart blocks, and aortic-valve disease.
Side effects are reported in 15% of patients but severe events are rare.24 Still, an estimated 10% to 20% of patients discontinue treatment because they cannot tolerate it.25 The more common complaints include fatigue, shortness of breath, sexual dysfunction, and sleep disorders.
Dosage
No specific starting dose of beta-blockers is agreed upon, but nadolol 20 to 40 mg once daily or long-acting propranolol 60 mg once daily can be used as initial therapy.25 Once-daily dosing increases the likelihood of compliance.
Since portal pressure progressively declines from 12 noon to 7 PM and then increases throughout the night and back to baseline by 9 AM,26 we recommend that the medication be taken in the evening to counteract increases in portal pressure that occur in the middle of the night.
ENDOSCOPIC VARICEAL LIGATION
Endoscopic variceal ligation has been investigated extensively for use as prophylaxis against first variceal hemorrhage. The procedure involves placing a rubber band around a varix aspirated into a cylinder on the tip of an endoscope.
Effectiveness of ligation
Lay et al,27 in a prospective, randomized trial in 126 cirrhotic patients endoscopically judged to be at high risk of hemorrhage, found that ligation significantly reduced the 2-year cumulative bleeding rate (19% with ligation vs 60% in an untreated control group) and the overall mortality rate (28% vs 58%). The lower risk of bleeding in the ligation group was attributed to a rapid reduction of variceal size; 60% of those in the ligation group had complete eradication of varices and 38% had varices reduced in size.
Imperiale and Chalasani28 performed a meta-analysis in 2001 that included 601 patients in five trials comparing prophylactic ligation with untreated controls and 283 patients in four trials comparing ligation with beta-blocker therapy. Compared with no treatment, ligation reduced the risk of first variceal hemorrhage, bleeding-related death, and death from any cause. Compared with propranolol, ligation reduced the risk of first-time bleeding but had no effect on the death rate.
Schepke et al,29 in a randomized controlled multicenter trial in 152 cirrhotic patients with two or more esophageal varices, found that neither bleeding incidence nor death rate differed significantly between ligation and propranolol.
Lui et al30 followed 172 cirrhotic patients with grade II or III esophageal varices for 6 years and found that ligation was equivalent to propranolol. However, many patients reported side effects with propranolol, and 30% of patients withdrew from propranolol treatment, making ligation a more attractive option.
Khuroo et al31 performed a meta-analysis of eight randomized controlled trials including 596 patients and found that ligation significantly reduced the rates of first gastrointestinal hemorrhage by 31% and of first variceal hemorrhage by 43%. In subgroup analysis, ligation had a significant advantage compared with beta-blockers in trials with patients with a high bleeding risk, ie, trials in which more than 30% of patients were in Child-Pugh class C and more than 50% of the patients had large varices.
Jutabha et al32 performed a multicenter, prospective trial (published in 2005) in 62 patients with high-risk esophageal varices randomized to propranolol or banding. The trial was ended early after an interim analysis showed that the failure rate of propranolol was significantly higher than that of banding (6/31 vs 0/31, P = .0098). Esophageal variceal hemorrhage occurred in 4 (12.9%) of the patients in the propranolol group compared with 0 in the ligation group. Similarly, 4 patients in the propranolol group died, compared with 0 in the ligation group. All the patients in this trial were liver transplant candidates and therefore all had severe liver disease.
In another trial favoring variceal banding over beta-blockers, Psilopoulos et al33 in 2005 followed 60 patients with cirrhosis and esophageal varices with no history of bleeding. Thirty percent of the patients in the propranolol group developed variceal bleeding compared with 6.7% in the ligation group (P = .043).
Lay et al34 followed 100 cirrhotic patients for 2 years and found comparable cumulative bleeding rates with ligation vs propranolol (18% vs 16%, respectively) and also comparable rates of death (28% vs 24%, respectively).
Sarin et al35 investigated the role of propranolol in addition to ligation in the prevention of first hemorrhage in 144 patients. Adding propranolol did not further decrease the incidence of initial bleeding (7% in the combination group vs 11% in the ligation-only group). Survival rates were similar at 20 months: 92% in the combination group vs 85% in the ligation-only group. However, the rate of variceal recurrence was lower with combination therapy: 6% in the combination group vs 15% with ligation alone.
Does esophageal variceal ligation increase gastric varices?
A less researched topic is whether variceal ligation results in gastric hemodynamic changes that increase the size of fundal varices and worsen portal hypertensive gastropathy.
Yuksel et al36 found that 37 of 85 patients had fundal varices before they underwent ligation of esophageal varices, increasing to 46 after the procedure, a statistically significant increment. The severity of portal hypertensive gastropathy also increased.
Further research is required regarding the long-term consequences of these findings.
ANGIOTENSIN II RECEPTOR ANTAGONISTS: ROLE UNKNOWN
Angiotensin II increases portal pressure, and angiotensin II levels are elevated in patients with cirrhosis, suggesting that this hormone plays a role in the pathogenesis of portal hypertension.
Losartan (Cozaar), an angiotensin II receptor antagonist, was found to decrease the HVPG significantly in patients with severe and moderate portal hypertension in a pilot study37 in 1999. However, in two subsequent studies,38,39 losartan only moderately reduced the HVPG and caused hypotension and a reduction in the glomerular filtration rate. The role of angiotensin II receptor blockers in primary prevention of variceal bleeding is still unknown.
SURGICAL PORTAL DECOMPRESSION HAS BEEN ABANDONED
The first method investigated to prevent variceal bleeding was surgical portal decompression.
A meta-analysis of four randomized controlled trials in 302 patients with varices of all sizes compared portocaval shunt surgery and medical therapy.11 Although shunt surgery was very effective in preventing variceal bleeding, the risk of chronic or recurrent encephalopathy was significantly increased (odds ratio 2.0), as was the risk of death (odds ratio 1.6).
These poor results, combined with advances in endoscopic procedures, led to the abandonment of surgical shunting for primary prophylaxis.
TIPS PROCEDURE: NO ROLE AT PRESENT
The transjugular intrahepatic portosystemic shunt (TIPS) procedure is used to treat the main consequences of portal hypertension, including ascites and variceal hemorrhage. The procedure entails accessing the hepatic vein via the right jugular vein and placing a stent to the portal vein, forming a low-resistance channel and allowing blood to return to the systemic circulation.
TIPS placement increases the risk of encephalopathy; liver failure is a rare complication, and procedural complications (ie, shunt dysfunction) also occur. Trials comparing the TIPS procedure with other forms of therapy to prevent first variceal hemorrhages have not been performed.40 Research to improve the outcome of the TIPS procedure is ongoing, but currently this procedure has no role in primary prevention of variceal bleeding.
ENDOSCOPIC SCLEROTHERAPY MAY INCREASE THE RISK OF DEATH
Numerous clinical trials evaluated sclerotherapy as prophylaxis against a first esophageal variceal hemorrhage. The procedure involves injecting a sclerosant in and around varices.
In a large Veterans Administration study,41 sclerotherapy was compared with sham treatment in 281 men with alcoholic liver disease who had documented varices but no history of bleeding. The trial was terminated after 22.5 months because the rate of all-cause mortality was significantly higher in the sclerotherapy group (32.5%) than in the sham therapy group (17.4%). The higher death rate did not persist after the treatment was discontinued, and it was speculated that, although sclerotherapy had reduced new episodes of variceal hemorrhage, the procedure might have caused bleeding from esophageal ulcers, leading to an increased mortality rate in that group.
The PROVA Study Group from Norway and Denmark found similar results when 286 cirrhotic patients were randomized to receive sclerotherapy, propranolol, combination sclerotherapy and propranolol, or no treatment to prevent a first variceal hemorrhage.42 The incidence of variceal bleeding was almost identical in the four groups, but the mortality rate with variceal bleeding was 2.75 times higher in the sclerotherapy groups than in the other groups (P = .002). It was speculated that repeated sclerotherapy sessions might be poorly tolerated by patients in Child-Pugh classes B and C and might have contributed to the precipitation of liver failure and other common complications of cirrhosis.
A meta-analysis by D’Amico et al11 evaluated 19 trials (1,630 patients) comparing sclerotherapy with nonactive treatment. Sclerotherapy tended to be favorable in trials with a high bleeding rate in the control patients and unfavorable in trials with a low bleeding rate. The benefit seen in patients at high risk is consistent with the efficacy of sclerotherapy for preventing rebleeding, whereas the harmful effect in the low-risk patients points towards side effects and complications exceeding the potential benefits.
In general, currently available evidence suggests that the benefits of prophylactic sclerotherapy are marginal, and therefore sclerotherapy is not recommended as primary prophylaxis for variceal hemorrhage.
NITRATES: NO LONGER USED AS MONOTHERAPY
Unlike vasoconstrictors, which decrease portal pressure by decreasing blood flow, vasodilators reduce hepatic pressure by decreasing intrahepatic and portocollateral vascular resistance.43 In addition, larger doses directly affect the arterial circulation, lowering systemic and therefore splanchnic perfusion pressure.44 Unfortunately, the systemic vasodilatory effects of nitrates exacerbate the hyperdynamic state that is characteristic of cirrhosis, thereby limiting their use and tolerability in many patients.
A trial comparing propranolol vs isosorbide mononitrate initially found that the groups did not differ significantly with regard to bleeding rates and 2-year survival rates,45 but a 6-year follow-up found the likelihood of death greater in patients older than 50 years in the nitrate group.46 In an additional study comparing isosorbide mononitrate vs placebo in patients with contraindications to or intolerance of beta-blockers, no difference in the relative risk of first variceal hemorrhage was found between the two groups.47 Therefore, nitrates are no longer used as monotherapy to prevent variceal bleeding.
Combination therapy with beta-blockers plus nitrates is controversial. In a trial in 1996, Merkel et al48 found the cumulative risk of variceal bleeding was 18% at 40 months with nadolol alone vs 7.5% with nadolol plus isosorbide mononitrate. However, in a later trial, Garcia-Pagán et al49 found no significant advantage to combination therapy. The incidence of variceal bleeding at 1 year was 8.3% in the group receiving propranolol plus placebo and 5% in the group receiving propranolol plus isosorbide mononitrate; at 2 years, the rates were 10.6% vs 12.5%.
RECOMMENDATIONS FOR SCREENING AND PROPHYLAXIS
- All patients with cirrhosis should be screened for varices at the time of diagnosis.
- The size of the varices, including small (≤ 5 mm) and large (> 5 mm), and the presence of red wale marks on the varices should be recorded.
- Patients who have no varices on screening endoscopy should be rescreened every 3 years if their liver function is stable or every year if their liver function deteriorates. (Varices grow at a rate proportional to the severity of the liver disease.)
- Patients with portal hypertension but without varices do not need treatment with nonselective beta-blockers. Endoscopy should be performed at the intervals suggested above.
- Those who are found to have small varices on screening endoscopy but who have well-compensated liver disease (Child-Pugh class A) and no red wale marks should be rescreened every other year because the development of large varices is greater in patients with small varices on initial endoscopy than in patients with no varices. Emerging data support the use of beta-blockers to prevent varices from increasing in size.
- Patients who have small varices with red wale signs or who are in Child-Pugh class B or C have an increased risk of bleeding and should be treated with beta-blockers. If beta-blockers are not used, endoscopy should be done every year to look for an increase in variceal size.
- Patients who have large varices without red wale signs or who are in Child-Pugh class B or C should be treated with nonselective beta-blockers. The dose should be adjusted to achieve maximal tolerable decrease in heart rate to a minimum of 55 beats per minute, and treatment should be continued indefinitely.
- Endoscopic variceal ligation is an acceptable alternative to beta-blocker treatment as first-line therapy in those who cannot tolerate beta-blockers or who have contraindications to their use, or in those who have red wale marking or who are in Child-Pugh class B or C.
- D’Amico G, De Franchis R Cooperative Study Group. Upper digestive bleeding in cirrhosis. Post-therapeutic outcome and prognostic indicators. Hepatology 2003; 38:599–612.
- D’Amico G, Luca A. Natural history. Clinicalhaemodynamic correlations. Prediction of the risk of bleeding. Baillieres Clin Gastroenterol 1997; 11:243–256.
- Kamath PS. Esophageal variceal bleeding: primary prophylaxis. Clin Gastroenterol Hepatol 2005; 3:90–93.
- Shah V, Toruner M, Haddad F, et al. Impaired endothelial nitric oxide synthase activity associated with enhanced caveolin binding in experimental cirrhosis in the rat. Gastroenterology 1999; 117:1222–1228.
- Morales-Ruiz M, Jimenez W, Perez-Sala D, et al. Increased nitric oxide synthase expression in arterial vessels of cirrhotic rats with ascites. Hepatology 1996; 24:1481–1486.
- De Franchis R, Dell’Era A, Iannuzzi F. Diagnosis and treatment of portal hypertension. Dig Liver Dis 2004; 36:787–798.
- Groszmann RJ, Wongcharatrawee S. The hepatic venous pressure gradient: anything worth doing should be done right. Hepatology 2004; 39:280–282.
- Garcia-Tsao G, Groszmann RJ, Fisher RL, Conn HO, Atterbury CE, Glickman M. Portal pressure, presence of gastroesophageal varices and variceal bleeding. Hepatology 1985; 5:419–424.
- Bosch J, Masti R, Kravetz D, et al. Effects of propranolol on azygos venous blood flow and hepatic and systemic hemodynamics in cirrhosis. Hepatology 1984; 4:1200–1205.
- Westaby D, Bihari DJ, Gimson AE, Crossley IR, Williams R. Selective and non-selective beta receptor blockade in the reduction of portal pressure in patients with cirrhosis and portal hypertension. Gut 1984; 25:121–124.
- D’Amico G, Pagliaro L, Bosch J. The treatment of portal hypertension: a meta-analytic review. Hepatology 1995; 22:332–354.
- Ideo G, Bellati G, Fesce E, Grimoldi D. Nadolol can prevent the first gastrointestinal bleeding in cirrhotics: a prospective, randomized study. Hepatology 1988; 8:6–9.
- Merkel C, Marin R, Angeli P, et al. Gruppo Triveneto per l’Ipertensione Portale. A placebo-controlled clinical trial of nadolol in the prophylaxis of growth of small esophageal varices in cirrhosis. Gastroenterology 2004; 127:476–484.
- Garcia-Tsao G, Grace ND, Groszmann RJ, et al. Short-term effects of propranolol on portal venous pressure. Hepatology 1986; 6:101–106.
- Kroeger RJ, Groszmann RJ. Increased portal venous resistance hinders portal pressure reduction during the administration of beta-adrenergic blocking agents in a portal hypertensive model. Hepatology 1985; 5:97–101.
- Merkel C, Bolognesi M, Sacerdoti D, et al. The hemodynamic response to medical treatment of portal hypertension as a predictor of clinical effectiveness in the primary prophylaxis of variceal bleeding in cirrhosis. Hepatology 2000; 32:930–934.
- Groszmann RJ, Bosch J, Grace ND, et al. Hemodynamic events in a prospective randomized trial of propranolol versus placebo in the prevention of a first variceal hemorrhage. Gastroenterology 1990; 99:1401–1407.
- Hicken BL, Sharara AI, Abrams GA, Eloubeidi M, Fallon MB, Arguedas MR. Hepatic venous pressure gradient measurements to assess response to primary prophylaxis in patients with cirrhosis: a decision analytical study. Aliment Pharmacol Ther 2003; 17:145–153.
- Grace ND. Diagnosis and treatment of gastrointestinal bleeding secondary to portal hypertension. American College of Gastroenterology Practice Parameters Committee. Am J Gastroenterol 1997; 92:1081–1091.
- Grace ND, Groszmann RJ, Garcia-Tsao G, et al. Portal hypertension and variceal bleeding: an AASLD single topic symposium. Hepatology 1998; 28:868–880.
- Feu F, Garcia-Pagán JC, Bosch J, et al. Relation between portal pressure response to pharmacotherapy and risk of recurrent variceal haemorrhage in patients with cirrhosis. Lancet 1995; 346:1056–1059.
- Abraczinskas DR, Ookubo R, Grace ND, et al. Propranolol for the prevention of first esophageal variceal hemorrhage: a lifetime commitment? Hepatology 2001; 34:1096–1102.
- Groszmann RJ, Garcia-Tsao G, Bosch J, et al Portal Hypertension Collaborative Group. Beta-blockers to prevent gastroesophageal varices in patients with cirrhosis. N Engl J Med 2005; 353:2254–2261.
- Garcia-Pagán JC, Bosch J. Pharmacological prevention of variceal bleeding. New developments. Baillier Clin Gastroenterol 1997; 11:271–287.
- Talwalkar JA, Kamath PS. An evidence-based medicine approach to beta-blocker therapy in patients with cirrhosis. Am J Med 2004; 116:759–766.
- Garcia-Pagan JC, Feu F, Castells A, et al. Circadian variations of portal pressure and variceal hemorrhage in patients with cirrhosis. Hepatology 1994; 19:595–601.
- Lay CS, Tsai YT, Teg CY, et al. Endoscopic variceal ligation in prophylaxis of first variceal bleeding in cirrhotic patients with high-risk esophageal varices. Hepatology 1997; 25:1346–1350.
- Imperiale TF, Chalasani N. A meta-analysis of endoscopic variceal ligation for primary prophylaxis of esophageal variceal bleeding. Hepatology 2001; 33:802–807.
- Schepke M, Kleber G, Nurnberg D, et al. Ligation versus propranolol for the primary prophylaxis of variceal bleeding in cirrhosis. Hepatology 2004; 40:65–72.
- Lui HF, Stanley AJ, Forrest EH, et al. Primary prophylaxis of variceal hemorrhage: a randomized controlled trial comparing band ligation, propranolol, and isosorbide mononitrate. Gastroenterology 2002; 123:735–744.
- Khuroo MS, Khuroo NS, Farahat KL, Khuroo YS, Sofi AA, Dahab ST. Meta-analysis: endoscopic variceal ligation for primary prophylaxis of oesophageal variceal bleeding. Aliment Pharmacol Ther 2005; 21:347–361.
- Jutabha R, Jensen DM, Martin P, Savides T, Han SH, Gornbein J. Randomized study comparing banding and propranolol to prevent initial variceal hemorrhage in cirrhotics with high-risk esophageal varices. Gastroenterology 2005; 128:870–881.
- Psilopoulos D, Galanis P, Goulas S, et al. Endoscopic variceal ligation vs. propranolol for prevention of first variceal bleeding: a randomized controlled trial. Eur J Gastroenterol Hepatol 2005; 17:1111–1117.
- Lay CS, Tsai YT, Lee FY, et al. Endoscopic variceal ligation versus propranolol in prophylaxis of first variceal bleeding in patients with cirrhosis. J Gastroenterol Hepatol 2006; 21:413–419.
- Sarin SK, Wadhawan M, Agarwal SR, Tyagi P, Sharma BC. Endoscopic variceal ligation plus propranolol versus endoscopic variceal ligation alone in primary prophylaxis of variceal bleeding. Am J Gastroenterol 2005; 100:797–804.
- Yuksel O, Koklu S, Arhan M, Yolcu OF, et al. Effects of esophageal variceal eradication on portal hypertensive gastropathy and fundal varices: a retrospective and comparative study. Dig Dis Sci 2006; 51:27–30.
- Schneider AW, Kalk JF, Klein CP. Effect of losartan, an angiotensin II receptor antagonist, on portal pressure in cirrhosis. Hepatology 1999; 29:334–339.
- Schepke M, Werner E, Biecker E, et al. Hemodynamic effects of the angiotensin II receptor antagonist irbesartan in patients with cirrhosis and portal hypertension. Gastroenterology 2001; 121:389–395.
- Gonzalez-Abraldes J, Albillos A, Banares R, et al. Randomized comparison of long-term losartan versus propranolol in lowering portal pressure in cirrhosis. Gastroenterology 2001; 121:382–388.
- Boyer TD, Haskal ZJ American Association for the Study of Liver Diseases. The role of transjugular intrahepatic portosystemic shunt in the management of portal hypertension. Hepatology 2005; 41:386–400.
- Prophylactic sclerotherapy for esophageal varices in men with alcoholic liver disease. A randomized, single-blind, multicenter clinical trial. The Veterans Affairs Cooperative Variceal Sclerotherapy Group. N Engl J Med 1991; 324:1779–1784.
- The PROVA Study Group. Prophylaxis of first hemorrhage from esophageal varices by sclerotherapy, propranolol or both in cirrhotic patients: a randomized multicenter trial. Hepatology 1991; 14:1016–1024.
- Escorsell A, Feu F, Bordas JM, et al. Effects of isosorbide-5-mononitrate on variceal pressure and systemic and splanchnic haemodynamics in patients with cirrhosis. J Hepatol 1996; 24:423–429.
- Hayes PC, Westaby D, Williams R. Effect and mechanism of action of isosorbide-5-mononitrate. Gut 1988; 29:752–755.
- Angelico M, Carli L, Piat C, et al. Isosorbide-5-mononitrate versus propranolol in the prevention of first bleeding in cirrhosis. Gastroenterology 1993; 104:1460–1465.
- Angelico M, Carli L, Piat C, Gentile S, Capocaccia L. Effects of isosorbide-5-mononitrate compared with propranolol on first bleeding and long-term survival in cirrhosis. Gastroenterology 1997; 113:1632–1639.
- Garcia-Pagan JC, Villanueva C, Vila MC, et al. MOVE Group. Mononitrato Varices Esofagicas. Isosorbide mononitrate in the prevention of first variceal bleed in patients who cannot receive beta-blockers. Gastroenterology 2001; 121:908–914.
- Merkel C, Marin R, Enzo E, et al. Randomised trial of nadolol alone or with isosorbide mononitrate for primary prophylaxis of variceal bleeding in cirrhosis. Gruppo-Triveneto per L’ipertensione portale (GTIP). Lancet 1996; 348:1677–1681.
- Garcia-Pagán JC, Morillas R, Banares R, et al Spanish Variceal Bleeding Study Group. Propranolol plus placebo versus propranolol plus isosorbide-5-mononitrate in the prevention of a first variceal bleed: a double-blind RCT. Hepatology 2003; 37:1260–1266.
- Garcia-Tsao G, Sanyal A, Grace N, et al. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology 2007; 46:922–938.
- D’Amico G, De Franchis R Cooperative Study Group. Upper digestive bleeding in cirrhosis. Post-therapeutic outcome and prognostic indicators. Hepatology 2003; 38:599–612.
- D’Amico G, Luca A. Natural history. Clinicalhaemodynamic correlations. Prediction of the risk of bleeding. Baillieres Clin Gastroenterol 1997; 11:243–256.
- Kamath PS. Esophageal variceal bleeding: primary prophylaxis. Clin Gastroenterol Hepatol 2005; 3:90–93.
- Shah V, Toruner M, Haddad F, et al. Impaired endothelial nitric oxide synthase activity associated with enhanced caveolin binding in experimental cirrhosis in the rat. Gastroenterology 1999; 117:1222–1228.
- Morales-Ruiz M, Jimenez W, Perez-Sala D, et al. Increased nitric oxide synthase expression in arterial vessels of cirrhotic rats with ascites. Hepatology 1996; 24:1481–1486.
- De Franchis R, Dell’Era A, Iannuzzi F. Diagnosis and treatment of portal hypertension. Dig Liver Dis 2004; 36:787–798.
- Groszmann RJ, Wongcharatrawee S. The hepatic venous pressure gradient: anything worth doing should be done right. Hepatology 2004; 39:280–282.
- Garcia-Tsao G, Groszmann RJ, Fisher RL, Conn HO, Atterbury CE, Glickman M. Portal pressure, presence of gastroesophageal varices and variceal bleeding. Hepatology 1985; 5:419–424.
- Bosch J, Masti R, Kravetz D, et al. Effects of propranolol on azygos venous blood flow and hepatic and systemic hemodynamics in cirrhosis. Hepatology 1984; 4:1200–1205.
- Westaby D, Bihari DJ, Gimson AE, Crossley IR, Williams R. Selective and non-selective beta receptor blockade in the reduction of portal pressure in patients with cirrhosis and portal hypertension. Gut 1984; 25:121–124.
- D’Amico G, Pagliaro L, Bosch J. The treatment of portal hypertension: a meta-analytic review. Hepatology 1995; 22:332–354.
- Ideo G, Bellati G, Fesce E, Grimoldi D. Nadolol can prevent the first gastrointestinal bleeding in cirrhotics: a prospective, randomized study. Hepatology 1988; 8:6–9.
- Merkel C, Marin R, Angeli P, et al. Gruppo Triveneto per l’Ipertensione Portale. A placebo-controlled clinical trial of nadolol in the prophylaxis of growth of small esophageal varices in cirrhosis. Gastroenterology 2004; 127:476–484.
- Garcia-Tsao G, Grace ND, Groszmann RJ, et al. Short-term effects of propranolol on portal venous pressure. Hepatology 1986; 6:101–106.
- Kroeger RJ, Groszmann RJ. Increased portal venous resistance hinders portal pressure reduction during the administration of beta-adrenergic blocking agents in a portal hypertensive model. Hepatology 1985; 5:97–101.
- Merkel C, Bolognesi M, Sacerdoti D, et al. The hemodynamic response to medical treatment of portal hypertension as a predictor of clinical effectiveness in the primary prophylaxis of variceal bleeding in cirrhosis. Hepatology 2000; 32:930–934.
- Groszmann RJ, Bosch J, Grace ND, et al. Hemodynamic events in a prospective randomized trial of propranolol versus placebo in the prevention of a first variceal hemorrhage. Gastroenterology 1990; 99:1401–1407.
- Hicken BL, Sharara AI, Abrams GA, Eloubeidi M, Fallon MB, Arguedas MR. Hepatic venous pressure gradient measurements to assess response to primary prophylaxis in patients with cirrhosis: a decision analytical study. Aliment Pharmacol Ther 2003; 17:145–153.
- Grace ND. Diagnosis and treatment of gastrointestinal bleeding secondary to portal hypertension. American College of Gastroenterology Practice Parameters Committee. Am J Gastroenterol 1997; 92:1081–1091.
- Grace ND, Groszmann RJ, Garcia-Tsao G, et al. Portal hypertension and variceal bleeding: an AASLD single topic symposium. Hepatology 1998; 28:868–880.
- Feu F, Garcia-Pagán JC, Bosch J, et al. Relation between portal pressure response to pharmacotherapy and risk of recurrent variceal haemorrhage in patients with cirrhosis. Lancet 1995; 346:1056–1059.
- Abraczinskas DR, Ookubo R, Grace ND, et al. Propranolol for the prevention of first esophageal variceal hemorrhage: a lifetime commitment? Hepatology 2001; 34:1096–1102.
- Groszmann RJ, Garcia-Tsao G, Bosch J, et al Portal Hypertension Collaborative Group. Beta-blockers to prevent gastroesophageal varices in patients with cirrhosis. N Engl J Med 2005; 353:2254–2261.
- Garcia-Pagán JC, Bosch J. Pharmacological prevention of variceal bleeding. New developments. Baillier Clin Gastroenterol 1997; 11:271–287.
- Talwalkar JA, Kamath PS. An evidence-based medicine approach to beta-blocker therapy in patients with cirrhosis. Am J Med 2004; 116:759–766.
- Garcia-Pagan JC, Feu F, Castells A, et al. Circadian variations of portal pressure and variceal hemorrhage in patients with cirrhosis. Hepatology 1994; 19:595–601.
- Lay CS, Tsai YT, Teg CY, et al. Endoscopic variceal ligation in prophylaxis of first variceal bleeding in cirrhotic patients with high-risk esophageal varices. Hepatology 1997; 25:1346–1350.
- Imperiale TF, Chalasani N. A meta-analysis of endoscopic variceal ligation for primary prophylaxis of esophageal variceal bleeding. Hepatology 2001; 33:802–807.
- Schepke M, Kleber G, Nurnberg D, et al. Ligation versus propranolol for the primary prophylaxis of variceal bleeding in cirrhosis. Hepatology 2004; 40:65–72.
- Lui HF, Stanley AJ, Forrest EH, et al. Primary prophylaxis of variceal hemorrhage: a randomized controlled trial comparing band ligation, propranolol, and isosorbide mononitrate. Gastroenterology 2002; 123:735–744.
- Khuroo MS, Khuroo NS, Farahat KL, Khuroo YS, Sofi AA, Dahab ST. Meta-analysis: endoscopic variceal ligation for primary prophylaxis of oesophageal variceal bleeding. Aliment Pharmacol Ther 2005; 21:347–361.
- Jutabha R, Jensen DM, Martin P, Savides T, Han SH, Gornbein J. Randomized study comparing banding and propranolol to prevent initial variceal hemorrhage in cirrhotics with high-risk esophageal varices. Gastroenterology 2005; 128:870–881.
- Psilopoulos D, Galanis P, Goulas S, et al. Endoscopic variceal ligation vs. propranolol for prevention of first variceal bleeding: a randomized controlled trial. Eur J Gastroenterol Hepatol 2005; 17:1111–1117.
- Lay CS, Tsai YT, Lee FY, et al. Endoscopic variceal ligation versus propranolol in prophylaxis of first variceal bleeding in patients with cirrhosis. J Gastroenterol Hepatol 2006; 21:413–419.
- Sarin SK, Wadhawan M, Agarwal SR, Tyagi P, Sharma BC. Endoscopic variceal ligation plus propranolol versus endoscopic variceal ligation alone in primary prophylaxis of variceal bleeding. Am J Gastroenterol 2005; 100:797–804.
- Yuksel O, Koklu S, Arhan M, Yolcu OF, et al. Effects of esophageal variceal eradication on portal hypertensive gastropathy and fundal varices: a retrospective and comparative study. Dig Dis Sci 2006; 51:27–30.
- Schneider AW, Kalk JF, Klein CP. Effect of losartan, an angiotensin II receptor antagonist, on portal pressure in cirrhosis. Hepatology 1999; 29:334–339.
- Schepke M, Werner E, Biecker E, et al. Hemodynamic effects of the angiotensin II receptor antagonist irbesartan in patients with cirrhosis and portal hypertension. Gastroenterology 2001; 121:389–395.
- Gonzalez-Abraldes J, Albillos A, Banares R, et al. Randomized comparison of long-term losartan versus propranolol in lowering portal pressure in cirrhosis. Gastroenterology 2001; 121:382–388.
- Boyer TD, Haskal ZJ American Association for the Study of Liver Diseases. The role of transjugular intrahepatic portosystemic shunt in the management of portal hypertension. Hepatology 2005; 41:386–400.
- Prophylactic sclerotherapy for esophageal varices in men with alcoholic liver disease. A randomized, single-blind, multicenter clinical trial. The Veterans Affairs Cooperative Variceal Sclerotherapy Group. N Engl J Med 1991; 324:1779–1784.
- The PROVA Study Group. Prophylaxis of first hemorrhage from esophageal varices by sclerotherapy, propranolol or both in cirrhotic patients: a randomized multicenter trial. Hepatology 1991; 14:1016–1024.
- Escorsell A, Feu F, Bordas JM, et al. Effects of isosorbide-5-mononitrate on variceal pressure and systemic and splanchnic haemodynamics in patients with cirrhosis. J Hepatol 1996; 24:423–429.
- Hayes PC, Westaby D, Williams R. Effect and mechanism of action of isosorbide-5-mononitrate. Gut 1988; 29:752–755.
- Angelico M, Carli L, Piat C, et al. Isosorbide-5-mononitrate versus propranolol in the prevention of first bleeding in cirrhosis. Gastroenterology 1993; 104:1460–1465.
- Angelico M, Carli L, Piat C, Gentile S, Capocaccia L. Effects of isosorbide-5-mononitrate compared with propranolol on first bleeding and long-term survival in cirrhosis. Gastroenterology 1997; 113:1632–1639.
- Garcia-Pagan JC, Villanueva C, Vila MC, et al. MOVE Group. Mononitrato Varices Esofagicas. Isosorbide mononitrate in the prevention of first variceal bleed in patients who cannot receive beta-blockers. Gastroenterology 2001; 121:908–914.
- Merkel C, Marin R, Enzo E, et al. Randomised trial of nadolol alone or with isosorbide mononitrate for primary prophylaxis of variceal bleeding in cirrhosis. Gruppo-Triveneto per L’ipertensione portale (GTIP). Lancet 1996; 348:1677–1681.
- Garcia-Pagán JC, Morillas R, Banares R, et al Spanish Variceal Bleeding Study Group. Propranolol plus placebo versus propranolol plus isosorbide-5-mononitrate in the prevention of a first variceal bleed: a double-blind RCT. Hepatology 2003; 37:1260–1266.
- Garcia-Tsao G, Sanyal A, Grace N, et al. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology 2007; 46:922–938.
KEY POINTS
- The hepatic vein pressure gradient (HVPG) correlates well with the portal pressure and is easier to measure. However, whether it is cost-effective to measure the HVPG in clinical practice is controversial.
- Nonselective beta-blockers are the mainstay of treatment; selective beta-blockers do not reduce portal pressure to the same degree and are not recommended for preventing variceal bleeding.
- Endoscopic variceal ligation is an acceptable alternative to beta-blocker therapy for patients who cannot tolerate these drugs and for patients with varices at high risk of bleeding.
- Nitrates are no longer used as monotherapy for preventing variceal hemorrhage, and their use in combination with beta-blockers is controversial. Surgical portal decompression, transjugular intrahepatic portosystemic shunting, and endoscopic sclerotherapy are not recommended.
Should patients on long-term warfarin take aspirin for heart disease?
The literature on this topic is limited, but it suggests that the decision to prescribe aspirin to patients already taking warfarin (Coumadin) should be individualized. On one hand, the cardiovascular benefit of starting or continuing aspirin in patients already on warfarin outweighs the increased risk of bleeding in patients presenting with an acute coronary syndrome or those with mechanical heart valves or coronary stents. However, for patients with stable coronary artery disease or at risk of coronary disease, the benefit of adding aspirin is not substantial, and continuing warfarin alone may be the preferred strategy.
In patients with coronary artery disease, aspirin has been shown to reduce the rate of death due to all causes by about 18% and the rate of vascular events by about 25% to 30%.1,2 Warfarin is at least as effective as aspirin in reducing the rate of future cardiovascular events (especially if the target international normalized ratio [INR] is greater than 2.5), albeit with a higher bleeding risk.3–6
The decision to prescribe or continue aspirin in patients with coronary artery disease who also need long-term anticoagulation with warfarin for an unrelated medical problem, such as pulmonary emboli, requires careful assessment of the individual patient’s bleeding risk and cardiovascular benefit.
ESTIMATING THE BLEEDING RISK FOR PATIENTS ON WARFARIN
In patients taking warfarin, the risk of major bleeding (defined in most studies as hospitalization because of bleeding and requiring transfusion of at least two units of packed red cells, or an intracranial, intraperitoneal, or fatal bleeding episode) is reported to be about 2.0% to 3.8% per person-year.7–11 The risk of major bleeding with aspirin alone is estimated to be 0.13% per person-year,12 but when aspirin is combined with warfarin, the risk increases significantly.13 In a meta-analysis of randomized controlled trials,14 the risk of major bleeding was calculated to be about 1.5 times higher with combination therapy with aspirin and warfarin than with warfarin alone.
The individual’s bleeding risk depends on specific risk factors and the intensity of anticoagulation.15 The outpatient Bleeding Risk Index (BRI) can be used to estimate the bleeding risk for patients on warfarin.16 The BRI includes four risk factors for major bleeding, each scored as 1 point:
- Age 65 or older
- History of gastrointestinal bleeding
- History of stroke
- One or more comorbid conditions—recent myocardial infarction, anemia (hematocrit < 30%), renal impairment (serum creatinine level > 1.5 mg/dL), or diabetes mellitus.
The risk is low if the score is 0, moderate if the score is 1 or 2, and high if the score is 3 or more. In a validation study of the BRI, the rate of major bleeding was found to be 0.8%, 2.5%, and 10.6% per person-year on warfarin in the low, intermediate, and high-risk groups, respectively.17 In addition, compared with patients with a target INR of 2.5, those with a target INR higher than 3.0 have a higher frequency of bleeding episodes.10,15
CONDITIONS IN WHICH ADDING ASPIRIN TO WARFARIN IS FAVORABLE
Acute coronary syndromes
Drugs that inhibit platelet function are the mainstay of medical treatment for acute coronary syndromes. The American College of Cardiology/American Heart Association (ACC/AHA) guidelines recommend that aspirin be started in patients who have an acute myocardial infarction even if they have been receiving warfarin long-term and their INR is in the therapeutic range, especially if a percutaneous coronary intervention is anticipated.4
After percutaneous coronary intervention
In patients who have undergone percutaneous coronary intervention with stent implantation, dual antiplatelet therapy with aspirin and a thienopyridine—ie, clopidogrel (Plavix) or ticlopidine (Ticlid)—is superior to aspirin or warfarin alone in reducing the risk of stent thrombosis and major adverse cardiovascular events such as myocardial infarction or urgent revascularization.18,19 If patients have an indication for long-term anticoagulation, triple therapy with aspirin, warfarin, and clopidogrel or ticlopidine may be considered in order to reduce the likelihood of stent thrombosis.4,20,21 In such patients the INR should be maintained between 2.0 and 3.0 to reduce the risk of bleeding.
The duration of triple therapy is guided by the type of stent used. For bare metal stents, aspirin, clopidogrel or ticlopidine, and warfarin should be given for at least 1 month, after which clopidogrel or ticlopidine may be discontinued. If drug-eluting stents are used, the duration of clopidogrel or ticlopidine therapy should be extended to 1 year or more.4,22
Mechanical heart valves
In patients with mechanical heart valves, the combination of aspirin and warfarin has been shown to decrease the frequency of thromboembolism.23 Guidelines recommend adding aspirin (75 to 100 mg per day) to warfarin in all patients with mechanical valves, especially in patients who have had an embolus while on warfarin therapy or who have a history of cerebrovascular or peripheral vascular disease, a hypercoagulable state, or coronary artery disease.24
CONDITIONS IN WHICH WARFARIN ALONE MAY BE SUFFICIENT
At risk of coronary artery disease
Aspirin therapy is generally recommended as primary prevention for patients whose estimated risk of coronary events is 1.5% per year or higher.25 However, warfarin has also been shown to be effective in the primary prevention of coronary artery disease in men,26 and for patients already taking warfarin, the possible benefit of adding aspirin for primary prevention is outweighed by the increased risk of major bleeding.14 The Medical Research Council directly compared low-intensity warfarin therapy (mean INR 1.47), aspirin, and placebo in a two-by-two factorial study of primary prevention of ischemic heart disease in men.26 Warfarin was more effective than aspirin, and men who received warfarin plus aspirin or warfarin plus placebo had a rate of ischemic heart disease that was 21% lower than those who received aspirin plus placebo or double placebo, and their rate of all-cause mortality was 17% lower. Combining aspirin and warfarin for patients at risk of coronary disease led to a higher rate of major bleeding but no difference in cardiovascular events or all-cause mortality (odds ratio 0.98; 95% confidence interval 0.77–1.25).14
Stable coronary artery disease without mechanical heart valves or stents
Large randomized trials have found warfarin to be effective in secondary prevention of coronary artery disease.4–6 For most patients with stable coronary artery disease (ie, who have had no ischemic events or coronary interventions in the last 6 months) who need anticoagulation because of atrial fibrillation or venous thromboembolism, warfarin alone (target INR 2.0–3.0) should provide satisfactory antithrombotic prophylaxis against both cerebral and myocardial ischemic events.27 The addition of an antiplatelet agent is not required unless a patient has a coronary stent, a mechanical valve, or an excessive thrombotic risk.4,24,27
TAKE-HOME POINTS
For patients receiving warfarin therapy, whether to add or continue aspirin to their treatment is a common clinical question. The risk of bleeding is greater with combination therapy than with warfarin alone. The cardiovascular benefit varies depending on the clinical situation:
- In patients who have had an acute coronary syndrome or who have a coronary stent or mechanical valve, combination therapy is usually recommended because the benefits outweigh the risks.
- In patients with stable coronary artery disease or those without coronary artery disease who are at risk of coronary events, the risks outweigh the benefits. Combination therapy is usually not indicated in these patients.
- Weisman SM, Graham DY. Evaluation of the benefits and risks of low-dose aspirin in the secondary prevention of cardiovascular and cerebrovascular events. Arch Intern Med 2002; 162:2197–2202.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002; 347:969–974.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:E1–E211.
- Van Es RF, Jonker JJ, Verheugt FW, et al. Antithrombotics in the Secondary Prevention of Events in Coronary Thrombosis-2 (ASPECT-2) Research Group. Aspirin and coumadin after acute coronary syndromes (the ASPECT-2 study): a randomised controlled trial. Lancet 2002; 360:109–113.
- Anand SS, Yusuf S. Oral anticoagulant therapy in patients with coronary artery disease: a meta-analysis. JAMA 1999; 282:2058–2067.
- Schulman S, Granqvist S, Holmstrom M, et al. The duration of oral anticoagulant therapy after a second episode of venous thromboembolism. The Duration of Anticoagulation Trial Study Group. N Engl J Med 1997; 336:393–398.
- Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999; 340:901–907.
- Agnelli G, Prandoni P, Santamaria MG, et al. Three months versus one year of oral anticoagulant therapy for idiopathic deep venous thrombosis. Warfarin Optimal Duration Italian Trial Investigators. N Engl J Med 2001; 345:165–169.
- Levine MN, Raskob G, Beyth RJ, Kearon C, Schulman S. Hemorrhagic complications of anticoagulant treatment: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl:287S–310S.
- Linkins LA, Choi PT, Douketis JD. Clinical impact of bleeding in patients taking oral anticoagulant therapy for venous thromboembolism: a meta-analysis. Ann Intern Med 2003; 139:893–900.
- McQuaid KR, Laine L. Systematic review and meta-analysis of adverse events of low-dose aspirin and clopidogrel in randomized controlled trials. Am J Med 2006; 119:624–638.
- Rothberg MB, Celestin C, Fiore LD, Lawler E, Cook JR. Warfarin plus aspirin after myocardial infarction or the acute coronary syndrome: meta-analysis with estimates of risk and benefit. Ann Intern Med 2005; 143:241–250.
- Dentali F, Douketis JD, Lim W, Crowther M. Combined aspirin-oral anticoagulant therapy compared with oral anticoagulant therapy alone among patients at risk for cardiovascular disease: a meta-analysis of randomized trials. Arch Intern Med 2007; 167:117–124.
- Hirsh J, Fuster V, Ansell J, Halperin JL. American Heart Association; American College of Cardiology Foundation. American Heart Association/American College of Cardiology Foundation guide to warfarin therapy. Circulation 2003; 107:1692–1711.
- Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med 1998; 105:91–99.
- Aspinall SL, DeSanzo BE, Trilli LE, Good CB. Bleeding Risk Index in an anticoagulation clinic. Assessment by indication and implications for care. J Gen Intern Med 2005; 20:1008–1013.
- Mehta SR, Yusuf S, Peters RJ, et al. Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCICURE study. Lancet 2001; 358:527–533.
- Bertrand ME, Legrand V, Boland J, et al. Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elective coronary stenting. The Full Anticoagulation versus Aspirin and Ticlopidine (FANTASTIC) study. Circulation 1998; 98:1597–1603.
- Kushner FG, Antman EM. Oral anticoagulation for atrial fibrillation after ST-elevation myocardial infarction: new evidence to guide clinical practice. Circulation 2005; 112:3212–3214.
- Porter A, Konstantino Y, Iakobishvili Z, Shachar L, Battler A, Hasdai D. Short-term triple therapy with aspirin, warfarin, and a thienopyridine among patients undergoing percutaneous coronary intervention. Catheter Cardiovasc Interv 2006; 68:56–61.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 Guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction—executive summary. A report of the ACC-AHA Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction). J Am Coll Cardiol 2007; 50:652–726.
- Turpie AG, Gent M, Laupacis A, et al. A comparison of aspirin with placebo in patients treated with warfarin after heart-valve replacement. N Engl J Med 1993; 329:524–529.
- Bonow RO, Carabello BA, Kanu C, et al. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the ACC/AHA Task Force on Practice Guidelines (writing committee to revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease). Circulation 2006; 114:e84–e231.
- Lauer MS. Clinical practice. Aspirin for primary prevention of coronary events. N Engl J Med 2002; 346:1468–1474.
- The Medical Research Council’s General Practice Research Framework. Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. Lancet 1998; 351:233–241.
- Fuster V, Ryden LE, Cannom DS, et al. ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation. Circulation 2006; 114:260–335.
The literature on this topic is limited, but it suggests that the decision to prescribe aspirin to patients already taking warfarin (Coumadin) should be individualized. On one hand, the cardiovascular benefit of starting or continuing aspirin in patients already on warfarin outweighs the increased risk of bleeding in patients presenting with an acute coronary syndrome or those with mechanical heart valves or coronary stents. However, for patients with stable coronary artery disease or at risk of coronary disease, the benefit of adding aspirin is not substantial, and continuing warfarin alone may be the preferred strategy.
In patients with coronary artery disease, aspirin has been shown to reduce the rate of death due to all causes by about 18% and the rate of vascular events by about 25% to 30%.1,2 Warfarin is at least as effective as aspirin in reducing the rate of future cardiovascular events (especially if the target international normalized ratio [INR] is greater than 2.5), albeit with a higher bleeding risk.3–6
The decision to prescribe or continue aspirin in patients with coronary artery disease who also need long-term anticoagulation with warfarin for an unrelated medical problem, such as pulmonary emboli, requires careful assessment of the individual patient’s bleeding risk and cardiovascular benefit.
ESTIMATING THE BLEEDING RISK FOR PATIENTS ON WARFARIN
In patients taking warfarin, the risk of major bleeding (defined in most studies as hospitalization because of bleeding and requiring transfusion of at least two units of packed red cells, or an intracranial, intraperitoneal, or fatal bleeding episode) is reported to be about 2.0% to 3.8% per person-year.7–11 The risk of major bleeding with aspirin alone is estimated to be 0.13% per person-year,12 but when aspirin is combined with warfarin, the risk increases significantly.13 In a meta-analysis of randomized controlled trials,14 the risk of major bleeding was calculated to be about 1.5 times higher with combination therapy with aspirin and warfarin than with warfarin alone.
The individual’s bleeding risk depends on specific risk factors and the intensity of anticoagulation.15 The outpatient Bleeding Risk Index (BRI) can be used to estimate the bleeding risk for patients on warfarin.16 The BRI includes four risk factors for major bleeding, each scored as 1 point:
- Age 65 or older
- History of gastrointestinal bleeding
- History of stroke
- One or more comorbid conditions—recent myocardial infarction, anemia (hematocrit < 30%), renal impairment (serum creatinine level > 1.5 mg/dL), or diabetes mellitus.
The risk is low if the score is 0, moderate if the score is 1 or 2, and high if the score is 3 or more. In a validation study of the BRI, the rate of major bleeding was found to be 0.8%, 2.5%, and 10.6% per person-year on warfarin in the low, intermediate, and high-risk groups, respectively.17 In addition, compared with patients with a target INR of 2.5, those with a target INR higher than 3.0 have a higher frequency of bleeding episodes.10,15
CONDITIONS IN WHICH ADDING ASPIRIN TO WARFARIN IS FAVORABLE
Acute coronary syndromes
Drugs that inhibit platelet function are the mainstay of medical treatment for acute coronary syndromes. The American College of Cardiology/American Heart Association (ACC/AHA) guidelines recommend that aspirin be started in patients who have an acute myocardial infarction even if they have been receiving warfarin long-term and their INR is in the therapeutic range, especially if a percutaneous coronary intervention is anticipated.4
After percutaneous coronary intervention
In patients who have undergone percutaneous coronary intervention with stent implantation, dual antiplatelet therapy with aspirin and a thienopyridine—ie, clopidogrel (Plavix) or ticlopidine (Ticlid)—is superior to aspirin or warfarin alone in reducing the risk of stent thrombosis and major adverse cardiovascular events such as myocardial infarction or urgent revascularization.18,19 If patients have an indication for long-term anticoagulation, triple therapy with aspirin, warfarin, and clopidogrel or ticlopidine may be considered in order to reduce the likelihood of stent thrombosis.4,20,21 In such patients the INR should be maintained between 2.0 and 3.0 to reduce the risk of bleeding.
The duration of triple therapy is guided by the type of stent used. For bare metal stents, aspirin, clopidogrel or ticlopidine, and warfarin should be given for at least 1 month, after which clopidogrel or ticlopidine may be discontinued. If drug-eluting stents are used, the duration of clopidogrel or ticlopidine therapy should be extended to 1 year or more.4,22
Mechanical heart valves
In patients with mechanical heart valves, the combination of aspirin and warfarin has been shown to decrease the frequency of thromboembolism.23 Guidelines recommend adding aspirin (75 to 100 mg per day) to warfarin in all patients with mechanical valves, especially in patients who have had an embolus while on warfarin therapy or who have a history of cerebrovascular or peripheral vascular disease, a hypercoagulable state, or coronary artery disease.24
CONDITIONS IN WHICH WARFARIN ALONE MAY BE SUFFICIENT
At risk of coronary artery disease
Aspirin therapy is generally recommended as primary prevention for patients whose estimated risk of coronary events is 1.5% per year or higher.25 However, warfarin has also been shown to be effective in the primary prevention of coronary artery disease in men,26 and for patients already taking warfarin, the possible benefit of adding aspirin for primary prevention is outweighed by the increased risk of major bleeding.14 The Medical Research Council directly compared low-intensity warfarin therapy (mean INR 1.47), aspirin, and placebo in a two-by-two factorial study of primary prevention of ischemic heart disease in men.26 Warfarin was more effective than aspirin, and men who received warfarin plus aspirin or warfarin plus placebo had a rate of ischemic heart disease that was 21% lower than those who received aspirin plus placebo or double placebo, and their rate of all-cause mortality was 17% lower. Combining aspirin and warfarin for patients at risk of coronary disease led to a higher rate of major bleeding but no difference in cardiovascular events or all-cause mortality (odds ratio 0.98; 95% confidence interval 0.77–1.25).14
Stable coronary artery disease without mechanical heart valves or stents
Large randomized trials have found warfarin to be effective in secondary prevention of coronary artery disease.4–6 For most patients with stable coronary artery disease (ie, who have had no ischemic events or coronary interventions in the last 6 months) who need anticoagulation because of atrial fibrillation or venous thromboembolism, warfarin alone (target INR 2.0–3.0) should provide satisfactory antithrombotic prophylaxis against both cerebral and myocardial ischemic events.27 The addition of an antiplatelet agent is not required unless a patient has a coronary stent, a mechanical valve, or an excessive thrombotic risk.4,24,27
TAKE-HOME POINTS
For patients receiving warfarin therapy, whether to add or continue aspirin to their treatment is a common clinical question. The risk of bleeding is greater with combination therapy than with warfarin alone. The cardiovascular benefit varies depending on the clinical situation:
- In patients who have had an acute coronary syndrome or who have a coronary stent or mechanical valve, combination therapy is usually recommended because the benefits outweigh the risks.
- In patients with stable coronary artery disease or those without coronary artery disease who are at risk of coronary events, the risks outweigh the benefits. Combination therapy is usually not indicated in these patients.
The literature on this topic is limited, but it suggests that the decision to prescribe aspirin to patients already taking warfarin (Coumadin) should be individualized. On one hand, the cardiovascular benefit of starting or continuing aspirin in patients already on warfarin outweighs the increased risk of bleeding in patients presenting with an acute coronary syndrome or those with mechanical heart valves or coronary stents. However, for patients with stable coronary artery disease or at risk of coronary disease, the benefit of adding aspirin is not substantial, and continuing warfarin alone may be the preferred strategy.
In patients with coronary artery disease, aspirin has been shown to reduce the rate of death due to all causes by about 18% and the rate of vascular events by about 25% to 30%.1,2 Warfarin is at least as effective as aspirin in reducing the rate of future cardiovascular events (especially if the target international normalized ratio [INR] is greater than 2.5), albeit with a higher bleeding risk.3–6
The decision to prescribe or continue aspirin in patients with coronary artery disease who also need long-term anticoagulation with warfarin for an unrelated medical problem, such as pulmonary emboli, requires careful assessment of the individual patient’s bleeding risk and cardiovascular benefit.
ESTIMATING THE BLEEDING RISK FOR PATIENTS ON WARFARIN
In patients taking warfarin, the risk of major bleeding (defined in most studies as hospitalization because of bleeding and requiring transfusion of at least two units of packed red cells, or an intracranial, intraperitoneal, or fatal bleeding episode) is reported to be about 2.0% to 3.8% per person-year.7–11 The risk of major bleeding with aspirin alone is estimated to be 0.13% per person-year,12 but when aspirin is combined with warfarin, the risk increases significantly.13 In a meta-analysis of randomized controlled trials,14 the risk of major bleeding was calculated to be about 1.5 times higher with combination therapy with aspirin and warfarin than with warfarin alone.
The individual’s bleeding risk depends on specific risk factors and the intensity of anticoagulation.15 The outpatient Bleeding Risk Index (BRI) can be used to estimate the bleeding risk for patients on warfarin.16 The BRI includes four risk factors for major bleeding, each scored as 1 point:
- Age 65 or older
- History of gastrointestinal bleeding
- History of stroke
- One or more comorbid conditions—recent myocardial infarction, anemia (hematocrit < 30%), renal impairment (serum creatinine level > 1.5 mg/dL), or diabetes mellitus.
The risk is low if the score is 0, moderate if the score is 1 or 2, and high if the score is 3 or more. In a validation study of the BRI, the rate of major bleeding was found to be 0.8%, 2.5%, and 10.6% per person-year on warfarin in the low, intermediate, and high-risk groups, respectively.17 In addition, compared with patients with a target INR of 2.5, those with a target INR higher than 3.0 have a higher frequency of bleeding episodes.10,15
CONDITIONS IN WHICH ADDING ASPIRIN TO WARFARIN IS FAVORABLE
Acute coronary syndromes
Drugs that inhibit platelet function are the mainstay of medical treatment for acute coronary syndromes. The American College of Cardiology/American Heart Association (ACC/AHA) guidelines recommend that aspirin be started in patients who have an acute myocardial infarction even if they have been receiving warfarin long-term and their INR is in the therapeutic range, especially if a percutaneous coronary intervention is anticipated.4
After percutaneous coronary intervention
In patients who have undergone percutaneous coronary intervention with stent implantation, dual antiplatelet therapy with aspirin and a thienopyridine—ie, clopidogrel (Plavix) or ticlopidine (Ticlid)—is superior to aspirin or warfarin alone in reducing the risk of stent thrombosis and major adverse cardiovascular events such as myocardial infarction or urgent revascularization.18,19 If patients have an indication for long-term anticoagulation, triple therapy with aspirin, warfarin, and clopidogrel or ticlopidine may be considered in order to reduce the likelihood of stent thrombosis.4,20,21 In such patients the INR should be maintained between 2.0 and 3.0 to reduce the risk of bleeding.
The duration of triple therapy is guided by the type of stent used. For bare metal stents, aspirin, clopidogrel or ticlopidine, and warfarin should be given for at least 1 month, after which clopidogrel or ticlopidine may be discontinued. If drug-eluting stents are used, the duration of clopidogrel or ticlopidine therapy should be extended to 1 year or more.4,22
Mechanical heart valves
In patients with mechanical heart valves, the combination of aspirin and warfarin has been shown to decrease the frequency of thromboembolism.23 Guidelines recommend adding aspirin (75 to 100 mg per day) to warfarin in all patients with mechanical valves, especially in patients who have had an embolus while on warfarin therapy or who have a history of cerebrovascular or peripheral vascular disease, a hypercoagulable state, or coronary artery disease.24
CONDITIONS IN WHICH WARFARIN ALONE MAY BE SUFFICIENT
At risk of coronary artery disease
Aspirin therapy is generally recommended as primary prevention for patients whose estimated risk of coronary events is 1.5% per year or higher.25 However, warfarin has also been shown to be effective in the primary prevention of coronary artery disease in men,26 and for patients already taking warfarin, the possible benefit of adding aspirin for primary prevention is outweighed by the increased risk of major bleeding.14 The Medical Research Council directly compared low-intensity warfarin therapy (mean INR 1.47), aspirin, and placebo in a two-by-two factorial study of primary prevention of ischemic heart disease in men.26 Warfarin was more effective than aspirin, and men who received warfarin plus aspirin or warfarin plus placebo had a rate of ischemic heart disease that was 21% lower than those who received aspirin plus placebo or double placebo, and their rate of all-cause mortality was 17% lower. Combining aspirin and warfarin for patients at risk of coronary disease led to a higher rate of major bleeding but no difference in cardiovascular events or all-cause mortality (odds ratio 0.98; 95% confidence interval 0.77–1.25).14
Stable coronary artery disease without mechanical heart valves or stents
Large randomized trials have found warfarin to be effective in secondary prevention of coronary artery disease.4–6 For most patients with stable coronary artery disease (ie, who have had no ischemic events or coronary interventions in the last 6 months) who need anticoagulation because of atrial fibrillation or venous thromboembolism, warfarin alone (target INR 2.0–3.0) should provide satisfactory antithrombotic prophylaxis against both cerebral and myocardial ischemic events.27 The addition of an antiplatelet agent is not required unless a patient has a coronary stent, a mechanical valve, or an excessive thrombotic risk.4,24,27
TAKE-HOME POINTS
For patients receiving warfarin therapy, whether to add or continue aspirin to their treatment is a common clinical question. The risk of bleeding is greater with combination therapy than with warfarin alone. The cardiovascular benefit varies depending on the clinical situation:
- In patients who have had an acute coronary syndrome or who have a coronary stent or mechanical valve, combination therapy is usually recommended because the benefits outweigh the risks.
- In patients with stable coronary artery disease or those without coronary artery disease who are at risk of coronary events, the risks outweigh the benefits. Combination therapy is usually not indicated in these patients.
- Weisman SM, Graham DY. Evaluation of the benefits and risks of low-dose aspirin in the secondary prevention of cardiovascular and cerebrovascular events. Arch Intern Med 2002; 162:2197–2202.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002; 347:969–974.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:E1–E211.
- Van Es RF, Jonker JJ, Verheugt FW, et al. Antithrombotics in the Secondary Prevention of Events in Coronary Thrombosis-2 (ASPECT-2) Research Group. Aspirin and coumadin after acute coronary syndromes (the ASPECT-2 study): a randomised controlled trial. Lancet 2002; 360:109–113.
- Anand SS, Yusuf S. Oral anticoagulant therapy in patients with coronary artery disease: a meta-analysis. JAMA 1999; 282:2058–2067.
- Schulman S, Granqvist S, Holmstrom M, et al. The duration of oral anticoagulant therapy after a second episode of venous thromboembolism. The Duration of Anticoagulation Trial Study Group. N Engl J Med 1997; 336:393–398.
- Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999; 340:901–907.
- Agnelli G, Prandoni P, Santamaria MG, et al. Three months versus one year of oral anticoagulant therapy for idiopathic deep venous thrombosis. Warfarin Optimal Duration Italian Trial Investigators. N Engl J Med 2001; 345:165–169.
- Levine MN, Raskob G, Beyth RJ, Kearon C, Schulman S. Hemorrhagic complications of anticoagulant treatment: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl:287S–310S.
- Linkins LA, Choi PT, Douketis JD. Clinical impact of bleeding in patients taking oral anticoagulant therapy for venous thromboembolism: a meta-analysis. Ann Intern Med 2003; 139:893–900.
- McQuaid KR, Laine L. Systematic review and meta-analysis of adverse events of low-dose aspirin and clopidogrel in randomized controlled trials. Am J Med 2006; 119:624–638.
- Rothberg MB, Celestin C, Fiore LD, Lawler E, Cook JR. Warfarin plus aspirin after myocardial infarction or the acute coronary syndrome: meta-analysis with estimates of risk and benefit. Ann Intern Med 2005; 143:241–250.
- Dentali F, Douketis JD, Lim W, Crowther M. Combined aspirin-oral anticoagulant therapy compared with oral anticoagulant therapy alone among patients at risk for cardiovascular disease: a meta-analysis of randomized trials. Arch Intern Med 2007; 167:117–124.
- Hirsh J, Fuster V, Ansell J, Halperin JL. American Heart Association; American College of Cardiology Foundation. American Heart Association/American College of Cardiology Foundation guide to warfarin therapy. Circulation 2003; 107:1692–1711.
- Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med 1998; 105:91–99.
- Aspinall SL, DeSanzo BE, Trilli LE, Good CB. Bleeding Risk Index in an anticoagulation clinic. Assessment by indication and implications for care. J Gen Intern Med 2005; 20:1008–1013.
- Mehta SR, Yusuf S, Peters RJ, et al. Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCICURE study. Lancet 2001; 358:527–533.
- Bertrand ME, Legrand V, Boland J, et al. Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elective coronary stenting. The Full Anticoagulation versus Aspirin and Ticlopidine (FANTASTIC) study. Circulation 1998; 98:1597–1603.
- Kushner FG, Antman EM. Oral anticoagulation for atrial fibrillation after ST-elevation myocardial infarction: new evidence to guide clinical practice. Circulation 2005; 112:3212–3214.
- Porter A, Konstantino Y, Iakobishvili Z, Shachar L, Battler A, Hasdai D. Short-term triple therapy with aspirin, warfarin, and a thienopyridine among patients undergoing percutaneous coronary intervention. Catheter Cardiovasc Interv 2006; 68:56–61.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 Guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction—executive summary. A report of the ACC-AHA Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction). J Am Coll Cardiol 2007; 50:652–726.
- Turpie AG, Gent M, Laupacis A, et al. A comparison of aspirin with placebo in patients treated with warfarin after heart-valve replacement. N Engl J Med 1993; 329:524–529.
- Bonow RO, Carabello BA, Kanu C, et al. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the ACC/AHA Task Force on Practice Guidelines (writing committee to revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease). Circulation 2006; 114:e84–e231.
- Lauer MS. Clinical practice. Aspirin for primary prevention of coronary events. N Engl J Med 2002; 346:1468–1474.
- The Medical Research Council’s General Practice Research Framework. Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. Lancet 1998; 351:233–241.
- Fuster V, Ryden LE, Cannom DS, et al. ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation. Circulation 2006; 114:260–335.
- Weisman SM, Graham DY. Evaluation of the benefits and risks of low-dose aspirin in the secondary prevention of cardiovascular and cerebrovascular events. Arch Intern Med 2002; 162:2197–2202.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002; 347:969–974.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:E1–E211.
- Van Es RF, Jonker JJ, Verheugt FW, et al. Antithrombotics in the Secondary Prevention of Events in Coronary Thrombosis-2 (ASPECT-2) Research Group. Aspirin and coumadin after acute coronary syndromes (the ASPECT-2 study): a randomised controlled trial. Lancet 2002; 360:109–113.
- Anand SS, Yusuf S. Oral anticoagulant therapy in patients with coronary artery disease: a meta-analysis. JAMA 1999; 282:2058–2067.
- Schulman S, Granqvist S, Holmstrom M, et al. The duration of oral anticoagulant therapy after a second episode of venous thromboembolism. The Duration of Anticoagulation Trial Study Group. N Engl J Med 1997; 336:393–398.
- Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999; 340:901–907.
- Agnelli G, Prandoni P, Santamaria MG, et al. Three months versus one year of oral anticoagulant therapy for idiopathic deep venous thrombosis. Warfarin Optimal Duration Italian Trial Investigators. N Engl J Med 2001; 345:165–169.
- Levine MN, Raskob G, Beyth RJ, Kearon C, Schulman S. Hemorrhagic complications of anticoagulant treatment: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl:287S–310S.
- Linkins LA, Choi PT, Douketis JD. Clinical impact of bleeding in patients taking oral anticoagulant therapy for venous thromboembolism: a meta-analysis. Ann Intern Med 2003; 139:893–900.
- McQuaid KR, Laine L. Systematic review and meta-analysis of adverse events of low-dose aspirin and clopidogrel in randomized controlled trials. Am J Med 2006; 119:624–638.
- Rothberg MB, Celestin C, Fiore LD, Lawler E, Cook JR. Warfarin plus aspirin after myocardial infarction or the acute coronary syndrome: meta-analysis with estimates of risk and benefit. Ann Intern Med 2005; 143:241–250.
- Dentali F, Douketis JD, Lim W, Crowther M. Combined aspirin-oral anticoagulant therapy compared with oral anticoagulant therapy alone among patients at risk for cardiovascular disease: a meta-analysis of randomized trials. Arch Intern Med 2007; 167:117–124.
- Hirsh J, Fuster V, Ansell J, Halperin JL. American Heart Association; American College of Cardiology Foundation. American Heart Association/American College of Cardiology Foundation guide to warfarin therapy. Circulation 2003; 107:1692–1711.
- Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med 1998; 105:91–99.
- Aspinall SL, DeSanzo BE, Trilli LE, Good CB. Bleeding Risk Index in an anticoagulation clinic. Assessment by indication and implications for care. J Gen Intern Med 2005; 20:1008–1013.
- Mehta SR, Yusuf S, Peters RJ, et al. Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCICURE study. Lancet 2001; 358:527–533.
- Bertrand ME, Legrand V, Boland J, et al. Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elective coronary stenting. The Full Anticoagulation versus Aspirin and Ticlopidine (FANTASTIC) study. Circulation 1998; 98:1597–1603.
- Kushner FG, Antman EM. Oral anticoagulation for atrial fibrillation after ST-elevation myocardial infarction: new evidence to guide clinical practice. Circulation 2005; 112:3212–3214.
- Porter A, Konstantino Y, Iakobishvili Z, Shachar L, Battler A, Hasdai D. Short-term triple therapy with aspirin, warfarin, and a thienopyridine among patients undergoing percutaneous coronary intervention. Catheter Cardiovasc Interv 2006; 68:56–61.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 Guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction—executive summary. A report of the ACC-AHA Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction). J Am Coll Cardiol 2007; 50:652–726.
- Turpie AG, Gent M, Laupacis A, et al. A comparison of aspirin with placebo in patients treated with warfarin after heart-valve replacement. N Engl J Med 1993; 329:524–529.
- Bonow RO, Carabello BA, Kanu C, et al. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the ACC/AHA Task Force on Practice Guidelines (writing committee to revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease). Circulation 2006; 114:e84–e231.
- Lauer MS. Clinical practice. Aspirin for primary prevention of coronary events. N Engl J Med 2002; 346:1468–1474.
- The Medical Research Council’s General Practice Research Framework. Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. Lancet 1998; 351:233–241.
- Fuster V, Ryden LE, Cannom DS, et al. ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation. Circulation 2006; 114:260–335.
What are the caveats to using sodium phosphate agents for bowel preparation?
Sodium phosphate (NaP) agents were introduced to provide a gentler alternative to polyethylene glycol (PEG) bowel preparations, which require patients to drink up to 4 liters of fluid over a few hours.
However, in May 2006 the US Food and Drug Administration (FDA) issued an alert that NaP products for bowel cleansing may, in some patients, pose a risk of acute phosphate nephropathy, a rare form of acute renal failure.
Although NaP preparations are generally safe and well tolerated, they can cause significant fluid shifts and electrolyte abnormalities. As such, they should not be used in patients with baseline electrolyte imbalances, renal or hepatic dysfunction, or a number of other comorbidities.
CURRENT BOWEL-CLEANSING OPTIONS
For many years the standard preparation for bowel cleansing was a 4-liter or a 2-liter PEG electrolyte solution plus a laxative (eg, magnesium citrate, bisacodyl, or senna).1–3 The most frequent complaint heard from patients was that “the preparation is worse than the colonoscopy,” attributable to the taste and volume of the fluid they had to consume. Thus, compliance was often a significant issue with patients presenting for colonoscopy. In fact, inadequate bowel preparation is one of the most common reasons polyps are missed during colonoscopy.
Aqueous and tablet forms of NaP (sometimes with a laxative) have become a widely used alternative to PEG solutions because they require much less volume and as a result are more palatable, thereby improving compliance.4,5
NaP agents cleanse the colon by osmotically drawing plasma water into the bowel lumen. The patient must drink significant amounts of water or other oral solutions to prevent dehydration.
NaP-based bowel-cleansing agents are available in two forms: aqueous solution and tablet. Aqueous NaP (such as Fleet Phospho-soda) is a low-volume hyperosmotic solution containing 48 g of monobasic NaP and 18 g of dibasic NaP per 100 mL.6 An oral tablet form (such as Visicol and OsmoPrep) was developed to improve patient tolerance.7 Each 2-g tablet of Visicol contains 1,500 mg of active ingredients (monobasic and dibasic NaP) and 460 mg of microcrystalline cellulose, an inert polymer. Each OsmoPrep tablet contains 1,500 mg of the same active ingredients as Visicol, but the inert ingredients include PEG and magnesium stearate.
At first, the regimen was 40 tablets such as Visicol to be taken with water and bisacodyl. Subsequent regimens such as OsmoPrep with fewer tablets have been shown to be as effective and better tolerated.8 Microcrystalline cellulose in the tablet can produce a residue that may obscure the bowel mucosa. Newer preparations contain lower amounts of this inert ingredient, allowing for improved visualization of the colonic mucosa during colonoscopy.9
ADVANTAGES OF SODIUM PHOSPHATE BOWEL CLEANSERS
In a recent review article, Burke and Church10 noted that NaP cleansing regimens have been shown to be superior to PEG-electrolyte lavage solution with respect to tolerability and acceptance by patients, improved quality of bowel preparation, better mucosal visualization, and more efficient endoscopic examination. In addition, the volume of the preparation may also help decrease the risk of aspiration in some patients.2,3
DISADVANTAGES OF SODIUM PHOSPHATE AGENTS
Despite their comparable or better efficacy and their better tolerability, NaP agents have certain disadvantages.
Effects on the colonic mucosa
In rare cases NaP agents have been shown to alter the microscopic and macroscopic features of the colonic mucosa, and they can induce aphthoid erosions that may mimic those seen in inflammatory bowel disease and enteropathy or colopathy associated with nonsteroidal anti-inflammatory drugs (NSAIDs).11–13 Therefore, NaP agents should not be used prior to the initial endoscopic evaluation of patients with suspected inflammatory bowel disease, microscopic colitis, or NSAID-induced colonopathy.
Fluid and electrolyte shifts
Because NaP acts by drawing plasma water into the bowel lumen, significant volume and electrolyte shifts may occur.14,15 These can cause hypokalemia, hyperphosphatemia, hypocalcemia, hyponatremia or hypernatremia, hypomagnesemia, elevated blood urea nitrogen levels, decreased exercise capacity, increased plasma osmolarity,15–17 seizures,18 and acute renal failure with or without nephrocalcinosis.17,19–21
Thus, patients with significant comorbidities—such as a recent history of myocardial infarction, renal or hepatic insufficiency, or malnutrition—should not use NaP agents.22
Pivotal study of adverse events
In May 2006, the FDA issued an alert outlining the concerns of using oral NaP in specific patient populations. Of note were documented cases of acute phosphate nephropathy in 21 patients who used aqueous NaP (Fleet Phospho-Soda or Fleet Accu-Prep), and in 1 patient who used NaP tablets (Visicol).23 Acute renal injury was not limited to patients with preexisting renal insufficiency. It is uncertain whether this means that otherwise healthy people suffered renal injury or had risk factors besides renal insufficiency, since the data cited by the FDA report do not elucidate the possible risk factors for the development of nephropathy in patients with no preexisting renal insufficiency. So far, no cases of acute phosphate nephropathy or acute renal failure have been reported with OsmoPrep, a NaP tablet bowel preparation recently approved by the FDA.24 The long-term safety of OsmoPrep needs further evaluation.
PROCEED WITH CAUTION
Certain situations such as advanced age and cardiac, renal, and hepatic dysfunction call for extreme caution in the use of NaP bowel preparation agents. Therefore, it is recommended that patients with the following conditions should avoid using NaP agents for colon preparation:
- Hepatic or renal insufficiency (there are no data as to the degree of hepatic or renal insufficiency)
- Congestive heart failure
- Over age 65
- Dehydration or hypercalcemia
- Chronic use of drugs that affect renal perfusion, such as NSAIDs, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, or diuretics for hypertension.
Patients who take diuretics should not take them while they are using NaP for bowel preparation because of the risk of electrolyte abnormalities such as hypokalemia. In patients who have no alternative but to proceed with NaP preparation, our recommendation would be that the patient hold off taking diuretics, ACE inhibitors, and angiotensin receptor blockers while using the NaP prep. Given the importance of these medications in controlling diseases such as hypertension, the physician and the patient should jointly determine whether the benefits of using an NaP agent justify holding these drugs. We believe that patients taking these drugs should try using a PEG solution before considering NaP.
TASK FORCE GUIDELINES
Guidelines for using NaP bowel preparation agents, published by a task force of the American Society of Colon and Rectal Surgeons, the American Society for Gastrointestinal Endoscopy, and the Society of American Gastrointestinal and Endoscopic Surgeons,25 include the following caveats:
- Aqueous and tablet NaP colonic preparations are an alternative to PEG solutions, except in pediatric populations, patients over age 65, and those with bowel obstruction or other structural intestinal disorder, gut dysmotility, renal or hepatic insufficiency, congestive heart failure, or seizure disorder.
- Dosing should be 45 mL in divided doses, 10 to 12 hours apart, with at least one dose taken on the morning of the procedure.25
- The significant volume contraction and resulting dehydration seen in some patients using NaP preparations may be lessened by encouraging patients to drink fluids liberally during the days leading up to their procedure, and especially during NaP bowel preparation.26
- NaP tablets should be dosed as 32 to 40 tablets. On the evening before the procedure the patient should take 20 tablets and then 12 to 20 tablets approximately 3 to 5 hours before undergoing endoscopy. The tablets should be taken four at a time every 15 minutes with approximately 8 oz of clear liquid.25

- Sharma VK, Chockalingham SK, Ugheoke EA, et al. Prospective, randomized, controlled comparison of the use of polyethylene glycol electrolyte lavage solution in four-liter versus two-liter volumes and pretreatment with either magnesium citrate or bisacodyl for colonoscopy preparation. Gastrointest Endosc 1998; 47:167–171.
- Frommer D. Cleansing ability and tolerance of three bowel preparations for colonoscopy. Dis Colon Rectum 1997; 40:100–104.
- Hsu CW, Imperiale TF. Meta-analysis and cost comparison of polyethylene glycol lavage versus sodium phosphate for colonoscopy preparation. Gastrointest Endosc 1998; 48:276–282.
- Poon CM, Lee DWH, Mak SK, et al. Two liters of polyethylene glycol-electrolyte solution versus sodium phosphate as bowel cleansing regimen for colonoscopy: a prospective randomized controlled trial. Endoscopy 2002; 34:560–563.
- Afridi SA, Barthel JS, King PD, et al. Prospective, randomized trial comparing a new sodium phosphate-bisacodyl regimen with conventional PEG-ES lavage for outpatient colonoscopy preparation. Gastrointest Endosc 1995; 41:485–489.
- Schiller LR. Clinical pharmacology and use of laxatives and lavage solutions. J Clin Gastroenterol 1988; 28:11–18.
- Kastenberg D, Chasen R, Choudhary C, et al. Efficacy and safety of sodium phosphate tablets compared with PEG solution in colon cleansing. Two identically designed, randomized, controlled, parallel group multicenter phase III trials. Gastrointest Endosc 2001; 54:705–713.
- Rex DK, Chasen R, Pushpin MB. Safety and efficacy of two reduced dosing regimens of sodium phosphate tablets for preparation prior to colonoscopy. Aliment Pharmacol Ther 2002; 16:937–944.
- Rex DK, Khashab M. Efficacy and tolerability of a new formulation of sodium phosphate tablets and a reduced sodium phosphate dose, in colon cleansing: a single-center open-label pilot trial. Aliment Pharmacol Ther 2005; 21:465–468.
- Burke CA, Church JM. Enhancing the quality of colonoscopy: the importance of bowel purgatives. Gastrointest Endosc 2007; 66:565–573.
- Rejchrt S, Bures J, Siroky M, et al. A prospective, observational study of colonic mucosal abnormalities associated with orally administered sodium phosphate for colon cleansing before colonoscopy. Gastrointest Endosc 2004; 59:651–654.
- Hixson LJ. Colorectal ulcers associated with sodium phosphate catharsis. Gastrointest Endosc 1995; 42:101–102.
- Zwas FR, Cirillo NW, El-Serag HB, Eisen RN. Colonic mucosal abnormalities associated with oral sodium phosphate solution. Gastrointest Endosc 1996; 43:463–466.
- Clarkston WK, Tsen TN, Dies DF, Schratz CL, Vaswani SK, Bjerregaard P. Oral sodium phosphate versus sulfate-free polyethylene glycol electrolyte lavage solution in outpatient preparation for colonoscopy: a prospective comparison. Gastrointest Endosc 1996; 43:42–48.
- Kolts BE, Lyles WE, Achem SR, et al. A comparison of the effectiveness and patient tolerance of oral sodium phosphate, castor oil, and standard electrolyte lavage for colonoscopy or sigmoidoscopy preparations. Am J Gastroenterol 1993; 88:1218–1223.
- Holte K, Neilsen KG, Madsen JL, Kehlet H. Physiologic effects of bowel preparation. Dis Colon Rectum 2004; 47:1397–1402.
- Clarkston WK, Tsen TN, Dies DF, et al. Oral sodium phosphate versus sulfate-free polyethylene glycol electrolyte lavage solution in outpatient preparation for colonoscopy: a prospective comparison. Gastrointest Endosc 1996; 43:42–48.
- Frizelle FA, Colls BM. Hyponatremia and seizures after bowel preparation: report of three cases. Dis Colon Rectum 2005; 48:393–396.
- Markowitz GS, Nasr SH, Klein P, et al. Renal failure due to acute nephrocalcinosis following oral sodium phosphate bowel cleansing. Hum Pathol 2004; 35:675–684.
- Lieberman DA, Ghormley J, Flora K. Effect of oral sodium phosphate colon preparation on serum electrolytes in patients with normal serum creatinine. Gastrointest Endosc 1996; 43:467–469.
- Gremse DA, Sacks AI, Raines S. Comparison of oral sodium phosphate to polyethylene-glycol-based solution for bowel preparation in children. J Pediatric Gastroenterol Nutr 1996; 23:586–590.
- Curran MP, Plosker GL. Oral sodium phosphate solution: a review of its use as a colonic cleanser. Drugs 2004; 64:1697–1714.
- Markowitz GS, Stokes MB, Radhakrishnan J, D’Agati VD. Acute phosphate nephropathy following oral sodium phosphate bowel purgative: an underrecognized cause of chronic renal failure. J Am Soc Nephrol 2005; 16:3389–3396.
- FDA Alert. Patient information sheet. Oral sodium phosphate (OSP) products for bowel cleansing. 2006 May, Accessed January 8, 2008. www.fda.gov/CDER/drug/InfoSheets/patient/OSP_solutionPIS.htm.
- Wexner SD, Beck DE, Baron TH, et al. A consensus document on bowel preparation before colonoscopy prepared by a task force from the American Society of Colon and Rectal Surgeons (ASCRS), the American Society for Gastrointestinal Endoscopy (ASGE), and the Society of American Gastrointestinal and Endoscopic Surgeons (SAGES). Gastrointest Endosc 2006; 63:894–909.
- Huynh T, Vanner S, Paterson W. Safety profile of 5-h oral sodium phosphate regimen for colonoscopy cleansing: lack of clinically significant hypocalcemia or hypovolemia. Am J Gastroenterol 1995; 90:104–107.
Sodium phosphate (NaP) agents were introduced to provide a gentler alternative to polyethylene glycol (PEG) bowel preparations, which require patients to drink up to 4 liters of fluid over a few hours.
However, in May 2006 the US Food and Drug Administration (FDA) issued an alert that NaP products for bowel cleansing may, in some patients, pose a risk of acute phosphate nephropathy, a rare form of acute renal failure.
Although NaP preparations are generally safe and well tolerated, they can cause significant fluid shifts and electrolyte abnormalities. As such, they should not be used in patients with baseline electrolyte imbalances, renal or hepatic dysfunction, or a number of other comorbidities.
CURRENT BOWEL-CLEANSING OPTIONS
For many years the standard preparation for bowel cleansing was a 4-liter or a 2-liter PEG electrolyte solution plus a laxative (eg, magnesium citrate, bisacodyl, or senna).1–3 The most frequent complaint heard from patients was that “the preparation is worse than the colonoscopy,” attributable to the taste and volume of the fluid they had to consume. Thus, compliance was often a significant issue with patients presenting for colonoscopy. In fact, inadequate bowel preparation is one of the most common reasons polyps are missed during colonoscopy.
Aqueous and tablet forms of NaP (sometimes with a laxative) have become a widely used alternative to PEG solutions because they require much less volume and as a result are more palatable, thereby improving compliance.4,5
NaP agents cleanse the colon by osmotically drawing plasma water into the bowel lumen. The patient must drink significant amounts of water or other oral solutions to prevent dehydration.
NaP-based bowel-cleansing agents are available in two forms: aqueous solution and tablet. Aqueous NaP (such as Fleet Phospho-soda) is a low-volume hyperosmotic solution containing 48 g of monobasic NaP and 18 g of dibasic NaP per 100 mL.6 An oral tablet form (such as Visicol and OsmoPrep) was developed to improve patient tolerance.7 Each 2-g tablet of Visicol contains 1,500 mg of active ingredients (monobasic and dibasic NaP) and 460 mg of microcrystalline cellulose, an inert polymer. Each OsmoPrep tablet contains 1,500 mg of the same active ingredients as Visicol, but the inert ingredients include PEG and magnesium stearate.
At first, the regimen was 40 tablets such as Visicol to be taken with water and bisacodyl. Subsequent regimens such as OsmoPrep with fewer tablets have been shown to be as effective and better tolerated.8 Microcrystalline cellulose in the tablet can produce a residue that may obscure the bowel mucosa. Newer preparations contain lower amounts of this inert ingredient, allowing for improved visualization of the colonic mucosa during colonoscopy.9
ADVANTAGES OF SODIUM PHOSPHATE BOWEL CLEANSERS
In a recent review article, Burke and Church10 noted that NaP cleansing regimens have been shown to be superior to PEG-electrolyte lavage solution with respect to tolerability and acceptance by patients, improved quality of bowel preparation, better mucosal visualization, and more efficient endoscopic examination. In addition, the volume of the preparation may also help decrease the risk of aspiration in some patients.2,3
DISADVANTAGES OF SODIUM PHOSPHATE AGENTS
Despite their comparable or better efficacy and their better tolerability, NaP agents have certain disadvantages.
Effects on the colonic mucosa
In rare cases NaP agents have been shown to alter the microscopic and macroscopic features of the colonic mucosa, and they can induce aphthoid erosions that may mimic those seen in inflammatory bowel disease and enteropathy or colopathy associated with nonsteroidal anti-inflammatory drugs (NSAIDs).11–13 Therefore, NaP agents should not be used prior to the initial endoscopic evaluation of patients with suspected inflammatory bowel disease, microscopic colitis, or NSAID-induced colonopathy.
Fluid and electrolyte shifts
Because NaP acts by drawing plasma water into the bowel lumen, significant volume and electrolyte shifts may occur.14,15 These can cause hypokalemia, hyperphosphatemia, hypocalcemia, hyponatremia or hypernatremia, hypomagnesemia, elevated blood urea nitrogen levels, decreased exercise capacity, increased plasma osmolarity,15–17 seizures,18 and acute renal failure with or without nephrocalcinosis.17,19–21
Thus, patients with significant comorbidities—such as a recent history of myocardial infarction, renal or hepatic insufficiency, or malnutrition—should not use NaP agents.22
Pivotal study of adverse events
In May 2006, the FDA issued an alert outlining the concerns of using oral NaP in specific patient populations. Of note were documented cases of acute phosphate nephropathy in 21 patients who used aqueous NaP (Fleet Phospho-Soda or Fleet Accu-Prep), and in 1 patient who used NaP tablets (Visicol).23 Acute renal injury was not limited to patients with preexisting renal insufficiency. It is uncertain whether this means that otherwise healthy people suffered renal injury or had risk factors besides renal insufficiency, since the data cited by the FDA report do not elucidate the possible risk factors for the development of nephropathy in patients with no preexisting renal insufficiency. So far, no cases of acute phosphate nephropathy or acute renal failure have been reported with OsmoPrep, a NaP tablet bowel preparation recently approved by the FDA.24 The long-term safety of OsmoPrep needs further evaluation.
PROCEED WITH CAUTION
Certain situations such as advanced age and cardiac, renal, and hepatic dysfunction call for extreme caution in the use of NaP bowel preparation agents. Therefore, it is recommended that patients with the following conditions should avoid using NaP agents for colon preparation:
- Hepatic or renal insufficiency (there are no data as to the degree of hepatic or renal insufficiency)
- Congestive heart failure
- Over age 65
- Dehydration or hypercalcemia
- Chronic use of drugs that affect renal perfusion, such as NSAIDs, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, or diuretics for hypertension.
Patients who take diuretics should not take them while they are using NaP for bowel preparation because of the risk of electrolyte abnormalities such as hypokalemia. In patients who have no alternative but to proceed with NaP preparation, our recommendation would be that the patient hold off taking diuretics, ACE inhibitors, and angiotensin receptor blockers while using the NaP prep. Given the importance of these medications in controlling diseases such as hypertension, the physician and the patient should jointly determine whether the benefits of using an NaP agent justify holding these drugs. We believe that patients taking these drugs should try using a PEG solution before considering NaP.
TASK FORCE GUIDELINES
Guidelines for using NaP bowel preparation agents, published by a task force of the American Society of Colon and Rectal Surgeons, the American Society for Gastrointestinal Endoscopy, and the Society of American Gastrointestinal and Endoscopic Surgeons,25 include the following caveats:
- Aqueous and tablet NaP colonic preparations are an alternative to PEG solutions, except in pediatric populations, patients over age 65, and those with bowel obstruction or other structural intestinal disorder, gut dysmotility, renal or hepatic insufficiency, congestive heart failure, or seizure disorder.
- Dosing should be 45 mL in divided doses, 10 to 12 hours apart, with at least one dose taken on the morning of the procedure.25
- The significant volume contraction and resulting dehydration seen in some patients using NaP preparations may be lessened by encouraging patients to drink fluids liberally during the days leading up to their procedure, and especially during NaP bowel preparation.26
- NaP tablets should be dosed as 32 to 40 tablets. On the evening before the procedure the patient should take 20 tablets and then 12 to 20 tablets approximately 3 to 5 hours before undergoing endoscopy. The tablets should be taken four at a time every 15 minutes with approximately 8 oz of clear liquid.25
Sodium phosphate (NaP) agents were introduced to provide a gentler alternative to polyethylene glycol (PEG) bowel preparations, which require patients to drink up to 4 liters of fluid over a few hours.
However, in May 2006 the US Food and Drug Administration (FDA) issued an alert that NaP products for bowel cleansing may, in some patients, pose a risk of acute phosphate nephropathy, a rare form of acute renal failure.
Although NaP preparations are generally safe and well tolerated, they can cause significant fluid shifts and electrolyte abnormalities. As such, they should not be used in patients with baseline electrolyte imbalances, renal or hepatic dysfunction, or a number of other comorbidities.
CURRENT BOWEL-CLEANSING OPTIONS
For many years the standard preparation for bowel cleansing was a 4-liter or a 2-liter PEG electrolyte solution plus a laxative (eg, magnesium citrate, bisacodyl, or senna).1–3 The most frequent complaint heard from patients was that “the preparation is worse than the colonoscopy,” attributable to the taste and volume of the fluid they had to consume. Thus, compliance was often a significant issue with patients presenting for colonoscopy. In fact, inadequate bowel preparation is one of the most common reasons polyps are missed during colonoscopy.
Aqueous and tablet forms of NaP (sometimes with a laxative) have become a widely used alternative to PEG solutions because they require much less volume and as a result are more palatable, thereby improving compliance.4,5
NaP agents cleanse the colon by osmotically drawing plasma water into the bowel lumen. The patient must drink significant amounts of water or other oral solutions to prevent dehydration.
NaP-based bowel-cleansing agents are available in two forms: aqueous solution and tablet. Aqueous NaP (such as Fleet Phospho-soda) is a low-volume hyperosmotic solution containing 48 g of monobasic NaP and 18 g of dibasic NaP per 100 mL.6 An oral tablet form (such as Visicol and OsmoPrep) was developed to improve patient tolerance.7 Each 2-g tablet of Visicol contains 1,500 mg of active ingredients (monobasic and dibasic NaP) and 460 mg of microcrystalline cellulose, an inert polymer. Each OsmoPrep tablet contains 1,500 mg of the same active ingredients as Visicol, but the inert ingredients include PEG and magnesium stearate.
At first, the regimen was 40 tablets such as Visicol to be taken with water and bisacodyl. Subsequent regimens such as OsmoPrep with fewer tablets have been shown to be as effective and better tolerated.8 Microcrystalline cellulose in the tablet can produce a residue that may obscure the bowel mucosa. Newer preparations contain lower amounts of this inert ingredient, allowing for improved visualization of the colonic mucosa during colonoscopy.9
ADVANTAGES OF SODIUM PHOSPHATE BOWEL CLEANSERS
In a recent review article, Burke and Church10 noted that NaP cleansing regimens have been shown to be superior to PEG-electrolyte lavage solution with respect to tolerability and acceptance by patients, improved quality of bowel preparation, better mucosal visualization, and more efficient endoscopic examination. In addition, the volume of the preparation may also help decrease the risk of aspiration in some patients.2,3
DISADVANTAGES OF SODIUM PHOSPHATE AGENTS
Despite their comparable or better efficacy and their better tolerability, NaP agents have certain disadvantages.
Effects on the colonic mucosa
In rare cases NaP agents have been shown to alter the microscopic and macroscopic features of the colonic mucosa, and they can induce aphthoid erosions that may mimic those seen in inflammatory bowel disease and enteropathy or colopathy associated with nonsteroidal anti-inflammatory drugs (NSAIDs).11–13 Therefore, NaP agents should not be used prior to the initial endoscopic evaluation of patients with suspected inflammatory bowel disease, microscopic colitis, or NSAID-induced colonopathy.
Fluid and electrolyte shifts
Because NaP acts by drawing plasma water into the bowel lumen, significant volume and electrolyte shifts may occur.14,15 These can cause hypokalemia, hyperphosphatemia, hypocalcemia, hyponatremia or hypernatremia, hypomagnesemia, elevated blood urea nitrogen levels, decreased exercise capacity, increased plasma osmolarity,15–17 seizures,18 and acute renal failure with or without nephrocalcinosis.17,19–21
Thus, patients with significant comorbidities—such as a recent history of myocardial infarction, renal or hepatic insufficiency, or malnutrition—should not use NaP agents.22
Pivotal study of adverse events
In May 2006, the FDA issued an alert outlining the concerns of using oral NaP in specific patient populations. Of note were documented cases of acute phosphate nephropathy in 21 patients who used aqueous NaP (Fleet Phospho-Soda or Fleet Accu-Prep), and in 1 patient who used NaP tablets (Visicol).23 Acute renal injury was not limited to patients with preexisting renal insufficiency. It is uncertain whether this means that otherwise healthy people suffered renal injury or had risk factors besides renal insufficiency, since the data cited by the FDA report do not elucidate the possible risk factors for the development of nephropathy in patients with no preexisting renal insufficiency. So far, no cases of acute phosphate nephropathy or acute renal failure have been reported with OsmoPrep, a NaP tablet bowel preparation recently approved by the FDA.24 The long-term safety of OsmoPrep needs further evaluation.
PROCEED WITH CAUTION
Certain situations such as advanced age and cardiac, renal, and hepatic dysfunction call for extreme caution in the use of NaP bowel preparation agents. Therefore, it is recommended that patients with the following conditions should avoid using NaP agents for colon preparation:
- Hepatic or renal insufficiency (there are no data as to the degree of hepatic or renal insufficiency)
- Congestive heart failure
- Over age 65
- Dehydration or hypercalcemia
- Chronic use of drugs that affect renal perfusion, such as NSAIDs, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, or diuretics for hypertension.
Patients who take diuretics should not take them while they are using NaP for bowel preparation because of the risk of electrolyte abnormalities such as hypokalemia. In patients who have no alternative but to proceed with NaP preparation, our recommendation would be that the patient hold off taking diuretics, ACE inhibitors, and angiotensin receptor blockers while using the NaP prep. Given the importance of these medications in controlling diseases such as hypertension, the physician and the patient should jointly determine whether the benefits of using an NaP agent justify holding these drugs. We believe that patients taking these drugs should try using a PEG solution before considering NaP.
TASK FORCE GUIDELINES
Guidelines for using NaP bowel preparation agents, published by a task force of the American Society of Colon and Rectal Surgeons, the American Society for Gastrointestinal Endoscopy, and the Society of American Gastrointestinal and Endoscopic Surgeons,25 include the following caveats:
- Aqueous and tablet NaP colonic preparations are an alternative to PEG solutions, except in pediatric populations, patients over age 65, and those with bowel obstruction or other structural intestinal disorder, gut dysmotility, renal or hepatic insufficiency, congestive heart failure, or seizure disorder.
- Dosing should be 45 mL in divided doses, 10 to 12 hours apart, with at least one dose taken on the morning of the procedure.25
- The significant volume contraction and resulting dehydration seen in some patients using NaP preparations may be lessened by encouraging patients to drink fluids liberally during the days leading up to their procedure, and especially during NaP bowel preparation.26
- NaP tablets should be dosed as 32 to 40 tablets. On the evening before the procedure the patient should take 20 tablets and then 12 to 20 tablets approximately 3 to 5 hours before undergoing endoscopy. The tablets should be taken four at a time every 15 minutes with approximately 8 oz of clear liquid.25
- Sharma VK, Chockalingham SK, Ugheoke EA, et al. Prospective, randomized, controlled comparison of the use of polyethylene glycol electrolyte lavage solution in four-liter versus two-liter volumes and pretreatment with either magnesium citrate or bisacodyl for colonoscopy preparation. Gastrointest Endosc 1998; 47:167–171.
- Frommer D. Cleansing ability and tolerance of three bowel preparations for colonoscopy. Dis Colon Rectum 1997; 40:100–104.
- Hsu CW, Imperiale TF. Meta-analysis and cost comparison of polyethylene glycol lavage versus sodium phosphate for colonoscopy preparation. Gastrointest Endosc 1998; 48:276–282.
- Poon CM, Lee DWH, Mak SK, et al. Two liters of polyethylene glycol-electrolyte solution versus sodium phosphate as bowel cleansing regimen for colonoscopy: a prospective randomized controlled trial. Endoscopy 2002; 34:560–563.
- Afridi SA, Barthel JS, King PD, et al. Prospective, randomized trial comparing a new sodium phosphate-bisacodyl regimen with conventional PEG-ES lavage for outpatient colonoscopy preparation. Gastrointest Endosc 1995; 41:485–489.
- Schiller LR. Clinical pharmacology and use of laxatives and lavage solutions. J Clin Gastroenterol 1988; 28:11–18.
- Kastenberg D, Chasen R, Choudhary C, et al. Efficacy and safety of sodium phosphate tablets compared with PEG solution in colon cleansing. Two identically designed, randomized, controlled, parallel group multicenter phase III trials. Gastrointest Endosc 2001; 54:705–713.
- Rex DK, Chasen R, Pushpin MB. Safety and efficacy of two reduced dosing regimens of sodium phosphate tablets for preparation prior to colonoscopy. Aliment Pharmacol Ther 2002; 16:937–944.
- Rex DK, Khashab M. Efficacy and tolerability of a new formulation of sodium phosphate tablets and a reduced sodium phosphate dose, in colon cleansing: a single-center open-label pilot trial. Aliment Pharmacol Ther 2005; 21:465–468.
- Burke CA, Church JM. Enhancing the quality of colonoscopy: the importance of bowel purgatives. Gastrointest Endosc 2007; 66:565–573.
- Rejchrt S, Bures J, Siroky M, et al. A prospective, observational study of colonic mucosal abnormalities associated with orally administered sodium phosphate for colon cleansing before colonoscopy. Gastrointest Endosc 2004; 59:651–654.
- Hixson LJ. Colorectal ulcers associated with sodium phosphate catharsis. Gastrointest Endosc 1995; 42:101–102.
- Zwas FR, Cirillo NW, El-Serag HB, Eisen RN. Colonic mucosal abnormalities associated with oral sodium phosphate solution. Gastrointest Endosc 1996; 43:463–466.
- Clarkston WK, Tsen TN, Dies DF, Schratz CL, Vaswani SK, Bjerregaard P. Oral sodium phosphate versus sulfate-free polyethylene glycol electrolyte lavage solution in outpatient preparation for colonoscopy: a prospective comparison. Gastrointest Endosc 1996; 43:42–48.
- Kolts BE, Lyles WE, Achem SR, et al. A comparison of the effectiveness and patient tolerance of oral sodium phosphate, castor oil, and standard electrolyte lavage for colonoscopy or sigmoidoscopy preparations. Am J Gastroenterol 1993; 88:1218–1223.
- Holte K, Neilsen KG, Madsen JL, Kehlet H. Physiologic effects of bowel preparation. Dis Colon Rectum 2004; 47:1397–1402.
- Clarkston WK, Tsen TN, Dies DF, et al. Oral sodium phosphate versus sulfate-free polyethylene glycol electrolyte lavage solution in outpatient preparation for colonoscopy: a prospective comparison. Gastrointest Endosc 1996; 43:42–48.
- Frizelle FA, Colls BM. Hyponatremia and seizures after bowel preparation: report of three cases. Dis Colon Rectum 2005; 48:393–396.
- Markowitz GS, Nasr SH, Klein P, et al. Renal failure due to acute nephrocalcinosis following oral sodium phosphate bowel cleansing. Hum Pathol 2004; 35:675–684.
- Lieberman DA, Ghormley J, Flora K. Effect of oral sodium phosphate colon preparation on serum electrolytes in patients with normal serum creatinine. Gastrointest Endosc 1996; 43:467–469.
- Gremse DA, Sacks AI, Raines S. Comparison of oral sodium phosphate to polyethylene-glycol-based solution for bowel preparation in children. J Pediatric Gastroenterol Nutr 1996; 23:586–590.
- Curran MP, Plosker GL. Oral sodium phosphate solution: a review of its use as a colonic cleanser. Drugs 2004; 64:1697–1714.
- Markowitz GS, Stokes MB, Radhakrishnan J, D’Agati VD. Acute phosphate nephropathy following oral sodium phosphate bowel purgative: an underrecognized cause of chronic renal failure. J Am Soc Nephrol 2005; 16:3389–3396.
- FDA Alert. Patient information sheet. Oral sodium phosphate (OSP) products for bowel cleansing. 2006 May, Accessed January 8, 2008. www.fda.gov/CDER/drug/InfoSheets/patient/OSP_solutionPIS.htm.
- Wexner SD, Beck DE, Baron TH, et al. A consensus document on bowel preparation before colonoscopy prepared by a task force from the American Society of Colon and Rectal Surgeons (ASCRS), the American Society for Gastrointestinal Endoscopy (ASGE), and the Society of American Gastrointestinal and Endoscopic Surgeons (SAGES). Gastrointest Endosc 2006; 63:894–909.
- Huynh T, Vanner S, Paterson W. Safety profile of 5-h oral sodium phosphate regimen for colonoscopy cleansing: lack of clinically significant hypocalcemia or hypovolemia. Am J Gastroenterol 1995; 90:104–107.
- Sharma VK, Chockalingham SK, Ugheoke EA, et al. Prospective, randomized, controlled comparison of the use of polyethylene glycol electrolyte lavage solution in four-liter versus two-liter volumes and pretreatment with either magnesium citrate or bisacodyl for colonoscopy preparation. Gastrointest Endosc 1998; 47:167–171.
- Frommer D. Cleansing ability and tolerance of three bowel preparations for colonoscopy. Dis Colon Rectum 1997; 40:100–104.
- Hsu CW, Imperiale TF. Meta-analysis and cost comparison of polyethylene glycol lavage versus sodium phosphate for colonoscopy preparation. Gastrointest Endosc 1998; 48:276–282.
- Poon CM, Lee DWH, Mak SK, et al. Two liters of polyethylene glycol-electrolyte solution versus sodium phosphate as bowel cleansing regimen for colonoscopy: a prospective randomized controlled trial. Endoscopy 2002; 34:560–563.
- Afridi SA, Barthel JS, King PD, et al. Prospective, randomized trial comparing a new sodium phosphate-bisacodyl regimen with conventional PEG-ES lavage for outpatient colonoscopy preparation. Gastrointest Endosc 1995; 41:485–489.
- Schiller LR. Clinical pharmacology and use of laxatives and lavage solutions. J Clin Gastroenterol 1988; 28:11–18.
- Kastenberg D, Chasen R, Choudhary C, et al. Efficacy and safety of sodium phosphate tablets compared with PEG solution in colon cleansing. Two identically designed, randomized, controlled, parallel group multicenter phase III trials. Gastrointest Endosc 2001; 54:705–713.
- Rex DK, Chasen R, Pushpin MB. Safety and efficacy of two reduced dosing regimens of sodium phosphate tablets for preparation prior to colonoscopy. Aliment Pharmacol Ther 2002; 16:937–944.
- Rex DK, Khashab M. Efficacy and tolerability of a new formulation of sodium phosphate tablets and a reduced sodium phosphate dose, in colon cleansing: a single-center open-label pilot trial. Aliment Pharmacol Ther 2005; 21:465–468.
- Burke CA, Church JM. Enhancing the quality of colonoscopy: the importance of bowel purgatives. Gastrointest Endosc 2007; 66:565–573.
- Rejchrt S, Bures J, Siroky M, et al. A prospective, observational study of colonic mucosal abnormalities associated with orally administered sodium phosphate for colon cleansing before colonoscopy. Gastrointest Endosc 2004; 59:651–654.
- Hixson LJ. Colorectal ulcers associated with sodium phosphate catharsis. Gastrointest Endosc 1995; 42:101–102.
- Zwas FR, Cirillo NW, El-Serag HB, Eisen RN. Colonic mucosal abnormalities associated with oral sodium phosphate solution. Gastrointest Endosc 1996; 43:463–466.
- Clarkston WK, Tsen TN, Dies DF, Schratz CL, Vaswani SK, Bjerregaard P. Oral sodium phosphate versus sulfate-free polyethylene glycol electrolyte lavage solution in outpatient preparation for colonoscopy: a prospective comparison. Gastrointest Endosc 1996; 43:42–48.
- Kolts BE, Lyles WE, Achem SR, et al. A comparison of the effectiveness and patient tolerance of oral sodium phosphate, castor oil, and standard electrolyte lavage for colonoscopy or sigmoidoscopy preparations. Am J Gastroenterol 1993; 88:1218–1223.
- Holte K, Neilsen KG, Madsen JL, Kehlet H. Physiologic effects of bowel preparation. Dis Colon Rectum 2004; 47:1397–1402.
- Clarkston WK, Tsen TN, Dies DF, et al. Oral sodium phosphate versus sulfate-free polyethylene glycol electrolyte lavage solution in outpatient preparation for colonoscopy: a prospective comparison. Gastrointest Endosc 1996; 43:42–48.
- Frizelle FA, Colls BM. Hyponatremia and seizures after bowel preparation: report of three cases. Dis Colon Rectum 2005; 48:393–396.
- Markowitz GS, Nasr SH, Klein P, et al. Renal failure due to acute nephrocalcinosis following oral sodium phosphate bowel cleansing. Hum Pathol 2004; 35:675–684.
- Lieberman DA, Ghormley J, Flora K. Effect of oral sodium phosphate colon preparation on serum electrolytes in patients with normal serum creatinine. Gastrointest Endosc 1996; 43:467–469.
- Gremse DA, Sacks AI, Raines S. Comparison of oral sodium phosphate to polyethylene-glycol-based solution for bowel preparation in children. J Pediatric Gastroenterol Nutr 1996; 23:586–590.
- Curran MP, Plosker GL. Oral sodium phosphate solution: a review of its use as a colonic cleanser. Drugs 2004; 64:1697–1714.
- Markowitz GS, Stokes MB, Radhakrishnan J, D’Agati VD. Acute phosphate nephropathy following oral sodium phosphate bowel purgative: an underrecognized cause of chronic renal failure. J Am Soc Nephrol 2005; 16:3389–3396.
- FDA Alert. Patient information sheet. Oral sodium phosphate (OSP) products for bowel cleansing. 2006 May, Accessed January 8, 2008. www.fda.gov/CDER/drug/InfoSheets/patient/OSP_solutionPIS.htm.
- Wexner SD, Beck DE, Baron TH, et al. A consensus document on bowel preparation before colonoscopy prepared by a task force from the American Society of Colon and Rectal Surgeons (ASCRS), the American Society for Gastrointestinal Endoscopy (ASGE), and the Society of American Gastrointestinal and Endoscopic Surgeons (SAGES). Gastrointest Endosc 2006; 63:894–909.
- Huynh T, Vanner S, Paterson W. Safety profile of 5-h oral sodium phosphate regimen for colonoscopy cleansing: lack of clinically significant hypocalcemia or hypovolemia. Am J Gastroenterol 1995; 90:104–107.
A report from the department of staph affairs

Once, we worried about penicillin-resistant staph, but we had methicillin and several other effective antibiotics. Then, in the 1960s, MRSA started to appear. Intravenous drug abusers and then the chronically ill were favored hosts. Some hospitals became nests for MRSA. It began to acquire a reputation as a particularly nasty invader, associated with necrotizing pneumonia and resistant endocarditis. By 2004 more than 60% of staph isolates from critical care units were resistant to methicillin. But we had vancomycin.
Now, the hospital bugs are being reinforced by their community brethren—new strains from the suburbs that carry toxins that can damage tissue via stimulation of apoptosis of host cells. Most community-associated MRSA species are still sensitive to vancomycin (as well as to trimethoprim-sulfamethoxazole and clindamycin). But not all are. A growing community of bugs is relatively resistant to vancomycin and carries toxins—microbial suicide bombers with body armor.
The clinical presentation of necrotizing cellulitis, first appearing as a “spider bite,” is now seen in emergency wards around the country. In some cities, the overwhelming majority of deep skin infections evaluated in emergency rooms are due to MRSA. And patients with these infections, as well as asymptomatic nasal carriers of MRSA, are bringing these bugs into our hospitals.
The trends of emerging antibiotic resistance, discovery (and then perhaps overprescription) of new antibiotics, and changing patterns of staph infections are intimately intertwined. The evolutionary pressure that we humans are putting on S aureus with our antibiotics is taking this bug to a whole new place.
Once, we worried about penicillin-resistant staph, but we had methicillin and several other effective antibiotics. Then, in the 1960s, MRSA started to appear. Intravenous drug abusers and then the chronically ill were favored hosts. Some hospitals became nests for MRSA. It began to acquire a reputation as a particularly nasty invader, associated with necrotizing pneumonia and resistant endocarditis. By 2004 more than 60% of staph isolates from critical care units were resistant to methicillin. But we had vancomycin.
Now, the hospital bugs are being reinforced by their community brethren—new strains from the suburbs that carry toxins that can damage tissue via stimulation of apoptosis of host cells. Most community-associated MRSA species are still sensitive to vancomycin (as well as to trimethoprim-sulfamethoxazole and clindamycin). But not all are. A growing community of bugs is relatively resistant to vancomycin and carries toxins—microbial suicide bombers with body armor.
The clinical presentation of necrotizing cellulitis, first appearing as a “spider bite,” is now seen in emergency wards around the country. In some cities, the overwhelming majority of deep skin infections evaluated in emergency rooms are due to MRSA. And patients with these infections, as well as asymptomatic nasal carriers of MRSA, are bringing these bugs into our hospitals.
The trends of emerging antibiotic resistance, discovery (and then perhaps overprescription) of new antibiotics, and changing patterns of staph infections are intimately intertwined. The evolutionary pressure that we humans are putting on S aureus with our antibiotics is taking this bug to a whole new place.
Once, we worried about penicillin-resistant staph, but we had methicillin and several other effective antibiotics. Then, in the 1960s, MRSA started to appear. Intravenous drug abusers and then the chronically ill were favored hosts. Some hospitals became nests for MRSA. It began to acquire a reputation as a particularly nasty invader, associated with necrotizing pneumonia and resistant endocarditis. By 2004 more than 60% of staph isolates from critical care units were resistant to methicillin. But we had vancomycin.
Now, the hospital bugs are being reinforced by their community brethren—new strains from the suburbs that carry toxins that can damage tissue via stimulation of apoptosis of host cells. Most community-associated MRSA species are still sensitive to vancomycin (as well as to trimethoprim-sulfamethoxazole and clindamycin). But not all are. A growing community of bugs is relatively resistant to vancomycin and carries toxins—microbial suicide bombers with body armor.
The clinical presentation of necrotizing cellulitis, first appearing as a “spider bite,” is now seen in emergency wards around the country. In some cities, the overwhelming majority of deep skin infections evaluated in emergency rooms are due to MRSA. And patients with these infections, as well as asymptomatic nasal carriers of MRSA, are bringing these bugs into our hospitals.
The trends of emerging antibiotic resistance, discovery (and then perhaps overprescription) of new antibiotics, and changing patterns of staph infections are intimately intertwined. The evolutionary pressure that we humans are putting on S aureus with our antibiotics is taking this bug to a whole new place.
Staphylococcus aureus: The new adventures of a legendary pathogen
Staphylococcus aureus is rearing its ugly head in new and interesting ways, both in the hospital and in the community.
Rates of invasive infections with methicillin-resistant S aureus (MRSA) have been increasing both in the hospital and in the community, a trend that has attracted considerable interest in the lay media. Curiously, the most common community-associated MRSA strain, which up to now has been distinct from hospital-associated MRSA strains, is invading our hospitals. Alarmingly, vancomycin (Vancocin), the drug of last resort for MRSA infections for the past 40 years, does not seem to be as effective as it used to be.
This paper summarizes the changing epidemiology of S aureus, particularly the emergence of MRSA outside of the hospital; reviews the difficulties associated with S aureus bacteremia and its treatment in view of; some changes in vancomycin susceptibility; and appraises the old and new treatment options.
MRSA IS ON THE RISE IN THE HOSPITAL
S aureus, a gram-positive, coagulase-positive bacterium, is one of the leading nosocomial bloodstream pathogens, second only to coagulase-negative staphylococci.1 And the incidence of S aureus infections is increasing. MRSA in particular is increasingly causing infections throughout hospitals, including intensive care units. As of 2004, nearly two-thirds of isolates of S aureus from intensive care units were MRSA.2
MRSA infections are worse than methicillin-susceptible S aureus (MSSA) infections in terms of the rates of death and other undesirable outcomes.3 Several factors may be responsible: MRSA infection may be a marker of severity of illness (sicker patients may be more likely to have MRSA), our treatment for MRSA may not be as effective as it is for MSSA, and the organism may be inherently more virulent.
METHICILLIN RESISTANCE IS ALSO ON THE RISE IN THE COMMUNITY
Community-associated MRSA began emerging clinically about 10 years ago. It was first described in a cohort of children with necrotizing pneumonia in Minnesota, but soon other populations at risk began to emerge, such as residents of correctional facilities, men who had sex with men, competitive athletes (eg, fencers, wrestlers, and football players), and Alaskan natives and other native populations. A common factor in all these groups was close proximity of the members to each other. Later, it began to spread beyond these traditional risk groups into the community at large.
Community-associated MRSA strains have a characteristic pattern of antimicrobial susceptibility (see below). In the laboratory, they grow somewhat faster than health-care-associated MRSA strains, but not as fast as MSSA. They have a strong association with skin and soft-tissue infections: when you see a skin or soft-tissue infection, be it in an outpatient or an inpatient, think about MRSA. Their virulence varies, but rapid onset and progression of illness are quite common. Their most common strain in the United States at present is USA 300.
Case 1: A young woman with necrotizing fasciitis
A 21-year-old college student presented to our service in May 2004 with high fever and severe arm pain, which had been worsening for several days. She had been previously healthy, had not had any contact with the health care system, and had not received any antibiotics.
Her blood cultures were positive for MRSA, as were cultures of the deep tissue of the deltoid muscle and fascia when she underwent emergency surgical debridement. The infection required several additional surgical debridements and removal of one head of her deltoid muscle, but she was fortunate: in the past, some patients with this problem might have undergone radical amputation of the arm or even more extensive surgery. This patient continued to have positive blood cultures 4 days postoperatively, but she ultimately recovered, completing 28 days of daptomycin (Cubicin) therapy at a dose of 6 mg/kg every 24 hours. The last 10 days of daptomycin therapy were given at home via a percutaneous intravenous central catheter.
Comment. The epidemiology of MRSA infections is changing. More patients who have no traditional risk factors, specifically health care contact, are getting MRSA infections. A recent report from the US Centers for Disease Control and Prevention (CDC) indicates that the proportion of patients with invasive disease due to MRSA has doubled since 2001–2002.4 Part of the reason undoubtedly is that MRSA, particularly community-associated MRSA, often carries specific virulence factors that make it more invasive. The CDC estimated that in 2005 there were nearly 100,000 cases of invasive MRSA infection in the United States, and nearly a fifth of these infections resulted in death.
Resistance and virulence factors in community-associated MRSA
Most community-associated MRSA strains carry a mobile genetic element called type IV SCCmec (staphylococcal chromosomal cassettemec) that enhances its antimicrobial resistance. This genetic component was probably borrowed from coagulase-negative staphylococci, in which it is quite common but does not cause as much of a problem. It is now present in a wide range of S aureus strains. Most of the S aureus strains that carry type IV SCCmec are MRSA, but a few MSSA strains do carry it as well.
The potent toxin Panton-Valentine leukocidin is an extracellular product that is detected in fewer than 5% of hospital strains but is more common in community-associated strains. It kills leukocytes by forming pores in the cell membrane and causing skin necrosis in cutaneous infections. It is associated with skin abscesses and rapidly progressive necrotizing pneumonia in MSSA or MRSA.
Epidemiologic differences between community- and health-care-associated MRSA
Patients with community-associated MRSA infections tend to be younger than those who traditionally get health-care-associated MRSA infections: in a study from Naimi et al in 2003, the mean ages were 23 vs 68 years.5 A greater proportion of patients with community-associated MRSA strains are nonwhite.4,5
Most community-associated MRSA infections are of the skin and soft tissue (75% in the series from Naimi et al5), but this pathogen causes other infections as well. Bacteremia of unknown origin has been seen, as has necrotizing pneumonia. Most of the skin and soft-tissue infections are relatively superficial, such as folliculitis or furunculosis, but deeper tissue infections such as necrotizing fasciitis and pyomyositis have also been seen.6
The incidence of community-associated MRSA infections varies greatly by geographic region.7 The northeastern United States has so far been relatively spared, but in Atlanta, Houston, and Los Angeles up to 80% of cases of characteristic skin or soft-tissue infections seen in emergency or outpatient departments are due to community-associated MRSA. Physicians at the Texas Children’s Hospital in Houston assume that all skin or soft-tissue infections are due to community-associated MRSA unless proven otherwise.8
Differences in antibiotic susceptibility
Community-associated MRSA is more susceptible to various antibiotics than health-care-associated MRSA,5 but not by much. Strains are usually susceptible to vancomycin, tetracyclines, trimethoprim-sulfamethoxazole (Bactrim, Septra), and rifampin (Rifadin). Unlike hospital strains, a fair number of community-acquired strains are susceptible to clindamycin (Cleocin) in the laboratory, but with a caveat: some of these clindamycin-susceptible strains actually may harbor the tools for inducible resistance. In fact, they can become resistant to clindamycin even without being exposed to it.
The laboratory test for inducible clindamycin resistance is called the D test. After coating an agar plate with S aureus, the technician places erythromycin and clindamycin disks. If the erythromycin induces clindamycin resistance, the plate is clear of growth around the clindamycin disk except for the portion nearest the erythromycin disk, leaving a characteristic D-shaped area of lucency.
Risk factors for MRSA
Moran et al7 analyzed the risk factors for community-associated MRSA in patients with skin or soft-tissue infections seen in the emergency department. The infection was more likely to be due to community-associated MRSA if the patient was black, had used any antibiotic in the past month, had a history of MRSA infection, or had close contact with a person with a similar infection. Many patients interpreted the infections as spider bites because the lesions tended to have a dark center surrounded by a tender area. These infections were not associated with underlying illness. In some cases, community-associated MRSA skin infections have been associated with tattooing and even manicuring.
However, it is very difficult to distinguish between community-associated MRSA and MSSA skin and soft-tissue infections on the basis of clinical and epidemiologic characteristics. Miller et al9 studied a large group of patients in Los Angeles who were hospitalized with community-associated skin and soft-tissue S aureus infections. All the patients were followed up for 30 days after hospital discharge. Regardless of whether they had MRSA or MSSA, they had similar outcomes. Close contacts of the patients also tended to develop infection.
A key point from this and many other studies: patients were more likely to remain infected if they did not undergo incision and drainage. This key intervention is indicated for any patient who has a skin and soft-tissue infection with an undrained focus of infection.
COMMUNITY-ASSOCIATED MRSA IS INVADING THE HOSPITAL
In a new development, community-associated MRSA strains are now appearing in the hospital. This is not only because patients are bacteremic when they come in: patients in the hospital are getting nosocomial infections due to community-associated MRSA strains.
Seybold et al10 analyzed 116 cases of MRSA bloodstream infections in Atlanta, GA. In 9 (8%) of the cases the patient had not had any contact with the health care system within the past year, and these cases were classified as truly community-associated. Of the remaining 107 cases, 49 (42%) were nosocomial, and the USA 300 strain—the predominant community-associated MRSA strain—accounted for 10 (20%) of the nosocomial cases.
In the recent CDC study of invasive MRSA infections, Klevens et al4 reported that nearly a third of cases of bacteremia were due to community-associated MRSA, and these strains accounted for a greater proportion of cases of cellulitis and endocarditis than did health-care-associated strains.
In a study of hospital-associated MRSA, Maree et al11 found that the percentage of cases in which the bacteria carried the SCCmec type IV marker had increased from less than 20% in 1999 to more than 50% in 2004.
Comment. Suffice it to say that we are surrounded by MRSA. Community-associated MRSA is here to stay. It is even invading our hospitals, and we need to consider this very carefully when choosing antimicrobial therapy.
NAGGING QUESTIONS ABOUT VANCOMYCIN
Case 2: Vancomycin-intermediate S aureus (VISA) bacteremia and endocarditis
In December 2006 we saw a very ill 60-year-old woman who was hospitalized with MRSA bacteremia, pacemaker endocarditis, and superior vena cava thrombosis. Although she was treated with vancomycin and rifampin, her condition worsened, she had a stroke, and she developed renal failure. In a difficult operation, the pacemaker was removed, but the bacteremia persisted. In early February 2007 she underwent another difficult operation in which the superior vena cava clot was debrided, a right atrial clot was removed, and her mitral valve was replaced. Less than 2 weeks later, and despite ongoing vancomycin and rifampin therapy, the MRSA bacteremia recurred.
During the approximately 6 weeks that the patient had been receiving these antibiotics, the minimal inhibitory concentration (MIC) of rifampin against the S aureus isolate increased from less than 1 μg/mL (susceptible) to 2 μg/mL (resistant). The MIC of vancomycin went from 2 μg/mL (susceptible) to 4 μg/mL (intermediately susceptible). Vancomycin and rifampin were discontinued, and daptomycin and gentamicin (Garamycin) therapy were started. (Her daptomycin MIC was 0.5 μg/mL). The patient’s condition stabilized, and she was discharged to a long-term nursing facility. She had no relapse of MRSA bacteremia, but she died in early April of that year.
Is vancomycin becoming less effective? Degrees of vancomycin resistance
Vancomycin has been our stalwart for treating MRSA infections for more than 40 years but it is not working as well as it used to, at least in certain situations.
VRSA (vancomycin-resistant S aureus) is rare. These fully resistant strains probably acquired a resistance mechanism (the vanA operon) from vancomycin-resistant enterococci. Infections tend to occur in patients simultaneously infected with both S aureus and vancomycin-resistant enterococci, giving the bacteria an opportunity to exchange genetic material.
VISA (vancomycin-intermediate S aureus) infections tend to occur in patients like the one described above who have had long-term vancomycin therapy. VISA strains appear to overproduce a matrix that captures vancomycin and keeps it from entering the cell. On electron microscopy, these bacteria have a very thick cell wall.13
Vancomycin tolerance is a state in which the bacteria are “stunned” or kept in check but not killed by vancomycin. That is manifested in the laboratory by a ratio of minimum bactericidal concentration to MIC greater than 32.
hVISA (heteroresistant VISA) is new and worrisome. These organisms have an overall MIC in the susceptible range, but within that population are individual isolates with an MIC that is much higher—in the intermediate or perhaps even in the resistant range.14
Reported rates of hVISA vary from less than 2% to as high as 76%, because the methods for detecting it are still very poorly standardized. The usual automated laboratory tests do not detect hVISA.
hVISA is probably clinically relevant, as evidence is emerging both in vitro and in vivo that the higher the MIC for vancomycin, the worse the clinical outcome.15 hVISA has been associated with failures of therapy in several situations, usually in cases of severe invasive or deep infection, endocarditis, and bacteremia with vertebral osteomyelitis where vancomycin concentrations at the site of infection may be suboptimal.16–19 While most hVISA strains that have been described were resistant to methicillin, some were susceptible.
The E test is emerging as the standard test for hVISA. This test uses a plastic strip that contains gradually increasing concentrations of vancomycin along its length. Placed in the culture dish, the strip inhibits growth of the organism at its high-concentration end but not at its low-concentration end. If the sample contains hVISA, the cutoff is not well defined, with a few colonies growing at higher concentrations.
New definition of vancomycin susceptibility
Recognizing that the MICs for vancomycin have been rising in the last few years, the Clinical and Laboratory Standards Institute last year changed the break points between susceptibility and resistance. The new definitions are:
- Susceptible—an MIC of 2.0 μg/mL or less (formerly 4.0 μg/mL or less)
- Intermediate—4.0 to 8.0 μg/mL (formerly 8.0 to 16 μg/mL)
- Resistant—16 μg/mL or greater (formerly 32 μg/mL or greater).
One should pay attention to the MIC numbers on the laboratory reports, not just to the words “susceptible” or “not susceptible.” If the number is, say, 0.5 μg/mL or less, the organism should really be susceptible. If the number is 1 or 2, it is still in the susceptible range, but those are the organisms that may cause problems later on.
Further, even if the vancomycin MIC is in the susceptible range, higher MICs may affect outcomes. The average duration of MRSA bacteremia on therapy is 8 to 9 days, vs 3 to 4 days with MSSA bacteremia.20,21 But Sakoulas et al15 found that, in MRSA bacteremia, the success rate with vancomycin therapy was 56% if the MIC was 0.5 or lower, compared with 10% if the MIC was 1.0 to 2.0 μg/mL. Examined in another way, the success rate was 50% if the logarithm of killing was 6.27 colony-forming units per mL or greater, 23% if 4.71 to 6.26, and zero if less than 4.71.
Case 3: Prolonged MRSA bacteremia
In the summer of 2006, a 66-year-old woman with a history of gastric bypass and cirrhosis underwent a long stay in the surgical intensive care unit because of a recurrent enterocutaneous fistula and chronic renal insufficiency. On November 5th, she had a positive blood culture for MRSA, which was treated appropriately with vancomycin for 4 weeks. She was discharged to subacute care but came back 2 days later, again with MRSA bacteremia. At that time her Hickman catheter, which had been inserted for total parenteral nutrition because of the enterocutaneous fistula, was removed.
Transthoracic echocardiography revealed no vegetations, but her bacteremia persisted. Her mental status was poor this entire time: she was mute and could barely be awakened. We looked for clots and infected clots; duplex ultrasonographic examinations of all four extremities were negative. Finally, magnetic resonance imaging of her back—performed empirically because of the persistent bacteremia—revealed vertebral osteomyelitis at level T12-L1. We also noticed on serial evaluations that the vancomycin MIC for her organism increased from 0.5 to 2.0 μg/mL, so therapy was changed from vancomycin to daptomycin.
Her bacteremia cleared. Follow-up echocardiography was negative, but she had two subsequent relapses of MRSA bacteremia, one in April 2007 and one before she died in the summer of 2007.
Prolonged bacteremia: Is it vancomycin resistance, or something else?
The MRSA isolates that cause prolonged bacteremia seem to have certain characteristics.22 Higher MICs are probably associated with longer periods of bacteremia. But some genetic components within some strains of S aureus give them a survival advantage. They have less susceptibility to the body’s thrombin-induced platelet microbicidal protein. These isolates are not only associated with prolonged bacteremia: they are also associated with osteomyelitis, deep abscesses, endocarditis, recurrent infection, and increased death rate.22 Clinical laboratories do not test for these genetic components. One wonders whether our patient may have had an isolate with these mutations that gave it a survival advantage.
Do not use vancomycin for MSSA
Avoid using vancomycin for MSSA infections. It has been shown time and time again that MSSA infections do not respond as well to vancomycin as they do to beta-lactam antibiotics, specifically to the semisynthetic penicillins such as oxacillin and nafcillin, and even some of the first-generation cephalosporins. Chang et al23 found that patients with MSSA bacteremia had higher rates of persistent infections, relapse, and bacteriologic failure if they received vancomycin than if they received nafcillin.
Do vancomycin trough levels affect toxicity?
The vancomycin trough levels that we aimed for in the past (5 to 10 μg/mL) were probably too low. Today, we aim for trough levels of 15 to 20 μg/mL, and many physicians are aiming for 20 to 25 μg/mL. Part of the reason is that vancomycin MICs are higher than they used to be: in order to keep the vancomycin level above the MIC for a longer period of time, the vancomycin trough level needs to be higher. In theory, keeping the vancomycin levels above the MIC for longer periods should improve outcomes. Yet Fowler et al22 found that vancomycin trough levels among patients who had persistent MRSA bacteremia were actually higher than trough levels among those in whom the bacteremia resolved, although the difference was not statistically significant.
We measure the vancomycin trough level to make sure it is high enough (and give larger doses if it is not); among adults, peak levels need not be monitored on a routine basis because of the predictable pharmacokinetics of vancomycin.
Vancomycin toxicity can be either idiosyncratic or synergistic. Idiosyncratic toxicity occurs when a patient who has been on vancomycin for a long time develops a fixed rash, not associated with infusion. This is an immunologic phenomenon. It is a rare and very serious situation and may require steroid therapy.
Synergistic toxicity occurs when vancomycin is given with other nephrotoxic agents, notably gentamicin. Vancomycin plus gentamicin equals nephrotoxicity. Vancomycin alone is usually not nephrotoxic, but close monitoring of renal function parameters is warranted with the use of higher doses.24
IN UNEXPLAINED BACTEREMIA, LOOK FOR ENDOCARDITIS
In blood cultures from patients with bacteremia, S aureus is never a contaminant. Even if just one blood culture is positive for S aureus, believe that S aureus is the culprit.
Reports in the 1950s suggested that at least half of patients who had S aureus bacteremia had endocarditis,25 leading to recommendations that all patients with S aureus bacteremia without an obvious primary source of infection should be evaluated for endocarditis. Subsequent estimates were lower, in the range of 15% to 25%.26,27 However, throughout the world S aureus endocarditis continues to have a very high mortality rate: at least a third of patients die.28
Clinical criteria (community acquisition, no primary focus, and metastatic sequelae) were developed to try to predict the risk of endocarditis in bacteremic patients.26 However, these criteria did not work very well. The clinical definition of endocarditis has evolved. The criteria of von Reyn et al29 from 1981 did not use echocardiography as part of the definition, but the 1994 Duke criteria,30 which were refined31 in 2000, use both clinical and echocardiographic parameters.
Stratton et al32 performed transthoracic echocardiography in 14 patients with bacteremia and found 1 patient with cryptic tricuspid infective endocarditis. Bayer et al33 subsequently reported that of 72 patients with bacteremia, 6 (18%) of those who had no clinical findings suggestive of infectious endocarditis had findings on echocardiography that led to changes in their regimen. Adding echocardiography to three clinical risk factors increased the sensitivity of diagnosing endocarditis from 70% to 85% with a specificity of 100% and predictive value of 96%.
The Duke criteria call for transesophageal echocardiography, which is not feasible in some patients, eg, those with cirrhosis and esophageal varices.
S aureus endocarditis has changed over the years as our patient population has changed, and MRSA endocarditis tends to hit some of our most vulnerable patients. In a study by Miro et al34 in 2005, MRSA was the leading pathogen in patients who were diagnosed with S aureus endocarditis in 1990 or later. We will only see these numbers go up. Patients with diabetes tend to have more MRSA, and of diabetic patients with MRSA endocarditis, 30% to 40% die in the hospital.
Indications for surgery
Certain conditions are indications for surgery among patients with endocarditis, and no antibiotic will cure the endocarditis if the patient has one of these conditions, eg:
- Persistent bacteremia during antibiotic therapy
- Recurrent emboli
- Heart failure that cannot be controlled
- Perivalvular or myocardial abscesses
- Large vegetations
- Early prosthetic valve infection
- Certain arrhythmias.
How long should S aureus bacteremia be treated?
In cases of bacteremia in which endocarditis has been ruled out and removable foci of infection (eg, intravascular catheters) have been removed, some evidence indicates that treatment for 2 weeks would be as effective as the 4 to 6 weeks that we would use for endocarditis or other severe or invasive infections.35 The issue is controversial. If the patient has had frequent hospitalizations or a chronic medical condition I would hesitate to treat for less than 4 weeks, even if the infection appears to be associated with a removable focus.
Treatment of endocarditis
In the guidelines for treatment of endocarditis from the American Heart Association and Infectious Diseases Society of America,36 all the recommendations are relatively old and many of them are somewhat empiric—they are not based on evidence from randomized clinical trials. Rather, they are best opinions based on clinical experience and some observational studies over the years.
For MSSA. In cases of native-valve endocarditis, oxacillin (Bactocill), nafcillin (Unipen), or another semisynthetic beta-lactam antibiotic is recommended. For penicillin-allergic patients, we have other options, such as cefazolin (Ancef, Kefzol).
Combination therapy is frequently recommended for native valve endocarditis as well as for prosthetic valve endocarditis, with either rifampin or gentamicin along with a primary agent. There is some evidence that one can clear staphylococcal bacteremia a day or two more quickly by use of combination therapy with nafcillin plus an aminoglycoside than with nafcillin alone.37,38 For MSSA-associated endocarditis, vancomycin does not work as well as beta-lactam antibiotics.39,40
Korzeniowski and Sande37 and Chambers et al38 reported that the mean duration of bacteremia was 3.4 days for patients treated with nafcillin alone and 2.9 days for those treated with nafcillin plus an aminoglycoside. These studies led to consideration of a short course of gentamicin to clear the bacteremia quickly.
With MRSA, bacteremia often requires a week or more to clear. Levine et al21 reported a study in 42 patients, mostly injection-drug users, with right-sided native-valve endocarditis. The median duration of bacteremia was 7 days in patients who received vancomycin alone vs 9 days in those who received vancomycin plus rifampin; however, some patients were bacteremic for up to 27 days. Fever persisted for a median of 7 days, probably partly due to septic pulmonary emboli. Three patients died, and three required valve replacement.
NEW ANTIBIOTICS
Several new antibiotics are active against gram-positive cocci.41–44 However, the majority of them have not been prospectively studied for treating bacteremia or endocarditis.
Quinupristin/dalfopristin (Synercid) has not been formally studied for treatment of MRSA bacteremia or endocarditis. There are a few case reports of its use in these conditions.45 Quinupristin/dalfopristin is bacteriostatic, and its use may be associated with phlebitis, myalgias, and arthralgias.46
Linezolid (Zyvox) is approved for treatment of complicated skin and soft-tissue infections and for hospital-acquired pneumonia. There have been no specific studies of linezolid in the treatment of S aureus bacteremia or endocarditis. However, Shorr et al47 retrospectively looked at the bacteremic patients in five previous studies of linezolid vs vancomycin and found 144 cases of S aureus bacteremia, half of which were due to MRSA. Of 53 assessable patients with MRSA bacteremia, the primary infection was cured in 14 (56%) of the linezolid patients and 13 (46%) of the vancomycin patients.
The oral form is 100% bioavailable. One should avoid concomitant use of serotonin-reuptake inhibitors because of the risk of serotonin syndrome. Adverse effects include altered taste sensation and peripheral neuropathy. There are other potential toxicities, including hematologic changes (thrombocytopenia, leukopenia) and metabolic effects (lactic acidosis), so clinical and laboratory monitoring is important.48 The role of linezolid in the treatment of patients with S aureus bacteremia or endocarditis remains to be defined.
Daptomycin is indicated for complicated skin and soft-tissue infections, bacteremia, and right-sided endocarditis due to S aureus. Fowler et al20 found that daptomycin was not inferior to beta-lactam antibiotics for treatment of MSSA bacteremia and right-sided endocarditis, and for MRSA infections it outperformed vancomycin, but the difference was not statistically significant.
The dosing interval should be increased from once every 24 hours to every 48 hours if the creatinine clearance is 30 mL/minute or less. Adverse effects include myalgia, rhabdomyolysis (rare), and elevations in creatine phosphokinase. Reports of rising MICs during daptomycin therapy, in some cases associated with persistent infection,49 suggest that careful attention be paid to dosing and clinical monitoring.
Tigecycline (Tygacil) is indicated for complicated skin and soft-tissue infections and complicated intra-abdominal infections due to susceptible organisms. It is active against both MSSA and MRSA, but clinical experience with its use in invasive infections is somewhat limited.50 The dose of tigecycline should be reduced in advanced cirrhosis. Adverse effects include nausea and vomiting.
Telavancin, dalbavancin, and oritavancin, investigational parenteral antibiotics that are derivatives of vancomycin, are in clinical trials. The pharmacokinetic activity of these agents is of interest: telavancin is being studied with a once-daily dosing interval and dalbavancin’s half-life allows once-weekly dosing. In a limited trial, dalbavancin was found to be safe and effective in the treatment of catheter-related bloodstream infections.51 None of the antibiotics in this group has been studied for treatment of S aureus endocarditis. Telavancin therapy has been associated with rash, hypokalemia, QT prolongation, and creatinine elevations. Gastrointestinal symptoms have been reported with the use of dalbavancin.
Ceftobiprole, another investigational agent, is the only cephalosporin antibiotic that is active against MRSA. It is given every 12 hours. Adverse effects include nausea and taste disturbance.
Iclaprim is a novel diaminopyrimidine and a dihydrofolate reductase inhibitor. In vitro, it is active against gram-positive bacteria, including MRSA, VISA, and VRSA; clinical investigations at this point are limited to the treatment of skin and soft-tissue infections.
- Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 2004; 39:309–371. Erratum in: Clin Infect Dis 2004; 39:1093.
- US Centers for Disease Control and Prevention. National Nosocomial Infections Surveillance (NNIS) System. Campaign to prevent antimicrobial resistance. www.cdc.gov/drugresistance/healthcare/ha/HASlideSet.ppt.
- Blot SI, Vandewoude KH, Hoste EA, Colardyn FA. Outcome and attributable mortality in critically ill patients with bacteremia involving methicillin-susceptible and methicillin-resistant Staphylococcus aureus. Arch Intern Med 2002; 162:2229–2235.
- Klevens RM, Morrison MA, Nadle J, et al; Active Bacterial Core surveillance (ABCs) MRSA Investigators. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007; 298:1763–1771.
- Naimi TS, LeDell KH, Como-Sabetti K, et al. Comparison of community- and health care-associated methicillin-resistant Staphylococcus aureus infection. JAMA 2003; 290:2976–2984.
- Miller LG, Perdreau-Remington F, Rieg G, et al. Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N Engl J Med 2005; 352:1445–1453.
- Moran GJ, Krishnadasan A, Gorwitz RJ, et al EMERGEncy ID Net Study Group. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med 2006; 355:666–674.
- Mishaan AM, Mason EO, Martinez-Aquilar G, et al. Emergence of a predominant clone of community-acquired Staphylococcus aureus among children in Houston, Texas. Pediatr Infect Dis J 2005; 24:201–206.
- Miller LG, Perdreau-Remington F, Bayer AS, et al. Clinical and epidemiologic characteristics cannot distinguish community-associated methicillin-resistant Staphylococcus aureus infection from methicillin-susceptible S. aureus infection: a prospective investigation. Clin Infect Dis 2007; 44:471–482.
- Seybold U, Kourbatova EV, Johnson JG, et al. Emergence of community-associated methicillin-resistant Staphylococcus aureus USA300 genotype as a major cause of health care-associated blood stream infections. Clin Infect Dis 2006; 42:647–656.
- Maree CL, Daum RS, Boyle-Vavra S, Matayoshi K, Miller LG. Community-associated methicillin-resistant Staphylococcus aureus isolates causing healthcare-associated infections. Emerg Infect Dis 2007; 13:236–242.
- Liu C, Chambers HF. Staphylococcus aureus with heterogeneous resistance to vancomycin: epidemiology, clinical significance, and critical assessment of diagnostic methods. Antimicrob Agents Chemother 2003; 47:3040–3045.
- Sieradzki K, Roberts RB, Haber SW, Tomasz A. The development of vancomycin resistance in a patient with methicillin-resistant Staphylococcus aureus infection. N Engl J Med 1999; 340:517–523.
- Schwaber MJ, Wright SB, Carmeli Y, et al. Clinical implications of varying degrees of vancomycin susceptibility in methicillin-resistant Staphylococcus aureus bacteremia. Emerg Infect Dis 2003; 9:657–664. Erratum in: Emerg Infect Dis 2004; 10:160.
- Sakoulas G, Moise-Broder PA, Schentag J, Forrest A, Moellering RC, Eliopoulos GM. Relationship of MIC and bactericidal activity to efficacy of vancomycin for treatment of methicillin-resistant Staphylococcus aureus bacteremia. J Clin Microbiol 2004; 42:2398–2402.
- Naimi TS, Anderson D, O’Boyle C, et al. Vancomycin-intermediate Staphylococcus aureus with phenotypic susceptibility to methicillin in a patient with recurrent bacteremia. Clin Infect Dis 2003; 36:1609–1612.
- Moore MR, Perdreau-Remington F, Chambers HF. Vancomycin treatment failure associated with heterogeneous vancomycin-intermediate Staphylococcus aureus in a patient with endocarditis and in the rabbit model of endocarditis. Antimicrob Agents Chemother 2003; 47:1262–1266.
- Charles PG, Ward PB, Johnson PD, Howden BP, Grayson ML. Clinical features associated with bacteremia due to heterogenous vancomycin-intermediate Staphylococcus aureus. Clin Infect Dis 2004; 38:448–451.
- Howden BP, Ward PB, Charles PG, et al. Treatment outcomes for serious infections caused by methicillin-resistant Staphylococcus aureus with reduced vancomycin susceptibility. Clin Infect Dis 2004; 38:521–528.
- Fowler VG, Boucher HW, Corey GR, et al. S. aureus Endocarditis and Bacteremia Study Group. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N Engl J Med 2006; 355:653–665.
- Levine DP, Fromm BS, Reddy BR. Slow response to vancomycin or vancomycin plus rifampin in methicillin-resistant Staphylococcus aureus endocarditis. Ann Intern Med 1991; 115:674–680.
- Fowler VG, Sakoulas G, McIntyre LM, et al. Persistent bacteremia due to methicillin-resistant Staphylococcus aureus infection is associated with agr dysfunction and low-level in vitro resistance to thrombin-induced platelet microbicidal protein. J Infect Dis 2004; 190:1140–1149.
- Chang FY, Peacock JE, Musher DM, et al. Staphylococcus aureus bacteremia: recurrence and the impact of antibiotic treatment in a prospective multicenter study. Medicine (Baltimore) 2003; 82:333–339.
- Hidayat LK, Hsu DI, Quist R, Shriner KA, Wong-Beringer A. High-dose vancomycin therapy for methicillin-resistant Staphylococcus aureus infections: efficacy and toxicity. Arch Intern Med 2006; 166:2138–2144.
- Wilson R, Hamburger M. Fifteen years’ experience with staphylococcus septicemia in a large city hospital; analysis of fifty-five cases in the Cincinnati General Hospital 1940 to 1954. Am J Med 1957; 22:437–457.
- Nolan CM, Beaty HN. Staphylococcus aureus bacteremia. Current clinical patterns. Am J Med 1976; 60:495–500.
- Shah M, Watanakunakorn C. Changing patterns of Staphylococcus aureus bacteremia. Am J Med Sci 1979; 278:115–121.
- Fowler VG, Miro JM, Hoen B, et al ICE Investigators. Staphylococcus aureus endocarditis: a consequence of medical progress. JAMA 2005; 293:3012–3021. Erratum in: JAMA 2005; 294:900.
- Von Reyn CF, Levy BS, Arbeit RD, Friedland G, Crumpacker CS. Infective endocarditis: an analysis based on strict case definition. Ann Intern Med 1981; 94:505–518.
- Durack DT, Lukes AS, Bright DK. New criteria for diagnosis of infective endocarditis: utilization of specific echocardiographic findings. Duke Endocarditis Service. Am J Med 1994; 96:200–209.
- Li JS, Sexton DJ, Mick N, et al. Proposed modifications to the Duke criteria for the diagnosis of infective endocarditis. Clin Infect Dis 2000; 30:633–638.
- Stratton JR, Werner JA, Pearlman AS, Janko CL, Kliman S, Jackson MC. Bacteremia and the heart. Serial echocardiographic findings in 80 patients with documented or suspected bacteremia. Am J Med 1982; 73:851–858.
- Bayer AS, Lam K, Ginzton L, Normal DC, Chiu CY, Ward JI. Staphylococcus aureus bacteremia. Clinical, serologic, and echocardiographic findings in patients with and without endocarditis. Arch Intern Med 1987; 147:457–462.
- Miro JM, Anguera I, Cabell CH, et al International Collaboration on Endocarditis Merged Database Study Group. Staphylococcus aureus native valve infective endocarditis: report of 566 episodes from the International Collaboration on Endocarditis Merged Database. Clin Infect Dis 2005; 41:507–514. Erratum in: Clin Infect Dis 2005; 41:1075–1077.
- Jernigan JA, Farr BM. Short-course therapy of catheter-related Staphylococcus aureus bacteremia: a meta-analysis. Ann Intern Med 1993; 119:304–311.
- Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis: diagnosis, antimicrobial therapy, and management of complications: a statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association: endorsed by the Infectious Diseases Society of America. Circulation 2005; 111:e394–e434. Erratum in: Circulation 2005; 112:2373. Circulation 2007; 115:e408.
- Korzeniowski O, Sande MA. Combination antimicrobial therapy for Staphylococcus aureus endocarditis in patients addicted to parenteral drugs and in nonaddicts: a prospective study. Ann Intern Med 1982; 97:496–503.
- Chambers HF, Korzeniowski OM, Sande MA. Staphylococcus aureus endocarditis: clinical manifestations in addicts and nonaddicts. Medicine (Baltimore) 1983; 62:170–177.
- Gentry CA, Rodvold KA, Novak RM, Hershow RC, Naderer OJ. Retrospective evaluation of therapies for Staphylococcus aureus endocarditis. Pharmacotherapy 1997; 17:990–997.
- Small PM, Chambers HF. Vancomycin for Staphylococcus aureus endocarditis in intravenous drug users. Antimicrob Agents Chemother 1990; 34:1227–1231.
- Eliopoulos GM. Quinupristin-dalfopristin and linezolid: evidence and opinion. Clin Infect Dis 2003; 36:473–481.
- Rybak MJ. Therapeutic options for Gram-positive infections. J Hosp Infect 2001; 49 suppl A:S25–S32.
- Micek ST. Alternatives to vancomycin for the treatment of methicillin-resistant Staphylococcus aureus infections. Clin Infect Dis 2007; 45 suppl 3:S184–S190.
- Appelbaum PC, Jacobs MR. Recently approved and investigational antibiotics for treatment of severe infections caused by Gram-positive bacteria. Curr Opin Microbiol 2005; 8:510–517.
- Drew RH, Perfect JR, Srinath L, Kirkimilis E, Dowzicky M, Talbot GH for the Synercid Emergency-Use Study Group. Treatment of methicillin-resistant Staphylococcus aureus infections with quinupristin-dalfopristin in patients intolerant of or failing prior therapy. J Antimicrob Chemother 2000; 46:775–784.
- Lamb HM, Figgitt DP, Faulds D. Quinupristin/dalfopristin: a review of its use in the management of serious gram-positive infections. Drugs 1999; 58:1061–1097.
- Shorr AF, Kunkel MJ, Kollef M. Linezolid versus vancomycin for Staphylococcus aureus bacteraemia: pooled analysis of randomized studies. J Antimicrob Chemother 2005; 56:923–929.
- Bishop E, Melvani S, Howden BP, Charles PG, Grayson ML. Good clinical outcomes but high rates of adverse reactions during linezolid therapy for serious infections: a proposed protocol for monitoring therapy in complex patients. Antimicrob Agents Chemother 2006; 50:1599–1602.
- Boucher HW, Sakoulas G. Perspectives on daptomycin resistance, with emphasis on resistance in Staphylococcus aureus. Clin Infect Dis 2007; 45:601–608.
- Munoz-Price LS, Lolans K, Quinn JP. Four cases of invasive methicillin-resistant Staphylococcus aureus (MRSA) infections treated with tigecycline. Scand J Infect Dis 2006; 38:1081–1084.
- Raad I, Darouiche R, Vazquez J, et al. Efficacy and safety of weekly dalbavancin therapy for catheter-related bloodstream infection caused by gram-positive pathogens. Clin Infect Dis 2005; 40:374–80.
Staphylococcus aureus is rearing its ugly head in new and interesting ways, both in the hospital and in the community.
Rates of invasive infections with methicillin-resistant S aureus (MRSA) have been increasing both in the hospital and in the community, a trend that has attracted considerable interest in the lay media. Curiously, the most common community-associated MRSA strain, which up to now has been distinct from hospital-associated MRSA strains, is invading our hospitals. Alarmingly, vancomycin (Vancocin), the drug of last resort for MRSA infections for the past 40 years, does not seem to be as effective as it used to be.
This paper summarizes the changing epidemiology of S aureus, particularly the emergence of MRSA outside of the hospital; reviews the difficulties associated with S aureus bacteremia and its treatment in view of; some changes in vancomycin susceptibility; and appraises the old and new treatment options.
MRSA IS ON THE RISE IN THE HOSPITAL
S aureus, a gram-positive, coagulase-positive bacterium, is one of the leading nosocomial bloodstream pathogens, second only to coagulase-negative staphylococci.1 And the incidence of S aureus infections is increasing. MRSA in particular is increasingly causing infections throughout hospitals, including intensive care units. As of 2004, nearly two-thirds of isolates of S aureus from intensive care units were MRSA.2
MRSA infections are worse than methicillin-susceptible S aureus (MSSA) infections in terms of the rates of death and other undesirable outcomes.3 Several factors may be responsible: MRSA infection may be a marker of severity of illness (sicker patients may be more likely to have MRSA), our treatment for MRSA may not be as effective as it is for MSSA, and the organism may be inherently more virulent.
METHICILLIN RESISTANCE IS ALSO ON THE RISE IN THE COMMUNITY
Community-associated MRSA began emerging clinically about 10 years ago. It was first described in a cohort of children with necrotizing pneumonia in Minnesota, but soon other populations at risk began to emerge, such as residents of correctional facilities, men who had sex with men, competitive athletes (eg, fencers, wrestlers, and football players), and Alaskan natives and other native populations. A common factor in all these groups was close proximity of the members to each other. Later, it began to spread beyond these traditional risk groups into the community at large.
Community-associated MRSA strains have a characteristic pattern of antimicrobial susceptibility (see below). In the laboratory, they grow somewhat faster than health-care-associated MRSA strains, but not as fast as MSSA. They have a strong association with skin and soft-tissue infections: when you see a skin or soft-tissue infection, be it in an outpatient or an inpatient, think about MRSA. Their virulence varies, but rapid onset and progression of illness are quite common. Their most common strain in the United States at present is USA 300.
Case 1: A young woman with necrotizing fasciitis
A 21-year-old college student presented to our service in May 2004 with high fever and severe arm pain, which had been worsening for several days. She had been previously healthy, had not had any contact with the health care system, and had not received any antibiotics.
Her blood cultures were positive for MRSA, as were cultures of the deep tissue of the deltoid muscle and fascia when she underwent emergency surgical debridement. The infection required several additional surgical debridements and removal of one head of her deltoid muscle, but she was fortunate: in the past, some patients with this problem might have undergone radical amputation of the arm or even more extensive surgery. This patient continued to have positive blood cultures 4 days postoperatively, but she ultimately recovered, completing 28 days of daptomycin (Cubicin) therapy at a dose of 6 mg/kg every 24 hours. The last 10 days of daptomycin therapy were given at home via a percutaneous intravenous central catheter.
Comment. The epidemiology of MRSA infections is changing. More patients who have no traditional risk factors, specifically health care contact, are getting MRSA infections. A recent report from the US Centers for Disease Control and Prevention (CDC) indicates that the proportion of patients with invasive disease due to MRSA has doubled since 2001–2002.4 Part of the reason undoubtedly is that MRSA, particularly community-associated MRSA, often carries specific virulence factors that make it more invasive. The CDC estimated that in 2005 there were nearly 100,000 cases of invasive MRSA infection in the United States, and nearly a fifth of these infections resulted in death.
Resistance and virulence factors in community-associated MRSA
Most community-associated MRSA strains carry a mobile genetic element called type IV SCCmec (staphylococcal chromosomal cassettemec) that enhances its antimicrobial resistance. This genetic component was probably borrowed from coagulase-negative staphylococci, in which it is quite common but does not cause as much of a problem. It is now present in a wide range of S aureus strains. Most of the S aureus strains that carry type IV SCCmec are MRSA, but a few MSSA strains do carry it as well.
The potent toxin Panton-Valentine leukocidin is an extracellular product that is detected in fewer than 5% of hospital strains but is more common in community-associated strains. It kills leukocytes by forming pores in the cell membrane and causing skin necrosis in cutaneous infections. It is associated with skin abscesses and rapidly progressive necrotizing pneumonia in MSSA or MRSA.
Epidemiologic differences between community- and health-care-associated MRSA
Patients with community-associated MRSA infections tend to be younger than those who traditionally get health-care-associated MRSA infections: in a study from Naimi et al in 2003, the mean ages were 23 vs 68 years.5 A greater proportion of patients with community-associated MRSA strains are nonwhite.4,5
Most community-associated MRSA infections are of the skin and soft tissue (75% in the series from Naimi et al5), but this pathogen causes other infections as well. Bacteremia of unknown origin has been seen, as has necrotizing pneumonia. Most of the skin and soft-tissue infections are relatively superficial, such as folliculitis or furunculosis, but deeper tissue infections such as necrotizing fasciitis and pyomyositis have also been seen.6
The incidence of community-associated MRSA infections varies greatly by geographic region.7 The northeastern United States has so far been relatively spared, but in Atlanta, Houston, and Los Angeles up to 80% of cases of characteristic skin or soft-tissue infections seen in emergency or outpatient departments are due to community-associated MRSA. Physicians at the Texas Children’s Hospital in Houston assume that all skin or soft-tissue infections are due to community-associated MRSA unless proven otherwise.8
Differences in antibiotic susceptibility
Community-associated MRSA is more susceptible to various antibiotics than health-care-associated MRSA,5 but not by much. Strains are usually susceptible to vancomycin, tetracyclines, trimethoprim-sulfamethoxazole (Bactrim, Septra), and rifampin (Rifadin). Unlike hospital strains, a fair number of community-acquired strains are susceptible to clindamycin (Cleocin) in the laboratory, but with a caveat: some of these clindamycin-susceptible strains actually may harbor the tools for inducible resistance. In fact, they can become resistant to clindamycin even without being exposed to it.
The laboratory test for inducible clindamycin resistance is called the D test. After coating an agar plate with S aureus, the technician places erythromycin and clindamycin disks. If the erythromycin induces clindamycin resistance, the plate is clear of growth around the clindamycin disk except for the portion nearest the erythromycin disk, leaving a characteristic D-shaped area of lucency.
Risk factors for MRSA
Moran et al7 analyzed the risk factors for community-associated MRSA in patients with skin or soft-tissue infections seen in the emergency department. The infection was more likely to be due to community-associated MRSA if the patient was black, had used any antibiotic in the past month, had a history of MRSA infection, or had close contact with a person with a similar infection. Many patients interpreted the infections as spider bites because the lesions tended to have a dark center surrounded by a tender area. These infections were not associated with underlying illness. In some cases, community-associated MRSA skin infections have been associated with tattooing and even manicuring.
However, it is very difficult to distinguish between community-associated MRSA and MSSA skin and soft-tissue infections on the basis of clinical and epidemiologic characteristics. Miller et al9 studied a large group of patients in Los Angeles who were hospitalized with community-associated skin and soft-tissue S aureus infections. All the patients were followed up for 30 days after hospital discharge. Regardless of whether they had MRSA or MSSA, they had similar outcomes. Close contacts of the patients also tended to develop infection.
A key point from this and many other studies: patients were more likely to remain infected if they did not undergo incision and drainage. This key intervention is indicated for any patient who has a skin and soft-tissue infection with an undrained focus of infection.
COMMUNITY-ASSOCIATED MRSA IS INVADING THE HOSPITAL
In a new development, community-associated MRSA strains are now appearing in the hospital. This is not only because patients are bacteremic when they come in: patients in the hospital are getting nosocomial infections due to community-associated MRSA strains.
Seybold et al10 analyzed 116 cases of MRSA bloodstream infections in Atlanta, GA. In 9 (8%) of the cases the patient had not had any contact with the health care system within the past year, and these cases were classified as truly community-associated. Of the remaining 107 cases, 49 (42%) were nosocomial, and the USA 300 strain—the predominant community-associated MRSA strain—accounted for 10 (20%) of the nosocomial cases.
In the recent CDC study of invasive MRSA infections, Klevens et al4 reported that nearly a third of cases of bacteremia were due to community-associated MRSA, and these strains accounted for a greater proportion of cases of cellulitis and endocarditis than did health-care-associated strains.
In a study of hospital-associated MRSA, Maree et al11 found that the percentage of cases in which the bacteria carried the SCCmec type IV marker had increased from less than 20% in 1999 to more than 50% in 2004.
Comment. Suffice it to say that we are surrounded by MRSA. Community-associated MRSA is here to stay. It is even invading our hospitals, and we need to consider this very carefully when choosing antimicrobial therapy.
NAGGING QUESTIONS ABOUT VANCOMYCIN
Case 2: Vancomycin-intermediate S aureus (VISA) bacteremia and endocarditis
In December 2006 we saw a very ill 60-year-old woman who was hospitalized with MRSA bacteremia, pacemaker endocarditis, and superior vena cava thrombosis. Although she was treated with vancomycin and rifampin, her condition worsened, she had a stroke, and she developed renal failure. In a difficult operation, the pacemaker was removed, but the bacteremia persisted. In early February 2007 she underwent another difficult operation in which the superior vena cava clot was debrided, a right atrial clot was removed, and her mitral valve was replaced. Less than 2 weeks later, and despite ongoing vancomycin and rifampin therapy, the MRSA bacteremia recurred.
During the approximately 6 weeks that the patient had been receiving these antibiotics, the minimal inhibitory concentration (MIC) of rifampin against the S aureus isolate increased from less than 1 μg/mL (susceptible) to 2 μg/mL (resistant). The MIC of vancomycin went from 2 μg/mL (susceptible) to 4 μg/mL (intermediately susceptible). Vancomycin and rifampin were discontinued, and daptomycin and gentamicin (Garamycin) therapy were started. (Her daptomycin MIC was 0.5 μg/mL). The patient’s condition stabilized, and she was discharged to a long-term nursing facility. She had no relapse of MRSA bacteremia, but she died in early April of that year.
Is vancomycin becoming less effective? Degrees of vancomycin resistance
Vancomycin has been our stalwart for treating MRSA infections for more than 40 years but it is not working as well as it used to, at least in certain situations.
VRSA (vancomycin-resistant S aureus) is rare. These fully resistant strains probably acquired a resistance mechanism (the vanA operon) from vancomycin-resistant enterococci. Infections tend to occur in patients simultaneously infected with both S aureus and vancomycin-resistant enterococci, giving the bacteria an opportunity to exchange genetic material.
VISA (vancomycin-intermediate S aureus) infections tend to occur in patients like the one described above who have had long-term vancomycin therapy. VISA strains appear to overproduce a matrix that captures vancomycin and keeps it from entering the cell. On electron microscopy, these bacteria have a very thick cell wall.13
Vancomycin tolerance is a state in which the bacteria are “stunned” or kept in check but not killed by vancomycin. That is manifested in the laboratory by a ratio of minimum bactericidal concentration to MIC greater than 32.
hVISA (heteroresistant VISA) is new and worrisome. These organisms have an overall MIC in the susceptible range, but within that population are individual isolates with an MIC that is much higher—in the intermediate or perhaps even in the resistant range.14
Reported rates of hVISA vary from less than 2% to as high as 76%, because the methods for detecting it are still very poorly standardized. The usual automated laboratory tests do not detect hVISA.
hVISA is probably clinically relevant, as evidence is emerging both in vitro and in vivo that the higher the MIC for vancomycin, the worse the clinical outcome.15 hVISA has been associated with failures of therapy in several situations, usually in cases of severe invasive or deep infection, endocarditis, and bacteremia with vertebral osteomyelitis where vancomycin concentrations at the site of infection may be suboptimal.16–19 While most hVISA strains that have been described were resistant to methicillin, some were susceptible.
The E test is emerging as the standard test for hVISA. This test uses a plastic strip that contains gradually increasing concentrations of vancomycin along its length. Placed in the culture dish, the strip inhibits growth of the organism at its high-concentration end but not at its low-concentration end. If the sample contains hVISA, the cutoff is not well defined, with a few colonies growing at higher concentrations.
New definition of vancomycin susceptibility
Recognizing that the MICs for vancomycin have been rising in the last few years, the Clinical and Laboratory Standards Institute last year changed the break points between susceptibility and resistance. The new definitions are:
- Susceptible—an MIC of 2.0 μg/mL or less (formerly 4.0 μg/mL or less)
- Intermediate—4.0 to 8.0 μg/mL (formerly 8.0 to 16 μg/mL)
- Resistant—16 μg/mL or greater (formerly 32 μg/mL or greater).
One should pay attention to the MIC numbers on the laboratory reports, not just to the words “susceptible” or “not susceptible.” If the number is, say, 0.5 μg/mL or less, the organism should really be susceptible. If the number is 1 or 2, it is still in the susceptible range, but those are the organisms that may cause problems later on.
Further, even if the vancomycin MIC is in the susceptible range, higher MICs may affect outcomes. The average duration of MRSA bacteremia on therapy is 8 to 9 days, vs 3 to 4 days with MSSA bacteremia.20,21 But Sakoulas et al15 found that, in MRSA bacteremia, the success rate with vancomycin therapy was 56% if the MIC was 0.5 or lower, compared with 10% if the MIC was 1.0 to 2.0 μg/mL. Examined in another way, the success rate was 50% if the logarithm of killing was 6.27 colony-forming units per mL or greater, 23% if 4.71 to 6.26, and zero if less than 4.71.
Case 3: Prolonged MRSA bacteremia
In the summer of 2006, a 66-year-old woman with a history of gastric bypass and cirrhosis underwent a long stay in the surgical intensive care unit because of a recurrent enterocutaneous fistula and chronic renal insufficiency. On November 5th, she had a positive blood culture for MRSA, which was treated appropriately with vancomycin for 4 weeks. She was discharged to subacute care but came back 2 days later, again with MRSA bacteremia. At that time her Hickman catheter, which had been inserted for total parenteral nutrition because of the enterocutaneous fistula, was removed.
Transthoracic echocardiography revealed no vegetations, but her bacteremia persisted. Her mental status was poor this entire time: she was mute and could barely be awakened. We looked for clots and infected clots; duplex ultrasonographic examinations of all four extremities were negative. Finally, magnetic resonance imaging of her back—performed empirically because of the persistent bacteremia—revealed vertebral osteomyelitis at level T12-L1. We also noticed on serial evaluations that the vancomycin MIC for her organism increased from 0.5 to 2.0 μg/mL, so therapy was changed from vancomycin to daptomycin.
Her bacteremia cleared. Follow-up echocardiography was negative, but she had two subsequent relapses of MRSA bacteremia, one in April 2007 and one before she died in the summer of 2007.
Prolonged bacteremia: Is it vancomycin resistance, or something else?
The MRSA isolates that cause prolonged bacteremia seem to have certain characteristics.22 Higher MICs are probably associated with longer periods of bacteremia. But some genetic components within some strains of S aureus give them a survival advantage. They have less susceptibility to the body’s thrombin-induced platelet microbicidal protein. These isolates are not only associated with prolonged bacteremia: they are also associated with osteomyelitis, deep abscesses, endocarditis, recurrent infection, and increased death rate.22 Clinical laboratories do not test for these genetic components. One wonders whether our patient may have had an isolate with these mutations that gave it a survival advantage.
Do not use vancomycin for MSSA
Avoid using vancomycin for MSSA infections. It has been shown time and time again that MSSA infections do not respond as well to vancomycin as they do to beta-lactam antibiotics, specifically to the semisynthetic penicillins such as oxacillin and nafcillin, and even some of the first-generation cephalosporins. Chang et al23 found that patients with MSSA bacteremia had higher rates of persistent infections, relapse, and bacteriologic failure if they received vancomycin than if they received nafcillin.
Do vancomycin trough levels affect toxicity?
The vancomycin trough levels that we aimed for in the past (5 to 10 μg/mL) were probably too low. Today, we aim for trough levels of 15 to 20 μg/mL, and many physicians are aiming for 20 to 25 μg/mL. Part of the reason is that vancomycin MICs are higher than they used to be: in order to keep the vancomycin level above the MIC for a longer period of time, the vancomycin trough level needs to be higher. In theory, keeping the vancomycin levels above the MIC for longer periods should improve outcomes. Yet Fowler et al22 found that vancomycin trough levels among patients who had persistent MRSA bacteremia were actually higher than trough levels among those in whom the bacteremia resolved, although the difference was not statistically significant.
We measure the vancomycin trough level to make sure it is high enough (and give larger doses if it is not); among adults, peak levels need not be monitored on a routine basis because of the predictable pharmacokinetics of vancomycin.
Vancomycin toxicity can be either idiosyncratic or synergistic. Idiosyncratic toxicity occurs when a patient who has been on vancomycin for a long time develops a fixed rash, not associated with infusion. This is an immunologic phenomenon. It is a rare and very serious situation and may require steroid therapy.
Synergistic toxicity occurs when vancomycin is given with other nephrotoxic agents, notably gentamicin. Vancomycin plus gentamicin equals nephrotoxicity. Vancomycin alone is usually not nephrotoxic, but close monitoring of renal function parameters is warranted with the use of higher doses.24
IN UNEXPLAINED BACTEREMIA, LOOK FOR ENDOCARDITIS
In blood cultures from patients with bacteremia, S aureus is never a contaminant. Even if just one blood culture is positive for S aureus, believe that S aureus is the culprit.
Reports in the 1950s suggested that at least half of patients who had S aureus bacteremia had endocarditis,25 leading to recommendations that all patients with S aureus bacteremia without an obvious primary source of infection should be evaluated for endocarditis. Subsequent estimates were lower, in the range of 15% to 25%.26,27 However, throughout the world S aureus endocarditis continues to have a very high mortality rate: at least a third of patients die.28
Clinical criteria (community acquisition, no primary focus, and metastatic sequelae) were developed to try to predict the risk of endocarditis in bacteremic patients.26 However, these criteria did not work very well. The clinical definition of endocarditis has evolved. The criteria of von Reyn et al29 from 1981 did not use echocardiography as part of the definition, but the 1994 Duke criteria,30 which were refined31 in 2000, use both clinical and echocardiographic parameters.
Stratton et al32 performed transthoracic echocardiography in 14 patients with bacteremia and found 1 patient with cryptic tricuspid infective endocarditis. Bayer et al33 subsequently reported that of 72 patients with bacteremia, 6 (18%) of those who had no clinical findings suggestive of infectious endocarditis had findings on echocardiography that led to changes in their regimen. Adding echocardiography to three clinical risk factors increased the sensitivity of diagnosing endocarditis from 70% to 85% with a specificity of 100% and predictive value of 96%.
The Duke criteria call for transesophageal echocardiography, which is not feasible in some patients, eg, those with cirrhosis and esophageal varices.
S aureus endocarditis has changed over the years as our patient population has changed, and MRSA endocarditis tends to hit some of our most vulnerable patients. In a study by Miro et al34 in 2005, MRSA was the leading pathogen in patients who were diagnosed with S aureus endocarditis in 1990 or later. We will only see these numbers go up. Patients with diabetes tend to have more MRSA, and of diabetic patients with MRSA endocarditis, 30% to 40% die in the hospital.
Indications for surgery
Certain conditions are indications for surgery among patients with endocarditis, and no antibiotic will cure the endocarditis if the patient has one of these conditions, eg:
- Persistent bacteremia during antibiotic therapy
- Recurrent emboli
- Heart failure that cannot be controlled
- Perivalvular or myocardial abscesses
- Large vegetations
- Early prosthetic valve infection
- Certain arrhythmias.
How long should S aureus bacteremia be treated?
In cases of bacteremia in which endocarditis has been ruled out and removable foci of infection (eg, intravascular catheters) have been removed, some evidence indicates that treatment for 2 weeks would be as effective as the 4 to 6 weeks that we would use for endocarditis or other severe or invasive infections.35 The issue is controversial. If the patient has had frequent hospitalizations or a chronic medical condition I would hesitate to treat for less than 4 weeks, even if the infection appears to be associated with a removable focus.
Treatment of endocarditis
In the guidelines for treatment of endocarditis from the American Heart Association and Infectious Diseases Society of America,36 all the recommendations are relatively old and many of them are somewhat empiric—they are not based on evidence from randomized clinical trials. Rather, they are best opinions based on clinical experience and some observational studies over the years.
For MSSA. In cases of native-valve endocarditis, oxacillin (Bactocill), nafcillin (Unipen), or another semisynthetic beta-lactam antibiotic is recommended. For penicillin-allergic patients, we have other options, such as cefazolin (Ancef, Kefzol).
Combination therapy is frequently recommended for native valve endocarditis as well as for prosthetic valve endocarditis, with either rifampin or gentamicin along with a primary agent. There is some evidence that one can clear staphylococcal bacteremia a day or two more quickly by use of combination therapy with nafcillin plus an aminoglycoside than with nafcillin alone.37,38 For MSSA-associated endocarditis, vancomycin does not work as well as beta-lactam antibiotics.39,40
Korzeniowski and Sande37 and Chambers et al38 reported that the mean duration of bacteremia was 3.4 days for patients treated with nafcillin alone and 2.9 days for those treated with nafcillin plus an aminoglycoside. These studies led to consideration of a short course of gentamicin to clear the bacteremia quickly.
With MRSA, bacteremia often requires a week or more to clear. Levine et al21 reported a study in 42 patients, mostly injection-drug users, with right-sided native-valve endocarditis. The median duration of bacteremia was 7 days in patients who received vancomycin alone vs 9 days in those who received vancomycin plus rifampin; however, some patients were bacteremic for up to 27 days. Fever persisted for a median of 7 days, probably partly due to septic pulmonary emboli. Three patients died, and three required valve replacement.
NEW ANTIBIOTICS
Several new antibiotics are active against gram-positive cocci.41–44 However, the majority of them have not been prospectively studied for treating bacteremia or endocarditis.
Quinupristin/dalfopristin (Synercid) has not been formally studied for treatment of MRSA bacteremia or endocarditis. There are a few case reports of its use in these conditions.45 Quinupristin/dalfopristin is bacteriostatic, and its use may be associated with phlebitis, myalgias, and arthralgias.46
Linezolid (Zyvox) is approved for treatment of complicated skin and soft-tissue infections and for hospital-acquired pneumonia. There have been no specific studies of linezolid in the treatment of S aureus bacteremia or endocarditis. However, Shorr et al47 retrospectively looked at the bacteremic patients in five previous studies of linezolid vs vancomycin and found 144 cases of S aureus bacteremia, half of which were due to MRSA. Of 53 assessable patients with MRSA bacteremia, the primary infection was cured in 14 (56%) of the linezolid patients and 13 (46%) of the vancomycin patients.
The oral form is 100% bioavailable. One should avoid concomitant use of serotonin-reuptake inhibitors because of the risk of serotonin syndrome. Adverse effects include altered taste sensation and peripheral neuropathy. There are other potential toxicities, including hematologic changes (thrombocytopenia, leukopenia) and metabolic effects (lactic acidosis), so clinical and laboratory monitoring is important.48 The role of linezolid in the treatment of patients with S aureus bacteremia or endocarditis remains to be defined.
Daptomycin is indicated for complicated skin and soft-tissue infections, bacteremia, and right-sided endocarditis due to S aureus. Fowler et al20 found that daptomycin was not inferior to beta-lactam antibiotics for treatment of MSSA bacteremia and right-sided endocarditis, and for MRSA infections it outperformed vancomycin, but the difference was not statistically significant.
The dosing interval should be increased from once every 24 hours to every 48 hours if the creatinine clearance is 30 mL/minute or less. Adverse effects include myalgia, rhabdomyolysis (rare), and elevations in creatine phosphokinase. Reports of rising MICs during daptomycin therapy, in some cases associated with persistent infection,49 suggest that careful attention be paid to dosing and clinical monitoring.
Tigecycline (Tygacil) is indicated for complicated skin and soft-tissue infections and complicated intra-abdominal infections due to susceptible organisms. It is active against both MSSA and MRSA, but clinical experience with its use in invasive infections is somewhat limited.50 The dose of tigecycline should be reduced in advanced cirrhosis. Adverse effects include nausea and vomiting.
Telavancin, dalbavancin, and oritavancin, investigational parenteral antibiotics that are derivatives of vancomycin, are in clinical trials. The pharmacokinetic activity of these agents is of interest: telavancin is being studied with a once-daily dosing interval and dalbavancin’s half-life allows once-weekly dosing. In a limited trial, dalbavancin was found to be safe and effective in the treatment of catheter-related bloodstream infections.51 None of the antibiotics in this group has been studied for treatment of S aureus endocarditis. Telavancin therapy has been associated with rash, hypokalemia, QT prolongation, and creatinine elevations. Gastrointestinal symptoms have been reported with the use of dalbavancin.
Ceftobiprole, another investigational agent, is the only cephalosporin antibiotic that is active against MRSA. It is given every 12 hours. Adverse effects include nausea and taste disturbance.
Iclaprim is a novel diaminopyrimidine and a dihydrofolate reductase inhibitor. In vitro, it is active against gram-positive bacteria, including MRSA, VISA, and VRSA; clinical investigations at this point are limited to the treatment of skin and soft-tissue infections.
Staphylococcus aureus is rearing its ugly head in new and interesting ways, both in the hospital and in the community.
Rates of invasive infections with methicillin-resistant S aureus (MRSA) have been increasing both in the hospital and in the community, a trend that has attracted considerable interest in the lay media. Curiously, the most common community-associated MRSA strain, which up to now has been distinct from hospital-associated MRSA strains, is invading our hospitals. Alarmingly, vancomycin (Vancocin), the drug of last resort for MRSA infections for the past 40 years, does not seem to be as effective as it used to be.
This paper summarizes the changing epidemiology of S aureus, particularly the emergence of MRSA outside of the hospital; reviews the difficulties associated with S aureus bacteremia and its treatment in view of; some changes in vancomycin susceptibility; and appraises the old and new treatment options.
MRSA IS ON THE RISE IN THE HOSPITAL
S aureus, a gram-positive, coagulase-positive bacterium, is one of the leading nosocomial bloodstream pathogens, second only to coagulase-negative staphylococci.1 And the incidence of S aureus infections is increasing. MRSA in particular is increasingly causing infections throughout hospitals, including intensive care units. As of 2004, nearly two-thirds of isolates of S aureus from intensive care units were MRSA.2
MRSA infections are worse than methicillin-susceptible S aureus (MSSA) infections in terms of the rates of death and other undesirable outcomes.3 Several factors may be responsible: MRSA infection may be a marker of severity of illness (sicker patients may be more likely to have MRSA), our treatment for MRSA may not be as effective as it is for MSSA, and the organism may be inherently more virulent.
METHICILLIN RESISTANCE IS ALSO ON THE RISE IN THE COMMUNITY
Community-associated MRSA began emerging clinically about 10 years ago. It was first described in a cohort of children with necrotizing pneumonia in Minnesota, but soon other populations at risk began to emerge, such as residents of correctional facilities, men who had sex with men, competitive athletes (eg, fencers, wrestlers, and football players), and Alaskan natives and other native populations. A common factor in all these groups was close proximity of the members to each other. Later, it began to spread beyond these traditional risk groups into the community at large.
Community-associated MRSA strains have a characteristic pattern of antimicrobial susceptibility (see below). In the laboratory, they grow somewhat faster than health-care-associated MRSA strains, but not as fast as MSSA. They have a strong association with skin and soft-tissue infections: when you see a skin or soft-tissue infection, be it in an outpatient or an inpatient, think about MRSA. Their virulence varies, but rapid onset and progression of illness are quite common. Their most common strain in the United States at present is USA 300.
Case 1: A young woman with necrotizing fasciitis
A 21-year-old college student presented to our service in May 2004 with high fever and severe arm pain, which had been worsening for several days. She had been previously healthy, had not had any contact with the health care system, and had not received any antibiotics.
Her blood cultures were positive for MRSA, as were cultures of the deep tissue of the deltoid muscle and fascia when she underwent emergency surgical debridement. The infection required several additional surgical debridements and removal of one head of her deltoid muscle, but she was fortunate: in the past, some patients with this problem might have undergone radical amputation of the arm or even more extensive surgery. This patient continued to have positive blood cultures 4 days postoperatively, but she ultimately recovered, completing 28 days of daptomycin (Cubicin) therapy at a dose of 6 mg/kg every 24 hours. The last 10 days of daptomycin therapy were given at home via a percutaneous intravenous central catheter.
Comment. The epidemiology of MRSA infections is changing. More patients who have no traditional risk factors, specifically health care contact, are getting MRSA infections. A recent report from the US Centers for Disease Control and Prevention (CDC) indicates that the proportion of patients with invasive disease due to MRSA has doubled since 2001–2002.4 Part of the reason undoubtedly is that MRSA, particularly community-associated MRSA, often carries specific virulence factors that make it more invasive. The CDC estimated that in 2005 there were nearly 100,000 cases of invasive MRSA infection in the United States, and nearly a fifth of these infections resulted in death.
Resistance and virulence factors in community-associated MRSA
Most community-associated MRSA strains carry a mobile genetic element called type IV SCCmec (staphylococcal chromosomal cassettemec) that enhances its antimicrobial resistance. This genetic component was probably borrowed from coagulase-negative staphylococci, in which it is quite common but does not cause as much of a problem. It is now present in a wide range of S aureus strains. Most of the S aureus strains that carry type IV SCCmec are MRSA, but a few MSSA strains do carry it as well.
The potent toxin Panton-Valentine leukocidin is an extracellular product that is detected in fewer than 5% of hospital strains but is more common in community-associated strains. It kills leukocytes by forming pores in the cell membrane and causing skin necrosis in cutaneous infections. It is associated with skin abscesses and rapidly progressive necrotizing pneumonia in MSSA or MRSA.
Epidemiologic differences between community- and health-care-associated MRSA
Patients with community-associated MRSA infections tend to be younger than those who traditionally get health-care-associated MRSA infections: in a study from Naimi et al in 2003, the mean ages were 23 vs 68 years.5 A greater proportion of patients with community-associated MRSA strains are nonwhite.4,5
Most community-associated MRSA infections are of the skin and soft tissue (75% in the series from Naimi et al5), but this pathogen causes other infections as well. Bacteremia of unknown origin has been seen, as has necrotizing pneumonia. Most of the skin and soft-tissue infections are relatively superficial, such as folliculitis or furunculosis, but deeper tissue infections such as necrotizing fasciitis and pyomyositis have also been seen.6
The incidence of community-associated MRSA infections varies greatly by geographic region.7 The northeastern United States has so far been relatively spared, but in Atlanta, Houston, and Los Angeles up to 80% of cases of characteristic skin or soft-tissue infections seen in emergency or outpatient departments are due to community-associated MRSA. Physicians at the Texas Children’s Hospital in Houston assume that all skin or soft-tissue infections are due to community-associated MRSA unless proven otherwise.8
Differences in antibiotic susceptibility
Community-associated MRSA is more susceptible to various antibiotics than health-care-associated MRSA,5 but not by much. Strains are usually susceptible to vancomycin, tetracyclines, trimethoprim-sulfamethoxazole (Bactrim, Septra), and rifampin (Rifadin). Unlike hospital strains, a fair number of community-acquired strains are susceptible to clindamycin (Cleocin) in the laboratory, but with a caveat: some of these clindamycin-susceptible strains actually may harbor the tools for inducible resistance. In fact, they can become resistant to clindamycin even without being exposed to it.
The laboratory test for inducible clindamycin resistance is called the D test. After coating an agar plate with S aureus, the technician places erythromycin and clindamycin disks. If the erythromycin induces clindamycin resistance, the plate is clear of growth around the clindamycin disk except for the portion nearest the erythromycin disk, leaving a characteristic D-shaped area of lucency.
Risk factors for MRSA
Moran et al7 analyzed the risk factors for community-associated MRSA in patients with skin or soft-tissue infections seen in the emergency department. The infection was more likely to be due to community-associated MRSA if the patient was black, had used any antibiotic in the past month, had a history of MRSA infection, or had close contact with a person with a similar infection. Many patients interpreted the infections as spider bites because the lesions tended to have a dark center surrounded by a tender area. These infections were not associated with underlying illness. In some cases, community-associated MRSA skin infections have been associated with tattooing and even manicuring.
However, it is very difficult to distinguish between community-associated MRSA and MSSA skin and soft-tissue infections on the basis of clinical and epidemiologic characteristics. Miller et al9 studied a large group of patients in Los Angeles who were hospitalized with community-associated skin and soft-tissue S aureus infections. All the patients were followed up for 30 days after hospital discharge. Regardless of whether they had MRSA or MSSA, they had similar outcomes. Close contacts of the patients also tended to develop infection.
A key point from this and many other studies: patients were more likely to remain infected if they did not undergo incision and drainage. This key intervention is indicated for any patient who has a skin and soft-tissue infection with an undrained focus of infection.
COMMUNITY-ASSOCIATED MRSA IS INVADING THE HOSPITAL
In a new development, community-associated MRSA strains are now appearing in the hospital. This is not only because patients are bacteremic when they come in: patients in the hospital are getting nosocomial infections due to community-associated MRSA strains.
Seybold et al10 analyzed 116 cases of MRSA bloodstream infections in Atlanta, GA. In 9 (8%) of the cases the patient had not had any contact with the health care system within the past year, and these cases were classified as truly community-associated. Of the remaining 107 cases, 49 (42%) were nosocomial, and the USA 300 strain—the predominant community-associated MRSA strain—accounted for 10 (20%) of the nosocomial cases.
In the recent CDC study of invasive MRSA infections, Klevens et al4 reported that nearly a third of cases of bacteremia were due to community-associated MRSA, and these strains accounted for a greater proportion of cases of cellulitis and endocarditis than did health-care-associated strains.
In a study of hospital-associated MRSA, Maree et al11 found that the percentage of cases in which the bacteria carried the SCCmec type IV marker had increased from less than 20% in 1999 to more than 50% in 2004.
Comment. Suffice it to say that we are surrounded by MRSA. Community-associated MRSA is here to stay. It is even invading our hospitals, and we need to consider this very carefully when choosing antimicrobial therapy.
NAGGING QUESTIONS ABOUT VANCOMYCIN
Case 2: Vancomycin-intermediate S aureus (VISA) bacteremia and endocarditis
In December 2006 we saw a very ill 60-year-old woman who was hospitalized with MRSA bacteremia, pacemaker endocarditis, and superior vena cava thrombosis. Although she was treated with vancomycin and rifampin, her condition worsened, she had a stroke, and she developed renal failure. In a difficult operation, the pacemaker was removed, but the bacteremia persisted. In early February 2007 she underwent another difficult operation in which the superior vena cava clot was debrided, a right atrial clot was removed, and her mitral valve was replaced. Less than 2 weeks later, and despite ongoing vancomycin and rifampin therapy, the MRSA bacteremia recurred.
During the approximately 6 weeks that the patient had been receiving these antibiotics, the minimal inhibitory concentration (MIC) of rifampin against the S aureus isolate increased from less than 1 μg/mL (susceptible) to 2 μg/mL (resistant). The MIC of vancomycin went from 2 μg/mL (susceptible) to 4 μg/mL (intermediately susceptible). Vancomycin and rifampin were discontinued, and daptomycin and gentamicin (Garamycin) therapy were started. (Her daptomycin MIC was 0.5 μg/mL). The patient’s condition stabilized, and she was discharged to a long-term nursing facility. She had no relapse of MRSA bacteremia, but she died in early April of that year.
Is vancomycin becoming less effective? Degrees of vancomycin resistance
Vancomycin has been our stalwart for treating MRSA infections for more than 40 years but it is not working as well as it used to, at least in certain situations.
VRSA (vancomycin-resistant S aureus) is rare. These fully resistant strains probably acquired a resistance mechanism (the vanA operon) from vancomycin-resistant enterococci. Infections tend to occur in patients simultaneously infected with both S aureus and vancomycin-resistant enterococci, giving the bacteria an opportunity to exchange genetic material.
VISA (vancomycin-intermediate S aureus) infections tend to occur in patients like the one described above who have had long-term vancomycin therapy. VISA strains appear to overproduce a matrix that captures vancomycin and keeps it from entering the cell. On electron microscopy, these bacteria have a very thick cell wall.13
Vancomycin tolerance is a state in which the bacteria are “stunned” or kept in check but not killed by vancomycin. That is manifested in the laboratory by a ratio of minimum bactericidal concentration to MIC greater than 32.
hVISA (heteroresistant VISA) is new and worrisome. These organisms have an overall MIC in the susceptible range, but within that population are individual isolates with an MIC that is much higher—in the intermediate or perhaps even in the resistant range.14
Reported rates of hVISA vary from less than 2% to as high as 76%, because the methods for detecting it are still very poorly standardized. The usual automated laboratory tests do not detect hVISA.
hVISA is probably clinically relevant, as evidence is emerging both in vitro and in vivo that the higher the MIC for vancomycin, the worse the clinical outcome.15 hVISA has been associated with failures of therapy in several situations, usually in cases of severe invasive or deep infection, endocarditis, and bacteremia with vertebral osteomyelitis where vancomycin concentrations at the site of infection may be suboptimal.16–19 While most hVISA strains that have been described were resistant to methicillin, some were susceptible.
The E test is emerging as the standard test for hVISA. This test uses a plastic strip that contains gradually increasing concentrations of vancomycin along its length. Placed in the culture dish, the strip inhibits growth of the organism at its high-concentration end but not at its low-concentration end. If the sample contains hVISA, the cutoff is not well defined, with a few colonies growing at higher concentrations.
New definition of vancomycin susceptibility
Recognizing that the MICs for vancomycin have been rising in the last few years, the Clinical and Laboratory Standards Institute last year changed the break points between susceptibility and resistance. The new definitions are:
- Susceptible—an MIC of 2.0 μg/mL or less (formerly 4.0 μg/mL or less)
- Intermediate—4.0 to 8.0 μg/mL (formerly 8.0 to 16 μg/mL)
- Resistant—16 μg/mL or greater (formerly 32 μg/mL or greater).
One should pay attention to the MIC numbers on the laboratory reports, not just to the words “susceptible” or “not susceptible.” If the number is, say, 0.5 μg/mL or less, the organism should really be susceptible. If the number is 1 or 2, it is still in the susceptible range, but those are the organisms that may cause problems later on.
Further, even if the vancomycin MIC is in the susceptible range, higher MICs may affect outcomes. The average duration of MRSA bacteremia on therapy is 8 to 9 days, vs 3 to 4 days with MSSA bacteremia.20,21 But Sakoulas et al15 found that, in MRSA bacteremia, the success rate with vancomycin therapy was 56% if the MIC was 0.5 or lower, compared with 10% if the MIC was 1.0 to 2.0 μg/mL. Examined in another way, the success rate was 50% if the logarithm of killing was 6.27 colony-forming units per mL or greater, 23% if 4.71 to 6.26, and zero if less than 4.71.
Case 3: Prolonged MRSA bacteremia
In the summer of 2006, a 66-year-old woman with a history of gastric bypass and cirrhosis underwent a long stay in the surgical intensive care unit because of a recurrent enterocutaneous fistula and chronic renal insufficiency. On November 5th, she had a positive blood culture for MRSA, which was treated appropriately with vancomycin for 4 weeks. She was discharged to subacute care but came back 2 days later, again with MRSA bacteremia. At that time her Hickman catheter, which had been inserted for total parenteral nutrition because of the enterocutaneous fistula, was removed.
Transthoracic echocardiography revealed no vegetations, but her bacteremia persisted. Her mental status was poor this entire time: she was mute and could barely be awakened. We looked for clots and infected clots; duplex ultrasonographic examinations of all four extremities were negative. Finally, magnetic resonance imaging of her back—performed empirically because of the persistent bacteremia—revealed vertebral osteomyelitis at level T12-L1. We also noticed on serial evaluations that the vancomycin MIC for her organism increased from 0.5 to 2.0 μg/mL, so therapy was changed from vancomycin to daptomycin.
Her bacteremia cleared. Follow-up echocardiography was negative, but she had two subsequent relapses of MRSA bacteremia, one in April 2007 and one before she died in the summer of 2007.
Prolonged bacteremia: Is it vancomycin resistance, or something else?
The MRSA isolates that cause prolonged bacteremia seem to have certain characteristics.22 Higher MICs are probably associated with longer periods of bacteremia. But some genetic components within some strains of S aureus give them a survival advantage. They have less susceptibility to the body’s thrombin-induced platelet microbicidal protein. These isolates are not only associated with prolonged bacteremia: they are also associated with osteomyelitis, deep abscesses, endocarditis, recurrent infection, and increased death rate.22 Clinical laboratories do not test for these genetic components. One wonders whether our patient may have had an isolate with these mutations that gave it a survival advantage.
Do not use vancomycin for MSSA
Avoid using vancomycin for MSSA infections. It has been shown time and time again that MSSA infections do not respond as well to vancomycin as they do to beta-lactam antibiotics, specifically to the semisynthetic penicillins such as oxacillin and nafcillin, and even some of the first-generation cephalosporins. Chang et al23 found that patients with MSSA bacteremia had higher rates of persistent infections, relapse, and bacteriologic failure if they received vancomycin than if they received nafcillin.
Do vancomycin trough levels affect toxicity?
The vancomycin trough levels that we aimed for in the past (5 to 10 μg/mL) were probably too low. Today, we aim for trough levels of 15 to 20 μg/mL, and many physicians are aiming for 20 to 25 μg/mL. Part of the reason is that vancomycin MICs are higher than they used to be: in order to keep the vancomycin level above the MIC for a longer period of time, the vancomycin trough level needs to be higher. In theory, keeping the vancomycin levels above the MIC for longer periods should improve outcomes. Yet Fowler et al22 found that vancomycin trough levels among patients who had persistent MRSA bacteremia were actually higher than trough levels among those in whom the bacteremia resolved, although the difference was not statistically significant.
We measure the vancomycin trough level to make sure it is high enough (and give larger doses if it is not); among adults, peak levels need not be monitored on a routine basis because of the predictable pharmacokinetics of vancomycin.
Vancomycin toxicity can be either idiosyncratic or synergistic. Idiosyncratic toxicity occurs when a patient who has been on vancomycin for a long time develops a fixed rash, not associated with infusion. This is an immunologic phenomenon. It is a rare and very serious situation and may require steroid therapy.
Synergistic toxicity occurs when vancomycin is given with other nephrotoxic agents, notably gentamicin. Vancomycin plus gentamicin equals nephrotoxicity. Vancomycin alone is usually not nephrotoxic, but close monitoring of renal function parameters is warranted with the use of higher doses.24
IN UNEXPLAINED BACTEREMIA, LOOK FOR ENDOCARDITIS
In blood cultures from patients with bacteremia, S aureus is never a contaminant. Even if just one blood culture is positive for S aureus, believe that S aureus is the culprit.
Reports in the 1950s suggested that at least half of patients who had S aureus bacteremia had endocarditis,25 leading to recommendations that all patients with S aureus bacteremia without an obvious primary source of infection should be evaluated for endocarditis. Subsequent estimates were lower, in the range of 15% to 25%.26,27 However, throughout the world S aureus endocarditis continues to have a very high mortality rate: at least a third of patients die.28
Clinical criteria (community acquisition, no primary focus, and metastatic sequelae) were developed to try to predict the risk of endocarditis in bacteremic patients.26 However, these criteria did not work very well. The clinical definition of endocarditis has evolved. The criteria of von Reyn et al29 from 1981 did not use echocardiography as part of the definition, but the 1994 Duke criteria,30 which were refined31 in 2000, use both clinical and echocardiographic parameters.
Stratton et al32 performed transthoracic echocardiography in 14 patients with bacteremia and found 1 patient with cryptic tricuspid infective endocarditis. Bayer et al33 subsequently reported that of 72 patients with bacteremia, 6 (18%) of those who had no clinical findings suggestive of infectious endocarditis had findings on echocardiography that led to changes in their regimen. Adding echocardiography to three clinical risk factors increased the sensitivity of diagnosing endocarditis from 70% to 85% with a specificity of 100% and predictive value of 96%.
The Duke criteria call for transesophageal echocardiography, which is not feasible in some patients, eg, those with cirrhosis and esophageal varices.
S aureus endocarditis has changed over the years as our patient population has changed, and MRSA endocarditis tends to hit some of our most vulnerable patients. In a study by Miro et al34 in 2005, MRSA was the leading pathogen in patients who were diagnosed with S aureus endocarditis in 1990 or later. We will only see these numbers go up. Patients with diabetes tend to have more MRSA, and of diabetic patients with MRSA endocarditis, 30% to 40% die in the hospital.
Indications for surgery
Certain conditions are indications for surgery among patients with endocarditis, and no antibiotic will cure the endocarditis if the patient has one of these conditions, eg:
- Persistent bacteremia during antibiotic therapy
- Recurrent emboli
- Heart failure that cannot be controlled
- Perivalvular or myocardial abscesses
- Large vegetations
- Early prosthetic valve infection
- Certain arrhythmias.
How long should S aureus bacteremia be treated?
In cases of bacteremia in which endocarditis has been ruled out and removable foci of infection (eg, intravascular catheters) have been removed, some evidence indicates that treatment for 2 weeks would be as effective as the 4 to 6 weeks that we would use for endocarditis or other severe or invasive infections.35 The issue is controversial. If the patient has had frequent hospitalizations or a chronic medical condition I would hesitate to treat for less than 4 weeks, even if the infection appears to be associated with a removable focus.
Treatment of endocarditis
In the guidelines for treatment of endocarditis from the American Heart Association and Infectious Diseases Society of America,36 all the recommendations are relatively old and many of them are somewhat empiric—they are not based on evidence from randomized clinical trials. Rather, they are best opinions based on clinical experience and some observational studies over the years.
For MSSA. In cases of native-valve endocarditis, oxacillin (Bactocill), nafcillin (Unipen), or another semisynthetic beta-lactam antibiotic is recommended. For penicillin-allergic patients, we have other options, such as cefazolin (Ancef, Kefzol).
Combination therapy is frequently recommended for native valve endocarditis as well as for prosthetic valve endocarditis, with either rifampin or gentamicin along with a primary agent. There is some evidence that one can clear staphylococcal bacteremia a day or two more quickly by use of combination therapy with nafcillin plus an aminoglycoside than with nafcillin alone.37,38 For MSSA-associated endocarditis, vancomycin does not work as well as beta-lactam antibiotics.39,40
Korzeniowski and Sande37 and Chambers et al38 reported that the mean duration of bacteremia was 3.4 days for patients treated with nafcillin alone and 2.9 days for those treated with nafcillin plus an aminoglycoside. These studies led to consideration of a short course of gentamicin to clear the bacteremia quickly.
With MRSA, bacteremia often requires a week or more to clear. Levine et al21 reported a study in 42 patients, mostly injection-drug users, with right-sided native-valve endocarditis. The median duration of bacteremia was 7 days in patients who received vancomycin alone vs 9 days in those who received vancomycin plus rifampin; however, some patients were bacteremic for up to 27 days. Fever persisted for a median of 7 days, probably partly due to septic pulmonary emboli. Three patients died, and three required valve replacement.
NEW ANTIBIOTICS
Several new antibiotics are active against gram-positive cocci.41–44 However, the majority of them have not been prospectively studied for treating bacteremia or endocarditis.
Quinupristin/dalfopristin (Synercid) has not been formally studied for treatment of MRSA bacteremia or endocarditis. There are a few case reports of its use in these conditions.45 Quinupristin/dalfopristin is bacteriostatic, and its use may be associated with phlebitis, myalgias, and arthralgias.46
Linezolid (Zyvox) is approved for treatment of complicated skin and soft-tissue infections and for hospital-acquired pneumonia. There have been no specific studies of linezolid in the treatment of S aureus bacteremia or endocarditis. However, Shorr et al47 retrospectively looked at the bacteremic patients in five previous studies of linezolid vs vancomycin and found 144 cases of S aureus bacteremia, half of which were due to MRSA. Of 53 assessable patients with MRSA bacteremia, the primary infection was cured in 14 (56%) of the linezolid patients and 13 (46%) of the vancomycin patients.
The oral form is 100% bioavailable. One should avoid concomitant use of serotonin-reuptake inhibitors because of the risk of serotonin syndrome. Adverse effects include altered taste sensation and peripheral neuropathy. There are other potential toxicities, including hematologic changes (thrombocytopenia, leukopenia) and metabolic effects (lactic acidosis), so clinical and laboratory monitoring is important.48 The role of linezolid in the treatment of patients with S aureus bacteremia or endocarditis remains to be defined.
Daptomycin is indicated for complicated skin and soft-tissue infections, bacteremia, and right-sided endocarditis due to S aureus. Fowler et al20 found that daptomycin was not inferior to beta-lactam antibiotics for treatment of MSSA bacteremia and right-sided endocarditis, and for MRSA infections it outperformed vancomycin, but the difference was not statistically significant.
The dosing interval should be increased from once every 24 hours to every 48 hours if the creatinine clearance is 30 mL/minute or less. Adverse effects include myalgia, rhabdomyolysis (rare), and elevations in creatine phosphokinase. Reports of rising MICs during daptomycin therapy, in some cases associated with persistent infection,49 suggest that careful attention be paid to dosing and clinical monitoring.
Tigecycline (Tygacil) is indicated for complicated skin and soft-tissue infections and complicated intra-abdominal infections due to susceptible organisms. It is active against both MSSA and MRSA, but clinical experience with its use in invasive infections is somewhat limited.50 The dose of tigecycline should be reduced in advanced cirrhosis. Adverse effects include nausea and vomiting.
Telavancin, dalbavancin, and oritavancin, investigational parenteral antibiotics that are derivatives of vancomycin, are in clinical trials. The pharmacokinetic activity of these agents is of interest: telavancin is being studied with a once-daily dosing interval and dalbavancin’s half-life allows once-weekly dosing. In a limited trial, dalbavancin was found to be safe and effective in the treatment of catheter-related bloodstream infections.51 None of the antibiotics in this group has been studied for treatment of S aureus endocarditis. Telavancin therapy has been associated with rash, hypokalemia, QT prolongation, and creatinine elevations. Gastrointestinal symptoms have been reported with the use of dalbavancin.
Ceftobiprole, another investigational agent, is the only cephalosporin antibiotic that is active against MRSA. It is given every 12 hours. Adverse effects include nausea and taste disturbance.
Iclaprim is a novel diaminopyrimidine and a dihydrofolate reductase inhibitor. In vitro, it is active against gram-positive bacteria, including MRSA, VISA, and VRSA; clinical investigations at this point are limited to the treatment of skin and soft-tissue infections.
- Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 2004; 39:309–371. Erratum in: Clin Infect Dis 2004; 39:1093.
- US Centers for Disease Control and Prevention. National Nosocomial Infections Surveillance (NNIS) System. Campaign to prevent antimicrobial resistance. www.cdc.gov/drugresistance/healthcare/ha/HASlideSet.ppt.
- Blot SI, Vandewoude KH, Hoste EA, Colardyn FA. Outcome and attributable mortality in critically ill patients with bacteremia involving methicillin-susceptible and methicillin-resistant Staphylococcus aureus. Arch Intern Med 2002; 162:2229–2235.
- Klevens RM, Morrison MA, Nadle J, et al; Active Bacterial Core surveillance (ABCs) MRSA Investigators. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007; 298:1763–1771.
- Naimi TS, LeDell KH, Como-Sabetti K, et al. Comparison of community- and health care-associated methicillin-resistant Staphylococcus aureus infection. JAMA 2003; 290:2976–2984.
- Miller LG, Perdreau-Remington F, Rieg G, et al. Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N Engl J Med 2005; 352:1445–1453.
- Moran GJ, Krishnadasan A, Gorwitz RJ, et al EMERGEncy ID Net Study Group. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med 2006; 355:666–674.
- Mishaan AM, Mason EO, Martinez-Aquilar G, et al. Emergence of a predominant clone of community-acquired Staphylococcus aureus among children in Houston, Texas. Pediatr Infect Dis J 2005; 24:201–206.
- Miller LG, Perdreau-Remington F, Bayer AS, et al. Clinical and epidemiologic characteristics cannot distinguish community-associated methicillin-resistant Staphylococcus aureus infection from methicillin-susceptible S. aureus infection: a prospective investigation. Clin Infect Dis 2007; 44:471–482.
- Seybold U, Kourbatova EV, Johnson JG, et al. Emergence of community-associated methicillin-resistant Staphylococcus aureus USA300 genotype as a major cause of health care-associated blood stream infections. Clin Infect Dis 2006; 42:647–656.
- Maree CL, Daum RS, Boyle-Vavra S, Matayoshi K, Miller LG. Community-associated methicillin-resistant Staphylococcus aureus isolates causing healthcare-associated infections. Emerg Infect Dis 2007; 13:236–242.
- Liu C, Chambers HF. Staphylococcus aureus with heterogeneous resistance to vancomycin: epidemiology, clinical significance, and critical assessment of diagnostic methods. Antimicrob Agents Chemother 2003; 47:3040–3045.
- Sieradzki K, Roberts RB, Haber SW, Tomasz A. The development of vancomycin resistance in a patient with methicillin-resistant Staphylococcus aureus infection. N Engl J Med 1999; 340:517–523.
- Schwaber MJ, Wright SB, Carmeli Y, et al. Clinical implications of varying degrees of vancomycin susceptibility in methicillin-resistant Staphylococcus aureus bacteremia. Emerg Infect Dis 2003; 9:657–664. Erratum in: Emerg Infect Dis 2004; 10:160.
- Sakoulas G, Moise-Broder PA, Schentag J, Forrest A, Moellering RC, Eliopoulos GM. Relationship of MIC and bactericidal activity to efficacy of vancomycin for treatment of methicillin-resistant Staphylococcus aureus bacteremia. J Clin Microbiol 2004; 42:2398–2402.
- Naimi TS, Anderson D, O’Boyle C, et al. Vancomycin-intermediate Staphylococcus aureus with phenotypic susceptibility to methicillin in a patient with recurrent bacteremia. Clin Infect Dis 2003; 36:1609–1612.
- Moore MR, Perdreau-Remington F, Chambers HF. Vancomycin treatment failure associated with heterogeneous vancomycin-intermediate Staphylococcus aureus in a patient with endocarditis and in the rabbit model of endocarditis. Antimicrob Agents Chemother 2003; 47:1262–1266.
- Charles PG, Ward PB, Johnson PD, Howden BP, Grayson ML. Clinical features associated with bacteremia due to heterogenous vancomycin-intermediate Staphylococcus aureus. Clin Infect Dis 2004; 38:448–451.
- Howden BP, Ward PB, Charles PG, et al. Treatment outcomes for serious infections caused by methicillin-resistant Staphylococcus aureus with reduced vancomycin susceptibility. Clin Infect Dis 2004; 38:521–528.
- Fowler VG, Boucher HW, Corey GR, et al. S. aureus Endocarditis and Bacteremia Study Group. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N Engl J Med 2006; 355:653–665.
- Levine DP, Fromm BS, Reddy BR. Slow response to vancomycin or vancomycin plus rifampin in methicillin-resistant Staphylococcus aureus endocarditis. Ann Intern Med 1991; 115:674–680.
- Fowler VG, Sakoulas G, McIntyre LM, et al. Persistent bacteremia due to methicillin-resistant Staphylococcus aureus infection is associated with agr dysfunction and low-level in vitro resistance to thrombin-induced platelet microbicidal protein. J Infect Dis 2004; 190:1140–1149.
- Chang FY, Peacock JE, Musher DM, et al. Staphylococcus aureus bacteremia: recurrence and the impact of antibiotic treatment in a prospective multicenter study. Medicine (Baltimore) 2003; 82:333–339.
- Hidayat LK, Hsu DI, Quist R, Shriner KA, Wong-Beringer A. High-dose vancomycin therapy for methicillin-resistant Staphylococcus aureus infections: efficacy and toxicity. Arch Intern Med 2006; 166:2138–2144.
- Wilson R, Hamburger M. Fifteen years’ experience with staphylococcus septicemia in a large city hospital; analysis of fifty-five cases in the Cincinnati General Hospital 1940 to 1954. Am J Med 1957; 22:437–457.
- Nolan CM, Beaty HN. Staphylococcus aureus bacteremia. Current clinical patterns. Am J Med 1976; 60:495–500.
- Shah M, Watanakunakorn C. Changing patterns of Staphylococcus aureus bacteremia. Am J Med Sci 1979; 278:115–121.
- Fowler VG, Miro JM, Hoen B, et al ICE Investigators. Staphylococcus aureus endocarditis: a consequence of medical progress. JAMA 2005; 293:3012–3021. Erratum in: JAMA 2005; 294:900.
- Von Reyn CF, Levy BS, Arbeit RD, Friedland G, Crumpacker CS. Infective endocarditis: an analysis based on strict case definition. Ann Intern Med 1981; 94:505–518.
- Durack DT, Lukes AS, Bright DK. New criteria for diagnosis of infective endocarditis: utilization of specific echocardiographic findings. Duke Endocarditis Service. Am J Med 1994; 96:200–209.
- Li JS, Sexton DJ, Mick N, et al. Proposed modifications to the Duke criteria for the diagnosis of infective endocarditis. Clin Infect Dis 2000; 30:633–638.
- Stratton JR, Werner JA, Pearlman AS, Janko CL, Kliman S, Jackson MC. Bacteremia and the heart. Serial echocardiographic findings in 80 patients with documented or suspected bacteremia. Am J Med 1982; 73:851–858.
- Bayer AS, Lam K, Ginzton L, Normal DC, Chiu CY, Ward JI. Staphylococcus aureus bacteremia. Clinical, serologic, and echocardiographic findings in patients with and without endocarditis. Arch Intern Med 1987; 147:457–462.
- Miro JM, Anguera I, Cabell CH, et al International Collaboration on Endocarditis Merged Database Study Group. Staphylococcus aureus native valve infective endocarditis: report of 566 episodes from the International Collaboration on Endocarditis Merged Database. Clin Infect Dis 2005; 41:507–514. Erratum in: Clin Infect Dis 2005; 41:1075–1077.
- Jernigan JA, Farr BM. Short-course therapy of catheter-related Staphylococcus aureus bacteremia: a meta-analysis. Ann Intern Med 1993; 119:304–311.
- Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis: diagnosis, antimicrobial therapy, and management of complications: a statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association: endorsed by the Infectious Diseases Society of America. Circulation 2005; 111:e394–e434. Erratum in: Circulation 2005; 112:2373. Circulation 2007; 115:e408.
- Korzeniowski O, Sande MA. Combination antimicrobial therapy for Staphylococcus aureus endocarditis in patients addicted to parenteral drugs and in nonaddicts: a prospective study. Ann Intern Med 1982; 97:496–503.
- Chambers HF, Korzeniowski OM, Sande MA. Staphylococcus aureus endocarditis: clinical manifestations in addicts and nonaddicts. Medicine (Baltimore) 1983; 62:170–177.
- Gentry CA, Rodvold KA, Novak RM, Hershow RC, Naderer OJ. Retrospective evaluation of therapies for Staphylococcus aureus endocarditis. Pharmacotherapy 1997; 17:990–997.
- Small PM, Chambers HF. Vancomycin for Staphylococcus aureus endocarditis in intravenous drug users. Antimicrob Agents Chemother 1990; 34:1227–1231.
- Eliopoulos GM. Quinupristin-dalfopristin and linezolid: evidence and opinion. Clin Infect Dis 2003; 36:473–481.
- Rybak MJ. Therapeutic options for Gram-positive infections. J Hosp Infect 2001; 49 suppl A:S25–S32.
- Micek ST. Alternatives to vancomycin for the treatment of methicillin-resistant Staphylococcus aureus infections. Clin Infect Dis 2007; 45 suppl 3:S184–S190.
- Appelbaum PC, Jacobs MR. Recently approved and investigational antibiotics for treatment of severe infections caused by Gram-positive bacteria. Curr Opin Microbiol 2005; 8:510–517.
- Drew RH, Perfect JR, Srinath L, Kirkimilis E, Dowzicky M, Talbot GH for the Synercid Emergency-Use Study Group. Treatment of methicillin-resistant Staphylococcus aureus infections with quinupristin-dalfopristin in patients intolerant of or failing prior therapy. J Antimicrob Chemother 2000; 46:775–784.
- Lamb HM, Figgitt DP, Faulds D. Quinupristin/dalfopristin: a review of its use in the management of serious gram-positive infections. Drugs 1999; 58:1061–1097.
- Shorr AF, Kunkel MJ, Kollef M. Linezolid versus vancomycin for Staphylococcus aureus bacteraemia: pooled analysis of randomized studies. J Antimicrob Chemother 2005; 56:923–929.
- Bishop E, Melvani S, Howden BP, Charles PG, Grayson ML. Good clinical outcomes but high rates of adverse reactions during linezolid therapy for serious infections: a proposed protocol for monitoring therapy in complex patients. Antimicrob Agents Chemother 2006; 50:1599–1602.
- Boucher HW, Sakoulas G. Perspectives on daptomycin resistance, with emphasis on resistance in Staphylococcus aureus. Clin Infect Dis 2007; 45:601–608.
- Munoz-Price LS, Lolans K, Quinn JP. Four cases of invasive methicillin-resistant Staphylococcus aureus (MRSA) infections treated with tigecycline. Scand J Infect Dis 2006; 38:1081–1084.
- Raad I, Darouiche R, Vazquez J, et al. Efficacy and safety of weekly dalbavancin therapy for catheter-related bloodstream infection caused by gram-positive pathogens. Clin Infect Dis 2005; 40:374–80.
- Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 2004; 39:309–371. Erratum in: Clin Infect Dis 2004; 39:1093.
- US Centers for Disease Control and Prevention. National Nosocomial Infections Surveillance (NNIS) System. Campaign to prevent antimicrobial resistance. www.cdc.gov/drugresistance/healthcare/ha/HASlideSet.ppt.
- Blot SI, Vandewoude KH, Hoste EA, Colardyn FA. Outcome and attributable mortality in critically ill patients with bacteremia involving methicillin-susceptible and methicillin-resistant Staphylococcus aureus. Arch Intern Med 2002; 162:2229–2235.
- Klevens RM, Morrison MA, Nadle J, et al; Active Bacterial Core surveillance (ABCs) MRSA Investigators. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007; 298:1763–1771.
- Naimi TS, LeDell KH, Como-Sabetti K, et al. Comparison of community- and health care-associated methicillin-resistant Staphylococcus aureus infection. JAMA 2003; 290:2976–2984.
- Miller LG, Perdreau-Remington F, Rieg G, et al. Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N Engl J Med 2005; 352:1445–1453.
- Moran GJ, Krishnadasan A, Gorwitz RJ, et al EMERGEncy ID Net Study Group. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med 2006; 355:666–674.
- Mishaan AM, Mason EO, Martinez-Aquilar G, et al. Emergence of a predominant clone of community-acquired Staphylococcus aureus among children in Houston, Texas. Pediatr Infect Dis J 2005; 24:201–206.
- Miller LG, Perdreau-Remington F, Bayer AS, et al. Clinical and epidemiologic characteristics cannot distinguish community-associated methicillin-resistant Staphylococcus aureus infection from methicillin-susceptible S. aureus infection: a prospective investigation. Clin Infect Dis 2007; 44:471–482.
- Seybold U, Kourbatova EV, Johnson JG, et al. Emergence of community-associated methicillin-resistant Staphylococcus aureus USA300 genotype as a major cause of health care-associated blood stream infections. Clin Infect Dis 2006; 42:647–656.
- Maree CL, Daum RS, Boyle-Vavra S, Matayoshi K, Miller LG. Community-associated methicillin-resistant Staphylococcus aureus isolates causing healthcare-associated infections. Emerg Infect Dis 2007; 13:236–242.
- Liu C, Chambers HF. Staphylococcus aureus with heterogeneous resistance to vancomycin: epidemiology, clinical significance, and critical assessment of diagnostic methods. Antimicrob Agents Chemother 2003; 47:3040–3045.
- Sieradzki K, Roberts RB, Haber SW, Tomasz A. The development of vancomycin resistance in a patient with methicillin-resistant Staphylococcus aureus infection. N Engl J Med 1999; 340:517–523.
- Schwaber MJ, Wright SB, Carmeli Y, et al. Clinical implications of varying degrees of vancomycin susceptibility in methicillin-resistant Staphylococcus aureus bacteremia. Emerg Infect Dis 2003; 9:657–664. Erratum in: Emerg Infect Dis 2004; 10:160.
- Sakoulas G, Moise-Broder PA, Schentag J, Forrest A, Moellering RC, Eliopoulos GM. Relationship of MIC and bactericidal activity to efficacy of vancomycin for treatment of methicillin-resistant Staphylococcus aureus bacteremia. J Clin Microbiol 2004; 42:2398–2402.
- Naimi TS, Anderson D, O’Boyle C, et al. Vancomycin-intermediate Staphylococcus aureus with phenotypic susceptibility to methicillin in a patient with recurrent bacteremia. Clin Infect Dis 2003; 36:1609–1612.
- Moore MR, Perdreau-Remington F, Chambers HF. Vancomycin treatment failure associated with heterogeneous vancomycin-intermediate Staphylococcus aureus in a patient with endocarditis and in the rabbit model of endocarditis. Antimicrob Agents Chemother 2003; 47:1262–1266.
- Charles PG, Ward PB, Johnson PD, Howden BP, Grayson ML. Clinical features associated with bacteremia due to heterogenous vancomycin-intermediate Staphylococcus aureus. Clin Infect Dis 2004; 38:448–451.
- Howden BP, Ward PB, Charles PG, et al. Treatment outcomes for serious infections caused by methicillin-resistant Staphylococcus aureus with reduced vancomycin susceptibility. Clin Infect Dis 2004; 38:521–528.
- Fowler VG, Boucher HW, Corey GR, et al. S. aureus Endocarditis and Bacteremia Study Group. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N Engl J Med 2006; 355:653–665.
- Levine DP, Fromm BS, Reddy BR. Slow response to vancomycin or vancomycin plus rifampin in methicillin-resistant Staphylococcus aureus endocarditis. Ann Intern Med 1991; 115:674–680.
- Fowler VG, Sakoulas G, McIntyre LM, et al. Persistent bacteremia due to methicillin-resistant Staphylococcus aureus infection is associated with agr dysfunction and low-level in vitro resistance to thrombin-induced platelet microbicidal protein. J Infect Dis 2004; 190:1140–1149.
- Chang FY, Peacock JE, Musher DM, et al. Staphylococcus aureus bacteremia: recurrence and the impact of antibiotic treatment in a prospective multicenter study. Medicine (Baltimore) 2003; 82:333–339.
- Hidayat LK, Hsu DI, Quist R, Shriner KA, Wong-Beringer A. High-dose vancomycin therapy for methicillin-resistant Staphylococcus aureus infections: efficacy and toxicity. Arch Intern Med 2006; 166:2138–2144.
- Wilson R, Hamburger M. Fifteen years’ experience with staphylococcus septicemia in a large city hospital; analysis of fifty-five cases in the Cincinnati General Hospital 1940 to 1954. Am J Med 1957; 22:437–457.
- Nolan CM, Beaty HN. Staphylococcus aureus bacteremia. Current clinical patterns. Am J Med 1976; 60:495–500.
- Shah M, Watanakunakorn C. Changing patterns of Staphylococcus aureus bacteremia. Am J Med Sci 1979; 278:115–121.
- Fowler VG, Miro JM, Hoen B, et al ICE Investigators. Staphylococcus aureus endocarditis: a consequence of medical progress. JAMA 2005; 293:3012–3021. Erratum in: JAMA 2005; 294:900.
- Von Reyn CF, Levy BS, Arbeit RD, Friedland G, Crumpacker CS. Infective endocarditis: an analysis based on strict case definition. Ann Intern Med 1981; 94:505–518.
- Durack DT, Lukes AS, Bright DK. New criteria for diagnosis of infective endocarditis: utilization of specific echocardiographic findings. Duke Endocarditis Service. Am J Med 1994; 96:200–209.
- Li JS, Sexton DJ, Mick N, et al. Proposed modifications to the Duke criteria for the diagnosis of infective endocarditis. Clin Infect Dis 2000; 30:633–638.
- Stratton JR, Werner JA, Pearlman AS, Janko CL, Kliman S, Jackson MC. Bacteremia and the heart. Serial echocardiographic findings in 80 patients with documented or suspected bacteremia. Am J Med 1982; 73:851–858.
- Bayer AS, Lam K, Ginzton L, Normal DC, Chiu CY, Ward JI. Staphylococcus aureus bacteremia. Clinical, serologic, and echocardiographic findings in patients with and without endocarditis. Arch Intern Med 1987; 147:457–462.
- Miro JM, Anguera I, Cabell CH, et al International Collaboration on Endocarditis Merged Database Study Group. Staphylococcus aureus native valve infective endocarditis: report of 566 episodes from the International Collaboration on Endocarditis Merged Database. Clin Infect Dis 2005; 41:507–514. Erratum in: Clin Infect Dis 2005; 41:1075–1077.
- Jernigan JA, Farr BM. Short-course therapy of catheter-related Staphylococcus aureus bacteremia: a meta-analysis. Ann Intern Med 1993; 119:304–311.
- Baddour LM, Wilson WR, Bayer AS, et al. Infective endocarditis: diagnosis, antimicrobial therapy, and management of complications: a statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association: endorsed by the Infectious Diseases Society of America. Circulation 2005; 111:e394–e434. Erratum in: Circulation 2005; 112:2373. Circulation 2007; 115:e408.
- Korzeniowski O, Sande MA. Combination antimicrobial therapy for Staphylococcus aureus endocarditis in patients addicted to parenteral drugs and in nonaddicts: a prospective study. Ann Intern Med 1982; 97:496–503.
- Chambers HF, Korzeniowski OM, Sande MA. Staphylococcus aureus endocarditis: clinical manifestations in addicts and nonaddicts. Medicine (Baltimore) 1983; 62:170–177.
- Gentry CA, Rodvold KA, Novak RM, Hershow RC, Naderer OJ. Retrospective evaluation of therapies for Staphylococcus aureus endocarditis. Pharmacotherapy 1997; 17:990–997.
- Small PM, Chambers HF. Vancomycin for Staphylococcus aureus endocarditis in intravenous drug users. Antimicrob Agents Chemother 1990; 34:1227–1231.
- Eliopoulos GM. Quinupristin-dalfopristin and linezolid: evidence and opinion. Clin Infect Dis 2003; 36:473–481.
- Rybak MJ. Therapeutic options for Gram-positive infections. J Hosp Infect 2001; 49 suppl A:S25–S32.
- Micek ST. Alternatives to vancomycin for the treatment of methicillin-resistant Staphylococcus aureus infections. Clin Infect Dis 2007; 45 suppl 3:S184–S190.
- Appelbaum PC, Jacobs MR. Recently approved and investigational antibiotics for treatment of severe infections caused by Gram-positive bacteria. Curr Opin Microbiol 2005; 8:510–517.
- Drew RH, Perfect JR, Srinath L, Kirkimilis E, Dowzicky M, Talbot GH for the Synercid Emergency-Use Study Group. Treatment of methicillin-resistant Staphylococcus aureus infections with quinupristin-dalfopristin in patients intolerant of or failing prior therapy. J Antimicrob Chemother 2000; 46:775–784.
- Lamb HM, Figgitt DP, Faulds D. Quinupristin/dalfopristin: a review of its use in the management of serious gram-positive infections. Drugs 1999; 58:1061–1097.
- Shorr AF, Kunkel MJ, Kollef M. Linezolid versus vancomycin for Staphylococcus aureus bacteraemia: pooled analysis of randomized studies. J Antimicrob Chemother 2005; 56:923–929.
- Bishop E, Melvani S, Howden BP, Charles PG, Grayson ML. Good clinical outcomes but high rates of adverse reactions during linezolid therapy for serious infections: a proposed protocol for monitoring therapy in complex patients. Antimicrob Agents Chemother 2006; 50:1599–1602.
- Boucher HW, Sakoulas G. Perspectives on daptomycin resistance, with emphasis on resistance in Staphylococcus aureus. Clin Infect Dis 2007; 45:601–608.
- Munoz-Price LS, Lolans K, Quinn JP. Four cases of invasive methicillin-resistant Staphylococcus aureus (MRSA) infections treated with tigecycline. Scand J Infect Dis 2006; 38:1081–1084.
- Raad I, Darouiche R, Vazquez J, et al. Efficacy and safety of weekly dalbavancin therapy for catheter-related bloodstream infection caused by gram-positive pathogens. Clin Infect Dis 2005; 40:374–80.
KEY POINTS
- Community-associated MRSA infections tend to affect patients younger than those who traditionally get hospital-associated MRSA infections. Most of these infections are of the skin and soft tissues, but this pathogen can also affect deeper tissues, and bacteremia and necrotizing pneumonia have been reported.
- For patients with skin and soft-tissue infections due to MRSA, incision and drainage rather than antibiotic therapy is often the key intervention.
- Vancomycin has been our stalwart for treating MRSA infections for more than 40 years, but it is not working as well as it used to, at least in certain situations. Vancomycin should not be used to treat infections due to methicillin-susceptible S aureus.
- Needed are better understanding of the factors that influence persistent S aureus bacteremia, well-controlled, prospective studies, and continued antibiotic development.
Infective endocarditis prophylaxis before dental procedures: New guidelines spark controversy
Many fewer people will need to receive antibiotics as prophylaxis against infective endocarditis before undergoing dental procedures, according to new guidelines released by the American Heart Association.1 Now, the only patients to receive antibiotics will be those at highest risk, ie, those with a prosthetic heart valve, a history of endocarditis, certain forms of congenital heart disease, or valvulopathy after heart transplantation, and only before certain dental procedures.
Unfortunately, these guidelines are still based largely on expert opinion, with very little hard evidence to show that antibiotic therapy actually prevents infective endocarditis. Nevertheless, the new guidelines appear reasonable, and we believe they should be followed.
A RARE BUT LIFE-THREATENING INFECTION
Infective endocarditis is a rare but life-threatening infection, with an incidence in the United States of 10,000 to 20,000 new cases per year. Mortality rates for both native-valve endocarditis and prosthetic-valve endocarditis range from 20% to 30%.2,3 For the past half-century, antibiotic prophylaxis for dental procedures has been recommended for patients judged to be at risk of infective endocarditis, in hopes of preventing this dreaded infectious disease.
ENDOCARDIAL INJURY, THEN BACTERIAL SEEDING
A combination of events must occur to cause infective endocarditis. First, injury to the endocardial surface induces focal adherence of platelets and fibrin. Then, a bacteremic event seeds this aggregate with microorganisms, attracting more platelets and fibrin, allowing uninhibited microbial growth and the development of an inflammatory plaque or vegetation.
The magnitude and duration of bacteremia that produces this cascade of events is uncertain. Transient bacteremia occurs commonly, not only during procedures that cause trauma to mucosal surfaces or tissue but also with daily activities such as brushing teeth and chewing. The reported incidence of bacteremia during dental intervention ranges from 10% to 100%, and with daily brushing and flossing, from 20% to 68%.1
STAPHYLOCOCCI OVERTAKING VIRIDANS STREPTOCOCCI AS CAUSE
While historically the viridans group of streptococci has been responsible for the largest percentage of cases of both native-valve endocarditis and late-onset prosthetic-valve endocarditis, times have changed. In more recently reported series, Staphylococcus aureus appears more common, and unlikely to be susceptible to antibiotics recommended for dental prophylaxis. Other causative pathogens include coagulase-negative staphylococci, enterococci, gram-negative microorganisms, and fungi.
PREVIOUS GUIDELINES—1997
Previous American Heart Association guidelines4 separated patients into three risk categories for infective endocarditis. High-risk patients were those with prosthetic heart valves, a history of infective endocarditis, complex cyanotic congenital heart disease, or surgically constructed systemic pulmonary shunts. Moderate-risk patients had other congenital cardiac defects, hypertrophic cardiomyopathy, or acquired valvular heart disease including mitral valve prolapse with regurgitation. Negligible-risk patients—ie, most patients—included those with coronary artery bypass grafts, a permanent pacemaker, or mitral valve prolapse without regurgitation. Antibiotic prophylaxis was recommended only for patients in the high-risk and moderate-risk groups.
THOUGHTS AND CHALLENGES
Although prophylaxis has been a standard practice for years, its efficacy and cost-effectiveness have never been proven, owing to a lack of prospective randomized controlled trials. A sequential relationship between dental procedures and infective endocarditis can be demonstrated in only 4% to 7.5% of cases.5 Most cases of infective endocarditis are not preceded by dental procedures.
Furthermore, the data are limited and insufficient to substantiate the efficacy of antibiotics in preventing endocarditis in patients with high-risk cardiac conditions who undergo dental procedures.6 Failures have occurred even when the infecting microorganism was susceptible to the antibiotic given for prophylaxis. Since bacteremia occurs also during brushing and flossing of teeth, why give prophylaxis just for dental procedures? Moreover, the risks of causing adverse or anaphylactic reactions from antibiotics, as well as contributing to the nationwide antibiotic resistance problem, are issues not to be taken lightly.
Poor compliance with prophylaxis has been documented. Studies by Duval et al7 and others have shown that practitioners adhere to recommended dental prophylaxis programs only about 40% of the time, while only 22% of patients with predisposing cardiac conditions could recall taking their prescribed prophylactic antibiotics before an indicated procedure, as recommended.8
NEW GUIDELINES—2007
To address many of these concerns, the American Heart Association1 released extensively revised guidelines in 2007. They are more pragmatic, narrowly focused for a selected group of patients who have a greater lifetime risk of illness and death from infective endocarditis.
The experts who wrote the guidelines agreed that evidence remains poor about which dental procedures increase the risk of infective endocarditis and the efficacy of antibiotic prophylaxis to prevent its development. They stress the importance of good oral hygiene and prevention of dental disease and argue persuasively that this will have a greater impact on decreasing the lifetime risk of infective endocarditis than will antibiotic prophylaxis.

Prophylaxis is now recommended only for patients with a prosthetic heart valve, a history of infective endocarditis, certain forms of congenital heart disease, and valvulopathy after cardiac transplantation (Table 1), and only before procedures that involve manipulation of gingival tissue or the periapical region of teeth, or perforation of the oral mucosa. Excluded are routine dental cleaning and anesthetic injections through noninfected tissue, dental radiography, placement and adjustment of appliances, shedding of deciduous teeth, and bleeding from trauma to the lips.
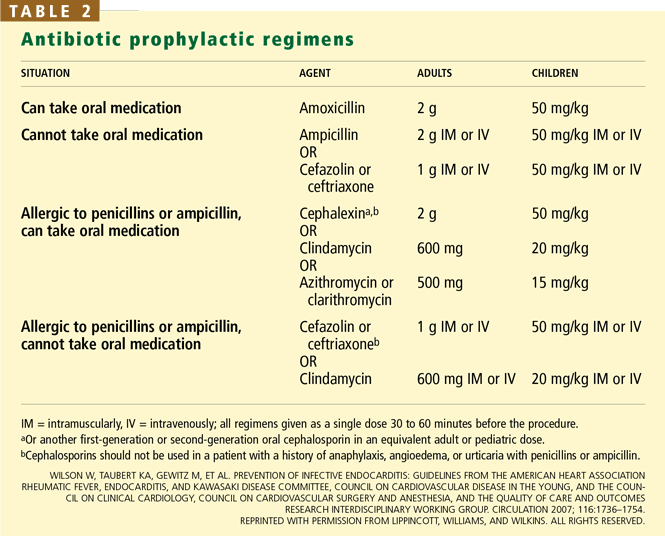
CONTROVERSY WILL CONTINUE
The new guidelines for dental prophylaxis have been extensively revised and simplified. They are now focused only on patients who have a greater lifetime risk of illness and death from infective endocarditis. But what about patients who had previously been advised to take prophylaxis, such as those with mitral valve prolapse with regurgitation, who will not receive prophylaxis any more?
These guidelines will likely stir emotions, not only for practitioners who have strong desires to practice preventive medicine, but also for patients who have been taking prophylaxis in good faith per previous guidelines. They may feel abandoned. Unfortunately, funding for a prospective randomized clinical trial large enough to prove that antibiotic prophylaxis for dental procedures benefits patients is unlikely. That leaves us with the current recommendations, which are based on scientific evidence that currently exists and on expert opinion.
The intention of the guidelines is laudable. Of course, there will continue to be controversies with the new rules. Nevertheless, we believe they should be followed until there is more persuasive evidence to the contrary.
- Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation 2007; 116:1736–1754.
- Hill EE, Herligers P, Claus P, Vanderschueren S, Herregods MC, Peetermans WE. Infective endocarditis: changing epidemiology and predictors of 6-month mortality: a prospective cohort study. Eur Heart J 2007; 28:196–203.
- Wang A, Athan E, Pappas PA, et al International Collaboration on Endocarditis-Prospective Cohort Study Investigators. Contemporary clinical profile and outcome of prosthetic valve endocarditis. JAMA 2007; 297:1354–1361.
- Dajani AS, Taubert KA, Wilson W, et al. Prevention of bacterial endocarditis. Recommendations by the American Heart Association. JAMA 1997; 277:1794–1801.
- Gendron R, Grenier D, Maheu-Robert LF. The oral cavity as a reservoir of bacterial pathogens for focal infections. Microbes Infect 2000; 2:897–906.
- Strom BL, Abrutyn E, Berlin JA, et al. Dental and cardiac risk factors for infective endocarditis. A population-based, case-control study. Ann Intern Med 1998; 129:761–769.
- Duval X, Alla F, Hoen B, et al. Estimated risk of endocarditis in adults with predisposing cardiac conditions undergoing dental procedures with or without antibiotic prophylaxis. Clin Infect Dis 2006; 42:e102–e107.
- van der Meer JT, van Wijk W, Thompson J, Valkenburg HA, Michel MF. Awareness of need and actual use of prophylaxis: lack of patient compliance in the prevention of bacterial endocarditis. J Antimicrob Chemother 1992; 29:187–194.
Many fewer people will need to receive antibiotics as prophylaxis against infective endocarditis before undergoing dental procedures, according to new guidelines released by the American Heart Association.1 Now, the only patients to receive antibiotics will be those at highest risk, ie, those with a prosthetic heart valve, a history of endocarditis, certain forms of congenital heart disease, or valvulopathy after heart transplantation, and only before certain dental procedures.
Unfortunately, these guidelines are still based largely on expert opinion, with very little hard evidence to show that antibiotic therapy actually prevents infective endocarditis. Nevertheless, the new guidelines appear reasonable, and we believe they should be followed.
A RARE BUT LIFE-THREATENING INFECTION
Infective endocarditis is a rare but life-threatening infection, with an incidence in the United States of 10,000 to 20,000 new cases per year. Mortality rates for both native-valve endocarditis and prosthetic-valve endocarditis range from 20% to 30%.2,3 For the past half-century, antibiotic prophylaxis for dental procedures has been recommended for patients judged to be at risk of infective endocarditis, in hopes of preventing this dreaded infectious disease.
ENDOCARDIAL INJURY, THEN BACTERIAL SEEDING
A combination of events must occur to cause infective endocarditis. First, injury to the endocardial surface induces focal adherence of platelets and fibrin. Then, a bacteremic event seeds this aggregate with microorganisms, attracting more platelets and fibrin, allowing uninhibited microbial growth and the development of an inflammatory plaque or vegetation.
The magnitude and duration of bacteremia that produces this cascade of events is uncertain. Transient bacteremia occurs commonly, not only during procedures that cause trauma to mucosal surfaces or tissue but also with daily activities such as brushing teeth and chewing. The reported incidence of bacteremia during dental intervention ranges from 10% to 100%, and with daily brushing and flossing, from 20% to 68%.1
STAPHYLOCOCCI OVERTAKING VIRIDANS STREPTOCOCCI AS CAUSE
While historically the viridans group of streptococci has been responsible for the largest percentage of cases of both native-valve endocarditis and late-onset prosthetic-valve endocarditis, times have changed. In more recently reported series, Staphylococcus aureus appears more common, and unlikely to be susceptible to antibiotics recommended for dental prophylaxis. Other causative pathogens include coagulase-negative staphylococci, enterococci, gram-negative microorganisms, and fungi.
PREVIOUS GUIDELINES—1997
Previous American Heart Association guidelines4 separated patients into three risk categories for infective endocarditis. High-risk patients were those with prosthetic heart valves, a history of infective endocarditis, complex cyanotic congenital heart disease, or surgically constructed systemic pulmonary shunts. Moderate-risk patients had other congenital cardiac defects, hypertrophic cardiomyopathy, or acquired valvular heart disease including mitral valve prolapse with regurgitation. Negligible-risk patients—ie, most patients—included those with coronary artery bypass grafts, a permanent pacemaker, or mitral valve prolapse without regurgitation. Antibiotic prophylaxis was recommended only for patients in the high-risk and moderate-risk groups.
THOUGHTS AND CHALLENGES
Although prophylaxis has been a standard practice for years, its efficacy and cost-effectiveness have never been proven, owing to a lack of prospective randomized controlled trials. A sequential relationship between dental procedures and infective endocarditis can be demonstrated in only 4% to 7.5% of cases.5 Most cases of infective endocarditis are not preceded by dental procedures.
Furthermore, the data are limited and insufficient to substantiate the efficacy of antibiotics in preventing endocarditis in patients with high-risk cardiac conditions who undergo dental procedures.6 Failures have occurred even when the infecting microorganism was susceptible to the antibiotic given for prophylaxis. Since bacteremia occurs also during brushing and flossing of teeth, why give prophylaxis just for dental procedures? Moreover, the risks of causing adverse or anaphylactic reactions from antibiotics, as well as contributing to the nationwide antibiotic resistance problem, are issues not to be taken lightly.
Poor compliance with prophylaxis has been documented. Studies by Duval et al7 and others have shown that practitioners adhere to recommended dental prophylaxis programs only about 40% of the time, while only 22% of patients with predisposing cardiac conditions could recall taking their prescribed prophylactic antibiotics before an indicated procedure, as recommended.8
NEW GUIDELINES—2007
To address many of these concerns, the American Heart Association1 released extensively revised guidelines in 2007. They are more pragmatic, narrowly focused for a selected group of patients who have a greater lifetime risk of illness and death from infective endocarditis.
The experts who wrote the guidelines agreed that evidence remains poor about which dental procedures increase the risk of infective endocarditis and the efficacy of antibiotic prophylaxis to prevent its development. They stress the importance of good oral hygiene and prevention of dental disease and argue persuasively that this will have a greater impact on decreasing the lifetime risk of infective endocarditis than will antibiotic prophylaxis.
Prophylaxis is now recommended only for patients with a prosthetic heart valve, a history of infective endocarditis, certain forms of congenital heart disease, and valvulopathy after cardiac transplantation (Table 1), and only before procedures that involve manipulation of gingival tissue or the periapical region of teeth, or perforation of the oral mucosa. Excluded are routine dental cleaning and anesthetic injections through noninfected tissue, dental radiography, placement and adjustment of appliances, shedding of deciduous teeth, and bleeding from trauma to the lips.
CONTROVERSY WILL CONTINUE
The new guidelines for dental prophylaxis have been extensively revised and simplified. They are now focused only on patients who have a greater lifetime risk of illness and death from infective endocarditis. But what about patients who had previously been advised to take prophylaxis, such as those with mitral valve prolapse with regurgitation, who will not receive prophylaxis any more?
These guidelines will likely stir emotions, not only for practitioners who have strong desires to practice preventive medicine, but also for patients who have been taking prophylaxis in good faith per previous guidelines. They may feel abandoned. Unfortunately, funding for a prospective randomized clinical trial large enough to prove that antibiotic prophylaxis for dental procedures benefits patients is unlikely. That leaves us with the current recommendations, which are based on scientific evidence that currently exists and on expert opinion.
The intention of the guidelines is laudable. Of course, there will continue to be controversies with the new rules. Nevertheless, we believe they should be followed until there is more persuasive evidence to the contrary.
Many fewer people will need to receive antibiotics as prophylaxis against infective endocarditis before undergoing dental procedures, according to new guidelines released by the American Heart Association.1 Now, the only patients to receive antibiotics will be those at highest risk, ie, those with a prosthetic heart valve, a history of endocarditis, certain forms of congenital heart disease, or valvulopathy after heart transplantation, and only before certain dental procedures.
Unfortunately, these guidelines are still based largely on expert opinion, with very little hard evidence to show that antibiotic therapy actually prevents infective endocarditis. Nevertheless, the new guidelines appear reasonable, and we believe they should be followed.
A RARE BUT LIFE-THREATENING INFECTION
Infective endocarditis is a rare but life-threatening infection, with an incidence in the United States of 10,000 to 20,000 new cases per year. Mortality rates for both native-valve endocarditis and prosthetic-valve endocarditis range from 20% to 30%.2,3 For the past half-century, antibiotic prophylaxis for dental procedures has been recommended for patients judged to be at risk of infective endocarditis, in hopes of preventing this dreaded infectious disease.
ENDOCARDIAL INJURY, THEN BACTERIAL SEEDING
A combination of events must occur to cause infective endocarditis. First, injury to the endocardial surface induces focal adherence of platelets and fibrin. Then, a bacteremic event seeds this aggregate with microorganisms, attracting more platelets and fibrin, allowing uninhibited microbial growth and the development of an inflammatory plaque or vegetation.
The magnitude and duration of bacteremia that produces this cascade of events is uncertain. Transient bacteremia occurs commonly, not only during procedures that cause trauma to mucosal surfaces or tissue but also with daily activities such as brushing teeth and chewing. The reported incidence of bacteremia during dental intervention ranges from 10% to 100%, and with daily brushing and flossing, from 20% to 68%.1
STAPHYLOCOCCI OVERTAKING VIRIDANS STREPTOCOCCI AS CAUSE
While historically the viridans group of streptococci has been responsible for the largest percentage of cases of both native-valve endocarditis and late-onset prosthetic-valve endocarditis, times have changed. In more recently reported series, Staphylococcus aureus appears more common, and unlikely to be susceptible to antibiotics recommended for dental prophylaxis. Other causative pathogens include coagulase-negative staphylococci, enterococci, gram-negative microorganisms, and fungi.
PREVIOUS GUIDELINES—1997
Previous American Heart Association guidelines4 separated patients into three risk categories for infective endocarditis. High-risk patients were those with prosthetic heart valves, a history of infective endocarditis, complex cyanotic congenital heart disease, or surgically constructed systemic pulmonary shunts. Moderate-risk patients had other congenital cardiac defects, hypertrophic cardiomyopathy, or acquired valvular heart disease including mitral valve prolapse with regurgitation. Negligible-risk patients—ie, most patients—included those with coronary artery bypass grafts, a permanent pacemaker, or mitral valve prolapse without regurgitation. Antibiotic prophylaxis was recommended only for patients in the high-risk and moderate-risk groups.
THOUGHTS AND CHALLENGES
Although prophylaxis has been a standard practice for years, its efficacy and cost-effectiveness have never been proven, owing to a lack of prospective randomized controlled trials. A sequential relationship between dental procedures and infective endocarditis can be demonstrated in only 4% to 7.5% of cases.5 Most cases of infective endocarditis are not preceded by dental procedures.
Furthermore, the data are limited and insufficient to substantiate the efficacy of antibiotics in preventing endocarditis in patients with high-risk cardiac conditions who undergo dental procedures.6 Failures have occurred even when the infecting microorganism was susceptible to the antibiotic given for prophylaxis. Since bacteremia occurs also during brushing and flossing of teeth, why give prophylaxis just for dental procedures? Moreover, the risks of causing adverse or anaphylactic reactions from antibiotics, as well as contributing to the nationwide antibiotic resistance problem, are issues not to be taken lightly.
Poor compliance with prophylaxis has been documented. Studies by Duval et al7 and others have shown that practitioners adhere to recommended dental prophylaxis programs only about 40% of the time, while only 22% of patients with predisposing cardiac conditions could recall taking their prescribed prophylactic antibiotics before an indicated procedure, as recommended.8
NEW GUIDELINES—2007
To address many of these concerns, the American Heart Association1 released extensively revised guidelines in 2007. They are more pragmatic, narrowly focused for a selected group of patients who have a greater lifetime risk of illness and death from infective endocarditis.
The experts who wrote the guidelines agreed that evidence remains poor about which dental procedures increase the risk of infective endocarditis and the efficacy of antibiotic prophylaxis to prevent its development. They stress the importance of good oral hygiene and prevention of dental disease and argue persuasively that this will have a greater impact on decreasing the lifetime risk of infective endocarditis than will antibiotic prophylaxis.
Prophylaxis is now recommended only for patients with a prosthetic heart valve, a history of infective endocarditis, certain forms of congenital heart disease, and valvulopathy after cardiac transplantation (Table 1), and only before procedures that involve manipulation of gingival tissue or the periapical region of teeth, or perforation of the oral mucosa. Excluded are routine dental cleaning and anesthetic injections through noninfected tissue, dental radiography, placement and adjustment of appliances, shedding of deciduous teeth, and bleeding from trauma to the lips.
CONTROVERSY WILL CONTINUE
The new guidelines for dental prophylaxis have been extensively revised and simplified. They are now focused only on patients who have a greater lifetime risk of illness and death from infective endocarditis. But what about patients who had previously been advised to take prophylaxis, such as those with mitral valve prolapse with regurgitation, who will not receive prophylaxis any more?
These guidelines will likely stir emotions, not only for practitioners who have strong desires to practice preventive medicine, but also for patients who have been taking prophylaxis in good faith per previous guidelines. They may feel abandoned. Unfortunately, funding for a prospective randomized clinical trial large enough to prove that antibiotic prophylaxis for dental procedures benefits patients is unlikely. That leaves us with the current recommendations, which are based on scientific evidence that currently exists and on expert opinion.
The intention of the guidelines is laudable. Of course, there will continue to be controversies with the new rules. Nevertheless, we believe they should be followed until there is more persuasive evidence to the contrary.
- Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation 2007; 116:1736–1754.
- Hill EE, Herligers P, Claus P, Vanderschueren S, Herregods MC, Peetermans WE. Infective endocarditis: changing epidemiology and predictors of 6-month mortality: a prospective cohort study. Eur Heart J 2007; 28:196–203.
- Wang A, Athan E, Pappas PA, et al International Collaboration on Endocarditis-Prospective Cohort Study Investigators. Contemporary clinical profile and outcome of prosthetic valve endocarditis. JAMA 2007; 297:1354–1361.
- Dajani AS, Taubert KA, Wilson W, et al. Prevention of bacterial endocarditis. Recommendations by the American Heart Association. JAMA 1997; 277:1794–1801.
- Gendron R, Grenier D, Maheu-Robert LF. The oral cavity as a reservoir of bacterial pathogens for focal infections. Microbes Infect 2000; 2:897–906.
- Strom BL, Abrutyn E, Berlin JA, et al. Dental and cardiac risk factors for infective endocarditis. A population-based, case-control study. Ann Intern Med 1998; 129:761–769.
- Duval X, Alla F, Hoen B, et al. Estimated risk of endocarditis in adults with predisposing cardiac conditions undergoing dental procedures with or without antibiotic prophylaxis. Clin Infect Dis 2006; 42:e102–e107.
- van der Meer JT, van Wijk W, Thompson J, Valkenburg HA, Michel MF. Awareness of need and actual use of prophylaxis: lack of patient compliance in the prevention of bacterial endocarditis. J Antimicrob Chemother 1992; 29:187–194.
- Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation 2007; 116:1736–1754.
- Hill EE, Herligers P, Claus P, Vanderschueren S, Herregods MC, Peetermans WE. Infective endocarditis: changing epidemiology and predictors of 6-month mortality: a prospective cohort study. Eur Heart J 2007; 28:196–203.
- Wang A, Athan E, Pappas PA, et al International Collaboration on Endocarditis-Prospective Cohort Study Investigators. Contemporary clinical profile and outcome of prosthetic valve endocarditis. JAMA 2007; 297:1354–1361.
- Dajani AS, Taubert KA, Wilson W, et al. Prevention of bacterial endocarditis. Recommendations by the American Heart Association. JAMA 1997; 277:1794–1801.
- Gendron R, Grenier D, Maheu-Robert LF. The oral cavity as a reservoir of bacterial pathogens for focal infections. Microbes Infect 2000; 2:897–906.
- Strom BL, Abrutyn E, Berlin JA, et al. Dental and cardiac risk factors for infective endocarditis. A population-based, case-control study. Ann Intern Med 1998; 129:761–769.
- Duval X, Alla F, Hoen B, et al. Estimated risk of endocarditis in adults with predisposing cardiac conditions undergoing dental procedures with or without antibiotic prophylaxis. Clin Infect Dis 2006; 42:e102–e107.
- van der Meer JT, van Wijk W, Thompson J, Valkenburg HA, Michel MF. Awareness of need and actual use of prophylaxis: lack of patient compliance in the prevention of bacterial endocarditis. J Antimicrob Chemother 1992; 29:187–194.
The STAR*D study: Treating depression in the real world
Depression can be treated successfully by primary care physicians under “real-world” conditions.
Furthermore, the particular drug or drugs used are not as important as following a rational plan: giving antidepressant medications in adequate doses, monitoring the patient’s symptoms and side effects and adjusting the regimen accordingly, and switching drugs or adding new drugs to the regimen only after an adequate trial.
These are among the lessons learned from the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study, the largest prospective clinical trial of treatment of major depressive disorder ever conducted. It was funded by the National Institutes of Health and directed by A. John Rush, MD.
WHAT WERE THE AIMS OF STAR*D?
Depression, a common and debilitating condition, affects approximately one in eight people in the United States.1 It is expected2 to be the second-leading cause of disability in the world by the year 2020; today, it is the second-leading cause of disability-adjusted life years in those 15 to 44 years old.3
Nevertheless, the available evidence base for treatment is limited, since most participants in clinical trials are recruited by advertisement rather than from representative practices, and they are often selected to have few comorbid disorders, either medical or psychiatric. In addition, those with chronic depression or current suicidal ideation are excluded.1,4 These uncomplicated and “pristine” participants are unlike typical patients seen by primary care physicians or psychiatrists.
Similarly, the protocols used in these trials do not represent usual clinic practice.Patients in clinical trials undergo more assessment and more frequent follow-up than in real-world practice, they have no say in treatment decisions, the doses are fixed, and the patients and physicians are blinded to the intervention. Consequently, how to translate the results of these efficacy trials into practice is unclear.5
Further, even in relatively uncomplicated cases, only about one-half of outpatients with nonpsychotic major depressive disorder initially treated with a single medication or with psychotherapy will experience a clinically significant improvement in symptoms (ie, a response) during the 8 to 12 weeks of acute-phase treatment,6–10 and only 20% to 35% of patients will reach remission,9 the aim of treatment.8,11 The remission rates are even lower in treatment-resistant depression.12 How to manage most patients—those whose depression does not remit with the first, second, or third step of treatment—is unclear.
Accordingly, the overall objective of STAR*D was to develop and evaluate feasible treatment strategies to improve clinical outcomes for real-world patients with treatment-resistant depression, who were identified prospectively from a pool of patients in a current major depressive episode.13–15 Specifically, STAR*D aimed to determine prospectively which of several treatments is the most effective “next step” for patients who do not reach remission with an initial or subsequent treatment or who cannot tolerate the treatment.
WHY IS STAR*D RELEVANT FOR PRIMARY CARE?
Nearly 10% of all primary care office visits are depression-related.16 Primary care physicians provide nearly half the outpatient care for depressed patients.17 Indeed, primary care physicians log approximately as many outpatient visits for depression as psychiatrists do.18 Medical comorbidity is especially common in primary care settings.19 When to refer to a psychiatrist is not clear.
KEY FEATURES OF THE STUDY DESIGN
STAR*D involved a national consortium of 14 university-based regional centers, which oversaw a total of 23 participating psychiatric and 18 primary care clinics. Enrollment began in 2000, with follow-up completed in 2004.
Entry criteria were broad and inclusive
Patients had to:
- Be between 18 and 75 years of age
- Have a nonpsychotic major depressive disorder, identified by a clinician and confirmed with a symptom checklist based on the Diagnostic and Statistical Manual, fourth edition revised,20 and for which antidepressant treatment is recommended
- Score at least 14 on the 17-item Hamilton Rating Scale for Depression (HAMD17)21
- Not have a primary diagnosis of bipolar disorder, obsessive-compulsive disorder, or an eating disorder, which would require a different treatment strategy, or a seizure disorder (which would preclude bupropion as a second-step treatment).
Dosing recommendations were flexible but vigorous
Medications often were increased to maximally tolerated doses. For example, citalopram (Celexa) was started at 20 mg/day and increased by 20 mg every 2 to 4 weeks if the patient was tolerating it but had not achieved remission, to a maximum dose of 60 mg/day. Treatment could be given for up to 14 weeks, during which side effects22 and clinical ratings23 were assessed by both patients and study coordinators.
Measurement-based care
We used a systematic approach to treatment called “measurement-based care,”24 which involves routinely measuring symptoms23 and side effects22 and using this information to modify the medication doses at critical decision points. This algorithmic approach provided flexible treatment recommendations to ensure that the dosage and duration of antidepressant drug treatment were adequate.25
The severity of depression was assessed by the clinician-rated, 16-item Quick Inventory of Depressive Symptomatology (QIDS-C16). The QIDS-SR16 (the self-report version) can substitute for the QIDS-C1623 to make this approach more feasible. Both tools are available at www.ids-qids.org.
This approach was easily worked into busy primary care and specialty care office workflows (clinic physicians, most with limited research experience, provided the treatment), and could be translated into primary care practice in the community as well.
Four-step protocol
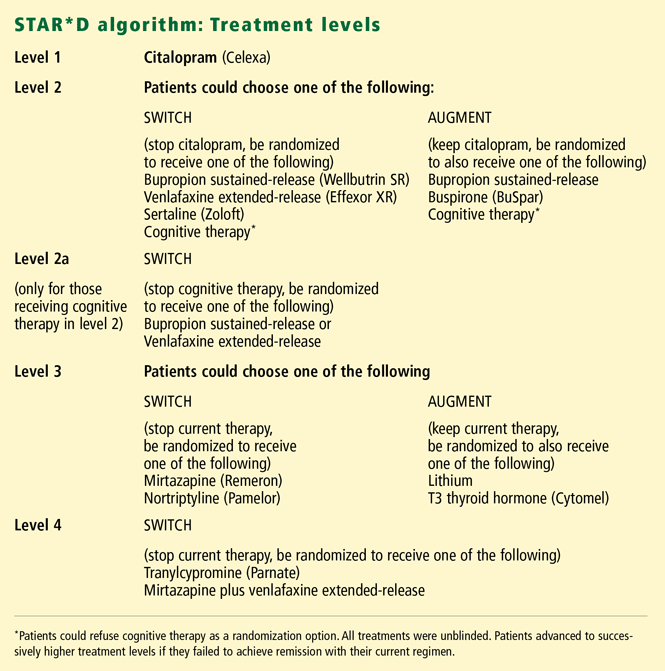
A unique feature of the study design was that the patients, in consultation with their physicians, had some choice in the treatments they received. In this “equipoise-stratified randomized design,”26 at levels 2 and 3 the patient could choose either to switch therapies (stop the current drug and be randomized to receive one of several different treatments) or to augment their current therapy (by adding one of several treatments in a randomized fashion). Patients could decline certain strategies as long as there were at least two possible options to which one might be randomized.
At level 2, one of the options for both switching and augmentation was cognitive therapy, although patients could decline that option. Conversely, if they definitely wanted cognitive therapy, they could choose to be randomized to either cognitive therapy alone or to cognitive therapy added to citalopram. Also, anyone who received cognitive therapy in level 2 and failed to enter remission was additionally randomized to either bupropion or venlafaxine (level 2a) to ensure that all patients had failed trials on two medications before entering level 3.
When switching to medications other than a monoamine oxidase inhibitor (MAOI), the clinician could choose either to stop the current medication and immediately begin the next one, or to decrease the current medication while starting the new one at a low dose and then tapering and titrating over 1 week. (Switching to an MAOI, used only in the final level of treatment, required a 7- to 10-day washout period.)
Outcomes measured
Remission (complete recovery from the depressive episode), the primary study outcome, was defined as a HAM-D17 score of 7 or less, as assessed by treatment-blinded raters.A secondary remission outcome was a QIDS-SR16 score of 5 or less. Of note, the HAM-D17 remission rates were slightly lower than the rates based on the QIDS-SR16, since patients who did not have a HAM-D17 score measured at exit were defined as not being in remission a priori. Thus, the QIDS-SR16 rates might have been a slightly better reflection of actual remission rates.
Response, a secondary outcome, was defined as a reduction of at least 50% in the QIDS-SR16 score from baseline at the last assessment.
FEW DIFFERENCES BETWEEN PSYCHIATRIC, PRIMARY CARE PATIENTS
The patients seen in primary care clinics were surprisingly similar to those seen in psychiatric clinics.27,28 The two groups did not differ in severity of depression, distribution of severity scores, the likelihood of presenting with any of the nine core criteria of a major depressive episode, or the likelihood of having a concomitant axis I psychiatric disorder in addition to depression (about half of participants in each setting had an anxiety disorder).
Recurrent major depressive disorders were common in both groups, though more so in psychiatric patients (78% vs 69%, P < .001), while chronic depression was more common in primary care than in psychiatric patients (30% vs 21%, P < .001). Having either a chronic index episode (ie, lasting > 2 years) or a recurrent major depressive disorder was common in both groups (86% vs 83%, P = .0067).
That said, primary care patients were older (44 years vs 39 years, P < .001), more of them were Hispanic (18% vs 9%, P < .001), and more of them had public insurance (23% vs 9%, P < .001). Fewer of the primary care patients had completed college (20% vs 28%, P < .001), and the primary care patients tended to have greater medical comorbidity. Psychiatric patients were more likely to have attempted suicide in the past and to have had their first depressive illness before age 18.
LEVEL 1: WHAT CAN WE EXPECT FROM INITIAL TREATMENT?
At level 1, all the patients received citalopram. The mean dose was 40.6 ± 16.6 mg/day in the primary care clinics and 42.5 ± 16.8 mg/day in the psychiatric clinics, which are adequate, middle-range doses and higher than the average US dose.29
Approximately 30% of patients achieved remission: 27% as measured on the HAM-D17 and 33% on the QIDS-SR16. The response rate (on the QIDS-SR16) was 47%. There were no differences between primary and psychiatric care settings in remission or response rates.
Patients were more likely to achieve remission if they were white, female, employed, more educated, or wealthier. Longer current episodes, more concurrent psychiatric disorders (especially anxiety disorders or drug abuse), more general medical disorders, and lower baseline function and quality of life were each associated with lower remission rates.
What is an adequate trial?
Longer times than expected were needed to reach response or remission. The average duration required to achieve remission was almost 7 weeks (44 days in primary care; 49 days in psychiatric care). Further, approximately one-third of those who ultimately responded and half of those who entered remission did so after 6 weeks.30 Forty percent of those who entered remission required 8 or more weeks to do so.
These results suggest that longer treatment durations and more vigorous medication dosing than generally used are needed to achieve optimal remission rates. It is imprudent to stop a treatment that the patient is tolerating in a robust dose if the patient reports only partial benefit by 6 weeks; indeed, raising the dose, if tolerated, may help a substantial number of patients respond by 12 or 14 weeks. Instruments to monitor depression severity (eg, self-report measures) can be useful. At least 8 weeks with at least moderately vigorous dosing is recommended.
LEVEL 2: IF THE FIRST TREATMENT FAILS
When switching to a new drug, does it matter which one?
No.
In level 2, if patients had not achieved remission on citalopram alone, they had the choice of switching: stopping citalopram and being randomized to receive either sertraline (Zoloft, another SSRI), venlafaxine extended-release (XR) (Effexor XR, a serotonin and norepinephrine reuptake inhibitor), or bupropion sustained-release (SR) (Wellbutrin SR, a norepinephrine and dopamine reuptake inhibitor). At the last visit the mean daily doses were bupropion SR 282.7 mg/day, sertraline 135.5 mg/day, and venlafaxine-XR 193.6 mg/day.
The remission rate was approximately one-fourth with all three drugs31:
- With bupropion SR—21.3% by HAM-D17, 25.5% by QIDS-SR16
- With sertraline—17.6% by HAM-D17, 26.6% by QIDS-SR16
- With venlafaxine-XR—24.8% by HAM-D17, 25.0% by QIDS-SR16. The remission rates were neither statistically nor clinically different by either measure.
Though the types of side effects related to specific medications may have varied, the overall side-effect burden and the rate of serious adverse events did not differ significantly.
When adding a new drug, does it matter which one?
Again, no.
Instead of switching, patients in level 2 could choose to stay on citalopram and be randomized to add either bupropion SR or buspirone (BuSpar) to the regimen (augmentation). The mean daily doses at the end of level 2 were bupropion SR 267.5 mg and buspirone 40.9 mg.
Rates of remission32:
- With bupropion SR—29.7% on the HAMD-D17, 39.0% on the QIDS-SR16
- With buspirone—30.1% on the HAM-D17, 32.9% on the QIDS-SR16.
However, the QIDS-SR16 scores declined significantly more with bupropion SR than with buspirone (25.3% vs 17.1%, P < .04). The mean total QIDS-SR16 score at the last visit was lower with bupropion SR (8.0) than with buspirone (9.1, P < .02), and augmentation with bupropion SR was better tolerated (the dropout rate due to intolerance was 12.5% with bupropion-SR vs 20.6% with buspirone 20.6%; P < .009).
Can we directly compare the benefits of switching vs augmenting?
No.
Patients could choose whether to switch from citalopram to another drug or to add another drug at the second treatment level.33 Consequently, we could not ensure that the patient groups were equivalent at the point of randomization at the beginning of level 2, and, indeed, they were not.
Those who benefitted more from citalopram treatment and who better tolerated it preferred augmentation, while those who benefitted little or who could not tolerate it preferred to switch. Consequently, those in the augmentation group at level 2 were somewhat less depressed than those who switched. Whether augmentation is better even if the initial treatment is minimally effective could not be evaluated in STAR*D.
What about cognitive therapy?
There was no difference between cognitive therapy (either as a switch or as augmentation) and medication (as a switch or as augmentation).34 Adding another drug was more rapidly effective than adding cognitive therapy. Switching to cognitive therapy was better tolerated than switching to a different antidepressant.
Of note, fewer patients accepted cognitive therapy as a randomization option than we expected, so the sample sizes were small. Possible reasons were that all patients had to receive a medication at study entry (which may have biased selection towards those preferring medication), and cognitive therapy entailed additional copayments and visiting still another provider at another site.
After two levels of treatment, how many patients reach remission?
About 30% of patients in level 1 achieved remission, and of those progressing to level 2, another 30% achieved remission. Together, this adds up to about 50% of patients achieving remission if they remained in treatment (30% in level 1 plus 30% of the roughly 70% remaining in level 2).
IF A SECOND TREATMENT FAILS
If switching again to another drug, does it matter which one?
No.
In level 3, patients could choose to stop the drug they had been taking and be randomized to receive either mirtazapine (Remeron) or nortriptyline (Pamelor).
Switching medications was not as effective as a third step as it was as a second step.35
Remission rates:
- With mirtazapine—12.3% on the HAM-D17, 8.0% on the QIDS-SR16
- With nortriptyline—19.8% on the HAM-D17, 12.4% on the QIDS-SR16.
Response rates were 13.4% with mirtazapine and 16.5% with nortriptyline. Statistically, neither the response nor the remission rates differed by treatment, nor did these two treatments differ in tolerability or side-effect burden.
Does choice of augmentation agent matter: Lithium vs T3?
Similarly, after two failed medication treatments, medication augmentation was less effective than it was at the second step.36 The two augmentation options tested, lithium and T3 thyroid hormone (Cytomel), are commonly considered by psychiatrists but less commonly used by primary care doctors.
Lithium is believed to increase serotonergic function, which may have a synergistic effect on the mechanism of action of antidepressants; a meta-analysis of placebo-controlled studies supports lithium’s effectiveness as adjunctive treatment.37 Its side effects, however, must be closely monitored.38 The primary monitoring concern is the small difference between the therapeutic blood level (0.6–1.2 mEq/L) and potentially toxic blood levels (> 1.5 mEq/L).
Lithium was started at 450 mg/day, and at week 2 it was increased to the recommended dose of 900 mg/day (a dose below the target dose for bipolar disorder). If patients could not tolerate 450 mg/day, the initial dose was 225 mg/day for 1 week before being increased to 450 mg/day, still with the target dose of 900 mg/day. The mean exit dose was 859.9 mg/day, and the median blood level was 0.6 mEq/L.
Thyroid hormone augmentation using T3 is believed to work through both direct and indirect effects on the hypothalamic-pituitary-thyroid axis, which has a strong relationship with depression. The efficacy of T3 augmentation is supported by a meta-analysis of eight studies,39 and T3 is effective whether or not thyroid abnormalities are present.
In STAR*D, T3 was started at 25 μg/day for 1 week, than increased to the recommended dose of 50 μg/day. The mean exit dose was 45.2 μg/day.
Remission rates:
- With lithium augmentation—15.9% by the HAM-D17, 13.2% by the QIDS-SR16
- With T3 augmentation—24.7% by both measures.
Response rates were 16.2% with lithium augmentation and 23.3% with T3 augmentation.
While neither response nor remission rates were statistically significantly different by treatment, lithium was more frequently associated with side effects (P = .045), and more participants in the lithium group left treatment because of side effects (23.2% vs 9.6%; P = .027). These results suggest that in cases in which a clinician is considering an augmentation trial, T3 has slight advantages over lithium in effectiveness and tolerability. T3 also offers the advantages of being easy to use and not necessitating blood level monitoring. These latter benefits are especially relevant to the primary care physician. However, T3’s potential for long-term side effects (eg, osteoporosis, cardiovascular effects) were not examined, and it is not clear when to discontinue it.
LEVEL 4: AFTER THREE FAILURES, HOW SHOULD A CLINICIAN PROCEED?
Switch to mirtazapine plus venlafaxine XR or tranylcypromine?
Patients who reached level 4 were considered to have a highly treatment-resistant depressive illness, so treatments at this level were, by design, more aggressive. Accordingly, at level 4 we investigated treatments that might be considered more demanding than those a primary care physician would use. Approximately 40% of patients in each treatment group were from primary care settings.
Remission rates40:
- With the combination of mirtazapine (mean dose 35.7 mg/day) and venlafaxine XR (mean dose 210.3 mg/day)—13.7% by the HAM-D17 and 15.7% by the QIDS-SR16
- With the MAOI tranylcypromine (Parnate, mean dose 36.9 mg/day)—6.9% by the HAM-D17 and 13.8% by the QIDS-SR16. Response rates were 23.5% with the combination and 12.1% with tranylcypromine. Neither remission nor response rates differed significantly.
However, the percentage reduction in QIDS-SR16 score between baseline and exit was greater with the combination than with tranylcypromine. Further, more patients dropped out of treatment with tranylcypromine because of side effects (P < .03). Tranylcypromine also has the disadvantage of necessitating dietary restrictions.
A significant limitation of this comparison is that patients were less likely to get an adequate trial of tranylcypromine, an MAOI, than of the combination. When the 2-week washout period (required before switching to an MAOI) is subtracted from the total time in treatment, approximately 30% of participants in the tranylcypromine group had less than 2 weeks of treatment, and nearly half had less than 6 weeks of treatment.
Therefore, even though the remission and response rates were similar between groups, the combination of venlafaxine-XR plus mirtazapine therapy might have some advantages over tranylcypromine. These results provided the first evidence of tolerability and at least modest efficacy of this combination for treatment-resistant cases.
Overall, what was the cumulative remission rate?
The theoretical cumulative remission rate after four acute treatment steps was 67%. Remission was more likely to occur during the first two levels of treatment than during the last two. The cumulative remission rates for the first four steps were:
- Level 1—33%
- Level 2—57%
- Level 3—63%
- Level 4—67%.
RESULTS FROM LONG-TERM FOLLOW-UP AFTER REMISSION OR RESPONSE
Patients with a clinically meaningful response or, preferably, remission at any level could enter into a 12-month observational follow-up phase. Those who had required more treatment levels had higher relapse rates during this phase.41 Further, if a patient achieved remission rather than just response to treatment, regardless of the treatment level, the prognosis at follow-up was better, confirming the importance of remission as the goal of treatment.
Results also provided a warning—the greater the number of treatment levels that a patient required, the more likely that patient and physician would settle for response. Whether the greater relapse rates reflect a harder-to-treat depression or the naturalistic design of the follow-up phase (with less control over dosing) is unclear.
WHAT DO THESE RESULTS MEAN FOR PRIMARY CARE PHYSICIANS?
- Measurement-based care is feasible in primary care. Primary care doctors can ensure vigorous but tolerable dosing using a self-report depression scale to monitor response, a side-effects tool to monitor tolerability, and medication adjustments at critical decision points guided by these two measures.
- Remission, ie, complete recovery from a depressive episode, rather than merely substantial improvement, is associated with a better prognosis and is the preferred goal of treatment.
- Pharmacologic differences between psychotropic medications did not translate into substantial clinical differences, although tolerability differed. These findings are consistent with a large-scale systematic evidence review recently completed by the Agency for Healthcare Research and Quality that compared the effectiveness of antidepressants.42 Given the difficulty in predicting what medication will be both efficacious for and tolerated by an individual patient, familiarity with a broad spectrum of antidepressants is prudent.
- Remission of depressive episodes will most likely require repeated trials of sufficiently sustained,vigorously dosed antidepressant medication. From treatment initiation, physicians should ensure maximal but tolerable doses for at least 8 weeks before deciding that an intervention has failed.
- If a first treatment doesn’t work, either switching or augmenting it is a reasonable choice. Augmentation may be preferred if the patient is tolerating and receiving partial benefit from the initial medication choice. While bupropion SR and buspirone were not different as augmenters by the primary remission outcome measure, secondary measures (eg, tolerability, depressive symptom change over the course of treatment, clinician-rated Quick Inventory of Depressive Symptomatology) recommended bupropion-SR over buspirone.
- If physicians switch, either a within-class switch (eg, citalopram to sertraline) or an out-of-class switch (eg, citalopram to bupropion SR) is effective, as is a switch to a dual-action agent (eg, venlafaxine XR).
- The likelihood of improvement after two aggressive medication trials is very low and likely requires more complicated medication regimens, and the existing evidence base is quite thin. These primary care patients should likely be referred to psychiatrists for more aggressive and intensive treatment.
- For patients who present with major depressive disorder, STAR*D suggests that with persistence and aggressive yet feasible care, there is hope: after one round, approximately 30% will have a remission; after two rounds, 50%; after three rounds, 60%; and after four rounds, 70%.
- While STAR*D excluded depressed patients with bipolar disorder, a depressive episode in a patient with bipolar disorder can be difficult to distinguish from a depressive episode in a patient with major depressive disorder. Primary care physicians need to consider bipolar disorder both in patients presenting with a depressive episode and in those who fail an adequate trial.43
FUTURE CONSIDERATIONS
Subsequent STAR*D analyses will compare in greater depth outcomes in primary care vs psychiatric settings at each level of treatment. Given the greater risk of depression persistence associated with more successive levels of treatment, subsequent research will focus on ways to more successfully treat depression in the earlier stages, possibly through medication combinations earlier in treatment (somewhat analogous to a “broad-spectrum antibiotic” approach for infections).
- Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003; 289:3095–3105.
- Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 1997; 349:1436–1442.
- Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet 1997; 349:1498–1504.
- Zimmerman M, Chelminski I, Posternak MA. Generalizability of antidepressant efficacy trials: differences between depressed psychiatric outpatients who would or would not qualify for an efficacy trial. Am J Psychiatry 2005; 162:1370–1372.
- Rothwell PM. External validity of randomised controlled trials: to whom do the results of this trial apply? Lancet 2005; 365:82–93.
- Depression Guideline Panel. Depression in primary care: Volume 1, diagnosis and detection. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Frank E, Karp J, Rush A. Efficacy of treatments for major depression. Psychopharmacol Bull 1993; 29:457–475.
- Depression Guideline Panel. Depression in primary care: Volume 2, Treatment of major depression. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Fava M, Davidson KG. Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am 1996; 19:179–200.
- Jarrett RB, Rush A. Short-term psychotherapy of depressive disorders: current status and future directions. Psychiatry: Interpers Biol Process 1994; 57:115–132.
- American Psychiatric Association. Practice guideline for the treatment of patients with major depression (revision). Am J Psychiatry 2000; 157(suppl 4):1–45.
- Dunner DL, Rush AJ, Russell JM, et al. Prospective, long-term, multicenter study of the naturalistic outcomes of patients with treatment-resistant depression. J Clin Psychiatry 2006; 67:688–695.
- Fava M, Rush A, Trivedi M, et al. Background and rationale for the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study. Psychiatr Clin North Am 2003; 26:457–494.
- Gaynes B, Davis L, Rush A, Trivedi M, Fava M, Wisniewski S. The aims and design of the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) Study. Prim Psychiatry 2005; 12:36–41.
- Rush A, Fava M, Wisniewski S, et al. Sequenced Treatment Alternatives to Relieve Depression (STAR*D): rationale and design. Control Clin Trials 2004; 25:119–142.
- Stafford RS, Ausiello JC, Misra B, Saglam D. National Patterns o fDepression Treatment in Primary Care. Prim Care Companion J Clin Psychiatry 2000; 2:211–216.
- Regier D, Narrow W, Rae D, Mandersheid R, Locke B, Goodwin F. The de facto US mental and addictive disorders service system: epidemiologic catchment area prospective 1-year prevalence rates of disorders and services. Arch Gen Psychiatry 1993; 50:85–94.
- Pincus H, Tanielian T, Marcus S, et al. Prescribing trends in psychotropic medications: primary care, psychiatry, and other medical specialities. JAMA 1998; 279:526–531.
- Vuorilehto M, Melartin T, Isometsa E. Depressive disorders in primary care: recurrent, chronic, and co-morbid. Psychol Med 2005; 35:673–682.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth edition, Text Revision. Washington, DC: American Psychiatric Association; 2000.
- Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23:56–61.
- Wisniewski SR, Rush AJ, Balasubramani GK, Trivedi MH, Nierenberg AA for the STAR*D Investigators. Self-rated global measure of the frequency, intensity, and burden of side effects. J Psychiatric Pract 2006; 12:71–79.
- Rush AJ, Bernstein IH, Trivedi MH, et al. An evaluation of the Quick Inventory of Depressive Symptomatology and the Hamilton Rating Scale for Depression: a Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial report. Biol Psychiatry 2006; 59:493–501.
- Trivedi MH, Rush AJ, Gaynes BN, et al. Maximizing the adequacy of medication treatment in controlled trials and clinical practice: STAR*D measurement-based care. Neuropsychopharmacology 2007/04/04/online 2007.
- Kawamoto K, Houlihan CA, Balas EA, Lobach DF. Improving clinical practice using clinical decision support systems: a systematic review of trials to identify features critical to success. BMJ 2005; 330:765 e-pub March 14 2005.
- Lavori P, Rush A, Wisniewski S, et al. Strengthening clinical effectiveness trials: equipoise-stratified randomization. Biol Psychiatry 2001; 50:792–801.
- Gaynes BN, Rush AJ, Trivedi MH, et al. Major depression symptoms in primary care and psychiatric care settings: a cross-sectional analysis. Ann Fam Med 2007; 5:126–134.
- Gaynes BN, Rush AJ, Trivedi M, et al. A direct comparison of presenting characteristics of depressed outpatients from primary vs. specialty care settings: preliminary findings from the STAR*D clinical trial. Gen Hosp Psychiatry 2005; 27:87–96.
- Sullivan PW, Valuck R, Saseen J, MacFall HM. A comparison of the direct costs and cost effectiveness of serotonin reuptake inhibitors and associated adverse drug reactions. CNS Drugs 2004; 18:911–932.
- Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006; 163:28–40.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med 2006; 354:1231–1242.
- Trivedi MH, Fava M, Wisniewski SR, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med 2006; 354:1243–1252.
- Wisniewski SR, Fava M, Trivedi MH, et al. Acceptability of second-step treatments to depressed outpatients: a STAR*D report. Am J Psychiatry 2007; 164:753–760.
- Thase ME, Friedman ES, Biggs MM, et al. Cognitive therapy versus medication in augmentation and switch strategies as second-step treatments: a STAR*D report. Am J Psychiatry 2007; 164:739–752.
- Fava M, Rush AJ, Wisniewski SR, et al. A comparison of mirtazapine and nortriptyline following two consecutive failed medication treatments for depressed outpatients: a STAR*D report. Am J Psychiatry 2006; 163:1161–1172.
- Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T3 augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry 2006; 163:1519–1530.
- Bschor T, Lewitzka U, Sasse J, Adli M, Koberle U, Bauer M. Lithium augmentation in treatment-resistant depression: clinical evidence, serotonergic and endocrine mechanisms. Pharmacopsychiatry 2003; 36(suppl 3):S230–S234.
- Freeman MP, Freeman SA. Lithium: clinical considerations in internal medicine. Am J Med 2006; 119:478–481.
- Aronson R, Offman HJ, Joffe RT, Naylor CD. Triiodothyronine augmentation in the treatment of refractory depression. A meta-analysis. Arch Gen Psychiatry 1996; 53:842–848.
- McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry 2006; 163:1531–1541.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 2006; 163:1905–1917.
- Gartlehner G, Hansen R, Thieda P, et al. Comparative Effectiveness of Second-generation Antidepressants in the Pharmacologic Treatment of Depression. Agency for Healthcare Research and Quality. http://effectivehealthcare.ahrq.gov/reports/topic.cfm?topic=8&sid=39&rType=3. Accessed December 12, 2007.
- Das AK, Olfson M, Gameroff MJ, et al. Screening for bipolar disorder in a primary care practice. JAMA 2005; 293:956–963.
Depression can be treated successfully by primary care physicians under “real-world” conditions.
Furthermore, the particular drug or drugs used are not as important as following a rational plan: giving antidepressant medications in adequate doses, monitoring the patient’s symptoms and side effects and adjusting the regimen accordingly, and switching drugs or adding new drugs to the regimen only after an adequate trial.
These are among the lessons learned from the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study, the largest prospective clinical trial of treatment of major depressive disorder ever conducted. It was funded by the National Institutes of Health and directed by A. John Rush, MD.
WHAT WERE THE AIMS OF STAR*D?
Depression, a common and debilitating condition, affects approximately one in eight people in the United States.1 It is expected2 to be the second-leading cause of disability in the world by the year 2020; today, it is the second-leading cause of disability-adjusted life years in those 15 to 44 years old.3
Nevertheless, the available evidence base for treatment is limited, since most participants in clinical trials are recruited by advertisement rather than from representative practices, and they are often selected to have few comorbid disorders, either medical or psychiatric. In addition, those with chronic depression or current suicidal ideation are excluded.1,4 These uncomplicated and “pristine” participants are unlike typical patients seen by primary care physicians or psychiatrists.
Similarly, the protocols used in these trials do not represent usual clinic practice.Patients in clinical trials undergo more assessment and more frequent follow-up than in real-world practice, they have no say in treatment decisions, the doses are fixed, and the patients and physicians are blinded to the intervention. Consequently, how to translate the results of these efficacy trials into practice is unclear.5
Further, even in relatively uncomplicated cases, only about one-half of outpatients with nonpsychotic major depressive disorder initially treated with a single medication or with psychotherapy will experience a clinically significant improvement in symptoms (ie, a response) during the 8 to 12 weeks of acute-phase treatment,6–10 and only 20% to 35% of patients will reach remission,9 the aim of treatment.8,11 The remission rates are even lower in treatment-resistant depression.12 How to manage most patients—those whose depression does not remit with the first, second, or third step of treatment—is unclear.
Accordingly, the overall objective of STAR*D was to develop and evaluate feasible treatment strategies to improve clinical outcomes for real-world patients with treatment-resistant depression, who were identified prospectively from a pool of patients in a current major depressive episode.13–15 Specifically, STAR*D aimed to determine prospectively which of several treatments is the most effective “next step” for patients who do not reach remission with an initial or subsequent treatment or who cannot tolerate the treatment.
WHY IS STAR*D RELEVANT FOR PRIMARY CARE?
Nearly 10% of all primary care office visits are depression-related.16 Primary care physicians provide nearly half the outpatient care for depressed patients.17 Indeed, primary care physicians log approximately as many outpatient visits for depression as psychiatrists do.18 Medical comorbidity is especially common in primary care settings.19 When to refer to a psychiatrist is not clear.
KEY FEATURES OF THE STUDY DESIGN
STAR*D involved a national consortium of 14 university-based regional centers, which oversaw a total of 23 participating psychiatric and 18 primary care clinics. Enrollment began in 2000, with follow-up completed in 2004.
Entry criteria were broad and inclusive
Patients had to:
- Be between 18 and 75 years of age
- Have a nonpsychotic major depressive disorder, identified by a clinician and confirmed with a symptom checklist based on the Diagnostic and Statistical Manual, fourth edition revised,20 and for which antidepressant treatment is recommended
- Score at least 14 on the 17-item Hamilton Rating Scale for Depression (HAMD17)21
- Not have a primary diagnosis of bipolar disorder, obsessive-compulsive disorder, or an eating disorder, which would require a different treatment strategy, or a seizure disorder (which would preclude bupropion as a second-step treatment).
Dosing recommendations were flexible but vigorous
Medications often were increased to maximally tolerated doses. For example, citalopram (Celexa) was started at 20 mg/day and increased by 20 mg every 2 to 4 weeks if the patient was tolerating it but had not achieved remission, to a maximum dose of 60 mg/day. Treatment could be given for up to 14 weeks, during which side effects22 and clinical ratings23 were assessed by both patients and study coordinators.
Measurement-based care
We used a systematic approach to treatment called “measurement-based care,”24 which involves routinely measuring symptoms23 and side effects22 and using this information to modify the medication doses at critical decision points. This algorithmic approach provided flexible treatment recommendations to ensure that the dosage and duration of antidepressant drug treatment were adequate.25
The severity of depression was assessed by the clinician-rated, 16-item Quick Inventory of Depressive Symptomatology (QIDS-C16). The QIDS-SR16 (the self-report version) can substitute for the QIDS-C1623 to make this approach more feasible. Both tools are available at www.ids-qids.org.
This approach was easily worked into busy primary care and specialty care office workflows (clinic physicians, most with limited research experience, provided the treatment), and could be translated into primary care practice in the community as well.
Four-step protocol
A unique feature of the study design was that the patients, in consultation with their physicians, had some choice in the treatments they received. In this “equipoise-stratified randomized design,”26 at levels 2 and 3 the patient could choose either to switch therapies (stop the current drug and be randomized to receive one of several different treatments) or to augment their current therapy (by adding one of several treatments in a randomized fashion). Patients could decline certain strategies as long as there were at least two possible options to which one might be randomized.
At level 2, one of the options for both switching and augmentation was cognitive therapy, although patients could decline that option. Conversely, if they definitely wanted cognitive therapy, they could choose to be randomized to either cognitive therapy alone or to cognitive therapy added to citalopram. Also, anyone who received cognitive therapy in level 2 and failed to enter remission was additionally randomized to either bupropion or venlafaxine (level 2a) to ensure that all patients had failed trials on two medications before entering level 3.
When switching to medications other than a monoamine oxidase inhibitor (MAOI), the clinician could choose either to stop the current medication and immediately begin the next one, or to decrease the current medication while starting the new one at a low dose and then tapering and titrating over 1 week. (Switching to an MAOI, used only in the final level of treatment, required a 7- to 10-day washout period.)
Outcomes measured
Remission (complete recovery from the depressive episode), the primary study outcome, was defined as a HAM-D17 score of 7 or less, as assessed by treatment-blinded raters.A secondary remission outcome was a QIDS-SR16 score of 5 or less. Of note, the HAM-D17 remission rates were slightly lower than the rates based on the QIDS-SR16, since patients who did not have a HAM-D17 score measured at exit were defined as not being in remission a priori. Thus, the QIDS-SR16 rates might have been a slightly better reflection of actual remission rates.
Response, a secondary outcome, was defined as a reduction of at least 50% in the QIDS-SR16 score from baseline at the last assessment.
FEW DIFFERENCES BETWEEN PSYCHIATRIC, PRIMARY CARE PATIENTS
The patients seen in primary care clinics were surprisingly similar to those seen in psychiatric clinics.27,28 The two groups did not differ in severity of depression, distribution of severity scores, the likelihood of presenting with any of the nine core criteria of a major depressive episode, or the likelihood of having a concomitant axis I psychiatric disorder in addition to depression (about half of participants in each setting had an anxiety disorder).
Recurrent major depressive disorders were common in both groups, though more so in psychiatric patients (78% vs 69%, P < .001), while chronic depression was more common in primary care than in psychiatric patients (30% vs 21%, P < .001). Having either a chronic index episode (ie, lasting > 2 years) or a recurrent major depressive disorder was common in both groups (86% vs 83%, P = .0067).
That said, primary care patients were older (44 years vs 39 years, P < .001), more of them were Hispanic (18% vs 9%, P < .001), and more of them had public insurance (23% vs 9%, P < .001). Fewer of the primary care patients had completed college (20% vs 28%, P < .001), and the primary care patients tended to have greater medical comorbidity. Psychiatric patients were more likely to have attempted suicide in the past and to have had their first depressive illness before age 18.
LEVEL 1: WHAT CAN WE EXPECT FROM INITIAL TREATMENT?
At level 1, all the patients received citalopram. The mean dose was 40.6 ± 16.6 mg/day in the primary care clinics and 42.5 ± 16.8 mg/day in the psychiatric clinics, which are adequate, middle-range doses and higher than the average US dose.29
Approximately 30% of patients achieved remission: 27% as measured on the HAM-D17 and 33% on the QIDS-SR16. The response rate (on the QIDS-SR16) was 47%. There were no differences between primary and psychiatric care settings in remission or response rates.
Patients were more likely to achieve remission if they were white, female, employed, more educated, or wealthier. Longer current episodes, more concurrent psychiatric disorders (especially anxiety disorders or drug abuse), more general medical disorders, and lower baseline function and quality of life were each associated with lower remission rates.
What is an adequate trial?
Longer times than expected were needed to reach response or remission. The average duration required to achieve remission was almost 7 weeks (44 days in primary care; 49 days in psychiatric care). Further, approximately one-third of those who ultimately responded and half of those who entered remission did so after 6 weeks.30 Forty percent of those who entered remission required 8 or more weeks to do so.
These results suggest that longer treatment durations and more vigorous medication dosing than generally used are needed to achieve optimal remission rates. It is imprudent to stop a treatment that the patient is tolerating in a robust dose if the patient reports only partial benefit by 6 weeks; indeed, raising the dose, if tolerated, may help a substantial number of patients respond by 12 or 14 weeks. Instruments to monitor depression severity (eg, self-report measures) can be useful. At least 8 weeks with at least moderately vigorous dosing is recommended.
LEVEL 2: IF THE FIRST TREATMENT FAILS
When switching to a new drug, does it matter which one?
No.
In level 2, if patients had not achieved remission on citalopram alone, they had the choice of switching: stopping citalopram and being randomized to receive either sertraline (Zoloft, another SSRI), venlafaxine extended-release (XR) (Effexor XR, a serotonin and norepinephrine reuptake inhibitor), or bupropion sustained-release (SR) (Wellbutrin SR, a norepinephrine and dopamine reuptake inhibitor). At the last visit the mean daily doses were bupropion SR 282.7 mg/day, sertraline 135.5 mg/day, and venlafaxine-XR 193.6 mg/day.
The remission rate was approximately one-fourth with all three drugs31:
- With bupropion SR—21.3% by HAM-D17, 25.5% by QIDS-SR16
- With sertraline—17.6% by HAM-D17, 26.6% by QIDS-SR16
- With venlafaxine-XR—24.8% by HAM-D17, 25.0% by QIDS-SR16. The remission rates were neither statistically nor clinically different by either measure.
Though the types of side effects related to specific medications may have varied, the overall side-effect burden and the rate of serious adverse events did not differ significantly.
When adding a new drug, does it matter which one?
Again, no.
Instead of switching, patients in level 2 could choose to stay on citalopram and be randomized to add either bupropion SR or buspirone (BuSpar) to the regimen (augmentation). The mean daily doses at the end of level 2 were bupropion SR 267.5 mg and buspirone 40.9 mg.
Rates of remission32:
- With bupropion SR—29.7% on the HAMD-D17, 39.0% on the QIDS-SR16
- With buspirone—30.1% on the HAM-D17, 32.9% on the QIDS-SR16.
However, the QIDS-SR16 scores declined significantly more with bupropion SR than with buspirone (25.3% vs 17.1%, P < .04). The mean total QIDS-SR16 score at the last visit was lower with bupropion SR (8.0) than with buspirone (9.1, P < .02), and augmentation with bupropion SR was better tolerated (the dropout rate due to intolerance was 12.5% with bupropion-SR vs 20.6% with buspirone 20.6%; P < .009).
Can we directly compare the benefits of switching vs augmenting?
No.
Patients could choose whether to switch from citalopram to another drug or to add another drug at the second treatment level.33 Consequently, we could not ensure that the patient groups were equivalent at the point of randomization at the beginning of level 2, and, indeed, they were not.
Those who benefitted more from citalopram treatment and who better tolerated it preferred augmentation, while those who benefitted little or who could not tolerate it preferred to switch. Consequently, those in the augmentation group at level 2 were somewhat less depressed than those who switched. Whether augmentation is better even if the initial treatment is minimally effective could not be evaluated in STAR*D.
What about cognitive therapy?
There was no difference between cognitive therapy (either as a switch or as augmentation) and medication (as a switch or as augmentation).34 Adding another drug was more rapidly effective than adding cognitive therapy. Switching to cognitive therapy was better tolerated than switching to a different antidepressant.
Of note, fewer patients accepted cognitive therapy as a randomization option than we expected, so the sample sizes were small. Possible reasons were that all patients had to receive a medication at study entry (which may have biased selection towards those preferring medication), and cognitive therapy entailed additional copayments and visiting still another provider at another site.
After two levels of treatment, how many patients reach remission?
About 30% of patients in level 1 achieved remission, and of those progressing to level 2, another 30% achieved remission. Together, this adds up to about 50% of patients achieving remission if they remained in treatment (30% in level 1 plus 30% of the roughly 70% remaining in level 2).
IF A SECOND TREATMENT FAILS
If switching again to another drug, does it matter which one?
No.
In level 3, patients could choose to stop the drug they had been taking and be randomized to receive either mirtazapine (Remeron) or nortriptyline (Pamelor).
Switching medications was not as effective as a third step as it was as a second step.35
Remission rates:
- With mirtazapine—12.3% on the HAM-D17, 8.0% on the QIDS-SR16
- With nortriptyline—19.8% on the HAM-D17, 12.4% on the QIDS-SR16.
Response rates were 13.4% with mirtazapine and 16.5% with nortriptyline. Statistically, neither the response nor the remission rates differed by treatment, nor did these two treatments differ in tolerability or side-effect burden.
Does choice of augmentation agent matter: Lithium vs T3?
Similarly, after two failed medication treatments, medication augmentation was less effective than it was at the second step.36 The two augmentation options tested, lithium and T3 thyroid hormone (Cytomel), are commonly considered by psychiatrists but less commonly used by primary care doctors.
Lithium is believed to increase serotonergic function, which may have a synergistic effect on the mechanism of action of antidepressants; a meta-analysis of placebo-controlled studies supports lithium’s effectiveness as adjunctive treatment.37 Its side effects, however, must be closely monitored.38 The primary monitoring concern is the small difference between the therapeutic blood level (0.6–1.2 mEq/L) and potentially toxic blood levels (> 1.5 mEq/L).
Lithium was started at 450 mg/day, and at week 2 it was increased to the recommended dose of 900 mg/day (a dose below the target dose for bipolar disorder). If patients could not tolerate 450 mg/day, the initial dose was 225 mg/day for 1 week before being increased to 450 mg/day, still with the target dose of 900 mg/day. The mean exit dose was 859.9 mg/day, and the median blood level was 0.6 mEq/L.
Thyroid hormone augmentation using T3 is believed to work through both direct and indirect effects on the hypothalamic-pituitary-thyroid axis, which has a strong relationship with depression. The efficacy of T3 augmentation is supported by a meta-analysis of eight studies,39 and T3 is effective whether or not thyroid abnormalities are present.
In STAR*D, T3 was started at 25 μg/day for 1 week, than increased to the recommended dose of 50 μg/day. The mean exit dose was 45.2 μg/day.
Remission rates:
- With lithium augmentation—15.9% by the HAM-D17, 13.2% by the QIDS-SR16
- With T3 augmentation—24.7% by both measures.
Response rates were 16.2% with lithium augmentation and 23.3% with T3 augmentation.
While neither response nor remission rates were statistically significantly different by treatment, lithium was more frequently associated with side effects (P = .045), and more participants in the lithium group left treatment because of side effects (23.2% vs 9.6%; P = .027). These results suggest that in cases in which a clinician is considering an augmentation trial, T3 has slight advantages over lithium in effectiveness and tolerability. T3 also offers the advantages of being easy to use and not necessitating blood level monitoring. These latter benefits are especially relevant to the primary care physician. However, T3’s potential for long-term side effects (eg, osteoporosis, cardiovascular effects) were not examined, and it is not clear when to discontinue it.
LEVEL 4: AFTER THREE FAILURES, HOW SHOULD A CLINICIAN PROCEED?
Switch to mirtazapine plus venlafaxine XR or tranylcypromine?
Patients who reached level 4 were considered to have a highly treatment-resistant depressive illness, so treatments at this level were, by design, more aggressive. Accordingly, at level 4 we investigated treatments that might be considered more demanding than those a primary care physician would use. Approximately 40% of patients in each treatment group were from primary care settings.
Remission rates40:
- With the combination of mirtazapine (mean dose 35.7 mg/day) and venlafaxine XR (mean dose 210.3 mg/day)—13.7% by the HAM-D17 and 15.7% by the QIDS-SR16
- With the MAOI tranylcypromine (Parnate, mean dose 36.9 mg/day)—6.9% by the HAM-D17 and 13.8% by the QIDS-SR16. Response rates were 23.5% with the combination and 12.1% with tranylcypromine. Neither remission nor response rates differed significantly.
However, the percentage reduction in QIDS-SR16 score between baseline and exit was greater with the combination than with tranylcypromine. Further, more patients dropped out of treatment with tranylcypromine because of side effects (P < .03). Tranylcypromine also has the disadvantage of necessitating dietary restrictions.
A significant limitation of this comparison is that patients were less likely to get an adequate trial of tranylcypromine, an MAOI, than of the combination. When the 2-week washout period (required before switching to an MAOI) is subtracted from the total time in treatment, approximately 30% of participants in the tranylcypromine group had less than 2 weeks of treatment, and nearly half had less than 6 weeks of treatment.
Therefore, even though the remission and response rates were similar between groups, the combination of venlafaxine-XR plus mirtazapine therapy might have some advantages over tranylcypromine. These results provided the first evidence of tolerability and at least modest efficacy of this combination for treatment-resistant cases.
Overall, what was the cumulative remission rate?
The theoretical cumulative remission rate after four acute treatment steps was 67%. Remission was more likely to occur during the first two levels of treatment than during the last two. The cumulative remission rates for the first four steps were:
- Level 1—33%
- Level 2—57%
- Level 3—63%
- Level 4—67%.
RESULTS FROM LONG-TERM FOLLOW-UP AFTER REMISSION OR RESPONSE
Patients with a clinically meaningful response or, preferably, remission at any level could enter into a 12-month observational follow-up phase. Those who had required more treatment levels had higher relapse rates during this phase.41 Further, if a patient achieved remission rather than just response to treatment, regardless of the treatment level, the prognosis at follow-up was better, confirming the importance of remission as the goal of treatment.
Results also provided a warning—the greater the number of treatment levels that a patient required, the more likely that patient and physician would settle for response. Whether the greater relapse rates reflect a harder-to-treat depression or the naturalistic design of the follow-up phase (with less control over dosing) is unclear.
WHAT DO THESE RESULTS MEAN FOR PRIMARY CARE PHYSICIANS?
- Measurement-based care is feasible in primary care. Primary care doctors can ensure vigorous but tolerable dosing using a self-report depression scale to monitor response, a side-effects tool to monitor tolerability, and medication adjustments at critical decision points guided by these two measures.
- Remission, ie, complete recovery from a depressive episode, rather than merely substantial improvement, is associated with a better prognosis and is the preferred goal of treatment.
- Pharmacologic differences between psychotropic medications did not translate into substantial clinical differences, although tolerability differed. These findings are consistent with a large-scale systematic evidence review recently completed by the Agency for Healthcare Research and Quality that compared the effectiveness of antidepressants.42 Given the difficulty in predicting what medication will be both efficacious for and tolerated by an individual patient, familiarity with a broad spectrum of antidepressants is prudent.
- Remission of depressive episodes will most likely require repeated trials of sufficiently sustained,vigorously dosed antidepressant medication. From treatment initiation, physicians should ensure maximal but tolerable doses for at least 8 weeks before deciding that an intervention has failed.
- If a first treatment doesn’t work, either switching or augmenting it is a reasonable choice. Augmentation may be preferred if the patient is tolerating and receiving partial benefit from the initial medication choice. While bupropion SR and buspirone were not different as augmenters by the primary remission outcome measure, secondary measures (eg, tolerability, depressive symptom change over the course of treatment, clinician-rated Quick Inventory of Depressive Symptomatology) recommended bupropion-SR over buspirone.
- If physicians switch, either a within-class switch (eg, citalopram to sertraline) or an out-of-class switch (eg, citalopram to bupropion SR) is effective, as is a switch to a dual-action agent (eg, venlafaxine XR).
- The likelihood of improvement after two aggressive medication trials is very low and likely requires more complicated medication regimens, and the existing evidence base is quite thin. These primary care patients should likely be referred to psychiatrists for more aggressive and intensive treatment.
- For patients who present with major depressive disorder, STAR*D suggests that with persistence and aggressive yet feasible care, there is hope: after one round, approximately 30% will have a remission; after two rounds, 50%; after three rounds, 60%; and after four rounds, 70%.
- While STAR*D excluded depressed patients with bipolar disorder, a depressive episode in a patient with bipolar disorder can be difficult to distinguish from a depressive episode in a patient with major depressive disorder. Primary care physicians need to consider bipolar disorder both in patients presenting with a depressive episode and in those who fail an adequate trial.43
FUTURE CONSIDERATIONS
Subsequent STAR*D analyses will compare in greater depth outcomes in primary care vs psychiatric settings at each level of treatment. Given the greater risk of depression persistence associated with more successive levels of treatment, subsequent research will focus on ways to more successfully treat depression in the earlier stages, possibly through medication combinations earlier in treatment (somewhat analogous to a “broad-spectrum antibiotic” approach for infections).
Depression can be treated successfully by primary care physicians under “real-world” conditions.
Furthermore, the particular drug or drugs used are not as important as following a rational plan: giving antidepressant medications in adequate doses, monitoring the patient’s symptoms and side effects and adjusting the regimen accordingly, and switching drugs or adding new drugs to the regimen only after an adequate trial.
These are among the lessons learned from the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study, the largest prospective clinical trial of treatment of major depressive disorder ever conducted. It was funded by the National Institutes of Health and directed by A. John Rush, MD.
WHAT WERE THE AIMS OF STAR*D?
Depression, a common and debilitating condition, affects approximately one in eight people in the United States.1 It is expected2 to be the second-leading cause of disability in the world by the year 2020; today, it is the second-leading cause of disability-adjusted life years in those 15 to 44 years old.3
Nevertheless, the available evidence base for treatment is limited, since most participants in clinical trials are recruited by advertisement rather than from representative practices, and they are often selected to have few comorbid disorders, either medical or psychiatric. In addition, those with chronic depression or current suicidal ideation are excluded.1,4 These uncomplicated and “pristine” participants are unlike typical patients seen by primary care physicians or psychiatrists.
Similarly, the protocols used in these trials do not represent usual clinic practice.Patients in clinical trials undergo more assessment and more frequent follow-up than in real-world practice, they have no say in treatment decisions, the doses are fixed, and the patients and physicians are blinded to the intervention. Consequently, how to translate the results of these efficacy trials into practice is unclear.5
Further, even in relatively uncomplicated cases, only about one-half of outpatients with nonpsychotic major depressive disorder initially treated with a single medication or with psychotherapy will experience a clinically significant improvement in symptoms (ie, a response) during the 8 to 12 weeks of acute-phase treatment,6–10 and only 20% to 35% of patients will reach remission,9 the aim of treatment.8,11 The remission rates are even lower in treatment-resistant depression.12 How to manage most patients—those whose depression does not remit with the first, second, or third step of treatment—is unclear.
Accordingly, the overall objective of STAR*D was to develop and evaluate feasible treatment strategies to improve clinical outcomes for real-world patients with treatment-resistant depression, who were identified prospectively from a pool of patients in a current major depressive episode.13–15 Specifically, STAR*D aimed to determine prospectively which of several treatments is the most effective “next step” for patients who do not reach remission with an initial or subsequent treatment or who cannot tolerate the treatment.
WHY IS STAR*D RELEVANT FOR PRIMARY CARE?
Nearly 10% of all primary care office visits are depression-related.16 Primary care physicians provide nearly half the outpatient care for depressed patients.17 Indeed, primary care physicians log approximately as many outpatient visits for depression as psychiatrists do.18 Medical comorbidity is especially common in primary care settings.19 When to refer to a psychiatrist is not clear.
KEY FEATURES OF THE STUDY DESIGN
STAR*D involved a national consortium of 14 university-based regional centers, which oversaw a total of 23 participating psychiatric and 18 primary care clinics. Enrollment began in 2000, with follow-up completed in 2004.
Entry criteria were broad and inclusive
Patients had to:
- Be between 18 and 75 years of age
- Have a nonpsychotic major depressive disorder, identified by a clinician and confirmed with a symptom checklist based on the Diagnostic and Statistical Manual, fourth edition revised,20 and for which antidepressant treatment is recommended
- Score at least 14 on the 17-item Hamilton Rating Scale for Depression (HAMD17)21
- Not have a primary diagnosis of bipolar disorder, obsessive-compulsive disorder, or an eating disorder, which would require a different treatment strategy, or a seizure disorder (which would preclude bupropion as a second-step treatment).
Dosing recommendations were flexible but vigorous
Medications often were increased to maximally tolerated doses. For example, citalopram (Celexa) was started at 20 mg/day and increased by 20 mg every 2 to 4 weeks if the patient was tolerating it but had not achieved remission, to a maximum dose of 60 mg/day. Treatment could be given for up to 14 weeks, during which side effects22 and clinical ratings23 were assessed by both patients and study coordinators.
Measurement-based care
We used a systematic approach to treatment called “measurement-based care,”24 which involves routinely measuring symptoms23 and side effects22 and using this information to modify the medication doses at critical decision points. This algorithmic approach provided flexible treatment recommendations to ensure that the dosage and duration of antidepressant drug treatment were adequate.25
The severity of depression was assessed by the clinician-rated, 16-item Quick Inventory of Depressive Symptomatology (QIDS-C16). The QIDS-SR16 (the self-report version) can substitute for the QIDS-C1623 to make this approach more feasible. Both tools are available at www.ids-qids.org.
This approach was easily worked into busy primary care and specialty care office workflows (clinic physicians, most with limited research experience, provided the treatment), and could be translated into primary care practice in the community as well.
Four-step protocol
A unique feature of the study design was that the patients, in consultation with their physicians, had some choice in the treatments they received. In this “equipoise-stratified randomized design,”26 at levels 2 and 3 the patient could choose either to switch therapies (stop the current drug and be randomized to receive one of several different treatments) or to augment their current therapy (by adding one of several treatments in a randomized fashion). Patients could decline certain strategies as long as there were at least two possible options to which one might be randomized.
At level 2, one of the options for both switching and augmentation was cognitive therapy, although patients could decline that option. Conversely, if they definitely wanted cognitive therapy, they could choose to be randomized to either cognitive therapy alone or to cognitive therapy added to citalopram. Also, anyone who received cognitive therapy in level 2 and failed to enter remission was additionally randomized to either bupropion or venlafaxine (level 2a) to ensure that all patients had failed trials on two medications before entering level 3.
When switching to medications other than a monoamine oxidase inhibitor (MAOI), the clinician could choose either to stop the current medication and immediately begin the next one, or to decrease the current medication while starting the new one at a low dose and then tapering and titrating over 1 week. (Switching to an MAOI, used only in the final level of treatment, required a 7- to 10-day washout period.)
Outcomes measured
Remission (complete recovery from the depressive episode), the primary study outcome, was defined as a HAM-D17 score of 7 or less, as assessed by treatment-blinded raters.A secondary remission outcome was a QIDS-SR16 score of 5 or less. Of note, the HAM-D17 remission rates were slightly lower than the rates based on the QIDS-SR16, since patients who did not have a HAM-D17 score measured at exit were defined as not being in remission a priori. Thus, the QIDS-SR16 rates might have been a slightly better reflection of actual remission rates.
Response, a secondary outcome, was defined as a reduction of at least 50% in the QIDS-SR16 score from baseline at the last assessment.
FEW DIFFERENCES BETWEEN PSYCHIATRIC, PRIMARY CARE PATIENTS
The patients seen in primary care clinics were surprisingly similar to those seen in psychiatric clinics.27,28 The two groups did not differ in severity of depression, distribution of severity scores, the likelihood of presenting with any of the nine core criteria of a major depressive episode, or the likelihood of having a concomitant axis I psychiatric disorder in addition to depression (about half of participants in each setting had an anxiety disorder).
Recurrent major depressive disorders were common in both groups, though more so in psychiatric patients (78% vs 69%, P < .001), while chronic depression was more common in primary care than in psychiatric patients (30% vs 21%, P < .001). Having either a chronic index episode (ie, lasting > 2 years) or a recurrent major depressive disorder was common in both groups (86% vs 83%, P = .0067).
That said, primary care patients were older (44 years vs 39 years, P < .001), more of them were Hispanic (18% vs 9%, P < .001), and more of them had public insurance (23% vs 9%, P < .001). Fewer of the primary care patients had completed college (20% vs 28%, P < .001), and the primary care patients tended to have greater medical comorbidity. Psychiatric patients were more likely to have attempted suicide in the past and to have had their first depressive illness before age 18.
LEVEL 1: WHAT CAN WE EXPECT FROM INITIAL TREATMENT?
At level 1, all the patients received citalopram. The mean dose was 40.6 ± 16.6 mg/day in the primary care clinics and 42.5 ± 16.8 mg/day in the psychiatric clinics, which are adequate, middle-range doses and higher than the average US dose.29
Approximately 30% of patients achieved remission: 27% as measured on the HAM-D17 and 33% on the QIDS-SR16. The response rate (on the QIDS-SR16) was 47%. There were no differences between primary and psychiatric care settings in remission or response rates.
Patients were more likely to achieve remission if they were white, female, employed, more educated, or wealthier. Longer current episodes, more concurrent psychiatric disorders (especially anxiety disorders or drug abuse), more general medical disorders, and lower baseline function and quality of life were each associated with lower remission rates.
What is an adequate trial?
Longer times than expected were needed to reach response or remission. The average duration required to achieve remission was almost 7 weeks (44 days in primary care; 49 days in psychiatric care). Further, approximately one-third of those who ultimately responded and half of those who entered remission did so after 6 weeks.30 Forty percent of those who entered remission required 8 or more weeks to do so.
These results suggest that longer treatment durations and more vigorous medication dosing than generally used are needed to achieve optimal remission rates. It is imprudent to stop a treatment that the patient is tolerating in a robust dose if the patient reports only partial benefit by 6 weeks; indeed, raising the dose, if tolerated, may help a substantial number of patients respond by 12 or 14 weeks. Instruments to monitor depression severity (eg, self-report measures) can be useful. At least 8 weeks with at least moderately vigorous dosing is recommended.
LEVEL 2: IF THE FIRST TREATMENT FAILS
When switching to a new drug, does it matter which one?
No.
In level 2, if patients had not achieved remission on citalopram alone, they had the choice of switching: stopping citalopram and being randomized to receive either sertraline (Zoloft, another SSRI), venlafaxine extended-release (XR) (Effexor XR, a serotonin and norepinephrine reuptake inhibitor), or bupropion sustained-release (SR) (Wellbutrin SR, a norepinephrine and dopamine reuptake inhibitor). At the last visit the mean daily doses were bupropion SR 282.7 mg/day, sertraline 135.5 mg/day, and venlafaxine-XR 193.6 mg/day.
The remission rate was approximately one-fourth with all three drugs31:
- With bupropion SR—21.3% by HAM-D17, 25.5% by QIDS-SR16
- With sertraline—17.6% by HAM-D17, 26.6% by QIDS-SR16
- With venlafaxine-XR—24.8% by HAM-D17, 25.0% by QIDS-SR16. The remission rates were neither statistically nor clinically different by either measure.
Though the types of side effects related to specific medications may have varied, the overall side-effect burden and the rate of serious adverse events did not differ significantly.
When adding a new drug, does it matter which one?
Again, no.
Instead of switching, patients in level 2 could choose to stay on citalopram and be randomized to add either bupropion SR or buspirone (BuSpar) to the regimen (augmentation). The mean daily doses at the end of level 2 were bupropion SR 267.5 mg and buspirone 40.9 mg.
Rates of remission32:
- With bupropion SR—29.7% on the HAMD-D17, 39.0% on the QIDS-SR16
- With buspirone—30.1% on the HAM-D17, 32.9% on the QIDS-SR16.
However, the QIDS-SR16 scores declined significantly more with bupropion SR than with buspirone (25.3% vs 17.1%, P < .04). The mean total QIDS-SR16 score at the last visit was lower with bupropion SR (8.0) than with buspirone (9.1, P < .02), and augmentation with bupropion SR was better tolerated (the dropout rate due to intolerance was 12.5% with bupropion-SR vs 20.6% with buspirone 20.6%; P < .009).
Can we directly compare the benefits of switching vs augmenting?
No.
Patients could choose whether to switch from citalopram to another drug or to add another drug at the second treatment level.33 Consequently, we could not ensure that the patient groups were equivalent at the point of randomization at the beginning of level 2, and, indeed, they were not.
Those who benefitted more from citalopram treatment and who better tolerated it preferred augmentation, while those who benefitted little or who could not tolerate it preferred to switch. Consequently, those in the augmentation group at level 2 were somewhat less depressed than those who switched. Whether augmentation is better even if the initial treatment is minimally effective could not be evaluated in STAR*D.
What about cognitive therapy?
There was no difference between cognitive therapy (either as a switch or as augmentation) and medication (as a switch or as augmentation).34 Adding another drug was more rapidly effective than adding cognitive therapy. Switching to cognitive therapy was better tolerated than switching to a different antidepressant.
Of note, fewer patients accepted cognitive therapy as a randomization option than we expected, so the sample sizes were small. Possible reasons were that all patients had to receive a medication at study entry (which may have biased selection towards those preferring medication), and cognitive therapy entailed additional copayments and visiting still another provider at another site.
After two levels of treatment, how many patients reach remission?
About 30% of patients in level 1 achieved remission, and of those progressing to level 2, another 30% achieved remission. Together, this adds up to about 50% of patients achieving remission if they remained in treatment (30% in level 1 plus 30% of the roughly 70% remaining in level 2).
IF A SECOND TREATMENT FAILS
If switching again to another drug, does it matter which one?
No.
In level 3, patients could choose to stop the drug they had been taking and be randomized to receive either mirtazapine (Remeron) or nortriptyline (Pamelor).
Switching medications was not as effective as a third step as it was as a second step.35
Remission rates:
- With mirtazapine—12.3% on the HAM-D17, 8.0% on the QIDS-SR16
- With nortriptyline—19.8% on the HAM-D17, 12.4% on the QIDS-SR16.
Response rates were 13.4% with mirtazapine and 16.5% with nortriptyline. Statistically, neither the response nor the remission rates differed by treatment, nor did these two treatments differ in tolerability or side-effect burden.
Does choice of augmentation agent matter: Lithium vs T3?
Similarly, after two failed medication treatments, medication augmentation was less effective than it was at the second step.36 The two augmentation options tested, lithium and T3 thyroid hormone (Cytomel), are commonly considered by psychiatrists but less commonly used by primary care doctors.
Lithium is believed to increase serotonergic function, which may have a synergistic effect on the mechanism of action of antidepressants; a meta-analysis of placebo-controlled studies supports lithium’s effectiveness as adjunctive treatment.37 Its side effects, however, must be closely monitored.38 The primary monitoring concern is the small difference between the therapeutic blood level (0.6–1.2 mEq/L) and potentially toxic blood levels (> 1.5 mEq/L).
Lithium was started at 450 mg/day, and at week 2 it was increased to the recommended dose of 900 mg/day (a dose below the target dose for bipolar disorder). If patients could not tolerate 450 mg/day, the initial dose was 225 mg/day for 1 week before being increased to 450 mg/day, still with the target dose of 900 mg/day. The mean exit dose was 859.9 mg/day, and the median blood level was 0.6 mEq/L.
Thyroid hormone augmentation using T3 is believed to work through both direct and indirect effects on the hypothalamic-pituitary-thyroid axis, which has a strong relationship with depression. The efficacy of T3 augmentation is supported by a meta-analysis of eight studies,39 and T3 is effective whether or not thyroid abnormalities are present.
In STAR*D, T3 was started at 25 μg/day for 1 week, than increased to the recommended dose of 50 μg/day. The mean exit dose was 45.2 μg/day.
Remission rates:
- With lithium augmentation—15.9% by the HAM-D17, 13.2% by the QIDS-SR16
- With T3 augmentation—24.7% by both measures.
Response rates were 16.2% with lithium augmentation and 23.3% with T3 augmentation.
While neither response nor remission rates were statistically significantly different by treatment, lithium was more frequently associated with side effects (P = .045), and more participants in the lithium group left treatment because of side effects (23.2% vs 9.6%; P = .027). These results suggest that in cases in which a clinician is considering an augmentation trial, T3 has slight advantages over lithium in effectiveness and tolerability. T3 also offers the advantages of being easy to use and not necessitating blood level monitoring. These latter benefits are especially relevant to the primary care physician. However, T3’s potential for long-term side effects (eg, osteoporosis, cardiovascular effects) were not examined, and it is not clear when to discontinue it.
LEVEL 4: AFTER THREE FAILURES, HOW SHOULD A CLINICIAN PROCEED?
Switch to mirtazapine plus venlafaxine XR or tranylcypromine?
Patients who reached level 4 were considered to have a highly treatment-resistant depressive illness, so treatments at this level were, by design, more aggressive. Accordingly, at level 4 we investigated treatments that might be considered more demanding than those a primary care physician would use. Approximately 40% of patients in each treatment group were from primary care settings.
Remission rates40:
- With the combination of mirtazapine (mean dose 35.7 mg/day) and venlafaxine XR (mean dose 210.3 mg/day)—13.7% by the HAM-D17 and 15.7% by the QIDS-SR16
- With the MAOI tranylcypromine (Parnate, mean dose 36.9 mg/day)—6.9% by the HAM-D17 and 13.8% by the QIDS-SR16. Response rates were 23.5% with the combination and 12.1% with tranylcypromine. Neither remission nor response rates differed significantly.
However, the percentage reduction in QIDS-SR16 score between baseline and exit was greater with the combination than with tranylcypromine. Further, more patients dropped out of treatment with tranylcypromine because of side effects (P < .03). Tranylcypromine also has the disadvantage of necessitating dietary restrictions.
A significant limitation of this comparison is that patients were less likely to get an adequate trial of tranylcypromine, an MAOI, than of the combination. When the 2-week washout period (required before switching to an MAOI) is subtracted from the total time in treatment, approximately 30% of participants in the tranylcypromine group had less than 2 weeks of treatment, and nearly half had less than 6 weeks of treatment.
Therefore, even though the remission and response rates were similar between groups, the combination of venlafaxine-XR plus mirtazapine therapy might have some advantages over tranylcypromine. These results provided the first evidence of tolerability and at least modest efficacy of this combination for treatment-resistant cases.
Overall, what was the cumulative remission rate?
The theoretical cumulative remission rate after four acute treatment steps was 67%. Remission was more likely to occur during the first two levels of treatment than during the last two. The cumulative remission rates for the first four steps were:
- Level 1—33%
- Level 2—57%
- Level 3—63%
- Level 4—67%.
RESULTS FROM LONG-TERM FOLLOW-UP AFTER REMISSION OR RESPONSE
Patients with a clinically meaningful response or, preferably, remission at any level could enter into a 12-month observational follow-up phase. Those who had required more treatment levels had higher relapse rates during this phase.41 Further, if a patient achieved remission rather than just response to treatment, regardless of the treatment level, the prognosis at follow-up was better, confirming the importance of remission as the goal of treatment.
Results also provided a warning—the greater the number of treatment levels that a patient required, the more likely that patient and physician would settle for response. Whether the greater relapse rates reflect a harder-to-treat depression or the naturalistic design of the follow-up phase (with less control over dosing) is unclear.
WHAT DO THESE RESULTS MEAN FOR PRIMARY CARE PHYSICIANS?
- Measurement-based care is feasible in primary care. Primary care doctors can ensure vigorous but tolerable dosing using a self-report depression scale to monitor response, a side-effects tool to monitor tolerability, and medication adjustments at critical decision points guided by these two measures.
- Remission, ie, complete recovery from a depressive episode, rather than merely substantial improvement, is associated with a better prognosis and is the preferred goal of treatment.
- Pharmacologic differences between psychotropic medications did not translate into substantial clinical differences, although tolerability differed. These findings are consistent with a large-scale systematic evidence review recently completed by the Agency for Healthcare Research and Quality that compared the effectiveness of antidepressants.42 Given the difficulty in predicting what medication will be both efficacious for and tolerated by an individual patient, familiarity with a broad spectrum of antidepressants is prudent.
- Remission of depressive episodes will most likely require repeated trials of sufficiently sustained,vigorously dosed antidepressant medication. From treatment initiation, physicians should ensure maximal but tolerable doses for at least 8 weeks before deciding that an intervention has failed.
- If a first treatment doesn’t work, either switching or augmenting it is a reasonable choice. Augmentation may be preferred if the patient is tolerating and receiving partial benefit from the initial medication choice. While bupropion SR and buspirone were not different as augmenters by the primary remission outcome measure, secondary measures (eg, tolerability, depressive symptom change over the course of treatment, clinician-rated Quick Inventory of Depressive Symptomatology) recommended bupropion-SR over buspirone.
- If physicians switch, either a within-class switch (eg, citalopram to sertraline) or an out-of-class switch (eg, citalopram to bupropion SR) is effective, as is a switch to a dual-action agent (eg, venlafaxine XR).
- The likelihood of improvement after two aggressive medication trials is very low and likely requires more complicated medication regimens, and the existing evidence base is quite thin. These primary care patients should likely be referred to psychiatrists for more aggressive and intensive treatment.
- For patients who present with major depressive disorder, STAR*D suggests that with persistence and aggressive yet feasible care, there is hope: after one round, approximately 30% will have a remission; after two rounds, 50%; after three rounds, 60%; and after four rounds, 70%.
- While STAR*D excluded depressed patients with bipolar disorder, a depressive episode in a patient with bipolar disorder can be difficult to distinguish from a depressive episode in a patient with major depressive disorder. Primary care physicians need to consider bipolar disorder both in patients presenting with a depressive episode and in those who fail an adequate trial.43
FUTURE CONSIDERATIONS
Subsequent STAR*D analyses will compare in greater depth outcomes in primary care vs psychiatric settings at each level of treatment. Given the greater risk of depression persistence associated with more successive levels of treatment, subsequent research will focus on ways to more successfully treat depression in the earlier stages, possibly through medication combinations earlier in treatment (somewhat analogous to a “broad-spectrum antibiotic” approach for infections).
- Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003; 289:3095–3105.
- Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 1997; 349:1436–1442.
- Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet 1997; 349:1498–1504.
- Zimmerman M, Chelminski I, Posternak MA. Generalizability of antidepressant efficacy trials: differences between depressed psychiatric outpatients who would or would not qualify for an efficacy trial. Am J Psychiatry 2005; 162:1370–1372.
- Rothwell PM. External validity of randomised controlled trials: to whom do the results of this trial apply? Lancet 2005; 365:82–93.
- Depression Guideline Panel. Depression in primary care: Volume 1, diagnosis and detection. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Frank E, Karp J, Rush A. Efficacy of treatments for major depression. Psychopharmacol Bull 1993; 29:457–475.
- Depression Guideline Panel. Depression in primary care: Volume 2, Treatment of major depression. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Fava M, Davidson KG. Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am 1996; 19:179–200.
- Jarrett RB, Rush A. Short-term psychotherapy of depressive disorders: current status and future directions. Psychiatry: Interpers Biol Process 1994; 57:115–132.
- American Psychiatric Association. Practice guideline for the treatment of patients with major depression (revision). Am J Psychiatry 2000; 157(suppl 4):1–45.
- Dunner DL, Rush AJ, Russell JM, et al. Prospective, long-term, multicenter study of the naturalistic outcomes of patients with treatment-resistant depression. J Clin Psychiatry 2006; 67:688–695.
- Fava M, Rush A, Trivedi M, et al. Background and rationale for the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study. Psychiatr Clin North Am 2003; 26:457–494.
- Gaynes B, Davis L, Rush A, Trivedi M, Fava M, Wisniewski S. The aims and design of the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) Study. Prim Psychiatry 2005; 12:36–41.
- Rush A, Fava M, Wisniewski S, et al. Sequenced Treatment Alternatives to Relieve Depression (STAR*D): rationale and design. Control Clin Trials 2004; 25:119–142.
- Stafford RS, Ausiello JC, Misra B, Saglam D. National Patterns o fDepression Treatment in Primary Care. Prim Care Companion J Clin Psychiatry 2000; 2:211–216.
- Regier D, Narrow W, Rae D, Mandersheid R, Locke B, Goodwin F. The de facto US mental and addictive disorders service system: epidemiologic catchment area prospective 1-year prevalence rates of disorders and services. Arch Gen Psychiatry 1993; 50:85–94.
- Pincus H, Tanielian T, Marcus S, et al. Prescribing trends in psychotropic medications: primary care, psychiatry, and other medical specialities. JAMA 1998; 279:526–531.
- Vuorilehto M, Melartin T, Isometsa E. Depressive disorders in primary care: recurrent, chronic, and co-morbid. Psychol Med 2005; 35:673–682.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth edition, Text Revision. Washington, DC: American Psychiatric Association; 2000.
- Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23:56–61.
- Wisniewski SR, Rush AJ, Balasubramani GK, Trivedi MH, Nierenberg AA for the STAR*D Investigators. Self-rated global measure of the frequency, intensity, and burden of side effects. J Psychiatric Pract 2006; 12:71–79.
- Rush AJ, Bernstein IH, Trivedi MH, et al. An evaluation of the Quick Inventory of Depressive Symptomatology and the Hamilton Rating Scale for Depression: a Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial report. Biol Psychiatry 2006; 59:493–501.
- Trivedi MH, Rush AJ, Gaynes BN, et al. Maximizing the adequacy of medication treatment in controlled trials and clinical practice: STAR*D measurement-based care. Neuropsychopharmacology 2007/04/04/online 2007.
- Kawamoto K, Houlihan CA, Balas EA, Lobach DF. Improving clinical practice using clinical decision support systems: a systematic review of trials to identify features critical to success. BMJ 2005; 330:765 e-pub March 14 2005.
- Lavori P, Rush A, Wisniewski S, et al. Strengthening clinical effectiveness trials: equipoise-stratified randomization. Biol Psychiatry 2001; 50:792–801.
- Gaynes BN, Rush AJ, Trivedi MH, et al. Major depression symptoms in primary care and psychiatric care settings: a cross-sectional analysis. Ann Fam Med 2007; 5:126–134.
- Gaynes BN, Rush AJ, Trivedi M, et al. A direct comparison of presenting characteristics of depressed outpatients from primary vs. specialty care settings: preliminary findings from the STAR*D clinical trial. Gen Hosp Psychiatry 2005; 27:87–96.
- Sullivan PW, Valuck R, Saseen J, MacFall HM. A comparison of the direct costs and cost effectiveness of serotonin reuptake inhibitors and associated adverse drug reactions. CNS Drugs 2004; 18:911–932.
- Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006; 163:28–40.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med 2006; 354:1231–1242.
- Trivedi MH, Fava M, Wisniewski SR, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med 2006; 354:1243–1252.
- Wisniewski SR, Fava M, Trivedi MH, et al. Acceptability of second-step treatments to depressed outpatients: a STAR*D report. Am J Psychiatry 2007; 164:753–760.
- Thase ME, Friedman ES, Biggs MM, et al. Cognitive therapy versus medication in augmentation and switch strategies as second-step treatments: a STAR*D report. Am J Psychiatry 2007; 164:739–752.
- Fava M, Rush AJ, Wisniewski SR, et al. A comparison of mirtazapine and nortriptyline following two consecutive failed medication treatments for depressed outpatients: a STAR*D report. Am J Psychiatry 2006; 163:1161–1172.
- Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T3 augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry 2006; 163:1519–1530.
- Bschor T, Lewitzka U, Sasse J, Adli M, Koberle U, Bauer M. Lithium augmentation in treatment-resistant depression: clinical evidence, serotonergic and endocrine mechanisms. Pharmacopsychiatry 2003; 36(suppl 3):S230–S234.
- Freeman MP, Freeman SA. Lithium: clinical considerations in internal medicine. Am J Med 2006; 119:478–481.
- Aronson R, Offman HJ, Joffe RT, Naylor CD. Triiodothyronine augmentation in the treatment of refractory depression. A meta-analysis. Arch Gen Psychiatry 1996; 53:842–848.
- McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry 2006; 163:1531–1541.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 2006; 163:1905–1917.
- Gartlehner G, Hansen R, Thieda P, et al. Comparative Effectiveness of Second-generation Antidepressants in the Pharmacologic Treatment of Depression. Agency for Healthcare Research and Quality. http://effectivehealthcare.ahrq.gov/reports/topic.cfm?topic=8&sid=39&rType=3. Accessed December 12, 2007.
- Das AK, Olfson M, Gameroff MJ, et al. Screening for bipolar disorder in a primary care practice. JAMA 2005; 293:956–963.
- Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 2003; 289:3095–3105.
- Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 1997; 349:1436–1442.
- Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet 1997; 349:1498–1504.
- Zimmerman M, Chelminski I, Posternak MA. Generalizability of antidepressant efficacy trials: differences between depressed psychiatric outpatients who would or would not qualify for an efficacy trial. Am J Psychiatry 2005; 162:1370–1372.
- Rothwell PM. External validity of randomised controlled trials: to whom do the results of this trial apply? Lancet 2005; 365:82–93.
- Depression Guideline Panel. Depression in primary care: Volume 1, diagnosis and detection. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Frank E, Karp J, Rush A. Efficacy of treatments for major depression. Psychopharmacol Bull 1993; 29:457–475.
- Depression Guideline Panel. Depression in primary care: Volume 2, Treatment of major depression. AHCPR publication No. 93-0550. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; 1993.
- Fava M, Davidson KG. Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am 1996; 19:179–200.
- Jarrett RB, Rush A. Short-term psychotherapy of depressive disorders: current status and future directions. Psychiatry: Interpers Biol Process 1994; 57:115–132.
- American Psychiatric Association. Practice guideline for the treatment of patients with major depression (revision). Am J Psychiatry 2000; 157(suppl 4):1–45.
- Dunner DL, Rush AJ, Russell JM, et al. Prospective, long-term, multicenter study of the naturalistic outcomes of patients with treatment-resistant depression. J Clin Psychiatry 2006; 67:688–695.
- Fava M, Rush A, Trivedi M, et al. Background and rationale for the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study. Psychiatr Clin North Am 2003; 26:457–494.
- Gaynes B, Davis L, Rush A, Trivedi M, Fava M, Wisniewski S. The aims and design of the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) Study. Prim Psychiatry 2005; 12:36–41.
- Rush A, Fava M, Wisniewski S, et al. Sequenced Treatment Alternatives to Relieve Depression (STAR*D): rationale and design. Control Clin Trials 2004; 25:119–142.
- Stafford RS, Ausiello JC, Misra B, Saglam D. National Patterns o fDepression Treatment in Primary Care. Prim Care Companion J Clin Psychiatry 2000; 2:211–216.
- Regier D, Narrow W, Rae D, Mandersheid R, Locke B, Goodwin F. The de facto US mental and addictive disorders service system: epidemiologic catchment area prospective 1-year prevalence rates of disorders and services. Arch Gen Psychiatry 1993; 50:85–94.
- Pincus H, Tanielian T, Marcus S, et al. Prescribing trends in psychotropic medications: primary care, psychiatry, and other medical specialities. JAMA 1998; 279:526–531.
- Vuorilehto M, Melartin T, Isometsa E. Depressive disorders in primary care: recurrent, chronic, and co-morbid. Psychol Med 2005; 35:673–682.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth edition, Text Revision. Washington, DC: American Psychiatric Association; 2000.
- Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23:56–61.
- Wisniewski SR, Rush AJ, Balasubramani GK, Trivedi MH, Nierenberg AA for the STAR*D Investigators. Self-rated global measure of the frequency, intensity, and burden of side effects. J Psychiatric Pract 2006; 12:71–79.
- Rush AJ, Bernstein IH, Trivedi MH, et al. An evaluation of the Quick Inventory of Depressive Symptomatology and the Hamilton Rating Scale for Depression: a Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial report. Biol Psychiatry 2006; 59:493–501.
- Trivedi MH, Rush AJ, Gaynes BN, et al. Maximizing the adequacy of medication treatment in controlled trials and clinical practice: STAR*D measurement-based care. Neuropsychopharmacology 2007/04/04/online 2007.
- Kawamoto K, Houlihan CA, Balas EA, Lobach DF. Improving clinical practice using clinical decision support systems: a systematic review of trials to identify features critical to success. BMJ 2005; 330:765 e-pub March 14 2005.
- Lavori P, Rush A, Wisniewski S, et al. Strengthening clinical effectiveness trials: equipoise-stratified randomization. Biol Psychiatry 2001; 50:792–801.
- Gaynes BN, Rush AJ, Trivedi MH, et al. Major depression symptoms in primary care and psychiatric care settings: a cross-sectional analysis. Ann Fam Med 2007; 5:126–134.
- Gaynes BN, Rush AJ, Trivedi M, et al. A direct comparison of presenting characteristics of depressed outpatients from primary vs. specialty care settings: preliminary findings from the STAR*D clinical trial. Gen Hosp Psychiatry 2005; 27:87–96.
- Sullivan PW, Valuck R, Saseen J, MacFall HM. A comparison of the direct costs and cost effectiveness of serotonin reuptake inhibitors and associated adverse drug reactions. CNS Drugs 2004; 18:911–932.
- Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006; 163:28–40.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med 2006; 354:1231–1242.
- Trivedi MH, Fava M, Wisniewski SR, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med 2006; 354:1243–1252.
- Wisniewski SR, Fava M, Trivedi MH, et al. Acceptability of second-step treatments to depressed outpatients: a STAR*D report. Am J Psychiatry 2007; 164:753–760.
- Thase ME, Friedman ES, Biggs MM, et al. Cognitive therapy versus medication in augmentation and switch strategies as second-step treatments: a STAR*D report. Am J Psychiatry 2007; 164:739–752.
- Fava M, Rush AJ, Wisniewski SR, et al. A comparison of mirtazapine and nortriptyline following two consecutive failed medication treatments for depressed outpatients: a STAR*D report. Am J Psychiatry 2006; 163:1161–1172.
- Nierenberg AA, Fava M, Trivedi MH, et al. A comparison of lithium and T3 augmentation following two failed medication treatments for depression: a STAR*D report. Am J Psychiatry 2006; 163:1519–1530.
- Bschor T, Lewitzka U, Sasse J, Adli M, Koberle U, Bauer M. Lithium augmentation in treatment-resistant depression: clinical evidence, serotonergic and endocrine mechanisms. Pharmacopsychiatry 2003; 36(suppl 3):S230–S234.
- Freeman MP, Freeman SA. Lithium: clinical considerations in internal medicine. Am J Med 2006; 119:478–481.
- Aronson R, Offman HJ, Joffe RT, Naylor CD. Triiodothyronine augmentation in the treatment of refractory depression. A meta-analysis. Arch Gen Psychiatry 1996; 53:842–848.
- McGrath PJ, Stewart JW, Fava M, et al. Tranylcypromine versus venlafaxine plus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry 2006; 163:1531–1541.
- Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 2006; 163:1905–1917.
- Gartlehner G, Hansen R, Thieda P, et al. Comparative Effectiveness of Second-generation Antidepressants in the Pharmacologic Treatment of Depression. Agency for Healthcare Research and Quality. http://effectivehealthcare.ahrq.gov/reports/topic.cfm?topic=8&sid=39&rType=3. Accessed December 12, 2007.
- Das AK, Olfson M, Gameroff MJ, et al. Screening for bipolar disorder in a primary care practice. JAMA 2005; 293:956–963.
KEY POINTS
- Remission (ie, complete relief from a depressive episode) rather than response (merely substantial improvement) should be the goal of treatment, as it is associated with a better prognosis and better function.
- Should the first treatment fail, either switching treat mentor augmenting the current treatment is reasonable.
- For most patients, remission will require repeated trials of sufficiently sustained, vigorously dosed antidepressant medication. Physicians should give maximal but tolerable doses for at least 8 weeks before deciding that an intervention has failed.
- After two well-delivered medication trials, the likelihood of remission substantially decreases. Such patients likely require more complicated regimens. Given the thin existing database, these patients are best referred to a psychiatrist for more complex treatments.
- With persistent and vigorous treatment, most patients will enter remission: about 33% after one step, 50% after two steps, 60% after three steps, and 70% after four steps (assuming patients stay in treatment).